JP5962760B2 - 重合性化合物、重合性組成物、高分子、光学異方体、及び重合性化合物の製造方法 - Google Patents
重合性化合物、重合性組成物、高分子、光学異方体、及び重合性化合物の製造方法 Download PDFInfo
- Publication number
- JP5962760B2 JP5962760B2 JP2014524688A JP2014524688A JP5962760B2 JP 5962760 B2 JP5962760 B2 JP 5962760B2 JP 2014524688 A JP2014524688 A JP 2014524688A JP 2014524688 A JP2014524688 A JP 2014524688A JP 5962760 B2 JP5962760 B2 JP 5962760B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- carbon atoms
- substituent
- compound
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 *C*1C2C1CC1C2C1 Chemical compound *C*1C2C1CC1C2C1 0.000 description 4
- KADSLELLEJWNKJ-UHFFFAOYSA-N C=CC(O)OCCCCCCOC(CC1)=CC=C1OC(C1CCCCC1)=O Chemical compound C=CC(O)OCCCCCCOC(CC1)=CC=C1OC(C1CCCCC1)=O KADSLELLEJWNKJ-UHFFFAOYSA-N 0.000 description 1
- OQDVHYTWSKDDHL-UHFFFAOYSA-N C=CC(OCCCCCCOC(CC1)=CC=C1OC(C1CCCCC1)O)=O Chemical compound C=CC(OCCCCCCOC(CC1)=CC=C1OC(C1CCCCC1)O)=O OQDVHYTWSKDDHL-UHFFFAOYSA-N 0.000 description 1
- JZNSHKDLZMXENA-UHFFFAOYSA-N C=CC(OCCCCCCOc(cc1)ccc1OC(C(CC1)CCC1C(Oc(cc1)cc(C=O)c1O)=O)=O)=O Chemical compound C=CC(OCCCCCCOc(cc1)ccc1OC(C(CC1)CCC1C(Oc(cc1)cc(C=O)c1O)=O)=O)=O JZNSHKDLZMXENA-UHFFFAOYSA-N 0.000 description 1
- DRIOMEVBVGXJPF-UHFFFAOYSA-N C=CC(OCCCCCCOc(cc1)ccc1OC(C1CCCCC1)=O)=O Chemical compound C=CC(OCCCCCCOc(cc1)ccc1OC(C1CCCCC1)=O)=O DRIOMEVBVGXJPF-UHFFFAOYSA-N 0.000 description 1
- UDLGUAULYYUUAH-HTFGYGFHSA-N CCCCC(CC)N(C1=NC2C=CC=CC2S1)NCc(cc(cc1)OC([C@H](CC2)CC[C@@H]2C(Oc(cc2)ccc2OCCCCCCOC(C=C)=O)=O)=O)c1O[IH](C(CC1)CCC1C(OC(CC1)=CC=C1OCCCCCCOC(C=C)=O)=O)=O Chemical compound CCCCC(CC)N(C1=NC2C=CC=CC2S1)NCc(cc(cc1)OC([C@H](CC2)CC[C@@H]2C(Oc(cc2)ccc2OCCCCCCOC(C=C)=O)=O)=O)c1O[IH](C(CC1)CCC1C(OC(CC1)=CC=C1OCCCCCCOC(C=C)=O)=O)=O UDLGUAULYYUUAH-HTFGYGFHSA-N 0.000 description 1
- BUNDNEPWTZLNCH-OGSAKLKBSA-N CN(c1nc(cccc2)c2[s]1)/N=C/c(cc(cc1)OC([C@H](CC2)CC[C@@H]2C(OC(CC2)=CC=C2OCCCCCCOC(C=C)=O)=O)=O)c1OC(C(CC1)CCC1C(OC(CC1)=CC=C1OCCCCCCOC(C=C)=O)=O)=O Chemical compound CN(c1nc(cccc2)c2[s]1)/N=C/c(cc(cc1)OC([C@H](CC2)CC[C@@H]2C(OC(CC2)=CC=C2OCCCCCCOC(C=C)=O)=O)=O)c1OC(C(CC1)CCC1C(OC(CC1)=CC=C1OCCCCCCOC(C=C)=O)=O)=O BUNDNEPWTZLNCH-OGSAKLKBSA-N 0.000 description 1
- OAFOSTYPDOWQFF-UHFFFAOYSA-N Cc1c2[s]c(NN)nc2nc(C)n1 Chemical compound Cc1c2[s]c(NN)nc2nc(C)n1 OAFOSTYPDOWQFF-UHFFFAOYSA-N 0.000 description 1
- GKUIEULNBCUAMZ-UHFFFAOYSA-N Cc1ccccc1N(C1=NC2C=CC=CC2S1)NCC(C=C(CC1)OC)=C1OC=O Chemical compound Cc1ccccc1N(C1=NC2C=CC=CC2S1)NCC(C=C(CC1)OC)=C1OC=O GKUIEULNBCUAMZ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F20/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride, ester, amide, imide or nitrile thereof
- C08F20/02—Monocarboxylic acids having less than ten carbon atoms, Derivatives thereof
- C08F20/10—Esters
- C08F20/34—Esters containing nitrogen, e.g. N,N-dimethylaminoethyl (meth)acrylate
- C08F20/36—Esters containing nitrogen, e.g. N,N-dimethylaminoethyl (meth)acrylate containing oxygen in addition to the carboxy oxygen, e.g. 2-N-morpholinoethyl (meth)acrylate or 2-isocyanatoethyl (meth)acrylate
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
- C07D277/68—Benzothiazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
- C07D277/82—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C243/00—Compounds containing chains of nitrogen atoms singly-bound to each other, e.g. hydrazines, triazanes
- C07C243/10—Hydrazines
- C07C243/12—Hydrazines having nitrogen atoms of hydrazine groups bound to acyclic carbon atoms
- C07C243/14—Hydrazines having nitrogen atoms of hydrazine groups bound to acyclic carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C243/00—Compounds containing chains of nitrogen atoms singly-bound to each other, e.g. hydrazines, triazanes
- C07C243/10—Hydrazines
- C07C243/12—Hydrazines having nitrogen atoms of hydrazine groups bound to acyclic carbon atoms
- C07C243/16—Hydrazines having nitrogen atoms of hydrazine groups bound to acyclic carbon atoms of an unsaturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C243/00—Compounds containing chains of nitrogen atoms singly-bound to each other, e.g. hydrazines, triazanes
- C07C243/10—Hydrazines
- C07C243/12—Hydrazines having nitrogen atoms of hydrazine groups bound to acyclic carbon atoms
- C07C243/16—Hydrazines having nitrogen atoms of hydrazine groups bound to acyclic carbon atoms of an unsaturated carbon skeleton
- C07C243/18—Hydrazines having nitrogen atoms of hydrazine groups bound to acyclic carbon atoms of an unsaturated carbon skeleton containing rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C243/00—Compounds containing chains of nitrogen atoms singly-bound to each other, e.g. hydrazines, triazanes
- C07C243/10—Hydrazines
- C07C243/20—Hydrazines having nitrogen atoms of hydrazine groups bound to carbon atoms of rings other than six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/74—Esters of carboxylic acids having an esterified carboxyl group bound to a carbon atom of a ring other than a six-membered aromatic ring
- C07C69/753—Esters of carboxylic acids having an esterified carboxyl group bound to a carbon atom of a ring other than a six-membered aromatic ring of polycyclic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/84—Naphthothiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F22/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides or nitriles thereof
- C08F22/10—Esters
- C08F22/12—Esters of phenols or saturated alcohols
- C08F22/24—Esters containing sulfur
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
- C08L33/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, which oxygen atoms are present only as part of the carboxyl radical
- C08L33/08—Homopolymers or copolymers of acrylic acid esters
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K19/00—Liquid crystal materials
- C09K19/04—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit
- C09K19/0403—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit the structure containing one or more specific, optionally substituted ring or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C249/00—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C249/16—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of hydrazones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C251/00—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C251/72—Hydrazones
- C07C251/86—Hydrazones having doubly-bound carbon atoms of hydrazone groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F20/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride, ester, amide, imide or nitrile thereof
- C08F20/62—Monocarboxylic acids having ten or more carbon atoms; Derivatives thereof
- C08F20/68—Esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F22/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides or nitriles thereof
- C08F22/10—Esters
- C08F22/1006—Esters of polyhydric alcohols or polyhydric phenols, e.g. ethylene glycol dimethacrylate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F22/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides or nitriles thereof
- C08F22/10—Esters
- C08F22/12—Esters of phenols or saturated alcohols
- C08F22/14—Esters having no free carboxylic acid groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F22/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides or nitriles thereof
- C08F22/10—Esters
- C08F22/12—Esters of phenols or saturated alcohols
- C08F22/20—Esters containing oxygen in addition to the carboxy oxygen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F22/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides or nitriles thereof
- C08F22/10—Esters
- C08F22/12—Esters of phenols or saturated alcohols
- C08F22/22—Esters containing nitrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F22/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides or nitriles thereof
- C08F22/10—Esters
- C08F22/26—Esters of unsaturated alcohols
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2500/00—Characteristics or properties of obtained polyolefins; Use thereof
- C08F2500/26—Use as polymer for film forming
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F26/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a single or double bond to nitrogen or by a heterocyclic ring containing nitrogen
- C08F26/06—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a single or double bond to nitrogen or by a heterocyclic ring containing nitrogen by a heterocyclic ring containing nitrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F28/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a bond to sulfur or by a heterocyclic ring containing sulfur
- C08F28/06—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a bond to sulfur or by a heterocyclic ring containing sulfur by a heterocyclic ring containing sulfur
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K19/00—Liquid crystal materials
- C09K19/04—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit
- C09K19/0403—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit the structure containing one or more specific, optionally substituted ring or ring systems
- C09K2019/0414—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit the structure containing one or more specific, optionally substituted ring or ring systems containing a heterocyclic ring
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K19/00—Liquid crystal materials
- C09K19/04—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit
- C09K2019/0477—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit characterized by the positioning of substituents on phenylene
- C09K2019/0481—Phenylene substituted in meta position
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B5/00—Optical elements other than lenses
- G02B5/30—Polarising elements
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F1/00—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics
- G02F1/01—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour
- G02F1/13—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on liquid crystals, e.g. single liquid crystal display cells
- G02F1/137—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on liquid crystals, e.g. single liquid crystal display cells characterised by the electro-optical or magneto-optical effect, e.g. field-induced phase transition, orientation effect, guest-host interaction or dynamic scattering
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F2202/00—Materials and properties
- G02F2202/02—Materials and properties organic material
