ES2913486T3 - Compuestos heterocíclicos para la utilización en el tratamiento de trastornos mediados por PI3K-gamma - Google Patents
Compuestos heterocíclicos para la utilización en el tratamiento de trastornos mediados por PI3K-gamma Download PDFInfo
- Publication number
- ES2913486T3 ES2913486T3 ES15713299T ES15713299T ES2913486T3 ES 2913486 T3 ES2913486 T3 ES 2913486T3 ES 15713299 T ES15713299 T ES 15713299T ES 15713299 T ES15713299 T ES 15713299T ES 2913486 T3 ES2913486 T3 ES 2913486T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- cancer
- compound
- heteroaryl
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
- A61K31/4725—Non-condensed isoquinolines, e.g. papaverine containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39558—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against tumor tissues, cells, antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39566—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against immunoglobulins, e.g. anti-idiotypic antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/12—Drugs for disorders of the metabolism for electrolyte homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2827—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against B7 molecules, e.g. CD80, CD86
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- Pulmonology (AREA)
- Neurology (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Dermatology (AREA)
- Oncology (AREA)
- Obesity (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Diabetes (AREA)
- Heart & Thoracic Surgery (AREA)
- Biochemistry (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Genetics & Genomics (AREA)
- Cardiology (AREA)
- Biophysics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Compuesto de fórmula (I'') o de fórmula (A''), o una forma farmacéuticamente aceptable del mismo, para la utilización en combinación con una terapia de punto de control inmunitario, en un método de tratamiento o prevención de un cáncer en un sujeto: **(Ver fórmula)** en la que: R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, - COOR3, o -CONR4R5, z es 0, 1, 2 o 3, cada aparición de R3a es, independientemente, hidrógeno, alquilo, alquenilo, alquinilo, alcoxilo, halógeno, ciano, amino, cicloalquilo, heterocicloalquilo, arilo o heteroarilo, B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6)3, en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo, R1c es hidrógeno, alquilo, alquenilo o alquinilo, R2c es hidrógeno, alquilo, alquenilo o alquinilo, Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y X es CR1a o N, en la que R1a es hidrógeno, halo, alquilo, alquenilo, alquinilo o CN, en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo)2, S(O)(alquilo), S(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo, en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo)2, S(O)(alquilo), o S(O)2(alquilo), en la que, en la fórmula (I''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es: **(Ver fórmula)** entonces R1 no es hidrógeno, Si(CH3)3, CH2Si(CH3)3, metilo, (CH2)NH2, (CH2)2NH2, (CH2)NHSO2CH3, o (CH2)nNHC(O)R1x; n es 1 o 2; R1x es metilo, alqueno C2, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en los que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituyen opcionalmente con uno o dos grupos seleccionados independientemente de entre oxo y ciano, en la que, en la fórmula (A''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es: **(Ver fórmula)** entonces R1 no es fenilo.
Description
DESCRIPCIÓN
Compuestos heterocíclicos para la utilización en el tratamiento de trastornos mediados por PI3K-gamma
Antecedentes
La actividad de las células puede estar regulada por señales externas que estimulan o inhiben sucesos intracelulares. El proceso por el que se transmiten las señales estimuladoras o inhibidoras hacia el interior y dentro de la célula, induciendo una respuesta intracelular, se denomina transducción de señales. Durante las últimas décadas, se han elucidado cascadas de sucesos de transducción de señales y se ha encontrado que desempeñan un papel crucial en una diversidad de respuestas biológicas. Se ha encontrado que defectos en diversos componentes de rutas de transducción de señales explican un amplio número de enfermedades, entre ellas numerosas formas de cáncer, trastornos inflamatorios, trastornos metabólicos, y enfermedades vasculares y neuronales (Gaestel et al., Current Medicinal Chemistry 14:2214-2234, 2007).
Las quinasas representan una clase de importantes moléculas de señalización. Las quinasas pueden clasificarse generalmente en proteína-quinasas y lípido-quinasas, y determinadas quinasas muestran especificidades dobles. Las proteína-quinasas son enzimas que fosforilan otras proteínas y/o a sí mismas (es decir, se autofosforilan). Las proteína quinasas pueden clasificarse generalmente en tres grupos principales basándose en su utilización de sustratos: tirosina quinasas, que fosforilan predominantemente los sustratos en los residuos de tirosina (p.ej., erb2, receptor de PDGF, receptor de EGF, receptor de VEGF, src, abl), las serina/treonina quinasas, que fosforilan predominantemente los sustratos en los residuos de serina y/o treonina (p.ej., mTorCI, mTorC2, ATM, a Tr , DNA-PK y Akt) y las quinasas de doble especificidad, que fosforilan los sustratos en los residuos de tirosina, serina y/o treonina.
Las lípido-quinasas son enzimas que catalizan la fosforilación de los lípidos. Dichos enzimas y los lípidos, y moléculas orgánicas biológicamente activas derivadas de lípidos, fosforilados resultantes desempeñan una función en muchos procesos fisiológicos diferentes, entre ellos la proliferación, migración, adhesión y diferenciación celular. Determinadas lípido quinasas están asociadas a membranas y catalizan la fosforilación de lípidos contenidos o asociados a las membranas celulares. Entre los ejemplos de dichos enzimas se incluyen las fosfoinositida(s) quinasas (p.ej., PI3-quinasas y PI4-quinasas), diacilglicerol quinasas y esfingosina quinasas.
La ruta de señalización de fosfoinositida-3-quinasas (PI3K) es uno de los sistemas más altamente mutados de entre los cánceres humanos. La señalización de PI3K también es un factor clave en muchas otras enfermedades humanas. La señalización de PI3K participa en muchos estados patológicos, incluyendo la dermatitis alérgica por contacto, la artritis reumatoide, la osteoartritis, las enfermedades intestinales inflamatorias, el trastorno pulmonar obstructivo crónico, la soriasis, la esclerosis múltiple, el asma, los trastornos relacionados con complicaciones diabéticas y las complicaciones inflamatorias del sistema cardiovascular, tales como el síndrome coronario agudo. En este sentido, el documento WO 2013/154878 A1 da a conocer compuestos y composiciones farmacéuticas que modulan la actividad de las quinasas, incluyendo la actividad de la quinasa PI3. Se dan a conocer además métodos de tratamiento de enfermedades y condiciones asociadas a la actividad de las quinasas, incluyendo la actividad de la quinasa PI3K.
Las PI3K son miembros de una familia única y conservada de lípido quinasas intracelulares que fosforilan el grupo 3'-OH en los fosfatidilinositoles o fosfoinositidas. La familia de PI3K comprende 15 quinasas con diferentes especificidades de sustrato, patrones de expresión y modos de regulación. Las PI3K de clase I (p110a p110p, p1105 y p110Y) típicamente resultan activadas por las tirosina-quinasas o los receptores acoplados a proteína G para generar PIP3, que se une a efectores situados corriente abajo, tales como los presentes en la ruta de Akt/PDK1, mTOR, las quinasas de la familia Tec y las GTPasas de la familia Rho. Las PI3K de clases II y III desempeñan un papel clave en el tráfico intracelular mediante la síntesis de PI(3)P y PI(3,4)P2. Las PI3K son proteína quinasas que controlan el crecimiento celular (mTORC1) o monitorizan la integridad del genoma (ATM, ATR, DNA-PK y hSmg-1).
La isoforma delta (8) de la PI3K de clase I se ha implicado, en particular, en varias enfermedades y procesos biológicos. La PI3K-8 se expresa principalmente en células hematopoyéticas, incluyendo leucocitos, tales como células T, células dendríticas, neutrófilos, mastocitos, células B y macrófagos. La PI3K-8 participa integralmente en funciones del sistema inmunitario de los mamíferos, tales como la función de las células T, la activación de las células B, la activación de los mastocitos, la función de las células dendríticas y la actividad de los neutrófilos. Debido a su papel central en el funcionamiento del sistema inmunitario, la PI3K-8 también participa en varias enfermedades relacionadas con una respuesta inmunitaria no deseable, tal como reacciones alérgicas, enfermedades inflamatorias, angiogénesis mediada por inflamación, artritis reumatoide y enfermedades autoinmunitarias, tales como el lupus, el asma, el enfisema y otras enfermedades respiratorias. Otra PI3K de clase I que participa en el funcionamiento del sistema inmunitario es PI3K-y, que desempeña un papel en la señalización de los leucocitos y ha sido relacionada con inflamación, artritis reumatoide y enfermedades autoinmunitarias tales como el lupus. Por ejemplo, PI3K-Y y PI3K-8 se expresan a nivel elevado en los leucocitos y se han asociado a inmunidad adaptativa e innata; de esta manera, estas isoformas de PI3K podrían ser mediadores importantes en trastornos inflamatorios y neoplasias malignas hematológicas.
La isoforma gamma (y) de la PI3K de clase I consiste en una la subunidad catalítica p100Y, que está asociada a la subunidad reguladora p101. La PI3K-Y está regulada por receptores acoplados a proteína G (GPCR) mediante la
asociación con las subunidades p/y de las proteínas G heterotriméricas. La PI3K-Y se expresa principalmente en las células hematopoyéticas y cardiomiocitos y participa en la inflamación, la respuesta inmunitaria innata, la diferenciación de las células mieloides, el tráfico de las células inmunitarias y el funcionamiento de los mastocitos. Los inhibidores de PI3K-Y resultan útiles para tratar una diversidad de enfermedades inflamatorias, alergias y enfermedades cardiovasculares, entre otras.
Al contrario que PI3K-6, la isoforma beta (p) de la PI3K de clase I aparentemente se expresa de forma ubicua. Se ha asociado la Pl3K-p principalmente en diversos tipos de cáncer, incluyendo el cáncer negativo para pTEN (Edgar et al. Cancer Research (2010) 70(3):1164-1172), y el cáncer sobreexpresante de HER2, tal como el cáncer de mama y el cáncer ovárico.
Sumario
En la presente memoria se describen compuestos capaces de inhibir selectivamente una o más isoformas de la PI3K de clase I sin afectar sustancialmente a la actividad de las isoformas restantes de la misma clase. Por ejemplo, se dan a conocer en la presente memoria inhibidores capaces de inhibir selectivamente PI3K-5 y/o PI3K-Y, aunque sin afectar sustancialmente la actividad de PI3K-a y/o PI3K-p. Los inhibidores dados a conocer en la presente memoria pueden resultar eficaces para mejorar condiciones patológicas asociadas a la actividad de PI3K-5 y/o PI3K-Y. Determinados compuestos dados a conocer en la presente memoria pueden ser capaces de inhibir selectivamente PI3-y con preferencia a PI3K-5.
En un primer aspecto de la presente invención, se proporciona un compuesto de fórmula (I'') o de fórmula (A''), o una forma farmacéuticamente aceptable del mismo, para la utilización en combinación con una terapia de punto de control inmunitario, en un método de tratamiento o prevención de un cáncer en un sujeto:

en las que:
R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
z es 0, 1, 2 o 3,
cada aparición de R3a es, independientemente, hidrógeno, alquilo, alquenilo, alquinilo, alcoxilo, halógeno, ciano, amino, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6)a,
en la que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
R1c es hidrógeno, alquilo, alquenilo o alquinilo,
R2c es hidrógeno, alquilo, alquenilo o alquinilo,
Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y
X es CR1a o N,
en la que R1a es hidrógeno, halo, alquilo, alquenilo, alquinilo o CN,
en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2 , NH(alquilo), N(alquilo)2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), s(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2 , S(O)(alquilo), o S(O)2(alquilo),
en las que, en la fórmula (I''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

R1 no es hidrógeno, Si(CHs)3 , CH2Si(CHs)3 , metilo, (CH2)NH2 , (C H )2NH2 , (CH2)NHSO2CH3 , o (CH2)nNHC(O)R1x; n es 1 o 2; R1x es metilo, alqueno C2, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en el que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituye opcionalmente con uno o dos grupos seleccionados independientemente de oxo y ciano,
en la que, en la fórmula (A''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

entonces R1 no es fenilo.
El compuesto o forma farmacéuticamente aceptable del mismo del primer aspecto de la invención puede ser el Compuesto 4 ("Compuesto BB") que presenta la fórmula:

El cáncer bajo tratamiento o que se previene según el primer aspecto de la invención puede ser un tumor sólido, particularmente carcinoma de células renales, cáncer de vejiga, cáncer de colon, glioblastoma, cáncer de mama (más particularmente, cáncer de mama triple negativo), cáncer de cabeza y cuello (más particularmente, carcinoma de células escamosas de cabeza y cuello), cáncer de pulmón (más particularmente, cáncer pulmonar no microcítico), melanoma, cáncer de glándulas adrenales, mesotelioma (más particularmente, mesotelioma epitelioide, mesotelioma sarcomatoide o mesotelioma bifásico), cáncer colorrectal, cáncer ovárico o cáncer de endometrio;
o puede seleccionarse de uno o más de entre un cáncer del sistema pulmonar, un cáncer cerebral, un cáncer del tracto gastrointestinal, un cáncer de piel, un cáncer genitourinario, cáncer de cabeza y cuello, un sarcoma, un carcinoma o un cáncer neuroendocrino;
o puede seleccionarse de uno o más de entre cáncer pancreático, cáncer de pulmón, cáncer de mama, cáncer de próstata, cáncer testicular, cáncer esofágico, cáncer hepático, cáncer gástrico, cáncer de colon, cáncer colorrectal, cáncer ovárico, cáncer cervical, cáncer uterino, cáncer de endometrio, cáncer de vejiga, cáncer renal, cáncer inducido por virus o carcinoma urotelial;
o puede seleccionarse de uno o más de entre meduloblastoma, carcinoma de células basales, glioma, cáncer hepatocelular, tumor estromal gastrointestinal (TEGI), melanoma, tumor neuroectodérmico, sarcoma de tejidos blandos, fibrosarcoma, mixosarcoma, liposarcoma, condrosarcoma, sarcoma osteogénico, cordoma, angiosarcoma, endoteliosarcoma, linfangiosarcoma, linfangioendoteliosarcoma, sinovioma, mesotelioma, leiomiosarcoma, carcinoma de vejiga, carcinoma epitelial, carcinoma de células escamosas, adenocarcinoma, carcinoma broncogénico, carcinoma de células renales, hepatoma, carcinoma del conducto biliar, tumor carcinoide, tumor de células gigantes de tipo difuso o glioblastoma;
o puede ser un cáncer hematológico, especialmente leucemia o linfoma, o leucemia linfocítica aguda (LLA), leucemia linfocítica crónica (LLC), leucemia prolinfocítica (LPL), leucemia de células pilosas (LCP), macroglobulinemia de Waldenstrom (MW), linfoma de células T periféricas (LCTP), leucemia/linfoma de células T adultas (LLTA), linfoma de células T cutáneo (LCTC), leucemia de linfocitos granulares grandes (LLGG), leucemia mielocítica aguda (LMA), linfoma de Hodgkin (LH), linfoma no de Hodgkin (LNH), linfoma folicular, linfoma de células B grandes difusas (LCBGD), linfoma de células del manto (LCM), mastocitosis, mieloma múltiple (MM), síndrome mielodisplásico (SMD) o trastorno mieloproliferativo (TMP);
o puede ser metastásico.
La terapia de punto de control inmunitario para la utilización según el primer aspecto de la invención puede ser un ligando de NKG2C, un inhibidor de CTLA-4, un inhibidor de PD-1, un inhibidor de PDL-1, un inhibidor de LAG3, un inhibidor de TIM-3, un inhibidor de BTLA, un inhibidor de VISTA, un inhibidor de TIGIT, un inhibidor de IDO, un agonista de ICOS, un agonista de CD137, un agonista de OX40, un agonista de GITR, un agonista de CD27, un agonista de HVEM, un ligando de TLR, un anticuerpo de B7-H3, un inhibidor de PDL-1 o un anticuerpo anti-PDL-1, preferentemente YW243.55.S70, MDPL3280A, MSB0010718C, MDX-1105 o MEDI-4736, un inhibidor de PD-1 o un anticuerpo anti-PD-1, preferentemente nivolumab, pembrolizumab, pidilizumab, AMP-224 o AMP-514, o un inhibidor de CTlA-4 o un anticuerpo anti-CTLA4, preferentemente tremelimumab o ipilimumab.
La terapia de punto de control inmunitario para la utilización según el primer aspecto de la invención puede ser:
a) una combinación de un inhibidor de PD-1 o un anticuerpo anti-PD-1 con un inhibidor de CTLA-4 o un anticuerpo anti-CTLA-4,
b) una combinación de un inhibidor de PD-1 o un anticuerpo anti-PD-1, un inhibidor de PDL-1 o un anticuerpo anti-PDL-1 y un inhibidor de CTLA-4, o un anticuerpo anti-CTLA-4, o
c) una combinación de un anticuerpo o fragmento del mismo, un ácido nucleico inhibidor, un ligando soluble o una fusión de PD-1 con una región Fc de una inmunoglobulina; un anticuerpo o fragmento del mismo, un ácido nucleico inhibidor, un ligando soluble o una fusión de PD-L1 con una región Fc de una inmunoglobulina; y un anticuerpo o fragmento del mismo, un ácido nucleico inhibidor, un ligando soluble o una fusión de un ligando de CTLA-4 con una región Fc de una inmunoglobulina.
En un segundo aspecto de la presente invención, se proporciona un compuesto que presenta la fórmula:

o una sal farmacéuticamente aceptable del mismo, para la utilización en combinación con un inhibidor de PD-1 o un anticuerpo anti-PD-1 en un método de tratamiento o prevención de un tumor sólido en un sujeto.
El tumor sólido bajo tratamiento o prevención según el segundo aspecto de la invención puede seleccionarse de uno o más de entre cáncer del sistema pulmonar, cáncer cerebral, cáncer del tracto gastrointestinal, cáncer de piel, cáncer genitourinario, cáncer de cabeza y cuello, sarcoma, carcinoma o cáncer neuroendocrino;
uno o más de entre cáncer pancreático, cáncer de pulmón, cáncer de mama, cáncer de próstata, cáncer testicular, cáncer esofágico, cáncer hepático, cáncer gástrico, cáncer de colon, cáncer colorrectal, cáncer ovárico, cáncer cervical, cáncer uterino, cáncer de endometrio, cáncer de vejiga, cáncer renal, cáncer inducido por virus, cáncer de glándulas adrenales y mesotelioma; o
uno o más de entre meduloblastoma, carcinoma de células basales, glioma, cáncer hepatocelular, tumor estromal gastrointestinal (TEGI), melanoma, tumor neuroectodérmico, sarcoma de tejidos blandos, fibrosarcoma, mixosarcoma, liposarcoma, condrosarcoma, sarcoma osteogénico, cordoma, angiosarcoma, endoteliosarcoma, linfangiosarcoma, linfangioendoteliosarcoma, sinovioma, mesotelioma, leiomiosarcoma, carcinoma de vejiga, carcinoma epitelial, carcinoma de células escamosas, adenocarcinoma, carcinoma broncogénico, carcinoma de células renales, hepatoma, carcinoma del conducto biliar, tumor carcinoide, tumor de células gigantes de tipo difuso y glioblastoma;
o el tumor sólido puede ser cáncer pulmonar, particularmente cáncer pulmonar no microcítico, melanoma, cáncer de colon o cáncer de cabeza y cuello.
El inhibidor de PD-1 o el anticuerpo anti-PD-1 para la utilización según el segundo aspecto de la invención puede ser nivolumab. El inhibidor de PD-1 o el anticuerpo anti-PD-1 para la utilización según el segundo aspecto de la invención puede ser pembrolizumab.
El compuesto o forma farmacéuticamente aceptable del mismo según el primer o segundo aspecto de la presente invención puede administrarse a una dosis de aproximadamente 1 a 100 mg al día, preferentemente de aproximadamente 1 a 50 mg al día, por ejemplo de 5 a 40 mg al día.
El compuesto o forma farmacéuticamente aceptable del mismo según el primer o segundo aspecto de la presente invención puede administrarse a una dosis con la que el nivel del compuesto en el sujeto, preferentemente medido en el plasma o tejidos del sujeto, es superior a la IC5o del compuesto de inhibición de PI3K-gamma durante por lo menos 70%, 80%, 90%, 95%, 97%, 98% o 99% de un periodo de tiempo seleccionado, p.ej., 6 horas, 12 horas, 24 horas o 48 horas inmediatamente después de la administración, y preferentemente puede ser superior a la IC90 del compuesto de inhibición de PI3K-gamma durante por lo menos 50%, 60%, 70%, 80%, 90%, 95%, 97%, 98% o 99% del periodo de tiempo seleccionado, y puede, más preferentemente, no aumentar por encima de la IC20 o IC50 del compuesto de inhibición de PI3K-delta dentro del periodo de tiempo seleccionado.
El compuesto según el primer o segundo aspecto de la presente invención puede administrarse a una dosis tal que proporciona una inhibición de por lo menos 50% de PI3K-gamma en el sujeto, aunque una inhibición inferior de 10% o 20% de PI3K-delta en el sujeto.
Según el primer o segundo aspecto de la presente invención, el sujeto puede ser un ser humano y el compuesto puede tener una semivida de aproximadamente 10 a 13 horas en el sujeto.
En un tercer aspecto de la presente invención, se proporciona un compuesto que presenta la fórmula:

o una sal farmacéuticamente aceptable del mismo, para la utilización en combinación con un inhibidor de PD-1 o un anticuerpo anti-PD-1 en un método de tratamiento o prevención de un carcinoma pulmonar no microcítico, tumor mesotelial (particularmente mesotelioma epitelioide, mesotelioma sarcomatoide o mesotelioma bifásico) o melanoma en un sujeto, en el que el compuesto o sal farmacéuticamente aceptables del mismo se administra a una dosis de 10 mg, 15 mg, 20 mg, 25 mg, 30 mg, 40 mg, 45 mg, 50 mg o 60 mg al día en el sujeto.
En un cuarto aspecto de la presente invención, se proporciona un compuesto que presenta la fórmula:

o una sal farmacéuticamente aceptable del mismo, para la utilización en un método de tratamiento o prevención de un cáncer en un sujeto, en el que el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia que es un tratamiento de punto de control.
Opcionalmente, el cáncer que se trata o previene según el cuarto aspecto de la presente invención es melanoma, y el sujeto ha sido pretratado o previamente tratado con uno o más tratamientos de inmunoterapia que son un tratamiento de punto de control.
Opcionalmente, el cáncer que se trata o previene según el cuarto aspecto de la presente invención es cáncer de cabeza y cuello, cáncer pulmonar (particularmente cáncer pulmonar no microcítico), carcinoma de células renales o
cáncer de vejiga, y el sujeto ha sido pretratado o previamente tratado con un tratamiento de inmunoterapia que es un tratamiento de punto de control.
El tratamiento de punto de control para la utilización según el cuarto aspecto de la presente invención puede ser un inhibidor de PD-1 o un anticuerpo anti-PD-1. El tratamiento de punto de control para la utilización según el cuarto aspecto de la presente invención puede ser nivolumab.
El compuesto o una forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de entre el primer a cuarto aspectos de la presente invención puede encontrarse en la forma de un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros.
En la presente memoria se proporcionan se ún la presente invención compuestos de fórmula (I1) o (A1):

o una forma farmacéuticamente aceptable de los mismos, en la que R1, X, B y Wd se definen en la presente memoria. El compuesto de fórmula (I') o (A') o una forma farmacéuticamente aceptable del mismo para la utilización según la presente invención puede encontrarse en la forma de un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros.
En la presente memoria se proporcionan según la presente invención compuestos de fórmula (I) o (A):

o una forma farmacéuticamente aceptable de los mismos, en la que R1, X, B y Wd se definen en la presente memoria. El compuesto de fórmula (I) o (A) o una forma farmacéuticamente aceptable del mismo para la utilización según la presente invención puede encontrarse en la forma de un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros.
En una realización, el compuesto de fórmula (I"), (I'), (I), (A"), (A') o (A) se encuentra predominantemente en una configuración estereoquímica (S). En una realización, el compuesto de fórmula (I"), (I'), (I), (A"), (A') o (A) es el enantiómero S que presenta un exceso enantiomérico seleccionado de entre superior a aproximadamente 25%, superior a aproximadamente 55%, superior a aproximadamente 80%, superior a aproximadamente 90% y superior a aproximadamente 95%. En una realización, el compuesto se encuentra presente en una composición farmacéutica que comprende el compuesto, o una sal farmacéuticamente aceptable del mismo, y uno o más excipientes farmacéuticamente aceptables.
En determinadas realizaciones, un compuesto dado a conocer en la presente memoria modula selectivamente la isoforma gamma de PI3K. En determinadas realizaciones, el compuesto inhibe selectivamente la isoforma gamma respecto a la isoforma alfa o beta. A título de ejemplo no limitativo, el índice de selectividad puede ser superior a un factor de aproximadamente 10, superior a un factor de aproximadamente 50, superior a un factor de aproximadamente 100, superior a un factor de aproximadamente 200, superior a un factor de aproximadamente 400, superior a un factor de aproximadamente 600, superior a un factor de aproximadamente 800, superior a un factor de aproximadamente 1000, superior a un factor de aproximadamente 1500, superior a un factor de aproximadamente 2000, superior a un factor de aproximadamente 5000, superior a un factor de aproximadamente 10.000, o superior a un factor de aproximadamente 20.000, en el que la selectividad puede medirse mediante una proporción de valores IC50, entre
otros medios. En una realización, la selectividad de isoterma gamma de PI3K respecto a la isoterma alfa o beta de PI3K se mide mediante la proporción entre el valor IC50 contra la isoterma alfa o beta de PI3K y el valor IC50 contra la isoforma gamma de PI3K.
En determinadas realizaciones, un compuesto dado a conocer en la presente memoria modula selectivamente la isoterma gamma de PI3K respecto a la isoterma delta. A título de ejemplo no limitativo, el índice de selectividad puede ser superior a un factor de aproximadamente 10, superior a un factor de aproximadamente 50, superior a un factor de aproximadamente 100, superior a un factor de aproximadamente 200, superior a un factor de aproximadamente 400, superior a un factor de aproximadamente 600, superior a un factor de aproximadamente 800, superior a un factor de aproximadamente 1000, superior a un factor de aproximadamente 1500, superior a un factor de aproximadamente 2000, superior a un factor de aproximadamente 5000, superior a un factor de aproximadamente 10.000, o superior a un factor de aproximadamente 20.000, en el que la selectividad puede medirse mediante una proporción de valores IC50, entre otros medios. En una realización, la selectividad de isoforma gamma de PI3K respecto a la isoforma alfa o beta de PI3K se mide mediante la proporción entre el valor IC50 contra la isoforma alfa o beta de PI3K y el valor IC50 contra la isoforma gamma de PI3K.
En determinadas realizaciones, un compuesto dado a conocer en la presente memoria modula selectivamente la isoforma delta de PI3K. En determinadas realizaciones, el compuesto inhibe selectivamente la isoforma delta respecto a la isoforma alfa o beta. A título de ejemplo no limitativo, el índice de selectividad puede ser superior a un factor de aproximadamente 10, superior a un factor de aproximadamente 50, superior a un factor de aproximadamente 100, superior a un factor de aproximadamente 200, superior a un factor de aproximadamente 400, superior a un factor de aproximadamente 600, superior a un factor de aproximadamente 800, superior a un factor de aproximadamente 1000, superior a un factor de aproximadamente 1500, superior a un factor de aproximadamente 2000, superior a un factor de aproximadamente 5000, superior a un factor de aproximadamente 10.000, o superior a un factor de aproximadamente 20.000, en el que la selectividad puede medirse mediante una proporción de valores IC50, entre otros medios. En una realización, la selectividad de isoforma delta de PI3K respecto a la isoforma alfa o beta de PI3K se mide mediante la proporción entre el valor IC50 contra la isoforma alfa o beta de PI3K y el valor IC50 contra la isoforma delta de PI3K.
En la presente memoria se proporciona para la utilización según la presente invención una composición (p.ej., una composición farmacéutica) que comprende un compuesto indicado en la presente memoria y un excipiente farmacéuticamente aceptable. Una quinasa de PI3 puede inhibirse mediante el contacto de la quinasa de PI3 con una cantidad eficaz de un compuesto o una composición farmacéutica indicada en la presente memoria. Dicha quinasa de PI3 puede encontrarse en una célula. La inhibición puede tener lugar en un sujeto que sufre de un cáncer. La presente exposición se refiere además a administrar un segundo agente terapéutico en el sujeto.
La isoforma gamma de la quinasa de PI3 puede inhibirse selectivamente respecto a una isoforma alfa o beta de la quinasa de PI3, en donde dicha inhibición tiene lugar dentro de la célula. La inhibición puede implicar poner en contacto la isoforma gamma de la quinasa de PI3 con una cantidad eficaz de un compuesto o una composición farmacéutica dada a conocer en la presente memoria. Dicho contacto puede producirse dentro de la célula.
Una isoforma gamma de la quinasa de PI3 puede inhibirse selectivamente respecto a la isoforma alfa o beta de la quinasa de PI3, en donde la inhibición tiene lugar en un sujeto que sufre de cáncer. La inhibición puede implicar la administración de una cantidad eficaz de un compuesto o una composición farmacéutica proporcionada en la presente memoria en dicho sujeto. Un sujeto que sufre de un trastorno asociada a quinasa de PI3 puede tratarse mediante la modulación selectiva de la isoforma gamma de la quinasa de PI3 respecto a la isoforma alfa o beta de la quinasa de PI3, mediante la administración de una cantidad de un compuesto o de una composición farmacéutica proporcionada en la presente memoria en dicho sujeto, en el que dicha cantidad resulta suficiente para la modulación selectiva de la isoforma gamma de la quinasa de PI3 respecto a la isoforma alfa o beta de la quinasa de PI3.
La isoforma delta de la quinasa de PI3 puede inhibirse selectivamente respecto a la isoforma alfa o beta de la quinasa de PI3, en donde dicha inhibición tiene lugar dentro de la célula. Lo anterior puede implicar poner en contacto la isoforma delta de la quinasa de PI3 con una cantidad eficaz de un compuesto o composición farmacéutica dada a conocer en la presente memoria. Dicho contacto puede producirse dentro de la célula.
La isoforma delta de la quinasa de PI3 puede inhibirse selectivamente respecto a la isoforma alfa o beta de la quinasa de PI3, en donde la inhibición tiene lugar en un sujeto que sufre de cáncer. La inhibición puede implicar la administración de una cantidad eficaz de un compuesto o una composición farmacéutica proporcionada en la presente memoria en dicho sujeto. Un sujeto que sufre de un trastorno asociada a quinasa de PI3 puede tratarse mediante la modulación selectiva de la isoforma delta de la quinasa de PI3 respecto a la isoforma alfa o beta de la quinasa de PI3, mediante la administración de una cantidad de un compuesto o de una composición farmacéutica proporcionada en la presente memoria en dicho sujeto, en el que dicha cantidad resulta suficiente para la modulación selectiva de la isoforma delta de la quinasa de PI3 respecto a la isoforma alfa o beta de la quinasa de PI3.
La isoforma gamma de la quinasa de PI3 puede inhibirse selectivamente respecto a la isoforma delta de la quinasa de PI3, en donde dicha inhibición tiene lugar dentro de la célula. La inhibición puede implicar poner en contacto la isoforma
gamma de la quinasa de PI3 con una cantidad eficaz de un compuesto o una composición farmacéutica dada a conocer en la presente memoria. Dicho contacto puede producirse dentro de la célula.
La isoterma gamma de la quinasa de PI3 puede inhibirse selectivamente respecto a la isoterma delta de la quinasa de PI3, en donde la inhibición tiene lugar en un sujeto que sufre de cáncer. La inhibición puede implicar la administración de una cantidad eficaz de un compuesto o una composición farmacéutica proporcionada en la presente memoria en dicho sujeto. Un sujeto que sufre de un trastorno asociada a quinasa de PI3 puede tratarse mediante la modulación selectiva de la isoforma gamma de la quinasa de PI3 respecto a la isoforma delta de la quinasa de PI3, mediante la administración de una cantidad de un compuesto o de una composición farmacéutica proporcionada en la presente memoria en dicho sujeto, en el que dicha cantidad resulta suficiente para la modulación selectiva de la isoforma gamma de la quinasa de PI3 respecto a la isoforma delta de la quinasa de PI3.
Una quinasa PI3 puede inhibirse en un sujeto que sufre de una enfermedad inflamatoria, una enfermedad inmunitaria o una enfermedad respiratoria, mediante la administración en el sujeto de una cantidad eficaz de un compuesto proporcionado en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., tal como se indica en la presente memoria). El compuesto puede ser un inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4. El sujeto puede ser un mamífero. El mamífero puede ser un ser humano. El sujeto puede ser un ser humano.
El trastorno tratado con los compuestos dados a conocer en la presente memoria es un cáncer. El cáncer puede ser un tumor sólido o de tejidos blandos (p.ej., un carcinoide, carcinoma o sarcoma), un tumor de tejido hematopoyético (p.ej., una neoplasia maligna hemática). El cáncer puede ser cáncer metastásico del hueso.
El cáncer tratado con los compuestos dados a conocer en la presente memoria puede ser un tumor de tejidos blandos, una neoplasia maligna hemática o un cáncer hemático. El cáncer puede ser leucemia mieloide aguda (LMA), leucemia mieloide crónica (LMC), síndrome mielodisplásico (SMD), trastornos mieloproliferativos, cáncer de mastocitos, enfermedad de Hodgkin, linfomas no de Hodgkin, linfoma de células B grandes difusas, leucemia/linfoma asociado a virus linfotrópico humano de tipo 1 (VLTH-1), linfoma relacionado con el SIDA, linfoma de células T adultas, leucemia linfoblástica aguda (LLA), leucemia linfoblástica aguda de células T, leucemia linfoblástica aguda de células B, leucemia linfocítica crónica o mieloma múltiple (MM). El cáncer puede ser leucemia o linfoma. La leucemia puede ser leucemia linfoblástica aguda de células B (LLA-B), leucemia mieloide aguda (LMA), leucemia linfoblástica aguda, leucemia mieloide crónica, leucemia de células pilosas, trastornos mieloproliferativos, leucemia mielógena aguda (LMA), leucemia mielógena crónica (LMC), leucemia linfocítica crónica (LLC), mieloma múltiple (MM), síndrome mielodisplásico (SMD) o cáncer de mastocitos. El linfoma puede ser linfoma de células B grandes difusas, linfoma inmunoblástico de células B, linfoma de células no hendidas pequeñas o de Burkitt, leucemia/linfoma asociado a virus linfotrópico humano de tipo 1 (VLTH-1), linfoma de células T adultas, enfermedad de Hodgkin o linfomas no de Hodgkin, o una lesión metastásica de los mismos.
El cáncer tratado con los compuestos dados a conocer en la presente memoria puede ser un tumor sólido (p.ej., un carcinoide, carcinoma o sarcoma) o una lesión metastásica de los mismos. El cáncer puede ser un cáncer pulmonar (p.ej., cáncer pulmonar no microcítico o cáncer pulmonar microcítico), un cáncer de piel, un melanoma, un cáncer de próstata, un glioblastoma, un cáncer de endometrio, un cáncer pancreático (p.ej., adenocarcinoma pancreático (p.ej., adenocarcinoma del conducto pancreático (ACP)), un carcinoma de células renales, un cáncer colorrectal, un cáncer de mama (p.ej., cáncer de mama triple negativo), un cáncer de tiroides, un sarcoma, un cáncer hepático o hepatocelular (CHC), un cáncer de cabeza y cuello, un cáncer de cuello uterino o vulvar, un cáncer esofágico, un cáncer gástrico, un cáncer adrenal, un cáncer ovárico o una lesión metastásica de los mismos. El tumor sólido puede ser cáncer de próstata, cáncer de mama o un glioblastoma, o una lesión metastásica de los mismos.
El cáncer o tumor tratado puede ser un tumor fibrótico sólido seleccionado de uno o más de cáncer pancreático (p.ej., adenocarcinoma pancreático o adenocarcinoma ductal pancreático), cáncer de mama, cáncer colorrectal, cáncer de colon, cáncer pulmonar (p.ej., cáncer pulmonar microcítico o no microcítico), cáncer de piel, cáncer ovárico, cáncer prostático, cáncer de cuello uterino, cáncer gastrointestinal (p.ej., carcinoide o estromal), cáncer de estómago, cáncer de cabeza y cuello, cáncer renal, cáncer cerebral o una lesión metastásica del mismo.
El cáncer o tumor tratado utilizando los métodos o compuestos dados a conocer en la presente memoria puede ser un cáncer o tumor seleccionado de uno o más de entre cánceres de cabeza, cuello, cavidad nasal, senos paranasales, nasofaríngeo, de cavidad oral, orofaringe, laringe, hipofaringe, glándulas salivales, paragangliomas, páncreas, estómago, piel, esófago, endometrio, hígado y árbol biliar, hueso, intestino, colon, recto, ovarios, próstata, mama, sistema linfático, sangre, médula ósea, sistema nervioso central, cerebro o una lesión metastásica de los mismos.
Los compuestos dados a conocer en la presente memoria pueden utilizarse en un método de inhibición de una quinasa de PI3 en un sujeto que sufre de un cáncer, que comprende administrar en el sujeto una cantidad eficaz de un compuesto proporcionado en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI), y (XVII), p.ej., tal como se indica en la presente memoria). El compuesto puede ser un inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4. El cáncer puede seleccionarse de entre leucemia mieloide aguda (LMA), leucemia mieloide crónica (LMC), síndrome mielodisplásico
(SMD), trastornos mieloproliferativos, cáncer de mastocitos, enfermedad de Hodgkin, linfomas no de Hodgkin, linfoma de células B grandes difusas, leucemia/linfoma asociada a virus linfotrópico humano de tipo 1 (VLTH-1), linfoma relacionado con el SIDA, linfoma de células T adultas, leucemia linfoblástica aguda (LLA), leucemia linfoblástica aguda de células B, leucemia linfoblástica aguda de células T, leucemia linfocítica crónica o mieloma múltiple (MM). El cáncer puede ser leucemia o linfoma. La leucemia puede seleccionarse de entre leucemia linfoblástica aguda de células B (LLA-B), leucemia linfocítica aguda, leucemia de células pilosas, mielodisplasia, trastornos mieloproliferativos, leucemia mielógena aguda (LMA), leucemia mielógena crónica (LMC), leucemia linfocítica crónica (l Lc ), mieloma múltiple (MM), síndrome mielodisplásico (SMD) o cáncer de mastocitos. El linfoma puede seleccionarse de entre linfoma de células B grandes difusas, linfoma inmunoblástico de células B, linfoma de células no hendidas pequeñas o de Burkitt, leucemia/linfoma asociado a virus linfotrópico humano de tipo 1 (VLTH-1), linfoma de células T adultas, enfermedad de Hodgkin o linfomas no de Hodgkin. El compuesto puede administrarse en combinación con uno o más agentes terapéuticos proporcionados en la presente memoria.
Los compuestos dados a conocer en la presente memoria pueden utilizarse en un método de inhibición de PI3K-Y en un sujeto, que comprende administrar en el sujeto una cantidad eficaz de un compuesto proporcionado en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (lI), (III), (iV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI), y (XVII), p.ej., tal como se indica en la presente memoria). El compuesto puede ser un inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4.
En la presente memoria se da a conocer además un método de preparación de un compuesto indicado en la presente memoria.
En la presente memoria se da a conocer además una mezcla de reacción que comprende un compuesto indicado en la presente memoria.
En la presente memoria se da a conocer además un kit que comprende un compuesto indicado en la presente memoria.
En algunas realizaciones, el compuesto para la utilización según la invención es un inhibidor de PI3K-gamma, p.ej., un inhibidor selectivo de PI3K-gamma, o una forma farmacéuticamente aceptable del mismo.
El compuesto para la utilización según la invención es un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), o una forma farmacéuticamente aceptable del mismo. En una realización, el compuesto es un compuesto de fórmula (XVII). En una realización, el compuesto o compuestos son inhibidores de PI3K gamma, p.ej., inhibidores selectivos de Pi3k gamma.
En algunas realizaciones, el compuesto se selecciona de uno o más de los compuestos 2, 4, 7, 9, 17, 19, 21,26, 27, 30, 32, 35, 37, 38, 40, 41,52, 60, 61, 63, 73, 75, 77, 79, 80, 81, o 88, o una forma farmacéuticamente aceptable de los mismos. En una realización, el compuesto o compuestos son inhibidores de PI3K gamma, p.ej., inhibidores selectivos de PI3K gamma.
En una realización, el compuesto es un compuesto de fórmula (I"), (I') o (I), o una forma farmacéuticamente aceptable de los mismos. En una realización, el compuesto es un compuesto de fórmula I o una forma farmacéuticamente aceptable del mismo,
En una realización, el compuesto es el Compuesto 4 (también denominado en la presente memoria, "Compuesto BB"):

o una forma farmacéuticamente aceptable del mismo. En algunas realizaciones, el Compuesto 4 es un inhibidor de PI3K gamma, p.ej., un inhibidor selectivo de PI3K-gamma.
En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma, p.ej., el inhibidor selectivo de PI3kgamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto
4), puede administrarse en un sujeto a una dosis (p.ej.; una dosis terapéuticamente eficaz) de aproximadamente 2 mg, de 1 a 3 mg, de 1 a 5 mg, de 1 a 10 mg, de 0,5 a 20 mg, o de 0,1 a 50 mg. La dosis (p.ej., una dosis terapéuticamente eficaz) puede ser de aproximadamente 2 mg, de 1 a 3 mg, de 1 a 5 mg, de 1 a 10 mg, de 0,5 a 20 mg, de 0,1 a 50 mg, de 0,1 a 75 mg, de 0,5 a 75 mg, de 1 a 75 mg, de 0,1 a 100 mg, de 0,5 a 100 mg, o de 1 a 100 mg. La dosis puede ser de aproximadamente 1 a 10 mg. La dosis puede ser de aproximadamente 1 a 50 mg. La dosis puede ser de aproximadamente 1 a 100 mg.
En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma, p.ej., el inhibidor selectivo de PI3K-gamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4), puede administrarse en un sujeto a una dosis (p.ej., a una dosis terapéuticamente eficaz) de aproximadamente 0,029 mg/kg, de 0,014 a 0,14 mg/kg, de 0,02 a 0,04 mg/kg, de 0,01 a 0,05 mg/kg, de 0,01 a 0,1, o de 0,01 a 0,5 mg/kg. La administración puede ser por vía intratraqueal.
En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma, p.ej., el inhibidor selectivo de PI3K-gamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4), puede administrarse en un sujeto en un programa de tratamiento seleccionado, p.ej., de entre una vez cada dos días, una vez al día o dos veces al día.
En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma, p.ej., el inhibidor selectivo de PI3K-gamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4), puede administrarse a una dosis con la que se inhiba selectivamente PI3K-gamma, aunque se consiga una inhibición inferior a 10% o 20% de PI3K-delta.
En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma, p.ej., el inhibidor selectivo de PI3K-gamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4), puede administrarse a una dosis con la que el nivel de compuesto en la sangre del sujeto no se eleve a más de un nivel predeterminado, p.ej., la IC50 de PI3K-delta, dentro de un periodo de tiempo seleccionado, p.ej., 24 horas. En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma o compuesto puede administrarse a una dosis con la que el nivel de compuesto en la sangre del sujeto no aumente por encima de un nivel predeterminado, p.ej., la IC20 de PI3K-delta, dentro de un periodo de tiempo seleccionado, p.ej., 24 horas.
En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma, p.ej., el inhibidor selectivo de PI3K-gamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4), puede administrarse en el sujeto a una dosis tal que el nivel de compuesto en la sangre del sujeto está por encima de la IC50 de PI3K-gamma durante por lo menos 70 %, 80 %, 90 %, 95 %, 97 %, 98 %, 99 % o 99 % del periodo de tiempo seleccionado, p.ej., 24 horas, inmediatamente después de la administración.
En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma, p.ej., el inhibidor selectivo de PI3K-gamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4), puede administrarse en el sujeto a una dosis tal que el nivel de compuesto en el cuerpo del sujeto está por encima de la IC90 de PI3K-gamma durante por lo menos 50 %, 60 %, 70 %, 80 %, 90 %, 95 %, 97 %, 98 %, 99 % o 99 % del periodo de tiempo seleccionado, p.ej., 24 horas, inmediatamente después de la administración.
En los usos dados a conocer en la presente memoria, el sujeto puede ser un humano y el inhibidor de PI3K gamma, p.ej., el inhibidor selectivo de PI3K-gamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4), puede presentar una semivida de aproximadamente 8 a 15 horas, o de aproximadamente 10 a 13 horas.
En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma, p.ej., el inhibidor selectivo de PI3K-gamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4), puede resultar más eficaz que el tratamiento con un inhibidor selectivo de PI3K delta, p.ej., en el caso de que ambos inhibidores se administren a la misma dosis y/o por la misma vía de administración.
En los usos dados a conocer en la presente memoria, el tratamiento con un inhibidor de PI3K gamma, p.ej., el inhibidor selectivo de PI3K-gamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4), puede resultar más eficaz (p.ej., por lo menos 10 %, 20 %, 40 %, 60 % o 80% más
eficaz) que el tratamiento con un inhibidor selectivo de PI3K delta, p.ej., en el caso de que ambos inhibidores se administren a la misma dosis y/o por la misma vía de administración.
En los usos dados a conocer en la presente memoria, el sujeto puede ser refractario o no responder a un inhibidor de PI3K-5.
Cáncer
Un cáncer puede tratarse o prevenirse en un sujeto mediante la utilización del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (M),(MI), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., tal como se indica en la presente memoria). El cáncer puede ser, o puede identificarse que es, un tumor sólido (p.ej., cáncer pulmonar, melanoma, cáncer de mama, sarcoma, cáncer hepatocelular, cáncer de cabeza y cuello, cáncer de cuello uterino o vulvar, cáncer esofágico, cáncer gástrico, cáncer adrenal, cáncer de colon o glioblastoma) o un cáncer hemático (p.ej., una leucemia linfocítica crónica (LLC)), p.ej., tal como se indica en la presente memoria). El cáncer puede ser melanoma, cáncer de vejiga, cáncer de cabeza y cuello, cáncer pulmonar (p.ej., cáncer pulmonar no microcítico), carcinoma de células renales, cáncer ovárico, cáncer de mama (p.ej., cáncer de mama triple negativo), cáncer de colon o glioblastoma.
El inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII),p.ej., el Compuesto 4, tal como se indica en la presente memoria), puede administrarse a una dosis tal que se inhiba selectivamente PI3K-gamma, pero que consiga una inhibición inferior a 10% o 20% de PI3K-delta.
El inhibidor de PI3K gamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4), puede administrarse a una dosis con la que el nivel de compuesto en la sangre del sujeto no aumente por encima de un nivel predeterminado, p.ej., la IC50 de PI3K-delta, dentro de un periodo de tiempo seleccionado, p.ej., 24 horas. El inhibidor de PI3K gamma, o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4, tal como se indica en la presente memoria), puede administrarse a una dosis con la que el nivel de compuesto en la sangre del sujeto no aumente por encima de un nivel predeterminado, p.ej., la IC20 de PI3K-delta, dentro de un periodo de tiempo seleccionado, p.ej., 24 horas.
En los usos dados a conocer en la presente memoria, el sujeto puede ser refractario o no responder a un inhibidor de PI3K-5. El sujeto puede no haber sido expuesto nunca al tratamiento inmunoterapéutico. El sujeto puede ser, o puede haber sido, sensible a un tratamiento de inmunoterapia. El tratamiento de inmunoterapia puede ser un tratamiento con un inhibidor de PD-1 o PD-L1.
La migración de células T CD3+ inducida por CXCL12 o la migración de macrófagos diferenciados inducida por CXCL12 al interior de un microambiente tumoral puede reducirse en un sujeto mediante la administración en el mismo de un inhibidor de PI3 gamma o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4, tal como se indica en la presente memoria), en una cantidad suficiente para reducir o inhibir la migración de células T CD3+ inducida por CXCL12, o la migración de macrófagos diferenciados inducida por CXCL12 en un microambiente tumoral en el sujeto.
En los usos dados a conocer en la presente memoria, el sujeto puede presentar, o puede haberse detectado que presenta, una reducción de los niveles de p-AKT tras la administración del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria.
En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma puede administrarse a una dosis con la que se inhiba selectivamente PI3K-gamma pero se consiga una inhibición inferior a 10% o 20% de PI3K-delta.
En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria puede administrarse a una dosis tal que el nivel de compuesto en la sangre del sujeto no aumente por encima de un nivel predeterminado, p.ej., la IC50 de PI3K-delta, dentro de un periodo de tiempo seleccionado, p.ej., 24 horas. En los usos dados a conocer en la presente memoria, el inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria puede administrarse a una dosis con la que el nivel de compuesto en la sangre del sujeto no aumente por encima de un nivel predeterminado, p.ej., la IC20 de PI3K-delta, dentro de un periodo de tiempo seleccionado, p.ej., 24 horas.
Una o más actividades de una célula inmunitaria proinmunitaria en un sujeto que presenta un cáncer pueden reducirse mediante la administración en el mismo de un inhibidor de PI3K-gamma o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII),
(IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4, tal como se indica en la presente memoria), en una cantidad suficiente para reducir o inhibir una o más actividades de la célula inmunitaria protumoral.
La célula inmunitaria protumoral puede ser una célula T, un macrófago M2, una célula estromal, una célula dendrítica, una célula endotelial o una célula mieloide. La célula mieloide puede ser una célula mieloide supresora asociada a tumor. La célula mieloide supresora asociada a tumor puede ser un macrófago asociado a tumor (MAT), una célula supresora de origen mieloide (CSOM), una célula mieloide monocítica inmadura (CMMi) o una CMMi/neutrófilo granulocítico.
El sujeto puede presentar, o haberse detectado que presenta, una reducción del número de células inmunitarias protumorales en un microambiente tumoral, en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria.
La cantidad administrada puede ser suficiente para producir una reducción del número de células inmunitarias protumorales en un microambiente tumoral, en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria.
El sujeto puede presentar, o haberse detectado que presenta, una actividad incrementada de las células inmunitarias antitumorales en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria.
La cantidad del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria puede resultar suficiente para producir una actividad incrementada de las células inmunitarias antitumorales en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o el compuesto tal como se indica en la presente memoria.
El sujeto puede presentar, o haberse detectado que presenta, una infiltración incrementada de las células inmunitarias antitumorales en un microambiente tumoral, en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria.
La cantidad de inhibidor de PI3K-gamma puede resultar suficiente para producir una infiltración incrementada de células inmunitarias antitumorales en un microambiente tumoral, en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria.
El sujeto puede presentar, o haberse detectado que presenta, un incremento del número de células inmunitarias antitumorales en un microambiente tumoral, en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria.
La cantidad de inhibidor de PI3K-gamma puede resultar suficiente para producir un incremento del número de células inmunitarias antitumorales en un microambiente tumoral, en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria.
El cáncer puede ser LLC. El microambiente tumoral puede ser un centro de proliferación de LLC.
El sujeto puede presentar, o haberse detectado que presenta, un volumen tumoral reducido en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria.
La cantidad del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria puede resultar suficiente para producir un volumen tumoral reducido en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o del compuesto tal como se indica en la presente memoria.
La cantidad del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria puede resultar suficiente para producir una reducción de por lo menos 10 %, 20 %, 30 %, 50 %, 60% o 60 % del volumen tumoral en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o el compuesto tal como se indica en la presente memoria.
El sujeto puede presentar, o haberse detectado que presenta, un nivel incrementado de apoptosis en las células de cáncer en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria.
La cantidad de inhibidor de PI3K-gamma puede resultar suficiente para producir un nivel incrementado de apóptosis en las células de cáncer en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria.
El sujeto puede presentar, o haberse detectado que presenta, un incremento de 10 %, 20 %, 30 %, 40 % o 50 % de
la apóptosis en las células de cáncer en comparación con un valor de referencia, tras la administración del inhibidor de Pl3K gamma o un compuesto tal como se indica en la presente memoria.
La cantidad del inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria puede resultar suficiente para producir un incremento de 10 %, 20 %, 30 %, 40% o 50 % de la apoptosis en las células de cáncer en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o el compuesto tal como se indica en la presente memoria.
La célula inmunitaria antitumoral puede ser un macrófago M1.
La actividad puede seleccionarse de uno o más de entre migración de la célula, o señalización de una célula inmunitaria antitumoral.
El sujeto puede presentar, o haberse detectado que presenta, niveles reducidos de p-AKT en la célula inmunitaria protumoral en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o el compuesto.
La cantidad puede resultar suficiente para reducir el nivel de p-AKT en la célula inmunitaria protumoral en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o el compuesto.
El sujeto puede presentar, o haberse detectado que presenta, una reducción de los niveles de p-AKT de aproximadamente 10 %, 20 %, 30 %, 40 %, 50 % o 60 % en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o el compuesto.
El sujeto puede presentar, o haberse detectado que presenta, una reducción de los niveles de p-AKT de aproximadamente 10 %, 20 %, 30 %, 40 %, 50 % o 60 % en comparación con un valor de referencia, tras la administración del inhibidor de PI3K gamma o el compuesto.
El sujeto puede presentar, o haberse detectado que presenta, cáncer pulmonar, cáncer de mama, cáncer de colon o glioblastoma. El cáncer de mama puede ser cáncer de mama triple negativo.
El nivel de macrófagos M2 en un microambiente tumoral en un sujeto que presenta un cáncer puede reducirse mediante la administración en el mismo de un inhibidor de PI3K gamma o un compuesto tal como se indica en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (x V iI), p.ej., el Compuesto 4, tal como se indica en la presente memoria), en una cantidad suficiente para reducir el nivel de macrófagos M2 en un microambiente tumoral.
La reducción del nivel de macrófagos M2 puede comprender la reducción o inhibición de la diferenciación de una célula mieloide asociada a tumor en un macrófago M2. La diferenciación en un macrófago M2 puede medirse como niveles reducidos de ARG1 en comparación con un valor de referencia, después de la administración del compuesto. El nivel de ARG1 puede reducirse en por lo menos 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 % o 90 % en comparación con un valor de referencia, después de la administración del compuesto.
La diferenciación en un macrófago M2 puede medirse como niveles reducidos de VEGF en comparación con un valor de referencia, después de la administración del compuesto.
El nivel de VEGF puede reducirse en por lo menos 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 % o 90 % en comparación con un valor de referencia, después de la administración del compuesto.
El sujeto puede presentar, o haberse detectado que presenta, un nivel normal de diferenciación de las células mieloides en macrófagos M1.
La cantidad puede ser tal que el compuesto no reduzca la diferenciación de las células mieloides en macrófagos M1. El sujeto puede presentar, o haberse detectado que presenta, un ataque inmunitario antitumoral incrementado por las células T efectoras, una vascularización reducida de un tumor, una degradación reducida de la MEC, un crecimiento tumoral reducido, o cualquier combinación de los mismos, en comparación con un valor de referencia, después de la administración del compuesto.
El cáncer puede ser, o puede haberse detectado que sea, un tumor sólido (p.ej., un cáncer seleccionado de entre cáncer pulmonar, cáncer de mama, cáncer de colon o glioblastoma).
El cáncer puede ser, o haberse detectado que es, un cáncer hemático.
Terapias de combinación adicionales:
La presente invención proporciona compuestos para la utilización en combinación con una terapia de punto de control inmunitario, en un método de tratamiento o prevención de un cáncer en un sujeto.
En una realización, el cáncer es de origen hematopoyético, p.ej., un cáncer seleccionado de entre una leucemia o un linfoma, p.ej., linfoma de células B, linfoma de células T, linfoma no de Hodgkin, linfoma de Hodgkin o linfoma de células grandes anaplásico. En una realización, el linfoma es linfoma de células B foliculares.
En otras realizaciones, el cáncer es un tumor sólido, p.ej., un cáncer seleccionado de entre un cáncer de mama, un cáncer pulmonar, un cáncer de colon, un melanoma, o un glioblastoma. En una realización, el cáncer es melanoma, cáncer de vejiga, cáncer de cabeza y cuello, cáncer pulmonar (p.ej., cáncer pulmonar no microcítico), carcinoma de células renales, cáncer ovárico, cáncer de mama (p.ej., cáncer de mama triple negativo), cáncer de colon o glioblastoma.
En algunas realizaciones, el sujeto nunca ha sido expuesto a tratamiento inmunoterapéutico. En algunas realizaciones, el sujeto es o ha sido respondedor a un tratamiento inmunoterapéutico. En una realización, el tratamiento de inmunoterapia es un tratamiento con un inhibidor de PD-1 o PD-L1.
La terapia de punto de control inmunitario puede ser una terapia de punto de control inmunitario seleccionado de entre un inhibidor de PD-1, PD-L1, PD-L2, CTLA4, TIM3, LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4, TGFR-beta, o IDO/TDO, o cualquier combinación de los mismos. En una realización, la terapia de punto de control inmunitario es un inhibidor de CTLA-4, PD-1 o PD-L1. La terapia de punto de control inmunitario puede seleccionarse de entre un anticuerpo o fragmento del mismo, un ácido nucleico inhibidor, un ligando soluble, o una fusión de una terapia de punto de control inmunitario (p.ej., CTLA-4, PD-1 o ligando de PD-1) con una región Fc de una inmunoglobulina.
En determinadas realizaciones, la terapia de punto de control inmunitario es un activador de una molécula coestimuladora. En una realización, el agonista de la molécula coestimuladora se selecciona de entre un agonista (p.ej., un anticuerpo agonista o fragmento de unión a antígeno del mismo, o una fusión soluble) de OX40, CD2, CD27, CDS, ICAM-1, LFA-1 (CD11a/CD18), ICOS (CD278), 4-1BB (CD137), GITR, CD30, CD40, BAFFR, HVEM, CD7, LIGHT, NKG2C, SLAMF7, NKp80, CD160, B7-H3 o ligando de CD83.
En determinadas realizaciones, la terapia de punto de control inmunitario se selecciona de entre un ligando coestimulador, un inhibidor de FECM/CSF-1R, un inmunoestimulador, un inhibidor de CXCR4/CXCL12, un inhibidor de CCL2 o un inhibidor de CCR2.
En determinadas realizaciones, el compuesto tal como se indica en la presente memoria se encuentra en una única forma de administración.
En determinadas realizaciones, el compuesto tal como se indica en la presente memoria y la terapia de punto de control inmunitario se encuentran en forma de administración separadas.
En determinadas realizaciones, el compuesto tal como se indica en la presente memoria y la terapia de punto de control inmunitario se administran concurrentemente.
En determinadas realizaciones, el compuesto tal como se indica en la presente memoria se administra después de la terapia de punto de control inmunitario.
En determinadas realizaciones, el compuesto tal como se indica en la presente memoria se administra antes de la terapia de punto de control inmunitario.
En determinadas realizaciones, la cantidad eficaz del compuesto tal como se indica en la presente memoria, la terapia de punto de control inmunitario, o ambos, que es una cantidad suficiente para causar una reducción del crecimiento tumoral de por lo menos 10 %, 20 %, 30 %, 40 % o 50 % en comparación con un valor de referencia, es reducida.
En determinadas realizaciones, el sujeto presenta, o se ha detectado que presenta, un crecimiento tumoral reducido de por lo menos 10 %, 20 %, 30 %, 40 % o 50 % en comparación con un valor de referencia, después de la administración del compuesto.
La terapia de punto de control inmunitario puede ser un inhibidor de PD-1 o PD-L1 (p.ej., un anticuerpo anti-PD-1 o un anticuerpo anti-PD-L1).
Puede administrarse en el sujeto una cantidad eficaz de un inhibidor de PI3K-8 o el compuesto.
En determinadas realizaciones, el nivel del compuesto en la sangre del sujeto es superior a un valor predeterminado, p.ej., la IC50 de PI3K-gamma, durante por lo menos 70 %, 80 %, 90 %, 95 %, 97 %, 98 %, 99 % o 99 % es un periodo de tiempo seleccionado, p.ej., 24 horas, inmediatamente después del tratamiento.
En determinadas realizaciones, el nivel del compuesto en la sangre del sujeto es inferior a un valor predeterminado, p.ej., la IC20 de PI3K-delta, durante por lo menos 70 %, 80 %, 90 %, 95 %, 97 %, 98 %, 99 % o 99 % de un periodo de tiempo seleccionado, p.ej., 24 horas, inmediatamente después del tratamiento.
La presente invención se refiere a composiciones que comprenden las combinaciones anteriormente indicadas del compuesto y la terapia de punto de control inmunitario. Las composiciones pueden proporcionarse en formas de administración iguales o separadas. Las composiciones pueden incluir además un excipiente farmacéuticamente aceptable. Las abreviaturas de las isoformas de PI3K se proporcionan intercambiablemente en toda la memoria, como la letra griega o el nombre correspondiente. Por ejemplo, los términos PI3K-Y y PI3K-gamma, o PI3K-8 y PI3K-delta se utilizan intercambiablemente en toda la memoria.
Breve descripción de los dibujos
La FIG. 1 muestra el efecto del compuesto BB en un modelo de artritis en la rata inducida con colágeno, medida como diámetro medio de tobillo respecto al tiempo.
La FIG. 2 muestra el efecto del compuesto BB en un modelo de artritis en la rata inducida con colágeno, medida mediante puntuaciones histopatológicas individuales de inflamación, pannus, daño en el cartílago, resorción ósea y formación de hueso periostial.
La FIG. 3 muestra el efecto del compuesto BB en un modelo de artritis en la rata inducida con colágeno, medida a partir de la medición del hueso periostial.
La FIG. 4 muestra el efecto del compuesto BB sobre la migración de neutrófilos inducida por IL-8 en un modelo de bolsa de aire en el ratón.
La FIG. 5 muestra el efecto del compuesto AA sobre la migración de neutrófilos inducida por IL-8 en un modelo de bolsa de aire en el ratón.
La FIG. 6 muestra un isobolograma que ilustra el efecto sinérgico de la combinación de un compuesto selectivo para PI3K delta, compuesto AA y un compuesto selectivo para PI3K gamma, el compuesto BB, en la línea celular TMD8.
La FIG. 7 muestra un isobolograma que ilustra el efecto sinérgico de la combinación de un compuesto selectivo para PI3K delta, el compuesto AA, y un compuesto selectivo para PI3K gamma, el compuesto BB, en la línea celular Farage.
La FIG. 8 muestra los efectos de un compuesto selectivo para PI3K delta, el compuesto AA, y un compuesto selectivo para PI3K, el compuesto BB, sobre la migración de las células T periféricas asociadas a leucemia linfocítica crónica (LLC).
La FIG. 9 muestra que pAKT inducida por CXCL12 en las células T es dependiente de PIK3-Y.
La FIG. 10 es un gráfico que muestra los recuentos celulares totales por mililitro en una muestra de lavado broncoalveolar (LBA) al exponer el modelo de rata de inflamación inducida con lipopolisacárido (LPS) a compuesto AA o compuesto BB.
La FIG. 11 es un gráfico que muestra los recuentos de neutrófilos por mililitro en una muestra de lavado broncoalveolar (LBA) al exponer el modelo de rata de inflamación inducida con lipopolisacárido (LPS) a compuesto AA o compuesto BB.
La FIG. 12 es un gráfico que muestra un pictograma de TNFa por mililitro en una muestra de lavado broncoalveolar (LBA) al exponer el modelo de rata de inflamación inducida con lipopolisacárido (LPS) a compuesto AA o compuesto BB.
La FIG. 13 muestra los efectos del compuesto AA y el compuesto BB sobre p-AKT inducida por CXCL 12 (SDF-1a) en macrófagos de fenotipo M2.
La FIG. 14 muestra que los cocultivos de macrófagos M2 con células de CPL condujeron a una supervivencia extendida de las células de CPL.
La FIG. 15 es un esquema de la diferenciación de las células progenitoras mieloides y las interacciones entre determinadas células T.
La FIG. 16 ilustra la diferenciación de una célula mieloide en un macrófago M1 o un macrófago M2.
La FIG. 17 muestra que el compuesto BB evita la diferenciación activada por RANKL de los osteoclastos a partir de macrófagos de la médula ósea.
La FIG. 18 muestra la concentración media de compuesto BB libre en plasma 1 o 5 horas después de la administración.
La FIG. 19 muestra un curso temporal de 24 horas de concentración libre en plasma de compuesto BB. Las cinco líneas horizontales representan las IC50 (y una IC90) de cuatro isoformas de PI3K. De parte superior a inferior, las líneas horizontales corresponden a la IC50 para PI3K-a, la IC50 para PI3K-p, la IC50 para PI3K-8" la IC90 para PI3K-Y y la IC50 para PI3K-Y.
La FIG. 20 muestra los perfiles de concentración media de compuesto 88 libre en plasma-tiempo para ratones C57 hembra con administración de 2, 7,5 o 15 mg/kg PO.
La FIG. 21 muestra que el compuesto AA es un inhibidor altamente selectivo de IFN-y en un modelo inducido con concanavalina-A de activación de las células T humanas. El compuesto BB presenta poco a ningún efecto en el presente ensayo a concentraciones selectivas de isoforma.
Las FIG. 22A y 22B muestran el nivel de ARG1 (arginasa-1) en macrófagos murinos polarizados a fenotipo M2 en respuesta a diferentes dosis de compuesto BB y compuesto AA, respectivamente.
La FIG. 23 muestra que la supervivencia de las células de CPL resulta potenciada con el cocultivo de macrófagos M2. El eje x corresponde al tiempo y el eje y corresponde al % de viabilidad de las CPL.
Las FIG. 24A y 24B muestran que el compuesto BB suprime VEGF y ARG1 en un ensayo de diferenciación celular de supresor de origen mieloide, respectivamente.
Las FIG. 25A y 25B muestran que el compuesto BB demuestra actividad de agente único en el modelo de carcinoma pulmonar de Lewis.
La FIG. 26 muestra las concentraciones medias de compuesto BB libre en el tumor y en plasma tras 12 días de administración oral diaria en solución de NMP.
Las FIG. 27A y 27B muestran el compuesto BB administrado junto con un inhibidor del punto de control anti-PD-L1 en el modelo de cáncer de colon CT26. La FIG. 27A muestra el efecto sobre el volumen tumoral respecto al tiempo. La FIG. 27B muestra el porcentaje de supervivencia respecto al tiempo.
La FIG. 28 muestra que la expansión de las CSOM tras el tratamiento con ciclofosfamida se redujo con compuesto BB en el modelo de cáncer de colon CT26.
Las FIG. 29A y 29B muestran la eficacia del compuesto BB solo y en combinación con 5-FU, respectivamente, en el modelo de cáncer de colon MC38. El eje x representa el tiempo y el eje y representa el tamaño tumoral, en mm3. La FIG. 30 muestra el análisis de IHQ de CD3 tras el tratamiento con compuesto BB en tumores de MC38.
La FIG. 31A muestra la correlación de CD3 y volumen tumoral para el vehículo. La FIG. 31B muestra la correlación de CD3 y volumen tumoral para el compuesto BB.
Las FIG. 32A y 32B muestran los efectos del compuesto BB y/o del compuesto AA en el modelo de linfoma de células B folicular humano DoHH2. La FIG. 32B es un estudio repetido.
La FIG. 33A muestra un crecimiento tumoral reducido con compuesto BB y/o la inhibición de PDL-1 en un modelo de cáncer de mama 4T1 ortotópico.
La FIG. 33B muestra un flujo reducido de luciferasa total con compuesto BB y/o la inhibición de PDL-1 en un modelo de cáncer de mama 4T1 ortotópico.
La FIG. 34 muestra que el compuesto BB demuestra actividad de agente único en el modelo de cáncer de mama 4T1 subcutáneo.
La FIG. 35 muestra que el compuesto BB por sí solo consigue una reducción del crecimiento tumoral, mientras que la coadministración del compuesto BB y 5-FU consigue una reducción adicional.
La FIG. 36 muestra que el compuesto BB demuestra actividad de agente único en el modelo de cáncer de mama 4T1 subcutáneo, mientras que la coadministración con docetaxel, paclitaxel o 5-FU consiguió una reducción adicional del crecimiento tumoral.
Las FIG. 37A, 37B, 37C, 37D, 37E, 37F, 37G y 37H muestran que las cantidades de células mieloides CD11b+ o CD68+ en tejido de cáncer procedente de ratones tratados con compuesto BB o ratones de control.
Las FIG. 38A y 38B muestran los niveles de diferentes células inmunitarias en tumores tratados y no tratados. Específicamente, la FIG. 38A muestra que los MAT están reducidos en un modelo de tumor de CPL brei tratado con compuesto BB. La FIG. 38B muestra que las CMMi monocíticas están reducidas en tumores subcutáneos de CT26 tratados con compuesto BB.
La FIG. 39 muestra que el compuesto BB reduce la cantidad de macrófagos supresores polarizados M2 CD206+ en un modelo de tumor de CPL brei.
La FIG. 40 muestra que la normalización respecto a la p-actina proporciona una expresión estable de los genes de GAPDH y 18S genes.
La FIG. 41 muestra el efecto del compuesto BB sobre los marcadores Cd8b1, Pdcd1/PD1, Cd4, Cd3e, Foxp3 y Clla4 w con o sin anticuerpo de PDL-1.
La FIG. 42A muestra que los marcadores de macrófago M2, VEGF y ARG-1 resultan reducidos por el compuesto BB con o sin anticuerpo de PDL-1. La FIG. 42B muestra el efecto del compuesto BB con o sin anticuerpo de PDL-1 sobre los marcadores de macrófago M2, ARG1, VEGFa y ADM.
La FIG. 43 muestra que la exposición oral a compuesto BB es elevada en la rata, perro y mono.
La FIG. 44 muestra que el compuesto BB muestra proporcionalidad de exposición a dosis.
La FIG. 45 muestra las concentraciones de compuesto BB libre en un estudio de toxicidad.
La FIG. 46 muestra la supervivencia de células de CPL humanas primarias con y sin células estromales en cocultivo.
Las FIG. 47A y la FIG. 47B muestran los efectos del compuesto BB QD sobre el volumen tumoral entre los días 9 y 20 después del implante y el flujo de luciferasa total en el modelo de glioma singénico GL-261. La FIG. 47C es un estudio repetido que muestra los efectos del compuesto BB QD sobre el volumen tumoral desde el día 9 al día 25 después del implante.
La FIG. 48 muestra el crecimiento tumoral tras el tratamiento con compuesto BB /- ciclofosfamida en el modelo de tumor de CPL brei.
Las FIG. 49A, 49B, 49C, 49D y 49E muestran gráficos de CD11b/Gr-1 tras el tratamiento con compuesto BB /-ciclofosfamida en el modelo de tumor de CPL brei. La FIG. 49A muestra el porcentaje de células que son CD3 respecto a las CD45+. La FIG. 49B muestra el porcentaje de células que son CD11b+Gr-1‘ respecto a las CD45+. La FIG. 49C muestra el porcentaje de células que son CD11b+Gr-1+ respecto a las CD45+. La FIG. 49D muestra el porcentaje de células que son CD11b'Gr-1‘ respecto a las CD45+. La FIG. 49E muestra el porcentaje de células que son CD11b'Gr-1 respecto a las CD45+.
Las FIG. 50A, 50B, 50C y 50D muestran el panel de CSOM de CD11b/Ly6C/Ly6G tras el tratamiento con compuesto BB /- ciclofosfamida en el modelo de tumor de CPL brei. La FIG. 50a muestra el porcentaje de células que son CD11b+ respecto a las CD45+. La FIG. 50B muestra el porcentaje de células que son CD11b+Ly6C+
respecto a las CD45+. La FIG. 50C muestra el porcentaje de células que son CD11b+Ly6C+Ly6G+ respecto a las CD45+. La FIG. 50D muestra el porcentaje de células que son CD11b+Ly6C-Ly6G- respecto a las CD45+.
Las FIG. 51A, 51B, 51C, 51D y 51E muestran gráficos de CD3/CD4/CD8 tras el tratamiento con compuesto BB /-ciclofosfamida en el modelo de tumor de CPL brei. La FIG. 51A muestra el porcentaje de células que son CD3+ respecto a las CD45+. La FIG. 51B muestra el porcentaje de células que son CD4+CD8- respecto a las CD45+. La FIG. 51C muestra el porcentaje de células que son CD4+CD8+ respecto a las CD45+. La FIG. 51D muestra el porcentaje de células que son CD4-CD8+ respecto a las CD45+. La f Ig .51E muestra el porcentaje de células que son CD4-CD8- respecto a las CD45+.
Las FIGS. 52A, 52B, 52C, 52D y 52E muestran el porcentaje de células que son CD45+, CD11b+ o CD206+, expresado como porcentaje del total de células, en un modelo de tumor de CPL brei tratado con compuesto BB /-ciclofosfamida. La FIG. 52A muestra el porcentaje de células que son CD45+ expresado como porcentaje del total de células. La FIG. 52B muestra el porcentaje de células que son CD11b expresado como porcentaje del total de células. La FIG.52C muestra el porcentaje de células que son CD206 expresado como porcentaje del total de células. La FIG.52D muestra el porcentaje de células que son CD11b expresado como porcentaje del total de células. La FIG.52E muestra el porcentaje de células que son CD206 expresado como porcentaje del total de células.
Las FIGS. 53A, 53B, 53C, 53D y 53E muestran el efecto del compuesto BB tras el tratamiento con temozolomida de tumores GL-261.
La FIG. 54A muestra el efecto del tratamiento TMZ respecto a vehículo. La FIG. 54B muestra los efectos del compuesto BB sobre el volumen tumoral después del tratamiento TMZ.
Las FIGS. 55A, 55B y 55C muestran el efecto del compuesto BB y anti-PDL-1 sobre la inhibición celular derivada del tumor de la proliferación de las células T frente a vehículo. La FIG. 55A muestra el efecto del tratamiento con vehículo sobre la proliferación de células T mediada por células derivadas de tumor. La FIG. 55B muestra el efecto del tratamiento con compuesto B sobre la proliferación de células T mediada por células derivadas de tumor. La FIG. 55C muestra el efecto del tratamiento con anti-PDL-1 sobre la proliferación de células T mediada por células derivadas de tumor.
Las FIGS. 56A y 56B muestran que las células aisladas a partir de tumores de compuesto BB y ratones tratados con anti-PDL-1 son menos inhibitorias de la proliferación de las células T de bazo que las células derivadas de tumor procedentes de ratones tratados con vehículo.
La FIG. 57 muestra que el compuesto BB reduce el medio inmunosupresor mediante un ensayo ex vivo de proliferación de células T.
La FIG. 58 muestra que el tratamiento con compuesto BB de ratones portadores de CPL inhibe la estimulación de sangre completa ex vivo de fosfo-AKT en las células T.
La FIG. 59 muestra que el tratamiento con compuesto BB de ratones portadores de CPL inhibe la estimulación de sangre completa ex vivo de fosfo-AKT en monocitos.
La FIG. 60 muestra el efecto del compuesto BB sobre pAKT inducido por CXCL12 o por anti-IgD, en células B. La FIG. 61 muestra perfiles de concentración libre en plasma de dosis únicas de compuesto BB frente al tiempo en perros de raza Beagle macho.
Descripción detallada
A menos que se indique lo contrario, todos los términos técnicos y científicos utilizados en la presente memoria presentan los mismos significados entendidos comúnmente por el experto en la materia a la que se refiere la presente especificación.
Tal como se utiliza en la especificación y en las reivindicaciones, las formas singulares “un”, “una” y “el” o “ la” incluyen los referentes plurales, a menos que el contexto indique claramente lo contrario.
Tal como se utiliza en la presente memoria, y a menos que se indique lo contrario, el término "aproximadamente" se refiere a un error aceptable para un valor particular determinado por el experto ordinario en la materia, que depende en parte de cómo se mide o determina el valor. En algunos casos, el término "aproximadamente" se refiere a menos de 1,2, 3 o 4 desviaciones estándar. En algunos casos, el término "aproximadamente" se refiere a 50 %, 20 %, 15 %, 10 %, 9 %, 8 %, 7 %, 6 %, 5 %, 4 %, 3 %, 2 %, 1 %, 0,5 % o 0,05 % de un valor o intervalo dado.
Tal como se utiliza en la presente memoria, "agente" o "agente biológicamente activo" o "segundo agente activo" se refiere a un compuesto u otra fracción de tipo biológico, farmacéutico o químico. Entre los ejemplos no limitativos se incluyen moléculas orgánicas o inorgánicas simples o complejas, un péptido, una proteína, un oligonucleótido, un anticuerpo, un derivado de anticuerpo, un fragmento de anticuerpo, una vitamina, un derivado de vitamina, un carbohidrato, una toxina, o un compuesto quimioterapéutico, y metabolitos de los mismos. Pueden sintetizarse diversos compuestos, por ejemplo, moléculas pequeñas y oligómeros (p.ej., oligopéptidos y oligonucleótidos), y compuestos orgánicos sintéticos basados en diversas estructuras básicas. Además, diversas fuentes naturales pueden proporcionar compuestos para el cribado, tales como extractos vegetales o animales, y similares. El experto en la materia podrá reconocer fácilmente que no hay límite a la naturaleza estructural de los agentes de la presente exposición.
El término "agonista" tal como se utiliza en la presente memoria se refiere a un compuesto o agente que presenta la
capacidad de iniciar o potenciar una función biológica de una proteína o polipéptido diana, tal como incrementar la actividad o expresión de la proteína o polipéptido diana. De acuerdo con lo anterior, el término "agonista" se define en el contexto de la función biológica de la proteína o polipéptido diana. Aunque algunos agonistas en la presente memoria interactúan específicamente con (p.ej., se unen a) la diana, los compuestos y/o agentes que inician o potencian una actividad biológica de la proteína o polipéptido diana mediante la interacción con otros elementos de la ruta de transducción de señales de la que es un miembro el polipéptido diana también se encuentran específicamente incluidos en dicha definición.
Los términos "antagonista" e "inhibidor" se utilizan intercambiablemente y se refieren a un compuesto o agente que presenta la capacidad de inhibir una función biológica de una proteína o polipéptido diana, tal como mediante la inhibición o la expresión de la proteína o polipéptido diana. De acuerdo con lo anterior, los términos "antagonista" e "inhibidor" se definen en el contexto de la función biológica de la proteína o polipéptido diana. Aunque algunos antagonistas en la presente memoria interactúan específicamente con (p.ej., se unen a) la diana, los compuestos que inhiben una actividad biológica de la proteína o polipéptido diana mediante la interacción con otros elementos de la ruta de transducción de señales de la que es un miembro la proteína o polipéptido diana también se encuentran específicamente incluidos en dicha definición. Entre los ejemplos no limitativos de actividad biológica inhibida por un antagonista se incluyen los asociados al desarrollo, crecimiento, o extensión de un tumor, o una respuesta inmunitaria no deseada que se manifiesta en una enfermedad autoinmune. El término "inhibición" o "inhibidor" tal como se utiliza en el presente contexto incluye una reducción de un parámetro determinado, p.ej., una actividad, de una molécula dada, p.ej., una isoforma de PI3K. Por ejemplo, la inhibición de una actividad, p.ej., una actividad de PI3K, de por lo menos 5 %, 10 %, 20 %, 30 %, 40 % o más se encuentra incluida en dicho término. De esta manera, la inhibición no es necesariamente de 100%.
Un "agente anticáncer", "agente antitumoral" o "agente quimioterapéutico" se refiere a cualquier agente útil en el tratamiento de una condición neoplásica. Una clase de agentes anticáncer comprende agentes quimioterapéuticos. La "quimioterapia" se refiere a la administración de uno o más fármacos quimioterapéuticos y/u otros agentes en un paciente de cáncer mediante diversos métodos, incluyendo la administración intravenosa, oral, intramuscular, intraperitoneal, intravesical, subcutánea, transdérmica o bucal, o por inhalación, o en la forma de un supositorio.
La expresión "proliferación celular" se refiere al fenómeno por el que cambia el número de células como resultado de la división. Dicha expresión comprende además el crecimiento celular, por el que cambia la morfología celular (p.ej., se incrementa de tamaño) en concordancia con una señal proliferativa.
El término “tumor” se refiere a cualquier crecimiento y proliferación celular neoplásica, sea maligna o benigna, y a todas las células y tejidos precancerosos y cancerosos. Tal como se utiliza en la presente memoria, el término "neoplásico" se refiere a cualquier forma de crecimiento celular desregulado o no regulado, sea maligno o benigno, que resulta en un crecimiento anormal de tejido. De esta manera, la expresión "células neoplásicas" incluye células malignas y benignas que presentan un crecimiento celular desregulado o no regulado.
El término "cáncer" incluye, aunque sin limitación, tumores sólidos y tumores hematológicos. El término "cáncer" se refiere a una enfermedad de tejidos de la piel, órganos, sangre y vasos, que incluye, aunque sin limitación, cánceres de vejiga, hueso o sangre, cerebro, mama, cuello uterino, tórax, colon, endometrio, esófago, ojo, cabeza, riñón, hígado, ganglios linfáticos, pulmón, boca, cuello, ovarios, páncreas, próstata, recto, estómago, testículo, garganta y útero.
El origen hematopoyético se refiere a que implica células generadas durante la hematopoyesis, un proceso por el que se generan elementos celulares de la sangre, tales como linfocitos, leucocitos, plaquetas, eritrocitos y células asesinas naturales. Los cánceres de origen hemopoyético incluyen el linfoma y la leucemia.
El término "resistente" o "refractario" se refiere a la situación en que un cáncer que ha reducido su sensibilidad a un tratamiento, p.ej., hasta el punto en que el cáncer no responde al tratamiento. El cáncer puede ser resistente al inicio del tratamiento o puede adquirir resistencia durante el tratamiento. El sujeto con cáncer puede presentar una o más mutaciones que causan que sea resistente al tratamiento, o el sujeto puede haber desarrollado tales mutaciones durante el tratamiento. En una realización, el cáncer o el sujeto no responde a un tratamiento terapéutico dado (p.ej., no responde en por lo menos 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 % o 100 % a un tratamiento dado). El fracaso del tratamiento puede medirse mediante, p.ej., un volumen tumoral o el periodo de tiempo durante el que se produce el nuevo crecimiento tumoral.
La expresión "enfermedad o trastorno canceroso hiperproliferativo" se refiere a todo crecimiento y proliferación de células neoplásicas, sean malignas o benignas, incluyendo todas las células y tejidos transformados y todas las células y tejidos cancerosos. Entre las enfermedades o trastornos hiperproliferativos se incluyen, aunque sin limitarse a ellos, lesiones precancerosas, crecimiento celular anormal, tumores benignos, tumores malignos y "cáncer".
La terapia de combinación o "en combinación con" se refiere a la utilización de más de un compuesto o agente para tratar un trastorno o condición particular. Por ejemplo, el Compuesto 4 puede administrarse en combinación con una terapia de punto de control inmunitario. La expresión "en combinación con" no pretende implicar que la terapia de punto de control inmunitario y el Compuesto 4 deben administrarse simultáneamente y/o formularse juntos para la
administración, aunque estos métodos de administración se encuentran comprendidos dentro del alcance de la presente exposición. El Compuesto 4 puede administrarse concurrentemente, antes (p.ej., 5 minutos, 15 minutos, 30 minutos, 45 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 12 horas, 24 horas, 48 horas, 72 horas, 96 horas, 1 semana, 2 semanas, 3 semanas, 4 semanas, 5 semanas, 6 semanas, 8 semanas, 12 semanas, o 16 semanas antes), o después (p.ej., 5 minutos, 15 minutos, 30 minutos, 45 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 12 horas, 24 horas, 48 horas, 72 horas, 96 horas, 1 semana, 2 semanas, 3 semanas, 4 semanas, 5 semanas, 6 semanas, 8 semanas, 12 semanas, o 16 semanas después), de otro u otros agentes adicionales de la terapia de punto de control inmunitario. En general, se administra cada agente terapéutico a una dosis y/o en un programa temporal determinado para ese agente particular. El otro agente terapéutico de la terapia de punto de control inmunitario puede administrarse con el Compuesto 4 en la presente memoria en una única composición o por separado en una composición diferente. Las combinaciones más altas, p.ej., la triple terapia, también se encuentran contempladas en la presente memoria.
Tal como se utiliza en la presente memoria, una "monoterapia" se refiere a la utilización de un agente individualmente (p.ej., como un único compuesto o agente), p.ej., sin un segundo ingrediente activo para tratar la misma indicación, p.ej., cáncer. Por ejemplo, en el presente contexto, el término monoterapia incluye la utilización del inhibidor de PI3K o el segundo agente individualmente para tratar el cáncer.
El término "sinergia" o "sinérgico" comprende un efecto más que aditivo de una combinación de dos o más agentes en comparación con sus efectos individualmente. En determinadas realizaciones, sinergia o efecto sinérgico se refiere a un efecto ventajoso de utilizar dos o más agentes en combinación, p.ej., en una composición farmacéutica, o en un método de tratamiento. En determinadas realizaciones, se consigue uno o más efectos ventajosos mediante la utilización de un inhibidor de PI3K en combinación con un segundo agente terapéutico (p.ej., uno o más segundos agentes terapéuticos) tal como se indica en la presente memoria.
En realizaciones, el efecto sinérgico es que se requiere una dosis inferior de uno o ambos agentes para conseguir un efecto. Por ejemplo, la combinación puede proporcionar un efecto seleccionado, p.ej., un efecto terapéutico, en el caso de que por lo menos uno de los agentes se administre a una dosis más baja que la dosis de ese agente que resultaría necesaria para conseguir el mismo efecto terapéutico administrando el agente como monoterapia. En determinadas realizaciones, la combinación de un inhibidor de PI3K (p.ej., el Compuesto 4) y un segundo agente (tal como se indica en la presente memoria) permite administrar el inhibidor de PI3K a una dosis inferior que la que resultaría necesaria para conseguir el mismo efecto terapéutico si se administrase el inhibidor de PI3K como monoterapia.
En realizaciones, un efecto sinérgico se refiere a la combinación de un inhibidor de PI3K (p.ej., el Compuesto 4 o una forma farmacéuticamente aceptable del mismo) y un segundo agente terapéutico (p.ej., uno o más agentes terapéuticos adicionales, o una forma farmacéuticamente aceptable de los mismos, tal como se indica en la presente memoria), resulta en un efecto terapéutico superior al efecto aditivo del inhibidor de PI3K y el segundo agente.
En realizaciones, un efecto sinérgico se refiere a que el valor del índice de combinación es inferior a un valor seleccionado, p.ej., para un efecto dado, p.ej., a un porcentaje seleccionado (p.ej., 50%) de inhibición o de inhibición del crecimiento, p.ej., tal como se indica en la presente memoria en los Ejemplos. En realizaciones, un efecto sinérgico se refiere a que la puntuación de sinergia es de 1 o superior. En determinadas realizaciones, la puntuación de sinergia es superior a 1. En determinadas realizaciones, la puntuación de sinergia es superior a 3.
El índice de combinación (IC) es una medida del cambio de potencia. El índice de combinación es conocido de la técnica y se describe en, p.ej., Chou et al., Adv Enzyme Regul 1984; 22: 27-55 y en la publicación de patente US n° 2013/0295102. Un valor de IC superior a 1 indica un efecto antagonista; un valor de IC de 1,0 es indicativo de un efecto aditivo y un valor de IC inferior a 1 es indicativo de un efecto sinérgico que resulta de la combinación. El valor de IC puede determinarse a diversos porcentajes de inhibición o inhibición del crecimiento.
El IC proporciona una estimación de la fracción de las dosis originales (monoterapia) de cada uno de los dos fármacos que se necesitarían en combinación respecto a las dosis de agente único requeridas para conseguir un nivel de efecto seleccionado. Por ejemplo, en el caso de que el índice de combinación presente un valor de 0,1, solo resulta necesario aproximadamente un décimo de las cantidades fraccionales totales de los agentes individuales (expresadas como una fracción de la cantidad de ese agente administrado como monoterapia para conseguir un efecto seleccionado) para que la combinación alcance el mismo nivel de efecto seleccionado. Por ejemplo, en el caso de que se requiera una dosis de 100 mg/kg de fármaco A individualmente o se requiera una dosis de 200 mg/kg de fármaco B individualmente para conseguir el efecto seleccionado, y el índice de combinación es 0,1, entonces aproximadamente 5 mg/kg de fármaco A y 10 mg/kg de fármaco B conseguirían el mismo efecto (un vigésimo de las dosis originales de cada uno de los agentes individuales suma un total de un décimo). Las dosis de los agentes individuales no necesita reducirse en el mismo valor fraccional con la condición de que la suma de sus valores fraccionales sume el índice de combinación; de esta manera, en el presente ejemplo, una dosis de aproximadamente 8 mg/kg de fármaco A y 4 mg/kg de fármaco B también conseguirían el mismo efecto seleccionado (es 0,08 veces la dosis original de fármaco A y 0,02 veces la dosis original de fármaco B; la suma de las cantidades fraccionales (0,08 0,02) es igual al índice de combinación, 0,1).
Según una realización, la puntuación de sinergia es una medida de los efectos en combinación en exceso de aditividad
de Loewe. En un ejemplo, la puntuación de sinergia es una medida escalar para caracterizar la potencia de la interacción sinérgica. La puntuación de sinergia puede calcularse de la manera siguiente:

