ES2812698T3 - Inhibidores de glutaminil ciclasa - Google Patents
Inhibidores de glutaminil ciclasa Download PDFInfo
- Publication number
- ES2812698T3 ES2812698T3 ES17194164T ES17194164T ES2812698T3 ES 2812698 T3 ES2812698 T3 ES 2812698T3 ES 17194164 T ES17194164 T ES 17194164T ES 17194164 T ES17194164 T ES 17194164T ES 2812698 T3 ES2812698 T3 ES 2812698T3
- Authority
- ES
- Spain
- Prior art keywords
- difluoropropoxy
- compound
- formula
- inhibitors
- azetidin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000003112 inhibitor Substances 0.000 title claims description 174
- 108010081484 glutaminyl-peptide cyclotransferase Proteins 0.000 title description 21
- 102000003642 glutaminyl-peptide cyclotransferase Human genes 0.000 title description 21
- 150000001875 compounds Chemical class 0.000 claims abstract description 148
- -1 1H-benzimidazole-5 -yl Chemical group 0.000 claims abstract description 102
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 53
- 239000001257 hydrogen Substances 0.000 claims abstract description 49
- 229910052736 halogen Chemical group 0.000 claims abstract description 41
- 150000003839 salts Chemical class 0.000 claims abstract description 36
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 34
- 150000002367 halogens Chemical group 0.000 claims abstract description 33
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 28
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 20
- 239000012453 solvate Substances 0.000 claims abstract description 12
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 3
- 239000003814 drug Substances 0.000 claims description 48
- 239000003795 chemical substances by application Substances 0.000 claims description 38
- 238000000034 method Methods 0.000 claims description 37
- 238000002360 preparation method Methods 0.000 claims description 29
- 102000004190 Enzymes Human genes 0.000 claims description 22
- 108090000790 Enzymes Proteins 0.000 claims description 22
- 229940088598 enzyme Drugs 0.000 claims description 22
- 230000015572 biosynthetic process Effects 0.000 claims description 21
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 18
- 108010090849 Amyloid beta-Peptides Proteins 0.000 claims description 16
- 102000013455 Amyloid beta-Peptides Human genes 0.000 claims description 16
- MNFORVFSTILPAW-UHFFFAOYSA-N azetidin-2-one Chemical compound O=C1CCN1 MNFORVFSTILPAW-UHFFFAOYSA-N 0.000 claims description 14
- 229910052731 fluorine Inorganic materials 0.000 claims description 14
- 239000011737 fluorine Substances 0.000 claims description 14
- 238000003786 synthesis reaction Methods 0.000 claims description 14
- 201000010099 disease Diseases 0.000 claims description 13
- 239000008194 pharmaceutical composition Substances 0.000 claims description 13
- 208000024827 Alzheimer disease Diseases 0.000 claims description 12
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 12
- 201000006417 multiple sclerosis Diseases 0.000 claims description 12
- 230000004770 neurodegeneration Effects 0.000 claims description 11
- 230000004075 alteration Effects 0.000 claims description 10
- 208000010877 cognitive disease Diseases 0.000 claims description 10
- 239000003085 diluting agent Substances 0.000 claims description 10
- 229940079593 drug Drugs 0.000 claims description 10
- 239000003540 gamma secretase inhibitor Substances 0.000 claims description 10
- 208000027061 mild cognitive impairment Diseases 0.000 claims description 10
- 239000002904 solvent Substances 0.000 claims description 10
- 201000001320 Atherosclerosis Diseases 0.000 claims description 9
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 9
- LZCDAPDGXCYOEH-UHFFFAOYSA-N adapalene Chemical compound C1=C(C(O)=O)C=CC2=CC(C3=CC=C(C(=C3)C34CC5CC(CC(C5)C3)C4)OC)=CC=C21 LZCDAPDGXCYOEH-UHFFFAOYSA-N 0.000 claims description 9
- 230000033228 biological regulation Effects 0.000 claims description 9
- 230000008569 process Effects 0.000 claims description 9
- 208000037803 restenosis Diseases 0.000 claims description 9
- 201000010374 Down Syndrome Diseases 0.000 claims description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 8
- 239000002439 beta secretase inhibitor Substances 0.000 claims description 8
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 8
- 206010028980 Neoplasm Diseases 0.000 claims description 7
- 210000004027 cell Anatomy 0.000 claims description 7
- MSYGAHOHLUJIKV-UHFFFAOYSA-N 3,5-dimethyl-1-(3-nitrophenyl)-1h-pyrazole-4-carboxylic acid ethyl ester Chemical compound CC1=C(C(=O)OCC)C(C)=NN1C1=CC=CC([N+]([O-])=O)=C1 MSYGAHOHLUJIKV-UHFFFAOYSA-N 0.000 claims description 6
- 206010012289 Dementia Diseases 0.000 claims description 6
- 229930012538 Paclitaxel Natural products 0.000 claims description 6
- 102000011017 Type 4 Cyclic Nucleotide Phosphodiesterases Human genes 0.000 claims description 6
- 108010037584 Type 4 Cyclic Nucleotide Phosphodiesterases Proteins 0.000 claims description 6
- 239000000164 antipsychotic agent Substances 0.000 claims description 6
- 239000000969 carrier Substances 0.000 claims description 6
- 229960001592 paclitaxel Drugs 0.000 claims description 6
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 claims description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 claims description 6
- NUKYPUAOHBNCPY-UHFFFAOYSA-N 4-aminopyridine Chemical compound NC1=CC=NC=C1 NUKYPUAOHBNCPY-UHFFFAOYSA-N 0.000 claims description 5
- 206010033645 Pancreatitis Diseases 0.000 claims description 5
- NITUEMISTORFON-PPFXTMJRSA-N (2S)-1-[(2S,3R)-2-[[(2S)-2-[[(2S)-1-[(2S,3R)-2-[[(2S)-2-[[(2S,3S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2R)-2-aminopropanoyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-3-methylbutanoyl]amino]-3-methylbutanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-methylpentanoyl]amino]-3-phenylpropanoyl]amino]propanoyl]amino]-4-oxobutanoyl]amino]-3-methylpentanoyl]amino]-3-methylbutanoyl]amino]-3-hydroxybutanoyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]-3-hydroxybutanoyl]pyrrolidine-2-carboxylic acid Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@@H](C)N)C(C)C)C(C)C)C1=CC=CC=C1 NITUEMISTORFON-PPFXTMJRSA-N 0.000 claims description 4
- QKUNGNBZEVCPSL-QGZVFWFLSA-N (4R)-1-(3H-benzimidazol-5-yl)-4-[4-(3,3-difluoropropoxy)-2-fluorophenyl]azetidin-2-one Chemical compound FC(CCOC1=CC(=C(C=C1)[C@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)F QKUNGNBZEVCPSL-QGZVFWFLSA-N 0.000 claims description 4
- QKUNGNBZEVCPSL-KRWDZBQOSA-N (4S)-1-(3H-benzimidazol-5-yl)-4-[4-(3,3-difluoropropoxy)-2-fluorophenyl]azetidin-2-one Chemical compound FC(CCOC1=CC(=C(C=C1)[C@@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)F QKUNGNBZEVCPSL-KRWDZBQOSA-N 0.000 claims description 4
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 4
- 108010022752 Acetylcholinesterase Proteins 0.000 claims description 4
- 102000012440 Acetylcholinesterase Human genes 0.000 claims description 4
- 208000035895 Guillain-Barré syndrome Diseases 0.000 claims description 4
- 206010049567 Miller Fisher syndrome Diseases 0.000 claims description 4
- 102100031674 Protein-L-isoaspartate(D-aspartate) O-methyltransferase Human genes 0.000 claims description 4
- 229940022698 acetylcholinesterase Drugs 0.000 claims description 4
- 230000003284 homeostatic effect Effects 0.000 claims description 4
- 239000003018 immunosuppressive agent Substances 0.000 claims description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 claims description 4
- 230000004044 response Effects 0.000 claims description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 4
- 108010018276 trimethylguanosine synthase Proteins 0.000 claims description 4
- 239000002452 tumor necrosis factor alpha inhibitor Substances 0.000 claims description 4
- 229940046728 tumor necrosis factor alpha inhibitor Drugs 0.000 claims description 4
- NWOYMJLBOFRVDM-CQSZACIVSA-N (4R)-1-(3H-benzimidazol-5-yl)-4-[4-(2,2-difluoropropoxy)-2,3-difluorophenyl]azetidin-2-one Chemical compound FC(COC1=C(C(=C(C=C1)[C@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)F)(C)F NWOYMJLBOFRVDM-CQSZACIVSA-N 0.000 claims description 3
- ZMZFBDJKSJXWHA-MRXNPFEDSA-N (4R)-1-(3H-benzimidazol-5-yl)-4-[4-(2,2-difluoropropoxy)-2,6-difluorophenyl]azetidin-2-one Chemical compound FC(COC1=CC(=C(C(=C1)F)[C@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)(C)F ZMZFBDJKSJXWHA-MRXNPFEDSA-N 0.000 claims description 3
- MNOJTEQXPIWHDF-QGZVFWFLSA-N (4R)-1-(3H-benzimidazol-5-yl)-4-[4-(2,2-difluoropropoxy)-2-fluorophenyl]azetidin-2-one Chemical compound FC(COC1=CC(=C(C=C1)[C@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)(C)F MNOJTEQXPIWHDF-QGZVFWFLSA-N 0.000 claims description 3
- IXAHWUXZBUZIHR-CQSZACIVSA-N (4R)-1-(3H-benzimidazol-5-yl)-4-[4-(3,3-difluoropropoxy)-2,3-difluorophenyl]azetidin-2-one Chemical compound FC(CCOC1=C(C(=C(C=C1)[C@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)F)F IXAHWUXZBUZIHR-CQSZACIVSA-N 0.000 claims description 3
- GURVAWDLVHIGNH-MRXNPFEDSA-N (4R)-1-(3H-benzimidazol-5-yl)-4-[4-(3,3-difluoropropoxy)-2,6-difluorophenyl]azetidin-2-one Chemical compound FC(CCOC1=CC(=C(C(=C1)F)[C@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)F GURVAWDLVHIGNH-MRXNPFEDSA-N 0.000 claims description 3
- NWOYMJLBOFRVDM-AWEZNQCLSA-N (4S)-1-(3H-benzimidazol-5-yl)-4-[4-(2,2-difluoropropoxy)-2,3-difluorophenyl]azetidin-2-one Chemical compound FC(COC1=C(C(=C(C=C1)[C@@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)F)(C)F NWOYMJLBOFRVDM-AWEZNQCLSA-N 0.000 claims description 3
- MNOJTEQXPIWHDF-KRWDZBQOSA-N (4S)-1-(3H-benzimidazol-5-yl)-4-[4-(2,2-difluoropropoxy)-2-fluorophenyl]azetidin-2-one Chemical compound FC(COC1=CC(=C(C=C1)[C@@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)(C)F MNOJTEQXPIWHDF-KRWDZBQOSA-N 0.000 claims description 3
- IXAHWUXZBUZIHR-AWEZNQCLSA-N (4S)-1-(3H-benzimidazol-5-yl)-4-[4-(3,3-difluoropropoxy)-2,3-difluorophenyl]azetidin-2-one Chemical compound FC(CCOC1=C(C(=C(C=C1)[C@@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)F)F IXAHWUXZBUZIHR-AWEZNQCLSA-N 0.000 claims description 3
- GURVAWDLVHIGNH-INIZCTEOSA-N (4S)-1-(3H-benzimidazol-5-yl)-4-[4-(3,3-difluoropropoxy)-2,6-difluorophenyl]azetidin-2-one Chemical compound FC(CCOC1=CC(=C(C(=C1)F)[C@@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)F GURVAWDLVHIGNH-INIZCTEOSA-N 0.000 claims description 3
- 102000009091 Amyloidogenic Proteins Human genes 0.000 claims description 3
- 108010048112 Amyloidogenic Proteins Proteins 0.000 claims description 3
- 206010057645 Chronic Inflammatory Demyelinating Polyradiculoneuropathy Diseases 0.000 claims description 3
- 208000030939 Chronic inflammatory demyelinating polyneuropathy Diseases 0.000 claims description 3
- 206010009944 Colon cancer Diseases 0.000 claims description 3
- 206010019375 Helicobacter infections Diseases 0.000 claims description 3
- 229940127523 NMDA Receptor Antagonists Drugs 0.000 claims description 3
- 201000004681 Psoriasis Diseases 0.000 claims description 3
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 3
- 229960002916 adapalene Drugs 0.000 claims description 3
- 229960000548 alemtuzumab Drugs 0.000 claims description 3
- 239000000935 antidepressant agent Substances 0.000 claims description 3
- 229940005513 antidepressants Drugs 0.000 claims description 3
- 239000002249 anxiolytic agent Substances 0.000 claims description 3
- 230000000949 anxiolytic effect Effects 0.000 claims description 3
- 230000037424 autonomic function Effects 0.000 claims description 3
- 210000001124 body fluid Anatomy 0.000 claims description 3
- 239000010839 body fluid Substances 0.000 claims description 3
- 229940112129 campath Drugs 0.000 claims description 3
- 201000011510 cancer Diseases 0.000 claims description 3
- 230000008021 deposition Effects 0.000 claims description 3
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 claims description 3
- 229940002658 differin Drugs 0.000 claims description 3
- 210000003038 endothelium Anatomy 0.000 claims description 3
- 230000037149 energy metabolism Effects 0.000 claims description 3
- 239000003623 enhancer Substances 0.000 claims description 3
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 claims description 3
- 235000012631 food intake Nutrition 0.000 claims description 3
- 206010017758 gastric cancer Diseases 0.000 claims description 3
- 239000003395 histamine H3 receptor antagonist Substances 0.000 claims description 3
- 230000003054 hormonal effect Effects 0.000 claims description 3
- 229940125721 immunosuppressive agent Drugs 0.000 claims description 3
- 230000028709 inflammatory response Effects 0.000 claims description 3
- 108700027921 interferon tau Proteins 0.000 claims description 3
- 230000001404 mediated effect Effects 0.000 claims description 3
- 201000001441 melanoma Diseases 0.000 claims description 3
- 238000013508 migration Methods 0.000 claims description 3
- 230000005012 migration Effects 0.000 claims description 3
- 229960005027 natalizumab Drugs 0.000 claims description 3
- 230000009826 neoplastic cell growth Effects 0.000 claims description 3
- 201000000980 schizophrenia Diseases 0.000 claims description 3
- 201000011549 stomach cancer Diseases 0.000 claims description 3
- QGZPXUVHMTXYLF-OAHLLOKOSA-N (4R)-1-(3H-benzimidazol-5-yl)-4-phenylazetidin-2-one Chemical compound N1C=NC2=C1C=C(C=C2)N1C(C[C@@H]1C1=CC=CC=C1)=O QGZPXUVHMTXYLF-OAHLLOKOSA-N 0.000 claims description 2
- QGZPXUVHMTXYLF-HNNXBMFYSA-N (4S)-1-(3H-benzimidazol-5-yl)-4-phenylazetidin-2-one Chemical compound N1C=NC2=C1C=C(C=C2)N1C(C[C@H]1C1=CC=CC=C1)=O QGZPXUVHMTXYLF-HNNXBMFYSA-N 0.000 claims description 2
- DLYKXVABTZKJEX-UHFFFAOYSA-N 1-(3H-benzimidazol-5-yl)-4-(2,6-difluoro-4-methoxyphenyl)azetidin-2-one Chemical compound N1C=NC2=C1C=CC(=C2)N1C(CC1C1=C(C=C(C=C1F)OC)F)=O DLYKXVABTZKJEX-UHFFFAOYSA-N 0.000 claims description 2
- 206010068597 Bulbospinal muscular atrophy congenital Diseases 0.000 claims description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 2
- 206010061825 Duodenal neoplasm Diseases 0.000 claims description 2
- 241000590002 Helicobacter pylori Species 0.000 claims description 2
- 208000023105 Huntington disease Diseases 0.000 claims description 2
- 102000004388 Interleukin-4 Human genes 0.000 claims description 2
- 108090000978 Interleukin-4 Proteins 0.000 claims description 2
- 208000027747 Kennedy disease Diseases 0.000 claims description 2
- 206010027476 Metastases Diseases 0.000 claims description 2
- 229940082332 Muscarinic M1 receptor antagonist Drugs 0.000 claims description 2
- 108010008881 NBI 5788 Proteins 0.000 claims description 2
- 208000028017 Psychotic disease Diseases 0.000 claims description 2
- 102100028656 Sigma non-opioid intracellular receptor 1 Human genes 0.000 claims description 2
- 101710104750 Sigma non-opioid intracellular receptor 1 Proteins 0.000 claims description 2
- 206010054184 Small intestine carcinoma Diseases 0.000 claims description 2
- 208000006269 X-Linked Bulbo-Spinal Atrophy Diseases 0.000 claims description 2
- 201000008629 Zollinger-Ellison syndrome Diseases 0.000 claims description 2
- 229940035678 anti-parkinson drug Drugs 0.000 claims description 2
- 239000003054 catalyst Substances 0.000 claims description 2
- 201000000312 duodenum cancer Diseases 0.000 claims description 2
- 239000002792 enkephalinase inhibitor Substances 0.000 claims description 2
- WBJINCZRORDGAQ-UHFFFAOYSA-N ethyl formate Chemical compound CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 claims description 2
- 201000000052 gastrinoma Diseases 0.000 claims description 2
- 229940037467 helicobacter pylori Drugs 0.000 claims description 2
- 230000002519 immonomodulatory effect Effects 0.000 claims description 2
- 208000000509 infertility Diseases 0.000 claims description 2
- 230000036512 infertility Effects 0.000 claims description 2
- 231100000535 infertility Toxicity 0.000 claims description 2
- 229940028885 interleukin-4 Drugs 0.000 claims description 2
- 210000000265 leukocyte Anatomy 0.000 claims description 2
- 230000003211 malignant effect Effects 0.000 claims description 2
- 229940121386 matrix metalloproteinase inhibitor Drugs 0.000 claims description 2
- 239000003771 matrix metalloproteinase inhibitor Substances 0.000 claims description 2
- 239000000234 muscarinic M1 receptor antagonist Substances 0.000 claims description 2
- 230000001067 neuroprotector Effects 0.000 claims description 2
- 230000001717 pathogenic effect Effects 0.000 claims description 2
- 239000003223 protective agent Substances 0.000 claims description 2
- 229950010980 tiplimotide Drugs 0.000 claims description 2
- ZMZFBDJKSJXWHA-INIZCTEOSA-N (4S)-1-(3H-benzimidazol-5-yl)-4-[4-(2,2-difluoropropoxy)-2,6-difluorophenyl]azetidin-2-one Chemical compound FC(COC1=CC(=C(C(=C1)F)[C@@H]1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)(C)F ZMZFBDJKSJXWHA-INIZCTEOSA-N 0.000 claims 1
- QGZPXUVHMTXYLF-UHFFFAOYSA-N 1-(3H-benzimidazol-5-yl)-4-phenylazetidin-2-one Chemical compound N1C=NC2=C1C=CC(=C2)N1C(CC1C1=CC=CC=C1)=O QGZPXUVHMTXYLF-UHFFFAOYSA-N 0.000 claims 1
- RVCKCEDKBVEEHL-UHFFFAOYSA-N 2,3,4,5,6-pentachlorobenzyl alcohol Chemical compound OCC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl RVCKCEDKBVEEHL-UHFFFAOYSA-N 0.000 claims 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims 1
- 206010062519 Poor quality sleep Diseases 0.000 claims 1
- 239000000939 antiparkinson agent Substances 0.000 claims 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 claims 1
- 210000000987 immune system Anatomy 0.000 claims 1
- 230000001771 impaired effect Effects 0.000 claims 1
- 208000015181 infectious disease Diseases 0.000 claims 1
- 210000003786 sclera Anatomy 0.000 claims 1
- 150000002431 hydrogen Chemical group 0.000 abstract description 8
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 45
- 239000000203 mixture Substances 0.000 description 45
- 235000002639 sodium chloride Nutrition 0.000 description 38
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 37
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 32
- 230000000694 effects Effects 0.000 description 32
- 238000004128 high performance liquid chromatography Methods 0.000 description 30
- 239000000243 solution Substances 0.000 description 25
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 24
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 24
- 230000002265 prevention Effects 0.000 description 24
- 108090000765 processed proteins & peptides Proteins 0.000 description 24
- 125000001153 fluoro group Chemical group F* 0.000 description 21
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 21
- 239000005557 antagonist Substances 0.000 description 20
- 125000005843 halogen group Chemical group 0.000 description 18
- 238000005160 1H NMR spectroscopy Methods 0.000 description 16
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 15
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 15
- 210000004556 brain Anatomy 0.000 description 15
- 238000006243 chemical reaction Methods 0.000 description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 15
- 239000008096 xylene Substances 0.000 description 15
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 14
- 239000003826 tablet Substances 0.000 description 14
- 102400000064 Neuropeptide Y Human genes 0.000 description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- 108010052343 Gastrins Proteins 0.000 description 12
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 12
- 238000003556 assay Methods 0.000 description 12
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 12
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 12
- 102000004196 processed proteins & peptides Human genes 0.000 description 12
- 102400000921 Gastrin Human genes 0.000 description 11
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 11
- 102400001103 Neurotensin Human genes 0.000 description 10
- 101800001814 Neurotensin Proteins 0.000 description 10
- 239000003480 eluent Substances 0.000 description 10
- 235000019441 ethanol Nutrition 0.000 description 10
- 238000002844 melting Methods 0.000 description 10
- 230000008018 melting Effects 0.000 description 10
- PCJGZPGTCUMMOT-ISULXFBGSA-N neurotensin Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 PCJGZPGTCUMMOT-ISULXFBGSA-N 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 239000000523 sample Substances 0.000 description 10
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 239000011230 binding agent Substances 0.000 description 9
- 239000002775 capsule Substances 0.000 description 9
- 238000004296 chiral HPLC Methods 0.000 description 9
- 230000005764 inhibitory process Effects 0.000 description 9
- 229940002612 prodrug Drugs 0.000 description 9
- 239000000651 prodrug Substances 0.000 description 9
- 108700021111 pyroglutamyl-glutamyl-proline amide Proteins 0.000 description 9
- 102000005962 receptors Human genes 0.000 description 9
- 108020003175 receptors Proteins 0.000 description 9
- 239000000758 substrate Substances 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- 102100021943 C-C motif chemokine 2 Human genes 0.000 description 8
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 8
- HYZBGWLLSXSYLX-GUBZILKMSA-N Pglu-glu-pro-amide Chemical compound NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCC(O)=O)NC(=O)[C@H]1NC(=O)CC1 HYZBGWLLSXSYLX-GUBZILKMSA-N 0.000 description 8
- 150000001413 amino acids Chemical group 0.000 description 8
- 239000002876 beta blocker Substances 0.000 description 8
- 239000012043 crude product Substances 0.000 description 8
- 239000003937 drug carrier Substances 0.000 description 8
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 8
- 125000006239 protecting group Chemical group 0.000 description 8
- 108090000623 proteins and genes Proteins 0.000 description 8
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 7
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 7
- 101710155857 C-C motif chemokine 2 Proteins 0.000 description 7
- 102000004127 Cytokines Human genes 0.000 description 7
- 108090000695 Cytokines Proteins 0.000 description 7
- 241000196324 Embryophyta Species 0.000 description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 7
- 229940123313 MCP-1 antagonist Drugs 0.000 description 7
- 241000124008 Mammalia Species 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 102000004316 Oxidoreductases Human genes 0.000 description 7
- 108090000854 Oxidoreductases Proteins 0.000 description 7
- 239000004480 active ingredient Substances 0.000 description 7
- 235000001014 amino acid Nutrition 0.000 description 7
- 229960005370 atorvastatin Drugs 0.000 description 7
- 229940097320 beta blocking agent Drugs 0.000 description 7
- 125000004925 dihydropyridyl group Chemical group N1(CC=CC=C1)* 0.000 description 7
- RCBVKBFIWMOMHF-UHFFFAOYSA-L hydroxy-(hydroxy(dioxo)chromio)oxy-dioxochromium;pyridine Chemical compound C1=CC=NC=C1.C1=CC=NC=C1.O[Cr](=O)(=O)O[Cr](O)(=O)=O RCBVKBFIWMOMHF-UHFFFAOYSA-L 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 210000002381 plasma Anatomy 0.000 description 7
- 235000018102 proteins Nutrition 0.000 description 7
- 102000004169 proteins and genes Human genes 0.000 description 7
- PKXWXXPNHIWQHW-RCBQFDQVSA-N (2S)-2-hydroxy-3-methyl-N-[(2S)-1-[[(5S)-3-methyl-4-oxo-2,5-dihydro-1H-3-benzazepin-5-yl]amino]-1-oxopropan-2-yl]butanamide Chemical compound C1CN(C)C(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@@H](O)C(C)C)C2=CC=CC=C21 PKXWXXPNHIWQHW-RCBQFDQVSA-N 0.000 description 6
- 102000004889 Interleukin-6 Human genes 0.000 description 6
- 108090001005 Interleukin-6 Proteins 0.000 description 6
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 6
- ZBBHBTPTTSWHBA-UHFFFAOYSA-N Nicardipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OCCN(C)CC=2C=CC=CC=2)C1C1=CC=CC([N+]([O-])=O)=C1 ZBBHBTPTTSWHBA-UHFFFAOYSA-N 0.000 description 6
- ODHCTXKNWHHXJC-GSVOUGTGSA-N Pyroglutamic acid Natural products OC(=O)[C@H]1CCC(=O)N1 ODHCTXKNWHHXJC-GSVOUGTGSA-N 0.000 description 6
- WKBAPRRMZYHPNB-BXBUPLCLSA-N QYNAD Chemical compound OC(=O)C[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CCC(N)=O)CC1=CC=C(O)C=C1 WKBAPRRMZYHPNB-BXBUPLCLSA-N 0.000 description 6
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 229920002472 Starch Polymers 0.000 description 6
- ODHCTXKNWHHXJC-UHFFFAOYSA-N acide pyroglutamique Natural products OC(=O)C1CCC(=O)N1 ODHCTXKNWHHXJC-UHFFFAOYSA-N 0.000 description 6
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 6
- 108060000200 adenylate cyclase Proteins 0.000 description 6
- 102000030621 adenylate cyclase Human genes 0.000 description 6
- 229940024606 amino acid Drugs 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- 239000000460 chlorine Substances 0.000 description 6
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- ADEBPBSSDYVVLD-UHFFFAOYSA-N donepezil Chemical compound O=C1C=2C=C(OC)C(OC)=CC=2CC1CC(CC1)CCN1CC1=CC=CC=C1 ADEBPBSSDYVVLD-UHFFFAOYSA-N 0.000 description 6
- 210000004188 enterochromaffin-like cell Anatomy 0.000 description 6
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 6
- 239000003102 growth factor Substances 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 229960002797 pitavastatin Drugs 0.000 description 6
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 6
- 229960002855 simvastatin Drugs 0.000 description 6
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 235000019698 starch Nutrition 0.000 description 6
- 239000003981 vehicle Substances 0.000 description 6
- 229940127291 Calcium channel antagonist Drugs 0.000 description 5
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 5
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 5
- 102100037838 Prolyl endopeptidase Human genes 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 239000000480 calcium channel blocker Substances 0.000 description 5
- 230000003197 catalytic effect Effects 0.000 description 5
- 210000003169 central nervous system Anatomy 0.000 description 5
- 229910052801 chlorine Inorganic materials 0.000 description 5
- 239000003086 colorant Substances 0.000 description 5
- 239000002552 dosage form Substances 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 238000003402 intramolecular cyclocondensation reaction Methods 0.000 description 5
- 239000000314 lubricant Substances 0.000 description 5
- 239000002808 molecular sieve Substances 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- 239000006187 pill Substances 0.000 description 5
- 239000002243 precursor Substances 0.000 description 5
- 239000003755 preservative agent Substances 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 5
- 125000001424 substituent group Chemical group 0.000 description 5
- 235000000346 sugar Nutrition 0.000 description 5
- 239000000375 suspending agent Substances 0.000 description 5
- XDHCBTYFHVDHFD-UHFFFAOYSA-N tert-butyl 5-aminobenzimidazole-1-carboxylate Chemical compound NC1=CC=C2N(C(=O)OC(C)(C)C)C=NC2=C1 XDHCBTYFHVDHFD-UHFFFAOYSA-N 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 4
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 4
- 102400000345 Angiotensin-2 Human genes 0.000 description 4
- 101800000733 Angiotensin-2 Proteins 0.000 description 4
- 241000416162 Astragalus gummifer Species 0.000 description 4
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- 101000585315 Homo sapiens Glutaminyl-peptide cyclotransferase Proteins 0.000 description 4
- CZGUSIXMZVURDU-JZXHSEFVSA-N Ile(5)-angiotensin II Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C=CC=CC=1)C([O-])=O)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=[NH2+])NC(=O)[C@@H]([NH3+])CC([O-])=O)C(C)C)C1=CC=C(O)C=C1 CZGUSIXMZVURDU-JZXHSEFVSA-N 0.000 description 4
- 108010005716 Interferon beta-1a Proteins 0.000 description 4
- 108010050904 Interferons Proteins 0.000 description 4
- 102000014150 Interferons Human genes 0.000 description 4
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 4
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 4
- 240000007472 Leucaena leucocephala Species 0.000 description 4
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 4
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- 229920001615 Tragacanth Polymers 0.000 description 4
- 229960000528 amlodipine Drugs 0.000 description 4
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- 229950006323 angiotensin ii Drugs 0.000 description 4
- 229950001863 bapineuzumab Drugs 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- 229960005110 cerivastatin Drugs 0.000 description 4
- SEERZIQQUAZTOL-ANMDKAQQSA-N cerivastatin Chemical compound COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 SEERZIQQUAZTOL-ANMDKAQQSA-N 0.000 description 4
- 229960002436 cladribine Drugs 0.000 description 4
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 description 4
- 238000007796 conventional method Methods 0.000 description 4
- 229920001577 copolymer Polymers 0.000 description 4
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 4
- 229960003957 dexamethasone Drugs 0.000 description 4
- 229960001259 diclofenac Drugs 0.000 description 4
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 4
- 208000035475 disorder Diseases 0.000 description 4
- 239000002934 diuretic Substances 0.000 description 4
- 230000035558 fertility Effects 0.000 description 4
- 229960003765 fluvastatin Drugs 0.000 description 4
- 235000003599 food sweetener Nutrition 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- ASUTZQLVASHGKV-JDFRZJQESA-N galanthamine Chemical compound O1C(=C23)C(OC)=CC=C2CN(C)CC[C@]23[C@@H]1C[C@@H](O)C=C2 ASUTZQLVASHGKV-JDFRZJQESA-N 0.000 description 4
- 229960002989 glutamic acid Drugs 0.000 description 4
- 235000013922 glutamic acid Nutrition 0.000 description 4
- 239000004220 glutamic acid Substances 0.000 description 4
- 125000000291 glutamic acid group Chemical group N[C@@H](CCC(O)=O)C(=O)* 0.000 description 4
- 229960001340 histamine Drugs 0.000 description 4
- 230000001976 improved effect Effects 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 239000003446 ligand Substances 0.000 description 4
- 229960004844 lovastatin Drugs 0.000 description 4
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 4
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 4
- 229920000609 methyl cellulose Polymers 0.000 description 4
- 239000001923 methylcellulose Substances 0.000 description 4
- 235000010981 methylcellulose Nutrition 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 210000001711 oxyntic cell Anatomy 0.000 description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 4
- 229960002965 pravastatin Drugs 0.000 description 4
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- 229960000672 rosuvastatin Drugs 0.000 description 4
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 4
- 230000019491 signal transduction Effects 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- 150000008163 sugars Chemical class 0.000 description 4
- 239000003765 sweetening agent Substances 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 4
- XFYDIVBRZNQMJC-UHFFFAOYSA-N tizanidine Chemical compound ClC=1C=CC2=NSN=C2C=1NC1=NCCN1 XFYDIVBRZNQMJC-UHFFFAOYSA-N 0.000 description 4
- 239000000196 tragacanth Substances 0.000 description 4
- 235000010487 tragacanth Nutrition 0.000 description 4
- 229940116362 tragacanth Drugs 0.000 description 4
- 229960005486 vaccine Drugs 0.000 description 4
- HMJIYCCIJYRONP-UHFFFAOYSA-N (+-)-Isradipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC(C)C)C1C1=CC=CC2=NON=C12 HMJIYCCIJYRONP-UHFFFAOYSA-N 0.000 description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 3
- SYTBZMRGLBWNTM-SNVBAGLBSA-N (R)-flurbiprofen Chemical compound FC1=CC([C@H](C(O)=O)C)=CC=C1C1=CC=CC=C1 SYTBZMRGLBWNTM-SNVBAGLBSA-N 0.000 description 3
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 3
- WFRXSXUDWCVSPI-UHFFFAOYSA-N 3h-benzimidazol-5-amine Chemical compound NC1=CC=C2NC=NC2=C1 WFRXSXUDWCVSPI-UHFFFAOYSA-N 0.000 description 3
- 239000005541 ACE inhibitor Substances 0.000 description 3
- 102000004400 Aminopeptidases Human genes 0.000 description 3
- 108090000915 Aminopeptidases Proteins 0.000 description 3
- 108050000824 Angiotensin II receptor Proteins 0.000 description 3
- 102000008873 Angiotensin II receptor Human genes 0.000 description 3
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 description 3
- XPCFTKFZXHTYIP-PMACEKPBSA-N Benazepril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N(CC(O)=O)C2=CC=CC=C2CC1)=O)CC1=CC=CC=C1 XPCFTKFZXHTYIP-PMACEKPBSA-N 0.000 description 3
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 3
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 3
- 240000006432 Carica papaya Species 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 229940097420 Diuretic Drugs 0.000 description 3
- 108010061435 Enalapril Proteins 0.000 description 3
- 108010008165 Etanercept Proteins 0.000 description 3
- 229940125373 Gamma-Secretase Inhibitor Drugs 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 108010072051 Glatiramer Acetate Proteins 0.000 description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 3
- 102100029865 Glutaminyl-peptide cyclotransferase-like protein Human genes 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 239000004471 Glycine Substances 0.000 description 3
- 102000015779 HDL Lipoproteins Human genes 0.000 description 3
- 108010010234 HDL Lipoproteins Proteins 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- ZJVFLBOZORBYFE-UHFFFAOYSA-N Ibudilast Chemical compound C1=CC=CC2=C(C(=O)C(C)C)C(C(C)C)=NN21 ZJVFLBOZORBYFE-UHFFFAOYSA-N 0.000 description 3
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 3
- BDABGOLMYNHHTR-UHFFFAOYSA-N Perzinfotel Chemical compound OP(O)(=O)CCN1CCCNC2=C1C(=O)C2=O BDABGOLMYNHHTR-UHFFFAOYSA-N 0.000 description 3
- 229940123934 Reductase inhibitor Drugs 0.000 description 3
- 108010052164 Sodium Channels Proteins 0.000 description 3
- 102000018674 Sodium Channels Human genes 0.000 description 3
- KPWYNAGOBXLMSE-UHFFFAOYSA-N Tipelukast Chemical compound CCCC1=C(O)C(C(C)=O)=CC=C1SCCCOC1=CC=C(C(C)=O)C(OCCCC(O)=O)=C1CCC KPWYNAGOBXLMSE-UHFFFAOYSA-N 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 229960005305 adenosine Drugs 0.000 description 3
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 3
- 239000000074 antisense oligonucleotide Substances 0.000 description 3
- 238000012230 antisense oligonucleotides Methods 0.000 description 3
- 230000006399 behavior Effects 0.000 description 3
- 229960004530 benazepril Drugs 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 235000012000 cholesterol Nutrition 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 238000002648 combination therapy Methods 0.000 description 3
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 3
- 229960004166 diltiazem Drugs 0.000 description 3
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 3
- 230000001882 diuretic effect Effects 0.000 description 3
- 229960003530 donepezil Drugs 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 229960000873 enalapril Drugs 0.000 description 3
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 239000002532 enzyme inhibitor Substances 0.000 description 3
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 229930195712 glutamate Natural products 0.000 description 3
- 229940049906 glutamate Drugs 0.000 description 3
- 235000004554 glutamine Nutrition 0.000 description 3
- 229960002003 hydrochlorothiazide Drugs 0.000 description 3
- 229960002491 ibudilast Drugs 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 229940047124 interferons Drugs 0.000 description 3
- 229940100601 interleukin-6 Drugs 0.000 description 3
- 229960004427 isradipine Drugs 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 230000010534 mechanism of action Effects 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 229960002237 metoprolol Drugs 0.000 description 3
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 3
- 210000004877 mucosa Anatomy 0.000 description 3
- OGZQTTHDGQBLBT-UHFFFAOYSA-N neramexane Chemical compound CC1(C)CC(C)(C)CC(C)(N)C1 OGZQTTHDGQBLBT-UHFFFAOYSA-N 0.000 description 3
- 229950004543 neramexane Drugs 0.000 description 3
- 208000015122 neurodegenerative disease Diseases 0.000 description 3
- 229960001783 nicardipine Drugs 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 230000001817 pituitary effect Effects 0.000 description 3
- 229940020573 plavix Drugs 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 3
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 3
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 3
- 229960003712 propranolol Drugs 0.000 description 3
- XNSAINXGIQZQOO-SRVKXCTJSA-N protirelin Chemical compound NC(=O)[C@@H]1CCCN1C(=O)[C@@H](NC(=O)[C@H]1NC(=O)CC1)CC1=CN=CN1 XNSAINXGIQZQOO-SRVKXCTJSA-N 0.000 description 3
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 3
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 3
- 229960002930 sirolimus Drugs 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 210000002784 stomach Anatomy 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 229940034199 thyrotropin-releasing hormone Drugs 0.000 description 3
- 229950004996 tipelukast Drugs 0.000 description 3
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 3
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 3
- 229960004699 valsartan Drugs 0.000 description 3
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 3
- 229960001722 verapamil Drugs 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- UZBODILCSLUHQR-JLMRSGIVSA-N zenvia Chemical compound C([C@@H]12)CCC[C@]11CCN(C)[C@H]2CC2=CC=C(OC)C=C21.C1C([C@H](C2)C=C)CCN2[C@H]1[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 UZBODILCSLUHQR-JLMRSGIVSA-N 0.000 description 3
- CEMAWMOMDPGJMB-UHFFFAOYSA-N (+-)-Oxprenolol Chemical compound CC(C)NCC(O)COC1=CC=CC=C1OCC=C CEMAWMOMDPGJMB-UHFFFAOYSA-N 0.000 description 2
- HQLHZNDJQSRKDT-QMMMGPOBSA-N (2s)-2-amino-4-(2-amino-4-chlorophenyl)-4-oxobutanoic acid Chemical compound OC(=O)[C@@H](N)CC(=O)C1=CC=C(Cl)C=C1N HQLHZNDJQSRKDT-QMMMGPOBSA-N 0.000 description 2
- PUOAETJYKQITMO-LANLRWRYSA-N (3e)-1-[(1s)-1-(4-fluorophenyl)ethyl]-3-[[3-methoxy-4-(4-methylimidazol-1-yl)phenyl]methylidene]piperidin-2-one Chemical compound C1([C@H](C)N2CCC\C(C2=O)=C/C=2C=C(C(=CC=2)N2C=C(C)N=C2)OC)=CC=C(F)C=C1 PUOAETJYKQITMO-LANLRWRYSA-N 0.000 description 2
- ABEJDMOBAFLQNJ-NHCUHLMSSA-N (3s,5s)-5-(3-cyclopentyloxy-4-methoxyphenyl)-3-[(3-methylphenyl)methyl]piperidin-2-one Chemical compound COC1=CC=C([C@@H]2C[C@@H](CC=3C=C(C)C=CC=3)C(=O)NC2)C=C1OC1CCCC1 ABEJDMOBAFLQNJ-NHCUHLMSSA-N 0.000 description 2
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 2
- UCTWMZQNUQWSLP-VIFPVBQESA-N (R)-adrenaline Chemical compound CNC[C@H](O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-VIFPVBQESA-N 0.000 description 2
- 229930182837 (R)-adrenaline Natural products 0.000 description 2
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 2
- PVHUJELLJLJGLN-INIZCTEOSA-N (S)-nitrendipine Chemical compound CCOC(=O)C1=C(C)NC(C)=C(C(=O)OC)[C@@H]1C1=CC=CC([N+]([O-])=O)=C1 PVHUJELLJLJGLN-INIZCTEOSA-N 0.000 description 2
- TWBNMYSKRDRHAT-RCWTXCDDSA-N (S)-timolol hemihydrate Chemical compound O.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1 TWBNMYSKRDRHAT-RCWTXCDDSA-N 0.000 description 2
- IRELROQHIPLASX-SEYXRHQNSA-N (z)-2-cyano-3-hydroxy-n-[4-(trifluoromethyl)phenyl]hept-2-en-6-ynamide Chemical compound C#CCCC(/O)=C(\C#N)C(=O)NC1=CC=C(C(F)(F)F)C=C1 IRELROQHIPLASX-SEYXRHQNSA-N 0.000 description 2
- UUUHXMGGBIUAPW-UHFFFAOYSA-N 1-[1-[2-[[5-amino-2-[[1-[5-(diaminomethylideneamino)-2-[[1-[3-(1h-indol-3-yl)-2-[(5-oxopyrrolidine-2-carbonyl)amino]propanoyl]pyrrolidine-2-carbonyl]amino]pentanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-methylpentanoyl]pyrrolidine-2-carbon Chemical compound C1CCC(C(=O)N2C(CCC2)C(O)=O)N1C(=O)C(C(C)CC)NC(=O)C(CCC(N)=O)NC(=O)C1CCCN1C(=O)C(CCCN=C(N)N)NC(=O)C1CCCN1C(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C1CCC(=O)N1 UUUHXMGGBIUAPW-UHFFFAOYSA-N 0.000 description 2
- YPTJKHVBDCRKNF-UHFFFAOYSA-N 2',6'-Dihydroxyacetophenone Chemical compound CC(=O)C1=C(O)C=CC=C1O YPTJKHVBDCRKNF-UHFFFAOYSA-N 0.000 description 2
- XEOSTBFUCNZKGS-UHFFFAOYSA-N 2-[4,6-bis(dimethylamino)-2-[[4-[[4-(trifluoromethyl)benzoyl]amino]phenyl]methyl]pyrimidin-5-yl]acetic acid Chemical compound CN(C)C1=C(CC(O)=O)C(N(C)C)=NC(CC=2C=CC(NC(=O)C=3C=CC(=CC=3)C(F)(F)F)=CC=2)=N1 XEOSTBFUCNZKGS-UHFFFAOYSA-N 0.000 description 2
- GSSLQBZZKKOMAF-UHFFFAOYSA-N 2-cyano-n-(4-cyanophenyl)-3-cyclopropyl-3-oxopropanamide Chemical compound C1CC1C(=O)C(C#N)C(=O)NC1=CC=C(C#N)C=C1 GSSLQBZZKKOMAF-UHFFFAOYSA-N 0.000 description 2
- SGUAFYQXFOLMHL-UHFFFAOYSA-N 2-hydroxy-5-{1-hydroxy-2-[(4-phenylbutan-2-yl)amino]ethyl}benzamide Chemical compound C=1C=C(O)C(C(N)=O)=CC=1C(O)CNC(C)CCC1=CC=CC=C1 SGUAFYQXFOLMHL-UHFFFAOYSA-N 0.000 description 2
- KPGXRSRHYNQIFN-UHFFFAOYSA-N 2-oxoglutaric acid Chemical compound OC(=O)CCC(=O)C(O)=O KPGXRSRHYNQIFN-UHFFFAOYSA-N 0.000 description 2
- SNKZJIOFVMKAOJ-UHFFFAOYSA-N 3-Aminopropanesulfonate Chemical compound NCCCS(O)(=O)=O SNKZJIOFVMKAOJ-UHFFFAOYSA-N 0.000 description 2
- XCGJIFAKUZNNOR-UHFFFAOYSA-N 3-[4-(4-chlorophenyl)sulfonyl-4-(2,5-difluorophenyl)cyclohexyl]propanoic acid Chemical compound C1CC(CCC(=O)O)CCC1(S(=O)(=O)C=1C=CC(Cl)=CC=1)C1=CC(F)=CC=C1F XCGJIFAKUZNNOR-UHFFFAOYSA-N 0.000 description 2
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 2
- UIAGMCDKSXEBJQ-IBGZPJMESA-N 3-o-(2-methoxyethyl) 5-o-propan-2-yl (4s)-2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound COCCOC(=O)C1=C(C)NC(C)=C(C(=O)OC(C)C)[C@H]1C1=CC=CC([N+]([O-])=O)=C1 UIAGMCDKSXEBJQ-IBGZPJMESA-N 0.000 description 2
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 2
- 125000004539 5-benzimidazolyl group Chemical group N1=CNC2=C1C=CC(=C2)* 0.000 description 2
- XKFPYPQQHFEXRZ-UHFFFAOYSA-N 5-methyl-N'-(phenylmethyl)-3-isoxazolecarbohydrazide Chemical compound O1C(C)=CC(C(=O)NNCC=2C=CC=CC=2)=N1 XKFPYPQQHFEXRZ-UHFFFAOYSA-N 0.000 description 2
- XDBHURGONHZNJF-UHFFFAOYSA-N 6-[2-(3,4-diethoxyphenyl)-1,3-thiazol-4-yl]pyridine-2-carboxylic acid Chemical compound C1=C(OCC)C(OCC)=CC=C1C1=NC(C=2N=C(C=CC=2)C(O)=O)=CS1 XDBHURGONHZNJF-UHFFFAOYSA-N 0.000 description 2
- ZXXHOPNSTZKWRI-UHFFFAOYSA-N 7-[2-[4-[3-(trifluoromethyl)phenyl]-3,6-dihydro-2h-pyridin-1-yl]ethyl]isoquinoline Chemical compound FC(F)(F)C1=CC=CC(C=2CCN(CCC=3C=C4C=NC=CC4=CC=3)CC=2)=C1 ZXXHOPNSTZKWRI-UHFFFAOYSA-N 0.000 description 2
- 101150059573 AGTR1 gene Proteins 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- UXOWGYHJODZGMF-QORCZRPOSA-N Aliskiren Chemical group COCCCOC1=CC(C[C@@H](C[C@H](N)[C@@H](O)C[C@@H](C(C)C)C(=O)NCC(C)(C)C(N)=O)C(C)C)=CC=C1OC UXOWGYHJODZGMF-QORCZRPOSA-N 0.000 description 2
- 102100023635 Alpha-fetoprotein Human genes 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- 102000002659 Amyloid Precursor Protein Secretases Human genes 0.000 description 2
- 206010003210 Arteriosclerosis Diseases 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 208000023275 Autoimmune disease Diseases 0.000 description 2
- 101000950981 Bacillus subtilis (strain 168) Catabolic NAD-specific glutamate dehydrogenase RocG Proteins 0.000 description 2
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 2
- 101800000285 Big gastrin Proteins 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- 102100031151 C-C chemokine receptor type 2 Human genes 0.000 description 2
- 101710149815 C-C chemokine receptor type 2 Proteins 0.000 description 2
- 102000017927 CHRM1 Human genes 0.000 description 2
- 102000055006 Calcitonin Human genes 0.000 description 2
- 108060001064 Calcitonin Proteins 0.000 description 2
- 108090000712 Cathepsin B Proteins 0.000 description 2
- 102000004225 Cathepsin B Human genes 0.000 description 2
- 101150015280 Cel gene Proteins 0.000 description 2
- 108010061846 Cholesterol Ester Transfer Proteins Proteins 0.000 description 2
- 102000012336 Cholesterol Ester Transfer Proteins Human genes 0.000 description 2
- 101150073075 Chrm1 gene Proteins 0.000 description 2
- WUFQLZTXIWKION-UHFFFAOYSA-N Deoxypeganine Chemical compound C1C2=CC=CC=C2N=C2N1CCC2 WUFQLZTXIWKION-UHFFFAOYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 2
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 2
- 101000930822 Giardia intestinalis Dipeptidyl-peptidase 4 Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 102000016901 Glutamate dehydrogenase Human genes 0.000 description 2
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 2
- 101000585397 Homo sapiens Glutaminyl-peptide cyclotransferase-like protein Proteins 0.000 description 2
- 101001129465 Homo sapiens Pyroglutamyl-peptidase 1 Proteins 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 102000013463 Immunoglobulin Light Chains Human genes 0.000 description 2
- 108010065825 Immunoglobulin Light Chains Proteins 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 108010041012 Integrin alpha4 Proteins 0.000 description 2
- 108010008212 Integrin alpha4beta1 Proteins 0.000 description 2
- 108010005714 Interferon beta-1b Proteins 0.000 description 2
- 108010047761 Interferon-alpha Proteins 0.000 description 2
- 102000006992 Interferon-alpha Human genes 0.000 description 2
- 108090000467 Interferon-beta Proteins 0.000 description 2
- 102000003996 Interferon-beta Human genes 0.000 description 2
- 108090000174 Interleukin-10 Proteins 0.000 description 2
- 102000010781 Interleukin-6 Receptors Human genes 0.000 description 2
- 108010038501 Interleukin-6 Receptors Proteins 0.000 description 2
- 108010010995 MART-1 Antigen Proteins 0.000 description 2
- 102000016200 MART-1 Antigen Human genes 0.000 description 2
- 102000010909 Monoamine Oxidase Human genes 0.000 description 2
- 108010062431 Monoamine oxidase Proteins 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 102000014413 Neuregulin Human genes 0.000 description 2
- 108050003475 Neuregulin Proteins 0.000 description 2
- 102000003797 Neuropeptides Human genes 0.000 description 2
- 108090000189 Neuropeptides Proteins 0.000 description 2
- FAIIFDPAEUKBEP-UHFFFAOYSA-N Nilvadipine Chemical compound COC(=O)C1=C(C#N)NC(C)=C(C(=O)OC(C)C)C1C1=CC=CC([N+]([O-])=O)=C1 FAIIFDPAEUKBEP-UHFFFAOYSA-N 0.000 description 2
- 108091034117 Oligonucleotide Proteins 0.000 description 2
- 102000002512 Orexin Human genes 0.000 description 2
- 108091007960 PI3Ks Proteins 0.000 description 2
- 108091093037 Peptide nucleic acid Proteins 0.000 description 2
- 102000004270 Peptidyl-Dipeptidase A Human genes 0.000 description 2
- 108090000882 Peptidyl-Dipeptidase A Proteins 0.000 description 2
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 2
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 2
- 244000113945 Pinus torreyana Species 0.000 description 2
- 235000006235 Pinus torreyana Nutrition 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 229920001710 Polyorthoester Polymers 0.000 description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 2
- 102000056251 Prolyl Oligopeptidases Human genes 0.000 description 2
- 102100031108 Pyroglutamyl-peptidase 1 Human genes 0.000 description 2
- 229940121991 Serotonin and norepinephrine reuptake inhibitor Drugs 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 2
- DHCOPPHTVOXDKU-UHFFFAOYSA-N Tofimilast Chemical compound C1CN2C(C=3SC=CC=3)=NN=C2C2=C1C(CC)=NN2C1CCCC1 DHCOPPHTVOXDKU-UHFFFAOYSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 102100040247 Tumor necrosis factor Human genes 0.000 description 2
- 240000008042 Zea mays Species 0.000 description 2
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 2
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 2
- PBHFNBQPZCRWQP-AZUAARDMSA-N [(3aS,8bR)-3,4,8b-trimethyl-2,3a-dihydro-1H-pyrrolo[2,3-b]indol-7-yl] N-phenylcarbamate Chemical compound CN([C@H]1[C@](C2=C3)(C)CCN1C)C2=CC=C3OC(=O)NC1=CC=CC=C1 PBHFNBQPZCRWQP-AZUAARDMSA-N 0.000 description 2
- OEWZGBLJCYAMEG-UHFFFAOYSA-N [2-(octadecoxymethyl)oxolan-2-yl]methyl 2-(trimethylazaniumyl)ethyl phosphate Chemical compound CCCCCCCCCCCCCCCCCCOCC1(COP([O-])(=O)OCC[N+](C)(C)C)CCCO1 OEWZGBLJCYAMEG-UHFFFAOYSA-N 0.000 description 2
- KFHYZKCRXNRKRC-MRXNPFEDSA-N abt-239 Chemical compound C[C@@H]1CCCN1CCC1=CC2=CC(C=3C=CC(=CC=3)C#N)=CC=C2O1 KFHYZKCRXNRKRC-MRXNPFEDSA-N 0.000 description 2
- 229960002122 acebutolol Drugs 0.000 description 2
- GOEMGAFJFRBGGG-UHFFFAOYSA-N acebutolol Chemical compound CCCC(=O)NC1=CC=C(OCC(O)CNC(C)C)C(C(C)=O)=C1 GOEMGAFJFRBGGG-UHFFFAOYSA-N 0.000 description 2
- 230000009858 acid secretion Effects 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 229960003767 alanine Drugs 0.000 description 2
- 125000006241 alcohol protecting group Chemical group 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 229960004601 aliskiren Drugs 0.000 description 2
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 2
- 125000006242 amine protecting group Chemical group 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- DZHSAHHDTRWUTF-SIQRNXPUSA-N amyloid-beta polypeptide 42 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O)[C@@H](C)CC)C(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(C)C)C1=CC=CC=C1 DZHSAHHDTRWUTF-SIQRNXPUSA-N 0.000 description 2
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 2
- 230000001028 anti-proliverative effect Effects 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 229940082992 antihypertensives mao inhibitors Drugs 0.000 description 2
- 208000011775 arteriosclerosis disease Diseases 0.000 description 2
- 229960002274 atenolol Drugs 0.000 description 2
- 210000003050 axon Anatomy 0.000 description 2
- 230000003376 axonal effect Effects 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 102000012740 beta Adrenergic Receptors Human genes 0.000 description 2
- 108010079452 beta Adrenergic Receptors Proteins 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 229960004324 betaxolol Drugs 0.000 description 2
- CHDPSNLJFOQTRK-UHFFFAOYSA-N betaxolol hydrochloride Chemical compound [Cl-].C1=CC(OCC(O)C[NH2+]C(C)C)=CC=C1CCOCC1CC1 CHDPSNLJFOQTRK-UHFFFAOYSA-N 0.000 description 2
- KUWBXRGRMQZCSS-HSZRJFAPSA-N bibp-3226 Chemical compound N([C@H](CCCN=C(N)N)C(=O)NCC=1C=CC(O)=CC=1)C(=O)C(C=1C=CC=CC=1)C1=CC=CC=C1 KUWBXRGRMQZCSS-HSZRJFAPSA-N 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- 229960002781 bisoprolol Drugs 0.000 description 2
- VHYCDWMUTMEGQY-UHFFFAOYSA-N bisoprolol Chemical compound CC(C)NCC(O)COC1=CC=C(COCCOC(C)C)C=C1 VHYCDWMUTMEGQY-UHFFFAOYSA-N 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 210000005013 brain tissue Anatomy 0.000 description 2
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 2
- BBBFJLBPOGFECG-VJVYQDLKSA-N calcitonin Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 BBBFJLBPOGFECG-VJVYQDLKSA-N 0.000 description 2
- 229960004015 calcitonin Drugs 0.000 description 2
- 125000006243 carbonyl protecting group Chemical group 0.000 description 2
- 125000006244 carboxylic acid protecting group Chemical group 0.000 description 2
- 238000012754 cardiac puncture Methods 0.000 description 2
- 229960001222 carteolol Drugs 0.000 description 2
- LWAFSWPYPHEXKX-UHFFFAOYSA-N carteolol Chemical compound N1C(=O)CCC2=C1C=CC=C2OCC(O)CNC(C)(C)C LWAFSWPYPHEXKX-UHFFFAOYSA-N 0.000 description 2
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 2
- 229960004195 carvedilol Drugs 0.000 description 2
- CFBUZOUXXHZCFB-OYOVHJISSA-N chembl511115 Chemical compound COC1=CC=C([C@@]2(CC[C@H](CC2)C(O)=O)C#N)C=C1OC1CCCC1 CFBUZOUXXHZCFB-OYOVHJISSA-N 0.000 description 2
- 230000001906 cholesterol absorption Effects 0.000 description 2
- 239000000544 cholinesterase inhibitor Substances 0.000 description 2
- 229950001653 cilomilast Drugs 0.000 description 2
- RRGUKTPIGVIEKM-UHFFFAOYSA-N cilostazol Chemical compound C=1C=C2NC(=O)CCC2=CC=1OCCCCC1=NN=NN1C1CCCCC1 RRGUKTPIGVIEKM-UHFFFAOYSA-N 0.000 description 2
- 208000019425 cirrhosis of liver Diseases 0.000 description 2
- 238000011260 co-administration Methods 0.000 description 2
- 238000013267 controlled drug release Methods 0.000 description 2
- 229940038717 copaxone Drugs 0.000 description 2
- 235000005822 corn Nutrition 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 239000002852 cysteine proteinase inhibitor Substances 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- SSQJFGMEZBFMNV-PMACEKPBSA-N dexanabinol Chemical compound C1C(CO)=CC[C@@H]2C(C)(C)OC3=CC(C(C)(C)CCCCCC)=CC(O)=C3[C@H]21 SSQJFGMEZBFMNV-PMACEKPBSA-N 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- GNGACRATGGDKBX-UHFFFAOYSA-N dihydroxyacetone phosphate Chemical compound OCC(=O)COP(O)(O)=O GNGACRATGGDKBX-UHFFFAOYSA-N 0.000 description 2
- 230000006806 disease prevention Effects 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- 230000004064 dysfunction Effects 0.000 description 2
- 238000000119 electrospray ionisation mass spectrum Methods 0.000 description 2
- 239000012055 enteric layer Substances 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 229960005139 epinephrine Drugs 0.000 description 2
- 229960003745 esmolol Drugs 0.000 description 2
- AQNDDEOPVVGCPG-UHFFFAOYSA-N esmolol Chemical compound COC(=O)CCC1=CC=C(OCC(O)CNC(C)C)C=C1 AQNDDEOPVVGCPG-UHFFFAOYSA-N 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 210000002744 extracellular matrix Anatomy 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 239000007850 fluorescent dye Substances 0.000 description 2
- 238000005194 fractionation Methods 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 229960003980 galantamine Drugs 0.000 description 2
- ASUTZQLVASHGKV-UHFFFAOYSA-N galanthamine hydrochloride Natural products O1C(=C23)C(OC)=CC=C2CN(C)CCC23C1CC(O)C=C2 ASUTZQLVASHGKV-UHFFFAOYSA-N 0.000 description 2
- 239000007897 gelcap Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-L glutamate group Chemical group N[C@@H](CCC(=O)[O-])C(=O)[O-] WHUUTDBJXJRKMK-VKHMYHEASA-L 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 150000002334 glycols Chemical class 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 239000003979 granulating agent Substances 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- WROHEWWOCPRMIA-UHFFFAOYSA-N gsk-189,254 Chemical compound N1=CC(C(=O)NC)=CC=C1OC1=CC=C(CCN(CC2)C3CCC3)C2=C1 WROHEWWOCPRMIA-UHFFFAOYSA-N 0.000 description 2
- 125000001188 haloalkyl group Chemical group 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- UQEAIHBTYFGYIE-UHFFFAOYSA-N hexamethyldisiloxane Chemical compound C[Si](C)(C)O[Si](C)(C)C UQEAIHBTYFGYIE-UHFFFAOYSA-N 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 239000000017 hydrogel Substances 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 2
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 229960001388 interferon-beta Drugs 0.000 description 2
- 229960002672 isocarboxazid Drugs 0.000 description 2
- CPVQJXZBSGXTGJ-TZDLBHCHSA-N juvenile hormone II Chemical compound CC[C@]1(C)O[C@@H]1CC\C(C)=C\CC\C(C)=C\C(=O)OC CPVQJXZBSGXTGJ-TZDLBHCHSA-N 0.000 description 2
- 229940043355 kinase inhibitor Drugs 0.000 description 2
- 229960001632 labetalol Drugs 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- TYZROVQLWOKYKF-ZDUSSCGKSA-N linezolid Chemical compound O=C1O[C@@H](CNC(=O)C)CN1C(C=C1F)=CC=C1N1CCOCC1 TYZROVQLWOKYKF-ZDUSSCGKSA-N 0.000 description 2
- 229960003907 linezolid Drugs 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 210000005171 mammalian brain Anatomy 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- BUGYDGFZZOZRHP-UHFFFAOYSA-N memantine Chemical compound C1C(C2)CC3(C)CC1(C)CC2(N)C3 BUGYDGFZZOZRHP-UHFFFAOYSA-N 0.000 description 2
- 229960004640 memantine Drugs 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- NSPJNIDYTSSIIY-UHFFFAOYSA-N methoxy(methoxymethoxy)methane Chemical compound COCOCOC NSPJNIDYTSSIIY-UHFFFAOYSA-N 0.000 description 2
- VKQFCGNPDRICFG-UHFFFAOYSA-N methyl 2-methylpropyl 2,6-dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OCC(C)C)C1C1=CC=CC=C1[N+]([O-])=O VKQFCGNPDRICFG-UHFFFAOYSA-N 0.000 description 2
- 239000002899 monoamine oxidase inhibitor Substances 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 210000003643 myeloid progenitor cell Anatomy 0.000 description 2
- VBMPTAUGUUBFJK-FGJQBABTSA-N n,n-dimethyl-2-[(2r)-6-[(4-phenylphenyl)methoxy]-1,2,3,4-tetrahydronaphthalen-2-yl]ethanamine;hydrate;hydrochloride Chemical compound O.Cl.C([C@H](CC1=CC=2)CCN(C)C)CC1=CC=2OCC(C=C1)=CC=C1C1=CC=CC=C1 VBMPTAUGUUBFJK-FGJQBABTSA-N 0.000 description 2
- OKFDRAHPFKMAJH-UHFFFAOYSA-N n-(3,5-dichloropyridin-4-yl)-4-(difluoromethoxy)-8-(methanesulfonamido)dibenzofuran-1-carboxamide Chemical compound C=12C3=CC(NS(=O)(=O)C)=CC=C3OC2=C(OC(F)F)C=CC=1C(=O)NC1=C(Cl)C=NC=C1Cl OKFDRAHPFKMAJH-UHFFFAOYSA-N 0.000 description 2
- UPSFMJHZUCSEHU-JYGUBCOQSA-N n-[(2s,3r,4r,5s,6r)-2-[(2r,3s,4r,5r,6s)-5-acetamido-4-hydroxy-2-(hydroxymethyl)-6-(4-methyl-2-oxochromen-7-yl)oxyoxan-3-yl]oxy-4,5-dihydroxy-6-(hydroxymethyl)oxan-3-yl]acetamide Chemical compound CC(=O)N[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1O[C@H]1[C@H](O)[C@@H](NC(C)=O)[C@H](OC=2C=C3OC(=O)C=C(C)C3=CC=2)O[C@@H]1CO UPSFMJHZUCSEHU-JYGUBCOQSA-N 0.000 description 2
- KGDFDVHLEYUZLN-AWEZNQCLSA-N n-[(4-chlorophenyl)methyl]-4-[(2s)-2-formylpyrrolidin-1-yl]-4-oxobutanamide Chemical compound C1=CC(Cl)=CC=C1CNC(=O)CCC(=O)N1[C@H](C=O)CCC1 KGDFDVHLEYUZLN-AWEZNQCLSA-N 0.000 description 2
- 229960004255 nadolol Drugs 0.000 description 2
- VWPOSFSPZNDTMJ-UCWKZMIHSA-N nadolol Chemical compound C1[C@@H](O)[C@@H](O)CC2=C1C=CC=C2OCC(O)CNC(C)(C)C VWPOSFSPZNDTMJ-UCWKZMIHSA-N 0.000 description 2
- 229960001597 nifedipine Drugs 0.000 description 2
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 2
- 229960005366 nilvadipine Drugs 0.000 description 2
- 229960000715 nimodipine Drugs 0.000 description 2
- 229960000227 nisoldipine Drugs 0.000 description 2
- 229960005425 nitrendipine Drugs 0.000 description 2
- 239000002767 noradrenalin uptake inhibitor Substances 0.000 description 2
- 229940127221 norepinephrine reuptake inhibitor Drugs 0.000 description 2
- 229950000175 oglemilast Drugs 0.000 description 2
- 108060005714 orexin Proteins 0.000 description 2
- OFNHNCAUVYOTPM-IIIOAANCSA-N orexin-a Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](C)NC(=O)CNC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H]1NC(=O)[C@H](CO)NC(=O)[C@@H]2CSSC[C@@H](C(=O)N[C@H](C(N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N2)[C@@H](C)O)=O)CSSC1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H]1NC(=O)CC1)C1=CNC=N1 OFNHNCAUVYOTPM-IIIOAANCSA-N 0.000 description 2
- 229960004570 oxprenolol Drugs 0.000 description 2
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 230000007310 pathophysiology Effects 0.000 description 2
- DOHVAKFYAHLCJP-UHFFFAOYSA-N peldesine Chemical compound C1=2NC(N)=NC(=O)C=2NC=C1CC1=CC=CN=C1 DOHVAKFYAHLCJP-UHFFFAOYSA-N 0.000 description 2
- 229960002035 penbutolol Drugs 0.000 description 2
- KQXKVJAGOJTNJS-HNNXBMFYSA-N penbutolol Chemical compound CC(C)(C)NC[C@H](O)COC1=CC=CC=C1C1CCCC1 KQXKVJAGOJTNJS-HNNXBMFYSA-N 0.000 description 2
- 108010067535 pentapeptide QYNAD Proteins 0.000 description 2
- 239000000813 peptide hormone Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 229940124531 pharmaceutical excipient Drugs 0.000 description 2
- 125000006245 phosphate protecting group Chemical group 0.000 description 2
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 2
- 229960002508 pindolol Drugs 0.000 description 2
- JZQKKSLKJUAGIC-UHFFFAOYSA-N pindolol Chemical compound CC(C)NCC(O)COC1=CC=CC2=C1C=CN2 JZQKKSLKJUAGIC-UHFFFAOYSA-N 0.000 description 2
- 239000013612 plasmid Substances 0.000 description 2
- 229920000747 poly(lactic acid) Polymers 0.000 description 2
- 229920001610 polycaprolactone Polymers 0.000 description 2
- 229920002721 polycyanoacrylate Polymers 0.000 description 2
- 239000004626 polylactic acid Substances 0.000 description 2
- 229920000656 polylysine Polymers 0.000 description 2
- 229920006324 polyoxymethylene Polymers 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- RZIMNEGTIDYAGZ-HNSJZBNRSA-N pro-gastrin Chemical compound N([C@@H](CC(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N1CCC[C@H]1C(=O)NCC(=O)NCC(=O)NCC(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N1CCC[C@H]1C(=O)NCC(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)NCC(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)C(=O)[C@@H]1CCC(=O)N1 RZIMNEGTIDYAGZ-HNSJZBNRSA-N 0.000 description 2
- 239000003649 prolyl endopeptidase inhibitor Substances 0.000 description 2
- 229940121649 protein inhibitor Drugs 0.000 description 2
- 239000012268 protein inhibitor Substances 0.000 description 2
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 2
- 208000005069 pulmonary fibrosis Diseases 0.000 description 2
- 229940038850 rebif Drugs 0.000 description 2
- 239000000018 receptor agonist Substances 0.000 description 2
- 229940044601 receptor agonist Drugs 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000002461 renin inhibitor Substances 0.000 description 2
- 229940086526 renin-inhibitors Drugs 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- MNDBXUUTURYVHR-UHFFFAOYSA-N roflumilast Chemical compound FC(F)OC1=CC=C(C(=O)NC=2C(=CN=CC=2Cl)Cl)C=C1OCC1CC1 MNDBXUUTURYVHR-UHFFFAOYSA-N 0.000 description 2
- 229960002586 roflumilast Drugs 0.000 description 2
- NEMGRZFTLSKBAP-LBPRGKRZSA-N safinamide Chemical compound C1=CC(CN[C@@H](C)C(N)=O)=CC=C1OCC1=CC=CC(F)=C1 NEMGRZFTLSKBAP-LBPRGKRZSA-N 0.000 description 2
- 229950002652 safinamide Drugs 0.000 description 2
- CDAISMWEOUEBRE-CDRYSYESSA-N scyllo-inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O CDAISMWEOUEBRE-CDRYSYESSA-N 0.000 description 2
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 description 2
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 description 2
- 210000000582 semen Anatomy 0.000 description 2
- 239000003775 serotonin noradrenalin reuptake inhibitor Substances 0.000 description 2
- 229960004034 sitagliptin Drugs 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 235000015424 sodium Nutrition 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 2
- 239000004299 sodium benzoate Substances 0.000 description 2
- 235000010234 sodium benzoate Nutrition 0.000 description 2
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 2
- 229960002370 sotalol Drugs 0.000 description 2
- ZBMZVLHSJCTVON-UHFFFAOYSA-N sotalol Chemical compound CC(C)NCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 ZBMZVLHSJCTVON-UHFFFAOYSA-N 0.000 description 2
- 230000004936 stimulating effect Effects 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 230000005062 synaptic transmission Effects 0.000 description 2
- 229960001685 tacrine Drugs 0.000 description 2
- YLJREFDVOIBQDA-UHFFFAOYSA-N tacrine Chemical compound C1=CC=C2C(N)=C(CCCC3)C3=NC2=C1 YLJREFDVOIBQDA-UHFFFAOYSA-N 0.000 description 2
- XOAAWQZATWQOTB-UHFFFAOYSA-N taurine Chemical compound NCCS(O)(=O)=O XOAAWQZATWQOTB-UHFFFAOYSA-N 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- FGTJJHCZWOVVNH-UHFFFAOYSA-N tert-butyl-[tert-butyl(dimethyl)silyl]oxy-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)O[Si](C)(C)C(C)(C)C FGTJJHCZWOVVNH-UHFFFAOYSA-N 0.000 description 2
- 229950002896 tetomilast Drugs 0.000 description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- 229960000278 theophylline Drugs 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 125000004001 thioalkyl group Chemical group 0.000 description 2
- 229960004605 timolol Drugs 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 229950003899 tofimilast Drugs 0.000 description 2
- 229960003570 tramiprosate Drugs 0.000 description 2
- LGSAOJLQTXCYHF-UHFFFAOYSA-N tri(propan-2-yl)-tri(propan-2-yl)silyloxysilane Chemical compound CC(C)[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)C(C)C LGSAOJLQTXCYHF-UHFFFAOYSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 230000001228 trophic effect Effects 0.000 description 2
- 229960004441 tyrosine Drugs 0.000 description 2
- 239000002691 unilamellar liposome Substances 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- 239000000230 xanthan gum Substances 0.000 description 2
- 229920001285 xanthan gum Polymers 0.000 description 2
- 235000010493 xanthan gum Nutrition 0.000 description 2
- 229940082509 xanthan gum Drugs 0.000 description 2
- ORZXYSPOAVJYRU-HOTGVXAUSA-N z-pro-prolinal Chemical compound O=C[C@@H]1CCCN1C(=O)[C@H]1N(C(=O)OCC=2C=CC=CC=2)CCC1 ORZXYSPOAVJYRU-HOTGVXAUSA-N 0.000 description 2
- 229930195724 β-lactose Natural products 0.000 description 2
- AHOUBRCZNHFOSL-YOEHRIQHSA-N (+)-Casbol Chemical compound C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-YOEHRIQHSA-N 0.000 description 1
- IGLYMJRIWWIQQE-QUOODJBBSA-N (1S,2R)-2-phenylcyclopropan-1-amine (1R,2S)-2-phenylcyclopropan-1-amine Chemical compound N[C@H]1C[C@@H]1C1=CC=CC=C1.N[C@@H]1C[C@H]1C1=CC=CC=C1 IGLYMJRIWWIQQE-QUOODJBBSA-N 0.000 description 1
- NVVSPGQEXMJZIR-BMIGLBTASA-N (1s,6r)-3-{[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8h)-yl]carbonyl}-6-(2,4,5-trifluorophenyl)cyclohex-3-en-1-amine Chemical compound C1([C@H]2CC=C(C[C@@H]2N)C(=O)N2CC=3N(C(=NN=3)C(F)(F)F)CC2)=CC(F)=C(F)C=C1F NVVSPGQEXMJZIR-BMIGLBTASA-N 0.000 description 1
- ZSTLCHCDLIUXJE-ZGBAEQJLSA-N (2S,5S)-2-methylspiro[1,3-oxathiolane-5,3'-1-azabicyclo[2.2.2]octane] hydrate dihydrochloride Chemical compound O.Cl.Cl.C1S[C@@H](C)O[C@@]21C(CC1)CCN1C2.C1S[C@@H](C)O[C@@]21C(CC1)CCN1C2 ZSTLCHCDLIUXJE-ZGBAEQJLSA-N 0.000 description 1
- CMIBUZBMZCBCAT-HZPDHXFCSA-N (2r,3r)-2,3-bis[(4-methylbenzoyl)oxy]butanedioic acid Chemical compound C1=CC(C)=CC=C1C(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(=O)C1=CC=C(C)C=C1 CMIBUZBMZCBCAT-HZPDHXFCSA-N 0.000 description 1
- RDEIXVOBVLKYNT-VQBXQJRRSA-N (2r,3r,4r,5r)-2-[(1s,2s,3r,4s,6r)-4,6-diamino-3-[(2r,3r,6s)-3-amino-6-(1-aminoethyl)oxan-2-yl]oxy-2-hydroxycyclohexyl]oxy-5-methyl-4-(methylamino)oxane-3,5-diol;(2r,3r,4r,5r)-2-[(1s,2s,3r,4s,6r)-4,6-diamino-3-[(2r,3r,6s)-3-amino-6-(aminomethyl)oxan-2-yl]o Chemical compound OS(O)(=O)=O.O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H](CC[C@@H](CN)O2)N)[C@@H](N)C[C@H]1N.O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H](CC[C@H](O2)C(C)N)N)[C@@H](N)C[C@H]1N.O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N RDEIXVOBVLKYNT-VQBXQJRRSA-N 0.000 description 1
- WORSVFBVUCBRIP-VNQPRFMTSA-N (2r,3s)-n-[(2s)-3,3-dimethyl-1-oxo-1-(pyridin-2-ylamino)butan-2-yl]-n'-hydroxy-3-methoxy-2-(2-methylpropyl)butanediamide Chemical compound ONC(=O)[C@@H](OC)[C@@H](CC(C)C)C(=O)N[C@@H](C(C)(C)C)C(=O)NC1=CC=CC=N1 WORSVFBVUCBRIP-VNQPRFMTSA-N 0.000 description 1
- HXEHCCYOTHDPPI-NBHSMZAVSA-N (2s)-2-[[(2s)-2-(3,5-difluorophenyl)-2-hydroxyacetyl]amino]-n-[(5s)-3-methyl-4-oxo-5h-2,3-benzodiazepin-5-yl]propanamide Chemical compound C1([C@H](O)C(=O)N[C@@H](C)C(=O)N[C@@H]2C(N(C)N=CC3=CC=CC=C32)=O)=CC(F)=CC(F)=C1 HXEHCCYOTHDPPI-NBHSMZAVSA-N 0.000 description 1
- NEHKZPHIKKEMAZ-ZFVKSOIMSA-N (2s)-2-[[(2s,3r)-2-[[(2s)-2-[[(2s,3s)-2-[[2-[[(2s,3s)-2-[[2-[[(2s)-2-[[(2s)-2-azaniumylpropanoyl]amino]propanoyl]amino]acetyl]amino]-3-methylpentanoyl]amino]acetyl]amino]-3-methylpentanoyl]amino]-4-methylpentanoyl]amino]-3-hydroxybutanoyl]amino]-3-methylb Chemical compound C[C@H](N)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(O)=O NEHKZPHIKKEMAZ-ZFVKSOIMSA-N 0.000 description 1
- IVBVTDXOGUNDHC-LURJTMIESA-N (2s)-2-amino-1-pyrrolidin-1-ylpropan-1-one Chemical compound C[C@H](N)C(=O)N1CCCC1 IVBVTDXOGUNDHC-LURJTMIESA-N 0.000 description 1
- YLOCGHYTXIINAI-XKUOMLDTSA-N (2s)-2-amino-3-(4-hydroxyphenyl)propanoic acid;(2s)-2-aminopentanedioic acid;(2s)-2-aminopropanoic acid;(2s)-2,6-diaminohexanoic acid Chemical compound C[C@H](N)C(O)=O.NCCCC[C@H](N)C(O)=O.OC(=O)[C@@H](N)CCC(O)=O.OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 YLOCGHYTXIINAI-XKUOMLDTSA-N 0.000 description 1
- VEPDWBJBPXVCEN-NSHDSACASA-N (2s)-2-amino-n-(4-methyl-2-oxochromen-7-yl)pentanediamide Chemical compound C1=C(NC(=O)[C@@H](N)CCC(N)=O)C=CC2=C1OC(=O)C=C2C VEPDWBJBPXVCEN-NSHDSACASA-N 0.000 description 1
- PHOZOHFUXHPOCK-QMMMGPOBSA-N (2s)-2-ethyl-8-methyl-1-thia-4,8-diazaspiro[4.5]decan-3-one Chemical compound N1C(=O)[C@H](CC)SC11CCN(C)CC1 PHOZOHFUXHPOCK-QMMMGPOBSA-N 0.000 description 1
- YZJSUQQZGCHHNQ-BYPYZUCNSA-N (2s)-6-amino-2-azaniumyl-6-oxohexanoate Chemical group OC(=O)[C@@H](N)CCCC(N)=O YZJSUQQZGCHHNQ-BYPYZUCNSA-N 0.000 description 1
- ARNUPLMOASAEAN-ASLNEKEESA-N (2s,3s)-2-amino-3-methyl-1-(1,3-thiazolidin-2-yl)pentan-1-one Chemical compound CC[C@H](C)[C@H](N)C(=O)C1NCCS1 ARNUPLMOASAEAN-ASLNEKEESA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- JSYGLDMGERSRPC-FQUUOJAGSA-N (2s,4s)-4-fluoro-1-[2-[[(1r,3s)-3-(1,2,4-triazol-1-ylmethyl)cyclopentyl]amino]acetyl]pyrrolidine-2-carbonitrile Chemical compound C1[C@@H](F)C[C@@H](C#N)N1C(=O)CN[C@H]1C[C@@H](CN2N=CN=C2)CC1 JSYGLDMGERSRPC-FQUUOJAGSA-N 0.000 description 1
- QAXRREHLPKHWBI-IJDFPQMLSA-N (3r)-3-(3-hexylsulfanylpyrazin-2-yl)oxy-1-azabicyclo[2.2.1]heptane;hydrochloride Chemical compound Cl.CCCCCCSC1=NC=CN=C1O[C@@H]1C(C2)CCN2C1 QAXRREHLPKHWBI-IJDFPQMLSA-N 0.000 description 1
- RICKQPKTSOSCOF-HKUYNNGSSA-N (3s)-2-[(2s)-2-amino-3-(1h-indol-3-yl)propanoyl]-3,4-dihydro-1h-isoquinoline-3-carboxylic acid Chemical compound C1C2=CC=CC=C2C[C@@H](C(O)=O)N1C(=O)[C@@H](N)CC1=CNC2=CC=CC=C12 RICKQPKTSOSCOF-HKUYNNGSSA-N 0.000 description 1
- DIWRORZWFLOCLC-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1,3-dihydro-1,4-benzodiazepin-2-one Chemical compound N([C@H](C(NC1=CC=C(Cl)C=C11)=O)O)=C1C1=CC=CC=C1Cl DIWRORZWFLOCLC-HNNXBMFYSA-N 0.000 description 1
- SVJMLYUFVDMUHP-XIFFEERXSA-N (4S)-2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylic acid O5-[3-(4,4-diphenyl-1-piperidinyl)propyl] ester O3-methyl ester Chemical compound C1([C@@H]2C(=C(C)NC(C)=C2C(=O)OC)C(=O)OCCCN2CCC(CC2)(C=2C=CC=CC=2)C=2C=CC=CC=2)=CC=CC([N+]([O-])=O)=C1 SVJMLYUFVDMUHP-XIFFEERXSA-N 0.000 description 1
- FNCPECYULVHYCJ-PAFCVIRNSA-N (4e,6e,8e,10z,12z,14e,16e)-3-[(3s,4s,5s,6r)-4-amino-3,5-dihydroxy-6-methyloxan-2-yl]oxy-19-[7-(4-aminophenyl)-5-hydroxy-7-oxoheptan-2-yl]-23,25,29,31,33,35,37-heptahydroxy-18-methyl-21-oxo-20,39-dioxabicyclo[33.3.1]nonatriaconta-4,6,8,10,12,14,16-heptaene Chemical compound O1C(=O)CC(O)CC(O)CCCC(O)CC(O)CC(O)CC(O2)(O)CC(O)C(C(O)=O)C2CC(OC2[C@H]([C@@H](N)[C@H](O)[C@@H](C)O2)O)\C=C\C=C\C=C\C=C/C=C\C=C\C=C\C(C)C1C(C)CCC(O)CC(=O)C1=CC=C(N)C=C1 FNCPECYULVHYCJ-PAFCVIRNSA-N 0.000 description 1
- 125000004400 (C1-C12) alkyl group Chemical group 0.000 description 1
- AFDXODALSZRGIH-QPJJXVBHSA-N (E)-3-(4-methoxyphenyl)prop-2-enoic acid Chemical compound COC1=CC=C(\C=C\C(O)=O)C=C1 AFDXODALSZRGIH-QPJJXVBHSA-N 0.000 description 1
- WSEQXVZVJXJVFP-HXUWFJFHSA-N (R)-citalopram Chemical compound C1([C@@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-HXUWFJFHSA-N 0.000 description 1
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 description 1
- NSMXQKNUPPXBRG-SECBINFHSA-N (R)-lisofylline Chemical compound O=C1N(CCCC[C@H](O)C)C(=O)N(C)C2=C1N(C)C=N2 NSMXQKNUPPXBRG-SECBINFHSA-N 0.000 description 1
- ZEUITGRIYCTCEM-KRWDZBQOSA-N (S)-duloxetine Chemical compound C1([C@@H](OC=2C3=CC=CC=C3C=CC=2)CCNC)=CC=CS1 ZEUITGRIYCTCEM-KRWDZBQOSA-N 0.000 description 1
- 0 *c(c(*)c1*)c(*)c(C(CC2=O)N2c2ccc3nc[n]c3c2)c1I Chemical compound *c(c(*)c1*)c(*)c(C(CC2=O)N2c2ccc3nc[n]c3c2)c1I 0.000 description 1
- OXFSTTJBVAAALW-UHFFFAOYSA-N 1,3-dihydroimidazole-2-thione Chemical class SC1=NC=CN1 OXFSTTJBVAAALW-UHFFFAOYSA-N 0.000 description 1
- NWOYMJLBOFRVDM-UHFFFAOYSA-N 1-(3H-benzimidazol-5-yl)-4-[4-(2,2-difluoropropoxy)-2,3-difluorophenyl]azetidin-2-one Chemical compound FC(COC1=C(C(=C(C=C1)C1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)F)(C)F NWOYMJLBOFRVDM-UHFFFAOYSA-N 0.000 description 1
- GURVAWDLVHIGNH-UHFFFAOYSA-N 1-(3H-benzimidazol-5-yl)-4-[4-(3,3-difluoropropoxy)-2,6-difluorophenyl]azetidin-2-one Chemical compound FC(CCOC1=CC(=C(C(=C1)F)C1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)F GURVAWDLVHIGNH-UHFFFAOYSA-N 0.000 description 1
- QKUNGNBZEVCPSL-UHFFFAOYSA-N 1-(3H-benzimidazol-5-yl)-4-[4-(3,3-difluoropropoxy)-2-fluorophenyl]azetidin-2-one Chemical compound FC(CCOC1=CC(=C(C=C1)C1CC(N1C1=CC2=C(NC=N2)C=C1)=O)F)F QKUNGNBZEVCPSL-UHFFFAOYSA-N 0.000 description 1
- NSMXQKNUPPXBRG-UHFFFAOYSA-N 1-(5-hydroxyhexyl)-3,7-dimethyl-3,7-dihydro-1H-purine-2,6-dione Chemical compound O=C1N(CCCCC(O)C)C(=O)N(C)C2=C1N(C)C=N2 NSMXQKNUPPXBRG-UHFFFAOYSA-N 0.000 description 1
- IWUCXVSUMQZMFG-RGDLXGNYSA-N 1-[(2s,3s,4r,5s)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2,4-triazole-3-carboxamide Chemical compound N1=C(C(=O)N)N=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 IWUCXVSUMQZMFG-RGDLXGNYSA-N 0.000 description 1
- WFFZHKKSIDENAJ-UHFFFAOYSA-N 1-[2-(4-hydroxyphenoxy)ethyl]-4-[(4-methylphenyl)methyl]piperidin-4-ol;hydrochloride Chemical compound Cl.C1=CC(C)=CC=C1CC1(O)CCN(CCOC=2C=CC(O)=CC=2)CC1 WFFZHKKSIDENAJ-UHFFFAOYSA-N 0.000 description 1
- STSYUQWYKFKDNG-UHFFFAOYSA-N 1-[3-fluoro-4-[4-(trifluoromethyl)phenyl]phenyl]cyclopropane-1-carboxylic acid Chemical compound C=1C=C(C=2C=CC(=CC=2)C(F)(F)F)C(F)=CC=1C1(C(=O)O)CC1 STSYUQWYKFKDNG-UHFFFAOYSA-N 0.000 description 1
- LIYLTQQDABRNRX-UHFFFAOYSA-N 1-[4-(3,4-dichlorophenyl)-3-fluorophenyl]cyclopropane-1-carboxylic acid Chemical compound C=1C=C(C=2C=C(Cl)C(Cl)=CC=2)C(F)=CC=1C1(C(=O)O)CC1 LIYLTQQDABRNRX-UHFFFAOYSA-N 0.000 description 1
- RRQYJINTUHWNHW-UHFFFAOYSA-N 1-ethoxy-2-(2-ethoxyethoxy)ethane Chemical class CCOCCOCCOCC RRQYJINTUHWNHW-UHFFFAOYSA-N 0.000 description 1
- SDTORDSXCYSNTD-UHFFFAOYSA-N 1-methoxy-4-[(4-methoxyphenyl)methoxymethyl]benzene Chemical compound C1=CC(OC)=CC=C1COCC1=CC=C(OC)C=C1 SDTORDSXCYSNTD-UHFFFAOYSA-N 0.000 description 1
- OPISVEPYALEQJT-UHFFFAOYSA-N 1-phenylazetidin-2-one Chemical group O=C1CCN1C1=CC=CC=C1 OPISVEPYALEQJT-UHFFFAOYSA-N 0.000 description 1
- MQBLKQOFJZXAOU-NSHDSACASA-N 1051ln7flm Chemical compound C1([C@@H](N2C3=C(C(=NC4=NC=NN43)C)CC2)C)=CC=C(F)C=C1 MQBLKQOFJZXAOU-NSHDSACASA-N 0.000 description 1
- IHWDSEPNZDYMNF-UHFFFAOYSA-N 1H-indol-2-amine Chemical compound C1=CC=C2NC(N)=CC2=C1 IHWDSEPNZDYMNF-UHFFFAOYSA-N 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- DCGKDDVUUMOTDH-UHFFFAOYSA-N 2,5-difluoro-4-methoxybenzaldehyde Chemical compound COC1=CC(F)=C(C=O)C=C1F DCGKDDVUUMOTDH-UHFFFAOYSA-N 0.000 description 1
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 1
- JHVHEDNLONERHY-UHFFFAOYSA-N 2-(2-chloro-5-methylsulfanylphenyl)-1-methyl-1-(3-methylsulfanylphenyl)guanidine Chemical compound CSC1=CC=CC(N(C)C(N)=NC=2C(=CC=C(SC)C=2)Cl)=C1 JHVHEDNLONERHY-UHFFFAOYSA-N 0.000 description 1
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical group O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 1
- IMSODMZESSGVBE-UHFFFAOYSA-N 2-Oxazoline Chemical group C1CN=CO1 IMSODMZESSGVBE-UHFFFAOYSA-N 0.000 description 1
- FGBGXESDYFKUFX-UHFFFAOYSA-N 2-[2,6-ditert-butyl-4-[2-(3,5-ditert-butyl-4-hydroxyphenyl)sulfanylpropan-2-ylsulfanyl]phenoxy]acetic acid Chemical compound CC(C)(C)C1=C(O)C(C(C)(C)C)=CC(SC(C)(C)SC=2C=C(C(OCC(O)=O)=C(C=2)C(C)(C)C)C(C)(C)C)=C1 FGBGXESDYFKUFX-UHFFFAOYSA-N 0.000 description 1
- GKGRZLGAQZPEHO-UHFFFAOYSA-N 2-[4-[(4-fluorophenyl)methyl]piperidin-1-yl]-2-oxo-n-(2-oxo-3h-1,3-benzoxazol-6-yl)acetamide Chemical compound C1=CC(F)=CC=C1CC1CCN(C(=O)C(=O)NC=2C=C3OC(=O)NC3=CC=2)CC1 GKGRZLGAQZPEHO-UHFFFAOYSA-N 0.000 description 1
- FIMRNLAKAARHPD-IRXDYDNUSA-N 2-[4-[[2-[(2s,5r)-2-cyano-5-ethynylpyrrolidin-1-yl]-2-oxoethyl]amino]-4-methylpiperidin-1-yl]pyridine-4-carboxylic acid Chemical compound C1CC(C)(NCC(=O)N2[C@@H](CC[C@@H]2C#C)C#N)CCN1C1=CC(C(O)=O)=CC=N1 FIMRNLAKAARHPD-IRXDYDNUSA-N 0.000 description 1
- XPRDUGXOWVXZLL-UHFFFAOYSA-N 2-[[2-fluoro-4-(3-methoxyphenyl)phenyl]carbamoyl]cyclopentene-1-carboxylic acid Chemical compound COC1=CC=CC(C=2C=C(F)C(NC(=O)C=3CCCC=3C(O)=O)=CC=2)=C1 XPRDUGXOWVXZLL-UHFFFAOYSA-N 0.000 description 1
- NSVFSAJIGAJDMR-UHFFFAOYSA-N 2-[benzyl(phenyl)amino]ethyl 5-(5,5-dimethyl-2-oxido-1,3,2-dioxaphosphinan-2-yl)-2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3-carboxylate Chemical compound CC=1NC(C)=C(C(=O)OCCN(CC=2C=CC=CC=2)C=2C=CC=CC=2)C(C=2C=C(C=CC=2)[N+]([O-])=O)C=1P1(=O)OCC(C)(C)CO1 NSVFSAJIGAJDMR-UHFFFAOYSA-N 0.000 description 1
- PPJUTIWEKWGPCG-UHFFFAOYSA-N 2-amino-5-bromo-6-(2,5-difluorophenyl)-1h-pyrimidin-4-one Chemical compound N1C(N)=NC(=O)C(Br)=C1C1=CC(F)=CC=C1F PPJUTIWEKWGPCG-UHFFFAOYSA-N 0.000 description 1
- HVRYWUNGKDMRNA-UHFFFAOYSA-N 2-amino-6-(2,5-dichlorophenyl)-5-iodo-1h-pyrimidin-4-one Chemical compound N1C(N)=NC(=O)C(I)=C1C1=CC(Cl)=CC=C1Cl HVRYWUNGKDMRNA-UHFFFAOYSA-N 0.000 description 1
- 125000001731 2-cyanoethyl group Chemical group [H]C([H])(*)C([H])([H])C#N 0.000 description 1
- 125000002774 3,4-dimethoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C(OC([H])([H])[H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- IPKFWLRXFSUTDZ-UHFFFAOYSA-N 3-ethyl-5-(1,4,5,6-tetrahydropyrimidin-5-yl)-1,2,4-oxadiazole Chemical compound CCC1=NOC(C2CN=CNC2)=N1 IPKFWLRXFSUTDZ-UHFFFAOYSA-N 0.000 description 1
- IZKNCYISUDTFAC-UHFFFAOYSA-N 4-(2,2-difluoropropoxy)-2,3-difluorobenzaldehyde Chemical compound FC(COC1=C(C(=C(C=O)C=C1)F)F)(C)F IZKNCYISUDTFAC-UHFFFAOYSA-N 0.000 description 1
- DVPQQLNKFWRYHT-UHFFFAOYSA-N 4-(2,2-difluoropropoxy)-2,6-difluorobenzaldehyde Chemical compound FC(COC1=CC(=C(C=O)C(=C1)F)F)(C)F DVPQQLNKFWRYHT-UHFFFAOYSA-N 0.000 description 1
- CDBMSJGONNALOZ-UHFFFAOYSA-N 4-(3,3-difluoropropoxy)-2,3-difluorobenzaldehyde Chemical compound FC(CCOC1=C(C(=C(C=O)C=C1)F)F)F CDBMSJGONNALOZ-UHFFFAOYSA-N 0.000 description 1
- YWDUSTXSGKKZAX-UHFFFAOYSA-N 4-(3,3-difluoropropoxy)-2,6-difluorobenzaldehyde Chemical compound FC(CCOC1=CC(=C(C=O)C(=C1)F)F)F YWDUSTXSGKKZAX-UHFFFAOYSA-N 0.000 description 1
- GBBJZZLUGBSILM-UHFFFAOYSA-N 4-(3,3-difluoropropoxy)-2-fluorobenzaldehyde Chemical compound FC(CCOC1=CC(=C(C=O)C=C1)F)F GBBJZZLUGBSILM-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- RURHILYUWQEGOS-VOTSOKGWSA-N 4-Methylcinnamic acid Chemical group CC1=CC=C(\C=C\C(O)=O)C=C1 RURHILYUWQEGOS-VOTSOKGWSA-N 0.000 description 1
- IZAOBRWCUGOKNH-OAHLLOKOSA-N 4-[2-[(1R)-1-(N-(4-chlorophenyl)sulfonyl-2,5-difluoroanilino)ethyl]-5-fluorophenyl]butanoic acid Chemical compound C=1C=C(Cl)C=CC=1S(=O)(=O)N([C@H](C)C=1C(=CC(F)=CC=1)CCCC(O)=O)C1=CC(F)=CC=C1F IZAOBRWCUGOKNH-OAHLLOKOSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- 125000005274 4-hydroxybenzoic acid group Chemical group 0.000 description 1
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- OJBLXSPBJMGZDN-UHFFFAOYSA-N 5-[3-(difluoromethyl)-4-fluorophenyl]-3-[(2-methylimidazol-1-yl)methyl]pyridazine;dihydrochloride Chemical compound Cl.Cl.CC1=NC=CN1CC1=CC(C=2C=C(C(F)=CC=2)C(F)F)=CN=N1 OJBLXSPBJMGZDN-UHFFFAOYSA-N 0.000 description 1
- WZJHGCSWOYSTGN-WBAQKLHDSA-N 5-[[(2r,3s)-3-[[(2s)-2-[[(2s)-2-[[(2s)-2-amino-3-(oxaloamino)propanoyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-2-hydroxy-4-phenylbutanoyl]amino]benzene-1,3-dicarboxylic acid Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@@H](N)CNC(=O)C(O)=O)C(C)C)CC(C)C)[C@@H](O)C(=O)NC=1C=C(C=C(C=1)C(O)=O)C(O)=O)C1=CC=CC=C1 WZJHGCSWOYSTGN-WBAQKLHDSA-N 0.000 description 1
- PSXOKXJMVRSARX-SCSAIBSYSA-N 5-chloro-n-[(2s)-4,4,4-trifluoro-1-hydroxy-3-(trifluoromethyl)butan-2-yl]thiophene-2-sulfonamide Chemical compound FC(F)(F)C(C(F)(F)F)[C@@H](CO)NS(=O)(=O)C1=CC=C(Cl)S1 PSXOKXJMVRSARX-SCSAIBSYSA-N 0.000 description 1
- RZTAMFZIAATZDJ-HNNXBMFYSA-N 5-o-ethyl 3-o-methyl (4s)-4-(2,3-dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound CCOC(=O)C1=C(C)NC(C)=C(C(=O)OC)[C@@H]1C1=CC=CC(Cl)=C1Cl RZTAMFZIAATZDJ-HNNXBMFYSA-N 0.000 description 1
- MJZJYWCQPMNPRM-UHFFFAOYSA-N 6,6-dimethyl-1-[3-(2,4,5-trichlorophenoxy)propoxy]-1,6-dihydro-1,3,5-triazine-2,4-diamine Chemical compound CC1(C)N=C(N)N=C(N)N1OCCCOC1=CC(Cl)=C(Cl)C=C1Cl MJZJYWCQPMNPRM-UHFFFAOYSA-N 0.000 description 1
- XVMSFILGAMDHEY-UHFFFAOYSA-N 6-(4-aminophenyl)sulfonylpyridin-3-amine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=N1 XVMSFILGAMDHEY-UHFFFAOYSA-N 0.000 description 1
- VNACOBVZDCLAEV-UHFFFAOYSA-N 6-[2-[[2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl]amino]ethylamino]pyridine-3-carbonitrile;dihydrochloride Chemical compound Cl.Cl.C1CCC(C#N)N1C(=O)CNCCNC1=CC=C(C#N)C=N1 VNACOBVZDCLAEV-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- UAWVRVFHMOSAPU-UHFFFAOYSA-N 7-chlorokynurenic acid Chemical compound C1=CC(Cl)=CC2=NC(C(=O)O)=CC(O)=C21 UAWVRVFHMOSAPU-UHFFFAOYSA-N 0.000 description 1
- 229940124596 AChE inhibitor Drugs 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 229940098747 AMPA receptor antagonist Drugs 0.000 description 1
- 239000000775 AMPA receptor antagonist Substances 0.000 description 1
- 229940080778 Adenosine deaminase inhibitor Drugs 0.000 description 1
- 102000009346 Adenosine receptors Human genes 0.000 description 1
- 108050000203 Adenosine receptors Proteins 0.000 description 1
- FHHHOYXPRDYHEZ-COXVUDFISA-N Alacepril Chemical compound CC(=O)SC[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 FHHHOYXPRDYHEZ-COXVUDFISA-N 0.000 description 1
- JBMKAUGHUNFTOL-UHFFFAOYSA-N Aldoclor Chemical class C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NC=NS2(=O)=O JBMKAUGHUNFTOL-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 108010043324 Amyloid Precursor Protein Secretases Proteins 0.000 description 1
- 101710137189 Amyloid-beta A4 protein Proteins 0.000 description 1
- 102100022704 Amyloid-beta precursor protein Human genes 0.000 description 1
- 101710151993 Amyloid-beta precursor protein Proteins 0.000 description 1
- 206010002383 Angina Pectoris Diseases 0.000 description 1
- 102000015427 Angiotensins Human genes 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 200000000007 Arterial disease Diseases 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 108010027740 BHT 3009 Proteins 0.000 description 1
- 102100021257 Beta-secretase 1 Human genes 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 102100023700 C-C motif chemokine 16 Human genes 0.000 description 1
- 102100023701 C-C motif chemokine 18 Human genes 0.000 description 1
- 102100032366 C-C motif chemokine 7 Human genes 0.000 description 1
- 102100034871 C-C motif chemokine 8 Human genes 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000002080 C09CA02 - Eprosartan Substances 0.000 description 1
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 1
- 239000002081 C09CA05 - Tasosartan Substances 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- ZXAVXYOUGKBIAC-UHFFFAOYSA-N CC(F)(F)COc1ccc(C=O)c(F)c1 Chemical compound CC(F)(F)COc1ccc(C=O)c(F)c1 ZXAVXYOUGKBIAC-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229940121926 Calpain inhibitor Drugs 0.000 description 1
- 102100035037 Calpastatin Human genes 0.000 description 1
- 108090000625 Cathepsin K Proteins 0.000 description 1
- 102000004171 Cathepsin K Human genes 0.000 description 1
- 229940122805 Cathepsin S inhibitor Drugs 0.000 description 1
- 108010067225 Cell Adhesion Molecules Proteins 0.000 description 1
- IFYLTXNCFVRALQ-OALUTQOASA-N Ceronapril Chemical compound O([C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)P(O)(=O)CCCCC1=CC=CC=C1 IFYLTXNCFVRALQ-OALUTQOASA-N 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- 108010089448 Cholecystokinin B Receptor Proteins 0.000 description 1
- GDLIGKIOYRNHDA-UHFFFAOYSA-N Clomipramine Chemical compound C1CC2=CC=C(Cl)C=C2N(CCCN(C)C)C2=CC=CC=C21 GDLIGKIOYRNHDA-UHFFFAOYSA-N 0.000 description 1
- 108010092897 Coat Protein Complex I Proteins 0.000 description 1
- 102000016726 Coat Protein Complex I Human genes 0.000 description 1
- 101000944206 Conus geographus Conantokin-G Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- MFYSYFVPBJMHGN-UHFFFAOYSA-N Cortisone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)(O)C(=O)CO)C4C3CCC2=C1 MFYSYFVPBJMHGN-UHFFFAOYSA-N 0.000 description 1
- 101710095468 Cyclase Proteins 0.000 description 1
- 229940123320 Cyclase inhibitor Drugs 0.000 description 1
- 102000001189 Cyclic Peptides Human genes 0.000 description 1
- 108010069514 Cyclic Peptides Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-UHFFFAOYSA-N D-alpha-Ala Natural products CC([NH3+])C([O-])=O QNAYBMKLOCPYGJ-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- URRAHSMDPCMOTH-LNLFQRSKSA-N Denagliptin Chemical compound C=1C=C(F)C=CC=1C([C@H](N)C(=O)N1[C@@H](C[C@H](F)C1)C#N)C1=CC=C(F)C=C1 URRAHSMDPCMOTH-LNLFQRSKSA-N 0.000 description 1
- VARHXCYGZKSOOO-UHFFFAOYSA-N Deoxyvasicinone Natural products C1=CC=C2C(=O)N(CCC3)C3=NC2=C1 VARHXCYGZKSOOO-UHFFFAOYSA-N 0.000 description 1
- HCYAFALTSJYZDH-UHFFFAOYSA-N Desimpramine Chemical compound C1CC2=CC=CC=C2N(CCCNC)C2=CC=CC=C21 HCYAFALTSJYZDH-UHFFFAOYSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- 102000016622 Dipeptidyl Peptidase 4 Human genes 0.000 description 1
- 229940124213 Dipeptidyl peptidase 4 (DPP IV) inhibitor Drugs 0.000 description 1
- 108090000194 Dipeptidyl-peptidases and tripeptidyl-peptidases Proteins 0.000 description 1
- 102000003779 Dipeptidyl-peptidases and tripeptidyl-peptidases Human genes 0.000 description 1
- YHFUPQNXMNMQAK-UHFFFAOYSA-N Elsibucol Chemical compound CC(C)(C)C1=C(O)C(C(C)(C)C)=CC(SC(C)(C)SC=2C=C(C(OCCCC(O)=O)=C(C=2)C(C)(C)C)C(C)(C)C)=C1 YHFUPQNXMNMQAK-UHFFFAOYSA-N 0.000 description 1
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 1
- 210000000712 G cell Anatomy 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 102100029846 Glutaminyl-peptide cyclotransferase Human genes 0.000 description 1
- 101710090339 Glutaminyl-peptide cyclotransferase-like protein Proteins 0.000 description 1
- NMJREATYWWNIKX-UHFFFAOYSA-N GnRH Chemical compound C1CCC(C(=O)NCC(N)=O)N1C(=O)C(CC(C)C)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)CNC(=O)C(NC(=O)C(CO)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C(CC=1NC=NC=1)NC(=O)C1NC(=O)CC1)CC1=CC=C(O)C=C1 NMJREATYWWNIKX-UHFFFAOYSA-N 0.000 description 1
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 1
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 101000894895 Homo sapiens Beta-secretase 1 Proteins 0.000 description 1
- 101000978375 Homo sapiens C-C motif chemokine 16 Proteins 0.000 description 1
- 101000978371 Homo sapiens C-C motif chemokine 18 Proteins 0.000 description 1
- 101000797758 Homo sapiens C-C motif chemokine 7 Proteins 0.000 description 1
- 101000946794 Homo sapiens C-C motif chemokine 8 Proteins 0.000 description 1
- 101000994375 Homo sapiens Integrin alpha-4 Proteins 0.000 description 1
- 101001095266 Homo sapiens Prolyl endopeptidase Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101000685817 Homo sapiens Solute carrier family 7 member 13 Proteins 0.000 description 1
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 1
- YZJSUQQZGCHHNQ-UHFFFAOYSA-N Homoglutamine Chemical compound OC(=O)C(N)CCCC(N)=O YZJSUQQZGCHHNQ-UHFFFAOYSA-N 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- ZRJBHWIHUMBLCN-SEQYCRGISA-N Huperzine A Natural products N1C(=O)C=CC2=C1C[C@H]1/C(=C/C)[C@]2(N)CC(C)=C1 ZRJBHWIHUMBLCN-SEQYCRGISA-N 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 235000003321 Ilex vomitoria Nutrition 0.000 description 1
- 241000209026 Ilex vomitoria Species 0.000 description 1
- 102100021042 Immunoglobulin-binding protein 1 Human genes 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102100032818 Integrin alpha-4 Human genes 0.000 description 1
- 102000012355 Integrin beta1 Human genes 0.000 description 1
- 108010022222 Integrin beta1 Proteins 0.000 description 1
- 101710144961 Interferon tau-1 Proteins 0.000 description 1
- 101710144950 Interferon tau-2 Proteins 0.000 description 1
- 101710144962 Interferon tau-3 Proteins 0.000 description 1
- 229940119524 Interleukin 12 antagonist Drugs 0.000 description 1
- 102000003814 Interleukin-10 Human genes 0.000 description 1
- 102100039068 Interleukin-10 Human genes 0.000 description 1
- 108010044467 Isoenzymes Proteins 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- 108010006916 KMI-008 Proteins 0.000 description 1
- QNAYBMKLOCPYGJ-UWTATZPHSA-N L-Alanine Natural products C[C@@H](N)C(O)=O QNAYBMKLOCPYGJ-UWTATZPHSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ULEBESPCVWBNIF-BYPYZUCNSA-N L-arginine amide Chemical compound NC(=O)[C@@H](N)CCCNC(N)=N ULEBESPCVWBNIF-BYPYZUCNSA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- GDBQQVLCIARPGH-UHFFFAOYSA-N Leupeptin Natural products CC(C)CC(NC(C)=O)C(=O)NC(CC(C)C)C(=O)NC(C=O)CCCN=C(N)N GDBQQVLCIARPGH-UHFFFAOYSA-N 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- 229940124761 MMP inhibitor Drugs 0.000 description 1
- 241000282567 Macaca fascicularis Species 0.000 description 1
- 241000282560 Macaca mulatta Species 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 1
- 208000007466 Male Infertility Diseases 0.000 description 1
- 102000003939 Membrane transport proteins Human genes 0.000 description 1
- 108090000301 Membrane transport proteins Proteins 0.000 description 1
- JEYCTXHKTXCGPB-UHFFFAOYSA-N Methaqualone Chemical compound CC1=CC=CC=C1N1C(=O)C2=CC=CC=C2N=C1C JEYCTXHKTXCGPB-UHFFFAOYSA-N 0.000 description 1
- 229920003091 Methocel™ Polymers 0.000 description 1
- CESYKOGBSMNBPD-UHFFFAOYSA-N Methyclothiazide Chemical compound ClC1=C(S(N)(=O)=O)C=C2S(=O)(=O)N(C)C(CCl)NC2=C1 CESYKOGBSMNBPD-UHFFFAOYSA-N 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 102000004868 N-Methyl-D-Aspartate Receptors Human genes 0.000 description 1
- 108090001041 N-Methyl-D-Aspartate Receptors Proteins 0.000 description 1
- QSQQPMHPCBLLGX-UHFFFAOYSA-N N-methyl-4-[2-(phenylmethyl)phenoxy]-1-butanamine Chemical compound CNCCCCOC1=CC=CC=C1CC1=CC=CC=C1 QSQQPMHPCBLLGX-UHFFFAOYSA-N 0.000 description 1
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 description 1
- 102000003945 NF-kappa B Human genes 0.000 description 1
- 108010057466 NF-kappa B Proteins 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical class O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 1
- 102000005781 Nogo Receptor Human genes 0.000 description 1
- 108020003872 Nogo receptor Proteins 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 239000005480 Olmesartan Substances 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108010015181 PPAR delta Proteins 0.000 description 1
- 108010016731 PPAR gamma Proteins 0.000 description 1
- 102000000536 PPAR gamma Human genes 0.000 description 1
- 108010044210 PPAR-beta Proteins 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- AHOUBRCZNHFOSL-UHFFFAOYSA-N Paroxetine hydrochloride Natural products C1=CC(F)=CC=C1C1C(COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-UHFFFAOYSA-N 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- RMUCZJUITONUFY-UHFFFAOYSA-N Phenelzine Chemical compound NNCCC1=CC=CC=C1 RMUCZJUITONUFY-UHFFFAOYSA-N 0.000 description 1
- 229940123932 Phosphodiesterase 4 inhibitor Drugs 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 229940122767 Potassium sparing diuretic Drugs 0.000 description 1
- MWQCHHACWWAQLJ-UHFFFAOYSA-N Prazepam Chemical compound O=C1CN=C(C=2C=CC=CC=2)C2=CC(Cl)=CC=C2N1CC1CC1 MWQCHHACWWAQLJ-UHFFFAOYSA-N 0.000 description 1
- 108010071690 Prealbumin Proteins 0.000 description 1
- IFFPICMESYHZPQ-UHFFFAOYSA-N Prenylamine Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)CCNC(C)CC1=CC=CC=C1 IFFPICMESYHZPQ-UHFFFAOYSA-N 0.000 description 1
- 101710178372 Prolyl endopeptidase Proteins 0.000 description 1
- 229940122210 Prolyl endopeptidase inhibitor Drugs 0.000 description 1
- 101710118538 Protease Proteins 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108090000783 Renin Proteins 0.000 description 1
- 102100028255 Renin Human genes 0.000 description 1
- XSVMFMHYUFZWBK-NSHDSACASA-N Rivastigmine Chemical compound CCN(C)C(=O)OC1=CC=CC([C@H](C)N(C)C)=C1 XSVMFMHYUFZWBK-NSHDSACASA-N 0.000 description 1
- 101150098533 SOST gene Proteins 0.000 description 1
- 102000012479 Serine Proteases Human genes 0.000 description 1
- 108010022999 Serine Proteases Proteins 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- 244000000231 Sesamum indicum Species 0.000 description 1
- 235000003434 Sesamum indicum Nutrition 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- ZRJBHWIHUMBLCN-UHFFFAOYSA-N Shuangyiping Natural products N1C(=O)C=CC2=C1CC1C(=CC)C2(N)CC(C)=C1 ZRJBHWIHUMBLCN-UHFFFAOYSA-N 0.000 description 1
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- 102100023135 Solute carrier family 7 member 13 Human genes 0.000 description 1
- 241000401348 Sotalia guianensis Species 0.000 description 1
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- RKSMVPNZHBRNNS-UHFFFAOYSA-N Succinobucol Chemical compound CC(C)(C)C1=C(O)C(C(C)(C)C)=CC(SC(C)(C)SC=2C=C(C(OC(=O)CCC(O)=O)=C(C=2)C(C)(C)C)C(C)(C)C)=C1 RKSMVPNZHBRNNS-UHFFFAOYSA-N 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 108010075383 Suppressor of Cytokine Signaling Proteins Proteins 0.000 description 1
- 102000008036 Suppressor of Cytokine Signaling Proteins Human genes 0.000 description 1
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 1
- ODKDMMTXTVCCLJ-UHFFFAOYSA-N TMC-2A Natural products C1=CC=C2C(CC(N)C(=O)N3C(CC4=CC(O)=C(C(=C4C3)O)OC)C(=O)NC(CC(CO)CO)C(O)=O)=CNC2=C1 ODKDMMTXTVCCLJ-UHFFFAOYSA-N 0.000 description 1
- JACAAXNEHGBPOQ-LLVKDONJSA-N Talampanel Chemical compound C([C@H](N(N=1)C(C)=O)C)C2=CC=3OCOC=3C=C2C=1C1=CC=C(N)C=C1 JACAAXNEHGBPOQ-LLVKDONJSA-N 0.000 description 1
- ZROUQTNYPCANTN-UHFFFAOYSA-N Tiapamil Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC1(C=2C=C(OC)C(OC)=CC=2)S(=O)(=O)CCCS1(=O)=O ZROUQTNYPCANTN-UHFFFAOYSA-N 0.000 description 1
- KJADKKWYZYXHBB-XBWDGYHZSA-N Topiramic acid Chemical compound C1O[C@@]2(COS(N)(=O)=O)OC(C)(C)O[C@H]2[C@@H]2OC(C)(C)O[C@@H]21 KJADKKWYZYXHBB-XBWDGYHZSA-N 0.000 description 1
- VXFJYXUZANRPDJ-WTNASJBWSA-N Trandopril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@H]2CCCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 VXFJYXUZANRPDJ-WTNASJBWSA-N 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 1
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 1
- 102400001359 Transforming growth factor beta-2 Human genes 0.000 description 1
- 101800000304 Transforming growth factor beta-2 Proteins 0.000 description 1
- 206010060872 Transplant failure Diseases 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- 102000009190 Transthyretin Human genes 0.000 description 1
- 229940123445 Tricyclic antidepressant Drugs 0.000 description 1
- 102000007537 Type II DNA Topoisomerases Human genes 0.000 description 1
- 108010046308 Type II DNA Topoisomerases Proteins 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical class NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 108010000134 Vascular Cell Adhesion Molecule-1 Proteins 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- 208000033774 Ventricular Remodeling Diseases 0.000 description 1
- FKCMADOPPWWGNZ-YUMQZZPRSA-N [(2r)-1-[(2s)-2-amino-3-methylbutanoyl]pyrrolidin-2-yl]boronic acid Chemical compound CC(C)[C@H](N)C(=O)N1CCC[C@H]1B(O)O FKCMADOPPWWGNZ-YUMQZZPRSA-N 0.000 description 1
- PBHFNBQPZCRWQP-QUCCMNQESA-N [(3ar,8bs)-3,4,8b-trimethyl-2,3a-dihydro-1h-pyrrolo[2,3-b]indol-7-yl] n-phenylcarbamate Chemical compound CN([C@@H]1[C@@](C2=C3)(C)CCN1C)C2=CC=C3OC(=O)NC1=CC=CC=C1 PBHFNBQPZCRWQP-QUCCMNQESA-N 0.000 description 1
- 229960003697 abatacept Drugs 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- AFCGFAGUEYAMAO-UHFFFAOYSA-N acamprosate Chemical compound CC(=O)NCCCS(O)(=O)=O AFCGFAGUEYAMAO-UHFFFAOYSA-N 0.000 description 1
- 229960004047 acamprosate Drugs 0.000 description 1
- 150000001241 acetals Chemical group 0.000 description 1
- FHEAIOHRHQGZPC-KIWGSFCNSA-N acetic acid;(2s)-2-amino-3-(4-hydroxyphenyl)propanoic acid;(2s)-2-aminopentanedioic acid;(2s)-2-aminopropanoic acid;(2s)-2,6-diaminohexanoic acid Chemical compound CC(O)=O.C[C@H](N)C(O)=O.NCCCC[C@H](N)C(O)=O.OC(=O)[C@@H](N)CCC(O)=O.OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 FHEAIOHRHQGZPC-KIWGSFCNSA-N 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 229940081735 acetylcellulose Drugs 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000030120 acrosome reaction Effects 0.000 description 1
- 230000036982 action potential Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 108091006088 activator proteins Proteins 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000002487 adenosine deaminase inhibitor Substances 0.000 description 1
- 239000000384 adrenergic alpha-2 receptor agonist Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 229950007884 alacepril Drugs 0.000 description 1
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- ZSBOMTDTBDDKMP-OAHLLOKOSA-N alogliptin Chemical compound C=1C=CC=C(C#N)C=1CN1C(=O)N(C)C(=O)C=C1N1CCC[C@@H](N)C1 ZSBOMTDTBDDKMP-OAHLLOKOSA-N 0.000 description 1
- 102000004305 alpha Adrenergic Receptors Human genes 0.000 description 1
- 108090000861 alpha Adrenergic Receptors Proteins 0.000 description 1
- 108010026331 alpha-Fetoproteins Proteins 0.000 description 1
- HWXBTNAVRSUOJR-UHFFFAOYSA-N alpha-hydroxyglutaric acid Natural products OC(=O)C(O)CCC(O)=O HWXBTNAVRSUOJR-UHFFFAOYSA-N 0.000 description 1
- 229940009533 alpha-ketoglutaric acid Drugs 0.000 description 1
- 229960004538 alprazolam Drugs 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- XSDQTOBWRPYKKA-UHFFFAOYSA-N amiloride Chemical compound NC(=N)NC(=O)C1=NC(Cl)=C(N)N=C1N XSDQTOBWRPYKKA-UHFFFAOYSA-N 0.000 description 1
- 229960002576 amiloride Drugs 0.000 description 1
- XXXHSQBVHSJQKS-UHFFFAOYSA-N amino benzoate Chemical compound NOC(=O)C1=CC=CC=C1 XXXHSQBVHSJQKS-UHFFFAOYSA-N 0.000 description 1
- 229960000836 amitriptyline Drugs 0.000 description 1
- KRMDCWKBEZIMAB-UHFFFAOYSA-N amitriptyline Chemical compound C1CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 KRMDCWKBEZIMAB-UHFFFAOYSA-N 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 108010064539 amyloid beta-protein (1-42) Proteins 0.000 description 1
- 108010087587 amyloid beta-protein (11-40) Proteins 0.000 description 1
- 108010087299 amyloid beta-protein (11-42) Proteins 0.000 description 1
- 206010002022 amyloidosis Diseases 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- PHFDAOXXIZOUIX-UHFFFAOYSA-N anipamil Chemical compound C=1C=CC(OC)=CC=1C(CCCCCCCCCCCC)(C#N)CCCN(C)CCC1=CC=CC(OC)=C1 PHFDAOXXIZOUIX-UHFFFAOYSA-N 0.000 description 1
- 229950011530 anipamil Drugs 0.000 description 1
- 230000001430 anti-depressive effect Effects 0.000 description 1
- 230000003276 anti-hypertensive effect Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000000561 anti-psychotic effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 239000003416 antiarrhythmic agent Substances 0.000 description 1
- 229940030600 antihypertensive agent Drugs 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 229940127218 antiplatelet drug Drugs 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 229960001164 apremilast Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 239000003693 atypical antipsychotic agent Substances 0.000 description 1
- 229940127236 atypical antipsychotics Drugs 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 229940003504 avonex Drugs 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 229960004067 benazeprilat Drugs 0.000 description 1
- MADRIHWFJGRSBP-ROUUACIJSA-N benazeprilat Chemical compound C([C@H](N[C@H]1CCC2=CC=CC=C2N(C1=O)CC(=O)O)C(O)=O)CC1=CC=CC=C1 MADRIHWFJGRSBP-ROUUACIJSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 150000001556 benzimidazoles Chemical class 0.000 description 1
- 229940049706 benzodiazepine Drugs 0.000 description 1
- 150000001557 benzodiazepines Chemical class 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- ORZXYSPOAVJYRU-UHFFFAOYSA-N benzyl 2-(2-formylpyrrolidine-1-carbonyl)pyrrolidine-1-carboxylate Chemical compound O=CC1CCCN1C(=O)C1N(C(=O)OCC=2C=CC=CC=2)CCC1 ORZXYSPOAVJYRU-UHFFFAOYSA-N 0.000 description 1
- 229950004221 besilate Drugs 0.000 description 1
- 229940030611 beta-adrenergic blocking agent Drugs 0.000 description 1
- 108091007737 beta-secretases Proteins 0.000 description 1
- 229960004933 bifemelane Drugs 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 230000001851 biosynthetic effect Effects 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 208000006752 brain edema Diseases 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- QIHLUZAFSSMXHQ-UHFFFAOYSA-N budipine Chemical compound C1CN(C(C)(C)C)CCC1(C=1C=CC=CC=1)C1=CC=CC=C1 QIHLUZAFSSMXHQ-UHFFFAOYSA-N 0.000 description 1
- 229960002452 budipine Drugs 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- QWCRAEMEVRGPNT-UHFFFAOYSA-N buspirone Chemical compound C1C(=O)N(CCCCN2CCN(CC2)C=2N=CC=CN=2)C(=O)CC21CCCC2 QWCRAEMEVRGPNT-UHFFFAOYSA-N 0.000 description 1
- 229960002495 buspirone Drugs 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000003491 cAMP production Effects 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 108010079785 calpain inhibitors Proteins 0.000 description 1
- 108010044208 calpastatin Proteins 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- SGZAIDDFHDDFJU-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SGZAIDDFHDDFJU-UHFFFAOYSA-N 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- KLOIYEQEVSIOOO-UHFFFAOYSA-N carbocromen Chemical compound CC1=C(CCN(CC)CC)C(=O)OC2=CC(OCC(=O)OCC)=CC=C21 KLOIYEQEVSIOOO-UHFFFAOYSA-N 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 102000008395 cell adhesion mediator activity proteins Human genes 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 229950005749 ceronapril Drugs 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- WUTYZMFRCNBCHQ-PSASIEDQSA-N cevimeline Chemical compound C1S[C@H](C)O[C@]21C(CC1)CCN1C2 WUTYZMFRCNBCHQ-PSASIEDQSA-N 0.000 description 1
- 229960001314 cevimeline Drugs 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 229960004782 chlordiazepoxide Drugs 0.000 description 1
- ANTSCNMPPGJYLG-UHFFFAOYSA-N chlordiazepoxide Chemical compound O=N=1CC(NC)=NC2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 ANTSCNMPPGJYLG-UHFFFAOYSA-N 0.000 description 1
- 229960002155 chlorothiazide Drugs 0.000 description 1
- JIVPVXMEBJLZRO-UHFFFAOYSA-N chlorthalidone Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C2(O)C3=CC=CC=C3C(=O)N2)=C1 JIVPVXMEBJLZRO-UHFFFAOYSA-N 0.000 description 1
- 239000003354 cholesterol ester transfer protein inhibitor Substances 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 229960005025 cilazapril Drugs 0.000 description 1
- HHHKFGXWKKUNCY-FHWLQOOXSA-N cilazapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N2[C@@H](CCCN2CCC1)C(O)=O)=O)CC1=CC=CC=C1 HHHKFGXWKKUNCY-FHWLQOOXSA-N 0.000 description 1
- 229960004588 cilostazol Drugs 0.000 description 1
- 229960001653 citalopram Drugs 0.000 description 1
- 229960001403 clobazam Drugs 0.000 description 1
- CXOXHMZGEKVPMT-UHFFFAOYSA-N clobazam Chemical compound O=C1CC(=O)N(C)C2=CC=C(Cl)C=C2N1C1=CC=CC=C1 CXOXHMZGEKVPMT-UHFFFAOYSA-N 0.000 description 1
- 229960004606 clomipramine Drugs 0.000 description 1
- 229960003120 clonazepam Drugs 0.000 description 1
- DGBIGWXXNGSACT-UHFFFAOYSA-N clonazepam Chemical compound C12=CC([N+](=O)[O-])=CC=C2NC(=O)CN=C1C1=CC=CC=C1Cl DGBIGWXXNGSACT-UHFFFAOYSA-N 0.000 description 1
- 229960003958 clopidogrel bisulfate Drugs 0.000 description 1
- 229960004362 clorazepate Drugs 0.000 description 1
- XDDJGVMJFWAHJX-UHFFFAOYSA-M clorazepic acid anion Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(C(=O)[O-])N=C1C1=CC=CC=C1 XDDJGVMJFWAHJX-UHFFFAOYSA-M 0.000 description 1
- 230000008045 co-localization Effects 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 230000000112 colonic effect Effects 0.000 description 1
- 108010002212 colostrinine Proteins 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- HTBKFGWATIYCSF-QGXIKSNHSA-N conantokin g Chemical compound NC(=O)C[C@@H](C(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC(C(O)=O)C(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C(O)=O)C(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C(O)=O)C(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C(O)=O)C(O)=O)NC(=O)[C@H](CC(C(O)=O)C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CN HTBKFGWATIYCSF-QGXIKSNHSA-N 0.000 description 1
- 238000011437 continuous method Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- GLNDAGDHSLMOKX-UHFFFAOYSA-N coumarin 120 Chemical compound C1=C(N)C=CC2=C1OC(=O)C=C2C GLNDAGDHSLMOKX-UHFFFAOYSA-N 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 230000001351 cycling effect Effects 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 229960002806 daclizumab Drugs 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical class CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 229960005227 delapril Drugs 0.000 description 1
- WOUOLAUOZXOLJQ-MBSDFSHPSA-N delapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N(CC(O)=O)C1CC2=CC=CC=C2C1)CC1=CC=CC=C1 WOUOLAUOZXOLJQ-MBSDFSHPSA-N 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- MUGNLPWYHGOJEG-UHFFFAOYSA-N delucemine Chemical compound C=1C=CC(F)=CC=1C(CCNC)C1=CC=CC(F)=C1 MUGNLPWYHGOJEG-UHFFFAOYSA-N 0.000 description 1
- 229950006926 delucemine Drugs 0.000 description 1
- 229950010300 denagliptin Drugs 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 229960003914 desipramine Drugs 0.000 description 1
- 238000004147 desorption mass spectrometry Methods 0.000 description 1
- 229960003529 diazepam Drugs 0.000 description 1
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 125000006001 difluoroethyl group Chemical group 0.000 description 1
- 239000003363 dihydroorotate dehydrogenase inhibitor Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000003603 dipeptidyl peptidase IV inhibitor Substances 0.000 description 1
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- QCHSEDTUUKDTIG-UHFFFAOYSA-L dipotassium clorazepate Chemical compound [OH-].[K+].[K+].C12=CC(Cl)=CC=C2NC(=O)C(C(=O)[O-])N=C1C1=CC=CC=C1 QCHSEDTUUKDTIG-UHFFFAOYSA-L 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 229940030606 diuretics Drugs 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 229960005426 doxepin Drugs 0.000 description 1
- ODQWQRRAPPTVAG-GZTJUZNOSA-N doxepin Chemical compound C1OC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 ODQWQRRAPPTVAG-GZTJUZNOSA-N 0.000 description 1
- HWXIGFIVGWUZAO-UHFFFAOYSA-N doxofylline Chemical compound C1=2C(=O)N(C)C(=O)N(C)C=2N=CN1CC1OCCO1 HWXIGFIVGWUZAO-UHFFFAOYSA-N 0.000 description 1
- 229960004483 doxofylline Drugs 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 229960002866 duloxetine Drugs 0.000 description 1
- 208000000718 duodenal ulcer Diseases 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 239000008157 edible vegetable oil Substances 0.000 description 1
- 229950003102 efonidipine Drugs 0.000 description 1
- 229940073621 enbrel Drugs 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 230000007515 enzymatic degradation Effects 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 230000008556 epithelial cell proliferation Effects 0.000 description 1
- 229960004563 eprosartan Drugs 0.000 description 1
- OROAFUQRIXKEMV-LDADJPATSA-N eprosartan Chemical compound C=1C=C(C(O)=O)C=CC=1CN1C(CCCC)=NC=C1\C=C(C(O)=O)/CC1=CC=CS1 OROAFUQRIXKEMV-LDADJPATSA-N 0.000 description 1
- 229960004341 escitalopram Drugs 0.000 description 1
- WSEQXVZVJXJVFP-FQEVSTJZSA-N escitalopram Chemical compound C1([C@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-FQEVSTJZSA-N 0.000 description 1
- CHDGAVDQRSPBTA-UHFFFAOYSA-N esuprone Chemical compound CC1=C(C)C(=O)OC2=CC(OS(=O)(=O)CC)=CC=C21 CHDGAVDQRSPBTA-UHFFFAOYSA-N 0.000 description 1
- 229950007673 esuprone Drugs 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- OPQRBXUBWHDHPQ-UHFFFAOYSA-N etazolate Chemical compound CCOC(=O)C1=CN=C2N(CC)N=CC2=C1NN=C(C)C OPQRBXUBWHDHPQ-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- UKDLORMZNPQILV-UHFFFAOYSA-N ethyl 3-hydroxypropanoate Chemical compound CCOC(=O)CCO UKDLORMZNPQILV-UHFFFAOYSA-N 0.000 description 1
- AKFNKZFJBFQFAA-DIOPXHOYSA-N ethyl 4-[[2-[(2s,4s)-2-cyano-4-fluoropyrrolidin-1-yl]-2-oxoethyl]amino]bicyclo[2.2.2]octane-1-carboxylate Chemical compound C1CC(C(=O)OCC)(CC2)CCC12NCC(=O)N1C[C@@H](F)C[C@H]1C#N AKFNKZFJBFQFAA-DIOPXHOYSA-N 0.000 description 1
- 229940062770 evoxac Drugs 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 229960004979 fampridine Drugs 0.000 description 1
- 229960003580 felodipine Drugs 0.000 description 1
- 230000004720 fertilization Effects 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 235000020375 flavoured syrup Nutrition 0.000 description 1
- 229960004930 fludiazepam Drugs 0.000 description 1
- ROYOYTLGDLIGBX-UHFFFAOYSA-N fludiazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1F ROYOYTLGDLIGBX-UHFFFAOYSA-N 0.000 description 1
- SMANXXCATUTDDT-QPJJXVBHSA-N flunarizine Chemical compound C1=CC(F)=CC=C1C(C=1C=CC(F)=CC=1)N1CCN(C\C=C\C=2C=CC=CC=2)CC1 SMANXXCATUTDDT-QPJJXVBHSA-N 0.000 description 1
- 229960000326 flunarizine Drugs 0.000 description 1
- 238000003682 fluorination reaction Methods 0.000 description 1
- 125000003709 fluoroalkyl group Chemical group 0.000 description 1
- 125000005817 fluorobutyl group Chemical group [H]C([H])(F)C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003784 fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 1
- 238000007421 fluorometric assay Methods 0.000 description 1
- 229960002464 fluoxetine Drugs 0.000 description 1
- 229960004038 fluvoxamine Drugs 0.000 description 1
- CJOFXWAVKWHTFT-XSFVSMFZSA-N fluvoxamine Chemical compound COCCCC\C(=N/OCCN)C1=CC=C(C(F)(F)F)C=C1 CJOFXWAVKWHTFT-XSFVSMFZSA-N 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000027119 gastric acid secretion Effects 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000002406 gastrin-secreting cell Anatomy 0.000 description 1
- OKGNKPYIPKMGLR-ZPCKCTIPSA-N gastrins Chemical class C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N1CCC[C@H]1C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)C(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H]1NC(=O)CC1)C1=CN=CN1 OKGNKPYIPKMGLR-ZPCKCTIPSA-N 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229960003776 glatiramer acetate Drugs 0.000 description 1
- 229960004666 glucagon Drugs 0.000 description 1
- 150000002307 glutamic acids Chemical class 0.000 description 1
- 150000002309 glutamines Chemical class 0.000 description 1
- LVASCWIMLIKXLA-LSDHHAIUSA-N halofuginone Chemical compound O[C@@H]1CCCN[C@H]1CC(=O)CN1C(=O)C2=CC(Cl)=C(Br)C=C2N=C1 LVASCWIMLIKXLA-LSDHHAIUSA-N 0.000 description 1
- 229950010152 halofuginone Drugs 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 208000002672 hepatitis B Diseases 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003707 hexyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 230000028996 humoral immune response Effects 0.000 description 1
- 230000008348 humoral response Effects 0.000 description 1
- ZRJBHWIHUMBLCN-YQEJDHNASA-N huperzine A Chemical compound N1C(=O)C=CC2=C1C[C@H]1\C(=C/C)[C@]2(N)CC(C)=C1 ZRJBHWIHUMBLCN-YQEJDHNASA-N 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 229960001195 imidapril Drugs 0.000 description 1
- KLZWOWYOHUKJIG-BPUTZDHNSA-N imidapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1C(N(C)C[C@H]1C(O)=O)=O)CC1=CC=CC=C1 KLZWOWYOHUKJIG-BPUTZDHNSA-N 0.000 description 1
- UTCSSFWDNNEEBH-UHFFFAOYSA-N imidazo[1,2-a]pyridine Chemical compound C1=CC=CC2=NC=CN21 UTCSSFWDNNEEBH-UHFFFAOYSA-N 0.000 description 1
- 229960004801 imipramine Drugs 0.000 description 1
- BCGWQEUPMDMJNV-UHFFFAOYSA-N imipramine Chemical compound C1CC2=CC=CC=C2N(CCCN(C)C)C2=CC=CC=C21 BCGWQEUPMDMJNV-UHFFFAOYSA-N 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- MNLULKBKWKTZPE-UHFFFAOYSA-N indantadol Chemical compound C1=CC=C2CC(NCC(=O)N)CC2=C1 MNLULKBKWKTZPE-UHFFFAOYSA-N 0.000 description 1
- 229950008308 indantadol Drugs 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229960004461 interferon beta-1a Drugs 0.000 description 1
- 229940076144 interleukin-10 Drugs 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 229960002198 irbesartan Drugs 0.000 description 1
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- PTKHFRNHJULJKT-UHFFFAOYSA-N jnj-5207852 Chemical compound C1CCCCN1CCCOC(C=C1)=CC=C1CN1CCCCC1 PTKHFRNHJULJKT-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 229960004340 lacidipine Drugs 0.000 description 1
- GKQPCPXONLDCMU-CCEZHUSRSA-N lacidipine Chemical compound CCOC(=O)C1=C(C)NC(C)=C(C(=O)OCC)C1C1=CC=CC=C1\C=C\C(=O)OC(C)(C)C GKQPCPXONLDCMU-CCEZHUSRSA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- FWUQWDCOOWEXRY-ZDUSSCGKSA-N lanicemine Chemical compound C([C@H](N)C=1C=CC=CC=1)C1=CC=CC=N1 FWUQWDCOOWEXRY-ZDUSSCGKSA-N 0.000 description 1
- 229960004577 laquinimod Drugs 0.000 description 1
- GKWPCEFFIHSJOE-UHFFFAOYSA-N laquinimod Chemical compound OC=1C2=C(Cl)C=CC=C2N(C)C(=O)C=1C(=O)N(CC)C1=CC=CC=C1 GKWPCEFFIHSJOE-UHFFFAOYSA-N 0.000 description 1
- 238000004989 laser desorption mass spectroscopy Methods 0.000 description 1
- 239000004816 latex Substances 0.000 description 1
- 229920000126 latex Polymers 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 229950007278 lenercept Drugs 0.000 description 1
- 230000023404 leukocyte cell-cell adhesion Effects 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- GDBQQVLCIARPGH-ULQDDVLXSA-N leupeptin Chemical compound CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C=O)CCCN=C(N)N GDBQQVLCIARPGH-ULQDDVLXSA-N 0.000 description 1
- 108010052968 leupeptin Proteins 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- CZRQXSDBMCMPNJ-ZUIPZQNBSA-N lisinopril dihydrate Chemical compound O.O.C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 CZRQXSDBMCMPNJ-ZUIPZQNBSA-N 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 229960004391 lorazepam Drugs 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 210000005265 lung cell Anatomy 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 150000002689 maleic acids Chemical class 0.000 description 1
- 238000001840 matrix-assisted laser desorption--ionisation time-of-flight mass spectrometry Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 229940115256 melanoma vaccine Drugs 0.000 description 1
- FJQXCDYVZAHXNS-UHFFFAOYSA-N methadone hydrochloride Chemical compound Cl.C=1C=CC=CC=1C(CC(C)N(C)C)(C(=O)CC)C1=CC=CC=C1 FJQXCDYVZAHXNS-UHFFFAOYSA-N 0.000 description 1
- GPKUICFDWYEPTK-UHFFFAOYSA-N methoxycyclohexatriene Chemical compound COC1=CC=C=C[CH]1 GPKUICFDWYEPTK-UHFFFAOYSA-N 0.000 description 1
- 229960003739 methyclothiazide Drugs 0.000 description 1
- KMYUINCCFSNYPC-VPTZPLTNSA-N methyl 6-[(1r,2s,3r)-3-hydroxy-2-[(e,5r)-3-hydroxy-5-methylnon-1-enyl]-5-oxocyclopentyl]sulfanylhexanoate Chemical compound CCCC[C@@H](C)CC(O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1SCCCCCC(=O)OC KMYUINCCFSNYPC-VPTZPLTNSA-N 0.000 description 1
- 150000004702 methyl esters Chemical group 0.000 description 1
- 150000005217 methyl ethers Chemical class 0.000 description 1
- CPZBTYRIGVOOMI-UHFFFAOYSA-N methylsulfanyl(methylsulfanylmethoxy)methane Chemical compound CSCOCSC CPZBTYRIGVOOMI-UHFFFAOYSA-N 0.000 description 1
- 229960001952 metrifonate Drugs 0.000 description 1
- 244000000010 microbial pathogen Species 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229960001785 mirtazapine Drugs 0.000 description 1
- RONZAEMNMFQXRA-UHFFFAOYSA-N mirtazapine Chemical compound C1C2=CC=CN=C2N2CCN(C)CC2C2=CC=CC=C21 RONZAEMNMFQXRA-UHFFFAOYSA-N 0.000 description 1
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 1
- ZAHQPTJLOCWVPG-UHFFFAOYSA-N mitoxantrone dihydrochloride Chemical compound Cl.Cl.O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO ZAHQPTJLOCWVPG-UHFFFAOYSA-N 0.000 description 1
- 229960004169 mitoxantrone hydrochloride Drugs 0.000 description 1
- 229960004644 moclobemide Drugs 0.000 description 1
- YHXISWVBGDMDLQ-UHFFFAOYSA-N moclobemide Chemical compound C1=CC(Cl)=CC=C1C(=O)NCCN1CCOCC1 YHXISWVBGDMDLQ-UHFFFAOYSA-N 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000002306 monocyte chemotactic protein 1 inhibitor Substances 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- CFRXVFRHMZLQBS-UHFFFAOYSA-N n-(3,5-dichloro-1-hydroxypyridin-4-ylidene)-6-(difluoromethoxy)-[1]benzofuro[3,2-c]pyridine-9-carboxamide Chemical compound ClC1=CN(O)C=C(Cl)C1=NC(=O)C1=CC=C(OC(F)F)C2=C1C1=CN=CC=C1O2 CFRXVFRHMZLQBS-UHFFFAOYSA-N 0.000 description 1
- DPHDSIQHVGSITN-UHFFFAOYSA-N n-(3,5-dichloropyridin-4-yl)-2-[1-[(4-fluorophenyl)methyl]-5-hydroxyindol-3-yl]-2-oxoacetamide Chemical compound C1=C(C(=O)C(=O)NC=2C(=CN=CC=2Cl)Cl)C2=CC(O)=CC=C2N1CC1=CC=C(F)C=C1 DPHDSIQHVGSITN-UHFFFAOYSA-N 0.000 description 1
- JERXUPDBWDWFCF-UHFFFAOYSA-N n-(3,5-dichloropyridin-4-yl)-2-[1-[(4-fluorophenyl)methyl]pyrrolo[2,3-b]pyridin-3-yl]-2-oxoacetamide Chemical compound C1=CC(F)=CC=C1CN1C2=NC=CC=C2C(C(=O)C(=O)NC=2C(=CN=CC=2Cl)Cl)=C1 JERXUPDBWDWFCF-UHFFFAOYSA-N 0.000 description 1
- WDZVWDXOIGQJIO-UHFFFAOYSA-N n-[4-(4-chlorophenyl)sulfonyl-4-(2,5-difluorophenyl)cyclohexyl]-1,1,1-trifluoromethanesulfonamide Chemical compound FC1=CC=C(F)C(C2(CCC(CC2)NS(=O)(=O)C(F)(F)F)S(=O)(=O)C=2C=CC(Cl)=CC=2)=C1 WDZVWDXOIGQJIO-UHFFFAOYSA-N 0.000 description 1
- UCBHICFFJKDMMR-AWEZNQCLSA-N n-[[(5s)-3-[3-fluoro-4-(2-methyl-4,6-dihydropyrrolo[3,4-c]pyrazol-5-yl)phenyl]-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide Chemical compound O=C1O[C@@H](CNC(=O)C)CN1C(C=C1F)=CC=C1N1CC2=NN(C)C=C2C1 UCBHICFFJKDMMR-AWEZNQCLSA-N 0.000 description 1
- 125000006606 n-butoxy group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000003935 n-pentoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003506 n-propoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229920001206 natural gum Polymers 0.000 description 1
- 229960001800 nefazodone Drugs 0.000 description 1
- VRBKIVRKKCLPHA-UHFFFAOYSA-N nefazodone Chemical compound O=C1N(CCOC=2C=CC=CC=2)C(CC)=NN1CCCN(CC1)CCN1C1=CC=CC(Cl)=C1 VRBKIVRKKCLPHA-UHFFFAOYSA-N 0.000 description 1
- 201000009925 nephrosclerosis Diseases 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 208000004296 neuralgia Diseases 0.000 description 1
- 210000004498 neuroglial cell Anatomy 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 208000021722 neuropathic pain Diseases 0.000 description 1
- 239000002658 neuropeptide Y receptor agonist Substances 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 1
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 1
- 229950010800 niguldipine Drugs 0.000 description 1
- VZWXXKDFACOXNT-UHFFFAOYSA-N niludipine Chemical compound CCCOCCOC(=O)C1=C(C)NC(C)=C(C(=O)OCCOCCC)C1C1=CC=CC([N+]([O-])=O)=C1 VZWXXKDFACOXNT-UHFFFAOYSA-N 0.000 description 1
- 229950000109 niludipine Drugs 0.000 description 1
- 239000002840 nitric oxide donor Substances 0.000 description 1
- DLWSRGHNJVLJAH-UHFFFAOYSA-N nitroflurbiprofen Chemical compound FC1=CC(C(C(=O)OCCCCO[N+]([O-])=O)C)=CC=C1C1=CC=CC=C1 DLWSRGHNJVLJAH-UHFFFAOYSA-N 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000002664 nootropic agent Substances 0.000 description 1
- 125000001736 nosyl group Chemical group S(=O)(=O)(C1=CC=C([N+](=O)[O-])C=C1)* 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid group Chemical group C(CCCCCCC\C=C/CCCCCCCC)(=O)O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- VTRAEEWXHOVJFV-UHFFFAOYSA-N olmesartan Chemical compound CCCC1=NC(C(C)(C)O)=C(C(O)=O)N1CC1=CC=C(C=2C(=CC=CC=2)C=2NN=NN=2)C=C1 VTRAEEWXHOVJFV-UHFFFAOYSA-N 0.000 description 1
- 229960005117 olmesartan Drugs 0.000 description 1
- 229950010444 onercept Drugs 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 150000002905 orthoesters Chemical group 0.000 description 1
- 229960004535 oxazepam Drugs 0.000 description 1
- ADIMAYPTOBDMTL-UHFFFAOYSA-N oxazepam Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(O)N=C1C1=CC=CC=C1 ADIMAYPTOBDMTL-UHFFFAOYSA-N 0.000 description 1
- ATYBXHSAIOKLMG-UHFFFAOYSA-N oxepin Chemical compound O1C=CC=CC=C1 ATYBXHSAIOKLMG-UHFFFAOYSA-N 0.000 description 1
- AFDXODALSZRGIH-UHFFFAOYSA-N p-coumaric acid methyl ether Natural products COC1=CC=C(C=CC(O)=O)C=C1 AFDXODALSZRGIH-UHFFFAOYSA-N 0.000 description 1
- KLAKIAVEMQMVBT-UHFFFAOYSA-N p-hydroxy-phenacyl alcohol Natural products OCC(=O)C1=CC=C(O)C=C1 KLAKIAVEMQMVBT-UHFFFAOYSA-N 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 230000003076 paracrine Effects 0.000 description 1
- 239000003182 parenteral nutrition solution Substances 0.000 description 1
- 229960002296 paroxetine Drugs 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000004963 pathophysiological condition Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 229950000039 peldesine Drugs 0.000 description 1
- 125000004115 pentoxy group Chemical group [*]OC([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 108010011903 peptide receptors Proteins 0.000 description 1
- 229940023041 peptide vaccine Drugs 0.000 description 1
- 229960002582 perindopril Drugs 0.000 description 1
- IPVQLZZIHOAWMC-QXKUPLGCSA-N perindopril Chemical compound C1CCC[C@H]2C[C@@H](C(O)=O)N(C(=O)[C@H](C)N[C@@H](CCC)C(=O)OCC)[C@H]21 IPVQLZZIHOAWMC-QXKUPLGCSA-N 0.000 description 1
- 229950006454 perzinfotel Drugs 0.000 description 1
- 239000000575 pesticide Substances 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000008024 pharmaceutical diluent Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960000964 phenelzine Drugs 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 239000002587 phosphodiesterase IV inhibitor Substances 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 230000006461 physiological response Effects 0.000 description 1
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 1
- 229940095638 pletal Drugs 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 239000003450 potassium channel blocker Substances 0.000 description 1
- 239000003286 potassium sparing diuretic agent Substances 0.000 description 1
- 229940097241 potassium-sparing diuretic Drugs 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 229960004856 prazepam Drugs 0.000 description 1
- 201000011461 pre-eclampsia Diseases 0.000 description 1
- 229960001989 prenylamine Drugs 0.000 description 1
- 238000004237 preparative chromatography Methods 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 229950003700 priliximab Drugs 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- TURAMGVWNUTQKH-UHFFFAOYSA-N propa-1,2-dien-1-one Chemical compound C=C=C=O TURAMGVWNUTQKH-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940023143 protein vaccine Drugs 0.000 description 1
- 208000002815 pulmonary hypertension Diseases 0.000 description 1
- 239000000784 purine nucleoside phosphorylase inhibitor Substances 0.000 description 1
- WQGWDDDVZFFDIG-UHFFFAOYSA-N pyrogallol Chemical class OC1=CC=CC(O)=C1O WQGWDDDVZFFDIG-UHFFFAOYSA-N 0.000 description 1
- 229960001455 quinapril Drugs 0.000 description 1
- JSDRRTOADPPCHY-HSQYWUDLSA-N quinapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2=CC=CC=C2C1)C(O)=O)CC1=CC=CC=C1 JSDRRTOADPPCHY-HSQYWUDLSA-N 0.000 description 1
- ZRJBHWIHUMBLCN-BMIGLBTASA-N rac-huperzine A Natural products N1C(=O)C=CC2=C1C[C@@H]1C(=CC)[C@@]2(N)CC(C)=C1 ZRJBHWIHUMBLCN-BMIGLBTASA-N 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- RUOKEQAAGRXIBM-GFCCVEGCSA-N rasagiline Chemical compound C1=CC=C2[C@H](NCC#C)CCC2=C1 RUOKEQAAGRXIBM-GFCCVEGCSA-N 0.000 description 1
- 229960000245 rasagiline Drugs 0.000 description 1
- 229960003770 reboxetine Drugs 0.000 description 1
- CBQGYUDMJHNJBX-RTBURBONSA-N reboxetine Chemical compound CCOC1=CC=CC=C1O[C@H](C=1C=CC=CC=1)[C@@H]1OCCNC1 CBQGYUDMJHNJBX-RTBURBONSA-N 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 108010003189 recombinant human tumor necrosis factor-binding protein-1 Proteins 0.000 description 1
- 229940077452 recombinant interferon beta-1b Drugs 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 230000029865 regulation of blood pressure Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 201000002793 renal fibrosis Diseases 0.000 description 1
- 102000027483 retinoid hormone receptors Human genes 0.000 description 1
- 108091008679 retinoid hormone receptors Proteins 0.000 description 1
- 229960004136 rivastigmine Drugs 0.000 description 1
- HJORMJIFDVBMOB-UHFFFAOYSA-N rolipram Chemical group COC1=CC=C(C2CC(=O)NC2)C=C1OC1CCCC1 HJORMJIFDVBMOB-UHFFFAOYSA-N 0.000 description 1
- 229950005741 rolipram Drugs 0.000 description 1
- IQWCBYSUUOFOMF-QTLFRQQHSA-N sabcomeline Chemical compound C1CC2[C@@H](C(/C#N)=N/OC)CN1CC2 IQWCBYSUUOFOMF-QTLFRQQHSA-N 0.000 description 1
- 229950000425 sabcomeline Drugs 0.000 description 1
- 108010033693 saxagliptin Proteins 0.000 description 1
- QGJUIPDUBHWZPV-SGTAVMJGSA-N saxagliptin Chemical compound C1C(C2)CC(C3)CC2(O)CC13[C@H](N)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QGJUIPDUBHWZPV-SGTAVMJGSA-N 0.000 description 1
- 229960004937 saxagliptin Drugs 0.000 description 1
- 210000004761 scalp Anatomy 0.000 description 1
- 230000000698 schizophrenic effect Effects 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 229950001900 semagacestat Drugs 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 229960002073 sertraline Drugs 0.000 description 1
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- MFFMDFFZMYYVKS-SECBINFHSA-N sitagliptin Chemical compound C([C@H](CC(=O)N1CC=2N(C(=NN=2)C(F)(F)F)CC1)N)C1=CC(F)=C(F)C=C1F MFFMDFFZMYYVKS-SECBINFHSA-N 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- KGZGMOFDZSCICN-YCBFMBTMSA-M sodium;(2s)-3-[4-[(7-fluoroquinolin-2-yl)methoxy]phenyl]-2-(quinoline-2-carbonylamino)propanoate Chemical compound [Na+].C1=CC=CC2=NC(C(=O)N[C@@H](CC=3C=CC(OCC=4N=C5C=C(F)C=CC5=CC=4)=CC=3)C(=O)[O-])=CC=C21 KGZGMOFDZSCICN-YCBFMBTMSA-M 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 229950009136 solimastat Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 229960002909 spirapril Drugs 0.000 description 1
- 108700035424 spirapril Proteins 0.000 description 1
- HRWCVUIFMSZDJS-SZMVWBNQSA-N spirapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2(C1)SCCS2)C(O)=O)CC1=CC=CC=C1 HRWCVUIFMSZDJS-SZMVWBNQSA-N 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 125000005415 substituted alkoxy group Chemical group 0.000 description 1
- WPLOVIFNBMNBPD-ATHMIXSHSA-N subtilin Chemical compound CC1SCC(NC2=O)C(=O)NC(CC(N)=O)C(=O)NC(C(=O)NC(CCCCN)C(=O)NC(C(C)CC)C(=O)NC(=C)C(=O)NC(CCCCN)C(O)=O)CSC(C)C2NC(=O)C(CC(C)C)NC(=O)C1NC(=O)C(CCC(N)=O)NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C1NC(=O)C(=C/C)/NC(=O)C(CCC(N)=O)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)CNC(=O)C(NC(=O)C(NC(=O)C2NC(=O)CNC(=O)C3CCCN3C(=O)C(NC(=O)C3NC(=O)C(CC(C)C)NC(=O)C(=C)NC(=O)C(CCC(O)=O)NC(=O)C(NC(=O)C(CCCCN)NC(=O)C(N)CC=4C5=CC=CC=C5NC=4)CSC3)C(C)SC2)C(C)C)C(C)SC1)CC1=CC=CC=C1 WPLOVIFNBMNBPD-ATHMIXSHSA-N 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 108010009573 talabostat Proteins 0.000 description 1
- 229950010637 talabostat Drugs 0.000 description 1
- 229950004608 talampanel Drugs 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- 229960000651 tasosartan Drugs 0.000 description 1
- ADXGNEYLLLSOAR-UHFFFAOYSA-N tasosartan Chemical compound C12=NC(C)=NC(C)=C2CCC(=O)N1CC(C=C1)=CC=C1C1=CC=CC=C1C=1N=NNN=1 ADXGNEYLLLSOAR-UHFFFAOYSA-N 0.000 description 1
- 229960003080 taurine Drugs 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- 229960004084 temocapril Drugs 0.000 description 1
- FIQOFIRCTOWDOW-BJLQDIEVSA-N temocapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N(CC(O)=O)C[C@H](SC1)C=1SC=CC=1)=O)CC1=CC=CC=C1 FIQOFIRCTOWDOW-BJLQDIEVSA-N 0.000 description 1
- 229960000331 teriflunomide Drugs 0.000 description 1
- UTNUDOFZCWSZMS-YFHOEESVSA-N teriflunomide Chemical compound C\C(O)=C(/C#N)C(=O)NC1=CC=C(C(F)(F)F)C=C1 UTNUDOFZCWSZMS-YFHOEESVSA-N 0.000 description 1
- 150000003505 terpenes Chemical class 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- SKQBOMOZPWGHAX-UHFFFAOYSA-N tert-butyl-[[tert-butyl(dimethyl)silyl]oxymethoxymethoxy]-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OCOCO[Si](C)(C)C(C)(C)C SKQBOMOZPWGHAX-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000012085 test solution Substances 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- 229960003433 thalidomide Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 239000003451 thiazide diuretic agent Substances 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 229950003137 tiapamil Drugs 0.000 description 1
- 229960000488 tizanidine Drugs 0.000 description 1
- ODKDMMTXTVCCLJ-BVSLBCMMSA-N tmc-2-a Chemical compound C1=CC=C2C(C[C@H](N)C(=O)N3[C@@H](CC4=CC(O)=C(C(=C4C3)O)OC)C(=O)N[C@@H](CC(CO)CO)C(O)=O)=CNC2=C1 ODKDMMTXTVCCLJ-BVSLBCMMSA-N 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 229960004394 topiramate Drugs 0.000 description 1
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 1
- 238000012549 training Methods 0.000 description 1
- 229960002051 trandolapril Drugs 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 239000012581 transferrin Substances 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 229940063648 tranxene Drugs 0.000 description 1
- 229960003741 tranylcypromine Drugs 0.000 description 1
- NFACJZMKEDPNKN-UHFFFAOYSA-N trichlorfon Chemical compound COP(=O)(OC)C(O)C(Cl)(Cl)Cl NFACJZMKEDPNKN-UHFFFAOYSA-N 0.000 description 1
- 239000003029 tricyclic antidepressant agent Substances 0.000 description 1
- 230000005748 tumor development Effects 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 150000004917 tyrosine kinase inhibitor derivatives Chemical class 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 229960003824 ustekinumab Drugs 0.000 description 1
- 108010084171 vanutide cridificar Proteins 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 229960004688 venlafaxine Drugs 0.000 description 1
- PNVNVHUZROJLTJ-UHFFFAOYSA-N venlafaxine Chemical compound C1=CC(OC)=CC=C1C(CN(C)C)C1(O)CCCCC1 PNVNVHUZROJLTJ-UHFFFAOYSA-N 0.000 description 1
- SYOKIDBDQMKNDQ-XWTIBIIYSA-N vildagliptin Chemical compound C1C(O)(C2)CC(C3)CC1CC32NCC(=O)N1CCC[C@H]1C#N SYOKIDBDQMKNDQ-XWTIBIIYSA-N 0.000 description 1
- 229960001254 vildagliptin Drugs 0.000 description 1
- 239000003039 volatile agent Substances 0.000 description 1
- 229940000119 zanaflex Drugs 0.000 description 1
- 229940051223 zetia Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Abstract
Compuesto de fórmula (I): **(Ver fórmula)** o una sal, solvato o polimorfo farmacéuticamente aceptable del mismo, incluyendo todos los tautómeros y estereoisómeros del mismo, en la que: A es heteroarilo seleccionado de 1H-benzimidazol-5-ilo o 1H-benzimidazol-6-ilo e imidazo[1,2-a]piridin-7-ilo; R1 representa hidrógeno, alquilo o halógeno; R2 representa hidrógeno, alquilo o halógeno; R3 representa hidrógeno, alquilo o alcoxilo; R4 representa hidrógeno o alquilo; y R5 representa hidrógeno, alquilo o halógeno; y en el que los grupos alquilo o alcoxilo anteriores se sustituyen opcionalmente por uno o más halógenos.
Description
DESCRIPCIÓN
Inhibidores de glutaminil ciclasa
Campo de la invención
La invención se refiere a derivados de azetidinona novedosos con propiedades farmacocinéticas mejoradas como inhibidores de glutaminil ciclasa (QC, EC 2.3.2.5). QC cataliza la ciclación intramolecular de residuos de glutamina del extremo N en ácido piroglutámico (5-oxo-prolilo, pGlu*) con liberación de amoniaco y la ciclación intramolecular de residuos de glutamato del extremo N en ácido piroglutámico con liberación de agua.
Antecedentes de la invención
La glutaminil ciclasa (QC, EC 2.3.2.5) cataliza la ciclación intramolecular de residuos de glutamina del extremo N en ácido piroglutámico (pGlu*) liberando amoniaco. Una QC se aisló por primera vez por Messer a partir del látex de la planta tropical Carica papaya en 1963 (Messer, M. 1963 Nature 4874, 1299). 24 años después se descubrió una actividad enzimática correspondiente en pituitaria de animal (Busby, W. H. J. et al. 1987 J Biol Chem 262, 8532-8536; Fischer, W. H. y Spiess, J. 1987 Proc Natl Acad Sci U S A 84, 3628-3632). Para la QC de mamífero, la conversión de Gln en pGlu por QC podría mostrarse para los precursores de TRH y GnRH (Busby, W. H. J. et al. 1987 J Biol Chem 262, 8532-8536; Fischer, W. H. y Spiess, J. 1987 Proc Natl Acad Sci U S A 84, 3628-3632). Además, los experimentos de localización inicial de QC revelaron una co-localización con sus productos putativos de catálisis en pituitaria bovina, mejorando adicionalmente la función sugerida en la síntesis de hormona peptídica (Bockers, T. M. et al. 1995 J Neuroendocrinol 7, 445-453). En cambio, la función fisiológica de la QC de planta es menos clara. En el caso de la enzima de C. papaya se sugirió una función en la defensa de planta contra microorganismos patógenos (EI Moussaoui, A. et al. 2001 Cell Mol Life Sci 58, 556-570). Las QC putativas de otras plantas se identificaron recientemente por comparaciones de secuencias (Dahl, S. W. et al.2000 Protein Expr Purif 20, 27-36). Sin embargo, la función fisiológica de estas enzimas todavía es ambigua.
Las QC conocidas de plantas y animales muestran una estricta especificidad por L-glutamina en la posición del extremo N de los sustratos y se encontró que su comportamiento cinético obedecía la ecuación de Michaelis-Menten (Pohl, T. et al. 1991 Proc Natl Acad Sci U S A 88, 10059-10063; Consalvo, A. P. et al. 1988 Anal Biochem 175, 131 138; Gololobov, M. Y. et al. 1996 Biol Chem Hoppe Seyler 377, 395-398). Sin embargo, una comparación de las estructuras primarias de las QC de C. papaya y las de las QC altamente conservadas de mamíferos no reveló ninguna homología de secuencias (Dahl, S. W. et al. 2000 Protein Expr Purif 20, 27-36). Mientras que las QC de planta parecen pertenecer a una nueva familia de enzimas (Dahl, S. W. et al. 2000 Protein Expr Purif 20, 27-36), se encontró que las QC de mamífero tenían una homología de secuencias pronunciada con aminopeptidasas bacterianas (Bateman, R. C. et al. 2001 Biochemistry 40, 11246-11250), conduciendo a la conclusión de que las QC de plantas y animales tienen diferentes orígenes evolutivos.
Recientemente se mostró que QC humana recombinante, además de actividad de QC de extractos de cerebro, catalizan ambas el glutaminilo del extremo N, además de la ciclación de glutamato Lo más llamativo es el hallazgo de que la conversión de Glu1 catalizada por ciclasa se favorece por un pH de aproximadamente 6,0 mientras que la conversión de G ln a derivados de pGlu se produce con un pH óptimo de aproximadamente 8,0. Como la formación de péptidos relacionados con pGlu-Ap puede suprimirse por la inhibición de QC humana recombinante y actividad de QC de extractos de pituitaria de cerdo, la enzima QC es una diana en el desarrollo de fármacos para el tratamiento de enfermedad de Alzheimer.
Los inhibidores de QC se describen en los documentos WO 2004/098625, WO 2004/098591, WO 2005/039548, WO 2005/075436, WO 2008/055945, WO 2008/055947, WO 2008/055950, WO 2008/065141, WO 2008/110523, WO 2008/128981, WO 2008/128982, WO 2008/128983, WO 2008/128984, WO 2008/128985, WO 2008/128986, WO 2008/128987, WO 2010/026212, WO 2011/029920,WO 2011/107530, WO 2011/110613, WO 2011/131748 y WO 2012/123563 y WO 2014/140279.
El documento EP 02 011 349.4 da a conocer polinucleótidos que codifican glutaminil ciclasa de insecto, además de polipéptidos así codificados y su uso en métodos de detección de agentes que reducen la actividad de glutaminil ciclasa. Tales agentes son útiles como pesticidas.
Definiciones
Los términos “ki” o “Ki” y “Kd” son constantes de unión, que describen la unión de un inhibidor a y la posterior liberación de una enzima. Otra medida es el valor de “CI50”, que refleja la concentración de inhibidor, que a una concentración de sustrato dada produce el 50 % de actividad enzimática.
El término “inhibidor de DP IV” o “inhibidor de dipeptidil peptidasa IV” es generalmente conocido para un experto en la técnica y significa inhibidores de enzima, que inhiben la actividad catalítica de DP IV o enzimas similares a DP IV.
La “actividad de DP IV” se define como la actividad catalítica de dipeptidil peptidasa IV (DP IV) y enzimas similares a DP IV. Estas enzimas son post-prolina (a un menor grado post-alanina, post-serina o post-glicina) que escinden serina proteasas encontradas en diversos tejidos del cuerpo de un mamífero incluyendo riñón, hígado e intestino, en los que eliminan dipéptidos del extremo N de péptidos biológicamente activos con una alta especificidad cuando la prolina o alanina forman los residuos que son adyacentes al aminoácido del extremo N en su secuencia.
El término “inhibidor de PEP” o “ inhibidor de prolil endopeptidasa” se conoce generalmente para un experto en la técnica y significa inhibidores de enzima, que inhiben la actividad catalítica de prolil endopeptidasa (PEP, prolil oligopeptidasa, POP).
Se define “actividad de PEP” como la actividad catalítica de una endoproteasa que es capaz de hidrolizar enlaces postprolina en péptidos o proteínas en los que la prolina está en la posición de aminoácido 3 o mayor contada desde el extremo N de un sustrato de péptido o proteína.
El término “QC” como se usa en el presente documento comprende glutaminil ciclasa (QC) y enzimas similares a QC. QC y enzimas similares a QC tienen actividad enzimática idéntica o similar, definida adicionalmente como actividad de Qc . A este respecto, enzimas similares a QC pueden diferenciarse fundamentalmente en su estructura molecular de QC. Ejemplos de enzimas similares a QC son las proteínas similares a glutaminil-péptido ciclotransferasa (QPCTL) de ser humano (GenBank NM_017659), ratón (GenBank BC058181), Macaca fascicularis (GenBank AB168255), Macaca mulatta (GenBank XM_001110995), Canis familiaris (GenBank XM_541552), Rattus norvegicus (GenBank XM_001066591), Mus musculus (GenBank BC058181) y Bos taurus (GenBank BT026254).
El término “actividad de QC” como se usa en el presente documento se define como ciclación intramolecular de residuos de glutamina del extremo N en ácido piroglutámico (pGlu*) o de L-homoglutamina o L-p-homoglutamina del extremo N en un derivado de piro-homoglutamina cíclico bajo liberación de amoniaco. Por tanto, véanse los esquemas 1 y 2.