- G02F2202/022—Materials and properties organic material polymeric
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F2202/00—Materials and properties
- G02F2202/40—Materials having a particular birefringence, retardation
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Materials Engineering (AREA)
- Engineering & Computer Science (AREA)
- Crystallography & Structural Chemistry (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Polarising Elements (AREA)
- Plural Heterocyclic Compounds (AREA)
- Liquid Crystal (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Liquid Crystal Substances (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Description
しかしながら、従来の位相差板には、位相差板を通過して出力される偏光が有色の偏光に変換されてしまうという問題があった。これは、位相差板を構成する材料が位相差について波長分散性を有し、可視光域の光線が混在する合成波である白色光に対して各波長ごとの偏光状態に分布が生じることから、全ての波長領域において正確な1/4λあるいは1/2λの位相差に調整することが不可能であることに起因する。
このような問題を解決するため、広い波長域の光に対して均一な位相差を与え得る広帯域位相差板、いわゆる逆波長分散性を有する位相差板が種々検討されている(例えば、特許文献1〜6)。
薄層化の方法としては、フィルム基材に低分子重合性化合物を含有する重合性組成物を塗布することにより位相差板を作成する方法が、近年では最も有効な方法とされている。優れた波長分散性を有する低分子重合性化合物又はそれを用いた重合性組成物の開発が多く行われている(例えば、特許文献7〜24)。
(1)下記式(I)
G1、G2はそれぞれ独立して、置換基を有していてもよい炭素数1〜20の二価の鎖状脂肪族基を表す。また、前記鎖状脂肪族基には、−O−、−S−、−O−C(=O)−、−C(=O)−O−、−O−C(=O)−O−、−NR2−C(=O)−、−C(=O)−NR2−、−NR2−、又は、−C(=O)−が介在していてもよい。ただし、−O−又は−S−がそれぞれ2以上隣接して介在する場合を除く。ここで、R2は、水素原子又は炭素数1〜6のアルキル基を表す。
Z1、Z2はそれぞれ独立して、ハロゲン原子で置換されていてもよい炭素数2〜10のアルケニル基を表す。
Axは、芳香族炭化水素環及び芳香族複素環からなる群から選ばれる少なくとも一つの芳香環を有する、炭素数2〜30の有機基を表す。
Ayは、水素原子、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、置換基を有していてもよい炭素数2〜20のアルキニル基、置換基を有していてもよい炭素数3〜12のシクロアルキル基、−C(=O)−R3、−SO2−R4、−C(=S)NH−R9、又は、芳香族炭化水素環及び芳香族複素環からなる群から選ばれる少なくとも一つの芳香環を有する、炭素数2〜30の有機基を表す。ここで、R3は、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、又は、置換基を有していてもよい炭素数3〜12のシクロアルキル基、炭素数5〜12の芳香族炭化水素基を表し、R4は、炭素数1〜20のアルキル基、炭素数2〜20のアルケニル基、フェニル基、又は、4−メチルフェニル基を表し、R9は置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、置換基を有していてもよい炭素数3〜12のシクロアルキル基、置換基を有していてもよい炭素数5〜20の芳香族基を表す。前記Ax及びAyが有する芳香環は置換基を有していてもよい。また、前記AxとAyは一緒になって、環を形成していてもよい。
A1は置換基を有していてもよい三価の芳香族基を表す。
A2、A3はそれぞれ独立して、置換基を有していてもよい炭素数3〜30の二価の脂環式炭化水素基を表し、
A4、A5はそれぞれ独立して、置換基を有していてもよい炭素数6〜30の二価の芳香族基を表す。
Q1は、水素原子、又は、置換基を有していてもよい炭素数1〜6のアルキル基を表す。〕で示される重合性化合物。
(3)前記A1が、置換基を有していてもよい、三価のベンゼン環基又は三価のナフタレン環基である(1)又は(2)に記載の重合性化合物。
(4)前記Y1〜Y8が、それぞれ独立して、化学的な単結合、−O−、−O−C(=O)−、−C(=O)−O−、又は、−O−C(=O)−O−である(1)〜(3)のいずれかに記載の重合性化合物。
(6)前記G1、G2がそれぞれ独立して、置換基を有していてもよい炭素数1〜20の二価の脂肪族基〔該脂肪族基には、−O−、−O−C(=O)−、−C(=O)−O−又は−C(=O)−が介在していてもよい。ただし、−O−が2以上隣接して介在する場合を除く。〕である(1)〜(5)のいずれかに記載の重合性化合物。
(7)前記G1、G2がそれぞれ独立して、炭素数1〜12のアルキレン基である(1)〜(6)のいずれかに記載の重合性化合物。
(9)前記(1)〜(7)のいずれかに記載の重合性化合物の少なくとも1種、及び重合開始剤を含有する重合性組成物。
(11)液晶性高分子である(10)に記載の高分子。
(12)前記(11)に記載の高分子を構成材料とする光学異方体。
(13)下記式(4)
G1、G2はそれぞれ独立して、置換基を有していてもよい炭素数1〜20の二価の鎖状脂肪族基を表す〔該鎖状脂肪族基には、−O−、−S−、−O−C(=O)−、−C(=O)−O−、−O−C(=O)−O−、−NR2−C(=O)−、−C(=O)−NR2−、−NR2−、又は、−C(=O)−が介在していてもよい。ただし、−O−又は−S−がそれぞれ2以上隣接して介在する場合を除く。ここで、R2は、水素原子又は炭素数1〜6のアルキル基を表す。〕。
Z1、Z2はそれぞれ独立して、無置換又はハロゲン原子で置換された炭素数2〜10のアルケニル基を表す。
A1は、置換基を有していてもよい三価の芳香族基を表す。
A2、A3はそれぞれ独立して、置換基を有していてもよい炭素数3〜30の二価の脂環式炭化水素基を表す。
A4、A5はそれぞれ独立して、置換基を有していてもよい炭素数6〜30の二価の芳香族基を表す。
Q1は、水素原子、又は、置換基を有していてもよい炭素数1〜6のアルキル基を表す。}で示されるカルボニル化合物。
(15)前記A2、A3がそれぞれ独立して、置換基を有していてもよい二価のシクロヘキシル基である(13)又は(14)に記載のカルボニル化合物。
(16)前記Z1、Z2が、それぞれ独立して、CH2=CH−、CH2=C(CH3)−、又は、CH2=C(Cl)−である(13)〜(15)のいずれかに記載のカルボニル化合物。
(17)(13)〜(16)のいずれかに記載のカルボニル化合物と、下記式
Ayは、水素原子、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、置換基を有していてもよい炭素数1〜20のアルキニル基、置換基を有していてもよい炭素数3〜12のシクロアルキル基、−C(=O)−R3、−SO2−R4、−C(=S)NH−R9、又は、芳香族炭化水素環及び芳香族複素環からなる群から選ばれる少なくとも一つの芳香環を有する、炭素数2〜30の有機基を表す。ここで、R3は、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、又は、置換基を有していてもよい炭素数3〜12のシクロアルキル基、炭素数5〜12の芳香族炭化水素基を表し、R4は、炭素数1〜20のアルキル基、炭素数2〜20のアルケニル基、フェニル基、又は、4−メチルフェニル基を表し、R9は置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、置換基を有していてもよい炭素数3〜12のシクロアルキル基、置換基を有していてもよい炭素数5〜20の芳香族基を表す。
前記Ax及びAyが有する芳香環は置換基を有していてもよい。
また、前記AxとAyは一緒になって、環を形成していてもよい。)
で示されるヒドラジン化合物を反応させることを特徴とする、下記式(I)
(18)(13)〜(16)のいずれかに記載のカルボニル化合物を、下記式(I)
本発明の光学異方体は、本発明の高分子を構成材料とするため、低コストで製造可能で、反射輝度が低く、かつ、広い波長域において一様の偏光変換が可能なものである。
本発明のフィルム状の光学異方体を偏光板と組み合わせることで反射防止フィルムを作製することができる。このものは、産業上、例えばタッチパネルや有機電界発光素子の反射防止に好適に使用することができる。
本発明の重合性化合物の製造方法によれば、本発明の重合性化合物を効率よく製造することができる。
本発明のカルボニル化合物を製造原料として使用することにより、本発明の重合性化合物を高収率で簡便に製造することができる。
本発明の重合性化合物は、前記式(I)で表される化合物である。
式中、Y1〜Y8はそれぞれ独立して、化学的な単結合、−O−、−S−、−O−C(=O)−、−C(=O)−O−、−O−C(=O)−O−、−NR1−C(=O)−、−C(=O)−NR1−、−O−C(=O)−NR1−、−NR1−C(=O)−O−、−NR1−C(=O)−NR1−、−O−NR1−、又は、−NR1−O−を表す。
R1の炭素数1〜6のアルキル基としては、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、sec−ブチル基、t−ブチル基、n−ペンチル基、n−へキシル基等が挙げられる。
R1としては、水素原子又は炭素数1〜4のアルキル基が好ましい。
炭素数1〜20の二価の脂肪族基としては、炭素数1〜20のアルキレン基、炭素数2〜20のアルケニレン基等の鎖状構造を有する二価の脂肪族基;炭素数3〜20のシクロアルカンジイル基、炭素数4〜20のシクロアルケンジイル基、炭素数10〜30の二価の脂環式縮合環基等の二価の脂肪族基;等が挙げられる。
前記脂肪族基に介在する基としては、−O−、−O−C(=O)−、−C(=O)−O−、−C(=O)−が好ましい。
該アルケニル基の炭素数としては、2〜6が好ましい。Z1及びZ2のアルケニル基の置換基であるハロゲン原子としては、フッ素原子、塩素原子、臭素原子等が挙げられ、塩素原子が好ましい。
本発明において、「芳香環」は、Huckel則に従う広義の芳香族性を有する環状構造、すなわち、π電子を(4n+2)個有する環状共役構造、及びチオフェン、フラン、ベンゾチアゾール等に代表される、硫黄、酸素、窒素等のヘテロ原子の孤立電子対がπ電子系に関与して芳香族性を示すものを意味する。
なお、Axの炭素数2〜30の有機基の「炭素数」は、置換基の炭素原子を含まない有機基全体の総炭素数を意味する(後述するAyにて同じである。)。
(1)芳香族炭化水素環基
(3)芳香族炭化水素環基及び芳香族複素環基からなる群から選ばれる少なくとも一つの芳香環を有する、アルキル基
なお、Axの炭素数2〜30の有機基の「炭素数」は、置換基の炭素原子を含まない有機基全体の総炭素数を意味する(後述するAyにて同じである。)。
置換基を有していてもよい炭素数2〜20のアルケニル基の炭素数は、2〜12であることが好ましい。
R4の、炭素数1〜20のアルキル基、及び炭素数2〜20のアルケニル基の具体例は、前記Ayの、炭素数1〜20のアルキル基、炭素数2〜20のアルケニル基の例として列記したものと同様のものが挙げられる。
Ayの、置換基を有していてもよい炭素数3〜12のシクロアルキル基、置換基を有してもよい炭素数6〜12の芳香族炭化水素基、置換基を有していてもよい炭素数3〜9の芳香族複素環基の置換基としては、フッ素原子、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、シアノ基が好ましい。
また、これらの環は置換基を有していてもよい。かかる置換基としては、Axが有する芳香環の置換基として例示したのと同様のものが挙げられる。
(α)Axが炭素数4〜30の、芳香族炭化水素基又は芳香族複素環基であり、Ayが水素原子、炭素数3〜8のシクロアルキル基、(ハロゲン原子、シアノ基、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、若しくは炭素数3〜8のシクロアルキル基)を置換基として有していてもよい炭素数6〜12の芳香族炭化水素基、(ハロゲン原子、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、シアノ基)を置換基として有していてもよい炭素数3〜9の芳香族複素環基、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数1〜20のアルケニル基、置換基を有していてもよい炭素数2〜20のアルキニル基であり、当該置換基が、ハロゲン原子、シアノ基、炭素数1〜20のアルコキシ基、炭素数1〜12のアルコキシ基で置換された炭素数1〜12のアルコキシ基、フェニル基、シクロヘキシル基、炭素数2〜12の環状エーテル基、炭素数6〜14のアリールオキシ基、水酸基、ベンゾジオキサニル基、ベンゼンスルホニル基、ベンゾイル基、−SR10のいずれかである組み合わせ、及び、
(β)AxとAyが一緒になって不飽和複素環又は不飽和炭素環を形成しているもの、
が挙げられる。ここで、R10は前記と同じ意味を表す。
(γ)Axが下記構造を有する基のいずれかであり、Ayが水素原子、炭素数3〜8のシクロアルキル基、(ハロゲン原子、シアノ基、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、若しくは炭素数3〜8のシクロアルキル基)を置換基として有していてもよい炭素数6〜12の芳香族炭化水素基、(ハロゲン原子、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、シアノ基)を置換基として有していてもよい炭素数3〜9の芳香族複素環基、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数1〜20のアルケニル基、置換基を有していてもよい炭素数2〜20のアルキニル基であり、当該置換基が、ハロゲン原子、シアノ基、炭素数1〜20のアルコキシ基、炭素数1〜12のアルコキシ基で置換された炭素数1〜12のアルコキシ基、フェニル基、シクロヘキシル基、炭素数2〜12の環状エーテル基、炭素数6〜14のアリールオキシ基、水酸基、ベンゾジオキサニル基、ベンゼンスルホニル基、ベンゾイル基、−SR10のいずれかである組み合わせである。ここで、R10は前記と同じ意味を表す。
AxとAyの特に好ましい組み合わせとしては、
(δ)Axが下記構造を有する基のいずれかであり、Ayが水素原子、炭素数3〜8のシクロアルキル基、(ハロゲン原子、シアノ基、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、若しくは炭素数3〜8のシクロアルキル基)を置換基として有していてもよい炭素数6〜12の芳香族炭化水素基、(ハロゲン原子、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、シアノ基)を置換基として有していてもよい炭素数3〜9の芳香族複素環基、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数1〜20のアルケニル基、置換基を有していてもよい炭素数2〜20のアルキニル基であり、当該置換基が、ハロゲン原子、シアノ基、炭素数1〜20のアルコキシ基、炭素数1〜12のアルコキシ基で置換された炭素数1〜12のアルコキシ基、フェニル基、シクロヘキシル基、炭素数2〜12の環状エーテル基、炭素数6〜14のアリールオキシ基、水酸基、ベンゾジオキサニル基、ベンゼンスルホニル基、ベンゾイル基、−SR10のいずれかである組合せである。下記式中、Xは前記と同じ意味を表す。ここで、R10は前記と同じ意味を表す。
なお、下記式においては、結合状態をより明確にすべく、置換基Y1、Y2を便宜上記載している(Y1、Y2は、前記と同じ意味を表す。以下にて同じ。)。
炭素数3〜30の二価の脂環式炭化水素基としては、炭素数3〜30のシクロアルカンジイル基、炭素数10〜30の二価の脂環式縮合環基等が挙げられる。
前記炭素数3〜30の二価の脂環式炭化水素基は、Y1、Y3(又はY2、Y4)と結合する炭素原子の立体配置の相違に基づく、シス型、トランス型の立体異性体が存在し得る。例えば、シクロヘキサン−1,4−ジイル基の場合には、下記に示すように、シス型の異性体(A32a)とトランス型の異性体(A32b)が存在し得る。
A4、A5の芳香族基は単環のものであっても、多環のものであってもよい。
A4、A5の好ましい具体例としては、下記のものが挙げられる。
置換基を有していてもよい炭素数1〜6のアルキル基としては、前記AXで例示したのと同様のものが挙げられる。
これらの中でも、Q1は、水素原子又は炭素数1〜6のアルキル基が好ましく、水素原子及びメチル基がより好ましい。
すなわち、式(3)で表されるヒドラジン化合物(ヒドラジン化合物(3))を、式(4)で表されるカルボニル化合物(カルボニル化合物(4))と、〔ヒドラジン化合物(3):カルボニル化合物(4)〕のモル比で、1:2〜2:1、好ましくは1:1.5〜1.5:1の割合で反応させることにより、高選択的かつ高収率で目的とする本発明の式(I)で示される重合性化合物を製造することができる。
これらの中でも、アルコール系溶媒、エーテル系溶媒、及びアルコール系溶媒とエーテル系溶媒の混合溶媒が好ましい。
この反応に用いる溶媒としては、反応に不活性なものであれば特に限定されない。例えば、メチルアルコール、エチルアルコール、n−プロピルアルコール、イソプロピルアルコール、n−ブチルアルコール、イソブチルアルコール、sec−ブチルアルコール、t−ブチルアルコール等のアルコール系溶媒;ジエチルエーテル、テトラヒドロフラン、1,2−ジメトキシエタン、1,4−ジオキサン、シクロペンチルメチルエーテル等のエーテル系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;n−ペンタン、n−ヘキサン、n−ヘプタン等の脂肪族炭化水素系溶媒;N,N−ジメチルホルムアミド、N−メチルピロリドン、ヘキサメチルリン酸トリアミド等のアミド系溶媒;ジメチルスルホキシド、スルホラン等の含硫黄系溶媒;及びこれらの2種以上からなる混合溶媒;等が挙げられる。
これらの中でも、アルコール系溶媒、エーテル系溶媒、及びアルコール系溶媒とエーテル系溶媒の混合溶媒が好ましい。
反応は、−10℃から用いる溶媒の沸点までの温度範囲で円滑に進行する。各反応の反応時間は、反応規模にもよるが、通常、数分から数時間である。
金属塩還元剤とは、一般に低原子価金属を含む化合物、もしくは金属イオンとヒドリド源からなる化合物である(「有機合成実験法ハンドブック」1990年社団法人有機合成化学協会編 丸善株式会社発行810ページを参照)。
金属塩還元剤としては、NaAlH4、NaAlHp(Or)q(p、qはそれぞれ独立して1〜3の整数を表し、p+q=4である。rは炭素数1〜6のアルキル基を表す。)、LiAlH4、iBu2AlH、LiBH4、NaBH4、SnCl2、CrCl2、TiCl3等が挙げられる。
また、ジアゾニウム塩(5)は、アニリン等の化合物から常法により製造することができる。
(i)式:D1−hal(halはハロゲン原子を表す。以下にて同じ。)で表される化合物と、式:D2−OMet(Metはアルカリ金属(主にナトリウム)を表す。以下にて同じ。)で表される化合物とを混合して縮合させる(ウイリアムソン合成)。なお、式中、D1及びD2は任意の有機基を表す(以下にて同じ。)
(ii)式:D1−halで表される化合物と、式:D2−OHで表される化合物とを水酸化ナトリウム、水酸化カリウム等の塩基存在下、混合して縮合させる。
(iii)式:D1−J(Jはエポキシ基を表す。)で表される化合物と、式:D2−OHで表される化合物とを水酸化ナトリウム、水酸化カリウム等の塩基存在下、混合して縮合させる。
(iv)式:D1−OFN(OFNは不飽和結合を有する基を表す。)で表される化合物と、式:D2−OMetで表される化合物を、水酸化ナトリウム、水酸化カリウム等の塩基存在下、混合して付加反応させる。
(v)式:D1−halで表される化合物と、式:D2−OMetで表される化合物とを、銅あるいは塩化第一銅存在下、混合して縮合させる(ウルマン縮合)。
(vi)式:D1−COOHで表される化合物と、式:D2−OH又はD2−NH2で表される化合物とを、脱水縮合剤(N,N−ジシクロヘキシルカルボジイミド等)の存在下に脱水縮合させる。
(vii)式:D1−COOHで表される化合物にハロゲン化剤を作用させることにより、式:D1−CO−halで表される化合物を得、このものと式:D2−OH又はD2−NH2で表される化合物とを、塩基の存在下に反応させる。
(viii)式:D1−COOHで表される化合物に酸無水物を作用させることにより、混合酸無水物を得た後、このものに、式:D2−OH又はD2−NH2で表される化合物を反応させる。
(ix)式:D1−COOHで表される化合物と、式:D2−OH又はD2−NH2で表される化合物とを、酸触媒あるいは塩基触媒の存在下に脱水縮合させる。
脱水縮合剤の使用量は、化合物(7)1モルに対し、通常1〜3モルである。
塩基の使用量は、化合物(7)1モルに対し、通常1〜3モルである。
この場合、前記式(7)中、L1がスルホニルオキシ基の化合物(混合酸無水物)を単離して次の反応を行ってもよい。
用いる塩基としては、トリエチルアミン、ピリジン等の有機塩基;水酸化ナトリウム、炭酸ナトリウム、炭酸水素ナトリウム等の無機塩基が挙げられる。
塩基の使用量は、化合物(7)1モルに対し、通常1〜3モルである。
溶媒の使用量は、特に限定されず、用いる化合物の種類や反応規模等を考慮して適宜定めることができるが、ヒドロキシ化合物(6)1gに対し、通常1〜50gである。
例えば、下記反応式に示す方法により製造することができる(WO2009/042544号、及び、The Journal of Organic Chemistry,2011,76,8082−8087等参照。)。化合物(6)として市販されているものを、所望により精製して用いることもできる。
すなわち、式(6a)で表されるジヒドロキシ化合物(1,4−ジヒドロキシベンゼン、1,4−ジヒドロキシナフタレン等)の水酸基をアルキル化して、式(6b)で表される化合物を得た後、OR’基のオルト位を、公知の方法により、ホルミル化又はアシル化することにより、式(6c)で表される化合物を得、このものを脱保護(脱アルキル化)することにより、目的とする化合物(6)を得ることができる。
また、化合物(6)として、市販されているものをそのまま、又は所望により精製して用いることもできる。
先ず、化合物(9’)に、式(10)で表されるスルホニルクロライドを、トリエチルアミン、4−(ジメチルアミノ)ピリジン等の塩基存在下で反応させる。
次いで、反応混合物に、化合物(8)と、トリエチルアミン、4−(ジメチルアミノ)ピリジン等の塩基を加えて反応を行う。