En el presente ejemplo, la inhibición fraccional de cada agente componente y el punto de combinación en la matriz se calculan respecto a la mediana de todos los pocillos de control tratados con vehículo. La ecuación del ejemplo de puntuación de sinergia integra el volumen de la actividad observada experimentalmente en cada punto en la matriz en exceso de una superficie modelo derivada numéricamente a partir de la actividad de los agentes componentes utilizando el modelo de Loewe de aditividad. Se utilizan términos adicionales en la ecuación de puntuación de sinergia (anteriormente) para normalizar diversos factores de dilución utilizados para agentes individuales y para permitir la comparación de las puntuaciones de sinergia a través de todo el experimento La inclusión de subgrupos de inhibición positiva o un multiplicador Idatos elimina el ruido en proximidad al nivel de efecto cero, y sesga los resultados para interacciones sinérgicas que se producen a niveles de actividad elevados. Según otras realizaciones, puede calcularse una puntuación de sinergia basándose en un enfoque de ajuste de la curva en el que la curvatura de la puntuación de sinergia se extrapola mediante la introducción de un valor de la mediana y un valor de origen (p.ej., un valor de dosis nula).
La medida de la puntuación de sinergia puede utilizarse para el análisis de autocruces. Las puntuaciones de sinergia de los autocruces se espera que sean aditivas por definición y, por lo tanto, mantengan una puntuación de sinergia de cero. Sin embargo, aunque algunas puntuaciones de sinergia de autocruce son prácticamente iguales a cero, muchas son mayores, lo que sugiere que el ruido experimental o un ajuste no óptimo a la curva de las respuestas a dosis de agentes individuales están contribuyendo a las ligeras perturbaciones en la puntuación. Esta estrategia está centrada en la línea celular, concentrándose en el comportamiento de autocruce en cada línea celular frente a una revisión global de la actividad del panel de líneas celulares. Las combinaciones en las que la puntuación de sinergia es superior al autocruce medio más dos desviaciones estándares o tres desviaciones estándares pueden considerarse sinergias candidatas a los niveles de confianza de 95 % y 99 %, respectivamente. La aditividad debería mantener una puntuación de sinergia de cero, y la puntuación de sinergia de dos o tres desviaciones estándares indica sinergia a los niveles estadísticamente significativos de 95 % y 99 %.
Puede utilizarse el volumen de Loewe (vol. Loewe) para evaluar la magnitud total de la interacción de la combinación en exceso del modelo de aditividad de Loewe. El volumen de Loewe resulta particularmente útil para distinguir los incrementos sinérgicos de una actividad fenotípica (volumen de Loewe positivo) frente a antagonismos sinérgicos (volumen de Loewe negativo). En el caso de que se observen antagonismos, el volumen de Loewe debería evaluarse para examinar si existe cualquier correlación entre el antagonismo y una actividad en una diana de fármaco o un genotipo celular particulares. Dicho modelo define la aditividad como una interacción de combinación no sinérgica en la que la superficie de la matriz de dosis de combinación debería ser indistinguible de cualquiera de los fármacos cruzado consigo mismo. El cálculo de la aditividad de Loewe se realiza de la manera siguiente:
iLoewe que cumple que (X/Xi) + (Y/Yi) = 1
en donde X y Yi son las concentraciones eficaces de agente individual para el efecto de combinación observado I. Por ejemplo, en el caso de que se alcance una inhibición de 50% por separado con 1 pM de fármaco A o 1 pM de fármaco B, una combinación de 0,5 pM de A y 0,5 pM de B también debería inhibir al 50%.
El término "coadministración", la expresión "administrado en combinación con", y sus equivalentes gramaticales, tal como se utilizan en la presente memoria, comprenden la administración de dos o más agentes en el sujeto de manera que ambos agentes y/o sus metabolitos se encuentran presentes en el sujeto simultáneamente. La coadministración incluye la administración simultánea en composiciones separadas, la administración en diferentes tiempos en composiciones separadas, o la administración en una composición en la que se encuentran presentes ambos agentes.
La expresión "cantidad eficaz" o "cantidad terapéuticamente eficaz" se refiere a aquella cantidad de un compuesto o composición farmacéutica tal como se indica en la presente memoria que resulta suficiente para producir la aplicación pretendida, incluyendo, aunque sin limitación, el tratamiento de la enfermedad, tal como se ilustra posteriormente. La cantidad terapéuticamente eficaz puede variar según la aplicación pretendida (in vitro o in vivo) o el sujeto y la condición de enfermedad bajo tratamiento, p.ej., el peso y la edad del sujeto, la gravedad de la condición de enfermedad, el modo de administración y similares, que pueden ser fácilmente determinados por el experto ordinario en la materia. La expresión se aplica además a una dosis que inducirá una respuesta particular en células diana, p.ej., una reducción de la adhesión plaquetaria y/o de la migración celular. La dosis específica variará según, por ejemplo, los compuestos particulares seleccionados, el régimen de administración que debe seguirse, si se administra en combinación con otros agentes, la planificación temporal de la administración, el tejido en el que se administra, y el sistema de administración física en el que está contenido.
Tal como se utiliza en la presente memoria, los términos "tratamiento", "tratar'1, "paliar" y "mejorar" se utilizan intercambiablemente en la presente memoria. Dichos términos se refieren a un enfoque para obtener resultados
beneficiosos o deseados, incluyendo, aunque sin limitación, un beneficio terapéutico. La expresión beneficio terapéutico se refiere a la erradicación o mejora del trastorno subyacente bajo tratamiento. Además, se consigue un beneficio terapéutico con la irradiación o mejora de uno o más de los síntomas fisiológicos asociados al trastorno subyacente, de manera que se observa una mejora en el paciente, aunque el paciente todavía puede encontrarse afectado por el trastorno subyacente.
Tal como se utiliza en la presente memoria, los términos "prevención" y "prevenir" se utilizan en la presente memoria para referirse a un enfoque para obtener resultados beneficiosos o deseados, incluyendo, aunque sin limitación, un beneficio profiláctico. Para el beneficio profiláctico, las composiciones farmacéuticas pueden administrarse en el paciente en riesgo de desarrollar una enfermedad particular, o en un paciente que informa de uno o más de los síntomas fisiológicos de una enfermedad, aunque podría no haberse llegado a un diagnóstico de dicha enfermedad.
Un "efecto terapéutico", tal como se utiliza esta expresión en la presente memoria; comprende un beneficio terapéutico y/o un beneficio profiláctico tal como se ha indicado anteriormente. Un efecto profiláctico incluye retrasar o eliminar la aparición de una enfermedad o condición, retrasar o eliminar la aparición de síntomas de una enfermedad o condición, retrasar, detener o revertir la progresión de una enfermedad o condición, o cualquier combinación de los mismos.
La expresión "transducción de señales" o "ruta de señalización" se refiere a un proceso durante el que se transmiten señales estimulatorias o inhibitorias hacia el interior o en el interior de una célula, induciendo una respuesta intracelular. Un "modulador" de una ruta de transducción de señales se refiere a un compuesto que modula la actividad de una o más proteínas celulares localizadas en la misma ruta específica de transducción de señales. Un modulador puede aumentar (agonista) o suprimir o inhibir (antagonista) la actividad de una molécula de señalización.
En determinadas realizaciones, la transducción de señales está mediada por una o más fosfoinositida 3-quinasas (PI3K). Las PI3K son miembros de una familia conservada de lípido quinasas que regulan numerosas funciones celulares, entre ellas la proliferación, la diferenciación, la supervivencia y el metabolismo celular. Existen varias clases de PI3K en las células de mamífero, incluyendo del subgrupo clase IA (p.ej., PI3K-a, p, 8), que se activan generalmente con las receptor-tirosina quinasas (RTK, por sus siglas en inglés); clase IB (p.ej., PI3K-y), que se activa con receptores acoplados a proteína G (GPCR, por sus siglas en inglés), entre otros. Las Pl3K ejercen sus actividades biológicas mediante una "ruta de señalización mediada por PI3K" que incluye varios componentes que transducen directa y/o indirectamente una señal inducida por una PI3K, incluyendo la generación del segundo mensajero fosfatidilinositol, 3,4,5-trifosfato (PIP3) en la membrana plasmática, la activación de la señalización de proteína G heterotrimérica y la generación de segundos mensajeros adicionales, tales como AMPc, DAG e IP3, la totalidad de los cuales conduce a una extensa cascada de activación de las proteína quinasas (revisión en Vanhaesebroeck, B. et al. (2001) Annu. Rev. Biochem. 70:535-602). En determinadas realizaciones, los compuestos dados a conocer en la presente memoria inhiben una quinasa PI3 o isoforma de PI3K, p.ej., uno, dos, tres o más de entre PI3K-a, p, 8 o -y.
En el contexto de las moléculas biológicas, "disminuir", "suprimir", "mejorar", "reducir", "inhibir" o similar, incluyen producir un decremento de un nivel o de una actividad (p.ej., una o más funciones) de una molécula dada. El nivel de una molécula dada, p.ej., el nivel de ARNm o proteína, o la actividad, pueden medirse en una muestra o utilizando los ensayos descritos en los Ejemplos en la presente memoria.
Los términos "disminuir", "mejorar", "reducir", "inhibir" (o similar) un trastorno o condición, o un síntoma asociado a un trastorno o condición, incluye reducir la gravedad y/o la frecuencia de uno o más síntomas del trastorno o condición, o reducir o retrasar la aparición del trastorno o condición y/o uno o más síntomas del trastorno o condición. En algunas realizaciones, el síntoma se reduce en por lo menos aproximadamente 2 %, en por lo menos aproximadamente 5 %, en por lo menos aproximadamente 10 %, en por lo menos aproximadamente 15 %, en por lo menos aproximadamente 20 %, en por lo menos aproximadamente 25 %, en por lo menos aproximadamente 30 %, en por lo menos aproximadamente 40 %, en por lo menos aproximadamente 50 %, en por lo menos aproximadamente 60 %, en por lo menos aproximadamente 70 %, en por lo menos aproximadamente 80 %, en por lo menos aproximadamente 90 %, o en por lo menos aproximadamente 95 % respecto a un nivel de control.
El término "inhibición" o "inhibir" tal como se utilizan en el presente contexto incluyen una reducción de un parámetro determinado, p.ej., una actividad, de una molécula dada, p.ej., una isoforma de Pl3K. Por ejemplo, la inhibición de una actividad, p.ej,. una actividad de PI3K, de por lo menos 5 %, 10 %, 20 %, 30 %, 40 % o más se encuentra incluida en dicho término. De esta manera, la inhibición no es necesariamente de 100%. En determinadas realizaciones, un inhibidor de PI3K tal como se da a conocer en la presente memoria inhibe una quinasa de PI3 de la isoforma gamma (una "isoforma PI3K-y).
La expresión "inhibición selectiva" o "inhibe selectivamente" tal como se aplica a un agente biológicamente activo se refiere a la capacidad del agente de reducir selectivamente la actividad de señalización de la diana en comparación con la actividad de señalización fuera de diana, mediante interacción directa o indirecta con la misma. Por ejemplo, un compuesto que inhibe selectivamente una isoforma de PI3K sobre otra isoforma de PI3K presenta una actividad por lo menos superior en aproximadamente 1X contra una primera isoforma en comparación con la actividad del compuesto contra la segunda isoforma (p.ej., por lo menos aproximadamente 2X, 3X, 5X, 10X, 20X, 50X, 100X, 200X, 500X, o 1000X). En determinadas realizaciones, dichas expresiones se refieren a (1) un compuesto tal como se indica
en la presente memoria que inhibe selectivamente la isoterma gamma sobre la isoterma alfa, beta o delta, o (2) un compuesto indicado en la presente memoria que inhibe selectivamente la isoterma delta sobre la isoterma alfa o beta. A título de ejemplo no limitativo, el índice de selectividad puede ser superior a un factor de aproximadamente 1, superior a un factor de aproximadamente 2, superior a un factor de aproximadamente 3, superior a un factor de aproximadamente 5, superior a un factor de aproximadamente 50, superior a un factor de aproximadamente 100, superior a un factor de aproximadamente 200, superior a un factor de aproximadamente 400, superior a un factor de aproximadamente 600, superior a un factor de aproximadamente 800, superior a un factor de aproximadamente 1000, superior a un factor de aproximadamente 1500, superior a un factor de aproximadamente 2000, superior a un factor de aproximadamente 5000, superior a un factor de aproximadamente 10.000, o superior a un factor de aproximadamente 20.000, en el que la selectividad puede medirse mediante una proporción de valores IC50, que a su vez pueden medirse mediante, p.ej., ensayos in vitro o in vivo tales como los descritos en los Ejemplos descritos en la presente memoria. En una realización, la selectividad de una primera isoforma de PI3K respecto a una segunda isoforma de PI3K se mide como la proporción entre el valor IC50 contra la segunda isoforma de PI3K y el valor IC50 contra la primera isoforma de PI3K gamma. Por ejemplo, puede medirse un índice de selectividad delta/gamma de un compuesto como la proporción entre la actividad inhibidora del compuesto contra la isoforma delta en términos de IC50 o similar y la actividad inhibidora del compuesto contra la isoforma gamma en términos de IC50 o similar. Si el índice de selectividad delta/gamma es superior a 1, el compuesto inhibe selectivamente la isoforma gamma sobre la isoforma delta. En determinadas realizaciones, la actividad IC50 de la isoforma PI3K gamma de un compuesto proporcionado en la presente memoria puede ser inferior a aproximadamente 1000 nM, inferior a aproximadamente 500 nM, inferior a aproximadamente 400 nM , inferior a aproximadamente 300 nM, inferior a aproximadamente 200 nM, inferior a aproximadamente 100 nM, inferior a aproximadamente 75 nM, inferior a aproximadamente 50 nM, inferior a aproximadamente 25 nM, inferior a aproximadamente 20 nM, inferior a aproximadamente 15 nM, inferior a aproximadamente 10 nM, inferior a aproximadamente 5 nM, o inferior a aproximadamente 1 nM. En determinadas realizaciones, la actividad IC50 de la isoforma PI3K delta de un compuesto proporcionado en la presente memoria puede ser inferior a aproximadamente 1000 nM, inferior a aproximadamente 500 nM, inferior a aproximadamente 400 nM , inferior a aproximadamente 300 nM, inferior a aproximadamente 200 nM, inferior a aproximadamente 100 nM, inferior a aproximadamente 75 nM, inferior a aproximadamente 50 nM, inferior a aproximadamente 25 nM, inferior a aproximadamente 20 nM, inferior a aproximadamente 15 nM, inferior a aproximadamente 10 nM, inferior a aproximadamente 5 nM, o inferior a aproximadamente 1 nM.
En determinadas realizaciones, un inhibidor de PI3K-Y inhibe selectivamente la isoforma gamma sobre la isoforma alfa, beta o delta (también denominado en la presente memoria "inhibidor selectivo de PI3K-y"). En una realización, el inhibidor de PI3K-Y inhibe selectivamente la isoforma gamma respecto a la isoforma alfa o beta. En una realización, el inhibidor de PI3K-Y inhibe selectivamente la isoforma gamma respecto a la isoforma alfa, beta o delta. En una realización, el inhibidor de PI3K-Y inhibe selectivamente la isoforma gamma respecto a las isoformas alfa y beta. En una realización, el inhibidor de PI3K-Y inhibe selectivamente la isoforma gamma respecto a las isoformas alfa y beta, aunque no la isoforma delta. A título de ejemplo no limitativo, el índice de selectividad puede ser superior a un factor de aproximadamente 10, superior a un factor de aproximadamente 50, superior a un factor de aproximadamente 100, superior a un factor de aproximadamente 200, superior a un factor de aproximadamente 400, superior a un factor de aproximadamente 600, superior a un factor de aproximadamente 800, superior a un factor de aproximadamente 1000, superior a un factor de aproximadamente 1500, superior a un factor de aproximadamente 2000, superior a un factor de aproximadamente 5000, superior a un factor de aproximadamente 10.000, o superior a un factor de aproximadamente 20.000, en el que la selectividad puede medirse mediante una proporción de valores IC50, entre otros medios. En una realización, la selectividad de la isoforma PI3K gamma respecto a otra isoforma de PI3K se mide como la proporción entre el valor IC50 contra la otra isoforma de PI3K y el valor IC50 contra la primera isoforma PI3K gamma. En determinadas realizaciones, la actividad IC50 de la isoforma PI3 quinasa gamma de un compuesto tal como se da a conocer en la presente memoria puede ser inferior a aproximadamente 1000 nM, inferior a aproximadamente 100 nM, inferior a aproximadamente 10 nM o inferior a aproximadamente 1 nM. Por ejemplo, un compuesto que inhibe selectivamente una isoforma de PI3K sobre otra isoforma de PI3K presenta una actividad por lo menos 2X contra una primera isoforma respecto a la actividad del compuesto contra la segunda isoforma (p.ej., por lo menos aproximadamente 3X, 5X, 10X, 20X, 50X, 100X, 200X, 500X, o 1000X).
En determinadas realizaciones, se utiliza o se administra un inhibidor selectivo de PI3K-Y en un sujeto a una dosis más baja (p.ej., en aproximadamente 10%, en aproximadamente 20%, en aproximadamente 30%, en aproximadamente 40%, en aproximadamente 50%, en aproximadamente 60%, en aproximadamente 70%, o en aproximadamente 80%) en comparación con el tratamiento con un inhibidor no selectivo o menos selectivo de PI3K-Y (p.ej., un inhibidor PI3Kpan, p.ej., mediante la inhibición de PI3K-a, p, 8 y y).
La expresión "terapia de radiación" se refiere a exponer un paciente a emisores de radiación, tales como, aunque sin limitación, radionucleidos emisores de partículas alfa (p.ej., radionucleidos de actinio y torio), emisores de radiación de baja transferencia de energía lineal (TEL) (p.ej., emisores beta), emisores de conversión de electrones (p.ej., estroncio-89 y samario-153-EDTMP) o radiación de alta energía, incluyendo, aunque sin limitación, rayos x, rayos gamma y neutrones. La terapia de radiación puede llevarse a cabo utilizando métodos rutinarios y composiciones conocidos por el médico.
Tal como se utiliza en la presente memoria, un "valor de referencia" se refiere a un valor con el que puede compararse
un valor dado. En algunas realizaciones, el valor de referencia se refiere a un control (p.ej., un control no tratado, p.ej., un sujeto no tratado o tratado con un placebo o una muestra no tratada); el curso de la enfermedad sin tratamiento; un sujeto sano o una media de sujetos sanos; un sujeto en un intervalo de tiempo diferente, p.ej., antes, durante o después del tratamiento).
El término "sujeto" en el que se contempla la administración incluye, aunque sin limitación, seres humanos (p.ej., un hombre o mujer de cualquier grupo de edad, p.ej., un sujeto pediátrico (p.ej., bebes, niños o adolescentes) o sujetos adultos (p.ej., adultos jóvenes, adultos de mediana edad o adultos de edad avanzada) y/o otros primates (p.ej., monos Cynomolgus y monos rhesus), mamíferos, incluyendo mamíferos comercialmente relevantes, tales como vacas, cerdos, caballos, ovejas, gatos y/o perros, y/o aves, incluyendo aves comercialmente relevantes, tales como pollos, patos, gansos, codornices y/o pavos.
La expresión "in vivo" se refiere a un suceso que tiene lugar en el cuerpo del sujeto.
El término "in vitro" se refiere a un suceso que tiene lugar fuera del cuerpo del sujeto. Por ejemplo, un ensayo in vitro comprende cualquier ensayo llevado a cabo fuera del sujeto. Los ensayos in vitro comprenden ensayos celulares en los que se utilizan células, vivas o muertas. Los ensayos in vitro comprenden además un ensayo sin células en el que no se utilizan células intactas.
Definiciones químicas
Tal como se utiliza en la presente memoria, la expresión "ésteres farmacéuticamente aceptables" incluye, aunque sin limitación, ésteres alquilo, alquenilo, alquinilo, arilo, aralquilo y cicloalquilo de grupos ácidos, incluyendo, aunque sin limitación, ácidos carboxílicos, ácidos fosfóricos, ácidos fosfínicos, ácidos sulfónicos, ácidos sulfínicos y ácidos borónicos.
Tal como se utiliza en la presente memoria, la expresión "éteres de enol farmacéuticamente aceptables" incluye, aunque sin limitación, derivados de fórmula -C=C(OR), en la que R puede seleccionarse de entre alquilo, alquenilo, alquinilo, arilo, aralquilo y cicloalquilo. Entre los ésteres de enol farmacéuticamente aceptables se incluyen, aunque sin limitación, derivados de fórmula -C=C(OC(OR), en la que R puede seleccionarse de entre hidrógeno, alquilo, alquenilo, alquinilo, arilo, aralquilo y cicloalquilo.
Tal como se utiliza en la presente memoria, una "forma farmacéuticamente aceptable" de un compuesto dado a conocer incluye, aunque sin limitación, sales, hidratos, solvatos e isómeros farmacéuticamente aceptables, y derivados isotópicamente marcados de compuestos dados a conocer. En una realización, una "forma farmacéuticamente aceptable" incluye, aunque sin limitación, sales e isómeros farmacéuticamente aceptables, y derivados isotópicamente marcados de compuestos dados a conocer.
En determinadas realizaciones, la forma farmacéuticamente aceptable es una sal farmacéuticamente aceptable. Tal como se utiliza en la presente memoria, la expresión "sal farmacéuticamente aceptable" se refiere a aquellas sales que resultan, dentro del alcance del criterio médico razonable, adecuadas para la utilización en contacto con los tejidos de los sujetos sin toxicidad indebida, irritación, respuesta alérgica y similares, y que son proporcionales a una relación de beneficio/riesgo razonable. Las sales farmacéuticamente aceptables son bien conocidas de la técnica. Por ejemplo, Berge et al. describe sales farmacéuticamente aceptables en detalle en J. Pharmaceutical Sciences (1977) 66:1-19. Entre las sales farmacéuticamente aceptables de los compuestos proporcionados en la presente memoria se incluyen las derivadas de ácidos y bases inorgánicos y orgánicos adecuados. Son ejemplos de sales de adición de ácido no tóxicas farmacéuticamente aceptables, las sales de un grupo amino formadas con ácidos inorgánicos, tales como ácido clorhídrico, ácido bromhídrico, ácido fosfórico, ácido sulfúrico y ácido perclórico, o con ácidos orgánicos, tales como ácido acético, ácido oxálico, ácido maleico, ácido tartárico, ácido cítrico, ácido succínico o ácido malónico o mediante la utilización de otros métodos utilizados en la técnica, tales como el intercambio iónico. Entre otras sales farmacéuticamente aceptables se incluyen las sales adipato, alginato, ascorbato, aspartato, bencenosulfonato, besilato, benzoato, bisulfato, borato, butirato, camforato, camforsulfonato, citrato, ciclopentanopropionato, digluconato, dodecilsulfato, etano-sulfonato, formato, fumarato, glucoheptonato, glicerofosfato, gluconato, hemisulfato, heptanoato, hexanoato, hidroyoduro, 2-hidroxi-etanosulfonato, lactobionato, lactato, laurato, lauril-sulfato, malato, maleato, malonato, metanosulfonato, 2-naftalenosulfonato, naftaleno-m,n-bissulfonatos, nicotinato, nitrato, oleato, oxalato, palmitato, pamoato, pectinato, persulfato, 3-fenilpropionato, fosfato, picrato, pivalato, propionato, estearato, succinato, sulfato, tartrato, tiocianato, p-toluenosulfonato, undecanoato, valerato, y similares. En algunas realizaciones, entre los ácidos orgánicos a partir de los que pueden derivarse sales se incluyen, por ejemplo, ácido acético, ácido propiónico, ácido glicólico, ácido pirúvico, ácido oxálico, ácido maleico, ácido malónico, ácido succínico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido cinámico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico, ácido salicílico, ácido naftalén-m,n-sulfónico, y similares.
Entre las sales farmacéuticamente aceptables derivadas de bases apropiadas se incluyen sales de metal alcalino, sales de metal alcalinotérreo, sales amónicas y sales de N+(alquilo C-m )4. Entre las sales de metal alcalino o alcalinotérreo representativas se incluyen sodio, litio, potasio, calcio, magnesio, hierro, cinc, cobre, manganeso, aluminio y similares. Entre las sales farmacéuticamente aceptables adicionales se incluyen, en caso apropiado,
cationes de amonio no tóxico, amonio cuaternario y amina, formadas utilizando contraiones, tales como haluro, hidróxido, carboxilato, sulfato, fosfato, nitrato, alquilo inferior-sulfonato y arilsulfonato. Entre las bases orgánicas a partir de las que pueden derivarse sales se incluyen, por ejemplo, aminas primarias, secundarias y terciarias, aminas sustituidas, incluyendo aminas sustituidas naturales, aminas cíclicas, resinas de intercambio iónico básicas, y similares, tales como isopropilamina, trimetilamina, dietilamina, trietilamina, tripropilamina y etanolamina. En algunas realizaciones, la sal de adición de base farmacéuticamente aceptable se selecciona de entre sales de amonio, potasio, sodio, calcio y magnesio.
En determinadas realizaciones, la forma farmacéuticamente aceptable es un solvato (p.ej., un hidrato). Tal como se utiliza en la presente memoria, el término "solvato" se refiere a compuestos que incluyen además una cantidad estequiométrica o no estequiométrica de solvente unidos mediante fuerzas intermoleculares no covalentes. El solvato puede ser de un compuesto dado a conocer o de una sal farmacéuticamente aceptable del mismo. En el caso de que el solvente sea agua, el solvato es un "hidrato". Los solvatos e hidratos farmacéuticamente aceptables son complejos que, por ejemplo, pueden incluir entre 1 y aproximadamente 100, o entre 1 y aproximadamente 10, o entre uno y aproximadamente 2, aproximadamente 3 o aproximadamente 4 moléculas de solvente o agua. Se entenderá que el término "compuesto" tal como se utiliza en la presente memoria comprende el compuesto y solvatos del compuesto, así como mezclas del mismo.
En un aspecto no reivindicado, el compuesto o una forma farmacéuticamente aceptable del compuesto puede administrarse como un constituyente de un profármaco y transformarse in vivo, rindiendo el compuesto dado a conocer o una forma farmacéuticamente aceptable del compuesto de la invención. Tal como se utiliza en la presente memoria, el término "profármaco" se refiere a compuestos que se transforman in vivo, rindiendo un compuesto dado a conocer o una forma farmacéuticamente aceptable del compuesto. Un profármaco puede estar inactivo al administrarlo en un sujeto, aunque se convierte in vivo en un compuesto activo, por ejemplo mediante hidrólisis (p.ej., hidrólisis en la sangre). En determinados casos, un profármaco presenta propiedades físicas y/o de administración mejoradas respecto al compuesto parental. Los profármacos están diseñados típicamente para potenciar las propiedades farmacéuticas y/o farmacocinéticas asociadas al compuesto parental. El compuesto profármaco con frecuencia ofrece ventajas de solubilidad, compatibilidad en los tejidos o liberación retardada en un organismo de mamífero (ver, p.ej., Bundgard, H., Design of Prodrugs (1985), páginas 7-9, 21-24 (Elsevier, Amsterdam). Se proporciona un análisis de los profármacos en Higuchi, T., et al., "Pro-drugs as Novel Delivery Systems," A.C.S. Symposium Series, vol. 14, y en Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
Entre las ventajas ejemplares de un profármaco pueden incluirse, aunque sin limitarse a ellas, sus propiedades físicas, tales como una solubilidad en agua mejorada para la administración parenteral a pH fisiológico en comparación con el compuesto parental, o potencia la absorción en el tracto digestivo, o puede potenciar la estabilidad del fármaco para el almacenamiento a largo plazo.
El término "profármaco" también pretende incluir cualesquiera portadores unidos covalentemente, que liberan el compuesto activo in vivo al administrar dicho profármaco en el sujeto. Los profármacos de un compuesto activo, tal como se indica en la presente memoria, pueden prepararse mediante modificación de grupos funcionales presentes en el compuesto activo de manera que las modificaciones se escindan, en una manipulación rutinaria o in vivo, formando el compuesto parental activo. Entre los profármacos se incluyen compuestos en los que un grupo hidroxi, amino o mercapto está unido a cualquier grupo que, al administrar el profármaco del compuesto activo en el sujeto, se escinde formando un grupo hidroxi libre, un grupo amino libre o un grupo mercapto libre, respectivamente. Entre los ejemplos de profármacos se incluyen, aunque sin limitarse a ellos, derivados acetato, formato y benzoato de un alcohol o derivados acetamida, formamida y benzamida de un grupo funcional amina en el compuesto activo, y similares. Entre otros ejemplos de profármacos se incluyen compuestos que comprenden fracciones -No , -NO2 , -ONO, o -ONO2. Pueden prepararse típicamente profármacos utilizando métodos bien conocidos, tales como los indicados en Burger's Medicinal Chemistry and Drug Discovery, 172-178, 949-982 (Manfred E. Wolff ed., 5a ed., 1995), y Design of Prodrugs (H. Bundgaard ed., Elsevier, New York, 1985).
Por ejemplo, en el caso de que un compuesto dado a conocer o una forma farmacéuticamente aceptable del compuesto contenga un grupo funcional ácido carboxílico, un profármaco puede comprender un éster farmacéuticamente aceptable formada mediante la sustitución del átomo de hidrógeno del grupo ácido por un grupo, tal como alquilo C-i-Cs, alcanoiloximetilo C2-C12, 1 -(alcanoiloxi)etilo con 4 a 9 átomos de carbono, 1-metil-1-(alcanoiloxi)-etilo con 5 a 10 átomos de carbono, alcoxicarboniloximetilo con 3 a 6 átomos de carbono, 1-(alcoxicarboniloxi)etilo con 4 a 7 átomos de carbono, 1-metil-1-(alcoxicarboniloxi)etilo con 5 a 8 átomos de carbono, N-(alcoxicarbonil)aminometilo con 3 a 9 átomos de carbono, 1-(N-(alcoxicarbonil)amino)etilo con 4 a 10 átomos de carbono, 3-ftalidilo, 4-crotonolactonilo, gamma-butirolacton-4-ilo, di-N,N-alquil C1-C2-aminoalquilo C2-C3 (tal como pdimetilaminoetilo), carbamoil-alquilo C1-C2, N,N-dialquil C1-C2-carbamoil-alquilo C1-C2 y piperidino-, pirrolidino- o morfolino-alquilo C2-C3.
De manera similar, en el caso de que un compuesto dado a conocer o una forma farmacéuticamente aceptable del compuesto contenga un grupo funcional alcohol, puede formarse un profármaco mediante la sustitución del átomo de hidrógeno del grupo alcohol por un grupo tal como alcanoiloximetilo C1-C6, 1-(alcanoiloxi C-i-C6)-etilo, 1 -metil-1(alcanoiloxi Ci-C6)etil-alcoxi(Ci-C6)-carboniloximetilo, N-alcoxi Ci-C6-carbonilaminometilo, succinoilo, alcanoilo Ci-C6, a-amino-alcanoilo C1-C4, arilacilo y a-aminoacilo, o a-aminoacil-a-aminoacilo, en el que cada grupo a-aminoacilo se selecciona independientemente de entre L-aminoácidos naturales, P(O)(OH)2, -P(O)(O-alquilo C1-C6)2 y glucosilo (el radical que resulta de la eliminación de un grupo hidroxilo en una forma hemiacetal de un carbohidrato).
En el caso de que un compuesto dado a conocer o una forma farmacéuticamente aceptable del compuesto incorpore un grupo funcional amina, puede formarse un profármaco mediante la sustitución de un átomo de hidrógeno en el grupo amina por un grupo tal como R-carbonilo, RO-carbonilo, NRR'-carbonilo, en los que R y R' son, cada uno independientemente, alquilo C1-C10, cicloalquilo C3-C7, bencilo, un a-aminoacilo natural o un a-aminoacilo natural- aaminoacilo natural, - C (0 h )C(O)OY1 en el que Y1 es H, alquilo C1-C6 o bencilo, -C(OY2)Y3 en el que Y2 es alquilo C1-C4 e Y3 es alquilo C1-C6, carboxi-alquilo C1-C6, amino-alquilo C1-C4 o mono-N- o di-N,N-alquil (C1-C6)-aminoalquilo, -C(Y4)Y5 en el que Y4 es H o metilo, e Y5 es mono-N- o di-N,N-alquil (C1-C6)-amino, morfolino, piperidín-1-ilo o pirrolidín-1-ilo.
En determinadas realizaciones, la forma farmacéuticamente aceptable es un isómero. Los "isómeros" son compuestos diferentes que presentan la misma fórmula molecular. Los "atropisómeros" son estereoisómeros procedentes de la rotación impedido en torno a enlaces sencillos y pueden resolverse o aislarse mediante métodos conocidos por el experto en la materia. Por ejemplo, determinados sustituyentes B de un compuesto de fórmula (I) proporcionada en la presente memoria con fenilo orto- o meta-sustituido pueden formar atropisómeros, a partir de donde pueden separarse y aislarse.
Los "estereoisómeros" son isómeros que difieren únicamente en el modo en que se disponen los átomos en el espacio. Tal como se utiliza en la presente memoria, el término "isómero" incluye todos y cada uno de los isómeros geométricos y estereoisómeros. Por ejemplo, los "isómeros" incluyen isómeros cis y trans de doble enlace o geométricos, también denominados isómeros E y Z, enantiómeros R y S, diastereómeros, (d)-isómeros y (/)-isómeros, mezclas racémicas de los mismos, y otras mezclas de los mismos, comprendidas dentro del alcance de la presente exposición.
En determinadas realizaciones, el símbolo ------ denota un enlace que puede ser sencillo o doble tal como se indica en la presente memoria.
En determinadas realizaciones, en la presente memoria se proporcionan diversos isómeros geométricos y mezclas de los mismos que resultan de la disposición de sustituyentes entorno a un doble enlace carbono-carbono o la disposición de sustituyentes en torno a un anillo carbocíclico. Los sustituentes en torno a un doble enlace carbono-carbono se designan como en la configuración "Z" o "E", en los que los términos "Z" y "E" se utilizan de acuerdo con normas de la IUPAC. A menos que se indique lo contrario, las estructuras que ilustran dobles enlaces comprenden isómeros tanto "E" como "Z".
Los sustituyentes en torno a un doble enlace carbono-carbono alternativamente pueden denominarse "cis" o "trans", en donde "cis" representa los sustituyentes en el mismo lado del doble enlace y "trans" representa sustituyentes en lados opuestos del doble enlace. La organización de los sustituyentes en torno a un anillo carbocíclico también puede denominarse "cis" o "trans". El término "cis" representa sustituyentes en el mismo lado del plano del anillo y el término "trans" representa sustituyentes en lados contrarios del plano del anillo. Las mezclas de compuestos en las que los sustituyentes están dispuestos tanto en el mismo lado como en lados contrarios del plano del anillo se denominan "cis/trans".
Los enantiómeros" son un par de estereoisómeros que son imágenes especulares no superponibles uno de otro. Una mezcla de un par de enantiómeros en cualquier proporción puede conocerse como una mezcla "racémica". El término "(±)" se utiliza para designar una mezcla rácemica, en su caso. Los "diastereoisómeros" son estereoisómeros que presentan por lo menos dos átomos asimétricos, pero que no son imágenes especulares uno de otro. La estereoquímica absoluta puede especificarse según el sistema R-S de Cahn-Ingold-Prelog. En el caso de que un compuesto sea un enantiómero, la estereoquímica en cada carbono quiral puede especificarse como R o S. Los compuestos resueltos cuya configuración absoluta sea desconocida pueden designarse como (+) o (-) dependiendo de la dirección (dextrorrotatoria o levorrotatoria) en la que hacen rotar el plano de la luz polarizada a la longitud de onda de la línea D del sodio. Determinados compuestos indicados en la presente memoria contienen uno o más centros asimétricos y, de esta manera, pueden dar lugar a enantiómeros, diastereómeros y otras formas estereoisoméricas que pueden definirse, en términos de estereoquímica absoluta en cada átomo asimétrico, como (R) o (S). Las presentes entidades químicas, composiciones farmacéuticas y métodos pretenden incluir la totalidad de dichos isómeros posibles, incluyendo mezclas racémicas, formas ópticamente puras sustancialmente y mezclas de intermediarios. Pueden prepararse isómeros (R) y (S) ópticamente activos, por ejemplo utilizando sintones quirales o reactivos quirales, o resolverse utilizando técnicas convencionales.
El "exceso enantiomérico" o "% de exceso enantiomérico" de una composición puede calcularse utilizando la ecuación mostrada a continuación. En el ejemplo mostrado posteriormente, una composición contiene 90 % de un enantiómmero, p.ej., un enantiómero S, y 10 % del otro enantiómero, p.ej., un enantiómero R.
ee = (90-10)/100 = 80%.
De esta manera, una composición que contiene 90 % de un enantiómero y 10 % del otro enantiómero se afirma que presenta un exceso enantiomérico de 80 %. Algunas composiciones indicadas en la presente memoria contienen un exceso enantiomérico de por lo menos aproximadamente 1 %, aproximadamente 5 %, aproximadamente 10 %, aproximadamente 20 %, aproximadamente 30 %, aproximadamente 40 %, aproximadamente 50 %, aproximadamente 75 %, aproximadamente 90 %, aproximadamente 95 %, o aproximadamente 99 % del enantiómero S. En otras palabras, las composiciones contienen un exceso enantiomérico del enantiómero S respecto al enantiómero R. En otras realizaciones, algunas composiciones indicadas en la presente memoria contienen un exceso enantiomérico de por lo menos aproximadamente 1 %, aproximadamente 5 %, aproximadamente 10 %, aproximadamente 20 %, aproximadamente 30 %, aproximadamente 40 %, aproximadamente 50 %, aproximadamente 75 %, aproximadamente 90 %, aproximadamente 95 %, o aproximadamente 99 % del enantiómero R. En otras palabras, las composiciones contienen un exceso enantiomérico del enantiómero R respecto al enantiómero S.
Por ejemplo, un isómero/enantiómero puede, en algunas realizaciones, proporcionarse sustancialmente libre del enantiómero correspondiente y también puede denominarse "ópticamente enriquecido", "enantioméricamente enriquecido", "enantioméricamente puro" y "no racémico", tal como se utilizan intercambiablemente en la presente memoria. Dichas expresiones se refieren a composiciones en las que la cantidad de un enantiómero es superior a la cantidad de ese enantiómero en una mezcla de control de la composición racémica (p.ej., superior a 1:1 en peso). Por ejemplo, una preparación enantioméricamente enriquecida en el enantiómero S, se refiere a una preparación del compuesto que presenta más de aproximadamente 50% en peso del enantiómero S respecto al peso total de la preparación (p.ej., peso total de isómeros S y R), tal como por lo menos aproximadamente 75% en peso, adicionalmente, tal como por lo menos aproximadamente 80% en peso. En algunas realizaciones, el enriquecimiento puede ser muy superior a aproximadamente 80%, proporcionando una preparación "sustancialmente enriquecida enantioméricamente", "sustancialmente pura enantioméricamente" o "sustancialmente no racémica", que se refiere a preparaciones de composiciones que presentan por lo menos aproximadamente 85% en peso de un enantiómero respecto al peso total de la preparación, tal como por lo menos aproximadamente 90% en peso, y adicionalmente, tal como por lo menos aproximadamente 95% en peso. En determinadas realizaciones, el compuesto proporcionado en la presente memoria está constituido de por lo menos aproximadamente 90% en peso de un enantiómero. En otras realizaciones, el compuesto está constituido de por lo menos aproximadamente 95%, aproximadamente 98% o aproximadamente 99% en peso de un enantiómero.
En algunas realizaciones, el compuesto es una mezcla racémica de isómeros (S) y (R). En otras realizaciones, en la presente memoria se proporciona una mezcla de compuestos en la que los compuestos individuales de la mezcla existen predominantemente en una configuración isomérica (S) o (R). Por ejemplo, en algunas realizaciones, la mezcla de compuestos presenta un exceso en enantiómero (S) superior a aproximadamente 10%, superior a aproximadamente 20%, superior a aproximadamente 30%, superior a aproximadamente 40%, superior a aproximadamente 50%, superior a aproximadamente 55%, superior a aproximadamente 60%, superior a aproximadamente 65%, superior a aproximadamente 70%, superior a aproximadamente 75%, superior a aproximadamente 80%, superior a aproximadamente 85%, superior a aproximadamente 90%, superior a aproximadamente 95%, superior a aproximadamente 96%, superior a aproximadamente 97%, superior a aproximadamente 98%, o superior a aproximadamente 99%. En algunas realizaciones, la mezcla de compuesto presenta un exceso de enantiómero (S) de aproximadamente 55 %, aproximadamente 60 %, aproximadamente 65 %, aproximadamente 70 %, aproximadamente 75 %, aproximadamente 80 %, aproximadamente 85 %, aproximadamente 90 %, aproximadamente 95 %, aproximadamente 96 %, aproximadamente 97 %, aproximadamente 98 %, aproximadamente 99 % o aproximadamente 99,5 % o más. En algunas realizaciones, la mezcla de compuestos presenta un exceso de enantiómero (S) de entre aproximadamente 55 % y aproximadamente 99,5 %, de entre aproximadamente 60 % y aproximadamente 99,5 %, de entre aproximadamente 65 % y aproximadamente 99,5 %, de entre aproximadamente 70 % y aproximadamente 99,5 %, de entre aproximadamente 75 % y aproximadamente 99,5 %, de entre aproximadamente 80 % y aproximadamente 99,5 %, de entre aproximadamente 85 % y aproximadamente 99,5 %, de entre aproximadamente 90 % y aproximadamente 99,5 %, de entre aproximadamente 95 % y aproximadamente 99,5 %, de entre aproximadamente 96 % y aproximadamente 99,5 %, de entre aproximadamente 97 % y aproximadamente 99,5 %, de entre aproximadamente 98 % y aproximadamente 99,5 %, o de entre aproximadamente 99 % y aproximadamente 99,5 %, o superior a aproximadamente 99,5 %.
En otras realizaciones, la mezcla de compuestos presenta un exceso en enantiómero (R) superior a aproximadamente 10%, superior a aproximadamente 20 %, superior a aproximadamente 30 %, superior a aproximadamente 40 %, superior a aproximadamente 50 %, superior a aproximadamente 55 %, superior a aproximadamente 60 %, superior a aproximadamente 65 %, superior a aproximadamente 70 %, superior a aproximadamente 75 %, superior a aproximadamente 80 %, superior a aproximadamente 85 %, superior a aproximadamente 90 %, superior a aproximadamente 95 %, superior a aproximadamente 96 %, superior a aproximadamente 97 %, superior a aproximadamente 98 %, o superior a aproximadamente 99 %. En algunas realizaciones, la mezcla de compuesto presenta un exceso de enantiómero (R) de aproximadamente 55 %, aproximadamente 60 %, aproximadamente 65 %, aproximadamente 70 %, aproximadamente 75 %, aproximadamente 80 %, aproximadamente 85 %, aproximadamente 90 %, aproximadamente 95 %, aproximadamente 96 %, aproximadamente 97 %, aproximadamente 98 %, aproximadamente 99 % o aproximadamente 99,5 % o superior. En algunas realizaciones, la mezcla de compuestos presenta un exceso de enantiómero (R) de entre aproximadamente 55 % y aproximadamente 99,5 %, de entre
aproximadamente 60 % y aproximadamente 99,5 %, de entre aproximadamente 65 % y aproximadamente 99,5 %, de entre aproximadamente 70 % y aproximadamente 99,5 %, de entre aproximadamente 75 % y aproximadamente 99,5 %, de entre aproximadamente 80 % y aproximadamente 99,5 %, de entre aproximadamente 85 % y aproximadamente 99.5 %, de entre aproximadamente 90 % y aproximadamente 99,5 %, de entre aproximadamente 95 % y aproximadamente 99,5 %, de entre aproximadamente 96 % y aproximadamente 99,5 %, de entre aproximadamente 97 % y aproximadamente 99,5 %, de entre aproximadamente 98 % y aproximadamente 99,5 %, o de entre aproximadamente 99 % y aproximadamente 99,5 %, o superior a aproximadamente 99,5 %.
En otras realizaciones, la mezcla de compuestos contiene entidades químicas idénticas excepto por sus orientaciones estereoquímicas, es decir, isómeros (S) o (R). Por ejemplo, en el caso de que un compuesto dado a conocer en la presente memoria presente una unidad -CH(R)- y R no sea hidrógeno, entonces el grupo -CH(R)- se encontrará en una orientación estereoquímica (S) o (R) en cada una de las entidades químicas idénticas (es decir, los estereoisómeros (S) o (R)). En algunas realizaciones, la mezcla de entidades químicas idénticas (es decir, la mezcla de estereoisómeros) es una mezcla racémica de isómeros (S) e isómeros (R). En otra realización, la mezcla de las entidades químicas idénticas (es decir, la mezcla de estereoisómeros) contiene predominantemente isómero (S) o predominantemente isómero (R). Por ejemplo, en algunas realizaciones, el isómero (S) en la mezcla de entidades químicas idénticas (es decir, la mezcla de estereoisómeros) se encuentra presente en una proporción de aproximadamente 55 %, aproximadamente 60 %, aproximadamente 65 %, aproximadamente 70 %, aproximadamente 75 %, aproximadamente 80 %, aproximadamente 85 %, aproximadamente 90 %, aproximadamente 95 %, aproximadamente 96 %, aproximadamente 97 %, aproximadamente 98 %, aproximadamente 99 %, o aproximadamente 99.5 % en peso, o superior, respecto al peso total de la mezcla de isómeros (S) y (R). En algunas realizaciones, el isómero (S) en la mezcla de entidades químicas idénticas (es decir, la mezcla de estereoisómeros) se encuentra presente en un exceso de enantiómero (S) de entre aproximadamente 10 % y aproximadamente 99,5 %, de entre aproximadamente 20 % y aproximadamente 99,5 %, de entre aproximadamente 30 % y aproximadamente 99.5 %, de entre aproximadamente 40 % y aproximadamente 99,5 %, de entre aproximadamente 50 % y aproximadamente 99,5 %, de entre aproximadamente 55 % y aproximadamente 99,5 %, de entre aproximadamente 60 % y aproximadamente 99,5 %, de entre aproximadamente 65 % y aproximadamente 99,5 %, de entre aproximadamente 70 % y aproximadamente 99,5 %, de entre aproximadamente 75 % y aproximadamente 99,5 %, de entre aproximadamente 80 % y aproximadamente 99,5 %, de entre aproximadamente 85 % y aproximadamente 99,5 %, de entre aproximadamente 90 % y aproximadamente 99,5 %, de entre aproximadamente 95 % y aproximadamente 99.5 %, de entre aproximadamente 96 % y aproximadamente 99,5 %, de entre aproximadamente 97 % y aproximadamente 99,5 %, de entre aproximadamente 98 % y aproximadamente 99,5 %, o de entre aproximadamente 99 % y aproximadamente 99,5 %, o superior a aproximadamente 99,5 %.
En otras realizaciones, el isómero (R) en la mezcla de entidades químicas idénticas (es decir, la mezcla de estereoisómeros) se encuentra presente en una proporción de aproximadamente 55 %, aproximadamente 60 %, aproximadamente 65 %, aproximadamente 70 %, aproximadamente 75 %, aproximadamente 80 %, aproximadamente 85 %, aproximadamente 90 %, aproximadamente 95 %, aproximadamente 96 %, aproximadamente 97 %, aproximadamente 98 %, aproximadamente 99 %, o aproximadamente 99.5 % en peso, o superior, respecto al peso total de la mezcla de isómeros (S) y (R). En algunas realizaciones, los isómeros (R) en la mezcla de entidades químicas idénticas (es decir, la mezcla de estereoisómeros) se encuentra presente en un exceso de enantiómero (R) de entre aproximadamente 10 % y aproximadamente 99,5 %, de entre aproximadamente 20 % y aproximadamente 99,5 %, de entre aproximadamente 30 % y aproximadamente 99,5 %, de entre aproximadamente 40 % y aproximadamente 99,5 %, de entre aproximadamente 50 % y aproximadamente 99,5 %, de entre aproximadamente 55 % y aproximadamente 99.5 %, de entre aproximadamente 60 % y aproximadamente 99,5 %, de entre aproximadamente 65 % y aproximadamente 99,5 %, de entre aproximadamente 70 % y aproximadamente 99,5 %, de entre aproximadamente 75 % y aproximadamente 99,5 %, de entre aproximadamente 80 % y aproximadamente 99,5 %, de entre aproximadamente 85 % y aproximadamente 99,5 %, de entre aproximadamente 90 % y aproximadamente 99,5 %, de entre aproximadamente 95 % y aproximadamente 99,5 %, de entre aproximadamente 96 % y aproximadamente 99,5 %, de entre aproximadamente 97 % y aproximadamente 99,5 %, de entre aproximadamente 98 % y aproximadamente 99.5 %, o de entre aproximadamente 99 % y aproximadamente 99,5 %, o superior a aproximadamente 99,5 %.
Pueden aislarse enantiómeros a partir de mezclas racémicas mediante cualquier método conocido por el experto en la materia, incluyendo cromatografía líquida de alta presión (HPLC) quiral y la formación y cristalización de sales quirales, o pueden prepararse mediante síntesis asimétricas. Ver, por ejemplo, Enantiomers, Racemates and Resolutions (Jacques, Ed., Wiley Interscience, New York, 1981);Wilen et al., Tetrahedron 33:2725 (1977); Stereochemistry of Carbon compuestos (E.L. Eliel, Ed., McGraw-Hill, NY, 1962) yTables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972).
En determinadas realizaciones, la forma farmacéuticamente aceptable es un tautómero. Tal como se utiliza en la presente memoria, el término "tautómero" es un tipo de isómero que incluye dos o más compuestos interconvertibles que resultan de por lo menos una migración formal de un átomo de hidrógeno y por lo menos un cambio de valencia (p.ej., un enlace sencillo por un doble enlace, un triple enlace por un doble enlace, o un triple enlace por un enlace sencillo, o viceversa). La "tautomerización" incluye la tautomerización protrópica o de desplazamiento de protones, que se considera un subgrupo de la química ácido-base. La "tautomerización prototrópica" o "tautomerización por desplazamiento de protones" implica la migración de un protón acompañada por cambios en el orden del enlace. La
proporción exacta de los tautómeros depende de varios factores, entre ellos la temperatura, el solvente y el pH. En donde resulta posible la tautomerización (p.ej., en solución), puede alcanzarse un equilibrio químico de los tautómeros. Las tautomerizaciones (es decir, la reacción que proporciona un par tautomérico) pueden catalizarse con ácido o base, o pueden producirse sin la acción o la presencia de un agente externo. Entre las tautomerizaciones ejemplares se incluyen, aunque sin limitación, las tautomerizaciones ceto-enol, amida-imida, lactamo-lactima, enamina-imina y enamina-enamina (una diferente). Un ejemplo específico de tautomerización ceto-enol es la interconversión de los tautómeros pentano-2,4-diona y 4-hidroxipent-3-en-2-ona. Otro ejemplo de tautomerización es la tautomerización fenol-ceto. Un ejemplo específico de tautomerización fenol-ceto es la interconversión de los tautómeros piridín-4-ol y piridín-4(1H)-ona.
A menos que se indique lo contrario, las estructuras ilustradas en la presente memoria también pretenden incluir compuestos que difieren únicamente en la presencia de uno o más átomos isotópicamente enriquecidos. Por ejemplo, los compuestos que presentan las estructuras presentes, excepto por la sustitución o enriquecimiento de un hidrógeno por deuterio o tritio en uno o más átomos en la molécula, o la sustitución o enriquecimiento de un carbono por 13C o 14C en uno o más átomos en la molécula, se encuentran comprendidos dentro del alcance de la presente exposición. En una realización, en la presente memoria se proporcionan compuestos marcados isotópicamente en los que se han sustituido o enriquecido uno o más átomos de hidrógeno por deuterio. En una realización, en la presente memoria se proporcionan compuestos marcados isotópicamente en los que se han sustituido o enriquecido uno o más átomos de hidrógeno por tritio. En una realización, en la presente memoria se proporcionan compuestos marcados isotópicamente en los que se han sustituido o enriquecido uno o más átomos de carbono por 13C. En una realización, en la presente memoria se proporcionan compuestos marcados isotópicamente en los que se han sustituido o enriquecido uno o más átomos de carbono por 14C.
La exposición comprende además compuestos marcados isotópicamente que son idénticos a los indicados en la presente memoria, excepto en que se ha sustituido uno o más átomos por un átomo que presenta una masa atómica o número másico diferente de la masa atómica o número másico habitualmente observado naturalmente. Entre los ejemplos de isótopos que pueden incorporarse en los compuestos dados a conocer se incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, azufre, flúor y cloro, tales como, p.ej., 2H, 3H, 13C, 14C, 15N, 18O, 170 , 31P, 32P, 35S, 18F y 36Cl, respectivamente. Determinados compuestos dados a conocer marcados isotópicamente (por ejemplo, los marcados con 3H y/o 14C) resultan útiles en ensayos de distribución en los tejidos del compuesto y/o el sustrato. Los isótopos tritiados (es decir, 3H) y carbono-14 (es decir, 14C) pueden permitir una fácil preparación y detectabilidad. Además, la sustitución con isótopos más pesados, tales como el deuterio (es decir 2H) puede proporcionar determinadas ventajas terapéuticas resultantes de la mayor estabilidad metabólica, por ejemplo semivida in vivo incrementada o necesidades de dosis menores. Los compuestos dados a conocer marcados isotópicamente pueden prepararse generalmente mediante sustitución de un reactivo marcado isotópicamente por un reactivo no marcado isotópicamente. En algunas realizaciones, en la presente memoria se proporcionan compuestos que también pueden contener proporciones no naturales de isótopos naturales en uno o más de los átomos que constituyen dichos compuestos. Todas las variantes isotópicas de los compuestos dados a conocer en la presente memoria, sean radioactivos o no, se encuentran comprendidos dentro del alcance de la presente exposición.
La expresión "portador farmacéuticamente aceptable" o "excipiente farmacéuticamente aceptable" incluye todos y cada uno de los solventes, medios de dispersión, recubrimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y retardantes de la absorción, y similares. La utilización de dichos medios y agentes para sustancias farmacéuticamente activas es bien conocida de la técnica. Excepto en la medida en que cualquier medio o agente convencional sea incompatible con el ingrediente activo, se encuentra contemplada la utilización en las composiciones terapéuticas tales como las dadas a conocer en la presente memoria. También pueden incorporarse ingredientes activos complementarios en las composiciones farmacéuticas.
Las definiciones de grupos funcionales específicos y términos químicos se describen en mayor detalle posteriormente. Los elementos químicos se identifican de acuerdo con la tabla periódica de los elementos, versión CAS, Handbook of Chemistry and Physics, 75a ed., tapa interior, y los grupos funcionales específicos se definen generalmente tal como se indica en dicha obra. Adicionalmente, se describen principios generales de química orgánica, así como fracciones funcionales específicas y reactividad, en Thomas Sorrell, Organic Chemistry, University Science Books, Sausalito, 1999; Smith y March, March's Advanced Organic Chemistry, 5a edición, John Wiley & Sons, Inc., New York, 2001; Larock, Comprehensive Organic Transformations, VCH Publishers, Inc., New York, 1989; y Carruthers, Some Modern métodos of Organic Synthesis, 3a edición, Cambridge University Press, Cambridge, 1987.
En el caso de que se indique un intervalo de valores, se pretende que se encuentren comprendidos cada valor y subintervalo dentro del intervalo. Por ejemplo, "alquilo C-i-a" se pretende que comprenda alquilo C1, C2 , C3 , C4, C5 , Ce, C1-6, C1-5, C1-4, C1-3, C1-2, C2-6, C2-5, C2-4, C2-3, C3-6, C3-5, C3-4, C4-6, C4-5 y C5-6.
"Alquilo" se refiere a un radical de cadena de hidrocarburo lineal o ramificado que consiste únicamente en átomos de carbono e hidrógeno, que no contiene insaturaciones, que presenta, en algunas realizaciones, entre uno y diez átomos de carbono (p.ej., alquilo C1-C10). Un alquilo lineal se refiere a un alquilo sin ramificaciones, p.ej., metilo, etilo y npropilo. En donde aparece en la presente memoria, un intervalo número tal como "1 a 10" se refiere a cada número entero en el intervalo dado, p.ej., "1 a 10 átomos de carbono" significa que el grupo alquilo puede consistir en 1 átomo
de carbono, 2 átomos de carbono, 3 átomos de carbono, 4 átomos de carbono, etc., hasta e incluyendo 10 átomos de carbono, aunque la presente definición incluye además la incidencia del término "alquilo" en donde no se indica ningún intervalo numérico. En algunas realizaciones, alquilo se refiere a un grupo alquilo C1-C6. En algunas realizaciones, los grupos alquilo presentan 1 a 10, 1 a 6, 1 a 4 o 1 a 3 átomos de carbono. Entre los alquilos de cadena lineal saturada representativos se incluyen, aunque sin limitarse a ellos, -metilo, -etilo, -n-propilo, -n-butilo, -n-pentilo y -n-hexilo, mientras que entre los alquilos ramificados saturados se incluyen, aunque sin limitarse a ellos, -isopropilo, -sec-butilo, -isobutilo, -terc-butilo, -isopentilo, 2-metilbutilo, 3-metilbutilo, 2-metilpentilo, 3-metilpentilo, 4-metilpentilo, 2-metilhexilo, 3-metilhexilo, 4-metilhexilo, 5-metilhexilo, 2,3-dimetilbutilo y similares. El alquilo se une a la molécula parental mediante un enlace sencillo. A menos que se indique lo contrario en la especificación, un grupo alquilo se encuentra sustituido opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3, -ORa, - SRa, -OC(O)-Ra, -N(Ra)2 , -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2 , -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2 , -N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria.
"Perhaloalquilo" se refiere a un grupo alquilo en el que la totalidad de los átomos de hidrógeno han sido sustituidos por un halógeno seleccionado de entre flúor, cloro, bromo y yodo. En algunas realizaciones, la totalidad de los átomos de hidrógeno se sustituye, cada uno, por flúor. En algunas realizaciones, la totalidad de los átomos de hidrógeno se sustituye, cada uno, por cloro. Entre los ejemplos de grupos perhaloalquilo se incluyen -CF3, -CF2CF3 , -CF2CF2CF3, -CCl3 , -CFCl2 , -CF2O y similares. El término "haloalquilo" se refiere a un grupo alquilo en el que uno o más de los átomos de hidrógeno ha sido sustituido por un halógeno seleccionado independientemente de entre flúor, cloro, bromo y yodo.
"Alquilcicloalquilo" se refiere a un radical -(alquil)cicloalquilo en el que alquilo y cicloalquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para alquilo y cicloalquilo, respectivamente. El "alquil-cicloalquilo" se une a la estructura molecular parental mediante el grupo alquilo. Los términos "alquenil-cicloalquilo" y "alquinil-cicloalquilo" reflejan la descripción anterior de "alquil-cicloalquilo", en la que el término "alquilo" se sustituye por "alquenilo" o "alquinilo", respectivamente, y "alquenilo" o "alquinilo" son tal como se indica en la presente memoria.
"Alquilarilo" se refiere a un radical -(alquil)arilo en el que arilo y alquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para arilo y alquilo, respectivamente. El "alquilarilo" se une a la estructura molecular parental mediante el grupo alquilo. Los términos "-(alquenil)arilo" y "-(alquinil)arilo" reflejan la descripción anterior de "-(alquil)arilo", en la que el término "alquilo" se sustituye por "alquenilo" o "alquinilo", respectivamente, y "alquenilo" o "alquinilo" son tal como se indica en la presente memoria.
"Alquilheteroarilo" se refiere a un radical -(alquil)heteroarilo en el que heteroarilo y alquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heteroarilo y alquilo, respectivamente. El "alquil-heteroarilo" se une a la estructura molecular parental mediante el grupo alquilo. Los términos "-(alquenil)heteroarilo" y "-(alquinil)heteroarilo" reflejan la descripción anterior de "-(alquil)heteroarilo", en la que el término "alquilo" se sustituye por "alquenilo" o "alquinilo", respectivamente, y "alquenilo" o "alquinilo" son tal como se indica en la presente memoria.
"Alquil-heterociclilo" se refiere a un radical -(alquil)heterociclilo en el que alquilo y heterociclilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heterociclilo y alquilo, respectivamente. El "alquil-heterociclilo" se une a la estructura molecular parental mediante el grupo alquilo. Los términos "-(alquenil)heterociclilo" y "-(alquinil)heterociclilo" reflejan la descripción anterior de "-(alquil)heterociclilo", en la que el término "alquilo" se sustituye por "alquenilo" o "alquinilo", respectivamente, y "alquenilo" o "alquinilo" son tal como se indica en la presente memoria.
"Alquenilo" se refiere a un radical de cadena de hidrocarburo lineal o ramificado que consiste únicamente en átomos de carbono e hidrógeno, que contiene por lo menos un doble enlace, y en algunas realizaciones, que presenta dos a diez átomos de carbono (es decir, alquenilo C2-C10). En donde se utiliza en la presente memoria, un intervalo numérico tal como "2 a 10" se refiere a cada número entero en el intervalo dado; p.ej., "2 a 10 átomos de carbono" significa que el grupo alquenilo puede consistir en 2 átomos de carbono, 3 átomos de carbono, 4 átomos de carbono, etc., hasta, e incluyendo, 10 átomos de carbono. En determinadas realizaciones, un alquenilo comprende dos a ocho átomos de carbono. En otras realizaciones, un alquenilo comprende dos a cinco átomos de carbono (p.ej., alquenilo C2-C5). El alquenilo se une a la estructura molecular parental mediante un enlace sencillo, por ejemplo, etenilo (es decir, vinilo), prop-1-enilo (es decir, alilo), but-1-enilo, pent-1-enilo, penta-1,4-dienilo, y similares. El enlace o enlaces dobles carbono-carbono pueden ser internos (tal como en 2-butenilo) o terminales (tal como en 1 -butenilo). Entre los ejemplos
de grupos alquenilo C2-4 se incluyen etenilo (C2), 1-propenilo (C3), 2-propenilo (C3), 1-butenilo (C4), 2-butenilo (C4), butadienilo (C4), y similares. Entre los ejemplos de grupos alquenilo C2-6 se incluyen los grupos alquenilo C2-4 anteriormente indicados, así como pentenilo (C5), pentadienilo (C5), hexenilo (Ca), y similares. Entre los ejemplos adicionales de alquenilo se incluyen heptenilo (C7), octenilo (C5), octatrienilo (C5), y similares. A menos que se indique lo contrario en la especificación, un grupo alquenilo se encuentra sustituido opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3, -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2 , -C(O)N(Ra)2 , -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2 , -N(Ra)C(NRa)N(Ra)2 , -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria.
"Alquinilo" se refiere a un radical de cadena de hidrocarburo lineal o ramificado que consiste únicamente en átomos de carbono e hidrógeno, que contiene por lo menos un triple enlace, y en algunas realizaciones, que presenta dos a diez átomos de carbono (es decir, alquinilo C2-C10). En donde se utiliza en la presente memoria, un intervalo numérico tal como "2 a 10" se refiere a cada número entero en el intervalo dado; p.ej., "2 a 10 átomos de carbono" significa que el grupo alquinilo puede consistir en 2 átomos de carbono, 3 átomos de carbono, 4 átomos de carbono, etc., hasta, e incluyendo, 10 átomos de carbono. En determinadas realizaciones, un alquinilo comprende dos a ocho átomos de carbono. En otras realizaciones, un alquinilo comprende dos a cinco átomos de carbono (p.ej., alquinilo C2-C5). El alquinilo se une a la estructura molecular parental mediante un enlace sencillo, por ejemplo, etinilo, propinilo, butinilo, pentinilo, hexinilo y similares. A menos que se indique lo contrario en la especificación, un grupo alquenilo se encuentra sustituido opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3, -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2, -N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria.
El término "alcoxi" se refiere al grupo -O-alquilo (en algunas realizaciones, que incluye 1 a 10 átomos de carbono), de una configuración lineal, ramificada o cíclica, y combinaciones de las mismas, unido a la estructura molecular parental mediante un oxígeno. Entre los ejemplos se incluyen metoxi, etoxi, propoxi, isopropoxi, ciclopropiloxi, ciclohexiloxi y similares. La expresión "alcoxi inferior" se refiere a grupos alcoxi que contienen uno a seis carbonos. En algunas realizaciones, alcoxi C1-C4 es un grupo alcoxi que comprende alquilos tanto de cadena lineal como ramificada, de entre 1 y 4 átomos de carbono. A menos que se indique lo contrario en la especificación, un grupo alcoxi se encuentra sustituido opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3, -ORa,-SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2, -N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria. Los términos "alquenoxi" y "alquinoxi" reflejan la descripción anterior de "alcoxi", en la que el prefijo "alqu" se sustituye por "alquen" o "alquin", respectivamente, y los términos parentales "alquenilo" o "alquinilo" son tal como se indica en la presente memoria.
El término "alcoxicarbonilo" se refiere a un grupo de fórmula (alcoxi)(C=O)- unida a la estructura molecular parental mediante el carbono del carbonilo (en algunas realizaciones, que presenta 1 a 10 átomos de carbono). De esta manera, un grupo alcoxicarbonilo C-i-Ca comprende un grupo alcoxi que presenta 1 a 6 átomos de carbono unidos mediante su oxígeno a un conector carbonilo. La denominación C1-Ca no incluye el carbono del carbonilo en el recuento de átomos. La expresión "alcoxicarbonilo inferior" se refiere a un grupo alcoxicarbonilo en el que la parte alquilo del grupo alcoxi es un grupo de alquilo inferior. En algunas realizaciones, alcoxicarbonilo C1-C4 comprende un grupo alcoxi que comprende grupos alcoxi tanto de cadena lineal como ramificada, de entre 1 y 4 átomos de carbono. A menos que se indique lo contrario en la especificación, un grupo alcoxicarbonilo se encuentra sustituido opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter,
mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)a, -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2 , -N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria. Los términos "alquenoxicarbonilo" y "alquinoxicarbonilo" reflejan la descripción anterior de "alcoxicarbonilo", en la que el prefijo "alqu" se sustituye por "alquen" o "alquin", respectivamente, y los términos parentales "alquenilo" o "alquinilo" son tal como se indica en la presente memoria.
"Acilo" se refiere a grupos R-C(O)-, tales como, aunque sin limitación, H, (alquil)-C(O)-, (alquenil)-C(O)-, (alquinil)-C(O)-, (aril)-C(O)-, (cicloalquil)-C(O)-, (heteroaril)-C(O)-, (heteroalquil)-C(O)- y (heterocicloalquil)-C(O)-, en los que el grupo está unido a la estructura molecular parental mediante la funcionalidad carbonilo. En algunas realizaciones, en la presente memoria se proporciona un radical acilo C1-C10 que se refiere al número total de átomos de la cadena o anillo de la parte, por ejemplo, alquilo, alquenilo, alquinilo, arilo, ciclohexilo, heteroarilo o heterocicloalquilo más el carbono de carbonilo del acilo. Por ejemplo, un acilo C4 presenta tres otros átomos de la cadena o anillo más carbonilo. En el caso de que el radical R sea heteroarilo o heterocicloalquilo, los heteroátomos del anillo o cadena contribuyen al número total de átomos de la cadena o anillo. A menos que se indique lo contrario en la especificación, la "R" de un grupo aciloxi puede sustituirse opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3 , -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2 , -N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria.
"Aciloxi" se refiere a un radical R(C=O)O- en el que "R" puede ser H, alquilo, alquenilo, alquinilo, heteroalquilo, heteroalquenilo, heteroalquinilo, arilo, ciclohexilo, heteroarilo o heterocicloalquilo, que son tal como se indica en la presente memoria. El grupo aciloxi se une a la estructura molecular parental mediante la funcionalidad oxígeno. En algunas realizaciones, un grupo aciloxi es un radical aciloxi C1-C4 que s erefier al número total de átomos de la cadena o anillo de la parte alquilo, alquenilo, alquinio, arilo, ciclohexilo, heteroarilo o heterocicloalquilo del grupo aciloxi más el carbono de carbonilo del acilo, p.ej., un aciloxi C4 presenta tres otros átomos de anillo o cadena más el carbonilo. En el caso de que el radical R sea heteroarilo o heterocicloalquilo, los heteroátomos del anillo o cadena contribuyen al número total de átomos de la cadena o anillo. A menos que se indique lo contrario en la especificación, la "R" de un grupo aciloxi puede sustituirse opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3 , -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2 , -N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria.
"Amino" o "amina" se refiere a un grupo radical -N(Rb)2, -N(Rb)Rb-, o -RbN(Rb)Rb-, en el que cada Rb se selecciona independientemente de entre hidrógeno, alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono de anillo), heterocicloalquilalquilo, heteroarilo (nido mediante un carbono de anillo) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria. En el caso de que un grupo -N(Rb)2 presente dos Rb diferentes de hidrógeno, pueden combinarse con el átomo de nitrógeno para formar un anillo de 3, 4, 5, 6, 7, u 8 elementos. Por ejemplo, -N(Rb)2 pretende incluir, aunque sin limitación, 1 -pirrolidinilo y 4-morfolinilo. A menos que se indique lo contrario en la especificación, un grupo amino se sustituye opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3 , -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2 , -N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo,
haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria.
Los términos "amina" y "amino" también pueden referirse a N-óxidos de los grupos -N+(H)(Ra)O' y - N+(Ra)(Ra)O', en los que Ra es tal como se ha indicado anteriormente, en donde el N-óxido está unido a la estructura molecular parental mediante el átomo de N. Pueden prepararse N-óxidos mediante tratamiento del grupo amino correspondiente con, por ejemplo, peróxido de hidrógeno o ácido m-cloroperoxibenzoico. Al experto en la materia le resultarán familiares las condiciones de reacción para llevar a cabo la N-oxidación.
"Amida" o "amido" se refiere a una fracción química con la fórmula -C(O)N(Rb)2 o -NRbC(O)Rb, en la que Rb se selecciona independientemente de entre hidrógeno, alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono de anillo), heterocicloalquilalquilo, heteroarilo (nido mediante un carbono de anillo) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria. En algunas realizaciones, un radical amido o amida es un radical amido o amida C1-C4 , que incluye el carbonilo de la amida en el número total de carbonos en el radical. En el caso de que un -C(O)N(Rb)2 presente dos Rb diferentes de hidrógeno, pueden combinarse con el átomo de nitrógeno para formar un anillo de 3, 4, 5, 6, 7 u 8 elementos. Por ejemplo, la parte N(Rb)2 de un radical -C(O)N(Rb)2 pretende incluir, aunque sin limitación, 1 -pirrolidinilo y 4-morfolinilo. A menos que se indique lo contrario en la especificación, un grupo Rb de amido se sustituye opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3 , -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2 , -N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria.
El término "amida" o "amido" también incluye la referencia a un aminoácido o a una molécula de péptido. Cualquier cadena lateral de amina, hidroxi o carboxilo en los compuestos indicados en la presente memoria puede transformarse en un grupo amida. Los procedimientos y grupos específicos para preparar dichas amidas son conocidos por el experto en la materia y pueden encontrarse fácilmente en fuentes de referencia, tales como Greene y Wuts, Protective Groups in Organic Synthesis, 4a dd., John Wiley & Sons, New York, NY, 2006.
"Amidino" se refiere a los radicales -C(=NRb)N(Rb)2 , -N(Rb)-C(=NRb)-Rb y -N(Rb)-C(=NRb)-, en los que cada Rb se selecciona independientemente de entre hidrógeno, alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono de anillo), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono de anillo) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria.
"Arilo" se refiere a un radical con seis a catorce átomos anulares (p.ej., arilo C6-C14 o arilo C6-C10) con por lo menos un anillo carbocíclico que presenta un sistema de electrones pi conjugados que es aromático (p.ej., que presenta 10 o 14 electrones n compartidos en una matriz cíclica) (p.ej., fenilo, fluorenilo y naftilo). En una realización, los radicales bivalentes formados a partir de derivados de benceno sustituidos y que presentan las valencias libres en átomos anulares se denominan radicales fenileno sustituidos. En otras realizaciones, los radicales bivalentes derivados de radicales hidrocarburo monocíclicos o policíclicos univalentes cuyos nombres terminan en "-ilo" por eliminación de un átomo de hidrógeno de un átomo de carbono con la valencia libre se nombran mediante la adición de "-ideno" al nombre del radical univalente correspondiente, p.ej., un grupo naftilo con dos puntos de unión se denomina naftilideno. En donde se utiliza en la presente memoria, un intervalo numérico tal como "arilo 6 a 10" se refiere a cada número entero en el intervalo dado; p.ej., "6 a 10 átomos anulares" significa que el grupo arilo puede consistir en 6 átomos anulares, 7 átomos anulares, etc., hasta, e incluyendo, 10 átomos anulares. El término incluye grupos monocíclicos o policíclicos de anillos fusionados. A menos que se indique lo contrario en la especificación, una fracción arilo puede sustituirse opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3, -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2, -N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas
fracciones puede sustituirse opcionalmente tal como se define en la presente memoria. En una realización, a menos que se indique lo contrario, "arilo" también incluye sistemas anulares en los que el anillo arilo, tal como se ha definido anteriormente, está fusionado con uno o más grupos cicloalquilo o heterociclilo, en los que el punto de unión a la estructura molecular parental se encuentra en el anillo arilo.
"Aralquilo" o "arilalquilo" se refiere a un radical -(aril)alquilo en el que arilo y alquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para arilo y alquilo, respectivamente. El "aralquilo" o "arilalquilo" se unen a la estructura molecular parental mediante el grupo alquilo. Los términos "aralquenilo/arilalquenilo" y "aralquinilo/arilalquinilo" reflejan la descripción anterior de "aralquilo/arilalquilo", en la que el término "alquilo" se sustituye por "alquenilo" o "alquinilo", respectivamente, y los términos "alquenilo" o "alquinilo" son tal como se indica en la presente memoria.
"Azido" se refiere al radical -N3.
"Carbamato" se refiere a cualquiera de los radicales siguientes: -O-(C=O)-N(Rb)-, -O-(C=O)-N(Rb)2, -N(Rb)-(C=O)-O-y -N(Rb)-(C=O)-ORb, en los que cada Rb se selecciona independientemente de entre H, alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en las que cada una de las fracciones puede ella misma estar opcionalmente sustituida tal como se indica en la presente memoria.
El término "carbonato" se refiere a un radical -O-(C=O)-O- o -O-(C=O)-OR, en el que R puede ser hidrógeno, alquilo, alquenilo, alquinilo, heteroalquilo, heteroalquenilo, heteroalquinilo, arilo, ciclohexilo, heteroarilo o heterocicloalquilo, que son tal como se indica en la presente memoria.
"Carbonilo" se refiere a un radical -(C=O)-.
"Carboxaldehído" se refiere a un radical -(C=O)H.
"Carboxilo" se refiere a un radical -(C=O)OH.
"Ciano" se refiere a un radical -CN.
"Cicloalquilo", o alternativamente, "carbociclilo", se refiere a un radical monocíclico o policíclico que contiene únicamente carbono e hidrógeno, y puede estar saturado o parcialmente insaturado. Los grupos cicloalquilo parcialmente insaturados pueden denominarse "cicloalquenilo" en el caso de que el carbociclo contenga por lo menos un doble enlace, o "cicloalquinilo" en el caso de que el carbociclo contenga por lo menos un triple enlace. Entre los grupos cicloalquilo se incluyen grupos con 3 a 10 átomos anulares (p.ej., cicloalquilo C3-C10). En donde se utiliza en la presente memoria, un intervalo numérico tal como "3 a 10" se refiere a cada número entero en el intervalo dado; p.ej., "3 a 10 átomos de carbono" significa que el grupo cicloalquilo puede consistir en 3 átomos de carbono, 4 átomos de carbono, 5 átomos de carbono, etc., hasta, e incluyendo, 10 átomos de carbono. El término "cicloalquilo" incluye además estructuras cíclicas puenteadas y fusionadas espiro que no contienen heteroátomos. El término incluye además grupos monocíclicos o policíclicos de anillos fusionados (es decir, anillos que comparten parejas contiguas de átomos anulares). En algunas realizaciones, es un radical cicloalquilo C3-C8. En algunas realizaciones, es un radical cicloalquilo C3-C5. Entre los ejemplos ilustrativos de grupos cicloalquilo se incluyen, aunque sin limitarse a ellas, las fracciones siguientes: entre los grupos carbociclilo C3-6 se incluyen, aunque sin limitarse a ellos, ciclopropilo (C3), ciclobutilo (C4), ciclopentilo (C5), ciclopentenilo (C5), ciclohexilo (Ce), ciclohexenilo (Ce), ciclohexadienilo (Ce) y similares. Entre los grupos carbociclilo C3-8 se incluyen los grupos carbociclilo C3-6 anteriormente indicados, así como cicloheptilo (C7), cicloheptadienilo (C7), cicloheptatrienilo (C7), ciclooctilo (C8), biciclo[2.2.1]heptanilo, biciclo[2.2.2]octanilo, y similares. Entre los ejemplos de grupos carbociclilo C3-10 se incluyen los grupos carbociclilo C3-8 anteriormente indicados, así como octahidro-1H-indenilo, decahidronaftalenilo, espiro[4.5]decanilo, y similares. A menos que se indique lo contrario en la especificación, un grupo cicloalquilo se sustituye opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3 , -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2, -N(Ra)C(NRa)N(Ra)2 , -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=o)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria. En una realización, a menos que se indique lo contrario, "cicloalquilo" o "carbociclilo" también incluye sistemas anulares en los que el anillo cicloalquilo o carbociclilo, tal como se ha definido anteriormente, está fusionado con uno o más grupos arilo o heteroarilo, en los que el punto de unión a la estructura molecular parental se encuentra en el anillo cicloalquilo o carbociclilo.
"Cidoalquilalquilo" se refiere a un radical -(cidoalquil)alquilo en el que cicloalquilo y alquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para cicloalquilo y alquilo, respectivamente. El "cicloalquil-alquilo" se une a la estructura molecular parental mediante el grupo cicloalquilo. Los términos "cicloalquilalquenilo" y "cicloalquilalquinilo" reflejan la descripción anterior de "cicloalquilalquilo", en la que el término "alquilo" se sustituye por "alquenilo" o "alquinilo", respectivamente, y "alquenilo" o "alquinilo" son tal como se indica en la presente memoria.
"Cicloalquil-heterocicloalquilo" se refiere a un radical -(cicloalquil)heterociclilalquilo en el que cicloalquilo y heterocicloalquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heterocicloalquilo y cicloalquilo, respectivamente. El "cicloalquil-heterocicloalquilo" se une a la estructura molecular parental mediante el grupo cicloalquilo.
"Cicloalquilheteroarilo" se refiere a un radical -(cicloalquil)heteroarilo en el que cicloalquilo y heteroarilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heteroarilo y cicloalquilo, respectivamente. El "cicloalquilheteroarilo" se une a la estructura molecular parental mediante el grupo cicloalquilo.
Tal como se utiliza en la presente memoria, un "enlace covalente" o "enlace directo" se refiere a un único enlace que une dos grupos.
"Éster" se refiere a un radical de fórmula -COOR, en la que R se selecciona de entre alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (nido mediante un carbono de cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo. Cualquier cadena lateral de amina, hidroxi o carboxilo en los compuestos indicados en la presente memoria puede esterificarse. Los procedimientos y grupos específicos para preparar dichos ésteres son conocidos por el experto en la materia y pueden encontrarse fácilmente en fuentes de referencia, tales como Greene y Wuts, Protective Groups in Organic Synthesis, 4a ed., John Wiley & Sons, New York, NY, 2006. A menos que se indique lo contrario en la especificación, un grupo éster se sustituye opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)s, -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2 , -C(O)N(Ra)2 , -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2 , -N(Ra)C(NRa)N(Ra)2 , -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria.
"Éter" se refiere a un radical -Rb-O-Rb, en el que cada Rb se selecciona independientemente de entre alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria.
"Halo", "haluro", o alternativamente, "halógeno" se refiere a flúor, cloro, bromo o yodo. Los términos "haloalquilo", "haloalquenilo", "haloalquinilo" y "haloalcoxi" incluyen estructuras de alquilo, alquenilo, alquinilo y alcoxi que se sustituyen con uno o más grupos halo o con combinaciones de los mismos. Por ejemplo, los términos "fluoroalquilo" y "fluoroalcoxi" incluyen grupos haloalquilo y haloalcoxi, respectivamente, en los que el halo es flúor, tal como, aunque sin limitación, trifluorometilo, difluorometilo, 2,2,2-trifluoroetilo, 1-fluorometil-2-fluoroetilo y similares. Cada uno de los grupos alquilo, alquenilo, alquinilo y alcoxi son tal como se define en la presente memoria y puede sustituirse opcionalmente de manera adicional tal como se define en la presente memoria.
"Heteroalquilo", "heteroalquenilo" y "heteroalquinilo" incluyen radicales alquilo, alquenilo y alquinilo, respectivamente, que presentan uno o más átomos de cadena de esqueleto seleccionados de un átomo diferente de carbono, p.ej., oxígeno, nitrógeno, azufre y fósforo, o combinaciones de los mismos. Puede proporcionarse un intervalo numérico, p.ej., heteroalquilo C1-C4, que se refiere a la longitud total de la cadena, que en el presente ejemplo puede ser de hasta 4 átomos de longitud. Por ejemplo, un radical -CH2OCH2CH3 se denomina heteroalquilo "C4", que incluye el centro heteroátomo en la descripción de la longitud de cadena de átomos. La conexión a la estructura molecular parental puede ser mediante un heteroátomo o un carbono en la cadena heteroalquilo. Por ejemplo, una fracción heteroalquilo que contiene N se refiere a un grupo en el que por lo menos uno de los átomos del esqueleto es un átomo de nitrógeno. Uno o más heteroátomos en el radical heteroalquilo pueden oxidarse opcionalmente. Uno o más átomos de nitrógeno, en caso de estar presentes, también pueden cuaternizarse opcionalmente. Por ejemplo, heteroalquilo incluye además cadenas de esqueleto sustituidas con uno o más sustituyentes de óxido de nitrógeno (
O-). Entre los grupos heteroalquilo ejemplares se incluyen, aunque sin limitarse a ellos, éteres, tales como metoxietanilo (-CH2CH2OCH3), etoximetanilo (-CH2OCH2CH3), (metoximetoxi)etanilo (-CH2CH2-OCH2OCH3), (metoximetoxi)metanilo (-CH2OCH2OCH3) y (metoxietoxi)metanilo (-CH2OCH2CH2OCH3) y similares; aminas, tales como -CH2CH2NHCH3 , -CH2CH2N(CH3)2, -CH2NHCH2CH3 , -CH2N(CH2CH3)(CH3), y similares. Cada uno de los grupos heteroalquilo, heteroalquenilo y heteroalquinilo puede sustituirse opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3, -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2 , -C(O)N(Ra)2 , -N(R N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2 , -N(Ra)C(NRa)N(Ra)2 , -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es
1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria.
"Heteroalquil-arilo" se refiere a un radical -(heteroalquil)arilo en el que heteroarilo y arilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heteroarilo y arilo, respectivamente. El "heteroalquilo-arilo" se une a la estructura molecular parental mediante un átomo del grupo heteroalquilo.
"Heteroalquil-heteroarilo" se refiere a un radical -(heteroalquil)heteroarilo en el que heteroalquilo y heteroarilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heteroalquilo y heteroarilo, respectivamente. El "heteroalquiloheteroarilo" se une a la estructura molecular parental mediante un átomo del grupo heteroalquilo.
"Heteroalquil-heterocicloalquilo" se refiere a un radical -(heteroalquil)heterocicloalquilo en el que heteroalquilo y heterocicloalquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heteroalquilo y heterocicloalquilo, respectivamente. El "heteroalquilo-heterocicloalquilo" se une a la estructura molecular parental mediante un átomo del grupo heteroalquilo.
"Heteroalquil-cicloalquilo" se refiere a un radical -(heteroalquil)cicloalquilo en el que heteroalquilo y cicloalquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heteroalquilo y cicloalquilo, respectivamente. El "heteroalquilocicloalquilo" se une a la estructura molecular parental mediante un átomo del grupo heteroalquilo.
"Heteroarilo", o alternativamente, "heteroaromático" se refiere a un radical de un sistema de anillos aromáticos monocíclico o policíclico (p.ej., bicíclico o tricíclico) de 5 a 18 elementos (p.ej., que presenta 6, 10 o 14 electrones n compartidos en una matriz cíclica) que presentan átomos de carbono anular y 1 a 6 heteroátomos anulares proporcionados en el sistema de anillos aromáticos, en el que cada heteroátomo se selecciona independientemente de nitrógeno, oxígeno, fósforo y azufre ("heteroarilo de 5 a 18 elementos"). Entre los sistemas de anillos policíclicos heteroarilo se pueden incluir uno o más heteroátomos en uno o más anillos. En donde se utiliza en la presente memoria, un intervalo numérico tal como "arilo 5 a 18" se refiere a cada número entero en el intervalo dado; p.ej., "5 a 18 átomos anulares" significa que el grupo heteroarilo puede consistir en 5 átomos anulares, 6 átomos anulares, 7 átomos anulares, 8 átomos anulares, 9 átomos anulares, 10 átomos anulares, etc., hasta, e incluyendo, 18 átomos anulares. En una realización, los radicales bivalentes derivados de radicales heteroarilo univalentes cuyos nombres terminan en "-ilo" por eliminación de un átomo de hidrógeno de un átomo con la valencia libre se nombran mediante la adición de "-ideno" al nombre del radical univalente correspondiente, p.ej., un grupo naftilo con dos puntos de unión es un piridilideno.
Por ejemplo, una fracción "heteroaromático" o "heteroarilo" que contiene N se refiere a un grupo aromático en el que por lo menos uno de los átomos del esqueleto del anillo es un átomo de nitrógeno. Uno o más heteroátomos en el radical heteroarilo pueden oxidarse opcionalmente. Uno o más átomos de nitrógeno, en caso de estar presentes, también pueden cuaternizarse opcionalmente. El término heteroarilo incluye además sistemas anulares sustituidos con uno o más sustituyentes de óxido de nitrógeno (-O-), tales como N-óxidos de piridinilo. El heteroarilo se une a la estructura molecular parental mediante cualquier átomo del anillo o anillos.
"Heteroarilo" incluye además sistemas anulares en los que el anillo heteroarilo, tal como se ha definido anteriormente, se fusiona con uno o más grupos arilo en los que el punto de unión a la estructura molecular parental se encuentra en el anillo arilo o heteroarilo, o en el que el anillo heteroarilo, tal como se ha definido anteriormente, se fusiona con uno o más grupos cicloalquilo o heterociclilo en el que el punto de unión a la estructura molecular parental se encuentra en el anillo heteroarilo. Para los grupos heteroarilo policíclicos en los que un anillo no contiene un heteroátomo (p.ej., indolilo, quinolinilo, carbazolilo y similares), el punto de unión a la estructura molecular parental puede encontrarse en el anillo portador de un heteroátomo (p.ej., 2-indolilo) o en el anillo que no contiene un heteroátomo (p.ej., 5-indolilo).
En algunas realizaciones, un grupo heteroarilo es un sistema de anillos aromáticos de 5 a 10 elementos que presentan
átomos de carbono anular y 1 a 4 heteroátomos anulares proporcionados en el sistema de anillos aromáticos, en el que cada heteroátomo se selecciona independientemente de entre nitrógeno, oxígeno, fósforo y azufre ("heteroarilo de 5 a 10 elementos"). En algunas realizaciones, un grupo heteroarilo es un sistema de anillos aromáticos de 5 a 8 elementos que presentan átomos de carbono anular y 1 a 4 heteroátomos anulares proporcionados en el sistema de anillos aromáticos, en el que cada heteroátomo se selecciona independientemente de entre nitrógeno, oxígeno, fósforo y azufre ("heteroarilo de 5 a 8 elementos"). En algunas realizaciones, un grupo heteroarilo es un sistema de anillos aromáticos de 5 a 6 elementos que presentan átomos de carbono anular y 1 a 4 heteroátomos anulares proporcionados en el sistema de anillos aromáticos, en el que cada heteroátomo se selecciona independientemente de entre nitrógeno, oxígeno, fósforo y azufre ("heteroarilo de 5 a 6 elementos"). En algunas realizaciones, el heteroarilo de 5 a 6 elementos presenta 1 a 3 heteroátomos anulares seleccionados independientemente de entre nitrógeno, oxígeno, fósforo y azufre. En algunas realizaciones, el heteroarilo de 5 a 6 elementos presenta 1 a 2 heteroátomos anulares seleccionados independientemente de entre nitrógeno, oxígeno, fósforo y azufre. En algunas realizaciones, el heteroarilo de 5 a 6 elementos presenta 1 heteroátomo anular seleccionado de entre nitrógeno, oxígeno, fósforo y azufre.
Entre los ejemplos de heteroarilo se incluyen, aunque sin limitarse a ellos, azepinilo, acridinilo, bencimidazolilo, bencindolilo, 1,3-benzodioxolilo, benzofuranilo, benzooxazolilo, benzo[d]tiazolilo, benzotiadiazolilo, benzo[b][1,4]dioxepinilo, benzo[b][1,4]oxazinilo, 1,4-benzodioxanilo, benzonaftofuranilo, benzoxazolilo, benzodioxolilo, benzodioxinilo, benzoxazolilo, benzopiranilo, benzopiranonilo, benzofuranilo, benzofuranonilo, benzofurazanilo, benzotiazolilo, benzotienilo (benzotiofenilo), benzotieno[3,2-d]pirimidinilo, benzotriazolilo, benzo[4,6]imidazo[1,2-a]piridinilo, carbazolilo, cinolinilo, ciclopenta[d]pirimidinilo, 6,7-dihidro-5H-ciclopenta[4,5]tieno[2,3-d]pirimidinilo, 5,6-dihidrobenzo[h]quinazolinilo, 5,6-dihidrobenzo[h]cinolinilo, 6,7-dihidro-5H-benzo[6,7]ciclohepta[1,2-c]piridazinilo, dibenzofuranilo, dibenzotiofenilo, furanilo, furazanilo, furanonilo, furo[3,2-c]piridinilo, 5,6,7,8,9,10-hexahidrocicloocta[d]pirimidinilo, 5,6,7,8,9,10-hexahidrocicloocta[d]piridazinilo, 5,6,7,8,9,10-hexahidrocicloocta[d]piridinilo,isotiazolilo, imidazolilo, indazolilo, indolilo, indazolilo, isoindolilo, indolinilo, isoindolinilo, isoquinolilo, indolizinilo, isoxazolilo, 5,8-metano-5,6,7,8-tetrahidroquinazolinilo, naftiridinilo, 1,6-naftiridinonilo, oxadiazolilo, 2-oxoazepinilo, oxazolilo, oxiranilo, 5,6,6a,7,8,9,10,10a-octahidrobenzo[h]quinazolinilo, 1-fenil-1H-pirrolilo, fenazinilo, fenotiazinilo, fenoxazinilo, ftalazinilo, pteridinilo, purinilo, piranilo, pirrolilo, pirazolilo, pirazolo[3,4-d]pirimidinilo, piridinilo, pirido[3,2-d]pirimidinilo, pirido[3,4-d]pirimidinilo, pirazinilo, pirimidinilo, piridazinilo, pirrolilo, quinazolinilo, quinoxalinilo, quinolinilo, isoquinolinilo, tetrahidroquinolinilo, 5,6,7,8-tetrahidroquinazolinilo, 5,6,7,8-tetrahidrobenzo[4,5]tieno[2,3-d]pirimidinilo, 6,7,8,9-tetrahidro-5H-ciclohepta[4,5]tieno[2,3-d]pirimidinilo, 5,6,7,8-tetrahidropirido[4,5-c]piridazinilo, tiazolilo, tiadiazolilo, tiapiranilo, triazolilo, tetrazolilo, triazinilo, tieno[2,3-d]pirimidinilo, tieno[3,2-d]pirimidinilo, tieno[2,3-c]pridinilo y tiofenilo (es decir, tienilo).
A menos que se indique lo contrario en la especificación, un grupo heteroarilo se sustituye opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)a, -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2, -N(Ra)C(NRa)N(Ra)2 , -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=o)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria.
"Heteroaril-alquilo" se refiere a un radical -(heteroaril)alquilo en el que heteroarilo y alquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heteroarilo y alquilo, respectivamente. El "heteroarilo-alquilo" se une a la estructura molecular parental mediante cualquier átomo del grupo heteroarilo.
"Heteroaril-heterocicloalquilo" se refiere a un radical -(heteroaril)heterocicloalquilo en el que heteroarilo y heterocicloalquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heteroalquilo y heterocicloalquilo, respectivamente. El "heteroarilo-heterocicloalquilo" se une a la estructura molecular parental mediante un átomo del grupo heteroarilo.
"Heteroaril-cicloalquilo" se refiere a un radical -(heteroaril)cicloalquilo en el que heteroarilo y cicloalquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heteroarilo y cicloalquilo, respectivamente. El "heteroarilo-cicloalquilo" se une a la estructura molecular parental mediante un átomo de carbono del grupo heteroarilo.
"Heterociclilo", "heterocicloalquilo" y "heterocarbociclilo" se refiere a cualquier fracción monocíclica o policíclica radical no aromática de 3 a 18 elementos que comprende por lo menos un heteroátomo anular seleccionado de entre nitrógeno, oxígeno, fósforo y azufre. Un grupo heterocicliilo puede ser un sistema de anillos monocíclico, bicíclico, tricíclico o tetracíclico, en el que los sistemas de anillos policíclicos pueden ser un sistema de anillos fusionados,
puenteados o espiro. Los sistemas de anillos policíclicos heterociclilo pueden incluir uno o más heteroátomos en uno o más anillos. Un grupo heterociclilo puede estar saturado o parcialmente insaturado. Los grupos heterocicloalquilo parcialmente insaturados pueden denominarse "heterocicloalquenilo" en el caso de que el heterociclilo contenga por lo menos un doble enlace, o "heterocicloalquinilo" en el caso de que el heterociclilo contenga por lo menos un triple enlace. En donde se utiliza en la presente memoria, un intervalo numérico tal como "5 a 18" se refiere a cada número entero en el intervalo dado; p.ej., "5 a 18 átomos anulares" significa que el grupo heterociclilo puede consistir en 5 átomos anulares, 6 átomos anulares, 7 átomos anulares, 8 átomos anulares, 9 átomos anulares, 10 átomos anulares, etc., hasta, e incluyendo, 18 átomos anulares. En una realización, los radicales bivalentes derivados de radicales heterociclilo univalentes cuyos nombres terminan en "-ilo" por eliminación de un átomo de hidrógeno de un átomo con la valencia libre se nombran mediante la adición de "-ideno" al nombre del radical univalente correspondiente, p.ej., un grupo piperidilo con dos puntos de unión es un piperidilideno.
Una fracción heterociclilo que contiene N se refiere a un grupo no aromático en el que por lo menos uno de los átomos anulares es un átomo de nitrógeno. Uno o más heteroátomos en el radical heterociclilo pueden oxidarse opcionalmente. Uno o más átomos de nitrógeno, en caso de estar presentes, pueden estar cuaternizados opcionalmente. El término heterociclilo incluye además sistemas anulares sustituidos con uno o más sustituyentes de óxido de nitrógeno (-O-), tales como N-óxidos de piperidinilo. El heterociclilo se une a la estructura molecular parental mediante cualquier átomo de cualquiera anillo o anillos.
"Heterociclilo" incluye además sistemas anulares en los que el anillo heterociclilo, tal como se ha definido anteriormente, se fusiona con uno o más grupos carbociclilo en los que el punto de unión se encuentra en el anillo carbociclilo o heterociclilo, en el que el anillo heterociclilo, tal como se ha definido anteriormente, se fusiona con uno 0 más grupos arilo o heteroarilo, en el que el punto de unión a la estructura molecular parental se encuentra en el anillo heterociclilo. En algunas realizaciones, un grupo heterociclilo es un sistema de anillos no aromáticos de 3 a 10 elementos que presenta átomos de carbono anular y 1 a 4 heteroátomos anulares, en el que cada heteroátomo se selecciona independientemente de entre nitrógeno, oxígeno, fósforo y azufre («heterociclilo de 3 a 10 elementos»). En algunas realizaciones, un grupo heterociclilo es un sistema de anillos no aromáticos de 5 a 8 elementos que presenta átomos de carbono anular y 1 a 4 heteroátomos anulares, en el que cada heteroátomo se selecciona independientemente de entre nitrógeno, oxígeno, fósforo y azufre ("heterociclilo de 5 a 8 elementos"). En algunas realizaciones, un grupo heterociclilo es un sistema de anillos no aromáticos de 5 a 6 elementos que presenta átomos de carbono anular y 1 a 4 heteroátomos anulares, en el que cada heteroátomo se selecciona independientemente de entre nitrógeno, oxígeno, fósforo y azufre ("heterociclilo de 5 a 6 elementos"). En algunas realizaciones, el heterociclilo de 5 a 6 elementos presenta 1 a 3 heteroátomos anulares seleccionados independientemente de entre nitrógeno, oxígeno, fósforo y azufre. En algunas realizaciones, el heterociclilo de 5 a 6 elementos presenta 1 a 2 heteroátomos anulares seleccionados independientemente de entre nitrógeno, oxígeno, fósforo y azufre. En algunas realizaciones, el heterociclilo de 5 a 6 elementos presenta 1 heteroátomo anular seleccionado de entre nitrógeno, oxígeno, fósforo y azufre.
Entre los grupos heterociclilo de 3 elementos ejemplares que contienen 1 heteroátomo se incluyen, aunque sin limitarse a ellos, aziridinilo, oxiranilo y tiorenilo. Entre los grupos heterociclilo de 4 elementos ejemplares que contienen 1 heteroátomo se incluyen, aunque sin limitarse a ellos, azetidinilo, oxetanilo y tietanilo. Entre los grupos heterociclilo de 5 elementos ejemplares que contienen 1 heteroátomo se incluyen, aunque sin limitarse a ellos, tetrahidrofuranilo, dihidrofuranilo, tetrahidrotiofenilo, dihidrotiofenilo, pirrolidinilo, dihidropirrolilo y pirrolil-2,5-diona. Entre los grupos heterociclilo de 5 elementos ejemplares que contienen 2 heteroátomos se incluyen, aunque sin limitarse a ellos, dioxolanilo, oxatiolanilo y ditiolanilo. Entre los heterociclilos de 5 elementos ejemplares que contienen 3 heteroátomos se incluyen, aunque sin limitarse a ellos, triazolinilo, oxadiazolinilo y tiadiazolinilo. Entre los grupos heterociclilo de 6 elementos ejemplares que contienen 1 heteroátomo se incluyen, aunque sin limitarse a ellos, piperidinilo, tetrahidropiranilo, dihidropiridinilo y tianilo. Entre los grupos heterociclilo de 6 elementos ejemplares que contiene 2 heteroátomos se incluyen, aunque sin limitarse a ellos, piperazinilo, morfolinilo, ditianilo, dioxanilo y triazinanilo. Entre los grupos heterociclilo de 7 elementos ejemplares que contienen 1 heteroátomo se incluyen, aunque sin limitarse a ellos, azepanilo, oxepanilo y tiepanilo. Entre los grupos heterociclilo de 8 elementos ejemplares que contienen 1 heteroátomo se incluyen, aunque sin limitarse a ellos, azocanilo, oxecanilo y tiocanilo. Entre los grupos heterociclilo bicíclicos ejemplares se incluyen, aunque sin limitarse a ellos, indolinilo, isoindolinilo, dihidrobenzofuranilo, dihidrobenzotienilo, tetrahidrobenzotienilo, tetrahidrobenzofuranilo, tetrahidroindolilo, tetrahidroquinolinilo, tetrahidroisoquinolinilo, decahidroquinolinilo, decahidroisoquinolinilo, octahidrocromenilo, octahidroisocromenilo, decahidronaftiridinilo, decahidro-1,8-naftiridinilo, octahidropirrolo[3,2-b]pirrol, indolinilo, ftalimidilo, naftalimidilo, cromanilo, cromenilo, 1H-benzo[e][1,4]diazepinilo, 1,4,5,7-tetrahidropirano[3,4-b]pirrolilo, 5,6-dihidro-4H-furo[3,2-b]pirrolilo, 6,7-dihidro-5H-furo[3,2-b]piranilo, 5,7-dihidro-4H-tieno[2,3-c]piranilo, 2,3-dihidro-1H-pirrolo[2,3-b]piridinilo, 2,3-dihidrofuro[2,3-b]piridinilo, 4,5,6,7-tetrahidro-1H-pirrolo[2,3-b]piridinilo, 4,5,6,7-tetrahidrofuro[3,2-c]piridinilo, 4,5,6,7-tetrahidro-tieno[3,2-b]piridinilo, 1,2,3,4-tetrahidro-1,6-naftiridinilo, y similares.
A menos que se indique lo contrario, las fracciones heterociclilo se sustituyen opcionalmente con uno o más sustituyentes entre los que se incluyen independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, amidino, imino, azida, carbonato, carbamato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, éter, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo,
sulfoxilo, sulfonato, urea, -Si(Ra)s, -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2 , -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2, -N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2), o -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria.
"Heterociclil-alquilo" se refiere a un radical -(heterociclil)alquilo en el que heterociclilo y alquilo son tal como se da a conocer en la presente memoria y que se sustituyen opcionalmente con uno o más de los sustituyentes indicados como sustituyentes adecuados para heterociclilo y alquilo, respectivamente. El "heterociclilo-alquilo" se une a la estructura molecular parental mediante cualquier átomo del grupo heterociclilo. Los términos "heterociclil-alquenilo" y "heterociclil-alquinilo" reflejan la descripción anterior de "heterociclil-alquilo", en la que el término "alquilo" se sustituye por "alquenilo" o "alquinilo", respectivamente, y "alquenilo" o "alquinilo" son tal como se indica en la presente memoria.
"Imino" se refiere a un radical "-C(=N-Rb)-Rb" en el que cada Rb se selecciona independientemente de entre hidrógeno, alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria.
"Fracción" se refiere a un segmento o grupo funcional específico de una molécula. Las fracciones químicas con frecuencia son entidades químicas reconocidas, incluidas o colgantes de una molécula.
"Nitro" se refiere al radical -NO2.
"Oxa" se refiere a un radical -O-.
"Oxo" se refiere a un radical =O.
"Fosfato" se refiere a un radical -O-P(=O)(ORb)2 en el que cada Rb se selecciona independientemente de entre hidrógeno, alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria. En algunas realizaciones, en el caso de que Ra sea hidrógeno y según el pH, el hidrógeno puede sustituirse por un contraión apropiadamente cargado.
"Fosfonato" se refiere a un radical -O-P(=O)(Rb)(ORb) en el que cada Rb se selecciona independientemente de entre hidrógeno, alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria. En algunas realizaciones, en el caso de que Ra sea hidrógeno y según el pH, el hidrógeno puede sustituirse por un contraión apropiadamente cargado.
"Fosfinato" se refiere a un radical -P(=O)(Rb)(ORb) en el que cada Rb se selecciona independientemente de entre hidrógeno, alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria. En algunas realizaciones, en el caso de que Ra sea hidrógeno y según el pH, el hidrógeno puede sustituirse por un contraión apropiadamente cargado.
Un "grupo o átomo saliente" es cualquier grupo o átomo que se escindirá, bajo las condiciones de reacción, respecto del material de partida, estimulando de esta manera la reacción en un sitio especificado. Entre los ejemplos no limitativos adecuados de dichos grupos, a menos que se especifique lo contrario, se incluyen átomos de halógeno, y los grupos mesiloxi, p-nitrobencenosulfoniloxi, trifluorometiloxi y tosiloxi.
"Grupo protector" presenta el significado asociado convencionalmente al mismo en la síntesis orgánica, p.ej., un grupo que bloquea selectivamente uno o más sitios reactivos en un compuesto multifuncional, de manera que puede llevarse a cabo una reacción química selectivamente en otro sitio reactivo no protegido y de manera que el grupo puede eliminarse fácilmente después de completar la reacción selectiva. Se da a conocer una diversidad de grupos protectores en, por ejemplo, T.H. Greene y P. G. M. Wuts, Protective Groups in Organic Synthesis, Cuarta edición, John Wiley & Sons, New York (2006). Por ejemplo, una forma protegida con hidroxi es una forma en que por lo menos
uno de los grupos hidroxi presentes en el compuesto está protegido con un grupo protector de hidroxi. De manera similar, pueden protegerse de manera similar aminas y otros grupos reactivos.
Tal como se utiliza en la presente memoria, los términos "sustituido" o "sustitución" se refieren a que por lo menos un hidrógeno presente en un átomo de grupo (p.ej., un átomo de carbono o nitrógeno) se sustituye por un sustituyente permisible, p.ej., un sustituyente que, tras la sustitución del hidrógeno, resulta en un compuesto estable, p.ej., un compuesto que no experimente espontáneamente una transformación, tal como la reorganización, ciclización, eliminación u otra reacción. A menos que se indique lo contrario, un grupo "sustituido" presenta un sustituyente en una o más posiciones sustituibles del grupo y, en el caso de que se sustituya más de una posición en cualquier estructura dada, el sustituyente es el mismo o uno diferente en cada posición. Entre los sustituyentes pueden incluirse uno o más grupos seleccionados individual e independientemente: acilo, alquilo, alquenilo, alquinilo, alcoxi, alquilarilo, cicloalquilo, aralquilo, arilo, ariloxi, amino, amido, azida, carbonato, carbonilo, heteroalquilo, heteroarilo, heteroarilalquilo, heterocicloalquilo, hidroxi, ciano, halo, haloalcoxi, haloalquilo, éster, mercapto, tio, alquiltio, ariltio, tiocarbonilo, nitro, oxo, fosfato, fosfonato, fosfinato, sililo, sulfinilo, sulfonilo, sulfonamidilo, sulfoxilo, sulfonato, urea, -Si(Ra)3, -ORa, - SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O) Ra, -N(Ra)C(O)N(Ra)2 , -N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (en el que t es 1 o 2), -S(O)tORa (en el que t es 1 o 2), -S(O)tN(Ra)2 (en el que t es 1 o 2) y -O-P(=O)(ORa)2, en el que cada Ra es, independientemente, hidrógeno, alquilo, haloalquilo, carbociclilo, carbociclilalquilo, arilo, aralquilo, heterocicloalquilo, heterocicloalquilalquilo, heteroarilo o heteroarilalquilo, y cada una de dichas fracciones puede sustituirse opcionalmente tal como se define en la presente memoria. Por ejemplo, un sustituyente cicloalquilo puede presentar un haluro sustituido en uno o más carbonos anulares, y similares. Los grupos protectores que pueden formar los derivados protectores de los sustituyentes anteriormente indicados son conocidos por el experto en la materia y pueden encontrarse en referencias, tales como Greene y Wuts, anteriormente.
"Sililo" se refiere a un radical -Si(Rb)3 en el que cada Rb se selecciona independientemente de entre alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria.
"Sulfanilo", "sulfuro" y "tio" se refiere cada uno a un radical -S-Rb, en el que Rb se selecciona de entre alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria. Por ejemplo, un "alquiltio" se refiere al radical "alquil-S-" y "ariltio" se refiere al radical "aril-S-", cada uno de los cuales está unido al grupo molecular parental mediante el átomo S. Cada uno de los términos "sulfuro", "tiol", "mercapto" y "mercaptano" también se refiere al grupo -RbSH.
"Sulfinilo" o "sulfóxido" se refiere a un radical -S(O)-Rb, en el que para "sulfInilo", Rb es H, y para "sulfóxido", Rb se selecciona de entre alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria.
"Sulfonilo" o "sulfona" se refiere a un radical -S(O2)-Rb en el que Rb se selecciona de entre hidrógeno, alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria.
"Suflonamidilo" o "sulfonamido" se refiere a los radicales siguientes: -S(=O)2-N(Rb)2 , -N(Rb)-S(=O)2-Rb, -S(=O)2-N(Rb)-o -N(Rb)-S(=O)2-, en los que cada Rb se selecciona independientemente de entre hidrógeno, alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, cada fracción de los cuales puede sustituirse opcionalmente tal como se indica en la presente memoria. Los grupos Rb en -S(=O)2-N(Rb)2 o - N(Rb)-S(=O)2-Rb pueden utilizarse junto con el nitrógeno al que se encuentran unidos para formar un anillo heterociclilo de 4, 5, 6, 7 o 8 elementos. En algunas realizaciones, el término designa un sulfonamido C1-C4 , en el que cada Rb en el sulfonamido contiene 1 carbono, 2 carbonos, 3 carbonos o 4 carbonos en total.
"Sulfoxilo" se refiere a un radical -S(=O)2OH.
"Sulfonato" se refiere a un radical -S(=O)2-ORb en el que Rb se selecciona de entre alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de la cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo,
heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalquilo, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede sustituirse opcionalmente tal como se indica en la presente memoria.
"Tiocarbonilo" se refiere a un radical -(C=S)-.
"Urea" se refiere a un radical -N(Rb)-(C=O)-N(Rb)2 o -N(Rb)-(C=O)-N(Rb)-, en los que cada Rb se seleccionados independientemente de entre hidrógeno, alquilo, alquenilo, alquinilo, haloalquilo, heteroalquilo (unido mediante un carbono de cadena), cicloalquilo, cicloalquilalquilo, arilo, aralquilo, heterocicloalquilo (unido mediante un carbono anular), heterocicloalquilalqulio, heteroarilo (unido mediante un carbono anular) y heteroarilalquilo, a menos que se indique lo contrario en la especificación, en los que cada fracción puede ella misma sustituirse opcionalmente tal como se indica en la presente memoria.
En donde se especifican grupos sustituyentes mediante sus fórmulas químicas convencionales, escritas de izquierda a derecha, igualmente comprenden los sustituyentes químicamente idénticos que resultarían de escribir la escritura de derecha a izquierda, p.ej., -CH2O- es equivalente a -OCH2-.
Compuestos
En la presente memoria se proporcionan según la presente invención compuestos de fórmula (I''') o fórmula (A''):

en las que:
R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
z es 0, 1, 2 o 3,
cada aparición de R3a es, independientemente, hidrógeno, alquilo, alquenilo, alquinilo, alcoxilo, halógeno, ciano, amino, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, -CONR4R5, o -Si(R6)3;
en los que R2, R3, R4, R5 y R6, son cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
R1c es hidrógeno, alquilo, alquenilo o alquinilo,
R2c es hidrógeno, alquilo, alquenilo o alquinilo,
Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y
X es CR1a o N,
en la que R1a es hidrógeno, halo, alquilo, alquenilo, alquinilo o CN,
en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), S(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, n H2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2 , S(O)(alquilo), o S(O)2(alquilo),
en la que, en la fórmula (I''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

R1 no es hidrógeno, Si(CH3)3, CH2Si(CH3)3, metilo, (CH2)NH2 , (CH2)2NH2 , (CH2)NHSO2CH3, o (CH2)nNHC(O)R1x;
n es 1 o 2; R1x es metilo, alqueno C2, cyclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en el que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituye opcionalmente con uno o dos grupos seleccionados independientemente de oxo y ciano,
en la que, en la fórmula (A''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

entonces R1 no es fenilo, o una forma farmacéuticamente aceptable del mismo.
En una realización, B es alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3 o -CONR4R5. En una realización, B es alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, o -CONR4R5, en el que los puntos de unión para el heterocicloalquilo y el heteroarilo es un átomo de carbono.
En una realización, R1c es alquilo, alquenilo o alquinilo.
En una realización, R1c es hidrógeno. En una realización, R1c es alquilo. En una realización, R1c es metilo o etilo. En una realización, R1c es metilo. En una realización, R1c es etilo.
En una realización, R2c es hidrógeno.
En una realización, en la fórmula (I''), X es N y R2c es hidrógeno.
En una realización, z es 0. En otra realización, z es 1. En otra realización, z es 2. En otra realización, z es 3.
En una realización, cada caso de R3aes, independientemente, hidrógeno, alquilo o halógeno. En una realización, cada caso de R3a es, independientemente, hidrógeno, metilo, flúor, cloro o bromo.
En determinadas realizaciones,
B es alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3 o -CONR4R5, en el que los puntos de unión para el heterocicloalquilo y el heteroarilo son un átomo de carbono,
en los que R2, R3, R4 y R5 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo.
En determinadas realizaciones, en la presente memoria se proporcionan para la utilización según la presente invención compuestos de fórmula (I') o fórmula (A'):

en las que:
R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6)3,
en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y
X es CR1a o N,
en la que R1a es hidrógeno, halo, alquilo, alquenilo, alquinilo o CN,
en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, haloalquilo, OH, alcoxi, NH2 , NH(alquilo), N(alquilo>2, COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2 , S(O)(alquilo), S(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, haloalquilo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2 , NH(alquilo), N(alquilo>2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo>2, S(O)(alquilo) o S(O>2(alquilo>,
en los que, en la fórmula (I">, en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

R1 no es hidrógeno, Si(CHs>s, CH2Si(CHs>s, metilo, (CH2>NH2 , (CH2>2NH2 , (CH2>NHSO2CHs, o (CH2>nNHC(O)R1x; n es 1 o 2; R1x es metilo, alqueno C2, cyclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en el que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituye opcionalmente con uno o dos grupos seleccionados independientemente de oxo y ciano,
en la que, en la fórmula (A''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

entonces R1 no es fenilo,
o una forma farmacéuticamente aceptable del mismo.
En determinadas realizaciones, en la presente memoria se proporcionan para la utilización según la presente invención compuestos de fórmula (I) o fórmula (A):

en las que:
R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6>3,
en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y
X es CR1a o N,
en la que R1a es hidrógeno, halo, alquilo, alquenilo, alquinilo o CN,
en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo>2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo>2, S(O)(alquilo), s(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo>2 , S(O)(alquilo), o S(O>2(alquilo>,
en los que, en la fórmula (I">, en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:
R1 no es hidrógeno, Si(CHs)3, CH2Si(CHs)3, metilo, (CH2)NH2 , (CH2)2NH2 , (CH2)NHSO2CH3, o (CH2)nNHC(O)R1x; n es 1 o 2; R1x es metilo, alqueno C2, cyclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en el que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituye opcionalmente con uno o dos grupos seleccionados independientemente de oxo y ciano,
en la que, en la fórmula (A''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

entonces R1 no es fenilo,
o una forma farmacéuticamente aceptable del mismo.
En determinadas realizaciones, en la presente memoria se proporciona para la utilización según la presente invención una mezcla de compuestos de fórmula (I"), (I'), (I), (A"), (A') o (A), en las que los compuestos individuales de la mezcla existen predominantemente en una configuración de isómero (S) o (R). Por ejemplo, la mezcla de compuestos puede presentar una pureza de enantiómero (S) superior a aproximadamente 55 %, aproximadamente 60 %, aproximadamente 65 %, aproximadamente 70 %, aproximadamente 75 %, aproximadamente 80 %, aproximadamente 85 %, aproximadamente 90 %, aproximadamente 95 %, aproximadamente 96 %, aproximadamente 97 %, aproximadamente 98 %, aproximadamente 99 %, aproximadamente 99,5 % o superior. En otras realizaciones, la mezcla de compuestos presenta una pureza de enantiómeros (S) de entre aproximadamente 55 % y aproximadamente 99.5 %, de entre aproximadamente 60 % y aproximadamente 99,5 %, de entre aproximadamente 65 % y aproximadamente 99,5 %, de entre aproximadamente 70 % y aproximadamente 99,5 %, de entre aproximadamente 75 % y aproximadamente 99,5 %, de entre aproximadamente 80 % y aproximadamente 99,5 %, de entre aproximadamente 85 % y aproximadamente 99,5 %, de entre aproximadamente 90 % y aproximadamente 99,5 %, de entre aproximadamente 95 % y aproximadamente 99,5 %, de entre aproximadamente 96 % y aproximadamente 99,5 %, de entre aproximadamente 97 % y aproximadamente 99,5 %, de entre aproximadamente 98 % y aproximadamente 99.5 %, o de entre aproximadamente 99 % y aproximadamente 99,5 %, o superior.
En otras realizaciones, la mezcla de compuestos puede presentar una pureza de enantiómero (R) superior a aproximadamente 55 %, aproximadamente 60 %, aproximadamente 65 %, aproximadamente 70 %, aproximadamente 75 %, aproximadamente 80 %, aproximadamente 85 %, aproximadamente 90 %, aproximadamente 95 %, aproximadamente 96 %, aproximadamente 97 %, aproximadamente 98 %, aproximadamente 99 %, aproximadamente 99.5 % o superior. En algunas otras realizaciones, la mezcla de compuestos presenta una pureza de enantiómeros (R) de entre aproximadamente 55 % y aproximadamente 99,5 %, de entre aproximadamente 60 % y aproximadamente 99.5 %, de entre aproximadamente 65 % y aproximadamente 99,5 %, de entre aproximadamente 70 % y aproximadamente 99,5 %, de entre aproximadamente 75 % y aproximadamente 99,5 %, de entre aproximadamente 80 % y aproximadamente 99,5 %, de entre aproximadamente 85 % y aproximadamente 99,5 %, de entre aproximadamente 90 % y aproximadamente 99,5 %, de entre aproximadamente 95 % y aproximadamente 99,5 %, de entre aproximadamente 96 % y aproximadamente 99,5 %, de entre aproximadamente 97 % y aproximadamente 99,5 %, de entre aproximadamente 98 % y aproximadamente 99,5 %, o de entre aproximadamente 99 % y aproximadamente 99,5 %, o superior.
En determinadas realizaciones, en la presente memoria se proporcionan para la utilización según la presente invención compuestos de fórmula (I'):

o una forma farmacéuticamente aceptable de los mismos, en los que R1, B, Wd y X se definen en la presente memoria.
En determinadas realizaciones, en la presente memoria se proporcionan para la utilización según la presente invención compuestos de fórmula (I):

o una forma farmacéuticamente aceptable de los mismos, en los que R1, B, W y X se definen en la presente memoria. En determinadas realizaciones, R1 es alquilo ramificado, arilo de 5 o 6 elementos, heteroarilo de 5 o 6 elementos, cicloalquilo de 5 o 6 elementos o heterocicloalquilo de 5 o 6 elementos,

ciclopropilo o metilo,
en los que RA es OH, alcoxi, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
x es 1,2, 3, 4, 5 o 6,
R7, R8 y R9 son, cada uno independientemente, hidrógeno, OH, alcoxi, NH2, NH(alquilo), N(alquilo)2, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo.
En algunas realizaciones, RA es hidroxilo, alcoxi o heterocicloalquilo. En algunas realizaciones, R7, R8 y R9 son, independientemente, alquilo de 1 a 4 carbonos, amino, hidroxilo o alcoxi de 1 a 4 carbonos.
En algunas realizaciones, R1 es un heteroarilo de 5 a 10 elementos. En determinadas realizaciones, R1 es un heteroarilo de 5 o 6 elementos. En determinadas realizaciones, R1 es un heteroarilo de 6 elementos. En determinadas realizaciones, R1 es un piridinilo. En determinadas realizaciones, R1 es un pirimidinilo. En determinadas realizaciones, R1 es un heteroarilo de 5 elementos. En determinadas realizaciones, R1 es un tiazolilo. En determinadas realizaciones, R1 es un pirazolilo. En determinadas realizaciones, R1 es un imidazolilo. En determinadas realizaciones, el heteroarilo se sustituye con uno o más alquilos.
En algunas realizaciones, R1 es metilo.
En algunas realizaciones, B es fenilo sustituido con 0, 1,2 o 3 apariciones de RZ. En algunas realizaciones, B es fenilo no sustituido. En algunas realizaciones, B es fenilo sustituido con 1 o 2 apariciones de RZ. En algunas realizaciones, B es fenilo sustituido opcionalmente en la posición para con RZ. En algunas realizaciones, B es fenilo monosustituido opcionalmente en la posición meta con RZ. En algunas realizaciones, B es fenilo monosustituido opcionalmente en la posición orto con RZ. En algunas realizaciones, B es fenilo disustituido opcionalmente en la posición meta con RZ. En algunas realizaciones, B es fenilo disustituido opcionalmente en la posición orto con RZ. En algunas realizaciones, B es fenilo disustituido opcionalmente en las posiciones meta y orto con RZ. En algunas realizaciones, B es fenilo disustituido opcionalmente en las posiciones meta y para con RZ. En algunas realizaciones, B es fenilo disustituido opcionalmente en las posiciones orto y para con RZ. En algunas realizaciones, B es fenilo no sustituido en las posiciones orto. En algunas realizaciones, RZ es halo o alquilo. En algunas realizaciones, B es metilo, isopropilo o ciclopropilo. En algunas realizaciones, B es ciclohexilo o alquilo sustituido opcionalmente. En algunas realizaciones, B es arilo, heteroarilo, cicloalquilo o heterocicloalquilo. En algunas realizaciones, B es arilo de 5 o 6 elementos o cicloalquilo de 3 a 6 elementos. En algunas realizaciones, B es

En algunas realizaciones, B es una de las fracciones siguientes: -CH3, -CH2CH3, -CH(CH3)2,

En algunas realizaciones, B se selecciona de entre las fracciones presentadas en la Tabla 1.
Tabla 1. Fracciones B ilustrativas de los compuestos indicados en la presente memoria.

Tabla 1 (continuación)

Tabla 1 (continuación)

Tabla 1 (continuación)

En algunas realizaciones, Wd es arilo (p.ej., un arilo monocíclico o un arilo bicíclico). En algunas realizaciones, Wd es fenilo sustituido o no sustituido. En algunas realizaciones, Wd es arilo bicíclico (p.ej., naftilo sustituido o no sustituido). En algunas realizaciones, Wd es:

En determinadas realizaciones, Wd es heteroarilo (p.ej., heteroarilo monocíclico, p.ej., un heteroarilo de 5 o 6 elementos monocíclico, o heteroarilo bicíclico, p.ej., un heteroarilo 5/6-bicíclico o un heteroarilo 6/6-bicíclico).
En algunas realizaciones, Wd es:

en las que:
Xi, X2 y X3 son, cada uno independientemente, C, CR13 o N,
X4, X5 y X6 son, cada uno independientemente, N, NR12, CR13, S o O, y
en el que cada uno de los grupos Wd se sustituye opcionalente con uno o más de entre R10, R11, R12 y R13, en los que R10, R11, R12 y R13 son, cada uno independientemente, hidrógeno, alquilo, heteroalquilo, alquenilo, alquinilo, cicloalquilo, heterociclilo, arilo, arilalquilo, heteroarilo, heteroarilalquilo, alcoxi, heterocicliloxi, amido, amino, acilo, aciloxi, alcoxicarbonilo, sulfonamido, halo, ciano, hidroxilo, nitro, fosfato, urea, carbonato o NR'R" en la que R' y R" junto con el nitrógeno al que se encuentran unidos forman una fracción cíclica, y
el punto de unión se encuentra en cualquier posición abierta en el grupo Wd.
En algunas realizaciones, Wd es:

en las que:
Xi, X2 y X3 son, cada uno independientemente, C, CR13 o N,
X4 , X5 y X6 son, cada uno independientemente, N, NR12, CR13, S o O, y
R10, R11, R12 y R13 son, cada uno independientemente, hidrógeno, alquilo, heteroalquilo, alquenilo, alquinilo,
cicloalquilo, heterociclilo, arilo, arilalquilo, heteroarilo, heteroarilalquilo, alcoxi, heterocicliloxi, amido, amino, acilo,
aciloxi, alcoxicarbonilo, sulfonamido, halo, ciano, hidroxilo, nitro, fosfato, urea, carbonato, o NR'R" en la que R' y
R" junto con el nitrógeno al que se encuentran unidos forman una fracción cíclica.
En determinadas realizaciones, X1 es  algunas realizaciones, X1 es CR13. En algunas realizaciones,
algunas realizaciones, X1 es CR13. En algunas realizaciones,
En determinadas realizaciones, X2 es  algunas realizaciones, X2 es CR13. En algunas realizaciones,
algunas realizaciones, X2 es CR13. En algunas realizaciones,
En determinadas realizaciones, X3 es  algunas realizaciones, X3 es CR13.
algunas realizaciones, X3 es CR13.
En determinadas realizaciones, X4 es  algunas realizaciones, X4 es CR13. En algunas realizaciones,
algunas realizaciones, X4 es CR13. En algunas realizaciones,
En determinadas realizaciones, X5 es NR12. En algunas realizaciones,  as realizaciones, X5 es En algunas realizaciones, X5 es S.
as realizaciones, X5 es En algunas realizaciones, X5 es S.
En determinadas realizaciones, X6 es N. En algunas realizaciones, X6 es NH. En algunas realizaciones, X6 es CR13.
En algunas realizaciones, X6 es NH. En algunas realizaciones, X6 es O.
En algunas realizaciones, cada R10 es, independientemente, hidrógeno, halo (p.ej., flúor, cloro o bromo), ciano,
hidroxilo, alquilo (p.ej., metilo o CF3), alcoxilo, amino (p.ej., cicloalquilamino (p.ej., ciclopropilamino), alquilamino (p.ej.,
metilamino o dimetilamino), o NH2), arilo (p.ej., fenilo sustituido o no sustituido), heteroarilo (p.ej., un heteroarilo de 5
o 6 elementos, p.ej., pirazolilo, piridinilo, entre otros), heterociclilo (p.ej., N-morfolinilo) o amido. En algunas
realizaciones, cada R10 es, independientemente, hidrógeno, alquilo (p.ej., metilo), amino (p.ej., ciclopropilamino,
metilamino o NH2), heterociclilo (p.ej., N-morfolinilo), heteroarilo (p.ej., 4-pirazolilo), amido o halo (p.ej., cloro). En una
realización, R10 es NH2. En una realización, R10 es H.
En determinadas realizaciones, cada R11 es, independientemente, hidrógeno, halo (p.ej., flúor, cloro o bromo), ciano,
hidroxilo, alquilo (p.ej., metilo o CF3), alcoxilo, amino (p.ej., cicloalquilamino (p.ej., ciclopropilamino), alquilamino (p.ej.,
metilamino o dimetilamino), o NH2), arilo (p.ej., fenilo sustituido o no sustituido), heteroarilo (p.ej., un heteroarilo de 5
o 6 elementos, p.ej., pirazolilo, piridinilo, entre otros), heterociclilo (p.ej., N-morfolinilo) o amido. En algunas
realizaciones, cada R11 es, independientemente, hidrógeno, amino, halo (p.ej., bromo), arilo (p.ej., fenilo) o alquilo
(p.ej., metilo). En una realización, R11 es H.
En determinadas realizaciones, cada R12 es, independientemente, hidrógeno, halo (p.ej., flúor, cloro o bromo), ciano,
hidroxilo, alquilo (p.ej., metilo o CF3), alcoxilo, amino (p.ej., cicloalquilamino (p.ej., ciclopropilamino), alquilamino (p.ej.,
metilamino o dimetilamino), o NH2), arilo (p.ej., fenilo sustituido o no sustituido), heteroarilo (p.ej., un heteroarilo de 5
o 6 elementos, p.ej., pirazolilo, piridinilo, entre otros), heterociclilo (p.ej., N-morfolinilo) o amido. En algunas
realizaciones, cada R12 es, independientemente, hidrógeno, amino o alquilo (p.ej., metilo). En una realización, R12 es
H.
En determinadas realizaciones, cada R13 es, independientemente, hidrógeno, halo (p.ej., flúor, cloro o bromo), ciano,
hidroxilo, alquilo (p.ej., metilo o CF3), alcoxilo, amino (p.ej., cicloalquilamino (p.ej., ciclopropilamino), alquilamino (p.ej.,
metilamino o dimetilamino), o NH2), arilo (p.ej., fenilo sustituido o no sustituido), heteroarilo (p.ej., un heteroarilo de 5
o 6 elementos, p.ej., pirazolilo, piridinilo, entre otros), heterociclilo (p.ej., N-morfolinilo) o amido. En algunas
realizaciones, cada R13 es, independientemente, hidrógeno, amino (p.ej., NH2), amido (p.ej., NH-C(=O)Me) o alquilo
(p.ej., metilo). En una realización, R13 es H.
,

en la que uno de entre Xi y X2 es C y el otro es N, y R10, R11, R12 y R13 son tal como se define en la presente memoria. En algunas realizaciones, R10 es hidrógeno, halo (p.ej., flúor, cloro o bromo), ciano, hidroxilo, alquilo (p.ej., metilo o CF3), alcoxilo, amino (p.ej., cicloalquilamino (p.ej., ciclopropilamino), alquilamino (p.ej., metilamino o dimetilamino), o NH2), arilo (p.ej., fenilo sustituido o no sustituido), heteroarilo (p.ej., un heteroarilo de 5 o 6 elementos, p.ej., pirazolilo, piridinilo, entre otros), heterociclilo (p.ej., N-morfolinilo) o amido. En algunas realizaciones, R10 es hidrógeno, alquilo (p.ej., metilo), amino (p.ej., ciclopropilamino, metilamino o NH2), heterociclilo (p.ej., N-morfolinilo), heteroarilo (p.ej., 4-pirazolilo), amido o halo (p.ej., cloro). En una realización, R10 es NH2. En una realización, R10 es H. En una realización específica, uno de entre X1 y X2 es C y el otro es N; R10 es H o NH2, y R11, R12 y R13 son tal como se define en la presente memoria. En realizaciones específicas, la totalidad de R11, R12 y R13 es H. En realizaciones específicas, dos de entre R11, R12 y R13son H, y uno de entre R11, R12 y R13 es alquilo (p.ej., metilo o CF3), halo, ciano, arilo (p.ej., fenilo) o heteroarilo (p.ej., un heteroarilo de 5 o 6 elementos, tal como piridinilo, pirimidinilo, pirazolilo, tiazolilo, imidazolilo, entre otros), y en algunas realizaciones, el arilo y el heteroarilo se sustituye opcionalmente con uno o más sustituyentes, tales como, por ejemplo, halo (p.ej., F o Cl), ciano, hidroxilo, alquilo (p.ej., metilo o CF3), alcoxilo (p.ej., metoxi, OCF3, etoxi o isopropiloxi), sulfonilo (p.ej., S(O)2Me), sulfonamidilo (p.ej., S(O)2Nh2, S(O)2NHMe, S(O)2N(Me)2, S(O)2NH-/-Pr, S(O)2NH-f-Bu, S(O)2 NH-c-Pr, S(O)2NHPh, S(O)2-N-pirrolidinilo, S(O)2-N-morfolinilo, S(O)2-N-piperazinilo, S(O)2-4-metil-N-piperazinilo, NHS(O)2Me, NHS(O)2Et, NHS(O)2-c-Pr), o sulfonilurea (p.ej., NHS(O)2N(Me)2).
En algunas realizaciones, Wd es:

en el que X3 es N o CR13, y R10, R11, R12 y R13 son tal como se define en la presente memoria. En realizaciones específicas, X3 es N o CR13; R10 es H o n H2, y R11, R12 y R13 son tal como se define en la presente memoria. En realizaciones específicas, R10 es NH2. En realizaciones específicas, X3 es N. En realizaciones específicas, uno de entre R11 y R12 es H, y el otro es alquilo (p.ej., metilo o CF3), halo, ciano, arilo (p.ej., fenilo) o heteroarilo (p.ej., un heteroarilo de 5 o 6 elementos, tal como piridinilo, pirimidinilo, pirazolilo, tiazolilo, imidazolilo, entre otros), y en algunas realizaciones, el arilo y heteroarilo se sustituyen opcionalmente con uno o más sustituyentes, tales como, por ejemplo, halo (p.ej., F o Cl), ciano, hidroxilo, alquilo (p.ej., metilo o CF3), alcoxilo (p.ej., metoxi, OCF3, etoxi o isopropiloxi), sulfonilo (p.ej., S(O)2Me), sulfonamidilo (p.ej., S(O)2NH2, S(O)2 NHMe, S(O)2 N(Me)2 , S(O)2NH-/-Pr, S(O)2 NH-f-Bu, S(O)2NH-c-Pr, S(O)2NHPh, S(O)2-N-pirrolidinilo, S(O)2-N-morfolinilo, S(O)2-N-piperazinilo, S(O)2-4-metil-N-piperazinilo, NHS(O)2Me, NHS(O)2-c-Pr), o sulfonilurea (p.ej., NHS(O)2 N(Me)2).
En algunas realizaciones, Wd es:

en el que uno de entre X1 y X2 es N y el otro es CR13, y R10, R11, R12 y R13 son tal como se define en la presente memoria. En una realización específica, uno de entre X1 y X2 es N y el otro es CR13; R10 es H o NH2, y R11, R12 y R13 son tal como se define en la presente memoria. En realizaciones específicas, X 1 es N y X2 es CR13. En realizaciones específicas, X1 es N y X2 es CH. En realizaciones específicas, R10 es NH2. En realizaciones específicas, R11, R12 y R13 son H. En realizaciones específicas, por lo menos uno de entre R11, R12 y R13 no es H. En realizaciones específicas, una aparición de R11, R12 y R13 no es H y las demás apariencias de R11, R12 y R13 son H, y una apariencia de R11, R12y R13 (que no es hidrógeno) es alquilo (p.ej., metilo o CF3), halo, ciano, arilo (p.ej., fenilo), o heteroarilo (p.ej., un heteroarilo de 5 o 6 elementos, tal como piridinilo, pirimidinilo, pirazolilo, tiazolilo, imidazolilo, entre otros), y en algunas realizaciones, el arilo y heteroarilo se sustituyen opcionalmente con uno o más sustituyentes, tales como, por ejemplo, halo (p.ej., F o Cl), ciano, hidroxilo, alquilo (p.ej., metilo o CF3), alcoxilo (p.ej., metoxi, OCF3, etoxi o isopropiloxi), sulfonilo (p.ej., S(O)2Me), sulfonamidilo (p.ej., S(O)2NH2, S(O)2NHMe, S(O)2N(Me)2, S(O)2NH-/-Pr,
S(O)2 NH-í-Bu, S(O)2NH-c-Pr, S(O)2NHPh, S ^-N -p irro lid im lo , S(O)2-A/-morfolimlo, S ^-N -p iperaz in ilo , S(O)2-4-metil-N-piperazinilo, NHS(O)2Me, NHS(O)2-c-Pr), o sulfonilurea (p.ej., NHS(O)2N(Me)2).
En realizaciones ejemplares, W es una de las fracciones siguientes:


en las que R11 y R12 son tal como se define en la presente memoria.
En algunas realizaciones, Wd es:

En algunas realizaciones, Wd es:

En algunas realizaciones, Wd es:

En algunas realizaciones, Wd es:

En algunas realizaciones, X es CH. En algunas realizaciones, X es N.
En algunas realizaciones, en la fórmula (I''), (I') o (I), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

entonces R1 no es hidrógeno, Si(CHs)s, CH2Si(CH3)3, metilo, (CH2)NH2 , (CH2)2NH2 , (CH2)NHSO2CH3, o (CH2)nNHC(O)R1x; n es 1 o 2; R1x es metilo, alqueno C2, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en los que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituye opcionalmente con uno o dos grupos seleccionados independientemente de entre oxo y ciano.
En algunas realizaciones, en la fórmula (I''), (I') o (I), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

entonces R1 no es (CH2)nNHC(O)R1x; n es 1; R1x es tetrahidrofuranilo o pirrolidinilo, en los que el tetrahidrofuranilo o pirrolidinilo se sustituye opcionalmente con oxo.
En algunas realizaciones, en la fórmula (A"), (A') o (A), en el caso de que X sea CH, B es fenilo no sustituido y W e s

’ R1 no es fenilo.
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula II:

en la que R1, B y X son tal como se define en la presente memoria. En algunas realizaciones, R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5, B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6)3,
en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
X es CR1a o N,
en la que R1a es hidrógeno, halo, alquilo, alquenilo, alquinilo o CN,
en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo>2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), s(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo>2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo>2, S(O)(alquilo), o S(O>2(alquilo>, y
en los que, en el caso de que X sea CH, y B sea fenilo no sustituido, R1 no es hidrógeno, Si(CH3>3, CH2Si(CH3>3, metilo, (CH2>NH2, (CH2>2NH2 , (CH2>NHSO2CH3, o (CH2>nNHC(O>R1x; n es 1 o 2; R1x es metilo, alqueno C2, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en los que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituyen opcionalmente con uno o dos grupos seleccionados independientemente de entre oxo y ciano.
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula

en la que R1 y B son tal como se define en la presente memoria. En algunas realizaciones, R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6>3 ,
en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo>2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), s(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo>2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo>2, S(O)(alquilo), o S(O>2(alquilo>, y
en los que, en el caso de que B sea fenilo no sustituido, R1 no es hidrógeno, Si(CH3>3, CH2Si(CH3>3, metilo, (CH2>NH2 , (CH2>2 NH2 , (CH2>NHSO2CH3 , o (CH2>nNHC(O>R1x; n es 1 o 2; R1x es metilo, alqueno C2 , ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en los que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituye opcionalmente con uno o dos grupos seleccionados independientemente de entre oxo y ciano.
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula IV:

en la que R1 y B son tal como se define en la presente memoria. En algunas realizaciones, R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6>3 ,
en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en las que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo>2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo>2, S(O)(alquilo), s(O>2(alquilo>, cicloalquilo, heterocicloalquilo, arilo o heteroarilo, y
en las que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo>2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo>2, S(O)(alquilo), o S(O>2(alquilo>.
En algunas realizaciones de fórmula II, III y IV, B es fenilo sustituido con 0, 1,2 o 3 apariciones de RZ. En algunas realizaciones, B es fenilo no sustituido. En algunas realizaciones, B es fenilo sustituido con 1 o 2 apariciones de RZ. En algunas realizaciones, RZ es halo o alquilo. En algunas realizaciones, B es metilo, isopropilo o ciclopropilo. En algunas realizaciones, B es ciclohexilo o alquilo sustituido opcionalmente. En algunas realizaciones, B es arilo, heteroarilo, cicloalquilo o heterocicloalquilo. En algunas realizaciones, B es arilo de 5 o 6 elementos o cicloalquilo de 3 a 6 elementos. En algunas realizaciones, B es

n agunas rea zacones, e compueso para a u zac n seg n a presene nvenc n es un compues o e rmua V:

en la que R1 y X son tal como se define en la presente memoria. En algunas realizaciones, R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
en los que R2, R3, R4 y R5 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
X es CR1a o N,
en la que R1a es hidrógeno, halo, alquilo, alquenilo, alquinilo o CN,
en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2 , NH(alquilo), N(alquilo)2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), S(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo)2, S(O)(alquilo), o S(O)2(alquilo), y
en los que, en el caso de que X sea CH, R1 no es hidrógeno, Si(CH3)3, CH2Si(CH3)3, metilo, (CH2)NH2, (CH2)2NH2, (CH2)NHSO2CH3, o (CH2)nNHC(O)R1x; n es 1 o 2; R1x es metilo, alqueno C2, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en los que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituyen opcionalmente con uno o dos grupos seleccionados independientemente de entre oxo y ciano.
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula

en la que R1 esal como se define en la presente memoria. En algunas realizaciones, R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
en los que R2, R3, R4 y R5 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2 , NH(alquilo), N(alquilo)2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), S(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo)2, S(O)(alquilo), o S(O)2(alquilo), y
en la que R1 no es hidrógeno, Si(CH3)3, CH2Si(CH3)3, metilo, (CH2)NH2, (CH2)2NH2, (CH2)NHSO2CH3, o (CH2)nNHC(O)R1x; n es 1 o 2; R1x es metilo, alqueno C2, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en los que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituye opcionalmente con uno o dos grupos seleccionados independientemente de entre oxo y ciano,
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula VII:

en la que R1 es tal como se define en la presente memoria. En algunas realizaciones, R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
en los que R2, R3, R4 y R5 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en las que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), S(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo, y
en las que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), o S(O)2(alquilo).
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula VIII:
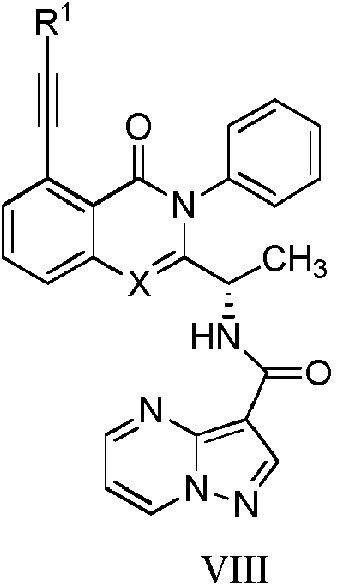
en la que X y R1 son tal como se define en la presente memoria. En algunas realizaciones, R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
en los que R2, R3, R4 y R5 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
X es CR1a o N,
en la que R1a es hidrógeno, halo, alquilo, alquenilo, alquinilo o CN,
en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2 , NH(alquilo), N(alquilo)2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), S(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en las que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), o S(O)2(alquilo).
En algunas realizaciones de fórmulas N-VNI, R1 es alquilo ramificado, arilo de 5 o 6 elementos, heteroarilo de 5 o 6 elementos, cicloalquilo de 5 o 6 elementos o heterocicloalquilo de 5 o 6 elementos,

en los que RA es OH, alcoxi, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
x es 1,2, 3, 4, 5 o 6,
R7, R8 y R9 son, cada uno independientemente, hidrógeno, OH, alcoxi, NH2, NH(alquilo), N(alquilo)2, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo.
En algunas realizaciones, RA es hidroxilo, alcoxi o heterocicloalquilo. En algunas realizaciones, R7, R8 y R9 son, independientemente, alquilo de 1 a 4 carbonos, amino, hidroxilo o alcoxi de 1 a 4 carbonos.
En determinadas realizaciones, R1 es un heteroarilo de 5 a 10 elementos. En determinadas realizaciones, R1 es un heteroarilo de 5 o 6 elementos. En determinadas realizaciones, R1 es un heteroarilo de 6 elementos. En determinadas realizaciones, R1 es un piridinilo. En determinadas realizaciones, R1 es un pirimidinilo. En determinadas realizaciones, R1 es un heteroarilo de 5 elementos. En determinadas realizaciones, R1 es un tiazolilo. En determinadas realizaciones, R1 es un pirazolilo. En determinadas realizaciones, R1 es un imidazolilo. En determinadas realizaciones, el heteroarilo se sustituye con uno o más alquilos.
En algunas realizaciones de fórmulas N-VNI, R1 es metilo,

En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula IX:

en la que R1, B y Wd son tal como se define en la presente memoria. En algunas realizaciones, R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6)3 ,
en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y
en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2 , NH(alquilo), N(alquilo>2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), s(O>2(alquilo>, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo>2, S(O)(alquilo), o S(O>2(alquilo>, y
en los que, en el caso de que B sea fenilo no sustituido, y Wd sea:

entonces R1 no es hidrógeno, Si(CH3>3, CH2Si(CH3>3, metilo, (CH2>NH2 , (CH2>2NH2 , (CH2>NHSO2CH3, o (CH2>nNHC(O>R1x; n es 1 o 2; R1x es metilo, alqueno C2, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en los que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituye opcionalmente con uno o dos grupos seleccionados independientemente de entre oxo y ciano.
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula X:

en la que R1, B y Wd son tal como se define en la presente memoria. En algunas realizaciones, R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5, B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6>3,
en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y
en las que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo>2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo>2, S(O)(alquilo), s(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo, y
en las que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo>2, S(O)(alquilo), o S(O>2(alquilo>.
En algunas realizaciones de fórmulas I-X, R1 es:


en la que RA y R7 a R9 son tal como se define en la presente memoria.
En determinadas realizaciones, R1 es un heteroarilo de 5 a 10 elementos. En determinadas realizaciones, R1 es un heteroarilo de 5 o 6 elementos. En determinadas realizaciones, R1 es un heteroarilo de 6 elementos. En determinadas realizaciones, R1 es un piridinilo. En determinadas realizaciones, R1 es un pirimidinilo. En determinadas realizaciones, R1 es un heteroarilo de 5 elementos. En determinadas realizaciones, R1 es un tiazolilo. En determinadas realizaciones,
R1 es un pirazolilo. En determinadas realizaciones, R1 es un imidazolilo. En determinadas realizaciones, el heteroarilo se sustituye con uno o más alquilos.
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula XI:

en la que R1 y Wd son tal como se define en la presente memoria. En algunas realizaciones, R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5, en los que R2, R3, R4 y R5 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y
en las que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo>2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), s(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo, y
en las que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo>2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo>2, S(O)(alquilo), o S(O>2(alquilo>.
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula XII:

en la que R1 y Wd son tal como se define en la presente memoria. En algunas realizaciones, R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,
en los que R2, R3, R4 y R5 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y
en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2 , NH(alquilo), N(alquilo>2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo>2, S(O)(alquilo), s(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo>2, S(O)(alquilo), o S(O>2(alquilo>, y
en la que, en el caso de que Wd sea:

entonces R1 no es hidrógeno, Si(CH3>3, CH2Si(CH3>3, metilo, (CH2>NH2 , (CH2>2NH2 , (CH2>NHSO2CH3, o (CH2>nNHC(O>R1x; n es 1 o 2; R1x es metilo, alqueno C2, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o
pirrolidinilo, en los que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituye opcionalmente con uno o dos grupos seleccionados independientemente de entre oxo y ciano.
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula XIII:

en la que B y Wd son tal como se define en la presente memoria. En algunas realizaciones, B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6)3, en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y
en las que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo>2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), s(O>2(alquilo>, cicloalquilo, heterocicloalquilo, arilo o heteroarilo, y
en las que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo>2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo>2 , S(O)(alquilo), o S(O>2(alquilo>.
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula XIV:
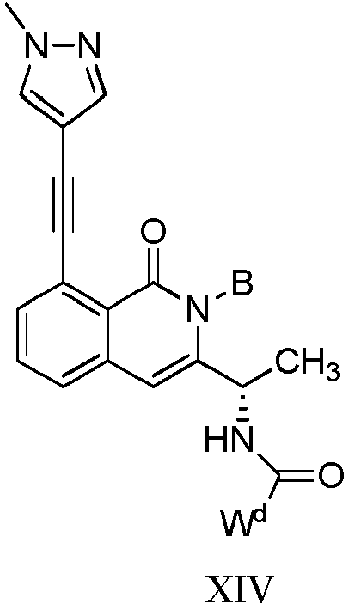
en la que B y Wd son tal como se define en la presente memoria. En algunas realizaciones, B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6>3 , en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y
en las que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo>2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo>2, S(O)(alquilo), s(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo, y
en las que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo>2 , S(O)(alquilo), o S(O>2(alquilo>.
,
XV:

en la que Wd es tal como se define en la presente memoria. En algunas realizaciones, Wd es cicloalquilo, heterocicloalquilo, arilo o heteroarilo, y en el que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo) o CON(alquilo)2.
En algunas realizaciones, el compuesto para la utilización según la presente invención es un compuesto de fórmula XVI:

en la que Wd es tal como se define en la presente memoria. En algunas realizaciones, Wd es cicloalquilo, heterocicloalquilo, arilo o heteroarilo, y en el que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más de entre halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo) o CON(alquilo)2.
En algunas realizaciones de fórmulas IX-XVI, Wd es:

En algunas realizaciones de fórmulas IX-XVI, Wd es:

En algunas realizaciones de fórmulas IX-XVI, Wd es:
En algunas realizaciones de fórmulas IX-XVI, Wd es:

En algunas realizaciones de fórmulas I-XII, R1 no es hidrógeno. En algunas realizaciones de fórmulas I-XII, R1 no es alquilo lineal o hidrógeno. En algunas realizaciones de fórmulas I-XII, R1 no es alquilo C1-C3 lineal o hidrógeno. En algunas realizaciones de fórmulas I-XII, R1 no es metilo o hidrógeno.
En determinadas realizaciones, en la presente memoria se proporcionan para la utilización según la presente invención compuestos de fórmula (A):


o una forma farmacéuticamente aceptable de los mismos, en los que R1, B, Wd y X son tal como se definen en la presente memoria. En determinadas realizaciones, R1 es alquilo o heteroarilo. En determinadas realizaciones, R1 es heteroarilo. En determinadas realizaciones, R1 es alquilo. En algunas realizaciones, B es fenilo. En determinadas realizaciones, X6 es CH o N. En determinadas realizaciones, X es CH.
En determinadas realizaciones, X es N. En determinadas realizaciones, Wd es:

En determinadas realizaciones, el compuesto de fórmula (A''), (A') o (A) es una mezcla de trans y cis (p.ej., en donde R1 es trans o cis). En determinadas realizaciones, R1 es trans. En determinadas realizaciones, R1 es cis. En determinadas realizaciones, el porcentaje de trans a cis es de aproximadamente 50 %, superior a aproximadamente 50 %, superior a aproximadamente 55 %, superior a aproximadamente 60 %, superior a aproximadamente 65 %, superior a aproximadamente 70 %, superior a aproximadamente 75 %, superior a aproximadamente 80 %, superior a aproximadamente 85 %, superior a aproximadamente 90 %, superior a aproximadamente 95 %, superior a aproximadamente 96 %, superior a aproximadamente 97 %, superior a aproximadamente 98 %, o superior a aproximadamente 99 %.
En una realización, en la presente memoria se proporciona para la utilización según la presente invención un compuesto de fórmula XVII:

en la que:
R1 y B son, cada uno independientemente, alquilo lineal o ramificado, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -c Oo R3, -CONR4R5, o -Si(R6)3,
en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, y
X es CH o N,
en los que, en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

y R1 es un alquilo lineal, entonces el alquilo lineal contiene por lo menos tres carbonos unidos consecutivamente, en los que, en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

entonces R1 no es Si(CH3)3, o una forma farmacéuticamente aceptable de los mismos.
En algunas realizaciones de fórmula XVII, R1 es alquilo ramificado, arilo de 5 o 6 elementos, heteroarilo de 5 o 6 elementos, cicloalquilo de 5 o 6 elementos o heterocicloalquilo de 5 o 6 elementos,

ciclopropilo o metilo, en los que RA es hidroxilo, alcoxi, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,
x es 1,2, 3, 4, 5 o 6,
R7, R8y R9 son, cada uno independientemente, hidrógeno, hidroxilo, alcoxi, amino, alquilo, alquenilo,
alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo, en los que por lo menos dos de entre R7, R8 y R9 no son hidrógenos.
En algunas realizaciones de fórmula XVII, RA es hidroxilo, alcoxi o heterocicloalquilo.
En algunas realizaciones de fórmula XVII, R7, R8 y R9 son, independientemente, alquilo de 1 a 4 carbonos, amino, hidroxilo o alcoxi de 1 a 4 carbonos.
En algunas realizaciones de fórmula XVII, Ri es metilo,

En algunas realizaciones de fórmula XVII, B es fenilo sustituido con 0, 1, 2 o 3 apariciones de RZ. En algunas realizaciones, B es fenilo no sustituido. En algunas realizaciones, B es fenilo sustituido con 1 o 2 apariciones de RZ. En algunas realizaciones, RZ es halo o alquilo. En algunas realizaciones, B es metilo, isopropilo o ciclopropilo. En algunas realizaciones, B es ciclohexilo o alquilo sustituido opcionalmente.
En algunas realizaciones de fórmula XVII, B es arilo, heteroarilo, cicloalquilo o heterocicloalquilo.
En algunas realizaciones de fórmula XVII, B es arilo de 5 o 6 elementos o cicloalquilo de 3 a 6 elementos.
En algunas realizaciones de fórmula XVII, B es:

En algunas realizaciones de fórmula XVII, Wd es:

En algunas realizaciones de fórmula XVII, Wd es:

En algunas realizaciones de fórmula XVII, Wd es:

En algunas realizaciones de fórmula XVII, X es CH. En algunas realizaciones, X es N.
En algunas realizaciones de fórmula XVII, en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:

y R1 es un alquilo lineal, entonces el alquilo lineal contiene por lo menos cuatro carbonos unidos consecutivamente. En algunas realizaciones de fórmula XVII, los compuestos presentan la fórmula siguiente:


En algunas realizaciones de fórmula XVII, el compuesto presenta la fórmula siguiente:

En algunas realizaciones, R1 es un heteroarilo, arilo, cicloalquilo o heterocicloalquilo. En determinadas realizaciones, R1 es heteroarilo. En determinadas realizaciones, R1 es un heteroarilo sustituido con un grupo iónico o polar. En algunas realizaciones, un grupo iónico o polar es una parte de un compuesto que incrementa la solubilidad, estabilidad o metabolismo o que reduce el efecto fuera de diana del compuesto. En algunas realizaciones, en la presente memoria se proporciona un compuesto de las fórmulas siguientes:

en las que X, B y Wd son tal como se define en la presente memoria,
Het es heteroarilo, y
R1b es halo, haloalquilo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2 , COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo)2, S(O)(alquilo), S(O)2(alquilo), S(O)2 NH2 , S(O)2NH(alquilo) o S(O)2 N(alquilo)2.
En determinadas realizaciones, alquilo es alquilo C1-C8. En determinadas realizaciones, alquilo es alquilo C1-C6. En otras realizaciones, alquilo es alquilo C1-C3. En determinadas realizaciones, alquenilo es alquenilo C2-C8. En determinadas realizaciones, alquenilo es alquenilo C2-C6. En determinadas realizaciones, alquenilo es alquenilo C2-C3. En determinadas realizaciones, alquinilo es alquinilo C2-C8. En determinadas realizaciones, alquinilo es alquinilo C2-C6. En determinadas realizaciones, alquinilo es alquinilo C2-C3.
En determinadas realizaciones, cicloalquilo es cicloalquilo C3-C8. En determinadas realizaciones, cicloalquilo es cicloalquilo C3-C6. En determinadas realizaciones, cicloalquilo es cicloalquilo C3-C4. En determinadas realizaciones, heterocicloalquilo es un ciclo saturado o parcialmente saturado de 3 a 14 elementos que contiene uno o más heteroátomos seleccionados de un grupo que consiste en N, O y S. En determinadas realizaciones, heterocicloalquilo es de 3 a 10 elementos. En otra realización, heterocicloalquilo es de 3 a 6 elementos. En otra realización, heterocicloalquilo es de 6 elementos. En determinadas realizaciones, arilo es un ciclo C6-C14 aromático. En determinadas realizaciones, arilo es C6-C10. En otra realización, arilo es C6. En determinadas realizaciones, heteroarilo
es un ciclo aromático de 5 a 14 elementos que contiene uno o más heteroátomos seleccionados de un grupo que consiste en N, O y S. En determinadas realizaciones, heteroarilo es de 5 a 10 elementos. En otra realización, heteroarilo es de 5 a 6 elementos. En otra realización, heteroarilo es de 6 elementos.
En determinadas realizaciones, el compuesto proporcionado en la presente memoria para la utilización según la presente invención no es un compuesto seleccionado de entre:


En determinadas realizaciones, el compuesto de fórmula (I"), (I'), (I), (A"), (A') o (A) se encuentra en una configuración estereoquímica (S).
En determinadas realizaciones, el compuesto de fórmula (I"), (I'), (I), (A"), (A') o (A) es el enantiómero (S) con una pureza enantiomérica superior a 75 %.
En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 o 14, o una forma farmacéuticamente aceptable del mismo.
En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 3, 4, 5 o 6, o una forma farmacéuticamente aceptable del mismo.
En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 3 o 4, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 3, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 5, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 7, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 9, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 11, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 13, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 4, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 6, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 8, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 10, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 12, o una forma farmacéuticamente aceptable del mismo. En determinadas realizaciones, el compuesto de fórmula (I') o (A') es un compuesto en la Tabla 14, o una forma farmacéuticamente aceptable del mismo.
Tabla 3








Tabla 4







Tabla 5








Tabla 6






Tabla 7








Tabla 8







Tabla 9








Tabla 10



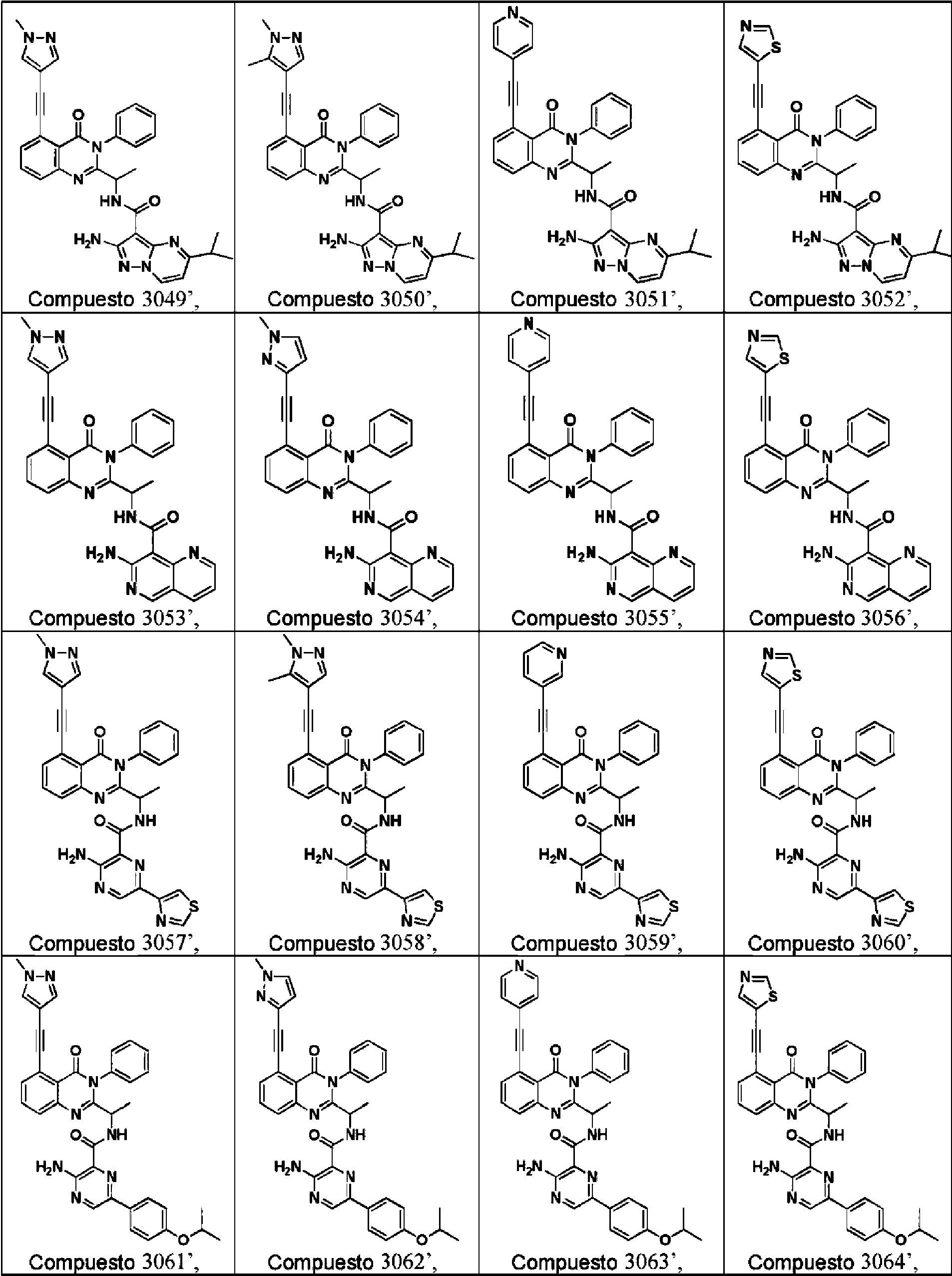


Tabla 11







Tabla 12







Tabla 13




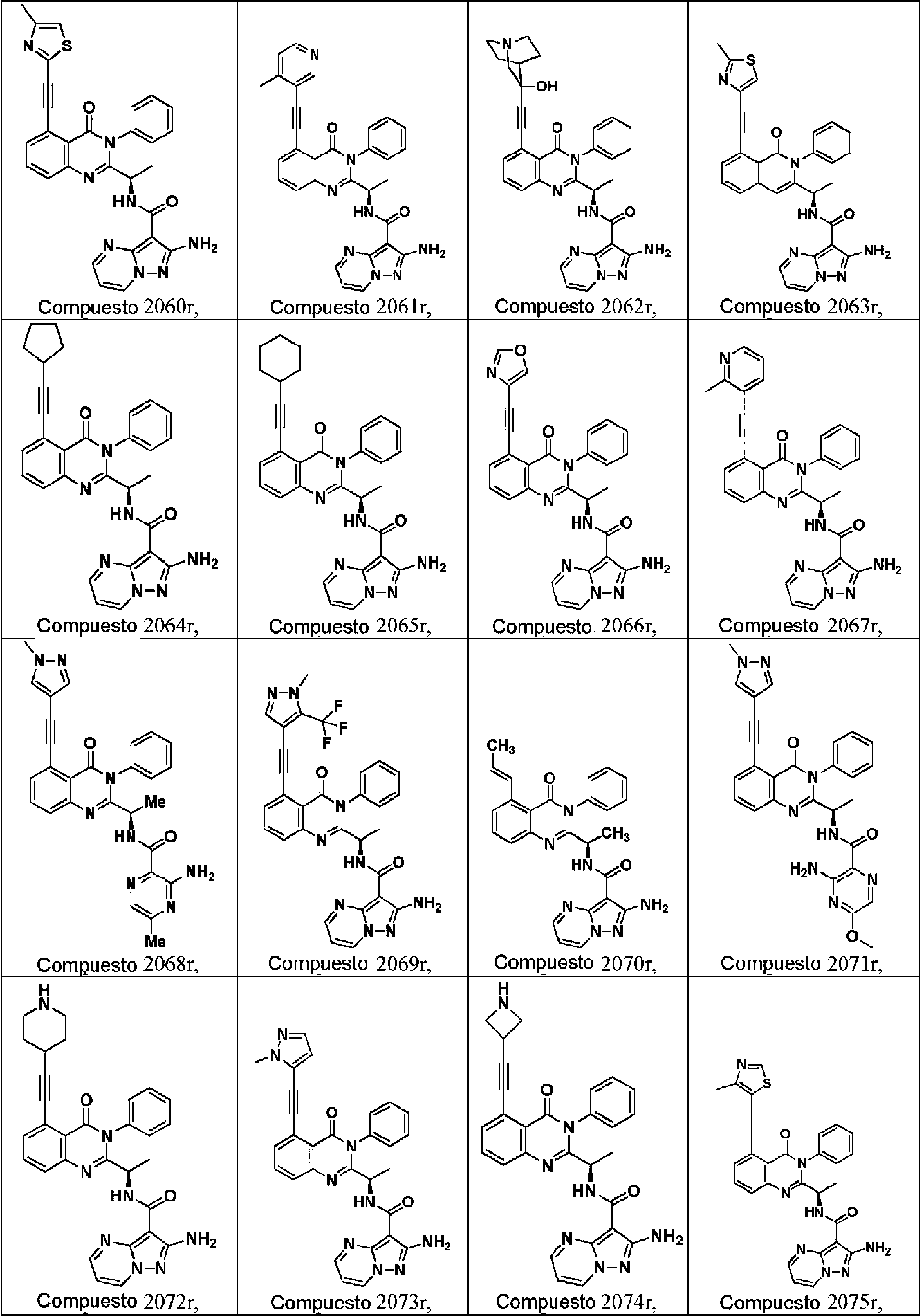



Tabla 14






En la presente memoria se da a conocer que el compuesto comparativo AA presenta la estructura:

El compuesto comparativo AA se da a conocer como Compuesto 359 en la solicitud de patente internacional n° WO2013/032591. Tal como se indica en la Tabla 4 de la solicitud de patente internacional n° WO2013/032591, el compuesto comparativo AA presenta una IC50 de PI3K-delta inferior a 100 nM, una IC50 de PI3K-gamma de entre 1 pM y 100 nM, una IC50 de PI3K-alfa de entre 1 pM y 10 pM y una IC50 de PI3K-beta de entre 1 pM y 10 pM.
En la presente memoria se proporciona para la utilización según la presente invención el compuesto BB, que es el Compuesto 4, de estructura:

En algunas realizaciones, uno o más compuestos indicados en la presente memoria se unen a una PI3 quinasa (p.ej., se unen selectivamente). En algunas realizaciones, uno o más compuestos indicados en la presente memoria se unen selectivamente a un subtipo y o 5 de una PI3 quinasa. En algunas realizaciones, uno o más compuestos indicados en la presente memoria se unen selectivamente a un subtipo y de una PI3 quinasa. En algunas realizaciones, uno o más compuestos indicados en la presente memoria se unen selectivamente a un subtipo 5 de una PI3 quinasa. En una realización, uno o más compuestos indicados en la presente memoria se unen selectivamente a 5 sobre y. En una realización, uno o más compuestos indicados en la presente memoria se unen selectivamente a y sobre 5.
La presente invención se refiere a compuestos para la utilización en el tratamiento o la prevención de un trastorno mediado por PI3K en un sujeto, en el que el trastorno es cáncer. En determinadas realizaciones, el trastorno mediado por PI3K es un trastorno mediado por PI3K-Y. En determinadas realizaciones, el trastorno mediado por PI3K es un trastorno mediado por PI3K-5.
En determinadas realizaciones, el compuesto para la utilización según la invención se selecciona de entre los compuestos n° 2, 4, 7, 9, 17, 19, 21,26, 27, 30, 32, 35, 37, 38, 40, 41,52, 60, 61,63, 73, 75, 77, 79, 80, 81, 88 y 89. En determinadas realizaciones, el compuesto se selecciona de entre los compuestos n° 1,3, 6 , 10, 11, 12, 16, 18, 20, 22, 25, 28, 34, 39, 42, 43, 53, 55, 59, 64, 65, 6 6 , 67, 70, 76, 78, 82, 83, 84, 85, 8 6 y 90. En determinadas realizaciones, el compuesto se selecciona de entre los compuestos n° 8 , 13, 15, 23, 29, 33, 45, 51, 54, 57 y 6 8. En determinadas realizaciones, el compuesto se selecciona de entre los compuestos n° 5, 14, 24, 31,36, 46, 50, 69, 72, 74 y 91. En la presente memoria se da a conocer métodos de síntesis de un compuesto proporcionado en la presente memoria. En la presente memoria se dan a conocer métodos de preparación de un compuesto selectivo de PI3K-Y que comprende sintetizar un compuesto que contiene tanto (a) un grupo heterocíclico bicíclico sustituido con alquino no terminal y (b) un grupo amido. El compuesto puede unirse selectivamente a PI3K-Y sobre PI3K-5.
En algunas realizaciones, la IC50 de un compuesto proporcionado en la presente memoria para p110a, p110p, p110Y, o p 1105 es inferior a aproximadamente 1 pM, inferior a aproximadamente 100 nM, inferior a aproximadamente 50 nM, inferior a aproximadamente 10 nM, inferior a 1 nM, o incluso inferior a aproximadamente 0,5 nM.
En algunas realizaciones, uno o más compuestos proporcionados en la presente memoria se unen específicamente a una PI3 quinasa. En algunas realizaciones, la IC50 de un compuesto proporcionado en la presente memoria para p110a, p110p, p110Y, o p1105 es inferior a aproximadamente 1 pM, inferior a aproximadamente 100 nM, inferior a
aproximadamente 50 nM, inferior a aproximadamente 10 nM, inferior a aproximadamente 1 nM, inferior a aproximadamente 0,5 nM, inferior a aproximadamente 100 pM o inferior a aproximadamente 50 pM.
En algunas realizaciones, uno o más de los compuestos proporcionados en la presente memoria puede inhibir selectivamente uno o más elementos de las fosfatidilinositol-3-quinasas (PI3 quinasa) de tipo I o clase I con un valor de IC50 de aproximadamente 100 nM, aproximadamente 50 nM, aproximadamente 10 nM, aproximadamente 5 nM, aproximadamente 100 pM, aproximadamente 10 pM, o aproximadamente 1 pM, o inferior, según medición en un ensayo de quinasa in vitro.
En algunas realizaciones, uno o más de los compuestos proporcionados en la presente memoria puede inhibir selectivamente uno o dos elementos de las fosfatidilinositol-3-quinasa (PI3-quinasa) de tipo I o clase I, tales como PI3-quinasa a, PI3-quinasa p, PI3-quinasa y y PI3-quinasa 8. En algunas realizaciones, algunos de los compuestos proporcionados en la presente memoria inhiben selectivamente la PI3-quinasa 8 en comparación con todas las demás PI3-quinasas de tipo I. En otras realizaciones, algunos de los compuestos proporcionados en la presente memoria inhiben selectivamente la PI3-quinasa 8 y la PI3-quinasa y en comparación con el resto de las PI3-quinasas de tipo I. En otras realizaciones, algunos de los compuestos proporcionados en la presente memoria inhiben selectivamente la PI3-quinasa y en comparación con todas las demás PI3-quinasas de tipo I.
En todavía otra realización, un inhibidor que inhibe selectivamente uno o más elementos de PI3-quinasas de tipo I, o un inhibidor que inhibe selectivamente una o más rutas de señalización mediadas por PI3-quinasa de tipo I, alternativamente puede entenderse que se refiere a un compuesto que muestra una concentración inhibidora al 50% (IC50) con respecto a una PI3-quinasa de tipo I dada, es decir, por lo menos aproximadamente 10 veces, por lo menos aproximadamente 20 veces, por lo menos aproximadamente 50 veces, por lo menos aproximadamente 100 veces, por lo menos aproximadamente 200 veces, por lo menos aproximadamente 500 veces, por lo menos aproximadamente 1000 veces, por lo menos aproximadamente 2000 veces, por lo menos aproximadamente 5000 veces, o por lo menos aproximadamente 10.000 veces inferior a la IC50 del inhibidor con respecto al resto de las demás PI3-quinasas de tipo I. En una realización, un inhibidor inhibe selectivamente la PI3-quinasa 8 en comparación con la PI3-quinasa p con una IC50 para la PI3-quinasa 8 por lo menos aproximadamente 10 veces inferior. En determinadas realizaciones, la IC50 para la PI3-quinasa 8 es inferior a aproximadamente 100 nM, mientras que la IC50 para la PI3-quinasa p es superior a aproximadamente 1000 nM. En determinadas realizaciones, la IC50 para la PI3-quinasa 8 es inferior a aproximadamente 50 nM, mientras que la IC50 para la PI3-quinasa p es superior a aproximadamente 5000 nM. En determinadas realizaciones, la IC50 para la PI3-quinasa 8 es inferior a aproximadamente 10 nM, mientras que la IC50 para la PI3-quinasa p es superior a aproximadamente 1000 nM, superior a aproximadamente 5.000 nM o superior a aproximadamente 10.000 nM. En una realización, un inhibidor inhibe selectivamente la PI3-quinasa y en comparación con la PI3-quinasa p con una IC50 para la PI3-quinasa y por lo menos aproximadamente 10 veces inferior. En determinadas realizaciones, la IC50 para la PI3-quinasa y es inferior a aproximadamente 100 nM, mientras que la IC50 para la PI3-quinasa p es superior a aproximadamente 1000 nM. En determinadas realizaciones, la IC50 para la PI3-quinasa y es inferior a aproximadamente 50 nM, mientras que la IC50 para la PI3-quinasa p es superior a aproximadamente 5000 nM. En determinadas realizaciones, la IC50 para la PI3-quinasa y es inferior a aproximadamente 10 nM, mientras que la IC50 para la PI3-quinasa p es superior a aproximadamente 1000 nM, superior a aproximadamente 5.000 nM o superior a aproximadamente 10.000 nM.
Un compuesto de PI3K-gamma, p.ej., un inhibidor de PI3K-gamma, puede inhibir la PI3K-gamma y opcionalmente puede inhibir además otras isoformas de PI3K, tales como la PI3K-delta. En una realización, un inhibidor de PI3K-gamma también es un inhibidor de PI3K-delta, es decir, un inhibidor doble de PI3K-gamma y PI3K-delta. En una realización, el inhibidor doble presenta una IC50 para la PI3K-gamma que es menos de 10 veces la IC50 para PI3K-delta.
Un compuesto selectivo para la PI3K-gamma, p.ej., un inhibidor selectivo para PI3K-gamma, puede mostrar una concentración inhibidora al 50% (IC50) con respecto a la PI3K-gamma que es por lo menos aproximadamente 10 veces inferior a la IC50 del compuesto con respecto a las demás PI3-quinasas de tipo I. En algunas realizaciones, el compuesto selectivo para PI3K-gamma muestra una concentración inhibidora al 50% (IC50) con respecto a PI3K-gamma que es por lo menos aproximadamente 20 veces, por lo menos aproximadamente 30 veces, por lo menos aproximadamente 40 veces, por lo menos aproximadamente 50 veces, por lo menos aproximadamente 60 veces, por lo menos aproximadamente 70 veces, por lo menos aproximadamente 80 veces, por lo menos aproximadamente 90 veces, por lo menos aproximadamente 100 veces, por lo menos aproximadamente 200 veces, por lo menos aproximadamente 500 veces, por lo menos aproximadamente 1000 veces, por lo menos aproximadamente 2000 veces, por lo menos aproximadamente 5.000 veces, o por lo menos aproximadamente 10.000 veces, inferior a la IC50 del compuesto con respecto a las demás PI3-quinasas de tipo I. En una realización, un inhibidor inhibe selectivamente la PI3-quinasa y en comparación con la PI3-quinasa 8 con una IC50 para la PI3-quinasa y por lo menos aproximadamente 10 veces inferior. En algunas realizaciones, un compuesto selectivo para PI3K-gamma presenta una IC50 de aproximadamente 1,8 nM para PI3K-gamma, una IC50 para PI3K-beta o PI3K-delta de aproximadamente 180 nM, un valor de IC90 para PI3K-gamma de aproximadamente 16 nM y un valor de IC20 para PI3K-beta o PI3K-delta de aproximadamente 45 nM.
Un compuesto de PI3K-delta, p.ej., un inhibidor de PI3K-delta, puede inhibir la PI3K-delta y opcionalmente puede
inhibir además otras isotermas de PI3K, tales como PI3K-gamma. En una realización, un inhibidor de PI3K-delta también es un inhibidor de PI3K-gamma.
Un compuesto selectivo para la PI3K-delta, p.ej., un inhibidor selectivo para PI3K-delta, puede mostrar una concentración inhibidora al 50% (IC50) con respecto a la PI3K-gamma que es por lo menos aproximadamente 10 veces inferior a la IC50 del compuesto con respecto a las demás PI3-quinasas de tipo I. En algunas realizaciones, el compuesto selectivo para PI3K-delta muestra una concentración inhibidora al 50% (IC50) con respecto a PI3K-delta que es por lo menos aproximadamente 20 veces, por lo menos aproximadamente 30 veces, por lo menos aproximadamente 40 veces, por lo menos aproximadamente 50 veces, por lo menos aproximadamente 60 veces, por lo menos aproximadamente 70 veces, por lo menos aproximadamente 80 veces, por lo menos aproximadamente 90 veces, por lo menos aproximadamente 100 veces, por lo menos aproximadamente 200 veces, por lo menos aproximadamente 500 veces, por lo menos aproximadamente 1000 veces, por lo menos aproximadamente 2000 veces, por lo menos aproximadamente 5000 veces, o por lo menos aproximadamente 10.000 veces, inferior a la IC50 del compuesto con respecto a las demás PI3-quinasas de tipo I. En una realización, un inhibidor inhibe selectivamente la PI3-quinasa 8 en comparación con la PI3-quinasa y con una IC50 para la PI3-quinasa 8 por lo menos aproximadamente 10 veces inferior.
Composiciones farmacéuticas
En algunas realizaciones, en la presente memoria se proporcionan composiciones farmacéuticas para la utilización según la presente invención, que comprenden un compuesto tal como se da a conocer en la presente memoria, o un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo (p.ej., sales, hidratos, solvatos, isómeros y derivados isotópicamente marcados farmacéuticamente aceptables) y un excipiente, diluyente o portador farmacéuticamente aceptable, incluyendo diluyentes y rellenos sólidos inertes, solución acuosa estéril y diversos solventes orgánicos, potenciadores de la permeación, solubilizadores y adyuvantes. En algunas realizaciones, una composición farmacéutica descrita en la presente memoria incluye un segundo agente activo, tal como un agente terapéutico adicional (p.ej., un quimioterapéutico).
Formulaciones
Las composiciones farmacéuticas pueden formularse especialmente para la administración en forma sólida o líquida, incluyendo las adaptadas para lo siguiente: administración oral, por ejemplo pócimas (soluciones o suspensiones acuosas o no acuosas), tabletas (p.ej., las destinadas a la absorción bucal, sublingual y sistémica), cápsulas, bolos, polvos, gránulos, pastas para la aplicación en la lengua y vías intraduodenales; administración parenteral, incluyendo intravenosa, intraarterial, subcutánea, intramuscular, intravascular, intraperitoneal o como infusión, tal como, por ejemplo, una solución o suspensión estéril, o formulación de liberación sostenida; aplicación tópica, por ejemplo como crema, pomada o un parche o spray de liberación controlada aplicado en la piel; intravaginal o intrarrectalmente, por ejemplo como pesario, crema, stent o espuma; por vía sublingual, ocular o pulmonar; administración local mediante catéter o stent; por vía intratecal o nasal.
Entre los ejemplos de portadores acuosos o no acuosos adecuados que pueden utilizarse en composiciones farmacéuticas se incluyen agua, etanol, polioles (tales como glicerol, propilenglicol, polietilenglicol y similares) y mezclas adecuadas de los mismos, aceites vegetales, tales como aceite de oliva, y ésteres orgánicos inyectables, tales como oleato de etilo. Puede mantenerse la fluidez apropiada, por ejemplo, mediante la utilización de materiales de recubrimiento, tales como lecitina, mediante el mantenimiento del tamaño de partícula requerido en el caso de dispersiones, y mediante la utilización de tensioactivos.
Dichas composiciones pueden contener además adyuvantes, tales como conservantes, agentes humectantes, agentes emulsionantes, agentes dispersantes, lubricantes y/o antioxidantes. La prevención de la acción de los microorganismos en los compuestos indicados en la presente memoria puede garantizarse mediante la inclusión de diversos agentes antibacterianos y antifúngicos, por ejemplo parabeno, clorobutanol, ácido fenolsórbico y similares. También puede resultar deseable incluir agentes isotónicos, tales como azúcares, cloruro sódico y similares en las composiciones. Además, la absorción prolongada de la forma farmacéutica inyectable puede producirse mediante la inclusión de agentes que retrasan la absorción, por ejemplo el monoestearato de aluminio y la gelatina.
Los métodos de preparación de dichas formulaciones o composiciones incluyen la etapa de asociar un compuesto indicado en la presente memoria y/o el quimioterapéutico con el portador y, opcionalmente, uno o más ingredientes accesorios. En general, las formulaciones se preparan asociando uniforme e íntimamente un compuesto tal como se da a conocer en la presente memoria con portadores líquidos, o portadores sólidos finamente divididos, o ambos, y después, en caso necesario, conformando el producto.
Las preparaciones para dichas composiciones farmacéuticas son bien conocidas de la técnica. Ver, p.ej., Anderson, Philip O.; Knoben, James E.; Troutman, William G, eds., Handbook of Clinical Drug Data, décima edición, McGraw-Hill, 2002; Pratt and Taylor, eds., Principles of Drug Action, tercera edición, Churchill Livingston, New York, 1990; Katzung, ed., Basic and Clinical Pharmacology, duodécima edición, McGraw Hill, 2011; Goodman y Gilman,
eds., The Pharmacological Basis of Therapeutics, décima edición, McGraw Hill, 2001; Remingtons Pharmaceutical Sciences, vigésima edición, Lippincott Williams & Wilkins., 2000; Martindale, The Extra Pharmacopoeia, trigésimo segunda edición (The Pharmaceutical Press, London, 1999). Excepto en la medida en que cualquier medio excipiente convencional sea incompatible con los compuestos proporcionados en la presente memoria, tal como por la producción de cualesquiera efectos biológico indeseable o de otro modo, mediante interacción de una manera perjudicial con cualquier otro componente o componentes de la composición farmacéuticamente aceptable, la utilización del excipiente se encuentra contemplada como comprendida dentro del alcance de la presente exposición.
En algunas realizaciones, entre las composiciones farmacéuticas proporcionadas en la presente memoria se incluyen formulaciones de nanopartículas con diana en CD44 (agregado de diferenciación 44) CD44 es una glucoproteína transmembranal que desempeña una función en las cascadas de señalización celular asociadas al inicio y progresión del cáncer. CD44 se sobreexpresa sobre la superficie de determinados cánceres, y el ácido hialurónico presenta una afinidad para dichos CD44 sobreexpresados. El sistema de administración dirigida de fármaco basado en ácido hialurónico (p.ej., nanopartículas de sílice mesoporoso modificado con ácido hialurónico) pueden ser un medio eficaz de administración de fármacos en células de cáncer. Ver, p.ej., S. Arpicco, et al., Molecules, 2014, 19, 3193-3230.
En algunas realizaciones, la concentración de uno o más de los compuestos proporcionados en las composiciones farmacéuticas dadas a conocer es inferior a aproximadamente 100 %, a aproximadamente 90 %, a aproximadamente 80 %, a aproximadamente 70 %, a aproximadamente 60 %, a aproximadamente 50 %, a aproximadamente 40 %, a aproximadamente 30 %, a aproximadamente 20 %, a aproximadamente 19 %, a aproximadamente 18 %, a aproximadamente 17 %, a aproximadamente 16 %, a aproximadamente 15 %, a aproximadamente 14 %, a aproximadamente 13 %, a aproximadamente 12 %, a aproximadamente 11 %, a aproximadamente 10 %, a aproximadamente 9 %, a aproximadamente 8 %, a aproximadamente 7 %, a aproximadamente 6 %, a aproximadamente 5 %, a aproximadamente 4 %, a aproximadamente 3 %, a aproximadamente 2 %, a aproximadamente 1 %, a aproximadamente 0,5 %, a aproximadamente 0,4 %, a aproximadamente 0,3 %, a aproximadamente 0,2 %, a aproximadamente 0,1 %, a aproximadamente 0,09 %, a aproximadamente 0,08 %, a aproximadamente 0,07 %, a aproximadamente 0,06 %, a aproximadamente 0,05 %, a aproximadamente 0,04 %, a aproximadamente 0,03 %, a aproximadamente 0,02 %, a aproximadamente 0,01 %, a aproximadamente 0,009 %, a aproximadamente 0,008 %, a aproximadamente 0,007 %, a aproximadamente 0,006 %, a aproximadamente 0,005 %, a aproximadamente 0,004 %, a aproximadamente 0,003 %, a aproximadamente 0,002 %, a aproximadamente 0,001 %, a aproximadamente 0,0009 %, a aproximadamente 0,0008 %, a aproximadamente 0,0007 %, a aproximadamente 0,0006 %, a aproximadamente 0,0005 %, a aproximadamente 0,0004 %, a aproximadamente 0,0003 %, a aproximadamente 0,0002 %, o a aproximadamente 0,0001 %, p/p, p/v o v/v.
En algunas realizaciones, la concentración de uno o más de los compuestos dados a conocer en la presente memoria es superior a aproximadamente 90 %, a aproximadamente 80 %, a aproximadamente 70 %, a aproximadamente 60 %, a aproximadamente 50 %, a aproximadamente 40 %, a aproximadamente 30 %, a aproximadamente 20 %, a aproximadamente 19,75 %, a aproximadamente 19,50 %, a aproximadamente 19,25 %, a aproximadamente 19 %, a aproximadamente 18,75 %, a aproximadamente 18,50 %, a aproximadamente 18,25 %, a aproximadamente 18 %, a aproximadamente 17,75 %, a aproximadamente 17,50 %, a aproximadamente 17,25 %, a aproximadamente 17 %, a aproximadamente 16,75 %, a aproximadamente 16,50 %, a aproximadamente 16,25 %, a aproximadamente 16 %, a aproximadamente 15,75 %, a aproximadamente 15,50 %, a aproximadamente 15,25 %, a aproximadamente 15 %, a aproximadamente 14,75 %, a aproximadamente 14,50 %, a aproximadamente 14,25 %, a aproximadamente 14 %, a aproximadamente 13,75 %, a aproximadamente 13,50 %, a aproximadamente 13,25 %, a aproximadamente 13 %, a aproximadamente 12,75 %, a aproximadamente 12,50 %, a aproximadamente 12,25 %, a aproximadamente 12 %, a aproximadamente 11,75 %, a aproximadamente 11,50 %, a aproximadamente 11,25 %, a aproximadamente 11 %, a aproximadamente 10,75 %, a aproximadamente 10,50 %, a aproximadamente 10,25 %, a aproximadamente 10 %, a aproximadamente 9,75 %, a aproximadamente 9,50 %, a aproximadamente 9,25 %, a aproximadamente 9 %, a aproximadamente 8,75 %, a aproximadamente 8,50 %, a aproximadamente 8,25 %, a aproximadamente 8 %, a aproximadamente 7,75 %, a aproximadamente 7,50 %, a aproximadamente 7,25 %, a aproximadamente 7 %, a aproximadamente 6,75 %, a aproximadamente 6,50 %, a aproximadamente 6,25 %, a aproximadamente 6 %, a aproximadamente 5,75 %, a aproximadamente 5,50 %, a aproximadamente 5,25 %, a aproximadamente 5 %, a aproximadamente 4,75 %, a aproximadamente 4,50 %, a aproximadamente 4,25 %, a aproximadamente 4 %, a aproximadamente 3,75 %, a aproximadamente 3,50 %, a aproximadamente 3,25 %, a aproximadamente 3 %, a aproximadamente 2,75 %, a aproximadamente 2,50 %, a aproximadamente 2,25 %, a aproximadamente 2 %, a aproximadamente 1,75 %, a aproximadamente 1,50 %, a aproximadamente 1,25 %, a aproximadamente 1 %, a aproximadamente 0,5 %, a aproximadamente 0,4 %, a aproximadamente 0,3 %, a aproximadamente 0,2 %, a aproximadamente 0,1 %, a aproximadamente 0,09 %, a aproximadamente 0,08 %, a aproximadamente 0,07 %, a aproximadamente 0,06 %, a aproximadamente 0,05 %, a aproximadamente 0,04 %, a aproximadamente 0,03 %, a aproximadamente 0,02 %, a aproximadamente 0,01 %, a aproximadamente 0,009 %, a aproximadamente 0,008 %, a aproximadamente 0,007 %, a aproximadamente 0,006 %, a aproximadamente 0,005 %, a aproximadamente 0,004 %, a aproximadamente 0,003 %, a aproximadamente 0,002 %, a aproximadamente 0,001 %, a aproximadamente 0,0009 %, a aproximadamente 0,0008 %, a aproximadamente 0,0007 %, a aproximadamente 0,0006 %, a aproximadamente 0,0005 %, a aproximadamente 0,0004 %, a aproximadamente 0,0003 %, a aproximadamente 0,0002 %, o a aproximadamente 0,0001 %, p/p, p/v, o v/v.
En algunas realizaciones, la concentración de uno o más de los compuestos dados a conocer en la presente memoria se encuentra comprendida en el intervalo de entre aproximadamente 0,0001 % y aproximadamente 50 %, de entre aproximadamente 0,001 % y aproximadamente 40 %, de entre aproximadamente 0,01 % y aproximadamente 30 %, de entre aproximadamente 0,02 % y aproximadamente 29 %, de entre aproximadamente 0,03 % y aproximadamente
28 %, de entre aproximadamente 0,04 % y aproximadamente 27 %, de entre aproximadamente 0,05 % y aproximadamente 26 %, de entre aproximadamente 0,06 % y aproximadamente 25 %, de entre aproximadamente
0,07 % y aproximadamente 24 %, de entre aproximadamente 0,08 % y aproximadamente 23 %, de entre aproximadamente 0,09 % y aproximadamente 22 %, de entre aproximadamente 0,1 % y aproximadamente 21 %, de entre aproximadamente 0,2 % y aproximadamente 20 %, de entre aproximadamente 0,3 % y aproximadamente 19 %, de entre aproximadamente 0,4 % y aproximadamente 18 %, de entre aproximadamente 0,5 % y aproximadamente 17
%, de entre aproximadamente 0,6 % y aproximadamente 16 %, de entre aproximadamente 0,7 % y aproximadamente
15 %, de entre aproximadamente 0,8 % y aproximadamente 14 %, de entre aproximadamente 0,9 % y aproximadamente 12 %, o de entre aproximadamente 1 % y aproximadamente 10 %, p/p, p/v o v/v.
En algunas realizaciones, la concentración de uno o más de los compuestos dados a conocer en la presente memoria se encuentra comprendida en el intervalo de entre aproximadamente 0,001 % y aproximadamente 10 %, de entre aproximadamente 0,01 % y aproximadamente 5 %, de entre aproximadamente 0,02 % y aproximadamente 4,5 %, de entre aproximadamente 0,03 % y aproximadamente 4 %, de entre aproximadamente 0,04 % y aproximadamente 3,5
%, de entre aproximadamente 0,05 % y aproximadamente 3 %, de entre aproximadamente 0,06 % y aproximadamente
2,5 %, de entre aproximadamente 0,07 % y aproximadamente 2 %, de entre aproximadamente 0,08 % y aproximadamente 1,5 %, de entre aproximadamente 0,09 % y aproximadamente 1 %, o de entre aproximadamente
0,1 % y aproximadamente 0,9 %, p/p, p/v o v/v.
En algunas realizaciones, la cantidad de uno o más de los compuestos dados a conocer en la presente memoria es igual o inferior a aproximadamente 10 g, a aproximadamente 9,5 g, a aproximadamente 9,0 g, a aproximadamente 8,5 g, a aproximadamente 8,0 g, a aproximadamente 7,5 g, a aproximadamente 7,0 g, a aproximadamente 6,5 g, a aproximadamente 6,0 g, a aproximadamente 5,5 g, a aproximadamente 5,0 g, a aproximadamente 4,5 g, a aproximadamente 4,0 g, a aproximadamente 3,5 g, a aproximadamente 3,0 g, a aproximadamente 2,5 g, a aproximadamente 2,0 g, a aproximadamente 1,5 g, a aproximadamente 1,0 g, a aproximadamente 0,95 g, a aproximadamente 0,9 g, a aproximadamente 0,85 g, a aproximadamente 0,8 g, a aproximadamente 0,75 g, a aproximadamente 0,7 g, a aproximadamente 0,65 g, a aproximadamente 0,6 g, a aproximadamente 0,55 g, a aproximadamente 0,5 g, a aproximadamente 0,45 g, a aproximadamente 0,4 g, a aproximadamente 0,35 g, a aproximadamente 0,3 g, a aproximadamente 0,25 g, a aproximadamente 0,2 g, a aproximadamente 0,15 g, a aproximadamente 0,1 g, a aproximadamente 0,09 g, a aproximadamente 0,08 g, a aproximadamente 0,07 g, a aproximadamente 0,06 g, a aproximadamente 0,05 g, a aproximadamente 0,04 g, a aproximadamente 0,03 g, a aproximadamente 0,02 g, a aproximadamente 0,01 g, a aproximadamente 0,009 g, a aproximadamente 0,008 g, a aproximadamente 0,007 g, a aproximadamente 0,006 g, a aproximadamente 0,005 g, a aproximadamente 0,004 g, a aproximadamente 0,003 g, a aproximadamente 0,002 g, a aproximadamente 0,001 g, a aproximadamente 0,0009 g, a aproximadamente 0,0008 g, a aproximadamente 0,0007 g, a aproximadamente 0,0006 g, a aproximadamente 0,0005 g, a aproximadamente 0,0004 g, a aproximadamente 0,0003 g, a aproximadamente 0,0002 g, o a aproximadamente
0,0001 g.
En algunas realizaciones, la cantidad de uno o más de los compuestos dados a conocer en la presente memoria es superior a aproximadamente 0,0001 g, a aproximadamente 0,0002 g, a aproximadamente 0,0003 g, a aproximadamente 0,0004 g, a aproximadamente 0,0005 g, a aproximadamente 0,0006 g, a aproximadamente 0,0007 g, a aproximadamente 0,0008 g, a aproximadamente 0,0009 g, a aproximadamente 0,001 g, a aproximadamente
0,0015 g, a aproximadamente 0,002 g, a aproximadamente 0,0025 g, a aproximadamente 0,003 g, a aproximadamente
0,0035 g, a aproximadamente 0,004 g, a aproximadamente 0,0045 g, a aproximadamente 0,005 g, a aproximadamente
0,0055 g, a aproximadamente 0,006 g, a aproximadamente 0,0065 g, a aproximadamente 0,007 g, a aproximadamente
0,0075 g, a aproximadamente 0,008 g, a aproximadamente 0,0085 g, a aproximadamente 0,009 g, a aproximadamente 0,0095 g, a aproximadamente 0,01 g, a aproximadamente 0,015 g, a aproximadamente 0,02 g, a aproximadamente
0,025 g, a aproximadamente 0,03 g, a aproximadamente 0,035 g, a aproximadamente 0,04 g, a aproximadamente
0,045 g, a aproximadamente 0,05 g, a aproximadamente 0,055 g, a aproximadamente 0,06 g, a aproximadamente
0,065 g, a aproximadamente 0,07 g, a aproximadamente 0,075 g, a aproximadamente 0,08 g, a aproximadamente
0,085 g, a aproximadamente 0,09 g, a aproximadamente 0,095 g, a aproximadamente 0,1 g, a aproximadamente 0,15 g, a aproximadamente 0,2 g, a aproximadamente 0,25 g, a aproximadamente 0,3 g, a aproximadamente 0,35 g, a aproximadamente 0,4 g, a aproximadamente 0,45 g, a aproximadamente 0,5 g, a aproximadamente 0,55 g, a aproximadamente 0,6 g, a aproximadamente 0,65 g, a aproximadamente 0,7 g, a aproximadamente 0,75 g, a aproximadamente 0,8 g, a aproximadamente 0,85 g, a aproximadamente 0,9 g, a aproximadamente 0,95 g, a aproximadamente 1 g, a aproximadamente 1,5 g, a aproximadamente 2 g, a aproximadamente 2,5 g, a aproximadamente 3 g, a aproximadamente 3,5 g, a aproximadamente 4 g, a aproximadamente 4,5 g, a aproximadamente 5 g, a aproximadamente 5,5 g, a aproximadamente 6 g, a aproximadamente 6,5 g, a aproximadamente 7 g, a aproximadamente 7,5 g, a aproximadamente 8 g, a aproximadamente 8,5 g, a aproximadamente 9 g, a aproximadamente 9,5 g, o a aproximadamente 10 g,
En algunas realizaciones, la cantidad de uno o más de los compuestos dados a conocer en la presente memoria se encuentra comprendida en el intervalo de entre aproximadamente 0,0001 y aproximadamente 10 g, de entre aproximadamente 0,0005 y aproximadamente 9 g, de entre aproximadamente 0,001 y aproximadamente 8 g, de entre aproximadamente 0,005 y aproximadamente 7 g, de entre aproximadamente 0,01 y aproximadamente 6 g, de entre aproximadamente 0,05 y aproximadamente 5 g, de entre aproximadamente 0,1 y aproximadamente 4 g, de entre aproximadamente 0,5 y aproximadamente 4 g, o de entre aproximadamente 1 y aproximadamente 3 g.
Formulaciones para la administración oral
En algunas realizaciones, en la presente memoria se proporcionan para la utilización según la presente invención composiciones farmacéuticas para la administración oral que contienen un compuesto tal como se da a conocer en la presente memoria y un excipiente farmacéutico adecuado para la administración oral. En algunas realizaciones, en la presente memoria se proporcionan composiciones farmacéuticas para la administración oral que contienen: (i) una cantidad eficaz de un compuesto dado a conocer, opcionalmente (ii) una cantidad eficaz de uno o más segundos agentes, y (iii) uno o más excipientes farmacéuticos adecuados para la administración oral. En algunas realizaciones, la composición farmacéutica contiene, además: (iv) una cantidad eficaz de un tercer agente.
En algunas realizaciones, la composición farmacéutica puede ser una composición farmacéutica líquida adecuada para el consumo oral. Las composiciones farmacéuticas adecuadas para la administración oral pueden presentarse como formas de administración discretas, tales como cápsulas, sellos o tabletas, o líquidos o pulverizadores de aerosol, cada uno de los cuales contiene una cantidad predeterminada de un ingrediente activo, tal como unos polvos o gránulos, una solución, o una suspensión en un líquido acuoso o no acuoso, una emulsión de aceite en agua o una emulsión líquida de agua en aceite.
Dichas formas de administración pueden prepararse mediante cualquiera de los métodos farmacéuticos, aunque todos los métodos incluyen la etapa de asociar el ingrediente activo con el portador, que constituye uno o más ingredientes. En general, las composiciones farmacéuticas se preparan asociando uniforme e íntimamente el ingrediente activo con portadores líquidos o portadores sólidos finamente divididos o ambos, y a continuación, en caso necesario, conformando el producto en la presentación deseada. Por ejemplo, puede prepararse una tableta mediante compresión o moldeo, opcionalmente con uno o más ingredientes accesorios. Pueden prepararse tabletas comprimidas mediante compresión en una máquina adecuada del ingrediente activo en una forma de flujo libre, tal como polvos o gránulos, opcionalmente mezclados con un excipiente, tal como, aunque sin limitación, un ligante, un lubricante, un diluyente inerte y/o un agente activo en superficie o dispersante. Pueden prepararse tabletas moldeadas mediante moldeo en una máquina adecuada de una mezcla del compuesto en polvo humectado con un diluyente líquido inerte.
La presente exposición comprende además composiciones farmacéuticas anhidras y formas de administración que comprenden un ingrediente activo, ya que el agua puede facilitar la degradación de algunos compuestos. Por ejemplo, puede añadirse agua (p.ej., aproximadamente 5 %) en las técnicas farmacéuticas como medio de simular el almacenamiento a largo plazo con el fin de determinar características tales como el tiempo de almacenamiento o la estabilidad de las formulaciones durante el tiempo. Pueden prepararse composiciones farmacéuticas y formas de administración anhidras mediante la utilización de ingredientes anhidros o que contienen baja humedad y condiciones de baja humedad. Por ejemplo, las composiciones farmacéuticas y formas de administración que contienen lactosa pueden convertirse en anhidras en caso de que se espere un contacto sustancial con humedad durante la preparación, envasado y/o almacenamiento. Puede prepararse una composición farmacéutica anhidra y almacenarse de manera que se mantenga su naturaleza anhidra. De acuerdo con lo anterior, pueden envasarse composiciones farmacéuticas anhidras utilizando materiales que es conocido que impiden la exposición al agua, de manera que pueden incluirse en kits de formulaciones adecuados. Entre los ejemplos de envases adecuados se incluyen, aunque sin limitarse a ellos, láminas selladas herméticamente, plástico o similar, envases de dosis unitaria, paquetes de blísters y paquetes de tiras.
Puede combinarse un ingrediente activo en una mezcla íntima con un portador farmacéutico según técnicas convencionales de preparación de composiciones farmacéuticas. El portador puede adoptar una amplia diversidad de formas según la forma de preparación deseada para la administración. Durante la preparación de las composiciones farmacéuticas para una forma de administración oral, pueden utilizarse cualesquiera de los medios farmacéuticos habituales como portadores, tales como, por ejemplo, agua, glicoles, aceites, alcoholes, agentes saborizantes, conservantes, agentes colorantes y similares en el caso de las preparaciones líquidas orales (tales como suspensiones, soluciones y elixires) o pueden utilizarse aerosoles, o portadores, tales como almidones, azúcares, celulosa microcristalina, diluyentes, agentes de granulación, lubricantes, ligantes y agentes desintegrantes en el caso de preparaciones sólidas orales, en algunas realizaciones sin utilizar lactosa. Por ejemplo, entre los portadores adecuados se incluyen polvos, cápsulas y tabletas, con las preparaciones orales sólidas. En algunas realizaciones, las tabletas pueden recubrirse mediante técnicas acuosas o no acuosas estándares.
Entre los ligantes adecuados para la utilización en composiciones farmacéuticas y formas de administración se incluyen, aunque sin limitarse a ellos, almidón de maíz, almidón de patata u otros almidones, gelatina, gomas naturales
y sintéticas, tales como acacia, alginato sódico, ácido algínico, otros alginatos, tragacanto en polvo, goma guar, celulosa y sus derivados (p.ej., etilcelulosa, acetato de celulosa, carboximetilcelulosa cálcica y carboximetilcelulosa sódica), polivinilpirrolidona, metilcelulosa, almidón pregelatinizado, hidroxipropilmetilcelulosa, celulosa, microcristalina y mezclas de los mismos.
Entre los ejemplos de rellenos adecuados para la utilización en las composiciones farmacéuticas y formas de administración dados a conocer en la presente memoria se incluyen, aunque sin limitarse a ellos, talco, carbonato cálcico (p.ej., gránulos o polvos), celulosa microcristalina, celulosa en polvo, dextratos, caolín, manitol, ácido silícico, sorbitol, almidón, almidón pregelatinizado y mezclas de los mismos.
Pueden utilizarse desintegrantes en las composiciones farmacéuticas tales como las proporcionadas en la presente memoria para proporcionar tabletas que se desintegran al exponerlas a un medio acuoso. Un exceso de desintegrante puede producir tabletas que pueden desintegrarse en la botella. Demasiado poco puede resultar insuficiente para que se produzca la desintegración y, de esta manera, puede alterar la velocidad y grado de liberación del ingrediente o ingredientes activos a partir de la forma de administración. De esta manera, una cantidad suficiente de desintegrante que no sea ni demasiado pequeña o excesiva para alterar perjudicialmente la liberación del ingrediente o ingredientes activos puede utilizarse para formar las formas de administración de los compuestos dados a conocer en la presente memoria. La cantidad de desintegrante utilizada puede variar según el tipo de formulación y el modo de administración, y puede ser fácil de discernir por el experto ordinario en la materia. Puede utilizarse entre aproximadamente 0,5 y aproximadamente 15 por ciento en peso de desintegrante, o entre aproximadamente 1 y aproximadamente 5 por ciento en peso de desintegrante en la composición farmacéutica. Entre los desintegrantes que pueden utilizarse para formar composiciones farmacéuticas y formas de administración se incluyen, aunque sin limitarse a ellos, agar-agar, ácido algínico, carbonato cálcico, celulosa microcristalina, croscarmelosa sódica, crospovidona, polacrilina potasio, glicolato de almidón sódico, almidón de patata o tapioca, otros almidones, almidón pregelatinizado, otros almidones, arcillas, otras alginas, otras celulosas, gomas o mezclas de los mismos.
Entre los lubricantes que pueden utilizarse para formar composiciones farmacéuticas y formas de administración se incluyen, aunque sin limitarse a ellos, estearato de calcio, estearato de magnesio, aceite mineral, aceite mineral ligero, glicerina, sorbitol, manitol, polietilenglicol, otros glicoles, ácido esteárico, laurilsulfato sódico, talco, aceite vegetal hidrogenado (p.ej., aceite de cacahuete, aceite de semilla de algodón, aceite de girasol, aceite de sésamo, aceite de oliva, aceite de maíz y aceite de soja), estearato de cinc, oleato de etilo, laurato de etilo, agar o mezclas de los mismos. Entre los lubricantes adicionales se incluyen, por ejemplo, gel de sílice siloide, un aerosol coagulado de sílice sintético 0 mezclas de los mismos. Puede añadirse opcionalmente un lubricante, en una cantidad inferior a aproximadamente 1 por ciento en peso de la composición farmacéutica.
En el caso de que se deseen suspensiones y/o elixires acuosos para la administración oral, el ingrediente activo en los mismos puede combinarse con diversos agentes edulcorantes o saborizantes, materias colorantes o pigmentos y, por ejemplo, agentes emulsionantes y/o de suspensión, junto con diluyentes tales como agua, etanol, propilenglicol, glicerina y diversas combinaciones de los mismos.
Las tabletas pueden ser no recubiertas o recubrirse mediante técnicas conocidas para retrasar la desintegración y absorción en el tracto gastrointestinal y proporcionar, de esta manera, una acción sostenida durante un periodo más largo. Por ejemplo, puede utilizarse un material de retardo temporal, tal como monoestearato de glicerina o diestearato de glicerilo. Las formulaciones para la utilización oral también pueden presentarse en forma de cápsulas de gelatina dura, en las que el ingrediente activo se mezcla con un diluyente sólido inerte, por ejemplo, carbonato cálcico, fosfato cálcico o caolín, o en forma de cápsulas de gelatina blanda, en las que el ingrediente activo se mezcla con agua o un medio aceitoso, por ejemplo, aceite de cacahuete, parafina líquida o aceite de oliva.
Entre los surfactantes que pueden utilizarse para formar composiciones farmacéuticas y formas de administración se incluyen, aunque sin limitarse a ellos, surfactantes hidrofílicos, surfactantes lipofílicos y mezclas de los mismos. Es decir, puede utilizarse una mezcla de surfactantes hidrofílicos; puede utilizarse una mezcla de surfactantes lipofílicos, o puede utilizarse una mezcla de por lo menos un surfactante hidrofílico y por lo menos un surfactante lipofílico.
Un surfactante hidrofílico adecuado puede presentar generalmente un valor de EHL de por lo menos aproximadamente 10, mientras que los surfactantes lipofílicos adecuados pueden presentar generalmente un valor de EHL inferior a aproximadamente 10. Un parámetro empírico utilizado para caracterizar la hidrofilicidad y la hidrofobicidad relativas de los compuestos anfifílicos no iónicos es el equilibrio hidrofílico-lipofílico (valor "EHL"). Los surfactantes con valores de EHL inferiores son más lipofílicos o hidrofóbicos, y presentan una mayor solubilidad en aceites, mientras que los surfactantes con valores de EHL más altos son más hidrofílicos y presentan una mayor solubilidad en soluciones acuosas. Los surfactantes hidrofílicos se consideran generalmente aquellos compuestos que presentan un valor de EHL superior a aproximadamente 10, así como compuestos aniónicos, catiónicos o zwiteriónicos para los que la escala de EHL no es generalmente aplicable. De manera similar, los surfactantes lipofílicos (es decir, hidrofóbicos) son compuestos que presentan un valor de EHL igual o inferior a aproximadamente 10. Sin embargo, el valor de EHL de un surfactante es una mera guía general que se utiliza generalmente para permitir la formulación de emulsiones industriales, farmacéuticas y cosméticas.
Los surfactantes hidrofílicos pueden ser iónicos o no iónicos. Entre los surfactantes iónicos adecuados se incluyen, aunque sin limitarse a ellos, sales de alquilamonio, sales de ácido fusídico, derivados de ácido graso de aminoácidos, oligopéptidos y polipéptidos, derivados de glicérido de aminoácidos, oligopéptidos y polipéptidos, lecitinas y lecitinas hidrogenadas, lisolecitinas y lisolecitinas hidrogenadas, fosfolípidos y derivados de los mismos, lisofosfolípidos y derivados de los mismos, sales de éster de ácido graso-carnitina, sales de alquilsulfatos, sales de ácido graso, docusato sódico, actilactilatos, ésteres de ácido tartárico monoacetilado y diacetilado de monoglicéridos y diglicéridos, monoglicéridos y diglicéridos succinilados, ésteres de ácido cítrico de monoglicéridos y diglicéridos, y mezclas de los mismos.
En el grupo anteriormente indicado, entre los surfactantes iónicos se incluyen, a título de ejemplo, lecitinas, lisolecitina, fosfolípidos, lisofosfolípidos y derivados de los mismos, sales de éster de ácido graso-carnitina, sales de alquilsulfatos, sales de ácido graso, docusato sódico, actilactilatos, ésteres de ácido tartárico monoacetilado y diacetilado de monoglicéridos y diglicéridos, monoglicéridos y diglicéridos succinilados, ésteres de ácido cítrico de monoglicéridos y diglicéridos, y mezclas de los mismos.
Los surfactantes iónicos pueden ser las formas ionizadas de lecitina, lisolecitina, fosfatidilcolina, fosfatidiletanolamina, fosfatidilglicerol, ácido fosfatídico, fosfatidilserina, lisofosfatidilcolina, lisofosfatidiletanolamina, lisofosfatidilglicerol, ácido lisofosfatídico, lisofosfatidilserina, PEG-fosfatidiletanolamina, PVP-fosfatidiletanolamina, ésteres lactílicos de ácidos grasos, estearoil-2-lactilato, lactilato de estearoilo, monoglicéridos succinilados, ésteres de ácido tartárico mono/diacetilado de mono/diglicéridos, ésteres de ácido cítrico de mono/diglicéridos, colilsarcosina, caproato, caprilato, caprato, laurato, miristato, palmitato, oleato, ricinoleato, linoleato, linolenato, estearato, laurilsulfato, sulfato de teracecilo, docusato, lauroilcarnitinas, palmitoilcarnitinas, miristoilcarnitinas, y sales y mezclas de los mismos.
Entre los surfactantes no iónicos hidrofílicos pueden incluirse, aunque sin limitarse a ellos, alquilglucósidos, alquilmaltósidos, alquiltioglucósidos, lauril-macrogolglicéridos, alquiléteres de polioxialquileno, tales como alquiléteres de polietilenglicol, alquilfenoles de polioxialquileno, tales como alquilfenoles de polietilenglicol, ésteres de ácido graso de alquilfenol de polioxialquileno, tales como monoésteres de ácidos grasos de polietilenglicol y diésteres de ácidos grasos de polietilenglicol, ésteres de ácido graso de glicerol de polietilenglicol, ésteres de ácido graso de poliglicerol, ésteres de ácido graso de polioxialquilén-sorbitán, tales como ésteres de ácido graso de sorbitán-polietilenglicol, productos de transesterificación hidrofílica de un poliol con por lo menos un miembro de entre glicéridos, aceites vegetales, aceites vegetales hidrogenados, ácidos grasos y esteroles, esteroles de polioxietileno, derivados y análogos de los mismos, vitaminas polioxietiladas y derivadas de las mismas, copolímeros en bloque de polioxietilenopolioxipropileno, y mezclas de los mismos, ésteres de ácido graso de polietilenglicol-sorbitán y productos de transesterificación hidrofílicos de un poliol con por lo menos un miembro de entre triglicéridos, aceites vegetales y aceites vegetales hidrogenados. El poliol puede ser glicerol, etilenglicol, polietilenglicol, sorbitol, propilenglicol, pentaeritritol o un sacárido.
Entre otros surfactantes no iónicos hidrofílicos se incluyen, aunque sin limitarse a ellos, laurato de PEG-10, laurato de PEG-12, laurato de PEG-20, laurato de PEG-32, dilaurato de PeG-32, oleato de PEG-12, oleato de PEG-15, oleato de PEG-20, dioleato de PEG-20, oleato de PEG-32, oleato de PEG-200, oleato de PEG-400, estearato de PEG-15, diestearato de PEG-32, estearato de PEG-40, estearato de PEG-100, dilaurato de PEG-20, trioleato de glicerilo de PEG-25, dioleato de PEG-32, gliceril-laurato de PEG-20, gliceril-laurato de PEG-30, gliceril-estearato de PEG-20, gliceril-oleato de PEG-20, gliceril-oleato de PEG-30, gliceril-laurato de PEG-30, gliceril-laurato de PEG-40, aceite de nuez de palma-PEG-40, aceite de ricino hidrogenado-PEG-50, aceite de ricino-PEG-40, aceite de ricino-PEG-35, aceite de ricino-PEG-60, aceite de ricino hidrogenado-PEG-40, aceite de ricino hidrogenado-PEG-60, aceite de maíz-PEG-60, caprato/caprilato glicéridos de PEG-6, caprato/caprilato glicéridos de PEG-8, laurato de poliglicerilo-10, colesterol-PEG-30, fitoesterol-PEG-25, esterol de soja-PEG-30, trioleato de PEG-20, oleato de sorbitán-PEG-40, laurato de sorbitán-PEG-80, polisorbato 20, polisorbato 80, éter laurílico-POE-9, éter laurílico-POE-23, éter oleílico-POE-10, éter oleílico-POE-20, éter estearílico-POE-20, succinato de tocoferil PEG-100, colesterol-PEG-24, oleato de poliglicerilo-10, Tween 40, Tween 60, monoestearato de sucrosa, monolaurato de sucrosa, monopalmitato de sucrosa, serie de nonilfenol PEG 10-100, serie de octilfenol PEG 15-100 y poloxámeros.
Entre los surfactantes lipofílicos adecuados se incluyen, únicamente a título de ejemplo: alcoholes grasos, ésteres de glicerol-ácido graso, ésteres de glicerol-ácido graso acetilados, ésteres de alcohol inferior de ácidos grasos, ésteres de ácido graso de propilenglicol, ésteres de ácido graso de sorbitán, ésteres de ácido graso de polietilenglicol-sorbitán, esteroles y derivados de esterol, esteroles polioxietilados y derivados de esterol, alquiléteres de polietilenglicol, ésteres de azúcares, éteres de azúcares, derivados de ácido láctico de monoglicéridos y diglicéridos, productos de transesterificación hidrofóbica de un poliol con por lo menos un miembro de glicéridos, aceites vegetales, aceites vegetales hidrogenados, ácidos grasos y esteroles, vitaminas liposolubles/derivados de vitaminas, y mezclas de los mismos. Dentro de dicho grupo, entre los ejemplos no limitativos de surfactantes lipofílicos se incluyen ésteres de glicerol-ácido graso, ésteres de propilenglicol-ácido graso y mezclas de los mismos, o son productos de transesterificación hidrofóbica de un poliol con por lo menos un miembro de entre aceites vegetales, aceites vegetales hidrogenados y triglicéridos.
En una realización, la composición farmacéutica puede incluir un solubilizador para garantizar una buena solubilización
y/o disolución de un compuesto tal como se proporciona en la presente memoria y para minimizar la precipitación del compuesto. Lo anterior puede resultar especialmente importante para las composiciones farmacéuticas para la utilización no oral, p.ej., composiciones farmacéuticas para inyección. También puede añadirse un solubilizador para incrementar la solubilidad del fármaco hidrofílico y/u otros componentes, tales como surfactantes, o para mantener la composición farmacéutica como solución o dispersión estable u homogénea.
Entre los ejemplos de solubilizadores adecuados se incluyen, aunque sin limitarse a ellos, los siguientes: alcoholes y polioles, tales como etanol, isopropanol, butanol, alcohol bencílico, etilenglicol, propilenglicol, butanodioles e isómeros de los mismos, glicerol, pentaeritritol, sorbitol, manitol, transcutol, isosórbido de dimetilo, polietilenglicol, polipropilenglicol, alcohol polivinílico, hidroxipropilmetilcelulosa y otros derivados de celulosa, ciclodextrinas y derivados de ciclodextrina, éteres de polietilenglicoles con un peso molecular medio de entre aproximadamente 200 y aproximadamente 6000, tales como éter de PEG de alcohol tetrahidrofurfurílico (glicofurol) o metoxi-PEG, amidas y otros compuestos que contienen nitrógeno, tales como 2-pirrolidona, 2-piperidona, £-caprolactamo, N-alquilpirrolidona, N-hidroxialquilpirrolidona, N-alquilpiperidona, N-alquilcaprolactamo, dimetilacetamida y polivinilpirrolidona; ésteres, tales como propionato de etilo, tributilcitrato, trietilcitrato de acetilo, tributilcitrato de acetilo, trietilcitrato, oleato de etilo, caprilato de etilo, butirato de etilo, triacetina, monoacetato de propilenglicol, diacetato de propilenglicol, £-caprolactona e isómeros de la misma, 8-valerolactona e isómeros de la misma, p-butirolactona e isómeros de la misma, y otros solubilizadores conocidos de la técnica, tales como dimetilacetamida, dimetilisosórbido, N-metilpirrolidonas, monooctanoína, monoetiléter de dietilenglicol y agua.
También pueden utilizarse mezclas de solubilizadores. Entre los ejemplos se incluyen, aunque sin limitarse a ellos, triacetina, trietilcitrato, oleato de etilo, caprilato de etilo, dimetilacetamida, N-metilpirrolidona, N-hidroxietilpirrolidona, polivinilpirrolidona, hidroxipropilmetilcelulosa, hidroxipropilciclodextrinas, etanol, polietilenglicol 200-100, glicofurol, transcutol, propilenglicol y isosórbido de dimetilo. En algunas realizaciones, entre los solubilizadores se incluyen sorbitol, glicerol, triacetina, alcohol etílico, PEG-400, glicofurol y propilenglicol.
La cantidad de solubilizador que puede incluirse no se encuentra particularmente limitada. La cantidad de un solubilizador dado puede limitarse a una cantidad bioaceptable, que puede ser fácilmente determinada por el experto en la materia. Bajo algunas circunstancias, puede resultar ventajoso incluir cantidades de solubilizadores muy en exceso de las cantidades bioaceptables, por ejemplo para maximizar la concentración del fármaco, eliminando el exceso de solubilizador antes de proporcionar la composición farmacéutica al sujeto, mediante técnicas convencionales, tales como la destilación o evaporación. De esta manera, en caso de estar presente, el solubilizador puede encontrarse en una proporción en peso de aproximadamente 10 %, 25 %, 50 %, 100 % o hasta de aproximadamente 200 % en peso, respecto al peso combinado del fármaco, y otros excipientes. Si se desea, también pueden utilizarse cantidades muy pequeñas de solubilizador, tales como aproximadamente 5 %, 2 %, 1 % o incluso menos. Típicamente, el solubilizador puede encontrarse presente en una cantidad de entre aproximadamente 1 % y aproximadamente 100 %, más típicamente de entre aproximadamente 5 % y aproximadamente 25 % en peso.
La composición farmacéutica puede incluir además uno o más aditivos o excipientes farmacéuticamente aceptables. Entre dichos aditivos y excipientes se incluyen, aunque sin limitarse a ellos, antiadherentes, agentes antiespumantes, agentes tamponadores, polímeros, antioxidantes, conservantes, agentes quelantes, viscomoduladores, tonificadores, saborizantes, colorantes, aceites, odorizantes, opacificadores, agentes de suspensión, ligantes, rellenos, plastificadores, lubricantes y mezclas de los mismos.
Entre los conservantes ejemplares pueden incluirse antioxidantes, agentes quelantes, conservantes antimcirobianos, conservantes antifúngicos, conservantes de alcohol, conservantes ácidos y otros conservantes. Entre los antioxidantes ejemplares se incluyen, aunque sin limitarse a ellos, alfa-tocoferol, ácido ascórbico, palmitato de ascorbilo, hidroxianisol butilado, hidroxitolueno butilado, monotioglicerol, metabisulfito potásico, ácido propiónico, galato de propilo, ascorbato sódico, bisulfito sódico, metabisulfito sódico y sulfito sódico. Entre los agentes quelantes ejemplares se incluyen ácido etilén-diamina-tetraacético (EDTA), monohidrato de ácido cítrico, edetato disódico, edetato dipotásico, ácido edético, ácido fumárico, ácido málico, ácido fosfórico, edetato sódico, ácido tartárico y edetato trisódico. Entre los conservantes antimicrobianos ejemplares se incluyen, aunque sin limitarse a ellos, cloruro de benzalconio, cloruro de bencetonio, alcohol bencílico, bronopol, cetrimida, cloruro de cetilpiridinio, clorhexidina, clorobutanol, clorocresol, cloroxilenol, cresol, alcohol etílico, glicerina, hexetidina, imidurea, fenol, fenoxietanol, alcohol feniletílico, nitrato fenilmercúrico, propilenglicol y timerosal. Entre los conservantes antifúngicos ejemplares se incluyen, aunque sin limitarse a ellos, butilparabeno, metilparabeno, etilparabeno, propilparabeno, ácido benzoico, ácido hidroxibenzoico, benzoato potásico, sorbato potásico, benzoato sódico, propionato sódico y ácido sórbico. Entre los conservantes alcohol ejemplares se incluyen, aunque sin limitarse a ellos, etanol, polietilenglicol, fenol, compuestos fenólicos, bisfenol, clorobutanol, hidroxibenzoato y alcohol feniletílico. Entre los conservantes ácidos ejemplares se incluyen, aunque sin limitarse a ellos, vitamina A, vitamina C, vitamina E, beta-caroteno, ácido cítrico, ácido acético, ácido deshidroacético, ácido ascórbico, ácido sórbico y ácido fítico. Entre otros conservantes se incluyen, aunque sin limitarse a ellos, tocoferol, acetato de tocoferol, mesilato de deteroxima, cetrimida, hidroxianisol butilado (BHA), hidroxitolueno butilado (BHT), etilendiamina, laurilsulfato sódico (SLS), sulfato de lauriléter sódico (SLES), bisulfito sódico, metabisulfito sódico, sulfito potásico, metabisulfito potásico, glidante Plus, Phenonip, metilparabeno, Germall 115, Germaben II, neolona, Kathon y euxilo. En determinadas realizaciones, el conservante es un antioxidante. En otras realizaciones, el conservante es un agente quelante.
Entre los aceites ejemplares se incluyen, aunque sin limitarse a ellos, aceites de almendra, aceite de hueso de albaricoque, aguacate, babasú, bergamota, semilla de grosella negra, borraja, cade, camomila, colza, alcaravea, carnauba, ricino, canela, manteca de cacao, hígado de bacalao, café, maíz, semilla de algodón, emú, eucalipto, onagra, pescado, semilla de lino, geraniol, calabaza común, semilla de uva, avellana, hisopo, miristato de isopropilo, jojoba, nuez de kukui, lavandín, lavanda, limón, Litsea cubeba, nuez de macadamian, malva, semilla de mango, pradera blanca, visón, nuez moscada, oliva, naranja, reloj anaranjado, palma, nuez de palma, nuez de melocotón, cacahuete, semilla de amapola, semilla de calabaza, semilla de colza, salvado de arroz, romero, cártamo, sándalo, sasanqua, ajedrea, espino amarillo, sésamo, manteca de karité, silicona, soja, girasol, árbol del té, cardo, camelia de Japón, vetiver, nuez y germen de trigo. Entre los aceites ejemplares se incluyen, aunque sin limitarse a ellos, estearato de butilo, triglicérido caprílico, triglicérido cáprico, ciclometicona, sebacato de dietilo, dimeticona 360, miristato de isopropilo, aceite mineral, octildodecanol, alcohol oleílico, aceite de silicona y combinaciones de los mismos.
Además, puede incorporarse un ácido o una base en la composición farmacéutica para facilitar el procesamiento, para potenciar la estabilidad o por otros motivos. Entre los ejemplos de bases farmacéuticamente aceptables se incluyen aminoácidos, ésteres de aminoácido, hidróxido amónico, hidróxido potásico, hidróxido sódico, hidrogenocarbonato sódico, hidróxido de aluminio, carbonato cálcico, hidróxido de magnesio, aluminosilicato de magnesio, aluminosilicato sintético, hidrocalcita sintética, hidróxido de magnesio-aluminio, diisopropiletilamina, etanolamina, etilendiamina, trietanolamina, trietilamina, triisopropanolamina, trimetilamina, tris(hidroximetil)aminometano (TRIS) y similares. También resultan adecuadas bases que son sales de un ácido farmacéuticamente aceptable, tal como ácido acético, ácido acrílico, ácido adípico, ácido algínico, ácido alcanosulfónico, aminoácidos, ácido ascórbico, ácido benzoico, ácido bórico, ácido butírico, ácido carbónico, ácido cítrico, ácidos grasos, ácido fórmico, ácido fumárico, ácido glucónico, ácido hidroquinosulfónico, ácido isoascórbico, ácido láctico, ácido maleico, ácido oxálico, ácido parabromofenilsulfónico, ácido propiónico, ácido p-toluenosulfónico, ácido salicílico, ácido esteárico, ácido succínico, ácido tánico, ácido tartárico, ácido tioglicólico, ácido toluenosulfónico, ácido úrico y similares. También pueden utilizarse sales de ácido polipróticos, tales como fosfato sódico, hidrogenofosfato disódico y dihidrogenofosfato sódico. En el caso de que la base sea una sal, el catión puede ser cualquier catión conveniente y farmacéuticamente aceptable, tal como amonio, metales alcalinos, metales alcalinotérreos, y similares. Entre los ejemplos pueden incluirse, aunque sin limitarse a ellos, sodio, potasio, litio, magnesio, calcio y amonio.
Son ácidos adecuados, los ácidos orgánicos o inorgánicos farmacéuticamente aceptables. Entre los ejemplos de ácidos inorgánicos adecuados se incluyen ácido clorhídrico, ácido bromhídrico, ácido yodhírico, ácido sulfúrico, ácido nítrico, ácido bórico, ácido fosfórico y similares. Entre los ejemplos de ácidos orgánicos adecuados se incluyen ácido acético, ácido acrílico, ácido adípico, ácido algínico, ácidos alcanosulfónicos, aminoácidos, ácido ascórbico, ácido benzoico, ácido bórico, ácido butírico, ácido carbónico, ácido cítrico, ácidos grasos, ácido fórmico, ácido fumárico, ácido glucónico, ácido hidroquinosulfónico, ácido isoascórbico, ácido láctico, ácido maleico, ácido metanosulfónico, ácido oxálico, ácido para-bromofenilsulfónico, ácido propiónico, ácido p-toluenosulfónico, ácido salicílico, ácido esteárico, ácido succínico, ácido tánico, ácido tartárico, ácido tioglicólico, ácido toluenosulfónico, ácido úrico y similares.
Formulaciones para la administración parenteral
En algunas realizaciones, en la presente memoria se proporcionan para la utilización según la presente invención composiciones farmacéuticas para la administración parenteral que contienen un compuesto tal como se da a conocer en la presente memoria y un excipiente farmacéutico adecuado para la administración parenteral. En algunas realizaciones, en la presente memoria se proporcionan composiciones farmacéuticas para la administración parenteral que contienen: (i) una cantidad eficaz de un compuesto dado a conocer, opcionalmente (ii) una cantidad eficaz de uno o más segundos agentes, y (iii) uno o más excipientes farmacéuticos adecuados para la administración parenteral. En algunas realizaciones, la composición farmacéutica contiene, además: (iv) una cantidad eficaz de un tercer agente.
Entre las formas en las que pueden incorporarse las composiciones farmacéuticas dadas a conocer para la administración mediante inyección se incluyen suspensiones acuosas o aceitosas, o emulsiones, con aceite de sésamo, aceite de maíz, aceite de semilla de algodón o aceite de cacahuete, así como elixires, manitol, dextrosa, o una solución acuosa estéril, y vehículos farmacéuticos similares.
Las soluciones acuosas en solución salina también se utilizan convencionalmente para la inyección. También puede utilizarse etanol, glicerol, propilenglicol, polietilenglicol líquido, y similares (y mezclas adecuadas de los mismos), derivados de ciclodextrina y aceites vegetales.
Las soluciones acuosas en solución salina también se utilizan convencionalmente para la inyección. También puede utilizarse etanol, glicerol, propilenglicol, polietilenglicol líquido, y similares (y mezclas adecuadas de los mismos), derivados de ciclodextrina y aceites vegetales. Puede mantenerse la fluidez apropiada, por ejemplo, mediante la utilización de un recubrimiento, tal como lecitina, para el mantenimiento del tamaño de partícula requerido en el caso de dispersiones, y mediante la utilización de sufactantes. El bloqueo de la acción de los microorganismos puede conseguirse mediante la inclusión de diversos agentes antibacterianos y antifúngicos, por ejemplo parabenos, clorobutanol, fenol, ácido sórbico, timerosal y similares.
Las soluciones inyectables estériles se preparan mediante la incorporación de un compuesto tal como se da a conocer en la presente memoria en la cantidad requerida en el solvente apropiado con otros ingredientes diversos tal como se han señalado anteriormente, según resulte apropiado, seguido de la esterilización mediante filtración. Generalmente, se preparan dispersiones mediante incorporación de los diversos ingredientes activos esterilizados en un vehículo estéril que contiene el medio de dispersión básico y los demás ingredientes apropiados de entre los señalados anteriormente. En el caso de polvos estériles para la preparación de soluciones inyectables estériles, ciertos métodos de preparación son las técnicas de secado al vacío o de liofilización, que rinden unos polvos del ingrediente activo más cualquier ingrediente deseado adicional a partir de una solución previamente esterilizada mediante filtración del mismo.
Las formulaciones inyectables pueden esterilizarse, por ejemplo, mediante filtración a través de un filtro de retención bacteriana, o mediante la incorporación de agentes esterilizantes en forma de composiciones sólidas estériles que pueden disolverse o dispersarse en agua estéril u otro medio inyectable estéril antes de la utilización. Las composiciones inyectables pueden contener entre aproximadamente 0,1 % y aproximadamente 5 % p/p de un compuesto tal como se da a conocer en la presente memoria.
Formulaciones para la administración tópica
En algunas realizaciones, en la presente memoria se proporcionan para la utilización según la presente invención composiciones farmacéuticas para la administración tópica (p.ej., transdérmica) que contienen un compuesto tal como se da a conocer en la presente memoria y un excipiente farmacéutico adecuado para la administración tópica. En algunas realizaciones, en la presente memoria se proporcionan composiciones farmacéuticas para la administración tópica que contienen: (i) una cantidad eficaz de un compuesto dado a conocer, opcionalmente (ii) una cantidad eficaz de uno o más segundos agentes, y (iii) uno o más excipientes farmacéuticos adecuados para la administración tópica. En algunas realizaciones, la composición farmacéutica contiene, además: (iv) una cantidad eficaz de un tercer agente.
Las composiciones farmacéuticas proporcionadas en la presente memoria pueden formularse en preparaciones en formas sólidas, semisólidas o líquidas adecuadas para la administración local o tópica, tales como geles, gelatinas solubles en agua, cremas, lociones, suspensiones, espumas, polvos, lodos, pomadas, soluciones, aceites, pastas, supositorios, pulverizaciones, emulsiones, soluciones salinas y soluciones a base de dimetilsulfóxido (DMSO). En general, los portadores con densidades más altas son capaces de proporcionar una zona con una exposición prolongada a los ingredientes activos. En contraste, una formulación de solución puede proporcionar una exposición más inmediata del ingrediente activo a la zona seleccionada.
Las composiciones farmacéuticas pueden comprender además portadores o excipientes sólidos o en fase gel, que son compuestos que permiten una penetración incrementada o que asisten en la administración de moléculas terapéuticas a través de la barrera de permeabilidad del estrato córneo de la piel. El experto en la técnica de la formulación tópica conocerá muchas de dichas moléculas potenciadoras de la penetración. Entre los ejemplos de dichos portadores y excipientes se incluyen, aunque sin limitarse a ellos, humectantes (p.ej., urea), glicoles (p.ej., propilenglicol), alcoholes (p.ej., etanol), ácidos grasos (p.ej., ácido oleico), surfactantes (p.ej., miristato de isopropilo y laurilsulfato sódico), pirrolidonas, monolaurato de glicerol, sulfóxidos, terpenos (p.ej., mentol), aminas, amidas, alcanos, alcanoles, agua, carbonato cálcico, fosfato cálcico, diversos azúcares, almidones, derivados de celulosa, gelatina y polímeros, tales como polietilenglicoles.
Otra formulación ejemplar para la utilización en los métodos dados a conocer utiliza dispositivos de administración transdérmica ("parches"). Dichos parches transdérmicos pueden utilizarse para proporcionar una infusión continua o discontinua de un compuesto tal como se proporciona en la presente memoria en cantidades controladas, con o sin otro agente.
La construcción y utilización de parches transdérmicos para la administración de agentes farmacéuticos es bien conocida de la técnica. Ver, p.ej., la patente US n° 5.023.252, n° 4.992.445 y n° 5.001.139. Dichos parches pueden construirse para la administración continua, pulsátil o a demanda de agentes farmacéuticos.
Entre los dispositivos adecuados para la utilización en la administración de composiciones intradérmicas farmacéuticamente aceptables en la presente memoria se incluyen dispositivos de aguja corta, tales como los indicados en las patentes US n° 4.886.499, n° 5.190.521, n° 5.328.483, n° 5.527.288, n° 4.270.537, n° 5.015.235, n° 5.141.496 y n° 5.417.662. Pueden administrarse composiciones intradérmicas mediante dispositivos que limitan la longitud de penetración efectiva de una aguja en la piel, tales como los indicados en la publicación de patente PCT n° WO 99/34850 y equivalentes funcionales de los mismos. Resultan adecuados los dispositivos de inyección de chorro que administran vacunas líquidas en la dermis mediante un inyector de chorro líquido y/o mediante una aguja que perfora el estrato córneo y produce un chorro que alcanza la dermis. Los dispositivos de inyección de chorro se describen en, por ejemplo, las patentes US n° 5.480.381, 5.599.302, 5.334.144, 5.993.412, 5.649.912, 5.569.189, 5.704.911, 5.383.851, 5.893.397, 5.466.220, 5 .339.163, 5.312.335, 5.503.627, 5.064.413, 5.520.639, 4.596.556, 4.790.824, 4.941.880, 4.940.460 y publicaciones de patente PCT n°WO 97/37705 y n°WO 97/13537. Resultan adecuados los dispositivos balísticos de
polvos/partículas que utilizan gas comprimido para acelerar la vacuna en forma de polvos a través de las capas externas de la piel hasta la dermis. Alternativa o adicionalmente, pueden utilizarse jeringas convencionales en el método clásico de Mantoux de administración intradérmica.
Las formulaciones administrables por vía tópica pueden comprender, por ejemplo, entre aproximadamente 1 % y aproximadamente 10 % (p/p) de un compuesto proporcionado en la presente memoria respecto al peso total de la formulación, aunque la concentración del compuesto proporcionada en la presente memoria en la formulación puede ser de hasta el límite de solubilidad del compuesto en el solvente. En algunas realizaciones, las formulaciones administrables por vía tópica pueden comprender, por ejemplo, entre aproximadamente 1 % y aproximadamente 9 % (p/p) de un compuesto proporcionado en la presente memoria, tal como de entre aproximadamente 1 % y aproximadamente 8 % (p/p), tal como de entre aproximadamente 1 % y aproximadamente 7 % (w/w), tal como de entre aproximadamente 1 % y aproximadamente 6 % (w/w), tal como de entre aproximadamente 1 % y aproximadamente 5 % (w/w), tal como de entre aproximadamente 1 % y aproximadamente 4% (w/w), tal como de entre aproximadamente 1 % y aproximadamente 3 % (p/p) y tal como de entre aproximadamente 1 % y aproximadamente 2 % (p/p) de un compuesto proporcionado en la presente memoria. Las formulaciones para administración tópica pueden comprender además uno o más excipientes farmacéuticamente aceptables adicionales indicados en la presente memoria.
Formulaciones para la administración mediante inhalación
En algunas realizaciones, en la presente memoria se proporcionan para la utilización según la presente invención composiciones farmacéuticas para la administración mediante inhalación que contienen un compuesto tal como se da a conocer en la presente memoria y un excipiente farmacéutico adecuado para la administración tópica. En algunas realizaciones, en la presente memoria se proporcionan composiciones farmacéuticas para la administración mediante inhalación que contienen: (i) una cantidad eficaz de un compuesto dado a conocer, opcionalmente (ii) una cantidad eficaz de uno o más segundos agentes, y (iii) uno o más excipientes farmacéuticos adecuados para la administración mediante inhalación. En algunas realizaciones, la composición farmacéutica contiene, además: (iv) una cantidad eficaz de un tercer agente.
Entre las composiciones farmacéuticas para inhalación o insuflado se incluyen soluciones y suspensiones en solventes acuosos u orgánicos farmacéuticamente aceptables, o mezclas de los mismos, y polvos. Las composiciones farmacéuticas líquidas o sólidas pueden contener excipientes farmacéuticamente aceptables adecuados tales como los indicados en la presente memoria. En algunas realizaciones, las composiciones farmacéuticas se administran por la vía oral o respiratoria nasal para un efecto local o sistémico. Las composiciones farmacéuticas en solventes farmacéuticamente aceptables pueden nebulizarse mediante la utilización de gases inertes. Las soluciones nebulizadas pueden inhalarse directamente a partir del dispositivo nebulizador o el dispositivo nebulizador puede unirse a una mascarilla de tipo tienda facial o un respirador de presión positiva intermitente. Pueden administrarse composiciones farmacéuticas en solución, suspensión o polvos, p.ej., por vía oral o nasal, a partir de dispositivos que administran la formulación de una manera apropiada.
En la presente memoria se proporcionan además formulaciones para la administración mediante inhalación. Pueden utilizarse todos los tipos de formulación inhalable conocidos de la técnica en relación a métodos proporcionados en la presente memoria.
En un inhalador de polvos secos, la dosis que debe administrarse se almacena en forma de unos polvos secos no presurizados y, mediante accionamiento del inhalador, las partículas de los polvos son inhaladas por el paciente. Los inhaladores de polvos secos pueden ser dispositivos "pasivos" en los que la respiración del paciente es la única fuente de gas que proporciona una fuerza motriz en el dispositivo, o dispositivos "activos" en los que se utiliza una fuente de gas comprimido o una fuente de energía alternativa. Las formulaciones proporcionadas en la presente memoria pueden administrarse con dispositivos inhaladores pasivos o activos.
Aunque resulta deseable administrar la proporción más elevada posible de las partículas de material activo en el pulmón profundo, habitualmente resulta preferible que el mínimo posible de los demás componentes penetre en el pulmón profundo. Por lo tanto, los polvos generalmente incluyen partículas de un material activo y las partículas portadoras para portar las partículas de material activo. Las partículas portadoras pueden estar compuestas de cualquier material o combinación de materiales farmacológicamente inerte que resulte aceptable para la inhalación. En algunas realizaciones, las partículas de portador están compuestas de uno o más azúcares cristalinos. En algunas realizaciones, las partículas de portador pueden estar compuestas de uno o más alcoholes de azúcar o polioles. En algunas realizaciones, las partículas de portador son partículas de dextrosa o lactosa. En algunas realizaciones, la cantidad de partículas de portador es de hasta 95 %, de hasta 90 %, de hasta 80 %, o de hasta 50 % en peso respecto al peso total de la composición.
También puede proporcionarse un material aditivo a una dosis que indique al paciente que se ha administrado la dosis (ver, p.ej., el documento n° WO 01/82906). El material aditivo, también denominado material indicador, puede encontrarse presente en los polvos como se han formulado para el inhalador de polvos secos, o puede encontrarse presente en una forma separada, tal como en una localización separada dentro del inhalador, de manera que el aditivo
resulte arrastrado por el flujo de aire generado con la inhalación, simultánea o secuencialmente con los polvos que contienen el material activo. De acuerdo con lo anterior, en la presente memoria se proporciona una formulación que comprende un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), por ejemplo en forma de un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo, en combinación con un material portador.
Las formulaciones proporcionadas en la presente memoria, al inhalarse, en algunas realizaciones muestran un tiempo hasta el efecto terapéutico inferior a 3 horas, 2 horas, 1 hora, 30 minutos, 15 minutos, 10 minutos o 5 minutos. En algunas realizaciones, las formulaciones proporcionadas en la presente memoria, al inhalarlas, presentarán una duración terapéutica de entre aproximadamente 1 y 48 horas.
En determinadas realizaciones de la presente invención, cada dosis se almacena en un "blíster" de un paquete de blísters. A este respecto, debido a que un agente activo puede ser susceptible de oxidación, en ocasiones resulta importante evitar (o limitar sustancialmente) la oxidación del agente activo antes de la administración. De esta manera, en algunas realizaciones, se impide la exposición al aire de la formulación antes de la administración mediante almacenamiento de cada dosis en un blíster sellado. En algunas realizaciones, la oxidación se bloquea (o limita) adicionalmente mediante la introducción de una pluralidad de blísteres en un recipiente sellado adicional, tal como una bolsa sellada realizada en, por ejemplo, una lámina, tal como papel de aluminio. En algunas realizaciones, la utilización de los blísters sellados (y bolsas selladas opcionales) puede minimizar la necesidad de incluir antioxidantes en la formulación.
En el caso de la administración mediante un inhalador de polvos secos de las partículas de ingrediente activo en el pulmón, en donde pueden absorberse, las características de tamaño de partícula de los polvos resultan particularmente importantes. En particular, para la administración efectiva del ingrediente activo profundamente dentro del pulmón, las partículas activas deben ser pequeñas y ser bien dispersadas al accionar el inhalador. En algunas realizaciones, se genera una fracción de partículas finas de por lo menos 35 % al accionar el dispositivo inhalador. En algunas realizaciones, al accionar se genera una fracción de partículas finas de por lo menos 60 %, de por lo menos 70 % o de por lo menos 80 %.
En determinadas realizaciones, la formulación puede contener además partículas finas de un material excipiente, que puede ser un material tal como uno de los indicados anteriormente como adecuados para la utilización como un material portador, por ejemplo un azúcar cristalino, tal como dextrosa o lactosa. El material excipiente fino puede ser del mismo material o un material diferente que las partículas de portador, en caso de hallarse ambas presentes. En determinadas realizaciones, en donde cualesquiera partículas de portador y/o cualquier material excipiente fino presente sea de un material que es capaz él mismo de inducir una sensación en la zona orofaríngea, las partículas de portador y/o el material excipiente fino también puede ser el material indicador. Por ejemplo, las partículas de portador y/o cualquier excipiente de partícula fina puede comprender manitol. En determinadas realizaciones, la cantidad de material excipiente fino, en caso de estar presente, puede ser de hasta 50 %, de hasta 30 %, o de hasta 20 % en peso respecto al peso total de la composición.
Las formulaciones proporcionadas en la presente memoria también pueden formularse con excipientes adicionales para asistir en la administración y liberación. En determinadas realizaciones, pueden formularse polvos con particulares de portador relativamente grandes que ayudan a las propiedades de flujo de los polvos. Entre los ejemplos de partículas de portador grandes se incluyen, aunque sin limitarse a ellos, partículas de lactosa con un diámetro aerodinámico medio superior a 90 micrómetros. En algunas realizaciones, las micropartículas hidrofóbicas pueden dispersarse en un material portador. Por ejemplo, las micropartículas hidrofóbicas pueden dispersarse en una matriz de polisacárido, formulando la composición global como micropartículas para la administración directa en el pulmón. El polisacárido actúa como una barrera adicional a la liberación inmediata del agente activo. Lo anterior puede ayudar adicionalmente al proceso controlado de liberación. Un ejemplo de un polisacárido adecuado es la goma xantana. Entre los ejemplos de materiales hidrofóbicos se incluyen, aunque sin limitarse a ellos, ácidos grasos de estado sólido, tales como ácido oleico, ácido láurico, ácido palmítico, ácido esteárico, ácido erúcico, ácido behénico o derivados (p.ej., esteres y sales) de los mismos. Entre los ejemplos específicos de dichos materiales se incluyen, aunque sin limitarse a ellos, fosfatidilcolinas, fosfatidilgliceroles y otros surfactantes pulmonares naturales y sintéticos. En algunas realizaciones, las formulaciones proporcionadas en la presente memoria contienen estearatos de metal, en particular estearato de magnesio, que ha sido autorizado para la administración por el pulmón.
Las formulaciones proporcionadas en la presente memoria pueden incluir además uno o más aditivos de control de flujo (ACF) además del portador y el ingrediente activo. En algunas realizaciones, los ACF pueden proporcionarse en una cantidad de entre aproximadamente 0,1 % y aproximadamente 10 % en peso, de entre aproximadamente 0,15 % y 5 % en peso, o de entre aproximadamente 0,5 % y aproximadamente 2 % en peso de la composición total. En algunas realizaciones, entre los ACF se incluyen, aunque sin limitarse a ellos, materiales antiadherentes. En algunas realizaciones, entre los ACF se incluyen, aunque sin limitarse a ellos, estearato de magnesio, leucina, lecitina y estearilfumarato sódico y los indicados en la patente US n° 6.153.224.
En determinadas realizaciones, las formulaciones proporcionadas en la presente memoria pueden ser una formulación
"sin portadores", que incluye únicamente el ingrediente activo y uno o más antiadherentes. Dichas formulaciones sin portador se describen en el documento n° WO 97/03649.
Tal como se utiliza en la presente memoria, a menos que se especifique lo contrario, la expresión "material antiadherente" se refiere a aquellos materiales aditivos que reducirán la cohesión entre las partículas de los polvos. Entre dichos materiales se incluirán, aunque sin limitarse a ellas, leucina y lecitina. En algunas realizaciones, el material antiadherente comprende un aminoácido. Se ha observado que los aminoácidos proporcionan, en caso de estar presentes como material antiadherente, una fracción respirable elevada del material activo y además buenas propiedades de flujo a los polvos. En algunas realizaciones, el aminoácido es la leucina, en particular la L-leucina. En algunas realizaciones, también pueden utilizarse las formas D- y DL-. El material antiadherente puede comprender uno o más de cualquiera de los aminoácidos siguientes: leucina, isoleucina, lisina, valina, metionina, cisteína y fenilalanina. En algunas realizaciones, el material antiadherente puede incluir estearato de magnesio o dióxido de silicio coloidal.
En algunas realizaciones, las formulaciones proporcionadas en la presente memoria son una formulación de aerosol. En algunas realizaciones, la formulación de aerosol puede estar contenida en un cartucho. Entre los ejemplos de formulación se aerosol se incluyen, aunque sin limitarse a ellos, una formulación de solución de aerosol y una formulación de suspensión de aerosol. En determinadas formulaciones, la formulación de aerosol puede contener un compuesto proporcionado en la presente memoria, opcionalmente en combinación con otro u otros ingredientes activos, en un propelente o en un sistema de propelente/solvente, y opcionalmente, un aditivo o excipiente farmacéuticamente aceptable adicional.
El propelente puede ser cualquier propelente licuado por presión y es preferentemente un hidrofluoroalcano (HFA) o una mezcla de diferentes HFA, incluyendo, aunque sin limitarse a ellos, HFA 134a (1,1,1,2-tetrafluoroetano), HFA 227 (1,1,1,2,3,3,3-heptafluoropropano) y mezclas de los mismos.
El solvente generalmente presenta una polaridad más elevada que la del propelente y puede incluir una o más sustancias, tales como un alcohol farmacéuticamente aceptable (p.ej., etanol), un poliol, tal como propilenglicol o polietilenglicol, o mezclas de los mismos. En algunas realizaciones, el solvente es un alcohol de alquilo inferior C1-C4 lineal o ramificado, tal como etanol y alcohol isopropílico. En una realización, el cosolvente es etanol.
En algunas realizaciones, el ingrediente activo de la formulación se disuelve de manera sustancialmente completa y homogénea en el sistema de propelente/solvente, es decir, la formulación es una formulación en solución.
Opcionalmente, la formulación puede comprender otros aditivos o excipientes farmacéuticamente aceptables, que son materiales sustancialmente inertes que son no tóxicos y no interactúan de manera negativa con otros componentes de la formulación. En algunas realizaciones, la formulación puede comprender uno o más cosolventes, surfactantes, carbohidratos, fosfolípidos, polímeros, agentes humectantes, estabilizadores, lubricantes o componentes de baja volatilidad.
En algunas realizaciones, puede utilizarse como estabilizador una cantidad adecuada de un ácido (ácido orgánico o inorgánico (ácidos minerales)). Entre los ejemplos se incluyen, aunque sin limitarse a ellos, ácido monoprótico o poliprótico farmacéuticamente aceptable, tal como ácido hidrácido (ácido clorhídrico, ácido bromhídrico, ácido yodhídrico, etc.), ácido fosfórico, ácido nítrico, ácido sulfúrico y oxoácidos de halógeno.
En algunas realizaciones, pueden utilizarse componentes de baja volatilidad con el fin de incrementar el diámetro aerodinámico de la mediana de la masa (DAMM) de las partículas de aerosol con el accionamiento del inhalador y/o para mejorar la solubilidad del ingrediente activo en el sistema de propelente/solvente. En algunas realizaciones, el componente de baja volatilidad presenta una presión de vapor a 25°C inferior a 0,1 kPa, o inferior a 0,05 kPa. Entre los ejemplos de componentes de baja volatilidad se incluyen, aunque sin limitarse a ellos, ésteres, tales como miristato de isopropilo, miristato de ascorbilo, ésteres de tocoferol; glicoles, tales como propilenglicol, polietilenglicol y glicerol, y agentes activos en superficie, tales como ácidos carboxílicos orgánicos saturados (p.ej., los ácidos láurico, mirístico y esteárico) y ácidos carboxílicos insaturados (p.ej., ácido oleico o ascórbico). La cantidad del componente de baja volatilidad puede variar entre 0,1 % y 10 % p/p, entre 0,5 % y 5 % (p/p) o entre 1 % y 2 % (p/p).
En algunas realizaciones, puede añadirse agua en una cantidad de entre 0,005 % y 0,3 % (p/p) a las formulaciones con el fin de afectar favorablemente la solubilidad del ingrediente activo sin incrementar la DAMM de las gotas de aerosol con el accionamiento.
Formulaciones para la administración ocular
En algunas realizaciones, en la presente memoria se proporciona para la utilización según la presente invención una composición farmacéutica para el tratamiento de trastornos oftálmicos. La composición farmacéutica puede contener una cantidad eficaz de un compuesto tal como se da a conocer en la presente memoria y un excipiente farmacéutico adecuado para la administración ocular. Las composiciones farmacéuticas adecuadas para la administración ocular pueden presentarse como formas de administración discretas, tales como gotas o pulverizaciones, cada uno de los
cuales contiene una cantidad predeterminada de un ingrediente activo, una solución, o una suspensión en un líquido acuoso o no acuoso, una emulsión de aceite en agua o una emulsión líquida de agua en aceite. Entre otras formas de administración se incluyen la inyección intraocular, la inyección intravítrea, la vía tópica, o mediante la utilización de un dispositivo de elución de fármaco, microcápsula, implante o dispositivo de microfluidos. En algunos casos, los compuestos tal como se dan a conocer en la presente memoria se administran con un portador o excipiente que incrementa la penetración intraocular del compuesto, tal como una emulsión de aceite y agua con partículas coloidales que presentan un núcleo aceitoso circundado por una película interfacial. Se encuentra contemplada la utilización de todas las vías locales en el ojo, entre ellas las vías tópica, subconjuntival, periocular, retrobulbar, subtenon, intracameral, intravítrea, intraocular, subretiniana, yuxtaescleral y supracoroidal. Puede ser viable la administración sistémica o parenteral, incluyendo, aunque sin limitación, la administración intravenosa, subcutánea y oral. Un método ejemplar de administración es la inyección intravítrea o subtenon de soluciones o suspensiones, o la colocación intravítrea o subtenon de dispositivos bioerosionales o no bioerosionables, o mediante la administración ocular tópica de soluciones o suspensiones, o la administración yuxtaescleral posterior de una formulación en gel o crema.
Pueden prepararse gotas oculares mediante disolución del ingrediente activo en una solución acuosa estéril, tal como solución salina fisiológica, solución tamponadora, etc., o mediante combinación de composiciones de polvos para la disolución antes del uso. Pueden seleccionarse otros vehículos, tal como es conocido de la técnica, incluyendo, aunque sin limitación, solución salina equilibrada, solución salina, poliéteres solubles en agua, tales como polietilenglicol; polivinilos, tales como alcohol polivinílico y povidona; derivados de celulosa, tales como metilcelulosa e hidroxipropilmetilcelulosa; derivados de petróleo, tales como aceite mineral y vaselina blanca; lípidos animales, tales como lanolina; polímeros de ácido acrílico, tales como gel de carboxipolimetileno; grasas vegetales, tales como aceite de cacahuete y polisacáridos, tales como dextranos, y glucosaminoglicanos, tales como hialuronato sódico. En algunas realizaciones, pueden añadirse aditivos habitualmente utilizados en las gotas oftálmicas. Entre dichos aditivos se incluyen agentes isotonificadores (p.ej., cloruro sódico, etc.), agente tamponador (p.ej., ácido bórico, monohidrogenofosfato sódico, dihidrogenofosfato sódico, etc.), conservantes (p.ej., cloruro de benzalconio, cloruro de bencetonio, clorobutanol, etc.), espesantes (p.ej., sacáridos, tales como lactosa, manitol, maltosa, etc.; p.ej., ácido hialurónico o sus sales, tales como hialuronato sódico, hialuronato potásico, etc.; p.ej., mucopolisacáridos, tales como condroitín sulfato, etc.; p.ej., poliacrilato sódico, polímero de carboxivinilo, poliacrilato entrecruzado, alcohol polivinílico, polivinilpirrolidona, metilcelulosa, hidroxipropilmetilcelulosa, hidroxietilcelulosa, carboximetilcelulosa, hidroxipropilcelulosa u otros agentes conocidos por el experto en la materia).
En algunos casos, entre las partículas coloidales se incluyen por lo menos un agente catiónico y por lo menos un surfactante no iónico, tal como poloxámero, tiloxapol, un polisorbato, un derivado de polioxietileno-aceite de ricino, un éster de sorbitán o un estearato de polioxilo. En algunos casos, el agente catiónico es una alquilamina, una alquilamina terciaria, un compuesto de amonio cuaternario, un lípido catiónico, un aminoalcohol, una sal de biguanidina, un compuesto catiónico o una mezcla de los mismos. En algunos casos, el agente catiónico es una sal biguanidina, tal como clorhexidina, biguanidina de poliaminopropilo, fenformina, alquilbiguanidina, o una mezcla de los mismos. En algunos casos, el compuesto de amino cuaternario es un haluro de benzalconio, haluro de lauralconio, cetrimida, haluro de hexadeciltrimetilamonio, haluro de tetradeciltrimetilamonio, haluro de dodeciltrimetilamonio, haluro de cetrimonio, haluro de bencetonio, haluro de behenalconio, haluro de cetalconio, haluro de cetetildimonio, haluro de cetilpiridinio, haluro de benzododecinio, haluro de cloralilmetenamina, haluro de miristilalconio, haluro de estearalconio o una mezcla de dos o más de los mismos. En algunos casos, el agente catiónico es un cloruro de benzalconio, cloruro de lauralconio, bromuro de benzododecinio, cloruro de bencetonio, bromuro de hexadeciltrimetilamonio, bromuro de tetradeciltrimetilamonio, bromuro de dodeciltrimetilamonio o una mezcla de dos o más de los mismos. En algunos casos, la fase aceite es aceite mineral y aceite mineral ligero, triglicéridos de cadena intermedia (TCI), aceite de coco; aceites hidrogenados que comprenden aceite de semilla de algodón hidrogenado, aceite de palma hidrogenado, aceite de ricino hidrogenado o aceite de soja hidrogenado; derivados de polioxietileno-aceite de ricino hidrogenado que comprenden polioxil-40-aceite de ricino hidrogenado, polioxil-60-aceite de ricino hidrogenado o polioxil-100-aceite de ricino hidrogenado.
Formulaciones para la administración de liberación controlada
En algunas realizaciones, en la presente memoria se proporcionan para la utilización según la presente invención composiciones farmacéuticas para la administración de liberación controlada que contiene un compuesto tal como se da a conocer en la presente memoria y un excipiente farmacéutico adecuado para la administración de liberación controlada. En algunas realizaciones, en la presente memoria se proporcionan composiciones farmacéuticas para la administración de liberación controlada, que contienen: (i) una cantidad eficaz de un compuesto dado a conocer, opcionalmente (ii) una cantidad eficaz de uno o más segundos agentes, y (iii) uno o más excipientes farmacéuticos adecuados para la administración de liberación controlada. En algunas realizaciones, la composición farmacéutica contiene además: (iv) una cantidad eficaz de un tercer agente.
Pueden administrarse agentes activos tales como los compuestos proporcionados en la presente memoria, mediante medios de liberación controlada o mediante dispositivos de administración que son bien conocidos por el experto ordinario en la materia. Entre los ejemplos, aunque sin limitarse a ellos, se incluyen los indicados en las patentes US n° 3.845.770, n° 3.916.899, n° 3.536.809, n° 3.598.123, y n° 4.008.719, n° 5.674.533, n° 5.059.595, n° 5.591.767, n° 5.120.548, n° 5.073.543, n° 5.639.476, n° 5.354.556, n° 5.639.480, n° 5.733.566, n° 5.739.108, n° 5.891.474, n°
5.922.356, n° 5.972.891, n° 5.980.945, n° 5.993.855, n° 6.045.830, n° 6.087.324, n° 6.113.943, n° 6.197.350, n° 6.248.363, n° 6.264.970, n° 6.267.981, n° 6.376.461, n° 6.419.961, n° 6.589.548, n° 6.613.358y n°6.699.500. Dichas formas de administración pueden utilizarse para proporcionar una liberación lenta o controlada de uno o más agentes activos utilizando, por ejemplo, hidroxipropilmetilcelulosa, otras matrices de polímero, geles, membranas permeables, sistemas osmóticos, recubrimientos multicapa, micropartículas, liposomas, microesferas o una combinación de los mismos, a fin de proporcionar el perfil de liberación deseada en proporciones variables. Las formulaciones de liberación controlada adecuadas conocidas por el experto ordinario en la materia, incluyendo las indicadas en la presente memoria, pueden seleccionarse fácilmente para la utilización con los agentes activos proporcionados en la presente memoria. De esta manera, las composiciones farmacéuticas proporcionadas comprenden formas de administración de dosis unitaria adecuadas para la administración oral, tales como, aunque sin limitación, tabletas, cápsulas, cápsulas de gelatina y comprimidos.
Todos los productos farmacéuticos de liberación controlada presentan el objetivo común de mejorar la terapia farmacológica respecto a la que se consigue con sus contrapartidas no controladas. En algunas realizaciones, la utilización de una preparación de liberación controlad en el tratamiento médico se caracteriza por la utilización de un mínimo de sustancia farmacológica para curar o controlar la enfermedad, trastorno o condición en un periodo de tiempo mínimo. Entre las ventajas de las formulaciones de liberación controlada se incluyen una actividad prolongada del fármaco, una frecuencia de dosis reducida y un mayor cumplimiento del sujeto. Además, pueden utilizarse formulaciones de liberación controlada para afectar al tiempo de inicio de acción u otras características, tales como los niveles sanguíneos del fármaco, y pueden, de esta manera, afectar a la aparición de efectos secundarios (p.ej., efectos adversos).
En algunas realizaciones, las formulaciones de liberación controlada están diseñadas para liberar inicialmente una cantidad de un compuesto tal como se da a conocer en la presente memoria que produce con rapidez el efecto terapéutico deseado, y liberar gradual y continuamente otras cantidades de los compuestos para mantener este nivel de efecto terapéutico o profiláctico durante un periodo de tiempo prolongado. Con el fin de mantener dicho nivel constante del compuesto en el cuerpo, éste debería liberarse a partir de la forma de administración a una velocidad que sustituya la cantidad de fármaco que resulta metabolizado y excretado del cuerpo. La liberación controlada de un agente activo puede estimularse mediante diversas condiciones, incluyendo, aunque sin limitarse a ellas, pH, temperatura, enzimas, agua u otras condiciones o compuestos fisiológicos.
En determinadas realizaciones, la composición farmacéutica puede administrarse utilizando la infusión intravenosa, una bomba osmótica implantable, un parche transdérmico, liposomas u otros modos de administración. En una realización, puede utilizarse una bomba (ver Sefton, CRC Crit. Ref. Biomed. Eng. 14:201 (1987); Buchwald et al., Surgery 88:507 (1980); Saudek et al., N. Engl. J. Med. 321:574 (1989)). En otra realización, pueden utilizarse materiales poliméricos. En todavía otra realización, puede introducirse un sistema de liberación controlada en un sujeto en un sitio apropiado que determina un profesional médico experto, p.ej., requiriendo de esta manera sólo una fracción de la dosis sistémica (ver, p.ej., Goodson, Medical Applications of Controlled Release, 115-138 (vol. 2, 1984). Se comentan otros sistemas de liberación controlada en la revisión de Langer, Science 249:1527-1533 (1990). El agente o agentes activos pueden dispersarse en una matriz interna sólida, p.ej., polimetilmetacrilato, polibutilmtacrilato, cloruro de polivinilo plastificado o no plastificado, nilón plastificado, tereftalato de polietileno plastificado, caucho natural, poliisopreno, poliisobutileno, polibutadieno, polietileno, copolímeros de etileno-vinilacetato, cauchos de silicona, polidimetilsiloxanos, copolímeros de silicona-carbonato, polímeros hidrofílicos, tales como hidrogeles de ésteres de ácido acrílico y ácido metacrílico, colágeno, alcohol polivinílico entrecruzado y acetato de polivinilo parcialmente hidrolizado entrecruzado, que se circunda por una membrana polimérica externa, p.ej., polietileno, polipropileno, copolímeros de etileno/propileno, copolímeros de etileno/acrilato de etilo, copolímeros de etileno/acetato de vinilo, cauchos de silicona, polidimetilsiloxanos, caucho de neopreno, polietileno clorado, cloruro de polivinilo, copolímeros de cloruro de vinilo con acetato de vinilo, cloruro de vinilideno, etileno y propileno, yonómero de tereftalato de polietileno, caucho de butilo, caucho de epiclorohidrina, copolímero de etileno/alcohol vinílico, terpolímero de etileno/acetato de vinilo/alcohol vinílico, y copolímero de etileno/viniloxietanol, que es insoluble en líquidos corporales. El agente o agentes activos a continuación se difunden a través de la membrana polimérica externa en una etapa de control de la velocidad de liberación. El porcentaje de agente activo en dichas composiciones parenterales es altamente dependiente de la naturaleza específica de las mismas, así como de las necesidades del sujeto.
Posología
Un compuesto indicado en la presente memoria (p.ej., un inhibidor de PI3K-Y, tal como el Compuesto 4) puede administrarse en la forma de composiciones farmacéuticamente aceptables que comprenden una cantidad terapéuticamente eficaz de uno o más compuestos indicados en la presente memoria y/o uno o más agentes terapéuticos adicionales, tales como un quimioterapéutico, formulados junto con uno o más excipientes farmacéuticamente aceptables. En algunos casos, el compuesto indicado en la presente memoria y el agente terapéutico adicional se administran en composiciones farmacéuticas separadas y pueden (p.ej., debido a las diferentes características físicas y/o químicas) administrarse por vías diferentes (p.ej., un terapéutico se administra por vía oral, mientras que el otro se administra por vía intravenosa). En otros casos, el compuesto indicado en la presente memoria y el agente terapéutico adicional pueden administrarse por separado, aunque por la misma vía
(p.ej., ambos por vía oral o ambos por vía intravenosa). En todavía otros casos, el compuesto indicado en la presente memoria y el agente terapéutico adicional pueden administrarse en la misma composición farmacéutica.
El nivel de dosis seleccionado dependerá de una diversidad de factores, incluyendo, por ejemplo, la actividad del compuesto particular utilizado, la vía de administración, el tiempo de administración, la tasa de excreción o metabolismo del compuesto particular que se utiliza, la velocidad y grado de absorción, la duración del tratamiento, otros fármacos, los compuestos y/o materiales utilizados en combinación con el compuesto particular utilizado, la edad, sexo, peso, condición, salud general y antecedentes médicos del paciente bajo tratamiento, y factores similares bien conocidos de las técnicas médicas.
En general, una dosis diaria adecuada de un compuesto indicado en la presente memoria y/o un quimioterapéutico será aquella cantidad del compuesto que, en algunas realizaciones, puede ser la dosis más baja eficaz para producir un efecto terapéutico. Dicha dosis eficaz generalmente dependerá de los factores indicados en la presente memoria. Generalmente, las dosis de los compuestos indicados en la presente memoria para un paciente, al utilizarlas para los efectos indicados, se encontrarán comprendidas entre aproximadamente 0,0001 mg y aproximadamente 100 mg al día, o entre aproximadamente 0,001 mg y aproximadamente 100 mg al día, o entre aproximadamente 0,01 mg y aproximadamente 100 mg al día, o entre aproximadamente 0,1 mg y aproximadamente 100 mg al día, o entre aproximadamente 0,0001 mg y aproximadamente 500 mg al día, o entre aproximadamente 0,001 mg y aproximadamente 500 mg al día, o entre aproximadamente 0,01 mg y 1000 mg, o entre aproximadamente 0,01 mg y aproximadamente 500 mg al día, o entre aproximadamente 0,1 mg y aproximadamente 500 mg al día, o entre aproximadamente 1 mg y 50 mg al día, o entre aproximadamente 5 mg y 40 mg al día. En algunas realizaciones, el intervalo es de entre aproximadamente 1 mg y aproximadamente 100 mg, entre aproximadamente 1 mg y aproximadamente 200 mg, entre aproximadamente 1 mg y aproximadamente 500 mg, entre aproximadamente 1 mg y aproximadamente 1000 mg, entre aproximadamente 100 mg y aproximadamente 200 mg, entre aproximadamente 100 mg y aproximadamente 500 mg, entre aproximadamente 100 y aproximadamente 750 mg, entre aproximadamente 100 mg y aproximadamente 1000 mg. Una dosis ejemplar es entre aproximadamente 10 y 30 mg al día. En algunas realizaciones, para un ser humano de 70 kg, una dosis adecuada sería de entre aproximadamente 0,05 y aproximadamente 7 g/día, tal como de entre aproximadamente 0,05 y aproximadamente 2,5 g/día. Los niveles de dosis reales de los ingredientes activos en las composiciones farmacéuticas indicadas en la presente memoria pueden modificarse de manera que se obtenga una cantidad del ingrediente activo que resulte eficaz para conseguir la respuesta terapéutica deseada para un paciente particular, composición y modo de administración, sin resultar tóxica para el paciente. En algunos casos, los niveles de dosis inferiores al límite inferior del intervalo anteriormente indicado pueden resultar más que adecuados, mientras que, en otros casos, pueden utilizarse dosis todavía mayores sin causar ningún efecto secundario perjudicial, p.ej., mediante la división de dichas dosis grandes en varias dosis pequeñas para la administración durante todo el día.
Los estudios farmacocinéticos en animales que se proporcionan en la presente memoria sugieren los intervalos de dosis eficaces para el Compuesto 4. En términos generales, para inhibir específicamente PI3K-Y, puede administrarse una dosis de un inhibidor específico de PI3K-Y que resulte en una concentración libre en plasma del fármaco que sea superior a un umbral predeterminado (p.ej., la IC50, ICa0, IC70, IC80 o IC90 para PI3K-y) durante un tiempo seleccionado (p.ej., 1 hora, 2 h, 3 h, 6 h, 12 h, 24 h, 2 días, 3 d, 5 d, o 7 d). Dicha dosis puede seleccionarse de manera que la concentración en plasma sea inferior a un segundo umbral predeterminado (p.ej., la IC20, IC30, IC40 o IC50 para PI3K-8, -a, o p) durante un tiempo seleccionado (p.ej., 1 hora, 2 h, 3 h, 6 h, 12 h, 24 h, 2 días, 3 d, 5 d, o 7 d). En algunas realizaciones, el inhibidor de PI3K-Y, p.ej., el Compuesto 4, se administra a una dosis que resulta en una concentración libre en plasma de Compuesto 4 que es superior a la IC90 de PI3K-Y durante como mínimo 1 hora, 2 h, 3 h, 6 h, 12 h 0 24 h. En algunas realizaciones, el inhibidor de PI3K-Y, p.ej., el Compuesto 4, se administra a una dosis que resulta en una concentración libre en plasma de Compuesto 4 que es superior a la IC50 de PI3K-y durante por lo menos 1 hora, 2 h, 3 h, 6 h, 12 h o 24 h.
Basándose en estudios en animales no seres humanos, una dosis humana predicha para conseguir la exposición a la IC90 para PI3K-Y es de aproximadamente 2 mg. De acuerdo con lo anterior, un inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4, puede administrarse en un ser humano, en el que cada dosis es de aproximadamente 2 mg, 1 a 3 mg, 1 a 5 mg, 1 a 10 mg, 0,5 a 20 mg, o 0,1 a 50 mg. En algunas realizaciones, la dosis (p.ej., una dosis terapéuticamente eficaz) es de aproximadamente 2 mg, 1 a 3 mg, 1 a 5 mg, 1 a 10 mg, 0,5 a 20 mg, 0,1 a 50 mg, 0,1 a 75 mg, 0,5 a 75 mg, 1 a 75 mg, 0,1 a 100 mg, 0,5 a 100 mg, o 1 a 100 mg. En algunas realizaciones, la dosis es de entre aproximadamente 1 y 10 mg. En algunas realizaciones, la dosis es de entre aproximadamente 1 y 50 mg. En algunas realizaciones, la dosis es de entre aproximadamente 1 y 100 mg. En un ser humano de 70 kg, una dosis de 2 mg corresponde a 0,029 mg/kg. De acuerdo con lo anterior, puede administrarse un inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4, en un ser humano, en el que cada dosis es de aproximadamente 0,029 mg/kg, de entre 0,014 y 0,14 mg/kg, de entre 0,02 y 0,04 mg/kg, de entre 0,01 y 0,05 mg/kg, de entre 0,01 y 0,1, o de entre 0,01 y 0,5 mg/kg.
Los compuestos pueden administrarse cada día, en días alternos, tres veces a la semana, dos veces a la semana, semanalmente o quincenalmente. El programa de administración puede incluir unas "vacaciones de los fármacos", p.ej., el fármaco puede administrarse durante dos semanas, una semana de pausa, o durante tres semanas, una semana de pausa, o durante cuatro semanas, una semana de pausa, etc., o continuamente, sin unas vacaciones de
los fármacos. Los compuestos pueden administrarse por vía oral, intravenosa, intraperitoneal, tópica, transdérmica, intramuscular, subcutánea, intranasal, sublingual o por cualquier otra vía.
Puede administrarse en múltiples dosis un compuesto tal como se proporciona en la presente memoria. La administración puede ser aproximadamente una vez, dos veces, tres veces, cuatro veces, cinco veces, seis veces o más de seis veces al día. La administración puede ser aproximadamente una vez al mes, aproximadamente una vez cada dos semanas, aproximadamente una vez a la semana o aproximadamente una vez en días alternos. Un compuesto tal como se da a conocer en la presente memoria y otro agente pueden administrarse juntos entre aproximadamente una vez al día y aproximadamente 6 veces al día. En otra realización, la administración de un compuesto tal como se proporciona en la presente memoria y un agente continúa durante menos de aproximadamente 7 días. La administración puede continuar durante más de aproximadamente 6 días, aproximadamente 10 días, aproximadamente 14 días, aproximadamente 28 días, aproximadamente dos meses, aproximadamente seis meses o aproximadamente un año. En algunos casos, la administración continua se consigue y se mantiene durante tanto tiempo como resulte necesario.
Basándose en estudios en animales no seres humanos proporcionados en la presente memoria, la semivida oral del Compuesto 4 en seres humanos se espera que sea de entre aproximadamente 10 y 13 horas. Dicho resultado informa de los tiempos de administración de un inhibidor de PI3K-Y, tal como el Compuesto 4. Por ejemplo, en algunas realizaciones, los tiempos se seleccionan de manera que la concentración libre en plasma del fármaco sea superior a un umbral predeterminado (p.ej., la IC50, ICao, IC70, iCso o IC90 para PI3-y) durante un tiempo seleccionado (p.ej., 1 hora, 2 h, 3 h, 6 h, 12 h, 24 h, 2 días, 3 d, 5 d o 7 d). Los tiempos de administración también pueden seleccionarse de manera que el nivel en plasma sea inferior a un segundo umbral predeterminado (p.ej., la 1C20, IC30, IC40 o IC50 para PI3K-5, a o p) durante un tiempo seleccionado (p.ej., 1 hora, 2 h, 3 h, 6 h, 12 h, 24 h, 2 días, 3 d, 5 d, o 7 d). El inhibidor de PI3K-Y, p.ej., el Compuesto 4, puede administrarse con unos tiempos que resulten en una concentración libre en plasma de Compuesto 4 que es superior a la IC90 de PI3K-Y durante como mínimo 1 hora, 2 h, 3 h, 6 h, 12 h o 24 h. El inhibidor de PI3K-Y, p.ej., el Compuesto 4, puede administrarse durante unos tiempos que resulten en una concentración libre en plasma de Compuesto 4 que es superior a la IC50 de PI3K-y durante por lo menos 1 hora, 2 h, 3 h, 6 h, 12 h o 24 h.
De acuerdo con lo anterior, un inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4, puede administrarse en un ser humano aproximadamente una vez al día. El inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4, puede administrarse en un ser humano una vez cada dos días. El inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4, puede administrarse en un ser humano dos o tres veces al día.
La administración de las composiciones farmacéuticas tal como se dan a conocer en la presente memoria puede continuar durante tanto tiempo como resulte necesario. Un agente tal como se da a conocer en la presente memoria puede administrarse durante más de aproximadamente 1, aproximadamente 2, aproximadamente 3, aproximadamente 4, aproximadamente 5, aproximadamente 6, aproximadamente 7, aproximadamente 14, o aproximadamente 28 días. Un agente tal como se da a conocer en la presente memoria puede administrarse durante menos de aproximadamente 28, aproximadamente 14, aproximadamente 7, aproximadamente 6, aproximadamente 5, aproximadamente 4, aproximadamente 3, aproximadamente 2, o aproximadamente 1 día. Un agente tal como se da a conocer en la presente memoria puede administrarse crónicamente de manera continua, p.ej., para el tratamiento de efectos crónicos.
Debido a que los compuestos indicados en la presente memoria pueden administrarse en combinación con otros tratamientos (tales como quimioterapéuticos, radiación o cirugía adicional), las dosis de cada agente o terapia pueden ser inferiores a la dosis correspondiente para la terapia de agente único. La dosis de terapia con un agente único puede estar comprendida entre, por ejemplo, aproximadamente 0,0001 y aproximadamente 200 mg, o entre aproximadamente 0,001 y aproximadamente 100 mg, o entre aproximadamente 0,01 y aproximadamente 100 mg, o entre aproximadamente 0,1 y aproximadamente 100 mg, o entre aproximadamente 1 y aproximadamente 50 mg por kilogramo de peso corporal al día. En algunas realizaciones, la dosis es de aproximadamente 1 mg/kg, aproximadamente 5 mg/kg, aproximadamente 7,5 mg/kg, aproximadamente 10 mg/kg, aproximadamente 15 mg/kg, aproximadamente 20 mg/kg, aproximadamente 25 mg/kg, aproximadamente 50 mg/kg, aproximadamente 75 mg/kg o aproximadamente 100 mg/kg al día. En algunas realizaciones, la dosis es de aproximadamente 1 mg/kg, aproximadamente 7,5 mg/kg, aproximadamente 20 mg/kg o aproximadamente 50 mg/kg al día.
En el caso de que un compuesto proporcionado en la presente memoria se administre en una composición farmacéutica que comprenda uno o más agentes, y el agente presente una semivida más corta que el compuesto proporcionado en la presente memoria, las formas de dosis unitaria del agente y del compuesto proporcionado en la presente memoria pueden ajustarse correspondientemente.
Actividades de PI3K-Y
La PI3K-Y es una PI3K de clase 1B que se asocia a las proteínas adaptadoras p101 y p84 (p87PIKAP) y señaliza canónicamente mediante los GPCR. Puede producirse la activación no canónica mediante los receptores de tirosina quinasa y RAS. La PI3K-Y activada conduce a la producción de PIP3, que sirve como un sitio de anclaje para las
proteínas efectoras posteriores, incluyendo AKT y BTK, llevando dichos enzimas a la membrana celular, en donde pueden ser activados. Se ha propuesto un papel de andamiaje para PI3K-Y y podría contribuir a la activación de la ruta de RAS/MEK/ERK. La interacción con la ruta de RAS explica actividades atribuidas a PI3K-Y sin actividad de quinasa en células o en animales. La PI3K-Y resulta esencial para el funcionamiento de una diversidad de células y rutas inmunitarias. Las respuestas de quimioquinas (incluyendo IL-8, fMLP y C5a), que conducen a la migración de neutrófilos, basófilos o monocitos, es dependiente de PI3K-Y (HIRSCH et al., "Central Role for G Protein-Coupled Phosphoinositide 3-Kinase y in Inflammation", Science 287:1049-1053 (2000); SASAKI et al., "Function of PI3Ky in Thymocyte Development, T Cell Activation, and Neutrophil Migration", Science 287:1040-1046 (2000); LI et al., "Roles of PLC-p2 and -p3 and PI3Ky in Chemoattractant-Mediated Signal Transduction", Science 287:1046-1049 (2000)). El requisito de migración de neutrófilos dependiente de PI3K-Y queda demostrado por el no desarrollo de artritis en el modelo de artritis de transferencia sérica de K/BXN en ratones con inactivación de PI3K-Y (Randis et al., Eur. J. Immunol., 2008, 38(5), 1215-24). De manera similar, los ratones no consiguen desarrollar inflamación celular e hiperreactividad de las vías respiratorias en el modelo de asma inducida por ovoalbúmina (Takeda et al., J. Allergy Clin. Immunol., 2009; 123, 805-12). Los ratones deficientes en PI3K-Y también presentan defectos en la función de las células T ayudantes. La producción de citoquinas y proliferación de las células T en respuesta a la activación es reducida, y la eliminación vírica dependiente de células T ayudantes es defectuosa (Sasaki et al., Science, 2000, 287, 1040-46). Los modelos de enfermedad inflamatoria dependiente de células T que incluyen EAE tampoco se desarrollan en ratones deficientes en PI3K-Y y tanto el defecto de activación de las células T como los defectos de migración celular podrían contribuir a la eficacia en dicho modelo (Comerfold, PLOS One, 2012, 7, e45095). El modelo de soriasis con imiquimod también se ha utilizado para demostrar la importancia de PI3K-Y en la respuesta inflamatoria. Mediante la utilización de ratones deficientes en PI3K-Y, se bloquea la acumulación de las células T Y§, así como la maduración y migración de las células dendríticas (ROLLER et al., "Blockade of Phosphatidylinositol 3-Kinase (PI3K)8 o PI3Ky Reduces IL-17 and Ameliorates Imiquimod-Induced Psoriasis-like Dermatitis", J. Immunol. 189:4612-4620, 2012). El papel de PI3K-Y en el tráfico celular también puede demostrarse en modelos oncológicos en los que la inflamación tumoral es importante para el crecimiento y metástasis de los cánceres. En el modelo de carcinoma pulmonar de Lewis, la activación, migración y diferenciación de los monocitos son defectuosas. Dichos defectos resultan en una reducción del crecimiento tumoral y en una supervivencia mayor en ratones deficientes en PI3K-Y (Schmid et al., Cancer Cell, 2011, 19, 715-27) o con el tratamiento con inhibidores que presentan PI3K-Y como diana. En el cáncer pancreático, PI3K-Y puede expresarse inapropiadamente y en este cáncer de tumor sólido o en otros en los que PI3K-Y desempeña un papel funcional, la inhibición de PI3K-Y podría resultar beneficiosa.
Por ejemplo, aunque sin respaldo teórico, PI3K-Y se expresa en células mieloides Gr1+CD11b+ y directamente estimula la invasión de las células mieloides y, en consecuencia, la inmunosupresión de los carcinomas ductales pancreáticos. Hardamon et. al., Proceedings: AACR 103rd Annual Meeting 2012, Cancer Research: 15 de abril 15, 2012; volumen 72, número 8, suplemento 1. La inhibición de PI3K-Y también resulta prometedora para el tratamiento de las neoplasias malignas hemáticas. En modelo de T-LLA utiliza una inactivación dirigida por las células T de pten, PI3K-8 y PI3K-Y resultan ambas esenciales para el desarrollo apropiado de la enfermedad, tal como muestra la deleción genética de ambos genes (Subramaniam et al., Cancer Cell 21, 459-472, 2012). Además, en dicho modelo de T-LLA, el tratamiento con un inhibidor de molécula pequeña de ambas quinasas conduce a una mayor supervivencia de dichos ratones. En el CPL, las redes de quimioquinas apoyan un microambiente pseudofolicular que incluye células de tipo nodriza, células estromales y células T ayudante. Las funciones de PI3K-y en la señalización de quimioquinas normal y la biología de las células T sugieren el valor de inhibir dicha diana en el CPL (Burger, "Inhibiting B-Cell Receptor Signaling Pathways in Chronic Lymphocytic Leukemia", Curr. Hematol. Malig. Rep. 7:26-33 (2012)). De acuerdo con lo anterior, los inhibidores de PI3K-Y resultan terapéuticamente interesantes para enfermedades del sistema inmunitario en las que el tráfico de células y la función de las células T o mieloides resulta importante. En oncología, los tumores sólidos que son dependientes de la inflamación tumoral, o los tumores con niveles elevados de expresión de PI3K-Y, pueden ser la diana. Para los cánceres hemáticos, un papel especial de las isoformas PI3K-Y y PI3K-8 en LLAT y potencialmente en CPL sugiere utilizar dichos PI3K como diana en dichas enfermedades.
Sin limitación a una teoría en particular, PI3K-Y ha demostrado que desempeña funciones en la inflamación, la artritis, el asma, la alergía, la escleros múltiple (EM) y el cáncer, entre otros (p.ej., Ruckle et al., Nature Rev., Drug Discovery, 2006, 5, 903-18; Schmid et al., "Myeloid cells in tumor inflammation," Vascular Cell, 2012, doi:10.1186/2045-824X-4-14). Por ejemplo, PI3K-Y funciona en múltiples rutas de señalización que participan en la activación y migración de leucocitos. Se ha mostrado que PI3K-Y induce la activación y la supervivencia de las células T CD4+ autorreactivas durante la encefalomielitis autoinmune experimental (EAE), un modelo de EM. En el caso de que se administre desde el inicio de EAE, se ha mostrado que un inhibidor de PI3K-Y causa la inhibición y reversión de la enfermedad clínica, y la reducción de la desmielinización y patología celular en el SNC (Comerford et al., PLOS One, 2012, 7, e45095). La PI3K-Y también regula el desarrollo de los timocitos, la activación de las células T, la migración de los neutrófilos y el estallido oxidativo (Sasaki et al., Science, 2000, 287, 1040-46). Además, se ha mostrado que la hiperreactividad alérgica de las vías respiratorias, la inflamación y el remodelado no se desarrollan en ratones deficientes en PI3K-Y (Takeda et al., J. Allergy Clin. Immunol., 2009; 123, 805-12). Se ha mostrado que PI3K-Y resulta necesaria para la producción inducida por quimioatrayentes del 3,4,5-trifosfato de fosfatidilinositol y presenta un importante papel en la producción de superóxido y la quimiotaxis inducidos por quimioatrayente en neutrófilos de ratón y en la producción de anticuerpos específicos de antígeno independientes de células T compuestos de la cadena ligera A de inmunoglobulina (Li et al., Science, 2000, 287, 1046-49). Se ha informado de que PI3K-Y es una molécula de señalización crucial necesaria para la acumulación de macrófagos en la inflamación (Hirsch et al., Science, 2000, 287, 1049-53). En los
cánceres, el bloqueo farmacológico o genético de p110Y suprime la inflamación, el crecimiento y la metástasis de los tumores implantados y espontáneos, sugiriendo que PI3K-Y puede ser una importante diana terapéutica en oncología (Schmid et al., Cancer Cell, 2011, 19, 715-27). Por ejemplo, se ha mostrado que PI3K-Y presenta una elevada acumulación específicamente en el tumor en el adenocarcinoma ductal pancreático (ACDP) en el ser humano, implicando un papel de la PI3K-Y en el cáncer pancreático (Edling et al., Human Cancer Biology, 2010, 16(2), 4928 37).
Un trastorno mediado por PI3K-gamma en un sujeto puede tratarse mediante la administración en el sujeto de una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (111), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., un inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4) o una forma farmacéuticamente aceptable del mismo.
La presente invención se refiere a compuestos para la utilización en el tratamiento o la prevención del cáncer. En una realización, la célula es un tumor sólido. En una realización, el cáncer se selecciona de uno o más de los siguientes: cáncer del sistema pulmonar, un cáncer cerebral, un cáncer del tracto gastrointestinal, un cáncer de piel, un cáncer genitourinario, un cáncer pancreático, un cáncer pulmonar, un meduloblastoma, un carcinoma de células basales, un glioma, un cáncer de mama, un cáncer de próstata, un cáncer testicular, un cáncer esofágico, un cáncer hepatocelular, un cáncer gástrico, un tumor estromal gastrointestinal (TEGI), un cáncer de colon, un cáncer colorrectal, un cáncer ovárico, un melanoma, un tumor neuroectodérmico, cáncer de cabeza y cuello, un sarcoma, un sarcoma de tejidos blandos, fibrosarcoma, mixosarcoma, liposarcoma, un condrosarcoma, un sarcoma osteogénico, un cordoma, un angiosarcoma, un endoteliosarcoma, un linfangiosarcoma, un linfangioendoteliosarcoma, un sinovioma, un mesotelioma, un leiomiosarcoma, un cáncer de cuello uterino, un cáncer uterino, un cáncer endometrial, un carcinoma, un carcinoma de vejiga, un carcinoma epitelial, un carcinoma de células escamosas, un adenocarcinoma, un carcinoma broncogénico, un carcinoma de células renales, un hepatoma, un carcinoma del conducto biliar, un cáncer neuroendocrino, un tumor carcinoide, tumor de células gigantes de tipo difuso y glioblastoma.
En una realización, el cáncer es un cáncer hemático.
En una realización, el sujeto es un ser humano. En una realización, el sujeto se identifica que presenta o está en riesgo de presentar un trastorno mediado por PI3K-gamma mediante la utilización de un biomarcador.
En una realización, la dosis terapéuticamente eficaz del compuesto para la utilización según la invención es de aproximadamente 2 mg, de aproximadamente 1 a 3 mg, de aproximadamente 1 a 5 mg, de aproximadamente 1 a 10 mg, de aproximadamente 0,5 a 20 mg, de aproximadamente 0,1 a 50 mg al día, de aproximadamente 0,1 a 75 mg al día, de aproximadamente 0,1 a 100 mg al día, de aproximadamente 0,1 a 250 mg al día, de aproximadamente 0,1 a 500 mg al día, de aproximadamente 0,1 a 1000 mg al día, de aproximadamente 1 a 50 mg al día, de aproximadamente 1 a 75 mg al día, de aproximadamente 1 a 100 mg al día, de aproximadamente 1 a 250 mg al día, de aproximadamente 1 a 500 mg al día, de aproximadamente 1 a 1000 mg al día, de aproximadamente 10 a 50 mg al día, de aproximadamente 10 a 75 mg al día, de aproximadamente 10 a 100 mg al día, de aproximadamente 10 a 250 mg al día, de aproximadamente 10 a 500 mg al día, de aproximadamente 10 a 1000 mg al día, de aproximadamente 100 a 500 mg al día, o de aproximadamente 100 a 1000 mg al día. En una realización, la dosis terapéuticamente eficaz es de aproximadamente 0,029 mg/kg, de aproximadamente 0,014 a 0,14 mg/kg, de aproximadamente 0,02 a 0,04 mg/kg, de aproximadamente 0,01 a 0,05 mg/kg, de aproximadamente 0,01 a 0,1, o de aproximadamente 0,01 a 0,5 mg/kg. En una realización, el compuesto es para la administración una vez cada dos días. En una realización, el compuesto es para la administración una vez al día. En una realización, el compuesto es para la administración dos veces al día.
En una realización, el compuesto es para la administración a una dosis con la que el nivel del compuesto en el sujeto es superior a la IC50 del compuesto de inhibición de PI3K-gamma durante por lo menos 70 %, 80 %, 90 %, 95 %, 97 %, 98 % o 99 % de un periodo de tiempo seleccionado, p.ej., 6 horas, 12 horas, 24 horas o 48 horas inmediatamente después de la administración. En una realización, el compuesto es para la administración a una dosis con la que el nivel del compuesto en el sujeto es superior a la IC90 del compuesto de inhibición de PI3K-gamma durante por lo menos 50 %, 60 %, 70 %, 80 %, 90 %, 95 %, 97 %, 98 % o 99 % de un periodo de tiempo seleccionado, p.ej., 6 horas, 12 horas, 24 horas o 48 horas inmediatamente después de la administración. En una realización, el compuesto es para la administración a una dosis con la que el nivel del compuesto en el sujeto no se incrementa hasta un nivel superior a la IC20 o IC50 del compuesto de inhibición de PI3K-delta en un periodo de tiempo seleccionado, p.ej., 6 horas, 12 horas, 24 horas o 48 horas inmediatamente después de la administración. El nivel del compuesto puede medirse a partir del plasma del sujeto. El nivel del compuesto puede medirse a partir de los tejidos del sujeto. El compuesto puede administrarse a una dosis con la que proporciona una inhibición de por lo menos 50% de PI3K-gamma en el sujeto, aunque una inhibición inferior de 10 % o 20 % de PI3K-delta en el sujeto.
En una realización, el sujeto es un ser humano y el compuesto presenta una semivida de aproximadamente 10 a 13 horas en el sujeto. En una realización, el segundo agente terapéutico que es un sustrato de P-gp también puede administrarse en el sujeto. En una realización, el segundo agente terapéutico es Norvir (ritonavir).
Actividades de PI3K-5 y/o PI3K-Y
La PI3K-8 presenta funciones en alteraciones de la señalización y desarrollo de las células B, producción de anticuerpos, función de las células T, diferenciación de Th1 y Th2 y degranulación de mastócitos y basófilos. Sin respaldo teórico, PI3K-Y presenta funciones en la función de las células T, el reclutamiento de neutrófilos y macrófagos, la activación de los macrófagos, el estallido oxidativo de neutrófilos y la migración de las células dendríticas. La inhibición de las isoformas PI3K-8 y/o PI3K-Y puede resultar en eficacia contra la inflamación y el cáncer, p.ej., en la artritis, esclerosis múltiple (EM) y en modelos tumorales. Por ejemplo, la deficiencia en PI3K-8 y/o PI3K-Y puede resultar en eficacia en modelos de artritis K/BxN (Kyburz et al., Springer Semin. Immunopathology, 2003, 25, 79-90) o modelo de transferencia sérica de K/BxN de artritis (Randis et al., Eur. J. Immunol., 2008, 38(5), 1215-24), en el que se ha mostrado que el reconocimiento de los complejos inmunitarios depende tanto de PI3K-8 como de PI3K-Y, mientras que la migración celular es dependiente de PI3K-Y. La deficiencia en PI3K-8 o PI3K-Y también puede resultar en eficacia en el modelo murino de asma alérgica inducida por ovoalbúmina (OVA) (Lee et al., FASEB J., 2006, 20, 455-65; Takeda et al., J. Allergy Clin. Immunol., 2009; 123, 805-12), en el que se muestra que la inhibición de PI3K-8 o PI3K-Y inhibe la infiltración pulmonar inducida por ovoalbúmina y mejora el grado de reactividad de las vías respiratorias. La deficiencia de PI3K-8 o PI3K-Y también puede resultar en eficacia en la encefalomielitis autoinmunitaria experimental murina (modelo de Em ), en el que se muestra que la deleción de PI3K-Y puede proporcionar una mejor eficacia que la deleción de PI3K-8 (Haylock-Jacob et al., J. Autoimmunity, 2011, 36, 278 87; Comerford et al., PLOS One, 2012, 7, e45095), incluyendo la reducción de la activación de las células T CD4+ inducida por receptores de células T, la infiltración leucocitaria, las respuestas de Th1/Th17 y la migración de células dendríticas (Comerfold, PLOS One, 2012, 7, e45095). Además, la inhibición de PI3K-Y también puede resultar en una inflamación y crecimiento tumoral reducidos (p.ej., el modelo de carcinoma pulmonar de Lewis, Schmid et al., Cancer Cell, 2011, 19(6), 715-27). La deleción de P|3k-y combinada con la deleción de PI3K-8 resulta en una supervivencia incrementada en la leucemia linfoblástica aguda de células T (LLA-T) (Subramaniam et al., Cancer Cell, 2012, 21, 459-72). Los inhibidores de tanto PI3K-8 como PI3K-Y también se ha mostrado que resultan eficaces en la línea celular de LLA-T con deleción de PTEN (MOLT-4). En ausencia de función supresora tumoral de fosfatasa PTEN, PI3K-8 o PI3K-Y sola puede apoyar el desarrollo de leucemia, mientras que la inactivación de ambas isoformas suprime la formación de tumor. De esta manera, los inhibidores de PI3K-8 y/o PI3K-Y pueden resultar útiles en el tratamiento de inflamación, tal como artritis, asma alérgica y EM, y en el tratamiento del cáncer, por ejemplo debido a efectos tales como reducciones en la inflamación asociada a tumores sólidos, la angiogénesis y la progresión tumoral.
La importancia de PI3K-8 en el desarrollo y función de las células B se ve apoyada por estudios de inhibidores y modelos genéticos. La PI3K-8 es un mediador importante de la señalización de los receptores de las células B (RCB) y se encuentra corriente arriba de AKT, el flujo de calcio, PLCy, la quinasa MAP, P70S6k y la activación de FOXO3a. La PI3K-8 también resulta importante en la señalización de IL4R, SIP y CXCR5, y se ha mostrado que modula las respuestas a los receptores de tipo Toll 4 y 9. Los inhibidores de PI3K-8 han mostrado la importancia de PI3K-8 en el desarrollo de las células B (células de zona marginal y células B1), la activación de las células B, la quimiotaxis, migración y migración dirigida ('homing') al tejido linfoide, y en el control de la conmutación de clase de las inmunoglobulinas que conduce a la producción de IgE. Clayton E. et al. (2002) J. Exp. Med. 196(6):753-63; Bilancio A, et al. (2006) Blood 107(2):642-50; Okkenhaug K. et al. (2002) Science 297(5583):1031-4; Al-Alwan MM et al. (2007) J. Immunol. 178(4):2328-35; Zhang TT, et al. (2008) J. Allergy Clin. Immunol. 2008;122(4):811-819.e2; Srinivasan L, et al. (2009) Cell 139(3):573-86).
En las células T, se ha demostrado que PI3K-8 presenta un papel en la señalización de receptores de células T y de citoquinas, y se encuentra corriente arriba de a Kt , PLCy y GSK3b. En ratones con deleción de PI3K-8 o inserción de la secuencia génica ('knock-in') sin actividad de quinasa, se han observado algunos defectos de las células T, incluyendo defectos de la proliferación, activación y diferenciación, que han conducido a una respuesta reducida de las células T ayudante 2 (TH2), defectos específicos de las células T de memoria (reducción de DTH), defectos en el tráfico celular dependiente de antígenos y defectos en la quimiotaxis/migración a quimioquinas (p.ej., SIP, CCR7 y CD62L). (Gargon F. et al. (2008) Blood 111(3): 1464-71; Okkenhaug K et al. (2006). J. Immunol. 177(8):5122-8; Soond DR, et al. (2010) Blood 115(11):2203-13; Reif K, (2004). J. Immunol. 2004;173(4):2236-40; Ji H. et al. (2007) Blood 110(8):2940-7; Webb LM, et al. (2005) J. Immunol. 175(5):2783-7; Liu D, et al. (2010) J. Immunol. 184(6):3098-105; Haylock-Jacobs S, et al. (2011) J. Autoimmun. 2011;36(3-4):278-87; Jarmin SJ, et al. (2008) J Clin Invest.
118(3):1154-64).
Numerosas publicaciones apoyan el papel de PI3K-8 y PI3K-Y en la diferenciación, mantenimiento y activación de las células inmunitarias y malignas, tal como se indica en mayor detalle en la presente memoria.
Las isoformas PI3K-8 y PI3K-Y se expresan preferentemente en leucocitos, en los que presentan papeles diferentes y no solapantes en el desarrollo y función de las células inmunitarias. Ver, p.ej., PURI and GOLD, "Selective inhibitors of phosphoinositide 3-kinase delta: modulators of B-cell function with potential for treating autoimmune inflammatory disease and B-cell malignancies", Front. Immunol. 3:256 (2012); BUITENHUIS et al., "The role of the PI3K-PKB signaling module in regulation of hematopoiesis", Cell Cycle 8(4):560-566 (2009); HOELLENRIEGEL and BURGER, "Phosphoinositide 3'-kinase delta: turning off BCR signaling in Chronic Lymphocytic Leukemia", Oncotarget 2(10):737-738 (2011); HIRSCH et al., "Central Role for G Protein-Coupled Phosphoinositide 3-Kinase y in Inflammation", Science 287:1049-1053 (2000); LI et al., "Roles of PLC-p2 and -p3 and PI3Ky in Chemoattractant-Mediated Signal Transduction", Science 287:1046-1049 (2000); SAsAk I et al., "Function of PI3Ky in Thymocyte Development, T Cell
Activation, and Neutrophil Migration", Science 287:1040-1046 (2000); CUSHING et al., "PI3K8 and PI3Ky as Targets for Autoimmune and Inflammatory Diseases", J. Med. Chem. 55:8559-8581 (2012); MAXWELL et al., "Attenuation of phosphoinositide 3-kinase 8 signaling restrains autoimmune disease", J. Autoimmun. 38:381-391 (2012); HAYLOCK-JACOBS et al., "PI3K8 drives the pathogenesis of experimental autoimmune encephalomyelitis by inhibiting effector T cell apoptosis and promoting Th17 differentiation", J. Autoimmun. 36:278-287 (2011); So OND et al., "PI3K p1108 regulates T-cell cytokine production during primary and secondary immune responses in mice and humans," Blood 115(11):2203-2213 (2010); ROLLER et al., "Blockade of Phosphatidylinositol 3-Kinase (PI3K)8 or PI3Ky Reduces IL-17 and Ameliorates Imiquimod-Induced Psoriasis-like Dermatitis", J. Immunol. 189:4612-4620 (2012); Ca MPS et al., "Blockade of PI3Ky suppresses joint inflammation and damage in mouse models of rheumatoid arthritis", Nat. Med.
11(9):936-943 (2005). Como enzimas clave en la señalización de los leucocitos, PI3K-8 y PI3K-Y facilitan las funciones normales de células B, células T y células mieloides, incluyendo la diferenciación, la activación y la migración. Ver, p.ej., HOELLENRIEGEL and Bu Rg ER, "Phosphoinositide 3'-kinase delta: turning off BCR signaling in Chronic Lymphocytic Leukemia", Oncotarget 2(10):737-738 (2011); CUSHING et al., "PI3K8 and PI3Ky as Targets for Autoimmune and Inflammatory Diseases", J. Med. Chem. 55:8559-8581 (2012). La actividad de PI3K-8 o PI3K-Y resulta crítica para los modelos preclínicos de enfermedades autoinmunitarias e inflamatorias. Ver, p.ej., HIRSCH et al., "Central Role for G Protein-Coupled Phosphoinositide 3-Kinase y in Inflammation", Science 287:1049-1053 (2000); LI et al., "Roles of PLC-p2 and -p3 and PI3Ky in Chemoattractant-Mediated Signal Transduction", Science 287:1046-1049 (2000); SASAKI et al., "Function of PI3Ky in Thymocyte Development, T Cell Activation, and Neutrophil Migration", Science 287:1040-1046 (2000); CUSHING et al., "PI3K8 and PI3Ky as Targets for Autoimmune and Inflammatory Diseases", J. Med. Chem. 55:8559-8581 (2012); MAXWELL et al., "Attenuation of phosphoinositide 3-kinase 8 signaling restrains autoimmune disease", J. Autoimmun. 38:381-391 (2012); HAYLo Ck -JACOBS et al., "PI3K8 drives the pathogenesis of experimental autoimmune encephalomyelitis by inhibiting effector T cell apoptosis and promoting Th17 differentiation", J. Autoimmun. 36:278-287 (2011); SOOND et al., "PI3K p1108 regulates T-cell cytokine production during primary and secondary immune responses in mice and humans," Blood 115(11):2203-2213 (2010); ROLLER et al., "Blockade of Phosphatidylinositol 3-Kinase (PI3K)8 or PI3Ky Reduces IL-17 and Ameliorates Imiquimod-Induced Psoriasis-like Dermatitis", J. Immunol. 189:4612-4620 (2012); CAMPS et al., "Blockade of PI3Ky suppresses joint inflammation and damage in mouse models of rheumatoid arthritis", Nat. Med. 11(9):936-943 (2005). Dado el papel clave de PI3K-8 y PI3K-Y en la función inmunitaria, los inhibidores de PI3K-8 y/o y presentan un potencial terapéutico en enfermedades inflamatorias o neoplásicas de tipo inmunitario.
La PI3K-8 y PI3K-Y son cruciales para el crecimiento y supervivencia de las neoplasias malignas de células B y T, y la inhibición de estas isoformas podría limitar eficazmente estas enfermedades. Ver, p.ej., SUBRAMANIAM et al., "Targeting Nonclassical Oncogenes for Therapy in T-ALL", Cancer Cell 21:459-472 (2012); LANNUTTI et al., "CAL-101 a p1108 selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability", Blood 117(2):591-594 (2011). La PI3K-8 y PI3K-Y apoyan el crecimiento y la supervivencia de determinadas neoplasias malignas de células B mediante la medicación en la señalización intracelular de RCB y las interacciones entre las células tumorales y su microentorno. Ver, p.ej., PURI y GOLD, "Selective inhibitors of phosphoinositide 3-kinase delta: modulators of B-cell function with potential for treating autoimmune inflammatory disease and B-cell malignancies", Front. Immunol. 3:256 (2012); HOELLENRIEGEL et al., "The phosphoinositide 3'-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leuckemia", Blood 118(13):3603-3612 (2011); BURg Er , "Inhibiting B-Cell Receptor Signaling Pathways in Chronic Lymphocytic Leukemia", Curr. Mematol. Malig. Rep. 7:26-33 (2012). Una señalización incrementada de los RCB es un mecanismo patológico crucial de las neoplasias malignas de células B y la activación de PI3K es una consecuencia directa de la activación de la ruta de los RCB. Ver, p.ej., BURGER, "Inhibiting B-Cell Receptor Signaling Pathways in Chronic Lymphocytic Leukemia," Curr. Mematol. Malig. Rep. 7:26-33 (2012); HERISHANU et al., "The lymph node microenvironment promotes B-cell receptor signaling, NF-kB activation, and tumor proliferation in chronic lymphocytic leukemia", Blood 117(2):563-574 (2011); DAVIS et al., "Chronic active B-cell-receptor signaling in diffuse large B-cell lymphoma", Nature 463:88-92 (2010); PIGHI et al., "Phospho-proteomic analysis of mantle cell lymphoma cells suggests a pro-survival role of B-cell receptor signaling", Cell Oncol. (Dordr) 34(2):141-153 (2011); RIZZATTI et al., "Gene expression profiling of mantle cell lymphoma cells reveals aberrant expression of genes from the PI3K-AKT, WNT and TGFp signaling pathways", Brit. J. Haematol. 130:516-526 (2005); MARTINEZ et al., "The Molecular Signature of Mantle Cell Lymphoma Reveals Multiple Signals Favoring Cell Survival", Cancer Res. 63:8226-8232 (2003). Las interacciones entre las células B malignas y las células de soporte (p.ej., células estromales y células de tipo nodriza) en el microambiente tumoral son importantes para la supervivencia, proliferación, migración dirigida y retención en los tejidos de las células tumorales. Ver, p.ej., BURGER, "Inhibiting B-Cell Receptor Signaling Pathways in Chronic Lymphocytic Leukemia", Curr. Mematol. Malig. Rep. 7:26-33 (2012); HERISHANU et al., "The lymph node microenvironment promotes B-cell receptor signaling, NF-kB activation, and tumor proliferation in chronic lymphocytic leukemia", Blood 117(2):563-574 (2011); KURTOVA et al., "Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance", Blood 114(20): 4441-4450 (2009); BURGER et al., "High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation", Blood 113(13) 3050-3058 (2009); q U iROGA et al., "B-cell antigen receptor signaling enhances chronic lymphocytic leukemia cell migration and survival: specific targeting with a novel spleen tyrosine kinase inhibitor, R406", Blood 114(5): 1029-1037, 2009). La inhibición de PI3K-8,y con un inhibidor en determinadas células B malignas puede bloquear la señalización de supervivencia intracelular mediada por RCB, así como interacciones clave con su microentorno que son críticas para su crecimiento.
La PI3K-8 y PI3K-Y también desempeñan un papel directo en la supervivencia y proliferación de determinadas neoplasias malignas de células T. Ver, p.ej., SUBRAMANIAM et al., "Targeting Nonclassical Oncogenes for Therapy in T-ALL", Cancer Cell 21:459-472 (20l2). Una actividad aberrante de PI3K-8 y PI3K-Y proporciona las señales necesarias para el desarrollo y crecimiento de determinadas neoplasias malignas de células T. Aunque la BTK se expresa en las células B, no se expresa en las células T, y por lo tanto, la BTK no es una diana viable para el tratamiento de las neoplasias malignas de células T. Ver, p.ej., NISITANI et al., "Posttranscriptional regulation of Bruton's tyrosine kinase expression in antigen receptor-stimulated splenic B cells", PNAS 97(6):2737-2742 (2000); DE WEEr S et al., "The Bruton's tyrosine kinase gene is expressed throughout B cell differentiation, from early precursor B cell stages preceding immunoglobulin gene rearrangement up to mature B cell stages", Eur. J. Immunol. 23:3109-3114 (1993); SMITH et al., "Expression of Bruton's Agammaglobulinemia Tyrosine Kinase Gene, BTK, Is Selectively Down-Regulated in T Lymphocytes and Plasma Cells", J. Immunol. 152:557-565 (1994). Los inhibidores de PI3K-8 y/o y pueden presentar un potencial terapéutico único en las neoplasias malignas de células T.
En los neutrófilos, la PI3K-8, junto con la PI3K-Y, contribuyen a las respuestas a los complejos inmunitarios, y a la señalización de FCyRII, incluyendo la migración y estallido respiratorio de neutrófilos. Los neutrófilos humanos experimentan una rápida inducción de Pip3 en respuesta al receptor de péptido formilo (FMLP, por sus siglas en inglés) o componente del complemento C5a (C5a) de una manera dependiente de la PI3K-Y, seguido de un periodo de producción de PIP3 más prolongado que es dependiente de PI3K-8 y resulta esencial para el estallido respiratorio. Las quinasas PI3K-8, PI3K-Y y PI3K-p contribuyen a la respuesta a los complejos inmunitarios, y son un importante mediador en el daño a los tejidos en modelos de enfermedad autoinmune (Randis TM et al., (2008) Eur. J. Immunol.
38(5):1215-24; Pinho V, (2007) J. Immunol. 179(11):7891-8; Sadhu C. et al. (2003) J. Immunol. 170(5):2647-54; Condliffe AM et al. (2005) Blood 106(4):1432-40). Se ha informado de que, en determinadas enfermedades autoinmunes, podría participar la activación preferente de PI3K-p (Kulkarni et al., Immunology (2011) 4(168) ra23: 1 11). También se ha informado de que los ratones deficientes en PI3K-p se encontraban altamente protegidos en un modelo dependiente de FcyR de la formación de ampollas en la piel inducida por autoanticuerpos y parcialmente protegidos en un modelo dependiente de FcyR de artritis inflamatoria, mientras que la deficiencia combinada de PI3K-p y PI3K-8 resultaba en una protección prácticamente completa en la artritis inflamatoria (Id.).
En macrófagos recogidos de pacientes con enfermedad pulmonar obstructiva crónica (EPOC), puede restaurarse la reactividad a glucocorticoides mediante el tratamiento de las células con inhibidores de PI3K-8. Los macrófagos también se basan en PI3K-8 y PI3K-Y para las respuestas a los complejos inmunitarios a través de la reacción de Arthus (señalización de FCyR y C5a) (Randis TM, et al. (2008) Eur. J. Immunol. 38(5):1215-24; Marwick JA et al. (2009) Am. J. Respir. Crit. Care. Med. 179(7):542-8; Konrad S, et al. (2008) J. Biol. Chem. 283(48):33296-303).
La teofilina incrementa la sensibilidad a histona desacetilasa-2 y a corticoesteroides in vitro y en ratones fumadores in vivo mediante inhibición de la quinasa PI3 (p.ej., PI3K-delta). La PI3K se activa en los pulmones con EOC y se ha mostrado que determinados inhibidores de PI3K imitan los efectos de la teofilina en la reversión de la resistencia a corticoesteroides. Yasuo, T., et al., Am. J. Respir. Crit. Care Med. 2010;182:897-904. Sin deseo de restringirse a ninguna teoría, una justificación para el uso de inhibidores de Pi3k (p.ej., compuestos proporcionados en la presente memoria) para tratar la EPOC es que un inhibidor de PI3K puede incrementar la sensibilidad a corticoesteroides en un sujeto.
En mastocitos, la proliferación, diferenciación y función dependientes de factores de célula madre (FCM) y dependiente de IL-3 son dependientes de PI3K-8, al igual que la quimiotaxis. El entrecruzamiento de alérgenos/IgE de f CyR1 que resulta en la liberación y degranulación de citoquinas de los mastocitos resulta gravemente inhibida por el tratamiento con inhibidores de PI3K-8, sugiriendo un papel para PI3K-8 en las enfermedades alérgicas (Ali K et al. (2004) Nature 431(7011):1007-11; Lee KS, et al. (2006) FASEB J. 20(3):455-65; Kim MS, et al. (2008) Trends Immunol. 29(10):493-501).
Las células asesinas naturales (NK) son dependientes tanto de PI3K-8 como de PI3K-Y para la migración eficiente hacia las quimioquinas, incluyendo CXCL10, CCL3, SIP y CXCL12, o en respuesta a LPS en el peritoneo (Guo H. et al. (2008) J. Exp. Med. 205(10):2419-35; Tassi I, et al. (2007) Immunity 27(2):214-27; Saudemont A, (2009) Proc Natl Acad Sci USA. 106(14):5795-800; Kim N, et al. (2007) Blood 110(9):3202-8).
Isoformas PI3K-5 y/o PI3K-Y en determinados cánceres
Las PI3K de clase I, particularmente las isoformas PI3K-8 y PI3K-Y, también estan asociadas a los cánceres (revisión en, p.ej., Vogt, PK et al. (2010) Curr. Top Microbiol. Immunol. 347:79-104; Fresno Vara, JA et al. (2004) Cancer Treat Rev. 30(2):193-204; Zhao, L. y Vogt, PK. (2008) Oncogene 27(41):5486-96). Los inhibidores de PI3K, p.ej., PI3K-8 y/o PI3K-Y, se ha mostrado que presentan actividad anticáncer (p.ej., Courtney, KD et al. (2010) J. Clin. Oncol. 28(6):1075-1083); Markman, B et al. (2010) Ann. Oncol. 21(4):683-91; Kong, D. y Yamori, T (2009) Curr Med Chem. 16(22):2839-54; Jimeno, A et al. (2009) J. Clin. Oncol. 27:156s (supl.; resumen n° 3542); Flinn, IW et al. (2009) J. Clin. Oncol.
27:156s (supl.; resumen n° 3543); Shapiro, G et al. (2009) J. Clin. Oncol. 27:146s (supl.; resumen n° 3500); Wagner, AJ etal. (2009) J. Clin. Oncol. 27:146s (supl.; resumen n° 3501); Vogt, PK et al. (2006) Virology 344(1):131-8; Ward, S
et al. (2003) Chem. Biol. 10(3):207-13; documentos n° WO 2011/041399; n° US 2010/0029693; n° US 2010/0305096 y n° US 2010/0305084).
En la presente memoria se describe un método de tratamiento del cáncer. En la presente memoria se da a conocer un método de tratamiento de un cáncer hemático que comprende administrar una cantidad farmacéuticamente eficaz de un compuesto proporcionado en la presente memoria en un sujeto que lo necesita. En la presente memoria se da a conocer un método de tratamiento de un tumor sólido que comprende administrar una cantidad farmacéuticamente eficaz de un compuesto proporcionado en la presente memoria en un sujeto que lo necesita. Entre los tipos de cáncer que pueden tratarse con un inhibidor de PI3k (particularmente, PI3K-8 y/o PI3K-y) se incluyen, p.ej., leucemia, leucemia linfocítica crónica, leucemia mieloide aguda y leucemia mieloide crónica (p.ej., Salmena L. et al. (2008) Cell 133:403-414; Chapuis, N et al. (2010) Clin. Cancer Res. 16(22):5424-35; Khwaja, A (2010) Curr. Top Microbiol. Immunol. 347:169-88); linfoma, p.ej., linfoma no de Hodgkin (p.ej., Salmena, L et al. (2008) Cell 133:403-414); cáncer pulmonar, p.ej., cáncer pulmonar no microcítico, cáncer pulmonar microcítico (p.ej., Herrera, VA et al. (2011) Anticancer Res. 31(3):849-54); melanoma (p.ej., Haluska, F et al. (2007) Semin Oncol. 34(6):546-54); cáncer de próstata (p.ej., Sarker, D et al. (2009) Clin. Cancer Res. 15(15):4799-805); glioblastoma (p.ej., Chen, JS et al. (2008) Mol. Cancer Ther. 7:841-850); cáncer endometrial (p.ej., Bansal, N et al. (2009) Cancer Control. 16(1):8-13); cáncer pancreático (p.ej., Furukawa, T (2008) J. Gastroenterol. 43(12):905-11); carcinoma de células renales (p.ej., Porta, C and Figlin, RA (2009) J Urol. 182(6):2569-77); cáncer colorrectal (p.ej., Saif, MW y Chu, E (2010) Cancer J. 16(3):196-201); cáncer de mama (p.ej., Torbett, NE et al. (2008) Biochem J. 415:97-100); cáncer tiroideo (p.ej., Brzezianska, E. y Pastuszak-Lewandoska, D. (2011) Front Biosci. 16:422-39) y cáncer ovárico (p.ej., Mazzoletti, M y Broggini, M (2010) Curr. Med. Chem. 17(36):4433-47).
Numerosas publicaciones apoyan un papel de PI3K-8 y PI3K-Y en el tratamiento de los cánceres hemáticos. PI3K-8 y PI3K-Y se expresan a nivel elevado en el compartimiento hemático, y en tumores sólidos, incluyendo tumores de próstata y mama, y los glioblastomas (Chen J.S. et al. (2008) Mol. Cancer Ther. 7(4):841-50; Ikeda H. et al. (2010) Blood 116(9):1460-8).
En los cánceres hemáticos, incluyendo la leucemia mieloide aguda (LMA), el mieloma múltiple (MM) y la leucemia linfocítica crónica (LLC), la sobreexpresión y activación constitutiva de PI3K-5 apoya el modelo de que la inhibición de PI3K-5 sea terapéutica Billottet C, et al. (2006) Oncogene 25(50):6648-59; Billottet C, et al. (2009) Cancer Res.
69(3):1027-36; Meadows, SA, 52nd Annual ASH Meeting and Exposition; 4-7 dic. de 2010; Orlando, Fl ; Ikeda H, et al. (2010) Blood 116(9):1460-8; Herman SE et al. (2010) Blood 116(12):2078-88; Herman SE et al. (2011). Blood 117(16):4323-7.
En la presente memoria se describe un método de tratamiento de cánceres hemáticos, incluyendo, aunque sin limitación, la leucemia mieloide aguda (LMA), el mieloma múltiple (MM) y la leucemia linfocítica crónica (LLC).
Se ha evaluado un inhibidor de PI3K-5 (CAL-101) en un ensayo de fase 1 en pacientes con neoplasias malignas hemáticas, y ha mostrado actividad en LLC en pacientes con características de mal pronóstico. En la LLC, la inhibición de PI3K-5 no solo afecta a las células tumorales directamente, sino que también afecta a la capacidad de las células tumorales de interactuar con su microentorno. Dicho microentorno incluye el contacto con, y los factores de, células estromales, células T, células de tipo nodriza, así como otras células tumorales. CAL-101 suprime la expresión de factores estromales y derivados de células T, incluyendo CCL3, CCL4 y CXCL13, así como la capacidad de las células tumorales LLC de responder a dichos factores. El tratamiento con CAl-101 en pacientes de LLC induce una rápida reducción de los ganglios linfáticos y la redistribución de los linfocitos en la circulación, y afecta a las señales de supervivencia tónicas a través de los RCB, conduciendo a una viabilidad celular reducida y a un incremento de la apoptosis. El tratamiento de agente único CAL-101 también es activo en el linfoma de células del manto y en el linfoma no de Hodgkin refractario (Furman, RR et al., 52nd Annual ASH Meeting and Exposition; 4-7 de dic. de 2010; Orlando, FL; Hoellenriegel, J, et al. 52nd Annual ASH Meeting and Exposition; 4-7 de dic. de 2010; Orlando, FL; Webb, HK, et al. 52nd Annual ASH Meeting and Exposition; 4-7 de dic. de 2010; Orlando, FL; Meadows, et al. 52nd Annual ASH Meeting and Exposition; 4-7 de dic. de 2010; Orlando, FL; Kahl, B, et al. 52nd Annual ASH Meeting and Exposition; 4 7 de dic. de 2010; Orlando, FL; Lannutti BJ, et al. (2011) Blood 117(2):591-4).
Los inhibidores de PI3K-5 han mostrado actividad contra los gliomas positivos para PI3K-5 in vitro (Kashishian A, et al. Póster presentado en: The American Association of Cancer Research 102nd Annual Meeting; 2-6 de abril de 2011; Orlando, FL). En dicho subgrupo de tumores, el tratamiento con el inhibidor de PI3K-5, solo o en combinación con un agente citotóxico, puede resultar eficaz.
Otro mecanismo para que los inhibidores de PI3K-5 tengan un efecto en los tumores sólidos implica la interacción de las células tumorales con su microentorno. La PI3K-5, PI3K-Y y PI3K-p se expresan en las células inmunitarias que infiltran los tumores, incluyendo linfocitos infiltrantes en tumores, macrófagos y neutrófilos. Los inhibidores de PI3K-5 pueden modificar la función de dichas células inmunitarias asociadas a tumor y cómo responden a señales del estroma, el tumor y entre ellas, y de esta manera afectan a las células y metástasis tumorales (Hoellenriegel, J, et al. 52nd Annual ASH Meeting and Exposition; 4 a 7 de dic. de 2010; Orlando, FL).
La PI3K-5 también se expresa en células endoteliales. Se ha mostrado que los tumores en ratones tratados con
inhibidores selectivos de PI3K-8 se eliminan más fácilmente mediante la terapia de radiación. En este mismo estudio, la formación de la red capilar se ve dificultada por el inhibidor de PI3K, y se plantea que este defecto contribuye a una mayor eliminación con la radiación. Los inhibidores de PI3K-8 pueden afectar a la manera en que los tumores interactúan con su microentorno, incluyendo las células estromales, las células inmunitarias y las células endoteliales, y pueden ser terapéuticos por si solos o junto con otras terapias (Meadows, SA et al., Artículo presentado en: 52nd Annual ASH Meeting and Exposition; 4 a 7 de dic. de 20l0; Orlando, FL; Geng L, et al. (2004) Cancer Res.
64(14):4893-9).
De acuerdo con lo anteriormente expuesto, en la presente memoria se da a conocer un método de tratamiento o prevención de un cáncer o enfermedad (incluyendo, aunque sin limitación, una neoplasia maligna hemática, o un tipo o subtipo específico de cáncer o enfermedad, tal como un tipo o subtipo específico de neoplasia maligna hemática) con un inhibidor selectivo de PI3K-Y, en el que se reducen los efectos adversos asociados a la administración de inhibidores para otra isoforma o isoformas de PI3k (p.ej., PI3K-a y/o PI3K-p). En la presente memoria se da a conocer un método de tratamiento o prevención del cáncer o de la enfermedad, con un inhibidor selectivo de PI3K-Y a una dosis más baja (p.ej., en aproximadamente 10 %, en aproximadamente 20 %, en aproximadamente 30 %, en aproximadamente 40 %, en aproximadamente 50 %, en aproximadamente 60 %, en aproximadamente 70 %, o en aproximadamente 80 %) en comparación con el tratamiento con un inhibidor no selectivo o menos selectivo de PI3K-Y (p.ej., un inhibidor PI3Kpan, p.ej., mediante la inhibición de PI3K-a, p, 8 y y).
La función de la ruta de PI3K-Y en la estimulación del tráfico de células mieloides a los tumores y el papel del bloqueo de p100Y en la supresión de la inflamación y crecimiento tumorales en el cáncer de mama, cáncer pancreático y cáncer pulmonar se informan en, por ejemplo, Schmid et al. (2011) Cancer Cell 19, 715-727. En la presente memoria se da a conocer un método de tratamiento o prevención del cáncer pancreático con un inhibidor de PI3K.
En la presente memoria se da a conocer un método de tratamiento o prevención del cáncer de mama con un inhibidor de PI3K. En la presente memoria se da a conocer un método de tratamiento o prevención del cáncer pulmonar con un inhibidor de PI3K. El inhibidor de PI3K puede ser un inhibidor de PI3K-Y, selectivo o no selectivo respecto a otra u otras isoformas de PI3K. El inhibidor de PI3K puede ser un inhibidor selectivo de PI3K-Y.
Aunque sin deseo de restringirse a ninguna teoría, se cree que el crecimiento tumoral está influenciado por dos clases de células inmunitarias en el microambiente tumoral: células efectoras, que incluyen células citotóxicas y macrófagos M1, y que presentan actividad antitumoral, y células supresoras, que incluyen macrófagos M2, MDSC (células supresoras derivadas de mieloides), Treg (células T reguladoras) y células dendríticas reguladoras, que presentan actividad protumoral porque inhiben las células efectoras. Una abundancia de células supresoras puede conducir a una tolerancia inmunitaria del tumor y la potenciación del crecimiento tumoral.
Se describen brevemente algunos de dichos tipos celulares. M1 denota un fenotipo proinflamatorio (antitumoral) de una CSDM o MAT. M2 denota un fenotipo proinflamatorio (protumoral) de un CSDM o MAT.
La PI3K-Y no se expresa en por lo menos algunos tipos de célula cancerosa. Schmid et al., 2011, Cancer Cell 19. De acuerdo con lo anterior, el inhibidor de PI3K-Y puede reducir el crecimiento de las células cancerosas sin presentar un efecto directo sustancial sobre la célula cancerosa misma. Por ejemplo, el inhibidor de PI3K-Y puede inhibir el crecimiento de las células cancerosas mediante cambios en el microentorno tumoral, p.ej., las células inmunitarias en estrecha proximidad a las células cancerosas.
Las pruebas proporcionadas en los Ejemplos en la presente memoria, en combinación con las pruebas en la literatura, apoyan la idea de que un inhibidor de PI3K-Y puede reducir las células mieloides asociadas a tumor. Por ejemplo, en ratones deficientes en PI3K-Y, se reducen las células mieloides asociadas a tumores. Schmid et al., 2011, Cancer Cell 19. Juntos, estos datos indican que una gran clase de inhibidores de PI3K-Y reducirían las células mieloides asociadas a los tumores, incrementando de esta manera la respuesta inmunitaria contra las células de cáncer y tratando el cáncer. Sin deseo de restringirse a ninguna teoría, una PI3K-Y podría funcionar mediante el mecanismo siguiente. La señalización de PI3K-Y podría inclinar el equilibro de las células inmunitarias hacia las células M2 protumorales y alejarlas de las células M1 antitumorales, mediante la inducción de la expresión de genes inmunosupresores de cicatrización de heridas, tales como la arginasa 1, TGFbeta1, PDGFBB, MMP9 y MMP13, y la supresión de factores proinflamatorios, tales como IL12, iNos e interferón gamma. El bloqueo de la señalización de PI3K-Y con un inhibidor inclina el equilibrio hacia las células M1 antitumorales mediante la estimulación de un programa de expresión de genes activadores de las células T. Kaneda et al. PI3-kinase gamma controls the macrophage M1-M2 switch, thereby promoting tumor immunosuppression and progression [La quinasa PI3-gamma controla el cambio M1-M2 de los macrófagos, estimulando de esta manera la inmunosupresión tumoral y la progresión] [Resumen]. en: Proceedings of the 105th Annual Meeting of the American Association for Cancer Research; 5 a 9 de abril de 2014; San Diego, CA. Philadelphia (PA): AACR; Cancer Res 2014;74(19 supl): Resumen n° 3650. doi:10.1158/1538-7445.AM2014-3650.
Puede administrarse un inhibidor de PI3K-Y en el paciente con el fin de bloquear una regulación negativa homeostática de la respuesta de células T. Aunque sin deseo de restringirse a ninguna teoría, lo anterior podría permitir al cuerpo generar una respuesta inmunitaria eficaz contra las células cancerosas. Entre los agentes ejemplares de este tipo se incluyen los terapéuticos de punto de control inmunitario, p.ej., agentes que actúan sobre CTLA-4, PD-1 o PD-L1, p.ej.,
anticuerpos que se unen a CTLA-4, PD-1 o PD-L1. Los terapéuticos de punto de control inmunitario se describen en mayor detalle posteriormente.
Puede administrarse un inhibidor de PI3K-Y en el paciente con el fin de eliminar las células inmunosupresoras en el microambiente tumoral. La célula inmunosupresora puede ser, p.ej., una célula T reguladora (p.ej., una célula que secreta mediadores que inducen la muerte de las células T citotóxicas CD8+), un macrófago asociado a tumor (mAt , p.ej., un MAT de M2 (protumoral) que bloquea la actividad de las células T y estimula la angiogénesis) o una célula supresora derivada de mieloide (CSDM, p.ej., una célula que secreta mediadores que inhiben la diferenciación y proliferación de las células T).
Puede administrarse un compuesto proporcionado en la presente memoria en un paciente con el fin de reducir la migración o la diferenciación de unas células mieloides asociadas a tumor. El compuesto puede ser un compuesto que muestra actividad de agente único en un sistema modelo singénico. El compuesto puede administrarse en combinación con un segundo terapéutico, tal como se comenta en la presente memoria. La administración puede resultar en una reducción del nivel de CSDM en el microambiente tumoral; el nivel de MAT M2 en el microambiente tumoral; el nivel de células T reguladoras en el microambiente tumoral, o cualquier combinación de los mismos. La administración puede resultar en un nivel sin cambios o incrementado de células T efectoras en el microambiente tumoral. La administración puede resultar en un incremento de la respuesta inmunitaria al tumor, p.ej., en un incremento de los niveles o actividad de ataque tumoral de las células T citotóxicas, MAT inflamatorios M1 o una combinación de los mismos.
Una CSDM puede presentar una o más de las propiedades siguientes: suprimir el ataque inmunitario antitumoral, inducir la vascularización del tumor, inducir la degradación de la MEC, p.ej., que puede contribuir a la metástasis y apoyar el crecimiento tumoral. De acuerdo con lo anterior, la administración de un inhibidor de PI3K-Y indicado en la presente memoria puede inhibir una o más de dichas funciones en una CSDM.
Los MAT (macrófagos asociados a tumor) también pueden presentar una o más de las propiedades siguientes: suprimir el ataque inmunitario antitumoral, inducir la vascularización del tumor, inducir la degradación de la MEC, p.ej., que puede contribuir a la metástasis y apoyar el crecimiento tumoral. De acuerdo con lo anterior, la administración de un inhibidor de PI3K-Y indicado en la presente memoria puede inhibir una o más de dichas funciones en un MAT.
Puede administrarse un inhibidor de PI3K-Y en un paciente que ha recibido quimioterapia y/o terapia de radiación. Sin deseo de restringirse a ninguna teoría, la quimioterapia o la terapia de radiación puede resultar en una respuesta de cicatrización de heridas que conduce a la repoblación del sitio canceroso, p.ej., tumoral, con MAT y CSDM. La administración del inhibidor de PI3K-Y puede reducir los niveles de MAT y CSDM en el microambiente, reduciendo su apoyo al crecimiento de las células tumorales y/o permitiendo que el sistema inmunitario ataque las células de cáncer. Ver Claire E. Lewis, "Imaging immune cell infiltrating tumors in zebrafish", AACR Annual Meeting (5 de abril, 2014).
Sin deseo de restringirse a ninguna teoría, una justificación para el uso de un inhibidor de PI3K-gamma es la terapia adicional a la radiación para evitar la acumulación de células mieloides de apoyo tumoral en el tumor radiado, que de esta manera dificultan el nuevo crecimiento tumoral tras la terapia de radiación. Lo anterior ha sido apoyado por el trabajo de Kioi et al. (2010) J. Clin. Invest. 120(3):694-705, que muestra que un inhibidor de la migración de las células mieloides al interior de los tumores postirradiación (p.ej., AMD3100) bloqueó la vasculogénesis y el nuevo crecimiento tumoral.
En la presente memoria se da a conocer un método de tratamiento de un trastorno o enfermedad indicado en la presente memoria, que comprende administrar un compuesto proporcionado en la presente memoria, p.ej., un inhibidor selectivo de PI3k-Y, un inhibidor selectivo de PI3K 8 o un inhibidor doble de PI3K y/8. Sin limitarse a ninguna teoría en particular, la inhibición selectiva de la isoforma PI3K-Y puede proporcionar un régimen de tratamiento en el que los efectos adversos asociados a la administración de un inhibidor no selectivo de PI3K se minimizan o se reducen. Sin limitarse a ninguna teoría en particular, la inhibición selectiva de la isoforma PI3K-8 puede proporcionar un régimen de tratamiento en el que los efectos adversos asociados a la administración de un inhibidor no selectivo de PI3K se minimizan o se reducen. Sin limitarse a ninguna teoría en particular, la inhibición selectiva de las isoformas PI3K-8 y Y puede proporcionar un régimen de tratamiento en el que los efectos adversos asociados a la administración de un inhibidor no selectivo de PI3K se minimizan o se reducen. Sin limitarse a ninguna teoría en particular, se cree que los efectos adversos pueden reducirse evitando la inhibición de otras isoformas (p.ej., a o p) de PI3K.
El efecto adverso puede ser la hiperglucemia. El efecto adverso pueden ser erupciones. El efecto adverso puede ser un deterioro de la fertilidad masculina que puede resultar de la inhibición de la isoforma p de PI3K (ver, p.ej., Ciraolo et al., Molecular Biology of the Cell, 21: 704-711 (2010)). El efecto adverso puede ser toxicidad testicular, que puede resultar la inhibición de PI3K-p (ver, p.ej., Wisler et al., Amgen SOT, resumen n° de ID 2334 (2012)). El efecto adverso puedes ser la letalidad embrionaria (ver, p.ej., Bi et al., J. Biol. Chem., 274: 10963-10968 (1999)). El efecto adverso puede ser una agregación plaquetaria defectuosa (ver, p.ej., Kulkarni et al., Science, 287: 1049-1053 (2000)). El efecto adverso pueden ser neutrófilos funcionalmente defectuosos (id.).
En la presente memoria se da a conocer un método de tratamiento o prevención del cáncer (p.ej., cáncer de colon,
melanoma, cáncer de vejiga, cáncer renal, de mama, cáncer pulmonar, glioblastoma, tumores sólidos y un cáncer de origen hematopoyético (p.ej., LCBGD, LLC, linterna de Hodgkin y linternas no de Hodgkin), que comprende administrar en el sujeto un inhibidor de PI3K (p.ej., un inhibidor de PI3K-Y, p.ej., un compuesto de cualquier fórmula de entre (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., el Compuesto 4, tal como se indica en la presente memoria).
Sin restringirse a ninguna teoría en particular, un argumento para la utilización de un inhibidor de PI3K en el tratamiento o prevención del cáncer es que las células derivadas de tumores (p.ej., tumores de ratón CT26) pueden suprimir la función de las células inmunitarias antitumorales, incluyendo la proliferación de las células T, tal como se muestra en los ejemplos proporcionados en la presente memoria, y el tratamiento con un compuesto proporcionado en la presente memoria puede liberar la supresión. El microambiente tumoral puede inhibir la activación y proliferación de las células efectoras inmunitarias debido a la presencia de células mieloides supresoras (p.ej., células supresoras derivadas de mieloides, o macrófagos CSDM y M2). Los compuestos proporcionados en la presente memoria pueden afectar al número y actividad de los macrófagos M2 en un microambiente tumoral, p.ej., reducir o inhibir el nivel de macrófagos protumorales M2. La reducción o inhibición de los macrófagos M2, que producen citoquinas antiinflamatorias y otros factores, conduciría a una inmunidad antitumoral incrementada, incluyendo la proliferación de las células T. Por lo tanto, un compuesto proporcionado en la presente memoria puede tratar o prevenir el cáncer, tal como cáncer de colon, melanoma, cáncer de vejiga, cáncer renal, cáncer de mama, cáncer pulmonar, glioblastoma, tumores sólidos y un cáncer de origen hematopoyético (p.ej., linfoma, LCBGD, LLC, enfermedad de Hodgkin y linfomas no de Hodgkin). Además, también se ha mostrado en los ejemplos proporcionados en la presente memoria que anti-PDL1 también puede liberar la supresión de la proliferación de las células T mediante el bloqueo de la interacción entre PD1 sobre las células T y PDL1 sobre las células tumorales y las células reguladoras. Las células T citotóxicas que resultan inducidas para proliferar y sobrevivir tanto por anti-PDL-1 como por el compuesto BB se plantea la hipótesis de que retrasan el crecimiento tumoral. Los compuestos proporcionados en la presente memoria pueden aliviar la inmunosupresión, lo que puede conducir a la proliferación y activación de las células T. Los compuestos proporcionados en la presente memoria pueden tratar o prevenir el cáncer, mediante la inducción de la inmunidad mediada por células T. En una realización, el compuesto proporcionado en la presente memoria puede reducir el volumen tumoral. En una realización, una combinación de un inhibidor de PI3K, tal como un compuesto proporcionado en la presente memoria y anti-PDL1, resultaría eficaz en el tratamiento o prevención del cáncer mediante la inducción de la inmunidad tumoral mediada por células T. En algunas realizaciones, el efecto de un compuesto proporcionado en la presente memoria sobre la función de las células T puede evaluarse mediante el análisis de los niveles de citoquinas proinflamatorias en tejidos tumorales y suero, p.ej., un panel proinflamatorio MSD. En otra realización, las citoquinas proinflamatorias se seleccionan de entre IFN-y, IL-1p, IL-10, IL-12 p70, IL-2, IL-4, IL-5, IL-6, KC/GRO y TNF-a. En una realización, el efecto de un compuesto proporcionado en la presente memoria sobre la función de las células T puede evaluarse mediante el análisis del nivel de IFN-y. Por ejemplo, los tejidos tumorales y suero tratados con un compuesto proporcionado en la presente memoria, p.ej., el Compuesto 4, pueden evaluarse mediante análisis del nivel de IFN-y.
Tratamiento de cánceres
La administración de una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., un inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4), o una forma farmacéuticamente aceptable del mismo, en un sujeto puede modular el microambiente tumoral de las células de cáncer en el sujeto.
Tal como se utiliza en la presente memoria y a menos que se especifique lo contrario, «microambiente tumoral» se refiere al medio celular y extracelular en el que se localizan los tumores. Dicha localización puede incluir vasos sanguíneos circundantes, células inmunitarias, fibroblastos, moléculas de señalización secretadas y la matriz extracelular. El microambiente tumoral incluye células estromales e inmunitarias, no neoplásicas, que proporcionan apoyo de crecimiento y supervivencia al tumor neoplásico.
Tal como se utiliza en la presente memoria y a menos que se especifique lo contrario, “ inmunoterapia” se refiere a tratamientos que estimulan, potencian o suprimen el propio sistema inmunitario del cuerpo en su ataque de la enfermedad. Entre las enfermedades que pueden resultar adecuadas para el tratamiento inmunoterapéutico se incluyen, aunque sin limitarse a ellas, cáncer, enfermedades inflamatorias y enfermedades infecciosas. La inmunoterapia incluye una diversidad de tratamientos que funcionan de maneras diferentes. Por ejemplo, algunos están destinados a reforzar las defensas del sistema inmunitario de una manera general; otros ayudan a entrenar al sistema inmunitario para reconocer y atacar específicamente las células de cáncer. Entre las inmunoterapias del cáncer se incluyen, aunque sin limitarse a ellas, terapias celulares (también conocidas como vacunas del cáncer), terapias de anticuerpos y terapias de citoquinas (p.ej., interleuquina-2 e interferón-a).
Es conocido que muchos cánceres son susceptibles de tratamiento con una o más inmunoterapias, incluyendo el tratamiento con diana en células efectoras en el microambiente tumoral (p.ej., terapia de punto de control inmunitario, tal como inhibidores de PD-1/PD-L1 e inhibidores de CTLA-4), el tratamiento con diana en células supresoras en el microambiente tumoral (p.ej., inhibidores de CSF-1R (que afectan a CSDM y MAT) e inhibidores IDO/TDO). Sin
restringirse a ninguna teoría en particular, un compuesto proporcionado en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., un inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4) puede afectar a las CSDM, MAT y otros componentes en el microambiente tumoral. El papel de MAT en el microambiente tumoral se describe en, p.ej., Lewis y Pollard, Cancer Res. 2006; 66: (2). 15 de enero de 2006.
El número de una o más células inmunitarias protumorales en el microambiente tumoral puede reducirse o la actividad de una o más células inmunitarias protumorales en el microambiente tumoral puede reducirse o inhibirse, después de la administración del compuesto. En algunas realizaciones, la célula inmunitaria protumoral es una célula T, un macrófago M2, una célula estromal, una célula dendrítica, una célula endotelial o una célula mieloide. En una realización, la célula mieloide es una célula mieloide supresora asociada a tumor. En una realización, la célula mieloide supresora asociada a tumor se identifica por (i) CD45+, CD11b+, Ly6C+ y Ly6G+, (ii) CD45+, CD11b+, Ly6C- y Ly6G-, (iii) CD45+, CD11b+, Ly6C- y Ly6G+, o (iv) CD45+, CD11b+, Ly6C+ y Ly6G-. En una realización, la célula mieloide supresora asociada a tumor es un macrófago asociado a tumor (MAT), una célula supresora derivada de mieloide (CSDM), una célula mieloide inmadura monocítica (CMi) o un iMc granulocítico/neutrófilo. En una realización, la MAT se identifica por CD45+, CD11b+, Ly6C- y Ly6G-. En una realización, la célula supresora derivada de mieloide (CSDM) se identifica por CD45+, CD11b+, Ly6C-y Ly6G+. En una realización, la célula mieloide inmadura (cMi) monocítica se identifica por CD45+, CD11b+, Ly6C+ y Ly6G-. En una realización, la CMi granulocítica/neutrófilo se identifica por CD45+, CD11b+, Ly6C+ y Ly6G+. Ver, p.ej., Coussens LM. et al., Cancer Discov. junio de 2011; 1(1):54-67.
La activación de los macrófagos M2 en el microambiente tumoral puede reducirse o inhibirse después de la administración del compuesto. El nivel de p-AKT en los macrófagos M2 puede reducirse después de la administración del compuesto. El número de células macrófagas M2 en el microambiente tumoral puede reducirse después de la administración del compuesto. La migración de las células macrófagas M2 en el microambiente tumoral puede reducirse o inhibirse después de la administración del compuesto. La diferenciación de las células mieloides en células macrófagas M2 en el microambiente tumoral puede reducirse o inhibirse después de la administración del compuesto. La diferenciación en células macrófagas M2 puede medirse a partir del nivel de arginasa-1 (ARG1) o el nivel de VEGF, y el nivel de ARG1 o el nivel de VEGF se reduce en por lo menos 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 % o 90 % en comparación con un valor de referencia.
El número de células supresoras derivadas de mieloides en el microambiente tumoral pueden reducirse después de la administración del compuesto. La diferenciación de células de la médula ósea en células supresoras derivadas de mieloides puede reducirse o inhibirse después de la administración del compuesto. La diferenciación en células supresoras derivadas de mieloides puede medirse a partir del nivel de arginasa-1 (ARG1), el nivel de VEGF o el nivel de iNOS, y el nivel de ARG1, el nivel de VEGF o el nivel de iNOS se reduce en por lo menos 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 % o 90 % en comparación con un valor de referencia.
La producción de factor proangiogénico puede reducirse o inhibirse después de la administración del compuesto. El factor proangiogénico puede reducirse o inhibirse mediante la reducción o inhibición de la diferenciación de macrófagos o CSDM. En una realización, el factor proangiogénico es VEGF.
El efecto de los compuestos proporcionados en la presente memoria (p.ej., el Compuesto 4) sobre la función de las CSDM (p.ej., las CSDm humanas) puede medirse a partir de la expresión de iNOS y arginasa, y de la producción de ROS e IL-10, medidas a partir de la función de supresión de las CSDM (p.ej., en ensayos en cocultivo con CD8+), mediante la activación de pAKT en respuesta a un estimulante (p.ej., CXCL12, IL-1b, TNF-a o CSF1), o mediante la medición en ensayos de quimiotaxis transpocillo (células T y CSDm ).
El efecto de los compuestos proporcionados en la presente memoria (p.ej., el Compuesto 4) sobre la función de las CSDM (p.ej., CSDM murinas) y la polarización de los macrófagos M2 pueden medirse mediante aislamiento de las células mieloides a partir de médula ósea, polarizando con IFNg o iL-4 y después sometiendo a ensayo para la secreción de TNF-a, IL-12, producción de ROS en M1 e IL-10, IL-1b, o VEGF, o mediante los métodos proporcionados en la presente memoria o en otro sitio.
El efecto de los compuestos proporcionados en la presente memoria (p.ej., el Compuesto 4) sobre las mieloides y CD8+ puede medirse mediante modelos in vivo (p.ej., MC38 y 4T1). El efecto puede medirse mediante TGI, infiltrados de CSDM y macrófagos, CD8+ y producción de IFN-gamma en CD8+.
El efecto de los compuestos proporcionados en la presente memoria (p.ej., el Compuesto 4) sobre las mieloides y CD8+ puede medirse mediante QT-PCR o FACS intracelular del infiltrado de las mieloides. El efecto puede medirse mediante la expresión de marcadores funcionales (p.ej., iNOS, arginasa o IL-10).
El número de una o más células inmunitarias antitumorales en el microambiente tumoral puede incrementarse, o la actividad de una o más células inmunitarias antitumorales en el microambiente tumoral puede incrementarse después de la administración del compuesto.
En una realización, el cáncer susceptible al tratamiento de una o más inmunoterapias es un cáncer hemático. En una
realización, el cáncer hemático es leucemia linfocítica crónica (LLC). En una realización, el microambiente tumoral es un centro de proliferación de LLC. En una realización, el cáncer hemático es linfoma.
En una realización, el cáncer susceptible de tratamiento de una o más inmunoterapias es un tumor sólido. En una realización, el tumor sólido es cáncer pulmonar, cáncer de mama, cáncer de colon o glioblastoma. En una realización, el cáncer se selecciona de uno o más de los siguientes: cáncer del sistema pulmonar, un cáncer cerebral, un cáncer del tracto gastrointestinal, un cáncer de piel, un cáncer genitourinario, un cáncer pancreático, un cáncer pulmonar, un meduloblastoma, un carcinoma de células basales, un glioma, un cáncer de mama, un cáncer de próstata, un cáncer testicular, un cáncer esofágico, un cáncer hepatocelular, un cáncer gástrico, un tumor estromal gastrointestinal (TEGI), un cáncer de colon, un cáncer colorrectal, un cáncer ovárico, un melanoma, un tumor neuroectodérmico, cáncer de cabeza y cuello, un sarcoma, un sarcoma de tejidos blandos, fibrosarcoma, mixosarcoma, liposarcoma, un condrosarcoma, un sarcoma osteogénico, un cordoma, un angiosarcoma, un endoteliosarcoma, un linfangiosarcoma, un linfangioendoteliosarcoma, un sinovioma, un mesotelioma, un leiomiosarcoma, un cáncer de cuello uterino, un cáncer uterino, un cáncer endometrial, un carcinoma, un carcinoma de vejiga, un carcinoma epitelial, un carcinoma de células escamosas, un adenocarcinoma, un carcinoma broncogénico, un carcinoma de células renales, un hepatoma, un carcinoma del conducto biliar, un cáncer neuroendocrino, un tumor carcinoide, tumor de células gigantes de tipo difuso y glioblastoma. En una realización, el tumor sólido es melanoma, cáncer de vejiga, cáncer de cabeza y cuello, cáncer pulmonar (p.ej., cáncer pulmonar no microcítico), carcinoma de células renales, cáncer ovárico, cáncer de mama (p.ej., cáncer de mama triple negativo), cáncer de colon o glioblastoma.
En una realización, el tumor sólido es melanoma. En una realización, el tumor sólido es cáncer pulmonar. En una realización, el tumor sólido es cáncer pulmonar no microcítico. En una realización, el tumor sólido es carcinomas de células renales. El melanoma, el cáncer pulmonar (p.ej., cáncer pulmonar no microcítico) y carcinoma de células renales es conocido que son sensibles a las inmunoterapias. Se ha informado de datos que asocian un mal pronóstico a recuentos altos de células MAT en cánceres de mama, próstata, endometrial, vejiga, riñón, esófago, superficial, carcinoma, melanoma y linfoma folicular. Ver, p.ej., Lewis y Pollard, Cancer Res. 2006; 66: (2). 15 de enero de 2006. Un fármaco anticuerpo anti-PD-1, el nivolumab, (Opdivo - Bristol Myers Squibb), ha producido respuestas completas o parciales en cáncer pulmonar no microcítico, melanoma y cáncer de células renales, en un ensayo clínico con un total de 296 pacientes.
En una realización, el tumo sólido es cáncer de cabeza y cuello. Los tumores de cabeza y cuello tienden a ser altamente inmunogénicos y presentan una fuerte eficacia anti-PD-1/PD-L1. En una realización, el tumor sólido es cáncer de vejiga. El cáncer de vejiga también presenta una fuerte eficacia anti-PD-1/PD-L1. Se ha asociado un número elevado de células MAT a un mal pronóstico y angiogénesis tumoral incrementada en el cáncer de vejiga.
En una realización, el tumor sólido es cáncer de mama. En una realización, el cáncer de mama es cáncer de mama triple negativo. Se ha asociado un número elevado de células MAT a un mal pronóstico en el cáncer de mama. Ver, p.ej., Lewis y Pollard, Cancer Res. 2006; 66: (2). 15 de enero de 2006. En una realización, el tumor sólido es cáncer ovárico. En una realización, el tumor sólido es cáncer de colon. El cáncer de mama, cáncer ovárico y cáncer de colon es conocido que son sensibles a las inmunoterapias (p.ej., bevacizumab y trastuzumab) y también pueden presentar eficacia anti-PD-1/PD-L1.
En una realización, el tumor sólido es glioblastoma. En una realización, el tumor sólido es glioblastoma multiforme Se ha informado de que la expresión de PI3K-gamma está regulada positivamente en la microglía cerebral. Aunque sin limitarse a ninguna teoría en particular, los inhibidores de PI3K-Y proporcionados en la presente memoria (p.ej., el compuesto BB) podrían presentar actividad inhibidora de la glucoproteína P y, de esta manera, podrían cruzar la barrera hematocefálica.
En una realización, el ataque inmunitario antitumoral por células T efectoras se encuentra incrementado, la vascularización del tumor se encuentra reducida, la degradación de la matriz extracelular (MEC) se encuentra reducida o el crecimiento tumoral se encuentra reducido, en comparación con un valor de referencia, después de la administración del compuesto.
En una realización, el volumen tumoral del cáncer se encuentra reducido después de la administración del compuesto. En una realización, el volumen tumoral del cáncer se encuentra reducido en por lo menos 10 %, 20 %, 30 %, 50 %, 60 % o 60 % en comparación con un valor de referencia.
El nivel de apoptosis de las células de cáncer puede incrementarse después de la administración del compuesto. El nivel de apoptosis de las células de cáncer puede incrementarse en por lo menos 10 %, 20 %, 30 %, 40 % o 50 % en comparación con un valor de referencia.
En algunas realizaciones, el sujeto nunca ha sido expuesto a tratamiento inmunoterapéutico. En algunas realizaciones, el sujeto nunca ha sido expuesto al tratamiento terapéutico de radiación. En algunas realizaciones, el sujeto nunca ha sido expuesto a tratamiento quimioterapéutico.
En algunas realizaciones, el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de
inmunoterapia. En una realización, el sujeto es respondedor al pretratamiento o tratamiento previo con la inmunoterapia. En una realización, el tratamiento de inmunoterapia es un tratamiento de punto de control, tal como un inhibidor de PD-1 o PD-L1. En una realización, el sujeto es un fumador. Se ha informado de que los pacientes fumadores podrían responder mejor a la inmunoterapia (p.ej., un inhibidor de PD-L1, MPDL3280A) que los pacientes no fumadores, en un estudio clínico de fase I de pacientes con melanoma o cáncer de pulmón, de riñón, de colon, de tracto GI o de cabeza y cuello.
En una realización, el cáncer es melanoma y el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia. En una realización, el sujeto ha sido pretratado o tratado previamente con dos o más tratamientos de inmunoterapia.
En una realización, el cáncer es cáncer de cabeza y cuello, cáncer pulmonar (p.ej., cáncer pulmonar no microcítico), carcinoma de células renales o cáncer de vejiga, y el sujeto ha sido pretratado o tratado previamente con un tratamiento de inmunoterapia.
En una realización, el cáncer es cáncer de mama (p.ej., cáncer de mama triple negativo), cáncer ovárico, glioblastoma o cáncer de colon, y el sujeto nunca ha sido sometido a tratamiento de inmunoterapia.
El melanoma puede tratarse, prevenirse o controlarse en un sujeto mediante la administración en el sujeto de una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4) o una forma farmacéuticamente aceptable del mismo, en el que el sujeto ha sido pretratado previamente tratado con uno o más tratamientos de inmunoterapia. El tratamiento de inmunoterapia puede ser ipilimumab (Yervoy), inteleuquina-2, vemurafenib, dabrafenib o trametinib.
El cáncer pulmonar (p.ej., el cáncer pulmonar no microcítico) puede tratarse, prevenirse o controlarse en un sujeto mediante la administración en el sujeto de una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4) o una forma farmacéuticamente aceptable del mismo, en el que el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia. En una realización, el tratamiento de inmunoterapia es bevacizumab, erlotinib, gefitinib, afatinib, o denosumab.
El carcinoma de células renales puede tratarse, prevenirse o controlarse en un sujeto mediante la administración en el sujeto de una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4) o una forma farmacéuticamente aceptable del mismo, en el que el sujeto ha sido pretratado o previamente tratado con uno o más tratamientos de inmunoterapia. El tratamiento de inmunoterapia puede ser bevacizumab, interleukin-2, axitinib, carfilzomib, everolimus, interferón-a, lenalidomide, pazopanib, sirolimus (rapamicina), sorafenib, sunitinib, temsirolimus, thalidomida o tivozanib.
El cáncer de vejiga puede tratarse, prevenirse o controlarse en un sujeto mediante la administración en el mismo de una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), o una forma farmacéuticamente aceptable del mismo, en el que el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia. El tratamiento de inmunoterpia puede ser Bacillus Calmette-Guérin (BCG).
El cáncer de cabeza y cuello puede tratarse, prevenirse o controlarse en un sujeto mediante la administración en el mismo de una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4) o una forma farmacéuticamente aceptable del mismo, en el que el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia. El tratamiento de inmunoterapia puede ser cetuximab, nimotuzumab, bevacizumab, o erlotinib.
El cáncer de mama (p.ej., el cáncer de mama triple negativo) puede tratarse, prevenirse o controlarse en un sujeto mediante la administración en el sujeto de una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), o una forma farmacéuticamente aceptable del mismo, en el que el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia. El tratamiento de inmunoterapia es bevacizumab o trastuzumab.
El cáncer ovárico puede tratarse, prevenirse o controlarse en un sujeto mediante la administración en el sujeto de una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4) o una forma farmacéuticamente aceptable del mismo, en el que el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia. El tratamiento de inmunoterapia puede ser bevacizumab.
El cáncer de colon puede tratarse, prevenirse o controlarse en un sujeto mediante la administración en el mismo de una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), o una forma farmacéuticamente aceptable del mismo, en el que el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia. El tratamiento de inmunoterapia puede ser bevacizumab, cetuximab, o panitumumab.
El cáncer al que se refiere la presente invención puede ser un cáncer de origen hematopoyético. En determinadas realizaciones, el cáncer de origen hematopoyético es linfoma o leucemia. En algunas realizaciones, el cáncer de origen hematopoyético se selecciona de entre leucemia linfocítica aguda (LLA), que incluye LLA de linaje B y LLA de linaje T, leucemia linfocítica crónica (LLC), leucemia prolinfocítica (LPL), leucemia de células pilosas (LCP) y macroglobulinemia de Waldenstrom (MW), linfomas de células T periféricas (LCTP), leucemia/linfoma de células T adultas (LCTA), linfoma de células T cutáneas (LCTC), leucemia linfocítica granular de células grandes (LGL), leucemia mielocítica aguda (LMA), linfoma de Hodgkin (LH), linfoma no de Hodgkin (LNH), linfoma folicular, linfoma de células B grandes difusas (LCBGD), linfoma de células del manto (LCM), mastocitosis (p.ej., mastocitosis sistémica), mieloma múltiple (MM), síndrome mielodisplásico (SMD), trastorno mieloproliferativo (TMP) (p.ej., policitemia vera (PV), trombocitosis esencial (TE), metaplasia mieloide agnogénica (MMA), también conocida como mielofibrosis (MF), mielofibrosis idiopática crónica, leucemia mielógena crónica (LMC), leucemia neutrofílica crónica (LNC) y síndrome hipereosinofílica (SHE)).
El cáncer al que se refiere la presente invención puede ser un tumor sólido. En algunas realizaciones, el tumor sólido se selecciona de entre cáncer ovárico, cáncer de colon, fibrosarcoma, cáncer pancreático, cáncer pulmonar, cáncer de mama, linfoma, melanoma y glioblastoma. En algunas realizaciones, el tumor sólido es un tumor del SNC. En una realización, el tumor del SNC es el glioblastoma. Los datos de ADME proporcionados en la presente memoria indican que un compuesto proporcionado en la presente memoria (p.ej., el compuesto BB) puede mostrar una buena permeabilidad a través de la barrera hematocefálica y puede conseguir una concentración eficaz en un tumor del SNC.
En una realización, un inhibidor de PI3K-Y, tal como el compuesto BB, puede ser un inhibidor de P-gp (glucoproteína P). La glucoproteína P impide la entrada de diversos fármacos que se utilizan en el tratamiento, por ejemplo, de enfermedades del sistema nervioso central. Sin restringirse a ninguna teoría en particular, el sustrato P-gp puede ayudar a mantener los niveles normales de actividad de P-gp en un paciente bajo tratamiento con un inhibidor de PI3K-Y. En algunas realizaciones, un inhibidor de PI3K-Y, tal como un compuesto BB, puede no fluir hacia el exterior del tumor y, de esta manera, puede mantener una concentración eficaz del inhibidor de PI3K-Y en el tumor. Por ejemplo, la concentración puede mantenerse durante aproximadamente por lo menos 6 horas, por lo menos 10 horas, por lo menos 12 horas, por lo menos 24 horas o por lo menos 48 horas. En una realización, un inhibidor de PI3K-Y, tal como el compuesto BB, puede administrarse una vez al día.
En algunas realizaciones, se administra un inhibidor de PI3K-Y, tal como el compuesto BB, en el paciente en combinación con un segundo terapéutico que es un sustrato P-gp. En otra realización, un inhibidor de PI3K-Y, tal como el compuesto BB, puede inhibir el flujo de salida del segundo terapéutico, tal como un fármaco de cáncer que es un sustrato P-gp. Por lo tanto, un inhibidor de PI3K-Y proporcionado en la presente memoria, tal como un compuesto BB, puede resultar eficaz para mantener la concentración del fármaco de cáncer coadministrado en un tumor. Por ejemplo, la concentración puede mantenerse durante aproximadamente por lo menos 6 horas, por lo menos 10 horas, por lo menos 12 horas, por lo menos 24 horas o por lo menos 48 horas. En una realización, un inhibidor de PI3K-Y, tal como el compuesto BB, puede administrarse una vez al día.
La glucoproteína P es un componente de la barrera hematoencefálica y se encuentra presente sobre la superficie de las células endoteliales de la barrera. Un inhibidor de PI3K-Y proporcionado en la presente memoria, tal como el compuesto BB, puede ser un inhibidor de glucoproteína P y, de esta manera, puede cruzar la barrera hematocefálica. En algunas realizaciones, un inhibidor de PI3K-Y proporcionado en la presente memoria, tal como el compuesto BB, puede mantener una concentración eficaz en tumores del SNC o en tumores cerebrales (p.ej., el glioblastoma).
Tal como se utiliza en la presente memoria, la expresión «tumor sólido» se refiere a una masa anormal de tejido. Los tumores sólidos pueden ser benignos o malignos. Un tumor sólido crece en un sitio anatómico fuera del torrente sanguíneo (en contraste, por ejemplo, con los cánceres de origen hematopoyético, tales como las leucemias) requiere la formación de pequeños vasos sanguíneos y capilares para suministrar nutrientes, etc. a la masa tumoral en crecimiento. Los tumores sólidos se denominan según los tipos de células que los forman. Son ejemplos no limitativos de tumores sólidos, los sarcomas, los carcinomas (tumores epiteliales), los melanomas y los glioblastomas.
En algunas realizaciones, la exposición se refiere a la utilización en la inhibición del crecimiento de un tumor. La expresión «inhibición del crecimiento de un tumor» se refiere a retrasar el crecimiento tumoral y/o a reducir el tamaño tumoral. La expresión «inhibición del crecimiento de un tumor» incluye, de esta manera, eliminar células tumorales, así como retrasar o detener el crecimiento de las células tumorales.
Entre los tumores sólidos ejemplares se incluyen, aunque sin limitarse a ellos, cáncer biliar (p.ej., colangiocarcinoma), cáncer de vejiga, cáncer de mama (p.ej., adenocarcinoma de mama, carcinoma papilar de mama, cáncer mamario y carcinoma medular de mama), cáncer cerebral (p.ej., meningioma, glioma, p.ej., astrocitoma, oligodendroglioma y meduloblastoma), cáncer de cuello uterino (p.ej., adenocarcinoma cervical), cáncer colorrectal (p.ej., cáncer de colon, cáncer rectal y adenocarcinoma colorrectal), cáncer gástrico (p.ej., adenocarcinoma de estómago), tumor estromal gastrointestinal (TEGI), cáncer de cabeza y cuello (p.ej., carcinoma de células escamosas de cabeza y cuello, cáncer oral (p.ej., carcinoma de células escamosas oral (CCEO)), cáncer renal (p.ej., nefroblastoma, también conocido como tumor de Wilms, y carcinoma de células renales), cáncer hepático (p.ej., cáncer hepatocelular (CHC) y hepatoma
maligno), cáncer pulmonar (p.ej., carcinoma broncogénico, cáncer pulmonar microcítico (CPM), cáncer pulmonar no microcítico (CPNM) y adenocarcinoma pulmonar), neuroblastoma, neurofibroma (p.ej., neurofibromatosis (NF) de tipo 1 o de tipo 2, y schwanomatosis), cáncer neuroendocrino (p.ej., tumor neuroendocrino gastroenteropancreático (TNE-GEP), y tumor carcinoide), osteosarcoma, cáncer ovárico (p.ej., cistadenocarcinoma, carcinoma embrionario ovárico y adenocarcinoma ovárico), cáncer pancreático (p.ej., adenocarcinoma pancreático, neoplasia papilar mucinosa intraductal (NPMI)), cáncer de próstata (p.ej., adenocarcinoma de próstata), cáncer piel (p.ej., carcinoma de células escamosas (CCE), queratoacantoma (A), melanoma, carcinoma de células basales (CCB)) y sarcoma de tejidos blandos p.ej., histiocitoma fibroso maligno (HFM), liposarcoma, tumor de vaina nerviosa periférica maligna (TVNPM), condrosarcoma, fibrosarcoma, mixosarcoma y osteosarcoma).
El cáncer al que se refiere la presente invención puede ser un cáncer de origen hematopoyético y puede administrarse en el sujeto un compuesto selectivo de gamma. En determinadas realizaciones, el cáncer de origen hematopoyético es linfoma o leucemia. En algunas realizaciones, el cáncer de origen hematopoyético se selecciona de entre leucemia linfocítica aguda (LLA), que incluye LLA de linaje B y LLA de linaje T, leucemia linfocítica crónica (LLC), leucemia prolinfocítica (LPL), leucemia de células pilosas (LCP) y macroglobulinemia de Waldenstrom (MW), linfomas de células T periféricas (LCTP), leucemia/linfoma de células T adultas (LCTA), linfoma de células T cutáneas (LCTC), leucemia linfocítica granular de células grandes (LGL), leucemia mielocítica aguda (LMA), linfoma de Hodgkin (LH), linfoma no de Hodgkin (LNH), linfoma folicular, linfoma de células B grandes difusas (LCBGD), linfoma de células del manto (LCM), mastocitosis (p.ej., mastocitosis sistémica), mieloma múltiple (MM), síndrome mielodisplásico (SMD), trastorno mieloproliferativo (TMP) (p.ej., policitemia vera (PV), trombocitosis esencial (TE), metaplasia mieloide agnogénica (MMA), también conocida como mielofibrosis (MF), mielofibrosis idiopática crónica, leucemia mielógena crónica (LMC), leucemia neutrofílica crónica (LNC) y síndrome hipereosinofílica (s He )).
El cáncer al que se refiere la presente invención puede ser un tumor sólido y puede administrarse en el sujeto un compuesto selectivo de gamma. En algunas realizaciones, el tumor sólido se selecciona de entre cáncer ovárico, cáncer de colon, fibrosarcoma, cáncer pancreático, cáncer pulmonar, cáncer de mama, linfoma, melanoma y glioblastoma.
En algunas realizaciones, el compuesto selectivo de gamma presenta una proporción de selectividad delta/gamma >1 a <10, 10 a <50, o 50 a <350 que puede combinarse con un compuesto que presenta una selectividad gamma/delta superior a un factor de aproximadamente 1, superior a un factor de aproximadamente 2, superior a un factor de aproximadamente 3, superior a un factor de aproximadamente 5, superior a un factor de aproximadamente 10, superior a un factor de aproximadamente 50, superior a un factor de aproximadamente 100, superior a un factor de aproximadamente 200, superior a un factor de aproximadamente 400, superior a un factor de aproximadamente 600, superior a un factor de aproximadamente 800, superior a un factor de aproximadamente 1000, superior a un factor de aproximadamente 1500, superior a un factor de aproximadamente 2000, superior a un factor de aproximadamente 5000, superior a un factor de aproximadamente 10.000, o superior a un factor de aproximadamente 20,000.
Entre los pacientes que pueden tratarse con un compuesto proporcionado en la presente memoria, o una forma farmacéuticamente aceptable (p.ej., sales, hidratos, solvatos, isómeros y derivados marcados isotópicamente farmacéuticamente aceptables) del mismo, o una composición farmacéutica tal como se proporciona en la presente memoria, según los métodos proporcionados en la presente memoria se incluyen pacientes que se ha diagnosticado que presentan soriasis, restenosis, ateroesclerosis, ictus isquémico, BPH; cáncer de mama, tal como carcinoma ductal, carcinoma lobular, carcinomas medulares, carcinomas coloides, carcinomas tubulares y cáncer de mama inflamatorio; cáncer ovárico, incluyendo tumores ováricos epiteliales, tales como adenocarcinoma en el ovario y un adenocarcinoma que ha migrado del ovario al interior de la cavidad abdominal; cáncer uterino; cáncer cervical, tal como adenocarcinoma en el epitelio del cérvix, incluyendo carcinoma de células escamosas y adenocarcinomas; cáncer de próstata, tal como un cáncer de próstata seleccionado de entre los siguientes: un adenocarcinoma o un adenocarcinoma que ha migrado al hueso; cáncer pancreático, tal como carcinoma epitelioide en el tejido ductal pancreático y un adenocarcinoma en un conducto pancreático; cáncer de vejiga, tal como un carcinoma de células transicionales en la vejiga urinaria, carcinoma uroteliales (carcinoma de células transicionales), tumores en las células uroteliales que revisten la vejiga, carcinomas de células escamosas, adenocarcinomas y cánceres de células pequeñas; leucemia, tal como leucemia linfoblástica aguda, leucemia mielógena crónica, leucemia de células pilosas, mielodisplasia, trastornos mieloproliferativos, leucemia de células NK (p.ej., neoplasia de células dendríticas plasmacitoides blásticas), leucemia mielógena aguda (LMA), leucemia mielógena crónica (LMC), mastocitosis, leucemia linfocítica crónica (LLC), mieloma múltiple (MM) y síndrome mielodisplásico (SMD); cáncer óseo; cáncer pulmonar, tal como cáncer pulmonar no microcítico (CPNM), que se divide en carcinomas de células escamosas, adenocarcinomas y carcinomas no diferenciados de células grandes, y cáncer pulmonar microcítico; cáncer de piel, tal como carcinoma de células basales, melanoma, carcinoma de células escamosas y queratosis actínica, que es una condición de la piel que se desarrolla ocasionalmente en el carcinoma de células escamosas; retinoblastoma ocular; melanoma cutáneo o intraocular (oftálmico); cáncer hepático primario; cáncer renal; cáncer tiroideo, tal como papilar, folicular, medular y anaplásico; linfoma, tal como linfoma de células B grandes difusas, linfoma inmunoblástico de células B, linfoma de células NK (p.ej., neoplasia de células dendríticas plasmacitoides blásticas) y linfoma de Burkitt, sarcoma de Kaposi; cánceres inducidos por virus, incluyendo el virus de la hepatitis B (VHB), virus de la hepatitis C (VHC) y carcinoma hepatocelular; virus linfotrópico humano de tipo 1 (VLTH-1) y leucemia/linfoma de células T adultas, y virus del papiloma humano (VPH) y cáncer de cuello uterino; cánceres del sistema nervioso central (SNC), tales como el tumor
cerebral primario, que incluye gliomas (astrocitoma, astrocitoma anaplásico o el glioblastoma multiforme), oligodendroglioma, ependimoma, meningioma, linfoma, schwanoma y meduloblastoma; cánceres del sistema nervioso periférico (SNP), incluyendo neurofibromas y schwanomas, fibrocitoma maligno, histiocitoma fibroso maligno, meningioma maligno, mesotelioma maligno y tumor Mülleriano mixto maligno; cánceres de la cavidad oral y orofaríngeos, tales como cáncer hipofaríngeo, cáncer laríngeo, cáncer nasofaríngeo y cáncer orofaríngeo; cánceres de estómago, tales como linfomas, tumores estromales gástricos y tumores carcinoides; cánceres testiculares, tales como tumores de células germinales (TCG), que incluyen seminomas y no seminomas, y tumores estromales gonadales, que incluye tumores de las células de Leydig y tumores de las células de Sertoli; cáncer de timo, tal como timomas, carcinomas tímicos, linfoma de Hodgkin, linfomas no de Hodgkin, carcinoides y tumores carcinoides, cáncer rectal y cáncer de colon.
Entre los pacientes que pueden tratarse con compuestos proporcionados en la presente memoria, o sal, éster, solvato, hidrato o derivado farmacéuticamente aceptable de dichos compuestos, según los métodos proporcionados en la presente memoria se incluyen, por ejemplo, pacientes que se ha diagnosticado que presentan condiciones entre las que se incluyen, aunque sin limitación, neuroma acústico, adenocarcinoma, cáncer de la glándula adrenal, cáncer anal, angiosarcoma (p.ej., linfangiosarcoma), linfangioendoteliosarcoma, hemangiosarcoma), gamopatía monoclonal benigna, cáncer biliar (p.ej., colangiocarcinoma), cáncer de vejiga, cáncer de mama (p.ej., adenocarcinoma de mama, carcinoma papilar de la mama, cáncer mamario y carcinoma medular de mama), cáncer cerebral (p.ej., meningioma, glioma, p.ej., astrocitoma, oligodendroglioma y meduloblastoma), cáncer bronquial, cáncer de cuello uterino (p.ej., adenocarcinoma cervical), coriocarcinoma, cordoma, craniofaringioma, cáncer colorrectal (p.ej., cáncer de colon, cáncer rectal y adenocarcinoma colorrectal), carcinoma epitelial, ependimoma, endoteliosarcoma (p.ej., sarcoma de Kaposi y sarcoma hemorrágico idiopático múltiple), cáncer endometrial, cáncer esofágico (p.ej., adenocarcinoma del esófago y adenocarcinoma de Barrett), sarcoma de Ewing, hipereosinofilia familiar, cáncer gástrico (p.ej., adenocarcinoma de estómago), tumor estromal gastrointestinal (TEG i), cáncer de cabeza y cuello (p.ej., carcinoma de células escamosas de cabeza y cuello, cáncer oral (p.ej., carcinoma oral de células escamosas (COCE)), enfermedad de cadena pesada (p.ej., enfermedad de cadena alfa, enfermedad de cadena gamma y enfermedad de cadena mu), hemangioblastoma, tumores miofibroblásticos inflamatorios, amiloidosis inmunocítica, cáncer renal (p.ej., nefroblastoma, también conocido como tumor de Wilms, carcinoma de células renales), cáncer hepático (p.ej., cáncer hepatocelular (HCC) y hepatoma maligno), cáncer pulmonar (p.ej., carcinoma broncogénico y cáncer pulmonar microcítico (CPM), cáncer pulmonar no microcítico (CPNM) y adenocarcinoma pulmonar), leucemia (p.ej., leucemia linfoblástica aguda (LLA), que incluye LLA de linaje B y LLA de linaje T, leucemia linfocítica crónica (LlC), leucemia prolinfocítica (LLP), leucemia de células pilosas (LCP) y macroglobulinemia de Waldenstrom (MW), linfomas de células T periféricas (LCTP), leucemia/linfoma de células T adultas (LCTA), linfoma de células T cutáneo (LCTC), leucemia de linfocitos granulares grandes (LLGG), leucemia mielocítica aguda (LMA), leucemia mielocítica crónica (LMC), leucemia linfocítica crónica (LLC)), linfoma (p.ej., linfoma de Hodgkin (LH), linfoma no de Hodgkin (LNH), linfoma folicular, linfoma de células B grandes difusas (LCBGD), linfoma de células del manto (LCM)), leiomiosarcoma (LMS), mastocitos (p.ej., mastocitosis sistémica), mieloma multiple (MM), síndrome mielodisplásico (SMD), mesotelioma, trastorno mieloproliferativo (TMP) (p.ej., policitemia vera (PV), trombocitosis esencial (TE), metaplasia mieloide agnogénica (m Ma ), también conocido como mielofibrosis (MF), mielofibrosis idiopática crónica, leucemia mielógena crónica (LMC), leucemia neutrofílica crónica (LNC), síndrome hipereosinofílico (SHE)), neuroblastoma, neurofibroma (p.ej., neurofibromatosis (NF) de tipo 1 o tipo 2, schwannomatosis), cáncer neuroendocrino (p.ej., tumor neuroendocrino gastroenteropancreático (TNE-GEP), oSTEOSArcoma, cáncer ovárico (p.ej., cistadenocarcinoma, carcinoma embrionario ovárico y adenocarcinoma ovárico), enfermedad de Paget de vulva, enfermedad de Paget del pene, adenocarcinoma papilar, cáncer pancreático (p.ej., adenocarcinoma pancreático, neoplasia mucinosa papilar intraductal (NMPI)), pinealoma, tumor neuroectodérmico primitivo (TNP), cáncer de próstata (p.ej., adenocarcinoma de próstata), rabdomiosarcoma, retinoblastoma, cáncer de glándulas salivales, cáncer de piel (p.ej., carcinoma de células escamosas (CCE), queratoacantoma (KA), melanoma, carcinoma de células basales (CCB)), cáncer de intestino delgado (p.ej., cáncer de apéndice), sarcoma de tejidos blandos (p.ej., histiocitoma fibroso maligno (HFM), liposarcoma, tumor de vaina nerviosa periférica maligno (TVNPM), condrosarcoma, fibrosarcoma y mixosarcoma), carcinoma de glándulas sebáceas, carcinoma de glándulas sudoríparas, sinovioma, cáncer testicular (p.ej., seminoma y carcinoma embrionario testicular), cáncer tiroideo (p.ej., carcinoma papilar del tiroides, carcinoma tiroideo papilar (CTP) y cáncer tiroideo medular) y macroglobulinemia de Waldenstrom.
Sin restringirse a ninguna teoría en particular, en una realización, el cáncer bajo tratamiento o prevención, tal como un trastorno sanguíneo o neoplasia maligna hemática, presenta un nivel de expresión elevado de una o más isoformas de PI3K (p.ej., PI3K-a, PI3K-p, PI3K-8, o PI3K-Y, o una combinación de las mismas). En una realización el cáncer que puede tratarse o prevenirse incluye un trastorno sanguíneo o una neoplasia maligna hemática, incluyendo, aunque sin limitación, un trastorno mieloide, trastorno linfoide, leucemia, linfoma, síndrome mielodisplásico (SMD), enfermedad mieloproliferativa (EMP), trastorno de mastocitos y mieloma (p.ej., mieloma múltiple), entre otros. En una realización, el trastorno sanguíneo o la neoplasia maligna hemática incluye, aunque sin limitación, leucemia linfoblástica aguda (LLA), LLA de células T (LLA-T), LLA de células B (LLA-B), leucemia mieloide aguda (LMA), leucemia linfocítica crónica (LLC), leucemia mielógena crónica (LMC), LMC de fase blástica, linfoma linfocítico pequeño (LLP), LLC/LLS, LLC transformado, síndrome de Richter, linfoma de Hodgkin (LH), linfoma no de Hodgkin (LNH), LNH de células B, LNH de células T, LNH indolente (LNHi), linfoma de células B grandes difusas (LCBGD), linfoma de células del manto (LCM), LNH de células B agresivo, linfoma de células B (LCB), síndrome de Richter (SR), linfoma de células T (LCT), linfoma de células T periféricas (LCTP), linfoma de células T cutáneo (LCTC), micosis fungoide transformado,
síndrome de Sézary, linterna de células grandes anaplásico (LCGA), linterna folicular (LF), macroglobulinemia de Waldenstrom (MW), linterna linfoplasmacítico , linterna de Burkitt, mieloma múltiple (MM), amiloidosis, TMP, trombocitosis esencial (TE), mielofibrosis (MF), policitemia vera (PV), leucemia mielomonocítica crónica (LMMC), síndrome mielodisplásico (SMD), linfoma angioinmunoblástico, SMD de riesgo alto y SMD de riesgo bajo. En una realización, la neoplasia maligna hemática es recidivante. En una realización, la neoplasia maligna es refractaria. En una realización, el cáncer o enfermedad ocurre en un paciente pediátrico (incluyendo un paciente infantil). En una realización, el cáncer o enfermedad ocurre en un paciente adulto. Se describen en otro sitio de la presente invención realizaciones adicionales de un cáncer o enfermedad bajo tratamiento o prevención mediante métodos, composiciones o kits proporcionados en la presente memoria.
En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LLC. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LLC/LLS. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LLC transformada o síndrome de Richter. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LLS. En una realización, sin limitarse a una teoría en particular, un compuesto proporcionado en la presente memoria (p.ej., un compuesto selectivo de PI3K-Y proporcionado en la presente memoria) inhibe la migración y/o la activación de las células T y las células mieloides (p.ej., macrófagos o macrófagos M2 polarizados), reduciendo la supervivencia y/o soporte proliferativo proporcionado por dichas células a las células de l Lc malignas en el microambiente tumoral (MAT). En una realización, aunque sin limitarse a una teoría en particular, la migración de las células T CD3+ a la quimioquina CXCL12 asociada a LLC resulta bloqueada por un compuesto proporcionado en la presente memoria (p.ej., un compuesto selectivo de PI3K-Y proporcionado en la presente memoria). En otra realización, aunque sin limitarse a una teoría en particular, un compuesto proporcionado en la presente memoria (p.ej., un compuesto selectivo de PI3K-Y proporcionado en la presente memoria) bloquea el nuevo crecimiento mediado por células mieloides de un cáncer tras la quimioterapia a través de sus efectos sobre la inhibición de la migración postquimioterapia de las células mieloides hacia el interior de un tumor.
En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LNHi. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LCBGD. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LNH de células B (p.ej., LNH de células B agresivo). En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LCM. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es SR. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LMA. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es MM. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LLA. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LLA-T. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LLA-B. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LCT. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LCGA. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es leucemia. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es linfoma. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es linfoma de células T. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es SMD (p.ej., SMD de grado bajo). En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es MPD. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es un trastorno de mastocitos. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es linfoma de Hodgkin (LH). En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es linfoma no de Hodgkin. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es PTCL. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LCTC (p.ej., micosis fungoide o síndrome de Sézary). En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es W m . En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es CML. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LF. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es micosis fungoide transformada. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es síndrome de Sézary. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es leucemia de células T aguda. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es leucemia de células B aguda. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es linfoma de Burkitt. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es neoplasia mieloproliferativa. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es linfoma esplénico de la zona marginal. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es linfoma nodal de la zona marginal. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es linfoma extranodal de la zona marginal.
En una realización, el cáncer o neoplasia maligna hemática es un linfoma de células B. En una realización, el linfoma de células B es LNHi. En otra realización, el linfoma de células B es linfoma folicular. En otra realización, el linfoma de células B es macroglobulinemia de Waldenstrom (linfoma linfoplasmacítico). En otra realización, el linfoma de células B es linfoma de la zona marginal (LZM). En otra realización, el linfoma de células B es LCM. En otra realización, el linfoma de células B es LH. En otra realización, el linfoma de células B es LNHa. En otra realización, el linfoma de células B es LCBGD. En otra realización, el linfoma de células B es linfoma de Richters.
En una realización, el cáncer o neoplasia maligna hemática es un linfoma de células T. En una realización, el linfoma de células T es linfoma de células T periféricas (LCTP). En otra realización, el linfoma de células T es linfoma de células T cutáneo (LCTC).
En una realización, el cáncer o neoplasia maligna hemática es síndrome de Sézary. Entre los síntomas asociados al síndrome de Sézary se incluyen, aunque sin limitarse a ellos, epidermotropismo por linfocitos CD4+ neoplásicos,
microabscesos de Pautrier, eritroderma, linfadenopatía, células T atípicas en la sangre periférica y hepatoesplenomegalia. En una realización, la cantidad terapéuticamente eficaz para el tratamiento o control del síndrome de Sézary es de entre aproximadamente 25 mg y 75 mg, administrados dos veces al día. En otras realizaciones, la cantidad terapéuticamente eficaz es de entre aproximadamente 50 mg y aproximadamente 75 mg, de entre aproximadamente 30 mg y aproximadamente 65 mg, de entre aproximadamente 45 mg y aproximadamente 60 mg, de entre aproximadamente 30 mg y aproximadamente 50 mg, o de entre aproximadamente 55 mg y aproximadamente 65 mg, cada uno de los cuales se administra dos veces al día. En una realización, la cantidad eficaz es de aproximadamente 60 mg, administrados dos veces al día.
En una realización, el cáncer o neoplasia maligna hemática es recidivante. En una realización, el cáncer o neoplasia maligna hemática es refractario. En determinadas realizaciones, el cáncer bajo tratamiento o prevención es un subtipo específico de cáncer indicado en la presente memoria. En determinadas realizaciones, la neoplasia maligna hemática bajo tratamiento o prevención es un subtipo específico de neoplasia maligna hemática indicado en la presente memoria. Determinadas clasificaciones de tipo o subtipo de cáncer o neoplasia maligna hemática proporcionado en la presente memoria son conocidas de la técnica. Sin restringirse a una teoría en particular, se cree que muchos de los cánceres que se vuelven recidivantes o refractarios, desarrollan resistencia a la terapia previa particular administrada para tratar los cánceres. De esta manera, sin limitarse a una teoría en particular, un compuesto proporcionado en la presente memoria puede proporcionar una terapia de segunda línea mediante la provisión de un mecanismo alternativo para tratar cánceres diferentes de los mecanismos utilizados por determinadas técnicas anteriores.
En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LNHi. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LLC refractario. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LLS refractario. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es refractario a la terapia de rituximab. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es refractario a la quimioterapia. En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es refractario a la radioinmunoterapia (RIT). En realizaciones ejemplares, el cáncer o neoplasia maligna hemática es LNHi, LF, linfoma esplénico de la zona marginal, linfoma nodal de la zona marginal, linfoma extranodal de la zona marginal o LLS; el cáncer o neoplasia maligna hemática es refractario a la terapia de rituximab, quimioterapia y/o RIT.
En otra realización ejemplar, el cáncer o neoplasia maligna hemática es linfoma, y el cáncer es recidivante o refractario al tratamiento con un inhibidor de BTK, tal como, aunque sin limitación, ibrutinib o ONO-4059. En otra realización ejemplar, el cáncer o neoplasia maligna hemática es LLC, y el cáncer es recidivante o refractario al tratamiento con un inhibidor de BTK, tal como, aunque sin limitación, ibrutinib o AVL-292.
Un tumor sólido puede tratarse o prevenirse mediante la administración en el sujeto de una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., un inhibidor selectivo de PI3K-Y, p.ej., el Compuesto 4) o una forma farmacéuticamente aceptable del mismo.
En una realización, el tumor sólido se selecciona de uno o más de los siguientes: cáncer del sistema pulmonar, un cáncer cerebral, un cáncer del tracto gastrointestinal, un cáncer de piel, un cáncer genitourinario, un cáncer pancreático, un cáncer pulmonar, un meduloblastoma, un carcinoma de células basales, un glioma, un cáncer de mama, un cáncer de próstata, un cáncer testicular, un cáncer esofágico, un cáncer hepatocelular, un cáncer gástrico, un tumor estromal gastrointestinal (TEGI), un cáncer de colon, un cáncer colorrectal, un cáncer ovárico, un melanoma, un tumor neuroectodérmico, cáncer de cabeza y cuello, un sarcoma, un sarcoma de tejidos blandos, fibrosarcoma, mixosarcoma, liposarcoma, un condrosarcoma, un sarcoma osteogénico, un cordoma, un angiosarcoma, un endoteliosarcoma, un linfangiosarcoma, un linfangioendoteliosarcoma, un sinovioma, un mesotelioma, un leiomiosarcoma, un cáncer de cuello uterino, un cáncer uterino, un cáncer endometrial, un carcinoma, un carcinoma de vejiga, un carcinoma epitelial, un carcinoma de células escamosas, un adenocarcinoma, un carcinoma broncogénico, un carcinoma de células renales, un hepatoma, un carcinoma del conducto biliar, un cáncer neuroendocrino, un tumor carcinoide, tumor de células gigantes de tipo difuso y glioblastoma.
En una realización, el compuesto, o una forma farmacéuticamente aceptable del mismo, se administra a una dosis suficiente para causar una reducción del crecimiento tumoral de por lo menos 10 %, 20 %, 30 %, 40 % o 50 % en comparación con un valor de referencia, después de la administración del compuesto.
La presente invención se refiere a compuestos para la utilización en combinación con una terapia de punto de control inmunitario. En una realización, la terapia de punto de control inmunitario es un inhibidor de PDL-1 o un anticuerpo anti-PDL-1.
En una realización, también puede administrarse en el sujeto un inhibidor de PI3K-delta.
En una realización, el compuesto, o una forma farmacéuticamente aceptable del mismo, es para la administración a una dosis con la que el nivel del compuesto en el sujeto es superior a la IC50 del compuesto de inhibición de PI3K-gamma durante por lo menos 70 %, 80 %, 90 %, 95 %, 97 %, 98 % o 99 % de un periodo de tiempo seleccionado, p.ej., 6 horas, l2 horas, 24 horas o 48 horas inmediatamente después de la administración. En una realización, el
compuesto, o una forma farmacéuticamente aceptable del mismo, es para la administración a una dosis con la que el nivel del compuesto en el sujeto es inferior a la IC50 del compuesto de inhibición de PI3K-delta durante por lo menos 70 %, 80 %, 90 %, 95 %, 97 %, 98 % o 99 % de un periodo de tiempo seleccionado, p.ej., 6 horas, 12 horas, 24 horas o 48 horas inmediatamente después de la administración. En una realización, el nivel del compuesto se mide a partir del plasma del sujeto. En una realización, el nivel del compuesto se mide a partir de un tejido del sujeto.
En una realización, el sujeto ha sido tratado anteriormente con ciclofosfamida, docetaxel, paclitaxel, 5-FU o temozolomida.
En una realización, el efecto antitumoral del compuesto se mantiene durante un periodo de tiempo después de la interrupción del tratamiento con el compuesto. En una realización, el periodo de tiempo es de por lo menos 1 día, 2 días, 3 días, 4 días, 5 días o 6 días.
Enfermedades pulmonares o respiratorias
La enfermedad pulmonar o respiratoria puede tratarse, prevenirse y/o controlarse utilizando un compuesto proporcionado en la presente memoria. Entre los ejemplos de enfermedad pulmonar o respiratoria se incluyen, aunque sin limitarse a ellos, mesotelioma, sarcoma, cáncer pulmonar, carcinoma pulmonar microcítico, carcinoma pulmonar no microcítico, tumores epiteliales, papilomas, adenomas, carcinoma de células escamosas, carcinoma microcítico, adenocarcinoma, carcinoma macrocítico, carcinoma adenoescamoso, tumor carcinoide, carcinoma de tipo glándula salival, tumores de tejidos blandos, tumor fibroso localizado, hemangioendotelioma epitelioide, blastoma pleuropulmonar, condroma, pseudotumor fibroso calcificante de la pleura, tumor miofibroblástico peribronquial congénito, linfangiomatosis pulmonar difuso, tumor de células redondas pequeñas desmoplásico, tumores mesoteliales, tumor adenomatoide, mesotelioma epitelioide, mesotelioma sarcomatoide, mesotelioma bifásico, hamartoma, hemangioma esclerosante, tumor de células claras, neoplasmas de células germinales, timoma, melanoma y tumor secundario. En determinadas realizaciones, en la presente memoria se proporciona un método de tratamiento, prevención y/o control de una enfermedad linfoproliferativa utilizando un compuesto proporcionado en la presente memoria. Entre los ejemplos de enfermedad linfoproliferativa se incluyen, aunque sin limitarse a ellos, granulomatosis linfomatoide.
En determinadas realizaciones, en el tratamiento, prevención y/o control de una enfermedad pulmonar o respiratoria indicada en la presente memoria, una cantidad terapéutica o profilácticamente eficaz de un compuesto proporcionado en la presente memoria para la utilización según la invención (p.ej., el Compuesto 4) o un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, de una forma farmacéuticamente aceptable del mismo, es de entre aproximadamente 0,005 y aproximadamente 1.000 mg al día, de entre aproximadamente 0,01 y aproximadamente 500 mg al día, de entre aproximadamente 0,01 y aproximadamente 250 mg al día, de entre aproximadamente 0,01 y aproximadamente 100 mg al día, de entre aproximadamente 0,1 y aproximadamente 100 mg al día, de entre aproximadamente 0.5 y aproximadamente 100 mg al día, de entre aproximadamente 1 y aproximadamente 100 mg al día, de entre aproximadamente 0.01 y aproximadamente 50 mg al día, de entre aproximadamente 0,1 y aproximadamente 50 mg al día, de entre aproximadamente 0.5 y aproximadamente 50 mg al día, de entre aproximadamente 1 y aproximadamente 50 mg al día, de entre aproximadamente 2 y aproximadamente 25 mg al día, o de entre aproximadamente 5 y aproximadamente 10 mg al día.
En determinadas realizaciones, la cantidad terapéutica o profilácticamente eficaz es de aproximadamente 0,1, aproximadamente 0,2, aproximadamente 0,5, aproximadamente 1, aproximadamente 2, aproximadamente 5, aproximadamente 10, aproximadamente 15, aproximadamente 20, aproximadamente 25, aproximadamente 30, aproximadamente 40, aproximadamente 45, aproximadamente 50, aproximadamente 60, aproximadamente 70, aproximadamente 80, aproximadamente 90, aproximadamente 100, o aproximadamente 150 mg al día.
En una realización, el intervalo de dosis diarias recomendado de un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4) o un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo, para las condiciones indicadas en la presente memoria se encuentra comprendido en el intervalo de entre aproximadamente 0,5 mg y aproximadamente 50 mg al día, en una única dosis de una vez al día o en dosis divididas durante todo un día. En algunas realizaciones, las dosis están comprendidas entre aproximadamente 1 mg y aproximadamente 50 mg al día. En algunas realizaciones, las dosis están comprendidas entre aproximadamente 0,5 mg y aproximadamente 5 mg al día. Entre las dosis específicas al día se incluyen 0,1, 0,2, 0,5, 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41,42, 43, 44, 45, 46, 47, 48, 49 o 50 mg al día.
En una realización, un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), o un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo, se administra a una dosis inferior a 0,1, de aproximadamente 0,1, inferior a 0,5, de aproximadamente 0,5, de entre aproximadamente 0,1 y aproximadamente 1,0, de entre aproximadamente 0,5 y aproximadamente 1,0, de aproximadamente 1 o de aproximadamente 2 mg al día.
En otra realización, un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), o un enantiómero,
una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo, se administra a una dosis inferior a 0,2, de aproximadamente 0,2, inferior a 1,0, de aproximadamente 1,0, de entre aproximadamente 0,2 y aproximadamente 2,0, de entre aproximadamente 1,0 y aproximadamente 2,0, de aproximadamente 2 o de aproximadamente 4 mg al día.
En una realización, la dosis es inferior a 0,1 mg al día.
En otra realización, la dosis es de aproximadamente 0,1 mg al día.
En otra realización, la dosis es inferior a 0,5 mg al día.
En otra realización, la dosis es de aproximadamente 0,5 mg al día.
En otra realización, la dosis es de entre aproximadamente 0,1 y aproximadamente 1,0 mg al día.
En otra realización, la dosis es de entre aproximadamente 0,5 y aproximadamente 1,0 mg al día.
En otra realización, la dosis es de aproximadamente 1 mg al día.
En otra realización, la dosis es de aproximadamente 2 mg al día.
En otra realización, la dosis es inferior a 0,2 mg al día.
En otra realización, la dosis es de aproximadamente 0,2 mg al día.
En otra realización, la dosis es inferior a 1,0 mg al día.
En otra realización, la dosis es de aproximadamente 1,0 mg al día.
En otra realización, la dosis es de entre aproximadamente 0,2 y aproximadamente 2,0 mg al día.
En otra realización, la dosis es de entre aproximadamente 1,0 y aproximadamente 2,0 mg al día.
En otra realización, la dosis es de aproximadamente 2 mg al día.
En otra realización, la dosis es de aproximadamente 4 mg al día.
En una realización específica, la dosis inicial recomendada puede ser de 0,5, 1,2, 3, 4, 5, 10, 15, 20, 25 o 50 mg al día. En otra realización, la dosis inicial recomendada puede ser de 0,5, 1, 2, 3, 4 o 5 mg al día. La dosis puede escalarse a 15, 20, 25, 30, 35, 40, 45 y 50 mg/día.
En determinadas realizaciones, la cantidad terapéutica o profilácticamente eficaz es de entre aproximadamente 0,001 y aproximadamente 100 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 50 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 25 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 10 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 9 mg/kg/día, 0,01 y aproximadamente 8 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 7 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 6 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 5 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 4 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 3 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 2 mg/kg/día, o de entre aproximadamente 0,01 y aproximadamente 1 mg/kg/día.
En determinadas realizaciones, la cantidad terapéutica o profilácticamente eficaz es de entre aproximadamente 0,01 y aproximadamente 25 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 20 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 15 mg/kg/día, de entre aproximadamente 0,01 y aproximadamente 10 mg/kg/día, de entre aproximadamente 0,05 y aproximadamente 25 mg/kg/día, de entre aproximadamente 0,05 y aproximadamente 20 mg/kg/día, de entre aproximadamente 0,05 y aproximadamente 15 mg/kg/día, o de entre aproximadamente 0,05 y aproximadamente 10 mg/kg/día.
La dosis administrada también puede expresarse en unidades diferentes de mg/kg/día. Por ejemplo, las dosis para la administración parenteral pueden expresarse en mg/m2/día. El experto ordinario en la materia conocerá fácilmente cómo convertir las dosis en mg/kg/día en mg/m2/día, dada la altura o peso del sujeto, o ambos (ver www.fda.gov/cder/cancer/animalframe.htm). Por ejemplo, una dosis de 1 mg/kg/día para un ser humano de 65 kg es aproximadamente igual a 38 mg/m2/día.
Un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), o un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del
mismo, puede administrarse una vez al día (QD), o en dosis divididas en múltiples dosis diarias, tal como dos veces al día (BID), tres veces al día (TID) y cuatro veces al día (QID). Además, la administración puede ser continua (es decir, diariamente durante días consecutivos o cada día), intermitente, p.ej., en ciclos (es decir, incluyendo días, semanas o meses de reposo sin fármaco). Tal como se utiliza en la presente memoria, el término "diario" pretende significar que un compuesto terapéutico, tal como el Compuesto 4, se administra una vez o más de una vez al día, por ejemplo, durante un periodo de tiempo. El término "continuo" pretende significar que un compuesto terapéutico, tal como el Compuesto 4, se administra diariamente durante un periodo sin interrupciones de por lo menos 10 días a 52 semanas. El término "intermitente" o "intermitentemente" tal como se utiliza en la presente memoria pretende significar detener e iniciar a intervalos regulares o irregulares. Por ejemplo, la administración intermitente de Compuesto 4 es la administración durante uno a seis días a la semana, la administración en ciclos (p.ej., la administración diaria durante dos a ocho semanas consecutivas, seguido de un periodo de reposo sin administración durante hasta una semana) o la administración en días alternos. El término "ciclado" tal como se utiliza en la presente memoria pretende significar que un compuesto terapéutico, tal como el Compuesto 4, se administra diariamente o continuamente, aunque con un periodo de reposo.
En algunas realizaciones, la frecuencia de administración se encuentra comprendida en el intervalo de entre aproximadamente una dosis diaria y aproximadamente una dosis mensual. En determinadas realizaciones, la administración es una vez al día, dos veces al día, tres veces al día, cuatro veces al día, una vez cada dos días, dos veces a la semana, una vez a la semana, una vez cada dos semanas, una vez cada tres semanas o una vez cada cuatro semanas. En una realización, el compuesto proporcionado en la presente memoria es para la administración una vez al día. En otra realización, el compuesto proporcionado en la presente memoria es para la administración dos veces al día. En todavía otra realización, el compuesto proporcionado en la presente memoria es para la administración tres veces al día. En todavía otra realización, el compuesto proporcionado en la presente memoria es para la administración cuatro veces al día.
En una realización, un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), que puede ser un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros, o una forma farmacéuticamente aceptable del mismo, es para la administración dos veces al día (BID). En una realización, la dosis es de aproximadamente 0,1, 0,2, 0,25, 0,5, 1,2, 2,5, 5, 10, 15, 20, 25 o 50 mg BID.
En una realización, un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), que puede ser un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo, se administra a una dosis inferior a 0,1, de aproximadamente 0,1, inferior a 0,5, de aproximadamente 0,5, de entre aproximadamente 0,1 y aproximadamente 1,0, de entre aproximadamente 0,5 y aproximadamente 1,0, de aproximadamente 1 o de aproximadamente 2 mg BID.
En otra realización, un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), que puede ser un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo, se administra a una dosis inferior a 0,2, de aproximadamente 0,2, inferior a 1,0, de aproximadamente 1,0, de entre aproximadamente 0,2 y aproximadamente 2,0, de entre aproximadamente 1,0 y aproximadamente 2,0, de aproximadamente 2 o de aproximadamente 4 mg BID.
En una realización, la dosis es inferior a 0,1 mg BID.
En otra realización, la dosis es de aproximadamente 0,1 mg BID.
En otra realización, la dosis es inferior a 0,5 mg BID.
En otra realización, la dosis es de aproximadamente 0,5 mg BID.
En otra realización, la dosis es de entre aproximadamente 0,1 y aproximadamente 1,0 mg BID.
En otra realización, la dosis es de entre aproximadamente 0,5 y aproximadamente 1,0 mg BID.
En otra realización, la dosis es de aproximadamente 1 mg BID.
En otra realización, la dosis es de aproximadamente 2 mg BID.
En otra realización, la dosis es inferior a 0,2 mg BID.
En otra realización, la dosis es de aproximadamente 0,2 mg BID.
En otra realización, la dosis es inferior a 1,0 mg BID.
En otra realización, la dosis es de aproximadamente 1,0 mg BID.
En otra realización, la dosis es de entre aproximadamente 0,2 y aproximadamente 2,0 mg BID.
En otra realización, la dosis es de entre aproximadamente 1,0 y aproximadamente 2,0 mg BID.
En otra realización, la dosis es de aproximadamente 2 mg BID.
En otra realización, la dosis es de aproximadamente 4 mg BID.
En una realización, un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), que puede ser un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo, es para la administración una vez al día (QD). En una realización, la dosis es de aproximadamente 0,1, 0,2, 0,25, 0,5, 1, 2, 2,5, 5, 10, 15, 20, 25 o 50 mg QD.
En una realización, un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), que puede ser un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo, se administra a una dosis inferior a 0,1, de aproximadamente 0,1, inferior a 0,5, de aproximadamente 0,5, de entre aproximadamente 0,1 y aproximadamente 1,0, de entre aproximadamente 0,5 y aproximadamente 1,0, de aproximadamente 1 o de aproximadamente 2 mg QD.
En otra realización, un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), que puede ser un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo, se administra a una dosis inferior a 0,2, de aproximadamente 0,2, inferior a 1,0, de aproximadamente 1,0, de entre aproximadamente 0,2 y aproximadamente 2,0, de entre aproximadamente 1,0 y aproximadamente 2,0, de aproximadamente 2 o de aproximadamente 4 mg QD.
En una realización, la dosis es inferior a 0,1 mg QD.
En otra realización, la dosis es de aproximadamente 0,1 mg QD.
En otra realización, la dosis es inferior a 0,5 mg QD.
En otra realización, la dosis es de aproximadamente 0,5 mg QD.
En otra realización, la dosis es de entre aproximadamente 0,1 y aproximadamente 1,0 mg QD.
En otra realización, la dosis es de entre aproximadamente 0,5 y aproximadamente 1,0 mg QD.
En otra realización, la dosis es de aproximadamente 1 mg QD.
En otra realización, la dosis es de aproximadamente 2 mg QD.
En otra realización, la dosis es inferior a 0,2 mg QD.
En otra realización, la dosis es de aproximadamente 0,2 mg QD.
En otra realización, la dosis es inferior a 1,0 mg QD.
En otra realización, la dosis es de aproximadamente 1,0 mg QD.
En otra realización, la dosis es de entre aproximadamente 0,2 y aproximadamente 2,0 mg QD.
En otra realización, la dosis es de entre aproximadamente 1,0 y aproximadamente 2,0 mg QD.
En otra realización, la dosis es de aproximadamente 2 mg QD.
En otra realización, la dosis es de aproximadamente 4 mg QD.
En una realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar del compuesto en estado estacionario, comprendida entre aproximadamente 0,005 y aproximadamente 100 |jM, de entre aproximadamente 0,005 y aproximadamente 10 jM , de entre aproximadamente 0,01 y aproximadamente 10 jM , de entre aproximadamente 0,01 y aproximadamente 5 jM , de entre aproximadamente 0,005 y aproximadamente 1 jM , de entre aproximadamente 0,005 y aproximadamente 0,5 jM , de entre aproximadamente 0,005 y aproximadamente 0,5 jM , de entre aproximadamente 0,01 y aproximadamente 0,2 jM , o de entre aproximadamente 0,01 y aproximadamente 0,1 jM . En una realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar en estado estacionario de entre aproximadamente 0,005 y
aproximadamente 100 pM. En otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar en estado estacionario de entre aproximadamente 0,005 y aproximadamente 10 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar en estado estacionario de entre aproximadamente 0,01 y aproximadamente 10 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar en estado estacionario de entre aproximadamente 0,01 y aproximadamente 5 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar en estado estacionario de entre aproximadamente 0,005 y aproximadamente 1 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar en estado estacionario de entre aproximadamente 0,005 y aproximadamente 0,5 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar del compuesto en estado estacionario de entre aproximadamente 0,01 y aproximadamente 0,2 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar del compuesto en estado estacionario de entre aproximadamente 0,01 y aproximadamente 0,1 pM. Tal como se utiliza en la presente memoria, la expresión "concentración pulmonar en estado estacionario" es la concentración alcanzada tras un periodo de administración de un compuesto. Una vez se ha alcanzado el estado estacionario, se producen picos y valles menores en la curva dependiente del tiempo de concentración pulmonar del compuesto.
En una realización, la cantidad administrada resulta suficiente para proporcionar una concentración pulmonar máxima (concentración pico) del compuesto, comprendida entre aproximadamente 0,005 y aproximadamente 100 pM, de entre aproximadamente 0,005 y aproximadamente 10 pM, de entre aproximadamente 0,01 y aproximadamente 10 pM, de entre aproximadamente 0,01 y aproximadamente 5 pM, de entre aproximadamente 0,005 y aproximadamente 1 pM, de entre aproximadamente 0,005 y aproximadamente 0,5 pM, de entre aproximadamente 0,01 y aproximadamente 0.2 pM, o de entre aproximadamente 0,01 y aproximadamente 0,1 pM. En una realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar máxima del compuesto de entre aproximadamente 0,005 y aproximadamente 100 pM. En otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar máxima del compuesto de entre aproximadamente 0,005 y aproximadamente 10 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar máxima del compuesto de entre aproximadamente 0,01 y aproximadamente 10 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar máxima del compuesto de entre aproximadamente 0,01 y aproximadamente 5 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar máxima del compuesto de entre aproximadamente 0,005 y aproximadamente 1 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar máxima del compuesto de entre aproximadamente 0,005 y aproximadamente 0,5 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar máxima del compuesto de entre aproximadamente 0,01 y aproximadamente 0,2 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar máxima del compuesto de entre aproximadamente 0,01 y aproximadamente 0,1 pM.
En una realización, la cantidad administrada resulta suficiente para proporcionar una concentración pulmonar mínima (concentración de valle) del compuesto, comprendida entre aproximadamente 0,005 y aproximadamente 100 pM, de entre aproximadamente 0,005 y aproximadamente 10 pM, de entre aproximadamente 0,01 y aproximadamente 10 pM, de entre aproximadamente 0,01 y aproximadamente 5 pM, de entre aproximadamente 0,005 y aproximadamente 1 pM, de entre aproximadamente 0,005 y aproximadamente 0,5 pM, de entre aproximadamente 0,01 y aproximadamente 0.2 pM, o de entre aproximadamente 0,01 y aproximadamente 0,1 pM, en el caso de que se administre más de una dosis. En una realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar mínima del compuesto de entre aproximadamente 0,005 y aproximadamente 100 pM. En otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar mínima del compuesto de entre aproximadamente 0,005 y aproximadamente 10 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar mínima del compuesto de entre aproximadamente 0,01 y aproximadamente 10 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar mínima del compuesto de entre aproximadamente 0,01 y aproximadamente 5 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar mínima del compuesto de entre aproximadamente 0,005 y aproximadamente 1 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar mínima del compuesto de entre aproximadamente 0,005 y aproximadamente 0,5 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar mínima del compuesto de entre aproximadamente 0,01 y aproximadamente 0,2 pM. En todavía otra realización, la cantidad del compuesto administrada resulta suficiente para proporcionar una concentración pulmonar mínima del compuesto de entre aproximadamente 0,01 y aproximadamente 0,1 pM.
En una realización, la cantidad administrada resulta suficiente para proporcionar un área bajo la curva (ABC) del compuesto, de entre aproximadamente 50 y aproximadamente 10.000 ng*h/ml, de entre aproximadamente 100 y aproximadamente 50.000 ng*h/ml, de entre aproximadamente 100 y 25.000 ng*h/ml, o de entre aproximadamente
10.000 y 25.000 ng*h/ml. En determinadas realizaciones, un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), o un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo, se administra una vez al día durante un día a seis meses, de una semana a tres meses, de una semana a cuatro semanas, de una semana a tres semanas, o de una semana a dos semanas.
En determinadas realizaciones, el compuesto proporcionado en la presente memoria es para la administración una vez al día durante una semana, dos semanas, tres semanas o cuatro semanas. En una realización, el compuesto proporcionado en la presente memoria es para la administración una vez al día durante una semana. En otra realización, el compuesto proporcionado en la presente memoria es para la administración una vez al día durante dos semanas. En todavía otra realización, el compuesto proporcionado en la presente memoria es para la administración una vez al día durante tres semanas. En todavía otra realización, el compuesto proporcionado en la presente memoria es para la administración una vez al día durante cuatro semanas.
En determinadas realizaciones, un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), que puede ser un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, o una forma farmacéuticamente aceptable del mismo, es para la administración dos veces al día durante un día a seis meses, durante una semana a tres meses, durante una semana a cuatro semanas, de una semana a tres semanas, o de una semana a dos semanas. En determinadas realizaciones, el compuesto proporcionado en la presente memoria es para la administración dos veces al día durante una semana, dos semanas, tres semanas o cuatro semanas. En una realización, el compuesto proporcionado en la presente memoria es para la administración dos veces al día durante una semana. En otra realización, el compuesto proporcionado en la presente memoria es para la administración dos veces al día durante dos semanas. En todavía otra realización, el compuesto proporcionado en la presente memoria es para la administración dos veces al día durante tres semanas. En todavía otra realización, el compuesto proporcionado en la presente memoria es para la administración dos veces al día durante cuatro semanas.
El compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4), que puede ser un enantiómero, una mezcla de enantiómeros, o una mezcla de dos o más diastereómeros del mismo, una forma farmacéuticamente aceptable del mismo, puede administrarse en forma de una sola dosis, tal como, p.ej., una inyección de un solo bolo, o tabletas o píldoras orales, o durante el tiempo, tal como, p.ej., la infusión continua durante el tiempo o en dosis de bolo divididas durante el tiempo. El compuesto puede administrarse repetidamente en caso necesario, por ejemplo, hasta que el paciente experimenta una enfermedad estable o la regresión, o hasta que el paciente experimente progresión de la enfermedad o toxicidad inaceptable.
Terapia de combinación
La presente exposición se refiere a terapias de combinación en las que un agente que es conocido que modula otras rutas, u otros componentes de la misma ruta, o incluso grupos solapantes de enzimas diana, se utilizan en combinación con un compuesto dado a conocer en la presente memoria, o una forma farmacéuticamente aceptable (p.ej., sales, hidratos, solvatos, isómeros, profármacos y derivados marcados isotópicamente farmacéuticamente aceptables) del mismo. Dicha terapia puede incluir la combinación del compuesto de la invención con agentes quimioterapéuticos, anticuerpos terapéuticos, y tratamiento de radiación, para proporcionar un efecto terapéutico sinérgico o aditivo.
La expresión "en combinación con" no pretende implicar que la otra terapia y el modulador de PI3K deben administrarse simultáneamente y/o formularse juntos para la administración juntos, aunque estos métodos de administración se encuentran comprendidos dentro del alcance de la presente exposición. El compuesto proporcionado en la presente memoria puede administrarse concurrentemente, antes (p.ej., 5 minutos, 15 minutos, 30 minutos, 45 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 12 horas, 24 horas, 48 horas, 72 horas, 96 horas, 1 semana, 2 semanas, 3 semanas, 4 semanas, 5 semanas, 6 semanas, 8 semanas, 12 semanas, o 16 semanas antes), o después (p.ej., 5 minutos, 15 minutos, 30 minutos, 45 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 12 horas, 24 horas, 48 horas, 72 horas, 96 horas, 1 semana, 2 semanas, 3 semanas, 4 semanas, 5 semanas, 6 semanas, 8 semanas, 12 semanas, o 16 semanas después), de otra u otras terapias (p.ej., otro u otros agentes adicionales). En general, se administra cada agente terapéutico a una dosis y/o en un programa temporal determinado para ese agente particular. El otro agente terapéutico puede administrarse con el compuesto proporcionado en la presente memoria en una única composición o por separado en una composición diferente. La triple terapia también se encuentra contemplada.
En general, se espera que los agentes terapéuticos adicionales utilizados en combinación se utilicen a niveles que no excedan los niveles a los que se utilizan individualmente. Los niveles utilizados en combinación pueden ser inferiores a los utilizados individualmente.
El compuesto proporcionado en la presente memoria puede ser un tratamiento de primera línea para cáncer o neoplasia maligna hemática, es decir, puede utilizarse en un sujeto en el que no se ha administrado anteriormente otro fármaco o terapia destinada al tratamiento de cáncer o neoplasia maligna hemática o uno o más síntomas del mismo.
Alternativamente, el compuesto proporcionado en la presente memoria puede ser un tratamiento de segunda línea
para cáncer o neoplasia maligna hemática, es decir, puede utilizarse en un sujeto en el que no se ha administrado anteriormente otro fármaco o terapia destinada al tratamiento de cáncer o neoplasia maligna hemática o uno o más síntomas del mismo.
Alternativamente, el compuesto proporcionado en la presente memoria puede ser un tratamiento de tercera o cuarta línea para cáncer o neoplasia maligna hemática, es decir, puede utilizarse en un sujeto en el que se han administrado dos o tres otros fármacos o terapias destinados al tratamiento de cáncer o neoplasia maligna hemática o uno o más síntomas del mismo.
En el caso de que se administren dos agentes, estos pueden administrarse en cualquier orden. Por ejemplo, pueden administrarse dos agentes concurrentemente (es decir, esencialmente al mismo tiempo, o dentro del mismo tratamiento) o secuencialmente (es decir, uno inmediatamente después del otro, o alternativamente, con un hueco entre las administraciones de los dos). El compuesto dado a conocer en la presente memoria puede administrarse secuencialmente (es decir, después del primer terapéutico).
Un compuesto dado a conocer en la presente memoria, o una forma farmacéuticamente aceptable del mismo, puede coadministrarse con otros agentes. En el caso de que se utilice en terapia de combinación, un compuesto dado a conocer en la presente memoria, o una forma farmacéuticamente aceptable del mismo, puede administrarse con un segundo agente simultánea o separadamente. Dicha administración en combinación puede incluir la administración simultánea de los dos agentes en la misma forma de administración, la administración simultánea en formas de administración separadas, y la administración separada. Es decir, un compuesto indicado en la presente memoria y uno o más agentes adicionales pueden formularse juntos en la misma forma de administración y administrarse simultáneamente. Alternativamente, un compuesto dado a conocer en la presente memoria y uno o más agentes adicionales pueden administrarse simultáneamente, en el que ambos agentes se encuentran presentes en formulaciones separadas. En otra alternativa, un compuesto dado a conocer en la presente memoria puede administrarse únicamente seguido de uno o más agentes adicionales, o viceversa. En el protocolo de administración separada, un compuesto dado a conocer en la presente memoria puede administrarse uno o más agentes adicionales separados por unos cuantos minutos, o por unas cuantas horas, o por unos cuantos días.
La administración de un compuesto dado a conocer en la presente memoria, o una forma farmacéuticamente aceptable del mismo, puede llevarse a cabo mediante cualquier método que permita la administración del compuesto en el sitio de acción. Una cantidad eficaz de un compuesto dado a conocer en la presente memoria, o una forma farmacéuticamente aceptable del mismo, puede administrarse en una sola dosis o en múltiples dosis mediante cualquiera de los modos aceptados de administración de agentes que presentan utilidades similares, incluyendo las vías rectal, bucal, intranasal y transdérmica, mediante inyección intraarterial, intravenosa, intraperitoneal, parenteral, intramuscular, subcutánea, oral, típica, como inhalante, o mediante un dispositivo impregnado o recubierto, tal como un stent, por ejemplo, o un polímero cilíndrico insertado en una arteria.
En el caso de que un compuesto dado a conocer en la presente memoria, o una forma farmacéuticamente aceptable del mismo, se administre en una composición farmacéutica que comprenda uno o más agentes, y el agente presente una semivida más corta que el compuesto proporcionado en la presente memoria, las formas de dosis unitaria del agente y el compuesto proporcionado en la presente memoria pueden ajustarse correspondientemente.
El compuesto dado a conocer en la presente memoria y el segundo agente pueden administrarse en composiciones separadas, p.ej., composiciones farmacéuticas. El modulador de PI3K y el agente pueden administrarse por separado, aunque por la misma vía (p.ej., ambos por vía oral o ambos por vía intravenosa). Alternativamente, el modulador de PI3K y el agente pueden administrarse en la misma composición, p.ej., la composición farmacéutica.
Combinaciones con inmunomoduladores
Aunque sin deseo de restringirse a ninguna teoría, se cree que el crecimiento tumoral está influenciado por como mínimo dos clases de células inmunitarias en el microambiente tumoral: células efectoras, (incluyendo células citotóxicas y macrófagos M1), que presentan actividad antitumoral, y células supresoras asociadas a tumor (incluyendo macrófagos M2, CSDM, Treg y células dendríticas reguladoras), que presentan actividad protumoral debido a que inhiben las células efectoras o proporcionan una estimulación directa del crecimiento a las células tumorales o la vasculatura tumoral. Una abundancia de células supresoras puede conducir a una tolerancia inmunitaria del tumor y la potenciación del crecimiento tumoral. Una terapia de combinación del cáncer puede diseñarse teniendo en consideración este mecanismo.
La presente invención proporciona compuestos (p.ej., el Compuesto 4) para la utilización en combinación con una terapia de punto de control inmunitario.
La presente invención proporciona compuestos para la utilización en combinación con una o más terapias de punto de control inmunitario. Un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (Xv II) (p.ej., el Compuesto 4, tal como se indica en la presente memoria) puede utilizarse en combinación con una o más terapias de punto de control inmunitario (p.ej., inhibidores de PD-1 o
PD-L1). En algunas realizaciones, la presente invención proporciona compuestos para la utilización en el tratamiento de un cáncer sólido en un sujeto que puede implicar la administración en el sujeto de Compuesto 4, o una forma farmacéuticamente aceptable del mismo, en combinación con uno o más inhibidores de PD-1 o PD-L1. En una realización, el cáncer es melanoma, cáncer de vejiga, cáncer de cabeza y cuello, cáncer pulmonar (p.ej., cáncer pulmonar no microcítico) o carcinoma de células renales. En una realización, el cáncer es melanoma. En una realización, el cáncer es cáncer de vejiga. En una realización, el cáncer es cáncer pulmonar. En una realización, el cáncer es cáncer pulmonar no microcítico. En una realización, la célula es un carcinoma de células renales. En una realización, el cáncer es cáncer de cabeza y cuello. En una realización, el cáncer es cáncer de mama. En una realización, el cáncer es cáncer de mama triple negativo. En una realización, el cáncer es cáncer de colon. En una realización, el cáncer es glioblastoma. En una realización, el cáncer es cáncer ovárico.
En algunas realizaciones, el sujeto nunca ha sido expuesto a tratamiento inmunoterapéutico. En algunas realizaciones, el sujeto nunca ha sido expuesto a tratamiento de radioterapia. En algunas realizaciones, el sujeto nunca ha sido expuesto a tratamiento quimioterapéutico.
En algunas realizaciones, el sujeto ha sido pretratado o previamente tratado con uno o más tratamientos de inmunoterapia. En una realización, el sujeto es reactivo al pretratamiento o el tratamiento anterior con la inmunoterapia. En una realización, el tratamiento de inmunoterapia es un tratamiento de punto de control, tal como inhibidor de PD-1 o PD-L1. En una realización, el sujeto es un fumador.
En una realización, el cáncer es melanoma, y el sujeto ha sido pretratado o previamente tratado con uno o más tratamientos de inmunoterapia. En una realización, el sujeto ha sido pretratado o previamente tratado con uno o más tratamientos de inmunoterapia.
En una realización, el cáncer es cáncer de cabeza y cuello, cáncer pulmonar (p.ej., cáncer pulmonar no microcítico), carcinoma de células renales o cáncer de vejiga, y el sujeto ha sido pretratado o previamente tratado con un tratamiento de inmunoterapia.
En una realización, el cáncer es cáncer de mama (p.ej., cáncer de mama triple negativo), cáncer ovárico, glioblastoma 0 cáncer de colon, y el sujeto nunca ha sido expuesto a tratamiento de inmunoterapia.
En algunas realizaciones, la terapia de punto de control inmunitario inhibe CTLA-4, PD-1 o PD-L1, o cualquier combinación de ellos. La terapia de punto de control inmunitario puede ser, p.ej., una molécula pequeña o un anticuerpo. En algunas realizaciones, la terapia de punto de control inmunitario es un anticuerpo que inhibe la proteína 1 de muerte celular programada (también conocida como PD-1). En otra realización, la terapia de punto de control inmunitario es nivolumab (también conocido como Opdivo). En algunas realizaciones, la terapia de punto de control inmunitario es anti-PD-L1 (ligando 1 de muerte celular programada, también conocida como agregado de diferenciación 274 (CD274)), o anticuerpo anti-PDL2 o anti-CTLA-4 (antígeno 4 de linfocito T citotóxico, también conocido como agregado de difrenciación (CD152)). Determinados anticuerpos anti-PD-1, anti-PD-L1 y anti-CTLA-4 presentan actividad en modelos tumorales preclínicos y clínicos. Cancer Res; 73(12), 15 de junio, 2013; Curran M A et al. PNAS 2010;107:4275-4280; Topalian et al. N Engl. J. Med. 2012; 366:2443-2454; Wolchok et al., 2013. NEJM 369.
Existen dos tipos principales de terapias de punto de control inmunitario: un activador de una molécula coestimuladora y un inhibidor de una molécula de punto de control inmunitario.
En el caso de que la terapia de punto de control inmunitario sea un activador de una molécula coestimuladora, puede seleccionarse, p.ej., de un agonista (p.ej., un anticuerpo agonista o fragmento de unión a antígeno del mismo, o una fusión soluble) de OX40, CD2, CD27, CDS, ICAM-1, LFA-1 (CD11a/CD18), ICOS (CD278), 4-1BB (CD137), GITR, CD30, CD40, BAFFR, HVEM, CD7, LIGHT, NKG2C, SLAMF7, NKp80, CD160, B7-H3 o ligando de CD83. En determinadas realizaciones, la terapia de punto de control inmunitario es un inhibidor de OX40 o anticuerpo anti-OX40.
En la segunda situación, la terapia de punto de control inmunitario es un inhibidor de una molécula de punto de control inmunitario, por ejemplo, un inhibidor de PD-1, PD-L1, PD-L2, CTLA4, TIM3, LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 y/o TGFR beta. Por ejemplo, el inhibidor de una molécula de punto de control inmunitario puede inhibir PD-1, PD-L1, LAG-3, TIM-3 o CTLÁ4, o cualquier combinación de los mismos.
La inhibición de una molécula inhibidora puede llevarse a cabo al nivel de ADN, ARN o proteínas. Por ejemplo, puede utilizarse un ácido nucleico inhibidor (p.ej., ARNdc, ARNmc o ARNhc) para inhibir la expresión de una molécula inhibidora. En otras realizaciones, el inhibidor de una señal inhibidora es un polipéptido, p.ej., un ligando soluble (p.ej., Ig de PD-1 o Ig de CTLA-4), o un anticuerpo o fragmento de unión a antígeno del mismo, que se une a la molécula inhibidora, p.ej., un anticuerpo o fragmento del mismo (también denominado en la presente memoria "molécula de anticuerpo") que se une a PD-1, PD-L1, PD-L2, CTLA4, TIM3, LAG3, VIstA, BTLA, TIGIT, LAIR1, CD160, 2B4 y/o TGFR-beta, o una combinación de los mismos.
La molécula de anticuerpo puede ser, p.ej., un anticuerpo completo o fragmento del mismo (p.ej., un Fab, F(ab')2, Fv,
0 fragmento Fv de cadena sencilla (scFv)). La molécula de anticuerpo puede encontrarse, p.ej., en la forma de una molécula de anticuerpo biespecífico. En una realización, la molécula de anticuerpo biespecífico presenta una primera especificidad de unión a PD-1 o PD-L1 y una segunda especificidad de unión, p.ej., una segunda especificidad de unión a TIM-3, LAG-3 o PD-L2. En determinadas realizaciones, la molécula de anticuerpo se administra mediante inyección (p.ej., subcutánea o intravenosa) a una dosis de entre aproximadamente 1 y 30 mg/kg, p.ej., de entre aproximadamente 5 y 25 mg/kg, de entre aproximadamente 10 y 20 mg/kg, de entre aproximadamente 1 y 5 mg/kg o de aproximadamente 3 mg/kg. El programa de administración puede variar entre, p.ej, una vez a la semana y una vez cada 2, 3 o 4 semanas.
En determinadas realizaciones, la terapia de punto de control inmunitario es un inhibidor de PD-1, p.ej., PD-1 humano. En otra realización, la terapia de punto de control inmunitario es un inhibidor de PD-L1, p.ej., PD-L1 humano. En una realización, el inhibidor de PD-1 o PD-L1 es una molécula de anticuerpo de PD-1 o PD-L1. El inhibidor de PD-1 o PD-L1 puede administrarse solo, o en combinación con otras terapias de punto de control inmunitario, p.ej., en combinación con un inhibidor de LAG-3, TIM-3 o CTLA4. En algunas realizaciones, el inhibidor de PD-1 o PD-L1, p.ej., la molécula de anticuerpo anti-PD-1 o PD-L1, se administra en combinación con un inhibidor de LAG-3, p.ej., una molécula de anticuerpo anti-LAG-3. En otra realización, el inhibidor de PD-1 o PD-L1, p.ej., la molécula de anticuerpo anti-PD-1 o PD-L1, se administra en combinación con un inhibidor de TIM-3, p.ej., una molécula de anticuerpo anti-TIM-3. En todavía otras realizaciones, el inhibidor de PD-1 o PD-L1, p.ej., la molécula de anticuerpo anti-PD-1, se administra en combinación con un inhibidor de LAG-3, p.ej., una molécula de anticuerpo anti-LAG-3, y un inhibidor de TIM-3, p.ej., una molécula de anticuerpo anti-TIM-3. Otras combinaciones de terapias de punto de control inmunitario con un inhibidor de PD-1 (p.ej., uno o más de entre PD-L2, CTLA4, TIM3, LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 y/o TGFR) también se encuentran comprendidas dentro de la presente invención. Puede utilizarse cualquiera de las moléculas de inhibidor de PI3K conocidas de la técnica o dadas a conocer en la presente memoria en las combinaciones anteriormente indicadas de inhibidores de molécula de punto de control.
En algunas realizaciones, el anticuerpo anti-PD-1 es el nivolumab. Entre los nombres alternativos para el nivolumab se incluyen M D X-1106, MDX-1106-04, ONO-4538, o BMS-936558. En algunas realizaciones, el anticuerpo anti-PD-1 es el nivolumab (número de registro CAS 946414-94-4). El nivolumab es un anticuerpo monoclonal IgG4 totalmente humano que bloquea específicamente PD1. El nivolumab (clon 5C4) y otros anticuerpos monoclonales humanos que se unen específicamente a PD1 se dan a conocer en la patente n° US 8.008.449 y en el documento n° WO2006/121168.
En otras realizaciones, el anticuerpo anti-PD-1 es el pembrolizumab. El pembrolizumab (nombre comercial: KEYTRUDA, anteriormente Lambrolizumab, también conocido como Merck 3745, MK-3475 o Sc H-900475) es un anticuerpo monoclonal IgG4 humanizado que se une a PD1. El pembrolizumab se da a conocer, p.ej., en Hamid, O. et al. (2013) New England Journal of Medicine 369 (2): 134-44, documento n° WO2009/114335 y patente n° US 8.354.509.
En algunas realizaciones, el anticuerpo anti-PD-1 es el pidilizumab. El pidilizumab (CT-011, Cure Tech) es un anticuerpo monoclonal IgGlk humanizado que se une a PD1. El pidilizumab y otros anticuerpos monoclonales anti-PD-1 humanizados se dan a conocer en el documento n° WO2009/101611. Se dan a conocer otros anticuerpos anti-PD1 en los documentos n° US 8.609.089, n° US 2010028330 y/o n° US 20120114649. Entre otros anticuerpos anti-PD1 se incluyen AMP 514 (Amplimmune).
En algunas realizaciones, el inhibidor de PD-1 es una inmunoadhesina (p.ej., una inmunoadhesina que comprende una parte extracelular o de unión a PD-1 de PD-L1 o PD-L2 fusionada con una región constante (p.ej., una región Fc de una secuencia de inmunoglobulina)). En algunas realizaciones, el inhibidor de PD-1 es AMP-224. En algunas realizaciones, el inhibidor de PI3K, p.ej., un inhibidor de PI3K-Y tal como se indica en la presente memoria (p.ej., el Compuesto 4), se administra junto con una inmunoadhesina (p.ej., una inmunoadhesina que comprende una parte extracelular o de unión a Pd-1 de PD-L1 o PD-L2 fusionada con una región constante (p.ej., una región Fc de una secuencia de inmunoglobulina)). La terapia de combinación es para la utilización en el tratamiento de un cáncer, tal como se indica en la presente memoria.
En algunas realizaciones, el inhibidor de PD-L1 es un anticuerpo anti-PD-L1. En algunas realizaciones, el inhibidor anti-PD-L1 se selecciona de entre YW243.55.S70, MPDL3280A, MEDI-4736, MSB-0010718C, o MDX-1105.
En una realización, el inhibidor de PD-1 es MDX-1105. MDX-1105, también conocido como BMS-936559, es un anticuerpo anti-PD-L1 descrito en el documento n° WO2007/005874.
En una realización, el inhibidor de PD-L1 es YW243.55.S70. El anticuerpo YW243.55.S70 es un anti-PD-L1 descrito en el documento n° WO 2010/077634 (secuencias de región variable de cadenas pesada y ligera mostradas en las SEC ID n° 20 y n° 21, respectivamente).
En una realización, el inhibidor de PD-L1 es MDPL3280A (Genentech/Roche). MDPL3280A es un anticuerpo monoclonal IgG1 de Fc optimizada humana que se une a PD-L1. MDPL3280A y otros anticuerpos monoclonales humanos de PD-L1 se dan a conocer en la patente US n° 7.943.743 y la publicación de patente US n° 20120039906.
En otras realizaciones, el inhibidor de PD-L2 es AMP-224. AMP-224 es un receptor soluble de fusión PD-L2 Fc que bloquea la interacción entre PD1 y B7-H1 (B7-DCIg; Amplimmune; p.ej., dado a conocer en los documentos n° WO2010/027827 y n° WO2011/066342).
En una realización, el inhibidor de LAG-3 es una molécula de anticuerpo anti-LAG-3. En una realización, el inhibidor de LAG-3 es BMS-986016.
En algunas realizaciones, el antagonista de unión anti-PD-LI se selecciona de entre YW243.55.S70, MPDL3280A, MEDI-4736, MSB-0010718C, o MDX-1105. MDX-1105, también conocido como BMS-936559, es un anticuerpo anti-PD-LI descrito en el documento n° WO2007/005874. El anticuerpo YW243.55.S70 (secuencias de región variable de cadenas pesada y ligera mostradas en las SEC id n° 20 y n° 21, respectivamente) es un anti-PD-LI descrito en el documento n° WO 2010/077634.
En algunas realizaciones, el anticuerpo anti-PD-1 es el nivolumab. Entre los nombres alternativos para el nivolumab se incluyen MDX-1106, MDX-1106-04, ONO-4538, o BMS-936558. En algunas realizaciones, el anticuerpo anti-PD-1 es el nivolumab (número de registro CAS 946414-94-4). El nivolumab (también denominado BMS-936558 o MDX1106; Bristol-Myers Squibb) es un anticuerpo monoclonal IgG4 totalmente humano que bloquea específicamente PD-1. El nivolumab (clon 5C4) y otros anticuerpos monoclonales humanos que se unen específicamente a PD1 se dan a conocer en las patentes n° US 8.008.449 y n° EP2161336 y en el documento n° WO2006/121168.
En algunas realizaciones, el anticuerpo anti-PD-1 es el pembrolizumab. El pembrolizumab (también denominado lambrolizumab, MK-3475, MK03475, SCH-900475 o KeYt RUDA®; Merck) es un anticuerpo monoclonal de IgG4 humanizado que se une a PD-1. El pembrolizumab y otros anticuerpos anti-PD-1 humanizados se dan a conocer en Hamid, O. et al. (2013) New England Journal of Medicine 369 (2): 134-44, patente n° US 8.354.509 y documento n° WO2009/114335.
El pidilizumab (CT-011, Cure Tech) es un anticuerpo monoclonal IgG1k humanizado que se une a PD1. El pidilizumab y otros anticuerpos monoclonales anti-PD-1 humanizados se dan a conocer en el documento n° WO2009/101611.
Entre otros anticuerpos anti-PD1 se incluyen AMP 514 (Amplimmune), entre otros, p.ej., anticuerpos anti-PD1 dados a conocer en la patente n° US 8.609.089, y en los documentos n° US 2010028330 y/o n° US 20120114649.
En algunas realizaciones, el anticuerpo anti-PD-L1 es MSB0010718C. MSB0010718C (también denominado A09-246-2; Merck Serono) es un anticuerpo monoclonal que se une a PD-L1. El pembrolizumab y otros anticuerpos anti-PD-L1 humanizados se dan a conocer en el documento n° WO2013/079174.
MDPL3280A (Genentech / Roche) es un anticuerpo monoclonal IgG1 de Fc optimizada humano que se une a PD-L1. MDPL3280A y otros anticuerpos monoclonales humanos de PD-L1 se dan a conocer en la patente US n° 7.943.743 y la publicación de patente US n° 20120039906. Entre otros agentes de unión anti-PD-L1 se incluyen YW243.55.S70 (las regiones variables de cadenas pesada y ligera se muestran en las SEC ID n° 20 y n° 21 en el documento n° WO2010/077634) y MDX-1105 (también denominado BMS-936559, y, p.ej., agentes de unión anti-PD-L1 dados a conocer en el documento n° WO2007/005874).
AMP-224 (B7-DCIg; Amplimmune; p.ej., dado a conocer en los documentos n° WO2010/027827 y n° WO2011/066342), es un receptor soluble de fusión PD-L2 Fc que bloquea la interacción entre PD1 y B7-H1.
En algunas realizaciones, el anticuerpo anti-LAG-3 es BMS-986016. BMS-986016 (también denominado BMS986016; Bristol-Myers Squibb) es un anticuerpo monoclonal que se une a LAG-3. BMS-986016 y otros anticuerpos anti-LAG-3 humanizados se dan a conocer en los documentos n° US 2011/0150892, n° WO2010/019570 y n° WO2014/008218.
En determinadas realizaciones, entre las terapias de combinación dadas a conocer en la presente memoria se incluyen un modulador de una molécula coestimuladora o una molécula inhibidora, p.ej., un ligando o receptor coinhibidor.
En una realización, el modulador coestimulador, p.ej., agonista, de una molécula coestimuladora se selecciona de un agonista (p.ej., un anticuerpo agonista o fragmento de unión a antígeno del mismo, o fusión soluble) de ligando de OX40, CD2, CD27, CDS, ICAM-1, LFA-1 (CDIIa/CD18), ICOS (CD278), 4-1BB (CD137), GITR, CD30, CD40, BAFFR, HVEM, CD7, LIGHT, NKG2C, SLAMF7, NKp80, CD160, B7-H3 o CD83.
En otra realización, entre las terapias de combinación dadas a conocer en la presente memoria se incluyen una molécula coestimuladora, p.ej., un agonista asociado a una señal positiva que incluye un dominio coestimulador de CD28, CD27, ICOS y GITR.
Entre los agonistas de GITR ejemplares se incluyen, p.ej., proteínas de fusión de GITR y anticuerpos anti-GITR (p.ej., anticuerpos anti-GITR bivalentes), tales como, una proteína de fusión de GITR descrito en la patente US n° 6.111.090, la patente europea n° 090505B1, la patente n° US 8.586.023, las publicaciones de patente p Ct n° WO 2010/003118 y
n° 2011/090754, o un anticuerpo anti-GITR descrito en, p.ej., la patente US n° 7.025.962, la patente europea n° 1947183B1, la patente US n° 7.812.135, la patente n° US 8.388.967, la patente n° US 8.591.886, la patente europea n° EP 1866339, la publicación de patente PCT n° WO 2011/028683, la publicación de patente PCT n° WO 2013/039954, la publicación de patente PCT n° W02005/007190, la publicación de patente PCT n° WO 2007/133822, la publicación de patente PCT n° WO2005/055808, la publicación de patente PCT n° WO 99/40196, la publicación de patente PCT n° WO 2001/03720, la publicación de patente PCT n° WO99/20758, la publicación de patente PCT n° WO2006/083289, la publicación de patente PCT n° WO 2005/115451, la patente US n° 7.618.632 y la publicación de patente PCT n° WO 2011/051726.
En una realización, el inhibidor es un ligando soluble (p.ej., un Ig-CTLA-4), o un anticuerpo o fragmento de anticuerpo que se une a PD-L1, PD-L2 o CTLA4. Por ejemplo, un compuesto dado a conocer en la presente memoria, p.ej., el Compuesto 4, puede administrarse en combinación con un anticuerpo anti-CTLA-4, p.ej., ipilimumab, por ejemplo para tratar un cáncer (p.ej., un cáncer seleccionado de entre: un melanoma, p.ej., un melanoma metastásico, un cáncer pulmonar, p.ej., carcinoma pulmonar no microcítico, o un cáncer de próstata). Entre los anticuerpos anti-CTAL4 ejemplares se incluyen tremelimumab (anticuerpo monoclonal IgG2 disponible de Pfizer, anteriormente conocido como ticilimumab, CP-675.206) e ipilimumab (anticuerpo de CTLA-4, también conocido como MDX-010, Yervoy, CAS n° 477202-00-9). En algunas realizaciones, un compuesto proporcionado en la presente memoria se administra en combinación con un inhibidor anti-PD-L1 (p.ej., nivolumab) y un anticuerpo de CTLA-4 (p.ej., ipilimumab). En algunas realizaciones, un compuesto proporcionado en la presente memoria se administra en combinación con nivolumab e ipilimumab.
En algunas realizaciones, un compuesto proporcionado en la presente memoria, o un derivado farmacéuticamente aceptable (p.ej., sal o solvato) del mismo, se administra en combinación con un anticuerpo anti-PD-L1 o anti-CTLA-4. En algunas realizaciones, un compuesto proporcionado en la presente memoria, o un derivado farmacéuticamente aceptable (p.ej., sal o solvato) del mismo, se administra en combinación con un anticuerpo anti-PD-L1. En otra realización, un compuesto proporcionado en la presente memoria, o un derivado farmacéuticamente aceptable (p.ej., sal o solvato) del mismo, se administra en combinación con un anticuerpo anti-CTLA-4. En algunas realizaciones, el anticuerpo anti-PD-L1 se selecciona de entre BMS-936559, MPDL3280A y MDX-1105. En algunas realizaciones, el anticuerpo anti-CTLA-4 se selecciona de entre ipilimumab y tremelimumab.
En algunas realizaciones, la presente invención proporciona compuestos para la utilización en el tratamiento de cáncer de mama, cáncer de colon, cáncer pancreático, melanoma, glioblastoma o cáncer pulmonar. Una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria (p.ej., los compuestos 2, 4, 7, 9, 17, 19, 21,26, 27, 30, 32, 35, 37, 38, 40, 41, 52, 60, 61,63, 73, 75, 77, 79, 80, 81 y 8 8 ), o un derivado farmacéuticamente aceptable (p.ej., sal o solvato) del mismo, puede administrarse en un paciente en combinación con un anticuerpo anti-PD-L1 o anti-CTLA-4. En otra realización, el cáncer se selecciona de entre un carcinoma (p.ej., carcinoma avanzado o metastásico), melanoma o un carcinoma pulmonar, p.ej., un carcinoma pulmonar no microcítico. En una realización, el cáncer es un cáncer pulmonar, p.ej., un cáncer pulmonar no microcítico. En una realización, el cáncer es un melanoma, p.ej., un melanoma avanzado. En una realización, el cáncer es un melanoma avanzado o no resecable que no responde a otras terapias. En otras realizaciones, el cáncer es un melanoma con una mutación BRAF (p.ej., una mutación BRAF V600E). En otra realización, el cáncer es un hepatocarcinoma, p.ej., un hepatocarcinoma avanzado, con o sin una infección vírica, p.ej., una hepatitis vírica crónica. En otra realización, el cáncer es un cáncer de próstata, p.ej., un cáncer de próstata avanzado. En todavía otra realización, el cáncer es un mieloma, p.ej., mieloma múltiple. En todavía otra realización, el cáncer es un cáncer renal, p.ej, un carcinoma de células renales (CCR) (p.ej., un CCR metastásico o carcinoma renal de células claras (CRCC)).
Por ejemplo, un compuesto proporcionado en la presente memoria para la utilización según la presente invención (p.ej., los compuestos 2, 4, 7, 9, 17, 19, 21, 26, 27, 30, 32, 35, 37, 38, 40, 41, 52, 60, 61, 63, 73, 75, 77, 79, 80, 81 y 8 8 ) puede administrarse en combinación con un anticuerpo anti-CTLA-4, p.ej., ipilimumab, por ejemplo para tratar un cáncer (p.ej., un cáncer seleccionado de: un melanoma, p.ej., un melanoma metastásico, un cáncer pulmonar, p.ej., un carcinoma pulmonar no microcítico, o un cáncer de próstata). En una realización, un compuesto proporcionado en la presente memoria (p.ej., los compuestos 2, 4, 7, 9, 17, 19, 21, 26, 27, 30, 32, 35, 37, 38, 40, 41, 52, 6 0 , 61,63, 73, 75, 77, 79, 80, 81 y 8 8 ) se administra después del tratamiento con un anticuerpo anti-CTLA4 (p.ej., el ipilimumab) con o sin un inhibidor de BRAF (p.ej., vemurafenib o dabrafenib).
En algunas realizaciones, la terapia de punto de control inmunitario es un ligando coestimulador. En algunas realizaciones, el ligando coestimulador es OX40L, 41BBL, CD153, ICOSL, o CD40L.
En algunas realizaciones, la terapia de punto de control inmunitario es un inhibidor de FECM/CSF-1R. Un anti-CSF-1R puede agotar las células TAM, resultando en la inhibición del crecimiento tumoral. Cancer Cell 25, 1-14, 16 de junio, 2014. En algunas realizaciones, el inhibidor de CSF-1R es BLZ945, GW2850, RO5509554, o PLX3397. En algunas realizaciones, el inhibidor de CSF-1R es BLZ945 o GW2850. En algunas realizaciones, el inhibidor de CSF-1R es PLX3397.
En algunas realizaciones, la terapia de punto de control inmunitario es un inmunoestimulante. En algunas realizaciones, el inmunoestimulante es FECMg , ligandos de TLR, 41BBL o ICOSL.
En algunas realizaciones, la terapia de punto de control inmunitario es un inhibidor de CXCR4/CXCL12. En algunas realizaciones, el inhibidor de CXCR4/CXCL12 es AMD3100, AMD11070, AMD12118, AMD11814, o AMD13073. En algunas realizaciones, el inhibidor de CXCR4/CXCL12 es AMD3100.
En algunas realizaciones, la inmunoterapia es un antagonista de CCL2 y/o CCR2. En algunas realizaciones, el antagonista de CCL2 y/o CCR2 es un anticuerpo anti-CCL2 o CCR2. CCL2 es una quimioquina y CCR2 es un receptor de quimioquina. CCL2 y CCR2, según una teoría no limitativa, desempeñan un papel en la migración de las CSDM.
En algunas realizaciones, un inhibidor de PI3K-Y dado a conocer en la presente memoria, p.ej., el Compuesto 4, es para la administración en combinación con un inhibidor de BTK. En una realización, el inhibidor de BTK es un inhibidor de BTK tal como ibrutinib, AVL-292, dasatinib, LFM-AI3, ONO-WG-307 y GDC-0834.
En algunas realizaciones, un inhibidor de PI3K-Y dado a conocer en la presente memoria, p.ej., el Compuesto 4, se administra en combinación con un inhibidor de iDo (indolamín-2,3-dioxigenasa) o un inhibidor de TDO (triptófano 2,3-dioxigenasa). En una realización, el inhibidor de IDO es indoximod, NLG919, INCB024360, F001287, norharmano, ácido rosmarínico o alfa-metiltriptófano. Aunque los inhibidores IDO actúan dentro del microambiente tumoral (MAT), no presentan como diana específica las CSDM. La sobreexpresión de IDO por las células dendríticas crea un microambiente tumoral inmunosupresor.
En algunas realizaciones, un inhibidor de PI3K-Y dado a conocer en la presente memoria, p.ej., el Compuesto 4, es para la administración en combinación con un inhibidor de uno o más elementos de la familia TAM, una subfamilia de receptor de tirosina quinasa (RTK) que comprende Tyro-3 (también denominado Sky), AxI y Mer. En una realización, el inhibidor de MAT es BGB324 (R428), S49076, TP0903, CEP-40783, ONO-9330547, bosutinib (SKI606, PF5208763), cabozantinib (XL184), sunitinib (SU11248), foretinib (XL880, GSK1363089), MGCD265, BMS777607 (ASLAN002), LY2801653, SGI7079, amuvatinib (SGI-0470-02, MP470), SNS314, PF-02341066, diaminopirimidina, espiroindolina, UNC569, UNC1062, UNC1666, UNC2025, o LDC1267. Entre los inhibidores de MAT adicionales se incluyen los indicados en Mollard et al., Med. Chem. Lett. 2011,2, 907-912 y en Feneyrolles et al., Mol. Cancer Ther.
13(9), publicado online previamente, el 19 de agosto, 2014.
En algunas realizaciones, un inhibidor de PI3K-Y dado a conocer en la presente memoria, p.ej., el Compuesto 4, es para la administración en un sujeto concurrentemente o antes de la administración de la terapia de punto de control inmunitario. En algunas realizaciones, se administra un inmunoestimulante en el sujeto concurrentemente o antes de la administración de terapia de punto de control inmunitario. En algunas realizaciones, se administra quimioterapia (p.ej., carboplatino, oxaliplatino o radiación) en el sujeto concurrentemente o antes de la administración de terapia de punto de control inmunitario.
En algunas realizaciones, un inhibidor de PI3K-Y dado a conocer en la presente memoria, p.ej., el Compuesto 4, es para la administración en combinación con un inhibidor de ARG1. Sin deseo de restringirse a ninguna teoría en particular, se ha informado de que las células mieloides asociadas a tumor establecen un microambiente inmunosupresor en tumores mediante la expresión de arginasa-1, que agota de arginina el microambiente tumoral, conduciendo de esta manera a la muerte o inhibición de las células inmunitarias antitumorales. Schmid et al., Proceedings: AACR 103rd Annual Meeting 2012, Cancer Research: 15 de abril, 2012; volumen 72, número 8, suplemento 1. Se ha informado de que la supresión de la expresión de PI3Kgamma o de arginasa-1 bloquea la muerte celular inducida por células mieloides de las células T in vitro. Id. Según la teoría no limitativa, la inhibición de PI3K-gamma bloquea la expresión de la arginasa-1, incrementando de esta manera el número de células T CD8+ en los tumores, estimulando la citotoxicidad mediada por células T de las células tumorales y suprimiendo el crecimiento y la metástasis de los mismos. Pueden diseñarse terapias de combinación de acuerdo con dicho mecanismo.
Por ejemplo, en algunas realizaciones, un inhibidor de PI3K-Y dado a conocer en la presente memoria, p.ej., el Compuesto 4, es para la administración en combinación con un inhibidor de ARG1. El inhibidor de ARG1 puede ser, p.ej., un ácido nucleico inhibidor, tal como un ARNip, un anticuerpo inhibidor anti-ARG-1, o un análogo de la arginina. Entre otros inhibidores ejemplares de ARG1 se incluyen N-hidroxi-guanidinio o N-hidroxi-nor-l-arginina, y derivados de ácido borónico, tales como ácido 2(S)-amino-6-boronohexanoico y S-(2-boronoetil)-l-cisteína, inhibidor de arginasa a base de aminoácido a-a-disustituido [tales como ácido (R)-2-amino-6-borono-2-(2-(piperidín-1-il)etil)hexanoico] y piceatanol-3'-O-p-d-glucopiranósido (PG). Steppan et al., "Development of novel arginase inhibitors for therapy of endothelial dysfunction.", Front Immunol. 17 de sept., 2013;4:278. doi: 10.3389/fimmu.2013.00278.
Los inhibidores de PI3K-Y dados a conocer en la presente memoria pueden presentar efectos mínimos sobre la activación de las células T en comparación con el efecto supresor de un inhibidor de PI3K-5 sobre la activación de las células T. El crecimiento tumoral del carcinoma pulmonar de Lewis puede reducir en ratones con inactivación de PI3K-Y y puede presentar menos infiltrados de células mieloides supresoras asociadas a tumor. Entre las células mieloides supresoras asociadas a tumor pueden incluirse, p.ej., células supresoras derivadas de mieloides (CSDM) y macrófagos asociados a tumor (MAT). Los ratones con inactivación de PI3K-Y presentan MAT en los que se ha perdido el fenotipo M2. Las células M2 son inmunosupresoras y apoyan el crecimiento tumoral. Los inhibidores de PI3k
proporcionados en la presente memoria pueden bloquear el fenotipo M2 (p.ej., en un sistema de diferenciación in vitro) y, de esta manera, pueden retrasar el crecimiento tumoral.
Por ejemplo, el efecto de los inhibidores de PI3K-Y y de los inhibidores de PI3K-8 sobre la activación de las células T medida como inhibición de IFN-y en respuesta a ConA ha mostrado que PI3K-8 desempeña un papel en la mediación de la activación de las células T, mientras que PI3K-Y presenta efectos mínimos sobre la activación de las células T. La IC50 para un inhibidor de PI3K 8 en el presente ensayo es de 3 nM, y la IC50 para un inhibidor de PI3K y es 2500 nM. La administración de inhibidores de PI3K-y puede conducir a un deterioro de la migración de las células T, aunque puede presentar efectos reducidos sobre la proliferación o la activación de las células T.
En algunas realizaciones, los inhibidores de PI3K-Y dados a conocer en la presente memoria pueden presentar potentes efectos sobre las células mieloides supresoras asociadas a tumor sin inhibir las células T efectoras. Los inhibidores de PI3K-Y dados a conocer en la presente memoria pueden presentar efectos potentes sobre las células mieloides supresoras asociadas a tumor sin bloquear los efectos antitumorales de células T y, de esta manera, pueden incrementar la actividad de las células T. En una realización, dicho efecto puede potenciarse mediante la administración de antagonistas de CTLA4 y/o antagonistas de PD-1 y PDL1. Los inhibidores de PI3K-Y dados a conocer en la presente memoria pueden incrementar la activación y proliferación de las células T. Las células mieloides supresoras asociadas a tumor pueden bloquearse sin inhibir los efectos sobre las células T antitumorales mediante la administración de una cantidad eficaz de un inhibidor de PI3K-Y dado a conocer en la presente memoria o una sal farmacéuticamente aceptable del mismo en un sujeto. Las células mieloides supresoras asociadas a tumor pueden bloquearse sin inhibir los efectos sobre las células T antitumorales mediante la administración de una cantidad eficaz de un compuesto dado a conocer en la presente memoria o una sal farmacéuticamente aceptable del mismo en un sujeto. El sujeto puede presentar cáncer pulmonar, cáncer de mama, glioblastoma o linfoma (p.ej., linfoma no de Hodgkin).
La actividad de quinasa puede modularse mediante la puesta en contacto de una quinasa con una cantidad de un compuesto proporcionado en la presente memoria que resulte suficiente para modular la actividad de la quinasa. Modular puede ser inhibir o activar la actividad de quinasa. La actividad de quinasa puede inhibirse mediante la puesta en contacto de una quinasa con una cantidad de un compuesto proporcionado en la presente memoria que resulte suficiente para modular la actividad de la quinasa. La actividad de quinasa puede inhibirse mediante la puesta en contacto de dicha solución con una cantidad de un compuesto proporcionado en la presente memoria que resulte suficiente para inhibir la actividad de la quinasa en dicha solución. La actividad de quinasa puede inhibirse en una célula mediante la puesta en contacto de dicha célula con una cantidad de un compuesto proporcionado en la presente memoria que resulte suficiente para inhibir la actividad de la quinasa en dicha célula. La actividad de quinasa puede inhibirse en un tejido mediante la puesta en contacto de dicho tejido con una cantidad de un compuesto proporcionado en la presente memoria que resulte suficiente para inhibir la actividad de la quinasa en dicho tejido. La actividad de quinasa puede inhibirse en un organismo mediante la puesta en contacto de dicho organismo con una cantidad de un compuesto proporcionado en la presente memoria que resulte suficiente para inhibir la actividad de la quinasa en dicho organismo. La actividad de quinasa puede inhibirse en un animal mediante la puesta en contacto de dicho animal con una cantidad de un compuesto proporcionado en la presente memoria que resulte suficiente para inhibir la actividad de la quinasa en dicho animal. La actividad de quinasa puede inhibirse en un mamífero mediante la puesta en contacto de dicho mamífero con una cantidad de un compuesto proporcionado en la presente memoria que resulte suficiente para inhibir la actividad de la quinasa en dicho mamífero. La actividad de quinasa puede inhibirse mediante la puesta en contacto de dicho ser humano con una cantidad de un compuesto dado a conocer en la presente memoria que resulte suficiente para inhibir la actividad de la quinasa en dicho ser humano. El porcentaje de actividad de quinasa después de la puesta en contacto de una quinasa con un compuesto proporcionado en la presente memoria puede ser inferior a 1, 5, 10, 20, 30, 40, 50, 60, 70, 8090, 95 o 99 % de la actividad de la quinasa en ausencia de dicha etapa de puesta en contacto.
En determinadas realizaciones, la presente invención se refiere a composiciones farmacéuticas que comprenden una cantidad terapéuticamente eficaz de un compuesto proporcionado en la presente memoria para la utilización según la invención (p.ej., un compuesto de cualquiera de las fórmulas (I"), (I'), (A'), (I), (A), (II), (III), (IV), (V), (VI), (VII), (IX), (X), (XI), (XII), (XIII), (XIV), (XV), (XVI) y (XVII), p.ej., un inhibidor selectivo de P i3k -y, p.ej., el Compuesto 4) o una forma farmacéuticamente aceptable del mismo, y un inmunomodulador.
En una realización, el inmunomodulador es un inhibidor de PD-1, PD-L1, LD-L2, CTLA-4, TIM3, LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4, TGFR-beta, o IDO/TDO, o una combinación de los mismos. En una realización, el inmunomodulador es un inhibidor de PD-L1. En una realización, el inmunomodulador es un anticuerpo o fragmento del mismo, un ácido nucleico inhibidor, un ligando soluble, o una fusión de un ligando de PD-1 con una región Fc de una inmunoglobulina. En una realización, el inmunomodulador es un ligando coestimulador, un inhibidor de FECM/CSF-1R, un inmunoestimulante, un inhibidor de CXCR4/CXCL12, un inhibidor de CCL2 o un inhibidor de CCR2. En una realización, el inmunomodulador es ciclofosfamida, docetaxel, paclitaxel, 5-FU o temozolomida.
Un trastorno mediado por PI3K en un sujeto puede tratarse mediante la administración en el sujeto de una cantidad terapéuticamente eficaz de la composición.
Los ejemplos y preparaciones proporcionados posteriormente ilustran y ejemplifican los compuestos proporcionados en la presente memoria y los métodos de preparación de dichos compuestos. En los ejemplos siguientes, las moléculas con un único centro quiral, a menos que se indique lo contrario, existen en forma de una mezcla racémica. Aquellas moléculas con dos o más centros quirales, a menos que se indique lo contrario, existen en forma de una mezcla racémica de diastereómeros. Pueden obtenerse enantiómeros/diastereómeros únicos mediante métodos conocidos por el experto en la materia.
Síntesis de compuestos
Los compuestos proporcionados en la presente memoria pueden prepararse según métodos conocidos de la técnica. Por ejemplo, los compuestos proporcionados en la presente memoria pueden sintetizarse según los esquemas, posteriormente. El Esquema 1 muestra la síntesis de amina A-30, F-50, X-40 y H50. El Esquema 2 muestra la síntesis de amida D-20 y la fórmula I.

Específicamente, en el Esquema 1 en el método A, la isoquinolinona amina, compuesto A-30, se genera en dos etapas. Por ejemplo, en la primera etapa, el compuesto A-10 se convierte en compuesto A-20. El compuesto A-20 se acopla con (1-(metoxi(metilamino)-1-oxopropán-2-il)carbamato de terc-butilo, proporcionando el compuesto A-30. Los compuestos isoquinolinona pueden prepararse opcionalmente según el método H. Por ejemplo, el compuesto H-10 se acopla con (1-(metoxi(metil)amino)-1-oxopropán-2-il)carbamato de terc-butilo para generar el compuesto H-20, que a continuación se convierte en H-30. El compuesto H-30 se hace reaccionar con B-NH2 , formando H-40, que a continuación se trata con, p.ej., un ácido, proporcionando H-50.
En el método F, se genera la quinazolinona F-50. Por ejemplo, el compuesto F-10 se convierte en compuesto F-20, que se acopla con ácido 2-((terc-butoxicarbonil)amino)propanoico, formando F-30. A continuación, el compuesto F-30 se convierte en F-40. El compuesto F-40 se desprotege, proporcionando el compuesto F-50. Alternativamente, puede prepararse la quinazolinona X-40 partiendo de ácido 2-amino-6-clorobenzoico, generando el compuesto X-10, que puede convertirse en compuesto X-20. El compuesto X-20 puede acoplarse con ácido 2-((tercbutoxicarbonil)amino)propanoico, generando el compuesto X-30, que puede convertirse en el compuesto deseado: X-40.
En el Esquema 2, el compuesto amina A-30, F50, X-40 o H50 se trata con Wd-C(O)OH, proporcionando la amida D20, que se trata con un alquino, generando un compuesto de fórmula (I).

EJEMPLOS
EJEMPLOS QUÍMICOS
Las entidades químicas indicadas en la presente memoria pueden sintetizarse según uno o más esquemas ilustrativos en la presente memoria y/o técnicas bien conocidas.
A menos que se especifique lo contrario, las reacciones indicadas en la presente memoria tienen lugar a presión atmosférica, generalmente en un intervalo de temperaturas de entre -10°C y 200°C. Además, excepto en caso de que se especifique lo contrario, los tiempos y condiciones de reacción pretenden ser aproximados, p.ej., tienen lugar a aproximadamente la presión atmosférica en un intervalo de temperaturas de entre aproximadamente -10°C y aproximadamente 110°C durante un periodo que es, por ejemplo, de entre aproximadamente 1 y aproximadamente 24 horas; las reacciones se dejan que se produzcan durante la noche, que puede ser un periodo medio de aproximadamente 16 horas.
Cada uno de los términos y expresiones "solvente", "solvente orgánico" y "solvente inerte" significa un solvente que es inerte bajo las condiciones de la reacción que se describe junto con ellos, incluyendo, por ejemplo, benceno, tolueno, acetonitrilo, tetrahidrofurano ("THF"), dimetilformamida ("DMF"), cloroformo, cloruro de metileno (o diclorometano), éter dietílico, metanol, N-metilpirrolidona ("NMP"), piridina y similares. A menos que se especifique lo contrario, los solventes utilizados en las reacciones indicadas en la presente memoria son solventes orgánicos inertes. A menos que se especifique lo contrario, por cada gramo del reactivo limitante, un cm3 (o ml) de solvente constituye un equivalente de volumen.
El aislamiento y la purificación de las entidades químicas e intermediarios indicados en la presente memoria pueden llevarse a cabo, si se desea, mediante cualquier procedimiento adecuado de separación o purificación, tal como, por ejemplo, filtración, extracción, cristalización, cromatografía de columna, cromatografía de capa fina o cromatografía de capa gruesa, o una combinación de dichos procedimientos. Se proporcionan ilustraciones específicas de procedimientos adecuados de separación y aislamiento mediante referencia a los ejemplos posteriormente en la presente memoria. Sin embargo, también pueden utilizarse otros procedimientos equivalentes de separación o aislamiento.
En caso de que se desee, los isómeros (R) y (S) de los compuestos ejemplares no limitativos, en caso de estar presentes, pueden resolverse mediante métodos conocidos por el experto en la materia, por ejemplo mediante formación de sales o complejos diastereoisoméricos que pueden separarse mediante, por ejemplo, cristalización, mediante formación de derivados diastereoisoméricos que pueden prepararse, por ejemplo mediante cristalización, cromatografía gas-líquida o líquida, reacción selectiva de un enantiómero con un reactivo específico de enantiómero, por ejemplo la oxidación o reducción enzimática, seguida de la separación de los enantiómeros modificados y no modificados, o cromatografía gas-líquido o líquida en un medio quiral, por ejemplo sobre un soporte quiral, tal como sílice con un ligando quiral unido o en presencia de un solvente quiral. Alternativamente, puede sintetizarse un enantiómero específico mediante síntesis asimétrica utilizando reactivos, sustratos, catalizadores o solventes ópticamente activos, o mediante conversión de un enantiómero en el otro mediante transformación asimétrica.
Además, los atropisómeros (es decir, estereoisómeros de la rotación impedida en torno a enlaces sencillos) de los compuestos proporcionados en la presente memoria pueden resolverse o aislarse mediante métodos conocidos por el experto en la materia. Por ejemplo, determinados sustituyentes B con fenilo sustituido en orto o meta pueden formar atropisómeros, a partir de donde pueden separarse y aislarse.
Los compuestos indicados en la presente memoria pueden ponerse en contacto opcionalmente con un ácido farmacéuticamente aceptable para formar las sales de adición de ácido correspondientes. Además, los compuestos indicados en la presente memoria pueden ponerse en contacto opcionalmente con una base farmacéuticamente aceptable para formar las sales de adición de base correspondientes.
Los compuestos dados a conocer en la presente memoria pueden sintetizarse generalmente mediante una combinación apropiada de métodos sintéticos generalmente bien conocidos. Las técnicas útiles para sintetizar dichas entidades químicas son fácilmente evidentes y accesibles para el experto en la materia relevante, basándose en la presente exposición. Muchos de los compuestos de partida opcionalmente sustituidos y otros reactivos se encuentran disponibles comercialmente, p.ej., de Aldrich Chemical Company (Milwaukee, WI) o pueden ser fácilmente preparados por el experto en la materia utilizando metodología sintética utilizada habitualmente.
La exposición de abajo se proporciona a fin de ilustrar algunos de la diversidad de métodos disponibles para la utilización en la preparación de los compuestos y no pretenden limitar el alcance de las reacciones o secuencias de reacción que pueden utilizarse en la preparación de los compuestos proporcionados en la presente memoria.
Métodos sintéticos generales
Los compuestos generalmente descritos en la presente memoria se entenderán más completamente haciendo referencia a los ejemplos siguientes.

Condiciones generales para la preparación de las (S)-3-(1-aminoetil)-isoquinolín-1(2H)-onas: A una mezcla bajo agitación de un ácido o-metilbenzoico dado (A-1) (1 eq., p.ej., 1,5 moles) y DMF (catalítico, p.ej., 2 ml) en DCM (1,2 M, p.ej., 1275 ml) a TA, se añadió cloruro de oxalilo (1,1 eq., p.ej., 1,65 moles) a lo largo de 5 min y la mezcla resultante se sometió a agitación a TA durante 2 h. A continuación, la mezcla se concentró al vacío. El residuo se disolvió en DCM (150 ml) y la solución resultante (solución A ) se utilizó directamente en la etapa siguiente.
A una mezcla bajo agitación de anilina (1,05 eq., p.ej., 1,58 moles) y trietilamina (2,1 eq, p.ej., 3,15 moles) en DCM (1,2 M, p.ej., 1350 ml), se añadió la solución anterior A (p.ej., 150 ml) gota a gota, manteniendo la temperatura de la reacción entre 25°C y 40°C con un baño de hielo-agua. La mezcla resultante se sometió a agitación a TA durante 2 h y después se añadió agua (p.ej., 1000 ml). Se separaron las capas orgánicas y se lavaron con agua (2x, p.ej., 1000 ml), se secaron sobre Na2SÜ4 y se filtraron. El filtrado se concentró al vacío. El producto se suspendió en n-heptanos (p.ej., 1000 ml) y se sometió a agitación a TA durante 30 min. Se recogió el precipitado mediante filtración, se enjuagó con heptanos (p.ej., 500 ml) y se secó adicionalmente al vacío, proporcionando la amida (A-2).
A una mezcla bajo agitación de amida (A-2) (1 eq., p.ej., 173 mmoles) en THF anhidro (p.ej., 250 ml) a -30°C bajo una
atmósfera de argón, se añadió una solución de n-butil-litio en hexanos (2,5 eq., 2,5 M, p.ej., 432 moles) gota agota durante 30 min, manteniendo simultáneamente la temperatura interna entre -30°C y -10°C. A continuación, la mezcla resultante se sometió a agitación a -30°C durante 30 min.
A una mezcla bajo agitación de 1-(metoxi(metil)amino)-1-oxopropán-2-ilcarbamato de (S)-terc-butilo (1,5 eq., p.ej., 260 mmoles) en THF anhidro (p.ej., 250 ml) a -30°C bajo una atmósfera de argón, se añadió una solución de cloruro de isopropilmagnesio en THF (1,65 eq., 1 M, p.ej., 286 mmoles) gota a gota durante 30 min, manteniendo simultáneamente la temperatura interna entre -30°C y -10°C. La mezcla resultante se sometió a agitación a -30°C durante 30 min. A continuación, dicha solución se añadió lentamente a la mezcla de reacción anteriormente indicada, manteniendo simultáneamente la temperatura interna entre -30°C y -10°C. La mezcla resultante se sometió a agitación a -15°C durante 1 h. La mezcla de reacción se desactivó con agua (p.ej. 50 ml) y después se acidificó con HCl con. a una temperatura de entre -10°C y 0°C para ajustar el pH a 1-3. Se dejó que la mezcla se calentase hasta la TA y se concentró al vacío. El residuo se disolvió en MeOH (p.ej., 480 ml) y después se añadió rápidamente HCl conc. (p.ej., 240 ml) a TA. La mezcla resultante se sometió a agitación bajo reflujo durante 1 h. La mezcla de reacción se concentró al vacío para reducir el volumen a aproximadamente 450 ml. El residuo se extrajo con una mezcla 2:1 de heptano y acetato de etilo (p.ej., 2 * 500 ml). La capa acuosa se basificó con hidróxido amónico concentrado para ajustar el valor del pH a 9-10, manteniendo simultáneamente la temperatura interna entre -10°C y 0°C. A continuación, la mezcla se extrajo con DCM (p.ej., 3x300 ml), se lavó con solución hipersalina, se secó sobre MgSO4 y se filtró. El filtrado se concentró al vacío y el residuo se disolvió en MeOH (p.ej., 1200 ml) a TA. A dicha solución se añadió ácido D-(-)-tartárico (0,8 eq., p.ej., 21 g, 140 mmoles) en una porción a TA. Tras someter a agitación a TA durante 30 min, precipitó un sólido blanco y la mezcla se mezcló en forma de suspensión a TA durante 10 h. Se recogió el sólido mediante filtración y se enjuagó con MeOH (p.ej., 3*50 ml). El sólido recogido se suspendió en agua (p.ej., 500 ml) y después se neutralizó con solución concentrada de hidróxido amónico a TA para ajustar el pH a 9-10. La mezcla se extrajo con DCM (p.ej., 3*200 ml). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre MgSO4 y se filtraron. El filtrado se concentró al vacío, proporcionando las (S)-3-(1-aminoetil)-isoquinolín-1(2H)-onas (A-3).

Método B:
condiciones generales para la preparación de 3-(aminometil)-isoquinolín-1(2H)-onas: Una mezcla de ácido benzoico (B-1) (1 eq., p.ej., 400 mmoles), cloruro de oxalilo (2 eq., p.ej., 101 g, 800 mmoles) y DMF (catalítico, p.ej., 0,2 ml) en Dcm (1 M, p.ej., 400 ml) se sometió a agitación a TA durante 2 h. La mezcla se concentró al vacío, proporcionando el cloruro de ácido (B-2). El producto obtenido se utilizó directamente en la etapa siguiente sin purificación adicional.
Una mezcla de amina R2NH2 (1,05 eq., p.ej., 420 mimóles) y trietilamina (1,7, p.ej., 700 mimóles) en DCM (1,4 M, p.ej., 300 ml) se sometió a agitación a TA durante 10 min. A esta mezcla se añadió cloruro de ácido (B-2) (1 eq., p.ej., 400 mmoles) gota a gota y la mezcla resultante se sometió a agitación a TA durante 30 min. La mezcla de reacción se vertió en agua (p.3j., 300 ml) y se extrajo con DCM (p.ej., 3x200 ml), se secó sobre Na2SO4 anhidro y se filtró. El filtrado se concentró al vacío, proporcionando el producto. El producto se suspendió en éter isopropílico (p.ej., 300 ml), se sometió a agitación bajo reflujo durante 30 min y después se enfrió a 0-5°C. El precipitado se recogió mediante filtración y se secó adicionalmente al vacío, proporcionando el producto amida (B-3).
A una solución bajo agitación de amida (B-3 (1,0 eQ:, p.ej., 0,1 moles) en THF anhidro (0,4 M, p.ej., 225 ml) a -78°C bajo una atmósfera de argón, se añadió una solución de n-butil-litio en hexanos (2,5 M, 3 eq., p.ej., 120 ml, 0,3 moles) gota a gota durante un periodo de tiempo de 1 h, manteniendo simultáneamente la temperatura interna entre -78°C y -50°C. La mezcla resultante se sometió a agitación a -70°C durante 1 h, y después se añadió rápidamente oxalato de dietilo (1,2 eq., p.ej., 17,5 g, 0,12 mmoles) (con un incremento de temperatura a -20°C con la adición). La mezcla se sometió a agitación a -50°C durante 10 min, y después se desactivó con agua (p.ej., 100 ml). Se separó la sal inorgánica mediante filtración y el filtrado se lavó con acetato de etilo (p.ej., 2x100 ml). Las capas orgánicas agrupadas se lavaron con solución hipersalina (p.ej., 100 ml), se secaron sobre MgSO4 y se filtraron. El filtrado se concentró al vacío, proporcionando el producto en forma de un semisólido. Se mezcló el producto en suspensión en éter isopropílico (p.ej., 100 ml) a TA durante 10 min. Se recogió el sólido mediante filtración y se secó adicionalmente al vacío, proporcionando el producto (B-4). El producto obtenido se utilizó directamente en la etapa siguiente.
El compuesto (B-4) (1 eq., p.ej., 88 mmoles) se disolvió a una concentración de 0,9 M eh HCl/MeOH (100 ml, p.ej., 10 M) y la mezcla resultante se sometió a agitación bajo reflujo durante 1 h. La mezcla de reacción se concentró al vacío y el residuo se mezcló en suspensión en acetato de etilo (100 ml) a TA durante 30 min. Se recogió el sólido mediante filtración, se enjuagó con acetato de etilo (3x50 ml) y se secó adicionalmente al vacío, proporcionando el producto (B-5).
A una suspensión bajo agitación de hidruro de litio-aluminio (3 eq., p.ej., 15,6 g, 410 mmoles) en THF anhidro (0,3 M, p.ej., 500 ml) a -78°C bajo una atmósfera de nitrógeno, se añadió lentamente B-5 (1 eq., 137 mmoles) durante un periodo de tiempo de 10 min. Se dejó que la mezcla resultante se calentase a -30°C y se sometió a agitación durante 30 min. A continuación, la mezcla se enfrió a -78°C, y se desactivó cuidadosamente con agua (p.ej., 100 ml). Se dejó que la mezcla se calentase hasta la TA, se filtró a través de gel de sílice (p.ej., 20 g) y el filtrado se concentró al vacío. La mezcla de productos se vertió en H2O (p.ej., 200 ml) y se extrajo con acetato de etilo (p.ej., 3*200 ml). Las capas orgánicas agrupadas se lavaron con solución hipersalina (p.ej., 100 ml), se secaron sobre Na2SO4 y se filtraron. El filtrado se concentró al vacío. El producto se suspendió en acetato de etilo (p.ej., 30 ml) y se sometió a agitación durante 10 min. Se recogió el sólido mediante filtración y se secó adicionalmente al vacío, proporcionando el producto (B-6).
Se disolvió tribromuro de fósforo (1,2 eq., p.ej., 3,42 g, 12,6 mmoles) y DMF (2,0 eq., p.ej., 1,6 g, 21,0 mmoles) en CH3CN (0,13 M, p.ej., 100 ml) y la mezcla resultante se sometió a agitación a -10°C durante 10 min. A esta mezcla se añadió alcohol (B-6) (1,0 eq, 10,5 mmoles) en partes. Se dejó que la mezcla resultante se calentase hasta la TA y se sometió a agitación durante 30 min adicionales. La mezcla de reacción se neutralizó con solución acuosa saturada de NaHCO3 a 0-5°C y después se filtró. El filtrado se extrajo con acetato de etilo (p.ej., 3*100 ml). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre Na2SO4 y se filtraron. El filtrado se concentró al vacío y el residuo se purificó mediante cromatografía flash de columna en gel de sílice (acetato de etilo al 20%-éter de petróleo), proporcionando el producto bromuro (B-7).
A una mezcla bajo agitación de ftalimida (1,1 eq., p.ej., 6,93 mmoles) en DMF (p.ej., 20 ml) a TA, se añadió en partes terc-butóxido potásico (1,5 eq., p.ej., 1,1 g, 9,45 mmoles) durante 10 min y después se añadió bromuro (B-7) (1,0 eq., p.ej., 6,3 mmoles). La mezcla resultante se sometió a agitación a 100°C durante 2 h. La mezcla de reacción se dejó enfriar hasta la TA y después se vertió en hielo-agua (p.ej., 30 ml). La mezcla se extrajo con acetato de etilo (p.ej., 3*20 ml). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre Na2SO4 y se filtraron. El filtrado se concentró al vacío y el residuo se purificó mediante cromatografía flash de columna en gel de sílice (acetato de etilo al 16%-éter de petróleo), proporcionando el producto diona (B-8).
Se disolvió la diona (B-8) (1,0 eq., p.ej., 1,5 mmoles) e hidrato de hidrazina (p.ej., 8,0 eq., 600 mg, 12 mmoles) en EtOH (p.ej., 20 ml) y la mezcla resultante se sometió a agitación bajo reflujo durante 1 h. Se dejó que la mezcla se enfriase hasta la TA y después se filtró. La torta de filtración se lavó con EtOH (p.ej., 10 ml). El filtrado agrupado se concentró al vacío y el residuo se purificó mediante cromatografía flash de columna en gel de sílice (p.ej., MeOH al 2,5%-DCM), proporcionando la amina (B-9).
(ii) Métodos generales de síntesis de amidas:

Método D:
A una mezcla de amina (D-1) (1,0 eq., p.ej., 0,5 mmoles), ácido carboxílico Wd-COOH (1,1 eq., p.ej., 0,55 mmoles) y N,N-diisopropiletilamina (2,0 eq., p.ej., 0,17 ml, 1,0 mmol) en DMF anhidro (p.ej., 5 ml), se añadieron secuencialmente hidrato de 1-hidroxibenzotriazol (1,3 eq., p.ej., 0,65 mmoles) e hidrocloruro de e Dc (1,3 eq., p.ej., 0,65 mmoles), y la mezcla resultante se sometió a agitación a TA durante 2 A 16 H. Se añadió hielo-agua o solución saturada de carbonato sódico a la mezcla de reacción y después se sometió a agitación durante 10 min. El precipitado se recogió mediante filtración, se enjuagó con agua y se secó al vacío. El sólido recogido se purificó adicionalmente mediante cromatografía flash de columna en gel de sílice (p.ej., MeOH a-10%-DCM), proporcionando el producto amida (D-2).
Método E:
Una solución de amina (D-1) (1 eq., p.ej., 0,25 mmoles), ácido carboxílico Wd-COOH (1,1 eq.) e hidrato de 1-hidroxibenzotriazol (1,3 eq.) en dimetilformamida (0,1 M) se trató con diisopropiletilamina (2 eq.), seguido de hidrocloruro de EDC (1,3 eq., p.ej.; 63 mg). La mezcla de reacción se sometió a agitación a temperatura ambiente durante la noche. La mezcla de mezcla de reacción se diluyó con agua (5x solvente) y ácido acético (1,5 eq.); después, la mezcla se sometió a agitación en un baño de hielo durante 40 min. El precipitado resultante se recogió mediante filtración y se lavó con agua (p.ej., 3x 3 ml). El sólido recogido se secó al vacío, proporcionando la amida (D-2).

Método F:
A una mezcla bajo agitación de ácido nitrobenzoico (F-1) (1,0 eq., 1,0 mol) y DMF (p.ej., 2,0 ml) en tolueno (p.ej., 800 ml), se añadió gota a gota cloruro de tionilo (4,0 eq., p.ej., 292 ml, 1,0 mol) (durante 15 min) y la mezcla resultante se sometió a agitación bajo reflujo durante 1,5 h. Se dejó que la mezcla se enfriase hasta la TA y después se concentró al vacío. El residuo se disolvió en DCM (p.ej., 100 ml), formando la solución A, que se utilizó directamente en la etapa siguiente.
A una mezcla bajo agitación de una amina dada R2-NH2 (1,1 eq, p.ej., 102,4 g, 1,1 moles) y trietilamina (2,0 eq., p.ej., 280 ml, 2,0 moles) en DCM (1,6 M, p.ej., 700 ml), se añadió solución A gota a gota, manteniendo simultáneamente una temperatura de reacción inferior a 10°C. Se dejó que la mezcla resultante se calentase hasta la TA y después se sometió a agitación a TA durante la noche. La mezcla de reacción se diluyó con hielo-agua (p.ej., 1,0 l) y se sometió a agitación durante 15 min. El precipitado se recogió mediante filtración, se enjuagó con éter isopropílico (p.ej., 3x100 ml) y éter de petróleo (p.ej., 3x100 ml) y después se secó al vacío, proporcionando el producto amida (F-2).
A una mezcla de nitrobenzamida (F-2) (1,0 eq., p.ej., 20,0 mmoles) y DMF (cat.) en tolueno (0,3 M, p.ej., 60 ml) a TA,
se añadió gota a gota (durante 5 min) cloruro de tionilo (8,2 eq., p.ej., 12 ml, 164 mmoles) y la mezcla resultante se sometió a agitación bajo reflujo durante 2 h. Se dejó que la mezcla se enfriase hasta la TA y después se concentró al vacío. El residuo se disolvió en DCM (p.ej., 100 ml), formando la solución B, que se utilizó directamente en la etapa siguiente.
A una mezcla bajo agitación of N-(terc-butoxicarbonil)-L-alanina (0,8 eq., p.ej., 16,0 mmoles) y N,N-diisopropiletilamina (1,5 eq., p.ej., 4,0 g, 31,0 moles) en DCM (0,8 M, p.ej., 20 ml), se añadió la solución B gota a gota, manteniendo simultáneamente la temperatura de reacción entre 0-10°C. La mezcla resultante se sometió a agitación a dicha temperatura durante 1 h y después se sometió a agitación a TA durante la noche. La mezcla de reacción se desactivó con hielo-agua (p.ej., 100 ml). Se separó la capa orgánica y se extrajo la capa acuosa con DCM (p.ej., 2 x 80 ml). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre Na2SO4 y se filtraron. El filtrado se concentró al vacío y el residuo se suspendió en éter isopropílico (p.ej., 100 ml9 durante 15 min. Se recogió el sólido mediante filtración y se secó al vacío, proporcionando el producto (F-3).
A una suspensión de polvos de cinc (10,0 eq., p.ej., 7,2 g, 110 mmoles) en ácido acético glacial (2,8 M, p.ej., 40 ml) a 15°C, se añadió una solución de F-3 (1,0 eq., p.ej., 11,0 mmoles) en ácido acético glacial (0,3 M, p.ej., 40 ml) y la mezcla resultante se sometió a agitación a Ta durante 4 h. La mezcla se vertió en hielo-agua (p.ej., 200 ml) y se neutralizó con solución acuosa saturada de NaHCO3 para ajustar el pH a 8. La mezcla resultante se extrajo con DCM (p.ej., 3x150 ml). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre Na2SO4 y se filtraron. El filtrado se concentró al vacío y el residuo se purificó mediante cromatografía flash de columna en gel de sílice (acetato de etilo al 7%-éter de petróleo), proporcionando el producto (F-4).
El compuesto (F-4) (1,0 eq., p.ej., 0,5 mmoles) se disolvió en solución hidroclórica de metanol (8 eq., p.ej., 2 N, 20 ml) y la mezcla resultante se sometió a agitación a TA durante 2 h. La mezcla se concentró al vacío. El residuo se diluyó con agua (30 ml) y después se neutralizó con solución acuosa saturada de NaHCO3 para ajustar el pH a 8, manteniendo simultáneamente una temperatura inferior a 5°C. La mezcla resultante se extrajo con DCM (p.ej., 3x30 ml). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre Na2SO4y se filtraron. El filtrado se concentró al vacío y el residuo se suspendió en éter de petróleo (p.ej., 10 ml). Se recogió el sólido mediante filtración y se secó al vacío, proporcionando el producto (F-5).
La quinazolinona (F-5) puede utilizarse para sintetizar compuestos indicados en la presente memoria, utilizando, por ejemplo, el método D para acoplar la amina a grupos Wd.

Método FF
Alternativamente, pueden prepararse compuestos con un núcleo quinazolinona siguiendo los procedimientos en la publicación de patente Pc T n° WO2013082540.
En el método FF, se disolvió el ácido 2-amino-6-clorobenzoico (63 mmoles, 1,0 equiv.) en acetonitrilo (60 ml) en un matraz de fondo redondo de 250 ml, sometido a una atmósfera de Ar y calentado a 50°C. Se añadió piridina (2,0 equiv.), seguido de la adición gota a gota de una solución de trifosgeno (0,34 equiv. en 30 ml de acetonitrilo), manteniendo simultáneamente una temperatura interna inferior a 60°C. A continuación, la mezcla se sometió a agitación a 50°C durante 2 h, punto en el que se eliminó el solvente al vacío. El residuo restante se dispersó en 50 ml de agua y se filtró. Se lavó el sólido resultante con una cantidad mínima de acetonitrilo para eliminar la decoloración y después se secó, proporcionando el anhídrido deseado X-1.
Se suspendió el anhídrido X-1 (25,5 mmoles, 1,0 equiv.) en dioxano (40 ml) bajo una atmósfera de Ar en un matraz de fondo redondo de 200 ml. Se añadió anilina (1,0 equiv.) gota a gota. Se inició el calentamiento a 40°C y se incrementó gradualmente a 100°C. Tras 4 h, se había consumido la mayor parte del material de partida, momento en
que se dejó que la reacción se enfriase. A continuación, se eliminó el solvente bajo vacío, proporcionando un aceite que se redisolvió en tolueno, seguido de la adición de hexanos hasta que aparece el solvente próximo a la repartición. La mezcla se sometió a agitación durante 14 h, punto en el que apareció un sólido en el matraz. Se aisló dicho sólido mediante filtración al vacío y se lavó con hexanos, proporcionando la amida X-2 deseada con un rendimiento elevado.
Se disolvió ácido (S)-2-((?erc-6ufox/carbon¡l)am¡no)propano¡co (33,0 mmoles, 2,0 equiv.) en tetrahidrofurano seco (70 ml) bajo una atmósfera de Ar, seguido de la adición gota a gota de N-metilmorfolina (2,2 equiv.). A continuación, se enfrió la mezcla a -17°C en un baño de acetona/hielo seco, seguido de la adición gota a gota de una solución de cloroformato de isobutilo (2,0 equiv. en 10 ml de tetrahidrofurano seco) a la mezcla, seguido de agitación durante 30 min. A continuación, se añadió una solución de amina X-2 (10 equiv. en 10 ml de tetrahidrofurano seco). Seguidamente se retiró el baño de hielo seco y la mezcla se sometió a agitación a TA durante 90 min. A continuación, se calentó a 60°C durante 2 h adicionales, punto en que se dejó enfriar. A continuación, se añadieron MTBE (150 ml) y agua (150 ml) sucesivamente bajo agitación vigorosa. Se separaron las fases y la fase orgánica se lavó con agua (2x50 ml) y solución hipersalina (50 ml), y se secaron sobre sulfato sódico. A continuación, la solución se concentró bajo presión reducida y el residuo en bruto se purificó mediante cromatografía flash en gel de sílice (gradiente 5-30 de acetato de etilo/hexanos), obteniendo X-3 como el producto acoplado.
A continuación, se suspendió el compuesto X-3 (4,9 mmoles, 1,0 equiv.) en acetonitrilo (100 ml). Seguidamente se añadió trietilamina (48 equiv.) bajo agitación, seguido de la adición gota a gota de clorotrimetilsilano (15 equiv.)- A continuación, se selló el matraz y se calentó a 90°C durante 3 días. Se dejó que la reacción se enfriase, seguido de la eliminación del solvente al vacío. A continuación, el residuo se disolvió en acetato de etilo (120 ml) y se lavó sucesivamente con carbonato sódico saturado (1x100 ml), agua (1x100 ml) y solución hipersalina (1*100 ml). A continuación, la capa orgánica se secó sobre sulfato sódico anhidro y se concentró bajo presión reducida, proporcionando el producto ciclizado X-4. El producto puede utilizarse directamente en reacciones posteriores o purificarse mediante cromatografía flash en gel de sílice.

Método G:
Condiciones generales para la preparación de (S)-3-(1-aminoetil)l-8-(trifluorometil)isoquinolín-1(2H)-onas: A una suspensión de ácido 2-amino-6-metilbenzoico (G-1) (20,0 g, 132,0 mmoles, 1,0 eq.) en H2O (55 ml) a 0-5°C, se añadió lentamente HCl conc. (al 36,5 %, 64 ml, 749 mmoles, 5,7 eq.). Tras someter a agitación durante 15 min, la mezcla se añadió gota a gota a una solución de nitrito sódico (12,02 g, 174,0 mmoles, 1,32 eq.) en H2O (36 ml) a 0-5°C, y la mezcla resultante se sometió a agitación durante 1 h. La solución resultante seguidamente se añadió a una solución de KI (60,5 g, 364,5 mmoles, 2,76 eq.) en H2O (150 ml) a 0-5°C. Se dejó que la mezcla de reacción se calentase hasta la TA y se sometió a agitación a TA durante la noche. La mezcla se extrajo con acetato de etilo (3*100 ml). Las capas orgánicas agrupadas se lavaron con agua (2*100 ml), se secaron sobre Na2SO4 anhidro y se filtraron. El filtrado se concentró al vacío y el residuo se purificó mediante cromatografía flash de columna en gel de sílice (0 20 % de acetato de etilo-éter de petróleo), proporcionando el producto ácido 2-yodo-6-metilbenzoico (G-2).
A una mezcla bajo agitación de ácido 2-yodo-6-metilbenzoico (G-2) (305,3 mmoles, 1,0 eq.) y DMF (0,3 ml) en DCM (350 ml) a TA, se añadió cloruro de oxalilo (466,4 mmoles, 1,5 eq.) gota a gota. La mezcla resultante se sometió a agitación a TA durante 3 h y después se concentró al vacío. El residuo se disolvió en DCM (150 ml) y la solución resultante (solución A) se utilizó directamente en la etapa siguiente.
A una mezcla bajo agitación of anilina R3-sustituida (335,7 mmoles, 1,1 eq.) y trietilamina (915,0 mmoles, 3,0 eq.) en
DCM (350 ml), se añadió solución A (150 ml) gota a gota, controlando simultáneamente la temperatura de reacción a un valor inferior a 30°C con un baño de hielo-agua. La mezcla de reacción se sometió a agitación a TA durante 1 h y después se desactivó con agua (200 ml). Se separó la capa orgánica, se lavó con agua (2x200 ml), se secó sobre Na2SO4 anhidro y se filtró. El filtrado se concentró al vacío. Se enjuagó el producto con éter isopropílico y se secó al vacío, proporcionando el producto amida (G-3).
Una mezcla de amida (G-3) (18,0 mmoles, 1,0 eq.), 2,2-difluoro-2-(fluorosulfonil)acetato de metilo (72,9 mmoles, 4,0 eq.) y CuI (3,63 mmoles, 0,2 eq.) en DMF (130 ml) se sometió a agitación a 70°C bajo una atmósfera de argón durante la noche. Se dejó que la mezcla se enfriase hasta la TA y después se concentró al vacío para eliminar el solvente. El residuo resultante se repartió entre acetato de etilo (60 ml) y agua (60 ml), y la capa acuosa se extrajo con acetato de etilo (2x60 ml). Las capas orgánicas agrupadas se lavaron con agua (2x60 ml), se secaron sobre Na2SO4 anhidro y se filtraron. El filtrado se concentró al vacío y el residuo se purificó mediante cromatografía flash de columna en gel de sílice, proporcionando el producto, trifluorometilamida (G-4).
A una mezcla bajo agitación de amida (G-4) (10,1 mmoles, 1,0 eq.) en THF anhidro (25 ml) a -40°C bajo una atmósfera de argón, se añadió gota a gota una solución de n-butil-litio en t Hf (2,5 M, 25,3 mmoles, 2,5 eq.) (durante 15 min) y se controló la temperatura interna entre -30°C y -20°C durante la adición. La mezcla resultante se sometió a agitación a -30°C durante 1 h adicional. A una mezcla bajo agitación de 1-(metoxi(metil)amino)-1-oxopropán-2-ilcarbamato de (S)-ferc-butilo (11,1 mmoles, 1,1 eq.) en THF anhidro (20 ml) a -30°C bajo una atmósfera de argón, se añadió gota a gota (durante 15 min) una solución de cloruro de isopropilmagnesio en THF (12,6 mmoles, 12,5 eq.) y se controló la temperatura interna en un valor inferior a -20°C durante la adición. La mezcla resultante se sometió a agitación a -15°C durante 1 h. A continuación, dicha solución se añadió lentamente a la mezcla de reacción anteriormente indicada a -30°C (durante 10 min) y la mezcla resultante se sometió a agitación a -30°C durante 30 min adicionales. La mezcla de reacción se desactivó con agua (50 ml) y después se acidificó con HCl conc. a -5°C para ajustar el pH a 5. Se dejó que la mezcla se calentase hasta la TA y se concentró al vacío. El residuo se disolvió en MeOH (10 ml) y después se añadió rápidamente HCl conc. (10 ml) a TA. La mezcla resultante se sometió a agitación bajo reflujo durante 2 h, se enfrió hasta la TA Y después se concentró al vacío. El residuo se suspendió en agua (15 ml), se basificó con hidróxido amónico concentrado para ajustar el pH a 9-10, manteniendo simultáneamente una temperatura interna inferior a 5°C y después se extrajo con DCM (3x15 ml). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre MgSO4 y se filtraron. El filtrado se concentró al vacío y el residuo se disolvió en MeOH (70 ml). A dicha solución, se añadió ácido D-(-)- tartárico (8,1 mmoles, 0,8 eq.) en una porción a TA. Tras someter a agitación a TA durante 30 min, precipitó un sólido y la mezcla se mezcló en forma de suspensión a TA durante 10 h. Se recogió el sólido mediante filtración y se enjuagó con MeOH (3x40 ml). El sólido recogido se suspendió en agua (30 ml) y después se neutralizó con solución concentrada de hidróxido amónico a TA para ajustar el pH a 9-10. La mezcla is se extrajo con DCM (3x15 ml). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre MgSO4y se filtraron. El filtrado se concentró al vacío, proporcionando el producto, (S)-3-(1-aminoetil)-8-(trifluorometil)isoquinolín-1(2H)-ona (G-5).

Método H
Condiciones generales para la preparación de (S)-3-(1-aminoetil)-isoquinolín-1(2H)-onas: Un ácido o-metilbenzoico (H-1) (1 eq., p.ej., 46,9 mmoles) en un matraz de fondo redondo seco a la llama, bajo nitrógeno se disolvió en t Hf (1 M, p.ej., 50 ml). La solución amarilla homogénea resultante se enfrió a -25°C y se añadió lentamente n-hexil-litio (4,3 eq, p.ej., 202 mmoles; 2,3 M en hexanos); en este punto la solución se tornó de color rojo oscuro y se sometió a agitación a -20°C durante 20 min.
Se cargó 1-(metoxi(metil)amino)-1-oxopropán-2-ilcarbamato de (S)-terc-butilo (1,3 eq., p.ej., 61,0 mmoles) en un segundo matraz de fondo redondo seco bajo N2 y se suspendió en 70 ml de THF seco y se enfrió a -10°C. Se añadió lentamente cloruro de isopropilmagnesio (2 M, 2,7 eq, p.ej., 127 mmoles), resultando en una solución amarillo transparente. A continuación, dicha solución se añadió lentamente gota a gota mediante una cánula al primer matraz de fondo redondo. Tras completar la adición, la solución oscura se calentó lentamente hasta la TAy se sometió a
agitación a Ta DURANTE 2 H. A continuación, la mezcla de reacción se enfrió nuevamente a -10°C y se introdujo mediante cánula rápidamente en otro matraz con acetato de etilo (p.ej., 15 ml) y ácido isobutírico (p.ej., 10 ml) a -10°C bajo N2. Durante este tiempo, la mezcla cambió de naranja y turbia a transparente y homogénea. Tras la adición, la mezcla se sometió a agitación durante 5 min, seguido de la adición rápida de agua (p.ej., 10 ml) y se sometió a agitación vigorosa durante 10 min a TA.
La mezcla a continuación se transfirió a un embudo de separación y se añadió agua (p.ej., 200 ml) para disolver las sales (pH ~ 9). La capa acuosa se extrajo con EtOAc (p.ej., 3 * 400 ml). A continuación, se acidificó la capa acuosa con HCl (2 M) a pH 3, y después se extrajo con EtOAc (p.ej., 3x500 ml), se secó sobre sulfato sódico y se concentró, proporcionando material en bruto, que se filtró al vacío a través de una almohadilla de gel de sílice utilizando MeOH/DCM (gradiente de 2-10 % MeOH), proporcionando el ácido H-2 tras la concentración.
Se llenó un matraz de fondo redondo de 50 ml con una barra de agitación con ácido benzoico H-2 (1 eq., p.ej., 14,63 mmoles) en anhídrido acético (1,5 M, p.ej., 10 ml) y después se sometió a agitación a 70°C durante 2,5 horas hasta la conversión completa en el producto, indicada por la CL/EM. Se evaporó el anhídrido acético bajo presión reducida y el residuo en bruto se purificó mediante Combiflash (gradiente de EtOAc/hexanos), proporcionando la lactona H-3.
Un matraz de fondo redondo seco de 50 ml con una barra de agitación se llenó con amina R2NH2 (5,1 eq., p.ej., 1,54 mmoles) en 2 ml de DCM (0,8 M), seguido de la adición de trimetilaluminio (5,1 eq., p.ej., 1,54 mmoles) a la solución y se sometió a agitación durante 15 min. A continuación, se añadió una solución de lactona H-3 (1,0 eq, p.ej., 0,31 mmoles) en DCM (1,5 M, p.ej., 2 ml). A continuación, la mezcla se sometió a agitación a TA durante 3 h hasta que el análisis de CL/EM mostró que se había completado la formación del producto deseado La mezcla de reacción se desactivó con 10 ml de sal de Rochelle y se sometió a agitación durante 2 h. La mezcla seguidamente se diluyó con DCM, se lavó con solución hipersalina, se secó sobre sulfato sódico y se evaporó, proporcionando un líquido pegajoso amarillo H-4 que se utilizó directamente en la etapa siguiente.
A la amida H-4 (1 eq., p.ej., 0,31 mmoles) en isopropanol (0,06 M, p.ej., 5 ml) se añadieron 3 ml de HCl concentrado (300 eq.). A continuación, se calentó la mezcla en un baño de hielo a 65°C durante 3 h hasta que la CL/EM mostró que no quedaba material de partida. A continuación, se retiró el matraz de la fuente de calor y se evaporaron los solventes bajo presión reducida, proporcionando un sólido amarillo H-5, que se utilizó directamente en transformaciones posteriores.
(iii) Métodos generales para la síntesis de alquinos:
Arilo Arilo

X = Br, I TMS H
1-1 I-2
Método I
Se cargó un recipiente sellado con PdCh(MeCN)2 y X-Phos (proporción 3:1 de X-Phos a PdCh(MeCN)2, 5-15 % molar de catalizador), carbonato de cesio (1,5-3,0 equiv.) y propionitrilo (0,5 M). La mezcla se sometió a agitación durante 5 min, seguido de la adición de sustrato de bromuro de arilo o yoduro de arilo. Tras 5 minutos adicionales de agitación, se añadió TMS-acetileno (3,0 equiv.) y el matraz se selló y se calentó a TA durante 10 min, seguido de 1 h de calentamiento a 95°C. Se dejó que se enfriase la reacción, seguido de su concentración directa sobre gel de sílice y purificación mediante cromatografía flash en gel de sílice (gradiente de acetato de etilo/hexanos), proporcionando el alquino I-1.
A continuación, se disolvió el alquino I-1 (1,0 equiv.) en tetrahidrofurano (0,13 M) y se cargó con TBAF (1,1 equiv., 1,0 M en tetrahidrofurano). La mezcla resultante se sometió a agitación a TA durante 6 h; a continuación, se vertió en solución saturada de bicarbonato y se extrajo con acetato de etilo. La capa orgánica se lavó con solución hipersalina y se concentró sobre gel de sílice, en donde se purificó directamente mediante cromatografía flash en gel de sílice (gradiente de acetato de etilo/hexanos), proporcionando el arilalquino I-2.
Método J

Se disolvió aldehido (1,0 equiv.) en metanol anhidro (0,2-0,5 mM) y se cargó con carbonato de cesio (1,0 equiv.) y se enfrió a 0-5°C. Se añadió gota a gota (1-diazo-2-oxopropil)fosfonato de dimetilo (1,0 equiv.), y después la reacción se dejó bajo agitación durante 1 a 18 h; a continuación, la mezcla en bruto se concentró sobre gel de sílice y se purificó directamente mediante cromatografía flash en gel de sílice, proporcionando el alquino deseado J-1.
Método K

K-1
Se disolvió una amina secundaria (1,0 equiv.) en acetonitrilo (0,42 M) y se añadió carbonato potásico (1,1 equiv.). La suspensión blanca se sometió a agitación a 0-5°C durante 5 min, punto en el que se añadió bromuro de propargilo (1,01 equiv.) gota a gota durante 3 min. A continuación, la reacción se sometió a agitación durante 15 min adicionales a 0-5°C y después a temperatura ambiente durante 15 h. A continuación, se filtró la mezcla heterogénea. El filtrado se concentró bajo presión reducida, se diluyó con MTBE y se lavó con agua (2x), solución hipersalina (1x), y se secó sobre sulfato sódico y después se filtró a través de Celite. El filtrado resultante se concentró y se purificó mediante cromatografía flash en gel de sílice, proporcionando el alquino deseado K-1.
Ejemplo 1

Se preparó el Compuesto 4 en 3 etapas a partir del compuesto A siguiendo los procedimientos siguientes: Se preparó el Compuesto A de acuerdo con el Método A. Se acopló con el ácido 2-((terc-butoxicarbonil)amino)pirazolo[1,5-a]pirimidín-3-carboxílico siguiendo el procedimiento siguiente: El Compuesto A (27,4 mmoles, 1,0 equiv.), hidrato de HOBt (1,2 equiv.), ácido 2-((terc-butoxicarbonyl)amino)pirazolo[1,5-a]pirimidín-3-carboxílico (1,05 equiv.) y EDC (1,25 equiv.) se añadieron a un matraz de fondo redondo de 200 ml con barra de agitación. Se añadió N,N-dimetilformamida (50 ml) y la suspensión se sometió a agitación a TA durante 2 min. Se añadió base de Hunig (4,0 equiv.), punto en el que la suspensión se tornó homogénea, y se sometió a agitación durante 22 h, resultando en la formación de una torta sólida en el matraz de reacción. Se añadió la mezcla sólida a agua (600 ml) y se sometió a agitación durante 3 h. El sólido de color crema resultante se filtró y se lavó con agua (2x100 ml) y se secó. A continuación, se disolvió el sólido en cloruro de metileno (40 ml), seguido de la adición de ácido trifluoroacético (10 equiv., 20 ml) y la reacción se sometió a agitación durante 30 min a TA, punto en el que no queda más material de partida según el análisis de CL/EM. A continuación, la solución se concentró y se coevaporó con una mezcla de cloruro de metileno/etanol (1:1 v/v) y después se secó bajo alto vacío durante la noche. El sólido resultante se trituró con 60 ml de etanol durante 1 h y después se recogió mediante filtración al vacío. A continuación, el sólido beige se neutralizó con solución de carbonato sódico (100 ml) y después se transfirió a un embudo de separación con cloruro de metileno (350 ml). La capa acuosa se extrajo con 100 ml adicionales de cloruro de metileno. Las capas orgánicas agrupadas se secaron sobre sulfato sódico, se filtraron y se concentraron al vacío, proporcionando un sólido amarillo pál(ido que se purificó mediante
cromatografía flash en gel de sílice (Combiflash, columna de 24 g, gradiente de 0-5 % de metanol/cloruro de metileno), proporcionando la amida B. EM-IEP m/z: 459,4 [M+H]+.
Se introdujo la amida B en un tubo sellado (0,67 mmoles, 1,0 equiv.), seguido de diclorobis(acetonitrilo)paladio (15 % molar), X-Phos (45 % molar) y carbonato de cesio (3,0 equiv.). Se añadió propionitrilo (5 ml) y la mezcla se burbujeó con Ar durante 1 min. Se añadió 4-etinil-1-metil-1H-pirazol (1,24 equiv.) y la mezcla naranja resultante se selló y se sometió a agitación en un baño de aceite a 85°C durante 1,5 h. La mezcla parduzca-negra resultante se dejó enfriar, punto en el que ya no quedaba material de partida según el análisis de c L/EM. A continuación, se filtró la mezcla a través de un tapón corto de algodón, utilizando acetonitrilo y cloruro de metileno. Los filtrados agrupados se concentraron sobre gel de sílice y se purificaron utilizando cromatografía flash en gel de sílice (columna Combiflash de 4 g, gradiente de 0-5 % cloruro de metileno/metanol). El material resultante se purificó adicionalmente mediante HPLC de fase inversa (15-90% acetonitrilo con ácido fórmico al 0,1 %/agua con ácido fórmico al 0,1 %), proporcionando el compuesto 4 deseado. EM-IEP m/z: 529,5 [M+H]+.
Se prepararon los compuestos siguientes de manera análoga. Los alquinos se encontraban disponibles comercialmente o se prepararon utilizando el método I, J o K tal como se describe en la presente memoria.

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

Ejemplo 2

Se preparó el compuesto A de acuerdo con el método F. A continuación, se convirtió en el compuesto AA1 mediante un procedimiento análogo al del compuesto B en el Ejemplo 1. A continuación, se preparó el compuesto 1 a partir del compuesto AA1 en dos etapas siguiendo los procedimientos siguientes: Se suspendió el compuesto a A 1 (0,55 mmoles, 1,0 equiv.), PdCl2(MeCN)2 (10 % molar), X-Phos (30 % molar) y carbonato de cesio (2,6 equiv.) en proprionitrilo (4 ml). La mezcla se burbujeó con Ar durante 25 min; a continuación, se añadió trimetil(propargil)silano (1,3 equiv.) y la reacción se selló y concentró a 90°C. Se dejó que se calentase la mezcla durante 4,5 h, y a continuación se enfrió y se repartió entre acetato de etilo y agua. Se separaron las capas y la capa acuosa se extrajo con acetato de etilo (2x). Se agruparon las capas orgánicas, se secaron sobre sulfato sódico y se concentraron sobre gel de sílice (2g). A continuación, el material en bruto se purificó mediante cromatografía flash en gel de sílice (ISCO Combiflash Si-12 g, gradiente de 10-55 % acetona/cloruro de metileno), proporcionando una mezcla de compuesto B y compuesto 1 desprotegido.
La mezcla (0,23 mmoles, 1,0 equiv.) se redisolvió en tetrahidrofurano anhidro (6 ml). Se añadió TBAF en THF (1,0 M, 1,2 equiv.) y la mezcla resultante se sometió a agitación a TA durante 45 min hasta la conversión completa en compuesto 1 según el análisis de CCF. A continuación, la reacción se concentró sobre gel de sílice (1 g) y se purificó mediante cromatografía flash en gel de sílice (Interchim Si-25g HP silicycle, gradiente de 14-45 % acetona/cloruro de metileno), proporcionando el compuesto 1. EM-IEP m/z 464,1 [M+H]+

Se preparó el compuesto kk a partir del compuesto A (Ejemplo 2) bajo condiciones de protección con Boc estándares. A continuación, se convirtió en el compuesto II utilizando un procedimiento de acoplamiento análogo para el compuesto B en el Ejemplo 2, excepto en que se utilizó 3,3-dimetilbut-1-ino en lugar de trietilsililacetileno, proporcionando el compuesto II.
Se disolvió el compuesto 11 (0,094 mmoles, 1,0 equiv.) en cloruro de metileno anhidro (2 ml). Se añadió ácido trifluoroacético (400 pl, 55 equiv.) y la reacción se dejó bajo agitación a TA durante 2 h, punto en el que no quedaba más material de partida según el análisis de CL/e M. La reacción se desactivó cuidadosamente con solución de bicarbonato sódico y la capa acuosa se extrajo con cloruro de metileno (2x). Las capas orgánicas agrupadas se secaron con sulfato sódico y se concentraron. El material en bruto se purificó mediante cromatografía de fase inversa (Interchim, gradiente de acetonitrilo y agua con ácido fórmico al 0,1 %), proporcionando la amina libre, que a continuación se acopló con ácido 2-((terc-butoxicarbonil)amino)pirazolo[1,5-a]pirimidín-3-carboxílico mediante el método D, seguido de la desprotección de Boc nuevamente bajo condiciones análogas a las del Ejemplo 11, proporcionando el compuesto deseado 3. EM-IEP m/z 505,1 [M+H]+
Ejemplo 4

Una solución de 3-butín-2-ol (10 ml, 128 mmoles) en N,N-dimetilformamida (20 ml) se añadió a lo largo de 30 minutos a una suspensión bajo agitación de hidruro sódico (dispersión al 60 % en aceite mineral (7,65 g, 2,5 equiv.) en N,N-dimetilformamida (100 ml) a 0°C bajo una atmósfera de argón. Tras 30 min, se añadió sulfato de dimetilo (1,5 equiv.) a lo largo de 30 min a 0°C. A continuación, la mezcla se sometió a agitación durante 30 min a 0°C, seguido de la adición lenta de ácido acético (1,05 equiv.) y se dejó que la reacción se calentase hasta la temperatura ambiente bajo agitación durante 2 h adicionales. Se aisló el producto mediante destilación fraccionada directamente a partir de la mezcla de reacción (58-63°C), proporcionando el éter 4-a, que se utilizó directamente en la etapa siguiente. A continuación, se acopló el compuesto 4-a con el compuesto A bajo condiciones de Sonogashira análogas a las del Ejemplo 1, generando el compuesto 22. EM (IEP) m/z: 507,5 [M+H]+
Ejemplo 5

Se preparó el compuesto 25 de manera análoga al compuesto B del Ejemplo 1. A continuación, se acopló con 4-etinil-1-metiMH-pirazol bajo condiciones de Sonogashira en el Ejemplo 1, proporcionando el compuesto 25. EM (IEP) m/z: 493,4 [M+H]+
Ejemplo 6

El compuesto 23 se preparó de manera análoga al compuesto 25 del Ejemplo 5, excepto en que se utilizó 5-etinil-1-metil-1H-imidazol en lugar de 4-etinil-1-metil-1H-pirazol. EM (IEP) m/z: 493,4 [M+H]+
Ejemplo 7

Se preparó el compuesto 24 de manera análoga al compuesto 25 del Ejemplo 5, excepto en que se utilizó etinilciclopropano en lugar de 4-etinil-1-metil-1H-pirazol. EM-IEP m/z 453,4 [M+H]+

Se aisló el compuesto 44 como producto secundario del Ejemplo 5. EM-IEP m/z 453,4 [M+H]+
Ejemplo 9

Se preparó el compuesto 21 a partir del compuesto AA1 utilizando condiciones de acoplamiento análogas para la preparación del compuesto 4 en el Ejemplo 1. EM-IEP m/z 530,2 [M+H]+
Ejemplo 10

Se acopló ácido 3-aminopirazín-2-carboxílico con el compuesto A mediante la utilización del método D, proporcionando el compuesto 10-a. A continuación, se convirtió en el compuesto 29 bajo condiciones de acoplamiento análogas a las utilizadas para la preparación del compuesto 4 del Ejemplo 1. EM-IEP m/z 490,3 [M+H]+
Ejemplo 11

Se acopló ácido pirazolo[1,5-a]pirimidín-3-carboxílico con el compuesto A mediante la utilización del método D, proporcionando el compuesto 11 -a. A continuación, se convirtió en el compuesto 39 bajo condiciones de acoplamiento análogas a las utilizadas para la preparación del compuesto 4 del Ejemplo 1. EM-IEP m/z 514,4 [M+H]+
Ejemplo 12

Se acopló el ácido 1,5-naftiridín-4-carboxílico con el compuesto A mediante la utilización del método D, proporcionando el compuesto 12a. A continuación, se convirtió en el compuesto 42 mediante la utilización de condiciones de acoplamiento análogas para la preparación de compuesto 4 del Ejemplo 1. EM-IEP m/z 525,3 [M+H]+
Ejemplo 13

Se disolvió el compuesto 13-a (0,058 mmoles, 1,0 equiv.) en acetonitrilo anhidro (2 ml). Se añadió yoduro sódico (1,5 equiv.), seguido de TMS-Cl (1,5 equiv), momento en que la solución se convirtió en una suspensión amarilla. A continuación, la mezcla se calentó a 65°C durante 5 h, y transcurrido este periodo ya no quedaba más material de partida según el análisis de CL/EM. Se dejó que se enfriase la reacción y se vertió en agua (4 ml) y se sometió a
agitación durante 15 min, seguido de la repartición entre agua y cloruro de metileno. A continuación, la capa orgánica se secó y se concentró. El material en bruto se purificó mediante HPLC de fase inversa (Interchim, gradiente de 10-90% acetonitrilo/agua con ácido fórmico al 0,1 %), proporcionando el compuesto 31 deseado. EM-IEP m/z 542,4 [M+H]+
Ejemplo 14

Se preparó el compuesto 33 a partir del compuesto 14-a mediante la utilización de condiciones análogas a las del Ejemplo 13. EM-IEP m/z 542,4 [M+H]+
Ejemplo 15

El compuesto 34 (0,47 mmoles, 1,0 equiv.) se disolvió en acetona (5 ml) y agua (4 ml). Se añadió ácido ptoluenosulfónico (al 25 % molar) y la mezcla turbia se calentó a 50°C. A continuación, se dejó que la mezcla se enfriase, seguido de la eliminación de la mayor parte del solvente al vacío. Seguidamente, se repartió el residuo entre cloruro de metileno y bicarbonato sódico saturado. Se separó la capa orgánica y se adsorbió sobre SiO2 (3 g), seguido de la purificación mediante cromatografía flash en gel de sílice (columna ISCO, 24 g de Si, gradiente de 25-100 % acetato de etilo/hexanos), proporcionando el aldehido 36 deseado. EM-IEP m/z: 477,2 [M+H]+.
Ejemplo 16
Se disolvió 5-etiniMH-pirazol (1,1 mmoles, 1,0 equiv.) en cloruro de metileno (10 ml). A continuación, se añadió trietilamina (3,0 equiv.) y anhídrido de Boc (1,0 equiv.) y la reacción se dejó bajo agitación durante 2 h. Se añadió agua (100 ml) y la mezcla se transfirió a un embudo de separación. Se separaron las capas y la capa acuosa se lavó con agua (2 x 20 ml). Se secaron las capas orgánicas sobre MgSO4 y se concentraron, proporcionando alquino 16-a, que se utilizó directamente en la etapa siguiente.
Se cargó un matraz presurizado (15 ml) con compuesto B (0,22 mmoles, 1,0 equiv.), X-Phos (45 % molar), diclorobis(acetonitrilo)Pd (15 % molar), y carbonato de cesio (1,1 equiv.) bajo un flujo de N2. Se añadió propionitrilo (3 ml) y la solución se burbujeó con Ar durante 1 min. A continuación, se añadió alquino 16-a (2,5 equiv), seguido de anhídrido de Boc (1,0 equiv.) y la reacción se selló y se concentró a 100°C durante 1 h. A continuación, la reacción se filtró y se concentró. El residuo se redisolvió en cloruro de metileno (3 ml), seguido de la adición de ácido trifluoroacético (800 ^l) y la mezcla se sometió a agitación durante 1 h. A continuación, la reacción se concentró sobre gel de sílice y se purificó mediante cromatografía flash en gel de sílice (gradiente de 0-30 % metanol/cloruro de metileno), proporcionando el compuesto 43. EM-IEP m/z 515,4 [M+H]+.
Ejemplo 17

Se preparó el compuesto 17-a de acuerdo con el método F. A continuación, se convirtió en el compuesto 55 de manera análoga al compuesto 21 en el Ejemplo 9. EM-IEP m/z 468,3 [M+H]+.
Ejemplo 18

Se cargó un tubo sellado (30 ml) con compuesto B (0,69 mmoles, 1,0 equiv.), diclorobis(acetonitrilo)paladio (10 % molar), X-Phos (30 % molar) y carbonato de cesio (1,5 equiv.). Se añadió acetonitrilo (10 ml) seguido de la adición de etiniltrimetilsilano (0,4 ml) y la mezcla se purgó con Ar durante 1 min. A continuación, la reacción se selló y se calentó en un baño de aceite a 85°C. Tras 45 min, se añadió una alícuota adicional de etiniltrimetilsilano (1,0 ml) y se calentó nuevamente a 75°C durante 14 h; transcurrido este periodo ya no había más material de partida según el análisis de CL/EM. Se filtró la mezcla y se concentró sobre gel de sílice y se purificó mediante cromatografía flash en gel de sílice (columna Combiflash, 12 g, gradiente de 0-5% metanol/cloruro de metileno), proporcionando el compuesto 18-a.
El compuesto 18-a (0,57 mmoles, 1,0 equiv.) se disolvió en tetrahidrofurano (4 ml). Se añadió una solución de TBAF en tetrahidrofurano (0,8 ml, 1,0 M) y la mezcla se sometió a agitación a TA durante 1 h, punto en el que se observó el producto desprotegido como el pico deseado según el análisis de CL/EM. Se filtró la mezcla y se concentró sobre gel de sílice y se purificó mediante cromatografía flash en gel de sílice (columna Combiflash, 12 g, gradiente de 0-5% metanol/cloruro de metileno), proporcionando el compuesto 18-b.
Se cargó un matraz de fondo redondo (MFR) secado al horno con barra de agitación con CuI (0,34 mmoles, 1,0 equiv.), 1,10-fenantrolina (1,0 equiv.) y KF (1,0 equiv.). Se añadió N,N-dimetilformamida seca (2 ml) y la mezcla se sometió a agitación durante 15 min bajo una atmósfera de aire. A continuación, se añadió trimetil(trifluorometil)silano (5,0 equiv.) y la mezcla se calentó a 100°C bajo una atmósfera de aire. Se añadió una solución de compuesto 18-b (1,0 equiv. en 2 ml de N,N-dimetilformamida) durante el curso de 4 h utilizando una bomba de jeringa. Tras completar la adición de compuesto 18-b, la reacción se sometió a agitación durante 1,5 h adicionales a 100°C. En este punto, se dejó que la reacción se enfriase y se añadió agua (100 ml) y la mezcla se extrajo con cloruro de metileno (3x). Las capas orgánicas agrupadas se lavaron con agua, se secaron sobre sulfato sódico y se concentraron sobre gel de sílice, seguido de la purificación del material mediante cromatografía flash en gel de sílice (columna Combiflash de 4 g, gradiente de 0-10 % de metanol/cloruro de metileno). El material en bruto se purificó adicionalmente mediante HPLC de fase inversa (Interchim, gradiente de 0-10 % acetonitrilo:agua con ácido fórmico al 0,1 %, proporcionando el alquino 58 deseado. EM-IEP m/z 517,5 [M+H]+.
Ejemplo 19

Se suspendió hidrocloruro de 3-quinuclidona (9,6 mmoles, 1,0 equiv.) en cloruro de metileno (30 ml) y se añadió solución de carbonato potásico (1,0 M, 16 ml). La mezcla se sometió a agitación durante 30 min, después se recogió la capa orgánica y se lavó la capa acuosa con cloruro de metileno (3x20 ml), se secó sobre sulfato sódico, se filtró y se concentró, proporcionando la base libre correspondiente.
Una solución de etiniltrimetilsilano (10,6 mmoles, 1,1 equiv.) en tetrahidrofurano (10 ml) se enfrió a -10°C. Se añadió nbutil-litio (2,5 M en THF, 1,15 equiv.) a lo largo de 7 min. La reacción se sometió a agitación a -10°C durante 30 min, seguido del enfriamiento a -78°C. Se añadió 3-quinuclidona (1,0 equiv. en 20 ml de THF) al matraz durante un periodo de 20 min, se sometió a agitación durante 15 min adicionales; después, se retiró el baño de enfriamiento y se dejó la reacción bajo agitación a 23°C durante 15 h. A continuación, la mezcla se desactivó con cloruro amónico saturado (50 ml) y se extrajo con acetato de etilo (5 x 25 ml). Seguidamente, las capas orgánicas agrupadas se lavaron con agua (1 x 20 ml) y solución hipersalina (1 * 20 ml), se secaron sobre sulfato sódico y se concentraron bajo presión reducida, proporcionando el alquino 19-a, que se utilizó directamente en la etapa siguiente.
Se disolvió el compuesto 19-a (7,7 mmoles, 1,0 equiv.) en metanol (17 ml) y se trató con carbonato potásico (1,05 equiv.). La reacción se dejó bajo agitación a temperatura ambiente durante 4 h, punto en el que se filtró a través de Celite, lavando con metanol al 10 % en cloruro de metileno. Los filtrados se concentraron bajo presión reducida a la mitad del volumen y se filtraron nuevamente, punto en el que se había concentrado por completo bajo presión reducida. A continuación, el material se redisolvió en cloroformo (30 ml) y se lavó con solución hipersalina saturada al 50 % (10 ml). La capa acuosa se extrajo con cloroformo (3x20 ml). A continuación, las capas orgánicas agrupadas se lavaron con solución hipersalina (5 ml), se secaron sobre sulfato sódico y se concentraron bajo presión reducida, proporcionando el compuesto 19-b.
Un tubo sellado y secado al horno se cargó con diclorobis(acetonitrio)paladio (15 % molar), X-Phos (45 % molar), y carbonato de cesio (1,2 equiv.) seguido de propionitrilo (5 ml). Se añadió el compuesto B (0,22 mmoles, 1,0 equiv.) y la reacción se desgasificó con Ar durante 15 min. Se añadió el alquino 19-b (3,0 equiv.) en forma de sólido y la mezcla se purgó durante 1 min adicional con Ar. A continuación, se selló el matraz y se calentó a 100°C durante 2,5 h, punto en el que no quedaba más material de partida según el análisis de CL/EM. Se filtró la mezcla a través de Celite y el filtrado se concentró bajo presión reducida y se adsorbió en proporción 1:4 de Si-Triamina y gel de sílice (1,5 g); a
continuación, se purificó mediante cromatografía flash en gel de sílice (Interchim, columna de Si de 12 g, gradiente de 0-20 %, amonio 1 M en metanol/cloruro de metileno), proporcionando el compuesto deseado 62. EM-IEP m/z 574,6 [M+H]+-Ejemplo 20

Se preparó el compuesto 20-a de acuerdo con el método F. A continuación, se acopló con ácido 2-((tercbutoxicarbonil)amino)pirazolo[1,5-a]pirimidín-3-carboxílico de acuerdo con el método D, proporcionando el compuesto 20-b. Se desprotegió el grupo Boc bajo condiciones estándares utilizando ácido trifluoroacético y siguiendo el procedimiento siguiente: Se disolvió el compuesto 20-b en cloruro de metileno 0,06 M. A continuación, se añadió ácido trifluoroacético (40 equiv.) y la reacción se dejó bajo agitación a temperatura ambiente durante 30 min. Seguidamente la mezcla se vertió en solución saturada de bicarbonato y se extrajo con cloruro de metileno (2x). Las capas orgánicas agrupadas se secaron sobre Na2SO4 y se concentraron, proporcionando el compuesto 20-c, que se utilizó directamente en la etapa siguiente.
A continuación, se cargó un vial con el compuesto 20-c (0,25 mmoles, 1,0 equiv.), carbonato de cesio (3,0 equiv.), PdCl2(CH3CN)2 (30 % molar), X-Phos (15 % molar), propionitrilo (3 ml) y Dm SO (0,5 ml). La mezcla se burbujeó con argón durante 10 min, seguido de la adición de TMS-acetileno (4,0 equiv.) y la reacción se selló y concentró a 100°C durante 2 h, hasta que ya no quedaba más material de partida según el análisis de CL/EM. A continuación, la reacción se repartió entre acetato de etilo y solución hipersalina. La capa acuosa se lavó con acetato de etilo (1x). Las capas orgánicas agrupadas se secaron sobre Na2SO4y se concentraron, proporcionando el compuesto en bruto 20-d, que se utilizó directamente en la etapa siguiente.
El compuesto 20-d (0,25 mmoles, 1,0 equiv.) se disolvió en tetrahidrofurano (10 ml) y a continuación TBAF 1 M en tetrahidrofurano (4,0 equiv., 989 ^l). Tras 15 min ya no quedaba más material de partida según el análisis de HPLC. A continuación, la reacción en bruto se repartió cloruro de metileno y agua. La capa acuosa en primer lugar se extrajo con cloruro de metileno (2x) and después se diluyó con HCl 1 N y se extrajo con acetato de etilo (2x). Se secaron todas las capas orgánicas sobre Na2SO4 y se concentraron, proporcionando material en bruto, que en primer lugar se purificó mediante cromatografía flash en gel de sílice (Interchim Si-25g HP silicycle, gradiente 30-100 de acetato de etilo/hexanos), proporcionando material que se purificó adicionalmente mediante HPLC (30-90 % de metanol/ácido trifluoroacético al 0,1 % en agua), proporcionando el compuesto 46. EM-IEP m/z 450,3 [M+H]+.
Ejemplo 21

Se preparó el compuesto 21-a de acuerdo con el método F. A continuación, se acopló con TES-acetileno siguiendo el procedimiento siguiente: A continuación, se cargó un vial con el compuesto 21-a (0,48 mmoles, 1,0 equiv.), carbonato de cesio (2,6 equiv.), PdCl2(CH3CN)2(10 % molar), X-Phos (30 % molar) y acetonitrilo (2 ml). La mezcla se burbujeó
con argón durante 10 min, seguido de la adición de TES-acetileno (1,3 equiv.) y el sellado de la reacción y concentración a 90°C durante 2 h hasta que ya no quedase más material de partida según indicase el análisis de CL/EM. A continuación, la reacción se repartió entre acetato de etilo y solución hipersalina. La capa acuosa se lavó con acetato de etilo (1x). Las capas orgánicas agrupadas se secaron sobre Na2SO4 y se concentraron, proporcionando el compuesto en bruto 21-b, que se purificó mediante cromatografía flash en gel de sílice (Interchim Si-25g HP silicycle, gradiente 30-100 de acetato de etilo/hexanos).
A continuación, se desprotegió de Boc el compuesto 21-b y se acopló con el ácido 3-amino-pirazín-2-carboxílico mediante el método D, proporcionando el compuesto 21-c. El compuesto 21-c (0,11 mmoles, 1,0 equiv.) se disolvió en tetrahidrofurano (4 ml) y se trató con TBAF 1 M en tetrahidrofurano (3,0 equiv., 320 pl). Tras 35 min ya no quedaba material de partida según el análisis de CL/EM. Se concentró la mezcla en crudo, se preadsorbió sobre gel de sílice y se purificó mediante cromatografía flash en gel de sílice (Interchim Si-12g HP silicycle, gradiente 40-100 de acetato de etilo/hexanos), proporcionando el compuesto 47 como el producto deseado. EM-IEP m/z 411,3 [M+H]+.
Ejemplo 22

Se preparó el compuesto 22-a de acuerdo con el método F. A continuación se cargó un vial de 2 drams con el compuesto 22-a (0,59 mmoles, 1,0 equiv.), carbonato de cesio (2,6 equiv), PdCl2(CH3CN)2 (10 % molar), X-Phos (30 % molar) y propionitrilo (2 ml). La mezcla se burbujeó con argón durante 25 min, seguido de la adición de TES-acetileno (2,0 equiv.) y el sellado de la reacción y concentración a 90°C durante 3 h hasta que ya no quedase más material de partida según indicase el análisis de CL/EM. A continuación, la reacción se repartió entre acetato de etilo y solución hipersalina. La capa acuosa se lavó con acetato de etilo (1x). Las capas orgánicas agrupadas se secaron sobre Na2SO4 y se concentraron, proporcionando el compuesto en bruto 32, que se purificó mediante cromatografía flash en gel de sílice (Interchim Si-25g HP silicycle, gradiente 0-30 de acetato de etilo/hexanos), proporcionando el material deseado.
Se eliminó el grupo TES y después se desprotegió de Boc, proporcionando la amina 22-b. A continuación, lo anterior se acopló con ácido 2-((terc-butoxicarbonil)amino)pirazolo[1,5-a]pirimidín-3-carboxílico mediante el método D, seguido de la desprotección de Boc, proporcionando el compuesto deseado 48. EM-IEP m/z 388,0 [M+H]+.
Ejemplo 23

Se preparó el compuesto 23-a de acuerdo con método F. A continuación, se convirtió en la amina. Lo anterior seguidamente se acopló con ácido 2-((terc-butoxicarbonil)amino)pirazolo[1,5-a]pirimidín-3-carboxílico mediante el método D, seguido de la desprotección de Boc, proporcionando el compuesto deseado 50. EM-IEP m/z 478,0 [M+H]+.
Ejemplo 24
El compuesto 23-b se acopló con ácido 3-amino-pirazín-2-carboxílico mediante el método D, preparando el compuesto 49. EM-IEP m/z 429,0 [M+H]+-Ejemplo 25

Se preparó el compuesto 45 de manera análoga al compuesto 49, utilizando 3,4-difluoroanilina en lugar de 4-fluoroanilina, y utilizando ácido 2-aminopirazolo[1,5-a]pirimidín-3-carboxílico en lugar de ácido 3-aminopirazín-2-carboxílico. EM-IEP m/z 486,1 [M+H]+-Ejemplo 26

Se preparó el compuesto A de acuerdo con el documento n° WO 2008118468.
A continuación, una mezcla de cloruro A (0,93 mmoles, 1,0 equiv.), ácido fenilborónico (1,5 equiv.), Pd(PPh3)4 (5 % molar) y carbonato sódico (2 equiv.) en dioxano/agua (4/1 v/v, 65 ml) se desgasificó con Ar durante 10 min. La mezcla resultante se calentó a 85 °C y se sometió a agitación durante 3 h. Se enfrió la suspensión resultante hasta la TA, se repartió entre acetato de etilo y una solución acuosa saturada de cloruro sódico. Se separó la fase orgánica, se secó con sulfató sódico, se preadsorbió sobre gel de sílice y se purificó mediante la utilización de cromatografía de gel de sílice con acetato de etilo y hexanos, proporcionando el compuesto B. EM-IEP m/z: 413,3 [M+H]+.
Una mezcla de ftalimida B (0,56 mmoles, 1,0 equiv.) e hidrazina (20 equiv.) en metanol (10 ml) se calentó a 75°C y se sometió a agitación durante 1 h. La mezcla resultante se concentró, se resuspendió en cloruro de metileno y se filtró. El filtrado se concentró a sequedad, proporcionando el compuesto C. EM-IEP m/z: 283,3 [M+H]+.
Se disolvió el compuesto C (1,3 mmoles, 1,0 equiv.) en N,N-dimetilformamida (5 ml) y se cargó con base de Hunig (2,0 equiv.) y anhídrido de Boc (1,1 equiv.). La mezcla se sometió a agitación a tA durante 1 h; transcurrido este periodo ya no quedaba material de partida según el análisis de HPLC. A continuación, la reacción se vertió en solución hipersalina y se extrajo con acetato de etilo. Se lavó la capa orgánica con solución hipersalina, se secó sobre sulfato sódico y se preadsorbió sobre gel de sílice (2 g). A continuación, se purificó el residuo mediante cromatografía flash en gel de sílice (Interchim, Si-25 g, gradiente 10-30 % de acetato de etilo/hexanos), proporcionando el compuesto D. EM-|Ep m/z: 383,1 [M+H]+.
Se añadió el compuesto D (0,52 mmoles, 1,0 equiv.) a un matraz de fondo redondo (MFR) de 25 ml que contenía una suspensión de PdCh(MeCN)2 (15 % molar), X-Phos (45 % molar) y carbonato de cesio (3,0 equiv.) en propionitrilo (5 ml). La mezcla se sometió a agitación durante 1 min, seguido de la adición de TMS-propargilsilano (3,0 equiv.). A continuación, la mezcla se sometió a agitación a TA durante 30 min, seguido de calentamiento a 95°C durante 1 h. El análisis de CL/EM mostró la conversión del material e partida a principalmente compuesto E; a continuación se dejó que se enfriase la reacción. A continuación, se repartió entre acetato de etilo y agua en un embudo de separación. Se separaron las capas y la capa acuosa se extrajo con acetato de etilo (1x). Las capas orgánicas agrupadas se secaron con sulfato sódico y se preadsorbieron sobre gel de sílice (2 g). El material resultante se purificó mediante cromatografía flash en gel de sílice (columna ISCO de 25 g, gradiente de 10-30 % acetato de etilo/hexanos), proporcionando el alquino E. EM-IEP m/z: 387,1 [M+H]+.
A continuación, el compuesto E se desprotegió de Boc siguiendo el procedimiento siguiente: El compuesto E (0,19 mmoles, 1,0 equiv.) se disolvió en cloruro de metileno (4 ml) seguido de la adición de ácido trifluoroacético (1 ml). La reacción se dejó bajo agitación a TA durante 90 min; transcurrido este periodo la totalidad del material de partida se había convertido, según el análisis de HPLC. La reacción se desactivó con solución saturada de bicarbonato sódico y se extrajo con cloruro de metileno. La capa orgánica se concentró sobre sulfato sódico y se concentró. A continuación, la amina resultante se convirtió en compuesto 51 mediante procedimientos análogos para la conversión del compuesto A en B en el Ejemplo 1. EM-IEP m/z 447,1 [M+H]+.
Ejemplo 27

Se preparó el compuesto 63 de manera análoga al compuesto 4 en el Ejemplo 1, excepto en que se utilizó compuesto AA1 como material de partida. EM-IEP m/z 547,2 [M+H]+.
Ejemplo 28

Se disolvió el compuesto 4 (0,12 mmoles, 1,0 equiv.) en una mezcla de etanol y acetato de etilo (20 ml, 3:1 v/v). Se añadió paladio sobre carbono (19 mg, Pd al 10 %) y la reacción se dejó bajo una atmósfera de H2. La mezcla se sometió a agitación a TA durante 41 h, seguido de la filtración a través de un disco de filtro, se concentró y se purificó mediante cromatografía flash en gel de sílice (columna Combiflash de 4 g de Si, gradiente de 0-5 % de metanol/cloruro de metileno), proporcionando el alqueno 53. Em -IEP m/z 531,6 [M+H]+.

Se agrupó 4-et¡ml-1-met¡l-'/H-p¡razol (1,8 mmoles, 1,0 equiv.) y pinacolborano (5,0 equiv.) en tolueno (8 ml) en un MFR bajo Ar. Se añadió carbonilclorohidridotris^rifenilfosfina^utenio (II) (10 % molar) y la reacción se calentó a 50°C durante 1,5 h; después de este periodo no quedaba más material de partida según el análisis de CL/EM. Se evaporó el solvente y el residuo en bruto se transfirió a un embudo de separación con acetato de etilo (10 ml) y se lavó con bicarbonato sódico saturado (10 ml), agua (10 ml) y solución hipersalina (10 ml). La capa orgánica se secó sobre sulfato de magnesio, se concentró y se purificó mediante cromatografía flash en gel de sílice (gradiente de 10-40 % de acetato de etilo/hexanos), proporcionando el alqueno 29-a.
El compuesto B (0,22 mmoles, 1,0 equiv.), PdCl2(Amphos)2 (10 % molar) y carbonato sódico (2,0 equiv.) se cargaron en un vial de 4 ml bajo una atmósfera de Ar. Se añadió una solución de compuesto 29-a en dioxano/agua (1,5 equiv., 2 ml de solvente, 4:1 v/v) y la reacción se sometió a agitación a TA durante 5 min bajo Ar antes de calentar a 85°C durante 1 h. A continuación, se dejó enfriar la reacción, se diluyó con cloruro de metileno (15 ml) y se lavó con agua (15 ml). Seguidamente, se lavó la capa acuosa con cloruro de metileno adicional (2 x 15 ml). Se agruparon las capas orgánicas y después se lavaron con agua (30 ml) y solución hipersalina (20 ml), se secaron sobre sulfato sódico y se concentraron, proporcionando el material en bruto, que en primer lugar se purificó mediante cromatografía flash de gel de sílice (Interchim Si-12 g, gradiente de 0-5 % de metanol/cloruro de metileno) seguido de la purificación mediante HPLC de fase inversa (columna Interchim C18-Sunfire, acetonitrilo/agua/ácido fórmico al 0,1 %), proporcionando el compuesto 52. EM-IEP m/z 531,4 [M+H]+.
Ejemplo 30

Se preparó el compuesto 68 según los métodos descritos en la presente memoria. EM-IEP m/z 504,2 [M+H]+.
Ejemplo 31
El compuesto B y el ácido frans-1-propén-1-ilborónico se acoplaron bajo condiciones de acoplamiento de Suzuki análogas a las del Ejemplo 29, proporcionando el compuesto 70. EM-IEP m/z 465,2 [M+H]+-Ejemplo 32

El compuesto B y terc-butil-éster de ácido 4-et¡n¡lp¡per¡dín-1-carboxíl¡co se acoplaron bajo las condiciones de acoplamiento de Sonogashira en el Ejemplo 1, proporcionando el compuesto 32a. A continuación, se disolvió el compuesto 32a en cloruro de metileno (0,007 M), seguido de la adición de ácido trifluoroacético (10 equiv.). La reacción se dejó bajo agitación durante 2 h a TA, seguido de la concentración al vacío. El residuo se trató con un exceso de bicarbonato sódico saturado. El residuo resultante se aisló mediante filtración al vacío y se lavó con exceso de agua, proporcionando el compuesto 72. EM-IEP m/z 532,6 [M+H]+.
Ejemplo 33

Se preparó el compuesto 74 en 3 etapas siguiendo los procedimientos siguientes: se convirtió 3-formilazetidín-1-carboxilato de terc-butilo en 3-et¡n¡lazet¡dín-1-carbox¡laot de terc-butilo según el método J. A continuación, se acopló con el compuesto B y después se desprotegió de manera análoga a la síntesis de compuesto 72 en el Ejemplo 32. EM-IEP m/z: 504,5 [M+H]+.
Ejemplo 34
Se preparó el compuesto 80 en 4 etapas a partir de 1H-pirazol-4-carbaldehído, siguiendo los procedimientos siguientes: Se disolvió 1H-pirazol-4-carbaldehído (2,1 mmoles, 1,0 equiv.) en 20 ml de cloruro de metileno seguido de la adición of trietilamina (3,0 equiv.) y cloruro de tritilo (1,0 equiv.). La reacción se sometió a agitación a TA durante 1 h, seguido de la desactivación con agua (1 ml) y la extracción con cloruro de metileno. Se concentraron las capas orgánicas y se purificaron mediante cromatografía flash en gel de sílice (gradiente de 0-30 % de metanol/cloruro de metileno con trietilamina al 0,5 %). A continuación, se convirtió 1-tritil-7H-pirazol-4-carbaldehído en su alquino correspondiente mediante el método J, seguido del acoplamiento con compuesto B utilizando condiciones de acoplamiento análogas en el Ejemplo 1. A continuación, el compuesto resultante se desprotegió bajo condiciones estándares de desprotección con ácido trifluoroacético en cloruro de metileno, seguido de la concentración y purificación mediante cromatografía flash en gel de sílice (ISCO, gradiente de 0-5 % de metanol/cloruro de metileno con trietilamina al 0,05 %, y después se purificó nuevamente mediante HPLC de fase inversa (columna Interchim C18-Sunfire, gradiente de acetonitrilo/agua con ácido fórmico al 0,01 %), proporcionando el compuesto 80. EM-IEP m/z 515,0 [M+H]+.
Ejemplo 35

Se preparó el compuesto 82 en 3 tapas, siguiendo los procedimientos siguientes: Se disolvió N-Boc-4-etinilpiperidina (3,8 mmoles) en dioxano (10 ml) y se añadió HCl en dioxano (4 M, 5,0 equiv.). La reacción se dejó bajo agitación a TA durante 22 h. La mezcla se concentró bajo presión reducida, se diluyó con 10 ml de dioxano y se reevaporó bajo presión reducida. A continuación, se añadió éter dietílico (20 ml) y la mezcla se reevaporó, proporcionando la sa1HCl, que se utilizó directamente en la etapa siguiente. Una suspensión de la sa1HCl (1,05 mmoles, 1,0 equiv.) en cloruro de metileno (1 ml) se enfrió a 0-5°C en un baño de hielo. Se añadió base de Hunig (3,0 equiv.) y después, tras un minuto de agitación, se añadió anhídrido acético (2,0 equiv.). La mezcla se sometió a agitación a TA durante 1 h; transcurrido este periodo ya no quedaba material de partida según el análisis de CCF. La reacción seguidamente se diluyó con cloruro de metileno (5 ml), se lavó con ácido cítrico al 5 % (1x2 ml), agua (1x2 ml), se secaron sobre sulfato sódico y se evaporaron bajo presión reducida. El residuo en bruto se purificó mediante cromatografía flash en gel de sílice (ISCO, columna de 4 g, 0-50 % de acetato de etilo en cloruro de metileno), proporcionando N-acetil-4-etinilpiperidina, que se acopló directamente con el compuesto B utilizando condiciones de acoplamiento de Sonogashira análogas a las del Ejemplo 1, proporcionando el compuesto 82. EM-IEP m/z 574,5 [M+H]+.
Ejemplo 36

Una suspensión de HCl de 4-etinilpiperidina (1,1 mmoles, 1,0 equiv.) se suspendió en cloruro de metileno (1 ml) y se
enfrió a 0-5°C en un baño de hielo. Se añadió base de Hunig (3,0 equiv. y después de un minuto de agitación, se añadió cloruro de metanosulfonilo (2,0 equiv.) y la reacción se dejó bajo agitación durante 1 h; transcurrido este periodo ya no quedaba más material de partida según el análisis de CL/Em . La mezcla seguidamente se diluyó con cloruro de metileno (5 ml), se lavó con ácido cítrico al 5 % (1x2 ml), agua (1x2 ml), se secó sobre sulfato sódico y se concentró. El residuo en bruto se purificó mediante cromatografía flash en gel de sílice (ISCO, columna de 12 g, 0-10% de acetato de etilo en cloruro de metileno), proporcionando N-acetil-4-etinilpiperidina, que se acopló directamente con el compuesto B bajo condiciones de acoplamiento de Sonogashira análogas a las del Ejemplo 1, proporcionando el compuesto 83. EM-IEP m/z 610,6 [M+H]+.
Ejemplo 37

El compuesto 88 se preparó de manera análoga al compuesto 21 en el Ejemplo 9, excepto en que se utilizó 4-etinil-1,5-dimetil-1H-pirazol en lugar de 4-etinil-1-metil-1H-pirazol. Una suspensión de (S)-2-amino-N-(1-(5-cloro-4-oxo-3-fenil-3,4-dihidroquinazolín-2-il)etil)pirazolo[1,5-a]pirimidín-3-carboxamida (146 mg, 0,317 mmoles), carbonato de cesio (198 mg, 0,608 mmoles, 2 eq.), diclorobis(acetonitrilo)paladio (II) (15 mg, 0,058 mmoles, 0,2 eq.) y Xphos (87 mg, 0,182, 0,6 eq.) en propionitrilo (2 ml) se burbujeó con argón durante 5 minutos. La mezcla se cargó con 4-etinil-1,5-dimetil-1H-pirazol (73 mg, 0,6 mmoles, 2 eq.), se calentó a 95°C y se sometió a agitación durante 2 h. La mezcla resultante se enfrió hasta la TA, se repartió entre acetato de etilo y agua. Se separó la fase orgánica, se lavó con solución acuosa saturada de cloruro sódico, se secó con sulfato sódico y se concentró. El residuo se purificó mediante cromatografía en gel de sílice utilizando un gradiente de DCM y MeoH, proporcionando (S)-2-amino-N-(1-(5 -((1,5 -dimetil- 1H-pirazol-4-il)etinil)-4-oxo-3-fenil-3,4-dihidroquinazolín-2-il)etil)pirazolo[1,5-a]pirimidín-3-carboxamida. EM-IEP m/z 544,2 [M+H]+.
Ejemplo 38
A una mezcla bajo agitación de ácido 2-metiM-naftoico 1 (2,5 g, 13,4 mmoles) y DMF (0,67 ml) en cloroformo anhidro se añadió cloruro de tionilo (1 ml, 13,6 mmoles) y la mezcla se calentó bajo reflujo durante 1 h. Se evaporaron los solventes, se disolvieron en 10 ml de DMC y se añadieron a una mezcla bifásica de anilina (2,5 ml, 27 mmoles) en 40 ml de DCM y 40 ml de solución acuosa de hidróxido sódico. La mezcla se sometió a agitación durante 30 min, La capa acuosa se extrajo with DCM (3x20 ml), se lavó con HCl 1 M frío (20 ml), agua (3x20 ml), solución hipersalina (20 ml), se secó y se evaporaron los solventes bajo presión reducida y el sólido en bruto (3,68 g, 92 %) se recristalizó a partir de DCM-hexanos, proporcionando 1,57 g de amida P2 pura. M+H 262.23; M-H 260.23. A una mezcla bajo agitación de amida P2 (1,05 g, 1 mmol, 1 eq.) en THF anhidro (8 ml) a -10°C bajo una atmósfera de argón, se añadió una solución de hexil-litio en hexanos (3,93 ml, 9,04 moles, 2,25 eq.) gota a gota durante 8 min, manteniendo simultáneamente la temperatura interna entre -10°C y -7°C. A continuación, la mezcla resultante se sometió a agitación a -10°C durante 30 min.
A una mezcla bajo agitación of 1-(metoxi(metil)amino)-1-oxopropán-2-ilcarbamate de (S)-terc-butilo (1,12 g, 4,82 mmoles, 1,2 eq.) en THF anhidro (8 ml) a -10°C bajo una atmósfera de argón, se añadió una solución de cloruro de isopropilmagnesio en THF (2,53 ml, 5,06 mmoles, 1,26 eq.) gota a gota durante 7 min, manteniendo simultáneamente la temperatura interna entre -10°C y -7°C. La mezcla resultante se sometió a agitación a -10°C durante 30 min. A continuación, dicha solución se añadió lentamente a la mezcla de reacción anteriormente indicada, manteniendo simultáneamente la temperatura interna entre -10°C y -13°C. La mezcla resultante se sometió a agitación a -10°C durante 1 h y después se calentó hasta la temperatura ambiente durante un periodo de 1 h. La mezcla de reacción se añadió a una mezcla bifásica de 20 ml de ácido cítrico 1 M y 30 ml de acetato de etilo a una temperatura de entre -5°C y 0°C. La capa acuosa se extrajo con acetato de etilo (3x20 ml), se lavó con agua y solución hipersalina (20 ml), se secó sobre sulfato sódico, se eliminaron al vacío los solventes y el residuo se purificó mediante cromatografía en gel de sílice (40 g, 0-50 % de EtOAc-Hexanos), proporcionando 1,353 g de P3 en forma de un sólido. M+H 432.42; M-H 431.43.
Una solución de 3 (1,1 g, 2,54 mmoles) en 9 ml de anisol se trató con ácido trifluoroacético (1,52 ml, 20,3 mmoles) y la mezcla se calentó a 50°C durante 18 h. La mezcla se enfrió, se trató con 25 ml de MTBE; se filtraron los sólidos precipitados, se lavaron con MTBE (3x10 ml) y se secaron, proporcionando 1,07 g (2,5 mmoles) de sal TFA de P4 en forma de un sólido.
Se suspendieron 200 mg de la sal TFA de 4 (0,467 mmoles) en 6 ml de DCM, se trataron con solución acuosa de hidróxido amónico (2 ml, ~6 %) durante 30 min. La mezcla se diluyó con agua (10 ml), se extrajo con DCM (2x5 ml), se lavó con agua (5 ml), se secó y los solventes se evaporaron al vacío, proporcionando 149 mg (0,467 mmoles) de P4 en bruto. El P4 en bruto (120 mg, 0,382 mmoles), ácido 2-aminopirazolo[1,5-a]pirimidín-carboxílico (75 mg, 0,42 mmoles), HOBt (70 mg, 0,46 mmoles), EDC (91 mg, 0,48 mmoles) y base de Hunig (0,27 ml, 1,53 mmoles) en 3 ml de DMF se sometió a agitación durante 19 h. La mezcla se diluyó lentamente con 6 ml de metanol, se calentó a 50°C y se enfrió hasta la temperatura ambiente. Se recogieron los sólidos precipitados, se lavaron con metanol y se secaron, proporcionando 89 en forma de un sólido (154 mg). EM-IEP m/z 475,46 [M+H]+-
Ejemplo 39

Se preparó el compuesto 90 de acuerdo con los métodos de formación de amida generalmente conocidos de la técnica. EM-IEP m/z 548,31 [M+H]+-
Ejemplo 40
Se prepararon los compuestos 91 y 92.

Ejemplo 41
Se prepararon los compuestos 93 a 108 de acuerdo con el procedimiento si uiente.

Una suspensión de cloruro de arilo (0,03 a 0,06 mmoles), carbonato de cesio (1,2 eq.), didorobis(acetonitrilo)paladio (II) (0,05 eq.) y Xphos (0,15 eq.) en acetonitrilo (2 ml) se burbujeó con argón durante 5 minutos. La mezcla se cargó con 4-etiniM-metiMH-pirazol (2 eq.), se calentó a 75°C y se sometió a agitación durante 6 h. La mezcla resultante se enfrió hasta la TA, se repartió entre acetato de etilo y agua. Se separó la fase orgánica, se lavó con solución acuosa saturada de cloruro sódico, se secó con sulfato sódico y se concentró. El residuo se purificó en HPLC semi-prep HPLC (C-18) utilizando un gradiente de ACN/agua/ácido fórmico (9,9/90/0,1 % a 49,9/50/0,1 %), proporcionando el compuesto deseado (confirmado mediante CL-EM).

(continuación)

(continuación)

(continuación)

(continuación)

Ejemplo 42

En un vial resistente a microondas, se disolvió (S)-3-(1-aminoetil)-8-doro-2-fenilisoquinolín-1(2H)-ona (700 mg, 2,343 mmoles), (4-metoxifenil)metanamina (3,2 g, 23,4 mmoles, 20 eq.) y diisopropiletilamina (1,6 ml, 9,4 mmoles, 4 eq.) en NMP (12 ml). El vial se selló y se concentró a 180°C bajo irradiación de microondas y se sometió a agitación durante 6 h. La mezcla de reacción se enfrió hasta la TA, se repartió entre acetato de etilo y agua. Se separó la fase orgánica, se lavó con solución acuosa saturada de cloruro sódico, se secó con sulfato sódico y se concentró. El residuo se purificó mediante cromatografía en gel de sílice utilizando un gradiente de DCM y MeOH, proporcionando (S)-3-(1-aminoetil)-8-((4-metoxibencil)amino)-2-fenilisoquinolín-1(2H)-ona. EM-IEP m/z: 400,1 [M+H]+. Se suspendió (s)-3-(laminoetil)-8-((4-metoxibencil)amino)-2-fenilisoquinolín-1(2H)-ona (720 mg, 1,8 mmoles), ácido 2-((tercbutoxicarbonil)amino)pirazolo[1,5-a]pirimidín-3-carboxílico (1,2 g, 4,31 mmoles, 2,4 eq.), HOBt (700 mg, 4,57 mmoles, 2,5 eq.) y EDC (800 mg, 4,17 mmoles, 2,3 eq.) en DMF (30 ml). La mezcla de reacción se cargó con diisopropiletilamina (2 ml, 11,45 mmoles, 6,4 eq.) y se sometió a agitación a tA durante 1 h. La mezcla de reacción se repartió entre acetato de etilo y agua. Se separó la fase orgánica, se lavó con solución acuosa saturada de cloruro sódico, se secó con sulfato sódico y se concentró. Se purificó el residuo mediante cromatografía en gel de sílice utilizando a gradiente de acetato de etilo y hexanos, y se trituró con MeOH, proporcionando (3-((1-(8-((4-metoxibencil)amino)-1-oxo-2-fenil-1,2-dihidroisoquinolín-3-il)etil)carbamoil)pirazolo[1,5-a]pirimidín-2-il)carbamato de (S)-terc-butilo. EM-IEP m/z: 660,3 [M+H]+. Se disolvió (3-((1-(8-((4-metoxibencil)amino)-1-oxo-2-fenil-1,2-dihidroisoquinolín-3-il)etil)carbamoil)pirazolo[1,5-a]pirimidín-2-il)carbamato de (S)-terc-butilo (360 mg, 0,546 mmoles) y anisol (238 pl, 2,183 mmoles, 4 eq.) en TFA (2 ml) y se sometió a agitación a 60°C durante 1 h. La mezcla de reacción se vertió en solución acuosa saturada de bicarbonato. La fase orgánica se secó con sulfato sódico y se concentró. Se purificó el residuo mediante cromatografía en gel de sílice utilizando a gradiente de DCM. Se purificó el residuo mediante HPLC
semiprep. (C-18) utilizando un gradiente de ACN/agua/ácido fórmico, proporcionando (S)-2-amino-N-(1-(8-amino-1-oxo-2-fenil-1,2-dihidroisoquinolín-3 -il)etil)pirazolo [1,5-a]pirimidm-3-carboxairiida. EM (iEp ) m/z: 440,2 [M+H]+ Evaluación de la actividad biológica
Tabla 15. Datos de IC
50
in vitro para compuestos seleccionados.

Tabla 15 (continuación)

Tabla 15 (continuación)

Los datos en la Tabla 15 están codificados de la manera siguiente.

Ejemplo 222: ensayo HTRF™ de PI3-quinasa
Se utilizó un kit de ensayo HTRF® de quinasa PI3 (n° de cat. 33-016) adquirido de Millipore Corporation, para cribar los compuestos proporcionados en la presente memoria. Dicho ensayo utiliza la unión específica de alta afinidad del dominio de homología de pleckstrina (PH) de GRP1 a PIP3, el producto de una quinasa PI31B de clase 1A que actúa sobre su sustrato fisiológico, PIP2. Durante la etapa de detección del ensayo, se generó un complejo entre el dominio PH etiquetado con GST y PIP3 de cadena corta biotinilado. El PIP3 biotinilado y el dominio PH etiquetado con GST reclutaron fluoróforos (estreptavidina-aloficocianina y anti-GST marcado con europio, respectivamente), formando la arquitectura de la transferencia de energía de resonancia fluorescente (FRET, por sus siglas en inglés), generando una señal de FRET con resolución temporal y estable. El complejo de FRET resultó alterado de manera competitiva por PIP3 no biotinilado, un producto que se forma en el ensayo de PI3 quinasa.
Se sometió a ensayo la actividad de quinasa PI3 a, p, y o 8 utilizando el kit de ensayo de quinasa PI3 HTRF® (n° de catálogo 33-016) adquirido de Millipore Corporation. Se obtuvo PI3Ka recombinante purificado (n° de catálogo 14-602-K), PI3Kp (n° de catálogo 14-603-K), PI3Ky (n° de catálogo 14-558-K) y PI3K8 (n° de catálogo 14-604-K) de Millipore Corporation. Se utilizó enzima PI3K recombinante purificado para catalizar la fosforilación del fosfatidilinositol-4,5-bifosfato (PIP2, 10 |jM) en fosfatidilinositol-3,4,5-trifosfato (PIP3) en presencia de ATP 10 j M. El ensayo se llevó a cabo en formato de 384 pocillos y se detectó utilizando un lector multimarcaje EnVision Xcite de Perkin Elmer. Las proporciones de emisión se convirtieron en porcentajes de inhibición y se importaron en el software GraphPad Prism. La concentración necesaria para alcanzar una inhibición de la actividad enzimática de 50 % (IC50) se calculó utilizando concentraciones de entre 20 j M y 0,1 nM (curva de 12 puntos). Se determinaron los valores de IC50 utilizndo un modelo de regresión no lineal disponible en GraphPad Prism 5.
Ejemplo 223: estabilidad química
Se determinó la estabilidad química de uno o más compuestos de la invención según procedimientos estándares conocidos de la técnica. A continuación, se detalla un procedimiento ejemplar para determinar la estabilidad química de un compuesto de la invención. El tampón por defecto utilizado para el ensayo de estabilidad química era solución salina tamponada con fosfato (PBS) a pH 7,4; pueden utilizarse otros tampones adecuados. Se añadió un compuesto de la invención a partir de una solución madre 100 j M a una alícuota de PBS (por duplicado9, proporcionando un volumen de ensayo final de 400 jl, que contenía 5 j M de compuesto de ensayo y DMSO al 1 % (para la determinación de la semivida, se preparó un volumen total de muestra de 700 jl). Se incubaron las reacciones, bajo agitación, durante 24 horas a 37°C; para la determinación de la semivida, las muestras se incubaron durante 0, 2, 4, 6 y 24 horas. Las reacciones se detuvieron mediante la adición inmediata de 100 j l de la mezcla de incubación a 100 j l de acetonitrilo
y la agitación con vórtex durante 5 minutos. A continuación, las muestras se almacenaron a -20°C hasta el análisis mediante HPLC-EM/EM. Si se desea, se somete a ensayo simultáneamente un compuesto de control o un compuesto de referencia, tal como clorambucilo (5 jM ) con un compuesto de la invención de interés, ya que dicho compuesto resulta hidrolizado en gran medida durante el curso de 24 horas. Las muestras se analizaron mediante (RP)HPLC-EM/EM utilizando la monitorización de reacciones seleccionadas (MRS). Las condiciones de HPLC consistieron en una bomba de CL binaria con automuestreador, una columna C12 de 2x20 mm de modo mixto, y un programa de gradiente. Las superficies de los picos correspondientes a los analitos se registraron mediante HPLC-EM/EM. La proporción de compuesto parental remanente tras 24 horas respecto a la cantidad remanente en el tiempo cero, expresada en porcentaje, se informa como la estabilidad química. En el caso de la determinación de la semivida, ésta se estimó a partir de la pendiente del intervalo lineal inicial de la curva logarítmica del compuesto remanente (%) frente al tiempo, suponiendo una cinética de primer orden.
Ejemplo 224: ensayos de expresión e inhibición de p110a/p85a, p110p/p85o, p1105/p85o y p110Y:
Las PI3-K de clase I pueden adquirirse (p110a/p85a, p110p/p85a, p1108/p85a de Upstate, y p110Y de Sigma) o expresarse tal como se ha descrito anteriormente (Knight et al., 2004). Los valores de IC50 se miden utilizando un ensayo de CCF estándar para la actividad de lípido quinasas (indicada posteriormente) o un ensayo de captura en membrana de alto rendimiento. Las reacciones de quinasa se llevaron a cabo mediante la preparación de una mezcla de reacción que contenía quinasa, inhibidor (concentración final de DMSO: 2 %), tampón (HEPES 25 mM, pH 7,4, MgCh 10 mM) y fosfatidilinositol recién sonicado (100 jg/ml). Las reacciones se iniciaron mediante la adición de ATP, que contenía l0 jC i de Y-32P-ATP hasta una concentración final de 10 o 100 jM y se dejó que transcurriesen durante 5 minutos a temperatura ambiente. Para el análisis de CCF, seguidamente se terminaron las reacciones mediante la adición de 105 j l de HCl 1 N, seguido de 160 j l de CHCh:MeOH (1:1). La mezcla bifásica se agitó con vórtex, se centrifugó brevemente y la fase orgánica se transfirió a un tubo nuevo utilizando una punta de pipeta de carga de gel prerrecubierta con CHCh. Dicho extracto se aplicó en puntos sobre placas de CCF y se reveló durante 3 a 4 horas en una solución 65:35 de n-propanol:ácido acético 1 M. A continuación, se secaron las placas de CCF, se expusieron a una pantalla Phosphorimager (Storm, Amersham) y se cuantificaron. Para cada compuesto se midió la actividad de quinasa a 10 a 12 concentraciones de inhibidor que representaban las diluciones de dos veces a partir de la concentración más alta sometida a ensayo (típicamente, 200 jM ). Para los compuestos que mostraban actividad significativa, se repitieron las determinaciones de IC50 dos a cuatro veces y el valor informado era la media de estas mediciones independientes.
Se encuentran disponibles otros kits o sistemas comerciales para el ensayo de las actividades de PI3-K. Los kits o sistemas disponibles comercialmente pueden utilizarse para cribar para inhibidores y/o agonistas de las PI3-K, incluyendo, aunque sin limitarse a ellos, las quinasas PI3 a, p, 8 y y. Un sistema ejemplar es el ensayo de quinasa PI3 (humana) HTRF™de Upstate. El ensayo puede llevarse a cabo siguiendo los procedimientos sugeridos por el fabricante. Brevemente, el ensayo es un ensayo FRET de resolución temporal que mide indirectamente el producto de PIP3 formado por la actividad de una PI3-K. La reacción de la quinasa se lleva a cabo en una placa de microtitulación (p.ej., una placa de microtitulación de 384 pocillos). El volumen total de reacción es de aproximadamente 20 j l en cada pocillo. En la primera etapa, cada pocillo recibe 2 j l de compuesto de ensayo en dimetilsulfóxido al 20 %, resultando en una concentración final de DMSO de 2 %. A continuación, se añadieron aproximadamente 14,5 j l de una mezcla de quinasa/PIP2 (diluida en tampón de reacción IX) en cada pocillo, para una concentración final de 0,25 a 0,3 jg/m l de quinasa y PIP2 10 jM . Se selló la placa y se incubó durante 15 minutos a temperatura ambiente. Para iniciar la reacción, se añadieron 3,5 j l de ATP (diluidos en tampón de reacción IX) a cada pocillo para una concentración final de ATP de 10 jM . Se selló la placa y se incubó durante 1 hora a temperatura ambiente. La reacción se detuvo mediante la adición de 5 j l de solución de parada en cada pocillo y después se añadieron 5 j l de mezcla de detección a cada pocillo. Se sella la placa y se incuba durante 1 hora a temperatura ambiente y después se lee en un lector de placas apropiado. Se analizaron los datos y se generaron las IC50 utilizando GraphPad Prism 5.
Ejemplo 225: ensayo de activación y proliferación de células B
Se determinó la capacidad de uno o más compuestos de la invención de inhibir la activación y proliferación de las células B según procedimientos estándares conocidos de la técnica. Por ejemplo, se estableció un ensayo in vitro de proliferación celular que mide la actividad metabólica de las células vivas. El ensayo se llevó a cabo en una placa de microtitulación de 96 pocillos utilizando reducción de azul Alamar. Se purificaron células B esplénicas de Balb/c en un gradiente de Ficoll-Paque™ PLUS seguido de la separación magnética de las células con un kit de aislamiento de células B MACS B (Miltenyi). Las células se sembraron en placas a razón de 90 jl, 50.000 células/pocillo en medio para células B (Rp M i FBS al 10 % pen./estrep. bME 50 jM HEPES 5 mM). Un compuesto proporcionado en la presente memoria se diluyó en medio para células B y se añadió en un volumen de 10 jl. Las placas se incubaron durante 30 min a 37°C y con 5 % de CO2 (concentración final de DMSO: 0,2 %). A continuación, se añadieron 50 j l de cóctel de estimulación de células B que contenía LPS 10 jg/m l o F(ab')2 de asno anti-IgM de ratón 5 jg/m l más lL4 de ratón recombinante 2 ng/ml en medio para células B. Las placas se incubaron durante 72 horas a 37°C y con 5 % de CO2. Se añadió a cada pocillo un volumen de 15 j l de reactivo de azul Alamar y las placas se incubaron durante 5 horas a 37°C y con 5 % de CO2. Se leyó la fluorescencia del azul Alamar a 560 Exc./590 Em. y se calcularon los valores de IC50 o EC50 utilizando GraphPad Prism 5.
Ejemplo 226: ensayo de proliferación de línea de células tumorales
La capacidad de uno o más compuestos de la invención de inhibir la proliferación de la línea de células tumorales puede determinarse según procedimientos estándares conocidos de la técnica. Por ejemplo, puede llevarse a cabo un ensayo in vitro de proliferación celular para medir la actividad metabólica de las células vivas. El ensayo se llevó a cabo en una placa de microtitulación de 96 pocillos utilizando reducción de azul Alamar. Se obtuvieron líneas de células tumorales humanas de ATCC (p.ej., m Cf 7, U-87 MG, MDA-MB-468 y PC-3), se cultivaron hasta la confluencia en matraces T75, se tripsinizaron con tripsina al 0,25 %, se lavaron una vez con medio para células tumorales (DMEM FBS al 10 %) y se sembraron en 90 pl a 5.000 células/pocillo en medio para células tumorales. Un compuesto proporcionado en la presente memoria se diluyó en medio para células tumorales y se añadió en un volumen de 10 pl. Las placas se incubaron durante 72 horas a 37°C y con 5 % de CO2. Se añadió a cada pocillo un volumen de 10 pl de reactivo de azul Alamar y las placas se incubaron durante 3 horas a 37°C y con 5 % de CO2. Se leyó la fluorescencia del azul Alamar a 560 Exc./590 Em. y se calcularon los valores de IC50 utilizando GraphPad Prism 5.
Ejemplo 227: actividad antitumoral in vivo
Los compuestos indicados en la presente memoria pueden evaluarse en un panel de modelos tumorales humanos y murinos. En un aspecto, los compuestos proporcionados en la presente memoria pueden evaluarse en los modelos siguientes según métodos conocidos de la técnica. Las dosis y régimen de administración pueden variarse según el modelo. Los resultados pueden evaluarse con los de inhibidores delta selectivos, y combinaciones de inhibidores delta y gamma, y/o con anticuerpos que bloquean receptores inhibitorios específicos.
Modelos de tumor refractario a paclitaxel
1. Modelo de carcinoma ovárico derivado clínicamente
Dicho modelo tumoral se estableció a partir de una biopsia tumoral de un paciente de cáncer ovárico. Se obtuvo una biopsia tumoral del paciente. Se administraron los compuestos indicados en la presente memoria en ratones desnudos portadores de tumores estadificados, utilizando un régimen de 2 días x 5.
2. Xenoinjerto de carcinoma ovárico humano A2780Tax (tubulina mutada)
A2780Tax es un modelo de carcinoma ovárico humano resistente al paclitaxel. Se deriva de la línea parental A2780 sensible de células con paclitaxel y verapamilo, un agente de inversión de MDR. Se ha mostrado que su mecanismo de resistencia no está relacionado con MDR y se atribuye a una mutación del gen codificante de la proteína betatubulina. Los compuestos indicados en la presente memoria en ratones desnudos portadores de tumores estadificados, utilizando un régimen de 2 días x 5.
3. Xenoinjerto de carcinoma de colon humano HCT116/VM46 (resistente a múltiples fármacos).
HCT116/VM46 es un carcinoma de colon resistente a MDR desarrollado a partir de la línea parenta1HCT116 sensible. In vivo, cultivado en ratones desnudos, HCT116/VM46 ha demostrado consistentemente una elevada resistencia al paclitaxel. Los compuestos indicados en la presente memoria en ratones desnudos portadores de tumores estadificados, utilizando un régimen de 2 días x 5.
Pueden utilizarse uno o más compuestos proporcionados en la presente memoria en combinación con otros agentes terapéuticos in vivo en los xenoinjertos de carcinoma de colon humano resistentes a múltiples fármacos HCT/VM46 o cualquier otro modelo conocido de la técnica, incluyendo los descritos en la presente memoria.
4. Modelo de sarcoma murino M5076
M5076 es un fibrosarcoma de ratón que es inherentemente refractario al paclitaxel in vivo. Los compuestos indicados en la presente memoria en ratones desnudos portadores de tumores estadificados, utilizando un régimen de 2 días x 5.
Modelos pancreáticos
El modelo KPC es un modelo de ratón transgénico de adenocarcinoma ductal pancreático (ADP), en el que se produce la expresión condicional de ambos alelos mutantes KrasG12D y p53R172H en las células pancreáticas. Los tumores se desarrollan espontáneamente en este ratón durante un periodo de 3 a 6 meses y pueden utilizarse para estudiar la eficacia profiláctica, así como terapéutica, de nuevos agentes. Las células de dichos tumores KPC también pueden transferirse adoptivamente a ratones híbridos B6.129 singénicos, creando un modelo con un periodo de latencia más corto y permitiendo establecer síncronamente un gran número de animales con tumores. Ver, p.ej., Cancer Cell 7:468 (2005). En algunas realizaciones, los compuestos proporcionados en la presente memoria pueden administrarse solos o en combinación con anti-PD-LI.
Modelo Pan02: La línea celular de adenocarcinoma pancreático murino Pan02 es una línea tumoral no metastásica, singénica a C57BL/6. Puede estudiarse siguiendo la inyección s.c. en el flanco u ortoépicamente tras la inyección directamente en el páncreas. Ver, p.ej., Cancer Res. 44: 717-726, 1984).
Modelo de carcinoma pulmonar de Lewis CPL:
modelo de carcinoma pulmonar de Lewis CPL: Las células de CPL se derivan de un tumor pulmonar espontáneo de ratones C57BL/6 y pueden estudiarse como tumor s.c. al inyectarse en el flanco, o en forma de tumor ortoépico en caso de inyectarse i.v., tras lo cual se localiza en el pulmón.
Las células de LLC también han sido modificadas para expresar un péptido a partir de ovoalbúmina (células LL2-OVA). La utilización de dichas células, tras la inyección s.c. o i.v., permite el seguimiento de los linfocitos CD8+ específicos de OVA y la medición de los efectos de la terapia sobre la respuesta inmunitaria adaptativa contra el tumor. Ver, p.ej., Science 330:827 (2010).
Modelo de mama
El carcinoma mamario 4T1 es una línea celular tumoral trasplantable que crece en ratones BALB/c singénicos. Es altamente tumorigénica e invasiva y, al contrario que la mayoría de modelos tumorales, puede metastatizar espontáneamente desde el tumor primario en la glándula mamaria a múltiples sitios distantes, incluyendo ganglios linfáticos, sangre, hígado, pulmón, cerebro y hueso. Ver, p.ej., Current Protocols in Immunology Unit 20.2 (2000). La línea celular 4T1 parental se ha modificado para expresar constitutivamente la luciferasa (4T1-luc). La línea celular 4T1-luc puede introducirse ortoépicamente en las almohadillas grasas mamarias de los ratones Balb/c. Al introducirse ortoépicamente, el modelo 4T-luc crece rápidamente en el sitio primario y forma metástasis en sitios distantes a lo largo de un periodo de 3 a 6 semanas. La metástasis rápida y eficiente a órganos afectados en el cáncer de mama humano lo convierte en un excelente modelo de ratón para el estudio de la progresión metastásica del cáncer de mama en el ser humano. Debido a que el modelo es singénico a los ratones Balb/c, puede utilizarse para estudiar el papel del sistema inmunitario en el crecimiento y metástasis tumorales. Ver, p.ej., Cancer Res. 15 de marzo, 1992;52(6):1399-405. Por ejemplo, las células de cáncer del modelo 4T1 pueden metastatizar al pulmón. Las células tumorales 4T1 son resistentes a 6-tioguanina, y las células metastásicas pueden detectarse y cuantificarse mediante el explante de órganos, el cultivo en placa de las células disociadas en medio complementado con 6-tioguanina, y el recuento del número de células tumorales clonogénicas resistentes a 6-TG. El modelo 4T1 puede ser un modelo de metástasis pulmonar. Ver, p.ej., Current Protocols in Immunology Unit 20.2 (2000) y Kerbel, R. S., The Breast 22 (2013), S57-S65.
Se cultiva 4T1-Luc, la línea celular 4T1 expresante de luciferina, en RPMI-1640 filtrado y complementado con suero de feto bovino inactivado por calor al 10 % a 37°C en aire atmosférico con 5 % de CO2. Se implantaron ortoépicamente 5000 células 4T1-Luc en 50 pl de solución salina tamponada con fosfato, en la almohadilla grasa mamaria de ratones Balb/c hembra de 6 a 8 semanas de edad. Una vez los tumores han alcanzado aproximadamente 50 a 100 mm3, los ratones se asignan aleatoriamente a grupos de tratamiento. Los ratones recibieron la administración oral de vehículo o un inhibidor de PI3Ky ejemplar dado a conocer en la presente memoria que presenta una proporción de selectividad delta/gamma superior a aproximadamente 50. Por ejemplo, el inhibidor de PI3Ky se administra por vía oral a una dosis de 3 mg/kg una vez al día durante 21 días consecutivos solo o en combinación con anticuerpos anti-PD-L1 (200 pg) o de isotipo de control (IgG2b k de rata; 200 pg) que se administran por vía intraperitoneal una vez cada 3 días en un total de 5 dosis. Se realizan mediciones tumorales y del peso corporal 3 veces a la semana. Se realizan mediciones de la luciferasa dos veces a la semana, con imágenes de IVIS 200. Al final del estudio, los ratones fueron eutanizados y se recolectaron los tumores para la evaluación de la inhibición de la ruta y la respuesta inmunitaria de supresión constante de la ruta de PI3Ky. Se recogió el plasma para el análisis farmacocinético (FC). Se determinó la eficacia, por ejemplo mediante mediciones del volumen tumoral.
Se describe a continuación otro modelo de mama. Antes de la inyección, se cultivaron células PyMT 8119 en F12K complementado con clon II fetal al 5 %, 50 pg/ml de gentamicina, 2,5 pg/ml de fugizona y 1 pl/ml de MITO. Se inyectaron células PyMT 8119 por vía subcutánea u ortoépica en la almohadilla grasa mamaria (1x 106 células/ratón) en ratones C57B16 o desnudos. Se registraron mediciones tumorales 3 veces a la semana desde el día d7 después de la inyección y se calcularon los volúmenes tumorales utilizando la fórmula siguiente: (longitud x anchura x anchura)/2=volumen en el que la longitud denota la dimensión más larga medida y la anchura denota la dimensión más corta.
Modelo de linfoma
EL4 es un timoma T de C57BL/6 y EG7 es un subclón expresante de OVA de EL4. La línea parental EL4 ha sido modificada para expresar constitutivamente la luciferasa, que permite la obtención no invasiva de imágenes del crecimiento tumoral en todo el animal utilizando la plataforma Xenogen de obtención de imágenes.
A20 es una línea celular de linfoma de células B de Balb/c derivada de una neoplasia espontánea observada en un
ratón Balb/cAnN de edad avanzada, que expresa las moléculas H-2d del CMH de clases I y II. La línea celular A20 parental se ha modificado para expresar constitutivamente la luciferasa (A20-Luc). La línea celular expresante de luciferasa A20-Luc se deriva de la línea celular A20 parental y puede implantarse en ratones Balb/c por vía subcutánea o intravenosa. El modelo singénico i.v. sistémico puede utilizarse para estudiar el papel del sistema inmunitario en el crecimiento y metástasis tumorales. Ver, p.ej., J. Clin Invest. 2013; 123(6):2447-2463.
Modelo de melanoma
Las células de melanoma murino B16 son singénicas a los ratones C57BL/6 y pueden estudiarse después de la inyección s.c., i.d. (intradérmica), o i.v. La implantación en cualquiera de los sitios resultará en metástasis a pulmón y otros órganos. Este modelo ha sido ampliamente estudiado en términos del papel que desempeñan los receptores inhibidores en la respuesta inmunitaria antitumoral. Ver, p.ej., PNAS 107:4275 (2010).
El modelo de tumor B16-F10 etiquetado con luc permite evaluar la eficacia y efectos farmacodinámicos de los inhibidores de PI3K-Y y otros agentes antitumorales. El modelo puede utilizarse de la manera siguiente. Se obtienen ratones C57 BLK (macho) de 5-6 semanas de Jackson Labs, n=40. Los ratones reciben dosis QD de un inhibidor de PI3K-Y. La dosis E3D de PD-L1 o vehículo se administra QD. Las mediciones de tumores y pesos corporales se realizan 3X/semana. Los tumores se recolectaron el último día o en el caso de que en el grupo de vehículo alcanzasen aproximadamente 2000 mm3. Los tumores se cortaron por la mitad. Una mitad se cortó nuevamente en mitades y una mitad se fijó en NBF al 10 % y la otra mitad se congeló en OCT para secciones congeladas. La mitad restante se procesó para generar una suspensión de células individuales y se evaluó mediante FACS utilizando dos paneles. Las dosis se indican en la Tabla 16, a continuación. La vacunación de dicho modelo puede potenciar la respuesta al tratamiento. Ver, p.ej., Duraiswamy, J. et al., Cancer Res, 73(12), 2013, 3591; Curran, M. A., et al., PNa S, 107(9), 2010, 4275.
Tabla 16

Es conocido que el melanoma es sensible a las inmunoterapias, y se ha informado de datos que asocian un mal pronóstico a elevados recuentos de células MAT en estos tumores. Sin limitarse a ninguna teoría en particular, un compuesto proporcionado en la presente memoria (p.ej., el Compuesto 4) puede afectar a los recuentos de células MAT en el microambiente tumoral y puede someterse a ensayo en uno o más modelos de melanoma conocidos de la técnica.
Modelo de cáncer de colon
CT26 es una línea celular de carcinoma de colon no diferenciado inducida por N-nitroso-N-metiluretano (NNMU). Se clonó para generar la línea celular denominada CT26.WT (ATCC n° CRL-2638). Se estableció el modelo de cáncer de colon CT26 singénico mediante implantación subcutánea de células CT26 en ratones BALB/c. Dicho modelo ha sido ampliamente utilizado para estudiar la actividad antitumoral de las inmunoterapias (Yu et al., Clinical Cancer Research, 2010; Daraiswamy et al., Cancer Research, 2013). Dicho modelo puede utilizarse para demostrar los efectos de los inhibidores de PI3K dados a conocer en la presente memoria. Un compuesto proporcionado en la presente memoria puede administrarse solo o en combinación con anti-PD-L1.
Otro modelo de cáncer de colon son los ratones C57BL/6 CR. Se prepararon ratones C57BL/6 CR hembra con 1x106 células MC38 en Matrigel al 0 % s.c. en el flanco. El volumen de inyección celular era de 0,05 ml en cada ratón. Una vez los tumores habían alcanzado un tamaño medio de 80 a 100 mm3, se emparejaron y se inició el tratamiento con un compuesto proporcionado en la presente memoria.
Modelo de glioblastoma
Puede utilizarse el modelo de xenoinjerto de ratón desnudo de glioma humano para estudiar los efectos de los compuestos proporcionados en la presente memoria sobre el glioblastoma. Se generaron tumores en ratones desnudos atímicos (Taconic Laboratories). Los animales recibieron una inyección subcutánea en el flanco derecho con 5x106 células de glioma humano U87 en 0,1 ml de PBS complementado con glucosa al 0,1 %. Se midieron los tumores con un calibrador externo, cada día del tratamiento, y se calculó el volumen. Una vez los tumores habían alcanzado un volumen de 200 mm3, los ratones fueron asignados aleatoriamente a diferentes grupos experimentales y tratados diariamente con vehículo del compuesto de control o con blanco o un compuesto proporcionado en la presente memoria a diversas dosis y en diversos regímenes. Los ratones fueron monitorizados diariamente para su estado de salud y para los volúmenes tumorales. Después de determinados días de tratamiento, los ratones fueron
sacrificados y se extrajeron los tumores y se midieron y pesaron. Ver, p.ej., Dolores Hernán Pérez de la Ossa, et al., PLOS ONE, 2013, vol. 8 (1), e54795.
En algunos casos se utilizó el modelo de glioblastoma GL261. Es un modelo multiforme de glioblastoma singénico utilizado subcutánea u ortotópicamente, y se describe en mayor detalle en los ejemplos proporcionados en la presente memoria. En el modelo puede haberse realizado previamente un tratamiento de radiación; sin embargo, se produce recurrencia del cáncer. Los compuestos proporcionados en la presente memoria pueden administrarse en modelos con una recurrencia o recaída del cáncer después del tratamiento previo.
Ejemplo 228: ensayo de estabilidad de microsomas
Se determinó la estabilidad química de uno o más compuestos de la invención según procedimientos estándares conocidos de la técnica. Por ejemplo, se estableció la estabilidad de uno o más compuestos de la invención mediante un ensayo in vitro. Por ejemplo, se estableció un ensayo de estabilidad in vitro de los microsomas, que mide la estabilidad de uno o más compuestos de la invención al reaccionar con microsomas de ratón, rata o ser humano de origen hepático. La reacción de los microsomas con los compuestos se lleva a cabo en tubos Eppendorf de 1,5 ml. Cada tubo contiene 0,1 pl de NADPH 10,0 mg/ml; 75 pl de microsomas hepáticos de ratón, rata o humanos 20,0 mg/ml; 0,4 pl de tampón fosfato 0,2 M y 425 pl de ddH2O. El tubo de control negativo (sin NADPH) contenía 75 pl de microsomas hepáticos de ratón, rata o humanos 20,0 mg/ml; 0,4 pl de tampón fosfato 0,2 M y 525 pl de ddH2O. La reacción se inició mediante la adición de 1,0 pl de compuesto sometido a ensayo 10,0 mM. Los tubos de reacción se incubaron a 37°C, se recogieron 100 pl de muestra en un nuevo tubo Eppendorf que contenía 300 pl de metanol frío a los 0, 5, 10, 15, 30 y 60 minutos de reacción. Las muestras se centrifugaron a 15.000 rpm para separar las proteínas. El sobrenadante de la muestra centrifugada se transfirió a un nuevo tubo. La concentración de compuesto estable después de la reacción con los microsomas en el sobrenadante se midió mediante cromatografía líquidaespectrometría de masas (CL-EM).
Ejemplo 229: ensayo de estabilidad en plasma
La estabilidad de uno o más compuestos de la invención en plasma se determinó según procedimientos estándares conocidos de la técnica. Ver, p.ej., Rapid Commun. Mass Spectrom., 10: 1019-1026, 1977. El procedimiento a continuación es un ensayo de HPLC-EM/EM con plasma humano; también se encuentran disponibles otras especies, entre ellas el mono, el perro, la rata y el ratón. Se descongeló plasma humano heparinizado congelado en un baño de agua fría y se centrifugó durante 10 minutos a 2000 rpm a 4°C antes de utilizarlo. Se añadió un compuesto de la invención a partir de una solución madre 400 pM a una alícuota de plasma precalentado, proporcionando un volumen de ensayo final de 400 pl (o 800 pl para la determinación de la semivida), que contenía compuesto de ensayo 5 pM y DMSO al 0,5 %. Se incubaron las reacciones, bajo agitación, durante 0 minutos y 60 minutos a 37°C, o durante 0, 15, 30, 45 y 60 minutos a 37°C para la determinación de la semivida. Las reacciones se detuvieron mediante la transferencia de 50 pl de la mezcla de incubación a 200 pl de acetonitrilo helado y se mezclaron mediante agitación durante 5 minutos. Las muestras se centrifugaron a 6000xg durante 15 minutos a 4°C y se extrajeron 120 pl de sobrenadante a tubos limpios. A continuación, las muestras se evaporaron a sequedad y se sometieron a análisis mediante HPLC-EM/EM.
Pueden someterse a ensayo uno o más compuestos de control o de referencia (5 pM) simultáneamente con los compuestos de ensayo: un compuesto, propoxicaína, con baja estabilidad en plasma y otros compuestos, propantelina, con estabilidad en plasma intermedia.
Las muestras se reconstituyen en acetonitrilo/metanol/agua (1/1/2 v/v/v) y se analizan mediante (RP)HPLC-EM/EM utilizando la monitorización de reacciones seleccionadas (MRS). Las condiciones de HPLC consistieron en una bomba de CL binaria con automuestreador, una columna C12 de 2x20 mm de modo mixto, y un programa de gradiente. Las superficies de los picos correspondientes a los analitos se registraron mediante HPLC-EM/EM. La proporción de compuesto parental remanente tras 60 minutos respecto a la cantidad remanente en el tiempo cero, expresada en porcentaje, se informa como la estabilidad en plasma. En el caso de la determinación de la semivida, ésta se estimó a partir de la pendiente del intervalo lineal inicial de la curva logarítmica del compuesto remanente (%) frente al tiempo, suponiendo una cinética de primer orden.
Ejemplo 230: señalización de quinasa en sangre
La señalización de PI3K/Akt/mTOR se mide en las células sanguíneas mediante el método 'phosflow' (Methods Enzymol. 434:131-54, 2007). Dicho método es por naturaleza un ensayo de células individuales, por lo que puede detectarse la heterogeneidad celular y no las medias poblacionales. Lo anterior permite una distinción concurrente entre los estados de señalización de diferentes poblaciones definidas por otros marcadores. El método 'phosflow' también es altamente cuantitativo. Con el fin de someter a ensayo uno o más compuestos proporcionados en la presente memoria, se estimularon esplenocitos no fraccionados, o células mononucleares de sangre periférica, con anti-CD3 para iniciar la señalización de receptores de células T. A continuación, se fijaron las células y se tiñeron para marcadores de superficie y fosfoproteínas intracelulares. Determinados inhibidores proporcionados en la presente memoria, p.ej., inhibidores de PI3K-5, inhiben la fosforilación mediada por anticuerpos anti-CD3 de Akt -S473 y S6,
mientras que la rapamicina inhibe la fosforilación de S6 y potencia la fosforilación de Akt bajo las condiciones sometidas a ensayo. Determinados inhibidores proporcionados en la presente memoria, p.ej., inhibidores de PI3K-Y, actúan sobre la estimulación de ligandos de GPCR (p.ej., CCL2, CXCL12 o IL8) de fosfo-KAT en las células sanguíneas. De acuerdo con lo anterior, para someter a ensayo el efecto de uno o más compuestos proporcionados en la presente memoria, se pusieron en contacto esplenocitos no fraccionados, o células mononucleares de sangre periférica, con un ligando de GPCR. A continuación, se fijaron las células y se tiñeron para marcadores de superficie y fosfoproteínas intracelulares.
De manera similar, se incubaron alícuotas de sangre completa durante 15 minutos con vehículo (p.ej., DMSO al 0,1%) o inhibidores de quinasa a diversas concentraciones, antes de la adición de estímulos para entrecruzar el receptor de células T (RCT) (p.ej., anti-CD3 con anticuerpo secundario) o el receptor de células B (RCB) utilizando anticuerpo anticadena ligera kappa (fragmentos Fab'2). Tras aproximadamente 5 y 15 minutos, se fijaron muestras (p.ej., con paraformaldehído al 4 % frío) y se utilizaron para el 'phosflow'. Se utilizó la tinción superficial para distinguir las células T y B utilizando anticuerpos dirigidos contra marcadores de superficie celular que son conocidos de la técnica. El nivel de fosforilación de los sustratos de quinasa, tales como Akt y S6, se midió a continuación mediante la incubación de las células fijadas con anticuerpos marcados específicos de las isoformas fosforiladas de dichas proteínas. A continuación, se analizó la población de células mediante citometría de flujo.
Ejemplo 231: ensayo de formación de colonias
Se sembraron en placa células de médula ósea murina recién transformadas con un retrovirus p190 BCR-AbI (en la presente memoria denominadas células transducidas con p190) en presencia de diversas combinaciones de fármacos en medio de metilcelulosa M3630 durante aproximadamente 7 días con IL-7 humano recombinante en aproximadamente 30 % de suero y se contó el número de colonias formadas mediante examen visual bajo un microscopio.
Alternativamente, se obtuvieron células mononucleares de sangre periférica humana de pacientes positivos para cromosoma Philadelphia (Ph+) y negativos para el mismo (Ph-) tras el diagnóstico inicial o una recaída. Se aislaron células vivas y se enriquecieron para progenitores de células B CD19+ CD34+. Tras el cultivo líquido durante la noche, las células se sembraron en placa en Methocult GF+ H4435 (Stem Cell Technologies), complementado con citoquinas (IL-3, IL-6, IL-7, G-CSF, GM-CSF, CF, ligando Flt3 y eritropoyetina) y diversas concentraciones de agentes quimioterapéuticos conocidos en combinación con compuestos de la presente exposición. Las colonias se contaron bajo el microscopio 12 a 14 días después. El presente método puede utilizarse para el ensayo para pruebas de actividad aditiva o sinérgica.
Ejemplo 232: efecto in vivo de los inhibidores de quinasa sobre las células leucémicas
Se irradiaron letalmente ratones receptores hembra a partir de una fuente de rayos y en dos dosis separadas por aproximadamente 4 h, con aproximadamente 5 Gy cada dosis. Aproximadamente 1 h después de la segunda dosis de radiación, los ratones recibieron inyecciones i.v. de aproximadamente 1x106 células leucémicas (p.ej., células humanas o murinas Ph+ o células de médula ósea transducidas con p190). Dichas células se administraron junto con una dosis radioprotectora de aproximadamente 5x106 células de médula ósea normal de ratones donantes de 3 a 5 semanas de edad. Los receptores recibieron antibióticos en el agua y fueron monitorizados diariamente. Los ratones que enfermaron tras aproximadamente 14 días fueron eutanizados y se recolectaron los órganos linfoides para el análisis. El tratamiento de inhibidor de quinasa se inició aproximadamente 10 días después de la inyección de células leucémicas y continuó diariamente hasta que enfermaron los ratones o un máximo de aproximadamente 35 días después del trasplante. Los inhibidores se administraron mediante irrigación oral.
Se recogieron células de sangre periférica aproximadamente el día 10 (pretratamiento) y tras la eutanización (post tratamiento), se pusieron en contacto con anticuerpos anti-hCD4 marcados y se contaron mediante citometría de flujo. Dicho método puede utilizarse para demostrar que el efecto sinérgico de uno o más compuestos proporcionados en la presente memoria en combinación con agentes quimioterapéuticos conocidos puede reducir los recuentos de células sanguíneas leucémicas en comparación con el tratamiento con agentes quimioterapéuticos conocidos (p.ej., Gleevec) solos bajo las condiciones sometidas a ensayo.
Ejemplo 233: tratamiento de modelo de ratón de enfermedad del lupus (Ejemplo de referencia)
Los ratones que no presentan el receptor inhibidor FcyRIIb que se opone a la señalización de PI3K en las células B desarrollan lupus con elevada penetrancia. Los ratones con inactivación de FcyRIIb (R2KO, Jackson labs) se consideran un modelo válido de la enfermedad humana ya que algunos pacientes de lupus muestran una expresión o función reducida de FcyRIIb (S. Bolland y J.V. Ravtech 2oo0. Immunity 12:277-285).
Los ratones R2KO desarrollan una enfermedad similar al lupus con anticuerpos antinucleares, glomerulonefritis y proteinuria aproximadamente dentro de los 4 a 6 primeros meses de vida. Para estos experimentos, se utilizó el análogo de rapamicina RAD001 (disponible de LC Laboratories) como compuesto de referencia, y se administró por
vía oral. Se ha demostrado que dicho compuesto alivia los síntomas del lupus en el modelo B6.Slelz Sle3z (T. Wu et al., J. Clin. Invest. 117:2186-2196).
Los ratones NZB/W F1 que desarrollan espontáneamente una enfermedad autoinmune sistémica son un modelo de lupus. El modelo de lupus NZB/W F1 murino presenta muchas características del lupus humano y se caracteriza por niveles elevados de autoanticuerpos antinucleares y anti-ADNdc; un papel crítico de las células dendríticas plasmacitoides e IFN-a, participación de células T, células B y macrófagos, anemia hemolítica, glomerulonefritis compleja inmune progresiva, proteinuria, gravedad e incidencia más pronunciadas en hembras, y supervivencia reducida. El tratamiento con un compuesto proporcionado en la presente memoria puede determinarse mediante evaluación de las puntuaciones de proteínas en la orina, pesos de los órganos, niveles plasmáticos de IgG anti-ADNdc e histopatología de los riñones. Los ratones se tratan a partir de las 20 semanas de edad para un modelo profiláctico y a las 23 semanas edad para un modelo terapéutico. Se obtuvieron muestras de sangre y orina durante todo el periodo de ensayo, y se sometieron a ensayo para anticuerpos antinucleares (en diluciones de suero) o para concentración de proteínas (en orina). También se sometió a ensayo el suero para anticuerpos anti-ADNmc y anti-ADNdc mediante ELISA. Se evaluó la glomerulonefritis en secciones de riñón teñidas con H+E al final del estudio, o el criterio de valoración puede ser la supervivencia. Por ejemplo, el inhibidor del proteosoma Bortezimib resulta eficaz para bloquear la enfermedad en el modelo NZB/W tanto en el modelo profiláctico como en el modelo terapéutico, con reducciones en la producción de autoanticuerpos, daños renales y mejoras de la supervivencia (Nature Medicine 14, 748-755 (2008)).
Se tratan ratones de modelo de enfermedad del lupus, tales como R2KO, BXSB o MLR/lpr a los 2 meses de edad aproximadamente, durante aproximadamente dos meses. Los ratones recibieron dosis de: vehículo, RAD001 a una dosis de aproximadamente 10 mg/kg, o compuestos proporcionados en la presente memoria a una dosis de entre aproximadamente 1 mg/kg y aproximadamente 500 mg/kg. Se obtuvieron muestras de sangre y orina durante todo el periodo de ensayo, y se sometieron a ensayo para anticuerpos antinucleares (en diluciones de suero) o para concentración de proteínas (en orina). También se sometió a ensayo el suero para anticuerpos anti-ADNmc y anti-ADNdc mediante ELISA. Los animales fueron eutanizados el día 60 y se recolectaron tejidos para medir el peso del bazo y la enfermedad renal. Se evaluó la glomerulonefritis en secciones de riñones tejidos con H+E. Se estudiaron otros animales durante aproximadamente dos meses tras cesar el tratamiento, utilizando los mismos criterios de valoración.
Este modelo establecido de la técnica puede utilizarse para demostrar que los inhibidores de quinasa proporcionados en la presente memoria pueden suprimir o retrasar la aparición de los síntomas de lupus en ratones de modelo de enfermedad de lupus.
Ejemplo 234: ensayo de trasplante de médula ósea murina
Los ratones receptores hembra se irradiaron letalmente a partir de una fuente de rayos y. Aproximadamente 1 hora después de la dosis de radiación, los ratones recibieron la inyección de aproximadamente 1x106 células leucémicas de cultivos transducidos con p190 de pase temprano (p.ej., tal como se indica en Cancer Genet. Cytogenet. agosto de 2005; 161(1):51-6). Dichas células se administraron junto con una dosis radioprotectora de aproximadamente 5x106 células de médula ósea normal de ratones donantes de 3 a 5 semanas de edad. Los receptores recibieron antibióticos en el agua y fueron monitorizados diariamente. Los ratones que enfermaron tras aproximadamente 14 días fueron eutanizados y se recolectaron los órganos linfoides para enriquecimiento mediante citometría de flujo y/o magnético El tratamiento se inició aproximadamente el día 10 y continuó diariamente hasta enfermar los ratones, o después de un máximo de aproximadamente 35 días después del trasplante. Los fármacos se administraron mediante sonda esofágica (p.o.). En un experimento piloto, se identificó una dosis de quimioterapéutico que no era curativa pero que retrasaba la aparición de leucemia en aproximadamente una semana o menos; los controles habían sido tratados con vehículo o tratados con agente quimioterapéutico, que previamente se había mostrado que retrasaba, pero no curaba la leucemogénesis en el presente modelo (p.ej., imatinib a una dosis aproximada de 70 mg/kg dos veces al día). Para la primera etapa, se utilizaron células p190 que expresaban eGFP, y el análisis postmórtem se limitó a la enumeración del porcentaje de células leucémicas en médula ósea, bazo y gánglios linfáticos (GN) mediante citometría de flujo. En la segunda etapa, se utilizaron células p190 que expresaban una forma sin cola de CD4 humana y el análisis postmórtem incluía la separación magnética de las células hCD4+ a partir de bazo seguido del análisis de inmunotransferencia de criterios de valoración de señalización clave: pAkt - T308 y S473, pS6 y p4EBP-1. Como controles para la detección mediante inmunotransferencia, las células separadas se incubaron en presencia o en ausencia de inhibidores de quinasa de entre los inhibidores de la presente exposición antes de la lisis. Opcionalmente, se utilizó 'phosflow' para detectar pAkt -S473 and pS6-S235/236 en las células acotadas según hCD4 sin separación previa. Dichos estudios de señalización resultan particularmente útiles en el caso de que, por ejemplo, los ratones tratados con fármaco no hayan desarrollado leucemia clínica en el punto temporal de los 35 días. Se generaron gráficos de Kaplan-Meier y se llevaron a cabo análisis estadísticos según métodos conocidos de la técnica. Los resultados de células p190 se analizaron por separado, así como acumuladamente.
Se obtuvieron muestras de sangre periférica (100 a 200 j l) cada semana de todos los ratones, desde el día 10 inmediatamente antes de iniciar el tratamiento. Se utilizó plasma para medir las concentraciones de fármaco y se
analizaron células para marcadores de leucemia (eGFP o hCD4) y biomarcadores de señalización tal como se indica en la presente memoria.
Dicho ensayo general conocido de la técnica puede utilizarse para demostrar que las dosis terapéuticas eficaces de los compuestos proporcionados en la presente memoria pueden utilizarse para inhibir la proliferación de las células leucémicas.
Ejemplo 235: ensayo de angiogénesis en tapón de Matrigel
Se inyectó Matrigel que contenía compuestos de ensayo por vía subcutánea o por vía intraocular, en donde solidifica formando un tapón. Se recuperó el tapón tras 7 a 21 días en el animal y se examinó histológicamente para determinar el grado en que han entrado vasos sanguíneos en el mismo. Se mide la angiogénesis mediante cuantificación de los vasos en secciones histológicas. Alternativamente, se lleva a cabo una medición de la fluorescencia del volumen de plasma utilizando dextrano 150 marcado con isotiocianato de fluoresceína (FITC). Se espera que los resultados indiquen uno o más compuestos proporcionados en la presente memoria que inhiben la angiogénesis y que, de esta manera, se espera que resulten útiles para tratar trastornos oculares relacionados con la angiogénesis y/o la permeabilidad vascular aberrantes.
Ejemplo 236: ensayo de angiogénesis corneal
Se realizó un bolsillo en la córnea y un tapón que contenía una formulación inductora de angiogénesis (p.ej., VEGF, FGF o células tumorales) introducido en dicho bolsillo indujo el crecimiento hacia el interior de nuevos vasos desde la vasculatura limbal periférica. Se utilizaron materiales de liberación lenta, tales como ELVAX (copolímero de etilenovinilo) o Hydron para introducir sustancias inductoras de angiogénesis en el bolsillo corneal. Alternativamente, se utiliza un material de esponja.
El efecto de inhibidores putativos sobre las reacciones angiogénicas inducidas localmente (p.ej., implante de esponja) en la córnea (p.ej., con EGF, VEGF o células tumorales). El compuesto de ensayo se administró por vía oral, sistémica o directamente en el ojo. La administración sistémica mediante inyección de bolo, o más eficazmente, mediante la utilización de un método de liberación sostenida, tal como la implantación de bombas osmóticas cargadas con el inhibidor de ensayo. La administración en el ojo se llevó a cabo mediante cualquiera de los métodos descritos en la presente memoria, aunque sin limitarse a gotas oculares, administración tópica de una crema, emulsión o gel, y la inyección intravítrea.
Se monitorizó la respuesta vascular mediante observación directa durante el curso del experimento utilizando un estereomicroscopio en ratones. Se consiguió la visualización definitiva de la vasculatura corneal mediante la administración de dextrano de alto peso molecular marcado con fluorocromo. La cuantificación se llevó a cabo mediante la medición de la superficie de penetración de los vasos, el avance de los vasos hacia el estímulo angiogénico durante el tiempo, o en el caso de la fluorescencia, el análisis de histogramas o recuentos de píxeles que superan un umbral específico (de fondo).
Los resultados pueden indicar que uno o más compuestos proporcionados en la presente memoria inhiben la angiogénesis y, de esta manera, pueden resultar útiles en el tratamiento de trastornos oculares relacionados con la angiogénesis aberrante y/o la permeabilidad vascular.
Ejemplo 237: ensayo de angiogénesis en placa de microtitulación
Se preparó la placa de ensayo introduciendo un tapón de colágeno en el fondo de cada pocillo con 5 a 10 esferoides celulares por cada tapón de colágeno, en el que cada esferoide contenía 400 a 500 células. Se cubrió cada tapón de colágeno con 1100 pl de medio de almacenamiento en cada pocillo y se almacenó para el uso en el futuro (1 a 3 días a 37°C, 5 % de CO2). Se selló la placa con sellante. Los compuestos de ensayo se disolvieron en 200 pl de medio de ensayo, en el que por lo menos un pocillo incluía un control positivo de VEGF y por lo menos un pocillo no presentaba VEGF o compuesto de ensayo a modo de control negativo. Se sacó la placa de ensayo del incubador y se aspiró cuidadosamente el medio de almacenamiento con una pipeta. El medio de ensayo que contenía los compuestos de ensayo se pipeteó sobre el tapón de colágeno. El tapón se introdujo en un incubador humidificado durante 24 a 48 horas (37°C, 5 % de CO2). Se cuantificó la angiogénesis mediante el recuento del número de brotes, midiendo la longitud media de brote o determinando la longitud acumulativa de brote. El ensayo puede preservarse para el análisis posterior mediante extracción del medio de ensayo, añadiendo 1 ml de paraformaldehído al 1 % en BSS de Hanks en cada pocillo y almacenando a 4°C. Se espera que los resultados identifiquen los compuestos que inhiben la angiogénesis en diversos tipos celulares sometidos a ensayo, incluyendo células de origen ocular.
Ejemplo 238: uso de combinación de inhibidores de PI3K-5 y agentes que inhiben la producción o actividad de IgE (Ejemplo de referencia)
Los compuestos tal como se proporcionan en la presente memoria pueden presentar una eficacia sinérgica o aditiva al administrarlos en combinación con agentes que inhiben la producción o actividad de IgE. Entre los agentes que
inhiben la producción de IgE se incluyen, por ejemplo, uno más de entre TEI-9874, ácido 2-(4-(6-ciclohexiloxi-2-naftiloxi)fenilacetamida)benzoico, rapamicina, análogos de rapamicina (es decir, rapálogos), inhibidores de TORC1, inhibidores de TORC2 y cualesquiera otros compuestos que inhiben mTORC1 y mTORC2. Entre los agentes que inhiben la actividad de IgE se incluyen, por ejemplo, anticuerpos anti-IgE tales como el omalizumab y TNX-901.
Uno o más de los compuestos de la invención capaces de inhibir PI3K-8 pueden resultar eficaces en el tratamiento de trastornos autoinmunes e inflamatorios (TAII), por ejemplo la artritis reumatoide. En el caso de que cualquiera de los compuestos cause un nivel no deseado de producción de IgE, puede seleccionarse la administración del mismo en combinación con un agente que inhibe la producción de IgE o la actividad de IgE. Además, la administración de inhibidores de PI3K-8 o PI3K-8/Y tal como se proporcionan en la presente memoria en combinación con inhibidores de mTOR también pueden mostrar sinergia a través de la inhibición potenciada de la ruta de PI3K. Pueden utilizarse diversos modelos in vivo e in vitro para establecer el efecto de dicho tratamiento de combinación sobre los TAII, incluyendo, aunque sin limitación: (a) Ensayo in vitro de producción de anticuerpos de células B, (b) ensayo de TNP in vivo y (c) modelo de artritis inducida con colágeno en roedores.
(a) Ensayo de células B
Los ratones fueron eutanizados y se extrajeron los bazos y se dispersaron a través de una malla de nilón para generar una suspensión de células individuales. Se lavaron los esplenocitos (tras la eliminación de los eritrocitos mediante choque osmótico) y se incubaron con microperlas conjugadas con anticuerpo anti-CD43 y anti-Mac-1 (Milteny Biotec). Las células unidas a perlas se separaron de las células no unidas utilizando un separador celular magnético. La columna magnetizada retiene las células no deseadas y las células B en reposo se recogen en el eluido. Las células B purificadas se estimulan con lipopolisacárido o un anticuerpo anti-CD40 e interleuquina 4. Las células B estimuladas se tratan con vehículo solo o con inhibidores de PI3K-8 tal como se proporcionan en la presente memoria con y sin inhibidores de mTOR, tales como rapamicina, rapálogos o inhibidores de mTORC1/C2. Se esperaba que los resultados mostrasen que, en presencia de inhibidores de mTOR (p.ej., rapamicina) solos, se producía un efecto pequeño o nulo sobre la respuesta de IgG e IgE. Sin embargo, en presencia de PI3K-8 o inhibidores de mTOR, se esperaba que las células B mostrasen una respuesta de IgG reducida en comparación con las células B tratadas con solo vehículo, y se esperaba que las células B mostrasen una respuesta de IgE reducida en comparación con la respuesta de las células B tratadas con inhibidores de PI3K-8 solos.
(b) Ensayo de TNP
Se inmunizaron los ratones con TNP-Ficoll o TNF-KHL y se trataron con: vehículo, un inhibidor de PI3K-8, un inhibidor de mTOR, por ejemplo rapamicina, o un inhibidor de PI3K-5 en combinación con un inhibidor de mTOR, tal como rapamicina. Se midió la IgE sérica específica de antígeno mediante ELISA utilizando placas recubiertas con TNP-BSA y anticuerpos marcados específicos de isotipo. Se esperaba que los ratones tratados con un inhibidor de mTOR solo mostrasen poco o ningún efecto sustancial sobre la respuesta de IgG3 específica de antígeno y una elevación no estadísticamente significativa de la respuesta de IgE en comparación con el control de vehículo. Se esperaba además que los ratones tratados con tanto inhibidor de PI3K-5 como inhibidor de mTOR mostrarían una reducción de la respuesta de IgG3 específica de antígeno en comparación con los ratones tratados con vehículo solo. Además, los ratones tratados con tanto inhibidor de PI3K-5 como inhibidor de mTOR mostrarían una reducción de la respuesta de IgE en comparación con los ratones tratados con inhibidor de PI3K-5 solo.
(c) Modelo de artritis inducida con colágeno en la rata
Se anestesiaron ratas de Lewis hembra y se les inyectó colágeno preparado y administrado tal como se ha indicado anteriormente, el día 0. El día 6, los animales fueron anestesiados y se les proporcionó una segunda inyección de colágeno. Se llevaron a cabo mediciones con calibrador de las articulaciones del tobillo derecho e izquierdo normales (preenfermedad) el día 9. Los días 10 y 11, típicamente se producía la artritis y las ratas se asignaban aleatoriamente a grupos de tratamiento. La aleatorización se llevó a cabo una vez se había establecido patentemente la inflamación articular de los tobillos y había pruebas sólidas de enfermedad bilateral.
Después de seleccionar un animal para la inclusión en el estudio, se inició el tratamiento. Los animales recibieron vehículo, inhibidor de PI3K-5 o inhibidor de PI3K-5 en combinación con rapamicina. Las dosis se administraron los días 1 a 6. Se pesaron las ratas los días 1 a 7 tras el establecimiento de artritis y se obtuvieron mediciones con calibrador de los tobillos cada día. Se obtuvieron los pesos corporales finales el día 7 y los animales fueron eutanizados.
El tratamiento de combinación con un compuesto proporcionado en la presente memoria y rapamicina puede proporcionar una mayor eficacia que el tratamiento con inhibidor de PI3K-5 solo.
Ejemplo 239: modelo de hipersensibilidad de tipo retardado (Ejemplo de referencia)
Se indujo DTH mediante sensibilización de 60 ratones BALB/c los días 0 y 1 con una solución de 2,4-dinitrofluorobenceno (DNFB) al 0,05 % en una mezcla 4:1 de acetona/aceite de oliva. Los ratones se restringieron
suavemente mientras se aplicaban 20 |jl de solución en las almohadillas de las patas traseras de cada ratón. Se utilizaron las almohadillas de las patas traseras de los ratones ya que representan un sitio anatómico que puede aislarse e inmovilizarse fácilmente sin anestesia. El día 5, los ratones recibieron la administración de una única dosis de vehículo, un compuesto proporcionado en la presente memoria a una dosis de 10, 3, 1 o 0,3 mg/kg, o dexametasona a una dosis de 5 mg/kg mediante sonda esofágica. Treinta minutos después, los ratones fueron anestesiados y se aplicó una solución de DNFB al 0,25 % en una solución 4:1 de acetona/aceite de oliva en las superficies interna y externa de la oreja izquierda. Dicha aplicación resulta en la inducción de la inflamación de la oreja izquierda y bajo estas condiciones, todos los animales respondieron a dicho tratamiento con inflamación de la oreja. Se aplicó una solución de control de vehículo 4:1 de acetona/aceite de oliva en las superficies interna y externa de la oreja derecha. Veinticuatro horas después, los ratones fueron anestesiados y se realizaron mediciones de las orejas izquierda y derecha utilizando un micrómetro digital. Se registró la diferencia entre las dos orejas como la cantidad de inflamación inducida por el reto de DNFB. Se compararon los grupos de tratamiento de fármaco con el control de vehículo a fin de generar el porcentaje de reducción de la inflamación de oreja. Se utiliza la dexametasona rutinariamente como control positivo, ya que presenta una amplia actividad antiinflamatoria.
Ejemplo 240: modelo de artritis de rata con peptidoglicano-polisacárido (Ejemplo de referencia)
(a) Modelo de artritis sistémica
Todas las inyecciones se llevaron a cabo bajo anestesia. Se anestesiaron 60 ratas de Lewis hembra (150 a 170) mediante inhalación de isoflurano utilizando un pequeño aparato de anestesia para animales. Se introdujeron los animales en la cámara de inducción hasta la anestesia mediante administración de isoflurano al 4-5 % en O2 y después se mantuvieron en ese estado con un cono nasal en la mesa de operaciones. El nivel de mantenimiento de isoflurano era de 1-2 %. Los animales recibieron una única inyección intraperitoneal (i.p.) de PG-PS 10S grupo A, cepa D58 (concentración: 25 jg /g de peso corporal) en suspensión en solución salina estéril al 0,85 %. Cada animal recibió un volumen total de 500 microlitros administrado en el cuadrante izquierdo inferior del abdomen utilizando una jeringa de 1 mililitro con una aguja de calibre 23. La colocación de la aguja es crítica para evitar inyectar el PG-PS 10S en el estómago o en el ciego. Los animales se encontraban bajo observación continua hasta su recuperación total de la anestesia y la recuperación de la movilidad. Una respuesta aguda de un incremento rápido de la medición del tobillo, típicamente 20 % superior a la medición de línea base, puede alcanzar un pico 3 a 5 días después de la inyección. El tratamiento con compuestos de ensayo puede ser PO, SC, IV o IP. Las ratas no recibieron más de dos dosis en un periodo de 24 horas. El tratamiento podía iniciarse el día 0 en cualquier día posterior hasta el día 30. Los animales se pesaron los días 0, 1, 2, 3, 4, 5, 6, 7 y nuevamente los días 12 a 30 o hasta terminar el estudio. Se midió el diámetro de pata/tobillo con un calibrador digital en los lados izquierdo y derecho el día 0 antes de la inyección y nuevamente los días 1,2, 3, 4, 5, 6 y 7. El día 12, se iniciaron nuevamente las mediciones y continuaron hasta el día 30. En este momento, los animales pueden anestesiarse con isoflurano, tal como se ha indicado anteriormente, y pueden obtenerse muestras de sangre terminal mediante extracciones de la vena de la cola para la evaluación de los niveles sanguíneos de compuesto, la química clínica o parámetros hemáticos. A continuación, los animales fueron eutanizados con una sobredosis de dióxido de carbono. Puede llevarse a cabo una toracotomía como medio de verificación de la muerte.
(b) Modelo de artritis monoarticular
Todas las inyecciones se llevaron a cabo bajo anestesia. Se anestesiaron 60 ratas de Lewis hembra (150 a 170) mediante inhalación de isoflurano utilizando un pequeño aparato de anestesia para animales. Se introdujeron los animales en la cámara de inducción hasta la anestesia mediante administración de isoflurano al 4-5 % en O2 y después se mantuvieron en ese estado con un cono nasal en la mesa de operaciones. El nivel de mantenimiento de isoflurano era de 1-2 %. Los animales recibieron una única inyección intraarticular (i.a.) de PG-PS 10S grupo A, cepa D58 (concentración: 500 jg/m l) en suspensión en solución salina estéril al 0,85 %. Cada rata recibió un volumen total de 10 microlitros administrado en el espacio articular tibiotalar utilizando una jeringa de 1 mililitro con una aguja de calibre 27. Los animales se encontraban bajo observación continua hasta su recuperación total de la anestesia y la recuperación de la movilidad. Los animales que respondieron 2 a 3 días después con un incremento agudo de la medición del tobillo, típicamente 20 % superior a la medición de línea base en la inyección i.a. inicial, fueron incluidos en el estudio. El día 14, todos los respondedores fueron nuevamente anestesiados utilizando el procedimiento anteriormente indicado. Los animales recibieron una inyección intravenosa (I.V.) de PG-PS (concentración: 250 jE/ml). Cada rata recibió un volumen total de 400 microlitros administrado lentamente en la vena lateral de la cola utilizando una jeringa de 1 mililitro con una aguja de calibre 27. Se obtuvieron mediciones de línea base del tobillo antes de la inyección I.V. y se continuaron realizando durante el curso de la inflamación o hasta el día 10. El tratamiento con compuestos de ensayo podía ser PO, SC, IV o IP. Las ratas no recibieron más de dos dosis en un periodo de 24 horas. El tratamiento podía iniciarse el día 0 en cualquier día posterior hasta el día 24. Los animales se pesaron los días 0, 1, 2, 3, 4 y 5, y nuevamente los días 14 a 24 o hasta terminar el estudio. Se midió el diámetro de pata/tobillo con un calibrador digital en los lados izquierdo y derecho el día 0 antes de la inyección y nuevamente los días 1,2, 3, 4 y 5, e iniciándose nuevamente los días 14 a 24, o hasta la terminación del estudio. En este momento, los animales pueden anestesiarse con isoflurano, tal como se ha indicado anteriormente, y pueden obtenerse muestras de sangre terminal mediante extracciones de la vena de la cola para la evaluación de los niveles sanguíneos de compuesto, la
química clínica o parámetros hemáticos. A continuación, los animales fueron eutanizados con una sobredosis de dióxido de carbono. Puede llevarse a cabo una toracotomía como medio de verificación de la muerte.
Ejemplo 241: modelos de ratón de asma (Ejemplo de referencia)
La eficacia de un compuesto proporcionado en la presente memoria en el tratamiento, prevención y/o control del asma puede evaluarse utilizando modelos animales convencionales, incluyendo diversos modelos de ratón descritos en, por ejemplo, Nials et al., Dis. Model Mech. 1(4-5): 213-220, 2008).
(a) Modelos de reto agudo con alérgeno
Varios modelos son conocidos de la técnica y puede utilizarse cualquiera de dichos modelos. Aunque pueden utilizarse diversos alérgenos para inducir condiciones similares al asma, el principio es consistente a lo largo de todos los métodos. Brevemente, se indujeron condiciones similares al asma mediante una administración sistémica múltiple del alérgeno (p.ej., huevas, extractos de ácaros del polvo domésticos y extractos de cucaracha) en presencia de un adyuvante, tal como hidróxido de aluminio. Alternativamente, puede utilizarse un sistema sin adyuvante, aunque habitualmente requiere un número más alto de exposiciones para conseguir una sensibilización adecuada. Una vez inducida, los animales muestran muchas características clave del asma clínico, tales como niveles elevados de IgE, inflamación de las vías respiratoria, hiperplasia de las células caliciformes, hipertrofia epitelial, hiperreactividad de las vías respiratorias a estímulos específicos y broncoconstricción de fase temprana y tardía. De esta manera, la eficacia potencial de un compuesto puede evaluarse mediante la determinación de si una o más de dichas características clínicas se revierte o se mitiga.
(b) Modelos de reto crónico de alérgeno
Los modelos de reto crónico de alérgeno tienen como objetivo reproducir más de las características del asma clínico, tales como el remodelado de las vías respiratorias y la hiperreactividad persistente de las vías respiratorias, que los modelos de reto agudo. Mientras que pueden utilizarse alérgenos similares a los utilizados en los modelos de reto agudo de alérgeno, en los modelos de reto crónico de alérgeno, los animales se someten a la exposición repetida de las vías respiratorias a niveles bajos de alérgeno durante un periodo de hasta 12 semanas. Una vez inducido, los animales muestran características clave del asma humano, tales como sensibilización dependiente de alérgeno, una inflamación alérgica dependiente de Th2 caracterizada por el flujo de entrada eosinofílico en la mucosa de las vías respiratorias, hiperreactividad y remodelado de las mismas, tal como pone de manifiesto la hiperplasia de las células caliciformes, la hipertrofia epitelial, y la fibrosis subepitelial o peribronquiolar. De esta manera, la eficacia potencial de un compuesto puede evaluarse mediante la determinación de si una o más de dichas características clínicas se revierte o se mitiga.
Ejemplo 242: modelos de soriasis (Ejemplo de referencia)
La eficacia de un compuesto proporcionado en la presente memoria en el tratamiento, prevención y/o control de la soriasis puede evaluarse utilizando modelos animales convencionales, incluyendo diversos modelos animales descritos en, por ejemplo, Boehncke et al., Clinics in Dermatology, 25: 596-605, 2007).
A título de ejemplo, puede proporcionarse el modelo de ratón basado en la transferencia adoptiva de células T CD4+CD45RBhi descrita en Hong et al., J. Immunol., 162: 7480-7491 (1999). Brevemente, se alojaron ratones BALB/cBY hembra (donantes) y C.B.-17/Prkdc scid/scid (receptores) en un medio libre de patógenos específicos y se utilizaron con edades de entre 6 y 8 semanas. Se enriqueció en células T CD4+ a partir de esplenocitos de BALB/cBy utilizando un kit de enriquecimiento de CD4 de ratón. A continuación, se marcaron las células con anticuerpo anti-CD4 conjugado con PE, anti-CD45RB conjugado con FITC y anti-CD25 conjugado con APC. Las células se separaron con un separador celular. Se recolectaron células CD4+CD45RBhiCD25. Las células se resuspendieron en solución salina y se inyectaron i.p. 4x108 células/ratón en ratones C.B.-17/Prkdc scid/scid. Los ratones recibieron dosis de LPS, citoquinas o anticuerpos, según resultase necesario. Los ratones fueron monitorizados para signos externos de lesiones en la piel dos veces a la semana. Tras la terminación, podían recogerse orejas, piel del lomo, ganglios linfáticos y bazo para estudios ex vivo adicionales.
Ejemplo 243: modelos de escleroderma (Ejemplo de referencia)
La eficacia de un compuesto en el tratamiento del escleroderma puede someterse a ensayo utilizando modelos animales. Un modelo animal ejemplar es un modelo de ratón de escleroderma inducido mediante inyecciones locales repetidas de bleomicina ("BLM") descritas en, por ejemplo, Yamamoto et al., J. Invest. Dermatol. 112: 456-462, 1999). Dicho modelo de ratón proporciona esclerosis dérmica que es estrechamente similar a la esclerosis sistémica tanto histológica como bioquímicamente. Entre los cambios escleróticos observados en el modelo se incluyen, aunque sin limitarse a ellos, haces de colágeno engrosados y homogéneos e infiltrados celulares, incremento gradual del número de mastocitos, degranulación de los mastocitos, liberación elevada de histaminas, incremento de la hidroxiprolina en la piel, presencia de anticuerpo antinuclear en el suero y fuerte expresión del ARNm de factor p-2 de crecimiento
transformante. Por lo tanto, la eficacia de un compuesto en el tratamiento del escleroderma puede evaluarse mediante la monitorización de la reducción de uno o más de dichos cambios.
Brevemente, pueden utilizarse los procedimientos ejemplares siguientes para generar el modelo de ratón de escleroderma: se adquieren ratones BALB/C hembra libres de patógenos específicos y ratones C3H de 6 semanas de edad, de aproximadamente 20 g de peso, y se mantienen con alimento y agua ad libitum. Se disuelve BLM en PBS a diferentes concentraciones y se esteriliza mediante filtración. Se inyectan por vía subcutánea alícuotas de cada concentración de BLM o PBS en el lomo afeitado de los ratones cada día durante 1 a 4 semanas utilizando una aguja. Alternativamente, los ratones reciben inyecciones en días alternos.
Los cambios histopatológicos y bioquímicos inducidos pueden evaluarse utilizando cualesquiera métodos puestos en práctica habitualmente en la técnica. Por ejemplo, los cambios histopatológicos pueden evaluarse utilizando una técnica estándar de avidina-biotina peroxidasa con anticuerpo monoclonal anti-L3T4, anticuerpo monoclonal anti-Lyt2, anticuerpo antimacrófagos de ratón fijados de múltiples tejidos, anticuerpo monoclonal antifactor de células madre, anticuerpo policlonal antifactor p de crecimiento transformante y anticuerpo antidecorina. La expresión de citoquinas en los infiltrados celulares puede evaluarse mediante la utilización de varios anticuerpos anticitoquina. El nivel de hidroxiprolina puede evaluarse mediante la hidrólisis de trozos de piel con ácido clorhídrico, neutralizando con hidróxido sódico y evaluando colorimétricamente en los hidrolizados a 560 nm con p-dimetilaminobenzaldehído. El colágeno resistente a pepsina puede evaluarse mediante tratamiento de la muestra de colágeno extraída de los tejidos biopsiados y análisis mediante electroforesis en gel acumulador de poliacrilamida. Los mastocitos pueden identificarse con azul de toluidina, y las células que contienen gránulos metacromáticos pueden contarse bajo una amplificación elevada de un microscopio óptico. Pueden evaluarse los niveles séricos de diversas citoquinas mediante ensayo de inmunosorción ligada a enzima y los niveles de ARNm de las citoquinas pueden evaluarse mediante reacción en cadena de la polimerasa-transcriptasa inversa. Los autoanticuerpos en suero pueden detectarse utilizando fibroblastos 3T3 como el sustrato para el cribado.
Ejemplo 244: modelos de miositis (Ejemplo de referencia)
La eficacia de un compuesto en el tratamiento de la miositis (p.ej., la dermatomiositis) puede someterse a ensayo utilizando modelos animales conocidos de la técnica. Un ejemplo de estos es el modelo de dermatomiositis canina familiar, descrito en Hargis et al., AJP 120(2): 323-325, 1985). Otro ejemplo es el modelo de ratón inducido con miosina de conejo, descrito en Phyanagi et al., Arthritis & Rheumatism, 60(10): 3118-3127, 2009).
Brevemente, se utilizaron ratones SJL/J macho de 5 semanas de edad. Se emulsionó miosina purificada de músculo esquelético de conejo (6,6 mg/ml) con una cantidad igual de adyuvante completo de Freund y 3,3 mg/ml de Mycobacterium butyricum. Los ratones fueron inmunizados repetidamente con miosina de conejo emulsionada. Una vez se hubo inducido la miositis, la infiltración de células inflamatorias y las fibras musculares necróticas deberían resultar evidentes en el modelo. En los músculos de los animales, las células T CD4+ se localizan principalmente en el perimisio y las células T CD8+ se localizan principalmente en el endomisio y circundando las fibras musculares no necróticas. TNFa, IPNy y perforina están reguladas positivamente y la molécula de adhesión intercelular 1 se encuentran incrementadas en los músculos.
Con el fin de evaluar la eficacia de un compuesto, tras la administración del compuesto por la vía adecuada a una dosis especificada, los ratones fueron eliminados y se recolectaron los tejidos musculares. El tejido muscular se congeló inmediatamente en isopentano frío, preenfriado en nitrógeno líquido, y después se prepararon secciones criostáticas. Las secciones se tiñeron con hematoxilina y eosina para el recuento del número de células infiltradas. Se prepararon tres secciones de cada ratón y se obtuvieron fotomicrografías. Para los ensayos inmunohistoquímicos, se secaron secciones criostáticas de músculo y se fijaron en acetona fría a -20°C. Los portaobjetos se rehidrataron en PBS y después se bloqueó la actividad endógena de peróxido mediante incubación en peróxido de hidrógeno al 1 %. Se incubaron las secciones durante la noche con anticuerpo monoclonal de rata anti-CD4 de ratón, anticuerpo monoclonal de rata anti-CD8 de ratón, anticuerpo monoclonal de rata anti-F4/80 de ratón o IgG de rata normal en diluyente de anticuerpo. Las muestras se lavaron con PBS y se incubaron con anticuerpo de conejo anti-IgG de rata conjugado con biotina y pretratado con suero de ratón normal al 5 %. Tras el lavado con PBS, las muestras se incubaron con estreptavidina- peroxidasa de rábano picante. Tras lavar con PBS, se utilizó diaminobencidina para la visualización.
Ejemplo 245: modelos de síndrome de Sjogren (Ejemplo de referencia)
La eficacia de un compuesto en el tratamiento del síndrome de Sjogren puede someterse a ensayo utilizando modelos animales conocidos de la técnica, por ejemplo los descritos en Chiorini et al., Journal of Autoimmunity 33: 190-196, 2009). Entre los ejemplos se incluyen: modelo de ratón desarrollado espontáneamente en primera generación filial de ratones NZB cruzados con ratones NZW (ver, p.ej., Jonsson et al., Clin. Immunol. Immunopathol. 42: 93-101 (1987); modelo ratón inducido mediante inyección i.p. de adyuvante de Freund incompleto (id.; Deshmukh et al., J Oral Pathol Med 38: 42-27 (2009)); modelos de ratón NOD en los que se desarrolla fenotipo de Sjogren en genotipos específicos (ver, p.ej., Cha et al., Arthritis Rheum 46: 1390-1398 (2002); Kong et al., Clin. Exp. Rheumatol. 16: 675-681 (1998); Podolin et al., J. Exp. Med. 178: 793-803 (1993); y Rasooly et al., Clin. Immunol. Immunopathol. 81: 287-292
(1996)); modelo de ratón desarrollado en mutación de Ipr espontánea; modelo de ratón desarrollado en ratones con inactivación de Id3 (ver, p.ej., Li et al., Immunity 21: 551-560 (2004)); modelo de ratón desarrollado en ratones con inactivación de PI3K (ver, p.ej., Oak et al., Proc. Natl. Acad. Sci. USA 103: 16882-16887 (2006)); modelo de ratón desarrollado en ratones transgénicos sobreexpresantes de BAFF (ver, p.ej., Groom et al., J Clin Invest 109: 59-68 (2002)); modelo de ratón inducido mediante inyección de antígeno Ro en ratones BALB/c (ver, p.ej., Oh-Hora et al., Nat. Immunol. 9: 432-443 (2008)); modelo de ratón inducido mediante inyección de anhidrasa carbónica II (ver, p.ej., Nishimori et al., J Immunol 154: 4865-4873 (1995); modelo de ratón desarrollado en ratones transgénicos sobreexpresantes de IL-14 (ver, p.ej., Shen et al., J. Immunol.177: 5676-5686 (2006)); y modelo de ratón desarrollado en ratones transgénicos expresantes de IL-12 (ver, p.ej., McGrath-Morrow et al., Am. J. Physiol. Lung Cell Mol. Physiol.
291: L837-846 (2006)).
Ejemplo 246: modelos de enfermedad mediada por complejo inmune (Ejemplo de referencia)
La reacción de Arthus es una respuesta inmunitaria de tipo 3 a complejos inmunitarios y, de esta manera, puede ser un modelo mecanístico que apoye las hipótesis terapéuticas para las enfermedades inmunitarias mediadas por complejos inmunes, tales como la artritis reumatoide, el lupus y otras enfermedades autoinmunes. Por ejemplo, pueden utilizarse ratones deficientes en PI3Ky y 8 como modelos experimentales de la reacción de Arthus y proporcionar una evaluación del potencial terapéutico de un compuesto respecto al tratamiento de las enfermedades mediadas por complejos inmunitarios. La reacción de Arthus puede inducirse utilizando los procedimientos ejemplares siguientes, tal como se indica en Konrad et al., Journal of Biological Chemistry (2008283(48): 33296-33303, 1977.
Se mantiene ratones deficientes en PI3Ky y PI3K8 bajo condiciones de barrera seca. Los ratones son anestesiados con ketamina y xilazina, y se canaliza la tráquea. Se aplica una cantidad apropiada de anticuerpo IgG anti-OVA purificado con proteína G y se administra por vía intravenosa una cantidad apropiada de antígeno OVA. Para los experimentos de bloqueo de PI3k, se administra por vía intratraqueal wortmanina junto con la aplicación de IgG anti OVA. Los ratones fueron sacrificados a las 2-4 horas tras el inicio de la inflamación y pueden llevarse a cabo las evaluaciones deseadas de seguimiento utilizando métodos conocidos de la técnica.
Ejemplo 247: ensayo de quinasa PI3 Promega™
Se utilizó el kit de ensayo Promega ADP-Glo Max (n° de cat. V7002) para determinar los valores de IC50 para las isoformas a, p, 8 y y de las quinasas PI3 de clase I humanas (Millipore). Se incubaron muestras de quinasa (20 nM isoforma a o 8, 40 nM isoforma p o y) con compuesto durante 15 minutos a temperatura ambiente en tampón de reacción (HEPES 15 mM, pH 7,4, NaCl 20 mM, Eg TA 1 mM, Tween 20 al 0,02 %, MgCh 10 mM, bovín-Y-globulinas 0,2 mg/ml) seguido de la adición de una mezcla de ATP/diC8-PtdInsP, proporcionando concentraciones finales de ATP 3 mM y diCa-PtdInsP 500 |jM. Las reacciones se incubaron a temperatura ambiente durante 2 horas, seguido de la adición de 25 j l de solución de parada. Tras una incubación de 40 minutos a temperatura ambiente, se añadieron 50 j l de mezcla de detección de Promega, seguido de la incubación durante 1 hora a temperatura ambiente. A continuación, se leyeron las placas en un lector de placas Envision en modo de luminiscencia. Se convirtieron los datos a % de inhibición utilizando la ecuación a continuación:

en la que S es la luminiscencia de la muestra, Pos es un control positivo sin PI3K añadido, Neg es el control negativo sin compuesto añadido. A continuación, se representaron gráficamente los datos como % de inhibición vs. concentración de compuesto. Ajuste de los datos a ecuación logística de 4 parámetros para determinar los valores de IC50:

Se sometieron a ensayo determinados compuestos proporcionados en la presente memoria en el ensayo de Promega de quinasas PI3 utilizando procedimientos tales como los indicados anteriormente para determinar los valores de IC50 para las isoformas a, p, 8 y/o Y. Se resumen los valores de IC50 en la Tabla 15.
Ejemplo 248: ensayos celulares selectivos para isoforma
(a) Ensayo selectivo de PI3K-5
La capacidad de un compuesto se inhibir selectivamente PI3K-8 puede evaluarse utilizando células RAJI, es decir, células linfocitos B derivados de pacientes de linfoma. Brevemente, se estimularon células RAJI sometidas a ayuno de suero con anticuerpo anti-IgM humano, causando de esta manera la señalización mediante los receptores de
células B, tal como se indica en, por ejemplo, He et al., Leukemia Research (2009) 33: 798-802. La señalización de receptores de células B resulta importante para la activación, diferenciación y supervivencia de las células B y determinados cánceres derivados de células B. La reducción de fosfo-AKT es indicativa de compuestos que podrían inhibir la proliferación y función de las células B en determinadas enfermedades. Mediante la monitorización de la reducción de fosfo-AKT en células RAJI estimuladas (utilizando, por ejemplo, anticuerpos de fosfo-AKT), puede evaluarse la eficacia potencial de un compuesto de inhibir selectivamente PI3K8.
Se sometieron a ensayo determinados compuestos proporcionados en la presente memoria en el modelo de células RAJI utilizando procedimientos descritos anteriormente. Los valores de IC50 de fosfo-AKT se resumen en la Tabla 15.
(b) Ensayo selectivo de PI3K-Y
La capacidad de un compuesto de inhibir selectivamente PI3K-Y puede evaluarse utilizando macrófagos RAW264.7. Brevemente, se estimulan células RAW264.7 sometidas a ayuno de suero con un agonista de GPCR C5a conocido. Ver, p.ej., Camps et al., Nature Medicine (2005) 11(9):936-943. Las células pueden tratarse con compuestos de ensayo antes, simultáneamente o después de la estimulación por C5a. Las células RAW 264.7 responden al fragmento C5a componente del complemento mediante la activación del receptor C5a, y el receptor C5a activa los macrófagos e induce la migración celular. La capacidad de los compuestos de ensayo de inhibir la fosforilación de AKT mediada por C5a es indicativa de inhibición selectiva de PI3K-Y. De esta manera, mediante la monitorización de la reducción de fosfo-AKT en células RAW 264.7 estimuladas (utilizando, por ejemplo, anticuerpos de fosfo-AKT), puede evaluarse la eficacia potencial de un compuesto de inhibir selectivamente P|3Ky.
Se sometieron a ensayo determinados compuestos proporcionados en la presente memoria en el modelo de células RAW 264.7 utilizando procedimientos descritos anteriormente. Los valores de IC50 de fosfo-AKT se resumen en la Tabla 15.
(c) Ensayo selectivo para PI3K-a
La capacidad de un compuesto de inhibir selectivamente PI3K-a puede evaluarse utilizando células SKOV-3, es decir, la línea celular de carcinoma ovárico humano. Brevemente, las células SKOV-3, en las que PI3a mutante se encuentra constitutivamente activo, pueden tratarse con compuestos de ensayo. La capacidad de los compuestos de ensayo de inhibir la fosforilación de AKT en células SKOV-3; por lo tanto, es indicativa de la inhibición selectiva de PI3Ka. De esta manera, mediante la monitorización de la reducción de fosfo-AKT en células SKOV -3 (utilizando, por ejemplo, anticuerpos de fosfo-AKT), puede evaluarse la eficacia potencial de un compuesto de inhibir selectivamente PI3Ka.
(b) Ensayo selectivo de PI3K-@
La capacidad de un compuesto de inhibir selectivamente PI3K-p puede evaluarse utilizando células 786-O, es decir, la línea celular de carcinoma renal humano. Brevemente, las células 786-O, en las que PI3p mutante se encuentra constitutivamente activo, pueden tratarse con compuestos de ensayo. La capacidad de los compuestos de ensayo de inhibir la fosforilación de AKT en células 786-O; por lo tanto, es indicativa de la inhibición selectiva de PI3Kp. De esta manera, mediante la monitorización de la reducción de fosfo-AKT en células 786-O (utilizando, por ejemplo, anticuerpos de fosfo-AKT), puede evaluarse la eficacia potencial de un compuesto de inhibir selectivamente PI3Kp.
Ejemplo 249: modelos de leucemia linfocítica crónica
El modelo de ratón transgénico TCL-1 es un modelo de leucemia linfocítica crónica (LLC) que se genera mediante la introducción del gen TCL1 humano bajo el control de un promotor Ig específico de células B. Los ratones transgénicos para TCL1 desarrollan una linfocitosis monoclonal de células B que es muy similar a la LLC humana en sus características inmunofenotípicas y clínicas. Los esplenocitos leucémicos de ratones enfermos también pueden transferirse adoptivamente a ratones C57BL/6 singénicos, creando un modelo que permite generar un gran número de animales con enfermedad establecida síncronamente. Ver, p.ej., Johnson, A. J., et al., Blood 108(4): 1334-8, 2006).
La inserción de tcl-1 bajo el control de un promotor de IgVH específico de células B y potenciador de IgH-E resulta en un fenotipo de células B similar en el que los ratones se desarrollan normalmente hasta la adultez, aunque después desarrollan bazos, hígados y ganglios linfáticos agrandados que se asocian a recuentos elevados de linfocitos sanguíneos. Los linfocitos transformados de los ratones TCL-1 se detienen en G0-1, son clonales y expresan CD19+/CD5+/IgM+, tal como se observa en la LLC humana. Se recolectó un bazo leucémico transgénico de un ratón C57BL/6 previamente implantado. Bajo condiciones asépticas, se disoció el bazo y se extrajeron los glóbulos rojos. Se trasplantaron por vía intravenosa 2x106 linfocitos de bazo leucémicos TCL1 a un ratón receptor C57BL/6 hembra de 6 a 8 semanas de edad. Dos semanas después de la injertación, los ratones se sangraron mediante sangrado submandibular dos veces a la semana para comprobar la sangre periférica para el porcentaje leucémico. Se evaluaron las muestras de sangre mediante citometría de flujo para la coexpresión de CD5 y CD19 y se informa la enfermedad como porcentaje de células doblemente positivas de la población parental CD45+. Una vez un animal había alcanzado 10 % a 20 % de tinción doble positiva en la sangre periférica, se asignaba a un grupo de tratamiento. Los animales se pesaron dos veces a la semana y se palparon para esplenomegalia. Al final del estudio, los animales fueron
eutanizados y se recolectaron los tejidos para medir el peso del bazo y la enfermedad orgánica sistémica. Se evaluó la enfermedad orgánica en secciones de tejido teñidas con H+E. En estudios ejemplares, se administró un compuesto proporcionado en la presente memoria solo o en combinación con un inhibidor de PI3K8. En un estudio ejemplar, un compuesto proporcionado en la presente memoria es un inhibidor de PI3Ky. En otro estudio ejemplar, el inhibidor de PI3K5 presenta una proporción de selectividad gamma/delta superior a aproximadamente 50. En todavía otro estudio ejemplar, el inhibidor de PI3Ky presenta una proporción de selectividad delta/gamma superior a aproximadamente 50.
Ejemplo 250: efecto sobre el modelo de artritis inducida con colágeno (Ejemplo de referencia)
La artritis inducida con colágeno en ratas es un modelo experimental de poliartritis que ha sido ampliamente utilizado para ensayos preclínicos de numerosos agentes antiartríticos que se encuentran en investigación preclínica o clínica o que se utilizan actualmente como terapéuticos para esta enfermedad. Las características diferenciadoras de dicho modelo son la aparición y progresión fiables de una inflamación poliarticular robusta y fácilmente medible, una marcada destrucción de los cartílagos asociada a la formación de pannus y leve a moderada resorción ósea y proliferación de hueso periostial.
Se administró compuesto BB (compuesto selectivo de PI3K-gamma dado a conocer en la presente memoria) en modelo de ratas de artritis inducida con colágeno (AIC). El compuesto BB es un compuesto ejemplar selectivo de PI3K-gamma proporcionado en la presente memoria que presenta una proporción de selectividad delta/gamma superior a aproximadamente 50. Se administró compuesto BB a una dosis de 0,5 mg/kg, 1,5 mg/kg y 5 mg/kg en grupos, tal como se identifica a continuación:
Tabla 17.

Puntuación de articulaciones: los tobillos con artritis inducida con colágeno recibieron puntuaciones de 0 a 5 según los criterios siguientes:
Para la inflamación, 0=normal, 0,5=inflamación focal mínima, 1=infiltración mínima de células inflamatorias en sinovio/tejido periarticular, 2=infiltración leve, 3=infiltración moderada con edema moderado, 4=infiltración marcada con edema marcado, y 5=infiltración grave con edema grave.
Para el pannus, 0=normal, 0,5=infiltración mínima de pannus en cartílago y hueso subcondral, afecta únicamente a zonas marginales y afecta únicamente a unas cuantas articulaciones, 1=infiltración mínima de pannus en cartílago y hueso subcondral, afecta principalmente zonas marginales, 2=infiltración leve (<1/4 de tibias o tarsianos en zonas marginales), 3=infiltración moderada (1/4 a 1/3 e tibias o tarsianos pequeños afectados en las zonas marginales), 4=infiltración marcada (1/2-3/4 de tibias o tarsianos afectados en zonas marginales) y 5=infiltración grave (>3/4 de tibias o tarsianos afectados en zonas marginales; distorsión grave de la arquitectura general).
Para el daño en los cartílagos (especialmente los tarsianos pequeños), 0=normal, 0,5=reducción mínima en la tinción de azul de T, afecta únicamente a zonas marginales y afecta únicamente a unas cuantas articulaciones, 1=mínima; pérdida mínima a leve de tinción de azul de toluidina sin pérdida evidente de condrocitos o disrupción del colágeno, 2=leve; pérdida leve de tinción de azul de toluidina con pérdida focal leve (superficial) de condrocitos y/o disrupción del colágeno, 3=moderada; pérdida moderada de tinción de azul de toluidina con pérdida multifocal moderada (profundidad hasta la zona intermedia) de condrocitos y/o disrupción del colágeno, afectación de tarsianos más pequeños hasta 1/2-3/4 de la profundidad con zonas infrecuentes de pérdida de grosor total, 4=marcada; pérdida marcada de tinción de azul de toluidina con pérdida multifocal marcada (profundidad hasta la zona profunda) de condrocitos y/o disrupción del colágeno, 1 o 2 superficies de tarsianos pequeños con pérdida de grosor total del cartílago, y 5=grave; pérdida difusa grave de tinción de azul de toluidina con pérdida multifocal grave (profundidad hasta la transición al hueso) de condrocitos y/o disrupción del colágeno que afecta a más de 2 superficies de cartílago.
Para la resorción ósea, 0=normal, 0,5=resorción mínima, afecta solo zonas marginales y afecta solo unas cuantas articulaciones; 1=mínima, pequeñas zonas de resorción, no inmediatamente evidente a baja magnificación, osteoclastos raros; 2=leve, zonas más numerosas de resorción, no inmediatamente evidente a baja magnificación, osteoclastos más numerosos, <1/4 de tibias o tarsianos resorbidos en zonas marginales; 3=moderada, resorción evidente de hueso trabecular medular y cortical sin defectos de grosor completo en el córtex, pérdida de algunas trabéculas medulares, lesión evidente a baja magnificación, osteoclastos más numerosos, 1/4 a 1/3 de tibias o tarsianos afectados en zonas marginales; 4=marcada, defectos de grosor completo en hueso cortical, con frecuencia con distorsión del perfil de la superficie cortical restante, pérdida marcada de hueso medular, numerosos osteoclastos, 1/2-3/4 de las tibias o tarsianos afectados en zonas marginales; 5=grave, defectos de grosor completo en hueso
cortical, con frecuencia con distorsión del perfil de la superficie cortical restante, pérdida marcada de hueso medular, numerosos osteoclastos, >3/4 de tibias o tarsianos afectados en zonas marginales, distorsión grave de la arquitectura general.
Formación de hueso nuevo periostial (medida a 16X): Los estudios que van más allá del estadio inflamatorio agudo con frecuencia muestran grados variables de formación de hueso nuevo periostial. Con el fin de comunicar el grado de formación de hueso nuevo periostial, se aplican las puntuaciones siguientes basadas en la distribución de la proliferación de hueso periostial y la medición de la anchura de formación de hueso nuevo periostial en la localización más amplia. 0=Normal, sin proliferación periostial; 0,5=prolfieración focal o multifocal mínima, medidas inferiores a 127 |jm de anchura (1-2) en cualquier sitio; 1,0=proliferación multifocal mínima, anchura en cualquier sitio mide 127 jm -252 jm (3-4 unidades); 2,0=multifocal level en tarsianos, difusa en algunos sitios, anchura en cualquier sitio de 253 jm-441 jm (5-7 unidades); 3,0=multifocal moderada en tarsianos, difusa en la mayoría de otros sitios, anchura en cualquier sitio mide 442 jm-630 jm (8-10 unidades); 4,0=multifocal marcada en tarsianos, difusa en la mayoría de otros sitios, anchura en cualquier sitio mide 630 jm-819 jm (11-13 unidades); 5,0=grave, multifocal en tarsianos, difusa en la mayoría de otros sitios, anchura en cualquier sitio mide >819 jm (>13 unidades).
Se analizaron los datos utilizando un ANOVA de un factor o una prueba de Kruskal-Wallis (ANOVA no paramétrico), junto con un post-test apropiado de comparaciones múltiples. A menos que se indique lo contrario, Bolder BioPATH, Inc. realiza análisis estadísticos de datos en bruto (no transformados) únicamente. Las pruebas estadísticas realizan determinadas suposiciones sobre la normalidad y homogeneidad de la varianza de los datos, y pueden requerirse análisis adicionales si las pruebas resultan en violaciones de estos supuestos. Se fijó la significancia para todas las pruebas en p<0,05.
La FIG. 1 muestra que el compuesto BB presenta efectos terapéuticos en el modelo de AIC de rata, en el que el diámetro medio del tobillo en pulgadas se mide entre los días 9 y 17. La FIG. 2 muestra las puntuaciones individuales de histopatología para inflamación, pannus, daños en el cartílago, resorción ósea y formación de hueso periostial. Más particularmente, en el experimento mostrado en la FIG. 2, se redujo la inflamación en 57 %, 36 % y 27 % al tratar los animales con compuesto BB a dosis de 5, 1,5 y 0,5 mg/kg, respectivamente. Se redujo el pannus en 71 %, 44 % y 28 % al tratar los animales con compuesto BB a dosis de 5, 1,5 y 0,5 mg/kg, respectivamente. Se redujo el daño a las articulaciones en 59 %, 45 % y 28 % al tratar los animales con compuesto BB a dosis de 5, 1,5 y 0,5 mg/kg, respectivamente. Se redujo la resorción ósea en 65 %, 44 % y 25 % al tratar los animales con compuesto BB a dosis de 5, 1,5 y 0,5 mg/kg, respectivamente. Se redujo la formación de hueso periostial en 82 %, 52 % y 52 % al tratar los animales con compuesto Bb a dosis de 5, 1,5 y 0,5 mg/kg, respectivamente. La FIG. 3 muestras las medidas del hueso periostial. Los animales que habían sido tratados con 5 mg/kg de compuesto BB presentaban reducciones de 57 % a 82 % en todas las puntuaciones que resultaron en una reducción significativa de 64 % de las puntuaciones sumadas. Las anchuras de hueso periostial también se redujeron significativamente en 82 %. Los animales que habían sido tratados con 1,5 mg/kg de compuesto BB presentaron puntuaciones significativamente reducidas de inflamación y daño en el cartílago (45 %), contribuyendo a una reducción significativa de 42 % de las puntuaciones sumadas. Aunque las puntuaciones de hueso periostial no se redujeron significativamente, las anchuras de hueso periostial se redujeron significativamente en 50 %. Los animales tratados con 0,5 mg/kg de compuesto BB presentaron anchuras de hueso periostial significativamente reducidas (47 %). Los parámetros puntuados se redujeron en 27 % a 52 %, aunque no significativamente, con una reducción no significativa de 31 % de las puntuaciones sumadas. Los resultados del presente estudio indican que el tratamiento con compuesto BB presenta efectos beneficiosos sobre las lesiones de la artritis inducida con colágeno en ratas que eran generalmente significativos a la dosis de 1,5 mg/kg y superior. El valor de EC50 para dicho tratamiento era de 2,132 mg/kg. Todas las dosis de compuesto BB mostraron en las pruebas una formación significativamente reducida de hueso periostial.
Ejemplo 251: efecto sobre el modelo murino de bolsa de aire inducida por IL-8 (Ejemplo de referencia)
El propósito era estudiar la evaluación de la respuesta a dosis del efecto de neutrófilos infiltrantes en bolsas de aire en ratones estimulados con IL-8_h recombinante tras el tratamiento con compuesto BB o compuesto AA. El compuesto AA es un compuesto selectivo de PI3K-delta que presenta una proporción de selectividad gamma/delta superior a aproximadamente 50. El compuesto BB es un compuesto selectivo de PI3K-gamma indicado en la presente memoria que presenta una proporción de selectividad delta/gamma superior a aproximadamente 50. Se adquirieron ratones Balb/c macho (stock n° 000651) de 6 semanas de edad de Jackson Labs (n=60).
Modelo/procedimiento:
Se utilizó el modelo de bolsa de aire como espacio pseudosinovial para investigar el papel de citoquinas específicas y/o el efecto de compuestos sobre sucesos de tipo inflamatorio. Las bolsas se estimularon con IL-8 humana recombinante, que es una quimioquina CXC proinflamatoria que puede señalar a través de los receptores CXCR1 y CXCR2. IL-8 quimioatrae y activa los neutrófilos.
Las bolsas se desarrollan anestesiando los ratones con isoflurano a un caudal de 3-4 %. Las bolsas de aire se producen mediante la administración de una inyección subcutánea de 5 ml de aire estéril. Lo anterior puede llevarse a cabo utilizando una jeringa de 5 ml con un microfiltro de 22 acoplado y una aguja 25G5/8" acoplada al filtro. Se aspiran 5 ml de aire con la jeringa; se coloca el filtro con la aguja en la jeringa. La zona intraescapular del lomo puede
frotarse con una almohadilla con alcohol al 70 %. Pinzando la piel entre la zona intraescapular, se crea un abombamiento en el que puede insertarse la aguja subcutáneamente entre los hombros del ratón. El aire se inyecta lentamente, utilizando la mano izquierda para guiar el aire para formar una bolsa cilíndrica alargada (aproximadamente 10 segundos), procurando no dejar que el aire baje por los brazos o sobre la cabeza del ratón. Una vez se ha administrado la totalidad de los 5 ml, se extrae la aguja y se pinza el sitio de inyección durante aproximadamente 5 s. A continuación, los ratones se devuelven a sus jaulas y se monitorizan durante hasta una hora, durante la recuperación. Los ratones se comprueban durante todo el día el día en que se forman bolsas y se comprueban diariamente durante los siguientes 3 días. El día 3, en los ratones se reinyectan 3 ml de aire estéril, siguiendo el procedimiento proporcionado anteriormente, utilizando una jeringa de 3 ml con una aguja 25G.
El día 6, se pesan los ratones y se asignan a un grupo y a continuación se administra compuesto. Una hora después de la administración, se extrae sangre de los ratones por vía submandibular. Se recogen aproximadamente 150 pl de sangre en un tubo microtainer con EDTA. Las muestras de sangre se dejan sobre hielo, se centrifugan durante l0 min a 10.000 rpm a 4 grados. Se introduce el plasma en un tubo Eppendorf y se almacena a -80°C hasta el análisis por el grupo de FC. Inmediatamente después de la recolección de sangre, los ratones son anestesiados y se inyecta 1 ml de estimulante IL-8 frío o PBS en la bolsa utilizando una jeringa de 1 ml con una aguja 25G. A continuación, se masajea la bolsa suavemente para garantizar que todo el revestimiento de la bolsa ha sido expuesto a la solución.
Recolección de muestras:
Cuatro horas después de la estimulación, los ratones se eutanizan con CO2. Se sangraron los ratones mediante punción cardiaca y se recogió la sangre igual que se ha indicado anteriormente. A continuación, se recogieron exudados de las bolsas de la manera siguiente: se inyectaron 3 ml de PBS libre de endotoxinas frío con EDTA 1 mM en la bolsa con una jeringa de 3 ml con aguja 20G. Se masajearon las bolsas para suspender adecuadamente las células en la bolsa, procurando que la aguja se mantuviese en la bolsa y evitar fugas. Se extrajeron 2 ml de líquido de lavado del interior de las bolsas y se introdujeron en un tubo de fondo redondo Falcon BD de 5 ml y se dejaron sobre hielo hasta el análisis. Se analizó el líquido de lavado en el Cell Dyn y se representaron gráficamente los resultados de las diferentes células, centrados en los recuentos de neutrófilos totales de cada muestra de lavado.
La línea temporal de ejemplo del procedimiento experimental se proporciona en la tabla a continuación:
Tabla 18:

Para las inyecciones en las bolsas, en modelos sin estimulación se inyectó 1 ml de PBS sin endotoxinas y en los modelos de estimulación, se inyectaron 10 pg de IL-8 en 1 ml de PBS sin endotoxinas. Los reactivos eran CXCL8/IL-8 humano recombinante - R&D Systems, n° de cat. 208-IL-050/CF, lote n° BA3313051; PBS sin endotoxinas-Teknova, n° de cat. P0300, y EDTA (0,5 M)- Sigma, n° de cat. E-7889EDTA (0,5 M)- Sigma, n° de cat. E-7889. Cada vial contenía 100 pg de IL-8, pueden utilizarse 6 para el presente experimento. La solución madre se reconstituyó a 0,1 mg/ml
mediante la adición de 1 ml de PBS sin endotoxinas, 10 pg/bolsa (5,5 ml de solución madre 50 ml de PBS). Se preparó EDTA utilizando una concentración de la solución madre de 0,5 M y añadiendo 500 pl de solución madre a 250 ml de PBS para una concentración final de EDTA de 1 mM.
Se sometieron a ensayo el compuesto BB y el compuesto AA en el modelo de bolsa de aire murino según los procedimientos descritos anteriormente. Se administró compuesto BB (compuesto selectivo de PI3K-gamma) en el modelo de neutrófilos inducidos por IL-8 a dosis de 0,1 mg/kg, 0,3 mg/kg, 1 mg/kg y 3 mg/kg. Se administró compuesto AA (compuesto selectivo de PI3K-delta) en el modelo de neutrófilos inducidos por IL-8 a dosis de 10 mg/kg, 25 mg/kg y aproximadamente 50 mg/kg. Se compararon los resultados con el vehículo. La FIG. 4 muestra que el compuesto BB (compuesto selectivo de PI3K-gamma) bloquea la migración de neutrófilos inducidos por IL-8 y la FIG. 5 muestra que el compuesto AA (compuesto selectivo de PI3K-delta) no inhibe la migración de neutrófilos inducidos por IL-8. El presente ejemplo de muestra que los compuestos selectivos de Pi3k-gamma proporcionados en la presente memoria pueden utilizarse para reducir y/o prevenir la inflamación.
Ejemplo 252: los compuestos selectivos para gamma muestran sinergia con los compuestos selectivos para delta
El propósito del presente estudio es evaluar los efectos anticáncer de combinaciones de compuestos selectivos de PI3K-gama proporcionados en la presente memoria y compuestos selectivos de PI3K-delta. Los compuestos selectivos de PI3K-gamma proporcionados en la presente memoria se combinan con un compuesto que inhibe selectivamente la isoforma delta de PI3K respecto a la isoforma gamma (p.ej., los compuestos selectivos de delta), proporcionando sinergia. Específicamente, los compuestos proporcionados en la presente memoria que presentan una proporción de selectividad delta/gamma entre superior a aproximadamente 1 y <10, entre superior a aproximadamente 10 y <50, o entre superior a aproximadamente 50 y <350 se combinan con una proporción de selectividad gamma/delta superior a aproximadamente un factor de aproximadamente 1, superior a un factor de aproximadamente 2, superior a un factor de aproximadamente 3, superior a un factor de aproximadamente 5, superior a un factor de aproximadamente 10, superior a un factor de aproximadamente 50, superior a un factor de aproximadamente 100, superior a un factor de aproximadamente 200, superior a un factor de aproximadamente 400, superior a un factor de aproximadamente 600, superior a un factor de aproximadamente 800, superior a un factor de aproximadamente 1000, superior a un factor de aproximadamente 1500, superior a un factor de aproximadamente 2000, superior a un factor de aproximadamente 5000, superior a un factor de aproximadamente 10.000, o superior a un factor de aproximadamente 20.000.
Procedimientos: Las células se descongelan a partir de un estado conservado en nitrógeno líquido. Una vez se han expandido las células y se han dividido en sus tiempos de duplicación esperados, se inicia el cribado. Las células se siembran en medio de crecimiento en placas tratadas de cultivo de tejidos negras de 1536 o 384 pocillos. A continuación, las células se equilibran en placas de ensayo mediante centrifugación y se introducen en incubadores incorporados en los módulos de administración a 37°C durante las 24 horas anteriores al tratamiento. En el momento del tratamiento, se recoge un grupo de placas de ensayo (que no reciben tratamiento) y se miden los niveles de ATP mediante la adición de ATPLite (Perkin Elmer). Dichas placas Tcero (T0) se leen utilizando luminiscencia ultrasensible en lectores de placa Envision (Perkin Elmer). Se incuban con compuesto durante 72 horas las placas de ensayo tratadas. Tras 72 horas, se revelan las placas para el análisis de valoración utilizando ATPLite. Todos los puntos de datos se recogen mediante procedimientos automatizados, se controla la calidad y se analizan utilizando el software Zalicus. Las placas de ensayo se aceptan si pasan los estándares de control de calidad siguientes: los valores relativos de luciferasa fueron consistentes durante todo el experimento, las puntuaciones de factor Z fueron superiores a 0,6, los controles no tratados/vehículos se comportaron consistentemente en las placas.
Se utilizó la inhibición del crecimiento (IC) como medida de la viabilidad celular. La viabilidad celular del vehículo se midió en el tiempo de administración (T0) y tras 72 horas (T72). Una lectura de IC de 0 % representa una inhibición nula del crecimiento - las señales de tratamiento de compuesto en T72 y de vehículo en T72 son equivalentes. Una lectura de IC de 100 % representa una inhibición completa del crecimiento - las señales de tratamiento de compuesto en T72 y de vehículo en T0 son equivalentes. No se incrementó el número de células durante el periodo de tratamiento en pocillos con IC de 100 % y podrían sugerir un efecto citostático de los compuestos que alcanzan una meseta en este nivel de efecto. Una lectura de IC de 200 % representa la muerte completa de todas las células en el pocillo de cultivo. Los compuestos que alcanzan una meseta de actividad de IC 200 % se consideran citotóxicos. Se calcula la IC mediante la aplicación de la prueba y ecuación siguientes:
SÍ T < V0 : 100
Si T > V0u : 100
en la que T es la medida de la señal para un artículo de ensayo, V es la medida de control tratado con vehículo y V0 es la medida de control de vehículo en el tiempo cero. Dicha fórmula se deriva del cálculo de inhibición del crecimiento utilizado en el cribado de alto rendimiento NCI-60 del National Cancer Institute.
La inhibición (I) se define como:

en donde T es el recuento celular tratado y V es el recuento celular no tratado (vehículo) (a las 72 horas). I se encuentra comprendido entre 0 % (si T=V) y 100 % (si T=0). El valor de IC50 se define como la concentración de fármaco necesaria para inhibir al 50 % el crecimiento celular en comparación con el crecimiento de las células tratadas con vehículo (la concentración de fármaco que proporciona I=50 %). La medida del efecto en el experimento puede ser la inhibición de la respuesta celular respecto al nivel no tratado (vehículo solo). Para el vehículo no tratado y los niveles tratados V y T, se calculó una inhibición fraccional I=1-TN. La inhibición se encontraba comprendida entre 0 % en el nivel no tratado y 100 % cuando T=0. Los niveles de inhibición son negativos para los agentes que realmente incrementan los niveles. Otras medidas de efecto, tales como una proporción de actividad r=T/V pueden resultar más apropiadas para algunos ensayos. En el caso de que se utilicen proporciones de actividad (p.ej., factor de incremento respecto al control estimulado), el efecto puede medirse utilizando una inducción I=ln(T/V). Con esta definición, todas las expresiones del efecto son iguales que para la inhibición.
Se recogieron los datos de análisis de combinación en una matriz de dosis 6x6. Se calculó la sinergia mediante la comparación de la respuesta de una combinación con las de los compuestos individuales, frente al modelo de referencia aditivo de dosis del fármaco consigo miso. Las desviaciones respecto a la aditividad de las dosis pueden evaluarse visualmente en un isobolograma o numéricamente con un índice de combinación (IndC). Ver la Tabla 3, posteriormente, para un IndC con 50 % de inhibición y un IndC con 50 % de inhibición del crecimiento. Una combinación proporciona un efecto aditivo en el caso de que el IndC=1,0; un efecto sinérgico en el caso de que IndC <1,0 y un efecto antagonista en el caso de que IndC>1,0.
Se evaluó el cambio de potencia utilizando un isobolograma, que demuestra cuanto menos fármaco se requiere en combinación para conseguir un nivel de efecto deseado, en comparación con las dosis de agente individual necesarias para alcanzar ese efecto. El isobolograma se representa mediante la identificación del locus de las concentraciones que corresponden al cruce del nivel de inhibición indicado. Lo anterior se lleva a cabo encontrando el punto de cruce para la concentración de cada agente individual en una matriz de dosis respecto a las concentraciones del otro agente individual. En la práctica, cada concentración vertical Cy se mantiene fija mientras se utiliza un algoritmo de bisección para identificar la concentración horizontal Cx en combinación con esa dosis vertical que proporciona el nivel de efecto seleccionado en la superficie de respuesta Z(CX,CY). A continuación, se conectan dichas concentraciones mediante interpolación lineal para generar el gráfico del isobolograma. Para las interacciones sinérgicas, la curva del isobolograma se encuentra bajo el umbral de aditividad y se aproxima al origen, y una interacción antagonista se encontraría por encima del umbral de aditividad. Las barras de error representan la incertidumbre que aparece de los puntos de datos individuales que se utilizan para generar el isobolograma. La incertidumbre de cada punto de cruce se estima a partir de los errores de respuesta utilizando la bisección para encontrar las concentraciones en las que Z-oz(Cx,Cy) y Z+oz(Cx,Cy) cruzan /corte, en donde o* es la desviación estándar del error residual en la escala del efecto.
Para medir los efectos de la combinación en exceso de la aditividad de Loewe, se crea una medida escalar para caracterizar la fuerza de la interacción sinérgica denominada puntuación de sinergia. La puntuación de sinergia se calcula de la manera siguiente:
Puntuación d e sinergia-- log / x log /Y

La inhibición fraccional de cada agente componente y el punto de combinación en la matriz se calculan respecto a la mediana de todos los pocillos de control tratados con vehículo. La ecuación de puntuación de sinergia integra el volumen de la actividad observada experimentalmente en cada punto en la matriz en exceso de una superficie modelo derivada numéricamente a partir de la actividad de los agentes componentes utilizando el modelo de Loewe de aditividad. Se utilizan términos adicionales en la ecuación de puntuación de sinergia (anteriormente) para normalizar diversos factores de dilución utilizados para agentes individuales y para permitir la comparación de las puntuaciones de sinergia a través de todo el experimento La inclusión de subgrupos de inhibición positiva o un multiplicador Idatos elimina el ruido en proximidad al nivel de efecto cero, y sesga los resultados para interacciones sinérgicas que se producen a niveles de actividad elevados.
La medida de la puntuación de sinergia se utiliza para el análisis de autocruces. Las puntuaciones de sinergia de los autocruces se espera que sean aditivas por definición y, por lo tanto, mantengan una puntuación de sinergia de cero. Sin embargo, aunque algunas puntuaciones de sinergia de autocruce son prácticamente iguales a cero, muchas son mayores, lo que sugiere que el ruido experimental o un ajuste no óptimo a la curva de las respuestas a dosis de agentes individuales están contribuyendo a las ligeras perturbaciones en la puntuación. Esta estrategia está centrada en la línea celular, concentrándose en el comportamiento de autocruce en cada línea celular frente a una revisión global de la actividad del panel de líneas celulares. La aditividad debería mantener una puntuación de sinergia de cero, y la puntuación de sinergia de dos o tres desviaciones estándares indica que la combinación es sinérgica a los niveles estadísticamente significativos de 95 % y 99 %, respectivamente.
Puede utilizarse el volumen de Loewe (vol. Loewe) para evaluar la magnitud total de la interacción de la combinación en exceso del modelo de aditividad de Loewe. El volumen de Loewe resulta particularmente útil para distinguir los incrementos sinérgicos de una actividad fenotípica (volumen de Loewe positivo) frente a antagonismos sinérgicos (volumen de Loewe negativo). En el caso de que se observen antagonismos, el volumen de Loewe debería evaluarse para examinar si existe cualquier correlación entre el antagonismo y una actividad en una diana de fármaco o un genotipo celular particulares. Dicho modelo define la aditividad como una interacción de combinación no sinérgica en la que la superficie de la matriz de dosis de combinación debería ser indistinguible de cualquiera de los fármacos cruzado consigo mismo. El cálculo de la aditividad de Loewe se realiza de la manera siguiente:
I Loewe que cumple que (X/Xi) + (Y/Yi) = 1
en donde XI y YI son las concentraciones eficaces de agente individual para el efecto de combinación observado I. Por ejemplo, en el caso de que se alcance una inhibición de 50% por separado con 1 pM de fármaco A o 1 pM de fármaco B, una combinación de 0,5 pM de A y 0,5 pM de B también debería inhibir al 50%.
Se sometieron a ensayo las combinaciones ejemplares de compuestos selectivos para PI3K-gamma y los compuestos selectivos para PI3K-delta siguiendo los procedimientos indicados anteriormente. En un estudio ejemplar, se estudiaron los efectos de combinación del compuesto AA con el compuesto BB a diversas concentraciones en diversas líneas celulares y se proporcionan los resultados en la Tabla 19, posteriormente. El compuesto AA es un inhibidor de PI3K8 (p.ej., un compuesto selectivo de delta) que presenta una proporción de selectividad gamma/delta superior a aproximadamente 50. El compuesto BB es un compuesto selectivo de PI3Ky (p.ej., un compuesto selectivo de gamma) indicado en la presente memoria que presenta una proporción de selectividad delta/gamma superior a aproximadamente 50. La proporción de selectividad gamma/delta se determina mediante división de la IC50 del inhibidor frente a la isoforma gamma de PI3K por la IC50 del inhibidor contra la isoforma delta de PI3K. La proporción de selectividad delta/gamma se determina mediante división de la IC50 contra la isoforma delta de PI3K por la IC50 del inhibidor contra la isoforma gamma de PI3K. Con fines ilustrativos, los isobologramas de la FIG. 6 (a una inhibición del crecimiento de 90 %) y la FIG. 7 (a una inhibición del crecimiento de 40 %) muestran que una combinación de compuesto AA y compuesto BB proporciona sinergia en el linfoma de células B grandes difusas (LCBGD) en las líneas celulares TMD8 y Farage. Los isobologramas de una combinación de compuesto AA y compuesto BB demuestran además efectos sinérgicos en una línea celular de linfoma de células B (línea celular Karpas-422) y en un linfoma de células T, linfoma no de Hodgkin, linfoma de Hodgkin y línea celular de linfoma de células grandes anaplásico (línea celular HH) (isobologramas no mostrados).
Los valores de IC50 para la inhibición del crecimiento y la inhibición en la Tabla 19 se clasifican de la manera siguiente: S=0,1 a <0,5, T=0,5 a <0,7, U=0,7 a <1, W=>1. Los valores de puntuación de sinergia para la inhibición del crecimiento y la inhibición se clasifican de la manera siguiente: A1=0,01 a <1, A2=1 a <3, y A3=>3. Los tipos de líneas celulares sometidos a ensayo son el linfoma de células B grandes difusas (LCBGD), similares a células B activadas (CBA), LCBGD similar a células B de centro germinal, linfoma folicular, linfoma de células del manto, mieloma múltiple y linfoma de células T. Los datos muestran que una combinación de un compuesto selectivo de gamma y un compuesto selectivo de delta proporciona un efecto sinérgico en diversos tipos de líneas celulares de cáncer.
Tabla 19

Tabla 19 (continuación)

Ejemplo 253: los compuestos selectivos de PI3K-gamma inhiben la migración de células T inducida por CXCL12 en un modelo in vitro de células B malignas
Los compuestos selectivos de PI3K-gamma proporcionados en la presente memoria pueden inhibir la migración de células T inducida por CXCL12. Específicamente, el material utilizado fue: medio (RPMI+suero de feto bovino al 0,5 %+Pen/Strep), Pb Mc de LLC totales (AllCells), placas de 24 transpocillos CoStar (inserto de 5 j M) n° 3421, tampón de fijación Bd Cytofix (n° 554655), tampón de tinción BD FBS (BD Biosciences, 554656), placa de 96 pocillos profundos (Axygen, P-2ML-SQ-C), rhCXCL12 (R&D, 350-NS-050), CD-19 APC Cy7 (BD n° 348794 1:20), CD3-PerCPCy5.5 (BD n° 560835, 1:20), CD5-PE (Biolegend n° 300608, 1:5), CD4-FITC (BD n° 561842, 1:20) y CD8-APC (BD n° 561953, 1:20).
Se preincubaron PBMC humanas de LLC totales con compuesto AA o BB en medio durante 45 minutos a 37°C. Se introdujeron 600 j l de medio basal o medio que contenía 300 ng/ml de CXCL12 /- DMSO o compuesto AA o compuesto BB en la cámara inferior del inserto de transpocillo. Tras 45 minutos de preincubación del compuesto, se introdujeron 500 a 750K de PBMC de LLC en la cámara superior del inserto de transpocillo en un volumen total de 100 jl. Se dejó que las células migrasen durante 2 a 4 horas a 37°C. Se sacaron los insertos y se transfirieron 550 j l de medio de la cámara inferior a una placa de 96 pocillos profundos. Las células se centrifugaron a 1280 rpm durante 5 minutos y se eliminó el medio. Las células se resuspendieron en 400 j l de tampón de citofijación BD y se incubaron a TA durante 10 minutos. Se añadió 1 ml de tampón de tinción BD FBS. Las células se centrifugaron a 1280 rpm y se desechó el tampón de fijación. Las células se resuspendieron en 100 j l de cóctel de anticuerpos CD3,4,8,5,19 constituido de tampón de tinción BD FBS, que a continuación se incubó a temperatura ambiente en la oscuridad durante 30 a 60 minutos. Se añadió 1 ml de tampón de tinción BD FBS a cada pocillo, que a continuación se centrifugó y volteó. Las células se resuspendieron en 150 j l de tampón de tinción FBS y se transfirieron a tubos con FACS que ya contenían 150 j l de tampón de tinción FBS. Cada muestra se leyó en el FACS durante 25 segundos. Se separaron las subpoblaciones CD3, 4, 8 y CD19/5 individualmente y se calculó el índice de migración medio a partir de muestras por triplicado. Ver, p.ej., Borge et al., Haematologica 2010, 95(5): 768-775; de Rooij et al., Blood 2012, 119: 2590 2594, 1977.
El compuesto AA es un compuesto selectivo de PI3K-delta indicado en la presente memoria que presenta una proporción de selectividad gamma/delta superior a aproximadamente 50. El compuesto BB es un compuesto selectivo de PI3K-gamma indicado en la presente memoria que presenta una proporción de selectividad delta/gamma superior a aproximadamente 50. La FIG. 8 muestra el porcentaje de inhibición celular de la migración de células T CD3+ inducida por CXCL12 a diversas concentraciones de compuesto AA (EC50=694 nM) y compuesto BB (EC50=10 nM). En un experimento similar, se determinó la EC50 para tres subgrupos de células T para un inhibidor de PI3K-8 (compuesto AA) y un inhibidor de PI3K-Y (compuesto BB). Se muestran los resultados en la tabla a continuación. El compuesto BB es un potente inhibidor de la migración de células T inducida por CXCL12.
Tabla 20

Los datos demuestran que el compuesto BB (un compuesto selectivo de gamma) es más potente que el compuesto AA (un compuesto selectivo de delta) en la inhibición de la migración de las células T CD3+ inducida por CXCL12 en las PBMC LLC. Pueden utilizarse compuestos selectivos de gamma para bloquear la migración de células T estimuladoras del crecimiento al nicho tumoral de células B, retrasando la progresión de la enfermedad. Lo anterior podría traducirse en una supervivencia libre de progresión incrementada o respuestas más profundas a nivel clínico con respecto a las neoplasias malignas de células B.
Se estudió el mecanismo de inhibición por parte del compuesto BB de la migración. Se encontró que el compuesto BB inhibió pAKT inducido por CXCL12 en células T más fuertemente que un inhibidor de PI3K-8 (compuesto a A) (la FIG.
9). Existe una correlación estrecha entre las EC50 observadas en el ensayo de pKAT inducido por CXCL12 y el ensayo de migración de células T. Este resultado sugiere que un inhibidor de PI3K-Y interfiere con la migración de las células T en las PBMC del paciente de LLC mediante el bloqueo de la señalización de pAKT. Aunque la inhibición de PI3K no anula la supervivencia de LLC inducida por IL4 (datos no mostrados), la inhibición dependiente de PI3K-Y de la migración de células T a los microambientes de los ganglios linfáticos con LLC puede prevenir indirectamente que las citoquinas derivadas de células T (p.ej., IL-4) proporcione señales de supervivencia de LLC independientes de PI3K.
Sin restringirse a ninguna teoría en particular, los datos sugieren el modelo siguiente de las neoplasias malignas de células B. Las células B malignas reciben señales de crecimiento y prosupervivencia de la activación de RCB y las células mesenquimales, transmitidas mediante las rutas de PI3K-8 y -y. El crecimiento y supervivencia de las células B malignas también se ve apoyado por las células T y las células mieloides mediante mecanismos dependientes e independientes de PI3K. La inhibición de PI3K-8 reduce la proliferación de las células B malignas; sin embargo, otras señales independientes de PI3K pueden potenciar la supervivencia de las células tumorales. La inhibición de PI3K-Y, p.ej., por el compuesto BB, puede bloquear la migración y/o la diferenciación de células T accesorias y células mieloides y anular dichas células de soporte clave dentro del microambiente tumoral. La falta de células de soporte clave dentro del microambiente tumoral como resultado de la inhibición de PI3K-Y, de esta manera, podría bloquear más completamente el crecimiento y supervivencia de las células B malignas. Sin restringirse a ninguna teoría en particular, el compuesto BB podría actuar inhibiendo el crecimiento y/o supervivencia celular en cualquiera de las etapas anteriormente indicadas.
Ejemplo 254: modelo de enfermedad intestinal inflamatoria (EII) (Ejemplo de referencia)
Puede llevarse a cabo un estudio para evaluar la eficacia potencial de un compuesto proporcionado en la presente memoria en ratones SCID hembra con enfermedad intestinal inflamatoria CD4+. En este modelo murino, en ratones C.B-17 SCID hembra se inyectaron por vía intraperitoneal (IP) células CD45RBhigh, un subgrupo de células T CD4+ obtenido de ratones BALB/c normales, para inducir una inflamación crónica espontánea en el intestino grueso. Los cambios macroscópicos e histopatológicos que resultan de dicho tratamiento son similares a los que ocurren en la enfermedad de Crohn y la colitis ulcerosa en el ser humano. Ver Leach et al., Inflammatory Bowel Disease in C.B-17 scid Mice Reconstituted with the CD45RBhigh Subset of CD4+ T Cells, American Journal of Pathology, 1996, 148 (5), 1503-1515.
El día de estudio 0, se sacrificaron los ratones Balb/C, y se obtuvieron los bazos para el aislamiento de las células CD45RBhigh según el protocolo de separación de células SCID IBD. Se pesaron los ratones SCID y recibieron inyecciones intraperitoneales (IP) de las células separadas (inyecciones de aprox. 4*10® células/ml, 100 pl/ratón). El día de estudio 2 1 , se pesaron los ratones y se aleatorizaron según pérdida de peso corporal en grupos de tratamiento, y se inició la administración diaria (QD) y oral (PO9. Se continuó la administración hasta el día de estudio 41 y se sacrificaron los ratones el día 42. Los ratones se dividieron en diversos grupos. El grupo 1 puede ser el grupo normal; el grupo 2 puede ser el control de vehículo (CMC al 0,5 %, Tween-80 al 0,05 % en H2O) y pueden administrarse diversas cantidades de un compuesto proporcionado en la presente memoria a grupos diferentes a modo de comparación.
Para cada animal, se recortó el colon entero (proximal y distal) en 8 trozos espaciados igualmente para el procesamiento e inclusión. Se tiñeron las secciones con hematoxilina y eosina (H+E). Para cada sección teñida con H+E, se cuantificó el edema submucosal mediante la medición de la distancia entre la mucosa muscular y el borde interno de la capa muscular externa en una zona no tangencial que se cree que representa mejor la gravedad de este cambio. Se midió además el grosor mucosal en una zona no tangencial de la sección que mejor representa el grosor mucosal global. Este parámetro es indicativo de la elongación de la glándula y la hiperplasia mucosal. Con el fin de incorporar este parámetro en la puntuación sumada, se obtuvo una puntuación de hiperplasia a partir de las mediciones, de la manera siguiente: 0=<250 pm; 1=250-349 pm; 2=350-449 pm; 3=450-599 pm; 4=600-699 pm y 5=>700 pm.
Se asignaron puntuaciones de gravedad a los grados de inflamación (macrófagos espumosos, linfocitos e infiltrado de PMN) siguiendo los criterios siguientes:
Normal=0
Mínima=1 (generalmente focal, afectando 1 % a 10 % de la mucosa o, en caso de ser difusa, una superficie mínima) Leve=2 (generalmente focal, afectando 11 % a 25% de la mucosa o, en caso de ser difusa, leve) Moderada=3 (26 % a 50 % de la mucosa afectada con zonas de pérdida glandular sustituida por infiltrado de células inflamatorias, más leve en zona restante de la mucosa).
Marcada=4 (51 % a 75 % de la mucosa afectada con zonas de pérdida glandular sustituida por infiltrado de células inflamatorias, más leve en zona restante de la mucosa).
Marcada=5 (51 % a 100% de la mucosa afectada con zonas de pérdida glandular sustituida por infiltrado de células inflamatorias, más leve en zona restante de la mucosa).
Los parámetros que reflejan la pérdida/daño de las células epiteliales se puntúan individualmente utilizando un método de puntuación que implica el porcentaje de superficie: Nulo=0; 1-10% de la mucosa afectada=1; 11-25% de la mucosa afectada=2; 26-50% de la mucosa afectada=3; 51-75% de la mucosa afectada=4; y 76-100% de la mucosa afectada=5.
Entre los parámetros puntuados con el porcentaje de afectación se incluyen: (1) pérdida de epitelio glandular del colon: incluye el epitelio de cripta, así como la pérdida epitelial glandular restante, y (2) erosión del colon: refleja la pérdida de epitelio superficial y se asocia generalmente a hemorragia mucosal (que refleja el sangrado que se observa clínicamente y en la necropsia).
Los 4 parámetros puntuados principales (inflamación, pérdida epitelial glandular, erosión e hiperplasia) finalmente se suman para llegar a una suma de puntuaciones histopatológicas, que indica el daño global y que presentaría una puntuación máxima de 20.
Los infiltrados de células inflamatorias en la mucosa colónica se evalúan para el % aproximado de neutrófilos en el infiltrado total utilizando los criterios a continuación. El % aproximado del total a continuación se multiplica por la puntuación de inflamación 0-5 en un intento de semicuantificar la infiltración relativa de PMN en diferentes secciones y animales: 0=aprox. 0 %; 10=aprox. 10 %; 25=aprox. 25 %; 50=aprox. 50 % y 75=75 % o superior. A continuación, dicho valor se multiplica por la puntuación de inflamación en un intento de semicuantificar la infiltración relativa de PMN en diferentes secciones y animales:
Ejemplo 255: modelo de encefalomielitis autoinmune (EAE) (Ejemplo de referencia)
Los efectos de un compuesto proporcionado en la presente memoria en el tratamiento de la inflamación pueden someterse a ensayo en un modelo de EAE de ratón semiterapéutico de 28 días. La EAE puede inducirse en 60 ratones utilizando el Hooke Kit™ MOG35-55/CFA Emulsion PTX (Hooke Laboratories, Lawrence MA), número de catálogo EK-0110, número de lote 0104, según el protocolo recomendado por el fabricante.
Los ratones recibieron inyecciones subcutáneas en dos sitios en el lomo, con el componente emulsión del kit (que contenía MOG35-55). Un sitio de inyección era la zona del lomo superior, aproximadamente 1 cm caudal respecto a la línea del cuello. El segundo sitio era la zona del lomo inferior, aproximadamente 2 cm en dirección craneal de la base de la cola. El volumen de inyección era de 0,1 ml en cada sitio. En las 2 horas siguientes a la inyección de emulsión y nuevamente 24 horas después de la inyección de emulsión, se administró por vía intraperitoneal el componente toxina pertussis del kit. El volumen de cada inyección era 0,1 ml.
Se indujo EAE en determinados ratones, que pueden dividirse en diversos grupos, p.ej., un grupo de control negativo y grupos tratados con diversas cantidades de un compuesto proporcionado en la presente memoria.
Las lecturas eran las puntuaciones de EAE y los cambios de peso corporal. Los ratones se puntuaron diariamente. Se puntuó la EAE en una escala de 0 a 5.
0: ningún cambio evidente de las funciones motoras del ratón en comparación con ratones no inmunizados. Al cogerles de la cola, la cola se tensa y está erecta. Las patas traseras habitualmente se abren. Al caminar el ratón, no hay desviación de la marcha o de la cabeza.
1: cola fláccida. Al coger el ratón por la cola, en lugar de estar erecta, toda la cola cae sobre el dedo.
2: cola fláccida y debilidad de las patas traseras. Al coger el ratón por la cola, las patas no se abren, sino que se mantienen más juntas. Al observar cómo camina el ratón, presenta una marcha claramente inestable.
3: cola fláccida y parálisis completa de las patas traseras (lo más común); o cola fláccida con parálisis de una pata delantera y una pata trasera, o la totalidad de: inclinación clara de la cabeza, marcha únicamente a lo largo de los bordes de la jaula, empujándose contra las paredes, y giro al cogerlo por la cola.
4: cola flácida, parálisis completa de patas traseras y parálisis parcial de pata delantera. El ratón se mueve mínimamente por la jaula, aunque tiene una apariencia alerta y se alimenta. Habitualmente, se recomienda la eutanasia después de que las puntuaciones del ratón estén a nivel 4 durante 2 días. Cuando el ratón resulta eutanizado por parálisis grave, se introduce una puntuación de 5 para ese ratón para el resto del experimento. 5: parálisis completa de patas traseras y completa de patas delanteras, no se mueve por la jaula o el ratón rueda espontáneamente en la jaula o se le encuentra muerto debido a parálisis.
Al final del estudio, pueden recogerse y analizarse los tejidos siguientes, p.ej., recolección de plasma para FC, recolección de suero, recolección de columnas para el análisis histológico, análisis histológico de columnas, recuento de focos inflamatorios, estimación de la zona de desmielinización o recuento de células apoptóticas.
Ejemplo 256: farmacocinética y reducción de la neutrofilia inducida por LPS - administración oral
Con el fin de evaluar el efecto del compuesto BB (inhibidor de PI3Ky (p.ej. compuesto selectivo de gamma proporcionado en la presente memoria)) y compuesto AA (inhibidor de Pl3K8 (p.ej., compuesto selectivo de delta)) sobre los parámetros inflamatorios inducidos por LPS, se administraron por vía oral compuesto BB y compuesto AA.
El compuesto AA presenta una proporción de selectividad gamma/delta superior a aproximadamente 50. El compuesto BB presenta una proporción de selectividad delta/gamma superior a aproximadamente 50.
El presente estudio incluye además la evaluación de la exposición a los compuestos en plasma y tejidos pulmonares. Los grupos y detalles de la administración se proporcionan en la tabla a continuación.
Tabla 21.

El resumen FC oral del compuesto AA y del compuesto BB en estudios de LPS en rata se proporcionan en la tabla, a continuación.
Tabla 22

El compuesto BB y el compuesto AA (p.o.) redujeron los neutrófilos en el líquido de lavado broncoalveolar (LLBA) 4 horas después del reto de LPS de una manera dependiente de la dosis. El compuesto BB presentó un efecto más profundo sobre el flujo de entrada de neutrófilos que el compuesto AA (FIG. 10, FIG. 11 y FIG. 12 (media con SEM, valor de p de la prueba t; los grupos se compararon individualmente con el grupo de LPS)). Estos datos muestran que PI3K-Y participa en la infiltración de neutrófilos en el pulmón, un tipo celular patológico clave en la enfermedad pulmonar obstructiva crónica (EPOC).
Los resultados de PK mostraron que niveles libres en plasma de tanto compuesto BB como compuesto AA a los niveles de dosis más altos eran iguales o superiores a la IC90 para gamma y delta, respectivamente, y sin embargo claramente inferiores a la IC50 para las siguientes isoformas de PI3K más próximas de cada compuesto.
En otro estudio ejemplar, se encontró que el incremento de p-AKT inducido por CXCL12 en células T CD3+ estaba mediado por PI3K-Y. En todavía otro estudio ejemplar se encontró que, dentro de la población de células B malignas, el incremento de p-AKT inducido por CXCL12 era dependiente de PI3K-8, lo que sugiere que las señales de CXCL12 ocurren a través de diferentes isoformas de PI3K en estos diferentes tipos celulares.
Ejemplo 257: efectos de la inducción de p-AKT inducida por CXCL12 en macrófagos diferenciados
Las células derivadas de mieloides y las células estromales mesenquimales pueden apoyar la supervivencia celular de LLC como componentes del microambiente tumoral (MAT). Algunos informes recientes sugieren que las células de tipo nodriza citoprotectoras de LLC podrían presentar una polarización de M2 y ser similares a las células mieloides asociadas a tumor inmunosupresoras que se observan en tumores sólidos. Giannoni et al., Haematologica 2014, 99(6), 1078-88. Un propósito del presente estudio es modelizar estos componentes del MAt y evaluar los efectos de los inhibidores selectivos de isoforma de PI3K sobre la inducción de p-AKT inducida por CXCL12 en macrófagos
diferenciados. El compuesto AA es un compuesto selectivo de PI3K-delta que presenta una proporción de selectividad gamma/delta superior a aproximadamente 50. El compuesto BB es un compuesto selectivo de PI3K-gamma indicado en la presente memoria que presenta una proporción de selectividad delta/gamma superior a aproximadamente 50.
Diferenciación y polarización de macrófagos: se recolectaron los huesos largos de ratones C57B6 jóvenes y se extrajo la médula ósea mediante lavado suave de las cavidades de la médula ósea con una aguja de calibre 28. Se extrajeron los glóbulos rojos utilizando tampón de lisis de glóbulos rojos (Sigma). Las células de médula ósea se diferenciaron en macrófagos utilizando FECM durante 6 días (RPMI y FCS al 20 %) con un cambio de medio (con eliminación de todas las células no adherentes) el día 3. Se polarizaron los macrófagos en M0 (FECM), M1 (FECM, IFNy y LPS durante 1 día) o M2 (FECM, IL4 durante 2 días).
Ensayo XCL12-iEnsayo de inducción de p-AKT inducida con CXCL12 Se levantaron los macrófagos polarizados con acutasa, y se sembraron en placa a razón de 0,5 M de células en cada pocillo (0,5 ml) en una placa no adherente de pocillos profundos (2 ml) en medio de polarización durante 1 hora. Se añadió DMSO o compuesto de ensayo en DMSO (concentración final de DMSO: 0,1 %, compuesto AA o compuesto BB) durante 40 minutos. Se añadió CXCL12 y se mezclaron las células en un agitador de placas durante 2 minutos. Se añadió 1 ml de tampón de lisis/fijación de fAc S a 37 grados (BD Bioscience) y la placa se mezcló durante 1 minuto. Se centrifugaron las placas y se decantaron y congelaron a -80°C. Se llevó a cabo la tinción con fosfo-AKT y anti-fosfo-AKT 473 marcado con PERCP (Cell Signaling Technologies) y se midió mediante citometría de flujo. Se utilizó el IMF celular total para calcular el efecto de los compuestos sometidos a ensayo sobre la inducción por CXCL12 de fosfo-AKT.
Resultados: El compuesto BB inhibió p-AKT inducido por CXCL12 en macrófagos diferenciados y polarizados en M1 con un valor de IC50 de aproximadamente 14 nM. El compuesto BB inhibió p-AKT inducido por CXCL12 en macrófagos diferenciados y polarizados en M2 con un valor de IC50 de aproximadamente 8,4 nM. El compuesto AA no inhibió significativamente p-AKT inducido por CXCL12 en macrófagos diferenciados y polarizados en M1 o M2. La FIG. 13 muestra el resultado de tinción de p-AKT para el fenotipo M2. Los resultados del presente estudio muestran que la activación de M2 mediada por CXCL12 es dependiente de PI3K-Y, ya que resulta más potentemente inhibida por el inhibidor selectivo de PI3K-Y (compuesto BB) que por el inhibidor selectivo de Pi3K-8 (compuesto AA).
Ejemplo 258: cocultivo de LLC con células macrófagas M2
Se obtuvieron células de LLC purificadas congeladas de un proveedor comercial (All Cells). Se polarizaron los macrófagos en M0 (FECM) o M2 (FECM, IL4 durante 2 días). Los macrófagos polarizados en M0 o m 2 se sembraron en una placa de cultivo de tejidos de 96 pocillos a la densidad indicada en RPMI FCS al 10 % con sus factores de polarización respectivos. Las células de LLC se sembraron en la placa de cultivo de tejidos a razón de 150 mil células en cada pocillo con medio o macrófagos M0 o M2. Se midió la viabilidad de las células de LLC mediante Guava viacount (Millipore). El cocultivo con macrófagos M2 de las células de LLC llevó a una supervivencia extendida de las células de LLC (FIG. 14). El resultado muestra que las células macrófagas M2 presentan la capacidad de proteger a las células de LLC de la muerte celular, que resulta potenciada sobre los efectos protectores de las células M0 a las 120 horas (datos no mostrados).
Los resultados indican que los compuestos selectivos de PI3K-Y pueden utilizarse para tratar las LLC mediante la inhibición potente de la activación de M2 y la reducción de la supervivencia de las células de LLC. La FIG. 15 es un esquema de la diferenciación de las células progenitoras mieloides y las interacciones entre determinadas células T. La FIG. 16 ilustra la diferenciación de una célula mieloide en un macrófago M1 o un macrófago M2.
Ejemplo 259: efecto sobre la diferenciación de los macrófagos en osteoclastos
Informes recientes sugieren que los ratones con inactivación de PI3K-Y presentan una actividad normal de los osteoblastos, aunque son deficientes en la actividad de los osteoclastos, lo que conduce a un incremento de la formación de hueso. La diferenciación de los osteoclastos a partir de las células de médula ósea es deficiente en estos ratones con inactivación de PI3K-Y. Kang et al., Proc. Natl. Acad. Sci. USA. 2010, 107(29): 12901-12906, 1977. Un objetivo del presente estudio era evaluar el efecto del inhibidor selectivo de PI3K-Y sobre la diferenciación de los osteoclastos a partir de macrófagos de la médula ósea. El compuesto BB es un compuesto selectivo de PI3K-gamma indicado en la presente memoria que presenta una proporción de selectividad delta/gamma superior a aproximadamente 50.
Macrófagos derivados de médula ósea como progenitores de osteoclastos: Se recolectaron los huesos largos de ratones C57B6 jóvenes y se extrajo la médula ósea mediante lavado suave de las cavidades de la médula ósea con una aguja de calibre 28. Se cribaron las células y se eliminaron los glóbulos rojos utilizando tampón de lisis de glóbulos rojos (Sigma). Las células de la médula ósea se diferenciaron en macrófagos utilizando 20 ng/ml de FECM de ratón (RnD Systems) durante 3 días (RPMI y FCS al 20 %). Se lavaron las células con PBS y se levantaron con acutasa durante 10 minutos.
Ensayo de diferenciación de osteoclastos: Las células macrófagas de la médula ósea se sembraron a razón de 150.000 células por pocillo en una placa de 96 pocillos en a MEM FCS al 10 %. Se trataron las células con DMSO o
compuesto BB en DMSO durante 40 minutos antes de la adición de citoquinas. A continuación, se añadieron 25 ng/ml de FECM de ratón con o sin RANKL de ratón (100 ng/ml, RnD Systems) durante 6 días. Se llevaron a cabo cambios de medio (incluyendo los fármacos) los días 3 y 5. A continuación, se fijaron las células y se tiñeron con el kit de ensayo de fosfatasa ácida resistente a tartrato (TRAP, por sus siglas en inglés) de Sigma siguiendo las instrucciones del fabricante. Se contaron manualmente las células positivas para TRAP y las células que experimentaban una diferenciación osteoclástica) en un transecto de los pocillos por triplicado (a una concentración de 20X) que cruzaba la parte más ancha de cada pocillo.
Resultados: Los resultados se ilustran en la FIG. 17, que muestra que el compuesto BB evita la diferenciación activada por RANKL de los osteoclastos a partir de macrófagos de la médula ósea. Los resultados indican que los compuestos selectivos de PI3K-Y pueden utilizarse para el tratamiento de las enfermedades óseas basadas en osteoclastos.
Ejemplo 260: farmacocinética
El objetivo del estudio era evaluar la farmacocinética del compuesto BB tras la administración de una sola dosis y la administración de dosis repetida en ratones CB6F1 hembra. Se administró compuesto BB por vía oral a una dosis de 2, 7,5 y 15 mg/kg una vez al día durante 3 días consecutivos en ratones hembra (ratones CB6F1 hembra, n=72 (5 a 6 semanas de edad)). Los animales se dividieron en tres grupos de administración: Compuesto BB a 2 mg/kg, PO en 5 % de NMP, 95 % de PEG400; compuesto BB a 7,5 mg/kg, PO en 5 % de NMP, 95 % de PEG400, y compuesto BB a 15 mg/kg, PO en 5 % de NMP, 95 % de PEG400.
El compuesto BB se administró por vía oral una vez al día durante un total de tres días a un volumen de dosis de 10 ml/kg basado en el peso corporal más reciente. Los animales se pesaron diariamente antes de la administración. El día de la primera dosis, los animales fueron monitorizados estrechamente durante la 1a hora después de la administración; los animales fueron observados por lo menos dos veces durante las 4 horas siguientes a la administración de la dosis, y al final del día. En los siguientes días de administración, se anotó el estado de los animales antes de la administración; los animales fueron observados en las 4 horas siguientes a la administración y al final del día. Podía realizarse una monitorización incrementada en el caso de que los animales mostrasen signos clínicos. En el caso de que un animal mostrase signos clínicos leves a moderados de toxicidad, podría suspenderse la administración, en consultas con el director del estudio, para permitir la recuperación del animal.
Se recogieron muestras en la predosis, y 0,25, 0,5, 1, 2, 4, 6 y 24 h después de la primera dosis el día 1 y predosis (valle), 0,25, 0,5, 1,2, 4, 6 y 24 h después de la dosis final el día 3. Los animales recibieron una única dosis oral diaria durante 3 días consecutivos. Se recogió sangre mediante sangrado retroorbital en todos los puntos temporales el día 1. El resto de extracciones de sangre se recogió mediante una punción cardiaca. La sangre se recogió en tubos K2EDTA y los tubos se dejaron sobre hielo húmedo. En los 15 min siguientes a la recolección, la muestra se centrifugó a 10.000 rpm a 4°C durante 10 min en una centrífuga de laboratorio y el plasma se transfirió a un nuevo tubo de microcentrífuga.
La FIG. 18 muestra la concentración media de compuesto BB libre en plasma 1 o 5 horas después de la administración. La FIG. 19 muestra los resultados del estudio, anteriormente, de medición de un curso temporal de 24 horas de concentración libre en plasma de compuesto BB. El presente experimento indica que el compuesto BB presenta una buena estabilidad en suero y que resulta adecuado para la administración, p.ej., una vez al día.
Se llevó a cabo un estudio similar con compuesto 88. La FIG. 20 muestra los perfiles de concentración media de compuesto 88 libre en plasma-tiempo para ratones C57 hembra con administración de 2, 7,5 o 15 mg/kg PO. A continuación, se proporciona una tabla que detalla los resultados para el compuesto 88:

Lo anterior muestra que el compuesto 88 también presenta una buena estabilidad en suero y resulta adecuado para la administración.
Ejemplo 261: efectos del compuesto BB sobre la activación de las células T
El objetivo del presente estudio es evaluar el efecto de diversos antígenos (Con-A) sobre la activación de las células T en sangre completa humana vs. sangre diluida 1:1 utilizando los compuestos de ensayo en el punto temporal de las 24 horas.
El medio de activación de las células T incluía RPMI-1000 ml, MEM NEAA-10 ml, piruvato sódico (100 mM)-10 ml, Pen-strep-10 ml, 2-mercaptoetanol-1 ml y FBS-100 ml. Se sembraron 100 pl de sangre completa en una placa de 96 pocillos. Se utilizaron solo los 60 pocillos internos y se llenaron los exteriores con medio o PBS. Sangre completa humana diluida con suero al 5 %-RPMI 16401:1 y siembra de 100 pl en placa de 96 pocillos. Se utilizaron solo los 60 pocillos internos y se llenaron los exteriores con medio o PBS. Se dejaron las muestras en reposo durante 45 minutos en el incubador y después se trataron con compuesto BB durante 45 minutos.
El compuesto se diluyó con DMSO (Sigma hybridmax). Para la concentración más alta, de 1000 nM, partiendo de una solución madre 10 mM, se añadieron 300 pl de DMSO a placas de pocillos profundos. A la concentración más alta, se añaden 10 pl de solución madre de compuesto 10 mM. Ver la tabla, posteriormente.

Se preparó una placa de medio a partir de la placa de DMSO añadiendo 100 pl a 900 pl de medio en una placa nueva de pocillos profundos, golpeando suavemente y dejando en un mezclador durante 5 minutos.
Las placas celulares reposadas se sacaron del incubador. Se añadieron 12 pl de compuesto diluido de la placa de medios (DMSO final: 1 %) y se incubaron durante 45 minutos.
Se preparó Con-A de la manera siguiente: 200 pl de solución madre en 1 ml de medio, 100 pl de lo anterior en 1 ml de medio, 100 pl de lo anterior en 1 ml de medio. Concentración más alta utilizada. Lo anterior se analizó tras 24 horas.
10x solución madre en medio, adición de 12 pl a cada pocillo. Incubación a 37°C en cámara húmeda durante 24 horas con 5 % de CO2. Se analizó mediante ELISA multiplex de MSD. Panel proinflamatorio humano 1 Kit V-Plex de IFNg.

La FIG. 21 muestra los efectos del compuesto BB y del compuesto AA sobre la activación de las células T, medida por la inhibición de IFN-y. Debido a que el compuesto BB es altamente selectivo para PI3K-Y y el compuesto AA es altamente selectivo para PI3K-6, las IC50 muy diferentes de los compuestos en el presente ensayo (3 nM para el compuesto AA y 2500 nM para el compuesto Bb ) indican que PI3K-6 es importante para la activación de las células T y PI3K-Y es menos importante.
Ejemplo 262: efecto del compuesto BB sobre la diferenciación de MAT M2 murinas
El compuesto BB, que inhibe selectivamente PI3K-Y, y el compuesto AA, que es altamente selectivo de PI3K-6, se sometieron a ensayo para su capacidad de bloquear la diferenciación de las células mieloides en MAT M2, y se muestran los resultados en la FIG. 22A y en la FIG. 22B, respectivamente. Los niveles elevados de ARG1 (arginasa-1) indican que las células mieloides se están diferenciando en macrófagos M2, por lo que niveles más bajos de ARG1 respecto al control indican interferencia con la diferenciación en macrófagos m 2. La expresión de arginasa-1 agota la arginina en el microambiente tumoral, estimulando de esta manera la muerte de las células T y la inhibición de las células NK. Schmid et al., Proceedings: AACR 103rd Annual Meeting 2012, Cancer Research: 15 de abril 15, 2012; volumen 72, número 8, suplemento 1. El compuesto BB inhibe la diferenciación de las MAT m2 mucho más eficazmente que el inhibidor de PI3K-6. Los MAT M2 estimulan la supervivencia de las células de cáncer (FIG. 23). Debido a que el compuesto BB evita la diferenciación en células M2 protumorales, el presente experimento indica que el compuesto BB se espera que incremente la respuesta inmunitaria antitumoral del sujeto.
Se llevó a cabo un experimento similar con células de médula ósea que se diferenciaban en CSDM. El compuesto BB suprime tanto VEGF (FIG. 24A) como ARG1 (FIG. 24B) en un ensayo de diferenciación de células supresoras derivadas de mieloides.
Se llevó a cabo un experimento similar sobre la diferenciación en M1. El compuesto BB no presentó un efecto observable sobre la diferenciación en macrófagos M1 (datos no mostrados). De acuerdo con lo anterior, el compuesto BB no se espera que interfiera con las actividades antitumorales de los macrófagos M1.
Ejemplo 263: actividad como agente individual del compuesto BB en el modelo de carcinoma pulmonar de Lewis
Se sometió a ensayo el compuesto BB en el modelo de carcinoma pulmonar de Lewis, que es conocido de la técnica y se ha descrito brevemente antes en la presente memoria.
Se preparó un modelo de tumor CPL brei y se inyectó por vía subcutánea en un ratón en la zona del flanco trasero. Se preparó el tumor brei de la manera siguiente. Se cultivaron células tumorales LL2-Luc en DMEM+FBS al 10 %. Las células se implantaron a razón de 1x106 células/100 pl s.c. en el flanco de ratones C57 Albino (macho). Una vez los tumores habían alcanzado 1000 mm3, se recolectaron. Solo se recolectó el tejido vivo blanco. Cualquier material que contuviese sangre o zona necrótica visible se aisló respecto del tejido tumoral. El tejido tumoral se introdujo en un homogeneizador de tejidos Dounce con 5 ml de DMEM (no FBS) caliente. Se recogieron aproximadamente 1 a 2 gramos de tejido tumoral, se homogeneizaron, se agruparon en un tubo Falcon de 50 ml y se contaron las células. Las células se centrifugaron a 12.000 rpm y se decantó el FBS. Las células se resuspendieron hasta una densidad de 1x107 células/ml en PBS estéril. Se implantaron 100 pl de células/ratón s.c. en la zona del flanco trasero. Se repitió este procedimiento una vez más antes de llevar a cabo el estudio de eficacia sobre el tumor brei.
Las FIGS. 25A y 25B muestran que el compuesto BB demuestra actividad de agente único en el modelo de carcinoma pulmonar de Lewis. La FIG. 26 muestra las concentraciones medias de compuesto BB libre en el tumor y en plasma tras 12 días de administración oral diaria en solución de NMP. A continuación, se proporciona una tabla con los detalles:

Los resultados indican que la exposición se incrementó con el incremento de la dosis. A las 2 h, los niveles de compuesto BB eran superiores a la IC50 gamma celular en todos los grupos. Para los grupos de 7,5 mg/kg y de 15 mg/kg, eran superiores a la IC50 gamma a las 24 h.
Ejemplo 264: efectos del compuesto BB sobre el crecimiento tumoral en modelos de cáncer de colon CT26 y MC38
El propósito del presente estudio era determinar la eficacia del compuesto BB como agente único y en combinación con anti-PD-L1 en el modelo de aloinjerto de CT26 de cáncer de colon de ratón. CT26 es una línea celular de carcinoma de colon no diferenciado inducido por N-nitroso-N-metiluretano (NNMU). Se clonó para generar la línea celular denominada CT26.WT (número de catálogo de ATCC n° CRL-2638). En ratones Balb/c hembra de 5-6 semanas de edad (Jackson Labs) se implantaron por vía subcutánea 5x105 células CT26 en 200 pl de PBS. Las células CT26 se cultivaron en DMEM complementado con FBS al 10 %. La administración se inició una vez los tumores habían alcanzado un volumen medio ~100 mm3. La administración de los compuestos se llevó a cabo según la tabla a continuación:
Tabla 23

Vehículo del compuesto BB: 5 % de NMP/95 % de PEG400
El compuesto BB se preparó a una dosis de 0,6 mg/ml en 5 % de NMP/95 % de PEG400
Control de isotipo (IgG2b de rata purificada Ultra-LEAF, control de isotipo k, clon RTK4530, lote n° B180477, Biolegend, n° de cat. 70640)
Anticuerpo anti-PD-L1 (anticuerpo B7H1 anti-CD274 de ratón purificado Ultra-LEAF, PD-L1-, clon 10F9G2, lote n° B178331, Biolegend, n° de cat. 124318)
Régimen de dosis:
Compuesto BB: QD durante 3 semanas a una dosis de 5 ml/kg
Anti-PD-LI: cada tercer día para un total de 4 dosis
Se midieron los pesos corporales y volúmenes tumorales tres veces a la semana.
La FIG. 27A y la FIG. 27B muestra el compuesto BB administrado junto con un inhibidor del punto de control anti-PD-L1 en el modelo CT26. La FIG. 27A muestra el efecto sobre el volumen tumoral respecto al tiempo. La FIG. 27B muestra el porcentaje de supervivencia respecto al tiempo.
La FIG.28 muestra que la expansión CSDM tras el tratamiento de ciclofosfamida se produce dentro del marco temporal de 5 a 9 días y que esta expansión fue reducida por el compuesto BB en el modelo de CT26.
Un segundo modelo de cáncer de colon, el modelo de M38, se utilizó para evaluar adicionalmente los efectos del compuesto BB. En dicho modelo, se aplica una línea celular de cáncer de colon singénico por vía subcutánea. El diseño experimental fue el siguiente.
Se prepararon ratones CR C57BL/6 hembra con 1x106 células MC38 en Matrigel al 0 % s.c. en el flanco. El volumen de inyección celular era de 0,05 ml en cada ratón. Una vez los tumores habían alcanzado un tamaño medio de 80 a 100 mm3, se emparejaron y se inició el tratamiento. Se midieron los tumores con un calibrador cada dos semanas.
Las dosis y formulaciones fueron las siguientes: 5-FU en D5W; compuesto BB en 5 % de NMP/95 % de PEG. El volumen de la dosis era de 5 ml/kg.
Para todos los animales, se recogió sangre mediante punción cardiaca terminal bajo anestesia con isoflurano. Se procesó la sangre para obtener el plasma. Además, se recogieron muestras tumorales.
La FIG. 29A muestra que la monoterapia de compuesto BB inhibió significativamente el crecimiento tumoral en el modelo de cáncer de colon MC38. La FIG. 29B muestra los efectos de compuesto BB y 5-FU utilizados por separado y en combinación en el modelo de MC38.
Al final del estudio, se aislaron los tumores, se fijaron en formalina tamponada neutra al 10% y se incluyeron en parafina para el análisis histológico. Se cortaron secciones tumorales para vehículo y monoterapia de compuesto BB para el análisis inmunohistoquímico. Se identificaron las células T dentro de las secciones tumorales mediante la utilización de un equipo de inmunohistoquímica automatizado de anticuerpos específicos de CD3 (Ventana). Después de la tinción, los portaobjetos se digitalizaron mediante un equipo de escaneo de portas Aperio. A continuación, se cuantificaron las secciones tumorales teñidas con el software Definians con el fin de evaluar el porcentaje de células positivas para CD3 dentro de cada tumor. La FIG. 30 muestra que los tumores de MC38 tratados con compuesto BB presentan significativamente más células T CD3+ intratumorales que el grupo de vehículo. El volumen tumoral final se representó gráficamente como una función del porcentaje de células T CD3+ en la muestra de vehículo (FIG. 31A) y la muestra de monoterapia de compuesto BB (FIG. 31B), respectivamente. Se añadió una línea de tendencia a los gráficos de vehículo y monoterapia de compuesto BB. Los volúmenes tumorales finales de vehículo no se correlacionaban con el porcentaje de células T CD3+, mientras que en la cohorte de monoterapia de compuesto BB, los volúmenes tumorales finales se correlacionaban fuertemente con el porcentaje intratumoral de células T CD3+ (R2=0,7873). Los resultados indican que el flujo de entrada significativo de células T CD3+ con la inhibición de PI3K-gamma conduce a reducciones del tamaño tumoral.
Ejemplo 265: efecto del compuesto BB en el modelo de linfoma folicular humano de células B DoHH2
El propósito del presente estudio era evaluar la actividad antitumoral del compuesto AA y del compuesto BB solos y en combinación en el modelo subcutáneo de linfoma folicular de células B transformado humano DoHH2 en ratones CB17.SCID hembra. DoHH2, una línea celular de linfoma folicular de células B humano, se cultivó en cultivo de tejido (RPMI-1640 filtrado, complementado con suero de feto bovino al 10%) y se implantó (5x106 células en 100 pl de medio RPMI-1640 y 100 pl de Matrigel (sin LDV)) por vía subcutánea en el flanco trasero de ratones CB17.SCID hembra. El tratamiento se inició una vez los tumores habían alcanzado 100 mm3 de volumen. En grupos de administración de 15 ratones por grupo se administró el fármaco de la manera siguiente: 1. Vehículo (5 % de NMP y 95 % de PEG400) Vehículo; 2. Compuesto AA (10 mg/kg) Vehículo; 3. Compuesto BB (15 mg/kg) Vehículo; 4. Compuesto AA Compuesto BB. Los animales recibieron dosis durante 21 días consecutivos Se determinaron comparaciones de eficacia mediante mediciones de los tumores con calibrador tres veces a la semana.
Las FIGS. 32A y 32B mostraron el resultado de inhibir PI3K-Y (compuesto BB) y/o PI3K-8 (compuesto AA) en el modelo DoHH2. La FIG. 32B es un estudio repetido. La inhibición doble de PI3K-8 y PI3K-Y fue superior a la inhibición de cualquiera de las isotermas por sí sola.
Ejemplo 266: efectos del compuesto BB y/o de la inhibición de PDL-1 sobre el crecimiento tumoral en un modelo ortotópico de cáncer de mama 4T1
El inhibidor selectivo de PI3K-Y, el compuesto BB, la terapia de punto de control inmunitario, PDL-1, se sometieron a ensayo separadamente y juntos para los efectos sobre el crecimiento tumoral en el modelo de cáncer de mama 4T1. Dicho sistema modelo se describe en mayor detalle en el Ejemplo 227. Brevemente, en dicho estudio se inició el tratamiento una vez los tumores habían alcanzado 100 mm3 El compuesto BB o vehículo se administró por vía oral una vez al día durante 3 semanas. Se administraron anticuerpos anti-PD-LI o de control de isotipo por vía intraperitoneal una vez cada 3 días durante un total de 5 dosis. En una muestra de 15 ratones, los grupos de administración fueron los siguientes. 1. Vehículo control de isotipo (IgG2bK de rata, 200 |jg); 2. Compuesto BB7 (7,5 mg/kg), PO control de isotipo (200 jg); 3. Vehículo anti-PD-L1 (200 jg); 4. Compuesto BB (7,5 mg/kg), PO anti-PD-L1 (200 jg). Durante el experimento, se midió el flujo de luciferina tumoral dos veces a la semana. Se determinaron comparaciones de eficacia mediante mediciones de los tumores con calibrador.
La FIG. 33A muestra un crecimiento tumoral reducido con compuesto BB y/o la inhibición de PDL-1 en un modelo de cáncer de mama 4T1 ortotópico. La FIG. 33B muestra un flujo reducido de luciferasa total con compuesto BB y/o la inhibición de PDL-1 en un modelo de cáncer de mama 4T1 ortotópico. La FIG. 34 muestra que el compuesto BB demuestra actividad de agente único en el modelo de cáncer de mama 4T1 subcutáneo y que el efecto antitumoral del compuesto BB se mantuvo durante por lo menos 6 días después de la interrupción del tratamiento con compuesto BB.
El modelo 4T1 también se utilizó para determinar la actividad anticáncer de compuesto BB y 5-FU, docetaxel o paclitaxel. Brevemente, el tratamiento se inició una vez los tumores habían alcanzado ~50 mm3 de volumen. El compuesto BB o vehículo se administró por vía oral una vez al día a una dosis de 15 mg/kg en la formulación de NMP en ratones Balb/c hembra durante 2 semanas. Se administraron por vía intraperitoneal docetaxel 33 mg/kg Q7D x2, paclitaxel 10 mg/kg Q5D x3, 5-FU 50 mg/kg QOD x7 o solución salina. Se determinó la eficacia según volumen tumoral y mediciones de la luciferina. Las mediciones de los tumores y de los pesos corporales se obtuvieron tres veces cada semana. Las mediciones de luciferina se realizaron dos veces a la semana.
La FIG. 35 muestra que el compuesto BB por sí solo consiguió una reducción del crecimiento tumoral, mientras que la coadministración del compuesto BB y 5-Fu consiguió una reducción adicional. La FIG. 36 muestra que el compuesto BB demuestra actividad de agente único en el modelo de cáncer de mama 4T1 subcutáneo, mientras que la coadministración con docetaxel, paclitaxel o 5-FU consiguió una reducción adicional del crecimiento tumoral.
En algunos casos, un tumor que está siendo tratado con una terapia inmunitaria puede experimentar un incremento transitorio de tamaño, ver ASCO 2008, resumen n° 3120 Wolchok. Sin embargo, dicho incremento de tamaño no se debe necesariamente al crecimiento de las células cancerosas. Por el contrario, podría deberse a la infiltración tumoral de linfocitos antitumorales.
En resumen, el compuesto BB mostró actividad en modelos de cáncer de mama, cáncer pulmonar, cáncer de colon y glioblastoma.
Ejemplo 267: estudios farmacodinámicos en modelos singénicos murinos de mama, pulmón y colon
Se caracterizó el efecto del compuesto BB sobre los infiltrados inmunitarios en tumor sólido.
Se llevó a cabo un análisis inmunohistoquímico para determinar la distribución de las células inmunitarias dentro del tumor. En particular, se examinaron las células mieloides CD11b, al igual que las células T reguladoras vs. las efectoras (CD3, CD4, CD8 y FoxP3). Brevemente, se llevó a cabo análisis IHQ para CD11b o CD68+ de la manera siguiente: los anticuerpos de CD11b se encuentran disponibles de Abcam, clon 1-70, n° de catálogo ab 8878. Los anticuerpos de CD68 se encuentran disponibles de Abcam, clon FA-11, n° de catálogo ab 53444. Se fijaron los tejidos en metanol frío, se secaron al aire y después se tiñeron utilizando un protocolo de IHQ Omni-UltraMap HRP XT
Como línea base, las células de cáncer de mama murino no tratadas mostraron niveles moderados de células mieloides CD11b+ (datos no mostrados). La FIG. 37A muestra que, con el tratamiento con compuesto BB, se produjo una reducción de la tinción de las células mieloides CD11b+ en zonas viables de los tumores 4T1 bajo tratamiento con compuesto BB en comparación con vehículo, el día 13 del estudio. Sin restringirse a ninguna teoría en particular, la reducción de las células mieloides asociadas a tumor tumorigénicas puede resultar en la activación de células T citotóxicas y en inmunidad antitumoral. La FIG. 37B muestra un análisis de imágenes de tejidos tumorales tratados con compuesto BB o vehículo, en las que el compuesto BB resultó en una reducción de las células CD11b+ en el tumor. La FIG. 37C muestra la cuantificación del porcentaje de células CD11b+ o de las células CD68+ en tejido
tumoral tratado con compuesto BB o vehículo. La FIG. 37E y la FIG. 37F muestran la cuantificación de los niveles de CD11b mediante IQC sobre células tumorales procedentes de tumores en almohadilla grasa mamaria 4TI-luc; el compuesto BB redujo el porcentaje observado de células tumorales. Al cuantificar las imágenes como células CD11b+ respecto al total de células viables (FIG. 37E), se obtuvo un valor de p en la prueba t de Student de 0,0355, en la comparación entre las células tratadas con el compuesto BB y los controles. Al cuantificar las imágenes como zona teñida CD11b+ respecto al total de zona visible (FIG. 37F), se obtuvo un valor de p en la prueba t de Student de 0,0441, en la comparación entre las células tratadas con el compuesto BB y los controles. De esta manera, el compuesto B causa una reducción estadísticamente significativa de las células CD11b+ en el presente ensayo. Las FIGs. 37g y 37H muestra un análisis IHQ adicional que muestra cómo el compuesto BB afecta al porcentaje de células teñidas con CD11b. Al cuantificar las imágenes como células CD11b+ respecto al total de células viables (FIG. 37G), se obtuvo un valor de p en la prueba t de Student de 0,0864 (d16) o de 0,3967 (d22), en la comparación entre las células tratadas con el compuesto Bb y los controles. Al cuantificar las imágenes como zona teñida CD11b+ respecto al total de zona visible (FiG. 37H), se obtuvo un valor de p en la prueba t de Student de 0,2074 (d16) o de 0,1538 (d22), en la comparación entre las células tratadas con el compuesto BB y los controles. Tal como indica la FIG. 37D, hay una fuerte correlación entre las células mieloides CD11b+ y CD68+ en los tumores 4T1 tratados con compuesto BB.
Se llevó a cabo citometría de flujo para obtener un análisis más amplio de subgrupos celulares. En particular, el estudio examinó células T vs. células mieloides, subgrupos de células mieloides, células T reguladoras vs. efectoras, y marcadores tumorales vs fórmula leucocitaria (p.ej., la expresión de PD-L1 sobre el tumor).
Pueden identificarse diferentes poblaciones de células mieloides asociadas a tumor mediante citometría de flujo, p.ej., células mieloides inmaduras monocíticas (CMi), CMi granulocíticas/neutrófilos, macrófagos asociados a tumor (Ma T) y CSDM granulocíticas. Las diferentes oblaciones y paneles para detectar las CSDM se indican en Cancer Discovery 2011;1:54-67 Los tumores pueden variar en su composición de células mieloides. En modelos de tumor LLC brei tratados con compuesto BB, se redujo el número de MAT Infiltrantes, tal como se muestra en la FIG. 38A. El presente experimento indica que el compuesto BB reduce con éxito un tipo de células inmunosupresoras en el microambiente tumoral, apoyando el modelo de que el compuesto BB trata el cáncer mediante el bloqueo de la inmunosupresión en el sitio tumoral o en proximidad al mismo. En un modelo de cáncer de colon (tumores subcutáneos CT26), el compuesto BB redujo el número de CMi monocíticas, tal como se muestra en la FIG. 38B. La FIG. 39 muestra que el compuesto BB redujo los macrófagos supresores polarizados en M2. CD206 es característico de los macrófagos M2 (protumorales) o las CSDM, y se redujo la tinción de CD206 en muestras de tumor LLC brei con el tratamiento con compuesto BB, en comparación con un control.
En conclusión, el compuesto BB muestra un efecto farmacodinámico sobre las células mieloides que es consistente con una actividad antitumoral. Se observó dicho efecto en células de cáncer de mama 4T1-luc, células de cáncer pulmonar LLC-luc y células de cáncer de colon CT26.
Además, se llevó a cabo el perfilado del ARN para determinar el estado de M1 vs. M2 de los macrófagos diferenciados, así como un panel inmunosupresor vs. inflamatorio más amplio en tumores completos.
Los macrófagos M2 pueden producirse de la manera siguiente. Se sacrifican los ratones y se recogen fémures y tibias. Se sumergen los huesos en una placa Petri en etanol al 70% durante 1 a 2 min, se sacan los huesos y se deja que se seque el etanol durante 3 a 4 min. Se recorta una porción muy pequeña del extremo de los huesos para facilitar el acceso por inserción de una aguja. Utilizando una aguja 27g llena de medio que contiene DMEM FBS al 10 % pen/strep al 1 % (DMEM completo), se extrae la médula ósea de ambos extremos de los huesos a una placa Petri de 15 ml. Se pipetea la médula ósea para disociar las células de médula ósea. Se centrifuga y se añade 1x tampón de lisis de glóbulos rojos al pellet. Se deja que los glóbulos rojos lisen durante 2 min y después se añaden cantidades iguales de DMEM completo y se centrifuga. La médula ósea de un ratón puede dividirse en 2 placas Petri que contienen 7 ml de medio BMM (Dm e M FBS al 20 % pen/strep al 1 % 50 ng/ml de M-CSF). El día en que se siembran en placa de Petri las células es el día uno. El día cuatro, se añaden 3 ml de medio BMM a cada placa. Se añade M-CSF inmediatamente antes del cultivo. Las células están listas para la polarización el día 6.
A continuación, pueden polarizarse las células. Para recolectar las células, eliminar el sobrenadante y lavar una vez con 1x PBS. Añadir 4 ml de solución de disociación a cada placa Petri. Dejar durante 5 a 10 min y recoger las células y lavar la placa Petri con DMEM completo para obtener todas las células sobre la placa. Un tiempo de recolección más largo puede incrementar el rendimiento. En este punto pueden sembrarse las células en las placas para los experimentos.
1. Añadir DMEM FBS al 10 % pen/strep al 1 % a los pellets celulares.
2. Sembrar las células en placas de 12 pocillo (1x106 células/2 ml de medio por pocillo) en DMEM completo que contiene 20 ng/ml de M-CSF.
3. Para la polarización en M1 añadir: 20 ng/ml de IFNg 100 ng/ml de LPS durante 24 h.
4. Para la polarización en M2 añadir: 20 ng/ml de IL-4 - 4 ng/ml durante 48 h.
5. Añadir los tratamientos/inhibidores simultáneamente.
6. Añadir el fármaco de ensayo (por ejemplo, concentraciones de ensayo del compuesto BB /- controles de DMSO)
7. Recolectar el ARN.
La cantidad y calidad del ARN pueden evaluarse para la normalización antes de la RT-PCR. Se lleva a cabo la qRT-PCR siguiendo las recomendaciones del fabricante, utilizando el kit 'RNA to CT one step' de los kits de cebadores ABI y ABI taqman para los genes relevantes. En el caso de que las células se analicen mediante FACS, las células diferenciadas se lisan/fijan (bd) después de la recolección y se congelan. La tinción para CD206 de ratón (Biolegend 161707) y ARG-1 (R & D Systems, IC5868F) se lleva a cabo en tampón de tinción b D con BSA.
Se llevó a cabo el perfilado de la expresión del ARN para el inmunofenotipado con una matriz de baja densidad Taqman (TLDA, por sus siglas en inglés). La Tabla 24, a continuación, muestra un panel de genes que pueden utilizarse para determinar el fenotipo de los infiltrados inmunitarios.
Tabla 24. Genes que pueden utilizarse para determinar el fenotipo de los infiltrados inmunitarios.

Se implantaron células de cáncer pancreático KPC en animales singénicos que seguidamente fueron tratados con vehículo (n=13), compuesto BB a una dosis de 3 mg/kg (n=7), anti-PDL1 (n=9) o una combinación de compuesto BB y anti-PDL1 (n=8). Para los tumores de ratón, se molieron las muestras congeladas instantáneamente, con un molino Genogrinder a temperaturas de nitrógeno líquido y se aisló el ARN con trizol. A continuación, se lavó la muestra con Qiashredder y después se utilizó un kit RNeasy.
Los datos se analizaron de la manera siguiente. Se utilizaron los valores de CT para el análisis. Para cada muestra Si y cada gen, se calcularon los valores siguientes.
dCT(Si,gen) = CT(Si,gen) - CT(Si,ACTB)
ddCT(Si,gen) = dCT(Si,gen) - mediana(dCT(muestras tratadas con vehículo, gen))
RQ(Si,gen) = 2(-ddCT(Si,aen))
Lo anterior generó una matriz de valores RQ: 48 genes (filas) x 39 muestras (cols.)
Se añadieron cuatro filas a la matriz RQ para representar el tratamiento con vehículo, compuesto BB, anti-PDL-1 o una combinación de compuesto BB y anti-PDL-1. El análisis se realizó con un paquete R 'Non-negative Matrix Factorization' (NMF) con rango=3 y el algoritmo "lee". La FIG. 40 muestra que la normalización respecto a la p-actina proporciona una expresión estable de los genes de GAPDH y 18S genes.
El análisis mostró que los marcadores de macrófago M2, VEGF y ARG-1, presentaban una tendencia a una reducción por el compuesto BB con o sin anticuerpo de PDL-1. Este resultado concuerda con la inhibición por compuesto BB de la diferenciación o la migración de los macrófagos M2.
El análisis indica que el compuesto BB por sí solo resultó en una tendencia de reducción de los genes de T reguladoras y T efectoras, y en una reducción de los marcadores de activación de las células dendríticas. Un anticuerpo de PDL-1 en combinación con compuesto BB no mostró dichos efectos. Indica además que el compuesto BB por sí solo o en combinación con anticuerpo de PDL1 rinde una reducción en los genes de M2 a RG-1 y VEGF, es decir, el compuesto BB aparentemente reduce el nivel de células M2 en la muestra, por ejemplo, mediante el bloqueo de la diferenciación de las células de médula ósea en células M2. A modo de control, el anticuerpo de PDL-1 resultó en la inducción esperada de genes proinflamatorios. La FIG. 41 muestra el efecto del compuesto BB sobre los marcadores Cd8b1, Pdcd1/PD1, Cd4, Cd3e, Foxp3 y Clla4 con o sin anticuerpo de PDL-1. La FIG. 42A muestra que los marcadores de macrófago M2, VEGF y ARG-1 resultan reducidos por el compuesto BB con o sin anticuerpo de PDL-1. La FIG. 42B muestra el efecto del compuesto BB con o sin anticuerpo de PDL-1 sobre los marcadores de macrófago M2, ARG1, VEGFa y ADM.
Ejemplo 268: propiedades físicas y farmacocinéticas del compuesto BB
Las propiedades fisicoquímicas del compuesto BB son de tipo fármaco, y adecuadas para la utilización del compuesto como un fármaco. Por ejemplo, el compuesto BB presenta un peso molecular de 528,6 Da, un cLog P de 4,0, una estabilidad en fluido gástrico simulado 38° >4 horas y una pKa que no muestra ionización entre pH 3 y 10.
Se examinaron las características ADME del compuesto BB y se muestran a continuación, en la Tabla 25.
Tabla 25. Características ADME del compuesto BB

El compuesto BB muestra una elevada permeabilidad de la membrana celular. Además, el compuesto no inhibe sustancialmente CYP. El compuesto BB consigue una biodisponibilidad oral elevada en especies animales de roedores y de no roedores. El compuesto BB alcanza un volumen elevado de distribución y parece distribuirse fácilmente en las células/tejidos. Presenta además una elevada estabilidad metabólica. Dicha estabilidad se traduce en un lavado reducido y una semivida in vivo prolongada. Estas propiedades indican la idoneidad del compuesto para la administración in vivo.
El compuesto BB parece ser un inhibidor de P-GP (la glucoproteína P 1A, inhibidor de PI3K-Y, tal como el compuesto BB, puede administrarse opcionalmente en el paciente en combinación con un segundo terapéutico que es un sustrato de P-gp. Sin restringirse a ninguna teoría en particular, el inhibidor de P-gp podría ayudar a mantener niveles del sustrato P-gp en tumores que sobreexpresan el transportador de P-gp.
La semivida oral del compuesto BB es de aproximadamente 3,3 horas en la rata; de aproximadamente 7,1 horas en el perro y de aproximadamente 10,2 horas en el mono. Basándose en estos datos, se espera que la semivida oral en el ser humano sea de 10 a 13 horas.
Existe un potencial bajo de inhibición reversible de las isoformas principales de citocromo P450. No se espera que el compuesto BB forme metabolitos reactivos.
En un estudio ejemplar, se midieron las concentraciones en plasma de compuesto BB en las 24 horas siguientes a la administración oral en ratas (7,5 mg/kg), perro (5 mg/kg) y mono (5 mg/kg) y se muestran los resultados en la FIG. 43. La exposición oral del compuesto BB es elevada en rata, perro y mono. La buena biodisponibilidad oral y el lavado reducido en especies animales de roedores y de no roedores resulta en una exposición oral elevada.
En otro estudio ejemplar, se midieron las exposiciones en plasma de compuesto BB en ratas después de la administración oral a dosis de 10, 25 y 100 mg/kg (mediante ABC0-último) y se muestran los resultados en la FIG. 44. El incremento de exposición del compuesto BB es superior o igual al incremento de la dosis. De esta manera, el compuesto muestra una buena proporcionalidad de dosis-exposición.
Ejemplo 269: estudios toxicológicos del compuesto BB
Los estudios de toxicología in vitro del compuesto BB mostraron que se espera que el compuesto presente una toxicidad muy baja. Se llevó a cabo un estudio de toxicidad en órganos diana (TOD) en ratas. Las ratas recibieron dosis orales de compuesto BB durante 10 días. El presente estudio evaluó observaciones clínicas/mortalidad, pesos corporales, patología clínica, toxicocinética e histopatología de tejidos seleccionados. Los tejidos estudiadores fueron: glándula adrenal, médula ósea (fémur), cerebro, intestino delgado (incluyendo GALT), corazón, intestino grueso, riñón, estómago, hígado, páncreas, bazo, testículo y timo. Se seleccionaron las dosis basándose en un estudio de tolerabilidad a las dosis repetidas, FC y unión de proteínas plasmáticas, y las IC50 celulares para las isoformas de PI3K. Las dosis seleccionadas fueron: dosis elevada: 50 mg/kg/día, que se esperaba que inhibiese las isoformas a, p, 8 y y de PI3K; dosis intermedia-alta: 20 mg/kg/día, que se esperaba que inhibiese las isoformas p, 8 y y de PI3K; dosis intermedia-baja: 7,5 mg/kg, que se esperaba que inhibiese la isoforma y de PI3K únicamente, y dosis baja: 1 mg/kg,
que se esperaba que inhibiese la isoterma y de PI3K únicamente. La formulación utilizada fue la Formulación: CMC al 0,5 %, Tween al 0,05 % en agua con compuesto BB.
El estudio TOD indicó que el compuesto BB resultaba bien tolerado durante 10 días. No se observó mortalidad o signos clínicos a ningún nivel de dosis. Se produjo una reducción mínima de los pesos corporales en los animales a dosis alta (5-8 %) en comparación con el vehículo. Respecto a la patología clínica, se observaron cambios mínimos en los neutrófilos, linfocitos y fibrinógeno (principalmente en los grupos de 20 y 50 mg/kg/día). En términos de histopatología, no se observaron cambios macroscópicos significativos. En el bazo, se produjo un agotamiento linfoide mínimo/leve de la zona marginal y de las vainas linfoides periarteriolares (VLPA) principalmente a dosis > 20 mg/kg/día, y en el timo, se produjo un agotamiento linfoide mínimo/leve en machos tratados a una dosis > 7,5 mg/kg/día, y en hembras a la dosis de 50 mg/kg/día.
La FIG. 45 muestra las concentraciones de compuesto BB libre en un estudio TOD. El compuesto BB libre alcanzaba concentraciones que se esperaba que inhibiesen las isoformas de PI3K. La evaluación de los datos de inhibición de isoformas de PI3K respecto a las concentraciones libres en plasma demuestra que las dosis que se administraron inhibieron las isoformas deseadas. Específicamente, las dosis de 1 y 7,5 mg/kg alcanzaron concentraciones suficientemente altas para inhibir selectivamente PI3K-Y sin inhibir sustancialmente PI3K a, p o 8.
Ejemplo 270: efecto del microambiente tumoral sobre la supervivencia de las células de cáncer
Las células estromales de médula ósea protegen a las células de LLC frente a la apoptosis espontánea in vitro. Lo anterior se observó en un experimento en que se añadieron células de LLC a estroma confluyente y después se evaluó la viabilidad de LLC durante el tiempo. La FIG.46 muestra que las células de LLC presentan una tasa de supervivencia más alta al cocultivarlas con células estromales. El presente experimento sugiere que las células estromales envían señales prosupervivencia a las células de LLC, y que estas señales están mediadas por PI3K. Los experimentos con inhibidores selectivos de PI3K-8 y PI3K-Y indican que la proliferación de LLC inducida por TME es dependiente de PI3K-8 (datos no mostrados).
Dicha señalización aparentemente también se produce in vivo, ya que se observa un incremento de la señalización de PI3K en centros de proliferación de los ganglios linfáticos de LLC en el ser humano, medida como niveles elevados de pS6. Balakrishnan et al., ASH 2013. Además de pS6, que es un indicador de la activación de AKT, dichos centros de proliferación presentan niveles elevados de células T, tal como indica la tinción de CD3 (datos no mostrados). La presencia de Ki-67 indica niveles elevados de proliferación.
Ejemplo 271: desarrollo de un panel de genes para identificar las CSDM
El objetivo del presente estudio es desarrollar un panel de células mieloides para células tumorales disociadas a fin de medir los infiltrados para MAT y CSDM. Se sometieron a ensayo tumores 4T1 mediante FACS para la presencia de diversas células inmunitarias. El estudio cuantificó el nivel de Ly6C y Ly6G en la población. El estudio indicó un enriquecimiento en células 4T1 mucho mayor en modelos de tumor brei granulocítico (Ly6C+, Ly6G+) que en LLC (datos no mostrados). El análisis de FACS también mostró que las células M2 (o M1) diferenciadas in vitro presentaban un fenotipo de CSDM "MAT" (datos no mostrados).
Entre los marcadores potenciales de un panel de células mieloides para células M1 se incluyen: M1 CD68 MHCII y CD68 iNOS (PLoS One, 2013; 8(12): e79769).
Entre los marcadores potenciales de un panel de células mieloides para células M2 se incluyen: CD68+ CD163+ o CD68+ VEGF+ (PLoS One, 2013; 8(12): e79769).
Entre los marcadores potenciales para un panel de células mieloides para miocitos se incluyen CD14 y CD16, teniendo en cuenta la relación entre los dos, p.ej., CD14+ CD16- es el patrón clásico, mientras que CD14low CD16bright es no clásico.
CSF1R es otro marcador potencial (Cancer Cell 25, 1-14, 16 de junio, 2014, a2014 Elsevier Inc.). La publicación Cancer Cell describe un ensayo de biomarcadores en tumores sólidos.
Entre otros marcadores potenciales para un panel se incluyen CD11b F4/80; CD11b LY6C/G; ARG1, CD206 y la expresión de MHC, que puede diferenciar entre las células M1 y M2 en el ratón), y VEGF (que se expresa a nivel elevado en un subgrupo de MAT).
Ejemplo 272: eficacia del compuesto BB en un modelo de glioblastoma
El objetivo del presente estudio es evaluar la actividad antitumoral y efectos inmunomoduladores del compuesto BB en el modelo de glioblastoma subcutáneo murino GL261-Luc. GL261-Luc era una línea celular murina de origen cerebral expresante de luciferasa que se transfectó establemente con gen de luciferasa de luciérnaga. Se cultivaron células adherentes GL261-Luc en cultivo de tejidos, en medio DMEM filtrado complementado con suero de feto bovino
al 10 % y pen/strep al 1 %. Se preparó el inóculo tumoral a una densidad de 1x107 células en 100 |jl de PBS estéril Matrigel. Las células se implantaron subcutáneamente en la zona del flanco posterior derecho en ratones C57BL/6 Albino marcho (stock n° 000058, edad: 6 semanas) de Jackson Laboratories. El tratamiento se inició una vez los tumores habían alcanzado ~100 a 200 mm3 de volumen. El compuesto BB o vehículo se administró por vía oral una vez al día a una dosis de 15 mg/kg en la formulación de NMP. Se determinó la eficacia según volumen tumoral y mediciones de la luciferina. Las mediciones de los tumores y de los pesos corporales se obtuvieron dos a tres veces cada semana. Las mediciones de luciferina se realizaron dos veces a la semana. Al final del estudio, se recolectaron células tumorales y se prepararon para el análisis mediante FACS, IHQ, análisis del ARN y otros análisis.
La FIG. 47A muestra que el compuesto BB redujo el volumen tumoral en el modelo de glioma singénico GL-261 a una dosis de 15 mg/kg QD PO. La FIG. 47B muestra que el compuesto BB redujo el flujo total en el mismo modelo. La FIG. 47C es un estudio de repetición del mismo modelo que muestra los efectos del compuesto BB QD sobre el volumen tumoral entre los días 9 y 25 después del implante.
Ejemplo 273: eficacia del compuesto BB con o sin ciclofosfamida en el modelo LLC
El objetivo del presente estudio es evaluar el efecto de la combinación de compuesto BB ciclofosfamida en el modelo de tumor brei LLC. En el presente experimento, el tratamiento se inició una vez los tumores habían alcanzado aprox.
150 mm3, aproximadamente el día 12. Se monitorizaron las imágenes Caliper y Xenogen. Grupos de tratamiento n=13-15/grupo Se utilizaron las dosis y formulaciones siguientes: vehículo (5 % de NMP/95 % de PEG 400 / PBS) PO/IP; Compuesto BB a 15mg/kg QD, PO; ciclofosfamida a 50 mg/kg EOD IP; Combinación (compuesto BB Ciclof.). Se recogieron muestras para el análisis de FACS el día 23 después del implante. Mediante FACS se sometieron a ensayo las células para el nivel de CD11b y Gr-1. Las células mieloides Gr1 CD11b+ son las células inflamatorias más prevalentes que se observan en los tumores, en donde estimulan directamente la angiogénesis tumoral y la inmunosupresión. Hardamon et. al., Proceedings: AACR 103rd Annual Meeting 2012, Cancer Research: 15 de abril 15, 2012; volumen 72, número 8, suplemento 1.
La FIG. 48 muestra los resultados del presente experimento. El compuesto BB por sí solo y la ciclofosfamida por sí sola redujeron cada uno el crecimiento tumoral en aproximadamente la misma medida. Al administrar juntos ambos compuestos, se retrasó adicionalmente el crecimiento tumoral. Las FIGS. 49A, 49B, 49C, 49D y 49E muestran los gráficos de CD11b/Gr-1 al administrar compuesto BB a una dosis de 15 mg/kg y ciclofosfamida a una dosis de 50 mg/kg. La FIG. 49A muestra el porcentaje de células que son CD3 respecto a las CD45+. La FIG. 49B muestra el porcentaje de células que son CD11b+Gr-1‘ respecto a las CD45+. La FIG. 49C muestra el porcentaje de células que son CD11b+Gr-1+ respecto a las CD45+. La FIG. 49D muestra el porcentaje de células que son CD11b'Gr-1‘ respecto a las CD45+. La FIG. 49E muestra el porcentaje de células que son CD11b-Gr-1+ respecto a las CD45+.
Las FIGS. 50A, 50B, 50C y 50D muestran el panel de CSDM CD11b/Ly6C/Ly6G al administrar compuesto B a una dosis de 15 mg/kg y ciclofosfamida a una dosis de 50 mg/kg. La FIG. 50a muestra el porcentaje de células que son CD11b+ respecto a las CD45+. La FIG. 50B muestra el porcentaje de células que son CD11b+Ly6C+ respecto a las CD45+. La FIG. 50C muestra el porcentaje de células que son CD11b+Ly6C+Ly6G+ respecto a las CD45+. La FIG. 50D muestra el porcentaje de células que son CD11b+Ly6C'Ly6G' respecto a las CD45+.
Las FIGS. 51A, 51B, 51C, 51D y 51C muestran gráficos de CD3/CD4/CD8 al administrar el compuesto BB a una dosis de 15 mg/kg y ciclofosfamida a una dosis de 50 mg/kg. La FIG. 51A muestra el porcentaje de células que son CD3+ respecto a las CD45+. La FIG. 51B muestra el porcentaje de células que son CD4+CD8- respecto a las CD45+. La FIG.
51C muestra el porcentaje de células que son CD4+CD8+ respecto a las CD45+. La FIG. 51D muestra el porcentaje de células que son CD4_c D8+ respecto a las CD45+. La FIG. 51E muestra el porcentaje de células que son CD4'CD8‘ respecto a las CD45+.
Ejemplo 274: Efecto del compuesto BB sobre los niveles de CD206 de los macrófagos en tumores LLC
El objetivo del presente estudio es evaluar los efectos del compuesto BB administrado por vía PO, QD, en paralelo o en combinación con ciclofosfamida. Se administró compuesto BB QD en ratones C57 Albino. Se administró ciclofosfamida EOD IP. Se realizaron mediciones de tumores y pesos corporales 2X/semana. Los tumores se recolectaron el último día o en el caso de que en los grupos de vehículo alcanzasen aproximadamente 3000 mm3. Los tumores se cortaron por la mitad. Una mitad se cortó nuevamente en mitades y una mitad se fijó en NBF al 10 % y la otra mitad se congeló en OCT para secciones congeladas. La mitad restante se procesó para generar una suspensión de células individuales y se evaluó mediante FACS utilizando dos paneles.
Los anticuerpos utilizados para la tinción para FACS fueron los siguientes:
Tinción con CD4 y CD8a: CD45-AF700 (BDB560510) a 1,25 jg/ml; CD3-FITC (BDB555274) a 1 jg/ml; CD4-PE (BDB553049) a 2,5 jg/m l y CD8a-PerCP (BDB553036) a 5 jg/ml.
Tinción con CD11b y GR-1: CD45-AF700 (BDB560510) a 1,25 jg/ml; CD3-FITC (BDB555274) a 1 jg/ml; CD11b-APC (BDB553312) a 2,5 jg/m l y GR-1-PE-CY7 (BDB552985) a 0,5 jg/ml.
Tinción con panel de CD206: CD45-AF700 (BDB560510) a 1,25 pg/ml; CD11b PE a 2,5 pg/ml y CD206 APC a 25 |jg/ml.
Tinción con panel de CSDM: CD45-AF700 (BDB560510) a 1,25 pg/ml; CD11b PE a 2,5 pg/ml; Ly6C APC a 2,5 pg/ml y Ly6G FITC a 2,5 pg/ml.
Las FIGS. 52A, 52B, 52C, 52D y 52E muestran el porcentaje de células que son CD45+, CD11b+ o CD206+, expresado como porcentaje del total de células, en un modelo de tumor de CPL brei tratado con compuesto BB /- ciclofosfamida. La FIG. 52A muestra el porcentaje de células que son CD45+ expresado como porcentaje del total de células. La FIG.
52B muestra el porcentaje de células que son CD11b expresado como porcentaje del total de células. La FIG. 52C muestra el porcentaje de células que son CD206 expresado como porcentaje del total de células. La FIG. 52D muestra el porcentaje de células que son CD11b expresado como porcentaje del total de células. La FIG. 52E muestra el porcentaje de células que son CD206 expresado como porcentaje del total de células.
Aparentemente se produce una reducción de la población de CD206 tanto con compuesto BB administrado solo como en la combinación con ciclofosfamida. Por lo tanto, el compuesto BB reduce el número de macrófagos M2 en el tumor, o bien evitando que estos migren hacia el interior del tumor, o bien impidiendo que las células mieloides inmaduras se diferencien en células M2, o ambos mecanismos.
Ejemplo 275: farmacocinética y reducción de la neutrofilia inducida por LPS - administración intratraqueal
El estudio de la administración intratraqueal (i.t.) de compuesto BB sobre el modelo de neutrofilia inducida por LPS se lleva a cabo utilizando métodos conocidos de la técnica. Por ejemplo, se administra compuesto BB por vía intratraqueal en ratas en una cantidad de, p.ej., 1 pg/kg, 10 pg/kg o 100 pg/kg. La administración de los controles se lleva a cabo como, p.ej.: (1) administración oral de compuesto BB a 10 mg/kg, (2) administración intratraqueal de solución salina (vehículo a 0,25 ml/rata, (3) administración intratraqueal de LSP (vehículo) a 1 pg de LPS/rata, y (4) administración intratraqueal de budenósido a 0,3 mg/kg. Una hora después de la administración se induce neutrofilia mediante tratamiento con LPS y se recogen las muestras mediante lavado broncoalveolar (LBA) a diversas horas después de la administración y se someten a examen adicional. El compuesto BB es un inhibidor de PI3Ky que presenta una proporción de selectividad delta/gamma superior a aproximadamente 50. A título comparativo, se llevan a cabo procedimientos similares utilizando un inhibidor de PI3K-8, que presenta una proporción de selectividad gamma/delta superior a aproximadamente 50.
Los niveles de compuesto BB en pulmón, líquido de lavado broncoalveolar (LLBA) y plasma se determinan utilizando métodos conocidos de la técnica.
Ejemplo 276: estudios con el modelo de ratones fumadores de enfermedad obstructiva pulmonar crónica (EPOC) (Ejemplo de referencia)
Los estudios del efecto de los compuestos proporcionados en la presente memoria sobre el modelo de ratón fumador de EPOC pueden llevarse a cabo mediante métodos conocidos de la técnica.
A. A continuación se proporciona un ejemplo ilustrativo de dicho método. Los ratones son expuestos a humo de tabaco durante 4 días en una caja de exposición de cuerpo completo. La exposición de cuerpo completo se lleva a cabo durante un cierto periodo de tiempo en una caja de metacrilato cilíndrica de 32 l construida al efecto (p.ej., Boehringer Ingelheim Pharma GmbH & Co. Kg , Biberach, Alemania) antes de la exposición a cada humo de tabaco o antes de la medición de la función pulmonar. Los animales se separan mediante espaciadores de acero inoxidable. El suelo de la caja se calefacta adicionalmente (p.ej. a 38°C) para mantener la temperatura corporal fisiológica de los animales. Los animales de control reciben solvente a modo de placebo. Los días uno y dos, los ratones son expuestos a la corriente principal de humo de tabaco. La exposición al humo de cada cigarrillo dura aproximadamente 15 min, seguido de una exposición de 8 min con aire limpio. Cada dos cigarrillos, se lleva a cabo una pausa adicional de 24 min de exposición a aire limpio. Se utiliza un encendedor de cigarrillos semiautomático y un generador de humo con un temporizador electrónico para controlar la exposición al tabaco (p.ej., Boehringer Ingelheim Pharma GmbH&Co. KG, Biberach, Alemania). Se monitorizó la concentración de partículas del humo de tabaco mediante un monitor de partículas ambientales en tiempo real (p.ej., MicroDustPro,Casella, Amherst, NH, EE.UU.). Los animales de control son expuestos a aire de la sala. Ver, p.ej., L. Wollin, et al., Pulmonary Pharmacology & Therapeutics 23 (2010) 345-354.
Los compuestos proporcionados en la presente memoria son administrados por vía oral o intratraqueal (i.t.) utilizando, p.ej., métodos conocidos de la técnica. Por ejemplo, un compuesto proporcionado en la presente memoria se administra por vía oral en forma de un sólido o una solución. Alternativamente, un compuesto proporcionado en la presente memoria se administra por vía intratraqueal mediante aerosolización de una solución del compuesto con un nebulizador de chorro. Se administra un compuesto proporcionado en la presente memoria a diversas concentraciones y regímenes. Por ejemplo, la exposición de cuerpo completo de un compuesto proporcionado en la presente memoria se administra durante 5 min y 1 h antes de la exposición a humo de tabaco.
Se determinan recuentos diferenciales de células en el líquido de lavado broncoalveolar (LLBA). Por ejemplo, se utilizan recuentos celulares totales de LBA y la cantidad de neutrófilos en las muestras para evaluar la eficacia de un compuesto proporcionado en la presente memoria. Los modelos de inflamación pulmonar inducida por humo de tabaco tienen un aumento del recuento de células totales en el LBA y también la cantidad de neutrófilos. De esta manera, una reducción del recuento de células totales en el LBA y la cantidad de neutrófilos en modelos en los que se ha administrado un compuesto proporcionado en la presente memoria comparados con un modelo de control (sin administración de un compuesto proporcionado en la presente memoria) ilustra la eficacia de un compuesto proporcionado en la presente memoria en el tratamiento de la inflamación pulmonar. Ver, p.ej., L. Wollin, et al., Pulmonary Pharmacology & Therapeutics 23 (2010) 345-354.
Además, se comparan secciones pulmonares teñidas con, p.ej., H+E o AB/PAS de los modelos de control (p.ej., modelos sin administración de un compuesto proporcionado en la presente memoria) con secciones de pulmón de modelos en los que se ha administrado un compuesto proporcionado en la presente memoria. La tinción de H+E muestra inflamación e infiltrados alveolares en los pulmones. La tinción de AB/PAS muestra el contenido de moco en las células caliciformes de las vías respiratorias grandes. Ver, p.ej., L. Wollin, et al., Pulmonary Pharmacology & Therapeutics 23 (2010) 345-354.
B. Se proporciona posteriormente un ejemplo de un modelo de EPOC por 10 días de humo de tabaco. El objetivo del presente estudio es determinar si el tratamiento con un compuesto proporcionado en la presente memoria puede evitar la inflamación pulmonar resistente a esteroides inducida por la exposición de los ratones a 10 días de humo de tabaco. Los ratones son expuestos en primer lugar durante 5 días a humo de tabaco para inducir resistencia a glucocorticoesteroides. Después, los animales son expuestos a 5 días adicionales de humo de tabaco junto con la administración de los compuestos de ensayo para investigar si puede reducirse la resistencia a glucocorticoesteroides.
Específicamente, ratones Balb/c byJ (peso: 24 a 26 g, 10 a 12 semanas de edad) son expuestos a aire estándar o humo de tabaco durante 10 días ("exposición de cuerpo completo"). Los ratones son expuestos en situación de agregación en una caja de metacrilato. Los cigarrillos utilizados son cigarrillos de investigación especiales estandarizados de Kentucky University: 3R4F sin filtro. La dosis y el tiempo se incrementan desde el 1° día hasta el 10° día de exposición (dos veces al día (=2 tandas); intervalo de exposición: 5 horas):
o 1° día: tanda 1=2 pares de cigarrillos y tanda 2=3 pares de cigarrillos. La dosis y tiempo de exposición al tabaco=10 a 15 min (dosis CO=150 a 300 ppm; concentración de 02=20,8 % medida con analizador PCA3 Baccharach).
o 2° día: tanda 1=4 pares de cigarrillos y tanda 2=5 pares de cigarrillos. La dosis y tiempo de exposición al tabaco=20 a 15 min (dosis C0=150 a 300 ppm; concentración de 02=20,8 %
o 3° día: tanda 1=6 pares de cigarrillos y tanda 2=7 pares de cigarrillos. La dosis y tiempo de exposición al tabaco=0 a 35 min (dosis C0=150 a 300 ppm; concentración de 02=20,8 %
o 4° día hasta el día 10°: tanda 1=7 pares de cigarrillos y tanda 2=7 pares de cigarrillos. La dosis y tiempo de exposición al tabaco=35 min (dosis c O=150 a 300 ppm; concentración de 0 2 =2 0 , 8 %.
Los animales son tratados con n compuesto proporcionado o solución de vehículo 1 o 2 por vía oral diariamente entre los días 6 y 10. Se administra dexametasona (5 mg/kg) por via intraperitoneal (IP) diariamente entre los días 6 y 10. Se monitoriza el peso corporal de los animales individuales diariamente entre los días 1 y 11.
Se sacrifican los ratones 1 día después de la última exposición a aire o humo de tabaco (HT) (día 10). Se extrae sangre mediante una punción cardiaca y el suero aislado se recoge y almacena a -30°C. Se lavan los pulmones, se aíslan las células del LBA, se cuentan y se diferencian. El fluido LBA se almacena para la determinación de citoquinas/quimioquinas.
A continuación, se proporcionan las tablas del diseño experimental:


C. A continuación se proporciona un modelo de fumador murino. Ver, p.ej., Yasuo, T., et al., Am. J. Respir. Crit. Care Med. 2010;182:897-904; Sato T, et al., Am. J. Respir. Crit. Care Med. 2006;174:530-537; and Kasagi S, et al., Am. J. Physiol. Lung Cell Mol. Physiol. 2006;290: 396-404, 1977.
Se lleva a cabo la inhalación de humo de tabaco utilizando cigarrillos de investigación sin filtro 1R1 (Tobacco Health Research Institute, Kentucky Univ., Lexington, KY) y el sistema experimental de inhalación de humo de tabaco para animales pequeños (modelo SIS-CS; Shibata Scientific Technology, Tokyo, Japón). El modelo SIS-CS consistía de tanto un generador de humo de tabaco (modelo SG-200) como una cámara de inhalación. El generador de humo es controlado mediante un ordenador portátil y generó automáticamente humo de tabaco mediante la fijación de un volumen de bomba de jeringa (10-50 cm3/aspiración) y varias aspiraciones/min (1a 12 aspiraciones).
El humo de tabaco generado se entrega en la cámara de inhalación, en la que se han dispuesto las sujeciones corporales para los ratones (se puede disponer un máximo de 12 sujeciones corporales simultáneamente) y en la que los ratones inhalan el humo de tabaco por el hocico. El humo de tabaco puede diluirse a una concentración deseada en el mezclador, en el que el humo de tabaco y aire comprimido se combinan antes de la administración a la cámara de inhalación. A las 12 semanas de edad, los ratones son sometidos al experimento de inhalación crónica de humo de tabaco. El diseño experimental es el siguiente: 15 ml de volumen de expulsión y 12 aspiraciones/min para generar humo de tabaco, humo de tabaco al 1,5 % diluido con aire comprimido. La concentración másica de materia particulada total (MPT) en el humo de tabaco al 1,5 % se determina mediante análisis gravimétrico de muestras de filtro obtenidas durante los periodos de exposición y era de 23,9 mg de MPT/m3.
En primer lugar, se entrena a los ratones para disponerse en una sujeción corporal durante 30 min/día sin fumar durante 5 días, seguido de la exposición a humo de tabaco al 1,5 % durante 15 min/día durante 5 días. Después de este periodo de introducción, los ratones continuan inhalando tabaco al 1, 5% durante 30 min/día, 5 días/semana y durante 8 semanas. Como control de la inhalación de humo de tabaco, los ratones son sometidos al mismo procedimiento experimental bajo las mismas condiciones indicadas anteriormente, aunque administrando aire en lugar de humo de tabaco.
Ejemplo 277: ensayo de inflamación pulmonar (Ejemplo de referencia)
Los compuestos proporcionados en la presente memoria pueden someterse a ensayo utilizando uno o ambos de entre el ensayo de inflamación pulmonar inducida por LPS y el ensayo de inflamación pulmonar inducido por ovoalbúmina.
Para llevar a cabo el ensayo de inflamación pulmonar inducida por LPS, los compuestos se administraron en dosis orales. Un grupo recibió vehículo únicamente y se utilizó dexametasona en otro grupo a modo de control positivo. Se determinó la inflamación pulmonar 6 h después de la instilación intranasal de LPS. Pueden evaluarse los parámetros siguientes: número total de leucocitos y número de neutrófilos en el lavado broncoalveolar (LBA).
Para llevar a cabo el ensayo de inflamación pulmonar inducida con ovoalbúmina, los compuestos se administran en dosis orales. Un grupo recibe dosis con vehículo únicamente y se utiliza dexametasona en otro grupo a modo de control positivo. Se determina la inflamación pulmonar 4 días después de instilaciones intranasales en 4 días consecutivos de ovoalbúmina. Los compuestos se administran mediante sonda esofágica 30 min antes de cada reto (4 retos) a las dosis indicadas. Pueden evaluarse los parámetros siguientes: número total de leucocitos y número de eosinófilos en el lavado broncoalveolar (LBA).
Ejemplo 278: eficacia del compuesto BB con o sin temozolomida en el modelo de glioma
El objetivo del presente modelo es evaluar el modelo de glioma GL-261 tratado continuamente con compuesto BB o con temoloamida (TMZ) seguido de vehículo o compuesto BB. Se administró la TMZ por vía IP 1X/día durante 7 días. A continuación, se asignó aleatoriamente el grupo a recibir compuesto BB a una dosis de 15 mg/kg QD por vía PO o vehículo, a fin de evaluar el retraso en el nuevo crecimiento tumoral.
Animales: ratones C57 albinos (macho) de Jackson Labs, n=80. Parámetros evaluados:
en vida: Los ratones recibieron dosis QD de compuesto BB. Se administró la temozolomida por vía IP QD durante 7 días. Se realizaron mediciones de tumores y pesos corporales 2X/semana, mediciones con calibrador y de Xenogen.
Análisis de criterios de valoración: Los tumores se cortaron por la mitad. Una mitad se cortó nuevamente en mitades y una mitad se fijó en NBF al 10 % y la otra mitad se congeló en OCT para secciones congeladas. La mitad restante se procesó para generar una suspensión de células individuales y se evaluó mediante FACS.
Grupo satélite PD - se sacrificaron 8 ratones de los ratones tratados con TMZ y 8 ratones del grupo tratado con vehículo el día 7 de tratamiento. Los tumores fueron procesados para generar una suspensión de células individuales y se evaluaron mediante FACS.
La vista general del estudio se muestra en la Tabla 26, a continuación:
Tabla 26.

Las FIGs. 53A, 53B, 53C, 53D y 53E muestran el efecto del compuesto BB tras el tratamiento con temozolomida de tumores GL-261. La FIG. 54A muestra el efecto del tratamiento con TMZ respecto a vehículo, y la FIG. 54B muestra los efectos del compuesto BB sobre el volumen tumoral después del tratamiento de TMZ. El compuesto BB resulta eficaz para reducir el volumen tumoral en tumores GL-261 tras el tratamiento de temozolomida.
El tratamiento con TMZ a una dosis de 1 mg/kg seguido del tratamiento con compuesto BB resultó en una reducción significativa del volumen tumoral en comparación con los ratones tratados con vehículo. El tratamiento de los ratones con compuesto BB desde el inicio del tratamiento también resultó en una reducción significativa del volumen tumoral en comparación con el tratamiento de vehículo de los ratones. Los dos grupos no eran significativamente diferentes, aunque el tratamiento y monitorización más largos podrían resultar en una ventana significativamente más grande.
Ejemplo 279: efecto del compuesto BB y anti-PDL-1 sobre la proliferación de las células T con tumores CT26
El objetivo del presente estudio es evaluar el efecto del compuesto BB sobre la proliferación de las células T en tumores CT26. CT26 es una línea celular de carcinoma de colon no diferenciado inducido por N-nitroso-N-metiluretano (NNMU). Se clonó para generar la línea celular denominada CT26.WT (ATCC n° CRL-2 6 3 8 ).
La duración del estudio fue de 1 semana de tratamiento. En ratones Balb/c hembra de 5-6 semanas de edad (Jackson Labs) se implantaron subcutáneamente 5x105 células CT26 en 100 pl de PBS. Las células CT26 se cultivaron en RPMI complementado con FBS al 10 %. La administración se inició una vez los tumores habían alcanzado un volumen medio de 500 a 600 mm3.
Los grupos se trataron de la manera siguiente:

En los grupos 1 y 2 (vehículo compuesto BB, respectivamente), los ratones recibieron dosis QD por vía PO durante 5 días a una dosis en volumen de 5 ml/kg. En el grupo 3 (aPD-L1), los ratones recibieron una dosis IP de 200 pg (200 pl de la solución madre 1 mg/ml).
Los ratones fueron monitorizados diariamente para efectos clínicos adversos. Se midieron los pesos corporales y
luminiscencia de los tumores dos veces a la semana. Al final del estudio, se recolectaron los tumores y se cortaron en 3 partes: un trozo para el ensayo de eliminación de células T citotóxicas (CTL), un trozo para el ensayo de proliferación de CSFE y un trozo (dividido en dos) para OCT/FFPE.
Los bazos de 3 ratones portadores de tumor tratados con vehículo se disociaron físicamente en medio para células T y se pasaron por un filtro de 70 micras. Después de la lisis de los glóbulos rojos, se agruparon las células, se resuspendieron en PBS y se tiñeron con CFSE (Invitrogen). Tras el lavado, dichas células se suspendieron en 20 ml de medio para células T. La mitad de las células recibió perlas Dynal con activador para ratones (entrecruzamiento de CD37CD28 a concentraciones 1:1). Las perlas con CD3/CD28 indujeron la proliferación en esplenocitos de ratones portadores de tumor (datos no mostrados).
Los tumores de ratones tratados con vehículo, los tratados con compuesto BB y los ratones tratados con anti-PDL1 se trituraron, se trataron con colagenasa y ADNasa durante 30 minutos a 37°C y se aislaron las células pasando el tumor digerido por un filtro de 70 micras. Las células se trataron con tampón de lisis de glóbulos rojos, se lavaron y se resuspendieron en 10 ml de medio para células T. Se sembraron 2 ml de cada suspensión de células tumorales en cada una de dos placas de 12 pocillos. A una de las placas se añadieron esplenocitos marcados con CSFE y activados con perlas Dynal y a la otra se añadieron esplenocitos marcados con CSFE y no activados. También se añadieron esplenocitos marcados con CSFE a pocillos que presentaban medio para células T sin células tumorales.
Después de cuatro días, se recolectaron las células de los pocillos y se tiñeron con anticuerpos para CD3, CD4 y CD8. El análisis se realizó mediante FACS, midiendo el porcentaje de células que se mantenía en un estado no proliferado (tinción CSFE sin diluir) después de 4 días. Las mediciones de la proliferación de los esplenocitos, activados o no activados, de los pocillos sin las células tumorales proporcionaron los controles positivo y negativo de la proliferación.
Las FIGS. 55A, 55B y 55C muestran el efecto del compuesto BB y anti-PDL-1 sobre la inhibición por células tumorales de la proliferación de las células T, en comparación con el tratamiento con vehículo. La FIG. 55A muestra el efecto de las células tumorales procedentes de ratones tratados con vehículo. La FIG. 55B muestra el efecto de las células tumorales procedentes de ratones tratados con compuesto BB. La FIG. 55C muestra el efecto de las células tumorales procedentes de ratones tratados con anti-PDL-1. Las FIGs. 56A y 56B muestran que las células tumorales procedentes de ratones tratados con compuesto BB y anti-PDL-1 son menos inhibitorias de la proliferación de las células T de bazo que las células tumorales procedentes de ratones tratados con vehículo.
La FIG. 57 muestra que el compuesto BB reduce el medio inmunosupresor mediante un ensayo ex vivo de proliferación de células T.
En resumen, la proliferación de las células T puede resultar suprimida por células derivadas de tumores de ratón CT26, pero el tratamiento con compuesto BB o anti-PDL1 puede eliminar la supresión.
Ejemplo 280: el compuesto BB inhibe la estimulación ex vivo por CXCL12 con sangre completa de fosfo-AKT en células T y monocitos
En ratones C57BL/6 Albino se implantaron 1x106 células en una inyección SC en el flanco posterior. El material de implantación celular se generó a partir de tumores brei que se habían pasado de ratón a ratón. El tratamiento se inició una vez los tumores habían llegado a 160 a 180 mm3. Los ratones recibieron dosis durante 12 días, QD, por vía PO de vehículo, 15, 7,5 o 3 mg/kg. El día 12, los ratones recibieron una administración y se recogió sangre terminal 2 h después de la dosis mediante varilla cardiaca. Se introdujo la sangre en tubos de recolección con EDTA K2 y se almacenaron a temperatura ambiente para el análisis FD.
La sangre de cada animal se dividió en alícuotas en 2 placas y se calentó a 37°C. Se añadió PBS a una placa como control no estimulado. Se añadió CXCL12/SDF-1 murino a la segunda placa a una concentración final de 800 ng/ml y la placa se devolvió al bloque de 37°C. Tras 2 minutos de incubación, se lisó la sangre y se fijó con paraformaldehído. Se lavaron las muestras y se congelaron hasta el análisis de citometría de flujo. Las células T se seleccionar utilizando un anticuerpo de CD3 y los monocitos, con LY6G'LY6Chigh. Se midió la pAKT S473 intracelular y se utilizó el porcentaje de positivas para la representación gráfica. La FIG. 58 muestra que los ratones portadores de LLC tratados con compuesto BB inhibe la estimulación de sangre completa ex vivo de fosfo-AKT en las células T. La FIG. 59 muestra que el tratamiento con compuesto BB de ratones portadores de LLC inhibe la estimulación de sangre completa ex vivo de fosfo-AKT en monocitos. La FIG. 60 muestra el efecto del compuesto BB sobre pAKt inducido por CXCL12 o por anti-IgD, en células B.
Ejemplo 281: FC de dosis únicas de compuesto BB en perros
Se midió la FC del compuesto BB en perros de raza Beagle macho a cuatro dosis diferentes: 2 mg/kg, 5 mg/kg, 10 mg/kg y 25 mg/kg. La FIG. 61 muestra perfiles de concentración libre en plasma de dosis únicas de compuesto BB frente al tiempo en perros de raza Beagle macho.
Ejemplo 282: ensayo de migración de macrófagos obtenidos de médula ósea (BMDM, por sus siglas en inglés)
Se obtuvieron las BMDM de médula ósea aislada a partir de fémures y tibias de ratones C57B1/6J. Se cultivaron las células de médula ósea en una placa Petri en presencia de RPMI con adición de medio acondicionado para células L al 30 % (sobrenadante del cultivo durante 5 días de células L929), FBS al 20 % y penicilina y estreptomicina. Tras aproximadamente 10 días, prácticamente todas las células se habían convertido en BMDM adheridas. Para el ensayo de migración, las BMDM fueron pretratadas con 6 concentraciones diferentes de un compuesto proporcionado en la presente memoria y se llevó a cabo el ensayo de migración mediante la utilización de una cámara de Boyden hacia la quimioquina C5a (50 nM) o CXCL12. Se contaron las BMDM migrantes, adheridas al policarbonato de la cámara Boyden, bajo el microscopio (5 campos/muestra) y se calculó el % de células migrantes mediante el establecimiento de la relación entre el número de células tratadas con el número tratado con el control de vehículo. Todos los valores se representaron gráficamente en una curva de respuesta a dosis y se definió la EC50 mediante la utilización de un análisis de regresión no lineal (programa GraphPad Prism). El compuesto BB puede someterse a ensayo para su efecto sobre la migración de las BMDM hacia C5a o CXCL12.
Ejemplo 283: combinación con temozolomida y terapia de radiación en la evaluación del crecimiento de tumores ortotópicos de células GL-261 en ratones C57BL/6
El objetivo del presente estudio es evaluar el efecto del compuesto proporcionado en la presente memoria en combinación con temozolomida y terapia de radiación en el crecimiento de tumores ortotópicos de células GL-261 en ratones C57BL/6.
Se anestesian sesenta (60) ratones C57BL/6 de 4 a 6 semanas de edad con xilazina (5 mg/kg) y ketamina (100 mg/kg) administrados mediante inyección intraperitoneal y la zona quirúrgica del cráneo se preparó con alcohol y Betadine. Se realiza una incisión diagonal desde detrás del ojo hasta la parte posterior de la cabeza. Se expone el bregma y las células se implantan 2 mm a la derecha y 1 mm anteriormente al bregma utilizando un pequeño taladro para generar el orificio, seguido de una jeringa Hamilton para administrar 1x105 células en un volumen total de 10 pl a una tasa de 1 pl por minuto. Se sutura la piel y los ratones se recuperan en una almohadilla térmica y después se devuelven a las jaulas. Todos los animales reciben medicación postoperatoria para el dolor cada 12 horas durante 72 horas.
Se captan imágenes IVIS de los animales 5 días después de la cirugía a fin de determinar el flujo total por animal para la aleatorización. Tras la aleatorización, los animales en el grupo 1 reciben dosis de vehículo (vía/régimen, dosis corporal total DCT) y los animales en el grupo 2 reciben dosis del compuesto proporcionado en la presente memoria, p.ej., compuesto BB a una dosis de 15 mg/kg. Los animales en el grupo 3 reciben dosis del compuesto proporcionado en la presente memoria, p.ej., el compuesto BB y 3 fracciones de radiación de 2 Gy cada uno centradas en la zona del tumor cerebral. Los animales en el grupo 4 reciben tratamiento de 25 mg/kg de temozolamida (i.p.) 5 veces a la semana en un programa los días 0, 1, 3, 5 y 6 y se administran tres (3) fracciones de 2 Gy de radiación enfocada en el tumor los días 0, 2 y 4 además del compuesto proporcionado en la presente memoria, p.ej., el compuesto BB a una dosis de 15 mg/kg. Los animales en los grupos 5 y 6 reciben tratamiento de 25 mg/kg de temozolamida (i.p.) 5 veces a la semana en un programa los días 0,1, 3, 5 y 6 y se administran tres (3) fracciones de 2 Gy de radiación enfocada en el tumor los días 0, 2 y 4.
Para la radiación enfocada, se coloca sobre el animal un protector de plomo con una ventana delgada recortada en la parte superior que expone únicamente la parte superior de la cabeza en la zona del tumor. Los días en que los ratones reciben radiación son anestesiados con xilazina (5 mg/kg) y ketamina (100 mgkg) administrados mediante inyección intraperitoneal. Los ratones se depositan sobre una placa de polimetilmetacrilato de 4 mm. Se genera la radiación con una fuente de rayos X de 160 kVp (15-ma) a una distancia focal de 25 cm, endurecida con un sistema de filtración de Cu de 0,35 mm a una tasa de 1 Gy/minuto. Los animales son pesados y monitorizados para su estado general de salud diariamente y se les asigna una puntuación de condición corporal. Los tumores se miden mediante imágenes IVIS dos veces a la semana desde el día 5 y se determina el flujo total para cada animal. Los datos experimentales se resumen en la tabla a continuación.

(continuación)

Se cultivan células GL-261 hasta subconfluencia en medio de cultivo DMEM que contenía FBS al 10 %. Las células se administran mediante inoculación intracraneal generando en primer lugar un orificio en el cráneo con un pequeño taladro, seguido de la inyección de las células 3 mm en el interior del cráneo con una jeringa Hamilton a razón de 1x105 células en un volumen de ~10 pl de medio sin suero a una tasa de 1 pl por minuto.
Los ratones son anestesiados con xilazina (5 mg/kg) y ketamina (100 mg/kg) administrados mediante inyección intraperitoneal. Los ratones se depositan sobre una placa de polimetilmetacrilato de 4 mm. Se coloca sobre el animal un protector de plomo con una ventana delgada recortada en la parte superior que expone únicamente la zona del cráneo que debía irradiarse. Los tumores se irradian directamente con 2 o 4 Gy de radiación los días 0, 2 y 4. Se genera la radiación con una fuente de rayos X de 160 kVp (15-ma) a una distancia focal de 25 cm, endurecida con un sistema de filtración de Cu de 0,35 mm a una tasa de 1,0 Gy/minuto.
Se evalúa la progresión en vida dos veces a la semana mediante imágenes IVIS. Los ratones reciben mediante inyección intraperitoneal 0,1 ml/20 g de peso corporal de sustrato bioluminiscente D-luciferin-K+ 15 mg/ml en PBS. Diez (10) minutos después de la inyección, los ratones son anestesiados con isoflurano e introducidos en el IVIS® Lumina a máxima sensibilidad durante 5 minutos de exposición como máximo para detectar la bioluminiscencia con un filtro de emisión abierto. Las imágenes guardadas se cargan en el software de análisis Living Image® y las escalas de color se hacen corresponder con las radiancias máxima y mínima (fotones/segundo/cm2/estreorradián). Se dibujan zonas de interés idénticas en torno de cada animal y se determina el flujo total en términos de fotones/segundo para cada zona de interés.
Además de medir los tumores en los animales vivos, se asigna una puntuación de condición corporal a cada animal 3 veces a la semana. Los animales con una condición corporal inferior a BC2 son sacrificados. Las condiciones se clasifican de la manera siguiente:
BC5: el animal es obeso, liso y voluminoso. No resulta posible identificar su estructura ósea bajo la carne y grasa.
Con frecuencia, los ratones en esta condición no pueden limpiarse bien y la capa de pelaje presenta una apariencia aceitosa y manchada.
BC4: el animal está sobrealimentado y las vértebras solo son palpables mediante presión firme.
BC3: el animal presenta una buena condición. Las vértebras y la pelvis son identificables mediante palpación.
BC2: el animal presenta una condición infraóptima. La segmentación de la columna vertebral es evidente y la pelvis es identificable mediante palpación.
BC1: el animal está demacrado; estructura esquelética muy prominente con poca cobertura de carne. Se percibe claramente la segmentación de las vértebras.
Todos los animales son pesados diariamente durante todo el presente estudio. El cambio de peso del grupo se expresa como porcentaje medio de cambio de peso. Las muertes de animales en este modelo generalmente se producen como consecuencia de la sobredosis de anestesia o la toxicidad de un fármaco. Los animales son monitorizados diariamente y los que muestran una pérdida de peso superior a 30 %, son incapaces de desplazarse, de procurarse alimento y agua y/o presentan una apariencia moribunda, son eutanizados. En el caso de que el tumor presente una apariencia ulcerada, el animal es eutanizado. Cualesquiera efectos adversos o muertes no previstas son comunicadas inmediatamente al veterinario.
Se determinan las diferencias estadísticas entre grupos de tratamiento utilizando técnicas estadísticas apropiadas. Se utiliza un ANOVA de un factor o ANOVA con rangos para evaluar la superficie bajo la curva para la ganancia de peso y la radiancia de los tumores.
Ejemplo 284: papel de las células T en la supresión del crecimiento tumoral
El objetivo es estudiar el papel de las células T en la supresión del crecimiento tumoral tras el tratamiento con un compuesto proporcionado en la presente memoria (p.ej., el compuesto BB).
Método A: células tumorales son implantadas en los ratones RAG-/-. Estos ratones presentan deficiencias en su capacidad de producir células T o B y no pueden conseguir memoria inmunológica. Se implantan células tumorales en ratones WT o RAG-/- en el fondo de cepa apropiado (para las células tumorales de colon CT26, BALB/C). Los animales portadores de tumor son aleatorizados a vehículo y esos tratados con un compuesto proporcionado en la presente memoria (p.ej., el compuesto BB), a grupos tanto WT como RAG-/-. Se evalúa la eficacia mediante la comparación de la cinética de crecimiento tumoral en WT en comparación con la misma en ratones RAG-/-.
Método B: El presente método utiliza anticuerpos contra los marcadores de células T CD4 y CD8 para agotar los subgrupos de células T. Se implantan células de CPL-Brei, de cáncer de colon CT26 o MC38 en la cepa huésped apropiada de ratón. Los ratones con tumores establecidos se aleatorizan en grupos de tratamiento tal como se indica a continuación.
Anticuerpos de agotamiento de CD4 y CD8
Control de isotipo aCD4/CD8: Isotipo de IgG2b de rata (clon BioXcell: LTF-2 Cat: BE0090-R005 Lote: TBFO) aCD4: diluido a 1 mg/ml en PBS sin endotoxinas (clon BioXcell: GK1.5 Cat: BE0003-1 Lote: TBFO) aCD8: diluido a 1 mg/ml en PBS sin endotoxinas (clon BioXcell: YTS 169.4 Cat: BE0117 Lote: TBFO) Régimen de dosis
Se administraron las terapias de aCD4/aCD8 i.p. cada 3 días (100 |jg cada vez)
Diseño del estudio
Vehículo; vehículo anti-CD4; vehículo anti-CD8; vehículo anti-CD4/CD8,
compuesto proporcionado en la presente memoria (p.ej., el compuesto BB),
compuesto proporcionado en la presente memoria (p.ej., el compuesto BB) anti-CD4,
compuesto proporcionado en la presente memoria (p.ej., el compuesto BB) anti-CD8,
compuesto proporcionado en la presente memoria (p.ej., el compuesto BB) anti-CD4/8.
Ejemplo 285: cáncer de hueso osteolítico en el modelo de cáncer óseo
El objetivo es evaluar el efecto del compuesto proporcionado en la presente memoria (p.ej., el compuesto BB) sobre el desarrollo de los osteoclastos y la posibilidad de evitar el daño óseo en el modelo NCTC-2472. Se propagó la línea celular tumoral NCTC-2472 en cultivo celular y se recolectó mientras se encontraba en crecimiento logarítmico. Se implantaron 100.000 células directamente en la cavidad medular del fémur distal el día de estudio 0.
El día de estudio 0, los ratones fueron pesados y aleatorizados según peso corporal en grupos de tratamiento. Los ratones fueron anestesiados con isoflurano y se llevó a cabo una artrotomía en la rodilla derecha. Se inyectaron las células en 20 j l con una aguja de calibre 29. El orificio de entrada en el hueso se llenó con cera ósea para evitar la extravasación de células tumorales.
Entre los días 2 y 22 se llevó a cabo el tratamiento con fármaco en los grupos siguientes: vehículo, compuesto BB (15 mg/kg) y resendronato como control. Se midió el peso dos veces a la semana. El día de estudio 22 los ratones fueron sangrados bajo isoflurano y se extrajeron muestras de fémur para el análisis de rayos X e histopatológico. Entre los criterios de valoración relevantes se incluyen la erosión ósea y la formación anormal del hueso.
Claims (1)
- REIVINDICACIONESCompuesto de fórmula (I'') o de fórmula (A''), o una forma farmacéuticamente aceptable del mismo, para la utilización en combinación con una terapia de punto de control inmunitario, en un método de tratamiento o prevención de un cáncer en un sujeto:
 R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,z es 0, 1, 2 o 3,cada aparición de R3a es, independientemente, hidrógeno, alquilo, alquenilo, alquinilo, alcoxilo, halógeno, ciano, amino, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6)3,en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,R1c es hidrógeno, alquilo, alquenilo o alquinilo,R2c es hidrógeno, alquilo, alquenilo o alquinilo,Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, yX es CR1a o N,en la que R1a es hidrógeno, halo, alquilo, alquenilo, alquinilo o CN,en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), S(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo)2, S(O)(alquilo), o S(O)2(alquilo),en la que, en la fórmula (I''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:
R1 es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, - COR2, -COOR3, o -CONR4R5,z es 0, 1, 2 o 3,cada aparición de R3a es, independientemente, hidrógeno, alquilo, alquenilo, alquinilo, alcoxilo, halógeno, ciano, amino, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,B es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, -COR2, -COOR3, -CONR4R5, o -Si(R6)3,en los que R2, R3, R4, R5 y R6 son, cada uno independientemente, hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo,R1c es hidrógeno, alquilo, alquenilo o alquinilo,R2c es hidrógeno, alquilo, alquenilo o alquinilo,Wd es heteroarilo, cicloalquilo, heterocicloalquilo o arilo, yX es CR1a o N,en la que R1a es hidrógeno, halo, alquilo, alquenilo, alquinilo o CN,en la que cada alquilo, alquenilo o alquinilo se sustituye opcionalmente con uno o más halo, OH, alcoxi, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2 , CONH(alquilo), CON(alquilo)2, S(O)(alquilo), S(O)2(alquilo), cicloalquilo, heterocicloalquilo, arilo o heteroarilo,en la que cada cicloalquilo, heterocicloalquilo, arilo o heteroarilo se sustituye opcionalmente con uno o más halo, alquilo, alquenilo, alquinilo, OH, alcoxi, oxo, NH2, NH(alquilo), N(alquilo)2, COH, CO(alquilo), COOH, COO(alquilo), CONH2, CONH(alquilo), CON(alquilo)2, S(O)(alquilo), o S(O)2(alquilo),en la que, en la fórmula (I''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es: entonces R1 no es hidrógeno, Si(CH3)3, CH2Si(CH3)3, metilo, (CH2)NH2 , (CH2)2NH2 , (CH2)NHSO2CH3, o (CH2)nNHC(O)R1x; n es 1 o 2; R1x es metilo, alqueno C2, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en los que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituyen opcionalmente con uno o dos grupos seleccionados independientemente de entre oxo y ciano, en la que, en la fórmula (A''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es:
entonces R1 no es hidrógeno, Si(CH3)3, CH2Si(CH3)3, metilo, (CH2)NH2 , (CH2)2NH2 , (CH2)NHSO2CH3, o (CH2)nNHC(O)R1x; n es 1 o 2; R1x es metilo, alqueno C2, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo, en los que el alqueno, ciclohexilo, ciclopentilo, tetrahidrofuranilo, furanilo o pirrolidinilo se sustituyen opcionalmente con uno o dos grupos seleccionados independientemente de entre oxo y ciano, en la que, en la fórmula (A''), en el caso de que X sea CH, B es fenilo no sustituido, y Wd es: entonces R1 no es fenilo.Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 1, en el que el compuesto es Compuesto 4 ("compuesto BB") de fórmula:
entonces R1 no es fenilo.Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 1, en el que el compuesto es Compuesto 4 ("compuesto BB") de fórmula: Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 1 o 2, en el que el cáncer es un tumor sólido, particularmente carcinoma de células renales, cáncer de vejiga, cáncer de colon, glioblastoma, cáncer de mama (más particularmente, cáncer de mama triple negativo), cáncer de cabeza y cuello (más particularmente, carcinoma de células escamosas de cabeza y cuello), cáncer de pulmón (más particularmente, cáncer pulmonar no microcítico), melanoma, cáncer de glándulas adrenales, mesotelioma (más particularmente, mesotelioma epitelioide, mesotelioma sarcomatoide o mesotelioma bifásico), cáncer colorrectal, cáncer ovárico o cáncer de endometrio,o se selecciona de uno o más de entre un cáncer del sistema pulmonar, un cáncer cerebral, un cáncer del tracto gastrointestinal, un cáncer de piel, un cáncer genitourinario, cáncer de cabeza y cuello, un sarcoma, un carcinoma o un cáncer neuroendocrino,o se selecciona de uno o más de entre un cáncer pancreático, un cáncer de pulmón, un cáncer de mama, un cáncer de próstata, un cáncer testicular, un cáncer esofágico, un cáncer hepático, un cáncer gástrico, un cáncer de colon, un cáncer colorrectal, un cáncer ovárico, un cáncer cervical, un cáncer uterino, un cáncer de endometrio, un cáncer de vejiga, un cáncer renal, un cáncer inducido por virus o un carcinoma urotelial, o se selecciona de uno o más de entre un meduloblastoma, un carcinoma de células basales, un glioma, un cáncer hepatocelular, un tumor estromal gastrointestinal (TEGI), un melanoma, un tumor neuroectodérmico, un sarcoma de tejidos blandos, un fibrosarcoma, un mixosarcoma, un liposarcoma, un condrosarcoma, un sarcoma osteogénico, un cordoma, un angiosarcoma, un endoteliosarcoma, un linfangiosarcoma, un linfangioendoteliosarcoma, un sinovioma, un mesotelioma, unleiomiosarcoma, un carcinoma de vejiga, un carcinoma epitelial, un carcinoma de células escamosas, un adenocarcinoma, un carcinoma broncogénico, un carcinoma de células renales, un hepatoma, un carcinoma del conducto biliar, un tumor carcinoide, tumor de células gigantes de tipo difuso o glioblastoma,o es un cáncer hematológico, especialmente leucemia o linfoma, o leucemia linfocítica aguda (LLA), leucemia linfocítica crónica (LLC), leucemia prolinfocítica (LPL), leucemia de células pilosas (LCP), macroglobulinemia de Waldenstrom (MW), linfoma de células T periféricas (LCTP), leucemia/linfoma de células T adultas (LLTA), linfoma de células T cutáneo (LCTC), leucemia de linfocitos granulares grandes (LLGG), leucemia mielocítica aguda (LMA), linfoma de Hodgkin (Lh ), linfoma no de Hodgkin (LNH), linfoma folicular, linfoma de células B grandes difusas (LCBGD), linfoma de células del manto (lCm ), mastocitosis, mieloma múltiple (MM), síndrome mielodisplásico (SMD) o trastorno mieloproliferativo (TMP),0 es metastásico.Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 3, en el que la terapia de punto de control inmunitario es un agonista de NKG2C, un inhibidor de CTLA-4, un inhibidor de PD-1, un inhibidor de PDL-1, un inhibidor de LAG3, un inhibidor de TIM-3, un inhibidor de BTLA, un inhibidor de VISTA, un inhibidor de TIGIT, un inhibidor de IDO, un agonista de ICOS, un agonista de CD137, un agonista de OX40, un agonista de GITR, un agonista de CD27, un agonista de HVEM, un ligando de TLR, un anticuerpo de B7-H3, un inhibidor de PDL-1 o un anticuerpo anti-PDL-1, preferentemente YW243.55.S70, MDPL3280A, MSB0010718C, MDX-1105 o MEDI-4736, un inhibidor de PD-1 o un anticuerpo anti-PD-1, preferentemente nivolumab, pembrolizumab, pidilizumab, AMP-224 o AMP-514, o un inhibidor de CTLA-4 o un anticuerpo anti-CTLA4, preferentemente tremelimumab o ipilimumab.Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 3, en el que la terapia de punto de control inmunitario es:una combinación de un inhibidor de PD-1 o un anticuerpo anti-PD-1 con un inhibidor de CTLA-4 o un anticuerpo anti-CTLA-4.Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 3, en el que la terapia de punto de control inmunitario es:una combinación de un inhibidor de p D-1 o un anticuerpo anti-PD-1, un inhibidor de PDL-1 o un anticuerpo anti-PDL-1 y un inhibidor de CTLA-4, o un anticuerpo anti-CTLA-4.7. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 3, en el que la terapia de punto de control inmunitario es:una combinación de un anticuerpo o fragmento del mismo, un ácido nucleico inhibidor, un ligando soluble o una fusión de PD-1 con una región Fc de una inmunoglobulina, un anticuerpo o fragmento del mismo, un ácido nucleico inhibidor, un ligando soluble o una fusión de PD-L1 con una región Fc de una inmunoglobulina, y un anticuerpo o fragmento del mismo, un ácido nucleico inhibidor, un ligando soluble o una fusión de un ligando de CTLA-4 con una región Fc de una inmunoglobulina.8. Compuesto de fórmula:
Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 1 o 2, en el que el cáncer es un tumor sólido, particularmente carcinoma de células renales, cáncer de vejiga, cáncer de colon, glioblastoma, cáncer de mama (más particularmente, cáncer de mama triple negativo), cáncer de cabeza y cuello (más particularmente, carcinoma de células escamosas de cabeza y cuello), cáncer de pulmón (más particularmente, cáncer pulmonar no microcítico), melanoma, cáncer de glándulas adrenales, mesotelioma (más particularmente, mesotelioma epitelioide, mesotelioma sarcomatoide o mesotelioma bifásico), cáncer colorrectal, cáncer ovárico o cáncer de endometrio,o se selecciona de uno o más de entre un cáncer del sistema pulmonar, un cáncer cerebral, un cáncer del tracto gastrointestinal, un cáncer de piel, un cáncer genitourinario, cáncer de cabeza y cuello, un sarcoma, un carcinoma o un cáncer neuroendocrino,o se selecciona de uno o más de entre un cáncer pancreático, un cáncer de pulmón, un cáncer de mama, un cáncer de próstata, un cáncer testicular, un cáncer esofágico, un cáncer hepático, un cáncer gástrico, un cáncer de colon, un cáncer colorrectal, un cáncer ovárico, un cáncer cervical, un cáncer uterino, un cáncer de endometrio, un cáncer de vejiga, un cáncer renal, un cáncer inducido por virus o un carcinoma urotelial, o se selecciona de uno o más de entre un meduloblastoma, un carcinoma de células basales, un glioma, un cáncer hepatocelular, un tumor estromal gastrointestinal (TEGI), un melanoma, un tumor neuroectodérmico, un sarcoma de tejidos blandos, un fibrosarcoma, un mixosarcoma, un liposarcoma, un condrosarcoma, un sarcoma osteogénico, un cordoma, un angiosarcoma, un endoteliosarcoma, un linfangiosarcoma, un linfangioendoteliosarcoma, un sinovioma, un mesotelioma, unleiomiosarcoma, un carcinoma de vejiga, un carcinoma epitelial, un carcinoma de células escamosas, un adenocarcinoma, un carcinoma broncogénico, un carcinoma de células renales, un hepatoma, un carcinoma del conducto biliar, un tumor carcinoide, tumor de células gigantes de tipo difuso o glioblastoma,o es un cáncer hematológico, especialmente leucemia o linfoma, o leucemia linfocítica aguda (LLA), leucemia linfocítica crónica (LLC), leucemia prolinfocítica (LPL), leucemia de células pilosas (LCP), macroglobulinemia de Waldenstrom (MW), linfoma de células T periféricas (LCTP), leucemia/linfoma de células T adultas (LLTA), linfoma de células T cutáneo (LCTC), leucemia de linfocitos granulares grandes (LLGG), leucemia mielocítica aguda (LMA), linfoma de Hodgkin (Lh ), linfoma no de Hodgkin (LNH), linfoma folicular, linfoma de células B grandes difusas (LCBGD), linfoma de células del manto (lCm ), mastocitosis, mieloma múltiple (MM), síndrome mielodisplásico (SMD) o trastorno mieloproliferativo (TMP),0 es metastásico.Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 3, en el que la terapia de punto de control inmunitario es un agonista de NKG2C, un inhibidor de CTLA-4, un inhibidor de PD-1, un inhibidor de PDL-1, un inhibidor de LAG3, un inhibidor de TIM-3, un inhibidor de BTLA, un inhibidor de VISTA, un inhibidor de TIGIT, un inhibidor de IDO, un agonista de ICOS, un agonista de CD137, un agonista de OX40, un agonista de GITR, un agonista de CD27, un agonista de HVEM, un ligando de TLR, un anticuerpo de B7-H3, un inhibidor de PDL-1 o un anticuerpo anti-PDL-1, preferentemente YW243.55.S70, MDPL3280A, MSB0010718C, MDX-1105 o MEDI-4736, un inhibidor de PD-1 o un anticuerpo anti-PD-1, preferentemente nivolumab, pembrolizumab, pidilizumab, AMP-224 o AMP-514, o un inhibidor de CTLA-4 o un anticuerpo anti-CTLA4, preferentemente tremelimumab o ipilimumab.Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 3, en el que la terapia de punto de control inmunitario es:una combinación de un inhibidor de PD-1 o un anticuerpo anti-PD-1 con un inhibidor de CTLA-4 o un anticuerpo anti-CTLA-4.Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 3, en el que la terapia de punto de control inmunitario es:una combinación de un inhibidor de p D-1 o un anticuerpo anti-PD-1, un inhibidor de PDL-1 o un anticuerpo anti-PDL-1 y un inhibidor de CTLA-4, o un anticuerpo anti-CTLA-4.7. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 3, en el que la terapia de punto de control inmunitario es:una combinación de un anticuerpo o fragmento del mismo, un ácido nucleico inhibidor, un ligando soluble o una fusión de PD-1 con una región Fc de una inmunoglobulina, un anticuerpo o fragmento del mismo, un ácido nucleico inhibidor, un ligando soluble o una fusión de PD-L1 con una región Fc de una inmunoglobulina, y un anticuerpo o fragmento del mismo, un ácido nucleico inhibidor, un ligando soluble o una fusión de un ligando de CTLA-4 con una región Fc de una inmunoglobulina.8. Compuesto de fórmula: 0 una sal farmacéuticamente aceptable del mismo, para la utilización en combinación con un inhibidor de PD-1 o un anticuerpo anti-PD-1 en un método de tratamiento o prevención de un tumor sólido en un sujeto. 9. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8, en el que el tumor se selecciona de uno o más de: un cáncer del sistema pulmonar, un cáncer cerebral, un cáncer del tracto gastrointestinal, un cáncer de piel, un cáncer genitourinario, un cáncer de cabeza y cuello, un sarcoma, un carcinoma o un cáncer neuroendocrino,uno o más de entre cáncer pancreático, cáncer de pulmón, cáncer de mama, cáncer de próstata, cáncer testicular, cáncer esofágico, cáncer hepático, cáncer gástrico, cáncer de colon, cáncer colorrectal, cáncer ovárico, cáncer cervical, cáncer uterino, cáncer de endometrio, cáncer de vejiga, cáncer renal, cáncer inducido por virus, cáncer de glándulas adrenales y mesotelioma, ouno o más de entre meduloblastoma, carcinoma de células basales, glioma, cáncer hepatocelular, tumor estromal gastrointestinal (TEGI), melanoma, tumor neuroectodérmico, sarcoma de tejidos blandos, fibrosarcoma, mixosarcoma, liposarcoma, condrosarcoma, sarcoma osteogénico, cordoma, angiosarcoma, endoteliosarcoma, linfangiosarcoma, linfangioendoteliosarcoma, sinovioma, mesotelioma, leiomiosarcoma, carcinoma de vejiga, carcinoma epitelial, carcinoma de células escamosas, adenocarcinoma, carcinoma broncogénico, carcinoma de células renales, hepatoma, carcinoma del conducto biliar, tumor carcinoide, tumor de células gigantes de tipo difuso y glioblastoma,o el tumor sólido es cáncer pulmonar, particularmente cáncer pulmonar no microcítico, melanoma, cáncer de colon o cáncer de cabeza y cuello.10. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8 o 9, en el que el inhibidor de PD-1 o el anticuerpo anti-PD-1 es nivolumab.11. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8 o 9, en el que el inhibidor de PD-1 o el anticuerpo anti-PD-1 es pembrolizumab.12. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 10, en el que el tumor sólido es cáncer de mama triple negativo.13. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 10, en el que el tumor sólido es cáncer pulmonar no microcítico.14. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 10, en el que el tumor sólido es melanoma.15. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 10, en el que el tumor sólido es carcinoma de células renales.16. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 10, en el que el tumor sólido es cáncer de vejiga.17. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8, en el que el tumor sólido es mesotelioma.18. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8, en el que el tumor sólido es carcinoma urotelial.19. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8, en el que el tumor sólido es carcinoma de células escamosas de cabeza y cuello.20. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8, en el que el tumor sólido es cáncer de la glándula adrenal.21. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 20, en el que el compuesto o forma farmacéuticamente aceptable se administra a una dosis de aproximadamente 1 a 100 mg al día, preferentemente de aproximadamente 1 a 50 mg al día, por ejemplo de 5 a 40 mg al día, o por ejemplo, de 10 a 30 mg al día.22. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 20, en el que el compuesto o forma farmacéuticamente aceptable se administra a una dosis con la que el nivel del compuesto en el sujeto, preferentemente medido en el plasma o tejidos del sujeto, es superior a la IC5o del compuesto de inhibición de PI3K-gamma durante por lo menos 70 %, 80 %, 90 %, 95 %, 97 %, 98 % o 99 % de un periodo de tiempo seleccionado, p.ej., 6 horas, 12 horas, 24 horas o 48 horas inmediatamente después de la administración, y preferentemente es superior a la IC90 del compuesto de inhibición de PI3K-gamma durante por lo menos 50 %, 60 %, 70 %, 80 %, 90 %, 95 %, 97 %, 98 % o 99 % del periodo de tiempo seleccionado, y más preferentemente, no aumenta por encima de la IC20 o IC50 del compuesto de inhibición de PI3K-delta dentro del periodo de tiempo seleccionado.23. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 20, en el que el compuesto se administra a una dosis con la que proporciona una inhibición de por lo menos 50 % de PI3K-gamma en el sujeto, aunque una inhibición inferior a 10 % o 20 % de PI3K-delta en el sujeto.24. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 23, en el que el sujeto es un ser humano y el compuesto presenta una semivida de entre aproximadamente 10 y 13 horas en el sujeto.25. Compuesto de fórmula:
0 una sal farmacéuticamente aceptable del mismo, para la utilización en combinación con un inhibidor de PD-1 o un anticuerpo anti-PD-1 en un método de tratamiento o prevención de un tumor sólido en un sujeto. 9. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8, en el que el tumor se selecciona de uno o más de: un cáncer del sistema pulmonar, un cáncer cerebral, un cáncer del tracto gastrointestinal, un cáncer de piel, un cáncer genitourinario, un cáncer de cabeza y cuello, un sarcoma, un carcinoma o un cáncer neuroendocrino,uno o más de entre cáncer pancreático, cáncer de pulmón, cáncer de mama, cáncer de próstata, cáncer testicular, cáncer esofágico, cáncer hepático, cáncer gástrico, cáncer de colon, cáncer colorrectal, cáncer ovárico, cáncer cervical, cáncer uterino, cáncer de endometrio, cáncer de vejiga, cáncer renal, cáncer inducido por virus, cáncer de glándulas adrenales y mesotelioma, ouno o más de entre meduloblastoma, carcinoma de células basales, glioma, cáncer hepatocelular, tumor estromal gastrointestinal (TEGI), melanoma, tumor neuroectodérmico, sarcoma de tejidos blandos, fibrosarcoma, mixosarcoma, liposarcoma, condrosarcoma, sarcoma osteogénico, cordoma, angiosarcoma, endoteliosarcoma, linfangiosarcoma, linfangioendoteliosarcoma, sinovioma, mesotelioma, leiomiosarcoma, carcinoma de vejiga, carcinoma epitelial, carcinoma de células escamosas, adenocarcinoma, carcinoma broncogénico, carcinoma de células renales, hepatoma, carcinoma del conducto biliar, tumor carcinoide, tumor de células gigantes de tipo difuso y glioblastoma,o el tumor sólido es cáncer pulmonar, particularmente cáncer pulmonar no microcítico, melanoma, cáncer de colon o cáncer de cabeza y cuello.10. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8 o 9, en el que el inhibidor de PD-1 o el anticuerpo anti-PD-1 es nivolumab.11. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8 o 9, en el que el inhibidor de PD-1 o el anticuerpo anti-PD-1 es pembrolizumab.12. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 10, en el que el tumor sólido es cáncer de mama triple negativo.13. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 10, en el que el tumor sólido es cáncer pulmonar no microcítico.14. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 10, en el que el tumor sólido es melanoma.15. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 10, en el que el tumor sólido es carcinoma de células renales.16. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 10, en el que el tumor sólido es cáncer de vejiga.17. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8, en el que el tumor sólido es mesotelioma.18. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8, en el que el tumor sólido es carcinoma urotelial.19. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8, en el que el tumor sólido es carcinoma de células escamosas de cabeza y cuello.20. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según la reivindicación 8, en el que el tumor sólido es cáncer de la glándula adrenal.21. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 20, en el que el compuesto o forma farmacéuticamente aceptable se administra a una dosis de aproximadamente 1 a 100 mg al día, preferentemente de aproximadamente 1 a 50 mg al día, por ejemplo de 5 a 40 mg al día, o por ejemplo, de 10 a 30 mg al día.22. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 20, en el que el compuesto o forma farmacéuticamente aceptable se administra a una dosis con la que el nivel del compuesto en el sujeto, preferentemente medido en el plasma o tejidos del sujeto, es superior a la IC5o del compuesto de inhibición de PI3K-gamma durante por lo menos 70 %, 80 %, 90 %, 95 %, 97 %, 98 % o 99 % de un periodo de tiempo seleccionado, p.ej., 6 horas, 12 horas, 24 horas o 48 horas inmediatamente después de la administración, y preferentemente es superior a la IC90 del compuesto de inhibición de PI3K-gamma durante por lo menos 50 %, 60 %, 70 %, 80 %, 90 %, 95 %, 97 %, 98 % o 99 % del periodo de tiempo seleccionado, y más preferentemente, no aumenta por encima de la IC20 o IC50 del compuesto de inhibición de PI3K-delta dentro del periodo de tiempo seleccionado.23. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 20, en el que el compuesto se administra a una dosis con la que proporciona una inhibición de por lo menos 50 % de PI3K-gamma en el sujeto, aunque una inhibición inferior a 10 % o 20 % de PI3K-delta en el sujeto.24. Compuesto o forma farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 1 a 23, en el que el sujeto es un ser humano y el compuesto presenta una semivida de entre aproximadamente 10 y 13 horas en el sujeto.25. Compuesto de fórmula: 0 una sal farmacéuticamente aceptable del mismo, para la utilización en combinación con un inhibidor de PD-1 o un anticuerpo anti-PD-1 en un método de tratamiento o prevención de un carcinoma pulmonar no microcítico, tumor mesotelial (particularmente mesotelioma epitelioide, mesotelioma sarcomatoide o mesotelioma bifásico) o melanoma en un sujeto, en el que el compuesto o sal farmacéuticamente aceptables del mismo se administra a una dosis de 10 mg, 15 mg, 20 mg, 25 mg, 30 mg, 40 mg, 45 mg, 50 mg o 60 mg al día en el sujeto.26. Compuesto de fórmula:
0 una sal farmacéuticamente aceptable del mismo, para la utilización en combinación con un inhibidor de PD-1 o un anticuerpo anti-PD-1 en un método de tratamiento o prevención de un carcinoma pulmonar no microcítico, tumor mesotelial (particularmente mesotelioma epitelioide, mesotelioma sarcomatoide o mesotelioma bifásico) o melanoma en un sujeto, en el que el compuesto o sal farmacéuticamente aceptables del mismo se administra a una dosis de 10 mg, 15 mg, 20 mg, 25 mg, 30 mg, 40 mg, 45 mg, 50 mg o 60 mg al día en el sujeto.26. Compuesto de fórmula: o una sal farmacéuticamente aceptable del mismo, para la utilización en un método de tratamiento o prevención de un cáncer en un sujeto, en el que el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia que es un tratamiento de punto de control.27. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es melanoma, y el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia que es un tratamiento de punto de control.28. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es cáncer de cabeza y cuello y el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia que es un tratamiento de punto de control.29. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es cáncer pulmonar y el sujeto ha sido pretratado o tratado previamente con un tratamiento de inmunoterapia que es un tratamiento de punto de control.30. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es cáncer pulmonar no microcítico y el sujeto ha sido pretratado o tratado previamente con un tratamiento de inmunoterapia que es un tratamiento de punto de control.31. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es carcinoma de células renales y el sujeto ha sido pretratado o tratado previamente con un tratamiento de inmunoterapia que es un tratamiento de punto de control.32. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es cáncer de vejiga y el sujeto ha sido pretratado o tratado previamente con un tratamiento de inmunoterapia que es un tratamiento de punto de control.33. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 26 a 32, en el que el tratamiento de punto de control es un inhibidor de PD-1 o un anticuerpo anti-PD-1.34. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 33, en el que el tratamiento de punto de control es nivolumab.
o una sal farmacéuticamente aceptable del mismo, para la utilización en un método de tratamiento o prevención de un cáncer en un sujeto, en el que el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia que es un tratamiento de punto de control.27. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es melanoma, y el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia que es un tratamiento de punto de control.28. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es cáncer de cabeza y cuello y el sujeto ha sido pretratado o tratado previamente con uno o más tratamientos de inmunoterapia que es un tratamiento de punto de control.29. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es cáncer pulmonar y el sujeto ha sido pretratado o tratado previamente con un tratamiento de inmunoterapia que es un tratamiento de punto de control.30. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es cáncer pulmonar no microcítico y el sujeto ha sido pretratado o tratado previamente con un tratamiento de inmunoterapia que es un tratamiento de punto de control.31. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es carcinoma de células renales y el sujeto ha sido pretratado o tratado previamente con un tratamiento de inmunoterapia que es un tratamiento de punto de control.32. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 26, en el que el cáncer es cáncer de vejiga y el sujeto ha sido pretratado o tratado previamente con un tratamiento de inmunoterapia que es un tratamiento de punto de control.33. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según cualquiera de las reivindicaciones 26 a 32, en el que el tratamiento de punto de control es un inhibidor de PD-1 o un anticuerpo anti-PD-1.34. Compuesto o sal farmacéuticamente aceptable del mismo para la utilización según la reivindicación 33, en el que el tratamiento de punto de control es nivolumab.
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461955717P | 2014-03-19 | 2014-03-19 | |
| US201461980484P | 2014-04-16 | 2014-04-16 | |
| US201462000923P | 2014-05-20 | 2014-05-20 | |
| US201462033008P | 2014-08-04 | 2014-08-04 | |
| US201462059766P | 2014-10-03 | 2014-10-03 | |
| US201462075173P | 2014-11-04 | 2014-11-04 | |
| US201562101980P | 2015-01-09 | 2015-01-09 | |
| PCT/US2015/021200 WO2015143012A1 (en) | 2014-03-19 | 2015-03-18 | Heterocyclic compounds for use in the treatment of pi3k-gamma mediated disorders |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2913486T3 true ES2913486T3 (es) | 2022-06-02 |
Family
ID=52780065
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15713299T Active ES2913486T3 (es) | 2014-03-19 | 2015-03-18 | Compuestos heterocíclicos para la utilización en el tratamiento de trastornos mediados por PI3K-gamma |
Country Status (16)
| Country | Link |
|---|---|
| US (4) | US9775844B2 (es) |
| EP (2) | EP3119397B1 (es) |
| JP (1) | JP6701088B2 (es) |
| CN (3) | CN113616656B (es) |
| AU (1) | AU2015231413B2 (es) |
| CA (1) | CA2943075C (es) |
| DK (1) | DK3119397T3 (es) |
| EA (1) | EA201691872A1 (es) |
| ES (1) | ES2913486T3 (es) |
| IL (1) | IL247841B (es) |
| MX (1) | MX382033B (es) |
| NZ (1) | NZ724368A (es) |
| PH (1) | PH12016501809A1 (es) |
| PT (1) | PT3119397T (es) |
| SG (2) | SG10201808053XA (es) |
| WO (1) | WO2015143012A1 (es) |
Families Citing this family (85)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2624554T3 (es) | 2005-12-28 | 2017-07-14 | Vertex Pharmaceuticals Incorporated | Formas sólidas de n- [2,4 - bis (1,1 - dimetiletil) - 5 - hidroxifenil] - 1,4 - dihidro - 4 - oxoquinolina - 3 - carboxamida |
| AU2009204487C1 (en) | 2008-01-04 | 2014-10-16 | Intellikine, Llc | Certain chemical entities, compositions and methods |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| CN103648499B (zh) | 2011-01-10 | 2017-02-15 | 无限药品股份有限公司 | 用于制备异喹啉酮的方法及异喹啉酮的固体形式 |
| EP2751093A1 (en) | 2011-08-29 | 2014-07-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| IL308613A (en) | 2012-10-15 | 2024-01-01 | Epizyme Inc | Cancer treatment methods |
| US10860683B2 (en) | 2012-10-25 | 2020-12-08 | The Research Foundation For The State University Of New York | Pattern change discovery between high dimensional data sets |
| TWI654206B (zh) | 2013-03-16 | 2019-03-21 | 諾華公司 | 使用人類化抗-cd19嵌合抗原受體治療癌症 |
| TWI657085B (zh) * | 2013-10-04 | 2019-04-21 | 英菲尼提製藥股份有限公司 | 雜環化合物及其用途 |
| WO2015051241A1 (en) | 2013-10-04 | 2015-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9775844B2 (en) | 2014-03-19 | 2017-10-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| CN111514283B (zh) * | 2014-04-07 | 2025-10-14 | 诺华股份有限公司 | 使用抗cd19嵌合抗原受体治疗癌症 |
| BR112017000132A2 (en) | 2014-07-04 | 2018-01-09 | Lupin Limited | compound, pharmaceutical composition and method of treating or preventing a disease responsive to inhibition of pi3k activity |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| CA2963945C (en) | 2014-10-07 | 2023-01-10 | Vertex Pharmaceuticals Incorporated | Co-crystals of modulators of cystic fibrosis transmembrane conductance regulator |
| GB201420859D0 (en) * | 2014-11-24 | 2015-01-07 | Cancer Res Inst Royal | Tumour analysis |
| LT3280729T (lt) | 2015-04-08 | 2022-08-10 | Novartis Ag | Terapijos cd20, terapijos cd22 ir kombinuotos terapijos su cd19 chimerinį antigeno receptorių (car) ekspresuojančia ląstele |
| AU2016257306B2 (en) | 2015-05-06 | 2019-06-27 | Snipr Technologies Limited | Altering microbial populations and modifying microbiota |
| KR20180042356A (ko) | 2015-08-24 | 2018-04-25 | 에피자임, 인코포레이티드 | 암 치료 방법 |
| CN114230571B (zh) | 2015-09-14 | 2025-07-08 | 无限药品股份有限公司 | 异喹啉酮的固体形式、其制备方法、包含其的组合物及其使用方法 |
| EA201890801A1 (ru) * | 2015-09-25 | 2018-09-28 | Эпизайм, Инк. | Способ лечения злокачественной рабдоидной опухоли яичников (mrto)/мелкоклеточного рака яичников гиперкальцемического типа (sccoht) с помощью ингибитора ezh2 |
| EA201891225A1 (ru) * | 2015-11-23 | 2018-11-30 | Грейс Терапьютикс Ллк | Пленкообразующий спрей для местного применения |
| WO2017106328A1 (en) | 2015-12-14 | 2017-06-22 | X4 Pharmaceuticals, Inc. | Methods for treating cancer |
| EP3389652B1 (en) | 2015-12-14 | 2022-09-28 | X4 Pharmaceuticals, Inc. | Methods for treating cancer |
| WO2017106312A1 (en) * | 2015-12-18 | 2017-06-22 | Drexel University | Methods of modulating levels of il-6 and pd-l1 |
| CN108883091A (zh) | 2015-12-22 | 2018-11-23 | X4 制药有限公司 | 用于治疗免疫缺陷病的方法 |
| US10898483B2 (en) | 2015-12-23 | 2021-01-26 | Moonshot Pharma Llc | Methods for inducing an immune response by promoting premature termination codon read-through |
| CN108883132A (zh) * | 2016-01-22 | 2018-11-23 | X4 制药有限公司 | 用于治疗癌症的方法 |
| US11951108B2 (en) | 2016-01-29 | 2024-04-09 | Epizyme, Inc. | Combination therapy for treating cancer |
| GB201604213D0 (en) * | 2016-03-11 | 2016-04-27 | Proximagen Ltd | Drug combination and its use in therapy |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| WO2017177230A1 (en) | 2016-04-08 | 2017-10-12 | X4 Pharmaceuticals, Inc. | Methods for treating cancer |
| US10947225B2 (en) | 2016-05-11 | 2021-03-16 | Emory University | Phosphotidylinositol 3-kinase inhibitors |
| TWI808055B (zh) | 2016-05-11 | 2023-07-11 | 美商滬亞生物國際有限公司 | Hdac 抑制劑與 pd-1 抑制劑之組合治療 |
| TWI794171B (zh) | 2016-05-11 | 2023-03-01 | 美商滬亞生物國際有限公司 | Hdac抑制劑與pd-l1抑制劑之組合治療 |
| CN109475558A (zh) * | 2016-05-25 | 2019-03-15 | 普渡研究基金会 | 通过靶向骨髓衍生的抑制细胞而治疗癌症的方法 |
| GB201609811D0 (en) | 2016-06-05 | 2016-07-20 | Snipr Technologies Ltd | Methods, cells, systems, arrays, RNA and kits |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2017223239A1 (en) | 2016-06-21 | 2017-12-28 | X4 Pharmaceuticals, Inc. | Cxcr4 inhibitors and uses thereof |
| JP7084624B2 (ja) | 2016-06-21 | 2022-06-15 | エックス4 ファーマシューティカルズ, インコーポレイテッド | Cxcr4阻害剤およびその使用 |
| EP3472129A4 (en) | 2016-06-21 | 2019-12-04 | X4 Pharmaceuticals, Inc. | CXCR4 INHIBITORS AND USES THEREOF |
| US20200179392A1 (en) * | 2016-08-05 | 2020-06-11 | Schepens Eye Research Institute | Idelalisib for Treating Proliferative Vitreoretinopathy and Abnormal Intraocular Neovascularization |
| US11542261B2 (en) | 2016-08-17 | 2023-01-03 | Children's Hospital Medical Center | Substituted Imidazo[1,2-a]-pyridines as IRAK 1/4 and FLT3 inhibitors |
| US11254667B2 (en) | 2016-08-17 | 2022-02-22 | Children's Hospital Medical Center | Substituted imidazo[1,2-A]pyridines as IRAK 1/4 and flt3 inhibitors |
| CA3044691A1 (en) | 2016-10-26 | 2018-05-03 | Genea Biocells USA (Holdings), Inc. | Improved generation of muscle lineage cells and therapeutic uses thereof |
| WO2018136625A2 (en) * | 2017-01-20 | 2018-07-26 | Children's Medical Center Corporation | Compositions and methods for treating diseases characterized by reactive microglia mediated synapse loss |
| US11654135B2 (en) | 2017-06-22 | 2023-05-23 | Moonshot Pharma Llc | Methods for treating colon cancer with compositions comprising amlexanox and immune checkpoint inhibitors |
| WO2019028395A1 (en) * | 2017-08-03 | 2019-02-07 | Fronthera U.S. Pharmaceuticals Llc | PI3 KINASE INHIBITORS AND USES THEREOF |
| JP2021028293A (ja) * | 2017-09-21 | 2021-02-25 | 第一三共株式会社 | 免疫抑制性ミエロイド細胞マーカー |
| US11117870B2 (en) | 2017-11-01 | 2021-09-14 | Drexel University | Compounds, compositions, and methods for treating diseases |
| US11485725B2 (en) | 2017-12-15 | 2022-11-01 | Auransa Inc. | Derivatives of piperlongumine and uses thereof |
| WO2019139388A1 (ko) * | 2018-01-10 | 2019-07-18 | 보령제약 주식회사 | Pi3 키나아제 억제제 및 면역관문 억제제를 포함하는 암의 예방 또는 치료용 약학적 조성물 |
| CA3083040A1 (en) | 2018-01-20 | 2019-07-25 | Sunshine Lake Pharma Co., Ltd. | Substituted aminopyrimidine compounds and methods of use |
| US10760075B2 (en) | 2018-04-30 | 2020-09-01 | Snipr Biome Aps | Treating and preventing microbial infections |
| CN108876772B (zh) * | 2018-06-05 | 2021-10-12 | 南华大学 | 一种基于大数据的肺癌图像诊断系统及方法 |
| CN109045067B (zh) * | 2018-08-08 | 2022-02-01 | 北京中台恒基生物技术有限公司 | 一种免疫抑制药物及其制备方法 |
| US10548889B1 (en) | 2018-08-31 | 2020-02-04 | X4 Pharmaceuticals, Inc. | Compositions of CXCR4 inhibitors and methods of preparation and use |
| WO2020069319A1 (en) * | 2018-09-27 | 2020-04-02 | New York University | Compositions and methods for use of cxcl 12 in treatment of bone disorders |
| US11851663B2 (en) | 2018-10-14 | 2023-12-26 | Snipr Biome Aps | Single-vector type I vectors |
| CN113395963B (zh) * | 2018-12-07 | 2024-06-25 | 马里兰大学巴尔的摩分校 | 非ATP/催化位点p38丝裂原活化蛋白激酶抑制剂 |
| GB201900724D0 (en) * | 2019-01-18 | 2019-03-06 | King S College London | Antibody |
| US12410170B2 (en) | 2019-06-04 | 2025-09-09 | Arcus Biosciences, Inc. | 2,3,5-trisubstituted pyrazolo[1,5-A]pyrimidine compounds |
| CN112194659A (zh) * | 2019-07-08 | 2021-01-08 | 浙江海正药业股份有限公司 | 炔类衍生物及其制备方法和用途 |
| US20220249389A1 (en) * | 2019-07-12 | 2022-08-11 | Oregon Health & Science University | Immunotherapeutic constructs and methods of their use |
| EP4017527A1 (en) | 2019-08-22 | 2022-06-29 | Juno Therapeutics, Inc. | Combination therapy of a t cell therapy and an enhancer of zeste homolog 2 (ezh2) inhibitor and related methods |
| EP4061371B1 (en) | 2019-11-22 | 2025-02-19 | AB Science | Masitinib for the treatment of sickle cell disease |
| CN115515585A (zh) | 2020-03-10 | 2022-12-23 | X4 制药有限公司 | 用于治疗中性粒细胞减少症的方法 |
| US12383547B2 (en) | 2020-07-01 | 2025-08-12 | Vanderbilt University | Methods of treatment for a kidney disease |
| US11357714B2 (en) | 2020-07-21 | 2022-06-14 | Chembeau LLC | Diester cosmetic formulations and uses thereof |
| CN111840297B (zh) * | 2020-08-24 | 2023-06-16 | 天津济坤医药科技有限公司 | 艾代拉里斯在制备用于治疗肝纤维化疾病的药物中的应用 |
| CN114569733B (zh) * | 2020-11-30 | 2023-09-19 | 浙江大学 | 一种吗替麦考酚酯的亚油酸偶联前药在抗纤维化或/和抗肿瘤药物中的应用 |
| CA3204091A1 (en) | 2020-12-08 | 2022-06-16 | Infinity Pharmaceuticals, Inc. | Eganelisib for use in the treatment of pd-l1 negative cancer |
| CN114452403A (zh) * | 2021-02-10 | 2022-05-10 | 同润生物医药(上海)有限公司 | 一种含有双功能免疫检查点/TGFβ抑制剂的药物组合 |
| CN117545479A (zh) * | 2021-04-20 | 2024-02-09 | 密歇根大学董事会 | Pi3k抑制剂、纳米制剂及其用途 |
| CA3220581A1 (en) * | 2021-05-19 | 2022-11-24 | Prescient Therapeutics Ltd | Methods of producing improved immune cell populations |
| CN113671064B (zh) * | 2021-07-15 | 2022-05-17 | 中国海洋大学 | 一种定量分析血浆中氨来占诺的血药浓度的检测方法 |
| CN113773361B (zh) * | 2021-10-25 | 2022-06-24 | 深圳弘汇生物医药有限公司 | 一种治疗高尿酸血症的化合物及其组合物、制备方法和医药用途 |
| WO2023096850A1 (en) * | 2021-11-24 | 2023-06-01 | Geode Therapeutics Inc. | Phosphoinositide 3 kinase gamma inhibitors and compositions and methods thereof |
| CN114272248B (zh) * | 2021-12-06 | 2023-08-01 | 上海市第十人民医院 | 一种化疗免疫凝胶及其制备方法与应用 |
| CA3255739A1 (en) * | 2022-04-08 | 2023-10-12 | Global Cancer Technology, Inc. | METHODS AND PRODUCTS FOR SCREENING AND TREATMENT OF GLIOBLASTOMA MULTIFORME AND OTHER CANCERS, INCLUDING BREAST CANCERS, USING A COMBINATION OF PI3KINASE INHIBITORS WITH CHECKPOINT INHIBITORS |
| GB202209518D0 (en) | 2022-06-29 | 2022-08-10 | Snipr Biome Aps | Treating & preventing E coli infections |
| WO2025096619A1 (en) * | 2023-10-30 | 2025-05-08 | The Regents Of The University Of Michigan | Dual functioning immune modulating compounds, formulations, and uses thereof |
| WO2025096405A1 (en) * | 2023-11-01 | 2025-05-08 | Dana-Farber Cancer Institute, Inc. | Methods of treating blood cancers |
| CN118440001B (zh) * | 2024-04-30 | 2024-10-25 | 江苏海洋大学 | 一种杜韦利西布中间体的合成方法 |
Family Cites Families (764)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US135A (en) | 1837-03-03 | Jesse j | ||
| GB812366A (en) | 1955-08-18 | 1959-04-22 | Wellcome Found | Improvements in and relating to derivatives of pyrimidine and the preparation thereof |
| US5458A (en) | 1848-02-22 | Bench-vise | ||
| US2517482A (en) | 1949-04-09 | 1950-08-01 | Sharp & Dohme Inc | Inhaler |
| GB937725A (en) | 1960-05-11 | 1963-09-25 | Ciba Ltd | Pyrazolo[3:4-d]pyrimidines |
| GB1182779A (en) | 1966-09-17 | 1970-03-04 | Fisons Pharmaceuticals Ltd | Inhalation Device |
| US3507277A (en) | 1966-09-17 | 1970-04-21 | Fisons Pharmaceuticals Ltd | Inhalation device |
| GB1268051A (en) | 1968-06-07 | 1972-03-22 | Fisons Pharmaceuticals Ltd | Inhalation device |
| US3536809A (en) | 1969-02-17 | 1970-10-27 | Alza Corp | Medication method |
| US3598123A (en) | 1969-04-01 | 1971-08-10 | Alza Corp | Bandage for administering drugs |
| US3831606A (en) | 1971-02-19 | 1974-08-27 | Alza Corp | Auto inhaler |
| IT941426B (it) | 1971-07-17 | 1973-03-01 | Isf Spa | Inalatore a camera di turbinio per sostanze medicamentose polveriformi |
| DE2139107A1 (de) | 1971-08-04 | 1973-02-15 | Merck Patent Gmbh | Heterocyclisch substituierte adenosinverbindungen |
| US3845770A (en) | 1972-06-05 | 1974-11-05 | Alza Corp | Osmatic dispensing device for releasing beneficial agent |
| US3795244A (en) | 1973-02-02 | 1974-03-05 | Syntex Corp | Inhalation device |
| US3916899A (en) | 1973-04-25 | 1975-11-04 | Alza Corp | Osmotic dispensing device with maximum and minimum sizes for the passageway |
| US3948264A (en) | 1975-05-21 | 1976-04-06 | Mead Johnson & Company | Inhalation device |
| US4008719A (en) | 1976-02-02 | 1977-02-22 | Alza Corporation | Osmotic system having laminar arrangement for programming delivery of active agent |
| US4270537A (en) | 1979-11-19 | 1981-06-02 | Romaine Richard A | Automatic hypodermic syringe |
| IT1153216B (it) | 1981-10-16 | 1987-01-14 | Schering Ag | Procedimento per la preparazione di composti cianoeterociclici |
| DE3244594A1 (de) | 1982-12-02 | 1984-06-07 | Hoechst Ag, 6230 Frankfurt | 1-phenylisochinolinderivate und verfahren zu ihrer herstellung, diese verbindung enthaltende pharmazeutische praeparate und deren anwendung |
| DE3406533A1 (de) | 1984-02-23 | 1985-08-29 | Boehringer Mannheim Gmbh, 6800 Mannheim | Verwendung von adenosin-derivaten als antiallergica und arzneimittel, die diese enthalten |
| US5310731A (en) | 1984-06-28 | 1994-05-10 | Whitby Research, Inc. | N-6 substituted-5'-(N-substitutedcarboxamido)adenosines as cardiac vasodilators and antihypertensive agents |
| KR890002631B1 (ko) | 1984-10-04 | 1989-07-21 | 몬산토 캄파니 | 생물학적으로 활성인 소마토트로핀을 지속적으로 유리하는 조성물 |
| US4795627A (en) | 1984-10-18 | 1989-01-03 | University Of Pittsburgh | Tritium labelled N-mustard type compounds and a process for their production |
| IE58110B1 (en) | 1984-10-30 | 1993-07-14 | Elan Corp Plc | Controlled release powder and process for its preparation |
| US4656159A (en) | 1984-10-31 | 1987-04-07 | Georgetown University | Galactose-C-6 nitrogen mustard compounds and their uses |
| JPS61109797A (ja) | 1984-11-01 | 1986-05-28 | Yuki Gosei Yakuhin Kogyo Kk | 標識化ヌクレオチドおよび標識化ポリヌクレオチド |
| US4596556A (en) | 1985-03-25 | 1986-06-24 | Bioject, Inc. | Hypodermic injection apparatus |
| US5023252A (en) | 1985-12-04 | 1991-06-11 | Conrex Pharmaceutical Corporation | Transdermal and trans-membrane delivery of drugs |
| US4886499A (en) | 1986-12-18 | 1989-12-12 | Hoffmann-La Roche Inc. | Portable injection appliance |
| GB8704027D0 (en) | 1987-02-20 | 1987-03-25 | Owen Mumford Ltd | Syringe needle combination |
| GB8707053D0 (en) | 1987-03-25 | 1987-04-29 | Ici Plc | Anti-tumour agents |
| US4992445A (en) | 1987-06-12 | 1991-02-12 | American Cyanamid Co. | Transdermal delivery of pharmaceuticals |
| US5001139A (en) | 1987-06-12 | 1991-03-19 | American Cyanamid Company | Enchancers for the transdermal flux of nivadipine |
| US4941880A (en) | 1987-06-19 | 1990-07-17 | Bioject, Inc. | Pre-filled ampule and non-invasive hypodermic injection device assembly |
| US4790824A (en) | 1987-06-19 | 1988-12-13 | Bioject, Inc. | Non-invasive hypodermic injection device |
| US4940460A (en) | 1987-06-19 | 1990-07-10 | Bioject, Inc. | Patient-fillable and non-invasive hypodermic injection device assembly |
| US5339163A (en) | 1988-03-16 | 1994-08-16 | Canon Kabushiki Kaisha | Automatic exposure control device using plural image plane detection areas |
| US5073543A (en) | 1988-07-21 | 1991-12-17 | G. D. Searle & Co. | Controlled release formulations of trophic factors in ganglioside-lipsome vehicle |
| WO1990003370A1 (en) | 1988-09-28 | 1990-04-05 | Microprobe Corporation | DERIVATIVES OF PYRAZOLO[3,4-d]PYRIMIDINE |
| FR2638359A1 (fr) | 1988-11-03 | 1990-05-04 | Tino Dalto | Guide de seringue avec reglage de la profondeur de penetration de l'aiguille dans la peau |
| IT1229203B (it) | 1989-03-22 | 1991-07-25 | Bioresearch Spa | Impiego di acido 5 metiltetraidrofolico, di acido 5 formiltetraidrofolico e dei loro sali farmaceuticamente accettabili per la preparazione di composizioni farmaceutiche in forma a rilascio controllato attive nella terapia dei disturbi mentali organici e composizioni farmaceutiche relative. |
| PH30995A (en) | 1989-07-07 | 1997-12-23 | Novartis Inc | Sustained release formulations of water soluble peptides. |
| US5442039A (en) | 1989-07-17 | 1995-08-15 | The Dow Chemical Company | Mesogenic polycyanates and thermosets thereof |
| US5428125A (en) | 1989-07-17 | 1995-06-27 | The Dow Chemical Company | Mesogenic polycyanates and thermosets thereof |
| US5763597A (en) | 1989-09-15 | 1998-06-09 | Metabasis Therapeutics, Inc. | Orally active adenosine kinase inhibitors |
| US5674998A (en) | 1989-09-15 | 1997-10-07 | Gensia Inc. | C-4' modified adenosine kinase inhibitors |
| US5763596A (en) | 1989-09-15 | 1998-06-09 | Metabasis Therapeutics, Inc. | C-4' modified adenosine kinase inhibitors |
| US5646128A (en) | 1989-09-15 | 1997-07-08 | Gensia, Inc. | Methods for treating adenosine kinase related conditions |
| US5795977A (en) | 1989-09-15 | 1998-08-18 | Metabasis Therapeutics, Inc. | Water soluble adenosine kinase inhibitors |
| US5721356A (en) | 1989-09-15 | 1998-02-24 | Gensia, Inc. | Orally active adenosine kinase inhibitors |
| US5120548A (en) | 1989-11-07 | 1992-06-09 | Merck & Co., Inc. | Swelling modulated polymeric drug delivery device |
| US5312335A (en) | 1989-11-09 | 1994-05-17 | Bioject Inc. | Needleless hypodermic injection device |
| US5064413A (en) | 1989-11-09 | 1991-11-12 | Bioject, Inc. | Needleless hypodermic injection device |
| GB9009542D0 (en) | 1990-04-27 | 1990-06-20 | Beecham Group Plc | Novel compounds |
| US5733566A (en) | 1990-05-15 | 1998-03-31 | Alkermes Controlled Therapeutics Inc. Ii | Controlled release of antiparasitic agents in animals |
| GB9113137D0 (en) | 1990-07-13 | 1991-08-07 | Ici Plc | Thioxo heterocycles |
| DE4026265A1 (de) | 1990-08-20 | 1992-02-27 | Boehringer Mannheim Gmbh | Neue phospholipid-derivate von nucleosiden, deren herstellung sowie deren verwendung als antivirale arzneimittel |
| US5563257A (en) | 1990-08-20 | 1996-10-08 | Boehringer Mannheim Gmbh | Phospholipid derivatives of nucleosides |
| US5190521A (en) | 1990-08-22 | 1993-03-02 | Tecnol Medical Products, Inc. | Apparatus and method for raising a skin wheal and anesthetizing skin |
| US5561134A (en) | 1990-09-25 | 1996-10-01 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Compounds having antihypertensive, cardioprotective, anti-ischemic and antilipolytic properties |
| DK0550631T3 (da) | 1990-09-25 | 1997-01-20 | Rhone Poulenc Rorer Int | Forbindelser med blodtrykssænkende virkning og virkning mod iskæmi |
| US5652366A (en) | 1990-09-25 | 1997-07-29 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | DI (1R)-(-)camphosulfonic acid) salt, preparation thereof and use thereof |
| US5527288A (en) | 1990-12-13 | 1996-06-18 | Elan Medical Technologies Limited | Intradermal drug delivery device and method for intradermal delivery of drugs |
| GB9103839D0 (en) | 1991-02-23 | 1991-04-10 | Smithkline Beecham Plc | Pharmaceuticals |
| AU658646B2 (en) | 1991-05-10 | 1995-04-27 | Rhone-Poulenc Rorer International (Holdings) Inc. | Bis mono-and bicyclic aryl and heteroaryl compounds which inhibit EGF and/or PDGF receptor tyrosine kinase |
| EP0592540B1 (en) | 1991-07-02 | 2000-01-26 | Inhale, Inc. | Method and device for delivering aerosolized medicaments |
| GB9118204D0 (en) | 1991-08-23 | 1991-10-09 | Weston Terence E | Needle-less injector |
| SE9102652D0 (sv) | 1991-09-13 | 1991-09-13 | Kabi Pharmacia Ab | Injection needle arrangement |
| US5272158A (en) | 1991-10-29 | 1993-12-21 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| US5580578A (en) | 1992-01-27 | 1996-12-03 | Euro-Celtique, S.A. | Controlled release formulations coated with aqueous dispersions of acrylic polymers |
| IL104369A0 (en) | 1992-01-13 | 1993-05-13 | Smithkline Beecham Corp | Novel compounds and compositions |
| US5916891A (en) | 1992-01-13 | 1999-06-29 | Smithkline Beecham Corporation | Pyrimidinyl imidazoles |
| DE4204031A1 (de) | 1992-02-12 | 1993-08-19 | Boehringer Mannheim Gmbh | Neue lipidphosphonsaeure-nucleosid-konjugate sowie deren verwendung als antivirale arzneimittel |
| DE4204032A1 (de) | 1992-02-12 | 1993-08-19 | Boehringer Mannheim Gmbh | Neue liponucleotide, deren herstellunmg sowie deren verwendung als antivirale arzneimittel |
| US5328483A (en) | 1992-02-27 | 1994-07-12 | Jacoby Richard M | Intradermal injection device with medication and needle guard |
| JPH05239036A (ja) | 1992-02-27 | 1993-09-17 | Nissan Chem Ind Ltd | 含窒素複素環化合物の製造方法 |
| WO1993018035A1 (en) | 1992-03-04 | 1993-09-16 | Abbott Laboratories | Angiotensin ii receptor antagonists |
| JP2737518B2 (ja) | 1992-03-16 | 1998-04-08 | 富士通株式会社 | 赤外線検知器の冷却構造 |
| US5294612A (en) | 1992-03-30 | 1994-03-15 | Sterling Winthrop Inc. | 6-heterocyclyl pyrazolo [3,4-d]pyrimidin-4-ones and compositions and method of use thereof |
| JPH07508711A (ja) | 1992-04-07 | 1995-09-28 | ザ リージェンツ オブ ザ ユニバーシティ オブ ミシガン | Cd28経路免疫調節 |
| GB9208135D0 (en) | 1992-04-13 | 1992-05-27 | Ludwig Inst Cancer Res | Polypeptides having kinase activity,their preparation and use |
| DE69333955D1 (de) | 1992-04-24 | 2006-02-02 | Stanford Res Inst Int | Targeting homologer sequenzen in eukaryotenzellen |
| JP3261617B2 (ja) | 1992-06-19 | 2002-03-04 | ハネウエル・インコーポレーテッド | 赤外線カメラ |
| US5383851A (en) | 1992-07-24 | 1995-01-24 | Bioject Inc. | Needleless hypodermic injection device |
| US6057305A (en) | 1992-08-05 | 2000-05-02 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Antiretroviral enantiomeric nucleotide analogs |
| US5569189A (en) | 1992-09-28 | 1996-10-29 | Equidyne Systems, Inc. | hypodermic jet injector |
| US5334144A (en) | 1992-10-30 | 1994-08-02 | Becton, Dickinson And Company | Single use disposable needleless injector |
| TW333456B (en) | 1992-12-07 | 1998-06-11 | Takeda Pharm Ind Co Ltd | A pharmaceutical composition of sustained-release preparation the invention relates to a pharmaceutical composition of sustained-release preparation which comprises a physiologically active peptide. |
| TW370529B (en) | 1992-12-17 | 1999-09-21 | Pfizer | Pyrazolopyrimidines |
| US5591767A (en) | 1993-01-25 | 1997-01-07 | Pharmetrix Corporation | Liquid reservoir transdermal patch for the administration of ketorolac |
| JPH08506343A (ja) | 1993-02-03 | 1996-07-09 | ジェンシア・インコーポレイテッド | リキソフラノシル誘導体を含むアデノシンキナーゼ阻害物質 |
| IL108523A0 (en) | 1993-02-03 | 1994-05-30 | Gensia Inc | Pharmaceutical compositions containing adenosine kinase inhibitors for preventing or treating conditions involving inflammatory responses and pain |
| GB9308957D0 (en) | 1993-04-30 | 1993-06-16 | Cancer Res Campaign Tech | Novel produgs |
| ES2240962T3 (es) | 1993-06-04 | 2005-10-16 | The United States Of America As Represented By The Secretary Of The Navy | Metodo para estimular selectivamente la proliferacion de celulas t. |
| US6087324A (en) | 1993-06-24 | 2000-07-11 | Takeda Chemical Industries, Ltd. | Sustained-release preparation |
| EP0634402A1 (en) | 1993-07-14 | 1995-01-18 | Takeda Chemical Industries, Ltd. | Isochinolinone derivatives, their production and use |
| US5504103A (en) | 1993-08-25 | 1996-04-02 | Eli Lilly And Company | Inhibition of phosphatidylinositol 3-kinase with 17 β-hydroxywortmannin and analogs thereof |
| US5525503A (en) | 1993-09-28 | 1996-06-11 | Dana-Farber Cancer Institute, Inc. | Signal transduction via CD28 |
| DE4345266C2 (de) | 1993-10-04 | 1996-12-19 | Luitpold Pharma Gmbh | Heterocyclische Carbamate, Verfahren zu ihrer Herstellung und Arzneimittel |
| WO1995012588A1 (en) | 1993-11-05 | 1995-05-11 | Biochem Pharma Inc. | Antineoplastic heteronaphthoquinones |
| US5679683A (en) | 1994-01-25 | 1997-10-21 | Warner-Lambert Company | Tricyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| IL112249A (en) | 1994-01-25 | 2001-11-25 | Warner Lambert Co | Pharmaceutical compositions containing di and tricyclic pyrimidine derivatives for inhibiting tyrosine kinases of the epidermal growth factor receptor family and some new such compounds |
| WO1995024176A1 (en) | 1994-03-07 | 1995-09-14 | Bioject, Inc. | Ampule filling device |
| US5466220A (en) | 1994-03-08 | 1995-11-14 | Bioject, Inc. | Drug vial mixing and transfer device |
| US6632789B1 (en) | 1994-04-29 | 2003-10-14 | The United States Of America As Represented By The Secretary Of The Navy | Methods for modulating T cell responses by manipulating intracellular signal transduction |
| DE4418690A1 (de) | 1994-05-28 | 1996-01-11 | Boehringer Mannheim Gmbh | Neue Lipidester von Nucleosid-Monophosphaten und deren Verwendung als immunsuppressive Arzneimittel |
| IT1270594B (it) | 1994-07-07 | 1997-05-07 | Recordati Chem Pharm | Composizione farmaceutica a rilascio controllato di moguisteina in sospensione liquida |
| US5756502A (en) | 1994-08-08 | 1998-05-26 | Warner-Lambert Company | Quinazolinone derivatives as cholyecystokinin (CCK) ligands |
| US6323201B1 (en) | 1994-12-29 | 2001-11-27 | The Regents Of The University Of California | Compounds for inhibition of ceramide-mediated signal transduction |
| US5599302A (en) | 1995-01-09 | 1997-02-04 | Medi-Ject Corporation | Medical injection system and method, gas spring thereof and launching device using gas spring |
| GB9501841D0 (en) | 1995-01-31 | 1995-03-22 | Co Ordinated Drug Dev | Improvements in and relating to carrier particles for use in dry powder inhalers |
| US6312894B1 (en) | 1995-04-03 | 2001-11-06 | Epoch Pharmaceuticals, Inc. | Hybridization and mismatch discrimination using oligonucleotides conjugated to minor groove binders |
| ATE195123T1 (de) | 1995-04-03 | 2000-08-15 | Novartis Erfind Verwalt Gmbh | Pyrazolderivate und verfahren zu deren herstellung |
| US5977061A (en) | 1995-04-21 | 1999-11-02 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | N6 - substituted nucleotide analagues and their use |
| JPH08295667A (ja) | 1995-04-27 | 1996-11-12 | Takeda Chem Ind Ltd | 複素環化合物、その製造法および剤 |
| US5593997A (en) | 1995-05-23 | 1997-01-14 | Pfizer Inc. | 4-aminopyrazolo(3-,4-D)pyrimidine and 4-aminopyrazolo-(3,4-D)pyridine tyrosine kinase inhibitors |
| WO1996037777A1 (en) | 1995-05-23 | 1996-11-28 | Nelson Randall W | Mass spectrometric immunoassay |
| US5730723A (en) | 1995-10-10 | 1998-03-24 | Visionary Medical Products Corporation, Inc. | Gas pressured needle-less injection device and method |
| US6403599B1 (en) | 1995-11-08 | 2002-06-11 | Pfizer Inc | Corticotropin releasing factor antagonists |
| WO1996040256A1 (en) | 1995-06-07 | 1996-12-19 | G.D. Searle & Co. | Method to treat cardiofibrosis with a combination of an angiotensin ii antagonist and spironolactone |
| ATE209047T1 (de) | 1995-06-07 | 2001-12-15 | Searle & Co | Epoxysteroide aldosteronantagonist und angiotensin ii rezeptor antagonist kombinationstherapie zur behandlung von congestivem herzversagen |
| US5665721A (en) | 1995-06-07 | 1997-09-09 | Abbott Laboratories | Heterocyclic substituted cyclopentane compounds |
| JPH11509838A (ja) | 1995-06-07 | 1999-08-31 | ジー.ディー.サール アンド カンパニー | 充血性心不全症を処置するためのスピロノラクトン化合物とアンギオテンシンii拮抗物質との組合わせ治療 |
| AU6242096A (en) | 1995-06-27 | 1997-01-30 | Takeda Chemical Industries Ltd. | Method of producing sustained-release preparation |
| GB9515182D0 (en) | 1995-07-24 | 1995-09-20 | Co Ordinated Drug Dev | Improvements in and relating to powders for use in dry powder inhalers |
| TW448055B (en) | 1995-09-04 | 2001-08-01 | Takeda Chemical Industries Ltd | Method of production of sustained-release preparation |
| WO1997010221A1 (en) | 1995-09-15 | 1997-03-20 | Torrey Pines Institute For Molecular Studies | Synthesis of quinazolinone libraries |
| JP2909418B2 (ja) | 1995-09-18 | 1999-06-23 | 株式会社資生堂 | 薬物の遅延放出型マイクロスフイア |
| US5763885A (en) | 1995-12-19 | 1998-06-09 | Loral Infrared & Imaging Systems, Inc. | Method and apparatus for thermal gradient stabilization of microbolometer focal plane arrays |
| JPH09143163A (ja) | 1995-11-29 | 1997-06-03 | Fuji Photo Film Co Ltd | 含窒素ヘテロ芳香族アミド類の製造方法 |
| US5893397A (en) | 1996-01-12 | 1999-04-13 | Bioject Inc. | Medication vial/syringe liquid-transfer apparatus |
| US5980945A (en) | 1996-01-16 | 1999-11-09 | Societe De Conseils De Recherches Et D'applications Scientifique S.A. | Sustained release drug formulations |
| US5747235A (en) | 1996-01-26 | 1998-05-05 | Eastman Kodak Company | Silver halide light sensitive emulsion layer having enhanced photographic sensitivity |
| CH690773A5 (de) | 1996-02-01 | 2001-01-15 | Novartis Ag | Pyrrolo(2,3-d)pyrimide und ihre Verwendung. |
| DE19603576A1 (de) | 1996-02-01 | 1997-08-07 | Bayer Ag | Acylierte 4-Amino und 4-Hydrazinopyrimidine |
| US5763608A (en) | 1996-02-05 | 1998-06-09 | Hoechst Celanese Corporation | Process for preparing pyrimidine derivatives |
| GB2310952B (en) | 1996-03-05 | 1998-08-19 | Mitsubishi Electric Corp | Infrared detector |
| US5872136A (en) | 1996-04-03 | 1999-02-16 | Merck & Co., Inc. | Arylheteroaryl inhibitors of farnesyl-protein transferase |
| EP0891361A1 (en) | 1996-04-03 | 1999-01-20 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| GB9607549D0 (en) | 1996-04-11 | 1996-06-12 | Weston Medical Ltd | Spring-powered dispensing device |
| GB9611460D0 (en) | 1996-06-01 | 1996-08-07 | Ludwig Inst Cancer Res | Novel lipid kinase |
| CA2258822A1 (en) | 1996-06-20 | 1997-12-24 | Sean Kerwin | Compounds and methods for providing pharmacologically active preparations and uses thereof |
| US6264970B1 (en) | 1996-06-26 | 2001-07-24 | Takeda Chemical Industries, Ltd. | Sustained-release preparation |
| FR2750862B1 (fr) | 1996-07-12 | 1998-10-16 | Dupin Jean Pierre | Utilisation d'heterocycles diazotes fusionnes avec un systeme aromatique ou heteroaromatique pour le traitement des maladies thrombo-emboliques |
| US6419961B1 (en) | 1996-08-29 | 2002-07-16 | Takeda Chemical Industries, Ltd. | Sustained release microcapsules of a bioactive substance and a biodegradable polymer |
| CA2217134A1 (en) | 1996-10-09 | 1998-04-09 | Sumitomo Pharmaceuticals Co., Ltd. | Sustained release formulation |
| US6153631A (en) | 1996-10-23 | 2000-11-28 | Zymogenetics, Inc. | Compositions and methods for treating bone deficit conditions |
| US5965573A (en) | 1996-10-23 | 1999-10-12 | Zymogenetics, Inc. | Compositions and methods for treating bone deficit conditions |
| US5994358A (en) | 1996-10-23 | 1999-11-30 | Zymogenetics, Inc. | Compositions and methods for treating bone deficit conditions |
| US6251901B1 (en) | 1996-10-23 | 2001-06-26 | Zymogenetics, Inc. | Compositions and methods for treating bone deficit conditions |
| US5948776A (en) | 1996-10-23 | 1999-09-07 | Zymogenetic, Inc. | Compositions and methods for treating bone deficit conditions |
| US5922753A (en) | 1996-10-23 | 1999-07-13 | Zymogenetics, Inc. | Methods for treating bone deficit conditions with benzothiazole |
| US5990169A (en) | 1996-10-23 | 1999-11-23 | Zymogenetics, Inc. | Compositions and methods for treating bone deficit conditions |
| US5919808A (en) | 1996-10-23 | 1999-07-06 | Zymogenetics, Inc. | Compositions and methods for treating bone deficit conditions |
| US6342514B1 (en) | 1996-10-23 | 2002-01-29 | Zymogenetics, Inc. | Compositions and methods for treating bone deficit conditions |
| PT839525E (pt) | 1996-10-31 | 2004-10-29 | Takeda Chemical Industries Ltd | Preparacao de libertacao prolongada |
| US5858753A (en) | 1996-11-25 | 1999-01-12 | Icos Corporation | Lipid kinase |
| US5977134A (en) | 1996-12-05 | 1999-11-02 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| AU7624798A (en) | 1996-12-06 | 1998-06-29 | Vertex Pharmaceuticals Incorporated | Inhibitors of interleukin-1 beta converting enzyme |
| AU7871298A (en) | 1996-12-20 | 1998-07-17 | Takeda Chemical Industries Ltd. | Method of producing a sustained-release preparation |
| US6093737A (en) | 1996-12-30 | 2000-07-25 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| JPH10206995A (ja) | 1997-01-21 | 1998-08-07 | Konica Corp | ハロゲン化銀写真感光材料 |
| US5891474A (en) | 1997-01-29 | 1999-04-06 | Poli Industria Chimica, S.P.A. | Time-specific controlled release dosage formulations and method of preparing same |
| AU755062B2 (en) | 1997-02-07 | 2002-12-05 | Princeton University | Engineered protein kinases which can utilize modified nucleotide triphosphate substrates |
| US7863444B2 (en) | 1997-03-19 | 2011-01-04 | Abbott Laboratories | 4-aminopyrrolopyrimidines as kinase inhibitors |
| SK125999A3 (en) | 1997-03-19 | 2000-05-16 | Knoll Ag | Pyrrolo[2,3d]pyrimidines and their use as tyrosine kinase inhibitors |
| US5993412A (en) | 1997-05-19 | 1999-11-30 | Bioject, Inc. | Injection apparatus |
| WO1998052611A1 (en) | 1997-05-23 | 1998-11-26 | Nippon Shinyaku Co., Ltd. | Medicinal composition for prevention or treatment of hepatopathy |
| US6207679B1 (en) | 1997-06-19 | 2001-03-27 | Sepracor, Inc. | Antimicrobial agents uses and compositions related thereto |
| ATE266636T1 (de) | 1997-09-09 | 2004-05-15 | Bristol Myers Squibb Pharma Co | Benzimidazolinone, benzoxazolinone, benzopiperazinone, indanone und deren derivate als faktor xa inhibitoren |
| DE69839887D1 (de) | 1997-10-02 | 2008-09-25 | Eisai R&D Man Co Ltd | Kondensierte pyridinderivate |
| US6649631B1 (en) | 1997-10-23 | 2003-11-18 | The Board Of Regents Of The University Of Texas System | Compositions and methods for treating bone deficit conditions |
| US6040321A (en) | 1997-11-12 | 2000-03-21 | Bristol-Myers Squibb Company | Aminothiazole inhibitors of cyclin dependent kinases |
| CA2309350C (en) | 1997-11-12 | 2007-04-03 | Mitsubishi Chemical Corporation | Purine derivatives and medicaments comprising the same as active ingredient |
| US6184377B1 (en) | 1997-12-15 | 2001-02-06 | Sepracor Inc. | Compositions containing N-amino- and N-hydroxy-quinazolinones and methods for preparing libraries thereof |
| IT1298087B1 (it) | 1998-01-08 | 1999-12-20 | Fiderm S R L | Dispositivo per il controllo della profondita' di penetrazione di un ago, in particolare applicabile ad una siringa per iniezioni |
| US6191170B1 (en) | 1998-01-13 | 2001-02-20 | Tularik Inc. | Benzenesulfonamides and benzamides as therapeutic agents |
| US6613358B2 (en) | 1998-03-18 | 2003-09-02 | Theodore W. Randolph | Sustained-release composition including amorphous polymer |
| EP0945441A3 (de) | 1998-03-27 | 2002-07-17 | Degussa AG | Verfahren zur Herstellung von cyclischen 4-Oxoamidinen |
| US6127121A (en) | 1998-04-03 | 2000-10-03 | Epoch Pharmaceuticals, Inc. | Oligonucleotides containing pyrazolo[3,4-D]pyrimidines for hybridization and mismatch discrimination |
| US7715989B2 (en) | 1998-04-03 | 2010-05-11 | Elitech Holding B.V. | Systems and methods for predicting oligonucleotide melting temperature (TmS) |
| US6432970B2 (en) | 1998-04-09 | 2002-08-13 | Johns Hopkins University School Of Medicine | Inhibitors of hedgehog signaling pathways, compositions and uses related thereto |
| KR19990085365A (ko) | 1998-05-16 | 1999-12-06 | 허영섭 | 지속적으로 약물 조절방출이 가능한 생분해성 고분자 미립구 및그 제조방법 |
| JP2000072773A (ja) | 1998-08-28 | 2000-03-07 | Zeria Pharmaceut Co Ltd | プリン誘導体 |
| AU752474B2 (en) | 1998-09-18 | 2002-09-19 | Abbott Gmbh & Co. Kg | 4-aminopyrrolopyrimidines as kinase inhibitors |
| NZ512870A (en) | 1998-12-19 | 2003-11-28 | Janssen Pharmaceutica Nv | Antihistaminic spiro compounds |
| JP4876239B2 (ja) | 1999-01-11 | 2012-02-15 | プリンストン ユニバーシティー | 標的確認のための高親和性阻害剤およびその使用 |
| CZ27399A3 (cs) | 1999-01-26 | 2000-08-16 | Ústav Experimentální Botaniky Av Čr | Substituované dusíkaté heterocyklické deriváty, způsob jejich přípravy, tyto deriváty pro použití jako léčiva, farmaceutická kompozice a kombinovaný farmaceutický přípravek tyto deriváty obsahující a použití těchto derivátů pro výrobu léčiv |
| US6248363B1 (en) | 1999-11-23 | 2001-06-19 | Lipocine, Inc. | Solid carriers for improved delivery of active ingredients in pharmaceutical compositions |
| US6596497B1 (en) | 1999-03-17 | 2003-07-22 | New York Blood Center, Inc. | Screening of antiviral compounds targeted to the HIV-1 gp41 core structure |
| EP1040831A3 (en) | 1999-04-02 | 2003-05-02 | Pfizer Products Inc. | Use of corticotropin releasing factor (CRF) antagonists to prevent sudden death |
| SE515856C2 (sv) | 1999-05-19 | 2001-10-22 | Ericsson Telefon Ab L M | Bärare för elektronikkomponenter |
| AU5179000A (en) | 1999-06-03 | 2000-12-28 | Basf Aktiengesellschaft | Benzothiazinone and benzoxazinone compounds |
| US6387894B1 (en) | 1999-06-11 | 2002-05-14 | Pfizer Inc. | Use of CRF antagonists and renin-angiotensin system inhibitors |
| PE20010306A1 (es) | 1999-07-02 | 2001-03-29 | Agouron Pharma | Compuestos de indazol y composiciones farmaceuticas que los contienen utiles para la inhibicion de proteina kinasa |
| TWI262914B (en) | 1999-07-02 | 2006-10-01 | Agouron Pharma | Compounds and pharmaceutical compositions for inhibiting protein kinases |
| GB9919588D0 (en) | 1999-08-18 | 1999-10-20 | Hoechst Schering Agrevo Gmbh | Fungicidal compounds |
| DE19940859A1 (de) | 1999-08-27 | 2001-03-01 | Bayer Ag | Selektive Herbizide auf Basis eines substituierten Phenylsulfonylaminocarbonyltriazolinons und Safenern |
| JP4831906B2 (ja) | 1999-08-27 | 2011-12-07 | ケモセントリックス, インコーポレイテッド | Cxcr3機能を調節するための複素環式化合物および方法 |
| DE19940860A1 (de) | 1999-08-27 | 2001-03-01 | Bayer Ag | Selektive Herbizide auf Basis eines substituierten Phenylsulfonyl aminocarbonyltriazolinons und Safenern II |
| EP1216234B1 (en) | 1999-09-16 | 2004-12-29 | Curis, Inc. | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| US20070021493A1 (en) | 1999-09-16 | 2007-01-25 | Curis, Inc. | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| US20050080138A1 (en) | 1999-09-16 | 2005-04-14 | Curis, Inc. | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| JP2003509428A (ja) | 1999-09-17 | 2003-03-11 | アボツト・ゲー・エム・ベー・ハー・ウント・コンパニー・カーゲー | 治療薬としてのピラゾロピリミジン |
| US6921763B2 (en) | 1999-09-17 | 2005-07-26 | Abbott Laboratories | Pyrazolopyrimidines as therapeutic agents |
| US6506769B2 (en) | 1999-10-06 | 2003-01-14 | Boehringer Ingelheim Pharmaceuticals, Inc. | Heterocyclic compounds useful as inhibitors of tyrosine kinases |
| DE60014130T2 (de) | 1999-10-06 | 2006-03-09 | Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield | Heterocyclische verbindungen verwendbar als tyrosinkinase inhibitoren |
| CA2386190C (en) | 1999-10-13 | 2009-04-14 | Johns Hopkins University School Of Medicine | Regulators of the hedgehog pathway, compositions and uses related thereto |
| WO2001026644A2 (en) | 1999-10-14 | 2001-04-19 | Curis, Inc. | Mediators of hedgehog signaling pathways, compositions and uses related thereto |
| US6472153B1 (en) | 1999-10-26 | 2002-10-29 | Epoch Biosciences, Inc. | Hybridization-triggered fluorescent detection of nucleic acids |
| US7230000B1 (en) | 1999-10-27 | 2007-06-12 | Cytokinetics, Incorporated | Methods and compositions utilizing quinazolinones |
| US6545004B1 (en) | 1999-10-27 | 2003-04-08 | Cytokinetics, Inc. | Methods and compositions utilizing quinazolinones |
| US7671200B2 (en) | 1999-10-27 | 2010-03-02 | Cytokinetics, Inc. | Quinazolinone KSP inhibitors |
| US6660845B1 (en) | 1999-11-23 | 2003-12-09 | Epoch Biosciences, Inc. | Non-aggregating, non-quenching oligomers comprising nucleotide analogues; methods of synthesis and use thereof |
| IL133809A0 (en) | 1999-12-30 | 2001-04-30 | Yeda Res & Dev | Steroidal alkaloids and pharmaceutical compositions comprising them |
| GB0002032D0 (en) | 2000-01-28 | 2000-03-22 | Zeneca Ltd | Chemical compounds |
| US7217722B2 (en) | 2000-02-01 | 2007-05-15 | Kirin Beer Kabushiki Kaisha | Nitrogen-containing compounds having kinase inhibitory activity and drugs containing the same |
| JP2001250689A (ja) | 2000-03-07 | 2001-09-14 | Toray Ind Inc | 発光素子 |
| WO2001072712A1 (en) | 2000-03-24 | 2001-10-04 | Cor Therapeutics, Inc. | ISOQUINOLONE INHIBITORS OF FACTOR Xa |
| DE60113823T3 (de) | 2000-03-30 | 2010-07-01 | Curis, Inc., Cambridge | Kleine organische moleküle als regulatoren der zellproliferation |
| US6683108B1 (en) | 2000-03-30 | 2004-01-27 | Curis, Inc. | Agonists of hedgehog signaling pathways and uses related thereto |
| US6683192B2 (en) | 2000-03-30 | 2004-01-27 | Curis, Inc. | Small organic molecule regulators of cell proliferation |
| US6613798B1 (en) | 2000-03-30 | 2003-09-02 | Curis, Inc. | Small organic molecule regulators of cell proliferation |
| US7115653B2 (en) | 2000-03-30 | 2006-10-03 | Curis, Inc. | Small organic molecule regulators of cell proliferation |
| US20050070578A1 (en) | 2000-03-30 | 2005-03-31 | Baxter Anthony David | Small organic molecule regulators of cell proliferation |
| US20020127625A1 (en) | 2000-03-31 | 2002-09-12 | Forskarpatent Is Syd Ab | Methods of diagnosing immune related diseases |
| US6667300B2 (en) | 2000-04-25 | 2003-12-23 | Icos Corporation | Inhibitors of human phosphatidylinositol 3-kinase delta |
| PT1278748E (pt) | 2000-04-25 | 2011-03-31 | Icos Corp | Inibidores de delta 3-cinase humana de fosfatidilinositol |
| GB0010709D0 (en) | 2000-05-03 | 2000-06-28 | Vectura Ltd | Powders for use a in dry powder inhaler |
| EP1289472A4 (en) | 2000-05-30 | 2004-09-08 | Advanced Res & Tech Inst | COMPOSITIONS AND METHODS FOR IDENTIFYING SUBSTANCES THAT MODULE THE PTEN FUNCTION AND THE PI-3 KINASE WAYS |
| EP1294713A2 (en) | 2000-06-27 | 2003-03-26 | Genelabs Technologies, Inc. | Compounds possessing antibacterial, antifungal or antitumor activity |
| US7080644B2 (en) | 2000-06-28 | 2006-07-25 | Microdose Technologies, Inc. | Packaging and delivery of pharmaceuticals and drugs |
| US6534691B2 (en) | 2000-07-18 | 2003-03-18 | E. I. Du Pont De Nemours And Company | Manufacturing process for α-olefins |
| JP4854909B2 (ja) | 2000-09-20 | 2012-01-18 | メルク・シャープ・エンド・ドーム・コーポレイション | イソキノリノン系カリウムチャンネル阻害剤 |
| AU2002213467A1 (en) | 2000-10-11 | 2002-04-22 | Chemocentryx, Inc. | Modulation of ccr4 function |
| US6811979B2 (en) | 2000-10-11 | 2004-11-02 | Applera Corporation | Fluorescent nucleobase conjugates having anionic linkers |
| JP2002131859A (ja) | 2000-10-19 | 2002-05-09 | Konica Corp | 撮影用赤外感光性ハロゲン化銀写真感光材料及び赤外感光性ハロゲン化銀乳剤 |
| US6890747B2 (en) | 2000-10-23 | 2005-05-10 | Warner-Lambert Company | Phosphoinositide 3-kinases |
| EP1343505A1 (en) | 2000-12-11 | 2003-09-17 | Tularik Inc. | Cxcr3 antagonists |
| CZ20031751A3 (cs) | 2000-12-21 | 2004-03-17 | Glaxo Group Limited | Makrolidy |
| CN100471838C (zh) | 2000-12-28 | 2009-03-25 | 第一制药株式会社 | Vla-4抑制剂 |
| EP2360166A1 (en) | 2001-01-22 | 2011-08-24 | Merck Sharp & Dohme Corp. | Nucleoside derivatives as inhibitors of RNA-dependent RNA viral polymerase |
| US7105499B2 (en) | 2001-01-22 | 2006-09-12 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of RNA-dependent RNA viral polymerase |
| GB0102239D0 (en) | 2001-01-29 | 2001-03-14 | Cancer Res Ventures Ltd | Methods of chemical synthisis |
| GB2373186A (en) | 2001-02-23 | 2002-09-18 | Astrazeneca Ab | Pharmaceutical combinations of a CCR3 antagonist and a compound which is usefulreatment of asthma, allergic disease or inflammation |
| MXPA03008560A (es) | 2001-03-22 | 2004-06-30 | Abbot Gmbh & Co Kg | Pirazolopirimidinas como agentes terapeuticos. |
| DE10118698A1 (de) | 2001-04-17 | 2002-11-07 | Jerini Ag | Verfahren zur Immobilisierung und damit hergestellte Anordnungen von Verbindungen auf einer planaren Oberfläche |
| US7250569B2 (en) | 2001-04-26 | 2007-07-31 | New York University School Of Medicine | Method for dissolving nanostructural materials |
| US7405235B2 (en) | 2001-05-04 | 2008-07-29 | Paratek Pharmaceuticals, Inc. | Transcription factor modulating compounds and methods of use thereof |
| ES2282410T3 (es) | 2001-05-08 | 2007-10-16 | Kudos Pharmaceuticals Limited | Derivados de isoquinolinona como inhibidores de parp. |
| US7144903B2 (en) | 2001-05-23 | 2006-12-05 | Amgen Inc. | CCR4 antagonists |
| US6794379B2 (en) | 2001-06-06 | 2004-09-21 | Tularik Inc. | CXCR3 antagonists |
| WO2002101007A2 (en) | 2001-06-13 | 2002-12-19 | Genesoft Pharmaceuticals, Inc | Antipathogenic benzamide compounds |
| US6825228B2 (en) | 2001-06-13 | 2004-11-30 | Genesoft Pharmaceuticals, Inc. | Benzothiophene compounds having antiinfective activity |
| US6777425B2 (en) | 2001-06-13 | 2004-08-17 | Genesoft Pharmaceuticals, Inc. | Isoquinoline compounds having antiinfective activity |
| WO2003000187A2 (en) | 2001-06-21 | 2003-01-03 | Ariad Pharmaceuticals, Inc. | Novel pyrazolo-and pyrrolo-pyrimidines and uses thereof |
| US6596723B1 (en) | 2001-07-16 | 2003-07-22 | Essential Therapeutics, Inc. | Fungal efflux pump inhibitors |
| US6689782B2 (en) | 2001-07-16 | 2004-02-10 | Essential Therapeutics, Inc. | Fungal efflux pump inhibitors |
| US20030220338A1 (en) | 2001-07-16 | 2003-11-27 | Watkins Will J. | Fungal efflux pump inhibitors |
| AU2002322585A1 (en) | 2001-07-20 | 2003-03-03 | Adipogenix, Inc. | Fat accumulation-modulating compounds |
| EA007339B1 (ru) | 2001-07-27 | 2006-08-25 | Кьюэрис, Инк. | Медиаторы путей передачи сигналов генами hedgehog, содержащие их композиции и способы применения указанных веществ |
| EP1414443B1 (en) | 2001-08-01 | 2006-11-15 | Merck & Co., Inc. | BENZIMIDAZO 4,5-f|ISOQUINOLINONE DERIVATIVES |
| WO2003020880A2 (en) | 2001-08-03 | 2003-03-13 | Abbott Laboratories | Method of identifying inhibitors of lck |
| WO2003016275A1 (fr) | 2001-08-10 | 2003-02-27 | Shionogi & Co., Ltd. | Agent antiviral |
| JP2003073357A (ja) | 2001-09-03 | 2003-03-12 | Mitsubishi Pharma Corp | アミド化合物を含有するRhoキナーゼ阻害剤 |
| DE60236605D1 (de) | 2001-09-05 | 2010-07-15 | Minerva Biotechnologies Corp | Zusammensetzungen und deren verwendung zur behandlung von krebs |
| US20030199516A1 (en) | 2001-09-13 | 2003-10-23 | Genesoft, Inc. | Methods of treating infection by drug resistant bacteria |
| AUPR769501A0 (en) | 2001-09-14 | 2001-10-11 | Biomolecular Research Institute Limited | Cytokine receptor 1 |
| US7101884B2 (en) | 2001-09-14 | 2006-09-05 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
| US7269663B2 (en) | 2001-09-28 | 2007-09-11 | Intel Corporation | Tagging packets with a lookup key to facilitate usage of a unified packet forwarding cache |
| TWI330183B (es) | 2001-10-22 | 2010-09-11 | Eisai R&D Man Co Ltd | |
| KR20040048920A (ko) | 2001-11-01 | 2004-06-10 | 얀센 파마슈티카 엔.브이. | 글리코겐 신타아제 키나제 3-베타 저해제(gsk저해제)로서의 헤테로아릴 아민 |
| US20030113828A1 (en) | 2001-11-09 | 2003-06-19 | Ginsberg Mark H. | Compositions and methods for modulating Syk function |
| US7053216B2 (en) | 2001-11-19 | 2006-05-30 | Iconix Pharmaceuticals, Inc. | Modulators of Rho C activity |
| US6753428B2 (en) | 2001-11-20 | 2004-06-22 | Cytokinetics, Inc. | Process for the racemization of chiral quinazolinones |
| MXPA04004982A (es) | 2001-11-27 | 2004-08-11 | Hoffmann La Roche | Derivados de benzotiazol. |
| WO2003048081A2 (en) | 2001-12-04 | 2003-06-12 | Bristol-Myers Squibb Company | Glycinamides as factor xa inhibitors |
| EP1551812B1 (en) | 2001-12-06 | 2009-03-04 | Merck & Co., Inc. | Mitotic kinesin inhibitors |
| DE60234278D1 (de) | 2001-12-06 | 2009-12-17 | Merck & Co Inc | Mitotische kinesin-hemmer |
| EP1465869B1 (en) | 2001-12-21 | 2013-05-15 | Exelixis Patent Company LLC | Modulators of lxr |
| US20030158195A1 (en) | 2001-12-21 | 2003-08-21 | Cywin Charles L. | 1,6 naphthyridines useful as inhibitors of SYK kinase |
| JP4085237B2 (ja) | 2001-12-21 | 2008-05-14 | 日本電気株式会社 | 携帯電話の利用契約システムと通信方法 |
| US7064218B2 (en) | 2001-12-26 | 2006-06-20 | Genelabs Technologies, Inc. | Aromatic compounds and poly(oxyalkylene) containing aromatic compounds possessing antibacterial, antifungal or antitumor activity |
| US7414036B2 (en) | 2002-01-25 | 2008-08-19 | Muscagen Limited | Compounds useful as A3 adenosine receptor agonists |
| US7009049B2 (en) | 2002-02-15 | 2006-03-07 | Cytokinetics, Inc. | Syntheses of quinazolinones |
| US20040043959A1 (en) | 2002-03-04 | 2004-03-04 | Bloom Laura A. | Combination therapies for treating methylthioadenosine phosphorylase deficient cells |
| ES2361403T3 (es) | 2002-03-07 | 2011-06-16 | X-Ceptor Therapeutics, Inc. | Moduladores de quinazolinona de receptores nucleares. |
| US6995144B2 (en) | 2002-03-14 | 2006-02-07 | Eisai Co., Ltd. | Nitrogen containing heterocyclic compounds and medicines containing the same |
| AU2003225933A1 (en) | 2002-03-22 | 2003-10-13 | Cellular Genomics, Inc. | AN IMPROVED FORMULATION OF CERTAIN PYRAZOLO(3,4-d) PYRIMIDINES AS KINASE MODULATORS |
| CA2480468C (en) | 2002-03-26 | 2012-03-13 | Biofrontera Discovery Gmbh | Fredericamycin derivatives |
| US7166293B2 (en) | 2002-03-29 | 2007-01-23 | Carlsbad Technology, Inc. | Angiogenesis inhibitors |
| US6900219B2 (en) | 2002-04-04 | 2005-05-31 | Cv Therapeutics, Inc. | ABCA-1 elevating compounds |
| DE10217046A1 (de) | 2002-04-17 | 2003-11-06 | Bioleads Gmbh | Fredericamycin-Derivate |
| EP1496905B1 (en) | 2002-04-22 | 2008-08-13 | Johns Hopkins University School of Medicine | Modulators of hedgehog signaling pathways, compositions and uses related thereto |
| WO2003090751A1 (en) | 2002-04-26 | 2003-11-06 | Pfizer Products Inc. | Pyrimidine-2, 4, 6-trione metallo-proteinase inhibitors |
| US6794562B2 (en) | 2002-05-01 | 2004-09-21 | Stine Seed Farm, Inc. | Soybean cultivar 0332143 |
| CA2485148A1 (en) | 2002-05-09 | 2003-11-20 | Cytokinetics, Inc. | Pyrimidinone compounds, compositions and methods |
| WO2003097053A1 (en) | 2002-05-09 | 2003-11-27 | Cytokinetics, Inc. | Compounds, compositions, and methods |
| TW200407143A (en) | 2002-05-21 | 2004-05-16 | Bristol Myers Squibb Co | Pyrrolotriazinone compounds and their use to treat diseases |
| AU2003265242A1 (en) | 2002-05-23 | 2003-12-22 | Cytokinetics, Inc. | Compounds, compositions, and methods |
| AU2003299517A1 (en) | 2002-05-23 | 2004-05-25 | Merck & Co., Inc. | Mitotic kinesin inhibitors |
| AU2003236527A1 (en) | 2002-06-14 | 2003-12-31 | Cytokinetics, Inc. | Compounds, compositions, and methods |
| US7265111B2 (en) | 2002-06-27 | 2007-09-04 | Sanofi-Aventis Deutschland Gmbh | Adenosine analogues and their use as pharmaceutical agents |
| KR101124245B1 (ko) | 2002-06-27 | 2012-07-02 | 노보 노르디스크 에이/에스 | 치료제로서 아릴 카르보닐 유도체 |
| MXPA05000130A (es) | 2002-06-27 | 2005-02-17 | Novo Nordisk As | Derivados de aril-carbonilo como agentes terapeuticos. |
| DE10230917A1 (de) | 2002-07-09 | 2004-02-05 | Bioleads Gmbh | Fredericamycin-Derivate |
| PL374718A1 (en) | 2002-07-10 | 2005-10-31 | Applied Research Systems Ars Holding N.V. | Azolidinone-vinyl fused-benzene derivatives |
| AU2003249244A1 (en) | 2002-07-15 | 2004-02-02 | Combinatorx, Incorporated | Methods for the treatment of neoplasms |
| CA2494048A1 (en) | 2002-08-13 | 2004-02-19 | Warner-Lambert Company Llc | 4-hydroxyquinoline derivatives as matrix metalloproteinase inhibitors |
| NZ538420A (en) | 2002-08-16 | 2007-06-29 | Kinacia Pty Ltd | Inhibition of phosphoinositide 3-kinase beta |
| JP2005536553A (ja) | 2002-08-21 | 2005-12-02 | サイトキネティクス・インコーポレーテッド | 化合物、組成物および方法 |
| US20030139427A1 (en) | 2002-08-23 | 2003-07-24 | Osi Pharmaceuticals Inc. | Bicyclic pyrimidinyl derivatives and methods of use thereof |
| WO2004020599A2 (en) | 2002-08-29 | 2004-03-11 | Curis, Inc. | Hedgehog antagonists, methods and uses related thereto |
| GB0220319D0 (en) | 2002-09-02 | 2002-10-09 | Cancer Res Campaign Tech | Enzyme activated self-immolative nitrogen mustard drugs |
| AU2003266668A1 (en) | 2002-09-30 | 2004-04-23 | Banyu Pharmaceutical Co., Ltd. | 2-aminobenzimidazole derivative |
| US20040132732A1 (en) | 2002-10-21 | 2004-07-08 | Wei Han | Quinazolinones and derivatives thereof as factor Xa inhibitors |
| JP2004161716A (ja) | 2002-11-15 | 2004-06-10 | Takeda Chem Ind Ltd | Jnk阻害剤 |
| CN1281590C (zh) | 2002-11-27 | 2006-10-25 | 南京凯衡科贸有限公司 | 具有抑制血管生成活性的六员氨基酰胺类衍生物 |
| US7265131B2 (en) | 2002-12-20 | 2007-09-04 | Exelixis, Inc. | Isoquinolinone derivatives and their use as therapeutic agents |
| US7247736B2 (en) | 2002-12-23 | 2007-07-24 | 4Sc Ag | Method of identifying inhibitors of DHODH |
| WO2004056746A1 (en) | 2002-12-23 | 2004-07-08 | 4Sc Ag | Cycloalkene dicarboxylic acid compounds as anti-inflammatory, immunomodulatory and anti-proliferatory agents |
| US7365094B2 (en) | 2002-12-23 | 2008-04-29 | 4Sc Ag | Compounds as anti-inflammatory, immunomodulatory and anti-proliferatory agents |
| PT1583562E (pt) * | 2003-01-06 | 2011-09-19 | Angiochem Inc | Angiopep-1, compostos relacionados, e suas utilizações |
| FR2850022B1 (fr) | 2003-01-22 | 2006-09-08 | Centre Nat Rech Scient | Nouvelle utilisation de la mifepristone et de ses derives comme modulateurs de la voie de signalisation des proteines hedgehog et ses applications |
| WO2004069145A2 (en) | 2003-02-07 | 2004-08-19 | Dr. Reddy's Laboratories Ltd. | Anticancer compounds, process for their preparation and pharmaceutical compositions containing them |
| US20060135790A1 (en) | 2003-03-06 | 2006-06-22 | Hyett David J | Process for the preparation of an alpha-amino carbonyl compound |
| AR050920A1 (es) | 2003-03-07 | 2006-12-06 | Astrazeneca Ab | Enantiomeros de heterociclos fusionados seleccionados y usos de los mismos |
| US20040242568A1 (en) | 2003-03-25 | 2004-12-02 | Syrrx, Inc. | Dipeptidyl peptidase inhibitors |
| GB0306907D0 (en) | 2003-03-26 | 2003-04-30 | Angiogene Pharm Ltd | Boireductively-activated prodrugs |
| US7217794B2 (en) | 2003-04-02 | 2007-05-15 | Daiamed, Inc. | Compounds and methods for treatment of thrombosis |
| WO2004092123A2 (en) | 2003-04-10 | 2004-10-28 | Microbia, Inc. | Inhibitors of fungal invasion |
| AU2004240754B9 (en) | 2003-05-20 | 2008-10-23 | Novartis Ag | N-acyl nitrogen heterocycles as ligands of Peroxisome Proliferator-Activated Receptors |
| US7022850B2 (en) | 2003-05-22 | 2006-04-04 | Bristol-Myers Squibb Co. | Bicyclicpyrimidones and their use to treat diseases |
| US7345046B2 (en) | 2003-05-30 | 2008-03-18 | Chiron Corporation | Heteroaryl-fused pyrimidinyl compounds as anticancer agents |
| EP1646615B1 (en) | 2003-06-06 | 2009-08-26 | Vertex Pharmaceuticals Incorporated | Pyrimidine derivatives as modulators of atp-binding cassette transporters |
| WO2004113307A1 (ja) | 2003-06-18 | 2004-12-29 | Ube Industries, Ltd. | ピリミジン−4−オン化合物の製造方法 |
| US7429596B2 (en) | 2003-06-20 | 2008-09-30 | The Regents Of The University Of California | 1H-pyrrolo [2,3-D] pyrimidine derivatives and methods of use thereof |
| CA2572324A1 (en) | 2003-07-02 | 2005-01-13 | Warner-Lambert Company Llc | Combination of an allosteric inhibitor of matrix metalloproteinase-13 and a ligand to an alpha-2-delta receptor |
| AU2004257167B2 (en) | 2003-07-03 | 2012-03-29 | The Trustees Of The University Of Pennsylvania | Inhibition of Syk kinase expression |
| EP1646628A1 (en) | 2003-07-08 | 2006-04-19 | Novartis AG | Benzenesulfonylamino compounds and pharmaceutical compositions containing these compounds |
| JP2005035933A (ja) | 2003-07-15 | 2005-02-10 | Takeda Chem Ind Ltd | 含窒素縮合複素環化合物の製造方法 |
| WO2005013800A2 (en) | 2003-07-15 | 2005-02-17 | The Johns Hopkins University | Elevated hedgehog pathway activity in digestive system tumors, and methods of treating digestive system tumors having elevated hedgehog pathway activity |
| GB0317951D0 (en) | 2003-07-31 | 2003-09-03 | Trigen Ltd | Compounds |
| WO2005014532A1 (en) | 2003-08-08 | 2005-02-17 | Transtech Pharma, Inc. | Aryl and heteroaryl compounds, compositions and methods of use |
| US7208601B2 (en) | 2003-08-08 | 2007-04-24 | Mjalli Adnan M M | Aryl and heteroaryl compounds, compositions, and methods of use |
| WO2005016349A1 (en) | 2003-08-14 | 2005-02-24 | Icos Corporation | Methods of inhibiting leukocyte accumulation |
| WO2005016348A1 (en) | 2003-08-14 | 2005-02-24 | Icos Corporation | Method of inhibiting immune responses stimulated by an endogenous factor |
| US20050124637A1 (en) | 2003-08-15 | 2005-06-09 | Irm Llc | Compounds and compositions as inhibitors of receptor tyrosine kinase activity |
| US7390820B2 (en) | 2003-08-25 | 2008-06-24 | Amgen Inc. | Substituted quinolinone derivatives and methods of use |
| WO2005044181A2 (en) | 2003-09-09 | 2005-05-19 | Temple University-Of The Commonwealth System Of Higher Education | Protection of tissues and cells from cytotoxic effects of ionizing radiation by abl inhibitors |
| GB0321710D0 (en) | 2003-09-16 | 2003-10-15 | Novartis Ag | Organic compounds |
| GB0322409D0 (en) | 2003-09-25 | 2003-10-29 | Astrazeneca Ab | Quinazoline derivatives |
| US8067608B2 (en) | 2003-09-29 | 2011-11-29 | The Johns Hopkins University | Hedgehog pathway antagonists |
| WO2005032343A2 (en) | 2003-10-01 | 2005-04-14 | The Johns Hopkins University | Hedgehog signaling in prostate regeneration neoplasia and metastasis |
| WO2005042700A2 (en) | 2003-10-20 | 2005-05-12 | The Johns Hopkins University | Use of hedgehog pathway inhibitors in small-cell lung cancer |
| TWI344364B (en) | 2003-11-10 | 2011-07-01 | Synta Pharmaceuticals Corp | Fused heterocyclic compounds |
| US20070135454A1 (en) | 2003-11-14 | 2007-06-14 | Tracy Bayliss | Bicyclic pyrimidin-4(3h)-ones and analogues and derivatives thereof which modulate the function of the vanilloid-1-receptor(vr1) |
| WO2005049838A2 (en) | 2003-11-14 | 2005-06-02 | Yale University | Syk-targeted nucleic acid interference |
| EP1687309A1 (en) | 2003-11-17 | 2006-08-09 | Pfizer Products Inc. | Pyrrolopyrimidine compounds useful in treatment of cancer |
| CA2546932A1 (en) | 2003-11-25 | 2005-06-09 | Chiron Corporation | Quinazolinone compounds as anticancer agents |
| WO2005054202A1 (en) | 2003-11-25 | 2005-06-16 | Eli Lilly And Company | 7-phenyl-isoquinoline-5-sulfonylamino derivatives as inhibitors of akt (proteinkinase b) |
| WO2005061460A1 (en) | 2003-12-08 | 2005-07-07 | Cytokinetics, Inc. | Compounds, compositions, and methods |
| AU2004308974A1 (en) | 2003-12-22 | 2005-07-14 | Gilead Sciences, Inc. | Kinase inhibitor phosphonate conjugates |
| PL1699800T3 (pl) | 2003-12-23 | 2010-07-30 | Novartis Ag | Bicykliczne heterocykliczne inhibitory kinazy p-38 |
| US20050152940A1 (en) | 2003-12-23 | 2005-07-14 | Medtronic Vascular, Inc. | Medical devices to treat or inhibit restenosis |
| JPWO2005061707A1 (ja) | 2003-12-24 | 2007-07-12 | 協和醗酵工業株式会社 | 癌細胞のEg5阻害剤に対する感受性を判定する方法 |
| US20050239809A1 (en) | 2004-01-08 | 2005-10-27 | Watts Stephanie W | Methods for treating and preventing hypertension and hypertension-related disorders |
| WO2005070457A1 (en) | 2004-01-23 | 2005-08-04 | Seattle Genetics, Inc. | Melphalan prodrugs |
| WO2005074603A2 (en) | 2004-02-03 | 2005-08-18 | Abbott Laboratories | Aminobenzoxazoles as therapeutic agents |
| KR20070026385A (ko) | 2004-02-13 | 2007-03-08 | 반유 세이야꾸 가부시끼가이샤 | 축합환 4-옥소피리미딘 유도체 |
| US7650848B2 (en) | 2004-02-17 | 2010-01-26 | University Of Florida Research Foundation, Inc. | Surface topographies for non-toxic bioadhesion control |
| US9016221B2 (en) | 2004-02-17 | 2015-04-28 | University Of Florida Research Foundation, Inc. | Surface topographies for non-toxic bioadhesion control |
| US7117807B2 (en) | 2004-02-17 | 2006-10-10 | University Of Florida Research Foundation, Inc. | Dynamically modifiable polymer coatings and devices |
| US20050187418A1 (en) | 2004-02-19 | 2005-08-25 | Small Brooke L. | Olefin oligomerization |
| DK1718353T3 (da) | 2004-02-24 | 2020-02-03 | Microdose Therapeutx Inc | Apparat til lægemiddelafgivelse på basis af en syntetisk strålestrøm |
| US20070049593A1 (en) | 2004-02-24 | 2007-03-01 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
| JP2007523202A (ja) | 2004-02-24 | 2007-08-16 | ビオアクソン・テラプティーク・インコーポレーテッド | 4置換ピペリジン誘導体 |
| CA2557575A1 (en) | 2004-02-27 | 2005-09-15 | F. Hoffmann-La Roche Ag | Fused derivatives of pyrazole |
| AU2005230818B2 (en) | 2004-04-02 | 2010-11-25 | Osi Pharmaceuticals, Inc. | 6,6-bicyclic ring substituted heterobicyclic protein kinase inhibitors |
| US8057815B2 (en) | 2004-04-19 | 2011-11-15 | Portola Pharmaceuticals, Inc. | Methods of treatment with Syk inhibitors |
| US8273743B2 (en) | 2004-04-30 | 2012-09-25 | Curis, Inc. | Quinoxaline inhibitors of the hedgehog signalling |
| US9212149B2 (en) | 2004-04-30 | 2015-12-15 | Takeda Pharmaceutical Company Limited | Substituted 2-amidoquinazol-4-ones as matrix metalloproteinase-13 inhibitors |
| DE102004022897A1 (de) | 2004-05-10 | 2005-12-08 | Bayer Cropscience Ag | Azinyl-imidazoazine |
| JP2007537291A (ja) | 2004-05-13 | 2007-12-20 | イコス・コーポレイション | ヒトホスファチジルイノシトール3−キナーゼデルタの阻害剤としてのキナゾリノン |
| WO2005112935A1 (en) | 2004-05-13 | 2005-12-01 | Vanderbilt University | Phosphoinositide 3-kinase delta selective inhibitors for inhibiting angiogenesis |
| JP2008500338A (ja) | 2004-05-25 | 2008-01-10 | イコス・コーポレイション | 造血細胞の異常増殖を治療及び/又は予防する方法 |
| WO2005120511A1 (en) | 2004-06-04 | 2005-12-22 | Icos Corporation | Methods for treating mast cell disorders |
| GB0412467D0 (en) | 2004-06-04 | 2004-07-07 | Astrazeneca Ab | Chemical compounds |
| US20050282834A1 (en) | 2004-06-09 | 2005-12-22 | Fady Malik | Compositions, devices and methods for treating cardiovascular disease |
| US7375102B2 (en) | 2004-06-28 | 2008-05-20 | Amgen Sf, Llc | Tetrahydroquinazolin-4(3H)-one-related and tetrahydropyrido[2,3-D]pyrimidin-4(3H)-one-related compounds, compositions and methods for their use |
| US7939538B2 (en) | 2004-06-28 | 2011-05-10 | Amgen Inc. | Compounds, compositions and methods for prevention and treatment of inflammatory and immunoregulatory disorders and diseases |
| WO2006023381A1 (en) | 2004-08-16 | 2006-03-02 | Taigen Biotechnology | Pyrimidinone compounds |
| US20060041128A1 (en) | 2004-08-18 | 2006-02-23 | Astrazeneca Ab | Selected fused heterocyclics and uses thereof |
| MX2007001953A (es) | 2004-08-18 | 2007-05-09 | Astrazeneca Ab | Enantiomeros de pirimidonas fusionadas seleccionadas y usos en el tratamiento y prevencion del cancer. |
| US7230004B2 (en) | 2004-08-27 | 2007-06-12 | Infinity Discovery, Inc. | Cyclopamine analogues and methods of use thereof |
| EP1786265A4 (en) | 2004-08-30 | 2009-08-19 | Smithkline Beecham Corp | NEW COMPOSITIONS AND PROCESSING METHODS |
| NZ553087A (en) | 2004-08-31 | 2010-12-24 | Astrazeneca Ab | Quinazolinone derivatives and their use as B-raf inhibitors |
| UA96565C2 (ru) | 2004-09-02 | 2011-11-25 | Дженентек, Инк. | Пиридильные ингибиторы хеджхоговской передачи сигнала |
| JP2008512457A (ja) | 2004-09-09 | 2008-04-24 | アメリカ合衆国 | A3及びa1アデノシン受容体作用薬としてのプリン誘導体 |
| GB0420722D0 (en) | 2004-09-17 | 2004-10-20 | Addex Pharmaceuticals Sa | Novel allosteric modulators |
| WO2006038865A1 (en) | 2004-10-01 | 2006-04-13 | Betagenon Ab | Nucleotide derivatives for the treatment of type 2 diabetes and other disorders |
| CN101083996A (zh) | 2004-10-28 | 2007-12-05 | Irm责任有限公司 | 作为hedgehog途径调节剂的化合物和组合物 |
| US7622451B2 (en) | 2004-11-03 | 2009-11-24 | University Of Kansas | Novobiocin analogues as neuroprotective agents and in the treatment of autoimmune disorders |
| US8212011B2 (en) | 2004-11-03 | 2012-07-03 | University Of Kansas | Novobiocin analogues |
| US8212012B2 (en) | 2004-11-03 | 2012-07-03 | University Of Kansas | Novobiocin analogues having modified sugar moieties |
| AU2005301957B2 (en) | 2004-11-03 | 2012-02-23 | Department Of Health And Human Services | Novobiocin analogues as anticancer agents |
| GB0425035D0 (en) | 2004-11-12 | 2004-12-15 | Novartis Ag | Organic compounds |
| WO2006068760A2 (en) | 2004-11-19 | 2006-06-29 | The Regents Of The University Of California | Anti-inflammatory pyrazolopyrimidines |
| WO2006065479A2 (en) | 2004-11-23 | 2006-06-22 | Ptc Therapeutics, Inc. | Substituted phenols as active agents inhibiting vegf production |
| US20060156485A1 (en) | 2005-01-14 | 2006-07-20 | The Procter & Gamble Company | Keratin dyeing compounds, keratin dyeing compositions containing them, and use thereof |
| JP2008527039A (ja) | 2005-01-19 | 2008-07-24 | メルク エンド カムパニー インコーポレーテッド | 有糸分裂キネシン阻害剤としてのフッ素化アミノアルキル−4−オキソ−3,4−ジヒドロピリド[3,4−d]ピリジン |
| WO2006078598A2 (en) | 2005-01-19 | 2006-07-27 | Merck & Co., Inc. | Mitotic kinesin inhibitors |
| US20070161644A1 (en) | 2005-01-25 | 2007-07-12 | Stockwell Brent R | Erastin analogs and uses thereof |
| EP1848698B1 (en) | 2005-01-25 | 2013-03-13 | Prolexys Pharmaceuticals, Inc. | Quinoxaline derivatives as antitumor agents |
| GB0501999D0 (en) | 2005-02-01 | 2005-03-09 | Sentinel Oncology Ltd | Pharmaceutical compounds |
| WO2006089106A2 (en) | 2005-02-17 | 2006-08-24 | Icos Corporation | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| US7579348B2 (en) | 2005-02-25 | 2009-08-25 | Pgxhealth, Llc | Derivatives of 8-substituted xanthines |
| EP1854793A4 (en) | 2005-02-28 | 2011-01-26 | Japan Tobacco Inc | NEW AMINOPYRIDEIN COMPOUND WITH SYK-HEMDERING EFFECT |
| US20090124654A1 (en) | 2005-03-01 | 2009-05-14 | Mjalli Adnan M M | Aryl and Heteroaryl Compounds, Compositions, Methods of Use |
| US7872050B2 (en) | 2005-03-14 | 2011-01-18 | Yaupon Therapeutics Inc. | Stabilized compositions of volatile alkylating agents and methods of using thereof |
| JP2006265107A (ja) | 2005-03-22 | 2006-10-05 | Kyowa Hakko Kogyo Co Ltd | 緑内障治療剤 |
| GB0506147D0 (en) | 2005-03-24 | 2005-05-04 | Merck Sharp & Dohme | Therapeutic agents |
| RU2405783C2 (ru) | 2005-03-25 | 2010-12-10 | Тиботек Фармасьютикалз Лтд | Гетеробициклические ингибиторы вируса гепатита с (hcv) |
| CN101189234B (zh) | 2005-03-25 | 2011-08-17 | 泰博特克药品有限公司 | Hcv的杂二环抑制剂 |
| JP2008534565A (ja) | 2005-03-31 | 2008-08-28 | アストラゼネカ アクチボラグ | Tie2阻害活性のあるアミノピリミジン誘導体 |
| RU2007140733A (ru) | 2005-04-06 | 2009-05-20 | Айрм Ллк (Bm) | Диариламинсодержащие соединения и композиции, и их применение в качестве модуляторов ядерных рецепторов стероидного гормона |
| KR100781704B1 (ko) | 2005-04-20 | 2007-12-03 | 에스케이케미칼주식회사 | 피리딘 유도체와 이의 제조방법, 및 이를 포함하는약제조성물 |
| AU2006239677A1 (en) | 2005-04-25 | 2006-11-02 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Use of compounds to enhance processivity of telomerase |
| US20090280153A1 (en) | 2005-05-10 | 2009-11-12 | Angiotech International Ag | electrical devices, anti-scarring agents, and therapeutic compositions |
| WO2006121522A2 (en) | 2005-05-10 | 2006-11-16 | Angiotech International Ag | Implantable sensors and pumps, anti-scarring agents, and therapeutic compositions |
| WO2006135479A2 (en) | 2005-05-10 | 2006-12-21 | Angiotech International Ag | Anti-scarring agents, therapeutic compositions, and use thereof |
| US20090298856A1 (en) | 2005-05-11 | 2009-12-03 | Rebecca Elizabeth Brown | 2,3 Substituted fused bicyclic pyrimidin-4(3H)-ones modulating the function of the vanilliod-1receptor (VR1) |
| GB0509573D0 (en) | 2005-05-11 | 2005-06-15 | Merck Sharp & Dohme | Therapeutic compounds |
| DE102005024017A1 (de) | 2005-05-25 | 2006-11-30 | Merck Patent Gmbh | Chinazolinone |
| CN100526315C (zh) | 2005-06-16 | 2009-08-12 | 浙江医药股份有限公司新昌制药厂 | N2-喹啉或异喹啉取代的嘌呤衍生物及其制备方法和其用途 |
| SG10201504801VA (en) | 2005-06-22 | 2015-07-30 | Chemocentryx Inc | Azaindazole compounds and methods of use |
| CA2612585A1 (en) | 2005-06-27 | 2007-01-04 | Amgen Inc. | Anti-inflammatory aryl nitrile compounds |
| WO2007008541A2 (en) | 2005-07-08 | 2007-01-18 | Kalypsys, Inc. | Cellular cholesterol absorption modifiers |
| JP2009500437A (ja) | 2005-07-11 | 2009-01-08 | デブジェン エヌブイ | キナーゼ阻害剤としてのアミド誘導体 |
| WO2007006546A1 (en) | 2005-07-11 | 2007-01-18 | Devgen N.V. | Amide derivatives as kinase inhibitors |
| WO2007015877A2 (en) | 2005-07-20 | 2007-02-08 | Kalypsys, Inc. | Inhibitors of p38 kinase and methods of treating inflammatory disorders |
| US20070017915A1 (en) | 2005-07-22 | 2007-01-25 | Weder Donald E | Collapsible and/or erectable substantially egg-shaped container |
| GB0516723D0 (en) | 2005-08-15 | 2005-09-21 | Novartis Ag | Organic compounds |
| MX2008002385A (es) | 2005-08-25 | 2008-03-18 | Hoffmann La Roche | Inhibidores de proteinas cinasas activads por mitogenos p38 y metodos para utilizarlos. |
| US20070049591A1 (en) | 2005-08-25 | 2007-03-01 | Kalypsys, Inc. | Inhibitors of MAPK/Erk Kinase |
| CA2620414A1 (en) | 2005-08-26 | 2007-03-01 | Methylgene Inc. | Benzodiazepine and benzopiperazine analog inhibitors of histone deacetylase |
| US20080318942A1 (en) | 2005-09-01 | 2008-12-25 | Bioagency Ag | Fredericamycin Derivatives |
| US20070072897A1 (en) | 2005-09-29 | 2007-03-29 | Wyeth | Phenylaminopropanol derivatives and methods of their use |
| WO2007056155A1 (en) | 2005-11-03 | 2007-05-18 | Chembridge Research Laboratories, Inc. | Heterocyclic compounds as tyrosine kinase modulators |
| US7893260B2 (en) | 2005-11-04 | 2011-02-22 | Hydra Biosciences, Inc. | Substituted quinazolin-4-one compounds for antagonizing TRPV3 function |
| WO2007054623A2 (en) | 2005-11-11 | 2007-05-18 | Licentia Oy | Mammalian hedgehog signaling inhiabitors |
| JP5225857B2 (ja) | 2005-11-14 | 2013-07-03 | ジェネンテック,インコーポレイティド | ヘッジホッグシグナル伝達のビスアミド阻害剤 |
| ES2365869T3 (es) | 2005-11-17 | 2011-10-11 | OSI Pharmaceuticals, LLC | COMPUESTOS BICÍCLICOS FUSIONADOS INHIBIDORES DE LA mTOR. |
| US20090088452A1 (en) | 2005-11-22 | 2009-04-02 | Coleman Paul J | Indole Orexin Receptor Antagonists |
| US20070265231A1 (en) | 2005-11-30 | 2007-11-15 | Michael Hofmann | Hydrazide Compounds for Combating Animal Pests |
| EP1956908A2 (en) | 2005-12-08 | 2008-08-20 | Cytokinetics, Inc. | Certain compositions and methods of treatment |
| JP2009520028A (ja) | 2005-12-19 | 2009-05-21 | オーエスアイ・ファーマスーティカルズ・インコーポレーテッド | Igfr抑制剤および抗癌剤の併用 |
| US7572809B2 (en) | 2005-12-19 | 2009-08-11 | Hoffmann-La Roche Inc. | Isoquinoline aminopyrazole derivatives |
| US20090170834A1 (en) | 2005-12-22 | 2009-07-02 | Prolexys Pharmaceuticals, Inc. | Fused Pyrimidones and Thiopyrimidones, and Uses Thereof |
| WO2007076055A2 (en) | 2005-12-22 | 2007-07-05 | Entremed, Inc. | Compositions and methods comprising proteinase activated receptor antagonists |
| EP1973917B1 (en) | 2005-12-29 | 2015-06-10 | AbbVie Inc. | Protein kinase inhibitors |
| WO2007087549A2 (en) | 2006-01-25 | 2007-08-02 | Smithkline Beecham Corporation | Chemical compounds |
| WO2007089669A2 (en) | 2006-01-26 | 2007-08-09 | Wyeth | Processes for the preparation of compounds which modulate cell proliferation |
| MX2008011220A (es) | 2006-03-07 | 2008-09-11 | Array Biopharma Inc | Compuestos de pirazol heterobiciclicos y metodos de uso. |
| JP2009530342A (ja) | 2006-03-20 | 2009-08-27 | エフ.ホフマン−ラ ロシュ アーゲー | Btkおよびsyk蛋白キナーゼを阻害する方法 |
| TW200800997A (en) | 2006-03-22 | 2008-01-01 | Astrazeneca Ab | Chemical compounds |
| HRP20160990T1 (hr) | 2006-03-24 | 2016-10-07 | Biogen Hemophilia Inc. | Pc5 kao enzim obrade faktor ix propeptida |
| EP2007373A4 (en) | 2006-03-29 | 2012-12-19 | Foldrx Pharmaceuticals Inc | INHIBITION OF ALPHA SYNUCLEINE TOXICITY |
| GB2453058A (en) | 2006-04-04 | 2009-03-25 | Univ California | Kinase antagonists |
| EP2007736A1 (en) | 2006-04-05 | 2008-12-31 | AstraZeneca AB | Substituted quinazolines with anti-cancer activity |
| US20090170849A1 (en) | 2006-04-05 | 2009-07-02 | Astrazeneca Ab | Quinazolinone derivatives having b-raf inhibitory activity |
| EP2010170A2 (en) | 2006-04-14 | 2009-01-07 | Novartis AG | Use of biarylcarboxamides in the treatment of hedgehog pathway-related disorders |
| WO2007121453A2 (en) | 2006-04-17 | 2007-10-25 | The Regents Of The University Of California | 2-hydroxy-1-oxo 1,2 dihydro isoquinoline chelating agents |
| EP2010504A1 (en) | 2006-04-18 | 2009-01-07 | AstraZeneca AB | Quinazolin-4-one derivatives, process for their preparation and pharmaceutical compositions containing them |
| NZ572072A (en) | 2006-04-20 | 2011-09-30 | Janssen Pharmaceutica Nv | Inhibitors of c-fms kinase |
| JP5373599B2 (ja) | 2006-04-21 | 2013-12-18 | ノバルティス アーゲー | アデノシンa2a受容体アゴニストとして使用するためのプリン誘導体 |
| GB0607950D0 (en) | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| GB0607948D0 (en) | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| JP2009536620A (ja) | 2006-04-25 | 2009-10-15 | アステックス、セラピューティックス、リミテッド | 医薬組み合わせ物 |
| JP2009536161A (ja) | 2006-04-25 | 2009-10-08 | アステックス、セラピューティックス、リミテッド | 医薬化合物 |
| DE102006020327A1 (de) | 2006-04-27 | 2007-12-27 | Bayer Healthcare Ag | Heterocyclisch substituierte, anellierte Pyrazol-Derivate und ihre Verwendung |
| UA93548C2 (uk) | 2006-05-05 | 2011-02-25 | Айерем Елелсі | Сполуки та композиції як модулятори хеджхогівського сигнального шляху |
| WO2007129226A2 (en) | 2006-05-09 | 2007-11-15 | New Era Biotech Ltd | Use of syk tyrosine kinase inhibitors for the treatment of cell proliferative disorders |
| KR100722686B1 (ko) | 2006-05-09 | 2007-05-30 | 주식회사 비에스이 | 부가적인 백 챔버를 갖고 기판에 음향홀이 형성된 실리콘콘덴서 마이크로폰 |
| CN101448827A (zh) | 2006-05-22 | 2009-06-03 | 阿斯利康(瑞典)有限公司 | 吲哚衍生物 |
| GB0610242D0 (en) | 2006-05-23 | 2006-07-05 | Novartis Ag | Organic compounds |
| GB0610317D0 (en) | 2006-05-24 | 2006-07-05 | Medical Res Council | Antiparasitic compounds and compositions |
| DE602006018877D1 (de) | 2006-05-24 | 2011-01-27 | Guardant S R L | Alkalisiertes Lokalanästhetikum im Beutel |
| EP2044086A2 (en) | 2006-06-30 | 2009-04-08 | Janssen Pharmaceutica N.V. | Thiazolopyrimidine modulators of trpv1 |
| WO2008011109A2 (en) | 2006-07-20 | 2008-01-24 | Amgen Inc. | Substituted pyridone compounds and methods of use |
| WO2008014373A2 (en) | 2006-07-27 | 2008-01-31 | Smithkline Beecham Corporation | Hspa1a as a marker for sensitivity to ksp inhibitors |
| WO2008013987A2 (en) | 2006-07-27 | 2008-01-31 | Prolexys Pharmaceuticals, Inc. | N-alkyl substituted piperazinylmethylquinazolinones and azepanylmethylquinazolinones |
| MX2009000864A (es) | 2006-07-28 | 2009-02-03 | Novartis Ag | Quinazolinas 2,4-sustituidas como inhibidores de cinasa de lipido. |
| EP2061764B1 (en) | 2006-08-22 | 2014-07-02 | Technion Research & Development Foundation LTD. | Heterocyclic derivatives binding to the peripheral-type benzodiazepine receptor (pbr) |
| JP2010501593A (ja) | 2006-08-24 | 2010-01-21 | セレネックス, インコーポレイテッド | イソキノリン、キナゾリンおよびフタラジン誘導体 |
| WO2008025755A1 (de) | 2006-09-01 | 2008-03-06 | Basf Se | Verwendung von n-haltigen heterozyklen in dermokosmetika |
| US7956064B2 (en) | 2006-09-01 | 2011-06-07 | Cylene Pharmaceuticals, Inc. | Fused tricyclic compounds as serine-threonine protein kinase and PARP modulators |
| EP1903044A1 (en) | 2006-09-14 | 2008-03-26 | Novartis AG | Adenosine Derivatives as A2A Receptor Agonists |
| JP5306818B2 (ja) | 2006-10-18 | 2013-10-02 | 武田薬品工業株式会社 | 縮合複素環化合物 |
| EP2431388A1 (en) | 2006-10-31 | 2012-03-21 | Government of the United States of America, Represented by the Secretary, Department of Health and Human Services | Smoothened polypeptides and methods of use |
| RU2333211C1 (ru) | 2006-11-01 | 2008-09-10 | Институт физиологически активных веществ Российской Академии наук | N,n`-замещенные 3, 7-диазабицикло[3.3.1]нонаны, обладающие фармакологической активностью, фармацевтические композиции на их основе и способ их применения |
| US7772180B2 (en) | 2006-11-09 | 2010-08-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US20100216820A1 (en) | 2006-11-13 | 2010-08-26 | White Stephen L | Thienopyrimidiones for treatment of inflammatory disorders and cancers |
| MY150216A (en) | 2006-11-20 | 2013-12-13 | Novartis Ag | Salts and crystal forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydroimidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| WO2008063625A2 (en) | 2006-11-20 | 2008-05-29 | Adolor Corporation | Pyridine compounds and methods of their use |
| US8658637B2 (en) | 2006-12-06 | 2014-02-25 | Boehringer Ingelheim International Gmbh | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions and uses thereof |
| EP2103619A4 (en) | 2006-12-14 | 2011-11-23 | Daiichi Sankyo Co Ltd | IMIDAZOTHIAZOLDERIVATE |
| JP5178738B2 (ja) | 2006-12-20 | 2013-04-10 | メルク・シャープ・アンド・ドーム・コーポレーション | 新規なjnk阻害剤 |
| CA2674525A1 (en) | 2006-12-22 | 2008-07-03 | Albert Einstein College Of Medicine Of Yeshiva University | Azetidine analogues of nucleosidase and phosphorylase inhibitors |
| TWI433674B (zh) | 2006-12-28 | 2014-04-11 | Infinity Discovery Inc | 環杷明(cyclopamine)類似物類 |
| US8389544B2 (en) | 2007-01-17 | 2013-03-05 | The Hong Kong University Of Science And Technology | Isoquinolone compounds as subtype-selective agonists for melatonin receptors MT1 and MT2 |
| US20130116277A1 (en) | 2007-01-22 | 2013-05-09 | James T. Dalton | Aldo-keto reductase subfamily 1c3 (akr1c3) inhibitors |
| US20170014401A1 (en) | 2007-01-22 | 2017-01-19 | Gtx, Inc. | Nuclear receptor binding agents |
| EA026578B1 (ru) | 2007-01-22 | 2017-04-28 | ДЖиТиЭкс, ИНК. | Вещества, связывающие ядерные рецепторы |
| EA200900983A1 (ru) | 2007-01-26 | 2010-02-26 | Айрм Ллк | Соединения и композиции в качестве ингибиторов киназы |
| CL2008000252A1 (es) | 2007-01-29 | 2008-03-14 | Xenon Pharmaceuticals Inc | Compuestos derivados de quinazolinona o pirimidinona; composicion farmaceutica que comprende a dichos compuestos; y su uso para tratar enfermedades mediadas por los canales de calcio, tales como dolor, depresion, enfermedades cardiovasculares, respir |
| RU2009137014A (ru) | 2007-03-07 | 2011-04-20 | Инфинити Дискавери, Инк. (Us) | Гетероциклические аналоги циклопамина и способы их применения |
| WO2008109829A1 (en) | 2007-03-07 | 2008-09-12 | Infinity Pharmaceuticals, Inc. | Cyclopamine lactam analogs and methods of use thereof |
| US20090306625A1 (en) | 2007-03-09 | 2009-12-10 | Angiodynamics, Inc. | Treatment systems and methods for renal-related diseases |
| CN101686952A (zh) | 2007-03-12 | 2010-03-31 | Vm生物医药公司 | 新型钙离子通道调节剂 |
| NZ598251A (en) | 2007-03-14 | 2013-06-28 | Exelixis Patent Co Llc | 2-Arylamino-pyridopyrimidines and their uses, particularly in the treatment of cancer |
| MX2009009873A (es) | 2007-03-15 | 2009-09-24 | Novartis Ag | Compuestos organicos y sus usos. |
| WO2008118454A2 (en) | 2007-03-23 | 2008-10-02 | Amgen Inc. | Derivatives of quinoline or benzopyrazine and their uses for the treatment of (inter alia) inflammatory diseases, autoimmune diseases or various kinds of cancer |
| PL2139882T3 (pl) | 2007-03-23 | 2014-05-30 | Amgen Inc | 3-podstawione pochodne chinoliny lub chinoksaliny i ich zastosowanie jako inhibitorów 3-kinazy fosfatydyloinozytolu (pi3k) |
| MX2009009968A (es) | 2007-03-23 | 2009-10-08 | Amgen Inc | Compuestos heterociclicos y sus usos. |
| WO2008156513A2 (en) | 2007-03-29 | 2008-12-24 | University Of Connecticut | Methods to protect skeletal muscle against injury |
| EP1977765A1 (en) | 2007-04-03 | 2008-10-08 | Diatos | Peptide prodrugs |
| CA2683578A1 (en) | 2007-04-13 | 2008-10-23 | Sanofi-Aventis | A transition metal catalyzed synthesis of n-aminoindoles |
| WO2008125014A1 (fr) | 2007-04-13 | 2008-10-23 | Institute Of Pharmacology And Toxicology Academy Of Military Medical Sciences P.L.A. | Composés d'urée, leurs procédés de préparation et leurs utilisations pharmaceutiques |
| WO2008131354A2 (en) | 2007-04-20 | 2008-10-30 | The Curators Of The University Of Missouri | Phytoestrogens as regulators of hedgehog signaling and methods of their use in cancer treatment |
| JP2010163361A (ja) | 2007-04-27 | 2010-07-29 | Dainippon Sumitomo Pharma Co Ltd | キノリン誘導体 |
| CA2685753A1 (en) | 2007-05-09 | 2008-11-20 | Neuromed Pharmaceuticals Ltd. | Bicyclic pyrimidine derivatives as calcium channel blockers |
| US7960353B2 (en) | 2007-05-10 | 2011-06-14 | University Of Kansas | Novobiocin analogues as neuroprotective agents and in the treatment of autoimmune disorders |
| CN101742911A (zh) | 2007-05-17 | 2010-06-16 | 科泰克斯药品公司 | 用于增强谷氨酸能突触反应的二取代的酰胺 |
| TW200902016A (en) | 2007-05-22 | 2009-01-16 | Taigen Biotechnology Co Ltd | Kinesin inhibitors |
| CN101743008A (zh) | 2007-05-23 | 2010-06-16 | 西佳技术公司 | 用于治疗或预防登革热感染的抗病毒药 |
| WO2008153701A1 (en) | 2007-05-24 | 2008-12-18 | Schering Corporation | Compounds for inhibiting ksp kinesin activity |
| US9603848B2 (en) | 2007-06-08 | 2017-03-28 | Senomyx, Inc. | Modulation of chemosensory receptors and ligands associated therewith |
| US7928111B2 (en) | 2007-06-08 | 2011-04-19 | Senomyx, Inc. | Compounds including substituted thienopyrimidinone derivatives as ligands for modulating chemosensory receptors |
| US20110217300A1 (en) | 2007-06-22 | 2011-09-08 | Arqule, Inc. | Quinazolinone Compounds and Methods of Use Thereof |
| AU2008268613A1 (en) | 2007-06-22 | 2008-12-31 | Arqule, Inc. | Quinazolinone compounds and methods of use thereof |
| US20110123486A1 (en) | 2007-06-25 | 2011-05-26 | Prolexys Pharmaceuticals, Inc. | Methods of treating multiple myeloma and resistant cancers |
| BRPI0812970A2 (pt) | 2007-06-25 | 2019-09-24 | Endocyte Inc | conjugados contendo espaçadores hidrofílicos |
| ATE538120T1 (de) | 2007-06-26 | 2012-01-15 | Sanofi Sa | Regioselektive, metallkatalysierte synthese annelierter benzimidazole und azabenzimidazole |
| EP2170274A1 (en) | 2007-07-02 | 2010-04-07 | Technion Research and Development Foundation, Ltd. | Compositions, articles and methods comprising tspo ligands for preventing or reducing tobacco-associated damage |
| WO2009006577A2 (en) | 2007-07-03 | 2009-01-08 | The Regents Of The University Of Michigan | Compositions and methods for inhibiting ezh2 |
| RU2345996C1 (ru) | 2007-07-17 | 2009-02-10 | Андрей Александрович Иващенко | Аннелированные азагетероциклические амиды, включающие пиримидиновый фрагмент, способ их получения и применения |
| JP2010535201A (ja) | 2007-07-31 | 2010-11-18 | シェーリング コーポレイション | 抗癌治療としての抗有糸分裂薬およびオーロラキナーゼ阻害薬 |
| WO2009018811A1 (de) | 2007-08-06 | 2009-02-12 | Schebo Biotech Ag | Neue pharmazeutika, verfahren zu ihrer herstellung und ihre verwendung in der medizinischen therapie |
| US20110081317A1 (en) | 2007-08-08 | 2011-04-07 | University Of Rochester | Enhancing Gene Transfer |
| EP2190466A4 (en) | 2007-08-10 | 2011-12-21 | Burnham Inst Medical Research | Tissue-specific alkaline phosphatase (TNAP) activators and their use |
| MX2010001784A (es) | 2007-08-13 | 2010-03-15 | Metabasis Therapeutics Inc | Activadores novedosos de la glucocinasa. |
| WO2009029617A1 (en) | 2007-08-27 | 2009-03-05 | Kalypsys, Inc. | Diarylamine-substituted quinolones useful as inducible nitric oxide synthase inhibitors |
| US9745278B2 (en) | 2007-09-10 | 2017-08-29 | Boston Biomedical, Inc. | Group of STAT3 pathway inhibitors and cancer stem cell pathway inhibitors |
| WO2009044707A1 (ja) | 2007-10-03 | 2009-04-09 | Riken | ニトロトリアゾール誘導体、およびそれを用いる化合物の製造方法 |
| KR20100089082A (ko) | 2007-10-15 | 2010-08-11 | 아스트라제네카 아베 | 조합 059 |
| JP2011503193A (ja) | 2007-11-13 | 2011-01-27 | アイコス、コーポレーション | ヒトホスファチジルイノシトール3−キナーゼδの阻害剤 |
| EP2225223B1 (de) | 2007-11-22 | 2017-01-11 | Boehringer Ingelheim International GmbH | Organische verbindungen |
| US20090163481A1 (en) | 2007-12-13 | 2009-06-25 | Murphy Brian J | Ppar-delta ligands and methods of their use |
| EP2070932A1 (en) | 2007-12-13 | 2009-06-17 | 4Sc Ag | Pyrazolotriazines |
| JP5620821B2 (ja) | 2007-12-13 | 2014-11-05 | シエナ ビオテク ソシエタ ペル アチオニSiena Biotech S.P.A. | ヘッジホッグ経路拮抗薬およびその治療用途 |
| ES2587738T3 (es) | 2007-12-21 | 2016-10-26 | Ucb Biopharma Sprl | Derivados de quinoxalina y quinolina como inhibidores de quinasa |
| JP2011507910A (ja) | 2007-12-21 | 2011-03-10 | ユニバーシティー オブ ロチェスター | 真核生物の寿命を変更するための方法 |
| US7960397B2 (en) | 2007-12-28 | 2011-06-14 | Institute Of Experimental Botany, Academy Of Sciences Of The Czech Republic | 6,9-disubstituted purine derivatives and their use as cosmetics and cosmetic compositions |
| US7894450B2 (en) | 2007-12-31 | 2011-02-22 | Nortel Network, Ltd. | Implementation of VPNs over a link state protocol controlled ethernet network |
| AU2009204487C1 (en) | 2008-01-04 | 2014-10-16 | Intellikine, Llc | Certain chemical entities, compositions and methods |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| JP2011509305A (ja) | 2008-01-09 | 2011-03-24 | ピージーエックスヘルス、リミテッド、ライアビリティー、カンパニー | A2arアゴニストによる神経障害性疼痛の髄腔内治療 |
| WO2009088086A1 (ja) | 2008-01-10 | 2009-07-16 | Asahi Glass Company, Limited | ガラス、発光装置用の被覆材および発光装置 |
| WO2009089598A2 (en) | 2008-01-18 | 2009-07-23 | Katholieke Universiteit Leuven | Msmb-gene methylation based diagnosis, staging and prognosis of prostate cancer |
| CN101925606A (zh) | 2008-01-24 | 2010-12-22 | Ucb医药有限公司 | 包含环丁氧基的化合物 |
| WO2009097233A1 (en) | 2008-01-28 | 2009-08-06 | Schering Corporation | Imidazopyrazines as protein kinase inhibitors |
| CA2714516A1 (en) | 2008-02-07 | 2009-08-13 | Gilead Palo Alto, Inc. | Abca-1 elevating compounds and the use thereof |
| WO2009100406A2 (en) | 2008-02-07 | 2009-08-13 | Synta Pharmaceuticals Corp. | Topical formulations for the treatment of psoriasis |
| TWI444384B (zh) | 2008-02-20 | 2014-07-11 | Gilead Sciences Inc | 核苷酸類似物及其在治療惡性腫瘤上的用途 |
| CN101531638B (zh) | 2008-03-13 | 2011-12-28 | 中国科学院广州生物医药与健康研究院 | 用作雌激素相关受体调节剂的化合物及其应用 |
| WO2009114874A2 (en) | 2008-03-14 | 2009-09-17 | Intellikine, Inc. | Benzothiazole kinase inhibitors and methods of use |
| JP5547099B2 (ja) | 2008-03-14 | 2014-07-09 | インテリカイン, エルエルシー | キナーゼ阻害剤および使用方法 |
| CA2716856C (en) | 2008-03-20 | 2013-02-19 | Amgen Inc. | Aurora kinase modulators and method of use |
| MX2010010525A (es) | 2008-03-24 | 2010-10-25 | Novartis Ag | Inhibidores de metaloproteasa de matriz basados en aril-sulfonamida. |
| MX2010010659A (es) | 2008-03-31 | 2010-10-26 | Genentech Inc | Compuestos de benzopirano y benzoxepina, inhibidores de fosfoinoinositida 3-cinasas y metodos para utilizarlos. |
| FR2929851B1 (fr) | 2008-04-09 | 2012-11-30 | Centre Nat Rech Scient | Molecules inhibant une voie metabolique impliquant la proteine tyrosine kinase syk et procede d'identification de ces molecules |
| ES2546502T3 (es) | 2008-04-16 | 2015-09-24 | Portola Pharmaceuticals, Inc. | 2,6-Diamino-pirimidin-5-il-carboxamidas como inhibidores de syk o JAK quinasas |
| GB0808282D0 (en) | 2008-05-07 | 2008-06-11 | Medical Res Council | Compounds for use in stabilizing p53 mutants |
| US8148408B2 (en) | 2008-05-09 | 2012-04-03 | Abbott Laboratories | Selective substituted pyridine ligands for neuronal nicotinic receptors |
| CA2721851A1 (en) | 2008-05-30 | 2009-12-03 | Genentech, Inc. | Purine pi3k inhibitor compounds and methods of use |
| CA2727196A1 (en) | 2008-06-10 | 2009-12-17 | Novartis Ag | Organic compounds |
| US20090312406A1 (en) | 2008-06-12 | 2009-12-17 | Hsing-Pang Hsieh | Coumarin compounds and their use for treating viral infection |
| WO2010006072A2 (en) | 2008-07-08 | 2010-01-14 | The Regents Of The University Of California | Mtor modulators and uses thereof |
| MX2011000216A (es) | 2008-07-08 | 2011-03-29 | Intellikine Inc | Inhibidores de cinasa y metodos para su uso. |
| CA2730610A1 (en) | 2008-07-16 | 2010-01-21 | Schering Corporation | Bicyclic heterocycle derivatives and their use as gpcr modulators |
| UY31984A (es) | 2008-07-16 | 2010-02-26 | Boehringer Ingelheim Int | DERIVADOS DE 1-(3,4-difluorobencil)-6-oxo-1,6-dihidropirimidin-5-carboxamidas N-sustituidas y de 2-(3,4-difluorobencil)-3-oxo-2,3-dihidro-1H-pirazol-4-carboxamidas N-sustituidas. |
| US8450344B2 (en) | 2008-07-25 | 2013-05-28 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| WO2010014739A2 (en) | 2008-07-29 | 2010-02-04 | Kalypsys, Inc. | Heterocyclic modulators of tgr5 |
| AU2009282480B2 (en) | 2008-08-11 | 2015-05-07 | Children's Medical Center Corporation | Halofuginone analogs for inhibition of tRNA synthetases and uses thereof |
| US20100099871A1 (en) | 2008-08-15 | 2010-04-22 | Miller Dwight W | Anti-psychotic compounds and pharmaceutical compositions thereof |
| US8513295B2 (en) | 2008-09-23 | 2013-08-20 | Georgetown University | Viral and fungal inhibitors |
| EP2346508B1 (en) | 2008-09-26 | 2016-08-24 | Intellikine, LLC | Heterocyclic kinase inhibitors |
| WO2010046780A2 (en) | 2008-10-22 | 2010-04-29 | Institut Pasteur Korea | Anti viral compounds |
| WO2010051550A1 (en) | 2008-10-31 | 2010-05-06 | University Of Rochester | Methods of diagnosing and treating fibrosis |
| BRPI0921433A2 (pt) | 2008-10-31 | 2017-06-06 | Abbott Biotherapeutics Corp | uso de anticorpos anti-cs1 para o tratamento de linfomas raros |
| US8476282B2 (en) | 2008-11-03 | 2013-07-02 | Intellikine Llc | Benzoxazole kinase inhibitors and methods of use |
| WO2010056758A1 (en) | 2008-11-12 | 2010-05-20 | Yangbo Feng | Quinazoline derivatives as kinase inhibitors |
| US20110269779A1 (en) | 2008-11-18 | 2011-11-03 | Intellikine, Inc. | Methods and compositions for treatment of ophthalmic conditions |
| EP2752189B1 (en) * | 2008-11-22 | 2016-10-26 | F. Hoffmann-La Roche AG | Use of anti-vegf antibody in combination with chemotherapy for treating breast cancer |
| WO2010065923A2 (en) | 2008-12-04 | 2010-06-10 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Phosphatidylinositol-3-kinase p110 delta-targeted drugs in the treatment of cns disorders |
| US8450321B2 (en) | 2008-12-08 | 2013-05-28 | Gilead Connecticut, Inc. | 6-(1H-indazol-6-yl)-N-[4-(morpholin-4-yl)phenyl]imidazo-[1,2-A]pyrazin-8-amine, or a pharmaceutically acceptable salt thereof, as a SYK inhibitor |
| ES2744541T3 (es) | 2008-12-08 | 2020-02-25 | Gilead Connecticut Inc | Inhibidores de imidazopirazina Syk |
| CN102292338B (zh) | 2008-12-08 | 2016-09-28 | 萌蒂制药国际有限公司 | 酪氨酸激酶蛋白受体拮抗剂 |
| CN103724328B (zh) | 2008-12-19 | 2015-10-14 | 贝林格尔.英格海姆国际有限公司 | 作为ccr2受体拮抗剂用于治疗炎症、哮喘和copd的环状嘧啶-4-甲酰胺 |
| WO2010075286A1 (en) | 2008-12-24 | 2010-07-01 | University Of Washington | MOLECULAR ACTIVATORS OF THE Wnt/β-CATENIN PATHWAY |
| CN102361640B (zh) | 2009-01-13 | 2016-10-19 | 范安德尔研究所 | 异噁唑吡啶酮在制备用于缓解或减轻温血哺乳动物关节炎和哮喘的症状的药物中的用途 |
| EP2376481B1 (en) | 2009-01-13 | 2013-08-07 | Glaxo Group Limited | Pyrimidinecarboxamide derivatives as inhibitors of syk kinase |
| GB2480028B (en) | 2009-02-04 | 2013-07-10 | Univ Georgia | Methods of inhibiting fibrogenesis and treating fibrotic disease |
| US8513284B2 (en) | 2009-02-13 | 2013-08-20 | Ucb Pharma, S.A. | Fused pyridine and pyrazine derivatives as kinase inhibitors |
| WO2010094695A1 (en) | 2009-02-17 | 2010-08-26 | Boehringer Ingelheim International Gmbh | Pyrimido [5,4-d] pyrimidine derivatives for the inhibition of tyrosine kinases |
| WO2010096680A2 (en) | 2009-02-20 | 2010-08-26 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | Protein structural biomarkers to guide targeted chemotherapies |
| WO2010118207A1 (en) | 2009-04-09 | 2010-10-14 | Schering Corporation | Pyrazolo [1, 5-a] pyrimidine derivatives as mtor inhibitors |
| WO2010127208A1 (en) | 2009-04-30 | 2010-11-04 | Forest Laboratories Holdings Limited | Inhibitors of acetyl-coa carboxylase |
| CA2760791C (en) | 2009-05-07 | 2017-06-20 | Intellikine, Inc. | Heterocyclic compounds and uses thereof |
| GB0908957D0 (en) | 2009-05-22 | 2009-07-01 | Ucb Pharma Sa | Therapeutic agents |
| SG182247A1 (en) | 2009-05-27 | 2012-08-30 | Hoffmann La Roche | Bicyclic indole-pyrimidine pi3k inhibitor compounds selective for p110 delta, and methods of use |
| BRPI1009022A2 (pt) | 2009-05-27 | 2016-03-08 | Hoffmann La Roche | "composto, composição farmacêutica, processo para produzir uma composição farmacêutica, uso de um composto, método para tratamento de uma doença ou transtorno e kit" |
| US20120065205A1 (en) | 2009-06-01 | 2012-03-15 | Mercer Swati P | Pyrazine carboxamide orexin receptor antagonists |
| WO2010147898A2 (en) | 2009-06-15 | 2010-12-23 | Rigel Pharmaceuticals, Inc. | Small molecule inhibitors of spleen tyrosine kinase (syk) |
| UA110324C2 (en) | 2009-07-02 | 2015-12-25 | Genentech Inc | Jak inhibitory compounds based on pyrazolo pyrimidine |
| CN101602768B (zh) | 2009-07-17 | 2012-05-30 | 河南省农科院农副产品加工研究所 | 一种芝麻素和芝麻林素的提纯方法 |
| EP2459558A2 (en) | 2009-07-30 | 2012-06-06 | Irm Llc | Compounds and compositions as syk kinase inhibitors |
| TW201105669A (en) | 2009-07-30 | 2011-02-16 | Irm Llc | Compounds and compositions as Syk kinase inhibitors |
| EP2286808A1 (en) | 2009-08-18 | 2011-02-23 | Rheinische Friedrich-Wilhelms Universität | Cytohesin inhibitors |
| TWI481601B (zh) | 2009-08-21 | 2015-04-21 | Otsuka Pharma Co Ltd | 含氮化合物及藥學組成物 |
| BR112012004333A2 (pt) | 2009-08-27 | 2015-09-08 | Merck Sharp & Dohme | composto, composição farmacêutica, e, método para o tratamento ou a prevenção de uma doença ou um distúrbio. |
| CN102834116B (zh) | 2009-09-21 | 2015-09-16 | 杜克大学 | Wnt/卷曲蛋白相关疾病的治疗 |
| US9079904B2 (en) | 2009-09-23 | 2015-07-14 | Medivation Technologies, Inc. | Pyrido[3,4-B]indoles and methods of use |
| CN103492391A (zh) | 2009-09-25 | 2014-01-01 | 沃泰克斯药物股份有限公司 | 用于制备用作蛋白激酶抑制剂的嘧啶衍生物的方法 |
| CA2775942A1 (en) | 2009-09-29 | 2011-04-07 | Xcovery Holding Company Llc | Pi3k (delta) selective inhibitors |
| US8106146B2 (en) | 2009-10-06 | 2012-01-31 | Medtronic, Inc. | Therapeutic polymers and methods of generation |
| US9095596B2 (en) | 2009-10-15 | 2015-08-04 | Southern Research Institute | Treatment of neurodegenerative diseases, causation of memory enhancement, and assay for screening compounds for such |
| GB0918249D0 (en) | 2009-10-19 | 2009-12-02 | Respivert Ltd | Compounds |
| US9062076B2 (en) | 2009-10-22 | 2015-06-23 | Fibrotech Therapeutics Pty Ltd | Fused ring analogues of anti-fibrotic agents |
| WO2011058109A1 (en) | 2009-11-12 | 2011-05-19 | Ucb Pharma S.A. | Fused bicyclic pyrrole and imidazole derivatives as kinase inhibitors |
| WO2011058108A1 (en) | 2009-11-12 | 2011-05-19 | Ucb Pharma S.A. | Quinoline and quinoxaline derivatives as kinase inhibitors |
| US8637543B2 (en) | 2009-11-12 | 2014-01-28 | Ucb Pharma S.A. | Quinoline derivatives as kinase inhibitors |
| CN102858767B (zh) | 2009-12-17 | 2015-08-19 | 默沙东公司 | 作为syk抑制剂的氨基嘧啶 |
| EP2512246B1 (en) | 2009-12-17 | 2015-09-30 | Merck Sharp & Dohme Corp. | Aminopyrimidines as syk inhibitors |
| JP2013514989A (ja) | 2009-12-18 | 2013-05-02 | アムジエン・インコーポレーテツド | 複素環式化合物およびその使用法 |
| WO2011087776A1 (en) | 2009-12-22 | 2011-07-21 | Vertex Pharmaceuticals Incorporated | Isoindolinone inhibitors of phosphatidylinositol 3-kinase |
| EP2516434B1 (en) | 2009-12-23 | 2015-05-27 | Takeda Pharmaceutical Company Limited | Fused heteroaromatic pyrrolidinones as syk inhibitors |
| WO2011103016A2 (en) | 2010-02-19 | 2011-08-25 | The Regents Of The University Of Michigan | Compositions and methods for inhibiting ezh2 |
| JP5770751B2 (ja) | 2010-02-22 | 2015-08-26 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | ピリド[3,2−d]ピリミジン系のPI3Kデルタ阻害化合物および使用方法 |
| US8247436B2 (en) | 2010-03-19 | 2012-08-21 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| DE102010013716A1 (de) | 2010-03-31 | 2011-10-06 | Rheinische Friedrich-Wilhelms-Universität Bonn Körperschaft des öffentlichen Rechts | Verwendung von Cytohesin-Inhibitoren |
| GB201007203D0 (en) | 2010-04-29 | 2010-06-16 | Glaxo Group Ltd | Novel compounds |
| CN110322924A (zh) | 2010-04-29 | 2019-10-11 | 加利福尼亚大学董事会 | 利用关于基因组模型的数据集成的途径识别方法(paradigm) |
| ES2593256T3 (es) | 2010-05-21 | 2016-12-07 | Infinity Pharmaceuticals, Inc. | Compuestos químicos, composiciones y métodos para las modulaciones de cinasas |
| WO2011150201A2 (en) | 2010-05-27 | 2011-12-01 | Ambit Biosciences Corporation | Azolyl amide compounds and methods of use thereof |
| WO2012009097A1 (en) | 2010-06-17 | 2012-01-19 | Cytokinetics, Inc. | Methods of treating lung disease |
| US20130195843A1 (en) | 2010-06-23 | 2013-08-01 | British Columbia Cancer Agency Branch | Biomarkers for Non-Hodgkin Lymphomas and Uses Thereof |
| EP2611300B1 (de) | 2010-09-03 | 2016-04-06 | Bayer Intellectual Property GmbH | Substituierte anellierte dihydropyrimidinonderivate |
| AR082799A1 (es) | 2010-09-08 | 2013-01-09 | Ucb Pharma Sa | Derivados de quinolina y quinoxalina como inhibidores de quinasa |
| US9175331B2 (en) | 2010-09-10 | 2015-11-03 | Epizyme, Inc. | Inhibitors of human EZH2, and methods of use thereof |
| DK2614369T3 (en) | 2010-09-10 | 2016-05-02 | Epizyme Inc | METHOD FOR DETERMINING THE SUITABILITY OF HUMAN EZH2 INHIBITORS TREATED |
| US9670212B2 (en) | 2010-09-14 | 2017-06-06 | Exelixis, Inc. | Inhibitors of PI3K-delta and methods of their use and manufacture |
| TWI617560B (zh) | 2010-09-14 | 2018-03-11 | 伊塞利克斯公司 | PI3Kδ 抑制劑以及其應用和生產方法 |
| CA2812467A1 (en) | 2010-09-24 | 2012-03-29 | Gilead Calistoga Llc | Atropisomers of pi3k-inhibiting compounds |
| WO2012054332A1 (en) | 2010-10-18 | 2012-04-26 | Millennium Pharmaceuticals, Inc. | Substituted hydroxamic acids and uses thereof |
| UY33337A (es) | 2010-10-18 | 2011-10-31 | Respivert Ltd | DERIVADOS SUSTITUIDOS DE 1H-PIRAZOL[ 3,4-d]PIRIMIDINA COMO INHIBIDORES DE LAS FOSFOINOSITIDA 3-QUINASAS |
| US8846928B2 (en) | 2010-11-01 | 2014-09-30 | Portola Pharmaceuticals, Inc. | Benzamides and nicotinamides as Syk modulators |
| MX2013005005A (es) | 2010-11-04 | 2013-10-25 | Amgen Inc | Derivados de ciano-4,6-diaminopirimidina o 6-aminopurina como inhibidores de pi3k-delta. |
| US8901133B2 (en) | 2010-11-10 | 2014-12-02 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2012065019A2 (en) | 2010-11-12 | 2012-05-18 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of p13k alpha |
| WO2012065057A2 (en) | 2010-11-12 | 2012-05-18 | Exelixis, Inc. | Phosphatidylinositol 3-kinase inhibitors and methods of their use |
| DK2649075T3 (en) | 2010-12-08 | 2018-07-30 | Us Health | SUBSTITUTED PYRAZOLOPYRIMIDINES AS GLUCOCEREBROSIDASE ACTIVATORS |
| GB201021104D0 (en) | 2010-12-13 | 2011-01-26 | Univ Leuven Kath | Novel compounds for the treatment of neurodegenerative diseases |
| CN103648499B (zh) | 2011-01-10 | 2017-02-15 | 无限药品股份有限公司 | 用于制备异喹啉酮的方法及异喹啉酮的固体形式 |
| JP2012184225A (ja) | 2011-02-18 | 2012-09-27 | Otsuka Pharmaceut Co Ltd | 医薬組成物 |
| JP5940562B2 (ja) | 2011-02-18 | 2016-06-29 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | イオンチャネルのモジュレーターとしてのクロマン−スピロ環式ピペリジンアミド |
| TW201242971A (en) | 2011-02-25 | 2012-11-01 | Takeda Pharmaceutical | N-substituted oxazinopteridines and oxazinopteridinones |
| JP5569437B2 (ja) | 2011-03-09 | 2014-08-13 | コニカミノルタ株式会社 | 光学フィルム及びそれを用いた偏光板、液晶表示装置 |
| KR20140032383A (ko) | 2011-03-21 | 2014-03-14 | 에프. 호프만-라 로슈 아게 | Pi3k p110 델타에 대하여 선택적인 벤즈옥사제핀 화합물 및 이의 사용 방법 |
| US9029389B2 (en) | 2011-04-21 | 2015-05-12 | Institut Pasteur Korea | Anti-inflammation compounds |
| AU2012250958B8 (en) | 2011-05-04 | 2017-02-09 | Merck Sharp & Dohme Corp. | Amino-pyridine-containing Spleen Tyrosine Kinase (Syk) inhibitors |
| PT2707361T (pt) | 2011-05-10 | 2017-11-28 | Gilead Sciences Inc | Compostos heterocíclicos fusionados como moduladores do canal de sódio |
| JP2013010734A (ja) | 2011-06-03 | 2013-01-17 | Sumitomo Chemical Co Ltd | N−置換ラクタム化合物の製造方法 |
| JP6061922B2 (ja) | 2011-06-22 | 2017-01-18 | ザ ジェネラル ホスピタル コーポレイション | プロテイノパチーの処置方法 |
| JP2014520855A (ja) | 2011-07-15 | 2014-08-25 | アッヴィ・インコーポレイテッド | 増殖性疾患の治療に有用な三環式キナーゼ阻害薬 |
| AU2012284091B2 (en) | 2011-07-19 | 2015-11-12 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| HK1198443A1 (en) | 2011-07-19 | 2015-04-24 | 无限药品股份有限公司 | Heterocyclic compounds and uses thereof |
| EP2751093A1 (en) | 2011-08-29 | 2014-07-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2013038381A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine/pyrazine amide derivatives |
| CN103842364A (zh) | 2011-09-30 | 2014-06-04 | 奇尼塔公司 | 抗病毒化合物 |
| JP2013087779A (ja) * | 2011-10-13 | 2013-05-13 | Aisin Ai Co Ltd | 自動変速機のシフト装置 |
| JP2015027951A (ja) | 2011-11-02 | 2015-02-12 | 日本農薬株式会社 | フタルアミド誘導体及び該誘導体を含有する農園芸用殺虫剤並びにその使用方法 |
| SG10201703249PA (en) | 2011-12-21 | 2017-05-30 | Novartis Ag | Compositions and methods for antibodies targeting factor p |
| NZ627963A (en) | 2012-03-13 | 2015-08-28 | Respivert Ltd | Dry powder pharmaceutical formulations for inhalation of a compound that inhibits pi3 kinase |
| ES2630062T3 (es) | 2012-04-10 | 2017-08-17 | Annji Pharmaceutical Co., Ltd. | Inhibidores de la histona desacetilasa (HDAC) |
| US8940742B2 (en) * | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2013188432A2 (en) | 2012-06-11 | 2013-12-19 | New York University | Asymmetric catalysts |
| EP2744830A4 (en) * | 2012-06-21 | 2015-06-17 | Compugen Ltd | LSR ANTIBODIES AND USES THEREOF FOR THE TREATMENT OF CANCER |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| BR112014033055B1 (pt) | 2012-07-04 | 2023-01-31 | Rhizen Pharmaceuticals S.A | Compostos inibidores seletivos de delta pi3k, composição farmacêutica contendo os mesmos, e uso dos referidos compostos |
| AR092380A1 (es) | 2012-08-30 | 2015-04-15 | Univ Tokyo | Agente de control de endoparasitos |
| US20140120060A1 (en) | 2012-11-01 | 2014-05-01 | Infinity Pharmaceuticals, Inc. | Treatment of rheumatoid arthritis and asthma using pi3 kinase inhibitors |
| US20140120083A1 (en) | 2012-11-01 | 2014-05-01 | Infinity Pharmaceuticals, Inc. | Treatment of cancers using pi3 kinase isoform modulators |
| BR112015014034A2 (pt) | 2013-01-10 | 2017-07-11 | Nimbus Iris Inc | inibidores de irak e usos dos mesmos |
| US20140275082A1 (en) | 2013-03-14 | 2014-09-18 | Abbvie Inc. | Apoptosis-inducing agents for the treatment of cancer and immune and autoimmune diseases |
| EP2981522A4 (en) | 2013-04-05 | 2016-08-31 | Salk Inst For Biological Studi | PPAR AGONISTS |
| UY35675A (es) | 2013-07-24 | 2015-02-27 | Novartis Ag | Derivados sustituidos de quinazolin-4-ona |
| RU2675814C2 (ru) | 2013-09-25 | 2018-12-25 | Вертекс Фармасьютикалз Инкорпорейтед | 6,7-ДИГИДРО-5Н-ПИРРОЛО[3,4-b]ПИРИДИН-5-ОН, ПОЛЕЗНЫЙ В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ-ГАММА |
| WO2015051241A1 (en) | 2013-10-04 | 2015-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| TWI657085B (zh) * | 2013-10-04 | 2019-04-21 | 英菲尼提製藥股份有限公司 | 雜環化合物及其用途 |
| KR20160093062A (ko) | 2013-12-05 | 2016-08-05 | 아세르타 파마. 비.브이. | Pi3k 억제제 및 btk 억제제의 치료적 조합 |
| TWI672297B (zh) | 2013-12-18 | 2019-09-21 | 義大利商吉斯藥品公司 | 作為磷脂肌醇-3-激酶抑制劑之異唏衍生物 |
| NZ720867A (en) | 2013-12-20 | 2018-01-26 | Gilead Calistoga Llc | Process methods for phosphatidylinositol 3-kinase inhibitors |
| US9775844B2 (en) | 2014-03-19 | 2017-10-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| KR20240038809A (ko) | 2014-04-14 | 2024-03-25 | 아비나스 오퍼레이션스, 인코포레이티드 | 단백질분해의 이미드-기초된 조절인자 및 연관된 이용 방법 |
| JP6752780B2 (ja) | 2014-09-24 | 2020-09-09 | メルク パテント ゲーエムベーハー | 有機エレクトロルミネッセンス素子のための材料 |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| WO2016057322A1 (en) | 2014-10-08 | 2016-04-14 | Salk Institute For Biological Studies | Ppar agonists and methods of use thereof |
| US10144724B2 (en) | 2015-07-22 | 2018-12-04 | Araxes Pharma Llc | Substituted quinazoline compounds and methods of use thereof |
| GB201513614D0 (en) | 2015-07-31 | 2015-09-16 | Univ College Cork Nat Univ Ie | Compositions, compounds and methods and uses relating thereto |
| CN114230571B (zh) | 2015-09-14 | 2025-07-08 | 无限药品股份有限公司 | 异喹啉酮的固体形式、其制备方法、包含其的组合物及其使用方法 |
| CN106632243B (zh) | 2015-10-28 | 2019-03-15 | 华领医药技术(上海)有限公司 | 吡咯烷衍生物 |
| JP7114076B2 (ja) | 2015-12-22 | 2022-08-08 | シャイ・セラピューティクス・エルエルシー | がん及び炎症性疾患の処置のための化合物 |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| SG11201809859VA (en) | 2016-05-06 | 2018-12-28 | Shanghai De Novo Pharmatech Co Ltd | Benzazepine derivative, preparation method, pharmaceutical composition and use thereof |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| TWI817929B (zh) | 2016-07-29 | 2023-10-11 | 美商瑞佩特治療公司 | 趨化介素受體調節劑及其用途 |
| KR102497742B1 (ko) | 2016-08-30 | 2023-02-10 | 다나-파버 캔서 인스티튜트 인크. | 약물 전달 조성물 및 그의 용도 |
| US10246462B2 (en) | 2016-09-09 | 2019-04-02 | Flx Bio, Inc. | Chemokine receptor modulators and uses thereof |
| WO2018106672A1 (en) | 2016-12-05 | 2018-06-14 | Old Dominion University Research Foundation | Methods and devices for treatment of tumors with nano-pulse stimulation |
| JP2020503289A (ja) | 2016-12-14 | 2020-01-30 | ターベダ セラピューティクス インコーポレイテッドTarveda Therapeutics,Inc. | Hsp90標的化コンジュゲート及びその製剤 |
| US20190070166A1 (en) | 2017-09-02 | 2019-03-07 | Richard Postrel | Optimized Method for Treating and Curing Arthritis, Diabetes, Multiple Sclerosis and Other Autoimmune Disease |
| CN108658937A (zh) | 2017-03-28 | 2018-10-16 | 中国海洋大学 | 一种双环生物碱类化合物及其制备方法和应用 |
| IL271230B2 (en) | 2017-06-21 | 2024-06-01 | SHY Therapeutics LLC | Compounds that interact with the ras superfamily for the treatment of cancers, inflammatory diseases, rasopathies, and fibrotic disease |
| US20200165257A1 (en) | 2017-06-22 | 2020-05-28 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Servic | Inhibitors of phosphoinositide 3-kinase and histone deacetylase for treatment of cancer |
| AU2018309739B2 (en) | 2017-07-31 | 2022-01-27 | The Trustees Of Columbia University In The City Of New York | Compounds, Compositions, and Methods for Treating T-cell Acute Lymphoblastic Leukemia |
| US12144241B2 (en) | 2017-09-05 | 2024-11-12 | Samsung Electronics Co., Ltd. | Organometallic compound, organic light-emitting device including the organometallic compound, and diagnostic composition including the organometallic compound |
| EP3687981A4 (en) | 2017-09-26 | 2021-03-31 | The Regents of the University of California | COMPOSITIONS AND METHODS OF TREATMENT OF CANCER |
| WO2019118830A1 (en) | 2017-12-14 | 2019-06-20 | Tarveda Therapeutics, Inc. | Hsp90-targeting conjugates and formulations thereof |
| CN108310384B (zh) | 2018-02-05 | 2021-06-15 | 苏州大学 | Pi3k抑制剂在制备治疗血小板数量减少相关疾病药物中的用途 |
| US20210069230A1 (en) | 2018-04-13 | 2021-03-11 | The Broad Institute, Inc. | Synergistic drug combinations predicted from genomic features and single-agent response profiles |
| CN112292374B (zh) | 2018-05-31 | 2023-01-03 | 信达生物制药(苏州)有限公司 | 一种新型磷酸肌醇3-激酶抑制剂及其制备方法和用途 |
| WO2019246315A1 (en) | 2018-06-19 | 2019-12-26 | Neon Therapeutics, Inc. | Neoantigens and uses thereof |
| JP7490925B2 (ja) | 2018-07-26 | 2024-05-28 | エータイアー ファーマ, インコーポレイテッド | Nrp2関連疾患を治療するための組成物および方法 |
| EP3836929A1 (en) | 2018-08-14 | 2021-06-23 | Camp4 Therapeutics Corporation | Methods of treating liver diseases |
| CN110833546B (zh) | 2018-08-17 | 2022-06-28 | 中国科学院分子细胞科学卓越创新中心 | 通佐溴胺在治疗胃癌中的用途 |
| CA3113080A1 (en) | 2018-09-28 | 2020-04-02 | Imaginab, Inc. | Cd8 imaging constructs and methods of use thereof |
| US20210338681A1 (en) | 2018-10-01 | 2021-11-04 | Secura Bio, Inc. | Combination therapies |
| WO2020081549A1 (en) | 2018-10-15 | 2020-04-23 | The Regents Of The University Of California | Methods and materials for assessing and treating cancer |
| BR112021008255A2 (pt) | 2018-10-31 | 2021-08-03 | Gilead Sciences, Inc. | compostos de 6-azabenzimidazol substituídos como inibidores de hpk1 |
| US11071730B2 (en) | 2018-10-31 | 2021-07-27 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| EP3903828A4 (en) | 2018-12-21 | 2022-10-05 | Daiichi Sankyo Company, Limited | ANTIBODY-DRUG CONJUGATE AND KINAS INHIBITOR COMBINATION |
| KR20220010469A (ko) | 2019-02-13 | 2022-01-25 | 바스진 테라퓨틱스, 인크. | 신규한 전립선암 치료용 마커로서 ephb4-에프린 b2 수용체 리간드 쌍 |
| US20220362381A1 (en) | 2019-02-21 | 2022-11-17 | Bambu Vault Llc | Remotely triggered therapy |
| US12440494B2 (en) | 2019-02-25 | 2025-10-14 | Albert Einstein College Of Medicine | Compounds useful for inhibiting RAF dimers |
| CN115677692A (zh) | 2019-04-10 | 2023-02-03 | 南京征祥医药有限公司 | 磷脂酰肌醇3-激酶抑制剂 |
| TWI826690B (zh) | 2019-05-23 | 2023-12-21 | 美商基利科學股份有限公司 | 經取代之烯吲哚酮化物及其用途 |
| US20220324880A1 (en) | 2019-06-10 | 2022-10-13 | Kymara Therapeutics, Inc. | Smarca inhibitors and uses thereof |
| WO2020257412A1 (en) | 2019-06-19 | 2020-12-24 | Orionis Biosciences, Inc. | Combination therapy with cd13-targeted chimeric proteins or chimeric protein complexes |
| CN112194659A (zh) | 2019-07-08 | 2021-01-08 | 浙江海正药业股份有限公司 | 炔类衍生物及其制备方法和用途 |
| US20210017607A1 (en) | 2019-07-18 | 2021-01-21 | The University Of Chicago | Methods of Predicting Responsiveness to Cancer Therapies |
-
2015
- 2015-03-18 US US14/661,656 patent/US9775844B2/en active Active
- 2015-03-18 AU AU2015231413A patent/AU2015231413B2/en active Active
- 2015-03-18 CN CN202110738772.5A patent/CN113616656B/zh active Active
- 2015-03-18 DK DK15713299.4T patent/DK3119397T3/da active
- 2015-03-18 EA EA201691872A patent/EA201691872A1/ru unknown
- 2015-03-18 WO PCT/US2015/021200 patent/WO2015143012A1/en not_active Ceased
- 2015-03-18 CN CN201580026102.3A patent/CN106456628A/zh active Pending
- 2015-03-18 JP JP2016557947A patent/JP6701088B2/ja active Active
- 2015-03-18 SG SG10201808053XA patent/SG10201808053XA/en unknown
- 2015-03-18 CN CN202110735999.4A patent/CN113620958A/zh active Pending
- 2015-03-18 PT PT157132994T patent/PT3119397T/pt unknown
- 2015-03-18 EP EP15713299.4A patent/EP3119397B1/en active Active
- 2015-03-18 SG SG11201607705XA patent/SG11201607705XA/en unknown
- 2015-03-18 ES ES15713299T patent/ES2913486T3/es active Active
- 2015-03-18 MX MX2016012021A patent/MX382033B/es unknown
- 2015-03-18 EP EP22155824.0A patent/EP4066834A1/en active Pending
- 2015-03-18 CA CA2943075A patent/CA2943075C/en active Active
- 2015-03-18 NZ NZ724368A patent/NZ724368A/en unknown
-
2016
- 2016-09-15 IL IL247841A patent/IL247841B/en unknown
- 2016-09-16 PH PH12016501809A patent/PH12016501809A1/en unknown
-
2017
- 2017-08-09 US US15/672,964 patent/US10675286B2/en active Active
-
2020
- 2020-03-11 US US16/816,132 patent/US11541059B2/en active Active
-
2022
- 2022-11-30 US US18/072,314 patent/US20240050437A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| AU2015231413B2 (en) | 2020-04-23 |
| DK3119397T3 (da) | 2022-03-28 |
| SG10201808053XA (en) | 2018-10-30 |
| EP3119397B1 (en) | 2022-03-09 |
| EP4066834A1 (en) | 2022-10-05 |
| JP2017507990A (ja) | 2017-03-23 |
| EP3119397A1 (en) | 2017-01-25 |
| IL247841A0 (en) | 2016-11-30 |
| MX2016012021A (es) | 2017-04-13 |
| US20210000837A1 (en) | 2021-01-07 |
| US10675286B2 (en) | 2020-06-09 |
| NZ724368A (en) | 2023-07-28 |
| CN106456628A (zh) | 2017-02-22 |
| US9775844B2 (en) | 2017-10-03 |
| EA201691872A1 (ru) | 2017-04-28 |
| CA2943075C (en) | 2023-02-28 |
| US11541059B2 (en) | 2023-01-03 |
| CN113616656B (zh) | 2023-02-17 |
| WO2015143012A1 (en) | 2015-09-24 |
| CA2943075A1 (en) | 2015-09-24 |
| US20150290207A1 (en) | 2015-10-15 |
| US20240050437A1 (en) | 2024-02-15 |
| CN113620958A (zh) | 2021-11-09 |
| US20180055852A1 (en) | 2018-03-01 |
| JP6701088B2 (ja) | 2020-05-27 |
| SG11201607705XA (en) | 2016-10-28 |
| PH12016501809A1 (en) | 2016-11-21 |
| PT3119397T (pt) | 2022-04-11 |
| CN113616656A (zh) | 2021-11-09 |
| AU2015231413A1 (en) | 2016-10-06 |
| IL247841B (en) | 2021-12-01 |
| MX382033B (es) | 2025-03-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2913486T3 (es) | Compuestos heterocíclicos para la utilización en el tratamiento de trastornos mediados por PI3K-gamma | |
| ES2900806T3 (es) | Compuestos heterocíclicos y usos de los mismos | |
| ES2848724T3 (es) | Compuestos dinucleotídicos cíclicos | |
| JP6980649B2 (ja) | イソキノリノン誘導体の固体形態、それを製造する方法、それを含む組成物、及びそれを使用する方法 | |
| JP6309505B2 (ja) | 複素環式化合物及びその使用 | |
| WO2016054491A1 (en) | Heterocyclic compounds and uses thereof | |
| AU2014329392A1 (en) | Heterocyclic compounds and uses thereof | |
| HK1233186B (en) | Heterocyclic compounds for use in the treatment of pi3k-gamma mediated disorders | |
| HK1233186A1 (en) | Heterocyclic compounds for use in the treatment of pi3k-gamma mediated disorders | |
| EA044635B1 (ru) | Гетероциклические соединения и их применения | |
| HK1227849B (en) | Heterocyclic compounds and uses thereof |