El término “EC” como se usa en el presente documento comprende la actividad de QC y enzimas similares a QC como glutamato ciclasa (EC), definida adicionalmente como actividad de EC.
El término “actividad de EC” como se usa en el presente documento se define como ciclación intramolecular de
residuos de glutamato del extremo N en ácido piroglutámico (pGlu*) por QC. Por tanto, véase el esquema 3.
Esquema 3: Ciclación del extremo N de glutamil péptidos sin carga por QC (EC)

El término “inhibidor de QC”, “ inhibidor de glutaminil ciclasa” se conoce generalmente para un experto en la técnica y significa inhibidores de enzima, que inhiben la actividad catalítica de glutaminil ciclasa (QC) o su actividad de glutamil ciclasa (EC).
Potencia de inhibición de QC
En vista de la correlación con la inhibición de QC, en realizaciones preferidas, el método objeto y uso médico utilizan un agente con una CI50 para la inhibición de QC de 10 pM o menos, más preferiblemente de 1 pM o menos, incluso más preferiblemente de 0,1 pM o menos o 0,01 pM o menos, o lo más preferiblemente 0,001 pM o menos. De hecho, se contemplan inhibidores con valores de Ki en el intervalo micromolar inferior, preferiblemente el nanomolar e incluso más preferiblemente el intervalo picomolar. De ese modo, aunque los agentes activos se describen en el presente documento, por comodidad, como “inhibidores de QC”, se entenderá que tal nomenclatura no se pretende que limite el objeto de la invención a un mecanismo de acción particular.
Peso molecular de inhibidores de QC
En general, los inhibidores de QC del método objeto o uso médico serán moléculas pequeñas, por ejemplo, con pesos moleculares de 500 g/mol o menos, 400 g/mol o menos, preferiblemente de 350 g/mol o menos, e incluso más preferiblemente de 300 g/mol o menos e incluso de 250 g/mol o menos.
El término “sujeto” como se usa en el presente documento se refiere a un animal, preferiblemente un mamífero, lo más preferiblemente un ser humano, que ha sido el objeto del tratamiento, observación o experimento.
El término “cantidad terapéuticamente eficaz” como se usa en el presente documento significa la cantidad de compuesto activo o agente farmacéutico que provoca la respuesta biológica o medicinal en un sistema de tejido, animal o humano, que se busca por un investigador, veterinario, doctor médico u otro profesional clínico, que incluye alivio de los síntomas de la enfermedad o trastorno que está tratándose.
Como se usa en el presente documento, el término “farmacéuticamente aceptable” engloba tanto uso humano como veterinario: Por ejemplo, el término “farmacéuticamente aceptable” engloba un compuesto veterinariamente aceptable o un compuesto aceptable en medicina humana y cuidado sanitario.
En toda la descripción y las reivindicaciones, el término “alquilo”, a menos que se limite específicamente, indica un grupo alquilo C1-12, adecuadamente un grupo alquilo C1-8, por ejemplo, grupo alquilo C1-6, por ejemplo, grupo alquilo C1-4. Los grupos alquilo pueden ser de cadena lineal o ramificada. Grupos alquilo adecuados incluyen, por ejemplo, metilo, etilo, propilo (por ejemplo, n-propilo e isopropilo), butilo (por ejemplo, n-butilo, iso-butilo, sec-butilo y tere-butilo), pentilo (por ejemplo, n-pentilo), hexilo (por ejemplo, n-hexilo), heptilo (por ejemplo, n-heptilo) y octilo (por ejemplo, noctilo). Los términos “alq” y “alc”, por ejemplo, en las expresiones “alcoxilo”, “haloalquilo” y “tioalquilo”, debe interpretarse según la definición de “alquilo”. Grupos alcoxilo a modo de ejemplo incluyen metoxilo, etoxilo, propoxilo (por ejemplo, n-propoxilo), butoxilo (por ejemplo, n-butoxilo), pentoxilo (por ejemplo, n-pentoxilo), hexoxilo (por ejemplo, n-hexoxilo), heptoxilo (por ejemplo, n-heptoxilo) y octoxilo (por ejemplo, n-octoxilo). Grupos tioalquilo a modo de ejemplo incluyen metiltio-. Grupos haloalquilo a modo de ejemplo incluyen fluoroalquilo, por ejemplo, CF3. fluoroetilo, fluropropilo, fluorobutilo, difluoroetilo, difluoropropilo y difluorobutilo.
El término “halógeno” o “halo” comprende flúor (F), cloro (Cl) y bromo (Br).
Cuando bencimidazolilo se muestra como bencimidazol-5-ilo, el cual se representa como:

es una estructura equivalente. Como se emplea en el presente documento, las dos formas de bencimidazolilo están cubiertas por el término “bencimidazol-5-ilo”.
Estereoisómeros:
Todos los posibles estereoisómeros de los compuestos reivindicados están incluidos en la presente invención.
Si los compuestos según esta invención tienen al menos un centro quiral, pueden, por consiguiente, existir como enantiómeros. Si los compuestos poseen dos o más centros quirales, pueden existir adicionalmente como diastereómeros Debe entenderse que todos aquellos isómeros y mezclas de los mismos están englobados dentro del alcance de la presente invención.
Preparación y aislamiento de estereoisómeros:
Si los procedimientos para la preparación de los compuestos según la invención dan lugar a una mezcla de estereoisómeros, estos isómeros pueden separarse por técnicas convencionales tales como cromatografía preparativa. Los compuestos pueden prepararse en forma racémica, o enantiómeros individuales pueden prepararse o bien por síntesis enantioespecífica o bien por resolución. Los compuestos pueden resolverse, por ejemplo, en sus componentes de enantiómeros por técnicas convencionales, tales como formación de pares diastereoméricos por formación de sales con un ácido ópticamente activo, tal como ácido (-)-di-p-toluoil-d-tartárico y/o ácido (+)-di-p-toluoil-I-tartárico, seguido de cristalización fraccionada y regeneración de la base libre. Los compuestos también pueden resolverse por formación de ésteres o amidas diastereoméricos, seguido de separación cromatográfica y eliminación del auxiliar quiral. Alternativamente, los compuestos pueden resolverse usando una columna de HPLC quiral.
Sales farmacéuticamente aceptables:
En vista de la estrecha relación entre los compuestos libres y los compuestos en la forma de sus sales o solvatos, siempre que un compuesto se cite en este contexto, también está previsto una sal, solvato o polimorfo correspondiente, siempre que sea posible o apropiado bajo las circunstancias.
Sales y solvatos de los compuestos de fórmula (I) y derivados fisiológicamente funcionales de los mismos que son adecuados para su uso en medicina son aquellos en los que el contraión o disolvente asociado es farmacéuticamente aceptable. Sin embargo, sales y solvatos que tienen contraiones farmacéuticamente no aceptables o disolventes asociados están dentro del alcance de la presente invención, por ejemplo, para su uso como productos intermedios en la preparación de otros compuestos y sus sales y solvatos farmacéuticamente aceptables.
Sales adecuadas según la invención incluyen aquellas formadas tanto con ácidos como con bases orgánicos o inorgánicos. Sales de adición de ácido farmacéuticamente aceptables incluyen aquellas formadas a partir de ácidos clorhídrico, bromhídrico, sulfúrico, nítrico, cítrico, tartárico, fosfórico, láctico, pirúvico, acético, trifluoroacético, trifenilacético, sulfámico, sulfanílico, succínico, oxálico, fumárico, maleico, málico, mandélico, glutámico, aspártico, oxaloacético, metanosulfónico, etanosulfónico, arilsulfónico (por ejemplo, p-toluenosulfónico, bencenosulfónico, naftalenosulfónico o naftalenodisulfónico), salicílico, glutárico, glucónico, tricarbalílico, cinámico, cinámico sustituido (por ejemplo, cinámico sustituido con fenilo, metilo, metoxilo o halo, incluyendo ácido 4-metil- y 4-metoxicinámico),
ascórbico, oleico, naftoico, hidroxinaftoico (por ejemplo, 1- o 3-hidroxi-2-naftoico), naftalenoacrílico (por ejemplo, naftaleno-2-acrílico), benzoico, 4- metoxibenzoico, 2- o 4-hidroxibenzoico, 4-clorobenzoico, 4-fenilbenzoico, bencenoacrílico (por ejemplo, 1,4-bencenodiacrílico), isetiónico, ácido perclórico, propiónico, glicólico, hidroxietanosulfónico, pamoico, ciclohexanosulfámico, salicílico, sacarínico y trifluoroacético. Sales de base farmacéuticamente aceptables incluyen sales de amonio, sales de metales alcalinos tales como aquellos de sodio y potasio, sales de metales alcalinotérreos tales como aquellos de calcio y magnesio, y sales con bases orgánicas tales como diciclohexilamina y W-metil-D-glucamina.
Todas las formas de sales de adición de ácido farmacéuticamente aceptables de los compuestos de la presente invención se pretende que estén englobadas por el alcance de esta invención.
Formas cristalinas polimorfas:
Además, algunas de las formas cristalinas de los compuestos pueden existir como polimorfos y como tales se pretende que se incluyan en la presente invención. Además, algunos de los compuestos pueden formar solvatos con agua (es decir, hidratos) o disolventes orgánicos comunes, y tales solvatos también se pretende que estén englobados dentro del alcance de esta invención. Los compuestos, incluyendo sus sales, también pueden obtenerse en forma de sus hidratos, o incluir otros disolventes usados para su cristalización.
Profármacos:
También se describen profármacos de los compuestos de esta invención. En general, tales profármacos serán derivados funcionales de los compuestos que son fácilmente convertibles in vivo en el compuesto terapéuticamente activo deseado. Así, en estos casos, los métodos de tratamiento de la presente invención, el término “administrar” debe englobar el tratamiento de los diversos trastornos descritos con versiones de profármaco de uno o más de los compuestos reivindicados, pero que se convierte en el compuesto anteriormente especificado in vivo después de la administración al sujeto. Procedimientos convencionales para la selección y preparación de derivados de profármacos adecuados se describen, por ejemplo, en “Design of Prodrugs”, ed. H. Bundgaard, Elsevier, 1985.
Grupos protectores:
Durante cualquiera de los procedimientos para la preparación de los compuestos de la presente invención, puede ser necesario y/o deseable proteger grupos sensibles o reactivos en cualquiera de las moléculas en cuestión. Esto puede lograrse por medio de grupos protectores convencionales, tales como aquellos descritos en Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973; y T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991, incorporado completamente en el presente documento por referencia. Los grupos de protección pueden eliminarse en una etapa posterior conveniente usando métodos conocidos de la técnica.
Un grupo protector o grupo de protección se introduce en una molécula mediante modificación química de un grupo funcional con el fin de obtener quimioselectividad en una reacción química subsecuente. Los grupos de protección son, por ejemplo, grupos de protección de alcohol, grupos de protección de amina, grupos de protección de carbonilo, grupos de protección de ácido carboxílico y grupos de protección de fosfato.
Ejemplos de grupos de protección de alcohol son acetil (Ac), benzoilo (Bz), el bencilo (Bn, Bnl) p-metoxietoximetil éter (Me M), mimetoxitritilo [bis-(4-metoxifenil)fenilmetil, Dm T], metoximetil éter (MOM), metoxitritilo [(4-metoxifenil)difenilmetilo, m Mt ), p-metoxibencil éter (PMB), metiltiometil éter, pivaloilo (Piv), tetrahidropiranilo (THP), tritilo (trifenilmetilo, Tr), éteres de sililo (tales como trimetilsililo éter (TMS), terc-butildimetilsililo éter(TBDMS), tercbutildimetilsililoximetil éter (TOM) y triisopropilsililo éter (TIPS)); metil éteres y etoxietil éteres (EE).
Se seleccionan grupos de protección de aminas adecuados a partir de carbobenciloxilo (Cbz), p-metoxibencilo carbonilo (Moz o MeOZ), terc-butiloxicarbonil (BOC), 9-fluorenilmetiloxicarbonilo (FMOC), acetilo (Ac), benzoilo (Bz), bencilo (Bn), p-metoxibencilo (PMB), 3,4-dimetoxibencilo (DMPB), p-metoxifenilo (PMP), tosilo (Ts) y otras sulfonamidas (Nosyl y Nps).
Se seleccionan grupos de protección de carbonilo adecuados a partir de acetales y cetales, acilales y ditianos.
Se seleccionan grupos de protección de ácido carboxílico adecuados a partir de metil ésteres, bencil ésteres, terc-butil ésteres, silil ésteres, ortoésteres y oxazolina.
Ejemplos de grupos de protección de fosfato son 2-cianoetilo y metilo (Me)
Como se usa en el presente documento, el término “composición” se pretende que englobe un producto que comprende los compuestos reivindicados en las cantidades terapéuticamente eficaces, así como cualquier producto que resulte, directa o indirectamente, de combinaciones de los compuestos reivindicados.
Vehículos y aditivos para formulaciones galénicas:
Por tanto, para preparaciones orales líquidas, tales como, por ejemplo, suspensiones, elixires y disoluciones, vehículos y aditivos adecuados pueden incluir ventajosamente agua, glicoles, aceites, alcoholes, agentes aromatizantes, conservantes, agentes colorantes y similares; para preparaciones orales sólidas tales como, por ejemplo, polvos, cápsulas, cápsulas de gel y comprimidos, vehículos y aditivos adecuados incluyen almidones, azúcares, diluyentes, agentes de granulación, lubricantes, aglutinantes, agentes disgregantes y similares.
Vehículos, que pueden añadirse a la mezcla, incluyen excipientes farmacéuticos necesarios e inertes, que incluyen, pero no se limitan a, aglutinantes, agentes de suspensión, lubricantes, aromatizantes, edulcorantes, conservantes, recubrimientos, agentes disgregantes, tintes y agentes colorantes adecuados.
Polímeros solubles como vehículos de fármaco elegibles como diana pueden incluir polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamida-fenol, polihidroxietilaspartamida-fenol, o poli(óxido de etileno)-polilisina sustituido con residuo de palmitoílo. Además, los compuestos de la presente invención pueden acoplarse a una clase de polímeros biodegradables útiles en alcanzar la liberación controlada de un fármaco, por ejemplo, ácido poliláctico, poli-épsiloncaprolactona, ácido polihidroxiburítico, poliortoésteres, poliacetales, polidihidropiranos, policianoacrilatos y copolímeros de bloques de hidrogeles reticulados o anfipáticos.
Aglutinantes adecuados incluyen, sin limitación, almidón, gelatina, azúcares naturales tales como glucosa o betalactosa, edulcorantes de maíz, gomas naturales y sintéticas tales como goma arábiga, tragacanto u oleato de sodio, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato sódico, cloruro sódico y similares.
Disgregantes incluyen, sin limitación, almidón, metilcelulosa, agar, bentonita, goma xantana y similares.
Sumario de la invención
Los inhibidores de glutaminil ciclasa se conocen en la técnica. Los documentos WO 2011/029920 y WO 2014/140279 dan a conocer, entre otras cosas, inhibidores de glutaminil ciclasa que comprende una fracción de oxazolidinona. Sin embargo, para su uso en medicina, es decir, la prevención y tratamiento de enfermedades, se necesitan compuestos adicionales, que han mejorado propiedades farmacocinéticas con el fin de reducir los niveles de dosificación y reducir de ese modo efectos secundarios no deseados y prevenir casos adversos después de la administración a un sujeto. En particular, para el tratamiento o la prevención de enfermedades del sistema nervioso central (SNC), por ejemplo, enfermedades neurodegenerativas como el deterioro cognitivo leve, la enfermedad de Alzheimer, la neurodegeneración en el síndrome de Down o las enfermedades familiares de Alzheimer, se necesitan nuevos compuestos, que muestren aumento de los niveles y aumento de la semivida en el SNC, por ejemplo, en el cerebro y el LCR.
Por tanto, ese fue el problema de la presente invención para proporcionar nuevos compuestos con propiedades farmacocinéticas mejoradas, en particular para el tratamiento de enfermedades relacionadas con el SNC.
Este problema se resolvió por la presente invención mediante la provisión de compuestos de fórmula (I). Según la invención se proporciona un compuesto de fórmula (I):

o una sal, solvato o polimorfo farmacéuticamente aceptable del mismo, incluyendo todos los tautómeros y estereoisómeros del mismo, en la que:
A es heteroarilo seleccionado de 1 H-benzimidazol-5-ilo o 1 H-benzimidazol-6-ilo e imidazo[1,2-a]piridin-7-ilo;
R1 representa hidrógeno, alquilo o halógeno;
R2 representa hidrógeno, alquilo o halógeno;
R3 representa hidrógeno, alquilo o alcoxilo;
R4 representa hidrógeno o alquilo; y
R5 representa hidrógeno, alquilo o halógeno;
y
en el que los grupos alquilo o alcoxilo anteriores se sustituyen opcionalmente por uno o más halógeno.
Descripción detallada de la invención
Sorprendentemente, los inventores encontraron que los compuestos, que comprenden un residuo de fenilazetidinona, resultan en inhibidores de glutaminil ciclasa, que tienen múltiples ventajas en comparación con los inhibidores de glutaminil ciclasa existentes en la técnica anterior. Estos compuestos son inhibidores potentes de la glutaminil ciclasa (QC), así como de su isoenzima, es decir, la proteína tipo glutaminil-péptido ciclotransferasa (QPCTL). Las constantes inhibidoras, tales como el valor Ki, de los compuestos están en el rango nanomolar bajo. La potencia de los compuestos podría mejorarse mediante la halogenación, en particular la fluoración del anillo de fenilo.
Cuando se sustituyen alquilo y alcoxilo, se sustituyen normalmente por 1 o más, como 1, 2 3, 4 o 5 sustituyentes. Preferiblemente, alquilo y alcoxilo se sustituyen por 1 o 2 sustituyentes, más preferiblemente por 2 sustituyentes. Normalmente, los sustituyentes son ambos halógenos. Más normalmente, los sustituyentes halógenos son flúor.
Cuando R1, R2, R3, R4 y R5 representan alquilo, ejemplos incluyen grupos alquilo ramificados o de cadena lineal C1-C12. Un alquilo adecuado es un alquilo C1-8, más adecuadamente, un alquilo C1-6, lo más adecuadamente un alquilo C1-4. Los grupos alquilo anteriormente mencionados no se sustituyen o se sustituyen por uno o más sustituyentes de halógeno, normalmente por 1 o 2 sustituyentes de halógeno. Adecuadamente, los sustituyentes de halógeno son cloro o flúor. Lo más adecuadamente, los sustituyentes de halógeno son flúor.
Cuando R3 representa alcoxilo, ejemplos incluyen grupos alcoxilo ramificados o de cadena lineal -O-C1-12. Un alcoxilo adecuado es un alquilo -O-C1-8, más adecuadamente un alquilo -O-C1-6, lo más adecuadamente un alquilo -O-C1-4. Ejemplos de alcoxilo incluyen grupos metoxilo, etoxilo, propoxilo y butoxilo. Los grupos alcoxilo anteriormente mencionados se sustituyen por uno o más sustituyentes halógenos, normalmente por 1 o 2 sustituyentes halógenos. Lo más adecuadamente, los sustituyentes halógenos son flúor. Ejemplos de grupos alcoxilo sustituidos incluyen diclorometoxilo, difluorometoxilo, dicloropropoxilo, por ejemplo, 2,2-dicloropropoxilo y 3,3- dicloropropoxilo, difluoropropo, por ejemplo, 2,2-difluoropropoxilo y 3,3-difluropropoxilo, diclorobutoxilo y difluorobutoxilo.
Se han obtenido compuestos particularmente ventajosos según la presente invención, cuando ambos son fluorados: (i) el anillo de fenilo, en el que al menos uno de R1, R2 y R5 es flúor, y (ii) el sustituyente en la posición R3
En una realización particular de la invención, se proporciona un compuesto de fórmula (I):

o una sal, solvato o polimorfo farmacéuticamente aceptable del mismo, que incluye todos los tautómeros y estereoisómeros del mismo, en el que:
R1 representa hidrógeno o halógeno;
R2 representa hidrógeno o halógeno;
R3 representa hidrógeno o alcoxilo;
R4 representa hidrógeno; y
R5 representa hidrógeno o halógeno;
y
en el que el grupo alcoxilo anterior se sustituye opcionalmente por uno o más halógenos.
En una realización preferida, A es 1H-benzoimidazolilo, especialmente 1H-benzimidazol-5-ilo o 1H-benzimidazol-6-ilo, y el compuesto de fórmula (I) es un compuesto de fórmula (la):

En una realización preferida, A es imidazo[1,2-a]piridina y el compuesto de fórmula (I) es un compuesto de fórmula (Ib):
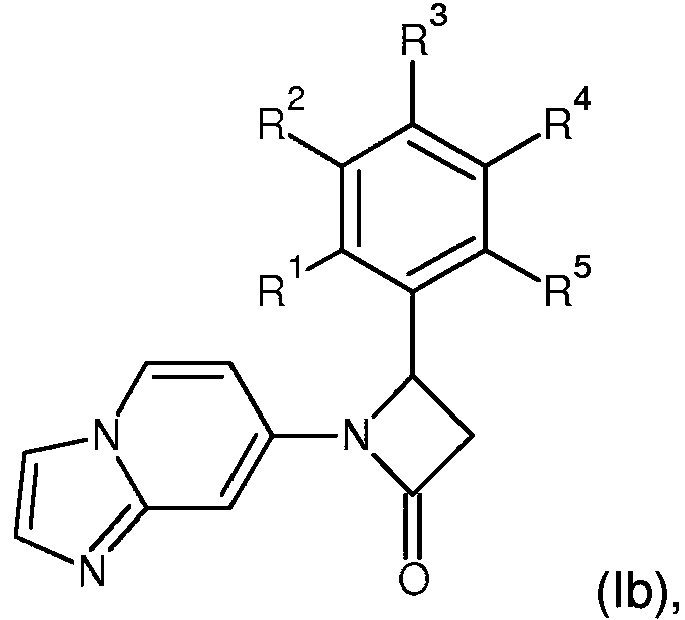
donde R1, R2, R3, R4 y R5 son tal y como se definen en el presente documento.
Cuando R1 representa halógeno, el halógeno es preferiblemente cloro o flúor. En una realización preferida, R1 es hidrógeno. En otra realización preferida, R1 es halógeno, lo más preferiblemente flúor.
Cuando R2 representa halógeno, el halógeno es preferiblemente cloro o flúor. En una realización preferida, R2 es hidrógeno. En otra realización preferida, R2 es halógeno, lo más preferiblemente flúor.
Más adecuadamente, R3 representa -O- alquilo C1-4, sustituido por uno o más halógenos como el flúor.
Preferiblemente, R3 representa metoxilo, difluoropropoxilo o difluorobutoxilo.
Más preferiblemente, R3 representa metoxilo o difluoropropoxilo.
En una realización más preferida, R3 es hidrógeno. En otra realización más preferida, R3 es metoxilo. En otra realización más preferida, R3 representa 2,2-difluoropropoxilo o 3,3-difluoropropoxilo.
R4 es preferiblemente hidrógeno.
Cuando R5 representa halógeno, el halógeno es preferiblemente cloro o flúor. En una realización preferida, R5 es hidrógeno. En otra realización preferida, R5 es halógeno, lo más preferiblemente flúor.
En una realización, R1, R2, R3, R4 y R5 son hidrógeno.
Se prefieren especialmente según la presente invención compuestos de fórmula (I), donde R3 es alcoxilo tal y como se describe en el presente documento.
Se prefieren adicionalmente según la presente invención compuestos en los que el anillo de fenilo en el compuesto de fórmula (I) se sustituye por al menos un halógeno, es decir, al menos uno de R1, R2, R4 y R5 es halógeno.
En una realización, R1 y R5 son halógeno y R2 y R4 son hidrógeno.
En una realización adicional, R1 es halógeno y R2, R4 y R5 son hidrógeno.
En una realización adicional, R1 y R2 son halógeno y R4 y R5 son hidrógeno.
Se prefieren según la presente invención compuestos de fórmula (I), donde al menos uno de R1, R2, R4 y R5 es flúor. Más preferiblemente, uno de R1, R2, R4 y R5 es flúor.
Más preferiblemente, adicionalmente, dos de R1, R2, R4 y R5 son flúor.
Lo más preferiblemente, R1 es flúor y R2, R4 y R5 son hidrógeno; o
R1 y R5 son flúor y R2 y R4 son hidrógeno; o
R1 y R2 son flúor y R4 y R5 son hidrógeno.
Compuestos preferidos de fórmula (I) se caracterizan por las siguientes realizaciones:
R1 es flúor;
R2 es hidrógeno;
R3 es metoxilo, 2,2-difluoropropoxilo o 3,3-difluoropropoxilo;
R4 es hidrógeno; y
R5 es flúor;
o
R1 es flúor,
R2 es hidrógeno,
R3 es 2,2-difluoropropoxilo o 3,3-difluoropropoxilo;
R4 es hidrógeno; y
R5 es hidrógeno;
o
R1 es flúor,
R2 es flúor,
R3 es 2,2-difluoropropoxilo o 3,3-difluoropropoxilo;
R4 es hidrógeno; y
R5 es hidrógeno;
o
R1 es flúor,
R2 es hidrógeno,
R3 es 2,2-difluoropropoxilo o 3,3-difluoropropoxilo;
R4 es hidrógeno; y
R5 es flúor;
Procedimientos
Según un aspecto adicional de la invención se proporciona un procedimiento para preparar un compuesto de fórmula (I) que comprende:
(a) preparar un compuesto de fórmula (I) a partir de un compuesto de fórmula (II):

en la que
R1 y R5 y se definen en el presente documento para los compuestos de fórmula (I).
El procedimiento normalmente implica hacer reaccionar un compuesto de fórmula (II) con ácido bromoacético en presencia de un catalizador adecuado como polvo de zinc, y un agente de protección, como cloruro de trimetilsililo (TMS-CI). En el método 1 en el presente documento se describe un ejemplo no limitativo de la metodología de procedimiento (a).
(b) preparar un compuesto de fórmula (I) a partir de un compuesto de fórmula (III):

en la que
R1, R2, R3, R4 y R5 son tal y como se definen en el presente documento para compuestos de fórmula (I).
El procedimiento (b) normalmente implica hacer reaccionar un compuesto de fórmula (III) con dietilazodicarboxilato en presencia de trifenilfosfina y un disolvente adecuado como tetrahidrofurano, y una etapa de desprotección. En el método 2 en el presente documento se describe un ejemplo no limitativo de la metodología de procedimiento (b).
Los compuestos de fórmula (I) y los compuestos intermedios también pueden prepararse usando técnicas análogas a aquellas conocidas para un experto, o descritas en el presente documento.
Productos intermedios novedosos se reivindican como un aspecto de la presente invención.
Usos terapéuticos
Sustratos fisiológicos de QC (EC) en mamíferos son, por ejemplo, péptidos beta-amiloides (3-40), (3-42), (11-40 y (11 42), ABri, ADan, gastrina, neurotensina, FPP, CCL 2, CCL 7, Cc L 8, CCL 16, CCL 18, fractalcina, orexina A, [Gln3]-glucagón (3-29), [Gln5]-sustancia P(5-11) y el péptido QYNAD. Para detalles adicionales véase la tabla 1. Los compuestos y/o combinaciones según la presente invención y composiciones farmacéuticas que comprenden al menos un inhibidor de QC (EC) son útiles para el tratamiento de afecciones que pueden tratarse por modulación de la actividad de QC.
Tabla 1: Secuencias de aminoácidos de péptidos activos fisiológicos con un residuo de glutamina del extremo N, que tienen tendencia a ciclarse a pGlu final




El glutamato se encuentra en las posiciones 3, 11 y 22 del péptido p-amiloide. Entre ellas, la mutación de ácido glutámico (E) a glutamina (Q) en la posición 22 (correspondiente a la proteína precursora de amiloide APP 693, Swissprot P05067) se ha descrito como la denominada mutación de amiloidosis cerebroarterial tipo holandesa.
Los péptidos p-amiloide con un residuo de ácido piroglutámico en la posición 3, 11 y/o 22 se han descrito que son más citotóxicos e hidrófobos que los péptidos p-amiloide 1-40(42/43) (Saido T.C. 2000 Medica1Hypotheses 54(3): 427 429).
Las múltiples variaciones del extremo N, por ejemplo, Abeta (3-40), Abeta (3-42), Abeta (11-40) y Abeta (11-42) pueden generarse por la enzima p-secretasa enzima escisora de proteína precursora de amiloide del sitio p (BACE) en diferentes sitios (Huse J.T. et al. 2002 J. Biol. Chem. 277 (18): 16278-16284), y/o por procesamiento de aminopeptidasa o dipeptidilaminopeptidasa de los péptidos de longitud completa Abeta (1-40) y Abeta (1-42). En todos los casos, la ciclación del residuo de ácido glutámico que luego se produce en el extremo N está catalizada por QC.
Células transductoras transepiteliales, particularmente la célula gastrina (G), coordinan la secreción de ácidos gástricos con la llegada de comida al estómago. Un trabajo reciente mostró que se generan múltiples productos activos del precursor de gastrina y que hay múltiples puntos de control en la biosíntesis de gastrina. Los precursores biosintéticos y productos intermedios (progastrina y Gly-gastrinas) son factores de crecimiento putativos; sus productos, las gastrinas amidadas, regulan la proliferación celular epitelial, la diferenciación de células parietales productoras de ácido y células similares a enterocromafines que secretan histamina (ECL), y la expresión de genes asociados a la síntesis de histaminas y almacenamiento en células ECL, así como de la secreción de ácido agudamente estimulante. La gastrina también estimula la producción de miembros de la familia del factor de crecimiento epidérmico (EGF), que a su vez inhiben la función de células parietales, pero estimulan el crecimiento de células epiteliales de la superficie. Las concentraciones de gastrina en plasma son elevadas en sujetos con Helicobacterpylori, que se sabe que tienen un riesgo elevado de enfermedad por úlcera duodenal y cáncer gástrico (Dockray, G.J. 1999 J Physiol 15315-324).
Se sabe que la hormona peptídica gastrina, liberada de células G antrales, estimula la síntesis y liberación de histamina de células ECL en la mucosa oxíntica mediante receptores de CCK-2. La histamina movilizada induce la secreción de ácido uniéndose a los receptores de H(2) localizados sobre células parietales. Estudios recientes sugieren que la gastrina, tanto en sus formas completamente amidadas como menos procesadas (progastrina y gastrina extendida a glicina), también es un factor de crecimiento para el tubo gastrointestinal. Se ha establecido que el principal efecto trófico de la gastrina amidada es para la mucosa oxíntica del estómago, en la que produce una elevada proliferación de citoblastos gástricos y células ECL, produciendo elevada masa de células parietales y ECL. Por otra parte, la principal diana trófica de la gastrina menos procesada (por ejemplo, gastrina extendida a glicina) parece ser la mucosa colónica (Koh, T.J. y Chen, D. 2000 Regul Pept 9337-44).
La neurotensina (NT) es un neuropéptido implicado en la patofisiología de la esquizofrenia que modula específicamente los sistemas neurotransmisores previamente demostrados que están regulados erróneamente en este trastorno. Estudios clínicos en los que se han medido las concentraciones de NT en líquido cefalorraquídeo (CSF) revelaron un subconjunto de pacientes esquizofrénicos con concentraciones de NT en CSF disminuidas que son restauradas por tratamiento con fármacos antipsicóticos eficaces. También existen pruebas considerables concordantes con la participación de sistemas de NT en el mecanismo de acción de fármacos antipsicóticos. Los efectos conductuales y bioquímicos de NT centralmente administrada se parecen sorprendentemente a aquellos de fármacos antipsicóticos sistémicamente administrados, y los fármacos antipsicóticos aumentan la neurotransmisión de NT. Esta concatenación de hallazgos condujo a la hipótesis de las funciones de NT como antipsicótico endógeno. Además, fármacos antipsicóticos típicos y atípicos alteran diferencialmente la neurotransmisión de NT en regiones terminales de dopamina nigroestriatal y mesolímbica, y estos efectos son predictivos de la sensibilidad y eficacia de efectos secundarios, respectivamente (Binder, E. B. et al. 2001 Biol Psychiatry 50856-872).
El péptido promotor de la fertilización (FPP), un tripéptido relacionado con la hormona liberadora de tirotrofina (TRH), se encuentra en plasma seminal. Pruebas recientes obtenidas in vitro e in vivo mostraron que FPP desempeña una función importante en la regulación de la fertilidad del esperma. Específicamente, FPP estimula inicialmente espermatozoides no fecundativos (incapacitados) a “encenderse” y volverse fértiles más rápidamente, pero entonces
se detiene la capacitación de manera que los espermatozoides no experimenten pérdida espontánea de acrosomas y, por tanto, no pierdan potencial fecundativo. Estas respuestas son imitadas, y de hecho aumentadas, por la adenosina, que se sabe que regula la ruta de transducción de señales de la adenilil ciclasa (AC)/AMPc. Se ha mostrado que tanto FPP como adenosina estimulan la producción de AMPc en células incapacitadas, pero la inhiben en células capacitadas, con receptores de FPP que interaccionan de alguna manera con receptores de adenosina y proteínas G para lograr la regulación de AC. Estos acontecimientos afectan el estado de fosforilación de tirosina de diversas proteínas, siendo algunas importantes en el “encendido” inicial, participando otros posiblemente en la propia reacción de acrosomas. La calcitonina y la angiotensina II, también encontradas en plasma seminal, tienen efectos similares in vitro sobre espermatozoides incapacitados y pueden aumentar respuestas a FPP. Estas moléculas tienen efectos similares in vivo, afectando a la fertilidad estimulando y luego manteniendo el potencial fecundativo. Tanto las reducciones en la disponibilidad de FPP, adenosina, calcitonina y angiotensina II como los defectos en sus receptores contribuyen a la infertilidad masculina (Fraser, L.R. y Adeoya-Osiguwa, S. A. 2001 Vitam Horm 63, 1-28).
CCL2 (MCP-1), CCL7, CCL8, CCL16, CCL18 y fractalcina desempeñan una función importante en afecciones patofisiológicas, tales como supresión de la proliferación de células progenitoras mieloides, neoplasia, respuestas inflamatorias del huésped, cáncer, psoriasis, artritis reumatoide, aterosclerosis, vasculitis, respuestas humorales e inmunitarias mediadas por células, procesos de adhesión y migración de leucocitos en el endotelio, enfermedad inflamatoria del intestino, reestenosis, fibrosis pulmonar, hipertensión pulmonar, fibrosis hepática, cirrosis hepática, nefroesclerosis, remodelación ventricular, insuficiencia cardíaca, arteriopatía después de trasplantes de órganos y fracaso de injertos de vena.
Varios estudios han enfatizado en particular la función crucial de MCP-1 para el desarrollo de aterosclerosis (Gu, L., et al., (1998) Mol. Cell 2, 275-281; Gosling, J., et al., (1999) J Clin.Invest 103, 773-778); artritis reumatoide (Gong, J. H., et al., (1997) J Exp.Med 186, 131-137; Ogata, H., et al., (1997) J Pathol. 182, 106-114); pancreatitis (Bhatia, M., et al., (2005) Am.J Physiol Gastrointest.Liver Physiol 288, G1259-G1265); enfermedad de Alzheimer (Yamamoto, M., et al., (2005) Am.J Pathol. 166, 1475-1485); fibrosis pulmonar (Inoshima, I., et al., (2004) Am.J Physiol Lung Cell Mol.Physiol 286, L1038-L1044); fibrosis renal (Wada, T., et al., (2004) J Am.Soc.Nephrol. 15, 940-948), y rechazo del injerto (Saiura, A., et al., (2004) Arterioscler. Thromb. Vasc. Biol. 24, 1886-1890). Además, MCP-1 también podría desempeñar una función en la gestosis (Katabuchi, H., et al., (2003) Med Electron Microsc. 36, 253-262), como factor paracrino en el desarrollo de tumores (Ohta, M., et al., (2003) Int. J Oncol. 22, 773-778; Li, S., et al., (2005) J Exp.Med 202, 617-624), dolor neuropático (White, F. A., et al., (2005) Proc. Natl. Acad.Sci.U.S.A) y AIDS (Park, I. W., Wang, J. F., y Groopman, J. E. (2001) Blood 97, 352-358; Coll, B., et al., (2006) Cytokine 34, 51-55).
Los niveles de MCP-1 son elevados en CSF de pacientes con EA y pacientes que muestran deterioro cognitivo leve (DCL) (Galimberti, D., et al., (2006) Arch.Neurol. 63, 538-543). Además, MCP-1 muestra un elevado nivel en suero de pacientes con DCL y EA precoz (Clerici, F., et al., (2006) Neurobiol. Aging 27, 1763-1768).
Recientemente se estudiaron varias vacunas basadas en péptidos de linfocitos T citotóxicos contra hepatitis B, virus de la inmunodeficiencia humana y melanoma en ensayos clínicos. Un candidato a vacuna para el melanoma interesante solo o en combinación con otros antígenos de tumor es el decapéptido ELA. Este péptido es un análogo del péptido inmunodominante de antígeno de Melan-A/MART-1, con un ácido glutámico del extremo N. Se ha informado que el grupo amino y el grupo gamma-carboxílico de ácidos glutámicos, así como el grupo amino y el grupo gamma-carboxamida de glutaminas, se condensan fácilmente para formar derivados piroglutámicos. Para vencer este problema de estabilidad se han desarrollado varios péptidos de interés farmacéutico con un ácido piroglutámico en lugar de glutamina o ácido glutámico del extremo N, sin pérdida de propiedades farmacológicas. Desafortunadamente en comparación con ELA, el derivado de ácido piroglutámico (PirELA) y también el derivado tapado con acetilo del extremo N (AcELA) fracasaron en provocar la actividad de linfocitos citotóxicos T (CTL). A pesar de las aparentes modificaciones menores introducidas en PirELA y AcELA, estos dos derivados probablemente tienen menor afinidad que ELA por el complejo de histocompatibilidad mayor de clase I específico. Por consiguiente, con el fin de conservar la actividad completa de ELA, la formación de PirELA debe evitarse (Beck A. et al. 2001, J Pept Res 57(6):528-38.).
La orexina A es un neuropéptido que desempeña una función significativa en la regulación del consumo de alimentos y sueño-vigilia, posiblemente coordinando las complejas respuestas conductuales y fisiológicas de estas funciones homeostáticas complementarias. También desempeña una función en la regulación homeostática del metabolismo de la energía, función autónoma, equilibrio hormonal y la regulación de fluidos corporales.
Recientemente, se identificaron elevados niveles del pentapéptido QYNAD en el líquido cefalorraquídeo (CSF) de pacientes que padecen esclerosis múltiple o síndrome de Guillain-Barré en comparación con individuos sanos (Brinkmeier H. et al. 2000, Nature Medicine 6, 808-811). Hay una gran controversia en la bibliografía sobre el mecanismo de acción del pentapéptido Gln-Tyr-Asn-Ala-Asp (QYNAD), especialmente su eficacia para interaccionar con y bloquear canales de sodio produciendo la promoción de disfunción axónica, que están implicados en enfermedades autoinmunitarias inflamatorias del sistema nervioso central. Pero recientemente, podría demostrarse que no QYNAD, sino su forma piroglutamada ciclada, pEYNAD, es la forma activa, que bloquea los canales de sodio produciendo la promoción de disfunción axónica. Los canales de sodio se expresan a alta densidad en axones mielinados y desempeñan una función obligatoria en realizar potenciales de acción a lo largo de axones dentro del cerebro y la médula espinal de mamífero. Por tanto, se especula que están implicados en varios aspectos de la
patofisiología de enfermedades autoinmunitarias inflamatorias, especialmente esclerosis múltiple, el síndrome de Guillain-Barré y polirradiculoneuropatía desmielinizante inflamatoria crónica.
Además, QYNAD es un sustrato de la enzima glutaminil ciclasa (QC, EC 2.3.2.5), que también está presente en el cerebro de mamíferos, especialmente en cerebro humano. La glutaminil ciclasa cataliza eficazmente la formación de pEYNAD a partir de su precursor QYNAD.
Por consiguiente, la presente invención proporciona el uso de los compuestos de fórmula (I) para la preparación de un medicamento para la prevención o alivio o tratamiento de una enfermedad seleccionada del grupo que consiste en deterioro cognitivo leve, enfermedad de Alzheimer, demencia familiar británica, demencia familiar danesa, neurodegeneración en síndrome de Down, enfermedad de Huntington, enfermedad de Kennedy, enfermedad ulcerosa, cáncer duodenal con o sin infecciones por Helicobacter pylori, cáncer colorrectal, síndrome de Zollinger-Ellison, cáncer gástrico con o sin infecciones por Helicobacter pylori, afecciones psicóticas patógenas, esquizofrenia, infertilidad, neoplasia, respuestas inflamatorias del huésped, cáncer, metástasis malignas, melanoma, psoriasis, artritis reumatoide, aterosclerosis, pancreatitis, reestenosis, alteración de respuestas inmunitarias humorales y mediadas por células, procesos de adhesión y migración de leucocitos en el endotelio, alteración del consumo de alimentos, alteración del sueño-vigilia, alteración de la regulación homeostática del metabolismo de la energía, alteración de la función autónoma, alteración del equilibrio hormonal o alteración de la regulación de fluidos corporales, esclerosis múltiple, el síndrome de Guillain-Barré y polirradiculoneuropatía desmielinizante inflamatoria crónica.
Además, por administración de un compuesto según la presente invención a un mamífero puede ser posible estimular la proliferación de células progenitoras mieloides.
Además, la administración de un inhibidor de QC según la presente invención puede conducir a supresión de fertilidad masculina.
En una realización preferida, la presente invención proporciona el uso de inhibidores de actividad de QC (EC) en combinación con otros agentes, especialmente para el tratamiento de enfermedades neuronales, arteriosclerosis y esclerosis múltiple.
La presente invención también proporciona un método de tratamiento de las enfermedades anteriormente mencionadas que comprende la administración de una cantidad terapéuticamente activa de al menos un compuesto de fórmula (I) a un mamífero, preferiblemente un ser humano.
Lo más preferiblemente, dicho método y usos correspondientes son para el tratamiento de una enfermedad seleccionada del grupo que consiste en deterioro cognitivo leve, enfermedad de Alzheimer, demencia familiar británica, demencia familiar danesa, neurodegeneración en síndrome de Down, enfermedad de Parkinson y enfermedad de Huntington, que comprende la administración de una cantidad terapéuticamente activa de al menos un compuesto de fórmula (I) a un mamífero, preferiblemente un ser humano.
Incluso preferiblemente, la presente invención proporciona un método de tratamiento y usos correspondientes para el tratamiento de artritis reumatoide, aterosclerosis, pancreatitis y reestenosis.
Combinaciones farmacéuticas
En una realización preferida, la presente invención proporciona una composición, preferiblemente una composición farmacéutica, que comprende al menos un inhibidor de QC opcionalmente en combinación con al menos otro agente seleccionado del grupo que consiste en agentes nootrópicos, neuroprotectores, fármacos antiparkinsonianos, inhibidores de la deposición de proteínas amiloides, inhibidores de la síntesis de amiloide beta, antidepresivos, fármacos ansiolíticos, fármacos antipsicóticos y fármacos contra la esclerosis múltiple.
Lo más preferiblemente, dicho inhibidor de QC es un compuesto de fórmula (I) de la presente invención.
Más específicamente, el otro agente anteriormente mencionado está seleccionado del grupo que consiste en anticuerpos para beta-amiloide, vacunas, inhibidores de cisteína proteasas, inhibidores de PEP, LiCl, inhibidores de acetilcolinesterasa (AChE), potenciadores de PIMT, inhibidores de beta-secretasas, inhibidores de gammasecretasas, inhibidores de aminopeptidasas, preferiblemente inhibidores de dipeptidil peptidasas, lo más preferiblemente inhibidores de DP IV; inhibidores de endopeptidasa neutra, inhibidores de fosfodiesterasa-4 (PDE-4), inhibidores de TNF-alfa, antagonistas de receptor muscarínico M1, antagonistas de receptores de NMDA, inhibidores de receptores sigma-1, antagonistas de histamina H3, agentes inmunomoduladores, agentes inmunosupresores, antagonistas de MCP-1 o un agente seleccionado del grupo que consiste en Antegren (natalizumab), Neurelan (fampridina-SR), Campath (alemtuzumab), IR 208, NBI 5788/Ms P 771 (tiplimotida), paclitaxel, Anergix.MS (AG 284), SH636, Differin (CD 271, adapaleno), BAY 361677 (interleucina-4), inhibidores de metaloproteinasas de matriz (por ejemplo, BB 76163), interferón-tau (trofoblastina) y SAlK-MS.
Además, el otro agente puede ser, por ejemplo, un ansiolítico o antidepresivo seleccionado del grupo que consiste en
(a) benzodiacepinas, por ejemplo, alprazolam, clordiazepóxido, clobazam, clonazepam, clorazepato, diazepam, fludiazepam, loflazepato, lorazepam, metaqualona, oxazepam, prazepam, tranxeno,
(b) inhibidores selectivos de la recaptación de serotonina (SSRI), por ejemplo, citalopram, fluoxetina, fluvoxamina, escitalopram, sertralina, paroxetina,
(c) antidepresivos tricíclicos, por ejemplo, amitriptilina, clomipramina, desipramina, doxepina, imipramina
(d) inhibidores de monoamina oxidasa (MAO),
(e) azapironas, por ejemplo, buspirona, tandopirona,
(f) inhibidores de la recaptación de serotonina-norepinefrina (SNRI), por ejemplo, venlafaxina, duloxetina,
(g) mirtazapina,
(h) inhibidores de la recaptación de norepinefrina (NRI), por ejemplo, reboxetina,
(i) bupropiona,
(j) nefazodona,
(k) beta-bloqueantes,
(l) ligandos de NPY-receptor: agonistas o antagonistas de NPY.
En una realización adicional, el otro agente puede ser, por ejemplo, un fármaco contra la esclerosis múltiple seleccionado del grupo que consiste en
a) inhibidores de dihidroorotato deshidrogenasa, por ejemplo, SC-12267, teriflunomida, MNA-715, HMR-1279 (sin. de HMR-1715, MNA-279),
b) supresor autoinmunitario, por ejemplo, laquinimod,
c) paclitaxel,
d) anticuerpos, por ejemplo, AGT-1, anticuerpo monoclonal anti-factor estimulante de colonias de granulocitos macrófagos (GM-CSF), moduladores de receptores Nogo, ABT-874, alemtuzumab (CAMPATH), anticuerpo anti-OX40, CNTO-1275, d N-1921, natalizumab (sin. de AN-100226, Antegren, VLA-4 Mab), daclizumab (sin. de Zenepax, Ro-34-7375, SMART anti-Tac), J-695, priliximab (sin. de Centara, CEN-000029, cM-T412), MRA, Dantes, anticuerpo anti-IL-12,
e) preparaciones de ácido nucleico peptídico (PNA), por ejemplo, reticulosa,
f) interferón alfa, por ejemplo, alfaferona, interferón alfa humano (sin. de Omniferon, Alfa Leucoferon),
g) interferón beta, por ejemplo, Frone, Avonex similar a interferón beta-1a, Betron (Rebif), análogos de interferón beta, proteína de fusión de interferón beta-transferrina, Betaseron similar a interferón beta-1b recombinante,
h) interferón tau,
i) péptidos, por ejemplo, AT-008, AnergiX.MS, inmunocina (alfa-inmunocina-NNSO3), péptidos cíclicos como ZD-7349, j) enzimas terapéuticas, por ejemplo, CD8 soluble (CD8),
k) plásmido que codifica autoantígeno específico para esclerosis múltiple y plásmido que codifica citocina, por ejemplo, BHT-3009;
l) inhibidor de TNF-alfa, por ejemplo, BLX-1002, talidomida, SH-636,
m) antagonistas de TNF, por ejemplo, solimastat, lenercept (sin. de RO-45-2081, Tenefuse), onercept (sTNFR1), CC-1069,
n) TNF alfa, por ejemplo, etanercept (sin. de Enbrel, TNR-001)
o) antagonistas de CD28, por ejemplo, abatacept,
p) inhibidores de tirosina cinasas Lck,
q) inhibidores de catepsina K,
r) análogos de la proteína transportadora de la membrana que elige neuronas como diana taurina y el inhibidor de calpaína derivado de plantas leupeptina, por ejemplo, Neurodur,
s) antagonista del receptor-1 de quimiocinas (CCR1), por ejemplo, BX-471,
t) antagonistas de CCR2,
u) antagonistas de receptores de AMPA, por ejemplo, ER-167288-01 y ER-099487, E-2007, talampanel,
v) bloqueantes de los canales de potasio, por ejemplo, fampridina,
w) antagonistas de molécula pequeña de tosil-prolina-fenilalanina de la interacción de VLA-4/VCAM, por ejemplo, TBC-3342,
x) inhibidores de molécula de adhesión a células, por ejemplo, TBC-772,
y) oligonucleótidos antisentido, por ejemplo, EN-101,
z) antagonistas de la cadena ligera de la inmunoglobulina libre (lgLC) que se unen a receptores de mastocitos, por ejemplo, F-991,
aa) antígenos inductores de la apoptosis, por ejemplo, Apogen EM,
bb) agonista de receptores adrenérgicos alfa-2, por ejemplo, tizanidina (sin. de Zanaflex, Ternelin, Sirdalvo, Sirdalud, Mionidine),
cc) copolímero de L-tirosina, L-lisina, ácido L-glutámico y L-alanina, por ejemplo, acetato de glatiramer (sin. de Copaxone, COP-1, copolímero-1),
dd) moduladores de la topoisomerasa II, por ejemplo, clorhidrato de mitoxantrona,
ee) inhibidor de adenosina desaminasa, por ejemplo, cladribina (sin. de Leustatin, Milinax, RWJ-26251),
ff) interleucina-10, por ejemplo, ilodecacina (sin. de Tenovil, Sch-52000, CSIF),
gg) antagonistas de interleucina-12, por ejemplo, lisofilina (sin. de CT-1501R, LSF, lisofilina),
hh) etanamina, por ejemplo, SRI-62-834 (sin. de CRC-8605, NSC-614383),
ii) inmunomoduladores, por ejemplo, SAIK-MS, PNU-156804, péptido de alfa-fetoproteína (AFP), IPDS,
jj) agonistas de receptores retinoides, por ejemplo, adapaleno (sin. de Differin, CD-271),
kk) TGF-beta, por ejemplo, GDF-1 (factor de crecimiento y diferenciación 1),
II) TGF-beta-2, por ejemplo, BetaKine,
mm) inhibidores de MMP, por ejemplo, glicomed,
nn) inhibidores de la fosfodiesterasa 4 (PDE4), por ejemplo, RPR-122818,
oo) inhibidores de purina nucleósido fosforilasa, por ejemplo, 9-(3-piridilmetil)-9-deazaguanina, peldesina (sin. de BCX-34, TO-200),
mm) antagonistas de integrina alfa-4/beta-1, por ejemplo, ISIS-104278,
qq) integrina alfa4 antisentido (CD49d), por ejemplo, ISIS-17044, ISIS-27104,
rr) agentes inductores de citocinas, por ejemplo, nucleósidos, ICN-17261,
ss) inhibidores de citocinas,
tt) vacunas de proteínas de choque térmico, por ejemplo, HSPPC-96,
uu) factores de crecimiento de neuregulina, por ejemplo, GGF-2 (sin. de neuregulina, factor de crecimiento de la glía 2),
vv) inhibidores de catepsina S,
ww) análogos de bropirimina, por ejemplo, PNU-56169, PNU-63693,
xx) inhibidores de la proteína-1 quimioatrayente de monocitos, por ejemplo, bencimidazoles como inhibidores de MCP-1, LKS-1456, PD-064036, PD-064126, PD-084486, PD-172084, PD-172386.
Además, la presente invención proporciona composiciones farmacéuticas, por ejemplo, para administración parenteral, enteral o por vía oral, que comprenden al menos un inhibidor de QC opcionalmente en combinación con al menos uno de los otros agentes anteriormente mencionados.
Estas combinaciones proporcionan un efecto particularmente beneficioso. Por tanto, se muestra que tales combinaciones son eficaces y útiles para el tratamiento de las enfermedades anteriormente mencionadas. Por consiguiente, la invención proporciona un método para el tratamiento de estas afecciones.
El método comprende tanto la administración de manera conjunta de al menos un inhibidor de QC como al menos uno de los otros agentes o la administración secuencial de los mismos.
La administración de manera conjunta incluye administración de una formulación, que comprende al menos un inhibidor de QC y al menos uno de los otros agentes o la administración esencialmente simultánea de formulaciones separadas de cada agente.
Los anticuerpos para beta-amiloide y composiciones que contienen los mismos se describen, por ejemplo, en los documentos WO/2009/065054, WO/2009/056490, WO/2009/053696, WO/2009/033743, WO/2007/113172, WO/2007/022416, WO 2006/137354, WO 2006/118959, WO 2006/103116, WO 2006/095041, WO 2006/081171, WO 2006/066233, WO 2006/066171, WO 2006/066089, WO 2006/066049, WO 2006/055178, WO 2006/046644, WO 2006/039470, WO 2006/036291, WO 2006/026408, WO 2006/016644, WO 2006/014638, WO 2006/014478, WO 2006/008661, WO 2005/123775, WO 2005/120571 , WO 2005/105998, WO 2005/081872, WO 2005/080435, WO 2005/028511, WO 2005/025616, WO 2005/025516, WO 2005/023858, WO 2005/018424, WO 2005/011599, WO 2005/000193, WO 2004/108895, WO 2004/098631, WO 2004/080419, WO 2004/071408, WO 2004/069182, WO 2004/067561, WO 2004/044204, WO 2004/032868, WO 2004/031400, WO 2004/029630, WO 2004/029629, WO 2004/024770, WO 2004/024090, WO 2003/104437, WO 2003/089460, WO 2003/086310, WO 2003/077858, WO 2003/074081, WO 2003/070760, WO 2003/063760, WO 2003/055514, WO 2003/051374, WO 2003/048204, WO 2003/045128, WO 2003/040183, WO 2003/039467, WO 2003/016466, WO 2003/015691, WO 2003/014162, WO 2003/012141, WO 2002/088307, WO 2002/088306, WO 2002/074240, WO 2002/046237, WO 2002/046222, WO 2002/041842, WO 2001/062801, WO 2001/012598, WO 2000/077178, WO 2000/072880, WO 2000/063250, WO 1999/060024, WO 1999/027944, WO 1998/044955, WO 1996/025435, WO 1994/017197, WO 1990/014840, WO 1990/012871, WO 1990/012870, WO 1989/006242.
Los anticuerpos para beta-amiloide pueden seleccionarse de, por ejemplo, anticuerpos policlonales, monoclonales, quiméricos o humanizados. Además, dichos anticuerpos pueden ser útiles para desarrollar terapias inmunitarias activas y pasivas, es decir, vacunas y anticuerpos monoclonales.
Ejemplos adecuados de anticuerpos para beta-amiloide son ACU-5A5, huC091 (Acumen/Merck); PF-4360365, RI-1014, RI-1219, RI-409, RN-1219 (Rinat Neuroscience Corp (Pfizer Inc)); los nanocuerpos terapéuticos de Ablynx/Boehringer Ingelheim; anticuerpos monoclonales humanizados específicos de beta-amiloide de Intellect Neurosciences/IBL; m266, m266.2 (Eli Lilly & Co.); AAB-02 (Elan); bapineuzumab (Elan); BAN-2401 (Bioarctic Neuroscience AB); ABP-102 (Abiogen Pharma SpA); BA-27, BC-05 (Takeda); R-1450 (Roche); ESBA-212 (ESBATech AG); AZD-3102 (AstraZeneca) y anticuerpos para beta-amiloide de Mindset BioPharmaceuticals Inc.
Especialmente se prefieren anticuerpos que reconocen el extremo N del péptido Ap. Un anticuerpo adecuado, que reconoce el extremo N del péptido Ap, es, por ejemplo, AcI-24 (AC Immune SA).
Anticuerpos monoclonales contra el péptido beta-amiloide se dan a conocer en los documentos WO 2007/068412, WO/2008/156621 y WO/2010/012004. Anticuerpos quiméricos y humanizados respectivos se dan a conocer en los documentos WO 2008/011348 y WO/2008/060364. Una composición de vacuna para tratar una enfermedad asociada a amiloide se da a conocer en los documentos w O/2002/096937, w O/2005/014041, WO 2007/068411, WO/2007/097251, WO/2009/029272, WO/2009/054537, WO/2009/090650, WO/2009/095857, WO/2010/016912, WO/2010/011947, WO/2010/011999, WO/2010/044464.
Vacunas adecuadas para tratar una enfermedad asociada al amiloide son, por ejemplo, Affitopes AD-01 y AD-02 (GlaxoSmith-Kline), ACC-01 y ACC-02 (Elan/Wyeth), CAD-106 (Novartis/Cytos Biotechnology).
Inhibidores de cisteínas proteasas adecuados son inhibidores de catepsina B. Los inhibidores de catepsina B y composiciones que contienen tales inhibidores se describen, por ejemplo, en los documentos WO/2008/077109, WO/2007/038772, WO 2006/060473, WO 2006/042103, WO 2006/039807, WO 2006/021413, WO 2006/021409, WO 2005/097103, WO 2005/007199, WO2004/084830, WO 2004/078908, WO 2004/026851, WO 2002/094881, WO 2002/027418, WO 2002/021509, WO 1998/046559, WO 1996/021655.
Ejemplos de potenciadores de PIMT adecuados son 10-aminoalifatil-dibenz[b,f]oxepinas descritas en los documentos W o 98/15647 y WO 03/057204, respectivamente. Adicionalmente útiles según la presente invención son los moduladores de la actividad de PIMT descritos en el documento WO 2004/039773.
Inhibidores de beta-secretasa y composiciones que contienen tales inhibidores se describen, por ejemplo, en los documentos WO/2010/094242, WO/2010/058333, WO/2010/021680, WO/2009/108550, WO/2009/042694, WO/2008/054698, WO/2007/051333, WO/2007/021793, W0/2007/019080, WO/2007/019078, WO/2007/011810, W003/059346, W02006/099352, W02006/078576, W02006/060109, W02006/057983, W02006/057945, WO2006/055434, WO2006/044497, WO2006/034296, WO2006/034277, WO2006/029850, WO2006/026204, W02006/014944, W02006/014762, W02006/002004, US 7.109.217, W02005/113484, W02005/103043, W02005/103020, W02005/065195, W02005/051914, W02005/044830, W02005/032471, W02005/018545, W02005/004803, W02005/004802, W02004/062625, W02004/043916, W02004/013098, W003/099202, W003/043987, W003/039454, US 6.562.783, W002/098849 y W002/096897.
Ejemplos adecuados de inhibidores de beta-secretasa con el fin de la presente invención son WY-25105 (Wyeth); posifeno, (+)-fenserina (TorreyPines / NIH); LSN-2434074, LY-2070275, LY-2070273, LY-2070102 (Eli Lilly & Co.); PNU-159775A, PNU-178025A, PNU-17820A, PNU-33312, PNU-38773, PNU-90530 (Elan / Pfizer); KMI-370, KMI-358, kmi-008 (Universidad de Kioto); OM-99-2, OM-003 (Athenagen Inc.); AZ-12304146 (AstraZeneca / Astex); GW-840736X (GlaxoSmithKline plc.), DNP-004089 (De Novo Pharmaceuticals Ltd.) y CT-21166 (CoMentis Inc.).
Inhibidores de gamma-secretasa y composiciones que contienen tales inhibidores se describen, por ejemplo, en los documentos W0/2010/090954, W0/2009/011851, W0/2009/008980, W0/2008/147800, W0/2007/084595, W02005/008250, W02006/004880, US 7.122.675, US 7.030.239, US 6.992.081, US 6.982.264, W02005/097768, W02005/028440, W02004/101562, US 6.756.511, US 6.683.091, W003/066592, W003/014075, W003/013527, W002/36555, W001/53255, US 7.109.217, US 7.101.895, US 7.049.296, US 7.034.182, US 6.984.626, W02005/040126, W02005/030731, W02005/014553, US 6.890.956, EP 1334085, EP 1263774, W02004/101538, W02004/00958, W02004/089911, W02004/073630, W02004/069826, W02004/039370, W02004/031139, W02004/031137, US 6.713.276, US 6.686.449, W003/091278, US 6.649.196, US 6.448.229, W001/77144 y W001/66564.
Inhibidores de gamma-secretasa adecuados con el fin de la presente invención son GSI-953, WAY-GSI-A, WAY-GSI-B (Wyeth); MK-0752, MRK-560, L-852505, L-685-458, L-852631, L-852646 (Merck & Co. Inc.); LY-450139, LY-411575, AN-37124 (Eli Lilly & Co.); BMS-299897, BMS-433796 (Bristol-Myers Squibb Co.); E-2012 (Eisai Co. Ltd.); EHT-0206, EHT-206 (ExonHit Therapeutics SA); NGX-555 (TorreyPines Therapeutics Inc.) y Semagacestat (Eli Lilly).
Inhibidores de DP IV y composiciones que contienen tales inhibidores se describen, por ejemplo, en los documentos US6.011.155; US6.107.317; US6.110.949; US6.124.305; US6.172.081; W099/61431, W099/67278, W099/67279, DE19834591, W097/40832, W095/15309, W098/19998, W000/07617, W099/38501, W099/46272, W099/38501, W001/68603, W001/40180, W001/81337, W001/81304, W001/55105, W002/02560, W001/34594, W002/38541, W002/083128, W003/072556, W003/002593, W003/000250, W003/000180, W003/000181, EP1258476, W003/002553, W003/002531, W003/002530, W003/004496, W003/004498, W003/024942, W003/024965, W003/033524, W003/035057, W003/035067, W003/037327, W003/040174, W003/045977, W003/055881, W003/057144, W003/057666, W003/068748, W003/068757, W003/082817, W003/101449, W003/101958, W003/104229, W003/74500, W02004/007446, W02004/007468, W02004/018467, W02004/018468, W02004/018469, W02004/026822, W02004/032836, W02004/033455, W02004/037169, W02004/041795, W02004/043940, W02004/048352, W02004/050022, W02004/052850, W02004/058266, W02004/064778, W02004/069162, W02004/071454, W02004/076433, W02004/076434, W02004/087053, W02004/089362, W02004/099185, W02004/103276, W02004/103993, W02004/108730, W02004/110436, W02004/111041, W02004/112701, W02005/000846, W02005/000848, W02005/011581, W02005/016911, W02005/023762, W02005/025554, W02005/026148, W02005/030751, W02005/033106, W02005/037828, W02005/040095, W02005/044195, W02005/047297, W02005/051950, W02005/056003, W02005/056013, W02005/058849, W02005/075426, W02005/082348, W02005/085246, W02005/087235, W02005/095339, W02005/095343, W02005/095381, W02005/108382, W02005/113510, W02005/116014, W02005/116029, W02005/118555, W02005/120494, W02005/121089, W02005/121131, W02005/123685, W02006/995613; W02006/009886; W02006/013104; W02006/017292; W02006/019965; W02006/020017; W02006/023750; W02006/039325; W02006/041976; W02006/047248; W02006/058064; W02006/058628; W02006/066747; W02006/066770 y
WO2006/068978.
Inhibidores de DP IV adecuados con el fin de la presente invención son, por ejemplo, sitagliptina, des-fluoro-sitagliptina (Merck & Co. Inc.); vildagliptina, DPP-728, SDZ-272-070 (Novartis); ABT-279, ABT-341 (Abbott Laboratories); denagliptina, TA-6666 (GlaxoSmithKline plc.); SYR-322 (Takeda San Diego Inc.); talabostat (Point Therapeutics Inc.); Ro-0730699, R-1499, R-1438 (Roche Holding AG); FE-999011 (Ferring Pharmaceuticals); TS-021 (Taisho Pharmaceutical Co. Ltd ); GRC-8200 (Glenmark Pharmaceuticals Ltd.); A l S-2-0426 (Alantos Pharmaceuticals Holding Inc.); ARI-2243 (Arisaph Pharmaceuticals Inc.); SSR-162369 (Sanofi-Synthelabo); MP-513 (Mitsubishi Pharma Corp.); DP-893, CP-867534-01 (Pfizer Inc.); TSL-225, TMC-2A (Tanabe Seiyaku Co. Ltd.); PHX-1149 (Phenomenix Corp.); saxagliptina (Bristol-Myers Squibb Co.); PSN-9301 ((OSI) Prosidion), S-40755 (Servier); KRP-104 (ActivX Biosciences Inc.); sulfostina (Zaidan Hojin); KR-62436 (Instituto Coreano de Investigación de Tecnología Química); P32/98 (Probiodrug AG); BI-A, BI-B (Boehringer Ingelheim Corp.); SK-0403 (Sanwa Kagaku Kenkyusho Co. Ltd.); y NNC-72-2138 (Novo Nordisk A/S).
Otros inhibidores de DP IV preferidos son
(i) compuestos similares a dipéptidos, desvelados en el documento WO 99/61431, por ejemplo, N-valilprolilo, O-benzoilhidroxilamina, alanilpirrolidina, isoleuciltiazolidina como L-alo-isoleucil-tiazolidina, L-treo-isoleucil-pirrolidina y sales de los mismos, especialmente las sales fumáricas, y L-alo-isoleucil-pirrolidina y sales de la misma;
(ii) estructuras de péptidos, dadas a conocer en el documento WO 03/002593, por ejemplo, tripéptidos;
(iii) peptidilcetonas, dadas a conocer en el documento WO 03/033524;
(vi) aminocetonas sustituidas, dadas a conocer en el documento WO 03/040174;
(v) inhibidores de DP IV tópicamente activos, dados a conocer en el documento WO 01/14318;
(vi) profármacos de inhibidores de DP IV, dados a conocer en los documentos WO 99/67278 y WO 99/67279; y
(v) inhibidores de DP IV basados en glutaminilo, dados a conocer en los documentos WO 03/072556 y WO 2004/099134.
Inhibidores de la síntesis de beta-amiloide adecuados con el fin de la presente invención son, por ejemplo, bisnorcimserina (Axonyx Inc.); (R)-flurbiprofeno (MCP-7869; Flurizan) (Myriad Genetics); nitroflurbiprofeno (NicOx); BGC-20-0406 (Sankyo Co. Ltd.) y BGC-20-0466 (BTG plc.), RQ-00000009 (RaQualia Pharma Inc).
Inhibidores de la deposición de proteína amiloide adecuados con el fin de la presente invención son, por ejemplo, SP-233 (Samaritan Pharmaceuticals); AZD-103 (Ellipsis Neurotherapeutics Inc.); AAB-001 (Bapineuzumab), a A b-002, ACC-001 (Elan Corp pic.); colostrinina (ReGen Therapeutics pic.); Tramiprosato (Neurochem); AdPEDI-(amiloidebeta1-6)11) (Vaxin Inc.); MPI-127585, m P i-423948 (Fundación Mayo); SP-08 (Universidad de Georgetown); ACU-5A5 (Acumen / Merck); transtirretina (Universidad del Estado de Nueva York); PTI-777, DP-74, DP 68, Exebryl (ProteoTech Inc.); m266 (Eli Lilly & Co.); EGb-761 (Dr. Willmar Schwabe GmbH); s P|-014 (Satori Pharmaceuticals Inc.); ALS-633, ALS-499 (Advanced Life Sciences Inc.); AGT-160 (ArmaGen Technologies Inc.); TAK-070 (Takeda Pharmaceutical Co. Ltd.); CHF-5022, CHF-5074, CHF-5096 y CHF-5105 (Chiesi Farmaceutici SpA.), SEN-1176 y SEN-1329 (Senexis Ltd.), AGT-160 (ArmaGen Technologies), Davunetida (Allon Therapeutics), ELND-005 (Elan Corp / Transition Therapeutics) y nilvadipina (Archer Pharmaceuticals).
Inhibidores de PDE-4 adecuados con el fin de la presente invención son, por ejemplo, doxofilina (Instituto Biologico Chemioterapica ABC SpA.) colirios idudilast, tipelukast, ibudilast (Kyorin Pharmaceutical Co. Ltd.); teofilina (Elan Corp.); cilomilast (GlaxoSmithKline pic.); Atopik (Barrier Therapeutics Inc.); tofimilast, CI-1044, Pd -189659, CP-220629, inhibidor de PDE 4d Bh N (Pfizer Inc.); arofilina, lAs -37779 (Almirall Prodesfarma SA.); roflumilast, hidroxipumafentrina (Altana AG), tetomilast (Otska Pharmaceutical Co. Ltd.); tipelukast, ibudilast (Kyorin Pharmaceutical), CC-10004 (Celgene Corp.); HT-0712, IPL-4088 (Inflazyme Pharmaceuticals Ltd.); MEM-1414, MEM-1917 (Memory Pharmaceuticals Corp.); oglemilast, GRC-4039 (Glenmark Pharmaceuticals Ltd.); AWD-12-281, ELB-353, ELB-526 (Elbion AG); EHT-0202 (ExonHit Therapeutics SA.); ND-1251 (Neuro3d SA.); 4AZA-PDE4 (4 AZA Bioscience NV.); AVE-8112 (Sanofi-Aventis); CR-3465 (Rottapharm SpA.); GP-0203, NCS-613 (Centre National de la Recherche Scientifique); KF-19514 (Kyowa Hakko Kogyo Co. Ltd.); On O-6126 (Ono Pharmaceutical Co. Ltd.); OS-0217 (Dainippon Pharmaceutical Co. Ltd.); IBFB-130011, IBFB-150007, IBFB-130020, IBFB-140301 (IBFB Pharma GmbH); IC-485 (ICOS Corp.); RBx-14016 y RBx-11082 (Ranbaxy Laboratories Ltd.). Un inhibidor de p De -4 preferido es rolipram.
Los inhibidores y composiciones de MAO que contienen tales inhibidores se describen, por ejemplo, en los documentos WO2006/091988, WO2005/007614, WO2004/089351, WO01/26656, WO01/12176, WO99/57120, WO99/57119, WO99/13878, WO98/40102, WO98/01157, WO96/20946, WO94/07890 y WO92/21333.
Inhibidores de MAO adecuados con el fin de la presente invención son, por ejemplo, linezolida (Pharmacia Corp.); RWJ-416457 (RW Johnson Pharmaceutical Research Institute); budipino (Altana AG); GPX-325 (BioResearch Ireland); isocarboxazida; fenelzina; tranilcipromina; indantadol (Chiesi Farmaceutici SpA.); moclobemida (Roche Holding AG); SL-25.1131 (Sanofi-Synthelabo); CX-1370 (Burroughs Wellcome Co.); CX-157 (Krenitsky Pharmaceuticals Inc.); desoxipeganina (HF Arzneimittelforschung GmbH & Co. KG); bifemelano (Mitsubishi-Tokyo Pharmaceuticals Inc.); RS-1636 (Sankyo Co. Ltd.); esuprona (BASF AG); rasagilina (Teva Pharmaceutical Industries Ltd ); ladostigil (Universidad hebrea de Jerusalén); safinamida (Pfizer), NW-1048 (Newron Pharmaceuticals SpA.), EVT-302 (Evotec).
Antagonistas de histamina H3 adecuados con el fin de la presente invención son, por ejemplo, ABT-239, ABT-834 (Abbott Laboratories); 3874-H1 (Aventis Pharma); UCL-2173 (Universidad Libre de Berlín), UCL-1470 (BioProjet, Societe Civile de Recherche); DWP-302 (Daewoong Pharmaceutical Co Ltd); GSK-189254A, GSK-207040A (GlaxoSmithKline Inc.); cipralisant, GT-2203 (Gliatech Inc.); Ciproxifan (INSERM), 1S,2S-2-(2-Aminoetil)-1-(1H-imidazol-4-yl)ciclopropano (Universidad de Hokkaido); JNJ-17216498, JNJ-5207852 (Johnson & Johnson); Nn C-0038-0000-1049 (Novo Nordisk A/S); y Sch-79687 (Schering-Plough).
Inhibidores de PEP y composiciones que contienen tales inhibidores se describen, por ejemplo, en los documentos JP 01042465, JP 03031298, JP 04208299, WO 00/71144, US 5.847.155; JP 09040693, JP 10077300, JP 05331072, JP 05015314, WO 95/15310, WO 93/00361, EP 0556482, JP 06234693, JP 01068396, EP 0709373, US 5.965.556, US 5.756.763, US 6.121.311, JP 63264454, JP 64000069, JP 63162672, EP 0268190, EP 0277588, EP 0275482, US 4.977.180, US 5.091.406, US 4.983.624, US 5.112.847, US 5.100.904, US 5.254.550, US 5.262.431, US 5.340.832, US 4.956.380, EP 0303434, JP 03056486, JP 01143897, JP 1226880, EP 0280956, US 4.857.537, EP 0461677, EP 0345428, JP 02275858, US 5,506,256, JP 06192298, EP 0618193, JP 03255080, EP 0468469, US 5.118.811, JP 05025125, WO 9313065, JP 05201970, WO 9412474, EP 0670309, EP 0451547, JP 06339390, US 5.073.549, US 4.999.349, EP 0268281, US 4.743.616, EP 0232849, EP 0224272, JP 62114978, JP 62114957, US 4.757.083, US 4.810.721, US 5.198.458, US 4.826.870, EP 0201742, EP 0201741, US 4.873.342, EP 0172458, JP 61037764, EP 0201743, US 4.772.587, EP 0372484, US 5.028.604, WO 91/18877, JP 04009367, JP 04235162, US 5.407.950, WO 95/01352, JP 01250370, JP 02207070, US 5.221.752, EP 0468339, JP 04211648, WO 99/46272, WO 2006/058720 any PCT/EP2006/061428.
Inhibidores de prolil endopeptidasa adecuados con el fin de la presente invención son, por ejemplo, Fmoc-Ala-Pirr-CN, Z-Phe-Pro-benzotiazol (Probiodrug), Z-321 (Zeria Pharmaceutical Co Ltd.); ONO-1603 (Ono Pharmaceutical Co Ltd); JTP-4819 (Japan Tobacco Inc.) y S-17092 (Servier).
Otros compuestos adecuados que pueden usarse según la presente invención en combinación con inhibidores de QC son NPY, un mimético de NPY o un agonista o antagonista de NPY o un ligando de los receptores de NPY.
Se prefieren según la presente invención antagonistas de los receptores de NPY.
Ligandos adecuados o antagonistas de los receptores de NPY son compuestos derivados de 3a, 4,5,9b-tetrahidro-1 hbenz[e]indol-2-il-amina como se da a conocer en el documento WO 00/68197.
Antagonistas de los receptores de NPY que pueden mencionarse incluyen los dados a conocer en las solicitudes de patente europea EP 0614911, EP 0747 357, EP 0747356 y EP 0747 378; solicitudes de patente internacional WO 94/17035, WO 97/19911, WO 97/19913, WO 96/12489, WO 97/19914, WO 96/22305, WO 96/40660, WO 96/12490, WO 97/09308, WO 97/20820, WO 97/20821, WO 97/20822, WO 97/20823, WO 97/19682, WO 97/25041, WO 97/34843, WO 97/46250, WO 98/03492, WO 98/03493, WO 98/03494 y WO 98/07420; WO 00/30674, patentes de EE.UU. n.° 5.552.411, 5.663.192 y 5.567.714; 6.114.336, solicitud de patente japonesa JP 09157253; solicitudes de patente internacional WO 94/00486, WO 93/12139, WO 95/00161 y WO 99/15498; patente de EE.UU. n.° 5.328.899; solicitud de patente alemana DE 393 97 97; solicitudes de patente europea EP 355 794 y EP 355 793; y solicitudes de patente japonesa JP 06116284 y JP 07267988. Antagonistas de NPY preferidos incluyen aquellos compuestos que se dan a conocer específicamente en estos documentos de patente. Compuestos más preferidos incluyen antagonistas de NPY basados en aminoácidos y en no péptidos. Los antagonistas de NPY basados en aminoácidos y en no péptidos que pueden mencionarse incluyen los dados a conocer en las solicitudes de patente europea EP 0 614 911, EP 0 747 357, EP 0 747 356 y EP 0 747 378; solicitudes de patente internacional WO 94/17035, WO 97/19911, WO 97/19913, WO 96/12489, WO 97/19914, WO 96/22305, WO 96/40660, WO 96/12490, WO 97/09308, WO 97/20820, WO 97/20821, WO 97/20822, WO 97/20823, WO 97/19682, WO 97/25041, WO 97/34843, WO 97/46250, WO 98/03492, WO 98/03493, WO 98/03494, WO 98/07420 y WO 99/15498; patentes de EE.UU. n.° 5.552.411, 5.663.192 y 5.567.714; y solicitud de patente japonesa JP 09157253. Antagonistas de NPY basados en aminoácidos y en no péptidos preferidos incluyen aquellos compuestos que se dan a conocer específicamente en estos documentos de patente.
Compuestos particularmente preferidos incluyen antagonistas de NPY basados en aminoácidos. Compuestos basados en aminoácidos que pueden mencionarse incluyen los dados a conocer en las solicitudes de patente internacional WO 94/17035, WO 97/19911, WO 97/19913, WO 97/19914 o, preferiblemente, WO 99/15498. Antagonistas de NPY basados en aminoácidos preferidos incluyen aquellos que se dan a conocer específicamente en estos documentos de
patente, por ejemplo, BIBP3226 y, especialmente, (R)-N2-(d¡fen¡lacet¡l)-(R)-N-[1-(4-h¡drox¡-fen¡l)et¡l]arg¡n¡na-am¡da (ejemplo 4 de la sol¡c¡tud de patente ¡nternac¡onal WO 99/15498).
Agon¡stas de receptores de M1 y compos¡c¡ones que cont¡enen tales ¡nh¡b¡dores se descr¡ben, por ejemplo, en los documentos WO2004/087158, WO91/10664.
Agon¡stas de receptores de M1 adecuados con el f¡n de la presente ¡nvenc¡ón son, por ejemplo, CDD-0102 (Cogn¡t¡ve Pharmaceut¡cals); cev¡mel¡na (Evoxac) (Snow Brand M¡lk Products Co. Ltd ); NGX-267 (TorreyP¡nes Therapeut¡cs); sabcomel¡na (GlaxoSm¡thKl¡ne); alvamel¡na (H Lundbeck A/S); LY-593093 (El¡ L¡lly & Co.); VRTX-3 (Vertex Pharmaceut¡cals Inc.); WAY-132983 (Wyeth), CI-101 7/ (PD-151832) (Pf¡zer Inc.) y MCD-386 (M¡tr¡d¡on Inc.).
Inh¡b¡dores de la acet¡lcol¡nesterasa y compos¡c¡ones que cont¡enen tales ¡nh¡b¡dores se descr¡ben, por ejemplo, en los documentos WO2006/071274, WO2006/070394, WO2006/040688, W02005/092009, WO2005/079789, W02005/039580, WO2005/027975, WO2004/084884, WO2004/037234, WO2004/032929, WO03/101458, WO03/091220, WO03/082820, WO03/020289, WO02/32412, WO01/85145, WO01/78728, WO01/66096, WO00/02549, WO01/00215, WO00/15205, WO00/23057, WO00/33840, WO00/30446, WO00/23057, WO00/15205, WO00/09483, WO00/07600, WO00/02549, WO99/47131, WO99/07359, WO98/30243, WO97/38993, WO97/13754, WO94/29255, WO94/20476, WO94/19356, WO93/03034 y WO92/19238.
Inh¡b¡dores de la acet¡lcol¡nesterasa adecuados con el f¡n de la presente ¡nvenc¡ón son, por ejemplo, donepez¡l (E¡sa¡ Co. Ltd.); r¡vast¡gm¡na (Novart¡s AG); (-)-fenser¡na (TorreyP¡nes Therapeut¡cs); ladost¡g¡l (Un¡vers¡dad Hebrea de Jerusalén); huperz¡na A (Fundac¡ón Mayo); galantam¡na (Johnson & Johnson); Memoqu¡na (Un¡vers¡dad de Bolon¡a); SP-004 (Samar¡tan Pharmaceut¡cals Inc.); BGC-20-1259 (Sankyo Co. Ltd.); f¡sost¡gm¡na (Forest Laborator¡es Inc.); NP-0361 (Neuropharma SA); ZT-1 (Deb¡opharm); tacr¡na (Warner-Lambert Co.); metr¡fonato (Bayer Corp.), INM-176 (Whanln), huperz¡na A (Neuro-H¡tech / Xel Pharmaceut¡cal), m¡mopez¡lo (Deb¡opharm) y D¡mebon (Med¡vat¡on/Pf¡zer).
Antagon¡stas de receptores de NMDA y compos¡c¡ones que cont¡enen tales ¡nh¡b¡dores se descr¡ben, por ejemplo, en los documentos WO2006/094674, WO2006/058236, WO2006/058059, WO2006/010965, WO2005/000216, WO2005/102390, WO2005/079779, WO2005/079756, WO2005/072705, WO2005/070429, WO2005/055996, WO2005/035522, WO2005/009421, WO2005/000216, WO2004/092189, WO2004/039371, WO2004/028522, WO2004/009062, WO03/010159, WO02/072542, WO02/34718, WO01/98262, WO01/94321, WO01/92204, WO01/81295, WO01/32640, WO01/10833, WO01/10831, WO00/56711, WO00/29023, WO00/00197, WO99/53922, WO99/48891, WO99/45963, WO99/01416, WO99/07413, WO99/01416, WO98/50075, WO98/50044, WO98/10757, WO98/05337, WO97/32873, WO97/23216, WO97/23215, WO97/23214, WO96/14318, WO96/08485, WO95/31986, WO95/26352, WO95/26350, WO95/26349, WO95/26342, WO95/12594, WO95/02602, WO95/02601, WO94/20109, WO94/13641, WO94/09016 y WO93/25534.
Antagon¡stas de receptores de NMDA adecuados con el f¡n de la presente ¡nvenc¡ón son, por ejemplo, memant¡na (Merz & Co. GmbH); top¡ramato (Johnson & Johnson); AVP-923 (Neurodex) (Centro de Estud¡o Neurológ¡co); EN-3231 (Endo Pharmaceut¡cals Hold¡ngs Inc.); neramexano (MRZ-2/579) (Merz and Forest); CNS-5161 (CeNeS Pharmaceut¡cals Inc.); dexanab¡nol (HU-211; S¡nnab¡dol; PA-50211) (Pharmos); Ep¡Cept Np -1 (Un¡vers¡dad de Dalhous¡e); ¡ndantadol (V-3381; CNP-3381) (Vernal¡s); perz¡nfotel (EAA-090, WAY-126090, EAA-129) (Wyeth); RGH-896 (Gedeon R¡chter Ltd.); traxoprod¡lo (CP-101606), besonprod¡lo (PD-196860, CI-1041) (Pf¡zer Inc.); CGX-1007 (Cognet¡x Inc.); delucem¡na (Np S-1506) (NPS Pharmaceut¡cals Inc.); EVT-101 (Roche Hold¡ng AG); acamprosato (Synchroneuron LLC.); CR-3991, CR- 2249, CR-3394 (Rottapharm SpA.); AV-101 (4-Cl-k¡nuren¡na (4-Cl-KYN)), ác¡do 7-cloro-k¡nurén¡co (7-Cl-KYNA) (V¡sta-Gen); NPS-1407 (NpS Pharmaceut¡cals Inc.); YT-1006 (Yaupon Therapeut¡cs Inc.); ED-1812 (Sose¡ R&D Ltd.); h¡mantano (clorh¡drato N-2-(adamantil)-hexametilenimina) (RAMs ); Lanc¡cem¡na (AR-R-15896) (AstraZeneca); EVT-102, Ro-25-6981 y Ro-63-1908 (Hoffmann-La Roche AG / Evotec), neramexano (Merz).
Además, la presente ¡nvenc¡ón se refiere a terap¡as de comb¡nac¡ón út¡les para el tratam¡ento de ateroscleros¡s, reestenos¡s o artr¡t¡s, adm¡n¡strando un ¡nh¡b¡dor de QC en comb¡nac¡ón con otro agente terapéut¡co selecc¡onado del grupo que cons¡ste en ¡nh¡b¡dores de la enz¡ma conversora de ang¡otens¡na (ACE); receptores bloqueantes de ang¡otens¡na II; d¡urét¡cos; bloqueantes de los canales de calc¡o (CCB); beta-bloqueantes; ¡nh¡b¡dores de la agregac¡ón plaquetar¡a; moduladores de la absorc¡ón del colesterol; ¡nh¡b¡dores de la HMG-Co-A reductasa; compuestos que aumentan la l¡poproteína de alta dens¡dad (HDL); ¡nh¡b¡dores de ren¡na; ¡nh¡b¡dores de IL-6; cort¡costero¡des ant¡¡nflamator¡os; agentes ant¡prol¡ferat¡vos; donantes de óx¡do nítr¡co; ¡nh¡b¡dores de la síntes¡s de matr¡z extracelular; ¡nh¡b¡dores de la transducc¡ón de señales de c¡toc¡nas o factores de crec¡m¡ento; antagon¡stas de MCP-1 e ¡nh¡b¡dores de t¡ros¡na c¡nasas que proporc¡onan efectos terapéut¡cos benef¡c¡osos o s¡nérg¡cos con respecto a cada componente de monoterap¡a solo.
Se ent¡ende que los bloqueantes de los receptores de la ang¡otens¡na II son aquellos agentes act¡vos que se unen al subt¡po de receptor AT1 del receptor de ang¡otens¡na II, pero no producen la act¡vac¡ón del receptor. Como consecuenc¡a del bloqueo del receptor de AT1, estos antagon¡stas pueden, por ejemplo, emplearse como agentes ant¡h¡pertensores.
Bloqueantes de los receptores de la angiotensina II adecuados que pueden emplearse en la combinación de la presente invención incluyen antagonistas de receptores de AT1 que tienen características estructurales diferentes, se prefieren aquellos con estructuras no peptídicas. Por ejemplo, puede hacerse mención de los compuestos que están seleccionados del grupo que consiste en valsartán (documento EP 443983), losartán (documento EP 253310), candesartán (documento Ep 459136), eprosartán (documento EP 403159), irbesartán (documento EP 454511), olmesartán (documento EP 503785), tasosartán (documento EP 539086), telmisartán (documento EP 522314), el compuesto con la designación E-41 77 de fórmula

el compuesto con la designación SC-52458 de la siguiente fórmula

y el compuesto con la designación del compuesto ZD-8731 de fórmula

o, en cada caso, una sal farmacéuticamente aceptable de los mismos.
Antagonistas de receptores de AT1 preferidos son aquellos agentes que han sido autorizados y llegaron al mercado, el más preferido es valsartán, o una sal farmacéuticamente aceptable del mismo.
La interrupción de la degradación enzimática de angiotensina a angiotensina II con inhibidores de ACE es una variante satisfactoria para la regulación de la tensión arterial y así también pone a disposición un método terapéutico para el tratamiento de hipertensión.
Un inhibidor de ACE adecuado para emplearse en la combinación de la presente invención es, por ejemplo, un compuesto seleccionado del grupo que consiste en alacepril, benazepril, benazeprilato; captopril, ceronapril, cilazapril, delapril, enalapril, enaprilato, fosinopril, imidapril, lisinopril, moveltopril, perindopril, quinapril, ramipril, espirapril, temocapril y trandolapril, o en cada caso, una sal farmacéuticamente aceptable de los mismos.
Inhibidores de ACE preferidos son aquellos agentes que han sido comercializados, los más preferidos son benazepril y enalapril.
Un diurético es, por ejemplo, un derivado de tiazida seleccionado del grupo que consiste en clorotiazida, hidroclorotiazida, metilclotiazida y clorotalidona. El diurético más preferido es hidroclorotiazida. Un diurético comprende además un diurético ahorrador de potasio tal como amilorida o triameterina, o una sal farmacéuticamente
aceptable de las mismas.
La clase de CCB comprende esencialmente dihidropiridinas (DHP) y no DHP, tales como CCB tipo diltiazem y tipo verapamilo.
Un CCB útil en dicha combinación es preferiblemente un representante de DHP seleccionado del grupo que consiste en amlodipino, felodipino, riosidina, isradipino, lacidipino, nicardipino, nifedipino, niguldipino, niludipino, nimodipino, nisoldipino, nitrendipino y nivaldipino, y es preferiblemente un representante de no DHP seleccionado del grupo que consiste en flunarizina, prenilamina, diltiazem, fendilina, galopamilo, mibefradilo, anipamilo, tiapamilo y verapamilo, y en cada caso, una sal farmacéuticamente aceptable de los mismos. Todos estos CCB se usan terapéuticamente, por ejemplo, como fármacos antihipertensores, contra la angina de pecho o antiarrítmicos.
CCB preferidos comprenden amlodipino, diltiazem, isradipino, nicardipino, nifedipino, nimodipino, nisoldipino, nitrendipino y verapamil o, por ejemplo, dependiente del CCB específico, una sal farmacéuticamente aceptable de los mismos Se prefiere especialmente como DHP amlodipino o una sal farmacéuticamente aceptable del mismo, especialmente el besilato. Un representante especialmente preferido de no DHP es verapamil o una sal farmacéuticamente aceptable, especialmente el clorhidrato, del mismo.
Beta-bloqueantes adecuados para su uso en la presente invención incluyen agentes bloqueantes beta-adrenérgicos (beta-bloqueantes) que compiten con la epinefrina por los receptores beta-adrenérgicos e interfieren con la acción de la epinefrina. Preferiblemente, los beta-bloqueantes son selectivos por el receptor beta-adrenérgico en comparación con los receptores alfa-adrenérgicos, y así no tienen un efecto alfa-bloqueante significativo. Beta-bloqueantes adecuados incluyen compuestos seleccionados de acebutolol, atenolol, betaxolol, bisoprolol, carteolol, carvedilol, esmolol, labetalol, metoprolol, nadolol, oxprenolol, penbutolol, pindolol, propranolol, sotalol y timolol. Si el betabloqueante es un ácido o base o de otro modo capaz de formar sales farmacéuticamente aceptables o profármacos, se considera que estas formas están englobadas en el presente documento, y se entiende que los compuestos pueden administrarse en forma libre o en forma de una sal farmacéuticamente aceptable o un profármaco, tal como un éster fisiológicamente hidrolizable y aceptable. Por ejemplo, el metoprolol se administra adecuadamente como su sal de tartrato, el propranolol se administra adecuadamente como la sal de clorhidrato y demás.
Los inhibidores de la agregación de plaquetas incluyen PLAVIX® (bisulfato de clopidogrel), PLETAL® (cilostazol) y aspirina.
Los moduladores de la absorción de colesterol incluyen ZETIA® (ezetimiba) y KT6-971 (Kotobuki Pharmaceutical Co. Japón).
Se entiende que los inhibidores de la HMG-Co-A reductasa (también denominados inhibidores de la beta-hidroxi-betametilglutaril-co-enzima-A reductasa o estatinas) son aquellos agentes activos que pueden usarse para reducir los niveles de lípidos que incluyen colesterol en sangre.
La clase de inhibidores de la HMG-Co-A reductasa comprende compuestos que tienen características estructurales diferentes. Por ejemplo, puede hacerse mención de los compuestos, que están seleccionados del grupo que consiste en atorvastatina, cerivastatina, fluvastatina, lovastatina, pitavastatina, pravastatina, rosuvastatina y simvastatina, o en cada caso, una sal farmacéuticamente aceptable de los mismos.
Inhibidores de la HMG-Co-A reductasa preferidos son aquellos agentes que han sido comercializados, el más preferido es atorvastatina, pitavastatina o simvastatina, o una sal farmacéuticamente aceptable de los mismos.
Los compuestos que aumentan HDL incluyen, pero no se limitan a, inhibidores de la proteína de transferencia de ésteres de colesterol (CETP). Ejemplos de inhibidores de CETP incluyen JTT7O5 dado a conocer en el ejemplo 26 de la patente de EE.UU. n.° 6.426.365 concedida el 30 de julio de 2002, y sales farmacéuticamente aceptables del mismo.
La inhibición de la inflamación mediada por interleucina 6 puede lograrse indirectamente mediante la regulación de la síntesis de colesterol endógeno y el agotamiento de isoprenoide o por inhibición directa de la vía de transducción de señales utilizando inhibidor/anticuerpo de interleucina-6, inhibidor/anticuerpo de receptores de interleucina-6, oligonucleótido antisentido de interleucina-6 (ASON), inhibidor/anticuerpo de la proteína gp130, inhibidores/anticuerpos de tirosina cinasas, inhibidores/anticuerpos de serina/treonina cinasas, inhibidores/anticuerpos de las proteínas cinasas activadas por mitógeno (MAP), inhibidores/anticuerpos de fosfatidilinositol 3-cinasa (Pl3K), inhibidores/anticuerpos del factor nuclear kappaB (NF-k B), inhibidores/anticuerpos de Ik B cinasa (IKK), inhibidores/anticuerpos de la proteína activadora-1 (AP-1), inhibidores/anticuerpos de factores de transcripción STAT, IL-6 alterada, péptidos parciales de IL-6 o receptor de IL-6, o proteína SOCs (supresores de la señalización de citocinas), activadores/ligandos de PPAR gamma y/o PPAR beta/delta o un fragmento funcional de los mismos.
Un corticosteroide antiinflamatorio adecuado es dexametasona.
Agentes antiproliferativos adecuados son cladribina, rapamicina, vincristina y taxol.
Un inhibidor adecuado de la síntesis de matriz extracelular es halofuginona.
Un inhibidor de la transducción de señales de factores de crecimiento o citocinas adecuado es, por ejemplo, el inhibidor ras R115777.
Un inhibidor de tirosina cinasas adecuado es tirfostina.
Inhibidores de renina adecuados se describen, por ejemplo, en el documento WO 2006/116435. Un inhibidor de renina preferido es aliskireno, preferiblemente en forma de la sal de hemi-fumarato del mismo.
Los antagonistas de MCP-1 pueden seleccionarse, por ejemplo, de anticuerpos anti-MCP-1, preferiblemente anticuerpos monoclonales o monoclonales humanizados, inhibidores de la expresión de MCP-1, antagonistas de CCR2, inhibidores de TNF-alfa, inhibidores de la expresión génica de VCAM-1 y anticuerpos monoclonales anti-C5a.
Antagonistas de MCP-1 y composiciones que contienen tales inhibidores se describen, por ejemplo, en los documentos WO02/070509, WO02/081463, WO02/060900, US2006/670364, US2006/677365, WO2006/097624, US2006/316449, WO2004/056727, WO03/053368, WO00/198289, WO00/157226, WO00/046195, WO00/046196, WO00/046199, WO00/046198, WO00/046197, WO99/046991, WO99/007351, WO98/006703, WO97/012615, WO2005/105133, WO03/037376, WO2006/125202, WO2006/085961, WO2004/024921, WO2006/074265.
Antagonistas de MCP-1 adecuados son, por ejemplo, C-243 (Telik Inc.); NOX-E36 (Noxxon Pharma AG); AP-761 (Actimis Pharmaceuticals Inc.); ABN-912, N iBR-177 (Novartis AG); CC-11006 (Celgene Corp.); SSR-150106 (Sanofi-Aventis); MLN-1202 (Millenium Pharmaceuticals Inc.); AGI-1067, AGIX-4207, AGI-1096 (AtherioGenics Inc.); PRS-211095, PRS-211092 (Pharmos Corp.); anticuerpos monoclonales anti-C5a, por ejemplo, neutrazumab (G2 Therapies Ltd ); AZD-6942 (AstraZeneca plc.); 2-mercaptoimidazoles (Johnson & Johnson); TEI-E00526, TEI-6122 (Deltagen); RS-504393 (Roche Holding AG); SB-282241, SB-380732, ADR-7 (GlaxoSmithKline); anticuerpos monoclonales anti-MCP-1 (Johnson & Johnson).
Combinaciones de inhibidores de QC con antagonistas de MCP-1 pueden ser útiles para el tratamiento de enfermedades inflamatorias en general, que incluyen enfermedades neurodegenerativas.
Se prefieren combinaciones de inhibidores de QC con antagonistas de MCP-1 para el tratamiento de enfermedad de Alzheimer.
Lo más preferiblemente, el inhibidor de QC se combina con uno o más compuestos seleccionados del siguiente grupo:
PF-4360365, m266, bapineuzumab, R-1450, posifeno, (+)-fenserina, MK-0752, LY-450139, E-2012, (R)-flurbiprofeno, AZD-103, AAB-001 (bapineuzumab), tramiprosato, EGb-761, TAK-070, doxofilina, teofilina, cilomilast, tofimilast, roflumilast, tetomilast, tipelukast, ibudilast, HT-0712, MEM-1414, oglemilast, linezolida, budipina, isocarboxazida, fenelzina, tranilcipromina, indantadol, moclobemida, rasagilina, ladostigil, safinamida, ABT-239, ABT-834, GSK-189254A, ciproxifan, JNJ-17216498, Fmoc-Ala-Pirr-CN, Z-Phe-Pro-benzotiazol, Z-321, ONO-1603, JTP-4819, S-17092, BIBP3226; amida de (R)-N2-(difenilacetil)-(R)-N-[1-(4-hidroxifenil)etil]arginina, cevimelina, sabcomelina, (PD-151832), donepezil, rivastigmina, (-)-fenserina, ladostigil, galantamina, tacrina, metrifonato, memantina, topiramato, AVP-923, EN-3231, neramexano, valsartán, benazeprilo, enalaprilo, hidroclorotiazida, amlodipino, diltiazem, isradipino, nicardipino, nifedipino, nimodipino, nisoldipino, nitrendipino, verapamil, amlodipino, acebutolol, atenolol, betaxolol, bisoprolol, carteolol, carvedilol, esmolol, labetalol, metoprolol, nadolol, oxprenolol, penbutolol, pindolol, propranolol, sotalol, timolol, PLAVIX® (bisulfato de clopidogrel), PLETAL® (cilostazol), aspirina, ZETIA® (ezetimiba) y KT6-971, estatinas, atorvastatina, pitavastatina o simvastatina; dexametasona, cladribina, rapamicina, vincristina, taxol, aliskireno, C-243, ABN-912, SSR-150106, MLN-1202 y betaferon.
En particular, se consideran las siguientes combinaciones:
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con atorvastatina para el tratamiento y/o la prevención de arteriosclerosis,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con agentes inmunosupresores, preferiblemente rapamicina, para la prevención y/o el tratamiento de reestenosis,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con agentes inmunosupresores, preferiblemente paclitaxel, para la prevención y/o el tratamiento de reestenosis,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC
seleccionado de uno cualquiera de los ejemplos 1,2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con inhibidores de AChE, preferiblemente donepezil, para la prevención y/o el tratamiento de enfermedad de Alzheimer,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con interferones, preferiblemente Aronex, para la prevención y/o el tratamiento de esclerosis múltiple,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con interferones, preferiblemente betaferon, para la prevención y/o el tratamiento de esclerosis múltiple,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con interferones, preferiblemente Rebif, para la prevención y/o el tratamiento de esclerosis múltiple
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con Copaxona, para la prevención y/o tratamiento de esclerosis múltiple,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de cualquiera de los ejemplos 1,2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con dexaetasona, para la prevención y/o tratamiento de reestenosis,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con dexametasona, para la prevención y/o tratamiento de aterosclerosis,
- un inhibidor QC, preferiblemente un inhibidor QC de la fórmula (I), más preferiblemente un inhibidor QC seleccionado de cualquiera de los ejemplos 1,2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con dexametasona, para la prevención y/o tratamiento de artritis reumatoide,
- un inhibidor de QC, preferiblemente un inhibidor de QC de la fórmula (I), más preferiblemente un inhibidor de QC seleccionado de cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con inhibidores de HMG-Co-A-reductasa, para la prevención y/o tratamiento de reestenosis, en el que el inhibidor de HMG-Co-A reductasa se selecciona de atorvastatina, cerivastatina, fluvastatina, lovastatina, pitavastatina, pravastatina, rosuvastatina y simvastatina,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1,2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con inhibidores de HMG-Co-A reductasa, para la prevención y/o el tratamiento de aterosclerosis, en el que el inhibidor de HMG-Co-A-reductasa se selecciona de atorvastatina, cerivastatina, fluvastatina, lovastatina, pitavastatina, pravastatina, rosuvastatina y simvastatina,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1,2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con inhibidores de HMG-Co-A reductasa, para la prevención y/o el tratamiento de artritis reumatoide, en el que el inhibidor de HMG-Co-A-reductasa se selecciona de atorvastatina, cerivastatina, fluvastatina, lovastatina, pitavastatina, pravastatina, rosuvastatina y simvastatina,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con anticuerpos para beta-amiloide para la prevención y/o el tratamiento de deterioro cognitivo leve, en el que el anticuerpo para betaamiloide es Acl-24,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con anticuerpos para beta-amiloide para la prevención y/o el tratamiento de enfermedad de Alzheimer, en el que el anticuerpo para beta-amiloide es Acl- 24,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1, 2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con anticuerpos para beta-amiloide para la prevención y/o el tratamiento de neurodegeneración en síndrome de Down, en el que el anticuerpo para beta-amiloide es Acl-24,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1,2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con inhibidores de
beta-secretasa para la prevención y/o el tratamiento de deterioro cognitivo leve, en el que el inhibidor de beta-secretasa se selecciona de WY-25105, GW-840736X y CTS-21166,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1,2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con inhibidores de beta-secretasa para la prevención y/o el tratamiento de enfermedad de Alzheimer, en el que el inhibidor de betasecretasa se selecciona de WY-25105, GW-840736X y CTS-21166,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1,2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con inhibidores de beta-secretasa para la prevención y/o el tratamiento de neurodegeneración en síndrome de Down, en el que el inhibidor de beta-secretasa se selecciona de WY-25105, GW-840736X y CTS-21166,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1,2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con inhibidores de gamma-secretasa para la prevención y/o el tratamiento de deterioro cognitivo leve, en el que el inhibidor de gammasecretasa se selecciona de LY-450139, LY-411575 y AN-37124,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1,2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con inhibidores de gamma-secretasa para la prevención y/o el tratamiento de enfermedad de Alzheimer, en el que el inhibidor de gammasecretasa se selecciona de LY-450139, LY-411575 y AN-37124,
- un inhibidor de QC, preferiblemente un inhibidor de QC de fórmula (I), más preferiblemente un inhibidor de QC seleccionado de uno cualquiera de los ejemplos 1,2, 4, 5, 7, 9, 11, 13, 15 y 17-29, en combinación con inhibidores de gamma-secretasa para la prevención y/o el tratamiento de neurodegeneración en síndrome de Down, en el que el inhibidor de gamma-secretasa se selecciona de LY-450139, LY-411575 y AN-37124.
Una terapia de combinación de este tipo es en particular útil para EA, FAD, FDD y neurodegeneración en síndrome de Down, así como aterosclerosis, artritis reumatoide, reestenosis y pancreatitis.
Tales terapias de combinación podrían producir un mejor efecto terapéutico (menos proliferación de placas además de menos inflamación, un estímulo para la proliferación) que el que se produciría con cualquier agente solo.
Con respecto a la combinación específica de inhibidores de QC y compuestos adicionales se remite en particular al documento WO 2004/098625 en este respecto.
Composiciones farmacéuticas
Para preparar las composiciones farmacéuticas de esta invención, al menos un compuesto de fórmula (I) opcionalmente en combinación con al menos uno de los otros agentes anteriormente mencionados puede usarse como principio(s) activo(s). El/Los principio(s) activo(s) se mezcla(n) íntimamente con un vehículo farmacéutico según técnicas de combinación farmacéutica convencionales, vehículo que puede tomar una amplia variedad de formas dependiendo de la forma de preparación deseada para administración, por ejemplo, oral o parenteral tal como intramuscular. En la preparación de las composiciones en forma de dosificación oral puede emplearse cualquiera de los medios farmacéuticos usuales. Por tanto, para preparaciones orales líquidas, tales como, por ejemplo, suspensiones, elixires y disoluciones, vehículos y aditivos adecuados incluyen agua, glicoles, aceites, alcoholes, aromatizantes, conservantes, agentes colorantes y similares; para preparaciones orales sólidas tales como, por ejemplo, polvos, cápsulas, cápsulas de gel y comprimidos, vehículos y aditivos adecuados incluyen almidones, azúcares, diluyentes, agentes de granulación, lubricantes, aglutinantes, disgregantes y similares. Debido a su facilidad de administración, los comprimidos y cápsulas representan la forma unitaria de dosificación oral más ventajosa, en cuyo caso se emplean obviamente vehículos farmacéuticos sólidos. Si se desea, los comprimidos pueden recubrirse de azúcar o recubrirse entéricamente por técnicas convencionales. Para parenterales, el vehículo comprenderá normalmente agua estéril, aunque pueden incluirse otros componentes, por ejemplo, para fines tales como ayudar en la solubilidad o para preservación.
También pueden prepararse suspensiones inyectables, en cuyo caso pueden emplearse vehículos líquidos apropiados, agentes de suspensión y similares. Las composiciones farmacéuticas en el presente documento contendrán, por unidad de dosificación, por ejemplo, comprimido, cápsula, polvo, inyección, cucharadita al ras y similares, una cantidad del/de los principio(s) activo(s) necesaria para administrar una dosis eficaz como se ha descrito anteriormente. Las composiciones farmacéuticas en el presente documento contendrán, por unidad de dosificación, por ejemplo, comprimido, cápsula, polvo, inyección, supositorio, cucharadita al ras y similares, de aproximadamente 0,03 mg a 100 mg/kg (preferida 0,1 - 30 mg/kg) y puede administrarse a una dosificación de aproximadamente 0,1 -300 mg/kg por día (preferida 1 - 50 mg/kg por día) de cada principio activo o combinación de los mismos. Sin embargo, pueden variarse las dosificaciones dependiendo del requisito de los pacientes, la gravedad de la afección que está tratándose y el compuesto que se emplea. Puede emplearse el uso de cualquier administración diaria o dosificación
post-periódica.
Preferiblemente, estas composiciones están en formas de dosificación unitaria tales como comprimidos, píldoras, cápsulas, polvos, gránulos, disoluciones o suspensiones parenterales estériles, aerosol dosificado o pulverizaciones líquidas, gotas, ampollas, dispositivos autoinyectores o supositorios; para administración parenteral oral, intranasal, sublingual o rectal, o para administración por inhalación o insuflación. Alternativamente, la composición puede presentarse en una forma adecuada para administración una vez a la semana o una vez al mes; por ejemplo, una sal insoluble del compuesto activo, tal como la sal de decanoato, puede adaptarse para proporcionar una preparación de liberación prolongada para inyección intramuscular. Para preparar composiciones sólidas tales como comprimidos, el principal principio activo se mezcla con un vehículo farmacéutico, por ejemplo, componentes de formación de comprimidos convencionales tales como almidón de maíz, lactosa, sacarosa, sorbitol, talco, ácido esteárico, estearato de magnesio, fosfato de dicalcio o gomas, y otros diluyentes farmacéuticos, por ejemplo, agua, para formar una composición de preformulación sólida que contiene una mezcla homogénea de un compuesto de la presente invención, o una sal farmacéuticamente aceptable del mismo. Cuando se hace referencia a estas composiciones de preformulación como homogéneas, se indica que el principio activo está disperso uniformemente por toda la composición de manera que la composición pueda subdividirse fácilmente en formas de dosificación igualmente eficaces tales como comprimidos, píldoras y cápsulas. Entonces, esta composición de preformulación sólida se subdivide en formas de dosificación unitarias del tipo descrito anteriormente que contienen de 0,1 a aproximadamente 500 mg de cada principio activo o combinaciones de los mismos de la presente invención.
Los comprimidos o píldoras de las composiciones de la presente invención pueden recubrirse o combinarse de otro modo para proporcionar una forma de dosificación que proporciona la ventaja de acción prolongada. Por ejemplo, el comprimido o píldora puede comprender un componente de dosificación interna y un componente de dosificación externa, estando el último en forma de una envoltura sobre el primero. Los dos componentes pueden separarse por una capa entérica que sirve para resistir a la disgregación en el estómago y permite que el componente interno pase intacto al duodeno o sea de liberación retardada. Puede usarse una variedad de materiales para tales capas o recubrimientos entéricos, incluyendo tales materiales varios ácidos poliméricos con materiales tales como goma laca, alcohol cetílico y acetato de celulosa.
Estas formas líquidas en las que las composiciones de la presente invención pueden incorporarse para administración por vía oral o por inyección incluyen disoluciones acuosas, jarabes adecuadamente aromatizados, suspensiones acuosas o aceitosas, y emulsiones aromatizadas con aceites comestibles tales como aceite de semilla de algodón, aceite de sésamo, aceite de coco o aceite de cacahuete, así como elixires y vehículos farmacéuticos similares. Agentes de dispersión o suspensión adecuados para suspensiones acuosas incluyen gomas sintéticas y naturales tales como tragacanto, goma arábiga, alginato, dextrano, carboximetilcelulosa de sodio, metilcelulosa, polivinilpirrolidona o gelatina.
La composición farmacéutica puede contener entre aproximadamente 0,01 mg y 100 mg, preferiblemente aproximadamente de 5 a 50 mg, de cada compuesto, y puede constituirse en cualquier forma adecuada para el modo de administración seleccionado. Vehículos incluyen excipientes farmacéuticos necesarios e inertes, que incluyen, pero no se limitan a, aglutinantes, agentes de suspensión, lubricantes, aromas, edulcorantes, conservantes, colorantes y recubrimientos. Composiciones adecuadas para administración por vía oral incluyen formas sólidas, tales como píldoras, comprimidos, comprimidos oblongos, cápsulas (incluyendo cada una formulaciones de liberación inmediata, liberación controlada y de liberación sostenida), gránulos y polvos, y formas líquidas, tales como disoluciones, jarabes, elixires, emulsiones y suspensiones. Formas útiles para administración parenteral incluyen suspensiones, emulsiones y disoluciones estériles.
Ventajosamente, los compuestos de la presente invención pueden administrarse en una única dosis diaria, o la dosificación diaria total puede administrarse en dosis divididas de dos, tres o cuatro veces al día. Además, los compuestos para la presente invención pueden administrarse en forma intranasal mediante uso tópico de vehículos intranasales adecuados, o mediante parches para la piel transdérmicos muy conocidos para aquellos expertos habituales en la técnica. Para administrarse en forma de sistema de administración transdérmica, la administración de dosificación será, por supuesto, continua en vez de intermitente durante toda la pauta de dosificación.
Por ejemplo, para administración por vía oral en forma de un comprimido o cápsula, el componente de fármaco activo puede combinarse con un vehículo inerte farmacéuticamente aceptable no tóxico oral tal como etanol, glicerol, agua y similares. Además, cuando se desee o sea necesario, también pueden incorporarse en la mezcla aglutinantes adecuados, lubricantes, agentes disgregantes y agentes colorantes. Aglutinantes adecuados incluyen, sin limitación, almidón, gelatina, azúcares naturales tales como glucosa o beta-lactosa, edulcorantes de maíz, gomas naturales y sintéticas tales como goma arábiga, tragacanto u oleato de sodio, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato sódico, cloruro sódico y similares. Disgregantes incluyen, sin limitación, almidón, metilcelulosa, agar, bentonita, goma xantana y similares.
Las formas líquidas en agentes de suspensión o dispersantes aromatizados adecuados tales como las gomas sintéticas y naturales, por ejemplo, tragacanto, goma arábiga, metilcelulosa y similares. Para administración parenteral se desean suspensiones y disoluciones estériles. Se emplean preparaciones isotónicas que generalmente contienen
conservantes adecuados cuando se desea administración intravenosa.
Los compuestos o combinaciones de la presente invención también pueden administrarse en forma de sistemas de administración de liposomas, tales como pequeñas vesículas unilaminares, vesículas unilaminares grandes y vesículas multilaminares. Los liposomas pueden formarse a partir de una variedad de fosfolípidos, tales como colesterol, estearilamina o fosfatidilcolinas.
Compuestos o combinaciones de la presente invención también pueden administrarse mediante el uso de anticuerpos monoclonales como vehículos individuales a los que se acoplan las moléculas de compuesto. Los compuestos de la presente invención también pueden acoplarse con polímeros solubles como vehículos de fármaco elegibles como diana. Tales polímeros pueden incluir polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamida-fenol, polihidroxietilaspartamida-fenol, o poli(óxido de etileno)-polilisina sustituido con residuo de palmitoílo. Además, los compuestos de la presente invención pueden acoplarse a una clase de polímeros biodegradables útiles en alcanzar la liberación controlada de un fármaco, por ejemplo, ácido poliláctico, poli-épsilon-caprolactona, ácido polihidroxiburítico, poliortoésteres, poliacetales, polidihidropiranos, policianoacrilatos y copolímeros de bloques de hidrogeles reticulados o anfipáticos.
Compuestos o combinaciones de la presente invención pueden administrarse en cualquiera de las anteriores composiciones y según pautas de dosificación establecidas en la técnica, siempre que se requiera el tratamiento de los trastornos tratados.
La dosificación diaria de los productos puede variar a lo largo de un amplio intervalo de 0,01 a 1.000 mg por mamífero por día. Para administración por vía oral, las composiciones se proporcionan preferiblemente en forma de comprimidos que contienen, 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 100, 150, 200, 250 y 500 miligramos de cada principio activo o combinaciones de los mismos para el ajuste sintomático de la dosificación al paciente que va a tratarse. Una cantidad eficaz del fármaco se suministra generalmente a un nivel de dosificación de aproximadamente 0,1 mg/kg a aproximadamente 300 mg/kg de peso corporal por día. Preferiblemente, el intervalo es de aproximadamente 1 a aproximadamente 50 mg/kg de peso corporal por día. Los compuestos o combinaciones pueden administrarse en una pauta de 1 a 4 veces por día. Dosificaciones óptimas que van a administrarse pueden determinarse fácilmente por aquellos expertos en la técnica, y variarán con el compuesto particular usado, el modo de administración, la concentración de la preparación, el modo de administración y el avance de la condición de enfermedad. Además, factores asociados al paciente particular que está tratándose, que incluyen edad del paciente, peso, dieta y tiempo de administración, producirán la necesidad de ajustar las dosis.
En un aspecto adicional, la invención también proporciona un procedimiento de preparación de una composición farmacéutica que comprende al menos un compuesto de fórmula (I), opcionalmente en combinación con al menos uno de los otros agentes anteriormente mencionados y un vehículo farmacéuticamente aceptable.
Las composiciones están preferiblemente en una forma de dosificación unitaria en una cantidad apropiada para la dosificación diaria relevante.
Dosis adecuadas, incluyendo especialmente dosis unitarias, de los compuestos de la presente invención incluyen las dosis conocidas, incluyendo las dosis unitarias para estos compuestos, tal como se describe o se menciona en el texto de referencia como las farmacopeas británica y estadounidense, Remington's Pharmaceutical Sciences (Mack Publishing Co.), Martindale The Extra Pharmacopoeia (Londres, The Pharmaceutical Press) (por ejemplo, véase la página 341 de la 31a edición y las páginas citadas en el mismo) o las publicaciones mencionadas anteriormente.
Ejemplos de la invención
Ejemplos de compuestos de fórmula (I)






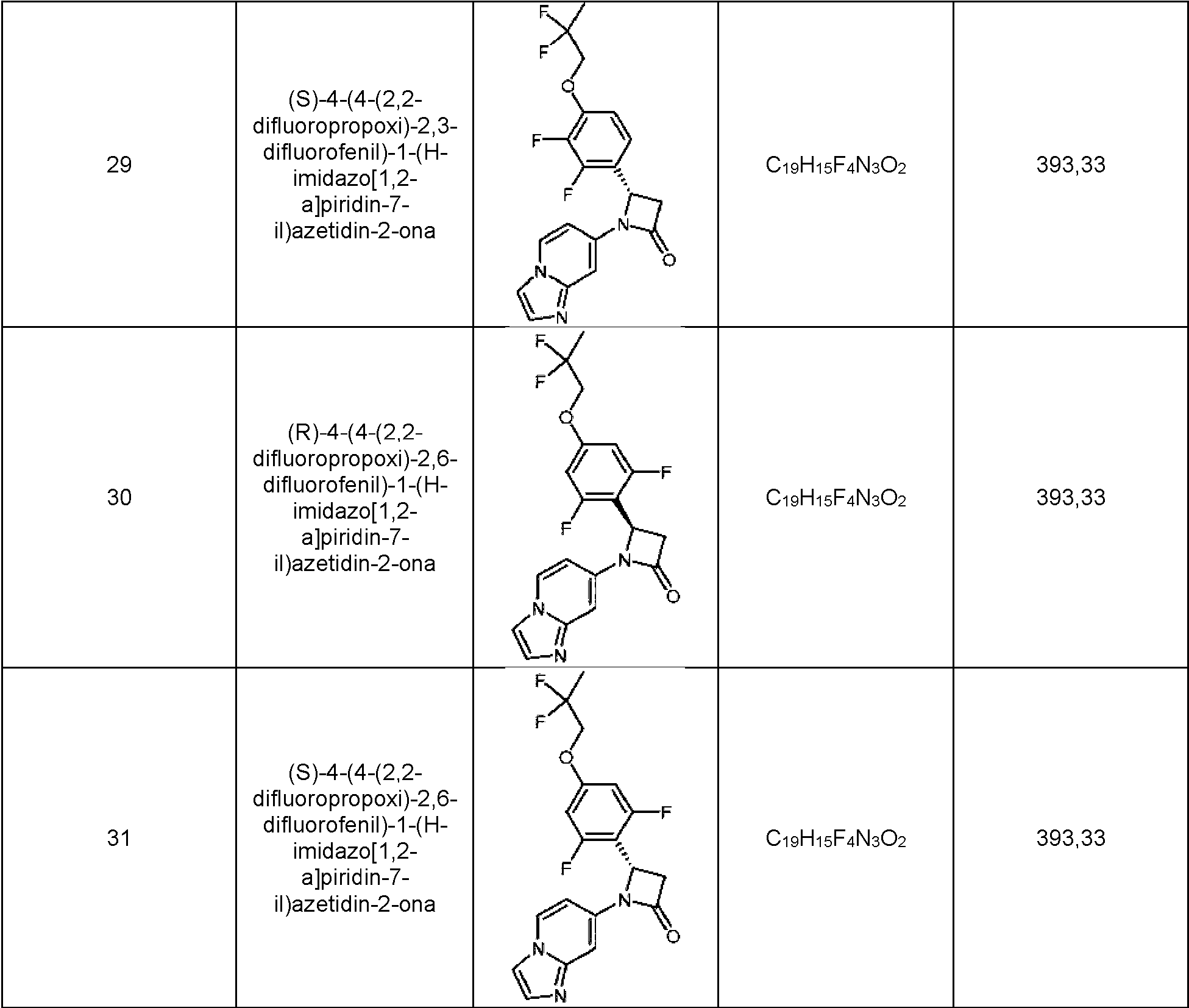
La invención además se refiere a los racematos, S-estereoisómeros y R-estereoisómeros de los compuestos de la presente invención como se muestra en la tabla anterior.
Síntesis de los compuestos de la invención
Descripción de síntesis general:
Método 1

Etapa A:
Se disolvieron 1,2 equivalentes de 5-amino-benzimidazol y 1,0 equivalentes del aldehído respectivo en tolueno y se calentaron a reflujo durante la noche. Después de la finalización de la reacción, se retiró el disolvente y se usó el aceite restante para etapas adicionales sin purificación adicional
Etapa B:
Se suspendieron 2,0 equivalentes de polvo de zinc en benceno. Se añadieron 0,7 equivalentes de TMS-Cl bajo una atmósfera de argón. La mezcla se agitó entonces a reflujo durante 15 min. Después de enfriar a temperatura ambiente, se añadieron 1 equivalente de la base de Schiff y 1,7 equivalentes de etil éster de ácido bromoacético. La mezcla se mantuvo a reflujo durante la noche. Posteriormente se retiró el disolvente y se tomaron los restos en poco cloruro de metileno, se extrajeron sobre gel de sílice y se sometieron a cromatografía en columna, aplicando un gradiente de CH2Cl2/MeOH.
Método 2

Se añadió cloruro de trimetilsilano (0,5 eq.) a una suspensión de polvo de zinc (1,5 eq.) en benceno seco (50 ml) y se sometió a reflujo durante 10min. La masa de reacción se enfrió a 0 °C, se añadieron de manera sucesiva bromoacetato de etilo (1,5 eq.) y una disolución de I (1 eq.) en benceno seco y se sometieron a reflujo durante 2 h. La masa de reacción se inactivó con disolución de cloruro de amonio saturada y se extrajo con acetato de etilo. La capa orgánica combinada se lavó de manera sucesiva con agua, salmuera; se secó sobre sulfato de sodio anhidro y se concentró en vacío para proporcionar un producto en bruto. Purificación por cromatografía en columna sobre gel de sílice (60 120 de malla) usando acetato de etilo al 20-25 % en éter de PET como eluyente proporcionadoproporcionado II.
Etapa B
Se añadió dicromato de piridinio (4 eq.), tamices moleculares a una disolución de II (1 eq.) en diclorometano (400 ml) a 0 °C y se agitó durante 4 h a temperatura ambiente. La masa de reacción se filtró sobre sílice diatomácea y se lavó con diclorometano. El filtrado combinado y lavado se concentró en vacío para proporcionar un producto en bruto. Purificación por cromatografía en columna sobre gel de sílice (60-120 de malla) usando acetato de etilo al 15 % en éter de PET como eluyente proporcionado III.
Etapa C
Se añadió una disolución de ferc-butil 5-amino-1H-benzimidazol-1-carboxilato (0,9 eq.) en xileno a una disolución de III (1 eq.) y piridina en xileno y se tomó en un matraz de fondo redondo equipado con un condensador de destilación a 140 °C. La masa de reacción además se calentó a 160 °C y se recogió xileno. El disolvente se evaporó en vacío para proporcionar un producto en bruto. Purificación por cromatografía en columna sobre gel de sílice (60-120 de malla) usando metanol al 5 % en DCM como eluyente proporcionado IV.
Etapa D
Se añadió borohidruro sódico (2 eq.) en un lote a una disolución de IV (1 eq.) en mezcla de tetrahidrofurano y metanol a 0 °C y se agitó durante 15min. La masa de reacción se inactivó en disolución saturada de cloruro de amonio y se extrajo con acetato de etilo. La capa orgánica combinada se lavó de manera sucesiva con agua, salmuera; se secó sobre sulfato de sodio anhidro y se concentró en vacío, se trituró con éter de PET y se secó a presión reducida para proporcionar V.
Etapa E
Se añadió dietilazodicarboxilato (1,5 eq.) a una disolución agitada de V (1 eq.) y trifenilfosfina (1,5 eq.) en tetrahidrofurano seco a 0 °C y se agitó a temperatura ambiente durante 30 min. La masa de reacción se inactivó con agua y se extrajo con acetato de etilo. La capa orgánica combinada se lavó con agua, salmuera, se secó sobre sulfato sódico anhidro y se concentró en vacío para proporcionar un producto en bruto. Purificación por cromatografía en columna sobre gel de sílice (60-120 de malla) usando acetato de etilo al 35 % en éter de PET como eluyente proporcionado VI.
Etapa F
Se añadió ácido trifluoroacético a una disolución de producto en bruto VI (1 eq.) en diclorometano a 0°C y se agitó durante 2 h a temperatura ambiente. Los volátiles se evaporaron en vacío y el residuo resultante se dividió entre disolución saturada de bicarbonato sódico y acetato de etilo. Se separó la capa orgánica y se extrajo la capa acuosa con acetato de etilo. La capa orgánica combinada se lavó de manera sucesiva con agua, salmuera; se secó sobre sulfato de sodio anhidro y se concentró en vacío para proporcionar un producto en bruto. Purificación por cromatografía en columna sobre alúmina neutra usando metanol al 5-8 % en diclorometano como eluyente proporcionado VII. Síntesis de compuestos de ejemplo 1 a 16:
Ejemplo 1
El compuesto se sintetizó según el método 1:
rac1-(1H-benzoimidazol-5-il)-4-fenilazetidin-2-ona:
Etapa A : 5-aminobenzimidazol (1,33 g, 10 mmol), benzaldehído (1,27 g, 1,22 ml, 12 mmol) en 80 ml de tolueno, produciendo 1,62 g de producto en bruto I;
Etapa B: I (0,433 g, 2 mmol), polvo de zinc (0,262 g, 4 mmol), TMS-Cl (0,180 ml, 1,4 mmol), etil éster de ácido bromoacético (0,377 ml, 3,4 mmol) .Rendimiento: 0,089 g (5,6 %); MS m/z 285,1 (M+H)+; 1H NMR (DMSO, 400 MHz): 8 0,82-0,91 (m, H); 0,97-1,16 (m, 4H); 1,39-1,42 (m, H); 1,52-1,69 (m, 5H); 3,24-3,27 (m, H); 3,42-3,46 (m, H); 4,48 4,52 (m, H); 6,92 (s, H); 7,56-7,59 (dd, H, 3J=9,1 Hz, 4J=2,1 Hz); 7,73-7,75 (d, H, 3J=9,1 Hz); 7,94-7,95 (d, H, 4J=2,1 Hz); 9,24 (s, H), HPLC 99 %.
Ejemplos 2 y 3
(R) -1-(1H-benzo[d]imidazol-6-il)-4-fenilazetidin-2-ona 2
El ejemplo 1 (0,089 g) se sometió a una HPLC de preparación quiral, proporcionando el compuesto 2. Rendimiento: 0,025 g, 1H-NMR (DMSO-d6): 812,25(d, 1H); 8,13(d, 1H); 7,53(d, 1H); 7,46-7,30(m, 6H); 7,12(d, 1H); 5,25(t, 1H); 3,63-3,57(m, 1H); 2,92-2,86(m, 1H); MS=264(M+1); HPLC~96,31 %: HPLC quiral~92,48 %
(S) -1-(1H-benzo[d]imidazol-6-il)-4-fenilazetidin-2-ona 3
El ejemplo 1 (0,089 g) se sometió a una HPLC de preparación quiral, proporcionando el compuesto 3. Rendimiento: 0,025 g, 1H-NMR (DMSO-d6): 812,25(d, 1H); 8,13(s, 1H); ,.46-7,30(m, 7H); 7,12(bs, 1H); 5,25(q, 1H); 3,63-3,57(m, 1H); 2,90-2,86(m, 1H); MS=264(M+1); HPLC~99,00 %: HPLC quiral~98,35 %.
Ejemplo 4
Rac 1-(1H-benzo[d]imidazol-5-il)-4-(2,6-difluoro-4-metoxifenil)azetidin-2-ona 4
Etapa A: 5-aminobenzimidazol (0,690g, 5,2 mmol), 2,5-difluoro-4metoxibenzaldehído (1,07 g, 6,2 mmol) en 80 ml de tolueno, produciendo 1,66 g de producto en bruto I;
Etapa B: I (1,15 g, 4 mmol), polvo de zinc (0,524 g, 8,31 mmol), TMS-Cl (0,360 ml, 2,8 mmol), etil éster de ácido bromoacético (0,754 ml, 6,8 mmol). Rendimiento: 0,022 g (1,2 %); MS m/z 330,4 (M+H)+; 1H NMR (DMSO-d6, 400
MHz): 83,25-3,29 (m, 1H), 3,63-3,67 (m, 1H), 3,76 (s, 3H), 5,49-5,51 8m, 1H), 6,80 (d, 2H, 3J=11,2 Hz), 7,34 (dd, 1H, 3J=8,8 Hz, 4J=2 Hz), 7,50 (d, 1H, 4J=2 Hz), 7,74 (d, 1H, 3J=8,8 Hz), 9,10 (s, 1H), HPLC [A]: 100 %)
Ejemplos 5 y 6
Los compuestos se prepararon según el método 2
rac-4-(4-(3,3-difluoropropoxi)-2-fluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona
Etapa A: Cloruro de trimetilsilano (1,2 ml, 9,174 mmol), polvo de zinc (1,8 g, 27,52 mmol), benceno (50 ml), bromoacetato de etilo (3,0 ml, 27,52 mmol), 4-(3,3-difluoropropoxi)-2-fluorobenzaldehído (4,0 g, 18,34 mmol) en benceno seco (20 ml), Rendimiento: 4 g (73 %)
Etapa B: Se añadieron dicromato de piridinio (19,6 g, 52,28 mmol), tamices moleculares (19,6 g) a una disolución de 3-(4-(3,3-difluoropropoxi)-2-fluorofenil)-3-hidroxipropanoato de etilo (4,0 g, 13,07 mmol) en diclorometano (400 ml), Rendimiento 3,5g (88 %)
Etapa C: Se añadió 5-amino-1H-benzo[d]imidazol-1-carboxilato de terc-butilo (1,7 g, 7,40 mmol) en xileno (20 ml) a una disolución de 3-(4-(3,3-difluoropropoxi)-2-fluorofenil)-3-oxopropanoato de etilo (2,5 g, 8,22 mmol) y piridina (0,5 ml) en xileno (40 ml, Rendimiento 2,2 g (50,7 %).
Etapa D: Borohidruro de sodio (340 mg, 8,944 mmol), N-(1-terc-butiloxicarbonil-1H-benzo[d]imidazol-5-il-)3-(4-(3,3-difluoropropoxi)-2-fluorofenil)-3-oxopropanamida (2,2 g, 4,47 mmol) en mezcla de tetrahidrofurano (35 ml) y metanol (15 ml) a 0°C, Rendimiento 1,8 g (77,6 %)
Etapa E: Dietilazodicarboxilato (1,2 ml, 7,59 mmol), N-(1-terc-butil-oxicarbonil-1H-benzo[d]imidazol-5-il-)3-(4-(3,3-difluoropropoxi)-2-fluorofenil)-3-hidroxipropanamida (2,5 g, 5,060 mmol) y trifenilfosfina (1,98 g, 7,59 mmol) Tetrahidrofurano (30 ml) a 0 °C, Rendimiento: 1,0 g (42 %)
Etapa F: Se añadió ácido trifluoroacético (2 ml) a una disolución de producto en bruto de 6-(2-(4-(3,3-difluoropropoxi)-2- fluorofenil)-4-oxoazetidin-1-il)-1H-benzo[d]imidazol-1-carboxilato de terc-butilo (1,0 g, 2,08 mmol) en diclorometano (20 ml) a 0°C, Rendimiento: 0,3 g (40 %).
Los restos se purificaron por HPLC de preparación quiral usando las siguientes condiciones:
Columna: Chiralcel-OX-H (250*30*5,0^); Fase móvil: Hexano (DEA al 0,1 %): Etanol (75:25);
Caudal: 30 ml/min; Diluyente: Fase móvil, las fracciones de preparación se evaporaron en vacío para proporcionar 80 mg de cada uno de los ejemplos:
(R) -4-(4-(3,3-difluoropropoxi)-2-fluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona (5)
Intervalo de fusión: 150-155°C; 1H-NMR (DMSO-d6): 812,36(d, 1H); 8,16(d, 1H); 7,57-7,26 (m, 3H); 6,90(dd, 1H); 6,79(d, 1H); 6,21(bt, 1H); 5,36(d, 1H); 4,11(t, 2H); 3,58(q, 1H); 2,97(dd, 1H); 2,35-2,20(m, 2H); MS=376(M+1); HPLC [B]~98,54 %: HPLC quiral~99,87 %.
(S) -4-(4-(3,3-difluoropropoxi)-2-fluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona (6)
Intervalo de fusión: 137-142°C; 1H-NMR (DMSO-d6): 812,39(s, 1H); 8,15(s, 1H); 7,53(d, 1H); 7,37(t, 2H); 7,20(bs, 1H); 6,93(dd, 1H); 6,79(dd, 1H); 6,21 (bt, 1H); 5,36(d, 1H); 4,11(t, 2H); 3,58(q, 1H); 2,98(d, 1H); 2,35-2,20(m, 2H); MS=376(M+1); HPLC [B]~99,14 %: HPLC quiral~99,03 %.
Ejemplos 7 y 8
rac4-(4-(3,3-difluoropropoxi)-2,3-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona
Etapa A: Cloruro de trimetilsilano (0,8 ml, 6,35 mmol), polvo de zinc (1,16 g, 7,79 mmol), benceno (80ml), bromoacetato de etilo (1,69 ml, 15,25 mmol), 4-(3,3-difluoropropoxi)-2,3-difluorobenzaldehído (3,0 g, 12,71 mmol) en benceno seco (20 ml), Rendimiento: 2 g (51 %)
Etapa B: Se añadieron dicromato de piridinio (9,28 g, 24,69 mmol), tamices moleculares (18,5 g) a una disolución de 3- (4-(3,3-difluoropropoxi)-2,3-fluorofenil)-3-hidroxipropanoato de etilo (2 g, 6,17mmol) en diclorometano (300 ml), Rendimiento 1,8 g (91 %)
Etapa C: Se añadió 5-amino-1H-benzo[d]imidazol-1-carboxilato de terc-butilo (1,17 g, 5,03 mmol) en xileno (30 ml) a una disolución de 3-(4-(3,3-difluoropropoxi)-2,3-difluorofenil)-3-oxopropanoato de etilo (1,8 g, 5,59 mmol) y piridina
(0,5 ml) en xileno (40 ml) Rendimiento 2,6 g (86,5 %).
Etapa D: Borohidruro de sodio (386 mg, 10,22 mmol)), N-(1-terc-butiloxicarbonil-1H-benzo[d]imidazol-5-il-)3-(4-(3,3-difluoropropoxi)-2,3-difluorofenil)-3-oxopropanamida (2,6 g, 5,11 mmol) en mezcla de tetrahidrofurano (35 ml) y metanol (15 ml) 0°C, Rendimiento 1,4 g (53,6 %)
Etapa E: Dietilazodicarboxilato (0,64 ml, 4,11 mmol), N-(1-terc-butiloxicarbonil-1H-benzo[d]imidazol-5-il-)3-(4-(3,3-difluoropropoxi)-2,3-fluorofenil)-3-hidroxipropanamida (1,4 g, 2,74 mmol) y trifenilfosfina (1,08 g, 4,11 mmol), Tetrahidrofurano (30 ml) a 0° C ,Rendimiento: 2,5 g de producto en bruto.
Etapa F: Se añadió ácido trifluoroacético (6 ml) a una disolución de 6-(2-(4-(3,3-difluoropropoxi)-2,3-difluorofenil)-4-oxoazetidin-1-il)-1H-benzo[d]imidazol-1-carboxilato de terc-butilo (2,5 g, 5,08 mmol) en diclorometano (30ml) a 0°C, Purificación por cromatografía en columna sobre alúmina neutra usando metanol al 1-1,5 % en diclorometano como eluyente proporcionado rac4-(4-(3,3-difluoropropoxi)-2,3-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona. Rendimiento: 0,45 g. Los restos se purificaron por HPLC de preparación quiral usando las siguientes condiciones: Columna: Chiralcel-OX-H (250*30*5,0p); Fase móvil: A: Hexano (DEA al 0,1 %); Etanol (75:25); Caudal: 30ml/min; Diluyente: Fase móvil.
Diluyente: Fase móvil, las fracciones de preparación se evaporaron en vacío para permitir obtener 80 mg de cada uno de los ejemplos:
(R) -4-(4-(3,3-difluoropropoxi)-2,3-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona (7)
Intervalo de fusión: 165-170°C; 1H-NMR (DMSO-d6): 812,40(d, 1H); 8,16(d, 1H); 7,58-7,42 (m, 2H); 7,34(t, 1H); 7,21(bs, 1H); 7,08(q, 2H); 6,21 (bt, 1H); 5,41(d, 1H); 4,21 (t, 2H); 3,67-3,60(q, 1H); 3,02(d, 1H); 2,39-2,24(m, 2H); MS=394(M+1); HPLC[B]~98,21 %: HPLC quiral~99,91 %.
(S) -4-(4-(3,3-difluoropropoxi)-2,3-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona (8)
Intervalo de fusión: 148-153°C; 1H-NMR (DMSO-d6): 812,40(d, 1H); 8,16(d, 1H); 7,58-7,42 (m, 2H); 7,34(t, 1H); 7,20(bs, 1H); 7,08(q, 2H); 6,21 (bt, 1H); 5,41(d, 1H); 4,21 (t, 2H); 3,67-3,60(q, 1H); 3,04(q, 1H); 2,36-2,26(m, 2H); MS=394(M+1); HPLC[B]~99,62 %: HPLC quiral~98,93 %.
Ejemplos 9 y 10
rac-4-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona
Etapa A: Cloruro de trimetilsilano (1,3 ml, 10,58 mmol), polvo de zinc (1,93 g, 29,66 mmol), benceno (30 ml), bromoacetato de etilo (2,8 ml, 25,14 mmol), 4-(3,3-difluoropropoxi)-2,6-difluorobenzaldehído (5 g, 21,18 mmol) en benceno seco (20 ml), Rendimiento: 4,5 g (66 %)
Etapa B: Dicromato de piridinio (15,67 g, 41,65 mmol) 3-(4-(3,3-difluoropropoxi)-2,6-fluorofenil)-3-hidroxipropanoato (4,5 g, 13,88 mmol) en diclorometano (400 ml), Rendimiento 1,2 g (28 %)
Etapa C: Se añadió 5-amino-1H-benzo[d]imidazol-1-carboxilato de terc-butilo (0,92 g, 3,975 mmol) en xileno (30 ml) a una disolución de 3-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-3-oxopropanoato de etilo (1,6 g, 4,968 mmol) y piridina (0,5 ml) en xileno (30 ml), Rendimiento 2,0 g de producto en bruto.
Etapa D: Borohidruro de sodio (297 mg, 7,858 mmol), N-(1-terc-butiloxicarbonil-1H-benzo[d]imidazol-5-il-) 3-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-3-oxopropanamida (2 g, 3,929 mmol) en mezcla de tetrahidrofurano (21 ml) y metanol (9 ml) a 0°C, Rendimiento 1,3 g (53,62 %)
Etapa E: Dietilazodicarboxilato (0,6 ml, 4,07 mmol), N-(1-terc-butiloxicarbonil-1H-benzo[d]imidazol-5-il-)3-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-3-hidroxipropanamida (1,3 g, 2,54 mmol) y trifenilfosfina (1 g, 4,07 mmol) tetrahidrofurano (30 ml) a 0°C, Rendimiento: 0,75g de producto en bruto.
Etapa F: Se añadió ácido trifluoroacético (1 ml) a una disolución de 6-(2-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-4-oxoazetidin-1-il)-1H-benzo[d]imidazol-1-carboxilato de terc-butilo (2,5 g, 5,08 mmol) en diclorometano (10 ml) a 0°C, Purificación mediante TLC preparativa permitió obtener 70 mg de rac4-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona como sólido amarillo pálido. Este se purificó por HPLC de preparación quiral usando las siguientes condiciones: Columna: Chiralcel-OX-H (250*30*5,0p); Fase móvil: A: Hexano (DEA al 0,1 %); Etanol (75:25); Caudal: 30 ml/min; Diluyente: Fase móvil. Las fracciones de preparación se evaporaron en vacío para proporcionar 28 mg y 18 mg de los enantiómeros respectivos
(R)-4-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona (9)
1H-NMR (DMSO-d6): 812,42(s, 1H); 8,18(s, 1H); 7,53(d, 1H); 7,33(s, 1H); 7,15(bs, 1H); 6,82(d, 2H); 6,19(bt, 1H); 5,49(d, 1H); 4,13(t, 2H); 3,62-3,58(m, 1H); 3,22(q, 1H); 2,33-2,22(t, 3H); MS=394(M+1); HPLC [B]~99,51 %: HPLC qu ira l-99,63 %.
(S)-4-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona (10)
1H-NMR (DMSO-d6): 812,42(s, 1H); 8,16(s, 1H); 7,53(d, 1H); 7,33(s, 1H); 7,15(bs, 1H); 6,82(d, 2H); 6,19(bt, 1H); 5,49(s, 1H); 4,13(t, 2H); 3,62-3,59(m, 1H); 2,30(q, 2H); MS=393,9(M+1); HPLC [B]-99,66 %: HPLC quiral-99,62 %. Ejemplos 11 y 12
rac4-(4-(2,2-difluoropropoxi)-2-fluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona
Etapa A: Cloruro de trimetilsilano (0,81 ml, 6,88 mmol), polvo de zinc (1,26 g, 19,26 mmol), benceno (30 ml), bromoacetato de etilo (1,9 ml, 16,51 mmol), 4-(2,2-difluoropropoxi)-2-fluorobenzaldehído (3 g, 13,76 mmol) en benceno seco (20 ml), Rendimiento: 3 g (75 %)
Etapa B: Se añadieron dicromato de piridinio (13,80 g, 36,72 mmol), tamices moleculares (15 g) a una disolución de 3-(4-(2,2-difluoropropoxi)-2-fluorofenil)-3-hidroxipropanoato de etilo (2,8 g, 9,18 mmol) en diclorometano (400 ml), Rendimiento 2,2 g (79 %)
Etapa C: Se añadió 5-amino-1H-benzo[d]imidazol-1-carboxilato de terc-butilo (830 mg, 3,56 mmol) en xileno (30 ml) a una disolución de 3-(4-(2,2-difluoropropoxi)-2-fluorofenil)-3-oxopropanoato de etilo (1,2 g, 3,96 mmol) y piridina (0,5 ml) en xileno (30 ml), Rendimiento 1,8 g de producto en bruto.
Etapa D: Borohidruro de sodio (280 mg, 7,33 mmol), N-(1-terc-butiloxicarbonil-1H-benzo[d]imidazol-5-il-)3-(4-(2,2-difluoropropoxi)-2-fluorofenil)-3-oxopropanamida (1,8 g, 3,66 mmol) en mezcla de tetrahidrofurano (21 ml) y metanol (9 ml) a 0°C, Rendimiento 0,75 g (42,2 %)
Etapa E: Dietilazodicarboxilato (0,36 ml, 2,27 mmol), N-(1-terc-butiloxicarbonil-1H-benzo[d]imidazol-5-il-)3-(4-(2,2-difluoropropoxi)-2-fluorofenil)-3-hidroxipropanamida (700 mg, 1,42 mmol) y trifenilfosfina (700 mg, 1,42 mmol), Tetrahidrofurano (30 ml) a 0°C, Rendimiento: 1,0 g de producto en bruto
Etapa F: Se añadió ácido trifluoroacético (4 ml) a una disolución de 6-(2-(4-(2,2-difluoropropoxi)-2-fluorofenil)-4-oxoazetidin-1-il)-1H-benzo[d]imidazol-1-carboxilato de terc-butilo producto en bruto (1,0 g, 2,08 mmol) en diclorometano (20 ml) a 0°C, Rendimiento: 0,13 g (40 %). Se repitió la misma síntesis (en las mismas cantidades) para proporcionar otros 120mg de rac4-(4-(2,2-difluoropropoxi)-2-fluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona. Ambos combinados de ~250mg y purificados por HPLC de preparación quiral usando las siguientes condiciones: Columna: Chiralcel-OX-H (250*30*5,0^); Fase móvil: A: Hexano (DEA al 0,1 %); Etanol (75:25); Caudal: 30 ml/min; Diluyente: Fase móvil.
Las fracciones de preparación se evaporaron en vacío y se trituraron con dietil éter para proporcionar 70 mg de cada isómero respectivamente.
(R) -4-(4-(2,2-difluoropropoxi)-2-fluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona (11)
Intervalo de fusión: 165-170°C; 1H-NMR (DMSO-d6): 812,35(d, 1H); 8,16(d, 1H); 7,55(d, 1H); 7,47-7,32(m, 2H); 7,07(q, 1H); 6,87(q, 1H); 5,37(d, 1H); 4,29(t, 2H); 3,63-3,58(m, 1H); 3,00(q, 1H); 1,71 (t, 3H); MS=376(M+1); HPLC [B ]-99,17 %: HPLC quiral-99,79 %.
(S) -4-(4-(2,2-difluoropropoxi)-2-fluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona (12)
Intervalo de fusión: 203-208°C; 1H-NMR (DMSO-d6): 812,37(d, 1H); 8,14(s, 1H); 7,59-7,37(m, 3H); 7,32(bs, 1H); 7,01(dd, 1H); 6,85(dd, 1H); 5,37(q, 1H); 4,29(t, 2H); 3,63-3,58(m, 1H); 3,00(q, 1H); 1,70(t, 3H); MS=376(M+1); HPLC [B ]-98,39 %: HPLC quiral-96,69 %.
Ejemplos 13 y 14
Rac-4-(4-(2,2-difluoropropoxi)-2,3-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona
Etapa A: Cloruro de trimetilsilano (0,79 ml, 6,35 mmol), polvo de zinc (1,2 g, 19,06 mmol), benceno (30 ml), bromoacetato de etilo (1,6 ml, 15,25 mmol), 4-(2,2-difluoropropoxi)-2,3-difluorobenzaldehído (3 g, 12,7 mmol) en benceno seco (20 ml), Rendimiento: 3,1 g (79 %)
Etapa B: Dicromato de piridinio (13,92 g, 37,03 mmol), se añadió a una disolución de 3-(4-(2,2-difluoropropoxi)-2,3-fluorofenil)-3-hidroxipropanoato (3 g, 9,25 mmol) en diclorometano (400 ml), Rendimiento 1,2 g (40 %)
Etapa C: Se añadió 5-amino-1H-benzo[d]imidazol-1-carboxilato de tere-butilo (0,72 g, 3,03 mmol) en xileno (30 ml) a una disolución de 3-(4-(2,2-difluoropropoxi)-2,3-difluorofenil)-3-oxopropanoato de etilo (1,2 g, 3,77 mmol) y piridina (0,5 ml) en xileno (30 ml, Rendimiento 1,8 g de producto en bruto.
Etapa D: Borohidruro de sodio (268 mg, 7,07 mmol), N-(1-tere-butiloxicarbonil-1H-benzo[d]imidazol-5-il-)3-(4-(2,2-difluoropropoxi)-2,3-difluorofenil)-3-oxopropanamida (1,8 g, 3,5 mmol) en mezcla de tetrahidrofurano (21 ml) y metanol (9 ml) a 0°C, Rendimiento 1,2 g (67,0 %)
Etapa E: Dietilazodicarboxilato (0,6 ml, 3,67 mmol), N-(1-tere-butiloxicarbonil-1H-benzo[d]imidazol-5-il-)3-(4-(2,2-difluoropropoxi)-2,3-fluorofenil)-3-hidroxipropanamida (1,2 g, 2,43 mmol) y trifenilfosfina (0,96 g, 3,67 mmol) tetrahidrofurano (30 ml) a 0°C, Rendimiento: 0,4 g de producto en bruto.
Etapa F: Se añadió ácido trifluoroacético (1 ml) a una disolución de 6-(2-(4-(2,2-difluoropropoxi)-2,3-difluorofenil)-4-oxoazetidin-1-il)-1H-benzo[d]imidazol-1-carboxilato de tere-butilo en bruto (400 mg, 0,811 mmol) en diclorometano (10 ml) a 0°C, Rendimiento: 0,07 g (22 %).
Esta se purificó por HPLC de preparación quiral usando las siguientes condiciones: Columna: Chiralcel-OX-H (250*30*5,0p); Fase móvil: A: Hexano (DEA al 0,1 %); Etanol (75:25); Caudal: 30 ml/min; Diluyente: Fase móvil. Las fracciones de preparación se evaporaron en vacío para proporcionar 30 mg y 34 mg de los enantiómeros respectivos. (R) -4-(4-(2,2-difluoropropoxi)-2,3-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona (13)
Intervalo de fusión: 173-177°C; 1H-NMR (DMSO-d6): 812,35(d, 1H); 8,15(s, 1H); 7,55-7,36(m, 2H); 7,22(t, 1H); 7,10(t, 1H); 5,44(d, 1H); 4,39(t, 2H); 3,66-3,61(m, 1H); 3,00(q, 1H); 1,71(t, 3H); MS=394(M+1); HPLC [B]~98,55 %: HPLC qu ira l-99,94 %.
(S) -4-(4-(2,2-difluoropropoxi)-2,3-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona (14)
Intervalo de fusión: 170-174°C; 1H-NMR (DMSO-d6): 812,35(d, 1H); 8,15(s, 1H); 7,55-7,36(m, 3H); 7,22(t, 1H); 7,10(t, 1H); 5,42(d, 1H); 4,40(t, 2H); 3,66-3,61 (m, 1H); 3,00(q, 1H); 1,71 (t, 3H); MS=394(M+1); HPLC [B]-99,15 %: HPLC qu ira l-98,09 %.
Ejemplos 15 y 16
Rae-4-(4-(2,2-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona
Etapa A: Cloruro de trimetilsilano (0,7 ml, 5,8 mmol), polvo de zinc (1 g, 16,3 mmol), benceno (30 ml), bromoacetato de etilo (2,33 g, 13,9 mmol), 4-(2,2-difluoropropoxi)-2,6-difluorobenzaldehído (2,7 g, 11,6 mmol) en benceno seco (20 ml), Rendimiento: 2,4 g (67 %).
Etapa B: Dicromato de piridinio (11 g, 29,6 mmol), tamices moleculares (11 g) 3-(4-(2,2-difluoropropoxi)-2,6-fluorofenil)-3-hidroxipropanoato (2,4 g, 7,40 mmol) en diclorometano (400 ml), Rendimiento 1,7 g (71,2 %)
Etapa C: Se añadió 5-amino-1H-benzo[d]imidazol-1-carboxilato de tere-butilo (984 mg, 4,2 mmol) en xileno (30 ml) a una disolución de 3-(4-(2,2-difluoropropoxi)-2,6-difluorofenil)-3-oxopropanoato de etilo (1,7 g, 5,2 mmol) y piridina (1,0 ml) en xileno (30 ml), Rendimiento 1,4 g de producto en bruto.
Etapa D: Borohidruro de sodio (209 mg, 5,5 mmol), N-(1-tere-butiloxicarbonil-1H-benzo[d]imidazol-5-il-)3-(4-(2,2-difluoropropoxi)-2,6-difluorofenil)-3-oxopropanamida (1,4 g, 2,7 mmol) en mezcla de tetrahidrofurano (14 ml) y metanol (6 ml) a 0°C, Rendimiento 1,3 g (94 %).
Etapa E: Dietilazodicarboxilato (0,5 ml, 3,2 mmol), N-(1-tere-butiloxicarbonil-1H-benzo[d]imidazol-5-il-)3-(4-(2,2-difluoropropoxi)-2,6-difluorofenil)-3-hidroxipropanamida (1,1 g, 2,15 mmol) y trifenilfosfina (845 mg, 3,2 mmol) tetrahidrofurano (30 ml) a 0°C, Rendimiento: 1,6 g de producto en bruto.
Etapa F: Se añadió ácido trifluoroacético (4 ml) a una disolución de 6-(2-(4-(2,2-difluoropropoxi)-2,6-difluorofenil)-4-oxoazetidin-1-il)-1H-benzo[d]imidazol-1-carboxilato de tere-butilo en bruto (1,6 g) en diclorometano (20 ml) a 0°C. Rendimiento: 0,3g de rac 4-(4-(2,2-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona. Esta se purificó por HPLC de preparación quiral usando las siguientes condiciones: Columna: Chiralcel-OX-H (250*30*5,0p); Fase móvil: A: Hexano (DEA al 0,1 %); Etanol (75:25); Caudal: 30ml/min; Diluyente: Fase móvil. Las fracciones de preparación se evaporaron en vacío y se trituraron con éter dietílico para proporcionar 70 mg de cada enantiómero respectivamente.
(R) -4-(4-(2,2-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona (15)
Intervalo de fusión: 158-162°C; 1H-NMR (DMSO-d6): 812,40(d, 1H); 8,15(d, 1H); 7,57-7,46(m, 1H); 7,38(s, 1H); 7,28(t, 1H); 6,91 (d, 2H); 5,45(d, 1H); 4,33(t, 2H); 3,62-3,58(m, 1H); 3,24(q, 1H); 1,69(t, 3H); MS=394(M+1); HPLC [B]~97,07 %: HPLC qu ira l-99,51 %.
(S) -4-(4-(2,2-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidi n-2-ona (16)
Intervalo de fusión: 163-166°C; 1H-NMR (DMSO-d6): 812,40(d, 1H); 8,14(bs, 1H); 7,55-7,26(m, 2H); 7,04(s, 1H); 6,90(d, 2H); 5,45(d, 1H); 4,32(t, 2H); 3,63-3,58(m, 1H); 3,24(d, 1H); 1,69(t, 3H); MS=394(M+1); HPLC [B]-98,92 %: HPLC qu ira l-99,69 %.
Cribado de actividad
Ensayos fluorom étricos
Todas las mediciones se realizaron con un BioAssay Reader HTS-7000Plus para microplacas (Perkin Elmer) a 30 °C. La actividad de QC se evaluó fluorométricamente usando H-Gln-/NA. Las muestras consistieron en sustrato fluorogénico 0,2 mM, 0,25 U de piroglutamil aminopeptidasa (Unizyme, Hcrsholm, Dinamarca) en Tris 0,2 M/HCl, pH 8.0 que contenía EDTA 20 mM y un alícuota apropiadamente diluida de QC en un volumen final de 250 ppl. Las longitudes de onda de excitación/emisión fueron 320/410 nm. Las reacciones de ensayo se iniciaron mediante la adición de glutaminil ciclasa. La actividad de QC se determinó a partir de una curva patrón de /-naftilamina bajo condiciones de ensayo. Una unidad se define como la cantidad de QC que cataliza la formación de 1 pmol de pGlu-/N A a partir de H-Gln-/NA por minuto bajo las condiciones descritas.
En un segundo ensayo fluorimétrico, la actividad de QC se determinó usando H-Gln-AMC como sustrato. Las reacciones se llevaron a cabo a 30 °C utilizando el lector NOVOStar para microplacas (BMG labtechnologies). Las muestras consistieron en concentraciones variables del sustrato fluorogénico, 0,1 U de piroglutamil aminopeptidasa (Qiagen) en Tris 0,05 M/HCl, pH 8,0 que contenía EDTA 5 mM y una alícuota apropiadamente diluida de QC en un volumen final de 250 pl. Las longitudes de onda de excitación/emisión fueron 380/460 nm. Las reacciones de ensayo se iniciaron mediante la adición de glutaminil ciclasa. La actividad de QC se determinó a partir de una curva patrón de 7-amino-4- metilcumarina bajo condiciones de ensayo. Los datos cinéticos se evaluaron usando el software GraFit.
Ensayo espectrofotométrico de QC
Este novedoso ensayo se usó para determinar los parámetros cinéticos para la mayoría de los sustratos de QC. La actividad de QC se analizó espectrofotométricamente usando un método continuo, que se derivó adaptando un ensayo discontinuo previo (Bateman, R. C. J. 1989 J Neurosci Methods 30, 23-28) utilizando glutamato deshidrogenasa como enzima auxiliar. Las muestras consistieron en el sustrato de QC respectivo, NADH 0,3 mM, ácido a-cetoglutárico 14 mM y 30 U/ml de glutamato deshidrogenasa en un volumen final de 250 pl. Las reacciones se iniciaron mediante la adición de QC y se siguieron monitorizando la disminución en la absorbancia a 340 nm durante 8-15 min.
Se evaluaron las velocidades iniciales y la actividad enzimática se determinó a partir de una curva patrón de amoniaco bajo condiciones de ensayo. Todas las muestras se midieron a 30 °C usando o bien el lector SPECTRAFluor Plus o bien Sunrise (ambos de TECAN) para microplacas. Los datos cinéticos se evaluaron usando el software GraFit.
Ensayo de inhibidor
Para el ensayo de inhibidor, la composición de muestra fue la misma que se ha descrito anteriormente, excepto por que se añadió el compuesto inhibidor putativo. Para una rápida prueba de inhibición de QC, las muestras contuvieron 4 mM del inhibidor respectivo y una concentración de sustrato a 1 Km. Para investigaciones detalladas de la inhibición y determinación de valores de Ki, primero se investigó la influencia del inhibidor sobre las enzimas auxiliares. En cada caso no hubo influencia sobre ninguna enzima detectada, permitiendo así la determinación fiable de la inhibición de QC. La constante inhibidora se evaluó ajustando el conjunto de curvas de progreso a la ecuación general para la inhibición competitiva usando el software GraFit. El ensayo del inhibidor se realizó a dos niveles de pH diferentes, pH 6.0 y pH 8,0: El valor de pH respectivo en la disolución de ensayo se ajustó usando métodos convencionales.
Parámetros farmacocinéticos
Métodos
Se administró a tres ratones (cepa CD-1) por vía oral 30 mg/kg de cada compuesto de ensayo disuelto en Methocel al 0,8 %. Las muestras se tomaron en los puntos de tiempo 10 min, 0,5, 1, 2, 4 y 8 h después de la administración del compuesto de ensayo para la recogida de plasma y cerebro.
Recogida de sangre
Los ratones se anestesiaron con isoflurano. Se recogieron aproximadamente 200 pl de cada muestra de sangre mediante punción cardíaca para sangrado terminal en tubos con K2EDTA. Las muestras de sangre se colocaron en hielo y se centrifugaron a 2000 g durante 5 min para obtener la muestra plasmática en 15 minutos. Recogida de LCR: Los animales se sacrificaron con inhalación de CO2 puro. Se realizó una incisión en la línea media en el cuello. El músculo bajo la piel se cortó para exponer la cisterna magna. La cisterna magna se penetró con el extremo afilado de un capilar y el LCR se recogió por capilaridad.
Recogida de cerebro
Después de la recogida de LCR, se realizó una perfusión con 7 veces el volumen total de sangre de ratón (aproximadamente 15 ml) de PBS enfriado con hielo (pH 7,4) mediante punción cardiaca antes de la recogida del cerebro. Se realizó una incisión en la línea media del cuero cabelludo del animal. Se extrajo el cerebro y se enjuagó con solución salina fría. Se colocó el cerebro en un tubo con tapa de rosca y se pesó. Se homogeneizaron las muestras de cerebro durante 2 min con 3 volúmenes (v/p) de PBS (pH 7,4) y luego se analizaron con CL-EM/EM. La concentración cerebral se corrigió con un factor de dilución de 4 de la siguiente manera:
Concentración cerebral = conc. de homogeneizado cerebral x 4, suponiendo que 1 g de tejido cerebral húmedo equivale a 1 ml.
Se almacenaron las muestras de plasma, cerebro y LCR a aproximadamente -80 °C hasta el análisis.
Preparación de muestras
Para muestras de plasma: se le añadieron a una alícuota de 20 pl de muestra 200 pI de IS (diclofenaco, 200 ng/ml) en ACN, se agitó la mezcla en vórtex durante 2 min y se centrifugó a 12.000 rpm durante 5 min. Se inyectó 1 pl de sobrenadante para análisis mediante CL-EM/EM.
Para muestras de plasma diluidas: se le añadieron a una alícuota de 4 pl de muestra 16 pl de plasma blanco, bien mezclado, al que se le añadieron 200 pl de IS (diclofenaco, 200 ng/ml) en ACN.
Para muestras de cerebro: se homogeneizó el tejido cerebral durante 2 min con 3 volúmenes (v/p) de PBS. Se le añadieron a una alícuota de 20 pl de muestra 200 pl de IS (diclofenaco, 200 ng/ml) en ACN, se agitó la mezcla en vórtex durante 2 min y se centrifugó a 12000 rpm durante 5 min. Se inyectó 1 pl de sobrenadante para análisis mediante CL-EM/EM.
Para muestras de LCR: a la muestra de LCR se le añadió un volumen correspondiente de 20 veces de IS (diclofenaco, 200 ng/ml) en ACN, se agitó la mezcla en vórtex durante 2 min. Se inyectaron 3 pl de sobrenadante para análisis mediante Cl-EM/EM.
Se calcularon los valores de Tmáx, T1/2, AUC y logBB utilizando métodos convencionales.
Resultados

Los compuestos de ejemplo 4, 5, 7, 9, 11, 13 y 15 mostraron un buen comportamiento de cruce de barrera hematoencefálica y valores de AUC en el cerebro, que apoyan su uso en el tratamiento de enfermedades neurodegenerativas.
Métodos analíticos
HPLC analítico
Método [A]: El sistema analítico de HPLC consistió en un Agilent MSD 1100 que utiliza una columna analítica columna analítica Waters SunFire RP 18 (2,5 pm) (longitud: 50 mm, diámetro: 2,1 mm) y detector de red de diodos (DAD) con
X = 254 nm como longitud de onda de notificación. Los compuestos se analizaron usando un gradiente a un caudal de 0,6 ml/min; mediante lo cual el eluyente (A) fue acetonitrilo, el eluyente (B) fue agua y el eluyente (C) ácido fórmico al 2 % en acetonitrilo aplicando el siguiente gradiente:

Las purezas de todos los compuestos notificados se determinaron por el porcentaje del área del pico a 214 nm.
Método [B]: El sistema analítico de HPLC consistió en un Agilent MSD 1100 que utiliza una columna analítica Waters SYMETRY RP 18 (3,5 pm) (longitud: 75 mm, diámetro: 4,6 mm) y un detector de red de diodos (DAD) con X = 254 nm como longitud de onda de notificación. Los compuestos se analizaron usando un gradiente a un caudal de 1,0 ml/min; en el que el eluyente (A) fue acetonitrilo, el eluyente (B) fue agua en acetato de amonio 0,01 M:

HPLC quiral
El sistema de HPLC quiral analítico consistió en un Agilent MSD 1100 que utiliza una columna analítica Waters Chiracel OX-H (5 pm) (longitud: 250 mm, diámetro: 4,6 mm) y un detector de red de diodos (DAD) con X = 254 nm como longitud de onda de notificación. Los compuestos se analizaron usando una mezcla isocrática de DEA al 0,1 % en hexano y ehanol (70/30) a un caudal de 1,0 ml/min.
Espectrometría de masas, espectroscopía de RMN:
Se obtuvieron los espectros de masas de ESI con un espectrómetro SCIEX API 365 (Perkin Elmer) utilizando el modo de ionización positiva.
Los espectros de RMN 1H (500 MHz) se registraron en un BRUKER AC 500. El disolvente fue DMSO-D6, a menos que se especifique de otro modo. Los desplazamientos químicos se expresan como partes por millón (ppm) a campo bajo de tetrametilsilano. Los patrones de fraccionamiento se han designado del siguiente modo: s (singlete), d (doblete), dd (doblete de doblete), t (triplete), m (multiplete) y bt (señal ancha).
Espectrometría de masas, espectroscopia RMN:
Se obtuvieron los espectros de masas de ESI con un espectrómetro SCIEX API 365 (Perkin Elmer) utilizando el modo de ionización positiva.
Los espectros de RMN 1H (500 MHz) se registraron en un BRUKER AC 500. El disolvente fue DMSO-D6, a menos que se especifique de otro modo. Los cambios químicos se expresan como partes por millón (ppm) por debajo del tetrametilsilano. Los patrones de fraccionamiento se han designado del siguiente modo: s (singlete), d (doblete), dd (doblete de doblete), t (triplete), m (multiplete) y bt (señal ancha).
Espectrometría de masas MALDI-TOF
La espectrometría de masas por desorción/ionización láser asistida por matriz se llevó a cabo usando el sistema Hewlett-Packard G2025 LD-TOf con un analizador de tiempo de vuelo lineal. El instrumento se equipó con un láser de nitrógeno de 337 nm, una fuente de aceleración de potencial (5 kV) y un tubo de vuelo de 1,0 m. El funcionamiento del detector fue en el modo positivo y las señales se registran y se filtran usando el osciloscopio de almacenamiento digital LeCroy 9350M conectado a un ordenador personal. Se mezclaron muestras (5 pl) con volúmenes iguales de la disolución de matriz. Para la disolución de matriz se usó DHAP/DAHC, preparado disolviendo 30 mg de 2',6'-dihidroxiacetofenona (Aldrich) y 44 mg de hidrogenocitrato de diamonio (Fluka) en 1 ml de acetonitrilo/ TFA al 0,1 % en agua (1/1, v/v). Un pequeño volumen (“ 1 pl) de la mezcla de matriz-analito se transfirió a una punta de la sonda y se evaporó inmediatamente en una cámara de vacío (accesorio prep de muestras Hewlett-Packard G2024A) para garantizar la rápida y homogénea cristalización de muestras.
Claims (18)
- REIVINDICACIONESi. Compuesto de fórmula (I):
 o una sal, solvato o polimorfo farmacéuticamente aceptable del mismo, incluyendo todos los tautómeros y estereoisómeros del mismo, en la que:A es heteroarilo seleccionado de 1H-benzimidazol-5-ilo o 1H-benzimidazol-6-ilo e imidazo[1,2-a]piridin-7-ilo; R1 representa hidrógeno, alquilo o halógeno;R2 representa hidrógeno, alquilo o halógeno;R3 representa hidrógeno, alquilo o alcoxilo;R4 representa hidrógeno o alquilo; yR5 representa hidrógeno, alquilo o halógeno;yen el que los grupos alquilo o alcoxilo anteriores se sustituyen opcionalmente por uno o más halógenos.
o una sal, solvato o polimorfo farmacéuticamente aceptable del mismo, incluyendo todos los tautómeros y estereoisómeros del mismo, en la que:A es heteroarilo seleccionado de 1H-benzimidazol-5-ilo o 1H-benzimidazol-6-ilo e imidazo[1,2-a]piridin-7-ilo; R1 representa hidrógeno, alquilo o halógeno;R2 representa hidrógeno, alquilo o halógeno;R3 representa hidrógeno, alquilo o alcoxilo;R4 representa hidrógeno o alquilo; yR5 representa hidrógeno, alquilo o halógeno;yen el que los grupos alquilo o alcoxilo anteriores se sustituyen opcionalmente por uno o más halógenos. - 2. Compuesto de fórmula (I) según la reivindicación 1, en el queR1 representa hidrógeno o halógeno;R2 representa hidrógeno o halógeno;R3 representa hidrógeno o alcoxilo;R4 representa hidrógeno; yR5 representa hidrógeno o halógeno;yen el que el grupo alcoxilo se sustituye opcionalmente por uno o más halógenos
- 3. Compuesto de fórmula (I) según la reivindicación 1 o 2, en el que A es 1H-benzoimidazol-5-ilo.
- 4. Compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 3, en el que R1 es hidrógeno.
- 5. Compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 3, en el que R1 es un halógeno, como el flúor.
- 6. Compuesto de la fórmula (I) según una cualquiera de las reivindicaciones anteriores, en el que R2 es hidrógeno.
- 7. Compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 5, en el que R2 es halógeno, como el flúor.
- 8. Compuesto de fórmula (I) según una cualquiera de las reivindicaciones anteriores, en el que R3 representa -O-alquilo C1-4, opcionalmente sustituido por uno o más halógenos, como el flúor.
- 9. Compuesto de fórmula (I) según una cualquiera de las reivindicaciones anteriores, en el que R3 representa metoxilo, difluoropropoxilo o difluorobutoxilo.
- 10. Compuesto de fórmula (I) según una cualquiera de las reivindicaciones anteriores, en el que R3 representa 2,2-difluoropropoxilo o 3,3-difluoropropoxilo.
- 11. Compuesto de fórmula (I) según una cualquiera de las reivindicaciones anteriores, en el que R5 es hidrógeno.
- 12. Compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 10, en el que R5 es halógeno, como el flúor.
- 13. Compuesto de fórmula (I) según una cualquiera de las reivindicaciones anteriores o una sal, solvato o polimorfo farmacéuticamente aceptable, incluyendo todos los tautómeros y estereoisómeros, en el que el compuesto de fórmula (I) es un compuesto seleccionado del grupo que consiste en1 1-(1H-benzo[d]imidazol-5-il)-4-fenilazetidin-2-ona;2 (R)-1-(1H-benzo[d]imidazol-6-il)-4-fenilazetidin-2-ona;3 (S)-1 -(1 H-benzo[d]imidazol-6-il)-4-fenilazetidin-2-ona;4 1-(1H-benzo[d]imidazol-5-il)-4-(2,6-difluoro-4-metoxifenil)azetidin-2-ona;5 (R)-4-(4-(3,3-difluoropropoxi)-2-fluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;6 (S)-4-(4-(3,3-difluoropropoxi)-2-fluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;7 (R)-4-(4-(3,3-difluoropropoxi)-2,3-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;8 (S)-4-(4-(3,3-difluoropropoxi)-2,3-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;9 (R)-4-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;10 (S)-4-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;11 (R)-4-(4-(2,2-difluoropropoxi)-2-fluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;12 (S)-4-(4-(2,2-difluoropropoxi)-2-fluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;13 (R)-4-(4-(2,2-difluoropropoxi)-2,3-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;14 (S)-4-(4-(2,2-difluoropropoxi)-2,3-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;15 (R)-4-(4-(2,2-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;16 (S)-4-(4-(2,2-difluoropropoxi)-2,6-difluorofenil)-1-(1H-benzo[d]imidazol-5-il)azetidin-2-ona;17 (R)-1-(H-imidazo[1,2-a]piridin-7-il)-4-fenilazetidin-2-ona;18 (S)-1 -(H-imidazo[1,2-a]piridin-7-il)-4-fenilazetidin-2-ona;19 4-(2,6-difluoro-4-metoxifenil)-1-(H-imidazo[1,2-a]piridin-7-il)azetidin-2-ona;20 (R)-4-(4-(3,3-difluoropropoxi)-2-fluorofenil)-1-(H-imidazo[1,2-a]piridin-7-il)azetidin-2-ona;21 (S)-4-(4-(3,3-difluoropropoxi)-2-fluorofenil)-1-(H-imidazo[1,2-a]piridin-7-il)azetidin-2-ona;22 (R)-4-(4-(3,3-difluoropropoxi)-2,3-difluorofenil)-1-(H-imidazo[1,2-a]piridin-7-il)azetidin-2-ona;23 (S)-4-(4-(3,3-difluoropropoxi)-2,3-difluorofenil)-1-(H-imidazo[1,2-a]piridin-7-il)azetidin-2-ona; 24 (R)-4-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-1-(H-imidazo[1,2-a]piridin-7-il)azetidin-2-ona;25 (S)-4-(4-(3,3-difluoropropoxi)-2,6-difluorofenil)-1-(H-imidazo[1,2-a]piridin-7-il)azetidin-2-ona;26 (R)-4-(4-(2,2-difluoropropoxi)-2-fluorofenil)-1-(H-imidazo[1,2-a]piridin-7-il)azetidin-2-ona;27 (S)-4-(4-(2,2-difluoropropoxi)-2-fluorofenil)-1-(H-imidazo[1,2-a]piridin-7-il)azetidin-2-ona;28 (R)-4-(4-(2,2-difluoropropoxi)-2,3-difluorofenil)-1-(H-imidazo[1,2-a]piridin-7-il)azetidin-2-ona; y29 (S)-4-(4-(2,2-difluoropropoxi)-2,3-difluorofenil)-1-(H-imidazo[1,2-a]piridin-7-il)azetidin-2-ona.
- 14. Composición farmacéutica que comprende un compuesto según una cualquiera de las reivindicaciones 1 a 13, opcionalmente en combinación con uno o más diluyentes o vehículos terapéuticamente aceptables.
- 15. Composición farmacéutica según la reivindicación 14, que comprende adicionalmente al menos un compuesto, seleccionado del grupo que consiste en neuroprotectores, fármacos antiparkinsonianos, inhibidores de la deposición de proteína amiloide, inhibidores de la síntesis de beta-amiloide, antidepresivos, fármacos ansiolíticos, fármacos antipsicóticos y fármacos contra la esclerosis múltiple.
- 16. Composición farmacéutica según la reivindicación 14 o 15, que comprende adicionalmente al menos un compuesto, seleccionado del grupo que consiste en inhibidores de PEP, LiCl, inhibidores de inhibidores de DP IV o enzimas similares a DP IV, inhibidores de acetilcolinesterasa (ACE), potenciadores de PIMT, inhibidores de beta-secretasas, inhibidores de gamma-secretasas, inhibidores de endopeptidasa neutra, inhibidores de fosfodiesterasa-4 (PDE-4), inhibidores de TNF-alfa, antagonistas de receptor muscarínico M1, antagonistas de receptores de NMDA, inhibidores de receptores sigma-1, antagonistas de histamina H3, agentes inmunomoduladores, agentes inmunosupresores o un agente seleccionado del grupo que consiste en Antegren (natalizumab), Neurelan (fampridina-SR), Campath (alemtuzumab), IR 208, NBI 5788/MSP 771 (tiplimotida), paclitaxel, Anergix.MS (AG 284), SH636, Differin (CD 271, adapaleno), BAY 361677 (interleucina-4), inhibidores de metaloproteinasas de matriz, interferón-tau (trofoblastina) y SAIK-MS.
- 17. Compuesto según una cualquiera de las reivindicaciones 1 a 13 o la composición farmacéutica según una cualquiera de las reivindicaciones 14 a 16 para su uso en el tratamiento de una enfermedad seleccionada del grupo que consiste en enfermedad de Kennedy, cáncer duodenal con o sin infecciones por Helicobacter pylori, cáncer colorrectal, síndrome de Zollinger-Ellison, cáncer gástrico con o sin infecciones por Helicobacter pylori, afecciones psicóticas patógenas, esquizofrenia, infertilidad, neoplasia, respuestas inflamatorias del huésped, cáncer, metástasis malignas, melanoma, psoriasis, alteración de respuestas inmunitarias humorales y mediadas por células, procesos de adhesión y migración de leucocitos en el endotelio, alteración del consumo de alimentos, alteración del sueño-vigilia, alteración de la regulación homeostática del metabolismo de la energía, alteración de la función autónoma, alteración del equilibrio hormonal o alteración de la regulación de fluidos corporales, esclerosis múltiple, el síndrome de Guillain-Barré, polirradiculoneuropatía desmielinizante inflamatoria crónica; deterioro cognitivo leve, enfermedad de Alzheimer, demencia familiar británica, demencia familiar danesa, neurodegeneración en síndrome de Down, enfermedad de Huntington; artritis reumatoide, aterosclerosis, pancreatitis y reestenosis.
- 18. Procedimiento para la preparación de un compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 13, que comprende:(a) preparar un compuesto de fórmula (I) a partir de un compuesto de fórmula (II):
 en el que R1 y R5 y son tal y como se definen en la reivindicación 1 para compuestos de fórmula (I), haciendo reaccionar un compuesto de fórmula (II) con ácido bromoacético en presencia de un catalizador adecuado, como el polvo de zinc, y un agente de protección, como cloruro de trimetiisililo (TMS-Cl)o(b) preparar un compuesto de fórmula (I) a partir de un compuesto de fórmula (III):
en el que R1 y R5 y son tal y como se definen en la reivindicación 1 para compuestos de fórmula (I), haciendo reaccionar un compuesto de fórmula (II) con ácido bromoacético en presencia de un catalizador adecuado, como el polvo de zinc, y un agente de protección, como cloruro de trimetiisililo (TMS-Cl)o(b) preparar un compuesto de fórmula (I) a partir de un compuesto de fórmula (III): en el que R1, R2, R3, R4 y R5 y son tal y como se definen en la reivindicación 1 para los compuestos de fórmula (I), haciendo reaccionar un compuesto de fórmula (III) con dietilazodicarboxilato en presencia de trifenilfosfina y un disolvente adecuado como tetrahidrofurano, y una etapa de desptrotección.
en el que R1, R2, R3, R4 y R5 y son tal y como se definen en la reivindicación 1 para los compuestos de fórmula (I), haciendo reaccionar un compuesto de fórmula (III) con dietilazodicarboxilato en presencia de trifenilfosfina y un disolvente adecuado como tetrahidrofurano, y una etapa de desptrotección.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17194164.4A EP3461819B1 (en) | 2017-09-29 | 2017-09-29 | Inhibitors of glutaminyl cyclase |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2812698T3 true ES2812698T3 (es) | 2021-03-18 |
Family
ID=59997286
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17194164T Active ES2812698T3 (es) | 2017-09-29 | 2017-09-29 | Inhibidores de glutaminil ciclasa |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US11279690B2 (es) |
| EP (1) | EP3461819B1 (es) |
| JP (2) | JP6821860B2 (es) |
| KR (2) | KR20210057206A (es) |
| CN (1) | CN111315738B (es) |
| AU (1) | AU2018340397B2 (es) |
| BR (1) | BR112020005625B1 (es) |
| CA (1) | CA3077314C (es) |
| DK (1) | DK3461819T3 (es) |
| EA (2) | EA037862B1 (es) |
| ES (1) | ES2812698T3 (es) |
| IL (2) | IL273214B (es) |
| MX (1) | MX2020003570A (es) |
| NZ (1) | NZ763312A (es) |
| PL (1) | PL3461819T3 (es) |
| SG (1) | SG11202002239VA (es) |
| WO (1) | WO2019063414A1 (es) |
| ZA (1) | ZA202001573B (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3221003A1 (en) * | 2021-06-24 | 2022-12-29 | Insilico Medicine Ip Limited | Beta-lactam derivatives for the treatment of diseases |
Family Cites Families (605)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5736816A (ja) | 1980-08-14 | 1982-02-27 | Toshiba Corp | Chodendokoirunoseizohoho |
| JPS57192573A (en) | 1981-05-25 | 1982-11-26 | Hochiki Co | Fire fighting agent |
| JPS60165712A (ja) | 1984-02-08 | 1985-08-28 | パイオニア株式会社 | 可変調整部品の自動調整方法 |
| JPS6126111A (ja) | 1984-07-16 | 1986-02-05 | Shin Meiwa Ind Co Ltd | 産業用ロボツト |
| JPS6137764A (ja) | 1984-07-31 | 1986-02-22 | Suntory Ltd | 抗プロリルエンドペプチダ−ゼ活性を有する新規生理活性化合物 |
| JPS6152021A (ja) | 1984-08-22 | 1986-03-14 | Fujitsu Ltd | スケルチ回路 |
| JPH0647573B2 (ja) * | 1984-08-29 | 1994-06-22 | メクト株式会社 | 2−アゼチジノン誘導体及びその製造方法 |
| JPS6172439A (ja) | 1984-09-18 | 1986-04-14 | Sanyo Electric Co Ltd | デ−タ転送方式 |
| EP0201741B1 (en) | 1985-04-16 | 1991-07-31 | Suntory Limited | Dipeptide derivatives, processes for preparing them, pharmaceutical composition and use |
| JPH0623190B2 (ja) | 1985-04-16 | 1994-03-30 | サントリー株式会社 | インヒビタ−活性を有するn−アシルピロリジン誘導体及びその製法並びに用途 |
| DE3684633D1 (de) | 1985-04-16 | 1992-05-07 | Suntory Ltd | Dipeptid-derivate von fettsaeure, verfahren zu ihrer herstellung, heilmittel, das sie enthaelt und anwendung. |
| JPS62114957A (ja) | 1985-11-13 | 1987-05-26 | Suntory Ltd | プロリルエンドペプチダ−ゼ阻害作用を有する新規ピロリジン誘導体およびその製法並びに用途 |
| JPH0714878B2 (ja) | 1985-11-14 | 1995-02-22 | サントリー株式会社 | ピロリジンアミドを有効成分とするプロリルエンドペプチダーゼ阻害剤 |
| JPH0764834B2 (ja) | 1985-11-29 | 1995-07-12 | サントリー株式会社 | プロリルエンドペプチダーゼ阻害活性を有する新規ピロリジンアミド誘導体およびその製法並びに用途 |
| US5198458A (en) | 1986-02-04 | 1993-03-30 | Suntory Limited | Pyrrolidineamide derivatives of acylamino acid and pharmaceutical composition containing the same |
| CA1320734C (en) | 1986-02-04 | 1993-07-27 | Suntory Limited | Pyrrolidineamide derivative of acylamino acid and pharmaceutical composition containing the same |
| CA1334092C (en) | 1986-07-11 | 1995-01-24 | David John Carini | Angiotensin ii receptor blocking imidazoles |
| JPS6386485A (ja) | 1986-09-30 | 1988-04-16 | Agency Of Ind Science & Technol | トンネル型ジヨセフソン接合素子 |
| US5223482A (en) | 1986-11-17 | 1993-06-29 | Scios Nova Inc. | Recombinant Alzheimer's protease inhibitory amyloid protein and method of use |
| JPH08806B2 (ja) | 1986-11-18 | 1996-01-10 | サントリー株式会社 | プロリルエンドペプチダ−ゼ阻害作用を有する新規ピロリジンアミド誘導体 |
| US5254550A (en) | 1986-11-20 | 1993-10-19 | Ono Pharmaceutical Co., Ltd. | Prolinal derivatives and pharmaceutical compositions thereof |
| DE3786229T2 (de) | 1986-11-20 | 1993-10-07 | Ono Pharmaceutical Co | Prolinalderivate. |
| JPS63162672A (ja) | 1986-12-25 | 1988-07-06 | Ono Pharmaceut Co Ltd | 新規なプロリナ−ル誘導体、それらの製造方法およびそれらを含有する抗健忘症剤 |
| DE3783754T2 (de) | 1986-12-29 | 1993-08-05 | Ono Pharmaceutical Co | Prolin-derivate. |
| JPH089591B2 (ja) | 1986-12-29 | 1996-01-31 | 小野薬品工業株式会社 | 新規なプロリナール誘導体 |
| US5262431A (en) | 1986-12-29 | 1993-11-16 | Ono Pharmaceutical Co., Ltd. | Prolinal derivatives and pharmaceutical compositions thereof |
| ES2054712T3 (es) | 1987-02-04 | 1994-08-16 | Ono Pharmaceutical Co | Un procedimiento para la preparacion de los derivados de prolinal. |
| ES2103287T3 (es) | 1987-02-23 | 1997-09-16 | Ono Pharmaceutical Co | Nuevos derivados de tiazolidina. |
| JPS6442465A (en) | 1987-08-07 | 1989-02-14 | Wakunaga Pharma Co Ltd | N-acylprolylpyrrolidine derivative, production and use thereof |
| JP2649237B2 (ja) | 1988-03-07 | 1997-09-03 | キッセイ薬品工業 株式会社 | チアゾリジン誘導体 |
| US4857524A (en) | 1987-08-08 | 1989-08-15 | Kissei Pharmaceutical Co., Ltd. | Thiazolidine compounds and therapeutic method |
| JP2515558B2 (ja) | 1987-09-10 | 1996-07-10 | 株式会社ヤクルト本社 | 新規なペプチドおよびそれを有効成分とする抗健忘症剤 |
| CA1339014C (en) | 1987-10-08 | 1997-03-25 | Ronald E. Majocha | Antibodies to a4 amyloid peptide |
| JPH0742309B2 (ja) | 1987-11-30 | 1995-05-10 | キッセイ薬品工業株式会社 | チアゾリジン誘導体 |
| JPH01250370A (ja) | 1987-12-23 | 1989-10-05 | Zeria Pharmaceut Co Ltd | 新規アミノ酸イミド誘導体、製法ならびに用途 |
| US5053414A (en) | 1988-04-08 | 1991-10-01 | Ono Pharmaceutical Co., Ltd. | Heterocyclic compounds |
| US5328899A (en) | 1988-07-15 | 1994-07-12 | The Salk Institute For Biological Studies | NPY peptide analogs |
| ZA896376B (en) | 1988-08-26 | 1990-05-30 | Merrell Dow Pharma | Neuropeptide y agonists |
| ZA896374B (en) | 1988-08-26 | 1990-05-30 | Merrell Dow Pharma | Neuropeptide y antagonists |
| JP2531989B2 (ja) | 1988-09-14 | 1996-09-04 | 吉富製薬株式会社 | ピリジン化合物 |
| CA2004028C (en) | 1988-12-08 | 1998-09-22 | Motoki Torizuka | Condensed benzene derivative |
| JPH02207070A (ja) | 1989-02-07 | 1990-08-16 | Zeria Pharmaceut Co Ltd | アミノ酸イミド誘導体、それを含有する医薬及び該化合物の製造中間体 |
| WO1990012005A1 (en) | 1989-04-13 | 1990-10-18 | Japan Tobacco Inc. | New amino acid derivatives having prolylendopeptidase inhibitor activity |
| AU5439790A (en) | 1989-04-14 | 1990-11-16 | Research Foundation For Mental Hygiene, Inc. | Cerebrovascular amyloid protein-specific monoclonal antibody sv17-6e10 |
| WO1990012870A1 (en) | 1989-04-14 | 1990-11-01 | Research Foundation For Mental Hygiene, Inc. | Monoclonal antibody to amyloid peptide |
| DE69034103T2 (de) | 1989-06-14 | 2004-07-15 | Smithkline Beecham Corp. | Imidazoalkensäure |
| DE3939797A1 (de) | 1989-12-01 | 1991-06-06 | Basf Ag | Neue vom neuropeptid y abgeleitete peptide |
| US4985560A (en) | 1990-01-12 | 1991-01-15 | American Home Products Corporation | Pyridazino(4,5-b)indolizines |
| DE19675036I2 (de) | 1990-02-19 | 2004-10-21 | Novartis Ag | Acylverbindungen. |
| JPH04211648A (ja) | 1990-07-27 | 1992-08-03 | Nippon Kayaku Co Ltd | ケト酸アミド誘導体 |
| JPH03255080A (ja) | 1990-03-05 | 1991-11-13 | Yoshitomi Pharmaceut Ind Ltd | ベンゼン化合物 |
| NZ237476A (en) | 1990-03-20 | 1994-01-26 | Sanofi Sa | N-substituted heterocyclic compounds and pharmaceutical compositions. |
| US5073549A (en) | 1990-03-22 | 1991-12-17 | Bristol-Myers Squibb Company | BU-4164E - A and B, prolyl endopeptidase inhibitors and their methods of use |
| US4999349A (en) | 1990-03-22 | 1991-03-12 | Bristol-Myers Squibb Co. | BU-4164E - A and B, prolyl endopeptidase inhibitors |
| US5196444A (en) | 1990-04-27 | 1993-03-23 | Takeda Chemical Industries, Ltd. | 1-(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[[2'-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate and compositions and methods of pharmaceutical use thereof |
| WO1991018891A1 (en) | 1990-06-04 | 1991-12-12 | Pfizer Inc. | Aromatic pyrrolidine and thiazolidine amides |
| ATE133407T1 (de) | 1990-06-07 | 1996-02-15 | Zeria Pharm Co Ltd | Neue arylalkanoylaminderivate und diese enthaltendes arzneimittel |
| EP0468469A2 (en) | 1990-07-27 | 1992-01-29 | Japan Tobacco Inc. | Proline derivatives |
| US5506256A (en) | 1990-07-27 | 1996-04-09 | Yoshitomi Pharmaceutical Industries, Ltd. | Proline derivatives possessing prolyl endopeptidase-inhibitory activity |
| JPH05186498A (ja) | 1991-12-27 | 1993-07-27 | Japan Tobacco Inc | プロリン誘導体 |
| JPH04235162A (ja) | 1990-08-09 | 1992-08-24 | Zeria Pharmaceut Co Ltd | 新規コハク酸アミド誘導体およびそれを含有する医薬 |
| JPH04208299A (ja) | 1990-11-30 | 1992-07-29 | Ajinomoto Co Inc | プロリルエンドペプチターゼ阻害ペプチド |
| IE920540A1 (en) | 1991-02-21 | 1992-08-26 | Sankyo Co | 1-biphenylmethylimidazole derivatives, their preparation and¹their therapetuic use |
| US5104880A (en) | 1991-05-01 | 1992-04-14 | Mayo Foundation For Medical Education And Research | Huperzine a analogs as acetylcholinesterase inhibitors |
| WO1992021333A2 (en) | 1991-05-24 | 1992-12-10 | Pharmavene, Inc. | Treatment of drug withdrawal symptoms and drug craving with type b monoamine oxidase inhibitors |
| DE69222142T2 (de) | 1991-06-20 | 1998-04-09 | Snow Brand Milk Products Co Ltd | Neuartige prolyl-endopeptidase-inhibitoren sna-115 und sna-115t, ihre herstellung, und hierbei verwendete stämme |
| DE4121975A1 (de) | 1991-07-03 | 1993-01-07 | Basf Ag | Thermoplastische formmassen auf der basis von polycarbonaten, styrol/acrylnitril-polymerisaten und polyolefinen |
| HUT66324A (en) | 1991-07-29 | 1994-11-28 | Warner Lambert Co | Quinazoline derivatives as acetylcholinesterase inhibitors and pharmaceutical compositions containing them |
| TW226375B (es) | 1991-10-24 | 1994-07-11 | American Home Prod | |
| CA2126212A1 (en) | 1991-12-19 | 1993-07-08 | Albert Tseng | A novel molecule which inhibits neuropeptide tyrosine biological function |
| JPH05301826A (ja) | 1991-12-24 | 1993-11-16 | Snow Brand Milk Prod Co Ltd | プロリルエンドペプチダーゼ阻害剤 |
| JPH05201970A (ja) | 1992-01-24 | 1993-08-10 | Japan Tobacco Inc | 新規プロリン誘導体 |
| JP3318622B2 (ja) | 1992-05-27 | 2002-08-26 | 独立行政法人産業技術総合研究所 | プロリルエンドペプチダーゼ阻害剤 |
| GB9212308D0 (en) | 1992-06-10 | 1992-07-22 | Ici Plc | Therapeutic compositions |
| GB9213215D0 (en) | 1992-06-20 | 1992-08-05 | Wellcome Found | Peptides |
| JPH06116284A (ja) | 1992-10-02 | 1994-04-26 | Yamanouchi Pharmaceut Co Ltd | 新規ペプチド |
| PT664291E (pt) | 1992-10-05 | 2000-11-30 | Ube Industries | Composto de pirimidina |
| US5260286A (en) | 1992-10-16 | 1993-11-09 | Japan Tobacco, Inc. | 2-piperidinecarboxylic acid derivatives useful as NMDA receptor antagonists |
| WO1994012474A1 (en) | 1992-11-20 | 1994-06-09 | Japan Tobacco Inc. | Compound with prolyl endopeptidase inhibitor activity and pharmaceutical use thereof |
| US5354758A (en) | 1992-12-16 | 1994-10-11 | Japan Tobacco Inc. | Benzomorphans useful as NMDA receptor antagonists |
| JPH06192298A (ja) | 1992-12-24 | 1994-07-12 | Mitsui Toatsu Chem Inc | 新規機能性ペプチド |
| DE4326465A1 (de) | 1993-01-20 | 1995-02-09 | Thomae Gmbh Dr K | Aminosäurederivate, diese Verbindungen enthaltende Arzneimittel und Verfahren zu ihrer Herstellung |
| US5750349A (en) | 1993-01-25 | 1998-05-12 | Takeda Chemical Industries Ltd. | Antibodies to β-amyloids or their derivatives and use thereof |
| JPH06234693A (ja) | 1993-02-09 | 1994-08-23 | Snow Brand Milk Prod Co Ltd | 新規イソテトラセノン系物質及びその製造法 |
| FR2701480B1 (fr) | 1993-02-15 | 1995-05-24 | Sanofi Elf | Composés à groupe sulfamoyle et amidino, leur procédé de préparation et les compositions pharmaceutiques les contenant. |
| EP0611769A1 (en) | 1993-02-16 | 1994-08-24 | Merrell Dow Pharmaceuticals Inc. | Silylated acetylcholinesterase inhibitors |
| AU4982293A (en) | 1993-03-02 | 1994-09-26 | Fujisawa Pharmaceutical Co., Ltd. | Novel heterocyclic compound |
| FR2702150B1 (fr) | 1993-03-03 | 1995-04-07 | Rhone Poulenc Rorer Sa | Application de dérivés de 2H-1,2-4-benzothiadiazine-3(4H)-one-1,1-dioxyde comme antagonistes non compétitifs du récepteur NMDA. |
| FR2703050B1 (fr) | 1993-03-24 | 1995-04-28 | Adir | Nouveaux dérivés bicycliques azotés, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent. |
| EP0627400A1 (en) | 1993-06-04 | 1994-12-07 | Merrell Dow Pharmaceuticals Inc. | Aromatic acetylcholinesterase inhibitors |
| EP0707490A1 (en) | 1993-06-18 | 1996-04-24 | University Of Cincinnati | Neuropeptide y antagonists and agonists |
| WO1995001352A1 (fr) | 1993-06-30 | 1995-01-12 | Zeria Pharmaceutical Co., Ltd. | Derive de thiazolidine et medicament le contenant |
| FR2707645B1 (fr) | 1993-07-16 | 1995-08-11 | Rhone Poulenc Rorer Sa | Dérivés d'imidazo[1,2-a]pirazine-4-one, leur préparation et les médicaments les contenant. |
| FR2707643B1 (fr) | 1993-07-16 | 1995-08-11 | Rhone Poulenc Rorer Sa | Dérivés d'imidazo[1,2-a]pyrazine-4-one, leur préparation et les médicaments les contenant. |
| DE69428703T2 (de) | 1993-07-23 | 2002-05-29 | Zaidan Hojin Biseibutsu | Pyrrolidin derivate |
| FR2711993B1 (fr) | 1993-11-05 | 1995-12-01 | Rhone Poulenc Rorer Sa | Médicaments contenant des dérivés de 7H-imidazol[1,2-a]pyrazine-8-one, les nouveaux composés et leur préparation. |
| ES2179093T3 (es) | 1993-12-02 | 2003-01-16 | Merrell Pharma Inc | Inhibidores de la prolilendopeptidasa. |
| IL111785A0 (en) | 1993-12-03 | 1995-01-24 | Ferring Bv | Dp-iv inhibitors and pharmaceutical compositions containing them |
| FR2717812B1 (fr) | 1994-03-28 | 1996-05-10 | Rhone Poulenc Rorer Sa | Indeno[1,2-e]pyrazine-4-ones, leur préparation et les médicaments les contenant. |
| FR2717811B1 (fr) | 1994-03-28 | 1996-04-26 | Rhone Poulenc Rorer Sa | Dérivés d'imidazo[1,2-a]pyrazine-4-one, leur préparation et les médicaments les contenant. |
| FR2717805B1 (fr) | 1994-03-28 | 1996-05-10 | Rhone Poulenc Rorer Sa | Dérivés de 5H-indeno[1,2-b]pyrazine-2,3-dione, leur préparation et les médicaments les contenant . |
| FR2717813B1 (fr) | 1994-03-28 | 1996-05-10 | Rhone Poulenc Rorer Sa | Dérivés d'imidazo[1,2-a]indeno[1,2-e]pyrazine-4-one, leur préparation et les médicaments les contenant . |
| JPH07267988A (ja) | 1994-03-31 | 1995-10-17 | Yamanouchi Pharmaceut Co Ltd | 新規ペプチド |
| GB9410320D0 (en) | 1994-05-24 | 1994-07-13 | Fisons Corp | Novel therapeutic method |
| GB9418443D0 (en) | 1994-09-13 | 1994-11-02 | Pfizer Ltd | Therapeutic agents |
| EP0716854A3 (en) | 1994-10-20 | 1996-08-28 | Lilly Co Eli | Compositions for inhibiting neuropeptide Y |
| US5663192A (en) | 1994-10-20 | 1997-09-02 | Eli Lilly And Company | Heterocyclic neuropeptide Y receptor antagonists |
| US6562862B1 (en) | 1994-10-20 | 2003-05-13 | Eli Lilly And Company | Methods of inhibiting physiological conditions associated with an excess of neuropeptide Y |
| FR2726275B1 (fr) | 1994-11-02 | 1996-12-06 | Rhone Poulenc Rorer Sa | Spiro heterocycle-imidazo(1,2-a)indeno(1,2-e)pyrazine)-4'- ones, leur preparation et les medicaments les contenants |
| IL116584A0 (en) | 1994-12-29 | 1996-03-31 | Res Dev Foundation | Novel flavin adenine dinucleotide analogue inhibitors of monoamine oxidase |
| US5691368A (en) | 1995-01-11 | 1997-11-25 | Hoechst Marion Roussel, Inc. | Substituted oxazolidine calpain and/or cathepsin B inhibitors |
| GB9500601D0 (en) | 1995-01-12 | 1995-03-01 | Wellcome Found | Modified peptides |
| US5786180A (en) | 1995-02-14 | 1998-07-28 | Bayer Corporation | Monoclonal antibody 369.2B specific for β A4 peptide |
| US5552411A (en) | 1995-05-26 | 1996-09-03 | Warner-Lambert Company | Sulfonylquinolines as central nervous system and cardiovascular agents |
| US5635503A (en) | 1995-06-07 | 1997-06-03 | Bristol-Myers Squibb Company | Dihydropyridine npy antagonists: piperazine derivatives |
| US5554621A (en) | 1995-06-07 | 1996-09-10 | Bristol-Myers Squibb Company | Dihydropyridine NPY antagonists: nitrogen heterocyclic derivatives |
| IL117997A0 (en) | 1995-06-07 | 1996-10-31 | Pfizer | Neuropeptide Y1 specific ligands |
| US5668151A (en) | 1995-06-07 | 1997-09-16 | Bristol-Myers Squibb Company | Dihydropyridine NPY antagonists: piperidine derivatives |
| JP3727383B2 (ja) | 1995-07-31 | 2005-12-14 | 月桂冠株式会社 | プロリルエンドペプチダーゼ阻害剤 |
| PL320010A1 (en) | 1995-09-01 | 1997-09-01 | Lilly Co Eli | Indolyllic antagonists of neuropeptide y receptors |
| AU6966696A (en) | 1995-10-05 | 1997-04-28 | Warner-Lambert Company | Method for treating and preventing inflammation and atherosclerosis |
| ES2100129B1 (es) | 1995-10-11 | 1998-02-16 | Medichem Sa | Nuevos compuestos aminopiridinicos policiclicos inhibidores de acetilcolinesterasa, procedimiento para su preparacion y su utilizacion. |
| DE19544685A1 (de) | 1995-11-30 | 1997-06-05 | Thomae Gmbh Dr K | Aminosäurederivate, diese Verbindungen enthaltende Arzneimittel und Verfahren zu ihrer Herstellung |
| DE19544687A1 (de) | 1995-11-30 | 1997-06-05 | Thomae Gmbh Dr K | Aminosäurederivate, diese Verbindungen enthaltende Arzneimittel und Verfahren zu ihrer Herstellung |
| DE19544686A1 (de) | 1995-11-30 | 1997-06-05 | Thomae Gmbh Dr K | Aminosäurederivate, diese Verbindungen enthaltende Arzneimittel und Verfahren zu ihrer Herstellung |
| AU7692896A (en) | 1995-12-01 | 1997-06-27 | Novartis Ag | Quinazolin-2,4-diazirines as NPY receptor antagonist |
| WO1997020821A1 (en) | 1995-12-01 | 1997-06-12 | Novartis Ag | Heteroaryl derivatives |
| WO1997020820A1 (en) | 1995-12-01 | 1997-06-12 | Novartis Ag | Heteroaryl compounds |
| WO1997020823A2 (en) | 1995-12-01 | 1997-06-12 | Novartis Ag | 2-amino quinazoline derivatives as npy receptor antagonists |
| WO1997019682A1 (en) | 1995-12-01 | 1997-06-05 | Synaptic Pharmaceutical Corporation | Aryl sulfonamide and sulfamide derivatives and uses thereof |
| JPH09157253A (ja) | 1995-12-12 | 1997-06-17 | Yamanouchi Pharmaceut Co Ltd | 新規アミノ酸誘導体 |
| ZA9610745B (en) | 1995-12-22 | 1997-06-24 | Warner Lambert Co | 4-Subsituted piperidine analogs and their use as subtype selective nmda receptor antagonists |
| ZA9610741B (en) | 1995-12-22 | 1997-06-24 | Warner Lambert Co | 4-Substituted piperidine analogs and their use as subtype selective nmda receptor antagonists |
| ZA9610736B (en) | 1995-12-22 | 1997-06-27 | Warner Lambert Co | 2-Substituted piperidine analogs and their use as subtypeselective nmda receptor antagonists |
| CA2242579A1 (en) | 1996-01-09 | 1997-07-17 | Eli Lilly And Company | Benzimidzolyl neuropeptide y receptor antagonists |
| GB9605027D0 (en) | 1996-03-09 | 1996-05-08 | Pfizer Ltd | Quinoxalinediones |
| US5662723A (en) | 1996-03-22 | 1997-09-02 | Libbey Glass Inc. | Apparatus and method for forming a decorative pattern on glassware having an edge |
| PT892796E (pt) | 1996-04-12 | 2002-03-28 | Aventis Pharma Inc | Derivados de isatina como inibidores de acetilcolinesterase e analgesicos |
| DE19616486C5 (de) | 1996-04-25 | 2016-06-30 | Royalty Pharma Collection Trust | Verfahren zur Senkung des Blutglukosespiegels in Säugern |
| EP1007073A4 (en) | 1996-06-04 | 2002-03-27 | Synaptic Pharma Corp | METHODS OF MODIFYING FOOD BEHAVIOR, COMPOUNDS USEFUL IN SAID METHODS, AND DNA ENCODING A HYPOTHALAMIC NEUROPEPTIDE Y / PEPTIDE YY Y5 RECEPTOR |
| CA2259010A1 (en) | 1996-07-05 | 1998-01-15 | The Wwk Trust | Compositions for the treatment of peripheral neuropathies containing antidepressants and/or monoamine oxidase inhibitors and/or vitamin b12 and/or precursors or inducers of a neurotransmitter |
| WO1998003492A1 (en) | 1996-07-23 | 1998-01-29 | Neurogen Corporation | Certain substituted benzylamine derivatives; a new class of neuropeptide y1 specific ligands |
| EP0918761B1 (en) | 1996-07-23 | 2003-05-02 | Neurogen Corporation | Certain amido- and amino-substituted benzylamine derivatives; a new class of neuropeptite y1 specific ligands |
| JP2000515151A (ja) | 1996-07-23 | 2000-11-14 | ニューロジェン・コーポレーション | 特定の置換ベンジルアミン誘導体;新規クラスの神経ペプチド―y1特異的リガンド |
| AU3967297A (en) | 1996-08-01 | 1998-02-25 | Cocensys, Inc. | Use of gaba and nmda receptor ligands for the treatment of migraine headache |
| JP2000516611A (ja) | 1996-08-14 | 2000-12-12 | ワーナー―ランバート・コンパニー | Mcp―1アンタゴニストとしての2―フェニルベンズイミダゾール誘導体 |
| ES2176776T3 (es) | 1996-08-23 | 2002-12-01 | Agouron Pharma | Ligandos del neuropeptido-y. |
| JP3880664B2 (ja) | 1996-09-04 | 2007-02-14 | 月桂冠株式会社 | プロリルエンドペプチダーゼ阻害ペプチド |
| AU4424697A (en) | 1996-09-11 | 1998-04-02 | Government Of The United States Of America, As Represented By The Secretary Of The Department Of Health And Human Services, The | The use of functional n-methyl-d-aspartate antagonists to ameliorate or prevent aminoglycoside-induced ototoxicity |
| WO1998015647A1 (en) | 1996-10-08 | 1998-04-16 | Novartis Ag | Modulation of apoptosis |
| FR2754709B1 (fr) | 1996-10-23 | 1999-03-05 | Sanofi Sa | Composition cosmetique contenant un antagoniste des recepteurs du neuropeptide gamma et alpha 2 antagonistes susceptibles d'etre incorpores dans une telle composition |
| TW492957B (en) | 1996-11-07 | 2002-07-01 | Novartis Ag | N-substituted 2-cyanopyrrolidnes |
| US6011155A (en) | 1996-11-07 | 2000-01-04 | Novartis Ag | N-(substituted glycyl)-2-cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| AU5716898A (en) | 1997-01-08 | 1998-08-03 | Warner-Lambert Company | Acetylcholinesterase inhibitors in combination with muscarinic agonists for the treatment of alzheimer's disease |
| US20030068316A1 (en) | 1997-02-05 | 2003-04-10 | Klein William L. | Anti-ADDL antibodies and uses thereof |
| JP2894445B2 (ja) | 1997-02-12 | 1999-05-24 | 日本たばこ産業株式会社 | Cetp活性阻害剤として有効な化合物 |
| CA2284001A1 (en) | 1997-03-13 | 1998-09-17 | Keith Tipton | Cytoprotective agents comprising monoamine oxidase inhibitors |
| ATE402717T1 (de) | 1997-04-09 | 2008-08-15 | Intellect Neurosciences Inc | Für die termini des beta-amyloids spezifische, rekombinante antikörper, dafür kodierende dna und verfahren zu ihrer verwendung |
| US8173127B2 (en) | 1997-04-09 | 2012-05-08 | Intellect Neurosciences, Inc. | Specific antibodies to amyloid beta peptide, pharmaceutical compositions and methods of use thereof |
| CA2286660A1 (en) | 1997-04-16 | 1998-10-22 | Arqule, Inc. | Synthesis and use of .alpha.-ketoamide derivatives and arrays |
| CA2289190A1 (en) | 1997-05-07 | 1998-11-12 | Algos Pharmaceutical Corporation | Composition and method combining an antidepressant with an nmda receptor antagonist, for treating neuropathic pain |
| WO1998050075A1 (en) | 1997-05-07 | 1998-11-12 | Algos Pharmaceutical Corporation | Cox-2 inhibitors in combination with nmda-blockers for treating pain |
| AU724974B2 (en) | 1997-06-30 | 2000-10-05 | Merz Pharma Gmbh & Co. Kgaa | 1-amino-alkylcyclohexane NMDA receptor antagonists |
| GB9716657D0 (en) | 1997-08-07 | 1997-10-15 | Zeneca Ltd | Chemical compounds |
| GB9716879D0 (en) | 1997-08-08 | 1997-10-15 | Shire Int Licensing Bv | Treatment of attention deficit disorders |
| AU7696098A (en) | 1997-08-11 | 1999-03-01 | Algos Pharmaceutical Corporation | Substance p inhibitors in combination with nmda-blockers for treating pain |
| SE9703376D0 (sv) | 1997-09-18 | 1997-09-18 | Astra Ab | A new combination |
| SE9703414D0 (sv) | 1997-09-23 | 1997-09-23 | Astra Ab | New compounds |
| US7964192B1 (en) | 1997-12-02 | 2011-06-21 | Janssen Alzheimer Immunotherapy | Prevention and treatment of amyloidgenic disease |
| US7179892B2 (en) | 2000-12-06 | 2007-02-20 | Neuralab Limited | Humanized antibodies that recognize beta amyloid peptide |
| TWI239847B (en) | 1997-12-02 | 2005-09-21 | Elan Pharm Inc | N-terminal fragment of Abeta peptide and an adjuvant for preventing and treating amyloidogenic disease |
| EP2574336A1 (en) | 1998-02-02 | 2013-04-03 | Trustees Of Tufts College | Use of dipeptidylpeptidase inhibitors to regulate glucose metabolism |
| AU3034299A (en) | 1998-03-09 | 1999-09-27 | Fondatech Benelux N.V. | Serine peptidase modulators |
| US6007841A (en) | 1998-03-13 | 1999-12-28 | Algos Pharmaceutical Corporation | Analgesic composition and method for treating pain |
| GB9805561D0 (en) | 1998-03-16 | 1998-05-13 | Merck Sharp & Dohme | A combination of therapeutic agents |
| US6541208B1 (en) | 1998-03-17 | 2003-04-01 | University Of Maryland Biotechnology Institute | Diagnostic method for distinguishing HIV-associated dementia from other forms of dementia |
| DE19812331A1 (de) | 1998-03-20 | 1999-09-23 | Merck Patent Gmbh | Piperidinderivate |
| US6054451A (en) | 1998-04-21 | 2000-04-25 | Algos Pharmaceutical Corporation | Analgesic composition and method for alleviating pain |
| WO1999057119A1 (en) | 1998-05-04 | 1999-11-11 | Neotherapeutics, Inc. | Novel dopamine-like 9-substituted hypoxanthine and methods of use |
| US6303617B1 (en) | 1998-05-04 | 2001-10-16 | Neotherapeutics, Inc. | Serotonin-like 9-substituted hypoxanthine and methods of use |
| JP2002515235A (ja) | 1998-05-21 | 2002-05-28 | ザ ユニヴァーシティ オブ テネシー リサーチ コーポレイション | 坑アミロイド抗体を用いるアミロイドの除去方法 |
| DE19823831A1 (de) | 1998-05-28 | 1999-12-02 | Probiodrug Ges Fuer Arzneim | Neue pharmazeutische Verwendung von Isoleucyl Thiazolidid und seinen Salzen |
| DE19828114A1 (de) | 1998-06-24 | 2000-01-27 | Probiodrug Ges Fuer Arzneim | Produgs instabiler Inhibitoren der Dipeptidyl Peptidase IV |
| DE19828113A1 (de) | 1998-06-24 | 2000-01-05 | Probiodrug Ges Fuer Arzneim | Prodrugs von Inhibitoren der Dipeptidyl Peptidase IV |
| PE20000728A1 (es) | 1998-06-26 | 2000-08-21 | Cocensys Inc | Heterociclos 4-bencil piperidina alquilsulfoxido y su uso como antagonistas receptores subtipo-selectivo nmda |
| US6262081B1 (en) | 1998-07-10 | 2001-07-17 | Dupont Pharmaceuticals Company | Composition for and method of treating neurological disorders |
| AU5027299A (en) | 1998-07-31 | 2000-02-28 | Novo Nordisk A/S | Use of glp-1 and analogues for preventing type ii diabetes |
| DE19834591A1 (de) | 1998-07-31 | 2000-02-03 | Probiodrug Ges Fuer Arzneim | Verfahren zur Steigerung des Blutglukosespiegels in Säugern |
| US6218383B1 (en) | 1998-08-07 | 2001-04-17 | Targacept, Inc. | Pharmaceutical compositions for the prevention and treatment of central nervous system disorders |
| IL125809A (en) | 1998-08-17 | 2005-08-31 | Finetech Lab Ltd | Process and intermediates for production of donepezil and related compounds |
| IT1304904B1 (it) | 1998-09-11 | 2001-04-05 | Eisai Co Ltd | Derivati anticolinesterasici per il trattamento delle sindromidolorose funzionali e/o organiche |
| EP1121131A2 (en) | 1998-10-16 | 2001-08-08 | Janssen Pharmaceutica N.V. | Atypical antipsychotic in combination with acetylcholinesterase inhibitor for improving cognition |
| AU1408699A (en) | 1998-11-12 | 2000-06-05 | Algos Pharmaceutical Corporation | Cox-2 inhibitors in combination with nmda-blockers for treating pain |
| EP2311441A1 (en) | 1998-11-23 | 2011-04-20 | Bonnie M. Davis | Dosage formulations for acetylcholinesterase inhibitors |
| WO2000030674A1 (en) | 1998-11-26 | 2000-06-02 | Ferring Bv | Neuropeptide y y4 agents in the treatment of reproductive disorders |
| CA2354035A1 (en) | 1998-12-11 | 2000-06-15 | Bonnie Davis | Use of acetylcholinesterase inhibitors on the modulation of the hypothalamic-pituitary-gonadal axis |
| GB9902461D0 (en) | 1999-02-05 | 1999-03-24 | Zeneca Ltd | Chemical compounds |
| GB9902459D0 (en) | 1999-02-05 | 1999-03-24 | Zeneca Ltd | Chemical compounds |
| GB9902453D0 (en) | 1999-02-05 | 1999-03-24 | Zeneca Ltd | Chemical compounds |
| GB9902455D0 (en) | 1999-02-05 | 1999-03-24 | Zeneca Ltd | Chemical compounds |
| GB9902452D0 (en) | 1999-02-05 | 1999-03-24 | Zeneca Ltd | Chemical compounds |
| US6495587B1 (en) | 1999-03-23 | 2002-12-17 | Sumitomo Pharmaceuticals Company, Limited | Tricyclic indole-2-carboxylic acid compound used as NMDA receptor antagonist |
| CA2363984A1 (en) | 1999-04-15 | 2000-10-26 | Merck Frosst Canada & Co. | Antibodies that recognize app cleaved by caspases and methods of use |
| US6121311A (en) | 1999-04-28 | 2000-09-19 | Japan Tobacco Inc. | Method for treating cocainism |
| CA2373035A1 (en) | 1999-05-05 | 2000-11-16 | Scott Dax | 3a,4,5,9b-tetrahydro-1h-benz[e]indol-2-yl amine-derived neuropeptide y receptors ligands useful in the treatment of obesity and other disorders |
| JP3148739B2 (ja) | 1999-05-19 | 2001-03-26 | ドーマー株式会社 | プロリルエンドペプチダーゼ阻害剤 |
| US6582945B1 (en) | 1999-06-16 | 2003-06-24 | Boston Biomedical Research Institute | Immunological control of β-amyloid levels in vivo |
| US6110949A (en) | 1999-06-24 | 2000-08-29 | Novartis Ag | N-(substituted glycyl)-4-cyanothiazolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| US6107317A (en) | 1999-06-24 | 2000-08-22 | Novartis Ag | N-(substituted glycyl)-thiazolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| US6172081B1 (en) | 1999-06-24 | 2001-01-09 | Novartis Ag | Tetrahydroisoquinoline 3-carboxamide derivatives |
| US6524616B1 (en) | 1999-06-25 | 2003-02-25 | Wake Forest University Health Services | Compositions and methods for treating or preventing neurodegeneration and cognitive decline and dysfunction associated with alzheimer's disease, aging, other dementia related disorders and estrogen deficiency related conditions |
| US6316449B1 (en) | 1999-07-08 | 2001-11-13 | Millennium Pharmaceuticals, Inc. | Chemokine receptor antagonists |
| DE19936521A1 (de) | 1999-08-06 | 2001-02-15 | Gruenenthal Gmbh | Substituierte Pyrrolidin-2,3,4-trion-3-oxim-Derivate |
| DE19936719A1 (de) | 1999-08-06 | 2001-02-15 | Gruenenthal Gmbh | Substituierte 1,5-Dihydropyrrol-2-on-Derivate |
| WO2001012598A2 (en) | 1999-08-13 | 2001-02-22 | The Trustees Of Columbia University In The City Of New York | METHODS OF INHIBITING BINDING OF β-SHEET FIBRIL TO RAGE AND CONSEQUENCES THEREOF |
| EP1078632A1 (en) | 1999-08-16 | 2001-02-28 | Sanofi-Synthelabo | Use of monoamine oxydase inhibitors for the manufacture of drugs intended for the treatment of obesity |
| DE19940130A1 (de) | 1999-08-24 | 2001-03-01 | Probiodrug Ges Fuer Arzneim | Neue Effektoren der Dipeptidyl Peptidase IV zur topischen Anwendung |
| TWI292316B (en) | 1999-10-11 | 2008-01-11 | Sod Conseils Rech Applic | Pharmaceutical composition of thiazole derivatives intended to inhibit mao and/or lipidic peroxidation and/or to act as modulators of sodium channels and the use thereof |
| UA72558C2 (uk) | 1999-11-01 | 2005-03-15 | Мерц Фарма Гмбх Унд Ко. Кгаа | 1-аміноалкілциклогексанові антагоністи рецептора nmda |
| WO2001034594A1 (en) | 1999-11-12 | 2001-05-17 | Guilford Pharmaceuticals, Inc. | Dipeptidyl peptidase iv inhibitors and methods of making and using dipeptidyl peptidase iv inhibitors |
| US20020094335A1 (en) | 1999-11-29 | 2002-07-18 | Robert Chalifour | Vaccine for the prevention and treatment of alzheimer's and amyloid related diseases |
| GB9928330D0 (en) | 1999-11-30 | 2000-01-26 | Ferring Bv | Novel antidiabetic agents |
| EP1254113A1 (en) | 2000-01-24 | 2002-11-06 | Novo Nordisk A/S | N-substituted 2-cyanopyroles and -pyrrolines which are inhibitors of the enzyme dpp-iv |
| JP2003520266A (ja) | 2000-01-24 | 2003-07-02 | メルク シャープ エンド ドーム リミテッド | γ−セクレターゼ阻害薬 |
| CA2399080C (en) | 2000-02-03 | 2013-05-21 | Millennium Pharmaceuticals, Inc. | Humanized anti-ccr2 antibodies and methods of use therefor |
| KR100767146B1 (ko) | 2000-02-24 | 2007-10-15 | 워싱톤 유니버시티 | Aβ 펩티드를 격리시키는 인간화 항체 |
| GB0005251D0 (en) | 2000-03-03 | 2000-04-26 | Merck Sharp & Dohme | Therapeutic compounds |
| US20030144255A1 (en) | 2000-03-06 | 2003-07-31 | Bain Allen I | Compositions for prevention and treatment of dementia |
| US6395767B2 (en) | 2000-03-10 | 2002-05-28 | Bristol-Myers Squibb Company | Cyclopropyl-fused pyrrolidine-based inhibitors of dipeptidyl peptidase IV and method |
| US6992081B2 (en) | 2000-03-23 | 2006-01-31 | Elan Pharmaceuticals, Inc. | Compounds to treat Alzheimer's disease |
| US6737420B2 (en) | 2000-03-23 | 2004-05-18 | Elan Pharmaceuticals, Inc. | Compounds to treat Alzheimer's disease |
| GB0008710D0 (en) | 2000-04-07 | 2000-05-31 | Merck Sharp & Dohme | Therapeutic compounds |
| EP1285656A4 (en) | 2000-04-13 | 2006-07-12 | Eisai Co Ltd | ACETYLCHOLINESTERASE HEMMER CONTAINING 1-BENZYLPYRIDINIUM SALT |
| GB0010188D0 (en) | 2000-04-26 | 2000-06-14 | Ferring Bv | Inhibitors of dipeptidyl peptidase IV |
| BR0110247A (pt) | 2000-04-26 | 2003-03-05 | Warner Lambert Co | Derivado de ciclohexilamina como antagonistas de receptor de nmda seletivos em subtipo |
| GB0010183D0 (en) | 2000-04-26 | 2000-06-14 | Ferring Bv | Inhibitors of dipeptidyl peptidase IV |
| US20010036949A1 (en) | 2000-05-09 | 2001-11-01 | Coe Jotham Wadsworth | Pharmaceutical composition and method of treatment of diseases of cognitive dysfunction in a mammal |
| AU2001261487A1 (en) | 2000-06-01 | 2001-12-11 | Warner Lambert Company | Cyclohexylamine derivatives as subtype selective nmda receptor antagonists |
| JP2003535851A (ja) | 2000-06-06 | 2003-12-02 | ワーナー−ランバート・カンパニー、リミテッド、ライアビリティ、カンパニー | 二環式シクロヘキシルアミン類およびそれらのnmda受容体アンタゴニストとしての使用 |
| NZ522349A (en) | 2000-06-22 | 2004-06-25 | Pharmos Corp | Non-psychotropic cannabinoids that afford neuroprotection by exhibiting anti-inflammatory and/or antioxidative and glutamate-receptor blocking mechanisms of action |
| GB0015488D0 (en) | 2000-06-23 | 2000-08-16 | Merck Sharp & Dohme | Therapeutic agents |
| US6713276B2 (en) | 2000-06-28 | 2004-03-30 | Scios, Inc. | Modulation of Aβ levels by β-secretase BACE2 |
| US6686449B2 (en) | 2000-06-30 | 2004-02-03 | Pharmacia & Upjohn Company | Mutant presenilin 1 polypeptides |
| WO2002002518A2 (en) | 2000-06-30 | 2002-01-10 | Elan Pharmaceuticals, Inc. | Compounds to treat alzheimer's disease |
| AU2001268958B2 (en) | 2000-07-04 | 2006-03-09 | Novo Nordisk A/S | Heterocyclic compounds, which are inhibitors of the enzyme dpp-iv |
| GB0016681D0 (en) | 2000-07-06 | 2000-08-23 | Merck Sharp & Dohme | Therapeutic compounds |
| US6556971B1 (en) | 2000-09-01 | 2003-04-29 | Snap-On Technologies, Inc. | Computer-implemented speech recognition system training |
| AU2001293056A1 (en) | 2000-09-25 | 2002-04-08 | Motorwiz, Inc. | Model-based machine diagnostics and prognostics using theory of noise and communications |
| US20020151591A1 (en) | 2000-10-17 | 2002-10-17 | Anabella Villalobos | Combination use of acetylcholinesterase inhibitors and GABAa inverse agonists for the treatment of cognitive disorders |
| HU227197B1 (en) | 2000-10-24 | 2010-10-28 | Richter Gedeon Nyrt | Nmda receptor antagonist carboxylic acid amide derivatives and pharmaceutical compositions containing them |
| JP3880051B2 (ja) | 2000-11-02 | 2007-02-14 | メルク シャープ エンド ドーム リミテッド | γ−セクレターゼ阻害剤としてのスルファミド |
| US20020150948A1 (en) | 2000-11-03 | 2002-10-17 | Gerardo Castillo | Antibody PTI-HS7 for treatment of alzheimer's disease and other amyloidoses and parkinson's disease |
| TWI243162B (en) | 2000-11-10 | 2005-11-11 | Taisho Pharmaceutical Co Ltd | Cyanopyrrolidine derivatives |
| PE20020574A1 (es) | 2000-12-06 | 2002-07-02 | Wyeth Corp | Anticuerpos humanizados que reconocen el peptido amiloideo beta |
| US6495335B2 (en) | 2000-12-07 | 2002-12-17 | Mario Chojkier | Compositions and methods for diagnosing alzheimer's disease |
| US6670364B2 (en) | 2001-01-31 | 2003-12-30 | Telik, Inc. | Antagonists of MCP-1 function and methods of use thereof |
| TWI245761B (en) | 2001-03-01 | 2005-12-21 | Telik Inc | Antagonists of MCP-1 function and methods of use thereof |
| EP1436258A4 (en) | 2001-03-08 | 2005-03-23 | Univ Emory | ANTAGONISTS OF THE NMDA RECEPTOR DEPENDENT OF PH |
| US6649196B2 (en) | 2001-03-12 | 2003-11-18 | Mayo Foundation For Medical Education And Research | Methods of reducing β-amyloid polypeptides |
| US6815175B2 (en) | 2001-03-16 | 2004-11-09 | Cornell Research Foundation, Inc. | Anti-amyloid peptide antibody based diagnosis and treatment of a neurological disease or disorder |
| US6677365B2 (en) | 2001-04-03 | 2004-01-13 | Telik, Inc. | Antagonists of MCP-1 function and methods of use thereof |
| US6573287B2 (en) | 2001-04-12 | 2003-06-03 | Bristo-Myers Squibb Company | 2,1-oxazoline and 1,2-pyrazoline-based inhibitors of dipeptidyl peptidase IV and method |
| EP1385545B1 (en) | 2001-04-30 | 2009-01-07 | Eli Lilly And Company | Humanized antibodies recognizing the beta-amyloid peptide |
| WO2002088307A2 (en) | 2001-04-30 | 2002-11-07 | Eli Lilly And Company | Humanized antibodies |
| FR2824825B1 (fr) | 2001-05-15 | 2005-05-06 | Servier Lab | Nouveaux derives d'alpha-amino-acides, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| SI20922A (sl) | 2001-05-18 | 2002-12-31 | Krka Tovarna Zdravil, D.D., Novo Mesto | Monoklonsko protitelo, ki nevtralizira aktivnost katepsina B, in njegove uporabe |
| US6562783B2 (en) | 2001-05-30 | 2003-05-13 | Neurologic, Inc. | Phosphinylmethyl and phosphorylmethyl succinic and glutauric acid analogs as β-secretase inhibitors |
| ES2307764T3 (es) | 2001-06-01 | 2008-12-01 | Elan Pharmaceuticals, Inc. | Derivados de hidroxialquilaminas como inhibidores de la beta-secretasa y su uso para el tratamiento de la enfermedad de alzheimer y enfermedades similares. |
| CA2450475A1 (en) | 2001-06-20 | 2003-01-03 | Linda Brockunier | Dipeptidyl peptidase inhibitors for the treatment of diabetes |
| WO2003000180A2 (en) | 2001-06-20 | 2003-01-03 | Merck & Co., Inc. | Dipeptidyl peptidase inhibitors for the treatment of diabetes |
| GB0115517D0 (en) | 2001-06-25 | 2001-08-15 | Ferring Bv | Novel antidiabetic agents |
| WO2003072556A1 (en) | 2001-06-27 | 2003-09-04 | Prosidion Ltd. | Glutaminyl based dpiv inhibitors |
| WO2003002553A2 (en) | 2001-06-27 | 2003-01-09 | Smithkline Beecham Corporation | Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
| DE10154689A1 (de) | 2001-11-09 | 2003-05-22 | Probiodrug Ag | Substituierte Aminoketonverbindungen |
| DE60223920T2 (de) | 2001-06-27 | 2008-11-13 | Smithkline Beecham Corp. | Pyrrolidine als dipeptidyl-peptidase-inhibitoren |
| CN1990469A (zh) | 2001-06-27 | 2007-07-04 | 史密丝克莱恩比彻姆公司 | 作为二肽酶抑制剂的氟代吡咯烷 |
| BR0210721A (pt) | 2001-06-27 | 2004-07-20 | Elan Pharm Inc | Composto, sal ou éster farmaceuticamente aceitável, método para fabricar um composto, e, método para tratar um paciente que tem, ou para evitar que o paciente adquira uma doença ou condição |
| DE10150203A1 (de) | 2001-10-12 | 2003-04-17 | Probiodrug Ag | Peptidylketone als Inhibitoren der DPIV |
| WO2003002593A2 (en) | 2001-06-27 | 2003-01-09 | Probiodrug Ag | Peptide structures useful for competitive modulation of dipeptidyl peptidase iv catalysis |
| ATE388951T1 (de) | 2001-07-03 | 2008-03-15 | Novo Nordisk As | Dpp-iv-inhibierende purin-derivative zur behandlung von diabetes |
| UA74912C2 (en) | 2001-07-06 | 2006-02-15 | Merck & Co Inc | Beta-aminotetrahydroimidazo-(1,2-a)-pyrazines and tetratriazolo-(4,3-a)-pyrazines as inhibitors of dipeptylpeptidase for the treatment or prevention of diabetes |
| KR100890676B1 (ko) | 2001-07-24 | 2009-03-26 | 리히데 게데온 베기에스제티 기아르 알티 | 신규한 카르복시산 아미드 화합물 |
| US20040234990A1 (en) | 2001-07-31 | 2004-11-25 | Hiroto Komano | Method of screening alzheimer's disease-associated gene |
| RU2004106540A (ru) | 2001-08-03 | 2005-07-27 | Шеринг Корпорейшн (US) | Новые ингибиторы гамма-секретазы |
| DE60226036T9 (de) | 2001-08-03 | 2016-09-29 | Medical & Biological Laboratories Co., Ltd. | ANTIKÖRPER, DER DAS GM1-GANGLIOSID-GEBUNDENE AMYLOID-b-PROTEIN ERKENNT, UND DNA, DIE FÜR DIESEN ANTIKÖRPER CODIERT |
| EP1411944A1 (en) | 2001-08-03 | 2004-04-28 | Schering Corporation | Sulfonamide derivatives as gamma secretase inhibitors |
| EP1519740A4 (en) | 2001-08-17 | 2005-11-09 | Lilly Co Eli | FASTER IMPROVEMENT OF COGNITION IN DISEASES ASSOCIATED WITH A-BETA |
| JP2005503789A (ja) | 2001-08-17 | 2005-02-10 | イーライ・リリー・アンド・カンパニー | 抗Aβ抗体 |
| CA2459146A1 (en) | 2001-08-30 | 2003-03-13 | Ortho-Mcneil Pharmaceutical, Inc. | Treatment of dementia and memory disorders with anticonvulsants and acetylcholinesterase inhibitors |
| KR20040033048A (ko) | 2001-09-14 | 2004-04-17 | 미츠비시 웰파마 가부시키가이샤 | 티아졸리딘 유도체 및 이의 약학적 용도 |
| EP1463727A2 (en) | 2001-09-19 | 2004-10-06 | Novo Nordisk A/S | Heterocyclic compounds that are inhibitors of the enzyme dpp-iv |
| GB0125446D0 (en) | 2001-10-23 | 2001-12-12 | Ferring Bv | Novel anti-diabetic agents |
| GB0125445D0 (en) | 2001-10-23 | 2001-12-12 | Ferring Bv | Protease Inhibitors |
| CA2464736A1 (en) | 2001-10-23 | 2003-05-15 | Oklahoma Medical Research Foundation | Beta-secretase inhibitors and methods of use |
| US6861440B2 (en) | 2001-10-26 | 2005-03-01 | Hoffmann-La Roche Inc. | DPP IV inhibitors |
| WO2003039467A2 (en) | 2001-11-02 | 2003-05-15 | Diagenics International Corporation | Monoclonal antibodies specific for beta-amyloid. |
| EP1447095B1 (en) | 2001-11-02 | 2012-06-13 | Kensuke Egashira | Preventives and/or remedies for post-transplant arteriosclerosis as rejection of organ transplant |
| WO2003040183A2 (en) | 2001-11-09 | 2003-05-15 | The Genetics Company, Inc | Compounds for the diagnosis/prevention/treatment of alzheimer's disease |
| IL162079A0 (en) | 2001-11-19 | 2005-11-20 | Upjohn Co | 3,4-Disubstituted, 3,5-disubstituted and substituted and 3,4,5-substituted piperidines and piperazines |
| WO2003045128A2 (en) | 2001-11-21 | 2003-06-05 | New York University | SYNTHETIC IMMUNOGENIC BUT NON-DEPOSIT-FORMING POLYPEPTIDES AND PEPTIDES HOMOLOGOUS TO AMYLOID β, PRION PROTEIN, AMYLIN, α-SYNUCLEIN, OR POLYGLUTAMINE REPEATS FOR INDUCTION OF AN IMMUNE RESPONSE THERETO |
| CA2468192A1 (en) | 2001-11-26 | 2003-06-05 | Trustees Of Tufts College | Peptidomimetic inhibitors of post-proline cleaving enzymes |
| AU2002354433A1 (en) | 2001-12-06 | 2003-06-17 | Japan As Represented By Secretary Of Chubu National Hospital | Alzheimer's disease-associated gene and protein and use thereof |
| US20050227941A1 (en) | 2001-12-17 | 2005-10-13 | Karen Duff | Sequestration of ass in the periphery in the absence of immunomodulating agent as a therapeutic approach for the treatment or prevention of beta-amyloid related diseases |
| RU2004121898A (ru) | 2001-12-19 | 2006-01-20 | Атеродженикс, Инк. (Us) | Производные халкона и их применение для лечения заболеваний |
| EP1467755A1 (en) | 2001-12-21 | 2004-10-20 | Antigenics Inc. | Compositions comprising immunoreactive reagents and saponins, and methods of use thereof |
| AU2002360732A1 (en) | 2001-12-26 | 2003-07-24 | Guilford Pharmaceuticals | Change inhibitors of dipeptidyl peptidase iv |
| US6727261B2 (en) | 2001-12-27 | 2004-04-27 | Hoffman-La Roche Inc. | Pyrido[2,1-A]Isoquinoline derivatives |
| WO2003057204A2 (en) | 2002-01-08 | 2003-07-17 | Nordic Bioscience A/S | Modulation of iamt (pimt or pcmt) in immune system |
| CA2473441A1 (en) | 2002-01-18 | 2003-07-24 | The Genetics Company Inc. | Beta-secretase inhibitors |
| WO2003063760A2 (en) | 2002-01-31 | 2003-08-07 | Tel Aviv University Future Technology Development L.P. | Peptides antibodies directed thereagainst and methods using same for diagnosing and treating amyloid-associated diseases |
| US20040171614A1 (en) | 2002-02-06 | 2004-09-02 | Schering-Plough Corporation | Novel gamma secretase inhibitors |
| TW200302717A (en) | 2002-02-06 | 2003-08-16 | Schering Corp | Novel gamma secretase inhibitors |
| CA2474578C (en) | 2002-02-13 | 2009-08-25 | F. Hoffmann-La Roche Ag | Novel pyridin- and pyrimidin-derivatives |
| AU2003206834B2 (en) | 2002-02-13 | 2006-10-19 | F. Hoffmann-La Roche Ag | Novel pyridine- and quinoline-derivatives |
| AR038568A1 (es) | 2002-02-20 | 2005-01-19 | Hoffmann La Roche | Anticuerpos anti-a beta y su uso |
| HUP0200849A2 (hu) | 2002-03-06 | 2004-08-30 | Sanofi-Synthelabo | N-aminoacetil-2-ciano-pirrolidin-származékok, e vegyületeket tartalmazó gyógyszerkészítmények és eljárás előállításukra |
| MY139983A (en) | 2002-03-12 | 2009-11-30 | Janssen Alzheimer Immunotherap | Humanized antibodies that recognize beta amyloid peptide |
| US7307164B2 (en) | 2002-03-25 | 2007-12-11 | Merck & Co., Inc. | β-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2003082820A1 (fr) | 2002-03-29 | 2003-10-09 | Eisai Co., Ltd. | Derive de (1-indanone)-(1,2,3,6-tetrahydropyridine) |
| WO2003086310A2 (en) | 2002-04-12 | 2003-10-23 | Ramot At Tel Aviv University Ltd. | Prevention of brain inflammation as a result of induced autoimmune response |
| CN100360555C (zh) | 2002-04-19 | 2008-01-09 | 多伦多大学董事局 | 治疗阿尔茨海默氏病的免疫学方法及组合物 |
| DE60329995D1 (de) | 2002-04-24 | 2009-12-24 | Hiroshi Mori | Gamma-sekretase-inhibitoren |
| MXPA04010552A (es) | 2002-04-26 | 2005-01-25 | Schering Corp | Antagonistas muscarinicos. |
| US7205336B2 (en) | 2002-05-17 | 2007-04-17 | Merck & Co., Inc. | β-secretase inhibitors |
| JP2005528431A (ja) | 2002-05-31 | 2005-09-22 | ハー・ルンドベック・アクチエゼルスカベット | アルツハイマー病を治療するためのnmda−アンタゴニスト及びアセチルコリンエステラーゼ阻害剤の組み合わせ物 |
| AU2003232405A1 (en) | 2002-06-04 | 2003-12-19 | Pfizer Products Inc. | Flourinated cyclic amides as dipeptidyl peptidase iv inhibitors |
| AU2003233010A1 (en) | 2002-06-04 | 2003-12-19 | Pfizer Products Inc. | Process for the preparation of 3,3,4,4-tetrafluoropyrrolidine and derivatives thereof |
| BR0311697A (pt) | 2002-06-06 | 2005-03-22 | Eisai Co Ltd | Novos derivados condensados de imidazol |
| US20050171300A1 (en) | 2002-06-19 | 2005-08-04 | Luc Moens | Semi-gloss powder coating compositions |
| AU2003248259A1 (en) | 2002-07-10 | 2004-02-02 | Yamanouchi Pharmaceutical Co., Ltd. | Novel azetidine derivative or salt thereof |
| DE60330485D1 (de) | 2002-07-15 | 2010-01-21 | Merck & Co Inc | Zur behandlung von diabetes |
| AU2003251993A1 (en) | 2002-07-19 | 2004-02-09 | Inge Grundke-Iqbal | NMDA RECEPTOR ANTAGONISTS AND THEIR USE IN INHIBITING ABNORMAL HYPERPHOSPHORYLATION OF MICROTUBULE ASSOCIATED PROTEIN tau |
| US7557137B2 (en) | 2002-08-05 | 2009-07-07 | Bristol-Myers Squibb Company | Gamma-lactams as beta-secretase inhibitors |
| EP1532149B9 (de) | 2002-08-21 | 2011-04-20 | Boehringer Ingelheim Pharma GmbH & Co. KG | 8-[3-amino-piperidin-1-yl] -xanthine, deren herstellung und deren verwendung als arzneimittel |
| DE10238477A1 (de) | 2002-08-22 | 2004-03-04 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Neue Purinderivate, deren Herstellung und deren Verwendung als Arzneimittel |
| DE10238470A1 (de) | 2002-08-22 | 2004-03-04 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Neue Xanthinderivate, deren Herstellung und deren Verwendung als Arzneimittel |
| DE60336848D1 (de) | 2002-09-12 | 2011-06-01 | Univ California | Immunogene und entsprechende antikörper, die spezifisch sind für häufige hochmolekulare aggregations-zwischenprodukte von amyloiden aus proteinen unterschiedlicher sequenz |
| ES2201929B1 (es) | 2002-09-12 | 2005-05-16 | Araclon Biotech, S.L. | Anticuerpos policlonales, metodo de preparacion y uso de los mismos. |
| WO2010012004A2 (en) | 2008-07-25 | 2010-01-28 | The Regents Of The University Of California | Monoclonal antibodies specific for pathological amyoid aggregates common to amyloids formed from proteins of differing sequence |
| US9535076B2 (en) | 2002-09-12 | 2017-01-03 | The Regents Of The University Of California | Methods and compositions for eliciting an amyloid-selective immune response |
| ATE469969T1 (de) | 2002-09-12 | 2010-06-15 | Chemo Sero Therapeut Res Inst | Menschlicher anti-mensch-mcp-1-antikorper sowie antikorperfragment davon |
| MXPA05003091A (es) | 2002-09-19 | 2005-05-27 | Abbott Lab | Composiciones farmaceuticas como inhibidores de peptidasa de dipeptidilo-iv de (dpp-iv). |
| AU2003282804A1 (en) | 2002-09-20 | 2004-04-08 | Axys Pharmaceuticals, Inc. | 3-(3,5-disubstituted-4-hydroxyphenyl)propionamide derivatives as cathepsin b inhibitors |
| US7273889B2 (en) | 2002-09-25 | 2007-09-25 | Innovative Drug Delivery Systems, Inc. | NMDA receptor antagonist formulation with reduced neurotoxicity |
| WO2004029629A1 (en) | 2002-09-27 | 2004-04-08 | Janssen Pharmaceutica N.V. | N-11 truncated amyloid-beta nomoclonal antibodies, compositions, methods and uses |
| JP2006508072A (ja) | 2002-10-01 | 2006-03-09 | ノースウエスタン ユニバーシティ | アミロイドベータ由来拡散性リガンド(ADDLs)、ADDL代替物、ADDL結合性分子、およびそれらの使用 |
| GB0223038D0 (en) | 2002-10-04 | 2002-11-13 | Merck Sharp & Dohme | Therapeutic compounds |
| GB0223039D0 (en) | 2002-10-04 | 2002-11-13 | Merck Sharp & Dohme | Therapeutic compounds |
| DE60315687T2 (de) | 2002-10-07 | 2008-06-05 | Merck & Co., Inc. | Antidiabetische heterocyclische beta-aminoverbindungen als inhibitoren von dipeptidylpeptidase |
| WO2004033455A2 (en) | 2002-10-08 | 2004-04-22 | Novo Nordisk A/S | Hemisuccinate salts of heterocyclic dpp-iv inhibitors |
| GB0223494D0 (en) | 2002-10-09 | 2002-11-13 | Neuropharma Sa | Dual binding site acetylcholinesterase inhibitors for the treatment of alzheimer's disease |
| PL377769A1 (pl) | 2002-10-09 | 2006-02-20 | Rinat Neuroscience Corp. | Sposób leczenia choroby Alzheimera z zastosowaniem przeciwciał skierowanych przeciw peptydowi beta amyloidu i ich kompozycje |
| NZ538897A (en) | 2002-10-18 | 2007-02-23 | Merck & Co Inc | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| AU2003274353B2 (en) | 2002-10-24 | 2007-04-05 | Merz Pharma Gmbh & Co. Kgaa | Combination therapy using 1-aminocyclohexane derivatives and acetylcholinesterase inhibitors |
| US20040082543A1 (en) | 2002-10-29 | 2004-04-29 | Pharmacia Corporation | Compositions of cyclooxygenase-2 selective inhibitors and NMDA receptor antagonists for the treatment or prevention of neuropathic pain |
| WO2004039773A2 (en) | 2002-10-30 | 2004-05-13 | Nordic Bioscience A/S | Coumpounds modulating the activity of gapdh and/or iamt |
| WO2004041795A1 (en) | 2002-10-30 | 2004-05-21 | Guilford Pharmaceuticals Inc. | Novel inhibitors of dipeptidyl peptidase iv |
| GB0225474D0 (en) | 2002-11-01 | 2002-12-11 | Merck Sharp & Dohme | Therapeutic agents |
| FR2846667B1 (fr) | 2002-11-06 | 2004-12-31 | Pasteur Institut | Fragments variables d'anticorps de camelides a chaine unique diriges contre le peptide beta-amyloide 1-42 et leurs applications pour le diagnostic et le traitement des maladies neuroagregatives |
| BR0315796A (pt) | 2002-11-07 | 2005-09-13 | Merck & Co Inc | Composto, composição farmacêutica, e, métodos para tratar diabetes, para tratar hiperglicemia, e para tratar obesidade em um mamìfero |
| US7109217B2 (en) | 2002-11-12 | 2006-09-19 | Merck & Co., Inc. | Phenylcarboxamide beta-secretase inhibitors for the treatment of Alzheimer's disease |
| AU2002952946A0 (en) | 2002-11-27 | 2002-12-12 | Fujisawa Pharmaceutical Co., Ltd. | Dpp-iv inhibitor |
| EP1578414A4 (en) | 2002-12-04 | 2007-10-24 | Merck & Co Inc | PHENYLALANINE DERIVATIVES ALSDIPEPTIDYL-PEPTIDASE INHIBITORS FOR THE TREATMENT OR PREVENTION OF DIABETES |
| US7420079B2 (en) | 2002-12-09 | 2008-09-02 | Bristol-Myers Squibb Company | Methods and compounds for producing dipeptidyl peptidase IV inhibitors and intermediates thereof |
| US20040181075A1 (en) | 2002-12-19 | 2004-09-16 | Weingarten M. David | Process of making chalcone derivatives |
| CA2508947A1 (en) | 2002-12-20 | 2004-07-15 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7371853B2 (en) | 2003-01-07 | 2008-05-13 | Merck & Co., Inc. | Macrocyclic β-secretase inhibitors for the treatment of Alzheimer's disease |
| US7265128B2 (en) | 2003-01-17 | 2007-09-04 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| DE10303974A1 (de) | 2003-01-31 | 2004-08-05 | Abbott Gmbh & Co. Kg | Amyloid-β(1-42)-Oligomere, Verfahren zu deren Herstellung und deren Verwendung |
| AU2004210149A1 (en) | 2003-01-31 | 2004-08-19 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US20060188512A1 (en) | 2003-02-01 | 2006-08-24 | Ted Yednock | Active immunization to generate antibodies to solble a-beta |
| NZ541324A (en) | 2003-02-04 | 2008-10-31 | Hoffmann La Roche | Malonamide derivatives as gamma-secretase inhibitors |
| ATE468886T1 (de) | 2003-02-10 | 2010-06-15 | Applied Molecular Evolution | Abeta-bindende moleküle |
| WO2004071454A2 (en) | 2003-02-13 | 2004-08-26 | Guilford Pharmaceuticals Inc. | Substituted azetidine compounds as inhibitors of dipeptidyl peptidase iv |
| CA2516259A1 (en) | 2003-02-18 | 2004-09-02 | Roskamp Research, Llc | Anti-angiogenic and anti-tumoral properties of beta and gamma secretase inhibitors |
| US8663650B2 (en) | 2003-02-21 | 2014-03-04 | Ac Immune Sa | Methods and compositions comprising supramolecular constructs |
| WO2004076434A1 (en) | 2003-02-28 | 2004-09-10 | Aic | Dipeptidyl peptidase inhibitors |
| WO2004076433A1 (en) | 2003-02-28 | 2004-09-10 | Aic | Dipeptidyl peptidase inhibitors |
| EP1454627A1 (en) | 2003-03-06 | 2004-09-08 | MyoContract Ltd. | Alpha-Keto carbonyl calpain inhibitors |
| WO2004084830A2 (en) | 2003-03-21 | 2004-10-07 | Buck Institute | Method for treating alzheimer’s dementia |
| US20040191334A1 (en) | 2003-03-24 | 2004-09-30 | Pang-Chui Shaw | Use of transhinone derivates as cholinesterase inhibitors in treating related diseases |
| JP4887139B2 (ja) | 2003-03-25 | 2012-02-29 | 武田薬品工業株式会社 | ジペプチジルペプチダーゼインヒビター |
| CA2520125A1 (en) | 2003-03-28 | 2004-10-14 | Acadia Pharmaceuticals Inc. | Muscarinic m1 receptor agonists for pain management |
| US20050129695A1 (en) | 2003-03-28 | 2005-06-16 | Marc Mercken | Anti-amyloid antibodies, compositions, methods and uses |
| ATE481103T1 (de) | 2003-04-09 | 2010-10-15 | Wyeth Llc | Derivate von 2-(8,9-dioxo-2,6- diazabicyclo(5.2.0)non-1(7)-en-2- yl)alkylphosphonsäure und deren verwendung als n- methyl-d-aspartat- (nmda-) rezeptorantagonisten |
| GB0308382D0 (en) | 2003-04-10 | 2003-05-21 | Univ Cambridge Tech | Therapeutic methods and means |
| GB0308318D0 (en) | 2003-04-10 | 2003-05-14 | Merck Sharp & Dohme | Therapeutic agents |
| WO2004089362A1 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | 2-cyanopyrroles and their analogues as ddp-iv inhibitors |
| US7371871B2 (en) | 2003-05-05 | 2008-05-13 | Probiodrug Ag | Inhibitors of glutaminyl cyclase |
| EP1622870A2 (en) | 2003-05-05 | 2006-02-08 | Prosidion Ltd. | Glutaminyl based dp iv-inhibitors |
| CA2524009C (en) | 2003-05-05 | 2014-04-29 | Probiodrug Ag | Use of effectors of glutaminyl and glutamate cyclases |
| ES2246105B1 (es) | 2003-05-08 | 2007-03-01 | Araclon Biotech, S.L. | Uso de anticuerpos para el tratamiento de enfermedades amiloideas. |
| AU2003902260A0 (en) | 2003-05-09 | 2003-05-29 | Fujisawa Pharmaceutical Co., Ltd. | Dpp-iv inhibitor |
| ES2308206T3 (es) | 2003-05-13 | 2008-12-01 | Schering Corporation | N-arilsulfonilpiperidinas con fuentes como inhibidores de gamma-secretasa. |
| WO2004103276A2 (en) | 2003-05-14 | 2004-12-02 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| EP1625122A1 (en) | 2003-05-14 | 2006-02-15 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| DE602004011767T2 (de) | 2003-05-16 | 2009-02-19 | Merck Sharp & Dohme Ltd., Hoddesdon | Cyclohexylsulfone als gamma-sekretase-inhibitoren |
| KR20070104480A (ko) | 2003-05-27 | 2007-10-25 | 포레스트 래보러토리즈, 인코포레이티드 | 우울증 및 그 밖의 기분장애의 치료를 위한 nmda수용체 길항제 및 선택적 세로토닌 재흡수 억제제의 배합물 |
| PE20050627A1 (es) | 2003-05-30 | 2005-08-10 | Wyeth Corp | Anticuerpos humanizados que reconocen el peptido beta amiloideo |
| AU2003902828A0 (en) | 2003-06-05 | 2003-06-26 | Fujisawa Pharmaceutical Co., Ltd. | Dpp-iv inhibitor |
| ATE463492T1 (de) | 2003-06-06 | 2010-04-15 | Merck Sharp & Dohme | Kondensierte indole als dipeptidyl-peptidase- hemmer zur behandlung oder prävention von diabetes |
| AU2003902946A0 (en) | 2003-06-12 | 2003-06-26 | Fujisawa Pharmaceutical Co., Ltd. | Dpp-iv inhibitor |
| US7456204B2 (en) | 2003-06-17 | 2008-11-25 | Merck & Co., Inc. | Cyclohexylglycine derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| RU2339636C2 (ru) | 2003-06-20 | 2008-11-27 | Ф.Хоффманн-Ля Рош Аг | Гексагидропиридоизохинолины в качестве ингибиторов дипептидилпептидазы iv (dpp-iv) |
| ES2355105T3 (es) | 2003-06-20 | 2011-03-22 | F. Hoffmann-La Roche Ag | Pirido(2,1-a)isoquinolina como inhibidores de la dpp-iv. |
| CN100430377C (zh) | 2003-06-30 | 2008-11-05 | 麦克公司 | 用于治疗阿尔茨海默病的N-烷基苯基甲酰胺β分泌酶抑制剂 |
| KR101215821B1 (ko) | 2003-06-30 | 2012-12-28 | 텔 아비브 유니버시티 퓨쳐 테크놀로지 디벨롭먼트 엘.피. | 아밀로이드 관련 질환을 진단 및 처치하기 위한 펩타이드, 그것에 대한 항체, 및 그 사용방법 |
| CN1909897A (zh) | 2003-07-01 | 2007-02-07 | 默克公司 | 用于治疗阿尔茨海默病的苯基羧化物β-分泌酶抑制剂 |
| WO2005007614A1 (en) | 2003-07-03 | 2005-01-27 | The Government Of The United States Of America As Represented By The Secretary, Department Of Health And Human Services 6011 | Monoamine oxidase inhibitors |
| ATE466595T1 (de) | 2003-07-16 | 2010-05-15 | Rvx Therapeutics Inc | Verbindungen und methoden für das downregulating der effekte von tgf-beta |
| WO2005008250A1 (en) | 2003-07-21 | 2005-01-27 | Angiogenetics Sweden Ab | Compounds and methods for promoting angiogenesis by using a gamma-secretase inhibitor or inhibiting the gamma-secretase pathway |
| WO2005014041A2 (en) | 2003-07-24 | 2005-02-17 | Novartis Ag | Use of an amyloid beta dna vaccine for the treatment and/or prevention of amyloid diseases |
| CA2529674A1 (en) | 2003-07-28 | 2005-02-03 | Merz Pharma Gmbh & Co. Kgaa | The use of 1-amino-alkylcyclohexane compounds in the treatment of pain hypersensitivity |
| EP1651623B1 (en) | 2003-07-31 | 2008-12-17 | Merck & Co., Inc. | Hexahydrodiazepinones as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2005011599A2 (en) | 2003-08-01 | 2005-02-10 | Northwestern University | Antibodies specific for toxic amyloid beta protein oligomers |
| GB0318447D0 (en) | 2003-08-05 | 2003-09-10 | Merck Sharp & Dohme | Therapeutic agents |
| ZA200602051B (en) | 2003-08-13 | 2007-10-31 | Takeda Pharmaceutical | 4-pyrimidone derivatives and their use as peptidyl peptidase inhibitors |
| WO2005018545A2 (en) | 2003-08-14 | 2005-03-03 | Merck & Co., Inc. | Macrocyclic beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2005018424A2 (en) | 2003-08-18 | 2005-03-03 | Research Foundation For Mental Hygiene, Inc. | Antibodies specific for fibrillar amyloid and a procedure to detect fibrillar amyloid deposits |
| WO2005023762A1 (en) | 2003-09-04 | 2005-03-17 | Abbott Laboratories | Pyrrolidine-2-carbonitrile derivatives and their use as inhibitors of dipeptidyl peptidase-iv (dpp-iv) |
| AU2003298171A1 (en) | 2003-09-05 | 2005-03-29 | Cellzome Ag | Protein complexes associated with app-processing |
| US7790734B2 (en) | 2003-09-08 | 2010-09-07 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US20050065144A1 (en) | 2003-09-08 | 2005-03-24 | Syrrx, Inc. | Dipeptidyl peptidase inhibitors |
| WO2005025616A1 (ja) | 2003-09-09 | 2005-03-24 | Takeda Pharmaceutical Company Limited | 抗体の用途 |
| TW200530157A (en) | 2003-09-09 | 2005-09-16 | Japan Tobacco Inc | Dipeptidyl peptidase iv inhibitor |
| JP2007527865A (ja) | 2003-09-12 | 2007-10-04 | ザ・レジェンツ・オブ・ザ・ユニバーシティ・オブ・カリフォルニア | 種々の配列を有するタンパク質から形成されたアミロイドに共通の高分子量凝集中間体に特異的なモノクローナル抗体 |
| GB0322140D0 (en) | 2003-09-22 | 2003-10-22 | Pfizer Ltd | Combinations |
| EP1667984B1 (en) | 2003-09-24 | 2011-05-18 | Merck Sharp & Dohme Ltd. | Gamma-secretase inhibitors |
| AU2004277981B2 (en) | 2003-10-03 | 2009-10-01 | Merck & Co., Inc. | Benzylether and benzylamino beta-secretase inhibitors for the treatment of Alzheimer's disease |
| AU2003269307A1 (en) | 2003-10-06 | 2005-04-21 | Torrent Pharmaceuticals Ltd. | Azolidinecarbonitriles and their use as dpp-iv inhibitors |
| RU2356895C2 (ru) | 2003-10-06 | 2009-05-27 | Ф.Хоффманн-Ля Рош Аг | ПРОИЗВОДНЫЕ ЗАМЕЩЕННОГО ДИБЕНЗОАЗЕПИНА И БЕНЗОДИАЗЕПИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ γ-СЕКРЕТАЗЫ |
| WO2005035522A1 (en) | 2003-10-08 | 2005-04-21 | Pfizer Japan, Inc. | 1-‘2-(4-hydroxyphenyl)-2-hydroxyethyl!-piperidin-4-ol compounds as nmda receptor antagonists |
| JP5707014B2 (ja) | 2003-10-15 | 2015-04-22 | プロビオドルグ エージー | グルタミニル、及びグルタミン酸シクラーゼのエフェクターの使用 |
| DK1675591T3 (da) | 2003-10-16 | 2011-11-14 | Neurosearch As | Farmaceutisk sammensætning omfattende en monoamin-neurotransmitter-genoptagelseshæmmer og en acetylcholinesterase-hæmmer |
| GB0324236D0 (en) | 2003-10-16 | 2003-11-19 | Astrazeneca Ab | Chemical compounds |
| TW200519105A (en) | 2003-10-20 | 2005-06-16 | Lg Life Science Ltd | Novel inhibitors of DPP-IV, methods of preparing the same, and pharmaceutical compositions containing the same as an active agent |
| CN1870984A (zh) | 2003-10-22 | 2006-11-29 | 莫茨药物股份两合公司 | 1-氨基环己烷衍生物用于调节淀粉样病变中原纤维生成Aβ肽的沉积的用途 |
| WO2006058720A2 (en) | 2003-11-03 | 2006-06-08 | Probiodrug Ag | Novel compounds for the treatment of neurological disorders |
| WO2005044195A2 (en) | 2003-11-04 | 2005-05-19 | Merck & Co., Inc. | Fused phenylalanine derivatives as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| AU2004287586A1 (en) | 2003-11-11 | 2005-05-19 | F. Hoffmann-La Roche Ag | Phosphinic acids derivatives, beta-secretase inhibitors for the treatment of Alzheimer's disease |
| EP1689757B1 (en) | 2003-11-12 | 2014-08-27 | Phenomix Corporation | Heterocyclic boronic acid compounds |
| WO2005051914A1 (en) | 2003-11-24 | 2005-06-09 | Merck & Co., Inc. | Benzylether and benzylamino beta-secretase inhibitors for the treatment of alzheimer's disease |
| DE10355304A1 (de) | 2003-11-27 | 2005-06-23 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Neue 8-(Piperazin-1-yl)-und 8-([1,4]Diazepan-1-yl)-xanthine, deren Herstellung und deren Verwendung als Arzneimittel |
| UY28650A1 (es) | 2003-12-05 | 2005-02-28 | Forest Laboratories | Memantina para la prevencion o disminucion de la conducta suicida y para el tratamiento de la depresion mayor asociada con esta conducta |
| EP1541143A1 (en) | 2003-12-09 | 2005-06-15 | Graffinity Pharmaceuticals Aktiengesellschaft | Dpp-iv inhibitors |
| EP1541148A1 (en) | 2003-12-09 | 2005-06-15 | Graffinity Pharmaceuticals Aktiengesellschaft | Dpp-iv inhibitors |
| WO2005058849A1 (en) | 2003-12-15 | 2005-06-30 | Glenmark Pharmaceuticals Ltd. | New dipeptidyl peptidase in inhibitors; process for their preparation and compositions containing them |
| EP1697308B1 (en) | 2003-12-19 | 2014-03-19 | Merck Sharp & Dohme Corp. | Phenylamide and pyridylamide beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2005070429A1 (en) | 2004-01-22 | 2005-08-04 | Neurosearch A/S | Pharmaceutical composition comprising a monoamine neurotransmitter re-uptake inhibitor and an n-methyl-d-aspartate (nmda) receptors antagonist |
| AU2005209310B2 (en) | 2004-01-29 | 2011-01-06 | Neuromolecular, Inc. | Combination of a NMDA receptor antagonist and a MAO-inhibitor or a GADPH-inhibitor for the treatment of central nervous system-related conditions |
| WO2005075426A1 (en) | 2004-02-03 | 2005-08-18 | Glenmark Pharmaceuticals Ltd. | Novel dipeptidyl peptidase iv inhibitors; processes for their preparation and compositions thereof |
| CN1918131B (zh) | 2004-02-05 | 2011-05-04 | 前体生物药物股份公司 | 谷氨酰胺酰基环化酶抑制剂 |
| US20050209218A1 (en) | 2004-02-13 | 2005-09-22 | Meyerson Laurence R | Methods and compositions for the treatment of psychiatric conditions |
| US20050182044A1 (en) | 2004-02-17 | 2005-08-18 | Bruinsma Gosse B. | Combinatorial therapy with an acetylcholinesterase inhibitor and (3aR)-1,3a,8-trimethyl-1,2,3,3a,8,8a-hexahydropyrrolo[2,3,-b]indol-5-yl phenylcarbamate |
| PL1758905T3 (pl) | 2004-02-18 | 2009-10-30 | Boehringer Ingelheim Int | 8-[3-Aminopiperydyn-1-ylo]-ksantyna, jej wytwarzanie i jej zastosowanie jako inhibitora DPP-IV |
| WO2005080435A1 (ja) | 2004-02-20 | 2005-09-01 | Immuno-Biological Laboratories Co., Ltd. | モノクローナル抗体およびその利用 |
| RU2379315C2 (ru) | 2004-02-23 | 2010-01-20 | Трастиз Оф Тафтс Колледж | Ингибиторы дипептидилпептидазы iv |
| EP1593671A1 (en) | 2004-03-05 | 2005-11-09 | Graffinity Pharmaceuticals AG | DPP-IV inhibitors |
| KR100844593B1 (ko) | 2004-03-09 | 2008-07-07 | 내셔날 헬스 리서치 인스티튜트 | 피롤리딘 화합물 |
| GEP20094679B (en) | 2004-03-15 | 2009-05-10 | Takeda Pharmaceuticals Co | Dipeptidyl peptidase inhibitors |
| JP2007529556A (ja) | 2004-03-19 | 2007-10-25 | アクソニクス インコーポレイテッド | 認知障害の処置に有用なアセチルコリンエステラーゼ阻害剤およびn−メチル−d−アスパラギン酸拮抗剤 |
| WO2005095339A1 (en) | 2004-03-31 | 2005-10-13 | Pfizer Products Inc. | Dicyanopyrrolidines as dipeptidyl peptidase iv inhibitors |
| WO2005097103A2 (en) | 2004-04-01 | 2005-10-20 | Axys Pharmaceuticals, Inc. | Diabetes and metabolic syndrome therapy utilizing cathepsin b inhibitors |
| CA2563033A1 (en) | 2004-04-05 | 2005-10-20 | Schering Corporation | Novel gamma secretase inhibitors |
| WO2005103020A1 (en) | 2004-04-20 | 2005-11-03 | Merck & Co., Inc. | 1,3,5-substituted phenyl derivative compounds useful as beta-secretase inhibitors for the treatment of alzheimer's disease |
| JP4764418B2 (ja) | 2004-04-20 | 2011-09-07 | メルク・シャープ・エンド・ドーム・コーポレイション | アルツハイマー病の治療のためのβ−セクレターゼ阻害剤として有用な2,4,6−置換ピリジル誘導体化合物 |
| WO2005102390A2 (en) | 2004-04-22 | 2005-11-03 | Pfizer Japan, Inc. | Combinations comprising alpha-2-delta ligands and nmda receptor antagonists |
| US9549895B2 (en) | 2004-04-23 | 2017-01-24 | Massachusetts Eye And Ear Infirmary | Methods and compositions for preserving the viability of photoreceptor cells |
| US7763249B2 (en) | 2004-04-27 | 2010-07-27 | Juridical Foundation The Chemo-Sero-Therapeutic Research Institute | Human anti-amyloid β peptide antibody and fragment of said antibody |
| CN1950349A (zh) | 2004-05-04 | 2007-04-18 | 默克公司 | 作为用于治疗或预防糖尿病的二肽基肽酶-ⅳ抑制剂的1,2,4-噁二唑衍生物 |
| RS51106B (sr) | 2004-05-12 | 2010-10-31 | Pfizer Products Inc. | Derivati prolina i njihova upotreba kao inhibitora dipeptidil peptidaze iv |
| US7449599B2 (en) | 2004-05-13 | 2008-11-11 | Merck + Co Inc. | Phenyl carboxamide compounds useful as beta-secretase inhibitors for the treatment of alzheimer's disease |
| JP2007538079A (ja) | 2004-05-18 | 2007-12-27 | メルク エンド カムパニー インコーポレーテッド | 糖尿病の治療又は予防のためのジペプチジルペプチダーゼ−iv阻害剤としてのシクロヘキシルアラニン誘導体 |
| EP1598341A1 (en) | 2004-05-21 | 2005-11-23 | Santhera Pharmaceuticals (Deutschland) Aktiengesellschaft | DPP-IV inhibitors |
| JP2008501714A (ja) | 2004-06-04 | 2008-01-24 | 武田薬品工業株式会社 | ジペプチジルペプチダーゼインヒビター |
| ATE476993T1 (de) | 2004-06-07 | 2010-08-15 | Univ Ramot | Verfahren zur passiven immunisierung gegen eine durch amyloidaggregation gekennzeichnete krankheit oder erkrankung mit vermindertem nervenentzündungsrisiko |
| EP1604662A1 (en) | 2004-06-08 | 2005-12-14 | Santhera Pharmaceuticals (Deutschland) Aktiengesellschaft | 1-[(3R)-Amino-4-(2-fluoro-phenyl)-butyl]-pyrrolidine-(2R)-carboxylic acid benzyl amine derivatives and related compounds as dipeptidyl peptidase IV (DPP-IV) inhibitors for the treatment of type 2 diabetes mellitus |
| EP1604980A1 (en) | 2004-06-08 | 2005-12-14 | Santhera Pharmaceuticals (Deutschland) Aktiengesellschaft | DPP-IV inhibitors |
| EP1604989A1 (en) | 2004-06-08 | 2005-12-14 | Santhera Pharmaceuticals (Deutschland) Aktiengesellschaft | DPP-IV inhibitors |
| AU2005257904A1 (en) | 2004-06-15 | 2006-01-05 | Merck Sharp & Dohme Corp. | Pyrrolidin-3-yl compounds useful as beta-secretase inhibitors for the treatment of Alzheimer's disease |
| GB0413389D0 (en) | 2004-06-16 | 2004-07-21 | Astrazeneca Ab | Chemical compounds |
| SE0401601D0 (sv) | 2004-06-21 | 2004-06-21 | Bioarctic Neuroscience Ab | Protofibril specific antibodies and uses thereof |
| WO2006009886A1 (en) | 2004-06-21 | 2006-01-26 | Merck & Co., Inc. | Aminocyclohexanes as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| US7371825B2 (en) | 2004-06-30 | 2008-05-13 | Centocor, Inc. | Anti-MCP-1 antibodies, compositions, methods and uses |
| CN101048393A (zh) | 2004-06-30 | 2007-10-03 | 先灵公司 | 作为γ-分泌酶抑制剂的取代的N-芳基磺酰基杂环胺 |
| CA2572602A1 (en) | 2004-07-02 | 2006-02-09 | Northwestern University | Monolocal antibodies that target pathological assemblies of amyloid .beta. (abeta) |
| CA2573848A1 (en) | 2004-07-12 | 2006-02-16 | Phenomix Corporation | Constrained cyano compounds |
| BRPI0513384A (pt) | 2004-07-14 | 2008-05-06 | Novartis Ag | combinação de inibidores de dpp-iv e compostos que modulam receptores de 5-ht3 e/ou 5-ht4 |
| WO2006019965A2 (en) | 2004-07-16 | 2006-02-23 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| WO2006008661A2 (en) | 2004-07-19 | 2006-01-26 | Neurochem (International) Limited | Diagnostic methods of multiple organ amyloidosis |
| US7955812B2 (en) | 2004-07-19 | 2011-06-07 | The General Hospital Corporation | Methods of diagnosing alzheimer's disease by detecting antibodies to cross-linked β-amyloid oligomers |
| CA2574218A1 (en) | 2004-07-22 | 2006-02-09 | Schering Corporation | Substituted amide beta secretase inhibitors |
| DE602005007245D1 (es) | 2004-07-28 | 2008-07-10 | Schering Corp | |
| HUP0401523A3 (en) | 2004-07-29 | 2007-05-02 | Richter Gedeon Vegyeszet | Indole-2-carboxamide derivatives, pharmaceutical compositions containing them and process for producing them |
| CA2575663C (en) | 2004-07-30 | 2013-04-23 | Rinat Neuroscience Corp. | Antibodies directed against amyloid-beta peptide and methods using same |
| EP1623983A1 (en) | 2004-08-05 | 2006-02-08 | Santhera Pharmaceuticals (Deutschland) Aktiengesellschaft | Heterocyclic compounds useful as DPP-IV inhibitors |
| CN101080421A (zh) | 2004-08-11 | 2007-11-28 | 三菱化学株式会社 | 抗体以及其利用 |
| EP1784188B1 (en) | 2004-08-23 | 2010-07-14 | Merck Sharp & Dohme Corp. | Fused triazole derivatives as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| US20080058324A1 (en) | 2004-08-25 | 2008-03-06 | Santhera Pharmaceuticals (Schweiz) Ag | Alpha-Keto Carbonyl Calpain Inhibitors |
| JP2008510759A (ja) | 2004-08-25 | 2008-04-10 | サンセラ ファーマシューティカルズ (シュバイツ) アーゲー | α−ケトカルボニルカルパイン阻害剤 |
| US7388007B2 (en) | 2004-08-26 | 2008-06-17 | Bristol-Myers Squibb Company | Gamma-lactams as beta-secretase inhibitors |
| TWI374935B (en) | 2004-08-27 | 2012-10-21 | Pfizer Ireland Pharmaceuticals | Production of α-abeta |
| CA2579472A1 (en) | 2004-09-14 | 2006-03-23 | The Genetics Company, Inc. | Hydrazone derivatives and their use as beta secretase inhibitors |
| JP2008513497A (ja) | 2004-09-17 | 2008-05-01 | コメンティス,インコーポレーテッド | メマプシン2βセクレターゼ活性を阻害するアミノ含有化合物およびその使用方法 |
| JP2008513495A (ja) | 2004-09-17 | 2008-05-01 | コメンティス,インコーポレーテッド | ベータ−セクレターゼ活性を阻害する二環式化合物およびその使用方法 |
| EP1799260A4 (en) | 2004-09-29 | 2011-09-28 | Centocor Inc | ANTI-AMYLOID ANTIBODIES, COMPOSITIONS, TECHNIQUES AND USES |
| CA2582447C (en) | 2004-10-01 | 2012-04-17 | Merck & Co., Inc. | Aminopiperidines as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2006042103A2 (en) | 2004-10-05 | 2006-04-20 | Axys Pharmaceuticals, Inc. | Reversible inhibitors of cathepsin b |
| KR20070099527A (ko) | 2004-10-08 | 2007-10-09 | 노파르티스 아게 | 유기 화합물의 조합물 |
| US9186345B2 (en) | 2004-10-12 | 2015-11-17 | Hakon Hakonarson | Method of treating skin diseases |
| US7256866B2 (en) | 2004-10-12 | 2007-08-14 | Asml Netherlands B.V. | Lithographic apparatus and device manufacturing method |
| CN101068545A (zh) | 2004-10-13 | 2007-11-07 | 默克公司 | 作为β-分泌酶抑制剂用于治疗阿尔茨海默病的螺哌啶化合物 |
| CA2626049A1 (en) | 2004-10-15 | 2006-04-20 | Biopharmacopae Design International Inc. | Methods and therapeutic compositions comprising plant extracts for the treatment of cancer |
| JP2008517921A (ja) | 2004-10-25 | 2008-05-29 | ノバルティス アクチエンゲゼルシャフト | Dpp−iv阻害剤、ppar抗糖尿病薬およびメトホルミンの組合わせ剤 |
| US7811563B2 (en) | 2004-10-25 | 2010-10-12 | Northwestern University | Anti-addl antibodies and uses thereof |
| BRPI0516674A (pt) | 2004-10-28 | 2008-09-16 | Sanko Junyaku Kk | método de análise de mal de alzheimer e reagente de diagnóstico |
| AU2005310239A1 (en) | 2004-10-29 | 2006-06-08 | Merck & Co., Inc. | 2-aminopyridine compounds useful as beta-secretase inhibitors for the treatment of Alzheimer's disease |
| JP2008520670A (ja) | 2004-11-17 | 2008-06-19 | メルク エンド カムパニー インコーポレーテッド | アルツハイマー病の治療のための大環状第三アミンβ−セクレターゼ阻害剤 |
| CA2587360A1 (en) | 2004-11-23 | 2006-06-01 | Merck & Co., Inc. | 2,3,4,6-substituted pyridyl derivative compounds useful as beta-secretase inhibitors for the treatment of alzheimer's disease |
| AU2005309601A1 (en) | 2004-11-23 | 2006-06-01 | Neuromolecular Pharmaceuticals, Inc. | Composition comprising a sustained release coating or matrix and an NMDA receptor antagonist, method for administration such NMDA antagonist to a subject |
| WO2006057983A1 (en) | 2004-11-23 | 2006-06-01 | Merck & Co., Inc. | Macrocyclic aminopyridyl beta-secretase inhibitors for the treatment of alzheimer's disease |
| CA2588296A1 (en) | 2004-11-24 | 2006-06-01 | Neuromolecular Pharmaceuticals, Inc. | Composition comprising an nmda receptor antagonist and levodopa and use thereof for treating neurological disease |
| CN101065357A (zh) | 2004-11-29 | 2007-10-31 | 默克公司 | 用于治疗或预防糖尿病的作为二肽基肽酶-ⅳ抑制剂的稠合氨基哌啶 |
| KR100917545B1 (ko) | 2004-11-30 | 2009-09-16 | 에프. 호프만-라 로슈 아게 | 당뇨병 치료를 위한 dpp-ⅳ 억제제로서의 치환된벤조퀴놀리진 |
| EP1827478A4 (en) | 2004-12-03 | 2009-08-05 | Mucosal Therapeutics Llc | METHODS OF TREATING JOINED OR SICK JOINTS |
| JP2008524247A (ja) | 2004-12-15 | 2008-07-10 | エラン ファーマ インターナショナル リミテッド | 認知の改善における使用のためのアミロイドβ抗体 |
| CA2590337C (en) | 2004-12-15 | 2017-07-11 | Neuralab Limited | Humanized amyloid beta antibodies for use in improving cognition |
| EP1838854B1 (en) | 2004-12-15 | 2012-10-31 | Janssen Alzheimer Immunotherapy | Antibodies that recognize Beta Amyloid Peptide |
| WO2006066233A1 (en) | 2004-12-15 | 2006-06-22 | Neuralab Limited | An immunoprecipitation-based assay for predicting in vivo efficacy of beta-amyloid antibodies |
| BRPI0519585A2 (pt) | 2004-12-20 | 2009-02-25 | Hoffmann La Roche | compostos de fàrmula i; processo para a fabricaÇço de compostos de fàrmula i; composiÇÕes farmacÊuticas; mÉtodo para o tratamento e/ou profilaxia de doenÇas que estço associadas com a dpp-iv e usos destes compostos |
| US7411093B2 (en) | 2004-12-20 | 2008-08-12 | Hoffman-La Roche Inc. | Aminocycloalkanes as DPP-IV inhibitors |
| EP1828192B1 (en) | 2004-12-21 | 2014-12-03 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| JP2008525439A (ja) | 2004-12-23 | 2008-07-17 | ボイジャー・ファーマシューティカル・コーポレーション | アルツハイマー病の治療のための酢酸ロイプロリド及びアセチルコリンエステラーゼインヒビターまたはnmdaレセプターアゴニスト |
| EP1831172B1 (en) | 2004-12-28 | 2009-02-18 | Council of Scientific and Industrial Research | Substituted carbamic acid quinolin-6-yl esters useful as acetylcholinesterase inhibitors |
| CN101098700A (zh) | 2005-01-06 | 2008-01-02 | 默克公司 | 治疗炎性疾病的药物组合疗法和药用组合物 |
| CA2594943A1 (en) | 2005-01-19 | 2006-07-27 | Merck & Co., Inc. | Aminomethyl beta-secretase inhibitors for the treatment of alzheimer's disease |
| EP1838669A1 (en) | 2005-01-19 | 2007-10-03 | Biolipox AB | Indoles useful in the treatment of inflammation |
| AU2006208226A1 (en) | 2005-01-24 | 2006-08-03 | Amgen Inc. | Humanized anti-amyloid antibody |
| US20060216331A1 (en) | 2005-02-28 | 2006-09-28 | Lines Thomas C | Composition for treating mental health disorders |
| WO2006094674A1 (en) | 2005-03-07 | 2006-09-14 | Michael Hermanussen | Nmda receptor antagonists in the medical intervention of metabolic disorders |
| ES2259270B1 (es) | 2005-03-09 | 2007-11-01 | Consejo Superior De Investigaciones Cientificas | Metodo de diagnostico in vitro de la enfermedad de alzheimer mediante un anticuerpo monoclonal. |
| WO2006099352A1 (en) | 2005-03-10 | 2006-09-21 | Bristol-Myers Squibb Company | Novel isophthalates as beta-secretase inhibitors |
| FR2883285B1 (fr) | 2005-03-17 | 2007-05-18 | Sanofi Aventis Sa | Sel besylate de la 7-(2-(4-(3-trifluoromethyl-phenyl) -1,2,3,6-tetrahudro-pyrid-1-yl)ethyl) isoquinoleine, sa preparation et son utilisation en therapeutique |
| ES2318918B1 (es) | 2005-04-01 | 2010-02-16 | Biotherapix Molecular Medicines, S.L.U. | Anticuerpos humanos con capacidad de union al peptido beta-amiloide y sus aplicaciones. |
| CA2605771A1 (en) | 2005-04-27 | 2006-11-02 | Novartis Ag | Methods of treating atherosclerosis |
| MY148086A (en) | 2005-04-29 | 2013-02-28 | Rinat Neuroscience Corp | Antibodies directed against amyloid-beta peptide and methods using same |
| PE20061444A1 (es) | 2005-05-19 | 2007-01-15 | Centocor Inc | Anticuerpo anti-mcp-1, composiciones, metodos y usos |
| US7741448B2 (en) | 2005-06-21 | 2010-06-22 | Medical & Biological Laboratories Co., Ltd. | Antibody having inhibitory effect on amyloid fibril formation |
| AU2006270084B2 (en) | 2005-07-18 | 2011-08-25 | Merck Sharp & Dohme Corp. | Spiropiperidine beta-secretase inhibitors for the treatment of Alzheimer's disease |
| US20090170830A1 (en) | 2005-08-03 | 2009-07-02 | Nantermet Philippe G | Tricyclic Beta-Secretase Inhibitors for the Treatment of Alzheimer's Disease |
| US20090182021A1 (en) | 2005-08-03 | 2009-07-16 | Nanternet Philippe G | Tricyclic Beta-Secretase Inhibitors for the Treatment of Alzheimer's Disease |
| US7338974B2 (en) | 2005-08-12 | 2008-03-04 | Bristol-Myers Squibb Company | Macrocyclic diaminopropanes as beta-secretase inhibitors |
| EP1937720B1 (en) | 2005-08-18 | 2014-04-09 | Ramot at Tel-Aviv University Ltd. | Single chain antibodies against beta-amyloid peptide |
| EP1931656B1 (en) | 2005-09-28 | 2011-02-09 | Robert Ternansky | Novel drugs for dementia |
| WO2007051333A1 (en) | 2005-11-02 | 2007-05-10 | Oncalis Ag | Triazine beta-secretase inhibitors |
| EP2808032B1 (en) | 2005-12-12 | 2018-08-01 | AC Immune S.A. | A beta 1-42 specific monoclonal antibodies with therapeutic properties |
| AU2006326283B2 (en) | 2005-12-12 | 2012-01-19 | Ac Immune S.A. | Therapeutic vaccine |
| MX2008009414A (es) | 2006-01-20 | 2008-10-01 | Schering Corp | Arilsulfonas carbociclicas y heterociclicas como inhibidores de gamma secretasa. |
| US8034353B2 (en) | 2006-02-22 | 2011-10-11 | Kabushiki Kaisha Hayashibara Seibutsu Kagaku Kenkyujo | Peptide vaccine for inducing the production of anti-amyloid β-peptide antibody |
| SI2177536T1 (sl) | 2006-03-30 | 2014-09-30 | Glaxo Group Limited | Protitelesa proti amiloidnemu peptidu beta |
| CA2657681C (en) | 2006-07-14 | 2019-03-19 | Ac Immune S.A. | Humanized antibodies against beta amyloid protein |
| SI2074145T1 (sl) | 2006-10-02 | 2017-12-29 | Ac Immune S.A. | Humanizirano protitelo proti amiloid beta |
| JP2010508272A (ja) | 2006-10-30 | 2010-03-18 | メルク エンド カムパニー インコーポレーテッド | アルツハイマー病を治療するためのスピロピペリジンβセクレターゼ阻害剤 |
| DK2086960T3 (da) | 2006-11-09 | 2014-06-10 | Probiodrug Ag | Nye inhibitorer af glutaminylcyclase. |
| DK2091945T3 (da) | 2006-11-09 | 2014-04-22 | Probiodrug Ag | Nye inhibitorer af glutaminylcyclase |
| EP2089383B1 (en) | 2006-11-09 | 2015-09-16 | Probiodrug AG | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| EP2091948B1 (en) | 2006-11-30 | 2012-04-18 | Probiodrug AG | Novel inhibitors of glutaminyl cyclase |
| US20100216875A1 (en) | 2006-12-20 | 2010-08-26 | Gregory Hook | Methods of treating alzheimer's disease |
| JP5612860B2 (ja) | 2007-03-09 | 2014-10-22 | プロビオドルグ エージー | グルタミニルシクラーゼ阻害剤としてのイミダゾ[1,5−a]ピリジン誘導体 |
| WO2008128982A1 (en) | 2007-04-18 | 2008-10-30 | Probiodrug Ag | Thioxoquinazolinone derivatives as glutaminyl cyclase inhibitors |
| WO2008128987A1 (en) | 2007-04-18 | 2008-10-30 | Probiodrug Ag | Novel inhibitors |
| ES2533484T3 (es) | 2007-04-18 | 2015-04-10 | Probiodrug Ag | Derivados de tiourea como inhibidores de la glutaminil ciclasa |
| US8227498B2 (en) | 2007-04-18 | 2012-07-24 | Probiodrug Ag | Inhibitors of glutaminyl cyclase |
| US9512082B2 (en) | 2007-04-18 | 2016-12-06 | Probiodrug Ag | Inhibitors of glutaminyl cyclase |
| US8772508B2 (en) | 2007-04-18 | 2014-07-08 | Probiodrug Ag | Inhibitors of glutaminyl cyclase |
| JP5676249B2 (ja) | 2007-04-20 | 2015-02-25 | プロビオドルグ エージー | グルタミニルシクラーゼ阻害剤としてのアミノピリジン誘導体 |
| ES2373253T3 (es) | 2007-05-25 | 2012-02-01 | Elan Pharmaceuticals Inc. | Pirazolopirrolidinos como inhibidores de gamma secretasa. |
| TWI516500B (zh) | 2007-06-12 | 2016-01-11 | Ac免疫公司 | 抗β類澱粉蛋白單株抗體 |
| AU2008275735B2 (en) | 2007-07-05 | 2014-03-27 | Merck Sharp & Dohme Corp. | Tetrahydropyranochromene gamma secretase inhibitors |
| WO2009011851A1 (en) | 2007-07-17 | 2009-01-22 | Schering Corporation | Benzenesulfonyl-chromane, thiochromane, tetrahydronaphthalene and related gamma secretase inhibitors |
| EA026787B1 (ru) | 2007-08-27 | 2017-05-31 | Общество С Ограниченной Ответственностью "Ниармедик Плюс" | Конструкция нуклеиновой кислоты и ее применение для лечения, предотвращения и сдерживания болезни альцгеймера |
| EP2527369A3 (en) | 2007-09-13 | 2012-12-19 | University Of Zurich Prorektorat Forschung | Monoclonal amyloid beta (abeta)-specific antibody and uses thereof |
| MX2010002938A (es) | 2007-09-24 | 2010-04-01 | Comentis Inc | Derivados de (3-hidroxi-4-amino-butan-2-il)-3-(2-tiazol-2-il-pirro lidin-1-carbonil)benzamida y compuestos relacionados como inhibidores de beta-secretasa para tratar enfermedad de alzheimer. |
| GB0720912D0 (en) | 2007-10-25 | 2007-12-05 | Univ Cardiff | Monoclonal Anitbody for APP |
| JP5219225B2 (ja) | 2007-10-25 | 2013-06-26 | 国立大学法人 鹿児島大学 | アミロイドβペプチドのミミック分子を用いたペプチドワクチン |
| AP2957A (en) | 2007-10-29 | 2014-08-31 | Inst Nat Sante Rech Med | New antibodies specific of the Beta-amyloid peptides and their uses as diagnostic agents or drugs |
| ES2639016T3 (es) | 2007-11-16 | 2017-10-25 | The Rockefeller University | Anticuerpos específicos para la forma protofibrilar de proteína beta-amiloide |
| WO2009090650A2 (en) | 2008-01-16 | 2009-07-23 | Ben-Gurion University Of The Negev Research And Development Authority | Vaccine for alzheimer's disease |
| ITNA20080006A1 (it) | 2008-01-28 | 2009-07-29 | Consiglio Nazionale Ricerche | Proteine chimeriche capaci di formare particelle virus-like, che contengono sequenze peptidiche del beta-amiloide e la componente e2 della alfachetoacido deidrogenasi di "geobacillus stearothermophilus", utili per l'induzione di una risposta anticorp |
| CA2714058A1 (en) | 2008-02-28 | 2009-09-03 | Ivory D. Hills | 2-aminoimidazole beta-secretase inhibitors for the treatment of alzheimer's disease |
| JP2011529084A (ja) | 2008-07-25 | 2011-12-01 | アボット・ラボラトリーズ | アミロイドβペプチド類似体、これらのオリゴマー、前記類似体またはオリゴマーを調製するためのプロセスおよび前記類似体またはオリゴマーを含む組成物ならびにこれらの使用 |
| CA2739318A1 (en) | 2008-08-07 | 2010-02-11 | Mercia Pharma, Llc | Immunotherapeutic compositions for the treatment of alzheimer's disease |
| WO2010021680A2 (en) | 2008-08-19 | 2010-02-25 | Vitae Pharmaceuticals, Inc. | Inhibitors of beta-secretase |
| SG193828A1 (en) | 2008-09-04 | 2013-10-30 | Probiodrug Ag | Novel inhibitors |
| KR101640147B1 (ko) | 2008-10-16 | 2016-07-18 | 잇빤 자이단호진 가가쿠오요비겟세이료호겐쿠쇼 | 변성된 아밀로이드 베타 펩티드 |
| KR20110086769A (ko) | 2008-11-23 | 2011-07-29 | 화이자 인코포레이티드 | 베타 세크레타제 억제제로서의 락탐 |
| EP2393776B1 (en) | 2009-02-06 | 2014-05-14 | Merck Sharp & Dohme Corp. | Novel trifluoromethylsulfonamide gamma secretase inhibitor |
| WO2010094242A1 (en) | 2009-02-20 | 2010-08-26 | Merck Sharp & Dohme Corp. | Spiropyrrolidine beta-secretase inhibitors for the treatment of alzheimer's disease |
| ES2548913T3 (es) * | 2009-09-11 | 2015-10-21 | Probiodrug Ag | Derivados heterocíclicos como inhibidores de glutaminil ciclasa |
| US9181233B2 (en) | 2010-03-03 | 2015-11-10 | Probiodrug Ag | Inhibitors of glutaminyl cyclase |
| EA022420B1 (ru) | 2010-03-10 | 2015-12-30 | Пробиодруг Аг | Гетероциклические ингибиторы глутаминилциклазы (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| DK2686313T3 (en) | 2011-03-16 | 2016-05-02 | Probiodrug Ag | Benzimidazole derivatives as inhibitors of glutaminyl cyclase |
| WO2014140279A1 (en) | 2013-03-15 | 2014-09-18 | Probiodrug Ag | Novel inhibitors |
| CN103524486B (zh) * | 2013-09-18 | 2016-03-02 | 中国科学院昆明植物研究所 | N-喹啉基取代的β-内酰胺类化合物及其药物组合物和合成方法与应用 |
-
2017
- 2017-09-29 ES ES17194164T patent/ES2812698T3/es active Active
- 2017-09-29 EP EP17194164.4A patent/EP3461819B1/en active Active
- 2017-09-29 PL PL17194164T patent/PL3461819T3/pl unknown
- 2017-09-29 DK DK17194164.4T patent/DK3461819T3/da active
-
2018
- 2018-09-20 JP JP2020517863A patent/JP6821860B2/ja active Active
- 2018-09-20 KR KR1020217013820A patent/KR20210057206A/ko not_active Application Discontinuation
- 2018-09-20 CA CA3077314A patent/CA3077314C/en active Active
- 2018-09-20 EA EA202090642A patent/EA037862B1/ru not_active IP Right Cessation
- 2018-09-20 MX MX2020003570A patent/MX2020003570A/es unknown
- 2018-09-20 WO PCT/EP2018/075494 patent/WO2019063414A1/en active Application Filing
- 2018-09-20 NZ NZ763312A patent/NZ763312A/en unknown
- 2018-09-20 KR KR1020207010866A patent/KR102251645B1/ko active IP Right Grant
- 2018-09-20 SG SG11202002239VA patent/SG11202002239VA/en unknown
- 2018-09-20 CN CN201880071623.4A patent/CN111315738B/zh active Active
- 2018-09-20 BR BR112020005625-1A patent/BR112020005625B1/pt active IP Right Grant
- 2018-09-20 AU AU2018340397A patent/AU2018340397B2/en active Active
- 2018-09-20 US US16/650,631 patent/US11279690B2/en active Active
- 2018-09-20 EA EA202190664A patent/EA202190664A1/ru unknown
-
2020
- 2020-03-11 IL IL273214A patent/IL273214B/en active IP Right Grant
- 2020-03-12 ZA ZA2020/01573A patent/ZA202001573B/en unknown
-
2021
- 2021-01-06 JP JP2021000850A patent/JP2021073202A/ja active Pending
- 2021-03-03 IL IL281216A patent/IL281216A/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CA3077314C (en) | 2021-07-13 |
| EP3461819A1 (en) | 2019-04-03 |
| CA3077314A1 (en) | 2019-04-04 |
| US20200223825A1 (en) | 2020-07-16 |
| NZ763312A (en) | 2023-04-28 |
| EA202090642A1 (ru) | 2020-05-14 |
| JP2021073202A (ja) | 2021-05-13 |
| IL273214B (en) | 2021-03-25 |
| KR20200074948A (ko) | 2020-06-25 |
| US11279690B2 (en) | 2022-03-22 |
| JP2020535186A (ja) | 2020-12-03 |
| WO2019063414A1 (en) | 2019-04-04 |
| KR102251645B1 (ko) | 2021-05-14 |
| CN111315738B (zh) | 2021-08-03 |
| BR112020005625B1 (pt) | 2022-05-24 |
| EA202190664A1 (ru) | 2021-05-27 |
| MX2020003570A (es) | 2021-11-19 |
| KR20210057206A (ko) | 2021-05-20 |
| EA037862B1 (ru) | 2021-05-28 |
| BR112020005625A2 (pt) | 2020-10-20 |
| EP3461819B1 (en) | 2020-05-27 |
| AU2018340397B2 (en) | 2020-06-11 |
| DK3461819T3 (da) | 2020-08-10 |
| AU2018340397A1 (en) | 2020-04-23 |
| PL3461819T3 (pl) | 2020-11-30 |
| ZA202001573B (en) | 2021-04-28 |
| JP6821860B2 (ja) | 2021-02-03 |
| CN111315738A (zh) | 2020-06-19 |
| IL281216A (en) | 2021-04-29 |
| SG11202002239VA (en) | 2020-04-29 |
| AU2018340397A8 (en) | 2020-04-30 |
| IL273214A (en) | 2020-04-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2478673T3 (es) | Derivados de tioxoquinazolinona como inhibidores de la glutaminil ciclasa | |
| US7803810B2 (en) | Inhibitors | |
| AU2011226074B2 (en) | Heterocyclic inhibitors of glutaminyl cyclase (QC, EC 2.3.2.5) | |
| ES2750645T3 (es) | Inhibidores novedosos | |
| EP2146968B1 (en) | Urea derivatives as glutaminyl cyclase inhibitors | |
| ES2812698T3 (es) | Inhibidores de glutaminil ciclasa | |
| BR112015023454B1 (pt) | Derivados de oxazolidinona halogenados, seu processo de preparação, composição farmacêutica, e seu uso |