スルホニルクロライドの使用量は、化合物(9’)1当量に対して、通常0.5〜0.7当量である。
化合物(8)の使用量は、化合物(9’)1当量に対して、通常0.5〜0.6当量である。
塩基の使用量は、化合物(3)1当量に対して、通常0.5〜0.7当量である。
反応温度は、20〜30℃であり、反応時間は反応規模等にもよるが、数分から数時間である。
溶媒の使用量は、特に限定されず、用いる化合物の種類や反応規模等を考慮して適宜定めることができるが、化合物(9’)1gに対し、通常1〜50gである。
本発明の第2は、本発明の重合性化合物、及び重合開始剤を含有する重合性組成物である。重合開始剤は本発明の重合性化合物の重合反応をより効率的に行う観点から配合される。
「水素供与体」とは、露光によりビイミダゾール系化合物から発生したラジカルに対して、水素原子を供与することができる化合物を意味する。水素供与体としては、下記で定義するメルカプタン系化合物、アミン系化合物等が好ましい。
これらの重合開始剤は一種単独で、又は二種以上を組み合わせて用いることができる。
本発明の重合性組成物において、重合開始剤の配合割合は、重合性化合物100重量部に対し、通常、0.1〜30重量部、好ましくは0.5〜10重量部である。
本発明の第3は、(1)本発明の重合性化合物を重合して得られる高分子、又は、(2)本発明の重合性組成物を重合して得られる高分子である。
ここで、「重合」とは、通常の重合反応のほか、架橋反応を含む広い意味での化学反応を意味するものとする。
本発明の重合性化合物を重合して得られる高分子としては、本発明の重合性化合物の単独重合体、本発明の重合性化合物の2種以上からなる共重合体、又は、本発明の重合性化合物と他の共重合可能な単量体との共重合体が挙げられる。
市販品としては、LC−242(BASF社製)等を用いることができる。また、特開2007−002208号公報、特開2009−173893号公報、特開2009−274984号公報、特開2010−030979号公報、特開2010−031223号公報、特開2011−006360号公報等に開示されている化合物等も用いることができる。
このような多官能単量体としては、1,2−ブタンジオールジアクリレート、1,3−ブタンジオールジアクリレート、1,4−ブタンジオールジアクリレート、ネオペンタンジオールジアクリレート、1,6−ヘキサンジオールジアクリレート等のアルカンジオールジアクリレート類;1,2−ブタンジオールジメタクリレート、1,3−ブタンジオールジメタクリレート、1,4−ブタンジオールジメタクリレート、ネオペンタンジオールジメタクリレート、1,6−ヘキサンジオールジメタリレート等のアルカンジオールジメタクリレート類;エチレングリコールジアクリレート、ジエチレングリコールジアクリレート、トリエチレングリコールジアクリレート、テトラエチレングリコールジアクリレート等のポリエチレングリコールジアクリレート類;プロピレングリコールジアクリレート、ジプロピレングリコールジアクリレート、トリプロピレングリコールジアクリレート、テトラプロピレングリコールジアクリレート等のポリプロピレングリコールジアクリレート類;エチレングリコールジメタクリレート、ジエチレングリコールジメタクリレート、トリエチレングリコールジメタクリレート、テトラエチレングリコールジメタクリレート等のポリエチレングリコールジメタクリレート類;プロピレングリコールジメタクリレート、ジプロピレングリコールジメタクリレート、トリプロピレングリコールジメタクリレート、テトラプロピレングリコールジメタクリレート等のポリプロピレングリコールジメタクリレート類;エチレングリコールジビニルエーテル、ジエチレングリコールジビニルエーテル、トリエチレングリコールジビニルエーテル、テトラエチレングリコールジビニルエーテル等のポリエチレングリコールジビニルエーテル類;エチレングリコールジアリルエーテル、ジエチレングリコールジアリルエーテル、トリエチレングリコールジアリルエーテル、テトラエチレングリコールジアリルエーテル等のポリエチレングリコールジアリルエーテル類;ビスフェノールFエトキシレートジアクリレート;ビスフェノールFエトキシレートジメタクリレート;ビスフェノールAエトキシレートジアクリレート;ビスフェノールAエトキシレートジメタクリレート;トリメチロールプロパントリアクリレート;トリメチロールプロパントリメタクリレート;トリメチロールプロパンエトキシレートトリアクリレート;トリメチロールプロパンエトキシレートトリメタクリレート;トリメチロールプロパンプロポキシレートトリアクリレート;トリメチロールプロパンプロポキシレートトリメタクリレート;イソシアヌル酸エトキシレートトリアクリレート;グリセロールエトキシレートトリアクリレート;グリセロールプロポキシレートトリアクリレート;ペンタエリスリトールエトキシレートテトラアクリレート;ジトリメチロールプロパンエトキリレートテトラアクリレート;ジペンタエリスリトールエトキシレートヘキサアクリレート等が挙げられる。
用いる重合開始剤としては、前記重合性組成物の成分として例示したのと同様のものが挙げられる。
また、用いる基板は、単層のものであっても、積層体であってもよい。
基板としては、有機材料が好ましく、この有機材料をフィルムとした樹脂フィルムが更に好ましい。
本発明の重合性組成物を重合することにより、本発明の高分子を容易に得ることができる。本発明においては、重合反応をより効率的に行う観点から、前記したような重合開始剤、特に光重合開始剤を含む重合性組成物を用いるのが好ましい。
本発明の高分子によれば、広い波長域において一様の偏光変換が可能な、性能面で満足のいく光学フィルムを低コストで得ることができる。
本発明の光学異方体は、本発明の高分子を構成材料とする。
本発明の光学異方体は、例えば、基板上に配向膜を形成し、該配向膜上に、さらに、本発明の高分子からなる液晶層を形成することによって、得ることができる。
配向膜は、ポリイミド、ポリビニルアルコール、ポリエステル、ポリアリレート、ポリアミドイミド、ポリエーテルイミド等のポリマーを含有する溶液(配向膜用組成物)を基板上に膜状に塗布し、乾燥させ、そして一方向にラビング処理等することで、得ることができる。
配向膜の厚さは0.001〜5μmであることが好ましく、0.001〜1μmであることがさらに好ましい。
また、ラビング処理する方法以外に、配向膜の表面に偏光紫外線を照射する方法によっても、面内で一方向に配向規制する機能を持たせることができる。
本発明の光学異方体としては、位相差板、液晶表示素子用配向膜、偏光板、視野角拡大板、カラーフィルター、ローパスフィルター、光偏光プリズム、各種光フィルター等が挙げられる。
本発明のカルボニル化合物は、前記式(4)で示される化合物である。
前述の通り、本発明のカルボニル化合物(4)は、本発明の重合性化合物(I)の製造中間体として好適に使用することができる。カルボニル化合物(4)の製造方法の詳細は、前記1)の重合性化合物の項で説明したとおりである。
本発明の、「重合性化合物の製造方法」は、本発明のカルボニル化合物(4)と、前記式(3)で表されるヒドラジン化合物とを反応させることを特徴とする、本発明の重合性化合物(I)の製造方法である。製造方法の詳細は、前記1)重合性化合物の項で説明したとおりである。
本発明の重合性化合物の製造方法によれば、本発明の重合性化合物(I)を、効率よく簡便に製造することができる。
本発明のカルボニル化合物(4)を製造原料として使用することにより、本発明の重合性化合物(I)を高収率で簡便に製造することができる。
得られた反応液に、4−(ジメチルアミノ)ピリジン 0.64g(5.22mmol)、及び、4−(6−アクリロイルオキシ−ヘクス−1−イルオキシ)フェノール(DKSH社製)13.80g(52.21mmol)を加え、再度反応器を水浴に浸して反応液内温を15℃とした。そこへ、トリエチルアミン 6.34g(62.65mmol)を、反応液内温を20〜30℃に保持しながら、10分間かけて滴下し、滴下終了後、全容を25℃でさらに2時間攪拌した。反応終了後、反応液に蒸留水1000mlと飽和食塩水100mlを加え、酢酸エチル400mlで2回抽出した。有機層を集め、無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から溶媒を蒸発除去した後、得られた残留物をシリカゲルカラムクロマトグラフィー(THF:トルエン=1:9(容積比、以下にて同じ。))により精製することで、中間体Aを白色固体として14.11g得た(収率:65%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、前記ステップ2で合成した中間体B 2.30g(2.45mmol)及びTHF25mlを入れ、均一な溶液とし、そこへ、濃塩酸 0.49ml(0.25mmol)を加えた。この溶液に、2−ヒドラジノベンゾチアゾール 0.40g(2.45mmol)のTHF5ml溶液を15分かけて滴下し、滴下終了後、全容を25℃にてさらに1時間撹拌した。反応終了後、反応液をメタノール400mlに投入して析出した固体をろ取した。ろ取した固体を真空乾燥機で乾燥させ、化合物1を淡黄色固体として2.4g得た(収率:90%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1のステップ2で合成した中間体B 1.20g(1.28mmol)及びTHF30mlを入れ、均一な溶液とした。そこへ、1N塩酸0.26ml(0.26mmol)を加え、前記合成ステップ3で合成した中間体E 0.48g(1.92mmol)のTHF5ml溶液を15分かけて滴下した。滴下終了後、全容を25℃にてさらに5時間撹拌した。反応液に、メタノール250mlに投入して、析出した固体をろ取した。得られた固体をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=97:3)により精製することで、化合物2を淡黄色固体として1.25g得た(収率:84%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、ステップ4で合成した中間体I 1.67g(1.69mmol)及びTHF30mlを入れ、均一な溶液とした。そこへ、1N塩酸 0.34ml(0.34mmol)を加え、実施例2のステップ3で合成した中間体E 0.85g(3.38mmol)のTHF5ml溶液を30分かけて滴下し、滴下終了後、全容を25℃にて5時間撹拌した。反応液をメタノール250mlに投入し、析出した固体をろ取した。ろ取した固体をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=97:3)により精製することで、化合物3を淡黄色固体として1.61g得た(収率:78%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた4つ口反応器に、窒素気流中、前記ステップ1で合成した中間体J 697mg(2.37mmol)、実施例1で合成した中間体B 2.00g(2.13mmol)、エタノール3ml及びTHF20mlを入れ、均一な溶液とした。この溶液に、(±)−10−カンファースルホン酸 55.1mg(0.24mmol)を加え、全容を40℃で5時間撹拌した。反応終了後、反応液を水150mlに投入し、酢酸エチル300mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、白色固体を得た。この白色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=90:10)により精製し、化合物4を白色固体として2.24g得た(収率:86.4%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMR、及び、13C−NMRで同定した。
13C−NMR(100MHz,CDCl3,TMS,δppm):164.6、149.7、134.7、132.3、129.5、129.2、128.4、128.1、127.5、124.0、118.7、42.8
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた4つ口反応器に、窒素気流中、前記ステップ4で合成した中間体N 588mg(1.40mmol)、実施例1で合成した中間体A 1.47g(3.50mmol)、4−(ジメチルアミノ)ピリジン 85.5mg(0.70mmol)、及びN−メチルピロリドン15mlを入れ、均一な溶液とした。この溶液に、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(WSC)805mg(4.20mmol)を加え、全容を25℃にて5時間攪拌した。反応終了後、反応液を水150mlに投入し、酢酸エチル300mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、白色固体を得た。この白色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=90:10)により精製し、化合物5を白色固体として1.12g得た(収率:65.5%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は、1H−NMR、及び、13C−NMRで同定した。
13C−NMR(125MHz,CDCl3,TMS,δppm):175.2、158.0、153.3、141.7、139.4、15.4
目的物の構造は1H−NMR、及び、13C−NMRで同定した。
13C−NMR(100MHz,DMSO−d6,TMS,δppm):175.5、160.4、150.8、140.7、135.3
温度計を備えた4つ口反応器に、窒素気流中、前記ステップ2で合成した中間体P 390mg(2.34mmol)、実施例1で合成した中間体B 2.08g(2.22mmol)、エタノール3ml、及びTHF15mlを入れ、均一な溶液とした。この溶液に、(±)−10−カンファースルホン酸54.4mg(0.23mmol)を加え、全容を40℃で5時間撹拌した。反応終了後、反応液を水150mlに投入し、酢酸エチル300mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ別した。ロータリーエバポレーターにて、ろ液から酢酸エチルを減圧留去して、淡黄色固体を得た。この淡黄色固体をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール=95:5)により精製し、化合物6を淡黄色固体として1.82g得た(収率:75.3%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた4つ口反応器に、窒素気流中、前記ステップ1で合成した中間体Q 338mg(1.34mmol)、実施例1で合成した中間体B 1.20g(1.28mmol)、エタノール3ml,及びTHF10mlを入れ、均一な溶液とした。この溶液に、(±)−10−カンファースルホン酸15.6mg(0.13mmol)を加え、全容を40℃で5時間撹拌した。反応終了後、反応液を水100mlに投入し、酢酸エチル300mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムを濾別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、白色固体を得た。この白色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=90:10)により精製し、化合物7を白色固体として1.21g得た(収率:79.4%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMR、及び、13C−NMRで同定した。
13C−NMR(125MHz,DMSO−d6,TMS,δppm):178.2、170.9、167.8、156.3、118.0、25.3、23.2
温度計を備えた4つ口反応器内において、窒素気流中、前記ステップ2で合成した中間体S 460mg(2.36mmol)及び、実施例1で合成した中間体B 2.00g(2.12mmol)を、エタノール3ml及びTHF20mlの混合溶媒に溶解させた。この溶液に、(±)−10−カンファースルホン酸54.8mg(0.24mmol)を加え、全容を40℃で3時間撹拌した。反応終了後、反応液を水150mlに投入し、酢酸エチル300mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール=90:10)により精製し、黄色固体として化合物8を1.02g得た(収率:43.1%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた4つ口反応器に、窒素気流中、中間体α 10.5g(15.3mmol)及びTHF80mlを入れ、均一な溶液とした。そこに、2−ヒドラジノベンゾチアゾール3.0g(18.3mmol)を加えて溶解させた。この溶液に、(±)−10−カンファースルホン酸18mg(0.08mmol)を加え、全容を25℃にて3時間攪拌した。反応終了後、反応液を10%の重曹水800mlに投入し、酢酸エチル100mlで2回抽出を行った。酢酸エチル層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、淡黄色固体を得た。この淡黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=8:2)により精製し、化合物Aを淡黄色固体として8.0g得た(収率:62.7%)。
目的物の構造は1H−NMR、マススペクトルで同定した。
LCMS(APCI)caled for C46H47N3O10S:833[M+];Found:833。
化合物1〜8、化合物A、化合物1r及び2rをそれぞれ10mg計量し、固体状態のまま、ラビング処理を施したポリイミド配向膜付きのガラス基板(E.H.C.Co.,Ltd.製、商品名:配向処理ガラス基板 以下にて同じ。)2枚に挟んだ。この基板をホットプレート上に載せ、40℃から200℃まで昇温した後、再び40℃まで降温した。昇温、降温する際の組織構造の変化を偏光光学顕微鏡(ニコン社製、ECLIPSE LV100POL型 以下にて同じ。)で観察した。但し、化合物4〜8については、40℃から250℃の範囲で、化合物Aについては、50℃〜200℃の範囲で相転移温度の測定を行った。
表1中、「C」はCrystal、「N」はNematic、「I」はIsotropicをそれぞれ表す。ここで、Crystalとは、試験化合物が固相にあることを、N
ematicとは、試験化合物がネマチック液晶相にあることを、Isotropicとは、試験化合物が等方性液体相にあることを、それぞれ示す(以下にて同じ。)。
実施例1で得た化合物1、参考例1の化合物1r、及び参考例2の化合物2rのそれぞれを1.0g、光重合開始剤A(ADEKA社製、アデカオプトマーN−1919、以下にて同じ。)を30mg、界面活性剤A(AGCセイミケミカル社製、KH−40、以下にて同じ。)の1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物1、1r、及び2rをそれぞれ得た。
実施例2、3で得た化合物2及び化合物3のそれぞれを1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン3.0g、ジメチルスルホキシド0.25gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物2及び3をそれぞれ得た。
実施例3で得た化合物3を0.5g、合成例1で合成した化合物Aを0.5g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物4を得た。
実施例3で得た化合物3を0.5g、化合物2rを0.5g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物5を得た。
実施例4で得た化合物4を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターで濾過し、重合性組成物6を得た。
実施例5で得た化合物5を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、クロロホルム2.9gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターで濾過し、重合性組成物7を得た。
実施例5で得た化合物5を0.5g、化合物Aを0.5g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.2g、クロロホルム1.7gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターで濾過し、重合性組成物8を得た。
実施例6で得た化合物6を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液200mgを、クロロホルム5.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターで濾過し、重合性組成物9を得た。
実施例6で得た化合物6を0.2g、化合物Aを0.8g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、クロロホルム3.7g、に溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターで濾過し、重合性組成物10を得た。
実施例7で得た化合物7を0.5g、化合物Aを0.5g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.2g、に溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターで濾過し、重合性組成物11を得た。
実施例7で得た化合物7を0.2g、化合物Aを0.8g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.2g、に溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターで濾過し、重合性組成物12を得た。
実施例8で得た化合物8を0.5g、化合物Aを0.5g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、クロロホルム8.0g、に溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターで濾過し、重合性組成物13を得た。
(i)重合性組成物による液晶層の形成1
ラビング処理されたポリイミド配向膜の付与された透明ガラス基板(E.H.C.Co.,Ltd.製、商品名:配向処理ガラス基板、以下にて同じ。)に、重合性組成物1〜8、10〜12、1r及び2rを、♯4のワイヤーバーを使用して塗布した。塗膜を、下記表2に示す温度で1分間乾燥した後、表2に示す温度で1分間配向処理し、液晶層を形成した。その後、液晶層の塗布面側から2000mJ/cm2の紫外線を表2に示す温度下にて照射して重合させ、波長分散測定用の試料とした。
ラビング処理されたポリイミド配向膜の付与された透明ガラス基板に、重合性組成物9及び重合性組成物13を♯6のワイヤーバーを使用して塗布した。塗膜を、下記表2に示す温度で1分間乾燥した後、表2に示す温度で1分間配向処理し、液晶層を形成した。その後、液晶層の塗布面側から2000mJ/cm2の紫外線を表2に示す温度下にて照射して重合させ、波長分散測定用の試料とした。
得られた試料につき、400nmから800nm間の位相差を、エリプソメーター(J.A.Woollam社製 M2000U型)を用いて測定した。
測定した位相差を用いて以下のように算出されるα、β値から波長分散を評価した。
即ち、αとβが同程度の値となるフラットな波長分散性が好ましく、αが1より小となり、βが1より大となる逆波長分散性が特に好ましい。
重合して得られた液晶性高分子膜の膜厚(μm)、波長548.5nmにおける位相差(Re)、α、βの値を、下記表2にまとめて示す。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B:0.70g(0.75mmol)をTHF15mlに溶解させた。この溶液に、1N塩酸0.15ml(0.15mmol)と前記ステップ1で合成した中間体T 0.27g(1.49mmol)を加え、全容を40℃で10時間撹拌した。その後、反応液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=98:2)により精製することで、淡黄色固体として化合物9を0.70g、収率85%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は、1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 0.70g(0.75mmol)をTHF15mlに溶解させた。この溶液に、1N塩酸0.15ml(0.15mmol)と前記ステップ1で合成した中間体U 0.29g(1.49mmol)を加え、全容を40℃で10時間撹拌した。反応終了後、反応液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=98:2)により精製することで、淡黄色固体として化合物10を0.67g、収率81%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.4g(1.49mmol)をTHF30mlに溶解させた。この溶液に、1N塩酸0.30ml(0.30mmol)と前記ステップ1で合成した中間体V 0.62g(2.98mmol)を加え、全容を40℃で10時間撹拌した。その後、反応液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=98:2)により精製することで、淡黄色固体として化合物11を1.40g、収率83%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B:1.40g(1.50mmol)をTHF30mlに溶解させた。この溶液に、1N塩酸0.30ml(0.30mmol)と前記ステップ1で合成した中間体W0.62g(2.98mmol)を加え、40℃で8時間撹拌した。その後、反応液をロータリーエバポレーターで濃縮した後、シリカゲルカラムクロマトグラフィー(クロロホルム:THF=98:2)により精製することで、淡黄色固体として化合物12を1.32g、収率78%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.00g(1.06mmol)をTHF30mlに溶解させた。この溶液に、1N塩酸0.22ml(0.22mmol)と前記ステップ1で合成した中間体X 0.38g(1.60mmol)を加え、全容を40℃で2時間攪拌した。反応終了後、反応液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=40:1)により精製することで、淡黄色固体として化合物13を1.14g、収率95%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.00g(1.06mmol)をTHF30mlに溶解させた。この溶液に、1N塩酸0.22ml(0.22mmol)と前記ステップ1で合成した中間体Y 0.32g(1.28mmol)を加え、全容を40℃で1時間攪拌した。反応終了後、反応液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=40:1)により精製することで、淡黄色固体として化合物14を1.16g、収率93%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.00g(1.06mmol)をTHF30mlに溶解させた。この溶液に、1N塩酸0.22ml(0.22mmol)と前記ステップ1で合成した中間体Z 0.34g(1.28mmol)を加え、全容を40℃で1時間攪拌した。反応終了後、反応液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=40:1)により精製することで、淡黄色固体として化合物15を1.18g、収率93%で得た。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 2.50g(2.66mmol)をTHF150mlに溶解させた。この溶液に、1N塩酸2.65ml(2.65mmol)と前記ステップ1で合成した中間体A1 4.0g(13.3mmol)を加え、全容を60℃で30時間攪拌した。反応終了後、反応液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=95:5)により精製することで、淡黄色固体として化合物16を1.40g、収率43%で得た。
目的物の構造は1H−NMRで同定した。
2.50−2.76(m,2H)、2.21−2.48(m,6H)、1.99−2.16(m,2H)、1.35−1.85(m,26H)
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、前記ステップ1で合成した中間体B1 2.00g(1.84mmol)と4−(ジメチルアミノ)ピリジン 0.02g(0.18mmol)をTHF100mlに溶解させた。この溶液に、n−オクタノイルクロリド 0.33g(2.03mmol)を加え、反応器を氷浴に浸して反応液内温を10℃とした。次いで、トリエチルアミン0.22g(2.21mmol)を10分間かけて滴下し、滴下終了後、全容を25℃で2時間攪拌した。反応終了後、反応液に蒸留水600ml及び飽和食塩水10mlを加え、クロロホルム200mlで2回抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=40:2)により精製することで、淡黄色固体として化合物17を1.72g、収率77%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.40g(1.49mmol)、前記ステップ1で合成した中間体C1 456mg(1.84mmol)、(±)−10−カンファスルホン酸38.6mg(0.166mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間攪拌した。反応終了後、反応液を水100mlに投入し、酢酸エチル200mlで抽出した。得られた酢酸エチル層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ別した。ロータリーエバポレーターにて、ろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=97:3)により精製し、淡黄色固体として化合物18を1.24g得た(収率:71.4%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体D1 438mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃で5時間攪拌した。反応終了後、反応液を水100mlに投入し、酢酸エチル200mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=85:15)により精製し、淡黄色固体として化合物19を1.31g得た(収率:70.2%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体E1 468mg(1.78mmol)、(±)−10−カンファスルホン酸 41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間攪拌した。反応終了後、反応液を水100mlに投入し、酢酸エチル200mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=9:1)により精製し、淡黄色固体として化合物20を1.46g得た(収率:77.5%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体F1 591mg(1.78mmol)、(±)−10−カンファスルホン酸 41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間攪拌した。反応終了後、反応液を水100mlに投入し、酢酸エチル200mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=9:1)により精製し、淡黄色固体として化合物21を1.44g得た(収率:71.9%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体G1 396mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間攪拌した。反応終了後、反応液を水100mlに投入し、酢酸エチル200mlで抽出した。得られた酢酸エチル層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=9:1)により精製し、淡黄色固体として化合物22を1.31g得た(収率:69.4%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、実施例30の化合物17合成のステップ1で合成した中間体B1 2.00g(1.84mmol)をTHF20mlに溶解させて、0℃に冷却した。この溶液に、クロロメチルシクロヘキシルエーテル412mg(2.76mmol)を加え、さらに、N、N−ジイソプロピルエチルアミン476mg(7.36mmol)のTHF5ml溶液を5分間かけて滴下した。滴下終了後、全容を25℃にて3時間撹拌した。反応終了後、反応液を水100mlに投入し、酢酸エチル200mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=95:5)により精製し、淡黄色固体として化合物24を1.54g得た(収率:70.0%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、前記ステップ4で合成した中間体K1 1.95g(1.96mmol)、前記実施例4の化合物4合成のステップ1で合成した中間体J 441mg(1.76mmol)、(±)−10−カンファスルホン酸 45.6mg(0.196mmol)、THF24ml、及びエタノール6mlを加え、均一な溶液とした。その後、全容を40℃にて5時間攪拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。クロロホルム層を無水硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液からクロロホルムを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=95:5)により精製し、淡黄色固体として化合物25を1.56g得た(収率:64.9%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた4つ口反応器内において、窒素気流中、前記ステップ1で合成した中間体L1 368mg(1.77mmol)及び、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)を、エタノール3ml及びTHF15mlの混合溶媒に溶解させた。この溶液に、(±)−10−カンファースルホン酸41.2mg(0.18mmol)を加え、全容を40℃で5時間撹拌した。反応終了後、反応液を水150mlに投入し、酢酸エチル300mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=90:10)により精製し、白色固体として化合物26を1.61g得た(収率:89.6%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた4つ口反応器内において、窒素気流中、前記ステップ1で合成した中間体M1 195mg(1.77mmol)、及び、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)を、エタノール3ml及びTHF15mlの混合溶媒に溶解させた。この溶液に、(±)−10−カンファースルホン酸41.2mg(0.18mmol)を加え、全容を40℃で8時間撹拌した。反応終了後、反応液を水150mlに投入し、酢酸エチル300mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=90:10)により精製し、白色固体として化合物27を1.26g得た(収率:69.3%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B:1.50g(1.60mmol)、前記ステップ1で合成した中間体N1:489mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間攪拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=9:1)により精製し、淡黄色固体として化合物28を1.47g得た(収率:77.2%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体O1 444mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間攪拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=92:8)により精製し、淡黄色固体として化合物29を1.35g得た(収率:72.4%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体P1 468mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間攪拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=9:1)により精製し、淡黄色固体として化合物30を1.44g得た(収率:76.3%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、前記ステップ4で合成した中間体T1 1.50g(1.36mmol)、前記実施例4の化合物4合成のステップ1で合成した中間体J 375mg(1.51mmol)、(±)−10−カンファスルホン酸35.1mg(0.151mmol)、THF24ml、及びエタノール6mlを加え、均一な溶液とした。その後、全容を40℃にて5時間攪拌した。反応終了後、反応液を水150mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、シリカゲルカラムクロマトグラフィー(クロロホルム:THF=97:3)により精製し、淡黄色固体として化合物31を1.54g得た(収率:84.6%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、前記ステップ2で合成した中間体V1 1.20g(1.28mmol)をTHF30mlに溶解させた。この溶液に、1N塩酸0.26ml(0.26mmol)と前記実施例4の化合物4合成のステップ1で合成した中間体J 0.48g(1.92mmol)を加え、全容を40℃で7時間攪拌した。反応終了後、反応液をロータリーエバポレーターで濃縮し、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=98:2)により精製することで、淡黄色固体として化合物32を1.23g、収率82%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内において、窒素気流中、前記ステップ2で合成した中間体X1:0.94g(1.00mmol)をTHF15mlに溶解させた。この溶液に、1N塩酸0.20ml(0.20mmol)と前記実施例4の化合物4合成のステップ1で合成した中間体J 0.37g(1.49mmol)を加え、全容を60℃で10時間攪拌した。反応終了後、反応液をロータリーエバポレーターで濃縮し、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=98:2)により精製することで、淡黄色固体として化合物33を0.92g、収率79%で得た。
目的物の構造は1H−NMRで同定した。
上記で得た化合物9〜33をそれぞれ10mg計量し、固体状態のままで、ラビング処理を施したポリイミド配向膜付きのガラス基板2枚に挟んだ。この基板をホットプレート上に載せ、40℃から250℃まで昇温した後、再び40℃まで降温した。昇温、降温する際の組織構造の変化を偏光光学顕微鏡で観察した。
測定した相転移温度を下記表3に示す。
実施例22、23で得た化合物9、10を、それぞれ1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3g、クロロホルム2.26gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物14、15を得た。
実施例24で得た化合物11を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物16を得た。
実施例25で得た化合物12を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3g、クロロホルム1.7gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物17を得た。
実施例26で得た化合物13を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物18を得た。
実施例27で得た化合物14を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3g、クロロホルム1.7gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物19を得た。
実施例28、29で得た化合物15、16を、それぞれ1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物20、21を得た。
実施例30で得た化合物17を0.5g、実施例4の化合物4を0.5g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、クロロホルム3.26gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物22を得た。
実施例31〜38で得た化合物18〜25を、それぞれ1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物23〜30を得た。
実施例39で得た化合物26を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3g、クロロホルム0.7gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物31を得た。
実施例40で得た化合物27を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物32を得た。
実施例41で得た化合物28を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3g、クロロホルム0.7gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物33を得た。
実施例42、43で得た化合物29、30を、それぞれ1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物34、35を得た。
実施例44で得た化合物31を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3g、クロロホルム0.7gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物36を得た。
実施例44、45で得た化合物32、33を、それぞれ0.3g、実施例4で得た化合物4を0.7g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物37、38をそれぞれ得た。
(i)重合性組成物による液晶層の形成1
ラビング処理されたポリイミド配向膜の付与された透明ガラス基板に、重合性組成物16〜18、20〜30、32及び34〜38のそれぞれを♯4のワイヤーバーを使用して塗布した。塗膜を、下記表4に示す温度で1分間乾燥した後、表4に示す温度で1分間配向処理し、液晶層を形成した。その後、液晶層の塗布面側から2000mJ/cm2の紫外線を表4に示す温度下にて照射して重合させ、波長分散測定用の試料とした。
ラビング処理されたポリイミド配向膜の付与された透明ガラス基板に、重合性組成物14、15、19、31及び33のそれぞれを♯6のワイヤーバーを使用して塗布した。塗膜を、下記表4に示す温度で1分間乾燥した後、表4に示す温度で1分間配向処理し、液晶層を形成した。その後、液晶層の塗布面側から2000mJ/cm2の紫外線を、表4に示す温度下にて照射して重合させ、波長分散測定用の試料とした。
得られた試料につき、位相差の測定、波長分散の評価を前記と同様の方法で行った。
重合して得られた液晶性高分子膜の膜厚(μm)、波長548.5nmにおける位相差(Re)、α、βの値を、下記表4にまとめて示す。
目的物の構造は1H−NMRで同定した。
温度計を備えた4つ口反応器において、窒素気流中、前記ステップ1で合成した中間体Y1 368mg(1.68mmol)及び、実施例1の化合物1合成のステップ2で合成した中間体B 1.0g(1.06mmol)を、エタノール3ml及びTHF15mlの混合溶媒に溶解させた。この溶液に、(±)−10−カンファースルホン酸49.2mg(0.21mmol)を加え、全容を40℃で5時間撹拌した。反応終了後、反応液を水150mlに投入し、酢酸エチル300mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=90:10)により精製し、白色固体として化合物34を1.07g得た(収率:88.5%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた4つ口反応器において、窒素気流中、前記ステップ1で合成した中間体Z1 518mg(2.34mmol)及び、実施例1の化合物1合成のステップ2で合成した中間体B 2.00g(2.12mmol)を、エタノール3ml及びTHF20mlの混合溶媒に溶解させた。この溶液に、(±)−10−カンファースルホン酸54.4mg(0.24mmol)を加え、全容を40℃で7時間撹拌した。反応終了後、反応液を水150mlに投入し、酢酸エチル300mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=90:10)により精製し、白色固体として化合物35を1.83g得た(収率:75.7%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた4つ口反応器において、窒素気流中、前記ステップ1で合成した中間体A2 627mg(2.13mmol)及び、実施例1の化合物1合成のステップ2で合成した中間体B 1.00g(1.06mmol)を、エタノール3ml及びTHF15mlの混合溶媒に溶解させた。この溶液に、(±)−10−カンファースルホン酸49.2mg(0.21mmol)を加え、全容を40℃で4時間撹拌した。反応終了後、反応液を水150mlに投入し、酢酸エチル300mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=90:10)により精製し、白色固体として化合物36を1.11g得た(収率:84.8%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた4つ口反応器において、窒素気流中、前記ステップ1で合成した中間体B2 312mg(1.77mmol)及び、実施例1の化合物1合成のステップ2で合成した中間体B 1.00g(1.06mmol)を、エタノール3ml及びTHF15mlの混合溶媒に溶解させた。この溶液に、(±)−10−カンファースルホン酸27.1mg(0.12mmol)を加え、全容を40℃で7時間撹拌した。反応終了後、反応液を水150mlに投入し、酢酸エチル300mlで抽出した。酢酸エチル層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ロータリーエバポレーターにてろ液から酢酸エチルを減圧留去して、黄色固体を得た。この黄色固体をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=90:10)により精製し、白色固体として化合物37を1.18g得た(収率:92.3%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体C2 534mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とし、全容を40℃にて5時間撹拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=9:1)により精製し、淡黄色固体として化合物38を1.67g得た(収率:86.9%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体D2 495mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とし、全容を40℃にて3時間撹拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=92:8)により精製し、淡黄色固体として化合物39を1.61g得た(収率:83.8%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体E2 447mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間撹拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を硫酸ナトリウムで乾燥した後、ロータリーエバポレーターで濃縮した後、シリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=85:15)により精製し、淡黄色固体として化合物40を1.60g得た(収率:85.7%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.35g(1.44mmol)、前記ステップ1で合成した中間体F2 654mg(1.59mmol)、(±)−10−カンファスルホン酸38.4mg(0.165mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とし、全容を40℃にて5時間撹拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=92:8)により精製し、淡黄色固体として化合物41を1.41g得た(収率:73.6%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.0g(1.06mmol)をTHF30mlに溶解させた。この溶液に、1N塩酸0.44ml(0.44mmol)と前記ステップ1で合成した中間体G2 1.12g(5.13mmol)を加え、全容を60℃で20時間撹拌した。反応終了後、反応液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=40:1)により精製することで、淡黄色固体として化合物42を0.55g、収率91%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.0g(1.06mmol)をTHF30mlに溶解させた。この溶液に、1N塩酸0.44ml(0.44mmol)と前記ステップ1で合成した中間体H2 0.74g(3.20mmol)を加え、全容を60℃で15時間撹拌した。反応終了後、反応液をロータリーエバポレーターで濃縮し、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=40:1)により精製することで、淡黄色固体として化合物43を1.04g、収率85%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.0g(1.06mmol)をTHF30mlに溶解させた。この溶液に、1N塩酸0.44ml(0.44mmol)と前記ステップ1で合成した中間体I2 0.64g(3.20mmol)を加え、全容を50℃で15時間撹拌した。反応終了後、反応液をロータリーエバポレーターで濃縮し、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=40:1)により精製することで、淡黄色固体として化合物44を1.10g、収率92%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器において、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.0g(1.06mmol)をTHF30mlに溶解させた。この溶液に、1N塩酸0.44ml(0.44mmol)と、前記ステップ1で合成した中間体J2:0.7g(3.20mmol)を加え、全容を50℃で15時間撹拌した。反応終了後、反応液をロータリーエバポレーターで濃縮し、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=40:1)により精製することで、淡黄色固体として化合物45を1.08g、収率89%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3口反応器において、窒素気流中、実施例1の化合物1合成のステップ1で合成した中間体A 10.4g(24.8mmol)をTHF150mlに溶解させた。この溶液に、メタンスルホニルクロリド2.9g(25.7mmol)を加え、反応器を水浴に浸して反応液内温を20℃とし、トリエチルアミン2.7g(26.5mmol)を10分間かけて滴下した。滴下終了後、水浴を除去し、全容を25℃で2時間撹拌した。次いで、4−(ジメチルアミノ)ピリジン 0.2g(1.7mmol)、前記ステップ4で合成した中間体N2 3.0g(8.3mmol)を加え、さらに、トリエチルアミン2.5g(24.8mmol)を10分間かけて滴下した。滴下終了後、全容を25℃で2時間撹拌した。反応終了後、反応液に蒸留水2000mlと飽和食塩水500mlを加え、クロロホルム1000mlで2回抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、シリカゲルカラムクロマトグラフィー(クロロホルム:THF=25:1)により精製することで、淡黄色固体として化合物46を1.8g、収率19%で得た。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3口反応器において、窒素気流中、実施例1の化合物1合成のステップ1で合成した中間体A 5.36g(12.78mmol)をTHF60mlに溶解させた。この溶液に、メタンスルホニルクロリド1.52g(13.22mmol)を加え、反応器を水浴に浸して反応液内温を20℃とし、トリエチルアミン1.38g(13.7mmol)を5分間かけて滴下した。次いで水浴を除去し、全容を25℃で2時間撹拌した。さらに、4−(ジメチルアミノ)ピリジン 0.1g(0.82mmol)、前記ステップ3で合成した中間体Q2 1.60g(4.26mmol)を加え、さらにトリエチルアミン1.30g(12.78mmol)を5分間かけて滴下した。滴下終了後、全容を25℃で2時間撹拌した。反応終了後、反応液に蒸留水400mlと飽和食塩水100mlを加え、クロロホルム200mlで2回抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(クロロホルム:THF=40:1)により精製することで、淡黄色固体として化合物47を1.04g、収率21%で得た。
目的物の構造は1H−NMRで同定した。
上記で得た化合物34〜47をそれぞれ10mg計量し、固体状態のままで、ラビング処理を施したポリイミド配向膜付きのガラス基板2枚に挟んだ。この基板をホットプレート上に載せ、40℃から250℃まで昇温した後、再び40℃まで降温した。昇温、降温する際の組織構造の変化を偏光光学顕微鏡で観察した。
測定した相転移温度を下記表5に示す。
実施例72〜74で得た化合物34〜36を、それぞれ1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物39〜41を得た。
実施例75で得た化合物37を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、クロロホルム3.0gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物42を得た。
実施例76〜79で得た化合物38〜41を、ぞれぞれ1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物43〜46を得た。
実施例80で得た化合物42を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、クロロホルム3.0gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物47を得た。
実施例81〜83で得た化合物43〜45を、それぞれ1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物48〜50を得た。
実施例84で得た化合物46を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、クロロホルム2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物51を得た。
実施例85で得た化合物47を1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物52を得た。
(i)重合性組成物による液晶層の形成
ラビング処理されたポリイミド配向膜の付与された透明ガラス基板に、重合性組成物39〜52のそれぞれを♯4のワイヤーバーを使用して塗布した。塗膜を、下記表6に示す温度で1分間乾燥した後、表6に示す温度で1分間配向処理し、液晶層を形成した。その後、液晶層の塗布面側から2000mJ/cm2の紫外線を表6に示す温度下にて照射して重合させ、波長分散測定用の試料とした。
得られた試料につき、位相差の測定、波長分散の評価を前記と同様の方法で行った。
重合して得られた液晶性高分子膜の膜厚(μm)、波長548.5nmにおける位相差(Re)、α、βの値を、下記表6にまとめて示す。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器内で、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体R2 387mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.165mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間撹拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=9:1)により精製し、淡黄色固体として化合物48を1.54g得た(収率:84.9%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体S2 534mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて3時間撹拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=92:8)により精製し、淡黄色固体として化合物49を1.62g得た(収率:83.8%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体T2 567mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間撹拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=9:1)により精製し、淡黄色固体として化合物50を1.53g得た(収率:77.3%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体U2 504mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間撹拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=85:15)により精製し、淡黄色固体として化合物51を1.59g得た(収率:82.9%)。
目的物の構造は1H−NMRで同定した。
目的物の構造は1H−NMRで同定した。
温度計を備えた3つ口反応器に、窒素気流中、実施例1の化合物1合成のステップ2で合成した中間体B 1.50g(1.60mmol)、前記ステップ1で合成した中間体V2 477mg(1.78mmol)、(±)−10−カンファスルホン酸41.4mg(0.178mmol)、THF16ml、及びエタノール4mlを加え、均一な溶液とした。その後、全容を40℃にて5時間撹拌した。反応終了後、反応液を水100mlに投入し、クロロホルム200mlで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、硫酸ナトリウムをろ別した。ろ液をロータリーエバポレーターで濃縮した後、濃縮物をシリカゲルカラムクロマトグラフィー(トルエン:酢酸エチル=92:8)により精製し、淡黄色固体として化合物52を1.56g得た(収率:82.3%)。
目的物の構造は1H−NMRで同定した。
化合物48〜52をそれぞれ10mg計量し、固体状態のままで、ラビング処理を施したポリイミド配向膜付きのガラス基板2枚に挟んだ。この基板をホットプレート上に載せ、40℃から250℃まで昇温した後、再び40℃まで降温した。昇温、降温する際の組織構造の変化を偏光光学顕微鏡で観察した。
測定した相転移温度を下記表7に示す。
実施例100〜104で得た化合物48〜52をそれぞれ1.0g、光重合開始剤Aを30mg、界面活性剤Aの1%シクロペンタノン溶液100mgを、シクロペンタノン2.3gに溶解させた。この溶液を0.45μmの細孔径を有するディスポーサブルフィルターでろ過し、重合性組成物53〜57を得た。
(i)重合性組成物による液晶層の形成
ラビング処理されたポリイミド配向膜の付与された透明ガラス基板に、重合性組成物53〜57のそれぞれを♯4のワイヤーバーを使用して塗布した。塗膜を、下記表8に示す温度で1分間乾燥した後、表8に示す温度で1分間配向処理し、液晶層を形成した。その後、液晶層の塗布面側から2000mJ/cm2の紫外線を表8に示す温度下にて照射して重合させ、波長分散測定用の試料とした。
得られた試料につき、位相差の測定、波長分散の評価を前記と同様の方法で行った。
重合して得られた液晶性高分子膜の膜厚(μm)、波長548.5nmにおける位相差(Re)、α、βの値を、下記表8にまとめて示す。
実施例4で得た化合物4を19.3部、光重合開始剤B(BASFジャパン社製、イルガキュアー379)を0.6部、界面活性剤B(AGCセイミケミカル社製、サーフロンS−420)の1%シクロペンタノン溶液5.8部を、シクロペンタノン74.2部に溶解し、0.6μmの細孔径を有するディスポーサブルフィルターで濾過して、重合性組成物58を得た。
実施例110において、実施例4で得た化合物4の代わりに、実施例26で得た化合物13を用いたほかは、実施例110と同様にして重合性組成物59を得た。
実施例110において、実施例4で得た化合物4の代わりに、実施例35で得た化合物22を用いたほかは、実施例110と同様にして重合性組成物60を得た。
実施例110において、実施例4で得た化合物4の代わりに、合成例1で得た化合物Aを用いたほかは、実施例110と同様にして重合性組成物61を得た。
得られた重合性組成物58〜61を、下記の方法で重合させて高分子を得た。得られた高分子のそれぞれについて、位相差の測定と反射輝度の評価を行った。
支持体(日本ゼオン社製、ゼオノアフィルム、商品名「ZF16」)の一方の面を、ラビングすることにより配向処理を行なった。かかる面上に、上記で得た重合性組成物58〜61のそれぞれを、スピンコーターで乾燥膜厚がそれぞれ2.5μm、1.9μm、1.9μm及び1.4μmになるように塗布した。オーブンにて130℃(重合性組成物60については、105℃)で2分間加熱することにより、重合性組成物の層を乾燥させた。これにより、支持体、及びその上に形成された乾燥した重合性組成物の層からなる複層物を得た。
得られた位相差板と直線偏光板(SANRITZ社製、製品名:HLC2−5618)を、光学用透明粘着材(日東電工社製、商品名:LUCIACS)で貼り合わせることにより、円偏光板を作製した。このとき、直線偏光板の吸収軸方向と位相差板の遅層軸方向(ラビング方向と平行方向)との相対角を45°とした。
得られた円偏光板の位相差板側に、前記光学用透明粘着材を用いてアルミニウム蒸着PETフィルム(東レフィルム加工社製、商品名:メタルミーTS#50)を貼り合わせて、測定用の試料を作製した。この試料の5°反射における反射スペクトルを分光光度計(日本分光社製、商品名:V7200)により測定した。測定波長は380nm〜780nmである。
Claims (26)
- 下記式(I)
G1、G2はそれぞれ独立して、置換基を有していてもよい炭素数1〜20の二価の鎖状脂肪族基を表す。前記鎖状脂肪族基には、−O−、−S−、−O−C(=O)−、−C(=O)−O−、−O−C(=O)−O−、−NR2−C(=O)−、−C(=O)−NR2−、−NR2−、又は、−C(=O)−が介在していてもよい。ただし、−O−又は−S−がそれぞれ2以上隣接して介在する場合を除く。ここで、R2は、水素原子又は炭素数1〜6のアルキル基を表す。
Z1、Z2はそれぞれ独立して、無置換又はハロゲン原子で置換された炭素数2〜10のアルケニル基を表す。
Axは、芳香族炭化水素環及び芳香族複素環からなる群から選ばれる少なくとも一つの芳香環を有する、炭素数2〜30の有機基を表す。
Ayは、水素原子、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、置換基を有していてもよい炭素数1〜20のアルキニル基、置換基を有していてもよい炭素数3〜12のシクロアルキル基、−C(=O)−R3、−SO2−R4、−C(=S)NH−R9、置換基を有していてもよい炭素数2〜30の芳香族炭化水素環基、又は、置換基を有していてもよい炭素数2〜30の芳香族複素環基を表す。ここで、R3は、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、又は、置換基を有していてもよい炭素数3〜12のシクロアルキル基、炭素数5〜12の芳香族炭化水素基を表し、R4は、炭素数1〜20のアルキル基、炭素数2〜20のアルケニル基、フェニル基、又は、4−メチルフェニル基を表し、R9は置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、置換基を有していてもよい炭素数3〜12のシクロアルキル基、置換基を有していてもよい炭素数5〜20の芳香族基を表す。
前記Ax及びAyが有する芳香環は置換基を有していてもよい。
A1は、置換基を有していてもよい三価の芳香族基を表す。
A2、A3はそれぞれ独立して、置換基を有していてもよい炭素数3〜30の二価の脂環式炭化水素基を表す。
A4、A5はそれぞれ独立して、置換基を有していてもよい炭素数6〜30の二価の芳香族基を表す。
Q1は、水素原子、又は、置換基を有していてもよい炭素数1〜6のアルキル基を表す。}で示される重合性化合物。 - 前記AxとAyに含まれるπ電子の総数が4以上24以下である請求項1に記載の重合性化合物。
- 前記A1が、置換基を有していてもよい、三価のベンゼン環基又は三価のナフタレン環基である請求項1又は2に記載の重合性化合物。
- 前記Y1〜Y8が、それぞれ独立して、化学的な単結合、−O−、−O−C(=O)−、−C(=O)−O−、又は、−O−C(=O)−O−である請求項1〜3のいずれかに記載の重合性化合物。
- 前記Z1、Z2が、それぞれ独立して、CH2=CH−、CH2=C(CH3)−、又は、CH2=C(Cl)−である請求項1〜4のいずれかに記載の重合性化合物。
- 前記G1、G2が、それぞれ独立して、置換基を有していてもよい炭素数1〜12の二価の脂肪族基〔該脂肪族基には、−O−、−O−C(=O)−、−C(=O)−O−又は−C(=O)−が介在していてもよい。ただし、−O−が2以上隣接して介在する場合を除く。〕である請求項1〜5のいずれかに記載の重合性化合物。
- 前記G1、G2がそれぞれ独立して、炭素数1〜12のアルキレン基である請求項1〜6のいずれかに記載の重合性化合物。
- Axが下記構造を有する基のいずれかであり、Xは、NR7、酸素原子、硫黄原子、−SO−、又は、−SO2−を表し、酸素原子、硫黄原子、−SO−、−SO2−が、それぞれ隣接する場合を除き、R7は水素原子;又は、メチル基、エチル基、プロピル基等の炭素数1〜6のアルキル基を表し、
Ayが、水素原子、炭素数3〜8のシクロアルキル基、(ハロゲン原子、シアノ基、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、若しくは炭素数3〜8のシクロアルキル基)を置換基として有していてもよい炭素数6〜12の芳香族炭化水素環基、(ハロゲン原子、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、シアノ基)を置換基として有していてもよい炭素数3〜9の芳香族複素環基、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、置換基を有していてもよい炭素数2〜20のアルキニル基であり、当該置換基が、ハロゲン原子、シアノ基、炭素数1〜20のアルコキシ基、炭素数1〜12のアルコキシ基で置換された炭素数1〜12のアルコキシ基、フェニル基、シクロヘキシル基、炭素数2〜12の環状エーテル基、炭素数6〜14のアリールオキシ基、水酸基、ベンゾジオキサニル基、ベンゼンスルホニル基、ベンゾイル基、−SR10のいずれかであり、
R10は炭素数1〜20のアルキル基、炭素数2〜20のアルケニル基、炭素数3〜12のシクロアルキル基、又は、炭素数6〜12の芳香族炭化水素基を表す、
請求項1〜7のいずれかに記載の重合性化合物。
- 前記A y が、置換基を有していてもよい、炭素数1〜20のアルキル基であり、
当該置換基が、ハロゲン原子、シアノ基、炭素数1〜20のアルコキシ基、炭素数1〜12のアルコキシ基で置換された炭素数1〜12のアルコキシ基、フェニル基、シクロヘキシル基、炭素数2〜12の環状エーテル基、炭素数6〜14のアリールオキシ基、水酸基、ベンゾジオキサニル基、フェニルスルホニル基、4−メチルフェニルスルホニル基、ベンゾイル基、−SR 10 のいずれかであり、
R 10 は、炭素数1〜20のアルキル基、炭素数2〜20のアルケニル基、炭素数3〜12のシクロアルキル基、又は、炭素数6〜12の芳香族炭化水素基を表す、
請求項8に記載の重合性化合物。 - 前記A y が、置換基を有していてもよい、炭素数2〜20のアルケニル基であり、
当該置換基が、ハロゲン原子、シアノ基、炭素数1〜20のアルコキシ基、炭素数1〜12のアルコキシ基で置換された炭素数1〜12のアルコキシ基、フェニル基、シクロヘキシル基、炭素数2〜12の環状エーテル基、炭素数6〜14のアリールオキシ基、水酸基、ベンゾジオキサニル基、フェニルスルホニル基、4−メチルフェニルスルホニル基、ベンゾイル基、−SR 10 のいずれかであり、
R 10 は、炭素数1〜20のアルキル基、炭素数2〜20のアルケニル基、炭素数3〜12のシクロアルキル基、又は、炭素数6〜12の芳香族炭化水素基を表す、
請求項8に記載の重合性化合物。 - 前記Ayが、置換基を有していてもよい、炭素数2〜20のアルキニル基であり、
当該置換基が、ハロゲン原子、シアノ基、炭素数1〜20のアルコキシ基、炭素数1〜12のアルコキシ基で置換された炭素数1〜12のアルコキシ基、フェニル基、シクロヘキシル基、炭素数2〜12の環状エーテル基、炭素数6〜14のアリールオキシ基、水酸基、ベンゾジオキサニル基、フェニルスルホニル基、4−メチルフェニルスルホニル基、ベンゾイル基、−SR10のいずれかであり、
R10は、炭素数1〜20のアルキル基、炭素数2〜20のアルケニル基、炭素数3〜12のシクロアルキル基、又は、炭素数6〜12の芳香族炭化水素基を表す、
請求項8に記載の重合性化合物。 - 前記A 2 およびA 3 が、それぞれ独立に、式(A32a)又は(A32b)で示される基である、
請求項1〜11のいずれかに記載の重合性化合物。
- 前記A 4 およびA 5 が、それぞれ独立に、式(A41)又は(A42)で示される基である、
請求項1〜12のいずれかに記載の重合性化合物。
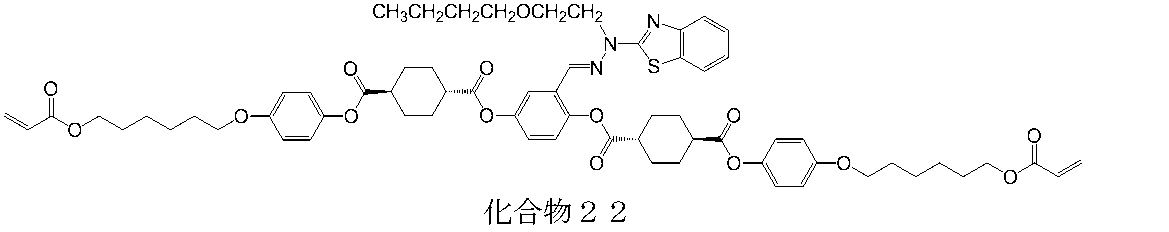
- 下記の化合物1〜52のいずれかで示される重合性化合物。
- 請求項1〜14のいずれかに記載の重合性化合物を少なくとも1種を含有する重合性組成物。
- 請求項1〜14のいずれかに記載の重合性化合物の少なくとも1種、及び重合開始剤を含有する重合性組成物。
- 請求項1〜14のいずれかに記載の重合性化合物、又は、請求項15若しくは請求項16に記載の重合性組成物を重合して得られる高分子。
- 液晶性高分子である請求項17に記載の高分子。
- 請求項18に記載の高分子を構成材料とする光学異方体。
- 下記式(4)
G1、G2はそれぞれ独立して、置換基を有していてもよい炭素数1〜20の二価の鎖状脂肪族基を表す。前記鎖状脂肪族基には、−O−、−S−、−O−C(=O)−、−C(=O)−O−、−O−C(=O)−O−、−NR2−C(=O)−、−C(=O)−NR2−、−NR2−、又は、−C(=O)−が介在していてもよい。ただし、−O−又は−S−がそれぞれ2以上隣接して介在する場合を除く。ここで、R2は、水素原子又は炭素数1〜6のアルキル基を表す。
Z1、Z2はそれぞれ独立して、無置換又はハロゲン原子で置換された炭素数2〜10のアルケニル基を表す。
A1は、置換基を有していてもよい三価の芳香族基を表す。
A2、A3はそれぞれ独立して、置換基を有していてもよい炭素数3〜30の二価の脂環式炭化水素基を表す。
A4、A5はそれぞれ独立して、置換基を有していてもよい炭素数6〜30の二価の芳香族基を表す。
Q1は、水素原子、又は、置換基を有していてもよい炭素数1〜6のアルキル基を表す。}で示されるカルボニル化合物。 - 前記A1が、置換基を有していてもよい、三価のベンゼン環基又は三価のナフタレン環基である請求項20に記載のカルボニル化合物。
- 前記A2、A3がそれぞれ独立して、置換基を有していてもよい二価のシクロヘキシル基である請求項20又は21に記載のカルボニル化合物。
- 前記Z1、Z2が、それぞれ独立して、CH2=CH−、CH2=C(CH3)−、又は、CH2=C(Cl)−である請求項20〜22のいずれかに記載のカルボニル化合物。
- 請求項20〜23のいずれかに記載のカルボニル化合物と、下記式
(式中、Axは、芳香族炭化水素環及び芳香族複素環からなる群から選ばれる少なくとも一つの芳香環を有する、炭素数2〜30の有機基を表す。
Ayは、水素原子、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、置換基を有していてもよい炭素数1〜20のアルキニル基、置換基を有していてもよい炭素数3〜12のシクロアルキル基、−C(=O)−R3、−SO2−R4、−C(=S)NH−R9、置換基を有していてもよい炭素数2〜30の芳香族炭化水素環基、又は、置換基を有していてもよい炭素数2〜30の芳香族複素環基を表す。ここで、R3は、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、又は、置換基を有していてもよい炭素数3〜12のシクロアルキル基、炭素数5〜12の芳香族炭化水素基を表し、R4は、炭素数1〜20のアルキル基、炭素数2〜20のアルケニル基、フェニル基、又は、4−メチルフェニル基を表し、R9は置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、置換基を有していてもよい炭素数3〜12のシクロアルキル基、置換基を有していてもよい炭素数5〜20の芳香族基を表す。
前記Ax及びAyが有する芳香環は置換基を有していてもよい。)
で示されるヒドラジン化合物を反応させることを特徴とする、下記式(I)
Y1〜Y8はそれぞれ独立して、化学的な単結合、−O−、−S−、−O−C(=O)−、−C(=O)−O−、−O−C(=O)−O−、−NR1−C(=O)−、−C(=O)−NR1−、−O−C(=O)−NR1−、−NR1−C(=O)−O−、−NR1−C(=O)−NR1−、−O−NR1−、又は、−NR1−O−を表す。ここで、R1は、水素原子又は炭素数1〜6のアルキル基を表す。
G1、G2はそれぞれ独立して、置換基を有していてもよい炭素数1〜20の二価の鎖状脂肪族基を表す。前記鎖状脂肪族基には、−O−、−S−、−O−C(=O)−、−C(=O)−O−、−O−C(=O)−O−、−NR2−C(=O)−、−C(=O)−NR2−、−NR2−、又は、−C(=O)−が介在していてもよい。ただし、−O−又は−S−がそれぞれ2以上隣接して介在する場合を除く。ここで、R2は、水素原子又は炭素数1〜6のアルキル基を表す。
Z1、Z2はそれぞれ独立して、無置換又はハロゲン原子で置換された炭素数2〜10のアルケニル基を表す。
A1は、置換基を有していてもよい三価の芳香族基を表す。
A2、A3はそれぞれ独立して、置換基を有していてもよい炭素数3〜30の二価の脂環式炭化水素基を表す。
A4、A5はそれぞれ独立して、置換基を有していてもよい炭素数6〜30の二価の芳香族基を表す。
Q1は、水素原子、又は、置換基を有していてもよい炭素数1〜6のアルキル基を表す。}
で示される重合性化合物の製造方法。 - 前記ヒドラジン化合物と前記カルボニル化合物をモル比で、1:2〜2:1の割合で反応させる、請求項24に記載の重合性化合物の製造方法。
- 請求項20〜23のいずれかに記載のカルボニル化合物を、下記式(I)
Ayは、水素原子、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、置換基を有していてもよい炭素数1〜20のアルキニル基、置換基を有していてもよい炭素数3〜12のシクロアルキル基、−C(=O)−R3、−SO2−R4、−C(=S)NH−R9、置換基を有していてもよい炭素数2〜30の芳香族炭化水素環基、又は、置換基を有していてもよい炭素数2〜30の芳香族複素環基を表す。ここで、R3は、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、又は、置換基を有していてもよい炭素数3〜12のシクロアルキル基、炭素数5〜12の芳香族炭化水素基を表し、R4は、炭素数1〜20のアルキル基、炭素数2〜20のアルケニル基、フェニル基、又は、4−メチルフェニル基を表し、R9は置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数2〜20のアルケニル基、置換基を有していてもよい炭素数3〜12のシクロアルキル基、置換基を有していてもよい炭素数5〜20の芳香族基を表す。前記Ax及びAyが有する芳香環は置換基を有していてもよい。)
Y1〜Y8はそれぞれ独立して、化学的な単結合、−O−、−S−、−O−C(=O)−、−C(=O)−O−、−O−C(=O)−O−、−NR1−C(=O)−、−C(=O)−NR1−、−O−C(=O)−NR1−、−NR1−C(=O)−O−、−NR1−C(=O)−NR1−、−O−NR1−、又は、−NR1−O−を表す。ここで、R1は、水素原子又は炭素数1〜6のアルキル基を表す。
G1、G2はそれぞれ独立して、置換基を有していてもよい炭素数1〜20の二価の鎖状脂肪族基を表す。前記鎖状脂肪族基には、−O−、−S−、−O−C(=O)−、−C(=O)−O−、−O−C(=O)−O−、−NR2−C(=O)−、−C(=O)−NR2−、−NR2−、又は、−C(=O)−が介在していてもよい。ただし、−O−又は−S−がそれぞれ2以上隣接して介在する場合を除く。ここで、R2は、水素原子又は炭素数1〜6のアルキル基を表す。〕。
Z1、Z2はそれぞれ独立して、無置換又はハロゲン原子で置換された炭素数2〜10のアルケニル基を表す。
A1は、置換基を有していてもよい三価の芳香族基を表す。
A2、A3はそれぞれ独立して、置換基を有していてもよい炭素数3〜30の二価の脂環式炭化水素基を表す。
A4、A5はそれぞれ独立して、置換基を有していてもよい炭素数6〜30の二価の芳香族基を表す。
Q1は、水素原子、又は、置換基を有していてもよい炭素数1〜6のアルキル基を表す。}で示される重合性化合物の製造原料として使用する方法。
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012153914 | 2012-07-09 | ||
| JP2012153914 | 2012-07-09 | ||
| JP2012232316 | 2012-10-19 | ||
| JP2012232316 | 2012-10-19 | ||
| JP2013064874 | 2013-03-26 | ||
| JP2013064874 | 2013-03-26 | ||
| JP2013075379 | 2013-03-29 | ||
| JP2013075379 | 2013-03-29 | ||
| PCT/JP2013/065040 WO2014010325A1 (ja) | 2012-07-09 | 2013-05-30 | 重合性化合物、重合性組成物、高分子、光学異方体、及び重合性化合物の製造方法 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015233875A Division JP6137284B2 (ja) | 2012-07-09 | 2015-11-30 | ヒドラジン化合物および光学異方体の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2014010325A1 JPWO2014010325A1 (ja) | 2016-06-20 |
| JP5962760B2 true JP5962760B2 (ja) | 2016-08-03 |
Family
ID=49915799
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014524688A Active JP5962760B2 (ja) | 2012-07-09 | 2013-05-30 | 重合性化合物、重合性組成物、高分子、光学異方体、及び重合性化合物の製造方法 |
| JP2015233875A Active JP6137284B2 (ja) | 2012-07-09 | 2015-11-30 | ヒドラジン化合物および光学異方体の製造方法 |
| JP2017091833A Active JP6439825B2 (ja) | 2012-07-09 | 2017-05-02 | 重合性化合物、重合性組成物、高分子、光学フィルム、光学異方体、フラットパネル表示装置および反射防止フィルム |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015233875A Active JP6137284B2 (ja) | 2012-07-09 | 2015-11-30 | ヒドラジン化合物および光学異方体の製造方法 |
| JP2017091833A Active JP6439825B2 (ja) | 2012-07-09 | 2017-05-02 | 重合性化合物、重合性組成物、高分子、光学フィルム、光学異方体、フラットパネル表示装置および反射防止フィルム |
Country Status (6)
| Country | Link |
|---|---|
| US (4) | US9586917B2 (ja) |
| EP (1) | EP2871192B1 (ja) |
| JP (3) | JP5962760B2 (ja) |
| KR (3) | KR101972064B1 (ja) |
| CN (2) | CN107253935B (ja) |
| WO (1) | WO2014010325A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20200067146A (ko) | 2017-10-13 | 2020-06-11 | 다이니폰 인사츠 가부시키가이샤 | 중합성 액정 화합물, 중합성 조성물, 중합체, 위상차 필름 및 그 제조 방법, 전사용 적층체, 광학 부재 및 그 제조 방법, 그리고 표시 장치 |
| JP2021103288A (ja) * | 2019-12-25 | 2021-07-15 | 住友化学株式会社 | 重合性液晶混合物、重合性液晶組成物 |
Families Citing this family (120)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012141245A1 (ja) * | 2011-04-15 | 2012-10-18 | 日本ゼオン株式会社 | 重合性化合物、重合性組成物、高分子、及び光学異方体 |
| KR101985943B1 (ko) * | 2011-04-27 | 2019-06-04 | 제온 코포레이션 | 중합성 화합물, 중합성 조성물, 고분자, 및 광학 이방체 |
| CN104470957B (zh) | 2012-05-30 | 2016-11-16 | 日本瑞翁株式会社 | 聚合性化合物、聚合性组合物、高分子、以及光学各向异性体 |
| US10227292B2 (en) | 2013-02-15 | 2019-03-12 | Zeon Corporation | Polymerizable compound, polymerizable composition, polymer, and optically anisotropic body |
| US10273322B2 (en) * | 2013-08-22 | 2019-04-30 | Zeon Corporation | Polymerizable compound, polymerizable composition, polymer, and optical anisotropic body |
| JP6427340B2 (ja) | 2013-09-11 | 2018-11-21 | 富士フイルム株式会社 | 光学異方性層とその製造方法、積層体とその製造方法、偏光板、液晶表示装置及び有機el表示装置 |
| KR102315630B1 (ko) | 2013-10-31 | 2021-10-20 | 제온 코포레이션 | 중합성 화합물, 중합성 조성물, 고분자, 및 광학 이방체 |
| JP6221771B2 (ja) * | 2014-01-27 | 2017-11-01 | 日本ゼオン株式会社 | エーテル化合物の製造方法、および重合性化合物の製造方法 |
| EP3106456B1 (en) * | 2014-02-12 | 2018-10-24 | Zeon Corporation | Polymerizable compound, polymerizable composition, polymer, and optical isomer |
| EP3106478B1 (en) | 2014-02-14 | 2019-12-25 | Zeon Corporation | Polymerizable compound, polymerizable composition, polymer, and optically anisotropic body |
| WO2015129654A1 (ja) * | 2014-02-28 | 2015-09-03 | 日本ゼオン株式会社 | 1,1-ジ置換ヒドラジン化合物の製造方法 |
| WO2015141784A1 (ja) * | 2014-03-19 | 2015-09-24 | 日本ゼオン株式会社 | 重合性化合物の製造方法 |
| JP6080884B2 (ja) | 2014-03-28 | 2017-02-15 | 富士フイルム株式会社 | 重合性化合物、ポリマー、重合性組成物、フィルム、および投映像表示用ハーフミラー |
| WO2015147243A1 (ja) * | 2014-03-28 | 2015-10-01 | 富士フイルム株式会社 | 重合性化合物、ポリマー、重合性組成物、フィルム、および投映像表示用ハーフミラー |
| JP6047604B2 (ja) * | 2014-03-31 | 2016-12-21 | 富士フイルム株式会社 | 液晶化合物および光学フィルム、ならびに光学フィルムの製造方法 |
| US10538477B2 (en) | 2014-06-30 | 2020-01-21 | Zeon Corporation | Production intermediate of polymerizable compound, production method for same, composition, and stabilization method |
| CN106573870B (zh) | 2014-09-05 | 2020-01-10 | 富士胶片株式会社 | 聚合性化合物、聚合物、聚合性组合物及薄膜 |
| WO2016047648A1 (ja) | 2014-09-25 | 2016-03-31 | 富士フイルム株式会社 | 重合性化合物を含む重合性組成物、フィルム、および投映像表示用ハーフミラー |
| KR20170068529A (ko) | 2014-10-09 | 2017-06-19 | 디아이씨 가부시끼가이샤 | 중합성 화합물 및 광학 이방체 |
| CN105524625B (zh) * | 2014-10-21 | 2020-10-09 | 富士胶片株式会社 | 光学各向异性层及其制造方法、层叠体、偏振片、显示装置、液晶化合物及其制造方法、羧酸化合物 |
| JP6476783B2 (ja) * | 2014-11-18 | 2019-03-06 | 日本ゼオン株式会社 | 重合性液晶組成物、高分子、光学異方体、及び偏光板 |
| KR102624959B1 (ko) * | 2014-12-04 | 2024-01-16 | 디아이씨 가부시끼가이샤 | 중합성 화합물, 조성물, 중합체, 광학 이방체, 액정 표시 소자 및 유기 el 소자 |
| US11261378B2 (en) | 2014-12-25 | 2022-03-01 | Dic Corporation | Polymerizable compound and optically anisotropic object |
| EP3246340B1 (en) * | 2015-01-16 | 2019-10-09 | DIC Corporation | Polymerizable composition and optically anisotropic body using same |
| US11186669B2 (en) | 2015-01-16 | 2021-11-30 | Dic Corporation | Polymerizable composition and optically anisotropic body using same |
| CN107108488A (zh) * | 2015-01-16 | 2017-08-29 | Dic株式会社 | 聚合性化合物和光学各向异性体 |
| WO2016114348A1 (ja) * | 2015-01-16 | 2016-07-21 | Dic株式会社 | 重合性組成物及び光学異方体 |
| JP6066252B2 (ja) * | 2015-01-16 | 2017-01-25 | Dic株式会社 | 重合性化合物及び光学異方体 |
| CN107108775B (zh) | 2015-01-16 | 2019-12-13 | Dic株式会社 | 聚合性组合物和使用该聚合性组合物的光学各向异性体 |
| US20180016502A1 (en) * | 2015-01-16 | 2018-01-18 | Dic Corporation | Polymerizable composition and optically anisotropic body using same |
| CN107207419B (zh) * | 2015-02-24 | 2021-03-09 | Dic株式会社 | 聚合性化合物和光学各向异性体 |
| KR20160106513A (ko) * | 2015-03-02 | 2016-09-12 | 제이엔씨 주식회사 | 중합성 액정 조성물 및 광학 이방성 필름 |
| WO2016143883A1 (ja) | 2015-03-12 | 2016-09-15 | 富士フイルム株式会社 | 重合性組成物、フィルム、および投映像表示用ハーフミラー |
| JP6669160B2 (ja) * | 2015-03-19 | 2020-03-18 | 日本ゼオン株式会社 | 液晶性組成物、位相差層の製造方法及び円偏光板 |
| EP3279181B1 (en) * | 2015-03-31 | 2021-08-04 | Zeon Corporation | Method for producing a mixture of a polymerizable compound |
| JP6618699B2 (ja) * | 2015-03-31 | 2019-12-11 | 日本ゼオン株式会社 | 1,1−ジ置換ヒドラジン化合物の製造方法 |
| JP2016190828A (ja) * | 2015-03-31 | 2016-11-10 | 日本ゼオン株式会社 | 重合性化合物の製造方法 |
| KR20170139535A (ko) * | 2015-04-24 | 2017-12-19 | 니폰 제온 가부시키가이샤 | 복층 필름의 제조 방법 및 복층 필름 |
| CN107533183B (zh) * | 2015-05-28 | 2020-09-25 | 日本瑞翁株式会社 | 圆偏振光分离膜及其制造方法 |
| WO2016194999A1 (ja) | 2015-06-03 | 2016-12-08 | 富士フイルム株式会社 | 光学フィルム、偏光板および画像表示装置 |
| WO2017038265A1 (ja) * | 2015-09-01 | 2017-03-09 | Dic株式会社 | 粉体混合物 |
| CN107924017A (zh) | 2015-09-03 | 2018-04-17 | Dic株式会社 | 包含具有介晶基团的化合物的组合物、将聚合性组合物聚合而得的聚合物、光学各向异性体及相位差膜 |
| US20180327668A1 (en) * | 2015-09-03 | 2018-11-15 | Dic Corporation | Compound containing mesogenic group and composition containing the compound, and polymer obtained by polymerizing polyermizable composition, optically anisotropic body, and phase difference film |
| WO2017057005A1 (ja) * | 2015-09-30 | 2017-04-06 | 日本ゼオン株式会社 | 光学フィルム及びその製造方法 |
| JP6718464B2 (ja) * | 2015-09-30 | 2020-07-08 | 富士フイルム株式会社 | 光学フィルム、偏光板および画像表示装置 |
| EP3357902A4 (en) * | 2015-10-02 | 2019-04-10 | Zeon Corporation | METHOD FOR PRODUCING AN ACID HALOGENIDE SOLUTION, MIXTURING SOLUTION AND METHOD FOR PRODUCING A MONOESTER COMPOUND |
| CN108137486B (zh) | 2015-10-23 | 2019-08-13 | Dic株式会社 | 聚合性化合物和光学各向异性体 |
| US10919870B2 (en) * | 2015-11-09 | 2021-02-16 | Dic Corporation | Polymerizable compound and optically anisotropic body |
| US20170145312A1 (en) * | 2015-11-25 | 2017-05-25 | Sumitomo Chemical Company, Limited | Liquid crystal composition |
| WO2017090644A1 (ja) | 2015-11-26 | 2017-06-01 | 富士フイルム株式会社 | 光学フィルム、偏光板、画像表示装置および重合性化合物ならびに1,4-シクロヘキサンジカルボン酸モノアリールエステルの製造方法 |
| WO2017098952A1 (ja) * | 2015-12-07 | 2017-06-15 | Dic株式会社 | 重合性化合物の製造方法 |
| US11046889B2 (en) | 2015-12-08 | 2021-06-29 | Dic Corporation | Polymerizable compound and optically anisotropic body |
| US10647920B2 (en) * | 2015-12-22 | 2020-05-12 | Zeon Corporation | Liquid crystalline composition, liquid crystal cured layer, method for producing same, and optical film |
| US20180370184A1 (en) * | 2015-12-25 | 2018-12-27 | Zeon Corporation | Optical anisotropic layer and manufacturing method therefor, optical anisotropic laminate, and circularly polarizing plate |
| WO2017130871A1 (ja) | 2016-01-26 | 2017-08-03 | 日本ゼオン株式会社 | 酸ハライド溶液の製造方法、及びモノエステル化合物の製造方法 |
| KR20180118133A (ko) * | 2016-03-08 | 2018-10-30 | 니폰 제온 가부시키가이샤 | 액정성 조성물, 액정 경화층 및 그 액정 경화층의 제조 방법 |
| WO2017154588A1 (ja) * | 2016-03-10 | 2017-09-14 | Dic株式会社 | エステル基を有する化合物の製造方法 |
| US10921672B2 (en) | 2016-03-23 | 2021-02-16 | Guardian Glass, LLC | Low haze switchable window |
| CN108495847B (zh) * | 2016-03-30 | 2022-07-01 | Dic株式会社 | 2-肼基苯并噻唑衍生物的制造方法 |
| JP6766870B2 (ja) * | 2016-03-30 | 2020-10-14 | 日本ゼオン株式会社 | 光学異方性層及びその製造方法、光学異方性積層体及びその製造方法、光学異方性転写体、偏光板、並びに画像表示装置 |
| EP3438713A4 (en) * | 2016-03-30 | 2019-11-20 | Zeon Corporation | OPTICALLY ANISOTROPIC LAMINATE, CIRCULAR POLARIZATION BLADE, AND IMAGE DISPLAY DEVICE |
| JP6055569B1 (ja) * | 2016-05-18 | 2016-12-27 | 日本ゼオン株式会社 | 重合性化合物、混合物、重合性液晶組成物、高分子、光学フィルム、光学異方体、偏光板、フラットパネル表示装置、有機エレクトロルミネッセンス表示装置および反射防止フィルム |
| JP6090514B1 (ja) | 2016-05-18 | 2017-03-08 | 日本ゼオン株式会社 | 重合性化合物の製造方法 |
| CN109477926B (zh) | 2016-08-08 | 2021-03-19 | 日本瑞翁株式会社 | 光学各向异性层叠体、偏振片及图像显示装置 |
| JP6146526B1 (ja) | 2016-10-06 | 2017-06-14 | 日本ゼオン株式会社 | 混合物、重合性組成物、高分子、光学フィルム、光学異方体、偏光板、フラットパネル表示装置、有機エレクトロルミネッセンス表示装置および反射防止フィルム、並びに重合性化合物の使用方法 |
| JP6191754B1 (ja) | 2016-11-22 | 2017-09-06 | 日本ゼオン株式会社 | 重合性化合物、混合物、重合性液晶組成物、高分子、光学フィルム、光学異方体、偏光板、表示装置および反射防止フィルム |
| KR20190078591A (ko) | 2016-11-22 | 2019-07-04 | 니폰 제온 가부시키가이샤 | 중합성 화합물, 중합성 조성물, 고분자, 광학 필름, 광학 이방체, 편광판, 플랫 패널 표시 장치, 유기 일렉트로루미네센스 표시 장치, 반사 방지 필름, 및 화합물 |
| CN110023800B (zh) | 2016-11-29 | 2021-06-15 | 富士胶片株式会社 | 聚合性液晶组合物、光学各向异性膜、光学膜、偏振片、图像显示装置及有机电致发光显示装置 |
| KR102210179B1 (ko) | 2016-11-29 | 2021-01-29 | 후지필름 가부시키가이샤 | 중합성 액정 조성물, 광학 이방성막, 광학 필름, 편광판, 화상 표시 장치 및 유기 일렉트로 루미네선스 표시 장치 |
| KR20190097027A (ko) | 2016-12-26 | 2019-08-20 | 니폰 제온 가부시키가이샤 | 중합성 화합물, 혼합물, 고분자, 광학 필름, 광학 이방체, 편광판, 표시 장치 및 반사 방지 필름 |
| WO2018123622A1 (ja) | 2016-12-26 | 2018-07-05 | 日本ゼオン株式会社 | 混合物、高分子、光学フィルム、光学異方体、偏光板、表示装置および反射防止フィルム、並びに、混合物の製造方法 |
| JP7067486B2 (ja) * | 2016-12-27 | 2022-05-16 | 日本ゼオン株式会社 | 重合性化合物、重合性液晶混合物、高分子、光学フィルム、光学異方体、偏光板、表示装置、反射防止フィルム、および化合物 |
| JP6880070B2 (ja) * | 2016-12-28 | 2021-06-02 | 富士フイルム株式会社 | 光学フィルムおよびその製造方法、偏光板、画像表示装置 |
| JP6418476B1 (ja) * | 2017-01-06 | 2018-11-07 | Dic株式会社 | 重合性化合物及び光学異方体 |
| JPWO2018155498A1 (ja) * | 2017-02-21 | 2019-11-14 | 富士フイルム株式会社 | 重合性液晶化合物、重合性液晶化合物の製造方法、重合性液晶組成物、光学異方性膜、光学フィルム、偏光板および画像表示装置 |
| EP3597643A4 (en) | 2017-03-17 | 2020-08-12 | Zeon Corporation | POLYMERIZABLE COMPOUND, POLYMERIZABLE LIQUID CRYSTAL MIXTURE, POLYMER, OPTICAL FILM, OPTICALLY ANISOTROPIC BODY, POLARIZING PLATE, DISPLAY DEVICE, ANTI-REFLECTIVE FILM AND COMPOUND |
| CN110392703B (zh) * | 2017-03-23 | 2022-02-01 | 日本瑞翁株式会社 | 聚合性化合物及其制造方法、聚合性组合物、高分子、光学膜、光学各向异性体 |
| US20200283399A1 (en) | 2017-03-27 | 2020-09-10 | Zeon Corporation | Method of producing polymerizable compound, and solution of polymerizable compound |
| JP6369594B2 (ja) * | 2017-04-27 | 2018-08-08 | 日本ゼオン株式会社 | 化合物、重合性化合物、混合物、重合性液晶組成物、高分子、光学フィルム、光学異方体、偏光板、フラットパネル表示装置、有機エレクトロルミネッセンス表示装置および反射防止フィルム |
| CN110891946B (zh) | 2017-07-19 | 2023-03-24 | 富士胶片株式会社 | 聚合性液晶化合物、聚合性液晶组合物、光学各向异性膜、光学膜、偏振片及图像显示装置 |
| WO2019017445A1 (ja) * | 2017-07-19 | 2019-01-24 | 富士フイルム株式会社 | 重合性液晶化合物、重合性液晶組成物、光学異方性膜、光学フィルム、偏光板および画像表示装置 |
| US11939510B2 (en) | 2017-08-15 | 2024-03-26 | Merck Patent Gmbh | Polymerisable liquid crystal material and polymerised liquid crystal film |
| US20200362245A1 (en) * | 2017-08-15 | 2020-11-19 | Merck Patent Gmbh | Polymerisable lc medium and polymer film with flat optical dispersion |
| US11492552B2 (en) | 2017-08-23 | 2022-11-08 | Zeon Corporation | Polymerizable liquid crystal material, polymerizable liquid crystal composition, polymer, optical film, optically anisotropic body, polarizing plate, anti-reflection film, display device, and method of producing polymerizable liquid crystal composition |
| TWI742155B (zh) * | 2017-09-08 | 2021-10-11 | 日商迪愛生股份有限公司 | 具有酯基之化合物之製造方法、其化合物及衍生物、以及使用該等之組成物、聚合物、光學各向異性體、顯示元件 |
| TWI828609B (zh) * | 2017-09-13 | 2024-01-11 | 日商迪愛生股份有限公司 | 2-肼基苯并噻唑衍生物之製造方法、從該衍生物衍生之化合物、組成物、聚合物、光學各向異性體及樹脂 |
| JP2019056069A (ja) * | 2017-09-21 | 2019-04-11 | 日本ゼオン株式会社 | 重合性液晶材料、重合性液晶組成物、高分子、光学フィルム、光学異方体、偏光板、反射防止フィルム、表示装置、並びに、重合性液晶組成物の製造方法 |
| KR102513064B1 (ko) | 2017-12-07 | 2023-03-22 | 후지필름 가부시키가이샤 | 장척 위상차 필름, 장척 적층체, 화상 표시 장치 |
| JP7293651B2 (ja) * | 2017-12-27 | 2023-06-20 | 大日本印刷株式会社 | 重合性液晶化合物、重合性組成物、重合体、位相差フィルム及びその製造方法、転写用積層体、光学部材及びその製造方法、並びに表示装置 |
| CN111655672B (zh) * | 2018-02-05 | 2023-05-12 | 日本瑞翁株式会社 | 1,1-二取代肼化合物的制造方法及聚合性化合物的制造方法 |
| KR102438545B1 (ko) | 2018-02-14 | 2022-08-30 | 후지필름 가부시키가이샤 | 혼정, 중합성 액정 조성물, 광학 이방성막, 광학 필름, 편광판 및 화상 표시 장치 |
| KR102351458B1 (ko) | 2018-02-14 | 2022-01-13 | 후지필름 가부시키가이샤 | 중합성 액정 조성물, 광학 이방성막, 광학 필름, 편광판 및 화상 표시 장치 |
| KR102442993B1 (ko) | 2018-02-14 | 2022-09-13 | 후지필름 가부시키가이샤 | 화상 표시 장치 및 감광성 접착제 포함 원 편광판 |
| WO2019160044A1 (ja) | 2018-02-14 | 2019-08-22 | 富士フイルム株式会社 | 光学異方性膜、光学フィルム、偏光板および画像表示装置 |
| JP7068436B2 (ja) | 2018-02-14 | 2022-05-16 | 富士フイルム株式会社 | 光学フィルム、偏光板および画像表示装置 |
| KR102426523B1 (ko) | 2018-02-14 | 2022-07-27 | 후지필름 가부시키가이샤 | 중합성 액정 조성물, 광학 이방성막, 광학 필름, 편광판 및 화상 표시 장치 |
| JP6916949B2 (ja) | 2018-02-14 | 2021-08-11 | 富士フイルム株式会社 | 光学フィルム、偏光板、画像表示装置 |
| WO2019160025A1 (ja) | 2018-02-14 | 2019-08-22 | 富士フイルム株式会社 | 重合性液晶組成物、重合性液晶組成物の製造方法、光学異方性膜、光学フィルム、偏光板および画像表示装置 |
| CN111727206B (zh) * | 2018-02-21 | 2022-06-14 | 富士胶片株式会社 | 聚合性液晶组合物、光学各向异性膜、光学膜、偏振片及图像显示装置 |
| WO2019167926A1 (ja) | 2018-02-28 | 2019-09-06 | 富士フイルム株式会社 | 積層体、有機電界発光装置、液晶表示装置 |
| CN111936897B (zh) | 2018-03-23 | 2022-09-06 | 富士胶片株式会社 | 胆甾醇型液晶层及其制造方法、层叠体、光学各向异性体、反射膜、防伪介质及判定方法 |
| CN111902749B (zh) | 2018-03-23 | 2022-09-20 | 富士胶片株式会社 | 胆甾醇型液晶层的制造方法、胆甾醇型液晶层、液晶组合物、固化物、光学各向异性体、反射层 |
| WO2019188495A1 (ja) * | 2018-03-30 | 2019-10-03 | 日本ゼオン株式会社 | 光学異方体及びその製造方法、1/4波長板、偏光板及び有機エレクトロルミネッセンス表示パネル |
| JP7081660B2 (ja) * | 2018-03-30 | 2022-06-07 | 日本ゼオン株式会社 | 液晶硬化フィルム及びその製造方法、第一硬化層、偏光板、並びに有機エレクトロルミネッセンス表示装置 |
| CN108640833A (zh) * | 2018-04-13 | 2018-10-12 | 上海皓元医药股份有限公司 | 4-(6-(丙烯酰氧基)已氧基)苯酚及其系列化合物的制备方法 |
| JP7225736B2 (ja) * | 2018-05-15 | 2023-02-21 | Jnc株式会社 | 化合物、液晶組成物、および液晶表示素子 |
| WO2019230848A1 (ja) * | 2018-06-01 | 2019-12-05 | 日本ゼオン株式会社 | 重合性化合物の製造方法 |
| JP7118153B2 (ja) * | 2018-07-25 | 2022-08-15 | 富士フイルム株式会社 | 重合性液晶組成物、光学異方性膜、光学フィルム、偏光板および画像表示装置 |
| JP7145955B2 (ja) | 2018-09-04 | 2022-10-03 | 富士フイルム株式会社 | 積層体、有機電界発光装置、液晶表示装置 |
| JPWO2020059768A1 (ja) * | 2018-09-21 | 2021-09-24 | 日本ゼオン株式会社 | 化合物およびその使用方法 |
| JP7251197B2 (ja) * | 2019-02-15 | 2023-04-04 | 大日本印刷株式会社 | 重合性液晶化合物、重合性組成物、重合体、位相差フィルム及びその製造方法、転写用積層体、光学部材及びその製造方法、並びに表示装置 |
| WO2021060428A1 (ja) | 2019-09-27 | 2021-04-01 | 富士フイルム株式会社 | 重合性液晶組成物、化合物、光学異方性膜、光学フィルム、偏光板および画像表示装置 |
| KR20220050169A (ko) | 2019-09-27 | 2022-04-22 | 후지필름 가부시키가이샤 | 중합성 액정 조성물, 광학 이방성막, 광학 필름, 편광판 및 화상 표시 장치 |
| WO2021124803A1 (ja) * | 2019-12-17 | 2021-06-24 | 富士フイルム株式会社 | 有機エレクトロルミネッセンス表示装置 |
| CN115280199A (zh) * | 2020-03-11 | 2022-11-01 | 住友化学株式会社 | 聚合性液晶组合物、相位差膜、椭圆偏光板及光学显示器 |
| EP4039776A3 (en) | 2020-11-20 | 2022-08-24 | Merck Patent GmbH | Polymerisable lc material and polymer film |
| JP2023031737A (ja) | 2021-08-25 | 2023-03-09 | 富士フイルム株式会社 | 液晶組成物、液晶硬化層、光学フィルム、偏光板および画像表示装置 |
| WO2023061903A1 (en) | 2021-10-11 | 2023-04-20 | Merck Patent Gmbh | Polymerisable compound, polymerisable lc material and polymer film |
| WO2023237572A1 (en) | 2022-06-10 | 2023-12-14 | Merck Patent Gmbh | Polymerisable liquid crystal medium and polymerised liquid crystal film |
| WO2024061796A1 (en) | 2022-09-21 | 2024-03-28 | Merck Patent Gmbh | Polymerisable liquid crystal medium and polymerised liquid crystal film |
Family Cites Families (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5512427B2 (ja) * | 1971-12-29 | 1980-04-02 | ||
| GB2116964B (en) * | 1981-10-16 | 1985-07-17 | Abbott Lab | (1-(2-benzoxazolyl)hydrazinojalkyl nitrile derivatives |
| DE3340932A1 (de) | 1983-11-11 | 1985-05-23 | Bayer Ag, 5090 Leverkusen | Verfahren zur herstellung von teilweise bekannten 2-hydrazino-benzimidazol-derivaten |
| JPH0619580B2 (ja) * | 1984-03-29 | 1994-03-16 | 東洋インキ製造株式会社 | 電子写真感光体 |
| JPS61296358A (ja) * | 1985-06-26 | 1986-12-27 | Toshiba Corp | 電子写真感光体 |
| JPS6217750A (ja) * | 1985-07-16 | 1987-01-26 | Toshiba Corp | 電子写真感光体 |
| DE3533331A1 (de) | 1985-09-18 | 1987-03-26 | Heumann Ludwig & Co Gmbh | Pyridothiazolderivate, verfahren zu ihrer herstellung und diese verbindungen enthaltende arzneimittel |
| EP0597128B1 (en) * | 1992-05-01 | 1999-09-15 | Idemitsu Kosan Company Limited | Ester polymer, production thereof, and electrophotographic photoreceptor made therefrom |
| FR2710644B1 (fr) * | 1993-10-01 | 1995-12-22 | Innothera Lab Sa | Dérivés d'indane cétoniques et de leurs analogues hétérocycliques et leur utilisation thérapeutique. |
| JP3579914B2 (ja) * | 1994-04-22 | 2004-10-20 | 大日本インキ化学工業株式会社 | 光学異方性を有する基板 |
| DE69419120T2 (de) | 1993-12-24 | 1999-10-28 | Dainippon Ink & Chemicals | Polymerisierbare Flüssigkristallzusammensetzung und optisch anisotroper Film, der eine solche Zusammensetzung enthält |
| US5567349A (en) | 1994-03-30 | 1996-10-22 | Hoffmann-La Roche Inc. | Photo cross-linkable liquid crystals |
| US6139771A (en) | 1997-04-04 | 2000-10-31 | Displaytech, Inc. | Mesogenic materials with anomalous birefringence dispersion and high second order susceptibility (X.sup.(2)). |
| JPH1090521A (ja) | 1996-07-24 | 1998-04-10 | Sumitomo Chem Co Ltd | 偏光軸回転積層位相差板およびこれを用いた投射型液晶表示装置 |
| JPH1068816A (ja) | 1996-08-29 | 1998-03-10 | Sharp Corp | 位相差板及び円偏光板 |
| JPH1152131A (ja) | 1997-08-01 | 1999-02-26 | Sumitomo Bakelite Co Ltd | 位相差板及びそれを用いた偏光素子 |
| EP1045261B1 (en) | 1998-10-30 | 2005-02-02 | Teijin Limited | Phase difference film and optical device using it |
| US6400433B1 (en) | 1998-11-06 | 2002-06-04 | Fuji Photo Film Co., Ltd. | Circularly polarizing plate comprising linearly polarizing membrane and quarter wave plate |
| JP3734211B2 (ja) | 1999-01-27 | 2006-01-11 | 富士写真フイルム株式会社 | 位相差板、円偏光板および反射型液晶表示装置 |
| DE19855757A1 (de) | 1998-12-03 | 2000-06-21 | Merck Patent Gmbh | Querverbrückte Cyclohexan-Derivate und flüssigkristallines Medium |
| JP2001004837A (ja) | 1999-06-22 | 2001-01-12 | Fuji Photo Film Co Ltd | 位相差板および円偏光板 |
| JP4666742B2 (ja) | 1999-12-14 | 2011-04-06 | 株式会社林原生物化学研究所 | 光吸収材とその用途 |
| JP4320125B2 (ja) | 2001-03-06 | 2009-08-26 | 富士フイルム株式会社 | 位相差膜 |
| JP4074155B2 (ja) | 2001-09-17 | 2008-04-09 | 富士フイルム株式会社 | 四員環化合物、それを用いた複屈折媒体および光学部材 |
| JP2005208416A (ja) | 2004-01-23 | 2005-08-04 | Nitto Denko Corp | 逆波長分散位相差フィルム、それを用いた偏光板及びディスプレイ装置 |
| JP2005208415A (ja) | 2004-01-23 | 2005-08-04 | Nitto Denko Corp | 逆波長分散位相差フィルム、それを用いた偏光板及びディスプレイ装置 |
| JP2005208414A (ja) | 2004-01-23 | 2005-08-04 | Nitto Denko Corp | 逆波長分散位相差フィルム、それを用いた偏光板及びディスプレイ装置 |
| JP4606195B2 (ja) | 2004-03-08 | 2011-01-05 | 富士フイルム株式会社 | 液晶化合物、液晶組成物、重合体、位相差板、及び楕円偏光板 |
| JP2005336103A (ja) | 2004-05-27 | 2005-12-08 | Fuji Photo Film Co Ltd | フェニルヒドラジン類の製造方法 |
| JP2005345781A (ja) * | 2004-06-03 | 2005-12-15 | Canon Inc | 電子写真感光体 |
| JP4186980B2 (ja) | 2004-11-11 | 2008-11-26 | 住友化学株式会社 | 光学フィルム |
| DE112005002754T5 (de) | 2004-11-11 | 2007-09-20 | Sumitomo Chemical Co., Ltd. | Optischer Film |
| WO2006118073A1 (ja) | 2005-04-28 | 2006-11-09 | Sumitomo Chemical Company, Limited | フィルム及びその製造方法 |
| JP5088769B2 (ja) | 2005-04-28 | 2012-12-05 | 住友化学株式会社 | フィルム及びその製造方法 |
| JP5401032B2 (ja) * | 2006-12-15 | 2014-01-29 | 富士フイルム株式会社 | 光学異方性膜、輝度向上フィルム、位相差板および液晶表示装置 |
| JP4283854B2 (ja) * | 2007-01-29 | 2009-06-24 | シャープ株式会社 | 芳香族ポリカーボネート、電子写真感光体および画像形成装置 |
| KR20100014882A (ko) * | 2007-03-01 | 2010-02-11 | 제온 코포레이션 | 중합성 액정 화합물, 중합성 액정 조성물, 액정 중합체 및 광학 이방체 |
| CN101687778B (zh) * | 2007-04-24 | 2013-09-25 | 日本瑞翁株式会社 | 聚合性液晶化合物、聚合性液晶组合物、液晶高分子及光学各向异性体 |
| JP5401822B2 (ja) * | 2007-04-24 | 2014-01-29 | 日本ゼオン株式会社 | 重合性液晶化合物、重合性液晶組成物、液晶性高分子および光学異方体 |
| EP2183337B1 (en) | 2007-09-03 | 2011-07-20 | Merck Patent GmbH | Fluorene derivatives |
| CN101796163B (zh) | 2007-09-03 | 2014-06-18 | 默克专利股份有限公司 | 棒状介晶化合物 |
| WO2009042542A1 (en) | 2007-09-26 | 2009-04-02 | Indiana University Research And Technology Corporation | Benzoquinone derivative e3330 in combination with chemotherapeutic agents for the treatment of cancer and angiogenesis |
| JP5453798B2 (ja) | 2007-12-28 | 2014-03-26 | 住友化学株式会社 | 化合物、光学フィルムおよび光学フィルムの製造方法 |
| JP5391682B2 (ja) | 2007-12-28 | 2014-01-15 | 住友化学株式会社 | 化合物、光学フィルム及び光学フィルムの製造方法 |
| JP5373293B2 (ja) | 2008-01-29 | 2013-12-18 | 富士フイルム株式会社 | 化合物、液晶組成物及び異方性材料 |
| JP2009276442A (ja) * | 2008-05-13 | 2009-11-26 | Konica Minolta Opto Inc | 1/4波長板、画像表示装置および液晶表示装置 |
| JP2009274984A (ja) | 2008-05-14 | 2009-11-26 | Sumitomo Chemical Co Ltd | 化合物、光学フィルムおよび光学フィルムの製造方法 |
| JP2010001284A (ja) * | 2008-05-20 | 2010-01-07 | Sumitomo Chemical Co Ltd | 化合物及び光学フィルム |
| JP5564773B2 (ja) * | 2008-09-19 | 2014-08-06 | 日本ゼオン株式会社 | 重合性液晶化合物、重合性液晶組成物、液晶性高分子及び光学異方体 |
| JP5453956B2 (ja) | 2009-06-26 | 2014-03-26 | 住友化学株式会社 | 化合物、光学フィルム及び光学フィルムの製造方法 |
| JP2011006360A (ja) | 2009-06-26 | 2011-01-13 | Sumitomo Chemical Co Ltd | 化合物、光学フィルム及び光学フィルムの製造方法 |
| JP5556078B2 (ja) * | 2009-07-30 | 2014-07-23 | 日本ゼオン株式会社 | キラル化合物の製造方法および製造中間体 |
| JP2011042606A (ja) | 2009-08-20 | 2011-03-03 | Sumitomo Chemical Co Ltd | 化合物、光学フィルム及び光学フィルムの製造方法 |
| JP5652011B2 (ja) * | 2010-06-10 | 2015-01-14 | 住友化学株式会社 | 光学フィルム |
| WO2012141245A1 (ja) | 2011-04-15 | 2012-10-18 | 日本ゼオン株式会社 | 重合性化合物、重合性組成物、高分子、及び光学異方体 |
| KR101985943B1 (ko) * | 2011-04-27 | 2019-06-04 | 제온 코포레이션 | 중합성 화합물, 중합성 조성물, 고분자, 및 광학 이방체 |
| US9150677B2 (en) * | 2011-06-24 | 2015-10-06 | Zeon Corporation | Polymerizable compounds, polymerizable composition, polymer, and optically anisotropic body |
| JP5804814B2 (ja) * | 2011-07-20 | 2015-11-04 | 富士フイルム株式会社 | 化合物、ヘイズ低下剤、液晶組成物、高分子材料およびフィルム |
| WO2013018526A1 (ja) * | 2011-07-29 | 2013-02-07 | 日本ゼオン株式会社 | 光学異方体の波長分散調整方法及び重合性組成物 |
| JP6123673B2 (ja) * | 2011-09-27 | 2017-05-10 | 日本ゼオン株式会社 | 重合性化合物の製造中間体及びその製造方法 |
| WO2013146633A1 (ja) * | 2012-03-30 | 2013-10-03 | 日本ゼオン株式会社 | 位相差フィルム積層体およびその製造方法、ならびに液晶表示装置 |
| JP5880226B2 (ja) * | 2012-04-03 | 2016-03-08 | 日本ゼオン株式会社 | 重合性化合物の製造方法 |
| CN104470957B (zh) * | 2012-05-30 | 2016-11-16 | 日本瑞翁株式会社 | 聚合性化合物、聚合性组合物、高分子、以及光学各向异性体 |
| CN104755512B (zh) * | 2012-10-19 | 2016-05-18 | 日本瑞翁株式会社 | 聚合性化合物、聚合性组合物、高分子以及光学各向异性体 |
| JP6476862B2 (ja) * | 2012-10-22 | 2019-03-06 | 日本ゼオン株式会社 | 位相差板、円偏光板、及び画像表示装置 |
| US9777096B2 (en) * | 2012-10-23 | 2017-10-03 | Zeon Corporation | Polymerizable compound, polymerizable composition, polymer, and optical anisotropic body |
| JP6270812B2 (ja) | 2013-02-28 | 2018-01-31 | 富士フイルム株式会社 | 位相差板、反射防止板、画像表示装置、および位相差板の製造方法 |
| KR102315630B1 (ko) * | 2013-10-31 | 2021-10-20 | 제온 코포레이션 | 중합성 화합물, 중합성 조성물, 고분자, 및 광학 이방체 |
| WO2015141784A1 (ja) * | 2014-03-19 | 2015-09-24 | 日本ゼオン株式会社 | 重合性化合物の製造方法 |
| JP6047604B2 (ja) * | 2014-03-31 | 2016-12-21 | 富士フイルム株式会社 | 液晶化合物および光学フィルム、ならびに光学フィルムの製造方法 |
| KR20170068529A (ko) * | 2014-10-09 | 2017-06-19 | 디아이씨 가부시끼가이샤 | 중합성 화합물 및 광학 이방체 |
| CN105524625B (zh) * | 2014-10-21 | 2020-10-09 | 富士胶片株式会社 | 光学各向异性层及其制造方法、层叠体、偏振片、显示装置、液晶化合物及其制造方法、羧酸化合物 |
| KR102624959B1 (ko) * | 2014-12-04 | 2024-01-16 | 디아이씨 가부시끼가이샤 | 중합성 화합물, 조성물, 중합체, 광학 이방체, 액정 표시 소자 및 유기 el 소자 |
-
2013
- 2013-05-30 CN CN201710233080.9A patent/CN107253935B/zh active Active
- 2013-05-30 KR KR1020157000771A patent/KR101972064B1/ko active IP Right Grant
- 2013-05-30 JP JP2014524688A patent/JP5962760B2/ja active Active
- 2013-05-30 US US14/413,787 patent/US9586917B2/en active Active
- 2013-05-30 KR KR1020197011175A patent/KR102094007B1/ko active IP Right Grant
- 2013-05-30 EP EP13816607.9A patent/EP2871192B1/en active Active
- 2013-05-30 WO PCT/JP2013/065040 patent/WO2014010325A1/ja active Application Filing
- 2013-05-30 CN CN201380046851.3A patent/CN104603165B/zh active Active
- 2013-05-30 KR KR1020207008292A patent/KR102212172B1/ko active IP Right Grant
-
2015
- 2015-11-30 JP JP2015233875A patent/JP6137284B2/ja active Active
-
2016
- 2016-06-10 US US15/179,145 patent/US10173992B2/en active Active
-
2017
- 2017-05-02 JP JP2017091833A patent/JP6439825B2/ja active Active
-
2018
- 2018-10-31 US US16/176,325 patent/US10487065B2/en not_active Expired - Fee Related
-
2019
- 2019-10-08 US US16/595,538 patent/US11091452B2/en active Active
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20200067146A (ko) | 2017-10-13 | 2020-06-11 | 다이니폰 인사츠 가부시키가이샤 | 중합성 액정 화합물, 중합성 조성물, 중합체, 위상차 필름 및 그 제조 방법, 전사용 적층체, 광학 부재 및 그 제조 방법, 그리고 표시 장치 |
| US11560518B2 (en) | 2017-10-13 | 2023-01-24 | Dai Nippon Printing Co., Ltd. | Polymerizable liquid crystal compound, polymerizable composition, polymer, retardation film, method for producing retardation film, transfer laminate, optical member, method for producing optical member, and display device |
| JP2021103288A (ja) * | 2019-12-25 | 2021-07-15 | 住友化学株式会社 | 重合性液晶混合物、重合性液晶組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN107253935A (zh) | 2017-10-17 |
| KR20150036047A (ko) | 2015-04-07 |
| EP2871192B1 (en) | 2018-06-20 |
| CN107253935B (zh) | 2020-10-09 |
| US10173992B2 (en) | 2019-01-08 |
| JP2017167553A (ja) | 2017-09-21 |
| US10487065B2 (en) | 2019-11-26 |
| EP2871192A1 (en) | 2015-05-13 |
| WO2014010325A1 (ja) | 2014-01-16 |
| US20190062289A1 (en) | 2019-02-28 |
| JP2016084349A (ja) | 2016-05-19 |
| US11091452B2 (en) | 2021-08-17 |
| KR102212172B1 (ko) | 2021-02-03 |
| EP2871192A4 (en) | 2015-12-23 |
| KR20190042777A (ko) | 2019-04-24 |
| US20150175564A1 (en) | 2015-06-25 |
| KR102094007B1 (ko) | 2020-03-26 |
| US9586917B2 (en) | 2017-03-07 |
| KR101972064B1 (ko) | 2019-04-24 |
| JPWO2014010325A1 (ja) | 2016-06-20 |
| KR20200034007A (ko) | 2020-03-30 |
| CN104603165A (zh) | 2015-05-06 |
| CN104603165B (zh) | 2017-04-26 |
| JP6137284B2 (ja) | 2017-05-31 |
| US20200048213A1 (en) | 2020-02-13 |
| US20160280672A1 (en) | 2016-09-29 |
| JP6439825B2 (ja) | 2018-12-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6439825B2 (ja) | 重合性化合物、重合性組成物、高分子、光学フィルム、光学異方体、フラットパネル表示装置および反射防止フィルム | |
| JP6183514B2 (ja) | 光学フィルム | |
| JP6206481B2 (ja) | 重合性化合物、重合性組成物、高分子、及び光学異方体 | |
| JP6206414B2 (ja) | 重合性化合物、重合性組成物、高分子、及び光学異方体 | |
| JP6485354B2 (ja) | 重合性化合物、重合性組成物、高分子、及び光学異方体 | |
| JP6222085B2 (ja) | 重合性化合物、重合性組成物、高分子、及び光学異方体 | |
| JP6206413B2 (ja) | 重合性化合物、重合性組成物、高分子、及び光学異方体 | |
| JPWO2015122384A1 (ja) | 重合性化合物、重合性組成物、高分子、及び光学異方体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160323 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20160531 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20160613 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5962760 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |