JP6860623B2 - キメラ抗原受容体を使用する癌の処置 - Google Patents
キメラ抗原受容体を使用する癌の処置 Download PDFInfo
- Publication number
- JP6860623B2 JP6860623B2 JP2019126002A JP2019126002A JP6860623B2 JP 6860623 B2 JP6860623 B2 JP 6860623B2 JP 2019126002 A JP2019126002 A JP 2019126002A JP 2019126002 A JP2019126002 A JP 2019126002A JP 6860623 B2 JP6860623 B2 JP 6860623B2
- Authority
- JP
- Japan
- Prior art keywords
- cells
- antigen
- car
- cell
- domain
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 title claims description 335
- 206010028980 Neoplasm Diseases 0.000 title claims description 238
- 201000011510 cancer Diseases 0.000 title claims description 65
- 238000011282 treatment Methods 0.000 title claims description 41
- 210000004027 cell Anatomy 0.000 claims description 381
- 108091007433 antigens Proteins 0.000 claims description 220
- 102000036639 antigens Human genes 0.000 claims description 220
- 239000000427 antigen Substances 0.000 claims description 217
- 210000001744 T-lymphocyte Anatomy 0.000 claims description 152
- -1 CDK4 / 6 inhibitors Substances 0.000 claims description 114
- 230000011664 signaling Effects 0.000 claims description 114
- 230000027455 binding Effects 0.000 claims description 112
- 230000014509 gene expression Effects 0.000 claims description 79
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 69
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 claims description 68
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 claims description 68
- 239000012642 immune effector Substances 0.000 claims description 68
- 229940121354 immunomodulator Drugs 0.000 claims description 68
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 68
- 108090000623 proteins and genes Proteins 0.000 claims description 67
- 229920001184 polypeptide Polymers 0.000 claims description 66
- 230000004068 intracellular signaling Effects 0.000 claims description 60
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 56
- 239000003795 chemical substances by application Substances 0.000 claims description 53
- 201000010099 disease Diseases 0.000 claims description 49
- 150000007523 nucleic acids Chemical class 0.000 claims description 45
- 230000002401 inhibitory effect Effects 0.000 claims description 43
- 102100040678 Programmed cell death protein 1 Human genes 0.000 claims description 42
- 101710089372 Programmed cell death protein 1 Proteins 0.000 claims description 42
- 239000003112 inhibitor Substances 0.000 claims description 42
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 39
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 claims description 36
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 claims description 36
- 102100024263 CD160 antigen Human genes 0.000 claims description 34
- 101000761938 Homo sapiens CD160 antigen Proteins 0.000 claims description 34
- 102000039446 nucleic acids Human genes 0.000 claims description 33
- 108020004707 nucleic acids Proteins 0.000 claims description 33
- 241000282414 Homo sapiens Species 0.000 claims description 32
- 230000000694 effects Effects 0.000 claims description 30
- 102000004169 proteins and genes Human genes 0.000 claims description 30
- 102100029215 Signaling lymphocytic activation molecule Human genes 0.000 claims description 28
- 230000000139 costimulatory effect Effects 0.000 claims description 28
- 230000003834 intracellular effect Effects 0.000 claims description 28
- 102000004127 Cytokines Human genes 0.000 claims description 27
- 108090000695 Cytokines Proteins 0.000 claims description 27
- 210000000822 natural killer cell Anatomy 0.000 claims description 27
- 229940043355 kinase inhibitor Drugs 0.000 claims description 26
- 239000003757 phosphotransferase inhibitor Substances 0.000 claims description 26
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 claims description 25
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 claims description 25
- 102000008394 Immunoglobulin Fragments Human genes 0.000 claims description 24
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 claims description 23
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 claims description 23
- 239000000203 mixture Substances 0.000 claims description 23
- 108010021625 Immunoglobulin Fragments Proteins 0.000 claims description 22
- 102100027207 CD27 antigen Human genes 0.000 claims description 21
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 claims description 21
- 101000633786 Homo sapiens SLAM family member 6 Proteins 0.000 claims description 21
- 102100032816 Integrin alpha-6 Human genes 0.000 claims description 21
- 102100029197 SLAM family member 6 Human genes 0.000 claims description 21
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 claims description 19
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 claims description 19
- 108091008874 T cell receptors Proteins 0.000 claims description 19
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 claims description 19
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 claims description 19
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 claims description 19
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 claims description 19
- 102000005962 receptors Human genes 0.000 claims description 19
- 108020003175 receptors Proteins 0.000 claims description 19
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 claims description 18
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 claims description 18
- 101000994375 Homo sapiens Integrin alpha-4 Proteins 0.000 claims description 18
- 101000935040 Homo sapiens Integrin beta-2 Proteins 0.000 claims description 18
- 102100032818 Integrin alpha-4 Human genes 0.000 claims description 18
- 102100025390 Integrin beta-2 Human genes 0.000 claims description 18
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 18
- 229940124302 mTOR inhibitor Drugs 0.000 claims description 18
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 claims description 18
- 101001078158 Homo sapiens Integrin alpha-1 Proteins 0.000 claims description 17
- 102100025323 Integrin alpha-1 Human genes 0.000 claims description 17
- 102100038083 Endosialin Human genes 0.000 claims description 16
- 101000851370 Homo sapiens Tumor necrosis factor receptor superfamily member 9 Proteins 0.000 claims description 16
- 102100036856 Tumor necrosis factor receptor superfamily member 9 Human genes 0.000 claims description 16
- 101000971538 Homo sapiens Killer cell lectin-like receptor subfamily F member 1 Proteins 0.000 claims description 15
- 102100022338 Integrin alpha-M Human genes 0.000 claims description 15
- 102100021458 Killer cell lectin-like receptor subfamily F member 1 Human genes 0.000 claims description 15
- 102100038082 Natural killer cell receptor 2B4 Human genes 0.000 claims description 15
- 102100027744 Semaphorin-4D Human genes 0.000 claims description 15
- 108010074687 Signaling Lymphocytic Activation Molecule Family Member 1 Proteins 0.000 claims description 15
- 101000884275 Homo sapiens Endosialin Proteins 0.000 claims description 14
- 101000994365 Homo sapiens Integrin alpha-6 Proteins 0.000 claims description 14
- 101001035237 Homo sapiens Integrin alpha-D Proteins 0.000 claims description 14
- 101000935043 Homo sapiens Integrin beta-1 Proteins 0.000 claims description 14
- 101000633780 Homo sapiens Signaling lymphocytic activation molecule Proteins 0.000 claims description 14
- 102100039904 Integrin alpha-D Human genes 0.000 claims description 14
- 102100022297 Integrin alpha-X Human genes 0.000 claims description 14
- 102100025304 Integrin beta-1 Human genes 0.000 claims description 14
- 239000003814 drug Substances 0.000 claims description 14
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 13
- 102100028785 Tumor necrosis factor receptor superfamily member 14 Human genes 0.000 claims description 13
- 102100022153 Tumor necrosis factor receptor superfamily member 4 Human genes 0.000 claims description 13
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 claims description 12
- 108010007712 Hepatitis A Virus Cellular Receptor 1 Proteins 0.000 claims description 12
- 102100034459 Hepatitis A virus cellular receptor 1 Human genes 0.000 claims description 12
- 101001046687 Homo sapiens Integrin alpha-E Proteins 0.000 claims description 12
- 101000971605 Homo sapiens Kita-kyushu lung cancer antigen 1 Proteins 0.000 claims description 12
- 101000633784 Homo sapiens SLAM family member 7 Proteins 0.000 claims description 12
- 108060003951 Immunoglobulin Proteins 0.000 claims description 12
- 102100022341 Integrin alpha-E Human genes 0.000 claims description 12
- 102100021533 Kita-kyushu lung cancer antigen 1 Human genes 0.000 claims description 12
- 102100032364 Pannexin-3 Human genes 0.000 claims description 12
- 101710165197 Pannexin-3 Proteins 0.000 claims description 12
- 102100029198 SLAM family member 7 Human genes 0.000 claims description 12
- 102000013532 Uroplakin II Human genes 0.000 claims description 12
- 108010065940 Uroplakin II Proteins 0.000 claims description 12
- 102000018358 immunoglobulin Human genes 0.000 claims description 12
- 102100026094 C-type lectin domain family 12 member A Human genes 0.000 claims description 11
- 102100038078 CD276 antigen Human genes 0.000 claims description 11
- 102100021197 G-protein coupled receptor family C group 5 member D Human genes 0.000 claims description 11
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 claims description 11
- 101001040713 Homo sapiens G-protein coupled receptor family C group 5 member D Proteins 0.000 claims description 11
- 101000878602 Homo sapiens Immunoglobulin alpha Fc receptor Proteins 0.000 claims description 11
- 102100038005 Immunoglobulin alpha Fc receptor Human genes 0.000 claims description 11
- 102000003812 Interleukin-15 Human genes 0.000 claims description 11
- 108090000172 Interleukin-15 Proteins 0.000 claims description 11
- 102100026181 Placenta-specific protein 1 Human genes 0.000 claims description 11
- 108050005093 Placenta-specific protein 1 Proteins 0.000 claims description 11
- 229940079593 drug Drugs 0.000 claims description 11
- 239000003446 ligand Substances 0.000 claims description 11
- 102100033793 ALK tyrosine kinase receptor Human genes 0.000 claims description 10
- 102100026423 Adhesion G protein-coupled receptor E5 Human genes 0.000 claims description 10
- 102100023003 Ankyrin repeat domain-containing protein 30A Human genes 0.000 claims description 10
- 102100031507 Fc receptor-like protein 5 Human genes 0.000 claims description 10
- 101000718243 Homo sapiens Adhesion G protein-coupled receptor E5 Proteins 0.000 claims description 10
- 101000757191 Homo sapiens Ankyrin repeat domain-containing protein 30A Proteins 0.000 claims description 10
- 101710196509 Leukocyte immunoglobulin-like receptor subfamily A member 2 Proteins 0.000 claims description 10
- 102100025586 Leukocyte immunoglobulin-like receptor subfamily A member 2 Human genes 0.000 claims description 10
- 102100032129 Lymphocyte antigen 6K Human genes 0.000 claims description 10
- 102100033486 Lymphocyte antigen 75 Human genes 0.000 claims description 10
- 101710157884 Lymphocyte antigen 75 Proteins 0.000 claims description 10
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 claims description 10
- 206010039491 Sarcoma Diseases 0.000 claims description 10
- 101710165473 Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 claims description 10
- 239000002253 acid Substances 0.000 claims description 10
- 108010003374 fms-Like Tyrosine Kinase 3 Proteins 0.000 claims description 10
- 201000001441 melanoma Diseases 0.000 claims description 10
- 101710109924 A-kinase anchor protein 4 Proteins 0.000 claims description 9
- 102100037982 Alpha-1,6-mannosylglycoprotein 6-beta-N-acetylglucosaminyltransferase A Human genes 0.000 claims description 9
- 101000846908 Homo sapiens Fc receptor-like protein 5 Proteins 0.000 claims description 9
- 101000795169 Homo sapiens Tumor necrosis factor receptor superfamily member 13C Proteins 0.000 claims description 9
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 claims description 9
- 101001047681 Homo sapiens Tyrosine-protein kinase Lck Proteins 0.000 claims description 9
- 101000814512 Homo sapiens X antigen family member 1 Proteins 0.000 claims description 9
- 102100025128 Olfactory receptor 51E2 Human genes 0.000 claims description 9
- 101710187841 Olfactory receptor 51E2 Proteins 0.000 claims description 9
- 101710120463 Prostate stem cell antigen Proteins 0.000 claims description 9
- 102100036735 Prostate stem cell antigen Human genes 0.000 claims description 9
- 102100027610 Rho-related GTP-binding protein RhoC Human genes 0.000 claims description 9
- 108010017842 Telomerase Proteins 0.000 claims description 9
- 102100021393 Transcriptional repressor CTCFL Human genes 0.000 claims description 9
- 102100029690 Tumor necrosis factor receptor superfamily member 13C Human genes 0.000 claims description 9
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 claims description 9
- 102100024036 Tyrosine-protein kinase Lck Human genes 0.000 claims description 9
- 102100039490 X antigen family member 1 Human genes 0.000 claims description 9
- 108010034034 alpha-1,6-mannosylglycoprotein beta 1,6-N-acetylglucosaminyltransferase Proteins 0.000 claims description 9
- 210000004698 lymphocyte Anatomy 0.000 claims description 9
- 108010073531 rhoC GTP-Binding Protein Proteins 0.000 claims description 9
- 108010088201 squamous cell carcinoma-related antigen Proteins 0.000 claims description 9
- 230000005945 translocation Effects 0.000 claims description 9
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 claims description 8
- 102100026402 Adhesion G protein-coupled receptor E2 Human genes 0.000 claims description 8
- 101710145634 Antigen 1 Proteins 0.000 claims description 8
- 102100025218 B-cell differentiation antigen CD72 Human genes 0.000 claims description 8
- 102100038080 B-cell receptor CD22 Human genes 0.000 claims description 8
- 108010056102 CD100 antigen Proteins 0.000 claims description 8
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 claims description 8
- 101000718211 Homo sapiens Adhesion G protein-coupled receptor E2 Proteins 0.000 claims description 8
- 101000934359 Homo sapiens B-cell differentiation antigen CD72 Proteins 0.000 claims description 8
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 claims description 8
- 101000608769 Homo sapiens Galectin-8 Proteins 0.000 claims description 8
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 claims description 8
- 101001109503 Homo sapiens NKG2-C type II integral membrane protein Proteins 0.000 claims description 8
- 101001109501 Homo sapiens NKG2-D type II integral membrane protein Proteins 0.000 claims description 8
- 101000589305 Homo sapiens Natural cytotoxicity triggering receptor 2 Proteins 0.000 claims description 8
- 101000824971 Homo sapiens Sperm surface protein Sp17 Proteins 0.000 claims description 8
- 108010061593 Member 14 Tumor Necrosis Factor Receptors Proteins 0.000 claims description 8
- 102100022683 NKG2-C type II integral membrane protein Human genes 0.000 claims description 8
- 102100022680 NKG2-D type II integral membrane protein Human genes 0.000 claims description 8
- 108010004217 Natural Cytotoxicity Triggering Receptor 1 Proteins 0.000 claims description 8
- 108010004222 Natural Cytotoxicity Triggering Receptor 3 Proteins 0.000 claims description 8
- 102100032870 Natural cytotoxicity triggering receptor 1 Human genes 0.000 claims description 8
- 102100032851 Natural cytotoxicity triggering receptor 2 Human genes 0.000 claims description 8
- 102100032852 Natural cytotoxicity triggering receptor 3 Human genes 0.000 claims description 8
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 claims description 8
- 108010003723 Single-Domain Antibodies Proteins 0.000 claims description 8
- 102100031989 Transmembrane protease serine 2 Human genes 0.000 claims description 8
- 210000004369 blood Anatomy 0.000 claims description 8
- 239000008280 blood Substances 0.000 claims description 8
- 239000012636 effector Substances 0.000 claims description 8
- 230000002062 proliferating effect Effects 0.000 claims description 8
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 claims description 7
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 7
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 7
- 102100032187 Androgen receptor Human genes 0.000 claims description 7
- 208000025324 B-cell acute lymphoblastic leukemia Diseases 0.000 claims description 7
- 108010017009 CD11b Antigen Proteins 0.000 claims description 7
- 102100038077 CD226 antigen Human genes 0.000 claims description 7
- 101150013553 CD40 gene Proteins 0.000 claims description 7
- 108010058905 CD44v6 antigen Proteins 0.000 claims description 7
- 102100027217 CD82 antigen Human genes 0.000 claims description 7
- 101710139831 CD82 antigen Proteins 0.000 claims description 7
- 101100518995 Caenorhabditis elegans pax-3 gene Proteins 0.000 claims description 7
- 102100027417 Cytochrome P450 1B1 Human genes 0.000 claims description 7
- 101100481408 Danio rerio tie2 gene Proteins 0.000 claims description 7
- 108010055196 EphA2 Receptor Proteins 0.000 claims description 7
- 102100030340 Ephrin type-A receptor 2 Human genes 0.000 claims description 7
- 101000585551 Equus caballus Pregnancy-associated glycoprotein Proteins 0.000 claims description 7
- 102000010449 Folate receptor beta Human genes 0.000 claims description 7
- 108050001930 Folate receptor beta Proteins 0.000 claims description 7
- 102000003817 Fos-related antigen 1 Human genes 0.000 claims description 7
- 108090000123 Fos-related antigen 1 Proteins 0.000 claims description 7
- 102100039554 Galectin-8 Human genes 0.000 claims description 7
- 101000884298 Homo sapiens CD226 antigen Proteins 0.000 claims description 7
- 101000884279 Homo sapiens CD276 antigen Proteins 0.000 claims description 7
- 101000725164 Homo sapiens Cytochrome P450 1B1 Proteins 0.000 claims description 7
- 101001046683 Homo sapiens Integrin alpha-L Proteins 0.000 claims description 7
- 101001046668 Homo sapiens Integrin alpha-X Proteins 0.000 claims description 7
- 101001015037 Homo sapiens Integrin beta-7 Proteins 0.000 claims description 7
- 101001043809 Homo sapiens Interleukin-7 receptor subunit alpha Proteins 0.000 claims description 7
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 claims description 7
- 101001051490 Homo sapiens Neural cell adhesion molecule L1 Proteins 0.000 claims description 7
- 101000873418 Homo sapiens P-selectin glycoprotein ligand 1 Proteins 0.000 claims description 7
- 101001124867 Homo sapiens Peroxiredoxin-1 Proteins 0.000 claims description 7
- 101000692259 Homo sapiens Phosphoprotein associated with glycosphingolipid-enriched microdomains 1 Proteins 0.000 claims description 7
- 101001136981 Homo sapiens Proteasome subunit beta type-9 Proteins 0.000 claims description 7
- 101000633778 Homo sapiens SLAM family member 5 Proteins 0.000 claims description 7
- 101000934346 Homo sapiens T-cell surface antigen CD2 Proteins 0.000 claims description 7
- 101000596234 Homo sapiens T-cell surface protein tactile Proteins 0.000 claims description 7
- 101000638154 Homo sapiens Transmembrane protease serine 2 Proteins 0.000 claims description 7
- 101000679857 Homo sapiens Tumor necrosis factor receptor superfamily member 3 Proteins 0.000 claims description 7
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 claims description 7
- 102100022339 Integrin alpha-L Human genes 0.000 claims description 7
- 108010041100 Integrin alpha6 Proteins 0.000 claims description 7
- 108010030465 Integrin alpha6beta1 Proteins 0.000 claims description 7
- 102100033016 Integrin beta-7 Human genes 0.000 claims description 7
- 102100021593 Interleukin-7 receptor subunit alpha Human genes 0.000 claims description 7
- 108700012912 MYCN Proteins 0.000 claims description 7
- 101150022024 MYCN gene Proteins 0.000 claims description 7
- 102000003735 Mesothelin Human genes 0.000 claims description 7
- 108090000015 Mesothelin Proteins 0.000 claims description 7
- 101100518997 Mus musculus Pax3 gene Proteins 0.000 claims description 7
- 101100481410 Mus musculus Tek gene Proteins 0.000 claims description 7
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 claims description 7
- 108700026495 N-Myc Proto-Oncogene Proteins 0.000 claims description 7
- 102100030124 N-myc proto-oncogene protein Human genes 0.000 claims description 7
- 101710141230 Natural killer cell receptor 2B4 Proteins 0.000 claims description 7
- 102100024964 Neural cell adhesion molecule L1 Human genes 0.000 claims description 7
- 102100034925 P-selectin glycoprotein ligand 1 Human genes 0.000 claims description 7
- 102100026066 Phosphoprotein associated with glycosphingolipid-enriched microdomains 1 Human genes 0.000 claims description 7
- 108091000080 Phosphotransferase Proteins 0.000 claims description 7
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 7
- 102100035764 Proteasome subunit beta type-9 Human genes 0.000 claims description 7
- 102100029216 SLAM family member 5 Human genes 0.000 claims description 7
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 claims description 7
- 102100035268 T-cell surface protein tactile Human genes 0.000 claims description 7
- 102100033733 Tumor necrosis factor receptor superfamily member 1B Human genes 0.000 claims description 7
- 101710187830 Tumor necrosis factor receptor superfamily member 1B Proteins 0.000 claims description 7
- 102100022156 Tumor necrosis factor receptor superfamily member 3 Human genes 0.000 claims description 7
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 claims description 7
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 claims description 7
- 102100022748 Wilms tumor protein Human genes 0.000 claims description 7
- 101710127857 Wilms tumor protein Proteins 0.000 claims description 7
- 108010080146 androgen receptors Proteins 0.000 claims description 7
- 208000035475 disorder Diseases 0.000 claims description 7
- 125000002446 fucosyl group Chemical group C1([C@@H](O)[C@H](O)[C@H](O)[C@@H](O1)C)* 0.000 claims description 7
- 230000004927 fusion Effects 0.000 claims description 7
- PFJKOHUKELZMLE-VEUXDRLPSA-N ganglioside GM3 Chemical compound O[C@@H]1[C@@H](O)[C@H](OC[C@@H]([C@H](O)/C=C/CCCCCCCCCCCCC)NC(=O)CCCCCCCCCCCCC\C=C/CCCCCCCC)O[C@H](CO)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@H](O)CO)C(O)=O)[C@@H](O)[C@@H](CO)O1 PFJKOHUKELZMLE-VEUXDRLPSA-N 0.000 claims description 7
- 102000020233 phosphotransferase Human genes 0.000 claims description 7
- 210000003289 regulatory T cell Anatomy 0.000 claims description 7
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 6
- 108010062802 CD66 antigens Proteins 0.000 claims description 6
- 102100024533 Carcinoembryonic antigen-related cell adhesion molecule 1 Human genes 0.000 claims description 6
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 6
- 102100035167 Coiled-coil domain-containing protein 54 Human genes 0.000 claims description 6
- 101000737052 Homo sapiens Coiled-coil domain-containing protein 54 Proteins 0.000 claims description 6
- 101000998120 Homo sapiens Interleukin-3 receptor subunit alpha Proteins 0.000 claims description 6
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 claims description 6
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 claims description 6
- 102100033493 Interleukin-3 receptor subunit alpha Human genes 0.000 claims description 6
- 206010025323 Lymphomas Diseases 0.000 claims description 6
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 6
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 6
- 241000700605 Viruses Species 0.000 claims description 6
- 201000003444 follicular lymphoma Diseases 0.000 claims description 6
- 239000003934 phosphoprotein phosphatase inhibitor Substances 0.000 claims description 6
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 5
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 claims description 5
- 101710188619 C-type lectin domain family 12 member A Proteins 0.000 claims description 5
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 claims description 5
- 102100039510 Cancer/testis antigen 2 Human genes 0.000 claims description 5
- 102100025137 Early activation antigen CD69 Human genes 0.000 claims description 5
- 108010021468 Fc gamma receptor IIA Proteins 0.000 claims description 5
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 claims description 5
- 102100029360 Hematopoietic cell signal transducer Human genes 0.000 claims description 5
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 claims description 5
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 claims description 5
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 claims description 5
- 101000990188 Homo sapiens Hematopoietic cell signal transducer Proteins 0.000 claims description 5
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 claims description 5
- 101000809875 Homo sapiens TYRO protein tyrosine kinase-binding protein Proteins 0.000 claims description 5
- 101000655352 Homo sapiens Telomerase reverse transcriptase Proteins 0.000 claims description 5
- 101000648507 Homo sapiens Tumor necrosis factor receptor superfamily member 14 Proteins 0.000 claims description 5
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 claims description 5
- 102100029204 Low affinity immunoglobulin gamma Fc region receptor II-a Human genes 0.000 claims description 5
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 claims description 5
- 201000003793 Myelodysplastic syndrome Diseases 0.000 claims description 5
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 claims description 5
- 101800001271 Surface protein Proteins 0.000 claims description 5
- 208000029052 T-cell acute lymphoblastic leukemia Diseases 0.000 claims description 5
- 102100038717 TYRO protein tyrosine kinase-binding protein Human genes 0.000 claims description 5
- 230000009977 dual effect Effects 0.000 claims description 5
- 230000031146 intracellular signal transduction Effects 0.000 claims description 5
- 230000003211 malignant effect Effects 0.000 claims description 5
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 claims description 4
- 101710185679 CD276 antigen Proteins 0.000 claims description 4
- 108010051152 Carboxylesterase Proteins 0.000 claims description 4
- 102000013392 Carboxylesterase Human genes 0.000 claims description 4
- 102100027816 Cytotoxic and regulatory T-cell molecule Human genes 0.000 claims description 4
- 101150029707 ERBB2 gene Proteins 0.000 claims description 4
- 108091006027 G proteins Proteins 0.000 claims description 4
- 102100022086 GRB2-related adapter protein 2 Human genes 0.000 claims description 4
- 102000030782 GTP binding Human genes 0.000 claims description 4
- 108091000058 GTP-Binding Proteins 0.000 claims description 4
- 208000017604 Hodgkin disease Diseases 0.000 claims description 4
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 4
- 101000856237 Homo sapiens Cancer/testis antigen 1 Proteins 0.000 claims description 4
- 101000889345 Homo sapiens Cancer/testis antigen 2 Proteins 0.000 claims description 4
- 101000900690 Homo sapiens GRB2-related adapter protein 2 Proteins 0.000 claims description 4
- 101000777628 Homo sapiens Leukocyte antigen CD37 Proteins 0.000 claims description 4
- 101001047640 Homo sapiens Linker for activation of T-cells family member 1 Proteins 0.000 claims description 4
- 101001090688 Homo sapiens Lymphocyte cytosolic protein 2 Proteins 0.000 claims description 4
- 101001014223 Homo sapiens MAPK/MAK/MRK overlapping kinase Proteins 0.000 claims description 4
- 101000578784 Homo sapiens Melanoma antigen recognized by T-cells 1 Proteins 0.000 claims description 4
- 101000702132 Homo sapiens Protein spinster homolog 1 Proteins 0.000 claims description 4
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 claims description 4
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 claims description 4
- 101001094545 Homo sapiens Retrotransposon-like protein 1 Proteins 0.000 claims description 4
- 108010031794 IGF Type 1 Receptor Proteins 0.000 claims description 4
- 102100034872 Kallikrein-4 Human genes 0.000 claims description 4
- 102100031586 Leukocyte antigen CD37 Human genes 0.000 claims description 4
- 102100024032 Linker for activation of T-cells family member 1 Human genes 0.000 claims description 4
- 102100034709 Lymphocyte cytosolic protein 2 Human genes 0.000 claims description 4
- 102100031520 MAPK/MAK/MRK overlapping kinase Human genes 0.000 claims description 4
- 108010010995 MART-1 Antigen Proteins 0.000 claims description 4
- 102000016200 MART-1 Antigen Human genes 0.000 claims description 4
- 102100022430 Melanocyte protein PMEL Human genes 0.000 claims description 4
- 102100028389 Melanoma antigen recognized by T-cells 1 Human genes 0.000 claims description 4
- 101100236305 Mus musculus Ly9 gene Proteins 0.000 claims description 4
- 102000048850 Neoplasm Genes Human genes 0.000 claims description 4
- 108700019961 Neoplasm Genes Proteins 0.000 claims description 4
- 208000006994 Precancerous Conditions Diseases 0.000 claims description 4
- 208000009052 Precursor T-Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 4
- 108010025832 RANK Ligand Proteins 0.000 claims description 4
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 claims description 4
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 claims description 4
- 102100021657 Tyrosine-protein phosphatase non-receptor type 6 Human genes 0.000 claims description 4
- 101710128901 Tyrosine-protein phosphatase non-receptor type 6 Proteins 0.000 claims description 4
- 101001038499 Yarrowia lipolytica (strain CLIB 122 / E 150) Lysine acetyltransferase Proteins 0.000 claims description 4
- 108010072917 class-I restricted T cell-associated molecule Proteins 0.000 claims description 4
- 230000004069 differentiation Effects 0.000 claims description 4
- 108010087914 epidermal growth factor receptor VIII Proteins 0.000 claims description 4
- 102000006815 folate receptor Human genes 0.000 claims description 4
- 108020005243 folate receptor Proteins 0.000 claims description 4
- 230000000968 intestinal effect Effects 0.000 claims description 4
- 108010024383 kallikrein 4 Proteins 0.000 claims description 4
- 208000011580 syndromic disease Diseases 0.000 claims description 4
- 210000001550 testis Anatomy 0.000 claims description 4
- 208000036170 B-Cell Marginal Zone Lymphoma Diseases 0.000 claims description 3
- 208000003950 B-cell lymphoma Diseases 0.000 claims description 3
- 102100032937 CD40 ligand Human genes 0.000 claims description 3
- 102100037904 CD9 antigen Human genes 0.000 claims description 3
- 229940124297 CDK 4/6 inhibitor Drugs 0.000 claims description 3
- 206010061850 Extranodal marginal zone B-cell lymphoma (MALT type) Diseases 0.000 claims description 3
- 102100026122 High affinity immunoglobulin gamma Fc receptor I Human genes 0.000 claims description 3
- 208000021519 Hodgkin lymphoma Diseases 0.000 claims description 3
- 101000738354 Homo sapiens CD9 antigen Proteins 0.000 claims description 3
- 101000913074 Homo sapiens High affinity immunoglobulin gamma Fc receptor I Proteins 0.000 claims description 3
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 claims description 3
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 claims description 3
- 101000934341 Homo sapiens T-cell surface glycoprotein CD5 Proteins 0.000 claims description 3
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 claims description 3
- 208000019758 Hypergammaglobulinemia Diseases 0.000 claims description 3
- 201000003791 MALT lymphoma Diseases 0.000 claims description 3
- 208000034578 Multiple myelomas Diseases 0.000 claims description 3
- 108091005804 Peptidases Proteins 0.000 claims description 3
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 3
- 239000004365 Protease Substances 0.000 claims description 3
- 229940116193 Protein phosphatase inhibitor Drugs 0.000 claims description 3
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 claims description 3
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 claims description 3
- 102100033019 Tyrosine-protein phosphatase non-receptor type 11 Human genes 0.000 claims description 3
- 101710116241 Tyrosine-protein phosphatase non-receptor type 11 Proteins 0.000 claims description 3
- 230000008556 epithelial cell proliferation Effects 0.000 claims description 3
- 201000009277 hairy cell leukemia Diseases 0.000 claims description 3
- 201000007924 marginal zone B-cell lymphoma Diseases 0.000 claims description 3
- 208000021937 marginal zone lymphoma Diseases 0.000 claims description 3
- 238000012737 microarray-based gene expression Methods 0.000 claims description 3
- 238000012243 multiplex automated genomic engineering Methods 0.000 claims description 3
- 206010000830 Acute leukaemia Diseases 0.000 claims description 2
- UCTWMZQNUQWSLP-UHFFFAOYSA-N Adrenaline Natural products CNCC(O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-UHFFFAOYSA-N 0.000 claims description 2
- 108091007065 BIRCs Proteins 0.000 claims description 2
- 108091033409 CRISPR Proteins 0.000 claims description 2
- 238000010354 CRISPR gene editing Methods 0.000 claims description 2
- 102000014914 Carrier Proteins Human genes 0.000 claims description 2
- 108010067225 Cell Adhesion Molecules Proteins 0.000 claims description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 claims description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 claims description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 claims description 2
- 208000017815 Dendritic cell tumor Diseases 0.000 claims description 2
- 108010042407 Endonucleases Proteins 0.000 claims description 2
- 101710144543 Endosialin Proteins 0.000 claims description 2
- 102100031547 HLA class II histocompatibility antigen, DO alpha chain Human genes 0.000 claims description 2
- 101100118545 Holotrichia diomphalia EGF-like gene Proteins 0.000 claims description 2
- 101000866278 Homo sapiens HLA class II histocompatibility antigen, DO alpha chain Proteins 0.000 claims description 2
- 102100039615 Inactive tyrosine-protein kinase transmembrane receptor ROR1 Human genes 0.000 claims description 2
- 102000055031 Inhibitor of Apoptosis Proteins Human genes 0.000 claims description 2
- 108010017521 Interleukin-11 Receptors Proteins 0.000 claims description 2
- 102000004553 Interleukin-11 Receptors Human genes 0.000 claims description 2
- 206010029260 Neuroblastoma Diseases 0.000 claims description 2
- KUIFHYPNNRVEKZ-VIJRYAKMSA-N O-(N-acetyl-alpha-D-galactosaminyl)-L-threonine Chemical compound OC(=O)[C@@H](N)[C@@H](C)O[C@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O KUIFHYPNNRVEKZ-VIJRYAKMSA-N 0.000 claims description 2
- 102100040891 Paired box protein Pax-3 Human genes 0.000 claims description 2
- 101710149060 Paired box protein Pax-3 Proteins 0.000 claims description 2
- 102100037504 Paired box protein Pax-5 Human genes 0.000 claims description 2
- 102100037891 Plexin domain-containing protein 1 Human genes 0.000 claims description 2
- 108050009432 Plexin domain-containing protein 1 Proteins 0.000 claims description 2
- 102100023832 Prolyl endopeptidase FAP Human genes 0.000 claims description 2
- 102100022441 Sperm surface protein Sp17 Human genes 0.000 claims description 2
- 108060008724 Tyrosinase Proteins 0.000 claims description 2
- 101710101493 Viral myc transforming protein Proteins 0.000 claims description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 2
- 108091008324 binding proteins Proteins 0.000 claims description 2
- 102000008395 cell adhesion mediator activity proteins Human genes 0.000 claims description 2
- 230000004663 cell proliferation Effects 0.000 claims description 2
- 229960004397 cyclophosphamide Drugs 0.000 claims description 2
- 230000003259 immunoinhibitory effect Effects 0.000 claims description 2
- 210000000265 leukocyte Anatomy 0.000 claims description 2
- 210000002569 neuron Anatomy 0.000 claims description 2
- 101710135378 pH 6 antigen Proteins 0.000 claims description 2
- 208000007525 plasmablastic lymphoma Diseases 0.000 claims description 2
- 108010013873 proacrosin Proteins 0.000 claims description 2
- 125000005630 sialyl group Chemical group 0.000 claims description 2
- 210000003556 vascular endothelial cell Anatomy 0.000 claims description 2
- 239000011701 zinc Substances 0.000 claims description 2
- 229910052725 zinc Inorganic materials 0.000 claims description 2
- 108010051118 Bone Marrow Stromal Antigen 2 Proteins 0.000 claims 2
- 102100037086 Bone marrow stromal antigen 2 Human genes 0.000 claims 2
- 102100029390 CMRF35-like molecule 1 Human genes 0.000 claims 2
- 102100038449 Claudin-6 Human genes 0.000 claims 2
- 108090000229 Claudin-6 Proteins 0.000 claims 2
- 101000990055 Homo sapiens CMRF35-like molecule 1 Proteins 0.000 claims 2
- 101000801234 Homo sapiens Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 claims 2
- 102100020793 Interleukin-13 receptor subunit alpha-2 Human genes 0.000 claims 2
- 101710112634 Interleukin-13 receptor subunit alpha-2 Proteins 0.000 claims 2
- 101710164680 Platelet-derived growth factor receptor beta Proteins 0.000 claims 2
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 claims 2
- 102100033728 Tumor necrosis factor receptor superfamily member 18 Human genes 0.000 claims 2
- BGFTWECWAICPDG-UHFFFAOYSA-N 2-[bis(4-chlorophenyl)methyl]-4-n-[3-[bis(4-chlorophenyl)methyl]-4-(dimethylamino)phenyl]-1-n,1-n-dimethylbenzene-1,4-diamine Chemical compound C1=C(C(C=2C=CC(Cl)=CC=2)C=2C=CC(Cl)=CC=2)C(N(C)C)=CC=C1NC(C=1)=CC=C(N(C)C)C=1C(C=1C=CC(Cl)=CC=1)C1=CC=C(Cl)C=C1 BGFTWECWAICPDG-UHFFFAOYSA-N 0.000 claims 1
- LKKMLIBUAXYLOY-UHFFFAOYSA-N 3-Amino-1-methyl-5H-pyrido[4,3-b]indole Chemical compound N1C2=CC=CC=C2C2=C1C=C(N)N=C2C LKKMLIBUAXYLOY-UHFFFAOYSA-N 0.000 claims 1
- 102000006942 B-Cell Maturation Antigen Human genes 0.000 claims 1
- 108010008014 B-Cell Maturation Antigen Proteins 0.000 claims 1
- 102100021663 Baculoviral IAP repeat-containing protein 5 Human genes 0.000 claims 1
- 108010071965 CD24 Antigen Proteins 0.000 claims 1
- 102000007645 CD24 Antigen Human genes 0.000 claims 1
- 101710178046 Chorismate synthase 1 Proteins 0.000 claims 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 claims 1
- 108010025464 Cyclin-Dependent Kinase 4 Proteins 0.000 claims 1
- 102100036252 Cyclin-dependent kinase 4 Human genes 0.000 claims 1
- 101710152695 Cysteine synthase 1 Proteins 0.000 claims 1
- 102000012804 EPCAM Human genes 0.000 claims 1
- 101150084967 EPCAM gene Proteins 0.000 claims 1
- 102000004533 Endonucleases Human genes 0.000 claims 1
- 101150032879 Fcrl5 gene Proteins 0.000 claims 1
- 102000044445 Galectin-8 Human genes 0.000 claims 1
- 101710088083 Glomulin Proteins 0.000 claims 1
- 102100022132 High affinity immunoglobulin epsilon receptor subunit gamma Human genes 0.000 claims 1
- 101000914324 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 5 Proteins 0.000 claims 1
- 101000914321 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 7 Proteins 0.000 claims 1
- 101000954709 Homo sapiens Doublecortin domain-containing protein 2 Proteins 0.000 claims 1
- 101000985516 Homo sapiens Hermansky-Pudlak syndrome 5 protein Proteins 0.000 claims 1
- 101000824104 Homo sapiens High affinity immunoglobulin epsilon receptor subunit gamma Proteins 0.000 claims 1
- 101000840267 Homo sapiens Immunoglobulin lambda-like polypeptide 1 Proteins 0.000 claims 1
- 101001103039 Homo sapiens Inactive tyrosine-protein kinase transmembrane receptor ROR1 Proteins 0.000 claims 1
- 101000614481 Homo sapiens Kidney-associated antigen 1 Proteins 0.000 claims 1
- 101001065550 Homo sapiens Lymphocyte antigen 6K Proteins 0.000 claims 1
- 101001103036 Homo sapiens Nuclear receptor ROR-alpha Proteins 0.000 claims 1
- 101000601724 Homo sapiens Paired box protein Pax-5 Proteins 0.000 claims 1
- 101000617725 Homo sapiens Pregnancy-specific beta-1-glycoprotein 2 Proteins 0.000 claims 1
- 101000880770 Homo sapiens Protein SSX2 Proteins 0.000 claims 1
- 101000665137 Homo sapiens Scm-like with four MBT domains protein 1 Proteins 0.000 claims 1
- 101000772267 Homo sapiens Thyrotropin receptor Proteins 0.000 claims 1
- 102000004867 Hydro-Lyases Human genes 0.000 claims 1
- 108090001042 Hydro-Lyases Proteins 0.000 claims 1
- 102100029616 Immunoglobulin lambda-like polypeptide 1 Human genes 0.000 claims 1
- 102000004556 Interleukin-15 Receptors Human genes 0.000 claims 1
- 108010017535 Interleukin-15 Receptors Proteins 0.000 claims 1
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 claims 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims 1
- 208000028018 Lymphocytic leukaemia Diseases 0.000 claims 1
- 101100341510 Mus musculus Itgal gene Proteins 0.000 claims 1
- 101710163270 Nuclease Proteins 0.000 claims 1
- 102100022019 Pregnancy-specific beta-1-glycoprotein 2 Human genes 0.000 claims 1
- 102100032831 Protein ITPRID2 Human genes 0.000 claims 1
- 102100037686 Protein SSX2 Human genes 0.000 claims 1
- 102000014128 RANK Ligand Human genes 0.000 claims 1
- 241001522306 Serinus serinus Species 0.000 claims 1
- 108010002687 Survivin Proteins 0.000 claims 1
- 101150057140 TACSTD1 gene Proteins 0.000 claims 1
- 102100029337 Thyrotropin receptor Human genes 0.000 claims 1
- 102100039094 Tyrosinase Human genes 0.000 claims 1
- MXKCYTKUIDTFLY-ZNNSSXPHSA-N alpha-L-Fucp-(1->2)-beta-D-Galp-(1->4)-[alpha-L-Fucp-(1->3)]-beta-D-GlcpNAc-(1->3)-D-Galp Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]1[C@H](O[C@H]2[C@@H]([C@@H](NC(C)=O)[C@H](O[C@H]3[C@H]([C@@H](CO)OC(O)[C@@H]3O)O)O[C@@H]2CO)O[C@H]2[C@H]([C@H](O)[C@H](O)[C@H](C)O2)O)O[C@H](CO)[C@H](O)[C@@H]1O MXKCYTKUIDTFLY-ZNNSSXPHSA-N 0.000 claims 1
- 230000001413 cellular effect Effects 0.000 claims 1
- 229910052804 chromium Inorganic materials 0.000 claims 1
- 239000011651 chromium Substances 0.000 claims 1
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 claims 1
- 239000005556 hormone Substances 0.000 claims 1
- 229940088597 hormone Drugs 0.000 claims 1
- 208000003747 lymphoid leukemia Diseases 0.000 claims 1
- 210000005075 mammary gland Anatomy 0.000 claims 1
- 210000005134 plasmacytoid dendritic cell Anatomy 0.000 claims 1
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 claims 1
- 101150047061 tag-72 gene Proteins 0.000 claims 1
- 230000002103 transcriptional effect Effects 0.000 claims 1
- 238000000034 method Methods 0.000 description 56
- 108091007741 Chimeric antigen receptor T cells Proteins 0.000 description 51
- 241000699670 Mus sp. Species 0.000 description 39
- 108020004999 messenger RNA Proteins 0.000 description 35
- 239000013598 vector Substances 0.000 description 31
- 235000018102 proteins Nutrition 0.000 description 27
- 108010002586 Interleukin-7 Proteins 0.000 description 25
- 102000000704 Interleukin-7 Human genes 0.000 description 25
- 239000012634 fragment Substances 0.000 description 25
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 23
- 238000005415 bioluminescence Methods 0.000 description 21
- 230000029918 bioluminescence Effects 0.000 description 21
- 229960005167 everolimus Drugs 0.000 description 21
- 230000001086 cytosolic effect Effects 0.000 description 19
- 238000004520 electroporation Methods 0.000 description 19
- 238000000684 flow cytometry Methods 0.000 description 19
- 230000000638 stimulation Effects 0.000 description 18
- 210000004881 tumor cell Anatomy 0.000 description 18
- 101001023379 Homo sapiens Lysosome-associated membrane glycoprotein 1 Proteins 0.000 description 16
- 102100035133 Lysosome-associated membrane glycoprotein 1 Human genes 0.000 description 16
- 230000004048 modification Effects 0.000 description 16
- 238000012986 modification Methods 0.000 description 16
- 229920002477 rna polymer Polymers 0.000 description 16
- 108010024755 CTL019 chimeric antigen receptor Proteins 0.000 description 15
- 241000699666 Mus <mouse, genus> Species 0.000 description 15
- 230000004044 response Effects 0.000 description 14
- 102100029822 B- and T-lymphocyte attenuator Human genes 0.000 description 13
- 102000011107 Diacylglycerol Kinase Human genes 0.000 description 13
- 108010062677 Diacylglycerol Kinase Proteins 0.000 description 13
- 101000864344 Homo sapiens B- and T-lymphocyte attenuator Proteins 0.000 description 13
- 241000571697 Icarus Species 0.000 description 13
- 230000006870 function Effects 0.000 description 13
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 12
- 229940045513 CTLA4 antagonist Drugs 0.000 description 12
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 12
- 101000831007 Homo sapiens T-cell immunoreceptor with Ig and ITIM domains Proteins 0.000 description 12
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 12
- 108700015968 Slam family Proteins 0.000 description 12
- 102100024834 T-cell immunoreceptor with Ig and ITIM domains Human genes 0.000 description 12
- 238000002347 injection Methods 0.000 description 12
- 239000007924 injection Substances 0.000 description 12
- 229940090044 injection Drugs 0.000 description 12
- 108010074708 B7-H1 Antigen Proteins 0.000 description 11
- 102100035932 Cocaine- and amphetamine-regulated transcript protein Human genes 0.000 description 11
- 102000010451 Folate receptor alpha Human genes 0.000 description 11
- 108050001931 Folate receptor alpha Proteins 0.000 description 11
- 101000715592 Homo sapiens Cocaine- and amphetamine-regulated transcript protein Proteins 0.000 description 11
- 102000003911 Thyrotropin Receptors Human genes 0.000 description 11
- 108090000253 Thyrotropin Receptors Proteins 0.000 description 11
- 229940068196 placebo Drugs 0.000 description 11
- 239000000902 placebo Substances 0.000 description 11
- 238000010361 transduction Methods 0.000 description 11
- 230000026683 transduction Effects 0.000 description 11
- 102100023458 C-type lectin-like domain family 1 Human genes 0.000 description 10
- 102100025278 Coxsackievirus and adenovirus receptor Human genes 0.000 description 10
- 102000050627 Glucocorticoid-Induced TNFR-Related Human genes 0.000 description 10
- 101000906643 Homo sapiens C-type lectin-like domain family 1 Proteins 0.000 description 10
- 101000666896 Homo sapiens V-type immunoglobulin domain-containing suppressor of T-cell activation Proteins 0.000 description 10
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 10
- 241000713666 Lentivirus Species 0.000 description 10
- 101710187882 Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 description 10
- 102100038282 V-type immunoglobulin domain-containing suppressor of T-cell activation Human genes 0.000 description 10
- 235000001014 amino acid Nutrition 0.000 description 10
- IJJVMEJXYNJXOJ-UHFFFAOYSA-N fluquinconazole Chemical compound C=1C=C(Cl)C=C(Cl)C=1N1C(=O)C2=CC(F)=CC=C2N=C1N1C=NC=N1 IJJVMEJXYNJXOJ-UHFFFAOYSA-N 0.000 description 10
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 10
- 206010022000 influenza Diseases 0.000 description 10
- 238000004519 manufacturing process Methods 0.000 description 10
- 101710168331 ALK tyrosine kinase receptor Proteins 0.000 description 9
- 229940124291 BTK inhibitor Drugs 0.000 description 9
- 102100037850 Interferon gamma Human genes 0.000 description 9
- 108010074328 Interferon-gamma Proteins 0.000 description 9
- 230000000875 corresponding effect Effects 0.000 description 9
- 210000002865 immune cell Anatomy 0.000 description 9
- 230000004936 stimulating effect Effects 0.000 description 9
- 238000002255 vaccination Methods 0.000 description 9
- 102000010956 Glypican Human genes 0.000 description 8
- 108050001154 Glypican Proteins 0.000 description 8
- 108050007237 Glypican-3 Proteins 0.000 description 8
- 229940024606 amino acid Drugs 0.000 description 8
- 150000001413 amino acids Chemical class 0.000 description 8
- 230000000259 anti-tumor effect Effects 0.000 description 8
- 238000000338 in vitro Methods 0.000 description 8
- 238000001727 in vivo Methods 0.000 description 8
- 238000001802 infusion Methods 0.000 description 8
- 230000019491 signal transduction Effects 0.000 description 8
- LTHJXDSHSVNJKG-UHFFFAOYSA-N 2-[2-[2-[2-(2-methylprop-2-enoyloxy)ethoxy]ethoxy]ethoxy]ethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCOCCOCCOCCOC(=O)C(C)=C LTHJXDSHSVNJKG-UHFFFAOYSA-N 0.000 description 7
- 241000282693 Cercopithecidae Species 0.000 description 7
- 102000053602 DNA Human genes 0.000 description 7
- 108020004414 DNA Proteins 0.000 description 7
- 101000894428 Homo sapiens Transcriptional repressor CTCFL Proteins 0.000 description 7
- 206010060862 Prostate cancer Diseases 0.000 description 7
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 7
- 102100038098 Protein-glutamine gamma-glutamyltransferase 5 Human genes 0.000 description 7
- 241000283984 Rodentia Species 0.000 description 7
- 125000000539 amino acid group Chemical group 0.000 description 7
- 238000003556 assay Methods 0.000 description 7
- 239000011324 bead Substances 0.000 description 7
- 230000002950 deficient Effects 0.000 description 7
- 108020001507 fusion proteins Proteins 0.000 description 7
- 102000037865 fusion proteins Human genes 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 238000011081 inoculation Methods 0.000 description 7
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 7
- 108010058721 transglutaminase 5 Proteins 0.000 description 7
- 101000912622 Homo sapiens C-type lectin domain family 12 member A Proteins 0.000 description 6
- 101000884271 Homo sapiens Signal transducer CD24 Proteins 0.000 description 6
- 101000714168 Homo sapiens Testisin Proteins 0.000 description 6
- 101100351020 Mus musculus Pax5 gene Proteins 0.000 description 6
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 6
- 108010051742 Platelet-Derived Growth Factor beta Receptor Proteins 0.000 description 6
- 102100038081 Signal transducer CD24 Human genes 0.000 description 6
- 102100036494 Testisin Human genes 0.000 description 6
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 6
- 102100040247 Tumor necrosis factor Human genes 0.000 description 6
- 101100351021 Xenopus laevis pax5 gene Proteins 0.000 description 6
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 6
- 238000012217 deletion Methods 0.000 description 6
- 230000037430 deletion Effects 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- XYFPWWZEPKGCCK-GOSISDBHSA-N ibrutinib Chemical compound C1=2C(N)=NC=NC=2N([C@H]2CN(CCC2)C(=O)C=C)N=C1C(C=C1)=CC=C1OC1=CC=CC=C1 XYFPWWZEPKGCCK-GOSISDBHSA-N 0.000 description 6
- 230000028993 immune response Effects 0.000 description 6
- 238000011068 loading method Methods 0.000 description 6
- 230000035755 proliferation Effects 0.000 description 6
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 230000028327 secretion Effects 0.000 description 6
- 229960002930 sirolimus Drugs 0.000 description 6
- 238000010186 staining Methods 0.000 description 6
- 229940083347 Cyclin-dependent kinase 4 inhibitor Drugs 0.000 description 5
- 102100031968 Ephrin type-B receptor 2 Human genes 0.000 description 5
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 5
- 108010038498 Interleukin-7 Receptors Proteins 0.000 description 5
- 102000010782 Interleukin-7 Receptors Human genes 0.000 description 5
- 108060001084 Luciferase Proteins 0.000 description 5
- 239000005089 Luciferase Substances 0.000 description 5
- 238000001994 activation Methods 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 238000003776 cleavage reaction Methods 0.000 description 5
- 230000001472 cytotoxic effect Effects 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- 229960003971 influenza vaccine Drugs 0.000 description 5
- 239000013642 negative control Substances 0.000 description 5
- 230000007017 scission Effects 0.000 description 5
- 238000007920 subcutaneous administration Methods 0.000 description 5
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 4
- RJBDSRWGVYNDHL-XNJNKMBASA-N (2S,4R,5S,6S)-2-[(2S,3R,4R,5S,6R)-5-[(2S,3R,4R,5R,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-2-[(2R,3S,4R,5R,6R)-4,5-dihydroxy-2-(hydroxymethyl)-6-[(E,2R,3S)-3-hydroxy-2-(octadecanoylamino)octadec-4-enoxy]oxan-3-yl]oxy-3-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-5-amino-6-[(1S,2R)-2-[(2S,4R,5S,6S)-5-amino-2-carboxy-4-hydroxy-6-[(1R,2R)-1,2,3-trihydroxypropyl]oxan-2-yl]oxy-1,3-dihydroxypropyl]-4-hydroxyoxane-2-carboxylic acid Chemical compound CCCCCCCCCCCCCCCCCC(=O)N[C@H](CO[C@@H]1O[C@H](CO)[C@@H](O[C@@H]2O[C@H](CO)[C@H](O[C@@H]3O[C@H](CO)[C@H](O)[C@H](O)[C@H]3NC(C)=O)[C@H](O[C@@]3(C[C@@H](O)[C@H](N)[C@H](O3)[C@H](O)[C@@H](CO)O[C@@]3(C[C@@H](O)[C@H](N)[C@H](O3)[C@H](O)[C@H](O)CO)C(O)=O)C(O)=O)[C@H]2O)[C@H](O)[C@H]1O)[C@@H](O)\C=C\CCCCCCCCCCCCC RJBDSRWGVYNDHL-XNJNKMBASA-N 0.000 description 4
- 102100035793 CD83 antigen Human genes 0.000 description 4
- 238000002965 ELISA Methods 0.000 description 4
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 4
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 description 4
- 101100005713 Homo sapiens CD4 gene Proteins 0.000 description 4
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 description 4
- 102100031413 L-dopachrome tautomerase Human genes 0.000 description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 4
- 239000002177 L01XE27 - Ibrutinib Substances 0.000 description 4
- 108010077524 Peptide Elongation Factor 1 Proteins 0.000 description 4
- 102000010292 Peptide Elongation Factor 1 Human genes 0.000 description 4
- 230000006052 T cell proliferation Effects 0.000 description 4
- 230000000735 allogeneic effect Effects 0.000 description 4
- 210000003719 b-lymphocyte Anatomy 0.000 description 4
- 229960002685 biotin Drugs 0.000 description 4
- 239000011616 biotin Substances 0.000 description 4
- 238000003501 co-culture Methods 0.000 description 4
- 239000002299 complementary DNA Substances 0.000 description 4
- 230000003013 cytotoxicity Effects 0.000 description 4
- 231100000135 cytotoxicity Toxicity 0.000 description 4
- 108010051081 dopachrome isomerase Proteins 0.000 description 4
- 229960001507 ibrutinib Drugs 0.000 description 4
- 208000032839 leukemia Diseases 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 210000004985 myeloid-derived suppressor cell Anatomy 0.000 description 4
- 239000002773 nucleotide Substances 0.000 description 4
- 125000003729 nucleotide group Chemical group 0.000 description 4
- AHJRHEGDXFFMBM-UHFFFAOYSA-N palbociclib Chemical compound N1=C2N(C3CCCC3)C(=O)C(C(=O)C)=C(C)C2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 AHJRHEGDXFFMBM-UHFFFAOYSA-N 0.000 description 4
- 229940126731 protein tyrosine phosphatase inhibitor Drugs 0.000 description 4
- 239000003806 protein tyrosine phosphatase inhibitor Substances 0.000 description 4
- 102000016914 ras Proteins Human genes 0.000 description 4
- 238000003753 real-time PCR Methods 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 238000013518 transcription Methods 0.000 description 4
- 230000035897 transcription Effects 0.000 description 4
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 4
- 229960004441 tyrosine Drugs 0.000 description 4
- 230000003827 upregulation Effects 0.000 description 4
- 238000001262 western blot Methods 0.000 description 4
- DWZAEMINVBZMHQ-UHFFFAOYSA-N 1-[4-[4-(dimethylamino)piperidine-1-carbonyl]phenyl]-3-[4-(4,6-dimorpholin-4-yl-1,3,5-triazin-2-yl)phenyl]urea Chemical compound C1CC(N(C)C)CCN1C(=O)C(C=C1)=CC=C1NC(=O)NC1=CC=C(C=2N=C(N=C(N=2)N2CCOCC2)N2CCOCC2)C=C1 DWZAEMINVBZMHQ-UHFFFAOYSA-N 0.000 description 3
- 108020005345 3' Untranslated Regions Proteins 0.000 description 3
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 3
- 241000282836 Camelus dromedarius Species 0.000 description 3
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 3
- 241001272567 Hominoidea Species 0.000 description 3
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 3
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 3
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 3
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 3
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 206010061535 Ovarian neoplasm Diseases 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- 230000006044 T cell activation Effects 0.000 description 3
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 3
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 3
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 3
- 102100024568 Tumor necrosis factor ligand superfamily member 11 Human genes 0.000 description 3
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 3
- HJSSPYJVWLTYHG-UHFFFAOYSA-N XL765 Chemical compound COC1=CC(OC)=CC(NC=2C(=NC3=CC=CC=C3N=2)NS(=O)(=O)C=2C=CC(NC(=O)C=3C=C(OC)C(C)=CC=3)=CC=2)=C1 HJSSPYJVWLTYHG-UHFFFAOYSA-N 0.000 description 3
- 229960005305 adenosine Drugs 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 231100000433 cytotoxic Toxicity 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 230000002489 hematologic effect Effects 0.000 description 3
- 230000002434 immunopotentiative effect Effects 0.000 description 3
- 230000006698 induction Effects 0.000 description 3
- PGHMRUGBZOYCAA-UHFFFAOYSA-N ionomycin Natural products O1C(CC(O)C(C)C(O)C(C)C=CCC(C)CC(C)C(O)=CC(=O)C(C)CC(C)CC(CCC(O)=O)C)CCC1(C)C1OC(C)(C(C)O)CC1 PGHMRUGBZOYCAA-UHFFFAOYSA-N 0.000 description 3
- PGHMRUGBZOYCAA-ADZNBVRBSA-N ionomycin Chemical compound O1[C@H](C[C@H](O)[C@H](C)[C@H](O)[C@H](C)/C=C/C[C@@H](C)C[C@@H](C)C(/O)=C/C(=O)[C@@H](C)C[C@@H](C)C[C@@H](CCC(O)=O)C)CC[C@@]1(C)[C@@H]1O[C@](C)([C@@H](C)O)CC1 PGHMRUGBZOYCAA-ADZNBVRBSA-N 0.000 description 3
- 229960000310 isoleucine Drugs 0.000 description 3
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 3
- 229920002521 macromolecule Polymers 0.000 description 3
- 238000010172 mouse model Methods 0.000 description 3
- 229960004390 palbociclib Drugs 0.000 description 3
- 230000002688 persistence Effects 0.000 description 3
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 238000011002 quantification Methods 0.000 description 3
- 230000035945 sensitivity Effects 0.000 description 3
- 210000000952 spleen Anatomy 0.000 description 3
- 210000002536 stromal cell Anatomy 0.000 description 3
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 230000004614 tumor growth Effects 0.000 description 3
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 3
- 239000004474 valine Substances 0.000 description 3
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 2
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 2
- QLUYMIVVAYRECT-OCCSQVGLSA-N 2-(2-chlorophenyl)-5,7-dihydroxy-8-[(2r,3s)-2-(hydroxymethyl)-1-methylpyrrolidin-3-yl]chromen-4-one Chemical compound OC[C@@H]1N(C)CC[C@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O QLUYMIVVAYRECT-OCCSQVGLSA-N 0.000 description 2
- XDLYKKIQACFMJG-UHFFFAOYSA-N 2-amino-8-[4-(2-hydroxyethoxy)cyclohexyl]-6-(6-methoxypyridin-3-yl)-4-methylpyrido[2,3-d]pyrimidin-7-one Chemical compound C1=NC(OC)=CC=C1C(C1=O)=CC2=C(C)N=C(N)N=C2N1C1CCC(OCCO)CC1 XDLYKKIQACFMJG-UHFFFAOYSA-N 0.000 description 2
- OVPNQJVDAFNBDN-UHFFFAOYSA-N 4-(2,6-dichlorobenzamido)-N-(piperidin-4-yl)-pyrazole-3-carboxamide Chemical compound ClC1=CC=CC(Cl)=C1C(=O)NC1=CNN=C1C(=O)NC1CCNCC1 OVPNQJVDAFNBDN-UHFFFAOYSA-N 0.000 description 2
- VVLHQJDAUIPZFH-UHFFFAOYSA-N 4-[4-[[5-fluoro-4-[3-(prop-2-enoylamino)anilino]pyrimidin-2-yl]amino]phenoxy]-n-methylpyridine-2-carboxamide Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC=3N=C(NC=4C=C(NC(=O)C=C)C=CC=4)C(F)=CN=3)=CC=2)=C1 VVLHQJDAUIPZFH-UHFFFAOYSA-N 0.000 description 2
- QYBGBLQCOOISAR-UHFFFAOYSA-N 5-(8-methyl-2-morpholin-4-yl-9-propan-2-ylpurin-6-yl)pyrimidin-2-amine Chemical compound N1=C2N(C(C)C)C(C)=NC2=C(C=2C=NC(N)=NC=2)N=C1N1CCOCC1 QYBGBLQCOOISAR-UHFFFAOYSA-N 0.000 description 2
- 108010013238 70-kDa Ribosomal Protein S6 Kinases Proteins 0.000 description 2
- BUROJSBIWGDYCN-GAUTUEMISA-N AP 23573 Chemical compound C1C[C@@H](OP(C)(C)=O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 BUROJSBIWGDYCN-GAUTUEMISA-N 0.000 description 2
- 208000004736 B-Cell Leukemia Diseases 0.000 description 2
- UDFAXHFPCDXHHH-UHFFFAOYSA-N C1=NC2=NN=CC2=C(N)N1NC1=CC=C(F)C=C1 Chemical compound C1=NC2=NN=CC2=C(N)N1NC1=CC=C(F)C=C1 UDFAXHFPCDXHHH-UHFFFAOYSA-N 0.000 description 2
- 206010008342 Cervix carcinoma Diseases 0.000 description 2
- 108091033380 Coding strand Proteins 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 208000036066 Hemophagocytic Lymphohistiocytosis Diseases 0.000 description 2
- 208000032672 Histiocytosis haematophagic Diseases 0.000 description 2
- 241000701806 Human papillomavirus Species 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- 102000000588 Interleukin-2 Human genes 0.000 description 2
- 108010043610 KIR Receptors Proteins 0.000 description 2
- 102100033627 Killer cell immunoglobulin-like receptor 3DL1 Human genes 0.000 description 2
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- UVSVTDVJQAJIFG-VURMDHGXSA-N LFM-A13 Chemical compound C\C(O)=C(/C#N)C(=O)NC1=CC(Br)=CC=C1Br UVSVTDVJQAJIFG-VURMDHGXSA-N 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- 102100029193 Low affinity immunoglobulin gamma Fc region receptor III-A Human genes 0.000 description 2
- 102000008135 Mechanistic Target of Rapamycin Complex 1 Human genes 0.000 description 2
- 108010035196 Mechanistic Target of Rapamycin Complex 1 Proteins 0.000 description 2
- 102000009308 Mechanistic Target of Rapamycin Complex 2 Human genes 0.000 description 2
- 108010034057 Mechanistic Target of Rapamycin Complex 2 Proteins 0.000 description 2
- OUSFTKFNBAZUKL-UHFFFAOYSA-N N-(5-{[(5-tert-butyl-1,3-oxazol-2-yl)methyl]sulfanyl}-1,3-thiazol-2-yl)piperidine-4-carboxamide Chemical compound O1C(C(C)(C)C)=CN=C1CSC(S1)=CN=C1NC(=O)C1CCNCC1 OUSFTKFNBAZUKL-UHFFFAOYSA-N 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- 239000012823 PI3K/mTOR inhibitor Substances 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 2
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 description 2
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 208000015634 Rectal Neoplasms Diseases 0.000 description 2
- 208000000453 Skin Neoplasms Diseases 0.000 description 2
- 102220497176 Small vasohibin-binding protein_T47D_mutation Human genes 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 238000010459 TALEN Methods 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 2
- 208000002495 Uterine Neoplasms Diseases 0.000 description 2
- 241001193851 Zeta Species 0.000 description 2
- 210000001015 abdomen Anatomy 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical compound OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 2
- 230000001093 anti-cancer Effects 0.000 description 2
- 230000006023 anti-tumor response Effects 0.000 description 2
- 210000000612 antigen-presenting cell Anatomy 0.000 description 2
- 239000012472 biological sample Substances 0.000 description 2
- 235000020958 biotin Nutrition 0.000 description 2
- 230000006037 cell lysis Effects 0.000 description 2
- 230000005859 cell recognition Effects 0.000 description 2
- 238000002659 cell therapy Methods 0.000 description 2
- 230000005754 cellular signaling Effects 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 201000010881 cervical cancer Diseases 0.000 description 2
- 238000002983 circular dichroism Methods 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 239000012228 culture supernatant Substances 0.000 description 2
- 230000016396 cytokine production Effects 0.000 description 2
- 206010052015 cytokine release syndrome Diseases 0.000 description 2
- JOGKUKXHTYWRGZ-UHFFFAOYSA-N dactolisib Chemical compound O=C1N(C)C2=CN=C3C=CC(C=4C=C5C=CC=CC5=NC=4)=CC3=C2N1C1=CC=C(C(C)(C)C#N)C=C1 JOGKUKXHTYWRGZ-UHFFFAOYSA-N 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 238000006471 dimerization reaction Methods 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 208000014752 hemophagocytic syndrome Diseases 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000010253 intravenous injection Methods 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 230000002147 killing effect Effects 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 201000007270 liver cancer Diseases 0.000 description 2
- 208000014018 liver neoplasm Diseases 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- 230000001589 lymphoproliferative effect Effects 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- MMNNTJYFHUDSKL-UHFFFAOYSA-N methyl n-[6-[2-(5-chloro-2-methylphenyl)-1-hydroxy-3-oxoisoindol-1-yl]-1h-benzimidazol-2-yl]carbamate Chemical compound C=1C=C2NC(NC(=O)OC)=NC2=CC=1C(C1=CC=CC=C1C1=O)(O)N1C1=CC(Cl)=CC=C1C MMNNTJYFHUDSKL-UHFFFAOYSA-N 0.000 description 2
- IJMHHZDBRUGXNO-UHFFFAOYSA-N n-[3-(8-anilinoimidazo[1,2-a]pyrazin-6-yl)phenyl]-4-tert-butylbenzamide Chemical compound C1=CC(C(C)(C)C)=CC=C1C(=O)NC1=CC=CC(C=2N=C(NC=3C=CC=CC=3)C3=NC=CN3C=2)=C1 IJMHHZDBRUGXNO-UHFFFAOYSA-N 0.000 description 2
- CDOOFZZILLRUQH-GDLZYMKVSA-N n-[3-[6-[4-[(2r)-1,4-dimethyl-3-oxopiperazin-2-yl]anilino]-4-methyl-5-oxopyrazin-2-yl]-2-methylphenyl]-4,5,6,7-tetrahydro-1-benzothiophene-2-carboxamide Chemical compound CN1CCN(C)C(=O)[C@H]1C(C=C1)=CC=C1NC1=NC(C=2C(=C(NC(=O)C=3SC=4CCCCC=4C=3)C=CC=2)C)=CN(C)C1=O CDOOFZZILLRUQH-GDLZYMKVSA-N 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 210000005259 peripheral blood Anatomy 0.000 description 2
- 239000011886 peripheral blood Substances 0.000 description 2
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 2
- 102000040430 polynucleotide Human genes 0.000 description 2
- 108091033319 polynucleotide Proteins 0.000 description 2
- 239000002157 polynucleotide Substances 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 206010038038 rectal cancer Diseases 0.000 description 2
- 201000001275 rectum cancer Diseases 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000001177 retroviral effect Effects 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 229960001153 serine Drugs 0.000 description 2
- 201000000849 skin cancer Diseases 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000011476 stem cell transplantation Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 229960002898 threonine Drugs 0.000 description 2
- 229960004799 tryptophan Drugs 0.000 description 2
- 230000010304 tumor cell viability Effects 0.000 description 2
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 2
- 206010046766 uterine cancer Diseases 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- YOVVNQKCSKSHKT-HNNXBMFYSA-N (2s)-1-[4-[[2-(2-aminopyrimidin-5-yl)-7-methyl-4-morpholin-4-ylthieno[3,2-d]pyrimidin-6-yl]methyl]piperazin-1-yl]-2-hydroxypropan-1-one Chemical compound C1CN(C(=O)[C@@H](O)C)CCN1CC1=C(C)C2=NC(C=3C=NC(N)=NC=3)=NC(N3CCOCC3)=C2S1 YOVVNQKCSKSHKT-HNNXBMFYSA-N 0.000 description 1
- SVNJBEMPMKWDCO-KCHLEUMXSA-N (2s)-2-[[(2s)-3-carboxy-2-[[2-[[(2s)-5-(diaminomethylideneamino)-2-[[4-oxo-4-[[4-(4-oxo-8-phenylchromen-2-yl)morpholin-4-ium-4-yl]methoxy]butanoyl]amino]pentanoyl]amino]acetyl]amino]propanoyl]amino]-3-hydroxypropanoate Chemical compound C=1C(=O)C2=CC=CC(C=3C=CC=CC=3)=C2OC=1[N+]1(COC(=O)CCC(=O)N[C@@H](CCCNC(=N)N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C([O-])=O)CCOCC1 SVNJBEMPMKWDCO-KCHLEUMXSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- YABJJWZLRMPFSI-UHFFFAOYSA-N 1-methyl-5-[[2-[5-(trifluoromethyl)-1H-imidazol-2-yl]-4-pyridinyl]oxy]-N-[4-(trifluoromethyl)phenyl]-2-benzimidazolamine Chemical compound N=1C2=CC(OC=3C=C(N=CC=3)C=3NC(=CN=3)C(F)(F)F)=CC=C2N(C)C=1NC1=CC=C(C(F)(F)F)C=C1 YABJJWZLRMPFSI-UHFFFAOYSA-N 0.000 description 1
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 1
- GZCWLCBFPRFLKL-UHFFFAOYSA-N 1-prop-2-ynoxypropan-2-ol Chemical compound CC(O)COCC#C GZCWLCBFPRFLKL-UHFFFAOYSA-N 0.000 description 1
- DNDJHEWLYGJJCY-UHFFFAOYSA-N 1-pyridin-3-ylpiperazine Chemical compound C1CNCCN1C1=CC=CN=C1 DNDJHEWLYGJJCY-UHFFFAOYSA-N 0.000 description 1
- DBXGGXLBTWZXBB-MRXNPFEDSA-N 2-(6-fluorobenzimidazol-1-yl)-9-[(4r)-8-fluoro-3,4-dihydro-2h-chromen-4-yl]-7h-purin-8-one Chemical compound C1COC2=C(F)C=CC=C2[C@@H]1N(C1=N2)C(=O)NC1=CN=C2N1C=NC2=CC=C(F)C=C21 DBXGGXLBTWZXBB-MRXNPFEDSA-N 0.000 description 1
- 125000004182 2-chlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(*)C([H])=C1[H] 0.000 description 1
- WJRRGYBTGDJBFX-UHFFFAOYSA-N 4-(2-methyl-3-propan-2-yl-4-imidazolyl)-N-(4-methylsulfonylphenyl)-2-pyrimidinamine Chemical compound CC(C)N1C(C)=NC=C1C1=CC=NC(NC=2C=CC(=CC=2)S(C)(=O)=O)=N1 WJRRGYBTGDJBFX-UHFFFAOYSA-N 0.000 description 1
- HHFBDROWDBDFBR-UHFFFAOYSA-N 4-[[9-chloro-7-(2,6-difluorophenyl)-5H-pyrimido[5,4-d][2]benzazepin-2-yl]amino]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1NC1=NC=C(CN=C(C=2C3=CC=C(Cl)C=2)C=2C(=CC=CC=2F)F)C3=N1 HHFBDROWDBDFBR-UHFFFAOYSA-N 0.000 description 1
- SEJLPXCPMNSRAM-GOSISDBHSA-N 6-amino-9-[(3r)-1-but-2-ynoylpyrrolidin-3-yl]-7-(4-phenoxyphenyl)purin-8-one Chemical compound C1N(C(=O)C#CC)CC[C@H]1N1C(=O)N(C=2C=CC(OC=3C=CC=CC=3)=CC=2)C2=C(N)N=CN=C21 SEJLPXCPMNSRAM-GOSISDBHSA-N 0.000 description 1
- YXHLJMWYDTXDHS-IRFLANFNSA-N 7-aminoactinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=C(N)C=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 YXHLJMWYDTXDHS-IRFLANFNSA-N 0.000 description 1
- 108700012813 7-aminoactinomycin D Proteins 0.000 description 1
- 102100029824 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 2 Human genes 0.000 description 1
- KVLFRAWTRWDEDF-IRXDYDNUSA-N AZD-8055 Chemical compound C1=C(CO)C(OC)=CC=C1C1=CC=C(C(=NC(=N2)N3[C@H](COCC3)C)N3[C@H](COCC3)C)C2=N1 KVLFRAWTRWDEDF-IRXDYDNUSA-N 0.000 description 1
- 208000006678 Abdominal Neoplasms Diseases 0.000 description 1
- 108010051457 Acid Phosphatase Proteins 0.000 description 1
- 102000013563 Acid Phosphatase Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 102100040024 Adhesion G-protein coupled receptor G5 Human genes 0.000 description 1
- 241000059559 Agriotes sordidus Species 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 108090000672 Annexin A5 Proteins 0.000 description 1
- 102000004121 Annexin A5 Human genes 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 102100027205 B-cell antigen receptor complex-associated protein alpha chain Human genes 0.000 description 1
- 102100027203 B-cell antigen receptor complex-associated protein beta chain Human genes 0.000 description 1
- YUXMAKUNSXIEKN-BTJKTKAUSA-N BGT226 Chemical compound OC(=O)\C=C/C(O)=O.C1=NC(OC)=CC=C1C1=CC=C(N=CC2=C3N(C=4C=C(C(N5CCNCC5)=CC=4)C(F)(F)F)C(=O)N2C)C3=C1 YUXMAKUNSXIEKN-BTJKTKAUSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 102000006734 Beta-Globulins Human genes 0.000 description 1
- 108010087504 Beta-Globulins Proteins 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 206010005949 Bone cancer Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 206010006143 Brain stem glioma Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 102100031151 C-C chemokine receptor type 2 Human genes 0.000 description 1
- 101710149815 C-C chemokine receptor type 2 Proteins 0.000 description 1
- 102100025238 CD302 antigen Human genes 0.000 description 1
- 102100036008 CD48 antigen Human genes 0.000 description 1
- 102100022436 CMRF35-like molecule 8 Human genes 0.000 description 1
- 241000282832 Camelidae Species 0.000 description 1
- 101710120600 Cancer/testis antigen 1 Proteins 0.000 description 1
- 101710120595 Cancer/testis antigen 2 Proteins 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 206010007953 Central nervous system lymphoma Diseases 0.000 description 1
- GEWLYFZWVLXQME-UHFFFAOYSA-N Cercosporamide Natural products CC12C(=O)C(C(=O)C)=C(O)C=C1OC1=C2C(O)=CC(O)=C1C(N)=O GEWLYFZWVLXQME-UHFFFAOYSA-N 0.000 description 1
- 102100035360 Cerebellar degeneration-related antigen 1 Human genes 0.000 description 1
- 101710181340 Chaperone protein DnaK2 Proteins 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 1
- 108010060385 Cyclin B1 Proteins 0.000 description 1
- 206010058314 Dysplasia Diseases 0.000 description 1
- 102100031334 Elongation factor 2 Human genes 0.000 description 1
- 208000001976 Endocrine Gland Neoplasms Diseases 0.000 description 1
- 206010014733 Endometrial cancer Diseases 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 102100031780 Endonuclease Human genes 0.000 description 1
- 102100023721 Ephrin-B2 Human genes 0.000 description 1
- 108010044090 Ephrin-B2 Proteins 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- 108700039887 Essential Genes Proteins 0.000 description 1
- 206010015548 Euthanasia Diseases 0.000 description 1
- 102100037362 Fibronectin Human genes 0.000 description 1
- 102000002090 Fibronectin type III Human genes 0.000 description 1
- 108050009401 Fibronectin type III Proteins 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- 102100032340 G2/mitotic-specific cyclin-B1 Human genes 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 102000001398 Granzyme Human genes 0.000 description 1
- 108060005986 Granzyme Proteins 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 206010069767 H1N1 influenza Diseases 0.000 description 1
- 102100030595 HLA class II histocompatibility antigen gamma chain Human genes 0.000 description 1
- 102100027529 Heat shock factor-binding protein 1 Human genes 0.000 description 1
- 102100031624 Heat shock protein 105 kDa Human genes 0.000 description 1
- 101710178419 Heat shock protein 70 2 Proteins 0.000 description 1
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 1
- 102100034458 Hepatitis A virus cellular receptor 2 Human genes 0.000 description 1
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 1
- 101000794082 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 2 Proteins 0.000 description 1
- 101000959600 Homo sapiens Adhesion G-protein coupled receptor G5 Proteins 0.000 description 1
- 101000914489 Homo sapiens B-cell antigen receptor complex-associated protein alpha chain Proteins 0.000 description 1
- 101000914491 Homo sapiens B-cell antigen receptor complex-associated protein beta chain Proteins 0.000 description 1
- 101000716130 Homo sapiens CD48 antigen Proteins 0.000 description 1
- 101000901669 Homo sapiens CMRF35-like molecule 8 Proteins 0.000 description 1
- 101001099051 Homo sapiens GPI inositol-deacylase Proteins 0.000 description 1
- 101001082627 Homo sapiens HLA class II histocompatibility antigen gamma chain Proteins 0.000 description 1
- 101001080298 Homo sapiens Heat shock factor-binding protein 1 Proteins 0.000 description 1
- 101000866478 Homo sapiens Heat shock protein 105 kDa Proteins 0.000 description 1
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 1
- 101001068133 Homo sapiens Hepatitis A virus cellular receptor 2 Proteins 0.000 description 1
- 101001043807 Homo sapiens Interleukin-7 Proteins 0.000 description 1
- 101001018097 Homo sapiens L-selectin Proteins 0.000 description 1
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 description 1
- 101000576894 Homo sapiens Macrophage mannose receptor 1 Proteins 0.000 description 1
- 101001014566 Homo sapiens Membrane-spanning 4-domains subfamily A member 3 Proteins 0.000 description 1
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 1
- 101000931590 Homo sapiens Prostaglandin F2 receptor negative regulator Proteins 0.000 description 1
- 101001095320 Homo sapiens Serine/threonine-protein phosphatase PP1-beta catalytic subunit Proteins 0.000 description 1
- 101000863900 Homo sapiens Sialic acid-binding Ig-like lectin 5 Proteins 0.000 description 1
- 101000868472 Homo sapiens Sialoadhesin Proteins 0.000 description 1
- 101000709256 Homo sapiens Signal-regulatory protein beta-1 Proteins 0.000 description 1
- 101000709188 Homo sapiens Signal-regulatory protein beta-1 isoform 3 Proteins 0.000 description 1
- 101000835093 Homo sapiens Transferrin receptor protein 1 Proteins 0.000 description 1
- 101000801255 Homo sapiens Tumor necrosis factor receptor superfamily member 17 Proteins 0.000 description 1
- 101000749631 Homo sapiens Uromodulin-like 1 Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 1
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 1
- 102100022516 Immunoglobulin superfamily member 2 Human genes 0.000 description 1
- 101710184277 Insulin-like growth factor 1 receptor Proteins 0.000 description 1
- 108010017511 Interleukin-13 Receptors Proteins 0.000 description 1
- 102100039078 Interleukin-4 receptor subunit alpha Human genes 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 208000005016 Intestinal Neoplasms Diseases 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 102100033467 L-selectin Human genes 0.000 description 1
- 239000002146 L01XE16 - Crizotinib Substances 0.000 description 1
- 102000017578 LAG3 Human genes 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 102100025354 Macrophage mannose receptor 1 Human genes 0.000 description 1
- 102100032517 Membrane-spanning 4-domains subfamily A member 3 Human genes 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 1
- 101100407308 Mus musculus Pdcd1lg2 gene Proteins 0.000 description 1
- 206010028570 Myelopathy Diseases 0.000 description 1
- MEKASOQEXYKAKM-UHFFFAOYSA-N N-[[5-[3-(4,6-difluoro-1H-benzimidazol-2-yl)-1H-indazol-5-yl]-4-methylpyridin-3-yl]methyl]ethanamine Chemical compound CCNCC1=CN=CC(C=2C=C3C(C=4NC5=CC(F)=CC(F)=C5N=4)=NNC3=CC=2)=C1C MEKASOQEXYKAKM-UHFFFAOYSA-N 0.000 description 1
- UQPMANVRZYYQMD-UHFFFAOYSA-N N3-(4-fluorophenyl)-2H-pyrazolo[3,4-d]pyrimidine-3,4-diamine Chemical compound C=12C(N)=NC=NC2=NNC=1NC1=CC=C(F)C=C1 UQPMANVRZYYQMD-UHFFFAOYSA-N 0.000 description 1
- 108010052419 NF-KappaB Inhibitor alpha Proteins 0.000 description 1
- 102100039337 NF-kappa-B inhibitor alpha Human genes 0.000 description 1
- 102000027581 NK cell receptors Human genes 0.000 description 1
- 108091008877 NK cell receptors Proteins 0.000 description 1
- 206010061309 Neoplasm progression Diseases 0.000 description 1
- 102100037589 OX-2 membrane glycoprotein Human genes 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 101710149067 Paired box protein Pax-5 Proteins 0.000 description 1
- 208000000821 Parathyroid Neoplasms Diseases 0.000 description 1
- 208000002471 Penile Neoplasms Diseases 0.000 description 1
- 206010034299 Penile cancer Diseases 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108010077519 Peptide Elongation Factor 2 Proteins 0.000 description 1
- 102000011755 Phosphoglycerate Kinase Human genes 0.000 description 1
- 208000007913 Pituitary Neoplasms Diseases 0.000 description 1
- 201000005746 Pituitary adenoma Diseases 0.000 description 1
- 206010061538 Pituitary tumour benign Diseases 0.000 description 1
- 108091036407 Polyadenylation Proteins 0.000 description 1
- 102100037935 Polyubiquitin-C Human genes 0.000 description 1
- 208000007541 Preleukemia Diseases 0.000 description 1
- 108700030875 Programmed Cell Death 1 Ligand 2 Proteins 0.000 description 1
- 101710094000 Programmed cell death 1 ligand 1 Proteins 0.000 description 1
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 description 1
- 102100020864 Prostaglandin F2 receptor negative regulator Human genes 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 108010006700 Receptor Tyrosine Kinase-like Orphan Receptors Proteins 0.000 description 1
- 101710148333 Regulator of G-protein signaling 13 Proteins 0.000 description 1
- 102100021035 Regulator of G-protein signaling 18 Human genes 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 108010034782 Ribosomal Protein S6 Kinases Proteins 0.000 description 1
- 102000009738 Ribosomal Protein S6 Kinases Human genes 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 102100037764 Serine/threonine-protein phosphatase PP1-beta catalytic subunit Human genes 0.000 description 1
- 102100029957 Sialic acid-binding Ig-like lectin 5 Human genes 0.000 description 1
- 102100032855 Sialoadhesin Human genes 0.000 description 1
- 102100032770 Signal-regulatory protein beta-1 isoform 3 Human genes 0.000 description 1
- 102000008115 Signaling Lymphocytic Activation Molecule Family Member 1 Human genes 0.000 description 1
- 108091027967 Small hairpin RNA Proteins 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 230000005867 T cell response Effects 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 210000000662 T-lymphocyte subset Anatomy 0.000 description 1
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical group C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- 206010057644 Testis cancer Diseases 0.000 description 1
- 101001099217 Thermotoga maritima (strain ATCC 43589 / DSM 3109 / JCM 10099 / NBRC 100826 / MSB8) Triosephosphate isomerase Proteins 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 101710128101 Transcriptional repressor CTCFL Proteins 0.000 description 1
- 102100026144 Transferrin receptor protein 1 Human genes 0.000 description 1
- 101710081844 Transmembrane protease serine 2 Proteins 0.000 description 1
- 102100033726 Tumor necrosis factor receptor superfamily member 17 Human genes 0.000 description 1
- 102000003425 Tyrosinase Human genes 0.000 description 1
- 108010056354 Ubiquitin C Proteins 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 208000006593 Urologic Neoplasms Diseases 0.000 description 1
- 102100040616 Uromodulin-like 1 Human genes 0.000 description 1
- BUBBEHCXSMCYNY-CVEARBPZSA-N [3-hydroxy-5-methyl-4-[(2S,3R)-2,3,4-trihydroxybutoxy]carbonylphenyl] 2,4-dihydroxy-6-methylbenzoate Chemical compound CC1=CC(O)=CC(O)=C1C(=O)OC1=CC(C)=C(C(=O)OC[C@H](O)[C@H](O)CO)C(O)=C1 BUBBEHCXSMCYNY-CVEARBPZSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 238000007792 addition Methods 0.000 description 1
- 201000005188 adrenal gland cancer Diseases 0.000 description 1
- 208000024447 adrenal gland neoplasm Diseases 0.000 description 1
- 235000019169 all-trans-retinol Nutrition 0.000 description 1
- 239000011717 all-trans-retinol Substances 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 229940125528 allosteric inhibitor Drugs 0.000 description 1
- 229950010817 alvocidib Drugs 0.000 description 1
- BIIVYFLTOXDAOV-YVEFUNNKSA-N alvocidib Chemical compound O[C@@H]1CN(C)CC[C@@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O BIIVYFLTOXDAOV-YVEFUNNKSA-N 0.000 description 1
- 230000002491 angiogenic effect Effects 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 238000002617 apheresis Methods 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000009702 cancer cell proliferation Effects 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- GEWLYFZWVLXQME-MRXNPFEDSA-N cercosporamide Chemical compound O=C([C@]12C)C(C(=O)C)=C(O)C=C1OC1=C2C(O)=CC(O)=C1C(N)=O GEWLYFZWVLXQME-MRXNPFEDSA-N 0.000 description 1
- XDLYKKIQACFMJG-WKILWMFISA-N chembl1234354 Chemical compound C1=NC(OC)=CC=C1C(C1=O)=CC2=C(C)N=C(N)N=C2N1[C@@H]1CC[C@@H](OCCO)CC1 XDLYKKIQACFMJG-WKILWMFISA-N 0.000 description 1
- JROFGZPOBKIAEW-HAQNSBGRSA-N chembl3120215 Chemical compound N1C=2C(OC)=CC=CC=2C=C1C(=C1C(N)=NC=NN11)N=C1[C@H]1CC[C@H](C(O)=O)CC1 JROFGZPOBKIAEW-HAQNSBGRSA-N 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 208000024207 chronic leukemia Diseases 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 description 1
- 229960005061 crizotinib Drugs 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 229960002433 cysteine Drugs 0.000 description 1
- 102000003675 cytokine receptors Human genes 0.000 description 1
- 108010057085 cytokine receptors Proteins 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 230000001461 cytolytic effect Effects 0.000 description 1
- 238000002784 cytotoxicity assay Methods 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- 229950006418 dactolisib Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 231100000517 death Toxicity 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- 230000001687 destabilization Effects 0.000 description 1
- 238000011038 discontinuous diafiltration by volume reduction Methods 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 230000007783 downstream signaling Effects 0.000 description 1
- BUBBEHCXSMCYNY-UHFFFAOYSA-N erythrin Natural products CC1=CC(O)=CC(O)=C1C(=O)OC1=CC(C)=C(C(=O)OCC(O)C(O)CO)C(O)=C1 BUBBEHCXSMCYNY-UHFFFAOYSA-N 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 108010072257 fibroblast activation protein alpha Proteins 0.000 description 1
- 238000002825 functional assay Methods 0.000 description 1
- 150000002270 gangliosides Chemical class 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 229950008209 gedatolisib Drugs 0.000 description 1
- 210000004392 genitalia Anatomy 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 229960002743 glutamine Drugs 0.000 description 1
- 239000005090 green fluorescent protein Substances 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 108091008039 hormone receptors Proteins 0.000 description 1
- 102000052622 human IL7 Human genes 0.000 description 1
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Substances C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 1
- 230000008004 immune attack Effects 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 201000002313 intestinal cancer Diseases 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 210000003292 kidney cell Anatomy 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 230000002101 lytic effect Effects 0.000 description 1
- 238000007433 macroscopic evaluation Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 208000029559 malignant endocrine neoplasm Diseases 0.000 description 1
- 208000020984 malignant renal pelvis neoplasm Diseases 0.000 description 1
- 208000026045 malignant tumor of parathyroid gland Diseases 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- PWDYHMBTPGXCSN-VCBMUGGBSA-N n,n'-bis[3,5-bis[(e)-n-(diaminomethylideneamino)-c-methylcarbonimidoyl]phenyl]decanediamide Chemical compound NC(N)=N/N=C(\C)C1=CC(C(=N/N=C(N)N)/C)=CC(NC(=O)CCCCCCCCC(=O)NC=2C=C(C=C(C=2)C(\C)=N\N=C(N)N)C(\C)=N\N=C(N)N)=C1 PWDYHMBTPGXCSN-VCBMUGGBSA-N 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- CGBJSGAELGCMKE-UHFFFAOYSA-N omipalisib Chemical compound COC1=NC=C(C=2C=C3C(C=4C=NN=CC=4)=CC=NC3=CC=2)C=C1NS(=O)(=O)C1=CC=C(F)C=C1F CGBJSGAELGCMKE-UHFFFAOYSA-N 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 210000001539 phagocyte Anatomy 0.000 description 1
- 229940043441 phosphoinositide 3-kinase inhibitor Drugs 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 208000021310 pituitary gland adenoma Diseases 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 208000016800 primary central nervous system lymphoma Diseases 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 108020001580 protein domains Proteins 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 201000007444 renal pelvis carcinoma Diseases 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229960001302 ridaforolimus Drugs 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- BTIHMVBBUGXLCJ-OAHLLOKOSA-N seliciclib Chemical compound C=12N=CN(C(C)C)C2=NC(N[C@@H](CO)CC)=NC=1NCC1=CC=CC=C1 BTIHMVBBUGXLCJ-OAHLLOKOSA-N 0.000 description 1
- 229950011005 semapimod Drugs 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 108091006024 signal transducing proteins Proteins 0.000 description 1
- 102000034285 signal transducing proteins Human genes 0.000 description 1
- 230000007781 signaling event Effects 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 239000004055 small Interfering RNA Substances 0.000 description 1
- YQDGWZZYGYKDLR-UZVLBLASSA-K sodium stibogluconate Chemical compound O.O.O.O.O.O.O.O.O.[Na+].[Na+].[Na+].O1[C@H]([C@H](O)CO)[C@H](O2)[C@H](C([O-])=O)O[Sb]21([O-])O[Sb]1(O)(O[C@H]2C([O-])=O)O[C@H]([C@H](O)CO)[C@@H]2O1 YQDGWZZYGYKDLR-UZVLBLASSA-K 0.000 description 1
- 229960001567 sodium stibogluconate Drugs 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 201000010740 swine influenza Diseases 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 206010042863 synovial sarcoma Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- 229960000235 temsirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-UHFFFAOYSA-N temsirolimus Natural products C1CC(O)C(OC)CC1CC(C)C1OC(=O)C2CCCCN2C(=O)C(=O)C(O)(O2)C(C)CCC2CC(OC)C(C)=CC=CC=CC(C)CC(C)C(=O)C(OC)C(O)C(C)=CC(C)C(=O)C1 QFJCIRLUMZQUOT-UHFFFAOYSA-N 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 206010043554 thrombocytopenia Diseases 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 230000002463 transducing effect Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 230000004565 tumor cell growth Effects 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 206010046885 vaginal cancer Diseases 0.000 description 1
- 208000013139 vaginal neoplasm Diseases 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
Images






























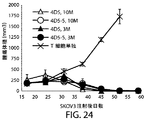


















































































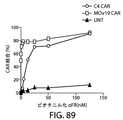


Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0636—T lymphocytes
- C12N5/0638—Cytotoxic T lymphocytes [CTL] or lymphokine activated killer cells [LAK]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/20—Interleukins [IL]
- A61K38/2086—IL-13 to IL-16
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001103—Receptors for growth factors
- A61K39/001104—Epidermal growth factor receptors [EGFR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001103—Receptors for growth factors
- A61K39/001106—Her-2/neu/ErbB2, Her-3/ErbB3 or Her 4/ErbB4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001103—Receptors for growth factors
- A61K39/001108—Platelet-derived growth factor receptors [PDGFR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001103—Receptors for growth factors
- A61K39/001109—Vascular endothelial growth factor receptors [VEGFR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001111—Immunoglobulin superfamily
- A61K39/001112—CD19 or B4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001111—Immunoglobulin superfamily
- A61K39/001113—CD22, BL-CAM, siglec-2 or sialic acid- binding Ig-related lectin 2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001116—Receptors for cytokines
- A61K39/001117—Receptors for tumor necrosis factors [TNF], e.g. lymphotoxin receptor [LTR] or CD30
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001116—Receptors for cytokines
- A61K39/001119—Receptors for interleukins [IL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001122—Ephrin Receptors [Eph]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001124—CD20
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001126—CD38 not IgG
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001129—Molecules with a "CD" designation not provided for elsewhere
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001148—Regulators of development
- A61K39/001149—Cell cycle regulated proteins, e.g. cyclin, CDC, CDK or INK-CCR
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001148—Regulators of development
- A61K39/00115—Apoptosis related proteins, e.g. survivin or livin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001148—Regulators of development
- A61K39/00115—Apoptosis related proteins, e.g. survivin or livin
- A61K39/001151—Apoptosis related proteins, e.g. survivin or livin p53
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001152—Transcription factors, e.g. SOX or c-MYC
- A61K39/001153—Wilms tumor 1 [WT1]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001154—Enzymes
- A61K39/001156—Tyrosinase and tyrosinase related proteinases [TRP-1 or TRP-2]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001154—Enzymes
- A61K39/001157—Telomerase or TERT [telomerase reverse transcriptase]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001154—Enzymes
- A61K39/001164—GTPases, e.g. Ras or Rho
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001166—Adhesion molecules, e.g. NRCAM, EpCAM or cadherins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001166—Adhesion molecules, e.g. NRCAM, EpCAM or cadherins
- A61K39/001168—Mesothelin [MSLN]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001169—Tumor associated carbohydrates
- A61K39/00117—Mucins, e.g. MUC-1
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001169—Tumor associated carbohydrates
- A61K39/001171—Gangliosides, e.g. GM2, GD2 or GD3
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001176—Heat shock proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/00118—Cancer antigens from embryonic or fetal origin
- A61K39/001182—Carcinoembryonic antigen [CEA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001184—Cancer testis antigens, e.g. SSX, BAGE, GAGE or SAGE
- A61K39/001188—NY-ESO
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/00119—Melanoma antigens
- A61K39/001191—Melan-A/MART
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/00119—Melanoma antigens
- A61K39/001192—Glycoprotein 100 [Gp100]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001193—Prostate associated antigens e.g. Prostate stem cell antigen [PSCA]; Prostate carcinoma tumor antigen [PCTA]; PAP or PSGR
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001193—Prostate associated antigens e.g. Prostate stem cell antigen [PSCA]; Prostate carcinoma tumor antigen [PCTA]; PAP or PSGR
- A61K39/001195—Prostate specific membrane antigen [PSMA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001196—Fusion proteins originating from gene translocation in cancer cells
- A61K39/001197—Breakpoint cluster region-abelson tyrosine kinase [BCR-ABL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/461—Cellular immunotherapy characterised by the cell type used
- A61K39/4611—T-cells, e.g. tumor infiltrating lymphocytes [TIL], lymphokine-activated killer cells [LAK] or regulatory T cells [Treg]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4631—Chimeric Antigen Receptors [CAR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4636—Immune checkpoint inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
- A61K39/464403—Receptors for growth factors
- A61K39/464404—Epidermal growth factor receptors [EGFR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
- A61K39/464403—Receptors for growth factors
- A61K39/464406—Her-2/neu/ErbB2, Her-3/ErbB3 or Her 4/ ErbB4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
- A61K39/464411—Immunoglobulin superfamily
- A61K39/464412—CD19 or B4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464466—Adhesion molecules, e.g. NRCAM, EpCAM or cadherins
- A61K39/464468—Mesothelin [MSLN]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464469—Tumor associated carbohydrates
- A61K39/46447—Mucins, e.g. MUC-1
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464469—Tumor associated carbohydrates
- A61K39/464471—Gangliosides, e.g. GM2, GD2 or GD3
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464493—Prostate associated antigens e.g. Prostate stem cell antigen [PSCA]; Prostate carcinoma tumor antigen [PCTA]; Prostatic acid phosphatase [PAP]; Prostate-specific G-protein-coupled receptor [PSGR]
- A61K39/464495—Prostate specific membrane antigen [PSMA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0636—T lymphocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/515—Animal cells
- A61K2039/5156—Animal cells expressing foreign proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55522—Cytokines; Lymphokines; Interferons
- A61K2039/55527—Interleukins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/10—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterized by the structure of the chimeric antigen receptor [CAR]
- A61K2239/21—Transmembrane domain
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/10—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterized by the structure of the chimeric antigen receptor [CAR]
- A61K2239/22—Intracellular domain
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/31—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterized by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/38—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the dose, timing or administration schedule
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/48—Blood cells, e.g. leukemia or lymphoma
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/49—Breast
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/58—Prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/59—Reproductive system, e.g. uterus, ovaries, cervix or testes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/7051—T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/22—Immunoglobulins specific features characterized by taxonomic origin from camelids, e.g. camel, llama or dromedary
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/569—Single domain, e.g. dAb, sdAb, VHH, VNAR or nanobody®
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/62—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising only variable region components
- C07K2317/622—Single chain antibody (scFv)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/03—Fusion polypeptide containing a localisation/targetting motif containing a transmembrane segment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/32—Fusion polypeptide fusions with soluble part of a cell surface receptor, "decoy receptors"
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Microbiology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Mycology (AREA)
- Oncology (AREA)
- Cell Biology (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Zoology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Biophysics (AREA)
- Hematology (AREA)
- General Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Developmental Biology & Embryology (AREA)
- Endocrinology (AREA)
- Dermatology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pregnancy & Childbirth (AREA)
- Inorganic Chemistry (AREA)
- Gynecology & Obstetrics (AREA)
- Vascular Medicine (AREA)
- Reproductive Health (AREA)
Description
本出願は、米国出願61/953,783号(2014年3月15日出願)、米国出願61/976,375号(2014年4月7日出願)、米国出願62/027,154(2014年7月21日出願)、米国出願62/076,146号(2014年11月6日出願)および米国出願62/097,286(2014年12月29日出願)に基づく優先権を主張する。これらの出願の内容の全体を引用により本明細書に包含させる。
本発明は、概して腫瘍抗原の発現と関係する疾患を処置するための、キメラ抗原受容体(CAR)を発現するように操作した免疫エフェクター細胞(例えば、T細胞、NK細胞)の使用に関する。
自己T細胞、特にキメラ抗原受容体(CAR)を形質導入したT細胞を用いる養子細胞移入(ACT)治療は、血液癌の治験で見込みが示されている。
本発明は、少なくとも一部、ここに記載する腫瘍抗原に結合するCARを発現するように操作した免疫エフェクター細胞(例えば、T細胞、NK細胞)の、該腫瘍抗原の発現と関係する癌の処置における使用に関する。
したがって、ある面において、本発明は、キメラ抗原受容体(CAR)をコードする単離核酸分子に関し、ここで、CARは、ここに記載する腫瘍抗原に結合する抗原結合ドメイン(例えば、抗体または抗体フラグメント、TCRまたはTCRフラグメント)、膜貫通ドメイン(例えば、ここに記載する膜貫通ドメイン)および細胞内シグナル伝達ドメイン(例えば、ここに記載する細胞内シグナル伝達ドメイン)(例えば、共刺激ドメイン(例えば、ここに記載する共刺激ドメイン)および/または一次シグナル伝達ドメイン(例えば、ここに記載する一次シグナル伝達ドメインを含む細胞内シグナル伝達ドメイン)を含む。ある態様において、腫瘍抗原は、CD19;CD123;CD22;CD30;CD171;CS−1(別名CD2サブセット1、CRACC、SLAMF7、CD319および19A24);C型レクチン様分子−1(CLL−1またはCLECL1);CD33;上皮細胞増殖因子受容体変異体III(EGFRvIII);ガングリオシドG2(GD2);ガングリオシドGD3(aNeu5Ac(2−8)aNeu5Ac(2−3)bDGalp(1−4)bDGlcp(1−1)Cer);TNF受容体ファミリーメンバーB細胞成熟抗原(BCMA);Tn抗原((Tn Ag)または(GalNAcα−Ser/Thr));前立腺特異的膜抗原(PSMA);受容体チロシンキナーゼ様オーファン受容体1(ROR1);Fms様チロシンキナーゼ3(FLT3);腫瘍関連糖タンパク質72(TAG72);CD38;CD44v6;癌胎児性抗原(CEA);上皮性細胞接着分子(EPCAM);B7H3(CD276);KIT(CD117);インターロイキン−13受容体サブユニットアルファ−2(IL−13Ra2またはCD213A2);メソテリン;インターロイキン11受容体アルファ(IL−11Ra);前立腺幹細胞抗原(PSCA);プロテアーゼセリン21(精巣inまたはPRSS21);血管内皮細胞増殖因子受容体2(VEGFR2);ルイス(Y)抗原;CD24;血小板由来増殖因子受容体ベータ(PDGFR−ベータ);ステージ特異的胚性抗原−4(SSEA−4);CD20;葉酸受容体アルファ;受容体チロシン−タンパク質キナーゼERBB2(Her2/neu);ムチン1、細胞表面関連(MUC1);上皮細胞増殖因子受容体(EGFR);神経細胞接着分子(NCAM);プロスターゼ;前立腺酸ホスファターゼ(PAP);伸長因子2変異型(ELF2M);エフリンB2;線維芽細胞活性化タンパク質アルファ(FAP);インシュリン様増殖因子1受容体(IGF−I受容体)、炭酸脱水酵素IX(CAIX);プロテアソーム(プロソーム、マクロパイン)サブユニット、ベータ型、9(LMP2);糖タンパク質100(gp100);易切断領域(BCR)およびエーベルソンマウス白血病ウイルス癌遺伝子ホモログ1(Abl)からなる癌遺伝子融合タンパク質(bcr−abl);チロシナーゼ;エフリンA型受容体2(EphA2);フコシルGM1;シアリルルイス接着分子(sLe);ガングリオシドGM3(aNeu5Ac(2−3)bDGalp(1−4)bDGlcp(1−1)Cer);トランスグルタミナーゼ5(TGS5);高分子量黒色腫関連抗原(HMWMAA);o−アセチル−GD2ガングリオシド(OAcGD2);葉酸受容体ベータ;腫瘍内皮マーカー1(TEM1/CD248);腫瘍内皮マーカー7関連(TEM7R);クローディン6(CLDN6);甲状腺刺激ホルモン受容体(TSHR);Gタンパク質共役受容体クラスCグループ5、メンバーD(GPRC5D);染色体Xオープンリーディングフレーム61(CXORF61);CD97;CD179a;未分化リンパ腫キナーゼ(ALK);ポリシアル酸;胎盤特異的1(PLAC1);globoHグリコセラミド(GloboH)の六糖部分;乳腺分化抗原(NY−BR−1);ウロプラキン2(UPK2);A型肝炎ウイルス細胞性受容体1(HAVCR1);アドレナリン受容体ベータ3(ADRB3);パンネキシン3(PANX3);Gタンパク質共役受容体20(GPR20);リンパ球抗原6複合体、座位K9(LY6K);嗅覚受容体51E2(OR51E2);TCRガンマ選択的リーディングフレームタンパク質(TARP);ウィルムス腫瘍タンパク質(WT1);癌/精巣抗原1(NY−ESO−1);癌/精巣抗原2(LAGE−1a);黒色腫関連抗原1(MAGE−A1);染色体12pに位置するETS転座変異体遺伝子6(ETV6−AML);精子タンパク質17(SPA17);X抗原ファミリー、メンバー1A(XAGE1);アンギオポエチン結合細胞表面受容体2(Tie 2);黒色腫癌精巣抗原−1(MAD−CT−1);黒色腫癌精巣抗原−2(MAD−CT−2);Fos関連抗原1;腫瘍タンパク質p53(p53);p53変異体;プロテイン;サバイビン(surviving);テロメラーゼ;前立腺癌腫瘍抗原−1(PCTA−1またはガレクチン8)、T細胞1により認識される黒色腫抗原(メランAまたはMART1);ラット肉腫(Ras)変異体;ヒトテロメラーゼ逆転写酵素(hTERT);肉腫転座切断点;アポトーシスの黒色腫阻害剤(ML−IAP);ERG(膜貫通型プロテアーゼ、セリン2(TMPRSS2)ETS融合遺伝子);N−アセチルグルコサミニル−トランスフェラーゼV(NA17);ペアードボックスタンパク質Pax−3(PAX3);アンドロゲン受容体;サイクリンB1;v−mycトリ骨髄細胞腫ウイルス癌遺伝子神経芽腫由来ホモログ(MYCN);RasホモログファミリーメンバーC(RhoC);チロシナーゼ関連タンパク質2(TRP−2);チトクロムP450 1B1(CYP1B1);CCCTC結合因子(亜鉛フィンガータンパク質)様(BORISまたはBrother of the Regulator of Imprinted Sites)、T細胞3により認識される扁平上皮細胞癌抗原(SART3);ペアードボックスタンパク質Pax−5(PAX5);プロアクロシン結合タンパク質p32(OY−TES1);リンパ球特異的タンパク質チロシンキナーゼ(LCK);Aキナーゼアンカータンパク質4(AKAP−4);滑膜肉腫、X切断点2(SSX2);終末糖化産物受容体(RAGE−1);腎臓ユビキタス1(RU1);腎臓ユビキタス2(RU2);レグマイン;ヒト乳頭腫ウイルスE6(HPV E6);ヒト乳頭腫ウイルスE7(HPV E7);腸管カルボキシルエステラーゼ;ヒートショックタンパク質70−2変異型(mut hsp70−2);CD79a;CD79b;CD72;白血球関連免疫グロブリン様受容体1(LAIR1);IgA受容体のFcフラグメント(FCARまたはCD89);白血球免疫グロブリン様受容体サブファミリーAメンバー2(LILRA2);CD300分子様ファミリーメンバー(CD300LF);C型レクチンドメインファミリー12メンバーA(CLEC12A);骨髄間質細胞抗原2(BST2);EGF様モジュール含有ムチン様ホルモン受容体様2(EMR2);リンパ球抗原75(LY75);グリピカン−3(GPC3);Fc受容体様5(FCRL5);および免疫グロブリンラムダ様ポリペプチド1(IGLL1)から選択される1以上である。
他の面において、本発明は、ここに記載するCARをコードする核酸配列を含むベクターに関する。ある態様において、ベクターは、DNAベクター、RNAベクター、プラスミド、レンチウイルスベクター、アデノウイルスベクターまたはレトロウイルスベクターから選択される。ある態様において、ベクターは、レンチウイルスベクターである。
他の面において、本発明は、抗原結合ドメイン、膜貫通ドメインおよび細胞内シグナル伝達ドメインを含む単離CARポリペプチド分子に関し、ここで、該抗原結合ドメインは、CD19、CD123、CD22、CD30、CD171、CS−1、CLL−1(CLECL1)、CD33、EGFRvIII、GD2、GD3、BCMA、Tn Ag、PSMA、ROR1、FLT3、TAG72、CD38、CD44v6、CEA、EPCAM、B7H3、KIT、IL−13Ra2、メソテリン、IL−11Ra、PSCA、PRSS21、VEGFR2、ルイスY、CD24、PDGFR−ベータ、SSEA−4、CD20、葉酸受容体アルファ、ERBB2(Her2/neu)、MUC1、EGFR、NCAM、プロスターゼ、PAP、ELF2M、エフリンB2、FAP、IGF−I受容体、CAIX、LMP2、gp100、bcr−abl、チロシナーゼ、EphA2、フコシルGM1、sLe、GM3、TGS5、HMWMAA、o−アセチル−GD2、葉酸受容体ベータ、TEM1/CD248、TEM7R、CLDN6、TSHR、GPRC5D、CXORF61、CD97、CD179a、ALK、ポリシアル酸、PLAC1、GloboH、NY−BR−1、UPK2、HAVCR1、ADRB3、PANX3、GPR20、LY6K、OR51E2、TARP、WT1、NY−ESO−1、LAGE−1a、MAGE−A1、MAGE A1、ETV6−AML、精子タンパク質17、XAGE1、Tie 2、MAD−CT−1、MAD−CT−2、Fos関連抗原1、p53、p53変異体、プロテイン、サバイビンおよびテロメラーゼ、PCTA−1/ガレクチン8、メランA/MART1、Ras変異体、hTERT、肉腫転座切断点、ML−IAP、ERG(TMPRSS2 ETS融合遺伝子)、NA17、PAX3、アンドロゲン受容体、サイクリンB1、MYCN、RhoC、TRP−2、CYP1B1、BORIS、SART3、PAX5、OY−TES1、LCK、AKAP−4、SSX2、RAGE−1、ヒトテロメラーゼ逆転写酵素、RU1、RU2、レグマイン、HPV E6,E7、腸管カルボキシルエステラーゼ、mut hsp70−2、CD79a、CD79b、CD72、LAIR1、FCAR、LILRA2、CD300LF、CLEC12A、BST2、EMR2、LY75、GPC3、FCRL5およびIGLL1から選択される1以上の腫瘍抗原に結合する。
他の面において、本発明は、ここに記載する核酸分子、CARポリペプチド分子またはベクターを含む、細胞、例えば、免疫エフェクター細胞(例えば、細胞集団、例えば、免疫エフェクター細胞集団)に関する。
他の面において、本発明は、CAR分子、例えば、ここに記載するCAR分子またはCAR分子、例えば、ここに記載するCAR分子をコードする核酸を含む細胞を投与することを含む、方法を提供する。ある態様において、対象はここに記載する障害を有し、例えば、対象は癌を有し、例えば、対象はここに記載する標的抗原を発現する癌を有する。ある態様において、対象はヒトである。
(i)CAR分子は、抗原結合ドメイン、膜貫通ドメインならびに共刺激ドメインおよび/または一次シグナル伝達ドメインを含む細胞内ドメインを含み、ここで、該抗原結合ドメインは疾患と関係する腫瘍抗原、例えばここに開示する腫瘍抗原と結合し;そして
(ii)免疫細胞の有効性を高める薬剤は、
(i)タンパク質ホスファターゼ阻害剤;
(ii)キナーゼ阻害剤;
(iii)サイトカイン;
(iv)免疫阻害分子の阻害剤;または
(v)TREG細胞のレベルまたは活性を低下させる薬剤
の1以上が選択される。
(i)CAR分子は、抗原結合ドメイン、膜貫通ドメインならびに共刺激ドメインおよび/または一次シグナル伝達ドメインを含む細胞内ドメインを含み、ここで、該抗原結合ドメインは疾患と関係する腫瘍抗原、例えば、ここに開示する腫瘍抗原と結合し;そして
(ii)CAR分子の抗原結合ドメインは、抗原結合ドメインが由来する抗体より少なくとも5倍低い結合親和性を有する。
(i)CAR分子は、抗原結合ドメイン、膜貫通ドメインならびに共刺激ドメインおよび/または一次シグナル伝達ドメインを含む細胞内ドメインを含み、ここで、該抗原結合ドメインは疾患と関係する腫瘍支持抗原、例えばここに記載する腫瘍支持抗原と結合し;そして
(ii)免疫細胞の有効性を高める薬剤は、
(i)タンパク質ホスファターゼ阻害剤;
(ii)キナーゼ阻害剤;
(iii)サイトカイン;
(iv)免疫阻害分子の阻害剤;または
(v)TREG細胞のレベルまたは活性を低下させる薬剤
の1以上が選択される。
(i)CAR分子は、抗原結合ドメイン、膜貫通ドメインならびに共刺激ドメインおよび/または一次シグナル伝達ドメインを含む細胞内ドメインを含み、ここで、該抗原結合ドメインは疾患と関係する腫瘍支持抗原、例えば、ここに記載する腫瘍支持抗原と結合し;そして
(ii)CAR分子の抗原結合ドメインは、抗原結合ドメインが由来する抗体より少なくとも5倍低い結合親和性を有する。
他の面において、本発明は、細胞、例えば、ここに記載するT細胞またはNK細胞に、CAR、例えば、ここに記載するCARをコードする核酸を含むベクター;またはCAR分子、例えば、ここに記載するCARをコードする核酸を導入(例えば、形質導入)することを含む、細胞(例えば、免疫エフェクター細胞またはその集団)を産生する方法に関する。
a. 免疫エフェクター細胞集団(例えば、T細胞またはNK細胞)を提供し;そして
b. 集団からT制御性細胞を除去し、それによりT制御性枯渇細胞集団を提供し;
ここで、工程a)およびb)を、CARをコードする核酸を集団に導入する前に行う。
CARをコードする核酸を集団に導入する前に、T制御性枯渇および腫瘍抗原枯渇細胞集団を提供するために、CD25を含まない腫瘍抗原を発現する細胞を、集団から除去することを含む。腫瘍抗原は、CD19、CD30、CD38、CD123、CD20、CD14またはCD11bまたはこれらの組み合わせから選択できる。
CARをコードする核酸を集団に導入する前に、T制御性枯渇および阻害分子枯渇細胞集団を提供するために、チェックポイント阻害剤を発現する細胞を集団から除去することを含む。チェックポイント阻害剤は、PD−1、LAG−3、TIM3、B7−H1、CD160、P1H、2B4、CEACAM(例えば、CEACAM−1、CEACAM−3および/またはCEACAM−5)、TIGIT、CTLA−4、BTLAおよびLAIR1から選択できる。
ある態様において、細胞集団は、8日またはそれ以下の期間増殖される。
定義
他に定義しない限り、ここで使用する全ての技術的および科学的用語は、本発明が属する技術分野の当業者に共通して理解されているのと同じ意味を有する。
を担う分子由来のものを含む。ある態様において、細胞内シグナル伝達ドメインは、共刺激性細胞内ドメインを含む。代表的共刺激性細胞内シグナル伝達ドメインは、共刺激シグナルまたは抗原非依存的刺激を担う分子由来のものを含む。例えば、CARTの場合、一次細胞内シグナル伝達ドメインはT細胞受容体の細胞質配列を含むことができ、共刺激性細胞内シグナル伝達ドメインは共受容体または共刺激分子からの細胞質配列を含むことができる。
例えば、記憶T細胞、例えば、記憶T細胞前駆体における、次のマーカーCD62L高、CD127高、CD27+およびBCL2の1以上の発現の増加;
例えば、記憶T細胞、例えば、記憶T細胞前駆体における、KLRG1の発現の減少;および
記憶T細胞前駆体、例えば、次のCD62L高増加、CD127高増加、CD27+増加、KLRG1減少およびBCL2増加の特性の任意の1つまたは組み合わせを有する細胞の数の増加;
ここで、上記変化のいずれかは、例えば、非処置対象と比較して、例えば、少なくとも一過性に生じる。
本発明のCARで操作された免疫エフェクター細胞(例えば、T細胞、NK細胞)を使用する癌のような疾患の処置に使用するための、組成物および方法がここで提供される。

本発明は、免疫エフェクター細胞を癌に向けさせる1以上のCARを含むように操作された免疫エフェクター細胞(例えば、T細胞、NK細胞)を提供する。これは、癌関連抗原に特異的なCAR上の抗原結合ドメインにより達成される。本発明のCARにより標的となり得る2クラスの癌関連抗原(腫瘍抗原)がある。(1)癌細胞の表面上に発現される癌関連抗原;および(2)癌関連抗原であって、それ自体細胞内であるが、しかしながら、このような抗原(ペプチド)のフラグメントがMHC(主要組織適合抗原複合体)により癌細胞の表面上に提示される抗原。
本発明のCARは、腫瘍支持抗原(例えば、腫瘍支持抗原)に結合する抗原結合ドメイン(例えば、抗体または抗体フラグメント、TCRまたはTCRフラグメント)を含み得る。ある態様において、腫瘍支持抗原は、間質細胞または骨髄球由来サプレッサー細胞(MDSC)上に存在する抗原である。間質細胞は、微小環境における細胞分裂を促進するために増殖因子を分泌できる。MDSC細胞はT細胞増殖および活性化を阻害できる。理論に縛られることを望まないが、ある態様において、CAR発現細胞は腫瘍支持細胞を破壊し、それにより間接的に腫瘍増殖または生存を阻害する。
本発明は、CARをコードする配列を含む組み換えDNA構築物を包含し、ここで、CARは、ここに記載する癌関連抗原に特異的に結合する抗原結合ドメイン(例えば、抗体または抗体フラグメント、TCRまたはTCRフラグメント)を含み、ここで、抗原結合ドメインの配列は、細胞内シグナル伝達ドメインをコードする核酸配列と隣接し、かつ同じリーディングフレーム内である。細胞内シグナル伝達ドメインは共刺激シグナル伝達ドメインおよび/または一次シグナル伝達ドメイン、例えば、ゼータ鎖を含み得る。共刺激シグナル伝達ドメインは、共刺激分子の細胞内ドメインの少なくとも一部を含むCARの部分をいう。
ある面において、本発明のCARは、抗原結合ドメインとも呼ぶ、標的特異的結合要素を含む。部分の選択は、標的細胞の表面を規定するリガンドのタイプおよび数による。例えば、抗原結合ドメインは、特定の疾患状態と関係する標的細胞上の細胞表面マーカーとして働くリガンドを認識するように選択し得る。それゆえに、本発明のCARにおける抗原結合ドメインのリガンドとして作用し得る細胞表面マーカーの例は、ウイルス、細菌および寄生虫感染、自己免疫性疾患および癌細胞と関係するものを含む。
ある態様において多特異的抗体分子は、二重特異的抗体分子である。二重特異的抗体は2を超える抗原に特異性を有しない。二重特異的抗体分子は、第一エピトープに結合特異性を有する第一免疫グロブリン可変ドメイン配列および第二エピトープに結合特異性を有する第二免疫グロブリン可変ドメイン配列により特徴付けられる。ある態様において、第一エピトープおよび第二エピトープは同じ抗原上、例えば、同じタンパク質(または多量体タンパク質のサブユニット)である。ある態様において、第一エピトープおよび第二エピトープは重複する。ある態様において、第一エピトープおよび第二エピトープは重複しない。ある態様において、第一エピトープおよび第二エピトープは、異なる抗原上、例えば、異なるタンパク質(または多量体タンパク質の異なるサブユニット)である。ある態様において、二重特異的抗体分子は、第一エピトープに結合特異性を有する重鎖可変ドメイン配列および軽鎖可変ドメイン配列ならびに第二エピトープに結合特異性を有する重鎖可変ドメイン配列および軽鎖可変ドメイン配列を含む。ある態様において、二重特異的抗体分子は、第一エピトープに結合特異性を有する半抗体および第二エピトープに結合特異性を有する半抗体を含む。ある態様において、二重特異的抗体分子は、第一エピトープに結合特異性を有する半抗体またはそのフラグメントおよび第二エピトープに結合特異性を有する半抗体またはそのフラグメントを含む。ある態様において、二重特異的抗体分子は、第一エピトープに結合特異性を有するscFvまたはそのフラグメントおよび第二エピトープに結合特異性を有するscFvまたはそのフラグメントを含む。
ここに記載する癌関連抗原に対する抗原結合ドメイン、例えば、scFv分子(例えば、可溶性scFv)の安定性を、従来の対照scFv分子または完全長抗体の生物物理学的特性(例えば、熱安定性)を参照して評価できる。ある態様において、ヒト化scFvは、記載するアッセイにおいて、対照結合分子(例えば従来のscFv分子)よりも約0.1℃、約0.25℃、約0.5℃、約0.75℃、約1℃、約1.25℃、約1.5℃、約1.75℃、約2℃、約2.5℃、約3℃、約3.5℃、約4℃、約4.5℃、約5℃、約5.5℃、約6℃、約6.5℃、約7℃、約7.5℃、約8℃、約8.5℃、約9℃、約9.5℃、約10℃、約11℃、約12℃、約13℃、約14℃または約15℃大きい熱安定性を有する。
抗原結合ドメインの安定性を、例えば、下記方法を使用して、評価し得る。このような方法は、協同的に変性する多ドメイン単位(例えば、単一変性転移を示す多ドメインタンパク質)で最も不安定なドメインが最初に変性するかまたは全体的安定性閾値を制限する、多重熱変性転移の決定を可能にする。最も不安定なドメインは、多数の付加的方法により決定できる。変異誘発は、どのドメインが全体的安定性を制限するかを探索するために実施できる。さらに、多ドメインタンパク質のプロテアーゼ耐性を、最も不安定なドメインがDSCまたは他のスペクトル方法により内因的に変性することが知られる条件下で実施できる(Fontana, et al., (1997) Fold. Des., 2: R17-26; Dimasi et al. (2009) J. Mol. Biol. 393: 672-692)。最も不安定なドメインが同定されたら、そのドメインをコードする配列(またはその一部)を、本方法における試験配列として用い得る。
組成物の熱安定性を、当分野で知られる多数の非限定的生物物理学的または生化学的技術を使用して分析し得る。ある態様において、熱安定性を、分析的スペクトロスコピーにより評価する。
組成物の安定性は、その凝集する能力の測定により決定できる。凝集は、多数の非限定的生化学的または生物物理学的技術により測定できる。例えば、組成物の凝集は、クロマトグラフィー、例えばサイズ排除クロマトグラフィー(SEC)を使用して、評価し得る。SECは、サイズに基づき分子を分ける。カラムに、イオンおよび小分子を内側に入れるが、大型のものは入れない重合ゲルの半固体ビーズを充填する。タンパク質組成物をカラムの上に載せたとき、小型の折りたたまれたタンパク質(すなわち非凝集タンパク質)は、大タンパク質凝集体よりも大量の溶媒に分散する。その結果、大凝集体はカラムをより迅速に移動し、この方法で混合物をその成分に分離または分画できる。各フラクションを、ゲルから溶出されるに従い、別々に定量化(例えば光散乱により)できる。したがって、組成物の凝集%を、フラクションの濃度とゲルに適用したタンパク質の総濃度の比較により決定できる。安定な組成物は、本質的に単一フラクションとしてカラムから溶出し、溶出プロファイルまたはクロマトグラムで本質的に単一ピークを有する。
組成物の安定性は、その標的結合親和性の決定により評価できる。結合親和性を決定するための多種多様な方法が当分野で知られる。代表的な結合親和性を測定するための方法は、表面プラズモン共鳴を用いる。表面プラズモン共鳴は、例えばBIAcore system(Pharmacia Biosensor AB, Uppsala, Sweden and Piscataway, N.J.)を使用する、バイオセンサーマトリクス内のタンパク質濃度の変化の検出により、実時間生体分子特異的相互作用の分析を可能にする光学現象である。さらなる記載について、Jonsson, U., et al. (1993) Ann. Biol. Clin. 51:19-26; Jonsson, U., i (1991) Biotechniques 11:620-627; Johnsson, B., et al. (1995) J. Mol. Recognit. 8:125-131; and Johnnson, B., et al. (1991) Anal. Biochem. 198:268-277参照。
膜貫通ドメインに関して、多様な態様において、CARは、CARの細胞外ドメインに結合する膜貫通ドメインを含むように設計できる。膜貫通ドメインは、膜貫通領域に隣接する1以上のさらなるアミノ酸、例えば、膜貫通ドメインが由来するタンパク質の細胞外領域と結合する1以上のアミノ酸(例えば、細胞外領域の1、2、3、4、5、6、7、8、9、10から最大15のアミノ酸)および/または膜貫通型タンパク質が由来するタンパク質の細胞内領域と結合する1以上のさらなるアミノ酸(例えば、細胞内領域の1、2、3、4、5、6、7、8、9、10から最大15のアミノ酸)を含むことができる。ある面において、膜貫通ドメインは、CARの他のドメインと結合しているもの、例えば、ある態様において、膜貫通ドメインは、シグナル伝達ドメイン、共刺激ドメインまたはヒンジドメインが由来するものと同じタンパク質に由来し得る。他の面において、膜貫通ドメインは、CARの他のいずれかのドメインが由来するのと同じタンパク質に由来しない。いくつかの例において、膜貫通ドメインは、例えば、受容体複合体の他のメンバーとの相互作用を最小化するため、このようなドメインの同じまたは異なる表面膜タンパク質の膜貫通ドメインへの結合を避けるため、選択できまたはアミノ酸置換により修飾できる。ある面において、膜貫通ドメインは、CAR発現細胞の細胞表面上の他のCARとホモ二量体化できる。異なる面において、膜貫通ドメインのアミノ酸配列は、同じCAR発現細胞に存在する天然の結合パートナーの結合ドメインとの相互作用を最小化するために、修飾または置換され得る。
ある面において、ヒンジまたはスペーサーは、KIR2DS2ヒンジを含む。
CARの細胞質ドメインまたは領域は、細胞内シグナル伝達ドメインを含む。細胞内シグナル伝達ドメインは、一般にCARが導入されている免疫細胞の正常エフェクター機能の少なくとも1つの活性化を担う。用語“エフェクター機能”は、細胞の専門化された機能をいう。T細胞のエフェクター機能は、例えば、サイトカインの分泌を含む細胞溶解性活性またはヘルパー活性であり得る。それゆえに、用語“細胞内シグナル伝達ドメイン”は、エフェクター機能シグナルを伝達し、細胞に専門化された機能の実施を指示するタンパク質の部分をいう。通常細胞内シグナル伝達ドメイン全体を用いることができるが、多くの場合、鎖全体を使用する必要はない。細胞内シグナル伝達ドメインの切断された部分を使用する限り、このような切断された部分は、エフェクター機能シグナルを伝達する限り、完全鎖の代わりに使用し得る。用語細胞内シグナル伝達ドメインは、それゆえに、エフェクター機能シグナルの伝達に十分な、細胞内シグナル伝達ドメインのあらゆる切断された部分を含むことを意図する。
ある態様において、CAR活性が制御できる調節可能なCAR(RCAR)が、CAR治療の安全性および有効性を最適化するために望まれる。CAR活性を制御できる多くの方法がある。例えば、二量体化ドメインに融合したカスパーゼを使用する、例えば、誘導性アポトーシス(例えば、Di et al., N Egnl. J. Med. 2011 Nov. 3; 365(18):1673-1683参照)を、本発明のCAR治療における安全スイッチとして使用できる。ある面において、RCARは、ここに記載する標準的CARの成分、例えば、抗原結合ドメインおよび細胞内シグナル伝達ドメインが別々のポリペプチドまたはメンバーに配置されている、一般に最も単純な態様において2つの、一連のポリペプチドを含む。ある態様において、一連のポリペプチドは、二量体化分子の存在下、ポリペプチドを互いに連結できる、例えば、抗原結合ドメインと細胞内シグナル伝達ドメインを連結できる、二量体化スイッチを含む。
二量体化スイッチは、非共有結合でも共有結合でもよい。非共有結合二量体化スイッチにおいて、二量体化分子は、スイッチドメイン間の非共有結合相互作用を促進する。において、共有結合二量体化スイッチ、二量体化分子は、スイッチドメイン間の共有結合相互作用を促進する。
スイッチドメイン間の結合は、二量体化分子により促進される。二量体化分子存在下、スイッチドメイン間の相互作用または結合は、第一スイッチドメインと結合した、例えば、融合したポリペプチドと第二スイッチドメインと結合した、例えば、融合したポリペプチドの間のシグナル伝達を可能にする。非律速レベルの二量体化分子存在下、シグナル伝達は、例えば、ここに記載する系において測定して、1.1倍、1.2倍、1.3倍、1.4倍、1.5倍、1.6倍、1.7倍、1.8倍、1.9倍、2倍、5倍、10倍、50倍、100倍増加する。
ある態様において、CAR発現細胞は、分裂CARを使用する。分裂CAR手法は、公開WO2014/055442号およびWO2014/055657号にさらに詳細に記載される。簡潔には、分裂CAR系は第一抗原結合ドメインおよび共刺激ドメイン(例えば、41BB)を有する第一CARを発現する細胞を含み、細胞はまた第二抗原結合ドメインおよび細胞内シグナル伝達ドメイン(例えば、CD3ゼータ)を有する第二CARを発現する。細胞が第一抗原と遭遇したとき、共刺激ドメインは活性化され、細胞が増殖する。細胞が第二抗原に遭遇したとき、細胞内シグナル伝達ドメインは活性化され、細胞死活性が開始する。それゆえに、CAR発現細胞は、両抗原の存在下でのみ完全活性化される。
インビトロ転写RNA CARを産生する方法がここに開示される。本発明はまた、細胞に直接トランスフェクトできるRNA構築物をコードするCARも含む。トランスフェクションに使用するためのmRNAを産生する方法は、一般に50〜2000塩基長(配列番号32)の、3’および5’非翻訳配列(“UTR”)、5’キャップおよび/または配列内リボソーム進入部位(IRES)、発現させるべき核酸およびポリAテイルを含む構築物を産生するための特別に設計されたプライマーを用いる鋳型のインビトロ転写(IVT)、続くポリA付加を含む。このように産生されたRNAは、異なる種の細胞を効率的にトランスフェクトできる。ある面において、鋳型はCARのための配列を含む。
ある面において、非ウイルス性の方法を使用して、ここに記載するCARをコードする核酸を細胞または組織または対象に送達できる。
本発明はまた、1以上のここに記載するCAR構築物をコードする核酸分子を提供する。ある面において、核酸分子は、メッセンジャーRNA転写物として提供される。ある面において、核酸分子はDNA構築物として提供される。
増殖および遺伝的修飾または他の修飾の前に、細胞、例えば、T細胞またはナチュラルキラー(NK)細胞の供給源を、対象から得ることができる。用語“対象”は、免疫応答が惹起できる生存生物(例えば、哺乳動物)を含むことを意図する。対象の例は、ヒト、サル、チンパンジー、イヌ、ネコ、マウス、ラットおよびそのトランスジェニック種を含む。T細胞は、末梢血単核細胞、骨髄、リンパ節組織、臍帯血、胸腺組織、感染部位からの組織、腹水、胸水、脾臓組織および腫瘍を含む、多数の供給源から得ることができる。
ここに記載する態様において、免疫エフェクター細胞は、同種免疫エフェクター細胞、例えば、T細胞またはNK細胞であり得る。例えば、細胞は、同種T細胞、例えば、機能的T細胞受容体(TCR)および/またはヒト白血球抗原(HLA)、例えば、HLAクラスIおよび/またはHLAクラスIIの発現を欠く同種T細胞である。
ある態様において、TCR発現および/またはHLA発現を、T細胞におけるTCRおよび/またはHLAをコードする核酸を標的とするsiRNAまたはshRNAを使用して、阻害できる。
ここで使用する“CRISPR”または“TCRおよび/またはHLAに対するCRISPR”または“TCRおよび/またはHLAを阻害するためのCRISPR”は、一連の群生性等間隔短回文反復配列またはこのような一連の反復を含む系をいう。ここで使用する“Cas”は、CRISPR関連タンパク質をいう。“CRISPR/Cas”系は、TCRおよび/またはHL遺伝子の発現抑制または変異に使用できる、CRISPRおよびCas由来の系をいう。
“TALEN”または“HLAおよび/またはTCRに対するTALEN”または“HLAおよび/またはTCRを阻害するためのTALEN”は、HLAおよび/またはTCR遺伝子の編集に使用できる人工ヌクレアーゼである、転写アクティベーター様エフェクターヌクレアーゼをいう。
“ZFN”または“亜鉛フィンガーヌクレアーゼ”または“HLAおよび/またはTCRに対するZFN”または“HLAおよび/またはTCRを阻害するためのZFN”は、HLAおよび/またはTCR遺伝子の編集に使用できる人工ヌクレアーゼである、亜鉛フィンガーヌクレアーゼをいう。
何らかの特定の理論に縛られることを望まないが、ある態様において、治療的T細胞は、T細胞における短縮されたテロメアのために、患者における残留性が短く、したがって、テロメラーゼ遺伝子を用いるトランスフェクションは、T細胞のテロメアを延長し、患者におけるT細胞の残留性を改善し得る。Carl June, “Adoptive T cell therapy for cancer in the clinic”, Journal of Clinical Investigation, 117:1466-1476 (2007)参照。それゆえに、ある態様において、免疫エフェクター細胞、例えば、T細胞、異所性にテロメラーゼサブユニットを、例えば、テロメラーゼの触媒的サブユニット、例えば、TERT、例えば、hTERT発現する。ある面において、本開示は、細胞とテロメラーゼサブユニット、例えば、テロメラーゼの触媒的サブユニット、例えば、TERT、例えば、hTERTをコードする核酸を接触させることを含む、CAR発現細胞を産生する方法を提供する。細胞を、CARをコードする構築物と接触させる前に、同時にまたは後に核酸と接触させ得る。
T細胞のような免疫エフェクター細胞は、例えば、米国特許6,352,694号;6,534,055号;6,905,680号;6,692,964号;5,858,358号;6,887,466号;6,905,681号;7,144,575号;7,067,318号;7,172,869号;7,232,566号;7,175,843号;5,883,223号;6,905,874号;6,797,514号;6,867,041号;および米国特許出願公開20060121005号に記載する方法を使用して、一般的に活性化および増殖できる。
ある面において、本発明は、ここに記載する癌関連抗原の発現と関係する疾患を処置する方法を提供する。
血液癌状態は、血液、骨髄およびリンパ系に影響する白血病、リンパ腫および悪性リンパ増殖性状態のようなタイプの癌である。
ここに記載するCAR発現細胞を、他の既知薬剤および治療と組み合わせて使用し得る。ここで使用する“組み合わせて”投与するは2(またはそれ以上)の異なる処置を、対象が障害に苦しんでいる過程の間に対象に送達することを意味し、例えば、2以上の処置を、対象が障害と診断された後にかつ障害が治癒または撲滅される前にまたは処置が他の理由で中止される前に送達される。ある態様において、投与期間の重複があるように、一方の処置の送達は、第二の処置の送達開始時にまだ行われている。これは、ここでは“同時の”または“一緒の送達”ということがある。他の態様において、一方の処置の送達は、他の処置の送達が始まる前に終わる。いずれかの場合のある態様において、処置は、組み合わせ投与のためにより有効である。例えば、第二処置はより有効である、例えば、等しい効果が少ない第二処置で見られるまたは第二処置が、第二処置を第一処置非存在下で投与したときに見られるよりも大きな程度で症状を軽減するまたは第一処置で同様な状況が見られる。ある態様において、送達は、障害と関係する症状または他のパラメーターの減少が、他方の処置の非存在下で一方の処置の送達で観察されるされるより大きい。2処置の効果は一部相加的、完全相加的または相加的より大きいものであり得る。送達は、送達した第一処置の効果が、第二剤の送達時になお検出可能であるようなものであり得る。
ある態様において、CAR分子を発現する細胞、例えば、ここに記載するCAR分子は、低い、免疫増強用量のmTOR阻害剤と組み合わせて投与される。
ここで使用する用語“mTOR阻害剤”は、細胞におけるmTORキナーゼを阻害する化合物またはリガンドまたはその薬学的に許容される塩をいう。ある態様において、mTOR阻害剤はアロステリック阻害剤である。ある態様において、mTOR阻害剤は触媒的阻害剤である。
mTORは、キナーゼP70 S6をリン酸化し、それによりP70 S6キナーゼを活性化し、それがその基質をリン酸化することを可能にする。mTOR阻害の程度はP70 S6キナーゼ阻害の程度で表すことができ、例えば、mTOR阻害の程度は、P70 S6キナーゼ活性レベルの減少、例えば、P70 S6キナーゼ基質のリン酸化の減少により決定できる。mTOR阻害のレベルを、阻害剤の非存在下、例えば、阻害剤の投与前にまたは阻害剤投与後にP70 S6キナーゼ活性(P70 S6キナーゼが基質をリン酸化する能力)を測定することにより決定できる。P70 S6キナーゼ阻害のレベルは、mTOR阻害のレベルを提供する。それゆえに、P70 S6キナーゼが40%阻害されるならば、mTOR活性は、P70 S6キナーゼ活性により測定して、40%阻害される。ここでいう阻害の程度またはレベルは、投与間隔の間の阻害の平均レベルである。例えば、阻害剤を週に1回与えるとき、阻害のレベルは、その間隔の間の、すなわち1週間の阻害の平均レベルを示す。
mTOR阻害のレベルはまた、PD1陰性T細胞対PD1陽性T細胞の比の変化によっても評価できる。末梢血からのT細胞を、当分野で知られる方法によりPD1陰性または陽性と同定できる。
ここに記載する方法は、低い、免疫増強用量のmTOR阻害剤、例えば、RAD001のようなラパログを含むアロステリックの複数用量を使用する。対照的に、mTOR経路を完全にまたはほぼ完全に阻害するレベルの阻害剤は免疫抑制性であり、例えば、臓器移植片拒絶の予防に使用される。さらに、mTORを完全に阻害する高用量のラパログも腫瘍細胞増殖を阻止し、多様な癌の処置に使用されている(例えば、Antineoplastic effects of mammalian target of rapamycine inhibitors. Salvadori M. World J Transplant. 2012 Oct 24;2(5):74-83; Current and Future Treatment Strategies for Patients with Advanced Hepatocellular Carcinoma: Role of mTOR Inhibition. Finn RS. Liver Cancer. 2012 Nov;1(3-4):247-256; Emerging Signaling Pathways in Hepatocellular Carcinoma. Moeini A, Cornella H, Villanueva A. Liver Cancer. 2012 Sep;1(2):83-93; Targeted cancer therapy - Are the days of systemic chemotherapy numbered? Joo WD, Visintin I, Mor G. Maturitas. 2013 Sep 20.; Role of natural and adaptive immunity in renal cell carcinoma response to VEGFR-TKIs and mTOR inhibitor. Santoni M, Berardi R, Amantini C, Burattini L, Santini D, Santoni G, Cascinu S. Int J Cancer. 2013 Oct 2参照)。
例えば、記憶T細胞、例えば、記憶T細胞前駆体における、次のマーカーCD62L高、CD127高、CD27+およびBCL2の1以上の発現の増加;
例えば、記憶T細胞、例えば、記憶T細胞前駆体における、KLRG1の発現の減少;および
記憶T細胞前駆体、例えば、次のCD62L高増加、CD127高増加、CD27+増加、KLRG1減少およびBCL2増加の特性の任意の1つまたは組み合わせを有する細胞の数の増加;
そして、ここで、上記のいずれかの変化は、例えば、非処置対象と比較して、例えば、少なくとも一過性に生じる(Araki, K et al. (2009) Nature 460:108-112)。記憶T細胞前駆体は、分化プログラムにおける初期にある記憶T細胞である。例えば、記憶T細胞は、次の1以上の特性を有する:CD62L高増加、CD127高増加、CD27+増加、KLRG1減少および/またはBCL2増加。
一つの面において、本発明は、ここに記載するCAR細胞と組み合わせて使用するために製剤された、mTOR阻害剤、例えば、ここに記載するmTOR阻害剤を含む医薬組成物に関する。
ここに開示するmTOR阻害剤、例えば、アロステリックmTOR阻害剤または触媒的mTOR阻害剤は、製品安定性要求を満たすおよび/または平均血漿ピーク濃度減少、薬物吸収の程度および血漿ピーク濃度の患者間および患者内ばらつき減少、Cmax/Cmin比減少および/または食効減少のような即時放出(IR)錠剤より都合よい薬物動態特性を有する、ここに開示するmTOR阻害剤、例えば、ラパマイシンまたはRAD001を含む経口固体投与形態の形の医薬製剤として提供できる。提供された医薬製剤は、より厳密な用量調節を可能にするおよび/または有害事象の頻度を減少させ、ここに開示するmTOR阻害剤、例えば、ラパマイシンまたはRAD001での患者のより安全な処置を提供することができる。
0.5時間:<45%または<40、例えば、<30%
1時間:20〜80%、例えば、30〜60%
2時間:>50%または>70%、例えば、>75%3時間:>60%または>65%、例えば、>85%、例えば、>90%。
ある態様において、ここに開示する1以上のCAR発現細胞を、バイオポリマー足場、例えば、バイオポリマーインプラントにより対象に投与または送達し得る。バイオポリマー足場は、ここに記載するCAR発現細胞の送達、増殖および/または分散を支持または増強できる。バイオポリマー足場は、天然に存在するまたは合成であり得る生体適合性(例えば、実質的に炎症性または免疫応答を誘発しない)および/または生分解性ポリマーを含む。
本発明の医薬組成物は、ここに記載のように、CAR発現細胞、例えば、複数のCAR発現細胞を、1以上の薬学的にまたは生理学的に許容される担体、希釈剤または添加物と組み合わせて含む。このような組成物は、中性緩衝化食塩水、リン酸緩衝化食塩水などのような緩衝液;グルコース、マンノース、スクロースまたはデキストラン、マンニトールのような炭水化物;タンパク質;グリシンのようなポリペプチドまたはアミノ酸;抗酸化剤;EDTAまたはグルタチオンのようなキレート剤;アジュバント(例えば、水酸化アルミニウム);および防腐剤を含み得る。本発明の組成物は、一つの面において静脈内投与用に製剤される。
ある態様において、阻害分子、例えば、プログラム細胞死1(PD−1)の細胞外ドメイン(ECD)を、4−1BBおよびCD3ゼータのような膜貫通ドメインおよび細胞内シグナル伝達ドメインと融合できる。ある態様において、PD−1 CARを単独で使用できる。ある態様において、PD−1 CARを、他のCAR、例えば、CD19CARと組み合わせて使用できる。ある態様において、PD−1 CARは、T細胞の持続を改善する。ある態様において、CARは、配列番号26で下線により示されるPD−1の細胞外ドメインを含むPD−1 CARである(PD−1ドメインを下線を引く)。
材料および方法:NFAT依存性プロモーター下にGFPを発言するJurkat T細胞(NF−GFP)に、メソテリン特異的活性化CAR(SS1−CAR)、CD19特異的活性化CAR(19−CAR)またはEGFRに特異的なラクダ類VHHドメインを使用して産生されたCAR(VHH−CAR)を形質導入した。活性化CARの形質導入後、細胞を、活性化および阻害性CAR(SS1+19PD1、19+19PD1またはVHH+19PD1)の両者を共発現する細胞を産生するため、CD19を認識するさらなる阻害性CAR(19−PD1)を形質導入した。形質導入したJurkat T細胞を、1)全標的抗原を欠く(K562)、2)メソテリン(K−meso)、CD19(K−19)またはEGFR(A431)のみを発現する、3)EGFRとメソテリン(A431−メソテリン)またはCD19(A431−CD19)の組み合わせを発現するまたは4)CD19とメソテリン(K−19/meso)の組み合わせを発現する、異なる細胞株と24時間共培養した。スティミュレーター細胞無し(no stim)またはK562と1μg/mLのOKT3(OKT3)を含むさらなる条件も、それぞれNFAT活性化の陰性および陽性対照として包含させた。NFAT活性化のマーカーとしてのGFP発現をフローサイトメトリーにより評価した。
葉酸受容体α(FRA)は、卵巣癌の約90%、ならびに子宮内膜癌、腎臓、乳房、肺、膵臓、結腸直腸の癌および中皮腫で過発現し、その発現は、化学療法剤の先の投与に影響されない(例えば、Despierre et al., Gynecol Oncol 130, 192-199 (2013)参照)。正常組織において、FRA発現は0であるかまたは低く、循環している抗FR剤には接触不可能であるように見える場所である、極性化上皮性細胞の尖端表面に制限される(Kelemen et al., International journal of cancer 119, 243-250 (2006))。
細胞株。レンチウイルスパッケージングを、ATCCから購入した不死化正常胎児腎臓293T細胞株で実施した。免疫ベースのアッセイに使用したヒト細胞株は、確立されたヒト卵巣癌細胞株SKOV3、A1847、OVCAR2、OVCAR3、OVCAR4、OVCAR5、A2780、A2008、C30およびPEO−1を含む。ヒトリンパ系細胞株SUP−T1をレンチウイルス力価分析に使用した。バイオルミネセンスアッセイのために、標的癌細胞株をホタルルシフェラーゼ(fLuc)を発現するようにトランスフェクトした。マウス悪性中皮腫細胞株、AE17(Steven Albelda, University of Pennsylvaniaから恵与)を陰性対照として使用した。全細胞株を、10%熱不活化FBS、100U/mLペニシリン、100mg/mL硫酸ストレプトマイシン、10mmol/L HEPES)を添加したRPMI−1640であるR10培地に維持した。
1. インビトロでのマウスMOv19 CARと比較したヒトC4 CARの増強された機能
上記産生および濃縮プロトコールを使用して、我々は、C4 CARコード化レンチウイルスが、恐らくヒトT細胞上でのヒトcFvの効率的発現の結果として、マウスMOv19 CARより高い有効力価を有することを発見した(図37a)。実際、我々は、C4 CARレンチウイルスの1ほど低い感染効率(MOI)が>20%ヒトT細胞の感染に十分であり、一方MOv19 CARレンチウイルスは5のMOIを必要とすることを観察した(図37b)。それゆえに、次の実験のために、T細胞を、それぞれ2および5のMOIの濃縮C4−27zおよびMOv19−27zベクターで感染させ、T細胞上のC4およびMOv19 CAR両者の表面発現を、組み換えFRAタンパク質染色により検出した(図34a)。
インビボでのC4 CAR T細胞とMOv19 CAR T細胞の抗腫瘍能力を比較するために、大きな、確立された皮下SKOV3腫瘍(約300mm3)を有するNSGマウスに、腫瘍接種40日および47日後に107 CAR+ T細胞を静脈内注射した。食塩水、非形質導入T細胞またはCD19−27z CAR T細胞で処置した動物における腫瘍は急速な増殖を続けた。対照的に、C4−27zまたはMOv19−27z CAR T細胞を受けたマウスは、最後の評価時点で全3対照群と比較して、腫瘍退縮を経験した(p<0.0001)。MOv19−27z CAR T細胞の抗腫瘍活性はC4−27z CAR T細胞よりわずかに良いように見えるが、有意レベルではなかった(p=0.058;図35a)。T細胞注射前および3週間後の腫瘍異種移植のBLIは、対照T細胞を受けた全動物における腫瘍の進行性増殖を示したが、CAR T細胞群ではなかった(図35b)。腫瘍BLI結果は、摘除した残留腫瘍のサイズと一致した(図35c)。次に、我々は、養子移入3週間後の末梢血における移入T細胞の持続を分析し、UNTおよびCD19−27z CAR T細胞処置群と比較して、C4およびMOv19 CAR T細胞の両者で処置された群のマウスで高いCD4+およびCD8+ T細胞数を検出し(図35d)、腫瘍抗原認識がインビボで養子移入T細胞の生存を駆動することが示唆された。これらの結果は、C4 CARの抗腫瘍活性が、先に十分に記載された(例えば、Song et al., Blood 119, 696-706 (2012); Song et al., Cancer Res 71, 4617-4627 (2011)参照)MOv19 CARと同等であることを示し、C4 CARは、その親和性が低下しているにも関わらず、インビボ適用に適することが確認された。
標的上毒性は、正常細胞で低レベルで発現される腫瘍結合抗原に特異的なCAR T細胞の臨床試験で観察されており、そして取り組むべき重大問題は、高親和性のCARが毒性のリスクを高め得るか否かである。低レベルのFRAを発現する正常細胞に対する、C4 CARおよびMOv19 CARで修飾された初代ヒトT細胞の機能的効果を試験するために、我々は、低いが、検出可能なレベルのFRAを発現するヒト胚性腎臓293T細胞または正常上皮性卵巣細胞株IOSE 6およびFRAPOS SKOV3細胞と共培養後、C4 CARおよびMOv19 CAR T細胞のサイトカイン産生を分析した(図36a)。C4およびMOv19 CAR T細胞はSKOV3に対して応答し、大きな活性が再びC4 CAR T細胞で観察された。しかしながら、大きなIFN−γサイトカイン産生は、低抗原発現細胞に対する応答においてMOv19 CAR T細胞で観察され、MOv19 CAR T細胞が低抗原により機能的に貪欲であり、感受性であることが示唆される(図36b)。一夜IFN−γ放出アッセイで我々が観察したものに類似して、5時間細胞内サイトカイン分泌アッセイは、C4 CAR T細胞と比較して、多くのMOv19 CAR T細胞がIFN−γおよびTNF−αを正常細胞上の低抗原に応答して産生し(図36c)、これは、“標的上”サイトカインストームに対する一つの主に提案されている寄与因子である28。これらのデータは、新たに記載したC4 CARが、安全かつ有効な操作されたT細胞治療の送達のためのより適切な親和性を有し得ることを示唆する。
ここに記載するインビトロ結果は、完全ヒトC4 CAR T細胞が、MOv19 CAR T細胞より機能的に優れ、CAR下方制御がMOv19 CARの抗腫瘍活性を障害するが、C4 CARを障害しない可能性を示唆した。MOv19 CAR T細胞による機能低下を説明する機構を理解するために、組み換えαFRタンパク質に対する相対的結合を測定する実験を実施した。C4およびMOv19 CAR T細胞にビオチン標識組み換えαFRタンパク質を予充填し、4℃または37℃で、10倍過剰の非ビオチニル化αFRコンペティター存在下、経時的に表面タンパク質解離を測定した。細胞表面上への抗原維持を、各培養終了後にフィコエリスリン(PE)結合ストレプトアビジン(SA)を添加することによりフローサイトメトリーで評価した。1時間以内に、いずれの温度でも、MOv19 CARと比較してC4 CAR T細胞の表面上で検出可能なαFRタンパク質は少なかった。解離レベルは時間および温度の両者に依存的であり、全試験条件下でC4 CAR T細胞で高かった(図88A、図88B)。類似する結果が、ビオチニル化αFRタンパク質のMOv19およびC4 CAR T細胞への結合に対する力価測定分析で得られた(図89)。活性化T細胞を、MOv19−27zまたはC4−27z−CARを発現するレンチウイルスベクターで形質導入し、14日目にCAR発現について評価した。10万非形質導入(UNT)またはCAR T細胞を、0.2nM/サンプル、0.5nM/サンプル、1nM/サンプル、2nM/サンプル、5nM/サンプル、10nM/サンプル、20nM/サンプル、50nM/サンプルまたは120nM/サンプルのビオチニル化αFRで染色した。T細胞を洗浄し、PE−SAで染色した。T細胞をフローサイトメーターを使用して分析し、データをFlowJoソフトウェアで分析した。これらの結果は、CAR構築物におけるC4が、可溶性αFR抗原に対してMOv19 CARより低い親和性を有することを示唆する。
CARの設計のために選択した完全ヒトC4 scFvの低下した親和性はT細胞認識に影響し得る。しかしながら、C4およびMOv19 CARで修飾したT細胞による腫瘍結合後のサイトカイン産生の直接比較は、低親和性を有するC4 CARが、E/T比1:1で優れることを示した。これは高レベルの抗原と遭遇する、高親和性のMOv19 CARの急速な内部移行によるものであり得る。我々がE/T比を上げるとき、C4 CAR T細胞の抗腫瘍活性はMOv19 CAR T細胞と同等である。インビボで抗腫瘍活性をさらに比較するために、我々は、高親和性MOv19 CARを発現するT細胞は、インビボでC4 CARと比較してわずかに優れる活性を仲介することを発見した。しかしながら、この差は統計学的有意ではなく、C4 CARの親和性がインビボ適用に適することを示唆する。
CAR操作されたT細胞での養子細胞治療(ACT)は、広範な悪性細胞を標的とし、殺すことができ、それによりある造血器悪性腫瘍の処置における耐久性の臨床応答を誘発する(Kochenderfer, J.N., et al. (2010) Blood 116:4099-4102; Porter, D.L., et al. (2011) N Engl J Med 365:725-733;およびBrentjens, R.J., et al. (2013) Sci Transl Med 5:177ra138)。しかしながら、多くの共通して標的とされる腫瘍抗原は健常組織によっても発現され、正常組織のT細胞介在破壊からの標的上腫瘍外毒性が、その他の点では有望なタイプの癌治療の開発および採用を制限している。CARまたはTCR操作Tリンパ球での患者の癌処置と関連した重篤な有害事象の最近の報告は、安全かつ効率的な治療のための標的選択の臨界的重要性をさらに説明する(Lamers et al., 2006, J Clin Oncol. 24:e20; Parkhurst et al., 2011, Molecular therapy: the journal of the American Society of Gene Therapy. 19:620-6; Morgan et al., 2013, J Immunotherapy. 36:133-151; Linette et al., 2013, Blood. 122:863-71)。具体的に、高親和性CARTでのErbB2(Her2/neuまたはCD340)のターゲティングは、正常心肺組織上の標的認識により、重篤な毒性をもたらし(Morgan et al., 2013, Mol Therapy. 18:843-851)、同様に、健常皮膚における相対的高レベルのEGFRの存在が、用量制限的皮膚毒性をもたらす(Perez-Soler et al., 2010, J Clin Oncol. 23:5235-46)。
細胞株および初代ヒトリンパ球
SK−BR3、SK−OV3、BT−474、MCF7、MDA231、MDA468、HCC2281、MDA−361、MDA−453、HCC−1419、HCC−1569、UACC−812、LnCap、MDA−175、MCF−10A、HCC38およびHG261細胞株をAmerican Type Culture Collectionから購入し、指示どおり培養した。7種の初代細胞株(ケラチン生成細胞、骨芽細胞、腎臓上皮性、肺動脈内皮細胞、肺動脈平滑筋、神経前駆細胞、CD34+富化PBMC)をPromocellから購入し、それらのプロトコールに従い培養した。初代リンパ球を、University of Pennsylvania Human Immunology Coreにより正常ドナーから単離し、R10培地(10%ウシ胎児血清を添加したRPMI 1640;Invitrogen)で培養した。初代リンパ球を、記載のとおり、CD3およびCD28刺激抗体で被覆したマイクロビーズ(Life Technologies, Grand Island, NY, Catalog)で刺激した(Barrett et al., 2009, Proc Nat Acad Sci USA, 106:3360)。T細胞を、90%ウシ胎児血清および10%ジメチルスルホキシド(DMSO)の溶液中、1×108細胞/バイアルで10日目に凍結保存した。
ErbB2またはEGFRに対するCAR scFvドメインを、関連する刊行物(Carter et al., 1992, Proc Nat Acad Sci USA, 89:4285; Zhou et al., 2007, J Mol Bio, 371:934)により提供される配列情報に基づき、PCRにより合成および/または増幅し、CD8膜貫通ドメインおよび4−1BBおよびCD3Z細胞内シグナル伝達ドメインに連結させ、pGEM.64A RNAベースのベクター(Zhao et al., 2010, Cancer Res, 70:9053)またはpTRPEレンチウイルスベクター(Carpenito et al., 2009, Proc Nat Acad Sci USA, 106:3360)にサブクローン化した。
ビオチニル化ErbB2を、200RUの密度でストレプトアビジン被覆センサーチップに動員した。親4D5抗体の結合親和性(Carter et al., 1992, Proc Nat Acad Sci USA, 89:4285)を組み換えscFvと比較した。scFvの純度および原子質量を液体クロマトグラフィー−マススペクトロメトリーにより確認した。ScFvサンプルを3倍連続希釈し、一定流速でチップ上に注入した。タンパク質複合体の結合および解離速度を、それぞれ270秒および400秒モニターした。二重参照をブランク固定化フローセルおよび緩衝液ブランクに対して行い、データを1:1 ラングミュアモデルまたは定常状態親和性を使用して、適宜Biacore T200評価ソフトウェアを用いてフィットさせた。
T7 mScript systems kit(CellScript)を使用して、IVT RNAを産生した。CD3/CD28ビーズ刺激T細胞を、先に記載のとおり(Zhao et al., 2010, Cancer Res, 70:9053)BTX EM830(Harvard Apparatus BTX)を使用してIVT RNAで電気穿孔した。簡潔にいうと、T細胞を3回洗浄し、OPTI−MEM(Invitrogen)に最終濃度1〜3×108細胞/mlで再懸濁させた。続いて、0.1mlの細胞を10μgのIVT RNA(または指示のとおり)と混合し、2mmキュベットで電気穿孔した。
抗体を次の業者から得た:抗ヒトCD3(BD Biosciences、555335)、抗ヒトCD8(BD Biosciences 555366)、抗ヒトCD107a(BD Biosciences 555801)、抗ヒトCD137(BD Biosciences 555956)。ErbB2の細胞表面発現をビオチン化抗ErbB2アフィボディ(Abcam、ab31890)により検出し、EGFRをFITC結合抗EGFRアフィボディ(Abcam、ab81872)により検出した。ErbB2、EGFRおよびCD19特異的CAR T細胞発現を、ErbB2−Fc融合タンパク質(R&D system、1129-ER)、EGFR−Fc融合タンパク質およびビオチン標識ポリクローナルヤギ抗マウスF(ab)2抗体(Jackson Immunoresearch、115-066-072)でそれぞれ検出し、4℃で25分インキュベートし、2回洗浄した(2%FBS含有PBS)。次いで、サンプルをPE結合抗ヒトIgG Fc Ab(eBioscience、12-4998-82)またはフィコエリスリン標識ストレプトアビジン(eBioscience、17-4317-82)で染色し、4℃で25分インキュベートし、1回洗浄した。フローサイトメトリー獲得をBD FACScaliburまたはAccuri C6 Cytometer(BD Biosciences)で行った。分析をFlowJoソフトウェア(Treestar)を使用して実施した。
標的細胞を洗浄し、1×106細胞/mlでR10培地(10%ウシ胎児血清を添加したRPMI 1640;Invitrogen)に懸濁した。100μlの各標的細胞型を96ウェル丸底プレート(Corning)にデュプリケートで添加した。エフェクターT細胞を洗浄し、1×106細胞/mlでR10培地に再懸濁し、100μlのT細胞を、記載するウェルにおいて標的細胞と合わせた。さらに、T細胞単独を含むウェルを調製した。プレートを37℃で18〜20時間インキュベートした。インキュベーション後、上清を回収し、ELISAアッセイ(eBioscience、88-7316-77; 88-7025-77)に付した。
細胞を、96ウェルプレート中、1:1のE:T(1×105エフェクター:1×105標的)で160μlの完全RPMI培地に播種した。20μlのフィコエリスリン標識抗CD107a Ab(BD Biosciences、555801)を添加し、プレートを37℃で1時間インキュベートし、Golgi Stop(3ml RPMI培地中2μlのGolgi Stop、20μl/ウェル;BD Biosciences、51−2092KZ)を添加し、さらに2.5時間インキュベートした。次いで5μlのFITC−抗CD8および5μlのAPC−抗CD3を添加し、37℃で30分インキュベートした。インキュベーション後、サンプルをFACS緩衝液で洗浄し、フローサイトメトリーで分析した。
休息しているCD4 T細胞を洗浄し、1×107細胞/mlの濃度でPBSに懸濁した。次いで120μlのCFSE作業溶液(25μM CFSE)を1×107細胞に3.5分、25℃で添加した。5%FBS(PBS中)で標識を停止させ、5%FBSで2回洗浄し、10IU/ml IL2含有R10で培養した。一夜培養後、CFSE標識T細胞を、多様な親和性ErbB2 CAR RNAで電気穿孔した。エレクトロポレーション2〜4時間後、T細胞を、1×106/mlの濃度でR10培地(10IU/ml IL2含有)に懸濁した。腫瘍またはK562細胞株を照射し、1×106/mLでR10培地に懸濁した。細胞を、48ウェルプレート中、1:1のE:T(5×105エフェクター:5×105標的)で1mlの完全RPMI培地に播種した。T細胞をついで計数し、3日目から2日毎に給餌した。CFSE希釈を、3日目、5日目および7日目にフローサイトメトリーでモニターした。
Nalm6−CBG腫瘍細胞を産生し、次のようなルシフェラーゼベースのCTLアッセイの修飾バージョンに用いた:クリックビートル緑色ルシフェラーゼ(CBG)をpELNSベクターにクローン化し、レンチウイルスに封入し、NALM6腫瘍細胞に形質導入し、CBG発現についてソートした。得られたNalm6−CBG細胞を洗浄し、1×105細胞/mlでR10培地に再懸濁し、100μlのCBG標識細胞を異なる比のT細胞(例えば30:1、15:1など)と、一夜、37℃でインキュベートした。100μlの混合物を、96ウェル白色ルミノメータープレートに移し、100μlの基質を添加し、発光を直ぐに決定した。
試験を、先に記載のものを少し修飾して実施した(Barrett et al., 2011, Human Gene Therapy, 22:1575; and Carpenito et al., 2009, PNAS, 106:336)。簡潔には、6〜10週齢NOD scid gamma(NSG)マウスに、1×106 PC3−CBG腫瘍細胞を、右側腹部に0日目に皮下注射し、同じマウスにSK−OV3−CBG腫瘍細胞(5×106細胞/マウス、s.c.)を左側腹部に5日目に与えた。マウスに、両腫瘍が約200mm3の体積となるように、PC3−CBG腫瘍接種23日目に尾静脈からT細胞で処置した。レンチウイルス形質導入T細胞を1×107細胞/マウス(10M)または3×106細胞/マウス(3M)で与えた。RNA電気穿孔T細胞を、最初の処置は5×107細胞/マウスで与え、続いて26日目、30日目および33日目の3処置を1×107 RNA電気穿孔T細胞/マウスの用量で与えた。
抗ErbB2 scFvの親和性低下は、インビトロでErbB2 CAR T細胞の治療指数を改善する
フローサイトメトリーで測定して広範なErbB2発現の一団の腫瘍株を収集した(図1)。SK−OV3(卵巣癌)、SK−BR3(乳癌)、BT−474(乳癌)はErbB2を過発現し、一方EM−Meso(中皮腫)、MCF7(乳癌)、293T(胚性腎臓293細胞)、A549(肺癌)、624mel(黒色腫)、PC3(前立腺癌)、MDA231(乳癌)はErbB2を低レベルで発現し、ErbB2はMDA468(乳癌)では検出されない。ErbB2 mRNAレベルも実時間PCRで測定し、二つの技術の間に強い相関があった(図2)。
上記結果に貢献するはずであるあらゆる腫瘍特異的効果を除外するために、一団のErbB2−BBζ CAR T細胞の活性を、多様なレベルのErbB2を発現する単一腫瘍株についてアッセイした(種々の量のErbB2 RNAで電気穿孔したK562細胞)。一致して、高親和性scFv(4D5および4D5−7)を発現するT細胞が、フローサイトメトリーにより検出可能なレベルの0.1μg mRNAより100倍低い0.001μgもの低用量のErbB2 RNAで電気穿孔したK562細胞を認識することが示された(図10、11Aおよび11B)。対照的に、低親和性scFv(4D5−5および4D5−3)を有するCARは、0.5μg(4D5−5;10)またはそれ以上の用量のErbB2 RNAで電気穿孔したK562しか認識せず、CAR T細胞感受性が、高親和性4D5 CAR T細胞と比較して2000倍(4D5−3)〜500倍(4D5−5)低下したことを示した。さらに、低親和性ErbB2 CAR(4D5−5および4D5−3;図11Aおよび11B)で観察された抗原用量関連反応性は、CFSEベースの増殖アッセイの実施により確認された(図12)。興味深いことに、CAR RNA用量の1/5への減少は(10μg RNA/100μl T細胞から2μg RNA/100μl T細胞)、サイトカイン分泌で評価してさらに低および高親和性CARを有するT細胞の抗原認識閾値を増加させ、T細胞表面上のCAR密度の微調整が、重要な変数であるか、または2μgのmRNAを超える量が全体的T細胞活性にいくぶん毒性を有し得ることを示唆する。
親4D5トラスツズマブ抗体からのscFvを取り込んだ高親和性ErbB2 CARの投与により生じる先の重篤な有害事象を考慮して(Morgan et al., 2010, Mol Therapy, 18:843)、親和性を低下させたErbB2 CAR T細胞の生理学的レベルのErbB2発現への反応性の可能性を評価することは最重要である。これに取り組むため、異なる臓器から単離した7種の初代細胞株を、ErbB2発現について試験した。初代株の大部分は検出可能なレベルの表面ErbB2を有し、神経前駆細胞株が、最高レベルのErbB2を発現した(図14)。高親和性4D5 CARを発現するT細胞は、CD107aレベルの上方制御により証明されるように、試験した全初代株に強く反応性であった(図15)。しかしながら、親和性を低下させたErbB2 CAR 4D5−5および4D5−3を発現するT細胞は、神経前駆細胞株への弱い反応性以外、初代株に反応性がなかった。これらの結果を、フローサイトメトリーにより低いまたは検出不可能なレベルのErbB2を有する細胞株の大きな一団の分析により確認した(図8および9)。
レンチウイルス形質導入により永続的にCARを発現するT細胞と、mRNA電気穿孔CAR T細胞の同等性を確立するために、一団の親和性調整CARを、レンチウイルス形質導入またはmRNAエレクトロポレーションのいずれかを使用して、同じ正常ドナーからのT細胞に発現させた(図16A)。T細胞を腫瘍細胞株(図16B)または種々の量のErbB2を発現するK562細胞(図16C)で刺激した。CAR T細胞認識および活性化を、CD107a上方制御(図17および18)、CD137上方制御(図19)およびIFN−γ分泌(図20および21)によりモニターした。先のErbB2 mRNA CAR T細胞結果に一致して、高親和性CARを構成的に発現するT細胞は、ErbB2を発現する全細胞株と強い反応性を示し、抗原発現レベルとT細胞活性の間に相関関係は見られなかった。対照的に、レンチウイルステクノロジーにより発現される低親和性CARを有するT細胞は、標的抗原発現と活性化の間に確固たる相関関係があった(図17、18、20および21)。これらの結果は、ErbB2抗原認識の感受性がmRNA電気穿孔およびレンチウイルス形質導入CAR T細胞の両者で使用したscFv親和性に依存的であることを確認する。
上記インビトロ結果を広げるために、一連の実験を、進行型血管新生化腫瘍異種移植したNSGマウスで実施した。図1における上記データに基づき、ヒト卵巣癌細胞株SK−OV3を、代表的ErbB2過発現腫瘍として選択し、ヒト前立腺癌株であるPC3を、正常組織ErbB2レベルのモデルのために選択した。NSGマウスにおける高親和性4D5 scFvまたは低親和性45D−5 scFvのいずれかを発現するErbB2 CAR T細胞の抗腫瘍有効性を、確立された側腹部SK−OV3腫瘍で18日目に比較した(図22)。連続的バイオルミネセンス造影は、高および低親和性CAR T細胞の両者が、腫瘍の迅速な除去をもたらすことを確認した。
scFvの親和性を微調整する戦略の広い適応性を試験するために、我々は、一団のEGFR CARを試験した。EGFR:BBζ CARを、親ヒト抗EGFR抗体C10由来のscFvから構築した(Heitner et al., 2001, J Immunol Methods, 248:17-30)。EGFR CARをコードする核酸配列を表4に提供する。
CAR T細胞の有効性が、一部腫瘍対正常組織における標的抗原の差次的発現により指示される。上記結果は、既知重篤標的上毒性を有するCARを、親和性調整により再操作でき、毒性を排除または減少しながら強力なインビボ有効性を維持することを示す。特に、トラスツズマブに基づく4D5 CARは、心肺組織において発現される生理学的レベルのErbB2の認識により(Press et al., 1990, Oncogene, 5:953)、致死的毒性を有した(Morgan et al., 2010, Mol Ther, 18:843)。CAR T細胞におけるscFvのKDの2〜3ログの減少により、治療指数における実質的な改善がErbB2およびEGFR CAR T細胞で示された。低親和性scFvを有するCAR T細胞は、高親和性CARと比較して同等に頑強な抗腫瘍活性をErbB2過発現腫瘍に対して示すが、生理学的レベルのErbB2に対してほとんど反応性を示さない。
加齢と最も明確に結びついている経路の一つは、mTOR経路である。mTOR阻害剤ラパマイシンがマウスで寿命を延長し、高齢マウスで多様な加齢関連状態を改善することが示されている(Harrison, DE et al. (2009) Nature 460:392-395; Wilkinson JE et al. (2012) Aging Cell 11:675-682;およびFlynn, JM et al. (2013) Aging Cell 12:851-862)。それゆえに、これらの知見は、mTOR阻害剤がヒトにおける加齢および加齢関連状態に有益な効果を有し得ることを示す。
ここに記載するとおり、ラパマイシン類似体であるmTOR阻害剤RAD001での6週間処置は、高齢ヒトボランティアにおけるインフルエンザワクチン接種に対する応答を改善した。
研究集団
不安定な基礎疾患のない65歳以上の高齢ボランティアが、ニュージーランドおよびオーストラリアの9箇所で参加した。スクリーニング時の除外基準はヘモグロビン<9.0g/dL、白血球数<3,500/mm3、好中球数<2,000/mm3または血小板数<125,000/mm3、未管理の糖尿病、不安定虚血性心疾患、臨床的に顕著な基礎肺疾患、免疫不全の病歴または免疫抑制性治療歴、凝固障害の病歴または長期抗凝固を必要とする 医学的状態、概算糸球体濾過速度<30ml/分、重度の未管理高コレステロール血症(>350mg/dL、9.1mmol/L)または高トリグリセリド血症(>500mg/dL、5.6mmol/L)の存在を含んだ。
*肥満度指数は、身長(m)の二乗で除した体重(kg)である
2011年12月から2012年4月まで、218名の高齢ボランティアが、無作為化、観察者盲検化、プラセボ対照試験に参加した。対象を、各処置アームにおけるRAD001対プラセボ比5:2で、検証された自動化無作為化系を使用して処置アームに無作為化した。処置アームは
RAD001 0.5mg連日またはプラセボ
RAD001 5mg毎週またはプラセボ
RAD001 20mg毎週またはプラセボ
であった。
有害事象評価ならびに血液学的および生化学的安全性評価のための採血は、来院日に実施した。有害事象情報は、対象が治験薬を受けている6週間の間、家で記載した日記からも集めた。全有害事象データを、インフォームド・コンセント時から最後の来院日から30日後まで集めた。事象は治験医により軽度、中程度または重度と分類された。
幾何平均力価比の一次解析を、無情報事前分布を用いる正規ベイズ回帰モデルを使用して行った。このモデルを、対数尺度で各抗体力価に適合させた。各モデルの主要評価項目は84日目測定値であった。63日目測定値をアウトカムベクターに包含させた。モデルを、先のステートメントと混ぜたSAS 9.2 procを使用してフィットさせた。マトリクス共分散構造は、非構造化(選択肢タイプ=UN)として考慮した。平らである事前分布を使用した。抗体陽転率の二次解析のために、ロジスティック回帰を使用した。
治療企図集団は、少なくとも1回の完全用量の治験薬を投与され、有効性データに影響する重大なプロトコール逸脱がない全対象と定義された。治験に参加した218名の対象中199名治験が治療企図集団であった。
末梢血単核細胞を、ベースライン、6週間の治験薬処置後および対象が6週間治験薬物から離れており、インフルエンザワクチン接種した4週間後である治験の最後の3時点に採血した全血から単離した。76PBMCサブセットを、先に記載されたように、Human Immune Monitoring Center at Stanford University, CA, USAで8色免疫表現型検査パネルを使用したフローサイトメトリーにより解析した(Maecker, HT et al. (2012) Nat Rev Immunol. 12:191-200)。76PBMCサブセットを、8色凍結乾燥免疫表現型検査パネルを使用するフローサイトメトリーにより解析した(BD Lyoplate、BD Biosciences, San Diego, CA)。生存能>80%および2×106細胞を超える収量のPBMCサンプルを解析に入れた。
一般に、RAD001は、特に0.5mg連日および5mg毎週投与レジメンで良好な耐容性であった。治験中に死亡例はなかった。3名の対象は、RAD001と無関係であると評価された4つの重篤な有害事象(SAE)を経験した。4つのSAEは、先に6週間5mg毎週RAD001の6週のコースを完了していた正常血小板数の対象の左眼の網膜出血とその後の失明;プラセボ処置対象における重度の背部痛およびプラセボ処置対象における重度の胃腸炎であった。あらゆる処置群における発生率>2%の処置関連有害事象(AE)の一覧を表5に提供する。最も一般的なRAD001関連AEは、大部分の症例で、軽度であった口腔内潰瘍であった。全体的に、RAD001を投与された対象と、プラセボ処置対照の重度のAE発生率は類似した。1つの重度のAEのみがRAD001と関連すると評価され、20mg毎週RAD001処置対象の口腔内潰瘍であった。
4週目はインフルエンザワクチン接種4週間後を示す
Nはコホートあたりの対象数である
GMTは幾何平均力価である
GMT比は、ワクチン接種4週目のGMT/ベースラインのGMTである
CV%は、変動係数を示す
サブグループ解析において、低ベースラインインフルエンザ力価(≦1:40)のサブセットの対象は、ITT集団より高い力価のRAD001関連増加を経験した(図41B)。これらのデータは、RAD001が、ベースラインで予防的(>1:40)力価を有さず、それゆえにインフルエンザ疾病の最高のリスクにある対象のインフルエンザワクチン応答の増強に特に有効であることを示す。
* RAD001とプラセボの間の抗体陽転のオッズ比は、有意に1より異なる(固定効果として処理したロジスティック回帰により得た両側p値<0.05)
* RAD001とプラセボの間の抗体陽転のオッズ比は、有意に1より異なる(固定効果として処理したロジスティック回帰により得た両側p値<0.05)
結論として、ここに提供したデータは、mTOR阻害剤RAD001が、インフルエンザワクチン接種に対する応答で評価して、ヒト高齢者の免疫学的機能における年齢関連減少を軽減し、この改善が許容されるリスク対効果バランスで得られることを示す。高齢マウスでの試験において、mTOR阻害剤ラパマイシンでの6週間処置はインフルエンザワクチン接種に対する応答を増加しただけでなく、寿命も延長し、免疫老化の改善は、加齢関連表現型に対するより広範な効果のマーカーであり得ることを示唆する。
免疫機能は高齢者で低下し、感染発生率増加およびワクチン接種に対する応答減少に至る。mTOR阻害がヒトにおいて抗加齢効果を有するかの決定の第一段階として、無作為化プラセボ対照治験を実施し、mTOR阻害剤RAD001が、高齢ボランティアにおけるワクチン接種に対する応答により評価された免疫機能の加齢関連低下を反転させるかを決定した。全例で、適切な患者同意を得て、治験は国の保健機関により承認された。
20mg毎週(トラフレベル:0.7ng/ml)
5mg毎週(トラフレベルは検出限界以下であった)
0.5mg連日(トラフレベル:0.9ng/ml)
前臨床モデルにおいて、ラパログであるラパマイシンでのmTOR阻害は、老齢マウスにおける自発的身体活動性を増加させる(Wilkinson et al. Rapamycin slows aging in mice. (2012) Aging Cell; 11:675-82)。興味深いことに、実施例2に記載した0.5mg連日投薬コホートの対象も、投与1年後のアンケートで、プラセボと比較してエネルギーおよび運動能の増加を報告した(図39)。これらのデータは、ラパログでの部分的mTOR阻害が、単なる免疫機能を超えて加齢関連罹病率に対する効果を有し得ることを示唆する。
モデリングおよびシミュレーションを実施し、mTOR活性を部分的に阻害すると予測されるRAD001の連日および毎週用量範囲を予測した。上記のとおり、P70 S6KはmTORによりリン酸化され、P70 S6Kのノックアウトが寿命を増加するため加齢とより密接に結びついたmTORの下流標的である。それゆえにモデリングを、P70 S6K活性を一部阻害するRAD001の用量で行った。≧0.1mgおよび<20mgの範囲の毎週投薬がP70 S6K活性の部分的阻害を達成すると予測される(図48)。
P70 S6Kを部分的にしか阻害しない用量のmTOR阻害剤を用いる、加齢関連罹患を処置するまたは一般に免疫応答を増強する方法。加齢適応症における低用量のRAD001での部分的mTOR阻害の有効性は予想外の発見である。≧0.1mg〜<20mg毎週および≧0.005mg〜<1.5mg連日のRAD001用量範囲が部分的mTOR阻害を達成し、それゆえに加齢関連罹患または免疫応答の増強に有効性を有することが期待される。
CART治療についての戦略は、腫瘍細胞上の標的細胞表面抗原の優先的発現または標的を発現する正常細胞のアブレーションが臨床的に耐容性であるときによる。B細胞ALLにおいて、CART治療によるCD19へのターゲティングは、臨床的に有効であり、実行可能であることが示されている。しかしながら、B細胞ALLを有する一部患者は、CD19を発現しないかもしくは低くまたはCAR19治療後CD19陰性疾患で再発する。さらに、T細胞ALLおよびAMLは、CART19細胞でのターゲティングに感受性ではない。それゆえに、種々の癌における標的表面抗原の欠如が、CARベースの手法の開発を妨害している。この実施例において、急性白血病(AL)、例えば、ALLおよびAMLにおける標的抗原発見の戦略を記載する。
(i)ALにおける発現既知(陽性対照として)、例えば、B細胞ALLに対するCD19またはAMLに対するCD−34;
(ii)ALが造血細胞の悪性腫瘍であると仮定して、造血中の発現(例えば、EMR2);
(iii)発現が標的細胞の表面上にある可能性が高い;
(iv)これらの抗原を担持する正常組織または細胞のアブレーションが臨床的に耐容性であると予測されるとき。
1. AMLおよびALL>NBMおよびA357
2. AMLまたはALL>NBMおよびA357
3. AMLおよびALL>A357、しかしNBMではない
4. AMLまたはALL>A357、しかしNBMではない
サイトカインは、T細胞増殖、分化、生存およびホメオスタシスに関連する重要な機能を有する。臨床使用のための最も重要なサイトカインファミリーの一つは、共通γ鎖(γc)ファミリーサイトカインであり、これはインターロイキン(IL)−2、IL−4、IL−7、IL−9、IL−15およびIL−21を含む(Liao et al., 2013, Immunity, 38:13-25)。IL−2は、癌の免疫治療剤として広範に使用されている。IL−2のサプリメントは、臨床試験で抗CD19 CAR−T細胞の抗腫瘍能を増強している(Xu et al., 2013, Lymphoma, 54:255-60)。しかしながら、IL−2の投与は、副作用および制御性T細胞の増殖の傾向および活性化誘発細胞死(AICD)の効果のために制限される(Malek et al., 2010, Immunity, 33:153-65;およびLenardo et al., 1999, Annu Rev Immunol, 17:221-53)。IL−7、IL−15およびIL−21は、各々養子性免疫治療の有効性を増強でき、IL−2と比較して低毒性であると考えられる(Alves et al., 2007, Immunol Lett, 108:113-20)。上記サイトカインの徹底的な前臨床および臨床試験にも関わらず、CAR−T細胞養子治療に対する多様な外来性γcサイトカインの役割の多パラメーター比較試験は欠如している。
CAR構築およびレンチウイルス製剤
pELNS−C4−27z CARベクターを先に記載のように構築し(レビュー中の原稿)、簡潔には、抗FRα C4/AFRA4 scFvを含むpHEN2プラスミドを、5'-ataggatcccagctggtggagtctgggggaggc-3'(配列番号52)および5'-atagctagcacctaggacggtcagcttggtccc-3'(配列番号53)(BamHIおよびNheIに下線を付した)のプライマーを使用するC4フラグメントのPCR増幅のための鋳型として使用した。PCR産物および第三世代自己不活性化レンチウイルス発現ベクターpELNSをBamHIおよびNheIで消化した。消化PCR産物を、次いで、導入遺伝子発現が伸長因子−1α(EF−1α)プロモーターにより駆動される、CD27−CD3z T細胞シグナル伝達ドメインを含むpELNSベクターに導入した。
末梢血リンパ球を、インフォームド・コンセント後、University Institutional Review Board at the University of Pennsylvaniaにより承認されたプロトコール下、健常ドナーから得た。初代T細胞を、陰性選択により精製後Human Immunology Coreから得た。T細胞を完全培地(10%FBS、100U/mLペニシリン、100μg/mL硫酸ストレプトマイシンを添加したRPMI 1640)で培養し、指示に従い、抗CD3および抗CD28 mAbs被覆ビーズ(Invitrogen)で、1:1比で刺激した。活性化24時間後、細胞を、5のMOIでレンチウイルスで形質導入した。記載するサイトカインを、翌日から最終濃度10ng/mLまで形質導入T細胞に添加した。サイトカインを3日毎に交換した。
フローサイトメトリーを、BD FACSCantoで実施した。抗ヒトCD45(HI30)、CD3(HIT3a)、CD8(HIT8a)、CD45RA(HI100)、CD62L(DREG−56)、CCR7(G043H7)、IL−7Rα(A019D5)、CD27(M−T271)、CD28(CD28.2)、CD95(DX2)、TNF−α(MAb11)、IFN−γ(4S.B3)、IL−2(MQ1−17H12)、パーフォリン(B−D48)、グランザイム−B(GB11)をBioLegendから得た。ビオチン−SP結合ウサギ抗ヒトIgG(H+L)をJackson Immunoresearchから購入し、APC結合ストレプトアビジンをBiolegandから購入した。抗ヒトBcl−xl(7B2.5)をSouhernBiotechから購入した。アポトーシスキットおよびTruCountチューブをBD Bioscienceから得た。末梢血T細胞数について、血液を後眼窩放血から得て、ヒトCD45、CD3、CD4およびCD8 T細胞の存在について染色した。ヒトCD45+−ゲート開閉、CD3+、CD4+およびCD8+サブセットを、製造業者の指示に従いTruCountチューブで定量化した。
雌非肥満糖尿病性/重症複合免疫不全/γ−鎖−/−(NSG)マウス8〜12週齢を、Stem Cell and Xenograft Core of the Abramson Cancer Center, University of Pennsylvaniaから得た。マウスに、0日目に3×106 fLuc+ SKOV3細胞を側腹部に皮下注射した。処置前に、群あたり4匹または5匹のマウスに無作為化した。触知可能となった後、ヒト初代T細胞を、先に記載のように活性化および形質導入した。T細胞を、IL−2(5ng/mL)の存在下約2週間増殖させた。腫瘍負荷が約250〜300mm3のとき、マウスに5×106 CAR−T細胞または100μl 食塩水を静脈内注射し、その後5μgのIL−2、IL−7、IL−15、IL−18、IL−21またはリン酸緩衝液(PBS)を連日7日間腹腔内注射した。腫瘍寸法をカリパスで測定し、腫瘍体積を次の式を用いて計算した:腫瘍体積=(長さ×幅2)/2。レシピエントマウス血液における移入T細胞の数および表現型を、後眼窩放血後にフローサイトメトリーにより決定した。腫瘍体積が2000mm3を超えたときマウスを安楽死させ、さらなる分析のために直ぐに腫瘍を摘除した。
統計解析をPrism 5(GraphPadソフトウェア)およびIBM SPSS Statistics 20.0ソフトウェアで行った。データは、明記しない限り、平均±SEMとして示した。対応サンプルt検定またはノンパラメトリックウィルコクソン順位検定を2群の比較に使用し、反復測定ANOVAまたはフリードマン検定を3以上の群の差の統計的有意の検定に使用した。0.05未満のP値のときに、実験値を統計学的有意とした。
抗FRα C4 CARの構築および発現
pELNS−C4−27z CARは、CD8αヒンジおよび膜貫通領域に連結した抗FRα C4 scFv、その後ろにCD27細胞内シグナル伝達モチーフと直列であるCD3ζシグナル伝達部分からなった(図59A)。初代ヒトT細胞を、C4 CARレンチウイルスベクターで効率的に形質導入し、形質導入48時間後に検出したとき、43%〜65%の形質導入効率であった(図59B)。CAR発現レベルは、CD4+およびCD8+ T細胞で同等であった(52.6±10.2%対49.5±17.1%、P=0.713)。
本試験は、CAR−T細胞注射と組み合わせたインビボサイトカイン投与がCAR−T細胞の抗腫瘍活性を増強できるかを試験した。皮下SKOV3腫瘍を担持するマウスは、食塩水または5×106 C4−27z CAR−T細胞を、39日に静脈内注射された(図60)。食塩水群と比較して、CAR−T細胞治療を受けたマウスは短期腫瘍退縮をし、腫瘍は56日目から反跳し始めた(図61)。多様なサイトカイン群の中で、IL−15およびIL−21注射を受けたマウスが最良の腫瘍抑制を示し、続いてIL−2およびIL−7であり、IL−18およびPBS処置マウスは、最も重い腫瘍負荷を有した。末梢血における移入T細胞の持続を、養子移入15日後および終了時に決定した。CD4+およびCD8+ T細胞の最高数が、IL−15、続いてIL−21で処置したマウスで見られた。+15日目CD4+およびCD8+ T細胞数は腫瘍退縮と一致し、最終腫瘍重量を予測した。マウスを腫瘍攻撃73日後に殺し、ヒトT細胞存在下の腫瘍を分析した(図63)。末梢血と同様、IL−15で処置したマウスが腫瘍における最高T細胞数を示し、続いてIL−21、IL−2、IL−7、PBSおよびIL−18であった(図64)。CD4対CD8比は種々のサイトカイン群で同等であり、全サイトカイン群で血液および腫瘍の両者でCD4+ T細胞が優性であった。CD8+ T細胞におけるCAR発現は上記群と同等であったが(45.1%〜62.4%)、IL−15およびIL−21群は、IL−2およびIL−18群よりCAR+ CD4 T細胞の比率が高かった(図65)。表現型に関して、腫瘍における全CAR−T細胞がCD62L−およびCCR7−であり、それらの35%〜60%がCD45RA+(Temra)を発現した(データは示していない)。CD8+ T細胞は、CD27発現を保持する可能性が高いが、CD28発現はCD4+とCD8+ T細胞で同等であった(図66)。
要約すると、これらの知見は、CAR−T細胞養子治療の有効性を増強するための外来性サイトカイン投与のための重要な含意を有する。全γcサイトカインは、CAR−T細胞注射と組み合わせて投与したとき、卵巣マウスモデルにおける抗腫瘍有効性を増強した。IL−15およびIL−21がインビボ追加のための最良のサイトカインであり、IL−7およびIL−2は、抗腫瘍結果の改善を示した。IL−18は、IL−1ファミリーに属する炎症誘発性サイトカインであり、これらのインビボ実験で抗腫瘍有効性に対する効果を示さなかった。
先の試験、例えば、実施例6に記載する実験は、CAR T細胞がインビボ(例えば、腫瘍微小環境)で経時的に有効性を失うことを示唆している。具体的に、腫瘍マウスモデルに注射したmesoCAR T細胞を、T細胞注入後腫瘍から単離し(例えば、39日後、以後、腫瘍浸潤性リンパ球、TILと称す)、新たに解凍したmesoCAR T細胞との比較において機能的活性を評価した。結果は、エクスビボ死滅アッセイおよびIFNγ放出アッセイにおいて、腫瘍から単離したmesoCAR T細胞は、抗原またはCD3/CD28刺激に対する応答において、腫瘍細胞を殺す能力が低下し(図67A)、IFNg産生が減少し(図67B)、ERKシグナル伝達が減少し(ウェスタンブロット分析におけるリン酸化により示す、図67C)、T細胞活性化の減少を示した。
CART細胞有効性に対するDGK阻害の効果を試験するために、DGK遺伝子DGKα、DGKζまたは両者が欠失したトランスジェニックマウスを使用した。野生型およびDGK欠損マウスからの脾臓T細胞を単離し、レトロウイルスを使用してmesoCAR(SS1 BBZ)を発現するように形質導入した。MIGR1 CARを対照として使用した。
次に、mesoCAR T細胞の治療有効性を、DGK阻害または欠損の状況下で試験した。AE17meso腫瘍細胞(中皮腫細胞)をC57BL/6マウスの皮下に注射した。腫瘍が100mm3に達したとき(約1週間後)、1000万mesoCAR T細胞を尾静脈に静脈内注射した。腫瘍体積を、少なくとも18日間追跡した。
CAR T細胞治療における一つの主要な障壁は、ジアシルグリセロールキナーゼ(DGK)のようなT細胞シグナル伝達の内因性の負のレギュレーターの上方制御である。実施例11に記載のように、CAR T細胞は、インビボで経時的に有効性を喪失することが示されている。DGKのようなT細胞機能の負のレギュレーターの阻害は、CAR発現T細胞の活性および機能を増強することが示された。
T細胞機能の他の重要な負のレギュレーターは転写因子イカロスである。近位TCRシグナル伝達に主に作用するDGKと異なり、イカロスは、Sin3A、CtBPおよびHDACのような染色質リモデリング複合体の動員を介して遺伝子発現を負に制御する亜鉛フィンガーDNA結合タンパク質である。イカロスは、CD4+ T細胞およびCD8+ T細胞においてサイトカイン産生および細胞溶解性機能に役割を有する。
この実施例において、イカロス発現が減少したレトロウイルス形質導入CAR T細胞の抗腫瘍有効性をインビトロおよびインビボで試験した。
細胞株。マウスAE17中皮腫細胞は、Jackman et al., J Immunol. 2003;171:5051-63に記載された。ヒトメソテリンをレンチウイルス形質導入によりAE17細胞に導入した。3T3Balb/C細胞をAmerican Type Culture Collectionから購入した。マウスFAP発現3T3BALB/C(3T3.FAP)細胞を、マウスFAPのFAP−3T3親株へのレンチウイルス形質導入により創製した。
イカロスのレベルが低下したCAR発現T細胞におけるサイトカイン産生および細胞溶解性メディエーター放出は増強される
イカロスが減少した細胞溶解性Tリンパ球(CTL)がインビトロおよびインビボでエフェクター機能が増強していることを考慮して(O'Brien, et al., J Immunol. 2014; 192:5118-29)、イカロスの枯渇がCAR T治療の有効性を改善するかを試験するために実験を実施した。C57BL/6背景の野生型C57BL/6およびイカロスハプロ欠損マウス(Ikzf1±)から単離したT細胞を、抗メソテリンCARを発現するようにレトロウイルスで形質導入した。エクスビボ活性化、形質導入およびIL2中の増殖後、野生型(WT)CAR T細胞と比較して、Ikzf1±CAR T細胞は、フローサイトメトリーおよびウェスタンブロットで少ないイカロスタンパク質を発現し続けることが確認された(図73A)。
イカロスレベルが低い細胞に加えて、マウスからのイカロスの一つのドミナントネガティブアレル(IkDN)を発現するT細胞を試験した。IkDNを発現するトランスジェニックマウスのリンパ系発達は正常であるが、イカロス DNA結合活性が90%減少した末梢T細胞を有する(Thomas et al., J Immunol. 2007; 179:7305-15;およびWinandy et al., Cell. 1995; 83:289-99)。WTおよびIkDNマウスの脾臓から単離したT細胞を、プレート結合抗CD3/CD28抗体で活性化し、メソテリンCARを形質導入し、続いてIL−2で増殖させた。IkDN CAR T細胞におけるイカロスのノックダウンがウェスタンで確認された。WTおよびIkDN CAR T細胞を、BSAまたはメソテリン被覆ビーズのいずれかで2:1 ビーズ:T細胞比で6時間再負荷し、CAR抗原に対してIFNおよびIL−2を産生する能力ならびに脱顆粒する能力を解析した。上記Ikzf1±データに類似して、ベースラインでいくぶんかの自己分泌IFN−γ産生が観察されたが(図74A)、IL−2は観察されなかった(BSA刺激;図74B)。CARとその標的抗原であるメソテリンとのライゲーションにより、IkDN T細胞は、WT T細胞より多くのIFN−γを産生した(メソテリン刺激;図74A)。CD107a上方制御で測定した脱顆粒も、野生型およびIkDN CAR T細胞両者で同様であった(図74D)。
イカロスの枯渇がCAR T細胞のサイトカイン放出を増強し、グランザイムBレベルおよびCD107a発現を増加させることを考慮して、可能性がある機構を探索した。エフェクター機能のこれらの変化が、野生型とIkzf1±形質導入T細胞の活性化の差異によるものである可能性は妥当である。それゆえに、CD69、CD25および4−1BB(T細胞活性化のマーカー)のレベルを、メソテリン被覆ビーズで6時間および24時間刺激後、フローサイトメトリーで測定した。早期活性化マーカーであるCD69は、野生型およびIkzf1±細胞の両者で同程度に上方制御された(図75A)。長い刺激で、野生型およびIkzf1±CAR発現T細胞は同様のレベルのCD69を発現し続けたが、Ikzf1±形質導入T細胞はCD25発現の増加を示した(図75B)。これは、T細胞活性化の差異を直接的には示さないが、しかしながら、Ikzf1±細胞によるIL−2が増加するに連れて(図75D)、IL−2RaであるCD25のポジティブ・フィードフォワード・ループで作用し得る(Depper et al., Proc Natl Acad of Sci USA. 1985; 82:4230-4;およびNakajima et al., Immunity. 1997; 7:691-701)。TNF受容体スーパーファミリーのメンバーである4−1BBも、活性化T細胞で発現され(Vinay et al., Seminars in Immunology. 1998; 10:481-9)、抗原刺激後CAR形質導入野生型およびIkzf1±T細胞で類似のレベルで発現された(図75C)。それゆえに、我々のWTおよびIkzf1±形質導入T細胞の間の機能的差異は、T細胞活性化の差異によるものではなかった。
イカロスが減少したCAR T細胞によるエフェクター因子産生の増加を考慮して、インビトロでのその標的腫瘍細胞に対する有効性を試験した。野生型、Ikzf1±およびIkDN T細胞発現メソCARを、異なる比で親腫瘍細胞株、AE17またはメソテリン発現細胞株、AE17メソと混合した。親細胞株と混合したとき、野生型、Ikzf1±およびIkDN T細胞のいずれも、AE17に応答したIFN−γ産生または細胞溶解ができなかった(図76A、76Bおよび76C)。対照的に、AE17メソと反応させたとき、野生型T細胞によるIFN−γ産生および細胞溶解は、E:T比が増加するに連れて増加した(図76Bおよび76C)。しかしながら、Ikzf1±およびIkDN T細胞の両者は、最低E:T比1.3:1でも、野生型T細胞より有意に多いIFN−γを産生し、腫瘍溶解が有意に増加した(図76Bおよび76C)。
マウスにおける確立されたAE17メソ腫瘍の増殖制御における、イカロスが減少したメソテリン特異的T細胞(Ikzf1±およびIkDN)の能力を試験した。200万のAE17メソ腫瘍細胞を、同質遺伝子的C57BL/6マウスの側腹部に注射し、大きな確立された腫瘍(約100〜150mm3)を形成させた。WTまたはイカロス欠損(Ikzf1±およびIkDN)マウスから調製した1000万のCAR T細胞を、次いで、この腫瘍担持マウスに養子移入し、腫瘍測定を続けた。野生型形質導入メソCAR T細胞により軽度の腫瘍増殖阻害が誘発されたが、Ikzf1±およびIkDN形質導入メソCAR T細胞の両者は、より顕著にAE17メソ腫瘍の増殖を阻止した(図77Aおよび77B)。
イカロス阻害CAR T細胞の有効性の増強を考慮して、Ikzf1±メソCAR T細胞を使用する可能性がある機構を探索した。これらのメソCAR T細胞がインビボで免疫抑制性腫瘍微小環境中どのように作動するかをさらに試験するために、養子移入3日後および9日後の腫瘍を収集し、これらの数および機能性を評価した。これらの二つの時点は、抗腫瘍免疫応答中の早期および後期時点でのそれらの活性の特徴づけを可能にした。
免疫抑制性腫瘍微小環境とメソCAR T細胞の相互作用をさらに特徴付けるために、インビトロ培養系を利用した。IDO、IL−10、アデノシンおよびTGF−βのような可溶性阻害因子(Wang et al., Oncoimmunology. 2013; 2:e26492)は、浸潤性腫瘍リンパ球の阻止に貢献することが示されている。イカロス欠損CAR T細胞における選択した阻害因子のインビトロの効果を試験して、免疫抑制性環境がその溶解性機能に影響するか否かを決定した。野生型CAR T細胞は、TGF−βおよびアデノシン存在下、IFN−γを製造する能力が50%減少し、溶解性機能が減少した(図79)。イカロスのレベルが減少したCAR T細胞(Ikzf1±およびIkDN)は、阻害剤非存在下で野生型カウンターパートより多くのIFN−γを製造し続け、TGF−βおよびアデノシン存在下で、わずかしか阻害されなかった(図79A)。野生型T細胞と比較したIkzf1±およびIkDN CAR T細胞の溶解性機能増加が観察された(図79B)。これらのデータは、イカロスが減少したT細胞は、TGF−βおよびアデノシン阻害に低感受性であることが示される。
本実施例において、腫瘍環境においてCAR発現T細胞を生存させ、そのエフェクター機能を増加させる新たな試みが同定された。T細胞機能に関与する多種多様な遺伝子、例えば、サイトカイン遺伝子(IL−2およびIFN−γ)、細胞溶解性メディエーター(グランザイムB)およびT細胞分化に影響する重要なT−box転写因子(R−BetおよびEomes)を阻害することが知られる、転写リプレッサーイカロスの不活性化である。
CAR T細胞の養子移入後、一部の患者は、最適以下のレベルの抗腫瘍活性をもたらし得る、CAR T細胞の限定的持続を経験する。この実施例において、外来性ヒトIL−7投与の効果を、CAR T細胞に対する初期最適以下応答が観察されているマウス異種移植モデルで評価する。
IL−7受容体CD127の発現を、まず異なる癌細胞株およびCAR発現細胞で評価した。2マントル細胞リンパ腫細胞株(RLおよびJeko−1)および1B−ALL細胞株(Nalm−6)を、CD127発現についてフローサイトメトリーで分析した。図80Aに示すように、試験した3癌細胞株試験中、RLはCD127の最高発現を有し、続いてJeko−1およびNalm−6であることが示された。CART19細胞をNSGマウスに注入し、CD127発現を、フローサイトメトリーにより循環CART19細胞において評価した。図80Bに示すように、CD127は全循環CART19細胞で均一に発現される。
IL−7受容体(CD127)発現を、フローサイトメトリーにより白血病細胞株(AML細胞株MOLM14およびB−ALL細胞株NALM6)および初代AML細胞で測定した(図83、上部パネル)。IL−7受容体発現は、B−ALL細胞株NALM6細胞で現れたが、AML細胞株MOLM14または初代AMLではなかった。フローサイトメトリー分析は、IL−7受容体発現が腫瘍細胞のみで検出されるようにゲート開閉した。
ここに引用する各および全ての特許、特許出願および刊行物の開示は、その全体を引用により本明細書に包含させる。本発明は、具体的面を参照して開示しているが、本発明の他の面および異形が、本発明の真の精神および範囲から逸脱することなく他の当業者により考案され得ることは明らかである。下記特許請求の範囲は、全てのこのような面および等価な異形を含むと解釈されることを意図する。
Claims (16)
- 腫瘍抗原の発現と関係する疾患を有する対象の処置においてサイトカインと組み合わせて投与される、キメラ抗原受容体(CAR)分子を包含する免疫エフェクター細胞(CAR発現細胞)を含む組成物であって、ここで、
CAR分子が、抗原結合ドメイン、膜貫通ドメインおよび細胞内ドメインを含み、
サイトカインが、インターロイキン−15(IL−15)、IL−21、IL−15およびIL−21の組合せ、またはIL−15ポリペプチドおよびIL−15受容体アルファ(IL−15Ra)ポリペプチドの組合せから選択される単離ポリペプチドを含み、
CAR発現細胞およびサイトカインが別個の組成物として逐次的に投与される、
組成物。 - IL−15ポリペプチドおよびIL−15Raポリペプチドの組合せが、hetIL−15を含む、請求項1に記載の組成物。
- 腫瘍抗原の発現と関係する疾患が腫瘍抗原の発現と関係する増殖性疾患、前癌状態、癌および非癌関連徴候からなる群より選択される、請求項1または2に記載の組成物。
- 癌が慢性リンパ性白血病(CLL)、急性白血病、急性リンパ性白血病(ALL)、B細胞性急性リンパ性白血病(B−ALL)、T細胞急性リンパ性白血病(T−ALL)、慢性骨髄性白血病(CML)、B細胞前リンパ球性白血病、芽球形質細胞様樹状細胞腫瘍、バーキットリンパ腫、汎発性大B細胞リンパ腫、濾胞性リンパ腫、ヘアリー細胞白血病、小細胞または大細胞濾胞性リンパ腫、悪性リンパ増殖性状態、MALTリンパ腫、マントル細胞リンパ腫、辺縁帯リンパ腫、多発性骨髄腫、骨髄異形成および骨髄異形成症候群、非ホジキンリンパ腫、ホジキンリンパ腫、形質芽球性リンパ腫、形質細胞様樹状細胞性腫瘍、ワルデンストレーム高ガンマグロブリン血症および前白血病状態の1以上から選択される血液の癌である、請求項3に記載の組成物。
- CAR発現細胞がまず投与され、サイトカインが次に投与される、請求項1に記載の組成物。
- サイトカインが、CAR発現細胞の投与の1日、2日、3日、4日、5日、6日または7日後に投与される、請求項5に記載の組成物。
- サイトカインが、CAR発現細胞の投与の少なくとも2週間、3週間、4週間、5週間、6週間、8週間、10週間後またはそれ以上後に投与される、請求項5に記載の組成物。
- サイトカインがまず投与され、CAR発現細胞が次に投与される、請求項1に記載の組成物。
- 対象がさらに、
(a)タンパク質ホスファターゼ阻害剤;
(b)キナーゼ阻害剤;
(c)免疫阻害分子の阻害剤;または
(d)TREG細胞のレベルもしくは活性を低下させる薬剤
から選択されるさらなる薬剤を投与される、請求項1から8のいずれか一項に記載の組成物。 - (i)タンパク質ホスファターゼ阻害剤が、SHP−1阻害剤および/またはSHP−2阻害剤であり;
(ii)キナーゼ阻害剤が、CDK4阻害剤、CDK4/6阻害剤、mTOR阻害剤、MNK阻害剤またはデュアルP13K/mTOR阻害剤の1以上から選択され;
(iii)免疫阻害分子を阻害する薬剤が、阻害分子の発現を阻害する抗体または抗体フラグメント、阻害性核酸、群生性等間隔短回文反復配列(CRISPR)、転写アクティベーター様エフェクターヌクレアーゼ(TALEN)または亜鉛フィンガーエンドヌクレアーゼ(ZFN)を含み;および/あるいは
(iv)TREG細胞のレベルまたは活性を低下させる薬剤が、シクロホスファミド、抗GITR抗体、CD25枯渇またはこれらの組み合わせから選択される、
請求項9に記載の組成物。 - 抗原結合ドメインが疾患と関係する腫瘍抗原と結合し、該腫瘍抗原が、CD19、TSHR、CD123、CD22、CD30、CD171、CS−1、C型レクチン様分子−1(CLL−1)、CD33、EGFRvIII、GD2、GD3、BCMA、Tn抗原(Tn Ag)、PSMA、ROR1、Fms様チロシンキナーゼ3(FLT3)、FAP、TAG72、CD38、CD44v6、CEA、EPCAM、B7H3(CD276)、KIT(CD117)、インターロイキン−13受容体サブユニットアルファ−2(IL−13Ra2)、メソテリン、インターロイキン11受容体アルファ(IL−11Ra)、前立腺幹細胞抗原(PSCA)、プロテアーゼセリン21(PRSS21)、血管内皮細胞増殖因子受容体2(VEGFR2)、ルイス Y 抗原、CD24、血小板由来増殖因子受容体ベータ(PDGFR−ベータ)、ステージ特異的胚性抗原−4(SSEA−4)、CD20、葉酸受容体アルファ、ERBB2(Her2/neu)、ムチン1細胞表面関連(MUC1)、上皮細胞増殖因子受容体(EGFR)、神経細胞接着分子(NCAM)、プロスターゼ、PAP、ELF2M、エフリンB2、IGF−I受容体、炭酸脱水酵素IX(CAIX)、LMP2、gp100、bcr−abl、チロシナーゼ、エフリンA型受容体2(EphA2)、フコシルGM1、シアリルルイス接着分子(sLe)、ガングリオシドGM3、TGS5、高分子量黒色腫関連抗原(HMWMAA)、o−アセチル−GD2ガングリオシド(OAcGD2)、葉酸受容体ベータ、腫瘍内皮マーカー1(TEM1/CD248)、腫瘍内皮マーカー7関連(TEM7R)、クローディン6(CLDN6)、Gタンパク質共役受容体クラスCグループ5メンバーD(GPRC5D)、染色体Xオープンリーディングフレーム61(CXORF61)、CD97、CD179a、未分化リンパ腫キナーゼ(ALK)、ポリシアル酸、胎盤特異的1(PLAC1)、GloboH、乳腺分化抗原(NY−BR−1)、ウロプラキン2(UPK2)、A型肝炎ウイルス細胞性受容体1(HAVCR1)、アドレナリン受容体ベータ3(ADRB3)、パンネキシン3(PANX3)、Gタンパク質共役受容体20(GPR20)、リンパ球抗原6複合体座位K9(LY6K)、嗅覚受容体51E2(OR51E2)、TCRガンマ選択的リーディングフレームタンパク質(TARP)、ウィルムス腫瘍タンパク質(WT1)、NY−ESO−1、LAGE−1a、MAGE−A1、レグマイン、HPV E6,E7、MAGE A1、染色体12pに位置するETS転座変異体遺伝子6(ETV6−AML)、精子タンパク質17(SPA17)、X抗原ファミリーメンバー1A(XAGE1)、アンギオポエチン結合細胞表面受容体2(Tie 2)、黒色腫癌精巣抗原−1(MAD−CT−1)、MAD−CT−2、Fos関連抗原1、p53、p53変異体、プロステイン、サバイビンおよびテロメラーゼ、PCTA−1/ガレクチン8、メランA/MART1、Ras変異体、ヒトテロメラーゼ逆転写酵素(hTERT)、肉腫転座切断点、アポトーシスの黒色腫阻害剤(ML−IAP)、ERG(膜貫通型プロテアーゼ、セリン2(TMPRSS2)ETS融合遺伝子)、N−アセチルグルコサミニル−トランスフェラーゼV(NA17)、ペアードボックスタンパク質Pax−3(PAX3)、アンドロゲン受容体、サイクリンB1、v−mycトリ骨髄細胞腫ウイルス癌遺伝子神経芽腫由来ホモログ(MYCN)、RasホモログファミリーメンバーC(RhoC)、TRP−2、チトクロムP450 1B1(CYP1B1)、BORIS、T細胞3により認識される扁平上皮細胞癌抗原(SART3)、PAX−5、プロアクロシン結合タンパク質p32(OY−TES1)、リンパ球特異的タンパク質チロシンキナーゼ(LCK)、Aキナーゼアンカータンパク質4(AKAP−4)、SSX2、RAGE−1、ヒトテロメラーゼ逆転写酵素、RU1、RU2、腸管カルボキシルエステラーゼ、mut hsp70−2、CD79a、CD79b、CD72、白血球関連免疫グロブリン様受容体1(LAIR1)、IgA受容体のFcフラグメント(FCAR)、白血球免疫グロブリン様受容体サブファミリーAメンバー2(LILRA2)、CD300分子様ファミリーメンバーf(CD300LF)、C型レクチンドメインファミリー12メンバーA(CLEC12A)、骨髄間質細胞抗原2(BST2)、EGF様モジュール含有ムチン様ホルモン受容体様2(EMR2)、リンパ球抗原75(LY75)、グリピカン−3(GPC3)、Fc受容体様5(FCRL5)および免疫グロブリンラムダ様ポリペプチド1(IGLL1)からなる群から選択される、請求項1から10のいずれか一項に記載の組成物。
- CAR分子の抗原結合ドメインが、抗体、抗体フラグメント、scFv、Fv、Fab、(Fab’)2、単一ドメイン抗体(SDAB)、VHまたはVLドメインまたはラクダ類VHHドメインを含む、請求項1から11のいずれか一項に記載の組成物。
- (i)膜貫通ドメインが、T細胞受容体のアルファ鎖、ベータ鎖またはゼータ鎖、CD28、CD3イプシロン、CD45、CD4、CD5、CD8、CD9、CD16、CD22、CD33、CD37、CD64、CD80、CD86、CD134、CD137、CD154、KIRDS2、OX40、CD2、CD27、LFA−1(CD11a、CD18)、ICOS(CD278)、4−1BB(CD137)、GITR、CD40、BAFFR、HVEM(LIGHTR)、SLAMF7、NKp80(KLRF1)、CD160、CD19、IL2Rベータ、IL2Rガンマ、IL7Rα、ITGA1、VLA1、CD49a、ITGA4、IA4、CD49D、ITGA6、VLA−6、CD49f、ITGAD、CD11d、ITGAE、CD103、ITGAL、CD11a、LFA−1、ITGAM、CD11b、ITGAX、CD11c、ITGB1、CD29、ITGB2、CD18、LFA−1、ITGB7、TNFR2、DNAM1(CD226)、SLAMF4(CD244、2B4)、CD84、CD96(Tactile)、CEACAM1、CRTAM、Ly9(CD229)、CD160(BY55)、PSGL1、CD100(SEMA4D)、SLAMF6(NTB−A、Ly108)、SLAM(SLAMF1、CD150、IPO−3)、BLAME(SLAMF8)、SELPLG(CD162)、LTBR、PAG/Cbp、NKp44、NKp30、NKp46、NKG2DおよびNKG2Cからなる群より選択されるたんぱく質の膜貫通ドメインを含むか;
(ii)膜貫通ドメインが、配列番号12のアミノ酸配列を含むか、または配列番号12のアミノ酸配列と95〜99%同一性を有するアミノ酸配列を含むか;あるいは
(iii)抗原結合ドメインがヒンジ領域により膜貫通ドメインに接続され、ここで、該ヒンジ領域は、配列番号2もしくは配列番号6のアミノ酸配列を含むか、または配列番号2もしくは配列番号6のアミノ酸配列と95〜99%同一性を有するアミノ酸配列を含む、
請求項1から12のいずれか一項に記載の組成物。 - 細胞内シグナル伝達ドメインが、(i)一次シグナル伝達ドメインを含むか;(ii)共刺激シグナル伝達ドメインを含むか;または、(iii) (i)および(ii)の両方を含む、請求項1から13のいずれか一項に記載の組成物。
- 細胞内シグナル伝達ドメインが、
(i)CD3ゼータ、CD3ガンマ、CD3デルタ、CD3イプシロン、共通FcRガンマ(FCER1G)、FcRベータ(FcイプシロンR1b)、CD79a、CD79b、FcガンマRIIa、DAP10またはDAP12のシグナル伝達ドメインを含む一次シグナル伝達ドメインを含むか、
(ii)CD27、CD28、4−1BB(CD137)、OX40、CD30、CD40、PD−1、ICOS、リンパ球機能関連抗原−1(LFA−1)、CD2、CD7、LIGHT、NKG2C、B7−H3、CD83と特異的に結合するリガンド、CDS、ICAM−1、GITR、BAFFR、HVEM(LIGHTR)、SLAMF7、NKp80(KLRF1)、CD160、CD19、CD4、CD8アルファ、CD8ベータ、IL2Rベータ、IL2Rガンマ、IL7Rアルファ、ITGA4、VLA1、CD49a、ITGA4、IA4、CD49D、ITGA6、VLA−6、CD49f、ITGAD、CD11d、ITGAE、CD103、ITGAL、CD11a、LFA−1、ITGAM、CD11b、ITGAX、CD11c、ITGB1、CD29、ITGB2、CD18、LFA−1、ITGB7、TNFR2、TRANCE/RANKL、DNAM1(CD226)、SLAMF4(CD244、2B4)、CD84、CD96(Tactile)、CEACAM1、CRTAM、Ly9(CD229)、CD160(BY55)、PSGL1、CD100(SEMA4D)、CD69、SLAMF6(NTB−A、Ly108)、SLAM(SLAMF1、CD150、IPO−3)、BLAME(SLAMF8)、SELPLG(CD162)、LTBR、LAT、GADS、SLP−76、PAG/Cbp、NKp44、NKp30、NKp46またはNKG2Dのシグナル伝達ドメインを含む共刺激シグナル伝達ドメインを含むか、
(iii)配列番号14または配列番号16のアミノ酸配列、配列番号14または配列番号16のアミノ酸配列と95〜99%同一性を有する配列、および/あるいは配列番号18または配列番号20のアミノ酸配列、配列番号18または配列番号20のアミノ酸配列と95〜99%同一性を有する配列を含むか、あるいは
(iv)配列番号15もしくは配列番号17の配列またはそれと95〜99%同一性を有する配列、および/あるいは配列番号19もしくは配列番号21の配列またはそれと95〜99%同一性を有する配列を含む、細胞内シグナル伝達ドメインをコードする核酸配列を含む、
請求項14に記載の組成物。 - 免疫エフェクター細胞が、ヒトT細胞またはヒトNK細胞である、請求項1から15のいずれか一項に記載の組成物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021053240A JP2021107395A (ja) | 2014-03-15 | 2021-03-26 | キメラ抗原受容体を使用する癌の処置 |
Applications Claiming Priority (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461953783P | 2014-03-15 | 2014-03-15 | |
| US61/953,783 | 2014-03-15 | ||
| US201461976375P | 2014-04-07 | 2014-04-07 | |
| US61/976,375 | 2014-04-07 | ||
| US201462027154P | 2014-07-21 | 2014-07-21 | |
| US62/027,154 | 2014-07-21 | ||
| US201462076146P | 2014-11-06 | 2014-11-06 | |
| US62/076,146 | 2014-11-06 | ||
| US201462097286P | 2014-12-29 | 2014-12-29 | |
| US62/097,286 | 2014-12-29 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016557287A Division JP2017513818A (ja) | 2014-03-15 | 2015-03-13 | キメラ抗原受容体を使用する癌の処置 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021053240A Division JP2021107395A (ja) | 2014-03-15 | 2021-03-26 | キメラ抗原受容体を使用する癌の処置 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2019206538A JP2019206538A (ja) | 2019-12-05 |
| JP6860623B2 true JP6860623B2 (ja) | 2021-04-21 |
Family
ID=52780057
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016557287A Pending JP2017513818A (ja) | 2014-03-15 | 2015-03-13 | キメラ抗原受容体を使用する癌の処置 |
| JP2019126002A Active JP6860623B2 (ja) | 2014-03-15 | 2019-07-05 | キメラ抗原受容体を使用する癌の処置 |
| JP2021053240A Pending JP2021107395A (ja) | 2014-03-15 | 2021-03-26 | キメラ抗原受容体を使用する癌の処置 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016557287A Pending JP2017513818A (ja) | 2014-03-15 | 2015-03-13 | キメラ抗原受容体を使用する癌の処置 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021053240A Pending JP2021107395A (ja) | 2014-03-15 | 2021-03-26 | キメラ抗原受容体を使用する癌の処置 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US20170335281A1 (ja) |
| EP (2) | EP3119423B1 (ja) |
| JP (3) | JP2017513818A (ja) |
| CN (1) | CN106163547A (ja) |
| ES (1) | ES2939760T3 (ja) |
| WO (1) | WO2015142675A2 (ja) |
Families Citing this family (392)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9724404B2 (en) | 2009-04-13 | 2017-08-08 | INSERM (Institut National de la Santé et de la Recherche Médicale) | HPV particles and uses thereof |
| DK2958943T3 (da) | 2013-02-20 | 2019-12-09 | Univ Pennsylvania | Behandling af cancer ved anvendelse af humaniseret anti-EGFRvIII kimær antigenreceptor |
| WO2014130635A1 (en) | 2013-02-20 | 2014-08-28 | Novartis Ag | Effective targeting of primary human leukemia using anti-cd123 chimeric antigen receptor engineered t cells |
| EP3623380A1 (en) * | 2013-03-15 | 2020-03-18 | Michael C. Milone | Targeting cytotoxic cells with chimeric receptors for adoptive immunotherapy |
| TWI654206B (zh) | 2013-03-16 | 2019-03-21 | 諾華公司 | 使用人類化抗-cd19嵌合抗原受體治療癌症 |
| KR102238226B1 (ko) | 2013-05-14 | 2021-04-09 | 보드 오브 리전츠, 더 유니버시티 오브 텍사스 시스템 | 가공된 키메라 항원 수용체 (car) t-세포의 인간 적용 |
| PL3046583T3 (pl) | 2013-09-18 | 2019-10-31 | Aura Biosciences Inc | Koniugaty cząstek wirusopodobnych do leczenia nowotworów |
| WO2015090230A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | Human mesothelin chimeric antigen receptors and uses thereof |
| JP6793902B2 (ja) | 2013-12-20 | 2020-12-02 | ノバルティス アーゲー | 調節可能キメラ抗原受容体 |
| EP4303229A3 (en) | 2014-01-21 | 2024-04-17 | Novartis AG | Enhanced antigen presenting ability of car t cells by co-introduction of costimulatory molecules |
| US10441654B2 (en) | 2014-01-24 | 2019-10-15 | Children's Hospital Of Eastern Ontario Research Institute Inc. | SMC combination therapy for the treatment of cancer |
| ES2939760T3 (es) | 2014-03-15 | 2023-04-26 | Novartis Ag | Tratamiento de cáncer utilizando un receptor quimérico para antígenos |
| MY191608A (en) | 2014-04-07 | 2022-07-01 | Novartis Ag | Treatment of cancer using anti-cd19 chimeric antigen receptor |
| US10865242B2 (en) | 2014-04-10 | 2020-12-15 | Seattle Children's Hospital | Method and compositions for cellular immunotherapy |
| GB201506423D0 (en) * | 2015-04-15 | 2015-05-27 | Tc Biopharm Ltd | Gamma delta T cells and uses thereof |
| AR101829A1 (es) * | 2014-07-21 | 2017-01-18 | Novartis Ag | Tratamiento de cáncer utilizando un receptor quimérico de antígeno cll-1 |
| EP3193915A1 (en) * | 2014-07-21 | 2017-07-26 | Novartis AG | Combinations of low, immune enhancing. doses of mtor inhibitors and cars |
| CN107109419B (zh) | 2014-07-21 | 2020-12-22 | 诺华股份有限公司 | 使用cd33嵌合抗原受体治疗癌症 |
| CN106687483B (zh) | 2014-07-21 | 2020-12-04 | 诺华股份有限公司 | 使用人源化抗-bcma嵌合抗原受体治疗癌症 |
| US11542488B2 (en) | 2014-07-21 | 2023-01-03 | Novartis Ag | Sortase synthesized chimeric antigen receptors |
| ES2767399T3 (es) * | 2014-08-19 | 2020-06-17 | Miltenyi Biotec Bv & Co Kg | Receptor de antígeno quimérico específico para el antígeno SSEA4 |
| JP7084138B2 (ja) | 2014-08-19 | 2022-06-14 | ノバルティス アーゲー | 癌処置に使用するための抗cd123キメラ抗原受容体(car) |
| KR20230149327A (ko) | 2014-09-17 | 2023-10-26 | 노파르티스 아게 | 입양 면역요법을 위한 키메라 수용체에 의한 세포독성 세포의 표적화 |
| KR20170068504A (ko) | 2014-10-08 | 2017-06-19 | 노파르티스 아게 | 키메라 항원 수용체 요법에 대한 치료 반응성을 예측하는 바이오마커 및 그의 용도 |
| WO2016090034A2 (en) | 2014-12-03 | 2016-06-09 | Novartis Ag | Methods for b cell preconditioning in car therapy |
| MX2017007138A (es) | 2014-12-03 | 2017-08-28 | Juno Therapeutics Inc | Metodos y composiciones para terapia celular adoptiva. |
| RU2020120613A (ru) | 2014-12-05 | 2021-12-06 | Мемориал Слоан-Кеттеринг Кэнсер Сентер | Антитела, нацеленные на рецептор, связанный с g-белками, и способы их применения |
| NZ732568A (en) | 2014-12-05 | 2023-03-31 | Memorial Sloan Kettering Cancer Center | Antibodies targeting b-cell maturation antigen and methods of use |
| JP7174522B2 (ja) * | 2014-12-05 | 2022-11-17 | メモリアル スローン ケタリング キャンサー センター | Fc受容体様5を標的とするキメラ抗原受容体およびその使用 |
| RS62870B1 (sr) | 2014-12-05 | 2022-02-28 | Memorial Sloan Kettering Cancer Center | Himerni antigenski receptori sa ciljanim delovanjem na receptore spregnute sa g-proteinom i njihove upotrebe |
| BR112017011909A2 (pt) | 2014-12-05 | 2018-02-27 | Eureka Therapeutics Inc | ?antígeno de maturação de célula b alvo de receptores de antígeno quimérico e usos do mesmo? |
| US20190054117A1 (en) * | 2014-12-19 | 2019-02-21 | Novartis Ag | Dimerization switches and uses thereof |
| EP3240803B1 (en) | 2014-12-29 | 2021-11-24 | Novartis AG | Methods of making chimeric antigen receptor-expressing cells |
| WO2016115482A1 (en) | 2015-01-16 | 2016-07-21 | Novartis Pharma Ag | Phosphoglycerate kinase 1 (pgk) promoters and methods of use for expressing chimeric antigen receptor |
| BR112017015453A2 (pt) | 2015-01-26 | 2018-01-23 | Cellectis | células imunes modificadas knockout para receptor de células t, dotadas de receptores quiméricos de antígeno, de ligação a cd123 para o tratamento de linfoma mieloide agudo recorrente/refratário ou neoplasia blástica de células dendríticas plasmacitoides |
| US11161907B2 (en) * | 2015-02-02 | 2021-11-02 | Novartis Ag | Car-expressing cells against multiple tumor antigens and uses thereof |
| EP3256492A4 (en) | 2015-02-09 | 2018-07-11 | University of Florida Research Foundation, Inc. | Bi-specific chimeric antigen receptor and uses thereof |
| EP3259352A4 (en) | 2015-02-19 | 2018-12-05 | University of Florida Research Foundation, Inc. | Chimeric antigen receptors and uses thereof |
| AU2016219785B2 (en) | 2015-02-20 | 2021-10-28 | Ohio State Innovation Foundation | Bivalent antibody directed against NKG2D and tumor associated antigens |
| WO2016138846A1 (en) | 2015-03-02 | 2016-09-09 | Shanghai Sidansai Biotechnology Co., Ltd | Reducing immune tolerance induced by pd‐l1 |
| CN107708710A (zh) | 2015-03-17 | 2018-02-16 | 嵌合体生物工程公司 | Smart CAR装置,DE CAR多肽,Side CAR及其使用 |
| CN106146666B (zh) * | 2015-03-26 | 2019-09-06 | 科济生物医药(上海)有限公司 | 靶向cldn6的免疫效应细胞及其制备方法和应用 |
| KR20180012749A (ko) | 2015-04-06 | 2018-02-06 | 지안후아 유 | 교아세포종에 대한 egfr-지시된 car 요법 |
| BR112017021500A2 (pt) | 2015-04-08 | 2018-09-25 | Novartis Ag | terapias com cd20, terapias com cd22 e terapias de combinação com uma célula que expressa (car) receptor de antígeno quimérico de cd19 |
| EP3283619B1 (en) | 2015-04-17 | 2023-04-05 | Novartis AG | Methods for improving the efficacy and expansion of chimeric antigen receptor-expressing cells |
| EP3466967A1 (en) | 2015-05-18 | 2019-04-10 | TCR2 Therapeutics Inc. | Compositions and methods for tcr reprogramming using fusion proteins |
| EP3325504A1 (en) | 2015-07-21 | 2018-05-30 | Novartis AG | Methods for improving the efficacy and expansion of immune cells |
| WO2017017184A1 (en) * | 2015-07-29 | 2017-02-02 | Onkimmune Limited | Modified natural killer cells and natural killer cell lines having increased cytotoxicity |
| WO2017023803A1 (en) | 2015-07-31 | 2017-02-09 | Regents Of The University Of Minnesota | Modified cells and methods of therapy |
| EP3331913A1 (en) | 2015-08-07 | 2018-06-13 | Novartis AG | Treatment of cancer using chimeric cd3 receptor proteins |
| EP3331920A4 (en) | 2015-08-07 | 2019-04-03 | Seattle Children's Hospital, dba Seattle Children's Research Institute | T CARRIER CARRIER CELLS FOR BISPECIFICATION OF SOLID TUMOR TARGETING |
| CN105384825B (zh) * | 2015-08-11 | 2018-06-01 | 南京传奇生物科技有限公司 | 一种基于单域抗体的双特异性嵌合抗原受体及其应用 |
| GB201514874D0 (en) * | 2015-08-20 | 2015-10-07 | Autolus Ltd | Cell |
| EP3340995A4 (en) * | 2015-08-28 | 2019-04-03 | The Trustees Of The University Of Pennsylvania | METHODS AND COMPOSITIONS FOR CELLS EXPRESSING A CHIMERIC INTRACELLULAR SIGNALING MOLECULE |
| JP6905163B2 (ja) | 2015-09-03 | 2021-07-21 | ザ トラスティーズ オブ ザ ユニバーシティ オブ ペンシルバニア | サイトカイン放出症候群を予測するバイオマーカー |
| JP2018532990A (ja) | 2015-09-04 | 2018-11-08 | オービーアイ ファーマ,インコーポレイテッド | グリカンアレイおよび使用の方法 |
| EP3352784A4 (en) * | 2015-09-23 | 2019-06-26 | Cytoimmune Therapeutics, LLC | FLT3-FACED CAR CELLS FOR IMMUNOTHERAPY |
| CN105567649A (zh) * | 2015-10-26 | 2016-05-11 | 哈尔滨新联合生物科技有限公司 | 一种修饰的增强型dc-cik靶向免疫细胞群的制备方法和用途 |
| US11059879B2 (en) | 2015-10-27 | 2021-07-13 | Board Of Regents, The University Of Texas System | Chimeric antigen receptor molecules and uses thereof |
| WO2017075465A1 (en) | 2015-10-28 | 2017-05-04 | The Broad Institute Inc. | Compositions and methods for evaluating and modulating immune responses by detecting and targeting gata3 |
| WO2017075451A1 (en) | 2015-10-28 | 2017-05-04 | The Broad Institute Inc. | Compositions and methods for evaluating and modulating immune responses by detecting and targeting pou2af1 |
| CA3003728A1 (en) * | 2015-10-30 | 2017-05-04 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Targeted cancer therapy |
| CA3001941A1 (en) | 2015-10-30 | 2017-05-04 | Nbe-Therapeutics Ag | Anti-ror1 antibodies |
| CA3003144A1 (en) | 2015-10-30 | 2017-05-04 | Aleta Biotherapeutics Inc. | Compositions and methods for tumor transduction |
| WO2017075537A1 (en) | 2015-10-30 | 2017-05-04 | Aleta Biotherapeutics Inc. | Compositions and methods for treatment of cancer |
| US10941381B2 (en) | 2015-11-19 | 2021-03-09 | Versiti Blood Research Institute Foundation, Inc. | Method of manufacturing dual-specific T-cells for use in cancer immunotherapy |
| AU2016363025B2 (en) * | 2015-12-03 | 2021-04-08 | Juno Therapeutics, Inc. | Modified chimeric receptors and related compositions and methods |
| CN116063508A (zh) | 2015-12-04 | 2023-05-05 | 纪念斯隆-凯特琳癌症中心 | 靶向Fc受体样5的抗体及使用方法 |
| US11052111B2 (en) * | 2015-12-08 | 2021-07-06 | Chimera Bioengineering, Inc. | Smart CAR devices and DE CAR polypeptides for treating disease and methods for enhancing immune responses |
| CN109069597A (zh) | 2015-12-22 | 2018-12-21 | 诺华股份有限公司 | 间皮素嵌合抗原受体(car)和抗pd-l1抗体抑制剂联用于抗癌治疗 |
| AU2016382512A1 (en) | 2015-12-30 | 2018-07-12 | Novartis Ag | Immune effector cell therapies with enhanced efficacy |
| CN116478293A (zh) | 2016-01-12 | 2023-07-25 | 克雷森多生物制剂有限公司 | 治疗分子 |
| BR112018014615A2 (pt) | 2016-01-20 | 2018-12-11 | The Scripps Research Institute | composições de anticorpo para ror1 e métodos relacionados |
| JOP20170017B1 (ar) | 2016-01-25 | 2021-08-17 | Amgen Res Munich Gmbh | تركيب صيدلي يتضمن تركيبات جسم مضاد ثنائي الاختصاص |
| CA3014001A1 (en) * | 2016-02-25 | 2017-08-31 | Cell Medica Switzerland Ag | Binding members to pd-l1 |
| BR112018067679A2 (pt) | 2016-03-04 | 2019-01-15 | Novartis Ag | células que expressam múltiplas moléculas do receptor de antígeno quimérico (car) e seu uso |
| CA3016552A1 (en) * | 2016-03-15 | 2017-09-21 | Genentech, Inc. | Methods of treating cancers using pd-1 axis binding antagonists and anti-gpc3 antibodies |
| US11325948B2 (en) | 2016-03-19 | 2022-05-10 | Exuma Biotech Corp. | Methods and compositions for genetically modifying lymphocytes to express polypeptides comprising the intracellular domain of MPL |
| US11111505B2 (en) | 2016-03-19 | 2021-09-07 | Exuma Biotech, Corp. | Methods and compositions for transducing lymphocytes and regulating the activity thereof |
| US11549099B2 (en) | 2016-03-23 | 2023-01-10 | Novartis Ag | Cell secreted minibodies and uses thereof |
| JP7250519B2 (ja) * | 2016-03-23 | 2023-04-03 | ヘルムホルツ ツェントゥルム ミュンヘン ドイチェス フォルシュングスツェントゥルム フューア ゲズントハイト ウント ウムヴェルト (ゲーエムベーハー) | Pd-1及び4-1bbの融合タンパク質 |
| US11041017B2 (en) | 2016-03-29 | 2021-06-22 | Obi Pharma, Inc. | Antibodies, pharmaceutical compositions and methods |
| CA3018382A1 (en) | 2016-03-29 | 2017-10-05 | University Of Southern California | Chimeric antigen receptors targeting cancer |
| US10980894B2 (en) | 2016-03-29 | 2021-04-20 | Obi Pharma, Inc. | Antibodies, pharmaceutical compositions and methods |
| US10654934B2 (en) | 2016-04-01 | 2020-05-19 | Innovative Cellular Therapeutics CO., LTD. | Use of chimeric antigen receptor modified cells to treat cancer |
| US10603380B2 (en) | 2016-04-01 | 2020-03-31 | Kite Pharma, Inc. | Chimeric antigen and T cell receptors and methods of use |
| KR20220054453A (ko) * | 2016-04-01 | 2022-05-02 | 암젠 인크 | Flt3에 대한 키메라 수용체 및 이의 사용 방법 |
| HUE060645T2 (hu) * | 2016-04-01 | 2023-04-28 | Kite Pharma Inc | Kiméra receptorok és felhasználásuk módszerei |
| WO2017167217A1 (en) | 2016-04-01 | 2017-10-05 | Innovative Cellular Therapeutics CO., LTD. | Use of chimeric antigen receptor modified cells to treat cancer |
| CU20180120A7 (es) | 2016-04-01 | 2019-05-03 | Kite Pharma Inc | Moléculas de unión a bcma |
| US20190161530A1 (en) * | 2016-04-07 | 2019-05-30 | Bluebird Bio, Inc. | Chimeric antigen receptor t cell compositions |
| EP3439685B1 (en) | 2016-04-08 | 2023-07-05 | Emory University | Methods of treating cancer and infectious diseases using cell based therapies |
| EP3443001A4 (en) | 2016-04-11 | 2020-04-29 | Obsidian Therapeutics, Inc. | REGULATED BIOCIRCUIT SYSTEMS |
| CA3044556C (en) * | 2016-04-12 | 2020-11-17 | Philogen S.P.A. | Combination therapy comprising an inflammatory immunocytokine and a chimeric antigen receptor (car)-t cell |
| CN106399255B (zh) * | 2016-04-13 | 2019-10-18 | 阿思科力(苏州)生物科技有限公司 | Pd-1 car-t细胞及其制备方法和应用 |
| CN109715808A (zh) | 2016-04-15 | 2019-05-03 | 诺华股份有限公司 | 用于选择性蛋白质表达的组合物和方法 |
| CN105907719B (zh) * | 2016-04-18 | 2019-10-18 | 阿思科力(苏州)生物科技有限公司 | Anti ROBO1 CAR-T细胞及其制备和应用 |
| KR20180128496A (ko) | 2016-04-22 | 2018-12-03 | 오비아이 파머 인코퍼레이티드 | 글로보 계열 항원을 통한 면역 활성화 또는 면역 조정에 의한 암 면역요법 |
| WO2017190096A1 (en) * | 2016-04-29 | 2017-11-02 | University Of Florida Research Foundation Incorporated | Chimeric antigen receptors and uses thereof |
| GB201607968D0 (en) * | 2016-05-06 | 2016-06-22 | Crescendo Biolog Ltd | Chimeric antigen receptor |
| US10222369B2 (en) | 2016-05-17 | 2019-03-05 | Chimera Bioengineering, Inc. | Methods for making novel antigen binding domains |
| KR101926166B1 (ko) * | 2016-05-24 | 2018-12-06 | 재단법인 아산사회복지재단 | 수용체 시너지 활성을 이용한 nk 세포의 활성도 검사 방법 및 이를 이용한 nk 세포의 활성도가 관련된 질환의 진단 방법 |
| JP2019519529A (ja) * | 2016-05-27 | 2019-07-11 | アメリカ合衆国 | Flt3特異的なキメラ抗原受容体及びそれを使用する方法 |
| SG11201810023QA (en) | 2016-05-27 | 2018-12-28 | Agenus Inc | Anti-tim-3 antibodies and methods of use thereof |
| WO2017210617A2 (en) | 2016-06-02 | 2017-12-07 | Porter, David, L. | Therapeutic regimens for chimeric antigen receptor (car)- expressing cells |
| JP7185530B2 (ja) | 2016-06-13 | 2022-12-07 | トルク セラピューティクス, インコーポレイテッド | 免疫細胞機能を促進するための方法および組成物 |
| WO2018012895A1 (ko) * | 2016-07-14 | 2018-01-18 | 주식회사 큐로셀 | 면역관문을 극복한 면역세포 및 상기 면역세포를 포함한 약제학적 조성물 |
| JP2019524700A (ja) | 2016-07-14 | 2019-09-05 | ミングサイト ファーマシューティカルズ,インク. | 癌の処置 |
| CN110461315A (zh) | 2016-07-15 | 2019-11-15 | 诺华股份有限公司 | 使用与激酶抑制剂组合的嵌合抗原受体治疗和预防细胞因子释放综合征 |
| CN106220736B (zh) * | 2016-07-20 | 2020-01-10 | 深圳市体内生物医药科技有限公司 | 一种嵌合抗原受体、表达其的细胞及其制备方法和用途 |
| SG11201900634VA (en) * | 2016-07-26 | 2019-02-27 | Tessa Therapeutics Pte Ltd | Chimeric antigen receptor |
| TWI752988B (zh) | 2016-07-27 | 2022-01-21 | 台灣浩鼎生技股份有限公司 | 免疫性/治療性聚醣組合物及其用途 |
| AU2017302668B9 (en) | 2016-07-28 | 2023-06-22 | Novartis Ag | Combination therapies of chimeric antigen receptors and PD-1 inhibitors |
| EP3491026A4 (en) | 2016-07-29 | 2020-07-29 | OBI Pharma, Inc. | HUMAN ANTIBODIES, PHARMACEUTICAL COMPOSITIONS AND METHODS |
| CA3032581A1 (en) | 2016-08-01 | 2018-02-08 | Novartis Ag | Treatment of cancer using a chimeric antigen receptor in combination with an inhibitor of a pro-m2 macrophage molecule |
| US11242376B2 (en) | 2016-08-02 | 2022-02-08 | TCR2 Therapeutics Inc. | Compositions and methods for TCR reprogramming using fusion proteins |
| WO2018027197A1 (en) * | 2016-08-04 | 2018-02-08 | Memorial Sloan-Kettering Cancer Center | Cancer antigen targets and uses thereof |
| CA3032838A1 (en) * | 2016-08-04 | 2018-02-08 | Memorial Sloan-Kettering Cancer Center | Compositions and methods for immunotherapy |
| EP3673911B1 (en) * | 2016-08-18 | 2024-03-20 | The UAB Research Foundation | Compositions and methods for cancer immunotherapy |
| CN109789164B (zh) | 2016-08-30 | 2023-04-28 | 亘喜生物科技(上海)有限公司 | 具有gitr胞内结构域作为共刺激结构域的嵌合抗原受体 |
| EP3858365B1 (en) | 2016-09-01 | 2024-01-31 | Chimera Bioengineering Inc. | Gold optimized car t-cells |
| CN107793482A (zh) * | 2016-09-06 | 2018-03-13 | 广州百尼夫生物科技有限公司 | 嵌合抗原受体及其基因和重组表达载体、car133‑nkt 细胞及其制备方法和应用 |
| WO2018049025A2 (en) | 2016-09-07 | 2018-03-15 | The Broad Institute Inc. | Compositions and methods for evaluating and modulating immune responses |
| EP3510156A1 (en) * | 2016-09-08 | 2019-07-17 | Dana-Farber Cancer Institute, Inc. | Compositions and methods of treating cancer |
| CN106434563B (zh) * | 2016-09-14 | 2017-09-22 | 深圳市默赛尔生物医学科技发展有限公司 | 转基因自然杀伤细胞及其制备方法和应用 |
| US11307205B2 (en) | 2016-09-19 | 2022-04-19 | University Of Southern California | Non-radioactive cytotoxicity assays |
| US20190307799A1 (en) | 2016-09-23 | 2019-10-10 | The Regents Of The University Of Michigan | Engineered lymphocytes |
| CN107868791B (zh) * | 2016-09-26 | 2021-11-23 | 阿思科力(苏州)生物科技有限公司 | 一种加强型Slit2 CAR-T和CAR-NK细胞制备方法和应用 |
| EP3519577A1 (en) | 2016-09-28 | 2019-08-07 | Novartis AG | Porous membrane-based macromolecule delivery system |
| CN106317228A (zh) * | 2016-09-28 | 2017-01-11 | 李华顺 | 一种嵌合抗原受体分子及其应用 |
| CN109804064A (zh) * | 2016-09-29 | 2019-05-24 | 南克维斯特公司 | 具有降低的免疫原性的hla i类缺陷的nk-92细胞 |
| US20190119636A1 (en) | 2017-10-23 | 2019-04-25 | Poseida Therapeutics, Inc. | Modified stem cell memory t cells, methods of making and methods of using same |
| JP2019528760A (ja) * | 2016-09-30 | 2019-10-17 | ポセイダ セラピューティクス, インコーポレイテッド | 改変された幹細胞メモリーt細胞、その作製方法およびその使用方法 |
| WO2018064921A1 (en) * | 2016-10-06 | 2018-04-12 | Innovative Cellular Therapeutics CO., LTD. | Use of chimeric antigen receptor modified cells to treat cancer |
| ES2875959T3 (es) | 2016-10-07 | 2021-11-11 | Tcr2 Therapeutics Inc | Composiciones y métodos para reprogramación de receptores de linfocitos T mediante el uso de proteínas de fusión |
| EP3523331A1 (en) | 2016-10-07 | 2019-08-14 | Novartis AG | Chimeric antigen receptors for the treatment of cancer |
| EP3522936A4 (en) * | 2016-10-10 | 2020-06-24 | The National Institute for Biotechnology in the Negev, Ltd. | MODIFIED NON-CYTOTOXIC CELLS AND THEIR USE |
| TWI773694B (zh) | 2016-10-11 | 2022-08-11 | 美商艾吉納斯公司 | 抗lag-3抗體及其使用方法 |
| KR20190084053A (ko) | 2016-10-13 | 2019-07-15 | 주노 쎄러퓨티크스 인코퍼레이티드 | 트립토판 대사 경로 조절인자 관련 면역 치료 방법 및 조성물 |
| CN110520530A (zh) | 2016-10-18 | 2019-11-29 | 明尼苏达大学董事会 | 肿瘤浸润性淋巴细胞和治疗方法 |
| EP3529353A4 (en) * | 2016-10-19 | 2020-05-13 | City of Hope | USE OF THE CMV TRIPLEX VACCINE IN T-CARBY LYMPHOCYTE THERAPY |
| CN107988164B (zh) | 2016-10-26 | 2020-07-07 | 阿思科力(苏州)生物科技有限公司 | 一种pd-1 car nk-92细胞及其制备方法与应用 |
| CN106591363A (zh) * | 2016-11-11 | 2017-04-26 | 广东万海细胞生物科技有限公司 | 一种通用型异体car‑t细胞制备方法及应用 |
| US11332713B2 (en) | 2016-11-16 | 2022-05-17 | KSQ Therapeutics, Inc. | Gene-regulating compositions and methods for improved immunotherapy |
| CA3044593A1 (en) | 2016-11-22 | 2018-05-31 | TCR2 Therapeutics Inc. | Compositions and methods for tcr reprogramming using fusion proteins |
| JP2020511462A (ja) * | 2016-12-03 | 2020-04-16 | ジュノー セラピューティクス インコーポレイテッド | キナーゼ阻害剤との組み合わせで治療用t細胞を使用するための方法および組成物 |
| EP3548083A1 (en) * | 2016-12-03 | 2019-10-09 | Juno Therapeutics, Inc. | Methods for modulation of car-t cells |
| CN106701827A (zh) * | 2016-12-05 | 2017-05-24 | 刘晓明 | 表达cd19和cd20抗体基因嵌合抗原受体t细胞的转化及扩增培养与保存方法 |
| EP3548030A4 (en) * | 2016-12-05 | 2020-08-12 | G1 Therapeutics, Inc. | PRESERVATION OF THE IMMUNE RESPONSE IN CHEMOTHERAPIES |
| DK3581190T3 (da) | 2016-12-09 | 2021-06-07 | Onk Therapeutics Ltd | Manipulerede naturlige dræberceller og anvendelser deraf |
| EP3551197B1 (en) | 2016-12-12 | 2023-11-22 | Seattle Children's Hospital (DBA Seattle Children's Research Institute) | Chimeric transcription factor variants with augmented sensitivity to drug ligand induction of transgene expression in mammalian cells |
| WO2018111340A1 (en) | 2016-12-16 | 2018-06-21 | Novartis Ag | Methods for determining potency and proliferative function of chimeric antigen receptor (car)-t cells |
| AU2017382902A1 (en) * | 2016-12-21 | 2019-07-18 | TCR2 Therapeutics Inc. | Engineered T cells for the treatment of cancer |
| JP2020501589A (ja) * | 2016-12-23 | 2020-01-23 | ウイルツ・バイオロジクス・リミテッド | がんの治療 |
| CN107286246B (zh) * | 2016-12-28 | 2019-12-17 | 时力生物科技(北京)有限公司 | 治疗脑胶质瘤的嵌合抗原受体修饰的树突状细胞及其制备方法 |
| CN108276493B (zh) * | 2016-12-30 | 2023-11-14 | 南京传奇生物科技有限公司 | 一种嵌合抗原受体及其应用 |
| US11814429B2 (en) | 2017-01-06 | 2023-11-14 | Crescendo Biologics Limited | Single domain antibodies to programmed cell death (PD-1) |
| IL267865B1 (en) | 2017-01-10 | 2024-03-01 | Intrexon Corp | Modulation of expression of polypeptides through a new system for changing gene expression |
| EP3568416A4 (en) * | 2017-01-13 | 2020-07-08 | Celdara Medical, LLC | TIM-1 TARGETED CHIMERIC ANTIGENIC RECEPTORS |
| CA3050668C (en) | 2017-01-17 | 2023-08-15 | Daiichi Sankyo Company, Limited | Anti-gpr20 antibody and anti-gpr20 antibody-drug conjugate |
| WO2018136606A1 (en) * | 2017-01-18 | 2018-07-26 | Thalia Papayannopoulou | Compositions and methods for transplant recipient conditioning |
| CN108330133B (zh) * | 2017-01-20 | 2020-06-30 | 上海恒润达生生物科技有限公司 | 靶向cd19嵌合抗原受体并对其双重修饰的方法及其用途 |
| WO2018140725A1 (en) | 2017-01-26 | 2018-08-02 | Novartis Ag | Cd28 compositions and methods for chimeric antigen receptor therapy |
| EP3577134A1 (en) * | 2017-01-31 | 2019-12-11 | Novartis AG | Treatment of cancer using chimeric t cell receptor proteins having multiple specificities |
| CN108395478A (zh) * | 2017-02-04 | 2018-08-14 | 上海恒润达生生物科技有限公司 | 靶向bcma的嵌合抗原受体并对其双重修饰的方法及其用途 |
| EP3581651A4 (en) * | 2017-02-07 | 2021-05-12 | Daiichi Sankyo Company, Limited | ANTI-GPRC5D ANTIBODY AND MOLECULE WITH IT |
| CN117357638A (zh) | 2017-02-17 | 2024-01-09 | 弗雷德哈钦森癌症中心 | 用于治疗bcma相关癌症和自身免疫性失调的联合疗法 |
| CN110753754A (zh) * | 2017-02-21 | 2020-02-04 | 优特力克斯有限公司 | Hla-dr car-t组合物以及制备方法和使用方法 |
| WO2018169922A2 (en) | 2017-03-13 | 2018-09-20 | Kite Pharma, Inc. | Chimeric antigen receptors for melanoma and uses thereof |
| CN110446489A (zh) * | 2017-03-20 | 2019-11-12 | 南加利福尼亚大学 | 具有表面缀合的载药纳米颗粒的car t细胞的过继转移及其用途 |
| US11851659B2 (en) | 2017-03-22 | 2023-12-26 | Novartis Ag | Compositions and methods for immunooncology |
| CN107271675B (zh) * | 2017-03-24 | 2020-06-02 | 郑猛 | 抗人adrb3单克隆抗体及其在疾病诊断和治疗中的应用 |
| SG11201908796XA (en) * | 2017-03-27 | 2019-10-30 | Hoffmann La Roche | Improved antigen binding receptors |
| WO2018183908A1 (en) | 2017-03-31 | 2018-10-04 | Dana-Farber Cancer Institute, Inc. | Compositions and methods for treating ovarian tumors |
| WO2018183921A1 (en) | 2017-04-01 | 2018-10-04 | The Broad Institute, Inc. | Methods and compositions for detecting and modulating an immunotherapy resistance gene signature in cancer |
| WO2018191553A1 (en) | 2017-04-12 | 2018-10-18 | Massachusetts Eye And Ear Infirmary | Tumor signature for metastasis, compositions of matter methods of use thereof |
| WO2018195339A1 (en) | 2017-04-19 | 2018-10-25 | Board Of Regents, The University Of Texas System | Immune cells expressing engineered antigen receptors |
| JOP20180042A1 (ar) | 2017-04-24 | 2019-01-30 | Kite Pharma Inc | نطاقات ربط مولد ضد متوافقة مع البشر وطرق الاستخدام |
| JP7295030B2 (ja) | 2017-04-26 | 2023-06-20 | ユーリカ セラピューティックス, インコーポレイテッド | グリピカン3を特異的に認識するコンストラクト及びその使用 |
| WO2018205926A1 (zh) * | 2017-05-08 | 2018-11-15 | 中国科学院动物研究所 | 经修饰的t细胞、其制备方法及用途 |
| KR20200005596A (ko) | 2017-05-12 | 2020-01-15 | 크리스퍼 테라퓨틱스 아게 | 세포를 엔지니어링하기 위한 물질 및 방법, 및 면역-종양학에서의 그의 용도 |
| US11166985B2 (en) | 2017-05-12 | 2021-11-09 | Crispr Therapeutics Ag | Materials and methods for engineering cells and uses thereof in immuno-oncology |
| WO2018210279A1 (zh) * | 2017-05-16 | 2018-11-22 | 科济生物医药(上海)有限公司 | Toll样受体激动剂与免疫效应细胞的联用 |
| CA3063905A1 (en) | 2017-05-16 | 2018-11-22 | The Johns Hopkins University | Manabodies and methods of using |
| WO2018219299A1 (en) * | 2017-05-31 | 2018-12-06 | Carsgen Therapeutics Co., Ltd. | Compositions and methods of cellular immunotherapy |
| US11566223B2 (en) * | 2017-06-01 | 2023-01-31 | Innovative Cellular Therapeutics Holdings, Ltd. | Chimeric antigen receptor cell preparation and uses thereof |
| CN108977453A (zh) * | 2017-06-02 | 2018-12-11 | 阿思科力(苏州)生物科技有限公司 | 一种以robo1为靶点的嵌合抗原受体细胞及其制备和应用 |
| CN110799640A (zh) * | 2017-06-07 | 2020-02-14 | 综合医院公司 | 表达嵌合抗原受体的t细胞 |
| CN110944652A (zh) * | 2017-06-12 | 2020-03-31 | 爱莫里大学 | T细胞抗原靶向的嵌合抗原受体(car)以及在细胞疗法中的用途 |
| WO2018232195A1 (en) | 2017-06-14 | 2018-12-20 | The Broad Institute, Inc. | Compositions and methods targeting complement component 3 for inhibiting tumor growth |
| CN109134660B (zh) * | 2017-06-16 | 2022-07-01 | 上海恒润达生生物科技股份有限公司 | 靶向Mesothelin的嵌合抗原受体并联合表达抗PD1抗体的方法及其用途 |
| JP2020525010A (ja) * | 2017-06-22 | 2020-08-27 | ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム | 制御性免疫細胞を産生するための方法及びその使用 |
| WO2019006418A2 (en) | 2017-06-30 | 2019-01-03 | Intima Bioscience, Inc. | ADENO-ASSOCIATED VIRAL VECTORS FOR GENE THERAPY |
| CN107164412B (zh) * | 2017-06-30 | 2020-10-23 | 山东兴瑞生物科技有限公司 | 一种安全型抗cea嵌合抗原受体修饰t细胞的制备方法及其应用 |
| EP3645036A1 (en) | 2017-06-30 | 2020-05-06 | Cellectis | Cellular immunotherapy for repetitive administration |
| GB201711068D0 (en) | 2017-07-10 | 2017-08-23 | Crescendo Biologics Ltd | Therapeutic molecules binding PSMA |
| CN109232742B (zh) * | 2017-07-11 | 2021-10-22 | 深圳市第二人民医院 | 一种嵌合抗原受体及其应用 |
| CN107353325B (zh) * | 2017-07-26 | 2020-08-11 | 中国药科大学 | 叶酸受体α特异性结合肽1及其应用 |
| CN107326014B (zh) * | 2017-07-31 | 2019-09-24 | 时力生物科技(北京)有限公司 | 一种双特异性嵌合抗原受体修饰的t淋巴细胞及其制备方法和应用 |
| SG11202000846WA (en) | 2017-08-07 | 2020-02-27 | Nbe Therapeutics Ag | Anthracycline-based antibody drug conjugates having high in vivo tolerability |
| CN111051330A (zh) | 2017-08-31 | 2020-04-21 | 第一三共株式会社 | 抗体-药物缀合物的改进制备方法 |
| WO2019044946A1 (ja) | 2017-08-31 | 2019-03-07 | 第一三共株式会社 | 抗体-薬物コンジュゲートの新規製造方法 |
| EP3678701A4 (en) | 2017-09-05 | 2021-12-01 | Torque Therapeutics, Inc. | THERAPEUTIC PROTEIN COMPOSITIONS AND METHOD FOR MANUFACTURING AND USING THEREOF |
| JP2020533289A (ja) * | 2017-09-06 | 2020-11-19 | フレッド ハッチンソン キャンサー リサーチ センター | 養子細胞治療を改善するための方法 |
| AU2018334279A1 (en) * | 2017-09-15 | 2020-04-02 | Kite Pharma, Inc. | Methods and systems for performing a patient-specific immunotherapy procedure with chain-of-custody and chain-of-identity biological sample tracking |
| EP3687569A1 (en) | 2017-09-29 | 2020-08-05 | Cell Design Labs, Inc. | Methods of making bispecific anti-cd307e and anti-bcma chimeric antigen receptors and uses of the same |
| CN107557341B (zh) * | 2017-09-30 | 2020-06-30 | 山东兴瑞生物科技有限公司 | 一种抗wt1增强型嵌合抗原受体修饰的免疫细胞及其应用 |
| WO2019070704A1 (en) * | 2017-10-02 | 2019-04-11 | Georgia Tech Research Corporation | METHODS AND COMPOSITIONS FOR ENGINEERING SYNTHESIS BIOLOGICAL SWITCHES FOR REMOTE CONTROL OF BIOLOGICAL ACTIVITY |
| KR20200063215A (ko) * | 2017-10-06 | 2020-06-04 | 오슬로 유니버시테시케후스 에이치에프 | 키메라 항원 수용체 |
| CA3078637A1 (en) * | 2017-10-12 | 2019-04-18 | Mcmaster University | T cell-antigen coupler with y182t mutation and methods and uses thereof |
| MX2020003856A (es) | 2017-10-13 | 2020-08-13 | Harpoon Therapeutics Inc | Proteinas de union a antigenos de maduracion de celulas b. |
| WO2019079569A1 (en) | 2017-10-18 | 2019-04-25 | Novartis Ag | COMPOSITIONS AND METHODS FOR SELECTIVE DEGRADATION OF A PROTEIN |
| US20210069241A1 (en) * | 2017-10-20 | 2021-03-11 | Fred Hutchinson Cancer Research Center | Compositions and methods of immunotherapy targeting tigit and/or cd112r or comprising cd226 overexpression |
| MX2020004229A (es) | 2017-10-25 | 2020-07-22 | Novartis Ag | Metodos de produccion de celulas que expresan receptores antigenicos quimericos. |
| MA49911A (fr) | 2017-11-01 | 2020-06-24 | Juno Therapeutics Inc | Anticorps et récepteurs antigéniques chimériques spécifiques de l'antigene de maturation des lymphocytes b |
| SG11202003866QA (en) | 2017-11-01 | 2020-05-28 | Juno Therapeutics Inc | Chimeric antigen receptors specific for b-cell maturation antigen (bcma) |
| EP3710039A4 (en) | 2017-11-13 | 2021-08-04 | The Broad Institute, Inc. | METHODS AND COMPOSITIONS FOR CANCER TREATMENT BY TARGETING THE CLEC2D-KLRB1 PATH |
| US11649294B2 (en) | 2017-11-14 | 2023-05-16 | GC Cell Corporation | Anti-HER2 antibody or antigen-binding fragment thereof, and chimeric antigen receptor comprising same |
| WO2019099707A1 (en) | 2017-11-16 | 2019-05-23 | Kite Pharma, Inc | Modified chimeric antigen receptors and methods of use |
| CN107916269B (zh) * | 2017-11-17 | 2020-12-11 | 山东兴瑞生物科技有限公司 | 靶向cd19的tcr基因、制备方法、具有该基因的质粒、试剂盒及应用 |
| AU2018370853A1 (en) * | 2017-11-21 | 2020-05-21 | Novartis Ag | Trispecific binding molecules against tumor-associated antigens and uses thereof |
| DK3720883T3 (da) | 2017-12-05 | 2023-07-24 | Medical Res Infrastructure & Health Services Fund Tel Aviv Medical Ct | T-celler omfattende kimære anti-cd38- og anti-cd138-antigenreceptorer og anvendelser deraf |
| US20210300986A1 (en) * | 2017-12-05 | 2021-09-30 | The Medical Research Infrastrutrure And Health Services Fund Of The Tel Aviv Medecal Center | T-cells comprising two different chimeric antigen receptors and uses thereof |
| MA51158A (fr) * | 2017-12-14 | 2020-10-21 | Bluebird Bio Inc | Récepteurs daric nkg2d |
| CN112218885A (zh) * | 2017-12-20 | 2021-01-12 | 波赛达治疗公司 | Vcar组合物和使用方法 |
| CN109609533B (zh) * | 2017-12-27 | 2020-07-10 | 赛德特生物科技开发有限公司 | 基于人源化cd276抗体的car慢病毒表达载体构建及其应用 |
| EP3731849A4 (en) * | 2017-12-28 | 2021-12-01 | Codiak BioSciences, Inc. | EXOSOME FOR IMMUNONCOLOGICAL AND ANTI-INFLAMMATORY THERAPY |
| CN109971712B (zh) * | 2017-12-28 | 2023-06-20 | 上海细胞治疗研究院 | 特异性靶向cd19抗原且高水平稳定表达pd-1抗体的car-t细胞及用途 |
| CN109971715A (zh) * | 2017-12-28 | 2019-07-05 | 深圳华大生命科学研究院 | 一种扩增特异性car-t细胞的培养方法 |
| CN109971722B (zh) * | 2017-12-28 | 2023-09-01 | 上海细胞治疗研究院 | 靶向cd19且高水平稳定表达cd40抗体的car-t细胞及其用途 |
| AU2018395272A1 (en) * | 2017-12-28 | 2020-07-09 | NEUGATE PHARMA, LLC aka NEUGATE THERANOSTICS | Compositions and formulations for treatment of malignancies |
| CN108103105A (zh) * | 2017-12-29 | 2018-06-01 | 深圳市茵冠生物科技有限公司 | 一种car-t细胞的制备方法、制得的car-t细胞及其应用 |
| EP3508855A1 (en) * | 2018-01-05 | 2019-07-10 | Allianz Pharmascience Ltd | Method for the selection of patients and kit |
| CN112218651A (zh) * | 2018-01-08 | 2021-01-12 | 诺华公司 | 用于与嵌合抗原受体疗法组合的免疫增强rna |
| MX2020007338A (es) * | 2018-01-09 | 2020-11-06 | H Lee Moffitt Cancer Ct & Res | Composiciones y métodos para el direccionamiento a tipos de cáncer que expresan clec12a. |
| US10561686B2 (en) * | 2018-01-12 | 2020-02-18 | Innovative Cellular Therapeutics CO., LTD. | Modified cell expansion and uses thereof |
| KR102327512B1 (ko) * | 2018-01-12 | 2021-11-17 | 주식회사 큐로셀 | 이원 shrna를 이용한 향상된 면역 세포 및 이를 포함하는 조성물 |
| WO2019143818A1 (en) * | 2018-01-17 | 2019-07-25 | Mingsight Pharmaceuticals, Inc. | Combination therapy for the treatment of cancer |
| EP3740569B1 (en) * | 2018-01-19 | 2023-07-26 | Miltenyi Biotec B.V. & Co. KG | Regulatory t cell expressing a chimeric antigen receptor |
| CN110093351B (zh) * | 2018-01-29 | 2023-06-23 | 华南生物医药研究院 | 可分离的核酸、多肽、重组载体、重组细胞及应用 |
| WO2019152660A1 (en) * | 2018-01-31 | 2019-08-08 | Novartis Ag | Combination therapy using a chimeric antigen receptor |
| CN111989108A (zh) | 2018-02-13 | 2020-11-24 | 嵌合体生物工程公司 | 利用rna去稳定元件协调基因表达 |
| GB201802573D0 (en) | 2018-02-16 | 2018-04-04 | Crescendo Biologics Ltd | Therapeutic molecules that bind to LAG3 |
| US20210040449A1 (en) | 2018-02-16 | 2021-02-11 | Kite Pharma, Inc. | Modified pluripotent stem cells and methods of making and use |
| US20210346431A1 (en) * | 2018-02-23 | 2021-11-11 | Endocyte, Inc. | Sequencing method for car t cell therapy |
| EP3762410A4 (en) * | 2018-03-06 | 2022-04-06 | The Trustees of the University of Pennsylvania | PROSTATE-SPECIFIC MEMBRANE ANTIGEN CARS AND METHODS OF USE |
| KR20210013013A (ko) * | 2018-03-09 | 2021-02-03 | 카르스젠 테라퓨틱스 리미티드 | 종양 치료 방법 및 조성물 |
| WO2019175328A1 (en) | 2018-03-14 | 2019-09-19 | Imba - Institut Für Molekulare Biotechnologie Gmbh | Bh4pathwayactivationandusethereoffortreatingcancer |
| BR112020018620A2 (pt) | 2018-03-15 | 2020-12-29 | KSQ Therapeutics, Inc. | Composições de regulação gênica e métodos para imunoterapia aprimorada |
| US20190284553A1 (en) | 2018-03-15 | 2019-09-19 | KSQ Therapeutics, Inc. | Gene-regulating compositions and methods for improved immunotherapy |
| EP3768700A1 (en) | 2018-03-23 | 2021-01-27 | Kite Pharma, Inc. | Chimeric transmembrane proteins and uses thereof |
| US10688182B2 (en) * | 2018-03-29 | 2020-06-23 | Cho Pharma Usa, Inc. | Monoclonal antibodies that bind to SSEA4 and uses thereof |
| EP3775883A1 (en) * | 2018-04-04 | 2021-02-17 | F. Hoffmann-La Roche AG | Diagnostic assays to detect tumor antigens in cancer patients |
| US20190328781A1 (en) * | 2018-04-06 | 2019-10-31 | Mustang Bio, Inc. | Manufacturing methods for cell-based therapeutic compositions |
| US10869888B2 (en) | 2018-04-17 | 2020-12-22 | Innovative Cellular Therapeutics CO., LTD. | Modified cell expansion and uses thereof |
| US11957695B2 (en) | 2018-04-26 | 2024-04-16 | The Broad Institute, Inc. | Methods and compositions targeting glucocorticoid signaling for modulating immune responses |
| WO2019210293A1 (en) * | 2018-04-27 | 2019-10-31 | Baylor College Of Medicine | Car t cells with one or more interleukins |
| WO2019209557A1 (en) | 2018-04-27 | 2019-10-31 | Mayo Foundation For Medical Education And Research | Foamy viruses and methods of use |
| AU2019260804A1 (en) * | 2018-04-27 | 2020-11-12 | Bayer Healthcare Llc | Methods and compositions of cytotoxic T cell depletion |
| WO2019213282A1 (en) | 2018-05-01 | 2019-11-07 | Novartis Ag | Biomarkers for evaluating car-t cells to predict clinical outcome |
| CN110438082B (zh) * | 2018-05-04 | 2024-03-08 | 恺兴生命科技(上海)有限公司 | 表达il-21r结合蛋白的免疫效应细胞 |
| EP3790629A1 (en) | 2018-05-11 | 2021-03-17 | CRISPR Therapeutics AG | Methods and compositions for treating cancer |
| JP7335272B2 (ja) | 2018-05-15 | 2023-08-29 | オートラス リミテッド | キメラ抗原受容体 |
| GB201807870D0 (en) * | 2018-05-15 | 2018-06-27 | Autolus Ltd | A CD79-specific chimeric antigen receptor |
| JOP20200292A1 (ar) * | 2018-05-16 | 2020-11-15 | Stichting Vumc | Bcma / cd3 و gprdc5d / cd3 مضادات غير محددة للاستخدام في علاج السرطان |
| WO2019227003A1 (en) | 2018-05-25 | 2019-11-28 | Novartis Ag | Combination therapy with chimeric antigen receptor (car) therapies |
| CN108623683B (zh) * | 2018-05-30 | 2021-06-04 | 福州迈新生物技术开发有限公司 | 抗Pax-5蛋白的单克隆抗体及其细胞株、制备方法和应用 |
| CN108715859B (zh) * | 2018-05-31 | 2021-08-03 | 中国医学科学院血液病医院(中国医学科学院血液学研究所) | 靶向cd22的嵌合抗原受体及其应用 |
| US20210371932A1 (en) | 2018-06-01 | 2021-12-02 | Massachusetts Institute Of Technology | Methods and compositions for detecting and modulating microenvironment gene signatures from the csf of metastasis patients |
| EP3801607A4 (en) * | 2018-06-01 | 2022-03-16 | OBI Pharma, Inc. | COMBINATION THERAPY USING ANTI-GLOBO-H OR ANTI-SSEA-4 ANTIBODIES AND ANTIBODIES AGAINST NEGATIVE IMMUNE CHECKPOINTS |
| CN110577604A (zh) * | 2018-06-07 | 2019-12-17 | 亘喜生物科技(上海)有限公司 | 携带gitr共刺激信号靶向egfr的嵌合抗原受体t细胞 |
| WO2019237035A1 (en) | 2018-06-08 | 2019-12-12 | Intellia Therapeutics, Inc. | Compositions and methods for immunooncology |
| BR112020025048A2 (pt) | 2018-06-13 | 2021-04-06 | Novartis Ag | Receptores de antígeno quimérico de bcma e usos dos mesmos |
| US20210268027A1 (en) * | 2018-06-22 | 2021-09-02 | The General Hospital Corporation | Chimeric antigen receptors targeting cd37 and cd19 |
| SG11202011751WA (en) | 2018-06-22 | 2021-01-28 | Kite Pharma Inc | Chimeric transmembrane proteins and uses thereof |
| CN110623980A (zh) * | 2018-06-25 | 2019-12-31 | 深圳宾德生物技术有限公司 | 靶向cd38的嵌合抗原受体t细胞在自身免疫性疾病中的应用 |
| CN108913709A (zh) * | 2018-06-26 | 2018-11-30 | 山东兴瑞生物科技有限公司 | 用于治疗hcc的核酸、其制备方法、具有该核酸的car-t细胞及细胞的制备方法 |
| CN108866004A (zh) * | 2018-06-26 | 2018-11-23 | 奥妙生物技术(广州)有限公司 | Shp-1敲除的t细胞及其构建方法 |
| TW202035444A (zh) | 2018-06-27 | 2020-10-01 | 台灣浩鼎生技股份有限公司 | 用於糖蛋白工程的糖苷合成酶變體及其使用方法 |
| US11001635B2 (en) * | 2018-06-29 | 2021-05-11 | Gensun Biopharma Inc. | Antitumor antagonists |
| WO2020000465A1 (zh) * | 2018-06-29 | 2020-01-02 | 深圳市博奥康生物科技有限公司 | 一种制备lightr基因敲除小鼠的方法 |
| CN112673093A (zh) * | 2018-07-04 | 2021-04-16 | 赛通免疫治疗公司 | 靶向flt3、pd-1和/或pd-l1的免疫疗法的组合物和方法 |
| US11952419B2 (en) * | 2018-07-10 | 2024-04-09 | University Of Connecticut | Reagents and methods for treating cancer and autoimmune disease |
| AR116109A1 (es) | 2018-07-10 | 2021-03-31 | Novartis Ag | Derivados de 3-(5-amino-1-oxoisoindolin-2-il)piperidina-2,6-diona y usos de los mismos |
| US10640562B2 (en) | 2018-07-17 | 2020-05-05 | Mcmaster University | T cell-antigen coupler with various construct optimizations |
| US20210308267A1 (en) * | 2018-08-07 | 2021-10-07 | Purdue Research Foundation | Rejuvenation of car t cell |
| KR20210057730A (ko) | 2018-08-09 | 2021-05-21 | 주노 쎄러퓨티크스 인코퍼레이티드 | 조작 세포 및 이의 조성물 생성 방법 |
| SG11202101415UA (en) * | 2018-08-10 | 2021-03-30 | Sangamo Therapeutics France | New car constructs comprising tnfr2 domains |
| CN112384231A (zh) * | 2018-08-26 | 2021-02-19 | 奥伊斯细胞生物技术公司 | 用于治疗胶质母细胞瘤的方法 |
| US20220348682A1 (en) | 2018-08-30 | 2022-11-03 | Innovative Cellular Therapeutics Holdings, Ltd. | Chimeric antigen receptor cells for treating solid tumor |
| SG11202101825QA (en) | 2018-08-31 | 2021-03-30 | Novartis Ag | Methods of making chimeric antigen receptor-expressing cells |
| EP3844265A2 (en) | 2018-08-31 | 2021-07-07 | Novartis AG | Methods of making chimeric antigen receptor-expressing cells |
| CA3114038A1 (en) | 2018-09-25 | 2020-04-02 | Harpoon Therapeutics, Inc. | Dll3 binding proteins and methods of use |
| CN109265561B (zh) * | 2018-09-25 | 2021-05-25 | 山东兴瑞生物科技有限公司 | 抗EGFRvⅢ安全型嵌合抗原受体、其制备方法、利用其修饰的NK细胞及应用 |
| EP3856779A1 (en) | 2018-09-28 | 2021-08-04 | Novartis AG | Cd22 chimeric antigen receptor (car) therapies |
| US20210347851A1 (en) | 2018-09-28 | 2021-11-11 | Novartis Ag | Cd19 chimeric antigen receptor (car) and cd22 car combination therapies |
| US20210382068A1 (en) | 2018-10-02 | 2021-12-09 | Dana-Farber Cancer Institute, Inc. | Hla single allele lines |
| WO2020081730A2 (en) | 2018-10-16 | 2020-04-23 | Massachusetts Institute Of Technology | Methods and compositions for modulating microenvironment |
| WO2020086989A1 (en) * | 2018-10-25 | 2020-04-30 | Innovative Cellular Therapeutics CO., LTD. | Increase or maintaining t-cell subpopulations in adoptive t-cell therapy |
| JP2022506781A (ja) * | 2018-11-09 | 2022-01-17 | フレッド ハッチンソン キャンサー リサーチ センター | メソテリンを標的とする免疫療法 |
| US20220033848A1 (en) * | 2018-11-19 | 2022-02-03 | Board Of Regents, The University Of Texas System | A modular, polycistronic vector for car and tcr transduction |
| US10918667B2 (en) * | 2018-11-20 | 2021-02-16 | Innovative Cellular Therapeutics CO., LTD. | Modified cell expressing therapeutic agent and uses thereof |
| WO2020113098A1 (en) | 2018-11-29 | 2020-06-04 | Vineti Inc. | Centralized and decentralized individualized medicine platform |
| MX2021006968A (es) | 2018-12-12 | 2021-10-13 | Kite Pharma Inc | Antígeno quimérico y receptores de las células t, y métodos de uso. |
| US20220062394A1 (en) | 2018-12-17 | 2022-03-03 | The Broad Institute, Inc. | Methods for identifying neoantigens |
| EP3898946B1 (en) * | 2018-12-19 | 2023-02-08 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Mucosal-associated invariant t (mait) cells expressing chimeric antigen receptors |
| WO2020128972A1 (en) | 2018-12-20 | 2020-06-25 | Novartis Ag | Dosing regimen and pharmaceutical combination comprising 3-(1-oxoisoindolin-2-yl)piperidine-2,6-dione derivatives |
| WO2020132557A1 (en) | 2018-12-21 | 2020-06-25 | Compass Therapeutics Llc | Transgenic mouse expressing common human light chain |
| CA3125345A1 (en) * | 2019-01-04 | 2020-07-09 | Marengo Therapeutics, Inc. | Anti-tcr antibody molecules and uses thereof |
| US11739156B2 (en) | 2019-01-06 | 2023-08-29 | The Broad Institute, Inc. Massachusetts Institute of Technology | Methods and compositions for overcoming immunosuppression |
| PE20211977A1 (es) * | 2019-01-18 | 2021-10-05 | Janssen Biotech Inc | Receptores de antigenos quimericos de gprc5d y celulas que los expresan |
| WO2020165834A1 (en) | 2019-02-15 | 2020-08-20 | Novartis Ag | Substituted 3-(1-oxoisoindolin-2-yl)piperidine-2,6-dione derivatives and uses thereof |
| CA3124935A1 (en) | 2019-02-15 | 2020-08-20 | Novartis Ag | 3-(1-oxo-5-(piperidin-4-yl)isoindolin-2-yl)piperidine-2,6-dione derivatives and uses thereof |
| US11321652B1 (en) | 2019-02-20 | 2022-05-03 | Vineti Inc. | Smart label devices, systems, and methods |
| US20220088075A1 (en) | 2019-02-22 | 2022-03-24 | The Trustees Of The University Of Pennsylvania | Combination therapies of egfrviii chimeric antigen receptors and pd-1 inhibitors |
| US20220152150A1 (en) | 2019-02-25 | 2022-05-19 | Novartis Ag | Mesoporous silica particles compositions for viral delivery |
| US20220154282A1 (en) | 2019-03-12 | 2022-05-19 | The Broad Institute, Inc. | Detection means, compositions and methods for modulating synovial sarcoma cells |
| US20220142948A1 (en) | 2019-03-18 | 2022-05-12 | The Broad Institute, Inc. | Compositions and methods for modulating metabolic regulators of t cell pathogenicity |
| US20220168389A1 (en) | 2019-04-12 | 2022-06-02 | Novartis Ag | Methods of making chimeric antigen receptor-expressing cells |
| US20220251152A1 (en) | 2019-04-24 | 2022-08-11 | Novartis Ag | Compositions and methods for selective protein degradation |
| CN114269777A (zh) * | 2019-04-26 | 2022-04-01 | 北卡罗来纳大学教堂山分校 | 嵌合抗原受体构建体及其在car-t细胞中的用途 |
| MX2021013359A (es) | 2019-04-30 | 2022-01-31 | Crispr Therapeutics Ag | Terapia de celulas alogénicas de neoplasias malignas de células b usando células t modificadas genéticamente dirigidas a cd19. |
| AU2020266841A1 (en) * | 2019-04-30 | 2021-11-25 | Memorial Sloan-Kettering Cancer Center | Combination therapies |
| WO2020223535A1 (en) | 2019-05-01 | 2020-11-05 | Juno Therapeutics, Inc. | Cells expressing a recombinant receptor from a modified tgfbr2 locus, related polynucleotides and methods |
| WO2020223571A1 (en) | 2019-05-01 | 2020-11-05 | Juno Therapeutics, Inc. | Cells expressing a chimeric receptor from a modified cd247 locus, related polynucleotides and methods |
| US20220235340A1 (en) | 2019-05-20 | 2022-07-28 | The Broad Institute, Inc. | Novel crispr-cas systems and uses thereof |
| WO2020243371A1 (en) | 2019-05-28 | 2020-12-03 | Massachusetts Institute Of Technology | Methods and compositions for modulating immune responses |
| WO2021009701A1 (en) * | 2019-07-17 | 2021-01-21 | Curocell Inc. | Enhanced immune cells using dual shrna and composition including the same |
| JP2022541315A (ja) * | 2019-07-24 | 2022-09-22 | エウレカ セラピューティクス インコーポレイテッド | キメラ抗原受容体及びキメラ刺激受容体を発現する細胞並びにその使用 |
| CA3145697A1 (en) * | 2019-07-30 | 2021-02-04 | Dendrocyte Biotech Pty Ltd | Anti-cd300f immunoconjugate |
| US20220282333A1 (en) | 2019-08-13 | 2022-09-08 | The General Hospital Corporation | Methods for predicting outcomes of checkpoint inhibition and treatment thereof |
| US20210046117A1 (en) | 2019-08-18 | 2021-02-18 | Chimera Bioengineering, Inc. | Combination Therapy with Gold Controlled Transgenes |
| CN110627909B (zh) * | 2019-08-28 | 2021-03-30 | 中国人民解放军第二军医大学 | 一种特异性活化nk细胞的嵌合抗原受体及其应用 |
| WO2021041922A1 (en) | 2019-08-30 | 2021-03-04 | The Broad Institute, Inc. | Crispr-associated mu transposase systems |
| EP4028413A1 (en) | 2019-09-10 | 2022-07-20 | Obsidian Therapeutics, Inc. | Ca2-il15 fusion proteins for tunable regulation |
| WO2021050656A1 (en) * | 2019-09-11 | 2021-03-18 | The Trustees Of The Univeristy Of Pennsylvania | Compositions and methods comprising prostate stem cell antigen (psca) chimeric antigen receptors (cars) |
| WO2021054789A1 (ko) * | 2019-09-18 | 2021-03-25 | 주식회사 에스엘바이젠 | 신규 키메라 항원 수용체 암호화 유전자가 형질도입된 유전자 변형 nk 세포주 및 그의 용도 |
| WO2021057906A1 (zh) * | 2019-09-25 | 2021-04-01 | 科济生物医药(上海)有限公司 | 表达il-15的免疫效应细胞 |
| US11793787B2 (en) | 2019-10-07 | 2023-10-24 | The Broad Institute, Inc. | Methods and compositions for enhancing anti-tumor immunity by targeting steroidogenesis |
| WO2021081052A1 (en) * | 2019-10-22 | 2021-04-29 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | High affinity nanobodies targeting b7h3 (cd276) for treating multiple solid tumors |
| EP4048402A4 (en) * | 2019-10-23 | 2023-11-22 | Cue Biopharma, Inc. | MODIFIED CYTOXIC T CELLS AND METHODS OF USE THEREOF |
| EP4048689A4 (en) * | 2019-10-23 | 2023-11-01 | Cue Biopharma, Inc. | T-CELL MODULATING CHIMERIC MOLECULES AND METHOD OF USE THEREOF |
| CN110627903B (zh) * | 2019-10-29 | 2021-09-21 | 河南大学 | 抗cd97单克隆抗体、其可变区与恒定区序列及应用 |
| WO2021092007A1 (en) * | 2019-11-04 | 2021-05-14 | Verastem, Inc. | Methods of making cellular therapies |
| KR20220105664A (ko) | 2019-11-26 | 2022-07-27 | 노파르티스 아게 | Bcma 및 cd19에 결합하는 키메라 항원 수용체 및 이의 용도 |
| EP4065157A1 (en) | 2019-11-26 | 2022-10-05 | Novartis AG | Cd19 and cd22 chimeric antigen receptors and uses thereof |
| AU2020394279A1 (en) * | 2019-11-28 | 2022-06-16 | CentricsBio, Inc. | Chimeric antigen receptor specifically binding to CD 300c antigen or receptor thereof |
| WO2021127428A1 (en) * | 2019-12-18 | 2021-06-24 | Albert Einstein College Of Medicine | Chimeric antigen receptors targeting b7-h3 (cd276) and associated methods |
| US20230056470A1 (en) | 2019-12-20 | 2023-02-23 | Novartis Ag | Uses of anti-tgf-beta antibodies and checkpoint inhibitors for the treatment of proliferative diseases |
| WO2021163618A1 (en) | 2020-02-14 | 2021-08-19 | Novartis Ag | Method of predicting response to chimeric antigen receptor therapy |
| CN115175695A (zh) | 2020-02-27 | 2022-10-11 | 诺华股份有限公司 | 制备表达嵌合抗原受体的细胞的方法 |
| MX2022010685A (es) | 2020-02-27 | 2022-09-23 | Novartis Ag | Metodos de produccion de celulas que expresan receptores de antigeno quimericos. |
| WO2021222944A1 (en) * | 2020-04-30 | 2021-11-04 | Board Of Regents, The University Of Texas System | Anti-cd79b antibodies and chimeric antigen receptors and methods of use thereof |
| WO2021249462A1 (zh) * | 2020-06-11 | 2021-12-16 | 南京北恒生物科技有限公司 | 表达nk抑制性分子的工程化免疫细胞及其用途 |
| US20230416359A1 (en) * | 2020-06-17 | 2023-12-28 | H. Lee Moffitt Cancer Center And Research Institute, Inc. | Olfactory receptors for use as targets for antigen binding molecules to detect and treat cancer |
| JP2023531676A (ja) | 2020-06-23 | 2023-07-25 | ノバルティス アーゲー | 3-(1-オキソイソインドリン-2-イル)ピぺリジン-2,6-ジオン誘導体を含む投与レジメン |
| CN116234558A (zh) | 2020-06-26 | 2023-06-06 | 朱诺治疗学有限公司 | 条件性地表达重组受体的工程化t细胞、相关多核苷酸和方法 |
| WO2022016119A1 (en) | 2020-07-17 | 2022-01-20 | Simurx, Inc. | Chimeric myd88 receptors for redirecting immunosuppressive signaling and related compositions and methods |
| CN116134027A (zh) | 2020-08-03 | 2023-05-16 | 诺华股份有限公司 | 杂芳基取代的3-(1-氧代异吲哚啉-2-基)哌啶-2,6-二酮衍生物及其用途 |
| WO2022031884A2 (en) * | 2020-08-05 | 2022-02-10 | Synthekine, Inc. | Il2rg binding molecules and methods of use |
| EP4192490A1 (en) * | 2020-08-05 | 2023-06-14 | Synthekine, Inc. | IL27Ra BINDING MOLECULES AND METHODS OF USE |
| CA3191161A1 (en) | 2020-08-14 | 2022-02-17 | Kite Pharma, Inc | Improving immune cell function |
| WO2022036495A1 (en) | 2020-08-17 | 2022-02-24 | Utc Therapeutics Inc. | Lymphocytes-antigen presenting cells co-stimulators and uses thereof |
| KR20230058427A (ko) | 2020-08-21 | 2023-05-03 | 노파르티스 아게 | Car 발현 세포의 생체내 생성을 위한 조성물 및 방법 |
| CN114369168A (zh) * | 2020-10-19 | 2022-04-19 | 南京卡提医学科技有限公司 | 包含dap12和共刺激信号分子信号域的嵌合受体及其使用方法 |
| CN114426583B (zh) * | 2020-10-29 | 2023-10-10 | 中国科学技术大学 | 用于急性髓系白血病的细胞疗法的嵌合抗原受体 |
| CN116635062A (zh) | 2020-11-13 | 2023-08-22 | 诺华股份有限公司 | 使用表达嵌合抗原受体(car)的细胞的组合疗法 |
| US20240002526A1 (en) * | 2020-11-17 | 2024-01-04 | Igm Biosciences, Inc. | Uses of effector cell engaging molecules with moieties of differing potencies |
| US11661459B2 (en) | 2020-12-03 | 2023-05-30 | Century Therapeutics, Inc. | Artificial cell death polypeptide for chimeric antigen receptor and uses thereof |
| TW202237638A (zh) | 2020-12-09 | 2022-10-01 | 日商武田藥品工業股份有限公司 | 烏苷酸環化酶c(gcc)抗原結合劑之組成物及其使用方法 |
| CN113234169B (zh) * | 2020-12-11 | 2022-11-01 | 广州百暨基因科技有限公司 | 靶向cll1嵌合抗原受体及其应用 |
| JP2024500847A (ja) | 2020-12-18 | 2024-01-10 | センチュリー セラピューティクス,インコーポレイテッド | 適合可能な受容体特異性を有するキメラ抗原受容体システム |
| WO2022147075A1 (en) * | 2020-12-30 | 2022-07-07 | St. Jude Children's Research Hospital, Inc. | Chimeric antigen receptors targeting splice variants of the extracellular matrix proteins tenascin c (tnc) and procollagen 11a1 (col11a1) |
| TW202241479A (zh) | 2020-12-30 | 2022-11-01 | 美商安迅生物製藥公司 | 遞送外來抗原之溶瘤病毒及表現靶向外來抗原之嵌合受體之經工程化免疫細胞之組合療法 |
| EP4284394A1 (en) | 2021-01-26 | 2023-12-06 | Cytocares (Shanghai) Inc. | Chimeric antigen receptor (car) constructs and nk cells expressing car constructs |
| JP2024504825A (ja) * | 2021-01-29 | 2024-02-01 | ミンフィ ファーマシューティカル (ハンチョウ) リミテッド | 抗原結合タンパク質およびその使用 |
| CN112851826B (zh) * | 2021-02-10 | 2023-08-11 | 上海煦顼技术有限公司 | Upk2嵌合抗原受体及其尿道癌的治疗 |
| KR20220124839A (ko) * | 2021-03-03 | 2022-09-14 | 주식회사 이뮤노로지컬디자이닝랩 | RANK 리간드(RANK ligand)에 특이적으로 결합하는 키메릭 항원 수용체 및 이의 용도 |
| KR20230155521A (ko) | 2021-03-11 | 2023-11-10 | 카이트 파마 인코포레이티드 | 면역 세포 기능의 향상 |
| TW202304979A (zh) | 2021-04-07 | 2023-02-01 | 瑞士商諾華公司 | 抗TGFβ抗體及其他治療劑用於治療增殖性疾病之用途 |
| WO2022229853A1 (en) | 2021-04-27 | 2022-11-03 | Novartis Ag | Viral vector production system |
| AU2022330406A1 (en) | 2021-08-20 | 2024-03-07 | Novartis Ag | Methods of making chimeric antigen receptor–expressing cells |
| EP4141028A1 (en) | 2021-08-31 | 2023-03-01 | Johann-Wolfgang-Goethe-Universität Frankfurt am Main | Personalized vnar-based chimeric antigen receptors (vnar-car) for the treatment of clonal b-and t cell malignancies, and methods for producing vnar-cars |
| US11615874B1 (en) | 2021-09-30 | 2023-03-28 | Vineti Inc. | Personalized medicine and therapies platform |
| CN114015674A (zh) | 2021-11-02 | 2022-02-08 | 辉二(上海)生物科技有限公司 | 新型CRISPR-Cas12i系统 |
| WO2023177954A1 (en) * | 2022-03-18 | 2023-09-21 | University Of Rochester | Combination therapy for treatment of cancer, methods and systems of delivery thereof |
| CN114805584B (zh) * | 2022-06-30 | 2022-09-09 | 上海优替济生生物医药有限公司 | 抗原结合蛋白及其用途 |
| WO2024014470A1 (ja) * | 2022-07-13 | 2024-01-18 | 国立大学法人大阪大学 | 腫瘍の予防および/または治療剤 |
| US20240091261A1 (en) | 2022-08-26 | 2024-03-21 | Kite Pharma, Inc. | Immune cell function |
| WO2024056809A1 (en) | 2022-09-15 | 2024-03-21 | Novartis Ag | Treatment of autoimmune disorders using chimeric antigen receptor therapy |
| WO2024077256A1 (en) | 2022-10-07 | 2024-04-11 | The General Hospital Corporation | Methods and compositions for high-throughput discovery ofpeptide-mhc targeting binding proteins |
| WO2024089639A1 (en) | 2022-10-26 | 2024-05-02 | Novartis Ag | Lentiviral formulations |
Family Cites Families (291)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US504048A (en) | 1893-08-29 | Sulky | ||
| ZA737247B (en) | 1972-09-29 | 1975-04-30 | Ayerst Mckenna & Harrison | Rapamycin and process of preparation |
| US4433059A (en) | 1981-09-08 | 1984-02-21 | Ortho Diagnostic Systems Inc. | Double antibody conjugate |
| US4444878A (en) | 1981-12-21 | 1984-04-24 | Boston Biomedical Research Institute, Inc. | Bispecific antibody determinants |
| DE3381783D1 (de) | 1982-03-03 | 1990-09-13 | Genentech Inc | Menschliches antithrombin iii, dns sequenzen dafuer, expressions- und klonierungsvektoren die solche sequenzen enthalten und damit transformierte zellkulturen, verfahren zur expression von menschlichem antithrombin iii und diese enthaltende pharmazeutische zusammensetzungen. |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4851332A (en) | 1985-04-01 | 1989-07-25 | Sloan-Kettering Institute For Cancer Research | Choriocarcinoma monoclonal antibodies and antibody panels |
| GB8607679D0 (en) | 1986-03-27 | 1986-04-30 | Winter G P | Recombinant dna product |
| US6548640B1 (en) | 1986-03-27 | 2003-04-15 | Btg International Limited | Altered antibodies |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| US5869620A (en) | 1986-09-02 | 1999-02-09 | Enzon, Inc. | Multivalent antigen-binding proteins |
| JPH021556A (ja) | 1988-06-09 | 1990-01-05 | Snow Brand Milk Prod Co Ltd | ハイブリッド抗体及びその作製方法 |
| US6905680B2 (en) | 1988-11-23 | 2005-06-14 | Genetics Institute, Inc. | Methods of treating HIV infected subjects |
| US5858358A (en) | 1992-04-07 | 1999-01-12 | The United States Of America As Represented By The Secretary Of The Navy | Methods for selectively stimulating proliferation of T cells |
| US6534055B1 (en) | 1988-11-23 | 2003-03-18 | Genetics Institute, Inc. | Methods for selectively stimulating proliferation of T cells |
| US6352694B1 (en) | 1994-06-03 | 2002-03-05 | Genetics Institute, Inc. | Methods for inducing a population of T cells to proliferate using agents which recognize TCR/CD3 and ligands which stimulate an accessory molecule on the surface of the T cells |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5703055A (en) | 1989-03-21 | 1997-12-30 | Wisconsin Alumni Research Foundation | Generation of antibodies through lipid mediated DNA delivery |
| US5399346A (en) | 1989-06-14 | 1995-03-21 | The United States Of America As Represented By The Department Of Health And Human Services | Gene therapy |
| DE3920358A1 (de) | 1989-06-22 | 1991-01-17 | Behringwerke Ag | Bispezifische und oligospezifische, mono- und oligovalente antikoerperkonstrukte, ihre herstellung und verwendung |
| US5585362A (en) | 1989-08-22 | 1996-12-17 | The Regents Of The University Of Michigan | Adenovirus vectors for gene therapy |
| WO1991003493A1 (en) | 1989-08-29 | 1991-03-21 | The University Of Southampton | Bi-or trispecific (fab)3 or (fab)4 conjugates |
| GB8928874D0 (en) | 1989-12-21 | 1990-02-28 | Celltech Ltd | Humanised antibodies |
| US5273743A (en) | 1990-03-09 | 1993-12-28 | Hybritech Incorporated | Trifunctional antibody-like compounds as a combined diagnostic and therapeutic agent |
| GB9012995D0 (en) | 1990-06-11 | 1990-08-01 | Celltech Ltd | Multivalent antigen-binding proteins |
| US5582996A (en) | 1990-12-04 | 1996-12-10 | The Wistar Institute Of Anatomy & Biology | Bifunctional antibodies and method of preparing same |
| EP0519596B1 (en) | 1991-05-17 | 2005-02-23 | Merck & Co. Inc. | A method for reducing the immunogenicity of antibody variable domains |
| DE4118120A1 (de) | 1991-06-03 | 1992-12-10 | Behringwerke Ag | Tetravalente bispezifische rezeptoren, ihre herstellung und verwendung |
| US5199942A (en) | 1991-06-07 | 1993-04-06 | Immunex Corporation | Method for improving autologous transplantation |
| US6511663B1 (en) | 1991-06-11 | 2003-01-28 | Celltech R&D Limited | Tri- and tetra-valent monospecific antigen-binding proteins |
| LU91067I2 (fr) | 1991-06-14 | 2004-04-02 | Genentech Inc | Trastuzumab et ses variantes et dérivés immuno chimiques y compris les immotoxines |
| US5637481A (en) | 1993-02-01 | 1997-06-10 | Bristol-Myers Squibb Company | Expression vectors encoding bispecific fusion proteins and methods of producing biologically active bispecific fusion proteins in a mammalian cell |
| CA2078539C (en) | 1991-09-18 | 2005-08-02 | Kenya Shitara | Process for producing humanized chimera antibody |
| ES2136092T3 (es) | 1991-09-23 | 1999-11-16 | Medical Res Council | Procedimientos para la produccion de anticuerpos humanizados. |
| US5932448A (en) | 1991-11-29 | 1999-08-03 | Protein Design Labs., Inc. | Bispecific antibody heterodimers |
| GB9125768D0 (en) | 1991-12-04 | 1992-02-05 | Hale Geoffrey | Therapeutic method |
| JP4157160B2 (ja) | 1991-12-13 | 2008-09-24 | ゾーマ テクノロジー リミテッド | 改変抗体可変領域の調製のための方法 |
| ATE151113T1 (de) | 1992-01-23 | 1997-04-15 | Merck Patent Gmbh | Fusionsproteine von monomeren und dimeren von antikörperfragmenten |
| DE69334255D1 (de) | 1992-02-06 | 2009-02-12 | Novartis Vaccines & Diagnostic | Marker für Krebs und biosynthetisches Bindeprotein dafür |
| GB9203459D0 (en) | 1992-02-19 | 1992-04-08 | Scotgen Ltd | Antibodies with germ-line variable regions |
| US5646253A (en) | 1994-03-08 | 1997-07-08 | Memorial Sloan-Kettering Cancer Center | Recombinant human anti-LK26 antibodies |
| ATE165113T1 (de) | 1992-05-08 | 1998-05-15 | Creative Biomolecules Inc | Mehrwertige chimäre proteine anologe und verfahren zu deren anwendungen |
| US6005079A (en) | 1992-08-21 | 1999-12-21 | Vrije Universiteit Brussels | Immunoglobulins devoid of light chains |
| ES2162823T5 (es) | 1992-08-21 | 2010-08-09 | Vrije Universiteit Brussel | Inmunoglobulinas desprovistas de cadenas ligeras. |
| US5350674A (en) | 1992-09-04 | 1994-09-27 | Becton, Dickinson And Company | Intrinsic factor - horse peroxidase conjugates and a method for increasing the stability thereof |
| US5639641A (en) | 1992-09-09 | 1997-06-17 | Immunogen Inc. | Resurfacing of rodent antibodies |
| JPH08504320A (ja) | 1992-09-25 | 1996-05-14 | コモンウエルス・サイエンティフィック・アンド・インダストリアル・リサーチ・オーガニゼーション | 標的結合性ポリペプチド |
| GB9221220D0 (en) | 1992-10-09 | 1992-11-25 | Sandoz Ag | Organic componds |
| GB9221657D0 (en) | 1992-10-15 | 1992-11-25 | Scotgen Ltd | Recombinant bispecific antibodies |
| WO1994009817A1 (en) | 1992-11-04 | 1994-05-11 | City Of Hope | Novel antibody construct |
| GB9323648D0 (en) | 1992-11-23 | 1994-01-05 | Zeneca Ltd | Proteins |
| JP3720353B2 (ja) | 1992-12-04 | 2005-11-24 | メディカル リサーチ カウンシル | 多価および多重特異性の結合タンパク質、それらの製造および使用 |
| US6476198B1 (en) | 1993-07-13 | 2002-11-05 | The Scripps Research Institute | Multispecific and multivalent antigen-binding polypeptide molecules |
| US5635602A (en) | 1993-08-13 | 1997-06-03 | The Regents Of The University Of California | Design and synthesis of bispecific DNA-antibody conjugates |
| WO1995009917A1 (en) | 1993-10-07 | 1995-04-13 | The Regents Of The University Of California | Genetically engineered bispecific tetravalent antibodies |
| US5843674A (en) | 1993-11-16 | 1998-12-01 | Pola Chemical Industries Inc. | Anti-human tyrosinase monoclonal antibody |
| CA2175215C (en) | 1993-11-19 | 2008-06-03 | Yat Sun Or | Semisynthetic analogs of rapamycin (macrolides) being immunomodulators |
| EP0734389B1 (en) | 1993-12-17 | 2000-03-29 | Novartis AG | Rapamycin derivatives useful as immunosuppressants |
| US5635388A (en) | 1994-04-04 | 1997-06-03 | Genentech, Inc. | Agonist antibodies against the flk2/flt3 receptor and uses thereof |
| US5362718A (en) | 1994-04-18 | 1994-11-08 | American Home Products Corporation | Rapamycin hydroxyesters |
| WO1995029193A2 (en) | 1994-04-22 | 1995-11-02 | The Government Of The United States Of America Represented By The Secretary, Department Of Health And Human Services | Melanoma antigens |
| US7175843B2 (en) | 1994-06-03 | 2007-02-13 | Genetics Institute, Llc | Methods for selectively stimulating proliferation of T cells |
| US5786464C1 (en) | 1994-09-19 | 2012-04-24 | Gen Hospital Corp | Overexpression of mammalian and viral proteins |
| WO1996013583A2 (en) | 1994-10-20 | 1996-05-09 | Morphosys Gesellschaft Für Proteinoptimierung Mbh | Targeted hetero-association of recombinant proteins to multi-functional complexes |
| WO1996022384A1 (en) | 1995-01-18 | 1996-07-25 | Boehringer Mannheim Gmbh | Anti-cd 30 antibodies preventing proteolytic cleavage and release of membrane-bound cd 30 antigen |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US6692964B1 (en) | 1995-05-04 | 2004-02-17 | The United States Of America As Represented By The Secretary Of The Navy | Methods for transfecting T cells |
| US7067318B2 (en) | 1995-06-07 | 2006-06-27 | The Regents Of The University Of Michigan | Methods for transfecting T cells |
| EP0827544B1 (en) | 1995-05-23 | 2004-08-18 | MorphoSys AG | Multimeric proteins |
| DE69624921T2 (de) | 1995-06-09 | 2003-09-11 | Novartis Ag | Rapamycinderivate |
| BE1009856A5 (fr) | 1995-07-14 | 1997-10-07 | Sandoz Sa | Composition pharmaceutique sous la forme d'une dispersion solide comprenant un macrolide et un vehicule. |
| WO1997014719A1 (en) | 1995-10-16 | 1997-04-24 | Unilever N.V. | A bifunctional or bivalent antibody fragment analogue |
| DE19608769C1 (de) | 1996-03-07 | 1997-04-10 | Univ Eberhard Karls | Antikörper BV10A4H2 |
| DK0894135T3 (da) | 1996-04-04 | 2004-12-06 | Unilever Nv | Multivalent og multispecifikt antigenbindingsprotein |
| EP0937082A2 (en) | 1996-07-12 | 1999-08-25 | Ariad Pharmaceuticals, Inc. | Materials and method for treating or preventing pathogenic fungal infection |
| US6111090A (en) | 1996-08-16 | 2000-08-29 | Schering Corporation | Mammalian cell surface antigens; related reagents |
| DK0920505T3 (da) | 1996-08-16 | 2008-09-08 | Schering Corp | Pattedyrcelleoverfladeantigener og tilhörende reagenser |
| US6114148C1 (en) | 1996-09-20 | 2012-05-01 | Gen Hospital Corp | High level expression of proteins |
| JP2001510987A (ja) | 1996-10-25 | 2001-08-07 | アメリカ合衆国 | 哺乳動物のCD97αサブユニットを含む炎症および脈管形成を阻害するための方法および組成物 |
| CA2288994C (en) | 1997-04-30 | 2011-07-05 | Enzon, Inc. | Polyalkylene oxide-modified single chain polypeptides |
| US20030207346A1 (en) | 1997-05-02 | 2003-11-06 | William R. Arathoon | Method for making multispecific antibodies having heteromultimeric and common components |
| US20020062010A1 (en) | 1997-05-02 | 2002-05-23 | Genentech, Inc. | Method for making multispecific antibodies having heteromultimeric and common components |
| CA2304254C (en) | 1997-06-11 | 2012-05-22 | Hans Christian Thogersen | Trimerising module |
| AU729035B2 (en) | 1997-06-12 | 2001-01-25 | Novartis Ag | Artificial antibody polypeptides |
| US6015815A (en) | 1997-09-26 | 2000-01-18 | Abbott Laboratories | Tetrazole-containing rapamycin analogs with shortened half-lives |
| TW557297B (en) | 1997-09-26 | 2003-10-11 | Abbott Lab | Rapamycin analogs having immunomodulatory activity, and pharmaceutical compositions containing same |
| EP1025228A4 (en) | 1997-10-21 | 2002-09-18 | Human Genome Sciences Inc | HUMAN PROTEIN TR11, TR11SV1 AND TR11SV2 SIMILAR TO THE TUMOR NECROSIS FACTOR RECEPTOR |
| AU2152299A (en) | 1997-10-27 | 1999-05-24 | Unilever Plc | Multivalent antigen-binding proteins |
| AU2719099A (en) | 1998-01-23 | 1999-08-09 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw | Multipurpose antibody derivatives |
| EP1053321A1 (en) | 1998-02-09 | 2000-11-22 | Genentech, Inc. | Novel tumor necrosis factor receptor homolog and nucleic acids encoding the same |
| HUP9900956A2 (hu) | 1998-04-09 | 2002-04-29 | Aventis Pharma Deutschland Gmbh. | Egyláncú, több antigéntkötőhely kialakítására képes molekulák, előállításuk és alkalmazásuk |
| WO1999052552A1 (en) | 1998-04-15 | 1999-10-21 | Brigham & Women's Hospital, Inc. | T cell inhibitory receptor compositions and uses thereof |
| DE19819846B4 (de) | 1998-05-05 | 2016-11-24 | Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts | Multivalente Antikörper-Konstrukte |
| GB9812545D0 (en) | 1998-06-10 | 1998-08-05 | Celltech Therapeutics Ltd | Biological products |
| US6803448B1 (en) | 1998-07-22 | 2004-10-12 | Vanderbilt University | GBS toxin receptor |
| ATE251181T1 (de) | 1998-07-28 | 2003-10-15 | Micromet Ag | Heterominikörper |
| US6333396B1 (en) | 1998-10-20 | 2001-12-25 | Enzon, Inc. | Method for targeted delivery of nucleic acids |
| US6528481B1 (en) | 1999-02-16 | 2003-03-04 | The Burnam Institute | NG2/HM proteoglycan-binding peptides that home to angiogenic vasculature and related methods |
| US7534866B2 (en) | 2005-10-19 | 2009-05-19 | Ibc Pharmaceuticals, Inc. | Methods and compositions for generating bioactive assemblies of increased complexity and uses |
| US7527787B2 (en) | 2005-10-19 | 2009-05-05 | Ibc Pharmaceuticals, Inc. | Multivalent immunoglobulin-based bioactive assemblies |
| IL147442A0 (en) | 1999-07-12 | 2002-08-14 | Genentech Inc | Promotion or inhibition of angiogenesis and cardiovscularization by tumor necrosis factor ligand/receptor homologs |
| BRPI0013391B8 (pt) | 1999-08-17 | 2021-05-25 | Apotech R&D S A | uso de polipeptídeos bcma na preparação de uma composição farmacêutica para tratar uma doença autoimune ou um distúrbio linfoproliferativo de célula b |
| AU783158B2 (en) | 1999-08-24 | 2005-09-29 | Ariad Pharmaceuticals, Inc. | 28-epirapalogs |
| ES2301491T3 (es) | 1999-09-30 | 2008-07-01 | Kyowa Hakko Kogyo Co., Ltd. | Anticuerpo humano de trasplante con region de determinacion de la complementariedad contra gangliosido gd3 y derivados del anticuerpo contra gangliosido gd3. |
| EP1235842A4 (en) | 1999-10-15 | 2003-04-23 | Univ Massachusetts | GENESIS OF THE RNA INTERFERENCE PATH AS AID OF TARGETED GENTIAN INTERFERENCE |
| US6326193B1 (en) | 1999-11-05 | 2001-12-04 | Cambria Biosciences, Llc | Insect control agent |
| ATE373078T1 (de) | 2000-02-24 | 2007-09-15 | Xcyte Therapies Inc | Gleichzeitige stimulation und konzentration von zellen |
| US6867041B2 (en) | 2000-02-24 | 2005-03-15 | Xcyte Therapies, Inc. | Simultaneous stimulation and concentration of cells |
| US6797514B2 (en) | 2000-02-24 | 2004-09-28 | Xcyte Therapies, Inc. | Simultaneous stimulation and concentration of cells |
| US7572631B2 (en) | 2000-02-24 | 2009-08-11 | Invitrogen Corporation | Activation and expansion of T cells |
| US20040002068A1 (en) | 2000-03-01 | 2004-01-01 | Corixa Corporation | Compositions and methods for the detection, diagnosis and therapy of hematological malignancies |
| HUP0300369A2 (hu) | 2000-04-11 | 2003-06-28 | Genentech, Inc. | Többértékű antitestek és alkalmazásuk |
| EP1299419A2 (en) | 2000-05-24 | 2003-04-09 | Imclone Systems, Inc. | Bispecific immunoglobulin-like antigen binding proteins and method of production |
| AU2001275474A1 (en) | 2000-06-12 | 2001-12-24 | Akkadix Corporation | Materials and methods for the control of nematodes |
| CA2410551A1 (en) | 2000-06-30 | 2002-01-10 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw (Vib) | Heterodimeric fusion proteins |
| CN1461344A (zh) | 2000-07-25 | 2003-12-10 | 免疫医疗公司 | 多价靶结合蛋白 |
| US20040242847A1 (en) | 2000-10-20 | 2004-12-02 | Naoshi Fukushima | Degraded agonist antibody |
| US7090843B1 (en) | 2000-11-28 | 2006-08-15 | Seattle Genetics, Inc. | Recombinant anti-CD30 antibodies and uses thereof |
| US7829084B2 (en) | 2001-01-17 | 2010-11-09 | Trubion Pharmaceuticals, Inc. | Binding constructs and methods for use thereof |
| WO2002072635A2 (en) | 2001-03-13 | 2002-09-19 | University College London | Specific binding members |
| CN1294148C (zh) | 2001-04-11 | 2007-01-10 | 中国科学院遗传与发育生物学研究所 | 环状单链三特异抗体 |
| JP4303105B2 (ja) | 2001-06-28 | 2009-07-29 | ドマンティス リミテッド | 二重特異性リガンドとその利用 |
| US6833441B2 (en) | 2001-08-01 | 2004-12-21 | Abmaxis, Inc. | Compositions and methods for generating chimeric heteromultimers |
| ES2337986T3 (es) | 2001-08-10 | 2010-05-03 | Aberdeen University | Dominios de union de antigenos de peces. |
| EP2383291B1 (en) | 2001-08-23 | 2019-04-17 | Rsr Limited | Epitope regions of a thyrotropin (TSH) receptor, uses thereof and antibodies thereto |
| DE60124912T2 (de) | 2001-09-14 | 2007-06-14 | Affimed Therapeutics Ag | Multimerische, einzelkettige, Tandem-Fv-Antikörper |
| EP1470159B1 (en) | 2001-12-04 | 2013-08-07 | Dana-Farber Cancer Institute, Inc. | Antibody to latent membrane proteins and uses thereof |
| US20030211078A1 (en) | 2001-12-07 | 2003-11-13 | Heavner George A. | Pseudo-antibody constructs |
| US7745140B2 (en) | 2002-01-03 | 2010-06-29 | The Trustees Of The University Of Pennsylvania | Activation and expansion of T-cells using an engineered multivalent signaling platform as a research tool |
| EA008379B1 (ru) | 2002-02-01 | 2007-04-27 | Ариад Джин Терапьютикс, Инк. | Фосфорсодержащие соединения и их применения |
| US20040018557A1 (en) | 2002-03-01 | 2004-01-29 | Immunomedics, Inc. | Bispecific antibody point mutations for enhancing rate of clearance |
| GB0208104D0 (en) | 2002-04-09 | 2002-05-22 | Univ Dundee | Method |
| AU2003227504A1 (en) | 2002-04-15 | 2003-10-27 | Chugai Seiyaku Kabushiki Kaisha | METHOD OF CONSTRUCTING scDb LIBRARY |
| US20040047858A1 (en) | 2002-09-11 | 2004-03-11 | Blumberg Richard S. | Therapeutic anti-BGP(C-CAM1) antibodies and uses thereof |
| US8501415B2 (en) | 2002-11-26 | 2013-08-06 | B.R.A.H.M.S. Gmbh | Identification of TSH receptor autoantibodies using affinity-purified antibodies |
| WO2004056875A1 (en) | 2002-12-23 | 2004-07-08 | Wyeth | Antibodies against pd-1 and uses therefor |
| GB0230203D0 (en) | 2002-12-27 | 2003-02-05 | Domantis Ltd | Fc fusion |
| GB0305702D0 (en) | 2003-03-12 | 2003-04-16 | Univ Birmingham | Bispecific antibodies |
| WO2004087758A2 (en) | 2003-03-26 | 2004-10-14 | Neopharm, Inc. | Il 13 receptor alpha 2 antibody and methods of use |
| AU2004232928A1 (en) | 2003-04-22 | 2004-11-04 | Ibc Pharmaceuticals | Polyvalent protein complex |
| CU23403A1 (es) | 2003-04-23 | 2009-08-04 | Centro Inmunologia Molecular | Anticuerpos recombinantes y fragmentos que reconocen el gangliósido n-glicolil gm3 y su uso para diagnóstico y tratamiento de tumores |
| US7618632B2 (en) | 2003-05-23 | 2009-11-17 | Wyeth | Method of treating or ameliorating an immune cell associated pathology using GITR ligand antibodies |
| US20050163782A1 (en) | 2003-06-27 | 2005-07-28 | Biogen Idec Ma Inc. | Modified binding molecules comprising connecting peptides |
| CA2530605A1 (en) | 2003-06-27 | 2005-05-26 | Diadexus, Inc. | Pro104 antibody compositions and methods of use |
| AU2004255216B2 (en) | 2003-07-01 | 2010-08-19 | Immunomedics, Inc. | Multivalent carriers of bi-specific antibodies |
| WO2005007190A1 (en) | 2003-07-11 | 2005-01-27 | Schering Corporation | Agonists or antagonists of the clucocorticoid-induced tumour necrosis factor receptor (gitr) or its ligand for the treatment of immune disorders, infections and cancer |
| US7696322B2 (en) | 2003-07-28 | 2010-04-13 | Catalent Pharma Solutions, Inc. | Fusion antibodies |
| EP2272566A3 (en) | 2003-08-18 | 2013-01-02 | MedImmune, LLC | Humanisation of antibodies |
| US20050042664A1 (en) | 2003-08-22 | 2005-02-24 | Medimmune, Inc. | Humanization of antibodies |
| WO2005035577A1 (ja) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | ガングリオシドgd3に特異的に結合する抗体組成物 |
| CA2542046A1 (en) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | Fused protein composition |
| US20130266551A1 (en) * | 2003-11-05 | 2013-10-10 | St. Jude Children's Research Hospital, Inc. | Chimeric receptors with 4-1bb stimulatory signaling domain |
| US7220755B2 (en) | 2003-11-12 | 2007-05-22 | Biosensors International Group, Ltd. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| EP1692318A4 (en) | 2003-12-02 | 2008-04-02 | Genzyme Corp | COMPOSITIONS AND METHODS FOR DIAGNOSIS AND TREATMENT OF LUNG CANCER |
| US20050136051A1 (en) | 2003-12-22 | 2005-06-23 | Bernard Scallon | Methods for generating multimeric molecules |
| GB0329825D0 (en) | 2003-12-23 | 2004-01-28 | Celltech R&D Ltd | Biological products |
| US20050266425A1 (en) | 2003-12-31 | 2005-12-01 | Vaccinex, Inc. | Methods for producing and identifying multispecific antibodies |
| US8383575B2 (en) | 2004-01-30 | 2013-02-26 | Paul Scherrer Institut | (DI)barnase-barstar complexes |
| SI2511297T1 (sl) | 2004-02-06 | 2015-07-31 | Morphosys Ag | Proti -CD38 humana protitelesa in njihova uporaba |
| GB0409799D0 (en) | 2004-04-30 | 2004-06-09 | Isis Innovation | Method of generating improved immune response |
| AU2005250408B2 (en) | 2004-05-27 | 2010-09-23 | The Trustees Of The University Of Pennsylvania | Novel artificial antigen presenting cells and uses therefor |
| EP1765402A2 (en) | 2004-06-04 | 2007-03-28 | Duke University | Methods and compositions for enhancement of immunity by in vivo depletion of immunosuppressive cell activity |
| JP2008512352A (ja) | 2004-07-17 | 2008-04-24 | イムクローン システムズ インコーポレイティド | 新規な四価の二重特異性抗体 |
| AU2005282700A1 (en) | 2004-09-02 | 2006-03-16 | Genentech, Inc. | Heteromultimeric molecules |
| MY146381A (en) | 2004-12-22 | 2012-08-15 | Amgen Inc | Compositions and methods relating relating to anti-igf-1 receptor antibodies |
| DK1866339T3 (da) | 2005-03-25 | 2013-09-02 | Gitr Inc | GTR-bindende molekyler og anvendelser heraf |
| EP3050963B1 (en) | 2005-03-31 | 2019-09-18 | Chugai Seiyaku Kabushiki Kaisha | Process for production of polypeptide by regulation of assembly |
| AU2006232920B2 (en) | 2005-04-06 | 2011-09-29 | Ibc Pharmaceuticals, Inc. | Methods for generating stably linked complexes composed of homodimers, homotetramers or dimers of dimers and uses |
| US9296816B2 (en) | 2005-04-15 | 2016-03-29 | Macrogenics, Inc. | Covalent diabodies and uses thereof |
| CA2607147C (en) | 2005-05-09 | 2018-07-17 | Ono Pharmaceutical Co., Ltd. | Human monoclonal antibodies to programmed death 1 (pd-1) and methods for treating cancer using anti-pd-1 antibodies alone or in combination with other immunotherapeutics |
| PT1899364T (pt) | 2005-05-17 | 2020-05-20 | Univ Connecticut | Composições e métodos para imunomodulação num organismo |
| GB0510390D0 (en) | 2005-05-20 | 2005-06-29 | Novartis Ag | Organic compounds |
| US20060263367A1 (en) | 2005-05-23 | 2006-11-23 | Fey Georg H | Bispecific antibody devoid of Fc region and method of treatment using same |
| EP1726650A1 (en) | 2005-05-27 | 2006-11-29 | Universitätsklinikum Freiburg | Monoclonal antibodies and single chain antibody fragments against cell-surface prostate specific membrane antigen |
| MX2007016306A (es) | 2005-06-15 | 2008-03-07 | Schering Corp | Formulaciones de anticuerpo anti-igf1r. |
| JP5252635B2 (ja) | 2005-07-01 | 2013-07-31 | メダレックス インコーポレーティッド | プログラム死リガンド1(pd−l1)に対するヒトモノクローナル抗体 |
| US20070036773A1 (en) | 2005-08-09 | 2007-02-15 | City Of Hope | Generation and application of universal T cells for B-ALL |
| US7612181B2 (en) | 2005-08-19 | 2009-11-03 | Abbott Laboratories | Dual variable domain immunoglobulin and uses thereof |
| MY169746A (en) | 2005-08-19 | 2019-05-14 | Abbvie Inc | Dual variable domain immunoglobulin and uses thereof |
| EP1757622B1 (en) | 2005-08-26 | 2009-12-23 | PLS Design GmbH | Bivalent IgY antibody constructs for diagnostic and therapeutic applications |
| DK1931645T3 (da) | 2005-10-07 | 2014-09-29 | Exelixis Inc | N-(3-aminoquinoxalin-2-yl)-sulfonamidderivater og deres anvendelse som inhibitorer af phosphatidylinositol-3-kinase |
| WO2007044887A2 (en) | 2005-10-11 | 2007-04-19 | Transtarget, Inc. | Method for producing a population of homogenous tetravalent bispecific antibodies |
| US8623356B2 (en) | 2005-11-29 | 2014-01-07 | The University Of Sydney | Demibodies: dimerization-activated therapeutic agents |
| EA017491B1 (ru) | 2005-12-08 | 2012-12-28 | Медарекс, Инк. | Человеческие моноклональные антитела к фукозил-gm1 и способы применения антифукозил-gm1 антител |
| EP1806365A1 (en) | 2006-01-05 | 2007-07-11 | Boehringer Ingelheim International GmbH | Antibody molecules specific for fibroblast activation protein and immunoconjugates containing them |
| CA2636111C (en) | 2006-01-13 | 2018-04-03 | The Government Of The United States, As Represented By The Secretary Of The Department Of Health And Human Services, National Institutes Of Health | Codon optimized il-15 and il-15r-alpha genes for expression in mammalian cells |
| US20110212086A1 (en) | 2006-01-19 | 2011-09-01 | Genzyme Corporation | GITR Antibodies For The Treatment of Cancer |
| BRPI0707824A2 (pt) | 2006-02-15 | 2011-05-10 | Imclone Systems Inc | proteÍna de ligaÇço a antÍgeno, e, mÉtodos de neutralizaÇço da ativaÇço de um receptor de tirosina quinase, de inibiÇço de angiogÊnese, de reduÇço de crescimento de tumor e de produÇço de uma proteÍna de ligaÇço a antÍgeno |
| KR101516823B1 (ko) | 2006-03-17 | 2015-05-07 | 바이오겐 아이덱 엠에이 인코포레이티드 | 안정화된 폴리펩티드 조성물 |
| EP1996716B1 (en) | 2006-03-20 | 2011-05-11 | The Regents of the University of California | Engineered anti-prostate stem cell antigen (psca) antibodies for cancer targeting |
| PT1999154E (pt) | 2006-03-24 | 2013-01-24 | Merck Patent Gmbh | Domínios proteicos heterodiméricos modificados |
| US8946391B2 (en) | 2006-03-24 | 2015-02-03 | The Regents Of The University Of California | Construction of a multivalent scFv through alkyne-azide 1,3-dipolar cycloaddition |
| US8603466B2 (en) | 2006-03-29 | 2013-12-10 | King's College London | Agonist antibodies against TSHR |
| EP4218801A3 (en) | 2006-03-31 | 2023-08-23 | Chugai Seiyaku Kabushiki Kaisha | Antibody modification method for purifying bispecific antibody |
| TWI395754B (zh) | 2006-04-24 | 2013-05-11 | Amgen Inc | 人類化之c-kit抗體 |
| JP5189082B2 (ja) | 2006-05-25 | 2013-04-24 | バイエル・ファルマ・アクチェンゲゼルシャフト | 二量体分子複合体 |
| US20070274985A1 (en) | 2006-05-26 | 2007-11-29 | Stefan Dubel | Antibody |
| MX2008015524A (es) | 2006-06-12 | 2009-01-13 | Trubion Pharmaceuticals Inc | Proteinas de union multivalentes monocatenarias con funcion efectora. |
| WO2008022349A2 (en) | 2006-08-18 | 2008-02-21 | Armagen Technologies, Inc. | Agents for blood-brain barrier delivery |
| WO2008027236A2 (en) | 2006-08-30 | 2008-03-06 | Genentech, Inc. | Multispecific antibodies |
| CA2977261A1 (en) | 2006-10-04 | 2008-04-10 | Kobenhavns Universitet | Generation of a cancer-specific immune response toward muc1 and cancer specific muc1 antibodies |
| FR2906808B1 (fr) | 2006-10-10 | 2012-10-05 | Univ Nantes | Utilisation d'anticorps monoclonaux specifiques de la forme o-acetylee du ganglioside gd2 dans le traitement de certains cancers |
| NZ576445A (en) | 2006-11-02 | 2012-03-30 | Daniel J Capon | Hybrid immunoglobulins with moving parts |
| WO2008101234A2 (en) | 2007-02-16 | 2008-08-21 | Sloan-Kettering Institute For Cancer Research | Anti ganglioside gd3 antibodies and uses thereof |
| US7635753B2 (en) | 2007-02-19 | 2009-12-22 | Wisconsin Alumni Research Foundation | Prostate cancer and melanoma antigens |
| CN101802015B (zh) | 2007-03-29 | 2015-05-06 | 根马布股份公司 | 双特异性抗体及其制造方法 |
| EP2514766A3 (en) | 2007-03-29 | 2013-06-05 | Technion Research & Development Foundation Ltd. | Antibodies, methods and kits for diagnosing and treating melanoma |
| CA2682605A1 (en) | 2007-04-18 | 2008-10-30 | Zymogenetics, Inc. | Single chain fc, methods of making and methods of treatment |
| JP2010190572A (ja) | 2007-06-01 | 2010-09-02 | Sapporo Medical Univ | IL13Ra2に対する抗体およびこれを含む診断・治療薬 |
| CN104945508B (zh) | 2007-06-18 | 2019-02-22 | 默沙东有限责任公司 | 针对人程序性死亡受体pd-1的抗体 |
| NZ703668A (en) | 2007-06-27 | 2016-07-29 | Us Sec Dep Of Health And Human Services | Complexes of il-15 and il-15ralpha and uses thereof |
| CA2693677C (en) | 2007-07-12 | 2018-02-13 | Tolerx, Inc. | Combination therapies employing gitr binding molecules |
| JP2010535032A (ja) | 2007-07-31 | 2010-11-18 | メディミューン,エルエルシー | 多重特異性エピトープ結合性タンパク質およびその用途 |
| CA2694990A1 (en) | 2007-07-31 | 2009-02-05 | Merck Sharp & Dohme Corp. | Igf-1r specific antibodies useful in the detection and diagnosis of cellular proliferative disorders |
| ES2628395T3 (es) | 2007-08-15 | 2017-08-02 | Bayer Pharma Aktiengesellschaft | Anticuerpo regulado por proteasa |
| JP2011500730A (ja) | 2007-10-26 | 2011-01-06 | ガバニング カウンセル オブ ザ ユニバーシティ オブ トロント | Tim−3を用いた治療および診断方法 |
| EP2215125A1 (en) | 2007-11-27 | 2010-08-11 | Ablynx N.V. | Method for obtaining polypeptide constructs comprising two or more single domain antibodies |
| US20110008345A1 (en) | 2007-11-30 | 2011-01-13 | Claire Ashman | Antigen-binding constructs |
| US8227577B2 (en) | 2007-12-21 | 2012-07-24 | Hoffman-La Roche Inc. | Bivalent, bispecific antibodies |
| US20090162359A1 (en) | 2007-12-21 | 2009-06-25 | Christian Klein | Bivalent, bispecific antibodies |
| US8242247B2 (en) | 2007-12-21 | 2012-08-14 | Hoffmann-La Roche Inc. | Bivalent, bispecific antibodies |
| US9266967B2 (en) | 2007-12-21 | 2016-02-23 | Hoffmann-La Roche, Inc. | Bivalent, bispecific antibodies |
| MX350962B (es) | 2008-01-07 | 2017-09-27 | Amgen Inc | Metodo para fabricar moleculas heterodimericas de fragmentos cristalizables de anticuerpo, utilizando efectos electrostaticos de direccion. |
| WO2009091826A2 (en) * | 2008-01-14 | 2009-07-23 | The Board Of Regents Of The University Of Texas System | Compositions and methods related to a human cd19-specific chimeric antigen receptor (h-car) |
| CN104548091A (zh) | 2008-02-11 | 2015-04-29 | 治疗科技公司 | 用于肿瘤治疗的单克隆抗体 |
| CA2715181A1 (en) | 2008-02-21 | 2009-08-27 | Astrazeneca Ab | Combination therapy 238 |
| US8168757B2 (en) | 2008-03-12 | 2012-05-01 | Merck Sharp & Dohme Corp. | PD-1 binding proteins |
| AR071891A1 (es) | 2008-05-30 | 2010-07-21 | Imclone Llc | Anticuerpos humanos anti-flt3 (receptor tirosina cinasa 3 tipo fms humano) |
| NZ590667A (en) | 2008-07-02 | 2013-01-25 | Emergent Product Dev Seattle | Tgf-b antagonist multi-target binding proteins |
| AR072999A1 (es) | 2008-08-11 | 2010-10-06 | Medarex Inc | Anticuerpos humanos que se unen al gen 3 de activacion linfocitaria (lag-3) y los usos de estos |
| BRPI0917891A2 (pt) | 2008-08-25 | 2015-11-24 | Amplimmune Inc | antagonistas de pd-1 e métodos de utilização dos mesmos |
| EP2350129B1 (en) | 2008-08-25 | 2015-06-10 | Amplimmune, Inc. | Compositions of pd-1 antagonists and methods of use |
| WO2010030002A1 (ja) | 2008-09-12 | 2010-03-18 | 国立大学法人三重大学 | 外来性gitrリガンド発現細胞 |
| CA2737758C (en) | 2008-09-19 | 2017-10-31 | Soldano Ferrone | Monoclonal antibodies for cspg4 for the diagnosis and treatment of basal breast carcinoma |
| US8476282B2 (en) | 2008-11-03 | 2013-07-02 | Intellikine Llc | Benzoxazole kinase inhibitors and methods of use |
| LT4209510T (lt) | 2008-12-09 | 2024-03-12 | F. Hoffmann-La Roche Ag | Anti-pd-l1 antikūnai ir jų panaudojimas t ląstelių funkcijos pagerinimui |
| EP2393835B1 (en) | 2009-02-09 | 2017-04-05 | Université d'Aix-Marseille | Pd-1 antibodies and pd-l1 antibodies and uses thereof |
| CA2950529C (en) | 2009-04-03 | 2019-04-30 | Verastem, Inc. | Pyrimidine substituted purine compounds as kinase (s) inhibitors |
| CA2759233C (en) | 2009-04-27 | 2019-07-16 | Oncomed Pharmaceuticals, Inc. | Method for making heteromultimeric molecules |
| ES2668874T3 (es) | 2009-04-30 | 2018-05-22 | Tel Hashomer Medical Research Infrastructure And Services Ltd. | Anticuerpos anti-CEACAM1 y métodos de uso de los mismos |
| EP3135294B1 (en) | 2009-08-14 | 2020-06-03 | The Government of the United States of America as represented by the Secretary of the Department of Health and Human Services | Use of il-15-il-15 receptor heterodimers to treat lymphopenia |
| CN102574924A (zh) | 2009-09-03 | 2012-07-11 | 先灵公司 | 抗-gitr抗体 |
| WO2011059836A2 (en) | 2009-10-29 | 2011-05-19 | Trustees Of Dartmouth College | T cell receptor-deficient t cell compositions |
| GB0919054D0 (en) | 2009-10-30 | 2009-12-16 | Isis Innovation | Treatment of obesity |
| US8956828B2 (en) | 2009-11-10 | 2015-02-17 | Sangamo Biosciences, Inc. | Targeted disruption of T cell receptor genes using engineered zinc finger protein nucleases |
| EP2504028A4 (en) | 2009-11-24 | 2014-04-09 | Amplimmune Inc | SIMULTANEOUS INHIBITION OF PD-L1 / PD-L2 |
| CN102753194B (zh) | 2009-12-02 | 2015-07-08 | 伊麦吉纳博公司 | 靶向人前列腺特异性膜抗原的j591微抗体和双抗体 |
| EA027502B1 (ru) | 2009-12-23 | 2017-08-31 | Зиниммуне Гмбх | Антитела против flt3 и способы их применения |
| WO2011090762A1 (en) | 2009-12-29 | 2011-07-28 | Emergent Product Development Seattle, Llc | Heterodimer binding proteins and uses thereof |
| IL290617B2 (en) | 2010-02-24 | 2023-11-01 | Immunogen Inc | Folate receptor 1 antibodies and immunoconjugates and uses thereof |
| SG10201800757TA (en) | 2010-04-20 | 2018-02-27 | Genmab As | Heterodimeric antibody fc-containing proteins and methods for production thereof |
| US9242014B2 (en) | 2010-06-15 | 2016-01-26 | The Regents Of The University Of California | Receptor tyrosine kinase-like orphan receptor 1 (ROR1) single chain Fv antibody fragment conjugates and methods of use thereof |
| CA2802344C (en) | 2010-06-18 | 2023-06-13 | The Brigham And Women's Hospital, Inc. | Bi-specific antibodies against tim-3 and pd-1 for immunotherapy in chronic immune conditions |
| ES2961381T3 (es) | 2010-06-19 | 2024-03-11 | Memorial Sloan Kettering Cancer Center | Anticuerpos anti-GD2 |
| AR084469A1 (es) | 2010-07-09 | 2013-05-22 | Exelixis Inc | Combinaciones de inhibidores de quinasas para el tratamiento del cancer |
| BR112013001088A2 (pt) | 2010-07-16 | 2016-05-24 | Piramal Entpr Ltd | "derivados substituídos como inibidores a quinase de imidazoquinolina" |
| CA2993567C (en) | 2010-07-21 | 2022-06-28 | Sangamo Biosciences, Inc. | Methods and compositions for modification of a t-cell receptor gene |
| WO2012033885A1 (en) | 2010-09-08 | 2012-03-15 | Baylor College Of Medicine | Immunotherapy of cancer using genetically engineered gd2-specific t cells |
| JP5947311B2 (ja) | 2010-12-09 | 2016-07-06 | ザ トラスティーズ オブ ザ ユニバーシティ オブ ペンシルバニア | 癌を治療するためのキメラ抗原受容体改変t細胞の使用 |
| JOP20210044A1 (ar) | 2010-12-30 | 2017-06-16 | Takeda Pharmaceuticals Co | الأجسام المضادة لـ cd38 |
| SG193956A1 (en) | 2011-04-01 | 2013-11-29 | Sloan Kettering Inst Cancer | T cell receptor-like antibodies specific for a wt1 peptide presented by hla-a2 |
| PL2694549T3 (pl) | 2011-04-08 | 2019-01-31 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Chimeryczne receptory antygenowe anty-wariant III receptora czynnika wzrostu naskórka i ich zastosowanie do leczenia raka |
| US20130071414A1 (en) * | 2011-04-27 | 2013-03-21 | Gianpietro Dotti | Engineered cd19-specific t lymphocytes that coexpress il-15 and an inducible caspase-9 based suicide gene for the treatment of b-cell malignancies |
| AR086044A1 (es) | 2011-05-12 | 2013-11-13 | Imclone Llc | Anticuerpos que se unen especificamente a un dominio extracelular de c-kit y usos de los mismos |
| DK3415531T3 (da) | 2011-05-27 | 2023-09-18 | Glaxo Group Ltd | Bcma (cd269/tnfrsf17)-bindende proteiner |
| WO2013006490A2 (en) | 2011-07-01 | 2013-01-10 | Cellerant Therapeutics, Inc. | Antibodies that specifically bind to tim3 |
| RU2636588C2 (ru) | 2011-08-11 | 2017-11-24 | ИНТЕЛЛАЙКИН, ЭлЭлСи | Полиморфы ингибитора киназы |
| US20130108641A1 (en) | 2011-09-14 | 2013-05-02 | Sanofi | Anti-gitr antibodies |
| WO2013040371A2 (en) | 2011-09-16 | 2013-03-21 | Baylor College Of Medicine | Targeting the tumor microenvironment using manipulated nkt cells |
| WO2013054320A1 (en) | 2011-10-11 | 2013-04-18 | Tel Hashomer Medical Research Infrastructure And Services Ltd. | Antibodies to carcinoembryonic antigen-related cell adhesion molecule (ceacam) |
| ITMO20110270A1 (it) | 2011-10-25 | 2013-04-26 | Sara Caldrer | Una cellula effettrice modificata per il trattamento di neoplasie esprimenti il disialonganglioside gd2 |
| AU2012328322A1 (en) | 2011-10-27 | 2014-06-12 | Genmab A/S | Production of heterodimeric proteins |
| WO2013074916A1 (en) | 2011-11-18 | 2013-05-23 | Board Of Regents, The University Of Texas System | Car+ t cells genetically modified to eliminate expression of t- cell receptor and/or hla |
| CA2892371C (en) | 2011-12-01 | 2021-01-19 | The Brigham And Women's Hospital, Inc. | Anti-ceacam1 recombinant antibodies for cancer therapy |
| US9439768B2 (en) | 2011-12-08 | 2016-09-13 | Imds Llc | Glenoid vault fixation |
| CN104185681A (zh) * | 2012-02-01 | 2014-12-03 | 卡姆普根有限公司 | C1orf32抗体及其用于治疗癌症的用途 |
| EP2814846B1 (en) | 2012-02-13 | 2020-01-08 | Seattle Children's Hospital d/b/a Seattle Children's Research Institute | Bispecific chimeric antigen receptors and therapeutic uses thereof |
| MX2014010183A (es) | 2012-02-22 | 2015-03-20 | Univ Pennsylvania | Composiciones y metodos para generar una poblacion persistente de celulas t utiles para el tratamiento de cancer. |
| AR090903A1 (es) | 2012-05-01 | 2014-12-17 | Genentech Inc | Anticuerpos e inmunoconjugados anti-pmel17 |
| LT2800811T (lt) | 2012-05-25 | 2017-09-11 | The Regents Of The University Of California | Būdai ir kompozicijos, skirti tikslinės dnr modifikavimui, panaudojant adresuotą rnr, ir transkripcijos moduliavimui, panaudojant adresuotą rnr |
| WO2013192294A1 (en) | 2012-06-20 | 2013-12-27 | Boston 3T Biotechnologies, Inc. | Cellular therapies for treating and preventing cancers and other immune system disorders |
| EP2879709B1 (en) | 2012-07-31 | 2020-01-08 | The Brigham and Women's Hospital, Inc. | Modulation of the immune response |
| WO2014055442A2 (en) | 2012-10-01 | 2014-04-10 | The Trustees Of The University Of Pennsylvania | Compositions and methods for targeting stromal cells for the treatment of cancer |
| US10117896B2 (en) | 2012-10-05 | 2018-11-06 | The Trustees Of The University Of Pennsylvania | Use of a trans-signaling approach in chimeric antigen receptors |
| CA2887528C (en) | 2012-10-12 | 2023-08-29 | The Brigham And Women's Hospital, Inc. | Enhancement of the immune response |
| KR20150105634A (ko) | 2012-12-12 | 2015-09-17 | 더 브로드 인스티튜트, 인코퍼레이티드 | 서열 조작을 위한 개선된 시스템, 방법 및 효소 조성물의 유전자 조작 및 최적화 |
| EP3825401A1 (en) | 2012-12-12 | 2021-05-26 | The Broad Institute, Inc. | Crispr-cas component systems, methods and compositions for sequence manipulation |
| US8697359B1 (en) | 2012-12-12 | 2014-04-15 | The Broad Institute, Inc. | CRISPR-Cas systems and methods for altering expression of gene products |
| WO2014110591A1 (en) | 2013-01-14 | 2014-07-17 | Fred Hutchinson Cancer Research Center | Compositions and methods for delivery of immune cells to treat un-resectable or non-resected tumor cells and tumor relapse |
| EP3623380A1 (en) * | 2013-03-15 | 2020-03-18 | Michael C. Milone | Targeting cytotoxic cells with chimeric receptors for adoptive immunotherapy |
| ES2939760T3 (es) | 2014-03-15 | 2023-04-26 | Novartis Ag | Tratamiento de cáncer utilizando un receptor quimérico para antígenos |
-
2015
- 2015-03-13 ES ES15712503T patent/ES2939760T3/es active Active
- 2015-03-13 US US15/126,036 patent/US20170335281A1/en not_active Abandoned
- 2015-03-13 EP EP15712503.0A patent/EP3119423B1/en active Active
- 2015-03-13 WO PCT/US2015/020606 patent/WO2015142675A2/en active Application Filing
- 2015-03-13 EP EP19169356.3A patent/EP3593812A3/en active Pending
- 2015-03-13 JP JP2016557287A patent/JP2017513818A/ja active Pending
- 2015-03-13 CN CN201580013987.3A patent/CN106163547A/zh active Pending
-
2019
- 2019-07-05 JP JP2019126002A patent/JP6860623B2/ja active Active
- 2019-10-11 US US16/599,988 patent/US20200283729A1/en active Pending
-
2021
- 2021-03-26 JP JP2021053240A patent/JP2021107395A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| US20200283729A1 (en) | 2020-09-10 |
| EP3593812A3 (en) | 2020-05-27 |
| JP2021107395A (ja) | 2021-07-29 |
| JP2019206538A (ja) | 2019-12-05 |
| WO2015142675A3 (en) | 2015-11-12 |
| WO2015142675A8 (en) | 2016-10-20 |
| CN106163547A (zh) | 2016-11-23 |
| EP3593812A2 (en) | 2020-01-15 |
| JP2017513818A (ja) | 2017-06-01 |
| ES2939760T3 (es) | 2023-04-26 |
| EP3119423B1 (en) | 2022-12-14 |
| US20170335281A1 (en) | 2017-11-23 |
| WO2015142675A2 (en) | 2015-09-24 |
| EP3119423A2 (en) | 2017-01-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6860623B2 (ja) | キメラ抗原受容体を使用する癌の処置 | |
| US20230312677A1 (en) | Cd28 compositions and methods for chimeric antigen receptor therapy | |
| US20230139800A1 (en) | Car t cell therapies with enhanced efficacy | |
| JP7082055B2 (ja) | 抗癌治療における組み合わせ使用のためのメソテリンキメラ抗原受容体(car)およびpd-l1阻害剤に対する抗体 | |
| AU2015301460B2 (en) | Treatment of cancer using GFR alpha-4 chimeric antigen receptor | |
| AU2015292755B2 (en) | Treatment of cancer using a CD33 chimeric antigen receptor | |
| US20180298068A1 (en) | Treatment of cancer using chimeric antigen receptor and protein kinase a blocker | |
| JP2023082071A (ja) | 増強された有効性を有するバイオマーカー及びcar t細胞療法 | |
| WO2018160731A1 (en) | Shp inhibitor compositions and uses for chimeric antigen receptor therapy | |
| JP2019513347A (ja) | 複数のキメラ抗原受容体(car)分子を発現する細胞およびその使用 | |
| US20180092968A1 (en) | Compositions to disrupt protein kinase a anchoring and uses thereof | |
| US20210179709A1 (en) | Anti-car compositions and methods | |
| US20200390811A1 (en) | Compositions to disrupt protein kinase a anchoring and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190731 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190731 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190802 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200519 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200814 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20210126 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20210224 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20210326 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6860623 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |