ES2939760T3 - Tratamiento de cáncer utilizando un receptor quimérico para antígenos - Google Patents
Tratamiento de cáncer utilizando un receptor quimérico para antígenos Download PDFInfo
- Publication number
- ES2939760T3 ES2939760T3 ES15712503T ES15712503T ES2939760T3 ES 2939760 T3 ES2939760 T3 ES 2939760T3 ES 15712503 T ES15712503 T ES 15712503T ES 15712503 T ES15712503 T ES 15712503T ES 2939760 T3 ES2939760 T3 ES 2939760T3
- Authority
- ES
- Spain
- Prior art keywords
- antigen
- cell
- car
- domain
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 246
- 108091007433 antigens Proteins 0.000 title claims abstract description 246
- 102000036639 antigens Human genes 0.000 title claims abstract description 246
- 239000000427 antigen Substances 0.000 title claims abstract description 242
- 201000011510 cancer Diseases 0.000 title claims abstract description 69
- 238000011282 treatment Methods 0.000 title description 39
- 108700010039 chimeric receptor Proteins 0.000 title description 3
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 claims abstract description 354
- 210000001744 T-lymphocyte Anatomy 0.000 claims abstract description 161
- 230000027455 binding Effects 0.000 claims abstract description 129
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 57
- 201000010099 disease Diseases 0.000 claims abstract description 53
- 239000013598 vector Substances 0.000 claims abstract description 34
- 210000004027 cell Anatomy 0.000 claims description 418
- 230000011664 signaling Effects 0.000 claims description 128
- -1 G 2 ganglioside Chemical class 0.000 claims description 121
- 239000012642 immune effector Substances 0.000 claims description 84
- 229940121354 immunomodulator Drugs 0.000 claims description 84
- 230000004068 intracellular signaling Effects 0.000 claims description 72
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 72
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 71
- 230000000139 costimulatory effect Effects 0.000 claims description 70
- 229920001184 polypeptide Polymers 0.000 claims description 69
- 108090000623 proteins and genes Proteins 0.000 claims description 60
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 claims description 58
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 claims description 58
- 150000007523 nucleic acids Chemical class 0.000 claims description 54
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 42
- 102000039446 nucleic acids Human genes 0.000 claims description 40
- 108020004707 nucleic acids Proteins 0.000 claims description 40
- 239000003112 inhibitor Substances 0.000 claims description 39
- 102100024263 CD160 antigen Human genes 0.000 claims description 34
- 101000761938 Homo sapiens CD160 antigen Proteins 0.000 claims description 34
- 241000282414 Homo sapiens Species 0.000 claims description 29
- 102000004169 proteins and genes Human genes 0.000 claims description 28
- 102000001301 EGF receptor Human genes 0.000 claims description 26
- 108060006698 EGF receptor Proteins 0.000 claims description 26
- 102100029215 Signaling lymphocytic activation molecule Human genes 0.000 claims description 26
- 230000003834 intracellular effect Effects 0.000 claims description 26
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 claims description 24
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 claims description 24
- 210000000822 natural killer cell Anatomy 0.000 claims description 23
- 230000004048 modification Effects 0.000 claims description 22
- 238000012986 modification Methods 0.000 claims description 22
- 102100027207 CD27 antigen Human genes 0.000 claims description 21
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 claims description 21
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 claims description 21
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 claims description 21
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 claims description 21
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 claims description 21
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 claims description 19
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 claims description 19
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 claims description 18
- 102100038083 Endosialin Human genes 0.000 claims description 18
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 claims description 18
- 101000994375 Homo sapiens Integrin alpha-4 Proteins 0.000 claims description 18
- 101000935040 Homo sapiens Integrin beta-2 Proteins 0.000 claims description 18
- 102100032818 Integrin alpha-4 Human genes 0.000 claims description 18
- 102100032816 Integrin alpha-6 Human genes 0.000 claims description 18
- 102100025390 Integrin beta-2 Human genes 0.000 claims description 18
- 102100029197 SLAM family member 6 Human genes 0.000 claims description 18
- 101001078158 Homo sapiens Integrin alpha-1 Proteins 0.000 claims description 17
- 101000633786 Homo sapiens SLAM family member 6 Proteins 0.000 claims description 17
- 102100025323 Integrin alpha-1 Human genes 0.000 claims description 17
- 101000884275 Homo sapiens Endosialin Proteins 0.000 claims description 16
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 16
- 108010074687 Signaling Lymphocytic Activation Molecule Family Member 1 Proteins 0.000 claims description 16
- 101000633784 Homo sapiens SLAM family member 7 Proteins 0.000 claims description 15
- 102100022338 Integrin alpha-M Human genes 0.000 claims description 15
- 102100038082 Natural killer cell receptor 2B4 Human genes 0.000 claims description 15
- 102100029198 SLAM family member 7 Human genes 0.000 claims description 15
- 102100027744 Semaphorin-4D Human genes 0.000 claims description 15
- 102100029822 B- and T-lymphocyte attenuator Human genes 0.000 claims description 14
- 102100026094 C-type lectin domain family 12 member A Human genes 0.000 claims description 14
- 101710188619 C-type lectin domain family 12 member A Proteins 0.000 claims description 14
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 claims description 14
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 claims description 14
- 101000864344 Homo sapiens B- and T-lymphocyte attenuator Proteins 0.000 claims description 14
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 claims description 14
- 101001035237 Homo sapiens Integrin alpha-D Proteins 0.000 claims description 14
- 101001046687 Homo sapiens Integrin alpha-E Proteins 0.000 claims description 14
- 101000935043 Homo sapiens Integrin beta-1 Proteins 0.000 claims description 14
- 101000971538 Homo sapiens Killer cell lectin-like receptor subfamily F member 1 Proteins 0.000 claims description 14
- 101000851370 Homo sapiens Tumor necrosis factor receptor superfamily member 9 Proteins 0.000 claims description 14
- 102000017182 Ikaros Transcription Factor Human genes 0.000 claims description 14
- 108010013958 Ikaros Transcription Factor Proteins 0.000 claims description 14
- 108060003951 Immunoglobulin Proteins 0.000 claims description 14
- 102100039904 Integrin alpha-D Human genes 0.000 claims description 14
- 102100022341 Integrin alpha-E Human genes 0.000 claims description 14
- 102100022297 Integrin alpha-X Human genes 0.000 claims description 14
- 102100025304 Integrin beta-1 Human genes 0.000 claims description 14
- 102100021458 Killer cell lectin-like receptor subfamily F member 1 Human genes 0.000 claims description 14
- 108091008874 T cell receptors Proteins 0.000 claims description 14
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 claims description 14
- 108090000253 Thyrotropin Receptors Proteins 0.000 claims description 14
- 102100028785 Tumor necrosis factor receptor superfamily member 14 Human genes 0.000 claims description 14
- 102100036856 Tumor necrosis factor receptor superfamily member 9 Human genes 0.000 claims description 14
- 102000018358 immunoglobulin Human genes 0.000 claims description 14
- 102100033793 ALK tyrosine kinase receptor Human genes 0.000 claims description 13
- 101710168331 ALK tyrosine kinase receptor Proteins 0.000 claims description 13
- 108090000229 Claudin-6 Proteins 0.000 claims description 13
- 102100036939 G-protein coupled receptor 20 Human genes 0.000 claims description 13
- 101710108873 G-protein coupled receptor 20 Proteins 0.000 claims description 13
- 101000994365 Homo sapiens Integrin alpha-6 Proteins 0.000 claims description 13
- 101000831007 Homo sapiens T-cell immunoreceptor with Ig and ITIM domains Proteins 0.000 claims description 13
- 101000655352 Homo sapiens Telomerase reverse transcriptase Proteins 0.000 claims description 13
- 102100022339 Integrin alpha-L Human genes 0.000 claims description 13
- 102100025128 Olfactory receptor 51E2 Human genes 0.000 claims description 13
- 101710187841 Olfactory receptor 51E2 Proteins 0.000 claims description 13
- 102100032364 Pannexin-3 Human genes 0.000 claims description 13
- 101710165197 Pannexin-3 Proteins 0.000 claims description 13
- 102100024834 T-cell immunoreceptor with Ig and ITIM domains Human genes 0.000 claims description 13
- 102000013532 Uroplakin II Human genes 0.000 claims description 13
- 108010065940 Uroplakin II Proteins 0.000 claims description 13
- 102100026181 Placenta-specific protein 1 Human genes 0.000 claims description 12
- 108050005093 Placenta-specific protein 1 Proteins 0.000 claims description 12
- 102100036494 Testisin Human genes 0.000 claims description 12
- 102100022153 Tumor necrosis factor receptor superfamily member 4 Human genes 0.000 claims description 12
- 239000003814 drug Substances 0.000 claims description 12
- 102000017918 ADRB3 Human genes 0.000 claims description 11
- 108060003355 ADRB3 Proteins 0.000 claims description 11
- 102100026423 Adhesion G protein-coupled receptor E5 Human genes 0.000 claims description 11
- 108010051118 Bone Marrow Stromal Antigen 2 Proteins 0.000 claims description 11
- 102100037086 Bone marrow stromal antigen 2 Human genes 0.000 claims description 11
- 102100023458 C-type lectin-like domain family 1 Human genes 0.000 claims description 11
- 102000010956 Glypican Human genes 0.000 claims description 11
- 108050001154 Glypican Proteins 0.000 claims description 11
- 108050007237 Glypican-3 Proteins 0.000 claims description 11
- 101000718243 Homo sapiens Adhesion G protein-coupled receptor E5 Proteins 0.000 claims description 11
- 101000906643 Homo sapiens C-type lectin-like domain family 1 Proteins 0.000 claims description 11
- 101000878602 Homo sapiens Immunoglobulin alpha Fc receptor Proteins 0.000 claims description 11
- 101001065550 Homo sapiens Lymphocyte antigen 6K Proteins 0.000 claims description 11
- 101000666896 Homo sapiens V-type immunoglobulin domain-containing suppressor of T-cell activation Proteins 0.000 claims description 11
- 102100038005 Immunoglobulin alpha Fc receptor Human genes 0.000 claims description 11
- 102100029616 Immunoglobulin lambda-like polypeptide 1 Human genes 0.000 claims description 11
- 101710107067 Immunoglobulin lambda-like polypeptide 1 Proteins 0.000 claims description 11
- 102100032129 Lymphocyte antigen 6K Human genes 0.000 claims description 11
- 102000003735 Mesothelin Human genes 0.000 claims description 11
- 108090000015 Mesothelin Proteins 0.000 claims description 11
- 108091000080 Phosphotransferase Proteins 0.000 claims description 11
- 206010039491 Sarcoma Diseases 0.000 claims description 11
- 102100038282 V-type immunoglobulin domain-containing suppressor of T-cell activation Human genes 0.000 claims description 11
- 239000002253 acid Substances 0.000 claims description 11
- IJJVMEJXYNJXOJ-UHFFFAOYSA-N fluquinconazole Chemical compound C=1C=C(Cl)C=C(Cl)C=1N1C(=O)C2=CC(F)=CC=C2N=C1N1C=NC=N1 IJJVMEJXYNJXOJ-UHFFFAOYSA-N 0.000 claims description 11
- 102000020233 phosphotransferase Human genes 0.000 claims description 11
- 102000005962 receptors Human genes 0.000 claims description 11
- 108020003175 receptors Proteins 0.000 claims description 11
- LTHJXDSHSVNJKG-UHFFFAOYSA-N 2-[2-[2-[2-(2-methylprop-2-enoyloxy)ethoxy]ethoxy]ethoxy]ethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCOCCOCCOCCOC(=O)C(C)=C LTHJXDSHSVNJKG-UHFFFAOYSA-N 0.000 claims description 10
- 102100037982 Alpha-1,6-mannosylglycoprotein 6-beta-N-acetylglucosaminyltransferase A Human genes 0.000 claims description 10
- 102100023003 Ankyrin repeat domain-containing protein 30A Human genes 0.000 claims description 10
- 108700012439 CA9 Proteins 0.000 claims description 10
- 102100038078 CD276 antigen Human genes 0.000 claims description 10
- 102100024423 Carbonic anhydrase 9 Human genes 0.000 claims description 10
- 102000012466 Cytochrome P450 1B1 Human genes 0.000 claims description 10
- 108050002014 Cytochrome P450 1B1 Proteins 0.000 claims description 10
- 102100039554 Galectin-8 Human genes 0.000 claims description 10
- 101000757191 Homo sapiens Ankyrin repeat domain-containing protein 30A Proteins 0.000 claims description 10
- 101000608769 Homo sapiens Galectin-8 Proteins 0.000 claims description 10
- 101001043809 Homo sapiens Interleukin-7 receptor subunit alpha Proteins 0.000 claims description 10
- 101000633780 Homo sapiens Signaling lymphocytic activation molecule Proteins 0.000 claims description 10
- 101000824971 Homo sapiens Sperm surface protein Sp17 Proteins 0.000 claims description 10
- 101000814512 Homo sapiens X antigen family member 1 Proteins 0.000 claims description 10
- 102100021593 Interleukin-7 receptor subunit alpha Human genes 0.000 claims description 10
- 108010069196 Neural Cell Adhesion Molecules Proteins 0.000 claims description 10
- 102100023616 Neural cell adhesion molecule L1-like protein Human genes 0.000 claims description 10
- 102100036735 Prostate stem cell antigen Human genes 0.000 claims description 10
- 101710120463 Prostate stem cell antigen Proteins 0.000 claims description 10
- 102100038098 Protein-glutamine gamma-glutamyltransferase 5 Human genes 0.000 claims description 10
- 102100027610 Rho-related GTP-binding protein RhoC Human genes 0.000 claims description 10
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 claims description 10
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 claims description 10
- 102100022748 Wilms tumor protein Human genes 0.000 claims description 10
- 101710127857 Wilms tumor protein Proteins 0.000 claims description 10
- 102100039490 X antigen family member 1 Human genes 0.000 claims description 10
- 108010034034 alpha-1,6-mannosylglycoprotein beta 1,6-N-acetylglucosaminyltransferase Proteins 0.000 claims description 10
- 201000001441 melanoma Diseases 0.000 claims description 10
- 108010073531 rhoC GTP-Binding Protein Proteins 0.000 claims description 10
- 108010058721 transglutaminase 5 Proteins 0.000 claims description 10
- 230000005945 translocation Effects 0.000 claims description 10
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 claims description 9
- 102100026402 Adhesion G protein-coupled receptor E2 Human genes 0.000 claims description 9
- 102100025218 B-cell differentiation antigen CD72 Human genes 0.000 claims description 9
- 102100031507 Fc receptor-like protein 5 Human genes 0.000 claims description 9
- 101150032879 Fcrl5 gene Proteins 0.000 claims description 9
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 claims description 9
- 101000718211 Homo sapiens Adhesion G protein-coupled receptor E2 Proteins 0.000 claims description 9
- 101000934359 Homo sapiens B-cell differentiation antigen CD72 Proteins 0.000 claims description 9
- 101001018034 Homo sapiens Lymphocyte antigen 75 Proteins 0.000 claims description 9
- 101000932478 Homo sapiens Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 claims description 9
- 102100033486 Lymphocyte antigen 75 Human genes 0.000 claims description 9
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 claims description 9
- 101710164680 Platelet-derived growth factor receptor beta Proteins 0.000 claims description 9
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 claims description 9
- 102100021393 Transcriptional repressor CTCFL Human genes 0.000 claims description 9
- 101710165473 Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 claims description 9
- 239000003446 ligand Substances 0.000 claims description 9
- 102100032187 Androgen receptor Human genes 0.000 claims description 8
- 102100038080 B-cell receptor CD22 Human genes 0.000 claims description 8
- 108010056102 CD100 antigen Proteins 0.000 claims description 8
- 108010058905 CD44v6 antigen Proteins 0.000 claims description 8
- 102100035167 Coiled-coil domain-containing protein 54 Human genes 0.000 claims description 8
- 108010060385 Cyclin B1 Proteins 0.000 claims description 8
- 101100481408 Danio rerio tie2 gene Proteins 0.000 claims description 8
- 102000003817 Fos-related antigen 1 Human genes 0.000 claims description 8
- 108090000123 Fos-related antigen 1 Proteins 0.000 claims description 8
- 102100032340 G2/mitotic-specific cyclin-B1 Human genes 0.000 claims description 8
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 claims description 8
- 101000737052 Homo sapiens Coiled-coil domain-containing protein 54 Proteins 0.000 claims description 8
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 claims description 8
- 101001051490 Homo sapiens Neural cell adhesion molecule L1 Proteins 0.000 claims description 8
- 101000873418 Homo sapiens P-selectin glycoprotein ligand 1 Proteins 0.000 claims description 8
- 101001136981 Homo sapiens Proteasome subunit beta type-9 Proteins 0.000 claims description 8
- 101000884271 Homo sapiens Signal transducer CD24 Proteins 0.000 claims description 8
- 101000714168 Homo sapiens Testisin Proteins 0.000 claims description 8
- 101000894428 Homo sapiens Transcriptional repressor CTCFL Proteins 0.000 claims description 8
- 101000638154 Homo sapiens Transmembrane protease serine 2 Proteins 0.000 claims description 8
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 claims description 8
- 101100481410 Mus musculus Tek gene Proteins 0.000 claims description 8
- 102100024964 Neural cell adhesion molecule L1 Human genes 0.000 claims description 8
- 102100034925 P-selectin glycoprotein ligand 1 Human genes 0.000 claims description 8
- 102100035764 Proteasome subunit beta type-9 Human genes 0.000 claims description 8
- 108010025832 RANK Ligand Proteins 0.000 claims description 8
- 102100038081 Signal transducer CD24 Human genes 0.000 claims description 8
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 claims description 8
- 230000004913 activation Effects 0.000 claims description 8
- 108010080146 androgen receptors Proteins 0.000 claims description 8
- 230000002950 deficient Effects 0.000 claims description 8
- 230000006870 function Effects 0.000 claims description 8
- 230000004927 fusion Effects 0.000 claims description 8
- 230000002381 testicular Effects 0.000 claims description 8
- 108010017009 CD11b Antigen Proteins 0.000 claims description 7
- 102100038077 CD226 antigen Human genes 0.000 claims description 7
- 108010062802 CD66 antigens Proteins 0.000 claims description 7
- 102100027217 CD82 antigen Human genes 0.000 claims description 7
- 101710139831 CD82 antigen Proteins 0.000 claims description 7
- 102100024533 Carcinoembryonic antigen-related cell adhesion molecule 1 Human genes 0.000 claims description 7
- 102100027816 Cytotoxic and regulatory T-cell molecule Human genes 0.000 claims description 7
- 101000884298 Homo sapiens CD226 antigen Proteins 0.000 claims description 7
- 101000884279 Homo sapiens CD276 antigen Proteins 0.000 claims description 7
- 101001046683 Homo sapiens Integrin alpha-L Proteins 0.000 claims description 7
- 101001046668 Homo sapiens Integrin alpha-X Proteins 0.000 claims description 7
- 101001015037 Homo sapiens Integrin beta-7 Proteins 0.000 claims description 7
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 claims description 7
- 101001109503 Homo sapiens NKG2-C type II integral membrane protein Proteins 0.000 claims description 7
- 101001109501 Homo sapiens NKG2-D type II integral membrane protein Proteins 0.000 claims description 7
- 101000589305 Homo sapiens Natural cytotoxicity triggering receptor 2 Proteins 0.000 claims description 7
- 101000633778 Homo sapiens SLAM family member 5 Proteins 0.000 claims description 7
- 101000596234 Homo sapiens T-cell surface protein tactile Proteins 0.000 claims description 7
- 101000795169 Homo sapiens Tumor necrosis factor receptor superfamily member 13C Proteins 0.000 claims description 7
- 101000648507 Homo sapiens Tumor necrosis factor receptor superfamily member 14 Proteins 0.000 claims description 7
- 102100033016 Integrin beta-7 Human genes 0.000 claims description 7
- 108010061593 Member 14 Tumor Necrosis Factor Receptors Proteins 0.000 claims description 7
- 101100236305 Mus musculus Ly9 gene Proteins 0.000 claims description 7
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 claims description 7
- 102100022683 NKG2-C type II integral membrane protein Human genes 0.000 claims description 7
- 102100022680 NKG2-D type II integral membrane protein Human genes 0.000 claims description 7
- 108010004217 Natural Cytotoxicity Triggering Receptor 1 Proteins 0.000 claims description 7
- 108010004222 Natural Cytotoxicity Triggering Receptor 3 Proteins 0.000 claims description 7
- 102100032870 Natural cytotoxicity triggering receptor 1 Human genes 0.000 claims description 7
- 102100032851 Natural cytotoxicity triggering receptor 2 Human genes 0.000 claims description 7
- 102100032852 Natural cytotoxicity triggering receptor 3 Human genes 0.000 claims description 7
- 101710141230 Natural killer cell receptor 2B4 Proteins 0.000 claims description 7
- 102100029216 SLAM family member 5 Human genes 0.000 claims description 7
- 102100035268 T-cell surface protein tactile Human genes 0.000 claims description 7
- 102100031989 Transmembrane protease serine 2 Human genes 0.000 claims description 7
- 102100029690 Tumor necrosis factor receptor superfamily member 13C Human genes 0.000 claims description 7
- 102100033733 Tumor necrosis factor receptor superfamily member 1B Human genes 0.000 claims description 7
- 101710187830 Tumor necrosis factor receptor superfamily member 1B Proteins 0.000 claims description 7
- 108010072917 class-I restricted T cell-associated molecule Proteins 0.000 claims description 7
- 150000002270 gangliosides Chemical class 0.000 claims description 7
- 239000002773 nucleotide Substances 0.000 claims description 7
- 125000003729 nucleotide group Chemical group 0.000 claims description 7
- 101150013553 CD40 gene Proteins 0.000 claims description 6
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 claims description 6
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 claims description 6
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 claims description 6
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 claims description 6
- 101000585551 Equus caballus Pregnancy-associated glycoprotein Proteins 0.000 claims description 6
- 102000010449 Folate receptor beta Human genes 0.000 claims description 6
- 108050001930 Folate receptor beta Proteins 0.000 claims description 6
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 claims description 6
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 claims description 6
- 101000998120 Homo sapiens Interleukin-3 receptor subunit alpha Proteins 0.000 claims description 6
- 101001124867 Homo sapiens Peroxiredoxin-1 Proteins 0.000 claims description 6
- 101000692259 Homo sapiens Phosphoprotein associated with glycosphingolipid-enriched microdomains 1 Proteins 0.000 claims description 6
- 101000934346 Homo sapiens T-cell surface antigen CD2 Proteins 0.000 claims description 6
- 101000679857 Homo sapiens Tumor necrosis factor receptor superfamily member 3 Proteins 0.000 claims description 6
- 102100033493 Interleukin-3 receptor subunit alpha Human genes 0.000 claims description 6
- 102100031413 L-dopachrome tautomerase Human genes 0.000 claims description 6
- 102100026066 Phosphoprotein associated with glycosphingolipid-enriched microdomains 1 Human genes 0.000 claims description 6
- 102100035703 Prostatic acid phosphatase Human genes 0.000 claims description 6
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 claims description 6
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 claims description 6
- 108010045108 Receptor for Advanced Glycation End Products Proteins 0.000 claims description 6
- 102000005622 Receptor for Advanced Glycation End Products Human genes 0.000 claims description 6
- 101800001271 Surface protein Proteins 0.000 claims description 6
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 claims description 6
- 108010017842 Telomerase Proteins 0.000 claims description 6
- 102100022156 Tumor necrosis factor receptor superfamily member 3 Human genes 0.000 claims description 6
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 claims description 6
- 210000003719 b-lymphocyte Anatomy 0.000 claims description 6
- 108010051081 dopachrome isomerase Proteins 0.000 claims description 6
- 208000032839 leukemia Diseases 0.000 claims description 6
- 108010043671 prostatic acid phosphatase Proteins 0.000 claims description 6
- 101710145634 Antigen 1 Proteins 0.000 claims description 5
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 claims description 5
- 102100025137 Early activation antigen CD69 Human genes 0.000 claims description 5
- 102100029360 Hematopoietic cell signal transducer Human genes 0.000 claims description 5
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 claims description 5
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 claims description 5
- 101000990188 Homo sapiens Hematopoietic cell signal transducer Proteins 0.000 claims description 5
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 claims description 5
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 claims description 5
- 102000006815 folate receptor Human genes 0.000 claims description 5
- 108020005243 folate receptor Proteins 0.000 claims description 5
- 230000002489 hematologic effect Effects 0.000 claims description 5
- BGFTWECWAICPDG-UHFFFAOYSA-N 2-[bis(4-chlorophenyl)methyl]-4-n-[3-[bis(4-chlorophenyl)methyl]-4-(dimethylamino)phenyl]-1-n,1-n-dimethylbenzene-1,4-diamine Chemical compound C1=C(C(C=2C=CC(Cl)=CC=2)C=2C=CC(Cl)=CC=2)C(N(C)C)=CC=C1NC(C=1)=CC=C(N(C)C)C=1C(C=1C=CC(Cl)=CC=1)C1=CC=C(Cl)C=C1 BGFTWECWAICPDG-UHFFFAOYSA-N 0.000 claims description 4
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 claims description 4
- 108010008014 B-Cell Maturation Antigen Proteins 0.000 claims description 4
- 102100021663 Baculoviral IAP repeat-containing protein 5 Human genes 0.000 claims description 4
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 claims description 4
- 102100039510 Cancer/testis antigen 2 Human genes 0.000 claims description 4
- 108010051152 Carboxylesterase Proteins 0.000 claims description 4
- 102000013392 Carboxylesterase Human genes 0.000 claims description 4
- 101150029707 ERBB2 gene Proteins 0.000 claims description 4
- 102100023721 Ephrin-B2 Human genes 0.000 claims description 4
- 108010044090 Ephrin-B2 Proteins 0.000 claims description 4
- 101000856237 Homo sapiens Cancer/testis antigen 1 Proteins 0.000 claims description 4
- 101000889345 Homo sapiens Cancer/testis antigen 2 Proteins 0.000 claims description 4
- 101000777628 Homo sapiens Leukocyte antigen CD37 Proteins 0.000 claims description 4
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 claims description 4
- 101000809875 Homo sapiens TYRO protein tyrosine kinase-binding protein Proteins 0.000 claims description 4
- 241000701806 Human papillomavirus Species 0.000 claims description 4
- 108010031794 IGF Type 1 Receptor Proteins 0.000 claims description 4
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 claims description 4
- 102100031586 Leukocyte antigen CD37 Human genes 0.000 claims description 4
- 108010010995 MART-1 Antigen Proteins 0.000 claims description 4
- 102000016200 MART-1 Antigen Human genes 0.000 claims description 4
- 102100022430 Melanocyte protein PMEL Human genes 0.000 claims description 4
- 108050008953 Melanoma-associated antigen Proteins 0.000 claims description 4
- 108700020796 Oncogene Proteins 0.000 claims description 4
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 claims description 4
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 claims description 4
- 108010002687 Survivin Proteins 0.000 claims description 4
- 102100038717 TYRO protein tyrosine kinase-binding protein Human genes 0.000 claims description 4
- 108050003829 Testisin Proteins 0.000 claims description 4
- 102000003425 Tyrosinase Human genes 0.000 claims description 4
- 108060008724 Tyrosinase Proteins 0.000 claims description 4
- 125000002446 fucosyl group Chemical group C1([C@@H](O)[C@H](O)[C@H](O)[C@@H](O1)C)* 0.000 claims description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 4
- 230000000968 intestinal effect Effects 0.000 claims description 4
- 210000000265 leukocyte Anatomy 0.000 claims description 4
- 230000003612 virological effect Effects 0.000 claims description 4
- 101710185679 CD276 antigen Proteins 0.000 claims description 3
- 102100032937 CD40 ligand Human genes 0.000 claims description 3
- 102100035793 CD83 antigen Human genes 0.000 claims description 3
- 102100037904 CD9 antigen Human genes 0.000 claims description 3
- 102100022086 GRB2-related adapter protein 2 Human genes 0.000 claims description 3
- 102100026122 High affinity immunoglobulin gamma Fc receptor I Human genes 0.000 claims description 3
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 claims description 3
- 101000738354 Homo sapiens CD9 antigen Proteins 0.000 claims description 3
- 101000900690 Homo sapiens GRB2-related adapter protein 2 Proteins 0.000 claims description 3
- 101000913074 Homo sapiens High affinity immunoglobulin gamma Fc receptor I Proteins 0.000 claims description 3
- 101001047640 Homo sapiens Linker for activation of T-cells family member 1 Proteins 0.000 claims description 3
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 claims description 3
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 claims description 3
- 101001090688 Homo sapiens Lymphocyte cytosolic protein 2 Proteins 0.000 claims description 3
- 101000702132 Homo sapiens Protein spinster homolog 1 Proteins 0.000 claims description 3
- 101000934341 Homo sapiens T-cell surface glycoprotein CD5 Proteins 0.000 claims description 3
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 claims description 3
- 102100024032 Linker for activation of T-cells family member 1 Human genes 0.000 claims description 3
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 claims description 3
- 102100034709 Lymphocyte cytosolic protein 2 Human genes 0.000 claims description 3
- 241001529936 Murinae Species 0.000 claims description 3
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 claims description 3
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 claims description 3
- 101001038499 Yarrowia lipolytica (strain CLIB 122 / E 150) Lysine acetyltransferase Proteins 0.000 claims description 3
- 230000004069 differentiation Effects 0.000 claims description 3
- 102100022907 Acrosin-binding protein Human genes 0.000 claims description 2
- 101710107749 Acrosin-binding protein Proteins 0.000 claims description 2
- 241000271566 Aves Species 0.000 claims description 2
- 108091007065 BIRCs Proteins 0.000 claims description 2
- 102100034159 Beta-3 adrenergic receptor Human genes 0.000 claims description 2
- 101100518995 Caenorhabditis elegans pax-3 gene Proteins 0.000 claims description 2
- 102100025933 Cancer-associated gene 1 protein Human genes 0.000 claims description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 claims description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 claims description 2
- 101710181340 Chaperone protein DnaK2 Proteins 0.000 claims description 2
- 101000827763 Drosophila melanogaster Fibroblast growth factor receptor homolog 1 Proteins 0.000 claims description 2
- 102100031334 Elongation factor 2 Human genes 0.000 claims description 2
- 101710144543 Endosialin Proteins 0.000 claims description 2
- 108010087819 Fc receptors Proteins 0.000 claims description 2
- 102000009109 Fc receptors Human genes 0.000 claims description 2
- 108010084795 Fusion Oncogene Proteins Proteins 0.000 claims description 2
- 102000005668 Fusion Oncogene Proteins Human genes 0.000 claims description 2
- 102000027583 GPCRs class C Human genes 0.000 claims description 2
- 108091008882 GPCRs class C Proteins 0.000 claims description 2
- 102000003886 Glycoproteins Human genes 0.000 claims description 2
- 108090000288 Glycoproteins Proteins 0.000 claims description 2
- 101710178419 Heat shock protein 70 2 Proteins 0.000 claims description 2
- 102100034458 Hepatitis A virus cellular receptor 2 Human genes 0.000 claims description 2
- 241000709721 Hepatovirus A Species 0.000 claims description 2
- 101100118545 Holotrichia diomphalia EGF-like gene Proteins 0.000 claims description 2
- 101000933825 Homo sapiens Cancer-associated gene 1 protein Proteins 0.000 claims description 2
- 101001068133 Homo sapiens Hepatitis A virus cellular receptor 2 Proteins 0.000 claims description 2
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 claims description 2
- 101001052849 Homo sapiens Tyrosine-protein kinase Fer Proteins 0.000 claims description 2
- 108010042653 IgA receptor Proteins 0.000 claims description 2
- 108010091135 Immunoglobulin Fc Fragments Proteins 0.000 claims description 2
- 102000018071 Immunoglobulin Fc Fragments Human genes 0.000 claims description 2
- 102000055031 Inhibitor of Apoptosis Proteins Human genes 0.000 claims description 2
- 102100023915 Insulin Human genes 0.000 claims description 2
- 108090001061 Insulin Proteins 0.000 claims description 2
- 102000004553 Interleukin-11 Receptors Human genes 0.000 claims description 2
- 108010017521 Interleukin-11 Receptors Proteins 0.000 claims description 2
- 102000007482 Interleukin-13 Receptor alpha2 Subunit Human genes 0.000 claims description 2
- 108010085418 Interleukin-13 Receptor alpha2 Subunit Proteins 0.000 claims description 2
- 102000017578 LAG3 Human genes 0.000 claims description 2
- 102000008840 Melanoma-associated antigen 1 Human genes 0.000 claims description 2
- 108050000731 Melanoma-associated antigen 1 Proteins 0.000 claims description 2
- 102100034256 Mucin-1 Human genes 0.000 claims description 2
- 108010008707 Mucin-1 Proteins 0.000 claims description 2
- 101100518997 Mus musculus Pax3 gene Proteins 0.000 claims description 2
- 101100351020 Mus musculus Pax5 gene Proteins 0.000 claims description 2
- 206010029260 Neuroblastoma Diseases 0.000 claims description 2
- KUIFHYPNNRVEKZ-VIJRYAKMSA-N O-(N-acetyl-alpha-D-galactosaminyl)-L-threonine Chemical compound OC(=O)[C@@H](N)[C@@H](C)O[C@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O KUIFHYPNNRVEKZ-VIJRYAKMSA-N 0.000 claims description 2
- 108700026244 Open Reading Frames Proteins 0.000 claims description 2
- 102000016978 Orphan receptors Human genes 0.000 claims description 2
- 108070000031 Orphan receptors Proteins 0.000 claims description 2
- 108091005804 Peptidases Proteins 0.000 claims description 2
- 108010077519 Peptide Elongation Factor 2 Proteins 0.000 claims description 2
- 239000004365 Protease Substances 0.000 claims description 2
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 claims description 2
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 claims description 2
- 101710100968 Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 claims description 2
- 102100022441 Sperm surface protein Sp17 Human genes 0.000 claims description 2
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 claims description 2
- 102100033254 Tumor suppressor ARF Human genes 0.000 claims description 2
- 101710102803 Tumor suppressor ARF Proteins 0.000 claims description 2
- 102100024537 Tyrosine-protein kinase Fer Human genes 0.000 claims description 2
- 101710101493 Viral myc transforming protein Proteins 0.000 claims description 2
- 210000001766 X chromosome Anatomy 0.000 claims description 2
- 101100351021 Xenopus laevis pax5 gene Proteins 0.000 claims description 2
- 108040005346 beta3-adrenergic receptor activity proteins Proteins 0.000 claims description 2
- 230000011748 cell maturation Effects 0.000 claims description 2
- 210000000349 chromosome Anatomy 0.000 claims description 2
- 230000003511 endothelial effect Effects 0.000 claims description 2
- 102000012803 ephrin Human genes 0.000 claims description 2
- 108060002566 ephrin Proteins 0.000 claims description 2
- 210000002950 fibroblast Anatomy 0.000 claims description 2
- 239000003102 growth factor Substances 0.000 claims description 2
- 108091008039 hormone receptors Proteins 0.000 claims description 2
- 229940125396 insulin Drugs 0.000 claims description 2
- 210000004698 lymphocyte Anatomy 0.000 claims description 2
- 210000005075 mammary gland Anatomy 0.000 claims description 2
- 101710135378 pH 6 antigen Proteins 0.000 claims description 2
- 230000001105 regulatory effect Effects 0.000 claims description 2
- 125000005630 sialyl group Chemical group 0.000 claims description 2
- 108010088201 squamous cell carcinoma-related antigen Proteins 0.000 claims description 2
- 206010042863 synovial sarcoma Diseases 0.000 claims description 2
- 239000000439 tumor marker Substances 0.000 claims description 2
- 102000003298 tumor necrosis factor receptor Human genes 0.000 claims description 2
- 108010064548 Lymphocyte Function-Associated Antigen-1 Proteins 0.000 claims 6
- 102100038449 Claudin-6 Human genes 0.000 claims 3
- 102100029337 Thyrotropin receptor Human genes 0.000 claims 3
- 101000801234 Homo sapiens Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 claims 2
- 102100040678 Programmed cell death protein 1 Human genes 0.000 claims 2
- 102000014128 RANK Ligand Human genes 0.000 claims 2
- 102100033728 Tumor necrosis factor receptor superfamily member 18 Human genes 0.000 claims 2
- 102000009840 Angiopoietins Human genes 0.000 claims 1
- 108010009906 Angiopoietins Proteins 0.000 claims 1
- 102000006942 B-Cell Maturation Antigen Human genes 0.000 claims 1
- 102100022132 High affinity immunoglobulin epsilon receptor subunit gamma Human genes 0.000 claims 1
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 claims 1
- 101000824104 Homo sapiens High affinity immunoglobulin epsilon receptor subunit gamma Proteins 0.000 claims 1
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 claims 1
- 101710089372 Programmed cell death protein 1 Proteins 0.000 claims 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 claims 1
- 101710185494 Zinc finger protein Proteins 0.000 claims 1
- 102100026497 Zinc finger protein 654 Human genes 0.000 claims 1
- 102100023597 Zinc finger protein 816 Human genes 0.000 claims 1
- 230000014509 gene expression Effects 0.000 abstract description 73
- 238000000034 method Methods 0.000 abstract description 37
- 239000000203 mixture Substances 0.000 abstract description 7
- 239000003795 chemical substances by application Substances 0.000 description 55
- 101100519207 Mus musculus Pdcd1 gene Proteins 0.000 description 46
- 108091007741 Chimeric antigen receptor T cells Proteins 0.000 description 44
- 230000002401 inhibitory effect Effects 0.000 description 39
- 241000699670 Mus sp. Species 0.000 description 37
- 230000000694 effects Effects 0.000 description 28
- 108020004999 messenger RNA Proteins 0.000 description 27
- 229940043355 kinase inhibitor Drugs 0.000 description 24
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 24
- 239000012634 fragment Substances 0.000 description 23
- 235000018102 proteins Nutrition 0.000 description 22
- 210000004881 tumor cell Anatomy 0.000 description 22
- 238000000684 flow cytometry Methods 0.000 description 21
- 102000004127 Cytokines Human genes 0.000 description 20
- 108090000695 Cytokines Proteins 0.000 description 20
- 230000007423 decrease Effects 0.000 description 20
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 19
- 238000005415 bioluminescence Methods 0.000 description 19
- 230000029918 bioluminescence Effects 0.000 description 19
- 230000001086 cytosolic effect Effects 0.000 description 19
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 18
- 230000000638 stimulation Effects 0.000 description 18
- 101001023379 Homo sapiens Lysosome-associated membrane glycoprotein 1 Proteins 0.000 description 16
- 102100035133 Lysosome-associated membrane glycoprotein 1 Human genes 0.000 description 16
- 230000004936 stimulating effect Effects 0.000 description 16
- 108010024755 CTL019 chimeric antigen receptor Proteins 0.000 description 14
- 230000002354 daily effect Effects 0.000 description 14
- 229940124302 mTOR inhibitor Drugs 0.000 description 14
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 14
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 13
- 229940045513 CTLA4 antagonist Drugs 0.000 description 13
- 241000699666 Mus <mouse, genus> Species 0.000 description 13
- 239000000902 placebo Substances 0.000 description 13
- 229940068196 placebo Drugs 0.000 description 13
- 229920002477 rna polymer Polymers 0.000 description 13
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 12
- 102000010451 Folate receptor alpha Human genes 0.000 description 12
- 108050001931 Folate receptor alpha Proteins 0.000 description 12
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 12
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 12
- 238000001802 infusion Methods 0.000 description 12
- 108010074708 B7-H1 Antigen Proteins 0.000 description 11
- 102000003911 Thyrotropin Receptors Human genes 0.000 description 11
- 238000004520 electroporation Methods 0.000 description 11
- 102000003859 Claudin-6 Human genes 0.000 description 10
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 10
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 10
- 206010022000 influenza Diseases 0.000 description 10
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 9
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 9
- 229940124291 BTK inhibitor Drugs 0.000 description 9
- 102100035932 Cocaine- and amphetamine-regulated transcript protein Human genes 0.000 description 9
- 102000050627 Glucocorticoid-Induced TNFR-Related Human genes 0.000 description 9
- 101000715592 Homo sapiens Cocaine- and amphetamine-regulated transcript protein Proteins 0.000 description 9
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 9
- 101710187882 Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 description 9
- 239000007924 injection Substances 0.000 description 9
- 238000002347 injection Methods 0.000 description 9
- 238000004519 manufacturing process Methods 0.000 description 9
- 230000004044 response Effects 0.000 description 9
- 238000002255 vaccination Methods 0.000 description 9
- 108700015968 Slam family Proteins 0.000 description 8
- 230000000259 anti-tumor effect Effects 0.000 description 8
- 238000003556 assay Methods 0.000 description 8
- 239000012636 effector Substances 0.000 description 8
- 210000002865 immune cell Anatomy 0.000 description 8
- 238000000338 in vitro Methods 0.000 description 8
- 238000001727 in vivo Methods 0.000 description 8
- 210000001266 CD8-positive T-lymphocyte Anatomy 0.000 description 7
- 102000053602 DNA Human genes 0.000 description 7
- 108020004414 DNA Proteins 0.000 description 7
- 210000004369 blood Anatomy 0.000 description 7
- 239000008280 blood Substances 0.000 description 7
- 238000012217 deletion Methods 0.000 description 7
- 230000037430 deletion Effects 0.000 description 7
- 238000002474 experimental method Methods 0.000 description 7
- 229940090044 injection Drugs 0.000 description 7
- 238000011081 inoculation Methods 0.000 description 7
- AHJRHEGDXFFMBM-UHFFFAOYSA-N palbociclib Chemical compound N1=C2N(C3CCCC3)C(=O)C(C(=O)C)=C(C)C2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 AHJRHEGDXFFMBM-UHFFFAOYSA-N 0.000 description 7
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 7
- 238000010186 staining Methods 0.000 description 7
- 102000003812 Interleukin-15 Human genes 0.000 description 6
- 108090000172 Interleukin-15 Proteins 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 6
- 208000006994 Precancerous Conditions Diseases 0.000 description 6
- 206010060862 Prostate cancer Diseases 0.000 description 6
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 6
- 102100024568 Tumor necrosis factor ligand superfamily member 11 Human genes 0.000 description 6
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 6
- 125000000539 amino acid group Chemical group 0.000 description 6
- 239000011324 bead Substances 0.000 description 6
- 108020001507 fusion proteins Proteins 0.000 description 6
- 102000037865 fusion proteins Human genes 0.000 description 6
- XYFPWWZEPKGCCK-GOSISDBHSA-N ibrutinib Chemical compound C1=2C(N)=NC=NC=2N([C@H]2CN(CCC2)C(=O)C=C)N=C1C(C=C1)=CC=C1OC1=CC=CC=C1 XYFPWWZEPKGCCK-GOSISDBHSA-N 0.000 description 6
- 230000002062 proliferating effect Effects 0.000 description 6
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 6
- 210000003289 regulatory T cell Anatomy 0.000 description 6
- 230000028327 secretion Effects 0.000 description 6
- 229960002930 sirolimus Drugs 0.000 description 6
- 238000010361 transduction Methods 0.000 description 6
- 230000026683 transduction Effects 0.000 description 6
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 5
- 229940083347 Cyclin-dependent kinase 4 inhibitor Drugs 0.000 description 5
- 102100031968 Ephrin type-B receptor 2 Human genes 0.000 description 5
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 5
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 5
- 108010041100 Integrin alpha6 Proteins 0.000 description 5
- 108010030465 Integrin alpha6beta1 Proteins 0.000 description 5
- 102100037850 Interferon gamma Human genes 0.000 description 5
- 108010074328 Interferon-gamma Proteins 0.000 description 5
- 108010038498 Interleukin-7 Receptors Proteins 0.000 description 5
- 102000010782 Interleukin-7 Receptors Human genes 0.000 description 5
- 108060001084 Luciferase Proteins 0.000 description 5
- 239000005089 Luciferase Substances 0.000 description 5
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 5
- 230000000735 allogeneic effect Effects 0.000 description 5
- 235000001014 amino acid Nutrition 0.000 description 5
- 230000001472 cytotoxic effect Effects 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 230000028993 immune response Effects 0.000 description 5
- 230000003259 immunoinhibitory effect Effects 0.000 description 5
- 229960003971 influenza vaccine Drugs 0.000 description 5
- 239000013642 negative control Substances 0.000 description 5
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 description 5
- 230000019491 signal transduction Effects 0.000 description 5
- 230000004083 survival effect Effects 0.000 description 5
- 238000002560 therapeutic procedure Methods 0.000 description 5
- 230000003827 upregulation Effects 0.000 description 5
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 4
- 241000282693 Cercopithecidae Species 0.000 description 4
- 238000002965 ELISA Methods 0.000 description 4
- 101100005713 Homo sapiens CD4 gene Proteins 0.000 description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 4
- 239000002177 L01XE27 - Ibrutinib Substances 0.000 description 4
- 241000713666 Lentivirus Species 0.000 description 4
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 4
- 208000007541 Preleukemia Diseases 0.000 description 4
- 229940116193 Protein phosphatase inhibitor Drugs 0.000 description 4
- 241000283984 Rodentia Species 0.000 description 4
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 4
- 102100040247 Tumor necrosis factor Human genes 0.000 description 4
- 102100033726 Tumor necrosis factor receptor superfamily member 17 Human genes 0.000 description 4
- 230000003013 cytotoxicity Effects 0.000 description 4
- 231100000135 cytotoxicity Toxicity 0.000 description 4
- 238000010586 diagram Methods 0.000 description 4
- 208000035475 disorder Diseases 0.000 description 4
- 201000005787 hematologic cancer Diseases 0.000 description 4
- 229960001507 ibrutinib Drugs 0.000 description 4
- 238000003384 imaging method Methods 0.000 description 4
- 230000036210 malignancy Effects 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 210000004985 myeloid-derived suppressor cell Anatomy 0.000 description 4
- 229960004390 palbociclib Drugs 0.000 description 4
- 239000003934 phosphoprotein phosphatase inhibitor Substances 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- 229940126731 protein tyrosine phosphatase inhibitor Drugs 0.000 description 4
- 239000003806 protein tyrosine phosphatase inhibitor Substances 0.000 description 4
- 102000016914 ras Proteins Human genes 0.000 description 4
- 238000003753 real-time PCR Methods 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 241000894007 species Species 0.000 description 4
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 4
- 238000001262 western blot Methods 0.000 description 4
- 108020005345 3' Untranslated Regions Proteins 0.000 description 3
- BUROJSBIWGDYCN-GAUTUEMISA-N AP 23573 Chemical compound C1C[C@@H](OP(C)(C)=O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 BUROJSBIWGDYCN-GAUTUEMISA-N 0.000 description 3
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 3
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 3
- 208000017604 Hodgkin disease Diseases 0.000 description 3
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 3
- 101000985516 Homo sapiens Hermansky-Pudlak syndrome 5 protein Proteins 0.000 description 3
- 101000665137 Homo sapiens Scm-like with four MBT domains protein 1 Proteins 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- 102000000588 Interleukin-2 Human genes 0.000 description 3
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 3
- 206010025323 Lymphomas Diseases 0.000 description 3
- 102000000440 Melanoma-associated antigen Human genes 0.000 description 3
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 3
- 206010061535 Ovarian neoplasm Diseases 0.000 description 3
- 108091007960 PI3Ks Proteins 0.000 description 3
- 108010077524 Peptide Elongation Factor 1 Proteins 0.000 description 3
- 102000010292 Peptide Elongation Factor 1 Human genes 0.000 description 3
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 3
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- 102100021657 Tyrosine-protein phosphatase non-receptor type 6 Human genes 0.000 description 3
- 101710128901 Tyrosine-protein phosphatase non-receptor type 6 Proteins 0.000 description 3
- HJSSPYJVWLTYHG-UHFFFAOYSA-N XL765 Chemical compound COC1=CC(OC)=CC(NC=2C(=NC3=CC=CC=C3N=2)NS(=O)(=O)C=2C=CC(NC(=O)C=3C=C(OC)C(C)=CC=3)=CC=2)=C1 HJSSPYJVWLTYHG-UHFFFAOYSA-N 0.000 description 3
- 229960005305 adenosine Drugs 0.000 description 3
- BIIVYFLTOXDAOV-YVEFUNNKSA-N alvocidib Chemical compound O[C@@H]1CN(C)CC[C@@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O BIIVYFLTOXDAOV-YVEFUNNKSA-N 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 208000035269 cancer or benign tumor Diseases 0.000 description 3
- 230000010261 cell growth Effects 0.000 description 3
- 230000002596 correlated effect Effects 0.000 description 3
- 230000009089 cytolysis Effects 0.000 description 3
- 231100000433 cytotoxic Toxicity 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 230000009977 dual effect Effects 0.000 description 3
- 229960005167 everolimus Drugs 0.000 description 3
- 201000003444 follicular lymphoma Diseases 0.000 description 3
- 230000001965 increasing effect Effects 0.000 description 3
- 230000006698 induction Effects 0.000 description 3
- PGHMRUGBZOYCAA-UHFFFAOYSA-N ionomycin Natural products O1C(CC(O)C(C)C(O)C(C)C=CCC(C)CC(C)C(O)=CC(=O)C(C)CC(C)CC(CCC(O)=O)C)CCC1(C)C1OC(C)(C(C)O)CC1 PGHMRUGBZOYCAA-UHFFFAOYSA-N 0.000 description 3
- PGHMRUGBZOYCAA-ADZNBVRBSA-N ionomycin Chemical compound O1[C@H](C[C@H](O)[C@H](C)[C@H](O)[C@H](C)/C=C/C[C@@H](C)C[C@@H](C)C(/O)=C/C(=O)[C@@H](C)C[C@@H](C)C[C@@H](CCC(O)=O)C)CC[C@@]1(C)[C@@H]1O[C@](C)([C@@H](C)O)CC1 PGHMRUGBZOYCAA-ADZNBVRBSA-N 0.000 description 3
- 238000010172 mouse model Methods 0.000 description 3
- 230000002688 persistence Effects 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 238000011002 quantification Methods 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 230000035945 sensitivity Effects 0.000 description 3
- 210000000952 spleen Anatomy 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 230000004614 tumor growth Effects 0.000 description 3
- YOVVNQKCSKSHKT-HNNXBMFYSA-N (2s)-1-[4-[[2-(2-aminopyrimidin-5-yl)-7-methyl-4-morpholin-4-ylthieno[3,2-d]pyrimidin-6-yl]methyl]piperazin-1-yl]-2-hydroxypropan-1-one Chemical compound C1CN(C(=O)[C@@H](O)C)CCN1CC1=C(C)C2=NC(C=3C=NC(N)=NC=3)=NC(N3CCOCC3)=C2S1 YOVVNQKCSKSHKT-HNNXBMFYSA-N 0.000 description 2
- DWZAEMINVBZMHQ-UHFFFAOYSA-N 1-[4-[4-(dimethylamino)piperidine-1-carbonyl]phenyl]-3-[4-(4,6-dimorpholin-4-yl-1,3,5-triazin-2-yl)phenyl]urea Chemical compound C1CC(N(C)C)CCN1C(=O)C(C=C1)=CC=C1NC(=O)NC1=CC=C(C=2N=C(N=C(N=2)N2CCOCC2)N2CCOCC2)C=C1 DWZAEMINVBZMHQ-UHFFFAOYSA-N 0.000 description 2
- YABJJWZLRMPFSI-UHFFFAOYSA-N 1-methyl-5-[[2-[5-(trifluoromethyl)-1H-imidazol-2-yl]-4-pyridinyl]oxy]-N-[4-(trifluoromethyl)phenyl]-2-benzimidazolamine Chemical compound N=1C2=CC(OC=3C=C(N=CC=3)C=3NC(=CN=3)C(F)(F)F)=CC=C2N(C)C=1NC1=CC=C(C(F)(F)F)C=C1 YABJJWZLRMPFSI-UHFFFAOYSA-N 0.000 description 2
- QLUYMIVVAYRECT-OCCSQVGLSA-N 2-(2-chlorophenyl)-5,7-dihydroxy-8-[(2r,3s)-2-(hydroxymethyl)-1-methylpyrrolidin-3-yl]chromen-4-one Chemical compound OC[C@@H]1N(C)CC[C@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O QLUYMIVVAYRECT-OCCSQVGLSA-N 0.000 description 2
- OVPNQJVDAFNBDN-UHFFFAOYSA-N 4-(2,6-dichlorobenzamido)-N-(piperidin-4-yl)-pyrazole-3-carboxamide Chemical compound ClC1=CC=CC(Cl)=C1C(=O)NC1=CNN=C1C(=O)NC1CCNCC1 OVPNQJVDAFNBDN-UHFFFAOYSA-N 0.000 description 2
- WJRRGYBTGDJBFX-UHFFFAOYSA-N 4-(2-methyl-3-propan-2-yl-4-imidazolyl)-N-(4-methylsulfonylphenyl)-2-pyrimidinamine Chemical compound CC(C)N1C(C)=NC=C1C1=CC=NC(NC=2C=CC(=CC=2)S(C)(=O)=O)=N1 WJRRGYBTGDJBFX-UHFFFAOYSA-N 0.000 description 2
- VVLHQJDAUIPZFH-UHFFFAOYSA-N 4-[4-[[5-fluoro-4-[3-(prop-2-enoylamino)anilino]pyrimidin-2-yl]amino]phenoxy]-n-methylpyridine-2-carboxamide Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC=3N=C(NC=4C=C(NC(=O)C=C)C=CC=4)C(F)=CN=3)=CC=2)=C1 VVLHQJDAUIPZFH-UHFFFAOYSA-N 0.000 description 2
- QYBGBLQCOOISAR-UHFFFAOYSA-N 5-(8-methyl-2-morpholin-4-yl-9-propan-2-ylpurin-6-yl)pyrimidin-2-amine Chemical compound N1=C2N(C(C)C)C(C)=NC2=C(C=2C=NC(N)=NC=2)N=C1N1CCOCC1 QYBGBLQCOOISAR-UHFFFAOYSA-N 0.000 description 2
- SEJLPXCPMNSRAM-GOSISDBHSA-N 6-amino-9-[(3r)-1-but-2-ynoylpyrrolidin-3-yl]-7-(4-phenoxyphenyl)purin-8-one Chemical compound C1N(C(=O)C#CC)CC[C@H]1N1C(=O)N(C=2C=CC(OC=3C=CC=CC=3)=CC=2)C2=C(N)N=CN=C21 SEJLPXCPMNSRAM-GOSISDBHSA-N 0.000 description 2
- 108010013238 70-kDa Ribosomal Protein S6 Kinases Proteins 0.000 description 2
- KVLFRAWTRWDEDF-IRXDYDNUSA-N AZD-8055 Chemical compound C1=C(CO)C(OC)=CC=C1C1=CC=C(C(=NC(=N2)N3[C@H](COCC3)C)N3[C@H](COCC3)C)C2=N1 KVLFRAWTRWDEDF-IRXDYDNUSA-N 0.000 description 2
- 206010000830 Acute leukaemia Diseases 0.000 description 2
- 102000030431 Asparaginyl endopeptidase Human genes 0.000 description 2
- 208000036170 B-Cell Marginal Zone Lymphoma Diseases 0.000 description 2
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 2
- 208000032568 B-cell prolymphocytic leukaemia Diseases 0.000 description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 2
- YUXMAKUNSXIEKN-BTJKTKAUSA-N BGT226 Chemical compound OC(=O)\C=C/C(O)=O.C1=NC(OC)=CC=C1C1=CC=C(N=CC2=C3N(C=4C=C(C(N5CCNCC5)=CC=4)C(F)(F)F)C(=O)N2C)C3=C1 YUXMAKUNSXIEKN-BTJKTKAUSA-N 0.000 description 2
- 208000011691 Burkitt lymphomas Diseases 0.000 description 2
- UDFAXHFPCDXHHH-UHFFFAOYSA-N C1=NC2=NN=CC2=C(N)N1NC1=CC=C(F)C=C1 Chemical compound C1=NC2=NN=CC2=C(N)N1NC1=CC=C(F)C=C1 UDFAXHFPCDXHHH-UHFFFAOYSA-N 0.000 description 2
- 208000016778 CD4+/CD56+ hematodermic neoplasm Diseases 0.000 description 2
- 229940124297 CDK 4/6 inhibitor Drugs 0.000 description 2
- 206010057248 Cell death Diseases 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 2
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 2
- 206010061850 Extranodal marginal zone B-cell lymphoma (MALT type) Diseases 0.000 description 2
- 108010021468 Fc gamma receptor IIA Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 208000036066 Hemophagocytic Lymphohistiocytosis Diseases 0.000 description 2
- 208000032672 Histiocytosis haematophagic Diseases 0.000 description 2
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 2
- 101000954709 Homo sapiens Doublecortin domain-containing protein 2 Proteins 0.000 description 2
- 101000614481 Homo sapiens Kidney-associated antigen 1 Proteins 0.000 description 2
- 101000578784 Homo sapiens Melanoma antigen recognized by T-cells 1 Proteins 0.000 description 2
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 2
- 101001094545 Homo sapiens Retrotransposon-like protein 1 Proteins 0.000 description 2
- 108010043610 KIR Receptors Proteins 0.000 description 2
- 102100034872 Kallikrein-4 Human genes 0.000 description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 description 2
- 102100033627 Killer cell immunoglobulin-like receptor 3DL1 Human genes 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- UVSVTDVJQAJIFG-VURMDHGXSA-N LFM-A13 Chemical compound C\C(O)=C(/C#N)C(=O)NC1=CC(Br)=CC=C1Br UVSVTDVJQAJIFG-VURMDHGXSA-N 0.000 description 2
- 208000031671 Large B-Cell Diffuse Lymphoma Diseases 0.000 description 2
- 102100029204 Low affinity immunoglobulin gamma Fc region receptor II-a Human genes 0.000 description 2
- 201000003791 MALT lymphoma Diseases 0.000 description 2
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 2
- 102000008135 Mechanistic Target of Rapamycin Complex 1 Human genes 0.000 description 2
- 108010035196 Mechanistic Target of Rapamycin Complex 1 Proteins 0.000 description 2
- 102000009308 Mechanistic Target of Rapamycin Complex 2 Human genes 0.000 description 2
- 108010034057 Mechanistic Target of Rapamycin Complex 2 Proteins 0.000 description 2
- 102100028389 Melanoma antigen recognized by T-cells 1 Human genes 0.000 description 2
- 208000034578 Multiple myelomas Diseases 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 206010035226 Plasma cell myeloma Diseases 0.000 description 2
- 208000035416 Prolymphocytic B-Cell Leukemia Diseases 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 208000015634 Rectal Neoplasms Diseases 0.000 description 2
- 206010038389 Renal cancer Diseases 0.000 description 2
- 208000000453 Skin Neoplasms Diseases 0.000 description 2
- 102220497176 Small vasohibin-binding protein_T47D_mutation Human genes 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 230000006044 T cell activation Effects 0.000 description 2
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 description 2
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- 102100033019 Tyrosine-protein phosphatase non-receptor type 11 Human genes 0.000 description 2
- 101710116241 Tyrosine-protein phosphatase non-receptor type 11 Proteins 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000002495 Uterine Neoplasms Diseases 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 2
- 229950010817 alvocidib Drugs 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000001093 anti-cancer Effects 0.000 description 2
- 230000006023 anti-tumor response Effects 0.000 description 2
- 210000000612 antigen-presenting cell Anatomy 0.000 description 2
- 108010055066 asparaginylendopeptidase Proteins 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 239000012472 biological sample Substances 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- 230000005859 cell recognition Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 230000005754 cellular signaling Effects 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 description 2
- 229960005061 crizotinib Drugs 0.000 description 2
- 239000012228 culture supernatant Substances 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 230000016396 cytokine production Effects 0.000 description 2
- 206010052015 cytokine release syndrome Diseases 0.000 description 2
- JOGKUKXHTYWRGZ-UHFFFAOYSA-N dactolisib Chemical compound O=C1N(C)C2=CN=C3C=CC(C=4C=C5C=CC=CC5=NC=4)=CC3=C2N1C1=CC=C(C(C)(C)C#N)C=C1 JOGKUKXHTYWRGZ-UHFFFAOYSA-N 0.000 description 2
- 229950006418 dactolisib Drugs 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 206010012818 diffuse large B-cell lymphoma Diseases 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 238000006471 dimerization reaction Methods 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 229940125436 dual inhibitor Drugs 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 description 2
- 201000009277 hairy cell leukemia Diseases 0.000 description 2
- 208000014829 head and neck neoplasm Diseases 0.000 description 2
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 2
- 208000014752 hemophagocytic syndrome Diseases 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- 108010024383 kallikrein 4 Proteins 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 201000010982 kidney cancer Diseases 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 230000003211 malignant effect Effects 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- 201000007924 marginal zone B-cell lymphoma Diseases 0.000 description 2
- 208000021937 marginal zone lymphoma Diseases 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- MMNNTJYFHUDSKL-UHFFFAOYSA-N methyl n-[6-[2-(5-chloro-2-methylphenyl)-1-hydroxy-3-oxoisoindol-1-yl]-1h-benzimidazol-2-yl]carbamate Chemical compound C=1C=C2NC(NC(=O)OC)=NC2=CC=1C(C1=CC=CC=C1C1=O)(O)N1C1=CC(Cl)=CC=C1C MMNNTJYFHUDSKL-UHFFFAOYSA-N 0.000 description 2
- 238000012737 microarray-based gene expression Methods 0.000 description 2
- 238000012243 multiplex automated genomic engineering Methods 0.000 description 2
- IJMHHZDBRUGXNO-UHFFFAOYSA-N n-[3-(8-anilinoimidazo[1,2-a]pyrazin-6-yl)phenyl]-4-tert-butylbenzamide Chemical compound C1=CC(C(C)(C)C)=CC=C1C(=O)NC1=CC=CC(C=2N=C(NC=3C=CC=CC=3)C3=NC=CN3C=2)=C1 IJMHHZDBRUGXNO-UHFFFAOYSA-N 0.000 description 2
- CDOOFZZILLRUQH-GDLZYMKVSA-N n-[3-[6-[4-[(2r)-1,4-dimethyl-3-oxopiperazin-2-yl]anilino]-4-methyl-5-oxopyrazin-2-yl]-2-methylphenyl]-4,5,6,7-tetrahydro-1-benzothiophene-2-carboxamide Chemical compound CN1CCN(C)C(=O)[C@H]1C(C=C1)=CC=C1NC1=NC(C=2C(=C(NC(=O)C=3SC=4CCCCC=4C=3)C=CC=2)C)=CN(C)C1=O CDOOFZZILLRUQH-GDLZYMKVSA-N 0.000 description 2
- CGBJSGAELGCMKE-UHFFFAOYSA-N omipalisib Chemical compound COC1=NC=C(C=2C=C3C(C=4C=NN=CC=4)=CC=NC3=CC=2)C=C1NS(=O)(=O)C1=CC=C(F)C=C1F CGBJSGAELGCMKE-UHFFFAOYSA-N 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 210000005259 peripheral blood Anatomy 0.000 description 2
- 239000011886 peripheral blood Substances 0.000 description 2
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 208000007525 plasmablastic lymphoma Diseases 0.000 description 2
- 210000005134 plasmacytoid dendritic cell Anatomy 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 206010038038 rectal cancer Diseases 0.000 description 2
- 201000001275 rectum cancer Diseases 0.000 description 2
- 230000001177 retroviral effect Effects 0.000 description 2
- 229960001302 ridaforolimus Drugs 0.000 description 2
- BTIHMVBBUGXLCJ-OAHLLOKOSA-N seliciclib Chemical compound C=12N=CN(C(C)C)C2=NC(N[C@@H](CO)CC)=NC=1NCC1=CC=CC=C1 BTIHMVBBUGXLCJ-OAHLLOKOSA-N 0.000 description 2
- 201000000849 skin cancer Diseases 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 210000000130 stem cell Anatomy 0.000 description 2
- 210000002536 stromal cell Anatomy 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 230000002463 transducing effect Effects 0.000 description 2
- 229960004799 tryptophan Drugs 0.000 description 2
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 2
- 206010046766 uterine cancer Diseases 0.000 description 2
- 239000004474 valine Substances 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- SVNJBEMPMKWDCO-KCHLEUMXSA-N (2s)-2-[[(2s)-3-carboxy-2-[[2-[[(2s)-5-(diaminomethylideneamino)-2-[[4-oxo-4-[[4-(4-oxo-8-phenylchromen-2-yl)morpholin-4-ium-4-yl]methoxy]butanoyl]amino]pentanoyl]amino]acetyl]amino]propanoyl]amino]-3-hydroxypropanoate Chemical compound C=1C(=O)C2=CC=CC(C=3C=CC=CC=3)=C2OC=1[N+]1(COC(=O)CCC(=O)N[C@@H](CCCNC(=N)N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C([O-])=O)CCOCC1 SVNJBEMPMKWDCO-KCHLEUMXSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 1
- DBXGGXLBTWZXBB-MRXNPFEDSA-N 2-(6-fluorobenzimidazol-1-yl)-9-[(4r)-8-fluoro-3,4-dihydro-2h-chromen-4-yl]-7h-purin-8-one Chemical compound C1COC2=C(F)C=CC=C2[C@@H]1N(C1=N2)C(=O)NC1=CN=C2N1C=NC2=CC=C(F)C=C21 DBXGGXLBTWZXBB-MRXNPFEDSA-N 0.000 description 1
- PIMQWRZWLQKKBJ-SFHVURJKSA-N 2-[(2S)-1-[3-ethyl-7-[(1-oxido-3-pyridin-1-iumyl)methylamino]-5-pyrazolo[1,5-a]pyrimidinyl]-2-piperidinyl]ethanol Chemical compound C=1C(N2[C@@H](CCCC2)CCO)=NC2=C(CC)C=NN2C=1NCC1=CC=C[N+]([O-])=C1 PIMQWRZWLQKKBJ-SFHVURJKSA-N 0.000 description 1
- XDLYKKIQACFMJG-UHFFFAOYSA-N 2-amino-8-[4-(2-hydroxyethoxy)cyclohexyl]-6-(6-methoxypyridin-3-yl)-4-methylpyrido[2,3-d]pyrimidin-7-one Chemical compound C1=NC(OC)=CC=C1C(C1=O)=CC2=C(C)N=C(N)N=C2N1C1CCC(OCCO)CC1 XDLYKKIQACFMJG-UHFFFAOYSA-N 0.000 description 1
- YXHLJMWYDTXDHS-IRFLANFNSA-N 7-aminoactinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=C(N)C=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 YXHLJMWYDTXDHS-IRFLANFNSA-N 0.000 description 1
- 108700012813 7-aminoactinomycin D Proteins 0.000 description 1
- 102100029824 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 2 Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 102100040024 Adhesion G-protein coupled receptor G5 Human genes 0.000 description 1
- 241000059559 Agriotes sordidus Species 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 102100034608 Angiopoietin-2 Human genes 0.000 description 1
- 108010048036 Angiopoietin-2 Proteins 0.000 description 1
- 108090000672 Annexin A5 Proteins 0.000 description 1
- 102000004121 Annexin A5 Human genes 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 102100027205 B-cell antigen receptor complex-associated protein alpha chain Human genes 0.000 description 1
- 102100027203 B-cell antigen receptor complex-associated protein beta chain Human genes 0.000 description 1
- 108010087504 Beta-Globulins Proteins 0.000 description 1
- 102000006734 Beta-Globulins Human genes 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 206010005949 Bone cancer Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 206010006143 Brain stem glioma Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 102100031151 C-C chemokine receptor type 2 Human genes 0.000 description 1
- 101710149815 C-C chemokine receptor type 2 Proteins 0.000 description 1
- 238000011357 CAR T-cell therapy Methods 0.000 description 1
- 102100025238 CD302 antigen Human genes 0.000 description 1
- 102100036008 CD48 antigen Human genes 0.000 description 1
- 102100022436 CMRF35-like molecule 8 Human genes 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 206010007953 Central nervous system lymphoma Diseases 0.000 description 1
- GEWLYFZWVLXQME-UHFFFAOYSA-N Cercosporamide Natural products CC12C(=O)C(C(=O)C)=C(O)C=C1OC1=C2C(O)=CC(O)=C1C(N)=O GEWLYFZWVLXQME-UHFFFAOYSA-N 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- 206010058314 Dysplasia Diseases 0.000 description 1
- 206010014733 Endometrial cancer Diseases 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 102100031780 Endonuclease Human genes 0.000 description 1
- 108010042407 Endonucleases Proteins 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- 206010015548 Euthanasia Diseases 0.000 description 1
- 102100037362 Fibronectin Human genes 0.000 description 1
- 102000002090 Fibronectin type III Human genes 0.000 description 1
- 108050009401 Fibronectin type III Proteins 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 102000001398 Granzyme Human genes 0.000 description 1
- 108060005986 Granzyme Proteins 0.000 description 1
- 206010069767 H1N1 influenza Diseases 0.000 description 1
- 102100030595 HLA class II histocompatibility antigen gamma chain Human genes 0.000 description 1
- 101150096895 HSPB1 gene Proteins 0.000 description 1
- 102100031624 Heat shock protein 105 kDa Human genes 0.000 description 1
- 102100039165 Heat shock protein beta-1 Human genes 0.000 description 1
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 1
- 102100028721 Hermansky-Pudlak syndrome 5 protein Human genes 0.000 description 1
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 1
- 108010088652 Histocompatibility Antigens Class I Proteins 0.000 description 1
- 101000794082 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 2 Proteins 0.000 description 1
- 101000959600 Homo sapiens Adhesion G-protein coupled receptor G5 Proteins 0.000 description 1
- 101000914489 Homo sapiens B-cell antigen receptor complex-associated protein alpha chain Proteins 0.000 description 1
- 101000914491 Homo sapiens B-cell antigen receptor complex-associated protein beta chain Proteins 0.000 description 1
- 101000716130 Homo sapiens CD48 antigen Proteins 0.000 description 1
- 101000901669 Homo sapiens CMRF35-like molecule 8 Proteins 0.000 description 1
- 101001099051 Homo sapiens GPI inositol-deacylase Proteins 0.000 description 1
- 101001082627 Homo sapiens HLA class II histocompatibility antigen gamma chain Proteins 0.000 description 1
- 101000866478 Homo sapiens Heat shock protein 105 kDa Proteins 0.000 description 1
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 1
- 101001043807 Homo sapiens Interleukin-7 Proteins 0.000 description 1
- 101001018097 Homo sapiens L-selectin Proteins 0.000 description 1
- 101000576894 Homo sapiens Macrophage mannose receptor 1 Proteins 0.000 description 1
- 101001014566 Homo sapiens Membrane-spanning 4-domains subfamily A member 3 Proteins 0.000 description 1
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 1
- 101000603882 Homo sapiens Nuclear receptor subfamily 1 group I member 3 Proteins 0.000 description 1
- 101000931590 Homo sapiens Prostaglandin F2 receptor negative regulator Proteins 0.000 description 1
- 101001095320 Homo sapiens Serine/threonine-protein phosphatase PP1-beta catalytic subunit Proteins 0.000 description 1
- 101000863900 Homo sapiens Sialic acid-binding Ig-like lectin 5 Proteins 0.000 description 1
- 101000868472 Homo sapiens Sialoadhesin Proteins 0.000 description 1
- 101000709256 Homo sapiens Signal-regulatory protein beta-1 Proteins 0.000 description 1
- 101000709188 Homo sapiens Signal-regulatory protein beta-1 isoform 3 Proteins 0.000 description 1
- 101000738335 Homo sapiens T-cell surface glycoprotein CD3 zeta chain Proteins 0.000 description 1
- 101000835093 Homo sapiens Transferrin receptor protein 1 Proteins 0.000 description 1
- 101000801255 Homo sapiens Tumor necrosis factor receptor superfamily member 17 Proteins 0.000 description 1
- 101000749631 Homo sapiens Uromodulin-like 1 Proteins 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 101150102264 IE gene Proteins 0.000 description 1
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 1
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 1
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 1
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 1
- 102100022516 Immunoglobulin superfamily member 2 Human genes 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 101100452750 Ipomoea batatas IPO gene Proteins 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- 102100033467 L-selectin Human genes 0.000 description 1
- 239000002146 L01XE16 - Crizotinib Substances 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 208000030289 Lymphoproliferative disease Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 102100025354 Macrophage mannose receptor 1 Human genes 0.000 description 1
- 208000032271 Malignant tumor of penis Diseases 0.000 description 1
- 102100032517 Membrane-spanning 4-domains subfamily A member 3 Human genes 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 1
- 101100407308 Mus musculus Pdcd1lg2 gene Proteins 0.000 description 1
- OUSFTKFNBAZUKL-UHFFFAOYSA-N N-(5-{[(5-tert-butyl-1,3-oxazol-2-yl)methyl]sulfanyl}-1,3-thiazol-2-yl)piperidine-4-carboxamide Chemical compound O1C(C(C)(C)C)=CN=C1CSC(S1)=CN=C1NC(=O)C1CCNCC1 OUSFTKFNBAZUKL-UHFFFAOYSA-N 0.000 description 1
- UQPMANVRZYYQMD-UHFFFAOYSA-N N3-(4-fluorophenyl)-2H-pyrazolo[3,4-d]pyrimidine-3,4-diamine Chemical compound C=12C(N)=NC=NC2=NNC=1NC1=CC=C(F)C=C1 UQPMANVRZYYQMD-UHFFFAOYSA-N 0.000 description 1
- 206010061309 Neoplasm progression Diseases 0.000 description 1
- 101150091206 Nfkbia gene Proteins 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 102100037589 OX-2 membrane glycoprotein Human genes 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 239000012823 PI3K/mTOR inhibitor Substances 0.000 description 1
- 208000000821 Parathyroid Neoplasms Diseases 0.000 description 1
- 208000002471 Penile Neoplasms Diseases 0.000 description 1
- 206010034299 Penile cancer Diseases 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 102100020739 Peptidyl-prolyl cis-trans isomerase FKBP4 Human genes 0.000 description 1
- 101710147152 Peptidyl-prolyl cis-trans isomerase FKBP4 Proteins 0.000 description 1
- 241000030360 Peromyscus truei Species 0.000 description 1
- 102000011755 Phosphoglycerate Kinase Human genes 0.000 description 1
- 108010004729 Phycoerythrin Proteins 0.000 description 1
- 208000007913 Pituitary Neoplasms Diseases 0.000 description 1
- 201000005746 Pituitary adenoma Diseases 0.000 description 1
- 206010061538 Pituitary tumour benign Diseases 0.000 description 1
- 108091036407 Polyadenylation Proteins 0.000 description 1
- 102100037935 Polyubiquitin-C Human genes 0.000 description 1
- 108700030875 Programmed Cell Death 1 Ligand 2 Proteins 0.000 description 1
- 101710094000 Programmed cell death 1 ligand 1 Proteins 0.000 description 1
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 102100020864 Prostaglandin F2 receptor negative regulator Human genes 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 101710148333 Regulator of G-protein signaling 13 Proteins 0.000 description 1
- 102100021035 Regulator of G-protein signaling 18 Human genes 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 108010034782 Ribosomal Protein S6 Kinases Proteins 0.000 description 1
- 102000009738 Ribosomal Protein S6 Kinases Human genes 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 102100038689 Scm-like with four MBT domains protein 1 Human genes 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 102100029957 Sialic acid-binding Ig-like lectin 5 Human genes 0.000 description 1
- 102100032855 Sialoadhesin Human genes 0.000 description 1
- 102100032770 Signal-regulatory protein beta-1 isoform 3 Human genes 0.000 description 1
- 108091027967 Small hairpin RNA Proteins 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- 208000032383 Soft tissue cancer Diseases 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 230000005867 T cell response Effects 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 102100037906 T-cell surface glycoprotein CD3 zeta chain Human genes 0.000 description 1
- 210000000662 T-lymphocyte subset Anatomy 0.000 description 1
- 238000010459 TALEN Methods 0.000 description 1
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- 206010057644 Testis cancer Diseases 0.000 description 1
- 101001099217 Thermotoga maritima (strain ATCC 43589 / DSM 3109 / JCM 10099 / NBRC 100826 / MSB8) Triosephosphate isomerase Proteins 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 102100026144 Transferrin receptor protein 1 Human genes 0.000 description 1
- 108010056354 Ubiquitin C Proteins 0.000 description 1
- 208000023915 Ureteral Neoplasms Diseases 0.000 description 1
- 206010046392 Ureteric cancer Diseases 0.000 description 1
- 206010046431 Urethral cancer Diseases 0.000 description 1
- 206010046458 Urethral neoplasms Diseases 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 102100040616 Uromodulin-like 1 Human genes 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 201000003761 Vaginal carcinoma Diseases 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 206010047741 Vulval cancer Diseases 0.000 description 1
- 208000016025 Waldenstroem macroglobulinemia Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 238000007792 addition Methods 0.000 description 1
- 208000024447 adrenal gland neoplasm Diseases 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 229940125528 allosteric inhibitor Drugs 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 238000002617 apheresis Methods 0.000 description 1
- 229950004111 apitolisib Drugs 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 238000003390 bioluminescence detection Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000009702 cancer cell proliferation Effects 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 238000002659 cell therapy Methods 0.000 description 1
- GEWLYFZWVLXQME-MRXNPFEDSA-N cercosporamide Chemical compound O=C([C@]12C)C(C(=O)C)=C(O)C=C1OC1=C2C(O)=CC(O)=C1C(N)=O GEWLYFZWVLXQME-MRXNPFEDSA-N 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 208000019065 cervical carcinoma Diseases 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- JROFGZPOBKIAEW-HAQNSBGRSA-N chembl3120215 Chemical compound N1C=2C(OC)=CC=CC=2C=C1C(=C1C(N)=NC=NN11)N=C1[C@H]1CC[C@H](C(O)=O)CC1 JROFGZPOBKIAEW-HAQNSBGRSA-N 0.000 description 1
- 208000024207 chronic leukemia Diseases 0.000 description 1
- 101150116749 chuk gene Proteins 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 230000000875 corresponding effect Effects 0.000 description 1
- 230000004940 costimulation Effects 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 230000001461 cytolytic effect Effects 0.000 description 1
- 238000002784 cytotoxicity assay Methods 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- 230000001687 destabilization Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 230000007783 downstream signaling Effects 0.000 description 1
- 210000000750 endocrine system Anatomy 0.000 description 1
- 201000003914 endometrial carcinoma Diseases 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 201000001343 fallopian tube carcinoma Diseases 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 230000003325 follicular Effects 0.000 description 1
- 238000002825 functional assay Methods 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 229950008209 gedatolisib Drugs 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 239000005090 green fluorescent protein Substances 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 102000052622 human IL7 Human genes 0.000 description 1
- 230000005931 immune cell recruitment Effects 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- SETFNECMODOHTO-UHFFFAOYSA-N indisulam Chemical compound C1=CC(S(=O)(=O)N)=CC=C1S(=O)(=O)NC1=CC=CC2=C1NC=C2Cl SETFNECMODOHTO-UHFFFAOYSA-N 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 230000001589 lymphoproliferative effect Effects 0.000 description 1
- 230000002101 lytic effect Effects 0.000 description 1
- 208000026037 malignant tumor of neck Diseases 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 210000002990 parathyroid gland Anatomy 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- CXWQAPHDSYDHBR-UHFFFAOYSA-N piperidine-4-carboxamide Chemical compound NC(=O)C1CCNCC1.NC(=O)C1CCNCC1 CXWQAPHDSYDHBR-UHFFFAOYSA-N 0.000 description 1
- 208000021310 pituitary gland adenoma Diseases 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 208000016800 primary central nervous system lymphoma Diseases 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 102220040233 rs79219465 Human genes 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 108091006024 signal transducing proteins Proteins 0.000 description 1
- 102000034285 signal transducing proteins Human genes 0.000 description 1
- 238000004088 simulation Methods 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 239000004055 small Interfering RNA Substances 0.000 description 1
- 201000002314 small intestine cancer Diseases 0.000 description 1
- YQDGWZZYGYKDLR-UZVLBLASSA-K sodium stibogluconate Chemical compound O.O.O.O.O.O.O.O.O.[Na+].[Na+].[Na+].O1[C@H]([C@H](O)CO)[C@H](O2)[C@H](C([O-])=O)O[Sb]21([O-])O[Sb]1(O)(O[C@H]2C([O-])=O)O[C@H]([C@H](O)CO)[C@@H]2O1 YQDGWZZYGYKDLR-UZVLBLASSA-K 0.000 description 1
- 229960001567 sodium stibogluconate Drugs 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 208000017572 squamous cell neoplasm Diseases 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 201000010740 swine influenza Diseases 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229960000235 temsirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-UHFFFAOYSA-N temsirolimus Natural products C1CC(O)C(OC)CC1CC(C)C1OC(=O)C2CCCCN2C(=O)C(=O)C(O)(O2)C(C)CCC2CC(OC)C(C)=CC=CC=CC(C)CC(C)C(=O)C(OC)C(O)C(C)=CC(C)C(=O)C1 QFJCIRLUMZQUOT-UHFFFAOYSA-N 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 230000004222 uncontrolled growth Effects 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 201000011294 ureter cancer Diseases 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 201000004916 vulva carcinoma Diseases 0.000 description 1
- 208000013013 vulvar carcinoma Diseases 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0636—T lymphocytes
- C12N5/0638—Cytotoxic T lymphocytes [CTL] or lymphokine activated killer cells [LAK]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/20—Interleukins [IL]
- A61K38/2086—IL-13 to IL-16
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001103—Receptors for growth factors
- A61K39/001104—Epidermal growth factor receptors [EGFR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001103—Receptors for growth factors
- A61K39/001106—Her-2/neu/ErbB2, Her-3/ErbB3 or Her 4/ErbB4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001103—Receptors for growth factors
- A61K39/001108—Platelet-derived growth factor receptors [PDGFR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001103—Receptors for growth factors
- A61K39/001109—Vascular endothelial growth factor receptors [VEGFR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001111—Immunoglobulin superfamily
- A61K39/001112—CD19 or B4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001111—Immunoglobulin superfamily
- A61K39/001113—CD22, BL-CAM, siglec-2 or sialic acid- binding Ig-related lectin 2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001116—Receptors for cytokines
- A61K39/001117—Receptors for tumor necrosis factors [TNF], e.g. lymphotoxin receptor [LTR] or CD30
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001116—Receptors for cytokines
- A61K39/001119—Receptors for interleukins [IL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001122—Ephrin Receptors [Eph]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001124—CD20
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001126—CD38 not IgG
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001102—Receptors, cell surface antigens or cell surface determinants
- A61K39/001129—Molecules with a "CD" designation not provided for elsewhere
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001148—Regulators of development
- A61K39/001149—Cell cycle regulated proteins, e.g. cyclin, CDC, CDK or INK-CCR
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001148—Regulators of development
- A61K39/00115—Apoptosis related proteins, e.g. survivin or livin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001148—Regulators of development
- A61K39/00115—Apoptosis related proteins, e.g. survivin or livin
- A61K39/001151—Apoptosis related proteins, e.g. survivin or livin p53
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001152—Transcription factors, e.g. SOX or c-MYC
- A61K39/001153—Wilms tumor 1 [WT1]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001154—Enzymes
- A61K39/001156—Tyrosinase and tyrosinase related proteinases [TRP-1 or TRP-2]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001154—Enzymes
- A61K39/001157—Telomerase or TERT [telomerase reverse transcriptase]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001154—Enzymes
- A61K39/001164—GTPases, e.g. Ras or Rho
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001166—Adhesion molecules, e.g. NRCAM, EpCAM or cadherins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001166—Adhesion molecules, e.g. NRCAM, EpCAM or cadherins
- A61K39/001168—Mesothelin [MSLN]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001169—Tumor associated carbohydrates
- A61K39/00117—Mucins, e.g. MUC-1
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001169—Tumor associated carbohydrates
- A61K39/001171—Gangliosides, e.g. GM2, GD2 or GD3
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001176—Heat shock proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/00118—Cancer antigens from embryonic or fetal origin
- A61K39/001182—Carcinoembryonic antigen [CEA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001184—Cancer testis antigens, e.g. SSX, BAGE, GAGE or SAGE
- A61K39/001188—NY-ESO
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/00119—Melanoma antigens
- A61K39/001191—Melan-A/MART
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/00119—Melanoma antigens
- A61K39/001192—Glycoprotein 100 [Gp100]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001193—Prostate associated antigens e.g. Prostate stem cell antigen [PSCA]; Prostate carcinoma tumor antigen [PCTA]; PAP or PSGR
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001193—Prostate associated antigens e.g. Prostate stem cell antigen [PSCA]; Prostate carcinoma tumor antigen [PCTA]; PAP or PSGR
- A61K39/001195—Prostate specific membrane antigen [PSMA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001196—Fusion proteins originating from gene translocation in cancer cells
- A61K39/001197—Breakpoint cluster region-abelson tyrosine kinase [BCR-ABL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/461—Cellular immunotherapy characterised by the cell type used
- A61K39/4611—T-cells, e.g. tumor infiltrating lymphocytes [TIL], lymphokine-activated killer cells [LAK] or regulatory T cells [Treg]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4631—Chimeric Antigen Receptors [CAR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4636—Immune checkpoint inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
- A61K39/464403—Receptors for growth factors
- A61K39/464404—Epidermal growth factor receptors [EGFR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
- A61K39/464403—Receptors for growth factors
- A61K39/464406—Her-2/neu/ErbB2, Her-3/ErbB3 or Her 4/ ErbB4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
- A61K39/464411—Immunoglobulin superfamily
- A61K39/464412—CD19 or B4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464466—Adhesion molecules, e.g. NRCAM, EpCAM or cadherins
- A61K39/464468—Mesothelin [MSLN]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464469—Tumor associated carbohydrates
- A61K39/46447—Mucins, e.g. MUC-1
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464469—Tumor associated carbohydrates
- A61K39/464471—Gangliosides, e.g. GM2, GD2 or GD3
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464493—Prostate associated antigens e.g. Prostate stem cell antigen [PSCA]; Prostate carcinoma tumor antigen [PCTA]; Prostatic acid phosphatase [PAP]; Prostate-specific G-protein-coupled receptor [PSGR]
- A61K39/464495—Prostate specific membrane antigen [PSMA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0636—T lymphocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/515—Animal cells
- A61K2039/5156—Animal cells expressing foreign proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55522—Cytokines; Lymphokines; Interferons
- A61K2039/55527—Interleukins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/10—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterized by the structure of the chimeric antigen receptor [CAR]
- A61K2239/21—Transmembrane domain
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/10—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterized by the structure of the chimeric antigen receptor [CAR]
- A61K2239/22—Intracellular domain
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/31—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterized by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/38—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the dose, timing or administration schedule
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/48—Blood cells, e.g. leukemia or lymphoma
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/49—Breast
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/58—Prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/59—Reproductive system, e.g. uterus, ovaries, cervix or testes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/7051—T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/22—Immunoglobulins specific features characterized by taxonomic origin from camelids, e.g. camel, llama or dromedary
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/569—Single domain, e.g. dAb, sdAb, VHH, VNAR or nanobody®
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/62—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising only variable region components
- C07K2317/622—Single chain antibody (scFv)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/03—Fusion polypeptide containing a localisation/targetting motif containing a transmembrane segment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/32—Fusion polypeptide fusions with soluble part of a cell surface receptor, "decoy receptors"
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
Abstract
La invención proporciona composiciones y métodos para tratar enfermedades asociadas con la expresión de un antígeno asociado al cáncer como se describe en el presente documento. La invención también se refiere al receptor de antígeno quimérico (CAR) específico para un antígeno asociado al cáncer como se describe en el presente documento, vectores que codifican el mismo y células T recombinantes que comprenden los CAR de la presente invención. La invención también incluye métodos para administrar un linfocito T genéticamente modificado que expresa un CAR que comprende un dominio de unión a antígeno que se une a un antígeno asociado al cáncer como se describe en el presente documento. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Tratamiento de cáncer utilizando un receptor quimérico para antígenos
Campo de la invención
SOLICITUDES RELACIONADAS
Esta solicitud reivindica la prioridad de la solicitud N° de serie de EE.UU. N° 61/953.783, presentada el 15 de marzo de 2014, de la solicitud N° de serie de EE.UU. 61/976.375, presentada el 7 de abril de 2014, de la solicitud N° de serie de EE.UU. 62/027.154, presentada el 21 de julio de 2014, de la solicitud N° de serie de EE.UU. 62/076.146, presentada el 6 de noviembre de 2014 y de la solicitud N° de serie de EE.UU. 62/097.286, presentada el 29 de diciembre de 2014. CAMPO DE LA INVENCIÓN
La presente invención se refiere en general al uso de células efectoras inmunitarias (p. ej., células T, células diseñadas para expresar un receptor quimérico para antígenos (CAR, por sus siglas en inglés) para tratar una enfermedad asociada a la expresión de un antígeno tumoral.
ANTECEDENTES DE LA INVENCIÓN
La terapia de transferencia celular adoptiva (ACT, por sus siglas en inglés) con células T autólogas, especialmente con células T transducidas con receptores quiméricos para antígenos (CAR), se ha mostrado prometedora en ensayos de cáncer hematológico. Jamnani et al., Biochimica et Biophysica Acta 1840 (2014):378-386 describe un CAR con VHH anti-HER2. Pirooznia et al., J Biomed Biotech (2011):578128 se refiere a modelos de CARs con VHH. Federov et al., Sci Transl Med (2013);5(215):215ra172 informa que los CAR inhibidores basados en PD-1 y CTLA-4 desvían las respuestas de inmunoterapia fuera del objetivo.
SUMARIO DE LA INVENCIÓN
La presente invención proporciona una célula efectora inmunitaria (p. ej., una población de células efectoras inmunitarias), que comprende:
(a) una primera molécula de receptor de antígeno quimérico (CAR) o ácido nucleico que la codifica, en donde dicha primera molécula de CAR es un polipéptido de CAR que comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio de señalización intracelular que comprende un dominio co-estimulador y/o un dominio de señalización primario, y en donde dicho dominio de unión a antígenos comprende un dominio VHH y se une a un antígeno tumoral; y
(b) una segunda molécula de CAR o ácido nucleico que la codifica, en donde dicha segunda molécula de CAR es un polipéptido CAR que tiene un dominio de unión a antígenos que comprende un scFv.
La presente invención también proporciona dicha célula efectora inmunitaria para uso como un medicamento.
La presente invención también proporciona dicha célula efectora inmunitaria para uso en el tratamiento de una enfermedad que expresa un antígeno tumoral.
Éstas y realizaciones adicionales se recogen en las reivindicaciones adjuntas.
DESCRIPCIÓN DETALLADA
La información técnica recogida a continuación puede, en algunos aspectos, ir más allá del alcance de la invención, que está definida por las reivindicaciones adjuntas. La información técnica adicional se proporciona para ubicar la invención real en un contexto técnico más amplio y para ilustrar posibles desarrollos técnicos relacionados.
Además, referencias incidentales a métodos para el tratamiento del cuerpo humano o animal mediante cirugía o terapia y los métodos de diagnóstico practicados en el cuerpo humano o animal no deben interpretarse como que constituyen reivindicación de protección para un método de este tipo por sí mismo, sino que deben ser considerados como una referencia a productos, en particular sustancias o composiciones, para uso en cualquiera de estos métodos.
La presente divulgación se refiere, al menos en parte, al uso de células efectoras inmunitarias (p. ej., células T, células NK) diseñadas para expresar un CAR que se une a un antígeno tumoral como se describe en esta memoria para tratar cáncer asociado con la expresión de dicho antígeno tumoral. La presente invención proporciona una célula efectora inmunitaria que comprende un primer CAR y un segundo CAR, o que comprende ácidos nucleicos que codifican los mismos y el uso terapéutico de una célula de este tipo, como se define en las reivindicaciones adjuntas.
Ácidos nucleicos que codifican CAR
Por consiguiente, en un aspecto, la invención emplea una molécula de ácido nucleico aislada que codifica un receptor de antígeno quimérico (CAR), en donde el CAR comprende un dominio de unión a antígenos que se une a un antígeno tumoral como se describe en esta memoria, un dominio transmembrana (p. ej., un dominio transmembrana descrito en esta memoria) y un dominio de señalización intracelular (p. ej., un dominio de señalización intracelular descrito en esta memoria) (p. ej., un dominio de señalización intracelular que comprende un dominio coestimulador (p. ej., un dominio coestimulador descrito en esta memoria) y/o un dominio de señalización primario (p. ej, un dominio de señalización primario descrito en esta memoria). ; En algunas realizaciones, el antígeno tumoral se elige entre uno o más de: CD19; CD123; CD22; CD30; CD171; CS-1 (también denominado subconjunto 1 de CD2, CRACC, SLAMF7, CD319 y 19A24); molécula 1 similar a lectina de tipo C (CLL-1 o CLECL1); CD33; variante III del receptor del factor de crecimiento epidérmico (EGFRvIII); gangliósido G2 (GD2); gangliósido GD3 (aNeu5Ac(2-8)aNeu5Ac(2-3)bDGalp(1-4)bDGlcp(1-1)Cer); maduración de células B de miembros de la familia de receptores de TNF (BCMA); antígeno Tn ((Tn Ag) o (GalNAca-Ser/Thr)); antígeno de membrana específico de próstata (PSMA); Receptor huérfano de tipo tirosina quinasa 1 (ROR1); Tirosina quinasa 3 de tipo Fms (FLT3); glucoproteína 72 asociada a tumores (TAG72); CD38; CD44v6; antígeno carcinoembrionario (CEA); molécula de adhesión de células epiteliales (EPCAM); B7H3 (CD276); KIT (CD117); subunidad alfa-2 del receptor de interleucina-13 (IL-13Ra2 o CD213A2); mesotelina; receptor alfa de interleucina 11 (IL-11Ra); antígeno de células madre prostáticas (PSCA); proteasa serina 21 (testisina o PRSS21); receptor 2 del factor de crecimiento endotelial vascular (VEGFR2); antígeno de Lewis(Y); CD24; receptor beta del factor de crecimiento derivado de plaquetas (PDGFR-beta); antígeno embrionario 4 específico de etapa (SSEA-4); CD20; receptor de folato alfa; tirosinaproteína quinasa receptora ERBB2 (Her2/neu); Mucina 1, asociada a la superficie celular (MUC1); receptor del factor de crecimiento epidérmico (EGFR); molécula de adhesión de células neurales (NCAM); prostasa; fosfatasa ácida prostática (PAP); factor de elongación 2 mutado (ELF2M); efrina B2; proteína alfa de activación de fibroblastos (FAP); receptor del factor insulínico de crecimiento 1 (receptor de IGF-I), anhidrasa carbónica IX (CAIX); subunidad de proteasoma (prosoma, macropaína), tipo beta, 9 (LMP2); glucoproteína 100 (gp100); proteína de fusión de oncogén que consiste en la región de agrupamiento de puntos de ruptura (BCR) y el homólogo 1 del oncogén vírico de la leucemia murina de Abelson (Abl) (bcr-abl); tirosinasa; receptor 2 de efrina de tipo A (EphA2); Fucosil GM1; molécula de adhesión de sialilo de Lewis (sLe); gangliósido GM3 (aNeu5Ac(2-3)bDGalp(1-4)bDGlcp(1-1)Cer); transglutaminasa 5 (TGS5); antígeno asociado a melanoma de alto peso molecular (HMWMAA); gangliósido o-acetil-GD2 (OAcGD2); receptor de folato beta; marcador endotelial tumoral 1 (TEM1/CD248); relacionado con el marcador endotelial tumoral 7 (TEM7R); claudina 6 (CLDN6); receptor de la hormona estimulante del tiroides (TSHR); receptor acoplado a proteína G clase C grupo 5, miembro D (GPRC5D); marco de lectura abierto 61 del cromosoma X (CXORF61); CD97; CD179a; quinasa de linfoma anaplásico (ALK); ácido polisiálico; específico de placenta 1 (PLAC1); porción de hexasacárido de glucoceramida globoH (GloboH); antígeno de diferenciación de glándula mamaria (NY-BR-1); uroplaquina 2 (UPK2); receptor celular 1 del virus de la hepatitis A (HAVCR1); adrenoceptor beta 3 (ADRB3); panexina 3 (PANX3); receptor acoplado a proteína G 20 (GPR20); complejo de antígeno de células 6, locus K 9 (LY6K); receptor olfativo 51E2 (OR51E2); Proteína de Marco de Lectura Alternativa TCR Gamma (TARP); proteína del tumor de Wilms (WT1); antígeno de cáncer/testículo 1 (NY-ESO-1); antígeno de cáncer/testículo 2 (LAGE-1a); antígeno 1 asociado al melanoma (MAGE-A1); gen 6 variante de translocación de ETS, ubicado en el cromosoma 12p (ETV6-AML); proteína de espermatozoide 17 (SPA17); familia de antígenos X, miembro 1A (XAGE1); receptor de superficie celular de unión a angiopoyetina 2 (Tie 2); antígeno de testículo y cáncer de melanoma 1 (MAD-CT-1); antígeno de testículo y cáncer de melanoma 2 (MAD-CT-2); antígeno 1 relacionado con Fos; proteína tumoral p53 (p53); p53 mutante; prosteína; survivina; telomerasa; antígeno tumoral de carcinoma de próstata-1 (PCTA-1 o Galectina 8), antígeno de melanoma reconocido por células T 1 (MelanA o MARTI); mutante de sarcoma de rata (Ras); transcriptasa inversa de telomerasa humana (hTERT); puntos de ruptura de translocación de sarcoma; inhibidor de melanoma de la apoptosis (ML-IAP); ERG (proteasa transmembrana, gen de fusión de ETS y serina 2 (TMPRSS2)); N-acetil glucosaminil-transferasa V (NA17); proteína de caja emparejada Pax-3 (PAX3); receptor de andrógenos; ciclina B1; homólogo derivado del neuroblastoma del oncogén vírico de la mielocitomatosis aviar v-myc (MYCN); miembro C de la familia de homólogos de Ras (RhoC); proteína 2 relacionada con la tirosinasa (TRP-2); citocromo P450 1B1 (CYP1B1); similar al factor de unión a CCCTC (proteína de dedo de zinc) (BORIS o hermano del regulador de sitios impresos), antígeno de carcinoma de células escamosas reconocido por células T 3 (SART3); proteína de caja emparejada Pax-5 (PAX5); proteína de unión a proacrosina sp32 (OY-TES1); proteína tirosina quinasa específica de células (LCK); proteína de anclaje de A quinasa 4 (AKAP-4); sarcoma sinovial, punto de ruptura 2 de X (SSX2); Receptor para productos finales de glucación avanzada (RAGE-1); renal ubicuo 1 (RU1); renal ubicuo 2 (RU2); legumaína; virus del papiloma humano E6 (HPV E6); virus del papiloma humano E7 (HPV E7); carboxil esterasa intestinal; proteína de choque térmico 70-2 mutada (mut hsp70-2); CD79a; CD79b; CD72; receptor 1 de tipo inmunoglobulina asociado a leucocitos (LAIR1); fragmento Fc del receptor de IgA (FCAR o CD89); miembro 2 de la subfamilia A del receptor de tipo inmunoglobulina leucocitaria (LILRA2); miembro f de la familia de tipo molécula CD300 (CD300LF); miembro A de la familia 12 de dominios de lectina de tipo C (CLEC12A); antígeno 2 de células estromales de la médula ósea (BST2); tipo receptor de hormona de tipo mucina que contiene módulo de tipo EGF 2 (EMR2); antígeno de células 75 (LY75); Glipicano-3 (GPC3); tipo receptor de Fc 5 (FCRL5); y el polipéptido 1 de tipo inmunoglobulina lambda (IGLL1).
En algunas realizaciones, el antígeno tumoral al que se une la molécula de CAR codificada se elige entre uno o más de: TSHR, CD171, CS-1, CLL-1, GD3, Tn Ag, FLT3, CD38, CD44v6, B7H3, KIT, IL-13Ra2, IL-11Ra, PSCA, PRSS21, VEGFR2, LewisY, CD24, PDGFR-beta, SSEA-4, MUC1, EGFR, NCAM, CAIX, LMP2, EphA2, Fucosil GM1, sLe, GM3,
TGS5, HMWMAA, o-acetil-GD2, receptor de folato beta, TEM1/CD248, TEM7R, CLDN6, GPRC5D, CXORF61, CD97, CD179a, ALK, ácido polisiálico, PLACI, GloboH, NY-BR-1, UPK2, HAVCR1, ADRB3, PANX3, GPR20, LY6K, OR51E2, TARP, WT1, ETV6-AML, proteína de espermatozoide 17, XAGE1, Tie 2, MAD- CT-1, MAD-CT-2, antígeno relacionado con Fos 1, p53 mutante, hTERT, puntos de corte de translocación de sarcoma, ML-IAP, ERG (gen de fusión de TMPRSS2 y ETS), NA17, PAX3, receptor de andrógenos, ciclina B1, MYCN, RhoC, CYP1B1, BORIS, SART3, PAX5, OY-TES1, LCK, AKAP-4, SSX2, CD79a, CD79b, CD72, LAIR1, FCAR, LILRA2, CD300LF, CLEC12A, BST2, EMR2, LY75, GPC3, FCRL5 e IGLL1.
En determinadas realizaciones, el antígeno tumoral al que se une la molécula de CAR codificada se elige entre uno o más de: TSHR, CLDN6, GPRC5D, CXORF61, CD97, CD179a, ALK, Polysialic acid, PLAC1, GloboH, NY-BR-1, UPK2, HAVCR1, ADRB3, PANX3, GPR20, LY6K y OR51E2.
En algunos casos, el dominio de unión al antígeno de la molécula de CAR codificada comprende un anticuerpo, un fragmento de anticuerpo, un scFv, un Fv, un Fab, un (Fab')2, un anticuerpo de dominio único (SDAB), un dominio VH o VL, o un dominio VHH de camélido. En la presente invención, el dominio de unión a antígenos del primer CAR comprende un dominio VHH y el dominio de unión a antígenos del segundo CAR comprende un scFv.
En algunas realizaciones, el dominio transmembrana de la molécula de CAR codificada comprende un dominio transmembrana elegido del dominio transmembrana de una cadena alfa, beta o zeta de un receptor de células T, CD28, CD3 épsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137, CD154, KIRDS2, OX40, CD2, CD27, LFA-1 (CD11a, CD18), ICOS (CD278), 4-1BB (CD137), GITR, CD40, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), CD160, CD19, IL2R beta, IL2R gamma, IL7R a, ITGA1, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, TNFR2, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), SLAMF6 (NTB-A, Ly108), SLAM (SLAMF1, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, PAG/Cbp, NKp44, NKp30, NKp46, NKG2D y/o NKG2C.
En determinadas realizaciones, el dominio transmembrana codificado comprende una secuencia de aminoácidos de un dominio transmembrana de CD8 que tiene al menos una, dos o tres modificaciones pero no más de 20, 10 o 5 modificaciones de una secuencia de aminoácidos de SEQ ID NO: 12, o una secuencia con una identidad del 95-99 % con una secuencia de aminoácidos de la SEQ ID NO: 12. En una realización, el dominio transmembrana codificado comprende la secuencia de la SEQ ID NO: 12.
En otras realizaciones, la molécula de ácido nucleico comprende una secuencia de nucleótidos de un dominio transmembrana CD8, p. ej., que comprende la secuencia de la SEQ ID NO: 13 o una secuencia con una identidad del 95 99 % de la misma.
En determinadas realizaciones, el dominio de unión a antígenos codificado está conectado al dominio transmembrana mediante una región de bisagra. En una realización, la región de bisagra codificada comprende la secuencia de aminoácidos de una bisagra de CD8, p. ej., la SEQ ID NO: 2; o la secuencia de aminoácidos de una bisagra de IgG4, p. ej., la SEQ ID NO: 6 o una secuencia con una identidad del 95-99 % con SEQ ID NO: 2 o 6. En otras realizaciones, la secuencia de ácido nucleico que codifica la región de bisagra comprende una secuencia de la SEQ ID NO: 3 o la SEQ ID NO: 7, correspondiente a una bisagra de CD8 o una bisagra de IgG4, respectivamente, o una secuencia con una identidad del 95-99 % con la SEQ ID NO: 3 o 7.
En otras realizaciones, la molécula de ácido nucleico codifica un dominio de señalización intracelular que comprende una secuencia que codifica un dominio de señalización primario y/o una secuencia que codifica un dominio de señalización coestimulador. En algunas realizaciones, el dominio de señalización intracelular comprende una secuencia que codifica un dominio de señalización primario. En algunas realizaciones, el dominio de señalización intracelular comprende una secuencia que codifica un dominio de señalización coestimulador. En algunas realizaciones, el dominio de señalización intracelular comprende una secuencia que codifica un dominio de señalización primario y una secuencia que codifica un dominio de señalización coestimulador.
En determinadas realizaciones, el dominio de señalización primario codificado comprende un dominio de señalización funcional de una proteína seleccionada del grupo que consiste en CD3 zeta, CD3 gamma, CD3 delta, CD3 épsilon, FcR gamma común (FCER1G), FcR beta (Fc Épsilon R1b), CD79a, CD79b, Fcgamma RIIa, DAP10 y DAP12.
En una realización, el dominio de señalización primario codificado comprende un dominio de señalización funcional de CD3 zeta. El dominio de señalización primario de CD3 zeta codificado puede comprender una secuencia de aminoácidos que tiene al menos una, dos o tres modificaciones, pero no más de 20, 10 o 5 modificaciones de una secuencia de aminoácidos de la SEQ ID NO: 18 o la SEQ ID NO: 20, o una secuencia con una identidad del 95-99 % con una secuencia de aminoácidos de la SEQ ID NO: 18 o la SEQ ID NO: 20. En algunas realizaciones, el dominio de señalización primario codificado comprende una secuencia de la SEQ ID NO: 18 o la SEQ ID NO: 20. En otras realizaciones, la secuencia de ácido nucleico que codifica el dominio de señalización primario comprende una secuencia de la SEQ ID NO: 19 o la SEQ ID NO: 21 o una secuencia con una identidad del 95-99 % de la misma.
En algunas realizaciones, el dominio de señalización intracelular codificado comprende una secuencia que codifica un dominio de señalización coestimulador. Por ejemplo, el dominio de señalización intracelular puede comprender una secuencia que codifica un dominio de señalización primario y una secuencia que codifica un dominio de señalización coestimulador. En algunas realizaciones, el dominio de señalización coestimulador codificado comprende un dominio de señalización funcional de una proteína elegida entre uno o más de CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, PD-1, ICOS, antígeno-1 asociado a la función de los célula (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-H3, un ligando que se une específicamente con CD83, CDS, ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), CD160, CD19, CD4, CD8alfa, CD8beta, IL2R beta, IL2R gamma, IL7R alfa, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, TNFR2, TRANCE/RANKL, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), CD69, SLAMF6 (NTB-A, Ly108), SLAM (SLAMF1, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, LAT, GADS, SLP-76, PAG/Cbp, NKp44, NKp30, NKp46 o NKG2D.
En determinadas realizaciones, el dominio de señalización coestimulador codificado comprende una secuencia de aminoácidos que tiene al menos una, dos o tres modificaciones pero no más de 20, 10 o 5 modificaciones de una secuencia de aminoácidos de la SEQ ID NO: 14 o la SEQ ID NO: 16, o una secuencia con una identidad del 95-99 % con una secuencia de aminoácidos de la SEQ ID NO: 14 o la SEQ ID NO: 16. En una realización, el dominio de señalización coestimulador codificado comprende una secuencia de la SEQ ID NO: 14 o la SEQ ID NO: 16. En otras realizaciones, la secuencia de ácido nucleico que codifica el dominio de señalización coestimulador comprende una secuencia de la SEQ ID NO: 15 o la SEQ ID NO: 17 o una secuencia con una identidad del 95-99 % de la misma.
En otras realizaciones, el dominio intracelular codificado comprende la secuencia de las SEQ ID NO: 14 o la SEQ ID NO: 16, y la secuencia de la SEQ ID NO: 18 o la SEQ ID NO: 20, en donde las secuencias que comprenden el dominio de señalización intracelular se expresan en el mismo marco y como una sola cadena polipeptídica.
En determinadas realizaciones, la secuencia de ácido nucleico que codifica el dominio de señalización intracelular comprende una secuencia de la SEQ ID NO: 15 o la SEQ ID NO: 17, o una secuencia con una identidad del 95-99 % de la misma, y una secuencia de la SEQ ID NO: 19 o la SEQ ID NO: 21 o una secuencia con una identidad del 95-99 % de la misma.
En algunas realizaciones, la molécula de ácido nucleico comprende además una secuencia líder. En una realización, la secuencia líder comprende la secuencia de la SEQ ID NO: 2.
En determinadas realizaciones, el dominio de unión a antígenos codificado tiene una KD de afinidad de unión de 10'4 M a 10-8 M.
En un caso, el dominio de unión a antígenos codificado es un dominio de unión a antígenos descrito en esta memoria, p. ej., un dominio de unión a antígenos descrito en esta memoria para un objetivo arriba proporcionado .
En una realización, la molécula de CAR codificada comprende un dominio de unión a antígenos que tiene una KD de afinidad de unión de 10'4 M a 10'8 M, p. ej., de 10'5 M a 10'7 M, p. ej., 10'6 M o 10'7 M, para el antígeno diana. En una realización, el dominio de unión a antígenos tiene una afinidad de unión que es al menos cinco veces, 10 veces, 20 veces, 30 veces, 50 veces, 100 veces o 1000 veces inferior a un anticuerpo de referencia, p. ej., un anticuerpo descrito en esta memoria. En una realización, el dominio de unión a antígenos codificado tiene una afinidad de unión al menos 5 veces menor que un anticuerpo de referencia (p. ej., un anticuerpo del que procede el dominio de unión a antígenos).
En un aspecto, la invención emplea una molécula de ácido nucleico aislada que codifica un receptor de antígeno quimérico (CAR), en donde el CAR comprende un dominio de unión a antígenos que se une a un antígeno de que sustenta el tumor (p. ej., un antígeno que sustenta el tumor como se describe en esta memoria) y un dominio de señalización intracelular (p. ej., un dominio de señalización intracelular descrito en esta memoria) (p. ej., un dominio de señalización intracelular que comprende un dominio coestimulador (p. ej., un dominio coestimulador descrito en esta memoria) y/o un dominio de señalización primario (p. ej, un dominio de señalización primario descrito en esta memoria). En algunas realizaciones, el antígeno que sustenta el tumor es un antígeno presente en una célula del estroma o una célula supresora de origen mieloide (MDSC).
Vectores
En otro aspecto, la invención emplea un vector que comprende una secuencia de ácido nucleico que codifica un CAR de acuerdo con las reivindicaciones. En una realización, el vector se selecciona de un vector de ADN o un vector de ARN, un plásmido, un vector lentivírico, un vector adenovírico o un vector retrovírico. En una realización, el vector es un vector lentivírico.
En una realización, el vector comprende una secuencia de ácido nucleico que codifica un CAR y una secuencia de ácido nucleico que codifica una molécula inhibidora que comprende: un dominio citoplasmático inhKIR; un dominio transmembrana, p. ej., un dominio transmembrana KIR; y un dominio citoplasmático inhibidor, p. ej., un dominio ITIM, p. ej., un dominio ITIM inhKIR. En una realización, la molécula inhibidora es un inhKIR de origen natural o una secuencia que comparte al menos un 50, 60, 70, 80, 85, 90, 95 o 99 % de homología con, o que no difiere en más de 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 15 o 20 residuos de, un inhKIR de origen natural.
En una realización, la secuencia de ácido nucleico que codifica una molécula inhibidora comprende: un dominio citoplasmático de la familia SLAM; un dominio transmembrana, p. ej., un dominio transmembrana de la familia SLAM; y un dominio citoplasmático inhibidor, p. ej., un dominio de la familia SLAM, p. ej., un dominio ITIM de la familia SLAM. En una realización, la molécula inhibidora es un SLAM de origen natural o una secuencia que comparte al menos un 50, 60, 70, 80, 85, 90, 95 o 99 % de homología con, o que no difiere en más de 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 15 o 20 residuos de, un SLAM de origen natural.
En una realización, el vector comprende, además, un promotor. En algunas realizaciones, el promotor se elige entre un promotor EF-1, un promotor del gen IE del CMV, un promotor EF-1a, un promotor de ubiquitina C o un promotor de fosfoglicerato quinasa (PGK). En una realización, el promotor es un promotor EF-1. En una realización, el promotor EF-1 comprende una secuencia de la SEQ ID NO: 1.
En una realización, el vector es un vector transcrito in vitro, p. ej., un vector que transcribe ARN de una molécula de ácido nucleico de acuerdo con las reivindicaciones. En una realización, la secuencia de ácido nucleico en el vector comprende, además, una cola de poli(A), p. ej., una cola de poli A descrita en esta memoria, p. ej., que comprende aproximadamente 150 bases de adenosina (SEQ ID NO: 33). En una realización, la secuencia de ácido nucleico en el vector comprende, además, una 3'UTR, p. ej., una 3'UTR descrita en esta memoria, p. ej., que comprende al menos una repetición de una 3'UTR procedente de beta-globulina humana. En una realización, la secuencia de ácido nucleico en el vector comprende además un promotor, p. ej., un promotor de T2A.
Polipéptidos CAR
En otro aspecto, la invención emplea una molécula de polipéptido CAR aislada que comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio de señalización intracelular, en donde dicho dominio de unión a antígenos se une a un antígeno tumoral elegido entre uno o más de: CD19, CD123, CD22, CD30, CD171, CS-1, CLL-1 (CLECL1), CD33, EGFRvIII, GD2, GD3, BCMA, Tn Ag, PSMA, ROR1, FLT3, TAG72, CD38, CD44v6, CEA, EPCAM, B7H3, KIT, IL-13Ra2, mesotelina, IL-11Ra, PSCA, PRSS21, VEGFR2, LewisY, CD24, PDGFR-beta, SSEA-4, CD20, receptor de folato alfa, ERBB2 (Her2/neu), MUC1, EGFR, NCAM, prostasa, PAP, ELF2M, efrina B2, FAP, receptor de IGF-I, CAIX, LMP2, gp100, bcr-abl, tirosinasa, EphA2, Fucosil GM1, sLe, GM3, TGS5, HMWMAA, o-acetil-GD2, receptor de folato beta, TEM1/CD248, TEM7R, CLDN6, TSHR, GPRC5D, CXORF61, CD97, CD179a, ALK, ácido polisiálico, PLAC1, GloboH, NY-BR-1, UPK2, HAVCR1, ADRB3, PANX3, GPR20, LY6K, OR51E2, TARP, WT1, NY-ESO-1, LAGE-1a, MAGE-A1, MAGE A1, ETV6-AML, proteína de espermatozoide 17, XAGE1, Tie 2, MAD-CT-1, MAD-CT-2, antígeno relacionado con Fos 1, p53, p53 mutante, prosteína, survivina y telomerasa, PCTA-1/Galectina 8, MelanA/MART1, Ras mutante, hTERT, puntos de corte de translocación de sarcoma, ML-IAP, ERG (gen de fusión de TMPRSS2 y ETS), NA17, PAX3, receptor de andrógenos, ciclina B1, MYCN, RhoC, TRP-2, CYP1B1, BORIS, SART3, PAX5, OY-TES1, LCK, AKAP-4, SSX2, RAGE-1, transcriptasa inversa de telomerasa humana, RU1, RU2, legumaína, HPV E6,E7, carboxil esterasa intestinal, mut hsp70-2, CD79a, CD79b, CD72, LAIR1, FCAR, LILRA2, CD300LF, CLEC12A, BST2, EMR2, LY75, GPC3, FCRL5 e IGLL1.
En algunas realizaciones, el dominio de unión a antígenos de la molécula polipeptídica CAR se une a un antígeno tumoral elegido entre uno o más de: TSHR, CD171, CS-1, CLL-1, GD3, Tn Ag, FLT3, CD38, CD44v6, B7H3, KIT, IL-13Ra2, IL-11Ra, PSCA, PRSS21, VEGFR2, LewisY, CD24, PDGFR-beta, SSEA-4, MUC1, EGFR, NCAM, CAIX, LMP2, EphA2, Fucosil GM1, sLe, GM3, TGS5, HMWMAA, o-acetil-GD2, receptor de folato beta, TEM1/CD248, TEM7R, CLDN6, GPRC5D, CXORF61, CD97, CD179a, ALK, ácido polisiálico, PLAC1, GloboH, NY-BR-1, UPK2, HAVCR1, ADRB3, PANX3, GPR20, LY6K, OR51E2, TARP, WT1, ETV6-AML, proteína de espermatozoide 17, XAGE1, Tie 2, MAD- CT-1, MAD-CT-2, antígeno relacionado con Fos 1, p53 mutante, hTERT, puntos de corte de translocación de sarcoma, ML-IAP, ERG (gen de fusión de TMPRSS2 y ETS), NA17, PAX3, receptor de andrógenos, ciclina B1, MYCN, RhoC, CYP1B1, BORIS, SART3, PAX5, OY-TES1, LCK, AKAP-4, SSX2, CD79a, CD79b, CD72, LAIR1, FCAR, LILRA2, CD300LF, CLEC12A, BST2, EMR2, LY75, GPC3, FCRL5 e IGLL1.
En algunas realizaciones, el dominio de unión a antígenos de la molécula polipeptídica CAR se une a un antígeno tumoral elegido entre uno o más de: TSHR, CLDN6, GPRC5D, CXORF61, CD97, CD179a, ALK, ácido polisiálico, PLAC1, GloboH, NY-BR-1, UPK2, HAVCR1, ADRB3, PANX3, GPR20, LY6K y OR51E2.
En algunos casos, el dominio de unión a antígenos de la molécula polipeptídica CAR comprende un anticuerpo, un fragmento de anticuerpo, un scFv, un Fv, un Fab, un (Fab')2, un anticuerpo de dominio único (SDAB), un dominio VH o VL, o un dominio VHH de camélido. En la presente invención, el dominio de unión a antígenos del primer CAR comprende un dominio VHH y el dominio de unión a antígenos del segundo CAR comprende un scFv.
En algunas realizaciones, el dominio de unión a antígenos de la molécula polipeptídica CAR comprende un dominio transmembrana de una proteína elegida de una cadena alfa, beta o zeta de un receptor de células T, CD28, CD3 épsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137, CD154, KIRDS2, OX40, CD2, CD27, LFA-1 (CD11a, CD18), ICOS (CD278), 4-1BB (CD137), GITR, CD40, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), CD160, CD19, IL2R beta, IL2R gamma, IL7R a, ITGA1, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, TNFR2, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), SLAMF6 (NTB-A, Ly108), SLAM (SLAMF1, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, PAG/Cbp, NKp44, NKp30, NKp46, NKG2D y/o NKG2C.
En otras realizaciones, el dominio transmembrana de la molécula polipeptídica CAR comprende una secuencia de aminoácidos que tiene al menos una, dos o tres modificaciones pero no más de 20, 10 o 5 modificaciones de una secuencia de aminoácidos de un dominio transmembrana de CD8, p. ej., SEQ ID NO: 12, 12, o una secuencia con una identidad del 95-99 % con una secuencia de aminoácidos de la SEQ ID NO: 12. En una realización, el dominio transmembrana comprende una secuencia de la SEQ ID NO: 12.
En otras realizaciones, el dominio de unión a antígenos,de la molécula polipeptídica CAR está conectado al dominio transmembrana mediante una región de bisagra. En una realización, la región de bisagra codificada comprende la secuencia de aminoácidos de una bisagra de CD8, p. ej., la SEQ ID NO: 2, o la secuencia de aminoácidos de una bisagra de IgG4, p. ej., la SEQ ID NO: 6 o una secuencia con una identidad del 95-99 % de la misma.
En otras realizaciones, el dominio de señalización intracelular de la molécula polipeptídica CAR comprende un dominio de señalización primario y/o un dominio de señalización coestimulador. En otras realizaciones, el dominio de señalización intracelular de la molécula polipeptídica CAR comprende un dominio de señalización primario. En otras realizaciones, el dominio de señalización intracelular de la molécula polipeptídica CAR comprende un dominio de señalización coestimulador. En aún otras realizaciones, el dominio de señalización intracelular de la molécula polipeptídica CAR comprende un dominio de señalización primario yo un dominio de señalización coestimulador.
En otras realizaciones, el dominio de señalización primario el dominio de señalización primario de la molécula polipeptídica CAR comprende un dominio de señalización funcional de una proteína seleccionada del grupo que consiste en CD3 zeta, CD3 gamma, CD3 delta, CD3 épsilon, FcR gamma común (FCER1G), FcR beta (Fc Épsilon R1b), CD79a, CD79b, Fcgamma RIIa, DAP10 y DAP12. En una realización, el dominio de señalización primario comprende un dominio de señalización funcional de CD3 zeta. El dominio de señalización primario de CD3 zeta puede comprender una secuencia de aminoácidos que tiene al menos una, dos o tres modificaciones, pero no más de 20, 10 o 5 modificaciones de una secuencia de aminoácidos de la SEQ ID NO: 18 o la SEQ ID NO: 20, o una secuencia con una identidad del 95-99 % con una secuencia de aminoácidos de la SEQ ID NO: 18 o la SEQ ID NO: 20. En algunas realizaciones, el dominio de señalización primario de la molécula polipeptídica CAR comprende una secuencia de la SEQ ID NO: 18 o la SEQ ID NO: 20.
En algunas realizaciones, el dominio de señalización intracelular de la molécula polipeptídica CAR comprende una secuencia que codifica un dominio de señalización coestimulador. Por ejemplo, el dominio de señalización intracelular puede comprender una secuencia que codifica un dominio de señalización primario y una secuencia que codifica un dominio de señalización coestimulador. En algunas realizaciones, el dominio de señalización coestimulador codificado comprende un dominio de señalización funcional de una proteína elegida entre uno o más de CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, PD-1, ICOS, antígeno-1 asociado a la función de los linfocitos (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-H3, un ligando que se une específicamente con CD83, CDS, ICAM-1, GITR, BaFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), CD160, CD19, CD4, CD8alfa, CD8beta, IL2R beta, IL2R gamma, IL7R alfa, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, TNFR2, TRANCE/RANKL, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), CD69, SLAMF6 (NTB-A, Ly108), SLAM (SLAMF1, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, LAT, GADS, SLP-76, PAG/Cbp, NKp44, NKp30, NKp46 o NKG2D.
En determinadas realizaciones, el dominio de señalización coestimulador de la molécula polipeptídica CAR comprende una secuencia de aminoácidos que tiene al menos una, dos o tres modificaciones pero no más de 20, 10 o 5 modificaciones de una secuencia de aminoácidos de la SEQ ID NO: 14 o la SEQ ID NO: 16, o una secuencia con una identidad del 95-99 % con una secuencia de aminoácidos de la SEQ ID NO: 14 o la SEQ ID NO: 16. En una realización, el dominio de señalización coestimulador codificado comprende una secuencia de la SEQ ID NO: 14 o la SEQ ID NO: 16. En otras realizaciones, el dominio intracelular de la molécula polipeptídica CAR comprende la secuencia de la SEQ ID NO: 14 o la SEQ ID NO: 16, y la secuencia de la SEQ ID nO: 18 o la SEQ ID NO: 20, en donde las secuencias que comprenden el dominio de señalización intracelular se expresan en el mismo marco y como una sola cadena polipeptídica.
En algunas realizaciones, la molécula polipeptídica CAR comprende además una secuencia líder. En una realización, la secuencia líder comprende la secuencia de la SEQ ID NO: 2.
En determinadas realizaciones, el dominio de unión a antígenos de la molécula polipeptídica CAR tiene una KD de afinidad de unión de 10-4 M a 10-8 M. En una realización, el dominio de unión a antígenos es un dominio de unión a antígenos descrito en esta memoria, p. ej., un dominio de unión a antígenos descrito en esta memoria para una diana proporcionada anteriormente. En una realización, la molécula de CAR comprende un dominio de unión a antígenos que tiene una KD de afinidad de unión de 10-4 M a 10-8 M, p. ej., de 10-5 M a 10-7 M, p. ej., 10-6 M o 10-7 M, para el antígeno diana. En una realización, el dominio de unión a antígenos tiene una afinidad de unión que es al menos cinco veces, 10 veces, 20 veces, 30 veces, 50 veces, 100 veces o 1000 veces inferior a un anticuerpo de referencia, p. ej., un anticuerpo descrito en esta memoria. En una realización, el dominio de unión a antígenos codificado tiene una afinidad de unión al menos 5 veces menor que un anticuerpo de referencia (p. ej., un anticuerpo del que procede el dominio de unión a antígenos).
En otro aspecto, la invención emplea una molécula polipeptídica CAR aislada que comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio de señalización intracelular, en donde dicho dominio de unión a antígenos se une a un antígeno tumoral como se describe en esta memoria). En algunos casos, el antígeno que sustenta el tumor es un antígeno presente en una célula del estroma o una célula supresora derivada de mieloide (MDSC).
Células que expresan CAR
En otro aspecto, la divulgación pertenece a una célula, p. ej., una célula efectora inmunitaria (p. ej., una población de células, p. ej., una población de células efectoras inmunitarias) que comprende una molécula de ácido nucleico, una molécula polipeptídica CAR o un vector como se describe en esta memoria. La presente invención proporciona una célula efectora inmunitaria de acuerdo con las reivindicaciones adjuntas.
En una realización, la célula es una célula T humana. En una realización, la célula es una célula descrita en el presente memoria, p. ej., una célula T humana, p. ej., una célula T humana descrita en esta memoria; o una célula NK humana, p. ej., una célula NK humana descrita o en esta memoria. En una realización, la célula T humana es una célula T CD8+. En una realización, la célula es una célula T y la célula T es deficiente en diaglicerol quinasa (DGK, por sus siglas en inglés). En una realización, la célula es una célula T y la célula T es deficiente en Ikaros. En una realización, la célula es una célula T y la célula T es deficiente tanto en DGK como en Ikaros.
En otra realización, una célula efectora inmunitaria que expresa CAR puede expresar además otro agente, p. ej., un agente que potencia la actividad de una célula que expresa CAR. Por ejemplo, en una realización, el agente puede ser un agente que inhibe una molécula inhibidora. Ejemplos de moléculas inhibidoras incluyen PD-1, PD-L1, CTLA-4, TIM-3, CEACAM (p. ej., CEACAM-1, CEACAM-3 y/o CEACAM-5), LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 y TGFR beta, p. ej., como se describe en esta memoria. En una realización, el agente que inhibe una molécula inhibidora comprende un primer polipéptido, p. ej., una molécula inhibidora, asociado con un segundo polipéptido que proporciona una señal positiva a la célula, p. ej., un dominio de señalización intracelular descrito en esta memoria. En una realización, el agente comprende un primer polipéptido, p. ej., de una molécula inhibidora tal como PD-1, PD-L1, CTLA-4, TIM-3, CEACAM (p. ej., CEACAM-1, CEACAM-3 y/o CEACAM-5), LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 o TGFR beta, o un fragmento de cualquiera de estos, y un segundo polipéptido que es un dominio de señalización intracelular descrito en esta memoria (p. ej., que comprende un dominio coestimulador (p. ej., 41BB, CD27 o CD28, p. ej., como se describe en esta memoria) y/o un dominio de señalización primario (p. ej., un dominio de señalización zeta de CD3 descrito en esta memoria). En una realización, el agente comprende un primer polipéptido de PD-1 o un fragmento del mismo, y un segundo polipéptido de un dominio de señalización intracelular descrito en esta memoria (p. ej., un dominio de señalización CD28, CD27, OX40 o 4-IBB descrito en esta memoria y/o un dominio de señalización CD3 zeta descrito en esta memoria).
En la presente invención, la célula efectora inmunitaria que expresa CAR comprende, además, un segundo CAR, p. ej., un segundo CAR que incluye un dominio de unión a antígenos diferente, p. ej., a la misma diana o a una diana diferente. En una realización, el segundo CAR incluye un dominio de unión de antígeno para una diana expresada en el mismo tipo de célula cancerosa que la diana del primer CAR. En una realización, la célula efectora inmunitaria que expresa CAR comprende un primer CAR que se dirige a un primer antígeno e incluye un dominio de señalización intracelular que tiene un dominio de señalización coestimulador, pero no un dominio de señalización primario, y un segundo CAR que se dirige a un segundo antígeno diferente e incluye un dominio de señalización intracelular que tiene un dominio de señalización primario, pero no un dominio de señalización coestimulador.
Sin pretender quedar ligados a la teoría, la colocación de un dominio de señalización coestimulador, p. ej., 4-1BB, CD28, CD27 u OX-40, en el primer CAR, y el dominio de señalización primario, p. ej., CD3 zeta, en el segundo CAR puede limitar la actividad del CAR a las células donde se expresan ambas dianas. En una realización, la célula efectora inmunitaria que expresa CAR comprende un primer CAR que incluye un dominio de unión a antígenos, un dominio transmembrana y un dominio coestimulador y un segundo CAR que fija como objetivo un antígeno distinto del antígeno fijado como objetivo por el primer CAR (p. ej., un antígeno expresado en el mismo tipo de célula cancerosa que la primera diana) e incluye un dominio de unión a antígenos, un dominio transmembrana y un dominio de señalización primario. En otra realización, la célula efectora inmunitaria que expresa CAR comprende un primer CAR que incluye un dominio de unión a antígenos, un dominio transmembrana y un dominio de señalización primario y un segundo CAR que fija como objetivo un antígeno
distinto del antígeno fijado como objetivo por el primer CAR (p. ej., un antígeno expresado en el mismo tipo de célula cancerosa que la primera diana) e incluye un dominio de unión al antígeno, un dominio transmembrana y un dominio de señalización coestimulador.
En una realización, la célula efectora inmunitaria que expresa CAR comprende una CAR, p. ej., un CAR para una diana arriba descrita, y un CAR inhibidor. En una realización, el CAR inhibidor comprende un dominio de unión a antígenos que se une a un antígeno presente en células normales pero no en células cancerosas, p. ej., células normales que también expresan la diana. En una realización, el CAR inhibidor comprende el dominio de unión a antígenos, un dominio transmembrana y un dominio intracelular de una molécula inhibidora. Por ejemplo, el dominio intracelular del CAR inhibidor puede ser un dominio intracelular de PD1, PD-L1, CTLA-4, TIM-3, c Ea CAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 o TGFR beta.
En una realización, una célula efectora inmunitaria (p. ej., célula T, célula NK) comprende un primer CAR que comprende un dominio de unión a antígenos que se une a un antígeno tumoral como se describe en esta memoria y un segundo CAR que comprende un dominio extracelular de PD1 o un fragmento del mismo.
En una realización, la célula comprende además una molécula inhibidora que comprende: un dominio citoplasmático inhKIR; un dominio transmembrana, p. ej., un dominio transmembrana KIR; y un dominio citoplasmático inhibidor, p. ej., un dominio ITIM, p. ej., un dominio ITIM inhKIR. En una realización, la molécula inhibidora es un inhKIR de origen natural o una secuencia que comparte al menos un 50, 60, 70, 80, 85, 90, 95 o 99 % de homología con, o que no difiere en más de 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15 o 20 residuos de, un inhKIR de origen natural.
En una realización, la célula comprende además una molécula inhibidora que comprende: un dominio citoplasmático de la familia SLAM; un dominio transmembrana, p. ej., un dominio transmembrana de la familia SLAM; y un dominio citoplasmático inhibidor, p. ej., un dominio de la familia SLAM, p. ej., un dominio ITIM de la familia SLAM. En una realización, la molécula inhibidora es un SLAM de origen natural o una secuencia que comparte al menos un 50, 60, 70, 80, 85, 90, 95 o 99 % de homología con, o que no difiere en más de 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 15 o 20 residuos de, un SLAM de origen natural.
En una realización, el segundo CAR en la célula es un CAR inhibidor, en donde el CAR inhibidor comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio intracelular de una molécula inhibidora. La molécula inhibidora se puede elegir entre una o más de: PD1, PD-L1, CTLA-4, TIM-3, LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4, TGFR beta, CEACAM-1, CEACAM-3 y CEACAM-5. En una realización, la segunda molécula de CAR comprende el dominio extracelular de PD1 o un fragmento del mismo.
En algunas realizaciones, la segunda molécula de CAR en la célula comprende además un dominio de señalización intracelular que comprende un dominio de señalización primario y/o un dominio de señalización intracelular.
En otras realizaciones, el dominio de señalización intracelular en la célula comprende un dominio de señalización primario que comprende el dominio funcional de CD3 zeta y un dominio de señalización coestimulador que comprende el dominio funcional de 4-1BB.
En una realización, la segunda molécula de CAR en la célula comprende la secuencia de aminoácidos de la SEQ ID NO: 26.
En determinadas realizaciones, el dominio de unión a antígenos de la segunda molécula de CAR comprende un scFv y el dominio de unión a antígenos de la primera molécula de CAR comprende un dominio VHH de camélido.
Métodos de tratamiento/Terapias combinadas
En otro aspecto, la presente divulgación proporciona un método que comprende administrar una molécula de CAR, p. ej., una molécula de c Ar descrita en esta memoria, o una célula que comprende un ácido nucleico que codifica una molécula de CAR, p. ej., una molécula de CAR descrita en esta memoria. En un caso, el sujeto tiene un trastorno descrito en esta memoria, p. ej., el sujeto tiene cáncer, p. ej., el sujeto tiene un cáncer que expresa un antígeno diana descrito en esta memoria. En un caso, el sujeto es un ser humano.
En otro aspecto, la divulgación pertenece a un método para tratar a un sujeto que tiene una enfermedad asociada con la expresión de un antígeno asociado con el cáncer como se describe en esta memoria, que comprende administrar al sujeto una cantidad eficaz de una célula que comprende una molécula de CAR, p. ej., una molécula de CAR descrita en esta memoria.
En aún otro aspecto, la divulgación presenta un método para tratar a un sujeto que tiene una enfermedad asociada con la expresión de un antígeno tumoral, que comprende administrar al sujeto una cantidad eficaz de una célula, p. ej., una célula efectora inmunitaria (p. ej., una población de células efectoras inmunitarias) que comprende una molécula de CAR, en donde la molécula de CAR comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio
intracelular, dicho dominio intracelular comprende un dominio coestimulador y/o un dominio de señalización primario, en donde dicho dominio de unión a antígenos se une al antígeno tumoral asociado con la enfermedad, p. ej., un antígeno tumoral como se describe en esta memoria.
En un aspecto relacionado, la divulgación presenta un método para tratar a un sujeto que tiene una enfermedad asociada con la expresión de un antígeno tumoral. El método comprende administrar al sujeto una cantidad eficaz de una célula, p. ej., una célula efectora inmunitaria (p. ej., una población de células efectoras inmunitarias) que comprende una molécula de CAR, en combinación con un agente que aumenta la eficacia de la célula inmunitaria, en donde:
(i) la molécula de CAR comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio intracelular que comprende un dominio coestimulador y/o un dominio de señalización primario, en donde dicho dominio de unión a antígenos se une al antígeno tumoral asociado con la enfermedad, p. ej., un antígeno tumoral como se describe en esta memoria; y
(ii) el agente que aumenta la eficacia de la célula inmunitaria se elige entre uno o más de:
(i) un inhibidor de proteína fosfatasa;
(ii) un inhibidor de quinasa
(iii) una citoquina;
(iv) un inhibidor de una molécula inmunoinhibidora; o
(v) un agente que disminuye el nivel o la actividad de una célula Treg.
En un aspecto relacionado, la divulgación presenta un método para tratar a un sujeto que tiene una enfermedad asociada con la expresión de un antígeno tumoral, que comprende administrar al sujeto una cantidad eficaz de una célula, p. ej., una célula efectora inmunitaria (p. ej., una población de células efectoras inmunitarias) que comprende una molécula de CAR, en el que:
(i) la molécula de CAR comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio intracelular que comprende un dominio coestimulador y/o un dominio de señalización primario, en donde dicho dominio de unión a antígenos se une al antígeno tumoral asociado con la enfermedad, p. ej., un antígeno tumoral como se describe en esta memoria; y
(ii) el dominio de unión a antígenos de la molécula de CAR tiene una afinidad de unión al menos 5 veces menor que un anticuerpo del que se deriva el dominio de unión a antígenos.
En otro aspecto, la divulgación presenta una composición que comprende una célula efectora inmunitaria (p. ej., una población de células efectoras inmunitarias) que comprende una molécula de CAR (p. ej., una molécula de CAR como se describe en esta memoria) para uso en el tratamiento de un sujeto que tiene una enfermedad asociada con la expresión de un antígeno tumoral, p. ej., un trastorno como se describe en esta memoria. La presente invención proporciona una célula efectora inmunitaria como se define en las reivindicaciones para uso como medicamento. La presente invención también proporciona una célula efectora inmunitaria como se define en las reivindicaciones para uso en el tratamiento de una enfermedad que expresa un antígeno tumoral.
En determinadas realizaciones de los usos antes mencionados, la enfermedad asociada con un antígeno tumoral, p. ej., un antígeno tumoral descrito en esta memoria, se selecciona de una enfermedad proliferativa tal como cáncer o tumor maligno o una afección precancerosa tal como mielodisplasia, un síndrome de mielodisplasia o una preleucemia, o es una indicación no relacionada con el cáncer asociada con la expresión de un antígeno tumoral descrito en esta memoria. En una realización, la enfermedad es un cáncer descrito en el presente memoria, p. ej., un cáncer descrito en el presente memoria como asociado con una diana descrita en el presente memoria. En una realización, la enfermedad es un cáncer hemático. En una realización, el cáncer hemático es leucemia. En una realización, el cáncer se selecciona del grupo que consiste en una o más leucemias agudas que incluyen, pero no se limitan a leucemia linfoide aguda de células B ("BALL", por sus siglas en inglés), leucemia linfoide aguda de células T ("TALL", por sus siglas en inglés), leucemia linfoide aguda (ALL, por sus siglas en inglés); una o más leucemias crónicas que incluyen, pero no se limitan a leucemia mielógena crónica (CML, por sus siglas en inglés), leucemia linfocítica crónica (CLL, por sus siglas en inglés); cánceres hematológicos adicionales o afecciones hematológicas que incluyen, pero no se limitan a leucemia prolinfocítica de células B, neoplasia de células dendríticas plasmocitoides blásticas, linfoma de Burkitt, linfoma difuso de células B grandes, linfoma folicular, leucemia de células pilosas, linfoma folicular de células pequeñas o de células grandes, enfermedades linfoproliferativas malignas, linfoma MALT, linfoma de células del manto, linfoma de la zona marginal, mieloma múltiple, mielodisplasia y síndrome mielodisplásico, linfoma no Hodgkin, linfoma de Hodgkin, linfoma plasmablástico, neoplasia de células dendríticas plasmocitoides, macroglobulinemia de Waldenstrom y "preleucemia", que son un diverso conjunto de afecciones hematológicas unidas por producción ineficaz (o displasia) de células sanguíneas mieloides, y a enfermedades
asociadas con la expresión de un antígeno tumoral descrito en esta memoria incluyen, pero no se limitan a cánceres atípicos y/o no clásicos, neoplasias malignas, afecciones precancerosas o enfermedades proliferativas que expresan un antígeno tumoral como se describe en esta memoria; y cualquier combinación de los mismos. En otra realización, la enfermedad asociada con un antígeno tumoral descrito en esta memoria es un tumor sólido.
En determinadas realizaciones de los usos antes mencionados, el antígeno tumoral asociado con la enfermedad se elige entre uno o más de: CD19, CD123, CD22, CD30, CD171, CS-1, CLL-1 (CLECL1), CD33, EGFRvIII, GD2, GD3, BCMA, Tn Ag, PSMA, ROR1, FLT3, TAG72, CD38, CD44v6, CEA, EPCAM, B7H3, KIT, IL-13Ra2, mesotelina, IL-11Ra, PSCA, PRSS21, VEGFR2, LewisY, CD24, PDGFR-beta, SSEA-4, CD20, receptor de folato alfa, ERBB2 (Her2/neu), MUC1, EGFR, NCAM, prostasa, PAP, ELF2M, efrina B2, FAP, receptor de IGF-I, CAIX, LMP2, gp100, bcr-abl, tirosinasa, EphA2, Fucosil GM1, sLe, GM3, TGS5, HMWMAA, o-acetil-GD2, receptor de folato beta, TEM1/CD248, TEM7R, CLDN6, TSHR, GPRC5D, CXORF61, CD97, CD179a, ALK, ácido polisiálico, PLAC1, GloboH, NY-BR-1, UPK2, HAVCR1, ADRB3, PANX3, GPR20, LY6K, OR51E2, TARP, WT1, NY-ESO-1, LAGE-1a, MAGE-A1, MAGE A1, ETV6-AML, proteína de espermatozoide 17, XAGE1, Tie 2, MAD-CT-1, MAD-CT-2, antígeno relacionado con Fos 1, p53, p53 mutante, prosteína, survivina y telomerasa, PCTA-1/Galectina 8, MelanA/MART1, Ras mutante, hTERT, puntos de corte de translocación de sarcoma, ML-IAP, ERG (gen de fusión de TMPRSS2 y ETS), NA17, PAX3, receptor de andrógenos, ciclina B1, MYCN, RhoC, TRP-2, CYP1B1, BORIS, SART3, PAX5, OY-TES1, LCK, AKAP-4, SSX2, RAGE-1, transcriptasa inversa de telomerasa humana, RU1, RU2, legumaína, HPV E6,E7, carboxil esterasa intestinal, mut hsp70-2, CD79a, CD79b, CD72, LAIR1, FCAR, LILRA2, CD300LF, CLEC12A, BST2, EMR2, LY75, GPC3, FCRL5 e IGLL1.
En otras realizaciones de los usos antes mencionados, el antígeno tumoral asociado con la enfermedad se elige entre uno o más de: TSHR, TSHR, CD171, CS-1, CLL-1, GD3, Tn Ag, FLT3, CD38, CD44v6, B7H3, KIT, IL-13Ra2, IL-11Ra, PSCA, PRSS21, VEGFR2, LewisY, CD24, PDGFR-beta, SSEA-4, MUC1, EGFR, NCAM, CAIX, LMP2, EphA2, Fucosil GM1, sLe, GM3, TGS5, HMWMAA, o-acetil-GD2, receptor de folato beta, TEM1/CD248, TEM7R, CLDN6, GPRC5D, CXORF61, CD97, CD179a, ALK, ácido polisiálico, PLAC1, GloboH, NY-BR-1, UPK2, HAVCR1, ADRB3, PANX3, GPR20, LY6K, OR51E2, TARP, WT1, ETV6-AML, proteína de espermatozoide 17, XAGE1, Tie 2, MAD- CT-1, MAD-CT-2, antígeno relacionado con Fos 1, p53 mutante, hTERT, puntos de corte de translocación de sarcoma, ML-IAP, ERG (gen de fusión de TMPRSS2 y ETS), NA17, PAX3, receptor de andrógenos, ciclina B1, MYCN, RhoC, CYP1B1, BORIS, SART3, PAX5, OY-TES1, LCK, AKAP-4, SSX2, CD79a, CD79b, CD72, LAIR1, FCAR, LILRA2, CD300LF, CLEC12A, BST2, EMR2, LY75, GPC3, FCRL5 e IGLL1.
En otras realizaciones de los usos antes mencionados, el antígeno tumoral asociado con la enfermedad se elige entre uno o más de: TSHR, CLDN6, GPRC5D, CXORF61, CD97, CD179a, ALK, Polysialic acid, PLAC1, GloboH, NY-BR-1, UPK2, HAVCR1, ADRB3, PANX3, GPR20, LY6K y OR51E2.
En determinadas realizaciones, los usos se llevan a cabo en combinación con un agente que aumenta la eficacia de la célula efectora inmunitaria, p. ej., un agente como se describe en esta memoria.
En cualquiera de los usos antes mencionados, la enfermedad asociada con la expresión del antígeno tumoral se selecciona del grupo que consiste en una enfermedad proliferativa, una afección precancerosa, un cáncer y una indicación no relacionada con el cáncer asociada con la expresión del antígeno tumoral.
El cáncer puede ser un cáncer hematológico, p. ej., un cáncer elegido entre uno o más de leucemia linfocítica crónica (CLL), leucemias agudas, leucemia linfoide aguda (ALL), leucemia linfoide aguda de células B (B-ALL), leucemia linfoide aguda de células T (T-ALL), leucemia mielógena crónica (CML), leucemia prolinfocítica de células B, neoplasia de células dendríticas plasmocitoides blásticas, linfoma de Burkitt, linfoma difuso de células B grandes, linfoma folicular, leucemia de células pilosas, leucemia de células pequeñas o un linfoma folicular de células grandes, afecciones linfoproliferativas malignas, linfoma MALT, linfoma de células del manto, linfoma de la zona marginal, mieloma múltiple, mielodisplasia y síndrome mielodisplásico, linfoma no Hodgkin, linfoma de Hodgkin, linfoma plasmablástico, neoplasia de células dendríticas plasmacitoides, macroglobulinemia de Waldenstrom o pre-leucemia.
El cáncer también puede elegirse entre cáncer de colon, cáncer de recto, carcinoma de células renales, cáncer de hígado, carcinoma no microcítico de pulmón, cáncer de intestino delgado, cáncer de esófago, melanoma, cáncer de huesos, cáncer de páncreas, cáncer de piel, cáncer de cabeza o cuello, melanoma maligno cutáneo o intraocular, cáncer de útero, cáncer de ovario, cáncer de recto, cáncer de la región anal, cáncer de estómago, cáncer de testículo, cáncer de útero, carcinoma de las trompas de Falopio, carcinoma de endometrio, carcinoma de cuello uterino, carcinoma de vagina, carcinoma de vulva, enfermedad de Hodgkin, linfoma no hodgkiniano, cáncer del sistema endocrino, cáncer de la glándula tiroides, cáncer de la glándula paratiroides, cáncer de la glándula suprarrenal, sarcoma de tejidos blandos, cáncer de uretra, cáncer de pene, tumores sólidos de la infancia, cáncer de vejiga, cáncer de riñón o de uréter, carcinoma de pelvis renal, neoplasia del sistema nervioso central (SNC), linfoma primario del SNC, angiogénesis tumoral, tumor del eje espinal, glioma del tronco encefálico, adenoma hipofisario, sarcoma de Kaposi, cáncer epidermoide, cáncer de células escamosas, linfoma de célula T, cánceres inducidos por el medio ambiente, combinaciones de dichos cánceres y lesiones metastásicas de dichos cánceres.
En determinadas realizaciones de los usos, la molécula de CAR se administra en combinación con un agente que aumenta la eficacia de la célula efectora inmunitaria, p. ej., uno o más de un inhibidor de proteína fosfatasa, un inhibidor de quinasa, una citoquina, un inhibidor de una molécula inmunoinhibidora; o un agente que disminuye el nivel o la actividad de una célula Treg.
En determinadas realizaciones, el inhibidor de proteína fosfatasa es un inhibidor de SHP-1 y/o un inhibidor de SHP-2.
En otras realizaciones, el inhibidor de quinasa se elige de uno o más de un inhibidor de CDK4, un inhibidor de CDK4/6 (p. ej., palbociclib), un inhibidor de BTK (p. ej., ibrutinib o RN-486), un inhibidor de mTOR (p. ej., rapamicina o everolimus (RAD001)), un inhibidor de MNK o un inhibidor dual de P13K/mTOR. En una realización, el inhibidor de BTK no reduce ni inhibe la actividad quinasa de la quinasa inducible por interleucina-2 (ITK).
En otras realizaciones, el agente que inhibe la molécula inmunoinhibidora comprende un anticuerpo o fragmento de anticuerpo, un ácido nucleico inhibidor, repeticiones palindrómicas cortas agrupadas regularmente interespaciadas (CRISPR, por sus siglas en inglés), una nucleasa efectora similar a un activador de la transcripción (TALEN, por sus siglas en inglés), o una endonucleasa con dedos de zinc (ZFN, por sus siglas en inglés) que inhibe la expresión de la molécula inhibidora.
En otras realizaciones, el agente que disminuye el nivel o la actividad de las células Treg se elige entre ciclofosfamida, anticuerpo anti-GITR, depleción de CD25 o una combinación de los mismos.
En determinadas realizaciones, la molécula inmunoinhibidora se selecciona del grupo que consiste en PD1, PD-L1, CTLA-4, TIM-3, LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4, TGFR beta, CEACAM-1, CEACAM-3 y CEACAM-5.
En otras realizaciones, el agente que inhibe la molécula inhibidora comprende un primer polipéptido que comprende una molécula inhibidora o un fragmento de la misma y un segundo polipéptido que proporciona una señal positiva a la célula, y en donde el primer y el segundo polipéptido se expresan en células inmunitarias que contienen CAR, en las que (i) el primer polipéptido comprende PD1, PD-L1, CTLA-4, TIM-3, LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4, TGFR beta, CEACAM-1, CEACAM-3 y CEACAM-5 o un fragmento del mismo; y/o (ii) el segundo polipéptido comprende un dominio de señalización intracelular que comprende un dominio de señalización primario y/o un dominio de señalización coestimulador. En una realización, el dominio de señalización primario comprende un dominio funcional de CD3 zeta; y/o el dominio de señalización coestimulador comprende un dominio funcional de una proteína seleccionada de 41BB, CD27 y CD28.
En otras realizaciones, la citoquina se elige entre IL-7, IL-15 o IL-21, o ambas.
En otras realizaciones, la célula efectora inmunitaria que comprende la molécula de CAR y una segunda, p. ej., cualquiera de las terapias de combinación desveladas en esta memoria (p. ej., el agente que aumenta la eficacia de la célula efectora inmunitaria) se administran sustancialmente de forma simultánea o secuencial.
En otras realizaciones, la célula inmunitaria que comprende la molécula de CAR se administra en combinación con una molécula que se dirige a GITR y/o modula la función de GITR. En determinadas realizaciones, la molécula que se dirige a GITR y/o modula la función de GITR se administra antes que la célula o población de células que expresan CAR, o antes de la aféresis.
En una realización, la infusión de célula, por ejemplo la infusión de células alogénicos, se utiliza en el tratamiento del cáncer, en donde la infusión de célula comprende al menos una célula que expresa CAR de la presente invención. En una realización, la infusión de células autólogos se utiliza en el tratamiento del cáncer, en donde la infusión de célula autólogos comprende al menos una célula que expresa CAR descrita en e esta memoria.
En una realización, la célula es una célula T y la célula T es deficiente en diaglicerol quinasa (DGK). En una realización, la célula es un célula T y el célula T es deficiente en Ikaros. En una realización, la célula es un célula T y el célula T es deficiente tanto en DGK como en Ikaros.
En una realización, una célula que expresa la molécula de CAR se administra en combinación con un agente que potencia la actividad de una célula que expresa CAR, en donde el agente es una citoquina, p. ej., IL-7, IL-15, IL- 21, o una combinación de los mismos. La citoquina se puede suministrar en combinación con, p. ej., simultáneamente o poco después de, la administración de la célula que expresa CAR. Como alternativa, la citoquina se puede suministrar después de un periodo prolongado de tiempo después de la administración de la célula que expresa CAR, p. ej., después de la evaluación de la respuesta del sujeto a la célula que expresa CAR. En una realización, la citoquina se administra al sujeto simultáneamente (p. ej., se administra el mismo día) con la administración o poco después de ella (p. ej., se administra 1 día, 2 días, 3 días, 4 días, 5 días, 6 días o 7 días después de la administración) de la célula o población de células de cualquiera de las reivindicaciones 61-80. En otras realizaciones, la citoquina se administra al sujeto después de un periodo de tiempo prolongado (p. ej., al menos 2 semanas, 3 semanas, 4 semanas, 6 semanas, 8 semanas, 10 semanas o más)
después de la administración de la célula o población de células de cualquiera de las reivindicaciones 61-80, o después de la evaluación de la respuesta del sujeto a la célula.
En otras realizaciones, las células que expresan una molécula de CAR se administran en combinación con un agente que mejora uno o más efectos secundarios asociados con la administración de una célula que expresa una molécula de c Ar . Los efectos secundarios asociados con la célula que expresa CAR pueden elegirse entre el síndrome de liberación de citoquinas (SLC) o la linfohistiocitosis hemofagocítica (LHH).
En realizaciones de cualquiera de los usos antes mencionados, las células que expresan la molécula de CAR se administran en combinación con un agente que trata la enfermedad asociada con la expresión del antígeno tumoral, p. ej., cualquiera de las segundas o terceras terapias descritas en esta memoria. Las combinaciones ilustrativas adicionales incluyen una o más de las siguientes.
En otra realización, la célula que expresa la molécula de CAR se puede administrar en combinación con otro agente, p. ej., un inhibidor de quinasa y/o un inhibidor de punto de control descrito en esta memoria. En una realización, una célula que expresa la molécula de CAR puede expresar además otro agente, p. ej., un agente que potencia la actividad de una célula que expresa CAR.
Por ejemplo, en una realización, el agente que potencia la actividad de una célula que expresa CAR puede ser un agente que inhibe una molécula inhibidora (p. ej., una molécula inhibidora inmunitaria). Ejemplos de moléculas inhibidoras incluyen PD1, PD-L1, CTLA-4, TIM-3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 y TGFR beta.
En una realización, el agente que inhibe la molécula inhibidora es un ácido nucleico inhibidor que es un ARNbc, un ARNip o un ARNhc. En algunas realizaciones, el ácido nucleico inhibidor está ligado al ácido nucleico que codifica un componente de la molécula de CAR. Por ejemplo, la molécula inhibidora se puede expresar en la célula que expresa CAR.
En otra realización, el agente que inhibe una molécula inhibidora, p. ej., es una molécula descrita en esta memoria, p. ej., un agente que comprende un primer polipéptido, p. ej., una molécula inhibidora, asociado con un segundo polipéptido que proporciona una señal positiva para la célula, p. ej., un dominio de señalización intracelular descrito en esta memoria. En una realización, el agente comprende un primer polipéptido, p. ej., de una molécula inhibidora tal como PD-1, PD-L1, CTLA-4, TIM-3, CEACAM (e.g., CEACAM-1, CEACAM-3 y/o CEACAM-5), LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 o TGFR beta, o un fragmento de cualquiera de estos (p. Ej., al menos una parte del dominio extracelular de cualquiera de estos), y un segundo polipéptido que es un dominio de señalización intracelular descrito en esta memoria (p. ej., que comprende un dominio coestimulador (p. ej., 41BB, CD27 o CD28, p. ej., como se describe en esta memoria) y/o un dominio de señalización primario (p. ej., un dominio de señalización zeta de CD3 descrito en esta memoria). En una realización, el agente comprende un primer polipéptido de PD1 o un fragmento del mismo (p. ej., al menos una parte del dominio extracelular de PD1) y un segundo polipéptido de un dominio de señalización intracelular descrito en esta memoria (p. ej., un dominio de señalización de CD28 descrito en esta memoria y/o un dominio de señalización de CD3 zeta descrito en esta memoria).
En una realización, la célula efectora inmunitaria que expresa CAR de la presente invención, p. ej., célula T o célula NK, se administra a un sujeto que ha recibido un trasplante previo de células madre, p. ej., un trasplante autólogo de células madre.
En una realización, la célula efectora inmunitaria que expresa CAR de la presente invención, p. ej., célula T o célula NK, se administra a un sujeto que ha recibido una dosis previa de melfalán.
En una realización, la célula que expresa una molécula de CAR se administra en combinación con un agente que aumenta la eficacia de una célula que expresa una molécula de CAR, p. ej., un agente descrito en esta memoria.
En una realización, las células que expresan una molécula de CAR se administran en combinación con una dosis baja potenciadora de la inmunidad de un inhibidor de mTOR. Si bien no se desea estar ligados por la teoría, se cree que el tratamiento con una dosis baja potenciadora del sistema inmunitario (p. ej., una dosis que es insuficiente para suprimir completamente el sistema inmunitario pero suficiente para mejorar la función inmunitaria) se acompaña de una disminución de células T PD-1 positivas o un aumento de células p D-1 negativas.. Las células T positivas para PD-1, pero no las células T negativas para PD-1, pueden agotarse mediante el acoplamiento con células que expresan un ligando PD-1, p. ej., PD-L1 o PD-L2.
En una realización, este enfoque puede utilizarse para optimizar el rendimiento de las células CAR en el sujeto. Aunque sin desear estar ligado por la teoría, se cree que, en una realización, se mejora el rendimiento de células efectoras inmunitarias no modificadas, endógenas, p. ej., células T o células NK. Sin desear estar ligados por la teoría, se cree que, en una realización, se mejora el rendimiento de una célula que expresa CAR de antígeno diana. En otras realizaciones, las células, p. ej., las células T o células NK, que tienen o se manipularán para expresar un CAR, pueden tratarse ex vivo por contacto con una cantidad de un inhibidor de mTOR que aumenta el número de células efectoras inmunitarias
negativas para PD1, p. ej., células T o aumenta la relación de células efectoras inmunitarias negativas para PD1, p. ej., células T/células efectoras inmunitarias positivas para PD1, p. ej., células T.
En una realización, la administración de una dosis baja, de potenciación inmunitaria, de un inhibidor de mTOR, p. ej., un inhibidor alostérico, p. ej., RAD001, o un inhibidor catalítico, se inicia antes de la administración de una célula que expresa CAR descrita en el presente memoria, p. ej., células T o células NK. En una realización, las células CAR se administran después de un tiempo suficiente, o una dosificación suficiente, de un inhibidor de mTOR, de modo que el nivel de células efectoras inmunitarias negativas para PD1, p. ej., células T o células NK, o la proporción de células efectoras inmunitarias negativas para PD1, p. ej., células T/células efectoras inmunitarias positivas para PD1, p. ej., células T, se ha incrementado, al menos transitoriamente.
La célula, p. ej., célula T o célula NK, que se ha de diseñar para expresar un CAR, se puede recolectar después de un tiempo suficiente, o después de una dosificación suficiente de la dosis baja potenciadora del sistema inmunitario de un inhibidor de mTOR, de modo que la el nivel de células efectoras inmunitarias PD1 negativas, p. ej., células T, o la relación de células efectoras inmunitarias PD1 negativas, p. ej., células T/células efectoras inmunitarias PD1 positivas, p. ej., células T, en el sujeto o recolectadas del sujeto ha aumentado, al menos transitoriamente.
En una realización, la célula que expresa una molécula de CAR se administra en combinación con un agente que mejora uno o más efectos secundarios asociados con la administración de una célula que expresa una molécula de CAR, p. ej., un agente descrito en esta memoria.
En una realización, la célula que expresa una molécula de CAR se administra en combinación con un agente que trata la enfermedad asociada con un antígeno asociado al cáncer como se describe en el presente memoria, p. ej., un agente descrito en esta memoria.
En una realización, una célula que expresa dos o más moléculas CAR se administra a un sujeto que lo necesita para tratar el cáncer. En una realización, se administra una población de células que incluye una célula que expresa c Ar a un sujeto que la necesita para tratar el cáncer.
En una realización, la célula que expresa una molécula de CAR se administra a una dosis y/o programa de dosificación descrito en esta memoria.
En una realización, la molécula de CAR se introduce en células efectoras inmunitarias (p. ej., células T, células NK), por ejemplo, utilizando la transcripción in vitro, y el sujeto (p. ej., ser humano) recibe una administración inicial de células que comprenden una molécula de CAR, y una o más administraciones posteriores de células que comprenden una molécula de CAR, en donde una o más administraciones posteriores se administran en menos de 15 días, p. ej., 14, 13, 12, 11, 10, 9, 8, 7, 6, 5 , 4 , 3 o 2 días después de la administración anterior. En una realización, se administra al sujeto más de una administración de células que comprenden una molécula de CAR (p. ej., ser humano) por semana, p. ej., se administran 2, 3 o 4 administraciones por semana de células que comprenden una molécula de CAR. En una realización, el sujeto (p. ej., sujeto humano) recibe más de una administración de células que comprenden una molécula de CAR por semana (p. ej., 2, 3 o 4 administraciones por semana) (también denominado en esta memoria un ciclo), seguido de una semana sin administración de células que comprenden una molécula de CAR y después se administran al sujeto una o más administraciones adicionales de células que comprenden una molécula de CAR (p. ej., más de una administración de las células que comprenden una molécula de CAR por semana). En otra realización, el sujeto (p. ej., sujeto humano) recibe más de un ciclo de células que comprenden una molécula de CAR y el tiempo entre cada ciclo es inferior a 10, 9, 8, 7, 6, 5, 4 o 3 días. En una realización, las células que comprenden una molécula de CAR se administran cada dos días para 3 administraciones por semana. En una realización, las células que comprenden una molécula de CAR se administran durante al menos dos, tres, cuatro, cinco, seis, siete, ocho o más semanas.
En una realización, las células que expresan una molécula de CAR se administran como tratamiento de primera línea para la enfermedad, p. ej., el cáncer, p. ej., el cáncer descrito en el presente memoria. En otra realización, las células que expresan una molécula de CAR se administran como un tratamiento de segunda, tercera y cuarta línea para la enfermedad, p. ej., el cáncer, p. ej., el cáncer descrito en el presente memoria.
En una realización, se administra una población de células de la invención.
En otro aspecto, la divulgación pertenece a la molécula de ácido nucleico aislada que codifica un CAR de la divulgación, la molécula polipeptídica aislada de un CAR de la divulgación, el vector que comprende un CAR de la divulgación y la célula que comprende un CAR de la divulgación para uso como medicamento.
En otro aspecto, la divulgación pertenece a la molécula de ácido nucleico aislada que codifica un CAR de la divulgación, la molécula polipeptídica aislada de un CAR de la divulgación, el vector que comprende un CAR de la divulgación y la célula que comprende un CAR de la divulgación para su uso en el tratamiento de una enfermedad que expresa un antígeno asociado al cáncer como se describe en esta memoria.
En otro aspecto, la invención pertenece a una célula que expresa una molécula de CAR como se define en las reivindicaciones para su uso como medicamento en combinación con una citoquina, p. ej., IL-7, IL-15 y/o IL-21 como se describe en esta memoria. En otro aspecto, la divulgación se refiere a una citoquina descrita en esta memoria para uso como un medicamento en combinación con una célula que expresa una molécula de CAR descrita en esta memoria. En otro aspecto, la invención se refiere a una célula que expresa una molécula de CAR como se define en las reivindicaciones para uso como un medicamento en combinación con un inhibidor de quinasa y/o un inhibidor de punto de control como se describe en esta memoria. En otro aspecto, la divulgación se refiere a un inhibidor de quinasa y/o un inhibidor de punto de control descrito en esta memoria para uso como un medicamento en combinación con una célula que expresa una molécula de CAR descrita en esta memoria.
En otro aspecto, la invención pertenece a una célula que expresa una molécula de CAR como se define en las reivindicaciones para uso en combinación con una citoquina, p. ej., IL-7, IL-15 y/o IL-21 como se describe en esta memoria, en el tratamiento de una enfermedad que expresa un antígeno tumoral fijado como objetivo por el CAR. . En otro aspecto, la divulgación se refiere a una citoquina descrita en esta memoria para uso en combinación con una célula que expresa una molécula de CAR descrita en esta memoria, en el tratamiento de una enfermedad que expresa un antígeno tumoral fijado como objetivo por el CAR
En otro aspecto, la invención se refiere a una célula que expresa una molécula de CAR como se define en las reivindicaciones para uso en combinación con un inhibidor de quinasa y/o un inhibidor de punto de control como se describe en esta memoria, en el tratamiento de una enfermedad que expresa un antígeno tumoral fijado como objetivo por el CAR. En otro aspecto, la divulgación se refiere a un inhibidor de quinasa y/o un inhibidor de punto de control descrito en esta memoria para uso en combinación con una célula que expresa una molécula de CAR descrita en esta memoria, en el tratamiento de una enfermedad que expresa un antígeno tumoral fijado como objetivo por el CAR.
En otro aspecto, la presente divulgación proporciona un método que comprende administrar una molécula de CAR, p. ej., una molécula de c Ar descrita en esta memoria, o una célula que comprende un ácido nucleico que codifica una molécula de CAR, p. ej., una molécula de CAR descrita en esta memoria. En un caso, el sujeto tiene un trastorno descrito en esta memoria, p. ej., el sujeto tiene cáncer, p. ej., el sujeto tiene un cáncer y tiene células que sustentan el tumor que expresan un antígeno que sustenta el tumor descrito en esta memoria. En un caso, el sujeto es un ser humano.
En otro aspecto, la divulgación pertenece a un método para tratar a un sujeto que tiene una enfermedad asociada con la expresión de un antígeno que sustenta el tumor como se describe en esta memoria, que comprende administrar al sujeto una cantidad eficaz de una célula que comprende una molécula de CAR, p. ej., una molécula de CAR descrita en esta memoria.
En aún otro aspecto, la divulgación presenta un método para tratar a un sujeto que tiene una enfermedad asociada con la expresión de un antígeno que sustenta el tumor, que comprende administrar al sujeto una cantidad eficaz de una célula, p. ej., una célula efectora inmunitaria (p. ej., una población de células efectoras inmunitarias) que comprende una molécula de CAR, en donde la molécula de c Ar comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio intracelular, dicho dominio intracelular comprende un dominio coestimulador y/o un dominio de señalización primario, en donde dicho dominio de unión a antígenos se une al antígeno que sustenta el tumor asociado con la enfermedad, p. ej., un antígeno que sustenta el tumor como se describe en esta memoria.
En un aspecto relacionado, la divulgación presenta un método para tratar a un sujeto que tiene una enfermedad asociada con la expresión de un antígeno que sustenta el tumor. El método comprende administrar al sujeto una cantidad eficaz de una célula, p. ej., una célula efectora inmunitaria (p. ej., una población de células efectoras inmunitarias) que comprende una molécula de CAR, en combinación con un agente que aumenta la eficacia de la célula inmunitaria, en donde:
(i) la molécula de CAR comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio intracelular que comprende un dominio coestimulador y/o un dominio de señalización primario, en donde dicho dominio de unión a antígenos se une al antígeno que sustenta el tumor asociado con la enfermedad, p. ej., un antígeno que sustenta el tumor como se describe en esta memoria; y
(ii) el agente que aumenta la eficacia de la célula inmunitaria se elige entre uno o más de:
(i) un inhibidor de proteína fosfatasa;
(ii) un inhibidor de quinasa
(iii) una citoquina;
(iv) un inhibidor de una molécula inmunoinhibidora; o
(v) un agente que disminuye el nivel o la actividad de una célula Treg.
En un aspecto relacionado, la divulgación presenta un método para tratar a un sujeto que tiene una enfermedad asociada con la expresión de un - antígeno que sustenta el tumor, que comprende administrar al sujeto una cantidad eficaz de una célula, p. ej., una célula efectora inmunitaria (p. ej.,, una población de células efectoras inmunitarias) que comprende una molécula de CAR, en el que:
(i) la molécula de CAR comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio intracelular que comprende un dominio coestimulador y/o un dominio de señalización primario, en donde dicho dominio de unión a antígenos se une al antígeno de soporte tumoral asociado con la enfermedad, p. ej., un antígeno de soporte tumoral como se describe en esta memoria; y
(ii) el dominio de unión a antígenos de la molécula de CAR tiene una afinidad de unión al menos 5 veces menor que un anticuerpo del que se deriva el dominio de unión a antígenos.
En otro aspecto, la invención presenta una composición que comprende una célula efectora inmunitaria (p. ej., una población de células efectoras inmunitarias) como se define en las reivindicaciones para uso en el tratamiento de un sujeto que tiene una enfermedad asociada con la expresión de un antígeno que sustenta el tumor, p. ej., un trastorno como se describe en esta memoria.
En cualquiera de los usos de la invención, la enfermedad asociada con la expresión del antígeno que sustenta el tumor se selecciona del grupo que consiste en una enfermedad proliferativa, una afección precancerosa, un cáncer y una indicación no relacionada con el cáncer asociada con la expresión del antígeno que sustenta el tumor. En una realización, la enfermedad asociada con un antígeno que sustenta el tumor descrito en esta memoria es un tumor sólido.
En una realización de los usos, la molécula de CAR se administra en combinación con otro agente. En una realización, el agente puede ser un inhibidor de quinasa, p. ej., un inhibidor de CDK4/6, un inhibidor de BTK, un inhibidor de mTOR, un inhibidor de MNK o un inhibidor doble de PI3K/mTOR, y combinaciones de los mismos. En una realización, el inhibidor de quinasa es un inhibidor de CDK4, p. ej., un inhibidor de CDK4 descrito en esta memoria, p. ej., un inhibidor de CD4/6 tal como, p. ej., hidrocloruro de 6-acetil-8-ciclopentil-5-metil-2-(5-piperazin-1-il-piridin-2-ilamino)-8H-pirido[2,3-d]pirimidin-7-ona (también denominado palbociclib o PD0332991). En una realización, el inhibidor de quinasa es un inhibidor de BTK, p. ej., un inhibidor de BTK descrito en esta memoria, tal como, p. ej., ibrutinib. En una realización, el inhibidor de quinasa es un inhibidor de mTOR, p. ej., un inhibidor de mTOR descrito en esta memoria, tal como, p. ej., rapamicina, un análogo de rapamicina, OSI-027. El inhibidor de mTOR puede ser, p. ej., un inhibidor de mTORC1 y/o un inhibidor de mTORC2, p. ej., un inhibidor de mTORC1 y/o un inhibidor de mTORC2 descrito en esta memoria. En una realización, el inhibidor de quinasa es un inhibidor de MNK, p. ej., un inhibidor de MNK descrito en esta memoria, tal como, p. ej., 4-amino-5-(4-fluoroanilino)-pirazolo [3,4-d] pirimidina. El inhibidor de MNK puede ser, p. ej., un inhibidor de MNK1a, MNK1b, MNK2a y/o MNK2b. El inhibidor doble de PI3K/mTOR puede ser, p. ej., PF-04695102.
En una realización de los usos, el inhibidor de quinasa es un inhibidor de CDK4 seleccionado de aloisina A; flavopiridol o HMR-1275, 2-(2-clorofenil)-5,7-dihidroxi-8-[(3S,4R)-3-hidroxi-1-metil-4-piperidinil]-4-cromenona; crizotinib (PF-02341066; hidrocloruro de 2-(2-clorofenil)-5,7-dihidroxi-8-[(2R,3S)-2-(hidroximetil)-1 -metil-3-pirrolidinil]- 4H-1-benzopiran-4-ona (P276-00); 1-metil-5-[[2-[5-(trifluorometil)-1H-imidazol-2-il]-4-piridinil]oxi]-N-[4-(trifluorometil)fenil]-1H-bencimidazol-2-amina (RAF265); indisulam (E7070); roscovitine (CYC202); palbociclib (PD0332991); dinaciclib (SCH727965); N-[5-[[(5-terc.-butiloxazol-2-il)metil]tio]tiazol-2-il]piperidina-4-carboxamida (BMS 387032); ácido 4-[[9-cloro-7-(2,6-difluorofenil)-5H-pirimido[5,4-d][2]benzazepin-2-il]amino]-benzoico (MLN8054); 5-[3-(4,6-difluoro-1H-bencimidazol-2-il)-1H-indazol-5-il]-N-etil-4-metil-3-piridinmetanamina (AG-024322); N-(piperidin-4-il)amida del ácido 4-(2,6-diclorobenzoilamino)-1H-pirazol-3-carboxílico (AT7519); 4-[2-metil-1-(1-metiletil)-1H-imidazol-5-il]-N-[4-(metilsulfonil)fenil]- 2-pirimidinamina (AZD5438); y XL281 (BMS908662).
En una realización de los usos, el inhibidor de quinasa es un inhibidor de CDK4, p. ej., palbociclib (PD0332991), y el palbociclib se administra a una dosis de aproximadamente 50 mg, 60 mg, 70 mg, 75 mg, 80 mg, 90 mg, 100 mg, 105 mg, 110 mg, 115 mg, 120 mg, 125 mg, 130 mg, 135 mg (p. ej., 75 mg, 100 mg o 125 mg) diariamente durante un período de tiempo, p. ej., diariamente durante 14 -21 días de un ciclo de 28 días, o diariamente durante 7-12 días de un ciclo de 21 días. En una realización, se administran 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 o más ciclos de palbociclib.
En una realización de los usos, el inhibidor de quinasa es un inhibidor de BTK seleccionado de ibrutinib (PCI-32765); GDC-0834; RN-486; CGI-560; CGI-1764; HM-71224; CC-292; ONO-4059; CNX-774; y LFM-A13. En una realización, el inhibidor de BTK no reduce ni inhibe la actividad quinasa de la quinasa inducible por interleucina-2 (ITK) y se selecciona de GDC-0834; RN-486; CGI-560; CGI-1764; HM-71224; CC-292; ONO-4059; CNX-774; y LFM-A13.
En una realización de los usos, el inhibidor de quinasa es un inhibidor de BTK, p. ej., ibrutinib (PCI-32765), y el ibrutinib se administra a una dosis de aproximadamente 250 mg, 300 mg, 350 mg, 400 mg, 420 mg, 440 mg, 460 mg, 480 mg, 500 mg, 520 mg, 540 mg, 560 mg, 580 mg, 600 mg (p. ej., 250 mg, 420 mg o 560 mg) diariamente durante un período de tiempo, p. ej., diariamente para un ciclo de 21 días o diariamente para un ciclo de 28 días. En una realización, se administran 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 o más ciclos de ibrutinib.
En una realización de los usos, el inhibidor de quinasa es un inhibidor de BTK que no inhibe la actividad de quinasa de ITK, p. ej., RN-486, y RN-486 se administra a una dosis de aproximadamente 1o0 mg, 110 mg, 120 mg, 130 mg, 140 mg, 150 mg, 160 mg, 170 mg, 180 mg, 190 mg, 200 mg, 210 mg, 220 mg, 230 mg, 240 mg, 250 mg (p. ej., 150 mg, 200 mg o 250 mg) diariamente durante un período de tiempo, p. ej., diariamente un ciclo de 28 días. . En una realización, se administran 1, 2, 3, 4, 5, 6, 7 o más ciclos de RN-486.
En una realización de los usos, el inhibidor de quinasa es un inhibidor de mTOR seleccionado de temsirolimus; ridaforolimus dimetilfosfinato de (1E,2E,4S)-4-[(2E)-2 [(1R,9S,12S,15R,16E,18R,19R,21R, 23S,24E,26E,28Z,30S,32S,35E)-1,18-dihidroxi-19,30-dimetoxi-15,17,21,23, 29,35-hexametil-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatriciclo[30.3.1.049] hexatriaconta-16,24,26,28-tetraen-12-il]propil]-2-metoxiciclohexilo, también conocido como AP23573 y MK8669; everolimus (RAD001); rapamicina (AY22989); simapimod; (5-{2,4-bis[(3S)-3-metilmorfolin-4-il]pirido[2,3-d]pirimidin-7-il}-2-metoxifenil)metanol (AZD8055); 2-amino-8-[frans-4-(2-hidroxietoxi)ciclohexil]-6-(6-metoxi-3-piridinil)-4-metil-pirido[2,3-d]pirimidin-7(8H)-ona (PF04691502); y W2-[1,4-dioxo-4-[[4-(4-oxo-8-fenil-4H-1-benzopiran-2-il)morfolinio-4-il]metoxi]butil]-L-arginilglicil-L-a-aspartil-L-serina-, sal interna (SF1126); y XL765.
En una realización de los usos, el inhibidor de quinasa es un inhibidor de mTOR, p. ej., rapamicina, y la rapamicina se administra a una dosis de aproximadamente 3 mg, 4 mg, 5 mg, 6 mg, 7 mg, 8 mg, 9 mg, 10 mg (p. ej., 6 mg) diariamente durante un período de tiempo, p. ej., diariamente durante un ciclo de 21 días o diariamente durante un ciclo de 28 días. En una realización, se administran 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 o más ciclos de rapamicina. En una realización, el inhibidor de quinasa es un inhibidor de mTOR, p. ej., everolimus, y el everolimus se administra en una dosis de aproximadamente 2 mg, 2,5 mg, 3 mg, 4 mg, 5 mg, 6 mg, 7 mg, 8 mg, 9 mg, 10 mg, 11 mg, 12 mg, 13 mg, 14 mg, 15 mg (p. ej., 10 mg) diariamente durante un periodo de tiempo, p. ej., diariamente durante un ciclo de 28 días. En una realización, se administran 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 o más ciclos de everolimus.
En una realización de los usos, el inhibidor de quinasa es un inhibidor de MNK seleccionado de CGP052088; 4-amino-3-(p-fluorofenilamino)-pirazolo [3,4-d] pirimidina (CGP57380); cercosporamida; ETC-1780445-2; y 4-amino-5-(4-fluoroanilino)-pirazolo [3,4-d] pirimidina.
En una realización de los usos, el inhibidor de quinasa es un inhibidor dual de fosfatidilinositol 3-quinasa (PI3K) y mTOR seleccionado de 2-amino-8-[frans-4-(2-hidroxietoxi)ciclohexil]-6i-(6-metoxi-3-piridinil)-4-metil-pirido[2,3-d]pirimidin-7(8H)-ona (PF-04691502); N-[4-[[4-(dimetilamino)-1-piperidinil]carbonil]fenil]-W-[4-(4,6-di-4-morfolinil-1,3,5-triazin-2-il)fenil]urea (PF-05212384, PKI-587); 2-metil-2-{4-[3-metil-2-oxo-8-(quinolin-3-il)-2,3-dihidro-1H-imidazo[4,5-c]quinolin-1-il]fenil}propanonitrilo (BEZ-235); apitolisib (Gd C-0980, RG7422); 2,4-difluoro-N-{2-(metiloxi)-5-[4-(4-piridazinil)-6-quinolinil]-3-piridinil}bencenesulfonamida (GSK2126458); 8-(6-metoxipiridin-3-il)-3-metil-1-(4-(piperazin-1-il)-3-(trifluorometil)fenil)-1H-imidazo[4,5-c]quinolin-2(3H)-ona ácido maleico (NVP-BGT226); 3-[4-(4-morfolinilpirido[3',2':4,5]furo[3,2-d]pirimidin-2-il]fenol (PI-103); 5-(9-isopropil-8-metil-2-morfolino-9H-purin-6-il)pirimidin-2-amina (VS-5584, SB2343); y N-[2-[(3,5-dimetoxifenil)amino]quinoxalin-3-il]-4-[(4-metil-3-metoxifenil)carbonil]aminofenilsulfonamida (XL765).
En una realización de los usos, una célula efectora inmunitaria que expresa un CAR descrita en esta memoria se administra a un sujeto en combinación con un inhibidor de la proteína tirosina fosfatasa, p. ej.,un inhibidor de la proteína tirosina fosfatasa descrito en esta memoria. En una realización, el inhibidor de proteína tirosina fosfatasa es un inhibidor de SHP-1, p. ej., un inhibidor de SHP-1 descrito en esta memoria, tal como, p. ej., estibogluconato de sodio. En una realización, el inhibidor de la proteína tirosina fosfatasa es un inhibidor de SHP-2.
En una realización de los usos, la molécula de CAR se administra en combinación con otro agente, y el agente es una citoquina.. La citoquina puede ser, p. ej., IL-7, IL-15, IL-21 o una combinación de las mismas. En otra realización, la molécula de CAR se administra en combinación con un inhibidor de puntos de control, p. ej., un inhibidor de puntos de control descrito en esta memoria. Por ejemplo, en una realización, el inhibidor del punto de control inhibe una molécula inhibidora seleccionada de PD-1, PD-L1, CTLA-4, TIM-3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 y TGFR beta.
Métodos para producir células que expresan CAR
En otro aspecto, la divulgación se refiere a un método para producir una célula (p. ej., una célula efectora inmunitaria o una población de la misma) que comprende introducir (p. ej., transducir) una célula, p. ej., una célula T o una célula NK descrita en esta memoria, con un vector que comprende un ácido nucleico que codifica un CAR, p. ej., un CAR descrito en esta memoria; o un ácido nucleico que codifica una molécula de CAR, p. ej., un CAR descrito en esta memoria. Los métodos para fabricar células que expresan CAR no se reivindican como tales.
La célula en los métodos es una célula efectora inmunitaria (p. ej., una célula T o una célula NK, o una combinación de las mismas). En algunos casos, la célula en los métodos es deficiente en diaglicerol quinasa (DGK) y/o Ikaros.
En algunos casos, la introducción de la molécula de ácido nucleico que codifica un CAR comprende transducir un vector que comprende la molécula de ácido nucleico que codifica un CAR, o transfectar la molécula de ácido nucleico que codifica un CAR, en donde la molécula de ácido nucleico es un ARN transcrito in vitro.
En algunos casos, el método comprende, además:
a. proporcionar una población de células efectoras inmunitarias (p. ej., células T o células NK); y
b. eliminar células T reguladoras de la población, proporcionando con ello una población de células T sin regulación; en el que las etapas a) y b) se realizan antes de introducir el ácido nucleico que codifica el CAR a la población.
En casos de los métodos, las células T reguladoras comprenden células T CD25+ y se eliminan de la población celular utilizando un anticuerpo anti-CD25, o un fragmento del mismo. El anticuerpo anti-CD25, o fragmento del mismo, puede conjugarse con un sustrato, p. ej., una perla.
En otros casos, la población de células T sin regulación proporcionada de la etapa (b) contiene menos del 30 %, 25 %, 20 %, 15 %, 10 %, 5 %, 4 %, 3 %, 2 %, 1% de células CD25+.
Todavía en otros casos, el método comprende, además:
eliminar células de la población que expresan un antígeno tumoral que no comprende CD25 para proporcionar una población de células sin células T reguladoras y sin antígeno tumoral antes de introducir el ácido nucleico que codifica un CAR en la población. El antígeno tumoral se puede seleccionar de CD19, CD30, CD38, CD123, CD20, CD14 o CD11b, o una combinación de los mismos.
En algunos casos, el método comprende, además
eliminar células de la población que expresan un inhibidor de punto de control, para proporcionar una población de células T sin regulador y sin moléculas inhibidoras antes de introducir el ácido nucleico que codifica un CAR en la población. El inhibidor de punto de control puede elegirse entre PD-1, LAG-3, TIM3, B7-H1, CD160, P1H, 2B4, CeAc AM (p. ej., CEACAM-1, CEACAM-3 y/o CEACAM-5), TIGIT, CTLA-4, BTLA y LAIR1.
Casos adicionales descritos en esta memoria abarcan proporcionar una población de células efectoras inmunitarias. La población de células efectoras inmunitarias proporcionadas puede seleccionarse basándose en la expresión de uno o más de CD3, CD28, CD4, CD8, CD45RA y/o CD45RO. En determinados casos la población de células efectoras inmunitarias proporcionadas son CD3+ y/o CD28+.
En determinados casos del método, el método comprende, además, expandir la población de células después de que se haya introducido la molécula de ácido nucleico que codifica un CAR.
En algunos casos, la población de células se expande durante un período de 8 días o menos.
En determinados casos, la población de células se expande en cultivo durante 5 días y las células resultantes son más potentes que las mismas células expandidas en cultivo durante 9 días bajo las mismas condiciones de cultivo.
En otros casos, la población de células que se expande en cultivo durante 5 días muestra al menos un aumento de una, dos, tres o cuatro veces en la duplicación de células tras la estimulación con antígeno en comparación con las mismas células expandidas en cultivo durante 9 días bajo las mismas condiciones de cultivo.
En aún otros casos, la población de células se expande en cultivo durante 5 días, y las células resultantes exhiben niveles proinflamatorios de If N-y y/o GM-CSF más altos, en comparación con las mismas células expandidas en cultivo durante 9 días. bajo las mismas condiciones de cultivo.
En otros casos, la población de células se expande cultivando las células en presencia de un agente que estimula una señal asociada al complejo CD3/TCR y/o un ligando que estimula una molécula coestimuladora en la superficie de las células. El agente puede ser una perla conjugada con un anticuerpo anti-CD3, o un fragmento del mismo, y/o un anticuerpo anti-CD28, o un fragmento del mismo.
En otros casos, la población de células se expande en un medio apropiado que incluye una o más interleuquinas que dan como resultado un aumento de al menos 200, 250, 300 o 350 veces en las células durante un período de expansión de 14 días, medido por citometría de flujo.
En otros casos, la población de células se expande en presencia de IL-15 y/o IL-7.
En determinados casos, el método incluye, además, crioconservar la población de células después del período de expansión apropiado.
En aún otros casos, el método de fabricación descrito en esta memoria comprende, además, poner en contacto la población de células efectoras inmunitarias con un ácido nucleico que codifica una subunidad de la telomerasa, p. ej., hTERT. El ácido nucleico que codifica la subunidad de telomerasa puede ser ADN.
La presente divulgación también proporciona un método para generar una población de células modificadas con ARN, p. ej., células descritas en esta memoria, p. ej., células efectoras inmunitarias (p. ej., células T, células NK), que expresan transitoriamente ARN exógeno. El método comprende introducir un ARN transcrito in vitro o un ARN sintético en una célula, en que el ARN comprende un ácido nucleico que codifica una molécula de CAR descrita en esta memoria.
En otro aspecto, la divulgación pertenece a un método para proporcionar una inmunidad anti-tumoral en un sujeto, que comprende administrar al sujeto una cantidad eficaz de una célula que comprende una molécula de CAR, p. ej., una célula que expresa una molécula de CAR descrita en esta memoria. En un caso, la célula es una célula T autóloga o una célula Nk. En un caso, la célula es una célula T alogénica o una célula NK. En un caso, el sujeto es un ser humano.
En un aspecto, la invención incluye una población de células autólogas que se transfectan o transducen con un vector que comprende una molécula de ácido nucleico que codifica una molécula de CAR, siendo dichas células de acuerdo con las reivindicaciones. En una realización, el vector es un vector retrovírico. En una realización, el vector es un vector lentivírico auto-activante como se describe en otra parte de esta memoria. En una realización, el vector se administra (p. ej., mediante transfección o electroporación) a una célula, p. ej., una célula T o una célula NK, en donde el vector comprende una molécula de ácido nucleico que codifica un CAR de acuerdo con las reivindicaciones, que se transcribe como una molécula de ARNm, y los CARs se traducen de la molécula de ARN y se expresan en la superficie de la célula.
En otro aspecto, la presente invención proporciona una población de células efectoras inmunitarias que expresan CAR (p. ej., células T o células NK) de acuerdo con las reivindicaciones. En algunas realizaciones, la población de células que expresan CAR comprende una mezcla de células que expresan CAR diferentes. Por ejemplo, en un caso, la población de células efectoras inmunitarias que expresan CAR (p. ej., células T o células NK) puede incluir una primera célula que expresa un CAR que tiene un dominio de unión a antígenos que se une a un primer antígeno tumoral como se describe en esta memoria, y una segunda célula que expresa un CAR que tiene un dominio de unión a antígenos diferente que se une a un segundo antígeno tumoral como se describe en esta memoria. Como otro ejemplo, la población de células que expresan CAR puede incluir una primera célula que expresa un CAR que incluye un dominio de unión a antígenos que se une a un antígeno tumoral como se describe en esta memoria y una segunda célula que expresa un CAR que incluye un dominio de unión a antígenos para una diana distinta de un antígeno tumoral descrito en esta memoria. En un caso, la población de células que expresan CAR incluye, por ejemplo, una primera célula que expresa un CAR que incluye un dominio de señalización intracelular primaria, y una segunda célula que expresa un CAR que incluye un dominio de señalización secundaria, por ejemplo, un dominio de señalización coestimulador.
En otro aspecto, la presente invención proporciona una población de células de acuerdo con las reivindicaciones, en donde al menos una célula de la población expresa un CAR que tiene un dominio de unión a antígenos que se une a un antígeno tumoral como se describe en esta memoria, y una segunda célula que expresa otro agente, p. ej., un agente que potencia la actividad de una célula que expresa CAR. Por ejemplo, en una realización, el agente puede ser un agente que inhibe una molécula inhibidora. Ejemplos de moléculas inhibidoras incluyen PD-1, PD-L1, CTLA-4, TIM-3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 y TGFR beta. En una realización, el agente que inhibe una molécula inhibidora, p. ej., es una molécula descrita en esta memoria, p. ej., un agente que comprende un primer polipéptido, p. ej., una molécula inhibidora, asociada con un segundo polipéptido que proporciona una señal positiva para la célula, p. ej., un dominio de señalización intracelular descrito en estamemoria. En una realización, el agente comprende un primer polipéptido, p. ej., de una molécula inhibidora tal como PD-1, LAG-3, CTLA-4, CD160, BTLA, LAIR1, TIM-3, CEACAM (p. ej., CEACAM-1, CEACAM-3 y/o CEACAM-5), 2B4 y TIGIT, o un fragmento de cualquiera de estos, y un segundo polipéptido que es un dominio de señalización intracelular descrito en esta memoria (p. ej., que comprende un dominio coestimulador (p. ej., 41BB, CD27 o CD28, p. ej., como se describe en esta memoria) y/o un dominio de señalización primario (p. ej., un dominio de señalización zeta de CD3 descrito en esta memoria). En una realización, el agente comprende un primer polipéptido de PD-1 o un fragmento del mismo, y un segundo polipéptido de un dominio de señalización intracelular descrito en esta memoria (p. ej., un dominio de señalización CD28, CD27, OX40 o 4-IBB descrito en esta memoria y/o un dominio de señalización CD3 zeta descrito en esta memoria).
En una realización, la molécula de ácido nucleico que codifica una molécula de CAR se expresa como una molécula de ARNm. En una realización, las células efectoras inmunitarias que expresan CAR modificadas genéticamente (p. ej., células T, células NK) pueden generarse transfectando o electroporando una molécula de ARN que codifica los CARs deseados (p. ej., sin una secuencia de vector) en la célula. En una realización, una molécula de CAR se traduce de la molécula de aRn una vez que se incorpora y se expresa en la superficie de la célula recombinante.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
Fig.1 : Un panel de imágenes que muestra la detección por citometría de flujo de la expresión en superficie de ErbB2 en tumores y líneas celulares. Las células se tiñeron con anti-ErbB2 Affibody-biotina y se detectaron con estreptavidina-aloficocianina (APC) (histogramas en blanco); las células incubadas con APC solo indican fondo (histogramas grises).
Fig. 2A, 2B y 2C: Correlación de la detección de ErbB2 por citometría de flujo y PCR cuantitativa. Número de copias de ErbB2 detectado por PCR cuantitativa (ErbB2/1E6 actina) (Fig. 2A). Intensidad de fluorescencia media de ErbB2 (ErbB2 MFI) como se muestra en los histogramas de la Fig. 1 (Fig. 2B). La correlación se representa gráficamente entre la expresión de ErbB2 detectada por citometría de flujo (MFI, eje x) y la PCR cuantitativa (eje y). Los cebadores directo e inverso y la sonda utilizados para la PCR cuantitativa de ErbB2 son los siguientes: ErbB2-1F, GCCTCCACTTCAACCACAGT (SEQ ID NO: 50); ErbB2-1R, TCAAACGTGTCTGTGTTGTAGGT; ErbB2-1M2, FAM-CAGTGCAGCTCACAGATG (SEQ ID NO: 51).
Fig. 3. Un panel de imágenes que muestra el análisis FACS de la expresión de CAR ajustada por afinidad en células T electroporadas con ARNm. Las células T se sometieron a electroporación con el ARNm de CAR indicado y, un día después de la electroporación, se detectó la expresión de CAR utilizando un anticuerpo Fab IgG anti-ratón (para CD19-BBZ) o ErbB2-Fc (para CAR ErbB2-BBZ). Se utilizaron células T sin electroporación como control negativo.
Fig. 4. Un panel de imágenes que muestra la inducción de la expresión de CD137 (4-1BB) en células CAR T después de que se midió la estimulación mediante células tumorales. Un día después de la electroporación, las diversas células CAR T (Kd, nM) se co-cultivaron con las líneas de células tumorales indicadas y se midió la expresión de CD137 después de 24 h.
Fig. 5. Se midió la secreción de citoquinas (ELISA) en sobrenadantes de cultivo. Las células T se sometieron a electroporación con 5 ug o 10 ug de ARNm de ErbB2 CAR ajustado por afinidad, como se indica. Un día después de la electroporación, las células CAR T se co-cultivaron con las líneas de células tumorales indicadas durante 24 h. El gráfico de barras muestra los resultados de un experimento representativo (los valores representan el promedio ± DE de duplicados) para IFN-gamma.
Fig. 6. Se midió la secreción de citoquinas (ELISA) en sobrenadantes de cultivo. Las células T se sometieron a electroporación con 5 ug o 10 ug de ARNm de ErbB2 CAR ajustado por afinidad, como se indica. Un día después de la electroporación, las células CAR T se co-cultivaron con las líneas de células tumorales indicadas durante 24 h. El gráfico de barras muestra los resultados de un experimento representativo (los valores representan el promedio ± DE de duplicados) para IL-2.
Fig. 7. Regulación positiva de CD107a en células CAR T estimulada por tumores. Las células T se sometieron a electroporación con 5 ug o 10 ug de ARNm de ErbB2 CAR que codifican el scFv indicado y, un día después, las células CAR T se co-cultivaron con la línea celular indicada durante 4 h. La expresión de CD107a se midió seleccionando células CD3+CD8+.
Fig. 8. Un panel de imágenes que muestra que se examinó la expresión de ErbB2 en líneas celulares tumorales adicionales mediante citometría de flujo utilizando tinción con biotina-ErbB2 Affibody (estreptavidina-PE) (histogramas en blanco). Las mismas células teñidas solo con Estreptavidina-PE se utilizaron como control negativo (histogramas grises).
Fig9. Las células T electroporadas con ARNm de ErbB2 CAR se estimularon con líneas tumorales testadas en C. Se informó que SK-OK3, BT-474, HCC2281, MDA-361, MDA-453, HCC-1419, HCC-1569, UACC-812 y LnCap eran tumores amplificados con ErbB2, mientras que se informó que MDA-175, MCF-10A, HCC38, HG261 eran líneas celulares bajas en ErbB2 o negativas. Después de 4 h de estimulación, se controló CD107a en las células T mediante tinción de citometría de flujo y se representó el % de células que expresaban CD107a.
Fig. 10. Un panel de imágenes que muestra que el reconocimiento de células K562 se sometió a electroporación con las cantidades indicadas de ARNm de ErbB2 y las células CAR T que expresan el scFv indicado (Kd, nM) se co-cultivaron con la diana durante 4 h y el % de expresión de CD107a se cuantificó en células CD3+CD8+.
Fig.11A. Un panel de imágenes que muestra la expresión de ErbB2 en células K562 después de la electroporación. Células K562 se sometieron a electroporación con la cantidad indicada de ARNm de ErbB2 y la tinción indica células con expresión de ErbB2 (histograma en blanco); las células incubadas con anticuerpo secundario solo indican fondo (histograma gris).
Fig. 11B. Secreción de IFN-gamma por el panel de células ErbB2 CART estimuladas por células K562 electroporadas con ARNm de ErbB2. Las células K562 se sometieron a electroporación con 2 ug o 10 ug de
ARNm de ErbB2 CAR como se indica. Células CAR T se co-cultivaron con dianas K562 indicadas y la secreción de IFN-gamma se midió mediante ELISA después de 24 h.
Fig. 12. Un panel de imágenes que muestra la proliferación del panel de células CAR T ajustadas por afinidad después de la estimulación por células K562 electroporadas con ARNm de ErbB2. Las células T en reposo se marcaron con CFSE y se sometieron a electroporación con 10 ug de ARNm de CAR. Las células K562 se sometieron a electroporación con la cantidad indicada de ARNm de ErbB2 o ARNm de CD19 de control (19BBBZ). Las células T y las dianas irradiadas se cultivaron (relación 1:1) durante 7 días y la dilución de CFSE se midió mediante citometría de flujo (controlado por CD3); se muestra el % de células T divididas.
Fig. 13A, 13B y 13C. Se midió la citotoxicidad del panel de células CAR T frente a células diana Nalm6-CBG electroporadas con ARNm de ErbB2. Las células T se sometieron a electroporación con ARNm de ErbB2 o CD19 CAR como se indica. Se sometieron a electroporación células diana CD19+ve Nalm6-CBG (escarabajo click verde) con ARNm de ErbB2 a la dosis indicada: 10 |jg de ARN de ErbB2 (Fig. 13A); 1 |jg de ARN de ErbB2 (Fig. 13B); y 0,1 jg de ARN de ErbB2 (Fig. 13C). Un día después de la electroporación, las células CAR T se co-cultivaron con células Nalm6-CBG en la relación E:T indicada y se calculó el % de lisis específica después de 8 h.
Fig. 14. Un panel de imágenes que muestra la expresión de ErbB2 en las líneas celulares primarias indicadas. Las líneas celulares primarias se tiñeron utilizando anti-ErBb2 Affibody-biotina y se detectaron utilizando estreptavidina-aloficocianina (APC) (histogramas en blanco); las células teñidas solo con APC se utilizaron como control (histogramas grises).
Fig. 15. Fijación selectiva de objetivo de ErbB2 en líneas celulares primarias. El panel de células CAR T se estimuló con las líneas celulares primarias acusadas durante 4 h y el % de células CAR T que expresan CD107a se midió seleccionando células CD3+CD8+.
Fig. 16A, 16B y 16C Paneles de imágenes que muestran que las células T se modificaron con ErbB2 CAR de afinidad alta (4D5) o baja (4D5-5) utilizando transducción lentiviral (LVV) o electroporación de ARNm (ARN) como se indica. El % de expresión CAR y el brillo se midió utilizando ErbB2-Fc (Fig. 16A). La expresión de ErbB2 en un panel de líneas tumorales y células K562 sometidas a electroporación con ARNm de ErbB2 se detectó mediante citometría de flujo (Figs. 16B y 16C); porcentaje de células células y (MFI) mostrado para células K562.
Fig.17. Un panel de imágenes que muestra el reconocimiento de células CAR T de las líneas tumorales indicadas. La regulación positiva de CD107a se midió en células CAR T transducidas con lentivirus o electroporadas con ARNm después de 4 h de estimulación con las líneas tumorales indicadas (activadas en células CD3+).
Fig.18. Panel de imágenes que muestran el reconocimiento de células CAR T de las células K562 electroporadas con las cantidades indicadas de ARNm de ErbB2. La inducción de la expresión de CD107a se midió en células CAR T transducidas con lentivirus o electroporadas con ARNm después de 4 h de estimulación con células K562 electroporadas con ErbB2 mediante activación de células CD3+.
Fig. 19. Panel de imágenes que muestran la regulación al alza dependiente de la diana ErbB2 de CD107a en células T transducidas por lentivirus o electroporadas con ARNm. Las células T como se muestra en el texto principal de la fig. 4A se estimularon durante 4 h con líneas de células tumorales que expresaban ErbB2 a niveles que variaban desde niveles sobre-expresados a bajos. La regulación al alza de CD107a se detectó mediante citometría de flujo (controlado por CD3+).
Fig.20. La secreción de IFN-gamma por células CAR T transducidas con lentivirus o electroporadas con ARN se midió mediante ELISA después de 18 h.
Fig. 21. La producción de IFN-gamma por células CAR T se midió 18 h después de la estimulación con células K562 sometidas a electroporación con la cantidad indicada de ARNm de ErbB2.
Fig. 22. Conjunto de imágenes que muestran la regresión de tumores vascularizados avanzados en ratones tratados con células ErbB2 CAR T ajustadas por afinidad. Los tumores de flanco se establecieron mediante la inyección de 5*10® SK-OV3-CBG (s.c.) en ratones NOD-SCID-y-/- (NSG) (n= 5). Dieciocho días después de la inoculación del tumor, los ratones se aleatorizaron para igualar la carga tumoral y se trataron con 1*107células T transducidas lentiviralmente que expresaban CAR de mayor afinidad (4D5.BBZ) o menor afinidad (4D5-5.BBZ). Los ratones tratados con células T no transducidas (Células T Solas) sirvieron como controles. Se tomaron imágenes de los animales en los puntos de tiempo indicados después de la inoculación del tumor.
Fig. 23. Conjunto de imágenes que muestran la discriminación in vivo de tumores que expresan ErbB2 alto (SK-OV3) y ErbB2 bajo (PC3) mediante CARs ajustados por afinidad. Células T modificadas con ErbB2 CARs de
diferente afinidad mediante transducción lentiviral se testaron en ratones NSG con injerto de tumor dual. A los ratones se les implantaron células tumorales PC3-CBG (1e6 células/ratón, s.c.) en el flanco derecho el día 0. El día 5, a los mismos ratones se les administraron células tumorales SK-OV3-CBG (5e6 células/ratón, s.c.) en el flanco izquierdo. Los ratones se trataron con células T (i.v.) el día 23 después de la inoculación del tumor PC3. Células CAR T se administraron como una única inyección de 10e6/ratón (10 M) o 3e6/ratón (3 M) según se indica. Los ratones tratados con células T no transducidas sirvieron como control. Se tomaron imágenes de los animales en el momento indicado después de la inoculación del tumor PC3.
Fig. 24. Tamaño del tumor SK-OV3 en ratones NSG injertados con doble tumor tratados con los ErbB2 CARs ajustados por afinidad indicados. Los tamaños de los tumores SK-OV3 se midieron a lo largo del tiempo (días, eje x) y el volumen del tumor se calculó y representó gráficamente (mm3, eje y).
Fig. 25. Tamaños de los tumores PC3 en ratones NSG injertados con doble tumor tratados con los ErbB2 CARs ajustados por afinidad indicados. Los tamaños de los tumores PC3 se midieron a lo largo del tiempo (días, eje x) y el volumen del tumor se calculó y representó gráficamente (mm3, eje y).
Fig. 26A y 26B. Conjuntos de imágenes que muestran la expresión de CAR en células T electroporadas con ARNm de EGFR CAR se tiñeron con un Fab de IgG anti-humano y se detectaron mediante tinción de citometría de flujo (Fig. 26A); se indica la afinidad del scFv (nM). Las líneas tumorales (Fig. 26B) se tiñeron con Affibody-FITC anti-EGFR (histogramas en blanco), las mismas células se tiñeron con IgG1-FITC de ratón como control de isotipo (histogramas grises).
Fig. 27. La sensibilidad de reconocimiento de EGFR CAR se correlaciona con la afinidad. Se estimuló un panel de células EGFR CAR T con la afinidad indicada del scFv (KD, nM) con el panel de tumores que expresan EGFR a la densidad mostrada en la Fig. 26B. Después de 4 h de estimulación, se detectó la regulación ascendente de CD107a en las células CAR T al ligar células CD3+.
Fig. 28. Un conjunto de imágenes que muestran la expresión de ErbB2 en células K562 sometidas a electroporación con la cantidad indicada de ARNm de EGFR. La expresión de EGFR se detectó utilizando tinción anti-EGFR Affibody-FITC 14 h después de la electroporación.
Fig. 29. La sensibilidad de reconocimiento de EGFR CAR se correlaciona con la afinidad. Células T se sometieron a electroporación con el panel de EGFR CARs con diferentes afinidades, como se indica, y se estimularon con K562 electroporadas con ARNm de EGFR a diferentes niveles, como se muestra en la Fig. 30. Después de 4 h de estimulación, se midió la expresión de CD107a en las células CAR T mediante ligamiento en células CD3+.
Fig. 30. Reconocimiento dependiente de la afinidad de líneas celulares primarias y células tumorales utilizando EGFR CARs ajustados por afinidad. Las células T se sometieron a electroporación con el ARNm de EGFR CAR indicado. Un día después de la electroporación, las células CAR T se estimularon con el panel de células durante 4 h y se cuantificó la inducción de la expresión de CD107a en las células CAR T (controlado por CD3+).
Fig. 31. Reconocimiento diferencial de líneas celulares primarias por células T modificados con EGFR CARs ajustados por afinidad. Se representó el porcentaje de células doblemente positivas CD8+CD107a+.
La Fig. 32. representa la actividad de luciferasa impulsada por el promotor inducible por NFAT de un PD1 CAR en comparación con el tratamiento de control por IgG1-Fc. La FIG. 22B representa la actividad de luciferasa impulsada por el promotor inducible por NFAT de un PD1 RCAR que incluye PD1-ECD-TM-FRB y FKBP-4 1BB-cD3 zeta en comparación con el tratamiento de control por IgG1-Fc.
Fig. 33A y 33B. Generación de células T del receptor de antígeno quimérico (CAR) completamente humano específico del receptor de folato alfa (FRA). (Fig. 33A) Representación esquemática de construcciones de CAR basadas en C4 que contienen el dominio citosólico de CD3Z solo (C4-z) o en combinación con el módulo coestimulador de CD27 (C4-27z). También se muestra el FRA Mov19-27z Car anti-humano murino. (Fig. 33B) Un conjunto de imágenes que muestran células T transducidas consistentes en células CD4- y CD8-positivas para, expresando ambos subconjuntos CARs C4. La expresión de CAR C4 (histogramas en blanco) se detectó a través de tinción con IgG anti-humana de conejo marcada con biotina (H+L), seguida de estreptavidinaficoeritrina después de la transducción con lentivirus en comparación con células T no transducidas (UNT) (histogramas grises rellenos). Las eficiencias de transducción se indican con el porcentaje de expresión de CAR entre paréntesis. ScFv, fragmento variable L de anticuerpo de cadena sencilla, enlazador; C4, scFv anti-FRA,VH, cadena H variable; VL, cadena L variable; TM, región transmembrana.
Fig. 34A, 34B, 34C, 34D y 34E. Comparación de la actividad antitumoral de los CARsC4 y MOv19 específicos para FR con el endodominio coestimulador de CD27 in vitro. (Fig. 34A) Conjunto de imágenes que muestran la expresión de CARs C4 y MOv19 en células T humanas primarias puede detectarse a través de la proteína FRA
recombinante marcada con biotina seguida de SA-PE. Como se muestra, tanto las células T CD8+ como las CD8- (CD4+) pueden expresar CARs de manera eficiente según lo medido por citometría de flujo. (Fig. 34B) Conjunto de gráficos que muestran células T transducidas con CARs C4 y MOv19 mostraron función lítica en un ensayo de exterminio bioluminiscente. Las células CAR-T mataron FR+ SKOV3 y A1847 en la relación E/T indicada durante más de 20 horas. Las células T no transducidas sirvieron como controles negativos. Se muestra la media y la DE de pocillos por triplicado de 1 de al menos 3 experimentos independientes. (Fig. 34C) Se co-cultivaron células CAR T C4 o MOv19 con células diana FRA+ (s Ko V3, A1847 y T47D) y FRA-(C30) en una relación E:T de 1:1. (Fig. 34D) Conjunto de imágenes que muestran células CAR T C4 o MOv19 estimuladas con células SKOV3 durante 5 horas en presencia de inhibidor de Golgi y analizadas mediante citometría de flujo para IFN-g intracelular, TNF-a e IL-2. (Fig. 34E) Ensayo de liberación de IFN-g de células CAR T C4 y MOv19 después del co-cultivo durante la noche con células tumorales FRA+ (a relaciones 1:10, 1:3, 1:1, 3:1 y 10:1).
Fig. 35A, 35B 35C y 35D. La actividad antitumoral de las células C4-CAR T es equiparable a la de las células CAR T MOv19. (Fig. 35A) Regresión del tumor mediada por células CAR T C4-27z y Mov19-27z. Ratones NSG que portaban un tumor subcutáneo establecido se trataron con inyecciones i.v. De 1*107 células CAR+ T C4-27z y MOv19-27z o células T CD19-27z y UNT de control o solución salina los días 40 y 45. El crecimiento del tumor se evaluó mediante la medición del calibre. Los tumores tratados con células CAR C4-27z o CAR T MOv19 (~ 60 % de expresión de CAR) retrocedieron (las flechas indican los días de infusión de células T); los tumores tratados con solución salina, UNT o células CAR T CD19-27z no retrocedieron 3 semanas después de la primera dosis de células T. (Fig. 35B) Conjunto de imágenes que muestran que la señal de bioluminiscencia de SKOV3 fLuc+ disminuyó en ratones tratados con células CAR T C4-27z y MOv19-27z en comparación con los grupos de tratamiento CD19-27z y control 3 semanas después de la primera dosis de células T. (Fig. 35C) Evaluación macroscópica de especímenes de tumores resecados después de la terapia con células T. Los tumores se recolectaron de ratones en el momento de la eutanasia, casi 45 días después de la primera inyección de células T. . (Fig. 35D) Persistencia estable de células CAR C4 y CAR T MOv19 in vivo. Se recogió sangre periférica 3 semanas después de la primera infusión de células T y se cuantificó el número absoluto de células T CD4+ y CD8+ humanas/pl de sangre. El recuento medio de células ± SEM se muestra con n = 5 para todos los grupos
Fig. 36A, 36B y 36C. Células CAR T C4 mostraron una actividad citotóxica mínima in vitro. . (Fig. 36A) Conjunto de imágenes que muestran células 293T de riñón embrionario humano y la línea celular de ovario epitelial normal IOSE6 expresan un nivel muy bajo de FRA. SKOV3 y C30 sirvieron como controles positivo y negativo, respectivamente. (Fig. 36B) Las células CAR T C4-27z secretan una cantidad mínima de IFN-y después de la incubación durante la noche con células 293T normales y líneas celulares IOSE 6 que expresan niveles bajos de FRA en superficie en comparación con las células CAR T MOvl9-27z. (Fig. 36C) Conjunto de imágenes que muestran células CAR T C4 y MOv19 estimuladas con células 293T o IOSE6 durante 5 horas en presencia de inhibidor de Golgi y analizadas mediante citometría de flujo para IFN-g intracelular y TNF-a.
Fig. 37A y 37B. CAR C4 completamente humano se expresa y detecta en la superficie de las células T. (Fig. 37A) Se determinó un conjunto de imágenes que muestran los títulos lentivirales (unidades de transducción, TU) utilizando células SupT1 en base a una dilución en serie de 3 veces de virus concentrado desde 1:3 hasta una dilución final de 1:6,561.Lentivirus que codifica CAR C4 tiene un título más alto cuando se compara con el título del lentivirus que codifica CAR MOv19, siguiendo los mismos protocolos de producción y concentración en paralelo. (Fig. 37B) Conjunto de imágenes que muestran que las células T humanas primarias se infectaron con lentivirus que codifica CAR C4 o CAR MOv19 a una multiplicidad de infección (MOI) de 1, 2 o 5. Estos datos representan uno de al menos tres experimentos independientes.
Fig. 38A, 38B, 38C. Las Figs.. 38A y 38B son conjuntos de imágenes que muestran células T no transducidas que fueron estimuladas con células FRA+ SKOV3 y células CAR T c 4 o Mov19 que fueron estimuladas con células FRA- C30 durante 5 horas en presencia de inhibidor de Golgi y analizadas mediante citometría de flujo para IFN-g intracelular, TNF-a e IL-2. La Fig. 38C muestra la expresión de aFR en las líneas celulares tumorales SKOV3, A1847 y T47D; la línea celular C30 se utilizó como control negativo.
Fig. 39A, 39B, 39C. Conjuntos de imágenes que muestran la modulación descendente de CAR pueden afectar a la actividad antitumoral de CAR MOv19 pero no de CAR C4. Células CAR T C4 y MOv19 se estimularon con células SKOV3 o C30 durante 5 horas en presencia de un inhibidor de Golgi y se analizaron mediante citometría de flujo para determinar la superficie de las células T de la expresión CAR e IFN-g intracelular, TNF-a e IL-2. (Fig. 39A) Análisis de citometría de flujo de los cambios en la expresión de CAR después de 4 h de co-cultivo con células tumorales aFR+ o aFR-. (Fig. 39B) Células CAR T MOv19 y C4 co-cultivadas con células tumorales aFR+ o aFR- y luego teñidas con anexina V y 7-AAD. Las células apoptóticas se indican como el porcentaje de células ligadas. En la Fig. 39C, las células se tiñeron para CD137.
Fig. 40. Conjunto de imágenes que muestran el análisis de citometría de flujo de los cambios en la expresión de CAR después del co-cultivo durante la noche con células tumorales FRA+ (a relaciones 1:10, 1:3, 1:1, 3:1 y 10:1).
La Fig. 41, que comprende Figs. 41A y 41B, son gráficos que muestran un aumento en los títulos de las cepas de la vacuna contra la gripe en comparación con el placebo. En la Fig. 41A, el aumento por encima del valor inicial en los títulos medios geométricos de la gripe para cada una de las 3 cepas de la vacuna contra la gripe (H1N1 A/California/07/2009, H3N2 A/Victoria/210/2009, B/Brisbane/60/2008) en relación con el aumento en la cohorte de placebo 4 semanas después de la vacunación se muestra para cada una de las cohortes de dosificación con RAD001 en la población por intención de tratar. La línea negra en negrita indica el aumento de 1,2 veces en los títulos en relación con el placebo que se requiere para que 2 de cada 3 cepas de la vacuna contra la gripe alcancen el criterio principal de valoración del estudio. El asterisco "*" indica que el aumento en el título de GMT en relación con el placebo supera 1 con una probabilidad posterior de al menos el 80 %. La FIG. 41B es un gráfico de los mismos datos que en la FIG. 41A para el subconjunto de sujetos con títulos iniciales de gripe <= 1:40.
La Fig. 42 muestra un conjunto de diagramas de dispersión de la concentración de RAD001 frente al aumento de veces en el título medio geométrico para cada una de las cepas de vacuna contra la gripe 4 semanas después de la vacunación. Las concentraciones de RAD001 (1 hora después de la dosis) se midieron después de que los sujetos hubieran recibido la dosis durante 4 semanas. Todos los sujetos que tenían mediciones farmacocinéticas se incluyeron en el conjunto de análisis. En el eje y se muestra el aumento de veces en los títulos medios geométricos a las 4 semanas después de la vacunación en relación con el valor de referencia.
La Fig. 43 es una representación gráfica que muestra un aumento en los títulos de las cepas de gripe heterólogas en comparación con el placebo. El aumento por encima del valor inicial en los títulos de la media geométrica de gripe para 2 cepas heterólogas de gripe (A/H1N1 cepa A/New Jersey/8/76 y A/H3N2 cepa A/Victoria/361/11) no contenidas en la vacuna contra la gripe en relación con el aumento en la cohorte de placebo 4 semanas después de la vacunación se muestra para cada una de las cohortes de dosificación de RAD001 en la intención de tratar la población. "*" indica que el aumento del título en relación con el placebo supera 1 con una probabilidad posterior de al menos el 80 %.
La Fig.44, que comprende las Figs. 44A y 44B, son representaciones gráficas de los niveles de IgG e IgM antes y después de la vacunación contra la gripe. Se midieron los niveles de IgG e IgM anti-A/H1N1/California/07/2009 gripe en suero obtenido de sujetos antes y 4 semanas después de la vacunación contra la gripe. No se detectaron diferencias significativas en el cambio del valor de referencia hasta 4 semanas después de la vacunación en los niveles de IgG e IgM contra la gripe H1N1 entre las cohortes RAD001 y placebo (todos los valores de p > 0,05 según la prueba de suma de rangos de Kruskal-Wallis).
La Fig. 45, que comprende las Figs. 45A, 45B y 45C son representaciones gráficas de la disminución en el porcentaje de CD4 y CD8 PD-1 positivos y el aumento de células T CD4 PD-1 negativas después del tratamiento con RAD001. El porcentaje de células T CD4, CD8 PD-1 positivas y PD-1 negativas se determinó mediante análisis FACS de muestras de PBMC al inicio del estudio, después de 6 semanas de tratamiento con el fármaco del estudio (Semana 6) y 6 semanas después de la interrupción del fármaco del estudio y 4 semanas después de la vacunación contra la gripe (Semana 12). La FIG. 45A muestra que hubo una disminución significativa ( 37,1 - -28,5 %) de células T CD4 PD-1 positivas en la semana 12 en cohortes que recibieron RAD001 a niveles de dosis de 0,5 mg/Día (n = 25), 5 mg/Semana (n = 29) y 20 mg/Semana (n = 30) en comparación con la cohorte de placebo (n = 25) con p = 0,002 (0,02), p = 0,003 (q = 0,03) y p = 0,01 (q = 0,05) respectivamente. La FIG.
45B muestra que hubo una disminución significativa (-43,3 - -38,5 %) de células T CD8 PD-1 positivas en la semana 12 en cohortes que recibieron RAD001 (n = 109) a niveles de dosis de 0,5 mg/Día (n = 25), 5 mg/Semana (n = 29) y 20 mg/Semana (n = 30) en comparación con la cohorte de placebo (n = 25) con p = 0,01 (0,05), p = 0,007 (q = 0,04) y p = 0,01 (q = 0,05), respectivamente. La FIG. 45C muestra un aumento significativo (3,0 -4,9 %) de células T CD4 PD-1 negativas en la semana 12 en cohortes que recibieron RAD001 (n = 109) a niveles de dosis de 0,5 mg/Día (n = 25), 5 mg/Semana (n = 29) y 20 mg/Semana (n = 30) en comparación con la cohorte de placebo (n = 25) con p = 0,0007 (0,02), p = 0,03 (q = 0,07) y p = 0,03 (q = 0,08) respectivamente.
La Fig. 46, que comprende las Figs. 46A y 46B son representaciones gráficas de la disminución en el porcentaje de CD4 y CD8 PD-1 positivas y el aumento de células T CD4 PD-1 negativas después del tratamiento con RAD001. El porcentaje de células T CD4, CD8 PD-1 positivas y PD-1 negativas se determinó mediante análisis FACS de muestras de PBMC al inicio del estudio, después de 6 semanas de tratamiento con el fármaco del estudio (Semana 6) y 6 semanas después de la interrupción del fármaco del estudio y 4 semanas después de la vacunación contra la gripe (Semana 12). La FIG. 46A muestra que hubo una disminución significativa (-37,1 - -28,5 %) de células T CD4 PD-1-positivas en la semana 12 en cohortes que recibieron RAD001 a niveles de dosis de 0,5 mg/Día (n = 25), 5 mg/Semana (n = 29) y 20 mg/Semana (n = 30) en comparación con la cohorte de placebo (n = 25) con p = 0,002 (0,02), p = 0,003 (q = 0,03) y p = 0,01 (q = 0,05) respectivamente. La FIG. 46B muestra que hubo una disminución significativa (-43,3 - -38,5 %) de células T CD8 PD-1 positivas en la semana 12 en cohortes que recibieron RAD001 (n = 109) a niveles de dosis de 0,5 mg/Día (n = 25), 5 mg/Semana (n = 29) y 20 mg/Semana (n = 30) en comparación con la cohorte de placebo (n = 25) con p = 0,01 (0,05), p = 0,007 (q = 0,04) y p = 0,01 (q = 0,05), respectivamente.
La Fig. 47 es un conjunto de gráficos que representan aumentos en el ejercicio y la energía en sujetos de edad avanzada en respuesta a RAD001.
La Fig. 48, que comprende las Figs. 48A y 48B, representan el efecto de RAD001 sobre la actividad de P70 S6K en líneas celulares. La FIG. 48A representa la inhibición de la quinasa P70 S6 con dosis más altas de RAD001 semanal y diaria; la FIG. 48B representa la inhibición de la quinasa P70 S6 con dosis más bajas de RAD001 semanal.
Fig. 49A, 49B, 49C, 49D, 49E y 49F. Los gráficos representan valores MFI normalizados para los siguientes genes en muestras individuales: CD79A (Fig. 49A), CCR2 (Fig. 49B), TNFRSF17 (Fig. 49C), HSPB1 (Fig. 49D), CD72 (Fig. 49E) y CD48 (Fig. 49F). El eje X representa el gen diana.
Fig. 50A, 50B, 50C, 50D, 50E y 50F. Los gráficos representan valores MFI normalizados para los siguientes genes en muestras individuales: TNFRSF13C (Fig. 50a ), IL3RA (Fig. 50B), SIGLEC1 (Fig. 50C), LAIR1 (Fig. 50D), FCAR (Fig. 50E) y CD79B (Fig. 50F). El eje X representa el gen diana.
Fig. 51A, 51B, 51C, 51D, 51E y 51F. Los gráficos representan valores MFI normalizados para los siguientes genes en muestras individuales: LILRA2 (Fig. 51A), CD37 (Fig. 51B), CD300LF (Fig. 51C), c Le C12A (Fig. 51D), BST2 (Fig. 51E) y CD276 (Fig. 51F). El eje X representa el gen diana.
Fig. 52A, 52B, 52C, 52D, 52E y 52F. Los gráficos representan valores MFI normalizados para los siguientes genes en muestras individuales: EMR2 (Fig. 52A), HSPH1 (Fig. 52B), RGS13 (Fig. 52C), CLECL1 (Fig. 52D), SPN (Fig. 52E) y CD200 (Fig. 52F). El eje X representa el gen diana.
Fig. 53A, 53B, 53C, 53D, 53E y 53F. Los gráficos representan valores MFI normalizados para los siguientes genes en muestras individuales: LY75 (Fig. 53A), SIRPB1 (Fig. 53B), FLT3 (Fig. 53C), CD22 (Fig. 53D), PTPRC (Fig. 53E) y GPRC5D (Fig. 53F). El eje X representa el gen diana.
Fig. 54A, 54B, 54C, 54D, 54E y 54F. Los gráficos representan valores MFI normalizados para los siguientes genes en muestras individuales: UMODL1 (Fig. 54A), CD74 (Fig. 54B), MS4A3 (Fig. 54C), CD302 (Fig. 54D), TNFRFSF13B (Fig. 54E) y MSN (Fig. 54F). El eje X representa el gen diana.
Fig. 55A, 55B, 55C, 55D, 55E y 55F. Los gráficos representan valores MFI normalizados para los siguientes genes en muestras individuales: KIT (Fig. 55A), GPC3 (Fig. 55B), CD101 (Fig. 55C), CD300A (Fig. 55D), SEMA4D (Fig. 55E) y CD86 (Fig. 55F). El eje X representa el gen diana.
Fig. 56A, 56B, 56C, 56D, 56E y 56F. Los gráficos representan valores MFI normalizados para los siguientes genes en muestras individuales: SIGLEC5 (Fig. 56A), GPR114 (Fig. 56B), FCRL5 (Fig. 56C), ROR1 (Fig. 56D), PTGFRN (Fig. 56E) y IGLL1 (Fig. 56F). El eje X representa el gen diana.
Fig. 57A, 57B, 57C, 57D y 57E. Los gráficos representan valores MFI normalizados para los siguientes genes en muestras individuales: CD244 (Fig. 57A), CD19 (Fig. 57B), CD34 (Fig. 57C), BST1 (Fig. 57D) y TFRC (Fig. 57E). El eje X representa el gen diana.
Fig. 58. Representación acumulativa de los valores de MFI normalizados promedio en relación con el gen de referencia PP1B de AML, ALL, médula ósea normal (NBM, por sus siglas en inglés) o línea celular OV357 (OV357). El eje Y representa MFI normalizado; El eje X representa los genes diana.
Fig. 59A y 59B. Generación de células T que expresan CAR que fijan como objetivo el Receptor de Folato a. La Fig. 59A es un diagrama esquemático del vector CAR C4-27z. La Fig. 59B es un conjunto de gráficos de histograma FACS representativos de la expresión de CAR en células T CD4+ y CD8+ 48 horas después de la transducción lentiviral.
Fig. 60. Diagrama esquemático del experimento in vivo para testar diferentes citoquinas en combinación con células CAR-T en un modelo de ratón con tumor de ovario.
Fig. 61. Curva de crecimiento del tumor de ratones tratados con diversas células CAR-T C4-27z expuestas a citoquinas, células CAR-T anti-CD19-27z y células T no transducidas. Los datos se presentan como valor medio ± SEM. La flecha indica el momento de la infusión de células T.
Fig. 62. Las imágenes de bioluminiscencia muestran tumores fLuc+ SKOV3 en ratones NSG inmediatamente antes (día 38), dos semanas (día 53) y cinco semanas (día 74) después de la primera inyección intravenosa de células CAR-T.
Fig. 63. Cuantificación de los recuentos de células T CD4+ y CD8+ humanas circulantes en sangre periférica de ratones 15 días después de la primera dosis de infusión de células CAR-T.
Fig. 64. Cuantificación de la expresión de CAR en células T CD4+ y CD8+ humanas circulantes en sangre de ratones.
Fig. 65. Distribución de subconjuntos de células T de células T humanas circulantes en sangre de ratones en base a la tinción de CD45RA y CD62L.
Fig. 66. Conjunto de gráficos que muestran la cuantificación de la expresión de CD27 y CD28 en células T CD4+ y CD8+ humanas circulantes en sangre de ratones.
Fig. 67A, 67B y 67C Pérdida de funcionalidad de las células mesoCAR T en el microentorno del tumor (TILs) a lo largo del tiempo en comparación con las células mesoCAR T frescas o descongeladas. A) Ensayo de citotoxicidad (ensayo de exterminio ex vivo EMMESO TIL (cosecha inmediata)); B) Ensayo de liberación de IFNy (nivel de IFNgamma después de 20 h de co-cultivo 50:1 con EMMESO 5K/células ffluc antes/después de 30 Ul/ml de IL2 durante la noche de reposo); y C) análisis de transferencia Western de la señalización de ERK (a través de la fosforilación).
Fig. 68. El efecto de la deleción de DGK en la citotoxicidad de las células mesoCAR T. El porcentaje de deleción de células diana se evalúa en diferentes relaciones de efector:diana.
Fig. 69 . El efecto de la deleción de DGK en la producción y liberación de IFNy de las células mesoCAR T. La concentración de IFNy se evalúa en diferentes relaciones de efector:diana.
Fig. 70. El efecto de la deleción de DGK en la señalización de ERK, o activación de células T, células mesoCAR T. B: albúmina, M: mesotelina, 3/28: células estimuladas con CD3/CD28.
Fig. 71. El efecto de la deleción de DGK sobre la sensibilidad a TGFp de células mesoCAR T con respecto a la actividad citotóxica.
Fig. 72A y 72B. El efecto de la deleción de DGK sobre la eficacia terapéutica de las células mesoCAR T en un modelo de ratón tumoral. A) El efecto sobre la actividad antitumoral se muestra por el volumen del tumor a lo largo del tiempo. B) Persistencia y proliferación de células tumorales infiltrantes.
La Fig. 73A, 73B, 73C, 73D, 73E y 73F muestran la producción de citoquinas y la liberación de mediadores citotóxicos en células T que expresan CAR con niveles reducidos de Ikaros. La Fig. 73A muestra la expresión de Ikaros en células CAR T tipo salvaje e Ikzf1+/- medida mediante citometría de flujo (panel izquierdo) y transferencia Western (panel derecho). Después de la estimulación con perlas recubiertas de mesotelina, PMA/Ionomicina (PMA/I) o perlas recubiertas con BSA (control), se determinó el porcentaje de células que producen IFN-y (Fig.73B), TNF-a (FIG. 73C) y la expresión de IL-2 (FIG. 73D), el mediador citotóxico granzima B (FIG. 73E) y CD107a (FIG. 73F).
Fig. 74A, 74B y 74C. Producción de citoquinas y liberación de mediadores citotóxicos en células T que expresan CAR con un alelo negativo dominante de Ikaros (IkDN). Después de la estimulación con perlas recubiertas de mesotelina, PMA/Ionomicina (PMA/I) o perlas recubiertas con BSA (control), se determinó el porcentaje de células que producen IFN-y (FIG. 74A), lL-2 (FIG. 74B) y la expresión de CDl07a (FIG. 74C).
Fig. 75A, 75B, 75C, 75D y 75E. El agotamiento de Ikaros no aumentó la activación ni la señalización de las células CAR T después de la estimulación con antígeno. Los niveles de CD69 (FIG. 75A), CD25 (FIG. 75B) y 4-1BB (FIG. 75C) se determinaron mediante citometría de flujo en los puntos de tiempo indicados en células CAR T Ikzf1+/-. En la Fig. 75D, las vías de señalización de RAS/ERK se examinaron en células de tipo salvaje (WT) y negativas dominantes de Ikaros (IkDN) después de la estimulación de TCR con anticuerpos CD3/CD28. Los niveles de proteínas de señalización de TCR fosforiladas, tales como PLCy fosforilada, Lck fosforilada, JNK fosforilada, Akt fosforilada, ERK fosforilada, IKKa fosforilada e iKBa, se evaluaron mediante transferencia Western. En la Fig. 75E, células WT e IkDN transducidas con mesoCAR se estimularon con BSA o perlas recubiertas de mesotelina, y las vías de señalización aguas abajo se examinaron mediante transferencia Western evaluando los niveles de ERK fosforilada y PLCy fosforilada.
Fig. 76A, 76B, 76C, 76D y 76E. La reducción de Ikaros en las células CAR T aumenta la respuesta contra las células diana AE17 o AE l7 que expresan mesotelina (AE17 meso) in vitro. La FIG. 76A representa la producción de IFNy en células WT y meso CART Ikzf1+/- en las relaciones indicadas de células efectoras:células diana. La citólisis de WT e Ikzf1+/-(FIG. 76B) e IkDN (FIG. 76C) que expresan meso CAR se midió en las relaciones indicadas de células efectoras:células diana. La producción de IFNy (FIG. 76D) y citólisis (FIG. 76E) de WT e
Ikzf1+/- transducidas con FAP-CAR se midió en las relaciones de células efectoras:células diana indicadas, en que las células diana eran células 3T3 que expresan FAP.
Fig. 77A, 77B y 77C. Eficacia de las células CAR T con agotamiento de Ikaros contra tumores establecidos in vivo. Se administraron células CAR T a ratones portadores de tumores AE17 que expresaban mesotelina establecidos. El volumen del tumor se midió después de la administración con WT e Ikzf1+/- que expresa mesoCAR (FIG. 77A) o IkDN (FIG. 77B). El volumen del tumor se midió después de la administración de WT que expresa FAP-CAR e Ikzf1+/- (FIG. 75C).
Fig. 78A, 78B, 78C, 78D, 78E y 78F. Mayor persistencia y resistencia de células CAR T Ikzf1+/- en el microentorno del tumor inmunosupresor en comparación con las células CAR T WT. El porcentaje de células WT o Ikzf1+/- que expresan CAR (GFP positivas se detectó mediante citometría de flujo a partir del bazo (FIG.
78A) y los tumores (FIG. 78B). La capacidad funcional de las células CAR T recolectadas 3 días después de la infusión del bazo o de los tumores se evaluó midiendo la producción de IFNy después de la estimulación con anticuerpos CD3/CD28 (FIG. 78C) o PMA/ionomicina (PmA/I) (FIG. 78D). Células T reguladoras (expresión de CD4+FoxP3+) y macrófagos (expresión de CD206) se evaluaron midiendo la expresión de Treg o marcadores de macrófagos en células CAR T recogidos 9 días después de la infusión del bazo o de los tumores.
Fig. 79A y 79B. Células T con niveles reducidos de Ikaros son menos sensibles a los factores inhibidores solubles TGFp y adenosina. Se ensayaron células WT, Ikzf1+/- e IkDN que expresan mesoCAR en cuanto a su capacidad para producir IFNy (FIG. 79A) y citotoxicidad (FIG. 79B) en respuesta a TGF-p o adenosina.
Las Fig. 80A y 80B muestran la expresión del receptor de IL-7 (CD127) en líneas de células cancerosas y células CART. La expresión de CD127 se midió mediante análisis de citometría de flujo en tres líneas celulares de cáncer: RL (linfoma de células del manto), JEKO (también conocido como Jeko-1, linfoma de células del manto) y Nalm-6 (LLA-B) (fig. 80A). La expresión de CD127 se midió mediante análisis de citometría de flujo en células positivas para CD3 (CART) que se habían infundido y circulaban en ratones NSG (fig. 80B).
Las Fig.81A, 81B y 81C muestran la respuesta antitumoral después del tratamiento con CART19 y el tratamiento posterior con IL-7. Los ratones NSG en los que se había injertado una línea celular de linfoma del manto que expresa luciferasa (RL-luc) el día 0 se trataron con dosis variables de células CART19 el día 6 y se supervisó la carga tumoral. Los ratones se dividieron en 4 grupos y recibieron células no CART19, 0,5*10® células CART19 (CART19 0,5E6), 1*10® células CART19 (CART19 1E6) o 2*10® células CART19 (CART19 2E6). La carga tumoral después del tratamiento con CART se midió mediante detección de bioluminiscencia (BLI media) (fig.
81A). Los ratones que recibieron 0.5*10® células CART19 (CART190,5E6) o 1*10® células CART19 (CART19 1E6) se aleatorizaron para recibir o no IL-7 humana recombinante (rhIL-7). La carga tumoral, representada aquí por bioluminiscencia media (BLI), se controló para los tres ratones (n° 3827, n° 3829 y n° 3815, que recibieron la dosis CART19 inicial indicada) de la Figura 81A que fueron tratados con IL-7 a partir del Día 85 (Figura 81B). La IL-7 se administró mediante inyección IP 3 veces por semana. La carga tumoral, representada aquí por la bioluminiscencia media (BLI) antes del día 85 (PRE) y después del día 115 (POST), se comparó entre ratones que no recibieron IL-7 (CTRL) y ratones que recibieron tratamiento con IL-7 (IL-7) (fig. 81C).
Las Fig. 82A y 82B muestran la dinámica de las células T después del tratamiento con IL-7. Se supervisó el nivel de células T humanas detectados en la sangre para cada uno de los ratones que recibieron IL-7 o ratones de control (fig. 82A). El nivel de células CART19 (células CD3+) detectados en la sangre se midió antes (PRE) y 14 días después (día 14) del inicio del tratamiento con IL-7 (fig. 82B).
La Fig. 83 es un conjunto de imágenes que muestra la expresión del receptor de IL-7 (IL-7R) detectada por citometría de flujo. Los paneles superiores muestran IL-7R en células de leucemia: línea celular AML MOLM14 (arriba a la izquierda), línea celular B-ALL NALM® (arriba en el centro) y AML primaria (arriba a la derecha). Los paneles inferiores muestran IL-7R en células de AML después de una recaída en modelos de ratones con AML que recibieron tratamiento con CART: células T no transducidas (UTD) (abajo a la izquierda), ratón tratado con CART33 (abajo en el centro) y un ratón tratado con CART123 (abajo a la derecha).
La Fig. 84 es un diagrama que muestra el esquema experimental para ratones NSG a los que se injertó luciferasa+ MOLM14 y se trataron con células no transducidas (UTD), CART33 o CART123, seguido de bLi en serie para evaluar la carga tumoral. Los ratones que experimentaron una recaída después de la respuesta inicial de la enfermedad se aleatorizaron para no recibir tratamiento (sin IL-7) o recibir tratamiento con IL-7200 ng IP tres veces por semana. Se realizó BLI en serie para evaluar la carga tumoral, la supervivencia y la expansión de células T.
Las Fig 85A, 85B y 85C muestran la respuesta antitumoral después del tratamiento con CART y el tratamiento posterior con IL-7. Los ratones NSG injertados con células MOLM14 que expresan luciferasa fueron tratados con células T no transducidas (UTD), células CART33 o células CART123. La carga tumoral después del tratamiento con CART se midió mediante formación de imágenes por bioluminiscencia (BLI) (Fig. 85A). Los ratones que
recibieron CART123 o CART33 inicialmente respondieron al tratamiento con células T pero recayeron 14 días después de las células T. Luego, estos ratones se asignaron al tratamiento con IL-7 o no el día 28. La carga tumoral después del tratamiento con IL-7 (IL-7) o de control (sin IL-7) se midió mediante imágenes de bioluminiscencia (BLI) (Fig. 85B) . Imágenes representativas de formación de imágenes de bioluminiscencia durante el tratamiento con IL-7 se muestran en la Figura 85C.
La Fig. 86A y 86B muestran la expansión y supervivencia de las células T después del tratamiento con IL-7 en el modelo de AML recidivante. El tratamiento con IL-7 resultó en una expansión de células T que se correlacionó con una reducción en la carga tumoral (Fig. 86A): la expansión de células T se evaluó midiendo las células T en la sangre (eje Y derecho) y se comparó con imágenes de bioluminiscencia (BLI, eje Y izquierdo). Se controló la supervivencia de los ratones que comparaban ratones que habían recibido tratamiento o control con IL-7 desde el momento de la inyección de MOLM14 (Fig. 86B).
Fig. 87. Este diagrama esquemático representa las estructuras de dos configuraciones RCAR ejemplares. Los miembros de unión a antígenos comprenden un dominio de unión a antígenos, un dominio transmembrana y un dominio de cambio. Los miembros de unión intracelular comprenden un dominio de conmutación, un dominio de señalización coestimulador y un dominio de señalización primario. Las dos configuraciones demuestran que el primer y segundo dominios de conmutación descritos en esta memoria pueden estar en diferentes orientaciones con respecto al miembro de unión a antígenos y al miembro de unión intracelular. Otras configuraciones de RCAR se describen con más detalle en esta memoria.
Las Fig.88A, 88B son un conjunto de gráficos que muestran los resultados de un ensayo de disociación de aFR. Se marcaron células CAR T C4-27z o MOv19-27z con proteína aFR recombinante biotinilada y luego se incubaron a 37 °C (Fig. 88A) o a 4 °C (Fig. 88B) en un ensayo de evolución temporal en presencia de exceso de aFR no biotinilado. El porcentaje de aFR retenido (eje y) se normalizó y calificó como intensidad de fluorescencia media (MFI) después de la incubación MFI de preincubación * 100.
La Fig.89 es un gráfico que muestra un análisis de titulación sobre la unión de proteína aFR biotinilada a células CAR T aFR. Se presenta el resultado de un experimento representativo de tres experimentos independientes.
Fig. 90A, 90B, 90C, 90D, 90E. Actividad antitumoral de células T humanas que expresan CAR C4 in vitro e in vivo. (A) Citotoxicidad de células tumorales SKOV3 que expresan aFR por células CAR T en un ensayo de bioluminiscencia de 18 horas en la relación E/T indicada. Las células T no transducidas (UNT) y las células CAR T CD19 sirvieron como controles efectores negativos. Las células C30 sirvieron como control de células diana negativas. El porcentaje de viabilidad de las células tumorales se calculó como la luminiscencia media de la muestra experimental menos el fondo dividido por la luminiscencia media del número de entrada de células diana utilizadas en el ensayo menos el fondo multiplicado por 100. Todos los datos se representan como una media de pocillos por triplicado. (B) Ratones NSG que tenían un tumor s.c. establecido fueron tratados con inyecciones intravenosas (i.v.) de 1*107 células CAR+ T el día 40 después de la inoculación del tumor. El crecimiento del tumor se evaluó mediante la medición de calibre [V = 1/2 (longitud * anchura 2)]. (C) La progresión del tumor fue seguida por imágenes de bioluminiscencia in vivo. La incorporación de señales CD27 potenció la actividad antitumoral in vivo; la terapia con células T C4-27z fue superior a la terapia con la célula CAR T C4-z que carece de un dominio de coestimulación CD27. (D) Los ratones NSG recibieron una inyección i.p. de 3 x 10 células tumorales SKOV3 fLuc y se aleatorizaron en 3 grupos antes de comenzar la terapia con células T UNT o células T que expresan C4-27z o CD19-27z CAR mediante infusión i.v. los días 21 y 25 después de la inoculación del tumor. Las imágenes de bioluminiscencia muestran tumores fLuc+ SKOV3 en ratones NSG inmediatamente antes y dos semanas después de la segunda inyección i.v. (E) Se cuantificó la emisión de fotones de las células tumorales fLuc y se determinó la señal de bioluminiscencia media ± DE antes y dos semanas después de la segunda inyección i.v. de 1 * 107 células CAR-T los días 21 y 25 después de la inoculación del tumor. Hubo una diferencia significativa en la carga tumoral entre las células CAR T C4-27z y los grupos de células T de control 14 días después de la inyección de la segunda dosis de células T (p = 0,002). Estos resultados sugieren que la actividad antitumoral mediada por las células T con CAR C4-27z era específica de antígeno in vivo.
DESCRIPCIÓN DETALLADA ADICIONAL
Definiciones
A menos que se defina de otro modo, todos las expresiones y términos técnicos y científicos utilizados en esta memoria tienen el mismo significado que entienden comúnmente los expertos en la técnica a la que pertenece la invención.
El término "un" y "uno/a" se refiere a uno o a más de uno (es decir, a al menos uno) del objeto gramatical del artículo. A modo de ejemplo, "un elemento" significa un elemento o más de un elemento.
Se entiende que el término "aproximadamente" cuando se refiere a un valor mensurable tal como una cantidad, una duración temporal y similares, abarca variaciones de ±20 % o en algunos casos ±10 %, o en algunos casos ±5 %, o en
algunos casos ±1 %, o en algunos casos ±0,1 % del valor especificado, ya que tales variaciones son adecuadas para realizar los métodos desvelados.
La expresión "receptor quimérico para antígenos" o, como alternativa, un "CAR" se refiere a un conjunto de polipéptidos, normalmente dos en las realizaciones más sencillas, que, cuando están en una célula efectora inmunitaria, proporcionan a la célula especificidad para una célula diana, normalmente una célula cancerosa, y con generación de señales intracelulares. En algunas realizaciones, un CAR comprende al menos un dominio de unión a antígenos extracelular, un dominio transmembrana y un dominio de señalización citoplasmático (al que también se alude en esta memoria como "un dominio de señalización intracelular") que comprende un dominio de señalización funcional derivado de una molécula estimulante y/o coestimulante como se define más adelante. En algunos aspectos, el conjunto de polipéptidos son contiguos entre sí. En algunas realizaciones, el conjunto de polipéptidos incluye un conmutador de la dimerización que, tras la presencia de una molécula de dimerización, puede acoplar los polipéptidos entre sí, p. ej., puede acoplar un dominio de unión a antígenos a un dominio de señalización intracelular. En un aspecto, la molécula estimulante es la cadena zeta asociada con el complejo receptor de células T. En un aspecto, el dominio de señalización citoplasmático comprende, además, uno o más dominios de señalización funcionales derivados de al menos una molécula coestimulante tal como se define más adelante. En un aspecto, la molécula coestimulante se elige entre las moléculas coestimulantes descritas en esta memoria, p. ej., 4-1BB (es decir, CD137), CD27 y/o CD28. En un aspecto, el CAR comprende una proteína de fusión quimérica que comprende un dominio de unión a antígenos extracelular, un dominio transmembrana y un dominio de señalización intracelular que comprende un dominio de señalización funcional derivado de una molécula estimulante. En un aspecto, el CAR comprende una proteína de fusión quimérica que comprende un dominio de unión a antígenos extracelular, un dominio transmembrana y un dominio de señalización intracelular que comprende un dominio de señalización funcional derivado de una molécula coestimulante y un dominio de señalización funcional derivado de una molécula estimulante. En un aspecto, el CAR comprende una proteína de fusión quimérica que comprende un dominio de unión a antígenos extracelular, un dominio transmembrana y un dominio de señalización intracelular que comprende dos dominios de señalización funcionales derivados de una o más moléculas coestimulantes y un dominio de señalización funcional derivado de una molécula estimulante. En un aspecto, el CAR comprende una proteína de fusión quimérica que comprende un dominio de unión a antígenos extracelular, un dominio transmembrana y un dominio de señalización intracelular que comprende al menos dos dominios de señalización funcionales derivados de una o más moléculas coestimulantes y un dominio de señalización funcional derivado de una molécula estimulante. En un aspecto, el CAR comprende una secuencia líder opcional en el extremo amino (N-ter) de la proteína de fusión CAR. En un aspecto, el CAR comprende, además, una secuencia líder en el extremo N del dominio de unión a antígenos extracelular, en donde la secuencia líder se escinde opcionalmente del dominio de unión a antígenos (p. ej., un scFv) durante el procesamiento celular y la localización del c Ar en la membrana celular.
Un CAR que comprende un dominio de unión a antígenos (p. ej., un scFv o TCR) que fija como objetivo un productor de tumor X específico, tal como los descritos en esta memoria, también se denomina XCAR. Por ejemplo, un CAR que comprende un dominio de unión a antígenos que fija como objetivo CD19 se denomina CD19CAR.
La expresión "dominio de señalización" se refiere a la parte funcional de una proteína que actúa mediante transmisión de la información dentro de la célula para regular la actividad celular a través de rutas de señalización definidas, mediante la generación de segundos mensajeros o funcionando como efectores al responder a dichos mensajeros.
El término "anticuerpo", como se usa en esta memoria, se refiere a una proteína o secuencia polipeptídica derivada de una molécula de inmunoglobulina que se une específicamente con un antígeno. Los anticuerpos pueden ser inmunoglobulinas policlonales o monoclonales, de cadena sencilla o múltiple, o intactas, y pueden proceder de fuentes naturales o de fuentes recombinantes. Los anticuerpos pueden ser tetrámeros de moléculas de inmunoglobulina.
La expresión "fragmento de anticuerpo" se refiere a al menos una porción de un anticuerpo, que retiene la capacidad de interactuar específicamente con (p. ej., mediante unión, impedimento estérico, estabilización/desestabilización, distribución espacial) un epítopo de un antígeno. Ejemplos de fragmentos de anticuerpos incluyen, pero no se limitan a fragmentos Fab, Fab', F(ab')2, Fv, fragmentos de anticuerpos scFv, Fv enlazados por disulfuro (sdFv), un fragmento Fd que consiste en los dominios VH y CH1, anticuerpos lineales, anticuerpos de un solo dominio tales como sdAb (ya sea Vl o VH), dominios VHH de camélidos, anticuerpos multiespecíficos formados a partir de fragmentos de anticuerpos, tal como un fragmento bivalente que comprende dos fragmentos Fab enlazados por un puente disulfuro en la región de bisagra, y un fragmento CDR aislado u otros fragmentos de unión a epítopos de un anticuerpo. Un fragmento de unión a antígenos también se puede incorporar en anticuerpos de un solo dominio, maxicuerpos, minicuerpos, nanocuerpos, intracuerpos, diacuerpos, tricuerpos, tetracuerpos, v-NAR y bis-scFv (véase, p. ej., Hollinger y Hudson, Nature Biotechnology 23: 1126-1136 , 2005). Los fragmentos de unión a antígenos también se pueden injertar en armazones basados en polipéptidos tales como una fibronectina de tipo III (Fn3) (véase la patente de EE. UU. N° 6.703.199, que describe minicuerpos polipeptídicos de fibronectina).
El término "scFv" se refiere a una proteína de fusión que comprende al menos un fragmento de anticuerpo que comprende una región variable de una cadena ligera y al menos un fragmento de anticuerpo que comprende una región variable de una cadena pesada, en donde las regiones variables de cadena ligera y pesada están ligadas de forma contigua, p. ej., a través de un enlazador sintético, p. ej., un enlazador polipeptídico flexible corto, y capaz de expresarse como un
polipéptido de cadena sencilla, y en donde el scFv conserva la especificidad del anticuerpo intacto del que procede. A menos que se especifique, tal como se utiliza en esta memoria, un scFv puede tener las regiones variables VL y VH en cualquier orden, p. ej., con respecto a los extremos N-terminal y C-terminal del polipéptido, el scFv puede comprender VL-enlazador-VH o puede comprender VH-enlazador-VL.
La porción del CAR de la divulgación que comprende un anticuerpo o un fragmento de anticuerpo del mismo puede existir en una diversidad de formas en las que el dominio de unión al antígeno se expresa como parte de una cadena polipeptídica contigua que incluye, por ejemplo, un fragmento de anticuerpo de dominio único (sdAb), un anticuerpo de cadena sencilla (scFv), un anticuerpo humanizado o un anticuerpo biespecífico (Harlow et al., 1999, en: Using Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, NY; Harlow et al., 1989, en: Antibodies: A Laboratory Manual, Cold Spring Harbor, Nueva York; Houston et al., 1988, Proc. Natl. Acad. Sci. USA 85:5879-5883; Bird et al., 1988, Science 242:423-426). En un aspecto, el dominio de unión a antígenos de una composición de CAR de la divulgación comprende un fragmento de anticuerpo. En un aspecto adicional, el CAR comprende un fragmento de anticuerpo que comprende un scFv. Los límites exactos de la secuencia de aminoácidos de una CDR dada pueden determinarse usando cualquiera de varios esquemas bien conocidos, incluidos los descritos por Kabat et al. (1991), "Sequences of Proteins of Immunological Interest", 5.a Ed. Public Health Service, Institutos Nacionales de la Salud, Bethesda, MD (esquema de numeración de "Kabat"), Al-Lazikani et al., (1997) JMB 273,927-948 (esquema de numeración de "Chothia") o una combinación de estos.
Tal como se utiliza en esta memoria, la expresión "dominio de unión" o "molécula de anticuerpo" se refiere a una proteína, p. ej., una cadena de inmunoglobulina o fragmento de la misma, que comprende al menos una secuencia de dominio variable de inmunoglobulina. La expresión "dominio de unión" o "molécula de anticuerpo" abarca anticuerpos y fragmentos de anticuerpos. En un caso, una molécula de anticuerpo es una molécula de anticuerpo multiespecífica, p. ej., comprende una pluralidad de secuencias de dominio variable de inmunoglobulina, en donde una primera secuencia de dominio variable de inmunoglobulina de la pluralidad tiene especificidad de unión para un primer epítopo y una segunda secuencia de dominio variable de inmunoglobulina de la pluralidad tiene especificidad de unión por un segundo epítopo. En un caso, una molécula de anticuerpo multiespecífica es una molécula de anticuerpo biespecífica. Un anticuerpo biespecífico tiene especificidad por no más de dos antígenos. Una molécula de anticuerpo biespecífico se caracteriza por una primera secuencia de dominio variable de inmunoglobulina que tiene especificidad de unión para un primer epítopo y una segunda secuencia de dominio variable de inmunoglobulina que tiene especificidad de unión para un segundo epítopo.
La porción del CAR de la divulgación que comprende un anticuerpo o fragmento de anticuerpo del mismo puede existir en una diversidad de formas en las que el dominio de unión al antígeno se expresa como parte de una cadena polipeptídica contigua que incluye, por ejemplo, un fragmento de anticuerpo de dominio único (sdAb), un anticuerpo de cadena sencilla (scFv), un anticuerpo humanizado o un anticuerpo biespecífico (Harlow et al., 1999, en: Using Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, NY; Harlow et al., 1989, en: Antibodies: A Laboratory Manual, Cold Spring Harbor, Nueva York; Houston et al., 1988, Proc. Natl. Acad. Sci. USA 85:5879-5883; Bird et al., 1988, Science 242:423-426). En un aspecto, el dominio de unión a antígenos de una composición de CAR de la divulgación comprende un fragmento de anticuerpo. En un aspecto adicional, el CAR comprende un fragmento de anticuerpo que comprende un scFv.
La expresión "cadena pesada del anticuerpo" se refiere al mayor de los dos tipos de cadenas polipeptídicas presentes en las moléculas de anticuerpo en sus conformaciones naturales y que normalmente determina la clase a la que pertenece el anticuerpo.
La expresión "cadena ligera de anticuerpo" se refiere al más pequeño de los dos tipos de cadenas polipeptídicas presentes en las moléculas de anticuerpo en sus conformaciones naturales. Las cadenas ligeras kappa (k) y lambda (A) se refieren a los dos isotipos principales de cadenas ligeras de anticuerpos.
La expresión "anticuerpo recombinante" se refiere a un anticuerpo que se genera utilizando tecnología de ADN recombinante, tal como, por ejemplo, un anticuerpo expresado por un bacteriófago o un sistema de expresión en levaduras. También se debe entender que la expresión se refiere a un anticuerpo que ha sido generado por la síntesis de una molécula de ADN que codifica el anticuerpo y donde la molécula de ADN expresa una proteína de anticuerpo, o una secuencia de aminoácidos que especifica el anticuerpo, en donde la secuencia de ADN o de aminoácidos se ha obtenido utilizando ADN recombinante o tecnología de secuencias de aminoácidos que está disponible y es muy conocida en la técnica.
El término "antígeno" o "Ag" se refiere a una molécula que provoca una respuesta inmunitaria. Esta respuesta inmunitaria puede implicar la producción de anticuerpos o la activación de células inmunocompetentes específicas, o ambas. El experto en la materia comprenderá que cualquier macromolécula, incluyendo prácticamente todas las proteínas o los péptidos, puede actuar como antígeno. Además, los antígenos pueden proceder de ADN recombinante o genómico. Un experto en la materia comprenderá que cualquier ADN, que comprenda una secuencia de nucleótidos o una secuencia de nucleótidos parcial que codifique una proteína que induzca una respuesta inmunitaria, codifica por lo tanto un "antígeno" tal como se utiliza ese término en esta memoria. Además, un experto en la materia comprenderá que no es necesario que un antígeno sea codificado únicamente por una secuencia de nucleótidos de longitud completa de un gen. Es muy evidente que la presente invención incluye, pero sin limitación, el uso de secuencias de nucleótidos parciales de
más de un gen y que estas secuencias de nucleótidos están dispuestas en diversas combinaciones para codificar polipéptidos que provocan la respuesta inmunitaria deseada. Además, un experto en la materia entenderá que no es necesario que un antígeno esté codificado por un "gen" en absoluto. Es fácilmente evidente que un antígeno se puede generar, sintetizar o derivar de una muestra biológica, o puede ser una macromolécula además de un polipéptido. Una muestra biológica de este tipo puede incluir, pero sin limitación, una muestra tisular, una muestra tumoral, una célula o un líquido con otros componentes biológicos.
La expresión "efecto anticancerígeno" se refiere a un efecto biológico que puede manifestarse por diversos medios, incluyendo, pero no limitados, p. ej., a una disminución en el volumen del tumor, una disminución en el número de células cancerosas, una disminución en el número de metástasis, un aumento en la expectativa de vida, una disminución en la proliferación de células cancerosas, una disminución en la supervivencia de células cancerosas o una mejora de diversos síntomas fisiológicos asociados con la afección cancerosa. Un "efecto anticancerígeno" también puede manifestarse por la capacidad de los péptidos, polinucleótidos, células y anticuerpos en la prevención de la aparición de cáncer en primer lugar. La expresión "efecto antitumoral" se refiere a un efecto biológico que se puede poner de manifiesto por diversos medios, que incluyen, pero sin limitación, p. ej., una disminución del volumen tumoral, una disminución del número de células tumorales, una disminución de la proliferación de células tumorales o una disminución de la supervivencia de células tumorales.
El término "autólogo" se refiere a cualquier material procedente del mismo individuo en el que luego se reintroducirá.
El término "alogénico" se refiere a cualquier material procedente de un animal diferente que es de la misma especie que el individuo en el que se introduce el material. Se dice que dos o más individuos son alogénicos entre sí cuando los genes en uno o más locus no son idénticos. En algunos aspectos, el material alogénico de individuos de la misma especie puede ser suficientemente diferente desde el punto de vista genético para interactuar de forma antigénica.
El término "xenogénico" se refiere a un injerto procedente de un animal de una especie diferente.
El término "cáncer" se refiere a una enfermedad caracterizada por el crecimiento descontrolado de células aberrantes. Las células cancerosas pueden diseminarse localmente o a través del torrente sanguíneo y el sistema linfático a otras partes del cuerpo. En esta memoria se describen ejemplos de diversos cánceres y estos incluyen, pero sin limitación, cáncer de mama, cáncer de próstata, cáncer de ovario, cáncer de cuello uterino, cáncer de piel, cáncer de páncreas, cáncer colorrectal, cáncer renal, cáncer de hígado, cáncer de cerebro, linfoma, leucemia, cáncer de pulmón y similares. Los términos "tumor" y "cáncer" se utilizan indistintamente en esta memoria, p. ej., ambos términos abarcan tumores sólidos y líquidos, p. ej., difusos o circulantes. Como se utiliza en esta memoria, el término "cáncer" o "tumor" incluye cánceres y tumores premalignos, así como malignos.
La expresión "derivado de", como se utiliza en esta memoria, indica una relación entre una primera y una segunda molécula. Se refiere de manera general a la similitud estructural entre la primera molécula y una segunda molécula, y no connota ni incluye una limitación, en cuanto a la fuente o el proceso, de una primera molécula que procede de una segunda molécula. Por ejemplo, en el caso de un dominio de señalización intracelular que procede de una molécula CD3zeta, el dominio de señalización intracelular mantiene suficiente estructura de CD3zeta para que tenga la función requerida, en concreto, la capacidad para generar una señal en las condiciones apropiadas. No connota ni incluye una limitación a un proceso particular para producir el dominio de señalización intracelular, p. ej., no significa que, para proporcionar el dominio de señalización intracelular, se deba comenzar con una secuencia de CD3zeta y eliminar la secuencia no deseada, o imponer mutaciones, para conseguir el dominio de señalización intracelular.
La expresión "enfermedad asociada con la expresión de un antígeno tumoral como se describe en esta memoria" incluye, pero sin limitación, una enfermedad asociada con la expresión de un antígeno tumoral como se describe en el presente memoria o una afección asociada con células que expresan un antígeno tumoral como se describe en esta memoria incluyendo, p. ej., una enfermedad proliferativa tal como un cáncer o una neoplasia maligna o una afección precancerosa, tal como una mielodisplasia, un síndrome mielodisplásico o una preleucemia; o una indicación no relacionada con el cáncer asociada con células que expresan un antígeno tumoral como se describe en esta memoria. En un aspecto, un cáncer asociado con la expresión de un antígeno tumoral como se describe en esta memoria es un cáncer hematológico. En un aspecto, un cáncer asociado con la expresión de un antígeno tumoral como se describe en esta memoria es un cáncer sólido. Enfermedades adicionales asociadas con la expresión de un antígeno tumoral como se describe en el presente memoria incluyen, pero sin limitación, p. ej., cánceres atípicos y/o no clásicos, neoplasias malignas, afecciones precancerosas o enfermedades proliferativas asociadas con la expresión de un antígeno tumoral como se describe en esta memoria. Indicaciones no relacionadas con el cáncer asociadas con la expresión de un antígeno tumoral como se describe en esta memoria incluyen, pero sin limitación, p. ej., enfermedad autoinmunitaria (p. ej., lupus), trastornos inflamatorios (alergia y asma) y trasplantes. En algunas realizaciones, las células que expresan el antígeno tumoral expresan, o han expresado en cualquier momento, el ARNm que codifica el antígeno tumoral. En una realización, las células que expresan el antígeno tumoral producen la proteína del antígeno tumoral (p. ej., de tipo salvaje o mutante), y la proteína del antígeno tumoral puede estar presente en niveles normales o niveles reducidos. En una realización, las células que expresan el antígeno tumoral produjeron niveles detectables de una proteína antigénica tumoral en un punto, y posteriormente produjeron una proteína antigénica tumoral sustancialmente no detectable.
La expresión "modificaciones de secuencia conservativas" se refiere a modificaciones de aminoácidos que no afectan o alteran significativamente las características de unión del anticuerpo o fragmento de anticuerpo que contiene la secuencia de aminoácidos. Modificaciones conservadoras de este tipo incluyen sustituciones, adiciones y supresiones de aminoácidos. Pueden introducirse modificaciones en un anticuerpo o fragmento de anticuerpo de la divulgación mediante técnicas convencionales conocidas en este campo, tales como mutagénesis dirigida y mutagénesis mediada por PCR. Las sustituciones de aminoácidos conservadoras son aquellas en las que el residuo de aminoácido se reemplaza por un residuo de aminoácido que tiene una cadena lateral similar. En la técnica se han definido familias de residuos de aminoácidos que tienen cadenas laterales similares. Estas familias incluyen aminoácidos con cadenas laterales de carácter básico (p. ej., lisina, arginina, histidina), cadenas laterales de carácter ácido (p. ej., ácido aspártico, ácido glutámico), cadenas laterales polares sin carga (p. ej., glicina, asparagina, glutamina, serina, treonina, tirosina , cisteína, triptófano), cadenas laterales no polares (p. ej., alanina, valina, leucina, isoleucina, prolina, fenilalanina, metionina), cadenas laterales ramificadas beta (p. ej., treonina, valina, isoleucina) y cadenas laterales aromáticas (p. ej., tirosina, fenilalanina, triptófano, histidina). Por lo tanto, uno o más residuos de aminoácidos dentro de un CAR utilizado en la invención pueden reemplazarse por otros residuos de aminoácidos de la misma familia de cadenas laterales y el CAR alterado puede analizarse utilizando los ensayos funcionales descritos en esta memoria.
El término "estimulación" se refiere a una respuesta primaria inducida por la unión de una molécula estimulante (p. ej., un complejo TCR/CD3 o CAR) con su ligando afín (o antígeno tumoral en el caso de un CAR) mediando de este modo en un acontecimiento de transducción de señales, tales como, pero sin limitación, la transducción de señales a través del complejo TCR/CD3 o la transducción de señales a través del receptor de NK adecuado o los dominios de señalización del CaR. La estimulación puede mediar en la expresión alterada de determinadas moléculas.
La expresión "molécula estimuladora" se refiere a una molécula expresada por una célula inmunitaria (p. ej., célula T, célula NK, célula B) que proporciona la o las secuencias de señalización citoplásmica que regulan la activación de la célula inmunitaria en un estímulo para al menos algún aspecto de la vía de señalización de las células inmunitarias. En un aspecto, la señal es una señal primaria que se inicia, p. ej., mediante la unión de un complejo TCR/CD3 con una molécula de MHC cargada con péptido, y que conduce a la mediación de una respuesta de células T, que incluye, pero sin limitación, proliferación, activación, diferenciación y similares. Una secuencia de señalización citoplasmática primaria (a la que también se alude como "dominio de señalización primario") que actúa de manera estimulante puede contener un motivo de señalización que se conoce como motivo de activación de inmunorreceptores basado en tirosina o ITAM. Los ejemplos de una secuencia de señalización citoplasmática que contiene ITAM que es de uso particular en la divulgación incluye, pero sin limitación, las derivadas de CD3 zeta, FcR gamma común (FCER1G), Fc gamma RIIa, FcR beta (Fc épsilon R1b), CD3 gamma, CD3 delta, CD3 épsilon, CD79a, CD79b, DAP10 y DAP12. En un CAR específico utilizado en la invención, el dominio de señalización intracelular en uno cualquiera o más CARS utilizados en la invención comprende una secuencia de señalización intracelular, p. ej., una secuencia de señalización primaria de CD3-zeta. En un CAR específico utilizado en la invención, la secuencia de señalización primaria de CD3-zeta es la secuencia proporcionada como la SEQ ID NO: 18 o los residuos equivalentes de una especie no humana, p. ej., ratón, roedor, mono, simio y similares. En un CAR específico utilizado en la invención, la secuencia de señalización primaria de CD3-zeta es la secuencia proporcionada en la SEQ ID NO: 20, o los residuos equivalentes de una especie no humana, p. ej., ratón, roedor, mono, simio y similares.
La expresión "célula presentadora de antígeno" o "APC" se refiere a una célula del sistema inmunitario tal como una célula accesoria (p. ej., una célula B, una célula dendrítica y similares) que muestra un antígeno extraño complejado con complejos de histocompatibilidad principal (MHC, por sus siglas en inglés) en su superficie. Las células T pueden reconocer estos complejos usando sus receptores de células T (TCR). Las APC procesan antígenos y los presentan a las células T.
Un "dominio de señalización intracelular", como se utiliza la expresión en el presente memoria, se refiere a una porción intracelular de una molécula. El dominio de señalización intracelular genera una señal que fomenta una función efectora inmunitaria de la célula que contiene el CAR, p. ej., un célula T-CAR. Ejemplos de la función efectora inmune, p. ej., en una célula CART, incluyen actividad citolítica y actividad auxiliar, incluida la secreción de citoquinas.
En una realización, el dominio de señalización intracelular puede comprender un dominio de señalización intracelular primaria. Los dominios de señalización intracelular primaria ilustrativos incluyen los procedentes de las moléculas responsables de la estimulación primaria o simulación dependiente de antígeno. En una realización, el dominio de señalización intracelular puede comprender un dominio intracelular coestimulante. Los dominios de señalización intracelular coestimulantes ilustrativos incluyen los procedentes de moléculas responsables de señales coestimulantes o estimulación independiente de antígeno. Por ejemplo, en el caso de un CART, un dominio de señalización intracelular primario puede comprender una secuencia citoplasmática de un receptor de células T y un dominio de señalización intracelular coestimulante puede comprender una secuencia citoplasmática del correceptor o molécula coestimulante.
Un dominio de señalización intracelular primario puede comprender un motivo de señalización que se conoce como motivo de activación de inmunorreceptores basado en tirosina o ITAM. Ejemplos de secuencias de señalización citoplasmáticas
primarias que contienen ITAM incluyen, pero no se limitan a los derivados de CD3 zeta, FcR gamma común (FCER1G), Fc gamma RIIa, FcR beta (Fc Épsilon R1b), CD3 gamma, CD3 delta, CD3 épsilon, CD79a, CD79b, DAP10 y DaP12.
El término "zeta" o alternativamente la expresión "cadena zeta", "CD3-zeta" o "TCR-zeta" se define como la proteína proporcionada como N° de Acceso a GenBank BAG36664.1, o los residuos equivalentes de una especie no humana, p. ej., ratón, roedor, mono, simio y similares, y un "dominio estimulador zeta" o alternativamente un "dominio estimulador CD3-zeta" o un "dominio estimulador TCR-zeta se define como los residuos de aminoácidos del dominio citoplásmico de la cadena zeta, o derivados funcionales del mismo, que son suficientes para transmitir funcionalmente una señal inicial necesaria para la activación de las células T. En un aspecto, el dominio citoplasmático de zeta comprende los residuos 52 a 164 del N° de Acceso a GenBank BAG36664.1 o los residuos equivalentes de una especie no humana, p. ej., ratón, roedor, mono, simio y similares, que son ortólogos funcionales de los mismos. En un aspecto, el «dominio estimulador zeta» o un «dominio estimulador CD3-zeta» es la secuencia proporcionada como la SEQ ID NO: 18. En un aspecto, el "dominio estimulador zeta" o un "dominio estimulador CD3-zeta" es la secuencia proporcionada como la SEQ ID NO:20.
La expresión "molécula coestimulante" se refiere a un compañero de unión afín en un célula T que se une específicamente con un ligando coestimulante, mediando de este modo en una respuesta coestimulante por el célula T, tal como, pero sin limitación, la proliferación. Las moléculas coestimulantes son moléculas de la superficie celular distintas de los receptores de antígenos o sus ligandos que contribuyen a una respuesta inmunitaria eficaz. Las moléculas coestimulantes incluyen, pero sin limitación, una molécula del MHC de clase I, BTLA y un receptor de ligando Toll, así como OX40, CD27, cD28, CDS, ICAM-1, LFA-1 (CD11a/CD18), ICOS (CD278) y 4-1BB (CD137). Otros ejemplos de tales moléculas coestimulantes incluyen CDS, ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, CD4, CD8alfa, CD8beta, IL2R beta, IL2R gamma, IL7R alfa, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, NKG2D, NKG2C, TNFR2, TRANCE/RANKL, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (táctil), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), CD69, SLAMF6 (NTB-A, Ly108), SLAM (SLAMF1, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, LAT, GADS, SLP-76, PAG/Cbp, CD19a y un ligando que se une específicamente con CD83.
Un dominio de señalización intracelular coestimulante puede ser la porción intracelular de una molécula coestimulante. Una molécula coestimulante puede representarse en las siguientes familias de proteínas: proteínas receptoras de TNF, proteínas de tipo inmunoglobulina, receptores de citocinas, integrinas, moléculas de activación linfocítica de señalización (proteínas SLAm) y receptores de células NK activadores. Los ejemplos de moléculas de este tipo incluyen CD27, CD28, 4-1BB (CD137), OX40, GITR, CD30, CD40, ICOS, BAFFR, HVEM, ICAM-1, antígeno asociado a la función linfocítica-1 (LFA-1), CD2, CDS, CD7, CD287, LIGHT, NKG2C, NKG2D, SLAMF7, NKp80, NKp30, NKp44, NKp46, CD160, B7-H3 y un ligando que se une específicamente con CD83, y similares.
El dominio de señalización intracelular puede comprender la porción intracelular completa, o el dominio de señalización intracelular nativo completo, de la molécula de la que procede, o un fragmento funcional o derivado de la misma.
El término "4-1BB" se refiere a un miembro de la superfamilia de TNFR con una secuencia de aminoácidos proporcionada como el n.° de ref. de GenBank AAA62478.2, o los residuos equivalentes de una especie no humana, p. ej., ratón, roedor, mono, simio y similares; y un "dominio coestimulante de 4-1BB" se define como los residuos de aminoácidos 214-255 del n.° de ref. de GenBank AAA62478.2, o los residuos equivalentes de una especie no humana, p. ej., ratón, roedor, mono, simio y similares. En un aspecto, el "dominio de co-estimulación 4-1BB" es la secuencia proporcionada como la SEQ ID NO: 14 o los residuos equivalentes de una especie no humana, p. ej., ratón, roedor, mono, simio y similares.
"Célula efectora inmunitaria", tal como se utiliza esa expresión en esta memoria, se refiere a una célula que está implicada en una respuesta inmunitaria, p. ej., en la promoción de una respuesta efectora inmunitaria. Ejemplos de células efectoras inmunitarias incluyen células T, p. ej., células T alfa/beta y células T gamma/delta, células B, células asesinas naturales (NK), células T asesinas naturales (NKT), mastocitos y fagocitos derivados de mieloide.
La "función efectora inmunitaria o respuesta efectora inmunitaria", tal como se utiliza la expresión en esta memoria, se refiere a una función o respuesta, p. ej., de una célula efectora inmunitaria, que potencia o fomenta un ataque inmunitario de una célula diana. Por ejemplo, una función o respuesta efectora inmunitaria se refiere a una propiedad de una célula T o NK que fomenta la muerte o la inhibición del crecimiento o proliferación, de una célula diana. En el caso de una célula T, la estimulación primaria y coestimulación son ejemplos de función o respuesta efectora inmunitaria.
El término "codificación" se refiere a la propiedad inherente de secuencias específicas de nucleótidos en un polinucleótido, tal como un gen, un ADNc o un ARNm, para servir como moldes para la síntesis de otros polímeros y macromoléculas en procesos biológicos que tienen una secuencia definida de nucleótidos (p. ej., ARNr, ARNt y ARNm) o una secuencia definida de aminoácidos y las propiedades biológicas resultantes de la misma. Por lo tanto, un gen, ADNc o ARN codifica una proteína si la transcripción y traducción del ARNm correspondiente a ese gen produce la proteína en una célula u otro sistema biológico. Tanto a la cadena codificante, cuya secuencia de nucleótidos es idéntica a la secuencia de ARNm y habitualmente se proporciona en listados de secuencias, como a la cadena no codificante, utilizada como molde para la transcripción de un gen o ADNc, se la puede aludir como codificante de la proteína u otro producto de ese gen o ADNc.
A menos que se especifique lo contrario, una "secuencia de nucleótidos que codifica una secuencia de aminoácidos" incluye todas las secuencias de nucleótidos que son versiones degeneradas entre sí y que codifican la misma secuencia de aminoácidos. La expresión secuencia de nucleótidos que codifica una proteína o un ARN también puede incluir intrones en la medida en que la secuencia de nucleótidos que codifica la proteína puede contener en alguna versión un intrón o intrones.
La expresión "cantidad eficaz" o "cantidad terapéuticamente eficaz" se utilizan indistintamente en el presente memoria y se refieren a una cantidad de un compuesto, formulación, material o composición, como se describe en esta memoria, eficaz para conseguir un resultado biológico particular.
El término "endógeno" se refiere a cualquier material de un organismo, célula, tejido o sistema o producido dentro de estos.
El término "exógeno" se refiere a cualquier material introducido desde fuera de un organismo, célula, tejido o sistema o producido fuera de estos.
El término "expresión" se refiere a la transcripción y/o traducción de una secuencia de nucleótidos particular impulsada por un promotor.
La expresión "vector de transferencia" se refiere a una composición de materia que comprende un ácido nucleico aislado y que se puede utilizar para suministrar el ácido nucleico aislado al interior de una célula. En la técnica se conocen numerosos vectores que incluyen, pero no se limitan a polinucleótidos lineales, polinucleótidos asociados con compuestos iónicos o anfifílicos, plásmidos y virus. Por tanto, la expresión "vector de transferencia" incluye un plásmido o un virus que se replica de forma autónoma. La expresión también debe interpretarse para incluir además compuestos no plasmídicos y no virales que facilitan la transferencia de ácido nucleico a las células, tales como, por ejemplo, un compuesto de polilisina, liposoma y similares. Ejemplos de vectores de transferencia viral incluyen, pero no se limitan a vectores adenovirales, vectores de virus adenoasociados, vectores retrovirales, vectores lentivirales y similares.
La expresión "vector de expresión" se refiere a un vector que comprende un polinucleótido recombinante que comprende secuencias de control de la expresión unidas operativamente a una secuencia de nucleótidos que se va a expresar. Un vector de expresión comprende suficientes elementos que actúan en cis para la expresión; otros elementos para la expresión pueden ser suministrados por la célula hospedadora o en un sistema de expresión in vitro. Los vectores de expresión incluyen todos los conocidos en la técnica, incluidos cósmidos, plásmidos (p. ej., desnudos o contenidos en liposomas) y virus (p. ej., lentivirus, retrovirus, adenovirus y virus adenoasociados) que incorporan el polinucleótido recombinante.
El término "lentivirus" se refiere a un género de la familia Retroviridae. Los lentivirus son únicos entre los retrovirus al ser capaces de infectar células que no se dividen; pueden suministrar una cantidad significativa de información genética en el ADN de la célula hospedadora, por lo que son uno de los métodos más eficientes de un vector de suministro de genes. El VIH, el VIS y el VIF son todos ejemplos de lentivirus.
La expresión "vector lentivírico" se refiere a un vector procedente de al menos una porción de un genoma de lentivirus, que incluye especialmente un vector lentivírico autoinactivante como se proporciona en Milone et al., Mol. Ther. 17(8): 1453-1464 (2009). Otros ejemplos de vectores de lentivirus que pueden utilizarse en la clínica incluyen, pero sin limitación, p. ej., la tecnología de suministro de genes LENTIVECTOR® de Oxford BioMedica, el sistema de vectores LENTIMAX™ de Lentigen y similares. También están disponibles tipos no clínicos de vectores lentivíricos y serán conocidos por un experto en la materia.
El término "homólogo" o "identidad" se refiere a la identidad de la secuencia subunitaria entre dos moléculas poliméricas, p. ej., entre dos moléculas de ácido nucleico, tales como dos moléculas de ADN o dos moléculas de ARN, o entre dos moléculas polipeptídicas. Cuando una posición subunitaria en ambas moléculas está ocupada por la misma subunidad monomérica; p. ej., si una posición en cada una de las dos moléculas de ADN está ocupada por adenina, entonces son homólogas o idénticas en esa posición. La homología entre dos secuencias es una función directa del número de posiciones coincidentes u homólogas; p. ej., si la mitad (p. ej., cinco posiciones en un polímero de diez subunidades de longitud) de las posiciones en dos secuencias son homólogas, las dos secuencias son 50 % homólogas; si el 90 % de las posiciones (p. ej., 9 de 10) coinciden o son homólogas, las dos secuencias son 90 % homólogas.
Las formas "humanizadas" de anticuerpos no humanos (p. ej., murinos) son inmunoglobulinas quiméricas, cadenas de inmunoglobulinas o fragmentos de las mismas (tales como Fv, Fab, Fab', F(ab')2 u otras subsecuencias de unión a antígenos de anticuerpos) que contienen secuencia mínima derivada de inmunoglobulina no humana. En su mayor parte, los anticuerpos humanizados y fragmentos de anticuerpos de los mismos son inmunoglobulinas humanas (anticuerpo o fragmento de anticuerpo receptor) en las que los residuos de una región determinante de complementariedad (CDR) del receptor son reemplazados por residuos de una CDR de una especie no humana (anticuerpo donante), tal como ratón, rata o conejo que tienen la especificidad, afinidad y capacidad deseadas. En algunos casos, los residuos de la región
marco conservada (FR) de Fv de la inmunoglobulina humana son reemplazados por residuos no humanos correspondientes. Además, un anticuerpo/fragmento de anticuerpo humanizado puede comprender residuos que no se encuentran ni en el anticuerpo receptor ni en la CDR importada o en las secuencias marco conservadas. Estas modificaciones pueden refinar y optimizar adicionalmente el rendimiento del anticuerpo o fragmento de anticuerpo. En general, el anticuerpo humanizado o fragmento de anticuerpo del mismo comprenderá sustancialmente todos de al menos uno, y normalmente dos dominios variables, en los que todas o sustancialmente todas las regiones CDR corresponden a las de una inmunoglobulina no humana y todas o una parte significativa de las regiones FR son las de una secuencia de inmunoglobulina humana. El anticuerpo o fragmento de anticuerpo humanizado también puede comprender al menos una parte de una región constante de inmunoglobulina (Fc), normalmente la de una inmunoglobulina humana. Para más detalles, véase Jones et al., Nature, 321: 522-525, 1986; Reichmann et al., Nature, 332: 323-329, 1988; Presta, Curr. Op. Struct. Biol., 2: 593-596, 1992.
"Totalmente humana" se refiere a una inmunoglobulina, tal como un anticuerpo o fragmento de anticuerpo, en donde la molécula completa es de origen humano o consiste en una secuencia de aminoácidos idéntica a una forma humana del anticuerpo o inmunoglobulina.
El término "aislado" significa alterado o eliminado del estado natural. Por ejemplo, un ácido nucleico o un péptido presente de forma natural en un animal vivo no está "aislado", pero el mismo ácido nucleico o péptido separado parcial o completamente de los materiales coexistentes de su estado natural está "aislado". Un ácido nucleico o una proteína aislada puede existir en forma sustancialmente purificada, o puede existir en un entorno no nativo tal como, p. ej., una célula hospedadora.
En el contexto de la presente invención, se utilizan las siguientes abreviaturas para las bases de ácido nucleico que se encuentran habitualmente. "A" se refiere a adenosina, "C" se refiere a citosina, "G" se refiere a guanosina, "T" se refiere a timidina y "U" se refiere a uridina.
La expresión "unido operativamente" o "control transcripcional" se refiere al enlace funcional entre una secuencia reguladora y una secuencia de ácido nucleico heteróloga que da lugar a la expresión de esta última. Por ejemplo, una primera secuencia de ácido nucleico está unida operativamente con una segunda secuencia de ácido nucleico cuando la primera secuencia de ácido nucleico se coloca en una relación funcional con la segunda secuencia de ácido nucleico. Por ejemplo, un promotor está unido operativamente a una secuencia codificante si el promotor afecta a la transcripción o expresión de la secuencia codificante. Las secuencias de ADN unidas operativamente pueden ser contiguas entre sí y, p. ej., cuando es necesario unir dos regiones codificantes de proteínas, están en el mismo marco de lectura.
La expresión administración "parenteral" de una composición inmunogénica incluye, p. ej., técnicas de inyección subcutánea (s.c.), intravenosa (i.v.), intramuscular (i.m.) o intraesternal, intra-tumoral o por infusión.
La expresión "ácido nucleico" o "polinucleótido" se refiere a ácidos desoxirribonucleicos (ADN) o ácidos ribonucleicos (ARN) y polímeros de los mismos en forma de cadena sencilla o doble. A menos que se limite específicamente, la expresión abarca ácidos nucleicos que contienen análogos conocidos de nucleótidos naturales que tienen propiedades de unión similares a las del ácido nucleico de referencia y se metabolizan de una manera similar a los nucleótidos que se producen de forma natural. A menos que se indique lo contrario, una secuencia particular de ácidos nucleicos también abarca implícitamente variantes modificadas de forma conservativa de la misma (p. ej., sustituciones de codones degenerados), alelos, ortólogos, SNPs y secuencias complementarias, así como la secuencia indicada explícitamente. Específicamente, las sustituciones de codones degenerados pueden lograrse generando secuencias en las que la tercera posición de uno o más codones seleccionados (o todos) se sustituye con residuos de base mixta y/o desoxiinosina (Batzer et al., Nucleic Acid Res. 19:5081 (1991); Ohtsuka et al., J. Biol. Chem. 260:2605-2608 (1985); y Rossolini et al., Mol. Cell. Probes 8:91-98 (1994)).
Los términos "péptido", "polipéptido" y "proteína" se utilizan indistintamente y se refieren a un compuesto compuesto por residuos de aminoácidos enlazados covalentemente por enlaces peptídicos. Una proteína o péptido debe contener al menos dos aminoácidos, y no se establece limitación alguna en el número máximo de aminoácidos que puede comprender una secuencia de proteína o péptido. Los polipéptidos incluyen cualquier péptido o proteína que comprenda dos o más aminoácidos unidos entre sí por enlaces peptídicos. Tal como se utiliza en esta memoria, el término se refiere tanto a cadenas cortas, que también se denominan comúnmente en la técnica como péptidos, oligopéptidos y oligómeros, por ejemplo, como a cadenas más largas, a las que generalmente se alude en la técnica como proteínas, de las cuales existen muchos tipos. Los "polipéptidos" incluyen, p. ej., fragmentos biológicamente activos, polipéptidos sustancialmente homólogos, oligopéptidos, homodímeros, heterodímeros, variantes de polipéptidos, polipéptidos modificados, derivados, análogos, proteínas de fusión, entre otros. Un polipéptido incluye un péptido natural, un péptido recombinante o una combinación de los mismos.
El término "promotor" se refiere a una secuencia de ADN reconocida por la maquinaria sintética de la célula, o maquinaria sintética introducida, necesaria para iniciar la transcripción específica de una secuencia polinucleotídica.
La expresión "secuencia promotora/reguladora" se refiere a una secuencia de ácido nucleico que es necesaria para la expresión de un producto génico unido operativamente a la secuencia promotora/reguladora. En algunos casos, esta secuencia puede ser la secuencia promotora central y, en otros casos, esta secuencia también puede incluir una secuencia potenciadora y otros elementos reguladores que son necesarios para la expresión del producto génico. La secuencia promotora/reguladora puede ser, por ejemplo, una que exprese el producto génico de una manera específica para el tejido.
La expresión promotor "constitutivo" se refiere a una secuencia de nucleótidos que, cuando se une operativamente con un polinucleótido que codifica o especifica un producto génico, hace que el producto génico se produzca en una célula en la mayoría o en todas las condiciones fisiológicas de la célula.
La expresión promotor "inducible" se refiere a una secuencia de nucleótidos que, cuando se une operativamente con un polinucleótido que codifica o especifica un producto génico, hace que el producto génico se produzca en una célula sustancialmente solo cuando un inductor que corresponde al promotor está presente en la célula.
La expresión promotor "específico para el tejido" se refiere a una secuencia de nucleótidos que, cuando se une operativamente con un polinucleótido codificado o especificado por un gen, hace que el producto génico se produzca en una célula sustancialmente solo si la célula es una célula del tipo de tejido correspondiente al promotor.
Las expresiones "antígeno asociado al cáncer" o "antígeno tumoral" se refieren indistintamente a una molécula (normalmente una proteína, carbohidrato o lípido) que se expresa en la superficie de una célula cancerosa, ya sea en su totalidad o como un fragmento (p. ej., MHC/péptido) y que es útil para el direccionamiento preferente de un agente farmacológico a la célula cancerosa. En algunas realizaciones, un antígeno tumoral es un marcador expresado tanto por células normales como por células cancerosas, p. ej., un marcador de linaje, p. ej., CD19 en células B. En algunas realizaciones, un antígeno tumoral es una molécula de la superficie celular que se sobre-expresa en una célula cancerosa en comparación con una célula normal, por ejemplo, sobre-expresión 1 vez, sobre-expresión 2 veces, sobreexpresión 3 veces o más en comparación con una célula normal. En algunas realizaciones, un antígeno tumoral es una molécula de la superficie celular que se sintetiza de manera inapropiada en la célula cancerosa, por ejemplo, una molécula que contiene deleciones, adiciones o mutaciones en comparación con la molécula expresada en una célula normal. En algunas realizaciones, un antígeno tumoral se expresará exclusivamente en la superficie celular de una célula cancerosa, en su totalidad o como un fragmento (p. ej., MHC/péptido), y no se sintetizará ni expresará en la superficie de una célula normal. En algunas realizaciones, los CARs de la presente divulgación incluyen CARs que comprenden un dominio de unión a antígenos (p. ej., anticuerpo o fragmento de anticuerpo) que se une a un péptido presentado por MHC. Normalmente, los péptidos derivados de proteínas endógenas llenan los bolsillos de las moléculas de clase I del complejo mayor de histocompatibilidad (MHC) y son reconocidos por los receptores de células T (TCR) en las células T CD8+. Los complejos del MHC de clase I son expresados de manera constitutiva por todas las células nucleadas. En el cáncer, los complejos péptido/MHC específicos del virus y/o específicos del tumor representan una clase única de dianas de la superficie celular para la inmunoterapia. Se han descrito anticuerpos similares a TCR que tienen como diana péptidos derivados de antígenos víricos o tumorales en el contexto del antígeno leucocitario humano (HLA)-A1 o HLA-A2 (véanse, p. ej., Sastry et al., J Virol. 2011 85(5): 1935-1942; Sergeeva et al., Blood, 2011 117(16):4262-4272; Verma et al., J Immunol 2010 184(4):2156-2165; Willemsen et al., Gene Ther 2001 8(21) 1601-1608; Dao et al., Sci Transl Med 2013 5(176) :176ra33 ; Tassev et al., Cancer Gene Ther 2012 19(2):84-100). Por ejemplo, el anticuerpo similar a TCR se puede identificar a partir de la selección de una colección tal como una colección presentada en fagos scFv humanos.
La expresión "antígeno de refuerzo de tumor" o "antígeno de refuerzo de cáncer" se refieren indistintamente a una molécula (normalmente una proteína, carbohidrato o lípido) que se expresa en la superficie de una célula que, de por sí, no es cancerosa, pero refuerza las células cancerosas, p. ej., fomentando su crecimiento o supervivencia, p. ej., resistencia a células inmunitarias. Células ilustrativas de este tipo incluyen células estromales y células supresoras de origen mieloide (MDSC, por sus siglas en inglés). No es necesario que el propio antígeno de refuerzo de tumor desempeñe una función en el refuerzo de las células tumorales siempre y cuando el antígeno esté presente en una célula que refuerce células cancerosas.
La expresión "enlazador polipeptídico flexible" o "enlazador", tal como se utiliza en el contexto de un scFv, se refiere a un enlazador peptídico constituido por aminoácidos tales como residuos de glicina y/o serina utilizados solos o combinados, para conectar regiones de la cadena pesada variables y de la cadena ligera variables. En una realización, el enlazador polipeptídico flexible es un enlazador Gly/Ser y comprende la secuencia de aminoácidos (Gly-Gly-Gly-Ser)n, donde n es un número entero positivo igual a o mayor que 1. Por ejemplo, n=1, n=2, n=3. n=4, n=5 y n=6, n=7, n=8, n=9 y n=10 (SEQ ID NO: 28). En una realización, los enlazadores polipeptídicos flexibles incluyen, pero sin limitación, (Gly4 Ser)4 (SEQ ID NO: 29) o (Gly4 Ser)3 (SEQ ID NO: 30). En otra realización, los enlazadores incluyen múltiples repeticiones de (Gly2Ser), (GlySer) o (GiyaSer) (SEQ ID NO: 31). También se incluyen dentro del alcance de la invención los enlazadores descritos en el documento WO2012/138475.
Tal como se utiliza en esta memoria, un casquete 5' (también denominado casquete de ARN, casquete de ARN 7-metilguanosina o casquete de ARN m7G) es un nucleótido de guanina modificado que se ha añadido a la parte "frontal" o extremo 5' de un ARN mensajero de un eucariota poco después del inicio de la transcripción. El casquete 5' consiste en
un grupo terminal que está ligado al primer nucleótido transcrito. Su presencia es crucial para el reconocimiento por parte del ribosoma y la protección contra las RNasas. La adición del casquete está acoplada a la transcripción y ocurre cotrancripcionalmente, de modo que cada uno influye en el otro. Poco después del comienzo de la transcripción, se une al extremo 5' del ARNm que se está sintetizando un complejo de síntesis del casquete asociado con la ARN polimerasa. Este complejo enzimático cataliza las reacciones químicas que son necesarias para la generación del casquete del ARNm. La síntesis prosigue como una reacción bioquímica de múltiples etapas. El resto de adición de la caperuza se puede modificar para modular la funcionalidad del ARNm tal como su estabilidad o eficiencia de traducción.
Como se utiliza en el presente memoria, el "ARN transcrito in vitro" se refiere a ARN, preferentemente ARNm, que se ha sintetizado in vitro. Generalmente, el ARN transcrito in vitro se genera a partir de un vector de transcripción in vitro. El vector de transcripción in vitro comprende un molde que se utiliza para generar el ARN transcrito in vitro.
Tal como se utiliza en esta memoria, un "poli(A)" es una serie de adenosinas unidas por poliadenilación al ARNm. En la realización preferida de una construcción para la expresión transitoria, el poliA está entre 50 y 5000 (SEQ ID NO: 34), preferentemente más de 64, más preferentemente más de 100, de la manera más preferente más de 300 o 400. Las secuencias de poli(A) se pueden modificar por medios químicos o enzimáticos para modular la funcionalidad del ARNm, tal como la ubicación, estabilidad o eficacia de la traducción.
Tal como se utiliza en esta memoria, el término "poliadenilación" se refiere al enlace covalente de un resto de poliadenililo, o su variante modificada, a una molécula de ARN mensajero. En los organismos eucariotas, la mayoría de las moléculas de ARN mensajero (ARNm) están poliadeniladas en el extremo 3'. La cola de poli(A) 3' es una secuencia larga de nucleótidos de adenina (a menudo varios cientos) añadidos al pre-ARNm a través de la acción de una enzima, la poliadenilato polimerasa. En los eucariotas superiores, la cola poli(A) se añade a las transcripciones que contienen una secuencia específica, la señal de poliadenilación. La cola de poli(A) y la proteína unida a ella ayudan a proteger el ARNm de la degradación por parte de exonucleasas. La poliadenilación también es importante para la terminación de la transcripción, la exportación del ARNm desde el núcleo y la traducción. La poliadenilación se produce en el núcleo inmediatamente después de la transcripción del ADN en a Rn , pero adicionalmente también se puede producir más tarde en el citoplasma. Una vez terminada la transcripción, la cadena de ARNm se escinde mediante la acción de un complejo de endonucleasa asociado con la ARN polimerasa. El sitio de escisión se caracteriza habitualmente por la presencia de la secuencia de bases AAUAAA cerca del sitio de escisión. Una vez que se ha escindido el ARNm, se añaden residuos de adenosina al extremo 3' libre en el sitio de escisión.
Tal como se utiliza en esta memoria, "transitoria" se refiere a la expresión de un transgén no integrado durante un periodo de horas, días o semanas, en donde el periodo de tiempo de expresión es menor que el periodo de tiempo para la expresión del gen si está integrado en el genoma o está contenido dentro de un replicón plasmídico estable en la célula huésped.
Tal como se utiliza en esta memoria, los términos "tratar", "tratamiento" y "tratando" se refieren a la reducción o mejora de la progresión, gravedad y/o duración de un trastorno proliferativo, o la mejora de uno o más síntomas (preferiblemente, uno o más síntomas discernibles) de un trastorno proliferativo resultante de la administración de una o más terapias (p. ej., uno o más agentes terapéuticos tales como un CAR de la divulgación). En realizaciones específicas, los términos "tratar", "tratamiento" y "tratando" se refieren a la mejora de al menos un parámetro físico mensurable de un trastorno proliferativo tal como el crecimiento de un tumor, no necesariamente perceptible por el paciente. En otras realizaciones los términos "tratar", "tratamiento" o "tratando" se refieren a la inhibición de la evolución de un trastorno proliferativo, ya sea de manera física, p. ej., mediante estabilización de un síntoma perceptible, fisiológica, p. ej., mediante estabilización de un parámetro físico, o ambas. En otras realizaciones, las expresiones "tratar", "tratamiento" y "tratando" se refieren a la reducción o estabilización del tamaño tumoral o recuento de células cancerosas.
La expresión "ruta de transducción de señales" se refiere a la relación bioquímica entre una diversidad de moléculas de transducción de señales que desempeñan una función en la transmisión de una señal de una parte de una célula a otra parte de una célula. La expresión "receptor de la superficie celular" incluye moléculas y complejos de moléculas capaces de recibir una señal y transmitir la señal a través de la membrana de una célula.
Se entiende que el término "sujeto" incluye organismos vivos en los que se puede inducir una respuesta inmunitaria (p. ej., mamíferos, seres humanos).
La expresión una célula "sustancialmente purificada" se refiere a una célula que está esencialmente exenta de otros tipos celulares. Una célula sustancialmente purificada también se refiere a una célula que se ha separado de otros tipos celulares con los que normalmente está asociada en su estado natural. En algunos casos, una población de células sustancialmente purificadas se refiere a una población homogénea de células. En otros casos, este término se refiere simplemente a células que han sido separadas de las células con las que están naturalmente asociadas en su estado natural. En algunos aspectos, las células se cultivan in vitro. En otros aspectos, las células no se cultivan in vitro.
El término "terapéutico", como se utiliza en esta memoria, significa un tratamiento. Se obtiene un efecto terapéutico mediante la reducción, supresión, remisión o erradicación de una patología.
El término "profilaxis", tal como se utiliza en esta memoria, significa la prevención o el tratamiento protector de una enfermedad o patología.
En el contexto de la presente invención, "antígeno tumoral" o "antígeno de un trastorno hiperproliferativo" o "antígeno asociado con un trastorno hiperproliferativo" se refiere a antígenos que son comunes a trastornos hiperproliferativos específicos. En ciertos aspectos, los antígenos del trastorno hiperproliferativo de la presente divulgación proceden de cánceres que incluyen, pero sin limitación, melanoma primario o metastásico, timoma, linfoma, sarcoma, cáncer de pulmón, cáncer de hígado, linfoma no hodgkiniano, linfoma de Hodgkin, leucemias, cáncer uterino, cáncer de cuello uterino, cáncer de vejiga, cáncer de riñón y adenocarcinomas tales como cáncer de mama, cáncer de próstata, cáncer de ovario, cáncer de páncreas, y similares.
El término "transfectado" o "transformado" o "transducido" se refiere a un proceso por el cual el ácido nucleico exógeno se transfiere o introduce en la célula hospedadora. Una célula "transfectada" o "transformada" o "transducida" es una que se ha transfectado, transformado o transducido con ácido nucleico exógeno. La célula incluye la célula objeto primaria y su progenie.
La expresión "se une específicamente" se refiere a un anticuerpo, o un ligando, que reconoce y se une a una proteína compañera de unión (p. ej., un antígeno tumoral) presente en una muestra, pero cuyo anticuerpo o ligando no reconoce sustancialmente ni se une a otras moléculas en la muestra.
El "receptor quimérico para antígenos regulable (RCAR)", como se usa esa expresión en esta memoria, se refiere a un conjunto de polipéptidos, normalmente dos en las realizaciones más sencillas, que cuando están en una célula RCARX, proporcionan a la célula RCARX especificidad para una célula diana, normalmente una célula cancerosa, y con generación o proliferación de señales intracelulares regulables, que pueden optimizar una propiedad efectora inmunitaria de la célula RCARX. Una célula RCARX se basa, al menos en parte, en un dominio de unión a antígenos para proporcionar especificidad para una célula diana que comprende el antígeno unido por el dominio de unión a antígenos. En una realización, un RCAR incluye un interruptor de dimerización que, en presencia de una molécula de dimerización, puede acoplar un dominio de señalización intracelular al dominio de unión al antígeno.
"Anclaje de membrana" o "dominio de anclaje de membrana", como se utiliza esta expresión en esta memoria, se refiere a un polipéptido o resto, p. ej., un grupo miristoílo, suficiente para anclar un dominio extracelular o intracelular a la membrana plasmática.
El "dominio de conmutación", como se utiliza esta expresión en esta memoria, p. ej., cuando se hace referencia a un RCAR, se refiere a una entidad, normalmente una entidad basada en polipéptidos, que, en presencia de una molécula de dimerización, se asocia con otro dominio de conmutación. La asociación da lugar a un acoplamiento funcional de una primera entidad ligada, p. ej., fusionada a, un primer dominio de conmutación, y una segunda entidad ligada a, p. ej., fusionada a, un segundo dominio de conmutación. A un primer y un segundo dominio de conmutación se les alude colectivamente como conmutador de la dimerización. En algunas realizaciones, los dominios de conmutación primero y segundo son iguales entre sí, p. ej., son polipéptidos que tienen la misma secuencia de aminoácidos primaria, y se les alude colectivamente como conmutador de la homodimerización. En algunas realizaciones, los dominios de conmutación primero y segundo son diferentes entre sí, p. ej., son polipéptidos que tienen diferentes secuencias de aminoácidos primarios, y se les alude colectivamente como conmutador de la heterodimerización. En algunas realizaciones, el conmutador es intracelular. En algunas realizaciones, el conmutador es extracelular. En algunas realizaciones, el dominio de conmutación es una entidad basada en polipéptidos, p. ej., basada en FKBP o FRB, y la molécula de dimerización es una molécula pequeña, p. ej., un rapálogo. En algunas realizaciones, el dominio de conmutación es una entidad basada en polipéptidos, p. ej., un scFv que se une a un péptido myc, y la molécula de dimerización es un polipéptido, un fragmento del mismo o un multímero de un polipéptido, p. ej., un ligando myc o multímeros de un ligando myc que se unen a uno o más scFv myc. En algunas realizaciones, el dominio de conmutación es una entidad basada en polipéptidos, p. ej., receptor myc, y la molécula de dimerización es un anticuerpo o fragmentos del mismo, p. ej., anticuerpo myc.
"Molécula de dimerización", tal se utiliza esta expresión en esta memoria, p. ej., cuando se alude a un RCAR, se refiere a una molécula que fomenta la asociación de un primer dominio de conmutación con un segundo dominio de conmutación. En algunas realizaciones, la molécula de dimerización no se produce de forma natural en el sujeto, o no se produce en concentraciones que darían lugar a una dimerización significativa. En realizaciones, la molécula de dimerización es una molécula pequeña, p. ej., rapamicina o un rapálogo, por ejemplo, RAD001.
El término "bioequivalente" se refiere una cantidad de un agente que no es el compuesto de referencia (por ejemplo, RAD001), necesaria para producir un efecto equivalente al efecto producido por la dosis de referencia o cantidad de referencia del compuesto de referencia (por ejemplo, RAD001). En una realización, el efecto es el nivel de inhibición de mTOR, p. ej., medido por la inhibición de la quinasa P70 S6, p. ej., evaluado en un ensayo in vivo o in vitro, p. ej., medido por un ensayo descrito en esta memoria, p. ej., el ensayo de Boulay. En una realización, el efecto es la alteración de la relación de células T PD-1 positivas/PD-1 negativas, medidos por clasificación celular. En una realización, una cantidad o dosis bioequivalente de un inhibidor de mTOR es la cantidad o dosis que alcanza el mismo nivel de inhibición de la
quinasa P70 S6 que la dosis de referencia o la cantidad de referencia de un compuesto de referencia. En una realización, una cantidad o dosis bioequivalente de un inhibidor de mTOR es la cantidad o dosis que logra el mismo nivel de alteración en la relación de célula T PD-1 positivos/PD-1 negativos que la dosis de referencia o la cantidad de referencia de un compuesto de referencia.
La expresión "dosis baja, de potenciación inmunitaria", cuando se utiliza junto con un inhibidor de mTOR, p. ej., un inhibidor de mTOR alostérico, p. ej., RAD001 o rapamicina, o un inhibidor de mTOR catalítico, se refiere a una dosis de inhibidor de mTOR que inhibe parcialmente, pero no completamente, la actividad de mTOR, p. ej., medida por la inhibición de la actividad quinasa P70 S6. Los métodos para evaluar la actividad de mTOR, p. ej., mediante la inhibición de la quinasa P70 S6, se analizan en esta memoria. La dosis es insuficiente para dar como resultado una inmunosupresión completa, pero es suficiente para potenciar la respuesta inmunitaria. En una realización, la dosis baja, de potenciación inmunitaria, de inhibidor de mTOR da como resultado una disminución en el número de célula T PD-1 positivos y/o un aumento en el número de célula T PD-1 negativos, o un aumento en la relación de células T PD-1 negativas/células T PD-1 positivas. En una realización, la dosis baja, de potenciación inmunitaria, de inhibidor de mTOR da como resultado un aumento en el número de células T vírgenes. En una realización, la dosis baja, de potenciación inmunitaria, de inhibidor de mTOR da como resultado uno o más de lo siguiente:
un aumento en la expresión de uno o más de los siguientes marcadores: CD62Lalto, CD127alto, CD27+ y BCL2, p. ej., en células T de la memoria, p. ej., precursores de células T de la memoria;
una disminución en la expresión de KLRG1, p. ej., en células T de la memoria, p. ej., precursores de células T de la memoria; y
un aumento en el número de precursores de células T de la memoria, p. ej., células con una cualquiera o una combinación de las siguientes características: CD62Lalto incrementado, CD127alto incrementado, CD27+ incrementado, KLRG1 reducido y BCL2 incrementado;
en donde cualquiera de los cambios descritos anteriormente se produce, p. ej., al menos de manera transitoria, p. ej., en comparación con un sujeto no tratado.
"Refractario", tal como se utiliza en esta memoria, se refiere a una enfermedad, p. ej., cáncer, que no responde a un tratamiento. En algunas realizaciones, un cáncer refractario puede ser resistente a un tratamiento antes o al comienzo del tratamiento. En otras realizaciones, el cáncer refractario se puede volver resistente durante un tratamiento. Un cáncer refractario también se denomina cáncer resistente.
"Recaída" tal como se utiliza en esta memoria se refiere al regreso de una enfermedad (p. ej., cáncer) o los signos y síntomas de una enfermedad tal como el cáncer después de un periodo de mejora, p. ej., después de un tratamiento previo de una terapia, p. ej., terapia contra el cáncer.
Intervalos: a lo largo de esta divulgación, se pueden presentar en un formato de intervalo diversos aspectos de la invención. Se debe entender que la descripción en formato de intervalo es meramente por conveniencia y brevedad y no se debe interpretar como una limitación inflexible en el alcance de la invención. Por consiguiente, se debe considerar que la descripción de un intervalo divulga específicamente todos los posibles subintervalos, así como también los valores numéricos individuales dentro de ese intervalo. Por ejemplo, se debe considerar que la descripción de un intervalo tal como de 1 a 6 divulga específicamente subintervalos tales como de 1 a 3, de 1 a 4, de 1 a 5, de 2 a 4, de 2 a 6, de 3 a 6, etc., así como también números individuales dentro de ese intervalo, por ejemplo, 1, 2, 2,7, 3, 4, 5, 5,3 y 6. Como otro ejemplo, un intervalo tal como un 95-99 % de identidad, incluye algo con una identidad de un 95 %, 96 %, 97 %, 98 % o 99 %, e incluye subintervalos tales como una identidad de un 96-99 %, 96-98 %, 96-97 %, 97-99 %, 97-98 % y 98-99 %. Esto se aplica independientemente de la amplitud del intervalo.
Descripción
En esta memoria se describen composiciones de materia y métodos de uso para el tratamiento de una enfermedad tal como el cáncer utilizando células efectoras inmunitarias (p. ej., células T, células NK) diseñadas con CARs de la divulgación.
En un aspecto, la divulgación proporciona un cierto número de receptores de antígenos quiméricos (CAR) que comprenden un dominio de unión a antígenos (p. ej., anticuerpo o fragmento de anticuerpo, TCR o fragmento de TCR) diseñado para la unión específica a un antígeno tumoral, p. ej., un antígeno tumoral descrito en esta memoria. En un aspecto, la divulgación proporciona una célula efectora inmunitaria (p. ej., célula T, célula NK) diseñada para expresar un c A r , en donde la célula efectora inmunitaria diseñada exhibe una propiedad anticancerígena. La presente invención proporciona una célula efectora inmunitaria que comprende un primer c A r y un segundo CAR, o que comprende ácidos nucleicos que codifican los mismos y el uso terapéutico de una célula de este tipo, tal como se define en las reivindicaciones adjuntas. En un aspecto, una célula se transforma con el CAR y el CAR se expresa en la superficie celular. En algunas realizaciones, la célula (p. ej., célula T, célula NK) se transduce con un vector vírico que codifica un CAR. En
algunas realizaciones, el vector vírico es un vector retrovírico. En algunas realizaciones, el vector vírico es un vector lentivírico. En algunas realizaciones de este tipo, la célula puede expresar de forma estable el CAR. En otra realización, la célula (p. ej., célula T, célula NK) se transfecta con un ácido nucleico, p. ej., ARNm, ADNc, ADN, que codifica un CAR. En algunas realizaciones de este tipo, la célula puede expresar de forma transitoria el CAR.
En un aspecto, el dominio de unión a antígenos de un CAR descrito en esta memoria es un fragmento de anticuerpo scFv. En un aspecto, fragmentos de anticuerpos de este tipo son funcionales, debido a que conservan la afinidad de unión equivalente, p. ej., se unen al mismo antígeno con una afinidad comparable a la del anticuerpo IgG del que proceden. En otras realizaciones, el fragmento de anticuerpo tiene una afinidad de unión más baja, p. ej., se une al mismo antígeno con una afinidad de unión más baja que el anticuerpo del que procede, pero es funcional, porque proporciona una respuesta biológica descrita en esta memoria. En una realización, la molécula de CAR comprende un fragmento de anticuerpo que tiene una KD de afinidad de unión de 10-4 M a 10-8 M, p. ej., 10-5 M a 10-7 M, p. ej., 10-6 M o 10-7 M, para el antígeno diana. En una realización, el fragmento de anticuerpo tiene una afinidad de unión que es al menos cinco veces, 10 veces, 20 veces, 30 veces, 50 veces, 100 veces o 1000 veces menor que un anticuerpo de referencia, p. ej., un anticuerpo descrito en esta memoria.
En un aspecto, dichos fragmentos de anticuerpos son funcionales porque proporcionan una respuesta biológica que puede incluir, pero sin limitación, la activación de una respuesta inmunitaria, la inhibición del origen de la transducción de señales a partir de su antígeno diana, la inhibición de la actividad quinasa y similares, como entenderá un experto en la materia.
En un aspecto, el dominio de unión a antígenos del CAR es un fragmento de anticuerpo scFv que se humaniza en comparación con la secuencia murina del scFv del que procede.
En un aspecto, el dominio de unión a antígenos de un CAR utilizado en la invención (p. ej., un scFv) es codificado por una molécula de ácido nucleico cuya secuencia se ha optimizado por codón para la expresión en una célula de mamífero. En un aspecto, la construcción de CAR completa utilizada en la invención está codificada por una molécula de ácido nucleico cuya secuencia completa se ha optimizado por codón para la expresión en una célula de mamífero. La optimización de codones se refiere al descubrimiento de que la frecuencia de aparición de codones sinónimos (es decir, codones que codifican el mismo aminoácido) en la codificación del ADN está sesgada en diferentes especies. . Una degeneración de codones de este tipo permite que un polipéptido idéntico sea codificado por una diversidad de secuencias de nucleótidos. Se conoce en la técnica una diversidad de métodos de optimización de codones, e incluyen, p. ej., métodos desvelados en al menos las patentes de los EE.UU. números 5.786.464 y 6.114.148.
En un aspecto, los CARs utilizados en la invención combinan un dominio de unión a antígenos de un anticuerpo específico con una molécula de señalización intracelular. Por ejemplo, en algunos aspectos, la molécula de señalización intracelular incluye, pero no se limita a módulos de señalización de cadena CD3-zeta, 4-1BB y CD28 y combinaciones de los mismos. En un aspecto, el dominio de unión a antígenos se une a un antígeno tumoral como se describe en el presente memoria.
Además, la presente invención proporciona células que expresan CAR y su uso en medicamentos o métodos para tratar, entre otras enfermedades, cáncer o cualquier tumor maligno o enfermedad autoinmune que involucre células o tejidos que expresan un antígeno tumoral como se describe en esta memoria.
En un aspecto, el CAR de la divulgación puede utilizarse para erradicar una célula normal que expresa un antígeno tumoral como se describe en esta memoria, por lo que es aplicable para su uso como terapia de acondicionamiento celular antes del trasplante de células. En un aspecto, la célula normal que expresa un antígeno tumoral como se describe en esta memoria es una célula madre normal y el trasplante de células es un trasplante de células madre.
En un aspecto, la divulgación proporciona una célula efectora inmunitaria (p. ej., célula T, célula NK) diseñada para expresar un receptor de antígeno quimérico (CAR), en donde la célula efectora inmunitaria diseñada exhibe una propiedad antitumoral. Un antígeno preferido es un antígeno asociado al cáncer (es decir, antígeno tumoral) descrito en esta memoria. En un aspecto, el dominio de unión a antígenos del CAR comprende un fragmento de anticuerpo parcialmente humanizado. En un aspecto, el dominio de unión a antígenos del CAR comprende un scFv parcialmente humanizado. Por consiguiente, la descripción proporciona CARs que comprenden un dominio de unión a antígenos humanizado y se modifican por ingeniería genética en una célula, p. ej., una célula T o una célula NK, y métodos para su uso para terapia adoptiva.
En un aspecto, los CAR utilizados en la invención comprenden al menos un dominio intracelular seleccionado del grupo de un dominio de señalización CD137 (4-1BB), un dominio de señalización CD28, un dominio de señalización CD27, un dominio de señalización CD3zeta y cualquier combinación de los mismos. En un aspecto, los CARs utilizados en la invención comprenden al menos un dominio de señalización intracelular procedente de una o más moléculas coestimuladoras distintas de CD137 (4-1BB) o CD28.
Secuencias de algunos ejemplos de diversos componentes de CARs utilizados en la presente invención se enumeran en la Tabla 1, en que aa representa aminoácidos y na representa ácidos nucleicos que codifican el péptido correspondiente.
Tabla 1. Secuencias de diversos componentes de CAR (aa - aminoácidos, na - ácidos nucleicos que codifican la proteína correspondiente














Antígenos asociados al cáncer
La presente divulgación proporciona células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para contener uno o más CARs que dirigen las células efectoras inmunitarias al cáncer. Esto se logra a través de un dominio de unión a antígenos en el CAR que es específico para un antígeno asociado al cáncer. Existen dos clases de antígenos asociados al cáncer (antígenos tumorales) que pueden ser fijados como objetivo por los CARs de la presente divulgación: (1) antígenos asociados al cáncer que se expresan en la superficie de las células cancerosas; y (2) antígenos asociados al cáncer que en sí mismos son intracelulares, sin embargo, un fragmento de dicho antígeno (péptido) se presenta en la superficie de las células cancerosas por MHC (complejo mayor de histocompatibilidad). La presente invención proporciona células efectoras inmunitarias de acuerdo con las reivindicaciones.
Por consiguiente, la presente invención emplea CARs que fijan como objetivo a los siguientes antígenos asociados al cáncer (antígenos tumorales): CD19, CD123, CD22, CD30, CD171, CS-1, CLL-1 (CLECL1), CD33, EGFRvIII, GD2, GD3, BCMA, Tn Ag, PSMA, ROR1, FLT3, FAP, TAG72, CD38, CD44v6, CEA, EPCAM, B7H3, KIT, IL-13Ra2, mesotelina, IL-11Ra, PSCA, VEGFR2, LewisY, CD24, PDGFR-beta, PRSS21, SSEA-4, CD20, receptor de folato alfa, ERBB2 (Her2/neu), MUC1, EGFR, NCAM, prostasa, PAP, ELF2M, efrina B2, receptor de IGF-I, CAIX, LMP2, gp100, bcr-abl, tirosinasa, EphA2, fucosil GM1, sLe, GM3, TGS5, HMWMAA, o-acetil-GD2, receptor de folato beta, TEM1/CD248, TEM7R, CLDN6, TSHR, GPRC5D, CXORF61, CD97, CD179a, ALK, ácido polisiálico, PLAC1, GloboH, NY-BR-1, UPK2, HAVCR1, ADRB3, PANX3, GPR20, LY6K, OR51E2, TARP, WT1, NY-ESO-1, LAGE-1a, legumaína, HPV E6,E7, MAGE-A1, MAGE A1, ETV6-AML, proteína de espermatozoide 17, XAGE1, Tie 2, MAD-CT-1, MAD-CT-2, antígeno 1 relacionado con Fos, p53, p53 mutante, prosteína, survivina y telomerasa, PCTA-1/Galectina 8, MelanA/MART1, Ras mutante, hTERT, puntos de corte de translocación de sarcoma, ML-IAP, ERG (gen de fusión de TMPRSS2 y ETS), NA17, PAX3, receptor de andrógenos, ciclina B1, MYCN, RhoC, TRP-2, CYP1B1, BORIS, SART3, PAX5, OY-TES1, LCK, AKAP-4, SSX2, RAGE-1, transcriptasa inversa de telomerasa humana, RU1, RU2, carboxil esterasa intestinal, mut hsp70-2, CD79a, CD79b, CD72, LAIR1, FcAR, LILRA2, CD300LF, CLEC12A, BST2, EMR2, LY75, GPC3, FCRL5 e IGLL1.
Antígenos de soporte tumoral
Un CAR descrito en esta memoria puede comprender un dominio de unión a antígenos (p. ej., anticuerpo o fragmento de anticuerpo, TCR o fragmento de TCR) que se une a un antígeno de que sustenta el tumor (p. ej., un antígeno de que sustenta el tumor como se describe en esta memoria). En algunas realizaciones, el antígeno que sustenta el tumor es un antígeno presente en una célula del estroma o una célula supresora de origen mieloide (MDSC). Las células del estroma pueden secretar factores de crecimiento para promover la división celular en el microentorno. Las células MDSC pueden inhibir la proliferación y activación de células T. Sin desear quedar ligados por la teoría, en algunas realizaciones, las células que expresan CAR destruyen las células que sustentan el tumor, inhibiendo de este modo indirectamente el crecimiento o la supervivencia del tumor.
En algunas realizaciones, el antígeno de células estromales se elige entre uno o más de: antígeno 2 de células estromales de la médula ósea (BST2), proteína de activación de fibroblastos (FAP) y tenascina. En una realización, el anticuerpo específico de FAP compite por la unión con, o tiene las mismas CDR que, sibrotuzumab. En algunas realizaciones, el antígeno de MDSC se elige entre uno o más de: CD33, CD11b, C14, CD15 y CD66b. Por consiguiente, en algunas realizaciones, el antígeno que sustenta el tumor se elige entre uno o más de: antígeno 2 de células estromales de la médula ósea (BST2), proteína de activación de fibroblastos (FAP) o tenascina, CD33, CD11b, C14, CD15, y CD66b.
Receptor quimérico para antígenos (CAR)
La presente invención puede emplear una construcción de ADN recombinante que comprende secuencias que codifican un CAR, en donde el CAR comprende un dominio de unión a antígenos que se une específicamente a un antígeno asociado al cáncer descrito en esta memoria, en donde la secuencia del dominio de unión al antígeno es contigua y en el mismo marco de lectura que una secuencia de ácido nucleico que codifica un dominio de señalización intracelular. El dominio de señalización intracelular puede comprender un dominio de señalización coestimulador y/o un dominio de señalización primario, p. ej., una cadena zeta. El dominio de señalización coestimulante se refiere a una parte del CAR que comprende al menos una parte del dominio intracelular de una molécula coestimulante.
En aspectos específicos, una construcción de CAR utilizada en la invención comprende un dominio scFv, en donde el scFv puede estar precedido por una secuencia conductora opcional como la proporcionada en la SEQ ID NO: 2, y seguido por una secuencia de bisagra opcional como la proporcionada en la SEQ ID NO: 4 o la SEQ ID NO: 6 o la SEQ ID NO: 8 o la SEQ ID NO: 10, una región transmembrana tal como se proporciona en la SEQ ID NO: 12, un dominio de señalización intracelular que incluye la SEQ ID NO: 14 o la SEQ ID NO: 16 y una secuencia CD3 zeta que incluye la SEQ ID NO: 18 o la SEQ ID NO: 20, p. ej., en donde los dominios son contiguos y están en el mismo marco de lectura para formar una única proteína de fusión.
En un aspecto, construcciones de CAR ejemplares comprenden una secuencia conductora opcional (p. ej., una secuencia conductora descrita en esta memoria), un dominio de unión a antígenos extracelular (p. ej., un dominio de unión a antígenos descrito en esta memoria), una bisagra (p. ej., una región de bisagra descrita en esta memoria), un dominio
transmembrana (p. ej., un dominio transmembrana descrito en esta memoria) y un dominio estimulador intracelular (p. ej., un dominio estimulador intracelular descrito en esta memoria). x En un aspecto, una construcción de CAR ilustrativa comprende una secuencia líder opcional (p. ej., una secuencia líder descrita en esta memoria), un dominio de unión a antígenos extracelular (p. ej., un dominio de unión a antígenos descrito en esta memoria), una bisagra (p. ej., una región de bisagra descrita en esta memoria), un dominio transmembrana (p. ej., un dominio transmembrana descrito en esta memoria), un dominio de señalización coestimulante intracelular (p. ej., un dominio de señalización coestimulante descrito en esta) y/o un dominio de señalización primario intracelular (p. ej., un dominio de señalización primario descrito en esta memoria).
Se proporciona una secuencia líder ilustrativa como la SEQ ID NO: 2. Se proporciona una secuencia de bisagra/espaciador ejemplar como SEQ ID NO: 4 o SEQ ID NO: 6 o SEQ ID NO: 8 o SEQ ID NO: 10. Se proporciona una secuencia ejemplar del dominio de transmembrana como SEQ ID NO: 12. Se proporciona una secuencia ejemplar del dominio de señalización intracelular de la proteína 4-1BB como SEQ ID NO: 14. Se proporciona una secuencia ejemplar del dominio de señalización intracelular de CD27 como SEQ ID NO: 16. Se proporciona una secuencia ilustrativa del dominio CD3zeta como la SEQ ID NO: 18 o la SEQ ID NO: 20.
En un aspecto, la presente invención emplea una construcción de ácido nucleico recombinante que comprende una molécula de ácido nucleico que codifica un CAR, en donde la molécula de ácido nucleico comprende la secuencia de ácido nucleico que codifica un dominio de unión a antígenos que es contiguo y está en el mismo marco de lectura que una secuencia de ácido nucleico que codifica un dominio de señalización intracelular.
En un aspecto, la presente invención emplea una construcción de ácido nucleico recombinante que comprende una molécula de ácido nucleico que codifica un CAR, en donde la molécula de ácido nucleico comprende la secuencia de ácido nucleico que codifica un dominio de unión a antígenos que es contigua y está en el mismo marco de lectura que una secuencia de ácido nucleico que codifica un dominio de señalización intracelular. Un dominio de señalización intracelular ejemplar que puede utilizarse en CAR incluye, pero no se limita a uno o más dominios de señalización intracelular de, p. ej., CD3-zeta, CD28, CD27, 4-1BB y similares. En algunos casos, el CAR puede comprender cualquier combinación de CD3-zeta, CD28, 4-1BB y similares.
Las secuencias de ácido nucleico que codifican las moléculas deseadas se pueden obtener utilizando métodos recombinantes conocidos en la técnica, tal como, por ejemplo, rastreando bibliotecas de células que expresan la molécula de ácido nucleico, obteniendo la molécula de ácido nucleico de un vector que se sabe que la incluye o aislando directamente de células y tejidos que la contienen, utilizando técnicas convencionales. Como alternativa, el ácido nucleico de interés se puede producir por vías sintéticas, en lugar de clonarse.
La presente invención puede emplear construcciones de vectores retrovirales y lentivirales que expresan un CAR que puede transducirse directamente en una célula.
La presente invención puede emplear una construcción de ARN que puede transfectarse directamente en una célula. Un método para generar ARNm para usar en la transfección implica la transcripción in vitro (IVT) de un molde con cebadores especialmente diseñados, seguida de la adición de poliA, para producir una construcción que contiene secuencias 3' y 5' no traducidas ("UTR") (p. ej., una UTR 3' y/o 5' descrita en esta memoria), un casquete 5' (p. ej., un casquete 5' descrito en esta memoria) y/o un Sitio de Entrada al Ribosoma Interno (IRES) (p. ej., un IREs descrito en esta memoria), el ácido nucleico que se ha de expresar y una cola de poliA, normalmente de 50-2000 bases de longitud (SEQ ID NO: 32). El ARN así producido puede transfectar eficientemente diferentes tipos de células. En un caso, el molde incluye secuencias para el CAR. En un caso, un vector CAR de ARN se transduce en una célula, p. ej., una célula T o una célula NK, mediante electroporación.
Dominio de unión a antígenos
El CAR empleado en la invención comprende un elemento de unión específico de la diana denominado de otro modo dominio de unión a antígenos. La elección del resto depende del tipo y número de ligandos que definen la superficie de una célula diana. Por ejemplo, el dominio de unión a antígenos puede elegirse para reconocer un ligando que actúa como marcador de la superficie celular en células diana asociadas con una patología particular. Por lo tanto, ejemplos de marcadores de la superficie celular que pueden actuar como ligandos para el dominio de unión a antígenos en un CAR de la divulgación incluyen aquellos asociados con infecciones virales, bacterianas y parasitarias, enfermedades autoinmunes y células cancerosas.
En un aspecto, la respuesta de células T mediada por CAR puede dirigirse a un antígeno de interés mediante la ingeniería de un dominio de unión a antígenos que se une específicamente a un antígeno deseado en el CAR
En un aspecto, la porción del CAR que comprende el dominio de unión a antígenos comprende un dominio de unión a antígenos que fija como objetivo un antígeno tumoral, p. ej., un antígeno tumoral descrito en esta memoria.
El dominio de unión a antígenos de un CAR de la divulgación puede ser cualquier dominio que se una al antígeno, incluyendo, pero no limitado a un anticuerpo monoclonal, un anticuerpo policlonal, un anticuerpo recombinante, un anticuerpo humano, un anticuerpo humanizado y un fragmento funcional del mismo, incluyendo, pero nop limitados a un anticuerpo de un solo dominio, tal como un dominio variable de cadena pesada (VH), un dominio variable de cadena ligera (VL) y un dominio variable (VHH) de nanocuerpos derivados de camélidos, y a un armazón alternativo conocido en la técnica para funcionar como dominio de unión a antígenos, tal como un dominio de fibronectina recombinante, un receptor de células T (TCR), o un fragmento del mismo, p. ej., TCR de cadena sencilla, y similares. En la presente invención, el dominio de unión a antígenos del primer CAR comprende un dominio VHH y el dominio de unión a antígenos del segundo CAR comprende un scFv. En algunos casos, es beneficioso que el dominio de unión al antígeno se derive de la misma especie en la que finalmente se utilizará el CAR. Por ejemplo, para uso en seres humanos, puede ser beneficioso que el dominio de unión al antígeno del CAR comprenda residuos humanos o humanizados para el dominio de unión a antígenos de un anticuerpo o fragmento de anticuerpo.
En una realización, un dominio de unión a antígenos contra CD22 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, por ejemplo, en Haso et al., Blood, 121 (7): 1165-1174 (2013); Wayne et al., Clin Cáncer Res 16 (6): 1894-1903 (2010); Kato et al., Leuk Res 37 (1): 83-88 (2013); Creative BioMart (creativebiomart.net): MOM-18047-S(P).
En una realización, un dominio de unión a antígenos contra CS-1 es una porción de unión a antígenos, p. ej., CDR, de Elotuzumab (BMS), véase, p. ej., Tai et al., 2008, Blood 112 (4): 1329-37; Tai et al., 2007, Blood. 110 (5): 1656-63.
En una realización, un dominio de unión a antígenos contra CLL-1 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo disponible de R&D, ebiosciences, Abcam, por ejemplo, PE-CLL1-hu n.° de cat. 353604 (BioLegend); y PE-CLL1 (CLEC12A) n.° de cat. 562566 (BD).
En una realización, un dominio de unión a antígenos contra CD33 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Bross et al., Clin Cancer Res 7 (6): 1490-1496 (2001) (Gemtuzumab Ozogamicina, hP67.6), Caron et al., Cancer Res 52 (24): 6761-6767 (1992) (Lintuzumab, HuM195), Lapusan et al., Invest New Drugs 30 (3): 1121-1131 (2012) (AVE9633), Aigner et al., Leukemia 27 (5): 1107-1115 (2013) (AMG330, CD33 BiTE), Dutour et al., Adv hematol 2012: 683065 (2012) y Pizzitola et al., Leukemia doi:10.1038/Lue.2014.62 (2014).
En una realización, un dominio de unión a antígenos contra GD2 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Mujoo et al., Cancer Res. 47 (4): 1098-1104 (1987); Cheung et al., Cancer Res 45 (6): 2642 2649 (1985), Cheung et al., J Clin Oncol 5 (9): 1430-1440 (1987), Cheung et al., J Clin Oncol 16 (9): 3053-3060 (1998), Handgretinger et al., Cancer Immunol Immunother 35 (3): 199-204 (1992). En algunas realizaciones, un dominio de unión a antígenos contra GD2 es una porción de unión a antígenos de un anticuerpo seleccionado de mAb 14.18, 14G2a, ch14.18, hu14.18, 3F8, hu3F8, 3G6, 8B6, 60C3, 10B8, ME36.1 y 8H9, véanse, p. ej., los documentos WO2012033885, WO2013040371, WO2013192294, WO2013061273, WO2013123061, WO2013074916 y WO201385552. En algunas realizaciones, un dominio de unión a antígenos contra GD2 es una porción de unión a antígenos de un anticuerpo descrito en la publicación de los EE. UU. n.°: 20100150910 o la publicación de PCT n.°: WO 2011160119.
En una realización, un dominio de unión a antígenos contra BCMA es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos WO2012163805, WO200112812 y WO2003062401.
En una realización, un dominio de unión a antígenos contra el antígeno Tn es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en el documento US 8.440.798, Brooks et al., PNAS 107 (22): 10056-10061 (2010) y Stone et al., OncoImmunology 1 (6): 863-873 (2012).
En una realización, un dominio de unión a antígenos contra PSMA es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Parker et al., Protein Expr Purif 89 (2): 136-145 (2013), US 20110268656 (J591 ScFv); Frigerio et al., European J Cancer 49 (9): 2223-2232 (2013) (scFvD2B); documento WO 2006125481 (mAb 3/A12, 3/E7 y 3/F11) y fragmentos de anticuerpos de cadena sencilla (scFv A5 y D7).
En una realización, un dominio de unión a antígenos contra ROR1 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Hudecek et al., Clin Cancer Res 19 (12): 3153-3164 (2013); documentos WO 2011159847; y US20130101607.
En una realización, un dominio de unión a antígenos contra FLT3 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos WO2011076922, US5777084, EP0754230, US20090297529, y varios anticuerpos de catálogos comerciales (R&D, ebiosciences, Abcam).
En una realización, un dominio de unión a antígenos contra TAG72 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Hombach e t al., G a s tro e n te ro lo g y 113 (4): 1163-1170 (1997); y Abcam ab691.
En una realización, un dominio de unión a antígenos contra FAP es una porción de unión a antígenos, p. ej., CDRs, de un anticuerpo descrito, p. ej., en Ostermann et al., Clinical Cancer Research 14: 4584-4592 (2008) (FAP5), patente de EE.UU. Publicación N° 2009/0304718; sibrotuzumab (véase, p. ej., Hofheinz et al., Oncology Research and Treatment 26(1), 2003 ); y Tran et al., J Exp Med 210(6):1125-1135 (2013 ).
En una realización, un dominio de unión a antígenos contra CD38 es una porción de unión a antígenos, p. ej., CDR, de daratumumab (véase, p. ej., Groen et al., Blood 116 (21): 1261-1262 (2010); MOR202 (véase, p. ej., el memoria US8,263,746); o anticuerpos descritos en el memoria Us 8,362,211.
En una realización, un dominio de unión a antígenos contra CD44v6 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Casucci et al., Blood 122 (20): 3461-3472 (2013).
En una realización, un dominio de unión a antígenos contra CEA es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Chmielewski et al., Gastoenterology 143 (4): 1095-1107 (2012).
En una realización, un dominio de unión a antígenos contra EPCAM es una porción de unión a antígenos, p. ej., CDRS, de un anticuerpo seleccionado de MT110, Ab biespecífico de EpCAM-CD3 (véase, p. ej., clinicaltrials.gov/ct2/show/NCT00635596); edrecolomab; 3622W94; ING-1; y adecatumumab (MT201).
En una realización, un dominio de unión a antígenos contra PRSS21 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito en la patente de los EE. UU. n.°: 8080650.
En una realización, un dominio de unión a antígenos contra B7H3 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo MGA271 (Macrogenics).
En una realización, un dominio de unión a antígenos contra KIT es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos US7915391, US20120288506 y varios anticuerpos de catálogos comerciales. En una realización, un dominio de unión a antígenos contra IL-13Ra2 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos WO2008/146911, WO2004087758, varios anticuerpos de catálogos comerciales y el memoria WO2004087758.
En una realización, un dominio de unión a antígenos contra CD30 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos US7090843 B1 y EP0805871.
En una realización, un dominio de unión a antígenos contra GD3 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos US7253263; US 8,207,308; US 20120276046; EP1013761; WO2005035577; y US6437098.
En una realización, un dominio de unión a antígenos contra CD171 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Hong et al., J Immunother 37 (2): 93-104 (2014).
En una realización, un dominio de unión a antígenos contra IL-11Ra es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo disponible de Abcam (n.° de cat ab55262) o Novus Biologicals (n.° de cat EPR5446). En otra realización, un dominio de unión a antígenos contra IL-11Ra es un péptido, véase, p. ej., Huang et al., Cancer Res 72 (1): 271-281 (2012).
En una realización, un dominio de unión a antígenos contra PSCA es una porción de unión a antígenos, por ejemplo, CDR, de un anticuerpo descrito, p. ej., en Morgenroth et al., Prostate 67 (10): 1121-1131 (2007) (scFv 7F5); Nejatollahi et al., J of Oncology 2013 (2013), artículo ID 839831 (scFv C5-II); y publicación de patente de los Ee . UU. n.° 20090311181. En una realización, un dominio de unión a antígenos contra VEGFR2 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Chinnasamy et al., J Clin Invest 120 (11): 3953-3968 (2010).
En una realización, un dominio de unión a antígenos contra LewisY es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Kelly et al., Cancer Biother Radiopharm 23 (4): 411-423 (2008) (Ab hu3s193 (scFv)); Dolezal et al., Protein Engineering 16 (1): 47-56 (2003) (scFv NC10).
En una realización, un dominio de unión a antígenos contra CD24 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Maliar et al., Gastroenterology 143 (5): 1375-1384 (2012).
En una realización, un dominio de unión a antígenos contra PDGFR-beta es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo Abcam ab32570.
En una realización, un dominio de unión a antígenos contra SSEA-4 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo MC813 (Cell Signaling), u otros anticuerpos disponibles en el mercado.
En una realización, un dominio de unión a antígenos contra CD20 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo Rituximab, Ofatumumab, Ocrelizumab, Veltuzumab o GA101.
En una realización, un dominio de unión a antígenos contra receptor de folato alfa es una porción de unión a antígenos, p. ej., CDR, del anticuerpo IMGN853, o un anticuerpo descrito en los documentos US20120009181; US4851332, LK26: US5952484.
En una realización, un dominio de unión a antígenos contra ERBB2 (Her2/neu) es una porción de unión a antígenos, p. ej., CDR, del anticuerpo trastuzumab o pertuzumab.
En una realización, un dominio de unión a antígenos contra MUC1 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo SAR566658.
En una realización, un dominio de unión a antígenos contra EGFR es una porción de unión a antígenos, p. ej., CDR, del anticuerpo cetuximab, panitumumab, zalutumumab, nimotuzumab o matuzumab.
En una realización, un dominio de unión a antígenos contra NCAM es una porción de unión a antígenos, p. ej., CDR, del clon de anticuerpo 2-2B: MAB5324 (EMD Millipore)
En una realización, un dominio de unión a antígenos contra efrina B2 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Abengozar et al., Blood 119 (19): 4565-4576 (2012).
En una realización, un dominio de unión a antígenos contra el receptor de IGF-I es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos US8344112 B2; EP2322550 A1; WO 2006/138315 o PCT/US2006/022995.
En una realización, un dominio de unión a antígenos contra CAIX es una porción de unión a antígenos, p. ej., CDRs, del clon de anticuerpos 303123 (R&D Systems).
En una realización, un dominio de unión a antígenos contra LMP2 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos US 7.410.640 o US20050129701.
En una realización, un dominio de unión a antígenos contra gp100 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo HMB45, NKIbetaB o un anticuerpo descrito en los documentos WO2013165940 o US20130295007
En una realización, un dominio de unión a antígenos contra tirosinasa es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos US5843674; o US19950504048.
En una realización, un dominio de unión a antígenos contra EphA2 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Yu et al., Mol Ther 22 (1): 102-111 (2014).
En una realización, un dominio de unión a antígenos contra GD3 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos US7253263; US 8,207,308; US 20120276046; EP1013761 A3; 20120276046; WO2005035577; o US6437098.
En una realización, un dominio de unión a antígenos contra fucosil GM1 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos US20100297138; o WO2007/067992.
En una realización, un dominio de unión a antígenos contra sLe es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo G193 (para Lewis Y), véase Scott AM et al., Cancer Res 60: 3254-61 (2000), también como se describe en Neeson et al, J Immunol mayo de 2013 190 (Meeting Abstract Supplement) 177.10.
En una realización, un dominio de unión a antígenos contra GM3 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo CA 2523449 (mAb 14F7).
En una realización, un dominio de unión a antígenos contra HMWMAA es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Kmiecik et al., Oncoimmunology 3 (1): e27185 (2014) (PMID: 24575382) (mAb9.2.27); documentos US6528481; WO2010033866; o US 20140004124.
En una realización, un dominio de unión a antígenos contra o-acetil-GD2 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo 8B6.
En una realización, un dominio de unión a antígenos contra TEM1/CD248 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Marty et al., Cancer Lett 235 (2): 298-308 (2006); Zhao et al., Immunol Methods 363 (2): 221-232 (2011).
En una realización, un dominio de unión a antígenos contra CLDN6 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo IMAB027 (Ganymed Pharmaceuticals), véase, p. ej., clinicaltrial.gov/show/NCT02054351.
En una realización, un dominio de unión a antígenos contra TSHR es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en los documentos US 8.603.466; US 8.501.415; o US 8.309.693.
En una realización, un dominio de unión a antígenos contra GPRC5D es una porción de unión a antígenos, p. ej., CDR, del anticuerpo FAB6300A (R&D Systems); o LS-A4180 (Lifespan Biosciences).
En una realización, un dominio de unión a antígenos contra CD97 es una porción de unión a antígenos, p. ej., CDRs, de un anticuerpo descrito, p. ej., en el documento US 6.846.911; de Groot et al., J Immunol 183(6):4127-4134 (2009); o un anticuerpo de R&D:Ma B3734..
En una realización, un dominio de unión a antígenos contra ALK es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Mino-Kenudson et al., Clin Cancer Res 16 (5): 1561-1571 (2010).
En una realización, un dominio de unión a antígenos contra ácido polisiálico es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Nagae et al., J Biol Chem 288 (47): 33784-33796 (2013).
En una realización, un dominio de unión a antígenos contra PLAC1 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Ghods et al., Biotechnol Appl Biochem 2013 doi:10.1002/bab.1177.
En una realización, un dominio de unión a antígenos contra GloboH es una porción de unión a antígenos del anticuerpo VK9; o un anticuerpo descrito, p. ej., en Kudryashov V et al., Glycoconj J.15 (3): 243-9 (1998), Lou et al., Proc Natl Acad Sci USA 111 (7): 2482-2487 (2014); MBr1: Bremer E-G et al. J Biol Chem 259: 14773-14777 (1984).
En una realización, un dominio de unión a antígenos contra NY-BR-1 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Jager et al., Appl Immunohistochem Mol Morphol 15 (1): 77-83 (2007).
En una realización, un dominio de unión a antígenos contra WT-1 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Dao et al., Sci Transl Med 5 (176): 176ra33 (2013); o documento WO2012/135854.
En una realización, un dominio de unión a antígenos contra MAGE-A1 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Willemsen et al., J Immunol 174 (12): 7853-7858 (2005) (scFv de tipo Tc R).
En una realización, un dominio de unión a antígenos contra la proteína de espermatozoide 17 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Song et al., Target Oncol 14 de agosto de 2013 (PMID:
23943313); Song et al., Med Oncol. 29 (4): 2923-2931 (2012).
En una realización, un dominio de unión a antígenos contra Tie 2 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo AB33 (Cell Signaling Technology).
En una realización, un dominio de unión a antígenos contra MAD-CT-2 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en PMID: 2450952; documento US7635753.
En una realización, un dominio de unión a antígenos contra antígeno 1 relacionado con Fos es una porción de unión a antígenos, p. ej., CDR, del anticuerpo 12F9 (Novus Biologicals).
En una realización, un dominio de unión a antígenos contra MelanA/MART1 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito en los documentos EP2514766 A2; o US 7.749.719.
En una realización, un dominio de unión a antígenos contra puntos de rotura de traslocación de sarcoma es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Luo et al., EMBO Mol Med. 4 (6): 453-461 (2012). En una realización, un dominio de unión a antígenos contra TRP-2 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Wang et al., J Exp Med. 184 (6): 2207-16 (1996).
Med. 4 (6): 453-461 (2012). En una realización, un dominio de unión a antígenos contra TRP-2 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Wang et al., J Exp Med. 184 (6): 2207-16 (1996).
En una realización, un dominio de unión a antígenos contra CYP1B1 es una porción de unión a antígenos, p. ej., CDR, de un anticuerpo descrito, p. ej., en Maecker et al., Blood 102 (9): 3287-3294 (2003).
En una realización, un dominio de unión a antígenos contra RAGE-1 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo MAB5328 (EMD Millipore).
En una realización, un dominio de unión a antígenos contra la transcriptasa inversa de telomerasa humana es una porción de unión a antígenos, p. ej., CDR, del anticuerpo con n.° de cat: LS-B95-100 (Lifespan Biosciences)
En una realización, un dominio de unión a antígenos contra carboxil esterasa intestinal es una porción de unión a antígenos, p. ej., CDR, del anticuerpo 4F12: n.° de cat: LS-B6190-50 (Lifespan Biosciences).
En una realización, un dominio de unión a antígenos contra hsp70-2 mut es una porción de unión a antígenos, p. ej., CDR, del anticuerpo Lifespan Biosciences: monoclonal: n.° de cat: LS-C133261-100 (Lifespan Biosciences).
En una realización, un dominio de unión a antígenos contra CD79a es una porción de unión a antígenos, p. ej., CDR, del anticuerpo anti-CD79a [HM47/A9] (ab3121), disponible de Abcam; anticuerpo CD79A n.° 3351 disponible de Cell Signaling Technology; o anticuerpo HPA017748 - Anticuerpo anti-CD79A producido en conejo, disponible de Sigma Aldrich.
En una realización, un dominio de unión a antígenos contra CD79b es una porción de unión a antígenos, p. ej., CDR, del anticuerpo polatuzumab vedotina, anti-CD79b descrito en Dornan et al., "Therapeutic potential of an anti-CD79b antibodydrug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma" Blood. 24 de septiembre de 2009; 114 (13): 2721-9. doi: 10.1182/blood-2009-02-205500. Epub 24 de julio de 2009, o el anticuerpo biespecífico anti-CD79b/CD3 descrito en "4507 Pre-Clinical Characterization of T Cell-Dependent Bispecific Antibody Anti-CD79b/CD3 As a Potential Therapy for B Cell Malignancies" Abstracts of 56th ASH Annual Meeting and Exposition, San Francisco, CA 6 9 de diciembre de 2014.
En una realización, un dominio de unión a antígenos contra CD72 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo J3-109 descrito en Myers y Uckun, "An anti-CD72 immunotoxin against therapy-refractory B-lineage acute lymphoblastic leukemia". Leuk Lymphoma. junio de 1995; 18 (1-2): 119-22, o anti-CD72 (10D6.8.1, mIgG1) descrito en Polson et al., "Antibody-Drug Conjugates for the Treatment of Non-Hodgkin's Lymphoma: Target and Linker-Drug Selection" Cancer Res 15 de marzo de 200969; 2358.
En una realización, un dominio de unión a antígenos contra LAIR1 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo ANT-301 LAIR1, disponible de ProSpec; o anticuerpo anti-CD305 humano (LAIR1), disponible de BioLegend. En una realización, un dominio de unión a antígenos contra FCAR es una porción de unión a antígenos, p. ej., CDR, del anticuerpo Cd89/anticuerpo FCAR (n.° de catálogo 10414-H08H), disponible de Sino Biological Inc.
En una realización, un dominio de unión a antígenos contra LILRA2 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo monoclonal LILRA2 (M17), clon 3C7 disponible de Abnova o anticuerpo de ratón anti-LILRA2, monoclonal (2D7), disponible de Lifespan Biosciences.
En una realización, un dominio de unión a antígenos contra CD300LF es una porción de unión a antígenos, p. ej., CDR, del anticuerpo de ratón anti molécula 1 de tipo CMRF35, monoclonal [UP-D2], disponible de BioLegend, o anticuerpo de rata anti-molécula 1 de tipo CMRF35, monoclonal [234903], disponible de R&D Systems.
En una realización, un dominio de unión a antígenos contra CLEC12A es una porción de unión a antígenos, p. ej., CDRs, del anticuerpo scFvanticuerpo biespecífico T cell Engager (BiTE) y ADC descrito en Noordhuis et al., "Targeting of CLEC12A In Acute Myeloid Leukemia by Antibody-Drug-Conjugates and Bispecific CLL-1xCD3 BiTE Antibody" 53a ASH Annual Meeting and Exposition, 10-13 de diciembre, 2011, y MCLA-117 (Merus).
En una realización, un dominio de unión a antígenos contra BST2 (también denominado CD317) es una porción de unión a antígenos, p. ej., CDRs, del anticuerpo Monoclonal[3H4] anti-CD317 de Ratón, disponible de Antibodies-Online o Anticuerpo Anti-CD317 de Ratón, monoclonal [696739], disponible de R&D Systems.
En una realización, un dominio de unión a antígenos contra EMR2 (también denominado CD312) es una porción de unión a antígenos, p. ej., CDRs, del anticuerpo Anti-CD312 de Ratón, Monoclonal[LS-B8033] disponible de Lifespan Biosciences o Anticuerpo Anti-CD312 de Ratón, Monoclonal [494025], disponible de R&D Systems.
En una realización, un dominio de unión a antígenos contra LY75 es una porción de unión a antígenos, p. ej., CDRs, del anticuerpo Anti-células antígeno 75 Monoclonal [HD30] disponible de EMD Millipore o anticuerpo Mouse Anti-células antígeno 75, Monoclonal [A15797] disponible de Life Technologies.
En una realización, un dominio de unión a antígenos contra GPC3 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo hGC33 descrito en Nakano K, Ishiguro T, Konishi H, et al. Generation of a humanized anti-glypican 3 antibody by CDR grafting and stability optimization. Anticancer Drugs. Nov de 2010; 21 (10): 907-916, o MDX-1414, HN3 o YP7,
los tres descritos en Feng et al., "Glypican-3 antibodies: a new therapeutic target for liver cáncer". FEBS Lett. 21 de enero de 2014; 588 (2): 377-82.
En una realización, un dominio de unión a antígenos contra FCRL5 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo anti-FcRL5 descrito en Elkins et al., "FcRL5 as a target of antibody-drug conjugates for the treatment of multiple myeloma" Mol Cancer Ther. octubre de 2012; 11 (10): 2222-32.
En una realización, un dominio de unión a antígenos contra IGLL1 es una porción de unión a antígenos, p. ej., CDR, del anticuerpo de ratón anti-polipéptido 1 de tipo inmunoglobulina lambda, monoclonal [AT1G4], disponible de Lifespan Biosciences, anticuerpo de ratón anti-polipéptido 1 de tipo inmunoglobulina lambda, monoclonal [HSL11], disponible de BioLegend.
En una realización, el dominio de unión a antígenos comprende una, dos, tres (p. ej., las tres) CDR de cadena pesada, CDR1 HC, CDR2 HC y CDR3 HC, de un anticuerpo enumerado anteriormente, y/o una, dos, tres (p. ej., las tres) CDR de cadena ligera, CDR1 LC, CDR2 LC y CDR3 LC, de un anticuerpo enumerado anteriormente. En una realización, el dominio de unión a antígenos comprende una región variable de cadena pesada y/o una región variable de cadena ligera de un anticuerpo enumerado anteriormente.
En otro aspecto, el dominio de unión a antígenos comprende un anticuerpo o un fragmento de anticuerpo humanizado. En algunos aspectos, se humaniza un anticuerpo no humano, donde las secuencias o regiones específicas del anticuerpo se modifican para aumentar la similitud con un anticuerpo producido de forma natural en un ser humano o un fragmento del mismo. En un aspecto, el dominio de unión a antígenos está humanizado.
Se puede producir un anticuerpo humanizado utilizando una diversidad de técnicas conocidas en este campo, incluidas, pero sin limitación, injerto de CDR (véase, p. ej., la Patente Europea N° EP 239.400; la Publicación Internacional N° WO 91/09967; y las Pat. de los EE. UU. N° 5.225.539, 5.530.101 y 5.585.089), rebarnizado o rechapado (véanse, p. ej., las Patentes Europeas N° EP 592.106 y EP 519.596; Padlan, 1991, Molecular Immunology, 28 (4/5): 489-498; Studnicka et al., 1994, Protein Engineering, 7 (6): 805-814; y Roguska et al., 1994, PNAS, 91: 969-973), reordenamiento de cadenas (véase, p. ej., la Pat. de los EE. UU. N° 5.565.332) y técnicas descritas, p. ej., en la publicación de solicitud de patente de EE. UU. N° US 2005/0042664, la publicación de solicitud de patente de EE. UU. N° US 2005/0048617, la patente de EE. UU. N° N° 6.407.213, la Pat. de los EE. UU. N° 5.766.886, Publicación Internacional N° WO 9317105, Tan et al., J. Immunol., 169: 1119-25 (2002), Caldas et al., Protein Eng., 13 (5): 353-60 (2000), Morea et al., Methods, 20 (3): 267-79 (2000), Baca et al., J. Biol. Chem., 272 (16): 10678-84 (1997), Roguska et al., Protein Eng., 9 (10): 895-904 (1996), Couto et al., Cancer Res., 55 (23 Sup): 5973s-5977s (1995), Couto et al., Cancer Res., 55 (8): 1717-22 (1995), Sandhu J S, Gene, 150 (2): 409-10 (1994), y Pedersen et al., J. Mol. Biol., 235 (3): 959-73 (1994). A menudo, los residuos de marco en las regiones de marco conservado se sustituirán con el residuo correspondiente del anticuerpo donante de CDR para alterar, por ejemplo, mejorar, la unión al antígeno. Estas sustituciones de marco conservado se identifican mediante métodos bien conocidos en la técnica, p. ej., modelando las interacciones de la CDR y los residuos de marco conservado para identificar residuos de marco conservado importantes para la unión a antígenos y comparación de secuencias para identificar residuos de marco conservado inusuales en posiciones particulares. (Véase, p. ej., Queen et al., patente de los EE. UU. n.° 5,585,089; y Riechmann et al., 1988, Nature, 332: 323).
Un anticuerpo o fragmento de anticuerpo humanizado tiene uno o más residuos aminoacídicos que permanecen en él de una fuente que no es humana. A estos residuos de aminoácidos no humanos se les alude a menudo como residuos "importados", que normalmente se toman de un dominio variable "importado". Como se proporciona en esta memoria, los anticuerpos o fragmentos de anticuerpo humanizados comprenden una o más CDR de moléculas de inmunoglobulina no humanas y regiones marco conservadas en donde los residuos de aminoácidos que comprenden el marco conservado proceden completa o principalmente de la línea germinal humana. Múltiples técnicas para la humanización de anticuerpos o fragmentos de anticuerpos son bien conocidas en la técnica y se pueden realizar esencialmente siguiendo el método de Winter y colaboradores (Jones et al., Nature, 321: 522-525 (1986); Riechmann et al. Nature, 332: 323-327 (1988); Verhoeyen et al., Science, 239: 1534-1536 (1988)), sustituyendo las secuencias correspondientes de un anticuerpo humano por las CDR o secuencias de CDR de roedores, es decir, injerto de CDR (memoria EP 239.400; publicación de PCT n.° WO 91/09967; y patentes de los EE. UU. n.° 4.816.567; 6.331.415; 5.225.539; 5.530.101; 5.585.089; 6.548.640). En anticuerpos y fragmentos de anticuerpos humanizados de este tipo, sustancialmente menos de un dominio variable humano intacto se ha sustituido por la secuencia correspondiente de una especie no humana. Los anticuerpos humanizados son, a menudo, anticuerpos humanos en los que algunos residuos de CDR y posiblemente algunos residuos de marco conservado (FR) están sustituidos por residuos de sitios análogos en anticuerpos de roedores. La humanización de anticuerpos y fragmentos de anticuerpos también se puede lograr mediante rebarnizado o rechapado (memorias EP 592.106; EP 519.596; Padlan, 1991, Molecular Immunology, 28 (4/5): 489-498; Studnicka et al., Protein Engineering, 7 (6): 805-814 (1994); y Roguska et al., PNAS, 91: 969-973 (1994)) o reordenamiento de cadena (patente de los EE. UU. N.° 5565332).
La elección de dominios variables humanos, tanto ligeros como pesados, que se pueden utilizar en la producción de los anticuerpos humanizados es para reducir la antigenicidad. De acuerdo con el llamado método de "mejor ajuste", la secuencia del dominio variable de un anticuerpo de roedor se analiza frente a la colección completa de secuencias de
dominio variable humanas conocidas. La secuencia humana más cercana a la del roedor se acepta entonces como el marco conservado (FR) humano para el anticuerpo humanizado (Sims et al., J. Immunol., 151: 2296 (1993); Chothia et al., J. Mol. Biol., 196: 901 (1987)). Otro método utiliza un marco conservado particular procedente de la secuencia consenso de todos los anticuerpos humanos de un subgrupo particular de cadenas ligeras o pesadas. Se puede utilizar el mismo armazón para varios anticuerpos humanizados diferentes (véanse, p. ej., Nicholson et al. Mol. Immun. 34 (16 17): 1157-1165 (1997); Carter et al., Proc. Natl. Acad. Sci. USA, 89: 4285 (1992); Presta et al., J. Immunol., 151: 2623 (1993)). En algunas realizaciones, la región marco, p. ej., las cuatro regiones marco, de la región variable de la cadena pesada se derivan de una secuencia de la línea germinal VH4)_4-59. En una realización, la región marco conservada puede comprender una, dos, tres, cuatro o cinco modificaciones, p. ej., sustituciones, p. ej., del aminoácido en la secuencia murina correspondiente. En una realización, la región marco, p. ej., las cuatro regiones marco de la región variable de la cadena ligera se derivan de una secuencia de la línea germinal VK3_1.25. En una realización, la región marco conservada puede comprender una, dos, tres, cuatro o cinco modificaciones, p. ej., sustituciones, p. ej., del aminoácido en la secuencia murina correspondiente.
En algunos aspectos, la porción de una composición de CAR de la invención que comprende un fragmento de anticuerpo se humaniza con retención de alta afinidad por el antígeno diana y otras propiedades biológicas favorables. De acuerdo con un aspecto de la divulgación, los anticuerpos humanizados y los fragmentos de anticuerpos se preparan mediante un proceso de análisis de las secuencias parentales y diversos productos humanizados conceptuales utilizando modelos tridimensionales de las secuencias parentales y humanizadas. Se puede disponer fácilmente de modelos tridimensionales de inmunoglobulina y los expertos en la técnica están familiarizados con ellos. Se puede disponer de programas informáticos que ilustran y presentan estructuras conformacionales tridimensionales probables de secuencias de inmunoglobulinas candidata seleccionadas. La inspección de estas presentaciones permite el análisis de la función probable de los residuos en el funcionamiento de la secuencia de inmunoglobulina candidata, por ejemplo, el análisis de residuos que influyen en la capacidad de la inmunoglobulina candidata para unirse al antígeno diana. De esta forma, los residuos Fr se pueden seleccionar y combinar de secuencias del receptor y de importación de modo que se logre la característica deseada de anticuerpo o fragmento de anticuerpo, tal como un incremento de la afinidad por el antígeno diana. En general, los residuos de CDR están directamente y muy sustancialmente implicados en influenciar la unión al antígeno.
Un anticuerpo o un fragmento de anticuerpo humanizado puede retener una especificidad antigénica similar a la del anticuerpo original, p. ej., en esta invención, la capacidad de unirse a un antígeno asociado al cáncer como se describe en la presente memoria. En algunas realizaciones, un anticuerpo o fragmento de anticuerpo humanizado puede tener mejor afinidad y/o especificidad de unión a un antígeno asociado al cáncer humano como se describe en esta memoria.
En un aspecto, el dominio de unión a antígenos empleado en la invención se caracteriza por características o propiedades funcionales particulares de un anticuerpo o fragmento de anticuerpo. Por ejemplo, en un aspecto, la porción de una composición de CAR de la invención que comprende un dominio de unión a antígenos se une específicamente a un antígeno tumoral como se describe en el presente memoria.
En un aspecto, el dominio de unión contra antígeno asociado al cáncer como se describe en el presente memoria es un fragmento, p. ej., un fragmento variable de cadena única (scFv). En un aspecto, el antígeno asociado contra el cáncer como se describe en esta memoria es un Fv, un Fab, un (Fab')2 o un anticuerpo híbrido bifuncional (p. ej., biespecífico) (p. ej., Lanzavecchia et al. , Eur. J. Immunol., 17, 105 (1987)). En la presente invención, el dominio de unión a antígenos del primer CAR comprende un dominio VHH y el dominio de unión a antígenos del segundo CAR comprende un scFv. En un aspecto, los anticuerpos y fragmentos de los mismos empleados en la invención se unen a un antígeno asociado al cáncer como se describe en esta memoria con afinidad de tipo salvaje o potenciada.
En algunos casos, los scFv se pueden preparar de acuerdo con el método conocido en la técnica (véase, por ejemplo, Bird et al., (1988) Science 242: 423-426 y Houston et al., (1988) Proc. Natl Acad. Sci. USA 85:5879-5883). Se pueden producir moléculas de ScFv ligando las regiones VH y VL entre sí utilizando enlazadores polipeptídicos flexibles. Las moléculas de scFv comprenden un enlazador (p. ej., un enlazador Ser-Gly) con una longitud y/o composición de aminoácidos optimizadas. La longitud del enlazador puede afectar en gran medida a cómo las regiones variables de un scFv se pliegan e interactúan. De hecho, si se emplea un enlazador polipeptídico corto (p. ej., entre 5-10 aminoácidos), se evita el plegamiento intracatenario. También se requiere el plegamiento intercatenario para reunir las dos regiones variables para formar un sitio de unión de epítopo funcional. Para consultar ejemplos de orientación y tamaño del enlazador, véanse, p. ej., Hollinger et al. 1993 Proc Natl Acad. Sci. U.S.A. 90: 6444-6448, publicaciones de solicitudes de patentes de los EE. UU. n.° 2005/0100543, 2005/0175606, 2007/0014794 y publicaciones de PCT n.° WO2006/020258 y WO2007/024715.
Un scFv puede comprender un enlazador de al menos 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, 50 o más residuos de aminoácidos entre sus regiones VL y VH. La secuencia de enlazador puede comprender cualquier aminoácido de origen natural. En algunas realizaciones, la secuencia de enlazador comprende los aminoácidos glicina y serina. En otra realización, la secuencia de enlazador comprende conjuntos de repeticiones de glicina y serina, tales como (Gly4Ser)n, en donde n es un número entero positivo igual a o mayor de 1 (SEQ ID NO: 22). En una realización, el enlazador puede ser (Gly4Ser)4 (SEQ ID NO: 29) o (Gly4Ser)3 (SEQ ID NO: 30). La variación en la
longitud del enlazador puede conservar o potenciar la actividad, lo que da lugar a una eficacia superior en los estudios de actividad.
En otro caso, el dominio de unión a antígenos es un receptor de células T ("TCR"), o un fragmento del mismo, por ejemplo, un TCR de cadena sencilla (scTCR). En la técnica se conocen métodos para fabricar tales TCR. Véase, p. ej., Willemsen RA et al., Gene Therapy 7: 1369-1377 (2000); Zhang T et al, Cancer Gene Ther 11: 487-496 (2004); Aggen et al, Gene Ther. 19 (4): 365-74 (2012). Por ejemplo, se puede diseñar scTCR que contenga los genes Va y Vp de un clon de célula T unido por un enlazador (p. ej., un péptido flexible). Este enfoque es muy útil para la diana asociada al cáncer que en sí mismo es intracelular, sin embargo, el MHC presenta un fragmento de dicho antígeno (péptido) en la superficie de las células cancerosas.
CAR biespecíficos
En una realización, una molécula de anticuerpo multiespecífico es una molécula de anticuerpo biespecífico. Un anticuerpo biespecífico tiene especificidad por dos antígenos como máximo. Una molécula de anticuerpo biespecífico está caracterizada por una primera secuencia del dominio variable de inmunoglobulina que tiene especificidad de unión por un primer epítopo y una segunda secuencia del dominio variable de inmunoglobulina que tiene especificidad de unión por un segundo epítopo. En una realización, los epítopos primero y segundo están en el mismo antígeno, p. ej., la misma proteína (o subunidad de una proteína multimérica). En una realización, el primer y segundo epítopos se solapan. En una realización, el primer y segundo epítopos no se solapan. En una realización, el primer y segundo epítopos se encuentran en diferentes antígenos, p. ej., diferentes proteínas (o diferentes subunidades de una proteína multimérica). En una realización, una molécula de anticuerpo biespecífico comprende una secuencia de dominio variable de cadena pesada y una secuencia de dominio variable de cadena ligera que tienen especificidad de unión para un primer epítopo y una secuencia de dominio variable de cadena pesada y una secuencia de dominio variable de cadena ligera que tienen especificidad de unión para un segundo epítopo. En una realización, una molécula de anticuerpo biespecífico comprende un medio anticuerpo que tiene especificidad de unión para un primer epítopo y un medio anticuerpo que tiene especificidad de unión para un segundo epítopo. En una realización, una molécula de anticuerpo biespecífico comprende un medio anticuerpo, o fragmento del mismo, que tiene especificidad de unión para un primer epítopo y un medio anticuerpo, o fragmento del mismo, que tiene especificidad de unión para un segundo epítopo. En una realización, una molécula de anticuerpo biespecífico comprende un scFv, o un fragmento del mismo, que tiene especificidad de unión para un primer epítopo y un scFv, o fragmento del mismo, que tiene especificidad de unión para un segundo epítopo.
En determinadas realizaciones, la molécula de anticuerpo es una molécula de anticuerpo multiespecífico (p. ej., uno biespecífico o uno triespecífico). Protocolos para generar moléculas de anticuerpos biespecíficos o heterodiméricos son conocidos en la técnica; incluyendo pero no limitado a, por ejemplo, el enfoque de "perilla en un agujero" descrito en, p. ej., el documento US 5731168; el emparejamiento Fc de dirección electrostática como se describe en, p. ej., los documentos WO 09/089004 , WO 06/106905 y WO 2010/129304; Strand Exchange Engineered Domains (SEED) formación de heterodímeros como se describe en, p. ej., el documento WO 07/110205; intercambio de brazo Fab como se describe en, p. ej., los documentos WO 08/119353 , WO 2011/131746 y WO 2013/060867; conjugado de doble anticuerpo, p. ej., por entrecruzamiento de anticuerpos para generar una estructura biespecífica usando un reactivo heterobifuncional que tiene un grupo reactivo con amina y un grupo reactivo con sulfhidrilo como se describe en, p. ej., el documento US 4433059; determinantes de anticuerpos biespecíficos generados mediante la recombinación de semianticuerpos (pares de cadenas ligeras y pesadas o Fabs) de diferentes anticuerpos a través del ciclo de reducción y oxidación de enlaces disulfuro entre las dos cadenas pesadas, como se describe en, p. ej., el documento US 4444878; anticuerpos trifuncionales, p. ej., tres fragmentos Fab' entrecruzados a través de grupos reactivos de sulfhidrilo, como se describe en, p. ej., el documento US5273743; proteínas de unión biosintéticas, p. ej., un par de scFv reticulados a través de colas C-terminales, preferiblemente a través de disulfuro o reticulación química reactiva con amina, como se describe en, p. ej., el documento US5534254; anticuerpos bifuncionales, p. ej., fragmentos Fab con diferentes especificidades de unión dimerizados a través de cremalleras de leucina (p. ej., c-fos y c-jun) que han reemplazado el dominio constante, como se describe en, p. ej., el documento US5582996; receptores mono y oligovalentes biespecíficos y oligoespecíficos, p. ej., regiones VH-CH1 de dos anticuerpos (dos fragmentos Fab) unidos a través de un espaciador polipeptídico entre la región CH1 de un anticuerpo y la región VH del otro anticuerpo típicamente con cadenas ligeras asociadas, como descrito en, p. ej., el documento US5591828; conjugados biespecíficos de ADN-anticuerpo, p. ej., entrecruzamiento de anticuerpos o fragmentos Fab a través de un trozo de ADN de doble cadena, como se describe en, p. ej., el documento US5635602; proteínas de fusión biespecíficas, p. ej., una construcción de expresión que contiene dos scFv con un enlazador peptídico helicoidal hidrofílico entre ellos y una región constante completa, como se describe en, p. ej., el documento US5637481; proteínas de unión multivalentes y multiespecíficas, p. ej., dímero de polipéptidos que tienen un primer dominio con una región de unión de la región variable de la cadena pesada de Ig y un segundo dominio con una región de unión de la región variable de la cadena ligera de Ig, generalmente denominados diacuerpos (también se incluyen estructuras de orden superior creadas para moléculas biespecíficas, triespecíficas o tetraespecíficas, como se describe en, p. ej., el documento US5837242; construcciones de minicuerpos con cadenas VL y VH unidas conectadas además con espaciadores peptídicos a una región de bisagra de anticuerpo y una región CH3, que pueden dimerizarse para formar moléculas biespecíficas/multivalentes, como se describe en, pp. ej., el documento US5837821; dominios VH y VL unidos con un enlazador peptídico corto (p. ej., 5 o 10 aminoácidos) o ningún enlazador en ninguna orientación, que puede formar dímeros para formar diacuerpos biespecíficos; trímeros y tetrámeros, como se describe en, p. ej., el documento
US5844094; cadena de dominios VH (o dominios VL en miembros de la familia) conectados por enlaces peptídicos con grupos entrecruzables en el extremo C asociados además con dominios VL para formar una serie de FV (o scFv), como se describe en, p. ej., el documento US5864019; y los polipéptidos de unión de cadena sencilla con dominios VH y VL unidos a través de un enlazador peptídico se combinan en estructuras multivalentes mediante entrecruzamiento no covalente o químico para formar, p. ej., estructuras homobivalentes, heterobivalentes, trivalentes y tetravalentes utilizando tanto formato scFV como tipo diacuerpo, como se describe en, p. ej., el documento US5869620. Moléculas multiespecíficas y biespecíficas adicionales y métodos para producir las mismas se encuentran, por ejemplo, en los documentos US5910573, US5932448, US5959083, US5989830, US6005079, US6239259, US6294353, US6333396, US6476198, US6511663, US6670453, US6743896, US6809185, US6833441, US7129330, US7183076, US7521056, US7527787, US7534866, US7612181, US2002004587A1, US2002076406A1, US2002103345A1, US2003207346A1, US2003211078A1, US2004219643A1, US2004220388A1, US2004242847A1, US2005003403A1, US2005004352A1, US2005069552A1, US2005079170A1, US2005100543A1, US2005136049A1, US2005136051A1, US2005163782A1, US2005266425A1, US2006083747A1, US2006120960A1, US2006204493A1, US2006263367A1, US2007004909A1, US2007087381A1, US2007128150A1, US2007141049A1, US2007154901A1, US2007274985A1, US2008050370A1, US2008069820A1, US2008152645A1, US2008171855A1, US2008241884A1, US2008254512A1, US2008260738A1, US2009130106A1, US2009148905A1, US2009155275A1, US2009162359A1, US2009162360A1, US2009175851A1, US2009175867A1, US2009232811A1, US2009234105A1, US2009263392A1, US2009274649A1, EP346087A2, WO0006605A2, WO02072635A2, WO04081051A1, WO06020258A2, WO2007044887A2, WO2007095338A2, WO2007137760A2, WO2008119353A1, WO2009021754A2, WO2009068630A1, WO9103493A1, WO9323537A1, WO9409131A1, WO9412625A2, WO9509917A1, WO9637621A2, WO9964460A1.
Dentro de cada anticuerpo o fragmento de anticuerpo (p. ej., scFv de una molécula de anticuerpo biespecífico), la VH puede estar aguas arriba o aguas abajo de la VL. En algunas realizaciones, el anticuerpo o fragmento de anticuerpo aguas arriba (p. ej., scFv) está dispuesto con su VH (VHi) aguas arriba de su VL (VLi) y el anticuerpo o fragmento de anticuerpo aguas abajo (p. ej., scFv) está dispuesto con su VL (VL2) aguas arriba de su VH (VL2), de modo que la molécula de anticuerpo biespecífico global tiene la disposición VH1-VL1-VL2-VH2. En otras realizaciones, el anticuerpo o fragmento de anticuerpo aguas arriba (p. ej., scFv) está dispuesto con su VL (VLi) aguas arriba de su VH (VHi) y el anticuerpo o fragmento de anticuerpo aguas abajo (p. ej., scFv) está dispuesto con su VH (VH2) aguas arriba de su VL (VH2), de modo que la molécula de anticuerpo biespecífico global tiene la disposición VL1-VH1-VH2-VL2. Opcionalmente, un enlazador está dispuesto entre los dos anticuerpos o fragmentos de anticuerpos (p. ej., scFvs), p. ej., entre VLi y VL2 si la construcción está dispuesta como VH1-VL1-VL2-VH2, o entre VHi y VH2 si la construcción está dispuesta como VL1-VH1-VH2-VL2. El enlazador puede ser un enlazador como se describe en esta memoria, p. ej., un enlazador (Gly4-Ser)n, en donde n es 1, 2, 3, 4, 5 o 6, preferentemente 4 (SEQ ID NO: 64). En general, el enlazador entre los dos scFv debe ser lo suficientemente largo para evitar emparejamientos erróneos entre los dominios de los dos scFv. Opcionalmente, se dispone un enlazador entre el VL y VH del primer scFv. Opcionalmente, se dispone un enlazador entre el VL y VH del segundo scFv. En construcciones que tienen múltiples enlazadores, dos o más cualesquiera de los enlazadores pueden ser idénticos o diferentes. En consecuencia, en algunas realizaciones, un CAR biespecífico comprende VL, VH y opcionalmente uno o más enlazadores en una disposición como se describe en esta memoria.
Estabilidad y mutaciones
La estabilidad de un dominio de unión a antígenos para un antígeno asociado al cáncer como se describe en esta memoria, p. ej., moléculas de scFv (p. ej., scFv soluble), puede evaluarse con referencia a las propiedades biofísicas (p. ej., estabilidad térmica) de una molécula scFv de control convencional o un anticuerpo de longitud completa. En una realización, el scFv humanizado tiene una estabilidad térmica que es mayor de aproximadamente 0.1, aproximadamente 0.25, aproximadamente 0.5, aproximadamente 0.75, aproximadamente 1, aproximadamente 1.25, aproximadamente 1.5, aproximadamente 1.75, aproximadamente 2, aproximadamente 2.5, aproximadamente 3, aproximadamente 3.5, aproximadamente 4, aproximadamente 4.5, aproximadamente 5, aproximadamente 5.5, aproximadamente 6, aproximadamente 6.5, aproximadamente 7, aproximadamente 7.5, aproximadamente 8, aproximadamente 8.5, aproximadamente 9, aproximadamente 9.5, aproximadamente 10 grados, aproximadamente 11 grados, aproximadamente 12 grados, aproximadamente 13 grados, aproximadamente 14 grados o aproximadamente 15 grados Celsius que una molécula de unión de control (p. ej., una molécula scFv convencional) en los ensayos descritos.
La estabilidad térmica mejorada del dominio de unión a antígenos para un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, se confiere posteriormente a la construcción de CAR completa, lo que conlleva propiedades terapéuticas mejoradas de la construcción de CAR. La estabilidad térmica mejorada del dominio de unión del antígeno a un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, se puede mejorar en al menos aproximadamente 2 °C o 3 °C en comparación con un anticuerpo convencional. En una realización, el dominio de unión al antígeno de un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, tiene una estabilidad térmica mejorada en 1 °C en comparación con un anticuerpo convencional. En otra realización, el dominio de unión a antígenos de un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, tiene una estabilidad térmica mejorada en 2 °C en comparación con un anticuerpo convencional. En otra realización, el scFv tiene una estabilidad térmica mejorada en 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 °C en comparación con un anticuerpo convencional. Se pueden hacer comparaciones, por ejemplo, entre las moléculas de scFv desveladas en esta memoria y las moléculas de scFv o fragmentos Fab de un anticuerpo del que procedieron los VH y VL de scFv. La estabilidad térmica se puede medir utilizando métodos conocidos en la técnica.
Por ejemplo, en un caso, se puede medir la Tf. Más adelante se describen en más detalle métodos para medir la Tm y otros métodos para determinar la estabilidad proteica.
Las mutaciones en scFv (que surgen a través de la humanización o mutagénesis directa del scFv soluble) pueden alterar la estabilidad del scFv y mejoran la estabilidad global del scFv y la construcción de CAR. La estabilidad del scFv humanizado se compara con el scFv murino utilizando mediciones tales como Tm, desnaturalización por temperatura y agregación por temperatura.
La capacidad de unión de los scFv mutantes se puede determinar utilizando ensayos conocidos en la técnica y descritos en esta memoria.
En una realización, el dominio de unión a antígenos de un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, comprende al menos una mutación que surge del proceso de humanización de manera que el scFv mutado confiera una estabilidad mejorada a la construcción de CAR. En otra realización, el dominio de unión a antígenos de un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, comprende al menos 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 mutaciones que surgen del proceso de humanización de manera que el scFv mutado confiere una estabilidad mejorada a la construcción CAR.
Métodos de evaluación de la estabilidad de las proteínas
La estabilidad de un dominio de unión a antígenos se puede evaluar utilizando, p. ej., los métodos descritos más adelante. Tales métodos permiten la determinación de múltiples transiciones de desplegamiento térmico donde el dominio menos estable se despliega primero o limita el umbral de estabilidad global de una unidad multidominio que se despliega cooperativamente (p. ej., una proteína multidominio que presenta una única transición de desplegamiento). El dominio menos estable se puede identificar de varias maneras adicionales. Se puede realizar mutagénesis para estudiar qué dominio limita la estabilidad global. Adicionalmente, la resistencia a la proteasa de una proteína multidominio se puede realizar en condiciones en las que se sabe que el dominio menos estable está intrínsecamente desplegado a través de DSC u otros métodos espectroscópicos (Fontana, et al., (1997) Fold. Des., 2: R17-26; Dimasi et al. (2009) J. Mol. Biol.
393: 672-692). Una vez que se identifica el dominio menos estable, la secuencia que codifica este dominio (o una porción de esta) se puede emplear como una secuencia de prueba en los métodos.
a) Estabilidad Térmica
La estabilidad térmica de las composiciones se puede analizar utilizando varias técnicas biofísicas o bioquímicas no limitantes conocidas en este campo. En ciertos casos, la estabilidad térmica se evalúa mediante espectroscopía analítica.
Un método de espectroscopia analítica ilustrativa es la calorimetría diferencial de barrido (DSC, por sus siglas en inglés). La DSC emplea un calorímetro que es sensible a las absorbancias de calor que acompañan el desplegamiento de la mayoría de las proteínas o dominios de proteínas (véase, p. ej., Sanchez-Ruiz, et al., Biochemistry, 27: 1648-52, 1988). Para determinar la estabilidad térmica de una proteína, se inserta una muestra de la proteína en el calorímetro y se eleva la temperatura hasta que se despliega el Fab o scFv. La temperatura a la que se despliega la proteína es indicativa de la estabilidad global de la proteína.
Otro método de espectroscopia analítica ilustrativa es la espectroscopia de dicroísmo circular (DC). La espectrometría de DC mide la actividad óptica de una composición en función de una temperatura creciente. La espectroscopia de dicroísmo circular (DC) mide las diferencias en la absorción de la luz polarizada a la izquierda frente a la luz polarizada a la derecha que surgen debido a la asimetría estructural. Una estructura desordenada o desplegada da lugar a un espectro de DC muy diferente al de una estructura ordenada o plegada. El espectro de DC refleja la sensibilidad de las proteínas a los efectos desnaturalizantes de la temperatura creciente y, por lo tanto, es indicativo de la estabilidad térmica de una proteína (véase van Mierlo y Steemsma, J. Biotechnol., 79 (3): 281-98, 2000).
Otro método de espectroscopia analítica ilustrativo para medir la estabilidad térmica es la espectroscopia de emisión de fluorescencia (véase van Mierlo y Steemsma, mencionado anteriormente). Otro método más de espectroscopia analítica ilustrativa para medir la estabilidad térmica es la espectroscopia de resonancia magnética nuclear (RMN) (véase, p. ej., van Mierlo y Steemsma, mencionado anteriormente).
La estabilidad térmica de una composición se puede medir por medios bioquímicos. Un método bioquímico ilustrativo para evaluar la estabilidad térmica es un ensayo de exposición térmica. En un "ensayo de exposición térmica", se somete una composición a un intervalo de temperaturas elevadas durante un periodo de tiempo establecido. Por ejemplo, en un caso, las moléculas scFv de prueba o las moléculas que comprenden moléculas de scFv se someten a un intervalo de temperaturas crecientes, por ejemplo, durante 1-1,5 horas. La actividad de la proteína se somete a ensayo a continuación con un ensayo bioquímico relevante. Por ejemplo, si la proteína es una proteína de unión (p. ej., un scFv o un polipéptido que contiene scFv), la actividad de unión de la proteína de unión se puede determinar mediante un ELISA funcional o cuantitativo.
Un ensayo de este tipo se puede realizar en un formato de alto rendimiento y en los divulgados en los ejemplos utilizando
E. coli y cribado de alto rendimiento. Se puede crear una biblioteca de dominios de unión a antígenos, p. ej., que incluya un dominio de unión a antígenos para un antígeno asociado al cáncer descrito en esta memoria, p. ej., variantes de scFv, utilizando métodos conocidos en la técnica. Se puede inducir la expresión del dominio de unión a antígenos, p. ej., para un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, y el dominio de unión a antígenos, p. ej., para un antígeno asociado al cáncer descrito en el presente memoria, p. ej., scFv, se puede someter a exposición térmica. Las muestras de prueba estimuladas pueden analizarse para determinar la unión y los dominios de unión a antígenos para un antígeno asociado al cáncer descritos en esta memoria, p. ej., scFv, que son estables, pueden ampliarse y caracterizarse adicionalmente.
La estabilidad térmica se evalúa midiendo la temperatura de fusión (Tm) de una composición utilizando cualquiera de las técnicas anteriores (p. ej., técnicas de espectroscopia analítica). La temperatura de fusión es la temperatura en el punto medio de una curva de transición térmica en donde el 50 % de las moléculas de una composición están en estado plegado (véase, p. ej., Dimasi et al. (2009) J. Mol Biol. 393: 672-692). En un caso, los valores de Tm para un dominio de unión a antígenos a un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv son aproximadamente 40 °C, 41 °C, 42 °C, 43 °C, 44 °C, 45 °C, 46 °C, 47 °C, 48 °C, 49 °C, 50 °C, 51 °C, 52 °C, 53 °C, 54 °C, 55 °C, 56 °C, 57
°C, 74 °C, 91 93 °C, 94 °C, 95 °C, 96 °C, 97 °C, 98 °C, 99 °C, 100 °C. En un caso, los valores de Tm para una IgG es aproximadamente
40 °C, 41 °C, 42 °C, 43 °C, 44 °C, 45 °C, 46 °C, 47 °C, 48 °C, 49 °C, 50 °C, 51 °C, 52 °C, 53 °C, 54 °C, 55 °C, 56 °C, 57 °C, 58 °C, 59 °C, 60 °C, 61 °C, 62 °C, 63 °C, 64 °C, 65 °C, 66 °C, 67 °C, 68 °C, 69 °C, 70 °C, 71 °C, 72 74 °C, 75 °C, 76 °C, 77 °C, 78 °C, 79 °C, 80 °C, 81 °C, 82 °C, 83 °C, 84 °C, 85 °C, 86 °C, 87 °C, 88 °C, 89 91 °C, 92 °C, 93 °C, 94 °C, 95 °C, 96 °C, 97 °C, 98 °C, 99 °C, 100 °C. En un caso, los valores de Tm para un anticuerpo multivalente es aproximadamente 40 °C, 41 °C, 42 °C, 43 °C, 44 °C, 45 °C, 46 °C, 47 °C, 48 °C, 49 °C, 50 °C, 51 °C, 52 °C, 53 °C, 54 °C, 55 °C, 56 °C, 57 °C, 58 °C, 59 °C, 60 °C, 61 °C, 62 °C, 63 °C, 64 °C, 65 °C, 66 °C, 67 69 °C, 70 °C, 71 °C, 72 °C, 73 °C, 74 °C, 75 °C, 76 °C, 77 °C, 78 °C, 79 °C, 80 °C, 81 °C, 82 °C, 83 °C, 84 86 °C, 87 °C, 88 °C, 89 °C, 90 °C, 91 °C, 92 °C, 93 °C, 94 °C, 95 °C, 96 °C, 97 °C, 98 °C, 99 °C, 100 °C.
La estabilidad térmica también se evalúa midiendo el calor específico o la capacidad calorífica (Cp) de una composición utilizando una técnica calorimétrica analítica (p. ej., DSC). El calor específico de una composición es la energía (p. ej., en kcal/mol) necesaria para aumentar en 1 °C la temperatura de 1 mol de agua. Como Cp grande es un rasgo característico de una composición proteica desnaturalizada o inactiva. El cambio en la capacidad calorífica (ACp) de una composición se mide determinando el calor específico de una composición antes y después de su transición térmica. La estabilidad térmica también se puede evaluar midiendo o determinando otros parámetros de estabilidad termodinámica, incluida la energía libre de Gibbs de desplegamiento (AG), la entalpía de desplegamiento (AH) o la entropía de desplegamiento (AS).
Uno o más de los ensayos bioquímicos anteriores (p. ej., un ensayo de desafío térmico) se utilizan para determinar la temperatura (es decir, el valor Tc ) en el que el 50% de la composición retiene su actividad (p. ej., actividad de unión).
Además, se pueden realizar mutaciones en el dominio de unión a antígenos de un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, para alterar la estabilidad térmica del dominio de unión a antígenos de un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, en comparación con el dominio de unión a antígenos no mutado de un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv. Cuando el dominio de unión a antígenos humanizado de un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, se incorpora a una construcción de CAR, el dominio de unión a antígenos del antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv humanizado, confiere estabilidad térmica a los CARs globales utilizados en la presente invención. En una realización, el dominio de unión a antígenos a un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, comprende una única mutación que confiere estabilidad térmica al dominio de unión a antígenos del antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv. En otra realización, el dominio de unión a antígenos para un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, comprende múltiples mutaciones que confieren estabilidad térmica al dominio de unión a antígenos al antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv. En una realización, las múltiples mutaciones en el dominio de unión a antígenos para un antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv, tienen un efecto aditivo sobre la estabilidad térmica del dominio de unión a antígenos para el dominio de unión a antígenos asociado al cáncer descrito en el presente memoria, p. ej., scFv.
b) % de Agregación
La estabilidad de una composición se puede determinar midiendo su propensión a agregarse. La agregación se puede medir mediante varias técnicas bioquímicas o biofísicas no limitantes. Por ejemplo, la agregación de una composición se puede evaluar utilizando cromatografía, p. ej., cromatografía de exclusión por tamaño (SEC, por sus siglas en inglés). La SEC separa moléculas en función del tamaño. Una columna se llena con perlas semisólidas de un gel polimérico que admitirá iones y moléculas pequeñas en su interior, pero no grandes. Cuando se aplica una composición proteica en la parte superior de la columna, las proteínas plegadas compactas (es decir, proteínas no agregadas) se distribuyen a través de un mayor volumen de disolvente que el que está disponible para los agregados de proteínas grandes. Por consiguiente, los agregados grandes se mueven con mayor rapidez a través de la columna, y de esta manera la mezcla se puede separar o fraccionar en sus componentes. Cada una de las fracciones se puede cuantificar por separado (p. ej., mediante
dispersión de la luz) a medida que se eluye del gel. Por consiguiente, el % de agregación de una composición se puede determinar comparando la concentración de una fracción con la concentración total de proteína aplicada al gel. Las composiciones estables eluyen de la columna como esencialmente una fracción única y aparecen esencialmente como un pico único en el perfil de elución o cromatograma.
c) Afinidad de unión
La estabilidad de una composición se puede evaluar determinando su afinidad de unión por la diana. En la técnica se conocen una amplia diversidad de métodos para determinar la afinidad de unión. Un método ilustrativo para determinar la afinidad de unión emplea resonancia de plasmones de superficie. La resonancia de plasmones de superficie es un fenómeno óptico que permite el análisis de interacciones bioespecíficas en tiempo real mediante la detección de alteraciones en las concentraciones de proteínas dentro de una matriz de biosensores, por ejemplo, utilizando el sistema BIAcore (Pharmacia Biosensor AB, Uppsala, Suecia y Piscataway, N.J.). Para descripciones más detalladas, véase Jonsson, U., et al. (1993) Ann. Biol. Clin. 51: 19-26; Jonsson, U., i (1991) Biotechniques 11: 620-627; Johnsson, B., et al. (1995) J. Mol. Recognit. 8: 125-131; y Johnnson, B., et al. (1991) Anal. Biochem. 198: 268-277.
En un aspecto, el dominio de unión a antígenos del CAR comprende una secuencia de aminoácidos que es homóloga de una secuencia de aminoácidos del dominio de unión a antígenos descrita en esta memoria y el dominio de unión a antígenos mantiene las propiedades funcionales deseadas del dominio de unión a antígenos descrito en esta memoria.
En un aspecto específico, la composición de CAR de la divulgación comprende un fragmento de anticuerpo. En un aspecto adicional, el fragmento de anticuerpo comprende un scFv.
En diversos aspectos, el dominio de unión a antígenos del CAR se altera modificando uno o más aminoácidos dentro de una o ambas regiones variables (p. ej., VH y/o VL), por ejemplo, dentro de una o más regiones CDR y/o dentro de una o más regiones marco conservadas. En un aspecto específico, la composición de CAR de la divulgación comprende un fragmento de anticuerpo. En un aspecto adicional, el fragmento de anticuerpo comprende un scFv.
Un experto ordinario en la técnica entenderá que el anticuerpo o el fragmento de anticuerpo utilizado en la invención puede modificarse adicionalmente de modo que varíen en la secuencia de aminoácidos (p. ej., del tipo salvaje), pero no en la actividad deseada. Por ejemplo, se pueden realizar sustituciones adicionales de nucleótidos en la proteína que dan lugar a sustituciones de aminoácidos en residuos aminoacídicos "no esenciales". Por ejemplo, un residuo de aminoácido no esencial en una molécula puede ser reemplazado por otro residuo de aminoácido de la misma familia de la cadena lateral. En otra realización, una cadena de aminoácidos se puede reemplazar por una cadena estructuralmente similar que difiere en orden y/o composición de los miembros de la familia de la cadena lateral, p. ej., puede realizarse una sustitución conservadora, en la que un residuo aminoacídico se reemplaza por un residuo aminoacídico que tiene una cadena lateral similar.
En la técnica se han definido las familias de residuos aminoacídicos que tienen cadenas laterales similares, que incluyen las cadenas laterales básicas (p. ej., lisina, arginina, histidina), cadenas laterales ácidas (p. ej., ácido aspártico, ácido glutámico), cadenas laterales polares sin carga (p. ej., glicina, asparagina, glutamina, serina, treonina, tirosina, cisteína), cadenas laterales no polares (p. ej., alanina, valina, leucina, isoleucina, prolina, fenilalanina, metionina, triptófano), cadenas laterales con ramificaciones en la posición beta (p. ej., treonina, valina, isoleucina) y cadenas laterales aromáticas (p. ej., tirosina, fenilalanina, triptófano, histidina).
El porcentaje de identidad en el contexto de dos o más ácidos nucleicos o secuencias polipeptídicas se refiere a dos o más secuencias que son iguales. Dos secuencias son "sustancialmente idénticas" si dos secuencias tienen un porcentaje especificado de residuos aminoácidos o nucleótidos que son iguales (p. ej., 60 % de identidad, opcionalmente 70 %, 71 %.
72 %. 73 %, 74 %, 75 %, 76 %, 77 %, 78 %, 79 %, 80 %, 81 %, 82 %, 83 %, 84 %, 85 %, 86 %, 87 %, 88 %, 89 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % de identidad a lo largo de una región específica o, cuando no se especifica, a lo largo de toda la secuencia), cuando se compara y se alinea para obtener la correspondencia máxima en una ventana de comparación, o región designada como medida utilizando uno de los siguientes algoritmos de comparación de secuencias o mediante alineamiento manual e inspección visual. Opcionalmente, la identidad existe en una región que tiene al menos aproximadamente 50 nucleótidos (o 10 aminoácidos) de longitud, o más preferiblemente en una región que tiene de 100 a 500 o 1000 o más nucleótidos (o 20, 50, 200 o más aminoácidos) de longitud.
Para la comparación de secuencias, habitualmente una secuencia actúa como secuencia de referencia respecto a la cual se comparan las secuencias de prueba. Cuando se utiliza un algoritmo de comparación de secuencias, las secuencias de prueba y de referencia se introducen en una computadora, se designan las coordenadas de la subsecuencia, si es necesario, y se designan los parámetros del programa del algoritmo de secuencia. Pueden usarse parámetros por defecto del programa o pueden indicarse parámetros alternativos. A continuación, el algoritmo de comparación de secuencias calcula el porcentaje de identidades de secuencia para las secuencias de prueba en relación con la secuencia de referencia, basándose en los parámetros del programa. Son bien conocidos en la técnica métodos de alineación de secuencias para comparación. El alineamiento óptimo de secuencias para comparación puede realizarse, p. ej., mediante el algoritmo de homología local de Smith y Waterman, (1970) Adv. Appl. Math. 2:482c, mediante el algoritmo de alineación
de homología de Wunsch, (1970) J. Mol. Biol. 48: 443, mediante el método de la búsqueda similitud de Pearson y Lipman (1988) Proc. Nat'l. Acad. Sci. USA 85: 2444, mediante implementaciones computarizadas de estos algoritmos (GAP, BESTFIT, FASTA y TFASTA en el paquete de programas informáticos de Wisconsin Genetics, Genetics Computer Group, 575 Science Dr., Madison, WI) o mediante alineación manual e inspección visual (véase, p. ej., Brent et al., (2003) Current Protocols in Molecular Biology).
Dos ejemplos de algoritmos que son adecuados para determinar el porcentaje de identidad secuencial y similitud secuencial son los algoritmos BLAST y BLAST 2.0, los cuales se describen en Altschul et al. (1977) Nuc. Acids Res. 25: 3389-3402; y Altschul et al., (1990) J. Mol. Biol. 215: 403-410, respectivamente. El programa informático para realizar análisis BLAST está a disposición del público a través del Centro Nacional de Información Biotecnológica.
El porcentaje de identidad entre dos secuencias de aminoácidos también se puede determinar utilizando el algoritmo de E. Meyers y W. Miller, (1988) Comput. Appl. Biosci. 4: 11-17) que se ha incorporado al programa ALIGN (versión 2.0), utilizando una tabla de pesos de residuos PAM120, una penalización de longitud de hueco de 12 y una penalización de hueco de 4. Además, el porcentaje de identidad entre dos secuencias de aminoácidos se puede determinar utilizando el algoritmo de Needleman y Wunsch (1970) J. Mol. Biol. 48: 444-453) que se ha incorporado al programa GAP en el paquete informático GCG (disponible en www.gcg.com), utilizando una matriz Blossom 62 o una matriz PAM250, y un peso de hueco de 16, 14, 12, 10, 8, 6 o 4 y un peso de longitud de 1, 2, 3, 4, 5 o 6.
En un aspecto, la presente invención contempla modificaciones de la secuencia de aminoácidos del anticuerpo o fragmento (p. ej., scFv) de partida que generan moléculas funcionalmente equivalentes. Por ejemplo, el VH o VL de un dominio de unión de antígeno a un antígeno asociado con cáncer descrito en esta memoria, p. ej., scFv, comprendido en el CAR puede modificarse para retener al menos aproximadamente 70 %, 71 %, 72%,. 73 %, 74 %, 75 %, 76 %, 77 %, 78 %, 79 %, 80 %, 81 %, 82 %, 83 %, 84 %, 85 %, 86 %, 87 %, 88 %, 89 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % de identidad de la región marco conservada inicial VH o VL del dominio de unión a antígenos con el antígeno asociado al cáncer descrito en esta memoria, p. ej., scFv. La presente invención contempla modificaciones de la construcción del CAR completa, p. ej., modificaciones en una o más secuencias de aminoácidos de los diversos dominios de la construcción de CAR para generar moléculas funcionalmente equivalentes. La construcción de CAR se puede modificar para mantener una identidad de al menos aproximadamente un 70 %, 71 %. 72 %. 73 %, 74 %, 75 %, 76 %, 77 %, 78 %, 79 %, 80 %, 81 %, 82 %, 83 %, 84 %, 85 %, 86 %, 87 %, 88 %, 89 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % de identidad de la construcción de CAR inicial.
Dominio transmembrana
Con respecto al dominio transmembrana, en diversas realizaciones, un CAR puede diseñarse para comprender un dominio transmembrana que está unido al dominio extracelular del CAR. Un dominio transmembrana puede incluir uno o más aminoácidos adicionales adyacentes a la región transmembrana, p. ej., uno o más aminoácidos asociados con la región extracelular de la proteína de la que procede la transmembrana (p. ej., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 hasta 15 aminoácidos de la región extracelular) y/o uno o más aminoácidos adicionales asociados con la región intracelular de la proteína de la que procede la proteína transmembrana (p. ej., 1,2, 3, 4, 5, 6, 7, 8, 9, 10 hasta 15 aminoácidos de la región intracelular). En un aspecto, el dominio transmembrana es uno que está asociado con uno de los otros dominios del CAR, p. ej., en una realización, el dominio transmembrana puede ser de la misma proteína de la que procede el dominio de señalización, el dominio coestimulante o el dominio bisagra. En otro aspecto, el dominio transmembrana no procede de la misma proteína de la que procede cualquier otro dominio del CAR. En algunos casos, el dominio transmembrana puede seleccionarse o modificarse mediante sustitución de aminoácidos para evitar la unión de dominios de este tipo a los dominios transmembrana de las mismas proteínas de membrana de superficie o diferentes, p. ej., para minimizar las interacciones con otros miembros del complejo receptor. En un aspecto, el dominio transmembrana tiene capacidad de homodimerización con otro CAR en la superficie celular de una célula que expresa CAR. En un aspecto diferente, la secuencia de aminoácidos del dominio transmembrana puede modificarse o sustituirse con el fin de minimizar las interacciones con los dominios de unión del compañero de unión nativo presente en la misma célula que expresa el CAR.
El dominio transmembrana puede proceder de una fuente natural o recombinante. Cuando la fuente es natural, el dominio puede derivar de cualquier proteína unida a la membrana o transmembrana. En un aspecto, el dominio transmembrana es capaz de señalizar al dominio o dominios intracelulares siempre que el CAR se haya unido a una diana. Un dominio transmembrana de uso particular en esta invención puede incluir al menos la o las regiones transmembrana de, p. ej., la cadena alfa, beta o zeta del receptor de célula T, CD28, CD27, CD3 épsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137, CD154. En algunas realizaciones, un dominio transmembrana puede incluir al menos la(s) región(regiones) transmembrana de, por ejemplo, KIRDS2, OX40, CD2, CD27, LFA-1 (CD11a, CD18), ICOS (CD278), 4-1BB (CD137), GITR, CD40, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, IL2R beta, IL2R gamma, IL7R a, ITGA1, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, TNFR2, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), SLAMF6 (NTB-A, Ly108), SLAM (SLAMF1, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, PAG/Cbp, NKG2D, NKG2C.
En algunos casos, el dominio transmembrana se puede unir a la región extracelular del CAR, por ejemplo, el dominio de unión al antígeno del CAR, a través de una bisagra, por ejemplo, una bisagra de una proteína humana. Por ejemplo, en una realización, la bisagra puede ser una bisagra de Ig (inmunoglobulina) humana (p. ej., una bisagra de IgG4, una bisagra de IgD), un enlazador GS (p. ej., un enlazador GS descrito en esta memoria), una bisagra KIR2DS2 o una bisagra CD8a. En una realización, la bisagra o el espaciador comprende (p. ej., consiste en) la secuencia de aminoácidos de la SEQ ID NO: 4. En un aspecto, el dominio transmembrana comprende (p. ej., consiste en) un dominio transmembrana de la SEQ ID NO: 12.
En un aspecto, la bisagra o el espaciador comprende una bisagra de IgG4. Por ejemplo, en una realización, la bisagra o el espaciador comprende una bisagra de la secuencia de aminoácidos ESKYGPPCPPCPAPEFLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQFN WYVDGVEVHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPS SIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQP ENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFSCSVMHEALHNHYTQKSLS LSLGKM (SEQ ID NO:6). En algunas realizaciones, la bisagra o el espaciador comprende una bisagra codificada por una secuencia de nucleótidos de GAGAGCAAGTACGGCCCTCCCTGCCCCCCTTGCCCTGCCCCCGAGTTCCTGGGC GGACCCAGCGTGTTCCTGTTCCCCCCCAAGCCCAAGGACACCCTGATGATCAGC CGGACCCCCGAGGTGACCTGTGTGGTGGTGGACGTGTCCCAGGAGGACCCCGA GGTCC AGTT C AACTGGT ACGT GGAC GGCGT GGAGGT GC AC AACGCC AAGACC A AGCCCCGGGAGGAGCAGTTCAATAGCACCTACCGGGTGGTGTCCGTGCTGACC GT GCTGC ACC AGGACTGGCTGAACGGC AAGGA AT AC AAGTGT AAGGT GTCC AA CAAGGGCCTGCCCAGCAGCATCGAGAAAACCATCAGCAAGGCCAAGGGCCAGC CTCGGGAGCCCCAGGTGTACACCCTGCCCCCTAGCCAAGAGGAGATGACCAAG AACCAGGTGTCCCTGACCTGCCTGGTGAAGGGCTTCTACCCCAGCGACATCGCC GTGGAGTGGGAGAGCAACGGCCAGCCCGAGAACAACTACAAGACCACCCCCCC TGTGCTGGACAGCGACGGCAGCTTCTTCCTGTACAGCCGGCTGACCGTGGACAA GAGCCGGT GGC AGGAGGGC AACGTCTTT AGCTGCTCCGT GAT GC ACGAGGCCC TGCACAACCACTACACCCAGAAGAGCCTGAGCCTGTCCCTGGGCAAGATG (SEQ
ID NO:7).
En un aspecto, la bisagra o el espaciador comprende una bisagra de IgD. Por ejemplo, en una realización, la bisagra o el espaciador comprende una bisagra de la secuencia de aminoácidos RWPESPKAQASSVPTAQPQAEGSLAKATTAPATTRNTGRGGEEKKKEKEKEEQEE RETKTPECPSHTQPLGVYLLTPAVQDLWLRDKATFTCFVVGSDLKDAHLTWEVAG KVPTGGVEEGLLERHSNGSQSQHSRLTLPRSLWNAGTSVTCTLNHPSLPPQRLMAL REPAAQAPVKLSLNLLASSDPPEAASWLLCEVSGFSPPNILLMWLEDQREVNTSGF APARPPPQPGSTTFWAWSVLRVPAPPSPQPATYTCVVSHEDSRTLLNASRSLEVSYV TDH (SEQ ID NO:8). En algunas realizaciones, la bisagra o el espaciador comprende una bisagra codificada por una secuencia de nucleótidos de AGGTGGCCCGAAAGTCCCAAGGCCCAGGCATCTAGTGTTCCTACTGCACAGCCC CAGGCAGAAGGCAGCCTAGCCAAAGCTACTACTGCACCTGCCACTACGCGCAA TACTGGCCGTGGCGGGGAGGAGAAGAAAAAGGAGAAAGAGAAAGAAGAACAG GAAGAGAGGGAGACCAAGACCCCTGAATGTCCATCCCATACCCAGCCGCTGGG CGTCTATCTCTTGACTCCCGCAGTACAGGACTTGTGGCTTAGAGATAAGGCCAC CTTTACATGTTTCGTCGTGGGCTCTGACCTGAAGGATGCCCATTTGACTTGGGA
GGTTGCCGGAAAGGTACCCACAGGGGGGGTTGAGGAAGGGTTGCTGGAGCGCC ATTCCAATGGCTCTCAGAGCCAGCACTCAAGACTCACCCTTCCGAGATCCCTGT GGAACGCCGGGACCTCTGTCACATGTACTCTAAATCATCCTAGCCTGCCCCCAC AGCGTCTGATGGCCCTTAGAGAGCCAGCCGCCCAGGCACCAGTTAAGCTTAGCC TGAATCTGCTCGCCAGTAGTGATCCCCCAGAGGCCGCCAGCTGGCTCTTATGCG AAGTGTCCGGCTTTAGCCCGCCCAACATCTTGCTCATGTGGCTGGAGGACCAGC GAGAAGTGAACACCAGCGGCTTCGCTCCAGCCCGGCCCCCACCCCAGCCGGGT TCTACCACATTCTGGGCCTGGAGTGTCTTAAGGGTCCCAGCACCACCTAGCCCC CAGCCAGCCACATACACCTGTGTTGTGTCCCATGAAGATAGCAGGACCCTGCTA AATGCTTCTAGGAGTCTGGAGGTTTCCTACGTGACTGACCATT (SEQ ID NO:9).
En un aspecto, el dominio transmembrana puede ser recombinante, en cuyo caso comprenderá predominantemente residuos hidrófobos, tales como leucina y valina. En un aspecto, se puede encontrar un triplete de fenilalanina, triptófano y valina en cada extremo de un dominio transmembrana recombinante.
Opcionalmente, un enlazador oligo o polipeptídico corto, de entre 2 y 10 aminoácidos de longitud, puede formar el enlace entre el dominio transmembrana y la región citoplasmática del CAR. Un doblete de glicina-serina proporciona un enlazador particularmente adecuado. Por ejemplo, en un aspecto, el enlazador comprende la secuencia de aminoácidos de GGGGSGGGGS (SEQ ID NO: 10). En algunas realizaciones, el enlazador está codificado por una secuencia de nucleótidos de GGTGGCGGAGGTTCTGGAGGTGGAGGTTCC (SEQ ID NO: 11).
En un aspecto, la bisagra o el espaciador comprende una bisagra de KIR2DS2.
Dominio citoplasmático
El dominio o la región citoplásmica del CAR incluye un dominio de señalización intracelular. Un dominio de señalización intracelular es generalmente responsable de la activación de al menos una de las funciones efectoras normales de la célula inmunitaria en la que se ha introducido el CAR. La expresión "función efectora" se refiere a una función especializada de una célula. La función efectora de una célula T, por ejemplo, puede ser actividad citolítica o actividad auxiliar, incluida la secreción de citoquinas. Por lo tanto, la expresión "dominio de señalización intracelular" se refiere a la porción de una proteína que transduce la señal de la función efectora y dirige a la célula para realizar una función especializada. Aunque habitualmente se puede emplear todo el dominio de señalización intracelular, en muchos casos no es necesario utilizar la cadena completa. En la medida en que se utilice una porción truncada del dominio de señalización intracelular, dicha porción truncada se puede utilizar en lugar de la cadena intacta, siempre que transduzca la señal de la función efectora. Por tanto, la expresión dominio de señalización intracelular pretende incluir cualquier parte truncada del dominio de señalización intracelular suficiente para transducir la señal de función efectora.
Ejemplos de dominios de señalización intracelular para uso en el CAR empleados en la invención incluyen las secuencias citoplásmicas del receptor de células T (TCR) y los co-receptores que actúan en conjunto para iniciar la transducción de señales después de la participación del receptor de antígeno, así como cualquier derivado o variante de estas secuencias y cualquier secuencia recombinante que tenga la misma capacidad funcional.
Se sabe que las señales generadas a través del TCR solo son insuficientes para la activación completa de la célula T y que también se requiere una señal secundaria y/o coestimuladora. Por lo tanto, se puede decir que la activación de células T está mediada por dos clases distintas de secuencias de señalización citoplasmáticas: las que inician la activación primaria dependiente de antígeno a través de los TCR (dominios de señalización intracelular primarios) y los que actúan de una manera independiente del antígeno para proporcionar una señal secundaria o coestimulante (dominio citoplasmático secundario, p. ej., un dominio coestimulante).
Un dominio de señalización primario regula la activación primaria del complejo TCR ya sea de una manera estimulante o de una manera inhibidora. Los dominios de señalización intracelular primarios que actúan de una manera estimulante pueden contener motivos de señalización que se conocen como motivos de activación de inmunorreceptores basados en tirosina o ITAM.
Ejemplos de dominios de señalización intracelular primarios que contienen ITAM que son de uso particular en la invención incluyen los de CD3 zeta, FcR gamma común (FCER1G), Fc gamma RIIa, FcR beta (Fc Épsilon R1b), CD3 gamma, CD3 delta, CD3 épsilon, CD79a, CD79b, DAP10 y DAP12. En una realización, un CAR empleado en la invención comprende un dominio de señalización intracelular, p. ej., un dominio de señalización primario de CD3-zeta.
En una realización, un dominio de señalización principal comprende un dominio de ITAM modificado, p. ej., un dominio de ITAM mutado que tiene actividad alterada (p. ej., aumentada o disminuida) en comparación con el dominio de ITAM nativo. En una realización, un dominio de señalización primario comprende un dominio de señalización intracelular primario que contiene ITAM modificado, p. ej., un dominio de señalización intracelular primario que contiene ITAM optimizado y/o truncado. En una realización, un dominio de señalización primario comprende uno, dos, tres, cuatro o más motivos ITAM.
El dominio de señalización intracelular del CAR puede comprender el dominio de señalización de CD3-zeta por sí mismo o puede combinarse con cualquier otro dominio de señalización intracelular deseado útil en el contexto de un CAR empleado en la invención. Por ejemplo, el dominio de señalización intracelular de CAR puede comprender una porción de cadena CD3 zeta y un dominio de señalización coestimulador. El dominio de señalización coestimulador se refiere a una porción del CAR que comprende el dominio intracelular de una molécula coestimulante. Una molécula coestimulante es una molécula de la superficie celular distinta de un receptor de antígeno o sus ligandos que se requiere para una respuesta eficaz de células frente a un antígeno. Los ejemplos de moléculas de este tipo incluyen CD27, CD28, 4-1BB (CD137), OX40, CD30, CD40, PD-1, ICOS, antígeno asociado a la función linfocítica-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-H3, y un ligando que se une específicamente con CD83, y similares. Por ejemplo, se ha demostrado que la coestimulación de CD27 potencia la expansión, función efectora y supervivencia de células CART humanos in vitro y aumenta la persistencia de célula T humanos y actividad antitumoral in vivo (Song et al. Blood. 2012; 119(3):696-706). Ejemplos adicionales de moléculas coestimuladoras de este tipo incluyen CDS, ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, CD4, CD8alpha, CD8beta, IL2R beta, IL2R gamma, IL7R alfa, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, TNFR2, TRANCE/RANKL, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), NKG2D, CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), CD69, SLAMF6 (NTB-A, Ly108), SLAM (SLAMF1, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, LAT, GADS, SLP-76, PAG/Cbp y CD19a.
Las secuencias de señalización intracelular dentro de la porción citoplásmica del CAR empleado en la invención se pueden unir entre sí en un orden aleatorio o especificado. Opcionalmente, un enlazador de oligopéptido o polipéptido corto, por ejemplo, entre 2 y 10 aminoácidos (p. ej., 2, 3, 4, 5, 6, 7, 8, 9 o 10 aminoácidos) de longitud puede formar el enlace entre secuencia de señalización intracelular. En una realización, se puede utilizar un doblete de glicina-serina como un enlazador adecuado. En una realización, se puede utilizar un único aminoácido, p. ej., una alanina, una glicina, como un enlazador adecuado.
En un aspecto, el dominio de señalización intracelular está diseñado para comprender dos o más, p. ej., 2, 3, 4, 5 o más, dominios de señalización coestimulantes. En una realización, los dos o más, p. ej., 2, 3, 4, 5 o más, dominios de señalización coestimulantes están separados por una molécula enlazadora, p. ej., una molécula enlazadora descrita en esta memoria. En una realización, el dominio de señalización intracelular comprende dos dominios de señalización coestimulantes. En algunas realizaciones, la molécula enlazadora es un residuo de glicina. En algunas realizaciones, el enlazador es un residuo de alanina.
En un aspecto, el dominio de señalización intracelular está diseñado para comprender el dominio de señalización de CD3-zeta y el dominio de señalización de CD28. En un aspecto, el dominio de señalización intracelular está diseñado para comprender el dominio de señalización de CD3-zeta y el dominio de señalización de 4-1BB. En un aspecto, el dominio de señalización de 4-1BB es un dominio de señalización de la SEQ ID NO: 14. En un aspecto, el dominio de señalización de CD3-zeta es un dominio de señalización de la SEQ ID NO: 18.
En un aspecto, el dominio de señalización intracelular está diseñado para comprender el dominio de señalización de CD3-zeta y el dominio de señalización de CD27. En un aspecto, el dominio de señalización de CD27 comprende una secuencia de aminoácidos de QRRKYRSNKGESPVEPAEPCRYSCPREEEGSTIPIQEDYRKPEPACSP (SEQ ID NO: 16). En un aspecto, el dominio de señalización de CD27 está codificado por una secuencia de ácido nucleico de AGGAGTAAGAGGAGCAGGCTCCTGCACAGTGACTACATGAACATGACTCCCCG CCGCCCCGGGCCCACCCGCAAGCATTACCAGCCCTATGCCCCACCACGCGACTT CGCAGCCTATCGCTCC (SEQ ID NO:17).
En la presente invención, la célula que expresa CAR comprende un segundo CAR que incluye un dominio de unión a antígenos diferente, p. ej., a la misma diana o a una diana diferente (p. ej., una diana que no sea un antígeno asociado al cáncer descrito en esta memoria o un antígeno diferente asociado al cáncer descrito en esta memoria). En una realización, el segundo CAR incluye un dominio de unión a antígenos para una diana que expresa el mismo tipo de célula cancerosa que el antígeno asociado al cáncer. En una realización, la célula que expresa CAR comprende un primer CAR que se dirige a un primer antígeno e incluye un dominio de señalización intracelular que tiene un dominio de señalización coestimulante, pero no un dominio de señalización primario, y un segundo CAR que se dirige a un segundo antígeno diferente e incluye un dominio de señalización intracelular que tiene un dominio de señalización primario, pero no un dominio de señalización coestimulante. Sin pretender quedar ligados a la teoría, la colocación de un dominio de señalización coestimulante, p. ej., 4-1BB, CD28, CD27 u OX-40, en el primer CAR, y el dominio de señalización primario,
p. ej., CD3 zeta, en el segundo CAR puede limitar la actividad del CAR a las células donde se expresan ambas dianas. En una realización, la célula que expresa CAR comprende un primer antígeno CAR asociado al cáncer que incluye un dominio de unión a antígenos que se une a un antígeno diana descrito en esta memoria, un dominio transmembrana y un dominio coestimulante y un segundo CAR que se dirige a un antígeno diana diferente (p. ej., un antígeno expresado en el mismo tipo de célula cancerosa que el primer antígeno diana) e incluye un dominio de unión a antígenos, un dominio transmembrana y un dominio de señalización primario. En otra realización, la célula que expresa CAR comprende un primer CAR que incluye un dominio de unión a antígenos que se une a un antígeno diana descrito en esta memoria, un dominio transmembrana y un dominio de señalización primario y un segundo CAR que se dirige a un antígeno distinto del primer antígeno diana (p. ej., un antígeno expresado en el mismo tipo de célula cancerosa que el primer antígeno diana) e incluye un dominio de unión a antígenos para el antígeno, un dominio transmembrana y un dominio de señalización coestimulante.
En una realización, la célula que expresa CAR comprende un XCAR descrito en esta memoria y un CAR inhibidor. En una realización, el CAR inhibidor comprende un dominio de unión a antígenos que se une a un antígeno presente en células normales pero no en células cancerosas, p. ej., células normales que también expresan LLC. En una realización, el CAR inhibidor comprende el dominio de unión a antígenos, un dominio transmembrana y un dominio intracelular de una molécula inhibidora. Por ejemplo, el dominio intracelular del CAR inhibidor puede ser un dominio intracelular de PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 o TGFR beta.
En la presente invención, la célula que expresa CAR comprende dos o más CARs diferentes y los dominios de unión a antígenos de los diferentes CARs pueden ser tales que los dominios de unión a antígenos no interactúen entre sí. Por ejemplo, una célula que expresa un primer y un segundo CAR puede tener un dominio de unión al antígeno del segundo CAR como un scFv que no forma una asociación con el dominio de unión al antígeno del primer CAR que es un VHH.
En algunos casos, el dominio de unión al antígeno comprende moléculas de unión al antígeno de dominio único (SDAB, por sus siglas en inglés) e incluye moléculas cuyas regiones determinantes de la complementariedad son parte de un polipéptido de dominio único. Los ejemplos incluyen, sin carácter limitante, dominios variables de la cadena pesada, moléculas de unión desprovistas de manera natural de cadenas ligeras, dominios únicos derivados de anticuerpos de 4 cadenas convencionales, dominios modificados y esqueletos de dominio único diferentes de aquellos derivados de anticuerpos. Las moléculas SDAB pueden ser cualesquiera de la técnica, o cualesquiera moléculas de dominio único futuras. Las moléculas SDAB pueden derivarse de cualquier especie que incluye, sin carácter limitante, ratón, ser humano, camello, llama, lamprea, pez, tiburón, cabra, conejo y bóvidos. El término también incluye moléculas de tipo anticuerpo de dominio único de especies diferentes de los tiburones y Camelidae.
En un caso, una molécula SDAB se puede derivar de una región variable de la inmunoglobulina que se encuentra en los peces, como, por ejemplo, la que se deriva del isotipo de inmunoglobulina conocido como Receptor de Antígeno Nuevo (NAR, por sus siglas en inglés) que se encuentra en el suero. de tiburón Métodos para producir moléculas de un solo dominio derivadas de una región variable de NAR ("IgNAR") se describen en el documento WO 03/014161 y Streltsov (2005) Protein Sci. 14:2901-2909 .
Según otro aspecto, una molécula SDAB es una molécula de unión al antígeno de dominio único de origen natural conocida como cadena pesada desprovista de cadenas ligeras. Moléculas de dominio único de este tipo se describen en el documento WO 9404678 y en Hamers-Casterman, C. et al. (1993) Nature 363: 446-448, por ejemplo. A efectos de claridad, este dominio variable derivado de una molécula de cadena pesada desprovista de manera natural de la cadena ligera se conoce en esta memoria como un VHH o nanocuerpo para distinguirlo del VH convencional de inmunoglobulinas de cuatro cadenas. Una molécula VHH de este tipo puede proceder de una especie de Camelidae, por ejemplo, en camello, llama, dromedario, alpaca y guanaco. Otras especies además de Camelidae pueden producir moléculas de cadena pesada desprovistas de forma natural de cadena ligera; las VHH de este tipo están dentro del alcance de la invención.
Las moléculas de SDAB pueden ser recombinantes, injertadas con CDR, humanizadas, camellizadas, desinmunizadas y/o generadas in vitro (p. ej., seleccionadas por presentación en fagos).
También se ha descubierto que las células que tienen una pluralidad de receptores quiméricos embebidos en la membrana que comprenden un dominio de unión a antígenos que interactúa entre el dominio de unión a antígenos de los receptores pueden ser indeseables, p. ej., porque inhiben la capacidad de uno o más de los dominios de unión de unirse a su antígeno afín. Por consiguiente, en esta memoria se describen células que tienen un primer y un segundo receptor quimérico incluido en la membrana de origen no natural que comprende dominios de unión a antígenos que minimizan interacciones de este tipo. También se divulgan en la presente ácidos nucleicos que codifican un primer y un segundo receptor integrado en la membrana quimérico de origen no natural que comprenden dominios de unión al antígeno que minimizan tales interacciones, así como también métodos para preparar y utilizar tales células y ácidos nucleicos. En un caso, el dominio de unión al antígeno de uno de dicho primer y dicho segundo receptor integrado en la membrana quimérico de origen no natural comprende un scFv y el otro comprende un dominio VH único, por ejemplo, un dominio Vh único de camélido, tiburón o lamprea, o un dominio VH único derivado de una secuencia humana (hu) o de ratón (ms).
En algunos casos, la divulgación comprende un primer y un segundo CAR, en donde el dominio de unión a antígenos de uno de dicho primer CAR y dicho segundo CAR no comprende un dominio ligero variable y un dominio pesado variable. En algunos casos, el dominio de unión al antígeno de uno de dicho primer CAR y dicho segundo CAR es un scFv y el otro no es un scFv. En algunos casos, el dominio de unión al antígeno de uno de dicho primer CAR y dicho segundo CAR comprende un dominio VH único, por ejemplo, un dominio VH único de camélido, tiburón o lamprea, o un dominio VH único derivado de una secuencia humana o de ratón. En algunos casos, el dominio de unión al antígeno de uno de dicho primer CAR y dicho segundo CAR comprende un nanocuerpo. En la presente invención, el dominio de unión a antígenos del primer cAr comprende un dominio VHH y el dominio de unión a antígenos del segundo CAR comprende un scFv. En algunas realizaciones, el dominio de unión a antígenos del primer CAR comprende un dominio VHH de camélido.
En algunos casos, el dominio de unión a antígenos de uno de dicho primer CAR, dicho segundo CAR comprende un scFv, y el otro comprende un único dominio VH, por ejemplo, un único dominio VH de camélido, tiburón o lamprea, o un único dominio VH derivado de una secuencia humana o de ratón. En algunos casos, el dominio de unión al antígeno de uno de dicho primer CAR y dicho segundo CAR es un scFv y el otro comprende un nanocuerpo. En algunas realizaciones, el dominio de unión a antígenos del segundo de dicho primer CAR y dicho segundo CAR comprende un scFv, y el otro comprende un dominio VHH de camélido.
En algunas realizaciones, cuando está presente en la superficie de una célula, la unión del dominio de unión a antígenos de dicho primer CAR a su antígeno afín no se ve reducida de manera sustancial por la presencia de dicho segundo CAR. En algunas realizaciones, la unión del dominio de unión al antígeno de dicho primer CAR a su antígeno afín en presencia de dicho segundo CAR es del 85 %, 90 %, 95 %, 96 %, 97 %, 98 % o 99 % de la unión del dominio de unión a antígenos de dicho primer CAR a su antígeno afín en ausencia de dicho segundo CAR
En algunas realizaciones, cuando están presentes en la superficie de una célula, los dominios de unión a antígenos de dicho primer CAR y dicho segundo CAR se asocian entre sí en menor medida que si ambos fueran dominios de unión a antígenos scFv. En algunas realizaciones, los dominios de unión a antígenos de dicho primer CAR y dicho segundo CAR se asocian entre sí un 85 %, 90 %, 95 %, 96 %, 97 %, 98 % o 99 % menos que si ambos fueran dominios de unión a antígenos scFv.
En otro aspecto, la célula que expresa CAR descrita en esta memoria puede expresar además otro agente, p. ej., un agente que potencia la actividad de una célula que expresa CAR. Por ejemplo, en una realización, el agente puede ser un agente que inhibe una molécula inhibidora. Las moléculas inhibidoras, p. ej., PD1, pueden, en algunas realizaciones, disminuir la capacidad de una célula que expresa CAR de montar una respuesta efectora inmunitaria. Ejemplos de moléculas inhibidoras incluyen PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEaCaM-5), LAG3, VISTA, BTLA, TIGiT, LAIR1, CD160, 2B4 y TGFR beta. En una realización, el agente que inhibe una molécula inhibidora, p. ej., es una molécula descrita en esta memoria, p. ej., un agente que comprende un primer polipéptido, p. ej., una molécula inhibidora, asociada con un segundo polipéptido que proporciona una señal positiva para la célula, p. ej., un dominio de señalización intracelular descrito en estamemoria. En una realización, el agente comprende un primer polipéptido, p. ej., de una molécula inhibidora tal como PD1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 y/o CeAcAm-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 o TGFR beta, o un fragmento de cualquiera de estos (p. ej., al menos una parte de un dominio extracelular de cualquiera de estos), y un segundo polipéptido que es un dominio de señalización intracelular descrito en esta memoria (p. ej., que comprende un dominio coestimulador (p. ej., 41BB, CD27 o CD28, p. ej., como se describe en esta memoria) y/o un dominio de señalización primario (p. ej., un dominio de señalización CD3 zeta descrito en esta memoria). En una realización, el agente comprende un primer polipéptido de PD1 o un fragmento del mismo (p. ej., al menos una parte de un dominio extracelular de PD1) y un segundo polipéptido de un dominio de señalización intracelular descrito en esta memoria (p. ej., un dominio de señalización de CD28 descrito en esta memoria y/o un dominio de señalización de CD3 zeta descrito en esta memoria). PD1 es un miembro inhibidor de la familia de receptores CD28 que también incluye CD28, CTLA-4, ICOS y BTLA. PD-1 se expresa en células B activados, células T y células mieloides (Agata et al. 1996 Int. Immunol 8:765-75). Se ha mostrado que dos ligandos para PD1, PD-L1 y PD-L2 regulan a la baja la activación de células T tras unirse a PD1 (Freeman et al. 2000 J Exp Med 192: 1027-34; Latchman et al. 2001 Nat Immunol 2: 261-8; Carter et al. 2002 Eur J Immunol 32: 634-43). PD-L1 es abundante en los cánceres humanos (Dong et al. 2003 J Mol Med 81: 281-7; Blank et al. 2005 Cancer Immunol. Immunother 54: 307-314; Konishi et al. 2004 Clin Cancer Res 10: 5094). La supresión inmunitaria puede invertirse inhibiendo la interacción local de PD1 con PD-L1.
En una realización, el agente comprende el dominio extracelular (ECD) de una molécula inhibidora, p. ej., Muerte Programada 1 (PD1), fusionada con un dominio transmembrana y dominios de señalización intracelular tales como 41BB y CD3 zeta (a los que también se alude en esta memoria como un CAR PD1). En una realización, el CAR PD1, cuando se utiliza en combinaciones con un XCAR descrito en esta memoria, mejora la persistencia del célula T. En una realización, el CAR es un CAR PD1 que comprende el dominio extracelular de PD1 indicado como subrayado en la SEQ ID NO: 26. En una realización, el CAR PD1 comprende la secuencia de aminoácidos de la SEQ ID NO: 26.
MalpvtalllplalllhaaiDPgwfldspdiDwnpptfspallvvtegdnatftcsfsntsesfvInwYrmspsn qtdklaafpedrsqpgqdcrfrvtqlpngrdfhmsvvrarrndsgtvlcgaislapkaqikeslraelrvterraevptahpspspr pagqfqtlvtttpaprpptpaptiasqplslrpeacrpaaggavhtrgldfacdiviwaplagtcgvlllslvitlvckrgrkkllvifk qpfmrpvqttqeedgcscrfpeeeeggcelrvkfsrsadapaykqgqnqlynelnlgrreeydvldkrrgrdpemggkprrkn pqeglynelqkdkmaeayseigmkgerrrgkghdglyqglstatkdtydalhmqalppr (SEQ ID NO:26).
En una realización, el CAR PD1 comprende la secuencia de aminoácidos proporcionada a continuación (SEQ ID NO: 39).
pgwfldspdrpwnpptfspallvvtegdnatftcsfsntsesfvlnwvrmspsnqtdklaafpedrsqpgqdcr frvtqlpngrdfhmsvvrarrndsgtvlcgaislapkaqikeslraelrvterraevptahpspsprpagqfqtlvtttpaprpptpa ptiasqplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitlyckrgrkkllyifkqpfmrpvqttqeedgcscrf peeeeggcelrvkfsrsadapaykqgqnqlynelnlgrreeydvldkrrgrdpemggkprrknpqeglynelqkdkmaeays eigmkgerrrgkghdglyqglstatkdtydalhmqalppr (SEQ ID NO:39).
En una realización, el agente comprende una secuencia de ácido nucleico que codifica el CAR PD1, p. ej., el CAR PD1 descrito en esta memoria. En una realización, la secuencia de ácido nucleico para el CAR PD1 se muestra a continuación, con el ECD PD1 subrayado a continuación en la SEQ ID NO: 27
atggccctccctgtcactgccctgcttctccccctcgcactcctgctccacgccgctagaccacccggatggtttctg gactctccggatcgcccgtggaatcccccaaccttctcaccggcactcttggttgtgactgagggcgataatgcgaccttcacgtgct cgttctccaacacctccgaatcattcgtgctgaactggtaccgcatgagcccgtcaaaccagaccgacaagctcgccgcgtttccgg aagatcggtcgcaaccgggacaggattgtcggttccgcgtgactcaactgccgaatggcagagacttccacatgagcgtggtccg cgctaggcgaaacgactccgggacctacctgtgcggagccatctcgctggcgcctaaggcccaaatcaaagagagcttgagggc cgaactgagagtgaccgagcgcagagctgaggtgccaactgcacatccatccccatcgcctcggcctgcggggcagtttcagacc ctggtcacgaccactccggcgccgcgcccaccgactccggccccaactatcgcgagccagcccctgtcgctgaggccggaagc atgccgccctgccgccggaggtgctgtgcatacccggggattggacttcgcatgcgacatctacatttgggctcctctcgccggaac ttgtggcgtgctccttctgtccctggtcatcaccctgtactgcaagcggggtcggaaaaagcttctgtacattttcaagcagcccttcat gaggcccgtgcaaaccacccaggaggaggacggttgctcctgccggttccccgaagaggaagaaggaggttgcgagctgcgcg tgaagttctcccggagcgccgacgcccccgcctataagcagggccagaaccagctgtacaacgaactgaacctgggacggcgg gaagagtacgatgtgctggacaagcggcgcggccgggaccccgaaatgggcgggaagcctagaagaaagaaccctcaggaag gcctgtataacgagctgcagaaggacaagatggccgaggcctactccgaaattgggatgaagggagagcggcggaggggaaa ggggcacgacggcctgtaccaaggactgtccaccgccaccaaggacacatacgatgccctgcacatgcaggcccttccccctcg
c (SEQ ID NO: 27).
En otro aspecto, la presente invención proporciona una población de células que expresan CAR, p. ej., células CART de acuerdo con las reivindicaciones. En algunos casos, la población de células que expresan CAR comprende una mezcla de células que expresan diferentes CAR. Por ejemplo, en un caso, la población de células CART puede incluir una primera célula que expresa un CAR que tiene un dominio de unión a antígenos a un antígeno asociado al cáncer descrito en esta memoria y una segunda célula que expresa un CAR que tiene un dominio de unión a antígenos diferente, p. ej., un dominio de unión a antígenos a un antígeno asociado al cáncer diferente descrito en esta memoria, p. ej., un dominio de unión a antígenos a un antígeno asociado al cáncer descrito en esta memoria que difiere del antígeno asociado al cáncer unido por el dominio de unión al antígeno del CAR expresado por la primera célula. Como otro ejemplo, la población de células que expresan CAR puede incluir una primera célula que expresa un CAR que incluye un dominio de unión a antígenos para un antígeno asociado al cáncer descrito en esta memoria y una segunda célula que expresa un CAR que incluye un dominio de unión de antígeno para una diana distinta de un antígeno asociado al cáncer descrito en esta memoria. En un caso, la población de células que expresan CAR incluye, p. ej., una primera célula que expresa un CAR que incluye un
dominio de señalización intracelular primario y una segunda célula que expresa un CAR que incluye un dominio de señalización secundario.
En otro aspecto, la presente invención proporciona una población de células de acuerdo con las reivindicaciones, en donde al menos una célula de la población expresa un CAR que tiene un dominio de unión a antígenos que se une a un antígeno asociado al cáncer como se describe en esta memoria, y una segunda célula que expresa otro agente, p. ej., un agente que potencia la actividad de una célula que expresa CAR. Por ejemplo, en una realización, el agente puede ser un agente que inhibe una molécula inhibidora. Las moléculas inhibidoras, p. ej., PD-1, pueden, en algunas realizaciones, disminuir la capacidad de una célula que expresa CAR de montar una respuesta efectora inmunitaria. Ejemplos de moléculas inhibidoras incluyen PD-1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEaCaM-5), LAG3, VISTA, BTLA, TIGiT, LAIR1, CD160, 2B4 y TGFR beta. En una realización, el agente que inhibe una molécula inhibidora, p. ej., es una molécula descrita en esta memoria, p. ej., un agente que comprende un primer polipéptido, p. ej., una molécula inhibidora, asociada con un segundo polipéptido que proporciona una señal positiva para la célula, p. ej., un dominio de señalización intracelular descrito en estamemoria. En una realización, el agente comprende un primer polipéptido, p. ej., de una molécula inhibidora tal como PD-1, PD-L1, CTLA4, TIM3, CEACAM (e.g., CEaCaM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 o TGFR beta, o un fragmento de cualquiera de estos, y un segundo polipéptido que es un dominio de señalización intracelular descrito en esta memoria (p. ej., que comprende un dominio coestimulador (p. ej., 41BB, CD27, OX40 o CD28, p. ej., como se describe en esta memoria) y/o un dominio de señalización primario (p. ej., un dominio de señalización zeta de CD3 descrito en esta memoria). En una realización, el agente comprende un primer polipéptido de PD-1 o un fragmento del mismo, y un segundo polipéptido de un dominio de señalización intracelular descrito en esta memoria (p. ej., un dominio de señalización de CD28 descrito en esta memoria y/o un dominio de señalización CD3 zeta descrito en esta memoria).
En un aspecto, la presente divulgación proporciona métodos que comprenden administrar una población de células que expresan CAR, p. ej., células CART, p. ej., una mezcla de células que expresan diferentes CAR, en combinación con otro agente, p. ej., un inhibidor de quinasa, tal como un inhibidor de quinasa descrito en esta memoria. En otro aspecto, la presente divulgación proporciona métodos que comprenden la administración de una población de células, en donde al menos una célula de la población expresa un CAR que tiene un dominio de unión a antígenos de un antígeno asociado al cáncer descrito en esta memoria, y una segunda célula que expresa otro agente, p. ej., un agente que potencia la actividad de una célula que expresa CAR, en combinación con otro agente, p. ej., un inhibidor de quinasa, tal como un inhibidor de quinasa descrito en esta memoria.
Receptores de antígenos quiméricos regulables
En algunas realizaciones, un CAR regulable (RCAR) donde la actividad CAR se puede controlar es deseable para optimizar la seguridad y eficacia de una terapia con CAR. Existen muchas formas de regular las actividades de CAR. Por ejemplo, la apoptosis inducible utilizando, p. ej., una caspasa fusionada con un dominio de dimerización (véase, p. ej., Di et al., N Egnl. J. Med. 3 de noviembre de 2011; 365 (18): 1673-1683), se puede utilizar como un interruptor de seguridad en la terapia con CAR de la presente invención. En un aspecto, un RCAR comprende un conjunto de polipéptidos, normalmente dos en las realizaciones más simples, en los que los componentes de un CAR convencional descrito en esta memoria, p. ej., un dominio de unión a antígenos y un dominio de señalización intracelular, se dividen en polipéptidos o miembros separados. En algunas realizaciones, el conjunto de polipéptidos incluye un conmutador de la dimerización que, tras la presencia de una molécula de dimerización, puede acoplar los polipéptidos entre sí, p. ej., puede acoplar un dominio de unión a antígenos a un dominio de señalización intracelular.
En un aspecto, un RCAR comprende dos polipéptidos o miembros: 1) un miembro de señalización intracelular que comprende un dominio de señalización intracelular, p. ej., un dominio de señalización intracelular primaria descrito en esta memoria, y un primer dominio de tipo interruptor; 2) un miembro de unión a antígenos que comprende un dominio de unión a antígenos, p. ej., que se dirige a un antígeno tumoral descrito en esta memoria, como se describe en esta memoria y un segundo dominio de tipo interruptor. Opcionalmente, el RCAR comprende un dominio transmembrana descrito en el presente memoria. En una realización, se puede disponer un dominio transmembrana en el miembro de señalización intracelular o el miembro de unión a antígenos, o en ambos. (A menos que se indique lo contrario, cuando se describen en esta memoria miembros o elementos de un RCAR, el orden puede ser según se proporciona, pero también se incluyen otros órdenes. Dicho de otra manera, en una realización el orden es como se expone en el texto, pero en otras realizaciones el orden puede ser diferente. P. ej., el orden de los elementos en un lado de la región transmembrana puede ser diferente del ejemplo, p. ej., la ubicación de un dominio de tipo interruptor respecto a un dominio de señalización intracelular puede ser diferente, p. ej., invertido).
En una realización, el primer y segundo dominios de tipo interruptor pueden formar un interruptor de dimerización intracelular o extracelular. En una realización, el interruptor de dimerización puede ser un interruptor de homodimerización, p. ej., donde el primer y segundo dominio de tipo interruptor son idénticos, o un interruptor de heterodimerización, p. ej., donde el primer y segundo dominios de tipo interruptor son diferentes entre sí.
En algunas realizaciones, un RCAR puede comprender un "multiinterruptor". Un multiinterruptor puede comprender dominios de tipo interruptor de heterodimerización o dominios de tipo interruptor de homodimerización. Un multiinterruptor
comprende una pluralidad de, p. ej., 2, 3, 4, 5, 6, 7, 8, 9 o 10, dominios de tipo interruptor, independientemente, en un primer miembro, p. ej., un miembro de unión a antígenos, y un segundo miembro, p. ej., un miembro de señalización intracelular. En una realización, el primer miembro puede comprender una pluralidad de primeros dominios de tipo interruptor, p. ej., dominios de tipo interruptor basados en FKBP, y el segundo miembro puede comprender una pluralidad de segundos dominios de tipo interruptor, p. ej., dominios de tipo interruptor basados en FRB. En una realización, el primer miembro puede comprender un primer y un segundo dominio de tipo interruptor, p. ej., un dominio de tipo interruptor basado en FKBP y un dominio de tipo interruptor basado en FRB, y el segundo miembro puede comprender un primer y un segundo dominio de tipo interruptor, p. ej., un dominio de tipo interruptor basado en FKBP y un dominio de tipo interruptor basado en FRB.
En una realización, el miembro de señalización intracelular comprende uno o más dominios de señalización intracelular, p. ej., un dominio de señalización intracelular primaria y uno o más dominios de señalización coestimulantes.
En una realización, el miembro de unión a antígenos puede comprender uno o más dominios de señalización intracelular, p. ej., uno o más dominios de señalización coestimulantes. En una realización, el miembro de unión a antígenos comprende una pluralidad, p. ej., 2 o 3 dominios de señalización coestimulantes descritos en esta memoria, p. ej., seleccionados entre 41BB, CD28, CD27, ICOS y OX40, y, en algunas realizaciones, ningún dominio de señalización intracelular primario. En una realización, el miembro de unión a antígenos comprende los siguientes dominios de señalización coestimulantes, desde la dirección extracelular a la intracelular: 41BB-CD27; 41BB-CD27; CD27-41BB; 41BB-CD28; CD28-41BB; OX40-CD28; CD28-OX40; CD28-41BB; o 41BB-CD28. En tales realizaciones, el miembro de unión intracelular comprende un dominio CD3zeta. En una realización de este tipo, el RCAR comprende (1) un miembro de unión a antígenos que comprende un dominio de unión a antígenos, un dominio transmembrana y dos dominios coestimulantes y un primer dominio de tipo interruptor; y (2) un dominio de señalización intracelular que comprende un dominio transmembrana o dominio de fijación a la membrana y al menos un dominio de señalización intracelular primario, y un segundo dominio de tipo interruptor.
Una realización proporciona RCAR en donde el miembro de unión a antígenos no está fijado a la superficie de la célula CAR. Esto permite que una célula que tiene un miembro de señalización intracelular se empareje convenientemente con uno o más dominios de unión a antígenos, sin transformar la célula con una secuencia que codifica el miembro de unión a antígenos. En tales realizaciones, el RCAR comprende: 1) un miembro de señalización intracelular que comprende: un primer dominio de tipo interruptor, un dominio transmembrana, un dominio de señalización intracelular, p. ej., un dominio de señalización intracelular primario, y un primer dominio de tipo interruptor; y 2) un miembro de unión a antígenos que comprende: un dominio de unión a antígenos y un segundo dominio de tipo interruptor, en donde el miembro de unión a antígenos no comprende un dominio transmembrana o dominio de fijación a la membrana y, opcionalmente, no comprende un dominio de señalización intracelular. En algunas realizaciones, el RCAR puede comprender además 3) un segundo miembro de unión a antígenos que comprende: un segundo dominio de unión a antígenos, p. ej., un segundo dominio de unión a antígenos que se une a un antígeno diferente del que se une al dominio de unión a antígenos; y un segundo dominio de tipo interruptor.
También se proporcionan en esta memoria RCARs en donde el miembro de unión a antígenos comprende capacidad de activación y direccionamiento biespecíficas. En esta realización, el miembro de unión a antígenos puede comprender una pluralidad, p. ej., 2, 3, 4 o 5 dominios de unión a antígenos, p. ej., scFv, en donde cada dominio de unión a antígenos se une a un antígeno diana, p. ej., antígenos diferentes o el mismo antígeno, p. ej., el mismo o diferentes epítopos en el mismo antígeno. En una realización, la pluralidad de dominios de unión a antígenos están en tándem y, opcionalmente, se dispone un enlazador o región de bisagra entre cada uno de los dominios de unión a antígenos. En esta memoria se describen enlazadores y regiones de bisagra adecuados.
Una realización proporciona RCAR que tienen una configuración que permite realizar un cambio de la proliferación. En esta realización, el RCAR comprende: 1) un miembro de señalización intracelular que comprende: opcionalmente, un dominio transmembrana o dominio de fijación a la membrana; uno o más dominios de señalización coestimulantes, p. ej., seleccionados entre 41BB, CD28, CD27, ICOS y OX40, y un dominio de tipo interruptor; y 2) un miembro de unión a antígenos que comprende: un dominio de unión a antígenos, un dominio transmembrana y un dominio de señalización intracelular primario, p. ej., un dominio de CD3zeta, en donde el miembro de unión a antígenos no comprende un dominio de tipo interruptor o no comprende un dominio de tipo interruptor que dimeriza con un dominio de tipo interruptor en el miembro de señalización intracelular. En una realización, el miembro de unión a antígenos no comprende un dominio de señalización coestimulante. En una realización, el miembro de señalización intracelular comprende un dominio de tipo interruptor de un interruptor de homodimerización. En una realización, el miembro de señalización intracelular comprende un primer dominio de tipo interruptor de un interruptor de heterodimerización y el RCAR comprende un segundo miembro de señalización intracelular que comprende un segundo dominio de tipo interruptor del interruptor de heterodimerización. En tales realizaciones, el segundo miembro de señalización intracelular comprende los mismos dominios de señalización intracelular que el miembro de señalización intracelular. En una realización, el interruptor de dimerización es intracelular. En una realización, el interruptor de dimerización es extracelular.
En cualquiera de las configuraciones RCAR descritas aquí, el primer y segundo dominios de tipo interruptor comprenden un interruptor basado en FKBP-FRB como se describe en esta memoria.
También se proporcionan en esta memoria células que comprenden un RCAR descrito en esta memoria. Como célula RCARX se puede utilizar cualquier célula que se haya modificado para expresar un RCAR. En una realización, la célula RCARX es un célula T y se denomina célula T-RCAR. En una realización, la célula RCARX es un célula NK y se denomina célula N-RCAR.
También se proporcionan en esta memoria ácidos nucleicos y vectores que comprenden secuencias que codifican RCAR. Las secuencias que codifican diversos elementos de un RCAR se pueden disponer en la misma molécula de ácido nucleico, p. ej., el mismo plásmido o vector, p. ej., vector vírico, p. ej., vector lentivírico. En una realización, (i) una secuencia que codifica un miembro de unión a antígenos y (ii) una secuencia que codifica un miembro de señalización intracelular pueden estar presentes en el mismo ácido nucleico, p. ej., vector. La producción de las correspondientes proteínas se puede lograr, p. ej., mediante el uso de promotores separados o mediante el uso de un producto de transcripción bicistrónico (que puede dar lugar a la producción de dos proteínas mediante la escisión de un único producto de traducción o mediante la traducción de dos productos proteicos separados). En una realización, se dispone entre (i) y (ii) una secuencia que codifica un péptido escindible, p. ej., secuencia P2A o F2A. En una realización, se dispone entre (i) y (ii) una secuencia que codifica un IRES, p. ej., un Em Cv o EV71 IRES. En estas realizaciones, (i) y (ii) se transcriben como un ARN único. En una realización, un primer promotor está ligado operativamente a (i) y un segundo promotor está ligado operativamente a (ii), de modo que (i) y (ii) se transcriben como ARNm separados.
Como alternativa, la secuencia que codifica diversos elementos de un RCAR se puede disponer en las diferentes moléculas de ácido nucleico, p. ej., diferentes plásmidos o vectores, p. ej., vector vírico, p. ej., vector lentivírico. P. ej., la (i) secuencia que codifica un miembro de unión a antígenos puede estar presente en un primer ácido nucleico, p. ej., un primer vector, y la (ii) secuencia que codifica un miembro de señalización intracelular puede estar presente en el segundo ácido nucleico, p. ej., el segundo vector.
Interruptores de dimerización
Los interruptores de dimerización pueden ser no covalentes o covalentes. En un interruptor de dimerización no covalente, la molécula de dimerización promueve una interacción no covalente entre los dominios de tipo interruptor. En un interruptor de dimerización covalente, la molécula de dimerización promueve una interacción covalente entre los dominios de tipo interruptor.
En una realización, el RCAR comprende un interruptor de dimerización basado en FKBP/FRB o FKBP/FRAP. FKBP12 (FKBP o proteína de unión a FK506) es una proteína citoplasmática abundante que actúa como la diana intracelular inicial para el fármaco inmunosupresor de tipo producto natural rapamicina. La rapamicina se une a FKBP y al homólogo de PI3K grande FRAP (RAFT, mTOR). FRB es una porción de 93 aminoácidos de FRAP que es suficiente para unirse al complejo FKBP-rapamicina (Chen, J., Zheng, X. F., Brown, E. J. y Schreiber, S. L. (1995) Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc Natl Acad Sci USA 92: 4947-51.)
En algunas realizaciones, un interruptor basado en FKBP/FRAP, p. ej., FKBP/FRB, puede usar una molécula de dimerización, p. ej., rapamicina o un análogo de rapamicina.
La secuencia de aminoácidos de FKBP es la siguiente:
D V P D Y A S L G G P S S P K K K R K V S R G V Q V E T I S P G D G R T F P K R G Q T C Y V H Y T G M L E D G K K F D S S R D R N K P F K F M L G K Q E V I R G W E E G Y A Q M S Y G Q R A K L T I S P D Y A Y G A T G H P G I I P P H A T L Y F D V E L L K L E T S Y (SEQ ID NO: 54)
En algunas realizaciones, un dominio de tipo interruptor FKBP puede comprender un fragmento de FKBP que tiene la capacidad de unirse a FRB, o un fragmento o análogo de este, en presencia de rapamicina o un rapálogo, p. ej., la porción subrayada de la SEQ ID NO: 54, que es:
V Q V E T I S P G D G R T F P K R G Q T C V V H Y T G M L E D G K K F D S S R D R N K P F K F M L G K Q E V I R G W E E G V A Q M S V G Q R A K L T I S P D Y A Y G A T G H P G I I P P H A T L V F D V E L L K L E T S (SEQ ID NO:55)
La secuencia de aminoácidos de FRB es la siguiente:
ILWHEMWHEG LEEASRLYFG ERNVKGMFEV LEPLHAMMER GPQTLKETSF
NQAYGRDLME AQEWCRKYMK SGNVKDLTQA WDLYYHVFRRISK (SEQ ID NO:
56)
"Interruptor basado en FKBP/FRAP, p. ej., FKBP/FRB", como se utiliza la expresión en esta memoria, se refiere a un interruptor de dimerización que comprende: un primer dominio de tipo interruptor, que comprende un fragmento de FKBP o un análogo de este que tiene la capacidad de unirse a FRB, o un fragmento o análogo de este, en presencia de rapamicina o un rapálogo, p. ej., RAD001, y que tiene una identidad de al menos un 70, 75, 80, 85, 90, 95, 96, 97, 98 o 99 % respecto a, o difiere en como máximo 30, 25, 20, 15, 10, 5, 4, 3, 2 o 1 residuos aminoacídicos de, la secuencia FKBP de la SEQ ID NO: 54 o 55; y un segundo dominio de tipo interruptor, que comprende un fragmento de FRB o un análogo de este que tiene la capacidad de unirse a FRB, o un fragmento o análogo de este, en presencia de rapamicina o un rapálogo, y que tiene una identidad de al menos un 70, 75, 80, 85, 90, 95, 96, 97, 98 o 99 % respecto a, o difiere en como máximo 30, 25, 20, 15, 10, 5, 4, 3, 2 o 1 residuos aminoacídicos de, la secuencia FRB de la SEQ ID NO: 56. En una realización, un RCAR descrito en esta memoria comprende un dominio de tipo interruptor que comprende residuos aminoacídicos desvelados en la SEQ ID NO: 54 (o la SEQ ID NO: 55) y un dominio de tipo interruptor comprende residuos aminoacídicos desvelados en la SEQ ID NO: 56.
En algunas realizaciones, el interruptor de dimerización FKBP/FRB comprende un dominio de tipo interruptor FRB modificado que muestra una formación de complejo alterada, p. ej., potenciada, entre un dominio de tipo interruptor basado en FRB, p. ej., el dominio de tipo interruptor FRB modificado, un dominio de tipo interruptor basado en FKBP y la molécula de dimerización, p. ej., rapamicina o un rapálogo, p. ej., RAD001. En una realización, el dominio de tipo interruptor FRB modificado comprende una o más mutaciones, p. ej., 2, 3, 4, 5, 6, 7, 8, 9, 10 o más, seleccionadas entre mutaciones en la o las posiciones aminoacídicas L2031, E2032, S2035, R2036, F2039, G2040, T2098, W2101, D2102, Y2105 y F2108, donde el aminoácido de tipo silvestre está mutado a cualquier otro aminoácido de origen natural. En una realización, un FRB mutante comprende una mutación en E2032, donde E2032 se muta a fenilalanina (E2032F), metionina (E2032M), arginina (E2032R), valina (E2032V), tirosina (E2032Y), isoleucina (E2032I), p. ej., la SEQ ID NO: 57, o leucina (E2032L), p. ej., la SEQ ID NO: 58. En una realización, un FRB mutante comprende una mutación en T2098, donde T2098 se muta a fenilalanina (T2098F) o leucina (T2098L), p. ej., la SEQ ID NO: 59. En una realización, un FRB mutante comprende una mutación en E2032 y en T2098, donde E2032 se muta a cualquier aminoácido, y donde T2098 se muta a cualquier aminoácido, p. ej., la SEQ ID NO: 60. En una realización, un FRB mutante comprende una mutación E2032I y una T2098L, p. ej., la SEQ ID NO: 61. En una realización, un FRB mutante comprende una mutación E2032L y una T2098L, p. ej., la SEQ ID NO: 62.
Tabla 10. FRB mutante ilustrativo que tiene una ma or afinidad por una molécula de dimerización.


Otros interruptores de dimerización adecuados incluyen el interruptor de dimerización basado en GyrB-GyrB, un interruptor de dimerización basado en giberelina, un interruptor de dimerización marcador/fijador y un interruptor de dimerización marcador-halo/marcador-SNAP. Siguiendo las directrices que se proporcionan en esta memoria, tales interruptores y moléculas de dimerización relevantes serán evidentes para un experto en la técnica.
Molécula de dimerización
La asociación entre los dominios de tipo interruptor está fomentada por la molécula de dimerización. En presencia de la molécula de dimerización la interacción o asociación entre dominios de tipo interruptor permite la transducción de señales entre un polipéptido asociado con, p. ej., fusionado con, un primer dominio de tipo interruptor, y un polipéptido asociado con, p. ej., fusionado con, un segundo dominio de tipo interruptor. En presencia de niveles no limitantes de la molécula de dimerización la transducción de señales aumenta en 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2, 5, 10, 50, 100 veces, p. ej., según se mide en un sistema descrito en esta memoria.
Se pueden utilizar rapamicina y análogos de rapamicina (denominados a veces rapálogos), p. ej., RAD001, como moléculas de dimerización en un interruptor de dimerización basado en FKBP/FRB descrito en esta memoria. En una realización, la molécula de dimerización se puede seleccionar entre rapamicina (sirolimus), RAD001 (everolimus), zotarolimus, temsirolimus, AP-23573 (ridaforolimus), biolimus y AP21967. En la sección titulada "Terapias combinadas" o en la subsección titulada "Inhibidor de mTOR ilustrativos" se describen adicionalmente más análogos de rapamicina adecuados para su uso con interruptores de dimerización basados en FKBP/FRB.
CAR dividido
En algunas realizaciones, la célula que expresa CAR utiliza un CAR dividido. El enfoque CAR dividido se describe con más detalle en las publicaciones WO2014/055442 y WO2014/055657 . En resumen, un sistema de CAR dividido comprende una célula que expresa un primer CAR que tiene un primer dominio de unión a antígenos y un dominio coestimulante (p. ej., 41BB), y la célula también expresa un segundo CAR que tiene un segundo dominio de unión a antígenos y un dominio de señalización intracelular (p. ej., CD3 zeta). Cuando la célula encuentra el primer antígeno, el dominio coestimulante se activa y la célula prolifera. Cuando la célula encuentra el segundo antígeno, el dominio de señalización intracelular se activa y comienza la actividad de destrucción celular. Por lo tanto, la célula que expresa CAR solo se activa completamente en presencia de ambos antígenos.
Transfección de ARN
En esta memoria se desvelan métodos para producir un ARN de CAR transcrito in vitro. La presente invención puede emplear una construcción de ARN que codifica CAR que puede transfectarse directamente en una célula. Un método para generar ARNm para su uso en la transfección puede implicar la transcripción in vitro (IVT) de un molde con cebadores especialmente diseñados, seguida de la adición de poliA, para producir una construcción que contenga una secuencia no traducida ("UTR") en 3' y 5', un casquete 5' y/o sitio de entrada del ribosoma interno (IRES), el ácido nucleico a expresar y una cola de poliA, típicamente de 50 a 2000 bases de longitud (SEQ ID NO: 32). El ARN así producido puede transfectar eficientemente diferentes tipos de células. En un aspecto, el molde incluye secuencias para el CAR.
En un aspecto, un CAR utilizado en la presente invención es codificado por un ARN mensajero (ARNm). En un aspecto, el ARNm que codifica un CAR descrito en esta memoria se introduce en una célula efectora inmunitaria, p. ej., un célula T o un célula NK, para la producción de una célula que expresa CAR, p. ej., un célula T-CAR o un célula Nk Car .
El ARN del CAR transcrito in vitro puede introducirse en una célula como una forma de transfección transitoria. El ARN se produce mediante transcripción in vitro utilizando un molde generado por la reacción en cadena de la polimerasa (PCR). El ADN de interés de cualquier fuente se puede convertir directamente mediante PCR en un molde para la síntesis de ARNm in vitro utilizando cebadores apropiados y ARN polimerasa. La fuente del ADN puede ser, por ejemplo, ADN genómico, ADN plasmídico, ADN de fago, ADNc, secuencia de ADN sintético o cualquier otra fuente apropiada de ADN. El molde deseado para la transcripción in vitro es un CAR descrito en esta memoria. Por ejemplo, el molde para el ARN del CAR comprende una región extracelular que comprende un dominio variable de cadena sencilla de un anticuerpo contra un antígeno asociado a un tumor descrito en esta memoria; una región de bisagra (p. ej., una región de bisagra descrita en esta memoria), un dominio transmembrana (p. ej., un dominio transmembrana descrito en esta memoria tal como un dominio transmembrana de CD8a); y una región citoplasmática que incluye un dominio de señalización intracelular, p. ej., un dominio de señalización intracelular descrito en esta memoria, p. ej., que comprende el dominio de señalización de CD3-zeta y el dominio de señalización de 4-1BB.
En un caso, el ADN que se va a utilizar para la PCR contiene un marco de lectura abierto. El ADN puede proceder de una secuencia de ADN que se produce de forma natural del genoma de un organismo. En un caso, el ácido nucleico puede incluir algunas o todas las regiones no traducidas (UTR) 5' y/o 3'. El ácido nucleico puede incluir exones e intrones. En un caso, el ADN que se va a utilizar para la PCR es una secuencia de ácido nucleico humano. En otro caso, el ADN que se va a utilizar para la PCR es una secuencia de ácido nucleico humano que incluye las UTR 5' y 3'. Como alternativa, el ADN puede ser una secuencia de ADN artificial que normalmente no se expresa en un organismo que se produce de forma natural. Una secuencia de ADN artificial ilustrativa es aquella que contiene porciones de genes que se ligan entre sí para formar un marco de lectura abierto que codifica una proteína de fusión. Las porciones de ADN que se ligan entre sí pueden ser de un solo organismo o de más de un organismo.
Se utiliza PCR para generar un molde para la transcripción in vitro de ARNm que se utiliza para la transfección. Los métodos para realizar la PCR son bien conocidos en la técnica. Los cebadores para su uso en la PCR están diseñados para tener regiones que sean sustancialmente complementarias a las regiones del ADN que se van a utilizar como un molde para la PCR. "Sustancialmente complementarias", tal como se utiliza en esta memoria, se refiere a secuencias de nucleótidos en las que la mayoría o todas las bases en la secuencia del cebador son complementarias, o una o más bases no son complementarias o están emparejadas erróneamente. Las secuencias sustancialmente complementarias son capaces de reasociarse o hibridarse con la diana de ADN pretendida en las condiciones de reasociación utilizadas para la PCR. Los cebadores pueden diseñarse para que sean sustancialmente complementarios a cualquier porción del molde de ADN. Por ejemplo, los cebadores pueden diseñarse para amplificar la porción de un ácido nucleico que normalmente se transcribe en las células (el marco de lectura abierto), incluyendo las UTR 5' y 3'. Los cebadores también pueden diseñarse para amplificar una porción de un ácido nucleico que codifica un dominio de interés particular. En un caso, los cebadores están diseñados para amplificar la región codificante de un ADNc humano, incluyendo todas o porciones de las UTR 5' y 3'. Se pueden generar cebadores útiles para la PCR mediante métodos sintéticos que son bien conocidos en la técnica. "Cebadores directos" son cebadores que contienen una región de nucleótidos que son sustancialmente complementarios a los nucleótidos en el molde de a Dn que están cadena arriba de la secuencia de ADN que se ha de amplificar. "Cadena arriba" se utiliza en esta memoria para referirse a una ubicación 5 con respecto a la secuencia de ADN que se ha de amplificar en relación con la cadena codificante. "Cebadores inversos" son cebadores que contienen una región de nucleótidos que son sustancialmente complementarios a un molde de ADN de doble cadena que se encuentran cadena abajo de la secuencia de ADN que se ha de amplificar. "Aguas abajo" se utiliza en esta memoria para referirse a una ubicación 3' de la secuencia de ADN que se ha de amplificar en relación con la cadena codificante.
Se puede utilizar cualquier ADN polimerasa útil para la PCR en los métodos desvelados en esta memoria. Los reactivos y la polimerasa están disponibles en el mercado de varios proveedores.
También se pueden utilizar estructuras químicas con la capacidad de fomentar la estabilidad y/o la eficacia de la traducción. El ARN tiene preferentemente UTR 5' y 3'. En una realización, la UTR 5' tiene una longitud de entre uno y 3000 nucleótidos. La longitud de las secuencias UTR 5' y 3' que se van a añadir a la región codificante se puede alterar mediante diferentes métodos, incluidos, pero sin limitación, diseño de cebadores para PCR que se hibridan con diferentes regiones de las UTR. Utilizando este enfoque, un experto en la técnica puede modificar la longitud de las UTR 5' y 3' necesaria para lograr una eficacia de traducción óptima después de la transfección con el ARN transcrito.
Las UTR 5' y 3' pueden ser las UTR 5' y 3' de origen natural, endógenas respecto al ácido nucleico de interés. Como alternativa, las secuencias UTR que no son endógenas respecto al ácido nucleico de interés se pueden añadir incorporando las secuencias UTR en los cebadores directo e inverso o mediante cualquier otra modificación del molde. El uso de secuencias UTR que no son endógenas respecto al ácido nucleico de interés puede ser útil para modificar la estabilidad y/o la eficacia de traducción del ARN. Por ejemplo, se sabe que elementos ricos en AU en las secuencias UTR 3' pueden disminuir la estabilidad del ARNm. Por lo tanto, las UTR 3' se pueden seleccionar o diseñar para aumentar la estabilidad del ARN transcrito en función de las propiedades de las UTRs que son bien conocidas en la técnica.
En una realización, la UTR 5' puede contener la secuencia de Kozak del ácido nucleico endógeno. Como alternativa, cuando se añade una UTR 5' que no es endógena respecto al ácido nucleico de interés por PCR, como se ha descrito anteriormente, se puede rediseñar una secuencia de Kozak consenso añadiendo la secuencia UTR 5'. Las secuencias de
Kozak pueden aumentar la eficiencia de la traducción de algunos transcritos de ARN, pero no parece que sean necesarias en todos los ARN para posibilitar una traducción eficiente. En la técnica se conoce la necesidad de secuencias de Kozak para muchos ARNm. En otras realizaciones, la UTR 5' puede ser una UTR 5' de un virus de ARN, cuyo genoma de ARN es estable en las células. En otras realizaciones, se pueden utilizar diversos análogos de nucleótidos en la UTR 3' o 5' para impedir la degradación por exonucleasa del ARNm.
Para permitir la síntesis de ARN a partir de un molde de ADN sin la necesidad de clonación génica, se debe unir un promotor de la transcripción al molde de ADN en dirección 5' respecto a la secuencia que se va a transcribir. Cuando se añade una secuencia que actúa como un promotor para una ARN polimerasa al extremo 5' del cebador directo, el promotor de ARN polimerasa se incorpora al producto de PCR en dirección 5' respecto al marco de lectura abierto que se va a transcribir. En un caso preferido, el promotor es un promotor de la polimerasa T7 tal como se describe en otra parte de esta memoria. Otros promotores útiles incluyen, pero no se limitan a promotores de ARN polimerasa T3 y SP6. Las secuencias de nucleótidos de consenso para los promotores T7, T3 y SP6 se conocen en la técnica.
En una realización preferida, el ARNm tiene tanto una caperuza en el extremo 5' como una cola de poli(A) 3' que determina la unión al ribosoma, el inicio de la traducción y la estabilidad del ARNm en la célula. En un molde de ADN circular, por ejemplo, ADN plasmídico, la ARN polimerasa produce un producto concatamérico largo que no es adecuado para la expresión en células eucariotas. La transcripción del ADN plasmídico linealizado en el extremo de la UTR 3' da lugar a un ARNm de tamaño normal que no es eficaz en la transfección eucariota, incluso si se poliadenila después de la transcripción.
En un molde de ADN lineal, la ARN polimerasa del fago T7 puede extender el extremo 3' del transcrito más allá de la última base del molde (Schenborn y Mierendorf, Nuc Acids Res., 13: 6223-36 (1985); Nacheva y Berzal-Herranz, Eur. J. Biochem., 270: 1485-65 (2003).
El método convencional de integración de tramos de poliA/T en un molde de ADN es la clonación molecular. Sin embargo, la secuencia de poliA/T integrada en el ADN plasmídico puede causar inestabilidad plasmídica, que es por lo que los moldes de ADN plasmídico obtenidos de células bacterianas están a menudo muy contaminados con supresiones y otras aberraciones. Esto hace que los procedimientos de clonación no solo sean laboriosos y lentos, sino que a menudo no sean fiables. Es por eso que un método que permite la construcción de moldes de ADN con un tramo de poliA/T 3' sin clonación es sumamente deseable.
El segmento de poliA/T del molde de ADN transcripcional se puede producir durante la PCR utilizando un cebador inverso que contiene una cola de poliT, tal como la cola 100T (SEQ ID NO: 35) (el tamaño puede ser 50-5000 T (SEQ ID NO: 36)) o después de la PCR por cualquier otro método, incluyendo, perro no limitados a la ligadura de ADN o la recombinación in vitro. Las colas de poli(A) también proporcionan estabilidad a los ARN y reducen su degradación. Generalmente, la longitud de una cola de poli(A) se correlaciona positivamente con la estabilidad del ARN transcrito. En una realización, la cola de poli(A) tiene entre 100 y 5000 adenosinas (SEQ ID NO: 37).
Las colas poli(A) de los ARN se pueden extender más después de la transcripción in vitro con el uso de una poli(A) polimerasa, tal como la poliA polimerasa de E. coli (E-PAP). En una realización, aumentar la longitud de una cola de poli(A) de 100 nucleótidos a entre 300 y 400 nucleótidos (SEQ ID NO: 38) da lugar a un aumento de aproximadamente dos veces en la eficiencia de traducción del ARN. Adicionalmente, la unión de diferentes grupos químicos al extremo 3' puede aumentar la estabilidad del ARNm. Una unión de este tipo puede contener nucleótidos modificados/artificiales, aptámeros y otros compuestos. Por ejemplo, los análogos de ATP se pueden incorporar en la cola de poli(A) utilizando poli(A) polimerasa. Los análogos de ATP pueden aumentar adicionalmente la estabilidad del ARN.
Las caperuzas 5' también proporcionan estabilidad a las moléculas de ARN. En una realización preferida, los ARN producidos por los métodos desvelados en esta memoria incluyen un casquete 5'. El casquete 5' se proporciona utilizando técnicas conocidas en la materia y descritas en el presente memoria (Cougot, et al., Trends in Biochem. Sci., 29: 436-444 (2001); Stepinski, et al., RNA, 7: 1468-95 (2001); Elango, et al., Biochim. Biophys. Res. Commun., 330: 958-966 (2005)).
Los ARN producidos por los métodos descritos en esta memoria también pueden contener una secuencia del sitio interno de entrada al ribosoma (IRES). La secuencia IRES puede ser cualquier secuencia vírica, cromosómica o diseñada por medios artificiales que inicie la unión del ribosoma independiente de la caperuza al ARNm y facilite el inicio de la traducción. Se pueden incluir cualquier soluto adecuado para la electroporación celular, que puede contener factores que facilitan la permeabilidad y la viabilidad celular, tales como azúcares, péptidos, lípidos, proteínas, antioxidantes y tensioactivos.
El ARN se puede introducir en células diana utilizando cualquiera de varios métodos diferentes, por ejemplo, métodos disponibles en el mercado que incluyen, pero sin limitación, electroporación (Amaxa Nucleofector-II (Amaxa Biosystems, Colonia, Alemania)), (ECM 830 (BTX) (Harvard Instruments, Boston, Mass.) o Gene Pulser II (BioRad, Denver, Colo.), Multiporator (Eppendort, Hamburgo Alemania), la transfección mediada por liposomas catiónicos utilizando lipofección, encapsulación de polímeros, transfección mediada por péptidos o sistemas de suministro biolístico de partículas, tales como "pistolas génicas" (véase, por ejemplo, Nishikawa, et al. Hum Gene Ther., 12 (8): 861-70 (2001).
Métodos de suministro no víricos
En algunos aspectos, se pueden utilizar métodos no víricos para suministrar un ácido nucleico que codifica un CAR descrito en esta memoria a una célula o tejido en un sujeto.
En algunos casos, el método no vírico incluye el uso de un transposón (también denominado elemento transponible). En algunos casos, un transposón es un trozo de ADN que puede insertarse en un lugar de un genoma, por ejemplo, un trozo de ADN que es capaz de autorreplicarse e insertar su copia en un genoma, o un trozo de ADN que se puede separar de un ácido nucleico más largo e insertarse en otro lugar del genoma. Por ejemplo, un transposón comprende una secuencia de ADN formada por repeticiones invertidas que flanquean los genes para la transposición.
Los métodos ilustrativos de suministro de ácidos nucleicos utilizando un transposón incluyen un sistema de transposones Sleeping Beauty (SBTS) y un sistema de transposones piggyBac (PB). Véase, p. ej., Aronovich et al. Hum. Mol. Genet.
20.R1 (2011): R14-20; Singh et al. Cancer Res. 15 (2008): 2961-2971; Huang et al. Mol. Ther. 16 (2008): 580-589; Grabundzija et al. Mol. Ther. 18 (2010): 1200-1209; Kebriaei et al. Blood. 122.21 (2013): 166; Williams. Molecular Therapy 16.9 (2008):1515-16; Bell et al. Nat. Protoc. 2.12 (2007): 3153-65; y Ding et al. Cell. 122.3 (2005): 473-83.
El SBTS incluye dos componentes: 1) un transposón que contiene un transgén y 2) una fuente de enzima transposasa. La transposasa puede transponer el transposón de un plásmido portador (u otro ADN donante) a un ADN diana tal como un cromosoma/genoma de una célula huésped. Por ejemplo, la transposasa se une al plásmido transportador/ADN donante, corta el transposón (incluidos el o los transgenes) del plásmido y lo inserta en el genoma de la célula hospedadora. Véase, p. ej., Aronovich et al., mencionado supra..
Los transposones ilustrativos incluyen un transposón basado en pT2. Véase, p. ej., Grabundzija et al. Nucleic Acids Res.
41.3 (2013): 1829-47; y Singh et al. Cancer Res. 68.8 (2008): 2961-2971. Transposones ejemplares incluyen una transposasa Tc1/tipo marinero, p. ej., la transposasa SB10 o la transposasa SB11 (una transposasa hiperactiva que puede expresarse, p. ej., a partir de un promotor de citomegalovirus). Véase, p. ej., Aronovich et al.; Kebriaei et al.; y Grabundzij a et al.
El uso de SBTS permite la integración y expresión eficientes de un transgén, p. ej., un ácido nucleico que codifica un CAR descrito en esta memoria. En esta memoria se proporcionan métodos para generar una célula, p. ej., un célula T o célula NK, que expresa de manera estable un CAR descrito en el presente memoria, p. ej., utilizando un sistema de transposón tal como SBTS.
De acuerdo con los métodos descritos en esta memoria, en algunos casos, uno o más ácidos nucleicos, p. ej., plásmidos, que contienen los componentes SBTS se administran a una célula (p. ej., célula T o NK). Por ejemplo, el o los ácidos nucleicos se suministran mediante métodos convencionales de administración de ácido nucleico (p. ej., ADN plasmídico), p. ej., métodos descritos en esta memoria, p. ej., electroporación, transfección o lipofección. En algunos casos, el ácido nucleico contiene un transposón que comprende un transgén, p. ej., un ácido nucleico que codifica un CAR descrito en esta memoria. En algunos casos, el ácido nucleico contiene un transposón que comprende un transgén (p. ej., un ácido nucleico que codifica un CAR descrito en esta memoria), así como una secuencia de ácido nucleico que codifica una enzima transposasa. En otros casos, se proporciona un sistema con dos ácidos nucleicos, p. ej., un sistema de plásmido dual, p. ej., en que un primer plásmido contiene un transposón que comprende un transgén, y un segundo plásmido contiene una secuencia de ácido nucleico que codifica una enzima transposasa. Por ejemplo, el primer y el segundo ácidos nucleicos se suministran conjuntamente a la célula huésped.
En algunos casos, se generan células, p. ej., células T o NK, que expresan un CAR descrito en esta memoria utiliozando una combinación de inserción de genes usando SBTS y edición genética utilizando una nucleasa (p. ej., nucleasas con dedos de zinc (ZFN, por sus siglas en inglés), Nucleasas Efectoras Similares a Activadores de la Transcripción (TALEN, por sus siglas en inglés), el sistema CRISPR/Cas o endonucleasas autodirigidas rediseñadas de meganucleasas).
En algunos casos, el uso de un método de suministro no vírico permite la reprogramación de células, p. ej., células T o NK, y la infusión directa de las células en un sujeto. Las ventajas de los vectores no víricos incluyen, pero sin limitación, la facilidad y el coste relativamente bajo de producir cantidades suficientes requeridas para satisfacer una población de pacientes, la estabilidad durante el almacenamiento y la falta de inmunogenicidad.
Construcciones de ácidos nucleicos que codifican un CAR
La presente invención también emplea moléculas de ácido nucleico que codifican una o más construcciones CAR de acuerdo con las reivindicaciones. En un aspecto, la molécula de ácido nucleico se proporciona como un transcrito de ARN mensajero. En un aspecto, la molécula de ácido nucleico se proporciona como una construcción de ADN.
Por consiguiente, en un aspecto, la invención emplea una molécula de ácido nucleico aislada que codifica un receptor de antígeno quimérico (CAR), en donde el CAR comprende un dominio de unión a antígenos que se une a un antígeno tumoral como se describe en esta memoria, un dominio transmembrana (p. ej., un dominio transmembrana descrito en esta memoria) y un dominio de señalización intracelular (p. ej., un dominio de señalización intracelular descrito en esta
memoria) que comprende un dominio coestimulador, p. ej., un dominio coestimulador descrito en esta memoria y/o un dominio de señalización primario (p. ej, un dominio de señalización primario descrito en esta memoria). En una realización, el dominio transmembrana es el dominio transmembrana de una proteína seleccionada del grupo que consiste en la cadena alfa, beta o zeta del receptor de células T, CD28, CD3 épsilon, CD45, CD4, CD5, CD8, Cd9, cD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137 y CD154. En algunas realizaciones, un dominio transmembrana puede incluir al menos la o las regiones transmembrana de, p. ej., KIRDS2, OX40, CD2, CD27, LFA-1 (CD11a, CD18), ICOS (CD278), 4-1BB (CD137), GITR, CD40, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, IL2R beta, IL2R gamma, IL7R a, ITGA1, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, NKG2D, NKG2C, TNFR2, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), SLAMF6 (NTB-A, Ly108), SLAM (SLAMF1, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, PAG/Cbp.
En una realización, el dominio transmembrana comprende una secuencia de la SEQ ID NO: 12, o una secuencia con un 95-99 % de identidad con la misma. En una realización, el dominio de unión a antígenos está conectado al dominio transmembrana por una región de bisagra, p. ej., una bisagra descrita en esta memoria. En una realización, la región de bisagra comprende las SEQ ID NO:4 o SEQ ID NO:6 o SEQ ID NO:8 o SEQ ID NO:10, o una secuencia con una identidad de un 95-99 % con las mismas. En una realización, la molécula de ácido nucleico aislada comprende, además, una secuencia que codifica un dominio coestimulante. En una realización, el dominio coestimulante es un dominio de señalización funcional de una proteína seleccionada del grupo que consiste en OX40, CD27, CD28, CDS, ICAM-1, LFA-1 (CD11a/CD18), ICOS (CD278) y 4-1BB (CD137). Ejemplos adicionales de moléculas coestimuladoras de este tipo incluyen CDS, ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, CD4, CD8alpha, CD8beta, IL2R beta, IL2R gamma, IL7R alfa, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, NKG2D, NKG2C, TNFR2, TRANCE/RANKL, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), CD69, SLAMF6 (NTB-A, Ly108), SLAM (SLAMF1, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, LAT, GADS, SLP-76, y PAG/Cbp. En una realización, el dominio coestimulante comprende una secuencia de la SEQ ID NO: 16, o una secuencia con 95-99 % de identidad con la misma. En una realización, el dominio de señalización intracelular comprende un dominio de señalización funcional de 4-1BB y un dominio de señalización funcional de CD3 zeta. En una realización, el dominio de señalización intracelular comprende la secuencia de la SEQ ID NO: 14 o la SEQ ID NO: 16, o una secuencia con 95 99 % de identidad con las mismas y la secuencia de la SEQ ID NO: 18 o la SEQ ID NO: 20, o una secuencia con una identidad de un 95-99 % con las mismas, en donde las secuencias que comprenden el dominio de señalización intracelular se expresan en el mismo marco y como una cadena polipeptídica única.
En otro aspecto, la invención emplea una molécula de ácido nucleico aislada que codifica una construcción de CAR que comprende una secuencia conductora de SEQ ID NO: 2, un dominio scFv como se describe en esta memoria, una región de bisagra de la SEQ ID NO: 4 o la SEQ ID NO :6 o la SEQ ID NO:8 o la SEQ ID NO: 10 (o una secuencia con un 95-99 % de identidad con la misma), un dominio transmembrana que tiene una secuencia de la SEQ ID NO: 12 (o una secuencia con 95-99 % de identidad de la misma), un dominio coestimulador 4-1BB que tiene una secuencia de la SEQ ID NO: 14 o un dominio coestimulador CD27 que tiene una secuencia de la SEQ ID NO: 16 (o una secuencia con una identidad de un 95-99% con esta), y un dominio estimulador de CD3 zeta que tiene una secuencia de la SEQ ID NO:18 o SEQ ID NO:20 (o una secuencia con una identidad de un 95-99% con estas).
En otro aspecto, la invención emplea una molécula de ácido nucleico que codifica una molécula de receptor de antígeno quimérico (CAR) que comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio de señalización intracelular que comprende un dominio estimulador, y en donde dicho dominio de unión a antígenos se une a un antígeno tumoral seleccionado de un grupo que consiste en: CD19, CD123, CD22, CD30, CD171, CS-1, CLL-1 (CLECL1), CD33, EGFRvIII, GD2, GD3, BCMA, Tn Ag, PSMA, ROR1, FLT3, FAP, TAG72, CD38, CD44v6, CEA, EPCAM, B7H3, KIT, IL-13Ra2, Mesotelina, IL-11Ra, PSCA, VEGFR2, LewisY, CD24, PDGFR-beta, SSEA-4, CD20, receptor de folato alfa, ERBB2 (Her2/neu), MUC1, EGFR, NCAM, Prostasa, PRSS21, PAP, ELF2M, Efrina B2, receptor de IGF-I, CAIX, LMP2, gp100, bcr-abl, tirosinasa, EphA2, Fucosil GM1, sLe, GM3, TGS5, HMWMAA, o-acetil-GD2, receptor de foilato beta, TEM1/CD248, TEM7R, CLDN6, TSHR, GPRC5D, CXORF61, CD97, CD179a, ALK, ácido polisiálico, PLAC1, GloboH, NY-BR-1, UPK2, HAVCR1, ADRB3, PANX3, GPR20, LY6K, OR51E2, TARP, WT1, NY-ESO-1, LAGE-1a, MAGE-A1, legumaina, HPV E6,E7, MAGE A1, ETV6-AML, proteína de esperma 17, XAGE1, Tie 2, MAD-CT-1, MAD-CT-2, antígeno 1 relacionado con Fos, p53, p53 mutante, prosteína, survivina y telomerasa, PCTA-1/Galectina 8, MelanA/MART1, Ras mutante, hTERT, puntos de ruptura de la translocación de sarcoma, ML-IAP, ERG (TMPRSS2 ETS gen de fusión), NA17, PAX3, receptor de andrógeno, Ciclina B1, MYCN, RhoC, TRP-2, CYP1B1, BORIS, SART3, PAX5, OY-TES1, LCK, AKAP-4, SSX2, RAGE-1, transcriptasa inversa de telomerasa humana, RU1, RU2, carboxil esterasa intestinal, mut hsp70-2, CD79a, CD79b, CD72, LAIR1, FCAR, LILRA2, CD300LF, CLEC12A, BST2, EMR2, LY75, GPC3, FCRL5 e IGLL1.
En una realización, la molécula de CAR codificada comprende además una secuencia que codifica un dominio coestimulador. En una realización, el dominio coestimulador es un dominio de señalización funcional de una proteína seleccionada del grupo que consiste en OX40, CD27, CD28, CDS, ICAM-1, LFA-1 (CD11a/CD18) y 4-1BB (CD137). En
una realización, el dominio coestimulador comprende una secuencia de la SEQ ID NO: 14. En una realización, el dominio transmembrana es un dominio transmembrana de una proteína seleccionada del grupo que consiste en la cadena alfa, beta o zeta del receptor de células T, CD28, CD3 épsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137 y CD154. En una realización, el dominio transmembrana comprende una secuencia de la SEQ ID NO: 12. En una realización, el dominio de señalización intracelular comprende un dominio de señalización funcional de 4-1BB y un dominio de señalización funcional de zeta. En una realización, el dominio de señalización intracelular comprende la secuencia de la SEQ ID NO: 14 y la secuencia de la SEQ ID NO: 18, en donde las secuencias que comprenden el dominio de señalización intracelular se expresan en el mismo marco y como una sola cadena polipeptídica. En una realización, el dominio de unión anti-antígeno asociado al cáncer a como se describe en esta memoria está conectado al dominio transmembrana mediante una región bisagra. En una realización, la región de bisagra comprende la SEQ ID NO: 4. En una realización, la región de bisagra comprende la SEQ ID NO: 6 o la SEQ ID NO: 8 o la SEQ ID NO: 10.
Las secuencias de ácido nucleico que codifican las moléculas deseadas se pueden obtener utilizando métodos recombinantes conocidos en la técnica tales como, por ejemplo, mediante la selección de colecciones de células que expresan el gen, derivando el gen de un vector que se sabe que incluye el mismo, o aislándolos directamente de células y tejidos que los contienen, utilizando técnicas estándares. Como alternativa, el gen de interés puede producirse sintéticamente, en lugar de clonarse.
La presente invención también emplea vectores en los que se inserta un ADN de acuerdo con las reivindicaciones. Vectores derivados de retrovirus tales como el lentivirus son herramientas adecuadas para lograr la transferencia de genes a largo plazo, ya que permiten la integración estable a largo plazo de un transgén y su propagación en células hijas. Los vectores lentivíricos tienen la ventaja añadida frente a los vectores procedentes de oncorretrovirus, tales como los virus de la leucemia murina, de que pueden transducir células no proliferantes, tales como hepatocitos. También tienen la ventaja añadida de una baja inmunogenicidad. Un vector retroviral también puede ser, p. ej., un vector gammaretroviral. Un vector gammarretrovírico puede incluir, p. ej., un promotor, una señal de empaquetamiento (y ), un sitio de unión del cebador (PBS), una o más (p. ej., dos) repeticiones terminales largas (LTR) y un transgén de interés, p. ej., un gen que codifica un CAR. Un vector gammarretrovírico puede carecer de genes estructurales víricos tales como gag, pol y env. Vectores gammaretrovirales ejemplares incluyen el virus de la leucemia murina (MLV), el virus que forma el foco del bazo (SFFV) y el virus del sarcoma mieloproliferativo (MPSV), y vectores derivados de los mismos. Otros vectores gammarretrovíricos se describen, p. ej., en Tobias Maetzig et al., "Gammaretroviral Vectors: Biology, Technology and Application" Viruses. Junio de 2011; 3 (6): 677-713.
En otra realización, el vector que comprende el ácido nucleico que codifica el CAR deseado es un vector adenoviral (A5/35). En otra realización, la expresión de ácidos nucleicos que codifican CARs se puede lograr utilizando transposones tales como nucleasas de dedos de zinc, CAS9 y bella durmiente. . Véase, a continuación, June et al. 2009 Nature Reviews Immunology 9.10: 704-716.
En resumen, la expresión de ácidos nucleicos naturales o sintéticos que codifican los CAR se logra normalmente ligando operativamente un ácido nucleico que codifica el polipéptido CAR o porciones del mismo a un promotor e incorporando la construcción en un vector de expresión. Los vectores pueden ser adecuados para la replicación y la integración en eucariotas. Los vectores típicos de clonación contienen terminadores de la transcripción y la traducción, secuencias de iniciación y promotores útiles para la regulación de la expresión de la secuencia deseada de ácido nucleico.
Las construcciones de expresión empleadas en la presente invención también se pueden utilizar para la inmunización con ácidos nucleicos y la terapia génica, utilizando protocolos estándares de administración de genes. Métodos para el suministro de genes son conocidos en la técnica. Véase, p. ej., Pat. de los EE. UU. N°s 5.399.346, 5.580.859, 5.589.466. En otro caso la divulgación proporciona un vector de terapia génica.
El ácido nucleico se puede clonar en un cierto número de vectores. Por ejemplo, el ácido nucleico puede clonarse en un vector que incluye, pero no se limita a un plásmido, un fagémido, un derivado de fago, un virus animal y un cósmido. Vectores de particular interés incluyen vectores de expresión, vectores de replicación, vectores de generación de sondas y vectores de secuenciación.
Además, el vector de expresión se puede proporcionar a una célula en forma de un vector viral. La tecnología de vectores virales es bien conocida en la técnica y se describe, por ejemplo, en Sambrook et al., 2012, MOLECULAR CLONING: A LABORATORY MANUAL, volúmenes 1-4, Cold Spring Harbor Press, NY) y en otros manuales de virología y biología molecular. Los virus que son útiles como vectores incluyen, pero sin limitación, retrovirus, adenovirus, virus adenoasociados, virus del herpes y lentivirus. En general, un vector adecuado contiene un origen de replicación funcional en al menos un organismo, una secuencia de promotor, sitios convenientes de endonucleasas de restricción y uno o más marcadores seleccionables (p. ej., documentos WO 01/96584; WO 01/29058; y patente de los EE. UU. N.° 6326 193).
Se han desarrollado varios sistemas basados en virus para la transferencia de genes a células de mamíferos. Por ejemplo, los retrovirus proporcionan una plataforma conveniente para los sistemas de suministro de genes. Un gen seleccionado puede insertarse en un vector y empaquetarse en partículas retrovirales utilizando técnicas conocidas en la técnica. A
continuación, el virus recombinante puede aislarse y suministrarse a las células del sujeto in vivo o ex vivo. En la técnica se conocen varios sistemas retrovíricos. En algunas realizaciones, se utilizan vectores adenovíricos. En la técnica se conocen varios vectores adenovíricos. En una realización, se utilizan vectores lentivíricos.
Elementos promotores adicionales, p. ej., potenciadores, regulan la frecuencia del inicio de la transcripción. Normalmente, estos se ubican en la región de 30-110 pb en dirección 5' respecto al sitio de inicio, aunque se ha mostrado que varios promotores contienen también elementos funcionales en dirección 3' respecto al sitio de inicio. El espacio entre los elementos promotores con frecuencia es flexible, de modo que la función del promotor se conserva cuando los elementos se invierten o se mueven uno con respecto al otro. En el promotor de timidina-quinasa (tk), el espacio entre los elementos promotores se puede aumentar a 50 pb de separación antes de que la actividad comience a disminuir. Dependiendo del promotor, parece ser que los elementos individuales pueden funcionar de manera cooperativa o independiente para activar la transcripción. Promotores ilustrativos incluyen promotores del gen IE de CMV, EF-1a, ubiquitina C o de la fosfogliceroquinasa (PGK).
Un ejemplo de un promotor que es capaz de expresar una molécula de ácido nucleico que codifica CAR en un célula T de mamífero es el promotor EFla. El promotor EFla nativo impulsa la expresión de la subunidad alfa del complejo del factor de elongación 1, que es responsable del suministro enzimático de los aminoacil-ARNt al ribosoma. El promotor EFla se ha utilizado exhaustivamente en los plásmidos de expresión de mamíferos y ha mostrado ser eficaz para impulsar la expresión de CAR a partir de moléculas de ácido nucleico clonadas en un vector lentivírico. Véase, p. ej., Milone et al., Mol. Ther. 17(8): 1453-1464 (2009). En un aspecto, el promotor EFla comprende la secuencia proporcionada como la SEQ ID NO:1.
Otro ejemplo de un promotor es la secuencia promotora del citomegalovirus (CMV) inmediata temprana. Esta secuencia promotora es una secuencia promotora constitutiva fuerte capaz de impulsar niveles elevados de expresión de cualquier secuencia polinucleotídica ligada operativamente a ella. Sin embargo, también se pueden utilizar otras secuencias de promotor constitutivas, que incluyen, pero no se limitan al promotor temprano del virus simio 40 (SV40), el virus del tumor mamario de ratón (MMTV), el promotor de repetición terminal larga (LTR) del virus de la inmunodeficiencia humana (VIH), promotor de MoMuLV, un promotor del virus de la leucemia aviar, un promotor temprano inmediato del virus de Epstein-Barr, un promotor del virus del sarcoma de Rous, así como promotores de genes humanos tales como, pero no limitados al promotor de actina, al promotor de miosina, al promotor de factor de elongación-la, al promotor de hemoglobina y al promotor de creatina quinasa. Además, la invención no debe limitarse al uso de promotores constitutivos. Los promotores inducibles también se contemplan como parte de la invención. El uso de un promotor inducible proporciona un conmutador molecular capaz de activar la expresión de la secuencia polinucleotídica que está ligada operativamente cuando se desea tal expresión, o capaz de desactivar la expresión cuando no se desea la expresión. Los ejemplos de promotores inducibles incluyen, pero sin limitación, un promotor de metalotionina, un promotor de glucocorticoides, un promotor de progesterona y un promotor de tetraciclina.
Un vector también puede incluir, p. ej., una secuencia señal para facilitar la secreción, una señal de poliadenilación y un terminador de la transcripción (p. ej., del gen de la hormona del crecimiento bovina (BGH)), un elemento que permite la replicación episómica y la replicación en procariotas (p. ej., origen SV40 y ColE1 u otros conocidos en la técnica) y/o elementos para permitir la selección (p. ej., gen de resistencia a ampicilina y/o marcador de zeocina).
Con el fin de evaluar la expresión de un polipéptido CAR o partes del mismo, el vector de expresión que se ha de introducir en una célula también puede contener un gen marcador seleccionable o un gen informador o ambos para facilitar la identificación y selección de células que expresan de la población de células que se busca transfectar o infectar a través de vectores virales. En otros aspectos, el marcador seleccionable puede transportarse en un trozo separado de ADN y utilizarse en un procedimiento de cotransfección. Tanto los marcadores seleccionables como los genes indicadores pueden estar flanqueados por secuencias reguladoras apropiadas para permitir la expresión en las células hospedadoras. Los marcadores seleccionables útiles incluyen, por ejemplo, genes de resistencia a antibióticos, tales como neo y similares.
Los genes indicadores se utilizan para identificar células potencialmente transfectadas y para evaluar la funcionalidad de secuencias reguladoras. En general, un gen indicador es un gen que no está presente en ni es expresado por el organismo o tejido receptor y que codifica un polipéptido cuya expresión se manifiesta por alguna propiedad fácilmente detectable, p. ej., la actividad enzimática. La expresión del gen indicador se ensaya en un momento adecuado después de que el ADN se haya introducido en las células receptoras. Los genes indicadores adecuados pueden incluir genes que codifican luciferasa, beta-galactosidasa, cloranfenicol acetil transferasa, fosfatasa alcalina secretada o el gen de la proteína verde fluorescente (p. ej., Ui-Tei et al., 2000 FEBS Letters 479: 79-82). Sistemas de expresión adecuados son bien conocidos y pueden prepararse utilizando técnicas conocidas u obtenerse comercialmente. En general, la construcción con la región flanqueante 5' mínima que muestra el nivel más alto de expresión del gen indicador se identifica como el promotor. Dichas regiones de promotor pueden ligarse a un gen indicador y utilizarse para evaluar la capacidad de los agentes para modular la transcripción impulsada por el promotor.
En la técnica se conocen métodos para introducir y expresar genes en una célula. En el contexto de un vector de expresión, el vector se puede introducir fácilmente en una célula hospedadora, p. ej., una célula de mamífero, bacteriana, de levadura
o de insecto mediante cualquier método en la técnica. Por ejemplo, el vector de expresión se puede transferir a una célula hospedadora por medios físicos, químicos o biológicos.
Los métodos físicos para introducir un polinucleótido en una célula hospedadora incluyen precipitación con fosfato cálcico, lipofección, bombardeo de partículas, microinyección, electroporación y similares. En la técnica son bien conocidos métodos para producir células que comprenden vectores y/o ácidos nucleicos exógenos. Véase, por ejemplo, Sambrook et al., 2012, MOLECULAR CLONING: A LABORATORY MANUAL, volúmenes 1-4, Cold Spring Harbor Press, NY). Un método preferido para la introducción de un polinucleótido en una célula hospedadora es la transfección con fosfato de calcio.
Los métodos biológicos para introducir un polinucleótido de interés en una célula hospedadora incluyen el uso de vectores de ADN y ARN. Los vectores víricos, y especialmente los vectores retrovíricos, se han convertido en el método más ampliamente utilizado para insertar genes en células de mamíferos, p. ej., humanas. Otros vectores víricos pueden proceder de lentivirus, poxvirus, virus del herpes simple I, adenovirus y virus adenoasociados y similares. Véanse, por ejemplo, las patentes de los EE. UU. n.° 5.350.674 y 5.585.362.
Los medios químicos para introducir un polinucleótido en una célula hospedadora incluyen sistemas de dispersión coloidal, tales como complejos de macromoléculas, nanocápsulas, microesferas, perlas y sistemas basados en lípidos que incluyen emulsiones de aceite en agua, micelas, micelas mixtas y liposomas. Un sistema coloidal ilustrativo para su uso como vehículo de suministro in vitro e in vivo es un liposoma (p. ej., una vesícula de membrana artificial). Se dispone de otros métodos de vanguardia de suministro dirigido de ácidos nucleicos tales como el suministro de polinucleótidos con nanopartículas dirigidas u otro sistema de suministro adecuado para tamaños submicrométricos.
En el caso en el que se utiliza un sistema de suministro no vírico, un vehículo de suministro ilustrativo es un liposoma. Se contempla el uso de formulaciones lipídicas para la introducción de los ácidos nucleicos en una célula hospedadora (in vitro, ex vivo o in vivo). En otro aspecto, el ácido nucleico puede estar asociado con un lípido. El ácido nucleico asociado con un lípido puede estar encapsulado en el interior acuoso de un liposoma, intercalado dentro de la bicapa lipídica de un liposoma, unido a un liposoma a través de una molécula de enlace que está asociada tanto con el liposoma como con el oligonucleótido, atrapado en un liposoma, formando complejo con un liposoma, disperso en una solución que contiene un lípido, mezclado con un lípido, combinado con un lípido, contenido como una suspensión en un lípido, contenido o formando complejo con una micela, o asociado de otro modo con un lípido. Composiciones asociadas a lípidos, lípidos/ADN o lípidos/vectores de expresión no se limitan a ninguna estructura particular en solución. Por ejemplo, pueden estar presentes en una estructura bicapa, como micelas, o con una estructura "colapsada". También pueden simplemente estar intercaladas en una solución, posiblemente formando agregados que no son uniformes en tamaño o forma. Los lípidos son sustancias grasas que pueden ser lípidos naturales o sintéticos. Por ejemplo, los lípidos incluyen las gotitas grasas que se producen de forma natural en el citoplasma, así como la clase de compuestos que contienen hidrocarburos alifáticos de cadena larga y sus derivados, tales como ácidos grasos, alcoholes, aminas, aminoalcoholes y aldehídos.
Se pueden obtener lípidos adecuados para su uso de fuentes comerciales. Por ejemplo, la dimiristilfosfatidilcolina ("DMPC") se puede obtener de Sigma, San Luis, MO; el fosfato de dicetilo ("DCP") se puede obtener de K & K Laboratories (Plainview, NY); el colesterol ("Choi") se puede obtener de Calbiochem-Behring; el dimiristilfosfatidilglicerol ("DMPG") y otros lípidos se pueden obtener de Avanti Polar Lipids, Inc. (Birmingham, AL.). Las soluciones madre de lípidos en cloroformo o cloroformo/metanol pueden almacenarse a aproximadamente -20 °C. El cloroformo se usa como el único disolvente ya que se evapora más fácilmente que el metanol. "Liposoma" es un término genérico que abarca una diversidad de vehículos lipídicos simples y multilaminares formados por la generación de bicapas o agregados lipídicos encerrados. Los liposomas se pueden caracterizar por tener estructuras vesiculares con una membrana de bicapa de fosfolípidos y un medio acuoso interno. Los liposomas multilaminares tienen múltiples capas de lípidos separadas por un medio acuoso. Se forman espontáneamente cuando los fosfolípidos se suspenden en un exceso de solución acuosa. Los componentes lipídicos se auto-reorganizan antes de la formación de estructuras cerradas y atrapan agua y solutos disueltos entre las bicapas lipídicas (Ghosh et al., 1991 Glycobiology 5: 505-10). Sin embargo, también se incluyen composiciones que tienen estructuras diferentes en solución que la estructura vesicular normal. Por ejemplo, los lípidos pueden asumir una estructura micelar o simplemente existir como agregados no uniformes de moléculas lipídicas. También se contemplan complejos de lipofectamina-ácido nucleico.
Independientemente del método utilizado para introducir ácidos nucleicos exógenos en una célula hospedadora o exponer de otro modo una célula al inhibidor de la presente divulgación, con el fin de confirmar la presencia de la secuencia de ADN recombinante en la célula hospedadora, se puede realizar una diversidad de ensayos. Ensayos de este tipo incluyen, por ejemplo, ensayos "moleculares biológicos" bien conocidos por los expertos en la técnica, tales como la transferencia Southern y Northern, RT-PCR y PCR; ensayos "bioquímicos", tales como detectar la presencia o ausencia de un péptido particular, p. ej., por medios inmunológicos (ELISA y transferencias Western) o mediante ensayos descritos en esta memoria para identificar agentes que caen dentro del alcance de la divulgación.
La presente invención también puede emplear un vector que comprende una molécula de ácido nucleico que codifica CAR. En un aspecto, un vector del CAR se puede transducir directamente en una célula, p. ej., un célula T o un célula NK. En un aspecto, el vector es un vector de clonación o de expresión, p. ej., un vector que incluye, pero sin limitación,
uno o más plásmidos (p. ej., plásmidos de expresión, vectores de clonación, minicírculos, minivectores, cromosomas de doble minuto), construcciones de vectores retrovíricos y lentivíricos. En un aspecto, el vector es capaz de expresar la construcción de CAR en células efectoras inmunitarias de mamífero, (p. ej., célula T, célula NK). En un aspecto, el célula T de mamífero es una célula T humana. En un aspecto, la célula NK de mamífero es una célula NK humana.
Fuentes de células
Antes de la expansión y modificación genética u otra modificación, puede obtenerse de un sujeto una fuente de células, p. ej., células T o células asesinas naturales (NK). Se entiende que el término "sujeto" incluye organismos vivos en los que se puede inducir una respuesta inmunitaria (p. ej., mamíferos). Los ejemplos de sujetos incluyen seres humanos, monos, chimpancés, perros, gatos, ratones, ratas y especies transgénicas de los mismos. Células T pueden obtenerse de varias fuentes, incluyendo células mononucleares de la sangre periférica, médula ósea, tejido de los ganglios linfáticos, sangre del cordón umbilical, tejido del timo, tejido de un sitio de infección, ascitis, derrame pleural, tejido del bazo y tumores.
En determinados aspectos de la presente divulgación, las células efectoras inmunitarias, p. ej., células T, pueden obtenerse de una unidad de sangre recogida de un sujeto utilizando cualquiera de varias técnicas conocidas por el experto en la materia, tales como la separación de Ficoll™. En un aspecto preferido, se obtienen células de la sangre circulante de un individuo por aféresis. El producto de aféresis contiene típicamente células, incluidas células T, monocitos, granulocitos, célula B, otros glóbulos blancos nucleados, glóbulos rojos y plaquetas. En un aspecto, las células recogidas por aféresis pueden lavarse para eliminar la fracción de plasma y, opcionalmente, colocar las células en un tampón o medio apropiado para las etapas de procesamiento posteriores. En un caso, las células se lavan con solución salina tamponada con fosfato (PBS). En un caso alternativo, la solución de lavado carece de calcio y puede carecer de magnesio 0 puede carecer de muchos, si no todos, los cationes divalentes.
Las etapas de activación iniciales en ausencia de calcio pueden conducir a una activación ampliada. Como apreciarán fácilmente los expertos habituales en la materia, una etapa de lavado puede llevarse a cabo por métodos conocidos por los expertos en la materia, tales como mediante el uso de una centrífuga de "flujo continuo" semiautomática (por ejemplo, el procesador de células Cobe 2991, el CytoMate de Baxter o el Cell Saver 5 de Haemonetics) según las instrucciones del fabricante. Después del lavado, las células se pueden resuspender en una diversidad de tampones biocompatibles, tales como, por ejemplo, PBS sin Ca, sin Mg, PlasmaLyte A u otra solución salina con o sin tampón. Como alternativa, se pueden eliminar los componentes indeseables de la muestra de aféresis y las células se resuspenden directamente en el medio de cultivo.
Se reconoce que los métodos de aplicación pueden utilizar condiciones de medios de cultivo que comprenden 5 % o menos, por ejemplo, 2 %, de suero AB humano, y pueden emplear condiciones y composiciones de medios de cultivo conocidas, por ejemplo, las descritas en Smith et al., "Ex vivo expansion of human T cells for adoptive immunotherapy using the novel Xeno-free CTS Immune Cell Serum Replacement" Clinical & Translational Immunology (2015) 4, e31; doi:.
10.1038/cti.2014.31.
En un aspecto, las células T se aíslan de células de la sangre periférica lisando los glóbulos rojos y agotando los monocitos, por ejemplo, mediante centrifugación a través de un gradiente PERCOLL™ o mediante elutriación centrífuga en contraflujo.
Los métodos descritos en esta memoria pueden incluir, p. ej., la selección de una subpoblación específica de células efectoras inmunitarias, p. ej., célula T, que son una población con agotamiento de célula T reguladores, células agotadas en CD25+, utilizando, p. ej., una técnica de selección negativa, p. ej., descrita en esta memoria. Preferentemente, la población de células con agotamiento de células T reguladores contiene menos de un 30 %, 25 %, 20 %, 15 %, 10 %, 5 %, 4 %, 3 %, 2 %, 1 % de células CD25+.
En un caso, las células T reguladoras, p. ej., célula T CD25+, se eliminan de la población utilizando un anticuerpo anti-CD25, o un fragmento del mismo, o un ligando de unión a CD25, IL-2. En un caso, el anticuerpo anti-CD25, o fragmento del mismo, o el ligando de unión a CD25 se conjuga con un sustrato, p. ej., una perla, o se aplica de otro modo sobre un sustrato, p. ej., una perla. En un caso, el anticuerpo anti-CD25, o un fragmento del mismo, se conjuga con un sustrato como se describe en esta memoria.
En un caso, las células T reguladoras, p. ej., las células T CD25+, se eliminan de la población utilizando el reactivo de agotamiento de CD25 de Miltenyi™. En un caso, la relación de células a reactivo de agotamiento de CD25 es 1e7 células a 20 uL, o 1e7 células a 15 uL, o 1e7 células a 10 uL, o 1e7 células a 5 uL, o 1e7 células a 2,5 uL, o 1e7 células a 1,25 uL. En un caso, p. ej., para células T reguladores, p. ej., el agotamiento de CD25+, se utilizan más de 500 millones de células/ml. En un aspecto adicional, se utiliza una concentración de células de 600, 700, 800 o 900 millones de células/ml.
En un caso, la población de células efectoras inmunitarias que se van a agotar incluye aproximadamente 6 x 109 células T CD25+. En otros aspectos, la población de células efectoras inmunitarias que va a depletar incluye de aproximadamente 1 x 109 a 1x 1010 células T CD25+, y cualquier valor entero intermedio. En un caso, la población resultante de células T reguladores empobrecidos tiene 2 x 109 células T reguladoras, p. ej., células CD25+, o menos (p. ej., 1 x 109, 5 x 108, 1 x 108, 5 x 107, 1 x 107, o menos células CD25+).
En un caso, las células T reguladoras, p. ej., células CD25+, se eliminan de la población utilizando el sistema CliniMAC con un conjunto de tubos de agotamiento, tal como, p. ej., el conjunto de tubos 162-01. En un caso, el sistema CliniMAC se ejecuta en una configuración de agotamiento tal como, p. ej., DEPLETION2.1.
Sin desear estar ligados por una teoría en particular, disminuir el nivel de reguladores negativos de las células inmunitarias (p. ej., disminuir el número de células inmunitarias no deseadas, p. ej., células Treg), en un sujeto antes de la aféresis o durante la fabricación de un producto celular que expresa CAR puede reducir el riesgo de recaída del sujeto. Por ejemplo, en la técnica se conocen métodos para agotar células Treg. Los métodos de disminución de células Treg incluyen, pero sin limitación, ciclofosfamida, anticuerpo anti-GITR (un anticuerpo anti-GITR descrito en esta memoria), agotamiento de CD25 y combinaciones de los mismos.
En algunos casos, los métodos de fabricación comprenden reducir (p. ej., agotar) el número de células Treg antes de la fabricación de la célula que expresa CAR. Por ejemplo, los métodos de producción comprenden poner en contacto la muestra, p. ej., la muestra de aféresis, con un anticuerpo anti-GITR y/o un anticuerpo anti-CD25 (o un fragmento del mismo, o un ligando de unión a CD25), p. ej., para agotar células Treg antes de producir el producto de tipo célula que expresa CAR (p. ej., célula T, célula NK).
En un caso, un sujeto se trata previamente con una o más terapias que reducen las células Treg antes de la recogida de células para la fabricación de productos celulares que expresan CAR, reduciendo con ello el riesgo de recaída del sujeto al tratamiento con células que expresan CAR. En un caso, los métodos para disminuir las células Treg incluyen, pero sin limitación, la administración al sujeto de uno o más de ciclofosfamida, anticuerpo anti-GITR, agotamiento de CD25 o una combinación de los mismos. La administración de uno o más de ciclofosfamida, anticuerpo anti-GITR, agotamiento de CD25, o una combinación de los mismos, puede realizarse antes, durante o después de una infusión del producto de tipo célula que expresa CAR.
En un caso, un sujeto se trata previamente con ciclofosfamida antes de la recogida de células para la fabricación de productos celulares que expresan CAR, reduciendo con ello el riesgo de recaída del sujeto al tratamiento con células que expresan CAR. En un caso, un sujeto se trata previamente con un anticuerpo anti-GITR antes de la recogida de células para la fabricación de productos celulares que expresan CAR, reduciendo con ello el riesgo de recaída del sujeto al tratamiento con células que expresan CAR.
En un caso, la población de células para eliminar no son las células T reguladoras ni las células tumorales, sino células que de otro modo afectan negativamente a la expansión y/o función de células CART, p. ej., células que expresan CD14, CD11b, CD33, CD15 u otros marcadores expresados por células potencialmente inmunosupresoras. En un caso, se prevé que este tipo de células se eliminen al mismo tiempo que las células T reguladoras y/o las células tumorales, o después de dicho agotamiento, o en otro orden.
Los métodos descritos en esta memoria pueden incluir más de una etapa de selección, p. ej., más de una etapa de agotamiento. El enriquecimiento de una población de células T por selección negativa se puede lograr, por ejemplo, con una combinación de anticuerpos dirigidos a marcadores de superficie exclusivos de las células seleccionadas negativamente. Un método es la clasificación y/o selección celular mediante inmunoadherencia magnética negativa o citometría de flujo que utiliza un cóctel de anticuerpos monoclonales dirigidos a marcadores de la superficie celular presentes en las células seleccionadas negativamente. Por ejemplo, para enriquecer células CD4+ mediante selección negativa, un cóctel de anticuerpos monoclonales puede incluir anticuerpos contra CD14, CD20, CD11b, CD16, HLA-DR y CD8.
Los métodos descritos en esta memoria pueden incluir, además, la eliminación de células de la población que expresan un antígeno tumoral, p. ej., un antígeno tumoral que no comprende CD25, p. ej., CD19, CD30, Cd 38, CD123, CD20, CD14 o CD11b, para proporcionar con ello una población de célula T reguladores agotados, p. ej., agotados en CD25+, y agotados en antígeno tumoral que son adecuados para la expresión de un CAR, p. ej., un CAR descrito en esta memoria. En un caso, las células que expresan un antígeno tumoral se eliminan simultáneamente con las células T reguladoras, p. ej., CD25+. Por ejemplo, un anticuerpo anti-CD25, o fragmento del mismo, y un anticuerpo antigénico antitumoral, o fragmento del mismo, se pueden fijar al mismo sustrato, p. ej., una perla, que se puede utilizar para eliminar las células o un anticuerpo anti-CD25, o fragmento del mismo, o el anticuerpo antigénico anti-tumoral, o fragmento del mismo, se pueden fijar a perlas separadas, una mezcla de las cuales se puede utilizar para eliminar las células. En otros casos, la eliminación de los células T reguladoras, p. ej., las células CD25+, y la eliminación de las células que expresan el antígeno tumoral es secuencial y se puede producir, p. ej., en cualquier orden.
También se proporcionan métodos que incluyen eliminar células de la población que expresen un inhibidor del punto de control, p. ej., un inhibidor del punto de control descrito en el presente memoria, p. ej., una o más de células PD1+, células LAG3+ y células TIM3+, para proporcionar con ello una población de células T reguladoras agotadas, p. ej., agotadas en CD25+ y células agotadas en inhibidor del punto de control, p. ej., células agotadas en PD1+, LAG3+ y/o TIM3+. Los inhibidores de puntos de control ilustrativos incluyen B7-H1, B7-1, CD160, P1H, 2B4, PD1, TIM3, c Ea CAM (p. ej., CEACAM-1, c Ea CAM-3 y/o CEACAM-5), LAG3, TIGIT, CTLA-4, BTLA y LAIR1. En un caso, las células que expresan
inhibidores de puntos de control se eliminan simultáneamente con las células T reguladoras, p. ej., CD25+. Por ejemplo, un anticuerpo anti-CD25, o un fragmento del mismo, y un anticuerpo anti-inhibidor del punto de control, o un fragmento del mismo, se pueden unir a la misma perla que se puede usar para eliminar las células, o un anticuerpo anti-CD25, o fragmento del mismo, y el anticuerpo anti-inhibidor del punto de control, o un fragmento del mismo, se pueden unir a perlas separadas, una mezcla de las cuales se puede utilizar para eliminar las células. En otros casos, la eliminación de las células T reguladoras, p. ej., células CD25+, y la eliminación de las células que expresan el inhibidor del punto de control es secuencial y se puede producir, p. ej., en cualquier orden.
Los métodos descritos en esta memoria pueden incluir una etapa de selección positiva. Por ejemplo, células T pueden aislarse mediante incubación con perlas conjugadas anti-CD3/anti-CD28 (p. ej., 3x28), tales como DYNABEADS® M-450 CD3/CD28 T, durante un periodo de tiempo suficiente para la selección positiva de las células T deseadas. En un caso, el período de tiempo es de aproximadamente 30 minutos. En otro caso, el período de tiempo varía de 30 minutos a 36 horas o más y todos los valores enteros entremedias. En un caso adicional, el período de tiempo es de al menos 1, 2, 3, 4, 5 o 6 horas. En aún otro caso, el período de tiempo es de 10 a 24 horas, p. ej., 24 horas. Se pueden utilizar tiempos de incubación más prolongados para aislar células T en cualquier situación en la que haya pocas células T en comparación con otros tipos de células, como en el aislamiento de células infiltrantes de tumores (TIL) del tejido tumoral o de individuos inmunocomprometidos. Además, el uso de tiempos de incubación más largos puede aumentar la eficiencia de captura de células T CD8+. Por tanto, simplemente acortando o alargando el tiempo en que se permite que las células T se unan a las perlas de CD3/CD28 y/o aumentando o disminuyendo la relación de perlas respecto a células T (como se describe más adelante en esta memoria), se puede seleccionar preferentemente a favor o en contra de subpoblaciones de células T en el inicio del cultivo o en otros puntos temporales durante el proceso. Adicionalmente, al aumentar o disminuir la relación de anticuerpos anti-CD3 y/o anti-CD28 en las perlas u otra superficie, se puede seleccionar preferentemente a favor o en contra de las subpoblaciones de células T en el inicio del cultivo o en otros puntos temporales deseados.
En un caso, se puede seleccionar una población de células T que expresa uno o más de IFN-Y, TNFa, IL-17A, IL-2, IL-3, IL-4, GM-CSF, IL-10, IL-13, granzima B y perforina u otras moléculas apropiadas, p. ej., otras citoquinas. Métodos para la selección de la expresión celular se pueden determinar, p. ej., mediante los métodos descritos en la publicación PCT N° WO 2013/126712.
Para el aislamiento de una población deseada de células mediante selección positiva o negativa, se puede variar la concentración de células y la superficie (p. ej., partículas tales como perlas). En determinados aspectos, puede ser deseable disminuir significativamente el volumen en el que se mezclan entre sí las perlas y las células (p. ej., aumentar la concentración de células), para garantizar el máximo contacto de las células y las perlas. Por ejemplo, en un aspecto, se utiliza una concentración de 10 mil millones de células/ml, 9 mil millones/ml, 8 mil millones/ml, 7 mil millones/ml, 6 mil millones/ml o 5 mil millones/ml. En un aspecto, se utiliza una concentración de mil millones de células/ml. En un aspecto más, se utiliza una concentración de células de 75, 80, 85, 90, 95 o 100 millones de células/ml. En aspectos adicionales, pueden utilizarse concentraciones de 125 o 150 millones de células/ml.
El uso de altas concentraciones puede dar lugar a un mayor rendimiento celular, activación celular y expansión celular. Además, el uso de altas concentraciones de células permite una captura más eficiente de las células que pueden expresar débilmente los antígenos diana de interés, tales como células T CD28-negativas, o de muestras en las que hay muchas células tumorales presentes (p. ej., sangre leucémica, tejido tumoral, etc.). Poblaciones de células de este tipo pueden tener un valor terapéutico y sería deseable obtenerlas. Por ejemplo, el uso de una alta concentración de células permite una selección más eficiente de células T CD8+ que normalmente tienen una expresión de CD28 más débil.
En un aspecto relacionado, puede ser deseable utilizar concentraciones más bajas de células. Al diluir significativamente la mezcla de células T y la superficie (p. ej., partículas tales como perlas), se minimizan las interacciones entre las partículas y las células. Esto selecciona las células que expresan grandes cantidades de antígenos deseados para unirse a las partículas. Por ejemplo, las células T CD4+ expresan niveles más altos de CD28 y se capturan de manera más eficiente que los célula T CD8+ en concentraciones diluidas. En un aspecto, la concentración de células utilizada es de 5 x 106/ml. En otros aspectos, la concentración utilizada puede ser de aproximadamente 1 x 105/ml a 1 x 106/ml, y cualquier valor entero intermedio.
En otros aspectos, las células se pueden incubar en un rotador durante periodos de tiempo variables a velocidades variables a 2-10 °C o a temperatura ambiente.
Las células T para la estimulación también se pueden congelar después de una etapa de lavado. Sin desear quedar ligado a la teoría, la etapa de congelación y descongelación posterior proporciona un producto más uniforme al eliminar los granulocitos y, en cierta medida, los monocitos de la población celular. Después de la etapa de lavado que elimina plasma y plaquetas, las células pueden suspenderse en una solución de congelación. Si bien se conocen en la técnica muchas soluciones y parámetros de congelación y serán útiles en este contexto, un método implica el uso de PBS que contiene 20 % de DMSO y 8 % de seroalbúmina humana, o medios de cultivo que contienen 10 % de dextrano 40 y 5 % de dextrosa, 20 % de seroalbúmina humana y 7,5 % de DMSO o 31,25 % de Plasmalyte-A, 31,25 % de dextrosa al 5 %, 0,45 % de NaCl, 10% de dextrano 40 y 5% de dextrosa, 20% de seroalbúmina humana y 7,5% de DMSO u otro medio de congelación celular adecuado que contiene, por ejemplo, Hespan y PlasmaLyte A, las células se congelan después a 80 °C a una velocidad de 1 ° por minuto y se almacenan en la fase de vapor de un tanque de almacenamiento de nitrógeno líquido. Pueden utilizarse otros métodos de congelación controlada, así como la congelación incontrolada inmediatamente a -20 °C o en nitrógeno líquido.
En determinados aspectos, las células crioconservadas se descongelan y lavan como se describe en esta memoria y se dejan reposar durante una hora a temperatura ambiente antes de la activación utilizando los métodos de la presente divulgación.
También se contempla en el contexto de la invención la recogida de muestras de sangre o producto de aféresis de un sujeto en un periodo de tiempo anterior al momento en que se podrían necesitar las células expandidas como se describen en esta memoria. Como tal, la fuente de las células que se van a expandir se puede recoger en cualquier momento necesario y las células deseadas, tales como células T, se pueden aislar y congelar para su uso posterior en la terapia de células efectoras inmunitarias para cualquiera de varias enfermedades o afecciones que se beneficiarían de la terapia con células efectoras inmunitarias, tales como las descritas en esta memoria. En un aspecto, se toma una muestra de sangre o una aféresis de un sujeto generalmente sano. En determinados aspectos, se toma una muestra de sangre o una aféresis de un sujeto generalmente sano que está en riesgo de desarrollar una enfermedad, pero que aún no ha desarrollado una enfermedad, y las células de interés se aíslan y congelan para su uso posterior. En determinados aspectos, las células T pueden expandirse, congelarse y utilizarse en un momento posterior. En determinados aspectos, las muestras se recogen de un paciente poco después del diagnóstico de una enfermedad particular, como se describe en esta memoria, pero antes de cualquier tratamiento. En un aspecto adicional, las células se aíslan de una muestra de sangre o una aféresis de un sujeto antes de cualquiera de varias modalidades de tratamiento relevantes, que incluyen, pero sin limitación, el tratamiento con agentes tales como natalizumab, efalizumab, agentes antivíricos, quimioterapia, radiación, agentes inmunosupresores, tales como ciclosporina, azatioprina, metotrexato, micofenolato y FK506, anticuerpos u otros agentes inmunoablativos, tales como CAMPATH, anticuerpos anti-CD3, citoxano, fludarabina, ciclosporina, FK506, rapamicina, ácido micofenólico, esteroides, FR901228 e irradiación.
En un aspecto adicional de la presente divulgación, las células T se obtienen de un paciente directamente después del tratamiento que deja al sujeto con células T funcionales. En este sentido, se ha observado que tras determinados tratamientos oncológicos, en particular tratamientos con fármacos que dañan el sistema inmunitario, poco después del tratamiento durante el periodo en el que los pacientes normalmente se estarían recuperando del tratamiento, la calidad de las células T obtenidas puede ser óptima o mejorada con respecto a su capacidad de expandirse ex vivo. Del mismo modo, después de la manipulación ex vivo utilizando los métodos descritos en esta memoria, estas células pueden estar en un estado preferido para un mejor prendimiento del injerto y expansión in vivo. Por tanto, se contempla dentro del contexto de la presente divulgación recoger células de la sangre, incluidos células T, células dendríticas u otras células del linaje hematopoyético, durante esta fase de recuperación. Además, en determinados aspectos, la movilización (por ejemplo, la movilización con GM-CSF) y los regímenes de acondicionamiento se pueden utilizar para crear una condición en un sujeto en la que se favorezca la repoblación, recirculación, regeneración y/o expansión de tipos de células particulares, especialmente durante una ventana de tiempo definida después de la terapia. Tipos de células ilustrativas incluyen células T, células B, células dendríticas y otras células del sistema inmunitario.
En un caso, las células efectoras inmunitarias que expresan una molécula de CAR, p. ej., una molécula de CAR descrita en esta memoria, se obtienen de un sujeto que ha recibido una dosis baja, potenciadora de la inmunidad, de un inhibidor de mTOR. En un caso, la población de células efectoras inmunitarias, p. ej., células T, que se han de manipular para expresar un CAR, se recoge después de un tiempo suficiente, o después de una dosificación suficiente de la dosis baja, potenciadora del sistema inmunológico, de un inhibidor de mTOR, de manera que el nivel de células efectoras inmunes PD1-negativas, p. ej., células T, o la relación de células efectoras inmunitarias PD1-negativas, p. ej., células T/células efectoras inmunitarias PD1-positivas, p. ej., células T, en el sujeto o recogido del sujeto aumentó, al menos transitoriamente.
En otros casos, la población de células efectoras inmunitarias, p. ej., las células T, que han sido o serán manipuladas para expresar un CAR, puede tratarse ex vivo por contacto con una cantidad de un inhibidor de mTOR que aumenta el número de células efectoras inmunitarias PD1-negativas, p. ej., células T o aumenta la relación de células efectoras inmunitarias PD1-negativas, p. ej., células T/células efectoras inmunitarias PD1-positivas, p. ej., células T.
En una realización, una población de células T es deficiente en diaglicerol quinasa (DGK, por sus siglas en inglés). Las células deficientes en d Gk incluyen células que no expresan ARN o proteína de DGK, o tienen una reducción o inhibición de la actividad de DGK. Las células deficientes en DGK se pueden generar mediante estrategias genéticas, por ejemplo, administración de agentes de interferencia por ARN, por ejemplo, ARNip, ARNhc, miARN, para reducir o prevenir el expresión de DGK. Como alternativa, las células deficientes en DGK se pueden generar mediante tratamiento con inhibidores de DGK descritos en la presente.
En una realización, una población de células T es deficiente en Ikaros. Las células deficientes en Ikaros incluyen células que no expresan el ARN o la proteína de Ikaros, o tienen una actividad de Ikaros reducida o inhibida, las células deficientes en Ikaros se pueden generar mediante enfoques genéticos, p. ej., administrando agentes de interferencia por ARN, p. ej.,
ARNip, ARNhc, miARN, para reducir o evitar la expresión de Ikaros. Como alternativa, las células deficientes en Ikaros se pueden generar mediante tratamiento con inhibidores de Ikaros, p. ej., lenalidomida.
En algunas realizaciones, una población de células T es deficiente en DGK y deficiente en Ikaros, p. ej., no expresa DGK ni Ikaros, o tiene una actividad de DGK e Ikaros reducida o inhibida. Células deficientes en DGK e Ikaros de este tipo se pueden generar mediante cualquiera de los métodos descritos en esta memoria.
En una realización, las células NK se obtienen del sujeto. En otra realización, las células NK son una línea de células NK, p. ej., línea celular NK-92 (Conkewest).
CAR alogénico
En una realización descrita en esta memoria, la célula efectora inmunitaria puede ser una célula efectora inmunitaria alogénica, p. ej., una célula T o una célula NK. Por ejemplo, la célula puede ser una célula T alogénica, p. ej., una célula T alogénica que carece de expresión de un receptor de célula T funcional (TCR) y/o antígeno leucocitario humano (HLA), p. ej., HLA de clase I y/o HLA de clase II.
Una célula T que carece de un TCR funcional puede, p. ej., manipularse de manera que no exprese ningún TCR funcional en su superficie, manipularse de manera que no exprese una o más subunidades que comprendan un TCR funcional o manipularse de manera que produzcan muy poco TCR funcional en su superficie. Como alternativa, la célula T puede expresar un TCR sustancialmente alterado, p. ej., mediante la expresión de formas mutadas o truncadas de una o más de las subunidades del TCR. La expresión "t Cr sustancialmente alterado" significa que este TCR no inducirá una reacción inmunitaria adversa en un hospedador.
Una célula T descrita en esta memoria puede, p. ej., manipularse de manera que no exprese un HLA funcional en su superficie. Por ejemplo, un célula T descrita en esta memoria puede manipularse de manera que la expresión de HLA en la superficie celular, p. ej., HLA de clase 1 y/o HLA de clase II, se regule a la baja.
En algunas realizaciones, la célula T puede carecer de un TCR funcional y un HLA funcional, p. ej., HLA de clase I y/o HLA de clase II.
Células T modificadas que carecen de expresión de un TCR y/o HLA funcionales pueden obtenerse por cualquier medio adecuado, incluida la inactivación o atenuación de una o más subunidades de TCR o HLA. Por ejemplo, la célula T puede incluir una atenuación de TCR y/o HLA utilizando ARNip, ARNhc, grupos de repeticiones palindrómicas cortas en intervalos regulares (CRISPR), nucleasa efectora de tipo activador de la transcripción (TALEN) o endonucleasa con dedos de zinc (ZFN).
En algunas realizaciones, la célula alogénica puede ser una célula que no expresa o expresa en niveles bajos una molécula inhibidora, p. ej., mediante cualquier método descrito en esta memoria. Por ejemplo, la célula puede ser una célula que no expresa o expresa en niveles bajos una molécula inhibidora, p. ej., que puede disminuir la capacidad de una célula que expresa CAR de montar una respuesta efectora inmunitaria. Ejemplos de moléculas inhibidoras incluyen PD1, PD-L1, CTLA4, TIM3, CEACAM (p. ej., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 y TGFR beta. La inhibición de una molécula inhibidora, p. ej., mediante inhibición a nivel de ADN, ARN o proteína, puede optimizar el rendimiento de una célula que expresa CAR. En algunas realizaciones, se puede utilizar un ácido nucleico inhibidor, p. ej., un ácido nucleico inhibidor, p. ej., un ARNbc, p. ej., un ARNip o ARNhc, grupos de repeticiones palindrómicas cortas en intervalos regulares (CRISPR), nucleasa efectora de tipo activador de la transcripción (TALEN) o una endonucleasa con dedos de zinc (ZFN), p. ej., como se describe en esta memoria.
ARNip y ARNhc para inhibir TCR o HLA
En algunas realizaciones, la expresión de TCR y/o la expresión de HLA se puede inhibir utilizando ARNip o ARNhc que se dirige a un ácido nucleico que codifica un TCR y/o HLA en una célula T.
La expresión de ARNip o ARNhc en células T se puede lograr utilizando cualquier sistema de expresión convencional, p. ej., tal como un sistema de expresión lentivírico.
Se describen ARNhc ilustrativos que regulan a la baja la expresión de componentes del TCR, p. ej., en la publicación de los EE. UU. n.°: 2012/0321667. Se describen ARNip y ARNhc ilustrativos que regulan a la baja la expresión de los genes de HLA de clase I y/o HLA de clase II, p. ej., en la publicación de los EE. UU. n.°: US 2007/0036773.
CRISPR para inhibir TCR o HLA
La expresión "CRISPR" o "CRISPR contra TCR y/o HLA" o "CRISPR para inhibir TCR y/o HLA", como se utiliza en esta memoria, se refiere a un conjunto de grupos de repeticiones palindrómicas cortas en intervalos regulares, o un sistema que comprende un conjunto de repeticiones de este tipo. El término "Cas", como se utiliza en esta memoria, se refiere a
una proteína asociada a CRISPR. Un sistema "CRISPR/Cas" se refiere a un sistema derivado de CRISPR y Cas que puede usarse para silenciar o mutar un gen de TCR y/o HLA.
Se observan sistemas CRISPR/Cas de origen natural en aproximadamente un 40 % de los genomas eubacterianos secuenciados y en un 90 % de las arqueas secuenciadas. Grissa et al. (2007) BMC Bioinformatics 8: 172. Este sistema es un tipo de sistema inmunitario procariótico que confiere resistencia a elementos genéticos exógenos tales como plásmidos y fagos y proporciona una forma de inmunidad adquirida. Barrangou et al. (2007) Science 315: 1709-1712; Marragini et al. (2008) Science 322: 1843-1845.
El sistema CRISPR/Cas se ha modificado para su uso en la edición génica (silenciamiento, potenciación o cambio de genes específicos) en eucariotas tales como ratones o primates. Wiedenheft et al. (2012) Nature 482: 331-8. Esto se logra introduciendo en la célula eucariota un plásmido que contiene un CRISPR diseñado específicamente y una o más Cas apropiadas.
La secuencia CRISPR, a veces denominada locus CRISPR, comprende repeticiones y espaciadores que se alternan. En un CRISPR de origen natural, los espaciadores comprenden normalmente secuencias exógenas respecto a la bacteria, tales como una secuencia de fago o plásmido; en el sistema CRISPR/Cas de TCR y/o HLA, los espaciadores proceden de la secuencia del gen de TCR o HLA.
El ARN del locus CRISPR se expresa de manera constitutiva y es procesado por proteínas Cas para obtener ARN pequeños. Estos comprenden un espaciador flanqueado por una secuencia repetida. Los ARN guían otras proteínas Cas para silenciar elementos genéticos exógenos a nivel de ARN o ADN. Horvath et al. (2010) Science 327: 167-170; Makarova et al. (2006) Biology Direct 1: 7. Los espaciadores sirven de este modo como moldes para moléculas de ARN, de manera análoga a los ARNip. Pennisi (2013) Science 341: 833-836.
Ya que estos ocurren de manera natural en muchos tipos diferentes de bacterias, las disposiciones exactas de los CRISPR y la estructura, función y número de genes Cas y su producto difieren algo entre especies. Haft et al. (2005) PLoS Comput. Biol. 1: e60; Kunin et al. (2007) Genome Biol. 8: R61; Mojica et al. (2005) J. Mol. Evol. 60: 174-182; Bolotin et al. (2005) Microbiol. 151: 2551-2561; Pourcel et al. (2005) Microbiol. 151: 653-663; y Stern et al. (2010) Trends. Genet. 28: 335-340. Por ejemplo, las proteínas Cse (subtipo Cas, E. coli) (p. ej., CasA) forman un complejo funcional, Cascade, que procesa los transcritos de ARN de CRISPR para obtener unidades de espaciador-repetición que retiene Cascade. Brouns et al. (2008) Science 321: 960-964. En otras procariotas, Cas6 procesa el transcrito CRISPR. La inactivación de fagos basada en CRISPR en E. coli requiere Cascade y Cas3, pero no Cas1 o Cas2. Las proteínas Cmr (módulo Cas RAMP) en Pyrococcus furiosus y otras procariotas forman un complejo funcional con ARN CRISPR pequeños que reconoce y escinde ARN diana complementarios. Un sistema CRISPR más simple depende de la proteína Cas9, que es una nucleasa con dos sitios de corte activos, uno para cada hebra de la doble hélice. La combinación de Cas9 y ARN del locus CRISPR modificado se puede utilizar en un sistema para la edición génica. Pennisi (2013) Science 341: 833-836.
El sistema CRISPR/Cas se puede utilizar, por lo tanto, para editar un gen de TCR y/o HLA (añadiendo o eliminando un par de bases) o introducir una parada prematura que disminuya de esta manera la expresión de TCR y/o HLA. Como alternativa, el sistema CRISPR/Cas se puede utilizar como un ARN de interferencia, desactivando el gen de TCR y/o HLA de manera reversible. En una célula de mamífero, por ejemplo, el ARN puede guiar la proteína Cas a un promotor de TCR y/o HLA, y bloquear estéricamente las ARN polimerasas.
Se pueden generar sistemas CRISPR/Cas artificiales que inhiben TCR y/o HLA, utilizando tecnología conocida en la técnica, p. ej., la descrita en la publicación de los EE. UU. n.° 20140068797 y Cong (2013) Science 339: 819-823. También se pueden generar otros sistemas CRISPR/Cas artificiales que se conocen en la técnica que inhiben TCR y/o HLA, p. ej., el descrito en Tsai (2014) Nature Biotechnol., 32:6569-576, patente de los EE. UU. n.°: 8,871,445; 8,865,406; 8,795,965; 8,771,945; y 8,697,359.
TALEN para inhibir TCR y/o HLA
"TALEN" o "TALEN contra HLA y/o TCR" o "TALEN para inhibir HLA y/o TCR" se refiere a una nucleasa efectora de tipo activador de la transcripción, una nucleasa artificial que se puede usar para editar el gen de HLA y/o TCR.
Los TALEN se producen de manera artificial fusionando un dominio de unión a ADN del efector TAL con un dominio de escisión de ADN. Los efectos de tipo activador de la transcripción (TALE) se pueden manipular para que se unan a cualquier secuencia de ADN deseada, incluida una porción del gen de HLA o TCR. Al combinar un TALE manipulado con un dominio de escisión de ADN, se puede producir una enzima de restricción que es específica para cualquier secuencia de ADN deseada, incluida una secuencia de HLA o TCR. Estas se pueden introducir posteriormente en una célula, en donde se pueden utilizar para la edición genómica. Boch (2011) Nature Biotech. 29: 135-6; y Boch et al. (2009) Science 326: 1509-12; Moscou et al. (2009) Science 326: 3501.
Los TALE son proteínas secretadas por bacterias Xanthomonas. El dominio de unión a ADN contiene una secuencia repetida de 33-34 aminoácidos muy conservados con excepción de los aminoácidos 12 y 13. Estas dos posiciones son
muy variables y muestran una sólida correlación con el reconocimiento de nucleótidos específicos. Por lo tanto, se pueden modificar para unirse a una secuencia de ADN deseada.
Para producir un TALEN, una proteína TALE se fusiona con una nucleasa (N), que es una endonucleasa Fokl de tipo salvaje o mutada. Se han realizado varias mutaciones de Fokl para su uso en TALEN; estos, por ejemplo, mejoran la especificidad o actividad de escisión. Cermak et al. (2011) Nucl. Acids Res. 39: e82; Miller et al. (2011) Nature Biotech.
29:: 143-8; Hockemeyer et al., (2011) Nature Biotech. 29: 731-734; Wood et al., (2011) Science 333: 307; Doyon et al., (2010) Nature Methods 8: 74-79; Szczepek et al., (2007) Nature Biotech. 25: 786-793; y Guo et al. (2010) J. Mol. Biol. 200: 96.
El dominio Fokl funciona como un dímero, lo que requiere dos construcciones con dominios de unión a ADN únicos para sitios en el genoma diana con orientación y espaciado adecuados. Tanto el número de residuos de aminoácidos entre el dominio de unión al ADN de TALE y el dominio de escisión de Fokl como el número de bases entre los dos sitios de unión de TALEN individuales parecen ser parámetros importantes para lograr altos niveles de actividad. Miller et al., (2011) Nature Biotech. 29: 143-8.
Se puede utilizar una TALEN para HLA o TCR dentro de una célula para producir una rotura bicatenaria (DSB, por sus siglas en inglés). Se puede introducir una mutación en el sitio de rotura si los mecanismos de reparación reparan de manera incorrecta la rotura mediante unión de extremos no homólogos. Por ejemplo, la reparación incorrecta puede introducir una mutación de desplazamiento del marco de lectura. Como alternativa, se puede introducir ADN exógeno en la célula junto con la TALEN; dependiendo de las secuencias del ADN exógeno y la secuencia cromosómica, este proceso se puede utilizar para corregir un defecto en el gen de HLA o TCR o introducir un defecto de este tipo en un gen de HLA o TCR ts, y reducir de este modo la expresión de HLA o TCR.
Se pueden construir TALEN específicas para secuencias en HLA o TCR utilizando cualquier método conocido en la técnica, incluidos varios esquemas que utilizan componentes modulares. Zhang et al., (2011) Nature Biotech. 29: 149-53; Geibler et al. (2011) PLoS ONE 6: e19509.
Nucleasa con dedos de zinc para inhibir HLA y/o TCR
"ZFN" o "nucleasa de dedos de zinc" o "ZFN contra HLA y/o TCR" o "ZFN para inhibir HLA y/o TCR" se refiere a una nucleasa de dedos de zinc, una nucleasa artificial que se puede usar para editar el gen de HLA y/o TCR.
Al igual que un TALEN, un ZFN comprende un dominio de nucleasa Fokl (o un derivado del mismo) fusionado con un dominio de unión a ADN. En el caso de una ZFN, el dominio de unión a ADN comprende uno o más dedos de zinc. Carroll et al., (2011) Genetics Society of America 188: 773-782; y Kim et al., (1996) Proc. Natl. Acad. Sci. USA 93: 1156-1160.
Un dedo de zinc es un pequeño motivo estructural proteico estabilizado por uno o más iones de zinc. Un dedo de zinc puede comprender, por ejemplo, Cys2His2, y puede reconocer una secuencia de aproximadamente 3 pb. Se pueden combinar diversos dedos de zinc de especificidad conocida para producir polipéptidos con múltiples dedos que reconocen secuencias de aproximadamente 6, 9, 12, 15 o 18 pb. Se puede disponer de diversas técnicas de selección y ensamblaje modular para generar dedos de zinc (y combinaciones de los mismos) que reconocen secuencias específicas, incluidas la presentación en fagos, sistemas de un único híbrido en levaduras, sistemas de un único híbrido y de doble híbrido bacterianos y células de mamífero.
Como una TALEN, una ZFN debe dimerizar para escindir ADN. Por lo tanto, se necesita un par de ZFN para dirigirse a sitios de ADN no palindrómicos. Las dos ZFN individuales deben unirse a hebras opuestas del ADN con sus nucleasas debidamente separadas. Bitinaite et al., (1998) Proc. Natl. Acad. Sci. USA 95: 10570-5.
También al igual que una TALEN, una ZFN puede crear una rotura bicatenaria en el ADN, que puede crear una mutación de desplazamiento del marco de lectura si se repara de forma incorrecta, lo que conlleva un descenso de la expresión y cantidad de HLA y/o TCR en una célula. Las ZFN también se pueden utilizar con recombinación homóloga para mutar en el gen de HLA o TcR.
Se pueden construir ZFN específicas para secuencias en HLA y/o TCR utilizando cualquier método conocido en la técnica. Véase, p. ej., Provasi (2011) Nature Med. 18: 807-815; Torikai (2013) Blood 122: 1341-1349; Cathomen et al., (2008) Mol. Ther. 16: 1200-7; Guo et al., (2010) J. Mol. Biol. 400: 96; publicación de patente de los EE. UU. 2011/0158957; y publicación de patente de los EE. UU. 2012/0060230.
Expresión de telomerasa
Sin desear quedar ligado a ninguna teoría particular, en algunas realizaciones, una célula T terapéutica tiene una persistencia poco prolongada en un paciente, debido a telómeros acortados en la célula T; en consecuencia, la transfección con un gen de telomerasa puede alargar los telómeros de la célula T y mejorar la persistencia de la célula T en el paciente. Véase Carl June, "Adoptive T cell therapy for cancer in the clinic", Journal of Clinical Investigation, 117:
1466-1476 (2007). Por lo tanto, en una realización, una célula efectora inmunitaria, p. ej., una célula T, expresa de manera ectópica una subunidad de telomerasa, p. ej., la subunidad catalítica de la telomerasa, p. ej., TERT, p. ej., hTERT. En algunos aspectos, esta divulgación proporciona un método para producir una célula que expresa CAR, que comprende poner en contacto una célula con un ácido nucleico que codifica una subunidad de telomerasa, p. ej., la subunidad catalítica de la telomerasa, p. ej., TERT, p. ej., hTERT. La célula se puede poner en contacto con el ácido nucleico antes, a la vez o después de ponerse en contacto con una construcción que codifica un CAR.
En un aspecto, la divulgación presenta un método para generar una población de células efectoras inmunitarias (p. ej., células T, células NK). En un caso, el método comprende: proporcionar una población de células efectoras inmunitarias (por ejemplo, células T o células NK), poner en contacto la población de células efectoras inmunitarias con un ácido nucleico que codifica un CAR; y poner en contacto la población de células efectoras inmunitarias con un ácido nucleico que codifica una subunidad de telomerasa, por ejemplo, hTERT, en condiciones que permiten la expresión de CAR y la telomerasa.
En un caso, el ácido nucleico que codifica la subunidad de telomerasa es ADN. En un caso, el ácido nucleico que codifica la subunidad de telomerasa comprende un promotor capaz de impulsar la expresión de la subunidad de telomerasa.
En una realización, la hTERT tiene la secuencia de aminoácidos de ID de proteína de GenBank AAC51724.1 (Meyerson et al., "hEST2, the Putative Human Telomerase Catalytic Subunit Gene, Is Up-Regulated in Tumor Cells and during Immortalization" Cell volumen 90, número 4, 22 de agosto de 1997, páginas 785-795) de la siguiente manera:
MPRAPRCR A VRSLLRSHYRE VLPL ATF VRRLGPQ GWRL V QRGDP A AFR AL V AQ CL Y C YP WD ARPPP AAP SFRQ Y S CLKEL V ARVLQRLCERGAKNVL AF GF ALLDGARGG PPEAFTTSVRSYLPNTVTDALRGSGAWGLLLRRVGDDVLVHLLARCALFVLVAPSC AYQVCGPPLYQLGAATQARPPPHASGPRRRLGCERAWNHSVREAGVPLGLPAPGA RRRGGS ASRSLPLPKRPRRGAAPEPERTP V GQGS W AHPGRTRGP SDRGF C VV SP AR P AEE AT SLEGAL S GTRHSHP S VGRQHH AGPP S T SRPPRPWDTPCPP V Y AETKHFL Y S SGDKEQLRPSFLLSSLRPSLTGARRLVETIFLGSRPWMPGTPRRLPRLPQRYWQMRP LFLELLGNHAQCPYGVLLKTHCPLRAAVTPAAGVCAREKPQGSVAAPEEEDTDPR RL VQLLRQHS SPW Q VY GF VRACLRRLVPPGLW GSRHNERRFLRNTKKFISLGKHA KLSLQELTWKMSVRGCAWLRRSPGVGCVPAAEHRLREEILAKFLHWLMSVYVVE LLRSFFYVTETTFQKNRLFFYRKSVWSKLQSIGIRQHLKRVQLRELSEAEVRQHREA RP ALLT SRLRFIPKPDGLRPIVNMD YVV GARTFRREKRAERLTSRVK ALF S VLNYER ARRPGLLGASVLGLDDIHRAWRTFVLRVRAQDPPPELYFVKYDVTGAYDTIPQDRL TEVIASIIKPQNTYCVRRYAVVQKAAHGHVRKAFKSHVSTLTDLQPYMRQFVAHL QETSPLRDAVVIEQSSSLNEASSGLFDVFLRFMCHHAVRIRGKSYVQCQGIPQGSILS TLLC SLC Y GDMENKLF AGIRRDGLLLRL VDDFLL VTPHLTHAKTFLRTL VRGVPE Y GC VVNLRKT VVNFP VEDEALGGT AF VQMP AHGLFPWCGLLLDTRTLE V Q SD Y S S Y ARTSIRASLTFNRGFKAGRNMRRKLFGVLRLKCHSLFLDLQVNSLQTVCTNIYKILL LQAYRFHACVLQLPFHQQVWKNPTFFLRVISDTASLCYSILKAKNAGMSLGAKGA AGPLP SE AVQWLCHQ AFLLKLTRHRVT YVPLLGSLRT AQTQL SRKLPGTTLTALEA AANPALPSDFKTILD (SEQ ID NO: 63)
En una realización, la hTERT tiene una secuencia al menos un 80 %, 85 %, 90 %, 95 %, 96 %, 97 %, 98 % o 99 % idéntica a la secuencia de la SEQ ID NO: 63. En una realización, la hTERT tiene una secuencia de la SEQ ID NO: 63. En una realización, la hTERT comprende una supresión (p. ej., de no más de 5, 10, 15, 20 o 30 aminoácidos) en el extremo N, el extremo C o ambos. En una realización, la hTERT comprende una secuencia de aminoácidos transgénica (p. ej., de no más de 5, 10, 15, 20 o 30 aminoácidos) en el extremo N, el extremo C o ambos.
En una realización, la hTERT está codificada por la secuencia de ácido nucleico de GenBank n.° de referencia AF018167 (Meyerson et al., "hEST2, the Putative Human Telomerase Catalytic Subunit Gene, Is Up-Regulated in Tumor Cells and during Immortalization" Cell volumen 90, número 4, 22 de agosto de 1997, páginas 785-795):
1 caggcagcgt ggtcctgctg cgcacgtggg aagccctggc cccggccacc cccgcgatgc
caggcagcgt ggtcctgctg cgcacgtggg aagccctggc cccggccacc cccgcgatgc
61 cgcgcgctcc ccgctgccga gccgtgcgct ccctgctgcg cagccactac cgcgaggtgc
121 tgccgctggc cacgttcgtg cggcgcctgg ggccccaggg ctggcggctg gtgcagcgcg
181 gggacccggc ggctttccgc gcgctggtgg cccagtgcct ggtgtgcgtg ccctgggacg
241 cacggccgcc ccccgccgcc ccctccttcc gccaggtgtc ctgcctgaag gagctggtgg
301 cccgagtgct gcagaggctg tgcgagcgcg gcgcgaagaa cgtgctggcc ttcggcttcg
361 cgctgctgga cggggcccgc gggggccccc ccgaggcctt caccaccagc gtgcgcagct
421 acctgcccaa cacggtgacc gacgcactgc gggggagcgg ggcgtggggg ctgctgttgc 481 gccgcgtggg cgacgacgtg ctggttcacc tgctggcacg ctgcgcgctc tttgtgctgg
541 tggctcccag ctgcgcctac caggtgtgcg ggccgccgct gtaccagctc ggcgctgcca
601 ctcaggcccg gcccccgcca cacgctagtg gaccccgaag gcgtctggga tgcgaacggg
661 cctggaacca tagcgtcagg gaggccgggg tccccctggg cctgccagcc ccgggtgcga
721 ggaggcgcgg gggcagtgcc agccgaagtc tgccgttgcc caagaggccc aggcgtggcg
781 ctgcccctga gccggagcgg acgcccgttg ggcaggggtc ctgggcccac ccgggcagga
841 cgcgtggacc gagtgaccgt ggtttctgtg tggtgtcacc tgccagaccc gccgaagaag
901 ccacctcttt ggagggtgcg ctctctggca cgcgccactc ccacccatcc gtgggccgcc
961 agcaccacgc gggcccccca tccacatcgc ggccaccacg tccctgggac acgccttgtc
1021 ccccggtgta cgccgagacc aagcacttcc tctactcctc aggcgacaag gagcagctgc
1081 ggccctcctt cctactcagc tctctgaggc ccagcctgac tggcgctcgg aggctcgtgg
1141 agaccatctt tctgggttcc aggccctgga tgccagggac tccccgcagg ttgccccgcc
1201 tgccccagcg ctactggcaa atgcggcccc tgtttctgga gctgcttggg aaccacgcgc
1261 agtgccccta cggggtgctc ctcaagacgc actgcccgct gcgagctgcg gtcaccccag
1321 cagccggtgt ctgtgcccgg gagaagcccc agggctctgt ggcggccccc gaggaggagg 1381 acacagaccc ccgtcgcctg gtgcagctgc tccgccagca cagcagcccc tggcaggtgt
1441 acggcttcgt gcgggcctgc ctgcgccggc tggtgccccc aggcctctgg ggctccaggc
1501 acaacgaacg ccgcttcctc aggaacacca agaagttcat ctccctgggg aagcatgcca
1561 agctctcgct gcaggagctg acgtggaaga tgagcgtgcg gggctgcgct tggctgcgca
1621 ggagcccagg ggttggctgt gttccggccg cagagcaccg tctgcgtgag gagatcctgg
1681 ccaagttcct gcactggctg atgagtgtgt acgtcgtcga gctgctcagg tctttctttt
1741 atgtcacgga gaccacgttt caaaagaaca ggctcttttt ctaccggaag agtgtctgga
1801 gcaagttgca aagcattgga atcagacagc acttgaagag ggtgcagctg cgggagctgt
1861 cggaagcaga ggtcaggcag catcgggaag ccaggcccgc cctgctgacg tccagactcc 1921 gcttcatccc caagcctgac gggctgcggc cgattgtgaa catggactac gtcgtgggag
1981 ccagaacgtt ccgcagagaa aagagggccg agcgtctcac ctcgagggtg aaggcactgt 2041 tcagcgtgct caactacgag cgggcgcggc gccccggcct cctgggcgcc tctgtgctgg
2101 gcctggacga tatccacagg gcctggcgca ccttcgtgct gcgtgtgcgg gcccaggacc
2161 cgccgcctga gctgtacttt gtcaaggtgg atgtgacggg cgcgtacgac accatccccc
2221 aggacaggct cacggaggtc atcgccagca tcatcaaacc ccagaacacg tactgcgtgc
2281 gtcggtatgc cgtggtccag aaggccgccc atgggcacgt ccgcaaggcc ttcaagagcc
2341 acgtctctac cttgacagac ctccagccgt acatgcgaca gttcgtggct cacctgcagg
2401 agaccagccc gctgagggat gccgtcgtca tcgagcagag ctcctccctg aatgaggcca
2461 gcagtggcct cttcgacgtc ttcctacgct tcatgtgcca ccacgccgtg cgcatcaggg
2521 gcaagtccta cgtccagtgc caggggatcc cgcagggctc catcctctcc acgctgctct
2581 gcagcctgtg ctacggcgac atggagaaca agctgtttgc ggggattcgg cgggacgggc
2641 tgctcctgcg tttggtggat gatttcttgt tggtgacacc tcacctcacc cacgcgaaaa
2701 ccttcctcag gaccctggtc cgaggtgtcc ctgagtatgg ctgcgtggtg aacttgcgga
2761 agacagtggt gaacttccct gtagaagacg aggccctggg tggcacggct tttgttcaga
2821 tgccggccca cggcctattc ccctggtgcg gcctgctgct ggatacccgg accctggagg
2881 tgcagagcga ctactccagc tatgcccgga cctccatcag agccagtctc accttcaacc
2941 gcggcttcaa ggctgggagg aacatgcgtc gcaaactctt tggggtcttg cggctgaagt
3001 gtcacagcct gtttctggat ttgcaggtga acagcctcca gacggtgtgc accaacatct
3061 acaagatcct cctgctgcag gcgtacaggt ttcacgcatg tgtgctgcag ctcccatttc
3121 atcagcaagt ttggaagaac cccacatttt tcctgcgcgt catctctgac acggcctccc
3181 tctgctactc catcctgaaa gccaagaacg cagggatgtc gctgggggcc aagggcgccg
3241 ccggccctct gccctccgag gccgtgcagt ggctgtgcca ccaagcattc ctgctcaagc
3301 tgactcgaca ccgtgtcacc tacgtgccac tcctggggtc actcaggaca gcccagacgc
3361 agctgagtcg gaagctcccg gggacgacgc tgactgccct ggaggccgca gccaacccgg
3421 cactgccctc agacttcaag accatcctgg actgatggcc acccgcccac agccaggccg
3481 agagcagaca ccagcagccc tgtcacgccg ggctctacgt cccagggagg gaggggcggc
3541 ccacacccag gcccgcaccg ctgggagtct gaggcctgag tgagtgtttg gccgaggcct
3601 gcatgtccgg ctgaaggctg agtgtccggc tgaggcctga gcgagtgtcc agccaagggc
3661 tgagtgtcca gcacacctgc cgtcttcact tccccacagg ctggcgctcg gctccacccc
3721 agggccagct tttcctcacc aggagcccgg cttccactcc ccacatagga atagtccatc
3781 cccagattcg ccattgttca cccctcgccc tgccctcctt tgccttccac ccccaccatc
3841 caggtggaga ccctgagaag gaccctggga gctctgggaa tttggagtga ccaaaggtgt
3901 gccctgtaca caggcgagga ccctgcacct ggatgggggt ccctgtgggt caaattgggg
3961 ggaggtgctg tgggagtaaa atactgaata tatgagtttt tcagttttga aaaaaaaaaa
4021 aaaaaaa (SEQ ID NO: 64)
En una realización, la hTERT está codificada por un ácido nucleico que tiene una secuencia al menos un 80%, 85%, 90 %, 95 %, 96 %, 97 %, 98 % o 99 % idéntica a la secuencia de la SEQ ID NO: 64. En una realización, el hTERT es codificado por un ácido nucleico de la SEQ ID NO: 64.
Activación y expansión de células efectoras inmunitarias (p. ej., célula T)
Las células efectoras inmunitarias tales como las células T pueden activarse y expandirse generalmente utilizando métodos como se describe, p. ej., en las patentes de los EE. UU. 6.352.694; 6.534.055; 6.905.680; 6.692.964; 5.858.358; 6.887.466; 6.905.681; 7.144.575; 7.067.318; 7.172.869; 7.232.566; 7.175.843; 5.883.223; 6.905.874; 6.797.514; 6.867.041; y la publicación de solicitud de patente de los EE. UU. N° 20060121005.
Generalmente, una población de células efectoras inmunitarias, p. ej., células con agotamiento de células T reguladoras, puede expandirse por contacto con una superficie que tiene unido a la misma un agente que estimula una señal asociada al complejo CD3/TCR y un ligando que estimula una molécula coestimulante en la superficie de las células T. En particular, las poblaciones de células T pueden estimularse como se describe en el presente memoria, tal como por contacto con un anticuerpo anti-CD3, o fragmento de unión a antígenos del mismo, o un anticuerpo anti-CD2 inmovilizado en una superficie, o por contacto con un activador de proteína quinasa C (p. ej., briostatina) junto con un ionóforo de calcio. Para la coestimulación de una molécula accesoria sobre la superficie de las células T se utiliza un ligando que se une a la molécula
accesoria. Por ejemplo, una población de células T puede ponerse en contacto con un anticuerpo anti-CD3 y un anticuerpo anti-CD28, en condiciones apropiadas para estimular la proliferación de las células T. Para estimular la proliferación de célula T CD4+ o célula T CD8+, se pueden utilizar un anticuerpo anti-CD3 y un anticuerpo anti-CD28. Los ejemplos de un anticuerpo anti-CD28 que se puede utilizar incluyen 9.3, B-T3, XR-CD28 (Diaclone, Besangon, Francia), al igual que otros métodos de uso común en la técnica (Berg et al., Transplant Proc. 30(8):3975-3977, 1998; Haanen et al., J. Exp. Med.
190(9):13191328, 1999; Garland et al., J. Immunol Meth. 227(1-2):53-63, 1999).
En determinados aspectos, la señal de estimulación primaria y la señal de coestimulación para la célula T pueden ser proporcionadas por diferentes protocolos. Por ejemplo, los agentes que proporcionan cada una de las señales pueden estar en solución o acoplados a una superficie. Cuando se acoplan a una superficie, los agentes pueden acoplarse a la misma superficie (es decir, en formación "cis") o a superficies separadas (es decir, en formación "trans"). Como alternativa, un agente puede estar acoplado a una superficie y el otro agente en solución. En un aspecto, el agente que proporciona la señal coestimuladora se une a una superficie celular y el agente que proporciona la señal de activación principal está en solución o acoplado a una superficie. En determinados aspectos, ambos agentes pueden estar en solución. En un aspecto, los agentes pueden estar en forma soluble y luego entrecruzarse con una superficie, como una célula que expresa receptores Fc o un anticuerpo u otro agente de unión que se unirá a los agentes. A este respecto, véanse, por ejemplo, las Publicaciones de Solicitud de Patente de los eE. UU. N°s 20040101519 y 20060034810 para células presentadoras de antígenos artificiales (aAPC) que se contemplan para su uso en la activación y expansión de células T en la presente divulgación.
En un aspecto, los dos agentes están inmovilizados en perlas, ya sea en la misma perla, es decir, "cis", o en perlas separadas, es decir, "trans". A modo de ejemplo, el agente que proporciona la señal de activación primaria es un anticuerpo anti-CD3 o un fragmento de unión a antígenos del mismo y el agente que proporciona la señal coestimulante es un anticuerpo anti-CD28 o un fragmento de unión a antígenos del mismo; y ambos agentes están co-inmovilizados en la misma perla en cantidades moleculares equivalentes. En un aspecto, se utiliza una relación 1:1 de cada uno de los anticuerpos unidos a las perlas para la expansión de las células T CD4+ y el crecimiento de las células T. En determinados aspectos de la presente divulgación, se utiliza una relación de anticuerpos anti CD3:CD28 unidos a las perlas de manera que se observe un aumento en la expansión de las células T en comparación con la expansión observada utilizando una relación de 1:1. En un aspecto particular, se observa un aumento de aproximadamente 1 a aproximadamente 3 veces en comparación con la expansión observada utilizando una relación de 1:1. En un aspecto, se utiliza una relación 1:1 de cada uno de los anticuerpos unido a las perlas para la expansión de células T CD4+ y el crecimiento de células T. En un aspecto, se une más anticuerpo anti-CD28 a las partículas que anticuerpo anti-CD3, es decir, la relación de CD3:CD28 es menor que uno. En determinados aspectos, la relación de anticuerpo anti-CD28 con respecto a anticuerpo anti-CD3 unido a las perlas es mayor que 2:1. En un aspecto particular, se utiliza una relación CD3:CD28 1:100 de anticuerpo unido a perlas. En un aspecto, se utiliza una relación CD3:CD28 1:75 de anticuerpo unido a perlas. En un aspecto adicional, se utiliza una relación CD3:CD28 1:50 de anticuerpo unido a perlas. En un aspecto, se utiliza una relación CD3:CD28 1:30 de anticuerpo unido a perlas. En un aspecto preferido, se utiliza una proporción de 1:10 de CD3:CD28 de anticuerpo unido a perlas. En un aspecto, se utiliza una relación de 1:3 CD3:CD28 de anticuerpo unido a las perlas. En un aspecto más, se utiliza una relación de 3:1 de CD3:CD28 de anticuerpo unido a las perlas.
Relaciones de partículas a células de 1:500 a 500:1 y cualquier valor entero intermedio pueden utilizarse para estimular las células T u otras células diana. Como pueden apreciar fácilmente los expertos ordinarios en la técnica, la relación de partículas con respecto a células puede depender del tamaño de partícula en relación con la célula diana. Por ejemplo, las perlas de tamaño pequeño solo podrían unirse a unas pocas células, mientras que las perlas más grandes podrían unirse a muchas. En determinados aspectos, la relación de células con respecto a partículas varía de 1:100 a 100:1 y cualquier valor entero intermedio y, en aspectos adicionales, la relación comprende de 1:9 a 9:1 y también se puede utilizar cualquier valor entero intermedio para estimular células T. La proporción de partículas acopladas anti-CD3 y anti-CD28 a células T que dan como resultado la estimulación de células T puede variar como se indicó anteriormente, sin embargo, determinados valores preferidos incluyen 1:100, 1:50, 1:40, 1:30, 1:20, 1:10, 1:9, 1:8, 1:7, 1:6, 1:5, 1:4, 1:3, 1:2, 1:1, 2:1, 3: 1,4:1, 5:1, 6:1, 7:1, 8:1, 9:1, 10:1 y 15:1 siendo una relación preferida al menos 1:1 de partículas por célula T. En un aspecto, se utiliza una relación de partículas a células de 1:1 o menos. En un aspecto particular, una relación de partículas:células preferida es 1:5. En aspectos adicionales, la relación de partículas con respecto a células puede variar dependiendo del día de estimulación. Por ejemplo, en un aspecto, la relación de partículas a células es de 1:1 a 10:1 el primer día y se agregan partículas adicionales a las células todos los días o cada dos días a partir de entonces hasta 10 días, en relaciones finales. de 1:1 a 1:10 (basado en recuentos de células el día de la adición). En un aspecto particular, la relación de partículas con respecto a células es 1:1 el primer día de estimulación y se ajusta a 1:5 el tercer y el quinto día de estimulación. En un aspecto, las partículas se añaden diariamente o cada dos días hasta una relación final de 1:1 el primer día y de 1:5 el tercer y el quinto días de estimulación. En un aspecto, la relación de partículas con respecto a células es 2:1 el primer día de estimulación y se ajusta a 1:10 el tercer y el quinto día de estimulación. En un aspecto, las partículas se añaden diariamente o cada dos días hasta una relación final de 1:1 el primer día y de 1:10 el tercer y el quinto días de estimulación. Un experto en la técnica apreciará que una diversidad de otras relaciones pueden ser adecuadas para su uso en la presente divulgación. En particular, las relaciones variarán dependiendo del tamaño de partícula y del tamaño y tipo de célula. En un aspecto, las relaciones más típicas de uso son del orden de 1:1,2:1 y 3:1 el primer día.
En aspectos adicionales, las células, tales como células T, se combinan con perlas recubiertas con agente, las perlas y las células se separan posteriormente y luego se cultivan las células. En un aspecto alternativo, antes del cultivo, las perlas recubiertas con agente y las células no se separan sino que se cultivan juntas. En un aspecto adicional, las perlas y las células se concentran primero mediante la aplicación de una fuerza, tal como una fuerza magnética, lo que da lugar a un aumento del ligamiento de los marcadores de la superficie celular, induciendo con ello la estimulación de las células.
A modo de ejemplo, las proteínas de la superficie celular pueden ligarse permitiendo que las perlas paramagnéticas a las que se unen anti-CD3 y anti-CD28 (3x28 perlas) entren en contacto con las células T. En un aspecto, las células (por ejemplo, 104 a 109 célula T) y las perlas (por ejemplo, perlas paramagnéticas DYNABEADS® M-450 CD3/CD28 T en una relación de 1:1) se combinan en un tampón, por ejemplo, PBS (sin cationes divalentes tales como calcio y magnesio). De nuevo, los expertos habituales en la materia pueden apreciar fácilmente que se puede utilizar cualquier concentración de células. Por ejemplo, la célula diana puede ser muy rara en la muestra y comprender solo el 0.01 % de la muestra o la muestra completa (es decir, el 100 %) puede comprender la célula diana de interés. Por consiguiente, cualquier número de células está dentro del contexto de la presente divulgación. En determinados aspectos, puede ser deseable disminuir significativamente el volumen en el que se mezclan entre sí partículas y células (es decir, aumenta la concentración de células), para garantizar el máximo contacto de las células y las partículas. Por ejemplo, en un aspecto, se utiliza una concentración de aproximadamente 10 billones de células/ml, 9 billones/ml, 8 billones/ml, 7 billones/ml, 6 billones/ml, 5 billones/ml o 2 billones de células/ml. En un aspecto, se utilizan más de 100 millones de células/ml. En un aspecto adicional, se utiliza una concentración de células de 10, 15, 20, 25, 30, 35, 40, 45 o 50 millones de células/ml. En un aspecto más, se utiliza una concentración de células de 75, 80, 85, 90, 95 o 100 millones de células/ml. En aspectos adicionales, pueden utilizarse concentraciones de 125 o 150 millones de células/ml. El uso de altas concentraciones puede dar lugar a un mayor rendimiento celular, activación celular y expansión celular. Además, el uso de altas concentraciones de células permite una captura más eficiente de las células que pueden expresar débilmente los antígenos diana de interés, tales como células T CD28-negativas. Poblaciones de células de este tipo pueden tener un valor terapéutico y sería deseable obtenerlas en determinados aspectos. Por ejemplo, el uso de una alta concentración de células permite una selección más eficiente de células T CD8+ que normalmente tienen una expresión de CD28 más débil.
En un caso, las células transducidas con un ácido nucleico que codifica un CAR, p. ej., un CAR descrito en esta memoria, se expanden, p. ej., mediante un método descrito en esta memoria. En un caso, las células se expanden en cultivo durante un periodo de varias horas (p. ej., aproximadamente 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 18, 21 horas) hasta aproximadamente 14 días (p. ej., 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 o 14 días). En un caso, las células se expanden durante un periodo de 4 a 9 días. En un caso, las células se expanden durante un periodo de 8 días o menos, p. ej., 7, 6 o 5 días. En un caso, las células, p. ej., una célula CAR CD19 descrita en esta memoria, se expanden en cultivo durante 5 días, y las células resultantes son más potentes que las mismas células expandidas en cultivo durante 9 días en las mismas condiciones de cultivo. La potencia se puede definir, p. ej., por diversas funciones de las células T, p. ej., proliferación, exterminio de células diana, producción de citoquinas, activación, migración o combinaciones de las mismas. En un caso, las células, p. ej., una célula CAR CD19 descrita en esta memoria, expandidas durante 5 días muestran al menos un aumento de una, dos, tres o cuatro veces en la duplicación de las células tras la estimulación del antígeno en comparación con las mismas células expandidas en cultivo durante 9 días en las mismas condiciones de cultivo. En un caso, las células, p. ej., las células que expresan una CAR CD19 descritas en esta memoria, se expanden en cultivo durante 5 días, y las células resultantes exhiben una mayor producción de citoquinas proinflamatorias, p. ej., niveles de IFN-y y/o GM-CSF, en comparación con las mismas células expandidas en cultivo durante 9 días en las mismas condiciones de cultivo. En un caso, las células, p. ej., una célula CAR CD19 descrita en esta memoria, expandida durante 5 días, muestran al menos un aumento de una, dos, tres, cuatro, cinco, diez veces o más en pg/ml de producción de citoquinas proinflamatorias, p. ej., niveles de IFN-y y/o GM-CSF, en comparación con las mismas células expandidas en cultivo durante 9 días bajo las mismas condiciones de cultivo.
También se pueden desear varios ciclos de estimulación, de modo que el tiempo de cultivo de las células T pueda ser de 60 días o más. Las condiciones apropiadas para el cultivo de células T incluyen un medio apropiado (p. ej., Medio Esencial Mínimo o Medio RPMI 1640 o X-vivo 15, (Lonza)) que puede contener factores necesarios para la proliferación y viabilidad, incluido suero (p. ej., suero bovino o humano fetal), interleucina-2 (IL-2), insulina, IFN-y, IL-4, IL-7, GM-CSF, IL-10, IL-12, IL-15, TGFp y TNF-a o cualquier otro aditivo para el crecimiento de células conocido por el experto en la materia. Otros aditivos para el crecimiento de células incluyen, pero sin limitación, tensioactivo, plasmanato y agentes reductores, tales como N-acetilcisteína y 2-mercaptoetanol. Los medios pueden incluir RPMI 1640, AIM-V, Dm Em , MEM, a-MEM, F-12, X-Vivo 15 y X-Vivo 20, Optimizer, con aminoácidos añadidos, piruvato de sodio y vitaminas, ya sea exentos de suero o complementados con una cantidad apropiada de suero (o plasma) o un conjunto definido de hormonas, y/o una cantidad de citoquina o citocinas suficiente para el crecimiento y la expansión de células T. Se incluyen antibióticos, p. ej., penicilina y estreptomicina, solo en cultivos experimentales, no en cultivos de células que se han de infundir en un sujeto. Las células diana se mantienen en las condiciones necesarias para sustentar el crecimiento, por ejemplo, una temperatura (p. ej., 37 °C) y una atmósfera (p. ej., aire más CO2 al 5 %) apropiadas.
En un caso, las células se expanden en un medio apropiado (p. ej., medio descrito en el presente memoria) que incluye una o más interleuquinas que dan como resultado un aumento de al menos 200 veces (p. ej., 200 veces, 250 veces, 300 veces, 350 veces) en las células durante un periodo de expansión de 14 días, p. ej., medido por un método descrito en
esta memoria tal como citometría de flujo. En un caso, las células se expanden en presencia de IL-15 y/o IL-7 (p. ej., IL-15 e IL-7).
En casos, métodos descritos en esta memoria, p. ej., métodos de fabricación de células que expresan CAR, comprenden eliminar las células T reguladoras, p. ej., las células T CD25+, de una población celular, p. ej., utilizando un anticuerpo anti-CD25, o un fragmento del mismo, o un ligando de unión a CD25, IL-2. En esta memoria se describen métodos para eliminar células T reguladoras, por ejemplo, células T CD25+, de una población de células. En casos, los métodos, por ejemplo, métodos de producción, comprenden además poner en contacto una población de células (por ejemplo, una población de células en las que se han agotado las células T reguladoras, tales como células T CD25+; o una población de células que se ha puesto en contacto previamente con un anticuerpo anti-CD25, o fragmento de este, o ligando de unión a CD25) con IL-15 y/o IL-7. Por ejemplo, la población de células (por ejemplo, que se ha puesto en contacto previamente con un anticuerpo anti-CD25, fragmento de este o ligando de unión a CD25) se expande en presencia de IL-15 y/o IL-7.
En casos, una célula que expresa CAR descrita en la presente se pone en contacto con una composición que comprende un polipéptido de tipo interleucina-15 (IL-15), un polipéptido de tipo receptor alfa de interleucina-15 (IL-15Ra), o una combinación de ambos, un polipéptido de tipo IL-15 y un polipéptido de tipo IL-15Ra, por ejemplo, hetIL-15, durante la producción de la célula que expresa CAR, por ejemplo, ex vivo. En casos, una célula que expresa CAR descrita en la presente se pone en contacto con una composición que comprende un polipéptido de tipo IL-15 durante la producción de la célula que expresa CAR, por ejemplo, ex vivo. En casos, una célula que expresa CAR descrita en la presente se pone en contacto con una composición que comprende una combinación de un polipéptido de tipo IL-15 y un polipéptido de tipo IL-15 Ra durante la producción de la célula que expresa CAR, por ejemplo, ex vivo. En casos, una célula que expresa CAR descrita en la presente se pone en contacto con una composición que comprende hetIL-15 durante la producción de la célula que expresa CAR, por ejemplo, ex vivo.
En un caso, la célula que expresa CAR descrita en la presente se pone en contacto con una composición que comprende hetIL-15 durante la expansión ex vivo. En un caso, la célula que expresa CAR descrita en la presente se pone en contacto con una composición que comprende un polipéptido de tipo IL-15 durante la expansión ex vivo. En un caso, la célula que expresa CAR descrita en la presente se pone en contacto con una composición que comprende un polipéptido de tipo IL-15 y un polipéptido de tipo IL-15Ra durante la expansión ex vivo. En un caso, la puesta en contacto da como resultado la supervivencia y proliferación de una subpoblación de células, por ejemplo, células T CD8+.
Las células T que han estado expuestas a tiempos de estimulación variados pueden presentar diferentes características. Por ejemplo, los productos de células mononucleares de la sangre periférica con aféresis o habituales de sangre tienen una población de célula T auxiliares (TH, CD4+) que es mayor que la población de células T citotóxicos o supresoras (TC, CD8+). La expansión ex vivo de células T mediante la estimulación de receptores CD3 y CD28 produce una población de células T que antes de aproximadamente los días 8-9 consiste predominantemente en célula TH, mientras que después de aproximadamente los días 8-9, la población de células T comprende una población cada vez mayor de células TC. En consecuencia, dependiendo del propósito del tratamiento, puede ser ventajoso infundir a un sujeto una población de células T que comprenda predominantemente células TH. De manera similar, si se ha aislado un subconjunto específico de antígeno de células TC, puede ser beneficioso expandir este subconjunto en mayor grado.
Asimismo, además de los marcadores CD4 y CD8, otros marcadores fenotípicos varían significativamente, pero, en gran parte, de forma reproducible durante el transcurso del proceso de expansión celular. Por lo tanto, una reproducibilidad de este tipo permite la capacidad de adaptar un producto de células T activadas para fines específicos.
Una vez que se construye un CAR descrito en esta memoria, se pueden utilizar diversos ensayos para evaluar la actividad de la molécula, tal como, pero sin limitación, la capacidad de expandir célula T después de la estimulación del antígeno, mantener la expansión de células T en ausencia de reestimulación y actividades antineoplásicas en modelos in vitro y en animales apropiados. Ensayos para evaluar los efectos de un cars de la presente divulgación se describen con mayor detalle a continuación.
El análisis de transferencia Western de la expresión de CAR en células T primarias puede utilizarse para detectar la presencia de monómeros y dímeros. Véase, p. ej., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Muy brevemente, células T (mezcla 1:1 de células T CD4+ y CD8+) que expresan las CARs se expanden in vitro durante más de 10 días seguido de lisis y SDS-PAGE en condiciones reductoras. Los CAR que contienen el dominio citoplásmico TCR-Z de longitud completa y la cadena TCR-Z endógena se detectan mediante transferencia de Western utilizando un anticuerpo contra la cadena TCR-Z. Los mismos subconjuntos de células T se utilizan para el análisis de SDS-PAGE en condiciones no reductoras para permitir la evaluación de la formación de dímeros covalentes.
La expansión in vitro de células CAR+ T después de la estimulación del antígeno puede medirse mediante citometría de flujo. Por ejemplo, se estimula una mezcla de células T CD4+ y CD8+ con aAPC aCD3/aCD28, lo cual va seguido de transducción con vectores lentivíricos que expresan GFP bajo el control de los promotores que se van a analizar. Promotores ilustrativos incluyen promotores del gen IE de CMV, EF-1a, ubiquitina C o de la fosfogliceroquinasa (PGK). La fluorescencia de GFP se evalúa el día 6 de cultivo en los subconjuntos de células T CD4+ y/o CD8+ mediante citometría
de flujo. Véase, p. ej., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Alternativamente, se estimula una mezcla de células T CD4+ y CD8+ con perlas magnéticas recubiertas de aCD3/aCD28 el día 0 y se transduce con CAR el día 1 utilizando un vector lentiviral bicistrónico que expresa CAR junto con eGFP utilizando una secuencia de salto ribosomal 2A . Los cultivos se vuelven a estimular con células K562 positivas para un antígeno asociado al cáncer como se describe en esta memoria+ (K562 que expresa un antígeno asociado al cáncer como se describe en esta memoria), células K562 de tipo silvestre (K562 de tipo silvestre) o células K562 que expresan hCD32 y 4-1BBL en presencia de anticuerpos antiCD3 y anti-CD28 (K562-BBL-3/28) después del lavado. Se añade IL-2 exógena a los cultivos cada dos días a razón de 100 UI/ml. Células T GFP+ se enumeran mediante citometría de flujo utilizando un recuento basado en perlas. Véase, p. ej., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009).
También se puede medir la expansión sostenida de célula T-CAR+ en ausencia de reestimulación. Véase, p. ej., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Brevemente, el volumen medio de células T (fl) se mide el día 8 de cultivo utilizando un contador de partículas Coulter Multisizer III, un celómetro Nexcelom Vision o Millipore Scepter, después de la estimulación con perlas magnéticas recubiertas de aCD3/aCD28 el día 0 y la transducción con el CAR indicado el día 1.
También se pueden utilizar modelos en animales para medir la actividad de un T-CAR. Por ejemplo, se puede utilizar un modelo de xenoinjerto utilizando células CAR+ T específicas para el antígeno asociado al cáncer descrito en esta memoria para tratar una LLA pre-B humana primaria en ratones inmunodeficientes. Véase, p. ej., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Muy brevemente, después del establecimiento de LLA, los ratones se distribuyen aleatoriamente en grupos de tratamiento. Se coinyectan diferentes números de células T modificadas con CAR específicas de antígeno asociado al cáncer en una relación de 1:1 en ratones NOD-SCID-y-/' portadores de LLA-B. El número de copias de un vector CAR específico de antígeno asociado al cáncer en el ADN del bazo de ratones se evalúa en diversos momentos después de la inyección de células T. Se evalúa la leucemia de los animales a intervalos semanales. Se miden los recuentos de blastocitos de B-ALL positivos para un antígeno asociado al cáncer de la sangre periférica como se describe en esta memoria en ratones a los que se inyectan célula CAR+ T con un antígeno asociado al cáncer descrito en esta memoria-Z o célula T con una transducción simulada. Las curvas de supervivencia de los grupos se comparan utilizando la prueba del orden logarítmico. Además, también se pueden analizar los recuentos absolutos de células T CD4+ y CD8+ de la sangre periférica 4 semanas después de la inyección de células T en ratones NOD-SCID-y-/'. Se inyectan en ratones células leucémicas y 3 semanas más tarde se les inyectan células T modificadas para expresar CAR mediante un vector lentivírico bicistrónico que codifica el CAR ligado a eGFP. Las células T se normalizan respecto a un 45-50 % de células GFP+ T de entrada mezclando células que han experimentado una transducción simulada antes de la inyección y se confirman mediante citometría de flujo. Se evalúa la leucemia en los animales en intervalos de 1 semana. Las curvas de supervivencia de los grupos de célula CAR+ T se comparan utilizando la prueba del orden logarítmico.
Se puede evaluar la respuesta al tratamiento con CAR dependiente de la dosis. Véase, p. ej., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Por ejemplo, la sangre periférica se obtiene 35-70 días después de establecer la leucemia en ratones a los que se inyectaron el día 21 con células CAR T, un número equivalente de células T transducidas de forma simulada o sin células T. Se extrae sangre de manera aleatoria de ratones de cada grupo para determinar los recuentos de blastos ALL positivos para un antígeno asociado al cáncer como se describe en esta memoria en la sangre periférica y posteriormente se sacrifican los días 35 y 49. Los animales restantes se evalúan los días 57 y 70.
La evaluación de la proliferación celular y la producción de citocinas se ha descrito previamente, p. ej., en Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Resumiendo, la evaluación de la proliferación mediada por CAR se realiza en placas de microtitulación mezclando células T lavadas con células K562 que expresan un antígeno asociado al cáncer descrito en esta memoria (K19) o CD32 y CD137 (KT32-BBL) para obtener una relación final de células T:K562 de 2:1. Las células K562 se irradian con radiación gamma antes de su uso. Se añaden anticuerpos monoclonales anti-CD3 (clon OKT3) y anti-CD28 (clon 9.3) a cultivos con células KT32-BBL para que actúen como control positivo para estimular la proliferación de células T, dado que estas señales propician la expansión a largo plazo de células T CD8+ ex vivo. Las células T se enumeran en cultivos utilizando perlas fluorescentes CountBright™ (Invitrogen, Carlsbad, CA) y citometría de flujo según lo descrito por el fabricante. Las células CAR+ T se identifican mediante la expresión de GFP utilizando células T que han sido modificadas con vectores lentivíricos que expresan CAR enlazado a eGFP-2A. Para células CAR+ T que no expresan GFP, las células CAR+ T se detectan con proteína de un antígeno asociado al cáncer recombinante biotinilado como se describe en esta memoria y un conjugado secundario de avidina-PE. La expresión de CD4+ y CD8+ en células T también se detecta simultáneamente con anticuerpos monoclonales específicos (BD Biosciences). Las mediciones de citocinas se realizan en sobrenadantes recogidos 24 horas después de la reestimulación utilizando el equipo de matriz de perlas citométricas de citocinas TH1/TH2 humanas (BD Biosciences, San Diego, CA) según las instrucciones del fabricante. La fluorescencia se evalúa utilizando un citómetro de flujo FACScalibur y los datos se analizan según las instrucciones del fabricante.
La citotoxicidad se puede evaluar mediante un ensayo convencional de liberación de 51Cr. Véase, p. ej., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Brevemente, células diana (líneas K562 y células pro-B-ALL primarias) se cargan con 51Cr (como NaCrO4, New England Nuclear, Boston, MA) a 37 °C durante 2 horas con agitación frecuente, se lavan dos veces en RPMI completo y se siembran en placas de microtitulación. Las células T efectoras se mezclan con las células diana en los pocillos en rPmI completo con relaciones variables de célula efectora:célula diana (E:D). También
se preparan pocilios adicionales que contienen solo medio (liberación espontánea, SR, por sus siglas en inglés) o una solución al 1 % de detergente triton-X 100 (liberación total, TR, por sus siglas en inglés). Después de 4 horas de incubación a 37 °C, se extrae el sobrenadante de cada uno de los pocillos. El 51Cr liberado se mide a continuación utilizando un contador de partículas gamma (Packard Instrument Co., Waltham, MA). Cada condición se realiza al menos por triplicado y el porcentaje de lisis se calcula utilizando la fórmula: % de lisis = (ER-SR) / (TR - SR), donde ER representa el 51Cr promedio liberado para cada condición experimental.
Se pueden utilizar tecnologías de obtención de imágenes para evaluar el tráfico y proliferación específicos de los CAR en modelos en animales portadores de tumores. Ensayos de este tipo se han descrito, por ejemplo, en Barrett et al., Human Gene Therapy 22:1575-1586 (2011). Resumiendo, se inyectan i.v. en ratones n Od /s C iD/yc7' (NSG) células Nalm-6, seguido 7 días después de células T 4 horas después de electroporación con las construcciones de CAR. Las células T están transfectadas de manera estable con una construcción lentivírica para expresar luciferasa de luciérnaga y se obtienen imágenes de los ratones por bioluminiscencia. Como alternativa, se pueden medir la eficacia terapéutica y especificidad de una única inyección de células CAR+ T en un modelo de xenoinjerto con Nalm-6 de la siguiente manera: a ratones NSG se les inyecta Nalm-6 transducido para expresar de manera estable la luciferasa de luciérnaga, seguido de una sola inyección en la vena de la cola de células T electroporadas con cars de la presente divulgación 7 días después. Se obtienen imágenes de los animales en varios momentos después de la inyección. Por ejemplo, se pueden generar mapas cromáticos de densidad fotónica de leucemia positiva para luciferasa de luciérnaga en ratones representativos el día 5 (2 días antes del tratamiento) y el día 8 (24 h después de PBL CAR+)
También se pueden utilizar otros ensayos, incluidos los descritos en la sección de ejemplos de esta memoria, así como los que se conocen en la técnica, para evaluar los CAR descritos en esta memoria.
Aplicación terapéutica
En un aspecto, la divulgación proporciona métodos para tratar una enfermedad asociada con la expresión de un antígeno asociado con el cáncer descrito en esta memoria.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un XCAR, en donde X representa un antígeno tumoral como se describe en esta memoria, y en el que las células cancerosas expresan dicho antígeno tumoral X.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer proporcionando al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que se modifican para expresar un XCAR descrito en esta memoria, en donde las células cancerosas expresan X. En un caso, X se expresa tanto en células normales como en células cancerosas, pero se expresa en niveles más bajos en células normales. En un caso, el método comprende, además, seleccionar un CAR que se une a X con una afinidad que permite que el XCAR se una y elimine las células cancerosas que expresan X pero menos del 30 %, 25 %, 20 %, 15 %, 10 %, 5 %. o menos de las células normales que expresan X mueren, p. ej., según se determina mediante un ensayo descrito en esta memoria. Por ejemplo, se puede utilizar el ensayo descrito en la Figura 13 o un ensayo de exterminio tal como citometría de flujo basada en Cr51 c Tl . En un caso, el CAR seleccionado un dominio de unión a antígenos que tiene una afinidad de unión KD 10'4 M a 10'8 M, p. ej., 10'5 M a 10'7 M, p. ej., 10'6 M o 10'7 M para el antígeno diana. En un caso, el dominio de unión a antígenos tiene una afinidad de unión que es al menos cinco veces, 10 veces, 20 veces, 30 veces, 50 veces, 100 veces o 1000 veces inferior a un anticuerpo de referencia, p. ej., un anticuerpo descrito en esta memoria.
En un caso, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c A r CD19, en donde las células cancerosas expresan CD19. En un caso, el cáncer a tratar es a Ll (leucemia linfoblástica aguda), CLL (leucemia linfocítica crónica), DLBCL (linfoma difuso de células B grandes), MCL (linfoma de células del manto) o MM (mieloma múltiple).
En un caso, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células n K) que están diseñadas para expresar un CAR EGFRvlII, en donde las células cancerosas expresan EGFRvlII. En un caso, el cáncer a tratar es glioblastoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de mesotelina, en donde las células cancerosas expresan mesotelina. En un caso, el cáncer a tratar es mesotelioma, cáncer de páncreas o cáncer de ovario.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD123, en donde las células cancerosas expresan CD123. En un caso, el cáncer a tratar es AML.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD22, en donde las células cancerosas expresan CD22. En un caso, el cáncer a tratar son tumores malignos de células B.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CS-1, en donde las células cancerosas expresan CS-1. En un caso, el cáncer a tratar es mieloma múltiple.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CLL-1, en donde las células cancerosas expresan CLL-1. En un caso, el cáncer a tratar es AML.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD33, en donde las células cancerosas expresan CD33. En un caso, el cáncer a tratar es AML.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de GD2, en donde las células cancerosas expresan GD2. En un caso, el cáncer a tratar es neuroblastoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de BCMA, en donde las células cancerosas expresan BCMA. En un caso, el cáncer a tratar es mieloma múltiple.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c A r de Tn, en donde las células cancerosas expresan antígeno Tn. En un caso, el cáncer a tratar es cáncer de ovario.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de PSMA, en donde las células cancerosas expresan PSMA. En un caso, el cáncer a tratar es cáncer de próstata.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de ROR1, en donde las células cancerosas expresan ROR1. En un caso, el cáncer a tratar son tumores malignos de células B. En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de FLT3, en donde las células cancerosas expresan FLT3. En un caso, el cáncer a tratar es AML.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de TAG72, en donde las células cancerosas expresan TAG72. En un caso, el cáncer a tratar es cáncer gastrointestinal.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD38, en donde las células cancerosas expresan CD38. En un caso, el cáncer a tratar es mieloma múltiple.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD44v6, en donde las células cancerosas expresan CD44v6. En un caso, el cáncer a tratar es cáncer cervical, AML o MM.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CEA, en donde las células cancerosas expresan CEA. En un caso, el cáncer a tratar es cáncer gastrointestinal o cáncer de pabcreático. En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de EPCAM, en donde las células cancerosas expresan EPCAM. En un caso, el cáncer a tratar es cáncer gastrointestinal.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de B7H3, en donde las células cancerosas expresan B7H3.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de KIT, en donde las células cancerosas expresan KIT. En un caso, el cáncer a tratar es cáncer gastrointestinal.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c A r de IL-13Ra2, en donde las células cancerosas expresan IL-13Ra2. En un caso, el cáncer a tratar es glioblastoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de PRSS21, en donde las células cancerosas expresan PRSS21. En un caso, el cáncer a tratar se selecciona de cáncer de ovario, pancreático, de pulmón y cáncer de mama..
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD30, en donde las células cancerosas expresan CD30. En un caso, el cáncer a tratar son linfomas o leucemias.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de GD3, en donde las células cancerosas expresan GD3. En un caso, el cáncer a tratar es melanoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD171, en donde las células cancerosas expresan CD171. En un caso, el cáncer a tratar es neuroblastoma, cáncer de ovario, melanoma, cáncer de mama, cáncer pancreático, cáncer de colon o NSCLC (cáncer de pulmón de células no pequeñas).
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de IL-11Ra, en donde las células cancerosas expresan IL-11Ra. En un caso, el cáncer a tratar es osteosarcoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de PSCA, en donde las células cancerosas expresan PSCA. En un caso, el cáncer a tratar es cáncer de próstata.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c Ar de VEGFR2, en donde las células cancerosas expresan VEGFR2. En un caso, el cáncer a tratar es un tumor sólido.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de LewisY, en donde las células cancerosas expresan LewisY. En un caso, el cáncer a tratar es cáncer de ovario o AML.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD24, en donde las células cancerosas expresan CD24. En un caso, el cáncer a tratar es cáncer pancreático.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de PDGFR, en donde las células cancerosas expresan PDGFR-. En un caso, el cáncer a tratar es cáncer de mama, cáncer de próstata, GIST (tumor del estroma gastrointestinal), CML, DFSP (dermatofibrosarcoma protuberans) o glioma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de SSEA-4, en donde las células cancerosas expresan SSEA-4. En un caso, el cáncer a tratar es glioblastoma, cáncer de mama, cáncer de pulmón o cáncer de células madre.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD20, en donde las células cancerosas expresan CD20. En un caso, el cáncer a tratar son tumores malignos de células B.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de receptor de folato alfa, en donde las células cancerosas expresan receptor de folato alfa. En un caso, el cáncer a tratar es cáncer de ovario, NSCLC, cáncer de endometrio, cáncer renal u otros tumores sólidos.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de ERBB2, en donde las células cancerosas expresan ERBB2 (Her2/neu).. En un caso, el cáncer a tratar es cáncer de mama, cáncer gástrico, cáncer colorrectal, cáncer de pulmón u otros tumores sólidos.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de MUC1, en donde las células cancerosas expresan MUC1. En un caso, el cáncer a tratar es cáncer de mama, cáncer de pulmón u otros tumores sólidos.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de EGFR, en donde las células cancerosas expresan EGFR. En un caso, el cáncer a tratar es glioblastoma, SCLC (cáncer de pulmón de células pequeñas), SCCHN (carcinoma de células escamosas de cabeza y cuello), NSCLC u otros tumores sólidos.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de NCAM, en donde las células cancerosas expresan NCAM. En un caso, el cáncer a tratar es un neuroblastoma u otros tumores sólidos.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CAIX, en donde las células cancerosas expresan CAIX. En un caso, el cáncer a tratar es cáncer renal, CRC, cáncer de cuello uterino u otros tumores sólidos.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de EphA2, en donde las células cancerosas expresan EphA2. En un caso, el cáncer a tratar es GBM.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de GD3, en donde las células cancerosas expresan GD3. En un caso, el cáncer a tratar es melanoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que se modifican para expresar un CAR de Fucosil GM1, en donde las células cancerosas expresan Fucosil GM.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de sLe, en donde las células cancerosas expresan sLe. En un caso, el cáncer a tratar es NSCLC o AML.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de GM3, en donde las células cancerosas expresan GM3.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de TGS5, en donde las células cancerosas expresan TGS5.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de HMWMAA, en donde las células cancerosas expresan HMWMAA. En un caso, el cáncer a tratar es melanoma, glioblastoma o cáncer de mama.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c A r de o-acetil-GD2, en donde las células cancerosas expresan o-acetil-GD2. En un caso, el cáncer a tratar es neuroblastoma o melanoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD19, en donde las células cancerosas expresan CD19. En un caso, el cáncer a tratar es AML con receptor de folato beta, mieloma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de TEM1/CD248, en donde las células cancerosas expresan TEM1/CD248. En un caso, el cáncer a tratar es un tuimor sólido.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de TEM7R, en donde las células cancerosas expresan TEM7R. En un caso, el cáncer tratar es tuimor sólido.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CLDN6, en donde las células cancerosas expresan CLDN6. En un caso, el cáncer a tratar es cáncer de ovario, cáncer de pulmón o cáncer de mama.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de TSHR, en donde las células cancerosas expresan TSHR. En un caso, el cáncer a tratar es cáncer de tiroides o mieloma múltiple.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c A r de GPRC5D, en donde las células cancerosas expresan GPRC5D. En un caso, el cáncer a tratar es mieloma múltiple.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CXORF61, en donde las células cancerosas expresan CXORF61.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD97, en donde las células cancerosas expresan CD97. En un caso, el cáncer a tratar es tumor maligno de células B, cáncer gástrico, cáncer pancreático, cáncer de esófago, glioblastoma, cáncer de mama o cáncer colorrectal.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD179a, en donde las células cancerosas expresan CD179a. En un caso, el cáncer a tratar son tumores malignos de células B.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de ALK, en donde las células cancerosas expresan ALK. En un caso, el cáncer a tratar es NSCLC, ALCL (linfoma anaplásico de células grandes), IMT (tumor miofibroblástico inflamatorio) o neuroblastoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de ácido polisiálico, en donde las células cancerosas expresan ácido polisiálico. En un caso, el cáncer a tratar es un cáncer de pulmón de células pequeñas.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de PLAC1, en donde las células cancerosas expresan PLAC1. En un caso, el cáncer a tratar es HCC (carcinoma hepatacelular).
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de GloboH, en donde las células cancerosas expresan GloboH. En un caso, el cáncer a tratar es cáncer de ovario, cáncer gástrico, cáncer de próstata, cáncer de pulmón, cáncer de mama o cáncer pancreático..
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c A r de NY-BR-1, en donde las células cancerosas expresan NY-BR-1. En un caso, el cáncer a tratar es cáncer de mama..
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de UPK2, en donde las células cancerosas expresan UPK2. En un caso, el cáncer a tratar es cáncer de vejiga.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c Ar de HAVCR1, en donde las células cancerosas expresan HAVCR1. En un caso, el cáncer a tratar es cáncer renal.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de ADRB3, en donde las células cancerosas expresan ADRB3. En un caso, el cáncer a tratar es sarcoma de Ewing.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de PANX3, en donde las células cancerosas expresan PANX3. En un caso, el cáncer a tratar es osteosarcoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de GPR20, en donde las células cancerosas expresan GPR20. En un caso, el cáncer a tratar es GIST.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de LY6K, en donde las células cancerosas expresan LY6K. En un caso, el cáncer a tratar es cáncer de mama, cáncer de pulmón, cáncer de ovario o cáncer de cuello uterino.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de OR51E2, en donde las células cancerosas expresan OR51E2. En un caso, el cáncer a tratar es cáncer de próstata.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de TARP, en donde las células cancerosas expresan TARP. En un caso, el cáncer a tratar es cáncer de próstata.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de WT1, en donde las células cancerosas expresan WT1.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de NY-ESO-1, en donde las células cancerosas expresan NY-ESO-1.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c Ar de LAGE-1a, en donde las células cancerosas expresan LAGE-1a.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de MAGE-A1, en donde las células cancerosas expresan MAGE-A1. En un caso, el cáncer a tratar es melanoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de MAGE A1, en donde las células cancerosas expresan MAGE-A1.
En un caso, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de ETV6-AML, en donde las células cancerosas expresan ETV6-AML.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar una proteína de esperma 17, en donde las células cancerosas expresan proteína de esperma 17. En un caso, el cáncer a tratar es cáncer de ovario, HCC o NSCLC.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de XAGE1, en donde las células cancerosas expresan XAGE1. En un caso, el cáncer a tratar es Ewings o cáncer de rabdo.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de Tie2, en donde las células cancerosas expresan Tie2. En un caso, el cáncer a tratar es un tuimor sólido.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de MAD-CT-1, en donde las células cancerosas expresan MAD-CT-1. En un caso, el cáncer a tratar es cáncer de próstata o melanoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de MAD-CT-2, en donde las células cancerosas expresan MAD-CT-2. En un caso, el cáncer a tratar es cáncer de próstata, melanoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de antígeno 1 relacionado con Fos, en donde las células cancerosas expresan antígeno 1 relacionado con Fos. En un caso, el cáncer a tratar es un glioma, un cáncer de células escamosas o un cáncer de páncreas.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de p53, en donde las células cancerosas expresan p53.
En un caso, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de prosteína, en donde las células cancerosas expresan prosteína.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de survivina y telomerasa, en donde las células cancerosas expresan survivina y telomerasa.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células n K) que están diseñadas para expresar un c Ar de PCTA-1/Galectina 8, en donde las células cancerosas expresan PCTA-1/Galectina 8.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de MelanA/MART1, en donde las células cancerosas expresan MelanA/MART1.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de RAS mutante, en donde las células cancerosas expresan RAS mutante.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c Ar de p53 mutante, en donde las células cancerosas expresan p53 mutante.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de hTERT, en donde las células cancerosas expresan hTERT.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de puntos de ruptura de translocación de sarcoma, en donde las células cancerosas expresan puntos de ruptura de translocación de sarcoma. En un caso, el cáncer a tratar es sarcoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de ML-IAP, en donde las células cancerosas expresan ML-IAP. En un caso, el cáncer a tratar es melanoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de ERG, en donde las células cancerosas expresan ERG (gen de fusión TMPRSS2 ETS)..
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de NA17, en donde las células cancerosas expresan NA17. En un caso, el cáncer a tratar es melanoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de PAX3, en donde las células cancerosas expresan PAX3. En un caso, el cáncer a tratar es rabdomiosarcoma alveolar.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de receptor de andrógeno, en donde las células cancerosas expresan receptor de andrógeno. En un caso, el cáncer a tratar es cáncer de próstata metastásico.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de Ciclina B1, en donde las células cancerosas expresan Ciclina B1.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de MYCN, en donde las células cancerosas expresan MYCN.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de RhoC, en donde las células cancerosas expresan RhoC.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de TRP-2, en donde las células cancerosas expresan TRP-2. En un caso, el cáncer a tratar es melanoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CYP1B1, en donde las células cancerosas expresan CYP1B1. En un caso, el cáncer a tratar es cáncer de mama, cáncer de colon, cáncer de pulmón, cáncer de esófago, cáncer de piel, cáncer de nódulo linfático, cáncer de cerebro o cáncer de testículo.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de BORIS, en donde las células cancerosas expresan BORIS. En un caso, el cáncer a tratar es cáncer de pulmón.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de SART3, en donde las células cancerosas expresan SART3
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de PAX5, en donde las células cancerosas expresan PAX5.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c Ar de OY-TES1, en donde las células cancerosas expresan OY-TES1.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de LCK, en donde las células cancerosas expresan LCK.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de AKAP-4, en donde las células cancerosas expresan AKAP-4.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de SSX2, en donde las células cancerosas expresan SSX2.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de RAGE-1, en donde las células cancerosas expresan RAGE-1. En un caso, el cáncer a tratar es cáncer RCC (cáncer de células renales) u otros tumores sólidos.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de una transcriptasa inversa de telomerasa humana, en donde las células cancerosas expresan transcriptasa inversa de telomerasa humana. En un caso, el cáncer tratar son tumores sólidos.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de RU1, en donde las células cancerosas expresan RU1.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de RU2, en donde las células cancerosas expresan RU2.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de carboxil esterasa intestinal, en donde las células cancerosas expresan carboxil esterasa intestinal. En un caso, el cáncer a tratar es cáncer de tiroides, RCC, CRC (cáncer colorrectal), cáncer de mama u otros tumores sólidos.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c A r de Prostasa, en donde las células cancerosas expresan Prostasa.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de PAP, en donde las células cancerosas expresan PAP.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de receptor de IGF-I, en donde las células cancerosas expresan receptor de IGF-I.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de gp100, en donde las células cancerosas expresan gp100.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de bcr-abl, en donde las células cancerosas expresan bcr-abl.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de tirosinasa, en donde las células cancerosas expresan tirosinasa.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c A r de Fucosil GM1, en donde las células cancerosas expresan Fucosil GM1.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un c A r de hsp70-2 mut, en donde las células cancerosas expresan hsp70-2 mut. En un caso, el cáncer a tratar es melanoma.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD79a, en donde las células cancerosas expresan CD79a.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD79b, en donde las células cancerosas expresan CD79b.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD72, en donde las células cancerosas expresan CD72.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de LAIR1, en donde las células cancerosas expresan LAIR1.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de FCAR, en donde las células cancerosas expresan FCAR.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de LILRA2, en donde las células cancerosas expresan LILRA2.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CD300LF, en donde las células cancerosas expresan CD300LF.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de CLEC12A, en donde las células cancerosas expresan CLEC12A.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de BST2, en donde las células cancerosas expresan BST2.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de EMR2, en donde las células cancerosas expresan EMR2.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de LY75, en donde las células cancerosas expresan LY75.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de GPC3, en donde las células cancerosas expresan GPC3.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de FCRL5, en donde las células cancerosas expresan FCRL5.
En un aspecto, la presente divulgación proporciona métodos para tratar el cáncer al proporcionar al sujeto que lo necesite células efectoras inmunitarias (p. ej., células T, células NK) que están diseñadas para expresar un CAR de IGLL1, en donde las células cancerosas expresan IGLL1.
En un aspecto, la presente divulgación se refiere al tratamiento de un sujeto in vivo utilizando un CAR de PD1 de tal manera que se inhibe el crecimiento de tumores cancerosos. Un CAR de PD1 puede usarse solo para inhibir el crecimiento de tumores cancerosos. Como alternativa, el CAR de PD1 puede usarse junto con otros CAR, agentes inmunogénicos, tratamientos convencionales contra el cáncer u otros anticuerpos. En un caso, el sujeto es tratado con un CAR de PD1 y un XCAR descritos en esta memoria. En un caso, un CAR de PD1 se utiliza junto con otro CAR, p. ej., un CAR descrito en esta memoria, y un inhibidor de quinasa, p. ej., un inhibidor de quinasa descrito en esta memoria.
En otro aspecto, se describe un método para tratar a un sujeto, p. ej., reducir o mejorar, una afección o trastorno hiperproliferativo (p. ej., un cáncer), p. ej., un tumor sólido, un tumor de tejido blando o una lesión metastásica en un sujeto. Tal como se utiliza en esta memoria, se entiende que el término "cáncer" incluye todos los tipos de crecimientos cancerosos o procesos oncogénicos, tejidos metastásicos o células, tejidos u órganos con transformación maligna, independientemente del tipo histopatológico o la etapa de invasividad. Ejemplos de tumores sólidos incluyen tumores malignos, p. ej., sarcomas, adenocarcinomas y carcinomas, de los diversos sistemas de órganos, tales como los que afectan al hígado, pulmón, mama, linfoide, tubo digestivo (p. ej., colon), aparato genitourinario (p. ej., células renales, uroteliales), próstata y faringe. Adenocarcinomas incluyen neoplasias malignas tales como la mayoría de los cánceres de colon, cáncer de recto, carcinoma de células renales, cáncer de hígado, carcinoma de pulmón no microcítico, cáncer de intestino delgado y cáncer de esófago. En una realización, el cáncer es un melanoma, p. ej., un melanoma en etapa avanzada. Lesiones metastásicas de los cánceres antes mencionados también se pueden tratar o prevenir utilizando los métodos de la divulgación y las composiciones de la invención. Ejemplos de otros cánceres que pueden tratarse incluyen cáncer de huesos, cáncer de páncreas, cáncer de piel, cáncer de cabeza o cuello, melanoma maligno cutáneo o intraocular, cáncer de útero, cáncer de ovario, cáncer de recto, cáncer de la región anal, cáncer de estómago, cáncer de testículo, cáncer de útero, carcinoma de las trompas de Falopio, carcinoma de endometrio, carcinoma de cuello uterino, carcinoma de vagina, carcinoma de vulva, enfermedad de Hodgkin, linfoma no hodgkiniano, cáncer de esófago, cáncer de intestino delgado, cáncer del sistema endocrino, cáncer de la glándula tiroides, cáncer de la glándula paratiroides, cáncer de la glándula suprarrenal, sarcoma de tejidos blandos, cáncer de uretra, cáncer de pene, leucemias crónicas o agudas incluidas leucemia mieloide aguda, leucemia mieloide crónica, leucemia linfoblástica aguda, leucemia linfocítica crónica, tumores sólidos de la infancia, linfoma linfocítico, cáncer de vejiga, cáncer de riñón o de uréter, carcinoma de pelvis renal, neoplasia del sistema nervioso central (SNC), linfoma primario del SNC, angiogénesis tumoral, tumor del eje espinal, glioma del tronco encefálico, adenoma hipofisario, sarcoma de Kaposi, cáncer epidermoide, cáncer de células escamosas, linfoma de célula T, cánceres inducidos por el medio ambiente, incluidos los inducidos por amianto y combinaciones de dichos cánceres. El tratamiento de cánceres metastásicos, p. ej., cánceres metastásicos que expresan PD-L1 (Iwai et al. (2005) Int. Immunol. 17: 133-144) puede efectuarse usando las moléculas de anticuerpo descritas en esta memoria.
Cánceres ilustrativos cuyo crecimiento puede inhibirse incluyen cánceres normalmente sensibles a la inmunoterapia. Ejemplos no limitantes de cánceres para el tratamiento incluyen melanoma (p. ej., melanoma maligno metastásico), cáncer renal (p. ej., carcinoma de células claras), cáncer de próstata (p. ej., adenocarcinoma de próstata refractario a hormonas), cáncer de mama, cáncer de colon y cáncer de pulmón (p. ej., cáncer de pulmón no microcítico). Además, las neoplasias malignas refractarias o recurrentes pueden tratarse usando las moléculas descritas en esta memoria.
En un aspecto, la invención emplea un vector que comprende un CAR enlazado operativamente al promotor para la expresión en células efectoras inmunitarias de mamíferos (p. ej., células T, células NK). En un aspecto, la invención proporciona una célula efectora inmunológica recombinante de acuerdo con las reivindicaciones para su uso en el tratamiento del cáncer que expresa un antígeno asociado al cáncer como se describe en esta memoria. En un aspecto, las células que expresan CAR de la invención son capaces de contactar una célula tumoral con al menos un antígeno asociado al cáncer expresado en su superficie de manera que la célula que expresa CAR se dirige a la célula cancerosa y se inhibe el crecimiento del cáncer.
En un aspecto, la divulgación se refiere a un método para inhibir el crecimiento de un cáncer, que comprende poner en contacto la célula cancerosa con una célula que expresa CAR de la presente invención de manera que el CART se active en respuesta al antígeno y se dirija a la célula cancerosa, en la que se inhibe el crecimiento del tumor.
En un aspecto, la divulgación se refiere a un método para tratar el cáncer en un sujeto. El método comprende administrar al sujeto una célula expresa CAR de la presente invención de manera que el cáncer se trate en el sujeto. En un aspecto, un cáncer asociado con la expresión de un antígeno asociado al cáncer como se describe en esta memoria es un cáncer hematológico. En un aspecto, el cáncer hemático es una leucemia o un linfoma. En un aspecto, el cáncer asociado con la expresión de un antígeno asociado al cáncer como se describe en esta memoria incluye cánceres y tumores malignos que incluyen, pero no se limitan, p. ej., a una o más leucemias agudas que incluyen, pero no se limitan, p. ej., a leucemia linfoide aguda de células B ( "BALL"), leucemia linfoide aguda de células T ("TALL"), leucemia linfoide aguda (ALL); una o más leucemias crónicas que incluyen pero no se limitan, p. ej., a leucemia mielógena crónica (CML), leucemia linfoide crónica (CLL). Cánceres o afecciones hematológicas adicionales asociados con la expresión de un antígeno asociado al cáncer como se describe en esta memoria incluyen, pero sin limitación, p. ej., leucemia prolinfocítica de célula B, neoplasia de células dendríticas plasmocitoides blásticas, linfoma de Burkitt, linfoma difuso de célula B grandes, linfoma folicular, tricoleucemia, linfoma folicular de células pequeñas o de células grandes, afecciones linfoproliferativas malignas, linfoma MALT, linfoma de células del manto, linfoma de la zona marginal, mieloma múltiple, mielodisplasia y síndrome mielodisplásico, linfoma no hodgkiniano, linfoma plasmablástico, neoplasia de células dendríticas plasmacitoides, macroglobulinemia de Waldenstrom y "preleucemia", que son una colección diversa de afecciones hematológicas unidas por la producción ineficaz (o displasia) de células mieloides de la sangre y similares. Además, una enfermedad asociada con un antígeno asociado al cáncer como se describe en esta memoria incluye, pero sin limitación, p. ej., cánceres atípicos y/o no clásicos, neoplasias malignas, afecciones precancerosas o enfermedades proliferativas asociadas con la expresión de un antígeno asociado al cáncer como se describe en esta memoria.
En algunas realizaciones, un cáncer que puede tratarse con la célula que expresa CAR de la presente invención es mieloma múltiple. El mieloma múltiple es un cáncer de la sangre que se caracteriza por la acumulación de un clon de células plasmáticas en la médula ósea. Las terapias actuales para el mieloma múltiple incluyen, pero sin limitación, tratamiento con lenalidomida, que es un análogo de la talidomida. La lenalidomida tiene actividades que incluyen actividad antitumoral, inhibición de la angiogénesis e inmunomodulación. En general, se cree que las células de mieloma son negativas para la expresión de un antígeno asociado al cáncer como se describe en esta memoria por citometría de flujo. Por tanto, un CAR CD19, p. ej., como se describe en esta memoria, puede utilizarse para dirigirse a células de mieloma. En algunas realizaciones, cars empleados en la terapia de la presente invención se pueden utilizar en combinación con una o más terapias adicionales, p. ej., el tratamiento con lenalidomida.
La invención incluye un tipo de terapia celular donde las células efectoras inmunitarias (p. ej., célula T, célula NK) se modifican genéticamente para expresar un receptor quimérico para antígeno (CAR) y el célula T o célula NK que expresa CAR se infunde en un receptor que lo necesite. La célula infundida puede destruir las células tumorales en el receptor. A diferencia de las terapias con anticuerpos, las células efectoras inmunitarias modificadas con CAR (p. ej., célula T, célula NK) son capaces de replicarse in vivo lo que da lugar a una persistencia a largo plazo que puede conllevar un control tumoral sostenido. En diversos aspectos, las células efectoras inmunitarias (p. ej., célula T, célula NK) que se administran al paciente, o su descendencia, persisten en el paciente durante al menos cuatro meses, cinco meses, seis meses, siete meses, ocho meses, nueve meses, diez meses, once meses, doce meses, trece meses, catorce meses, quince meses, dieciséis meses, diecisiete meses, dieciocho meses, diecinueve meses, veinte meses, veintiún meses, veintidós meses, veintitrés meses, dos años, tres años, cuatro años o cinco años después de la administración de la célula T o célula NK al paciente.
La invención también incluye un tipo de terapia celular donde las células efectoras inmunitarias (p. ej., célula T, célula NK) están modificadas, p. ej., mediante ARN transcrito in vitro, para expresar de manera transitoria un receptor quimérico para antígenos (CAR) y la célula T o célula NK que expresa CAR se infunde a un receptor que lo necesite. La célula infundida puede destruir las células tumorales en el receptor. Por tanto, en diversos aspectos, las células efectoras inmnunitarias
(p. ej., célula T o célula NK) administradas al paciente están presentes durante menos de un mes, p. ej., tres semanas, dos semanas, una semana, después de la administración de la célula T o célula NK al paciente.
Sin desear quedar limitados a ninguna teoría en particular, la respuesta inmunitaria antitumoral inducida por las células efectoras inmunitarias modificadas con CAR (p. ej., células T, células NK) puede ser una respuesta inmunitaria activa o pasiva, o, como alternativa, puede deberse a una respuesta inmunitaria directa frente a indirecta. En un aspecto, las células efectoras inmunitarias transducidas con CAR (p. ej., células T, células NK) presentan una secreción de citocinas proinflamatorias específica y una potente actividad citolítica en respuesta a las células cancerosas humanas que expresan un antígeno asociado al cáncer como se describe en el presente memoria, resisten la inhibición de un antígeno asociado al cáncer soluble como se describe en el presente memoria, median en la destrucción de células cercanas y median en la regresión de un tumor humano establecido. Por ejemplo, las células tumorales sin antígeno dentro de un campo heterogéneo de un tumor que expresa un antígeno asociado al cáncer como se describe en el presente memoria pueden ser susceptibles de destrucción indirecta por parte de células efectoras inmunitarias redirigidas a un antígeno asociado al cáncer como se describe en el presente memoria (p. ej., células T, células NK) que han reaccionado previamente contra células cancerosas positivas para el antígeno adyacentes.
En un aspecto, las células efectoras inmunitarias modificadas con CAR totalmente humanas (p. ej., células T, células NK) de la invención pueden ser un tipo de vacuna para inmunización ex vivo y/o terapia in vivo en un mamífero. En un aspecto, el mamífero es un ser humano.
Con respecto a la inmunización ex vivo, ocurre al menos una de las siguientes in vitro antes de administrar la célula a un mamífero: i) expansión de las células, ii) introducción de un ácido nucleico que codifica un CAR en las células o iii) crioconservación de las células.
Los procedimientos ex vivo son bien conocidos en la técnica y se analizan con más detalle más adelante. En resumen, las células se aíslan de un mamífero (p. ej., un ser humano) y se modifican genéticamente (es decir, se transducen o transfectan in vitro) con un vector que expresa un CAR descrito en esta memoria. La célula modificada con CAR se puede administrar a un receptor mamífero para proporcionar un beneficio terapéutico. El receptor mamífero puede ser un ser humano y la célula modificada con cAr puede ser autóloga con respecto al receptor. Como alternativa, las células pueden ser alogénicas, singénicas o xenogénicas con respecto al receptor.
El procedimiento para la expansión ex vivo de células progenitoras y células madre hematopoyéticas se describe en la patente de los EE. UU. N° 5.199.942 se puede aplicar a las células de la presente invención. En la técnica existe constancia de otros métodos adecuados, por lo tanto, la presente invención no se limita a ningún método particular de expansión ex vivo de las células. En resumen, el cultivo ex vivo y la expansión de células efectoras inmunitarias (p. ej., células T, células NK) comprende: 1) recoger células madre y progenitoras hematopoyéticas CD34+ de un mamífero a partir de explantes de sangre periférica o médula ósea; y (2) expandir dichas células ex vivo. Además de los factores de crecimiento celular descritos en la patente de los EE. uU. n.° 5.199.942, se pueden utilizar otros factores tales como flt3-L, IL-1, IL-3 y ligando c-kit, para el cultivo y la expansión de las células.
Además de utilizar una vacuna basada en células en lo que se refiere a la inmunización ex vivo, la presente divulgación también proporciona composiciones y métodos para la inmunización in vivo para inducir una respuesta inmunitaria dirigida contra un antígeno en un paciente.
Generalmente, las células activadas y expandidas como se describe en esta memoria se pueden utilizar en el tratamiento y la prevención de enfermedades que surgen en individuos inmunodeprimidos. En particular, las células efectoras inmunitarias modificadas con CAR (p. ej., células T, células NK) de la invención se utilizan en el tratamiento de enfermedades, trastornos y afecciones asociadas con la expresión de un antígeno asociado al cáncer como se describe en esta memoria. En determinados aspectos, las células de la invención se utilizan en el tratamiento de pacientes con riesgo de desarrollar enfermedades, trastornos y afecciones asociados con la expresión de un antígeno asociado al cáncer como se describe en esta memoria. Por lo tanto, la presente divulgación proporciona métodos para el tratamiento o la prevención de enfermedades, trastornos y afecciones asociados con la expresión de un antígeno asociado con el cáncer como se describe en esta memoria, que comprende administrar a un sujeto que lo necesite una cantidad terapéuticamente eficaz de las células efectoras inmunitarias modificadas con CAR. (p. ej., células T, células NK) de la invención.
En un aspecto, las células que expresan CAR de las invenciones se pueden utilizar para tratar una enfermedad proliferativa tal como un cáncer o neoplasia maligna o es una afección precancerosa tal como mielodisplasia, un síndrome mielodisplásico o una preleucemia. Además, una enfermedad asociada con un antígeno asociado al cáncer como se describe en esta memoria incluye, pero sin limitación, p. ej., cánceres atípicos y/o no clásicos, neoplasias malignas, afecciones precancerosas o enfermedades proliferativas que expresan un antígeno asociado al cáncer como se describe en el presente memoria. Las indicaciones no relacionadas con el cáncer asociadas con la expresión de un antígeno asociado al cáncer como se describe en esta memoria incluyen, pero sin limitación, p. ej., enfermedad autoinmunitaria (p. ej., lupus), trastornos inflamatorios (alergia y asma) y trasplante.
Las células efectoras inmunitarias modificadas con CAR (p. ej., células T, células NK) de la presente invención pueden administrarse solas o como una composición farmacéutica en combinación con diluyentes y/o con otros componentes tales como IL-2 u otras citocinas o poblaciones celulares.
Cáncer hematológico
Las afecciones de cáncer hematológico son los tipos de cáncer tales como leucemia, linfoma y afecciones linfoproliferativas malignas que afectan a la sangre, la médula ósea y el sistema linfático.
La leucemia se puede clasificar como leucemia aguda y leucemia crónica. La leucemia aguda se puede clasificar además como leucemia mielógena aguda (AML) y leucemia linfoide aguda (ALL). La leucemia crónica incluye leucemia mielógena crónica (CML) y leucemia linfoide crónica (CLL). Otras afecciones relacionadas incluyen los síndromes mielodisplásicos (SMD, anteriormente conocidos como "preleucemia") que son una colección diversa de afecciones hematológicas unidas por la producción ineficaz (o displasia) de células mieloides de la sangre y el riesgo de transformación a AML.
El linfoma es un grupo de tumores de células sanguíneas que se desarrolla a partir de linfocitos. Linfomas ilustrativos incluyen linfoma no hodgkiniano y linfoma de Hodgkin.
La presente invención proporciona composiciones para tratar el cáncer. En un aspecto, el cáncer es un cáncer hematológico que incluye, pero no se limita a un cáncer hematológico que es una leucemia o un linfoma. En un aspecto,las células que expresan CAR de la invención pueden utilizarse para tratar cánceres y tumores malignos tales como, pero no limitados a leucemias agudas que incluyen, pero no se limitan a, p. ej., leucemia linfoide aguda de células B ("BALL"), leucemia linfoide aguda de células T ("t Al L"), leucemia linfoide aguda (ALL); una o más leucemias crónicas que incluyen pero no se limitan, p. ej., a leucemia mielógena crónica (CML), leucemia linfocítica crónica (CLL); cánceres hematológicos o afecciones hematológicas adicionales que incluyen, pero no se limitan a leucemia prolinfocítica de células B, neoplasia de células dendríticas plasmocitoides blásticas, linfoma de Burkitt, linfoma difuso de células B grandes, linfoma folicular, leucemia de células pilosas, linfoma folicular de células pequeñas o células grandes, afecciones linfoproliferativas malignas, linfoma MALt , linfoma de células del manto, linfoma de la zona marginal, mieloma múltiple, mielodisplasia y síndrome mielodisplásico, linfoma no Hodgkin, linfoma plasmablástico, neoplasia de células dendríticas plasmocitoides, macroglobulinemia de Waldenstrom y "preleucemia", que son una diversa colección de afecciones hematológicas unidas por producción ineficaz (o displasia) de células sanguíneas mieloides, y similares. Además, una enfermedad asociada con un antígeno asociado al cáncer como se describe en esta memoria incluye, pero sin limitación, p. ej., cánceres atípicos y/o no clásicos, tumores malignos, afecciones precancerosas o enfermedades proliferativas que expresan un antígeno asociado al cáncer como se describe en esta memoria.
La presente divulgación también proporciona métodos para inhibir la proliferación o reducir un antígeno asociado con el cáncer como se describe en esta memoria que expresa una población de células, que comprenden poner en contacto una población de células que comprende un antígeno asociado con el cáncer como se describe en esta memoria que expresa una célula con una célula T o célula NK que expresa CAR de la invención que se une a un antígeno asociado al cáncer como se describe en esta memoria que expresa la célula. En un aspecto específico, la presente divulgación proporciona métodos para inhibir la proliferación o reducir la población de células cancerosas que expresan un antígeno asociado al cáncer como se describe en esta memoria, comprendiendo los métodos poner en contacto un antígeno asociado al cáncer como se describe en esta memoria que expresa la población de células cancerosas con células T o células NK que expresan un CAR de la invención que se unen a un antígeno asociado al cáncer como se describe en esta memoria que expresan la célula. En un aspecto, la presente divulgación proporciona métodos para inhibir la proliferación o reducir la población de células cancerosas que expresan un antígeno asociado a un cáncer como se describe en esta memoria, comprendiendo los métodos poner en contacto un antígeno asociado con el cáncer como se describe en esta memoria que expresa una población de células cancerosas con una célula T o célula NK que expresa CAR de la invención que se une a un antígeno asociado al cáncer como se describe en esta memoria. En determinados aspectos, un célula T o célula NK que expresa CAR de la invención reduce la magnitud, el número, la cantidad o el porcentaje de células y/o células cancerosas en al menos un 25 %, al menos un 30 %, al menos un 40 %, al menos un 50 %, al menos un 65 %, al menos un 75 %, al menos un 85 %, al menos un 95 % o al menos un 99 % en un sujeto con o modelo animal para leucemia mieloide u otro cáncer asociado con células que expresan un antígeno asociado al cáncer como se describe en el presente memoria en relación con un control negativo. En un aspecto, el sujeto es un ser humano.
La presente divulgación también proporciona métodos para prevenir, tratar y/o gestionar una enfermedad asociada con un antígeno asociado con el cáncer como se describe en esta memoria que expresa células (p. ej., un cáncer hematológico o un cáncer atípico que expresa un antígeno asociado con el cáncer como se describe en esta memoria), comprendiendo los métodos administrar a un sujeto que lo necesite una célula CAR T o una célula NK de la invención que se une a un antígeno asociado al cáncer como se describe en esta memoria que expresa la célula. En un aspecto, el sujeto es un ser humano. Ejemplos no limitativos de trastornos asociados con un antígeno asociado al cáncer como se describe en esta memoria que expresan células incluyen trastornos autoinmunitarios (tales como lupus), trastornos inflamatorios (tales como alergias y asma) y cánceres (tales como cánceres hematológicos o cánceres atípicos que expresan un antígeno asociado al cáncer como se describe en esta memoria).
La presente divulgación también proporciona métodos para prevenir, tratar y/o gestionar una enfermedad asociada con un antígeno asociado con el cáncer como se describe en esta memoria que expresa células, comprendiendo los métodos administrar a un sujeto que lo necesite una célula CAR T o una célula NK de la invención que se une a un antígeno asociado al cáncer como se describe en esta memoria que expresa la célula. En un aspecto, el sujeto es un ser humano.
La presente divulgación proporciona métodos para prevenir la recaída del cáncer asociado con un antígeno asociado al cáncer como se describe en esta memoria que expresa células, comprendiendo los métodos administrar a un sujeto que lo necesite una célula CAR T o una célula NK de la invención que se une a un antígeno asociado al cáncer como se describe en esta memoria que expresa la célula. En un aspecto, los métodos comprenden administrar al sujeto que lo necesite una cantidad eficaz de una célula T o célula NK que expresa CAR descrita en esta memoria que se une a un antígeno asociado al cáncer como se describe en esta memoria que expresa la célula en combinación con una cantidad eficaz de otra terapia ..
Terapias combinadas
Se puede utilizar una célula que expresa CAR descrita en esta memoria en combinación con otros agentes y terapias conocidos. Administrado "en combinación", tal como se utiliza en esta memoria, significa que se administran dos (o más) tratamientos diferentes al sujeto durante el curso de la aflicción del sujeto por el trastorno, p. ej., los dos o más tratamientos se administran después de que el sujeto haya sido diagnosticado. con el trastorno y antes de que el trastorno haya sido curado o eliminado o el tratamiento haya cesado por otras razones. En algunas realizaciones, el suministro de un tratamiento todavía se está produciendo cuando comienza el suministro del segundo, de modo que hay solapamiento en términos de administración. Esto se denomina, en ocasiones, en esta memoria "suministro simultáneo" o "concurrente". En otras realizaciones, el suministro de un tratamiento termina antes de que comience el suministro del otro tratamiento. En algunas realizaciones de cualquier caso, el tratamiento es más eficaz debido a la administración combinada. Por ejemplo, el segundo tratamiento es más eficaz, es decir, se observa un efecto equivalente con menos del segundo tratamiento, o el segundo tratamiento reduce los síntomas en mayor medida que lo que se vería si el segundo tratamiento se administrara en ausencia del primer tratamiento, o la situación análoga se ve con el primer tratamiento. En algunas realizaciones, el suministro es tal que la reducción de un síntoma u otro parámetro relacionado con el trastorno es mayor de lo que se observaría con un tratamiento suministrado en ausencia del otro. El efecto de los dos tratamientos puede ser parcialmente aditivo, totalmente aditivo o mayor que aditivo. El suministro puede ser tal que un efecto del primer tratamiento suministrado todavía sea detectable cuando se suministra el segundo.
Una célula que expresa CAR descrita en esta memoria y el al menos un agente terapéutico adicional se pueden administrar simultáneamente, en las mismas composiciones o en composiciones separadas, o secuencialmente. Para la administración secuencial, la célula que expresa el CAR descrita en esta memoria se puede administrar primero y el agente adicional puede administrarse en segundo lugar, o el orden de administración se puede invertir.
La terapia con CAR y/u otros agentes, procedimientos o modalidades terapéuticos se pueden administrar durante periodos de trastorno activo o durante un periodo de remisión o enfermedad menos activa. La terapia con CAR se puede administrar antes del otro tratamiento, a la vez que el tratamiento, después del tratamiento o durante la remisión del trastorno.
Cuando se administran combinados, la terapia con CAR y el agente adicional (p. ej., segundo o tercer agente), o todos, se pueden administrar en una cantidad o dosis que es superior, inferior o igual a la cantidad o dosificación de cada agente que se utiliza de manera individual, p. ej., como una monoterapia. En determinadas realizaciones, la cantidad o dosificación administrada de la terapia con CAR, el agente adicional (p. ej., segundo o tercer agente), o todos, es inferior (p. ej., al menos un 20 %, al menos un 30 %, al menos un 40 % o al menos un 50 %) a la cantidad o dosificación de cada agente utilizado de manera individual, p. ej., como una monoterapia. En otras realizaciones, la cantidad o dosificación de la terapia con CAR, el agente adicional (p. ej., segundo o tercer agente), o todos, que da como resultado un efecto deseado (p. ej., el tratamiento del cáncer) es inferior (p. ej., al menos un 20 %, al menos un 30 %, al menos un 40 % o al menos un 50 % inferior) a la cantidad o dosificación de cada agente utilizado de manera individual, p. ej., como una monoterapia, requerida para lograr el mismo efecto terapéutico.
En aspectos adicionales, una célula que expresa CAR descrita en esta memoria puede utilizarse en una pauta de tratamiento en combinación con cirugía, quimioterapia, radiación, agentes inmunosupresores, tales como ciclosporina, azatioprina, metotrexato, micofenolato y FK506, anticuerpos u otros agentes inmunoablativos, tales como CAMPATH, anticuerpos anti-CD3 u otras terapias con anticuerpos, citoxina, fludarabina, ciclosporina, FK506, rapamicina, ácido micofenólico, esteroides, FR901228, citocinas e irradiación. Vacuna peptídica, tal como la descrita en Izumoto et al., 2008 J Neurosurg 108: 963-971.
En una realización, una célula que expresa CAR puede utilizarse en combinación con un agente quimioterapéutico. Agentes quimioterapéuticos ejemplares incluyen una antraciclina (p. ej., doxorrubicina (p. ej., doxorrubicina liposomal)). un alcaloide de la vinca (p. ej., vinblastina, vincristina, vindesina, vinorelbina), un agente alquilante (p. ej., ciclofosfamida, descarbacina, melfalán, ifosfamida, temozolomida), un anticuerpo de células inmunitarias (p. ej., alemtuzamab, gemtuzumab, rituximab, ofatumumab, tositumomab, brentuximab), un antimetabolito (incluyendo, p. ej., antagonistas del ácido fólico, análogos de pirimidina, análogos de purina e inhibidores de la adenosina desaminasa (p. ej., fludarabina)),
un inhibidor de mTOR, un agonista de la proteína relacionada con el TNFR (GITR) inducida por glucocorticoides TNFR, un inhibidor del proteasoma (p. ej,, aclacinomicina A, gliotoxina o bortezomib), un inmunomodulador tal como la talidomida o un derivado de la talidomida (p. ej., lenalidomida).
Los agentes quimioterápicos generales considerados para su uso en terapias combinadas incluyen anastrozol (Arimidex®), bicalutamida (Casodex®), sulfato de bleomicina (Blenoxane®), busulfán (Myleran®), busulfán para inyección (Busulfex®), capecitabina (Xeloda®), N4-pentoxicarbonil-5-desoxi-5-fluorocitidina, carboplatino (Paraplatin®), carmustina (BiCNU®), clorambucilo (Leukeran®), cisplatino (Platinol®), cladribina (Leustatin®), ciclofosfamida (Cytoxan® o Neosar®), citarabina, arabinósido de citosina (Cytosar-U®), citarabina en liposomas para inyección (DepoCyt®), dacarbazina (DTIC-Dome®), dactinomicina (dactinomicina, Cosmegan), clorhidrato de daunorubicina (Cerubidine®), citrato de daunorubicina en liposomas para inyección (DaunoXome®), dexametasona, docetaxel (Taxotere®), clorhidrato de doxorubicina (Adriamycin®, Rubex®), etopósido (Vepesid®), fosfato de fludarabina (Fludara®), 5-fluorouracilo (Adrucil®, Efudex®), flutamida (Eulexin®), tezacitibina, Gemcitabina (difluorodesoxicitidina), hidroxiurea (Hydrea®), Idarubicina (Idamycin®), ifosfamida (IFEX®), irinotecán (Camptosar®), L-asparaginasa (ELSPAR®), leucovorina de calcio, melfalán (Alkeran®), 6-mercaptopurina (Purinethol®), metotrexato (Folex®), mitoxantrona (Novantrone®), mylotarg, paclitaxel (Taxol®), phoenix (Itrio90/MX-DTPA), pentostatina, polifeprosán 20 con implante de carmustina (Gliadel®), citrato de tamoxifeno (Nolvadex®), tenipósido (Vumon®), 6-tioguanina, tiotepa, tirapazamina (Tirazone®), clorhidrato de topotecán para inyección (Hycamptin®), vinblastina (Velban®), vincristina (Oncovin®) y vinorelbina (Navelbine®).
Agentes alquilantes ejemplares incluyen, sin limitación, mostazas nitrogenadas, derivados de etilenimina, sulfonatos de alquilo, nitrosoureas y triazenos): mostaza de uracilo (Aminouracil Mustard®, Chlorethaminacil®, Demethyldopan®, Desmethyldopan®, Haemanthamine®, Nordopan®, Uracil nitrogen mustard®, Uracillost®, Uracilmostaza®, Uramustin®, Uramustine®), clormetina (Mustargen®), ciclofosfamida (Cytoxan®, Neosar®, Clafen®, Endoxan®, Procitox®, Revimmune™), ifosfamida (Mitoxana®), melfalán (Alkeran®), clorambucilo (Leukeran®), pipobromán (Amedel®, Vercyte®), trietilenmelamina (Hemel®, Hexalen®, Hexastat®), trietilentiofosforamina, temozolomida (Temodar®), tiotepa (Thioplex®), busulfán (Busilvex®, Myleran®), carmustina (BiCNU®), lomustina (CeeNU®), estreptozocina (Zanosar®) y Dacarbazina (DTIC-Dome®). Agentes alquilantes ilustrativos adicionales incluyen, sin limitación, oxaliplatino (Eloxatin®); Temozolomida (Temodar® y Temodal®); Dactinomicina (también conocida como actinomicina-D, Cosmegen®); Melfalán (también conocido como L-PAM, L-sarcolisina y mostaza de fenilalanina, Alkeran®); Altretamina (también conocida como hexametilmelamina (HMM), Hexalen®); Carmustina (BiCNU®); Bendamustina (Treanda®); Busulfán (Busulfex® y Myleran®); carboplatino (Paraplatin®); Lomustina (también conocida como CCNU, CeeNU®); Cisplatino (también conocido como CDDP, Platinol® y Platinol®-AQ); Clorambucilo (Leukeran®); Ciclofosfamida (Cytoxan® y Neosar®); Dacarbazina (también conocida como DTIC, DIC e imidazol carboxamida, DTIC-Dome®); Altretamina (también conocida como hexametilmelamina (HMM), Hexalen®); Ifosfamida (Ifex®); prednumustina; Procarbazina (Matulane®); Mecloretamina (también conocida como mostaza nitrogenada, mustina y clorhidrato de mecloretamina, Mustargen®); Estreptozocina (Zanosar®); Tiotepa (también conocido como tiofosfamida, TESPA y TSPA, Thioplex®); Ciclofosfamida (Endoxan®, Cytoxan®, Neosar®, Procitox®, Revimmune®); y bendamustina HCl (Treanda®).
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con fludarabina, ciclofosfamida y/o rituximab. En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con fludarabina, ciclofosfamida y rituximab (FCR). En algunas realizaciones, el sujeto tiene LLC. Por ejemplo, el sujeto tiene una supresión en la rama corta del cromosoma 17 (del(17p), p. ej., en una célula leucémica). En otros ejemplos, el sujeto no tiene un del(17p). En algunas realizaciones, el sujeto comprende una célula leucémica que comprende una mutación en el gen de la región variable de cadena pesada de inmunoglobulina (IgVH). En otras realizaciones, el sujeto no comprende una célula leucémica que comprende una mutación en el gen de la región variable de cadena pesada de inmunoglobulina (IgVH). En algunas realizaciones, la fludarabina se administra a una dosis de aproximadamente 10-50 mg/m2 (p. ej., de aproximadamente 10-15, 15-20, 20-25, 25-30, 30-35, 35-40, 40-45 o 45-50 mg/m2), p. ej., por vía intravenosa. En algunas realizaciones, la ciclofosfamida se administra a una dosis de aproximadamente 200-300 mg/m2 (p. ej., de aproximadamente 200-225, 225-250, 250-275 o 275-300 mg/m2), p. ej., por vía intravenosa. En algunas realizaciones, el rituximab se administra a una dosis de aproximadamente 400-600 mg/m2 (p. ej., 400-450, 450-500, 500-550 o 550 600 mg/m2), p. ej., por vía intravenosa.
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con bendamustina y rituximab. En algunas realizaciones, el sujeto tiene LLC. Por ejemplo, el sujeto tiene una supresión en la rama corta del cromosoma 17 (del(17p), p. ej., en una célula leucémica). En otros ejemplos, el sujeto no tiene un del(17p). En algunas realizaciones, el sujeto comprende una célula leucémica que comprende una mutación en el gen de la región variable de cadena pesada de inmunoglobulina (IgVH). En otras realizaciones, el sujeto no comprende una célula leucémica que comprende una mutación en el gen de la región variable de cadena pesada de inmunoglobulina (IgVH). En algunas realizaciones, la bendamustina se administra a una dosis de aproximadamente 70-110 mg/m2 (p. ej., 70-80, 80-90, 90-100 o 100-110mg/m2), p. ej., por vía intravenosa. En algunas realizaciones, el rituximab se administra a una dosis de aproximadamente 400-600 mg/m2 (p. ej., 400-450, 450-500, 500-550 o 550-600 mg/m2), p. ej., por vía intravenosa.
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con rituximab, ciclofosfamida, doxorrubicina, vincristina y/o un corticosteroide (p. ej., prednisona). En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona (R-CHOP).
En realizaciones, el sujeto tiene linterna difuso de célula B grandes (LDLBG). En casos, el sujeto tiene LDLBG en estadio limitado no voluminoso (p. ej., comprende un tumor que tiene un tamaño/diámetro de menos de 7 cm). En algunas realizaciones, se trata al sujeto con radiación en combinación con R-CHOP. Por ejemplo, al sujeto se le administra R-CHOP (p. ej., 1-6 ciclos, p. ej., 1, 2, 3, 4, 5 o 6 ciclos de R-CHOP), seguido de radiación. En algunos casos, al sujeto se le administra R-CHOP (p. ej., 1-6 ciclos, p. ej., 1, 2, 3, 4, 5 o 6 ciclos de R-CHOP) después de la radiación.
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con etopósido, prednisona, vincristina, ciclofosfamida, doxorrubicina y/o rituximab. En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con etopósido, prednisona, vincristina, ciclofosfamida, doxorrubicina y rituximab (EPOCH-R). En algunas realizaciones, una célula que expresa CAR descrita en esta memoria se administra a un sujeto combinada con EPOCH-R ajustado a la dosis (DA-EPOCH-R). En algunas realizaciones, el sujeto tiene un linfoma de célula B, p. ej., un linfoma agresivo de células B con reordenamiento de Myc.
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con rituximab y/o lenalidomida.. Lenalidomida ((RS)-3-(4-amino-1-oxo 1,3-dihidro-2H-isoindol- 2-il)piperidina-2,6-diona) es un inmunomodulador. En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con rituximab y lenalidomida. En algunas realizaciones, el sujeto tiene linfoma folicular (FL) o linfoma de células del manto (MCL). En algunas realizaciones, el sujeto tiene FL y no se ha tratado previamente con una terapia contra el cáncer. En algunas realizaciones, se administra lenalidomida a una dosis de aproximadamente 10-20 mg (p. ej., 10-15 o 15-20 mg), p. ej., diariamente. En algunas realizaciones, se administra rituximab a una dosis de aproximadamente 350-550 mg/m2 (p. ej., 350-375, 375-400, 400 425, 425-450, 450-475 o 475-500 mg/m2), p. ej., por vía intravenosa.
Inhibidores de mTOR ejemplares incluyen, p. ej., temsirolimus; ridaforolimus (anteriormente conocido como deferolimus, dimetilfosfinato de (1R,2R,4S)-4-[(2R)-2 [(1R,9S,12S,15R,16E,18R,19R,21R, 23S,24E,26E,28Z,30S,32S,35R)-1,18-dihidroxi-19,30-dimetoxi-15,17,21,23, 29,35-hexametil-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatriciclo[30.3.1.049] hexatriaconta-16,24,26,28-tetraen-12-il]propil]-2-metoxiciclohexilo, también conocido como AP23573 y MK8669, y descrito en la Publicación PCT N° WO 03/064383); everolimus (Afinitor® o RAD001); rapamicina (AY22989, Sirolimus®); simapimod (CAS 164301-51-3); emsirolimus, (5-{2,4-Bis[(3S)-3-metilmorfolin-4-il]pirido[2,3-d]pirimidin-7-il}-2-metoxifenil)metanol (AZD8055); 2-amino-8-[frans-4-(2-hidroxietoxi)ciclohexil]-6-(6-metoxi-3-piridinil)-4-metil-pirido[2,3-d]pirimidin-7(8H)-ona (PF04691502, CAS 1013101-36-4) y N2-[1,4-dioxo-4-[[4-(4-oxo-8-fenil-4H-1-benzopiran-2-il)morfolinio-4-il]metoxi]butil]-L-arginilglicil-L-a-aspartilL-serina-, sal interna (SF1126, Cas 936487-67-1) y XL765.
Los inmunomoduladores ilustrativos incluyen, p. ej., afutuzumab (disponible de Roche®); pegfilgrastim (Neulasta®); lenalidomida (CC-5013, Revlimid®); talidomida (Thalomid®), actimid (CC4047); e IRX-2 (mezcla de citocinas humanas que incluyen interleucina 1, interleucina 2 e interferón y, CAS 951209-71-5, disponible de IRX Therapeutics).
Las antraciclinas ilustrativas incluyen, p. ej., doxorubicina (Adriamycin® y Rubex®); bleomicina (lenoxano®); daunorubicina (clorhidrato de dauorubicina, daunomicina y clorhidrato de rubidomicina, Cerubidine®); daunorubicina liposómica (liposoma de citrato de daunorubicina, DaunoXome®); mitoxantrona (DHAD, Novantrone®); epirubicina (Ellence™); idarubicina (Idamycin®, Idamycin PFS®); mitomicina C (Mutamycin®); geldanamicina; herbimicina; ravidomicina; y desacetilravidomicina.
Los alcaloides de la vinca ilustrativos incluyen, p. ej., tartrato de vinorelbina (Navelbine®), vincristina (Oncovin®) y vindesina (Eldisine®)); vinblastina (también conocida como sulfato de vinblastina, vincaleucoblastina y VLB, Alkaban-AQ®y Velban®); y vinorelbina (Navelbine®).
Los inhibidores ilustrativos del proteasoma incluyen bortezomib (Velcade®); carfilzomib (PX-171-007, (S)-4-metil-W-((S)-1-(((S)-4-metil-1-((R)-2-metiloxiran-2-il)-1-oxopentan-2-il)amino)-1-oxo-3-fenilpropan-2-il)-2-((S)-2-(2-morfolinoacetamido)-4-fenilbutanamido)pentanamida); marizomib (NPI-0052); citrato de ixazomib (MLN-9708); delanzomib (CEP-18770); y 0-metil-W-[(2-metil-5-tiazolil)carbonil]-L-seril-0-metil-W-[(1S)-2-[(2R)-2-metil-2-oxiranil]-2-oxo-1-(fenilmetil)etil]-L-serinamida (ONX-0912).
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con bendamustina y brentuximab. Brentuximab es un conjugado de anticuerpo-fármaco de un anticuerpo anti-CD30 y monometil-auristatina E. En algunas realizaciones, el sujeto padece linfoma de Hodgkin (LH), p. ej., LH recurrente o refractario. En algunas realizaciones, el sujeto comprende Lh CD30+. En algunas realizaciones, el sujeto se ha sometido a un autotrasplante de células madre (ASCT). En algunas realizaciones, el sujeto no se ha sometido a un ASTC. En algunas realizaciones, se administra brentuximab a una dosis de aproximadamente 1-3 mg/kg (p. ej., aproximadamente 1-1.5, 1.5-2, 2-2.5 o 2.5-3 mg/kg), p. ej., por vía intravenosa, p. ej., cada 3 semanas.
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con brentuximab y dacarbazina o en combinación con brentuximab y bendamustina. Dacarbazina es un agente alquilante con el nombre químico de 5-(3,3-dimetil-1-triazenil)imidazol-4-carboxamida. La bendamustina es un agente alquilante con un nombre químico de ácido 4-[5-[bis(2-cloroetil)amino]-1-metilbencimidazol-2-il]butanoico. En algunas realizaciones, el sujeto padece linfoma de Hodgkin (LH). En algunas realizaciones, el sujeto no se ha tratado previamente con una terapia contra el cáncer. En
algunas realizaciones, el sujeto tiene al menos 60 años de edad, p. ej., 60, 65, 70, 75, 80, 85 o más. En algunas realizaciones, se administra dacarbazina a una dosis de aproximadamente 300-450 mg/m2 (p. ej., 300-325, 325-350, 350 375, 375-400, 400-425 o 425-450 mg/m2), p. ej., por vía intravenosa. En realizaciones, la bendamustina se administra a una dosificación de aproximadamente 75-125 mg/m2 (por ejemplo, 75-100 o 100-125 mg/m2, por ejemplo, aproximadamente 90 mg/m2), p. ej., por vía intravenosa. En algunas realizaciones, se administra brentuximab a una dosis de aproximadamente 1-3 mg/kg (p. ej., aproximadamente 1-1.5, 1.5-2, 2-2.5 o 2.5-3 mg/kg), p. ej., por vía intravenosa, p. ej., cada 3 semanas.
En algunas realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con un inhibidor de CD20, p. ej., un anticuerpo anti-CD20 (p. ej., un anticuerpo mono o biespecífico anti-CD20) o un fragmento del mismo. Ejemplos de anticuerpos anti-CD20 incluyen, pero no se limitan a rituximab, ofatumumab, ocrelizumab, veltuzumab, obinutuzumab, TRU-015 (Trubion Pharmaceuticals), ocaratuzumab y Pro131921 (Genentech). Véase, p. ej., Lim et al. Haematologica. 95,1 (2010): 135-43.
En algunas realizaciones, el anticuerpo anti-CD20 comprende rituximab. Rituximab es un anticuerpo monoclonal IgG1 kappa quimérico de ratón/ser humano que se une a CD20 y provoca la citólisis de una célula que expresa CD20, p. ej., como se describe en www.accessdata.fda.gov/drugsatfda_docs/label/2010/103705s5311lbl.pdf. En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con rituximab. En algunas realizaciones, el sujeto tiene LLC o LLCP.
En algunas realizaciones, se administra rituximab por vía intravenosa, p. ej., como una infusión intravenosa. Por ejemplo, cada infusión proporciona aproximadamente 500-2000 mg (p. ej., aproximadamente 500-550, 550-600, 600-650, 650-700, 700-750, 750-800, 800-850, 850-900, 900-950, 950-1000, 1000-1100, 1100-1200, 1200-1300, 1300-1400, 1400-1500, 1500-1600, 1600-1700, 1700-1800, 1800-1900 o 1900-2000 mg) de rituximab. En algunos casos, rituximab se administra a una dosis de 150 mg/m2 a 750 mg/m2, p. ej., aproximadamente 150-175 mg/m2, 175-200 mg/m2, 200-225 mg/m2, 225 250 mg/m2, 250-300 mg/m2, 300-325 mg/m2, 325-350 mg/m2, 350-375 mg/m2, 375-400 mg/m2, 400-425 mg/m2, 425 450 mg/m2, 450-475 mg/m2, 475-500 mg/m2, 500-525 mg/m2, 525-550 mg/m2, 550-575 mg/m2, 575-600 mg/m2, 600 625 mg/m2, 625-650 mg/m2, 650-675 mg/m2 o 675-700 mg/m2, donde m2 indica el área de superficie corporal del sujeto. En algunas realizaciones, se administra rituximab en un intervalo de dosificaciones de al menos 4 días, p. ej., 4, 7, 14, 21, 28, 35 días o más. Por ejemplo, se administra rituximab en un intervalo de dosificaciones de al menos 0,5 semanas, p. ej., 0,5, 1, 2, 3, 4, 5, 6, 7, 8 semanas o más. En algunas realizaciones, se administra rituximab a una dosis y un intervalo de dosificaciones descritos en esta memoria durante un periodo de tiempo, p. ej., al menos 2 semanas, p. ej., al menos 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 semanas o más. Por ejemplo, se administra rituximab a una dosis y un intervalo de dosificaciones descritos en esta memoria durante un total de al menos 4 dosis por ciclo de tratamiento (p. ej., al menos 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 o más dosis por ciclo de tratamiento).
En algunas realizaciones, el anticuerpo anti-CD20 comprende ofatumumab. El ofatumumab es un anticuerpo monoclonal humano IgG1K anti-CD20 con un peso molecular de aproximadamente 149 kDa. Por ejemplo, se genera ofatumumab utilizando tecnología de ratón transgénico e hibridoma y se expresa y purifica a partir de una línea celular murina recombinante (NS0). Véase, p. ej., www.accessdata.fda.gov/drugsatfda_docs/label/2009/125326lbl.pdf; y los números de identificador de ensayos clínicos NCT01363128, NCT01515176, NCT01626352 y NCT01397591. En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con ofatumumab. En algunas realizaciones, el sujeto tiene LLC o LLCP.
En algunas realizaciones, se administra ofatumumab como una infusión intravenosa. Por ejemplo, cada infusión proporciona aproximadamente 150-3000 mg (p. ej., aproximadamente 150-200, 200-250, 250-300, 300-350, 350-400, 400-450, 450-500, 500-550, 550-600, 600-650, 650-700, 700-750, 750-800, 800-850, 850-900, 900-950, 950-1000, 1000 1200, 1200-1400, 1400-1600, 1600-1800, 1800-2000, 2000-2200, 2200-2400, 2400-2600, 2600-2800 o 2800-3000 mg) de ofatumumab. En algunas realizaciones, se administra ofatumumab a una dosis de partida de aproximadamente 300 mg, seguida de 2000 mg, p. ej., durante aproximadamente 11 dosis, p. ej., durante 24 semanas. En algunas realizaciones, se administra ofatumumab en un intervalo de dosificaciones de al menos 4 días, p. ej., 4, 7, 14, 21, 28, 35 días o más. Por ejemplo, se administra ofatumumab en un intervalo de dosificaciones de al menos 1 semana, p. ej., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 24, 26, 28, 20, 22, 24, 26, 28, 30 semanas o más. En algunas realizaciones, se administra ofatumumab a una dosis y un intervalo de dosificaciones descritos en esta memoria durante un periodo de tiempo, p. ej., al menos 1 semana, p. ej., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 22, 24, 26, 28, 30, 40, 50, 60 semanas o más, o 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 meses o más, o 1,2, 3, 4, 5 años o más. Por ejemplo, se administra ofatumumab a una dosis y un intervalo de dosificaciones descritos en esta memoria durante un total de al menos 2 dosis por ciclo de tratamiento (p. ej., al menos 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 18, 20 o más dosis por ciclo de tratamiento).
En algunos casos, el anticuerpo anti-CD20 comprende ocrelizumab. Ocrelizumab es un anticuerpo monoclonal anti-CD20 humanizado, p. ej., como se describe en los identificadores de ensayos clínicos n.° NCT00077870, NCT01412333, NCT00779220, NCT00673920, NCT01194570 y Kappos et al. Lancet. 19.378(2011):1779-87.
En algunos casos, el anticuerpo anti-CD20 comprende veltuzumab. Veltuzumab es un anticuerpo monoclonal humanizado contra CD20. Véanse, p. ej., los N°s de identificador de ensayos clínicos NCT00547066, NCT00546793, NCT01101581, y Goldenberg et al. Leuk Lymphoma. 51 (5) (2010): 747-55.
En algunos casos, el anticuerpo anti-CD20 comprende GA101. GA101 (también llamado obinutuzumab o RO5072759) es un anticuerpo monoclonal anti-CD20 humanizado y modificado por glicoingeniería. Véase, p. ej., Robak. Curr. Opin. Investig. Drugs. 10.6(2009):588-96; Números de Identificadores de Ensayos Clínicos: NCT01995669, NCT01889797, NCT02229422 y NCT014M205; y www.accessdata.fda.gov/drugsatfda_docs/label/2013/125486s000lbl.pdf.
En algunos casos, el anticuerpo anti-CD20 comprende AME-133v. AME-133v (también denominado LY2469298 u ocaratuzumab) es un anticuerpo monoclonal IgG1 humanizado contra CD20 con mayor afinidad por el receptor FcYRIIIa y una actividad mejorada de citotoxicidad celular dependiente de anticuerpos (AdCc) en comparación con rituximab. Véase, p. ej., Robak et al. BioDrugs 25.1 (2011): 13-25; y Forero-Torres et al. Clin Cancer Res. 18.5 (2012): 1395-403. En algunos casos, el anticuerpo anti-CD20 comprende PRO 131921. PRO131921 es un anticuerpo monoclonal anti-CD20 humanizado diseñado para tener una mejor unión a FcYRIIIa y ADCC mejorada en comparación con rituximab. Véase, p. ej., Robak et al. BioDrugs 25.1 (2011): 13-25; y Casulo et al. Clin Immunol. 154.1 (2014): 37-46; y el n.° de identificador de ensayos clínicos NCT00452127.
En algunos casos, el anticuerpo anti-CD20 comprende TRU-015. El TRU-015 es una proteína de fusión anti-CD20 derivada de dominios de un anticuerpo contra cD20. El TRU-015 es más pequeño que los anticuerpos monoclonales, pero conserva las funciones efectoras mediadas por Fc. Véase, p. ej., Robak et al. BioDrugs 25.1 (2011): 13-25. TRU-015 contiene un fragmento variable de cadena sencilla anti-CD20 (scFv) ligado a los dominios de bisagra, CH2 y CH3 de IgG1 humana, pero carece de los dominios CH1 y CL.
En algunas realizaciones, un anticuerpo anti-CD20 descrito en esta memoria se conjuga o se une de otro modo a un agente terapéutico, p. ej., un agente quimioterapéutico (p. ej., citoxano, fludarabina, inhibidor de histona desacetilasa, agente desmetilante, vacuna peptídica, antibiótico antitumoral, inhibidor de tirosina quinasa, agente alquilante, agente antimicrotúbulos o antimitótico), agente antialérgico, agente antináuseas (o antiemético), analgésico o agente citoprotector descrito en esta memoria.
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con un inhibidor del linfoma 2 de células B (BCL-2) (p. ej., venetoclax, también llamado ABT-199 o GDC-0199;) y/o rituximab. En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con venetoclax y rituximab. Venetoclax es una pequeña molécula que inhibe la proteína antiapoptótica BCL-2. La estructura de venetoclax (4-(4-{[2-(4-clorofenil)-4,4-dimetilciclohex-1-en-1-il]metil}piperazin-1-il)-N-({3-nitro-4-[(tetrahidro-2H-piran-4-ilmetil)amino]fenil}sulfonil)-2-(1H-pirrolo[2,3-£>]piridin-5-iloxi)benzamida) se muestra a continuación.

En algunas realizaciones, el sujeto tiene LLC. En algunas realizaciones, el sujeto tiene LLC recidivante, p. ej., al sujeto se le ha administrado previamente una terapia contra el cáncer. En algunas realizaciones, se administra venetoclax a una dosis de aproximadamente 15-600 mg (p. ej., 15-20, 20-50, 50-75, 75-100, 100-200, 200-300, 300-400, 400-500 o 500 600 mg), p. ej., diariamente. En algunas realizaciones, se administra rituximab a una dosis de aproximadamente 350 550 mg/m2 (p. ej., 350-375, 375-400, 400-425, 425-450, 450-475 o 475-500 mg/m2), p. ej., por vía intravenosa, p. ej., mensualmente.
En una realización, las células que expresan un CAR se administran a un sujeto en combinación con una molécula que disminuye la población de células Treg. Métodos que disminuyen el número de (p. ej., agotan) las células Treg son conocidos en la técnica e incluyen, p. ej., el agotamiento de CD25, la administración de ciclofosfamida, la modulación de la función GITR. Sin desear ceñirse a ninguna teoría, se cree que la reducción del número de célula Treg en un sujeto antes de la aféresis o antes de la administración de una célula que expresa CAR descrita en el presente memoria reduce el número de células inmunitarias no deseadas (p. ej., Treg) en el microentorno del tumor y reduce el riesgo de que el sujeto recaiga. En una realización, las células que expresan un CAR se administran a un sujeto en combinación con una molécula que fija como objetivo GITR y/o que modula las funciones de GITR tal como un agonista de GITR y/o un anticuerpo de GITR que agota las células T reguladoras (Tregs). En realizaciones, las células que expresan un CAR se administran a un sujeto en combinación con ciclofosfamida. En una realización, las moléculas de unión a GITR y/o
moléculas que modulan las funciones de GITR (p. ej., agonista de GITR y/o anticuerpos de GITR con agotamiento de Treg) se administran antes de la administración de la célula que expresa CAR. Por ejemplo, en una realización, el agonista de GITR se puede administrar antes de la aféresis de las células. En realizaciones, la ciclofosfamida se administra al sujeto antes de la administración (p. ej., infusión o reinfusión) de la célula que expresa CAR o antes de la aféresis de las células. En realizaciones, la ciclofosfamida y un anticuerpo anti-GITR se administran al sujeto antes de la administración (p. ej., infusión o reinfusión) de la célula que expresa c Ar o antes de la aféresis de las células. En una realización, el sujeto tiene cáncer (p. ej., un cáncer sólido o un cáncer hematológico tal como LLA o LLC). En una realización, el sujeto tiene LLC. En realizaciones, el sujeto tiene LLA. En realizaciones, el sujeto tiene un cáncer sólido, p. ej., un cáncer sólido descrito en esta memoria. Los agonistas de GITR ilustrativos incluyen, p. ej., proteínas de fusión de GITR y anticuerpos anti-GITR (p. ej., anticuerpos anti-GITR bivalentes), tales como, p. ej., una proteína de fusión de GITR descrita en la patente de los EE. UU. n.°: 6.111.090, patente europea n.°: 090505B1, patente de los EE. UU. n.°: 8.586.023, publicaciones PCT n.°: WO 2010/003118 y 2011/090754, o un anticuerpo anti-GITR descrito, p. ej., en la patente de los EE. UU. n.°: 7.025.962, patente europea n.°: 1947183B1, patente de los EE. UU. n.°: 7.812.135, patente de los EE. UU. n.°: 8.388.967, patente de los EE. UU. n.°: 8.591.886, patente europea n.°: EP 1866339, publicación PCT n.°: WO 2011/028683, publicación PCT n °.:WO 2013/039954, publicación PCT n ° W02005/007190, Publicación de PCT N.°: WO 2007/133822, Publicación de PCT N.°: WO2005/055808, publicación PCT n.°: WO 99/40196, publicación PCT n.°: WO 2001/03720, publicación PCT n.°: WO99/20758, publicación PCT n.°: WO2006/083289, publicación PCT n.°: WO 2005/115451, patente de los EE. UU. n.°: 7.618.632 y publicación PCT n.°: WO 2011/051726.
En una realización, una célula que expresa CAR se administra a un sujeto en combinación con un inhibidor de mTOR, p. ej., un inhibidor de mTOR descrito en esta memoria, p. ej., un rapálogo tal como everolimus. En una realización, el inhibidor de mTOR se administra antes que la célula que expresa CAR. Por ejemplo, en una realización, el inhibidor de mTOR se puede administrar antes de la aféresis de las células. En una realización, el sujeto tiene LLC.
En una realización, una célula que expresa CAR se administra a un sujeto en combinación con un agonista de GITR, p. ej., un agonista de GITR descrito en esta memoria. En una realización, el agonista de GITR se administra antes que la célula que expresa CAR. Por ejemplo, en una realización, el agonista de GITR se puede administrar antes de la aféresis de las células. En una realización, el sujeto tiene LLC.
En una realización, una célula que expresa CAR puede utilizarse en combinación con un inhibidor de quinasa. En una realización, el inhibidor de quinasa es un inhibidor de CDK4, p. ej., un inhibidor de CDK4 descrito en esta memoria, p. ej., un inhibidor de CD4/6, tal como, p. ej., 6-acetil-8-ciclopentil-5-metil-2-(5-piperazin-1-il-piridin-2-ilamino)-8H-pirido[2,3-d]pirimidin-7-ona, clorhidrato (al que también se alude como palbociclib o PD0332991). En una realización, el inhibidor de quinasa es un inhibidor de BTK, p. ej., un inhibidor de BTK descrito en esta memoria, tal como, p. ej., ibrutinib. En una realización, el inhibidor de quinasa es un inhibidor de mTOR, p. ej., un inhibidor de mTOR descrito en esta memoria, tal como, p. ej., rapamicina, un análogo de rapamicina, OSI-027. El inhibidor de mTOR puede ser, p. ej., un inhibidor de mTORC1 y/o un inhibidor de mTORC2, p. ej., un inhibidor de mTORC1 y/o un inhibidor de mTORC2 descrito en esta memoria. En una realización, el inhibidor de quinasa es un inhibidor de MNK, p. ej., un inhibidor de MNK descrito en esta memoria, tal como, p. ej., 4-amino-5-(4-fluoroanilino)-pirazolo [3,4-d] pirimidina. El inhibidor de MNK puede ser, p. ej., un inhibidor de MNK1a, MNK1b, MNK2a y/o MNK2b. En una realización, el inhibidor de quinasa es un inhibidor doble de PI3K/mTOR descrito en esta memoria, tal como, p. ej., PF-04695102.
En una realización el inhibidor de quinasa es un inhibidor de CDK4 seleccionado de aloisina A; flavopiridol o HMR-1275, 2-(2-clorofenil)-5,7-dihidroxi-8-[(3S,4R)-3-hidroxi-1-metil-4-piperidinil]-4-cromenona; crizotinib (p F-02341066; 2-(2-clorofenil)-5,7-dihidroxi-8-[(2R,3S)-2-(hidroximetil)-1-metil-3-pirrolidinil]- 4H-1-benzopiran-4-ona, hidrocloruro (P276-00); 1-metil-5-[[2-[5-(trifluorometil)-1H-imidazol-2-il]-4-piridinil]oxi]-W-[4-(trifluorometil)fenil]-1H-bencimidazol-2-amina (RAF265); indisulam (E7070); roscovitina (CYC202); palbociclib (PD0332991); dinaciclib (SCH727965); N-[5-[[(5-terc.-butiloxazol-2-il)metil]tio]tiazol-2-il]piperidina-4-carboxamida (BMS 387032); ácido 4-[[9-cloro-7-(2,6-difluoroíenil)-5H-pirimido[5,4-d][2]benzazepin-2-il]amino]-benzoico (MLN8054); N-(piperidin-4-il)amida del ácido 5-[3-(4,6-difluoro-1H-bencimidazol-2-il)-1H-indazol-5-il]-N-etil-4-metil-3-piridinametanamina (AG-024322); 4-(2,6-diclorobenzoilamino)-1H-pirazol-3-carboxílico (AT7519); 4-[2-metil-1-(1-metiletil)-1H-imidazol-5-il]-A/-[4-(metilsulfonil)fenil]-2-pirimidinamina (AZD5438); y XL281 (BMS908662).
En una realización, el inhibidor de quinasa es un inhibidor de CDK4, p. ej., palbociclib (PD0332991), y el palbociclib se administra en una dosis de aproximadamente 50 mg, 60 mg, 70 mg, 75 mg, 80 mg, 90 mg, 100 mg, 105 mg, 110 mg, 115 mg, 120 mg, 125 mg, 130 mg, 135 mg (p. ej., 75 mg, 100 mg o 125 mg) al día durante un periodo de tiempo, p. ej., diariamente durante 14- 21 días de un ciclo de 28 días, o diariamente durante 7-12 días de un ciclo de 21 días. En una realización, se administran 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 o más ciclos de palbociclib.
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con un inhibidor 4 o 6 de quinasa dependiente de ciclina (CDK), p. ej., un inhibidor de CDK4 o un inhibidor de CDK6 descrito en esta memoria. En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con un inhibidor de CDK4/6 (p. ej., un inhibidor que fija como objetivo tanto a CDK4 como a CDK6), p. ej., un inhibidor de CDK4/6 descrito en esta memoria. En una realización, el sujeto tiene LCM. El LCM es un cáncer agresivo que responde poco a las terapias actualmente disponibles, es decir, es esencialmente incurable. En muchos casos de lCm , la ciclina D1 (un regulador de CDK4/6) se
expresa (p. ej., debido a la translocación cromosómica que implica genes de inmunoglobulina y ciclina D1) en células de LCM. Por lo tanto, sin estar ligados a la teoría, se cree que las células de LCM son muy sensibles a la inhibición de CDK4/6 con alta especificidad (es decir, efecto mínimo sobre las células inmunitarias normales). Los inhibidores de CDK4/6 solos han tenido cierta eficacia en el tratamiento del LCM, pero solo han logrado una remisión parcial con una alta tasa de recaída. Un inhibidor de CDK4/6 ilustrativo es LEE011 (también denominado ribociclib), cuya estructura se muestra a continuación.
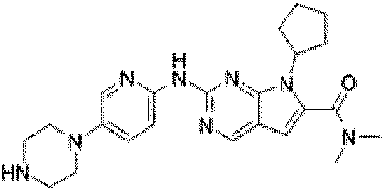
Sin quedar ligados a la teoría, se cree que la administración de una célula que expresa CAR descrita en esta memoria con un inhibidor de CDK4/6 (p. ej., LEE011 u otro inhibidor de CDK4/6 descrito en esta memoria) puede logar una respuesta elevada, p. ej., con tasas de remisión más elevadas y/o tasas de recaídas menores, p. ej., en comparación con un inhibidor de CDK4/6 solo.
En una realización, el inhibidor de quinasa es un inhibidor de BTK seleccionado de ibrutinib (PCI-32765); GDC-0834; RN-486; CGI-560; CGI-1764; HM-71224; CC-292; ONO-4059; CNX-774; y LFM-A13. En una realización preferida, el inhibidor de BTK no reduce ni inhibe la actividad quinasa de la quinasa inducible por interleucina-2 (ITK) y se selecciona de GDC-0834; RN-486; CGI-560; CGI-1764; HM-71224; CC-292; ONO-4059; CNX-774; y LFM-A13.
En una realización, el inhibidor de quinasa es un inhibidor de BTK, p. ej., ibrutinib (PCI-32765). En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con un inhibidor de BTK (p. ej., ibrutinib). En algunas realizaciones, una célula que expresa CAR descrita en esta memoria se administra a un sujeto combinada con ibrutinib (también denominado PCI-32765). La estructura de ibrutinib (1-[(3R)-3-[4-amino-3-(4-fenoxifenil)-1H-pirazolo[3,4-d]pirimidin-1-il]piperidin-1-il]prop-2-en-1-ona) se muestra a continuación.

En algunas realizaciones, el sujeto padece LLC, linfoma de las células del manto (LCM) o linfoma linfocítico de células pequeñas (LLCP). Por ejemplo, el sujeto tiene una supresión en la rama corta del cromosoma 17 (del(17p), p. ej., en una célula leucémica). En otros ejemplos, el sujeto no tiene un del(17p). En algunas realizaciones, el sujeto padece LLC o LLCP recurrente, p. ej., al sujeto se le ha administrado previamente una terapia contra el cáncer (p. ej., se le han administrado previamente una, dos, tres o cuatro terapias contra el cáncer anteriores). En algunas realizaciones, el sujeto padece LLC o LLCP refractario. En otras realizaciones, el sujeto padece linfoma folicular, p. ej., linfoma folicular refractario o recurrente. En algunas realizaciones, se administra ibrutinib a una dosis de aproximadamente 300-600 mg/día (p. ej., aproximadamente 300-350, 350-400, 400-450, 450-500, 500-550 o 550-600 mg/día, p. ej., aproximadamente 420 mg/día o aproximadamente 560 mg/día), p. ej., por vía oral. En realizaciones, el ibrutinib se administra a una dosis de aproximadamente 250 mg, 300 mg, 350 mg, 400 mg, 420 mg, 440 mg, 460 mg, 480 mg, 500 mg, 520 mg, 540 mg, 560 mg, 580 mg, 600 mg (p. ej., 250 mg, 420 mg o 560 mg) diariamente durante un período de tiempo, p. ej., diariamente durante un ciclo de 21 días o diariamente durante un ciclo de 28 días. En una realización, se administran 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 o más ciclos de ibrutinib. Sin estar ligados a la teoría, se cree que la adición de ibrutinib mejora la respuesta proliferativa de los célula T y puede cambiar los célula T de un fenotipo T-auxiliar-2 (Th2) a T-auxiliar-1 (Th1). Th1 y Th2 son fenotipos de los célula T auxiliares, donde Th1 frente a Th2 dirigen diferentes rutas de respuesta inmunitaria. Un fenotipo Th1 se asocia con respuestas proinflamatorias, p. ej., para provocar la destrucción de células, tales como virus/patógenos intracelulares o células cancerosas, o para perpetuar respuestas autoinmunitarias. Un fenotipo Th2 se asocia con respuestas antiinflamatorias y de acumulación de eosinófilos.
En una realización, el inhibidor de quinasa es un inhibidor de mTOR seleccionado de temsirolimus; ridaforolimus dimetilfosfinato de (1R,2R,4S)-4-[(2R)-2 [(1R,9S,12S,15R,16E,18R,19R,21R, 23S,24E,26E,28Z,30S,32S,35R)-1,18-dihidroxi-19,30-dimetoxi-15,17,21,23, 29,35-hexametil-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatriciclo[30.3.1.049] hexatriaconta-16,24,26,28-tetraen-12-il]propil]-2-metoxiciclohexilo, también conocido como AP23573 y MK8669; everolimus (RAD001); rapamician (AY22989); simapimod; (5-{2,4-bis[(3S)-3-metilmorfolin-4-il]pirido[2,3-d]pirimidin-7-il}-2-metoxifenil)metanol (AZD8055); 2-amino-8-[frans-4-(2-hidroxietoxi)ciclohexil]-6-(6-metoxi-3-piridinil)-4-metil-pirido[2,3cf]p¡rim¡d¡n-7(8H)-ona (PF04691502); y ^ 2-[1,4-dioxo-4-[[4-(4-oxo-8-fenil-4H-1-benzopiran-2-il)morfolinio-4-il]metoxi]butilj-L-arginilglicil-L-a-aspartilL-serina-, sal interna (SF1126); y XL765.
En una realización, el inhibidor de quinasa es un inhibidor de mTOR, p. ej., rapamicina, y la rapamicina se administra a una dosis de aproximadamente 3 mg, 4 mg, 5 mg, 6 mg, 7 mg, 8 mg, 9 mg, 10 mg (p. ej., 6 mg) diariamente durante un período de tiempo, p. ej., diariamente durante un ciclo de 21 días o diariamente durante un ciclo de 28 días. En una realización, se administran 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 o más ciclos de rapamicina. En una realización, el inhibidor de quinasa es un inhibidor de mTOR, p. ej., everolimus, y el everolimus se administra en una dosis de aproximadamente 2 mg, 2,5 mg, 3 mg, 4 mg, 5 mg, 6 mg, 7 mg, 8 mg, 9 mg, 10 mg, 11 mg, 12 mg, 13 mg, 14 mg, 15 mg (p. ej., 10 mg) diariamente durante un periodo de tiempo, p. ej., diariamente durante un ciclo de 28 días. En una realización, se administran 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 o más ciclos de everolimus.
En una realización, el inhibidor de quinasa es un inhibidor de MNK seleccionado entre CGP052088; 4-amino-3-(pfluorofenilamino)pirazolo [3,4-d] pirimidina (CGP57380); cercosporamida; ETC-1780445-2; y 4-amino-5-(4-fluoroanilino)pirazolo [3,4-d] pirimidina.
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con un inhibidor de fosfoinositida 3-quinasa (PI3K) (p. ej., un inhibidor de PI3K descrito en esta memoria, p. ej., idelalisib o duvelisib) y/o rituximab. En algunas realizaciones, una célula que expresa CAR descrita en el presente memoria se administra a un sujeto combinada con idelalisib y rituximab. En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con duvelisib y rituximab. Idelalisib (también denominado GS-1101 o CAL-101; Gilead) es una molécula pequeña que bloquea la isoforma delta de PI3K. A continuación se muestra la estructura de la estructura de idelalisib (5-fluoro-3-fenil-2-[(1S)-1-(7H-purin-6-¡lam¡no)prop¡l]-4(3H)-qu¡nazol¡nona).
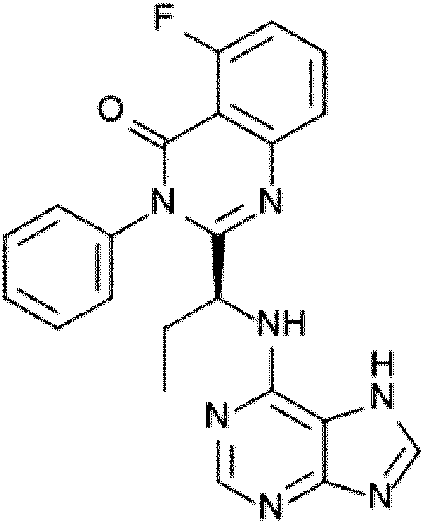
Duvelisib (también denominado IPI-145; Infinity Pharmaceuticals y Abbvie) es una molécula pequeña que bloquea PI3K-5,y. La estructura de duvelisib (8-cloro-2-fen¡l-3-[(1S)-1-(9H-pur¡n-6-¡lam¡no)et¡l]-1(2H)-¡soqu¡nol¡nona) se muestra a continuación.

En algunas realizaciones, el sujeto tiene LLC. En algunas realizaciones, el sujeto tiene LLC recidivante, p. ej., al sujeto se le ha administrado previamente una terapia contra el cáncer (p. ej., se le ha administrado previamente un anticuerpo anti-CD20 o se le ha administrado previamente ibrutinib). Por ejemplo, el sujeto tiene una supresión en la rama corta del cromosoma 17 (del(17p), p. ej., en una célula leucémica). En otros ejemplos, el sujeto no tiene un del(17p). En algunas realizaciones, el sujeto comprende una célula leucémica que comprende una mutación en el gen de la región variable de cadena pesada de inmunoglobulina (IgVH). En otras realizaciones, el sujeto no comprende una célula leucémica que comprende una mutación en el gen de la región variable de cadena pesada de inmunoglobulina (IgVH). En realizaciones, el sujeto tiene una deleción en el brazo largo del cromosoma 11 (del(l 1q)). En otras realizaciones, el sujeto no tiene una del(11q). En algunas realizaciones, se administra idelalisib en una dosificación de aproximadamente 100-400 mg (p. ej., 100-125, 125-150, 150-175, 175-200, 200-225, 225-250, 250-275, 275-300, 325-350, 350-375 o 375-400 mg), p. ej., BID. En algunas realizaciones, se administra duvelisib a una dosis de aproximadamente 15-100 mg (p. ej., aproximadamente 15-25, 25-50, 50-75 o 75-100 mg), p. ej., dos veces al día. En algunas realizaciones, se administra rituximab a una dosis de aproximadamente 350-550 mg/m2 (p. ej., 350-375, 375-400, 400-425, 425-450, 450-475 o 475-500 mg/m2), p. ej., por vía intravenosa.
En una realización, el inhibidor de quinasa es un inhibidor dual de fosfatidilinositol 3-quinasa (PI3K) y mTOR seleccionado de 2-amino-8-[frans-4-(2-hidroxietoxi)cclohexil]-6i-(6-metoxi-3-piridinil)-4-metil-pirido[2,3-d]pirimidin-7(8H)-ona (PF-04691502); N-[4-[[4-(dimetilamino)-1-piperidinil]carbonil]fenil]-W-[4-(4,6-di-4-morfolinil-1,3,5-triazin-2-il)fenil]urea (PF-05212384, PKI-587); 2-metil-2-{4-[3-metil-2-oxo-8-(quinolin-3-il)-2,3-dihidro-1H-imidazo[4,5-c]quinolin-1-il]fenil}propanenitrilo (BEZ-235); apitolisib (Gd C-0980, RG7422); 2,4-difluoro-N-{2-(metiloxi)-5-[4-(4-piridazinil)-6-quinolinil]-3-piridinil}bencenesulfonamida (GSK2126458); 8-(6-metoxipiridin-3-il)-3-metil-1-(4-(piperazin-1-il)-3-(trifluorometil)fenil)-1H-imidazo[4,5-c]quinolin-2(3H)-ona ácido maleico (NVP-BGT226); 3-[4-(4-morfolinilpirido[3',2':4,5]furo[3,2-d]pirimidin-2-il]fenol (PI-103); 5-(9-isopropil-8-metil-2-morfolino-9H-purin-6-il)pirimidin-2-amina (VS-5584, SB2343); y N-[2-[(3,5-dimetoxifenil)amino]quinoxalin-3-il]-4-[(4-metil-3-metoxifenil)carbonil]aminofenilsulfonamida (XL765).
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con un inhibidor de lñinfoma quinasa anaplásico (ALK). Las quinasas ALK ilustrativas incluyen, pero sin limitación, crizotinib (Pfizer), ceritinib (Novartis), alectinib (Chugai), brigatinib (también denominado AP26113; Ariad), entrectinib (Ignyta), PF-06463922 (Pfizer), TSR-011 (Tesaro) (véase, p. ej., el n.° de identificador de ensayos clínicos NCT02048488), CEP-37440 (Teva) y X-396 (Xcovery). En algunas realizaciones, el sujeto tiene un cáncer sólido, p. ej., un cáncer sólido descrito en esta memoria, p. ej., cáncer de pulmón.
El nombre químico del crizotinib es 3-[(1R)-1-(2,6-dicloro-3-fluorofenil)etoxi]-5-(1-piperidin-4-ilpirazol-4-il)piridin-2-amina. El nombre químico de ceritinib es 5-cloro-W2-[2-isopropoxi-5-metil-4-(4-piperidinil)fenil]-W4-[2-(isopropilsulfonil)fenil]-2,4-pirimidinadiamina. El nombre químico de alectinib es 9-etil-6,6-dimetil-8-(4-morfolinopiperidin-1-il)-11-oxo-6,11-dihidro-5H-benzo[b]carbazol-3-carbonitrilo. El nombre químico de brigatinib es 5-cloro-N2-{4-[4-(dimetilamino)-1-piperidinil]-2-metoxifenil}-N4-[2-(dimetilfosforil)fenil]-2,4-pirimidinodiamina. El nombre químico de entrectinib es N-(5-(3,5-difluorobencil)-1H-indazol-3-il)-4-(4-metilpiperazin-1-il)-2-((tetrahidro-2H-piran-4-il)amino)benzamida. El nombre químico de PF-06463922 es (10R)-7-amino-12-fluoro-2,10,16-trimetil-15-oxo-10,l5,16,17-tetrahidro-2H-8,4-(meteno)pirazolo[4,3-h][2,5,11]-benzoxadiazaciclotetradecino-3-carbonitrilo. La estructura química de CEP-37440 es (S)-2-((5-cloro-2-((6-(4-(2-hidroxietil)piperazin-1-il)-1-metoxi-6,7,8,9-tetrahidro-5H-benzo[7] annul en-2-il)amino)pirimidin-4-il)amino)-N-metilbenzamida. El nombre químico de X-396 es (R)-6-amino-5-(1-(2,6-dicloro-3-fluorofenil)etoxi)-N-(4-(4-metilpiperazina-1-carbonil)fenil)piridazina-3-carboxamida.
Fármacos que inhiben la fosfatasa calcineurina dependiente de calcio (ciclosporina y FK506) o inhibir la p70S6 quinasa que es importante para la señalización inducida por el factor de crecimiento (rapamicina). (Liu et al., Cell 66:807-815, 1991; Henderson et al., Immun. 73:316-321, 1991; Bierer et al., Curr. Opin. Immun. 5:763-773, 1993). En un aspecto adicional, las composiciones de células de la presente invención se pueden administrar a un paciente junto con (p. ej., antes, simultáneamente o después) de un trasplante de médula ósea, terapia de ablación de células T utilizando agentes de quimioterapia tales como fludarabina, radiación de haz externo (XRT), ciclofosfamida y/o anticuerpos como OKT3 o CAMPATH. En un aspecto, las composiciones celulares de la presente invención se administran después de una terapia ablativa de células B tal como agentes que reaccionan con CD20, p. ej., Rituxan. Por ejemplo, en una realización, los sujetos se pueden someter a un tratamiento convencional con una dosis elevada de quimioterapia seguida del trasplante de células madre de la sangre periférica. En determinadas realizaciones, después del trasplante, los sujetos reciben una infusión de las células inmunitarias expandidas de la presente invención. En una realización adicional, las células expandidas se administran antes o después de la cirugía.
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con un inhibidor de indoleamina 2,3-dioxigenasa (IDO). IDO es una enzima que cataliza la degradación del aminoácido L-triptófano a quinurenina. Muchos cánceres sobre-expresan IDO, p. ej., cáncer de próstata, colorrectal, pancreático, del cuello uterino, gástrico, ovárico, de cabeza y de pulmón. Los pCD, macrófagos y células dendríticas (CD) pueden expresar IDO. Sin estar ligados por la teoría, se cree que una disminución de L-triptófano (p. ej., catalizada por IDO) da lugar a un medio inmunosupresor mediante la inducción de anergia de células T y la apoptosis. Por lo tanto, sin estar ligados por la teoría, se cree que un inhibidor de IDO puede potenciar la eficacia de una célula que expresa CAR descrita en esta memoria, p. ej., disminuyendo la supresión o destrucción de una célula inmunitaria que expresa CAR. En algunas realizaciones, el sujeto tiene un tumor sólido, p. ej., un tumor sólido descrito en esta memoria, p. ej., cáncer de próstata, colorrectal, pancreático, del cuello uterino, gástrico, ovárico, de cabeza o de pulmón. Inhibidores ejemplares de IDO incluyen, pero no se limitan a 1-metiltriptófano, indoximod (NewLink Genetics) (véase, p. ej., Identificadores de Ensayos Clínicos N°s NCT01191216; NCT01792050) e INCB024360 (Incyte Corp.) (véase, p. ej., Clinical Trial Identifier N°s NCT01604889; NCT01685255)
En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con un modulador de células supresoras derivadas de mieloides (MDSCs). MDSCs se acumulan en la periferia y en el sitio del tumor de muchos tumores sólidos. Estas células suprimen las respuestas de células T, impidiendo de este modo la eficacia de la terapia con células que expresan CAR. Sin estar ligados por la teoría, se cree que la administración de un modulador de MDSC potencia la eficacia de una célula que expresa CAR descrita en esta memoria. En una realización, el sujeto tiene un tumor sólido, p. ej., un tumor sólido descrito en esta memoria, p. ej., glioblastoma. Los moduladores ilustrativos de CSOM incluyen, pero sin limitación, MCS110 y BLZ945. MCS110 es un anticuerpo monoclonal (mAb) contra el factor estimulante de colonias de macrófagos (M-CSF). Véase, p. ej., Identificador de Ensayos Clínicos N° NCT00757757. BLZ945 es una
molécula de bajo peso molecular inhibidora del receptor del factor 1 estimulante de colonias (CSF1R, por sus siglas en inglés). Véase, p. ej., Pyonteck et al. Nat. Med. 19(2013):1264-72. La estructura de BLZ945 se muestra a continuación.

En realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con una célula CART CD19 (p. ej., CTL019, p. ej., como se describe en el documento WO2012/079000). En realizaciones, el sujeto padece un linfoma CD19+, p. ej., un linfoma no hodgkiniano (LNH) CD19+, un LF CD19+ o un LDLBG CD19+. En realizaciones, el sujeto padece un linfoma CD19+ recurrente o refractario. En realizaciones, se administra quimioterapia linforreductora al sujeto antes, a la vez o después de la administración (p. ej., infusión) de células CAR T c D19. En un ejemplo, la quimioterapia linforreductora se administra al sujeto antes de la administración de células CAR T CD19. Por ejemplo, la quimioterapia linfoagotadora finaliza 1-4 días (p. ej., 1, 2, 3 o 4 días) antes de la infusión de células cA rT CD19. En algunas realizaciones, se administran múltiples dosis de células CAR T CD19, p. ej., como se describe en esta memoria. Por ejemplo, una sola dosis comprende aproximadamente 5 * 108 célula CAR T CD19. En realizaciones, se administra al sujeto una quimioterapia que agota los linfocitos antes, simultáneamente o después de la administración (p. ej., infusión) de una célula que expresa CAR descrita en esta memoria, p. ej., una célula que no expresa CAR CD19. En algunas realizaciones, se administra un CART CD19 al sujeto antes, al mismo tiempo o después de la administración (p. ej., infusión) de una célula que no expresa CAR de CD19, p. ej., una célula que no expresa CAR de CD19 descrita en esta memoria.
En algunas realizaciones, una célula que expresa CAR se administra a un sujeto en combinación con un polipéptido de interleuquina-15 (IL-15), un polipéptido del receptor alfa de interleuquina-l5 (IL-15Ra), o una combinación tanto de polipéptido IL-15 como de polipéptido IL-15Ra, p. ej., hetIL-15 (Admune Therapeutics, LLC). hetIL-15 es un complejo heterodimérico no covalente de IL-15 e IL-15Ra. hetIL-15 se describe, p. ej., en los documentos US 8.124.084, US 2012/0177598, US 2009/0082299, US 2012/0141413 y US 2011/0081311 . En algunas realizaciones, se administra het-IL-15 por vía subcutánea. En algunas realizaciones, el sujeto tiene un cáncer, p. ej., un cáncer sólido, p. ej., melanoma o cáncer de colon. En algunas realizaciones, el sujeto tiene un cáncer metastásico.
En una realización, al sujeto se le puede administrar un agente que reduce o mejora un efecto secundario asociado con la administración de una célula que expresa CAR. Los efectos secundarios asociados con la administración de una célula que expresa CAR incluyen, pero sin limitación, SLC y la linfohistiocitosis hemofagocítica (LHH), también denominada síndrome de activación macrofágica (SAM). Los síntomas de SLC incluyen fiebre alta, náuseas, hipotensión transitoria, hipoxia y similares. El SLC puede incluir signos y síntomas clínicos inespecíficos tales como fiebre, fatiga, anorexia, mialgias, artralgias, náuseas, vómitos y cefaleas. El SLC puede incluir signos y síntomas clínicos cutáneos tales como sarpullido. CRS puede incluir señales y síntomas clínicos gastrointestinales tales como náuseas, vómitos y diarrea. El SLC puede incluir signos y síntomas clínicos respiratorios tales como taquipnea e hipoxemia. CRS puede incluir señales y síntomas clínicos cardiovasculares tales como taquicardia, presión arterial diferencial amplia, hipotensión, aumento del gasto cardiaco (temprano) y potencialmente disminución del gasto cardiaco (tardío). CRS puede incluir señales y síntomas clínicos de coagulación tales como dímero D elevado, hipofibrinogenemia con o sin hemorragia. El SLC puede incluir signos y síntomas clínicos renales tales como azotemia. El SLC puede incluir signos y síntomas clínicos hepáticos tales como la elevación de las transaminasas e hiperbilirrubinemia. El SLC puede incluir signos y síntomas clínicos neurológicos tales como cefaleas, cambios del estado mental, confusión, delirio, dificultad para encontrar palabras o afasia clara, alucinaciones, temblores, dismetría, marcha alterada y convulsiones.
Por consiguiente, los métodos descritos en esta memoria pueden comprender administrar una célula que expresa CAR descrita en esta memoria a un sujeto y administrar adicionalmente uno o más agentes para gestionar niveles elevados de un factor soluble resultante del tratamiento con una célula que expresa CAR. En un caso, el factor soluble elevado en el sujeto es uno o más de IFN-y, TNFa, IL-2 e IL-6. En un caso, el factor elevado en el sujeto es uno o más de IL-1, GM-c Sf , IL-10, IL-8, IL-5 y fraktalkina. Por lo tanto, un agente administrado para tratar este efecto secundario puede ser un agente que neutralice uno o más de estos factores solubles. En una realización, el agente que neutraliza uno o más de estas formas solubles es un anticuerpo o fragmento de unión a antígenos del mismo. Los ejemplos de agentes de este tipo incluyen, pero sin limitación, un esteroide (p. ej., corticosteroide), un inhibidor de TNFa y un inhibidor de IL-6. Un ejemplo de un inhibidor de TNFa es una molécula de anticuerpo anti-TNFa tal como infliximab, adalimumab, certolizumab pegol y golimumab. Otro ejemplo de un inhibidor de TNFa es una proteína de fusión tal como entanercept. Los inhibidores de moléculas pequeñas de TNFa incluyen, pero sin limitación, derivados de xantina (p. ej., pentoxifilina) y bupropión. Un ejemplo de un inhibidor de IL-6 es una molécula de anticuerpo anti-IL-6 o una molécula de anticuerpo antirreceptor de IL-6, tal como tocilizumab (toc), sarilumab, elsilimomab, CNTO 328, ALD518/BMS-945429, CNTO 136, CPSI-2364, CDP6038, VX30, ARGX-109, FE301 y f M101. En una realización, la molécula de anticuerpo anti-receptor de IL-6 es tocilizumab. Un ejemplo de un inhibidor basado en IL-1R es anakinra.
En una realización, al sujeto se le puede administrar un agente que potencia la actividad de una célula que expresa CAR. Por ejemplo, en una realización, el agente puede ser un agente que inhibe una molécula inhibidora. Las moléculas inhibidoras, p. ej., muerte programada 1 (PD-1), pueden, en algunas realizaciones, disminuir la capacidad de una célula que expresa CAR de montar una respuesta efectora inmunitaria. Ejemplos de moléculas inhibidoras incluyen PD-1, PD-L1, CTLA-4, TIM-3, CEACAM (p. ej., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 y TGFR beta. La inhibición de una molécula inhibidora, p. ej., mediante inhibición a nivel de ADN, ARN o proteína, puede optimizar el rendimiento de una célula que expresa CAR. En algunas realizaciones, se puede utilizar un ácido nucleico inhibidor, p. ej., un ácido nucleico inhibidor, p. ej., un ARNbc, p. ej., un ARNip o ARNhc, grupos de repeticiones palindrómicas cortas en intervalos regulares (CRISPR), una nucleasa efectora de tipo activador de la transcripción (TALEN) o una endonucleasa con dedos de zinc (ZFN), p. ej., como se describe en el presente memoria, para inhibir la expresión de una molécula inhibidora en la célula que expresa CAR. En una realización, el inhibidor es un ARNhc. En una realización, la molécula inhibidora está inhibida dentro de una célula que expresa CAR. En estas realizaciones, una molécula de ARNbc que inhibe la expresión de la molécula inhibidora está ligada al ácido nucleico que codifica un componente, p. ej., todos los componentes, del CAR. En una realización, el inhibidor de una señal inhibidora puede ser, p. ej., un anticuerpo o fragmento de anticuerpo que se une a una molécula inhibidora. Por ejemplo, el agente puede ser un anticuerpo o un fragmento de anticuerpo que se une a PD-1, PD-L1, PD-L2 o CTLA4 (p. ej., ipilimumab (al que también se alude como MDX-010 y MDX-101, y comercializado como Yervoy®; Bristol-Myers Squibb; Tremelimumab (anticuerpo monoclonal IgG2 disponible de Pfizer, anteriormente conocido como ticilimumab, CP-675,206)). En una realización, el agente es un anticuerpo o fragmento de anticuerpo que se une a TIM3. En una realización, el agente es un anticuerpo o fragmento de anticuerpo que se une a CEACAM (CEACAM-1, CEACAM-3 y/o CEACAM-5). En una realización, el agente es un anticuerpo o fragmento de anticuerpo que se une a LAG3.
PD-1 es un miembro inhibidor de la familia de receptores CD28 que también incluye CD28, CTLA-4, ICOS y BTLA. PD-1 se expresa en células B activados, células T y células mieloides (Agata et al. 1996 Int. Immunol 8:765-75). Se ha mostrado que dos ligandos para PD-1, PD-L1 y PD-L2 regulan a la baja la activación de células T tras unirse a PD-1 (Freeman et al. 2000 J Exp Med 192: 1027-34; Latchman et al. 2001 Nat Immunol 2: 261-8; Carter et al. 2002 Eur J Immunol 32: 634 43). PD-L1 es abundante en los cánceres humanos (Dong et al. 2003 J Mol Med 81: 281-7; Blank et al. 2005 Cancer Immunol. Immunother 54: 307-314; Konishi et al. 2004 Clin Cancer Res 10: 5094). La supresión inmunitaria puede invertirse inhibiendo la interacción local de PD-1 con PD-L1. Anticuerpos, fragmentos de anticuerpos y otros inhibidores de PD-1, PD-L1 y PD-L2 están disponibles en la técnica y se pueden utilizar en combinación con los cars utilizados en la presente invención. Por ejemplo, nivolumab (al que también se alude como BMS-936558 o MDX1106; Bristol-Myers Squibb) es un anticuerpo monoclonal IgG4 completamente humano que bloquea de manera específica PD-1. Nivolumab (clon 5C4) y otros anticuerpos monoclonales humanos que se unen de manera específica a PD-1 se divulgan en los documentos US 8.008.449 y WO2006/121168. El pidilizumab (CT-011; Cure Tech) es un anticuerpo monoclonal IgGlk humanizado que se une a PD-1. El pidilizumab y otros anticuerpos monoclonales anti-PD-1 humanizados se describen en el documento WO2009/101611. El pembrolizumab (conocido anteriormente como lambrolizumab y también denominado MK03475; Merck) es un anticuerpo monoclonal IgG4 humanizado que se une a PD-1. Pembrolizumab y otros anticuerpos anti-PD-1 humanizados se describen en los documentos US 8.354.509 y WO2009/114335. El MeDi4736 (Medimmune) es un anticuerpo monoclonal humano que se une a PDL1 e inhibe la interacción del ligando con PD1. El MDPL3280A (Genentech / Roche) es un anticuerpo monoclonal IgG1 optimizado para Fc humano que se une a PD-L1. El MDPL3280A y otros anticuerpos monoclonales humanos contra PD-L1 se desvelan en la patente de los EE. UU. n.°: 7.943.743 y publicación de los EE. UU. n.°: 20120039906. Otros agentes de unión anti-PD-L1 incluyen YW243.55.S70 (las regiones variables de la cadena pesada y ligera se muestran en las SEQ ID NO 20 y 21 en el documento WO2010/077634) y MDX-1 105 (también denominado BMS-936559 y, p. ej., agentes de unión anti-PD-L1 descritos en el documento WO2007/005874). AmP-224 (B7-DCIg; Amplimmune; p. ej., descrito en los documentos WO2010/027827 y WO2011/066342), es un receptor soluble de fusión de PD-L2 Fc que bloquea la interacción entre PD-1 y B7-H1. Otros anticuerpos anti-PD-1 incluyen AMP 514 (Amplimmune), entre otros, p. ej., anticuerpos anti-PD-1 descritos en los documentos US 8.609.089, US 2010028330 y/o US 20120114649.
La TIM-3 (inmunoglobulina-3 de célula T) también regula negativamente la función de los célula T, en particular en las células T auxiliares 1 CD4+ y T citotóxicas 1 CD8+ secretoras de IFN-g, y desempeña una función fundamental en el agotamiento de las células T. La inhibición de la interacción entre TIM3 y sus ligandos, p. ej., galectina-9 (Gal9), fosfatidilserina (PS) y HMGB1, puede incrementar la respuesta inmunitaria. En la técnica se puede disponer de anticuerpos, fragmentos de anticuerpo y otros inhibidores de TIM3 y sus ligandos, y estos se pueden utilizar combinados con un CAR CD19 descrito en esta memoria. Por ejemplo, los anticuerpos, fragmentos de anticuerpo, moléculas pequeñas o inhibidores peptídicos que se dirigen a TIM3 se unen al dominio IgV de TIM3 para inhibir la interacción con sus ligandos. En los documentos WO2013/006490 y US20100247521 se describen anticuerpos y péptidos que inhiben TIM3. Otros anticuerpos anti-TIM3 incluyen versiones humanizadas de RMT3-23 (descrito en Ngiow et al., 2011, Cancer Res, 71: 3540-3551) y el clon 8B.2C12 (desvelado en Monney et al., 2002, Nature, 415: 536-541). En el documento US20130156774 se describen anticuerpos biespecíficos que inhiben TIM3 y PD-1.
En otras realizaciones, el agente que potencia la actividad de una célula que expresa CAR es un inhibidor de CEACAM (p. ej., inhibidor de CEACAM-1, CEAcAm-3 y/o CEACAM-5). En una realización, el inhibidor de CEACAM es una molécula de anticuerpo anti-CEACAM. Se describen anticuerpos anti-CEACAM-1 ilustrativos en los documentos WO 2010/125571, WO 2013/082366 WO 2014/059251 y WO 2014/022332, p. ej., un anticuerpo monoclonal 34B1, 26H7 y 5F4; o una forma
recombinante de los mismos, como se describe, p. ej., en los documentos US 2004/0047858, US 7.132.255 y WO 99/052552. En otras realizaciones, el anticuerpo anti-CEACAM se une a CEACAM-5 como se describe, p. ej., en Zheng et al. PLoS One. 2 de septiembre de 2010; 5 (9). pii: e12529 (DOI:10:1371/journal.pone.0021146), o presenta reactividad cruzada con CEACAM-1 y CEACAM-5 como se describe, p. ej., en los documentos WO 2013/054331 y US 2014/0271618.
Sin desear estar limitados por la teoría, se cree que las moléculas de adhesión celular del antígeno carcinoembrionario (CEACAM), tales como CEACAM-1 y CEACAM-5, median, al menos en parte, en la inhibición de una respuesta inmune antitumoral (véase, p. ej., Markel et al. J Immunol. 15 de marzo de 2002;168(6):2803-10; Markel et al. J Immunol. 1 de noviembre de 2006;177(9):6062-71; Markel et al. Immunology. febrero de 2009;126(2):186-200; Markel et al. Cancer Immunol Immunother. febrero de 2010;59(2):215-30; Ortenberg et al. Mol Cancer Ther. junio de 2012; 11(6):1300-10; Stern et al. J Immunol. 1 de junio de 2005;174(11):6692-701; Zheng et al. PLoS One. 2 de septiembre de 2010;5(9). pii: e12529). Por ejemplo, CEACAM-1 se ha descrito como un ligando heterófilo para TIM-3 y desempeña una función en la tolerancia y el agotamiento de los célula T mediados por TIM-3 (véase, p. ej., el documento WO 2014/022332; Huang, et al. (2014) Nature doi:10.1038/nature13848). En algunas realizaciones, se ha mostrado que el bloqueo conjunto de CEACAM-1 y TIM-3 potencia una respuesta inmunitaria antitumoral en modelos de xenoinjerto de cáncer colorrectal (véase, p. ej., el documento WO 2014/022332; Huang, et al. (2014), supra). En otras realizaciones, el bloqueo conjunto de CEACAM-1 y PD-1 reduce la tolerancia de los célula T como se ha descrito, p. ej., en el documento WO 2014/059251. Por lo tanto, los inhibidores de CEACAM se pueden utilizar con los otros inmunomoduladores descritos en esta memoria (p..ej.,, inhibidores de anti-PD-1 y/o anti-TIM-3) para mejorar una respuesta inmunitaria contra un cáncer, p. ej., un melanoma, un cáncer de pulmón cáncer (por ejemplo, NSCLC), un cáncer de vejiga, un cáncer de colon, un cáncer de ovario y otros cánceres como se describe en esta memoria.
El LAG-3 (gen-3 de activación de célula o CD223) es una molécula de la superficie celular expresada en células T y células B activadas que se ha mostrado que desempeña una función en el agotamiento de las células T CD8+. En la técnica se puede disponer de anticuerpos, fragmentos de anticuerpo y otros inhibidores de LAG-3 y sus ligandos, y estos se pueden utilizar combinados con un CAR CD19 descrito en esta memoria. Por ejemplo, BMS-986016 (Bristol-Myers Squib) es un anticuerpo monoclonal que se dirige a LAG3. IMP701 (Immutep) es un anticuerpo antagonista contra LAG-3 e IMP731 (Immutep y GlaxoSmithKline) es un anticuerpo contra LAG-3 de agotamiento. Otros inhibidores de LAG-3 incluyen IMP321 (Immutep), que es una proteína de fusión recombinante de una porción soluble de LAG3 e Ig que se une a moléculas del MHC de clase II y activa las células presentadoras de antígenos (APC). Se describen otros anticuerpos, p. ej., en el documento WO2010/019570.
En algunas realizaciones, el agente que potencia la actividad de una célula que expresa CAR puede ser, por ejemplo, una proteína de fusión que comprende un primer dominio y un segundo dominio, donde el primer dominio es una molécula inhibidora, o fragmento de esta, y el segundo dominio es un polipéptido que está asociado con una señal positiva, por ejemplo, un polipéptido que comprende un dominio de señalización intracelular como se describe en la presente. En algunas realizaciones, el polipéptido que se asocia con una señal positiva puede incluir un dominio coestimulante de CD28, CD27, ICOS, p. ej., un dominio de señalización intracelular de CD28, CD27 y/o ICOS, y/o un dominio de señalización primaria, p. ej., de CD3 zeta, p. ej., descrito en esta memoria. En una realización, la proteína de fusión es expresada por la misma célula que expresó el CAR. En otra realización, la proteína de fusión es expresada por una célula, p. ej., una célula T que no expresa un CAR de la presente divulgación.
En una realización, el agente que potencia la actividad de una célula que expresa CAR descrita en esta memoria es miR-17-92.
En una realización, el agente que potencia la actividad de un CAR descrito en esta memoria es una citoquina. Las citoquinas tienen importantes funciones relacionadas con la expansión, la diferenciación, la supervivencia y la homeostasis de las células T. Citoquinas que se pueden administrar al sujeto que recibe una célula que expresa CAR descritas en esta memoria incluyen: IL-2, IL-4, IL-7, IL-9, IL-15, IL-18 e IL-21, o una combinación de las mismas. En realizaciones preferidas, la citocina administrada es IL-7, IL-15 o IL-21, o una combinación de las mismas. La citocina se puede administrar una vez al día o más de una vez al día, p. ej., dos veces al día, tres veces al día o cuatro veces al día. La citocina se puede administrar durante más de un día, p. ej., la citocina se administra durante 2 días, 3 días, 4 días, 5 días, 6 días, 1 semana, 2 semanas, 3 semanas o 4 semanas. Por ejemplo, la citocina se administra una vez al día durante 7 días.
En realizaciones, la citoquina se administra en combinación con células T que expresan CAR. La citoquina se puede administrar de manera simultánea o a la vez que las células T que expresan CAR, p. ej., administrarse el mismo día. La citoquina se puede preparar en la misma composición farmacéutica que las células T que expresan CAR, o se puede preparar en una composición farmacéutica separada. Como alternativa, la citoquina se puede administrar poco después de la administración de las células T que expresan CAR, p. ej., 1 día, 2 días, 3 días, 4 días, 5 días, 6 días o 7 días después de la administración de las células T que expresan CAR. En realizaciones donde la citoquina se administra en un régimen de dosificación que se produce a lo largo de más de un día, el primer día del régimen de dosificación de citoquinas puede ser el mismo día de la administración con los célula T que expresan CAR o el primer día del régimen de dosificación de citoquinas puede ser 1 día, 2 días, 3 días, 4 días, 5 días, 6 días o 7 días después de la administración de las células T que expresan CAR. En una realización, el primer día, las células T que expresan CAR se administran al sujeto, y el
segundo día se administra una citoquina una vez al día durante los siguientes 7 días. En una realización preferida, la citoquina que se ha de administrar combinada con células T que expresan CAR es IL-7, IL-15 o IL-21.
En otras realizaciones, la citocina se administra un periodo de tiempo después de la administración de células que expresan CAR, p. ej., al menos 2 semanas, 3 semanas, 4 semanas, 6 semanas, 8 semanas, 10 semanas, 12 semanas, 4 meses, 5 meses, 6 meses, 7 meses, 8 meses, 9 meses, 10 meses, 11 meses o 1 año o más después de la administración de células que expresan CAR. En una realización, la citocina se administra después de la evaluación de la respuesta del sujeto a las células que expresan CAR. Por ejemplo, al sujeto se le administran células que expresan CAR según la dosificación y los regímenes descritos en esta memoria. La respuesta del sujeto a la terapia con células que expresan CAR se evalúa 2 semanas, 3 semanas, 4 semanas, 6 semanas, 8 semanas, 10 semanas, 12 semanas, 4 meses, 5 meses, 6 meses, 7 meses, 8 meses, 9 meses, 10 meses, 11 meses o 1 año o más después de la administración de células que expresan CAR, utilizando cualquiera de los métodos descritos en esta memoria, incluidas la inhibición del crecimiento tumoral, reducción de las células tumorales circulantes o regresión tumoral. A los sujetos que no muestran una respuesta suficiente a la terapia con células que expresan CAR se les puede administrar una citocina. La administración de la citocina al sujeto que tiene una respuesta subóptima a la terapia con células que expresan CAR mejora la eficacia de las células que expresan CAR o la actividad anticancerosa. En una realización preferida, la citocina administrada después de la administración de células que expresan CAR es IL-7.
Combinación con una dosis baja de un inhibidor de mTOR
En una realización, las células que expresan una molécula de CAR se administran en combinación con una dosis baja potenciadora de la inmunidad de un inhibidor de mTOR.
En una realización, una dosis de un inhibidor de mTOR está asociada con, o proporciona, una inhibición de mTOR de al menos el 5 pero no más del 90 %, al menos el 10 pero no más del 90 %, al menos el 15, pero no más del 90 %, al menos el 20 pero no más del 90 %, al menos el 30 pero no más del 90 %, al menos el 40 pero no más del 90 %, al menos el 50 pero no más del 90 %, al menos el 60 pero no más del 90 % o al menos el 70 pero no más del 90 %.
En una realización, una dosis de un inhibidor de mTOR está asociada con, o proporciona, una inhibición de mTOR de al menos el 5 pero no más del 80 %, al menos el 10 pero no más del 80 %, al menos el 15, pero no más del 80 %, al menos el 20 pero no más del 80 %, al menos el 30 pero no más del 80 %, al menos el 40 pero no más del 80 %, al menos el 50 pero no más del 80 % o al menos el 60 pero no más del 80 %.
En una realización, una dosis de un inhibidor de mTOR está asociada con, o proporciona, una inhibición de mTOR de al menos el 5 pero no más del 70 %, al menos el 10 pero no más del 70 %, al menos el 15, pero no más del 70 %, al menos el 20 pero no más del 70 %, al menos el 30 pero no más del 70 %, al menos el 40 pero no más del 70 % o al menos el 50 pero no más del 70 %.
En una realización, una dosis de un inhibidor de mTOR está asociada con, o proporciona, una inhibición de mTOR de al menos el 5 pero no más del 60 %, al menos el 10 pero no más del 60 %, al menos el 15, pero no más del 60 %, al menos el 20 pero no más del 60 %, al menos el 30 pero no más del 60 % o al menos el 40 pero no más del 60 %.
En una realización, una dosis de un inhibidor de mTOR está asociada con, o proporciona, una inhibición de mTOR de al menos el 5 pero no más del 50 %, al menos el 10 pero no más del 50 %, al menos el 15, pero no más del 50 %, al menos el 20 pero no más del 50 %, al menos el 30 pero no más del 50 % o al menos el 40 pero no más del 50 %.
En una realización, una dosis de un inhibidor de mTOR está asociada con, o proporciona, una inhibición de mTOR de al menos el 5 pero no más del 40 %, al menos el 10 pero no más del 40 %, al menos el 15, pero no más del 40 %, al menos el 20 pero no más del 40 %, al menos el 30 pero no más del 40 % o al menos el 35 pero no más del 40 %.
En una realización, una dosis de un inhibidor de mTOR está asociada con, o proporciona, una inhibición de mTOR de al menos el 5 pero no más del 30 %, al menos el 10 pero no más del 30 %, al menos el 15, pero no más del 30 %, al menos el 20 pero no más del 30 % o al menos el 25 pero no más del 30 %.
En una realización, una dosis de un inhibidor de mTOR está asociada con, o proporciona, una inhibición de mTOR de al menos el 1, 2, 3, 4 o 5 pero no más del 20 %, al menos el 1, 2, 3, 4 o 5 pero no más del 30 %, al menos el 1, 2, 3, 4 o 5 pero no más del 35 %, al menos el 1, 2, 3, 4 o 5 pero no más del 40 % o al menos el 1, 2, 3, 4 o 5 pero no más del 45 %.
En una realización, una dosis de un inhibidor de mTOR está asociada con, o proporciona, una inhibición de mTOR de al menos 1, 2, 3, 4 o 5 pero no más del 90 %.
Como se analiza en esta memoria, el grado de inhibición de mTOR puede expresarse como el grado de inhibición de la quinasa P70 S6, p. ej., el grado de inhibición de mTOR puede determinarse por el nivel de disminución de la actividad de la quinasa P70 s 6, p. ej., por la disminución de la fosforilación de un sustrato de quinasa P70 S6. El nivel de inhibición de
mTOR puede evaluarse mediante un método descrito en esta memoria, p. ej., por el ensayo de Boulay, o la medición de los niveles de S6 fosforilada por transferencia Western.
INHIBIDORES DE MTOR EJEMPLARES
Como se utiliza en esta memoria, la expresión "inhibidor de mTOR" se refiere a un compuesto o ligando, o una sal farmacéuticamente aceptable del mismo, que inhibe la quinasa mTOR en una célula. En una realización, un inhibidor de mTOR es un inhibidor alostérico. En una realización, un inhibidor de mTOR es un inhibidor catalítico.
Los inhibidores de mTOR alostéricos incluyen el compuesto tricíclico neutro rapamicina (sirolimus), compuestos relacionados con rapamicina, es decir, compuestos que tienen similitud estructural y funcional con rapamicina, incluidos, p. ej., derivados de rapamicina, análogos de rapamicina (también denominados rapálogos) y otros compuestos macrólidos que inhiben actividad de mTOR.
La rapamicina es un antibiótico macrólido conocido, producido por Streptomyces hygroscopicus, que tiene la estructura mostrada en la fórmula A.

Véase, p. ej., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) 113: 7433; patente de los EE. UU. n.° 3.929.992. Existen diversos esquemas de numeración propuestos para rapamicina. Para evitar confusiones, cuando se nombran análogos de rapamicina específicos en esta memoria, los nombres se dan con referencia a rapamicina utilizando el esquema de numeración de fórmula A.
Análogos de rapamicina útiles en la invención son, por ejemplo, análogos sustituidos en O, en los que el grupo hidroxilo en el anillo ciclohexilo de la rapamicina está reemplazado por ORi, en el que Ri es hidroxialquilo, hidroxialcoxialquilo, acilaminoalquilo o aminoalquilo; p. ej., RAD001, también conocido como everolimus como se describe en los documentos US 5.665.772 y WO94/09010. Otros análogos de rapamicina adecuados incluyen los sustituidos en la posición 26 o 28. El análogo de rapamicina puede ser un epímero de un análogo mencionado anteriormente, particularmente un epímero de un análogo sustituido en la posición 40, 28 o 26, y opcionalmente se puede hidrogenar adicionalmente, por ejemplo, como se describe en los documentos US 6015815, WO95/14023 y WO99/15530, p. ej., ABT578 también conocido como zotarolimus o un análogo de rapamicina descrito en los documentos US 7091 213, WO98/02441 y WO01/14387, p. ej., AP23573 también conocido como ridaforolimus.
Ejemplos de análogos de rapamicina adecuados para su uso en la presente invención del documento US 5.665.772 incluyen, pero sin limitación, 40-O-bencil-rapamicina, 40-O-(4’-hidroximetil)bencil-rapamicina, 40-O-[4’-(1,2-dihidroxietil)]bencil-rapamicina, 40-O-alil-rapamicina, 40-O-[3’-(2,2-dimetil-1,3-dioxolan-4(S)-il)-prop-2’-en-1’-il]-rapamicina, (2’E,4’S)-40-O-(4’,5’-dihidroxipent-2’-en-1’-il)-rapamicina, 40-O-(2-hidroxi)etoxicarbonilmetil-rapamicina, 40-O-(2-hidroxi)etil-rapamicina, 40-O-(3-hidroxi)propil-rapamicina, 40-O-(6-hidroxi)hexil-rapamicina, 40-O-[2-(2-hidroxi)etoxi]etil-rapamicina, 40-O-[(3S)-2,2-dimetildioxolan-3-il]metil-rapamicina, 40-O-[(2S)-2,3-dihidroxiprop-1-il]-rapamicina, 40-O-(2-acetoxi)etil-rapamicina, 40-O-(2-nicotinoiloxi)etil-rapamicina, 40-O-[2-(N-morfolino)acetoxi]etilrapamicina, 40-O-(2-N-imidazolilacetoxi)etil-rapamicina, 40-O-[2-(N-metil-N’-piperazinil)acetoxi]etil-rapamicina, 39-O-desmetil-39,40-O,O-etileno-rapamicina, (26R)-26-dihidro-40-O-(2-hidroxi)etil-rapamicina, 40-O-(2-aminoetil)-rapamicina, 40-O-(2-acetaminoetil)-rapamicina, 40-O-(2-nicotinamidoetil)-rapamicina, 40-O-(2-(N-metil-imidazo-2’ilcarbetoxamido)etil)-rapamicina, 40-O-(2-etoxicarbonilaminoetil)-rapamicina, 40-O-(2-tolilsulfonamidoetil)-rapamicina y 40-O-[2-(4',5'-dicarboetoxi-1',2',3'-triazol-1'-il)-etil]-rapamicina.
Se conocen otros análogos de rapamicina útiles en la presente invención que son análogos en donde el grupo hidroxilo del anillo de ciclohexilo de rapamicina y/o el grupo hidroxi en la posición 28 es reemplazado por un grupo hidroxiéster, por ejemplo, análogos de rapamicina que se encuentran en el documento US RE44.768, p. ej., temsirolimus.
Otros análogos de rapamicina útiles en la presente invención incluyen aquellos donde el grupo metoxi en la posición 16 es reemplazado por otro sustituyente, preferentemente alquiniloxi, bencilo, ortometoxibencilo o clorobencilo (opcionalmente hidroxi-sustituido) y/o donde el grupo mextoxi en la posición 39 se elimina junto con el carbono 39 de manera que el anillo de ciclohexilo de la rapamicina se convierte en un anillo de ciclopentilo que carece del grupo metioxi de la posición 39; p. ej., como se describe en los documentos WO95/16691 y WO96/41807. Los análogos se pueden modificar adicionalmente de modo que el hidroxi en la posición 40 de rapamicina esté alquilado y/o el carbonilo-32 esté reducido.
Análogos de rapamicina del documento WO95/16691 incluyen, pero no se limitan a 16-demetoxi-16-(pent-2-inil)oxirapamicina, 16-demetoxi-16-(but-2-inil)oxi-rapamician, 16-demetoxi-16-(propargil)oxi-rapamicina, 16-demetoxi-16-(4-hidroxi-but-2-inil)oxi-rapamicina, 16-demetoxi-16-benciloxi-40-O-(2-hidroxietil)-rapamicina, 16-demtoxi-16-benciloxirapamicina, 16-demetoxi-16-orto-metoxibencil-rapamicina, 16-demetoxi-40-O-(2-metoxietil)-16-pent-2-inil)oxi-rapamicina, 3 9-demetoxi-40-desoxi-39-formil-42-nor-rapamicina, 39-demetoxi-40-desoxi-39-hidroximetil-42-nor-rapamicina, 39-demetoxi-40-desoxi-39-carboxi-42-nor-rapamicina, 39-demetoxi-40-desoxi-39-(4-metil-piperazin-1-il)carbonil-42-norrapamicina, 39-demetoxi-40-desoxi-39-(morfolin-4-il)carbonil-42-nor-rapamicina, 39-demetoxi-40-desoxi-39-[N-metil, N-(2-piridin-2-il-etil)]carbamoil-42-nor-rapamicina y 39-demetoxi-40-desoxi-39-(p-toluenosulfonilhidrazonometil)-42-norrapamicina.
Análogos de rapamicina del documento WO96/41807 incluyen, pero sin limitación, 32-desoxo-rapamicina, 16-O-pent-2-inil-32-desoxo-rapamicina, 16-O-pent-2-inil-32-desoxo- 40-O-(2-hidroxi-etil)-rapamicina, 16-O-pent-2-inil-32-(S)-dihidro-40-O-(2-hidroxietil)-rapamicina, 32(S)-dihidro-40-O-(2-metoxi)etil-rapamicina y 32(S)-dihidro-40-O-(2-hidroxietil)-rapamicina.
Otro análogo de rapamicina adecuado es umirolimus tal como se describe en el documento US2005/0101624.
RAD001, también conocido como everolimus (Afinitor®), tiene el nombre químico (1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18-dihidroxi-12-{(1R)-2-[(1S,3R,4R)-4-(2-hidroxi etoxi)-3-metoxiciclohexil]-1-metiletil}-19,30-dimetoxi-15,17,21,23,29,35-hexametil-11,36-dioxa-4-azatriciclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraeno-2,3,10,14,20-pentaona
Ejemplos adicionales de inhibidores alostéricos de mTOR incluyen sirolimus (rapamicina, AY-22989), 40-[3-hidroxi-2-(hidroximetil)-2-metilpropanoato]-rapamicina (también denominado temsirolimus o CCI-779) y ridaforolimus (AP-23573/MK-8669). Otros ejemplos de inhibidores alostéricos de mTor incluyen zotarolimus (ABT578) y umirolimus.
Como alternativa o adicionalmente, se ha encontrado que inhibidores catalíticos de mTOR competitivos con ATP se fijan directamente como objetivo el dominio de la quinasa mTOR y fijan como objetivo tanto a mTORC1 como a mTORC2. Estos también son inhibidores más eficaces de mTORC1 que los inhibidores de mTOR alostéricos tales como rapamicina, porque modulan las salidas de mTORC1 resistente a rapamicina tales como la fosforilación de 4EBP1-T37/46 y la traducción dependiente de la caperuza.
Inhibidores catalíticos incluyen: BEZ235 o 2-metil-2-[4-(3-metil-2-oxo-8-quinolin-3-il-2,3-dihidro-imidazo[4,5-c]quinolin-1-il)-fenil]-propionitrilo o la forma de sal monotosilato. La síntesis de BEZ235 se describe en el documento WO2006/122806; CCG168 (de otro modo conocido como AZD-8055, Chresta, C.M., et al., Cancer Res, 2010, 70(1), 288-298), que tiene el nombre químico {5-[2,4-bis-((S)-3-metil-morfolin-4-il)-pirido[2,3d]pirimidin-7-il]-2-metoxi-fenil}-metanol; 3-[2,4-bis[(3S)-3-metilmorfolin-4-il]pirido[2,3-d]pirimidin-7-il]-N-metilbenzamida (documento WO09104019); 3-(2-aminobenzo[d]oxazol-5-il)-1-isopropil-1H-pirazolo[3,4-d]pirimidin-4-amina (documentos WO10051043 y WO2013023184); A N-(3-(N-(3-((3,5-dimetoxifenil)amino)quinoxalina-2-il)sulfamoil)fenil)-3-metoxi-4-metilbenzamida (documentos WO07044729 y WO12006552); PKI-587 (Venkatesan, A.M., J. Med.Chem., 2010, 53, 2636-2645) que tiene el nombre químico 1-[4-[4-(dimetilamino)piperidina-1-carbonil]fenil]-3-[4-(4,6-dimorflino-1,3,5-triazin-2-il)fenil]urea; GSK-2126458 (ACS Med. Chem. Lett., 2010 , 1 , 39-43), que tiene el nombre químico 2,4-difluoro-N-{2-metoxi-5-[4-(4-piridazinil)-6-quinolinil]-3-piridinil}bencenosulfonamida; 5-(9-isopropil-8-metil-2-morfolino-9H-purin-6-il)pirimidin-2-amina (documento WO10114484); (E)-N-(8-(6-amino-5-(trifluorometil)piridin-3-il)-1-(6-(2-cianopropan-2-il)piridin-3-il)-3-metil-1H-imidazo[4,5-c]quinolin-2(3H)-ilideno)cianamida (documento wO12007926).
Ejemplos adicionales de inhibidores catalíticos de mTOR incluyen 8-(6-metoxi-piridin-3-il)-3-metil-1-(4-piperazin-1-il-3-trifluorometil-fenil)-1,3-dihidro-imidazo[4,5-c]quinolin-2-ona (documento WO2006/122806) y Ku-0063794 (Garcia-Martinez
JM, et al.,Biochem J., 2009, 421(1), 29-42. Ku-0063794 es un inhibidor específico de la diana mamífero de rapamicina (mTOR). El WYE-354 es otro ejemplo de un inhibidor de mTOR catalítico (Yu K, et al. (2009). Biochemical, Cellular, and In vivo Activity of Novel ATP-Competitive and Selective Inhibitors of the Mammalian Target of Rapamycin. Cancer Res.
69(15): 6232-6240).
Los inhibidores de mTOR útiles según la presente invención también incluyen profármacos, derivados, sales farmacéuticamente aceptables o análogos de los mismos de cualquiera de los anteriores.
Inhibidores de mTOR, tales como RAD001, se pueden formular para el suministro basándose en métodos muy consolidados en la técnica en función de las dosificaciones particulares descritas en esta memoria. En particular, la patente de los EE. UU. 6.004.973 proporciona ejemplos de formulaciones que se pueden utilizar con los inhibidores de mTOR descritos en esta memoria.
EVALUACIÓN DE LA INHIBICIÓN DE MTOR
mTOR fosforila la quinasa P70 S6, activando de este modo la quinasa P70 S6 y permitiéndole fosforilar su sustrato. El grado de inhibición de mTOR puede expresarse como el grado de inhibición de la quinasa P70 S6, p. ej., el grado de inhibición de mTOR puede determinarse por el nivel de disminución de la actividad de la quinasa P70 s6, p. ej., por la disminución de la fosforilación de un sustrato de quinasa P70 S6. Se puede determinar el nivel de inhibición de mTOR midiendo la actividad de la quinasa P70 S6 (la capacidad de la quinasa P70 S6 para fosforilar un sustrato), en ausencia de inhibidor, p. ej., antes de la administración de inhibidor y en presencia de inhibidor, o después de la administración del inhibidor. El nivel de inhibición de la quinasa P70 S6 proporciona el nivel de inhibición de mTOR. Por tanto, si la quinasa P70 S6 se inhibe en un 40 %, la actividad de mTOR, medida por la actividad de la quinasa P70 S6, se inhibe en un 40 %. El grado o nivel de inhibición al que se hace referencia en esta memoria es el nivel promedio de inhibición durante el intervalo de dosificación. A modo de ejemplo, si el inhibidor se administra una vez a la semana, el nivel de inhibición viene dado por el nivel promedio de inhibición durante ese intervalo, es decir, una semana.
Boulay et al., Cancer Res, 2004, 64:252-61 enseña un ensayo que se puede utilizar para evaluar el nivel de inhibición de mTOR (denominado en esta memoria ensayo de Boulay). En un caso, el ensayo se basa en la medición de la actividad de la quinasa P70 S6 de muestras biológicas antes y después de la administración de un inhibidor de mTOR, p. ej., RAD001. Las muestras se pueden tomar en momentos preseleccionados después del tratamiento con un inhibidor de mTOR, p. ej., 24, 48 y 72 horas después del tratamiento. Se pueden usar muestras biológicas, p. ej., de piel o células mononucleares de sangre periférica (PBMC). Se preparan extractos de proteínas totales a partir de las muestras. La quinasa P70 S6 se aísla de los extractos de proteínas mediante inmunoprecipitación usando un anticuerpo que reconoce específicamente la quinasa P70 S6. La actividad de la quinasa P70 S6 aislada se puede medir en un ensayo de quinasa in vitro. La quinasa aislada se puede incubar con sustratos de la subunidad ribosómica 40S (que es un sustrato endógeno de la quinasa P70 S6) y gamma-32P en condiciones que permitan la fosforilación del sustrato. A continuación, la mezcla de reacción se puede resolver en un gel de SDS-PAGE y la señal 32P se analiza usando un PhosphorImager. Una señal de 32P correspondiente al tamaño de la subunidad ribosómica 40S indica sustrato fosforilado y la actividad de la quinasa P70 S6. Los aumentos y disminuciones de la actividad de la quinasa se pueden calcular cuantificando el área y la intensidad de la señal de 32P del sustrato fosforilado (p. ej., utilizando ImageQuant, Molecular Dynamics), asignando valores unitarios arbitrarios a la señal cuantificada y comparando los valores posteriores a la administración con los valores anteriores a la administración o con un valor de referencia. Por ejemplo, el porcentaje de inhibición de la actividad quinasa se puede calcular con la siguiente fórmula: 1-(valor obtenido tras la administración/valor obtenido antes de la administración) X 100. Como se ha descrito anteriormente, el alcance o nivel de inhibición al que se hace referencia en esta memoria es el nivel promedio de inhibición durante el intervalo de dosificación.
Métodos para la evaluación de la actividad quinasa, p. ej., la actividad quinasa P70 S6, también se proporcionan en el documento US 7.727.950.
El nivel de inhibición de mTOR también puede evaluarse mediante un cambio en la relación de células T PD1 negativos con respecto a PD1 positivas. Células T de sangre periférica pueden identificarse como PD1 negativas o positivas mediante métodos conocidos en la técnica.
Inhibidores de mTOR de dosis baja
Los métodos descritos en esta memoria utilizan dosis bajas, de potenciación inmunitaria, de inhibidores de mTOR, p. ej., inhibidores de mTOR alostéricos, incluidos rapálogos tales como RAD001. Por el contrario, los niveles de inhibidor que inhiben completamente o casi completamente la ruta de mTOR son inmunosupresores y se usan, p. ej., para prevenir el rechazo de trasplantes de órganos. Además, las altas dosis de rapálogos que inhiben completamente mTOR también inhiben el crecimiento de células tumorales y se usan para tratar diversos cánceres (véase, p. ej., Antineoplastic effects of mammalian target of rapamycine inhibitors. Salvadori M. World J Transplant. 24 de octubre de 2012; 2 (5): 74-83; Current and Future Treatment Strategies for Patients with Advanced Hepatocellular Carcinoma: Role of mTOR Inhibition. Finn RS. Liver Cancer. noviembre de 2012; 1 (3-4): 247-256; Emerging Signaling Pathways in Hepatocellular Carcinoma. Moeini A, Cornella H, Villanueva A. Liver Cancer. septiembre de 2012; 1 (2): 83-93; Targeted cancer therapy - Are the
days of systemic chemotherapy numbered? Joo WD, Visintin I, Mor G. Maturitas. 20 de septiembre de 2013; Role of natural and adaptive immunity in renal cell carcinoma response to VEGFR-TKIS and mTOR inhibitor. Santoni M, Berardi R, Amantini C, Burattini L, Santini D, Santoni G, Cascinu S. Int J Cancer. 2 de octubre de 2013).
La presente divulgación se basa, al menos en parte, en el hallazgo sorprendente de que las dosis de inhibidores de mTOR muy por debajo de las utilizadas en los entornos clínicos actuales tuvieron un efecto superior en el aumento de la respuesta inmunitaria en un sujeto y aumentaron la relación de células T P D -1 negativas/células T PD-1 positivas. Fue sorprendente que las dosis bajas de inhibidores de mTOR, que producen solo una inhibición parcial de la actividad de mTOR, pudieran mejorar de manera efectiva las respuestas inmunitarias en sujetos humanos y aumentar la proporción de células T PD-1 negativas/células T PD-1 positivas.
Como alternativa, o además, sin desear quedar ligado por teoría alguna, se cree que una dosis baja, potenciadora del sistema inmunitario, de un inhibidor de mTOR puede aumentar el número de células T vírgenes, p. ej., al menos de forma transitoria, p. ej., en comparación con un sujeto sin tratar. Como alternativa o adicionalmente, nuevamente sin desear quedar ligado a la teoría, se cree que el tratamiento con un inhibidor de mTOR después de una cantidad de tiempo suficiente o una dosificación suficiente da lugar a uno o más de los siguientes:
un aumento en la expresión de uno o más de los siguientes marcadores: CD62Lalto, CD127alto, CD27+ y BCL2, p. ej., en células T de la memoria, p. ej., precursores de células T de la memoria;
una disminución en la expresión de KLRG1, p. ej., en células T de la memoria, p. ej., precursores de células T de la memoria; y
un aumento en el número de precursores de células T de la memoria, p. ej., células con una cualquiera o una combinación de las siguientes características: CD62Lalto incrementado, CD127alto incrementado, CD27+ incrementado, KLRG1 reducido y BCL2 incrementado;
y en donde cualquiera de los cambios descritos anteriormente se produce, p. ej., al menos de manera transitoria, p. ej., en comparación con un sujeto sin tratar (Araki, K et al. (2009) Nature 460: 108-112). Precursores de células T de la memoria son células T de la memoria que se encuentran en las primeras etapas del programa de diferenciación. Por ejemplo, las células T de la memoria tienen una o más de las siguientes características: CD62Lalto incrementado, CD127alto incrementado, CD27+ incrementado, KLRG1 reducido y/o BCL2 incrementado.
En una realización, la invención puede emplear una composición o forma de dosificación de un inhibidor de mTOR, p. ej., un inhibidor de mTOR alostérico, p. ej., un rapálogo, rapamicina o RAD001, o un inhibidor catalítico de mTOR, que, cuando se administra en un régimen de dosificación seleccionado, p. ej., una vez al día o una vez a la semana, está asociado con: un nivel de inhibición de mTOR que no está asociado con una supresión inmunitaria completa o significativa, pero está asociado con una mejora de la respuesta inmunitaria.
Un inhibidor de mTOR, p. ej., un inhibidor de mTOR alostérico, p. ej., un rapálogo, rapamicina o RAD001, o un inhibidor catalítico de mTOR, puede proporcionarse en una formulación de liberación sostenida. Cualquiera de las composiciones o formas de dosificación unitaria descritas en esta memoria se puede proporcionar en una formulación de liberación sostenida. En algunas realizaciones, una formulación de liberación sostenida tendrá una biodisponibilidad más baja que una formulación de liberación inmediata. P. ej., en realizaciones, para lograr un efecto terapéutico similar de una formulación de liberación inmediata, una formulación de liberación sostenida tendrá de aproximadamente 2 a aproximadamente 5, de aproximadamente 2,5 a aproximadamente 3,5 o aproximadamente 3 veces la cantidad de inhibidor proporcionada en la formulación de liberación inmediata.
En una realización, se proporcionan formas de liberación inmediata, p. ej., de RAD001, normalmente utilizadas para una administración por semana, que tienen de 0.1 a 20, de 0.5 a 10, de 2.5 a 7.5, de 3 a 6 o aproximadamente 5 mg por forma de dosificación unitaria. Para administraciones una vez a la semana, estas formulaciones de liberación inmediata corresponden a formas de liberación sostenida, que tienen, respectivamente, 0.3 a 60, 1.5 a 30, 7.5 a 22.5, 9 a 18 o aproximadamente 15 mg de un inhibidor de mTOR, p. ej., un inhibidor de mTOR alostérico, p. ej., rapamicina o RAD001. En realizaciones, ambas formas se administran una vez por semana.
En una realización, las formas de liberación inmediata, p. ej., de RAD001, se utilizan típicamente para una administración por día, que tienen 0,005 a 1,5, 0,01 a 1,5, 0,1 a 1,5, 0,2 a 1,5, 0,3 a 1,5, 0,4 a 1,5, Se proporcionan 0,5 a 1,5, 0,6 a 1,5, 0,7 a 1,5, 0,8 a 1,5, 1,0 a 1,5, 0,3 a 0,6 o aproximadamente 0,5 mg por forma de dosificación unitaria. Para administraciones una vez al día, estas formulaciones de liberación inmediata corresponden a formas de liberación sostenida, que tienen, respectivamente, de 0.015 a 4.5, 0.03 a 4.5, 0.3 a 4.5, 0.6 a 4.5, 0.9 a 4.5, 1.2 a 4.5, 1.5 a 4.5, 1.8 a 4.5, 2.1 a 4.5, 2.4 a 4.5, 3.0 a 4.5, 0.9 a 1.8 o aproximadamente 1.5 mg de un inhibidor de mTOR, p. ej., un inhibidor de mTOR alostérico, p. ej., rapamicina o RAD001. Para administraciones una vez a la semana, estas formulaciones de liberación inmediata corresponden a formas de liberación sostenida, que tienen, respectivamente, de 0.1 a 30, de 0.2 a 30, de 2 a 30, de 4 a 30, de 6 a 30, de 8 a 30, de 10 a 30, de 1.2 a 30, de 14 a 30, de 16 a 30, de 20 a 30, de 6 a 12 o aproximadamente 10 mg de un inhibidor de mTOR, p. ej., un inhibidor de mTOR alostérico, p. ej., rapamicina o RAD001.
En una realización, se proporcionan formas de liberación inmediata, p. ej., de RAD001, típicamente utilizadas para una administración por día, que tienen de 0,01 a 1,0 mg por forma de dosificación unitaria. Para administraciones una vez al día, estas formas de liberación inmediata corresponden a formas de liberación sostenida, que tienen, respectivamente, de 0.03 a 3 mg de un inhibidor de mTOR, p. ej., un inhibidor alostérico de mTOR, p. ej., rapamicina o RAD001. Para administraciones una vez a la semana, estas formas de liberación inmediata corresponden a formas de liberación sostenida, que tienen, respectivamente, de 0.2 a 20 mg de un inhibidor de mTOR, p. ej., un inhibidor alostérico de mTOR, p. ej., rapamicina o RAD001.
En una realización, se proporcionan formas de liberación inmediata, p. ej., de RAD001, típicamente utilizadas para una administración a la semana, que tienen de 0,5 a 5,0 mg por forma de dosificación unitaria. Para administraciones una vez a la semana, estas formas de liberación inmediata corresponden a formas de liberación sostenida, que tienen, respectivamente, de 1.5 a 15 mg de un inhibidor de mTOR, p. ej., un inhibidor alostérico de mTOR, p. ej., rapamicina o RAD001.
Como se ha descrito anteriormente, una diana de la ruta de mTOR es la quinasa P70 S6. Por tanto, las dosis de inhibidores de mTOR que son útiles en los métodos y composiciones descritos en esta memoria son las que son suficientes para lograr una inhibición no superior al 80 % de la actividad de la quinasa P70 S6 con respecto a la actividad de la quinasa P70 S6 en ausencia de un inhibidor de mTOR, p. ej., medido mediante un ensayo descrito en esta memoria, p. ej., el ensayo de Boulay. En un aspecto adicional, la invención puede emplear una cantidad de un inhibidor de mTOR suficiente para lograr una inhibición no superior al 38 % de la actividad de la quinasa P70 S6 en relación con la actividad de la quinasa P70 S6 en ausencia de un inhibidor de mTOR.
En un aspecto, la dosis de inhibidor de mTOR útil en los métodos y las composiciones de la invención es suficiente para lograr, p. ej., cuando se administra a un sujeto humano, 90 /-5 % (es decir, 85-95 %), 89+/-5 %, 88+/-5 %, 87+/-5 %, 86+/-5 %, 85+/-5 %, 84+/-5 %, 83+/-5 %, 82+/-5 %, 81+/-5 %, 80+/-5 %, 79+/-5 %, 78+/-5 %, 77+/-5 %, 76+/-5 %, 75+/-5 %, 74+/-5 %, 73+/-5 %, 72 /-5 %, 71 /-5 %, 70 /-5 %, 69 /-5 %, 68 /-5 %, 67 /-5 %, 66 /-5 %, 65 /-5 %, 64 /-5 %, 63 /-5 %, 62 /-5 %, 61 /-5 %, 60 /-5 %, 59 /-5 %, 58 /-5 %, 57 /-5 %, 56 /-5 %, 55 /-5 %, 54 /-5 %, 54 /-5 %, 53 /-5 %, 52 /-5 %, 51 /-5 %, 50 /-5 %, 49 /-5 %, 48 /-5 %, 47 /-5 %, 46 /-5 %, 45 /-5 %, 44 /-5 %, 43 /-5 %, 42 /-5 %, 41 /-5 %, 40 /-5 %, 39 /-5 %, 38 /-5 %, 37 /-5 %, 36 /-5 %, 35 /-5 %, 34 /-5 %, 33 /-5 %, 32 /-5 %, 31 /-5 %, 30 /-5 %, 29 /-5 %, 28 /-5 %, 27 /-5 %, 26 /-5 %, 25 /-5 %, 24 /-5 %, 23 /-5 %, 22 /-5 %, 21 /-5 %, 20 /-5 %, 19 /-5 %, 18 /-5 %, 17 /-5 %, 16 /-5 %, 15 /-5 %, 14 /-5 %, 13 /-5 %, 12 /-5 %, 11 /-5 % o 10 /-5 % de inhibición de la actividad de la quinasa P70 S6, p. ej., medida mediante un ensayo descrito en esta memoria, p. ej., el ensayo de Boulay.
La actividad de la quinasa P70 S6 en un sujeto puede medirse utilizando métodos conocidos en la técnica, tales como, p. ej., según los métodos descritos en la patente de los EE. UU. 7.727.950, mediante análisis de inmunotransferencia de los niveles de fosfoP70 S6K y/o niveles de fosfoP70 S6 o mediante ensayos de actividad quinasa in vitro.
Como se usa en esta memoria, el término "aproximadamente" en referencia a una dosis de inhibidor de mTOR se refiere a una variabilidad de hasta /-10 % en la cantidad de inhibidor de mTOR, pero puede no incluir variabilidad en torno a la dosis indicada.
En algunos casos, la divulgación proporciona métodos que comprenden administrar a un sujeto un inhibidor de mTOR, p. ej., un inhibidor alostérico, p. ej., RAD001, en una dosis dentro de un nivel mínimo objetivo. En algunos casos, el nivel mínimo es significativamente más bajo que los niveles mínimos asociados con los regímenes de dosificación utilizados en trasplantes de órganos y pacientes con cáncer. En un caso, se administra un inhibidor de mTOR, p. ej., RAD001 o rapamicina, para dar como resultado un nivel mínimo que es menos de ^ , 1/4, 1/10 o 1/20 del nivel mínimo que resulta en inmunosupresión o un efecto anticancerígeno. En un caso, se administra un inhibidor de mTOR, p. ej., RAD001 o rapamicina, para obtener un nivel mínimo que es menos de ^ , 1/4, 1/10 o 1/20 del nivel mínimo proporcionado en el prospecto aprobado por la FDA. para uso en inmunosupresión o indicaciones contra el cáncer.
En un caso, un método descrito en esta memoria comprende administrar a un sujeto un inhibidor de mTOR, p. ej., un inhibidor alostérico, p. ej., RAD001, en una dosis que proporciona un nivel mínimo objetivo de 0,1 a 10 ng/ml, 0,1 a 5 ng /ml, 0,1 a 3 ng/ml, 0,1 a 2 ng/ml o 0,1 a 1 ng/ml.
En un caso, un método descrito en esta memoria comprende administrar a un sujeto un inhibidor de mTOR, p. ej., un inhibidor alostérico, p. ej., RAD001, en una dosis que proporciona un nivel mínimo objetivo de 0,2 a 10 ng/ml, 0,2 a 5 ng /ml, 0,2 a 3 ng/ml, 0,2 a 2 ng/ml o 0,2 a 1 ng/ml.
En un caso, un método descrito en esta memoria comprende administrar a un sujeto un inhibidor de mTOR, p. ej., un inhibidor alostérico, p. ej., RAD001, en una dosis que proporciona un nivel mínimo objetivo de 0,3 a 10 ng/ml, 0,3 a 5 ng /ml, 0,3 a 3 ng/ml, 0,3 a 2 ng/ml o 0,3 a 1 ng/ml.
En un caso, un método descrito en esta memoria comprende administrar a un sujeto un inhibidor de mTOR, p. ej., un inhibidor alostérico, p. ej., RAD001, en una dosis que proporciona un nivel mínimo objetivo de 0,4 a 10 ng/ml, 0,4 a 5 ng /ml, 0,4 a 3 ng/ml, 0,4 a 2 ng/ml o 0,4 a 1 ng/ml.
En un caso, un método descrito en esta memoria comprende administrar a un sujeto un inhibidor de mTOR, p. ej., un inhibidor alostérico, p. ej., RAD001, en una dosis que proporciona un nivel mínimo objetivo de 0,5 a 10 ng/ml, 0,5 a 5 ng /ml, 0,5 a 3 ng/ml, 0,5 a 2 ng/ml o 0,5 a 1 ng/ml.
En un caso, un método descrito en esta memoria comprende administrar a un sujeto un inhibidor de mTOR, p. ej., un inhibidor alostérico, p. ej., RAD001, en una dosis que proporciona un nivel mínimo objetivo de 1 a 10 ng/ml, 1 a 5 ng /ml, 1 a 3 ng/ml, o 1 a 2 ng/ml.
Tal como se utiliza en esta memoria, la expresión "nivel mínimo" se refiere a la concentración de un fármaco en plasma justo antes de la siguiente dosis o la concentración mínima de fármaco entre dos dosis.
En algunos casos, un nivel mínimo objetivo de RAD001 está en un intervalo de entre aproximadamente 0,1 y 4,9 ng/ml. En un caso, el nivel mínimo objetivo está por debajo de 3 ng/ml, p. ej., está entre 0,3 o menos y 3 ng/ml. En un caso, el nivel mínimo objetivo está por debajo de 3 ng/ml, p. ej., está entre 0,3 o menos y 1 ng/ml.
En otro aspecto, la invención puede utilizar un inhibidor de mTOR distinto de RAD001 en una cantidad que esté asociada con un nivel mínimo diana que sea bioequivalente al nivel mínimo diana especificado para RAD001. En una realización, el nivel mínimo diana para un inhibidor de mTOR distinto de RAD001, es un nivel que proporciona el mismo nivel de inhibición de mTOR (p. ej., medido mediante un método descrito en el presente memoria, p. ej., la inhibición de P70 S6) que un nivel mínimo de RAD001 descrito en esta memoria.
Composiciones farmacéuticas: inhibidores de mTOR
En un aspecto, la presente invención puede emplear composiciones farmacéuticas que comprenden un inhibidor de mTOR, p. ej., un inhibidor de mTOR como se describe en esta memoria, formulado para uso en combinación con células CAR de acuerdo con las reivindicaciones.
En algunas realizaciones, el inhibidor de mTOR se formula para la administración en combinación con uno adicional, p. ej., como se describe en el presente memoria.
En general, los compuestos de la divulgación se administrarán en cantidades terapéuticamente eficaces como se describe anteriormente a través de cualquiera de los modos habituales y aceptables conocidos en la técnica, solos o en combinación con uno o más agentes terapéuticos.
Las formulaciones farmacéuticas se pueden preparar utilizando procedimientos convencionales de disolución y mezcla. Por ejemplo, la sustancia farmacológica a granel (p. ej., un inhibido de mTOR o forma estabilizada del compuesto (p. ej., complejo con un derivado de ciclodextrina u otro agente de formación de complejo conocido) se disuelve en un disolvente adecuado en presencia de uno o más de los excipientes descritos en el presente memoria. El inhibidor de mTOR se formula normalmente en formas de dosificación farmacéuticas para proporcionar una dosis fácilmente controlable del fármaco y para dar al paciente un producto elegante y fácilmente manejable.
Compuestos de la presente invención se pueden administrar como composiciones farmacéuticas mediante cualquier vía convencional, en particular por vía enteral, p. ej., por vía oral, p. ej., en forma de comprimidos o cápsulas, o por vía parenteral, p. ej., en forma de soluciones o suspensiones inyectables, por vía tópica, p. ej., en forma de lociones, geles, ungüentos o cremas, o en una forma nasal o de supositorio. Cuando un inhibidor de mTOR se administra en combinación con (ya sea simultáneamente o por separado) otro agente como se describe en esta memoria, en un aspecto, ambos componentes pueden administrarse por la misma vía (p. ej., por vía parenteral). Como alternativa, se puede administrar otro agente por una vía diferente en relación con el inhibidor de mTOR. Por ejemplo, un inhibidor de mTOR se puede administrar por vía oral y el otro agente se puede administrar por vía parenteral.
LIBERACIÓN SOSTENIDA
Los inhibidores de mTOR, p. ej., inhibidores de mTOR alostéricos o inhibidores de mTOR catalíticos, descritos en esta memoria se pueden proporcionar como formulaciones farmacéuticas en forma de formas de dosificación sólidas orales que comprenden un inhibidor de mTOR desvelados en esta memoria, p. ej., rapamicina o RAD001, que satisfacen los requisitos de estabilidad del producto y/o tienen propiedades farmacocinéticas favorables con respecto a los comprimidos de liberación inmediata (LI), tales como concentraciones máximas plasmáticas promedio reducidas, variabilidad entre pacientes y dentro del paciente reducida en el grado de absorción del fármaco y en la concentración máxima plasmática, relación Cmáx / Cmín reducida y/o reducción de los efectos de los alimentos. Formulaciones farmacéuticas proporcionadas pueden permitir un ajuste de dosis más preciso y/o reducir la frecuencia de acontecimientos adversos, proporcionando
así tratamientos más seguros para pacientes con un inhibidor de mTOR desvelado en esta memoria, p. ej., rapamicina o RAD001.
En algunos casos, la presente divulgación proporciona formulaciones estables de liberación prolongada de un inhibidor de mTOR descrito en esta memoria, p. ej., rapamicina o RAD001, que son sistemas multiparticulados y pueden tener capas y recubrimientos funcionales.
La expresión "formulación multiparticulada de liberación prolongada, como se usa en esta memoria, se refiere a una formulación que permite la liberación de un inhibidor de mTOR descrito en esta memoria, p. ej., rapamicina o RAD001, durante un periodo de tiempo prolongado, p. ej., durante al menos 1, 2, 3, 4, 5 o 6 horas. La formulación de liberación prolongada puede contener matrices y recubrimientos hechos de excipientes especiales, p. ej., como se describe en esta memoria, que se formulan de manera que el principio activo esté disponible durante un periodo de tiempo prolongado después de la ingestión.
La expresión "liberación extendida" puede usarse indistintamente con las expresiones "liberación sostenida" (LS) o "liberación prolongada". El término "liberación extendida" se refiere a una formulación farmacéutica que no libera el principio activo inmediatamente después de la dosificación oral, sino durante un periodo prolongado de acuerdo con la definición de las farmacopeas Ph. Eur. (7.a edición) Monografía para comprimidos y cápsulas y capítulo general de la USP <1151> para formas farmacéuticas. La expresión "Liberación inmediata" (LI), tal como se utiliza en esta memoria, se refiere a una formulación farmacéutica que libera el 85 % del principio activo en menos de 60 minutos de acuerdo con la definición de "Guidance for Industry: "Dissolution Testing of Immediate Release Solid Oral Dosage Forms" (FDA CDER, 1997). En algunas realizaciones, la expresión "liberación inmediata" significa la liberación de everolismus de los comprimidos en un tiempo de 30 minutos, p. ej., según se mide en el ensayo de disolución descrito en esta memoria. Las formulaciones estables de liberación prolongada de un inhibidor de mTOR descrito en esta memoria, p. ej., rapamicina o RAD001, se pueden caracterizar por un perfil de liberación in vitro utilizando ensayos conocidos en la técnica, tales como un ensayo de disolución como se describe en esta memoria: un recipiente de disolución lleno con 900 mL de tampón fosfato pH 6.8 que contiene dodecilsulfato de sodio al 0.2 % a 37 °C y la disolución se realiza utilizando un método de paleta a 75 rpm según la monografía de ensayos 711 de la USP y la monografía de ensayos 2.9.3. de la Ph. Eur., respectivamente.
En algunas realizaciones, las formulaciones estables de liberación prolongada de un inhibidor de mTOR descritas en esta memoria, p. ej., rapamicina o RAD001, liberan el inhibidor de mTOR en el ensayo de liberación in vitro según las siguientes especificaciones de liberación:
0.5 h: <45 % o <40, p. ej., <30 %
1 h: 20-80 %, p. ej., 30-60 %
2 h: >50 % o >70 %, p. ej., >75 %
3 h: >60 % o >65 %, p. ej., >85 %, p. ej., >90 %.
En algunas realizaciones, las formulaciones estables de liberación prolongada de un inhibidor de mTOR desveladas en esta memoria, p. ej., rapamicina o RAD001, liberan el 50 % del inhibidor de mTOR no antes de los 45, 60, 75, 90, 105 min o 120 min en el ensayo de disolución in vitro.
Métodos de suministro de biopolímeros
En algunas realizaciones, una o más células que expresan CAR descritas en esta memoria se pueden administrar o suministrar al sujeto mediante un armazón biopolimérico, p. ej., un implante biopolimérico. Los armazones biopoliméricos pueden respaldar o potenciar el suministro, la expansión y/o la dispersión de las células que expresan CAR descritas en esta memoria. Un armazón de biopolímero comprende un polímero biocompatible (p. ej., no induce sustancialmente una respuesta inflamatoria o inmunitaria) y/o un polímero biodegradable que se puede producir de forma natural o sintética. Ejemplos de biopolímeros adecuados incluyen, pero no se limitan a agar, agarosa, alginato, alginato/cemento de fosfato de calcio (CPC), beta-galactosidasa (p-GAL), (1,2,3,4,6-pentaacetil a-D-galactosa), celulosa, quitina, quitosano, colágeno, elastina, gelatina, ácido hialurónico colágeno, hidroxiapatita, poli(3-hidroxibutirato-co-3-hidroxi-hexanoato) (PHBHHx), poli(lactida), poli(caprolactona) (PCL), poli(lactida-co-glicolida) (PLG), óxido de polietileno (PEO), poli(ácido láctico-coglicólico) (PLGA), poli(óxido de propileno) (PPO), poli(alcohol vinílico) (PVA) , seda, proteína de soja y aislado de proteína de soja, solos o en combinación con cualquier otra composición polimérica, en cualquier concentración y en cualquier proporción. El biopolímero se puede aumentar o modificar con moléculas que fomentan la adhesión o migración, p. ej., péptidos que mimetizan colágeno que se unen al receptor de colágeno de las células, y/o moléculas estimuladoras para potenciar el suministro, expansión o función, p. ej., actividad antineoplásica, de las células que se van a suministrar. El armazón biopolimérico puede ser inyectable, p. ej., un gel, o una composición semisólida o sólida.
En algunas realizaciones, las células que expresan CAR descritas en esta memoria se siembran en el armazón biopolimérico antes del suministro al sujeto. En algunas realizaciones, el armazón biopolimérico comprende además uno o más agentes terapéuticos adicionales descritos en esta memoria (p. ej., otra célula que expresa CAR, un anticuerpo o una molécula pequeña) o agentes que potencian la actividad de una célula que expresa c Ar , p. ej., incorporada a los biopolímeros del armazón o conjugada con estos. En algunas realizaciones, el armazón biopolimérico se inyecta, p. ej., por vía intra-tumoral, o se implanta quirúrgicamente en el tumor o a una distancia del tumor lo suficientemente próxima para mediar en un efecto antitumoral. Se describen ejemplos adicionales de composiciones biopoliméricas y métodos para su suministro en Stephan et al., Nature Biotechnology, 2015, 33: 97-101; y el documento WO2014/110591.
Composiciones farmacéuticas y tratamientos
Las composiciones farmacéuticas empleadas en la presente invención pueden comprender una célula que expresa CAR, p. ej., una pluralidad de células que expresan CAR, en combinación con uno o más soportes, diluyentes o excipientes farmacéutica o fisiológicamente aceptables. Composiciones de este tipo pueden comprender tampones, tales como solución salina tamponada neutra, solución salina tamponada con fosfato y similares; hidratos de carbono, tales como glucosa, manosa, sacarosa o dextranos, manitol; proteínas; polipéptidos o aminoácidos tales como glicina; antioxidantes; agentes quelantes tales como EDTA o glutatión; adyuvantes (p. ej., hidróxido de aluminio); y conservantes. Las composiciones empleadas en la presente invención están, en un aspecto, formuladas para administración intravenosa.
Las composiciones farmacéuticas empleadas en la presente invención se pueden administrar de una manera apropiada para la enfermedad a tratar (o prevenir). La cantidad y la frecuencia de administración estarán determinadas por factores tales como el estado del paciente y el tipo y la gravedad de la enfermedad del paciente, aunque las dosis apropiadas pueden determinarse mediante ensayos clínicos.
En una realización, la composición farmacéutica está sustancialmente exenta de, p. ej., no hay niveles detectables de un contaminante, p. ej., seleccionado del grupo constituido por endotoxina, micoplasma, lentivirus competente para la replicación (RCL), p24, ácido nucleico de VSV-G, gag de HIV, perlas residuales recubiertas con anti-CD3/anti-CD28, anticuerpos de ratón, suero humano combinado, seroalbúmina bovina, suero bovino, componentes de medios de cultivo, componentes de plásmidos o células de empaquetamiento de vectores, una bacteria y un hongo. En una realización, la bacteria es al menos una seleccionada del grupo que consiste en Alcaligenes faecalis, Candida albicans, Escherichia coli, Haemophilus gripe, Neisseria meningitides, Pseudomonas aeruginosa, Staphylococcus aureus, Streptococcus pneumonia y grupo A de Streptococcus pyogenes.
Cuando se indica "una cantidad inmunológicamente eficaz", "una cantidad antitumoral eficaz", "una cantidad eficaz inhibidora de tumores" o "cantidad terapéutica", la cantidad precisa de las composiciones empleadas en la presente invención a administrar puede ser determinada por un médico considerando las diferencias individuales en edad, peso, tamaño del tumor, extensión de la infección o metástasis y estado del paciente (sujeto). Generalmente se puede afirmar que una composición farmacéutica que comprende las células efectoras inmunitarias (p. ej., células T, células NK) descritas en esta memoria se puede administrar a una dosis de 104 a 109 células/kg de peso corporal, en algunos casos de 105 a 106 células/kg de peso corporal, incluidos todos los valores enteros dentro de esos intervalos. Las composiciones de células T también se pueden administrar múltiples veces a estas dosis. Las células se pueden administrar utilizando técnicas de infusión de uso común en inmunoterapia (véase, p. ej., Rosenberg et al., New Eng. J. of Med. 319: 1676, 1988).
En determinados aspectos, puede ser deseable administrar células efectoras inmunitarias activadas (p. ej., células T, células NK) a un sujeto y luego volver a extraer sangre (o realizar una aféresis), activar células efectoras inmunitarias (p. ej., células T, células NK) de las mismas de acuerdo con la presente divulgación, y reinfundir al paciente estas células efectoras inmunitarias activadas y expandidas (p. ej., células T, células NK). Este proceso se puede llevar a cabo varias veces cada pocas semanas. En determinados aspectos, las células efectoras inmunitarias (p. ej., célula T, célula NK) pueden activarse a partir de extracciones de sangre de 10 cm3 a 400 cm3. En determinados aspectos, las células efectoras inmunitarias (p. ej., célula T, célula NK) se activan a partir de extracciones de sangre de 20 cm3, 30 cm3, 40 cm3, 50 cm3, 60 cm3, 70 cm3, 80 cm3, 90 cm3 o 100 cm3.
La administración de las composiciones objeto se puede llevar a cabo de cualquier manera conveniente, incluyendo mediante inhalación de aerosol, inyección, ingestión, transfusión, implantación o trasplante. Las composiciones descritas en el presente memoria pueden administrarse a un paciente por vía transarterial, subcutánea, intradérmica, intra-tumoral, intranodal, intramedular, intramuscular, por inyección intravenosa (i.v.) o intraperitoneal. En un aspecto, las composiciones de células T de la presente invención se administran a un paciente mediante inyección intradérmica o subcutánea. En un aspecto, las composiciones de células T de la presente invención se administran mediante inyección i.v. Las composiciones de células efectoras inmunitarias (p. ej., células T, células NK) se pueden inyectar directamente en un tumor, ganglio linfático o sitio de infección.
En un aspecto particular ilustrativo, los sujetos pueden someterse a leucaféresis, en donde los leucocitos se recogen, enriquecen o agotan ex vivo para seleccionar y/o aislar las células de interés, p. ej., células T. Estos aislados de células
T pueden expandirse mediante métodos conocidos en la técnica y tratarse de manera que se puedan introducir una o más construcciones CAR empleadas en la invención, creando así una célula CAR T de la invención. Los sujetos que lo necesiten pueden someterse posteriormente a un tratamiento estándar con dosis altas de quimioterapia seguido de un trasplante de células madre de sangre periférica. En determinados aspectos, después o de forma concomitante con el trasplante, los sujetos reciben una infusión de célula CAR T expandidas de la presente invención. En un aspecto adicional, las células expandidas se administran antes o después de la cirugía.
La dosis de los tratamientos anteriores que se va a administrar a un paciente variará según la naturaleza precisa de la afección que se esté tratando y el receptor del tratamiento. El cambio de escala de las dosificaciones para la administración a seres humanos se puede realizar de acuerdo con las prácticas aceptadas en la técnica. La dosis de CAMPATH, por ejemplo, estará generalmente en el intervalo de 1 a aproximadamente 100 mg para un paciente adulto, normalmente administrada a diario durante un periodo de entre 1 y 30 días. La dosis diaria preferida es de 1 a 10 mg al día aunque en algunos casos se pueden utilizar dosis mayores de hasta 40 mg al día (descritas en la patente de EE. UU. n.° 6.120.766).
En una realización, el CAR se introduce en células efectoras inmunitarias (p. ej., células T, células NK), p. ej., utilizando transcripción in vitro, y el sujeto (p. ej., ser humano) recibe una administración inicial de células efectoras inmunitarias CAR (p. ej., células T, células nK) de la invención, y una o más administraciones posteriores de las células efectoras inmunitarias CAR (p. ej., células T, células NK) de la invención, en donde la una o más administraciones posteriores se administran menos de 15 días, p. ej., 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3 o 2 días después de la administración previa. En una realización, se administra más de una administración de las células efectoras inmunitarias CAR (p. ej., células T, células NK) de la invención al sujeto (p. ej., ser humano) a la semana, p. ej., se administran 2, 3 o 4 administraciones de las células efectoras inmunitarias CAR (p. ej., células T, células NK) de la invención a la semana. En una realización, el sujeto (p. ej., sujeto humano) recibe más de una administración de las células efectoras inmunitarias CAR (p. ej., células T, células Nk) a la semana (p. ej., 2, 3 o 4 administraciones a la semana) (también denominadas en el presente memoria un ciclo), seguidas de una semana sin administraciones de células efectoras inmunitarias CAR (p. ej., células T, células NK), y después se administran al sujeto una o más administraciones adicionales de las células efectoras inmunitarias CAR (p. ej., células T, células NK) (p. ej., más de una administración de las células efectoras inmunitarias CAR (p. ej., células T, células NK) a la semana). En otra realización, el sujeto (p. ej., sujeto humano) recibe más de un ciclo de células efectoras inmunitarias CAR (p. ej., células T, células NK) y el tiempo entre cada ciclo es inferior a 10, 9, 8, 7, 6, 5, 4 o 3 días. En una realización, las células efectoras inmunitarias CAR (p. ej., células T, células NK) se administran en días alternos para tener 3 administraciones a la semana. En una realización, las células efectoras inmunitarias CAR (p. ej., células T, células NK) de la invención se administran durante al menos dos, tres, cuatro, cinco, seis, siete, ocho o más semanas.
En un aspecto, las células que expresan CAR de la presente invención se generan usando vectores víricos lentivíricos, tales como lentivirus. Las células, p. ej., T-CAR, generadas de esa manera tendrán una expresión de CAR estable.
En un aspecto, las células que expresan CAR, p. ej., CARTs, se generan utilizando un vector vírico tal como un vector gammarretrovírico, p. ej., un vector gammarretrovírico descrito en esta memoria. CARTs generados utilizando estos vectores pueden tener una expresión de CAR estable.
En un aspecto, los CARTs expresan transitoriamente vectores CAR durante 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 días después de la transducción. La expresión transitoria de CARs puede efectuarse mediante la administración del vector CAR de ARN. En un aspecto, el ARN de CAR se transduce en la célula T mediante electroporación.
Un problema potencial que puede surgir en los pacientes tratados con células efectoras inmunitarias que expresan CAR de forma transitoria (p. ej., células T, células NK) (particularmente con CARTs que portan scFv murino) es la anafilaxia después de múltiples tratamientos.
Sin quedar ligados a esta teoría, se cree que una respuesta anafiláctica de este tipo podría ser causada por un paciente que desarrolla una respuesta humoral anti-CAR, es decir, anticuerpos anti-CAR que tienen un isotipo anti-IgE. Se cree que células productoras de anticuerpos de un paciente experimentan una conmutación de clase de isotipo IgG (que no provoca anafilaxia) a isotipo IgE cuando hay una pausa de diez a catorce días de la exposición al antígeno.
Si un paciente tiene un alto riesgo de generar una respuesta de anticuerpos anti-CAR durante el transcurso de la terapia con CAR transitoria (tal como las generadas por las transducciones de ARN), las pausas de infusión de CART no deberían durar más de diez a catorce días.
EJEMPLOS
La invención se describe adicionalmente en más detalle haciendo referencia a los siguientes ejemplos experimentales. Estos ejemplos se proporcionan únicamente con fines ilustrativos y no pretenden ser limitativos a menos que se defina de otro modo. La invención se define mediante las reivindicaciones adjuntas.
Ejemplo 1: CAR de PD-1
En una realización, el dominio extracelular (ECD, por sus siglas en inglés) de una molécula inhibidora, p. ej., Muerte Programada 1 (PD-1), se puede fusionar con un dominio transmembrana y dominios de señalización intracelular tales como 4-1BB y CD3 zeta. En un caso, el CAR de PD-1 se puede utilizar solo. En un caso, el CAR de PD-1 se puede utilizar en combinación con otro CAR, p. ej., CAR de CD19. En una realización, el CAR de de PD-1 mejora la persistencia de la célula T. En una realización, el CAR es un CAR de PD-1 que comprende el dominio extracelular de pD-1 indicado como subrayado en la SEQ ID NO: 26 (el dominio de PD-1 está subrayado)
MalpvtalllplalllhaaiDPgwfldspdiDwnpptfspallvvtegdnatftcsfsntsesfvInwvrmspsn qtdklaafpedrsqpgqdcrfrvtqlpngrdfhmsvvrarrndsgtvlcgaislapkaqikeslraelrvterraevptahpspspr pagqfqtlvtttpaprpptpaptiasqplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitlyckrgrkkllyifk qpfmrpvqttqeedgcscrfpeeeeggcelrvkfsrsadapaykqgqnqlynelnlgrreeydvldkrrgrdpemggkprrkn pqeglynelqkdkmaeayseigmkgerrrgkghdglyqglstatkdtydalhmqalppr SEQ ID NO: 26
La secuencia de nucleótidos correspondiente para el CAR de PD-1 se muestra a continuación, con el ECD de PD-1 subrayado a continuación en SEQ ID NO: 27 (el dominio de PD-1 está subrayado)
Atggccctccctgtcactgccctgcttctccccctcgcactcctgctccacgccgctagaccacccggatggtttct ggactctccggatcgcccgtggaatcccccaaccttctcaccggcactcttggttgtgactgagggcgataatgcgaccttcacgtg ctcgttctccaacacctccgaatcattcgtgctgaactggtaccgcatgagcccgtcaaaccagaccgacaagctcgccgcgtttcc ggaagatcggtcgcaaccgggacaggattgtcggttccgcgtgactcaactgccgaatggcagagacttccacatgagcgtggtc cgcgctaggcgaaacgactccgggacctacctgtgcggagccatctcgctggcgcctaaggcccaaatcaaagagagcttgagg gccgaactgagagtgaccgagcgcagagctgaggtgccaactgcacatccatccccatcgcctcggcctgcggggcagtttcag accctggtcacgaccactccggcgccgcgcccaccgactccggccccaactatcgcgagccagcccctgtcgctgaggccgga agcatgccgccctgccgccggaggtgctgtgcatacccggggattggacttcgcatgcgacatctacatttgggctcctctcgccgg aacttgtggcgtgctccttctgtccctggtcatcaccctgtactgcaagcggggtcggaaaaagcttctgtacattttcaagcagccctt catgaggcccgtgcaaaccacccaggaggaggacggttgctcctgccggttccccgaagaggaagaaggaggttgcgagctgc gcgtgaagttctcccggagcgccgacgcccccgcctataagcagggccagaaccagctgtacaacgaactgaacctgggacgg cgggaagagtacgatgtgctggacaagcggcgcggccgggaccccgaaatgggcgggaagcctagaagaaagaaccctcag gaaggcctgtataacgagctgcagaaggacaagatggccgaggcctactccgaaattgggatgaagggagagcggcggaggg gaaaggggcacgacggcctgtaccaaggactgtccaccgccaccaaggacacatacgatgccctgcacatgcaggcccttcccc
ctcgc SEQ ID NO: 27
Otros ejemplos de moléculas inhibidoras incluyen PD1, CTLA4, TIM3, LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 y TGFR beta. PD1 es un miembro inhibidor de la familia de receptores CD28 que también incluye CD28, CTLA-4, ICOS y BTLA. PD-1 se expresa en células B activados, células T y células mieloides (Agata et al. 1996 Int. Immunol 8:765-75). Se ha mostrado que dos ligandos para PD1, PD-L1 y PD-L2 regulan a la baja la activación de células T tras unirse a PD1 (Freeman et al. 2000 J Exp Med 192: 1027-34; Latchman et al. 2001 Nat Immunol 2: 261-8; Carter et al. 2002 Eur J Immunol 32: 634-43). PD-L1 es abundante en los cánceres humanos (Dong et al. 2003 J Mol Med 81: 281-7; Blank et al.
2005 Cáncer Immunol. Immunother 54: 307-314; Konishi et al. 2004 Clin Cáncer Res 10: 5094). La supresión inmunitaria puede invertirse inhibiendo la interacción local de PD1 con PD-L1.
Se cultivaron células Jurkat con informador NFAT-LUC (JNL) a una densidad de 0.5 * 106/ml en medio de cultivo de células Jurkat con puromicina a 0,5 pg/ml. Para cada transfección , se centrifugaron 3 * 106 células a 100 g durante 10 minutos. Se utilizaron cuatro pg de ADN por construcción por transfección. Se mezcló la solución V de Amaxa Nucleofector y el complemento I y se añadieron 100 pl al tubo con la construcción de ADN. A continuación, la mezcla se añadió a las células y se transfirió a la cubeta de electroporación. La electroporación se realizó con el ajuste X-001 utilizando el dispositivo Amaxa Nucleofactor II. Se añadieron 0,5 ml de medio de crecimiento inmediatamente después de la electroporación y la mezcla se transfirió a 2 ml de medio de crecimiento en un pocillo de la placa de 6 pocillos. Después de dos horas, se añadió a las células el compuesto rapálogo en diversas concentraciones. Las células se aplicaron a pocillos de placas de cultivo de tejidos que estaban recubiertos por la diana. La placa de cultivo de tejidos se recubrió con 5 pg/ml de PDL1-Fc o IgG1-Fc o cualquier diana durante 2 h a 37 °C, luego se bloqueó con el tampón de
bloqueo (DPBS con suero al 5 %) durante 30 minutos. Las células transfectadas se añadieron a la placa diana con 100 |j| por pocillo y se incubaron durante 16 horas adicionales. Se añadieron 100 j l de reactivo Luciferase One Glo por pocillo. Las muestras se incubaron durante 5 min a 37 °C y luego se midió la luminiscencia utilizando el lector de placas Envision.
La construcción CAR de PD1 comprende PD1-ECD-TM-4-1BB-CD3zeta. Esta construcción puede mejorar la persistencia de las células transfectadas con la construcción, p. ej., células CART transfectadas con CAR de PD1.
Como se muestra en la FIG. 32: CAR de PD1 mostró una activación significativa inducida por PD1 de la actividad de luciferasa impulsada por el promotor inducible por NFAT, en comparación con el tratamiento de control por IgG1- Fc. Esto sugiere que la interacción de PD1 con PDL-1 es suficiente para provocar el agrupamiento de PD1 en la superficie de células Jurkat y desencadena la fuerte activación de la vía NFAT.
Ejemplo 2: Un CAR basado en un único dominio VHH de camélido se puede expresar en una superficie de células T en combinación con un CAR basado en scFv sin interacción apreciable con el receptor
Material y Método: células T Jurkat que expresan GFP bajo un promotor dependiente de NFAT (NF-GFP) se transdujeron con un CAR activador específico de mesotelina (SS1-CAR), un activador específico de CD19 (19-CAR) o un CAR generado utilizando un dominio VHH de camélido específico para EGFR (VHH-CAR). Después de la transducción con el CAR activador, las células se transdujeron con un CAR inhibidor adicional que reconoce CD19 (19-PD1) para generar células que co-expresan tanto el CAR activador como el inhibidor (SS1 19PDl, 19+19PD1 o VHH+19PD1). Las células T Jurkat transducidas se co-cultivaron durante 24 horas con diferentes líneas celulares que 1) carecen de todos los antígenos diana (K562), 2) expresan mesotelina (K-meso), CD19 (K-19) o EGFR (A431) únicamente, 3) expresan una combinación de EGFR y mesotelina (A431-mesotelina) o CD19 (A431-CD19) o 4) expresan una combinación de CD19 y mesotelina (K-19/meso). Condiciones adicionales que incluyen células sin estimulador (sin estímulo) o K562 con 1 ug/mL de OKT3 (OKT3) también se incluyeron como controles negativos y positivos para la activación de NFAT, respectivamente. La expresión de GFP, como marcador de la activación de NFAT, se evaluó mediante citometría de flujo.
Resultado: Los camellos y especies relacionadas (p. ej., llama) producen de forma natural anticuerpos que tienen un solo dominio variable similar a una cadena pesada. Este dominio, conocido como dominio VHH de camélido, ha evolucionado para existir sin aparearse con un dominio variable de cadena ligera. Se encontró que la posibilidad de que dos moléculas scFv heterólogas puedan disociarse y reasociarse entre sí cuando se muestran en la superficie de una célula como lo demuestra la interrupción en la unión de scFv al ligando afín durante la co-expresión del receptor. El presente ejemplo mostró la interacción reducida esperada entre un CAR de scFv que se muestra en la superficie de una célula en combinación con un CAR basado en el dominio VHH. Se encontró que la co-expresión de dos CAR basados en scFv (SS1-z CAR activador y CAR inhibidor de CD19-PD1) en la superficie de un Jurkat conduce a la incapacidad del CAR activador (SS1-z) de reconocer su ligando afín en la célula diana y desencadenar la activación de las células T a pesar de la ausencia del ligando del receptor inhibidor. Esto es consistente con la unión de ligando reducida observada en la superficie. Por el contrario, la coexpresión del mismo CAR inhibidor (CD19-PD1) con un CAR activador basado en VHH de camélido (VHH-z) no tiene impacto en la capacidad del CAR activador basado en VHH de reconocer su ligando afín. Estos datos respaldan el modelo de que un CAR activador basado en VHH puede expresarse con un CAR basado en scFv sin una interacción significativa entre los receptores debido a la capacidad reducida de los dominios scFv y VHH de interactuar.
Ejemplo 3: CART que fija como objetivo un tumor que expresa el receptor alfa de folato
El receptor de folato a (FRA) se sobre-expresa en aproximadamente el 90 % de los carcinomas de ovario, así como en los cánceres de endometrio, riñón, mama, pulmón, páncreas, cáncer colorrectal y mesotelioma, y su expresión no se ve afectada por la administración de quimioterapia (véase, p. ej., Despierre et al., Gynecol Oncol 130, 192-199 (2013)). En tejidos normales, la expresión de FRA es nula o baja y está restringida a la superficie apical de las células epiteliales polarizadas (Kelemen et al., International journal of cancer 119, 243-250 (2006)), en que parece ser inaccesible a los fármacos anti-FR circulantes.
La terapia con células CAR-T en oncología se testó por primera vez en el cáncer de ovario, en donde se demostró que la administración de células T diseñadas para expresar un CAR anti-FRA compuesto por scFv MOv18 murino y un endodominio CD3z era factible pero no inducía la regresión del tumor debido a la escasa persistencia de las células T modificadas genéticamente (Kershaw et al, Clin Cancer Res 12, 6106-6115 (2006)).
En este ejemplo, los autores de la invención construyeron un CAR anti-FRA C4 completamente humano para reducir el riesgo de inmunogenicidad potencial del transgén c A r . Aunque la afinidad de unión del fragmento Fab C4 humano (2 *107 M'1) es aproximadamente cinco veces menor que la del anticuerpo MOv19 murino de alta afinidad (Figini, M. et al. (1998) Cancer Res 58, 991-996), conserva su especificidad para FRA y se predice que su K(d) de <108 M_1 conferirá la activación exclusiva de CAR al encontrarse con células tumorales que portan grandes cantidades de FRA en la superficie. El dominio de fijación de objetivo está vinculado a una cadena de señalización intracelular combinada de CD27 y CD3z para mejorar adicionalmente la eficacia de este receptor (al que se alude en lo sucesivo como "C4-27z"). Además, el CAR C4-27z tiene una actividad reducida contra las células normales que portan un nivel bajo de antígeno y disminuye el
riesgo potencial de toxicidad en el antígeno y fuera del tumor. Estos resultados conducen a una terapia de células CAR T C4 completamente humanas para el tratamiento seguro y eficaz de un amplio espectro de tumores malignos que expresan FRA.
Materiales y métodos
Líneas celulares. El empaquetamiento de lentivirus se realizó en la línea de células 293T renales fetales normales inmortalizadas adquiridas de ATCC. Las líneas celulares humanas utilizadas en los ensayos de base inmunitaria incluyen las líneas celulares de cáncer de ovario humano establecidas SKOV3, A1847, OVCAR2, OVCAR3, OVCAR4, OVCAR5, A2780, A2008, C30 y PEO-1. Las líneas de células linfoides humanas SUP-T1 se utilizaron para el análisis del título de lentivirus. Para los ensayos de bioluminiscencia, se transfectaron líneas celulares de cáncer diana para expresar luciferasa de luciérnaga (fLuc). La línea celular de mesotelioma maligno de ratón, AE17 (amablemente proporcionada por Steven Albelda, Universidad de Pensilvania) se utilizó como control negativo. Todas las líneas celulares se mantuvieron en medio R10: RPMI-1640 complementado con FBS inactivado por calor al 10 %, penicilina 100 U/mL, sulfato de estreptomicina 100 mg/mL, HEPES 10 mmol/L).
Construcción de CAR y producción de lentivirus El plásmido pHEN2 que contiene el scFv anti-FRa C4/AFRA4 proporcionado amablemente por la Dra. Silvana Canevari (Figini, M. et al. (1998) Cancer Res 58, 991-996) se utilizó como molde para la amplificación por PCR de un fragmento C4 de 729 pb utilizando los siguientes cebadores: 5'-ataggatcccagctggtggagtctgggggaggc-3' (BamHI está subrayado) y 5'-atagctagcacctaggacggtcagcttggtccc-3' (Nhel está subrayado). Los vectores de expresión lentiviral auto-inactivantes de tercera generación pELNS descritos anteriormente se digirieron con BamHI y Nhel y se purificaron en gel. Los productos de PCR digeridos se insertaron luego en el vector pELNS que contenía dominios de señalización de células T CD3z o CD27-CD3z en los que la expresión del transgén está impulsada por el promotor del factor de elongación-la (EF-1a). La construcción resultante se denominó pELNS-C4-z o C4-27z. Se produjeron y concentraron vectores lentivirales defectuosos en la replicación de alto título como se describió previamente en Parry, R.V., et al. (2003) The Journal of Immunology 171, 166-174. Brevemente, se sembraron células 293T en matraces de 150 cm2y se transfectaron utilizando Express In (Open Biosystems) de acuerdo con las instrucciones del fabricante. Se cotransfectaron quince microgramos de plásmido del transgén CAR específico para FR con 7 ug de pVSV-G (plásmido de expresión de glicoproteína VSV), 18 ug de pRSV.REV (plásmido de expresión de Rev) y 18 ug de pMDLg/p.RRE (plásmido de expresión de Gag/Pol) con 174 ul de Express In (1ug/ul) por matraz. Los sobrenadantes se recogieron a las 24 h y 48 h después de la transfección, se concentraron 10 veces mediante ultracentrifugación durante 2 horas a 28.000 rpm con un rotor Beckman SW32Ti (Beckman Coulter). Alternativamente, se realizó una sola recolección de los medios 30 h después del cambio de medios. Los medios que contienen virus se utilizan alternativamente sin concentrar o concentrados mediante el concentrador Lenti-X (Clontech, Cat n° 631232). Los virus se dividieron en partes alícuotas en tubos y se almacenaron a -80 °C hasta que estuvieran listos para usar para la titulación o los experimentos. Todos los lentivirus utilizados en los experimentos procedían de materiales concentrados.
Determinación del título lentiviral. Los títulos de vectores lentivirales concentrados que codifican FRA CAR se determinaron diluyendo en serie (3 veces) preparados de vectores en medio R10 y transduciendo células SUP-T1. Brevemente, se sembraron células SUP-T1 (20.000 células/100 ul/pocillo) en un único pocillo de una placa de 96 pocillos y se transfirieron 50 ul de sobrenadante de vector diluido 3 veces y se incubaron durante la noche. Al día siguiente, las células se alimentaron con 100 ul de medio R10 precalentado. Dos días después de la transducción, se determinaron los títulos de los vectores mediante citometría de flujo aplicando métodos de citometría de flujo estándares para el análisis de la expresión de CAR. Los títulos (unidades de transducción [TU] = (% positivo/100) x 2E4 x 20 x dilución. Todos los experimentos se repitieron al menos tres veces y los títulos promedio obtenidos de los experimentos se utilizaron para el análisis de datos.
Células T humanas y transfección . Células T CD4+ y CD8+ humanas primarias, adquiridas de Human Immunology Core de la Universidad de Pensilvania, se aislaron de donantes voluntarios sanos después de la leucaféresis mediante selección negativa. Todos los especímenes fueron recolectados bajo un protocolo aprobado por una Junta de Revisión Institucional de la Universidad, y se obtuvo el consentimiento informado por escrito de cada uno de los donantes. Las células T se cultivaron en medio R10 y se estimularon con perlas recubiertas de anticuerpos monoclonales (mAb) anti-CD3 y anti-CD28 (Invitrogen). Dieciocho a 24 horas después de la activación, las células T humanas se transdujeron utilizando un procedimiento de espinoculación. Brevemente, se infectaron 0,5*106 células T con una multiplicidad de infección (MOI) de 2 y 5 del vector concentrado C4-27z y MOv19-27z, respectivamente. Las mezclas de células y vectores se centrifugaron a temperatura ambiente durante 90 min (2500 rpm) en una centrífuga de mesa (Sorvall ST 40). Se añadió interleuquina-2 recombinante humana (IL-2; Novartis) cada 2-3 días hasta una concentración final de 100 UI/mL y se mantuvo una densidad celular de 0,5 ><106 a 1 *106 células/mL. Una vez que los cultivos de células T diseñados parecieron descansar, según lo determinado tanto por la disminución de la cinética de crecimiento como por el tamaño celular determinado con el contador Multisizer 3 Coulter (Beckman Coulter), un celómetro Nexcelom Vision o Millipore Scepter, las células T se utilizaron para el análisis funcional.
Análisis de citometría de flujo. Se utilizaron los siguientes anticuerpos monoclonales para el análisis fenotípico: APC-Cy7 anti-CD3 humano; FITC antihumano CD4; APC anti-humano c D8; PE-anti-CD45 humano; PE antihumano CD137. Se utilizó 7-aminoactinomicina D (7-AAD) para la tinción de viabilidad. Todos los anticuerpos monoclonales se adquirieron
de BD Biosciences. En los experimentos de transferencia de células T, se obtuvo sangre periférica mediante sangrado retroorbital y se tiñó para determinar la presencia de células T CD45, CD4 y CD8 humanas. Después de seleccionar la población humana de CD45+, los subconjuntos de CD4+ y CD8+ se cuantificaron utilizando tubos TruCount (BD Biosciences) con números conocidos de perlas fluorescentes, tal como se describe en las instrucciones del fabricante. La expresión de FRa en la superficie de la célula tumoral se realizó utilizando el mAb MOv18 seguido del Ab anti-ratón de cabra marcado con APC. La expresión en la superficie de las células T de CAR tanto C4 como MOv19 se evaluó utilizando proteína FRa recombinante marcada con biotina (R&D Systems, Inc) seguido de estreptavidina-APC (eBioscience, Inc.) o IgG anti-humana de conejo marcada con biotina e y fragmento F(ab')2 de IgG anti-ratón de cabra, seguido de estreptavidina-APC, respectivamente. Para la tinción de citoquinas intracelulares, las células se estimularon en medio de cultivo que contenía ácido fosfomolíbdico (PMA) (30 ng/mL) (Sigma-Aldrich), ionomicina (500 ng/mL) (Sigma-Aldrich) y monensina (GolgiStop) (1 pl/mL) (BD Biosciences) en una incubadora de células con 10 % de CO2 a 37 °C durante 4 h. Para determinar la producción de citoquinas en las células CAR T, las células se co-cultivaron con células de cáncer de ovario FRpos durante 5 h. Después de teñir los marcadores de superficie, las células se fijaron y permeabilizaron utilizando tampón Cytofix/Cytoperm y Perm/Wash (BD Biosciences) de acuerdo con las instrucciones del fabricante. Luego, las células se tiñeron con anticuerpos de citoquinas conjugados con fluorescencia que incluyen PE anti-IFN-Y humano, azul del Pacífico anti-TNF-a humano o FITC anti-IL-2 humana antes del análisis. La citometría de flujo se realizó con un citómetro de flujo BD FACSCanto II (BD Biosciences) y los datos de citometría de flujo se analizaron con el software FlowJo versión 7.2.5 (Tree Star, Ashland, OR).
Ensayos de liberación de citoquinas. Los ensayos de liberación de citoquinas se realizaron mediante co-cultivo de 1*105 células T con 1*105 células diana por pocillo por triplicado en placas de fondo plano de 96 pocillos en un volumen de 200 ul de medio R10. Después de 20-24 horas, se analizó la presencia de IFN-y en los sobrenadantes del co-cultivo utilizando un kit ELISA, de acuerdo con las instrucciones del fabricante (Biolegend, San Diego, CA). Los valores representan la media de pocillos por triplicado.
Ensayos de citotoxicidad. Para los ensayos de bioluminiscencia basados en células, se cultivaron 5 * 104 células tumorales que expresan luciferasa de luciérnaga (fLuc) con medio R10 en presencia de diferentes relaciones de células T transducidas con el uso de una microplaca de 96 pocillos (BD Biosciences). Después de la incubación durante ~20 horas a 37 °C, cada uno de los pocillos se llenó con 50 uL de DPBS resuspendido con 1 ul de D-luciferina (0,015 g/mL) y se tomaron imágenes con Xenogen IVIS Spectrum. . El porcentaje de viabilidad de las células tumorales se calculó como la luminiscencia media de la muestra experimental menos el fondo dividido por la luminiscencia media del número de entrada de células diana utilizadas en el ensayo menos el fondo multiplicado por 100. Todos los datos se representan como una media de pocillos por triplicado.
Células CAR T (5 * 105) se co-cultivaron con 5 * 105 células cancerosas A1847 FRpos o células AE17 FRneg en 1 ml en una placa de 24 pocillos. Se añadió GolgiStop (BD Biosciences) después del co-cultivo. A continuación, las células se cultivaron durante 4 h adicionales. Los cultivos se tiñeron para MOv19 o C4 scFv, seguido de CD3 y CD8. . A continuación, las células permeabilizadas se tiñeron intracelularmente para determinar la producción de IFN-g, TNF-a e IL-2. Las células T se controlaron en la expresión de CD3 y CD8 y se analizaron adicionalmente para la expresión de citoquinas utilizando una plataforma de puerta booleana para evaluar todos los patrones posibles de respuestas de citoquinas.
Modelo de xenoinjerto de cáncer de ovario Todos los animales se obtuvieron del Stem Cell and Xenograft Core del Abramson Cancer Center, Universidad de Pensilvania. Se criaron, trataron y mantuvieron ratones NOD/SCID/y-chain-/-(NSG) de seis a 12 semanas de edad en condiciones libres de patógenos internamente según los protocolos aprobados por IACUC de la Universidad de Pensilvania. Para un modelo de cáncer de ovario establecido, se inocularon por vía s.c. ratones NSG hembra de 6 a 12 semanas de edad con 3 * 106 células SKOV3 fLuc+ en el flanco el día 0. Después de que los tumores se vuelven palpables alrededor de 1 mes, las células T primarias humanas (CD4+ y las células T CD8+ utilizadas se mezclaron en una proporción de 1:1), se activaron y se transdujeron como se describe arriba. Después de 2 semanas de expansión de células T, cuando la carga tumoral era ~200-300 mm3, los ratones fueron tratados con células T. La vía, la dosis y el momento de las inyecciones de células T se indican en las leyendas individuales de las figuras. Las dimensiones de los tumores se midieron con calibradores y los volúmenes de los tumores se calcularon utilizando la fórmula V = 1/2 (longitud * anchura 2), en que la longitud es el mayor diámetro longitudinal y la anchura es el mayor diámetro transversal. Se tomaron imágenes de los animales antes de la transferencia de células T y aproximadamente cada semana a partir de entonces para evaluar el crecimiento del tumor. La emisión de fotones de las células fLuc+ se cuantificó utilizando el software "Living Image" (Xenogen) para todos los experimentos in vivo. Los tumores se resecaron inmediatamente después de la eutanasia aproximadamente 40 días después de la primera dosis de células T para la medición del tamaño y la inmunohistoquímica.
Para el modelo intraperitoneal de cáncer de ovario, se inyectaron i.p. A ratones NSG 5 * 106 células SKOV3 fLuc+. Veinte días después de la inoculación peritoneal, los ratones que portaban tumores SKOV3 bien establecidos se dividieron en grupos y se trataron. Los ratones fueron sacrificados y sometidos a necropsia cuando los ratones estaban angustiados y moribundos. Para controlar el grado de progresión del tumor, se tomaron imágenes de los ratones semanal o quincenalmente y se midió el peso corporal de los ratones. En todos los modelos, se aleatorizaron 4-5 ratones por grupo antes del tratamiento.
Formación de imágenes por bioluminiscencia. El crecimiento del tumor también se controló mediante formación de imágenes bioluminiscentes (BLI). BLI se realizó utilizando el sistema de formación de imágenes Xenogen IVIS y los fotones emitidos por las células que expresan fLuc dentro del cuerpo del animal se cuantificaron utilizando el software Living Image (Xenogen). Brevemente, a los ratones que portaban células tumorales SKOV3 fLuc+ se les inyectó por vía intraperitoneal D-luciferina (150 mg/kg de material, 100 pL de D-luciferina por cada 10 gramos de peso corporal del ratón) suspendida en PBS y se tomaron imágenes bajo anestesia con isoflurano después de 5~10 minutos. Se generó una imagen de pseudocolor que representa la intensidad de la luz (azul, menos intenso; rojo, más intenso) utilizando Living Image. Los hallazgos de BLI se confirmaron en la necropsia.
Análisis estadístico. Los datos se presentan como medias y DE. El análisis estadístico se realizó mediante el uso de ANOVA de medidas repetidas de 2 vías para la carga tumoral (volumen del tumor, recuento de fotones). Se utilizó el test t de Student para evaluar las diferencias en los números absolutos de células T transferidas, la secreción de citoquinas y la citólisis específica. Se utilizó GraphPad Prism 5.0 (GraphPad Software) para los cálculos estadísticos. P<0.05 se consideró significativo.
Resultados
1. Función potenciada del CAR C4 humano en comparación con el CAR MOvl9 murino in vitro
Utilizando los protocolos de producción y concentración arriba descritos, los autores de la invención encontraron que el lentivirus que codifica CAR C4 tiene un título efectivo más alto que el CAR MOv19 murino, posiblemente como resultado de una expresión más eficiente del scFv humano en las células T humanas (Figura 37a). De hecho, observaron que una multiplicidad de infección (MOI) de lentivirus C4 CAR tan baja como 1 es suficiente para infectar >20 % de células T humanas, mientras que el lentivirus MOv19 CAR requirió una MOI de 5 (Figura 37b). Por lo tanto, para los siguientes experimentos, las células T se infectaron con una MOI de 2 y 5 del vector concentrado C4-27z y MOv19-27z, respectivamente, y se detectó la expresión de la superficie de CAR tanto C4 como MOv19 en las células T mediante la tinción de la proteína FRA recombinante (Figura 34a).
Los ScFv utilizados para la construcción de CAR requieren una afinidad antigénica mínima para alcanzar el umbral de activación para la célula T diseñada, sin embargo, los scFv de mayor afinidad no inducen necesariamente una activación más potente de las células CAR T que los scFv de baja afinidad. Dado que la afinidad de unión del fragmento Fab de C4 humano (2 *107 M'1) es aproximadamente cinco veces más débil que la del MOv19 murino, los autores de la invención examinaron si la afinidad más baja del scFv de C4 utilizado para construir el CAR C4 completamente humano podría influir en la función de células T redirigida a través de la comparación con el MOv19 CAR que contiene un scFv anti-FRA de mayor afinidad. Las células T modificadas para expresar C4-27z o MOv19-27z CAR lisaron específicamente células tumorales SKOV3 y A1847 FRApos con una eficacia aproximadamente equivalente en co-cultivos durante la noche (Figura 34b). Sin embargo, el análisis de producción de citoquinas in vitro mostró que las células CAR T MOv19-27z secretaron significativamente menos IFN-y que las células CAR T C4 en una relación equivalente de 1:1 E:T después del co-cultivo durante la noche (Figura 34c). Este resultado fue validado por ensayos de producción de citoquinas intracelulares de 5 horas. Los gráficos representativos del clasificador de células activadas por fluorescencia (FACS) de la expresión de citoquinas intracelulares de 5 horas por células CAR T activadas por tumores muestran que las células CAR T tanto C4 como MOv19 producen citoquinas IFN-y, TNF-a e IL-2 cuando se incuban durante la noche con células de cáncer de ovario SKOV3 FRApos, pero las células CAR T MOv19 produjeron menos de estas citoquinas que las células CAR T C4 (Figura 34d). La frecuencia de células CAR T C4 que expresan citoquinas fue 5,6 veces mayor para IFN-y, 6,1 veces mayor para TNF-a y 9 veces mayor para IL-2, que la observada en células CAR T MOv19 in vitro. Las células T no transducidas co-cultivadas con células cancerosas FRApos o FRA negativas, o las células CAR T co-cultivadas con células cancerosas FRA negativas, no produjeron citoquinas proinflamatorias (Figura 38).
Los resultados obtenidos in vitro por los autores de la invención sugirieron que células CAR T C4 con una afinidad intermedia por FRA pueden ser funcionalmente superiores a las células CAR T MOv19 con una mayor afinidad por scFv. . Para comprender los mecanismos que explican la función reducida de las células CAR T MOv19 de alta afinidad, los autores de la invención analizaron cuidadosamente la expresión de CAR en células T después de la co-incubación con células tumorales que expresan antígenos. La estimulación con células cancerosas SKOV3, que expresan un alto nivel de FRA, indujo una modulación descendente rápida y marcada de la expresión de CAR MOv19 en superficie después de la interacción con el antígeno (Figura 39). Cinco horas después de la exposición a las células tumorales, la frecuencia de CAR Mov19 se redujo rápidamente desde aproximadamente el 65 % de las células T hasta ~1 %. Este hallazgo también se confirmó mediante el uso de células A1847 FRApos y la línea celular de cáncer de mama T47D, que también expresan altos niveles de FRA (datos no mostrados). En comparación, el CAR C4 no se modulaba notablemente a la baja (Figura 39). El análisis de expresión de citoquinas intracelulares demostró que células T con expresión en superficie de CAR C4 mantenida produjeron IFN-y, TNF-a e IL-2, mientras que la producción de citoquinas se detectó exclusivamente en la fracción CAR negativa del grupo MOv19, lo que indica que la modulación a la baja de CAR y la producción de citoquinas había ocurrido después del encuentro con el antígeno.
Hubo una frecuencia similar de Anexina V+/7-AAD+ medida por tinción de apoptosis en células T modificadas con C4 en comparación con CARs MOv19 después de la estimulación con células SKOV3, respectivamente. CARs con dominio CD28 tenían un AICD más bajo en comparación con 4-1BB (R12: 16,4 %/18,4 % frente a 2A238,1 %/39,6 %).
La avidez general entre CAR y la molécula diana puede explicar esta diferencia observada en la expresión de CAR. Los autores de la invención evaluaron el impacto de la relación de células T a células diana en la expresión relativa de CAR por células CAR T C4 o MOv19 después del co-cultivo con células SKOV3. En relaciones E:T más bajas de 1:10, 1:3 y 1:1, las células CAR T MOv19 mostraron una modulación a la baja marcada y dependiente de la dosis en la expresión de CAR en comparación con CAR C4, que mantuvo ~50 % de la expresión CAR inicial en la relación E:T más baja testada. Sin embargo, a relaciones E:T altas de 3:1 y 10:1 en que el antígeno tumoral es más limitante, las células T que portaban CAR C4 o MOv19 mantuvieron una expresión CAR alta (Figura 40). De acuerdo con los cambios en la expresión de CAR después de la estimulación con antígeno, las células CAR T C4 liberaron más IFN-y que las células cAr T MOv19 en relaciones E:T de 1:10, 1:3 y 1:1, pero cantidades similares en relaciones E:T de 3:1 y 10:1 (Figura 34e). Por lo tanto, la modulación a la baja de CAR se produce de una manera dependiente de la dosis de antígeno, siendo las células CAR T anti-FRA que portan el MOv19 scFv de alta afinidad más sensibles al bajo nivel de antígeno.
2. Actividad antitum oral com parable de células CAR T C4 y MOv19 in vivo
Para comparar la capacidad antitumoral de células CAR T C4 con células CAR T MOv19 in vivo, ratones NSG con tumores SKOV3 subcutáneos grandes establecidos (~300 mm3) recibieron inyecciones intravenosas de 107 células CAR+ T los días 40 y 47 de la inoculación post-tumoral. Los tumores en animales tratados con solución salina, células T no transducidas o células T CAR CD19-27z continuaron creciendo rápidamente. Por el contrario, los ratones que recibieron células CAR T C4-27z o MOv19-27z experimentaron una regresión del tumor (p < 0,0001), en comparación con los 3 grupos de control en el último momento evaluado. La actividad antitumoral de las células CAR T MOv19-27z parecía ligeramente mejor que la de las células CAR T C4-27z, pero no a un nivel significativo (p = 0,058; Figura 35a). La BLI de xenoinjertos tumorales antes y 3 semanas después de la inyección de células T mostró un crecimiento progresivo de tumores en todos los animales que recibieron células T de control, pero no en los grupos de células CAR T (Figura 35b). Los resultados del BLI del tumor fueron consistentes con el tamaño de los tumores residuales resecados (Figura 35c). A continuación, los autores de la invención analizaron la persistencia de las células T transferidas en la sangre periférica 3 semanas después de la transferencia adoptiva y detectamos un mayor número de células T CD4+ y CD8+ en ratones tratados con los grupos de células CAR T C4 y MOv19 en comparación con el grupo de tratamiento con células CAR T UNT y CD19- 27z (Figura 35d), lo que sugiere que el reconocimiento del antígeno tumoral impulsa la supervivencia de las células T transferidas adoptivamente in vivo. Estos resultados demostraron que la actividad antitumoral de CAR C4 es comparable a CAR MOv19, que se describió bien previamente (véase, p. ej., Song et al., Blood 119, 696-706 (2012); Song et al., Cancer Res 71 , 4617-4627 (2011)) y confirman que el CAR C4, a pesar de su afinidad disminuida, es adecuado para la aplicación in vivo.
3. CAR anti-FRA con m enor afinidad puede d ism inuir el riesgo de toxicidad "en la diana"
Se han observado toxicidades en la diana en ensayos clínicos con células CAR T específicas para antígenos asociados a tumores que se expresan en niveles bajos en células normales, y un problema crítico que se debe abordar es si los CARs con mayor afinidad pueden aumentar el riesgo. de toxicidad. Para investigar el efecto funcional de las células T humanas primarias modificadas con CAR C4 y CAR Mov19 en células normales que expresan niveles bajos de FRA, los autores de la invención analizaron la producción de citoquinas de células CAR T C4 y MOv19 CAR después del co-cultivo con células 293T de riñón embrionario humano o línea celular epitelial de ovario normal IOSE 6, que expresa niveles bajos pero detectables de células SKOV3 FRA y FRApos (Figura 36a). Las células CAR T C4 y MOv19 respondieron contra SKOV3 con una mayor actividad observada nuevamente a partir de las células CAR T C4. Sin embargo, se observó una mayor producción de citoquinas IFN-y en las células CAR T MOv19 en respuesta a las células con baja expresión de antígeno, lo que sugiere que las células CAR T MOv19 son funcionalmente más ávidas y sensibles al antígeno bajo (Figura 36b). De manera similar a lo que los autores de la invención observaron en los ensayos de liberación de IFN-y durante la noche, los ensayos de secreción de citoquinas intracelulares de 5 horas demostraron que más células CAR T MOv19 produjeron IFN-y y TNF-a en respuesta al bajo nivel de antígenos en las células normales (Figura 36c), que es uno de los principales contribuyentes propuestos a la tormenta de citoquinas 28 "en el objetivo", en comparación con las células CAR T C4. Estos datos sugieren que el nuevo CAR C4 descrito puede tener una afinidad más apropiada para la administración de una terapia de células T segura y eficaz.
4. CAR C4 con m enor afinidad por el antígeno aFR soluble que CAR MOv19
Los resultados in vitro descritos en esta memoria sugirieron que las células CAR T C4 completamente humanas pueden ser funcionalmente superiores a las células CAR T MOv19 y que la modulación a la baja de CAR puede afectar la actividad antitumoral de CAR MOv19 pero no de CAR C4. Para comprender los mecanismos que explican la función reducida de las células CAR T MOv19, se realizaron experimentos para medir la unión relativa a la proteína aFR recombinante. Células CAR T C4 y MOv19 se cargaron previamente con proteína aFR recombinante marcada con biotina y se midió la disociación de la proteína de superficie a lo largo del tiempo a 4 o 37 °C en presencia de un competidor aFR no biotinilado con un exceso de diez veces. La retención de antígenos en la superficie celular se evaluó mediante citometría de flujo
mediante la adición de estreptavidina (SA) conjugada con ficoeritrina (PE) después del final de cada período de cultivo. En una hora, se detectó menos proteína aFR en la superficie de las células CAR T C4, en comparación con CAR MOv19, a cualquier temperatura. El nivel de disociación dependía tanto del tiempo como de la temperatura y era mayor en las células CAR T C4 en todas las condiciones testadas (Fig. 88A, Fig. 88B). Se obtuvieron resultados similares en un análisis de titulación sobre la unión de proteína aFR biotinilada a células CAR T MOv19 y C4 (Fig. 89). Las células T activadas se transdujeron con un vector lentiviral que expresa MOv19-27z o C4-27z-CAR y se analizaron para determinar la expresión de CAR el día 14. Cien mil células T sin transducir (UNT) o CAR se tiñeron con 0,2, 0,5, 1,2, 5 , 10, 20, 50 o 120 nM/muestra de aFR biotinilado. A continuación, las células T se lavaron y tiñeron con PE-SA. Las células T se analizaron utilizando un citómetro de flujo y los datos se analizaron con el software FlowJo. Estos resultados sugieren que C4 en la construcción CAR tenía una menor afinidad por el antígeno aFR soluble que CAR MOv19.
Conclusión:
La afinidad disminuida del scFv C4 completamente humano seleccionado para diseñar un CAR podría afectar el reconocimiento de células T. Sin embargo, una comparación directa de la producción de citoquinas después de la participación del tumor por parte de células T modificadas con los CAR C4 y MOv19 mostró que el CAR C4 con menor afinidad fue superior en una relación E/T de 1:1. Esto puede deberse a la rápida internalización de CAR MOv19 con mayor afinidad al encontrarse con altos niveles de antígeno. Cuando aumentamos las relaciones E/T, la actividad antitumoral de las células CAR T C4 es equiparable a las células CAR T MOv19. Para comparar adicionalmente la actividad antitumoral in vivo, los autores de la invención encontraron que las células T que expresan el CAR MOv19 de alta afinidad mediaron una actividad ligeramente superior in vivo en comparación con el CAR C4. Sin embargo, esta diferencia no es estadísticamente significativa, lo que sugiere que la afinidad de CAR C4 es adecuada para la aplicación in vivo.
Es necesario considerar las posibles toxicidades en la diana, fuera del tumor resultantes de la expresión de TAA en tejidos normales en la aplicación del enfoque CAR. El desarrollo de CAR o TCR de alta afinidad con gran actividad antitumoral puede conducir a una toxicidad grave. El estudio de los autores de la invención demostró que las células CAR T C4 liberan una cantidad mínima de citoquinas en comparación con las células CAR T MOv19 cuando se encuentran con células normales que expresan niveles bajos de FRA. Por lo tanto, el CAR C4 de afinidad relativamente más baja podría disminuir el riesgo de toxicidad en la diana, mientras que el CAR MOv19 de afinidad más alta podría aumentar este riesgo in vivo.
Ejemplo 4: La disminución de la afinidad de CAR aumenta la eficacia terapéutica
La terapia celular adoptiva (ACT, por sus siglas en inglés) con células T modificadas con CAR puede fijar como objetivo y eliminar células malignas generalizadas, lo que induce respuestas clínicas duraderas en el tratamiento de algunas neoplasias hematopoyéticas (Kochenderfer, J.N., et al. (2010) Blood 116:4099-4102; Porter, D.L., et al. (2011) N Engl J Med 365:725-733; y Brentjens, R.J., et al. (2013) Sci Transl Med 5:177ra138). Sin embargo, muchos antígenos tumorales comúnmente fijados como objetivo también se expresan en tejidos sanos y la toxicidad del tumor en la diana de la destrucción del tejido normal mediada por células T ha limitado el desarrollo y la adopción de este tipo prometedor de terapia contra el cáncer. Informes recientes sobre eventos adversos graves asociados con el tratamiento de pacientes con cáncer con células T modificadas con CAR o TCR ilustran adicionalmente la importancia crítica de la selección de dianas para una terapia segura y eficiente (Lamers et al., 2006, J Clin Oncol. 24:e20; Parkhurst et al., 2011, Molecular therapy: the journal of the American Society of Gene Therapy. 19:620-6; Morgan et al., 2013, J Immunotherapy. 36:133-151; Linette et al., 2013, Blood. 122:863-71). En concreto, la fijación como objetivo de ErbB2 (Her2/neu o CD340) con CARTs de alta afinidad condujo a una toxicidad grave debido al reconocimiento de la diana en tejido cardiopulmonar normal (Morgan et al., 2013, Mol Therapy. 18: 843-851), y de forma similar, la presencia de niveles relativamente altos de EGFR en piel sana conduce a una toxicidad cutánea que limita la dosis (Perez-Soler et al., 2010, J Clin Oncol. 23:5235-46).
La selección de antígenos altamente restringidos a tejidos, antígenos de cáncer de testículo, productos de genes mutados o proteínas virales como dianas podría mejorar significativamente el perfil de seguridad del uso de células CART. Sin embargo, ninguno de estos antígenos está presente con alta frecuencia en cánceres comunes, constitutivamente expresados exclusivamente por células malignas, funcionalmente importantes para el crecimiento del tumor y susceptibles de ser fijados como objetivo por CART. La mayoría de los antígenos diana mejor clasificados que podrían ser fijados como objetivo por CART se expresan en tejidos normales potencialmente importantes, tales como ErbB2, EGFR, MUC1, PSMA y GD2 (Cheever et al., 2009, Clinical Cancer Research. 15:5323-37). Las estrategias actuales para generar CARs consisten en seleccionar scFv con alta afinidad, ya que estudios previos han demostrado que el umbral de activación se correlaciona inversamente con la afinidad del scFv (Chmielewski et al., 2004, J Clin Oncol. 173:7647-53; y Hudecek et al., 2013, Clincial Cancer Research.. 19:3153-64. Estudios indican que el dominio coestimulador de los CARs no influye en el umbral de activación (Chmielewski et al., 2011, Gene Therapy. 18:62-72). Después de la estimulación con TCR existe una estrecha ventana de afinidad para la activación óptima de células T, y el aumento de la afinidad de los TCR no mejora necesariamente la eficacia del tratamiento (Zhong et al., 2013, Proc Natl Acad Sci USA. 110:6973-8; y Schmid et al., 2010, J Immunol. 184:4936-46).
En este ejemplo, se determinó si equipar a las células T con scFv de alta afinidad puede limitar la utilidad de los CARs, debido a la mala discriminación del CART para tumores y tejidos normales que expresan el mismo antígeno a niveles más bajos. También se determinó si el ajuste fino de la afinidad del scFv podría aumentar la capacidad de las células CART de discriminar los tumores de los tejidos normales que expresan el mismo antígeno a niveles más bajos. CARs con afinidades contra dos dianas validadas, ErbB2 y EGFR, que se amplifican o sobre-expresan en una diversidad de cánceres pero que también se expresan en niveles más bajos en tejidos normales, se testaron ampliamente contra múltiples líneas tumorales, así como líneas celulares primarias de tejidos normales y órganos Se descubrió que la disminución de la afinidad del scFv podría aumentar significativamente el índice terapéutico de los CARs al tiempo que se mantiene una sólida eficacia antitumoral.
Se usaron los siguientes materiales y métodos en los experimentos descritos en este ejemplo:
Líneas celulares y linfocitos hum anos primarios
Las líneas celulares SK-BR3, SK-OV3, BT-474, MCF7, MDA231, MDA468, HCC2281, MDA-361, MDA-453, HCC-1419, HCC-1569, UACC-812, LnCap, MDA-175, MCF-10A, HCC38 y HG261 se adquirieron de la American Type Culture Collection y se cultivaron según las instrucciones. Siete líneas celulares primarias (queratinocitos, osteoblastos, células epiteliales renales, células endoteliales de la arteria pulmonar, músculo liso de la arteria pulmonar, progenitor neural, PBMC enriquecidas con CD34+) se obtuvieron de Promocell y se cultivaron de acuerdo con sus protocolos. Los linfocitos primarios fueron aislados de donantes normales por el Centro de Inmunología Humana de la Universidad de Pensilvania y fueron cultivados en medio R10 (RPMI 1640 complementado con suero de ternera fetal al 10 %; Invitrogen). Los linfocitos primarios se estimularon con microperlas recubiertas con anticuerpos estimulantes CD3 y CD28 (Life Technologies, Grand Island, NY, Catalog) como se describe (Barrett et al., 2009, Proc Nat Acad Sci USA, 106:3360). Las células T se crioconservaron el día 10 en una solución de suero de ternera fetal al 90 % y dimetilsulfóxido (DMSO) al 10 % a razón de 1 * 108 células/vial.
G eneración de construcciones CAR para e lectroporación de A R Nm y transducción lentiviral.
Los dominios CAR scFv contra ErbB2 o EGFR se sintetizaron y/o amplificaron mediante PCR, basándose en la información de secuenciación proporcionada por las publicaciones relevantes (Carter et al., 1992, Proc Nat Acad Sci USA, 89:4285; Zhou et al., 2007, J Mol Bio, 371:934), vinculado al dominio transmembrana CD8 y dominios de señalización intracelular 4-1BB y CD3Z, y subclonado en el vector basado en ARN pGEM.64A (Zhao et al., 2010, Cancer Res, 70:9053) o vectores lentivirales pTRPE (Carpenito et al., 2009, Proc Nat Acad Sci USA, 106:3360.
Ensayo Biacore
Se movilizó ErbB2 biotinilado a un chip sensor revestido con estreptavidina a una densidad de 200 RU. La afinidad de unión del anticuerpo 4D5 original (Carter et al., 1992, Proc Nat Acad Sci USA, 89:4285) se comparó con el scFv recombinante. La pureza y la masa atómica del scFv se verificaron mediante cromatografía líquida-espectrometría de masas. Las muestras de ScFv se diluyeron en serie 3 veces y se inyectaron sobre el chip a un caudal constante. Las tasas de asociación y disociación del complejo proteico se controlaron durante 270 s y 400 s, respectivamente. Se realizó una referencia doble contra una celda de flujo inmovilizada en blanco y un tampón en blanco y los datos se ajustaron utilizando un modelo de Langmuir 1:1 o afinidad de estado estacionario cuando correspondía con el software de evaluación Biacore T200.
Transcripción in vitro de AR Nm y e lectroporación de células T
Se utilizó el kjt de sistemas T7 mScript (CellScript) para generar ARN de IVT. Se sometieron a electroporación células T estimuladas con perlas CD3/CD28 con ARN de IVT utilizando BTX EM830 (Harvard Apparatus BTX) como se describió previamente (Zhao et al., 2010, Cancer Res, 70:9053). Brevemente, las células T se lavaron tres veces y se resuspendieron en OPTI-MEM (Invitrogen) a una concentración final de 1-3 * 108células/ml. Posteriormente, se mezclaron 0,1 ml de células con 10 ug de ARN de IVT (o como se indique) y se sometieron a electroporación en una cubeta de 2 mm.
A nálisis de citom etría de flujo
Los anticuerpos se obtuvieron de los siguientes proveedores: CD3 anti-humano (BD Biosciences, 555335), CD8 anti humano (BD Biosciences 555366), CD107a anti-humano (BD Biosciences 555801), CD137 anti-humano (BD Biosciences 555956). La expresión de ErbB2 en la superficie celular se detectó mediante aficuerpo anti-ErbB2 biotinilado (Abcam, ab31890), y EGFr mediante aficuerpo anti-EGFR conjugado con FITC (Abcam, ab81872). La expresión de células CAR T específicas para ErbB2, EGFR y CD19 se detectó mediante la proteína de fusión ErbB2-Fc (R&D system, 1129-ER), la proteína de fusión EGFR-Fc y los anticuerpos policlonales anti-ratón de cabra F(ab)2 marcados con biotina (Jackson Immunoresearch , 115-066-072) respectivamente, se incubaron a 4 °C durante 25 minutos y se lavaron dos veces (PBS con FBS al 2 %). A continuación, las muestras se tiñeron con Fc Ab IgG anti-humano conjugado con PE (eBioscience, 12-4998-82) o estreptavidina marcada con ficoeritrina (eBioscience, 17-4317-82), se incubaron a 4 °C durante 25 minutos
y se lavaron una vez. La adquisición de citometría de flujo se realizó en un citómetro BD FacsCalibur o Accuri C6 (BD Biosciences). El análisis se realizó utilizando el software FlowJo (Treestar).
Ensayos ELISA
Las células diana se lavaron y suspendieron a razón de 1* 106 células/ml en medio R10 (RPMI 1640 complementado con suero de ternera fetal al 10 %; Invitrogen). Se añadieron 100 ul de cada uno de los tipos de célula diana por duplicado a una placa de fondo redondo de 96 pocillos (Corning). Células T efectoras se lavaron y se resuspendieron a razón de 1* 106 células/ml en medio R10 y luego se combinaron 100 ul de células T con células diana en los pocillos indicados. Además, se prepararon pocillos que contenían células T solas. Las placas se incubaron a 37°C durante 18 a 20 horas. Después de la incubación, se recogió el sobrenadante y se sometió a un ensayo ELISA (eBioscience, 88-7316-77; 88 7025-77).
Tinción con CD107a
Las células se sembraron en placas a una E:T de 1: 1 (1 * 105 efectoras: 1 * 105 dianas) en 160 pl de medio RPMI completo en una placa de 96 pocillos. Se añadieron 20 pl de Ab anti-CD107a marcado con ficoeritrina (BD Biosciences, 555801) y la placa se incubó a 37 °C durante 1 hora antes de añadir Golgi Stop (2 ul de Golgi Stop en 3 ml de medio RPMI, 20 ul/pocillo; BD Biosciences, 51 -2092KZ) y se incubó durante otras 2,5 horas. Luego se añadieron 5 pl de FITC-anti-CD8 y 5 ul de APC-anti-CD3 y se incubaron a 37 °C durante 30 min. Después de la incubación, las muestras se lavaron con tampón FACS y se analizaron por citometría de flujo.
Ensayo de proliferación de células T basado en CFSE
Células T CD4 en reposo se lavaron y suspendieron a una concentración de 1 * 107 células/ml en PBS. A continuación, se añadieron 120 ul de solución de trabajo de CFSE (25 pM de CFSE) a razón de 1 * 107 células durante 3,5 min a 25 °C. El marcaje se detuvo con FBS al 5 % (en PBS), se lavó dos veces con FBS al 5 % y se cultivó en R10 con 10 UI/ml de IL2. Después del cultivo durante la noche, las células T marcadas con CFSE se sometieron a electroporación con ARN CAR de ErbB2 de diferente afinidad. De dos a cuatro horas después de la electroporación, las células T se suspendieron a una concentración de 1 * 106/ml en medio R10 (con 10 UI/ml de IL2). Las líneas celulares tumorales o K562 se irradiaron y suspendieron a razón de 1 * 106/mL en medio R10. Las células se sembraron en una E:T de 1:1 (5 * 105 efectoras: 5 * 105 dianas) en 1 ml de medio RPMI completo en una placa de 48 pocillos. Luego se contaron las células T y se alimentaron cada 2 días a partir del día 3. La dilución de CFSE se controló mediante citometría de flujo el día 3, el día 5 y el día 7.
Ensayo CTL basado en luciferasa.
Se generaron células tumorales Nalm6-CBG y se emplearon en una versión modificada de un ensayo CTL basado en luciferasa de la siguiente manera: se clonó luciferasa de escarabajo click verde (CBG) en el vector pELNS, se empaquetó en lentivirus, se transdujo en células tumorales NALM6 y se clasificó para la expresión de CBG. Las células Nalm6-CBG resultantes se lavaron y resuspendieron a razón de 1 * 105 células/ml en medio R10, y se incubaron 100 ul de células marcadas con CBG con diferentes relaciones de células T (p. ej., 30:1, 15:1, etc.) durante la noche a 37 °C. Se transfirieron 100 pl de la mezcla a una placa luminométrica blanca de 96 pocillos, se añadieron 100 pl de sustrato y se determinó inmediatamente la luminiscencia.
Estudios de xenoin jerto de ratón
Los estudios se realizaron como se describió previamente con determinadas modificaciones (Barrett et al., 2011, Human Gene Therapy, 22:1575 ; y Carpenito et al., 2009, PNAS, 106:336). Brevemente, a ratones No D scid gamma (NSG) de 6-10 semanas de edad se les inyectaron por vía subcutánea 1 * 106 células tumorales PC3-CBG en el flanco derecho el día 0 y a los mismos ratones se les administraron células tumorales SK-OV3-CBG (5 * 106 células/ratón, s.c.) en el flanco izquierdo el día 5. Los ratones fueron tratados con células T a través de la vena de la cola el día 23 después de la inoculación del tumor con PC3-CBG de manera que ambos tumores tenían un volumen de aproximadamente 200 mm3. Las células T transducidas lentiviralmente se administraron a razón de 1 * 107 células/ratón (10 M), o 3 * 106 células/ratón (3 M). Las células T electroporadas con ARN se administraron a razón de 5 * 107 células/ratón para el primer tratamiento, seguido de 3 tratamientos los días 26, 30 y 33 en la dosis de 1 * 107 células T electroporadas con ARN/ratón.
RESULTADOS
La reducción de la afinidad del scFv anti-ErbB2 m ejora el índice terapéutico de las células CA R T ErbB2 in vitro
Se compiló un panel de líneas tumorales con un amplio intervalo de expresión de ErbB2 medida por citometría de flujo (Fig. 1). SK-OV3 (cáncer de ovario), SK-BR3 (cáncer de mama), BT-474 (cáncer de mama) sobre-expresan ErbB2, mientras que EM-Meso (mesotelioma), MCF7 (cáncer de mama), 293T (célula embrionaria de riñón 293), A549 (cáncer de pulmón), 624mel (melanoma), PC3 (cáncer de próstata), MDA231 (cáncer de mama) expresan ErbB2 a niveles más
bajos y no se detectó ErbB2 en MDA468 (cáncer de mama). Los niveles de ARNm de ErbB2 también se midieron mediante PCR en tiempo real y hubo una fuerte correlación entre las dos técnicas (Fig. 2).
Se construyó un panel de CARs de ErbB2 haciendo uso de scFv derivado de las mutaciones publicadas del anticuerpo 4D5 parental (Carter et al. (1992) Proc Natl Acad Sci USA 89:4285-4289). Las secuencias que codifican los CARs contra ErbB2 se proporcionan en la Tabla 2.
Tabla 2 Secuencias de ácidos nucleicos que codifican CARs contra ErbB2




Las afinidades monovalentes de los scFv de ErbB2 variaron en aproximadamente 3 órdenes de magnitud (Tabla 3), en contraste con los anticuerpos mutantes correspondientes que retuvieron afinidades de unión dentro de 10 veces entre sí (Carter, P., et al.1992).
T l . m r i n l fini m i l ni r 4D i lv m n l Fv rr n i n

Los CARs se construyeron uniendo los diversos scFv al dominio transmembrana y bisagra CD8 alfa seguido de los dominios de señalización intracelular 4-1BB y CD3Z. Los CARs se expresaron mediante tecnología de vector lentiviral o
mediante clonación en un vector basado en ARN (Zhao et al., 2010, Cáncer Res, 70:9053). Después de la producción de ARNm mediante transcripción in vitro y electroporación en células T, la expresión en superficie del panel de CARs de ARN de ErbB2 modificado por afinidad fue similar (Fig. 3). Para comparar los umbrales de reconocimiento, el panel de células CAR T ErbB2 se estimuló con líneas celulares tumorales de expresión alta (SK-BR3, SK-OV3 y BT-474) o de expresión baja de ErbB2 (MCF7, 293T, A549, 624Mel, PC3, MDA231 y MDA468) y la activación de las células T se evaluó mediante la regulación al alza de CD137 (4-1BB; Fig. 4), la secreción de IFN-y (Fig. 5) e IL-2 (Fig. 6) y la inducción de la expresión de CD107a en la superficie (Fig. 7). Las células T que expresan un CAR específico para CD19 sirvieron como control para la reactividad alogénica. Las células CAR T de afinidad más baja (4D5-5 y 4D5-3) fueron fuertemente reactivas a los tumores con expresión amplificada de ErbB2 y exhibieron una reactividad indetectable o baja a las líneas tumorales que expresaban ErbB2 a niveles más bajos. Por el contrario, las células CAR T de mayor afinidad (4D5 y 4D5-7) mostraron una fuerte reactividad a las líneas tumorales que expresan niveles altos y bajos de ErbB2, como lo demuestra la regulación positiva de CD137, la secreción de citoquinas y la translocación de CDl07a. Estos resultados se ampliaron analizando líneas celulares adicionales que expresan ErbB2 (Fig. 8 y 9). Curiosamente, las células CAR T de mayor afinidad secretaron niveles más altos de IFN-y e IL-2 cuando se expusieron a dianas que expresaban niveles bajos de ErbB2, mientras que las células CAR T de afinidad más baja secretaron más citoquinas cuando se expusieron a células que expresaban niveles altos de diana (Fig. 5.y 6). Como era de esperar, el CAR CD19-BBZ no fue reactivo contra las líneas celulares que expresan ErbB2. En resumen, las células T 405-BBZ de mayor afinidad reconocieron todas las líneas que expresan ErbB2 analizadas, mientras que los CARs con scFv de menor afinidad, 405-5-BBZ o 4D5-3-BBZ, fueron altamente reactivos a todas las líneas tumorales con ErbB2 sobre-expresado, pero mostraron reactividad insignificante a líneas celulares que expresan niveles bajos o indetectables de ErbB2.
Los CA R de ErbB2 con scFv de m enor afinidad discrim inan entre células tum orales que expresan niveles bajos y altos de ErbB2.
Para excluir cualquier efecto específico del tumor que pudiera contribuir a los resultados anteriores, se ensayó la actividad del panel de células CAR T ErbB2-BBZ frente a una única línea tumoral que expresaba niveles variables de ErbB2 (células K562 electroporadas con cantidades variables de ARN de ErbB2). De acuerdo, se observó que células T que expresan scFv de mayor afinidad (4D5 y 4D5-7) reconocieron las células K562 electroporadas con ARN de ErbB2 a dosis tan bajas como 0,001 |jg, que es 100 veces menor que el nivel detectable por citometría de flujo de 0,1 |jg de ARNm. (Fig. 10, 11A y 11B). Por el contrario, los CARs con scFv de menor afinidad (4D5-5 y 4D5-3) solo reconocieron el ARN de ErbB2 electroporado con K562 en dosis de 0,5 jg (4D5-5; 10) o más, lo que indica que la sensibilidad de las células CAR T disminuyó en 2000 (4D5-3) a 500 veces (4D5-5) en comparación con las células CAR T 4D5 de alta afinidad. Además, la reactividad asociada a la dosis de antígeno observada con los CARs de ErbB2 de menor afinidad (4D5-5 y 4D5-3; Fig. 11A y 11B) se confirmó realizando un ensayo de proliferación basado en CFSE (Fig. 12). Curiosamente, al disminuir 5 veces la dosis de ARN de CAR (de 10 jg de ARN/100 j l de células T a 2 jg de ARn /100 j l de células T), aumentó adicionalmente el umbral de reconocimiento de antígenos de las células CAR T de baja y alta afinidad según lo evaluado por la secreción de citoquinas, lo que sugiere que el ajuste fino de la densidad de CAR en la superficie de las células T es una variable importante, o que las dosis superiores a 2 jg de ARNm pueden tener cierta toxicidad en la actividad general de las células T.
Se utilizó un ensayo de células T citolíticas (CTL, por sus siglas en inglés) basado en luciferasa para determinar si las células T con CARs disminuidos por afinidad podían mantener una potente actividad exterminadora contra dianas que sobre-expresan ErbB2 mientras se preservan las células que expresan niveles más bajos de ErbB2. Cuando las células diana Nalm6 se transfectaron con 10 jg de ARN de ErbB2, las células T con CARs ErbB2 de mayor o menor afinidad lisaron eficazmente las células diana (Fig. 13A). CARs con scFv de mayor afinidad (4D5 y 4D5-7) exhiben una potente actividad lítica contra las células diana transfectadas con 1 jg de ARN de ErbB2, pero los scFv de menor afinidad (4D5-5 y 4D5-3) mostraron una actividad letal disminuida (Fig. 13B). Finalmente, solo los CARs con scFvs de mayor afinidad pudieron matair las células diana que expresaban cantidades muy bajas de diana después de la electroporación con 0,1 jg de ARN de ErbB2 (Fig. 13C). Dado que Nalm6 es una línea celular positiva para c D19, CART19 mantuvo la actividad citolítica independientemente de los niveles de ARN de ErbB2 transfectado. Estos datos indican que el ajuste fino de la afinidad de las células CAR T ErbB2 potencia la discriminación del tumor que sobre-expresa ErbB2 de las células tumorales que tienen niveles bajos o indetectables de expresión de ErbB2.
Dism inución de la afinidad de células CAR T de ErbB2 C por reconocer niveles fisiológicos de ErbB2
Dado el evento adverso grave anterior que se produjo tras la administración de CAR de ErbB2 de alta afinidad que incorporó el scFv del anticuerpo trastuzumab 4D5 original (Morgan et al., 2010, Mol Therapy, 18:843), es de suma importancia para evaluar la reactividad potencial de las células CAR T de ErbB2 de afinidad reducida a niveles fisiológicos de expresión de ErbB2. Para solucionar este problema, se analizó la expresión de ErbB2 en siete líneas celulares primarias aisladas de diferentes órganos. La mayoría de las líneas primarias tenían niveles detectables de ErbB2 en superficie, y la línea progenitora neural expresaba los niveles más altos de ErbB2 (Fig. 14). Las células T que expresan CAR de 4D5 de alta afinidad fueron fuertemente reactivas con todas las líneas primarias testadas, como lo demuestran los niveles de regulación positiva de CD107a (Fig. 15). Sin embargo, las células T que expresan la afinidad disminuida de CARs de ErbB2 4D5-5 y 4D5-3 no exhibieron reactividad a las líneas primarias con la excepción de una reactividad
débil a la línea progenitora neural. Estos resultados se confirmaron mediante el análisis de un panel más grande de líneas celulares que tenían niveles bajos o indetectables de ErbB2 mediante citometría de flujo (Figs. 8 y 9).
Efectos com parables con CARs de ErbB2 a justados por afinidad expresados m ediante transducción lentiviral o electroporación de ARN
Para establecer la comparabilidad entre las células T que expresan CARs de forma permanente mediante transducción lentiviral con células CAR T electroporadas con ARNm, el panel de CARs ajustados por afinidad se expresó en células T del mismo donante normal utilizando transducción lentiviral o electroporación de ARNm (Fig. 16A). Las células T se estimularon con líneas de células tumorales (Fig. 16B) o células K562, que expresaban cantidades variables de ErbB2 (Fig. 16C). El reconocimiento y la activación de las células CAR T se controlaron mediante la regulación al alza de CD107a (Figs. 17 y 18), la regulación al alza de CD137 (Fig. 19) y la secreción de IFN-y (Figs. 20 y 21). De acuerdo con los resultados anteriores de células CAR T con ARNm de ErbB2, las células T que expresaban constitutivamente CAR de alta afinidad mostraron una fuerte reactividad con todas las líneas celulares que expresan ErbB2; no se observó correlación entre los niveles de expresión del antígeno y la actividad de las células T. Por el contrario, las células T con CARs de baja afinidad expresadas mediante tecnología lentiviral demostraron una sólida correlación entre la expresión y la activación del antígeno diana (Figs. 17, 18, 20 y 21). Estos resultados confirman que la sensibilidad del reconocimiento del antígeno ErbB2 depende de la afinidad de scFv utilizando células CAR T transducidas con ARNm tanto electroporadas como lentivirales.
Dism inución de la afinidad de las células C AR T de ErbB2 que elim inan el tum or in vivo e ignoran los tejidos que expresan niveles fis iológicos de ErbB2
Para ampliar los resultados in vitro anteriores, se realizaron una serie de experimentos en ratones NSG con xenoinjertos tumorales vascularizados avanzados. Basándose en los datos anteriores en la Figura 1, la línea celular de cáncer de ovario humano SK-OV3 se seleccionó como un tumor representativo que sobre-expresa ErbB2 y PC3, una línea de cáncer de próstata humano, se eligió para modelar los niveles de ErbB2 en tejido normal. La eficacia antitumoral de las células CAR T de ErbB2 que expresan el scFv 4D5 de alta afinidad o el scFv 45D-5 de baja afinidad en ratones NSG se comparó con los tumores del flanco SK-OV3 establecidos el día 18 (Fig. 22). Las imágenes de bioluminiscencia en serie revelaron que las células CAR T de alta y baja afinidad dieron como resultado la rápida eliminación de los tumores.
Para evaluar adicionalmente el índice terapéutico de las células CAR T ErbB2 de baja afinidad in vivo, se diseñó un modelo de ratón para comparar simultáneamente la eficacia y la toxicidad tisular normal de los CARs de ErbB2 de alta afinidad (4D5:BBZ) y baja afinidad (4D5-5 :BBZ). Las líneas de células tumorales SK-OV3 y PC3 se inyectaron por vía subcutánea en los flancos opuestos del mismo ratón NSG y se administraron células T cuando los volúmenes del los tumores alcanzaron aproximadamente 200 mm3. A los ratones se les inyectaron (i.v.) 3*10® o 1*107 células CAR T el día 22 y se realizaron evaluaciones de tamaño del tumor y formación de imágenes de bioluminiscencia en serie. Los ratones tratados con cualquiera de las dosis de las células c A r T exhibieron una regresión casi completa del tumor SK-OV3 que sobre-expresa ErbB2 (Figs. 23, 24 y 25). Además, también se observó una regresión casi completa del tumor PC3 que expresa ErbB2 a niveles bajos en el flanco opuesto para los ratones tratados con células CAR T basadas en 4D5 de alta afinidad. Por el contrario, el crecimiento progresivo del tumor de PC3 se observó en los ratones tratados con células CAR T basadas en 4D5-5 de baja afinidad, lo que indica que mientras que las células CAR T de menor afinidad fueron eficaces contra el tumor que sobre-expresa ErbB2, muestran una reactividad limitada o no detectable contra células que expresan ErbB2 a niveles fisiológicos. Además, se observó la eliminación selectiva del tumor en ratones tratados con dosis altas y bajas de células CAR T. Los efectos anteriores no se debieron al alorreconocimiento porque se observó un crecimiento progresivo de ambos tumores en ratones tratados con células T transducidas de forma simulada.
El ajuste de afinidad de scFv aum enta el índice terapéutico de las células CA R T de EGFR
Para testar la aplicabilidad más amplia de la estrategia para ajustar la afinidad del scFv, los autores de la invención evaluaron un panel de CARs de EGFR. Los CARs de EGFR:BBZ se construyeron a partir de scFv derivado del anticuerpo C10 anti-EGFR humano parental (Heitner et al., 2001, J Immunol Methods, 248:17-30). Las secuencias de ácido nucleico que codifican los CARs de EGFR se proporcionan en la Tabla 4.
Tabla 4 Secuencias de Ácido Nucleico de CARs de EGFR Eem lares

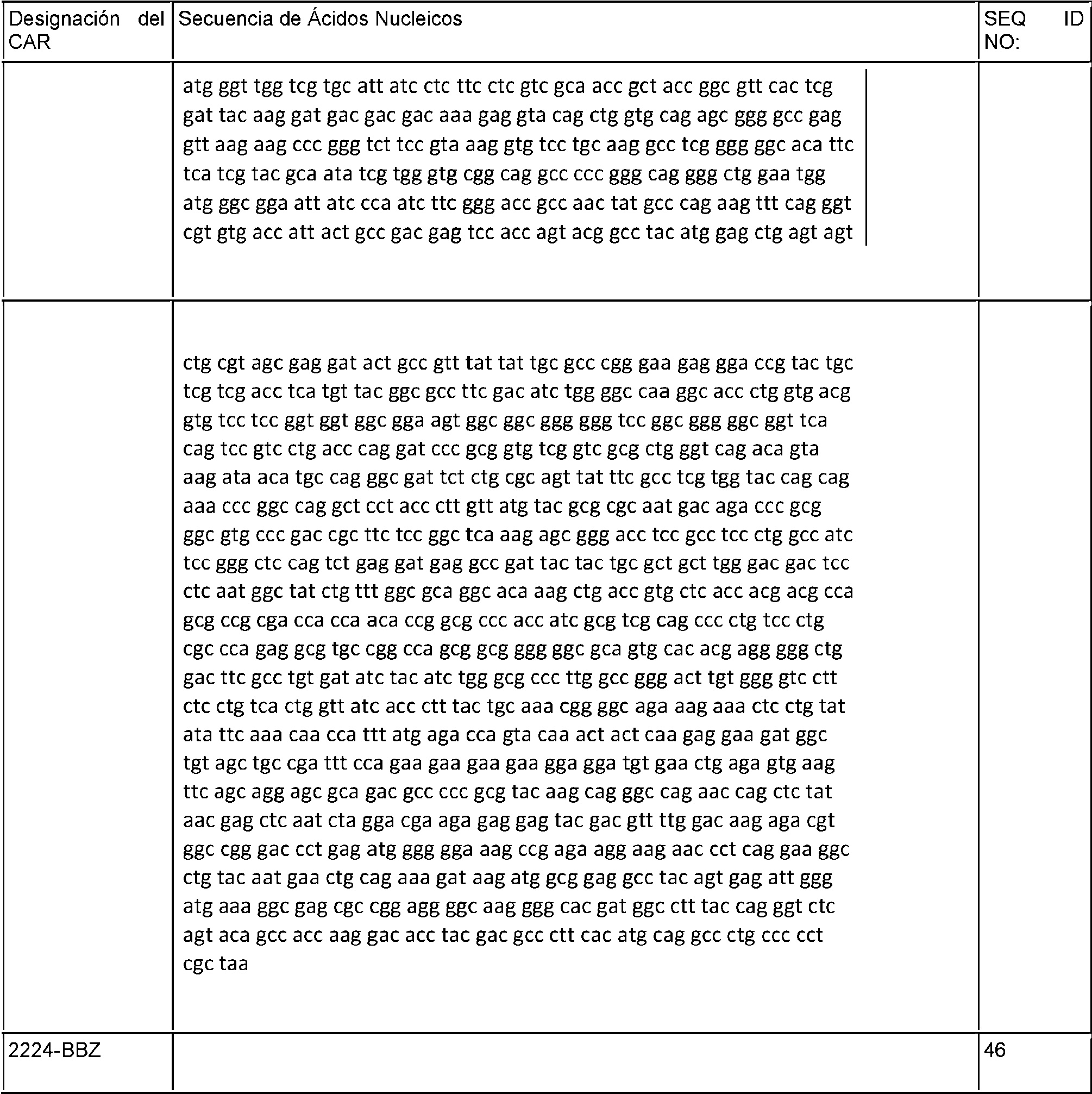



Las afinidades monovalentes del panel de scFvs específicos para EGFR variaron en un intervalo de aproximadamente 300 veces (Zhoe et al., 2007, J Mol Biol, 371:934). Los scFvs 2224, P2-4, P3-5 y C10 se clonaron en un vector basado
en ARN y se transcribieron in vitro para electroporación de ARNm de células T. Se ensayaron los niveles de expresión en superficie de CAR y se encontró que eran similares entre las construcciones de EGFR (Fig. 26A). Para comparar las reactividades del panel de CARs de EGFR, se estimularon células CAR T con líneas de células tumorales que expresan EGFR que tienen una amplia gama de expresión de EGFR en la superficie celular (Fig. 26B). La activación de las células CAR T se evaluó mediante los niveles de regulación al alza de CD107a; los datos se resumen en la Fig. 27. . Los CARs de EGFR de mayor afinidad (2224:BBZ y P2-4:BBZ) respondieron a todas las líneas tumorales positivas para EGFR (MDA468, MDA231 y SK-OV3) independientemente de los niveles de expresión de EGFR (Fig. 27). Sin embargo, la reactividad exhibida por los CARs de EGFR de menor afinidad (P3-5.BBZ y C10.BBZ) contra las líneas tumorales que expresan EGFR se correlacionó con los niveles de expresión de EGFR. Además, los CARs de EGFR de menor afinidad mostraron una reactividad más potente al tumor que sobre-expresa EGFR, MDA468, que los CARs de EGFR de mayor afinidad, mientras que provocaban una respuesta mucho más débil a las células de expresión baja de EGFR (Fig. 27).. Ninguna de las células CAR T de EGFR reaccionó a la línea tumoral negativa para EGFR K562
Para confirmar que el nivel de respuesta estaba relacionado con la afinidad de scFv y el nivel de expresión de EGFR, y para excluir los efectos específicos del tumor, el panel de células CAR T de EGFR se co-cultivó con células K562 que expresan niveles variables de EGFR después electroporación con ARNm de EGFR (Fig. 28). Los CARs de EGFR de mayor afinidad no discriminaron entre células diana con diferentes niveles de expresión de EGFR (Fig. 29). Por ejemplo, células T que expresan CAR 2224 respondieron igualmente bien a las células K562 sometidas a electroporación con una diferencia de 200 veces en el ARNm de EGFR (0,1 |jg a 20 |jg). Sin embargo, de acuerdo con los resultados de CAR de ErbB2 anteriores, los CARs de EGFR de menor afinidad (P3-5 y C10) exhibieron una alta correlación entre las respuestas de las células T y los niveles de expresión de EGFR; los datos se resumen en la Fig. 29.
Para confirmar el perfil de seguridad incrementado de los CARs de EGFR de menor afinidad, los autores de la invención testaron las reactividades de los CARs de EGFR frente a células primarias derivadas de diferentes órganos. Se analizaron cinco líneas de células primarias y cinco líneas de células tumorales tanto para los niveles en superficie de EGFR (Fig. 30) como para la capacidad de desencadenar la reactividad de las células CAR T (Fig. 31). Tres de las líneas celulares primarias examinadas expresan niveles detectables de EGFR y dos no (músculo liso de la arteria pulmonar y PBMC). Dos de las líneas de células tumorales (MCF7 y Raji) no expresaron EGFR detectable en la superficie celular. Al comparar las células CAR T de EGFR con las células c A r T de Cd 19, las células CAR T de mayor afinidad por EGFR (2224 y P2-4) reaccionaron con todas las líneas primarias analizadas y todos los tumores excepto Raji (Fig. 31). Sin embargo, las células T con la afinidad disminuida de las células CAR T de EGFR P3-5 y C10 no fueron reactivas a ninguna de las cinco células primarias analizadas (Fig. 31). Células CAR T específicas para CD19 reaccionaron con la línea Raji CD19+ y con las PBMCs, presumiblemente con las células B en PBMC, pero no respondieron a ninguna de las líneas tumorales ni a otras líneas celulares primarias. Estos datos demuestran que el ajuste de la afinidad de scFv puede aumentar el índice terapéutico para las células CAR T que fijan como objetivo ErbB2 o EGFR.
ANÁLISIS
La eficacia de las células CAR T está dictada en parte por la expresión diferencial del antígeno diana en tejido tumoral frente a tejido normal. Los resultados arriba descritos demuestran que los CARs con toxicidades severas conocidas en la diana pueden rediseñarse mediante el ajuste de afinidad, conservando una potente eficacia in vivo, al tiempo que eliminan o reducen la toxicidad. En particular, el CAR de 4D5 basado en trastuzumab tuvo toxicidad letal (Morgan et al., 2010, Mol Ther, 18:843) debido al reconocimiento de niveles fisiológicos de ErbB2 expresados en tejidos cardiopulmonares (Press et al., 1990, Oncogene, 5:953). Se demostró que al reducir la Kd de scFv empleada en las células CAR T en 2 a 3 log, se mostró una mejora sustancial en el índice terapéutico para las células CAR T de ErbB2 y EGFR. Las células CAR T con scFv de menor afinidad mostraron una actividad antitumoral igualmente sólida contra los tumores que sobre-expresan ErbB2 en comparación con los CARs de alta afinidad, pero mostraron poca reactividad contra los niveles fisiológicos de ErbB2.
CARs específicos para los antígenos CD19 y CD20 del linaje de células B han sido testados por una diversidad de grupos y han mostrado una potente eficacia en los tumores malignos de células B (Maus et al., 2014, Blood, 123:2625). Sin embargo, en tumores sólidos, con la excepción de isoformas específicas para el tumor como EGFRviii (Morgan et al., 2012, Human Gene Therapy), se prevé que la toxicidad en la diana sea una limitación grave para las células CAR T. Se espera que esta limitación sea más grave con los CARs que con las terapias con anticuerpos que utilizan anticuerpos intactos o conjugados de anticuerpos y fármacos, debido al límite más bajo de sensibilidad a la diana para las células CAR T en comparación con las terapias basadas en anticuerpos que difieren en varios órdenes de magnitud. Los estudios actuales que utilizan células diana electroporadas con ARNm de ErbB2 o EGFR son consistentes con estudios previos que indican que las células CAR T pueden reconocer células tumorales con ~100 dianas por célula (Stone et al., 2012, Oncoimmunology, 1:863). Por el contrario, la amplificación de ErbB2 se produce en aproximadamente el 20 % al 25 % de los cánceres de mama humanos primarios y normalmente da como resultado una sobre-expresión de la proteína ErbB2 en > 1 millón de copias por célula (Robertson et al., 1996, Cancer Res, 56:3823; y Vogel et al., 2002, J Clin Oncol, 20:719). Actualmente, los datos disponibles indican que las células cancerosas no pierden la expresión de ErbB2 cuando se vuelven refractarias a las terapias dirigidas por ErbB2 (Ritter et al., 2007, Clin Cancer Res, 13:4909).
Estos hallazgos respaldan el trabajo previo de Chmielewski (Chmielewski et al., 2004, J Immunol, 173:7647), lo que sugiere que los CARs de alta afinidad exhiben menos discriminación entre células diana con niveles de expresión diana altos o bajos. Sin embargo, los resultados actuales difieren de los de Chmielewski y colaboradores en que ninguno de los CARs de mayor afinidad (con una Kd que varía de 15 pM a 16 nM) en su informe fueron reactivos a las células con bajo nivel de expresión de ErbB2 y su CAR de menor afinidad que solo reconoció tumores. con ErbB2 amplificado mostró una reducción sustancial en la eficacia de las células T en comparación con los CARs de mayor afinidad. Por el contrario, se encontró que el CAR de ErbB2 utilizando el scFv 4DF5 con una Kd a 0,3 nM era fuertemente reactivo con los queratinocitos e incluso con las líneas celulares transfectadas con cantidades extremadamente bajas de ARNm de ErbB2 que estaban 100 veces por debajo de los niveles detectables, mientras que las células CAR T ajustadas por afinidad conservaron la reactividad a los tumores amplificados con ErbB2 que fue al menos tan potente como el CAR de alta afinidad, tanto in vitro como en modelos de tumores de ratón agresivos. Algunas variables que pueden explicar estas diferencias incluyen el uso de diferentes scFvs (C5.6 frente a 4D5) que pueden reconocer diferentes epítopos, una configuración distinta del dominio de señalización CAR (zeta solo frente a 4-1BB-zeta) y diferentes enfoques de transferencia de genes (transducción retroviral) frente a electroporación de ARN o transducción lentiviral) que afectan los niveles de expresión de la superficie CAR en las células T. Juntos, esto sugiere que cada uno de estos factores debe tenerse en cuenta al seleccionar la afinidad de un CAR en situaciones clínicas relevantes.
Los hallazgos descritos en este ejemplo también demostraron la importancia de seleccionar la afinidad correcta para un CAR que fija como objetivo un antígeno asociado a tumor (TAA) particular. Los resultados in vitro e in vivo fueron consistentes entre sí, lo que demuestra que los CARs que tienen menor afinidad eran al menos tan potentes como los CARs de alta afinidad (tanto para las células CAR T transducidas lentiviralmente como electroporadas con ARN), pero tenían un impacto mínimo en las células que tenía una baja expresión de un TAA (ErbB2), que representa tejido normal. Por el contrario, los CARs con alta afinidad eran más reactivos con las células TAA de baja expresión que representan el tejido normal. Por lo tanto, los CARs con alta afinidad pueden no ser los preferidos para los cánceres en los que el TAA también se expresa en tejido normal, ya que estos resultados demuestran que los CARs con alta afinidad también pueden fijar como objetivo tejidos normales y, por lo tanto, provocarían efectos secundarios adversos. Tomados en conjunto, estos resultados indican que la afinidad de los CARs debe considerarse con respecto a la naturaleza del cáncer (p. ej., si el TAA se expresa solo en células cancerosas o si el TAA se expresa más en células cancerosas, pero también se expresa en un nivel bajo en tejido normal) por razones de potencia y seguridad.
El advenimiento de estrategias de transferencia adoptiva más potentes ha impulsado una re-evaluación de los objetivos considerados previamente como seguros utilizando estrategias inmunoterapéuticas más débiles (Hinrichs et al., 2013, Nature Biotechnology, 31:999). Las estrategias para maximizar el índice terapéutico de las células CAR T incluyen la selección de objetivos, el diseño de CAR, la fabricación de células y las técnicas de transferencia de genes. Además del ajuste de la afinidad, otras estrategias que se están desarrollando para controlar la toxicidad en la diana incluyen el uso de enfoques de células CAR T duales (Kloss et al., 2013, Nat Biotech, 31:999; y Lanitis et al., 2013, Cancer Immunol Res, 1:43), deleción condicional y sistemas suicidas (Di Stasi et al., 2011, NEjM, 365:1673; y Wang et al., 2011, Blood, 118:1255) e infusiones repetidas de células T que tienen CARs de ARNm que tienen expresión transitoria y toxicidad autolimitante (Beatty et al., 2014, Cancer Immunol Res, 2:112).
Estos resultados demuestran que el ajuste de afinidad puede aumentar el índice terapéutico para ErbB2 y EGFR. Además de la afinidad de scFv, otras variables que requieren examen para aumentar el índice terapéutico para otras dianas incluyen la ubicación del epítopo diana, la longitud de la bisagra y la naturaleza del dominio de señalización (Hudecek et al., 2013, Clin Cancer Res, 15:5323; y Guedan et al., 2014, Blood, 124:1070).
En resumen, ErbB2 y EGFR se han considerado previamente como dianas no susceptibles de tratamiento para las células CAR T. Dado que la desregulación de la expresión de ErbB2 y EGFR ocurre con frecuencia en múltiples carcinomas humanos, incluyendo los de mama, glioblastoma, pulmón, páncreas, ovario, cáncer de células escamosas de cabeza y cuello y cáncer de colon, estos hallazgos tienen una importancia clínica considerable. Esta estrategia de ajuste de afinidad tiene el potencial no solo de mejorar el perfil de seguridad y el resultado clínico de los CARs dirigidos contra dianas validadas, sino también de ampliar el panorama a dianas que antes no eran susceptibles de tratamiento con células CAR T debido a las toxicidades en la diana. De manera más general, estos hallazgos sugieren que el ajuste de afinidad sugiere que se pueden diseñar CARs más seguros y potentes empleando scFvs de afinidad reducida para una diversidad de carcinomas comunes.
Ejemplo 5: Efectos de la inhibición de mTOR sobre la inmunosenescencia en ancianos
Una de las vías más claramente relacionadas con el envejecimiento es la vía mTOR. Se ha demostrado que el inhibidor de mTOR rapamicina prolonga la vida útil en ratones y mejora una diversidad de afecciones relacionadas con el envejecimiento en ratones viejos (Harrison, DE et al. (2009) Nature 460:392-395; Wilkinson JE et al. (2012) Aging Cell 11:675-682 y Flynn, JM et al (2013) Aging Cell 12:851-862). Por lo tanto, estos hallazgos indican que los inhibidores de mTOR pueden tener efectos beneficiosos sobre el envejecimiento y las afecciones relacionadas con el envejecimiento en seres humanos.
Un fenotipo relacionado con la edad que se puede estudiar en un breve período de tiempo de ensayo clínico es la inmunosenescencia. La inmunosenescencia es la disminución de la función inmunitaria que se produce en los ancianos, lo que conduce a una mayor susceptibilidad a las infecciones y una menor respuesta a la vacunación, incluida la vacunación contra la gripe. La disminución de la función inmunitaria con la edad se debe a una acumulación de defectos inmunitarios, incluyendo una disminución de la capacidad de las células madre hematopoyéticas (HSCs) de generar linfocitos que no han sido tratados, y un aumento en el número de linfocitos PD-1 positivos agotados que tienen respuestas defectuosas a la estimulación antigénica (Boraschi, D et al. (2013) Sci. Transl. Med.5:185ps8; Lages, CS et al. (2010) Aging Cell 9:785-798 ; y Shimatani, K et al., ( 2009) Proc. Natl. Acad. Sci. USA 106:15807-15812). Estudios en ratones de edad avanzada demostraron que 6 semanas de tratamiento con el inhibidor de mTOR, rapamicina, rejuvenecieron la función de las HSC, lo que condujo a una mayor producción de linfocitos que no han sido tratados, una mejor respuesta a la vacunación contra la gripe y una mayor esperanza de vida (Chen, C et al. (2009) Sci. Signal. 2 .2:ra75).
Para evaluar los efectos de la inhibición de mTOR en los fenotipos humanos relacionados con el envejecimiento y si el inhibidor de mTOR RAD001 mejora la inmunosenescencia, se evaluó la respuesta a la vacuna contra la gripe en voluntarios de edad avanzada que recibieron RAD001 o placebo. Los hallazgos presentados aquí sugieren que RAD001 potenció la respuesta a la vacuna contra la gripe en voluntarios de edad avanzada en dosis que fueron bien toleradas. RAD001 también redujo el porcentaje de linfocitos T CD4 y CD8 positivos de muerte programada (PD)-1 que se acumulan con la edad. Estos resultados demuestran que la inhibición de mTOR tiene efectos beneficiosos sobre la inmunosenescencia en voluntarios de edad avanzada.
Como se describe en esta memoria, un tratamiento de 6 semanas con el inhibidor de mTOR RAD001, un análogo de la rapamicina, mejoró la respuesta a la vacunación contra la gripe en voluntarios humanos de edad avanzada.
Métodos
Población de estudio
Voluntarios mayores >= 65 años de edad sin enfermedades médicas subyacentes inestables se inscribieron en 9 lugares en Nueva Zelanda y Australia. Los criterios de exclusión en la selección incluyeron hemoglobina < 9,0 g/dL, recuento de glóbulos blancos < 3500/mm3, recuento de neutrófilos < 2000/mm3 o recuento de plaquetas < 125000/mm3, diabetes no controlada, cardiopatía isquémica inestable, enfermedad subyacente clínicamente significativa enfermedad pulmonar, antecedentes de inmunodeficiencia o tratamiento inmunosupresor, antecedentes de coagulopatía o afección médica que requiera anticoagulación a largo plazo, tasa de filtración glomerular estimada < 30 ml/min, presencia de hipercolesterolemia grave no controlada (>350 mg/dL, 9,1 mmol/L) o hipertrigliceridemia (>500 mg/dL, 5,6 mmol/L).
La demografía inicial entre los brazos de tratamiento fue similar (Tabla 4). De los 218 sujetos inscritos, 211 completaron el estudio. Siete sujetos se retiraron del estudio. Cinco sujetos se retiraron debido a eventos adversos (AE), un sujeto retiró el consentimiento y un sujeto abandonó el estudio como resultado de una violación del protocolo.
Tabla 4 Características demográficas y Basales de los Pacientes del Estudio
Población RAD001 0,5 mg RAD001 5 mg RAD001 20 mg Placebo diarios N=53 semanales N=53 semanales N=53 agrupado

N=59 Edad (Años) Media (DE) 70,8 (5,0) 72,0 (5,3) 71,4 (5,2) 71,1 (5,1)  71,3 (5,2) Género Masculino- n 34 (64%) 27 (51%) 32 (60%) 31 (53%) 124 (57%)
71,3 (5,2) Género Masculino- n 34 (64%) 27 (51%) 32 (60%) 31 (53%) 124 (57%)
(%)

BMI* (kg/m2) Media (DE) 27,4 (4,2) 28,8 (5,0) 28,0 (4,1) 28,0 (4,2) 28,0 (4,4)
Raza-n (%) Caucásica 48 (91%)  50 (94%) 46 (87%)
50 (94%) 46 (87%)  54 (92%) 198 (91%)
54 (92%) 198 (91%)
Otras 5(9%) 3 (6%) 7 (13%) 5 (8%)  20 (9%)
20 (9%)
* El índice de masa corporal es el peso en kilogramos dividido por el cuadrado de la altura en metros 
Diseño y Realización del Estudio
Desde diciembre de 2011 hasta abril de 2012, 218 voluntarios de edad avanzada se inscribieron en un ensayo aleatorizado, ciego para el observador y controlado con placebo. Los sujetos fueron aleatorizados a los brazos de tratamiento utilizando un sistema de aleatorización automatizado validado con una relación de RAD001 a placebo de 5:2 en cada brazo de tratamiento. Los brazos de tratamiento fueron:
RAD001 0,5 mg diarios o placebo
RAD001 5 mg semanales o placebo
RAD001 20 mg semanales o placebo
El ensayo fue ciego para el observador porque el placebo en las cohortes de 0,5 mg diarios y 20 mg semanales de RAD001 difería ligeramente de los comprimidos de RAD001 en esas cohortes. El personal del estudio que evaluó a los sujetos no vio el medicamento del estudio y, por lo tanto, estaba totalmente cegado. La duración del tratamiento para todas las cohortes fue de 6 semanas durante las cuales los sujetos se sometieron a evaluaciones de seguridad en la clínica cada 2 semanas. Después de que los sujetos habían recibido la dosis durante 4 semanas, se midieron los niveles de estado estacionario de RAD001 antes de la dosis y una hora después de la dosis. Después de completar el curso de 6 semanas del fármaco del estudio, los sujetos recibieron un descanso de 2 semanas sin fármaco para revertir cualquier posible inmunosupresión inducida por RAD001, y luego recibieron una vacuna contra la gripe estacional de 2012 (Agrippal®, Novartis Vaccines and Diagnostics, Siena, Italia) que contenía las cepas H1N1 A/California/07/2009, H3N2 A/Victoria/210/2009, B/Brisbane/60/2008. Cuatro semanas después de la vacunación contra la gripe, se recogió suero a los sujetos para medir los títulos de gripe. Los títulos de anticuerpos para las 3 cepas de vacunas contra la gripe, así como para 2 cepas heterólogas (A/H1N1 cepa A/New Jersy/8/76 y A/H3N2 cepa A/Victoria/361/11) se midieron mediante un ensayo estándar de inhibición de la hemaglutinación (Kendal , A p et al. (1982) Concepts and procedures for laboratorybased influenza surveillance. . Atlanta: Centers for Disease Control and Prevention B17-B35). Los niveles de IgG e IgM específicos para A/H1N1/California/07/2009 se midieron en muestras de suero tomadas antes y 4 semanas después de la vacunación contra la gripe como se describió previamente (Spensieri, F. et al. (2013) Proc. Natl. Acad . Sci. USA 110:14330-14335). Los resultados se expresaron como intensidad de fluorescencia.
Todos los sujetos dieron su consentimiento informado por escrito. El estudio se realizó de acuerdo con los principios de Buenas Prácticas Clínicas y fue aprobado por los Comités de Ética y las Agencias Reguladoras correspondientes.
Seguridad
La evaluación de eventos adversos y la recogida de sangre para las evaluaciones de seguridad hematológica y bioquímica se realizaron durante las visitas del estudio. La información sobre eventos adversos también se recopiló en diarios que los sujetos completaron en casa durante las 6 semanas que estuvieron tomando el fármaco del estudio. Los datos sobre todos los eventos adversos se recopilaron desde el momento del consentimiento informado hasta 30 días después de la última visita del estudio. Los investigadores clasificaron los eventos como leves, moderados o graves.
Análisis Estadístico
El análisis principal de las relaciones de títulos de la media geométrica se realizó utilizando un modelo de regresión bayesiano normal con antecedentes no informativos. Este modelo se ajustó a cada uno de los títulos de anticuerpo en la escala logarítmica. El resultado primario en cada modelo fue la medición del Día 84. La medición del Día 63 se incluyó en el vector de resultados. El modelo se ajustó con el proceso SAS 9.2 mezclado con la declaración anterior. La estructura de covarianza de la matriz se consideró como no estructurada (tipo de opción=UN). Se utilizó un plano anterior. Para el análisis secundario de las tasas de seroconversión se utilizó la regresión logística.
La población con intención de tratar se definió como todos los sujetos que recibieron al menos una dosis completa del fármaco del estudio y que no tuvieron desviaciones importantes del protocolo que afectaran los datos de eficacia. 199 del total de 218 sujetos inscritos en el estudio estaban en la población por intención de tratar.
Inmunofenotipado
Se aislaron células mononucleares de sangre periférica a partir de sangre total recogida en 3 momentos: inicio; después de 6 semanas de tratamiento con el fármaco del estudio; y al final del estudio cuando los sujetos habían estado sin el fármaco del estudio durante 6 semanas y 4 semanas después de la vacunación contra la gripe. Setenta y seis subconjuntos de PBMC se analizaron mediante citometría de flujo utilizando paneles de inmunofenotipado de 8 colores en el Human Immune Monitoring Center de la Universidad de Stanford, CA, EE. UU. como se describe previamente (Maecker, HT et al. (2012) Nat Rev Immunol. 12:191-200). Setenta y seis subconjuntos de PBMC se analizaron mediante citometría de flujo utilizando paneles de inmunofenotipificación liofilizados de 8 colores (BD Lyoplate, BD Biosciences, San Diego, CA). Se incluyeron en el análisis muestras de PBMC con una viabilidad > 80 % y un rendimiento de 2*106 células o más.
Se calcularon los cambios relativos de los inmunofenotipos desde el inicio hasta la semana 6 del tratamiento con el fármaco del estudio y desde el inicio hasta el final del estudio (Semana 12) para cada una de las cohortes de dosificación de RAD001. Se realizó el test T de Student para examinar si el cambio relativo de los inmunofenotipos desde el inicio hasta los dos puntos de muestreo de sangre fue significativamente diferente de cero, respectivamente, dentro de cada grupo de dosificación después de ajustar el efecto placebo. No se realizó la imputación de datos que faltaban en el análisis del efecto del tratamiento. Por lo tanto, si a un paciente le faltan datos de fenotipo al inicio del estudio, este
paciente no se incluirá en el análisis de este fenotipo. Si a un paciente le faltaban datos de fenotipo a las 6 o 12 semanas, entonces este paciente no contribuyó al análisis de este fenotipo para el momento afectado.
Se realizaron 608 pruebas en 76 fenotipos en 3 grupos de dosificación para comparar el efecto del tratamiento con el efecto placebo. Se implemento la metodología de control de la tasa de descubrimiento falso estratificado (FDR, por sus siglas en inglés) para controlar la aparición de falsos positivos asociados con múltiples pruebas y, al mismo tiempo, proporcionar una potencia considerablemente mejor. El grupo de tipo celular se tomó como factor de estratificación y se realizó el cálculo de FDR (valor q) dentro de cada uno de los estratos, respectivamente. Todas las hipótesis nulas se rechazaron a un nivel de significancia de 0,05 con un valor q correspondiente < 0,1. La estrategia de ajuste de pruebas múltiples con rechazo a un nivel de significancia de 0,05 y q < 0,1 correspondiente aseguró que menos del 10 % de los resultados fueran falsos.
En un segundo análisis, el inmunofenotipo cambia entre los grupos de tratamiento combinado y placebo, donde se combinaron los tres grupos de dosificación de RAD001. Para determinar qué cambios en el inmunofenotipo diferían entre los grupos tratados y de placebo, se calcularon las proporciones de recuento de células dentro del paciente para cada fenotipo medido entre el inicio y la semana 6 del tratamiento con el fármaco del estudio y entre el inicio y el final del estudio (Semana 12). Las proporciones se transformaron logarítmicamente y se analizaron mediante análisis de covarianza en cada momento para detectar una diferencia entre los grupos de tratamiento agrupado y placebo. Se realizaron 152 pruebas en 76 fenotipos para comparar el efecto combinado del tratamiento con el efecto placebo. Se implementó la metodología de control de la tasa de falsos descubrimientos (FDR) estratificada para controlar la aparición de falsos positivos asociados con pruebas múltiples y, al mismo tiempo, proporcionar una potencia considerablemente mejor (Benjamini, Y. et al. (1995) J. Roy. Statist.57:289-300; y Sun, L. et al. (2006) Genet. Epidemiol.30:519-530). El grupo de tipo celular se tomó como factor de estratificación y el cálculo de FDR (valor q) se realizó dentro de cada uno de los estratos, respectivamente. Se rechazaron todas las hipótesis nulas con un nivel de significancia de 0,05 y un valor de q inferior al 20 %. Esto puede interpretarse como el rechazo solo de aquellas hipótesis con valores de P inferiores a 0,05 y menos del 20 % de probabilidad de que cada resultado significativo observado se deba a múltiples pruebas.
Resultados
En general, RAD001 fue bien tolerado, particularmente los regímenes de dosificación de 0,5 mg diarios y 5 mg semanales. No se produjeron muertes durante el estudio. Tres sujetos experimentaron cuatro eventos adversos graves (SAEs, por sus siglas en inglés) que se evaluaron como no relacionados con RAD001. Los 4 SAEs fueron hemorragia retiniana del ojo izquierdo con ceguera posterior en un sujeto con recuentos de plaquetas normales que había completado un ciclo de 6 semanas de 5 mg semanales de RAD0016 semanas antes; dolor de espalda severo en un sujeto tratado con placebo y gastroenteritis severa en un sujeto tratado con placebo. En la Tabla 5 se proporciona una lista de eventos adversos (AEs) relacionados con el tratamiento con una incidencia >2 % en cualquier grupo de tratamiento. El AE más común relacionado con RAD001 fue la úlcera bucal que, en la mayoría de los casos, fue de gravedad leve. En general, los sujetos que recibieron RAD001 tuvieron una incidencia similar de AEs graves que los tratados con placebo. Solo se evaluó un AE grave relacionado con las úlceras bucales de RAD001 en un sujeto tratado con 20 mg semanales de RAD001.
Tabla 5 Incidencia de AEs relacionados con el tratamiento > 2% en cual uier ru o de tratamiento or término referido


La capacidad de RAD001 de mejorar la función inmunitaria en voluntarios de edad avanzada se evaluó midiendo la respuesta serológica a la vacuna contra la gripe estacional de 2012. Los títulos medios geométricos (GMT) de la inhibición de la hemaglutinación (HI) para cada una de las 3 cepas de la vacuna contra la gripe al inicio y 4 semanas después de la vacunación contra la gripe se proporcionan en la Tabla 6. La variable de análisis principal fue la relación Hi GMT (4 semanas después de la vacunación/valor inicial). El estudio fue diseñado para poder demostrar que en al menos 2 de las 3 cepas de la vacuna contra la gripe hubo 1) un aumento de GMT > 1,2 veces en relación con el placebo; y 2) una probabilidad posterior no inferior al 80 % de que la proporción de GMT corregida con placebo supere 1. Se eligió este criterio de valoración porque un aumento de 1,2 veces en la proporción de GMT de la gripe inducida por el adyuvante de la vacuna MF-59 se asoció con una disminución de enfermedad gripal (lob, A et al. (2005) Epidemiol Infect 133:687-693).
Tabla 6. GMT de HI para cada una de las cepas de la vacuna contra la gripe al inicio y 4 semanas después de la vacunación contra la ri e

Relación
GMT (CV %) 2,6 (302,5) 2,5 (214,3) 1,8 (201,5) 2,0 (132,7)

Inicio indica 2 semanas antes de la vacunación contra la gripe

En la población con intención de tratar (ITT), las cohortes de dosis bajas de RAD001 (0,5 mg diarios o 5 mg semanales) que potencian el sistema inmunitario, pero no las cohortes de dosis más altas (20 mg semanales), cumplieron con el criterio principal de valoración del estudio.(Figura 41A). Esto demuestra que existe un mecanismo inmunomodulador distinto de RAD001 en las dosis más bajas, y que en la dosis más alta pueden entrar en juego los efectos inmunosupresores conocidos de la inhibición de mTOR. Además, los resultados sugieren una tendencia hacia una función inmunológica mejorada en los ancianos después de un tratamiento con dosis bajas de RAD001 que mejoran el sistema inmunológico.
En un análisis de subgrupos, el subconjunto de sujetos con títulos de gripe de referencia bajos (< 1:40) experimentó un mayor aumento de títulos asociado con RAD001 que la población ITT (Figura 41B). Estos datos demuestran que RAD001 es particularmente eficaz para potenciar la respuesta a la vacuna contra la gripe de sujetos que no tenían títulos protectores (>1:40) al inicio y, por lo tanto, tenían el mayor riesgo de enfermedad por gripe.
Los diagramas de dispersión de la concentración de RAD001 frente al aumento en el título para cada cepa de vacuna contra la gripe muestran una relación inversa de exposición/respuesta (Figura 42). El modelado y la simulación basados en la fosforilación de la quinasa S6 (S6K) mediada por mTOR predice que el régimen de dosificación semanal de 20 mg inhibe casi por completo la actividad de S6K mediada por mTOR, el régimen de dosificación semanal de 5 mg inhibe la actividad de S6K en más del 50 % y el régimen de dosificación de 0,5 mg diarios inhibe la fosforilación de S6K en aproximadamente un 38% durante el intervalo de dosificación (Tanaka, C et al. (2008) J. Clin. Oncol 26:1596-1602). Por lo tanto, la inhibición parcial de mTOR, p. ej., la fosforilación de S6K mediada por mTOR, con dosis bajas de RAD001 potenciadoras del sistema inmunitario puede ser tan eficaz, si no más, que la inhibición casi completa de mTOR con dosis altas de RAD001 para potenciar la respuesta inmunitaria de los ancianos.
También se evaluaron las tasas de seroconversión 4 semanas después de la vacunación contra la gripe. La seroconversión se definió como el cambio de un título negativo anterior a la vacunación (es decir, título HI <1:10) a un título HI posterior a la vacunación > 1:40 o un aumento de al menos 4 veces de un título HI no negativo (> 1:10 ) previo a la vacunación. En la población con intención de tratar, las tasas de seroconversión para las cepas H3N2 y B aumentaron en el RAD001 en comparación con las cohortes de placebo, aunque los aumentos no alcanzaron la significancia estadística (Tabla 7). En la subpoblación de sujetos con títulos iniciales de gripe <= 1:40, el tratamiento con RAD001 también aumentó las tasas de seroconversión a las cepas H3N2 y B, y estos resultados alcanzaron significancia estadística para la cepa B en la cohorte de dosificación diaria de 0,5 mg. Estos datos demuestran, además, que RAD001 mejoró la respuesta serológica a la vacunación contra la gripe en los ancianos.
Tabla 7 Porcentae de suetos con seroconversión a la ripe 4 semanas después de la vacunación


Las vacunas contra la gripe estacional actuales proporcionan a menudo una protección inadecuada contra cepas de gripe que surgen continuamente y que se presentan como variantes de virus que circulaban previamente. Sin embargo, los ratones vacunados contra la gripe en presencia del inhibidor mTOR rapamicina, en comparación con el placebo, desarrollaron una respuesta serológica más amplia a la gripe. La respuesta serológica más amplia incluía anticuerpos contra epítopos conservados expresados por múltiples subtipos de gripe que proporcionaban protección contra la infección con cepas heterólogas de influenza no contenidas en la vacuna (Keating, R et al. (2013) Nat Immunology 14:2166-2178). Para determinar si RAD001 amplió la respuesta serológica a la gripe en los voluntarios de edad avanzada, se midieron los títulos de HI a 2 cepas heterólogas de gripe no contenidas en la vacuna contra la gripe (A/H1N1 cepa A/New Jersey/8/76 y A/H3N2 cepa A/Victoria /361/11). El aumento en las relaciones de HI Gm T para las cepas heterólogas fue mayor en el RAD001 en comparación con las cohortes de placebo (Figura 43). Además, las tasas de seroconversión para las cepas heterólogas fueron más altas en el RAD001 en comparación con las cohortes de placebo. El aumento en las tasas de seroconversión en las cohortes de dosificación semanal de RAD001 de 5 y 20 mg fue estadísticamente significativo para la cepa heteróloga H3N2 (Tabla 8). La tasa de seroconversión H3N2 para las cohortes combinadas de RAD001 fue del 39 % frente al 20 % para la cohorte de placebo (p = 0,007). Los resultados presentados en esta memoria sugieren que la inhibición de mTOR amplía la respuesta serológica de los voluntarios de edad avanzada a la vacunación contra la gripe y aumenta los títulos de anticuerpos contra las cepas heterólogas de gripe que no están contenidas en la vacuna contra la gripe estacional.
La respuesta serológica ampliada a las cepas heterólogas de gripe en ratones tratados con rapamicina se ha asociado con una inhibición del cambio de clase en las células B y un aumento en los niveles de IgM contra la gripe (Keating , R. et al. (2013) Nat Immunol 14:2166-2178). Sin embargo, la inhibición del cambio de clase puede no estar involucrada en la respuesta serológica ampliada en seres humanos tratados con RAD001 porque los niveles de IgM e IgG contra la gripe después de la vacunación no difirieron entre las cohortes tratadas con RAD001 y con placebo (Figura 44).
Tabla 8 Porcentaje de sujetos que seroconvierten a cepas heterólogas de gripe 4 semanas después de la vacunación contra la ripe estacional

Para abordar el mecanismo por el cual RAD001 mejoró la función inmunitaria en voluntarios de edad avanzada, se realizó un inmunofenotipado en muestras de PBMC obtenidas de sujetos al inicio, después de 6 semanas de tratamiento con el fármaco del estudio y 4 semanas después de la vacunación contra la gripe (6 semanas después de la interrupción del fármaco del estudio). Aunque el porcentaje de la mayoría de los subconjuntos de PBMC no difirió entre las cohortes RAD001 y placebo, el porcentaje de células CD4 y CD8 PD-1 positivas fue menor en las cohortes RAD001 en comparación con las cohortes placebo (Figura 45). Las células CD4 y CD8 PD-1 positivas se acumulan con la edad y tienen respuestas defectuosas a la estimulación del antígeno porque PD-1 inhibe la proliferación de células T inducida por el receptor de células T, la producción de citoquinas y la función citolítica (Lages, CS et al. (2010) Aging Cell 9 :785-798). Hubo un aumento en el porcentaje de células T PD-1 positivas a lo largo del tiempo en la cohorte de placebo. En la semana 12 (4 semanas después de la vacunación), este aumento puede deberse a la vacunación contra la gripe, ya que se ha demostrado que el virus de la gripe aumenta las células T PD-1 positivas (Erikson, JJ et al. (2012) JCI 122: 2967-2982). Sin embargo, el porcentaje de células T CD4 PD-1 positivas disminuyó desde el valor inicial en las semanas 6 y 12 en todas las cohortes RAD001 (Figura 45A). El porcentaje de células CD8 PD-1 positivas también disminuyó desde el valor inicial tanto en la semana 6 como en la semana 12 en las dos cohortes de RAD001 de dosis más baja (Figura 45B). El porcentaje de células T CD4 PD-1 negativas se evaluó y aumentó en las cohortes de RAD001 en comparación con las cohortes de placebo (Figura 45C).
Bajo un análisis estadístico más estricto, en que los resultados de las cohortes RAD001 se agruparon y ajustaron según las diferencias en la expresión inicial de PD-1, hubo una disminución estadísticamente significativa del 30,2 % en las células T CD4 PD-1 positivas en la semana 6 en la cohorte de RAD agrupada (n=84) en comparación con la cohorte de placebo (n = 25) con p = 0,03 (q = 0,13) (Figura 46A). La disminución de células T CD4 PD-1 positivas en la semana 12 en la RAD agrupada en comparación con la cohorte de placebo es del 32,7 % con p = 0,05 (q = 0,19). La Figura 46B muestra una disminución estadísticamente significativa del 37,4% en células T CD8 PD-1 positivas en la semana 6 en la
cohorte RAD001 agrupada (n = 84) en comparación con la cohorte de placebo (n=25) con p = 0,008 (q = 0,07). La disminución de células T CD8 PD-1 positivas en la semana 12 en el RAD001 agrupado en comparación con la cohorte de placebo es del 41,4 % con p = 0,066 (q = 0,21). Por lo tanto, los resultados de las Figuras 45 y 46 juntos sugieren que la disminución asociada con RAD001 en el porcentaje de células T CD4 y CD8 PD-1 positivas puede contribuir a potenciar la función inmune.
Conclusión
En conclusión, los datos presentados en esta memoria demuestran que el inhibidor de mTOR RAD001 mejora la disminución relacionada con la edad en la función inmunológica de los seres humanos de edad avanzada según lo evaluado por la respuesta a la vacunación contra la gripe, y que esta mejora se obtiene con un balance riesgo/beneficio aceptable. En un estudio de ratones de edad avanzada, el tratamiento de 6 semanas con el inhibidor de mTOR rapamicina no solo potenció la respuesta a la vacuna contra la gripe, sino que también prolongó la vida útil, lo que sugiere que la mejora de la inmunosenescencia puede ser un marcador de un efecto más amplio en los fenotipos relacionados con el envejecimiento.
Dado que la dosificación de RAD001 se interrumpió 2 semanas antes de la vacunación, los efectos de mejora inmunitaria de RAD001 pueden estar mediados por cambios en una población celular relevante que persiste después de la interrupción del tratamiento farmacológico. Los resultados presentados en este documento demuestran que RAD001 disminuyó el porcentaje de células T CD4 y CD8 PD-1 positivas agotadas en comparación con el placebo. La expresión de PD-1 es inducida por la señalización de TCR y permanece alta en el contexto de estimulación persistente de antígenos, incluida la infección viral crónica. Si bien no se desea estar ligados por la teoría, es posible que RAD001 redujera la activación inmunitaria crónica en voluntarios de edad avanzada y, por lo tanto, condujera a una disminución en la expresión de PD-1. RAD001 también puede inhibir directamente la expresión de PD-1 como se ha informado para la inmunofilina ciclosporina A (Oestreich, KJ et al. (2008) J Immunol. 181:4832-4839). Es probable que una reducción inducida por RAD001 en el porcentaje de células T PD-1 positivas mejore la calidad de las respuestas de las células T. Esto es consistente con estudios previos que demuestran que la inhibición de mTOR mejoró la calidad de la respuesta de las células T CD8 de la memoria a la vacunación en ratones y primates (Araki, K et al. (2009) Nature 460:108-112). En ratones de edad avanzada, también se ha demostrado que la inhibición de mTOR aumenta el número de células madre hematopoyéticas, lo que conduce a una mayor producción de linfocitos que no han sido tratados (Chen, C et al. (2009) Sci Signal 2:ra75). Aunque en este ejemplo no se detectaron diferencias significativas en los porcentajes de linfocitos que no han sido tratados en las cohortes RAD001 frente a placebo, este posible mecanismo puede investigarse más a fondo.
El mecanismo por el cual RAD001 amplió la respuesta serológica a cepas heterólogas de la gripe puede investigarse más a fondo. También se ha demostrado que rapamicina inhibe el cambio de clase en las células B después de la vacunación contra la gripe. Como resultado, se generó un repertorio único de anticuerpos anti-gripe que promovió la protección de cepas cruzadas contra la infección letal con subtipos del virus de la gripe no contenidos en la vacuna contra la gripe (Keating, R et al. (2013) Nat Immunol. 14:2166- 2178). Los resultados descritos en esta memoria no demostraron que RAD001 alterara el cambio de clase de células B en los sujetos de edad avanzada que habían interrumpido RAD0012 semanas antes de la vacunación contra la gripe. Aunque el mecanismo subyacente requiere una mayor aclaración, la mayor respuesta serológica a las cepas de la gripe heterólogas descritas en esta memoria puede conferir una mayor protección contra la enfermedad de la gripe en los años en que hay poca coincidencia entre la vacuna estacional y las cepas de la gripe circulantes en la comunidad.
El efecto de RAD001 en los títulos de anticuerpos contra la gripe fue equiparable al efecto del adyuvante de la vacuna MF59 que está aprobado para mejorar la respuesta de los ancianos a la vacunación contra la gripe (Podda, A (2001) Vaccine 19:2673-2680). Por lo tanto, la potenciación impulsada por RAD001 de la respuesta de anticuerpos a la vacunación contra la gripe puede traducirse en un beneficio clínico como se demostró con la vacuna contra la gripe con adyuvante MF59 en ancianos (lob, A et al. (2005) Epidemiol Infect. 133:687-693). Sin embargo, RAD001 también se utiliza para suprimir la respuesta inmunitaria de los pacientes de trasplante de órganos. Estos hallazgos aparentemente paradójicos plantean la posibilidad de que los efectos inmunomoduladores de los inhibidores de mTOR puedan depender de la dosis y/o del antígeno (Ferrer, IR et al. (2010) J Immunol. 185:2004-2008). En esta memoria se observó una tendencia hacia una relación inversa de exposición a RAD001/respuesta a la vacunación. Es posible que la inhibición completa de mTOR suprima la función inmunitaria a través del mecanismo ciclofilina-rapamicina normal, mientras que la inhibición parcial de mTOR, al menos en los ancianos, mejora la función inmunitaria debido a una inhibición del fenotipo distinta relacionada con el envejecimiento. De interés, la actividad de mTOR aumenta en una diversidad de tejidos, incluyendo las células madre hematopoyéticas en modelos animales de envejecimiento (Chen C. et al. (2009) Sci Signal 2:ra75 y Barns, M. et al. (2014) Int J Biochem Cell Biol. 53:174-185). Por lo tanto, reducir la actividad de mTOR a los niveles observados en el tejido joven, en lugar de una supresión más completa de la actividad de mTOR, puede ser de beneficio clínico en las indicaciones de envejecimiento.
El perfil de seguridad de los inhibidores de mTOR como RAD001 en el tratamiento de indicaciones relacionadas con el envejecimiento ha sido motivo de preocupación. La toxicidad de RAD001 en dosis utilizadas en oncología o indicaciones de trasplante de órganos incluye tasas de estomatitis, diarrea, náuseas, citopenias, hiperlipidemia e hiperglucemia que serían inaceptables para muchas indicaciones relacionadas con el envejecimiento. Sin embargo, estos AEs están
relacionados con los niveles mínimos de RAD001 en la sangre. Por lo tanto, los regímenes de dosificación de RAD001 utilizados en este estudio se eligieron para minimizar los niveles mínimos. Los niveles valle promedio de RAD001 de las cohortes de dosis de 0,5 mg diarios, 5 mg semanales y 20 mg semanales fueron de 0,9 ng/ml, por debajo de 0,3 ng/ml (el límite inferior de cuantificación) y 0,7 ng/ml, respectivamente. . Estos niveles mínimos son significativamente más bajos que los niveles mínimos asociados con los regímenes de dosificación utilizados en trasplantes de órganos y pacientes con cáncer. Además, el curso de tratamiento limitado de 6 semanas disminuyó el riesgo de eventos adversos. Estos hallazgos sugieren que los regímenes de dosificación utilizados en este estudio pueden tener un riesgo/beneficio aceptable para algunas afecciones de los ancianos. No obstante, un número significativo de sujetos en los experimentos descritos en esta memoria desarrollaron úlceras bucales incluso cuando se les administró una dosis tan baja como 0,5 mg al día. Por lo tanto, el perfil de seguridad de la dosis baja de RAD001, potenciador del sistema inmunitario, merece un estudio adicional. El desarrollo de inhibidores de mTOR con perfiles de seguridad más limpios que los rapálogos actualmente disponibles puede proporcionar mejores opciones terapéuticas en el futuro para las afecciones asociadas con el envejecimiento.
Ejemplo 6: Mejora de la Respuesta Inmunitaria a la Vacuna en Sujetos Ancianos
La función inmunitaria disminuye en los ancianos, lo que conduce a un aumento de la incidencia de infecciones y a una disminución de la respuesta a la vacunación. Como primer paso para determinar si la inhibición de mTOR tiene efectos contra el envejecimiento en seres humanos, se realizó un ensayo aleatorizado controlado con placebo para determinar si el inhibidor de mTOR RAD001 revierte la disminución de la función inmunológica relacionada con el envejecimiento según lo evaluado por la respuesta a la vacunación en voluntarios de edad avanzada. En todos los casos, se obtuvieron los consentimientos de patente apropiados y el estudio fue aprobado por las Autoridades Sanitarias Nacionales.
En el estudio se utilizaron los siguientes 3 regímenes de dosificación de RAD001:
20 mg semanales (nivel mínimo: 0,7 ng/ml)
5 mg semanales (el nivel mínimo estaba por debajo de los límites de detección)
0,5 mg diarios (nivel mínimo: 0,9 ng/ml)
Se eligieron estos regímenes de dosificación porque tienen niveles mínimos más bajos que las dosis de RAD001 aprobadas para indicaciones oncológicas y de trasplantes. El nivel mínimo es el nivel más bajo de un fármaco en el cuerpo. El nivel mínimo de RAD001 asociado con el régimen de dosificación oncológico diario de 10 mg es de aproximadamente 20 ng/ml. El nivel mínimo asociado con el régimen de dosificación de trasplante de 0,75-1,5 mg dos veces al día es de aproximadamente 3 ng/ml. Por el contrario, el nivel mínimo asociado con los regímenes de dosificación utilizados en este estudio de inmunización fue de 3 a 20 veces menor.
Dado que los AEs relacionados con RAD001 están asociados con niveles mínimos, se predijo que los 3 regímenes de dosificación tendrían una seguridad adecuada para los voluntarios normales. Además, se predijo que las 3 dosis proporcionarían un intervalo de inhibición de mTOR. La quinasa P70 S6 (P70 S6K) es una diana posterior que es fosforilada por mTOR. Los niveles de fosforilación de P70 S6K sirven como medida de la actividad de mTOR. Según el modelo y la simulación de los datos de fosforilación de P70 S6K obtenidos en estudios preclínicos y clínicos de RAD001, se predijo que 20 mg a la semana inhibirían casi por completo la actividad de mTOR durante una semana completa, mientras que se predijo que 5 mg a la semana y 0,5 mg al día inhibirían parcialmente la actividad de mTOR .
Voluntarios de edad avanzada >= 65 años de edad fueron aleatorizados a uno de los 3 grupos de tratamiento RAD001 (50 sujetos por brazo) o placebo (20 sujetos por brazo). Los sujetos fueron tratados con el fármaco del estudio durante 6 semanas, se les dio un descanso de 2 semanas y luego recibieron vacunas contra la gripe (Aggrippal, Novartis) y neumocócica (Pneumovax 23, Merck). La respuesta a la vacunación contra la gripe se evaluó midiendo los títulos medios geométricos (GMT) mediante el ensayo de inhibición de la hemaglutinación para las 3 cepas de gripe (subtipos de gripe H1N1, H3N2 y B) en la vacuna contra la gripe 4 semanas después de la vacunación. Los criterios de valoración principales del estudio fueron (1) seguridad y tolerabilidad y (2) un aumento de 1,2 veces en los títulos de la gripe en comparación con el placebo en 2/3 de las cepas de la vacuna contra la gripe 4 semanas después de la vacunación. Se eligió este criterio de valoración porque un aumento de 1,2 veces en los títulos de gripe se asocia con una disminución de la enfermedad de la gripe después de la vacunación y, por lo tanto, es clínicamente relevante. Las dosis semanales de 5 mg y diarias de 0,5 mg fueron bien toleradas y, a diferencia de la dosis semanal de 20 mg, cumplieron el criterio principal de valoración GMT (Figura 41A). RAD001 no solo mejoró la respuesta a la vacunación contra la gripe, sino que también mejoró la respuesta a la vacunación antineumocócica en comparación con el placebo en voluntarios de edad avanzada. La vacuna neumocócica contiene antígenos de 23 serotipos neumocócicos. Los títulos de anticuerpos contra 7 de los serotipos se midieron en sujetos de los autores de la invención. Los títulos de anticuerpos contra 6/7 serotipos aumentaron en las 3 cohortes de RAD en comparación con el placebo.
Los datos de títulos combinados de la gripe y neumococo sugieren que la inhibición parcial (menos del 80-100 %) de mTOR es más eficaz para revertir la disminución de la función inmunitaria relacionada con el envejecimiento que una inhibición más completa de mTOR.
Ejemplo 7: La inhibición de dosis bajas de mTOR aumenta la energía y el ejercicio
En modelos preclínicos, la inhibición de mTOR con el rapálogo rapamicina aumenta la actividad física espontánea en ratones viejos (Wilkinson et al. Rapamycin slows aging in mice. (2012 ) Aging Cell; 11:675-82). De interés, los sujetos en la cohorte de dosificación diaria de 0,5 mg descrita en el Ejemplo 6 también informaron un aumento de energía y capacidad de ejercicio en comparación con el placebo en cuestionarios administrados un año después de la dosificación (Figura 47). Estos datos sugieren que la inhibición parcial de mTOR con rapálogos puede tener efectos beneficiosos sobre la morbilidad relacionada con el envejecimiento más allá de la función inmunológica.
Ejemplo 8: Inhibición de la quinasa P70 S6 con RAD001
Se realizaron modelos y simulaciones para predecir intervalos de dosis diarias y semanales de RAD001 que se predice que inhiben parcialmente la actividad de mTOR. Como se señaló anteriormente, P70 S6K es fosforilado por mTOR y es la diana posterior de mTOR que está más estrechamente relacionada con el envejecimiento porque la eliminación de P70 S6K aumenta la vida útil. Por lo tanto, se modelaron dosis de RAD001 que inhiben parcialmente la actividad de P70 S6K. Se predice que la dosificación semanal en el intervalo de >= 0,1 mg y < 20 mg logrará una inhibición parcial de la actividad de P70 S6K (Figura 48).
Para la dosificación diaria, las concentraciones de RAD001 de 30 pM a 4 nM inhibieron parcialmente la actividad de P70 S6K en las líneas celulares (Tabla 9). Se prevé que estas concentraciones séricas se alcancen con dosis de RAD001 >= 0,005 mg a < 1,5 mg al día.
Tabla 9 Porcentae de inhibición de la actividad de P70 S6K en células HeLa in vitro

Conclusión
Métodos para tratar la morbilidad relacionada con el envejecimiento o, en general, potenciar una respuesta inmunitaria, con dosis de inhibidores de mTOR que solo inhiben parcialmente P70 S6K. La eficacia de la inhibición parcial de mTOR con dosis bajas de RAD001 en indicaciones de envejecimiento es un hallazgo inesperado. Los intervalos de dosis de RAD001 entre >= 0,1 mg y < 20 mg por semana y >= 0,005 mg y < 1,5 mg al día lograrán una inhibición parcial de mTOR y, por lo tanto, se espera que tenga eficacia en la morbilidad relacionada con el envejecimiento o en el potenciamiento de la respuesta inmunitaria.
Ejemplo 9: Identificación de nuevos antígenos diana para la terapia con CART
La estrategia para la terapia con CART depende de la expresión preferente de un antígeno de la superficie celular diana en células tumorales o cuando la ablación de células normales que expresan la diana es clínicamente tolerable. En la ALL de células B, la terapia con CART que fija como objetivo CD19 ha demostrado ser eficaz y factible clínicamente. Sin embargo, algunos pacientes con ALL de células B tienen una expresión baja o nula de CD19, o recaen después de la terapia con CAR19 con enfermedad CD19 negativa. Además, la ALL y la AML de células T no son susceptibles de ser fijadas como objetivo con células CART19. Por lo tanto, la falta de antígenos de superficie diana en diferentes tipos de cáncer ha impedido el desarrollo de enfoques basados en CAR. En este ejemplo, se describe una estrategia para el descubrimiento de antígenos diana en leucemias agudas (AL), p. ej., ALL y AML.
Se utilizaron ensayos QuantiGene (Affymetrix) para medir el nivel de ARN de 53 genes candidatos para antígenos diana en leucemias agudas. Los ensayos QuantiGene utilizan un ensayo de ADN ramificado (ADNb), que es un método de hibridación de ácido nucleico en sándwich que utiliza moléculas de ADNb para amplificar la señal del ARN objetivo capturado. El ARN se mide directamente desde la fuente de la muestra, sin purificación ni manipulación enzimática. QuantiGene es, por tanto, un ensayo robusto y reproducible con un amplio intervalo dinámico.
Se seleccionaron 53 genes candidatos para someterlos a prueba en función de uno de cuatro criterios:
(i) expresión conocida en AL (como controles positivos), p. ej., CD19 para ALL de células B o CD-34 para AML;
(ii) expresión durante la hematopoyesis, suponiendo que AL es un tumor maligno de las células hematopoyéticas (p.
ej., EMR2);
(iii) es probable que la expresión esté en la superficie de las células diana;
(iv) cuando se espera que la ablación de tejidos normales o células portadoras de estos antígenos sea clínicamente tolerable.
El ARN se preparó a partir de un panel de 33 muestras de pacientes con AML, 7 muestras ALL, 3 controles de médula ósea sana (NBM), así como una línea celular de ALL, una línea celular de AML y una neoplasia no hematopoyética (melanoma A357) para servir como control negativo. Todas las muestras se analizaron por duplicado en el Ensayo QuantiGene. El análisis se realizó como sigue. Se calculó la lectura de fondo promedio y la desviación estándar para cada uno de los genes. Se excluyeron las lecturas que no excedan el promedio 3xSD para ese gen y los puntos de datos duplicados que fallan en el control de calidad (p. ej., en que el CV para los duplicados es > 10%). Los genes domésticos (p. ej., PP1B o GUSB) se normalizaron. Se expresa la intensidad de fluorescencia media (MFI) para cada uno de los genes en relación con el gen de referencia. Cuando había más de puntos de datos disponibles para los controles de médula ósea normal, se realizó el cálculo estadístico de la significancia utilizando ANOVA seguido de la prueba posterior de Dunnett. Los genes candidatos se ordenaron por preferencia para la investigación posterior de la siguiente manera:
1. AML y ALL > NBM y A357
2. AML o ALL > NBM y A357
3. AML y ALL > A357, pero no NBM
4. AML o ALL > A357, pero no NBM
Los valores de MFI normalizados calculados para cada uno de los genes candidatos e muestran en las Figuras 49-57. La Figura 58 es una representación acumulativa de los valores de MFI normalizados promedio en relación con el gen de mantenimiento PP1B para AML y ALL, médula ósea normal o la línea celular A357. Según el análisis arriba descrito, se identificaron los siguientes antígenos diana novedosos para las leucemias agudas: C79a, CD72, LAIR1, FCAR, CD79b, LILRA2, CD300LF, CLEC12A, BST2, EMR2, CLECL1 (CLL-1), LY75, FLT3, CD22, KIT, GPC3, FCRL5 e IGLL1.
La investigación posterior de los genes diana incluye citometría de flujo de muestras de pacientes para confirmar la expresión del nivel de proteína en la membrana celular, inmunohistoquímica en micromatrices de tejido sano para excluir la presencia del antígeno diana en tejidos normales importantes.
Ejemplo 10: Optimización de la terapia con CAR con la administración de citoquinas exógenas
Las citoquinas tienen funciones importantes relacionadas con la expansión, diferenciación, supervivencia y homeostasis de las células T. Una de las familias de citoquinas más importantes para uso clínico es la familia de citoquinas de cadena Y común (Yc), que incluye interleuquina (IL)-2, IL-4, IL-7, IL-9, IL-15 e IL- 21 (Liao et al., 2013, Immunity, 38:13-25). La IL-2 se ha estudiado ampliamente como un agente inmunoterapéutico para el cáncer. El complemento de IL-2 potenció la capacidad antitumoral de las células CAR-T anti-CD19 en los ensayos clínicos (Xu et al., 2013, Lymphoma, 54:255-60). Sin embargo, la administración de IL-2 está limitada por los efectos secundarios y la propensión a la expansión de las células T reguladoras y el efecto de la muerte celular inducida por activación (AICD) (Malek et al., 2010, Immunity, 33:153-65; y Lenardo et al., 1999, Annu Rev Immunol, 17:221-53). iL-7, IL-15 e IL-21 pueden potenciar cada una la eficacia de las inmunoterapias adoptivas y parecen ser menos tóxicos en comparación con IL-2 (Alves et al., 2007, Immunol Lett, 108:113-20). A pesar de los extensos estudios preclínicos y clínicos sobre el papel de las citoquinas anteriores, faltan estudios comparativos de parámetros múltiples sobre los roles de varias citoquinas Yc exógenas en la terapia adoptiva de células CAR-T.
Además de las citoquinas de cadena y, IL-18 es otra citoquina inmunoestimuladora que regula las respuestas inmunitarias, lo que aumenta la producción de IFN-y por parte de las células T y aumenta la actividad citolítica de los CTL (Srivastava et al., 2010, Curr Med Chem , 17:3353-7). La administración de iL-18 es segura y bien tolerada, incluso cuando la dosis alcanza los 1000 pg/kg (Robertson et al., 2006, Clin Cancer Res, 12:4265-73). Por tanto, IL-18 podría ser otro candidato utilizado para potenciar el efecto antitumoral de las células CAR-T.
Para potenciar adicionalmente la eficacia de la terapia adoptiva con células T modificadas con CAR (CAR-T), se examinó la optimización de la terapia CAR con la administración de citoquinas exógenas. Para comparar las funciones de diferentes citoquinas administradas de forma exógena durante la inmunoterapia con células CAR-T y encontrar la citoquina óptima para uso clínico, se probó la capacidad antitumoral in vivo de las células CAR-T utilizando modelos animales de cáncer de ovario.
Se utilizaron los siguientes materiales y métodos en los experimentos descritos en este ejemplo.
Construcción de CAR y preparación de lentivirus
El vector pELNS-C4-27z CAR se construyó como se describió previamente (manuscrito en revisión). Brevemente, el plásmido pHEN2 que contiene el scFv anti-FRa C4/AFRA4 se utilizó como molde para la amplificación por PCR del fragmento C4 utilizando los cebadores de 5'-ataggatcccagctggtggagtctgggggaggc-3' (SEQ ID NO: 52) y 5'-atagctagcacctaggacggtcagcttggtccc-3' (SEQ ID NO: 53) (BamHI y NheI estaban subrayados). El producto de la PCR y los vectores de expresión lentivirales autoinactivantes pELNS de tercera generación se digirieron con BamHI y NheI. Los productos de PCR digeridos se insertaron luego en el vector pELNS que contenía el dominio de señalización de células T CD27-CD3z en el que la expresión del transgén está impulsada por el promotor del factor de elongación-la (EF-1a).
Se generó un lentivirus defectuoso en la replicación de título alto mediante la transfección de células de la línea celular de riñón embrionario humano 293T (293T) con cuatro plásmidos (pVSV-G, pRSV.REV, pMDLg/p.RRE y pELNS-C4-27z CAR) mediante el uso de Express In (Open Biosystems) como se describió previamente (manuscrito en proceso de revisión). Los sobrenadantes se recogieron y filtraron a las 24 h y 48 h después de la transfección. El medio se concentró por ultracentrifugación. Alternativamente, se realizó una única recolección 3o h después del cambio de medio. Los medios que contenían virus se utilizaron alternativamente sin concentrar o concentrados mediante el concentrador Lenti-X (Clontech, Cat n° 631232). Los títulos de virus se determinaron en función de la eficiencia de transducción de lentivirus a células SupTI utilizando el método de dilución limitante.
Células T y líneas celulares
Linfocitos de sangre periférica se obtuvieron de donantes sanos después del consentimiento informado según un protocolo aprobado por la Junta de Revisión Institucional Universitaria de la Universidad de Pensilvania. Las células T primarias se adquirieron de Human Immunology Core después de purificarlas mediante selección negativa. Los linfocitos T se cultivaron en medio completo (RPMI 1640 complementado con FBS al 10 %, 100 U/mL de penicilina, 100 pg/mL de sulfato de estreptomicina) y se estimularon con microesferas recubiertas de mAbs anti-CD3 y anti-CD28 (Invitrogen) en una relación de 1: 1 siguiendo las instrucciones. Veinticuatro horas después de la activación, las células se transdujeron con lentivirus a una MOI de 5. Se añadieron las citoquinas indicadas a las células T transducidas a partir del día siguiente con una concentración final de 10 ng/mL. Las citoquinas fueron reemplazadas cada 3 días.
La célula 293T utilizada para el empaquetamiento de lentivirus y la célula SupT1 utilizada para la titulación lentiviral se obtuvieron de la ATCC. Las líneas celulares de cáncer de ovario establecidas SKOV3 (FRa+) y C30 (FRa-) se utilizaron como células diana para el ensayo de citotoxicidad y secreción de citoquinas. Para los ensayos de bioluminiscencia, se transdujo SKOV3 con lentivirus para expresar luciferasa de luciérnaga (fLuc).
Análisis de citometría de flujo y clasificación de células
La citometría de flujo se realizó en un BD FACSCanto. CD45 (HI30), CD3 (HIT3a), CD8 (HIT8a), CD45RA (HI100), CD62L (DREG-56), CCR7 (G043H7), IL-7Ra (A019D5), CD27 (M-T271), CD28 (CD28.2), CD95 (DX2), TNF-a (MAb11), IFN-y (4S.B3), IL-2 (MQ1-17H12), perforina (B-D48), granzima-B (g B11) anti-humano se obtuvieron de Biolegend La IgG antihumana de conejo conjugada con biotina-SP (H+L) se adquirió de Jackson Immunoresearch y la estreptavidina conjugada con APC se adquirió de Biolegand. La Bcl-xl antihumana (7B2.5) se adquirió de SouhernBiotech. El kit de apoptosis y los tubos TruCount se obtuvieron de BD Bioscience. Para el recuento de células T en sangre periférica, se obtuvo sangre mediante sangrado retroorbital y se tiñó para detectar la presencia de células T CD45, CD3, CD4 y CD8 humanas. Los subconjuntos CD45+ controlados, CD3+, CD4+ y CD8+ humanos se cuantificaron con los tubos TruCount siguiendo las instrucciones del fabricante.
Estudio in vivo de terapia celular adoptiva
Se obtuvieron ratones hembra no obesos diabéticos/de inmunodeficiencia combinada grave/de cadena y _/' (NSG) de 8 a 12 semanas de edad del Stem Cell and Xenograft Core del Abramson Cancer Center, Universidad de Pensilvania. Los ratones se inocularon por vía subcutánea con 3*10® células SKOV3 fLuc+ en el flanco el día 0. Cuatro o cinco ratones se aleatorizaron por grupo antes del tratamiento. Después de que los tumores se hicieron palpables, las células T primarias humanas se activaron y transdujeron como se describió previamente. Las células T se expandieron en presencia de IL-2 (5 ng/mL) durante aproximadamente 2 semanas. Cuando la carga tumoral era de ~250-300 mm3, a los ratones se les inyectaron 5*10® células CAR-T o 100 pl de solución salina por vía intravenosa y luego recibieron una inyección intraperitoneal diaria de 5 pg de IL-2, IL-7, IL-15, IL-18, IL-21 o solución tampón fosfato (PBS) durante 7 días. Las dimensiones del tumor se midieron con calibradores y los volúmenes del tumor se calcularon con la siguiente fórmula: volumen del tumor = (longitud x anchura 2)/2. El número y el fenotipo de las células T transferidas en la sangre del ratón receptor se determinaron mediante citometría de flujo después de una hemorragia retroorbitaria. Los ratones se sacrificaron cuando los volúmenes de los tumores eran superiores a 2000 mm3 y los tumores se extirparon inmediatamente para su posterior análisis.
Análisis estadístico.
El análisis estadístico se realizó con Prism 5 (software GraphPad) y el software IBM SPSS Statistics 20.0. Los datos se mostraron como media ± SEM a menos que se aclare. Se utilizaron tests t de muestras pareadas o pruebas de rango de Wilcoxon no paramétricas para la comparación de dos grupos y medidas repetidas ANOVA o test de Friedman para testar la significancia estadística de las diferencias entre tres o más grupos. Los hallazgos se consideraron estadísticamente significativos cuando los valores de P fueron inferiores a 0,05.
RESULTADOS
Construcción y expresión de CAR C4 anti-FRa
El CAR pELNS-C4-27z constaba del scFv C4 anti-FRa enlazado a una región transmembrana y de bisagra de CD8a, seguido de un resto de señalización de CD3Z junto con el motivo de señalización intracelular de CD27 (Figura 59A). Las células T humanas primarias se transdujeron eficientemente con vectores lentivirales CAR C4 con eficiencias de transducción del 43 %~65 % cuando se detectaron 48 h después de la transducción (Figura 59B). Los niveles de expresión de CAR fueron equiparables entre las células T CD4+ y CD8+ (52,6 ± 10,2 % frente a 49,5 ± 17,1 %, P = 0,713).
Diferente eficacia anti-tumoral de diversas citoquinas en modelos animales
Este estudio examinó si la administración de citoquinas in vivo en combinación con la inyección de células CAR-T podría mejorar la actividad anti-tumoral de células CAR-T. Ratones que portaban tumores SKOV3 subcutáneos recibieron solución salina o 5*10® células CAR-T C4-27z por vía intravenosa el día 39 (Figura 60). En comparación con el grupo de solución salina, los ratones que recibieron terapia con células CAR-T experimentaron una regresión del tumor de corta duración y el tumor comenzó a recuperarse a partir del día 56 (Figura ®1). De los diversos grupos de citoquinas, los ratones que recibieron la inyección de IL-15 e IL-21 presentaron la mejor supresión del tumor, seguidos de lL-2 e IL-7, mientras que los ratones tratados con IL-18 y PBS tuvieron la mayor carga tumoral. La persistencia de las células T transferidas en la sangre periférica se determinó 15 días después de la transferencia adoptiva y cuando terminó. Los números más altos de células T CD4+ y CD8+ se detectaron en ratones tratados con IL-15, seguido de IL-21. El recuento de células T CD4+ y CD8+ del día 15 fue consistente con la regresión del tumor y predijo el peso final del tumor. Se sacrificaron los ratones 73 días después de la exposición al tumor y se analizó la presencia de células T humanas en los tumores (Figura 63). De manera similar con la sangre periférica, los ratones tratados con IL-15 presentaron el mayor número de células T en el tumor, seguidos de IL-21, IL-2, IL-7, PBS e IL-18 (Figura 64). Las relaciones de CD4 a CD8 fueron equiparables entre los diferentes grupos de citoquinas, con un predominio de células T CD4+ tanto en sangre como en el tumor en todos los grupos de citoquinas. La expresión de CAR en las células T CD8+ fue equiparable entre los grupos anteriores (45,1 % ~ 62,4 %), mientras que los grupos de IL-15 e IL-21 tenían relaciones más altas de células CAR+ T CD4 que los grupos de IL-2 e IL-18 (Figura 65). En cuanto al fenotipo, todas las células CAR-T en el tumor eran CD62L' y CCR7', mientras que el 35 %~60 % de ellas expresaban CD45RA+ (Temra) (datos no mostrados). Las células T CD8+ tenían más probabilidades de retener la expresión de CD27, mientras que la expresión de CD28 era equiparable entre las células T CD4+ y CD8+ (Figura 66).
ANÁLISIS
En resumen, estos hallazgos tienen implicaciones importantes para la administración de citoquinas exógenas para potenciar la eficacia de la terapia adoptiva con células CAR-T. Todas las citoquinas Yc cuando se administran en combinación con la inyección de células CAR-T potencian la eficacia anti-tumoral en el modelo de ratón de ovario. IL-15 e IL-21 fueron las mejores citoquinas para el complemento in vivo, e IL-7 e IL-2 mostraron evidencia de mejorar el resultado anti-tumoral. IL-18 es una citoquina proinflamatoria que pertenece a la familia de IL-1 y, en estos experimentos in vivo, no mostró un efecto potenciado sobre la eficacia antit-umoral.
Ejemplo 11: La Inhibición de DGK Aumenta la Eficacia de CART
Estudios previos, por ejemplo, los experimentos discutidos en el Ejemplo 6, han sugerido que las células CAR T pierden eficacia con el tiempo in vivo (p. ej., en el microentorno del tumor). Específicamente, las células mesoCAR T que se inyectaron en un modelo de ratón tumoral se aislaron de los tumores después de la infusión de células T (p. ej., 39 días después, en lo sucesivo denominados linfocitos infiltrantes de tumores, TIL, por sus siglas en inglés) y se evaluaron en cuanto a su actividad funcional en comparación con células células mesoCAR T recién descongeladas. Los resultados demostraron que en ensayos de eliminación ex vivo y ensayos de liberación de IFNy, las células mesoCAR T aisladas del tumor tenían una capacidad reducida para eliminar células tumorales (Fig. 67A), una producción reducida de IFNg (Fig. 67B) y una señalización reducida de ERK (como se muestra por fosforilación en análisis de transferencia Western, Fig. 67C) en respuesta a estímulos de antígeno o CD3/CD28 (lo que indica una activación reducida de células T).
Mecanismos inhibidores que posiblemente explican la disminución de la actividad de las células CAR T in vivo con el tiempo incluyen: factores solubles (TGFb, PGE2, adenosina, IL10, ligandos RAGE, etc.), contacto de célula a célula (PD-1, Lag3, CTLA4, TIM3, CD160, etc.), y sistemas de retroalimentación negativa intracelular inducida por activación intrínseca (diaglicerolquinasas: isoformas a y Z, Egrs (2 y 3), SHP-1, NFAT2, BLIMP-1, Itch, GRAIL, Cbl -b, Ícaros, etc.). La activación de células T puede inducir factores tales como DGK. DGK, a su vez, inhibe la señalización de DAG al
fosforilar DAG. Esto limita la activación inducida por DAG de la vía RAS-ERK-AP1 que conduce a la activación de las células T. Estudios anteriores han demostrado que ratones deficientes en DGKa o DGKZ dan como resultado células T CD4 que demuestran una transducción de señal mejorada y parecen más resistentes a los estímulos que inducen anergia.
Ensayos de Liberación de Citoquinas y Citotoxicidad in vitro
Para investigar el efecto de la inhibición de DGK sobre la eficacia de las células CART, se utilizaron ratones transgénicos con deleciones en los genes DGK DGKa, DGKZ o ambos. Se aislaron células T esplénicas de ratones de tipo salvaje y deficientes en DGK y se transdujeron para expresar mesoCAR (SS1 BBZ) utilizando retrovirus. MIGR1 CAR se utilizó como control.
Se realizó un ensayo de citotoxicidad utilizando métodos similares a los descritos en los Ejemplos anteriores. Se incubaron células que expresan mesoCAR de tipo salvaje y deficientes en DGK (KO) en diversas relaciones efector:diana y se cuantificó la citotoxicidad (% de células diana muertas) (Fig. 68). Como se muestra en la Figura 68, la deleción de DGK mejoró notablemente la función efectora de las células CAR T, especialmente en relaciones bajas de efector:diana.
De forma similar, se examinó la liberación de IFNy en respuesta a las células diana en relaciones efector:diana variables después de 18 horas. Como se muestra en la Figura 69, se descubrió que la deleción de DGKs potencia notablemente la función efectora de las células mesoCAR T, especialmente en relaciones bajas de efector:diana.
El análisis de transferencia Western de las células mesoCAR T deficientes en DGK en comparación con las células mesoCAR T de tipo salvaje mostró un aumento de la fosforilación de ERK (Fig. 70), lo que indica que la presencia de DGK suprime la señalización de ERK, mientras que la deleción de DGK da como resultado un aumento de la señalización de ERK. El aumento en la señalización de ERK en el fondo eliminado de DGK sugiere que la inhibición de DGK da como resultado la activación de la vía Ras-FRK-API y, por lo tanto, la activación de células T.
Estudios recientes han demostrado que TGFp modula la funcionalidad de las células CD8 T infiltrantes de tumores aunque interfiere con la transducción de señales RAS/ERK, las mismas moléculas de señalización por las que la deficiencia de DGK confiere efectos aumentados de células T. Se examinó la sensibilidad de las células mesoCAR T deficientes en DGK a TGFp. Se incubaron células mesoCAR T WT y deficientes en DGK con células tumorales AE17 que expresan mesotelina o -10 ng/ml de TGFp durante 18 horas. Se midió la citotoxicidad y la producción de IFNg por estas células T. Como se muestra en la Figura 71, el TGFp inhibió la destrucción en un 50 % en las células CAR T W t (flechas). Sin embargo, esta inhibición inducida por TGFp no se observó en células CAR T con deleción de DGK, lo que demuestra que las células deficientes en DGK no son sensibles a la modulación de TGFp y son más resistentes a los estímulos inhibidores como TGFp, lo que puede contribuir al aumento de T actividad celular.
Eficacia terapéutica de la inhibición de mesoCAR y DGK in vivo
A continuación, se examinó la eficacia terapéutica de las células mesoCAR T en el contexto de la inhibición o deficiencia de DGK. Se inyectaron por vía subcutánea células tumorales AE17meso (células de mesotelioma) en ratones C57BL/6. Cuando los tumores alcanzaron los 100 mm3 (aproximadamente una semana más tarde), se inyectaron 10 millones de células mesoCAR T por vía intravenosa a través de la vena de la cola. A continuación, se siguieron los volúmenes tumorales durante al menos 18 días.
Las células mesoCAR T deficientes en DGK demostraron una actividad antitumoral mejorada y prolongada en comparación con las células mesoCAR de tipo salvaje (WT) y no tratadas (Fig. 72A). Específicamente, se demostró que cada una de las tres células mesoCAR T deficientes en DGK inhibe el crecimiento tumoral por volumen en comparación con las células WT y no tratadas hasta 18 días después de la inyección. También se demostró que las células deficientes en DGKz que expresan mesoCAR persisten y proliferan mejor que las células mesoCAR T de tipo salvaje en ratones (Fig. 72B).
Estos resultados tomados en conjunto demuestran que la inhibición de DGK en combinación con el tratamiento con células mesoCAR T puede mejorar la activación de células mesoCAR T y la actividad anti-tumoral en la terapia.
Ejemplo 12: La inhibición de Ikaros aumenta la capacidad anti-tumoral de células CAR-T
Uno de los principales obstáculos en la terapia de células CAR T es la regulación al alza de los reguladores negativos intrínsecos de la señalización de las células T, tales como diacilglicerol quinasa (DGK). Como se describe en el Ejemplo 11, se ha demostrado que las células CAR T pierden eficacia in vivo con el tiempo. Se demostró que la inhibición de los reguladores negativos de la función de las células T, tales como DGK, potencia la actividad y la función de las células T que expresan CAR.
Otro importante regulador negativo de la función de las células T es el factor de transcripción Ikaros. A diferencia de las DGK, que actúan principalmente en la señalización de TCR proximal, Ikaros es una proteína de unión al ADN con dedos de zinc que regula negativamente la expresión génica a través del reclutamiento de complejos de remodelación de la
cromatina, tales como Sin3A, CtBP y HDAC. Ikaros juega un papel en la regulación de la producción de citoquinas y la función citolítica en las células T CD4+ y las células T CD8+,
En este ejemplo, se examinó in vitro e in vivo. la eficacia antitumoral de células CAR T transducidas retroviralmente con expresión reducida de Ikaros .
Materiales y Métodos
Líneas celulares. Se describieron células de mesotelioma AE17 de ratón en Jackman et al., J Immunol. 2003; 171:5051-63). Se introdujo mesotelina humana en células AE17 mediante transducción lentiviral. Las células 3T3Balb/C se adquirieron de la American Type Culture Collection. Se crearon células 3T3BALB/C (3T3.FAP) que expresan FAP de ratón mediante transducción lentiviral de la línea parental FAP-3T3 con FAP murina.
Animales . Se adquirieron ratones C57BL/6 libres de patógenos de Charles River Laboratories Inc. (Wilmington, MA). Ratones Ikaros DN+/- contienen un alelo Ikaros de tipo salvaje y un alelo Ikaros con una deleción de un dominio de unión al ADN (Winandy et al., Cell. 1995; 83:289-99). Los ratones Ikzf1+/- tienen un alelo Ikaros de tipo salvaje y un alelo con deleción del exón 7 (Avitahl et al., Immunity. 1999; 10:333-43). Los animales utilizados para todos los experimentos fueron ratones hembra de entre 6 y 12 semanas de edad y se alojaron en instalaciones para animales libres de patógenos.
Aislamiento, Transducción y Expansión de Linfocitos TPrimarios de Ratón. Células T esplénicas murinas primarias se aislaron utilizando el kit "Pan T cell Negative Selection" según lo sugerido por el fabricante (Miltenyi Biotec), y se activaron en placas de 24 pocillos (4*10® células/pocillo en 2 mL de RPMI-1640 complementado con 100 U/mL de IL-2) recubierto previamente con -CD3 (1 pg/mL) y -CD28 (2 pg/mL). . Después de 48 horas, las células (1*10® células/pocillo) se mezclaron con retrovirus (1 mL de sobrenadante viral bruto) en una placa de 24 pocillos recubierta con Retronectina (50 pg/mL; Clontech) y se centrifugaron, sin freno, a temperatura ambiente durante 45 minutos a 1200g. Después de la incubación durante la noche, las células se expandieron con 50 U/mL de IL-2 durante 48 horas adicionales
Perlas recubiertas de antígeno o anticuerpo. La proteína de dominio extracelular de mesotelina recombinante, albúmina sérica bovina (Fisher Scientific) o anticuerpos anti-CD3/anti-CD28 (eBioscience) se entrecruzaron químicamente con Dynabeads de 4,5 pm activadas con tosilo (Invitrogen, n° 140-13) según las instrucciones del fabricante.
Inmunotransferencia. Se incubaron células T transducidas con anti-mesotelina-CAR con perlas recubiertas con BSA, mesotelina o anticuerpo anti-CD3 (a una relación de 2:1 de perlas a células T) durante 5 y 20 min. A continuación, se prepararon lisados celulares totales y se sometieron a inmunotransferencia para ERK fosforilada, AKT fosforilada, IKK fosforilada, JNK fosforilada, Lck fosforilada, PKC fosforilada, PLC fosforilada o ZAP70 fosforilada. Todos los anticuerpos anti-fosfo-proteína se adquirieron de Cell Signaling, con excepción del anti-fosfo-Lck, que se adquirió de Sigma Aldrich. Un anticuerpo anti-ratón de cabra frente a Ikaros (SC-9861) reactivo en el extremo C y un anticuerpo de actina anti-ratón de cabra (SC-1615) se adquirieron de Santa Cruz. Se determinaron los niveles de expresión de p-actina para normalizar las diferencias en la carga.
Citotoxicidad y ELISA de IFN Se transdujeron células AE17, AE17.meso, 3T3 y 3T3.FAP con luciferasa como se describe (Moon et al., Clinical Cancer Research. 2011; 17:4719-30). Células T y células diana se co-cultivaron en las relaciones indicadas, por triplicado, en placas de fondo redondo de 9® pocillos. Después de 18 horas, los sobrenadantes del cultivo se recogieron para el análisis de IFN utilizando un ELISA (IFN de ratón, BDOpEIA). La citotoxicidad de las células T transducidas se determinó detectando la actividad de luciferasa restante del lisado celular utilizando un ensayo descrito previamente (Riese et al., Cancer Res. 2013; 73:3566-77).
Transferencia de células CAR T a ratones portadores de tumores establecidos. A ratones se les inyectaron por vía subcutánea 2 * 10® células tumorales AE17.meso en el flanco dorsal-lateral de ratones C57BL/6. Los ratones portadores de grandes tumores establecidos (100-150 mm3) se asignaron al azar para recibir células CAR T de tipo salvaje, células CAR T deficientes en Ikaros o permanecieron sin tratar (mínimo, cinco ratones por grupo, cada experimento repetido al menos una vez). Se administraron 1*107 células T a través de la vena de la cola. El tamaño del tumor se midió con balanzas electrónicas y calibradores, respectivamente.
Para la evaluación de la actividad de las células T del Día 9, el bazo y los tumores se procesaron en suspensiones de células individuales como se describió anteriormente (Moon et al., Clinical Cancer Research. 2014; 20(1®):42®2-73). Los esplenocitos y las suspensiones de células individuales tumorales se reestimularon con anticuerpos anti-CD3/CD28 solubles (1,0 pg/ml) o con éster de forbol/ionomicina (PMA/I: 30 ng/ml, 1 uM) durante 4-6 horas en presencia de Golgi Stop (BD Biosciences, 0,66 pl/ml) y luego se recolectó para análisis de citometría de flujo.
Ensayos de citometría de flujo. Anticuerpos conjugados con fluorocromo contra IFN-y anti-ratón (XMG1), CD25 anti ratón (PC61), IL-2 anti-ratón (JES6-1A12), CD8 anti-ratón (53-6.7), CD44 anti-ratón (IM7) y CD4 anti-ratón (GK1.5) se adquirieron de Biolegend. El colorante Live/Dead Aqua fijable (L34957) se adquirió de Invitrogen. El anticuerpo fluorocromo contra Granzima B anti-ratón (NGZB) y FoxP3 (FJK-16s) se adquirió de eBioscience. Los anticuerpos de fluorocromo contra TNF-a anti-ratón (MP6-XT22) y CD69 anti-ratón (H1.2F3) se adquirieron de BD Biosciences. Para la
tinción de citoquinas intracelulares, las células se trataron con Golgi Stop (BD Biosciences, 0,66 |jg/ml) durante 4-6 horas. Después de la recolección, las células se fijaron con paraformaldehído al 1 % durante 30 minutos, se centrifugaron y se lavaron una vez con tampón FACS. Luego, las células se lavaron con BD Perm Wash (BD Biosciences) 2 veces y luego se tiñeron con anticuerpos de citoquina durante 45 minutos a temperatura ambiente. Las células se lavaron 2 veces en BD Perm Wash y luego se resuspendieron en tampón FACS. Para la tinción del factor de transcripción, las células se tiñeron en la superficie con anticuerpos primarios marcados con fluorocromo durante 20 minutos en hielo. Después de lavar en tampón FACS, las células se fijaron con tampón Fix/Perm de eBioscience. Después de la fijación, las células se permeabilizaron y se tiñeron con APC anti-FoxP3 de ratón. Para la tinción de Ikaros, se utilizó Ikaros anti-ratón de conejo (Abcam, ab26083, 1:2000) después de la fijación y permeabilización con el kit eBioscience FoxP3. Después de la tinción con el anticuerpo Ikaros, las células se lavaron y luego se tiñeron con un anticuerpo secundario anti-conejo marcado con PE (1:2000). Después de completar las tinciones, las células se procesaron en un CyanADP (Beckman Coulter) para el análisis de citometría de flujo.
Análisis estadístico. Todas las pruebas estadísticas se realizaron con GraphPad Prism. Se realizó ANOVA de dos vías con pruebas post-hoc, con * p < 0,05, ** p < 0,01, *** p < 0,001 y **** p < 0,0001. Los datos se presentan como media /-SEM.
Resultados
Se aumenta la producción de citoquinas y la liberación de mediadores citolíticos en las células T que expresan CAR con niveles reducidos de Ikaros
Dado que los linfocitos T citolíticos (CTLs, por sus siglas en inglés) con Ikaros reducido tienen una función efectora mejorada in vitro e in vivo (O'Brien, et al., J Immunol. 2014; 192:5118-29), se realizaron experimentos para testar si el agotamiento de Ikaros podría mejorar la eficacia de la terapia con CAR T. Células T aisladas de C57BL/6 de tipo salvaje y ratones haplodeficientes en Ikaros (Ikzf1+/-) en el fondo C57BL/6 se transdujeron retroviralmente para expresar un CAR anti-mesotelina. Después de la activación, transducción y expansión ex vivo en IL2, se confirmó que, en comparación con las células CAR T de tipo salvaje (WT), las células CAR T Ikzf1+/- continuaron expresando menos proteína Ikaros mediante citometría de flujo y transferencia Western (Fig. 73A).
Dado que Ikaros es un represor transcripcional para loci de genes de múltiples citoquinas (Thomas et al., J Immunol. 2007; 179:7305-15; Bandyopadhyay et al., Blood. 2006; 109:2878-2886; Thomas et al., J of Biological Chemistry. 2010; 285:2545-53; and O'Brien et al., J Immunol. 2014; 192:5118-29), se examinó a continuación si la reducción de Ikaros resultó en la producción de citoquinas autocrinas por parte de células CAR T y también si las células CAR T haplodeficientes de Ikaros respondieron mejor que sus contrapartes WT a su antígeno diana. Se estimularon células CAR T WT e Ikzf1+/- con microesferas recubiertas con BSA-(control) o mesotelina (el antígeno CAR) en una proporción de perlas:células T de 2:1 durante 6 horas, y se analizó su capacidad para producir IFNy, TNFp e IL2 por citometría de flujo. Al inicio del estudio (estimulación con b Sa ), hubo un aumento de ~3 veces en las células CAR T Ikzf1+/- productoras de IFN en comparación con las células CAR T WT (4,35 % frente a 1,4 %, Fig. 73B), pero no hubo una diferencia significativa en el % de células productoras de IL2 (Fig. 73D). Después de la estimulación con perlas recubiertas de mesotelina, hubo un aumento espectacular en el % de células CAR T Ikzf1+/- productoras de citoquinas IFN-y (25 %), mientras que la respuesta fue modesta en las células CAR T WT (7 %). También se observó un aumento en la producción de TNFa (Fig. 73C). Para investigar si este aumento en la producción de citoquinas se generalizó a través de diferentes estímulos o se limitó al antígeno CAR, se trataron células CAR T tanto WT como Ikzf+/- con PMA e ionomicina durante 6 horas. En este caso, se observaron más células productoras de citoquinas IFNy, TNF-p e IL-2 en la célula CAR T Ikzf1+/- en comparación con sus contrapartes WT (Figs. 73B-73D). Estos datos respaldan la hipótesis de que Ikaros es uno de los factores limitantes que suprime la producción de citoquinas de las células T o células CAR T.
Se demostró que un mediador citotóxico importante, granzima B, estaba regulado al alza en células OT-I deficientes en Ikaros activadas con CD3/CD28, y esto aumentó su actividad citolítica contra las células tumorales EL4 que expresan OVA (O'Brien et al . al., J Immunol. 2014; 192:5118-29). Se planteó la hipótesis de que la producción de granzima B también se potenciaría en las células T Ikzf+/- que portan CAR. Tanto las células CAR T WT como Ikzfl +/- se estimularon con perlas recubiertas con BSA (inicio del estudio) o mesotelina (antígeno CAR) en una relación de 2:1 de perlas:células T durante 6 horas. Se utilizó PMA/ionomicina como control positivo para el ensayo. De manera similar a los datos anteriores, el nivel de granzima B fue mayor en las células CAR T Ikzf+/- que en las células CAR T WT al inicio del estudio (estimulación con BSA; Fig. 73E) . Después de la estimulación con perlas recubiertas de mesotelina o PMA/ionomicina, el nivel de granzima B aumentó en las células CAR T WT e Ikzf+/-, pero la producción fue mucho mayor en las células CAR T Ikzf+/- (Fig. 73E). Para determinar si también había una diferencia en la desgranulación de las células CAR T con Ikaros reducido, se evaluó la expresión de CD107a después de la estimulación con antígeno. Las células T transducidas de tipo salvaje tenían niveles moderados de expresión de CD107a después de la reestimulación del antígeno, sin embargo, las células T con Ikaros reducido demostraron una regulación positiva mejorada de CD107a (Fig. 73F). Por lo tanto, en respuesta a la reestimulación, los CTLs con Ikaros reducido se desgranulan más y liberan más mediadores citotóxicos en comparación con sus contrapartes de tipo salvaje.
El agotamiento de Ikaros con un alelo negativo dominante potencia la función de las células CAR T
Además de las células con niveles más bajos de Ikaros, se estudiaron las células T de ratones que expresaban un alelo negativo dominante de Ikaros (IkDN). Ratones transgénicos que expresan IkDN tienen un desarrollo linfoide normal pero tienen células T periféricas con una actividad de unión al ADN de Ikaros reducida en un 90 % (Thomas et al., J Immunol.
2007; 179:7305-15; y Winandy et al., Cell. 1995; 83: 289-99). Células T aisladas de bazos de ratones WT e IkDN se activaron con anticuerpos anti-CD3/CD28 unidos a placa, se transdujeron con CAR anti-mesotelina, seguido de expansión con IL2. La reducción de Ikaros en las células CAR T IkDN fue confirmado por transferencia western. Células CAR T WT e IkDN se volvieron a enfrentar a microesferas recubiertas de BSA o mesotelina en una relación de microesferas:células T de 2:1 durante 6 horas, y se analizó su capacidad de producir IFN e IL2, así como de desgranular en respuesta al antígeno CAR. De manera similar a los datos de Ikzf1+/- anteriores, se observó una cierta producción de IFNy autocrino al inicio del estudio (Fig. 74A), pero no con IL2 (estimulación con BSA; Fig. 74B). Tras la unión de CAR con su antígeno diana, mesotelina, las células T IkDN produjeron más IFNy que las células T WT (estimulación con mesotelina; Fig. 74A). La desgranulación, medida por la regulación al alza de CD107a, también fue similar en las células CAR T de tipo salvaje e IkDN (Fig. 74D).
El agotamiento de Ikaros no aumentó la activación ni la señalización de las células CAR T después de la estimulación con antígeno
Dado que el agotamiento en Ikaros aumentó la liberación de citoquinas y aumentó los niveles de granzima B y la expresión de CD107a de las células CAR T, se exploraron posibles mecanismos. Es plausible que estos cambios en la función efectora se deban a diferencias en la activación de las células T de tipo salvaje y transducidas con Ikzf1+/-. Así, los niveles de CD69, CD25 y 4-1BB (marcadores de activación de células T) se midieron por citometría de flujo después de estimular con perlas recubiertas de mesotelina durante 6 y 24 horas. CD69, un marcador de activación temprano, se regulaba al alza en la misma medida tanto por las células de tipo salvaje como por las células Ikzf1+/- (Fig. 75a ). Con una estimulación más prolongada, las células T que expresan CAR de tipo salvaje e Ikzf1+/- continuaron expresando niveles similares de CD69, pero las células T transducidas con Ikzf1+/- exhibieron una mayor expresión de CD25 (Fig. 75B). Sin embargo, es posible que esto no indique directamente una diferencia en la activación de las células T, ya que el aumento de IL-2 por parte de las células Ikzf1+/- (Fig. 71D) puede actuar en un bucle positivo de alimentación hacia adelante en CD25, la IL-2Ra (Depper et al., Proc Natl Acad of Sci USA. 1985; 82:4230-4; y Nakajima et al., Immunity.
1997; 7:691-701). 4-1BB, un miembro de la superfamilia de receptores TNF también se expresa en células T activadas (Vinay et al., Seminars in Immunology. 1998; 10:481-9) y se expresó a niveles similares por células T de tipo salvaje e Ikzf1+/- transducidas con CAR después de la estimulación con antígeno (Fig. 75C). Por lo tanto, las diferencias funcionales entre las células T WT e Ikzf1+/- transducidas no se debieron a diferencias en la activación de las células T.
Los experimentos descritos en el Ejemplo 11 y Riese et al., Cancer Research. 2013; 73:3566-77, demuestran que el agotamiento de la enzima diacilglicerol quinasa (DGK) en las células CAR T dio como resultado un aumento en la señalización de RAS/ERK, que se correlacionó bien con una activación potenciada de las células CAR T. Se examinaron algunas vías de señalización en células T WT y deficientes en Ikaros después de la estimulación de TCR con anticuerpos CD3/CD28. Se prepararon lisados de células T estimuladas y se sometieron a inmunotransferencia para diversas fosfoproteínas implicadas en la señalización proximal (PLC y Lck) y distal (ERK1/2, JNK, AKT e IKKa) del TCR. Hubo una activación basal constitutiva de bajo nivel de algunas proteínas de señalización de TCR en células T deficientes en Ikaros, incluidas Lck, ERK y AKT (Fig. 75D). Con la estimulación con TCR/CD28, todas las proteínas estudiadas se fosforilaron al mismo nivel al comparar las células T WT y las células T deficientes en Ikaros, con la excepción de fosfo-IKK, que fue ligeramente superior en las células T deficientes en Ikaros 20 minutos después de la estimulación. Para determinar si la vía de NFB se mejoró en las células T con un nivel reducido de Ikaros, se volvió a sondear la misma transferencia para IB, la diana aguas abajo para IKK. No hubo diferencia en la degradación de IB entre las células T WT y las células T empobrecidas en Ikaros. Para estudiar la señalización de CAR, se reestimularon células T transducidas con CAR anti-mesotelina WT e IkDN con perlas recubiertas de mesotelina. De manera similar a los datos con la estimulación con CD3/CD28, no se encontraron diferencias en la fosforilación de PLC y ERK (Fig. 75E). Juntos, estos datos indican que el agotamiento en Ikaros no altera la señalización mediada por TCR/CAR.
La reducción de Ikaros en las células CAR T aumenta su respuesta contra sus células diana.
Dada la mayor producción de factores efectores por parte de las células CAR T con Ikaros reducido, se testó su eficacia contra las células tumorales diana in vitro. Células T de tipo salvaje, Ikzf1+/- e IkDN que expresan mesoCAR se mezclaron en diferentes relaciones con la línea celular tumoral parental, AE17 o la línea celular que expresa mesotelina, AE17meso. Cuando se mezclaron con la línea celular parental, las células T de tipo salvaje, Ikzf1+/- e IkDN no produjeron IFN-y ni lisaron células en respuesta a AE17 (Figs. 76A, 76B y 76C). Por el contrario, cuando se hizo reaccionar con AE17meso, la producción de IFN-y y la citólisis por parte de las células T de tipo salvaje aumentaron a medida que aumentaba la relación E:T (Figs. 76B y 76C). Sin embargo, tanto las células T Ikzf1+/- como IkDN produjeron significativamente más IFN-y y tuvieron una lisis tumoral significativamente mayor que las células T de tipo salvaje, incluso con la relación E:T más baja de 1,3:1 (Figs. 76B y 76C).
Para estudiar la generalización de este efecto, se examinaron las células T que expresan una construcción CAR diferente, que fija como objetivo la proteína de activación de fibroblastos (FAP-CAR) y tiene el mismo dominio de señalización
intracelular que la CAR anti-mesotelina utilizada anteriormente. La eficacia de células T esplénicas FAP-CAR transducidas de forma equiparable aisladas de WT C57BL/6 se comparó con las de ratones Ikzf1+/-. Las células FAP-CAR T Ikzf1+/- fueron más eficientes para lisar células 3T3.FAP (Fig. 76D) y para secretar más IFN (Fig. 76E) que las células CAR T WT FAP, con retención de especificidad in vitro.
E l agotam iento de Ikaros potencia la eficacia de las células CAR T contra tum ores establecidos
A continuación, se examinó la capacidad de las células T específicas para mesotelina con Ikaros reducido (Ikzf1+/- e IkDN) de controlar el crecimiento de tumores AE17meso establecidos en ratones. Se inyectaron dos millones de células tumorales AE17meso en los flancos de ratones C57BL/6 singénicos y se permitió que formaran grandes tumores establecidos (~100-150 mm3). Diez millones de células CAR T preparadas a partir de ratones WT o deficientes en Ikaros (Ikzf1+/- e IkDN) se transfirieron luego de manera adoptiva a esos ratones portadores de tumores y se siguieron las mediciones de los tumores. La inhibición leve del crecimiento del tumor fue inducida por células mesoCAR T transducidas de tipo salvaje, mientras que las células mesoCAR T tanto transducidas con Ikzf1+/-como IkDN inhibían significativamente más el crecimiento de tumores AE17meso (Figs. 77A y 77B).
También se estudió si la reducción de Ikaros podría mejorar el potencial terapéutico de las células FAP-CAR T. Se transfirieron de forma adoptiva ratones con tumores AE17meso establecidos (100-150 mm3) con 10 millones de células CAR T anti-FAP de ratón de tipo salvaje o transducidas con Ikzf1+/-. Los ratones que recibieron células transducidas de tipo salvaje proporcionaron un retraso tumoral mínimo y los tumores AE17meso continuaron creciendo (Fig. 77). Por el contrario, las células T transducidas con Ikzf1+/- fueron capaces de retrasar significativamente el crecimiento del tumor.
Células CAR T Ikzf1+/- persisten m ás tiem po y son m ás resistentes a l m icroentorno de l tum or inm unosupresor que las células CAR T W T
Dada la eficacia potenciada de las células CAR T inhibidas por Ikaros, se exploraron los posibles mecanismos que utilizan las células mesoCAR T Ikzf1+/-. Para investigar más a fondo cómo funcionan estas células mesoCAR T en un microentorno del tumor inmunosupresor in vivo, se recolectaron tumores a los 3 y 9 días después de la transferencia adoptiva y se evaluó su número y funcionalidad. Estos dos momentos permitieron la caracterización de su actividad en momentos tempranos y tardíos durante la respuesta inmune anti-tumoral.
El Día 3 después de la transferencia, los autores de la invención observaron una frecuencia similar de células mesoCAR T de tipo salvaje e Ikzf1+/- tanto en el bazo (Fig. 78A) como en los tumores (Fig. 78B). Estos niveles similares indican que tanto las células mesoCAR T de tipo salvaje como Ikzf1+/- inicialmente transitan igualmente bien hacia el tumor. Al evaluar el momento del día 9, el número tanto de células mesoCAR T de tipo salvaje como células mesoCAR T Ikzf1+/-disminuyó en el bazo. Sin embargo, cuando se examinaron los tumores en este momento, hubo un aumento significativo en el número de células mesoCAR T Ikzf1+/- en comparación con las células mesoCAR T WT (Fig. 78B). Estos datos demuestran que las células mesoCAR T Ikzf1+/- persisten o proliferan mejor que las células mesoCAR T WT en el microentorno inmunosupresor.
Los linfocitos infiltrantes de tumores (TILs) se vuelven hipofuncionales en respuesta a sus antígenos afines dentro del microentorno del tumor inmunosupresor, y es un fenómeno clave asociado con la progresión del tumor (Prinz et al., J Immunol. 2012; 188:5990-6000; y Kerkar et al., Cancer Res. 2012, 72:3125-30). Aunque había más células mesoCAR T Ikzf1+/- en los tumores el Día 9, aún podrían verse afectadas negativamente por el microentorno del tumor. Para evaluar la funcionalidad, se utilizaron anticuerpos CD3/CD28 para estimular TILS aislados de células mesoCAR T de tipo salvaje e Ikzf1+/- el Día 9 después de la transferencia y se caracterizaron las diferencias en la producción de mediadores líticos. El Día 9 después de la transferencia, las células mesoCAR T de tipo salvaje en el bazo continuaron produciendo algunos niveles moderados de IFN (Fig. 78C). Como era de esperar, las células T de tipo salvaje aisladas de los tumores produjeron mucho menos de esta citoquina en comparación con las células T de tipo salvaje aisladas del bazo. Esto indica que los TILs de tipo salvaje comienzan a volverse hipofuncionales el Día 9 posterior a la transferencia. Por el contrario, las células mesoCAR T Ikzf1+/- esplénicas continuaron produciendo más IFN- al inicio del estudio y tras la estimulación (Fig. 78C). En comparación con los TILs de tipo salvaje, los TILs Ikzf1+/- produjeron mayores cantidades de IFNy. Estos datos indican que los TILs Ikzf1+/- podrían ser menos sensibles al microentorno del tumor inmunosupresor.
Se puede evitar el defecto proximal de la señalización de TCR que se observa a menudo en los TILs mediante el uso de PMA/Ionomicina (PMA/I) (Prinz et al., J Immunol. 2012; 188:5990-6000). Los linfocitos mesoCAR T de tipo salvaje e Ikzf1+/- se reestimularon con PMA/I para determinar si los TILs de tipo salvaje e Ikzf1+/- insensibles a la estimulación por TCR todavía eran capaces de producir citoquinas en respuesta a otros estímulos. El Día 9 después de la transferencia, los TILs de tipo salvaje demostraron una caída notable en la producción de IFN-y (Fig. 78C), y esto se restauró parcialmente a través de la estimulación con PMA/I (Fig. 78D). Sin embargo, la estimulación de las células mesoCAR T Ikzf1+/- esplénicas y aisladas del bazo todavía dio como resultado un aumento de los niveles de IFN-y en comparación con las células transferidas de tipo salvaje. Al omitir cualquier defecto en la señalización de TCR a través de la estimulación con PMA/I, estos resultados demuestran que la producción de IFNy difiere en el nivel de cromatina y probablemente se deba a la función diferencial de Ikaros.
A la vista del aumento de la actividad anti-tumoral de las células T Ikzf1+/- transferidas, también se examinaron los impactos en la composición del microentorno del tumor inmunosupresor y la cantidad de células T reguladoras (Tregs) y células supresoras derivadas de mieloides. (MDSC) se evaluó el Día 9. El Día 9 después de la transferencia, se observaron niveles similares de Tregs en huéspedes que recibieron células mesoCAR T de tipo salvaje o Ikzfl +/-(Fig. 78E). Se determinó la presencia de macrófagos Ly6G-/CD11b+/CD206+, que típicamente se caracterizan como macrófagos M2 inmunosupresores y pro-tumorigénicos. En los grupos tratados el Día 9, esa expresión de CD206 fue similar en los 3 grupos (sin tratar, de tipo salvaje e Ikzf1+/-) (Fig. 78F).
Células T con Ikaros reducidos son menos sensibles a los factores inhibidores solubles TGF y adenosina
Para caracterizar adicionalmente la interacción del microentorno del tumor inmunosupresor con las células mesoCAR T, se utilizó un sistema de cultivo in vitro. Se ha demostrado que factores inhibidores solubles, tales como IDO, IL-10, adenosina y TGF-p (Wang et al., Oncoimmunology. 2013; 2:e26492) contribuyen a inhibir la infiltración de linfocitos tumorales. Se testaron los efectos de factores inhibidores selectos in vitro en las células CAR T deficientes en Ikaros para determinar si el entorno inmunosupresor podría afectar a su función lítica. Células CAR T de tipo salvaje tenían una reducción del 50 % en su capacidad para producir IFN-y y tenían una reducción en su función lítica en presencia de TGF-p y adenosina (Fig. 79). Las células CAR T con niveles reducidos de Ikaros (Ikzf1+/- e IkDN) continuaron produciendo más IFN y que sus contrapartes de tipo salvaje en ausencia de inhibidores y solo se inhibieron marginalmente en presencia de TGF p y adenosina (Fig. 79A). Se observó un aumento de la función lítica en las células CAR T Ikzf1+/- e IkDN en comparación con las células T de tipo salvaje (Fig. 79B). Estos datos demuestran que las células T con Ikaros reducido son menos sensibles a la inhibición de TGFp y adenosina.
Análisis
En este ejemplo, se ha identificado un nuevo enfoque para permitir que células T que expresan CAR sobrevivan y potencien sus funciones efectoras en el entorno del tumor: la inactivación del represor transcripcional Ikaros, que se sabe que inhibe una diversa gama de genes implicados. en la función de las células T, p. ej., genes de citoquinas (IL2 e IFNy), mediadores citolíticos (granzima B) y los factores de transcripción clave de la caja T que influyen en la diferenciación de las células T (R-Bet y Eomes).
Un hallazgo importante de los experimentos descritos anteriormente es que las células CAR T que eran deficientes en la función Ikaros fueron significativamente mejores que las células CAR T de tipo salvaje para restringir el crecimiento del tumor (Fig. 77). Estos resultados se observaron en múltiples modelos de tumores y utilizando dos construcciones CAR diferentes. Debido a la gran cantidad de genes que regula Ikaros, es plausible que Ikaros pueda regular muchas vías que normalmente son sensibles a la inmunosupresión. El aumento de la producción de IFN-g al reducir el nivel de Ikaros en las células CAR T puede dar como resultado una regulación positiva de la expresión de MHC de clase I en el tumor y, por lo tanto, mejorar su inmunogenicidad, mejorar la actividad antiangiogénica e impulsar la función mediada por STATl de las células Th1. Un posible efecto del aumento de IFNy podría haber sido una alternancia del fenotipo de macrófagos dentro de los tumores; sin embargo, no se observaron diferencias en el número total de macrófagos ni en la proporción de macrófagos similares a M2 (medidas por la expresión de CD206). El aumento de la producción de IL-2 también podría haber aumentado la formación de células Treg CD4, sin embargo, no se observaron diferencias al comparar los tumores tratados con células CAR T WT con células CAR T deficientes en Ikaros.
Se observó un aumento en el número de linfocitos infiltrantes en el tumor nueve días después de la inyección. Esto probablemente no se debió a un mayor tráfico, ya que el número de TIL WT frente a los deficientes en Ikaros fue similar el Día 3. En cambio, estos resultados sugieren que los TIL deficientes en Ikaros mostraron una proliferación incrementada o una disminución de la muerte celular inducida por antígeno (AICD, por sus siglas en inglés). Estudios In vitro sugieren que la AICD fue similar entre los dos tipos de células T, lo que hace más probable que la diferencia se deba a una proliferación incrementada. Esto sería consistente con el aumento de IL-2 producida por estas células. Además de una mayor persistencia, el TIL deficiente en Ikaros parecía ser menos hipofuncional. Cuando los linfocitos CAR T que infiltran el tumor se volvieron a estimular con anticuerpo anti-CD3 o PMA e ionomicina, los CAR TILs con deficiencia de Ikaros pudieron producir más IFNy que sus contrapartes de tipo salvaje (Fig. 78C y 78D).
Los estudios in vitro permitieron realizar más estudios sobre los fundamentos mecánicos de la mayor eficacia anti-tumoral observada in vivo. De acuerdo con las funciones inhibidoras conocidas de Ikaros, la eliminación de un alelo de Ikaros (Ikzf+l-) o el reemplazo de uno de sus alelos por una construcción negativa dominante de Ikaros (IkDN) dio como resultado células T que tenían un aumento en la producción inicial de IFNy autocrino y granzima B. (Fig. 73), pero lo que es más importante, mostró una secreción de citoquinas y una liberación de gránulos marcadamente aumentada después de la estimulación con TCR o CAR in vitro.. Esto estuvo acompañado por un aumento del exterminio de células tumorales in vitro.. Para testar si las células CAR T no deficientes en Ikaros tienen o no un umbral de activación más bajo que las células CAR T WT, las Dynabeads se recubrieron con 10 veces menos de proteína de mesotelina y se demostró que las células CAR T Ikzf+/- aún podían responder al antígeno de mesotelina a baja densidad para producir IFNy y TNF-a, pero las células CAR T WT no pudieron. Las células CAR T inhabilitadas para Ikaros también eran más resistentes a la inhibición por factores inmunosupresores conocidos, tales como TGF-p y adenosina (Fig. 79). Esto puede deberse al hecho de que las citoquinas (es decir, IL2 e IFNy y los genes efectores de células T (es decir, granzima B) son más
accesibles para la transcripción en células T deficientes en Ikaros después de la activación de TCR/CAR. Esto parece ayudar a compensar la activación de células T dentro del microentorno del tumor inmunosupresor.
En estudios anteriores, p. ej., el Ejemplo 11, se observó un fenotipo similar (es decir, un aumento en la citólisis y la producción de IFN) en las células Ca R T a través del agotamiento de las DGKs, enzimas que metabolizan el segundo mensajero diacilglicerol y limitan la activación de RAS/ERK. Sin embargo, con la eliminación de DGK, se observaron cambios claros en la vía de señalización CAR/TCR. Específicamente, la activación de RAS/ERK mejoró drásticamente después de la activación de TCR y CAR. Esto dio como resultado una activación mejorada, medida por una mayor regulación positiva de CD69, sin embargo, la producción de moléculas efectoras como TRAIL, FasL e IFNy. Perforina y Granzima B fueron similares entre las células CAR T WT y deficientes en DGK. Un fenotipo muy diferente en este estudio con células CAR T deficientes en Ikaros. En contraste con las células CAR T deficientes en DGK, las células CAR T deficientes en Ikaros tenían una activación de CAR/TCR similar, como lo demuestra la regulación al alza de CD69 y CD25 (Fig. 75), y una señalización similar de CAR/TCR medida por la fosforilación de múltiples moléculas de señalización TCR (Fig. 75) y señalización de calcio. A diferencia de las células T con deficiencia de DGK, las células CAR T sin Ikaros tenían niveles más altos de granzima B e IFN al inicio (Figs. 73 y 74), así como una activación constitutiva de bajo nivel de algunas cascadas de señalización de TCR, tales como ERK y Akt (Fig. 75). Esta activación inicial puede deberse a la inducción de T-bet (Thomas et al., J of Biol Chemistry. 2010; 285:2545-53), que coopera con otros factores de transcripción tales como Eomes para transactivar la expresión génica de IFN y granzima B (Pearce et al., Science. 2003; 302:1041-3; e Intelkofer et al., Nat Immunol. 2005; 6:1236-44). 2003; 302:1041-3; and Intelkofer et al., Nat Immunol. 2005; 6:1236-44).
Estos hallazgos plantean la posibilidad de que fijar terapéuticamente como objetivo Ikaros en células T humanas transducidas (en ensayos clínicos) podría ser beneficiosa utilizando enfoques genéticos o bioquímicos. Los enfoques genéticos podrían imitar estos resultados en ratones e incluir la eliminación de Ikaros en las células CAR T utilizando ARNsh o el uso de una construcción negativa dominante para competir con los Ikaros endógenos. Otra opción sería utilizar un inhibidor farmacológico para reducir los niveles de Ikaros de forma transitoria. Informes recientes han indicado que el fármaco inmunomodulador, lenalidomida fija como objetivo a Ikaros para la degradación mediada por ubiquitina por el complejo de ligasa E3 CRL4CRBN (Gandhi et al., Br J Haematol. 2013; 164:811-21; Kronke et al, Science. 2014; 343:301-5; y Sakamaki et al., Leukemia. 2013; 28:329-37). Células T humanas estimuladas con CD3 tratadas con lenalidomida producen más IL-2 (Gandhi et al., Br J Haematol. 2013; 164:811-21), un rasgo clave de las células T con niveles reducidos de Ikaros. En estudios preliminares, las PBMC humanas transducidas con mesotelina-CAR estimuladas con TCR/CD28 in vitro produjeron más IL-2 e IFNy después del pretratamiento con lenalidomida. Por lo tanto, la combinación de la terapia con células CAR T con la administración in vivo de lenalidomida puede proporcionar una estrategia terapéutica para revertir la hipofunción de las células T a través de la inhibición de Ikaros.
En conclusión, este ejemplo demuestra por primera vez que fijar como objetivo un represor transcripcional puede potenciar la inmunidad antitumoral mediada por CAR. Los mecanismos implicaron una función mejorada de citoquinas y efectores sin alteraciones en la transducción de señales. La traducción de este enfoque a la clínica se puede llevar a cabo mediante el uso de ARNsh, una construcción negativa dominante o un inhibidor farmacológico (tal como lenalidomida) para fijar como objetivo a Ikaros en las células T que expresan CAR.
Ejemplo 14: La IL-7 exógena mejora la función de los célula CAR T
Después de la transferencia adoptiva de célula CAR T, algunos pacientes experimentan una persistencia limitada de los célula CAR T, lo que puede dar lugar a niveles subóptimos de actividad antitumoral. En este ejemplo, los efectos de la administración de IL-7 humana exógena se evalúan en modelos de xenoinjerto de ratón en los que se ha observado una respuesta subóptima inicial a los célula CAR T.
Tratamiento con IL-7 exógena en un modelo de linfoma
La expresión del receptor CD127 de IL-7 se evaluó primero en diferentes líneas celulares de cáncer y en células que expresan CAR. Se analizaron dos líneas celulares de linfoma de células del manto (RL y Jeko-1) y una línea celular l La -B (Nalm-6) mediante citometría de flujo para determinar la expresión de CD127. Como se muestra en la Figura 80A, de las tres líneas de células cancerosas testadas, se demostró que RL tenía la expresión más alta de CD127, seguida de Jeko-1 y Nalm-6. Se infundieron célula CART19 en ratones NSG y se evaluó la expresión de CD127 en las células CART19 circulantes mediante citometría de flujo. Como se muestra en la Figura 80B, CD127 se expresa uniformemente en todas las células CART19 circulantes.
A continuación, se evaluó el efecto del tratamiento con IL-7 exógena sobre la actividad antitumoral de las células CART19 en un modelo animal de linfoma. A los ratones NSG se les injertó una línea celular del manto que expresa luciferasa (RL luc) el día 0 (D0), seguido de un tratamiento con células CART19 el Día 6. Los ratones NSG se dividieron en grupos, en donde un grupo no recibió células CART19, un el segundo grupo recibió 0,5 *106 células CART19, un tercer grupo recibió 1*106 células CART19 y un cuarto grupo recibió 2*106 células CART19. El tamaño del tumor se controló midiendo la bioluminiscencia media de los tumores injertados durante más de 80 días. Solo los ratones que recibieron 2*106 células CART19 demostraron rechazo del tumor e inhibición del crecimiento del tumor (Figura 81A). Los ratones de los dos
grupos que recibieron 0,5 ><106 células CART19 o 1 x106 células CART19 mostraron una respuesta antitumoral subóptima. Luego, los ratones de estos dos grupos se aleatorizaron, en donde tres ratones (ratón n° 3827 y n° 3829 que recibieron 0,5^10® células CART19, y el ratón n° 3815 que recibió 1 x io6 células CART19) recibieron IL-7 humana recombinante exógena a una dosis de 200 ng/ratón mediante inyección intraperitoneal tres veces por semana a partir del Día 85, y dos ratones no lo hicieron. La carga tumoral de ratones que recibieron IL-7 exógena desde el Día 85-125, detectada por bioluminiscencia media, se muestra en la Figura 81B. Todos los ratones que recibieron IL-7 mostraron una respuesta considerable de 1-3 log de reducción de la carga tumoral. Los ratones que originalmente recibieron una dosis más alta de células CART19 (ratón n.° 3815 que recibió 1x106 células CART19) mostraron una respuesta más profunda. Al comparar la carga tumoral de los ratones que recibieron tratamiento con IL-7 con el control, antes y después del tratamiento con IL-7, la reducción del tumor en la carga tumoral solo se observó en los ratones que habían recibido tratamiento con IL-7 (Figura 81C).
También se examinó la dinámica de células T después del tratamiento con IL-7 en el modelo animal de linfoma. Células CART19 humanas no eran detectables en la sangre antes del tratamiento con IL-7. Tras el tratamiento con IL-7, hubo un aumento rápido, pero variable, en el número de células T en los ratones tratados (Figura 82A). El grado de expansión de células T observado en los ratones que recibieron IL-7 también se correlacionó con la respuesta del tumor. El ratón con el mayor número de células T detectadas en la sangre en la expansión máxima durante el tratamiento con IL-7 (ratón n° 3815) tuvo la reducción más sólida en la carga tumoral (véase la Figura 81B). Por otra parte, el momento de la expansión máxima se correlacionó con la dosis de células T inyectada como valor de referencia. El número/nivel de células que expresan CD3 en la sangre también se midió antes y después del tratamiento con IL-7. En los ratones de control, se detectaron muy pocas células que expresaban CD3, mientras que los ratones tratados con IL-7 mostraron un aumento significativo de células CD3+ después del tratamiento con IL-7 (Figura 82B).
Tratamiento con IL-7 exógena en un modelo de leucemia
La expresión del receptor de IL-7 (CD127) se midió en líneas celulares de leucemia (línea celular AML MOLM14 y línea celular B-ALL NALM6) y células AML primarias mediante citometría de flujo (Figura 83, paneles superiores). La expresión del receptor de IL-7 se expresa en las células de la línea celular B-ALL NALM6, pero no en la línea celular AML MOLM14 o AML primaria. El análisis de citometría de flujo se controló de tal manera que la expresión del receptor de IL-7 se detectó solo en las células tumorales.
A continuación, se evaluó el efecto del tratamiento con IL-7 exógena sobre la actividad antitumoral de las células CART33 o CART123 en un modelo animal de leucemia de recaída de AML después de un tratamiento inicial con CART (Figura 84). Se inyectaron células MOLM14 que expresan luciferasa en ratones NSG y los ratones desarrollaron AML. Se inició el tratamiento con CART33 o CART123 y se controló la carga tumoral mediante imágenes de bioluminiscencia en serie. Se inyectaron células T no transducidas como control. Los ratones que recibieron tratamiento con CART33 o CART123 respondieron inicialmente al tratamiento con células T, pero recayeron 14 días después de la infusión de células T (Figura 85A). La expresión del receptor de IL-7 se midió en células de AML mediante citometría de flujo en los ratones que presentaban una recaída de AML. La expresión del receptor de IL-7 no se detectó en las células de AML recidivantes, tanto si fueron tratadas con CART33 como con CART123 (Figura 83, paneles inferiores).
El Día 28, los ratones que habían recaído se aleatorizaron y se asignaron para no recibir tratamiento (control) o recibir tratamiento con IL-7 a una dosis de 200 ng/ratón mediante inyección intraperitoneal tres veces por semana. La carga tumoral después del tratamiento con IL-7 o el tratamiento de control se controló mediante imágenes de bioluminiscencia semanales y también se evaluó la respuesta, la expansión de las células T y la supervivencia general. La Figura 85B muestra la mejor respuesta después de que se mostró el tratamiento con IL-7 para los grupos de tratamiento y control con IL-7, según lo determinado por formación de imágenes por bioluminiscencia (BLI). Las imágenes de bioluminiscencia representativas se muestran en la Figura 85C durante los 28 días de tratamiento con IL-7 o de control, lo que muestra que la respuesta antitumoral aumentó en ratones que recibieron tratamiento con IL-7. La expansión de células T (p. ej., CART33 y CART123) se cuantificó a partir de la sangre de los ratones, y el aumento del número de células T en la sangre durante el tratamiento con IL-7 se correlacionó con la reducción de la carga tumoral (Figura 86A). Los ratones que recibieron tratamiento con IL-7 también demostraron una mayor supervivencia (Figura 86B).
Juntos, los resultados de este ejemplo demuestran que el tratamiento con IL-7 exógena aumenta la proliferación de células T y la actividad antitumoral in vivo,, lo que indica que el uso de IL-7 en pacientes con resultados subóptimos después de la terapia con CAR o recaída puede mejorar la respuesta anti-tumoral en estos pacientes.
Sin una descripción adicional, se cree que un experto en la técnica puede, utilizando la descripción anterior y los siguientes ejemplos ilustrativos, preparar y utilizar los compuestos de la presente descripción y practicar los métodos descritos.
EQUIVALENTES
Claims (1)
- REIVINDICACIONES1. Una célula efectora inmunitaria (p. ej. una población de células efectoras inmunitarias), que comprende:(a) una primera molécula de receptor de antígeno quimérico (CAR) o ácido nucleico que la codifica, en donde dicha primera molécula de CAR es un polipéptido de CAR que comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio de señalización intracelular que comprende un dominio co-estimulador y/o un dominio de señalización primario, y en donde dicho dominio de unión a antígenos comprende un dominio VHH y se une a un antígeno tumoral; y(b) una segunda molécula de CAR o ácido nucleico que la codifica, en donde dicha segunda molécula de CAR es un polipéptido CAR que tiene un dominio de unión a antígenos que comprende un scFv.2. La célula efectora inmunitaria de la reivindicación 1, en donde el ácido nucleico que codifica la primera molécula de CAR y el ácido nucleico que codifica la segunda molécula de CAR están presentes cada uno en un vector.3. La célula efectora inmunitaria de la reivindicación 1 o 2, en donde el dominio VHH es un dominio VHH de camélido.4. La célula efectora inmunitaria de una cualquiera de las reivindicaciones 1-3, en donde el antígeno tumoral se selecciona de un grupo que consiste en: CD19; mesotelina; receptor del factor de crecimiento epidérmico (EGFR); CD123; CD22; CD30; CD171; CS-1 (también denominado subconjunto 1 de CD2, CRACC, SLAMF7, CD319 y 19A24); molécula 1 similar a lectina de tipo C (CLL-1 o CLECL1); CD33; variante III del receptor del factor de crecimiento epidérmico (EGFRvIII); gangliósido G2 (GD2); gangliósido GD3 (aNeu5Ac(2-8)aNeu5Ac(2-3)bDGalp(1-4)bDGlcp(1-1)Cer); maduración de células B de miembros de la familia de receptores de TNF (BCMA); antígeno Tn ((Tn Ag) o (GalNAca-Ser/Thr)); antígeno de membrana específico de próstata (PSMA); Receptor huérfano de tipo tirosina quinasa 1 (ROR1); Tirosina quinasa 3 de tipo Fms (FLT3); glucoproteína 72 asociada a tumores (TAG72); CD38; CD44v6; antígeno carcinoembrionario (CEA); molécula de adhesión de células epiteliales (EPCAM); b 7h 3 (CD276); KIT (CD117); subunidad alfa-2 del receptor de interleucina-13 (IL-13Ra2 o CD213A2); mesotelina; receptor alfa de interleucina 11 (IL-11Ra); antígeno de células madre prostáticas (PSCA); proteasa serina 21 (testisina o PRSS21); receptor 2 del factor de crecimiento endotelial vascular (VEGFR2); antígeno de Lewis(Y); CD24; receptor beta del factor de crecimiento derivado de plaquetas (PDGFR-beta); antígeno embrionario 4 específico de etapa (SSEA-4); CD20; receptor de folato alfa; tirosina-proteína quinasa receptora ERBB2 (Her2/neu); Mucina 1, asociada a la superficie celular (MUC1); receptor del factor de crecimiento epidérmico (EGFR); molécula de adhesión de células neurales (NCAM); prostasa; fosfatasa ácida prostática (PAP); factor de elongación 2 mutado (ELF2M); efrina B2; proteína alfa de activación de fibroblastos (FAP); receptor del factor insulínico de crecimiento 1 (receptor de IGF-I), anhidrasa carbónica IX (CAIX); subunidad de proteasoma (prosoma, macropaína), tipo beta, 9 (LMP2); glucoproteína 100 (gp100); proteína de fusión de oncogén que consiste en la región de agrupamiento de puntos de ruptura (BCR) y el homólogo 1 del oncogén vírico de la leucemia murina de Abelson (Abl) (bcr-abl); tirosinasa; receptor 2 de efrina de tipo A (EphA2); Fucosil GM1; molécula de adhesión de sialilo de Lewis (sLe); gangliósido GM3 (aNeu5Ac(2-3)bDGalp(1-4)bDGlcp(1-1)Cer); transglutaminasa 5 (TGS5); antígeno asociado a melanoma de alto peso molecular (HMWMAA); gangliósido o-acetil-GD2 (OAcGD2); receptor de folato beta; marcador endotelial tumoral 1 (TEM1/CD248); relacionado con el marcador endotelial tumoral 7 (TEM7R); claudina 6 (CLDN6); receptor de la hormona estimulante del tiroides (TSHR); receptor acoplado a proteína G clase C grupo 5, miembro D (GPRC5D); marco de lectura abierto 61 del cromosoma X (CXORF61); CD97; CD179a; quinasa de linfoma anaplásico (ALK); ácido polisiálico; específico de placenta 1 (PLAC1); porción de hexasacárido de glucoceramida globoH (GloboH); antígeno de diferenciación de glándula mamaria (NY-BR-1); uroplaquina 2 (UPK2); receptor celular 1 del virus de la hepatitis A (HAVCR1); adrenoceptor beta 3 (ADRB3); panexina 3 (PANX3); receptor acoplado a proteína G 20 (GPR20); complejo de antígeno de células 6, locus K 9 (LY6K); receptor olfativo 51E2 (OR51E2); Proteína de Marco de Lectura Alternativa TCR Gamma (TARP); proteína del tumor de Wilms (WT1); antígeno de cáncer/testículo 1 (NY-ESO-1); antígeno de cáncer/testículo 2 (LAGE-1a); antígeno 1 asociado al melanoma (MAGE-A1); gen 6 variante de translocación de ETS, ubicado en el cromosoma 12p (ETV6-AML); proteína de espermatozoide 17 (SPA17); familia de antígenos X, miembro 1A (XAGE1); receptor de superficie celular de unión a angiopoyetina 2 (Tie 2); antígeno de testículo y cáncer de melanoma 1 (MAD-CT-1); antígeno de testículo y cáncer de melanoma 2 (MAD-CT-2); antígeno 1 relacionado con Fos; proteína tumoral p53 (p53); p53 mutante; prosteína; survivina; telomerasa; antígeno tumoral de carcinoma de próstata-1 (PCTA-1 o Galectina 8), antígeno de melanoma reconocido por células T 1 (MelanA o MARTI); mutante de sarcoma de rata (Ras); transcriptasa inversa de telomerasa humana (hTERT); puntos de ruptura de translocación de sarcoma; inhibidor de melanoma de la apoptosis (ML-IAP); ERG (proteasa transmembrana, gen de fusión de ETS y serina 2 (TMPRSS2)); N-acetil glucosaminil-transferasa V (NA17); proteína de caja emparejada Pax-3 (PAX3); receptor de andrógenos; ciclina B1; homólogo derivado del neuroblastoma del oncogén vírico de la mielocitomatosis aviar v-myc (MYCN); miembro C de la familia de homólogos de Ras (RhoC); proteína 2 relacionada con la tirosinasa (TRP-2); citocromo P450 1B1 (CYP1B1); similar al factor de unión a CCCTC (proteína de dedo de zinc) (BORIS o hermano del regulador de sitios impresos), antígeno de carcinoma de células escamosas reconocido por células T 3 (SART3); proteína de caja emparejada Pax-5 (PAX5); proteína de unión a proacrosina sp32 (OY-TES1); proteína tirosina quinasa específica de células (LCK); proteína de anclaje de A quinasa 4 (AKAP-4); sarcoma sinovial, punto de ruptura 2 de X (SSX2); Receptor para productos finales de glucación avanzada (RAGE-1); renal ubicuo 1 (RU1); renal ubicuo 2 (RU2); legumaína; virus del papiloma humano E6 (HPV E6); virus del papiloma humano E7 (HPV E7); carboxil esterasa intestinal; proteína de choque térmico 70-2 mutada (mut hsp70-2); CD79a; CD79b; CD72; receptor 1 de tipo inmunoglobulina asociado a leucocitos (LAIR1); fragmento Fc del receptor de IgA (FCAR o CD89); miembro 2 de la subfamilia A del receptor de tipo inmunoglobulina leucocitaria (LILRA2); miembro f de la familia de tipo molécula CD300 (CD300LF); miembro A de la familia 12 de dominios de lectina de tipo C (CLEC12A); antígeno 2 de células estromales de la médula ósea (BST2); tipo receptor de hormona de tipo mucina que contiene módulo de tipo EGF 2 (EMR2); antígeno de células 75 (LY75); Glipicano-3 (GPC3); tipo receptor de Fc 5 (FCRL5); y el polipéptido 1 de tipo inmunoglobulina lambda (IGLL1).5. La célula efectora inmunitaria de una cualquiera de las reivindicaciones 1-4, en donde:(i) el dominio transmembrana comprende un dominio transmembrana de la cadena alfa, beta o zeta de un receptor de células T, CD28, CD3 épsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137, CD154, KIRDS2, OX40, CD2, CD27, LFA-1 (CD11a, CD18), ICOS (CD278), 4-1BB (CD137), GITR, CD40, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), CD160, CD19, IL2R beta, IL2R gamma, IL7R a, ITGA1, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, TNFR2, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), SLAMF6 (NTB-A, Ly108), SLAM (SLAMF1, CD150, IPO-3), BLAME (SLAMF8), SELPLG (162 CD162), LTBR, PAG/Cbp, NKp44, NKp30, NKp46, NKG2D o NKG2C.(ii) el dominio transmembrana comprende:(a) una secuencia de aminoácidos que tiene al menos una, dos o tres modificaciones, pero no más de 20, 10 o 5 modificaciones de una secuencia de aminoácidos de la SEQ ID NO: 12, o una secuencia con una identidad del 95-99 % con una secuencia de aminoácidos de la SEQ ID NO: 12; o(b) la secuencia de aminoácidos de la SEQ ID NO: 12;(iii) la secuencia de ácido nucleico que codifica el dominio transmembrana comprende la secuencia de nucleótidos de la SEQ ID NO: 13 o una secuencia con una identidad del 95-99 % con la misma;(iv) el dominio de unión al antígeno está conectado al dominio transmembrana por una región de bisagra, en donde dicha región de bisagra comprende la secuencia de aminoácidos de SEQ ID NO: 4 o SEQ ID NO: 6, o una secuencia con 95-99% de identidad con la misma; o dicha región de bisagra es codificada por la secuencia de nucleótidos de SEQ ID NO: 5 o SEQ ID NO: 7, o una secuencia de nucleótidos con una identidad de un 95-99% respecto a la SEQ ID NO: 7; y/o (v) el CAR comprende además una secuencia conductora que comprende la SEQ ID NO:2.6. La célula efectora inmunitaria de una cualquiera de las reivindicaciones 1-5, en donde:(i) el dominio de señalización intracelular comprende un dominio de señalización primario que comprende:(a) un dominio de señalización funcional de CD3 zeta, CD3 gamma, CD3 delta, CD3 épsilon, FcR gamma común (FCER1G), FcR beta (Fc Épsilon R1b), CD79a, CD79b, Fcgamma RIIa, DAP10, or DAP12; y/o(b) una secuencia de aminoácidos que tiene:(1) al menos una, dos o tres modificaciones, pero no más de 20, 10 o 5 modificaciones de una secuencia de aminoácidos de la SEQ ID NO: 18 o SEQ ID NO: 20, o una secuencia con una identidad del 95-99 % con una secuencia de aminoácidos de la SEQ ID NO: 18 o SEQ ID NO: 20; o(2) la secuencia de aminoácidos de la SEQ ID NO: 18 o SEQ ID NO: 20; y/o(ii) el dominio de señalización intracelular comprende un dominio de señalización coestimulador que comprende un dominio de señalización funcional de CD27, CD28 , 4-1BB (CD137), OX40, CD30, CD40, PD-1, ICOS, antígeno-1 asociado a la función de los linfocitos (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-H3, un ligando que se une específicamente con CD83, CDS, ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), CD160, CD19, CD4, CD8alfa, CD8beta, IL2R beta, IL2R gamma, IL7R alfa, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD11d, ITGAE, CD103, ITGAL, CD11a, LFA-1, ITGAM, CD11b, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, TNFR2, TRANCE/RANKL, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CEACAM1, CRTAM, Ly9 (CD229), CD160 (BY55), PSGL1, CD100 (SEMA4D), CD69, SLAMF6 (NTB-A, Ly108), SLAM (SLAMF1, CD150, IPO-3), BLAME (SLAMF8), SELPLG (CD162), LTBR, LAT, GADS, SLP-76, PAG/Cbp, NKp44, NKp30, NKp46 o NKG2D.7. La célula efectora inmunitaria de una cualquiera de las reivindicaciones 1-6, en donde:(a) el dominio de señalización coestimulador comprende:(i) una secuencia de aminoácidos que tiene al menos una, dos o tres modificaciones, pero no más de 20, 10 o 5 modificaciones de la secuencia de aminoácidos de la SEQ ID NO: 14 o la SEQ ID NO: 16, o una secuencia con una identidad del 95-99 % con la secuencia de aminoácidos de la SEQ ID NO: 14 o la SEQ ID NO: 16; o(ii) la secuencia de la SEQ ID NO: 14 o SEQ ID NO: 16; o(b) la secuencia de ácido nucleico que codifica el dominio de señalización coestimulador comprende la secuencia de la sEq ID NO: 15 o SEQ ID NO: 17 o una secuencia con una identidad del 95-99 % con la misma.8. La célula efectora inmunitaria de una cualquiera de las reivindicaciones 1-7, en donde:(i) el dominio de señalización intracelular comprende la secuencia de la SEQ ID NO: 14 o la SEQ ID NO: 16, y la secuencia de la SEQ ID NO: 18 o SEQ ID NO: 20, en donde las secuencias que comprenden el dominio de señalización intracelular se expresan en el mismo marco y como una sola cadena polipeptídica; o(ii) la secuencia de ácido nucleico que codifica el dominio de señalización intracelular comprende la secuencia de la SEQ ID NO: 15 o la SEQ ID NO: 17, o una secuencia con una identidad del 95-99 % con la misma, y una secuencia de la SEQ ID NO: 19 o la SEQ ID NO: 21 o una secuencia con una identidad del 95-99 % con la misma.9. La célula efectora inmunitaria de una cualquiera de las reivindicaciones 1-8, en donde:(i) la segunda molécula de CAR fija como objetivo al mismo antígeno que la primera molécula de CAR, y en donde el dominio de unión a antígenos de la segunda molécula de CAR es diferente del dominio de unión a antígenos de la primera molécula de CAR; o(ii) el dominio de unión a antígenos de la segunda molécula de CAR fija como objetivo a un antígeno diferente que el dominio de unión a antígenos de la primera molécula de CAR, opcionalmente en donde el dominio de unión a antígenos de la segunda molécula de CAR fija como objetivo a un antígeno tumoral definido en la reivindicación 4.10. La célula efectora inmunitaria de una cualquiera de las reivindicaciones 1-9, en donde la primera molécula de CAR comprende un dominio de señalización intracelular que comprende: un dominio de señalización coestimulador, pero no un dominio de señalización primario; y en donde la segunda molécula de CAR comprende un dominio de señalización intracelular que comprende: un dominio de señalización primario, pero no un dominio de señalización coestimulador; además, dicho dominio de unión a antígenosde la primera molécula de CAR se une a un antígeno tumoral seleccionado de un grupo que consiste en: TSHR, CD171, CS-1, CLL-1, GD3, Tn Ag, FLT3, CD38, CD44v6, B7H3, KIT, IL-13Ra2, IL-11Ra, PSCA, PRSS21, VEGFR2, LewisY, CD24, PDGFR-beta, SSEA-4, MUC1, EGFR, NCAM, CAIX, LMP2, EphA2, Fucosil GM1, sLe, GM3, TGS5, HMWMAA, o-acetil-GD2, receptor de folato beta, TEM1/CD248, TEM7R, CLDN6, GPRC5D, CXORF61, CD97, CD179a, ALK, ácido polisiálico, PLAC1, GloboH, NY-BR-1, UPK2, HAVCR1, ADRB3, PANX3, GPR20, LY6K, OR51E2, TARP, WT1, ETV6-AML, proteína de espermatozoide 17, XAGE1, Tie 2, MAD-CT-1, MAD-CT-2, antígeno relacionado con Fos 1, p53 mutante, hTERT, puntos de corte de translocación de sarcoma, ML-IAP, ERG (gen de fusión de TMPRSS2 y ETS), NA17, PAX3, receptor de andrógenos, ciclina B1, MYCN, RhoC, CYP1B1, BORIS, SART3, PAX5, OY-TES1, LCK, AKAP-4, SSX2, CD79a, CD79b, CD72, LAIR1, FCAR, LILRA2, CD300LF, CLEC12A, BST2, EMR2, LY75, GPC3, FCRL5 e IGLL1.11. La célula efectora inmunitaria de una cualquiera de las reivindicaciones 1-10, en donde:(i) la primera molécula de CAR comprende un dominio de señalización coestimulador que comprende un dominio de señalización funcional de 4-1BB, c D28, CD27 u OX-40; y la segunda molécula de c Ar comprende un dominio de señalización primario que comprende un dominio de señalización funcional de CD3 zeta;(ii) la segunda molécula de CAR es un CAR inhibidor, en donde el CAR inhibidor comprende un dominio de unión a antígenos, un dominio transmembrana y un dominio intracelular de una molécula inhibidora, en donde la molécula inhibidora se elige entre: PD1, PD-L1, CTLA4, TIM3, LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4, TGFR beta, CEACAM-1, CEACAM-3 o CEACAM-5;(iii) la segunda molécula de CAR comprende, además, un dominio de señalización intracelular que comprende un dominio de señalización primario y/o un dominio de señalización intracelular, en donde el dominio de señalización intracelular comprende un dominio de señalización primario que comprende el dominio funcional de CD3 zeta y un dominio de señalización coestimulador que comprende el dominio funcional de 4-1BB; y/o(iv) la segunda molécula de CAR comprende la secuencia de aminoácidos de la SEQ ID NO: 26.12. La célula efectora inmunitaria de una cualquiera de las reivindicaciones 1-11, en donde la célula efectora inmunitaria es una célula T humana (p. ej., célula T CD8+) o una célula NK humana, opcionalmente, en donde la célula T es deficiente en diaglicerol quinasa (DGK) y/o Ikaros.13. La célula efectora inmunitaria de una cualquiera de las reivindicaciones 1-12, para uso como un medicamento.14. La célula efectora inmunitaria de una cualquiera de las reivindicaciones 1-12, para uso en el tratamiento de una enfermedad que expresa un antígeno tumoral.15. La célula efectora inmunitaria para uso de la reivindicación 14, en donde la enfermedad es cáncer, p. ej., un cáncer hematológico.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461953783P | 2014-03-15 | 2014-03-15 | |
| US201461976375P | 2014-04-07 | 2014-04-07 | |
| US201462027154P | 2014-07-21 | 2014-07-21 | |
| US201462076146P | 2014-11-06 | 2014-11-06 | |
| US201462097286P | 2014-12-29 | 2014-12-29 | |
| PCT/US2015/020606 WO2015142675A2 (en) | 2014-03-15 | 2015-03-13 | Treatment of cancer using chimeric antigen receptor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2939760T3 true ES2939760T3 (es) | 2023-04-26 |
Family
ID=52780057
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15712503T Active ES2939760T3 (es) | 2014-03-15 | 2015-03-13 | Tratamiento de cáncer utilizando un receptor quimérico para antígenos |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US20170335281A1 (es) |
| EP (2) | EP3593812A3 (es) |
| JP (3) | JP2017513818A (es) |
| CN (1) | CN106163547A (es) |
| ES (1) | ES2939760T3 (es) |
| WO (1) | WO2015142675A2 (es) |
Families Citing this family (384)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9724404B2 (en) | 2009-04-13 | 2017-08-08 | INSERM (Institut National de la Santé et de la Recherche Médicale) | HPV particles and uses thereof |
| ES2760023T3 (es) | 2013-02-20 | 2020-05-12 | Univ Pennsylvania | Tratamiento del cáncer utilizando receptor de antígeno quimérico anti-EGFRvIII humanizado |
| TW201446794A (zh) | 2013-02-20 | 2014-12-16 | Novartis Ag | 利用抗-cd123嵌合抗原受體工程化t細胞之初級人類白血病有效靶向 |
| WO2014145252A2 (en) | 2013-03-15 | 2014-09-18 | Milone Michael C | Targeting cytotoxic cells with chimeric receptors for adoptive immunotherapy |
| TWI654206B (zh) | 2013-03-16 | 2019-03-21 | 諾華公司 | 使用人類化抗-cd19嵌合抗原受體治療癌症 |
| KR102452767B1 (ko) | 2013-05-14 | 2022-10-12 | 더 보드 오브 리젠츠 오브 더 유니버시티 오브 텍사스 시스템 | 가공된 키메라 항원 수용체 (car) t-세포의 인간 적용 |
| JP6444417B2 (ja) | 2013-09-18 | 2018-12-26 | オーラ バイオサイエンシーズ, インコーポレイテッド | 腫瘍を診断および処置するためのウイルス様粒子結合体 |
| EP3083964B1 (en) | 2013-12-19 | 2022-01-26 | Novartis AG | Human mesothelin chimeric antigen receptors and uses thereof |
| EP3087101A4 (en) | 2013-12-20 | 2017-12-06 | Novartis AG | Regulatable chimeric antigen receptor |
| ES2963718T3 (es) | 2014-01-21 | 2024-04-01 | Novartis Ag | Capacidad presentadora de antígenos de células CAR-T potenciada mediante introducción conjunta de moléculas co-estimuladoras |
| US10441654B2 (en) | 2014-01-24 | 2019-10-15 | Children's Hospital Of Eastern Ontario Research Institute Inc. | SMC combination therapy for the treatment of cancer |
| ES2939760T3 (es) | 2014-03-15 | 2023-04-26 | Novartis Ag | Tratamiento de cáncer utilizando un receptor quimérico para antígenos |
| KR102487608B1 (ko) | 2014-04-07 | 2023-01-12 | 노파르티스 아게 | 항-cd19 키메라 항원 수용체를 사용한 암의 치료 |
| MX2016013158A (es) | 2014-04-10 | 2017-04-27 | Seattle Children's Hospital (Dba Seattle Children's Res Institute) | Productos de celulas t modificadas de genes de composicion definida. |
| GB201506423D0 (en) * | 2015-04-15 | 2015-05-27 | Tc Biopharm Ltd | Gamma delta T cells and uses thereof |
| CA2955386A1 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Treatment of cancer using humanized anti-bcma chimeric antigen receptor |
| WO2016014553A1 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Sortase synthesized chimeric antigen receptors |
| JP2017528433A (ja) * | 2014-07-21 | 2017-09-28 | ノバルティス アーゲー | 低い免疫増強用量のmTOR阻害剤とCARの組み合わせ |
| TWI719942B (zh) | 2014-07-21 | 2021-03-01 | 瑞士商諾華公司 | 使用cd33嵌合抗原受體治療癌症 |
| EP3171882A1 (en) | 2014-07-21 | 2017-05-31 | Novartis AG | Treatment of cancer using a cll-1 chimeric antigen receptor |
| CN112410363A (zh) | 2014-08-19 | 2021-02-26 | 诺华股份有限公司 | 抗cd123嵌合抗原受体(car)用于癌症治疗 |
| CN106661129B (zh) * | 2014-08-19 | 2021-02-05 | 美天施生物科技有限两合公司 | 对ssea4抗原具有特异性的嵌合抗原受体 |
| ES2891332T3 (es) | 2014-09-17 | 2022-01-27 | Novartis Ag | Direccionamiento a células citotóxicas con receptores quiméricos para la inmunoterapia adoptiva |
| EP3204777A1 (en) | 2014-10-08 | 2017-08-16 | Novartis AG | Biomarkers predictive of therapeutic responsiveness to chimeric antigen receptor therapy and uses thereof |
| WO2016090034A2 (en) | 2014-12-03 | 2016-06-09 | Novartis Ag | Methods for b cell preconditioning in car therapy |
| KR20170087514A (ko) | 2014-12-03 | 2017-07-28 | 주노 쎄러퓨티크스 인코퍼레이티드 | 입양 세포 치료를 위한 방법 및 조성물 |
| CN107206077B (zh) | 2014-12-05 | 2021-06-08 | 纪念斯隆-凯特琳癌症中心 | 靶向g-蛋白偶联受体的抗体和使用方法 |
| PL3227432T3 (pl) | 2014-12-05 | 2024-03-11 | Memorial Sloan Kettering Cancer Center | Chimeryczne receptory antygenowe ukierunkowane na antygen dojrzewania komórek b i ich zastosowania |
| NZ732570A (en) | 2014-12-05 | 2022-12-23 | Memorial Sloan Kettering Cancer Center | Chimeric antigen receptors targeting fc receptor-like 5 and uses thereof |
| LT3227339T (lt) | 2014-12-05 | 2022-01-10 | Memorial Sloan-Kettering Cancer Center | Chimeriniai antigeno receptoriai, nukreipti į receptorių su prijungtu g baltymu ir jų naudojimo būdai |
| KR20170108944A (ko) | 2014-12-05 | 2017-09-27 | 메모리얼 슬로안-케터링 캔서 센터 | B-세포 성숙화 항원을 표적화하는 항체 및 사용 방법 |
| US20190054117A1 (en) * | 2014-12-19 | 2019-02-21 | Novartis Ag | Dimerization switches and uses thereof |
| US10273300B2 (en) * | 2014-12-29 | 2019-04-30 | The Trustees Of The University Of Pennsylvania | Methods of making chimeric antigen receptor-expressing cells |
| US11459390B2 (en) | 2015-01-16 | 2022-10-04 | Novartis Ag | Phosphoglycerate kinase 1 (PGK) promoters and methods of use for expressing chimeric antigen receptor |
| ES2842212T3 (es) | 2015-01-26 | 2021-07-13 | Cellectis | Receptores de antígenos quiméricos monocatenarios específicos anti-CLL1 (scCAR) para inmunoterapia contra el cáncer |
| WO2016126608A1 (en) * | 2015-02-02 | 2016-08-11 | Novartis Ag | Car-expressing cells against multiple tumor antigens and uses thereof |
| WO2016130598A1 (en) | 2015-02-09 | 2016-08-18 | University Of Florida Research Foundation, Inc. | Bi-specific chimeric antigen receptor and uses thereof |
| WO2016134284A1 (en) | 2015-02-19 | 2016-08-25 | University Of Florida Research Foundation, Inc. | Chimeric antigen receptors and uses thereof |
| EP3258967A4 (en) | 2015-02-20 | 2018-10-03 | Ohio State Innovation Foundation | Bivalent antibody directed against nkg2d and tumor associated antigens |
| BR112017018770A2 (pt) | 2015-03-02 | 2018-04-17 | Innovative Cellular Therapeutics CO., LTD. | redução da tolerância imunológica induzida por pd-l1 |
| EP3270936A4 (en) | 2015-03-17 | 2018-08-08 | Chimera Bioengineering Inc. | Smart car devices, de car polypeptides, side cars and uses thereof |
| CN106146666B (zh) * | 2015-03-26 | 2019-09-06 | 科济生物医药(上海)有限公司 | 靶向cldn6的免疫效应细胞及其制备方法和应用 |
| KR20180012749A (ko) | 2015-04-06 | 2018-02-06 | 지안후아 유 | 교아세포종에 대한 egfr-지시된 car 요법 |
| HUE059218T2 (hu) | 2015-04-08 | 2022-11-28 | Novartis Ag | CD20-terápiák, CD22-terápiák és kombinációs terápiák CD19 kiméra antigénreceptort (CAR-t) expresszáló sejttel |
| ES2948133T3 (es) | 2015-04-17 | 2023-08-31 | Novartis Ag | Métodos para mejorar la eficacia y expansión de células que expresan un receptor de antígeno quimérico |
| CA2986254A1 (en) | 2015-05-18 | 2016-11-24 | TCR2 Therapeutics Inc. | Compositions and methods for tcr reprogramming using fusion proteins |
| AU2016297014B2 (en) | 2015-07-21 | 2021-06-17 | Novartis Ag | Methods for improving the efficacy and expansion of immune cells |
| CA2993796C (en) * | 2015-07-29 | 2024-02-20 | Onkimmune Limited | Modified natural killer cells and natural killer cell lines having increased cytotoxicity |
| WO2017023803A1 (en) | 2015-07-31 | 2017-02-09 | Regents Of The University Of Minnesota | Modified cells and methods of therapy |
| US11667691B2 (en) | 2015-08-07 | 2023-06-06 | Novartis Ag | Treatment of cancer using chimeric CD3 receptor proteins |
| CN108174604B (zh) | 2015-08-07 | 2023-06-23 | 西雅图儿童医院(Dba西雅图儿童研究所) | 用于实体瘤靶向的双特异性car t细胞 |
| CN105384825B (zh) | 2015-08-11 | 2018-06-01 | 南京传奇生物科技有限公司 | 一种基于单域抗体的双特异性嵌合抗原受体及其应用 |
| GB201514874D0 (en) * | 2015-08-20 | 2015-10-07 | Autolus Ltd | Cell |
| EP3340995A4 (en) * | 2015-08-28 | 2019-04-03 | The Trustees Of The University Of Pennsylvania | METHODS AND COMPOSITIONS FOR CELLS EXPRESSING A CHIMERIC INTRACELLULAR SIGNALING MOLECULE |
| US11747346B2 (en) | 2015-09-03 | 2023-09-05 | Novartis Ag | Biomarkers predictive of cytokine release syndrome |
| AR105910A1 (es) | 2015-09-04 | 2017-11-22 | Obi Pharma Inc | Matrices de glicano y método de uso |
| EP3569244A1 (en) * | 2015-09-23 | 2019-11-20 | CytoImmune Therapeutics, LLC | Flt3 directed car cells for immunotherapy |
| CN105567649A (zh) * | 2015-10-26 | 2016-05-11 | 哈尔滨新联合生物科技有限公司 | 一种修饰的增强型dc-cik靶向免疫细胞群的制备方法和用途 |
| US11059879B2 (en) | 2015-10-27 | 2021-07-13 | Board Of Regents, The University Of Texas System | Chimeric antigen receptor molecules and uses thereof |
| WO2017075465A1 (en) | 2015-10-28 | 2017-05-04 | The Broad Institute Inc. | Compositions and methods for evaluating and modulating immune responses by detecting and targeting gata3 |
| WO2017075451A1 (en) | 2015-10-28 | 2017-05-04 | The Broad Institute Inc. | Compositions and methods for evaluating and modulating immune responses by detecting and targeting pou2af1 |
| AU2016343805A1 (en) | 2015-10-30 | 2018-06-07 | Aleta Biotherapeutics Inc. | Compositions and methods for tumor transduction |
| KR20180095511A (ko) * | 2015-10-30 | 2018-08-27 | 더 유나이티드 스테이츠 오브 어메리카, 애즈 리프리젠티드 바이 더 세크러테리, 디파트먼트 오브 헬쓰 앤드 휴먼 서비씨즈 | 표적화된 암 요법 |
| US10508143B1 (en) * | 2015-10-30 | 2019-12-17 | Aleta Biotherapeutics Inc. | Compositions and methods for treatment of cancer |
| SG11201803098VA (en) | 2015-10-30 | 2018-05-30 | Nbe Therapeutics Ag | Anti-ror1 antibodies |
| JP2019500028A (ja) | 2015-11-19 | 2019-01-10 | ブラッドセンター リサーチ ファウンデーション | 癌免疫療法で使用するための二重特異性t細胞を製造する方法 |
| WO2017096329A1 (en) * | 2015-12-03 | 2017-06-08 | Juno Therapeutics, Inc. | Modified chimeric receptors and related compositions and methods |
| SG11201804656PA (en) | 2015-12-04 | 2018-06-28 | Memorial Sloan Kettering Cancer Center | Antibodies targeting fc receptor-like 5 and methods of use |
| US11052111B2 (en) | 2015-12-08 | 2021-07-06 | Chimera Bioengineering, Inc. | Smart CAR devices and DE CAR polypeptides for treating disease and methods for enhancing immune responses |
| MA44140A (fr) | 2015-12-22 | 2021-05-19 | Dana Farber Cancer Inst Inc | Récepteur d'antigène chimérique (car) contre la mésothéline et anticorps contre l'inhibiteur de pd-l1 pour une utilisation combinée dans une thérapie anticancéreuse |
| RU2018127657A (ru) | 2015-12-30 | 2020-01-31 | Новартис Аг | Виды терапии на основе иммуноэффекторных клеток с улучшенной эффективностью |
| JP2019506155A (ja) | 2016-01-12 | 2019-03-07 | クレッシェンド、バイオロジックス、リミテッドCrescendo Biologics Limited | 治療分子 |
| BR112018014615A2 (pt) | 2016-01-20 | 2018-12-11 | The Scripps Research Institute | composições de anticorpo para ror1 e métodos relacionados |
| TWI797073B (zh) * | 2016-01-25 | 2023-04-01 | 德商安美基研究(慕尼黑)公司 | 包含雙特異性抗體建構物之醫藥組合物 |
| EP3420001B1 (en) * | 2016-02-25 | 2021-12-01 | Cell Medica Switzerland AG | Binding members to pd-l1 |
| SG11201807489PA (en) | 2016-03-04 | 2018-09-27 | Novartis Ag | Cells expressing multiple chimeric antigen receptor (car) molecules and uses therefore |
| BR112018067923A2 (pt) * | 2016-03-15 | 2019-02-05 | Chugai Pharmaceutical Co Ltd | métodos de tratamento de cânceres usando antagonistas de ligação de eixo pd-1 e anticorpos anti-gpc3 |
| US11325948B2 (en) | 2016-03-19 | 2022-05-10 | Exuma Biotech Corp. | Methods and compositions for genetically modifying lymphocytes to express polypeptides comprising the intracellular domain of MPL |
| US11111505B2 (en) | 2016-03-19 | 2021-09-07 | Exuma Biotech, Corp. | Methods and compositions for transducing lymphocytes and regulating the activity thereof |
| EP3432924A1 (en) | 2016-03-23 | 2019-01-30 | Novartis AG | Cell secreted minibodies and uses thereof |
| WO2017162797A1 (en) * | 2016-03-23 | 2017-09-28 | Helmholtz Zentrum München - Deutsches Forschungszentrum für Gesundheit und Umwelt (GmbH) | Fusion proteins of pd-1 and 4-1bb |
| US10980894B2 (en) | 2016-03-29 | 2021-04-20 | Obi Pharma, Inc. | Antibodies, pharmaceutical compositions and methods |
| AU2017244108B2 (en) | 2016-03-29 | 2021-03-18 | University Of Southern California | Chimeric antigen receptors targeting cancer |
| TWI780045B (zh) | 2016-03-29 | 2022-10-11 | 台灣浩鼎生技股份有限公司 | 抗體、醫藥組合物及方法 |
| WO2017173256A1 (en) | 2016-04-01 | 2017-10-05 | Kite Pharma, Inc. | Chimeric antigen and t cell receptors and methods of use |
| CN113082201A (zh) * | 2016-04-01 | 2021-07-09 | 上海斯丹赛生物技术有限公司 | 刺激t细胞介导的对表达抗原的细胞群的免疫应答的方法 |
| UA123276C2 (uk) * | 2016-04-01 | 2021-03-10 | Кайт Фарма, Інк. | Химерний рецептор та спосіб лікування пухлини або злоякісного новоутворення |
| KR102591930B1 (ko) | 2016-04-01 | 2023-10-24 | 카이트 파마 인코포레이티드 | Bcma 결합 분자 및 그의 사용 방법 |
| EA201892193A1 (ru) * | 2016-04-01 | 2019-06-28 | Амген Инк. | Химерные рецепторы к flt3 и способы их применения |
| US10654934B2 (en) | 2016-04-01 | 2020-05-19 | Innovative Cellular Therapeutics CO., LTD. | Use of chimeric antigen receptor modified cells to treat cancer |
| WO2017177137A1 (en) * | 2016-04-07 | 2017-10-12 | Bluebird Bio, Inc. | Chimeric antigen receptor t cell compositions |
| EP3439685B1 (en) * | 2016-04-08 | 2023-07-05 | Emory University | Methods of treating cancer and infectious diseases using cell based therapies |
| EP3443001A4 (en) | 2016-04-11 | 2020-04-29 | Obsidian Therapeutics, Inc. | REGULATED BIOCIRCUIT SYSTEMS |
| US20190125840A1 (en) * | 2016-04-12 | 2019-05-02 | Philogen S.P.A. | Combination therapy comprising an inflammatory immunocytokine and a chimeric antigen receptor (car)-t cell |
| CN106399255B (zh) * | 2016-04-13 | 2019-10-18 | 阿思科力(苏州)生物科技有限公司 | Pd-1 car-t细胞及其制备方法和应用 |
| WO2017181119A2 (en) | 2016-04-15 | 2017-10-19 | Novartis Ag | Compositions and methods for selective protein expression |
| CN105907719B (zh) * | 2016-04-18 | 2019-10-18 | 阿思科力(苏州)生物科技有限公司 | Anti ROBO1 CAR-T细胞及其制备和应用 |
| KR20230110820A (ko) | 2016-04-22 | 2023-07-25 | 오비아이 파머 인코퍼레이티드 | 글로보 계열 항원을 통한 면역 활성화 또는 면역 조정에의한 암 면역요법 |
| WO2017190096A1 (en) * | 2016-04-29 | 2017-11-02 | University Of Florida Research Foundation Incorporated | Chimeric antigen receptors and uses thereof |
| GB201607968D0 (en) * | 2016-05-06 | 2016-06-22 | Crescendo Biolog Ltd | Chimeric antigen receptor |
| EP3458077A4 (en) | 2016-05-17 | 2020-04-01 | Chimera Bioengineering Inc. | METHODS OF MANUFACTURING NEW AREAS OF ANTIGEN BINDING |
| KR101926166B1 (ko) * | 2016-05-24 | 2018-12-06 | 재단법인 아산사회복지재단 | 수용체 시너지 활성을 이용한 nk 세포의 활성도 검사 방법 및 이를 이용한 nk 세포의 활성도가 관련된 질환의 진단 방법 |
| CA3024508A1 (en) | 2016-05-27 | 2017-11-30 | Agenus Inc. | Anti-tim-3 antibodies and methods of use thereof |
| WO2017205747A1 (en) * | 2016-05-27 | 2017-11-30 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Flt3-specific chimeric antigen receptors and methods using same |
| US20210177896A1 (en) | 2016-06-02 | 2021-06-17 | Novartis Ag | Therapeutic regimens for chimeric antigen receptor (car)- expressing cells |
| US11472856B2 (en) | 2016-06-13 | 2022-10-18 | Torque Therapeutics, Inc. | Methods and compositions for promoting immune cell function |
| WO2018012895A1 (ko) * | 2016-07-14 | 2018-01-18 | 주식회사 큐로셀 | 면역관문을 극복한 면역세포 및 상기 면역세포를 포함한 약제학적 조성물 |
| CN115381832A (zh) | 2016-07-14 | 2022-11-25 | 深圳明赛瑞霖药业有限公司 | 癌症的治疗 |
| CA3030837A1 (en) | 2016-07-15 | 2018-01-18 | Novartis Ag | Treatment and prevention of cytokine release syndrome using a chimeric antigen receptor in combination with a kinase inhibitor |
| CN106220736B (zh) * | 2016-07-20 | 2020-01-10 | 深圳市体内生物医药科技有限公司 | 一种嵌合抗原受体、表达其的细胞及其制备方法和用途 |
| AU2017301826A1 (en) * | 2016-07-26 | 2019-03-14 | Tessa Therapeutics Ltd. | Chimeric antigen receptor |
| CA3032049C (en) | 2016-07-27 | 2023-11-07 | Obi Pharma, Inc. | Immunogenic/therapeutic glycan compositions and uses thereof |
| SG11201900677SA (en) | 2016-07-28 | 2019-02-27 | Novartis Ag | Combination therapies of chimeric antigen receptors adn pd-1 inhibitors |
| EP3491026A4 (en) | 2016-07-29 | 2020-07-29 | OBI Pharma, Inc. | HUMAN ANTIBODIES, PHARMACEUTICAL COMPOSITIONS AND METHODS |
| US20190161542A1 (en) | 2016-08-01 | 2019-05-30 | Novartis Ag | Treatment of cancer using a chimeric antigen receptor in combination with an inhibitor of a pro-m2 macrophage molecule |
| JP7109789B2 (ja) | 2016-08-02 | 2022-08-01 | ティーシーアール2 セラピューティクス インク. | 融合タンパク質を使用したtcrの再プログラム化のための組成物及び方法 |
| WO2018027197A1 (en) * | 2016-08-04 | 2018-02-08 | Memorial Sloan-Kettering Cancer Center | Cancer antigen targets and uses thereof |
| CA3032838A1 (en) * | 2016-08-04 | 2018-02-08 | Memorial Sloan-Kettering Cancer Center | Compositions and methods for immunotherapy |
| AU2017313828B2 (en) * | 2016-08-18 | 2019-12-05 | IN8bio, Inc. | Compositions and methods for cancer immunotherapy |
| CN109789164B (zh) * | 2016-08-30 | 2023-04-28 | 亘喜生物科技(上海)有限公司 | 具有gitr胞内结构域作为共刺激结构域的嵌合抗原受体 |
| CN110121352B (zh) | 2016-09-01 | 2020-12-11 | 嵌合体生物工程公司 | Gold优化的car t-细胞 |
| CN107793482A (zh) * | 2016-09-06 | 2018-03-13 | 广州百尼夫生物科技有限公司 | 嵌合抗原受体及其基因和重组表达载体、car133‑nkt 细胞及其制备方法和应用 |
| WO2018049025A2 (en) | 2016-09-07 | 2018-03-15 | The Broad Institute Inc. | Compositions and methods for evaluating and modulating immune responses |
| WO2018049187A1 (en) * | 2016-09-08 | 2018-03-15 | Dana-Farber Cancer Institute, Inc. | Compositions and methods of treating cancer |
| CN106434563B (zh) * | 2016-09-14 | 2017-09-22 | 深圳市默赛尔生物医学科技发展有限公司 | 转基因自然杀伤细胞及其制备方法和应用 |
| US11307205B2 (en) | 2016-09-19 | 2022-04-19 | University Of Southern California | Non-radioactive cytotoxicity assays |
| US20190307799A1 (en) | 2016-09-23 | 2019-10-10 | The Regents Of The University Of Michigan | Engineered lymphocytes |
| CN107868791B (zh) * | 2016-09-26 | 2021-11-23 | 阿思科力(苏州)生物科技有限公司 | 一种加强型Slit2 CAR-T和CAR-NK细胞制备方法和应用 |
| CN106317228A (zh) * | 2016-09-28 | 2017-01-11 | 李华顺 | 一种嵌合抗原受体分子及其应用 |
| EP3519577A1 (en) | 2016-09-28 | 2019-08-07 | Novartis AG | Porous membrane-based macromolecule delivery system |
| EP3519562A4 (en) * | 2016-09-29 | 2020-03-25 | Nantkwest, Inc. | NK-92 HLA CLASS I DEFICIENT CELLS WITH REDUCED IMMUNOGENICITY |
| JP2019528760A (ja) * | 2016-09-30 | 2019-10-17 | ポセイダ セラピューティクス, インコーポレイテッド | 改変された幹細胞メモリーt細胞、その作製方法およびその使用方法 |
| US20190119636A1 (en) | 2017-10-23 | 2019-04-25 | Poseida Therapeutics, Inc. | Modified stem cell memory t cells, methods of making and methods of using same |
| WO2018064921A1 (en) * | 2016-10-06 | 2018-04-12 | Innovative Cellular Therapeutics CO., LTD. | Use of chimeric antigen receptor modified cells to treat cancer |
| MX2019003899A (es) | 2016-10-07 | 2019-08-14 | Tcr2 Therapeutics Inc | Composiciones y metodos para reprogramacion de receptores de linfocitos t mediante el uso de proteinas de fusion. |
| EP3523331A1 (en) | 2016-10-07 | 2019-08-14 | Novartis AG | Chimeric antigen receptors for the treatment of cancer |
| CN110022907A (zh) * | 2016-10-10 | 2019-07-16 | 国家生物技术研究所公司 | 非细胞毒性的修饰细胞及其用途 |
| MA46529A (fr) | 2016-10-11 | 2019-08-21 | Agenus Inc | Anticorps anti-lag-3 et leurs procédés d'utilisation |
| JP2019533676A (ja) | 2016-10-13 | 2019-11-21 | ジュノー セラピューティクス インコーポレイテッド | トリプトファン代謝経路調節剤を含む免疫療法の方法および組成物 |
| GB2605540B (en) | 2016-10-18 | 2022-12-21 | Univ Minnesota | Tumor infiltrating lymphocytes and methods of therapy |
| WO2018075794A1 (en) * | 2016-10-19 | 2018-04-26 | City Of Hope | Use of triplex cmv vaccine in car t cell therapy |
| CN107988164B (zh) * | 2016-10-26 | 2020-07-07 | 阿思科力(苏州)生物科技有限公司 | 一种pd-1 car nk-92细胞及其制备方法与应用 |
| CN106591363A (zh) * | 2016-11-11 | 2017-04-26 | 广东万海细胞生物科技有限公司 | 一种通用型异体car‑t细胞制备方法及应用 |
| US11332713B2 (en) | 2016-11-16 | 2022-05-17 | KSQ Therapeutics, Inc. | Gene-regulating compositions and methods for improved immunotherapy |
| CN110177803A (zh) | 2016-11-22 | 2019-08-27 | T细胞受体治疗公司 | 用于使用融合蛋白进行tcr重新编程的组合物和方法 |
| CN110248678A (zh) * | 2016-12-03 | 2019-09-17 | 朱诺治疗学股份有限公司 | 调节car-t细胞的方法 |
| US11590167B2 (en) | 2016-12-03 | 2023-02-28 | Juno Therapeutic, Inc. | Methods and compositions for use of therapeutic T cells in combination with kinase inhibitors |
| KR20190092478A (ko) | 2016-12-05 | 2019-08-07 | 쥐원 쎄라퓨틱스, 인크. | 화학요법 레지멘 동안의 면역 반응의 보존 |
| CN106701827A (zh) * | 2016-12-05 | 2017-05-24 | 刘晓明 | 表达cd19和cd20抗体基因嵌合抗原受体t细胞的转化及扩增培养与保存方法 |
| DK3581190T3 (da) | 2016-12-09 | 2021-06-07 | Onk Therapeutics Ltd | Manipulerede naturlige dræberceller og anvendelser deraf |
| MX2019006631A (es) | 2016-12-12 | 2019-11-12 | Seattle Childrens Hospital Dba Seattle Childrens Res Inst | Variantes quimericas de factores de transcripcion con sensibilidad aumentada a induccion por ligando de farmaco de expresion transgenica en celulas mamiferas. |
| WO2018111340A1 (en) | 2016-12-16 | 2018-06-21 | Novartis Ag | Methods for determining potency and proliferative function of chimeric antigen receptor (car)-t cells |
| EP3558348A1 (en) * | 2016-12-21 | 2019-10-30 | TCR2 Therapeutics Inc. | Engineered t cells for the treatment of cancer |
| JP2020501589A (ja) * | 2016-12-23 | 2020-01-23 | ウイルツ・バイオロジクス・リミテッド | がんの治療 |
| CN107286246B (zh) * | 2016-12-28 | 2019-12-17 | 时力生物科技(北京)有限公司 | 治疗脑胶质瘤的嵌合抗原受体修饰的树突状细胞及其制备方法 |
| CN108276493B (zh) * | 2016-12-30 | 2023-11-14 | 南京传奇生物科技有限公司 | 一种嵌合抗原受体及其应用 |
| CN110121510A (zh) | 2017-01-06 | 2019-08-13 | 克雷森多生物制剂有限公司 | 程序性细胞死亡(pd-1)的单结构域抗体 |
| IL310711A (en) | 2017-01-10 | 2024-04-01 | Intrexon Corp | Modulation of expression of polypeptides through a new system for changing gene expression |
| US11541076B2 (en) * | 2017-01-13 | 2023-01-03 | Celdara Medical, Llc | Chimeric antigen receptors targeting TIM-1 |
| EP3572428A4 (en) | 2017-01-17 | 2020-12-30 | Daiichi Sankyo Company, Limited | ANTI-GPR20 ANTIBODY AND ANTI-GPR20 ANTIBODY MEDICINAL CONJUGATE |
| EP3570850A4 (en) * | 2017-01-18 | 2020-10-14 | Thalia Papayannopoulou | COMPOSITIONS AND PROCEDURES FOR CONDITIONING TRANSPLANT RECIPIENTS |
| CN108330133B (zh) * | 2017-01-20 | 2020-06-30 | 上海恒润达生生物科技有限公司 | 靶向cd19嵌合抗原受体并对其双重修饰的方法及其用途 |
| ES2912408T3 (es) | 2017-01-26 | 2022-05-25 | Novartis Ag | Composiciones de CD28 y métodos para terapia con receptores quiméricos para antígenos |
| US20190375815A1 (en) * | 2017-01-31 | 2019-12-12 | Novartis Ag | Treatment of cancer using chimeric t cell receptor proteins having multiple specificities |
| CN108395478A (zh) * | 2017-02-04 | 2018-08-14 | 上海恒润达生生物科技有限公司 | 靶向bcma的嵌合抗原受体并对其双重修饰的方法及其用途 |
| KR20190133160A (ko) * | 2017-02-07 | 2019-12-02 | 다이이찌 산쿄 가부시키가이샤 | 항-gprc5d 항체 및 항-gprc5d 항체를 포함하는 분자 |
| ES2949364T3 (es) | 2017-02-17 | 2023-09-28 | Fred Hutchinson Cancer Center | Terapias de combinación para el tratamiento de cánceres y trastornos autoinmunitarios relacionados con BCMA |
| JP2020508080A (ja) * | 2017-02-21 | 2020-03-19 | ユーティレックス カンパニー リミテッド | Hla−dr car−t組成物ならびにその作製方法および使用方法 |
| TW201837175A (zh) | 2017-03-13 | 2018-10-16 | 美商凱特製藥公司 | 用於黑色素瘤之嵌合抗原受體及其用途 |
| CN110446489A (zh) * | 2017-03-20 | 2019-11-12 | 南加利福尼亚大学 | 具有表面缀合的载药纳米颗粒的car t细胞的过继转移及其用途 |
| BR112019018124A2 (pt) | 2017-03-22 | 2020-04-07 | Intellia Therapeutics Inc | composições e métodos para imunooncologia |
| CN107271675B (zh) * | 2017-03-24 | 2020-06-02 | 郑猛 | 抗人adrb3单克隆抗体及其在疾病诊断和治疗中的应用 |
| RU2019133202A (ru) | 2017-03-27 | 2021-04-28 | Ф. Хоффманн-Ля Рош Аг | Улучшенные антигенсвязывающие рецепторы |
| US11963966B2 (en) | 2017-03-31 | 2024-04-23 | Dana-Farber Cancer Institute, Inc. | Compositions and methods for treating ovarian tumors |
| EP3606518A4 (en) | 2017-04-01 | 2021-04-07 | The Broad Institute, Inc. | METHODS AND COMPOSITIONS FOR DETECTION AND MODULATION OF IMMUNOTHERAPY RESISTANCE GENE SIGNATURE IN CANCER |
| US20200071773A1 (en) | 2017-04-12 | 2020-03-05 | Massachusetts Eye And Ear Infirmary | Tumor signature for metastasis, compositions of matter methods of use thereof |
| US11344578B2 (en) | 2017-04-19 | 2022-05-31 | Board Of Regents, The University Of Texas System | Immune cells expressing engineered antigen receptors |
| JOP20180042A1 (ar) | 2017-04-24 | 2019-01-30 | Kite Pharma Inc | نطاقات ربط مولد ضد متوافقة مع البشر وطرق الاستخدام |
| CA3059820A1 (en) * | 2017-04-26 | 2018-11-01 | Eureka Therapeutics, Inc. | Constructs specifically recognizing glypican 3 and uses thereof |
| WO2018205926A1 (zh) * | 2017-05-08 | 2018-11-15 | 中国科学院动物研究所 | 经修饰的t细胞、其制备方法及用途 |
| EP3621981A2 (en) | 2017-05-12 | 2020-03-18 | CRISPR Therapeutics AG | Materials and methods for engineering cells and uses thereof in immuno-oncology |
| US11166985B2 (en) | 2017-05-12 | 2021-11-09 | Crispr Therapeutics Ag | Materials and methods for engineering cells and uses thereof in immuno-oncology |
| WO2018210279A1 (zh) * | 2017-05-16 | 2018-11-22 | 科济生物医药(上海)有限公司 | Toll样受体激动剂与免疫效应细胞的联用 |
| JP7381345B2 (ja) | 2017-05-16 | 2023-11-15 | ザ・ジョンズ・ホプキンス・ユニバーシティ | Manaボディおよび使用方法 |
| EP3630846A4 (en) * | 2017-05-31 | 2021-02-24 | Carsgen Therapeutics Co., Ltd. | COMPOSITIONS AND METHODS OF CELLULAR IMMUNOTHERAPY |
| JP2020521479A (ja) * | 2017-06-01 | 2020-07-27 | イノベイティブ セルラー セラピューティクス シーオー.,エルティディ.Innovative Cellular Therapeutics Co.,Ltd. | キメラ抗原受容体細胞の作製及びその使用 |
| CN108977453A (zh) | 2017-06-02 | 2018-12-11 | 阿思科力(苏州)生物科技有限公司 | 一种以robo1为靶点的嵌合抗原受体细胞及其制备和应用 |
| WO2018226958A1 (en) * | 2017-06-07 | 2018-12-13 | The General Hospital Corporation | T cells expressing a chimeric antigen receptor |
| CA3067244A1 (en) * | 2017-06-12 | 2018-12-20 | Emory University | T-cell antigen targeted chimeric antigen receptor (car) and uses in cell therapies |
| EP3638218A4 (en) | 2017-06-14 | 2021-06-09 | The Broad Institute, Inc. | COMPOSITIONS AND METHOD OF TARGETING COMPLEMENTING COMPONENT 3 FOR INHIBITION OF TUMOR GROWTH |
| CN109134660B (zh) * | 2017-06-16 | 2022-07-01 | 上海恒润达生生物科技股份有限公司 | 靶向Mesothelin的嵌合抗原受体并联合表达抗PD1抗体的方法及其用途 |
| EP4302768A3 (en) * | 2017-06-22 | 2024-05-01 | Board Of Regents, The University Of Texas System | Methods for producing regulatory immune cells and uses thereof |
| JP2020530307A (ja) | 2017-06-30 | 2020-10-22 | インティマ・バイオサイエンス,インコーポレーテッド | 遺伝子治療のためのアデノ随伴ウイルスベクター |
| CN107164412B (zh) * | 2017-06-30 | 2020-10-23 | 山东兴瑞生物科技有限公司 | 一种安全型抗cea嵌合抗原受体修饰t细胞的制备方法及其应用 |
| US20220233588A1 (en) | 2017-06-30 | 2022-07-28 | Cellectis | Cellular immunotherapy for repetitive administration |
| GB201711068D0 (en) | 2017-07-10 | 2017-08-23 | Crescendo Biologics Ltd | Therapeutic molecules binding PSMA |
| CN109232742B (zh) * | 2017-07-11 | 2021-10-22 | 深圳市第二人民医院 | 一种嵌合抗原受体及其应用 |
| CN107353325B (zh) * | 2017-07-26 | 2020-08-11 | 中国药科大学 | 叶酸受体α特异性结合肽1及其应用 |
| CN107326014B (zh) * | 2017-07-31 | 2019-09-24 | 时力生物科技(北京)有限公司 | 一种双特异性嵌合抗原受体修饰的t淋巴细胞及其制备方法和应用 |
| CA3071212C (en) | 2017-08-07 | 2023-12-12 | Nbe-Therapeutics Ag | Anthracycline-based antibody drug conjugates having high in vivo tolerability |
| EP3677589A4 (en) | 2017-08-31 | 2021-04-21 | Daiichi Sankyo Company, Limited | IMPROVED PROCESS FOR PREPARING ANTIBODY-ACTIVE CONJUGATE |
| JP7248578B2 (ja) | 2017-08-31 | 2023-03-29 | 第一三共株式会社 | 抗体-薬物コンジュゲートの新規製造方法 |
| AU2018328209A1 (en) | 2017-09-05 | 2020-04-23 | Torque Therapeutics, Inc. | Therapeutic protein compositions and methods of making and using the same |
| WO2019051135A1 (en) * | 2017-09-06 | 2019-03-14 | Fred Hutchinson Cancer Research Center | METHODS FOR IMPROVING ADOPTIVE CELL THERAPY |
| EP4035674A1 (en) * | 2017-09-15 | 2022-08-03 | Kite Pharma, Inc. | Methods and systems for performing a patient-specific immunotherapy procedure with chain-of-custody and chain-of-identity biological sample tracking |
| WO2019067677A1 (en) | 2017-09-29 | 2019-04-04 | Cell Design Labs, Inc. | METHODS OF MANUFACTURING RECEPTORS OF ANTI-CD307E ANTI-BACTERIAL CHIMERIC ANTIGENS AND USES THEREFOR |
| CN107557341B (zh) * | 2017-09-30 | 2020-06-30 | 山东兴瑞生物科技有限公司 | 一种抗wt1增强型嵌合抗原受体修饰的免疫细胞及其应用 |
| US20200299686A1 (en) * | 2017-10-02 | 2020-09-24 | Georgia Tech Research Corporation | Methods and Compositions for Engineering Synthetic Bioswitches for Remote Control of Biological Activity |
| AU2018346719A1 (en) * | 2017-10-06 | 2020-04-23 | Oslo Universitetssykehus Hf | Chimeric antigen receptors |
| CN111629749A (zh) | 2017-10-18 | 2020-09-04 | 诺华股份有限公司 | 用于选择性蛋白质降解的组合物和方法 |
| EP3697435A1 (en) * | 2017-10-20 | 2020-08-26 | Fred Hutchinson Cancer Research Center | Compositions and methods of immunotherapy targeting tigit and/or cd112r or comprising cd226 overexpression |
| RU2020116579A (ru) | 2017-10-25 | 2021-11-25 | Новартис Аг | Способы получения клеток, экспрессирующих химерный антигенный рецептор |
| KR20200116077A (ko) | 2017-11-01 | 2020-10-08 | 주노 쎄러퓨티크스 인코퍼레이티드 | B 세포 성숙 항원에 특이적인 키메라 항원 수용체 및 암호화 폴리뉴클레오타이드 |
| EP3703688A2 (en) | 2017-11-01 | 2020-09-09 | Juno Therapeutics, Inc. | Antibodies and chimeric antigen receptors specific for b-cell maturation antigen |
| US20210363260A1 (en) | 2017-11-13 | 2021-11-25 | The Broad Institute, Inc. | Methods and compositions for treating cancer by targeting the clec2d-klrb1 pathway |
| US11649294B2 (en) | 2017-11-14 | 2023-05-16 | GC Cell Corporation | Anti-HER2 antibody or antigen-binding fragment thereof, and chimeric antigen receptor comprising same |
| WO2019099707A1 (en) | 2017-11-16 | 2019-05-23 | Kite Pharma, Inc | Modified chimeric antigen receptors and methods of use |
| CN107916269B (zh) * | 2017-11-17 | 2020-12-11 | 山东兴瑞生物科技有限公司 | 靶向cd19的tcr基因、制备方法、具有该基因的质粒、试剂盒及应用 |
| WO2019104075A1 (en) * | 2017-11-21 | 2019-05-31 | Novartis Ag | Trispecific binding molecules against tumor-associated antigens and uses thereof |
| DK3720883T3 (da) | 2017-12-05 | 2023-07-24 | Medical Res Infrastructure & Health Services Fund Tel Aviv Medical Ct | T-celler omfattende kimære anti-cd38- og anti-cd138-antigenreceptorer og anvendelser deraf |
| WO2019111250A1 (en) * | 2017-12-05 | 2019-06-13 | The Medical Research Infrastructure And Health Services Fund Of The Tel Aviv Medical Center | T-cells comprising two different chimeric antigen receptors and uses thereof |
| CA3085210A1 (en) * | 2017-12-14 | 2019-06-20 | Bluebird Bio, Inc. | Nkg2d daric receptors |
| US20210139557A1 (en) * | 2017-12-20 | 2021-05-13 | Poseida Therapeutics, Inc. | Vcar compositions and methods for use |
| CN109609533B (zh) * | 2017-12-27 | 2020-07-10 | 赛德特生物科技开发有限公司 | 基于人源化cd276抗体的car慢病毒表达载体构建及其应用 |
| CN109971715A (zh) * | 2017-12-28 | 2019-07-05 | 深圳华大生命科学研究院 | 一种扩增特异性car-t细胞的培养方法 |
| CN109971712B (zh) * | 2017-12-28 | 2023-06-20 | 上海细胞治疗研究院 | 特异性靶向cd19抗原且高水平稳定表达pd-1抗体的car-t细胞及用途 |
| AU2018395272A1 (en) * | 2017-12-28 | 2020-07-09 | NEUGATE PHARMA, LLC aka NEUGATE THERANOSTICS | Compositions and formulations for treatment of malignancies |
| CN109971722B (zh) * | 2017-12-28 | 2023-09-01 | 上海细胞治疗研究院 | 靶向cd19且高水平稳定表达cd40抗体的car-t细胞及其用途 |
| EA202091134A1 (ru) * | 2017-12-28 | 2020-11-20 | Кодиак Байосайенсес, Инк. | Экзосомы для иммуноонкологической и противовоспалительной терапии |
| CN108103105A (zh) * | 2017-12-29 | 2018-06-01 | 深圳市茵冠生物科技有限公司 | 一种car-t细胞的制备方法、制得的car-t细胞及其应用 |
| EP3508855A1 (en) * | 2018-01-05 | 2019-07-10 | Allianz Pharmascience Ltd | Method for the selection of patients and kit |
| WO2019136432A1 (en) * | 2018-01-08 | 2019-07-11 | Novartis Ag | Immune-enhancing rnas for combination with chimeric antigen receptor therapy |
| WO2019139888A1 (en) | 2018-01-09 | 2019-07-18 | H. Lee Moffitt Cancer Center And Research Institute Inc. | Compositions and methods for targeting clec12a-expressing cancers |
| US10561686B2 (en) * | 2018-01-12 | 2020-02-18 | Innovative Cellular Therapeutics CO., LTD. | Modified cell expansion and uses thereof |
| CA3088234A1 (en) * | 2018-01-12 | 2019-07-18 | Curocell Inc. | Enhanced immune cells using dual shrna and composition including the same |
| TWI753229B (zh) * | 2018-01-17 | 2022-01-21 | 美商明塞特製藥公司 | 用於治療癌症之組合療法 |
| CA3088832A1 (en) * | 2018-01-19 | 2019-07-25 | Miltenyi Biotec B.V. & Co. KG | Regulatory t cell expressing a chimeric antigen receptor |
| CN110093351B (zh) * | 2018-01-29 | 2023-06-23 | 华南生物医药研究院 | 可分离的核酸、多肽、重组载体、重组细胞及应用 |
| AU2019215031A1 (en) * | 2018-01-31 | 2020-08-20 | Novartis Ag | Combination therapy using a chimeric antigen receptor |
| JP2021518747A (ja) | 2018-02-13 | 2021-08-05 | キメラ・バイオエンジニアリング,インコーポレーテッド | Rna不安定化エレメントを使用した遺伝子発現の調整 |
| GB201802573D0 (en) | 2018-02-16 | 2018-04-04 | Crescendo Biologics Ltd | Therapeutic molecules that bind to LAG3 |
| JP2021513839A (ja) | 2018-02-16 | 2021-06-03 | カイト ファーマ インコーポレイテッドKite Pharma, Inc | 改変された多能性幹細胞並びに製造方法及び使用方法 |
| WO2019165237A1 (en) * | 2018-02-23 | 2019-08-29 | Endocyte, Inc. | Sequencing method for car t cell therapy |
| CA3093078A1 (en) | 2018-03-06 | 2019-09-12 | The Trustees Of The University Of Pennsylvania | Prostate-specific membrane antigen cars and methods of use thereof |
| KR20210013013A (ko) * | 2018-03-09 | 2021-02-03 | 카르스젠 테라퓨틱스 리미티드 | 종양 치료 방법 및 조성물 |
| WO2019175328A1 (en) | 2018-03-14 | 2019-09-19 | Imba - Institut Für Molekulare Biotechnologie Gmbh | Bh4pathwayactivationandusethereoffortreatingcancer |
| JP2021518160A (ja) | 2018-03-15 | 2021-08-02 | ケーエスキュー セラピューティクス, インコーポレイテッド | 免疫療法の改善のための遺伝子調節組成物及び遺伝子調節方法 |
| US20190284553A1 (en) | 2018-03-15 | 2019-09-19 | KSQ Therapeutics, Inc. | Gene-regulating compositions and methods for improved immunotherapy |
| US20210015864A1 (en) | 2018-03-23 | 2021-01-21 | Kite Pharma, Inc. | Chimeric transmembrane proteins and uses thereof |
| US10688182B2 (en) * | 2018-03-29 | 2020-06-23 | Cho Pharma Usa, Inc. | Monoclonal antibodies that bind to SSEA4 and uses thereof |
| JP2021520209A (ja) * | 2018-04-04 | 2021-08-19 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | 癌患者における腫瘍抗原を検出するための診断アッセイ |
| EP3773631A4 (en) * | 2018-04-06 | 2022-02-09 | Mustang Bio, Inc. | MANUFACTURING PROCESSES FOR CELL-BASED THERAPEUTIC COMPOSITIONS |
| US10869888B2 (en) | 2018-04-17 | 2020-12-22 | Innovative Cellular Therapeutics CO., LTD. | Modified cell expansion and uses thereof |
| US11957695B2 (en) | 2018-04-26 | 2024-04-16 | The Broad Institute, Inc. | Methods and compositions targeting glucocorticoid signaling for modulating immune responses |
| CN112752767A (zh) * | 2018-04-27 | 2021-05-04 | 克里斯珀医疗股份公司 | 细胞毒性t细胞耗竭的方法和组合物 |
| WO2019209557A1 (en) | 2018-04-27 | 2019-10-31 | Mayo Foundation For Medical Education And Research | Foamy viruses and methods of use |
| EP3784256A4 (en) * | 2018-04-27 | 2022-06-29 | Baylor College of Medicine | Car t cells with one or more interleukins |
| EP3788369A1 (en) | 2018-05-01 | 2021-03-10 | Novartis Ag | Biomarkers for evaluating car-t cells to predict clinical outcome |
| CN110438082B (zh) * | 2018-05-04 | 2024-03-08 | 恺兴生命科技(上海)有限公司 | 表达il-21r结合蛋白的免疫效应细胞 |
| MX2020012028A (es) | 2018-05-11 | 2021-03-29 | Crispr Therapeutics Ag | Metodos y composiciones para tratar el cancer. |
| EP3794034A1 (en) | 2018-05-15 | 2021-03-24 | Autolus Limited | Chimeric antigen receptor |
| GB201807870D0 (en) * | 2018-05-15 | 2018-06-27 | Autolus Ltd | A CD79-specific chimeric antigen receptor |
| CN112368019A (zh) * | 2018-05-16 | 2021-02-12 | 詹森生物科技公司 | 治疗癌症并增强t细胞重定向治疗剂的功效的方法 |
| EP3801769A1 (en) | 2018-05-25 | 2021-04-14 | Novartis AG | Combination therapy with chimeric antigen receptor (car) therapies |
| CN108623683B (zh) * | 2018-05-30 | 2021-06-04 | 福州迈新生物技术开发有限公司 | 抗Pax-5蛋白的单克隆抗体及其细胞株、制备方法和应用 |
| CN108715859B (zh) * | 2018-05-31 | 2021-08-03 | 中国医学科学院血液病医院(中国医学科学院血液学研究所) | 靶向cd22的嵌合抗原受体及其应用 |
| WO2019232542A2 (en) | 2018-06-01 | 2019-12-05 | Massachusetts Institute Of Technology | Methods and compositions for detecting and modulating microenvironment gene signatures from the csf of metastasis patients |
| US20190389963A1 (en) * | 2018-06-01 | 2019-12-26 | Obi Pharma, Inc. | Combination therapy by using anti-globo h or anti-ssea-4 antibody with anti-negative immune check points antibody |
| CN110577604A (zh) * | 2018-06-07 | 2019-12-17 | 亘喜生物科技(上海)有限公司 | 携带gitr共刺激信号靶向egfr的嵌合抗原受体t细胞 |
| EP3802825A1 (en) | 2018-06-08 | 2021-04-14 | Intellia Therapeutics, Inc. | Compositions and methods for immunooncology |
| CA3100724A1 (en) | 2018-06-13 | 2019-12-19 | Novartis Ag | B-cell maturation antigen protein (bcma) chimeric antigen receptors and uses thereof |
| WO2019246563A1 (en) | 2018-06-22 | 2019-12-26 | Kite Pharma, Inc. | Chimeric transmembrane proteins and uses thereof |
| WO2019246546A1 (en) * | 2018-06-22 | 2019-12-26 | The General Hospital Corporation | Chimeric antigen receptors targeting cd37 and cd19 |
| CN110623980A (zh) * | 2018-06-25 | 2019-12-31 | 深圳宾德生物技术有限公司 | 靶向cd38的嵌合抗原受体t细胞在自身免疫性疾病中的应用 |
| CN108866004A (zh) * | 2018-06-26 | 2018-11-23 | 奥妙生物技术(广州)有限公司 | Shp-1敲除的t细胞及其构建方法 |
| CN108913709A (zh) * | 2018-06-26 | 2018-11-30 | 山东兴瑞生物科技有限公司 | 用于治疗hcc的核酸、其制备方法、具有该核酸的car-t细胞及细胞的制备方法 |
| WO2020006176A1 (en) | 2018-06-27 | 2020-01-02 | Obi Pharma, Inc. | Glycosynthase variants for glycoprotein engineering and methods of use |
| WO2020000465A1 (zh) * | 2018-06-29 | 2020-01-02 | 深圳市博奥康生物科技有限公司 | 一种制备lightr基因敲除小鼠的方法 |
| US20210301024A1 (en) * | 2018-07-04 | 2021-09-30 | Cytoimmune Therapeutics, Inc. | Compositions and methods for immunotherapy targeting flt3, pd-1, and/or pd-l1 |
| EP3820571A1 (en) * | 2018-07-10 | 2021-05-19 | University of Connecticut | Reagents and methods for treating cancer and autoimmune disease |
| AR116109A1 (es) | 2018-07-10 | 2021-03-31 | Novartis Ag | Derivados de 3-(5-amino-1-oxoisoindolin-2-il)piperidina-2,6-diona y usos de los mismos |
| US20210308267A1 (en) * | 2018-08-07 | 2021-10-07 | Purdue Research Foundation | Rejuvenation of car t cell |
| JP2021533746A (ja) | 2018-08-09 | 2021-12-09 | ジュノー セラピューティクス インコーポレイテッド | 操作された細胞およびその組成物を生成するための方法 |
| CN112888481A (zh) * | 2018-08-10 | 2021-06-01 | 桑格摩生物治疗法国公司 | 包含tnfr2结构域的新型car构建体 |
| WO2020046766A1 (en) * | 2018-08-26 | 2020-03-05 | Oaiscell Biotechnologies | Method for treating glioblastoma |
| US20220348682A1 (en) | 2018-08-30 | 2022-11-03 | Innovative Cellular Therapeutics Holdings, Ltd. | Chimeric antigen receptor cells for treating solid tumor |
| US20210171909A1 (en) | 2018-08-31 | 2021-06-10 | Novartis Ag | Methods of making chimeric antigen receptor?expressing cells |
| BR112021003305A2 (pt) | 2018-08-31 | 2021-05-25 | Novartis Ag | métodos para produzir células que expressam receptor de antígeno quimérico |
| CN109265561B (zh) * | 2018-09-25 | 2021-05-25 | 山东兴瑞生物科技有限公司 | 抗EGFRvⅢ安全型嵌合抗原受体、其制备方法、利用其修饰的NK细胞及应用 |
| JP7425049B2 (ja) | 2018-09-25 | 2024-01-30 | ハープーン セラピューティクス,インク. | Dll3結合タンパク質および使用方法 |
| US20210347851A1 (en) | 2018-09-28 | 2021-11-11 | Novartis Ag | Cd19 chimeric antigen receptor (car) and cd22 car combination therapies |
| EP3856779A1 (en) | 2018-09-28 | 2021-08-04 | Novartis AG | Cd22 chimeric antigen receptor (car) therapies |
| WO2020072700A1 (en) | 2018-10-02 | 2020-04-09 | Dana-Farber Cancer Institute, Inc. | Hla single allele lines |
| US20210379057A1 (en) | 2018-10-16 | 2021-12-09 | Massachusetts Institute Of Technology | Nutlin-3a for use in treating a mycobacterium tuberculosis infection |
| WO2020086989A1 (en) * | 2018-10-25 | 2020-04-30 | Innovative Cellular Therapeutics CO., LTD. | Increase or maintaining t-cell subpopulations in adoptive t-cell therapy |
| JP2022513076A (ja) * | 2018-11-19 | 2022-02-07 | ボード オブ リージェンツ,ザ ユニバーシティ オブ テキサス システム | Carおよびtcr形質導入用のモジュール式ポリシストロニックベクター |
| US10918667B2 (en) * | 2018-11-20 | 2021-02-16 | Innovative Cellular Therapeutics CO., LTD. | Modified cell expressing therapeutic agent and uses thereof |
| CA3118511A1 (en) | 2018-11-29 | 2020-06-04 | Vineti Inc. | Centralized and decentralized individualized medicine platform |
| BR112021012172A2 (pt) | 2018-12-12 | 2021-08-31 | Kite Pharma, Inc. | Receptores de antígenos quiméricos e de célula t e métodos de uso |
| WO2020131586A2 (en) | 2018-12-17 | 2020-06-25 | The Broad Institute, Inc. | Methods for identifying neoantigens |
| WO2020127513A1 (en) * | 2018-12-19 | 2020-06-25 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Mucosal-associated invariant t (mait) cells expressing chimeric antigen receptors |
| BR112021011874A2 (pt) | 2018-12-20 | 2021-09-08 | Novartis Ag | Regime de dosagem e combinação farmacêutica compreendendo derivados de 3-(1-oxoisoindolin-2-il)piperidina-2,6-diona |
| US20220090125A1 (en) | 2018-12-21 | 2022-03-24 | Compass Therapeutics Llc | Transgenic mouse expressing common human light chain |
| EP3906057A4 (en) * | 2019-01-04 | 2022-09-14 | Marengo Therapeutics, Inc. | ANTI-TCR ANTIBODY MOLECULES AND THEIR USES |
| US11739156B2 (en) | 2019-01-06 | 2023-08-29 | The Broad Institute, Inc. Massachusetts Institute of Technology | Methods and compositions for overcoming immunosuppression |
| MX2021009763A (es) | 2019-02-15 | 2021-09-08 | Novartis Ag | Derivados de 3-(1-oxo-5-(piperidin-4-il)isoindolin-2-il)piperidina -2,6-diona y usos de los mismos. |
| KR20210129672A (ko) | 2019-02-15 | 2021-10-28 | 노파르티스 아게 | 치환된 3-(1-옥소이소인돌린-2-일)피페리딘-2,6-디온 유도체 및 이의 용도 |
| US11321652B1 (en) | 2019-02-20 | 2022-05-03 | Vineti Inc. | Smart label devices, systems, and methods |
| US20220088075A1 (en) | 2019-02-22 | 2022-03-24 | The Trustees Of The University Of Pennsylvania | Combination therapies of egfrviii chimeric antigen receptors and pd-1 inhibitors |
| EP3930763A1 (en) | 2019-02-25 | 2022-01-05 | Novartis AG | Mesoporous silica particles compositions for viral delivery |
| WO2020186101A1 (en) | 2019-03-12 | 2020-09-17 | The Broad Institute, Inc. | Detection means, compositions and methods for modulating synovial sarcoma cells |
| EP3942023A1 (en) | 2019-03-18 | 2022-01-26 | The Broad Institute, Inc. | Compositions and methods for modulating metabolic regulators of t cell pathogenicity |
| WO2020210678A1 (en) | 2019-04-12 | 2020-10-15 | Novartis Ag | Methods of making chimeric antigen receptor-expressing cells |
| EP3959320A1 (en) | 2019-04-24 | 2022-03-02 | Novartis AG | Compositions and methods for selective protein degradation |
| CN114269777A (zh) * | 2019-04-26 | 2022-04-01 | 北卡罗来纳大学教堂山分校 | 嵌合抗原受体构建体及其在car-t细胞中的用途 |
| CA3138633A1 (en) | 2019-04-30 | 2020-11-05 | Crispr Therapeutics Ag | Allogeneic cell therapy of b cell malignancies using genetically engineered t cells targeting cd19 |
| CN114025775A (zh) * | 2019-04-30 | 2022-02-08 | 纪念斯隆-凯特琳癌症中心 | 联合疗法 |
| WO2020223535A1 (en) | 2019-05-01 | 2020-11-05 | Juno Therapeutics, Inc. | Cells expressing a recombinant receptor from a modified tgfbr2 locus, related polynucleotides and methods |
| JP2022531577A (ja) | 2019-05-01 | 2022-07-07 | ジュノー セラピューティクス インコーポレイテッド | 改変されたcd247遺伝子座からキメラ受容体を発現する細胞、関連ポリヌクレオチド、および方法 |
| WO2020236967A1 (en) | 2019-05-20 | 2020-11-26 | The Broad Institute, Inc. | Random crispr-cas deletion mutant |
| US20220226464A1 (en) | 2019-05-28 | 2022-07-21 | Massachusetts Institute Of Technology | Methods and compositions for modulating immune responses |
| KR20220081972A (ko) * | 2019-07-17 | 2022-06-16 | 주식회사 큐로셀 | 이중 shrna를 이용하는 향상된 면역 세포 및 이를 포함하는 조성물 |
| WO2021016609A1 (en) * | 2019-07-24 | 2021-01-28 | Eureka Therapeutics, Inc. | Cells expressing chimeric antigen receptors and chimeric stimulating receptors and uses thereof |
| JP2022543779A (ja) * | 2019-07-30 | 2022-10-14 | デンドロサイト バイオテック ピーティーワイ リミテッド | イムノコンジュゲート |
| US20220282333A1 (en) | 2019-08-13 | 2022-09-08 | The General Hospital Corporation | Methods for predicting outcomes of checkpoint inhibition and treatment thereof |
| AU2020334884A1 (en) | 2019-08-18 | 2022-02-17 | Chimera Bioengineering, Inc. | Combination therapy with gold controlled transgenes |
| CN110627909B (zh) * | 2019-08-28 | 2021-03-30 | 中国人民解放军第二军医大学 | 一种特异性活化nk细胞的嵌合抗原受体及其应用 |
| WO2021041922A1 (en) | 2019-08-30 | 2021-03-04 | The Broad Institute, Inc. | Crispr-associated mu transposase systems |
| MX2022002747A (es) | 2019-09-10 | 2022-04-06 | Obsidian Therapeutics Inc | Proteinas de fusion de ca2-il15 para regulacion ajustable. |
| JP2022547552A (ja) * | 2019-09-11 | 2022-11-14 | ザ トラスティーズ オブ ザ ユニバーシティ オブ ペンシルバニア | 前立腺幹細胞抗原(psca)キメラ抗原受容体(car)を含む組成物および方法 |
| EP4032972A1 (en) * | 2019-09-18 | 2022-07-27 | Slbigen Inc. | Genetically modified nk cell line transduced with gene encoding novel chimeric antigen receptor and use thereof |
| WO2021057906A1 (zh) * | 2019-09-25 | 2021-04-01 | 科济生物医药(上海)有限公司 | 表达il-15的免疫效应细胞 |
| US11793787B2 (en) | 2019-10-07 | 2023-10-24 | The Broad Institute, Inc. | Methods and compositions for enhancing anti-tumor immunity by targeting steroidogenesis |
| JP2022552875A (ja) * | 2019-10-22 | 2022-12-20 | ザ ユナイテッド ステイツ オブ アメリカ, アズ リプレゼンテッド バイ ザ セクレタリー, デパートメント オブ ヘルス アンド ヒューマン サービシーズ | 多様な固形腫瘍を処置するためのb7h3(cd276)を標的とする高親和性ナノボディ |
| WO2021081239A1 (en) * | 2019-10-23 | 2021-04-29 | Cue Biopharma, Inc. | T-cell modulatory chimeric molecules and methods of use thereof |
| EP4048402A4 (en) * | 2019-10-23 | 2023-11-22 | Cue Biopharma, Inc. | MODIFIED CYTOXIC T CELLS AND METHODS OF USE THEREOF |
| CN110627903B (zh) * | 2019-10-29 | 2021-09-21 | 河南大学 | 抗cd97单克隆抗体、其可变区与恒定区序列及应用 |
| EP4054601A1 (en) * | 2019-11-04 | 2022-09-14 | Secura Bio, Inc. | Methods of making cellular therapies |
| AU2020393912A1 (en) | 2019-11-26 | 2022-06-09 | Novartis Ag | Chimeric antigen receptors binding BCMA and CD19 and uses thereof |
| AU2020394279A1 (en) * | 2019-11-28 | 2022-06-16 | CentricsBio, Inc. | Chimeric antigen receptor specifically binding to CD 300c antigen or receptor thereof |
| WO2021127428A1 (en) * | 2019-12-18 | 2021-06-24 | Albert Einstein College Of Medicine | Chimeric antigen receptors targeting b7-h3 (cd276) and associated methods |
| CA3165399A1 (en) | 2019-12-20 | 2021-06-24 | Novartis Ag | Uses of anti-tgf-beta antibodies and checkpoint inhibitors for the treatment of proliferative diseases |
| WO2021163618A1 (en) | 2020-02-14 | 2021-08-19 | Novartis Ag | Method of predicting response to chimeric antigen receptor therapy |
| MX2022010604A (es) | 2020-02-27 | 2022-09-09 | Novartis Ag | Metodos de produccion de celulas que expresan receptores antigenicos quimericos. |
| JP2023515211A (ja) | 2020-02-27 | 2023-04-12 | ノバルティス アーゲー | キメラ抗原受容体発現細胞を作製する方法 |
| WO2021222944A1 (en) * | 2020-04-30 | 2021-11-04 | Board Of Regents, The University Of Texas System | Anti-cd79b antibodies and chimeric antigen receptors and methods of use thereof |
| CN113801238A (zh) * | 2020-06-11 | 2021-12-17 | 南京北恒生物科技有限公司 | 表达nk抑制性分子的工程化免疫细胞及其用途 |
| WO2021257905A2 (en) * | 2020-06-17 | 2021-12-23 | H. Lee Moffitt Cancer Center And Research Institute, Inc. | Olfactory receptors for use as targets for antigen binding molecules to detect and treat cancer |
| BR112022026202A2 (pt) | 2020-06-23 | 2023-01-17 | Novartis Ag | Regime de dosagem compreendendo derivados de 3-(1-oxoisoindolin-2-il)piperidina-2,6-diona |
| WO2021260186A1 (en) | 2020-06-26 | 2021-12-30 | Juno Therapeutics Gmbh | Engineered t cells conditionally expressing a recombinant receptor, related polynucleotides and methods |
| US20230310606A1 (en) | 2020-07-17 | 2023-10-05 | Simurx, Inc. | Chimeric myd88 receptors for redirecting immunosuppressive signaling and related compositions and methods |
| CN116134027A (zh) | 2020-08-03 | 2023-05-16 | 诺华股份有限公司 | 杂芳基取代的3-(1-氧代异吲哚啉-2-基)哌啶-2,6-二酮衍生物及其用途 |
| CA3190415A1 (en) * | 2020-08-05 | 2022-02-10 | Synthekine, Inc. | Il2rb/il2rg synthetic cytokines |
| MX2023001491A (es) * | 2020-08-05 | 2023-03-08 | Synthekine Inc | Moleculas de union a gp130 y metodos de uso. |
| CN116113689A (zh) | 2020-08-14 | 2023-05-12 | 凯德药业股份有限公司 | 改善免疫细胞功能 |
| WO2022036495A1 (en) | 2020-08-17 | 2022-02-24 | Utc Therapeutics Inc. | Lymphocytes-antigen presenting cells co-stimulators and uses thereof |
| EP4199960A2 (en) | 2020-08-21 | 2023-06-28 | Novartis AG | Compositions and methods for in vivo generation of car expressing cells |
| CN114369168A (zh) * | 2020-10-19 | 2022-04-19 | 南京卡提医学科技有限公司 | 包含dap12和共刺激信号分子信号域的嵌合受体及其使用方法 |
| CN114426583B (zh) * | 2020-10-29 | 2023-10-10 | 中国科学技术大学 | 用于急性髓系白血病的细胞疗法的嵌合抗原受体 |
| MX2023005609A (es) | 2020-11-13 | 2023-05-29 | Novartis Ag | Terapias de combinacion con celulas que expresan receptores quimericos para el antigeno (car). |
| WO2022109023A1 (en) * | 2020-11-17 | 2022-05-27 | Igm Biosciences, Inc. | Uses of effector cell engaging molecules with moieties of differing potencies |
| US11661459B2 (en) | 2020-12-03 | 2023-05-30 | Century Therapeutics, Inc. | Artificial cell death polypeptide for chimeric antigen receptor and uses thereof |
| TW202237638A (zh) | 2020-12-09 | 2022-10-01 | 日商武田藥品工業股份有限公司 | 烏苷酸環化酶c(gcc)抗原結合劑之組成物及其使用方法 |
| CN113234169B (zh) * | 2020-12-11 | 2022-11-01 | 广州百暨基因科技有限公司 | 靶向cll1嵌合抗原受体及其应用 |
| US11883432B2 (en) | 2020-12-18 | 2024-01-30 | Century Therapeutics, Inc. | Chimeric antigen receptor system with adaptable receptor specificity |
| TW202241479A (zh) | 2020-12-30 | 2022-11-01 | 美商安迅生物製藥公司 | 遞送外來抗原之溶瘤病毒及表現靶向外來抗原之嵌合受體之經工程化免疫細胞之組合療法 |
| WO2022147075A1 (en) * | 2020-12-30 | 2022-07-07 | St. Jude Children's Research Hospital, Inc. | Chimeric antigen receptors targeting splice variants of the extracellular matrix proteins tenascin c (tnc) and procollagen 11a1 (col11a1) |
| US20240115607A1 (en) | 2021-01-26 | 2024-04-11 | Cytocares (Shanghai) Inc. | Chimeric antigen receptor (car) constructs and nk cells expressing car constructs |
| AU2022213745A1 (en) * | 2021-01-29 | 2023-09-07 | Minghui Pharmaceutical (Hangzhou) Limited | Antigen binding protein and use thereof |
| CN112851826B (zh) * | 2021-02-10 | 2023-08-11 | 上海煦顼技术有限公司 | Upk2嵌合抗原受体及其尿道癌的治疗 |
| KR20220124839A (ko) * | 2021-03-03 | 2022-09-14 | 주식회사 이뮤노로지컬디자이닝랩 | RANK 리간드(RANK ligand)에 특이적으로 결합하는 키메릭 항원 수용체 및 이의 용도 |
| KR20230155521A (ko) | 2021-03-11 | 2023-11-10 | 카이트 파마 인코포레이티드 | 면역 세포 기능의 향상 |
| TW202304979A (zh) | 2021-04-07 | 2023-02-01 | 瑞士商諾華公司 | 抗TGFβ抗體及其他治療劑用於治療增殖性疾病之用途 |
| WO2022229853A1 (en) | 2021-04-27 | 2022-11-03 | Novartis Ag | Viral vector production system |
| WO2023021477A1 (en) | 2021-08-20 | 2023-02-23 | Novartis Ag | Methods of making chimeric antigen receptor–expressing cells |
| EP4141028A1 (en) | 2021-08-31 | 2023-03-01 | Johann-Wolfgang-Goethe-Universität Frankfurt am Main | Personalized vnar-based chimeric antigen receptors (vnar-car) for the treatment of clonal b-and t cell malignancies, and methods for producing vnar-cars |
| US11615874B1 (en) | 2021-09-30 | 2023-03-28 | Vineti Inc. | Personalized medicine and therapies platform |
| CN114015674A (zh) | 2021-11-02 | 2022-02-08 | 辉二(上海)生物科技有限公司 | 新型CRISPR-Cas12i系统 |
| WO2023177954A1 (en) * | 2022-03-18 | 2023-09-21 | University Of Rochester | Combination therapy for treatment of cancer, methods and systems of delivery thereof |
| CN114805584B (zh) * | 2022-06-30 | 2022-09-09 | 上海优替济生生物医药有限公司 | 抗原结合蛋白及其用途 |
| WO2024014470A1 (ja) * | 2022-07-13 | 2024-01-18 | 国立大学法人大阪大学 | 腫瘍の予防および/または治療剤 |
| WO2024044670A1 (en) | 2022-08-26 | 2024-02-29 | Kite Pharma, Inc. | Improving immune cell function |
| WO2024056809A1 (en) | 2022-09-15 | 2024-03-21 | Novartis Ag | Treatment of autoimmune disorders using chimeric antigen receptor therapy |
| WO2024077256A1 (en) | 2022-10-07 | 2024-04-11 | The General Hospital Corporation | Methods and compositions for high-throughput discovery ofpeptide-mhc targeting binding proteins |
Family Cites Families (291)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US504048A (en) | 1893-08-29 | Sulky | ||
| ZA737247B (en) | 1972-09-29 | 1975-04-30 | Ayerst Mckenna & Harrison | Rapamycin and process of preparation |
| US4433059A (en) | 1981-09-08 | 1984-02-21 | Ortho Diagnostic Systems Inc. | Double antibody conjugate |
| US4444878A (en) | 1981-12-21 | 1984-04-24 | Boston Biomedical Research Institute, Inc. | Bispecific antibody determinants |
| DE3381783D1 (de) | 1982-03-03 | 1990-09-13 | Genentech Inc | Menschliches antithrombin iii, dns sequenzen dafuer, expressions- und klonierungsvektoren die solche sequenzen enthalten und damit transformierte zellkulturen, verfahren zur expression von menschlichem antithrombin iii und diese enthaltende pharmazeutische zusammensetzungen. |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4851332A (en) | 1985-04-01 | 1989-07-25 | Sloan-Kettering Institute For Cancer Research | Choriocarcinoma monoclonal antibodies and antibody panels |
| GB8607679D0 (en) | 1986-03-27 | 1986-04-30 | Winter G P | Recombinant dna product |
| US6548640B1 (en) | 1986-03-27 | 2003-04-15 | Btg International Limited | Altered antibodies |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| US5869620A (en) | 1986-09-02 | 1999-02-09 | Enzon, Inc. | Multivalent antigen-binding proteins |
| JPH021556A (ja) | 1988-06-09 | 1990-01-05 | Snow Brand Milk Prod Co Ltd | ハイブリッド抗体及びその作製方法 |
| US6352694B1 (en) | 1994-06-03 | 2002-03-05 | Genetics Institute, Inc. | Methods for inducing a population of T cells to proliferate using agents which recognize TCR/CD3 and ligands which stimulate an accessory molecule on the surface of the T cells |
| US5858358A (en) | 1992-04-07 | 1999-01-12 | The United States Of America As Represented By The Secretary Of The Navy | Methods for selectively stimulating proliferation of T cells |
| US6905680B2 (en) | 1988-11-23 | 2005-06-14 | Genetics Institute, Inc. | Methods of treating HIV infected subjects |
| US6534055B1 (en) | 1988-11-23 | 2003-03-18 | Genetics Institute, Inc. | Methods for selectively stimulating proliferation of T cells |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5703055A (en) | 1989-03-21 | 1997-12-30 | Wisconsin Alumni Research Foundation | Generation of antibodies through lipid mediated DNA delivery |
| US5399346A (en) | 1989-06-14 | 1995-03-21 | The United States Of America As Represented By The Department Of Health And Human Services | Gene therapy |
| DE3920358A1 (de) | 1989-06-22 | 1991-01-17 | Behringwerke Ag | Bispezifische und oligospezifische, mono- und oligovalente antikoerperkonstrukte, ihre herstellung und verwendung |
| US5585362A (en) | 1989-08-22 | 1996-12-17 | The Regents Of The University Of Michigan | Adenovirus vectors for gene therapy |
| WO1991003493A1 (en) | 1989-08-29 | 1991-03-21 | The University Of Southampton | Bi-or trispecific (fab)3 or (fab)4 conjugates |
| GB8928874D0 (en) | 1989-12-21 | 1990-02-28 | Celltech Ltd | Humanised antibodies |
| US5273743A (en) | 1990-03-09 | 1993-12-28 | Hybritech Incorporated | Trifunctional antibody-like compounds as a combined diagnostic and therapeutic agent |
| GB9012995D0 (en) | 1990-06-11 | 1990-08-01 | Celltech Ltd | Multivalent antigen-binding proteins |
| US5582996A (en) | 1990-12-04 | 1996-12-10 | The Wistar Institute Of Anatomy & Biology | Bifunctional antibodies and method of preparing same |
| EP0519596B1 (en) | 1991-05-17 | 2005-02-23 | Merck & Co. Inc. | A method for reducing the immunogenicity of antibody variable domains |
| DE4118120A1 (de) | 1991-06-03 | 1992-12-10 | Behringwerke Ag | Tetravalente bispezifische rezeptoren, ihre herstellung und verwendung |
| US5199942A (en) | 1991-06-07 | 1993-04-06 | Immunex Corporation | Method for improving autologous transplantation |
| US6511663B1 (en) | 1991-06-11 | 2003-01-28 | Celltech R&D Limited | Tri- and tetra-valent monospecific antigen-binding proteins |
| DE122004000008I1 (de) | 1991-06-14 | 2005-06-09 | Genentech Inc | Humanisierter Heregulin Antikörper. |
| US5637481A (en) | 1993-02-01 | 1997-06-10 | Bristol-Myers Squibb Company | Expression vectors encoding bispecific fusion proteins and methods of producing biologically active bispecific fusion proteins in a mammalian cell |
| AU669124B2 (en) | 1991-09-18 | 1996-05-30 | Kyowa Hakko Kirin Co., Ltd. | Process for producing humanized chimera antibody |
| US5565332A (en) | 1991-09-23 | 1996-10-15 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| US5932448A (en) | 1991-11-29 | 1999-08-03 | Protein Design Labs., Inc. | Bispecific antibody heterodimers |
| GB9125768D0 (en) | 1991-12-04 | 1992-02-05 | Hale Geoffrey | Therapeutic method |
| CA2103887C (en) | 1991-12-13 | 2005-08-30 | Gary M. Studnicka | Methods and materials for preparation of modified antibody variable domains and therapeutic uses thereof |
| AU676150B2 (en) | 1992-01-23 | 1997-03-06 | Merck Patent Gmbh | Monomeric and dimeric antibody-fragment fusion proteins |
| ATE295420T1 (de) | 1992-02-06 | 2005-05-15 | Chiron Corp | Marker für krebs und biosynthetisches bindeprotein dafür |
| GB9203459D0 (en) | 1992-02-19 | 1992-04-08 | Scotgen Ltd | Antibodies with germ-line variable regions |
| US5646253A (en) | 1994-03-08 | 1997-07-08 | Memorial Sloan-Kettering Cancer Center | Recombinant human anti-LK26 antibodies |
| ATE165113T1 (de) | 1992-05-08 | 1998-05-15 | Creative Biomolecules Inc | Mehrwertige chimäre proteine anologe und verfahren zu deren anwendungen |
| PT1498427E (pt) | 1992-08-21 | 2010-03-22 | Univ Bruxelles | Imunoglobulinas desprovidas de cadeias leves |
| US6005079A (en) | 1992-08-21 | 1999-12-21 | Vrije Universiteit Brussels | Immunoglobulins devoid of light chains |
| US5350674A (en) | 1992-09-04 | 1994-09-27 | Becton, Dickinson And Company | Intrinsic factor - horse peroxidase conjugates and a method for increasing the stability thereof |
| US5639641A (en) | 1992-09-09 | 1997-06-17 | Immunogen Inc. | Resurfacing of rodent antibodies |
| EP1550729B1 (en) | 1992-09-25 | 2009-05-27 | Avipep Pty Limited | Target binding polypeptide comprising an IG-like VL domain linked to an IG-like VH domain |
| GB9221220D0 (en) | 1992-10-09 | 1992-11-25 | Sandoz Ag | Organic componds |
| GB9221657D0 (en) | 1992-10-15 | 1992-11-25 | Scotgen Ltd | Recombinant bispecific antibodies |
| EP0627932B1 (en) | 1992-11-04 | 2002-05-08 | City Of Hope | Antibody construct |
| GB9323648D0 (en) | 1992-11-23 | 1994-01-05 | Zeneca Ltd | Proteins |
| EP0672142B1 (en) | 1992-12-04 | 2001-02-28 | Medical Research Council | Multivalent and multispecific binding proteins, their manufacture and use |
| US6476198B1 (en) | 1993-07-13 | 2002-11-05 | The Scripps Research Institute | Multispecific and multivalent antigen-binding polypeptide molecules |
| US5635602A (en) | 1993-08-13 | 1997-06-03 | The Regents Of The University Of California | Design and synthesis of bispecific DNA-antibody conjugates |
| WO1995009917A1 (en) | 1993-10-07 | 1995-04-13 | The Regents Of The University Of California | Genetically engineered bispecific tetravalent antibodies |
| AU7863794A (en) | 1993-11-16 | 1995-06-06 | Pola Chemical Industries Inc. | Antihuman tyrosinase monoclonal antibody |
| JP4105761B2 (ja) | 1993-11-19 | 2008-06-25 | アボット・ラボラトリーズ | ラパミシン(マクロライド)の半合成類似体免疫調節剤 |
| PT734389E (pt) | 1993-12-17 | 2000-09-29 | Novartis Ag | Derivados de rapamicina uteis como imunossupressores |
| US5635388A (en) | 1994-04-04 | 1997-06-03 | Genentech, Inc. | Agonist antibodies against the flk2/flt3 receptor and uses thereof |
| US5362718A (en) | 1994-04-18 | 1994-11-08 | American Home Products Corporation | Rapamycin hydroxyesters |
| CA2188432C (en) | 1994-04-22 | 2011-02-01 | Yutaka Kawakami | Melanoma antigens |
| US7175843B2 (en) | 1994-06-03 | 2007-02-13 | Genetics Institute, Llc | Methods for selectively stimulating proliferation of T cells |
| US5786464C1 (en) | 1994-09-19 | 2012-04-24 | Gen Hospital Corp | Overexpression of mammalian and viral proteins |
| WO1996013583A2 (en) | 1994-10-20 | 1996-05-09 | Morphosys Gesellschaft Für Proteinoptimierung Mbh | Targeted hetero-association of recombinant proteins to multi-functional complexes |
| ES2141467T5 (es) | 1995-01-18 | 2006-07-16 | Roche Diagnostics Gmbh | Anticuerpos anti-cd30 que evitan la escision proteolitica y liberacion del antigeno cd30 fijado a la membrana. |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US6692964B1 (en) | 1995-05-04 | 2004-02-17 | The United States Of America As Represented By The Secretary Of The Navy | Methods for transfecting T cells |
| US7067318B2 (en) | 1995-06-07 | 2006-06-27 | The Regents Of The University Of Michigan | Methods for transfecting T cells |
| JPH11508126A (ja) | 1995-05-23 | 1999-07-21 | モルフォシス ゲゼルシャフト ファー プロテインオプティマイルング エムベーハー | 多量体タンパク質 |
| KR100400620B1 (ko) | 1995-06-09 | 2004-02-18 | 노파르티스 아게 | 라파마이신유도체 |
| BE1009856A5 (fr) | 1995-07-14 | 1997-10-07 | Sandoz Sa | Composition pharmaceutique sous la forme d'une dispersion solide comprenant un macrolide et un vehicule. |
| AU6873396A (en) | 1995-10-16 | 1997-05-07 | Unilever N.V. | A bifunctional or bivalent antibody fragment analogue |
| DE19608769C1 (de) | 1996-03-07 | 1997-04-10 | Univ Eberhard Karls | Antikörper BV10A4H2 |
| DK0894135T3 (da) | 1996-04-04 | 2004-12-06 | Unilever Nv | Multivalent og multispecifikt antigenbindingsprotein |
| US6258823B1 (en) | 1996-07-12 | 2001-07-10 | Ariad Pharmaceuticals, Inc. | Materials and method for treating or preventing pathogenic fungal infection |
| US6111090A (en) | 1996-08-16 | 2000-08-29 | Schering Corporation | Mammalian cell surface antigens; related reagents |
| EP1947183B1 (en) | 1996-08-16 | 2013-07-17 | Merck Sharp & Dohme Corp. | Mammalian cell surface antigens; related reagents |
| US6114148C1 (en) | 1996-09-20 | 2012-05-01 | Gen Hospital Corp | High level expression of proteins |
| WO1998017796A2 (en) | 1996-10-25 | 1998-04-30 | The Government Of The United States Of America As Represented By The Secretary, Department Of Health And Human Services | Methods and compositions for inhibiting inflammation and angiogenesis comprising a mammalian cd97 alpha subunit |
| WO1998049198A1 (en) | 1997-04-30 | 1998-11-05 | Enzon, Inc. | Single-chain antigen-binding proteins capable of glycosylation, production and uses thereof |
| US20030207346A1 (en) | 1997-05-02 | 2003-11-06 | William R. Arathoon | Method for making multispecific antibodies having heteromultimeric and common components |
| US20020062010A1 (en) | 1997-05-02 | 2002-05-23 | Genentech, Inc. | Method for making multispecific antibodies having heteromultimeric and common components |
| PT1012280E (pt) | 1997-06-11 | 2005-02-28 | Borean Pharma As | Modulo de trimerizacao |
| JP3614866B2 (ja) | 1997-06-12 | 2005-01-26 | リサーチ コーポレイション テクノロジーズ,インコーポレイティド | 人工抗体ポリペプチド |
| US6015815A (en) | 1997-09-26 | 2000-01-18 | Abbott Laboratories | Tetrazole-containing rapamycin analogs with shortened half-lives |
| TW557297B (en) | 1997-09-26 | 2003-10-11 | Abbott Lab | Rapamycin analogs having immunomodulatory activity, and pharmaceutical compositions containing same |
| US6509173B1 (en) | 1997-10-21 | 2003-01-21 | Human Genome Sciences, Inc. | Human tumor necrosis factor receptor-like proteins TR11, TR11SV1, and TR11SV2 |
| ATE461282T1 (de) | 1997-10-27 | 2010-04-15 | Bac Ip Bv | Multivalente antigenbindende proteine |
| DK1049787T3 (da) | 1998-01-23 | 2005-04-04 | Vlaams Interuniv Inst Biotech | Antistofderivater med flere anvendelsesmuligheder |
| JP2002502607A (ja) | 1998-02-09 | 2002-01-29 | ジェネンテク・インコーポレイテッド | 新規な腫瘍壊死因子レセプター相同体及びそれをコードする核酸 |
| HUP9900956A2 (hu) | 1998-04-09 | 2002-04-29 | Aventis Pharma Deutschland Gmbh. | Egyláncú, több antigéntkötőhely kialakítására képes molekulák, előállításuk és alkalmazásuk |
| DE69925909T2 (de) | 1998-04-15 | 2006-05-11 | Brigham & Women's Hospital, Inc., Boston | T-zell inhibierende rezeptorzusammensetzungen sowie deren verwendung |
| DE19819846B4 (de) | 1998-05-05 | 2016-11-24 | Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts | Multivalente Antikörper-Konstrukte |
| GB9812545D0 (en) | 1998-06-10 | 1998-08-05 | Celltech Therapeutics Ltd | Biological products |
| US6803448B1 (en) | 1998-07-22 | 2004-10-12 | Vanderbilt University | GBS toxin receptor |
| JP2002521053A (ja) | 1998-07-28 | 2002-07-16 | マイクロメット アーゲー | ヘテロミニ体 |
| US6333396B1 (en) | 1998-10-20 | 2001-12-25 | Enzon, Inc. | Method for targeted delivery of nucleic acids |
| US6528481B1 (en) | 1999-02-16 | 2003-03-04 | The Burnam Institute | NG2/HM proteoglycan-binding peptides that home to angiogenic vasculature and related methods |
| US7534866B2 (en) | 2005-10-19 | 2009-05-19 | Ibc Pharmaceuticals, Inc. | Methods and compositions for generating bioactive assemblies of increased complexity and uses |
| US7527787B2 (en) | 2005-10-19 | 2009-05-05 | Ibc Pharmaceuticals, Inc. | Multivalent immunoglobulin-based bioactive assemblies |
| EP1196186B1 (en) | 1999-07-12 | 2007-10-31 | Genentech, Inc. | Promotion or inhibition of angiogenesis and cardiovascularization by tumor necrosis factor ligand/receptor homologs |
| RS51157B (sr) | 1999-08-17 | 2010-10-31 | Biogen Idec Ma Inc. | Baff receptor (bcma), imunoregulatorni agens |
| ATE264863T1 (de) | 1999-08-24 | 2004-05-15 | Ariad Gene Therapeutics Inc | 28-epirapaloge |
| EP1238985B1 (en) | 1999-09-30 | 2008-03-05 | Kyowa Hakko Kogyo Co., Ltd. | Human type complementarity determining region transplantation antibody against ganglioside gd3 and derivatives of antibody against ganglioside gd3 |
| AU1086501A (en) | 1999-10-15 | 2001-04-30 | Carnegie Institution Of Washington | Rna interference pathway genes as tools for targeted genetic interference |
| US6326193B1 (en) | 1999-11-05 | 2001-12-04 | Cambria Biosciences, Llc | Insect control agent |
| US6797514B2 (en) | 2000-02-24 | 2004-09-28 | Xcyte Therapies, Inc. | Simultaneous stimulation and concentration of cells |
| US6867041B2 (en) | 2000-02-24 | 2005-03-15 | Xcyte Therapies, Inc. | Simultaneous stimulation and concentration of cells |
| KR20030032922A (ko) | 2000-02-24 | 2003-04-26 | 싸이트 테라피스 인코포레이티드 | 세포의 동시 자극 및 농축 |
| US7572631B2 (en) | 2000-02-24 | 2009-08-11 | Invitrogen Corporation | Activation and expansion of T cells |
| US20040002068A1 (en) | 2000-03-01 | 2004-01-01 | Corixa Corporation | Compositions and methods for the detection, diagnosis and therapy of hematological malignancies |
| ES2637801T3 (es) | 2000-04-11 | 2017-10-17 | Genentech, Inc. | Anticuerpos multivalentes y usos de los mismos |
| WO2001090192A2 (en) | 2000-05-24 | 2001-11-29 | Imclone Systems Incorporated | Bispecific immunoglobulin-like antigen binding proteins and method of production |
| WO2001096584A2 (en) | 2000-06-12 | 2001-12-20 | Akkadix Corporation | Materials and methods for the control of nematodes |
| CA2410551A1 (en) | 2000-06-30 | 2002-01-10 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw (Vib) | Heterodimeric fusion proteins |
| CA2417185A1 (en) | 2000-07-25 | 2002-01-31 | Shui-On Leung | Multivalent target binding protein |
| JP4261907B2 (ja) | 2000-10-20 | 2009-05-13 | 中外製薬株式会社 | 低分子化アゴニスト抗体 |
| US7090843B1 (en) | 2000-11-28 | 2006-08-15 | Seattle Genetics, Inc. | Recombinant anti-CD30 antibodies and uses thereof |
| US7829084B2 (en) | 2001-01-17 | 2010-11-09 | Trubion Pharmaceuticals, Inc. | Binding constructs and methods for use thereof |
| WO2002072635A2 (en) | 2001-03-13 | 2002-09-19 | University College London | Specific binding members |
| CN1294148C (zh) | 2001-04-11 | 2007-01-10 | 中国科学院遗传与发育生物学研究所 | 环状单链三特异抗体 |
| AU2002319402B2 (en) | 2001-06-28 | 2008-09-11 | Domantis Limited | Dual-specific ligand and its use |
| US6833441B2 (en) | 2001-08-01 | 2004-12-21 | Abmaxis, Inc. | Compositions and methods for generating chimeric heteromultimers |
| CA2457636C (en) | 2001-08-10 | 2012-01-03 | Aberdeen University | Antigen binding domains |
| JP2005509412A (ja) | 2001-08-23 | 2005-04-14 | アールエスアール リミテッド | チロトロフィン(tsh)受容体の抗原決定領域、その使用およびその抗体 |
| ES2276735T3 (es) | 2001-09-14 | 2007-07-01 | Affimed Therapeutics Ag | Anticuerpos fv multimericos de cadena sencilla en tandem. |
| JP4716350B2 (ja) | 2001-12-04 | 2011-07-06 | ダナ−ファーバー キャンサー インスティテュート インク. | 潜伏期膜タンパク質に対する抗体およびそれらの使用 |
| WO2003049684A2 (en) | 2001-12-07 | 2003-06-19 | Centocor, Inc. | Pseudo-antibody constructs |
| US7745140B2 (en) | 2002-01-03 | 2010-06-29 | The Trustees Of The University Of Pennsylvania | Activation and expansion of T-cells using an engineered multivalent signaling platform as a research tool |
| IL162734A0 (en) | 2002-02-01 | 2005-11-20 | Ariad Gene Therapeutics Inc | Phosphorus-containing compounds & uses thereof |
| EP1487879B1 (en) | 2002-03-01 | 2012-12-26 | Immunomedics, Inc. | Bispecific antibody point mutations for enhancing rate of clearance |
| GB0208104D0 (en) | 2002-04-09 | 2002-05-22 | Univ Dundee | Method |
| WO2003087163A1 (fr) | 2002-04-15 | 2003-10-23 | Chugai Seiyaku Kabushiki Kaisha | Procede d'elaboration d'une banque scdb |
| US20040047858A1 (en) | 2002-09-11 | 2004-03-11 | Blumberg Richard S. | Therapeutic anti-BGP(C-CAM1) antibodies and uses thereof |
| EP1565492B1 (de) | 2002-11-26 | 2006-12-20 | B.R.A.H.M.S Aktiengesellschaft | Nachweis von tsh-rezeptor-autoantikörpern mit affinitätsgereinigten antikörpern |
| US7521051B2 (en) | 2002-12-23 | 2009-04-21 | Medimmune Limited | Methods of upmodulating adaptive immune response using anti-PD-1 antibodies |
| GB0230203D0 (en) | 2002-12-27 | 2003-02-05 | Domantis Ltd | Fc fusion |
| GB0305702D0 (en) | 2003-03-12 | 2003-04-16 | Univ Birmingham | Bispecific antibodies |
| WO2004087758A2 (en) | 2003-03-26 | 2004-10-14 | Neopharm, Inc. | Il 13 receptor alpha 2 antibody and methods of use |
| AU2004232928A1 (en) | 2003-04-22 | 2004-11-04 | Ibc Pharmaceuticals | Polyvalent protein complex |
| CU23403A1 (es) | 2003-04-23 | 2009-08-04 | Centro Inmunologia Molecular | Anticuerpos recombinantes y fragmentos que reconocen el gangliósido n-glicolil gm3 y su uso para diagnóstico y tratamiento de tumores |
| MXPA05012475A (es) | 2003-05-23 | 2006-05-25 | Wyeth Corp | Anticuerpos y moleculas relacionadas con el ligando para el receptor tnf inducido por glucorticoides (gitr) y el ligando (gitr) y usos de los mismos. |
| US7479546B2 (en) | 2003-06-27 | 2009-01-20 | Diadexus, Inc. | Pro104 antibody compositions and methods of use |
| CA2529945A1 (en) | 2003-06-27 | 2005-01-06 | Biogen Idec Ma Inc. | Use of hydrophobic-interaction-chromatography or hinge-region modifications for the production of homogeneous antibody-solutions |
| US20050100543A1 (en) | 2003-07-01 | 2005-05-12 | Immunomedics, Inc. | Multivalent carriers of bi-specific antibodies |
| EP1660126A1 (en) | 2003-07-11 | 2006-05-31 | Schering Corporation | Agonists or antagonists of the clucocorticoid-induced tumour necrosis factor receptor (gitr) or its ligand for the treatment of immune disorders, infections and cancer |
| US7696322B2 (en) | 2003-07-28 | 2010-04-13 | Catalent Pharma Solutions, Inc. | Fusion antibodies |
| AU2004286198C1 (en) | 2003-08-18 | 2011-02-24 | Medimmune, Llc | Humanization of antibodies |
| WO2005035575A2 (en) | 2003-08-22 | 2005-04-21 | Medimmune, Inc. | Humanization of antibodies |
| EP1688439A4 (en) | 2003-10-08 | 2007-12-19 | Kyowa Hakko Kogyo Kk | HYBRID PROTEIN COMPOSITION |
| JPWO2005035577A1 (ja) | 2003-10-08 | 2007-11-22 | 協和醗酵工業株式会社 | ガングリオシドgd3に特異的に結合する抗体組成物 |
| US20130266551A1 (en) * | 2003-11-05 | 2013-10-10 | St. Jude Children's Research Hospital, Inc. | Chimeric receptors with 4-1bb stimulatory signaling domain |
| US7220755B2 (en) | 2003-11-12 | 2007-05-22 | Biosensors International Group, Ltd. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| JP2007518399A (ja) | 2003-12-02 | 2007-07-12 | ジェンザイム コーポレイション | 肺癌を診断および治療する組成物並びに方法 |
| WO2005062916A2 (en) | 2003-12-22 | 2005-07-14 | Centocor, Inc. | Methods for generating multimeric molecules |
| GB0329825D0 (en) | 2003-12-23 | 2004-01-28 | Celltech R&D Ltd | Biological products |
| US20050266425A1 (en) | 2003-12-31 | 2005-12-01 | Vaccinex, Inc. | Methods for producing and identifying multispecific antibodies |
| US8383575B2 (en) | 2004-01-30 | 2013-02-26 | Paul Scherrer Institut | (DI)barnase-barstar complexes |
| US8263746B2 (en) | 2004-02-06 | 2012-09-11 | Morphosys Ag | Anti-CD38 human antibodies and uses thereof |
| WO2006107617A2 (en) | 2005-04-06 | 2006-10-12 | Ibc Pharmaceuticals, Inc. | Methods for generating stably linked complexes composed of homodimers, homotetramers or dimers of dimers and uses |
| GB0409799D0 (en) | 2004-04-30 | 2004-06-09 | Isis Innovation | Method of generating improved immune response |
| US7754482B2 (en) | 2004-05-27 | 2010-07-13 | The Trustees Of The University Of Pennsylvania | Artificial antigen presenting cells and uses therefor |
| WO2006083289A2 (en) | 2004-06-04 | 2006-08-10 | Duke University | Methods and compositions for enhancement of immunity by in vivo depletion of immunosuppressive cell activity |
| JP2008512352A (ja) | 2004-07-17 | 2008-04-24 | イムクローン システムズ インコーポレイティド | 新規な四価の二重特異性抗体 |
| AU2005282700A1 (en) | 2004-09-02 | 2006-03-16 | Genentech, Inc. | Heteromultimeric molecules |
| MY146381A (en) | 2004-12-22 | 2012-08-15 | Amgen Inc | Compositions and methods relating relating to anti-igf-1 receptor antibodies |
| PT2343320T (pt) | 2005-03-25 | 2018-01-23 | Gitr Inc | Anticorpos anti-gitr e as suas utilizações |
| AU2006232287B2 (en) | 2005-03-31 | 2011-10-06 | Chugai Seiyaku Kabushiki Kaisha | Methods for producing polypeptides by regulating polypeptide association |
| ES2707152T3 (es) | 2005-04-15 | 2019-04-02 | Macrogenics Inc | Diacuerpos covalentes y usos de los mismos |
| AU2006244885B2 (en) | 2005-05-09 | 2011-03-31 | E. R. Squibb & Sons, L.L.C. | Human monoclonal antibodies to programmed death 1(PD-1) and methods for treating cancer using anti-PD-1 antibodies alone or in combination with other immunotherapeutics |
| US8124084B2 (en) * | 2005-05-17 | 2012-02-28 | University Of Connecticut | Compositions and methods for immunomodulation in an organism using IL-15 and soluble IL-15Ra |
| GB0510390D0 (en) | 2005-05-20 | 2005-06-29 | Novartis Ag | Organic compounds |
| US20060263367A1 (en) | 2005-05-23 | 2006-11-23 | Fey Georg H | Bispecific antibody devoid of Fc region and method of treatment using same |
| EP1726650A1 (en) | 2005-05-27 | 2006-11-29 | Universitätsklinikum Freiburg | Monoclonal antibodies and single chain antibody fragments against cell-surface prostate specific membrane antigen |
| JP2008546699A (ja) | 2005-06-15 | 2008-12-25 | シェーリング コーポレイション | 抗igf1r抗体処方物 |
| HUE026039T2 (en) | 2005-07-01 | 2016-05-30 | Squibb & Sons Llc | Human monoclonal antibodies programmed for death ligand 1 (PD-L1) |
| US20070036773A1 (en) | 2005-08-09 | 2007-02-15 | City Of Hope | Generation and application of universal T cells for B-ALL |
| WO2007024715A2 (en) | 2005-08-19 | 2007-03-01 | Abbott Laboratories | Dual variable domain immunoglobin and uses thereof |
| US7612181B2 (en) | 2005-08-19 | 2009-11-03 | Abbott Laboratories | Dual variable domain immunoglobulin and uses thereof |
| EP1757622B1 (en) | 2005-08-26 | 2009-12-23 | PLS Design GmbH | Bivalent IgY antibody constructs for diagnostic and therapeutic applications |
| GEP20115199B (en) | 2005-10-07 | 2011-04-11 | Exelixis Inc | Phosphatidylinositol 3-kinase inhibitors and their use |
| WO2007044887A2 (en) | 2005-10-11 | 2007-04-19 | Transtarget, Inc. | Method for producing a population of homogenous tetravalent bispecific antibodies |
| WO2007062466A1 (en) | 2005-11-29 | 2007-06-07 | The University Of Sydney | Demibodies: dimerisation-activated therapeutic agents |
| CA2638902C (en) | 2005-12-08 | 2014-09-23 | Medarex, Inc. | Human monoclonal antibodies to fucosyl-gm1 and methods for using anti-fucosyl-gm1 antibodies |
| EP1806365A1 (en) | 2006-01-05 | 2007-07-11 | Boehringer Ingelheim International GmbH | Antibody molecules specific for fibroblast activation protein and immunoconjugates containing them |
| AU2007207785B2 (en) | 2006-01-13 | 2013-11-14 | The Government Of The United States, As Represented By The Secretary Of The Department Of Health And Human Services, National Institutes Of Health | Codon optimized IL- 15 and IL- 15R-alpha genes for expression in mammalian cells |
| EP1981969A4 (en) | 2006-01-19 | 2009-06-03 | Genzyme Corp | ANTI-GITRANT ANTIBODIES FOR THE TREATMENT OF CANCER |
| JP2009526857A (ja) | 2006-02-15 | 2009-07-23 | イムクローン・リミテッド・ライアビリティ・カンパニー | 機能性抗体 |
| CA2646508A1 (en) | 2006-03-17 | 2007-09-27 | Biogen Idec Ma Inc. | Stabilized polypeptide compositions |
| ES2363891T3 (es) | 2006-03-20 | 2011-08-18 | The Regents Of The University Of California | Anticuerpos contra el antígeno de células troncales de la próstata (psca) modificados genéticamente para el direccionamiento al cáncer. |
| PL1999154T3 (pl) | 2006-03-24 | 2013-03-29 | Merck Patent Gmbh | Skonstruowane metodami inżynierii heterodimeryczne domeny białkowe |
| WO2007112362A2 (en) | 2006-03-24 | 2007-10-04 | The Regents Of The University Of California | Construction of a multivalent scfv through alkyne-azide 1,3-dipolar cycloaddition |
| JP5165672B2 (ja) | 2006-03-29 | 2013-03-21 | キングス カレッジ ロンドン | Tshrに対するアゴニスト抗体 |
| JP5144499B2 (ja) | 2006-03-31 | 2013-02-13 | 中外製薬株式会社 | 二重特異性抗体を精製するための抗体改変方法 |
| TWI395754B (zh) | 2006-04-24 | 2013-05-11 | Amgen Inc | 人類化之c-kit抗體 |
| EP2799449A1 (en) | 2006-05-25 | 2014-11-05 | Bayer Intellectual Property GmbH | Dimeric molecular complexes |
| US20070274985A1 (en) | 2006-05-26 | 2007-11-29 | Stefan Dubel | Antibody |
| NZ573646A (en) | 2006-06-12 | 2012-04-27 | Wyeth Llc | Single-chain multivalent binding proteins with effector function |
| US8759297B2 (en) | 2006-08-18 | 2014-06-24 | Armagen Technologies, Inc. | Genetically encoded multifunctional compositions bidirectionally transported between peripheral blood and the cns |
| WO2008027236A2 (en) | 2006-08-30 | 2008-03-06 | Genentech, Inc. | Multispecific antibodies |
| WO2008040362A2 (en) | 2006-10-04 | 2008-04-10 | Københavns Universitet | Generation of a cancer-specific immune response toward muc1 and cancer specific muc1 antibodies |
| FR2906808B1 (fr) | 2006-10-10 | 2012-10-05 | Univ Nantes | Utilisation d'anticorps monoclonaux specifiques de la forme o-acetylee du ganglioside gd2 dans le traitement de certains cancers |
| MX2009004664A (es) | 2006-11-02 | 2009-10-12 | Daniel J Capon | Inmunoglobulinas hibridas con partes moviles. |
| WO2008101234A2 (en) | 2007-02-16 | 2008-08-21 | Sloan-Kettering Institute For Cancer Research | Anti ganglioside gd3 antibodies and uses thereof |
| WO2008103645A2 (en) | 2007-02-19 | 2008-08-28 | Wisconsin Alumni Research Foundation | Prostate cancer and melanoma antigens |
| US9212230B2 (en) | 2007-03-29 | 2015-12-15 | Genmab A/S | Bispecific antibodies and methods for production thereof |
| EP2144935A2 (en) | 2007-03-29 | 2010-01-20 | Technion Research & Development Foundation Ltd. | Antibodies, methods and kits for diagnosing and treating melanoma |
| CA2682605A1 (en) | 2007-04-18 | 2008-10-30 | Zymogenetics, Inc. | Single chain fc, methods of making and methods of treatment |
| JP2010190572A (ja) | 2007-06-01 | 2010-09-02 | Sapporo Medical Univ | IL13Ra2に対する抗体およびこれを含む診断・治療薬 |
| SI2170959T1 (sl) | 2007-06-18 | 2014-04-30 | Merck Sharp & Dohme B.V. | Protitelesa proti receptorjem pd-1 za humano programirano smrt |
| EP2173378B1 (en) | 2007-06-27 | 2014-03-19 | Admune Therapeutics LLC | Complexes of il-15 and il-15ralpha and uses thereof |
| EP3124046B1 (en) | 2007-07-12 | 2019-12-25 | GITR, Inc. | Combination therapies employing gitr binding molecules |
| WO2009017679A2 (en) | 2007-07-31 | 2009-02-05 | Merck & Co., Inc. | Igf-1r specific antibodies useful in the detection and diagnosis of cellular proliferative disorders |
| EP2069401A4 (en) | 2007-07-31 | 2011-02-23 | Medimmune Llc | MULTISPECIENT EPITOP BINDING PROTEINS AND THEIR USE |
| EP2178914A2 (en) | 2007-08-15 | 2010-04-28 | Bayer Schering Pharma Aktiengesellschaft | Monospecific and multispecific antibodies and method of use |
| CA2703947C (en) | 2007-10-26 | 2018-12-04 | Governing Council Of The University Of Toronto | Therapeutic and diagnostic methods using tim-3 |
| JP2011504740A (ja) | 2007-11-27 | 2011-02-17 | アブリンクス エン.ヴェー. | ヘテロ二量体サイトカイン及び/又はこれらの受容体に指向性を有するアミノ酸配列、並びにこれを含むポリペプチド |
| CA2706419A1 (en) | 2007-11-30 | 2009-06-04 | Glaxo Group Limited | Antigen-binding constructs binding il-13 |
| US8242247B2 (en) | 2007-12-21 | 2012-08-14 | Hoffmann-La Roche Inc. | Bivalent, bispecific antibodies |
| US20090162359A1 (en) | 2007-12-21 | 2009-06-25 | Christian Klein | Bivalent, bispecific antibodies |
| US8227577B2 (en) | 2007-12-21 | 2012-07-24 | Hoffman-La Roche Inc. | Bivalent, bispecific antibodies |
| US9266967B2 (en) | 2007-12-21 | 2016-02-23 | Hoffmann-La Roche, Inc. | Bivalent, bispecific antibodies |
| SI2235064T1 (sl) | 2008-01-07 | 2016-04-29 | Amgen Inc. | Metoda za izdelavo heterodimernih molekul - protitelesa fc z uporabo elektrostatičnih usmerjevalnih učinkov |
| WO2009091826A2 (en) * | 2008-01-14 | 2009-07-23 | The Board Of Regents Of The University Of Texas System | Compositions and methods related to a human cd19-specific chimeric antigen receptor (h-car) |
| US8747847B2 (en) | 2008-02-11 | 2014-06-10 | Curetech Ltd. | Monoclonal antibodies for tumor treatment |
| EP2262504A1 (en) | 2008-02-21 | 2010-12-22 | AstraZeneca AB | Combination therapy 238 |
| EP2262837A4 (en) | 2008-03-12 | 2011-04-06 | Merck Sharp & Dohme | PD-1 BINDING PROTEINS |
| AR071891A1 (es) | 2008-05-30 | 2010-07-21 | Imclone Llc | Anticuerpos humanos anti-flt3 (receptor tirosina cinasa 3 tipo fms humano) |
| EP2310508A1 (en) | 2008-07-02 | 2011-04-20 | Emergent Product Development Seattle, LLC | Tgf-b antagonist multi-target binding proteins |
| AR072999A1 (es) | 2008-08-11 | 2010-10-06 | Medarex Inc | Anticuerpos humanos que se unen al gen 3 de activacion linfocitaria (lag-3) y los usos de estos |
| PE20110435A1 (es) | 2008-08-25 | 2011-07-20 | Amplimmune Inc | Composiciones antagonistas del pd-1 |
| AU2009288289B2 (en) | 2008-08-25 | 2012-11-08 | Amplimmune, Inc. | PD-1 antagonists and methods of use thereof |
| WO2010030002A1 (ja) | 2008-09-12 | 2010-03-18 | 国立大学法人三重大学 | 外来性gitrリガンド発現細胞 |
| CA2737758C (en) | 2008-09-19 | 2017-10-31 | Soldano Ferrone | Monoclonal antibodies for cspg4 for the diagnosis and treatment of basal breast carcinoma |
| US8476431B2 (en) | 2008-11-03 | 2013-07-02 | Itellikine LLC | Benzoxazole kinase inhibitors and methods of use |
| CN114835812A (zh) | 2008-12-09 | 2022-08-02 | 霍夫曼-拉罗奇有限公司 | 抗-pd-l1抗体及它们用于增强t细胞功能的用途 |
| EP3192811A1 (en) | 2009-02-09 | 2017-07-19 | Université d'Aix-Marseille | Pd-1 antibodies and pd-l1 antibodies and uses thereof |
| CA2950529C (en) | 2009-04-03 | 2019-04-30 | Verastem, Inc. | Pyrimidine substituted purine compounds as kinase (s) inhibitors |
| CN102459346B (zh) | 2009-04-27 | 2016-10-26 | 昂考梅德药品有限公司 | 制造异源多聚体分子的方法 |
| SI2424896T1 (sl) | 2009-04-30 | 2015-12-31 | Tel Hashomer Medical Research Infrastructure And Services Ltd. | Protitelesa anti-ceacam1 in postopki za njihovo uporabo |
| EP2464377B1 (en) | 2009-08-14 | 2016-07-27 | The Government of the United States of America as represented by The Secretary of the Department of Health and Human Services | Use of il-15 to increase thymic output and to treat lymphopenia |
| ES2788869T3 (es) | 2009-09-03 | 2020-10-23 | Merck Sharp & Dohme | Anticuerpos anti-GITR |
| US9181527B2 (en) | 2009-10-29 | 2015-11-10 | The Trustees Of Dartmouth College | T cell receptor-deficient T cell compositions |
| GB0919054D0 (en) | 2009-10-30 | 2009-12-16 | Isis Innovation | Treatment of obesity |
| US8956828B2 (en) | 2009-11-10 | 2015-02-17 | Sangamo Biosciences, Inc. | Targeted disruption of T cell receptor genes using engineered zinc finger protein nucleases |
| EP2504028A4 (en) | 2009-11-24 | 2014-04-09 | Amplimmune Inc | SIMULTANEOUS INHIBITION OF PD-L1 / PD-L2 |
| EP3511023A1 (en) | 2009-12-02 | 2019-07-17 | Imaginab, Inc. | J591 minibodies and cys-diabodies for targeting human prostate specific membrane antigen (psma) and methods for their use |
| HUE028629T2 (en) | 2009-12-23 | 2016-12-28 | Synimmune Gmbh | Anti-FLT3 antibodies and ways of their use |
| EP2519544A1 (en) | 2009-12-29 | 2012-11-07 | Emergent Product Development Seattle, LLC | Polypeptide heterodimers and uses thereof |
| KR20230044026A (ko) | 2010-02-24 | 2023-03-31 | 이뮤노젠 아이엔씨 | 엽산염 수용체 1 항체와 면역접합체 및 이들의 용도 |
| EA201201435A1 (ru) | 2010-04-20 | 2013-04-30 | Генмаб А/С | ГЕТЕРОДИМЕРНЫЕ АНТИТЕЛО-Fc-СОДЕРЖАЩИЕ БЕЛКИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ |
| WO2011159847A2 (en) | 2010-06-15 | 2011-12-22 | The Regents Of The University Of California | Receptor tyrosine kinase-like orphan receptor 1 (ror1) single chain fv antibody fragment conjugates and methods of use thereof |
| CA2802344C (en) | 2010-06-18 | 2023-06-13 | The Brigham And Women's Hospital, Inc. | Bi-specific antibodies against tim-3 and pd-1 for immunotherapy in chronic immune conditions |
| EP2582722A4 (en) | 2010-06-19 | 2013-12-18 | Sloan Kettering Inst Cancer | ANTIBODIES AGAINST GD2 |
| AU2011274510A1 (en) | 2010-07-09 | 2013-01-24 | Exelixis, Inc. | Combinations of kinase inhibitors for the treatment of cancer |
| AR084706A1 (es) | 2010-07-16 | 2013-06-05 | Piramal Life Sciences Ltd | Derivados sustituidos de imidazoquinolinas como inhibidores de quinasa y proceso para su preparacion |
| WO2012012667A2 (en) | 2010-07-21 | 2012-01-26 | Sangamo Biosciences, Inc. | Methods and compositions for modification of a hla locus |
| WO2012033885A1 (en) | 2010-09-08 | 2012-03-15 | Baylor College Of Medicine | Immunotherapy of cancer using genetically engineered gd2-specific t cells |
| PT3214091T (pt) | 2010-12-09 | 2019-01-11 | Univ Pennsylvania | Utilização de células t modificadas por recetor de antigénio quimérico para tratar o cancro |
| JOP20210044A1 (ar) | 2010-12-30 | 2017-06-16 | Takeda Pharmaceuticals Co | الأجسام المضادة لـ cd38 |
| EP3323833B1 (en) | 2011-04-01 | 2019-12-04 | Memorial Sloan-Kettering Cancer Center | T cell receptor-like bispecific antibodies specific for a wt1 peptide presented by hla-a2 |
| CN103596981B (zh) | 2011-04-08 | 2017-06-16 | 美国卫生和人力服务部 | 抗‑表皮生长因子受体变体iii嵌合抗原受体及其用于治疗癌症的用途 |
| US20130071414A1 (en) * | 2011-04-27 | 2013-03-21 | Gianpietro Dotti | Engineered cd19-specific t lymphocytes that coexpress il-15 and an inducible caspase-9 based suicide gene for the treatment of b-cell malignancies |
| AR086044A1 (es) | 2011-05-12 | 2013-11-13 | Imclone Llc | Anticuerpos que se unen especificamente a un dominio extracelular de c-kit y usos de los mismos |
| EP4338754A2 (en) | 2011-05-27 | 2024-03-20 | Glaxo Group Limited | Antigen binding proteins |
| WO2013006490A2 (en) | 2011-07-01 | 2013-01-10 | Cellerant Therapeutics, Inc. | Antibodies that specifically bind to tim3 |
| SG10201606288TA (en) | 2011-08-11 | 2016-09-29 | Intellikine Llc | Kinase inhibitor polymorphs |
| US20130108641A1 (en) | 2011-09-14 | 2013-05-02 | Sanofi | Anti-gitr antibodies |
| US20140255363A1 (en) | 2011-09-16 | 2014-09-11 | Baylor College Of Medicine | Targeting the tumor microenvironment using manipulated nkt cells |
| WO2013054320A1 (en) | 2011-10-11 | 2013-04-18 | Tel Hashomer Medical Research Infrastructure And Services Ltd. | Antibodies to carcinoembryonic antigen-related cell adhesion molecule (ceacam) |
| ITMO20110270A1 (it) | 2011-10-25 | 2013-04-26 | Sara Caldrer | Una cellula effettrice modificata per il trattamento di neoplasie esprimenti il disialonganglioside gd2 |
| LT2771364T (lt) | 2011-10-27 | 2019-09-10 | Genmab A/S | Heterodimerinių baltymų gamyba |
| US10391126B2 (en) | 2011-11-18 | 2019-08-27 | Board Of Regents, The University Of Texas System | CAR+ T cells genetically modified to eliminate expression of T-cell receptor and/or HLA |
| CA2892371C (en) | 2011-12-01 | 2021-01-19 | The Brigham And Women's Hospital, Inc. | Anti-ceacam1 recombinant antibodies for cancer therapy |
| US9439768B2 (en) | 2011-12-08 | 2016-09-13 | Imds Llc | Glenoid vault fixation |
| MX2014009289A (es) * | 2012-02-01 | 2015-09-08 | Compugen Ltd | Anticuerpos c1orf32 y usos de los mismos para el tratamiento del cancer. |
| EP3594245A1 (en) | 2012-02-13 | 2020-01-15 | Seattle Children's Hospital d/b/a Seattle Children's Research Institute | Bispecific chimeric antigen receptors and therapeutic uses thereof |
| SG11201404285VA (en) | 2012-02-22 | 2014-10-30 | Univ Pennsylvania | Compositions and methods for generating a persisting population of t cells useful for the treatment of cancer |
| AR090903A1 (es) | 2012-05-01 | 2014-12-17 | Genentech Inc | Anticuerpos e inmunoconjugados anti-pmel17 |
| PE20190844A1 (es) | 2012-05-25 | 2019-06-17 | Emmanuelle Charpentier | Modulacion de transcripcion con arn de direccion a adn generico |
| WO2013192294A1 (en) | 2012-06-20 | 2013-12-27 | Boston 3T Biotechnologies, Inc. | Cellular therapies for treating and preventing cancers and other immune system disorders |
| WO2014022332A1 (en) | 2012-07-31 | 2014-02-06 | The Brigham And Women's Hospital, Inc. | Modulation of the immune response |
| US9365641B2 (en) | 2012-10-01 | 2016-06-14 | The Trustees Of The University Of Pennsylvania | Compositions and methods for targeting stromal cells for the treatment of cancer |
| WO2014055657A1 (en) | 2012-10-05 | 2014-04-10 | The Trustees Of The University Of Pennsylvania | Use of a trans-signaling approach in chimeric antigen receptors |
| KR102229873B1 (ko) | 2012-10-12 | 2021-03-19 | 더 브리검 앤드 우먼즈 하스피털, 인크. | 면역 반응의 향상 |
| EP3252160B1 (en) | 2012-12-12 | 2020-10-28 | The Broad Institute, Inc. | Crispr-cas component systems, methods and compositions for sequence manipulation |
| US8697359B1 (en) | 2012-12-12 | 2014-04-15 | The Broad Institute, Inc. | CRISPR-Cas systems and methods for altering expression of gene products |
| SG10201801969TA (en) | 2012-12-12 | 2018-04-27 | Broad Inst Inc | Engineering and Optimization of Improved Systems, Methods and Enzyme Compositions for Sequence Manipulation |
| WO2014110591A1 (en) | 2013-01-14 | 2014-07-17 | Fred Hutchinson Cancer Research Center | Compositions and methods for delivery of immune cells to treat un-resectable or non-resected tumor cells and tumor relapse |
| WO2014145252A2 (en) * | 2013-03-15 | 2014-09-18 | Milone Michael C | Targeting cytotoxic cells with chimeric receptors for adoptive immunotherapy |
| ES2939760T3 (es) | 2014-03-15 | 2023-04-26 | Novartis Ag | Tratamiento de cáncer utilizando un receptor quimérico para antígenos |
-
2015
- 2015-03-13 ES ES15712503T patent/ES2939760T3/es active Active
- 2015-03-13 US US15/126,036 patent/US20170335281A1/en not_active Abandoned
- 2015-03-13 EP EP19169356.3A patent/EP3593812A3/en active Pending
- 2015-03-13 CN CN201580013987.3A patent/CN106163547A/zh active Pending
- 2015-03-13 WO PCT/US2015/020606 patent/WO2015142675A2/en active Application Filing
- 2015-03-13 JP JP2016557287A patent/JP2017513818A/ja active Pending
- 2015-03-13 EP EP15712503.0A patent/EP3119423B1/en active Active
-
2019
- 2019-07-05 JP JP2019126002A patent/JP6860623B2/ja active Active
- 2019-10-11 US US16/599,988 patent/US20200283729A1/en active Pending
-
2021
- 2021-03-26 JP JP2021053240A patent/JP2021107395A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| CN106163547A (zh) | 2016-11-23 |
| EP3593812A3 (en) | 2020-05-27 |
| US20200283729A1 (en) | 2020-09-10 |
| WO2015142675A3 (en) | 2015-11-12 |
| JP2019206538A (ja) | 2019-12-05 |
| US20170335281A1 (en) | 2017-11-23 |
| WO2015142675A2 (en) | 2015-09-24 |
| WO2015142675A8 (en) | 2016-10-20 |
| EP3119423B1 (en) | 2022-12-14 |
| JP2017513818A (ja) | 2017-06-01 |
| EP3593812A2 (en) | 2020-01-15 |
| JP6860623B2 (ja) | 2021-04-21 |
| JP2021107395A (ja) | 2021-07-29 |
| EP3119423A2 (en) | 2017-01-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230312677A1 (en) | Cd28 compositions and methods for chimeric antigen receptor therapy | |
| JP6860623B2 (ja) | キメラ抗原受容体を使用する癌の処置 | |
| US20230139800A1 (en) | Car t cell therapies with enhanced efficacy | |
| US20200339651A1 (en) | Methods for b cell preconditioning in car therapy | |
| ES2805475T3 (es) | Tratamiento del cáncer utilizando un receptor antigénico quimérico de CD33 | |
| ES2791248T3 (es) | Receptor antigénico quimérico (CAR) anti-CD123 para su uso en el tratamiento del cáncer | |
| US20200048359A1 (en) | Shp inhibitor compositions and uses for chimeric antigen receptor therapy | |
| ES2876263T3 (es) | Tratamiento del cáncer usando receptor de antígeno quimérico anti-cd19 | |
| US20180298068A1 (en) | Treatment of cancer using chimeric antigen receptor and protein kinase a blocker | |
| US20200281973A1 (en) | Cells expressing multiple chimeric antigen receptor (car) molecules and uses therefore | |
| US20170274014A1 (en) | Combinations of low, immune enhancing, doses of mtor inhibitors and cars | |
| WO2019210153A1 (en) | Car t cell therapies with enhanced efficacy | |
| WO2016126608A1 (en) | Car-expressing cells against multiple tumor antigens and uses thereof | |
| EP3193915A1 (en) | Combinations of low, immune enhancing. doses of mtor inhibitors and cars | |
| US20180092968A1 (en) | Compositions to disrupt protein kinase a anchoring and uses thereof | |
| US20210179709A1 (en) | Anti-car compositions and methods | |
| US20200390811A1 (en) | Compositions to disrupt protein kinase a anchoring and uses thereof | |
| US20240139244A1 (en) | Cells expressing multiple chimeric antigen receptor (car) molecules and uses therefore |