-
Die
vorliegende Erfindung betrifft im Allgemeinen das Gebiet der Molekularbiologie
und befasst sich mit einem Verfahren zur Verbesserung von Ertragsmerkmalen
und/oder Verbesserung verschiedener Pflanzenwachstumseigenschaften
durch Modulation der Expression einer Nukleinsäure, die
ein GRP (Wachstums-Regulations-Protein, Growth Regulating Protein)
kodiert, in einer Pflanze. Das GRP ist ausgewählt aus einem
die LOB-Domäne (LOB: Lateral Organ Boundaries, Laterale
Organgrenzen) umfassenden Protein, hierin abgekürzt als
LBD Polypeptid, einem JMJC (JUMONJI-C) Polypeptid, einem CKI (Casein
Kinase I) Polypeptid, einem bHLH11-ähnlichem (basic Helix-Loop-Helix
11) Protein, einem Pflanzen-Hömöodomänen-Finger-Homöodomäne-(plant
homeodomain fingerhomeodomain (PHDf-HD))-Polypeptid, einem ASR-(Abscisinsäure-,
stress- und reifungsinduzierten, (abscisic acid-, stress-, and ripening-induced))-Polypeptid
und/oder einem Squamosa-Promotor Bindungsprotein-ähnlichem
11 (SPL11) Transkriptionsfaktorpolypeptid. Die vorliegende Erfindung
betrifft auch Pflanzen mit modulierter Expression einer Nukleinsäure,
die ein GRP kodiert, wobei die Pflanzen verbesserte Wachstumseigenschaften
im Vergleich zu entsprechenden Wildtyp-Pflanzen oder anderen Kontrollpflanzen
aufweisen. Die Erfindung stellt auch neue GRP-Nukleinsäuren
und GRP-Polypeptide, sowie Konstrukte bereit, die sich für
die erfindungsgemäßen Verfahren eignen.
-
Da
die Weltbevölkerung ständig zunimmt und immer
weniger Kulturfäche für die Landwirtschaft verfügbar
ist, muss die Forschung gezwungenermaßen die Effizienz
der Landwirtschaft verbessern. Herkömmliche Maßnahmen
zur kulturpflanzen- und gartenbaulichen Verbesserung nutzen Selektionszüchtungstechniken,
um Pflanzen mit wünschenswerten Eigenschaften zu identifizieren.
Diese Selektionszüchtungstechniken weisen jedoch mehrere
Nachteile auf, nämlich, dass diese Techniken gewöhnlich
arbeitsintensiv sind und Pflanzen ergeben, die häufig heterogene
genetische Komponenten enthalten, die nicht immer dazu führen, dass
das wünschenswerte Merkmal von den Elternpflanzen weiter
vererbt wird. Durch Fortschritte in der Molekularbiologie ist es
der Menschheit nun möglich, das Protoplasma von Tieren
und Pflanzen zu modifizieren. Die pflanzliche Gentechnik beinhaltet
die Isolation und Manipulation von genetischem Material (gewöhnlich
in Form von DNA oder RNA) und die anschließende Einführung
von diesem genetischen Material in eine Pflanze. Mit dieser Technologie
kann man Kulturpflanzen oder Pflanzen mit verschiedenen verbesserten
wirtschaftlichen, agronomischen oder gartenbaulichen Merkmalen produzieren.
-
Ein
Merkmal von besonderem wirtschaftlichem Interesse ist erhöhter
Ertrag. Ertrag wird normalerweise als messbares Kulturpflanzenprodukt
mit wirtschaftlichem Wert definiert. Dies kann bezüglich
Quantität und/oder Qualität definiert werden.
Der Ertrag hängt direkt von verschiedenen Faktoren ab,
wie zum Beispiel Anzahl und Größe der Organe,
der Pflanzenarchitektur (zum Beispiel die Anzahl an Verzweigungen),
der Samenproduktion, der Blattalterung und anderen. Weitere wichtige
Faktoren, die den Ertrag bestimmen, sind Wurzelentwicklung, Nährstoffaufnahme,
Stresstoleranz und Jungpflanzenvitalität. Eine Optimierung
der oben genannten Faktoren kann daher zur Erhöhung des
Kulturpflanzenertrags beitragen.
-
Der
Samenertrag ist deshalb ein besonders wichtiges Merkmal, weil die
Samen vieler Pflanzen für die menschliche und tierische
Ernährung wichtig sind. Kulturpflanzen wie Mais, Reis,
Weizen, Canola-Raps und Sojabohne machen mehr als die Hälfte
der Gesamtkalorienaufnahme des Menschen aus, und zwar entweder durch
direkten Verzehr der Samen selbst oder durch den Verzehr von Fleischprodukten,
die mit verarbeiteten Samen erzeugt wurden. Sie bilden weiterhin
eine Quelle für Zucker, Öle und vielen Arte von
Stoffwechselprodukten, die in industriellen Verfahren eingesetzt
werden. Samen enthalten einen Embryo (den Ursprung für neue
Sprosse und Wurzeln) und ein Endosperm (die Nährstoffquelle
für das Embryowachstum während der Keimung und
des frühen Wachstums der Keimlinge). An der Entwicklung
eines Samens sind viele Gene beteiligt; sie erfordert den Transfer
von Stoffwechselprodukten von den Wurzeln, Blättern und
Stängeln in den wachsenden Samen. Insbesondere das Endosperm
assimiliert die Stoffwechselvorstufen von Kohlenhydraten, Ölen
und Proteinen und synthetisiert daraus Speichermakromoleküle,
die das Korn ausfüllen.
-
Ein
weiteres wichtiges Merkmal für viele Pflanzen ist die Jungpflanzenvitalität.
Die Verbesserung der Jungpflanzenvitalität ist eine wichtige
Aufgabe bei modernen Reiszüchtungsprogrammen bei gemäßigten
sowie tropischen Reis-Kultivaren. Lange Wurzeln sind für
eine korrekte Verankerung in wassergesetztem Reis wichtig. Wird
Reis direkt auf geflutete Felder gesät, und müssen
die Pflanzen rasch durch das Wasser auftauchen, sind längere
Stängel mit Wüchsigkeit gepaart. Bei der Ausführung
von Drill- bzw. Reihensaat sind längere Mesokotyle und
Koleoptile wichtig für ein gutes Auflaufen des Keimlings.
Die Fähigkeit zu gentechnischen Einbringung von Jungpflanzenvitalität
in Pflanzen ist in der Landwirtschaft von großer Bedeutung.
Eine schlechte Jungpflanzenvitalität war beispielsweise
eine Einschränkung bei der Einführung von Mais(Zea
mays L.)-Hybriden auf der Basis von dem im Maisgürtel vorherrschenden
Protoplasmas im Europäischen Atlantik.
-
Ein
weiteres wichtiges Merkmal ist die verbesserte abiotische Stresstoleranz.
Abiotischer Stress ist eine Hauptursache für weltweite
Ernteverluste und reduziert die durchschnittlichen Erträge
bei den meisten Hauptkulturpflanzenarten um mehr als 50% (Wang
et al., Planta (2003), 218: 1–14). Abiotischer
Stress kann durch Trockenheit, Versalzung, Temperaturextrems, chemische
Toxizität und oxidativen Stress verursacht werden. Die
Fähigkeit, die Pflanzentoleranz gegenüber abiotischen
Stress zu verbessern, wäre weltweit von großem
wirtschaftlichem Vorteil für die Landwirte und würde
den Anbau von Kulturpflanzen unter ungünstigen Bedingungen
und in Gebieten, wo ein Anbau von Kulturpflanzen sonst nicht möglich
wäre, gestatten.
-
Der
Kulturpflanzenertrag kann somit durch Optimierung von einem der
vorstehend genannten Faktoren gesteigert werden.
-
Je
nach dem Endgebrauch kann die Modifikation bestimmter Ertragsmerkmale
gegenüber anderen bevorzugt sein. Für Anwendungen
wie Viehfutter- oder Holzproduktion oder für den Rohstoff
Biokraftstoff kann eine Steigerung der vegetativen Pflanzenteile
gewünscht sein, und für Anwendungen, wie die Mehl-,
Stärke- oder Ölproduktion kann eine Steigerung
der Samenparameter besonders gewünscht sein. Sogar unter
den Samenparametern können je nach der Anwendung einige
anderen gegenüber bevorzugt sein. Verschiedene Mechanismen
können zur Steigerung des Samenertrags beitragen, und zwar
in der Form einer erhöhten Samengröße
oder einer gesteigerten Samenanzahl.
-
Ein
Ansatz zur Steigerung des Ertrags (Samenertrag und/oder Biomasse)
in Pflanzen kann über die Modifikation interner Wachstumsmechanismen
einer Pflanze erfolgen, wie Zellzyklus oder verschiedene Signalwege,
die an dem Pflanzenwachstum oder Verteidigungsmechanismen beteiligt
sind.
-
Es
wurde nun überraschend gefunden, dass die Modulation der
Expression einer Nukleinsäure, die ein GRP-Polypeptid kodiert,
ausgewählt aus einem die LOB-Domäne (LOB: Lateral
Organ Boundaries, Laterale Organgrenzen) umfassenden Protein, hierin
abgekürzt als LBD-Polypeptid, einem JMJC-(JUMONJI-C)-Polypeptid,
einem Casein Kinase 1-Polypeptid, einem Pflanzen-Hömöodomänen-Finger-Homöodomänen-(plant
homeodomain fingerhomeodomain (PHDf-HD))-Polypeptid, einem bHLH11-ähnlichem
(basic Helix-Loop-Helix 11) Protein, einem ASR-(Abscisinsäure-,
stress- und reifungsinduzierten (abscisic acid-, stress-, and ripening-induced))-Polypeptid
und/oder einem Squamosa-Promotor Bindungsproteinähnlichem
11 (SPL11)-Transkriptionsfaktorpolypeptid, Pflanzen mit verbesserten
Ertragsmerkmalen und/oder verschiedenen verbesserten Pflanzenwachstumseigenschaften
gegenüber Kontrollpflanzen ergibt.
-
Gemäß einer
Ausführungsform wird ein Verfahren zur Verbesserung oder
Steigerung von Ertragsmerkmalen und/oder zur Verbesserung verschiedener
Pflanzenwachstumseigenschaften einer Pflanze gegenüber
Kontrollpflanzen bereitgestellt, umfassend die Modulation der Expression
einer Nukleinsäure, die ein GRP-Polypeptid kodiert, ausgewählt
aus einem die LOB-Domäne (LOB: Lateral Organ Boundaries,
Laterale Organgrenzen) umfassenden Protein, hierin abgekürzt
als LBD-Polypeptid, einem JMJC-(JUMONJI-C)Polypeptid, einem Casein
Kinase 1-Polypeptid, einem Pflanzen-Hömöodomänen-Finger-Homöodomänen-(plant homeodomain
finger-homeodomain (PHDf-HD))-Polypeptid, einem bHLH11-ähnlichem
(basic Helix-Loop-Helix 11) Protein, einem ASR- (Abscisinsäure-,
stress- und reifungsinduzierten (abscisic acid-, stress-, and ripening-induced))-Polypeptid
und/oder einem Squamosa-Promotor Bindungsprotein-ähnlichem
11 (SPL11) Transkriptionsfaktorpolypeptid, in einer Pflanze.
-
Stand der Technik
-
I. LOB-Domäne umfassendes Protein
(LOB: Lateral Organ Boundaries, Laterale Organgrenzen)
-
LBD-Proteine
(oder LOB-Domänenproteine) weisen eine gemeinsame konservierte
Domäne in der N-terminalen Region auf, die als LOB-Domäne
bezeichnet wird. LOB-Domänenproteine (
Shuai et
al., Plant Physiol. 129, 747–761, 2002) werden
in verschiedenen Pflanzenarten gefunden und machen eine große
Genfamilie aus: Arabidopsis besitzt Berichten zufolge mehr als 40
Gene, die LOB-Domänenproteine kodieren, mindestens 35 Gene
werden in Reis gefunden und mindestens 15 in Mais. LBD-Polypeptide
können Regulatoren von Transkriptionsfaktoren sein (zu
denen die KNOX Transkriptionsfaktoren gehören) und spielen
Postulationen zufolge eine Rolle bei der männlichen Blütenstands-
und Kolbenverzweigung bei Mais (
Bortiri et al., Plant Cell
18, 574–587, 2006), Adventivwurzelbildung (
Liu
et al., Plant J. 43, 47–56, 2005;
Inukai
et al., Plant Cell 17, 1387–1396, 2005), bei der
Proliferation weiblicher Gametophyten (
Evans et al., Plant
Cell 19, 46–62, 2007), bei der Proximal-Distal-Mustererzeugung
in den Petalen, (
Chalfun-Junior et al., Plant Mol. Biol.
57, 559–575, 2005); Blatt-Morphologie und Aderung
(
Iwakawa et al., Plant Cell Physiol. 43, 467–478,
2002).
Yang et al. (Molecular Phylogenetics and
Evolution 39, 248–262, 2006) unterschieden drei
Klassen von LBD-Proteinen in Reis, und zwar auf der Grundlage der
Klassifizierung von
Iwakawa et al. (2005) and
Shuai
et al. (2002). Die Modulation der Klasse I-LOB-Genexpression
(Auf- oder Abwärts-Regulation der Expression) führt
oft zu pleiotropen Effekten, was eine anomale Pflanzenform und Unfruchtbarkeit
mit sich bringt.
US20060218674 offenbart
beispielsweise ein Verfahren zur Steigerung der Größe
des Endosperms in einem Pflanzensamen durch Expression eines Klasse
I-LOP-Polypeptids, jedoch sank die Größe des Embryos
proportional.
-
Es
wurde überraschend gefunden, dass die Modulation der Expression
einer Nukleinsäure, die ein LBD-Polypeptid kodiert, Pflanzen
mit verbesserten Ertragsmerkmalen ergibt, insbesondere gesteigerter
Biomasse und gesteigertem Samenertrag im Vergleich zu Kontrollpflanzen.
-
Gemäß einer
Ausführungsform wird ein Verfahren zur Verbesserung von
Ertragsmerkmalen einer Pflanze gegenüber Kontrollpflanzen
bereitgestellt, umfassend die Modulation der Expression einer Nukleinsäure,
die ein LBD-Polypeptid kodiert, in einer Pflanze. Die verbesserten
Ertragsmerkmale umfassten eine gesteigerte Biomasse und einen gesteigerten
Samenertrag.
-
II. JMJC(JUMONJI-C)-Polypeptid
-
Das
erstmals beschriebene JUMONJI (was auf Japanisch kreuzförmig
bedeutet) Gen wurde identifiziert durch Gen-Trapping in Mäusen,
wo es bei der Entwicklung multipler Gewebe eine wesentliche Rolle spielt.
Bisher machen JUMONJI-Polypeptide eine eigene Klasse von Proteinen
aus, die man sowohl in Prokaryoten als auch Eukaryoten findet, einschließlich
Bakterien, Pilzen und Pflanzen.
-
Die
meisten Mitglieder der JUMONJI Polypeptid-Familie sind durch das
Vorhandensein einer JmjC- oder JUMONJI-C-Domäne charakterisiert.
Man nimmt an, dass während der Evolution alte Proteine,
die nur auf der JmjC-Domäne erworbene zusätzliche
Domänen umfassen, das Spektrum der JMJC Polypeptide, die man
in der Natur vorfindet, erheblich erweitert. Besonders interessant
sind solche JMJC-Polypeptide, die erworbene konservierte Domänen
aufweisen, die an der DNA, RNA und Protein-Bindung beteiligt sind,
wie Zinkfinger, FY-reiche RING-Finger-Protein, und F-Box-Domänen,
die nahelegen, dass Polypeptide der JUMONJI Familie die Transkription,
Chromatinfunktion und/oder Proteinumsatz regulieren können.
Folglich können JUMONJI-Proteine bei Tieren Berichten zufolge
die Entwicklung durch Steuerung der Genexpression und Chromatinaktivität
beeinträchtigen. Ein Maus-JUMONJI-Gen reprimiert beispielsweise
Cyclin D1 in Embryos, und diese Aktivität ist für
die normale Kardiogenese erforderlich (Toyoda et al. 2003
Dev Cell. 5 (1): 85–97.).
-
Hinsichtlich
des Verständnisses der Wirkungsweise von Proteinen, die
die JmjC-Domäne enthalten, wurden große Fortschritte
erzielt. Entscheidend für ihre biologische Funktion können
einige der neuer entdeckten Enzymaktivitäten bei JMJC-Polypeptiden
sein, beispielsweise JHDM1, ein JMJC Polypeptid menschlichen Ursprungs
(Tsukada, Nature. 2006; 439 (7078): 811–6)
hat eine Histondimethylase-Aktivität, und Asparaginylhydroxylase-Aktivität
wurde bei FIH (HIF-lalpha hemmender Faktor), einem Transkriptionsfaktor,
der bei der zellulären Reaktion gegen Hypoxie beteiligt
ist, beschrieben (Linke et al. (2004) J. Biol. Chem., Vol.
279, 14391–14397).
-
Die
Strukturen der JmjC-Domänen ähneln gewöhnlich
denen einiger Metalloenzyme. Neuere Strukturanalysen der im JUMONJI-Protein
FIH vorhanden JmjC-Domäne enthüllten die Aminosäurereste,
die an der Bindung an die Cofaktoren 2-Oxoglutarat und Eisen Fe(II)
beteiligt sind (Dann et al; Proc Natl Acad Sci U S A. 2002;
99 (24): 15351–15356). FIH hat das HXD/E-Eisenbindungsmotiv,
das für die meisten der 2-Oxoglutaratoxygenasen charakteristisch
ist. Zusätzlich zu und in Übereinstimmung mit
der Hydrolaseaktivität besteht die FIH-Sekundärstruktur
aus einem Betastrang-Jellyroll-Kern, der die Fe(II)-Bindungsstelle
umgibt. Diese Eigenschaften sind unter den JmjC-Domäne
enthaltenden Proteinen konserviert (Trewick et al. EMBO
Rep. 2005 (4): 15–20).
-
Bei
Pflanzen sind zwei JMJC-Polypeptide den Berichten zufolge an der
Steuerung der Blühdauer beteiligt. Sie wirken beide als
Repressoren der Blühwege in Arabidopsis thaliana (Noh
et al. Plant Cell. 2004. 16 (10): 2601–13). Die
Enzymaktivität für die Pflanzenproteine wurde
bisher nicht experimentell bestimmt.
-
Es
wurde nun überraschend gefunden, dass die Modulation der
Expression einer Nukleinsäure, die das JUMONJI-C- oder
JMJC-Polypeptid kodiert, in einer Pflanze, Pflanzen mit gesteigerten
Ertragsmerkmalen, insbesondere einen erhöhten Ertrag gegenüber
Kontrollpflanzen ergibt.
-
Gemäß einer
Ausführungsform wird ein Verfahren zum Verbessern der Ertragsmerkmale
einer Pflanze gegenüber Kontrollpflanzen bereitgestellt,
umfassend die Modulation der Expression einer Nukleinsäure,
die ein JMJC-Polypeptid kodiert, in einer Pflanze. Die verbesserten
Ertragsmerkmale umfassen eine oder mehrere von Gesamt-Samengewicht
pro Pflanze, Tausendkorn (1000-Samen) Gewicht, Samenfüllrate,
Harvest-Index, Jungpflanzenvitalität und Wurzel/Stängel-Index,
und zwar und optimalen Wachstumsbedingungen und suboptimalen milden
trockenen Bedingungen.
-
III. Casein-Kinase I
-
Proteinkinasen
veranschaulichen eine Superfamilie, und die Mitglieder dieser Superfamilie
katalysieren die reversible Übertragung einer Phosphatgruppe
von ATP auf Serin, Threonin und Tyrosin-Aminosäure-Seitenketten
auf Ziel-Polypeptiden. Insbesondere die Caseinkinase 1 (CKI) Familie
(EC 2.7.11.1) der Proteinkinasen wirken als Regulatoren der Signaltransduktionswege
in den meisten eukaryotischen Organismen. In Hefe ist CKI an der
Regulation der DNA-Reparatur und Zellzyklusprogression beteiligt
(Hoekstra MF et al., Science 253: Brockman JL et al., Proc.
Natl Acad Sci. USA 89: 9454–9458, 1992; Dhillon
N und Hoekstra MF, EMBO J 13: 2777–2788, 1994).
Caseinkinase I Proteine sind monomere Proteinkinasen vom Serin/Threonin-Typ,
die eine hochkonservierte zentrale Kinasedomäne aufweisen.
Mitglieder dieser Familie haben divergente N-terminale and C-terminale
Extensionen. Die N-terminale Region ist für die Substraterkennung
verantwortlich, und die C-terminale Extension ist für die
Wechselwirkung der Kinase mit Substraten wichtig. Die C-terminale
Extension ist wahrscheinlich auch wichtig zur Vermittlung der Regulation
durch Autophosphorylierung (Gross and Anderson, 1998 Cell
Signal 10: 699–711; Craves und Roach,
1995, J Biol Chem 270: 21689–21694).
-
In
Pflanzen wurden mehrere Mitglieder der CKI-Proteinfamilie kloniert
und biochemisch charakterisiert (Klimczak und Cashmore AR,
1993. Biochem. J. 293: 283–288; Liu et
al. 2003, Plant Journal. 36, 189–202; Lee
et al. 2005 Plant Cell. 17 (10): 2817–2831). Das
Arabidopsis-Genom enthielt Befunden zufolge mindestens 14 Casein
Kinase I-artige (CKL) Gene. In den konservierten Kinasedomänen
wiesen die Polypeptide 89% Sequenzähnlichkeit auf der Aminosäureebene
zueinander auf. Die 14 Arabidopsis CKL Isoformen wurden zudem auf
der Basis der subzellulären Lokalisierung in drei Gruppen
unterteilt. Die Gruppe 1 befindet sich vorwiegend am Zellrand; die
Gruppe 2 im Kern; die Gruppe 3 im Cytoplasma. Ein Nicotiana tabacum
CKI wurde in den Plasmodesmen lokalisiert und scheint eine Rolle
bei der Kommunikation zwischen den Zellen zu spielen. (Lee et
al. 2005). Andere vorgeschlagene Rollen für Pflanzen-CKI
ist die Signaltransduktion in Reaktion auf Umweltstimuli, Wurzelentwicklung
und Pflanzenhormon-Empfindlichkeit (Liu et al.).
-
Überraschend
wurde es jetzt entdeckt, dass die Modulation der Expression einer
Nukleinsäure, die ein CKI-Polypeptid kodiert, Pflanzen
mit gesteigerten Ertragsmerkmalen gegenüber Kontrollpflanzen
ergibt.
-
Gemäß einer
Ausführungsform wird ein Verfahren zur Verbesserung der
Ertragsmerkmale einer Pflanze gegenüber Kontrollpflanzen
bereitgestellt, umfassend die Modulation der Expression einer Nukleinsäure, die
ein CKI-Polypeptid kodiert, in einer Pflanze. Die verbesserten Ertragsmerkmale
umfassten einen oder mehrere von gesteigerter Biomasse, gesteigerter
Not-Vitaliät und gesteigertem Samenertrag.
-
IV. Pflanzen-Homöodomänen-Finger-Homöodomänen-(Plant
homeodomain finger-homeodomain (PHDf-HD))-Polypeptid
-
Transkriptionsfaktoren
werden gewöhnlich als Proteine definiert, die eine sequenzspezifische DNA-Bindungsaffinität
zeigen, und die die Transkription aktivieren und/oder reprimieren
können. Das Arabidopsis thaliana Genom kodiert mindestens
1533 Transkriptionsregulatoren, die ~5,9% der geschätzten
Gesamtzahl der Gene ausmachen (Riechmann et al. (2000) Science
290: 2105–2109). Die Datenbank der Reis-Transkriptionsfaktoren
(DRTF) ist eine Sammlung bekannter und vorhergesagter Transkriptionsfaktoren von
Oryza sativa L. ssp. indica und Oryza sativa L. ssp. japonica, und
enthält derzeit 2025 mutmaßliche Genmodelle für
Transkriptionsfakten (TF) in indica and 2,384 in japonica, die in
63 Familien verteilt sind (Gao et al. (2006) Bioinformatics
2006, 22 (10): 1286–7).
-
Eine
dieser Familien ist die Superfamilie der Homöodömänen-(HD)-Transkriptionsfaktoren,
die bei vielen Aspekten der Entwicklungsprozesse beteiligt sind.
HD-Transkriptionsfaktoren sind durch die Anwesenheit einer Homöodomäne
(HD) charakterisiert, bei der es sich um eine 60 Aminosäure
DNA-bindende Domäne (BD) handelt. Arabidopsis thaliana
und Reis enthalten etwa 100 HD-Transkriptionsfaktoren, die auf der
Basis der Aminosäuresequenz-Identität in Subfamilien
unterteilt werden können (Richardt et al. (2007)
Plant Phys 143 (4): 1452–1466). Einige dieser
Subfamilien sind durch die Anwesenheit zusätzlicher konservierter
Domänen charakterisiert, die die DNA-Bindung und/oder Protein-Protein-Wechselwirkungen
erleichtern.
-
Eine
dieser Domänen ist der PHD-Finger, der als Pflanzen-Homöodomänen-Finger
(plant homeodomain finger (PHDf)) bezeichnet wird, aufgrund seiner
Assoziation auf einem gleichen Polypeptid mit einer DNA-bindenden
HD, die ursprünglich durch Aminosäuresequenzidentität
zwischen einem Mais-Homöobox-Transkriptionsfaktor ZmHOX1a
(Bellman & Werr
(1992) EMBO J 11: 3367–3374) und seinem Arabidopsis-Verwandten
ATHAT3.1 (Schindler et al. (1993) Plant J 4: 137–150)
identifiziert wurde. Der PHDf ist ein Cys4-His-Cys3 Zinkinger-artiges Motiv, das mit zwei Zinkionen
Chelate bilden kann. PHDFs findet man in nukleären Proteinen
und sind wahrscheinlich an der chromatinvermittelten Transkriptionsregulation
beteiligt (Halbach et al. (2000) Nucleic Acid Res 28 (18):
3542–3550).
-
Transkriptionsfaktoren,
die einen PHDf und eine HD kombinieren, werden daher als PHDf-HDs
bezeichnet (Halbach et al. (2000) siehe oben).
In Pflanzen, sind diese PHDf-HDs weiter gekennzeichnet durch die
Anwesenheit eines Leucin-Zippers (ZIP) stromaufwärts des
PHDf. Beide Domänen (ZIP und der PHDf) bilden zusammen
eine stark konservierte 180 Aminosäure-Region, die als
ZIP/PHDf Domäne bezeichnet wird (Halbach et al.
(2000) siehe oben).
-
Transgene
Tabakpflanzen, die eines der beiden Mais-PHDf-HD Polypeptide (ZmHOX1a
oder ZmHOX1b) stark überexprimieren, und zwar mit einem
Blumenkohlmosaikvirus 35S Promotor in Kombination mit einem Omega-Enhancer,
zeigten identische morphologische Änderungen: Größenreduktion,
Adventivwurzelbildung und homöotische Blütentransformationen
(Uberlacker et al. (1996) Plant Cell 8: 349–362).
Transgene Reis und Tabakpflanzen, die Oryza sativa HAZ1 PHDF-HD-Polypeptid
mit dem Blumenkohl-Mosaikvirus 35S-Promotor stark überexprimieren,
zeigten kein anormales Wachstum oder Phänotypänderung
im Vergleich mit den Wildtypen (Ito et al. (2004) Gene 331:
9–15).
-
In
US-Patent 7 196 245 , wurde
ein Arabidopsis thaliana PHDf-HD-Polypeptid (identifiziert als G416)
in Arabidopsis transformiert und zeigte ein frühes Blühen
in den transgenen Pflanzen im Vergleich mit Kontrollpflanzen ohne
Auswirkung auf den Samenertrag.
-
Es
wurde überraschend entdeckt, dass die Modulation, vorzugsweise
die Steigerung der Expression einer Nukleinsäuresequenz,
die ein PHDf-HD-Polypeptid ergibt, Pflanzen mit gesteigerten Ertragsmerkmalen, vorzugsweise
gesteigerten Samen-Ertragsmerkmalen, im Vergleich mit Kontrollpflanzen
ergibt.
-
Gemäß einer
Ausführungsform wird ein Verfahren zur Steigerung der Ertragsmerkmale,
vorzugsweise der Samen-Ertragsmerkmale, in Pflanzen im Vergleich
zu Kontrollpflanzen bereitgestellt, umfassend die Modulation, vorzugsweise
die Erhöhung der Expression einer Nukleinsäuresequenz,
die ein PHDf-HD-Polypeptid kodiert, in einer Pflanze. Die gesteigerten
Ertragsmerkmale, vorzugsweise gesteigerten Samen-Ertragsmerkmale,
umfassen eine oder mehr von: einer gesteigerten Anzahl von Primärrispen,
einem gesteigerten Gesamtsamenertrag pro Pflanze, einer gesteigerten
Anzahl gefüllter Samen, einem gesteigerten Tausendkorngewicht (TKG),
einem gesteigerten Harvest-Index.
-
V. bHLH11-artiges(basic Helix-Loop-Helix
11)-Protein
-
Transkriptionsfaktoren
werden gewöhnlich als Proteine definiert, die eine sequenzspezifische DNA-Bindung
zeigen, und die die Transkription aktivieren und/oder reprimieren
können. Die Superfamilie der basischen Helix-Loop-Helix-Transkriptionsfaktoren
ist eine der größten Familien der Transkriptionsfaktoren, die
in Arabidopsis thaliana (Toledo-Ortiz et al., Plant Cell
15, 1749–1770, 2003; Bailey et al., Plant
Cell 15, 2497–2501, 2003) und in Reis (Li
et al Plant Physiol. 141, 1167–1184, 2006) charakterisiert
wurde. Die unterscheidende Eigenschaft der bHLH-Transkriptionsfaktor-Famile
ist die Anwesenheit einer zweiteiligen Domäne, die aus
etwa 60 Aminosäuren besteht. Diese zweiteilige Domäne
umfasst eine DNA-bindende basische Region, die an eine Konsensus-Hexanucleotid-E-box
bindet, und zwei a-Helices, die durch eine variable Loop-Region
getrennt ist, die sich C-terminal von der basischen Domäne
befindet. Die beiden a-Helices fördern die Dimerisierung,
was die Bildung von Homo- und Heterodimeren zwischen verschiedenen
Familienmitgliedern ermöglicht. Die bHLH-Domäne
ist zwar evolutionär konserviert, jedoch gibt es nur eine
geringe Sequenzähnlichkeit zwischen den Stämmen
außerhalb der Domäne. Li et al. (2006) klassifizieren
die bHLH-Transkriptionsfaktoren von Reis und Arabidopsis auf der
Basis der Sequenz der bHLH-Domänen in 22 Subfamilien.
-
Über
die Funktion der bHLH11-artigen Polypeptide in Pflanzen ist wenig
bekannt. Bisher wurde nur ein bHLH11-artiges Polypeptid, OsPTF1
aus Reis, charakterisiert. OsPTF1 ist Berichten zufolge an der Toleanz gegenüber
Phosphat-Hungern beteiligt (Vi et al., Plant Physiol. 138,
2087–2096). Reispflanzen, die dieses Gen unter
der Kontrolle des 35S-Promotors überexprimieren, zeigten
keinerlei verschiedenen Phänotyp im Vergleich mit Kontrollpflanzen,
wenn sie unter normalen Bedingungen gewachsen waren, aber unter
Phosphatmangelbedingungen hatten die Pflanzen eine verbesserte Phosphataufnahme.
Unter Phosphatmange zeigten die transgenen Pflanzen einen Anstieg
der Biomasse, Phosphatgehalt, verstärkte Ausläuferbildung
und gesteigerten Samenertrag.
-
Es
wurde überraschend gefunden, dass die Modulation der Expression
I in einer Pflanze von einer Nukleinsäure, die ein bHLH11-artiges
Polypeptid kodiert, Pflanzen mit gesteigerten Ertragsmerkmalen,
insbesondere einen gesteigerten Ertrag im Vergleich mit Kontrollpflanzen
ergibt. Diese Wirkungen wurden unter Wachstumsbedingungen gezeigt,
bei denen Phosphat nicht limitiert war.
-
Gemäß einer
Ausführungsform wird ein Verfahren zur Verbesserung von
Ertragsmerkmalen einer Pflanze gegenüber Kontrollpflanzen
bereitgestellt, umfassend die Modulation der Expression einer Nukleinsäure,
die ein bHLH11-artiges Polypeptid kodiert, in einer Pflanze. Die
verbesserten Ertragsmerkmale umfassten einen gesteigerten Samenertrag.
-
VI. ASR(abscisinsäure-, stress-,
und reifungsinduziertes)Protein
-
ASR-(abscisinsäure-,
stress-, und reifungsinduzierte)-Polypeptide wurden zuerst in Tomate
(Iusem et al 1993 Plant Physiol 102: 1353–1354)
als kleine stark geladene hydrophile Proteine identifiziert, die
sich auf den Kernen der Zellen befinden und an Chromatin gebunden
sind. Die Asr-Genfamilie, die ASR-Polypeptide kodiert, ist in höheren
Pflanzen weitverbreitet, und ASR-Homologa wurden aus einer großen
Zahl dikotyler und monokotyler Pflanzen kloniert (Carrai
et al. 2004 Trends Plant Sci 9: 57–59). Die meisten
Asr-Gene werden unter verschiedenen Umweltstressbedingungen während
der Fruchtreife und bei der Zellbehandlung mit dem Hormon ABA aufwärts
reguliert. ASR-Polypeptide zeigen einen hohen Grad von Sequenzkonservierung
(Frankel et al. 2006. Gene Pages 74–83).
Alle bekannten Asr-Gene enthalten zwei hochkonservierte Regionen.
Die erste Region enthält einen Bereich von His-Resten am
N-Terminus, und besitzt eine sequenzspezifische Zn2+-abhängige
DNA-Aktivität (Kalifa et al., 2004a Biochem J 381:
373–378). Der zweite Bereich ist ein großer Teil
der C-terminalen Sequenz, der oft ein Kernlokalisierungssignal NLS
besitzt (Cakir et al., 2003 Plant Cell 15: 2165–2180).
Das ASR1-Protein von Tomate ist ein intrinsisch unstrukturiertes
Protein, das bei der Bindung von Zinkionen geordnet (gefaltet) wird
und Dimere bildet (Goldgur et al. Plant Physiol. 2007 Feb;
143 (2): 617–28).
-
Eine
mutmaßliche Rolle der ASR-Polypeptide bei der Regulation
der Gentranskription wurde vorgeschlagen. Berichten zufolge ergaben
Hefe-Ein-Hybrid-Experimente, dass ein ASR von Wein an den Promotor eines
Hexose-Transporter-Gens (VvHT1) bindet. Übereinstimmend
wurde eine Rolle für Asr1 bei der Kontrolle der Hexose-Aufnahme
bei heterotrophen Organen, wie Kartoffel-Knollen vorgeschlagen (Frankel
et al. Plant Mol Biol. 2007 Mar; 63 (5): 719–30).
Die zinkabhängige DNA-Bindungsaktivität eines
Proteinmitglieds der ASR-Familie wurde beschrieben (Kalifa
et al. 2004 Biochem J. 2004 Jul 15; 381 (Pt 2): 373–8).
DNA- and Zinkbindende Domänen des ASR1-Proteins wurden
kartiert (Rom et al. 2006. Biochimie. 88 (6): 621–8; Goldgur
et al. 2007. Plant Physiol. Feb; 143 (2): 617–28).
-
Die
Verwendung der ASR-Polypeptide zur Verbesserung agronomischer Merkmale
in Pflanzen wurde offenbart als Verfahren zur Steigerung der Toleranz
der Pflanze gegen bestimmten abiotischen Stress. Die Überexpression
in Arabidopsis (Arabidopsis thaliana) des ASR1 orthologen LLA23
Gens aus der Lilie (Lilium longiflorum) steigert beispielsweise
die Pflanzentoleranz gegenüber Trockenheit und Versalzung
(
Yang et al. 2005. Plant Physiol 139: 836–846).
Das
US-Patent 7 154 025 offenbart
zudem Verfahren zur Steigerung der Resistenz gegenüber
Wassermangelstress durch Steigern der Menge der ASR-Proteine in
einer Pflanze.
-
Überraschend
wurde es gefunden, dass eine Modulation der Expression in einer
Pflanze einer Nukleinsäure, die ein ASR-Polypeptid kodiert,
Pflanzen mit gesteigerten Ertragsmerkmalen unter Nicht-Stress-Wachstumsbedingungen
im Vergleich zu Kontrollpflanzen ergibt. Gemäß einer
ersten Ausführungsform stellt die vorliegende Erfindung ein
Verfahren zur Steigerung der Ertragsmerkmale unter Nicht-Stress-Wachstumsbedingungen
im Vergleich zu Kontrollpflanzen bereit, umfassend die Modulation
der Expression von einer Nukleinsäure, die ein ASR-Polypeptid
kodiert, in einer Pflanze.
-
VII. Squamosa-Promotorbindungsprotein-artiges
11 (SPL11)
-
Transkriptionsfaktor-Polypeptide
werden gewöhnlich als Proteine definiert, die eine sequenzspezifische
DNA-Bindungsaffinität zeigen, und die die Transkription
aktivieren und/oder reprimieren können. Die Squamosa-Promotorbindungsprotein-artigen
(Squamosa Promoter binding Protein-like (SPL)) Transkriptionsfaktor-Polypeptide
sind strukturell verschiedene Proteine, die ein hochkonservierte
DNA-Bindungsdomäne (DBD) mit etwa 80 Aminosäuren
Länge miteinander teilen (Klein et al. (1996) Mol
Gen Genet 259: 7–16; Cardon et al. (1999)
Gene 237: 91–104). Die Bindungsstelle der SPL
Transkriptionsfaktor-DNA Konsensussequenz im Promotor der Zielgene
ist 5'-TNCGTACAA-3', wobei N für eine beliebige Base steht.
In der SPL-DBD befinden sich 10 konservierte Cystein (Cys) oder
Histidin-(His)-Reste (siehe 28),
von denen 8 zinkkoordinierende Reste sind, die zwei Zinkionen binden,
die zur Bildung einer SPL-spezifischen Zinkfinger-Tertiärstruktur
notwendig sind. (Yamasaki et al. (2004) J Mol Biol 337:
49–63). Ein zweites konserviertes Merkmal in der SPL
DBD ist ein zweigeteiltes Kernlokalisierungssignal. Außerhalb
der DBD wird ein Mikro-RNA(miRNA)-Ziel-Motiv, spezifisch angezielt
durch die miR156-Familie der miRNAs in den meisten der Nukleinsäuresequenzen
gefunden, die SPL-Transkriptionsfaktor-Polypeptide (entweder in
der kodierenden Region oder in der 3'-UTR) im gesamten Pflanzenreich
kodieren. (Rhoades et al. (2002) Cell 110: 513–520).
miRNAs steuern die SPL-Genexpression posttranskriptionell durch
Anzielen SPL-kodierender mRNAs zum Abbau oder durch translationale
Repression.
-
Das
Arabidopsis-Genom kodiert mindestens 1533 Transkriptionsregulatoren,
die ~5,9% der geschätzten Gesamtgenzahl ausmachen (Riechmann
et al. (2000) Science 290: 2105–2109). Die Autoren
berichten über 16 SPL Transkriptionsfaktor-Polypeptide
in Arabidopsis thaliana, mit einer geringen Sequenzähnlichkeit zwischen
ihnen (abgesehen von den vorstehend genannten Merkmalen), wobei
die Größe des abgeleiteten SPL-Polypeptids von
131 bis 927 Aminosäuren reicht. Trotzdem wurden Paare der
SPL-Transkriptionsfaktor-Polypeptide, die eine höhere Sequenzhomologie
teilen, in der SPL-Familie dieser Pflanze entdeckt (Cardon et
al. (1999)).
-
Die
SPL-Transkriptionsfaktor-Polypeptide (die nur in Pflanzen gefunden
werden), die bisher charakterisiert wurden, funktionieren in der
Pflanzenentwicklung, insbesondere in der Blütenentwicklung.
Transgene Pflanzen, die ein SPL3-Transkriptionsfaktor-Polypeptid überexprimieren,
blühen Berichten zufolge früher (
Cardon
et al. (1997) Plant J 12: 367–377). In der Europäischen
Patentanmeldung
EP1033405 ,
sind die Nukleinsäure und die abgeleiteten Polypeptidsequenzen
des SPL11 Transkriptionsfaktorpolypeptids offenbart.
-
Es
wurde überraschend gefunden, dass die Modulierung der Expression
I in eine Pflanze von einer Nukleinsäure, die ein SPL11-Polypeptid
kodiert, Pflanzen mit gesteigerten Ertragsmerkmalen, insbesondere gesteigertem
Ertrag im Vergleich zu Kontrollpflanzen ergibt.
-
Gemäß einer
Ausführungsform wird ein Verfahren zur Steigerung der Ertragsmerkmale
einer Pflanze im Vergleich mit Kontrollpflanzen bereitgestellt,
umfassend die Expression einer Nukleinsäure, die ein SPL11-Polypeptid
kodiert, in einer Pflanze. Das gesteigerte Ertragsmerkmal umfasst
eine oder mehrere von gesteigerter Auflaufvitalität (verbesserte
Jungpflanzenvitalität des Keimlings), Gesamtsamenertrag
(Samengewicht), Samenfüllrate (Samenfüllrate),
Anzahl der gefüllten Samen, Anzahl der Blüten
(Samen) pro Rispe, Harvest-Index und Tausendkorn (1000-Korn)-Gewicht,
wobei ein solcher Anstieg unter optimalen und unter suboptimalen
Wachstumsbedingungen, vorzugsweise milden Trockenbedingungen, auftreten
kann.
-
Gemäß einer
Ausführungsform werden neue SPL11-Nukleinsäuren
und SPL11-Polypeptide sowie Konstrukte bereitgestellt, die SPL11-kodierende
Nukleinsäuren umfassen, die sich bei der Durchführung
der erfindungsgemäßen Verfahren eignen.
-
Definitionen
-
Polypeptid(e)/Protein(e)
-
Die
Begriffe ”Polypeptid” und ”Protein” werden
im vorliegenden Text austauschbar eingesetzt und beziehen sich auf
Aminosäuren in polymerer Form mit beliebiger Länge,
welche über Peptidbindungen miteinander verbunden sind.
-
Polynukleotid(e)/Nukleinsäure(n)/Nukleinsäuresequenz(en)/Nukleotidsequenz(en)
-
Die
Begriffe ”Polynukleotid(e)”, ”Nukleinsäuresequenz(en)”, ”Nukleotidsequenz(en)”, ”Nukleinsäure(n)” werden
im vorliegenden Text austauschbar eingesetzt und beziehen sich auf
Nukleotide, und zwar entweder Ribonukleotide oder Desoxyribonukleotide
oder eine Kombination davon, in einer polymeren unverzweigten Form
mit einer beliebigen Länge.
-
Kontroilpflanze(n)
-
Die
Wahl von geeigneten Kontrollpflanzen ist ein Routinebestandteil
eines Versuchsaufbaus und kann entsprechende Wildtyppflanzen oder
entsprechende Pflanzen ohne das interessierende Gen beinhalten.
Gewöhnlich gehört die Kontrollpflanze zur selben
Pflanzenart oder sogar derselben Sorte wie die zu beurteilende Pflanze.
Bei der Kontrollpflanze kann es sich auch um eine Nullizygote der
zu beurteilenden Pflanze handeln. Nullizygoten sind Individuen,
denen das Transgen aufgrund von Segregation fehlt. Eine ”Kontrollpflanze” steht im
vorliegenden Zusammenhang nicht nur für ganze Pflanzen,
sondern auch Pflanzenteile, darunter Samen und Samenteile.
-
Homologon/Homologa
-
”Homologa” eines
Proteins umfassen Peptide, Oligopeptide, Polypeptide, Proteine und
Enzyme mit Aminosäuresubstitutionen, -deletionen und/oder
-insertionen im Vergleich zu dem jeweiligen nichtmodifizierten Protein,
sie weisen eine ähnliche biologische und funktionelle Aktivität
wie das nichtmodifizierte Protein, von dem sie abstammen, auf.
-
Eine
Deletion bezieht sich auf die Entfernung von einer oder mehreren
Aminosäuren aus einem Protein.
-
Eine
Insertion bezieht sich auf einen oder mehrere Aminosäurereste,
die in eine vorbestimmte Stelle in ein Protein eingeführt
werden. Insertionen können N-terminale und/oder C-terminale
Fusionen sowie Insertionen von einzelnen oder multiplen Aminosäuren
in eine Sequenz hinein umfassen. Im Allgemeinen sind Insertionen
innerhalb der Aminosäuresequenz kleiner als N- oder C-terminale
Fusionen, und zwar in einer Größenordnung von
ungefähr 1 bis 10 Resten. Zu Beispielen für N-
oder C-terminale Fusionsproteine oder -peptide gehören
die Bindungsdomäne oder Aktivierungsdomäne eines
Transkriptionsaktivators, wie er im Hefe-Zwei-Hybrid-System verwendet
wird, Phagen-Hüllproteine, (Histidin)-6-Tag, Glutathion-S-Transferase-Tag, Protein
A, Maltose-Bindungsprotein, Dihydrofolatreduktase, Tag·100-Epitop,
c-myc-Epitop, FLAG®-Epitop, lacZ,
CMP (Calmodulin-bindendes Peptid), HA-Epitop, Protein-C-Epitop und
VSV-Epitop.
-
Eine
Substitution bezieht sich auf den Austausch von Aminosäuren
des Proteins durch andere Aminosäuren mit ähnlichen
Eigenschaften (wie ähnlicher Hydrophobie, Hydrophilie,
Antigenität, Neigung zur Bildung oder zum Bruch von α-Helixstrukturen
oder β-Faltblattstrukturen). Aminosäuresubstitutionen
sind typischerweise solche von einzelnen Resten, sie können
jedoch je nach den funktionellen Zwängen, die dem Polypeptid auferlegt
werden, in Form von Clustern vorliegen; Insertionen liegen üblicherweise
in der Größenordnung von ungefähr 1 bis
10 Aminosäureresten vor. Bei den Aminosäuresubstitutionen
handelt es sich vorzugsweise um konservative Aminosäuresubstitutionen.
Konservative Substitutionstabellen sind dem Fachmann bestens bekannt
(siehe zum Beispiel
Creighton (1984), Proteins. W. H. Freeman
and Company (Hrsg.) und Tabelle 1 unten). Tabelle 1: Beispiele für konservierte
Aminosäuresubstitutioren
| Rest | konservative
Substitutionen | Rest | konservative
Substitutionen |
| Ala | Ser | Leu | Ile;
Val |
| Arg | Lys | Lys | Arg;
Gln |
| Asn | Gln;
His | Met | Leu;
Ile |
| Asp | Glu | Phe | Met;
Leu; Tyr |
| Gln | Asn | Ser | Thr;
Gly |
| Cys | Ser | Thr | Ser;
Val |
| Glu | Asp | Trp | Tyr |
| Gly | Pro | Tyr | Trp;
Phe |
| His | Asn;
Gln | Val | Ile;
Leu |
| Ile | Leu,
Val | | |
-
Aminosäuresubstitutionen,
-deletionen und/oder -insertionen lassen sich leicht mit Hilfe von
dem Fachmann bekannten Peptidsynthese-Techniken, wie Festphasen-Peptidsynthese
und dergleichen, oder durch rekombinante DNA-Manipulation, herstellen.
Verfahren für die Manipulation von DNA-Sequenzen zur Herstellung
von Substitutions-, Insertions- oder Deletionsvarianten eines Proteins
sind im Stand der Technik bestens bekannt. So kennt der Fachmann
zum Beispiel Techniken zur Herstellung von Substitutionsmutationen
an vorbestimmten Stellen in DNA; wozu M13-Mutagenese, T7-Gen-in-vitro-Mutagenese
(USB, Cleveland, OH), QuickChange ortsgerichtete Mutagenese (Stratagene,
San Diego, CA), PCR-vermittelte ortsgerichtete Mutagenese oder andere
Protokolle für die ortsgerichtete Mutagenese gehören.
-
Derivate
-
”Derivate” beinhalten
Peptide, Oligopeptide, Polypeptide, die im Vergleich zu der Aminosäuresequenz der
natürlich vorkommenden Form des Proteins, wie das interessierende
Protein, Substitutionen von Aminosäuren durch nicht natürlich
vorkommende Aminosäurereste oder Additionen von nicht natürlich
vorkommenden Aminosäureresten umfassen. ”Derivate” eines
Proteins umfassen auch Peptide, Oligopeptide, Polypeptide, die natürlich
vorkommende veränderte (glykosylierte, acylierte, prenylierte,
phosphorylierte, myristoylierte, sulfatierte usw.) oder nicht natürlich
veränderte Aminosäurereste im Vergleich zu der
Aminosäuresequenz einer natürlich vorkommenden
Form des Polypeptids umfassen. Ein Derivat kann auch eine(n) oder
mehrere Nicht-Aminosäure-Substituenten oder Additionen
im Vergleich zu der Aminosäuresequenz, von der es abstammt,
umfassen, zum Beispiel ein Reportermolekül oder einen anderen
Liganden, das bzw. der kovalent oder nichtkovalent an die Aminosäuresequenz
gebunden ist, wie ein Reportermolekül, das gebunden ist,
damit es leichter nachgewiesen werden kann, und nicht natürlich
vorkommende Aminosäurereste im Vergleich zu der Aminosäuresequenz
eines natürlich vorkommenden Proteins. ”Derivate” umfassen
darüber hinaus auch Fusionen der natürlich vorkommenden
Form des Proteins mit Tagging-Peptiden, wie FLAG, HIS6 oder Thioredoxin
(für einen Überblick über Tagging-Peptide,
siehe Terpe, Appl. Microbiol. Biotechnol. 60, 523–533,
2003).
-
Ortholog(a)/Paralog(a)
-
Orthologa
und Paraloga umfassen Evolutionskonzepte, mit denen die Stammbeziehungen
von Genen beschrieben werden. Paraloga sind Gene innerhalb derselben
Art, die durch Duplikation eines Urgens entstanden sind, und Orthologa
sind Gene von unterschiedlichen Organismen, die durch Artbildung
entstanden sind, und die auch von einem gemeinsamen Urgen abstammen.
-
Domäne
-
Der
Begriff ”Domäne” steht für einen
Satz von Aminosäuren, die an spezifischen Positionen entlang eines
Alignments von Sequenzen von evolutionsmäßig verwandten
Proteinen konserviert sind. Während Aminosäuren
an anderen Positionen zwischen Homologa variieren können,
zeigen Aminosäuren, die an spezifischen Positionen stark
konserviert sind, Aminosäuren an, die wahrscheinlich für
die Struktur, Stabilität oder Funktion eines Proteins essentiell
sind. Sie werden durch ihren hohen Konservierungsgrad in als Alignment dargestellten
Sequenzen einer Familie von Proteinhomologa identifiziert und können
als Identifikatoren verwendet werden, um zu bestimmen, ob ein bestimmtes
Polypeptid zu einer bereits identifizierten Polypeptidfamilie gehört.
-
Motiv/Konsensussequenz/Signatur
-
Der
Begriff ”Motiv” oder ”Konsensussequenz” oder ”Signatur” steht
für eine kurze konservierte Region in der Sequenz von evolutionsmäßig
verwandten Proteinen. Bei Motiven handelt es sich häufig
um hochkonservierte Teile von Domänen, sie können
jedoch auch nur einen Teil der Domäne beinhalten oder außerhalb einer
konservierten Domäne lokalisiert sein (wenn alle Aminosäuren
des Motivs außerhalb einer definierten Domäne
liegen).
-
Hybridisierung
-
Der
Begriff ”Hybridisierung” ist wie hier definiert
ein Vorgang, bei dem im Wesentlichen homologe komplementäre
Nukleotidsequenzen Basenpaarungen eingehen. Der Hybridisierungsvorgang
kann vollständig in Lösung stattfinden, d. h.
beide komplementären Nukleinsäuren liegen in Lösung
vor. Der Hybridisierungsvorgang kann auch stattfinden, wenn eine
der komplementären Nukleinsäuren auf einer Matrix,
wie magnetischen Perlen, Sepharose-Perlen oder einem anderen Harz
immobilisiert ist. Der Hybridisierungsvorgang kann außerdem
stattfinden, wenn eine der komplementären Nukleinsäuren
an einen festen Träger, wie eine Nitrozellulose- oder Nylonmembran,
immobilisiert ist oder mittels z. B. Photolithographie auf z. B.
einen Quarzglasträger immobilisiert ist (wobei letzteres
als Nukleinsäure-Arrays oder ”micorarrays” oder
Nukleinsäure-Chips bekannt ist). Die Nukleinsäuremoleküle
werden im Allgemeinen thermisch oder chemisch denaturiert, um einen
Doppelstrang zu zwei Einzelsträngen aufzuschmelzen und/oder
um ”hairpins” oder andere Sekundärstrukturen aus
einzelsträngigen Nukleinsäuren zu entfernen, so
dass die Hybridisierung stattfinden kann.
-
Der
Begriff ”Stringenz” steht für diejenigen
Bedingungen, unter denen eine Hybridisierung stattfindet. Die Stringenz
der Hybridisierung wird durch Bedingungen, wie Temperatur, Salzkonzentration,
Ionenstärke und Zusammensetzung des Hybridisierungspuffers
beeinflusst. Im Allgemeinen werden niedrige Stringenzbedingungen
so gewählt, dass sie ungefähr 30°C niedriger
sind als die Schmelztemperatur (Tm) für
die spezifische Sequenz bei einer definierten Ionenstärke
und einem definierten pH-Wert. Mittlere Stringenzbedingungen liegen
dann vor, wenn die Temperatur 20°C unter Tm liegt,
und hohe Stringenzbedingungen dann, wenn die Temperatur 10°C
unter Tm liegt. Hochstringente Hybridisierungsbedingungen
werden gewöhnlich zur Isolation von hybridisierenden Sequenzen,
die eine hohe Sequenzähnlichkeit zur Zielnukleinsäuresequenz
aufweisen, eingesetzt. Die Sequenzen der Nukleinsäuren
können jedoch aufgrund der Degeneration des genetischen
Codes voneinander abweichen, und dennoch kodieren die Nukleinsäuren
ein im Wesentlichen identisches Polypeptid. Zum Identifizieren von
solchen Nukleinsäuremolekülen können
daher manchmal Hybridisierungsbedingungen mit mittlerer Stringenz
erforderlich sein.
-
Bei
dem Tm-Wert handelt es sich um diejenige
Temperatur bei definierter Ionenstärke und definiertem pH-Wert,
bei der 50% der Zielsequenz mit einer perfekt passenden Sonde hybridisiert.
Der Tm-Wert hängt von den Lösungsbedingungen
und der Basenzusammensetzung und der Länge der Sonde ab.
Längere Sequenzen hybridisieren zum Beispiel spezifisch
bei höheren Temperaturen. Die maximale Hybridisierungsrate
wird bei 16°C bis 32°C unter dem Tm-Wert
erzielt. Das Vorliegen von einwertigen Kationen in der Hybridisierungslösung
reduziert die elektrostatische Abstoßung zwischen den beiden
Nukleinsäuresträngen und fördert so die Hybridbildung;
dieser Effekt wird für Natriumkonzentrationen von bis zu
0,4 M beobachtet (bei höheren Konzentrationen kann dieser
Effekt außer Acht gelassen werden). Formamid verringert
die Schmelztemperatur von DNA-DNA- und DNA-RNA-Doppelsträngen
mit 0,6 bis 0,7°C pro Prozent Formamid, und die Zugabe
von 50% Formamid ermöglicht eine Hybridisierung bei 30
bis 45°C, obwohl die Hybridisierungsrate gesenkt wird. Basenpaar-Fehlpaarungen
verringern die Hybridisierungsrate und die Hitzestabilität
der Doppelstränge. Durchschnittlich, und für lange Sonden
sinkt der Tm-Wert um ungefähr 1°C
pro Prozent Basen-Fehlpaarung. Der Tm-Wert
kann je nach der Art der Hybride mit den folgenden Gleichungen berechnet
werden:
- 1) DNA-DNA-Hybride (Meinkoth
und Wahl, Anal. Biochem., 138: 267–284, 1984): Tm = 81,5°C
+ 16,6 × log10[Na+]a + 0,41 × %[G/Cb] – 500 × [Lc]–1 – 0,61 × %
Formamid
- 2) DNA-RNA- oder RNA-RNA-Hybride: Tm = 79,8 + 18,5(log10[Na+]a) + 0,58(%G/Cb) + 11,8(%G/Cb)2 – 820/Lc
- 3) oligo-DNA- oder oligo-RNAd-Hybride:
Für < 20 Nukleotide:
Tm = 2 (Ia)
Für
20–35 Nukleotide: Tm = 22 + 1,46
(Ia)
- a
- oder für
ein anderes einwertiges Kation, jedoch nur im Bereich von 0,01–0,4
M genau.
- b
- nur für %GC
im Bereich von 30% bis 75% genau.
- b
- L = Länge
des Doppelstrangs in Basenpaaren.
- d
- oligo, Oligonukleotid;
In, effektive Länge des Primers =
2 × (Anz. G/C) + (Anz. A/T).
-
Eine
unspezifische Bindung kann dadurch bekämpft werden, dass
man eine von mehreren bekannten Techniken einsetzt, wie zum Beispiel
Blockieren der Membran mit proteinhaltigen Lösungen, Zusätze
von heterologer RNA, DNA und SDS zum Hybridisierungspuffer und Behandlung
mit RNase. Für nichthomologe Sonden können eine
Reihe von Hybridisierungen durchgeführt werden, und zwar
dadurch, dass man entweder (i) die Anlagerungstemperatur nach und
nach senkt (zum Beispiel von 68°C auf 42°C) oder
(ii) dass man die Formamidkonzentration nach und nach senkt (zum
Beispiel von 50% auf 0%). Der Fachmann ist mit verschiedenen Parametern
vertraut, die während der Hybridisierung verändert
werden können und die die Stringenzbedingungen entweder
aufrechterhalten oder verändern.
-
Neben
den Hybridisierungsbedingungen hängt die Spezifität
der Hybridisierung gewöhnlich auch von der Funktion der
Waschvorgänge nach der Hybridisierung ab. Um einen Hintergrund,
der durch unspezifische Hybridisierung entsteht, zu entfernen, werden
die Proben mit verdünnten Salzlösungen gewaschen.
Zu entscheidenden Faktoren solcher Waschvorgänge gehören
die Ionenstärke und Temperatur der letzten Waschlösung:
Je niedriger die Salzkonzentration und je höher die Waschtemperatur
ist, desto höher ist die Stringenz des Waschvorgangs. Die
Waschbedingungen werden gewöhnlich bei oder unter Hybridisierungsstringenz durchgeführt.
Eine positive Hybridisierung ergibt ein Signal, das mindestens zweimal
so stark wie der Hintergrund ist. Im Allgemeinen sind geeignete
Stringenzbedingungen für Nukleinsäurehybridisierungs-Assays
oder Genamplifizierungsnachweisverfahren wie oben dargestellt. Es
können auch Bedingungen höherer oder niedrigerer
Stringenz ausgewählt werden. Dem Fachmann sind die verschiedenen
Parameter geläufig, die während des Waschens verändert
werden können und die die Stringenzbedingungen entweder
aufrechterhalten oder verändern.
-
Übliche
hochstringente Hybridisierungsbedingungen für DNA-Hybride,
die länger als 50 Nukleotide sind, umfassen zum Beispiel
die Hybridisierung bei 65°C in 1 × SSC oder bei
42°C in 1 × SSC und 50% Formamid, wonach Waschen
bei 65°C in 0,3 × SSC erfolgt. Beispiele für
Hybridisierungsbedingungen mittlerer Stringenz für DNA-Hybride,
die länger als 50 Nukleotide sind, umfassen Hybridisierung
bei 50°C in 4 × SSC oder bei 40°C in
6 × SSC und 50% Formamid, und anschließendes Waschen
bei 50°C in 2 × SSC. Die Länge des Hybrids
ist die erwartete Länge für die hybridisierende
Nukleinsäure. Werden Nukleinsäuren mit einer bekannten
Sequenz hybridisiert, kann die Hybridlänge dadurch bestimmt
werden, dass man mit den Sequenzen ein Alignment durchführt
und die darin beschriebenen konservierten Regionen identifiziert.
1 × SSC ist 0,15 M NaCl und 15 mM Natriumcitrat; die Hybridisierungslösung
und die Waschlösungen können zusätzlich
5 × Denhardt-Reagens, 0,5–– 1,0% SDS,
100 μg/ml denaturierte, fragmentierte Lachssperma-DNA und
0,5% Natriumpyrophosphat beinhalten.
-
Zur
Bestimmung des Stringenzausmaßes, kann Sambrook
et al., (2001), Molecular Cloning: a laboratory manual, 3. Ausgabe,
Cold Spring Harbor Laboratory Press, CSH, New York, oder Current
Protocols in Molecular Biology, John Wiley & Sons, N. Y. (1989 und jährliche
Neufassungen) herangezogen werden.
-
Spleißvariante
-
Der
Begriff ”Spleißvariante”, wie er hier
verwendet wird, umfasst Varianten einer Nukleinsäuresequenz,
in der ausgewählte Introns und/oder Exons ausgeschnitten,
ersetzt, versetzt oder zugefügt wurden, oder in der Introns
verkürzt oder verlängert wurden. Bei solchen Varianten
bleibt die biologische Aktivität des Proteins im Wesentlichen
erhalten; dies lässt sich erzielen, indem man funktionelle
Segmente des Proteins selektiv beibehält. Solche Spleißvarianten
findet man in der Natur oder künstlich hergestellt. Verfahren
zur Vorhersage und Isolation dieser Spleißvarianten sind
im Stand der Technik bestens bekannt (siehe beispielsweise Foissac
und Schiex (2005) BMC Bioinformatics 6: 25).
-
Allelvariante
-
Allele
oder Allelvarianten sind alternative Formen eines bestimmten Gens,
die sich an der gleichen Chromosomenposition befinden. Allelvarianten
umfassen Einzelnukleotid-Polymorphismus (Single Nucleotide Polymorphisms
(SNPs)), sowie Kleine Insertions/Deletions-Polymorphismen (Small
Insertion/Deletion Polymorphisms (INDELs)). Die Größe
der INDELs ist gewöhnlich kleiner als 100 bp. SNPs und
INDELs bilden den größten Satz an Sequenzvarianten
in natürlich vorkommenden polymorphen Stämmen
der meisten Organismen.
-
Genshuffling/gerichtete Evolution
-
Genshuffling
oder gerichtete Evolution besteht aus iterativem DNA-Shuffling und
anschließendem geeigneten Screening und/oder Selektieren,
um Varianten von Nukleinsäuren oder Teilen davon zu erzeugen,
die Proteine mit modifizierter biologischer Aktivität kodieren
(
Castle et al., (2004), Science 304 (5674): 1151–4;
US-Patente 5,811,238 und
6,395,547 ).
-
Regulationselement/Kontrollsequenz/Promotor
-
Die
Begriffe ”Regulationselement”, ”Kontrollsequenz” und ”Promotor” werden
im vorliegenden Text jeweils austauschbar verwendet und sollen dahingehend
breit interpretiert werden, dass sie regulatorische Nukleinsäuresequenzen
bedeuten, die diejenigen Sequenzen, mit denen sie ligiert sind,
exprimieren können. Der Begriff ”Promotor” bezieht
sich gewöhnlich auf eine Nukleinsäurekontrollsequenz,
die sich stromaufwärts vom Transkriptionsstart eines Gens
befindet und die an der Erkennung und an der Bindung der RNA-Polymerase und
anderer Proteine beteiligt ist, wodurch die Transkription einer
funktionsfähig verknüpften Nukleinsäure
gesteuert wird. Die oben genannten Begriffe umfassen auch Transkriptionsregulationssequenzen,
die sich von einem klassischen eukaryontischen genomischen Gen ableiten
(darunter auch die TATA-Box, die für eine präzise
Initiation der Transkription erforderlich ist, mit oder ohne CCAAT-Box-Sequenz)
sowie zusätzliche Regulationselemente (d. h. stromaufwärts
aktivierende Sequenzen, Enhancer und Silencer), die die Genexpression auf
Umweltreize und/oder von außen wirkende Reize hin oder
auf gewebespezifische Art und Weise verändern. Der Begriff
beinhaltet auch eine Transkriptionsregulationssequenz eines klassischen
prokaryontischen Gens, der in diesem Fall eine -35-Box-Sequenz und/oder
-10-Box-Transkriptionsregulationssequenzen beinhaltet. Der Begriff ”Regulationselement” umfasst
auch ein synthetisches Fusionsmolekül oder Derivat, das
die Expression eines Nukleinsäuremoleküls in einer
Zelle, einem Gewebe oder einem Organ vermittelt, aktiviert oder
verbessert.
-
Ein ”pflanzlicher
Promotor” umfasst Regulationselemente, die die Expression
eines kodierenden Sequenzabschnitts in pflanzlichen Zellen vermitteln.
Ein pflanzlicher Promotor muss daher nicht pflanzlichen Ursprungs
sein, sondern kann von Viren oder Mikroorganismen, zum Beispiel
von Viren, die Pflanzenzellen angreifen, abstammen. Der ”pflanzliche
Promotor” kann auch von einer Pflanzenzelle abstammen,
z. B. von der Pflanze, die mit der bei dem erfindungsgemäßen
Verfahren zu exprimierenden und hier beschriebenen Nukleinsäuresequenz
transformiert wird. Dies trifft auch auf andere ”pflanzliche” Regulationssignale,
wie ”pflanzliche” Terminatoren zu. Die stromaufwärts
der bei den Verfahren der vorliegenden Erfindung geeigneten Nukleotidsequenzen
gelegenen Promotor können durch eine oder mehrere Nukleotidsubstitution(en),
-insertion(en) und/oder -deletion(en) modifiziert werden, ohne dass
die Funktionsfähigkeit oder Aktivität der Promotoren,
des offenen Leserasters (ORF) oder der 3'-Regulationsregion wie
Terminatoren oder anderen 3'-Regulationsregionen, die vom ORF beabstandet
liegen, gestört wird. Weiterhin kann man auch die Aktivität
der Promotoren durch Modifizieren ihrer Sequenz steigern oder sie
vollständig durch aktivere Promotoren ersetzen, sogar durch
Promotoren von heterologen Organismen. Für die Expression
in Pflanzen muss das Nukleinsäuremolekül wie oben
beschrieben funktionsfähig mit einem geeigneten Promotor
verbunden sein oder einen geeigneten Promotor umfassen, der das
Gen zum richtigen Zeitpunkt und mit dem erforderlichen räumlichen
Expressionsmuster exprimiert.
-
Zur
Identifikation funktionell äquivalenter Promotoren kann
die Promotorstärke und/oder das Expressionsmuster eines
Kandidaten-Promotors analysiert werden, indem man den Promotor funktionsfähig
mit einem Reporter-Gen verknüpft und das Expressionsausmaß und/oder
das Muster des Reportergens in verschiedenen Geweben der Pflanze
untersucht. Geeignete bestens bekannte Reportergene umfassen beispielsweise Betaglucuronidase
oder Betagalactosidase. Die Promotor-Aktivität wird untersucht,
indem man die Enzymaktivität der Betaglucuronidase oder
Betagalactosidase misst. Die Promotorstärke und/oder das
Expressionsmuster können dann mit der- bzw. demjenigen
eines Referenz-Promotors verglichen werden (wie er beispielsweise
in den erfindungsgemäßen Verfahren verwendet wird).
Alternativ kann die Promotorstärke durch Quantifizieren
der mRNA-Spiegel oder durch Vergleich der mRNA-Spiegel der in den
erfindungsgemäßen Verfahren verwendeten Nukleinsäure
mit mRNA-Spiegeln von Housekeeping-Genen, wie 18S rRNA, untersucht
werden, wobei Verfahren des Standes der Technik, wie Northern-Blotting
mit densitometrischer Analyse der Autoradiogramme, quantitative
Echtzeit-PCR oder RT-PCR verwendet werden (Neid et al.,
1996 Genome Methods 6: 986–994). Ein ”schwacher
Promotor” ist im Allgemeinen ein Promotor, der die Expression
einer kodierenden Sequenz auf einem niedrigen Spiegel steuert. ”Niedrige
Spiegel” sind Spiegel von etwa 1/10000 Transkripten bis
zu etwa 1/100000 Transkripte, bis etwa 1/500000 Transkripte pro
Zelle. Ein ”starker Promotor” steuert die Expression
einer kodierenden Sequenz dagegen auf hohem Spiegel, oder bei etwa
1/10 Transkripten bis etwa 1/100 Transkripten bis etwa 1/1000 Transkripten
pro Zelle. Ein ”mittelstarker Promotor” ist im
Allgemeinen ein Promotor, der die Expression einer kodierenden Sequenz
bei einem niedrigeren Spiegel als ein starker Promotor steuert,
insbesondere auf einem Spiegel, der in allen Fällen unter
demjenigen Spiegel liegt, der sich unter der Kontrolle eines 35S
CaMV-Promotors ergibt.
-
Funktionsfähig verknüpft
-
Der
Begriff ”funktionsfähig verknüpft” bedeutet
im vorliegenden Zusammenhang eine funktionelle Verknüpfung
zwischen der Promotorsequenz und dem interessierenden Gen, so dass
die Promotorsequenz die Transkription des interessierenden Gens
einleiten kann.
-
Konstitutiver Promotor
-
Ein ”konstitutiver
Promotor” steht für einen Promotor, der während
der meisten, jedoch nicht unbedingt allen, Wachstums- und Entwicklungsphasen
und unter den meisten Umweltbedingungen in mindestens einer Zelle,
einem Gewebe oder einem Organ transkriptionell aktiv ist. Beispiele
für konstitutive Promotoren finden sich in Tabelle 2a unten. Tabelle 2a: Beispiele für konstitutive
Promotoren
| Herkunftsgen | Literaturangabe |
| Actin | McElroy
et al., Plant Cell, 2: 163–171, 1990 |
| HMGP | WO 2004/070039 |
| CAMV
35S | Odell
et al., Nature, 313: 810–812, 1985 |
| CaMV
19S | Nilsson
et al., Physiol. Plant. 100: 456–462, 1997 |
| GOS2 | de
Pater et al., Plant J. Nov.; 2 (6): 837–44, 1992, WO 2004/065596 |
| Ubiquitin | Christensen
et al., Plant Mol. Biol. 18: 675–689, 1992 |
| Reis-Cyclophilin | Buchholz
et al., Plant Mol. Biol. 25 (5): 837–43, 1994 |
| Mais-H3-Histon | Lepetit
et al., Mol. Gen. Genet. 231: 276–285, 1992 |
| Luzerne-H3-Histon | Wu
et al., Plant Mol. Biol. 11: 641–649, 1988 |
| Actin
2 | An
et al., Plant J. 10 (1); 107–121, 1996 |
| 34S
FMV | Sanger
et al., Plant. Mol. Biol., 14, 1990: 433–443 |
| Kleine
Rubisco-Untereinheit | US 4,962,028 |
| OCS | Leisner
(1988), Proc. Natl. Acad. Sci. USA 85 (5): 2553 |
| SAD1 | Jain
et al., Crop Science, 39 (6), 1999: 1696 |
| SAD2 | Jain
et al., Crop Science, 39 (6), 1999: 1696 |
| Nos | Shaw
et al., (1984), Nucleic Acids Res. 12 (20): 7831–7846 |
| V-ATPase | WO 01/14572 |
| Superpromotor | WO 95/14098 |
| G-Box-Proteine | WO 94/12015 |
-
Ubiquitärer Promotor
-
Ein
Ubiquitärer Promotor ist in im Wesentlichen allen Geweben
oder Zellen eines Organismus aktiv.
-
Entwicklungsregulierter Promotor
-
Ein
entwicklungsregulierter Promotor ist während gewisser Entwicklungsstadien
oder in Teilen der Pflanze, bei denen entwicklungsrelevante Veränderungen
auftreten, aktiv.
-
Induzierbarer Promotor
-
Ein
induzierbarer Promotor weist eine induzierte oder erhöhte
Transkriptionsinitiation auf einen chemischen Reiz (siehe Übersichtsartikel
von Gatz 1997, Annu. Rev. Plant Physiol. Plant Mol. Biol.,
48: 89–108), einen Umweltreiz oder einen physikalischen
Reiz hin auf, oder kann ”stressinduzierbar” sein,
d. h. dann aktiviert werden, wenn eine Pflanze verschiedenen Stressbedingungen
ausgesetzt ist, oder ”pathogen induzierbar”, d.
h. wird dann aktiviert, wenn eine Pflanze verschiedenen Pathogenen
ausgesetzt ist.
-
Organspezifischer/gewebespezifischer Promotor
-
Ein
organspezifischer oder gewebespezifischer Promotor ist ein Promotor,
der die Transkription bevorzugt in bestimmten Organen oder Geweben,
wie den Blättern, Wurzeln, dem Samengewebe usw. initiieren kann.
Ein ”wurzelspezifischer Promotor” ist beispielsweise
ein Promotor, der in erster Linie in Pflanzenwurzeln transkriptionell
aktiv ist, und zwar im Wesentlichen unter Ausschluss von jeglichen
anderen Teilen einer Pflanze, obwohl trotzdem noch ”leaky”-Expression
in diesen anderen Pflanzenteilen möglich ist. Promotoren,
die die Transkription nur in bestimmten Zellen initiieren können,
werden im vorliegenden Text als ”zellspezifisch” bezeichnet.
-
Beispiele
für wurzelspezifische Promotoren sind in Tabelle 2b unten
angeführt: Tabelle 2b: Beispiele für wurzelspezifische
Promotoren
| Herkunftsgen | Literaturangabe |
| RCc3 | Plant
Mol. Biol. 1995 Jan; 27 (2): 237–48 |
| Arabidopsis-PHT1 | Kovama
et al., 2005; Mudge et al. (2002, Plant J. 31: 341) |
| Phosphattransporter
aus Medicago | Xiao
et al., 2006 |
| Arabidopsis-Pyk10 | Nitz
et al. (2001) Plant Sci. 161 (2): 337–346 |
| in
Wurzeln exprimierbare Gene | Tingey
et al., EMBO J. 6: 1, 1987 |
| auxininduzierbares
Gen des Tabaks | Van
der Zaal et al., Plant Mol. Biol. 16, 983, 1991 |
| β-Tubulin | Oppenheimer
et al., Gene 63: 87, 1988 |
| wurzelspezifische
Gene aus Tabak | Conkling
et al., Plant Physiol. 93: 1203, 1990 |
| G1-3b-Gen
aus B. napus | United States Patent No. 5,401,836 |
| SbPRP1 | Suzuki
et al., Plant Mol. Biol. 21: 109–119, 1993 |
| LRX1 | Baumberger
et al., 2001, Genes & Dev.
15: 1128 |
| BTG-26
aus Brassica napus | US 20050044585 |
| LeAMT1
(Tomate) | Lauter
et al. (1996, PNAS 3: 8139) |
| Das
LeNRT1-1 (Tomate) | Lauter
et al. (1996, PNAS 3: 8139) |
| Klasse-I-Patatingen
(Kartoffel) | Liu
et al., Plant Mol. Biol. 153: 386–395, 1991 |
| KDC1
(Daucus carota) | Downey
et al. (2000, J. Biol. Chem. 275: 39420) |
| TobRB7-Gen | W
Song (1997) PhD Thesis, North Carolina State University, Raleigh,
NC USA |
| OsRAB5a
(Reis) | Wang
et al., 2002, Plant Sci. 163: 273 |
| ALF5
(Arabidopsis) | Diener
et al. (2001, Plant Cell 13: 1625) |
| NRT2;1Np
(N. plumbaginifolia) | Quesada
et al. (1997, Plant Mol. Biol. 34: 265) |
-
Ein
samenspezifischer Promotor ist in erster Linie in Samengewebe, jedoch
nicht unbedingt ausschließlich in Samengewebe (bei ”leaky”-Expression)
transkriptionell aktiv. Der samenspezifische Promotor kann während
der Samenentwicklung und/oder während der Keimung aktiv
sein. Der samenspezifische Promotor kann endosperm- und/oder aleuron-
und/oder embryospezifisch sein. Beispiele für samenspezifische Promotoren
(endosperm-/aleuron-/embryospezifisch) sind in Tabelle 2d, e, f
unten angegeben. Weitere Beispiele für samenspezifische
Promotoren sind bei
Qing Qu und Takaiwa (Plant Biotechnol.
J. 2, 113–125, 2004) angegeben, und diese Offenbarung
ist hiermit durch Bezugnahme aufgenommen als wäre sie hier
vollständig beschrieben. Tabelle 2c: Beispiele für samenspezifische
Promotoren
| Herkunftsgen | Literaturangabe |
| Samenspezifische
Gene | Simon
et al., Plant Mol. Biol. 5: 191, 1985; |
| | Scofield
et al., J. Biol. Chem. 262: 12202, 1987; |
| | Baszczynski
et al., Plant Mol. Biol. 14: 633, 1990 |
| Paranuss-Albumin | Pearson
et al., Plant Mol. Biol. 18: 235–245, 1992 |
| Legumin | Ellis
et al., Plant Mol. Biol. 10: 203–214, 1988 |
| Glutelin
(Reis) | Takaiwa
et al., Mol. Gen. Genet. 208: 15–22, 1986; |
| | Takaiwa
et al., FEBS Letts. 221: 43–47, 1987 |
| Zein | Matzke
et al., Plant Mol. Biol., 14 (3): 323–32, 1990 |
| napA | Stalberg
et al., Planta 199: 515–519, 1996 |
| LMW-
und HMW-Glutenin-1 aus Weizen | Mol.
Gen. Genet. 216: 81–90, 1989; NAR 17: 461–2, 1989 |
| Weizen-SPA | Albani
et al., Plant Cell, 9: 171–184, 1997 |
| α, β, γ-Gliadine
aus Weizen | EMBO
J. 3: 1409–15, 1984 |
| Itr1-Promotor
aus Gerste | Diaz
et al., (1995), Moll Gen. Genet. 248 (5): 592–8 |
| B1,
C, D, Hordein aus Gerste | Theor.
Appl. Gen. 98: 1253–62, 1999; Plant J.
4: 343–55, 1993; Mol. Gen. Genet. 250:
750–60, 1996 |
| Gerste-DOF | Mena
et al., The Plant Journal, 116 (1): 53–62, 1998 |
| blz2 | EP99106056.7 |
| synthetischer
Promotor | Vicente-Carbajosa
et al., Plant J. 13: 629–640, 1998 |
| Reis-Prolamin
NRP33 | Wu
et al., Plant Cell Physiology 39 (8) 885–889, 1998 |
| a-Globulin
Glb-1 aus Reis | Wu
et al., Plant Cell Physiology 39 (8) 885–889, 1998 |
| Reis-OSH1 | Sato
et al., Proc. Natl. Acad. Sci. USA, 93: 8117–8122, 1996 |
| a-Globulin
REB/OHP-1 aus Reis | Nakase
et al., Plant Mol. Biol. 33: 513–522, 1997 |
| Reis-ADP-Glukosepyrophosphorylase | Trans.
Res. 6: 157–68, 1997 |
| Mais-ESR-Genfamilie | Plant
J. 12: 235–46, 1997 |
| a-Kafirin
aus Sorghumhirse | DeRose
et al., Plant Mol. Biol. 32: 1029–35, 1996 |
| KNOX | Postma-Haarsma
et al., Plant Mol. Biol. 39: 257–71, 1999 |
| Reis-Oleosin | Wu
et al., J. Biochem. 123: 386, 1998 |
| Sonnenblumen-Oleosin | Cummins
et al., Plant Mol. Biol. 19: 873–876, 1992 |
| PRO0117,
mutmaßliches 40S-Ribosomenprotein aus Reis | WO 2004/070039 |
| PRO0136,
Reis-Alaninaminotransferase | unveröffentlicht |
| PRO0147,
Trypsinhemmer ITR1 (Gerste) | unveröffentlicht |
| PRO0151,
Reis-WSI18 | WO 2004/070039 |
| PRO0175,
Reis-RAB21 | WO 2004/070039 |
| PRO005 | WO 2004/070039 |
| PRO0095 | WO 2004/070039 |
| a-Amylase
(Amy32b) | Lanahan
et al., Plant Cell 4: 203–211, 1992; Skriver et
al., Proc. Natl. Acad. Sci., USA 88: 7266–7270, 1991 |
| Cathepsin β-artiges
Gen | Cejudo
et al., Plant Mol. Biol. 20: 849–856, 1992 |
| Gerste-Ltp2 | Kalla
et al., Plant J. 6: 849–60, 1994 |
| Chi26 | Leah
et al., Plant J. 4: 579–89, 1994 |
| Mais
B-Peru | Selinger
et al., Genetics 149; 1125–38, 1998 |
Tabelle 2d: Beispiele für endospermspezifische
Promotoren
| Herkunftsgen | Literaturangabe |
| Glutelin
(Reis) | Takaiwa
et al., (1986), Mol. Gen. Genet. 208: 15–22; Takaiwa
et al., (1987), FERS Letts. 221: 43–47 |
| Zein | Matzke
et al., (1990), Plant Mol. Biol. 14 (3): 323–32 |
| LMW-
und HMW-Glutenin-1 aus Weizen | Colot
et al., (1989), Mol. Gen. Genet. 216: 81–90, Anderson
et al., (1989), NAR 17: 461–2 |
| Weizen-SPA | Albani
et al., (1997), Plant Cell 9: 171–184 |
| Weizen-Gliadine | Rafalski
et al., (1984), EMBO 3: 1409–15 |
| Itr1-Promotor
aus der Gerste | Diaz
et al., (1995), Mol. Gen. Genet. 248 (5): 592–8 |
| B1,
C, D, Hordein aus der Gerste | Cho
et al., (1999), Theor. Appl. Genet. 98: 1253–62; Muller
et al., (1993), Plant J. 4: 343–55; Sorenson
et al., (1996), Mol. Gen. Genet. 250: 750–60 |
| Gersten-DOF | Mena
et al., (1998), Plant J. 116 (1): 53–62 |
| blz2 | Onate
et al., (1999), J. Biol. Chem. 274 (14): 9175–82 |
| Synthetischer
Promotor | Vicente-Carbajosa
et al., (1998), Plant J. 13: 629–640 |
| Reis-Prolamin
NRP33 | Wu
et al., (1998), Plant Cell Physiol. 39 (8) 885–889 |
| Reis-Globulin
Glb-1 | Wu
et al., (1998), Plant Cell Physiol. 39 (8) 885–889 |
| Reis-Globulin
REB/OHP-1 | Nakase
et al., (1997), Plant Molec. Biol. 33: 513–522 |
| Reis-ADP-Glukosepyrophosphorylase | Russell
et al., (1997), Trans. Res. 6: 157–68 |
| Mais-ESR-Genfamilie | Opsahl-Ferstad
et al., (1997), Plant J. 12: 235–46 |
| Sorghumhirse-Kafirin | DeRose
et al., (1996), Plant Mol. Biol. 32: 1029–35 |
Tabelle 2e: Beispiele für embryospezifische
Promotoren:
| Herkunftsgen | Literaturangabe |
| Reis-OSH1 | Sato
et al., Proc. Natl. Acad. Sci. USA, 93: 8117–8122, 1996 |
| KNOX | Postma-Haarsma
et al., Plant Mol. Biol. 39: 257–71, 1999 |
| PRO0151 | WO 2004/070039 |
| PRO0175 | WO 2004/070039 |
| PRO005 | WO 2004/070039 |
| PRO0095 | WO 2004/070039 |
Tabelle 2f: Beispiele für aleuronspezifische
Promotoren:
| Herkunftsgen | Literaturangabe |
| a-Amylase
(Amy32b) | Lanahan
et al., Plant Cell 4: 203–211, 1992; Skriver et
al., Proc. Natl. Acad. Sci. USA 88: 7266–7270, 1991 |
| Cathepsin-β-artiges
Gen | Cejudo
et al., Plant Mol. Biol. 20: 849–856, 1992 |
| Gersten-Ltp2 | Kalla
et al., Plant J. 6: 849–60, 1994 |
| Chi26 | Leah
et al., Plant J. 4: 579–89, 1994 |
| Mais
B-Peru | Selinger
et al., Genetics 149; 1125–38, 1998 |
-
Ein
für grünes Gewebe spezifischer Promotor wie im
vorliegenden Text definiert ist ein Promotor, der vorwiegend in
grünem Gewebe transkriptionell aktiv ist, und zwar im Wesentlichen
unter Ausschluss von jeglichen anderen Teilen einer Pflanze, obwohl
trotzdem noch ”leaky”-Expression in diesen anderen
Pflanzenteilen möglich ist.
-
Beispiele
für Promotoren, die für das grüne Gewebe
spezifisch sind und die zur Durchführung der erfindungsgemäßen
Verfahren verwendet werden können, sind in der Tabelle
2g dargestellt. Tabelle 2g: Beispiele für die
Promotoren, die für das grüne Gewebe spezifisch
sind
| Gen | Expression | Literaturangabe |
| Orthophosphatdikinase
aus Mais | Blattspezifisch | Fukavama
et al., 2001 |
| Phosphoenolpyruvatcarboxylase
aus Mais | Blattspezifisch | Kausch
et al., 2001 |
| Phosphoenolpyruvatcarboxylase
aus Reis | Blattspezifisch | Liu
et al., 2003 |
| Kleine
Rubisco-Untereinheit aus Reis | Blattspezifisch | Nomura
et al., 2000 |
| Beta-Expansin
EXBP9 aus Reis | Sprossspezifisch | WO 2004/070039 |
| Kleine
Rubisco-Untereinheit aus Straucherbse | Blattspezifisch | Panguluri
et al., 2005 |
| RBCS3A
aus Erbse | Blattspezifisch | |
-
Ein
weiteres Beispiel für einen gewebespezifischen Promotor
ist ein meristemspezifischer Promotor, der in erster Linie in Meristemgewebe
transkriptionell aktiv ist, und zwar im Wesentlichen unter Ausschluss
von jeglichen anderen Teilen einer Pflanze, obwohl trotzdem noch ”leaky”-Expression
in diesen anderen Pflanzenteilen möglich ist. Beispiele
für Promotoren, die für das grüne Meristem
spezifisch sind und die für die Durchführung der
erfindungsgemäßen Verfahren verwendet werden können,
sind in Tabelle 2h unten dargestellt. Tabelle 2h: Beispiele für meristemspezfische
Promotoren
| Herkunftsgen | Expressionsmuster | Literaturangabe |
| Reis-OSH1 | Sprossapikalmeristem,
von globulärem Embryostadium bis zum Keimlingsstadium | Sato
et al. (1996) Proc. Natl. Acad. Sci. USA, 93: 8117–8122 |
| Reis-Metallothionein | Meristemspezifisch | BAD87835.1 |
| WAK1 & WAK2 | Spross-
und Wurzelapikalmeristeme, sowie in sich ausbreitenden Blättern
und Kelchblättern | Wagner & Kohorn (2001)
Plant Cell 13 (2): 303–318 |
-
Terminator
-
Der
Begriff ”Terminator” umfasst eine Kontrollsequenz,
bei der es sich um eine DNA-Sequenz am Ende einer Transkriptionseinheit
handelt, die die 3'-Prozessierung und Polyadenylierung eines primären
Transkripts und die Transkriptionstermination signalisiert. Der
Terminator kann von dem natürlichen Gen, von verschiedenen
anderen pflanzlichen Genen oder von T-DNA abstammen. Der hinzuzufügende
Terminator kann zum Beispiel vom Nopalinsynthasegen oder vom Octopinsynthasegen
oder auch von einem anderen pflanzlichen Gen oder, was weniger bevorzugt
ist, von einem beliebigen anderen eukaryontischen Gen abstammen.
-
Modulation
-
Der
Begriff ”Modulation” steht in Bezug auf die Expression
oder Genexpression für ein Verfahren, bei dem das Expressionsausmaß durch
die Genexpression im Vergleich zu der Kontrollpflanze dahingehend
geändert ist, dass das Expressionsausmaß erhöht
oder gesenkt ist. Die ursprüngliche unmodulierte Expression kann
eine Art Expression einer strukturellen RNA (rRNA, tRNA) oder mRNA
mit nachfolgender Translation sein. Der Begriff ”Modulation
der Aktivität” bedeutet eine Änderung
der Expression der erfindungsgemäßen Nukleinsäuresequenzen
oder der kodierten Proteine, die einen erhöhten Ertrag
und/oder ein gesteigertes Wachstum der Pflanzen ergibt.
-
Expression
-
Der
Begriff ”Expression” oder ”Genexpression” steht
für die Transkription eines spezifischen Gens oder spezifischer
Gene oder eines spezifischen genetischen Konstrukts. Der Begriff ”Expression” oder ”Genexpression” steht
insbesondere für die Transkription eines Gens oder mehrerer
Gene oder eines genetischen Konstrukts in strukturelle RNA (rRNA, tRNA)
oder mRNA mit oder ohne darauffolgende Translation des letzteren
in ein Protein. Der Prozess beinhaltet die Transkription der DNA
und die Prozessierung des resultierenden mRNA-Produkts.
-
Erhöhte Expression/Überexpression
-
Der
Begriff ”erhöhte Expression” oder ”Überexpression”,
wie es hier verwendet wird steht für eine beliebige Form
von Expression, die über den Spiegel des ursprünglichen
Wildtyp-Spiegel hinaus geht.
-
Verfahren
zur Erhöhung der Expression von Genen oder Genprodukten
sind im Stand der Technik gut dokumentiert und beinhalten beispielsweise
die Überexpression, die durch geeignete Promotoren getrieben wird,
die Verwendung von Transkriptions-Enhancern oder Translations-Enhancern.
Isolierte Nukleinsäuren, die als Promotor- oder Enhancer-Elemente
dienen, können in einer geeigneten Position (gewöhnlich
stromaufwärts) einer nicht-heterologen Form eines Polynukleotids
eingebracht werden, so dass die Expression einer Nukleinsäure,
die das interessierende Polypeptid kodiert, aufwärtsreguliert
wird. Endogene Promotoren können beispielsweise in vivo
durch Mutation, Deletion und/oder Substitution verändert
werden (siehe Kmiec,
US 5,565,350 ;
Zarling et al.,
WO9322443 ),
oder isolierte Promotoren können in der korrekten Orientierung
und im korrekten Abstand von einem erfindungsgemäßen
Gen in eine Pflanzenzelle eingebracht werden, so dass die Expression
des Gens gesteuert wird.
-
Ist
eine Polypeptidexpression gewünscht, wird wünschenswerterweise
eine Polyadenylierungsregion am 3'-Ende einer Polynukleotid-kodierenden
Region aufgenommen. Die Polyadenylierungsregion kann von dem natürlichen
Gen, von einer Anzahl anderer Pflanzengene oder von T-DNA hergeleitet
sein. Die hinzuzufügende 3'-Endsequenz kann beispielsweise
hergeleitet sein von Nopalinsynthase- oder Octopinsynthase-Genen
oder alternativ von einem anderen Pflanzengen oder weniger bevorzugt
von einem anderen eukaryotischen Gen.
-
Eine
Intronsequenz kann ebenfalls an der 5'-untranslatierten Region (UTR)
oder der kodierenden Sequenz der partiellen kodierenden Sequenz
zugegeben werden, um die Menge der reifen Botschaft zu steigern, die
sich im Cytosol ansammelt. Die Aufnahme eines spleißbaren
Introns in die Transkriptionseinheit in Pflanzen- und Tier-Expressionskonstrukten
steigert Befunden zufolge die Genexpression auf mRNA- und Protein-Ebenen
bis zu 1000fach (Buchman and Berg (1988) Mol. Cell biol.
8: 4395–4405; Callis et al. (1987) Genes Dev
1: 1183–1200). Eine solche Intron-Steigerung der
Genexpression ist gewöhnlich am größten,
wenn es sich nahe dem 5'-Ende der Transkriptionseinheit befindet.
Die Verwendung der Mais-Introns Adh1-S-Intron 1, 2 und 6, das Bronze-1-Intron,
sind im Stand der Technik bekannt. Für eine allgemeine
Information siehe: The Maize Handbook, Chapter 116, Freeling
and Walbot, Eds., Springer, N. Y. (1994).
-
Endogenes Gen
-
Eine
Bezugnahme im vorliegenden Text auf ein ”endogenes” Gen
betrifft nicht nur das fragliche Gen, wie es in einer Pflanze in
seiner natürlichen Form gefunden wird (d. h. ohne dass
ein menschlicher Eingriff vorliegt), sondern betrifft auch das gleiche
Gen, (oder eine im Wesentlichen homologe Nukleinsäure bzw.
Gen) in einer isolierten Form, das anschließend in eine
Pflanze (wieder) eingebracht wird (ein Transgen). Eine transgene
Pflanze, die ein solches Transgen enthält, kann eine wesentliche
Reduktion der Transgenexpression und/oder eine wesentliche Reduktion
der Expression des endogenen Gens erfahren. Das isolierte Gen kann aus
einem Organismus isoliert werden oder von Menschenhand hergestellt
werden, beispielsweise durch chemische Synthese.
-
Verringerte Expression
-
Eine
Bezugnahme im vorliegenden Text auf ”verringerte Expression” oder ”Reduktion
oder wesentliche Eliminierung” der Expression soll eine
Abnahme der endogenen Genexpression und/oder der Polypeptid-Spiegel
und/oder Polypeptid-Aktivität im Vergleich zu Kontrollpflanzen
bedeuten. Die Reduktion oder wesentliche Eliminierung ist in steigender
Reihenfolge der Bevorzugung mindestens 10%, 20%, 30%, 40% oder 50%,
60%, 70%, 80%, 85%, 90%, oder 95%, 96%, 97%, 98%, 99% oder noch
mehr im Vergleich zu Kontrollpflanzen.
-
Für
die Reduktion oder wesentliche Eliminierung der Expression eines
endogenen Gens in einer Pflanze ist eine hinreichende Länge
der im Wesentlichen durchgehenden Nukleotide einer Nukleinsäuresequenz erforderlich.
Zur Durchführung von Gensilencing können diese
nur 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10 oder weniger Nukleotide
betragen (u. a. mit der 5'- und/oder 3'-UTR, und zwar entweder teilweise
oder ganz). Der Bereich der im Wesentlichen zusammenhängenden
Nukleotide kann von der Nukleinsäure, die das interessierende
Protein (Zielgen) kodiert, oder von einer Nukleinsäure
hergeleitet werden, die ein Orthologon, Paralogon oder Homologon
des interessierenden Proteins kodieren kann. Der Bereich der im
Wesentlichen zusammenhängenden Nukleotide kann Wasserstoffbrückenbindungen
mit dem Zielgen eingehen (und zwar entweder mit dem Sense- oder
Antisense-Strang), stärker bevorzugt hat der Bereich der
im Wesentlichen zusammenhängenden Nukleotide in steigender
Reihenfolge der Bevorzugung 50%, 60%, 70%, 80%, 85%, 90%, 95%, 96%,
97%, 98%, 99%, 100% Sequenzidentität zu dem Zielgen (entweder
Sense- oder Antisense-Strang). Eine Nukleinsäure, die ein
(funktionelles) Polypeptid kodiert, ist keine Anforderung für
die verschiedenen hier erörterten Verfahren für
die Reduktion oder wesentliche Eliminierung der Expression eines
endogenen Gens.
-
Diese
Reduktion oder wesentliche Eliminierung der Expression kann mit
Routine- Werkzeugen und -Techniken erzielt werden. Ein bevorzugtes
Verfahren zur Reduktion oder wesentlichen Eliminierung der endogenen
Genexpression erfolgt durch Einbringen und Exprimieren eines Genkonstruktes,
in das die Nukleinsäure (in diesem Fall ein Bereich von
im Wesentlichen zusammenhängenden Nukleotiden, die von
dem interessierenden Gen abstammen, oder von einer beliebigen Nukleinsäure,
die ein Orthologon, Paralogon oder Homologon von einem der interessierenden
Proteine kodieren kann) als inverted Repeat (teilweise oder ganz), getrennt
durch einen Spacer (nicht-kodierende DNA) kloniert wird, in einer
Pflanze.
-
In
einem solchen bevorzugten Verfahren wird die Expression des endogenen
Gens reduziert oder im Wesentlichen eliminiert durch RNA-vermitteltes
Silencing mit einem inverted Repeat einer Nukleinsäure
oder einem Teil davon (in diesem Fall einem Bereich von im Wesentlichen
zusammenhängenden Nukleotiden, die von dem interessierenden
Gen hergeleitet sind, oder von einer beliebigen Nukleinsäure,
die ein Orthologon, Paralogon oder Homologon des interessierenden
Proteins kodieren kann), der eine Hairpin-Struktur bilden kann.
Der inverted repeat wird in einen Expressionsvektor kloniert, der
Kontrollsequenzen umfasst. Eine nichtkodierende DNA-Nukleinsäuresequenz
(ein Spacer, beispielsweise ein Fragment der Matrixbindungsregion (matrix
attachment region fagment (MAR)), ein Intron, ein Polylinker, etc.)
befindet sich zwischen den beiden invertierten Nukleinsäuren,
die den inverted Repeat bilden. Nach der Transkription des inverted
repeats wird eine chimäre RNA mit einer selbstkomplementären
Struktur gebildet (teilweise oder vollständig). Diese doppelsträngige
RNA-Struktur wird als Hairpin-RNA (hpRNA) bezeichnet. Die hpRNA
wird durch die Pflanze in siRNAs prozessiert, die in einen RNA-induzierten
Silencing-Komplex eingebracht sind (RISC). Der RISC spaltet zudem
die mRNA-Transkripte, wodurch im Wesentlichen die Anzahl der mRNA-Transkripte
in Polypeptide translatiert wird. Für weitere allgemeine
Einzelheiten können beispielsweise Grierson et al. (1998)
WO 98/53083 ; Waterhouse
et al. (1999)
WO 99/53050 )
herangezogen werden.
-
Die
Leistungsfähigkeit der erfindungsgemäßen
Verfahren beruht nicht auf der Einbringung und Expression eines
Genkonstrukts, in das die Nukleinsäure als inverted Repeat
kloniert wurde, in eine bzw. einer Pflanze, aber eine oder mehrere
einiger bekannter ”Gen-Silencing”-Verfahren kann
zur Erzielung der gleichen Effekte verwendet werden.
-
Ein
solches Verfahren zur Reduktion der endogenen Genexpression ist
das RNA-vermittelte Silencing der Genexpression (Abwärtsregulation).
Das Silencing wird in diesem Fall in einer Pflanze durch eine doppelsträngige
RNA-Sequenz (dsRNA) getriggert, die dem endogenen Zielgen im Wesentlichen ähnelt.
Diese dsRNA wird weiter durch die Pflanze in etwa 20 bis etwa 26
Nukleotide prozessiert, die als kurze interferierende RNAs (short
interfering RNAs (siRNAs)) bezeichnet werden. Die siRNAs werden
in einen RNA-induzierten Silencing-Komplex (RISC) eingebracht, der
das mRNA-Transkript des endogenen Zielgens spaltet, wodurch die Anzahl
der zu einem Polypeptid zu translatierenden mRNA-Transkripte verringert
wird. Die doppelsträngige RNA-Sequenz entspricht einem
Zielgen.
-
Ein
weiteres Beispiel für ein RNA-Silencing-Verfahren beinhaltet
die Einbringung von Nukleinsäure-Sequenzen oder Teilen
davon (in diesem Fall ein Bereich von im Wesentlichen zusammenhängenden
Nukleotiden, die von dem interessierenden Gen hergeleitet sind,
oder von einer beliebigen Nukleinsäure, die ein Orthologon,
Paralogon oder Homologon des interessierenden Proteins kodieren
kann) in einer Sense-Orientierung in eine Pflanze. ”Sense
Orientierung” betrifft eine DNA-Sequenz, die zu einem mRNA-Transkript
davon homolog ist. In eine Pflanze wird daher mindestens eine Kopie
der Nukleinsäuresequenz eingebracht. Die zusätzliche
Nukleinsäure verringert die Expression des endogenen Gens,
was ein Phänomen erzeugt, das als Co-Suppression bezeichnet
wird. Die Reduktion der Genexpression wird ausgeprägter,
wenn mehrere zusätzliche Kopien einer Nukleinsäuresequenz
in die Pflanze eingebracht werden, da es eine positive Korrelation
zwischen hohen Transkriptspiegeln und dem Triggern der Co-Suppression
gibt.
-
Ein
weiteres Beispiel für ein RNA-Silencing-Verfahren beinhaltet
die Verwendung von Antisense-Nukleinsäuresequenzen. Eine ”Antisense”-Nukleinsäuresequenz
umfasst eine Nukleotid-Sequenz, die zu einer ”Sense”-Nukleinsäuresequenz,
die ein Protein kodiert, komplementär ist, d. h. komplementär
zum kodierenden Strang eines doppelsträngigen cDNA-Moleküls
oder komplementär zu einer mRNA-Transkriptsequenz. Die Antisense-Nukleinsäuresequenz
ist vorzugsweise komplementär zu dem zu silencenden endogenen
Gen. Die Komplementarität kann sich in der ”kodierenden
Region” und/oder in der ”nichtkodierenden Region” eines Gens
befinden. Der Begriff ”kodierende Region” betrifft
eine Region der Nukleinsäuresequenz, die Codons umfasst,
die zu Aminosäureresten translatiert werden. Der Begriff ”nicht
kodierende Region” betrifft 5'- und 3'-Sequenzen, die die
kodierende Region flankieren, die transkribiert, aber nicht zu Aminosäuren
translatiert werden (und die auch als 5'- und 3'-untranslatierte
Regionen bezeichnet werden).
-
Antisense-Nukleinsäuresequenzen
können gemäß der Basenpaarungsregeln
von Watson und Crick entwickelt werden. Die Antisense-Nukleinsäuresequenz
kann komplementär zur gesamten Nukleinsäuresequenz
sein (in diesem Fall ein Bereich von im Wesentlichen zusammenhängenden
Nukleotiden, die von dem interessierenden Gen hergeleitet sind,
oder von einer beliebigen Nukleinsäure, die ein Orthologon,
Paralogon oder Homologon des interessierenden Proteins kodieren
kann), kann aber auch ein Oligonukleotid sein, das nur zu einem
Teil der Nukleinsäuresequenz Antisense-Orientierung aufweist
(wie auch mRNA 5' und 3' UTR). Die Antisense-Oligonukleotidsequenz
kann beispielsweise komplementär zu der Region sein, die
die Translationsstartstelle eines mRNA-Transkripts umgibt, das ein
Polypeptid kodiert. Die Länge einer geeigneten Antisense-Oligonukleotid-Sequenz
ist im Stand der Technik bekannt und kann bei etwa 50, 45, 40, 35,
30, 25, 20, 15 oder 10 Nukleotiden Länge oder weniger beginnen.
Eine erfindungsgemäße Antisense-Nukleinsäuresequenz
kann durch chemische Synthese und enzymatische Ligationsreaktionen
mit Verfahren des Standes der Technik konstruiert werden. Eine Antisense-Nukleinsäuresequenz
(beispielsweise eine Antisense-Oligonukleotid-Sequenz) kann beispielsweise
chemisch synthetisiert werden, wobei natürlich vorkommende
Nukloeotide oder verschiedene modifizierte Nukleotide verwendet
werden, die dazu ausgelegt sind, dass die biologische Stabilität
der Moleküle oder die physikalische Stabilität
des Doppelstrangs erhöht wird, der zwischen den Antisense-
und den Sense-Nukleinsäuresequenzen gebildet wird, beispielsweise
können Phosphorthioat-Derivate und acridinsubstituierte
Nukleotide verwendet werden. Beispiele für modifizierte
Nukleotide, die sich zur Erzeugung von Antisense-Nukleinsäuresequenzen
verwenden lassen, sind im Stand der Technik bestens bekannt. Bekannte
Nukleotid-Modifikationen umfassen, Methylierung, Cyclisierung und
'Caps' sowie die Substitution von einer oder mehreren der natürlich
vorkommenden Nukleotide mit einem Analogon, wie Inosin. Andere Modifikationen
von Nukleotiden sind im Stand der Technik bekannt.
-
Die
Antisense-Nukleinsäuresequenz kann biologisch produziert
werden mit einem Expressionsvektor, in den eine Nukleinsäuresequenz
in Antisense-Orientierung subkloniert wurde (d. h. RNA, die aus
der eingefügten Nukleinsäure transkribiert wurde,
ist zu einer interessierenden Ziel-Nukleinsäure in Antisense-Orientierung).
Die Produktion der Antisense-Nukleinsäuresequenzen in Pflanzen
erfolgt vorzugsweise durch ein stabil integriertes Nukleinsäure-Konstrukt,
das einen Promotor, ein funktionsfähig verknüpftes
Antisense-Oligonukleotid und einen Terminator umfasst.
-
Die
Nukleinsäuremoleküle, die in den erfindungsgemäßen
Verfahren zum Silencen verwendet werden (unabhängig davon,
ob sie in eine Pflanze eingebracht wurden, oder in situ erzeugt
wurden), hybridisieren mit oder binden an mRNA-Transkripte und/oder
genomische DNA, die ein Polypeptid kodiert, wodurch die Expression
des Proteins gehemmt wird, beispielsweise durch Hemmen der Transkription
und/oder Translation. Die Hybridisierung kann durch konventionelle
Nukleotid-Komplementarität erfolgen, so dass man einen
stabilen Doppelstrang erhält, oder beispielsweise im Falle
einer Antisense-Nukleinsäuresequenz, die an DNA-Doppelstränge
bindet, über spezifische Wechselwirkungen in der großen
Furche der Doppelhelix. Antisense-Nukleinsäuresequenzen
können durch Transformation oder direkte Injektion an einer
bestimmten Gewebestelle in eine Pflanze eingebracht werden. Alternativ
können die Antisense-Nukleinsäuresequenzen zum
Anzielen ausgewählter Zellen modifiziert werden und dann
systemisch verabreicht werden. Zur systemischen Verabreichung können
die Antisense-Nukleinsäuresequenzen derart modifiziert
werden, dass sie spezifisch an Rezeptoren oder Antigene binden,
die auf einer ausgewählten Zelloberfläche exprimiert
werden, beispielsweise durch Verknüpfen der Antisense-Nukleinsäuresequenz
an Peptide oder Antikörper, die an Zelloberflächenrezeptoren
oder Antigene binden. Die Antisense- Nukleinsäuresequenzen
können auch auf Zellen übertragen werden, und
zwar mit den hier beschriebenen Vektoren.
-
Gemäß einem
weiteren Aspekt ist die Antisense-Nukleinsäuresequenz eine
a-anomere Nukleinsäuresequenz. Eine a-anomere Nukleinsäuresequenz
bildet spezifische doppelständige Hybride mit komplementärer
RNA, in der im Gegensatz zu den üblichen b-Einheiten die
Stränge parallel zueinander verlaufen (Gaultier et
al. (1987) Nucl Ac Res 15: 6625–6641). Die Antisense-Nukleinsäuresequenz
kann auch ein 2'-o-Methylribonukleotid (Inoue et al. (1987)
Nucl Ac Res 15, 6131–6148) oder ein chimäres
RNA-DNA-Analogon umfassen (Inoue et al. (1987) FERS Lett.
215, 327–330).
-
Die
Reduktion oder wesentliche Eliminierung der endogenen Genexpression
kann auch mit Ribozymen durchgeführt werden. Die Ribozyme
sind katalytische RNA-Moleküle mit Ribonuklease-Aktivität,
die eine einzelsträngige Nukleinsäuresequenz spalten
können, wie eine mRNA, zu der sie eine komplementäre
Region aufweisen. Somit können Ribozyme (beispielsweise
Hammerkopf-Ribozyme (beschrieben in
Haselhoff and Gerlach
(1988) Nature 334, 585–591) zur katalytischen
Spaltung von mRNA-Transkripten verwendet werden, die in ein Polypeptid
translatiert werden, wodurch die Anzahl der mRNA-Transkripte, die
in ein Polypeptid translatiert werden sollen, erheblich verringert
wird. Ein Ribozym mit Spezifität für eine Nukleinsäuresequenz kann
entwickelt werden (siehe beispielsweise: Cech et al.
U.S. Patent No. 4,987,071 ; und Cech
et al.
U.S. Patent No. 5,116,742 ).
Alternativ können mRNA-Transkripte, die einer Nukleinsäuresequenz
entsprechen, zur Auswahl einer katalytischen mRNA mit einer spezifischen
Ribonukleaseaktivität aus einem Pool von RNA-Molekülen
ausgewählt werden (
Bartel and Szostak (1993) Science
261, 1411–1418). Die Verwendung von Ribozymen
zum Gen-Silencing ist im Stand der Technik bekannt (beispielsweise
Atkins et al. (1994)
WO 94/00012 ; Lenne
et al. (1995)
WO 95/03404 ;
Lutziger et al. (2000)
WO 00/00619 ;
Prinsen et al. (1997)
WO 97/13865 und Scott
et al. (1997)
WO 97/38116 ).
-
Das
Gensilencing kann ebenfalls durch Insertionsmutagenese erzielt (beispielsweise
T-DNA-Insertion oder Transposon-Insertion) oder durch Strategien,
wie sie u. a. von
Angell and Baulcombe ((1999) Plant J 20 (3):
357–62), (Amplicon VIGS
WO 98/36083 ), oder Baulcombe (
WO 99/15682 ) beschrieben
wurden.
-
Das
Gensilencing kann auch erfolgen, wenn eine Mutation an einem endogenem
Gen und/oder eine Mutation an einem isolierten Gen bzw. einer isolierten
Nukleinsäure vorliegt, die anschließend in eine
Pflanze eingeführt wurde. Die Reduktion oder wesentliche
Eliminierung kann durch ein nicht-funktionelles Polypeptid verursacht
werden. Das Polypeptid kann beispielsweise an verschiedene wechselwirkende
Proteine binden; eine oder mehrere Mutationen und/oder Verkürzungen
können daher ein Polypeptid bereitstellen, das noch an wechselwirkende
Proteine binden kann (wie Rezeptorproteine), die aber ihre normale
Funktion nicht ausüben können (beispielsweise
als signalgebender Ligand).
-
Ein
weiterer Ansatz zum Gensilencing ist durch Anzielen von Nukleinsäuresequenzen,
die zur regulatorischen Region des Gens komplementär sind
(beispielsweise Promotor und/oder Enhancer), so dass dreifach helikale
Strukturen erzeugt werden, die die Transkription des Gens in Zielzellen
verhindern. Siehe Helene, C., Anticancer Drug Res. 6, 569–84,
1991; Helene et al., Ann. N. Y. Acad. Sci. 660, 27–36 1992;
und Maher, L. J. Bioassays 14, 807–15, 1992.
-
Andere
Verfahren, wie die Verwendung von Antikörpern gegen ein
endogenes Polypeptid zur Hemmung seiner Funktion in Pflanzen oder
die Interferenz in dem Signalweg, an dem ein Polypeptid beteiligt
ist, sind dem Fachmann bestens bekannt. Es kann insbesondere angestrebt
werden, dass sich von Menschenhand gemachte Moleküle zur
Hemmung der biologischen Funktion eines Ziel-Polypeptids oder zum
Eingreifen in den Signalweg, an dem das Ziel-Polypeptid beteiligt
ist, eignet.
-
Alternativ
kann ein Screening-Programm aufgestellt werden, mit dem man in einer
Pflanze die natürlichen Genvarianten identifiziert, wobei
die Varianten Polypeptide mit reduzierter Aktivität kodieren.
Solche natürlichen Varianten können auch beispielsweise
zur Durchführung einer homologen Rekombination verwendet werden.
-
Künstliche
und/oder natürliche mikroRNAs (mRNAs) können zum
Knockout der Genexpression und/oder mRNA-Translation verwendet werden.
Endogene miRNAs sind einzelsträngige kleine RNAs mit gewöhnlich
19 bis 24 Nukleotiden Länge. In erster Linie regulieren
sie die Genexpression und/oder mRNA-Translation. Die meisten Pflanzen-mikroRNAs
(miRNAs) haben eine perfekte oder nahezu perfekte Komplementarität
zu ihren Zielsequenzen. Es gibt jedoch natürliche Ziele
mit bis zu 5 Mismatches. Sie werden aus längeren nicht-kodierenden
RNAs mit charakteristischen Rückfaltungsstrukturen durch
doppelstrangspezifische RNAsen der Dicer-Familie prozessiert. Bei
der Prozessierung werden sie durch Bindung an ihre Hauptkomponente, ein
Argonaut-Protein, in den RNA-induzierten Silencing-Komplex (RISC)
eingebracht. MiRNAs wirken als Spezifitäts-Komponenten
von RISC), da sie mit Ziel-Nukleinsäuren, meist mRNAs im
Cytoplasma Basenpaare eingehen. Nachfolgende Regulationsereignisse
umfassen Ziel-mRNA-Spaltung und die Zerstörung und/oder translationale
Hemmung. Die Effekte der miRNA-Überexpression spiegeln
sich somit oft in verringerten mRNA-Spiegeln der Zielgene wider.
-
Künstliche
mikroRNAs (amiRNAs), die gewöhnlich 21 Nukleotide lang
sind, können genetisch modifiziert werden, so dass sie
die Genexpression einzelner oder multipler interessierender Gene
negativ regulieren. Determinanten der Pflanzen-mikroRNA-Zielselektion
sind im Stand der Technik bestens bekannt. Empirische Parameter
zur Zielerkennung wurden definiert und können zur Unterstützung
der Aufmachung spezifischer amiRNAs verwendet werden (Schwab
et al., Dev. Cell 8, 517–527, 2005). Herkömmliche
Werkzeuge zur Aufmachung und Erzeugung von amiRNAs und ihrer Vorstufen
sind ebenfalls öffentlich zugänglich (Schwab
et al., Plant Cell 18, 1121–1133, 2006).
-
Für
eine optimale Leistung erfordern die Gensilencing-Techniken, die
zur Reduktion der Expression in einer Pflanze eines endogenen Gens
verwendet werden, die Verwendung von Nukleinsäuresequenzen
aus monokotylen Pflanzen zur Transformation monokotyler Pflanzen
und aus dikotylen Pflanzen zur Transformation dikotyler Pflanzen.
Vorzugsweise wird eine Nukleinsäuresequenz aus einer gegebenen
Pflanzenart in die gleiche Pflanzenart eingebracht. Beispielsweise
wird eine Nukleinsäuresequenz aus Reis in eine Reispflanze transformiert.
Es ist jedoch kein absolutes Erfordernis, dass die einzubringende
Nukleinsäuresequenz aus der gleichen Pflanzenart stammt
wie die Pflanze, in die sie eingebracht wird. Es reicht, wenn eine
wesentliche Homologie zwischen endogenem Zielgen und der einzubringenden
Nukleinsäure vorliegt.
-
Vorstehend
sind Beispiele für verschiedene Verfahren zur Verringerung
oder wesentlichen Eliminierung der Expression eines endogenen Gens
in einer Pflanze beschrieben. Der Fachmann kann leicht die vorstehend
genannten Verfahren zum Silencing so anpassen, dass die Reduktion
der Expression eines endogenen Gens in einer vollständigen
Pflanze oder in Teilen davon durch die Verwendung beispielsweise
eines geeigneten Promotors erzielt wird.
-
Selektionsmarker(gen)/Reportergen
-
”Selektionsmarker”, ”Selektionsmarkergen” oder ”Reportergen” beinhaltet
jegliches Gen, das einer Zelle, in der es exprimiert wird, um die
Identifikation und/oder Selektion von Zellen, die mit einem erfindungsgemäßen
Nukleinsäurekonstrukt transfiziert sind oder transformiert
sind, zu erleichtern, einen Phänotyp verleiht. Mit diesen
Markergenen kann ein erfolgreicher Transfer der Nukleinsäuremoleküle
mittels einer Reihe von unterschiedlichen Prinzipien identifiziert
werden. Geeignete Marker können aus Markern ausgewählt
werden, die Antibiotika- oder Herbizidresistenz verleihen, die ein
neues Stoffwechselmerkmal einführen oder die eine visuelle
Selektion gestatten. Zu Selektionsmarkergenen gehören zum
Beispiel Gene, die Resistenz gegen Antibiotika (wie nptII, das Neomycin
und Kanamycin phosphoryliert, oder hpt, das Hygromycin phosphoryliert, oder
Gene, die Resistenz gegen zum Beispiel Bleomycin, Streptomycin,
Tetracyclin, Chloramphenicol, Ampicillin, Gentamycin, Geneticin
(G418), Spectinomycin oder Blasticidin verleihen), gegen Herbizide
(zum Beispiel bar, das Resistenz gegen Basta® vermittelt;
aroA oder gox, die Resistenz gegen Glyphosat vermitteln, oder die Gene,
die Resistenz gegen z. B. Imidazolinon, Phosphinothricin oder Sulfonylharnstoff
verleihen), oder Gene, die ein Stoffwechselmerkmal bereitstellen
(wie manA, das es Pflanzen ermöglicht, Mannose als einzige
Kohlenstoffquelle zu verwerten, oder Xyloseisomerase für
die Verwertung von Xylose, oder Antinährstoffmarker, wie
Resistenz gegen 2-Desoxyglucose), vermitteln. Die Expression visueller
Markergene führt zur Bildung von Farbe (zum Beispiel β-Glucuronidase,
GUS oder β-Galactosidase mit ihren gefärbten Substraten,
zum Beispiel X-Gal), Lumineszenz (wie das Luziferin/Luziferase-System)
oder Fluoreszenz (Green Fluorescent Protein, GFP, und seine Derivate).
Diese Aufzählung stellt nur eine kleine Anzahl von möglichen
Markern dar. Der Fachmann ist mit solchen Markern vertraut. Je nach
dem Organismus und dem Selektionsverfahren werden unterschiedliche
Marker bevorzugt.
-
Bei
stabiler oder transienter Integration von Nukleinsäuren
in Pflanzenzellen nimmt bekanntlich nur ein kleiner Teil der Zellen
die fremde DNA auf, und integriert sie wenn gewünscht je
nach dem verwendeten Expressionsvektor und der verwendeten Transfektionstechnik
in ihr Genom. Zur Identifikation und Selektion dieser Integranten
wird gewöhnlich ein Gen, das einen selektierbaren Marker
kodiert (wie diejenigen, die oben beschrieben sind) in die Wirtszelle
zusammen mit dem interessierenden Gen eingesetzt. Diese Marker können beispielsweise
in Mutanten verwendet werden, in denen diese Gene nicht funktionell
sind, beispielsweise durch Deletion durch herkömmliche
Verfahren. Darüber hinaus können Nukleinsäuremoleküle,
die einen Selektionsmarker kodieren, in eine Wirtszelle auf dem
gleichen Vektor eingebracht werden, der die Sequenz umfasst, die
die erfindungsgemäßen Polypeptide kodiert, oder
in den erfindungsgemäßen Verfahren verwendet werden,
oder ansonsten in einem gesonderten Vektor. Zellen, die stabil mit
der eingebrachten Nukleinsäure transfiziert wurden, können
beispielsweise durch Selektion identifiziert werden (beispielsweise
Zellen, die den Selektionsmarker integriert haben, überleben,
wohingegen andere Zellen sterben).
-
Da
die Markergene, insbesondere Gene für die Resistenz gegen
Antibiotika und Herbizide, in der transgenen Wirtszelle nicht länger
erforderlich sind oder ungewünscht sind, sobald die Nukleinsäuren
erfolgreich eingebracht worden sind, setzt das erfindungsgemäße
Verfahren zum Einbringen von Nukleinsäuren erfolgreich
Techniken ein, die die Entfernung oder Abspaltung dieser Markergene
ermöglichen. Ein solches Verfahren wird als Cotransformation
bezeichnet. Die Cotransformation setzt 2 Vektoren ein, die simultan
zur Transformation eingesetzt werden, wobei ein Vektor die erfindungsgemäße
Nukleinsäure trägt und der zweite das oder die
Markergene trägt. Ein großer Anteil der Transformanten
erhält, oder umfasst bei Pflanzen (bis zu 40% oder mehr
Transformanten) beide Vektoren. Bei der Transformation mit Agrobakterien,
erhalten die Transformanten gewöhnlich nur einen Teil des
Vektors, d. h. die Sequenz, die von der T-DNA flankiert wird, die gewöhnlich
die Expressions-Kassette veranschaulicht. Die Markergene können
anschließend aus der transformierten Pflanze durch Kreuzungen
entfernt werden. Bei einem anderen Verfahren werden die in ein Transposon
integrierten Markergene zur Transformation zusammen mit der gewünschten
Nukleinsäure verwendet (was als Ac/Ds-Technologie bekannt
ist). Die Transformanten können mit einer Quelle für
Transposase gekreuzt werden, oder die Transformanten werden mit
einem Nukleinsäure-Konstrukt transformiert, das die transiente
oder stabile Expression einer Transposase vermittelt. In einigen
Fällen (etwa 10%) springt das Transposon aus dem Genom
der Wirtszelle, sobald die Transformation erfolgreich stattgefunden
hat und verloren geht. Bei einer weiteren Anzahl von Fällen,
springt das Transposon an eine andere Stelle. In diesen Fällen muss
das Markergen mit Hilfe von Kreuzungen eliminiert werden. In der
Mikrobiologie wurden Techniken entwickelt, die den Nachweis solcher
Ereignisse ermöglichen oder erleichtern. Ein weiteres vorteilhaftes
Verfahren beruht auf sogenannten Rekombinationssystemen, deren Vorteil
ist, dass auf die Eliminierung durch Kreuzung verzichtet werden
kann. Das am besten bekannte System dieser Art ist als Cre/Iox-System
bekannt. Cre1 ist eine Rekombinase, die die zwischen den IoxP-Sequenzen
befindlichen Sequenzen entfernt. Ist der Marker zwischen den IoxP-Sequenzen
integriert, wird er entfernt, sobald die Transformation erfolgreich
stattgefunden hat, durch Expression der Rekombinase. Weitere Rekombinationssysteme
sind das HIN/HIX-, FLP/FRT- und REP/STB-System (Tribble
et al., J. Biol. Chem., 275, 2000: 22255–22267; Velmurugan
et al., J. Cell Biol., 149, 2000: 553–566). Die
erfindungsgemäßen Nukleinsäuresequenzen
können stellenspezifisch in das Pflanzengenom integriert
werden. Natürlich können diese Verfahren auch
auf Mikroorganismen angewendet werden, wie Hefe, Pilze oder Bakterien.
-
Transgen/transgen/rekombinant
-
Im
Zusammenhang mit der vorliegenden Erfindung bedeutet ”transgen”, ”Transgen” oder ”rekombinant” eine
Nukleinsäuresequenz, eine Expressionskassette, ein Genkonstrukt
oder einen Vektor, umfassend die Nukleinsäuresequenz oder
einen Organismus, der mit den erfindungsgemäßen
Nukleinsäuresequenzen, Expressionskassetten oder Vektoren
transformiert ist, wobei alle diese Konstrukte mittels rekombinanter
Verfahren entstehen, in denen entweder
- (a)
die Nukleinsäuresequenzen, die Proteine kodieren, die bei
den erfindungsgemäßen Verfahren geeignet sind,
oder
- (b) genetische Kontrollsequenz(en) in funktionsfähiger
Verknüpfung mit der erfindungsgemäßen
Nukleinsäuresequenz, zum Beispiel einem Promotor, oder
- (c) a) und b)
sich nicht in ihrer natürlichen
genetischen Umgebung befinden oder durch rekombinante Verfahren
modifiziert worden sind, wobei es sich bei der Modifikation zum
Beispiel um eine Substitution, Addition, Deletion, Inversion oder
Insertion von einem oder mehreren Nukleotidresten handeln kann.
Unter der natürlichen genetischen Umgebung versteht man
den natürlichen genomischen oder chromosomalen Locus in
der Ursprungspflanze oder das Vorhandensein in einer genomischen
Bank. Bei einer genomischen Bank wird die natürliche genetische Umgebung
der Nukleinsäuresequenz vorzugsweise zumindest teilweise beibehalten.
Die Umgebung flankiert die Nukleinsäuresequenz zumindest
auf einer Seite und weist eine Sequenzlänge von mindestens
50 Bp, vorzugsweise mindestens 500 Bp, besonders bevorzugt mindestens
1000 Bp, am stärksten bevorzugt mindestens 5000 Bp, auf.
Eine natürlich vorkommende Expressionskassette – zum
Beispiel die natürlich vorkommende Kombination des natürlichen
Promotors der Nukleinsäuresequenzen mit der entsprechenden
Nukleinsäuresequenz, die ein Polypeptid kodiert, das bei
den Verfahren der vorliegenden Erfindung geeignet ist, wie oben definiert – wird
zu einer transgenen Expressionskassette, wenn diese Expressionskassette
durch unnatürliche, synthetische (”künstliche”)
Verfahren wie zum Beispiel mutagene Behandlung modifiziert wird.
Geeignete Verfahren sind zum Beispiel in US 5,565,350 oder WO 00/15815 beschrieben.
-
Unter
einen transgenen Pflanze versteht man für erfindungsgemäße
Zwecke wie oben, dass die bei dem erfindungsgemäßen
Verfahren verwendeten Nukleinsäuren nicht an ihrem natürlichen
Locus im Genom dieser Pflanze vorliegen, wobei die Nukleinsäuren
homolog oder heterolog exprimiert werden können. Wie erwähnt
bedeutet transgen jedoch auch, dass die erfindungsgemäßen
Nukleinsäuren oder die Nukleinsäuren, die in dem
erfindungsgemäßen Verfahren verwendet werden,
zwar in ihrer natürlichen Position im Genom einer Pflanze
vorliegen, die Sequenz jedoch in Bezug auf die natürliche
Sequenz modifiziert worden ist und/oder dass die Regulationssequenzen
der natürlichen Sequenzen modifiziert worden sind. Transgen
bedeutet vorzugsweise die Expression der erfindungsgemäßen
Nukleinsäuren an einem unnatürlichen Locus in
dem Genom, d. h. dass eine homologe, oder vorzugsweise, heterologe
Expression der Nukleinsäuren stattfindet. Bevorzugte transgene
Pflanzen werden im vorliegenden Text erwähnt.
-
Transformation
-
Der
Begriff ”Einführung” oder ”Transformation” umfasst
im vorliegenden Zusammenhang den Transfer eines Fremdpolynukleotids
in eine Wirtszelle, und zwar unabhängig von dem für
den Transfer eingesetzten Verfahren. Pflanzengewebe, das entweder
durch Organogenese oder Embryogenese zu einer anschließenden
klonalen Vermehrung befähigt ist, kann mit einem erfindungsgemäßen
Genkonstrukt transformiert werden, und daraus kann eine ganze Pflanze
regeneriert werden. Das jeweils ausgewählte Gewebe hängt
von den jeweiligen klonalen Vermehrungssytemen ab, die für
die jeweilige zu transformierende Art verfügbar und am
besten geeignet sind. Zu Zielgeweben gehören zum Beispiel
Blattscheiben, Pollen, Embryonen, Kotyledonen, Hypokotyle, Megagametophyten,
Kallusgewebe, existierendes Meristemgewebe (z. B. Apikalmeristem,
Achelknospen und Wurzelmeristeme) und induziertes Meristemgewebe
(z. B. Kotyledonenmeristem und Hypokotylmeristem). Das Polynukleotid
kann in eine Wirtszelle transient oder stabil eingeführt
werden und kann in nichtintegrierter Form, zum Beispiel als Plasmid,
aufrechterhalten werden. Es kann jedoch auch in das Wirtsgenom integriert
werden. Mit den erhaltenen transformierten Pflanzenzellen kann man
dann eine transformierte Pflanze auf dem Fachmann bekannte Art und
Weise regenerieren.
-
Der
Transfer von Fremdgenen in das Genom einer Pflanze wird als Transformation
bezeichnet. Die Transformation von Pflanzenarten ist eine Technik,
die heutzutage relativ routinemäßig erfolgt. Um
das interessierende Gen in eine geeignete Vorfahrenzelle einzuführen,
kann vorteilhafterweise irgendeine von verschiedenen Transformationsmethoden
eingesetzt werden. Die für die Transformation und Regeneration
von Pflanzen aus Pflanzengeweben oder Pflanzenzellen beschriebenen
Methoden können für die transiente oder für
die stabile Transformation eingesetzt werden. Zu Transformationsmethoden
gehören die Verwendung von Liposomen, die Elektroporation,
Chemikalien, die die freie DNA-Aufnahme verstärken, die
Injektion der DNA direkt in die Pflanze, der Teilchenkanonenbeschuss,
die Transformation mit Viren oder Pollen und die Mikroprojektion.
Die Verfahren können aus den folgenden ausgewählt
werden: Calcium/Polyethylenglycol-Methode für Protoplasten
(
Krens, F. A. et al., (1982), Nature 296, 72–74;
Negrutiu
I et al., (1987), Plant Mol. Biol. 8: 363–373);
Elektroporation von Protoplasten (
Shillito R. D. et al.,
(1985), Bio/Technol. 3, 1099–1102); Mikroinjektion
in Pflanzenmaterial (
Crossway A et al., (1986), Mol. Gen.
Genet. 202: 179–185); Beschuss mit DNA- oder RNA-beschichteten
Partikeln (
Klein TM et al., (1987), Nature 327: 70),
Infektion mit (nichtintegrierenden) Viren und dergleichen. Transgene
Pflanzen, darunter auch transgene Kulturpflanzen, werden vorzugsweise mittels
Agrobacterium-vermittelter Transformation erzeugt. Eine vorteilhafte
Transformationsmethode ist die Transformation in die Pflanze. Zu
diesem Zweck kann man zum Beispiel die Agrobakterien auf Pflanzensamen einwirken
lassen oder das Pflanzenmeristem mit Agrobakterien beimpfen. Erfindungsgemäß hat
sich besonders günstig erwiesen, eine Suspension von transformierten
Agrobakterien auf die intakte Pflanze oder zumindest die Blütenprimordien
einwirken zu lassen. Die Pflanze wird anschließend weiter
herangezogen, bis man die Samen der behandelten Pflanze erhält
(
Clough und Bent, Plant J. (1998), 16, 735–743).
Zu den Verfahren für die Agrobacterium-vermittelte Transformation
des Reises gehören gut bekannte Verfahren für
die Reistransformation, wie diejenigen, die in einer der folgenden
Schriften beschrieben sind: Europäische Patentanmeldung
EP 1198985 A1 ,
Aldemita
und Hodges (Planta 199: 612–617, 1996);
Chan
et al., (Plant Mol. Biol. 22 (3): 491–506, 1993),
Hiei
et al., (Plant J. 6 (2): 271–282, 1994), die hiermit
vollinhaltlich durch Bezugnahme aufgenommen sind. Bei der Transformation
von Mais ist das bevorzugte Verfahren entweder wie bei
Ishida
et al., (Nat. Biotechnol. 14 (6): 745–50, 1996)
oder bei
Frame et al. (Plant Physiol. 129 (1): 13–22,
2002) beschrieben, die hiermit vollinhaltlich durch Bezugnahme
aufgenommen sind. Diese Verfahren sind weiterhin zum Beispiel bei
B.
Jenes et al., Techniques for Gene Transfer, in: Transgenic Plants,
Band 1, Engineering and Utilization, Hrsg. S. D. Kung und R. Wu,
Academic Press (1993), 128–143 und in
Potrykus
Annu. Rev. Plant Physiol. Plant Molec. Biol. 42 (1991) 205–225)
beschrieben. Die zu exprimierenden Nukleinsäuren bzw. das
zu exprimierende Konstrukt werden/wird vorzugsweise in einen Vektor
kloniert, der sich für die Transformation von Agrobacterium
tumefaciens eignet, zum Beispiel pBin19 (
Bevan et al., Nucl.
Acids Res. 12 (1984) 8711). Agrobakterien, die mit solch
einem Vektor transformiert wurden, können dann auf bekannte
Weise für die Transformation von Pflanzen eingesetzt werden,
wie Pflanzen, die als Modell verwendet werden, wie Arabidopsis (Arabidopsis
thaliana wird innerhalb des Schutzbereichs der vorliegenden Erfindung
nicht als Kulturpflanze angesehen), oder Kulturpflanzen, wie zum
Beispiel Tabakpflanzen, und zwar zum Beispiel dadurch, dass man
verletzte Blätter oder zerkleinerte Blätter in
eine Agrobakterienlösung taucht und sie dann in geeigneten
Medien kultiviert. Die Transformation von Pflanzen mittels Agrobacterium
tumefaciens wird zum Beispiel bei
Höfgen und Willmitzer
in Nucl. Acid Res. (1988), 16, 9877 beschrieben oder ist
unter anderem aus
F. F. White, Vectors for Gene Transfer
in Higher Plants; in Transgenic Plants, Band 1, Engineering and
Utilization, Hrsg. S. D. Kung und R. Wu, Academic Press, 1993, S.
15–38 bekannt.
-
Zusätzlich
zu der Transformation von somatischen Zellen, die dann zu intakten
Pflanzen regeneriert werden müssen, kann man auch Zellen
von pflanzlichen Meristemen transformieren, insbesondere solche
Zellen, die sich zu Gameten entwickeln. In diesem Fall folgen die
transformierten Gameten der natürlichen Pflanzenentwicklung
und ergeben transgene Pflanze. So werden zum Beispiel Samen von
Arabidopsis mit Agrobakterien behandelt, und man erhält
von den sich entwickelnden Pflanzen, von denen ein gewisser Teil
transformiert und daher transgen ist, Samen [Feldman, KA
und Marks MD (1987). Mol. Gen. Genet. 208: 274–289; Feldmann
K (1992). In: C Koncz, N-H Chua und J Shell, Hrsg., Methods in Arabidopsis
Research. Word Scientific, Singapur, S. 274–289].
Alternative Verfahren beruhen auf der wiederholten Entfernung der
Infloreszenzen und Inkubation der Exzisionsstelle in der Mitte der
Rosette mit transformierten Agrobakterien, wodurch ebenfalls später
transformierte Samen erhalten werden können (Chang
(1994). Plant J. 5: 551–558; Katavic (1994). Mol. Gen.
Genet., 245: 363–370). Ein besonders wirksames
Verfahren ist jedoch die Vakuuminfiltrationsmethode mit ihren Modifikationen,
wie der ”floral dip”-Methode. Bei der Vakuuminfiltration
von Arabidopsis werden intakte Pflanzen unter verringertem Druck
mit einer Agrobakteriensuspension behandelt [Bechthold,
N (1993). C. R. Acad. Sci. Paris Life Sci., 316: 1194–1199],
während bei der ”floral dip”-Methode
das sich entwickelnde Blütengewebe kurz mit einer mit Tensid
behandelten Agrobakteriensuspension inkubiert wird [Clough, SJ
und Bent AF (1998), The Plant J. 16, 735–743].
Ein gewisser Anteil transgener Samen wird in beiden Fällen geerntet,
und diese Samen lassen sich von nichttransgenen Samen dadurch unterscheiden,
dass man sie unter den oben beschriebenen Selektionsbedingungen
heranzieht. Außerdem ist die stabile Transformation von Plastiden
vorteilhaft, da Plastide bei den meisten Kulturpflanzen mütterlich
vererbt werden, wodurch die Gefahr des Transgenflusses durch Pollen
reduziert oder eliminiert wird. Die Transformation des Chloroplastengenoms erfolgt
im Allgemeinen durch ein Verfahren, das schematisch bei Klaus
et al., 2004, [Nature Biotechnology 22 (2), 225–229] gezeigt
wurde. Kurz gesagt werden die zu transformierenden Sequenzen gemeinsam
mit einem Selektionsmarkergen zwischen flankierende Sequenzen, die
zu dem Chloroplastengenom homolog sind, kloniert. Diese homologen
flankierenden Sequenzen steuern die ortsgerichtete Integration in
das Plastom. Die Plastidentransformation ist für viele
unterschiedliche Pflanzenarten beschrieben worden, und eine Übersicht findet
sich bei Bock (2001) Transgenic plastids in basic research
and plant biotechnology. J. Mol. Biol. 2001 Sept. 21; 312 (3): 425–38 oder Maliga,
P (2003), Progress towards commercialization of plastid transformation technology.
Trends Biotechnol. 21, 20–28. Über einen
weiteren Fortschritt in der Biotechnologie wurde in jüngster
Zeit in Form von markerfreien Plastidentransformanten, die durch
ein transientes cointegriertes Markergen erzeugt werden können,
berichtet (Klaus et al., 2004, Nature Biotechnology 22 (2),
225–229).
-
T-DNA-Aktivierungstagging
-
T-DNA-Aktivierungstagging
(Hayashi et al. Science (1992) 1350–1353)
beinhaltet die Einführung von T-DNA, die gewöhnlich
einen Promotor enthält (es kann auch ein Translations-Enhancer
oder ein Intron sein) in der genomischen Region des interessierenden
Gens von bis zu 10 kb stromaufwärts oder stromabwärts
der kodierenden Region eines Gens in einer Konfiguration, so dass
der Promotor die Expression des angezielten Gens steuert. Die Regulation
der Expression des angezielten Gens durch seinen natürlichen
Promotor wird gewöhnlich unterbrochen und das Gen gerät
unter die Kontrolle des neu eingeführten Promotors. Der
Promotor ist gewöhnlich in eine T-DNA eingebettet. Diese
T-DNA wird statistisch in das Pflanzengenom eingeführt, beispielsweise
durch Agrobacterium Infektion und führt zu modifizierter
Expression der Gene nahe der eingeführten T-DNA. Die resultierenden
transgenen Pflanzen zeigen dominante Phänotypen aufgrund
der modifizierten Expression der Gene nahe dem eingeführten
Promotor.
-
TILLING
-
Der
Begriff TILLING ist eine Abkürzung für ”Targeted
Induced Local Lesions In Genomes” und steht für
eine Mutagenesetechnologie, die sich dazu eignet, Nukleinsäuren,
die Proteine mit modifizierter Expression und/oder Aktivität
kodieren, zu erzeugen und/oder zu identifizieren. TILLING ermöglicht
auch die Selektion von Pflanzen, die solche Mutantenvarianten tragen.
Diese Mutantenvarianten können eine modifizierte Expression aufweisen,
und zwar entweder bezüglich der Stärke oder der
Lokalisierung oder des Zeitpunkts (wenn die Mutationen zum Beispiel
den Promotor betreffen). Diese Mutantenvarianten können
eine höhere Aktivität aufweisen als diejenige
des Gens in seiner natürlichen Form. Beim TILLING wird
Mutagenese in hoher Dichte mit Screening-Methoden mit hohem Durchsatz
kombiniert. Die Schritte, die beim TILLING gewöhnlich erfolgen, sind:
(a) EMS-Mutagenese (Redei GP und Koncz C (1992), In Methods
in Arabidopsis Research, Koncz C, Chua NH, Schell J, Hrsg. Singapur,
World Scientific Publishing Co, S. 16–82; Feldmann
et al., (1994) In Meyerowitz EM, Somerville CR, Hrsg, Arabidopsis.
Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, S.
137–172; Lightner J und Caspar T (1998),
In J Martinez-Zapater, J Salinas, Hrsg., Methods an Molecular Biology,
Band 82. Humana Press, Totowa, NJ, S. 91–104);
(b) DNA-Herstellung und Poolen von Individuen; (c) PCR-Amplifikation
einer interessierenden Region; (d) Denaturieren und Annealing, um
die Bildung von Heteroduplices zu ermöglichen; (e) DHPLC,
wo das Vorhandensein eines Heteroduplex in einem Pool als zusätzlicher
Peak im Chromatogramm nachgewiesen wird; (f) Identifizieren des
mutierten Individuums; und (g) Sequenzieren des PCR-Produkts der
Mutante. TILLING-Methoden sind im Stand der Technik bestens bekannt (McCallum
et al., (2000), Nat. Biotechnol. 18: 455–457; in einem Übersichtsartikel
von Stemple (2004), Nat. Rev. Genet. 5 (2): 145–50)
erwähnt.
-
Homologe Rekombination
-
Homologe
Rekombination ermöglicht die Einführung einer
ausgewählten Nukleinsäure in ein Genom, an einer
definierten ausgewählten Position. Die homologe Rekombination
ist eine Standard-Technik, die routinemäßig in
den biologischen Wissenschaften für niedere Organismen
verwendet wird, wie Hefe und das Moos Physcomitrella. Verfahren
zur Durchführung der homologen Rekombination in Pflanzen
wurden nicht nur für Pflanzenmodelle beschrieben (Offringa
et al. (1990) EMBO J 9 (10): 3077–84) sondern
auch für Kulturpflanzen, wie beispielsweise Reis (Terada
et al. (2002) Nat Biotech 20 (10): 1030–4; Iida
and Terada (2004) Curr Opin Biotech 15 (2): 132–8),
und es gibt Ansätze, die gewöhnlich anwendbar
sind, ungeachtet des Zielorganismus (Miller et al, Nature
Biotechnol. 25, 778–785, 2007).
-
Ertrag
-
Der
Begriff ”Ertrag” bedeutet im Allgemeinen ein messbares
Produkt von wirtschaftlichem Wert, das gewöhnlich mit einer
bezeichneten Kulturpflanze, einem Ort und einem Zeitraum einher
geht. Die einzelnen Pflanzenteile tragen direkt aufgrund ihrer Anzahl,
Größe und/oder ihres Gewichts zum Ertrag bei,
oder der tatsächliche Ertrag ist derjenige Ertrag pro m2 für eine Kulturpflanze und ein
Jahr, der dadurch bestimmt wird, dass man die Gesamtproduktion (was
sowohl geerntete als auch geschätzte Produktion beinhaltet)
durch die bewirtschafteten m2 dividiert.
Der Begriff ”Ertrag” einer Pflanze kann die vegetative
Biomasse (Wurzel und/oder Spross-Biomasse), Fortpflanzungsorgane
und/oder Verbreitungseinheiten (wie Samen) dieser Pflanze betreffen.
-
Jungpflanzenvitalität
-
”Jungpflanzenvitalität” steht
für das aktive gesunde gut balancierte Wachstum, insbesondere
während früher Stadien des Pflanzenwachstums,
und kann sich aus einer gesteigerten Pflanzen-Fitness ergeben, beispielsweise
wenn die Pflanzen besser an ihre Umgebung angepasst sind (d. h.
durch Optimierung der Verwendung von Energiequellen und der Verteilung
zwischen Spross und Wurzel). Pflanzen mit eine Jungpflanzenvitalität
zeigen auch ein gesteigertes Überleben des Keimlings und
eine bessere Entwicklung der Kulturpflanze, was häufig
zu sehr einheitlichen Feldern (wobei die Kulturpflanze einheitlich
wächst, d. h. die Mehrheit der Pflanzen erreicht im Wesentlichen
gleichzeitig verschiedene Entwicklungsstadien) und oft einem besseren
und höheren Ertrag führt. Daher kann die Jungpflanzenvitalität
durch Messen verschiedener Faktoren bestimmt werden, wie Tausendkorngewicht,
Prozent Keimung, Prozent Auflaufen, Wachstum des Keimlings, Höhe
des Keimlings, Wurzellänge, Biomasse von Wurzel und Spross
und vielen anderen.
-
Erhöhen/Verbessern/Fördern
-
Die
Begriffe ”erhöhen”, ”verbessern” oder ”steigern” sind
untereinander austauschbar und sollen im Rahmen der vorliegenden
Erfindung mindestens 3%, 4%, 5%, 6%, 7%, 8%, 9% oder 10%, vorzugsweise
mindestens 15% oder 20%, stärker bevorzugt 25%, 30%, 35%
oder 40% mehr Ertrag und/oder Wachstum im Vergleich zu Kontrollpflanzen
wie im vorliegenden Text definiert bedeuten.
-
Samenertrag
-
Ein
erhöhter Samenertrag kann sich als eine oder mehrere der
folgenden Erscheinungen äußern: a) Erhöhung
der Samenbiomasse (Samengesamtgewicht), entweder auf Grundlage der
einzelnen Samen und/oder pro Pflanze und/oder oder pro m2; b) erhöhte Anzahl Blüten
pro Pflanze; c) erhöhte Anzahl (gefüllter) Samen;
d) erhöhte Samenfüllungsrate (die als Verhältnis
zwischen der Anzahl gefüllter Samen dividiert durch die
Gesamtzahl Samen ausgedrückt wird); e) erhöhter
Harvest Index, der als Verhältnis des Ertrags von Erntegut
wie Samen dividiert durch die Gesamtbiomasse ausgedrückt
wird; sowie f) erhöhtes Tausendkorngewicht (TKG), das aufgrund
der Anzahl der gezählten gefüllten Samen und ihres
Gesamtgewichts extrapoliert wird. Ein erhöhtes TKG kann
das Ergebnis einer erhöhten Samengröße
und/oder eines erhöhten Samengewichts sein und kann auch
das Ergebnis einer Erhöhung der Embryo- und/oder Endospermgröße
sein.
-
Eine
Erhöhung des Samenertrags kann sich auch als Erhöhung
der Samengröße und/oder des Samenvolumens äußern.
Eine Erhöhung des Samenertrags kann sich auch als Vergrößerung
der Samenfläche und/oder Samenlänge und/oder Samenbreite
und/oder des Samenumfangs äußern. Ein erhöhter
Ertrag kann auch zu einer modifizierten Architektur führen
oder kann aufgrund einer modifizierten Architektur vorliegen.
-
Greenness Index
-
Der ”Greenness
Index”, wie hier verwendet, wird aus digitalen Bildern
von Pflanzen berechnet. Für jeden zu dem Pflanzenobjekt
auf dem Bild gehörigen Pixel wird das Verhältnis
des Grünwertes gegenüber dem Rotwert (im RGB-Modell
für Farbkodierung) berechnet. Der Greenness Index wird
ausgedrückt als Prozent der Pixel, für die das
Grün:Rot-Verhältnis eine bestimmte Schwelle überschreitet.
Unter normalen Wachstumsbedingungen, unter Salzstress-Wachstumsbedingungen,
und unter den Wachstumsbedingungen einer reduzierten Nährstoffverfügbarkeit
wird der Greenness Index der Pflanzen bei der letzten Bildnahme
vor dem Blühen gemessen. Unter Trockenstress-Wachstumsbedingungen,
wird der Greenness Index der Pflanzen in der erste Bildnahme nach
dem Trockenstress gemessen.
-
Pflanze
-
Der
Begriff ”Pflanze” umfasst im vorliegenden Zusammenhang
ganze Pflanzen, Vorfahren und Nachfahren der Pflanzen und Pflanzenteile,
wozu Samen, Sprosse, Stängel, Blätter, Wurzeln
(einschließlich Knollen), Blüten und Gewebe und
Organe gehören, wobei jedes der genannten Objekte das interessierende
Gen bzw. die interessierende Nukleinsäure umfasst. Der
Begriff ”Pflanze” umfasst auch Pflanzenzellen,
Suspensionskulturen, Kallusgewebe, Embryonen, meristematische Regionen,
Gametophyten, Sporophyten, Pollen und Mikrosporen, wobei wiederum
jedes der genannten Objekte das interessierende Gen bzw. die interessierende Nukleinsäure
umfasst.
-
Zu
Pflanzen, die sich für die erfindungsgemäßen
Verfahren besonders eignen, gehören all diejenigen Pflanzen,
die zu der Überfamilie Viridiplantae gehören,
insbesondere monokotyle und dikotyle Pflanzen, darunter Futterleguminosen,
Zierpflanzen, Nahrungsmittelpflanzen, Bäume oder Sträucher
aus der Liste, die unter Anderem die folgenden Pflanzen umfasst:
Acer spp., Actinidia spp., Abelmoschus spp., Agave sisalana, Agropyron
spp., Agrostis stolonifera, Allium spp., Amaranthus spp., Ammophila
arenaria, Ananas comosus, Annona spp., Apium graveolens, Arachis
spp, Artocarpus spp., Asparagus officinalis, Avena spp. (bspw. Avena sativa,
Avena fatua, Avena byzantina, Avena fatua var. sativa, Avena hybrida),
Averrhoa carambola, Bambusa sp., Benincasa hispida, Bertholletia
excelsea, Beta vulgaris, Brassica spp. (bspw. Brassica napus, Brassica rapa
ssp. [Canola-Raps, normaler Raps, Rübsen]), Cadaba farinosa,
Camellia sinensis, Canna indica, Cannabis sativa, Capsicum spp.,
Carex elata, Carica papaya, Carissa macrocarpa, Carya spp., Carthamus
tinctorius, Castanea spp., Ceiba pentandra, Cichorium endivia, Cinnamomum
spp., Citrullus lanatus, Citrus spp., Cocos spp., Coffea spp., Colocasia
esculenta, Cola spp., Corchorus sp., Coriandrum sativum, Corylus
spp., Crataegus spp., Crocus sativus, Cucurbita spp., Cucumis spp.,
Cynara spp., Daucus carota, Desmodium spp., Dimocarpus longan, Dioscorea
spp., Diospyros spp., Echinochloa spp., Elaeis (bspw. Elaeis guineensis,
Elaeis oleifera), Eleusine coracana, Eragrostis tef, Erianthus sp.,
Eriobotrya japonica, Eucalyptus sp., Eugenia uniflora, Fagopyrum
spp., Fagus spp., Festuca arundinacea, Ficus carica, Fortunella
spp., Fragaria spp., Ginkgo biloba, Glycine spp. (bspw. Glycine
max, Soja hispida oder Soja max), Gossypium hirsutum, Helianthus
spp. (bspw. Helianthus annuus), Hemerocallis fulva, Hibiscus spp.,
Hordeum spp. (bspw. Hordeum vulgare), Ipomoea batatas, Juglans spp.,
Lactuca sativa, Lathyrus spp., Lens culinaris, Linum usitatissimum,
Litchi chinensis, Lotus spp., Luffa acutangula, Lupinus spp., Luzula
sylvatica, Lycopersicon spp. (bspw. Lycopersicon esculentum, Lycopersicon
lycopersicum, Lycopersicon pyriforme), Macrotyloma spp., Malus spp.,
Malpighia emarginata, Mammea americana, Mangifera indica, Manihot
spp., Manilkara zapota, Medicago sativa, Melilotus spp., Mentha
spp., Miscanthus sinensis, Momordica spp., Morus nigra, Musa spp.,
Nicotiana spp., Olea spp., Opuntia spp., Ornithopus spp., Oryza
spp. (bspw. Oryza sativa, Oryza latifolia), Panicum miliaceum, Panicum
virgatum, Passiflora edulis, Pastinaca sativa, Pennisetum sp., Persea
spp., Petroselinum crispum, Phalaris arundinacea, Phaseolus spp.,
Phleum pratense, Phoenix spp., Phragmites australis, Physalis spp.,
Pinus spp., Pistacia vera, Pisum spp., Poa spp., Populus spp., Prosopis
spp., Prunus spp., Psidium spp., Punica granatum, Pyrus communis,
Quercus spp., Raphanus sativus, Rheum rhabarbarum, Ribes spp., Ricinus
communis, Rubus spp., Saccharum spp., Salix sp., Sambucus spp.,
Secale cereale, Sesamum spp., Sinapis sp., Solanum spp. (bspw. Solanum
tuberosum, Solanum integrifolium oder Solanum lycopersicum), Sorghum
bicolor, Spinacia spp., Syzygium spp., Tagetes spp., Tamarindus
indica, Theobroma cacao, Trifolium spp., Tripsacum dactyloides,
Triticosecale rimpaui, Triticum spp. (bspw. Triticum aestivum, Triticum
durum, Triticum turgidum, Triticum hybernum, Triticum macha, Triticum
sativum, Triticum monococcum oder Triticum vulgare), Tropaeolum
minus, Tropaeolum majus, Vaccinium spp., Vicia spp., Vigna spp.,
Viola odorata, Vitis spp., Zea mays, Zizania palustris, Ziziphus
spp.
-
Genaue Beschreibung der Erfindung
-
I. Eine LOB-Domäne enthaltendes
Protein (LOB: Lateral Organ Boundaries, laterale Organgrenzen)
-
Es
wurde jetzt überraschenderweise gefunden, dass die Modulation
der Expression einer Nukleinsäure, die ein LBD-Polypeptid
kodiert, in einer Pflanze zu Pflanzen mit gesteigerten Ertragsmerkmalen
im Vergleich zu Kontrollpflanzen führt. In einer ersten
Ausführungsform stellt die vorliegende Erfindung ein Verfahren zur
Steigerung der Ertragsmerkmale in Pflanzen im Vergleich zu Kontrollpflanzen
bereit, umfassend das Modulieren der Expression der Nukleinsäure,
die ein LBD-Polypeptid kodiert, in einer Pflanze.
-
Ein
bevorzugtes Verfahren zum Modulieren (vorzugsweise Erhöhen)
der Expression einer Nukleinsäure, die ein LBD-Polypeptid
kodiert, besteht darin, dass man eine Nukleinsäure, die
für ein LBD-Polypeptid kodiert, in eine(r) Pflanze einbringt
und exprimiert.
-
Wird
im folgenden Text auf ein ”Protein, das für die
erfindungsgemäßen Verfahren geeignet ist” Bezug genommen,
soll darunter ein LBD-Polypeptid, wie im vorliegenden Text definiert,
verstanden werden. Wird im folgenden Text auf eine ”Nukleinsäure,
die für die erfindungsgemäßen Verfahren
geeignet ist” Bezug genommen, soll dies eine Nukleinsäure
bedeuten, die ein solches LBD-Polypeptid kodieren kann. Die Nukleinsäure, die
in eine Pflanze eingeführt werden soll (und daher zur Durchführung
der erfindungsgemäßen Verfahren geeignet ist),
ist jede Nukleinsäure, die für den Typ Protein
kodiert, der im Folgenden beschrieben wird, und wird im Folgenden
auch als ”LBD-Nukleinsäure” oder ”LBD-Gen” bezeichnet.
-
Ein
LBD-Polypeptid, wie hier definiert, betrifft jedes Polypeptid, das
eine DUF260-Domäne (Pfam-Zugangsnummer PF03195, Interpro-Zugangsnummer
IPR004883, siehe 1) umfasst. Vorzugsweise bildet die
LBD-Polypeptidsequenz, wenn sie zur Konstruktion eines Stammbaums,
wie dem in 3 dargestellten (oder wie von Yang
et al., 2006, unter Verwendung des HKY-Verfahrens (Hasegawa
et al., J. Mol. Evol. 22, 160–174, 1985) definiert)
verwendet wird, eher Cluster mit der Gruppe der Klasse II-LOB-Domäne-Polypeptide,
welche die durch SEQ ID NO: 2 dargestellte Aminosäuresequenz
umfassen, als mit jeder anderen Gruppe.
-
Weiterhin
umfasst das LBD-Protein vorzugsweise mindestens eines der folgenden
konservierten Motive:
- Motiv 1: MSCNGCRXLRKGCX (SEQ ID NO:
5); wobei X an Position 8 jede Aminosäure, aber vorzugsweise V
oder I sein kann und wobei X an Position 14 jede Aminosäure
sein kann, aber vorzugsweise eine Aminosäure aus S, G oder
N ist.
- Motiv 2: QXXATXFXAKFXGR (SEQ ID NO: 6), wobei X an Position
2 jede Aminosäure sein kann, aber vorzugsweise eine Aminosäure
aus A, S, oder G ist; wobei X an Position 3 jede Aminosäure,
aber vorzugsweise eine Aminosäure aus N, Q oder H sein
kann; wobei X an Position 6 jede Aminosäure, aber vorzugsweise
eine Aminosäure aus V, L oder I sein kann; wobei X an Position
8 jede Aminosäure, aber vorzugsweise eine Aminosäure
aus L, V, A oder I sein kann; und wobei X an Position 12 jede Aminosäure,
aber vorzugsweise eine Aminosäure aus Y oder F sein kann.
- Motiv 3: FXSLLXEAXG (SEQ ID NO: 7); wobei X an Position 2 jede
Aminosäure, aber vorzugsweise eine Aminosäure
aus R, S, K oder Q sein kann; wobei X an Position 6 jede Aminosäure,
aber vorzugsweise eine Aminosäure aus Y, H oder F sein
kann; und wobei X an Position 9 jede Aminosäure, aber vorzugsweise
C oder A sein kann.
-
Weiterhin
bevorzugt umfasst das LBD-Protein mehr als sieben, stärker
bevorzugt mindestens neun Cys-Reste und umfasst nicht die DP(V/I)YG-Signatur
(SEQ ID NO: 8).
-
Die
DUF260-Domäne ist gekennzeichnet durch das Vorliegen eines
C-x(2)-C-x(6)-C-x(3)-C-Motivs (SEQ ID NO: 9), wobei X jede Aminosäure
sein kann.
-
Der
Begriff ”Domäne” und ”Motiv” wird
hier im Abschnitt ”Definitionen” definiert. Für
die Identifikation von Domänen gibt es Spezialdatenbanken,
zum Beispiel SMART (Schultz et al., (1998), Proc. Natl.
Acad. Sci. USA 95, 5857–5864; Letunic
et al., (2002), Nucleic Acids Res. 30, 242–244,
InterPro (Mulder et al., (2003), Nucl. Acids Res. 31, 315–318,
Prosite (Sucher und Bairoch (1994), A generalized Profile
syntax for biomolecular sequences motifs and its function in automatic
sequence interpretation. (In) ISMB-94; Proceedings 2nd International
Conference an Intelligent Systems for Molecular Biology. Altman
R., Brutlag D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61,
AAAI Press, Menlo Park; Hulo et al., Nucl. Acids
Res. 32: D134–D137, (2004), oder Pfam (Bateman
et al., Nucleic Acids Research 30 (1): 276–280 (2002).
Ein Satz von Werkzeugen für die In-silico-Analyse von Proteinsequenzen
ist auf dem ExPASY-Proteomics-Server verfügbar (Swiss Institute
of Bioinformatics (Gasteiger et al., ExPASy: the proteomics
server for in-depth Protein knowledge and analysis, Nucleic Acids
Res. 31: 3784–3788 (2003)). Domänen können
auch unter Verwendung von Routinetechniken, wie Sequenz-Alignment,
identifiziert werden.
-
Verfahren
für das Alignment von Sequenzen für Vergleichszwecke
sind in der Fachwelt gut bekannt, dazu zählen GAP, BESTFIT,
BLAST, FASIA und TFASTA. Bei GAP wird der Algorithmus von Needleman
und Wunsch ((1970), J. Mol. Biol. 48: 443–453)
dazu verwendet, das globale Alignment (d. h. das Alignment, das sich über
die vollständigen Sequenzen erstreckt) von zwei Sequenzen
zu finden, das die Anzahl der ”matches” maximiert
und die Anzahl der ”gaps” minimiert. Beim BLAST-Algorithmus
(Altschul et al., (1990), J. Mol. Biol. 215: 403–10)
wird die Sequenzidentität in Prozent berechnet, und es
wird eine statistische Analyse der Ähnlichkeit zwischen
den beiden Sequenzen durchgeführt. Die Software für
die Durchführung einer BLAST-Analyse ist über
das National Centre for Biotechnology Information (NCBI) öffentlich
zugänglich. Homologe können zum Beispiel unter
Verwendung des multiplen Sequenz-Alignment-Algorithmus ClustalW
(Version 1.83) leicht identifiziert werden, und zwar mit den Default-Parametern
für paarweises Alignment und einer Scoring-Methode in Prozent.
Die Gesamtprozent an Ähnlichkeit und Identität
können auch unter Verwendung eines der im MatGAT-Software-Paket
verfügbaren Verfahren bestimmt werden (Campanella
et al., BMC Bioinformatics. 2003, Juli, 10; 4: 29. MatGAT: an application
that generstes similarity/identity matrices using Protein or DNA sequences.).
Für eine Optimierung des Alignments zwischen konservierten
Motiven können kleinere Änderungen per Hand vorgenommen
werden, wie dem Fachmann klar ist. Weiterhin können anstelle
von Volllängensequenzen auch spezifische Domänen
für die Identifikation von Homologen verwendet werden.
Die Sequenzidentitätswerte können mit den oben
erwähnten Programmen unter Verwendung der Default-Parameter über die
gesamte Nukleinsäure- oder Aminosäuresequenz oder über
ausgewählte Domänen oder ein konservierte(s) Motiv(e)
bestimmt werden.
-
Außerdem
können LBD-Polypeptide (zumindest in ihrer nativen Form)
als Transkriptionsfaktoren in der Regel DNA-Bindungsaktivität
aufweisen. Werkzeuge und Techniken zum Messen der DNA-Bindungsaktivität
sind im Stand der Technik bekannt und beinhalten zum Beispiel Gelretardationsassays.
Experimentelle Ansätze zur Charakterisierung der Aktivität
von Transkriptionsfaktoren lassen sich zum Beispiel in Sambrook et
al. (2001) Molecular Cloning: a laboratory manual, 3. Ausgabe Cold
Spring Harbor Laboratory Press, CSH, New York, oder in
den Bänden 1 und 2 des (jährlich aktualisierten) Ausubel
et al., Current Protocols in Molecular Biology, Current Protocols,
finden.
-
Die
vorliegende Erfindung wird dadurch veranschaulicht, dass man Pflanzen
mit der Nukleinsäuresequenz gemäß SEQ
ID NO: 1, welche die Polypeptidsequenz gemäß SEQ
ID NO: 2 kodiert, transformiert. Die Durchführung der Erfindung
ist jedoch nicht auf diese Sequenzen beschränkt; die erfindungsgemäßen
Verfahren können vorteilhaft unter Verwendung jeder beliebigen
Nukleinsäure, die ein LBD-Polypeptid kodiert, oder des
LBD-Polypeptids, wie hier definiert, durchgeführt werden.
-
Beispiele
für Nukleinsäuren, die LBD-Polypeptide kodieren,
sind hier in Tabelle A1 von Beispiel 1 angegeben. Solche Nukleinsäuren
eignen sich zur Durchführung der erfindungsgemäßen
Verfahren. Die in Tabelle A1 von Beispiel genannten Aminosäuresequenzen
sind beispielhafte Sequenzen von Orthologen und Paralogen des LBD-Polypeptids
gemäß SEQ ID NO: 2, wobei die Begriffe ”Orthologe” und ”Paraloge” wie
hier definiert sind. Weitere Orthologe und Paraloge können
leicht mittels Durchführen einer so genannten reziproken
Blast-Suche identifiziert werden. Dies beinhaltet üblicherweise
eine erste BLAST, bei der mit einer Abfragesequenz ein ”BLASTing” gegen
eine beliebige Sequenzdatenbank, wie die öffentlich zugängliche
NCBI-Datenbank, durchgeführt wird (zum Beispiel unter Verwendung
von einer der in Tabelle A1 von Beispiel 1 aufgelisteten Sequenzen).
BLASTN oder TBLASTX (unter Verwendung von Standard-Default-Werten)
werden im Allgemeinen verwendet, wenn man von einer Nukleotidsequenz
ausgeht, und BLASTP oder TBLASTN (unter Verwendung von Standard-Default-Werten),
wenn man von einer Proteinsequenz ausgeht. Die BLAST-Ergebnisse
können gegebenenfalls gefiltert werden. Mit den Volllängen-Sequenzen
der gefilterten oder ungefilterten Ergebnisse wird anschließend
ein zweites BLASTing gegen Sequenzen des Organismus, aus dem die
Abfragesequenz stammt, durchgeführt (ist die Abfragesequenz
die SEQ ID NO: 1 oder SEQ ID NO: 2, dann wäre das zweite
BLASTing daher gegen Sequenzen aus Arabidopsis). Die Ergebnisse
des ersten und des zweiten BLASTing werden dann verglichen. Ein
Paralog wird identifiziert, wenn ein hochrangiger Hit von dem ersten BLASTing
aus derselben Art stammt wie die Abfragesequenz, in diesem Fall
führt ein zweites BLASTing idealerweise zu der Abfragesequenz
unter den höchstrangigen Hits; ein Ortholog wird identifiziert,
wenn ein hochrangiger Hit in dem ersten BLASTing nicht von derselben
Art stammt wie die Abfragesequenz, und führt vorzugsweise
beim zweiten BLASTing dazu, dass die Abfragesequenz unter den höchstrangigen
Hits ist.
-
Hochrangige
Hits sind solche mit niedrigem E-Wert. Je niedriger der E-Wert,
desto signifikanter der ”Score” (anders ausgedrückt,
desto niedriger die Wahrscheinlichkeit, dass der Hit durch Zufall
gefunden wurde). Die Berechnung des E-Werts ist in der Fachwelt
gut bekannt. Das ”Scoring” der Vergleiche erfolgt
nicht nur mittels E-Werten, sondern auch anhand der Prozent Identität.
Prozent Identität bezieht sich auf die Anzahl an identischen
Nukleotiden (oder Aminosäuren) zwischen den zwei verglichenen
Nukleinsäure-(oder Polypeptid-)Sequenzen über
eine bestimmte Länge. Bei großen Familien kann
man ClustalW und anschließend einen Neighbour-Joining-Tree
verwenden, um die Bildung von Clustern verwandter Gene leichter
sichtbar zu machen und Orthologe und Paraloge zu identifizieren.
-
Zur
Durchführung der erfindungsgemäßen Verfahren
können auch Nukleinsäurevarianten geeignet sein.
Zu solchen Varianten zählen beispielsweise Nukleinsäuren,
die für Homologe und Derivate von einer der in Tabelle
A1 von Beispiel 1 genannten Aminosäuresequenzen kodieren,
wobei die Begriffe ”Homolog” und ”Derivat” wie
hier definiert sind. Für die erfindungsgemäßen
Verfahren sind auch Nukleinsäuren geeignet, die Homologe
und Derivate von Orthologen oder Paralogen von einer der in Tabelle
A1 von Beispiel 1 genannten Aminosäuresequenzen kodieren.
Homologe und Derivate, die für die erfindungsgemäßen
Verfahren geeignet sind, weisen im Wesentlichen dieselbe biologische
und funktionelle Aktivität auf wie das unmodifizierte Protein, von
dem sie stammen.
-
Zu
weiteren Nukleinsäurevarianten, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet sind,
zählen Abschnitte von Nukleinsäuren, die für
LBD-Polypeptide kodieren, Nukleinsäuren, die mit Nukleinsäuren,
die für LBD-Polypeptide kodieren, hybridisieren, Spleißvarianten
von Nukleinsäuren, die für LBD-Polypeptide kodieren,
allelische Varianten von Nukleinsäuren, die für
LBD-Polypeptide kodieren, und Varianten von Nukleinsäuren,
die für LBD-Polypeptide kodieren, die mittels ”gene
shuffling” erhalten wurden. Die Begriffe hybridisierende
Sequenz, Spleißvariante, allelische Variante und ”gene
shuffling” sind wie hier definiert.
-
Vorteilhafterweise
stellt die vorliegende Erfindung bisher unbekannte LBD-Nukleinsäure-
und Polypeptidsequenzen bereit.
-
Gemäß einer
weiteren Ausführungsform der vorliegenden Erfindung wird
ein isoliertes Nukleinsäuremolekül bereitgestellt,
das Folgendes umfasst:
- (i) eine durch SEQ ID
NO: 69 dargestellte Nukleinsäure;
- (ii) eine Nukleinsäure oder ein Fragment davon, die/das
zu einer der unter (i) angegebenen SEQ ID NO: komplementär
ist;
- (iii) eine Nukleinsäure, die ein LBD-Polypeptid kodiert
und, in steigender Reihenfolge der Präferenz, mindestens
60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder
mehr Sequenzidentität mit der SEQ ID NO: 70 besitzt;
- (iv) eine Nukleinsäure, die unter stringenten Bedingungen
an eine der unter (i), (ii) oder (iii) oben angegebenen Nukleinsäuren
hybridisieren kann.
-
Gemäß einer
weiteren Ausführungsform der vorliegenden Erfindung wird
deshalb ein isoliertes Polypeptid bereitgestellt, das Folgendes
umfasst:
- (i) eine Aminosäuresequenz,
die, in steigender Reihenfolge der Präferenz, mindestens
60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder
100% Sequenzidentität mit der unter SEQ ID NO: 70 angegebenen
Aminosäuresequenz hat;
- (ii) Derivate von einer der unter (i) angegebenen Aminosäuresequenzen.
-
Nukleinsäuren,
die LBD-Polypeptide kodieren, müssen keine Volllängen-Nukleinsäuren
sein, da die Durchführung der erfindungsgemäßen
Verfahren nicht auf die Verwendung von Volllängen-Nukleinsäuresequenzen
angewiesen ist. Erfindungsgemäß wird daher ein
Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen bereitgestellt,
bei dem man in eine(r) Pflanze einen Abschnitt von einer der in
Tabelle A1 von Beisipiel 1 angegebenen Nukleinsäuresequenzen
oder einen Abschnitt einer Nukleinsäure, die ein Ortholog,
Paralog oder Homolog von einer der in Tabelle A1 von Beispiel 1
genannten Aminosäuresequenzen kodiert, einführt
und exprimiert.
-
Ein
Abschnitt einer Nukleinsäure kann zum Beispiel dadurch
hergestellt werden, dass man eine oder mehrere Deletionen an der
Nukleinsäure durchführt. Die Abschnitte können
in isolierter Form verwendet werden oder können an andere
kodierende (oder nichtkodierende) Sequenzen fusioniert werden, um
zum Beispiel ein Protein herzustellen, das mehrere Aktivitäten
in sich vereinigt. Wenn es an andere kodierende Sequenzen fusioniert
ist, kann das so erhaltene, nach der Translation hergestellte Polypeptid
größer sein als für den Proteinabschnitt
vorhergesagt wurde.
-
Abschnitte,
die für die erfindungsgemäßen Verfahren
geeignet sind, kodieren ein LBD-Polypeptid, wie hier definiert,
und haben im Wesentlichen dieselbe biologische Aktivität
wie die in Tabelle A1 von Beispiel 1 angegebenen Aminosäuresequenzen.
Vorzugsweise handelt es sich bei dem Abschnitt um einen Abschnitt von
einer der in Tabelle A1 von Beispiel 1 genannten Nukleinsäuren
oder um einen Abschnitt einer Nukleinsäure, die ein Ortholog
oder Paralog von einer der in Tabelle A1 von Beispiel 1 genannten
Aminosäuresequenzen kodiert. Vorzugsweise ist der Abschnitt
mindestens 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000 aufeinander
folgende Nukleotide lang, wobei es sich bei den aufeinander folgenden
Nukleotiden um eine der in Tabelle A1 von Beispiel 1 genannten Nukleinsäuresequenzen
oder um eine Nukleinsäure handelt, die ein Ortholog oder
Paralog von einer der in Tabelle A1 von Beispiel 1 genannten Aminosäuresequenzen
kodiert. Am stärksten bevorzugt ist der Abschnitt ein Abschnitt
der Nukleinsäure gemäß SEQ ID NO: 1.
Vorzugsweise kodiert der Abschnitt eine Aminosäuresequenz,
die, wenn sie zur Konstruktion eines Stammbaums, wie dem in 3 dargestellten
(oder von Yang et al., 2006, unter Verwendung des
HKY-Verfahrens (Hasegawa et al., J. Mol. Evol. 22, 160–174,
1985) definierten), verwendet wird, eher mit der Gruppe
der Klasse II-LOB-Domäne-Polypeptide, welche die Aminosäuresequenz
gemäß SEQ ID NO: 2 umfassen, als mit irgendeiner
anderen Gruppe Cluster bildet.
-
Eine
andere Nukleinsäurevariante, die für die erfindungsgemäßen
Verfahren geeignet ist, ist eine Nukleinsäure, die unter
Bedingungen einer verringerten Stringenz, vorzugsweise unter stringenten
Bedingungen, mit einer Nukleinsäure, die ein LBD-Polypeptid,
wie hier definiert, kodiert, oder mit einem Abschnitt, wie hier definiert,
hybridisieren kann.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Nukleinsäure,
die mit einer der in Tabelle A1 von Beispiel 1 angegebenen Nukleinsäuren
hybridisieren kann, einführt und exprimiert oder bei dem
man in eine(r) Pflanze eine Nukleinsäure, die mit einer
Nukleinsäure hybridisieren kann, die für ein Ortholog,
Paralog oder Homolog von einer der in Tabelle A1 von Beispiel 1
genannten Nukleinsäuresequenzen kodiert, einführt
und exprimiert.
-
Hybridisierende
Sequenzen, die für die erfindungsgemäßen
Verfahren geeignet sind, kodieren ein LBD-Polypeptid, wie hier definiert,
und weisen im Wesentlichen dieselbe biologische Aktivität
wie die in Tabelle A1 von Beispiel genannten Aminosäuresequenzen
auf. Vorzugsweise kann die hybridisierende Sequenz mit einer der
in Tabelle A1 von Beispiel 1 genannten Nukleinsäuren oder
mit einem Abschnitt von einer dieser Sequenzen hybridisieren, wobei
ein Abschnitt wie oben definiert ist, oder wobei die hybridisierende
Sequenz mit einer Nukleinsäuresequenz, die ein Ortholog
oder Paralog von einer der in Tabelle A1 von Beispiel 1 genannten
Aminosäuresequenzen kodiert, hybridisieren kann. Am stärksten
bevorzugt kann die hybridisierende Sequenz mit einer Nukleinsäure
gemäß SEQ ID NO: 1 oder einem Abschnitt davon
hybridisieren.
-
Vorzugsweise
kodiert die hybridisierende Sequenz eine Aminosäuresequenz,
die, wenn sie zur Konstruktion eines Stammbaums, wie dem in 3 dargestellten
(oder von Yang et al., 2006, unter Verwendung des
HKY-Verfahrens (Hasegawa et al., J. Mol. Evol. 22, 160–174,
1985) definierten), verwendet wird, eher mit der Gruppe
der Klasse II-LOB-Domäne-Polypeptide, welche die Aminosäuresequenz
gemäß SEQ ID NO: 2 umfassen, als mit irgendeiner
anderen Gruppe Cluster bildet.
-
Eine
andere Nukleinsäurevariante, die für die erfindungsgemäßen
Verfahren geeignet ist, ist eine Spleißvariante, die ein
LBD-Polypeptid, wie hier vorstehend definiert, kodiert, wobei eine
Spleißvariante wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen bereitgestellt,
bei dem man in eine(r) Pflanze eine Spleißvariante von
einer der in Tabelle A1 von Beispiel angegebenen Nukleinsäuresequenzen
oder eine Spleißvariante einer Nukleinsäure, die
ein Ortholog, Paralog oder Homolog von einer der in Tabelle A1 von
Beispiel 1 genannten Aminosäuresequenzen kodiert, einführt
und exprimiert.
-
Bei
bevorzugten Spleißvarianten handelt es sich um Spleißvarianten
einer Nukleinsäure gemäß SEQ ID NO: 1
oder eine Spleißvariante einer Nukleinsäure, die
ein Ortholog oder Paralog der SEQ ID NO: 2 kodiert. Vorzugsweise
bildet die Aminosäuresequenz, die von der Spleißvariante
kodiert wird, wenn sie zur Konstruktion eines Stammbaums, wie dem
in 3 dargestellten (oder von Yang et al.,
2006, unter Verwendung des HKY-Verfahrens (Hasegawa
et al., J. Mol. Evol. 22, 160–174, 1985) definierten),
verwendet wird, eher mit der Gruppe der Klasse II-LOB-Domäne-Polypeptide,
welche die Aminosäuresequenz gemäß SEQ
ID NO: 2 umfassen, als mit irgendeiner anderen Gruppe Cluster.
-
Eine
andere Nukleinsäurevariante, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet ist,
ist eine allelische Variante einer Nukleinsäure, die ein
LBD-Polypeptid, wie hier vorstehend definiert, kodiert, wobei eine
allelische Variante wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine allelische Variante
von einer der in Tabelle A1 von Beispiel 1 angegebenen Nukleinsäuresequenzen
einführt und exprimiert oder bei dem man in eine(r) Pflanze
eine allelische Variante einer Nukleinsäure, die für
ein Ortholog, Paralog oder Homolog von einer der in Tabelle A1 von
Beispiel 1 genannten Aminosäuresequenzen kodiert, einführt
und exprimiert.
-
Die
allelischen Varianten, die für die erfindungsgemäßen
Verfahren geeignet sind, haben im Wesentlichen dieselbe biologische
Aktivität wie das LBD-Polypeptid gemäß SEQ
ID NO: 2 und irgendeine der in Tabelle A1 von Beispiel 1 dargestellten
Aminosäuren. Allelische Varianten kommen in der Natur vor
und von den erfindungsgemäßen Verfahren wird die
Verwendung dieser natürlichen Allele mit umfasst. Vorzugsweise
handelt es sich bei der allelischen Variante um eine allelische
Variante gemäß SEQ ID NO: 1 oder eine allelische Variante
einer Nukleinsäure, die ein Ortholog oder Paralog der SEQ
ID NO: 2 kodiert. Vorzugsweise bildet die Aminosäuresequenz,
die von der allelischen Variante kodiert wird, wenn sie zur Konstruktion
eines Stammbaums, wie dem in 3 dargestellten
(oder von Yang et al., 2006, unter Verwendung des
HKY-Verfahrens (Hasegawa et al., J. Mol. Evol. 22, 160–174,
1985) definierten), verwendet wird, eher mit der Gruppe
der Klasse II-LOB-Domane-Polypeptide, welche die Aminosäuresequenz
gemäß SEQ ID NO: 2 umfassen, als mit irgendeiner
anderen Gruppe Cluster.
-
”Gene
shuffling” oder gerichtete Evolution kann ebenfalls dazu
verwendet werden, Varianten von Nukleinsäuren herzustellen,
die LBD-Polypeptide, wie oben definiert, kodieren, wobei der Begriff ”gene
shuffling” wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren für die Verbesserung von Ertragsmerkmalen
bei Pflanzen bereitgestellt, bei dem man in eine(r) Pflanze eine
Variante von einer der in Tabelle A1 von Beispiel 1 angegebenen Nukleinsäuresequenzen
einführt und exprimiert oder bei dem man in eine(r) Pflanze
eine Variante einer Nukleinsäure, die ein Ortholog, Paralog
oder Homolog von einer der in Tabelle A1 von Beispiel 1 angegebenen Aminosäuresequenzen
kodiert, einführt und exprimiert, wobei die Nukleinsäurevariante
mittels ”gene shuffling” erhalten wird.
-
Vorzugsweise
bildet die Aminosäuresequenz, die von der durch gene shuffling
erhaltenen Nukleinsäurevariante kodiert wird, wenn sie
zur Konstruktion eines Stammbaums, wie dem in 3 dargestellten
(oder von Yang et al., 2006, unter Verwendung des
HKY-Verfahrens (Hasegawa et al., J. Mol. Evol. 22, 160–174, 1985)
definierten), verwendet wird, eher mit der Gruppe der Klasse II-LOB-Domäne-Polypeptide,
welche die Aminosäuresequenz gemäß SEQ
ID NO: 2 umfassen, als mit irgendeiner anderen Gruppe Cluster.
-
Nukleinsäurevarianten
können weiterhin auch durch ortsgerichtete Mutagenese erhalten
werden. Zur Erzielung einer ortsgerichteten Mutagenese stehen mehrere
Verfahren zur Verfügung, von denen die üblichsten
Verfahren auf PCR-Basis sind (Current Protocols in Molecular
Biology. Wiley Hrsg.).
-
Nukleinsäuren,
die LBD-Polypeptide kodieren, können aus einer beliebigen
natürlichen oder künstlichen Quelle stammen. Die
Nukleinsäure kann von ihrer nativen Form in ihrer Zusammensetzung
und/oder genomischen Umgebung durch gezielte Manipulation durch
den Menschen modifiziert werden. Vorzugsweise stammt die Nukleinsäure,
die ein LBD-Polypeptid kodiert, aus einer Pflanze, weiterhin bevorzugt
aus einer zweikeimblättrigen Pflanze, stärker
bevorzugt aus der Familie Brassicaceae, am stärksten bevorzugt
stammt die Nukleinsäure aus Arabidopsis thaliana.
-
Die
Durchführung der erfindungsgemäßen Verfahren
führt zu Pflanzen mit verbesserten Ertragsmerkmalen. Insbesondere
führt die Durchführung der erfindungsgemäßen
Verfahren zu Pflanzen mit erhöhtem Ertrag, insbesondere
erhöhtem Samenertrag, im Vergleich zu Kontrollpflanzen.
Die Begriffe ”Ertrag” und ”Samenertrag” sind
hier im Abschnitt ”Definitionen” genauer beschrieben.
-
Werden
hier verbesserte Ertragsmerkmale erwähnt, so soll dies
eine Erhöhung der Biomasse (des Gewichts) von einem oder
mehreren Teilen einer Pflanze bedeuten, was oberirdische (erntbare)
Teile und/oder unterirdische (erntbare) Teile beinhalten kann. Insbesondere
sind solche oberirdischen erntbaren Teile die Sprossbiomasse und
Samen, und die Durchführung der erfindungsgemäßen
Verfahren führt zu Pflanzen mit erhöhter Sprossbiomasse
und Samenausbeute im Vergleich zu der Sprossbiomasse und der Samenausbeute von
Kontrollpflanzen.
-
Wählt
man Mais als Beispiel, kann sich eine Ertragserhöhung als
eines oder mehrere der folgenden Merkmale äußern:
Unter anderem erhöhte Anzahl an Pflanzen, die pro Hektar
oder Acre wachsen, eine Erhöhung der Anzahl an Ähren
pro Pflanze, eine Erhöhung der Anzahl an Reihen, der Anzahl
an Körnern pro Reihe, des Korngewichts, Tausendkorngewichts,
der Ährenlänge/des Ährendurchmessers,
eine Erhöhung der Samenfüllungsrate (d. h. der
Anzahl gefüllter Samen dividiert durch die Gesamtzahl der
Samen und multipliziert mit 100). Wählt man Reis als Beispiel,
kann sich eine Ertragserhöhung als eine Erhöhung
eines oder mehrerer der folgenden Merkmale ausdrücken:
Unter anderem Anzahl der Pflanzen pro Hektar oder Acre, Anzahl der Rispen
pro Pflanze, Anzahl der Ährchen pro Rispe, Anzahl der Blüten
(Einzelblüten) pro Rispe (ausgedrückt als Verhältnis
der Anzahl an gefüllten Samen zu der Anzahl an Primärrispen),
erhöhte Samenfüllungsrate (d. h. erhöhte
Anzahl gefüllter Samen dividiert durch die Gesamtzahl an
Samen und multipliziert mit 100), erhöhtes Tausendkorngewicht.
-
Die
vorliegende Erfindung stellt ein Verfahren zur Erhöhung
des Ertrags, insbesondere des Sprossbiomasse- und/oder Samenertrags
von Pflanzen, im Vergleich zu Kontrollpflanzen bereit, wobei man
bei dem Verfahren in einer Pflanze die Expression einer Nukleinsäure,
die ein LBD-Polypeptid, wie hier definiert, kodiert, moduliert,
vorzugsweise erhöht.
-
Weil
die erfindungsgemäßen transgenen Pflanzen einen
erhöhten Ertrag aufweisen, ist es wahrscheinlich, dass
diese Pflanzen (zumindest während eines Teils ihres Lebenszyklus)
eine erhöhte Wachstumsrate aufweisen als Kontrollpflanzen
in einem entsprechenden Stadium ihres Lebenszyklus.
-
Die
erhöhte Wachstumsrate kann für einen oder mehrere
Teile einer Pflanze (darunter Samen) spezifisch sein oder kann im
Wesentlichen in der ganzen Pflanze stattfinden. Pflanzen mit einer
erhöhten Wachstumsrate können einen kürzeren
Lebenszyklus aufweisen. Unter dem Lebenszyklus einer Pflanze kann
man diejenige Zeit verstehen, die die Pflanze benötigt,
um von einem trockenen reifen Samen bis zu dem Stadium heranzuwachsen,
in dem die Pflanze trockene reife Samen ähnlich dem Ausgangsmaterial
produziert hat. Dieser Lebenszyklus kann von Faktoren, wie Jungpflanzenvitalität,
Wachstumsrate, ”Greenness Index”, Blütezeit und
Geschwindigkeit der Samenreifung, beeinflusst werden. Die Erhöhung
der Wachstumsrate kann in einem Stadium oder in mehreren Stadien
im Lebenszyklus einer Pflanze oder im Wesentlichen während
des gesamten Lebenszyklus der Pflanze erfolgen. Eine erhöhte
Wachstumsrate während der Frühstadien im Lebenszyklus
einer Pflanze kann verbesserte Vitalität widerspiegeln.
Die Erhöhung der Wachstumsrate kann den Erntezyklus einer
Pflanze verändern, so dass die Pflanzen später
gesät werden können und/oder früher geerntet werden
können als dies sonst möglich wäre (ein ähnlicher
Effekt lässt sich mit einer früheren Blütezeit
erzielen). Ist die Wachstumsrate ausreichend erhöht, kann
dies weiteres Aussäen von Samen derselben Pflanzenart ermöglichen
(zum Beispiel Aussäen und Ernten von Reispflanzen und anschließendes
Aussäen und Ernten von weiteren Reispflanzen sogar innerhalb
einer herkömmlichen Wachstumsperiode). Wenn die Wachstumsrate
ausreichend erhöht ist, kann dies ebenso das weitere Aussäen
von Samen anderer Pflanzenarten ermöglichen (zum Beispiel
das Aussäen und Ernten von Maispflanzen und anschließend
zum Beispiel Aussäen und gegebenenfalls Ernten von Sojabohne,
Kartoffel oder einer anderen geeigneten Pflanze). Auch mehrmalige weitere
Ernten von demselben Wurzelstock können bei einigen Kulturpflanzen
möglich sein. Eine Veränderung des Erntezyklus
einer Pflanze kann zu einer Erhöhung der jährlichen
Biomasseproduktion pro Acre führen (weil man eine bestimmte
Pflanze öfter (z. B. pro Jahr) heranziehen und ernten kann).
Eine erhöhte Wachstumsrate kann auch dazu führen,
dass man transgene Pflanzen in einem breiteren geographischen Bereich
als ihre Wildtyp-Gegenstücke anbauen kann, da die räumlichen
Beschränkungen für den Anbau einer Kultur häufig von
ungünstigen Umweltbedingungen entweder während
der Pflanzzeit (früh in der Saison) oder während
der Erntezeit (spät in der Saison) bestimmt werden. Solche
ungünstigen Bedingungen können vermieden werden, wenn
der Erntezyklus verkürzt ist. Die Wachstumsrate kann durch
Ableiten verschiedener Parameter aus Wachstumskurven bestimmt werden,
wobei die Parameter u. a. folgende sein können: T-Mid (Zeitdauer,
die die Pflanzen benötigen, um 50% ihrer Maximalgröße
zu erreichen) und T-90 (Zeitdauer, die die Pflanzen benötigen,
um 90% ihrer Maximalgröße zu erreichen).
-
Gemäß einem
bevorzugten Merkmal der vorliegenden Erfindung führt die
Durchführung der erfindungsgemäßen Verfahren
zu Pflanzen mit einer im Vergleich zu Kontrollpflanzen erhöhten
Wachstumsrate. Erfindungsgemäß wird daher ein
Verfahren zur Erhöhung der Wachstumsrate von Pflanzen bereitgestellt,
wobei man bei dem Verfahren in einer Pflanze die Expression einer
Nukleinsäure, die ein LBD-Polypeptid, wie hier definiert,
kodiert, moduliert, vorzugsweise erhöht. Eine Erhöhung
der Wachstumsrate wurde insbesondere während der frühen
Wachstumsstadien der Pflanze beobachtet (Jungpflanzenvitalität).
-
Eine
Erhöhung des Ertrags und/oder der Wachstumsrate tritt unabhängig
davon auf, ob die Pflanze unter Nichtstressbedingungen steht oder
ob die Pflanze im Vergleich zu Kontrollpflanzen verschiedenen Stressbedingungen
unterworfen wird. Üblicherweise reagieren Pflanzen auf
Stress dadurch, dass sie langsamer wachsen. Unter starken Stressbedingungen
kann die Pflanze sogar ihr Wachstum völlig einstellen.
Leichter Stress ist dagegen hier als jeglicher Stress definiert,
dem eine Pflanze ausgesetzt ist und der nicht dazu führt,
dass die Pflanze völlig aufhört zu wachsen, ohne
dass sie die Fähigkeit besitzt, ihr Wachstum wieder aufzunehmen.
Leichter Stress im Sinne der Erfindung führt zu einer Verringerung
des Wachstums der gestressten Pflanzen von weniger als 40%, 35%
oder 30%, vorzugsweise weniger als 25%, 20% oder 15%, stärker
bevorzugt weniger als 14%, 13%, 12%, 11% oder 10% oder weniger im
Vergleich zu der Kontrollpflanze unter Nichtstressbedingungen. Aufgrund
von Fortschritten bei den landwirtschaftlichen Praktiken (Bewässerung,
Düngung, Pestizidbehandlungen) findet man bei angebauten
Kulturpflanzen nicht oft starke Stressbedingungen. Infolgedessen
ist das durch leichten Stress induzierte geschwächte Wachstum
häufig ein unerwünschtes Merkmal in der Landwirtschaft.
Leichte Stressbedingungen sind die alltäglichen biotischen
und/oder abiotischen (Umwelt-)Stressbedingungen, denen eine Pflanze
ausgesetzt ist. Abiotischer Stress kann durch Trockenheit oder Wasserüberschuss,
anaeroben Stress, Salzstress, chemische Toxizität, oxidativen
Stress und heiße, kalte oder Gefriertemperaturen verursacht
werden. Bei dem abiotischen Stress kann es sich um einen osmotischen
Stress handeln, der durch Wasserstress (insbesondere aufgrund von
Trockenheit), Salzstress, oxidativen Stress oder innenbedingten
Stress verursacht wird. Unter biotischem Stress versteht man gewöhnlich
Stressbedingungen, die durch Pathogene, wie Bakterien, Viren, Pilze
und Insekten, verursacht werden.
-
Insbesondere
können die erfindungsgemäßen Verfahren
unter Nichtstressbedingungen oder unter leichten Trockenheitsbedingungen
durchgeführt werden, wobei man Pflanzen erhält,
deren Ertrag im Vergleich zu Kontrollpflanzen erhöht ist.
Wie von Wang et al. (Planta (2003), 218: 1–14)
beschrieben, führt abiotischer Stress zu einer Reihe von
morphologischen, physiologischen, biochemischen und molekularen
Veränderungen, die das Pflanzenwachstum und die Pflanzenproduktivität
negativ beeinflussen. Es ist bekannt, dass Trockenheit, Salinität,
extreme Temperaturen und oxidativer Stress miteinander in Verbindung
stehen und über ähnliche Mechanismen Wachstums-
und Zellschäden induzieren können. Rabbani
et al., (Plant Physiol. (2003), 133: 1755–1767)
beschreibt ein besonders hohes Ausmaß an gegenseitiger
Beeinflussung (”Cross-Talk”) von Trockenheitsstress
und durch hohe Salinität verursachtem Stress. Zum Beispiel äußern
sich Trockenheit und/oder Versalzung in erster Linie als osmotischer
Stress, was zur Störung der Homöostase und der
Ionenverteilung in der Zelle führt. Oxidativer Stress,
der häufig hohe oder niedrige Temperaturen, Salinität oder
Trockenheitsstress begleitet, kann zur Denaturierung von funktionellen
und strukturellen Proteinen führen. Infolgedessen aktivieren
diese verschiedenen Umweltstressbedingungen häufig ähnliche
Zellsignalwege und Zellreaktionen, wie die Produktion von Stressproteinen,
die Hinaufregulation von Antioxidantien, die Akkumulation von kompatiblen
gelösten Stoffen und ein Einstellen des Wachstums. Der
Begriff ”Nichtstress”-Bedingungen, wie hier verwendet,
steht für diejenigen Umweltbedingungen, die ein optimales
Wachstum von Pflanzen gestatten. Der Fachmann ist mit normalen Bodenbedingungen
und Klimabedingungen für einen bestimmten Standort vertraut.
Pflanzen unter optimalen Wachstumsbedingungen (die unter Nichtstressbedingungen
herangezogen werden) liefern oft einen Ertrag von, in der Reihenfolge
der zunehmenden Präferenz, mindestens 90%, 87%, 85%, 83%,
80%, 77% oder 75% der durchschnittlichen Produktion einer solchen
Pflanze in einer gegebenen Umgebung. Die durchschnittliche Produktion
kann bezogen auf die Ernte und/oder die Saison berechnet werden.
Der Fachmann ist mit den durchschnittlichen Erträgen einer
Kultur vertraut.
-
Die
Durchführung der erfindungsgemäßen Verfahren
führt zu Pflanzen, die bei Wachstum unter Nichtstressbedingungen
oder unter leichten Trockenheitsbedingungen einen im Vergleich zu
Kontrollpflanzen, die unter vergleichbaren Bedingungen herangezogen
werden, erhöhten Ertrag aufweisen. Erfindungsgemäß wird daher
ein Verfahren zur Erhöhung des Ertrags von Pflanzen, die
unter Nichtstressbedingungen oder unter leichten Trockenheitsbedingungen
herangezogen werden, bereitgestellt, wobei man bei dem Verfahren
in einer Pflanze die Expression einer Nukleinsäure, die
ein LBD-Polypeptid kodiert, erhöht.
-
Die
Durchführung der erfindungsgemäßen Verfahren
verleiht Pflanzen, die unter Nährstoffmangelbedingungen,
insbesondere unter Stickstoffmangelbedingungen, herangezogen werden,
eine erhöhte Jungpflanzenvitalität und einen erhöhten
Ertrag im Vergleich zu Kontrollpflanzen, die unter vergleichbaren
Bedingungen herangezogen werden. Erfindungsgemäß wird
daher auch ein Verfahren zur Erhöhung der Jungpflanzenvitalität
und des Ertrags in Pflanzen, die unter Nährstoffmangelbedingungen
herangezogen werden, bereitgestellt, wobei man bei dem Verfahren
die Expression einer Nukleinsäure, die ein LBD-Polypeptid
kodiert, in einer Pflanze erhöht. Ein Nährstoffmangel
kann durch ein Fehlen oder einen Überschuss von Nährstoffen,
wie unter anderem Stickstoff, Phosphaten und anderen phosphorhaltigen
Verbindungen, Kalium, Calcium, Cadmium, Magnesium, Mangan, Eisen
und Bor hervorgerufen werden.
-
Die
vorliegende Erfindung umfasst Pflanzen oder Teile davon (einschließlich
Samen), die durch die erfindungsgemäßen Verfahren
erhältlich sind. Die Pflanzen oder Teile davon umfassen
ein Nukleinsäuretransgen, das ein LBD-Polypeptid, wie oben
definiert, kodiert.
-
Die
Erfindung stellt auch Genkonstrukte und Vektoren bereit, um die
Einführung und/oder Expression von Nukleinsäuren,
die LBD-Polypeptide kodieren, in Pflanzen zu erleichtern. Die Genkonstrukte
können in Vektoren eingefügt werden, die im Handel
erhältlich sein können und für die Transformation
in Pflanzen und für die Expression des interessierenden
Gens in den transformierten Zellen geeignet sind. Die Erfindung
stellt auch die Verwendung eines Genkonstrukts, wie hier definiert,
bei den erfindungsgemäßen Verfahren bereit.
-
Genauer
gesagt, stellt die vorliegende Erfindung ein Konstrukt bereit, das
Folgendes umfasst:
- (a) eine Nukleinsäure,
die ein LBD-Polypeptid kodiert, wie oben definiert;
- (b) eine oder mehrere Kontrollsequenzen, die die Expression
der Nukleinsäuresequenz unter (a) vorantreiben können,
und gegebenenfalls
- (c) eine Transkriptionsterminationssequenz.
-
Vorzugsweise
ist die Nukleinsäure, die ein LBD-Polypeptid kodiert, wie
vorstehend definiert. Die Begriffe ”Kontrollsequenz” und ”Terminationssequenz” sind
wie hier definiert.
-
Pflanzen
werden mit einem Vektor transformiert, der eine der oben beschriebenen
Nukleinsäuren umfasst. Der Fachmann kennt die genetischen
Elemente, die in einem Vektor vorhanden sein müssen, damit Wirtszellen,
die die interessierende Sequenz enthalten, erfolgreich transformiert,
selektiert und vermehrt werden können. Die interessierende
Sequenz ist mit einer oder mehreren Kontrollsequenzen (mindestens
mit einem Promoter) funktionsfähig verbunden.
-
Jede
Art von Promoter, ob natürlich oder synthetisch, kann vorteilhaft
für das Vorantreiben der Expression der Nukleinsäuresequenz
verwendet werden. Ein konstitutiver Promoter ist für die
Verfahren besonders geeignet. Die Definitionen der verschiedenen
Promotertypen siehe hier im Abschnitt ”Definitionen”.
-
Es
sollte selbstverständlich sein, dass die Anwendbarkeit
der vorliegenden Erfindung weder auf die erfindungsgemäße,
ein LBD-Poylpeptid kodierende Nukleinsäure gemäß SEQ
ID NO: 1 noch auf die Expression einer ein LBD-Polypeptid kodierenden
Nukleinsäure, die von einem konstitutiven Promotor angetrieben wird,
beschränkt ist.
-
Der
konstitutive Promotor ist vorzugsweise ein GOS2-Promotor, vorzugsweise
ein GOS2-Promotor aus Reis. Weiterhin bevorzugt wird der konstitutive
Promotor durch eine Nukleinsäuresequenz wiedergegeben,
die im Wesentlichen der SEQ ID NO: 10 gleicht, am stärksten
bevorzugt wird der Protomotor durch die SEQ ID NO: 10 dargestellt.
Weitere Beispiele für konstitutive Promotoren siehe hier
im Abschnitt ”Definitionen.
-
Gegebenenfalls
kann/können eine oder mehrere Terminatorsequenzen in dem
Konstrukt, das in eine Pflanze eingeführt wird, verwendet
werden. Weitere Regulationselemente können u. a. Transkriptionsenhancer
und Translationsenhancer sein. Der Fachmann ist mit Terminator-
und Enhancersequenzen vertraut, die für die Durchführung
der Erfindung geeignet sein können. Es kann auch eine Intronsequenz
zu der 5'-untranslatierten Region (UTR) oder der kodierenden Sequenz
hinzugefügt werden, um die Menge an reifer Botschaft, die
im Cytosol akkumuliert, zu erhöhen, wie im Abschnitt Definitionen
beschrieben ist. Weitere Kontrollsequenzen (neben Promotor-, Enhancer-,
Silencer-, Intronsequenzen, 3'UTR- und/oder 5'UTR-Regionen) können Protein-
und/oder RNA-stabilisierende Elemente sein. Solche Sequenzen sind
dem Fachmann bekannt oder können von ihm leicht erhalten
werden.
-
Die
erfindungsgemäßen Genkonstrukte können
weiterhin eine Replikationsursprungssequenz beinhalten, die für
die Aufrechterhaltung und/oder die Replikation in einem bestimmten
Zelltyp erforderlich ist. Ein Beispiel ist, wenn ein Genkonstrukt
in einer Bakterienzelle als episomales genetisches Element (z. B.
ein Plasmid- oder Cosmidmolekül) aufrechterhalten werden
muss. Bevorzugte Replikationsursprünge sind u. a. der f1-ori
und colE1, sie sind jedoch nicht hierauf beschränkt.
-
Für
den Nachweis eines erfolgreichen Transfers der Nukleinsäuresequenzen,
wie sie bei den erfindungsgemäßen Verfahren verwendet
werden, und/oder für die Selektion transgener Pflanzen,
die diese Nukleinsäuren umfassen, ist es vorteilhaft, Markergene
(oder Reportergene) zu verwenden. Daher kann das Genkonstrukt gegebenenfalls
ein Selektionsmarkergen umfassen. Selektionsmarker sind hier im
Abschnitt ”Definitionen” eingehender beschrieben.
-
Bekanntlich
nimmt je nach dem verwendeten Expressionsvektor und der verwendeten
Transfektionstechnik nach der stabilen oder transienten Integration
von Nukleinsäuren in Pflanzenzellen nur eine Minderheit
der Zellen die Fremd-DNA auf und integriert sie in ihr Genom, falls
gewünscht. Um diese Integranten zu identifizieren und zu
selektieren wird üblicherweise ein Gen, das für
einen Selektionsmarker (wie die oben beschriebenen) kodiert, zusammen
mit dem interessierenden Gen in die Wirtszellen eingeführt.
Diese Marker können zum Beispiel in Mutanten verwendet
werden, in denen diese Gene, zum Beispiel durch Deletion durch herkömmliche
Verfahren, nicht funktionell sind. Weiterhin können Nukleinsäuremoleküle,
die einen Selektionsmarker kodieren, in eine Wirtszelle auf demselben
Vektor, der die Sequenz umfasst, die die erfindungsgemäßen
Polypeptide oder die Polypeptide, die bei den Verfahren der Erfindung
verwendet werden, kodiert, oder auch auf einem separaten Vektor
eingeführt werden. Zellen, die mit der eingeführten
Nukleinsäure stabil transfiziert worden sind, können
zum Beispiel durch Selektion identifiziert werden (zum Beispiel überleben
Zellen, die den Selektionsmarker integriert haben, während
die anderen Zellen absterben). Die Markergene können aus
der transgenen Zelle entfernt oder ausgeschnitten werden, wenn sie
nicht mehr gebraucht werden. Techniken zur Entfernung von Markergenen
sind im Stand der Technik bekannt, geeignete Techniken sind vorstehend
im Abschnitt Definitionen beschrieben.
-
Die
Erfindung stellt auch ein Verfahren zur Herstellung transgener Pflanzen
mit verbesserten Ertragsmerkmalen im Vergleich zu Kontrollpflanzen
bereit, bei dem man in eine(r) Pflanze eine Nukleinsäure,
die ein LBD-Polypeptid, wie oben definiert, kodiert, einführt
und exprimiert.
-
Genauer
gesagt stellt die vorliegende Erfindung ein Verfahren für
die Herstellung transgener Pflanzen mit erhöhten verbesserten
Ertragsmerkmalen, insbesondere erhöhter Sprossbiomasse
und/oder erhöhtem Samenertrag bereit, wobei das Verfahren
Folgendes umfasst:
- (i) Einführen und
Exprimieren einer Nukleinsäure, die für ein LBD-Polypeptid
kodiert, in eine(r) Pflanze oder Pflanzenzelle und
- (ii) Kultivieren der Pflanzenzelle unter Bedingungen, die das
Pflanzenwachstum und die Pflanzenentwicklung fördern.
-
Bei
der Nukleinsäure unter (i) kann es sich um irgendeine der
Nukleinsäuren handeln, die ein LBD-Polypeptid, wie hier
definiert, kodieren können.
-
Die
Nukleinsäure kann direkt in eine Pflanzenzelle oder in
die Pflanze selbst eingeführt werden (was das Einführen
in ein Gewebe, Organ oder einen irgendeinen anderen Teil einer Pflanze
beinhaltet). Gemäß einem bevorzugten Merkmal der
vorliegenden Erfindung wird die Nukleinsäure vorzugsweise
mittels Transformation in eine Pflanze eingeführt. Der
Begriff ”Transformation” ist hier im Abschnitt ”Definitionen” genauer
beschrieben.
-
Die
genetisch modifizierten Pflanzenzellen können durch alle
Verfahren regeneriert werden, mit denen der Fachmann vertraut ist.
Geeignete Verfahren finden sich in den oben genannten Veröffentlichungen
von S. D. Kung und R. Wu, Potrykus oder Höfgen
und Willmitzer.
-
Im
Allgemeinen werden nach der Transformation die Pflanzenzellen oder
Zellgruppierungen hinsichtlich des Vorhandenseins von einem oder
mehreren Markern selektiert, die von Genen kodiert werden, die in Pflanzen
exprimierbar sind und die mit dem interessierenden Gen cotransferiert
wurden, wonach das transformierte Material zu einer ganzen Pflanze
regeneriert wird. Für die Selektion von transformierten
Pflanzen wird das bei der Transformation erhaltene Pflanzenmaterial
in der Regel derartigen Selektionsbedingungen unterworfen, dass
man transformierte Pflanzen von untransformierten Pflanzen unterscheiden
kann. So können zum Beispiel Samen, die auf die oben beschriebene
Weise erhalten wurden, ausgepflanzt werden und nach einer anfänglichen
Wachstumsphase einer geeigneten Selektion durch Spritzen unterworfen
werden. Eine weitere Möglichkeit besteht darin, dass man
die Samen, gegebenenfalls nach Sterilisieren, auf Agarplatten unter
Verwendung eines geeigneten Selektionsmittels heranzieht, so dass
nur die transformierten Samen zu Pflanzen heranwachsen können.
Alternativ werden die transformierten Pflanzen auf das Vorhandensein
eines Selektionsmarkers, wie die oben beschriebenen, gescreent.
-
Nach
DNA-Transfer und Regeneration können mutmaßlich
transformierte Pflanzen auch zum Beispiel unter Verwendung der Southern-Analyse
hinsichtlich des Vorhandenseins des interessierenden Gens, der Kopienzahl
und/oder der Genomorganisation untersucht werden. Alternativ oder
zusätzlich können die Expressionsniveaus der neu
eingeführten DNA unter Verwendung von Northern- und/oder
Western-Analyse verfolgt werden, wobei beide Techniken dem Durchschnittsfachmann
bekannt sind.
-
Die
erzeugten transformierten Pflanzen können durch eine Vielzahl
von Mitteln vermehrt werden, wie durch klonale Vermehrung oder klassische
Züchtungstechniken. So kann zum Beispiel eine transformierte Pflanze
der ersten Generation (T1-Pflanze) geselbstet werden, und homozygote
Transformanten der zweiten Generation (T2-Pflanzen) können
selektiert werden, und die T2-Pflanzen können dann unter
Verwendung klassischer Züchtungstechniken weiter vermehrt
werden. Die erzeugten transformierten Organismen können
in einer Vielzahl von Formen vorliegen. So kann es sich zum Beispiel
um Chimären von transformierten Zellen und untransformierten
Zellen, klonale Transformanten (z. B. dass alle Zellen so transformiert
wurden, dass sie die Expressionskassette enthalten), Pfropfmaterial
von transformiertem und untransformiertem Gewebe (z. B. bei Pflanzen
ein transformierter Wurzelstock, der auf ein untransformiertes Edelreis
gepfropft wurde) handeln.
-
Die
vorliegende Erfindung erstreckt sich eindeutig auf jede Pflanzenzelle
oder Pflanze, die durch eines der hier beschriebenen Verfahren hergestellt
wurde, und auf alle Pflanzenteile und sämtliches Vermehrungsmaterial
davon. Die vorliegende Erfindung umfasst weiterhin die Nachkommenschaft
eine(r/s) primär transformierten oder transfizierten Zelle,
Gewebes, Organs oder ganzen Pflanze, die/das durch eines der oben
genannten Verfahren hergestellt wurde, wobei die einzige Voraussetzung
ist, dass die Nachkommenschaft dasselbe/dieselben genotypische(n)
und/oder phänotypische(n) Merkmal(e) aufweist, wie es/sie
der Elter bei den erfindungsgemäßen Verfahren
zeigt.
-
Die
Erfindung beinhaltet auch Wirtszellen, die eine isolierte Nukleinsäure
enthalten, die ein LBD-Polypeptid, wie oben definiert, kodiert.
Bevorzugte erfindungsgemäße Wirtszellen sind Pflanzenzellen.
Wirtspflanzen für die Nukleinsäuren oder den Vektor,
die bei dem erfindungsgemäßen Verfahren verwendet
werden, die Expressionskassette oder das Konstrukt oder den Vektor
sind im Prinzip vorteilhafterweise alle Pflanzen, welche die bei
dem erfindungsgemäßen Verfahren verwendeten Polypeptide
synthetisieren können.
-
Die
erfindungsgemäßen Verfahren lassen sich vorteilhaft
auf jede beliebige Pflanze anwenden. Zu den Pflanzen, die für
die erfindungsgemäßen Verfahren besonders geeignet
sind, gehören alle Pflanzen, die zur Superfamilie der Viridiplantae
gehören, insbesondere monokotyle und dikotyle Pflanzen,
einschließlich Futter- oder Weidepflanzenleguminosen, Zierpflanzen,
Nahrungsmittelkulturen, Bäume oder Sträucher.
Gemäß einer bevorzugten Ausführungsform
der vorliegenden Erfindung handelt es sich bei der Pflanze um eine
Kulturpflanze. Beispiele für Kulturpflanzen sind zum Beispiel
u. a. Sojabohne, Sonnenblume, Canola-Raps, Luzerne, Raps, Baumwolle,
Tomate, Kartoffel und Tabak. Weiterhin bevorzugt handelt es sich
bei der Pflanze um eine monokotyle Pflanze. Beispiele für
monokotyle Pflanzen sind zum Beispiel u. a. Zuckerrohr. Stärker
bevorzugt handelt es sich bei der Pflanze um ein Getreide. Beispiele
für Getreide sind zum Beispiel u. a. Reis, Mais, Weizen,
Gerste, Hirse, Roggen, Triticale, Sorghum und Hafer.
-
Die
Erfindung erstreckt sich auch auf erntbare Teile einer Pflanze,
wie zum Beispiel, aber nicht beschränkt auf Samen, Blätter,
Früchte, Blüten, Stängel, Rhizome, Knollen
und Zwiebeln. Die Erfindung betrifft weiterhin Produkte, die, vorzugsweise
direkt, von einem erntbaren Teil einer solchen Pflanze stammen,
wie trockene Pellets oder Pulver, Öl, Fett und Fettsäuren,
Stärke oder Proteine.
-
Gemäß einem
bevorzugten Merkmal der Erfindung handelt es sich bei der modulierten
Expression um erhöhte Expression. Verfahren zur Erhöhung
der Expression von Nukleinsäuren oder Genen oder Genprodukten
sind in der Fachwelt gut bekannt und Beispiele werden im Abschnitt
Definitionen gegeben.
-
Wie
vorstehend erwähnt, ist ein bevorzugtes Verfahren zum Modulieren
(vorzugsweise Erhöhen) der Expression einer Nukleinsäure,
die ein LBD-Polypeptid kodiert, durch Einführen und Exprimieren
einer Nukleinsäure, die ein LBD-Polypeptid kodiert, in
eine(r) Pflanze; die Wirkungen der Durchführung des Verfahren,
d. h. das Erhöhen der Ertragsmerkmale, können
jedoch auch unter Verwendung anderer bekannter Techniken erzielt
werden, einschließlich, aber nicht beschränkt
auf T-DNA-Activation-Tagging, TILLING, homologe Rekombination. Eine
Beschreibung dieser Techniken findet sich im Abschnitt Definitionen.
-
Die
vorliegende Erfindung umfasst auch die Verwendung von Nukleinsäuren,
die LBD-Polypeptide, wie hier beschrieben, kodieren, und die Verwendung
dieser LBD-Polypeptide zur Verbesserung eines der oben genannten
Ertragsmerkmale in Pflanzen.
-
Hier
beschriebene Nukleinsäuren, die LBD-Polypeptid kodieren,
oder die LBD-Polypeptide selbst können Verwendung in Züchtungsprogrammen
finden, in denen ein DNA-Marker identifiziert wird, der mit einem Gen,
das ein LBD-Polpeptid kodiert, genetisch verknüpft sein
kann. Die Nukleinsäuren/Gene oder die LBD-Polypeptide selbst
können dazu verwendet werden, einen molekularen Marker
zu bestimmen. Dieser DNA- oder Protein-Marker kann dann in Züchtungsprogrammen
zur Selektion von Pflanzen mit erhöhten Ertragsmerkmalen
verwendet werden, wie hier vorstehend bei den erfindungsgemäßen
Verfahren definiert.
-
Allelische
Varianten einer Nukleinsäure/eines Gens, die/das für
ein LBD-Polypeptid kodiert, können auch Verwendung in Marker-unterstützten
Züchtungsprogrammen finden. Bei solchen Züchtungsprogrammen ist
manchmal das Einführen von allelischer Variation durch
Mutagenbehandlung der Pflanzen erforderlich, wobei zum Beispiel
EMS-Mutagenese verwendet wird; alternativ kann das Programm von
einer Sammlung von allelischen Varianten so genannten ”natürlichen” Ursprungs
ausgehen, die nicht mit Absicht hervorgerufen wurden. Dann erfolgt
die Identifikation allelischer Varianten, zum Beispiel mittels PCR.
Darauf folgt ein Schritt zur Selektion besserer allelischer Varianten
der in Frage kommenden Sequenz, die bessere Erträge liefern. Die
Selektion wird gewöhnlich durchgeführt, indem
man die Wachstumsleistung von Pflanzen verfolgt, die verschiedene
allelische Varianten der in Frage kommenden Sequenz enthalten. Die
Wachstumsleistung kann im Gewächshaus oder auf dem Feld
verfolgt werden. Weitere Schritte sind gegebenenfalls u. a. das
Kreuzen der Pflanzen, in denen die bessere allelische Variante identifiziert
wurde, mit einer anderen Pflanze.
-
Dies
kann zum Beispiel zur Herstellung einer Kombination interessanter
phänotypischer Merkmale verwendet werden.
-
Nukleinsäuren,
die LBD-Polypeptide kodieren, können auch als Sonden zur
genetischen und physikalischen Kartierung der Gene, von denen sie
ein Teil sind, und als Marker für Merkmale, die mit diesen
Genen verknüpft sind, verwendet werden. Diese Information
kann für die Pflanzenzüchtung nützlich
sein, um Linien mit gewünschten Phänotypen zu
entwickeln. Eine solche Verwendung von Nukleinsäuren, die
LBD-Polypeptide kodieren, erfordert nur eine Nukleinsäuresequenz
mit einer Länge von mindestens 15 Nukleotiden. Die Nukleinsäuren,
die ein LBD-Polypeptid kodieren, können als Marker für
Restriktionsfragmentlängenpolymorphismus (RFLP) verwendet
werden. Southern-Blots (Sambrook J, Fritsch EF und Maniatis
T (1989) Molecular Cloning, A Laboratory Manual) von restriktionsgespaltener
genomischer Pflanzen-DNA können mit den LBD kodierenden
Nukleinsäuren sondiert werden. Die erhaltenen Bandenmuster
können dann genetischen Analysen unter Verwendung von Computerprogrammen,
wie MapMaker (Lander et al. (1987) Genomics 1: 174–181),
unterworfen werden, wodurch eine Genkarte erstellt wird. Außerdem
können die Nukleinsäuren zum Sondieren von Southern-Blots
verwendet werden, die restriktionsendonukelasebehandelte genomische
DNAs von einem Satz von Individuen enthalten, die den Elter und
die Nachkommen einer bestimmten genetischen Kreuzung darstellen.
Die Segregation der DNA-Polymorphismen wird ermittelt und dazu verwendet,
die Position der Nukleinsäure, die ein LBD-Polypeptid kodiert,
in der zuvor unter Verwendung dieser Population erhaltenen Genkarte
zu berechnen (Botstein et al. (1980) Am. J. Hum. Genet.
32: 314–331).
-
Die
Herstellung und Verwendung von Sonden, die von Pflanzengenen stammen,
für die Verwendung bei der genetischen Kartierung ist in Bernatzky
und Tanksley (1986) Plant Mol. Biol. Reporter 4: 37–41 beschrieben.
Zahlreiche Veröffentlichungen beschreiben die genetische
Kartierung spezifischer cDNA-Klone, wobei die vorstehend erläuterte
Methodik oder Varianten davon verwendet werden. Für die
Kartierung können zum Beispiel F2-Inzuchtpopulationen,
Rückkreuzungspopulationen, zufallsgemäß gepaarte
Populationen, nahezu isogene Linien und andere Sätze von
Individuen verwendet werden.
-
Die
Nukleinsäuresonden können auch zur physikalischen
Kartierung (d. h. zum Anordnen von Sequenzen in physikalischen Karten;
siehe Hoheisel et al. In: Non-mammalian Genomic Analysis:
A Practical Guide, Academic Press 1996, S. 319–346 und
darin genannte Bezugsstellen) verwendet werden.
-
In
einer anderen Ausführungsform können die Nukleinsäuresonden
bei der Kartierung mittels direkter Fluoreszenz-in-situ-Hybridisierung
(FISH) (Trask (1991) Trends Genet. 7: 149–154)
verwendet werden. Zwar bevorzugen derzeitige FISH-Kartierungsverfahren
die Verwendung großer Klone (mehrere kb bis mehrere Hundert
kb; siehe Laan et al. (1995) Genome Res. 5: 13–20),
aber durch Verbesserungen in der Empfindlichkeit kann die FISH-Kartierung
auch unter Verwendung kürzerer Sonden durchgeführt
werden.
-
Eine
Reihe von Verfahren auf Basis von Nukleinsäureamplifikation
für die genetische und physikalische Kartierung kann unter
Verwendung der Nukleinsäuren durchgeführt werden.
Beispiele sind u. a. die allelspezifische Amplifikation (Kazazian
(1989) J. Lab. Clin. Med 11: 95–96), Polymorphismus
PCR-amplifizierter Fragmente (CAPS; Sheffield et al. (1993)
Genomics 16: 325–332), allelspezifische Ligation
(Landegren et al. (1988) Science 241: 1077–1080),
Nukleotidverlängerungsreaktionen (Sokolov (1990)
Nucleic Acid Res. 18: 3671), Radiation-Hybrid-Kartierung
(Walter et al. (1997) Nat. Genet. 7: 22–28)
und Happy-Kartierung (Dear und Cook (1989) Nucleic Acid
Res. 17: 6795–6807). Bei diesen Verfahren wird
die Sequenz einer Nukleinsäure dazu verwendet, Primer-Paare
zu gestalten und herzustellen, die bei der Amplifikationsreaktion
oder bei Primerverlängerungsreaktionen verwendet werden.
Die Gestaltung dieser Primer ist dem Fachmann bekannt. Bei Verfahren,
die eine genetische Kartierung auf PCR-Basis einsetzen, kann es
notwendig sein, dass man DNA-Sequenz-Unterschiede zwischen den Eltern
der Kartierungskreuzung in dem Bereich identifiziert, welcher der
vorliegenden Nukleinsäuresequenz entspricht. Dies ist jedoch
für Kartierungsverfahren nicht allgemein erforderlich.
-
Die
erfindungsgemäßen Verfahren führen zu
Pflanzen mit verbesserten Ertragsmerkmalen, wie hier vorstehend
beschrieben. Diese Merkmale können auch mit anderen ökonomisch
vorteilhaften Merkmalen, wie weiteren Ertragsmerkmalen, Toleranz
gegenüber anderen abiotischen und biotischen Stressbedingungen, Merkmalen,
die verschiedene architektonische Merkmale und/oder biochemisch
und/oder physiologische Merkmale modifizieren, kombiniert werden.
-
II. JMJC-(JUMONJI-C-)Polypeptid
-
Es
wurde jetzt überraschenderweise gefunden, dass die Modulation
der Expression einer Nukleinsäure, die ein JMJC-Polypeptid
kodiert, in einer Pflanze zu Pflanzen mit gesteigerten Ertragsmerkmalen
im Vergleich zu Kontrollpflanzen führt. In einer ersten
Ausführungsform stellt die vorliegende Erfindung ein Verfahren zur
Steigerung von Ertragsmerkmalen in Pflanzen im Vergleich zu Kontrollpflanzen
bereit, wobei man die Expression einer Nukleinsäure, die
ein JMJC-Polypeptid kodiert, in einer Pflanze moduliert.
-
Ein
bevorzugtes Verfahren zum Modulieren (vorzugsweise Erhöhen)
der Expression einer Nukleinsäure, die ein JMJC-Polypeptid
kodiert, besteht darin, dass man eine Nukleinsäure, die
ein JMJC-Polypeptid kodiert, in eine Pflanze einbringt und in dieser
exprimiert.
-
Wird
im folgenden Text auf ein ”Protein, das für die
erfindungsgemäßen Verfahren geeignet ist” Bezug genommen,
soll darunter ein JMJC-Polypeptid, wie hier definiert, verstanden
werden. Wird hier im Folgenden auf eine ”Nukleinsäure,
die für die erfindungsgemäßen Verfahren
geeignet ist” Bezug genommen, bedeutet dies eine Nukleinsäure,
die ein solches JMJC-Polypeptid kodieren kann. Die Nukleinsäure,
die in eine Pflanze eingeführt werden soll (und die daher
zur Durchführung der erfindungsgemäßen
Verfahren geeignet ist), ist jede beliebige Nukleinsäure,
welche den Typ Protein, der im Folgenden beschrieben wird, kodiert
und wird im Folgenden auch als ”JMJC-Nukleinsäure” oder ”JMJC-Gen” bezeichnet.
-
Ein
JMJC-Polypeptid, wie hier definiert, betrifft ein Polypeptid, das
mindestens eine JmjC-Domäne umfasst.
-
Bei
der JmjC-Domäne handelt es sich um eine konservierte Sequenz,
die man in Proteinen prokaryotischer und eukaryotischer Organismen
findet. JmjC-Domänen sind durchschnittlich 111 Aminosäuren
lang. Die Länge einer JmjC-domäne kann gewöhnlich
von 25 bis 200 Aminosäuren reichen und längere
Versionen können möglich sein. Für JmjC-Domänen
wird vorhergesagt, dass es sich um Metalloenzyme handelt, welche die
Cupin-Faltung annehmen, und sie sind Kandidaten für Enzyme,
welche die Chromatinumgestaltung regulieren (Clissold et
al. Trends Biochem Sci. 2001 Jan; 26 (1): 7–9).
-
JMJC-Polypeptide
findet man in speziellen Datenbanken, wie Pfam (Finn et
al. Nucleic Acids Research (2006) Database Issue 34: D247–D251).
Pfam listet eine große Sammlung an mehrfachen Sequenz-Alignments
und Hidden-Markov-Modellen (HMM) auf, die viele häufige
Proteindomänen und -familien abdeckt und über
das Sanger Institute in Großbritannien verfügbar
ist.
-
Die
Erfassungsausschlussgrenze für die JmjC-Domäne
im Pfam HMM_fs-Modell beträgt 16,0 und –8,0 im
HMM_Is-Modell. In der Pfam-Datenbank gelten als zuverlässige Übereinstimmungen
solche mit einem Score oberhalb der Erfassungsausschlussgrenze.
Mögliche Übereinstimmungen, die echte JmjC-Domänen umfassen,
können jedoch unter den Erfassungsausschluss fallen. Vorzugsweise
ist ein JMJC-Polypeptid ein Protein, das in seiner Sequenz eine
oder mehrere Domänen aufweist, die den Erfassungsausschluss
der Pfam- Proteindomänenfamilie PF02373, jumonji, jmjC, überschreiten.
-
Alternativ
kann eine JmjC-Domäne in einem Polypeptid identifiziert
werden, indem man einen Sequenzvergleich mit bekannten Polypeptiden,
die eine JmjC-domäne umfassen, durchführt und
die Ähnlichkeit im Bereich der JmjC-Domäne feststellt.
Das Alignment der Sequenzen kann durch jedes im Stand der Technik bekannte
Verfahren durchgeführt werden, wie Blast-Algorithmen, und
die Wahrscheinlichkeit, dass ein Alignment mit einer gegebenen Sequenz
auftritt, wird als Basis für die Identifikation ähnlicher
Polypeptide verwendet. Ein Parameter, der üblicherweise
dazu verwendet wird, diese Wahrscheinlichkeit in paarweisen Vergleichen
darzustellen, wird als e-Wert bezeichnet. Der e-Wert beschreibt,
wie oft das zufallsgemäße Auftreten von einem
bestimmten Score erwartet wird. Die e-Wert-Ausschlussgrenze kann
bis zu 1,0 betragen. Üblicherweise haben Alignments, bei
denen die Wahrscheinlichkeit, dass sie auftreten, hoch ist, einen
e-Wert unter 0,1, 0,01, 0,001, 1·e–05, 1·e–10,
1·e–15, 1·e–20, 1·e–25,
1·e–50, 1·e–75, 1·e–100
und 1·e–200. Vorzugsweise umfassen erfindungsgemäße
JMJC-Polypeptide eine Sequenz, die in steigender Reihenfolge der
Präferenz einen e-Wert unter 0,1, 0,01, 0,001, 1·e–05,
1·e–10, 1·e–15, 1·e–20,
1·e–25, 1·e–50, 1·e–75,
1·e–100 und 1·e–200 bei einem Alignment
mit einer JmjC-Domäne aufweist, die man in einem bekannten
JMJC-Polypeptid findet.
-
Beispiele
für JMJC-Polypeptide sind in Tabelle B1 angegeben. Die
Aminosäurekoordinaten der JmjC-Domänen in beispielhaften
JMJC-Polypeptiden aus Dikotyledonen sind in Tabelle B4 angegeben.
Ein Beispiel für eine JmjC-Domäne zeigt SEQ ID
NO: 78.
-
Die
Sequenzidentität zwischen JmjC-Domänen ist gewöhnlich
klein, durchschnittlich 23%, und kann sogar nur 10% betragen. Bevorzugte
erfindungsgemäße JMJC-Polypeptide sind solche,
die, in steigender Reihenfolge der Präferenz, mindestens
50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% oder mehr Sequenzidentität
mit der SEQ ID NO: 78 oder mit einer der JmjC-Domänen aufweisen,
die in den JMJC-Polypeptiden enthalten sind, die durch SEQ ID NO:
84; SEQ ID NO: 86; SEQ ID NO: 96; SEQ ID NO: 98; SEQ ID NO: 104;
SEQ ID NO: 108; SEQ ID NO: 110; SEQ ID NO: 112; SEQ ID NO: 114;
SEQ ID NO: 116; SEQ ID NO: 118; SEQ ID NO: 120; SEQ ID NO: 122;
SEQ ID NO: 124; SEQ ID NO: 128; SEQ ID NO: 130; SEQ ID NO: 132 und SEQ
ID NO: 134 dargestellt werden und deren Aminosäurekoordinaten
in Tabelle B4 angegeben sind.
-
Zusätzlich
zu der JmjC-Domäne können JMJC-Polypeptide gegebenenfalls
andere hochkonservierte Sequenzmotive umfassen, wie sie in SEQ ID
NO: 79, SEQ ID NO: 80, SEQ ID NO: 81 dargestellt sind, die man in
SEQ ID NO: 74 findet. Weiterhin können die erfindungsgemäßen
Polypeptide eine konservierte Sequenz umfassen, die man in Oxygenasen
findet und die an der Koordination von Eisenkationen beteiligt ist,
hier als HXD(V) oder EXnH dargestellt (SEQ ID NO: 82), wobei ”X” für
eine beliebige Aminosäure steht und ”n” für
die Anzahl der X-Reste steht, die von 1–5 einschließlich
der Grenzen reicht.
-
Ein
bevorzugtes erfindungsgemäßes JMJC-Polypeptid
betrifft jedes Polypeptid, das eine JmjC-Domäne umfasst
und, in steigender Reihenfolge der Präferenz, mindestens
50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% oder mehr Sequenzidentität
mit einer oder mehreren der konservierten Sequenzmotive aufweist,
die durch SEQ ID NO: 79, SEQ ID NO: 80, SEQ ID NO: 81 und SEQ ID
NO: 82 dargestellt werden.
-
Über
Befunde für Domänenaustausch (domain swapping)
in der JUMONJI-Proteinfamilie wurde berichtet (Balciunas
und Ronne. Trends Biochem Sci 2000; 25: 274–276).
Folglich enthalten JMJC-Polypeptide zusätzlich zu der JmjC-Domäne üblicherweise
eine oder mehrere verschiedene Arten anderer bekannter konservierter
Domänen. Relevant für die erfindungsgemäßen
Polypeptide sind JMJC-Polypeptide, die zusätzlich zu der
JmjC-Domäne andere konservierte Domänen umfassen,
die mutmaßlich an DNA-Eindungs- und Transkriptionsaktivitäten
beteiligt sind, wie Zinkfingerdomänen, oder an der Proteinwechselwirkung
und am Protein-Turnover, wie F-Box und Zinkfinger-RING-Domänen.
-
Daher
umfasst das für die erfindungsgemäßen
Verfahren geeignete JMJC-Polypeptid eine JmjC-Domäne und
gegebenenfalls eine oder mehrere der folgenden konservierten Domänen:
JmJN (pfam-Zugangsnummer PF02375); C5HC2 Zinkfinger (pfam-Zugangsnummer:
PF02928), FY-reiche Domäne, N-terminal (InterPro-Zugangsnummer:
IPR003888) und FY-reiche Domäne, C-terminal (InterPro-Zugangsnummer: IPR003889),
eine Zinkfinger-FYVE/PHD-Zn-Typ (InterPro-Zugangsnummer: IPR011011),
eine Zinkfinger-C2H2-Typ- (pfam-Zugangsnummer: PF00096), ein Zinkfinger–RING-Typ-
(zf-C3HC4) (pfam-Zugangsnummer PF00097) und eine F-Box-Domäne
(pfam-Zugangsnummer: PF00646).
-
Vorzugsweise
umfasst das erfindungsgemäße JMJC-Polypeptid eine
Sequenz mit, in steigender Reihenfolge der Präferenz, 50%,
60%, 70%, 75%, 60%, 85%, 90%, 92%, 94%, 96%, 98% oder mehr Sequenzidentität
mit der SEQ ID NO: 74, SEQ ID NO: 86, SEQ ID NO: 94, SEQ ID NO:
104, SEQ ID NO: 122, SEQ ID NO: 134, SEQ ID NO: 136, SEQ ID NO:
140, SEQ ID NO: 142 und SEQ ID NO: 148.
-
Vorzugsweise
die Polypeptidsequenz, die, wenn sie zur Konstruktion eines Stammbaums,
wie dem in 8 dargestellten, verwendet
wird, eher mit der Gruppe der JMJC-Polypeptide (Takeuchi
et al. Dev Dyn. 2006 235 (9): 2449–59), welche
die in SEQ ID NO: 74 dargestellte Aminosäuresequenz umfassen,
als mit jeder anderen Gruppe Cluster bildet.
-
Der
Begriff ”Domäne” und ”Motiv” wird
hier im Abschnitt ”Definitionen” definiert. Für
die Identifikation von Domänen gibt es Spezialdatenbanken,
zum Beispiel SMART (Schultz et al., (1998), Proc. Natl.
Acad. Sci. USA 95, 5857–5864; Letunic
et al., (2002), Nucleic Acids Res. 30, 242–244,
InterPro (Mulder et al., (2003), Nucl. Acids Res. 31, 315–318,
Prosite (Sucher und Bairoch (1994), A generalized Profile
syntax for biomolecular sequences motives und its function in automatic
sequence interpretation. (In) ISMB-94; Proceedings 2nd International
Conference an Intelligent Systems for Molecular Biology. Altman
R., Brutlag D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61,
AAAIPress, Menlo Park; Hulo et al., Nucl. Acids
Res. 32: D134–D137, (2004), oder Pfam (Bateman
et al., Nucleic Acids Research 30 (1): 276–280 (2002).
Ein Satz von Werkzeugen für die In-silico-Analyse von Proteinsequenzen
ist auf dem ExPASy-Proteomics-Server verfügbar (Swiss Institute
of Bioinformatics (Gasteiger et al., ExPASy: the proteomics
server for in-depth Protein knowledge and analysis, Nucleic Acids
Res. 31: 3784–3788 (2003)). Domänen können
auch unter Verwendung von Routinetechniken, wie Sequenz-Alignment,
identifiziert werden.
-
Verfahren
für das Alignment von Sequenzen für Vergleichszwecke
sind in der Fachwelt gut bekannt, dazu zählen GAP, BESTFIT,
BLAST, FASIA und TFASTA. Bei GAP wird der Algorithmus von Needleman
und Wunsch ((1970), J. Mol. Biol. 48: 443–453)
dazu verwendet, das globale Alignment (d. h. das Alignment, das sich über
die vollständigen Sequenzen erstreckt) von zwei Sequenzen
zu finden, das die Anzahl der Übereinstimmungen (”Matches”)
maximiert und die Anzahl der Lücken (”Gaps”)
minimiert. Beim BLAST-Algorithmus (Altschul et al., (1990),
J. Mol. Biol. 215: 403–10) wird die Sequenzidentität
in Prozent berechnet, und es wird eine statistische Analyse der Ähnlichkeit
zwischen den beiden Sequenzen durchgeführt. Die Software
für die Durchführung einer BLAST-Analyse ist über
das National Centre for Biotechnology Information (NCBI) öffentlich
zugänglich. Homologe können zum Beispiel unter
Verwendung des multiplen Sequenz-Alignment-Algorithmus ClustalW
(Version 1.83) leicht identifiziert werden, und zwar mit den Default-Parametern
für paarweises Alignment und einer Scoring-Methode in Prozent.
Die Gesamtprozent an Ähnlichkeit und Identität
können auch unter Verwendung eines der in dem MatGAT-Software-Paket
verfügbaren Verfahren bestimmt werden (Campanella
et al., BMC Bioinformatics. Juli 2003, 10; 4:29. MatGAT: an application
that generstes similarity/identity matrices using Protein or DNA
sequences.). Für eine Optimierung des Alignments
zwischen konservierten Motiven können kleinere Änderungen
per Hand vorgenommen werden, wie dem Fachmann bekannt ist. Weiterhin können
für die Identifikation von Homologen auch spezifische Domänen
anstelle von Volllängensequenzen verwendet werden. Die
Sequenzidentitätswerte können mit den oben erwähnten
Programmen unter Verwendung der Default-Parameter über
die gesamte Nukleinsäure- oder Aminosäuresequenz
oder über ausgewählte Domänen oder ein
konservierte(s) Motiv(e) bestimmt werden.
-
Außerdem
können JMJC-Polypeptide gewöhnlich Proteinhydrolaseaktivität,
insbesondere Peptid-aspartat-Beta-dioxygenase-(PABD-)Aktivität
(EC 1.14.11.16) besitzen. Folgende Reaktion wird katalysiert: Peptid-L-aspartat
+ 2-Oxoglutarat + O(2) <=> Peptid-3-hydroxy-L-aspartat
+ Succinat + CO(2). Die Cofaktoren bei der Reaktion sind Eisen und
2-Oxogluatarat. Werkzeuge und Techniken für die Messung
der PABD-Aktivität sind im Stand der Technik bekannt (siehe
zum Beispiel Lavaissiere et al. J Clin Invest. 1996; 98
(6): 1313–23; Linke et al. J Biol Chem.
2004; 279 (14): 14391–7; Lee, et al. J.
Biol. Chem. 278: 7558–7563; 2003; Lando
et al. Genes Dev. 16: 1466–1471; 2002). Eisen
(II)/2-Oxoglutarat-(2-OG)-abhängige Oxygenasen katalysieren oxidative
Reaktionen bei verschiedenen Stoffwechselreaktionen. Vor kurzem
wurde vorgeschlagen, dass JMJC-Polypeptide auch eine Histon-Demethylase-Aktivität
besitzen können (Trewick et al. 2005).
-
Die
vorliegende Erfindung wird dadurch veranschaulicht, dass man Pflanzen
mit der Nukleinsäuresequenz gemäß SEQ
ID NO: 73, welche die Polypeptidsequenz gemäß SEQ
ID NO: 74 kodiert, transformiert. Die Durchführung der
Erfindung ist jedoch nicht auf diese Sequenzen beschränkt;
die erfindungsgemäßen Verfahren können
vorteilhaft unter Verwendung jeder JMJC-kodierenden Nukleinsäure
oder jedes JMJC-Polypeptids, wie hier definiert, durchgeführt
werden.
-
Beispiele
für Nukleinsäuren, die JMJC-Polypeptide kodieren,
sind hier in Tabelle B1 von Beispiel 12 angegeben. Solche Nukleinsäuren
eignen sich zur Durchführung der erfindungsgemäßen
Verfahren. Die in Tabelle B1 von Beispiel 12 angegebenen Aminosäuresequenzen
sind beispielhafte Sequenzen von Orthologen und Paralogen des JMJC-Polypeptids
gemäß SEQ ID NO: 74, wobei die Begriffe ”Orthologe” und ”Paraloge” wie
hier definiert sind. Weitere Orthologe und Paraloge können
leicht identifiziert werden, indem man eine so genannte reziproke
Blast-Suche durchführt. Dies beinhaltet üblicherweise
eine erste BLAST, bei der mit einer Abfragesequenz ein ”BLASTing” gegen
eine beliebige Sequenzdatenbank, wie die öffentlich zugängliche
NCBI-Datenbank, durchgeführt wird (zum Beispiel unter Verwendung
von einer der in Tabelle B1 von Beispiel 12 aufgelisteten Sequenzen).
BLASTN oder TBLASTX (unter Verwendung von Standard-Default-Werten)
werden im Allgemeinen dann verwendet, wenn man von einer Nukleotidsequenz
ausgeht, und BLASTP oder TBLASTN (unter Verwendung von Standard-Default-Werten),
wenn man von einer Proteinsequenz ausgeht. Die BLAST-Ergebnisse
können gegebenenfalls gefiltert werden. Mit den Volllängen-Sequenzen
der gefilterten oder ungefilterten Ergebnisse wird anschließend
ein zweites BLASTing gegen Sequenzen des Organismus, von dem die
Abfragesequenz stammt, durchgeführt (ist die Abfragesequenz
die SEQ ID NO: 73 oder SEQ ID NO: 74, dann wäre das zweite
BLASTing daher gegen Sequenzen aus Arabidopsis). Die Ergebnisse
des ersten und des zweiten BLASTing werden dann verglichen. Ein
Paralog wird identifiziert, wenn ein hochrangiger Hit von dem ersten
BLASTing aus derselben Art stammt wie die Abfragesequenz, in diesem
Fall führt ein zweites BLASTing idealerweise zu der Abfragesequenz
unter den höchstrangigen Hits; ein Ortholog wird identifiziert,
wenn ein hochrangiger Hit in dem ersten BLASTing nicht von derselben
Art stammt wie die Abfragesequenz und vorzugsweise beim zweiten
BLASTing dazu führt, dass die Abfragesequenz unter den
höchstrangigen Hits ist.
-
Hochrangige
Hits sind solche mit niedrigem E-Wert. Je niedriger der E-Wert,
desto signifikanter der ”Score” (anders ausgedrückt,
desto niedriger die Wahrscheinlichkeit, dass der Hit durch Zufall
gefunden wurde). Die Berechnung des E-Werts ist in der Fachwelt
gut bekannt. Das ”Scoring” der Vergleiche erfolgt
nicht nur mittels E-Werten, sondern auch anhand der Prozent Identität.
Prozent Identität bezieht sich auf die Anzahl an identischen
Nukleotiden (oder Aminosäuren) zwischen den zwei verglichenen
Nukleinsäure-(oder Polypeptid-)Sequenzen über
eine bestimmte Länge. Bei großen Familien kann
man ClustalW und anschließend einen Neighbour-Joining-Tree
verwenden, um die Bildung von Clustern verwandter Gene leichter
sichtbar zu machen und Orthologe und Paraloge zu identifizieren.
-
Zur
Durchführung der erfindungsgemäßen Verfahren
können auch Nukleinsäurevarianten geeignet sein.
Zu solchen Varianten zählen beispielsweise Nukleinsäuren,
die für Homologe und Derivate von einer der in Tabelle
B1 von Beispiel 12 genannten Aminosäuresequenzen kodieren,
wobei die Begriffe ”Homolog” und ”Derivat” wie
hier definiert sind. Für die erfindungsgemäßen
Verfahren sind auch Nukleinsäuren geeignet, die für
Homologe und Derivate von Orthologen oder Paralogen von einer der
in Tabelle B1 von Beispiel 12 genannten Aminosäuresequenzen
kodieren. Homologe und Derivate, die für die erfindungsgemäßen
Verfahren geeignet sind, weisen im Wesentlichen dieselbe biologische
und funktionelle Aktivität auf wie das unmodifizierte Protein,
von dem sie stammen.
-
Zu
weiteren Nukleinsäurevarianten, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet sind,
zählen Abschnitte von Nukleinsäuren, die JMJC-Polypeptide
kodieren, Nukleinsäuren, die mit Nukleinsäuren,
die JMJC-Polypeptide kodieren, hybridisieren, Spleißvarianten
von Nukleinsäuren, die JMJC-Polypeptide kodieren, allelische
Varianten von Nukleinsäuren, die JMJC-Polypeptide kodieren,
und Varianten von Nukleinsäuren, die JMJC-Polypeptide kodieren,
die mittels ”gene shuffling” erhalten wurden.
Die Begriffe hybridisierende Sequenz, Spleißvariante, allelische
Variante und ”gene shuffling” sind wie hier definiert.
-
Nukleinsäuren,
die JMJC-Polypeptide kodieren, müssen keine Volllängen-Nukleinsäuren
sein, da die Durchführung der erfindungsgemäßen
Verfahren nicht auf die Verwendung von Volllängen-Nukleinsäuresequenzen
angewiesen ist. Erfindungsgemäß wird daher ein
Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen bereitgestellt,
bei dem man in eine(r) Pflanze einen Abschnitt einer beliebigen
der in Tabelle B1 von Beisipiel 12 angegebenen Nukleinsäuresequenzen
oder einen Abschnitt einer Nukleinsäure, die ein Ortholog,
Paralog oder Homolog von einer der in Tabelle B1 von Beispiel 12
genannten Aminosäuresequenzen kodiert, einführt
und exprimiert.
-
Ein
Abschnitt einer Nukleinsäure kann zum Beispiel dadurch
hergestellt werden, dass man eine oder mehrere Deletionen an der
Nukleinsäure durchführt. Die Abschnitte können
in isolierter Form verwendet werden oder können an andere
kodierende (oder nichtkodierende) Sequenzen fusioniert werden, um
zum Beispiel ein Protein herzustellen, das mehrere Aktivitäten
in sich vereinigt. Wenn es an andere kodierende Sequenzen fusioniert
ist, kann das so erhaltene, nach der Translation hergestellte Polypeptid
größer sein als für den Proteinabschnitt
vorhergesagt wurde.
-
Abschnitte,
die für die erfindungsgemäßen Verfahren
geeignet sind, kodieren ein JMJC-Polypeptid, wie hier definiert,
und weisen im Wesentlichen dieselbe biologische Aktivität
wie die in Tabelle B1 von Beispiel 12 angegebenen Aminosäuresequenzen
auf. Vorzugsweise handelt es sich bei dem Abschnitt um einen Abschnitt
einer der in Tabelle B1 von Beispiel 12 genannten Nukleinsäuren
oder um einen Abschnitt einer Nukleinsäure, die ein Ortholog
oder Paralog von einer der in Tabelle B1 von Beispiel 12 genannten
Aminosäuresequenzen kodiert. Vorzugsweise ist der Abschnitt
mindestens 100, 200, 500, 550, 600, 650, 700, 750, 800, 850, 900,
950, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 5000 aufeinander
folgende Nukleotide lang, wobei es sich bei den aufeinander folgenden
Nukleotiden um eine der in Tabelle B1 von Beispiel 12 genannten
Nukleinsäuresequenzen oder um eine Nukleinsäure
handelt, die ein Ortholog oder Paralog von einer der in Tabelle
B1 von Beispiel 12 genannten Aminosäuresequenzen kodiert.
Am stärksten bevorzugt ist der Abschnitt ein Abschnitt
der Nukleinsäure gemäß SEQ ID NO: 73.
Vorzugsweise kodiert der Abschnitt eine Aminosäuresequenz,
die, wenn sie zur Konstruktion eines Stammbaums, wie dem in 8 dargestellten,
verwendet wird, eher mit der Gruppe der JMJC-Polypeptide (Takeuchi
et al. Dev Dyn. 2006 235 (9): 2449–59), welche
die in SEQ ID NO: 74 dargestellte Aminosäuresequenz umfassen,
als mit jeder anderen Gruppe Cluster bildet.
-
Eine
andere Nukleinsäurevariante, die für die erfindungsgemäßen
Verfahren geeignet ist, ist eine Nukleinsäure, die unter
Bedingungen einer verringerten Stringenz, vorzugsweise unter stringenten
Bedingungen, mit einer Nukleinsäure, die ein JMJC-Polypeptid
kodiert, wie hier definiert, oder mit einem Abschnitt, wie hier definiert,
hybridisieren kann.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Nukleinsäure,
die mit einer der in Tabelle B1 von Beispiel 12 angegebenen Nukleinsäuren
hybridisieren kann, einführt und exprimiert, oder bei dem
man in eine(r) Pflanze eine Nukleinsäure, die mit einer
Nukleinsäure hybridisieren kann, die ein Ortholog, Paralog
oder Homolog von einer der in Tabelle B1 von Beispiel 12 genannten
Nukleinsäuresequenzen kodiert, einführt und exprimiert.
-
Hybridisierende
Sequenzen, die für die erfindungsgemäßen
Verfahren geeignet sind, kodieren ein JMJC-Polypeptid, wie hier
definiert, und weisen im Wesentlichen dieselbe biologische Aktivität
wie die in Tabelle B1 von Beispiel 12 genannten Aminosäuresequenzen
auf. Vorzugsweise kann die hybridisierende Sequenz mit einer der
in Tabelle B1 von Beispiel 12 genannten Nukleinsäuren oder
mit einem Abschnitt von einer dieser Sequenzen hybridisieren, wobei
ein Abschnitt wie oben definiert ist, oder wobei die hybridisierende
Sequenz mit einer Nukleinsäuresequenz, die ein Ortholog
oder Paralog von einer der in Tabelle B1 von Beispiel 12 genannten
Aminosäuresequenzen kodiert, hybridisieren kann. Am stärksten
bevorzugt kann die hybridisierende Sequenz mit einer Nukleinsäure
gemäß SEQ ID NO: 73 oder einem Abschnitt davon
hybridisieren.
-
Vorzugsweise
kodiert die hybridisierende Sequenz eine Aminosäuresequenz,
die, wenn sie zur Konstruktion eines Stammbaums, wie dem in 8 dargestellten,
verwendet wird, eher mit der Gruppe der JMJC-Polypeptide (Takeuchi
et al. Dev Dyn. 2006 235 (9): 2449–59), welche
die in SEQ ID NO: 74 dargestellte Aminosäuresequenz umfassen,
als mit jeder anderen Gruppe Cluster bildet.
-
Eine
andere Nukleinsäurevariante, die für die erfindungsgemäßen
Verfahren geeignet ist, ist eine Spleißvariante, die ein
JMJC-Polypeptid, wie hier vorstehend definiert, kodiert, wobei eine
Spleißvariante wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Spleißvariante
von einer der in Tabelle B1 von Beispiel 12 angegebenen Nukleinsäuresequenzen
oder eine Spleißvariante von einer Nukleinsäure,
die ein Ortholog, Paralog oder Homolog von einer der in Tabelle
B1 von Beispiel 12 genannten Aminosäuresequenzen kodiert,
einführt und exprimiert. Ein Beispiel für eine
gespleißte Variante des Gens, das SEQ ID NO: 74 kodiert,
wird durch SEQ ID NO: 73 und SEQ ID NO: 83 (die SEQ ID NO: 84 kodiert)
dargestellt.
-
Bevorzugte
Spleißvarianten sind Spleißvarianten einer Nukleinsäure
gemäß SEQ ID NO: 73 oder eine Spleißvariante
einer Nukleinsäure, die ein Ortholog oder Paralog der SEQ
ID NO: 74 kodiert. Vorzugsweise bildet die Aminosäuresequenz,
die von der Spleißvariante kodiert wird, wenn sie zur Konstruktion
eines Stammbaums, wie dem in 8 dargestellten,
verwendet wird, eher mit der Gruppe der JMJC-Polypeptide (Takeuchi
et al. Dev Dyn. 2006 235 (9): 2449–59), welche
die in SEQ ID NO: 74 dargestellte Aminosäuresequenz umfassen,
als mit jeder anderen Gruppe Cluster.
-
Eine
andere Nukleinsäurevariante, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet ist,
ist eine allelische Variante einer Nukleinsäure, die ein
JMJC-Polypeptid, wie hier vorstehend definiert, kodiert, wobei eine
allelische Variante wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine allelische Variante
von einer der in Tabelle B1 von Beispiel 12 angegebenen Nukleinsäuresequenzen
einführt und exprimiert oder bei dem man in eine(r) Pflanze
eine allelische Variante einer Nukleinsäure, die ein Ortholog,
Paralog oder Homolog von einer der in Tabelle B1 von Beispiel 12
genannten Aminosäuresequenzen kodiert, einführt
und exprimiert.
-
Die
allelischen Varianten, die für die erfindungsgemäßen
Verfahren geeignet sind, haben im Wesentlichen dieselbe biologische
Aktivität wie das JMJC-Polypeptid gemäß SEQ
ID NO: 74 und jede der in Tabelle B1 von Beispiel 12 dargestellten
Aminosäuren. Allelische Varianten kommen in der Natur vor
und von den erfindungsgemäßen Verfahren wird die
Verwendung dieser natürlichen Allele mit umfasst. Vorzugsweise
handelt es sich bei der allelischen Variante um eine allelische
Variante gemäß SEQ ID NO: 73 oder eine allelische
Variante einer Nukleinsäure, die ein Ortholog oder Paralog
der SEQ ID NO: 74 kodiert. Vorzugsweise bildet die Aminosäuresequenz,
die von der allelischen Variante kodiert wird, wenn sie zur Konstruktion
eines Stammbaums, wie dem in 8 dargestellten,
verwendet wird, eher mit der Gruppe der JMJC-Polypeptide (Takeuchi et
al. Dev Dyn. 2006 235 (9): 2449–59), welche die
in SEQ ID NO: 74 dargestellte Aminosäuresequenz umfassen,
als mit jeder anderen Gruppe Cluster.
-
Man
kann auch ”Gene shuffling” oder gerichtete Evolution
dazu verwenden, Varianten von Nukleinsäuren zu erzeugen,
die JMJC-Polypeptide, wie vorstehend definiert, kodieren, wobei
der Begriff ”gene shuffling” wie hier definiert
ist.
-
Erfindungsgemäß wird
ein Verfahren für die Verbesserung von Ertragsmerkmalen
bei Pflanzen bereitgestellt, bei dem man in eine(r) Pflanze eine
Variante von einer der in Tabelle B1 von Beispiel 12 angegebenen Nukleinsäuresequenzen
einführt und exprimiert oder bei dem man eine Variante
einer Nukleinsäure, die ein Ortholog, Paralog oder Homolog
von einer der in Tabelle B1 von Beispiel 12 angegebenen Aminosäuresequenzen
kodiert, einführt und exprimiert, wobei die Nukleinsäurevariante
mittels ”gene shuffling” erhalten wird.
-
Vorteilhafterweise
stellt die vorliegende Erfindung bisher unbekannte JMJC-Nukleinsäure-
und Polypeptidsequenzen bereit.
-
Gemäß einer
weiteren Ausführungsform der vorliegenden Erfindung wird
ein isoliertes Nukleinsäuremolekül bereitgestellt,
das Folgendes umfasst:
- (i) eine durch SEQ ID
NO: 169 dargestellte Nukleinsäure;
- (ii) eine Nukleinsäure oder ein Fragment davon, die/das
zu der SEQ ID NR: 169 komplementär ist;
- (ii) eine Nukleinsäure, die ein JMJC-Polypeptid kodiert
und, in steigender Reihenfolge der Präferenz, mindestens
70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
mit der in SEQ ID NO: 170 angegebenen Aminosäuresequenz
besitzt;
- (iv) eine Nukleinsäure, die unter stringenten Bedingungen
an eine der unter (i), (ii) oder (iii) oben angegebenen Nukleinsäuren
hybridisieren kann.
-
Gemäß einer
weiteren Ausführungsform der vorliegenden Erfindung wird
deshalb ein isoliertes Polypeptid bereitgestellt, das Folgendes
umfasst:
- (i) eine Aminosäuresequenz,
die, in steigender Reihenfolge der Präferenz, mindestens
70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder 100% oder
mehr Sequenzidentität mit der in SEQ ID NO: 170 angegebenen
Aminosäuresequenz hat;
- (ii) Derivate von einer der unter (i) angegebenen Aminosäuresequenzen.
-
Vorzugsweise
bildet die Aminosäuresequenz, die von der durch gene shuffling
erhaltenen Nukleinsäurevariante kodiert wird, wenn sie
zur Konstruktion eines Stammbaums, wie dem in 8 dargestellten,
verwendet wird, eher mit der Gruppe der JMJC-Polypeptide (Takeuchi
et al. Dev Dyn. 2006 235 (9): 2449–59), welche
die in SEQ ID NO: 74 dargestellte Aminosäuresequenz umfassen,
als mit jeder anderen Gruppe Cluster.
-
Nukleinsäurevarianten
können weiterhin auch durch ortsgerichtete Mutagenese erhalten
werden. Zur Erzielung einer ortsgerichteten Mutagenese sind verschiedene
Verfahren verfügbar, von denen die häufigsten Verfahren
auf PCR-Basis sind (Current Protocols in Molecular Biology.
Wiley Hrsg.).
-
Nukleinsäuren,
die JMJC-Polypeptide kodieren, können von einer beliebigen
natürlichen oder künstlichen Quelle stammen. Die
Nukleinsäure kann von ihrer nativen Form in ihrer Zusammensetzung
und/oder genomischen Umgebung durch gezielte Manipulation durch
den Menschen modifiziert werden. Vorzugsweise stammt die Nukleinsäure,
die das JMJC-Polypeptid kodiert, aus einer Pflanze, weiter bevorzugt
aus einer zweikeimblättrigen Pflanze, stärker
bevorzugt aus der Familie Brassicaceae, am stärksten bevorzugt
stammt die Nukleinsäure aus Arabidopsis thaliana.
-
Die
Durchführung der erfindungsgemäßen Verfahren
führt zu Pflanzen mit verstärkten Ertragsmerkmalen.
Insbesondere führt die Durchführung der erfindungsgemäßen
Verfahren zu Pflanzen mit erhöhtem Ertrag, insbesondere
erhöhtem Samenertrag, im Vergleich zu Kontrollpflanzen.
Die Begriffe ”Ertrag” und ”Samenertrag” werden
hier im Abschnitt ”Definitionen” genauer beschrieben.
-
Werden
hier verbesserte Ertragsmerkmale erwähnt, so soll dies
eine Erhöhung der Biomasse (des Gewichts) von einem oder
mehreren Teilen einer Pflanze bedeuten, was oberirdische (erntbare)
Teile und/oder unterirdische (erntbare) Teile beinhalten kann. Insbesondere
sind solche oberirdischen erntbaren Teile Samen, und die Durchführung
der erfindungsgemäßen Verfahren führt
zu Pflanzen mit höherem Ertrag im Vergleich zu Kontrollpflanzen.
-
Wählt
man Mais als Beispiel, kann sich eine Ertragserhöhung als
eines oder mehrere der folgenden Merkmale äußern:
Unter anderem erhöhte Anzahl an Pflanzen, die pro Hektar
oder Acre wachsen, eine Erhöhung der Anzahl an Ähren
pro Pflanze, eine Erhöhung der Anzahl an Reihen, der Anzahl
an Körnern pro Reihe, des Korngewichts, Tausendkorngewichts,
der Ährenlänge/des Ährendurchmessers,
eine Erhöhung der Samenfüllungsrate (d. h. der
Anzahl gefüllter Samen dividiert durch die Gesamtzahl der
Samen und multipliziert mit 100). Wählt man Reis als Beispiel,
kann sich eine Ertragserhöhung als Erhöhung bezüglich
eines oder mehrerer der folgenden Merkmale ausdrücken:
Unter anderem Anzahl der Pflanzen pro Hektar oder Acre, Anzahl der
Rispen pro Pflanze, Anzahl der Ährchen pro Rispe, Anzahl
der Blüten (Einzelblüten) pro Rispe (ausgedrückt
als Verhältnis der Anzahl an gefüllten Samen zu
der Anzahl an Primärrispen), Erhöhung der Samenfüllungsrate
(d. h. Anzahl gefüllter Samen dividiert durch die Gesamtzahl
an Samen und multipliziert mit 100), erhöhtes Tausendkorngewicht.
-
Die
vorliegende Erfindung stellt ein Verfahren zur Erhöhung
des Ertrags, insbesondere des Samenertrags von Pflanzen, verglichen
mit Kontrollpflanzen bereit, wobei das Verfahren die Modulation,
vorzugsweise die Erhöhung, der Expression einer Nukleinsäure,
die ein JMJC-Polypeptid, wie hier definiert, kodiert, in einer Pflanze
umfasst.
-
Weil
die erfindungsgemäßen transgenen Pflanzen einen
erhöhten Ertrag aufweisen, ist es wahrscheinlich, dass
diese Pflanzen (zumindest während eines Teils ihres Lebenszyklus)
eine höhere Wachstumsrate aufweisen als Kontrollpflanzen
in einem entsprechenden Stadium ihres Lebenszyklus.
-
Die
erhöhte Wachstumsrate kann für einen oder mehrere
Teile einer Pflanze (darunter Samen) spezifisch sein oder kann im
Wesentlichen in der ganzen Pflanze stattfinden. Pflanzen mit einer
erhöhten Wachstumsrate können einen kürzeren
Lebenszyklus aufweisen. Unter dem Lebenszyklus einer Pflanze kann
man diejenige Zeit verstehen, die die Pflanze benötigt,
um von einem trockenen reifen Samen bis zu dem Stadium heranzuwachsen,
in dem die Pflanze trockene reife Samen ähnlich dem Ausgangsmaterial
produziert hat. Dieser Lebenszyklus kann von Faktoren, wie Jungpflanzenvitalität,
Wachstumsrate, ”Greenness Index”, Blütezeit und
Geschwindigkeit der Samenreifung, beeinflusst werden. Die Erhöhung
der Wachstumsrate kann in einem Stadium oder in mehreren Stadien
im Lebenszyklus einer Pflanze oder im Wesentlichen während
des gesamten Lebenszyklus der Pflanze erfolgen. Eine erhöhte
Wachstumsrate während der Frühstadien im Lebenszyklus
einer Pflanze kann verbesserte Vitalität widerspiegeln.
Die erhöhte Wachstumsrate kann den Erntezyklus einer Pflanze
verändern, so dass die Pflanzen später gesät
werden können und/oder früher geerntet werden können
als dies sonst möglich wäre (ein ähnlicher
Effekt lässt sich mit einer früheren Blütezeit
erzielen). Ist die Wachstumsrate ausreichend erhöht, kann
dies weiteres Aussäen von Samen derselben Pflanzenart ermöglichen
(zum Beispiel Aussäen und Ernten von Reispflanzen und anschließendes
Aussäen und Ernten von weiteren Reispflanzen sogar innerhalb
einer herkömmlichen Wachstumsperiode). Wenn die Wachstumsrate
ausreichend erhöht ist, kann dies ebenso das weitere Aussäen
von Samen anderer Pflanzenarten ermöglichen (zum Beispiel
das Aussäen und Ernten von Maispflanzen und anschließend
zum Beispiel Aussäen und gegebenenfalls Ernten von Sojabohne,
Kartoffel oder einer anderen geeigneten Pflanze). Auch mehrmalige
zusätzliche Ernten von demselben Wurzelstock können
bei einigen Kulturpflanzen möglich sein. Eine Veränderung des
Erntezyklus einer Pflanze kann zu einer Erhöhung der jährlichen
Biomasseproduktion pro Acre führen (weil man eine bestimmte
Pflanze öfter (z. B. pro Jahr) heranziehen und ernten kann).
Eine erhöhte Wachstumsrate kann auch den Anbau transgener
Pflanzen in einem breiteren geographischen Bereich als ihre Wildtyp-Gegenstücke
ermöglichen, da die räumlichen Beschränkungen
für den Anbau einer Kultur häufig von ungünstigen Umweltbedingungen
entweder während der Pflanzzeit (früh in der Saison)
oder während der Erntezeit (spät in der Saison)
bestimmt werden. Solche ungünstigen Bedingungen können
vermieden werden, wenn der Erntezyklus verkürzt ist. Die
Wachstumsrate kann durch Ableiten verschiedener Parameter aus Wachstumskurven bestimmt
werden, wobei die Parameter u. a. folgende sein können:
T-Mid (Zeitdauer, die die Pflanzen benötigen, um 50% ihrer
Maximalgröße zu erreichen) und T-90 (Zeitdauer,
die die Pflanzen benötigen, um 90% ihrer Maximalgröße
zu erreichen).
-
Gemäß einem
bevorzugten Merkmal der vorliegenden Erfindung führt die
Durchführung der erfindungsgemäßen Verfahren
zu Pflanzen mit einer im Vergleich zu Kontrollpflanzen erhöhten
Wachstumsrate. Erfindungsgemäß wird daher ein
Verfahren zur Erhöhung der Wachstumsrate von Pflanzen bereitgestellt,
wobei das Verfahren die Modulation, vorzugsweise die Erhöhung,
der Expression einer Nukleinsäure, die ein JMJC-Polypeptid,
wie hier definiert, kodiert, in einer Pflanze umfasst.
-
Eine
Erhöhung des Ertrags und/oder der Wachstumsrate tritt unabhängig
davon auf, ob die Pflanze unter Nichtstressbedingungen steht oder
ob die Pflanze im Vergleich zu Kontrollpflanzen verschiedenen Stressbedingungen
unterworfen wird. Typischerweise reagieren Pflanzen auf Stress dadurch,
dass sie langsamer wachsen. Unter starken Stressbedingungen kann
die Pflanze sogar ihr Wachstum völlig einstellen. Leichter
Stress ist dagegen hier als jeglicher Stress definiert, dem eine
Pflanze ausgesetzt ist und der nicht dazu führt, dass die
Pflanze völlig aufhört zu wachsen, ohne dass sie
die Fähigkeit besitzt, ihr Wachstum wieder aufzunehmen.
Leichter Stress im Sinne der Erfindung führt zu einer Verringerung
des Wachstums der gestressten Pflanzen von weniger als 40%, 35%
oder 30%, vorzugsweise weniger als 25%, 20% oder 15%, stärker
bevorzugt weniger als 14%, 13%, 12%, 11% oder 10% oder weniger im
Vergleich zu der Kontrollpflanze unter Nichtstressbedingungen. Aufgrund
von Fortschritten bei den landwirtschaftlichen Praktiken (Bewässerung,
Düngung, Pestizidbehandlungen) findet man bei angebauten
Kulturpflanzen nicht oft starke Stressbedingungen. Daher ist das
durch leichten Stress induzierte geschwächte Wachstum häufig
ein unerwünschtes Merkmal in der Landwirtschaft. Leichte
Stressbedingungen sind die alltäglichen biotischen und/oder
abiotischen (Umwelt-)Stressbedingungen, denen eine Pflanze ausgesetzt
ist. Abiotischer Stress kann durch Trockenheit oder Wasserüberschuss,
anaeroben Stress, Salzstress, chemische Toxizität, oxidativen
Stress und heiße, kalte oder Gefriertemperaturen verursacht
werden. Bei dem abiotischen Stress kann es sich um einen osmotischen
Stress handeln, der durch Wasserstress (insbesondere aufgrund von
Trockenheit), Salzstress, oxidativen Stress oder ionenbedingten
Stress verursacht wird. Unter biotischem Stress versteht man gewöhnlich
Stressbedingungen, die durch Pathogene, wie Bakterien, Viren, Pilze
und Insekten, verursacht werden.
-
Insbesondere
können die erfindungsgemäßen Verfahren
unter Nichtstressbedingungen oder unter leichten Trockenheitsbedingungen
durchgeführt werden, wobei man Pflanzen erhält,
deren Ertrag im Vergleich zu Kontrollpflanzen erhöht ist.
Wie von Wang et al. (Planta (2003), 218: 1–14)
beschrieben, führt abiotischer Stress zu einer Reihe von
morphologischen, physiologischen, biochemischen und molekularen
Veränderungen, die das Pflanzenwachstum und die Pflanzenproduktivität
negativ beeinflussen. Es ist bekannt, dass Trockenheit, Salinität,
extreme Temperaturen und oxidativer Stress miteinander in Verbindung
stehen und über ähnliche Mechanismen Wachstums-
und Zellschäden induzieren können. Rabbani
et al. (Plant Physiol. (2003), 133: 1755–1767)
beschreibt ein besonders hohes Ausmaß an gegenseitiger
Beeinflussung (”Cross-Talk”) von Trockenheitsstress
und durch hohe Salinität verursachtem Stress. Zum Beispiel äußern
sich Trockenheit und/oder Versalzung in erster Linie als osmotischer
Stress, was zur Störung der Homöostase und der
Ionenverteilung in der Zelle führt. Oxidativer Stress,
der häufig hohe oder niedrige Temperatur, Salinität
oder Trockenheitsstress begleitet, kann zur Denaturierung funktioneller
und struktureller Proteine führen. Infolgedessen aktivieren
diese verschiedenen Umweltstressbedingungen häufig ähnliche
Zellsignalwege und Zellreaktionen, wie die Produktion von Stressproteinen,
die Hinaufregulation von Antioxidantien, die Akkumulation von kompatiblen
gelösten Stoffen und ein Einstellen des Wachstums. Der
Begriff ”Nichtstress”-Bedingungen, wie hier verwendet,
steht für diejenigen Umweltbedingungen, die ein optimales
Wachstum von Pflanzen gestatten. Der Fachmann ist mit normalen Bodenbedingungen
und Klimabedingungen für einen bestimmten Standort vertraut.
Pflanzen unter optimalen Wachstumsbedingungen (die unter Nichtstressbedingungen
herangezogen werden) liefern oft einen Ertrag von, in der Reihenfolge
der zunehmenden Präferenz, mindestens 90%, 87%, 85%, 83%,
80%, 77% oder 75% der durchschnittlichen Produktion einer solchen
Pflanze in einer gegebenen Umgebung. Die durchschnittliche Produktion
kann bezogen auf die Ernte und/oder die Saison berechnet werden.
Der Fachmann ist mit den durchschnittlichen Erträgen einer
Kultur vertraut.
-
Die
Durchführung der erfindungsgemäßen Verfahren
führt zu Pflanzen, die bei Wachstum unter Nichtstressbedingungen
oder unter leichten Trockenheitsbedingungen einen im Vergleich zu
Kontrollpflanzen, die unter vergleichbaren Bedingungen herangezogen
werden, erhöhten Ertrag aufweisen. Erfindungsgemäß wird daher
ein Verfahren zur Erhöhung des Ertrags von Pflanzen, die
unter Nichtstressbedingungen oder unter leichten Trockenheitsbedingungen
herangezogen werden, bereitgestellt, wobei das Verfahren die Erhöhung der
Expression einer Nukleinsäure, die ein JMJC-Polypeptid
kodiert, in einer Pflanze umfasst.
-
Die
Durchführung der erfindungsgemäßen Verfahren
verleiht Pflanzen, die unter Nährstoffmangelbedingungen,
insbesondere unter Stickstoffmangelbedingungen, herangezogen werden,
einen erhöhten Ertrag im Vergleich zu Kontrollpflanzen,
die unter vergleichbaren Bedingungen herangezogen werden. Erfindungsgemäß wird
daher auch ein Verfahren zur Erhöhung des Ertrags in Pflanzen,
die unter Nährstoffmangelbedingungen herangezogen werden,
bereitgestellt, wobei man bei dem Verfahren die Expression einer
Nukleinsäure, die ein JMJC-Polypeptid kodiert, in einer
Pflanze erhöht. Ein Nährstoffmangel kann durch
ein Fehlen oder einen Überschuss von Nährstoffen,
wie unter anderem Stickstoff, Phosphate und andere phosphorhaltige
Verbindungen, Kalium, Calcium, Cadmium, Magnesium, Mangan, Eisen
und Bor, hervorgerufen werden.
-
Die
vorliegende Erfindung umfasst Pflanzen oder Teile davon (einschließlich
Samen), die durch die erfindungsgemäßen Verfahren
erhältlich sind. Die Pflanzen oder Teile davon umfassen
ein Nukleinsäuretransgen, das ein JMJC-Polypeptid kodiert,
wie oben definiert.
-
Die
Erfindung stellt auch Genkonstrukte und Vektoren bereit, um die
Einführung und/oder Expression von Nukleinsäuren,
die JMJC-Polypeptide kodieren, in Pflanzen zu erleichtern. Die Genkonstrukte
können in Vektoren eingefügt werden, die im Handel
erhältlich sein können und die für die
Transformation des interessierenden Gens in Pflanzen und für
die Expression des interessierenden Gens in den transformierten
Zellen geeignet sind. Die Erfindung stellt auch die Verwendung eines
Genkonstrukts, wie hier definiert, bei den erfindungsgemäßen
Verfahren bereit.
-
Genauer
gesagt, stellt die vorliegende Erfindung ein Konstrukt bereit, das
folgendes umfasst:
- (a) eine Nukleinsäure,
die ein JMJC-Polypeptid kodiert, wie oben definiert;
- (b) eine oder mehrere Kontrollsequenzen, welche die Expression
der Nukleinsäuresequenz unter (a) vorantreiben können,
und gegebenenfalls
- (c) eine Transkriptionsterminationssequenz.
-
Vorzugsweise
ist die Nukleinsäure, die ein JMJC-Polypeptid kodiert,
wie vorstehend definiert.
-
Die
Begriffe ”Kontrollsequenz” und ”Terminationssequenz” sind
wie hier definiert.
-
Pflanzen
werden mit einem Vektor transformiert, der eine der oben beschriebenen
Nukleinsäuren umfasst. Der Fachmann kennt die genetischen
Elemente, die in einem Vektor vorhanden sein müssen, damit Wirtszellen
erfolgreich transformiert, Wirtszellen, die die interessierende
Sequenz enthalten, selektiert und vermehrt werden können.
Die interessierende Sequenz ist mit einer oder mehreren Kontrollsequenzen
(mindestens mit einem Promoter) funktionsfähig verbunden.
-
Jede
Art von Promoter, ob natürlich oder synthetisch, kann vorteilhaft
für das Vorantreiben der Expression der Nukleinsäuresequenz
verwendet werden. Ein konstitutiver Promoter ist für die
Verfahren besonders geeignet. Die Definitionen der verschiedenen
Promotertypen siehe hier im Abschnitt ”Definitionen”.
-
Es
sollte selbstverständlich sein, dass die Anwendbarkeit
der vorliegenden Erfindung weder auf die durch SEQ ID NO: 73 dargestellte,
ein JMJC-Poylpeptid kodierende Nukleinsäure noch auf die
Expression einer ein JMJC-Polypeptid kodierenden Nukleinsäure,
wenn sie von einem konstitutiven Promotor angetrieben wird, beschränkt
ist.
-
Der
konstitutive Promotor ist vorzugsweise ein GOS2-Promotor, vorzugsweise
ein GOS2-Promotor aus Reis. Weiterhin bevorzugt wird der konstitutive
Promotor durch eine Nukleinsäuresequenz wiedergegeben,
die im Wesentlichen der SEQ ID NO: 75 gleicht, am stärksten
bevorzugt wird der Protomotor durch die SEQ ID NO: 75 dargestellt.
Weitere Beispiele für konstitutive Promotoren siehe hier
im Abschnitt ”Definitionen”.
-
Gegebenenfalls
kann/können eine oder mehrere Terminatorsequenzen in dem
Konstrukt, das in eine Pflanze eingeführt wird, verwendet
werden. Weitere Regulationselemente können u. a. Transkriptionsenhancer
und Translationsenhancer sein. Der Fachmann ist mit Terminator-
und Enhancersequenzen vertraut, die für die Durchführung
der Erfindung geeignet sein können. Es kann auch eine Intronsequenz
zu der 5'-untranslatierten Region (UTR) oder der kodierenden Sequenz
hinzugefügt werden, um die Menge an reifer Botschaft, die
im Cytosol akkumuliert, zu erhöhen, wie im Abschnitt Definitionen
beschrieben ist. Weitere Kontrollsequenzen (neben Promotor-, Enhancer-,
Silencer-, Intronsequenzen, 3'UTR- und/oder 5'UTR-Regionen) können Protein-
und/oder RNA-stabilisierende Elemente sein. Solche Sequenzen sind
dem Fachmann bekannt oder können von ihm leicht erhalten
werden.
-
Die
erfindungsgemäßen Genkonstrukte können
weiterhin eine Replikationsursprungssequenz beinhalten, die für
die Aufrechterhaltung und/oder die Replikation in einem bestimmten
Zelltyp erforderlich ist. Ein Beispiel ist, wenn ein Genkonstrukt
in einer Bakterienzelle als episomales genetisches Element (z. B.
als Plasmid- oder Cosmidmolekül) aufrechterhalten werden
muss. Bevorzugte Replikationsursprünge sind u. a. der f1-ori
und colE1, sie sind jedoch nicht hierauf beschränkt.
-
Für
den Nachweis eines erfolgreichen Transfers der Nukleinsäuresequenzen,
wie sie bei den erfindungsgemäßen Verfahren verwendet
werden, und/oder für die Selektion von transgenen Pflanzen,
die diese Nukleinsäuren umfassen, ist es vorteilhaft, Markergene
(oder Reportergene) zu verwenden. Daher kann das Genkonstrukt gegebenenfalls
ein Selektionsmarkergen umfassen. Selektionsmarker sind hier im
Abschnitt ”Definitionen” eingehender beschrieben.
-
Bekanntlich
nimmt abhängig von dem verwendeten Expressionsvektor und
der verwendeten Transfektionstechnik nach einer stabilen oder transienten
Integration von Nukleinsäuren in Pflanzenzellen nur eine
Minderheit der Zellen die Fremd-DNA auf und integriert sie in ihr
Genom, falls dies gewünscht ist. Um diese Integranten zu
identifizieren und zu selektieren, wird üblicherweise ein
Gen, das einen Selektionsmarker (wie die oben beschriebenen) kodiert,
zusammen mit dem interessierenden Gen in die Wirtszellen eingeführt.
Diese Marker können zum Beispiel in Mutanten verwendet
werden, in denen diese Gene, zum Beispiel durch Deletion durch herkömmliche
Verfahren, nicht funktionell sind. Weiterhin können Nukleinsäuremoleküle,
die einen Selektionsmarker kodieren, in eine Wirtszelle auf demselben
Vektor, der die Sequenz umfasst, die die erfindungsgemäßen
Polypeptide oder die Polypeptide, die bei den erfindungsgemäßen
Verfahren verwendet werden, kodiert, oder auch auf einen separaten
Vektor eingeführt werden. Zellen, die mit der eingeführten
Nukleinsäure stabil transfiziert worden sind, können
zum Beispiel durch Selektion identifiziert werden (zum Beispiel überleben
Zellen, die den Selektionsmarker integriert haben, während
die anderen Zellen absterben). Die Markergene können aus
der transgenen Zelle entfernt oder ausgeschnitten werden, wenn sie
nicht mehr gebraucht werden. Techniken zur Entfernung von Markergenen
sind im Stand der Technik bekannt, geeignete Techniken sind vorstehend
im Abschnitt Definitionen beschrieben.
-
Die
Erfindung stellt auch ein Verfahren zur Herstellung von transgenen
Pflanzen mit verbesserten Ertragsmerkmalen im Vergleich zu Kontrollpflanzen
bereit, bei dem man in eine(r) Pflanze eine Nukleinsäure,
die ein JMJC-Polypeptid, wie oben definiert, kodiert, einführt
und exprimiert.
-
Genauer
gesagt stellt die vorliegende Erfindung ein Verfahren für
die Herstellung von transgenen Pflanzen mit erhöhten verbesserten
Ertragsmerkmalen, insbesondere erhöhtem (Samen-)Ertrag
bereit, wobei das Verfahren Folgendes umfasst:
- (i)
Einführen und Exprimieren einer Nukleinsäure,
die ein JMJC-Polypeptid kodiert, in eine(r) Pflanze oder Pflanzenzelle
und
- (ii) Kultivieren der Pflanzenzelle unter Bedingungen, die das
Pflanzenwachstum und die Pflanzenentwicklung fördern.
-
Bei
der Nukleinsäure gemäß (i) kann es sich
um eine der Nukleinsäuren handeln, die ein JMJC-Polypeptid,
wie hier definiert, kodieren können.
-
Die
Nukleinsäure kann direkt in eine Pflanzenzelle oder in
die Pflanze selbst eingeführt werden (was das Einführen
in ein Gewebe, Organ oder irgendeinen anderen Teil einer Pflanze
beinhaltet). Gemäß einem bevorzugten Merkmal der
vorliegenden Erfindung wird die Nukleinsäure vorzugsweise
mittels Transformation in eine Pflanze eingeführt. Der
Begriff ”Transformation” ist hier im Abschnitt ”Definitionen” genauer
beschrieben.
-
Die
genetisch modifizierten Pflanzenzellen können durch alle
Verfahren regeneriert werden, mit denen der Fachmann vertraut ist.
Geeignete Verfahren finden sich in den oben genannten Veröffentlichungen
von S. D. Kung und R. Wu, Potrykus oder Höfgen und Willmitzer.
-
Im
Allgemeinen werden nach der Transformation die Pflanzenzellen oder
Zellgruppierungen hinsichtlich des Vorhandenseins von einem oder
mehreren Markern selektiert, die von in Pflanzen exprimierbaren
Genen kodiert werden, die mit dem interessierenden Gen cotransferiert
wurden, wonach das transformierte Material zu einer ganzen Pflanze
regeneriert wird. Für die Selektion von transformierten
Pflanzen wird das bei der Transformation erhaltene Pflanzenmaterial
in der Regel derartigen Selektionsbedingungen unterworfen, dass man
transformierte Pflanzen von untransformierten Pflanzen unterscheiden
kann. So können zum Beispiel Samen, die auf die oben beschriebene
Weise erhalten wurden, ausgepflanzt werden und nach einer anfänglichen Wachstumsphase
einer geeigneten Selektion durch Spritzen unterworfen werden. Eine
weitere Möglichkeit besteht darin, dass man die Samen,
gegebenenfalls nach Sterilisieren, auf Agarplatten unter Verwendung
eines geeigneten Selektionsmittels heranzieht, so dass nur die transformierten
Samen zu Pflanzen heranwachsen können. Alternativ werden
die transformierten Pflanzen auf das Vorhandensein eines Selektionsmarkers,
wie die oben beschriebenen, gescreent.
-
Nach
DNA-Transfer und Regeneration können mutmaßlich
transformierte Pflanzen auch zum Beispiel unter Verwendung der Southern-Analyse
auf das Vorhandensein des interessierenden Gens, die Kopienzahl und/oder
die Genomorganisation untersucht werden. Alternativ oder zusätzlich
können die Expressionsniveaus der neu eingeführten
DNA unter Verwendung von Northern- und/oder Western-Analyse verfolgt
werden, wobei beide Techniken dem Durchschnittsfachmann bekannt
sind.
-
Die
erzeugten transformierten Pflanzen können durch eine Vielzahl
von Mitteln vermehrt werden, wie durch klonale Vermehrung oder klassische
Züchtungstechniken. So kann zum Beispiel eine transformierte Pflanze
der ersten Generation (T1-Pflanze) geselbstet werden, und homozygote
Transformanten der zweiten Generation (T2-Pflanzen) können
selektiert werden, und die T2-Pflanzen können dann mit
Hilfe von klassischen Züchtungstechniken weiter vermehrt
werden. Die erzeugten transformierten Organismen können
in einer Vielzahl von Formen vorliegen. So kann es sich zum Beispiel
um Chimären von transformierten Zellen und untransformierten
Zellen, klonale Transformanten (bei denen z. B. alle Zellen so transformiert
wurden, dass sie die Expressionskassette enthalten), Pfropfmaterial
von transformiertem und untransformiertem Gewebe (z. B. bei Pflanzen
ein transformierter Wurzelstock, der auf ein untransformiertes Edelreis
gepfropft wurde) handeln.
-
Die
vorliegende Erfindung erstreckt sich eindeutig auf jede Pflanzenzelle
oder Pflanze, die durch eines der hier beschriebenen Verfahren hergestellt
wurde, und auf alle Pflanzenteile und sämtliches Vermehrungsmaterial
davon. Die vorliegende Erfindung umfasst weiterhin die Nachkommenschaft
eine(r/s) primär transformierten oder transfizierten Zelle,
Gewebes, Organs oder ganzen Pflanze, die/das nach einem der oben
genannten Verfahren erzeugt wurde, wobei die einzige Voraussetzung
ist, dass die Nachkommenschaft dasselbe/dieselben genotypische(n)
und/oder phänotypische(n) Merkmal(e) aufweist, wie es/sie
der Elter bei den erfindungsgemäßen Verfahren
aufweist.
-
Die
Erfindung beinhaltet auch Wirtszellen, die eine isolierte Nukleinsäure
enthalten, die ein JMJC-Polypeptid, wie hier vorstehend definiert,
kodiert. Bevorzugte erfindungsgemäße Wirtszellen
sind Pflanzenzellen. Wirtspflanzen für die Nukleinsäuren
oder den Vektor, die bei dem erfindungsgemäßen
Verfahren verwendet werden, die Expressionskassette oder das Konstrukt
oder den Vektor sind im Prinzip vorteilhafterweise alle Pflanzen,
welche die bei dem erfindungsgemäßen Verfahren
verwendeten Polypeptide synthetisieren können.
-
Die
erfindungsgemäßen Verfahren lassen sich vorteilhaft
auf jede beliebige Pflanze anwenden. Zu den Pflanzen, die für
die erfindungsgemäßen Verfahren besonders geeignet
sind, gehören alle Pflanzen, die zur Superfamilie der Viridiplantae
gehören, insbesondere monokotyle und dikotyle Pflanzen,
einschließlich Futter- oder Weidepflanzenleguminosen, Zierpflanzen,
Nahrungsmittelkulturen, Bäume oder Sträucher.
Gemäß einer bevorzugten Ausführungsform
der vorliegenden Erfindung handelt es sich bei der Pflanze um eine
Kulturpflanze. Beispiele für Kulturpflanzen sind zum Beispiel
u. a. Sojabohne, Sonnenblume, Canola-Raps, Luzerne, Raps, Baumwolle,
Tomate, Kartoffel und Tabak. Weiterhin bevorzugt handelt es sich
bei der Pflanze um eine monokotyle Pflanze. Beispiele für
monokotyle Pflanzen sind zum Beispiel u. a. Zuckerrohr. Stärker
bevorzugt handelt es sich bei der Pflanze um ein Getreide. Beispiele
für Getreide sind zum Beispiel u. a. Reis, Mais, Weizen,
Gerste, Hirse, Roggen, Triticale, Sorghum und Hafer.
-
Die
Erfindung erstreckt sich auch auf erntbare Teile einer Pflanze,
wie zum Beispiel, aber nicht beschränkt auf Samen, Blätter,
Früchte, Blüten, Stängel, Rhizome, Knollen
und Zwiebeln. Die Erfindung betrifft weiterhin Produkte, die, vorzugsweise
direkt, von einem erntbaren Teil einer solchen Pflanze stammen,
wie trockene Pellets oder Pulver, Öl, Fett und Fettsäuren,
Stärke oder Proteine.
-
Gemäß einem
bevorzugten Merkmal der Erfindung handelt es sich bei der modulierten
Expression um erhöhte Expression. Verfahren zur Erhöhung
der Expression von Nukleinsäuren oder Genen oder Genprodukten
sind in der Fachwelt gut bekannt und Beispiele werden im Abschnitt
Definitionen gegeben.
-
Wie
vorstehend erwähnt, besteht ein bevorzugtes Verfahren zum
Modulieren (vorzugsweise Erhöhen) der Expression einer
Nukleinsäure, die ein JMJC-Polypeptid kodiert, darin, dass
man in eine(r) Pflanze eine Nukleinsäure, die ein JMJC-Polypeptid
kodiert, einführt und exprimiert; die Wirkungen der Durchführung
des Verfahren, d. h. das Erhöhen der Ertragsmerkmale, können
jedoch auch unter Verwendung anderer bekannter Techniken erzielt
werden, einschließlich, aber nicht beschränkt
auf T-DNA-Activation-Tagging, TILLING, homologe Rekombination. Eine
Beschreibung dieser Techniken findet sich im Abschnitt Definitionen.
-
Die
vorliegende Erfindung umfasst auch die Verwendung von Nukleinsäuren,
die JMJC-Polypeptide, wie hier beschrieben, kodieren, und die Verwendung
dieser JMJC-Polypeptide zur Verbesserung eines der oben genannten
Ertragsmerkmale in Pflanzen.
-
Hier
beschriebene Nukleinsäuren, die JMJC-Polypeptide kodieren,
oder die JMJC-Polypeptide selbst können Verwendung in Züchtungsprogrammen
finden, in denen ein DNA-Marker identifiziert wird, der mit einem
ein JMJC-Polpeptid kodierenden Gen genetisch verknüpft
sein kann. Die Nukleinsäuren/Gene oder die JMJC-Polypeptide
selbst können dazu verwendet werden, einen molekularen
Marker zu bestimmen. Dieser DNA- oder Protein-Marker kann dann in
Züchtungsprogrammen zur Selektion von Pflanzen mit erhöhten
Ertragsmerkmalen verwendet werden, wie hier vorstehend unter den
erfindungsgemäßen Verfahren definiert.
-
Allelische
Varianten einer Nukleinsäure/eines Gens, die/das ein JMJC-Polypeptid
kodiert, können auch Verwendung in Marker-unterstützten
Züchtungsprogrammen finden. Bei solchen Züchtungsprogrammen ist
manchmal das Einführen von allelischer Variation durch
Mutagenbehandlung der Pflanzen erforderlich, wobei zum Beispiel
EMS-Mutagenese verwendet wird; alternativ kann das Programm von
einer Sammlung von allelischen Varianten so genannten ”natürlichen” Ursprungs
ausgehen, die nicht absichtlich hervorgerufen wurden. Dann erfolgt
die Identifikation allelischer Varianten, zum Beispiel mittels PCR.
Darauf folgt ein Schritt zur Selektion besserer allelischer Varianten
der in Frage kommenden Sequenz, die bessere Erträge liefern.
Die Selektion wird gewöhnlich durchgeführt, indem
man die Wachstumsleistung von Pflanzen verfolgt, die verschiedene
allelische Varianten der in Frage kommenden Sequenz enthalten. Die
Wachstumsleistung kann im Gewächshaus oder auf dem Feld
verfolgt werden. Weitere Schritte beinhalten gegebenenfalls das
Kreuzen der Pflanzen, in denen die bessere allelische Variante identifiziert
wurde, mit einer anderen Pflanze. Dies kann zum Beispiel zur Herstellung
einer Kombination interessanter phänotypischer Merkmale
verwendet werden.
-
Nukleinsäuren,
die JMJC-Polypeptide kodieren, können auch als Sonden zur
genetischen und physikalischen Kartierung der Gene, von denen sie
ein Teil sind, und als Marker für Merkmale, die mit diesen
Genen verknüpft sind, verwendet werden. Diese Information
kann für die Pflanzenzüchtung nützlich
sein, um Linien mit gewünschten Phänotypen zu
entwickeln. Eine solche Verwendung von Nukleinsäuren, die
JMJC-Polypeptide kodieren, erfordert nur eine Nukleinsäuresequenz
mit einer Länge von mindestens 15 Nukleotiden. Die Nukleinsäuren,
die ein JMJC-Polypeptid kodieren, können als Marker für
Restriktionsfragmentlängenpolymorphismus (RFLP) verwendet
werden. Southern-Blots (Sambrook J, Fritsch EF und Maniatis
T (1989) Molecular Cloning, A Laboratory Manual) von restriktionsgespaltener
genomischer Pflanzen-DNA können mit den JMJC-kodierenden
Nukleinsäuren sondiert werden. Die erhaltenen Bandenmuster
können dann genetischen Analysen unter Verwendung von Computerprogrammen,
wie MapMaker (Lander et al. (1987) Genomics 1: 174–181),
unterworfen werden, wodurch eine Genkarte erstellt wird. Außerdem
können die Nukleinsäuren zum Sondieren von Southern-Blots
verwendet werden, die Restriktionsendonuklease-behandelte genomische DNAs
von einem Satz von Individuen enthalten, die den Elter und die Nachkommen
einer bestimmten genetischen Kreuzung wiedergeben. Die Segregation
der DNA-Polymorphismen wird festgestellt und zur Berechnung der
Position der Nukleinsäure, die das JMJC-Polypeptid kodiert,
in der zuvor unter Verwendung dieser Population erhaltenen Genkarte
verwendet (Botstein et al. (1980) Am. J. Hum. Genet. 32:
314–331).
-
Die
Herstellung und Verwendung von Sonden, die von Pflanzengenen stammen,
für die Verwendung bei der genetischen Kartierung ist in Bernatzky
und Tanksley (1986) Plant Mol. Biol. Reporter 4: 37–41 beschrieben.
Zahlreiche Veröffentlichungen beschreiben die genetische
Kartierung spezifischer cDNA-Klone, wobei die vorstehend erläuterte
Methodik oder Varianten davon verwendet werden. Für die
Kartierung können zum Beispiel F2-Inzuchtpopulationen,
Rückkreuzungspopulationen, zufallsgemäß gepaarte
Populationen, nahezu isogene Linien und andere Sätze von
Individuen verwendet werden. Diese Methodik ist dem Fachmann bekannt.
-
Die
Nukleinsäuresonden können auch zur physikalischen
Kartierung (d. h. zum Anordnen von Sequenzen in physikalischen Karten;
siehe Hoheisel et al. In: Non-mammalian Genomic Analysis:
A Practical Guide, Academic Press 1996, S. 319–346 und
darin genannte Bezugsstellen) verwendet werden.
-
In
einer anderen Ausführungsform können die Nukleinsäuresonden
zur Kartierung mittels direkter Fluoreszenz-in-situ-Hybridisierung
(FISH) (Trask (1991) Trends Genet. 7: 149–154)
verwendet werden. Zwar bevorzugen derzeitige FISH-Kartierungsverfahren
die Verwendung großer Klone (mehrere kb bis mehrere Hundert
kb; siehe Laan et al. (1995) Genome Res. 5: 13–20),
aber durch Verbesserungen in der Empfindlichkeit kann die FISH-Kartierung
auch unter Verwendung kürzerer Sonden durchgeführt
werden.
-
Eine
Reihe von Verfahren auf Basis von Nukleinsäureamplifikation
für die genetische und physikalische Kartierung kann unter
Verwendung der Nukleinsäuren durchgeführt werden.
Beispiele sind u. a. die allelspezifische Amplifikation (Kazazian
(1989) J. Lab. Clin. Med 11: 95–96), Polymorphismus
PCR-amplifizierter Fragmente (CAPS; Sheffield et al. (1993)
Genomics 16: 325–332), allelspezifische Ligation
(Landegren et al. (1988) Science 241: 1077–1080),
Nukleotidverlängerungsreaktionen (Sokolov (1990)
Nucleic Acid Res. 18: 3671), Radiation-Hybrid-Kartierung
(Walter et al. (1997) Nat. Genet. 7: 22–28)
und Happy-Kartierung (Dear und Cook (1989) Nucleic Acid
Res. 17: 6795–6807). Bei diesen Verfahren wird
die Sequenz einer Nukleinsäure dazu verwendet, Primer-Paare
zu gestalten und herzustellen, die in der Amplifikationsreaktion
oder in Primerverlängerungsreaktionen verwendet werden.
Die Gestaltung dieser Primer ist dem Fachmann bekannt. Bei Verfahren,
die eine genetische Kartierung auf PCR-Basis einsetzen, kann es
notwendig sein, dass man DNA-Sequenz- Unterschiede zwischen den Eltern
der Kartierungskreuzung in dem Bereich identifiziert, welcher der
vorliegenden Nukleinsäuresequenz entspricht. Dies ist jedoch
für Kartierungsverfahren nicht allgemein erforderlich.
-
Die
erfindungsgemäßen Verfahren führen zu
Pflanzen mit verbesserten Ertragsmerkmalen, wie hier vorstehend
beschrieben. Diese Merkmale können auch mit anderen ökonomisch
vorteilhaften Merkmalen kombiniert werden, wie weiteren Ertragsmerkmalen,
Toleranz gegenüber anderen abiotischen und biotischen Stressbedingungen,
Merkmalen, die verschiedene architektonische Merkmale und/oder biochemische und/oder
physiologische Merkmale modifizieren.
-
III. CKI(Casein-Kinase 1)-Polypeptid
-
Es
wurde überraschend entdeckt, dass die Modulation der Expression
einer Nukleinsäure, die ein CKI-Polypeptid kodiert, in
einer Pflanze, Pflanzen mit verbesserten Ertragsmerkmalen im Vergleich
zu Kontrollpflanzen ergibt. Gemäß einer ersten
Ausführungsform stellt die vorliegende Erfindung ein Verfahren
zur Verbesserung von Ertragsmerkmalen bei Pflanzen im Vergleich
zu Kontrollpflanzen bereit, bei dem die Expression einer Nukleinsäure,
die ein CKI-Polypeptid kodiert, in einer Pflanze moduliert wird.
-
Ein
bevorzugtes Verfahren zur Modulation (vorzugsweise Steigerung) der
Expression einer Nukleinsäure, die ein CKI-Polypeptid kodiert,
erfolgt durch Einführen und Expression einer Nukleinsäure,
die ein CKI-Polypeptid kodiert, in eine(r) Pflanze.
-
Jede
Bezugnahme auf ein ”Protein, das sich in den erfindungsgemäßen
Verfahren eignet” soll ein CKI-Polypeptid bedeuten, wie
es hier definiert ist. Jede im erfindungsgemäßen
Zusammenhang erfolgende Bezugnahme auf eine ”Nukleinsäure,
die sich in den erfindungsgemäßen Verfahren eignet” soll
eine Nukleinsäure bedeuten, die ein solches CKI-Polypeptid
kodieren kann. Die in eine Pflanze einzubringende (und daher bei
der Durchführung der erfindungsgemäßen
Verfahren geeignete) Nukleinsäure ist eine beliebige Nukleinsäure,
die den nachstehend beschriebenen Proteintyp kodiert, der auch als ”CKI-Nukleinsäure” oder ”CKI-Gen” bezeichnet
wird.
-
Ein ”CKI-Polypeptid” wie
es hier definiert ist, bezeichnet die in der SEQ ID NO: 174 dargestellten
Proteine und Homologa (Orthologa und Paraloga) davon. Die Homologa
der SEQ ID NO: 174 haben vorzugsweise eine Caseinkinasedomäne.
-
Die
Begriffe ”Domäne” und ”Motiv” sind
im vorliegenden Text in dem Abschnitt ”Definitionen” definiert. Für
die Identifikation von Domänen gibt es Spezialdatenbanken,
zum Beispiel SMART (Schultz et al. (1998), Proc. Natl. Acad.
Sci. USA 95, 5857–5864; Letunic et al.,
(2002), Nucleic Acids Res. 30, 242–244, InterPro (Mulder
et al., (2003), Nucl. Acids Res. 31, 315–318,
Prosite (Sucher und Bairoch (1994), A generalized Profile syntax
for biomolecular sequences Motivs and its function in automatic
sequence interpretation. (In) ISMB-94; Proceedings 2nd International
Conference an Intelligent Systems for Molecular Biology. Altman
R., Brutlag D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61,
AAAI Press, Menlo Park; Hulo et al., Nucl. Acids
Res. 32: D134–D137, (2004), oder Pfam (Bateman
et al., Nucleic Acids Research 30 (1): 276–280 (2002).
Ein Satz Werkzeuge für die In-silico-Analyse von Proteinsequenzen
findet sich auf dem ExPASY-Proteomics-Server (Swiss Institute of
Bioinformatics (Gasteiger et al., ExPASy: the Proteomics
server for in-depth Protein knowledge and analysis, Nucleic Acids
Res. 31: 3784–3788 (2003)). Domänen oder
Motive können auch unter Verwendung von Routinetechniken,
wie Sequenz-Alignment, identifiziert werden.
-
Verfahren
für das Alignment von Sequenzen für Vergleichszwecke
sind in der Fachwelt gut bekannt, dazu zählen GAP, BESTFIT,
BLAST, FASIA und TFASTA. Bei GAP wird der Algorithmus von Needleman
und Wunsch ((1970), J. Mol. Biol. 48: 443–453) verwendet,
um das globale Alignment (d. h. das Alignment, das sich über
die vollständigen Sequenzen erstreckt) von zwei Sequenzen
zu finden, das die Anzahl der ”matches” maximiert
und die Anzahl der ”gaps” minimiert. Beim BLAST-Algorithmus
(Altschul et al., (1990), J. Mol. Biol. 215: 403–10) wird
die Sequenzidentität in Prozent berechnet, und es wird
eine statistische Analyse der Ähnlichkeit zwischen den
beiden Sequenzen durchgeführt. Die Software für
die Durchführung einer BLAST-Analyse ist der Öffentlichkeit über
das National Centre for Biotechnology Information (NCBI) zugänglich.
Homologa können leicht unter Verwendung von zum Beispiel
dem multiplen Sequenz-Alignment-Algorithmus ClustalW (Version 1.83)
identifiziert werden, und zwar mit den Default-Parametern für
paarweises Alignment und einer Scoring-Methode in Prozent. Die Gesamtprozentsätze
der Ähnlichkeit und der Identität können
auch unter Verwendung von einer der in dem MatGAT-Software-Paket
verfügbaren Methoden bestimmt werden (Campanella et
al., BMC Bioinformatics. 2003, Juli, 10; 4: 29. MatGAT: an application
that generstes similarity/identity matrices using Protein or DNA
sequences.). Für eine Optimierung des Alignments
zwischen konservierten Motiven können, wie es dem Fachmann
ersichtlich wird, kleine manuelle Veränderungen vorgenommen
werden. Darüber hinaus können statt Volllängensequenzen
für die Identifikation von Homologa auch spezifische Domänen verwendet
werden. Die Sequenzidentitätswerte können mit
den oben erwähnten Programmen unter Einstellung der Default-Parameter über
die gesamte Nukleinsäure- oder Aminosäuresequenz
oder über ausgewählte Domänen oder konservierte
Motiv(e) bestimmt werden.
-
Darüber
hinaus haben CKI-Polypeptide, und soweit SEQ ID NO: 174 und seine
Homologa betroffen sind, die in den erfindungsgemäßen
Verfahren geeigneten CKI-Proteine, gewöhnlich Kinase-Aktivität.
Verfahren zum Messen der Kinase-Aktivität sind im Stand
der Technik bekannt.
-
Die
vorliegende Erfindung wird dadurch, dass man Pflanzen mit der Nukleinsäuresequenz
gemäß SEQ ID NO: 173, die die Polypeptidsequenz
gemäß SEQ ID NO: 174 kodiert, transformiert, dargestellt.
Die Durchführung der Erfindung ist jedoch nicht auf diese
Sequenzen beschränkt; die erfindungsgemäßen
Verfahren können vorteilhaft mit jeder CKI-kodierenden
Nukleinsäure oder mit jedem CKI-Polypeptid, wie hier definiert,
durchgeführt werden.
-
Beispiele
für Nukleinsäuren, die CKI-Polypeptide kodieren,
finden sich in den Datenbanken, die im Stand der Technik bekannt
sind. Solche Nukleinsäuren sind zur Durchführung
der erfindungsgemäßen Verfahren geeignet. Orthologa
und Paraloga, wobei die Begriffe ”Orthologa” und ”Paraloga” die
hier definierte Bedeutung haben, können leicht durch Durchführen
einer so genannten reziproken Blast-Suche identifiziert werden. Dies
beinhaltet typischerweise einen ersten BLAST, bei der mit einer
Abfragesequenz (zum Beispiel SEQ ID NO: 174) ein ”BLASTing” gegen
eine beliebige Sequenzdatenbank, wie die öffentlich zugängliche
NCBI-Datenbank, durchgeführt wird. BLASTN oder TBLASTX
(unter Verwendung von Standard-Default-Werten) werden im Allgemeinen
dann verwendet, wenn man von einer Nukleotidsequenz ausgeht, und
BLASTP oder TBLASTN (unter Verwendung von Standard-Default-Werten),
wenn man von einer Proteinsequenz ausgeht. Die BLAST-Ergebnisse
können gegebenenfalls gefiltert werden. Mit den Volllängen-Sequenzen
der gefilterten oder ungefilterten Ergebnisse wird anschließend
ein Rück-BLASTing (zweiter BLAST) gegen Sequenzen des Organismus,
von dem die Abfragesequenz stammt, durchgeführt (ist die
Abfragesequenz SEQ ID NO: 173 oder SEQ ID NO: 174, erfolgt das zweite
BLASTing daher gegen Sequenzen aus Nicotiana tabacum). Die Ergebnisse
des ersten und des zweiten BLASTing werden dann verglichen. Ein
Paralogon wird dann identifiziert, wenn ein hochrangiger Hit von
dem ersten BLASTing von derselben Art ist, von der die Abfragesequenz stammt,
in diesem Fall führt ein Rück-BLASTing idealerweise
zu der Abfragesequenz unter den höchstrangigen Hits; ein
Orthologon wird dann identifiziert, wenn ein hochrangiger Hit in
dem ersten BLASTing nicht von derselben Art wie derjenigen, von
der die Abfragesequenz stammt, ist, und führt vorzugsweise
beim Rück-BLASTing dazu, dass die Abfragesequenz unter
den höchstrangigen Hits ist.
-
Hochrangige
Hits sind solche mit niedrigem E-Wert. Je niedriger der E-Wert,
desto signifikanter der ”Score” (anders ausgedrückt,
desto niedriger die Wahrscheinlichkeit, dass der Hit durch Zufall
gefunden wurde). Die Berechnung des E-Werts ist Stand der Technik.
Das ”Scoring” der Vergleiche erfolgt nicht nur
mittels E-Werten, sondern auch mit dem Prozentsatz der Identität.
Der Prozentsatz der Identität bezieht sich auf die Anzahl
der identischen Nukleotide (oder Aminosäuren) zwischen
den beiden verglichenen Nukleinsäure-(oder Polypeptid-)Sequenzen über
eine bestimmte Länge. Bei großen Familien kann
man ClustalW und anschließend einen Neighbour-Joining-Tree
verwenden, um die Cluster der verwandten Gene leichter sichtbar
zu machen und um Orthologa und Paraloga zu identifizieren.
-
Zur
Durchführung der erfindungsgemäßen Verfahren
können sich auch Nukleinsäurevarianten eignen, die
Homologa und Derivate von SEQ ID NO: 174 kodieren, wobei die Begriffe ”Homologon” und ”Derivat” die hier
definierte Bedeutung haben. Für die erfindungsgemäßen
Verfahren eignen sich auch Nukleinsäuren, die Homologa
und Derivate von Orthologa oder Paraloga von SEQ ID NO: 174 kodieren.
Homologa und Derivate, die sich bei den erfindungsgemäßen
Verfahren eignen, weisen im Wesentlichen die gleiche biologische
und funktionelle Aktivität wie das unmodifizierte Protein,
von dem sie abstammen, auf.
-
Zu
weiteren Nukleinsäurevarianten, die sich zur Durchführung
der erfindungsgemäßen Verfahren eignen, gehören
Abschnitte von Nukleinsäuren, die CKI-Polypeptide kodieren,
Nukleinsäuren, die mit Nukleinsäuren, die CKI-Polypeptide
kodieren, hybridisieren, Spleißvarianten von Nukleinsäuren,
die CKI-Polypeptide kodieren, allelische Varianten von Nukleinsäuren,
die CKI-Polypeptide kodieren und Varianten von Nukleinsäuren,
die CKI-Polypeptide kodieren, die durch ”Gen-Shuffling” erhalten
wurden. Die Begriffe hybridisierende Sequenz, Spleißvariante,
allelische Variante und ”Gen-Shuffling” sind wie
im vorliegenden Text definiert.
-
Nukleinsäuren,
die CKI-Polypeptide kodieren, müssen nicht unbedingt Volllängennukleinsäuren
sein, da die Durchführung der erfindungsgemäßen
Verfahren nicht auf die Verwendung von Volllängen-Nukleinsäuresequenzen
angewiesen ist. Erfindungsgemäß wird daher ein
Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen bereitgestellt,
bei dem man in eine(r) Pflanze einen Abschnitt von SEQ ID NO: 173
oder einen Abschnitt von einer Nukleinsäure, die ein Orthologon,
Paralogon oder Homologon von SEQ ID NO: 174 kodiert, einführt
und exprimiert.
-
Ein
Abschnitt einer Nukleinsäure kann zum Beispiel dadurch
hergestellt werden, dass man eine oder mehrere Deletionen bei der
Nukleinsäure durchführt. Die Abschnitte können
in isolierter Form verwendet werden oder können mit anderen
kodierenden Sequenzen (oder nichtkodierenden Sequenzen) fusioniert
werden, um zum Beispiel ein Protein herzustellen, bei dem mehrere
Aktivitäten kombiniert sind. Bei der Fusion mit anderen
kodierenden Sequenzen kann das bei der Translation hergestellte
Polypeptid größer sein, als für den Proteinabschnitt
vorhergesagt wurde.
-
Abschnitte,
die sich bei den erfindungsgemäßen Verfahren eignen,
kodieren ein CKI-Polypeptid wie hier definiert und weisen im Wesentlichen
die gleiche biologische Aktivität wie die in SEQ ID NO:
174 angegebene Aminosäuresequenzen auf. Vorzugsweise handelt
es sich bei dem Abschnitt um einen Abschnitt von einer der in SEQ
ID NO: 173 angegebenen Nukleinsäuren oder um einen Abschnitt
einer Nukleinsäure, die ein Orthologon oder Paralogon von
einer der in SEQ ID NO: 173 angegebenen Aminosäuresequenzen
kodiert. Vorzugsweise ist der Abschnitt mindestens 500, 550, 600,
650, 700, 750, 800, 850, 900, 950, 1000, 1200, 1300, 1400 aufeinanderfolgende
Nukleotide lang, wobei es sich bei den aufeinander folgenden Nukleotiden um
SEQ ID NO: 173 handelt oder um eine Nukleinsäure, die ein
Orthologon oder Paralogon von SEQ ID NO: 174 kodiert, handelt. Am
stärksten bevorzugt ist der Abschnitt ein Abschnitt der
Nukleinsäure gemäß SEQ ID NO: 173.
-
Eine
weitere Nukleinsäurevariante, die sich bei den erfindungsgemäßen
Verfahren eignet, ist eine Nukleinsäure, die unter reduzierten
Stringenzbedingungen, vorzugsweise unter stringenten Bedingungen,
mit einer Nukleinsäure, die ein CKI-Polypeptid wie hier
definiert kodiert, oder mit einem Abschnitt wie im vorliegenden
Text hybridisieren kann.
-
Gemäß der
vorliegenden Erfindung wird ein Verfahren zur Verbesserung von Ertragsmerkmalen
bei Pflanzen bereitgestellt, bei dem man in eine(r) Pflanze eine
Nukleinsäure, die mit SEQ ID NO: 173 hybridisieren kann,
einführt und exprimiert, oder bei dem man in eine(r) Pflanze
eine Nukleinsäure, die mit einer Nukleinsäure,
die ein Orthologon, Paralogon oder Homologon von SEQ ID NO: 173
kodiert, hybridisieren kann, einführt und exprimiert.
-
Hybridisierende
Sequenzen, die sich bei den erfindungsgemäßen
Verfahren eignen, kodieren ein CKI-Polypeptid wie hier definiert,
und weisen im Wesentlichen die gleiche biologische Aktivität
wie die in SEQ ID NO: 174 angegebenen Aminosäuresequenzen
auf. Die hybridisierende Sequenz kann vorzugsweise mit SEQ ID NO:
173 oder einem Abschnitt von einer dieser Sequenzen hybridisieren,
wobei ein Abschnitt wie oben definiert ist, oder die hybridisierende
Sequenz kann mit einer Nukleinsäure, die ein Orthologon
oder Paralogon von SEQ ID NO: 174 kodiert, hybridisieren.
-
Eine
weitere Nukleinsäurevariante, die sich in den erfindungsgemäßen
Verfahren eignet, ist eine Spleißvariante, die ein CKI-Polypeptid
wie vorstehend definiert kodiert, wobei eine Spleißvariante
die hier definierte Bedeutung hat.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung der Ertragsmerkmale bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Spleißvariante
von SEQ ID NO: 173 einführt und exprimiert, oder bei dem
man in eine(r) Pflanze eine Spleißvariante einer Nukleinsäure,
die ein Orthologon, Paralogon oder Homologon von SEQ ID NO: 174
kodiert, einführt und exprimiert.
-
Eine
weitere Nukleinsäurevariante, die sich bei den erfindungsgemäßen
Verfahren eignet ist eine allelische Variante einer Nukleinsäure,
die ein CKI-Polypeptid wie vorstehend definiert kodiert, wobei eine
allelische Variante die hier definierte Bedeutung hat.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung der Ertragsmerkmale bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine allelische Variante
von SEQ ID NO: 173 einführt und exprimiert, oder bei dem
man in eine(r) Pflanze eine allelische Variante einer Nukleinsäure,
die ein Orthologon, Paralogon oder Homologon von SEQ ID NO: 174
kodiert, einführt und exprimiert.
-
Die
in den erfindungsgemäßen Verfahren geeigneten
allelischen Varianten haben im Wesentlichen die gleiche biologische
Aktivität, wie das CKI-Polypeptid von SEQ ID NO: 174. Allelische
Varianten kommen in der Natur vor, und in den erfindungsgemäßen
Verfahren ist die Verwendung dieser natürlichen Allele
mit einbezogen. Gen-Shuffling oder gerichtete Evolution können
ebenfalls zur Erzeugung von Varianten von Nukleinsäuren
verwendet werden, die CKI-Polypeptide wie vorstehend definiert kodieren,
wobei der Begriff ”Gen-Shuffling” die hier definierte
Bedeutung hat.
-
Gemäß der
vorliegenden Erfindung wird ein Verfahren zur Verbesserung von Ertragsmerkmalen
bei Pflanzen bereitgestellt, bei dem man in eine(r) Pflanze eine
Variante von SEQ ID NO: 173 einführt und exprimiert, oder
bei dem man in eine(r) Pflanze eine Variante einer Nukleinsäure,
die ein Orthologon, Paralogon oder Homologon von SEQ ID NO: 174
kodiert, einführt und exprimiert, wobei die Nukleinsäurevariante
durch Gen-Shuffling erhalten wird.
-
Nukleinsäurevarianten
können weiterhin auch durch ortsgerichtete Mutagenese erhalten
werden. Zur Erzielung einer ortsgerichteten Mutagenese sind verschiedene
Verfahren verfügbar, von denen die häufigsten Verfahren
auf PCR-Basis sind (Current Protocols in Molecular Biology.
Wiley Hrsg.).
-
Nukleinsäuren,
die CKI-Polypeptide kodieren, können von einer beliebigen
natürlichen oder künstlichen Quelle abstammen.
Die Nukleinsäure kann von ihrer nativen Form in ihrer Zusammensetzung
und/oder genomischen Umgebung durch überlegtes menschliches
Eingreifen modifiziert werden. Vorzugsweise stammt die Nukleinsäure,
die das CKI-Polypeptid kodiert, aus einer Pflanze. Bei SEQ ID NO:
173, ist die CKI-Polypeptid-kodierende Nukleinsäure vorzugsweise
aus einer dikotylen Pflanze, stärker bevorzugt aus der
Familie der Solanaceae, am stärksten bevorzugt stammt die
Nukleinsäure aus Nicotiana tabacum.
-
Die
Durchführung der erfindungsgemäßen Verfahren
ergibt Pflanzen mit verstärkten Ertragsmerkmalen. Insbesondere
ergibt die Durchführung der erfindungsgemäßen
Verfahren Pflanzen mit erhöhter Jungpflanzenvitalität
und erhöhtem Ertrag, insbesondere erhöhtem Biomasse-
und/oder erhöhtem Samenertrag, im Vergleich zu Kontrollpflanzen.
Die Begriffe ”Ertrag” und ”Samenertrag” sind
im vorliegenden Text im Abschnitt ”Definitionen” genauer
beschrieben.
-
Werden
hier verbesserte Ertragsmerkmale erwähnt, soll dies eine
Erhöhung der Jungpflanzenvitalität und/oder der
Biomasse (Gewicht) von einem oder mehreren Teilen einer Pflanze
bedeuten, was oberirdische (erntefähige) Teile und/oder
unterirdische (erntefähige) Teile beinhalten kann. Insbesondere
sind solche erntefähige Teile Biomasse und/oder Samen,
und die Durchführung der erfindungsgemäßen
Verfahren führt zu Pflanzen mit erhöhter Jungpflanzenvitalität,
gesteigertem Biomasse- und/oder Samenertrag in Bezug auf die Jungpflanzenvitalität,
Biomasse und/oder Samenausbeute von Kontrollpflanzen.
-
Wählt
man Mais als Beispiel, kann sich eine Ertragserhöhung unter
Anderem als eines oder mehrere der folgenden Merkmale äußern:
Erhöhung der Anzahl Pflanzen, die sich pro Hektar oder
Acre entwickeln, Erhöhung der Anzahl Ähren pro
Pflanze, Erhöhung der Anzahl Reihen, der Anzahl Körner
pro Reihe, des Korngewichts, Tausendkorngewichts, Ährenlänge/durchmessers,
Erhöhung der Samenfüllungsrate (Anzahl gefüllter
Samen dividiert durch die Gesamtzahl Samen und multipliziert mit
Hundert). Wählt man Reis als Beispiel, kann sich eine Ertragssteigerung
unter Anderem als Erhöhung von einem oder mehreren der
folgenden Merkmale ausdrücken: Anzahl Pflanzen pro Hektar
oder Acre, Anzahl Rispen pro Pflanze, Anzahl Ährchen pro
Rispe, Anzahl Blüten (Blütchen) pro Rispe (ausgedrückt
als Verhältnis der Anzahl der gefüllten Samen
zu der Anzahl der Primärrispen), erhöhte Samenfüllungsrate
(Anzahl gefüllter Samen dividiert durch die Gesamtzahl
Samen und multipliziert mit Hundert), erhöhtes Tausendkorngewicht.
-
Die
vorliegende Erfindung stellt ein Verfahren zur Erhöhung
des Ertrags, insbesondere Biomasse- und/oder Samenertrags von Pflanzen
im Vergleich zu Kontrollpflanzen bereit, wobei man bei dem Verfahren in
einer Pflanze die Expression einer Nukleinsäure, die ein
CKI-Polypeptid wie hier definiert kodiert, moduliert, vorzugsweise
erhöht.
-
Da
die erfindungsgemäßen transgenen Pflanzen einen
erhöhten Ertrag aufweisen zeigen diese Pflanzen (zumindest
während eines Teils ihres Lebenszyklus) wahrscheinlich
eine im Vergleich zu der Wachstumsrate von Kontrollpflanzen in einem
entsprechenden Stadium ihres Lebenszyklus erhöhte Wachstumsrate.
-
Die
erhöhte Wachstumsrate kann für einen oder mehrere
Teile einer Pflanze (einschließlich Samen) spezifisch sein
oder kann im Wesentlichen in der ganzen Pflanze stattfinden. Pflanzen
mit einer erhöhten Wachstumsrate können einen
kürzeren Lebenszyklus aufweisen. Unter dem Lebenszyklus
einer Pflanze kann man diejenige Zeit verstehen, die die Pflanze
braucht, um von einem reifen trockenen Samen bis zu dem Stadium
heranzuwachsen, in dem die Pflanze reife trockene Samen ähnlich
dem Ausgangsmaterial produziert hat. Dieser Lebenszyklus kann von
Faktoren wie Jungpflanzenvitalität, Wachstumsrate, ”Greenness
Index”, Blütezeit und Geschwindigkeit der Samenreifung
beeinflusst werden. Die erhöhte Wachstumsrate kann in einem
Stadium oder in mehreren Stadien im Lebenszyklus einer Pflanze oder
im Wesentlichen während des gesamten Lebenszyklus der Pflanze
stattfinden. Eine erhöhte Wachstumsrate während
der Frühstadien im Lebenszyklus einer Pflanze kann verbesserte
Vitalität widerspiegeln. Die erhöhte Wachstumsrate
kann den Erntezyklus einer Pflanze verändern, so dass die
Pflanzen später gesät werden können und/oder
früher geerntet werden können, als dies sonst
möglich wäre (ein ähnlicher Effekt lässt
sich mit einer früheren Blütezeit erzielen). Ist
die Wachstumsrate ausreichend erhöht, kann dies ein weiteres
Aussäen von Samen derselben Pflanzenart ermöglichen
(zum Beispiel Aussäen und Ernten von Reispflanzen und anschließendes
Aussäen und Ernten von weiteren Reispflanzen innerhalb einer
traditionellen Wacnstumsperiode). Entsprechend kann dies bei ausreichender
Steigerung der Wachstumsrate ein weiteres Aussäen von Samen
von unterschiedlichen Pflanzenarten ermöglichen (zum Beispiel
das Aussäen und Ernten von Maispflanzen und anschließend
z. B. Aussäen und gegebenenfalls Ernten von Sojabohnen,
Kartoffeln oder sonstigen geeigneten Pflanzen). Bei manchen Kulturpflanzen
kann man auch zusätzlich mehrmals von demselben Wurzelstock
ernten. Eine Veränderung des Erntezyklus einer Pflanze
kann zu einer Erhöhung der jährlichen Biomasseproduktion
pro Acre führen (und zwar aufgrund einer Erhöhung
der Häufigkeit (z. B. pro Jahr), mit der eine bestimmte
Pflanze herangezogen und geerntet werden kann). Eine erhöhte
Wachstumsrate kann auch den Anbau von transgenen Pflanzen in einem
weiteren geographischen Bereich als ihre Wildtyp-Gegenstücke
ermöglichen, da die räumlichen Begrenzungen für
den Anbau einer Kulturpflanze häufig von ungünstigen
Umweltbedingungen entweder während der Pflanzzeit (früh
in der Saison) oder während der Erntezeit (spät
in der Saison) bestimmt wird. Solche ungünstigen Bedingungen
können vermieden werden, wenn der Erntezyklus verkürzt
ist. Die Wachstumsrate kann durch Ableiten von verschiedenen Parametern
von Wachstumskurven bestimmt werden, wobei die Parameter unter Anderem
folgendes sein können: T-Mid (Zeitdauer, die die Pflanzen
benötigen, um 50% ihrer Maximalgröße
zu erreichen) und T-90 (Zeitdauer, die die Pflanzen benötigen,
um 90% ihrer Maximalgröße zu erreichen).
-
Gemäß einem
bevorzugten Merkmal der vorliegenden Erfindung ergibt die Durchführung
der erfindungsgemäßen Verfahren Pflanzen mit einer
im Vergleich zu Kontrollpflanzen erhöhten Wachstumsrate.
Die vorliegende Erfindung stellt daher ein Verfahren zur Erhöhung
der Wachstumsrate von Pflanzen bereit, wobei man bei dem Verfahren
in einer Pflanze die Expression einer Nukleinsäure, die
ein CKI-Polypeptid wie hier definiert kodiert, moduliert, vorzugsweise
erhöht. In einer bestimmten Ausführungsform ergibt
die Durchführung der erfindungsgemäßen
Verfahren insbesondere Pflanzen mit erhöhter Jungpflanzenvitalität.
-
Eine
Erhöhung des Ertrags und/oder der Wachstumsrate findet
unabhängig davon statt, ob die Pflanze unter Nichtstressbedingungen
steht oder ob die Pflanze im Vergleich zu Kontrollpflanzen verschiedenen Stressfaktoren
unterworfen wird. Typischerweise reagieren Pflanzen auf Stress durch
langsameres Wachstum. Unter starken Stressbedingungen kann die Pflanze
sogar ihr Wachstum einstellen. Leichter Stress wiederum wird im
vorliegenden Zusammenhang als jeglicher Stress definiert, dem eine
Pflanze ausgesetzt ist und der nicht dazu führt, dass die
Pflanze ihr Wachstum völlig einstellt, ohne ihr Wachstum
wieder aufnehmen zu können. Leichter Stress im Sinne der
Erfindung führt zu einer Verringerung des Wachstums der
gestressten Pflanzen von weniger als 40%, 35% oder 30%, vorzugsweise
weniger als 25%, 20% oder 15%, stärker bevorzugt weniger
als 14%, 13%, 12%, 11% oder 10% oder weniger im Vergleich zu der
Kontrollpflanze unter Nichtstressbedingungen. Aufgrund der Fortschritte
bei den landwirtschaftlichen Kulturmaßnahmen (Bewässerung, Düngung,
Pestizidbehandlungen) findet man bei angebauten Kulturpflanzen nicht
oft starken Stress. Daher ist das durch leichten Stress induzierte
geschwächte Wachstum häufig ein unerwünschtes
Merkmal in der Landwirtschaft. Leichter Stress ist der alltägliche
biotische und/oder abiotische Stress (Umweltstress), dem eine Pflanze
ausgesetzt ist. Abiotischer Stress kann durch Trockenheit oder Wasserüberschuss,
anaeroben Stress, Salzstress, chemische Toxizität, oxidativen
Stress und Hitze, Kälte oder Gefriertemperaturen verursacht
werden. Bei dem abiotischen Stress kann es sich um einen osmotischen
Stress handeln, der durch Wasserstress (insbesondere aufgrund von
Trockenheit), Salzstress, oxidativen Stress oder ionenbedingten
Stress verursacht wird. Biotischer Stress wird gewöhnlich
durch Pathogene, wie Bakterien, Viren, Pilze und Insekten verursacht.
-
Insbesondere
können die erfindungsgemäßen Verfahren
unter Nichtstressbedingungen oder unter leichten Trockenheitsbedingungen
durchgeführt werden, wodurch man Pflanzen mit einem im
Vergleich zu Kontrollpflanzen erhöhten Ertrag erhält.
Wie von Wang et al. (Planta (2003), 218: 1–14)
beschrieben, führt abiotischer Stress zu einer Reihe von
morphologischen, physiologischen, biochemischen und molekularen
Veränderungen, die das Pflanzenwachstum und die Produktivität
negativ beeinflussen. Trockenheit, Salinität, extreme Temperaturen
und oxidativer Stress stehen bekanntlich miteinander in Verbindung
und können über ähnliche Mechanismen
Wachstums- und Zellschäden induzieren. Rabbani
et al. (Plant Physiol. (2003), 133: 1755–1767)
beschreiben ein besonders hohes Ausmaß an gegenseitiger
Beeinflussung von Trockenheitsstress und durch hohe Salinität
verursachtem Stress. Trockenheit und/oder Versalzung äußern
sich beispielsweise in erster Linie als osmotischer Stress, was
zur Störung der Homöostase und der Ionenverteilung
in der Zelle führt. Oxidativer Stress, der häufig
Hoch- oder Niedertemperatur-, Salinitäts- oder Trockenheitsstress
begleitet, kann zur Denaturierung funktioneller und struktureller
Proteinen führen. Demzufolge aktivieren diese verschiedenen
Umweltstressfaktoren häufig ähnliche Signalleitungswege
der Zelle und Zellreaktionen, wie die Produktion von Stressproteinen,
die Aufwärtsregulation von Antioxidantien, die Anreicherung
kompatibler gelöster Stoffe und ein Einstellen des Wachstums.
Der Begriff ”Nichtstress”-Bedingungen bedeutet
im vorliegenden Zusammenhang diejenigen Umweltbedingungen, die ein
optimales Wachstum der Pflanzen ermöglichen. Der Fachmann
ist mit den normalen Bodenbedingungen und Klimabedingungen für
einen bestimmten Standort vertraut. Pflanzen mit optimalen Wachstumsbedingungen
(die unter Nicht-Stress-Bedingungen gezüchtet wurden) ergeben
gewöhnlich in steigender Reihenfolge der Präferenz
mindestens 90%, 87%, 85%, 83%, 80%, 77% oder 75% der durchschnittlichen
Produktion einer solchen Pflanze bei einer gegebenen Umgebung. Die durchschnittliche
Produktion kann auf der Basis der Ernte und/oder der Jahreszeit
berechnet werden. Dem Fachmann sind Produktionen einer Kulturpflanze
mit durchschnittlichem Ertrag geläufig.
-
Die
Durchführung der erfindungsgemäßen Verfahren
ergibt Pflanzen, die bei Wachstum unter Nichtstressbedingungen oder
unter leichten Trockenheitsbedingungen einen im Vergleich zu Kontrollpflanzen,
die unter vergleichbaren Bedingungen wachsen, einen erhöhten
Ertrag und/oder eine erhöhte Jungpflanzenvitalität
aufweisen. Gemäß der vorliegenden Erfindung wird
daher ein Verfahren zur Erhöhung des Ertrags und/oder der
Jungpflanzenvitalität von Pflanzen, die unter Nichtstressbedingungen
oder unter leichten Trockenheitsbedingungen heranwachsen, bereitgestellt,
wobei man bei dem Verfahren in einer Pflanze die Expression einer Nukleinsäure,
die ein CKI-Polypeptid kodiert, erhöht.
-
Die
Durchführung der erfindungsgemäßen Verfahren
ergibt Pflanzen, die unter Nährstoffmangelbedingungen gezüchtet
wurden, insbesondere unter Stickstoffmangelbedingungen, mit besserem
Ertrag im Vergleich zu unter vergleichbaren Bedingungen gezüchteten
Kontrollpflanzen. Daher wird erfindungsgemäß ein Verfahren
zur Steigerung des Ertrags bei Pflanzen bereitgestellt, die unter
Nährstoffmangelbedingungen gezüchtet wurden, bei
dem man die Expression einer Nukleinsäure, die ein CKI-Polypeptid
kodiert, in einer Pflanze steigert. Nährstoffmangel kann
sich unter Anderem aus einem Fehlen von Nährstoffen, wie
Stickstoff, Phosphaten und anderen phosphorhaltigen Verbindungen,
Kalium, Calcium, Cadmium, Magnesium, Mangan, Eisen und Bor, ergeben.
-
Die
vorliegende Erfindung umfasst Pflanzen oder Teile davon (einschließlich
Samen), die durch die erfindungsgemäßen Verfahren
erhältlich sind. Die Pflanzen oder Teile davon umfassen
ein Nukleinsäuretransgen, das ein CKI-Polypeptid wie oben
definiert, kodiert.
-
Die
Erfindung stellt auch Genkonstrukte und Vektoren bereit, um die
Einführung und/oder Expression von Nukleinsäuren,
die CKI-Polypeptide kodieren, in Pflanzen zu erleichtern. Die Genkonstrukte
können in handelsübliche Vektoren eingeführt
werden, die für die Transformation in Pflanzen und für
die Expression des interessierenden Gens in den transformierten
Zellen geeignet sind. Die Erfindung stellt auch die Verwendung eines
wie im vorliegenden Text definierten Genkonstrukts in den erfindungsgemäßen
Verfahren bereit.
-
Genauer
ausgedrückt stellt die vorliegende Erfindung ein Konstrukt
bereit, umfassend
- (a) eine Nukleinsäure,
die ein CKI-Polypeptid wie oben definiert kodiert;
- (b) eine oder mehrere Kontrollsequenzen, die die Expression
der Nukleinsäuresequenz gemäß (a) steuern kann
bzw. können; sowie gegebenenfalls
- (c) eine Transkriptionsterminationssequenz.
-
Vorzugsweise
ist die Nukleinsäure, die eine CKI-Polypeptid kodiert,
wie oben definiert. Der Begriff ”Kontrollsequenz” und ”Terminationssequenz” sind
wie hier definiert.
-
Die
Pflanzen werden mit einem Vektor transformiert, der eine beliebige
der oben beschriebenen Nukleinsäuren umfasst. Der Fachmann
ist mit den genetischen Elementen, die in einem Vektor vorhanden
sein müssen, um Wirtszellen, die die interessierende Sequenz
enthalten, erfolgreich zu transformieren, zu selektieren und zu
vermehren, bestens vertraut. Die interessierende Sequenz ist funktionsfähig
mit einer oder mehreren Kontrollsequenzen (mindestens mit einem
Promotor) verbunden.
-
Jede
Art von Promotor, sowohl natürlich als auch synthetisch,
kann vorteilhaft für das Steuern der Expression der Nukleinsäuresequenz
verwendet werden. Ein samenspezifischer Promotor ist besonders bei
den erfindungsgemäßen Verfahren geeignet, der
Promotor ist vorzugsweise ein embryospezifischer Promotor. Für die
Definitionen der verschiedenen Promotortypen siehe Abschnitt ”Definitionen” im
vorliegenden Text. Bei den erfindungsgemäßen Verfahren
eignet sich auch ein konstitutiver Promotor.
-
Man
muss sich darüber im Klaren sein, dass die Anwendbarkeit
der vorliegenden Erfindung weder auf die CKI-Polypeptid-kodierende
Nukleinsäure, die durch SEQ ID NO: 173 veranschaulicht
wird, noch auf die Expression einer CKI-Polypeptid-kodierenden Nukleinsäure,
die von einem samenspezifischen Promotor gesteuert wird, beschränkt
ist.
-
Der
samenspezifische Promotor ist vorzugsweise ein WSI18-Promotor, vorzugsweise
ein WSI18-Promotor aus Reis. Weiter bevorzugt ist der samenspezifische
Promotor durch eine Nukleinsäuresequenz veranschaulicht,
die SEQ ID NO: 175 ähnelt, am stärksten bevorzugt
ist der samenspezifische Promotor durch SEQ ID NO: 175 veranschaulicht.
Im Abschnitt ”Defintionen” sind weitere Beispiele
für samenspezifische Promotoren beschrieben.
-
Gegebenenfalls
können eine oder mehrere Terminatorsequenzen in dem in
eine Pflanze eingebrachten Konstrukt verwendet werden. Zusätzliche
regulatorische Elemente können Transkriptions- und Translations-Enhancer
umfassen. Der Fachmann kennt Terminator- und Enhancer-Sequenzen,
die sich zur Durchführung der Erfindung eignen. Eine Intronsequenz
kann ebenfalls zu der 5'-untranslatierten Region (UTR) oder in der
kodierenden Sequenz hinzugefügt werden, damit die Menge
der reifen Botschaft gesteigert wird, die sich im Cytosol anreichert,
wie es im Abschnitt Definitionen beschrieben ist. Andere Kontrollsequenzen
(neben Promotor, Enhancer, Silencer, Intronsequenzen, 3'UTR- und/oder
5'UTR-Regionen) können Protein- und/oder RNA-stabilisierende
Elemente sein. Solche Sequenzen sind dem Fachmann bekannt oder können
leicht von ihm erhalten werden.
-
Die
erfindungsgemäßen Genkonstrukte können
weiterhin eine Replikationsursprungssequenz beinhalten, die zur
Aufrechterhaltung und/oder Replikation in einem bestimmten Zelltyp
erforderlich ist. Bei einem Beispiel muss ein Genkonstrukt in einer
Bakterienzelle als episomales genetisches Element aufrechterhalten werden
(z. B. Plasmid- oder Cosmidmolekül). Bevorzugte Replikationsursprünge
umfassen f1-ori und colE1, sind jedoch nicht darauf beschränkt.
-
Für
den Nachweis des erfolgreichen Transfers der Nukleinsäuresequenzen,
wie sie bei den erfindungsgemäßen Verfahren verwendet
wurden, und/oder für die Selektion der transgenen Pflanzen,
die diese Nukleinsäuren umfassen, werden vorteilhafterweise
Markergene (oder Reportergene) verwendet. Das Genkonstrukt kann
daher gegebenenfalls ein Selektionsmarkergen umfassen. Selektionsmarker
sind hier eingehender im Abschnitt ”Definitionen” beschrieben.
Die Markergene können aus der transgenen Zelle entfernt oder
gespalten werden, sobald sie nicht mehr vonnöten sind.
Techniken zur Markerentfernung sind Stand der Technik, geeignete
Techniken sind oben im Abschnitt Definitionen beschrieben.
-
Die
Erfindung stellt auch ein Verfahren zur Produktion von transgenen
Pflanzen mit verbesserten Ertragsmerkmalen gegenüber Kontrollpflanzen
bereit, wobei eine beliebige Nukleinsäure, die ein CKI-Polypeptid wie
oben definiert, in eine(r) Pflanze eingeführt und exprimiert
wird.
-
Insbesondere
stellt die vorliegende Erfindung ein Verfahren zur Produktion von
transgenen Pflanzen mit verbesserten Ertragsmerkmalen, insbesondere
gesteigerter Jungpflanzenvitalität und/oder gesteigertem Ertrag
bereit, wobei das Verfahren umfasst:
- (i) Einbringen
und Exprimieren einer CKI Polypeptid-kodierenden Nukleinsäure
in eine(r) Pflanze oder Pflanzenzelle; und
- (ii) Züchten der Pflanzenzelle unter Bedingungen, die
das Pflanzenwachstum und die Entwicklung fördern.
-
Die
Nukleinsäure von (i) kann eine beliebige Nukleinsäure
sein, die ein CKI-Polypeptid wie hier definiert kodieren kann.
-
Die
Nukleinsäure kann in eine Pflanzenzelle oder in die Pflanze
selbst direkt eingeführt werden (was das Einführen
in ein Gewebe, Organ oder einen beliebigen anderen Teil einer Pflanze
beinhaltet). Gemäß einem bevorzugten Merkmal der
vorliegenden Erfindung wird die Nukleinsäure vorzugsweise
mittels Transformation in eine Pflanze eingeführt. Der
Begriff ”Transformation” wird im vorliegenden
Text im Abschnitt ”Definitionen” genauer beschrieben.
-
Die
genetisch modifizierten Pflanzenzellen können nach allen
Verfahren, mit denen der Fachmann vertraut ist, regeneriert werden.
Geeignete Verfahren finden sich in den oben genannten Veröffentlichungen von
S. D. Kung und R. Wu, Potrykus oder Höfgen und Willmitzer.
-
Im
Allgemeinen werden nach der Transformation die Pflanzenzellen oder
Zellgruppen auf das Vorhandensein von einem oder mehreren Markern
selektiert, die von den mit dem interessierenden Gen gemeinsam transferierten,
in Pflanzen exprimierbaren Genen kodiert werden, wonach das transformierte
Material zu einer ganzen Pflanze regeneriert wird. Für
die Selektion von transformierten Pflanzen wird das bei der Transformation
erhaltene Pflanzenmaterial im Allgemeinen Selektionsbedingungen
unterworfen, so dass man transformierte Pflanzen von untransformierten
Pflanzen unterscheiden kann. So können zum Beispiel Samen,
die auf die oben beschriebene Art und Weise erhalten wurden, ausgepflanzt
werden und nach einer anfänglichen Wachstumsphase durch
Spritzen einer geeigneten Selektion unterworfen werden. Eine weitere
Möglichkeit besteht darin, dass man die Samen, gegebenenfalls
nach Sterilisieren, auf Agarplatten heranzieht, wobei man ein geeignetes
Selektionsmittel verwendet, so dass nur die transformierten Samen
zu Pflanzen heranwachsen können. Alternativ werden die
transformierten Pflanzen auf das Vorhandensein eines Selektionsmarkers
wie die oben beschriebenen gescreent.
-
Nach
dem DNA-Transfer und der Regeneration können mutmaßlich
transformierte Pflanzen auch zum Beispiel unter Verwendung der Southern-Analyse
auf das Vorhandensein des interessierenden Gens, die Kopienzahl
und/oder die Genomorganisation ausgewertet werden. Alternativ dazu
oder zusätzlich können die Expressionsniveaus
der neu eingeführten DNA mittels Northern- und/oder Western-Analyse überwacht
werden; wobei beide Techniken dem Durchschnittsfachmann gut bekannt
sind.
-
Die
erzeugten transformierten Pflanzen können mit einer Reihe
von Mitteln vermehrt werden, wie durch klonale Vermehrung oder durch
klassische Züchtungstechniken. So kann zum Beispiel eine
transformierte Pflanze der ersten Generation (T1-Pflanze) geselbstet
werden, und homozygote Transformanten der zweiten Generation (T2-Pflanze)
können selektiert werden, und die T2-Pflanzen können
dann mit Hilfe von klassischen Züchtungstechniken weiter
vermehrt werden. Die erzeugten transformierten Organismen können in
verschiedener Form vorliegen. Sie können beispielsweise
Chimären von transformierten Zellen und untransformierten
Zellen sein; klonale Transformanten (z. B. alle Zellen wurden dahingehend
transformiert, dass sie die Expressionskassette enthalten); Pfropfmaterial
von transformiertem und untransformiertem Gewebe (z. B. bei Pflanzen
ein transformierter Wurzelstock, der auf ein untransformiertes Edelreis
gepfropft wurde).
-
Die
vorliegende Erfindung erstreckt sich eindeutig auf eine jede Pflanzenzelle
oder Pflanze, die nach einem der hier beschriebenen Verfahren erzeugt
wurde, und auf sämtliche Pflanzenteile und sämtliches
Vermehrungsmaterial davon. Die vorliegende Erfindung umfasst weiterhin
die Nachkommenschaft einer/eines primär transformierten
oder transfizierten Zelle, Gewebes, Organs oder ganzen Pflanze,
die/das nach einem der oben genannten Verfahren erzeugt wurde, wobei
die einzige Voraussetzung ist, dass die Nachkommenschaft dasselbe/dieselben
genotypische(n) und/oder phänotypische(n) Merkmal(e) aufweist,
wie es/sie von dem Elter bei den erfindungsgemäßen
Verfahren gezeigt wird.
-
Die
Erfindung beinhaltet auch Wirtszellen, die eine isolierte Nukleinsäure,
die ein wie oben definiertes CKI-Polypeptid kodiert, enthalten.
Bevorzugte erfindungsgemäße Wirtszellen sind Pflanzenzellen.
Wirtspflanzen für die bei dem erfindungsgemäßen
Verfahren verwendeten Nukleinsäuren oder den Vektor, für
die Expressionskassette oder das Konstrukt oder den Vektor, sind
im Prinzip vorteilhafterweise alle Pflanzen, die die bei dem erfindungsgemäßen
Verfahren verwendeten Polypeptide synthetisieren können.
-
Die
erfindungsgemäßen Verfahren lassen sich vorteilhaft
auf jede beliebige Pflanze anwenden. Zu den Pflanzen, die bei den
erfindungsgemäßen Verfahren besonders geeignet sind,
gehören alle Pflanzen, die zu der Superfamilie Viridiplantae
gehören, insbesondere monokotyle und dikotyle Pflanzen,
einschließlich Futter- oder Weidepflanzenleguminosen, Zierpflanzen,
Nahrungsmittelkulturpflanzen, Bäume oder Sträucher.
Gemäß einer bevorzugten Ausführungsform
der vorliegenden Erfindung handelt es sich bei der Pflanze um eine
Kulturpflanze. Zu den Kulturpflanzen gehören zum Beispiel
Sojabohne, Sonnenblume, Canola-Raps, Luzerne, Raps, Baumwolle, Tomate,
Kartoffel und Tabak. Weiter bevorzugt handelt es sich bei der Pflanze
um eine monokotyle Pflanze. Zu den monokotylen Pflanzen gehört
zum Beispiel Zuckerrohr. Stärker bevorzugt handelt es sich
bei der Pflanze um ein Getreide. Zu den Getreiden gehören
zum Beispiel Reis, Mais, Weizen, Gerste, Hirse, Roggen, Triticale,
Sorghum und Hafer.
-
Die
Erfindung betrifft auch erntefähige Teile einer Pflanze
wie zum Beispiel, jedoch nicht ausschließlich, Samen, Blätter,
Früchte, Blüten, Stängel, Wurzeln, Rhizome,
Knollen und Zwiebeln. Die Erfindung betrifft weiterhin Produkte,
die von einem erntefähigen Teil einer solchen Pflanze abstammen,
vorzugsweise direkt abstammen, wie trockene Pellets oder Pulver, Öl,
Fett und Fettsäuren, Stärke oder Proteine.
-
Gemäß einem
bevorzugten Merkmal der Erfindung handelt es sich bei der modulierten
Expression um erhöhte Expression. Verfahren zur Erhöhung
der Expression von Nukleinsäuren oder Genen oder Genprodukten,
sind im Fachgebiet gut beschrieben; und Beispiele sind im Abschnitt
Definitionen gegeben.
-
Wie
vorstehend erwähnt, ist ein bevorzugtes Verfahren zum Modulieren
(vorzugsweise Erhöhen) der Expression einer Nukleinsäure,
die ein CKI-Polypeptid kodiert, durch Einführen und Exprimieren
einer Nukleinsäure, die ein CKI-Polypeptid kodiert, in
eine(r) Pflanze; die Wirkungen der Durchführung des Verfahren,
d. h. das Erhöhen der Ertragsmerkmale, können
auch unter Verwendung anderer bekannter Techniken erzielt werden,
einschließlich, aber nicht beschränkt auf T-DNA-Activation-Tagging,
TILLING, homologe Rekombination. Eine Beschreibung dieser Techniken
findet sich im Abschnitt Definitionen.
-
Die
vorliegende Erfindung umfasst auch die Verwendung von Nukleinsäuren,
die CKI-Polypeptide, wie hier beschrieben, kodieren, und die Verwendung
dieser CKI-Polypeptide zur Verbesserung eines der oben genannten
Ertragsmerkmale in Pflanzen.
-
Hier
beschriebene Nukleinsäuren, die das CKI-Polypeptid kodieren,
oder die CKI-Polypeptide selbst können Verwendung in Züchtungsprogrammen
finden, in denen ein DNA-Marker identifiziert wird, der mit einem
für ein CKI-Polpeptid kodierenden Gen genetisch verknüpft
sein kann. Die Nukleinsäuren/Gene oder die CKI-Polypeptide
selbst können dazu verwendet werden, einen molekularen
Marker zu bestimmen. Dieser DNA- oder Protein-Marker kann dann in
Züchtungsprogrammen zur Selektion von Pflanzen mit verbesserten Ertragsmerkmalen
verwendet werden, wie hier unter den erfindungsgemäßen
Verfahren definiert.
-
Allelische
Varianten einer Nukleinsäure/eines Gens, die/das ein CKI-Polypeptid
kodiert, können auch Verwendung in Marker-unterstützten
Züchtungsprogrammen finden. Bei solchen Züchtungsprogrammen
ist manchmal das Einführen von allelischer Variation durch
Mutagenbehandlung der Pflanzen erforderlich, wobei zum Beispiel
EMS-Mutagenese verwendet wird; alternativ kann das Programm von
einer Sammlung von allelischen Varianten so genannten ”natürlichen” Ursprungs
ausgehen, die ohne Absicht hervorgerufen wurden. Dann erfolgt die
Identifikation allelischer Varianten, zum Beispiel mittels PCR.
Darauf folgt ein Schritt zur Selektion besserer allelischer Varianten
der in Frage kommenden Sequenz, die bessere Erträge liefern.
Die Selektion wird gewöhnlich durchgeführt, indem
man die Wachstumsleistung von Pflanzen verfolgt, die verschiedene
allelische Varianten der in Frage kommenden Sequenz enthalten. Die
Wachstumsleistung kann im Gewächshaus oder auf dem Feld
verfolgt werden. Weitere Schritte sind gegebenenfalls u. a. das
Kreuzen der Pflanzen, in denen die bessere allelische Variante identifiziert
wurde, mit einer anderen Pflanze. Dies kann zum Beispiel zur Herstellung
einer Kombination interessanter phänotypischer Merkmale
verwendet werden.
-
CKI-Polypeptide
kodierende Nukleinsäuren können auch als Sonden
zur genetischen und physikalischen Kartierung der Gene, von denen
sie ein Teil sind, und als Marker für Merkmale, die mit
diesen Genen verknüpft sind, verwendet werden. Diese Information
kann für die Pflanzenzüchtung nützlich
sein, um Linien mit gewünschten Phänotypen zu
entwickeln. Eine solche Verwendung der CKI-Polypeptid-kodierenden
Nukleinsäuren benötigt nur eine Nukleinsäuresequenz
von mindestens 15 Nukleotiden Länge. Die CKI-Polypeptid-kodierenden
Nukleinsäuren können als Restriktionsfragmentlängenpolymorphismus
(RFLP)-Marker verwendet werden. Southern-Blots (Sambrook
J, Fritsch EF and Maniatis T (1989) Molecular Cloning, A Laboratory
Manual) von restriktions-gespaltener genomischer Pflanzen-DNA
kann mit den CKI-kodierenden Nukleinsäuren sondiert werden.
Die erhaltenen Bandenmuster können dann genetischen Analysen
mit Computerprogrammen unterworfen werden, wie MapMaker (Lander
et al. (1987) Genomics 1: 174–181), so dass man
eine genetische Karte erhält. Die Nukleinsäuren
können zudem zum Sondieren von Southern-Blots verwendet
werden, die Restriktionsendonuklease behandelte genomische DNAs
von einem Satz von Individuen enthalten, die die Eltern und die
Nachfahren einer definierten genetischen Kreuzung repräsentieren.
Die Segregation der DNA-Polymorphismen wird festgestellt und zur
Berechnung der Position der CKI-Polypeptid-kodierenden Nukleinsäure
in der genetischen Karte verwendet, die mit dieser Population erhalten
wurde (Botstein et al. (1980) Am. J. Hum. Genet. 32: 314–331).
-
Die
Herstellung und Verwendung von Sonden, die von Pflanzengenen stammen,
für die Verwendung bei der genetischen Kartierung ist in Bernatzky
und Tanksley (1986) Plant Mol. Biol. Reporter 4: 37–41 beschrieben.
Zahlreiche Veröffentlichungen beschreiben die genetische
Kartierung spezifischer cDNA-Klone, wobei die vorstehend erläuterte
Methodik oder Varianten davon verwendet werden. Für die
Kartierung können zum Beispiel F2-Inzuchtpopulationen,
Rückkreuzungspopulationen, zufallsgemäß gepaarte
Populationen, nahezu isogene Linien und andere Sätze von
Individuen verwendet werden. Diese Methoden sind dem Fachmann bestens
bekannt.
-
Die
Nukleinsäuresonden können auch zur physikalischen
Kartierung (d. h. zum Anordnen von Sequenzen in physikalischen Karten;
siehe Hoheisel et al. In: Non-mammalian Genomic Analysis:
A Practical Guide, Academic Press 1996, S. 319–346 und
darin genannte Literaturstellen) verwendet werden.
-
In
einer anderen Ausführungsform können die Nukleinsäuresonden
zur Kartierung durch direkte Fluoreszenz-in-situ-Hybridisierung
(FISH) (Trask (1991) Trends Genet. 7: 149–154)
verwendet werden. Zwar bevorzugen derzeitige FISH-Kartierungsverfahren
die Verwendung großer Klone (mehrere kb bis mehrere Hundert
kb; siehe Laan et al. (1995) Genome Res. 5: 13–20),
aber durch Verbesserungen in der Empfindlichkeit kann die FISH-Kartierung
auch unter Verwendung kürzerer Sonden durchgeführt
werden.
-
Eine
Reihe von Verfahren auf der Basis von Nukleinsäureamplifikation
für die genetische und physikalische Kartierung kann mit
Hilfe der Nukleinsäuren durchgeführt werden. Beispiele
sind u. a. die allelspezifische Amplifikation (Kazazian
(1989) J. Lab. Clin. Med 11: 95–96), Polymorphismus
PCR-amplifizierter Fragmente (CAPS; Sheffield et al. (1993)
Genomics 16: 325–332), allelspezifische Ligation
(Landegren et al. (1988) Science 241: 1077–1080),
Nukleotidverlängerungsreaktionen (Sokolov (1990)
Nucleic Acid Res. 18: 3671), Radiation-Hybrid-Kartierung
(Walter et al. (1997) Nat. Genet. 7: 22–28)
und Happy-Kartierung (Dear und Cook (1989) Nucleic Acid
Res. 17: 6795–6807). Bei diesen Verfahren wird
die Sequenz einer Nukleinsäure dazu verwendet, Primer-Paare
zu gestalten und zu erzeugen, die bei der Amplifikationsreaktion
oder Primerverlängerungsreaktionen verwendet werden. Die
Gestaltung dieser Primer ist dem Fachmann bekannt. Bei Verfahren, die
eine genetische Kartierung auf PCR-Basis einsetzen, kann es notwendig
sein, dass man DNA-Sequenz-Unterschiede zwischen den Eltern der
Kartierungskreuzung in dem Bereich identifiziert, welcher der vorliegenden
Nukleinsäuresequenz entspricht. Dies ist jedoch für
Kartierungsverfahren nicht allgemein erforderlich.
-
Die
erfindungsgemäßen Verfahren ergeben Pflanzen mit
verbesserten Ertragsmerkmalen, wie hier vorstehend beschrieben.
Diese Merkmale können auch mit anderen ökonomisch
vorteilhaften Merkmalen kombiniert werden, wie weiteren Ertragsmerkmalen,
Toleranz gegenüber anderen abiotischen und biotischen Stressbedingungen,
Merkmalen, die verschiedene architektonische Merkmale und/oder biochemisch
und/oder physiologische Merkmale modifizieren.
-
IV. Pflanzen-Homöodomänen-Finger-Homöodomänen-(Plant
homeodomain finger-homeodomain (PHDf-HD))-Polypeptid
-
Es
wurde überraschend entdeckt, dass die Modulation, vorzugsweise
Steigerung, der Expression einer Nukleinsäuresequenz, die
ein PHDf-HD-Polypeptid kodiert, in einer Pflanze Pflanzen mit verbesserten
Ertragsmerkmalen, vorzugsweise verbesserten Samenertragsmerkmalen,
im Vergleich zu Kontrollpflanzen ergibt. Gemäß einer
ersten Ausführungsform stellt die vorliegende Erfindung
ein Verfahren zur Verbesserung von Ertragsmerkmalen, vorzugsweise
Verbesserung der Samenertragsmerkmale, bei Pflanzen, im Vergleich zu
Kontrollpflanzen bereit, bei dem die Expression einer Nukleinsäure,
die ein PHDf-HD-Polypeptid kodiert, in einer Pflanze moduliert,
vorzugsweise erhöht wird.
-
Ein
bevorzugtes Verfahren zur Modulation, vorzugsweise Steigerung, der
Expression einer Nukleinsäure, die ein PHDf-HD-Polypeptid
kodiert, erfolgt durch Einführen und Expression einer Nukleinsäure,
die ein PHDf-HD-Polypeptid kodiert, in eine(r) Pflanze.
-
Jede
Bezugnahme auf ein ”Protein, das sich in den erfindungsgemäßen
Verfahren eignet” soll ein PHDf-HD-Polypeptid bedeuten,
wie es hier definiert ist. Jede im erfindungsgemäßen
Zusammenhang erfolgende Bezugnahme auf eine ”Nukleinsäuresequenz,
die sich in den erfindungsgemäßen Verfahren eignet” soll eine
Nukleinsäuresequenz bedeuten, die ein solches PHDf-HD-Polypeptid
kodieren kann. Die in eine Pflanze einzubringende (und daher bei
der Durchführung der erfindungsgemäßen
Verfahren geeignete) Nukleinsäuresequenz ist eine beliebige
Nukleinsäuresequenz, die den nachstehend beschriebenen
Proteintyp kodiert, der auch als ”PHDf-HD-Nukleinsäuresequenz” oder ”PHDf-HD-Gen” bezeichnet
wird.
-
Ein
PHDf-HD-Polypeptid, wie es hier definiert ist betrifft ein Polypeptid,
umfassend (i) eine Domäne mit mindestens 25%, 30%, 35%,
40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%,
99% oder mehr Aminosäuresequenz-Identität zu einer
Leucin-Zipper/Pflanzenhomöodomänenfinger-(ZIP/PHDf)-Domäne
wie sie durch SEQ ID NO: 233 veranschaulicht ist; und (ii) eine
Domäne mit mindestens 25%, 30%, 35%, 40%, 45%, 50%, 55%,
60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99% oder mehr Aminosäuresequenz-Identität
zu einer Homöodomäne (HD) wie sie durch SEQ ID
NO: 234 veranschaulicht wird.
-
Alternativ
oder zusätzlich betrifft ein ”PHDf-HD-Polypeptid” wie
es hier definiert ist, ein Polypeptid, umfassend: (i) eine PHD-Domäne,
wie sie durch PFAM00628 versanschaulicht wird; und (ii) eine HD
wie sie durch PFAM00046 veranschaulicht wird.
-
Alternativ
oder zusätzlich betrifft ein ”PHDf-HD-Polypeptid” wie
es hier definiert ist eine Polypeptid-Sequenz, die bei der Verwendung
bei der Konstruktion eines HD-Stammbaums wie in 13 gezeigt, eher mit der PHDf-HD Gruppe von Polypeptiden,
die die in SEQ ID NO: 180 gezeigte Polypeptid-Sequenz umfassen, statt
mit einer anderen HD Gruppe, Cluster bildet.
-
Alternativ
oder zusätzlich betrifft ein ”PHDf-HD-Polypeptid” wie
es hier definiert ist ein Polypeptid, das in steigender Reihenfolge
der Präferenz mindestens 25%, 30%, 35%, 40%, 45%, 50%,
55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99% oder mehr
Aminosäuresequenz-Identität zu dem PHDf-HD-Polypeptid wie
es in SEQ ID NO: 180 gezeigt ist, oder zu einer der in Tabelle D1
hier angegebenen Polypeptid-Sequenzen aufweist.
-
Die
Begriffe ”Domäne” und ”Motiv” sind
im vorliegenden Text in dem Abschnitt ”Definitionen” definiert. Für
die Identifikation von Domänen gibt es Spezialdatenbanken,
zum Beispiel SMART (Schultz et al. (1998), Proc. Natl. Acad.
Sci. USA 95, 5857–5864; Letunic et al.
(2002), Nucleic Acids Res. 30, 242–244, InterPro (Mulder
et al. (2003), Nucl. Acids Res. 31, 315–318, Prosite
(Sucher und Bairoch (1994), A generalized Profile syntax
for biomolecular sequences motifs and its function in automatic
sequence interpretation. (In) ISMB-94; Proceedings 2nd International
Conference an Intelligent Systems for Molecular Biology. Altman
R., Brutlag D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61, AAAI
Press, Menlo Park; Hulo et al., Nucl. Acids Res. 32: D134–D137,
(2004), oder Pfam (Bateman et al., Nucleic Acids
Research 30 (1): 276–280 (2002). Ein Satz Werkzeuge
für die In-silico-Analyse von Proteinsequenzen findet sich
auf dem ExPASY-Proteomics-Server (Swiss Institute of Bioinformatics
(Gasteiger et al., ExPASy: the proteomics server for in-depth
Protein knowledge and analysis, Nucleic Acids Res. 31: 3784–3788
(2003)). Domänen können auch unter Verwendung
von Routinetechniken, wie Sequenz-Alignment, identifiziert werden.
Die Analyse der Polypeptid-Sequenz von SEQ ID NO: 180 ist nachstehend
in den Beispielen 30 und 32 hier angegeben. Ein PHDf-HD-Polypeptid,
wie es beispielsweise durch SEQ ID NO: 180 angegeben ist, umfasst
einen PHD-Finger mit einem Pfam-Eintrag PF00628, und eine HD mit
Pfam-Eintrag PF00046.
-
Verfahren
für das Alignment von Sequenzen für Vergleichszwecke
sind in der Fachwelt gut bekannt, dazu zählen GAP, BESTFIT,
BLAST, FASIA und TFASTA. Bei GAP wird der Algorithmus von Needleman
und Wunsch ((1970), J. Mol. Biol. 48: 443–453)
verwendet, um das globale Alignment (d. h. das Alignment, das sich über
die vollständigen Sequenzen erstreckt) von zwei Sequenzen,
das die Anzahl der ”matches” maximiert und die
Anzahl der ”gaps” minimiert. Beim BLAST-Algorithmus
(Altschul et al. (1990), J. Mol. Biol. 215: 403–10)
wird die Sequenzidentität in Prozent berechnet, und es
wird eine statistische Analyse der Ähnlichkeit zwischen
den beiden Sequenzen durchgeführt. Die Software für
die Durchführung einer BLAST-Analyse ist der Öffentlichkeit über
das National Centre for Biotechnology Information (NCBI) zugänglich.
Homologa können leicht unter Verwendung von zum Beispiel
dem multiplen Sequenz-Alignment-Algorithmus ClustalW (Version 1.83)
identifiziert werden, und zwar mit den Default-Parametern für
paarweises Alignment und einer Scoring-Methode in Prozent. Die Gesamtprozentsätze
der Ähnlichkeit und der Identität können
auch unter Verwendung von einer der in dem MatGAT-Software-Paket
verfügbaren Methoden bestimmt werden ((Campanella
et al., BMC Bioinformatics. 2003, Juli, 10; 4: 29. MatGAT: an application
that generstes similarity/identity matrices using Protein or DNA
sequences.). Für eine Optimierung des Alignments
zwischen konservierten Motiven können, wie es dem Fachmann
ersichtlich wird, kleine manuelle Veränderungen vorgenommen
werden. Darüber hinaus können statt Volllängensequenzen
für die Identifikation von Homologa auch spezifische Domänen
verwendet werden. Die Sequenzidentitätswerte können
mit den oben erwähnten Programmen unter Einstellung der
Default-Parameter über die gesamte Nukleinsäure-
oder Polypeptid-Sequenz oder über ausgewählte
Domänen oder konservierte Motiv(e) bestimmt werden. Das
hierin aufgeführte Beispiel 31 beschreibt in der Tabelle
D2 die Prozent Identität zwischen ZIP/PHDf wie es durch
SEQ ID NO: 233 veranschaulicht wird and ZIP/PHDf der in Tabelle
D1 aufgeführten PHDf-HD-Polypeptide, und in Tabelle D3
die Prozent Identität zwischen der HD wie sie durch SEQ
ID NO: 234 veranschaulicht wird, und der HD der in Tabelle D1 von
Beispiel 29 gezeigten PHDf-HD-Polypeptide.
-
Darüber
hinaus können die Aminosäuresequenzen, die an
basischen (Lys und Arg) oder sauren (Glu und Asp) Aminosäuren,
angereichert sind, und die als basische bzw saure Abschnitte bezeichnet
werden, leicht durch visuelle Untersuchung identifiziert werden
(17). Alternativ kann die primäre Aminosäure-Zusammensetzung
(in %) berechnet werden, um zu bestimmen, ob eine Polypeptid-Domäne
reich am spezifischen Aminosäuren ist, wobei Software-Programme
vom ExPASy-Server verwendet werden, insbesondere dem ProtParam Werkzeug
(Gasteiger E et al. (2003) ExPASy: the proteomics server
for in-depth Protein knowledge and analysis. Nucleic Acids Res 31:
3784–3788). Die Zusammensetzung des interessierenden
Proteins kann dann mit der durchschnittlichen Aminosäure-Zusammensetzung
(in %) in der Swiss-Prot Protein Sequenz-Datenbank verglichen werden.
-
Coiled
Coils sind zur Identifikation von Protein-Protein-Wechselwirkungen,
wie Oligomerisierung, entweder von identischen Proteinen, Proteinen
der gleichen Familie oder von unverwandten Proteinen wichtig. Ein
PHDf-HD-Polypeptid zeigt mindestens eine vorhergesagte Coiled-Coil-Region.
Neuerdings wurde ein großer Fortschritt hinsichtlich der
mittels Computer errechneten Vorhersage von Coiled Coils aus Sequenzdaten gemacht.
An der ExPASy sind dem Fachmann bestens bekannte Algorithmen zugänglich,
wie die Proteomics-Werkzeuge COILS, PAIRCOIL, PAIRCOIL2, MULTICOIL,
oder MARCOIL, die vom Schweizer Institut für Bioinformatik
gehosted werden. Im Beispiel 33 und in der 16 sind
jeweils die numerischen und graphischen Ergebnis von SEQ ID NO:
180 gezeigt, wie sie bei der Analyse mit dem COILS-Algorithmus erhalten werden.
Zwei vorhergesagte N-terminale Coiled Coil-Domänen werden
in einer PHDf-HD Polypeptid-Sequenz, wie sie durch SEQ ID NO: 180
veranschaulicht ist, mit einer hohen Wahrscheinlicheit identifiziert.
Ein C-terminaler Coiled Coil wird ebenfalls mit niedriger Wahrscheinlichkeit
vorhergesagt.
-
Die
Aufgabe der Vorhersage der subzellulären Proteinlokalisation
ist wichtig und gut untersucht. Das Wissen über die Lokalisierung
eines Proteins unterstützt die Aufklärung seiner
Funktion. Experimentelle Verfahren zur Proteinlokalisierung reichen
von der Immunlokalisierung bis zum Tagging von Proteinen mit grün fluoreszierendem
Protein (GFP). Solche Verfahren sind zwar präzise aber
verglichen mit Computer-Verfahren arbeitsintensiv. Neuerdings wurde
ein großer Fortschritt hinsichtlich der mittels Computer
errechneten Vorhersage der Proteinlokalisation aus Sequenzdaten
gemacht. An der ExPASy sind dem Fachmann bestens bekannte Algorithmen
zugänglich, wie u. a beispielsweise die Proteomics-Werkzeuge
PSort, TargetP, ChloroP, LocTree, Predotar, LipoP, MITOPROT, PATS,
PTS1, Signale, die vom Schweizer Institut für Bioinformatik
gehosted werden. Die Identifikation der subzellulären Lokalisierung
des erfindungsgemäßen Polypeptids ist in Beispiel
34 gezeigt. Insbesondere SEQ ID NO: 180 der vorliegenden Erfindung
wird dem Kern-Kompartiment eukaryotischer Zellen zugeordnet.
-
PHDf-HD-Polypeptide,
die sich in den erfindungsgemäßen Verfahren eignen
(zumindest in ihrer nativen Form), haben gewöhnlich, aber
nicht unbedingt transkriptionsregulatorische Aktivität
und die Fähigkeit, mit anderen Proteinen Wechselwirkungen
einzugehen. Daher können PHDf-HD-Polypeptide mit reduzierter
transkriptionsregulatorischer Aktivität, ohne transkriptionsregulatorische
Aktivität, mit reduzierter Protein-Protein-Wechselwirkungs-Kapazität
oder ohne Protein-Protein-Wechselwirkungs-Kapazität gleichermaßen
für die erfindungsgemäßen Verfahren geeignet
sein. Die DNA-Bindungs-Aktivität und die Protein-Protein-Wechselwirkungen
können leicht in vitro oder in vivo mittels Techniken des
Standes der Technik bestimmt werden (beispielsweise in Current
Protocols in Molecular Biology, Bd. 1 und 2, Ausubel et al. (1994),
Current Protocols). Zur Bestimmung der DNA-Bindungs-Aktivität
von PHDf-HD-Polypeptiden stehen mehrere Tests zur Verfügung, wie
die DNA-Bindungs-Gelshift-Tests (oder Gelretardations-Tests; Korfhage
et al. (1994) Plant C 6: 695–708), in vitro DNA
Bindungstests (Schindler et al. (1993) Plant J 4 (1): 137–150),
oder Transkriptionsaktivierung von PHDf-HD-Polypeptiden in Hefe-,
Tier- und Pflanzenzellen (Halbach et al. (2000) Nucleic
Acid Res 28 (18): 3542–3550). Spezifische DNA-Bindungssequenzen
können mit der statistischen Oligonukleotid-Selektionstechnik
bestimmt werden (Viola & Gonzalez
(May 26, 2007) Biochemistry).
-
Die
vorliegende Erfindung wird anhand der Transformation von Pflanzen
mit der in SEQ ID NO: 179 gezeigten Nukleinsäuresequenz
gezeigt, die die Polypeptid-Sequenz von SEQ ID NO: 180 kodiert.
Die Durchführung der Erfindung ist jedoch nicht auf diese
Sequenzen eingeschränkt; die erfindungsgemäßen
Verfahren können vorteilhaft mit einer jeden Nukleinsäuresequenz
durchgeführt werden, die ein PHDf-HD oder PHDf-HD-Polypeptid
wie hier definiert kodiert.
-
Beispiele
für Nukleinsäuren, die PHDf-HD-Polypeptide kodieren,
sind in der Tabelle D1 des Beispiels 29 hierin beschrieben. Solche
Nukleinsäuresequenzen sind zur Durchführung der
erfindungsgemäßen Verfahren geeignet. Die in Tabelle
D1 von Beispiel 29 angegebenen Polypeptid-Sequenzen sind Beispiel-Sequenzen für
Orthologa und Paraloga des durch SEQ ID NO: 180 veranschaulichten
PHDf-HD-Polypeptids, wobei die Begriffe ”Orthologa” und ”Paraloga” die
hier definierte Bedeutung haben. Weitere Orthologa und Paraloga
können leicht durch Durchführen einer so genannten
reziproken Blast-Suche identifiziert werden. Dies beinhaltet typischerweise
einen ersten BLAST, bei der mit einer Abfragesequenz (zum Beispiel
eine der in Tabelle D1 von Beispiel 29 aufgeführten Sequenzen)
ein ”BLASTing” gegen eine beliebige Sequenzdatenbank,
wie die öffentlich zugängliche NCBI-Datenbank,
durchgeführt wird. BLASTN oder TBLASTX (unter Verwendung
von Standard-Default-Werten) werden im Allgemeinen dann verwendet,
wenn man von einer Nukleotidsequenz ausgeht, und BLASTP oder TBLASTN
(unter Verwendung von Standard-Default-Werten), wenn man von einer Proteinsequenz
ausgeht. Die BLAST-Ergebnisse können gegebenenfalls gefiltert
werden. Mit den Volllängen-Sequenzen der gefilterten oder
ungefilterten Ergebnisse wird anschließend ein Rück-BLASTing
(zweiter BLAST) gegen Sequenzen des Organismus, von dem die Abfragesequenz
stammt, durchgeführt (ist die Abfragesequenz SEQ ID NO:
179 oder SEQ ID NO: 180, dann erfolgt das zweite BLASTing daher
gegen Reis-Sequenzen). Die Ergebnisse des ersten und des zweiten
BLASTing werden dann verglichen. Ein Paralogon wird dann identifiziert,
wenn ein hochrangiger Hit von dem ersten BLASTing von derselben
Art ist, von der die Abfragesequenz stammt, in diesem Fall führt
ein Rück-BLASTing idealerweise zu der Abfragesequenz unter
den höchstrangigen Hits; ein Orthologon wird dann identifiziert,
wenn ein hochrangiger Hit in dem ersten BLASTing nicht von der gleichen
Art wie derjenigen, von der die Abfragesequenz stammt, ist, und
führt vorzugsweise beim Rück-BLASTing dazu, dass
die Abfragesequenz unter den höchstrangigen Hits ist.
-
Hochrangige
Hits sind solche mit niedrigem E-Wert. Je niedriger der E-Wert,
desto signifikanter der ”Score” (anders ausgedrückt,
desto niedriger die Wahrscheinlichkeit, dass der Hit durch Zufall
gefunden wurde). Die Berechnung des E-Werts ist Stand der Technik.
Das ”Scoring” der Vergleiche erfolgt nicht nur
mittels E-Werten, sondern auch mit dem Prozentsatz der Identität.
Der Prozentsatz der Identität bezieht sich auf die Anzahl
der identischen Nukleotide (oder Aminosäuren) zwischen
den beiden verglichenen Nukleinsäure-(oder Polypeptid-)Sequenzen über
eine bestimmte Länge. Bei großen Familien kann
man ClustalW und anschließend einen Neighbour-Joining-Tree
verwenden, um die Cluster der verwandten Gene leichter sichtbar
zu machen und um Orthologa und Paraloga zu identifizieren. Ein jeder
Sequenz-Cluster innerhalb der Gruppe, die SEQ ID NO: 180 umfasst
(in 29 eingekreist) liegt in der
vorstehend genannten Definition eines PHDf-HD-Polypeptids, und wird
zur Verwendung bei den erfindungsgemäßen Verfahren
als geeignet angesehen.
-
Zur
Durchführung der erfindungsgemäßen Verfahren
können sich auch Nukleinsäurevarianten eignen. Beispiele
für solche Varianten umfassen die Nukleinsäuresequenzen,
die Homologa und Derivate von einer der in Tabelle D1 von Beispiel
29 hier angegebenen Polypeptid-Sequenzen kodieren, wobei die Begriffe ”Homologon” und ”Derivat” die
hier definierte Bedeutung haben. Für die erfindungsgemäßen
Verfahren eignen sich auch Nukleinsäuresequenzen, die Homologa
und Derivate von Orthologa oder Paraloga von einer der in Tabelle
D1 von Beispiel 29 hier angegebenen Polypeptid-Sequenzen kodieren.
Homologa und Derivate, die sich bei den erfindungsgemäßen
Verfahren eignen, weisen im Wesentlichen die gleiche biologische
und funktionelle Aktivität wie das unmodifizierte Protein,
von dem sie abstammen, auf.
-
Zu
weiteren Nukleinsäurevarianten, die sich zur Durchführung
der erfindungsgemäßen Verfahren eignen, gehören
Abschnitte von Nukleinsäuresequenzen, die PHDf-HD-Polypeptide
kodieren, Nukleinsäuresequenzen, die mit Nukleinsäuresequenzen,
die PHDf-HD-Polypeptide kodieren, hybridisieren, Spleißvarianten von
Nukleinsäuresequenzen, die PHDf-HD-Polypeptide kodieren,
allelische Varianten von Nukleinsäuresequenzen, die PHDf-HD-Polypeptide
kodieren, und Varianten von Nukleinsäuresequenzen, die
PHDf-HD-Polypeptide kodieren, die durch ”Gen-Shuffling” erhalten
wurden. Die Begriffe hybridisierende Sequenz, Spleißvariante,
allelische Variante und ”Gen-Shuffling” sind wie
hier Text beschrieben.
-
Nukleinsäuresequenzen,
die PHDf-HD-Polypeptide kodieren, müssen nicht unbedingt
Volllängennukleinsäuren sein, da die Durchführung
der erfindungsgemäßen Verfahren nicht auf die
Verwendung von Volllängen-Nukleinsäuresequenzen
angewiesen ist. Erfindungsgemäß wird ein Verfahren
zur Verbesserung der Ertragsmerkmale, vorzugsweise Verbesserung
der Samenertragsmerkmale, bei Pflanzen bereitgestellt, bei dem man
in eine(r) Pflanze einen Abschnitt von einer der in Tabelle D1 von
Beispiel 29 angegebenen Nukleinsäuresequenzen oder einen
Abschnitt von einer Nukleinsäuresequenz, die ein Orthologon,
Paralogon oder Homologon von einer der in Tabelle D1 von Beispiel
29 angegebenen Nukleinsäuresequenzen kodiert, einführt
und exprimiert.
-
Ein
Abschnitt einer Nukleinsäuresequenz kann zum Beispiel dadurch
hergestellt werden, dass man eine oder mehrere Deletionen bei der
Nukleinsäuresequenz durchführt. Die Abschnitte
können in isolierter Form verwendet werden oder können
mit anderen kodierenden (oder nichtkodierenden) Sequenzen fusioniert werden,
um zum Beispiel ein Protein herzustellen, bei dem mehrere Aktivitäten
kombiniert sind. Bei der Fusion mit anderen kodierenden Sequenzen
kann das bei der Translation hergestellte Polypeptid größer
sein, als für den Proteinabschnitt vorhergesagt wurde.
-
Abschnitte,
die sich bei den erfindungsgemäßen Verfahren eignen,
kodieren ein PHDf-HD-Polypeptid wie im vorliegenden Text definiert
und weisen im Wesentlichen die gleiche biologische Aktivität
wie die in Tabelle D1 von Beispiel 29 angegebenen Polypeptidsequenzen
auf. Vorzugsweise handelt es sich bei dem Abschnitt um einen Abschnitt
von einer der in Tabelle D1 von Beispiel 29 angegebenen Nukleinsäuresequenzen oder
um einen Abschnitt einer Nukleinsäuresequenz, die ein Orthologon
oder Paralogon von einer der in Tabelle D1 von Beispiel 29 angegebenen
Polypeptidsequenzen kodiert. Vorzugsweise ist der Abschnitt mindestens
in steigender Reihenfolge der Präferenz mindestens 600,
800, 1000, 1200, 1400, 1600, 1800, 2000, 2200, 2400, 2600, 2800
aufeinanderfolgende Nukleotide lang, wobei es sich bei den aufeinander
folgenden Nukleotiden um eine der in Tabelle D1 von Beispiel 29
angegebenen Nukleinsäuresequenzen oder um eine Nukleinsäuresequenz,
die ein Orthologon oder Paralogon von einer der in Tabelle D1 von
Beispiel 29 angegebenen Polypeptidsequenzen kodiert, handelt. Am
stärksten bevorzugt ist der Abschnitt ein Abschnitt der
Nukleinsäuresequenz gemäß SEQ ID NO:
179. Der Abschnitt kodiert vorzugsweise eine Polypeptidsequenz,
die bei der Verwendung bei der Konstruktion eines HD-Stammbaums,
wie er in der 13 gezeigt ist, eher mit der
Gruppe der PHDf-HD-Polypeptide, umfassend die durch SEQ ID NO: 180
veranschaulichte Polypeptidsequenz, Cluster bildet, als mit einer
anderen HD-Gruppe.
-
Eine
weitere Nukleinsäuresequenzvariante, die sich in den erfindungsgemäßen
Verfahren eignet, ist eine Nukleinsäuresequenz, die unter
reduzierten Stringenzbedingungen, vorzugsweise unter stringenten
Bedingungen mit einer Nukleinsäuresequenz, die ein PHDf-HD-Polypeptid
kodiert, wie hier definiert, oder mit einem Abschnitt, wie hier
definiert, hybridisieren kann.
-
Gemäß der
vorliegenden Erfindung wird ein Verfahren zur Verbesserung von Ertragsmerkmalen,
vorzugsweise Verbesserung von Samenertragsmerkmalen, bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Nukleinsäuresequenz,
die an eine der in Tabelle D1 von Beispiel 29 angegebenen Nukleinsäuresequenzen
hybridisieren kann, einführt oder exprimiert, oder bei
der man in eine(r) Pflanze eine Nukleinsäuresequenz, die
an eine Nukleinsäuresequenz, die ein Orthologon, Paralogon
oder Homologon von einer der in Tabelle D1 von Beispiel 29 angegebenen
Nukleinsäuresequenzen kodiert, hybridisieren kann, einführt
oder exprimiert.
-
Hybridisierende
Sequenzen, die sich bei den erfindungsgemäßen
Verfahren eignen, kodieren ein PHDf-HD-Polypeptid wie hier definiert
und haben im Wesentlichen die gleiche biologische Aktivität
wie die in der Tabelle D1 von Beispiel 29 angegebenen Polypeptidsequenzen.
Die hybridisierende Sequenz kann vorzugsweise an eine der in Tabelle
D1 von Beispiel 29 angegebenen Nukleinsäuresequenzen oder
an einen Abschnitt von einer dieser Sequenzen hybridisieren, wobei
ein Abschnitt die vorstehend definierte Bedeutung hat, oder wobei
die hybridisierende Sequenz an eine Nukleinsäuresequenz
hybridisieren kann, die ein Orthologon, oder Paralogon von einer
der in Tabelle D1 von Beispiel 29 aufgeführten Polypeptidsequenzen
kodiert. Am stärksten bevorzugt kann die hybridisierende
Sequenz an eine Nukleinsäuresequenz hybridisieren, wie
sie durch SEQ ID NO: 179 veranschaulicht wird, oder an einen Abschnitt
davon.
-
Die
hybridisierende Sequenz kodiert vorzugsweise eine Polypeptidsequenz,
die bei der Verwendung bei der Konstruktion eines HD-Stammbaums,
wie er in der 13 gezeigt ist, eher mit der
Gruppe der PHDf-HD-Polypeptide, umfassend die durch SEQ ID NO: 180
veranschaulichte Polypeptidsequenz, Cluster bildet, als mit einer
anderen HD-Gruppe.
-
Eine
weitere Nukleinsäuresequenzvariante, die sich bei den erfindungsgemäßen
Verfahren eignet ist eine Spleißvariante, die ein PHDf-HD-Polypeptid
wie hier definiert kodiert, wobei eine Spleißvariante die
vorstehend definierte Bedeutung hat.
-
Gemäß der
vorliegenden Erfindung wird ein Verfahren zur Verbesserung von Ertragsmerkmalen,
vorzugsweise Verbesserung von Samenertragsmerkmalen, bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Spleißvariante
von einer der in Tabelle D1 von Beispiel 29 angegebenen Nukleinsäuresequenzen oder
eine Spleißvariante einer Nukleinsäuresequenz,
die ein Orthologon, Paralogon oder Homologon von einer der in Tabelle
D1 von Beispiel 29 angegebenen Polypeptidsequenzen kodiert, einführt
oder exprimiert.
-
Bevorzugte
Spleißvarianten sind Spleißvarianten einer Nukleinsäuresequenz,
die durch SEQ ID NO: 179 veranschaulicht ist, oder ist eine Spleißvariante
einer Nukleinsäuresequenz, die ein Orthologon oder Paralogon
von SEQ ID NO: 180 kodiert. Das von der Spleißvariante
kodierte Polypeptid bildet bei der Verwendung bei der Konstruktion
eines HD-Stammbaums, wie er in der 13 gezeigt
ist, eher mit der Gruppe der PHDf-HD-Polypeptide, umfassend die
durch SEQ ID NO: 180 veranschaulichte Polypeptidsequenz, Cluster als
mit einer anderen HD-Gruppe.
-
Eine
weitere Nukleinsäuresequenzvariante, die sich bei den erfindungsgemäßen
Verfahren eignet ist eine allelische Variante, die ein PHDf-HD-Polypeptid
wie hier definiert kodiert, wobei eine allelische Variante die vorstehend
definierte Bedeutung hat.
-
Gemäß der
vorliegenden Erfindung wird ein Verfahren zur Verbesserung von Ertragsmerkmalen
bei Pflanzen, vorzugsweise zur Verbesserung von Samenertragsmerkmalen,
bereitgestellt, bei dem man in eine(r) Pflanze eine allelische Variante
von einer der in Tabelle D1 von Beispiel 29 angegebenen Nukleinsäuresequenzen
einführt oder exprimiert oder bei der man in eine(r) Pflanze
eine allelische Variante einer Nukleinsäuresequenz, die
ein Orthologon, Paralogon oder Homologon von einer der in Tabelle
D1 von Beispiel 29 angegebenen Polypeptidsequenzen kodiert, einführt
oder exprimiert.
-
Die
allelischen Varianten, die bei den erfindungsgemäßen
Verfahren geeignet sind, weisen im Wesentlichen die gleiche biologische
Aktivität wie das Polypeptid von SEQ ID NO: 180 und eine
der Polypeptidsequenzen in Tabelle D1 von Beispiel 29 auf. Allelische
Varianten kommen in der Natur vor, und die Verwendung dieser natürlichen
Allele ist von den Verfahren der vorliegenden Erfindung umfasst.
Vorzugsweise handelt es sich bei der allelischen Variante um eine
allelische Variante von SEQ ID NO: 179 oder eine allelische Variante
einer Nukleinsäuresequenz, die ein Orthologon oder Paralogon
von SEQ ID NO: 180 kodiert. Vorzugsweise bildet die von der allelischen
Variante kodierte Polypeptidsequenz bei Verwendung bei der Konstruktion
eines HD-Stammbaums wie er in 13 gezeigt
ist eher mit den PHDf-HD-Polypeptiden, umfassend die Polypeptidsequenz
gemäß SEQ ID NO: 180, Cluster, als mit einer anderen
HD-Gruppe.
-
”Gen-Shuffling” oder
gerichtete Evolution können ebenfalls eingesetzt werden,
um Varianten von Nukleinsäuresequenzen, die PHDf-HD-Polypeptide
wie oben definiert kodieren, zu erzeugen, wobei der Begriff ”Gen-Shuffling” die
vorstehend definierte Bedeutung hat.
-
Gemäß der
vorliegenden Erfindung wird ein Verfahren zur Verbesserung von Ertragsmerkmalen,
vorzugsweise zur Verbesserung von Samenertragsmerkmalen in Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Variante von
einer der in Tabelle D1 von Beispiel 29 angegebenen Nukleinsäuresequenzen
einführt und exprimiert, oder bei dem man in eine(r) Pflanze
eine Variante einer Nukleinsäuresequenz, die ein Orthologon,
Paralogon oder Homologon von einer der in Tabelle D1 von Beispiel
29 angegebenen Polypeptidsequenzen kodiert, wobei diese Nukleinsäuresequenzvariante
durch ”Gen-Shuffling” erhalten wurde, einführt und
exprimiert.
-
Die
vorliegende Erfindung stellt vorteilhafterweise bisher unbekannte
PHDf-HD- und Polypeptid-Sequenzen bereit.
-
Gemäß einer
weiteren Ausführungsform der vorliegenden Erfindung wird
ein isoliertes Nukleinsäuremolekül bereitgestellt,
umfassend:
- (i) eine durch SEQ ID NO: 242 dargestellte
Nukleinsäure;
- (ii) eine Nukleinsäure oder ein Fragment davon, die/das
zu SEQ ID NO: 242 komplementär ist;
- (iii) eine Nukleinsäure, die ein JMJC Polypeptid kodiert,
mit in steigender Reihenfolge der Präferenz 70%, 75%, 80%,
85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
zu der in SEQ ID NO: 243 angegebenen Aminosäuresequenz;
- (iv) eine Nukleinsäure, die unter stringenten Bedingungen
an eine der in (i), (ii) oder (iii) oben angegebenen Nukleinsäuren
hybridisieren kann.
-
Gemäß einer
weiteren Ausführungsform der vorliegenden Erfindung wird
ein isoliertes Polypeptid bereitgestellt, umfassend:
- (i) eine durch SEQ ID NO: 242 dargestellte Nukleinsäure;
- (ii) eine Nukleinsäure oder ein Fragment davon, die/das
zu SEQ ID NO: 242 komplementär ist;
- (iii) eine Nukleinsäure, die ein JMJC Polypeptid kodiert,
mit in steigender Reihenfolge der Präferenz 70%, 75%, 80%,
85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
zu der in SEQ ID NO: 243 angegebenen Aminosäuresequenz;
- (iv) eine Nukleinsäure, die unter stringenten Bedingungen
an eine der in (i), (ii) oder (iii) oben angegebenen Nukleinsäuren
hybridisieren kann.
-
Vorzugsweise
bildet die von der durch Gen-Shuffling erhaltene Nukleinsäurevariante
kodierte Polypeptidsequenz bei Verwendung bei der Konstruktion eines
HD-Stammbaums wie er in 13 gezeigt
ist eher mit den PHDf-HD-Polypeptiden, umfassend die Polypeptidsequenz
gemäß SEQ ID NO: 180, Cluster, als mit einer anderen
HD-Gruppe.
-
Darüber
hinaus können Nukleinsäurevarianten auch durch
ortsgerichtete Mutagenese erhalten werden. Es gibt verschiedene
Verfahren zur Erzielung von ortsgerichteter Mutagenes, wobei die üblichsten
Verfahren auf PCR-Basis sind (Current Protocols in Molecular
Biology. Wiley Eds.).
-
Nukleinsäuresequenzen,
die PHDf-HD-Polypeptide kodieren, können von einer beliebigen
natürlichen oder künstlichen Quelle stammen. Die
Nukleinsäuresequenz kann aus ihrer nativen Form in ihrer
Zusammensetzung und/oder genomischen Umgebung durch gezielten menschlichen
Eingriff modifiziert werden. Die das PHDf-HD-Polypeptid kodierende
Nukleinsäuresequenz stammt vorzugsweise aus einer Pflanze,
vorzugsweise aus einer monokotylen Pflanze, stärker bevorzugt
aus der Familie der Poaceae, am stärksten bevorzugt stammt
die Nukleinsäuresequenz aus Oryza sativa.
-
Die
Durchführung der erfindungsgemäßen Verfahren
ergibt Pflanzen mit verbesserten Ertragsmerkmalen. Die Durchführung
der erfindungsgemäßen Verfahren ergibt insbesondere
Pflanzen mit erhöhtem Ertrag, insbesondere erhöhtem
Samenertrag im Vergleich zu Kontrollpflanzen. Die Begriffe ”Ertrag” und ”Samenertrag” sind
eingehender im Abschnitt ”Definitionen” beschrieben.
-
Werden
hier verbesserte Ertragsmerkmale erwähnt, soll dies eine
Erhöhung der Biomasse (Gewicht) von einem oder mehreren
Teilen einer Pflanze bedeuten, was oberirdische (erntefähige)
Teile und/oder unterirdische (erntefähige) Teile beinhalten
kann. Insbesondere sind solche erntefähigen Teile Samen,
und die Durchführung der erfindungsgemäßen
Verfahren ergibt Pflanzen mit erhöhtem Samenertrag im Vergleich
zu Kontrollpflanzen.
-
Wählt
man Mais als Beispiel, kann sich eine Ertragserhöhung unter
Anderem als eines oder mehrere der folgenden Merkmale äußern:
Erhöhte Anzahl Pflanzen, die sich pro Hektar oder Acre
entwickeln, Erhöhung der Anzahl Ähren pro Pflanze,
Erhöhung der Anzahl Reihen, der Anzahl Körner
pro Reihe, des Korngewichts, Tausendkorngewichts, Ährenlänge/durchmessers,
Erhöhung der Samenfüllungsrate (Anzahl gefüllter
Samen dividiert durch die Gesamtzahl Samen und multipliziert mit
Hundert). Wählt man Reis als Beispiel, kann sich eine Ertragserhöhung
unter Anderem als Erhöhung von einem oder mehreren der
folgenden Merkmale ausdrücken: Anzahl Pflanzen pro Hektar
oder Acre, Anzahl Rispen pro Pflanze, Anzahl Ährchen pro
Rispe, Anzahl Blüten (Blütchen) pro Rispe (ausgedrückt
als Verhältnis der Anzahl der gefüllten Samen
zu der Anzahl der Primärrispen), erhöhte Samenfüllungsrate
(Anzahl gefüllter Samen dividiert durch die Gesamtzahl
Samen und multipliziert mit Hundert), erhöhtes Tausendkorngewicht.
-
Die
vorliegende Erfindung stellt ein Verfahren zur Erhöhung
des Ertrags, insbesondere Samenertrags von Pflanzen im Vergleich
zu Kontrollpflanzen bereit, wobei man bei dem Verfahren in einer
Pflanze die Expression einer Nukleinsäuresequenz, die ein
PHDf-HD-Polypeptid wie im vorliegenden Text definiert kodiert, moduliert,
vorzugsweise erhöht.
-
Da
die erfindungsgemäßen transgenen Pflanzen verbesserte
Ertragsmerkmale, vorzugsweise verbesserte Samenertragsmerkmale,
aufweisen, zeigen diese Pflanzen (zumindest während eines
Teils ihres Lebenszyklus) wahrscheinlich eine im Vergleich zu der
Wachstumsrate von Kontrollpflanzen in einem entsprechenden Stadium
ihres Lebenszyklus erhöhte Wachstumsrate.
-
Die
erhöhte Wachstumsrate kann für einen oder mehrere
Teile einer Pflanze (einschließlich Samen) spezifisch sein
oder kann im Wesentlichen in der ganzen Pflanze stattfinden. Pflanzen
mit einer erhöhten Wachstumsrate können einen
kürzeren Lebenszyklus aufweisen. Unter dem Lebenszyklus
einer Pflanze kann man diejenige Zeit verstehen, die die Pflanze
braucht, um von einem reifen trockenen Samen bis zu dem Stadium
heranzuwachsen, in dem die Pflanze reife trockene Samen ähnlich
dem Ausgangsmaterial produziert hat. Dieser Lebenszyklus kann von
Faktoren wie Jungpflanzenvitalität, Wachstumsrate, ”Greenness
Index”, Blütezeit und Geschwindigkeit der Samenreifung
beeinflusst werden. Die erhöhte Wachstumsrate kann in einem
Stadium oder in mehreren Stadien im Lebenszyklus einer Pflanze oder
im Wesentlichen während des gesamten Lebenszyklus der Pflanze
stattfinden. Eine erhöhte Wachstumsrate während
der Frühstadien im Lebenszyklus einer Pflanze kann verbesserte
(Jungpflanzen)-Vitalität widerspiegeln. Die erhöhte
Wachstumsrate kann den Erntezyklus einer Pflanze verändern,
so dass die Pflanzen später gesät werden können
und/oder früher geerntet werden können, als dies
sonst möglich wäre (ein ähnlicher Effekt
lässt sich mit einer früheren Blütezeit
erzielen). Ist die Wachstumsrate ausreichend erhöht, kann
dies ein weiteres Aussäen von Samen derselben Pflanzenart
ermöglichen (zum Beispiel Aussäen und Ernten von
Reispflanzen und anschließendes Aussäen und Ernten
von weiteren Reispflanzen innerhalb einer traditionellen Wachstumsperiode).
Entsprechend kann dies bei ausreichender Steigerung der Wachstumsrate
ein weiteres Aussäen von Samen von unterschiedlichen Pflanzenarten
ermöglichen (zum Beispiel das Aussäen und Ernten
von Maispflanzen und anschließendes z. B. Aussäen
und gegebenenfalls Ernten von Sojabohnen, Kartoffeln oder sonstigen
geeigneten Pflanzen). Bei manchen Kulturpflanzen kann man auch zusätzlich
mehrmals von demselben Wurzelstock ernten. Eine Veränderung
des Erntezyklus einer Pflanze kann zu einer Erhöhung der
jährlichen Biomasseproduktion pro Acre führen
(und zwar aufgrund einer Erhöhung der Häufigkeit
(z. B. pro Jahr), mit der eine bestimmte Pflanze herangezogen und
geerntet werden kann). Eine erhöhte Wachstumsrate kann
auch den Anbau von transgenen Pflanzen in einem weiteren geographischen
Bereich als ihre Wildtyp-Gegenstücke ermöglichen, da
die räumlichen Begrenzungen für den Anbau einer
Kulturpflanze häufig von ungünstigen Umweltbedingungen
entweder während der Pflanzzeit (früh in der Saison)
oder während der Erntezeit (spät in der Saison)
bestimmt wird. Solche ungünstigen Bedingungen können
vermieden werden, wenn der Erntezyklus verkürzt ist. Die
Wachstumsrate kann durch Ableiten von verschiedenen Parametern von
Wachstumskurven bestimmt werden, wobei die Parameter unter Anderem
folgendes sein können: T-Mid (Zeitdauer, die die Pflanzen
benötigen, um 50% ihrer Maximalgröße
zu erreichen) und T-90 (Zeitdauer, die die Pflanzen benötigen,
um 90% ihrer Maximalgröße zu erreichen). Die hier
definierten Wachstumsraten sollen kein früheres Blühen
bedeuten.
-
Gemäß einem
bevorzugten Merkmal der vorliegenden Erfindung ergibt die Durchführung
der erfindungsgemäßen Verfahren Pflanzen mit einer
im Vergleich zu Kontrollpflanzen erhöhten Wachstumsrate.
Die vorliegende Erfindung stellt daher ein Verfahren zur Erhöhung
der Wachstumsrate von Pflanzen bereit, wobei man bei dem Verfahren
in einer Pflanze die Expression einer Nukleinsäure, die
ein PHDf-HD-Polypeptid wie im vorliegenden Text definiert kodiert,
moduliert, vorzugsweise erhöht.
-
Verbesserte
Ertragsmerkmale, vorzugsweise verbesserte Samenertragsmerkmale,
treten auf unabhängig davon, ob die Pflanze unter Nichtstressbedingungen
steht oder ob die Pflanze im Vergleich zu Kontrollpflanzen, die
unter vergleichbaren Bedingungen gewachsen waren, verschiedenen
Stressfaktoren unterworfen wird. Typischerweise reagieren Pflanzen
auf Stress durch langsameres Wachstum. Unter starken Stressbedingungen
kann die Pflanze sogar ihr Wachstum einstellen. Leichter Stress
wiederum wird im vorliegenden Zusammenhang als jeglicher Stress
definiert, dem eine Pflanze ausgesetzt ist und der nicht dazu führt,
dass die Pflanze ihr Wachstum völlig einstellt, ohne ihr
Wachstum wieder aufnehmen zu können. Leichter Stress im Sinne
der Erfindung führt zu einer Verringerung des Wachstums
der gestressten Pflanzen von weniger als 40%, 35% oder 30%, vorzugsweise
weniger als 25%, 20% oder 15%, stärker bevorzugt weniger
als 14%, 13%, 12%, 11% oder 10% oder weniger im Vergleich zu der
Kontrollpflanze unter Nichtstressbedingungen. Aufgrund der Fortschritte
bei den landwirtschaftlichen Kulturmaßnahmen (Bewässerung,
Düngung, Pestizidbehandlungen) findet man bei angebauten
Kulturpflanzen nicht oft starken Stress. Daher ist das durch leichten
Stress induzierte geschwächte Wachstum häufig
ein unerwünschtes Merkmal in der Landwirtschaft. Leichter
Stress ist der alltägliche biotische und/oder abiotische
Stress (Umweltstress), dem eine Pflanze ausgesetzt ist. Abiotischer
Stress kann durch Trockenheit oder Wasserüberschuss, anaeroben
Stress, Salzstress, chemische Toxizität, oxidativen Stress
und Hitze, Kälte oder Gefriertemperaturen verursacht werden.
Bei dem abiotischen Stress kann es sich um einen osmotischen Stress
handeln, der durch Wasserstress (insbesondere aufgrund von Trockenheit),
Salzstress, oxidativen Stress oder ionenbedingten Stress verursacht
wird. Biotischer Stress wird gewöhnlich durch Pathogene,
wie Bakterien, Viren, Pilze, Nematoden und Insekten verursacht.
Der Begriff der ”Nichtstress”-Bedingungen wie
hier verwendet, beinhaltet solche Umweltbedingungen, die ein optimales
Wachstum der Pflanzen ermöglichen. Der Fachmann ist mit
den normalen Bodenund Klimabedingungen für einen bestimmten
Standort vertraut.
-
Die
Durchführung der erfindungsgemäßen Verfahren
ergibt Pflanzen, die bei Wachstum unter Nichtstressbedingungen oder
unter milden Trockenheitsbedingungen im Vergleich zu Kontrollpflanzen,
die unter vergleichbaren Stressbedingungen wachsen, verbesserte
Ertragsmerkmale, vorzugsweise verbesserte Samenertragsmerkmale aufweisen.
Gemäß der vorliegenden Erfindung wird daher ein
Verfahren zur Verbesserung von Ertragsmerkmalen von Pflanzen, die
unter Nichtstressbedingungen oder unter milden Trockenheitsbedingungen
heranwachsen, bereitgestellt, wobei man bei dem Verfahren in einer
Pflanze die Expression einer Nukleinsäuresequenz, die ein
PHDf-HD-Polypeptid wie oben definiert kodiert, moduliert, vorzugsweise
erhöht. Pflanzen mit optimalen Wachstumsbedingungen, die
unter Nicht-Stressbedingungen herangezogen wurden, ergeben in steigender
Reihenfolge der Präferenz mindestens 90%, 87%, 85%, 83%,
80%, 77% oder 75% der durchschnittlichen Produktion einer solchen
Pflanze in einer bestimmten Umgebung. Die durchschnittliche Produktion
kann für einen bestimmten Standort auf Ernte und/oder Jahreszeit-Basis
berechnet werden. Der Fachmann ist mit der durchschnittlichen Ertragsproduktionen
einer Kulturpflanze vertraut.
-
Die
Durchführung der erfindungsgemäßen Verfahren
ergibt unter abiotischen Stressbedingungen gewachsene Pflanzen mit
verbesserten Ertragsmerkmalen, vorzugsweise verbesserten Samenertragsmerkmalen,
im Vergleich zu Kontrollpflanzen, die unter vergleichbaren Stressbedingungen
gewachsen waren. Wie von Wang et al., (Planta (2003), 218:
1–14) beschrieben, führt abiotischer
Stress zu einer Reihe von morphologischen, physiologischen, biochemischen
und molekularen Veränderungen, die das Pflanzenwachstum
und die Produktivität negativ beeinflussen. Trockenheit,
Salinität, extreme Temperaturen und oxidativer Stress stehen bekanntlich
miteinander in Verbindung und können über ähnliche
Mechanismen Wachstums- und Zellschäden induzieren. Rabbani
et al. (Plant Physiol. (2003), 133: 1755–1767)
beschreiben ein besonders hohes Ausmaß an gegenseitiger
Beeinflussung von Trockenheitsstress und durch hohe Salinität
verursachtem Stress. Trockenheit und/oder Versalzung äußern
sich beispielsweise in erster Linie als osmotischer Stress, was
zur Störung der Homöostase und der Ionenverteilung
in der Zelle führt. Oxidativer Stress, der häufig
Hoch- oder Niedertemperatur-, Salinitäts- oder Trockenheitsstress
begleitet, kann zur Denaturierung funktioneller und struktureller
Proteine führen. Demzufolge aktivieren diese verschiedenen Umweltstressfaktoren
häufig ähnliche Signalleitungswege der Zelle und
Zellreaktionen, wie die Produktion von Stressproteinen, die Aufwärtsregulation von
Antioxidantien, die Anreicherung kompatibler gelöster Stoffe
und ein Einstellen des Wachstums. Da verschiedene Umweltstressfaktoren ähnliche
Stoffwechselwege aktivieren, sollte die beispielhafte Erklärung
von Trockenheitsstress in der vorliegenden Erfindung nicht als Einschränkung
auf Trockenheitsstress angesehen werden, sondern soll lediglich
die Beteiligung der PHDf-HD-Polypeptide wie vorstehend definiert
bei der Verbesserung der Ertragsmerkmale, vorzugsweise bei der Verbesserung
der Samenertragsmerkmale, im Vergleich zu Kontrollpflanzen, die
bei vergleichbaren Stressbedingungen, insbesondere bei abiotischem
Stress, gewachsen waren, anzeigen.
-
Der
Begriff „abiotischer Stress” wie im vorliegenden
Text definiert soll einen oder mehrere der folgenden Stressfaktoren
bedeuten: Wasserstress (aufgrund von Trockenheit oder Wasserüberschuss),
anaerober Stress, Salzstress, Temperaturstress (aufgrund von Hitze,
Kälte oder Gefriertemperaturen), chemischer Toxizitätsstress
und oxidativer Stress. Gemäß einem Aspekt der
Erfindung handelt es sich bei dem abiotischen Stress um einen osmotischen
Stress, ausgewählt aus Wasserstress, Salzstress, oxidativem
Stress und ionenbedingtem Stress. Vorzugsweise handelt es sich bei
dem Wasserstress um Trockenheitsstress. Der Begriff Salzstress ist
nicht auf Natriumchlorid (NaCl) beschränkt, sondern es
kann sich dabei unter Anderem um eines der folgenden Salze handeln:
NaCl, KCl, LiCl, MgCl2, CaCl2.
-
Die
Durchführung der erfindungsgemäßen Verfahren
unter abiotischen Stressbedingungen ergibt Pflanzen mit verbesserten
Ertragsmerkmalen, vorzugsweise verbesserten Samenertragsmerkmalen,
im Vergleich zu Kontrollpflanzen, die unter vergleichbaren Stressbedingungen
gewachsen waren. Daher wird erfindungsgemäß ein
Verfahren zur Verbesserung der Ertragsmerkmale, vorzugsweise Samenertragsmerkmale
in Pflanzen, die unter abiotischen Stressbedingungen gewachsen waren,
bereitgestellt, wobei man bei dem Verfahren in einer Pflanze die
Expression einer Nukleinsäuresequenz, die ein PHDf-HD-Polypeptid
kodiert, moduliert, vorzugsweise erhöht. Gemäß einem
Aspekt der Erfindung ist der abiotische Stress ein osmotischer Stress,
ausgewählt aus einem oder mehreren der Folgenden: Wasserstress,
Salzstress, oxidativem Stress und ionenbedingtem Stress.
-
Ein
weiteres Beispiel für einen abiotischen Umweltstress ist
die reduzierte Verfügbarkeit von einem oder mehreren Nährstoffen,
die von der Pflanze für ihr Wachstum und ihre Entwicklung
assimiliert werden müssen. Aufgrund des starken Einflusses,
den die Nährstoffverwertungseffizienz einer Pflanze auf
den Ertrag und die Produktqualität ausübt, wird
eine enorm hohe Menge Dünger auf die Felder gestreut, um
das Pflanzenwachstum und die Pflanzenqualität zu optimieren.
Die Produktivität von Pflanzen wird üblicherweise
von den drei Hauptnährstoffen Phosphor, Kalium und Stickstoff
begrenzt, wobei von diesen drei Nährstoffen üblicherweise
der letztgenannte das limitierende Element der Pflanzenwachstumsrate
ist. Das wichtigste Nährstoffelement, das für
das Pflanzenwachstum erforderlich ist, ist daher der Stickstoff
(N). Er ist ein Bestandteil von zahlreichen wichtigen Verbindungen,
die man in lebenden Zellen findet, wie u. a. von Aminosäuren,
Proteinen (Enzymen), Nukleinsäuren und Chlorophyll. 1,5%
bis 2% der pflanzlichen Trockenmasse ist Stickstoff und ungefähr
16% des gesamten pflanzlichen Proteins. Die Verfügbarkeit
von Stickstoff ist daher ein wesentlicher limitierender Faktor für
das Kulturpflanzenwachstum und die Kulturpflanzenproduktion (Frink
et al. (1999), Proc. Natl. Acad. Sci. USA 96 (4): 1175–1180)
und hat auch eine wichtige Auswirkung auf die Proteinakkumulation und
auf die Aminosäurezusammensetzung. Kulturpflanzen, die
bei Wachstum unter Stickstoffmangelbedingungen verbesserte Ertragsmerkmale,
vorzugsweise verbesserte Samenertragsmerkmale, aufweisen, sind daher
von großem Interesse.
-
Die
Durchführung der erfindungsgemäßen Verfahren
ergibt Pflanzen, die bei Wachstum unter Nährstoffmangelbedingungen,
insbesondere unter Stickstoffmangelbedingungen, verbesserte Ertragsmerkmale, vorzugsweise
verbesserte Samenertragsmerkmale, im Vergleich zu unter vergleichbaren
Bedingungen gezüchteten Kontrollpflanzen aufweisen. Daher
wird erfindungsgemäß ein Verfahren zur Verbesserung
der Ertragsmerkmale, vorzugsweise Verbesserung der Samenertragsmerkmale,
bei Pflanzen bereitgestellt, die unter Nährstoffmangelbedingungen
gezüchtet wurden, wobei man bei dem Verfahren in einer
Pflanze die Expression einer Nukleinsäuresequenz, die ein
PHDf-HD-Polypeptid kodiert, moduliert, vorzugsweise steigert. Nährstoffmangel
kann sich unter Anderem aus einem Fehlen von Nährstoffen,
wie Stickstoff, Phosphaten und anderen phosphorhaltigen Verbindungen,
Kalium, Calcium, Cadmium, Magnesium, Mangan, Eisen und Bor, ergeben.
-
Die
vorliegende Erfindung umfasst Pflanzen oder Teile davon (einschließlich
Samen), die durch die erfindungsgemäßen Verfahren
erhältlich sind. Die Pflanzen oder Teile davon umfassen
ein Nukleinsäuretransgen, das ein PHDf-HD-Polypeptid wie
oben definiert, kodiert.
-
Die
Erfindung stellt auch Genkonstrukte und Vektoren bereit, um die
Einführung und/oder modulierte, vorzugsweise erhöhte
Expression von Nukleinsäuresequenzen, die PHDf-HD-Polypeptide
kodieren, in Pflanzen zu erleichtern. Die Genkonstrukte können
in handelsübliche Vektoren eingeführt werden,
die für die Transformation in Pflanzen und für
die Expression des interessierenden Gens in den transformierten
Zellen geeignet sind. Die Erfindung stellt auch die Verwendung eines
wie im vorliegenden Text definierten Genkonstrukts in den erfindungsgemäßen
Verfahren bereit.
-
Genauer
ausgedrückt stellt die vorliegende Erfindung ein Konstrukt
bereit, umfassend
- (a) eine Nukleinsäure,
die ein PHDf-HD-Polypeptid wie oben definiert kodiert;
- (b) eine oder mehrere Kontrollsequenzen, die die Expression
der Nukleinsäuresequenz gemäß (a) modulieren,
vorzugsweise steigern kann bzw. können; sowie gegebenenfalls
- (c) eine Transkriptionsterminationssequenz.
-
Vorzugsweise
ist die Nukleinsäuresequenz, die ein PHDf-HD-Polypeptid
kodiert, wie oben definiert. Der Begriff ”Kontrollsequenz” und ”Terminationssequenz” sind
wie hier definiert.
-
Eine
der Kontrollsequenzen eines Konstrukts ist vorzugsweise ein konstitutiver
Promotor, der aus einem Pflanzengenom isoliert wurde. Ein Beispiel
für einen pflanzlichen konstitutiven Promotor ist ein GOS2-Promotor,
vorzugsweise ein GOS2-Promotor aus Reis, stärker bevorzugt
ein GOS2-Promotor, der durch die SEQ ID NO: 235 veranschaulicht
ist.
-
Pflanzen
werden mit einem Vektor transformiert, der eine der vorstehend beschriebenen
Nukleinsäuresequenzen umfasst. Dem Fachmann sind die genetischen
Elemente, die auf einem Vektor zur erfolgreichen Transformation,
Selektion und Vermehrung der Wirtszellen, die die interessierende
Sequenz enthalten, vorhanden sein müssen, geläufig.
Die interessierende Sequenz ist funktionsfähig mit einer
oder mehreren Kontrollsequenzen (mindestens mit einem Promotor)
verknüpft.
-
Jede
Art von Promotor kann unabhängig davon, ob er natürlich
oder synthetisch ist, vorteilhafterweise zur Modulation, vorzugsweise
Steigerung, der Expression der Nukleinsäuresequenz, verwendet
werden. Ein konstitutiver Promotor ist bei den Verfahren besonders
geeignet, vorzugsweise ein aus einem Pflanzengenom isolierter konstitutiver
Promotor. Der konstitutive Pflanzenpromotor steuert die Expression
einer kodierenden Sequenz auf einem Niveau, das in allen Fällen
unter demjenigen ist, das unter der Kontrolle eines 35S CaMV-Promotors
erhalten wird.
-
Andere
organspezifische Promotoren, beispielsweise zur bevorzugten Expression
in Blättern, Stängeln, Knollen, Meristemen, Samen
(Embryo und/oder Endosperm), eignen sich zur Durchführung
der erfindungsgemäßen Verfahren. Für
die Definitionen der verschiedenen Promotortypen siehe Abschnitt ”Definitionen” im
vorliegenden Text.
-
Es
sollte klar sein, dass die Anwendbarkeit der vorliegenden Erfindung
nicht auf die PHDf-HD-Polypeptide-kodierende Nukleinsäuresequenz
eingeschränkt ist, die durch SEQ ID NO: 179 veranschaulicht
ist, entsprechend ist die Anwendbarkeit der vorliegenden Erfindung
nicht auf die Expression einer PHDf-HD-Polypeptid-kodierenden Nukleinsäuresequenz
eingeschränkt, wenn sie durch einen konstitutiven Promotor
gesteuert wird.
-
Bei
dem Konstrukt, das in eine Pflanze eingeführt wird, kann/können
gegebenenfalls eine oder mehrere Terminatorsequenzen verwendet werden.
Zusätzliche Regulationselemente können Transkriptionsenhancer
sowie Translationsenhancer umfassen. Der Fachmann ist mit Terminator-
und Enhancer-Sequenzen, die zur Durchführung der Erfindung
geeignet sein können, vertraut. Zur Steigerung der Menge
der reifen Botschaft, die sich im Cytosol anreichert, kann auch
eine Introsequenz zu der 5'-untranslatierten Region (UTR) oder in
der kodierenden Sequenz hinzugefügt werden, wie es im Abschnitt
Definitionen beschrieben wurde. Andere Kontrollsequenzen (neben
Promotor, Enhancer, Silencer, Intronsequenzen, 3'UTR- und/oder 5'UTR-Regionen)
können Protein- und/oder RNA-stabilisierende Elemente sein.
Solche Sequenzen sind dem Fachmann bekannt oder leicht zugänglich.
-
Die
erfindungsgemäßen Genkonstrukte können
weiterhin eine Replikationsursprungssequenz beinhalten, die zur
Aufrechterhaltung und/oder die Replikation in einem bestimmten Zelltyp
erforderlich ist. Ein Beispiel ist, wenn ein Genkonstrukt in einer
Bakterienzelle als episomales genetisches Element aufrechterhalten werden
muss (z. B. Plasmid- oder Cosmidmolekül). Zu bevorzugten
Replikationsursprüngen gehören der f1-ori und
colE1, sind jedoch nicht hierauf beschränkt.
-
Für
den Nachweis des erfolgreichen Transfers der Nukleinsäuresequenzen,
wie sie bei den erfindungsgemäßen Verfahren verwendet
wurden, und/oder für die Selektion der transgenen Pflanze,
die diese Nukleinsäuresequenzen umfassen, ist es vorteilhaft,
Markergene (oder Reportergene) zu verwenden. Das Genkonstrukt kann
daher gegebenenfalls ein Selektionsmarkergen umfassen. Selektionsmarker
sind genauer im Abschnitt ”Definitionen” im vorliegenden
Text beschrieben.
-
Es
ist bekannt, dass je nach dem verwendeten Expressionsvektor und
der verwendeten Transfektionstechnik bei der stabilen oder transienten
Integration von Nukleinsäuresequenzen in Pflanzenzellen
nur ein kleiner Teil der Zellen die Fremd-DNA aufnimmt und falls
gewünscht in ihr Genom integriert. Um diese Integranten
zu identifizieren und zu selektieren, wird üblicherweise
ein Gen, das einen Selektionsmarker (wie die oben beschriebenen
Selektionsmarker) kodiert, zusammen mit dem interessierenden Gen
in die Wirtszellen eingeführt. Diese Marker können
zum Beispiel in Mutanten, in denen diese Gene nicht funktionell
sind, verwendet werden, und zwar zum Beispiel durch Deletion nach
herkömmlichen Verfahren. Weiterhin können Nukleinsäuresequenzmoleküle,
die einen Selektionsmarker kodieren, in eine Wirtszelle auf demselben
Vektor eingeführt werden, der die Sequenz, die die erfindungsgemäßen
Polypeptide oder die Polypeptide, die bei den Verfahren der Erfindung
verwendet werden, umfasst, oder auch auf einen separaten Vektor.
Zellen, die mit der eingeführten Nukleinsäure
stabil transfiziert worden sind, können zum Beispiel durch
Selektion identifiziert werden (zum Beispiel Zellen, die den Selektionsmarker
integriert haben, überleben, während die anderen
Zellen absterben). Die Markergene können aus der transgenen
Zelle entfernt oder gespalten werden, sobald sie nicht mehr benötigt
werden. Techniken zur Entfernung des Markergens sind im Stand der
Technik bekannt, geeignete Techniken sind oben im Abschnitt Definitionen
beschrieben.
-
Die
Erfindung stellt auch ein Verfahren zur Produktion von transgenen
Pflanzen mit verbesserten Ertragsmerkmalen vorzugsweise verbesserten
Samenertragsmerkmalen gegenüber Kontrollpflanzen bereit,
wobei eine beliebige Nukleinsäuresequenz, die ein PHDf-HD-Polypeptid
wie oben definiert, in eine(r) Pflanze eingeführt und exprimiert
wird.
-
Insbesondere
stellt die vorliegende Erfindung ein Verfahren zur Produktion von
transgenen Pflanzen mit verbesserten Ertragsmerkmalen, vorzugsweise
verbesserten Samenertragsmerkmalen bereit, wobei das Verfahren umfasst:
- (i) Einbringen und Exprimieren einer PHDf-HD-Polypeptid-kodierenden
Nukleinsäuresequenz in eine(r) Pflanze oder Pflanzenzelle
unter der Kontrolle eines konstitutiven Pflanzenpromotors; und
- (ii) Züchten der Pflanzenzelle unter Bedingungen, die
das Pflanzenwachstum und die Entwicklung fördern.
-
Die
Nukleinsäure von (i) kann eine beliebige Nukleinsäure
sein, die ein PHDf-HD-Polypeptid wie hier definiert kodieren kann.
-
Die
Nukleinsäure kann in eine Pflanzenzelle oder in die Pflanze
selbst direkt eingeführt werden (was das Einführen
in ein Gewebe, Organ oder einen beliebigen anderen Teil einer Pflanze
beinhaltet). Gemäß einem bevorzugten Merkmal der
vorliegenden Erfindung wird die Nukleinsäure vorzugsweise
mittels Transformation in eine Pflanze eingeführt. Der
Begriff ”Transformation” wird im vorliegenden
Text im Abschnitt ”Definitionen” genauer beschrieben.
-
Die
genetisch modifizierten Pflanzenzellen können nach allen
Verfahren, mit denen der Fachmann vertraut ist, regeneriert werden.
Geeignete Verfahren finden sich in den oben genannten Veröffentlichungen von
S. D. Kung und R. Wu, Potrykus oder Höfgen und Willmitzer.
-
Im
Allgemeinen werden nach der Transformation die Pflanzenzellen oder
Zellgruppen auf das Vorhandensein von einem oder mehreren Markern
selektiert, die von den mit dem interessierenden Gen gemeinsam transferierten,
in Pflanzen exprimierbaren Genen kodiert werden, wonach das transformierte
Material zu einer ganzen Pflanze regeneriert wird. Für
die Selektion von transformierten Pflanzen wird das bei der Transformation
erhaltene Pflanzenmaterial im Allgemeinen Selektionsbedingungen
unterworfen, so dass man transformierte Pflanzen von untransformierten
Pflanzen unterscheiden kann. So können zum Beispiel Samen,
die auf die oben beschriebene Art und Weise erhalten wurden, ausgepflanzt
werden und nach einer anfänglichen Wachstumsphase durch
Spritzen einer geeigneten Selektion unterworfen werden. Eine weitere
Möglichkeit besteht darin, dass man die Samen, gegebenenfalls
nach Sterilisieren, auf Agarplatten heranzieht, wobei man ein geeignetes
Selektionsmittel verwendet, so dass nur die transformierten Samen
zu Pflanzen heranwachsen können. Alternativ werden die
transformierten Pflanzen auf das Vorhandensein eines Selektionsmarkers
wie die oben beschriebenen gescreent.
-
Nach
dem DNA-Transfer und der Regeneration können mutmaßlich
transformierte Pflanzen auch zum Beispiel unter Verwendung der Southern-Analyse
auf das Vorhandensein des interessierenden Gens, die Kopienzahl
und/oder die Genomorganisation ausgewertet werden. Alternativ dazu
oder zusätzlich können die Expressionsniveaus
der neu eingeführten DNA mittels Northern- und/oder Western-Analyse überwacht
werden; wobei beide Techniken dem Durchschnittsfachmann gut bekannt
sind.
-
Die
erzeugten transformierten Pflanzen können mit unterschiedlichen
Mitteln vermehrt werden, wie durch klonale Vermehrung oder durch
klassische Züchtungstechniken. So kann zum Beispiel eine
transformierte Pflanze der ersten Generation (T1-Pflanze) geselbstet
werden, und homozygote Transformanten der zweiten Generation (T2-Pflanze)
können selektiert werden, und die T2-Pflanzen können
dann mit Hilfe von klassischen Züchtungstechniken weiter
vermehrt werden. Die erzeugten transformierten Organismen können in
verschiedener Form vorliegen. Sie können beispielsweise
Chimären von transformierten Zellen und untransformierten
Zellen sein; klonale Transformanten (z. B. alle Zellen wurden dahingehend
transformiert, dass sie die Expressionskassette enthalten); Pfropfmaterial
von transformiertem und untransformiertem Gewebe (z. B. bei Pflanzen
ein transformierter Wurzelstock, der auf ein untransformiertes Edelreis
gepfropft wurde).
-
Die
vorliegende Erfindung erstreckt sich eindeutig auf eine jede Pflanzenzelle
oder Pflanze, die nach einem der hier beschriebenen Verfahren erzeugt
wurde, und auf sämtliche Pflanzenteile und sämtliches
Vermehrungsmaterial davon. Die vorliegende Erfindung umfasst weiterhin
die Nachkommenschaft einer/eines primär transformierten
oder transfizierten Zelle, Gewebes, Organs oder ganzen Pflanze,
die/das nach einem der oben genannten Verfahren erzeugt wurde, wobei
die einzige Voraussetzung ist, dass die Nachkommenschaft dasselbe/dieselben
genotypische(n) und/oder phänotypische(n) Merkmal(e) aufweist,
wie es/sie von dem Elter bei den erfindungsgemäßen
Verfahren gezeigt wird.
-
Die
Erfindung beinhaltet auch Wirtszellen, die eine isolierte Nukleinsäuresequenz,
die ein wie oben definiertes PHDf-HD-Polypeptid kodiert, enthalten,
die funktionsfähig mit einem konstitutiven Pflanzenpromotor verknüpft
ist. Bevorzugte erfindungsgemäße Wirtszellen sind
Pflanzenzellen. Wirtspflanzen für die bei dem erfindungsgemäßen
Verfahren verwendeten Nukleinsäuren oder den Vektor, für
die Expressionskassette oder das Konstrukt oder den Vektor, sind
im Prinzip vorteilhafterweise alle Pflanzen, die die bei dem erfindungsgemäßen
Verfahren verwendeten Polypeptide synthetisieren können.
-
Die
erfindungsgemäßen Verfahren lassen sich vorteilhaft
auf jede beliebige Pflanze anwenden. Zu den Pflanzen, die bei den
erfindungsgemäßen Verfahren besonders geeignet
sind, gehören alle Pflanzen, die zu der Superfamilie Viridiplantae
gehören, insbesondere monokotyle und dikotyle Pflanzen,
einschließlich Futter- oder Weidepflanzenleguminosen, Zierpflanzen,
Nahrungsmittelkulturpflanzen, Bäume oder Sträucher.
Gemäß einer bevorzugten Ausführungsform
der vorliegenden Erfindung handelt es sich bei der Pflanze um eine Kulturpflanze.
Zu den Kulturpflanzen gehören zum Beispiel Sojabohne, Sonnenblume,
Canola-Raps, Luzerne, Raps, Baumwolle, Tomate, Kartoffel und Tabak.
Weiter bevorzugt handelt es sich bei der Pflanze um eine monokotyle
Pflanze. Zu den monokotylen Pflanzen gehört zum Beispiel
Zuckerrohr. Weiter bevorzugt handelt es sich bei der Pflanze um
ein Getreide. Zu den Getreiden gehören zum Beispiel Reis,
Mais, Weizen, Gerste, Hirse, Roggen, Triticale, Sorghum und Hafer.
-
Die
Erfindung betrifft auch erntefähige Teile einer Pflanze,
die eine funktionsfähig mit einem konstitutiven Pflanzenpromotor
verknüpfte isolierte Nukleinsäuresequenz umfasst,
die eine PHDf-HD (wie vorstehend definiert) kodiert, wie zum Beispiel,
jedoch nicht ausschließlich, Samen, Blätter, Früchte,
Blüten, Stängel, Rhizome, Knollen und Zwiebeln.
Die Erfindung betrifft weiterhin Produkte, die von einem erntefähigen
Teil einer solchen Pflanze abstammen, vorzugsweise direkt abstammen,
wie trockene Pellets oder Pulver, Öl, Fett und Fettsäuren,
Stärke oder Proteine.
-
Gemäß einem
bevorzugten Merkmal der Erfindung handelt es sich bei der modulierten
Expression um erhöhte Expression. Verfahren zur Erhöhung
der Expression von Nukleinsäuren oder Genen oder Genprodukten,
sind im Fachgebiet gut beschrieben; und Beispiele sind im Abschnitt
Definitionen gegeben.
-
Wie
vorstehend erwähnt, ist ein bevorzugtes Verfahren zum Modulieren,
vorzugsweise Erhöhen, der Expression einer Nukleinsäuresequenz,
die ein PHDf-HD-Polypeptid kodiert, durch Einführen und
Exprimieren einer Nukleinsäuresequenz, die ein PHDf-HD-Polypeptid
kodiert, in eine(r) Pflanze; die Wirkungen der Durchführung
des Verfahrens, d. h. das Erhöhen der Ertragsmerkmale können
auch unter Verwendung anderer bekannter Techniken erzielt werden,
einschließlich, aber nicht beschränkt auf T-DNA-Activation-Tagging,
TILLING, homologe Rekombination. Eine Beschreibung dieser Techniken
findet sich im Abschnitt Definitionen.
-
Die
vorliegende Erfindung umfasst auch die Verwendung von Nukleinsäuresequenzen,
die PHDf-HD-Polypeptide, wie hier beschrieben, kodieren, und die
Verwendung dieser PHDf-HD-Polypeptide zur Verbesserung eines der
oben genannten Ertragsmerkmale, vorzugsweise Samenertragsmerkmale,
in Pflanzen.
-
Hier
beschriebene Nukleinsäuren, die das PHDf-HD-Polypeptid
kodieren, oder die PHDf-HD-Polypeptide selbst können Verwendung
in Züchtungsprogrammen finden, in denen ein DNA-Marker
identifiziert wird, der mit einem für ein PHDf-HD-Polpeptid
kodierenden Gen genetisch verknüpft sein kann. Die Gene/Nukleinsäuresequenzen
oder die PHDf-HD-Polypeptide selbst können dazu verwendet
werden, einen molekularen Marker zu bestimmen. Dieser DNA- oder
Protein-Marker kann dann in Züchtungsprogrammen zur Selektion von
Pflanzen mit erhöhten Ertragsmerkmalen, vorzugsweise erhöhten
Samenertragsmerkmalen, verwendet werden, wie hier unter den erfindungsgemäßen
Verfahren definiert.
-
Allelische
Varianten eines Gens/einer Nukleinsäure, das/die ein PHDf-HD-Polypeptid
kodiert, können auch Verwendung in Marker-unterstützten
Züchtungsprogrammen finden. Bei solchen Züchtungsprogrammen ist
manchmal das Einführen von allelischer Variation durch
Mutagenbehandlung der Pflanzen erforderlich, wobei zum Beispiel
EMS-Mutagenese verwendet wird; alternativ kann das Programm von
einer Sammlung von allelischen Varianten so genannten ”natürlichen” Ursprungs
ausgehen, die ohne Absicht hervorgerufen wurden. Dann erfolgt die
Identifikation allelischer Varianten, zum Beispiel mittels PCR.
Darauf folgt ein Schritt zur Selektion besserer allelischer Varianten
der in Frage kommenden Sequenz, die bessere Ertragsmerkmale liefern.
Die Selektion wird gewöhnlich durchgeführt, indem
man die Wachstumsleistung von Pflanzen verfolgt, die verschiedene
allelische Varianten der in Frage kommenden Sequenz enthalten. Die
Wachstumsleistung kann im Gewächshaus oder auf dem Feld
verfolgt werden. Weitere Schritte sind gegebenenfalls u. a. das
Kreuzen der Pflanzen, in denen die bessere allelische Variante identifiziert
wurde, mit einer anderen Pflanze. Dies kann zum Beispiel zur Herstellung
einer Kombination interessanter phänotypischer Merkmale
verwendet werden.
-
PHDf-HD-Polypeptide
kodierende Nukleinsäuresequenzen können auch als
Sonden zur genetischen und physikalischen Kartierung der Gene, von
denen sie ein Teil sind, und als Marker für Merkmale, die
mit diesen Genen verknüpft sind, verwendet werden. Diese
Information kann für die Pflanzenzüchtung nützlich
sein, um Linien mit gewünschten Phänotypen zu
entwickeln. Eine solche Verwendung der PHDf-HD-Polypeptid-kodierenden
Nukleinsäuresequenzen benötigt nur eine Nukleinsäuresequenz
von mindestens 15 Nukleotiden Länge. Die PHDf-HD-Polypeptid-kodierenden
Nukleinsäuresequenzen können als Restriktionsfragmentlängenpolymorphismus
(RFLP)-Marker verwendet werden. Southern-Blots (Sambrook
J, Fritsch EF and Maniatis T (1989) Molecular Cloning, A Laboratory
Manual) von restriktions-gespaltener genomischer Pflanzen-DNA kann
mit den PHDf-HD-kodierenden Nukleinsäuren sondiert werden.
Die erhaltenen Bandenmuster können dann genetischen Analysen
mit Computerprogrammen unterworfen werden, wie MapMaker (Lander
et al. (1987) Genomics 1: 174–181), so dass man
eine genetische Karte erhält. Die Nukleinsäuren
können zudem zum Sondieren von Southern-Blots verwendet
werden, die Restriktionsendonuklease behandelte genomische DNAs
von einem Satz von Individuen enthalten, die die Eltern und die
Nachfahren einer definierten genetischen Kreuzung repräsentieren.
Die Segregation der DNA-Polymorphismen wird festgestellt und zur
Berechnung der Position der PHDf-HD-Polypeptidkodierenden Nukleinsäure
in der genetischen Karte verwendet, die mit dieser Population erhalten
wurde (Botstein et al. (1980) Am. J. Hum. Genet. 32: 314–331).
-
Die
Herstellung und Verwendung von Sonden, die von Pflanzengenen stammen,
für die Verwendung bei der genetischen Kartierung ist in Bernatzky
und Tanksley (1986) Plant Mol. Biol. Reporter 4: 37–41 beschrieben.
Zahlreiche Veröffentlichungen beschreiben die genetische
Kartierung spezifischer cDNA-Klone, wobei die vorstehend erläuterte
Methodik oder Varianten davon verwendet werden. Für die
Kartierung können zum Beispiel F2-Inzuchtpopulationen,
Rückkreuzungspopulationen, zufallsgemäß gepaarte
Populationen, nahezu isogene Linien und andere Sätze von
Individuen verwendet werden. Diese Verfahren sind dem Fachmann sehr
geläufig.
-
Die
Nukleinsäuresequenzsonden können auch zur physikalischen
Kartierung (d. h. zum Anordnen von Sequenzen in physikalischen Karten;
siehe Hoheisel et al. In: Non-mammalian Genomic Analysis:
A Practical Guide, Academic Press 1996, S. 319–346 und
darin genannte Literaturstellen) verwendet werden.
-
In
einer anderen Ausführungsform können die Nukleinsäuresonden
zur Kartierung durch direkte Fluoreszenz-in-situ-Hybridisierung
(FISH) (Trask (1991) Trends Genet. 7: 149–154)
verwendet werden. Zwar bevorzugen derzeitige FISH-Kartierungsverfahren
die Verwendung großer Klone (mehrere kb bis mehrere Hundert
kb; siehe Laan et al. (1995) Genome Res. 5: 13–20),
aber durch Verbesserungen in der Empfindlichkeit kann die FISH-Kartierung
auch unter Verwendung kürzerer Sonden durchgeführt
werden.
-
Eine
Reihe von Verfahren auf Basis von Nukleinsäuresequenzamplifikation
für die genetische und physikalische Kartierung kann unter
Verwendung der Nukleinsäuresequenzen durchgeführt
werden. Beispiele sind u. a. die allelspezifische Amplifikation
(Kazazian (1989) J. Lab. Clin. Med 11: 95–96),
Polymorphismus PCR-amplifizierter Fragmente (CAPS; Sheffield
et al. (1993) Genomics 16: 325–332), allelspezifische
Ligation (Landegren et al. (1988) Science 241: 1077–1080),
Nukleotidverlängerungsreaktionen (Sokolov (1990)
Nucleic Acid Res. 18: 3671), Radiation-Hybrid-Kartierung
(Walter et al. (1997) Nat. Genet. 7: 22–28)
und Happy-Kartierung (Dear und Cook (1989) Nucleic Acid
Res. 17: 6795–6807). Bei diesen Verfahren wird
die Sequenz einer Nukleinsäure dazu verwendet, Primer-Paare
zu gestalten und zu erzeugen, die bei der Amplifikationsreaktion oder
Primerverlängerungsreaktionen verwendet werden. Die Gestaltung
dieser Primer ist dem Fachmann bekannt. Bei Verfahren, die eine
genetische Kartierung auf PCR-Basis einsetzen, kann es notwendig
sein, dass man DNA-Sequenz-Unterschiede zwischen den Eltern der
Kartierungskreuzung in dem Bereich identifiziert, welcher der vorliegenden
Nukleinsäuresequenz entspricht. Dies ist jedoch für
Kartierungsverfahren nicht allgemein erforderlich.
-
Die
erfindungsgemäßen Verfahren ergeben Pflanzen mit
verbesserten Ertragsmerkmalen, vorzugsweise verbesserten Samenertragsmerkmalen
wie hier vorstehend beschrieben. Diese Merkmale können
auch mit anderen ökonomisch vorteilhaften Merkmalen kombiniert
werden, wie weiteren Ertragsmerkmalen, Toleranz gegenüber
anderen abiotischen und biotischen Stressbedingungen, Merkmalen,
die verschiedene architektonische Merkmale und/oder biochemisch
und/oder physiologische Merkmale modifizieren.
-
V. bHLH11-ähnlich (basic Helix-Loop-Helix
11)
-
Es
wurde überraschend entdeckt, dass die Modulation der Expression
in einer Pflanze einer Nukleinsäure, die ein bHLH11-ähnliches
Polypeptid kodiert, Pflanzen ergibt, die verbesserte Ertragsmerkmale
im Vergleich zu Kontrollpflanzen aufweisen. Gemäß einer
ersten Ausführungsform stellt die vorliegende Erfindung ein
Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen bereit,
bei dem die Expression einer Nukleinsäure, die ein bHLH11-ähnliches
Polypeptid kodiert, in einer Pflanze moduliert wird.
-
Ein
bevorzugtes Verfahren zur Modulation (vorzugsweise Steigerung) der
Expression einer Nukleinsäure, die ein bHLH11-ähnliches
Polypeptid kodiert, erfolgt durch Einführen und Expression
einer Nukleinsäure, die ein bHLH11-ähnliches Polypeptid
kodiert, in eine(r) Pflanze.
-
Jede
Bezugnahme auf ein ”Protein, das sich in den erfindungsgemäßen
Verfahren eignet”, soll ein bHLH11-ähnliches Polypeptid
bedeuten, wie es hier definiert ist. Jede im erfindungsgemäßen
Zusammenhang erfolgende Bezugnahme auf eine ”Nukleinsäure,
die sich in den erfindungsgemäßen Verfahren eignet” soll eine
Nukleinsäure bedeuten, die ein solches bHLH11-ähnliches
Polypeptid kodieren kann. Die in eine Pflanze einzubringende (und
daher bei der Durchführung der erfindungsgemäßen
Verfahren geeignete) Nukleinsäure ist eine beliebige Nukleinsäure,
die den nachstehend beschriebenen Proteintyp kodiert, der auch als ”bHLH11-ähnliche
Nukleinsäure” oder” bHLH11-ähnliches
Gen” bezeichnet wird.
-
Ein ”bHLH11-ähnliches
Polypeptid” wie es hier definiert ist, betrifft ein Polypeptid,
das eine basische Domäne und danach eine HLH-Domäne
umfasst (HMMPFam PF00010, ProfileScan PS50888, SMART SM00353) wodurch
eine basische Helix-Loop-Helix-Domäne (bHLH) (Interpro
IPR001092) gebildet wird. Das bHLH11-ähnliche Polypeptid
umfasst vorzugsweise mindestens eine, vorzugsweise zwei, stärker
bevorzugt drei, am stärksten bevorzugt vier oder mehr der
folgenden Motive:
- Motiv 1 (SEQ ID NO: 246): (E/D)(D/S/E)(F/M)(L/F)(D/E/Q/L)(Q/H/E)
- Motiv 2 (SEQ ID NO: 247): RA(R/I/Q)RG(Q/H)ATDPHSIAER
- Motiv 3 (SEQ ID NO: 248): (M/I/V/L)(K/R)(A/S/Q/D/N)LQ(E/D/V)LVP
- Motiv 4 (SEQ ID NO: 249): (M/I)(L/I)DEI(I/V/L)(D/E/G)Y(V/L/I)(K/R)FL(Q/R)LQ(V/I)K
- Motiv 5 (SEQ ID NO: 250): (V/I)LSMSR(L/V)G
- Motiv 6 (SEQ ID NO: 251): V(A/V/L/I)(K/R)(L/M)(M/L)(E/D)(E/D/S/K/T)(D/N/S)(M/V/I)(G/T/I)XAMQ(Y/L/F)L
wobei
X eine beliebige Aminosäure sein kann, jedoch vorzugsweise
eine von S, T, A, M, K, N Motiv 7 (SEQ ID NO: 252): (M/V)(P/S)(I/V)(S/T/A)LA
-
Alternativ
hat das Homologon eines bHLH11-ähnlichen Proteins in steigender
Reihenfolge der Präferenz mindestens 25%, 26%, 27%, 28%,
29%, 30%, 31%, 32%, 33%, 34%, 35%, 36%, 37%, 38%, 39%, 40%, 41%,
42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, 50%, 51%, 52%, 53%, 54%,
55%, 56%, 57%, 58%, 59%, 60%, 61%, 62%, 63%, 64%, 65%, 66%, 67%,
68%, 69%, 70%, 71%, 72%, 73%, 74%, 75%, 76%, 77%, 78%, 79%, 80%,
81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%,
94%, 95%, 96%, 97%, 98%, oder 99% Gesamt-Sequenzidentität,
wie sie durch SEQ ID NO: 245 veranschaulicht sind, vorausgesetzt das
homologe Proteine umfasst die konservierten Motive wie oben erwähnt.
Die Gesamtsequenzidentität wird bestimmt mit einem allgemeinen
Alignment-Algorithmus, wie dem Algorithmus von Needleman Wunsch
in dem Programm GAP (GCG Wisconsin Package, Accelrys), vorzugsweise
mit Default-Parametern. Verglichen mit der Gesamt-Sequenzidentität
ist die Sequenzidentität im Allgemeinen höher,
wenn nur konservierte Domänen oder Motive berücksichtigt
werden. Die Sequenzkonservierung ist viel höher in dem
Bereich der bHLH Domäne (siehe Tabelle E3 in Beispiel 45
und 21). Daher ist die bHLH-Domäne
ein gutes Kriterium zur Definition der Gruppe der bHLH11-ähnlichen
Proteine. Das bHLH11-ähnliche Polypeptid umfasst die Sequenz
von Motiv 8 (SEQ ID NO: 253): SIAERLRRERIAERMRALQELVPNTNKTDRAVMLDEILDYVKFLRLQVKVL,
oder eine Sequenz, die in steigender Reihenfolge der Präferenz
mindestens 75%, 76%, 77%, 78%, 79%, 80%, 81%, 82%, 83%, 84%, 85%,
86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%,
oder 99% Sequenzidentität zu SEQ ID NO: 253 aufweist. Die
HLH-Domäne wie durch SMART bestimmt, überspannt
den Rest 132 bis 181 in SEQ ID NO: 245 und ist in Motiv 8 enthalten.
-
Die
Polypeptid-Sequenz, die bei der Verwendung in der Konstruktion eines
Stammbaums verwendet wird, wie er in der 22 veranschaulicht
ist, bildet eher in der Gruppe der bHLH11-ähnlichen Proteine
Cluster als mit anderen bHLH-Proteinen. Entsprechend bildet das
bHLH11-ähnliche Protein der Wahl eher Cluster in der Subgruppe
C, wenn ein Stammbaum gemäß 6 in Li
et al. (2006), konstruiert wird als mit irgendeiner anderen
Gruppe.
-
Die
Begriffe ”Domäne”, ”Signatur” und ”Motiv” sind
im vorliegenden Text in dem Abschnitt ”Definitionen” definiert.
Für die Identifikation von Domänen gibt es Spezialdatenbanken,
zum Beispiel SMART (Schultz et al. (1998), Proc. Natl. Acad.
Sci. USA 95, 5857–5864; Letunic et al.
(2002), Nucleic Acids Res. 30, 242–244, InterPro
(Mulder et al. (2003), Nucl. Acids Res. 31, 315–318,
Prosite (Sucher und Bairoch (1994), A generalized Profile
syntax for biomolecular sequences motifs and its function in automatic
sequence interpretation. (In) ISMB-94; Proceedings 2nd International
Conference an Intelligent Systems for Molecular Biology. Altman
R., Brutlag D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61,
AAAI Press, Menlo Park; Hulo et al., Nucl. Acids Res.
32: D134–D137, (2004), oder Pfam (Bateman
et al., Nucleic Acids Research 30 (1): 276–280 (2002).
Ein Satz Werkzeuge für die In-silico-Analyse von Proteinsequenzen
findet sich auf dem ExPASY-Proteomics-Server (Swiss Institute of
Bioinformatics (Gasteiger et al., ExPASy: the proteomics
server for in-depth Protein knowledge and analysis, Nucleic Acids
Res. 31: 3784–3788 (2003)). Domänen können
auch unter Verwendung von Routinetechniken, wie Sequenz-Alignment,
identifiziert werden.
-
Verfahren
für das Alignment von Sequenzen für Vergleichszwecke
sind in der Fachwelt gut bekannt, dazu zählen GAP, BESTFIT,
BLAST, FASIA und TFASTA. Bei GAP wird der Algorithmus von Needleman
und Wunsch ((1970), J. Mol. Biol. 48: 443–453)
verwendet, um das globale Alignment (d. h. das Alignment, das sich über
die vollständigen Sequenzen erstreckt) von zwei Sequenzen,
das die Anzahl der ”matches” maximiert und die
Anzahl der ”gaps” minimiert. Beim BLAST-Algorithmus
(Altschul et al., (1990), J. Mol. Biol. 215: 403–10)
wird die Sequenzidentität in Prozent berechnet, und es
wird eine statistische Analyse der Ähnlichkeit zwischen
den beiden Sequenzen durchgeführt. Die Software für
die Durchführung einer BLAST-Analyse ist der Öffentlichkeit über
das National Centre for Biotechnology Information (NCBI) zugänglich.
Homologa können leicht unter Verwendung von zum Beispiel
dem multiplen Sequenz-Alignment-Algorithmus ClustalW (Version 1.83)
identifiziert werden, und zwar mit den Default-Parametern für
paarweises Alignment und einer Scoring-Methode in Prozent. Die Gesamtprozentsätze
der Ähnlichkeit und der Identität können
auch unter Verwendung von einer der in dem MatGAT-Software-Paket
verfügbaren Methoden bestimmt werden ((Campanella
et al., BMC Bioinformatics. 2003, Juli, 10;4: 29. MatGAT: an application
that generstes similarity/identity matrices using Protein or DNA
sequences.). Für eine Optimierung des Alignments
zwischen konservierten Motiven können, wie es dem Fachmann
ersichtlich wird, kleine manuelle Veränderungen vorgenommen
werden. Darüber hinaus können statt Volllängensequenzen
für die Identifikation von Homologa auch spezifische Domänen
verwendet werden. Die Sequenzidentitätswerte können
mit den oben erwähnten Programmen unter Einstellung der
Default-Parameter über die gesamte Nukleinsäure-
oder Aminosäuresequenz oder über ausgewählte
Domänen oder konservierte Motiv(e) bestimmt werden. Für
lokale Alignments ist der Smith-Waterman-Algorithmus besonders geeignet
(Smith TF, Waterman MS (1981) J. Mol. Biol 147 (1); 195–7).
-
Die
bHLH11-ähnlichen Polypeptide (zumindest in ihrer nativen
Form) haben gewöhnlich DNA Bindungsaktivität.
Werkzeuge und Techniken zum Messen der DNA-Bindungsaktivität
sind im Stand der Technik bestens bekannt. Zudem ergibt wie erfindungsgemäß gezeigt
ein bHLH11-ähnliches Protein, wie SEQ ID NO: 245, bei Überexpression
in Reis, Pflanzen mit verbesserten Ertragsmerkmalen, insbesondere
gesteigerter Füllrate. Weitere Einzelheiten sind im Abschnitt
Beispiele gezeigt.
-
Die
vorliegende Erfindung wird anhand der Transformation von Pflanzen
mit der in SEQ ID NO: 244 gezeigten Nukleinsäuresequenz,
die die Polypeptidsequenz von SEQ ID NO: 245 kodiert, veranschaulicht.
Die Durchführung der Erfindung ist nicht auf diese Sequenzen
eingeschränkt; die erfindungsgemäßen
Verfahren können vorteilhafterweise durch Verwendung einer
beliebigen bHLH11-ähnlichen kodierenden Nukleinsäure oder
bHLH11-ähnlichen Polypeptid, wie hier definiert durchgeführt
werden.
-
Beispiele
für Nukleinsäuren, die bHLH11-ähnliche
Polypeptide kodieren, sind in der Tabelle E1 von Beispiel 43 hierin
beschrieben. Solche Nukleinsäuresequenzen eignen sich zur
Durchführung der erfindungsgemäßen Verfahren.
Die in Tabelle E1 von Beispiel 43 angegebenen Aminosäuresequenzen
sind Beispiel-Sequenzen für Orthologa und Paraloga des
durch SEQ ID NO: 245 veranschaulichten bHLH11-ähnlichen
Polypeptids, wobei die Begriffe ”Orthologa” und ”Paraloga” die
hier definierte Bedeutung haben. Weitere Orthologa und Paraloga
können leicht durch Durchführen einer so genannten
reziproken Blast-Suche identifiziert werden. Dies beinhaltet typischerweise
einen ersten BLAST, bei der mit einer Abfragesequenz (zum Beispiel
eine der in Tabelle E1 von Beispiel 43 aufgeführten Sequenzen)
ein ”BLASTing” gegen eine beliebige Sequenzdatenbank,
wie die öffentlich zugängliche NCBI-Datenbank,
durchgeführt wird. BLASTN oder TBLASTX (unter Verwendung
von Standard-Default-Werten) werden im Allgemeinen dann verwendet,
wenn man von einer Nukleotidsequenz ausgeht, und BLASTP oder TBLASTN
(unter Verwendung von Standard-Default-Werten), wenn man von einer
Proteinsequenz ausgeht. Die BLAST-Ergebnisse können gegebenenfalls
gefiltert werden. Mit den Volllängen-Sequenzen der gefilterten
oder ungefilterten Ergebnisse wird anschließend ein Rück-BLASTing
(zweiter BLAST) gegen Sequenzen des Organismus, von dem die Abfragesequenz
stammt, durchgeführt (ist die Abfragesequenz SEQ ID NO:
244 oder SEQ ID NO: 245, dann wäre das zweite BLASTing
daher gegen Triticum aestivum-Sequenzen). Die Ergebnisse des ersten
und des zweiten BLASTing werden dann verglichen. Ein Paralogon wird
dann identifiziert, wenn ein hochrangiger Hit von dem ersten BLASTing
von der gleichen Art ist, von der die Abfragesequenz stammt, in
diesem Fall führt ein Rück-BLASTing idealerweise
zu der Abfragesequenz unter den höchstrangigen Hits; ein
Orthologon wird dann identifiziert, wenn ein hochrangiger Hit in
dem ersten BLASTing nicht von der gleichen Art wie derjenigen, von
der die Abfragesequenz stammt, ist, und führt vorzugsweise
beim Rück-BLASTing dazu, dass die Abfragesequenz unter
den höchstrangigen Hits ist.
-
Hochrangige
Hits sind solche mit niedrigem E-Wert. Je niedriger der E-Wert,
desto signifikanter der ”Score” (anders ausgedrückt,
desto niedriger die Wahrscheinlichkeit, dass der Hit durch Zufall
gefunden wurde). Die Berechnung des E-Werts ist Stand der Technik.
Das ”Scoring” der Vergleiche erfolgt nicht nur
mittels E-Werten, sondern auch mit dem Prozentsatz der Identität.
Der Prozentsatz der Identität bezieht sich auf die Anzahl
der identischen Nukleotide (oder Aminosäuren) zwischen
den beiden verglichenen Nukleinsäure-(oder Polypeptid-)Sequenzen über
eine bestimmte Länge. Bei großen Familien kann
man ClustalW und anschließend einen Neighbour-Joining-Tree
verwenden, um die Cluster der verwandten Gene leichter sichtbar
zu machen und um Orthologa und Paraloga zu identifizieren.
-
Zur
Durchführung der erfindungsgemäßen Verfahren
können sich auch Nukleinsäurevarianten eignen. Beispiele
für solche Varianten umfassen die Nukleinsäuresequenzen,
die Homologa und Derivate von einer der in Tabelle E1 von Beispiel
43 hier angegebenen Polypeptid-Sequenzen kodieren, wobei die Begriffe ”Homologon” und ”Derivat” die
hier definierte Bedeutung haben. Für die erfindungsgemäßen
Verfahren eignen sich auch Nukleinsäuren, die Homologa
und Derivate von Orthologa oder Paraloga von einer der in Tabelle
E1 von Beispiel 43 hier angegebenen Aminosäuresequenzen
kodieren. Homologa und Derivate, die sich bei den erfindungsgemäßen
Verfahren eignen, weisen im Wesentlichen dieselbe biologische und
funktionelle Aktivität wie das unmodifizierte Protein,
von dem sie abstammen, auf.
-
Zu
weiteren Nukleinsäurevarianten, die sich zur Durchführung
der erfindungsgemäßen Verfahren eignen, gehören
Abschnitte von Nukleinsäuren, die bHLH11-ähnliche
Polypeptide kodieren, Nukleinsäuren, die mit Nukleinsäuren,
die bHLH11-ähnliche Polypeptide kodieren, hybridisieren,
Spleißvarianten von Nukleinsäuren, die bHLH11-ähnliche
Polypeptide kodieren, allelische Varianten von Nukleinsäuren,
die bHLH11-ähnliche Polypeptide kodieren, und Varianten
von Nukleinsäuren, die bHLH11-ähnliche Polypeptide
kodieren, die durch ”Gen-Shuffling” erhalten wurden.
Die Begriffe hybridisierende Sequenz, Spleißvariante, allelische
Variante und ”Gene-Shuffling” sind wie im vorliegenden
Text definiert.
-
Nukleinsäuren,
die bHLH11-ähnliche Polypeptide kodieren, müssen
nicht unbedingt Volllängennukleinsäuren sein,
da die Durchführung der erfindungsgemäßen
Verfahren nicht auf die Verwendung von Volllängen-Nukleinsäuresequenzen
angewiesen ist. Erfindungsgemäß wird daher ein
Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen bereitgestellt,
bei dem man in eine(r) Pflanze einen Abschnitt von einer der in
Tabelle E1 von Beispiel 43 angegebenen Nukleinsäuresequenzen
oder einen Abschnitt von einer Nukleinsäure, die ein Orthologon,
Paralogon oder Homologon von einer der in Tabelle E1 von Beispiel
43 angegebenen Nukleinsäuresequenzen kodiert, einführt
und exprimiert.
-
Ein
Abschnitt einer Nukleinsäure kann zum Beispiel dadurch
hergestellt werden, dass man eine oder mehrere Deletionen bei der
Nukleinsäure durchführt. Die Abschnitte können
in isolierter Form verwendet werden oder können mit anderen
kodierenden (oder nichtkodierenden) Sequenzen fusioniert werden,
um zum Beispiel ein Protein herzustellen, bei dem mehrere Aktivitäten
kombiniert sind. Bei der Fusion mit anderen kodierenden Sequenzen
kann das bei der Translation hergestellte Polypeptid größer
sein, als für den Proteinabschnitt vorhergesagt wurde.
-
Abschnitte,
die für die erfindungsgemäßen Verfahren
geeignet sind, kodieren für ein bHLH11-ähnliches
Polypeptid, wie hier definiert, und weisen im Wesentlichen dieselbe
biologische Aktivität wie die in Tabelle E1 von Beispiel
43 angegebenen Aminosäuresequenzen auf. Vorzugsweise handelt
es sich bei dem Abschnitt um einen Abschnitt von einer der in Tabelle
E1 von Beispiel 43 genannten Nukleinsäuren oder um einen
Abschnitt einer Nukleinsäure, die ein Ortholog oder Paralog
von einer der in Tabelle E1 von Beispiel 43 genannten Aminosäuresequenzen
kodiert. Vorzugsweise ist der Abschnitt mindestens 500, 550, 600,
650, 700, 750, 800, 850, 900, 950, 1000, 1100, 1200, 1300, 1400,
1500, 1600, 1700, 1800, 1900, 2000, 2100, 2200, 2300, 2400, 2500
aufeinander folgende Nukleotide lang, wobei es sich bei den aufeinander
folgenden Nukleotiden um eine der in Tabelle E1 von Beispiel 43
genannten Nukleinsäuresequenzen oder um eine Nukleinsäure
handelt, die ein Ortholog oder Paralog von einer der in Tabelle
E1 von Beispiel 43 genannten Aminosäuresequenzen kodiert.
Am stärksten bevorzugt ist der Abschnitt ein Abschnitt
der Nukleinsäure gemäß SEQ ID NO: 244.
Vorzugsweise kodiert der Abschnitt ein Fragment von einer Aminosäuresequenz,
die, wenn sie zur Konstruktion eines Stammbaums, wie dem in 22 dargestellten, verwendet wird, eher innerhalb
der Gruppe der bHLH11-ähnlichen Polypeptide als mit anderen
bHLH-Proteinen Cluster bildet. Ebenso bildet das gewählte bHLH11-ähnliche
Protein eher innerhalb der Subgruppe C als mit irgendeiner anderen
Gruppe Cluster, wenn ein Baum gemäß 6 in Li
et al. (2006) konstruiert wird.
-
Eine
andere Nukleinsäurevariante, die für die erfindungsgemäßen
Verfahren geeignet ist, ist eine Nukleinsäure, die unter
Bedingungen einer verringerten Stringenz, vorzugsweise unter stringenten
Bedingungen, mit einer Nukleinsäure, die ein bHLH11-ähnliches
Polypeptid kodiert, wie hier definiert, oder mit einem Abschnitt,
wie hier definiert, hybridisieren kann.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Nukleinsäure,
die mit einer der in Tabelle E1 von Beispiel 43 angegebenen Nukleinsäuren
hybridisieren kann, einführt und exprimiert, oder bei dem
man in eine(r) Pflanze eine Nukleinsäure, die mit einer
Nukleinsäure hybridisieren kann, die ein Ortholog, Paralog
oder Homolog von einer der in Tabelle E1 von Beispiel 43 genannten
Nukleinsäuresequenzen kodiert, einführt und exprimiert.
-
Hybridisierende
Sequenzen, die für die erfindungsgemäßen
Verfahren geeignet sind, kodieren ein bHLH11-ähnliches
Polypeptid, wie hier definiert, das im Wesentlichen dieselbe biologische
Aktivität wie die in Tabelle E1 von Beispiel 43 genannten
Aminosäuresequenzen aufweist. Vorzugsweise kann die hybridisierende
Sequenz mit einer der in Tabelle E1 von Beispiel 43 genannten Nukleinsäuren
oder mit einem Abschnitt von einer dieser Sequenzen hybridisieren,
wobei ein Abschnitt wie oben definiert ist, oder wobei die hybridisierende Sequenz
mit einer Nukleinsäuresequenz, die ein Ortholog oder Paralog
von einer der in Tabelle E1 von Beispiel 43 genannten Aminosäuresequenzen
kodiert, hybridisieren kann. Am stärksten bevorzugt kann
die hybridisierende Sequenz mit einer Nukleinsäure gemäß SEQ
ID NO: 244 oder einem Abschnitt davon hybridisieren.
-
Vorzugsweise
kodiert die hybridisierende Sequenz ein Polypeptid mit einer Aminosäuresequenz,
die in voller Länge, wenn sie zur Konstruktion eines Stammbaums,
wie dem in 22 dargestellten, verwendet wird,
eher innerhalb der Gruppe der bHLH11-ähnlichen Polypeptide
als mit anderen bHLH-Proteinen Cluster bildet. Ebenso bildet das
gewählte bHLH11-ähnliche Protein eher innerhalb
der Subgruppe C als mit irgendeiner anderen Gruppe Cluster, wenn
ein Baum gemäß 6 in Li
et al. (2006) konstruiert wird.
-
Eine
andere Nukleinsäurevariante, die für die erfindungsgemäßen
Verfahren geeignet ist, ist eine Spleißvariante, die ein
bHLH11-ähnliches Polypeptid, wie hier vorstehend definiert,
kodiert, wobei eine Spleißvariante wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Spleißvariante
von einer der in Tabelle E1 von Beispiel 43 angegebenen Nukleinsäuresequenzen
oder eine Spleißvariante von einer Nukleinsäure,
die ein Ortholog, Paralog oder Homolog von einer der in Tabelle
E1 von Beispiel 43 genannten Aminosäuresequenzen kodiert,
einführt und exprimiert.
-
Bevorzugte
Spleißvarianten sind Spleißvarianten einer Nukleinsäure
gemäß SEQ ID NO: 244 oder eine Spleißvariante
einer Nukleinsäure, die ein Ortholog oder Paralog der SEQ
ID NO: 245 kodiert. Vorzugsweise bildet die Aminosäuresequenz,
die von der Spleißvariante kodiert wird, wenn sie zur Konstruktion
eines Stammbaums, wie dem in 22 dargestellten,
verwendet wird, eher innerhalb der Gruppe der bHLH11-ähnlichen
Polypeptide als mit anderen bHLH-Proteinen Cluster. Ebenso bildet
das gewählte bHLH11-ähnliche Protein eher innerhalb
der Subgruppe C als mit irgendeiner anderen Gruppe Cluster, wenn
ein Baum gemäß 6 in Li
et al. (2006) konstruiert wird.
-
Eine
andere Nukleinsäurevariante, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet ist,
ist eine allelische Variante einer Nukleinsäure, die ein
bHLH11-ähnliches Polypeptid, wie hier vorstehend definiert,
kodiert, wobei eine allelische Variante wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine allelische Variante
von einer der in Tabelle E1 von Beispiel 43 angegebenen Nukleinsäuresequenzen
einführt und exprimiert oder bei dem man in eine(r) Pflanze
eine allelische Variante einer Nukleinsäure, die ein Ortholog, Paralog
oder Homolog von einer der in Tabelle E1 von Beispiel 43 genannten
Aminosäuresequenzen kodiert, einführt und exprimiert.
-
Die
von allelischen Varianten, die für die erfindungsgemäßen
Verfahren geeignet sind, kodierten Polypeptide haben im Wesentlichen
dieselbe biologische Aktivität wie das bHLH11-ähnliche
Polypeptid gemäß SEQ ID NO: 244 und eine der in
Tabelle E1 von Beispiel 43 dargestellten Aminosäuren. Allelische
Varianten kommen in der Natur vor und von den erfindungsgemäßen
Verfahren wird die Verwendung dieser natürlichen Allele
mit umfasst. Vorzugsweise handelt es sich bei der allelischen Variante
um eine allelische Variante gemäß SEQ ID NO: 244
oder eine allelische Variante einer Nukleinsäure, die ein
Ortholog oder Paralog der SEQ ID NO: 245 kodiert. Vorzugsweise bildet
die Aminosäuresequenz, die von der allelischen Variante
kodiert wird, wenn sie zur Konstruktion eines Stammbaums, wie dem
in 22 dargestellten, verwendet wird, eher innerhalb
der Gruppe der bHLH11-ähnlichen Polypeptide als mit anderen
bHLH-Proteinen Cluster. Ebenso bildet das gewählte bHLH11-ähnliche
Protein eher innerhalb der Subgruppe C als mit irgendeiner anderen
Gruppe Cluster, wenn ein Baum gemäß 6 in Li
et al. (2006) konstruiert wird.
-
Man
kann auch ”Gene shuffling” oder gerichtete Evolution
dazu verwenden, Varianten von Nukleinsäuren zu erzeugen,
die bHLH11-ähnliche Polypeptide, wie oben definiert, kodieren,
wobei der Begriff ”gene shuffling” wie hier definiert
ist.
-
Erfindungsgemäß wird
ein Verfahren für die Verbesserung von Ertragsmerkmalen
bei Pflanzen bereitgestellt, bei dem man in eine(r) Pflanze eine
Variante von einer der in Tabelle E1 von Beispiel 43 angegebenen Nukleinsäuresequenzen
einführt und exprimiert oder bei dem man in eine(r) Pflanze
eine Variante einer Nukleinsäure, die ein Ortholog, Paralog
oder Homolog von einer der in Tabelle E1 von Beispiel 43 angegebenen Aminosäuresequenzen
kodiert, einführt und exprimiert, wobei die Nukleinsäurevariante
mittels ”gene shuffling” erhalten wird.
-
Vorzugsweise
bildet die Aminosäuresequenz, die von der durch gene shuffling
erhaltenen Nukleinsäurevariante kodiert wird, wenn sie
zur Konstruktion eines Stammbaums, wie dem in 22 dargestellten, verwendet wird, eher innerhalb
der Gruppe der bHLH11-ähnlichen Polypeptide als mit anderen
bHLH-Proteinen Cluster. Ebenso bildet das gewählte bHLH11-ähnliche
Protein eher innerhalb der Subgruppe C als mit irgendeiner anderen
Gruppe Cluster, wenn ein Baum gemäß 6 in Li
et al. (2006) konstruiert wird.
-
Nukleinsäurevarianten
können weiterhin auch durch ortsgerichtete Mutagenese erhalten
werden. Zur Erzielung einer ortsgerichteten Mutagenese sind verschiedene
Verfahren verfügbar, von denen die häufigsten Verfahren
auf PCR-Basis sind (Current Protocols in Molecular Biology.
Wiley Hrsg.).
-
Nukleinsäuren,
die bHLH11-ähnliche Polypeptide kodieren, können
von einer beliebigen natürlichen oder künstlichen
Quelle stammen. Die Nukleinsäure kann von ihrer nativen
Form in ihrer Zusammensetzung und/oder genomischen Umgebung durch
gezielte Manipulation durch den Menschen modifiziert werden. Vorzugsweise
stammt die Nukleinsäure, die das bHLH11-ähnliche
Polypeptid kodiert, aus einer Pflanze, weiter bevorzugt aus einer
einkeimblättrigen Pflanze, stärker bevorzugt aus
der Familie Poaceae, am stärksten bevorzugt stammt die
Nukleinsäure aus Triticum aestivum.
-
Die
Durchführung der erfindungsgemäßen Verfahren
führt zu Pflanzen mit verstärkten Ertragsmerkmalen.
Insbesondere führt die Durchführung der erfindungsgemäßen
Verfahren zu Pflanzen mit erhöhtem Ertrag, insbesondere
erhöhtem Samenertrag, im Vergleich zu Kontrollpflanzen.
Die Begriffe ”Ertrag” und ”Samenertrag” werden
hier im Abschnitt ”Definitionen” genauer beschrieben.
-
Werden
hier verbesserte Ertragsmerkmale erwähnt, so soll dies
eine Erhöhung der Biomasse (des Gewichts) von einem oder
mehreren Teilen einer Pflanze bedeuten, was oberirdische (erntbare)
Teile und/oder unterirdische (erntbare) Teile beinhalten kann. Insbesondere
sind solche oberirdischen erntbaren Teile Samen, und die Durchführung
der erfindungsgemäßen Verfahren führt
zu Pflanzen mit höherem Samenertrag im Vergleich zu dem
Samenertrag von Kontrollpflanzen.
-
Wählt
man Mais als Beispiel, kann sich eine Ertragserhöhung als
eines oder mehrere der folgenden Merkmale äußern:
Unter anderem erhöhte Anzahl an Pflanzen, die pro Quadratmeter
wachsen, eine Erhöhung der Anzahl an Ähren pro
Pflanze, eine Erhöhung der Anzahl an Reihen, der Anzahl
an Körnern pro Reihe, des Korngewichts, Tausendkorngewichts,
der Ährenlänge/des Ährendurchmessers,
eine Erhöhung der Samenfüllungsrate (d. h. der
Anzahl gefüllter Samen dividiert durch die Gesamtzahl der
Samen und multipliziert mit 100). Wählt man Reis als Beispiel,
kann sich eine Ertragserhöhung als Erhöhung bezüglich
eines oder mehrerer der folgenden Merkmale ausdrücken:
Unter anderem Anzahl der Pflanzen pro Quadratmeter, Anzahl der Rispen pro
Pflanze, Anzahl der Ährchen pro Rispe, Anzahl der Blüten
(Einzelblüten) pro Rispe (ausgedrückt als Verhältnis
der Anzahl an gefüllten Samen zu der Anzahl an Primärrispen),
erhöhte Samenfüllungsrate (d. h. der Anzahl gefüllter
Samen dividiert durch die Gesamtzahl an Samen und multipliziert
mit 100), erhöhtes Tausendkorngewicht.
-
Die
vorliegende Erfindung stellt ein Verfahren zur Erhöhung
des Ertrags, insbesondere des Samenertrags von Pflanzen, verglichen
mit Kontrollpflanzen bereit, wobei das Verfahren die Modulation
der Expression einer Nukleinsäure, die ein bHLH11-ähnliches
Polypeptid, wie hier definiert, kodiert, in einer Pflanze umfasst.
-
Weil
die erfindungsgemäßen transgenen Pflanzen einen
erhöhten Ertrag aufweisen, ist es wahrscheinlich, dass
diese Pflanzen (zumindest während eines Teils ihres Lebenszyklus)
eine höhere Wachstumsrate aufweisen als Kontrollpflanzen
in einem entsprechenden Stadium ihres Lebenszyklus.
-
Die
erhöhte Wachstumsrate kann für einen oder mehrere
Teile einer Pflanze (darunter Samen) spezifisch sein oder kann im
Wesentlichen in der ganzen Pflanze stattfinden. Pflanzen mit einer
erhöhten Wachstumsrate können einen kürzeren
Lebenszyklus aufweisen. Unter dem Lebenszyklus einer Pflanze kann
man diejenige Zeit verstehen, die die Pflanze benötigt,
um von einem trockenen reifen Samen bis zu dem Stadium heranzuwachsen,
in dem die Pflanze trockene reife Samen ähnlich dem Ausgangsmaterial
produziert hat. Dieser Lebenszyklus kann von Faktoren, wie Jungpflanzenvitalität,
Wachstumsrate, ”Greenness Index”, Blütezeit und
Geschwindigkeit der Samenreifung, beeinflusst werden. Die Erhöhung
der Wachstumsrate kann in einem Stadium oder in mehreren Stadien
im Lebenszyklus einer Pflanze oder im Wesentlichen während
des gesamten Lebenszyklus der Pflanze erfolgen. Eine erhöhte
Wachstumsrate während der Frühstadien im Lebenszyklus
einer Pflanze kann verbesserte Vitalität widerspiegeln.
Die erhöhte Wachstumsrate kann den Erntezyklus einer Pflanze
verändern, so dass die Pflanzen später gesät
werden können und/oder früher geerntet werden können
als dies sonst möglich wäre (ein ähnlicher
Effekt lässt sich mit einer früheren Blütezeit
erzielen). Ist die Wachstumsrate ausreichend erhöht, kann
dies weiteres Aussäen von Samen derselben Pflanzenart ermöglichen
(zum Beispiel Aussäen und Ernten von Reispflanzen und anschließendes
Aussäen und Ernten von weiteren Reispflanzen sogar innerhalb
einer herkömmlichen Wachstumsperiode). Wenn die Wachstumsrate
ausreichend erhöht ist, kann dies ebenso das weitere Aussäen
von Samen anderer Pflanzenarten ermöglichen (zum Beispiel
das Aussäen und Ernten von Maispflanzen und anschließend
zum Beispiel Aussäen und gegebenenfalls Ernten von Sojabohne,
Kartoffel oder einer anderen geeigneten Pflanze). Auch mehrmalige
zusätzliche Ernten von demselben Wurzelstock können
bei einigen Kulturpflanzen möglich sein. Eine Veränderung des
Erntezyklus einer Pflanze kann zu einer Erhöhung der jährlichen
Biomasseproduktion pro Quadratmeter führen (weil man eine
bestimmte Pflanze öfter (z. B. pro Jahr) heranziehen und
ernten kann). Eine erhöhte Wachstumsrate kann auch den
Anbau transgener Pflanzen in einem breiteren geographischen Bereich
als ihre Wildtyp-Gegenstücke ermöglichen, da die
räumlichen Beschränkungen für den Anbau
einer Kultur häufig von ungünstigen Umweltbedingungen
entweder während der Pflanzzeit (früh in der Saison)
oder während der Erntezeit (spät in der Saison)
bestimmt werden. Solche ungünstigen Bedingungen können
vermieden werden, wenn der Erntezyklus verkürzt ist. Die
Wachstumsrate kann durch Ableiten verschiedener Parameter aus Wachstumskurven
bestimmt werden, wobei die Parameter u. a. folgende sein können:
T-Mid (Zeitdauer, die die Pflanzen benötigen, um 50% ihrer
Maximalgröße zu erreichen) und T-90 (Zeitdauer,
die die Pflanzen benötigen, um 90% ihrer Maximalgröße
zu erreichen).
-
Gemäß einem
bevorzugten Merkmal der vorliegenden Erfindung führt die
Durchführung der erfindungsgemäßen Verfahren
zu Pflanzen mit einer im Vergleich zu Kontrollpflanzen erhöhten
Wachstumsrate. Erfindungsgemäß wird daher ein
Verfahren zur Erhöhung der Wachstumsrate von Pflanzen bereitgestellt,
wobei das Verfahren die Modulation der Expression einer Nukleinsäure,
die ein bHLH11-ähnliches Polypeptid, wie hier definiert,
kodiert, in einer Pflanze umfasst.
-
Eine
Erhöhung des Ertrags und/oder der Wachstumsrate tritt unabhängig
davon auf, ob die Pflanze unter Nichtstressbedingungen steht oder
ob die Pflanze im Vergleich zu Kontrollpflanzen verschiedenen Stressbedingungen
unterworfen wird. Typischerweise reagieren Pflanzen auf Stress dadurch,
dass sie langsamer wachsen. Unter starken Stressbedingungen kann
die Pflanze sogar ihr Wachstum völlig einstellen. Leichter
Stress ist dagegen hier als jeglicher Stress definiert, dem eine
Pflanze ausgesetzt ist und der nicht dazu führt, dass die
Pflanze völlig aufhört zu wachsen, ohne dass sie
die Fähigkeit besitzt, ihr Wachstum wieder aufzunehmen.
Leichter Stress im Sinne der Erfindung führt zu einer Verringerung
des Wachstums der gestressten Pflanzen von weniger als 40%, 35%
oder 30%, vorzugsweise weniger als 25%, 20% oder 15%, stärker
bevorzugt weniger als 14%, 13%, 12%, 11% oder 10% oder weniger im
Vergleich zu der Kontrollpflanze unter Nichtstressbedingungen. Aufgrund
von Fortschritten bei den landwirtschaftlichen Praktiken (Bewässerung,
Düngung, Pestizidbehandlungen) findet man bei angebauten
Kulturpflanzen nicht oft starke Stressbedingungen. Daher ist das
durch leichten Stress induzierte geschwächte Wachstum häufig
ein unerwünschtes Merkmal in der Landwirtschaft. Leichte
Stressbedingungen sind die alltäglichen biotischen und/oder
abiotischen (Umwelt-)Stressbedingungen, denen eine Pflanze ausgesetzt
ist. Abiotischer Stress kann durch Trockenheit oder Wasserüberschuss,
anaeroben Stress, Salzstress, chemische Toxizität, oxidativen
Stress und heiße, kalte oder Gefriertemperaturen verursacht
werden. Bei dem abiotischen Stress kann es sich um einen osmotischen
Stress handeln, der durch Wasserstress (insbesondere aufgrund von
Trockenheit), Salzstress, oxidativen Stress oder ionenbedingten
Stress verursacht wird. Unter biotischem Stress versteht man gewöhnlich
Stressbedingungen, die durch Pathogene, wie Bakterien, Viren, Pilze,
Nematoden und Insekten, verursacht werden.
-
Insbesondere
können die erfindungsgemäßen Verfahren
unter Nichtstressbedingungen oder unter leichten Trockenheitsbedingungen
durchgeführt werden, wobei man Pflanzen mit einem im Vergleich
zu Kontrollpflanzen erhöhten Ertrag erhält. Wie
von Wang et al. (Planta (2003), 218: 1–14)
beschrieben, führt abiotischer Stress zu einer Reihe von
morphologischen, physiologischen, biochemischen und molekularen
Veränderungen, die das Pflanzenwachstum und die Pflanzenproduktivität
negativ beeinflussen. Es ist bekannt, dass Trockenheit, Salinität,
extreme Temperaturen und oxidativer Stress miteinander in Verbindung
stehen und über ähnliche Mechanismen Wachstums-
und Zellschäden induzieren können. Rabbani
et al. (Plant Physiol. (2003), 133: 1755–1767)
beschreibt ein besonders hohes Ausmaß an gegenseitiger
Beeinflussung (”Cross-Talk”) von Trockenheitsstress
und durch hohe Salinität verursachtem Stress. Zum Beispiel äußern
sich Trockenheit und/oder Versalzung in erster Linie als osmotischer
Stress, was zur Störung der Homöostase und der
Ionenverteilung in der Zelle führt. Oxidativer Stress,
der häufig hohe oder niedrige Temperatur, Salinität
oder Trockenheitsstress begleitet, kann zur Denaturierung funktioneller
und struktureller Proteine führen. Infolgedessen aktivieren
diese verschiedenen Umweltstressbedingungen häufig ähnliche
Zellsignalwege und Zellreaktionen, wie die Produktion von Stressproteinen,
die Hinaufregulation von Antioxidantien, die Akkumulation von kompatiblen
gelösten Stoffen und ein Einstellen des Wachstums. Der
Begriff ”Nichtstress”-Bedingungen, wie hier verwendet,
steht für diejenigen Umweltbedingungen, die ein optimales
Wachstum von Pflanzen gestatten. Der Fachmann ist mit normalen Bodenbedingungen
und Klimabedingungen für einen bestimmten Standort vertraut.
Pflanzen unter optimalen Wachstumsbedingungen (die unter Nichtstressbedingungen
herangezogen werden) liefern oft einen Ertrag von, in der Reihenfolge
der zunehmenden Präferenz, mindestens 90%, 87%, 85%, 83%,
80%, 77% oder 75% der durchschnittlichen Produktion einer solchen
Pflanze in einer gegebenen Umgebung. Die durchschnittliche Produktion
kann bezogen auf die Ernte und/oder die Saison berechnet werden.
Der Fachmann ist mit den durchschnittlichen Erträgen einer
Kultur vertraut.
-
Die
Durchführung der erfindungsgemäßen Verfahren
verleiht Pflanzen, die unter Nichtstressbedingungen oder unter leichten
Trockenheitsbedingungen wachsen, einen erhöhten Ertrag
im Vergleich zu Kontrollpflanzen, die unter vergleichbaren Bedingungen
herangezogen werden. Erfindungsgemäß wird daher
ein Verfahren zur Erhöhung des Ertrags von Pflanzen, die
unter Nichtstressbedingungen oder unter leichten Trockenheitsbedingungen
herangezogen werden, bereitgestellt, wobei das Verfahren die Modulation
der Expression einer Nukleinsäure, die ein bHLH11-ähnliches
Polypeptid kodiert, in einer Pflanze umfasst.
-
Die
Durchführung der erfindungsgemäßen Verfahren
verleiht Pflanzen, die unter Nährstoffmangelbedingungen,
insbesondere unter Stickstoffmangelbedingungen, herangezogen werden,
einen erhöhten Ertrag im Vergleich zu Kontrollpflanzen,
die unter vergleichbaren Bedingungen herangezogen werden. Erfindungsgemäß wird
daher auch ein Verfahren zur Erhöhung des Ertrags in Pflanzen,
die unter Nährstoffmangelbedingungen herangezogen werden,
bereitgestellt, wobei man bei dem Verfahren die Expression einer
Nukleinsäure, die ein bHLH11-ähnliches Polypeptid
kodiert, in einer Pflanze moduliert, vorausgesetzt, dass es sich
bei dem Nährstoffmangel nicht um einen Phosphatmangel handelt.
Ein Nährstoffmangel kann durch ein Fehlen von Nährstoffen,
wie unter anderem Stickstoff, Phosphate und andere phosphorhaltige
Verbindungen, Kalium, Calcium, Cadmium, Magnesium, Mangan, Eisen
und Bor, hervorgerufen werden. Der Begriff ”Nährstoffmandel”, wie
er im Kontext der vorliegenden Erfindung verwendet wird, umfasst
jedoch keinen Phosphatmangel.
-
Die
vorliegende Erfindung umfasst Pflanzen oder Teile davon (einschließlich
Samen), die durch die erfindungsgemäßen Verfahren
erhältlich sind. Die Pflanzen oder Teile davon umfassen
ein Nukleinsäuretransgen, das ein bHLH11-ähnliches
Polypeptid kodiert, wie oben definiert.
-
Die
Erfindung stellt auch Genkonstrukte und Vektoren bereit, um die
Einführung und/oder Expression von Nukleinsäuren,
die für bHLH11-ähnliche Polypeptide kodieren,
in Pflanzen zu erleichtern. Die Genkonstrukte können in
Vektoren eingefügt werden, die im Handel erhältlich
sein können und die für die Transformation des
interessierenden Gens in Pflanzen und für die Expression
des interessierenden Gens in den transformierten Zellen geeignet
sind. Die Erfindung stellt auch die Verwendung eines Genkonstrukts,
wie hier definiert, bei den erfindungsgemäßen
Verfahren bereit.
-
Genauer
gesagt, stellt die vorliegende Erfindung ein Konstrukt bereit, das
folgendes umfasst:
- (a) eine Nukleinsäure,
die ein bHLH11-ähnliches Polypeptid kodiert, wie oben definiert;
- (b) eine oder mehrere Kontrollsequenzen, die die Expression
der Nukleinsäuresequenz unter (a) vorantreiben können,
und gegebenenfalls
- (c) eine Transkriptionsterminationssequenz.
-
Vorzugsweise
ist die Nukleinsäure, die ein bHLH11-ähnliches
Polypeptid kodiert, wie vorstehend definiert. Die Begriffe ”Kontrollsequenz” und ”Terminationssequenz” sind
wie hier definiert.
-
Pflanzen
werden mit einem Vektor transformiert, der eine der oben beschriebenen
Nukleinsäuren umfasst. Der Fachmann kennt die genetischen
Elemente, die in einem Vektor vorhanden sein müssen, damit Wirtszellen
erfolgreich transformiert, Wirtszellen, die die interessierende
Sequenz enthalten, erfolgreich selektiert und vermehrt werden können.
Die interessierende Sequenz ist mit einer oder mehreren Kontrollsequenzen
(mindestens mit einem Promoter) funktionsfähig verbunden.
-
Jede
Art von Promoter, ob natürlich oder synthetisch, kann vorteilhaft
für das Vorantreiben der Expression der Nukleinsäuresequenz
verwendet werden, aber vorzugsweise stammt der Promotor aus einer
Pflanze. Ein konstitutiver Promoter ist für die Verfahren
besonders geeignet. Vorzugsweise ist der konstitutive Promoter außerdem
ein ubiquitärer Promotor mittlerer Stärke. Die
Definitionen der verschiedenen Promotertypen siehe hier im Abschnitt ”Definitionen”.
-
Es
sollte selbstverständlich sein, dass die Anwendbarkeit
der vorliegenden Erfindung weder auf die durch SEQ ID NO: 1 dargestellte,
ein bHLH11-ähnliches Polypeptid kodierende Nukleinsäure
noch auf die Expression einer Nukleinsäure, die ein bHLH11-ähnliches
Polypeptid kodiert, wenn sie von einem konstitutiven Promotor angetrieben
wird, beschränkt ist.
-
Der
konstitutive Promotor ist vorzugsweise ein Promotor mittlerer Stärke,
wie der GOS2-Promotor, vorzugsweise handelt es sich bei dem Promotor
um einen GOS2-Promotor aus Reis. Weiterhin bevorzugt wird der konstitutive
Promotor durch eine Nukleinsäuresequenz wiedergegeben,
die im Wesentlichen der SEQ ID NO: 256 gleicht, am stärksten
bevorzugt wird der Protomotor durch die SEQ ID NO: 256 dargestellt.
Weitere Beispiele für konstitutive Promotoren siehe hier
im Abschnitt ”Definitionen”.
-
Gegebenenfalls
kann/können eine oder mehrere Terminatorsequenzen in dem
Konstrukt, das in eine Pflanze eingeführt wird, verwendet
werden. Vorzugsweise umfasst das Konstrukt eine Expressionskassette, die
den GOS2-Promotor, der im Wesentlichen gleich der SEQ ID NO: 256
ist, und die Nukleinsäure, die das bHLH11-ähnliche
Polypeptid kodiert, umfasst.
-
Zu
weiteren Regulationselementen können Transkriptionsenhancer
sowie Translationsenhancer gehören. Der Fachmann ist mit
Terminator- und Enhancersequenzen vertraut, die für die
Durchführung der Erfindung geeignet sein können.
Es kann auch eine Intronsequenz zu der 5'-untranslatierten Region
(UTR) oder in der kodierenden Sequenz hinzugefügt werden,
um die Menge an reifer Botschaft, die im Cytosol akkumuliert, zu
erhöhen, wie im Abschnitt Definitionen beschrieben ist.
Weitere Kontrollsequenzen (neben Promotor-, Enhancer-, Silencer-,
Intronsequenzen, 3'UTR- und/oder 5'UTR-Regionen) können
Protein- und/oder RNA-stabilisierende Elemente sein. Solche Sequenzen
sind dem Fachmann bekannt oder können von ihm leicht erhalten werden.
-
Die
erfindungsgemäßen Genkonstrukte können
weiterhin eine Replikationsursprungssequenz beinhalten, die für
die Aufrechterhaltung und/oder die Replikation in einem bestimmten
Zelltyp erforderlich ist. Ein Beispiel ist, wenn ein Genkonstrukt
in einer Bakterienzelle als episomales genetisches Element (z. B.
als Plasmid- oder Cosmidmolekül) aufrechterhalten werden
muss. Bevorzugte Replikationsursprünge sind u. a. der f1-ori
und colE1, sie sind jedoch nicht hierauf beschränkt.
-
Für
den Nachweis eines erfolgreichen Transfers der Nukleinsäuresequenzen,
wie sie bei den erfindungsgemäßen Verfahren verwendet
werden, und/oder für die Selektion von transgenen Pflanzen,
die diese Nukleinsäuren umfassen, ist es vorteilhaft, Markergene
(oder Reportergene) zu verwenden. Daher kann das Genkonstrukt gegebenenfalls
ein Selektionsmarkergen umfassen. Selektionsmarker sind hier im
Abschnitt ”Definitionen” eingehender beschrieben.
Die Markergene können aus der transgenen Zelle entfernt
oder ausgeschnitten werden, wenn sie nicht mehr gebraucht werden.
Techniken zur Entfernung von Markern sind im Stand der Technik bekannt,
geeignete Techniken sind vorstehend im Abschnitt Definitionen beschrieben.
-
Die
Erfindung stellt auch ein Verfahren zur Herstellung von transgenen
Pflanzen mit verbesserten Ertragsmerkmalen im Vergleich zu Kontrollpflanzen
bereit, welches das Einführen und Exprimieren einer Nukleinsäure,
die ein bHLH11-ähnliches Polypeptid, wie oben definiert,
kodiert, in eine(r) Pflanze umfasst.
-
Genauer
gesagt stellt die vorliegende Erfindung ein Verfahren für
die Herstellung von transgenen Pflanzen mit erhöhten verbesserten
Ertragsmerkmalen, insbesondere erhöhtem (Samen-)Ertrag
bereit, wobei das Verfahren Folgendes umfasst:
- (i)
Einführen und Exprimieren einer Nukleinsäure,
die ein bHLH11-ähnliches Polypeptid kodiert, in eine(r) Pflanze
oder Pflanzenzelle und
- (ii) Kultivieren der Pflanzenzelle unter Bedingungen, die das
Pflanzenwachstum und die Pflanzenentwicklung fördern.
-
Bei
der Nukleinsäure gemäß (i) kann es sich
um eine der Nukleinsäuren handeln, die ein bHLH11-ähnliches
Polypeptid, wie hier definiert, kodieren können.
-
Die
Nukleinsäure kann direkt in eine Pflanzenzelle oder in
die Pflanze selbst eingeführt werden (was das Einführen
in ein Gewebe, Organ oder irgendeinen anderen Teil einer Pflanze
beinhaltet). Gemäß einem bevorzugten Merkmal der
vorliegenden Erfindung wird die Nukleinsäure vorzugsweise
mittels Transformation in eine Pflanze eingeführt. Der
Begriff ”Transformation” ist hier im Abschnitt ”Definitionen” genauer
beschrieben.
-
Die
genetisch modifizierten Pflanzenzellen können durch alle
Verfahren regeneriert werden, mit denen der Fachmann vertraut ist.
Geeignete Verfahren finden sich in den oben genannten Veröffentlichungen
von S. D. Kung und R. Wu, Potrykus oder Höfgen und Willmitzer.
-
Im
Allgemeinen werden nach der Transformation die Pflanzenzellen oder
Zellgruppierungen hinsichtlich des Vorhandenseins von einem oder
mehreren Markern selektiert, die von in Pflanzen exprimierbaren
Genen kodiert werden, die mit dem interessierenden Gen cotransferiert
wurden, wonach das transformierte Material zu einer ganzen Pflanze
regeneriert wird. Für die Selektion von transformierten
Pflanzen wird das bei der Transformation erhaltene Pflanzenmaterial
in der Regel derartigen Selektionsbedingungen unterworfen, dass man
transformierte Pflanzen von untransformierten Pflanzen unterscheiden
kann. So können zum Beispiel Samen, die auf die oben beschriebene
Weise erhalten wurden, ausgepflanzt werden und nach einer anfänglichen Wachstumsphase
einer geeigneten Selektion durch Spritzen unterworfen werden. Eine
weitere Möglichkeit besteht darin, dass man die Samen,
gegebenenfalls nach Sterilisieren, auf Agarplatten unter Verwendung
eines geeigneten Selektionsmittels heranzieht, so dass nur die transformierten
Samen zu Pflanzen heranwachsen können. Alternativ werden
die transformierten Pflanzen auf das Vorhandensein eines Selektionsmarkers,
wie die oben beschriebenen, gescreent.
-
Nach
DNA-Transfer und Regeneration können mutmaßlich
transformierte Pflanzen auch zum Beispiel unter Verwendung der Southern-Analyse
auf das Vorhandensein des interessierenden Gens, die Kopienzahl und/oder
die Genomorganisation untersucht werden. Alternativ oder zusätzlich
können die Expressionsniveaus der neu eingeführten
DNA unter Verwendung von Northern- und/oder Western-Analyse verfolgt
werden, wobei beide Techniken dem Durchschnittsfachmann bekannt
sind.
-
Die
erzeugten transformierten Pflanzen können durch eine Vielzahl
von Mitteln vermehrt werden, wie durch klonale Vermehrung oder klassische
Züchtungstechniken. So kann zum Beispiel eine transformierte Pflanze
der ersten Generation (T1-Pflanze) geselbstet werden, und homozygote
Transformanten der zweiten Generation (T2-Pflanzen) können
selektiert werden, und die T2-Pflanzen können dann mit
Hilfe von klassischen Züchtungstechniken weiter vermehrt
werden. Die erzeugten transformierten Organismen können
in einer Vielzahl von Formen vorliegen. So kann es sich zum Beispiel
um Chimären von transformierten Zellen und untransformierten
Zellen, klonale Transformanten (bei denen z. B. alle Zellen so transformiert
wurden, dass sie die Expressionskassette enthalten), Pfropfmaterial
von transformiertem und untransformiertem Gewebe (z. B. bei Pflanzen
ein transformierter Wurzelstock, der auf ein untransformiertes Edelreis
gepfropft wurde) handeln.
-
Die
vorliegende Erfindung erstreckt sich eindeutig auf jede Pflanzenzelle
oder Pflanze, die durch eines der hier beschriebenen Verfahren hergestellt
wurde, und auf alle Pflanzenteile und sämtliches Vermehrungsmaterial
davon. Die vorliegende Erfindung umfasst weiterhin die Nachkommenschaft
eine(r/s) primär transformierten oder transfizierten Zelle,
Gewebes, Organs oder ganzen Pflanze, die/das nach einem der oben
genannten Verfahren hergestellt wurde, wobei die einzige Voraussetzung
ist, dass die Nachkommenschaft dasselbe/dieselben genotypische(n)
und/oder phänotypische(n) Merkmal(e) aufweist, wie es/sie
der Elter bei den erfindungsgemäßen Verfahren
aufweist.
-
Die
Erfindung beinhaltet auch Wirtszellen, die eine isolierte Nukleinsäure
enthalten, die ein bHLH11-ähnliches Polypeptid, wie hier
vorstehend definiert, kodiert. Bevorzugte erfindungsgemäße
Wirtszellen sind Pflanzenzellen. Wirtspflanzen für die
Nukleinsäuren oder den Vektor, die bei dem erfindungsgemäßen Verfahren
verwendet werden, die Expressionskassette oder das Konstrukt oder
den Vektor sind im Prinzip vorteilhafterweise alle Pflanzen, welche
die bei dem erfindungsgemäßen Verfahren verwendeten
Polypeptide synthetisieren können.
-
Die
erfindungsgemäßen Verfahren lassen sich vorteilhaft
auf jede beliebige Pflanze anwenden. Zu den Pflanzen, die für
die erfindungsgemäßen Verfahren besonders geeignet
sind, gehören alle Pflanzen, die zur Superfamilie der Viridiplantae
gehören, insbesondere monokotyle und dikotyle Pflanzen,
einschließlich Futter- oder Weidepflanzenleguminosen, Zierpflanzen,
Nahrungsmittelkulturen, Bäume oder Sträucher.
Gemäß einer bevorzugten Ausführungsform
der vorliegenden Erfindung handelt es sich bei der Pflanze um eine
Kulturpflanze. Beispiele für Kulturpflanzen sind zum Beispiel
u. a. Sojabohne, Sonnenblume, Canola-Raps, Luzerne, Raps, Baumwolle,
Tomate, Kartoffel und Tabak. Weiterhin bevorzugt handelt es sich
bei der Pflanze um eine monokotyle Pflanze. Beispiele für
monokotyle Pflanzen beinhalten Zuckerrohr. Stärker bevorzugt
handelt es sich bei der Pflanze um ein Getreide. Beispiele für
Getreide sind u. a. Reis, Mais, Weizen, Gerste, Hirse, Roggen, Triticale,
Sorghum, Emmer, Dinkel, Secale, Einkorn, Zwerghirse, Mohrenhirse
und Hafer.
-
Die
Erfindung erstreckt sich auch auf erntbare Teile einer Pflanze,
wie zum Beispiel, aber nicht beschränkt auf Samen, Blätter,
Früchte, Blüten, Stängel, Wurzeln, Rhizome,
Knollen und Zwiebeln, wobei die erntbaren Teile eine rekombinante
Nukleinsäure, die ein bHLH11-ähnliches Polypeptid
kodiert, umfassen. Die Erfindung betrifft weiterhin Produkte, die,
vorzugsweise direkt, von einem erntbaren Teil einer solchen Pflanze stammen,
wie trockene Pellets oder Pulver, Öl, Fett und Fettsäuren,
Stärke oder Proteine.
-
Gemäß einem
bevorzugten Merkmal der Erfindung handelt es sich bei der modulierten
Expression um erhöhte Expression. Verfahren zur Erhöhung
der Expression von Nukleinsäuren oder Genen oder Genprodukten
sind in der Fachwelt gut bekannt und Beispiele werden im Abschnitt
Definitionen gegeben.
-
Wie
vorstehend erwähnt, besteht ein bevorzugtes Verfahren zum
Modulieren der Expression einer Nukleinsäure, die ein bHLH11-ähnliches
Polypeptid kodiert, darin, dass man eine Nukleinsäure,
die ein bHLH11-ähnliches Polypeptid kodiert, in eine(r)
Pflanze einführt und exprimiert; die Wirkungen der Durchführung
des Verfahrens, d. h. das Erhöhen der Ertragsmerkmale,
können jedoch auch unter Verwendung anderer bekannter Techniken
erzielt werden, einschließlich, aber nicht beschränkt
auf T-DNA-Activation-Tagging, TILLING, homologe Rekombination. Eine
Beschreibung dieser Techniken findet sich im Abschnitt Definitionen.
-
Die
vorliegende Erfindung umfasst auch die Verwendung von Nukleinsäuren,
die bHLH11-ähnliche Polypeptide, wie hier beschrieben,
kodieren, und die Verwendung dieser bHLH11-ähnlichen Polypeptide
zur Verbesserung eines der oben genannten Ertragsmerkmale in Pflanzen.
-
Hier
beschriebene Nukleinsäuren, die ein bHLH11-ähnliches
Polypeptid kodieren, oder die bHLH11-ähnlichen Polypeptide
selbst können Verwendung in Züchtungsprogrammen
finden, in denen ein DNA-Marker identifiziert wird, der mit einem
Gen, das ein bHLH11-ähnliches Polpeptid kodiert, genetisch
verknüpft sein kann. Die Nukleinsäuren/Gene oder
die bHLH11-ähnlichen Polypeptide selbst können
dazu verwendet werden, einen molekularen Marker zu bestimmen. Dieser
DNA- oder Protein-Marker kann dann in Züchtungsprogrammen
zur Selektion von Pflanzen mit erhöhten Ertragsmerkmalen
verwendet werden, wie hier vorstehend in den erfindungsgemäßen
Verfahren definiert.
-
Allelische
Varianten einer Nukleinsäure/eines Gens, die/das ein bHLH11-ähnliches
Polypeptid kodiert, können auch Verwendung in Marker-unterstützten
Züchtungsprogrammen finden. Bei solchen Züchtungsprogrammen
ist manchmal das Einführen von allelischer Variation durch
Mutagenbehandlung der Pflanzen erforderlich, wobei zum Beispiel
EMS-Mutagenese verwendet wird; alternativ kann das Programm von
einer Sammlung von allelischen Varianten so genannten ”natürlichen” Ursprungs
ausgehen, die nicht mit Absicht hervorgerufen wurden. Dann erfolgt
die Identifikation allelischer Varianten, zum Beispiel mittels PCR.
Darauf folgt ein Schritt zur Selektion besserer allelischer Varianten
der in Frage kommenden Sequenz, die bessere Erträge liefern.
Die Selektion wird gewöhnlich durchgeführt, indem
man die Wachstumsleistung von Pflanzen verfolgt, die verschiedene
allelische Varianten der in Frage kommenden Sequenz enthalten. Die
Wachstumsleistung kann im Gewächshaus oder auf dem Feld
verfolgt werden. Weitere Schritte beinhalten gegebenenfalls das
Kreuzen der Pflanzen, in denen die bessere allelische Variante identifiziert
wurde, mit einer anderen Pflanze. Dies kann zum Beispiel zur Herstellung
einer Kombination interessanter phänotypischer Merkmale
verwendet werden.
-
Nukleinsäuren,
die bHLH11-ähnliche Polypeptide kodieren, können
auch als Sonden zur genetischen und physikalischen Kartierung der
Gene, von denen sie ein Teil sind, und als Marker für Merkmale,
die mit diesen Genen verknüpft sind, verwendet werden.
Diese Information kann für die Pflanzenzüchtung
nützlich sein, um Linien mit gewünschten Phänotypen
zu entwickeln. Eine solche Verwendung von Nukleinsäuren,
die ein bHLH11-ähnliches Polypeptid kodieren, erfordert
nur eine Nukleinsäuresequenz mit einer Länge von
mindestens 15 Nukleotiden. Die Nukleinsäuren, die ein bHLH11-ähnliches
Polypeptid kodieren, können als Marker für Restriktionsfragmentlängenpolymorphismus
(RFLP) verwendet werden. Southern-Blots (Sambrook J, Fritsch EF
und Maniatis T (1989) Molecular Cloning, A Laboratory Manual)
von restriktionsgespaltener genomischer Pflanzen-DNA können
mit den Nukleinsäuren, die bHLH11-ähnliches Polypeptid
kodieren, sondiert werden. Die erhaltenen Bandenmuster können
dann genetischen Analysen unter Verwendung von Computerprogrammen,
wie MapMaker (Lander et al. (1987) Genomics 1: 174–181),
unterworfen werden, wodurch eine Genkarte erstellt wird. Außerdem
können die Nukleinsäuren zum Sondieren von Southern-Blots
verwendet werden, die Restriktionsendonuklease-behandelte genomische
DNAs von einem Satz von Individuen enthalten, die den Elter und
die Nachkommen einer bestimmten genetischen Kreuzung wiedergeben.
Die Segregation der DNA-Polymorphismen wird festgestellt und zur
Berechnung der Position der Nukleinsäure, die das bHLH11-ähnliche
Polypeptid kodiert, in der zuvor unter Verwendung dieser Population
erhaltenen Genkarte verwendet (Botstein et al. (1980) Am.
J. Hum. Genet. 32: 314–331).
-
Die
Herstellung und Verwendung von Sonden, die von Pflanzengenen stammen,
für die Verwendung bei der genetischen Kartierung ist in Bernatzky
und Tanksley (1986) Plant Mol. Biol. Reporter 4: 37–41 beschrieben.
Zahlreiche Veröffentlichungen beschreiben die genetische
Kartierung spezifischer cDNA-Klone, wobei die vorstehend erläuterte
Methodik oder Varianten davon verwendet werden. Für die
Kartierung können zum Beispiel F2-Inzuchtpopulationen,
Rückkreuzungspopulationen, zufallsgemäß gepaarte
Populationen, nahezu isogene Linien und andere Sätze von
Individuen verwendet werden. Diese Methodik ist dem Fachmann bekannt.
-
Die
Nukleinsäuresonden können auch zur physikalischen
Kartierung (d. h. zum Anordnen von Sequenzen in physikalischen Karten;
siehe Hoheisel et al. In: Non-mammalian Genomic Analysis:
A Practical Guide, Academic Press 1996, S. 319–346 und
darin genannte Bezugsstellen) verwendet werden.
-
In
einer anderen Ausführungsform können die Nukleinsäuresonden
zur Kartierung mittels direkter Fluoreszenz-in-situ-Hybridisierung
(FISH) (Trask (1991) Trends Genet. 7: 149–154)
verwendet werden. Zwar bevorzugen derzeitige FISH-Kartierungsverfahren
die Verwendung großer Klone (mehrere kb bis mehrere Hundert
kb; siehe Laan et al. (1995) Genome Res. 5: 13–20),
aber durch Verbesserungen in der Empfindlichkeit kann die FISH-Kartierung
auch unter Verwendung kürzerer Sonden durchgeführt
werden.
-
Eine
Reihe von Verfahren auf Basis von Nukleinsäureamplifikation
für die genetische und physikalische Kartierung kann unter
Verwendung der Nukleinsäuren durchgeführt werden.
Beispiele sind u. a. die allelspezifische Amplifikation (Kazazian
(1989) J. Lab. Clin. Med 11: 95–96), Polymorphismus
PCR-amplifizierter Fragmente (CAPS; Sheffield et al. (1993)
Genomics 16: 325–332), allelspezifische Ligation
(Landegren et al. (1988) Science 241: 1077–1080),
Nukleotidverlängerungsreaktionen (Sokolov (1990)
Nucleic Acid Res. 18: 3671), Radiation-Hybrid-Kartierung
(Walter et al. (1997) Nat. Genet. 7: 22–28)
und Happy-Kartierung (Dear und Cook (1989) Nucleic Acid
Res. 17: 6795–6807). Bei diesen Verfahren wird
die Sequenz einer Nukleinsäure dazu verwendet, Primer-Paare
zu gestalten und herzustellen, die in der Amplifikationsreaktion
oder in Primerverlängerungsreaktionen verwendet werden.
Die Gestaltung dieser Primer ist dem Fachmann bekannt. Bei Verfahren,
die eine genetische Kartierung auf PCR-Basis einsetzen, kann es
notwendig sein, dass man DNA-Sequenz-Unterschiede zwischen den Eltern
der Kartierungskreuzung in dem Bereich identifiziert, welcher der
vorliegenden Nukleinsäuresequenz entspricht. Dies ist jedoch
für Kartierungsverfahren nicht allgemein erforderlich.
-
Die
erfindungsgemäßen Verfahren führen zu
Pflanzen mit verbesserten Ertragsmerkmalen, wie hier vorstehend
beschrieben. Diese Merkmale können auch mit anderen ökonomisch
vorteilhaften Merkmalen kombiniert werden, wie weiteren Ertragsmerkmalen,
Toleranz gegenüber anderen abiotischen und biotischen Stressbedingungen,
Merkmalen, die verschiedene architektonische Merkmale und/oder biochemische und/oder
physiologische Merkmale modifizieren.
-
III. ASR-(Abszisinsäure-, Stress-
und Reifungs-induziertes)Polypeptid
-
Ein
bevorzugtes Verfahren zum Modulieren (vorzugsweise Erhöhen)
der Expression einer Nukleinsäure, die ein ASR-Polypeptid
kodiert, besteht darin, dass man eine Nukleinsäure, die
ein ASR-Polypeptid kodiert, in eine Pflanze einbringt und in dieser
exprimiert.
-
Wird
im folgenden Text auf ein ”Protein, das für die
erfindungsgemäßen Verfahren geeignet ist” Bezug genommen,
soll darunter ein ASR-Polypeptid, wie hier definiert, verstanden
werden. Wird hier im Folgenden auf eine ”Nukleinsäure,
die für die erfindungsgemäßen Verfahren
geeignet ist” Bezug genommen, bedeutet dies eine Nukleinsäure,
die ein solches ASR-Polypeptid kodieren kann. Die Nukleinsäure,
die in eine Pflanze eingeführt werden soll (und die daher
zur Durchführung der erfindungsgemäßen
Verfahren geeignet ist), ist jede beliebige Nukleinsäure,
welche den Typ Protein kodiert, der im Folgenden beschrieben wird,
und wird im Folgenden auch als ”ASR-Nukleinsäure” oder ”ASR-Gen” bezeichnet.
-
Ein ”ASR-Polypeptid”,
wie hier definiert, betrifft die durch SEQ ID NO: 397 dargestellten
Proteine und Homologe (Orthologe und Paraloge) davon. Vorzugsweise
haben die Homologe der SEQ ID NO: 397 eine ABA-WDS-Domäne
(Pfam-Eintrag PF02496).
-
Weiterhin
bevorzugt sind erfindungsgemäße ASR-Polypeptide
diejenigen, die, in steigender Reihenfolge der Präferenz,
mindestens 50%, 55%, 60%, 65%, 70%, 75%, 80%, 83%, 85%, 87%, 90%,
91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
mit einem oder mehreren der in Tabelle F1 angegebenen Polypeptide
aufweisen.
-
Der
Begriff ”Domäne” und ”Motiv” wird
hier im Abschnitt ”Definitionen” definiert. Für
die Identifikation von Domänen gibt es Spezialdatenbanken,
zum Beispiel SMART (Schultz et al., (1998), Proc. Natl.
Acad. Sci. USA 95, 5857–5864; Letunic
et al., (2002), Nucleic Acids Res. 30, 242–244,
InterPro (Mulder et al., (2003), Nucl. Acids Res. 31, 315–318,
Prosite (Sucher und Bairoch (1994), A generalized Profile
syntax for biomolecular sequences motives and its function in automatic
sequence interpretation. (In) ISMB-94; Proceedings 2nd International
Conference an Intelligent Systems for Molecular Biology. Altman
R., Brutlag D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61,
AAAIPress, Menlo Park; Hulo et al., Nucl. Acids
Res. 32: D134–D137, (2004), oder Pfam (Bateman
et al., Nucleic Acids Research 30 (1): 276–280 (2002)).
Ein Satz von Werkzeugen für die In-silico-Analyse von Proteinsequenzen
ist auf dem ExPASY-Proteomics-Server verfügbar (Swiss Institute
of Bioinformatics (Gasteiger et al., ExPASy: the Proteomics
server for in-depth Protein knowledge and analysis, Nucleic Acids
Res. 31: 3784–3788 (2003)). Domänen können
auch unter Verwendung von Routinetechniken, wie durch Sequenz-Alignment,
identifiziert werden.
-
Verfahren
für das Alignment von Sequenzen für Vergleichszwecke
sind in der Fachwelt gut bekannt, dazu zählen GAP, BESTFIT,
BLAST, FASIA und TFASTA. Bei GAP wird der Algorithmus von Needleman
und Wunsch ((1970), J. Mol. Biol. 48: 443–453)
dazu verwendet, das globale Alignment (d. h. das Alignment, das sich über
die vollständigen Sequenzen erstreckt) von zwei Sequenzen
zu finden, das die Anzahl der Übereinstimmungen (”Matches”)
maximiert und die Anzahl der Lücken (”Gaps”)
minimiert. Beim BLAST-Algorithmus (Altschul et al., (1990),
J. Mob. Biol. 215: 403–10) wird die Sequenzidentität
in Prozent berechnet, und es wird eine statistische Analyse der Ähnlichkeit
zwischen den beiden Sequenzen durchgeführt. Die Software
für die Durchführung einer BLAST-Analyse ist über
das National Centre for Biotechnology Information (NCBI) öffentlich
zugänglich. Homologe können zum Beispiel unter
Verwendung des multiplen Sequenz-Alignment-Algorithmus ClustalW
(Version 1.83) leicht identifiziert werden, und zwar mit den Default-Parametern
für paarweises Alignment und einer Scoring-Methode in Prozent.
Die Gesamtprozent an Ähnlichkeit und Identität
können auch unter Verwendung eines der in dem MatGAT-Software-Paket
verfügbaren Verfahren bestimmt werden (Campanella
et al., BMC Bioinformatics. Juli 2003, 10; 4: 29. MatGAT: an application
that generstes similarity/identity matrices using Protein or DNA
sequences.). Für eine Optimierung des Alignments
zwischen konservierten Motiven können kleinere Änderungen
per Hand vorgenommen werden, wie dem Fachmann bekannt ist. Weiterhin können
anstelle von Volllängensequenzen für die Identifikation
von Homologen auch spezifische Domänen verwendet werden.
Die Sequenzidentitätswerte können mit den oben
erwähnten Programmen unter Verwendung der Default-Parameter über
die gesamte Nukleinsäure- oder Aminosäuresequenz
oder über ausgewählte Domänen oder ein
konservierte(s) Motiv(e) bestimmt werden.
-
Weiterhin
haben ASR-Polypeptide (zumindest in ihrer nativen Form), soweit
es SEQ ID NO: 397 und ihre Homologe betrifft, üblicherweise
die Fähigkeit, die Salzstressresistenz von Pflanzen zu
erhöhen. Werkzeuge und Techniken für die Expression
der ASR in Pflanzen und das Testen auf eine erhöhte Salzstressresistenz
sind in der Fachwelt bekannt.
-
Die
vorliegende Erfindung wird dadurch veranschaulicht, dass man Pflanzen
mit der Nukleinsäuresequenz gemäß SEQ
ID NO: 396, welche die Polypeptidsequenz gemäß SEQ
ID NO: 397 kodiert, transformiert. Die Durchführung der
Erfindung ist jedoch nicht auf diese Sequenzen beschränkt;
die erfindungsgemäßen Verfahren können
vorteilhaft unter Verwendung jeder ASR-kodierenden Nukleinsäure
oder jedes ASR-Polypeptids, wie hier definiert, durchgeführt
werden.
-
Beispiele
für Nukleinsäuren, die ASR-Polypeptide kodieren,
lassen sich in Datenbanken finden, die in der Fachwelt bekannt sind.
Solche Nukleinsäuren eignen sich zur Durchführung
der erfindungsgemäßen Verfahren. Orthologe und
Paraloge, wobei die Begriffe ”Orthologe” und ”Paraloge” wie
hier definiert sind, können leicht identifiziert werden,
indem man eine so genannte reziproke Blast-Suche durchführt.
Dies beinhaltet üblicherweise eine erste BLAST, bei der
mit einer Abfragesequenz (zum Beispiel unter Verwendung der SEQ
ID NO: 397) ein ”BLASTing” gegen eine beliebige
Sequenzdatenbank, wie die öffentlich zugängliche
NCBI-Datenbank, durchgeführt wird. BLASTN oder TBLASTX
(unter Verwendung von Standard-Default-Werten) werden im Allgemeinen
dann verwendet, wenn man von einer Nukleotidsequenz ausgeht, und
BLASTP oder TBLASTN (unter Verwendung von Standard-Default-Werten),
wenn man von einer Proteinsequenz ausgeht. Die BLAST-Ergebnisse
können gegebenenfalls gefiltert werden. Mit den Volllängen-Sequenzen
der gefilterten oder ungefilterten Ergebnisse wird anschließend
ein zweites BLASTing gegen Sequenzen des Organismus, von dem die
Abfragesequenz stammt, durchgeführt (ist die Abfragesequenz
die SEQ ID NO: 396 oder SEQ ID NO: 397, dann wäre das zweite
BLASTing daher gegen Sequenzen aus Oryza sativa). Die Ergebnisse
des ersten und des zweiten BLASTing werden dann verglichen. Ein
Paralog wird identifiziert, wenn ein hochrangiger Hit von dem ersten
BLASTing aus derselben Art stammt wie die Abfragesequenz, in diesem
Fall führt ein zweites BLASTing idealerweise zu der Abfragesequenz
unter den höchstrangigen Hits; ein Ortholog wird identifiziert,
wenn ein hochrangiger Hit in dem ersten BLASTing nicht von derselben
Art stammt wie die Abfragesequenz und vorzugsweise beim zweiten
BLASTing dazu führt, dass die Abfragesequenz unter den
höchstrangigen Hits ist.
-
Hochrangige
Hits sind solche mit niedrigem E-Wert. Je niedriger der E-Wert,
desto signifikanter der ”Score” (anders ausgedrückt,
desto niedriger die Wahrscheinlichkeit, dass der Hit durch Zufall
gefunden wurde). Die Berechnung des E-Werts ist in der Fachwelt
gut bekannt. Das ”Scoring” der Vergleiche erfolgt
nicht nur mittels E-Werten, sondern auch anhand der Prozent Identität.
Prozent Identität bezieht sich auf die Anzahl an identischen Nukleotiden
(oder Aminosäuren) zwischen den zwei verglichenen Nukleinsäure-(oder
Polypeptid-)Sequenzen über eine bestimmte Länge.
Bei großen Familien kann man ClustalW und anschließend
einen Neighbour-Joining-Tree verwenden, um die Bildung von Clustern
verwandter Gene leichter sichtbar zu machen und Orthologe und Paraloge
zu identifizieren.
-
Zur
Durchführung der erfindungsgemäßen Verfahren
können auch Nukleinsäurevarianten geeignet sein,
die Homologe und Derivate der SEQ ID NO: 397 kodiere, wobei die
Begriffe ”Homolog” und ”Derivat” hier definiert
sind. Für die erfindungsgemäßen Verfahren
sind auch Nukleinsäuren geeignet, die Homologe und Derivate
von Orthologen oder Paralogen der SEQ ID NO: 397 kodieren. Homologe
und Derivate, die für die erfindungsgemäßen
Verfahren geeignet sind, weisen im Wesentlichen dieselbe biologische
und funktionelle Aktivität wie das unmodifizierte Protein,
von dem sie stammen, auf.
-
Zu
weiteren Nukleinsäurevarianten, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet sind,
zählen Abschnitte von Nukleinsäuren, die ASR-Polypeptide
kodieren, Nukleinsäuren, die mit Nukleinsäuren,
die ASR-Polypeptide kodieren, hybridisieren, Spleißvarianten
von Nukleinsäuren, die ASR-Polypeptide kodieren, allelische
Varianten von Nukleinsäuren, die ASR-Polypeptide kodieren,
und Varianten von Nukleinsäuren, die ASR-Polypeptide kodieren,
die mittels ”gene shuffling” erhalten wurden.
Die Begriffe hybridisierende Sequenz, Spleißvariante, allelische
Variante und ”gene shuffling” sind hier definiert.
-
Nukleinsäuren,
die ASR-Polypeptide kodieren, müssen keine Volllängen-Nukleinsäuren
sein, da die Durchführung der erfindungsgemäßen
Verfahren nicht auf die Verwendung von Volllängen-Nukleinsäuresequenzen
angewiesen ist. Erfindungsgemäß wird ein Verfahren
zur Verbesserung von Ertragsmerkmalen bei Pflanzen bereitgestellt,
bei dem man in eine(r) Pflanze einen Abschnitt der SEQ ID NO: 396
oder einen Abschnitt einer Nukleinsäure, die ein Ortholog,
Paralog oder Homolog der SEQ ID NO: 397 kodiert, einführt
und exprimiert.
-
Ein
Abschnitt einer Nukleinsäure kann zum Beispiel dadurch
hergestellt werden, dass man eine oder mehrere Deletionen an der
Nukleinsäure vornimmt. Die Abschnitte können in
isolierter Form verwendet werden oder können an andere
kodierende (oder nichtkodierende) Sequenzen fusioniert werden, um
zum Beispiel ein Protein herzustellen, das mehrere Aktivitäten
in sich vereinigt. Wenn es an andere kodierende Sequenzen fusioniert
ist, kann das so erhaltene, nach der Translation hergestellte Polypeptid
größer sein als für den Proteinabschnitt
vorhergesagt wurde.
-
Abschnitte,
die für die erfindungsgemäßen Verfahren
geeignet sind, kodieren ein ASR- Polypeptid, wie hier definiert,
und weisen im Wesentlichen dieselbe biologische Aktivität
wie die in SEQ ID NO: 397 angegebenen Aminosäuresequenzen
auf. Vorzugsweise handelt es sich bei dem Abschnitt um einen Abschnitt
von einer der in SEQ ID NO: 396 angegebenen Nukleinsäuren
oder um einen Abschnitt einer Nukleinsäure, die ein Ortholog
oder Paralog von einer der in der SEQ ID NO: 396 angegebenen Aminosäuresequenzen
kodiert. Vorzugsweise ist der Abschnitt mindestens 400, 450, 500,
550, 600, 650, 700, 750 aufeinander folgende Nukleotide lang, wobei
die aufeinander folgenden Nukleotide von der SEQ ID NO: 396 oder
von einer Nukleinsäure, die ein Ortholog oder Paralog der
SEQ ID NO: 397 kodiert, stammen. Am stärksten bevorzugt
ist der Abschnitt ein Abschnitt der Nukleinsäure gemäß SEQ
ID NO: 396.
-
Eine
andere Nukleinsäurevariante, die für die erfindungsgemäßen
Verfahren geeignet ist, ist eine Nukleinsäure, die unter
Bedingungen einer verringerten Stringenz, vorzugsweise unter stringenten
Bedingungen, mit einer Nukleinsäure, die ein ASR-Polypeptid
kodiert, wie hier definiert, oder mit einem Abschnitt, wie hier definiert,
hybridisieren kann.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Nukleinsäure,
die mit der SEQ ID NO: 396 hybridisieren kann, einführt
und exprimiert, oder bei dem man in eine(r) Pflanze eine Nukleinsäure,
die mit einer Nukleinsäure hybridisieren kann, die ein
Ortholog, Paralog oder Homolog der SEQ ID NO: 396 kodiert, einführt
und exprimiert.
-
Hybridisierende
Sequenzen, die für die erfindungsgemäßen
Verfahren geeignet sind, kodieren ein ASR-Polypeptid, wie hier definiert,
das im Wesentlichen dieselbe biologische Aktivität wie
die in SEQ ID NO: 397 angegebenen Aminosäuresequenzen aufweist.
Vorzugsweise kann die hybridisierende Sequenz mit der SEQ ID NO:
396 oder mit einem Abschnitt von einer dieser Sequenzen hybridisieren,
wobei ein Abschnitt wie oben definiert ist, oder wobei die hybridisierende
Sequenz mit einer Nukleinsäuresequenz, die ein Ortholog oder
Paralog der SEQ ID NO: 397 kodiert, hybridisieren kann.
-
Eine
andere Nukleinsäurevariante, die für die erfindungsgemäßen
Verfahren geeignet ist, ist eine Spleißvariante, die ein
ASR-Polypeptid, wie hier vorstehend definiert, kodiert, wobei eine
Spleißvariante wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Spleißvariante
der SEQ ID NO: 396 oder eine Spleißvariante einer Nukleinsäure,
die ein Ortholog, Paralog oder Homolog der SEQ ID NO: 397 kodiert,
einführt und exprimiert.
-
Eine
andere Nukleinsäurevariante, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet ist,
ist eine allelische Variante einer Nukleinsäure, die ein
ASR-Polypeptid, wie hier vorstehend definiert, kodiert, wobei eine
allelische Variante wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine allelische Variante
der SEQ ID NO: 396 einführt und exprimiert oder bei dem
man in eine(r) Pflanze eine allelische Variante einer Nukleinsäure,
die ein Ortholog, Paralog oder Homolog der durch SEQ ID NO: 397
dargestellten Aminosäuresequenzen kodiert, einführt
und exprimiert.
-
Die
allelischen Varianten, die für die erfindungsgemäßen
Verfahren geeignet sind, haben im Wesentlichen dieselbe biologische
Aktivität wie das ASR-Polypeptid gemäß SEQ
ID NO: 397. Allelische Varianten kommen in der Natur vor und von
den erfindungsgemäßen Verfahren wird die Verwendung
dieser natürlichen Allele mit umfasst. Man kann auch ”Gene
shuffling” oder gerichtete Evolution dazu verwenden, Varianten
von Nukleinsäuren zu erzeugen, die ASR-Polypeptide, wie
oben definiert, kodieren, wobei der Begriff ”gene shuffling” wie
hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren für die Verbesserung von Ertragsmerkmalen
bei Pflanzen bereitgestellt, bei dem man in eine(r) Pflanze eine
Variante der SEQ ID NO: 396 einführt und exprimiert oder
bei dem man in eine(r) Pflanze eine Variante einer Nukleinsäure,
die ein Ortholog, Paralog oder Homolog der SEQ ID NO: 397 kodiert,
einführt und exprimiert, wobei die Nukleinsäurevariante
mittels ”gene shuffling” erhalten wird.
-
Nukleinsäurevarianten
können weiterhin auch durch ortsgerichtete Mutagenese erhalten
werden. Zur Erzielung einer ortsgerichteten Mutagenese sind verschiedene
Verfahren verfügbar, von denen die häufigsten Verfahren
auf PCR-Basis sind (Current Protocols in Molecular Biology. Wiley
Hrsg.).
-
Nukleinsäuren,
die ASR-Polypeptide kodieren, können von einer beliebigen
natürlichen oder künstlichen Quelle stammen. Die
Nukleinsäure kann von ihrer nativen Form in ihrer Zusammensetzung
und/oder genomischen Umgebung durch gezielte Manipulation durch
den Menschen modifiziert werden. Vorzugsweise stammt die Nukleinsäure,
die ein ASR-Polypeptid kodiert, aus einer Pflanze. Im Fall der SEQ
ID NO: 396 stammt die Nukleinsäure, die ein ASR-Polypeptid
kodiert, vorzugsweise aus einer einkeimblättrigen Pflanze, stärker
bevorzugt aus der Familie Poaceae, am stärksten bevorzugt
stammt die Nukleinsäure aus Oryza sativa.
-
Die
Erfindung stellt auch bisher unbekannte ASR-kodierende Nukleinsäuren
und ASR-Polypeptide bereit.
-
Gemäß einer
weiteren Ausführungsform der vorliegenden Erfindung wird
daher ein isoliertes Nukleinsäuremolekül bereitgestellt,
das ausgewählt ist aus:
- (i) einer
Nukleinsäure, die durch eine der SEQ ID NO: 401, 403, 405,
407, 409, 411, 413, 415 und 417 dargestellt wird;
- (ii) dem Komplement einer Nukleinsäure, die durch eine
der SEQ ID NO: 401, 403, 405, 407, 409, 411, 413, 415 und 417 dargestellt
wird;
- (iii) einer Nukleinsäure, die ein ASR-Polypeptid kodiert
und, in steigender Reihenfolge der Präferenz, mindestens
50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%,
99% oder mehr Sequenzidentität mit der Aminosäuresequenz
aufweist, die durch eine der SEQ ID NO: 402, 404, 406, 408, 410,
412, 414, 416 und 418 dargestellt wird.
-
Gemäß einer
weiteren Ausführungsform der vorliegenden Erfindung wird
auch ein isoliertes Polypeptid bereitgestellt, das ausgewählt
ist aus:
- (i) einer Aminosäuresequenz,
die durch eine der SEQ ID NO: 402, 404, 406, 408, 410, 412, 414,
416 und 418 dargestellt wird;
- (ii) einer Aminosäuresequenz, die, in steigender Reihenfolge
der Präferenz, mindestens 50%, 55%, 60%, 65%, 70%, 75%,
80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
mit der Aminosäuresequenz hat, die durch eine der SEQ ID
NO: 402, 404, 406, 408, 410, 412, 414, 416 und 418 dargestellt wird;
- (iii) Derivaten von einer der vorstehend unter (i) oder (ii)
angegebenen Aminosäuresequenzen.
-
Die
Durchführung der erfindungsgemäßen Verfahren
führt zu Pflanzen mit verstärkten Ertragsmerkmalen.
Insbesondere führt die Durchführung der erfindungsgemäßen
Verfahren zu Pflanzen mit erhöhter Jungpflanzenvitalität
und erhöhtem Ertrag, insbesondere erhöhter Biomasse
und erhöhtem Samenertrag, im Vergleich zu Kontrollpflanzen.
Die Begriffe ”Ertrag” und ”Samenertrag” werden
hier im Abschnitt ”Definitionen” genauer beschrieben.
-
Werden
hier verbesserte Ertragsmerkmale erwähnt, so soll dies
eine Erhöhung der Jungpflanzenvitalität und/oder
der Biomasse (des Gewichts) von einem oder mehreren Teilen einer
Pflanze bedeuten, was oberirdische (erntbare) Teile und/oder unterirdische
(erntbare) Teile beinhalten kann. Insbesondere sind solche oberirdischen
erntbaren Teile Biomasse und/oder Samen, und die Durchführung
der erfindungsgemäßen Verfahren führt
zu Pflanzen mit höherer Jungpflanzenvitalität,
höherer Biomasse und/oder höherem Samenertrag im
Vergleich zu der Jungpflanzenvitalität, der Biomasse oder
dem Samenertrag von Kontrollpflanzen.
-
Wählt
man Mais als Beispiel, kann sich eine Ertragserhöhung als
eines oder mehrere der folgenden Merkmale äußern:
Unter anderem erhöhte Anzahl an Pflanzen, die pro Hektar
oder Acre wachsen, eine Erhöhung der Anzahl an Ähren
pro Pflanze, eine Erhöhung der Anzahl an Reihen, der Anzahl
an Körnern pro Reihe, des Korngewichts, Tausendkorngewichts,
der Ährenlänge/des Ährendurchmessers,
eine Erhöhung der Samenfüllungsrate (d. h. der
Anzahl gefüllter Samen dividiert durch die Gesamtzahl der
Samen und multipliziert mit 100). Wählt man Reis als Beispiel,
kann sich eine Ertragserhöhung als Erhöhung bezüglich
eines oder mehrerer der folgenden Merkmale ausdrücken:
Unter anderem Anzahl der Pflanzen pro Hektar oder Acre, Anzahl der
Rispen pro Pflanze, Anzahl der Ährchen pro Rispe, Anzahl
der Blüten (Einzelblüten) pro Rispe (ausgedrückt
als Verhältnis der Anzahl an gefüllten Samen zu
der Anzahl an Primärrispen), erhöhte Samenfüllungsrate
(d. h. der Anzahl gefüllter Samen dividiert durch die Gesamtzahl
an Samen und multipliziert mit 100), erhöhtes Tausendkorngewicht.
-
Die
vorliegende Erfindung stellt ein Verfahren zur Erhöhung
des Ertrags, insbesondere des Biomasse- und/oder des Samenertrags
von Pflanzen, verglichen mit Kontrollpflanzen bereit, wobei das
Verfahren die Modulation der Expression, vorzugsweise die Erhöhung
der Expression, einer Nukleinsäure, die ein ASR-Polypeptid,
wie hier definiert, kodiert, in einer Pflanze umfasst.
-
Weil
die erfindungsgemäßen transgenen Pflanzen einen
erhöhten Ertrag aufweisen, ist es wahrscheinlich, dass
diese Pflanzen (zumindest während eines Teils ihres Lebenszyklus)
eine höhere Wachstumsrate aufweisen als Kontrollpflanzen
in einem entsprechenden Stadium ihres Lebenszyklus.
-
Die
erhöhte Wachstumsrate kann für einen oder mehrere
Teile einer Pflanze (darunter Samen) spezifisch sein oder kann im
Wesentlichen in der ganzen Pflanze stattfinden. Pflanzen mit einer
erhöhten Wachstumsrate können einen kürzeren
Lebenszyklus aufweisen. Unter dem Lebenszyklus einer Pflanze kann
man diejenige Zeit verstehen, die die Pflanze benötigt,
um von einem trockenen reifen Samen bis zu dem Stadium heranzuwachsen,
in dem die Pflanze trockene reife Samen ähnlich dem Ausgangsmaterial
produziert hat. Dieser Lebenszyklus kann von Faktoren, wie Jungpflanzenvitalität,
Wachstumsrate, ”Greenness Index”, Blütezeit und
Geschwindigkeit der Samenreifung, beeinflusst werden. Die Erhöhung
der Wachstumsrate kann in einem Stadium oder in mehreren Stadien
im Lebenszyklus einer Pflanze oder im Wesentlichen während
des gesamten Lebenszyklus der Pflanze erfolgen. Eine erhöhte
Wachstumsrate während der Frühstadien im Lebenszyklus
einer Pflanze kann verbesserte Vitalität widerspiegeln.
Die erhöhte Wachstumsrate kann den Erntezyklus einer Pflanze
verändern, so dass die Pflanzen später gesät
werden können und/oder früher geerntet werden können
als dies sonst möglich wäre (ein ähnlicher
Effekt lässt sich mit einer früheren Blütezeit
erzielen). Ist die Wachstumsrate ausreichend erhöht, kann
dies weiteres Aussäen von Samen derselben Pflanzenart ermöglichen
(zum Beispiel Aussäen und Ernten von Reispflanzen und anschließendes
Aussäen und Ernten von weiteren Reispflanzen sogar innerhalb
einer herkömmlichen Wachstumsperiode). Wenn die Wachstumsrate
ausreichend erhöht ist, kann dies ebenso das weitere Aussäen
von Samen anderer Pflanzenarten ermöglichen (zum Beispiel
das Aussäen und Ernten von Maispflanzen und anschließend
zum Beispiel Aussäen und gegebenenfalls Ernten von Sojabohne,
Kartoffel oder einer anderen geeigneten Pflanze). Auch mehrmalige
zusätzliche Ernten von demselben Wurzelstock können
bei einigen Kulturpflanzen möglich sein. Eine Veränderung des
Erntezyklus einer Pflanze kann zu einer Erhöhung der jährlichen
Biomasseproduktion pro Acre führen (weil man eine bestimmte
Pflanze öfter (z. B. pro Jahr) heranziehen und ernten kann).
Eine erhöhte Wachstumsrate kann auch den Anbau transgener
Pflanzen in einem breiteren geographischen Bereich als ihre Wildtyp-Gegenstücke
ermöglichen, da die räumlichen Beschränkungen
für den Anbau einer Kultur häufig von ungünstigen Umweltbedingungen
entweder während der Pflanzzeit (früh in der Saison)
oder während der Erntezeit (spät in der Saison)
bestimmt werden. Solche ungünstigen Bedingungen können
vermieden werden, wenn der Erntezyklus verkürzt ist. Die
Wachstumsrate kann durch Ableiten verschiedener Parameter aus Wachstumskurven bestimmt
werden, wobei die Parameter u. a. folgende sein können:
T-Mid (Zeitdauer, die die Pflanzen benötigen, um 50% ihrer
Maximalgröße zu erreichen) und T-90 (Zeitdauer,
die die Pflanzen benötigen, um 90% ihrer Maximalgröße
zu erreichen).
-
Gemäß einem
bevorzugten Merkmal der vorliegenden Erfindung führt die
Durchführung der erfindungsgemäßen Verfahren
zu Pflanzen mit einer im Vergleich zu Kontrollpflanzen erhöhten
Wachstumsrate. Erfindungsgemäß wird daher ein
Verfahren zur Erhöhung der Wachstumsrate von Pflanzen bereitgestellt,
wobei das Verfahren die Modulation der Expression, vorzugsweise
die Erhöhung der Expression, einer Nukleinsäure, die
ein ASR-Polypeptid, wie hier definiert, kodiert, in einer Pflanze
umfasst. In einer bestimmten Ausführungsform ergibt die
Durchführung der erfindungsgemäßen Verfahren
Pflanzen mit erhöhter Jungpflanzenvitalität.
-
Eine
Erhöhung des Ertrags und/oder der Wachstumsrate tritt unabhängig
davon auf, ob die Pflanze unter Nichtstressbedingungen steht oder
ob die Pflanze im Vergleich zu Kontrollpflanzen verschiedenen Stressbedingungen
unterworfen wird. Typischerweise reagieren Pflanzen auf Stress dadurch,
dass sie langsamer wachsen. Unter starken Stressbedingungen kann
die Pflanze sogar ihr Wachstum völlig einstellen. Leichter
Stress ist dagegen hier als jeglicher Stress definiert, dem eine
Pflanze ausgesetzt ist und der nicht dazu führt, dass die
Pflanze völlig aufhört zu wachsen, ohne dass sie
die Fähigkeit besitzt, ihr Wachstum wieder aufzunehmen.
Leichter Stress im Sinne der Erfindung führt zu einer Verringerung
des Wachstums der gestressten Pflanzen von weniger als 40%, 35%
oder 30%, vorzugsweise weniger als 25%, 20% oder 15%, stärker
bevorzugt weniger als 14%, 13%, 12%, 11% oder 10% oder weniger im
Vergleich zu der Kontrollpflanze unter Nichtstressbedingungen. Aufgrund
von Fortschritten bei den landwirtschaftlichen Praktiken (Bewässerung,
Düngung, Pestizidbehandlungen) findet man bei angebauten
Kulturpflanzen nicht oft starke Stressbedingungen. Daher ist das
durch leichten Stress induzierte geschwächte Wachstum häufig
ein unerwünschtes Merkmal in der Landwirtschaft. Leichte
Stressbedingungen sind die alltäglichen biotischen und/oder
abiotischen (Umwelt-)Stressbedingungen, denen eine Pflanze ausgesetzt
ist. Abiotischer Stress kann durch Trockenheit oder Wasserüberschuss,
anaeroben Stress, Salzstress, chemische Toxizität, oxidativen
Stress und heiße, kalte oder Gefriertemperaturen verursacht
werden. Bei dem abiotischen Stress kann es sich um einen osmotischen
Stress handeln, der durch Wasserstress (insbesondere aufgrund von
Trockenheit), Salzstress, oxidativen Stress oder ionenbedingten
Stress verursacht wird. Unter biotischem Stress versteht man gewöhnlich
Stressbedingungen, die durch Pathogene, wie Bakterien, Viren, Pilze
und Insekten, verursacht werden.
-
Insbesondere
können die erfindungsgemäßen Verfahren
unter Nichtstressbedingungen oder unter leichten Trockenheitsbedingungen
durchgeführt werden, wobei man Pflanzen mit einem im Vergleich
zu Kontrollpflanzen erhöhten Ertrag erhält. Wie
von Wang et al. (Planta (2003), 218: 1–14)
beschrieben, führt abiotischer Stress zu einer Reihe von
morphologischen, physiologischen, biochemischen und molekularen
Veränderungen, die das Pflanzenwachstum und die Pflanzenproduktivität
negativ beeinflussen. Es ist bekannt, dass Trockenheit, Salinität,
extreme Temperaturen und oxidativer Stress miteinander in Verbindung
stehen und über ähnliche Mechanismen Wachstums-
und Zellschäden induzieren können. Rabbani
et al. (Plant Physiol. (2003), 133: 1755–1767)
beschreibt ein besonders hohes Ausmaß an gegenseitiger
Beeinflussung (”Cross-Talk”) von Trockenheitsstress
und durch hohe Salinität verursachtem Stress. Zum Beispiel äußern
sich Trockenheit und/oder Versalzung in erster Linie als osmotischer
Stress, was zur Störung der Homöostase und der
Ionenverteilung in der Zelle führt. Oxidativer Stress,
der häufig hohe oder niedrige Temperatur, Salinität
oder Trockenheitsstress begleitet, kann zur Denaturierung funktioneller
und struktureller Proteine führen. Infolgedessen aktivieren
diese verschiedenen Umweltstressbedingungen häufig ähnliche
Zellsignalwege und Zellreaktionen, wie die Produktion von Stressproteinen,
die Hinaufregulation von Antioxidantien, die Akkumulation von kompatiblen
gelösten Stoffen und ein Einstellen des Wachstums. Der
Begriff ”Nichtstress”-Bedingungen, wie hier verwendet,
steht für diejenigen Umweltbedingungen, die ein optimales
Wachstum von Pflanzen gestatten. Der Fachmann ist mit normalen Bodenbedingungen
und Klimabedingungen für einen bestimmten Standort vertraut.
Pflanzen unter optimalen Wachstumsbedingungen (die unter Nichtstressbedingungen
herangezogen werden) liefern üblicherweise einen Ertrag
von, in der Reihenfolge der zunehmenden Präferenz, mindestens 90%,
87%, 85%, 83%, 80%, 77% oder 75% der durchschnittlichen Produktion
einer solchen Pflanze in einer gegebenen Umgebung. Die durchschnittliche
Produktion kann bezogen auf die Ernte und/oder die Saison berechnet
werden. Der Fachmann ist mit den durchschnittlichen Erträgen
einer Kultur vertraut.
-
Die
Durchführung der erfindungsgemäßen Verfahren
verleiht Pflanzen bei Wachstum unter Nichtstressbedingungen oder
unter leichten Trockenheitsbedingungen einen im Vergleich zu Kontrollpflanzen,
die unter vergleichbaren Bedingungen herangezogen werden, erhöhten
Ertrag und/oder erhöhte Jungpflanzenvitalität.
Erfindungsgemäß wird daher ein Verfahren zur Erhöhung
des Ertrags und/oder der Jungpflanzenvitalität von Pflanzen,
die unter Nichtstressbedingungen oder unter leichten Trockenheitsbedingungen
herangezogen werden, bereitgestellt, wobei das Verfahren die Erhöhung
der Expression einer Nukleinsäure, die ein ASR-Polypeptid
kodiert, in einer Pflanze umfasst.
-
Die
Durchführung der erfindungsgemäßen Verfahren
verleiht Pflanzen, die unter Nährstoffmangelbedingungen,
insbesondere unter Stickstoffmangelbedingungen, herangezogen werden,
einen erhöhten Ertrag im Vergleich zu Kontrollpflanzen,
die unter vergleichbaren Bedingungen herangezogen werden. Erfindungsgemäß wird
daher auch ein Verfahren zur Erhöhung des Ertrags in Pflanzen,
die unter Nährstoffmangelbedingungen herangezogen werden,
bereitgestellt, wobei man bei dem Verfahren die Expression einer
Nukleinsäure, die ein ASR-Polypeptid kodiert, in einer
Pflanze erhöht. Ein Nährstoffmangel kann durch
ein Fehlen von Nährstoffen, wie unter anderem Stickstoff,
Phosphate und andere phosphorhaltige Verbindungen, Kalium, Calcium, Cadmium,
Magnesium, Mangan, Eisen und Bor, hervorgerufen werden.
-
Die
vorliegende Erfindung umfasst Pflanzen oder Teile davon (einschließlich
Samen), die durch die erfindungsgemäßen Verfahren
erhältlich sind. Die Pflanzen oder Teile davon umfassen
ein Nukleinsäuretransgen, das ein ASR-Polypeptid kodiert,
wie oben definiert.
-
Die
Erfindung stellt auch Genkonstrukte und Vektoren bereit, um die
Einführung und/oder Expression von Nukleinsäuren,
die ASR-Polypeptide kodieren, in Pflanzen zu erleichtern. Die Genkonstrukte
können in Vektoren eingefügt werden, die im Handel
erhältlich sein können und die für die
Transformation des interessierenden Gens in Pflanzen und für
die Expression des interessierenden Gens in den transformierten
Zellen geeignet sind. Die Erfindung stellt auch die Verwendung eines
Genkonstrukts, wie hier definiert, bei den erfindungsgemäßen
Verfahren bereit.
-
Genauer
gesagt, stellt die vorliegende Erfindung ein Konstrukt bereit, das
folgendes umfasst:
- (a) eine Nukleinsäure,
die ein ASR-Polypeptid kodiert, wie oben definiert;
- (b) eine oder mehrere Kontrollsequenzen, die die Expression
der Nukleinsäuresequenz unter (a) vorantreiben können,
und gegebenenfalls
- (c) eine Transkriptionsterminationssequenz.
-
Vorzugsweise
ist die Nukleinsäure, die ein ASR-Polypeptid kodiert, wie
vorstehend definiert. Die Begriffe ”Kontrollsequenz” und ”Terminationssequenz” sind
wie hier definiert.
-
Pflanzen
werden mit einem Vektor transformiert, der eine der oben beschriebenen
Nukleinsäuren umfasst. Der Fachmann kennt die genetischen
Elemente, die in einem Vektor vorhanden sein müssen, damit Wirtszellen
erfolgreich transformiert, Wirtszellen, die die interessierende
Sequenz enthalten, selektiert und vermehrt werden können.
Die interessierende Sequenz ist mit einer oder mehreren Kontrollsequenzen
(mindestens mit einem Promoter) funktionsfähig verbunden.
-
Jede
Art von Promoter, ob natürlich oder synthetisch, kann vorteilhaft
für das Vorantreiben der Expression der Nukleinsäuresequenz
verwendet werden. Ein konstitutiver Promoter ist für die
erfindungsgemäßen Verfahren besonders geeignet.
Die Definitionen der verschiedenen Promotertypen siehe hier im Abschnitt ”Definitionen”.
-
Es
sollte selbstverständlich sein, dass die Anwendbarkeit
der vorliegenden Erfindung weder auf die durch SEQ ID NO: 396 dargestellte
Nukleinsäure, die ein ASR-Poylpeptid kodiert, noch auf
die Expression einer Nukleinsäure, die ein ASR-Polypeptid
kodiert, wenn sie von einem konstitutiven Promotor angetrieben wird,
beschränkt ist.
-
Der
konstitutive Promotor ist vorzugsweise ein GOS2-Promotor, vorzugsweise
ein GOS2-Promotor aus Reis. Weiterhin bevorzugt wird der konstitutive
Promotor durch eine Nukleinsäuresequenz wiedergegeben,
die im Wesentlichen der SEQ ID NO: 398 gleicht, am stärksten
bevorzugt wird der Protomotor durch die SEQ ID NO: 398 dargestellt.
Weitere Beispiele für konstitutive Promotoren siehe hier
im Abschnitt ”Definitionen”.
-
Gegebenenfalls
kann/können eine oder mehrere Terminatorsequenzen in dem
Konstrukt, das in eine Pflanze eingeführt wird, verwendet
werden. Zu weiteren Regulationselementen können Transkriptionsenhancer
sowie Translationsenhancer gehören. Der Fachmann ist mit
Terminator- und Enhancersequenzen vertraut, die für die
Durchführung der Erfindung geeignet sein können.
Es kann auch eine Intronsequenz zu der 5'-untranslatierten Region
(UTR) oder in der kodierenden Sequenz hinzugefügt werden,
um die Menge an reifer Botschaft, die im Cytosol akkumuliert, zu
erhöhen, wie im Abschnitt Definitionen beschrieben ist.
Weitere Kontrollsequenzen (neben Promotor-, Enhancer-, Silencer-,
Intronsequenzen, 3'UTR- und/oder 5'UTR-Regionen) können
Protein- und/oder RNA-stabilisierende Elemente sein. Solche Sequenzen
sind dem Fachmann bekannt oder können von ihm leicht erhalten
werden.
-
Die
erfindungsgemäßen Genkonstrukte können
weiterhin eine Replikationsursprungssequenz beinhalten, die für
die Aufrechterhaltung und/oder die Replikation in einem bestimmten
Zelltyp erforderlich ist. Ein Beispiel ist, wenn ein Genkonstrukt
in einer Bakterienzelle als episomales genetisches Element (z. B.
als Plasmid- oder Cosmidmolekül) aufrechterhalten werden
muss. Bevorzugte Replikationsursprünge sind u. a. der f1-ori
und colE1, sie sind jedoch nicht hierauf beschränkt.
-
Für
den Nachweis eines erfolgreichen Transfers der Nukleinsäuresequenzen,
wie sie bei den erfindungsgemäßen Verfahren verwendet
werden, und/oder für die Selektion von transgenen Pflanzen,
die diese Nukleinsäuren umfassen, ist es vorteilhaft, Markergene
(oder Reportergene) zu verwenden. Daher kann das Genkonstrukt gegebenenfalls
ein Selektionsmarkergen umfassen. Selektionsmarker sind hier im
Abschnitt ”Definitionen” eingehender beschrieben.
Die Markergene können aus der transgenen Zelle entfernt
oder ausgeschnitten werden, wenn sie nicht mehr gebraucht werden.
Techniken zur Entfernung von Markern sind im Stand der Technik bekannt,
geeignete Techniken sind vorstehend im Abschnitt Definitionen beschrieben.
-
Die
Erfindung stellt auch ein Verfahren zur Herstellung von transgenen
Pflanzen mit verbesserten Ertragsmerkmalen im Vergleich zu Kontrollpflanzen
bereit, welches das Einführen und Exprimieren einer Nukleinsäure,
die für ein ASR-Polypeptid, wie hier oben definiert, kodiert,
in eine(r) Pflanze umfasst.
-
Genauer
gesagt, stellt die vorliegende Erfindung ein Verfahren für
die Herstellung von transgenen Pflanzen mit erhöhten verbesserten
Ertragsmerkmalden, insbesondere erhöhter Jungpflanzenvitalität und/oder
erhöhtem Ertrag bereit, wobei das Verfahren Folgendes umfasst:
- (i) Einführen und Exprimieren einer
Nukleinsäure, die ein ASR-Polypeptid kodiert, in eine(r)
Pflanze oder Pflanzenzelle und
- (ii) Kultivieren der Pflanzenzelle unter Bedingungen, die das
Pflanzenwachstum und die Pflanzenentwicklung fördern.
-
Bei
der Nukleinsäure gemäß (i) kann es sich
um eine der Nukleinsäuren handeln, die ein ASR-Polypeptid,
wie hier definiert, kodieren können.
-
Die
Nukleinsäure kann direkt in eine Pflanzenzelle oder in
die Pflanze selbst eingeführt werden (was das Einführen
in ein Gewebe, Organ oder irgendeinen anderen Teil einer Pflanze
beinhaltet). Gemäß einem bevorzugten Merkmal der
vorliegenden Erfindung wird die Nukleinsäure vorzugsweise
mittels Transformation in eine Pflanze eingeführt. Der
Begriff ”Transformation” ist hier im Abschnitt ”Definitionen” genauer
beschrieben.
-
Die
genetisch modifizierten Pflanzenzellen können durch alle
Verfahren regeneriert werden, mit denen der Fachmann vertraut ist.
Geeignete Verfahren finden sich in den oben genannten Veröffentlichungen
von S. D. Kung und R. Wu, Potrykus oder Höfgen und Willmitzer.
-
Im
Allgemeinen werden nach der Transformation die Pflanzenzellen oder
Zellgruppierungen hinsichtlich des Vorhandenseins von einem oder
mehreren Markern selektiert, die von in Pflanzen exprimierbaren
Genen kodiert werden, die mit dem interessierenden Gen cotransferiert
wurden, wonach das transformierte Material zu einer ganzen Pflanze
regeneriert wird. Für die Selektion von transformierten
Pflanzen wird das bei der Transformation erhaltene Pflanzenmaterial
in der Regel derartigen Selektionsbedingungen unterworfen, dass man
transformierte Pflanzen von untransformierten Pflanzen unterscheiden
kann. So können zum Beispiel Samen, die auf die oben beschriebene
Weise erhalten wurden, ausgepflanzt werden und nach einer anfänglichen Wachstumsphase
einer geeigneten Selektion durch Spritzen unterworfen werden. Eine
weitere Möglichkeit besteht darin, dass man die Samen,
gegebenenfalls nach Sterilisieren, auf Agarplatten unter Verwendung
eines geeigneten Selektionsmittels heranzieht, so dass nur die transformierten
Samen zu Pflanzen heranwachsen können. Alternativ werden
die transformierten Pflanzen auf das Vorhandensein eines Selektionsmarkers,
wie die oben beschriebenen, gescreent.
-
Nach
DNA-Transfer und Regeneration können mutmaßlich
transformierte Pflanzen auch zum Beispiel unter Verwendung der Southern-Analyse
auf das Vorhandensein des interessierenden Gens, die Kopienzahl und/oder
die Genomorganisation untersucht werden. Alternativ oder zusätzlich
können die Expressionsniveaus der neu eingeführten
DNA unter Verwendung von Northern- und/oder Western-Analyse verfolgt
werden, wobei beide Techniken dem Durchschnittsfachmann bekannt
sind.
-
Die
erzeugten transformierten Pflanzen können durch eine Vielzahl
von Mitteln vermehrt werden, wie durch klonale Vermehrung oder klassische
Züchtungstechniken. So kann zum Beispiel eine transformierte Pflanze
der ersten Generation (T1-Pflanze) geselbstet werden, und homozygote
Transformanten der zweiten Generation (T2-Pflanzen) können
selektiert werden, und die T2-Pflanzen können dann mit
Hilfe von klassischen Züchtungstechniken weiter vermehrt
werden. Die erzeugten transformierten Organismen können
in einer Vielzahl von Formen vorliegen. So kann es sich zum Beispiel
um Chimären von transformierten Zellen und urtransformierten
Zellen, klonale Transformanten (bei denen z. B. alle Zellen so transformiert
wurden, dass sie die Expressionskassette enthalten), Pfropfmaterial
von transformiertem und untransformiertem Gewebe (z. B. bei Pflanzen
ein transformierter Wurzelstock, der auf ein untransformiertes Edelreis
gepfropft wurde) handeln.
-
Die
vorliegende Erfindung erstreckt sich eindeutig auf jede Pflanzenzelle
oder Pflanze, die durch eines der hier beschriebenen Verfahren erzeugt
wurde, und auf alle Pflanzenteile und sämtliches Vermehrungsmaterial
davon. Die vorliegende Erfindung umfasst weiterhin die Nachkommenschaft
eine(r/s) primär transformierten oder transfizierten Zelle,
Gewebes, Organs oder ganzen Pflanze, die/das durch eines der oben
genannten Verfahren erzeugt wurde, wobei die einzige Voraussetzung
ist, dass die Nachkommenschaft dasselbe/dieselben genotypische(n)
und/oder phänotypische(n) Merkmal(e) aufweist, wie es/sie
der Elter bei den erfindungsgemäßen Verfahren
aufweist.
-
Die
Erfindung beinhaltet auch Wirtszellen, die eine isolierte Nukleinsäure
enthalten, die ein ASR-Polypeptid, wie hier vorstehend definiert,
kodiert. Bevorzugte erfindungsgemäße Wirtszellen
sind Pflanzenzellen. Wirtspflanzen für die Nukleinsäuren
oder den Vektor, die bei dem erfindungsgemäßen
Verfahren verwendet werden, die Expressionskassette oder das Konstrukt
oder den Vektor sind im Prinzip vorteilhafterweise alle Pflanzen,
welche die bei dem erfindungsgemäßen Verfahren
verwendeten Polypeptide synthetisieren können.
-
Die
erfindungsgemäßen Verfahren lassen sich vorteilhaft
auf jede beliebige Pflanze anwenden. Zu den Pflanzen, die für
die erfindungsgemäßen Verfahren besonders geeignet
sind, gehören alle Pflanzen, die zur Superfamilie der Viridiplantae
gehören, insbesondere monokotyle und dikotyle Pflanzen,
einschließlich Futter- oder Weidepflanzenleguminosen, Zierpflanzen,
Nahrungsmittelkulturen, Bäume oder Sträucher.
Gemäß einer bevorzugten Ausführungsform
der vorliegenden Erfindung handelt es sich bei der Pflanze um eine
Kulturpflanze. Beispiele für Kulturpflanzen sind zum Beispiel
u. a. Sojabohne, Sonnenblume, Canola-Raps, Luzerne, Raps, Baumwolle,
Tomate, Kartoffel und Tabak. Weiterhin bevorzugt handelt es sich
bei der Pflanze um eine monokotyle Pflanze. Beispiele für
monokotyle Pflanzen beinhalten u. a. Zuckerrohr. Stärker
bevorzugt handelt es sich bei der Pflanze um ein Getreide. Beispiele
für Getreide sind u. a. Reis, Mais, Weizen, Gerste, Hirse, Roggen,
Triticale, Sorghum und Hafer.
-
Die
Erfindung erstreckt sich auch auf erntbare Teile einer Pflanze,
wie zum Beispiel, aber nicht beschränkt auf Samen, Blätter,
Früchte, Blüten, Stängel, Wurzeln, Rhizome,
Knollen und Zwiebeln. Die Erfindung betrifft weiterhin Produkte,
die, vorzugsweise direkt, von einem erntbaren Teil einer solchen
Pflanze stammen, wie trockene Pellets oder Pulver, Öl,
Fett und Fettsäuren, Stärke oder Proteine.
-
Gemäß einem
bevorzugten Merkmal der Erfindung handelt es sich bei der modulierten
Expression um erhöhte Expression. Verfahren zur Erhöhung
der Expression von Nukleinsäuren oder Genen oder Genprodukten
sind in der Fachwelt gut bekannt und Beispiele werden im Abschnitt
Definitionen gegeben.
-
Wie
vorstehend erwähnt, besteht ein bevorzugtes Verfahren zum
Modulieren (vorzugsweise Erhöhen) der Expression einer
Nukleinsäure, die ein ASR-Polypeptid kodiert, darin, dass
man eine Nukleinsäure, die ein ASR-Polypeptid kodiert,
in eine Pflanze einführt und darin exprimiert; die Wirkungen
der Durchführung des Verfahrens, d. h. das Erhöhen
der Ertragsmerkmale, können jedoch auch unter Verwendung
anderer bekannter Techniken erzielt werden, einschließlich,
aber nicht beschränkt auf T-DNA-Activation-Tagging, TILLING,
homologe Rekombination. Eine Beschreibung dieser Techniken findet
sich im Abschntt Definitionen.
-
Die
vorliegende Erfindung umfasst auch die Verwendung von Nukleinsäuren,
die ASR-Polypeptide, wie hier beschrieben, kodieren, und die Verwendung
dieser ASR-Polypeptide zur Verbesserung eines der oben genannten
Ertragsmerkmale in Pflanzen.
-
Hier
beschriebene Nukleinsäuren, die ASR-Polypeptide kodieren,
oder die ASR-Polypeptide selbst können Verwendung in Züchtungsprogrammen
finden, in denen ein DNA-Marker identifiziert wird, der mit einem
Gen genetisch verknüpft sein kann, das ein ASR-Polpeptid kodiert.
Die Nukleinsäuren/Gene oder die ASR-Polypeptide selbst
können dazu verwendet werden, einen molekularen Marker
zu bestimmen. Dieser DNA- oder Protein-Marker kann dann in Züchtungsprogrammen
zur Selektion von Pflanzen mit erhöhten Ertragsmerkmalen
verwendet werden, wie hier vorstehend unter den erfindungsgemäßen
Verfahren definiert.
-
Allelische
Varianten einer Nukleinsäure/eines Gens, die/das ein ASR-Polypeptid
kodiert, können auch Verwendung in Marker-unterstützten
Züchtungsprogrammen finden. Bei solchen Züchtungsprogrammen
ist manchmal das Einführen von allelischer Variation durch
Mutagenbehandlung der Pflanzen erforderlich, wobei zum Beispiel
EMS-Mutagenese verwendet wird; alternativ kann das Programm von
einer Sammlung von allelischen Varianten so genannten ”natürlichen” Ursprungs
ausgehen, die nicht mit Absicht hervorgerufen wurden. Dann erfolgt
die Identifikation allelischer Varianten, zum Beispiel mittels PCR.
Darauf folgt ein Schritt zur Selektion besserer allelischer Varianten
der in Frage kommenden Sequenz, die bessere Erträge liefern.
Die Selektion wird gewöhnlich durchgeführt, indem
man die Wachstumsleistung von Pflanzen verfolgt, die verschiedene
allelische Varianten der in Frage kommenden Sequenz enthalten. Die
Wachstumsleistung kann im Gewächshaus oder auf dem Feld
verfolgt werden. Weitere Schritte beinhalten gegebenenfalls das
Kreuzen der Pflanzen, in denen die bessere allelische Variante identifiziert
wurde, mit einer anderen Pflanze. Dies kann zum Beispiel zur Herstellung
einer Kombination interessanter phänotypischer Merkmale
verwendet werden.
-
Nukleinsäuren,
die ASR-Polypeptide kodieren, können auch als Sonden zur
genetischen und physikalischen Kartierung der Gene, von denen sie
ein Teil sind, und als Marker für Merkmale, die mit diesen
Genen verknüpft sind, verwendet werden. Diese Information
kann für die Pflanzenzüchtung nützlich
sein, um Linien mit gewünschten Phänotypen zu
entwickeln. Eine solche Verwendung von Nukleinsäuren, die
ASR-Polypeptide kodieren, erfordert nur eine Nukleinsäuresequenz
mit einer Länge von mindestens 15 Nukleotiden. Die ein ASR-Polypeptid
kodierenden Nukleinsäuren können als Marker für
Restriktionsfragmentlängenpolymorphismus (RFLP) verwendet
werden. Southern-Blots (Sambrook J, Fritsch EF und Maniatis
T (1989) Molecular Cloning, A Laboratory Manual) von restriktionsgespaltener
genomischer Pflanzen-DNA können mit den ASR-kodierenden
Nukleinsäuren sondiert werden. Die erhaltenen Bandenmuster
können dann genetischen Analysen unter Verwendung von Computerprogrammen,
wie MapMaker (Lander et al. (1987) Genomics 1: 174–181),
unterworfen werden, wodurch eine Genkarte erstellt wird. Außerdem
können die Nukleinsäuren zum Sondieren von Southern-Blots
verwendet werden, die Restriktionsendonuklease-behandelte genomische
DNAs von einem Satz von Individuen enthalten, die den Elter und
die Nachkommen einer bestimmten genetischen Kreuzung wiedergeben.
Die Segregation der DNA-Polymorphismen wird festgestellt und zur
Berechnung der Position der Nukleinsäure, die das ASR-Polypeptid
kodiert, in der zuvor unter Verwendung dieser Population erhaltenen
Genkarte verwendet (Botstein et al. (1980) Am. J. Hum. Genet.
32: 314–331).
-
Die
Herstellung und Verwendung von Sonden, die von Pflanzengenen stammen,
für die Verwendung bei der genetischen Kartierung ist in Bernatzky
und Tanksley (1986) Plant Mol. Biol. Reporter 4: 37–41 beschrieben.
Zahlreiche Veröffentlichungen beschreiben die genetische
Kartierung spezifischer cDNA-Klone, wobei die vorstehend erläuterte
Methodik oder Varianten davon verwendet werden. Für die
Kartierung können zum Beispiel F2-Inzuchtpopulationen,
Rückkreuzungspopulationen, zufallsgemäß gepaarte
Populationen, nahezu isogene Linien und andere Sätze von
Individuen verwendet werden. Diese Methodik ist dem Fachmann bekannt.
-
Die
Nukleinsäuresonden können auch zur physikalischen
Kartierung (d. h. zum Anordnen von Sequenzen in physikalischen Karten;
siehe Hoheisel et al. In: Non-mammalian Genomic Analysis:
A Practical Guide, Academic Press 1996, S. 319–346 und
darin genannte Bezugsstellen) verwendet werden.
-
In
einer anderen Ausführungsform können die Nukleinsäuresonden
zur Kartierung durch direkte Fluoreszenz-in-situ-Hybridisierung
(FISH) (Trask (1991) Trends Genet. 7: 149–154)
verwendet werden. Zwar bevorzugen derzeitige FISH-Kartierungsverfahren
die Verwendung großer Klone (mehrere kb bis mehrere Hundert
kb; siehe Laan et al. (1995) Genome Res. 5: 13–20),
aber durch Verbesserungen in der Empfindlichkeit kann die FISH-Kartierung
auch unter Verwendung kürzerer Sonden durchgeführt
werden.
-
Eine
Reihe von Verfahren auf Basis von Nukleinsäureamplifikation
für die genetische und physikalische Kartierung kann unter
Verwendung der Nukleinsäuren durchgeführt werden.
Beispiele sind u. a. die allelspezifische Amplifikation (Kazazian
(1989) J. Lab. Clin. Med 11: 95–96), Polymorphismus
PCR-amplifizierter Fragmente (CAPS; Sheffield et al. (1993)
Genomics 16: 325–332), allelspezifische Ligation
(Landegren et al. (1988) Science 241: 1077–1080),
Nukleotidverlängerungsreaktionen (Sokolov (1990)
Nucleic Acid Res. 18: 3671), Radiation-Hybrid-Kartierung
(Walter et al. (1997) Nat. Genet. 7: 22–28)
und Happy-Kartierung (Dear und Cook (1989) Nucleic Acid
Res. 17: 6795–6807). Bei diesen Verfahren wird
die Sequenz einer Nukleinsäure dazu verwendet, Primer-Paare
zu gestalten und zu erzeugen, die in der Amplifikationsreaktion
oder in Primerverlängerungsreaktionen verwendet werden.
Die Gestaltung dieser Primer ist dem Fachmann bekannt. Bei Verfahren,
die eine genetische Kartierung auf PCR-Basis einsetzen, kann es
notwendig sein, dass man DNA-Sequenz-Unterschiede zwischen den Eltern
der Kartierungskreuzung in dem Bereich identifiziert, welcher der
vorliegenden Nukleinsäuresequenz entspricht. Dies ist jedoch
für Kartierungsverfahren im Allgemeinen nicht erforderlich.
-
Die
erfindungsgemäßen Verfahren führen zu
Pflanzen mit verbesserten Ertragsmerkmalen, wie hier vorstehend
beschrieben. Diese Merkmale können auch mit anderen ökonomisch
vorteilhaften Merkmalen kombiniert werden, wie weiteren Ertragsmerkmalen,
Toleranz gegenüber anderen abiotischen und biotischen Stressbedingungen,
Merkmalen, die verschiedene architektonische Merkmale und/oder biochemische und/oder
physiologische Merkmale modifizieren.
-
VII. Squamosa-Promotor-Bindungsprotein-ähnlich-11-(SPL11-)Transkriptionsfaktor-Polypeptid
-
Es
wurde jetzt überraschenderweise gefunden, dass die Modulation
der Expression einer Nukleinsäure, die ein SPL11-Polypeptid
kodiert, in einer Pflanze zu Pflanzen mit gesteigerten Ertragsmerkmalen
im Vergleich zu Kontrollpflanzen führt. In einer ersten
Ausführungsform stellt die vorliegende Erfindung ein Verfahren zur
Steigerung der Ertragsmerkmale in Pflanzen im Vergleich zu Kontrollpflanzen
bereit, umfassend das Modulieren der Expression einer Nukleinsäure,
die ein SPL11-Polypeptid kodiert, in einer Pflanze.
-
Die
vorliegende Erfindung stellt auch bisher unbekannte SPL11-kodierende
Nukleinsäuren und SPL11-Polypeptide bereit. Diese Sequenzen
sind ebenfalls zur Durchführung der erfindungsgemäßen
Verfahren geeignet.
-
Daher
wird gemäß einer weiteren Ausführungsform
der vorliegenden Erfindung ein isoliertes Nukleinsäuremolekül
bereitgestellt, das Folgendes umfasst:
- (i)
eine durch SEQ ID NO: 448 dargestellte Nukleinsäure;
- (ii) ein Komplement einer durch SEQ ID NO: 448 dargestellten
Nukleinsäure;
- (iii) eine Nukleinsäure, die für ein SPL11-Polypeptid
kodiert und, in steigender Reihenfolge der Präferenz, mindestens
70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
mit der in SEQ ID NO: 449 angegebenen Aminosäuresequenz
besitzt und, in steigender Reihenfolge der Präferenz, mindestens
70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
mit der SEQ ID NO: 465: SYCQVEGCRTDLSSAKDYHRKHRVCEPHSKAPKVVVAGLERRFC
QQCSRFHGLAEFDQKKKSCRRRLNDHNARRRKPQPEAL (die die SBP-Domäne
in der SEQ ID NO: 449 darstellt) besitzt;
- (iv) eine Nukleinsäure, die unter stringenten Bedingungen
an SEQ ID NO: 448 hybridisiert.
-
Weiterhin
wird auch ein isoliertes Polypeptid bereitgestellt, das Folgendes
umfasst:
- (i) eine Aminosäuresequenz
gemäß SEQ ID NO: 449;
- (ii) eine Aminosäuresequenz, die, in steigender Reihenfolge
der Präferenz, mindestens 70%, 75%, 80%, 85%, 90%, 95%,
96%, 97%, 98%, 99% oder mehr Sequenzidentität mit der in
SEQ ID NO: 449 angegebenen Aminosäuresequenz besitzt und,
in steigender Reihenfolge der Präferenz, mindestens 70%,
75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
mit der SEQ ID NO: 465: SYCQVEGCRTDLSSAKDYHRKHRVCEPHSKAPKVVVAGLERRFC
QQCSRFHGLAEFDQKKKSCRRRLNDHNARRRKPQPEAL (die die SBP-Domäne
in der SEQ ID NO: 449 darstellt) besitzt;
- (iii) Derivate von einer der vorstehend unter (i) oder (ii)
angegebenen Aminosäuresequenzen.
-
Ein
bevorzugtes Verfahren zum Modulieren (vorzugsweise Erhöhen)
der Expression einer Nukleinsäure, die ein SPL11-Polypeptid
kodiert, besteht darin, dass man eine Nukleinsäure, die
ein SPL11-Polypeptid kodiert, in eine Pflanze einbringt und in dieser
exprimiert.
-
Wird
im folgenden Text auf ein ”Protein, das für die
erfindungsgemäßen Verfahren geeignet ist” Bezug genommen,
versteht man darunter ein SPL11-Polypeptid, wie hier definiert.
Wird hier im Folgenden auf eine ”Nukleinsäure,
die für die erfindungsgemäßen Verfahren
geeignet ist” Bezug genommen, bedeutet dies eine Nukleinsäure,
die ein solches SPL11-Polypeptid kodieren kann. Die Nukleinsäure,
die in eine Pflanze eingeführt werden soll (und die daher
zur Durchführung der erfindungsgemäßen
Verfahren geeignet ist), ist jede beliebige Nukleinsäure,
die den Typ Protein kodiert, der im Folgenden beschrieben wird,
und wird im Folgenden auch als ”SPL11-Nukleinsäure” oder ”SPL11-Gen” bezeichnet.
-
Ein
SPL11-Polypeptid, wie hier definiert, betrifft ein Polypeptid, das
eine Squamosa-Bindungsprotein-(SBP-)Domäne umfasst, wobei
eine solche Domäne, in steigender Reihenfolge der Präferenz,
mindestens 70%, 75%, 80%, 85%, 90%, 95%, 97% oder mehr Sequenzidentität
mit einer der durch SEQ ID NO: 456 bis SEQ ID NO: 468 oder SEQ ID
NO: 478 dargestellten SBP-Domänen aufweist.
-
Ein ”SPL11-Polypeptid”,
wie hier definiert, umfasst das Protein gemäß SEQ
ID NO: 448, die identisch mit der SEQ ID NO: 172 ist, und Homologe
(Orthologe und Paraloge) davon.
-
Vorzugsweise
haben die Homologe der SEQ ID NO: 172 eine DNA-Bindungsdomäne.
-
SPL11-Polypeptide
lassen sich in speziellen Datenbanken finden, wie Pfam (Finn
et al. Nucleic Acids Research (2006) Database Issue 34: D247–D251).
Pfam listet eine große Sammlung an mehrfachen Sequenz-Alignments
und Hidden-Markov-Modellen (HMM) auf, die viele häufige
Proteindomänen und -familien abdeckt, und ist über
das Sanger Institute in Großbritannien verfügbar.
-
Die
Erfassungsausschlussgrenze für die SBP-Domäne
im Pfam HMM_fs-Modell und im Pfam HMM_Is-Modell beträgt
25,0. In der Pfam-Datenbank gelten als zuverlässige Übereinstimmungen
solche mit einem Score oberhalb der Erfassungsausschlussgrenze.
Mögliche Übereinstimmungen, die echte SBP-Domänen
umfassen, können jedoch unter den Erfassungsausschluss
fallen. Vorzugsweise ist ein SPL11-Polypeptid ein Protein mit einer
oder mehreren Domänen in seiner Sequenz, welche den Erfassungsausschluss
der Pfam-Proteindomänenfamilie PF03110, die als SBP-Domäne
bekannt ist, überschreiten.
-
Alternativ
kann eine SBP-Domäne in einem Polypeptid identifiziert
werden, indem man einen Sequenzvergleich mit bekannten Polypeptiden,
die eine SBP-Domäne umfassen, durchführt und die Ähnlichkeit im
Bereich der SBP-Domäne feststellt. Das Alignment der Sequenzen
kann unter Verwendung jedes im Stand der Technik bekannten Verfahrens
durchgeführt werden, wie Blast-Algorithmen. Die Wahrscheinlichkeit,
dass ein Alignment mit einer gegebenen Sequenz auftritt, wird als
Basis für die Identifikation ähnlicher Polypeptide verwendet.
Ein Parameter, der üblicherweise dazu verwendet wird, diese
Wahrscheinlichkeit darzustellen, wird als e-Wert bezeichnet. Der
E-Wert ist ein Maß für die Zuverlässigkeit
des S-Score. Der S-Score ist ein Maß für die Ähnlichkeit
der Abfragesequenz mit der gezeigten Sequenz. Der e-Wert beschreibt,
wie oft das zufallsgemäße Auftreten eines bestimmten
S-Score erwartet wird. Die e-Wert-Ausschlussgrenze kann bis zu 1,0
betragen. Die übliche Grenze für einen guten e-Wert
bei der Ausgabe einer BLAST-Suche, bei der ein SPL11-Polypeptid
als Abfragesequenz verwendet wird, kann kleiner als e5 (=
10–5), 1·e–10,
1·e–15, 1·e–20, 1·e–25,
1·e–50, 1·e–75 1·e–100 1·e–200 1·e–300,
1·e–400 1·e–500, 1·e–600 1·e–700 und 1·e–800 sein.
Erfindungsgemäße SPL11-Polypeptide umfassen vorzugsweise
eine Sequenz mit einem e-Wert von, in steigender Reihenfolge der
Präferenz, unter e–5 (=
10–5), 1·e–10,
1·e–15, 1·e–20, 1·e–25,
1·e–50, 1·e–75 1·e–100 1·e–200 1·e–300,
1·e–400 1·e–500, 1·e–600 1·e–700 und 1·e–800 bei einem
Alignment mit einer SBP-Domäne, die man in einem bekannten
SPL11-Polypeptid findet.
-
Beispiele
für SPL11-Polypeptide, die für die erfindungsgemäßen
Verfahren geeignet sind, sind in Tabelle G1 angegeben. Die Aminosäurekoordinaten
der SBP-Domäne in dem beispielhaften SPL11-Protein von Tabelle
G1 sind in Tabelle G4 angegeben und die Domänensequenz
wird durch SEQ ID NO: 456 bis SEQ ID NO: 468 dargestellt. Eine Konsensussequenz
der SBP-Domänen, die in SPL11-Polypeptiden vorliegen, zeigt SEQ
ID NO: 478.
-
Bevorzugte
erfindungsgemäße SPL11-Polypeptide sind solche,
die, in steigender Reihenfolge der Präferenz, mindestens
70%, 75%, 80%, 85%, 90%, 95% oder mehr Sequenzidentität
mit einem der in Tabelle G2 angegebenen Polypeptiden aufweisen.
-
SPL11-Polypeptide
können gewöhnlich zusätzlich zu der SBP-Domäne
eines oder mehrere der folgenden konservierten Motive an konservierten
Positionen in der Sequenz relativ zu der SBP-Domäne umfassen:
(i) Motif 1, das durch SEQ ID NO: 469 dargestellt wird, (ii) Motif
2 ist eine Serin-reiche Region, die man üblicherweise am
N-Terminus der SBP-Domäne findet und kann durch SEQ ID
NO: 470 dargestellt werden; (iii) Motif 3 gemäß SEQ
ID: 471, das in der Regel von Nukleotiden kodiert wird, die in der
SPL11-Polynukleotidregion enthalten sind, die von Mitgliedern der
miR156-MicroRNA-Familie angesteuert wird; (iv) Motif 4 gemäß SEQ
ID NO: 472. 27 zeigt die konservierten
Motive und ihre relative Position in der SPL11-Polypeptidsequenz,
die durch SEQ ID NO: 428 dargestellt wird.
-
Daher
umfassen bevorzugte SPL11-Polypeptide, die für das erfindungsgemäße
Verfahren geeignet sind, zusätzlich zu der SBP-Domäne
ein oder mehrere der folgenden konservierten Motive:
- (i) Motiv 1 gemäß SEQ ID NO: 469, wobei eine
beliebige konservative Aminosäuresubstitution und/oder
1 oder 2 nicht-konservative Substitutionen erlaubt sind,
- (ii) Motiv 2 gemäß SEQ ID NO: 470, wobei eine
beliebige Aminosäuresubstitution erlaubt ist unter der
Voraussetzung, dass mindestens 4 Aminosäuren eine polare
Seitenkette haben, vorzugsweise Serin oder Threonin, und unter der
Voraussetzung, dass sich das Motiv am N-Terminus der SBP-Domäne
befindet;
- (iii) Motiv 3 gemäß SEQ ID: 471, wobei 1 oder
2 Unterschiede erlaubt sind;
- (iv) Motiv 4 gemäß SEQ ID: 472, wobei 1, 2
oder 3 Unterschiede erlaubt sind.
-
Beispiele
für konservative Aminosäuresubstitutionen werden
im Abschnitt Hintergrund genannt. Üblicherweise sind die
in Polypeptiden enthaltenen Aminosäuren alpha-Aminosäuren
mit einer Amino- und einer Carboxylgruppe, die an das gleiche Kohlenstoffatom
gebunden sind, wobei die Aminosäuremoleküle oft
eine Seitenkette umfassen, die an das alpha-Kohlenstoffatom gebunden
ist. Tabelle 3 zeigt die Einteilung von Aminosäuren auf
der Basis der physikalischen und biochemischen Eigenschaften der
Seitenkette. Tabelle 3. Einteilung von Aminosäuren
anhand der Seitenketteneigenschaften.
| Aminosäure | 3-Buchstaben | 1-Buchstaben | Seitenkettenpolarität | Seitenkettenazidität
oder basizität | Hydropathie-Index |
| Arginin | Arg | R | polar | basisch | –4,5 |
| Asparagin | Asn | N | polar | neutral | –3,5 |
| Asparaginsäure | Asp | D | polar | sauer | –3,5 |
| Cystein | Cys | C | polar | neutral | 2,5 |
| Glutaminsäure | Glu | E | polar | sauer | –3,5 |
| Glutamin | Gin | Q | polar | neutral | –3,5 |
| Histidin | His | H | polar | basisch | –3,2 |
| Lysin | Lys | K | polar | basisch | –3,9 |
| Serin | Ser | S | polar | neutral | –0,8 |
| Threonin | Thr | T | polar | neutral | –0,7 |
| Tyrosin | Tyr | Y | polar | neutral | –1,3 |
| Alanin | Ala | A | unpolar | neutral | 1,8 |
| Glycin | Gly | G | unpolar | neutral | –0,4 |
| Isoleucin | Ile | I | unpolar | neutral | 4,5 |
| Leucin | Leu | L | unpolar | neutral | 3,8 |
| Methionin | Met | M | unpolar | neutral | 1,9 |
| Phenylalanin | Phe | F | unpolar | neutral | 2,8 |
| Prolin | Pro | P | unpolar | neutral | –1,6 |
| Tryptophan | Trp | W | unpolar | neutral | –0,9 |
| Valin | Val | V | unpolar | neutral | 4,2 |
-
Beispiele
für SPL11-Polypeptide, die ein oder mehrere der konservierten
Motive Motiv 1 bis Motiv 4 umfassen, werden in Beispiel 62 genannt. 28 zeigt die Position der konservierten Motive
in diesen SPL11-Polypeptiden.
-
Wenn
es zur Konstruktion eines Stammbaums, wie dem in 29 dargestellten, verwendet wird, bildet das erfindungsgemäße
SPL11-Polypeptid vorzugsweise eher mit der S3-Gruppe, die die Aminosäuresequenz
gemäß SEQ ID NO: 428 umfasst, (in 29 als AtSPL11 bezeichnet) als mit jeder anderen
Gruppe Cluster.
-
Der
Begriff ”Domäne” und ”Motiv” wird
hier im Abschnitt ”Definitionen” definiert. Für
die Identifikation von Domänen gibt es Spezialdatenbanken,
zum Beispiel SMART (Schultz et al., (1998), Proc. Natl.
Acad. Sci. USA 95, 5857–5864; Letunic
et al., (2002), Nucleic Acids Res. 30, 242–244,
InterPro (Mulder et al., (2003), Nucl. Acids Res. 31, 315–318,
Prosite (Sucher und Bairoch (1994), A generalized Profile
syntax for biomolecular sequences motifs and its function in automatic
sequence interpretation. (In) ISMB-94; Proceedings 2nd International
Conference an Intelligent Systems for Molecular Biology. Altman
R., Brutlag D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61,
AAAIPress, Menlo Park; Hulo et al., Nucl. Acids
Res. 32: D134–D137, (2004), oder Pfam (Bateman
et al., Nucleic Acids Research 30 (1): 276–280 (2002).
Ein Satz von Werkzeugen für die In-si/ico-Analyse von Proteinsequenzen
ist auf dem ExPASy-Proteomics-Server verfügbar (Swiss Institute
of Bioinformatics (Gasteiger et al., ExPASy: the proteomics
server for in-depth Protein knowledge and analysis, Nucleic Acids
Res. 31: 3784–3788 (2003)). Domänen können
auch unter Verwendung von Routinetechniken, wie Sequenz-Alignment,
identifiziert werden.
-
Verfahren
für das Alignment von Sequenzen für Vergleichszwecke
sind in der Fachwelt gut bekannt, dazu zählen GAP, BESTFIT,
BLAST, FASIA und TFASTA. Bei GAP wird der Algorithmus von Needleman
und Wunsch ((1970), J. Mol. Biol. 48: 443–453)
dazu verwendet, das globale Alignment (d. h. das Alignment, das sich über
die vollständigen Sequenzen erstreckt) von zwei Sequenzen
zu finden, das die Anzahl der Übereinstimmungen (”Matches”)
maximiert und die Anzahl der Lücken (”Gaps”)
minimiert. Beim BLAST-Algorithmus (Altschul et al., (1990),
J. Mob. Biol. 215: 403–10) wird die Sequenzidentität
in Prozent berechnet, und es wird eine statistische Analyse der Ähnlichkeit
zwischen den beiden Sequenzen durchgeführt. Die Software
für die Durchführung einer BLAST-Analyse ist über
das National Centre for Biotechnology Information (NCBI) öffentlich
zugänglich. Homologe können zum Beispiel unter
Verwendung des multiplen Sequenz-Alignment-Algorithmus ClustalW
(Version 1.83) leicht identifiziert werden, und zwar mit den Default-Parametern
für paarweises Alignment und einer Scoring-Methode in Prozent.
Die Gesamtprozent an Ähnlichkeit und Identität
können auch unter Verwendung eines der in dem MatGAT-Software-Paket
verfügbaren Verfahren bestimmt werden (Campanella
et al., BMC Bioinformatics. Juli 2003, 10;4: 29. MatGAT: an application
that generstes similarity/identity matrices using Protein or DNA
sequences.). Für eine Optimierung des Alignments
zwischen konservierten Motiven können kleinere Änderungen
per Hand vorgenommen werden, wie dem Fachmann bekannt ist. Weiterhin können
anstelle von Volllängensequenzen für die Identifikation
von Homologen auch spezifische Domänen verwendet werden.
Die Sequenzidentitätswerte können mit den oben
erwähnten Programmen unter Verwendung der Default-Parameter über
die gesamte Nukleinsäure- oder Aminosäuresequenz
oder über ausgewählte Domänen oder ein
konservierte(s) Motiv(e) bestimmt werden.
-
Weiterhin
können SPL11-Polypeptide (zumindest in ihrer nativen Form)
gewöhnlich DNA-Bindungsaktivität besitzen, insbesondere
können sie DNA-Fragmente binden, welche die SBP-Domänen-DNA-Bindungsbox
umfassen, wie in SEQ ID NO: 49 dargestellt. In der Regel findet
man die SBP-Domänen-DNA-Bindungsbox in Genpromotoren pflanzlichen
Ursprungs, wie im SQAMOSA-Gen. Fragmente, welche die SBP-Domänen-DNA-Bindungsbox
umfassen, sind vorzugsweise mehr als 10, 15, 20, 25, 100, 200, 500,
1000, 2000 Basenpaare lang. Verfahren zur Ermittlung der DNA-Bindung
von Proteinen, die eine SBP-Domäne enthalten, lassen sich
auf SPL11-Polypeptide anwenden und sind im Stand der Technik bekannt
(Klein at al. Mol Gen Genet. 1996, 15; 250 (1): 7–16; Yamasaki
et al. 2004 J Mol Biol. 2004, 12; 337 (1): 49–63).
-
Bevorzugte
erfindungsgemäße SPL11-Polypeptide sind solche
mit DNA-Bindungsaktivität, stärker bevorzugt solche,
die ein DNA-Fragment binden, das die SEQ ID NO: 475 umfasst.
-
Die
vorliegende Erfindung wird dadurch veranschaulicht, dass man Pflanzen
mit der Nukleinsäuresequenz gemäß SEQ
ID NO: 427, die die Polypeptidsequenz gemäß SEQ
ID NO: 428 kodiert, transformiert. Die Durchführung der
Erfindung ist jedoch nicht auf diese Sequenzen beschränkt;
die erfindungsgemäßen Verfahren können
vorteilhaft unter Verwendung jeder SPL11-kodierenden Nukleinsäure
oder mit SPL11-Polypeptid, wie hier definiert, durchgeführt
werden.
-
Beispiele
für Nukleinsäuren, die SPL11-Polypeptide kodieren,
sind hier in Tabelle G1 von Beispiel 62 angegeben. Solche Nukleinsäuren
eignen sich zur Durchführung der erfindungsgemäßen
Verfahren. Die in Tabelle G1 von Beispiel 62 angegebenen Aminosäuresequenzen
sind beispielhafte Sequenzen von Orthologen und Paralogen des SPL11-Polypeptids
gemäß SEQ ID NO: 428, wobei die Begriffe ”Orthologe” und ”Paraloge” wie
hier definiert sind. Weitere Orthologe und Paraloge können
leicht identifiziert werden, indem man eine so genannte reziproke
Blast-Suche durchführt. Dies beinhaltet üblicherweise
eine erste BLAST, bei der mit einer Abfragesequenz ein ”BLASTing” gegen
eine beliebige Sequenzdatenbank, wie die öffentlich zugängliche
NCBI-Datenbank, durchgeführt wird (zum Beispiel unter Verwendung
von einer der in Tabelle G1 von Beispiel 62 aufgelisteten Sequenzen).
BLASTN oder TBLASTX (unter Verwendung von Standard-Default-Werten)
werden im Allgemeinen dann verwendet, wenn man von einer Nukleotidsequenz
ausgeht, und BLASTP oder TBLASTN (unter Verwendung von Standard-Default-Werten),
wenn man von einer Proteinsequenz ausgeht. Die BLAST-Ergebnisse
können gegebenenfalls gefiltert werden. Mit den Volllängen-Sequenzen
der gefilterten oder ungefilterten Ergebnisse wird anschließend
ein zweites BLASTing gegen Sequenzen des Organismus, von dem die
Abfragesequenz stammt, durchgeführt (ist die Abfragesequenz
die SEQ ID NO: 427 oder SEQ ID NO: 428, dann wäre das zweite
BLASTing daher gegen Sequenzen aus Arabidopsis). Die Ergebnisse
des ersten und des zweiten BLASTing werden dann verglichen. Ein
Paralog wird identifiziert, wenn ein hochrangiger Hit von dem ersten
BLASTing aus derselben Art stammt wie die Abfragesequenz, in diesem
Fall führt ein zweites BLASTing idealerweise zu der Abfragesequenz
unter den höchstrangigen Hits; ein Ortholog wird identifiziert,
wenn ein hochrangiger Hit in dem ersten BLASTing nicht von derselben
Art stammt wie die Abfragesequenz und vorzugsweise beim zweiten
BLASTing dazu führt, dass die Abfragesequenz unter den
höchstrangigen Hits ist.
-
Hochrangige
Hits sind solche mit niedrigem E-Wert. Je niedriger der E-Wert,
desto signifikanter der ”Score” (anders ausgedrückt,
desto niedriger die Wahrscheinlichkeit, dass der Hit durch Zufall
gefunden wurde). Die Berechnung des E-Werts ist in der Fachwelt
gut bekannt. Das ”Scoring” der Vergleiche erfolgt
nicht nur mittels E-Werten, sondern auch anhand der Prozent Identität.
Prozent Identität bezieht sich auf die Anzahl an identischen
Nukleotiden (oder Aminosäuren) zwischen den zwei verglichenen
Nukleinsäure-(oder Polypeptid-)Sequenzen über
eine bestimmte Länge. Bei großen Familien kann
man ClustalW und anschließend einen Neighbour-Joining-Tree
verwenden, um die Bildung von Clustern verwandter Gene leichter
sichtbar zu machen und Orthologe und Paraloge zu identifizieren.
-
Zur
Durchführung der erfindungsgemäßen Verfahren
können auch Nukleinsäurevarianten geeignet sein.
Zu solchen Varianten zählen beispielsweise Nukleinsäuren,
die Homologe und Derivate von einer der in Tabelle G1 von Beispiel
62 genannten Aminosäuresequenzen kodieren, wobei die Begriffe ”Homolog” und ”Derivat” wie
hier definiert sind. Für die erfindungsgemäßen
Verfahren sind auch Nukleinsäuren geeignet, die Homologe
und Derivate von Orthologen oder Paralogen von einer der in Tabelle
G1 von Beispiel 62 genannten Aminosäuresequenzen kodieren.
Homologe und Derivate, die für die erfindungsgemäßen
Verfahren geeignet sind, weisen im Wesentlichen dieselbe biologische
und funktionelle Aktivität wie das unmodifizierte Protein, von
dem sie stammen, auf.
-
Zu
weiteren Nukleinsäurevarianten, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet sind,
zählen Abschnitte von Nukleinsäuren, die SPL11-Polypeptide
kodieren, Nukleinsäuren, die mit Nukleinsäuren,
die SPL11-Polypeptide kodieren, hybridisieren, Spleißvarianten
von Nukleinsäuren, die SPL11-Polypeptide kodieren, allelische
Varianten von Nukleinsäuren, die SPL11-Polypeptide kodieren,
und Varianten von Nukleinsäuren, die SPL11-Polypeptide
kodieren, die mittels ”gene shuffling” erhalten
wurden.
-
Die
Begriffe hybridisierende Sequenz, Spleißvariante, allelische
Variante und ”gene shuffling” sind wie hier beschrieben.
-
Eine
bevorzugte Nukleinsäurevariante, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet ist,
ist eine Nukleinsäure, die ein SPL11-Polypeptid kodiert,
das mikroRNA-unempfindlich ist, weiterhin bevorzugt ist die Nukleinsäure
unempfindlich gegenüber mikroRNAs, die zur miR156-Familie
gehören. MikroRNAs steuern Nukleinsäuren (insbesondere
RNA) an, um sie zu zerstören, wodurch es in der Regel zu
einer Verringerung oder Hemmung der Akkumulation der angesteuerten
RNA kommt. Für die Ansteuerung ist eine Hybridisierung
zwischen der mikroRNA und der angesteuerten Nukleinsäure
(RNA) in einer ganz spezifischen Region erforderlich, die man als
miR-(mikroRNA-)Zielstelle bezeichnet und die eine Sequenz umfasst, die
zu einem Abschnitt des reifen mikroRNA-Gens komplementär
ist. Üblicherweise umfassen die mikroRNA-unempfindlichen
Nukleinsäuren eine Sequenz, die, in steigender Reihenfolge
der Präferenz, 1, 2, 3, 4, 5 oder mehr Fehlpaarungen bei
einem Alignment mit dem entsprechenden mikroRNA-Molekül
in der miR-Zielstelle aufweist. MikroRNA-unempfindliche Nukleinsäuren
können in einer Zelle bis auf akkumulieren Niveaus, die,
in steigender Reihenfolge der Präferenz, das 5-, 10-, 20-,
30-, 40-, 50-Fache oder mehr der Nukleinsäure sind, die
von der entsprechenden mikroRNA angesteuert wird, die im Allgemeinen
100% Sequenzkomplementarität in der miR-Zielstelle aufweist.
-
Die
miR156-Familie wurde bereits beschrieben und eine Auflistung der
mikroRNAs einschließlich der Mitglieder der miR156-Familie
ist unter miRBase-Datenbank (Griffiths-Jones et al. 2006
Nucleic Acids Research, 2006, Bd. 34, Database issue D140–D144)
zu finden. Die miRBase-Datenbank wird am The Wellcome Trust Sanger
Institute in Cambridge, GB, gepflegt. Die miR156-Zielstelle in einer
SPL11-Nukleinsäure ist komplementär zu der reifen
Sequenz von miR156-mikroRNAs. Ein Beispiel für reife Sequenzen
der Mitglieder der miR156-Familie aus Reis und ihre entsprechenden
Zielstellen auf Reis-SPL-Nukleinsäuren sind in 30A angegeben. 31B zeigt
ein Alignment beispielhafter SPL11-Nukleinsäuren (in der
DNA-Form), in dem die Zielstelle für miR156 in den entsprechenden
Ribonukleinsäuren angegeben ist. Ein Beispiel für
eine miR156-unempfindliche SPL11-Nukleinsäure, welche die
SEQ ID NO: 428 kodiert, wird durch SEQ ID NO: 431 wiedergegeben.
Weitere Beispiele für miR156-unempfindliche SPL11-Nukleinsäuren
werden durch SEQ ID NO: 440 und SEQ ID NO: 454 wiedergegeben, wobei
in der Letzteren die miR156-Zielstelle fehlt.
-
Eine
SPL11-Nukleinsäure, die für die erfindungsgemäßen
Verfahren geeignet ist, hat vorzugsweise 1, 2, 3, 4 oder mehr Fehlpaarungen
in der miR156-Zielstelle oder ihr fehlt die Zielstelle des miR156-Gens.
Beispiele für solche SPL11-Nukleinsäuren sind
in 31B angegeben. Weiterhin bevorzugt
ist eine Nukleinsäure, wie sie durch SEQ ID NO: 431, SEQ
ID 440 und SEQ ID NO: 454 dargestellt wird.
-
Nukleinsäuren,
die SPL11-Polypeptide kodieren, müssen keine Volllängen-Nukleinsäuren
sein, da die Durchführung der erfindungsgemäßen
Verfahren nicht auf die Verwendung von Volllängen-Nukleinsäuresequenzen
angewiesen ist. Erfindungsgemäß wird daher ein
Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen bereitgestellt,
bei dem man in eine Pflanze einen Abschnitt einer beliebigen der
in Tabelle G1 von Beispiel 62 angegebenen Nukleinsäuresequenzen
oder einen Abschnitt einer Nukleinsäure, die ein Ortholog, Paralog
oder Homolog von einer der in Tabelle G1 von Beispiel 62 genannten
Aminosäuresequenzen kodiert, einführt und darin
exprimiert.
-
Ein
Abschnitt einer Nukleinsäure kann zum Beispiel dadurch
hergestellt werden, dass man eine oder mehrere Deletionen an der
Nukleinsäure durchführt. Die Abschnitte können
in isolierter Form verwendet werden oder können an andere
kodierende (oder nichtkodierende) Sequenzen fusioniert werden, um
zum Beispiel ein Protein herzustellen, das mehrere Aktivitäten
in sich vereinigt. Wenn es an andere kodierende Sequenzen fusioniert
ist, kann das so erhaltene, nach der Translation hergestellte Polypeptid
größer sein als für den Proteinabschnitt
vorhergesagt wurde.
-
Abschnitte,
die für die erfindungsgemäßen Verfahren
geeignet sind, kodieren ein SPL11-Polypeptid, wie hier definiert,
und weisen im Wesentlichen dieselbe biologische Aktivität
wie die in Tabelle G1 von Beispiel 62 angegebenen Aminosäuresequenzen
auf. Vorzugsweise handelt es sich bei dem Abschnitt um einen Abschnitt
einer der in Tabelle G1 von Beispiel 62 genannten Nukleinsäuren
oder um einen Abschnitt einer Nukleinsäure, die ein Ortholog
oder Paralog von einer der in Tabelle G1 von Beispiel 62 genannten
Aminosäuresequenzen kodiert. Vorzugsweise ist der Abschnitt
mindestens 70, 100, 200, 500, 550, 600, 650, 700, 750, 800, 850,
900, 950, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 5000 aufeinander
folgende Nukleotide lang, wobei es sich bei den aufeinander folgenden
Nukleotiden um eine der in Tabelle G1 von Beispiel 62 genannten
Nukleinsäuresequenzen oder um eine Nukleinsäure
handelt, die ein Ortholog oder Paralog von einer der in Tabelle
G1 von Beispiel 62 genannten Aminosäuresequenzen kodiert.
Vorzugsweise kodiert der Abschnitt mindestens eine SBP-Domäne.
Am stärksten bevorzugt ist der Abschnitt ein Abschnitt
der Nukleinsäure gemäß SEQ ID NO: 427.
Vorzugsweise kodiert der Abschnitt eine Aminosäuresequenz,
die, wenn sie zur Konstruktion eines Stammbaums, wie dem in 29 dargestellten, verwendet wird, eher mit der
Gruppe der SPL11-Polypeptide, welche die in SEQ ID NO: 428 dargestellte
Aminosäuresequenz (AtSPL 11) umfassen, als mit jeder anderen
Gruppe Cluster bildet.
-
Eine
andere Nukleinsäurevariante, die für die erfindungsgemäßen
Verfahren geeignet ist, ist eine Nukleinsäure, die unter
Bedingungen einer verringerten Stringenz, vorzugsweise unter stringenten
Bedingungen, mit einer Nukleinsäure, die ein SPL11-Polypeptid
kodiert, wie hier definiert, oder mit einem Abschnitt, wie hier definiert,
hybridisieren kann.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine Pflanze eine Nukleinsäure,
die mit einer der in Tabelle G1 von Beispiel 62 angegebenen Nukleinsäuren
hybridisieren kann, einführt und darin exprimiert, oder
bei dem man in eine Pflanze eine Nukleinsäure, die mit
einer Nukleinsäure hybridisieren kann, die ein Ortholog,
Paralog oder Homolog von einer der in Tabelle G1 von Beispiel 62
genannten Nukleinsäuresequenzen kodiert, einführt
und darin exprimiert.
-
Hybridisierende
Sequenzen, die für die erfindungsgemäßen
Verfahren geeignet sind, kodieren ein SPL11-Polypeptid, wie hier
definiert, und weisen im Wesentlichen dieselbe biologische Aktivität
wie die in Tabelle G1 von Beispiel 62 genannten Aminosäuresequenzen
auf. Vorzugsweise kann die hybridisierende Sequenz mit einer der
in Tabelle G1 von Beispiel 62 genannten Nukleinsäuren oder
mit einem Abschnitt von einer dieser Sequenzen hybridisieren, wobei
ein Abschnitt wie oben definiert ist, oder wobei die hybridisierende
Sequenz mit einer Nukleinsäuresequenz, die ein Ortholog
oder Paralog von einer der in Tabelle G1 von Beispiel 62 genannten
Aminosäuresequenzen kodiert, hybridisieren kann. Am stärksten
bevorzugt kann die hybridisierende Sequenz mit einer Nukleinsäure
gemäß SEQ ID NO: 427 oder einem Abschnitt davon
hybridisieren.
-
Vorzugsweise
kodiert die hybridisierende Sequenz eine Aminosäuresequenz,
die, wenn sie zur Konstruktion eines Stammbaums, wie dem in 29 dargestellten, verwendet wird, eher mit der
Gruppe der SPL11-Polypeptide, welche die in SEQ ID NO: 428 dargestellte
Aminosäuresequenz (AtSPL 11) umfassen, als mit jeder anderen
Gruppe Cluster bildet.
-
Eine
andere Nukleinsäurevariante, die für die erfindungsgemäßen
Verfahren geeignet ist, ist eine Spleißvariante, die ein
SPL11-Polypeptid, wie hier vorstehend definiert, kodiert, wobei
eine Spleißvariante wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine Spleißvariante
einer der in Tabelle G1 von Beispiel 62 angegebenen Nukleinsäuresequenzen
oder eine Spleißvariante von einer Nukleinsäure,
die ein Ortholog, Paralog oder Homolog von einer der in Tabelle
G1 von Beispiel 62 genannten Aminosäuresequenzen kodiert,
einführt und exprimiert. Beispiele für gespleißte
Varianten des Gens, das SEQ ID NO: 428 kodiert, werden durch SEQ
ID NO: 427; SEQ ID NO: 429 and SEQ ID NO: 430 dargestellt.
-
Bevorzugte
Spleißvarianten sind Spleißvarianten einer Nukleinsäure
gemäß SEQ ID NO: 427 oder eine Spleißvariante
einer Nukleinsäure, die ein Ortholog oder Paralog der SEQ
ID NO: 428 kodiert. Vorzugsweise bildet die von der Spleißvariante
kodierte Aminosäuresequenz, wenn sie zur Konstruktion eines
Stammbaums, wie dem in 29 dargestellten,
verwendet wird, eher mit der Gruppe der SPL11-Polypeptide, welche die
in SEQ ID NO: 428 dargestellte Aminosäuresequenz (AtSPL
11) umfassen, als mit jeder anderen Gruppe Cluster.
-
Eine
andere Nukleinsäurevariante, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet ist,
ist eine allelische Variante einer Nukleinsäure, die ein
SPL11-Polypeptid, wie hier vorstehend definiert, kodiert, wobei
eine allelische Variante wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren zur Verbesserung von Ertragsmerkmalen bei Pflanzen
bereitgestellt, bei dem man in eine(r) Pflanze eine allelische Variante
von einer der in Tabelle G1 von Beispiel 62 angegebenen Nukleinsäuresequenzen
einführt und exprimiert oder bei dem man in eine(r) Pflanze
eine allelische Variante einer Nukleinsäure, die ein Ortholog,
Paralog oder Homolog von einer der in Tabelle G1 von Beispiel 62
genannten Aminosäuresequenzen kodiert, einführt
und exprimiert.
-
Die
allelischen Varianten, die für die erfindungsgemäßen
Verfahren geeignet sind, haben im Wesentlichen dieselbe biologische
Aktivität wie das SPL11-Polypeptid gemäß SEQ
ID NO: 428 und eine der in Tabelle G1 von Beispiel 62 dargestellten
Aminosäuren. Allelische Varianten kommen in der Natur vor
und von den erfindungsgemäßen Verfahren wird die
Verwendung dieser natürlichen Allele mit umfasst. Vorzugsweise
handelt es sich bei der allelischen Variante um eine allelische
Variante gemäß SEQ ID NO: 427 oder eine allelische Variante
einer Nukleinsäure, die ein Ortholog oder Paralog der SEQ
ID NO: 428 kodiert. Vorzugsweise bildet die Aminosäuresequenz,
die von der allelischen Variante kodiert wird, wenn sie zur Konstruktion
eines Stammbaums, wie dem in 29 dargestellten,
verwendet wird, eher mit der Gruppe der SPL11-Polypeptide, welche die
in SEQ ID NO: 428 dargestellte Aminosäuresequenz (AtSPL
11) umfassen, als mit jeder anderen Gruppe Cluster.
-
Man
kann auch ”Gene shuffling” oder gerichtete Evolution
dazu verwenden, Varianten von Nukleinsäuren zu erzeugen,
die SPL11-Polypeptide, wie oben definiert, kodieren, wobei der Begriff ”gene
shuffling” wie hier definiert ist.
-
Erfindungsgemäß wird
ein Verfahren für die Verbesserung von Ertragsmerkmalen
bei Pflanzen bereitgestellt, bei dem man in eine(r) Pflanze eine
Variante von einer der in Tabelle G1 von Beispiel 62 angegebenen Nukleinsäuresequenzen
einführt und exprimiert oder bei dem man in eine(r) Pflanze
eine Variante einer Nukleinsäure, die ein Ortholog, Paralog
oder Homolog von einer der in Tabelle G1 von Beispiel 62 angegebenen Aminosäuresequenzen
kodiert, einführt und exprimiert, wobei die Nukleinsäurevariante
mittels ”gene shuffling” erhalten wird.
-
Vorzugsweise
bildet die Aminosäuresequenz, die von der durch gene shuffling
erhaltenen Nukleinsäurevariante kodiert wird, wenn sie
zur Konstruktion eines Stammbaums, wie dem in 29 dargestellten, verwendet wird, eher mit der
Gruppe der SPL11-Polypeptide, welche die in SEQ ID NO: 428 dargestellte
Aminosäuresequenz (AtSPL 11) umfassen, als mit jeder anderen
Gruppe Cluster.
-
Nukleinsäurevarianten
können weiterhin auch durch ortsgerichtete Mutagenese erhalten
werden. Zur Erzielung einer ortsgerichteten Mutagenese sind verschiedene
Verfahren verfügbar, von denen die häufigsten Verfahren
auf PCR-Basis sind (Current Protocols in Molecular Biology. Wiley
Hrsg.).
-
Nukleinsäuren,
die SPL11-Polypeptide kodieren, können von einer beliebigen
natürlichen oder künstlichen Quelle stammen. Die
Nukleinsäure kann von ihrer nativen Form in ihrer Zusammensetzung
und/oder genomischen Umgebung durch gezielte Manipulation durch
den Menschen modifiziert werden. Vorzugsweise stammt die Nukleinsäure,
die das SPL11-Polypeptid kodiert, aus einer Pflanze, weiter bevorzugt
aus einer zweikeimblättrigen Pflanze, stärker
bevorzugt aus der Familie Brassicaceae, am stärksten bevorzugt
stammt die Nukleinsäure aus Arabidopsis thaliana.
-
Die
Durchführung der erfindungsgemäßen Verfahren
führt zu Pflanzen mit verstärkten Ertragsmerkmalen.
Insbesondere führt die Durchführung der erfindungsgemäßen
Verfahren zu Pflanzen mit erhöhtem Ertrag, insbesondere
erhöhtem Samenertrag, im Vergleich zu Kontrollpflanzen.
Die Begriffe ”Ertrag” und ”Samenertrag” werden
hier im Abschnitt ”Definitionen” genauer beschrieben.
-
Werden
hier verbesserte Ertragsmerkmale erwähnt, so soll dies
eine Erhöhung der Biomasse (des Gewichts) von einem oder
mehreren Teilen einer Pflanze bedeuten, was oberirdische (erntbare)
Teile und/oder unterirdische (erntbare) Teile beinhalten kann. Insbesondere
sind solche oberirdischen erntbaren Teile Samen, und die Durchführung
der erfindungsgemäßen Verfahren führt
zu Pflanzen mit höherem Ertrag im Vergleich zu kontrollpflanzen.
-
Wählt
man Mais als Beispiel, kann sich eine Ertragserhöhung unter
Anderem als eines oder mehrere der folgenden Merkmale äußern:
Erhöhung der Anzahl Pflanzen, die sich pro Hektar oder
Acre entwickeln, Erhöhung der Anzahl Ähren pro
Pflanze, Erhöhung der Anzahl Reihen, der Anzahl Körner
pro Reihe, des Korngewichts, Tausendkorngewichts, Ährenlänge/durchmessers,
Erhöhung der Samenfüllungsrate (Anzahl gefüllter
Samen dividiert durch die Gesamtzahl Samen und multipliziert mit
Hundert). Wählt man Reis als Beispiel, kann sich eine Ertragssteigerung
unter Anderem als Erhöhung von einem oder mehreren der
folgenden Merkmale ausdrücken: Anzahl Pflanzen pro Hektar
oder Acre, Anzahl Rispen pro Pflanze, Anzahl Ährchen pro
Rispe, Anzahl Blüten (Blütchen) pro Rispe (ausgedrückt
als Verhältnis der Anzahl der gefüllten Samen
zu der Anzahl der Primärrispen), erhöhte Samenfüllungsrate
(Anzahl gefüllter Samen dividiert durch die Gesamtzahl
Samen und multipliziert mit Hundert), erhöhtes Tausendkorngewicht.
-
Die
vorliegende Erfindung stellt ein Verfahren zur Steigerung des Ertrags,
insbesondere Samenertrags von Pflanzen im Vergleich zu Kontrollpflanzen
bereit, wobei man bei dem Verfahren in einer Pflanze die Expression
einer Nukleinsäure, die ein SPL11-Polypeptid wie hier definiert
kodiert, moduliert, vorzugsweise erhöht.
-
Da
die erfindungsgemäßen transgenen Pflanzen einen
erhöhten Ertrag aufweisen zeigen diese Pflanzen (zumindest
während eines Teils ihres Lebenszyklus) wahrscheinlich
eine im Vergleich zu der Wachstumsrate von Kontrollpflanzen in einem
entsprechenden Stadium ihres Lebenszyklus erhöhte Wachstumsrate.
-
Die
erhöhte Wachstumsrate kann für einen oder mehrere
Teile einer Pflanze (einschließlich Samen) spezifisch sein
oder kann im Wesentlichen in der ganzen Pflanze stattfinden. Pflanzen
mit einer erhöhten Wachstumsrate können einen
kürzeren Lebenszyklus aufweisen. Unter dem Lebenszyklus
einer Pflanze kann man diejenige Zeit verstehen, die die Pflanze
braucht, um von einem reifen trockenen Samen bis zu dem Stadium
heranzuwachsen, in dem die Pflanze reife trockene Samen ähnlich
dem Ausgangsmaterial produziert hat. Dieser Lebenszyklus kann von
Faktoren wie Jungpflanzenvitalität, Wachstumsrate, ”Greenness
Index”, Blütezeit und Geschwindigkeit der Samenreifung
beeinflusst werden. Die erhöhte Wachstumsrate kann in einem
Stadium oder in mehreren Stadien im Lebenszyklus einer Pflanze oder
im Wesentlichen während des gesamten Lebenszyklus der Pflanze
stattfinden. Eine erhöhte Wachstumsrate während
der Frühstadien im Lebenszyklus einer Pflanze kann verbesserte
Vitalität widerspiegeln. Die erhöhte Wachstumsrate
kann den Erntezyklus einer Pflanze verändern, so dass die
Pflanzen später gesät werden können und/oder
früher geerntet werden können, als dies sonst
möglich wäre (ein ähnlicher Effekt lässt
sich mit einer früheren Blütezeit erzielen). Ist
die Wachstumsrate ausreichend erhöht, kann dies ein weiteres
Aussäen von Samen der gleichen Pflanzenart ermöglichen
(zum Beispiel Aussäen und Ernten von Reispflanzen und anschließendes
Aussäen und Ernten von weiteren Reispflanzen innerhalb
einer traditionellen Wachstumsperiode). Entsprechend kann dies bei
ausreichender Steigerung der Wachstumsrate ein weiteres Aussäen
von Samen von unterschiedlichen Pflanzenarten ermöglichen
(zum Beispiel das Aussäen und Ernten von Maispflanzen und
anschließend z. B. Aussäen und gegebenenfalls
Ernten von Sojabohnen, Kartoffeln oder sonstigen geeigneten Pflanzen). Bei
manchen Kulturpflanzen kann man auch zusätzlich mehrmals
von demselben Wurzelstock ernten. Eine Veränderung des
Erntezyklus einer Pflanze kann zu einer Erhöhung der jährlichen
Biomasseproduktion pro Acre führen (und zwar aufgrund einer
Erhöhung der Häufigkeit (z. B. pro Jahr), mit
der eine bestimmte Pflanze herangezogen und geerntet werden kann).
Eine erhöhte Wachstumsrate kann auch den Anbau von transgenen
Pflanzen in einem weiteren geographischen Bereich als ihre Wildtyp-Gegenstücke
ermöglichen, da die räumlichen Begrenzungen für
den Anbau einer Kulturpflanze häufig von ungünstigen
Umweltbedingungen entweder während der Pflanzzeit (früh
in der Saison) oder während der Erntezeit (spät
in der Saison) bestimmt wird. Solche ungünstigen Bedingungen
können vermieden werden, wenn der Erntezyklus verkürzt
ist. Die Wachstumsrate kann durch Ableiten von verschiedenen Parametern
von Wachstumskurven bestimmt werden, wobei die Parameter unter Anderem
folgendes sein können: T-Mid (Zeitdauer, die die Pflanzen
benötigen, um 50% ihrer Maximalgröße
zu erreichen) und T-90 (Zeitdauer, die die Pflanzen benötigen,
um 90% ihrer Maximalgröße zu erreichen).
-
Gemäß einem
bevorzugten Merkmal der vorliegenden Erfindung ergibt die Durchführung
der erfindungsgemäßen Verfahren Pflanzen mit einer
im Vergleich zu Kontrollpflanzen erhöhten Wachstumsrate.
Die vorliegende Erfindung stellt daher ein Verfahren zur Erhöhung
der Wachstumsrate von Pflanzen bereit, wobei man bei dem Verfahren
in einer Pflanze die Expression einer Nukleinsäure, die
ein SPL11-Polypeptid wie hier definiert kodiert, moduliert, vorzugsweise
erhöht.
-
Eine
Erhöhung des Ertrags und/oder der Wachstumsrate findet
unabhängig davon statt, ob die Pflanze unter Nichtstressbedingungen
steht oder ob die Pflanze im Vergleich zu Kontrollpflanzen verschiedenen Stressfaktoren
unterworfen wird. Typischerweise reagieren Pflanzen auf Stress durch
langsameres Wachstum. Unter starken Stressbedingungen kann die Pflanze
sogar ihr Wachstum einstellen. Leichter Stress wiederum wird im
vorliegenden Zusammenhang als jeglicher Stress definiert, dem eine
Pflanze ausgesetzt ist und der nicht dazu führt, dass die
Pflanze ihr Wachstum völlig einstellt, ohne ihr Wachstum
wieder aufnehmen zu können. Leichter Stress im Sinne der
Erfindung führt zu einer Verringerung des Wachstums der
gestressten Pflanzen von weniger als 40%, 35% oder 30%, vorzugsweise
weniger als 25%, 20% oder 15%, stärker bevorzugt weniger
als 14%, 13%, 12%, 11% oder 10% oder weniger im Vergleich zu der
Kontrollpflanze unter Nichtstressbedingungen. Aufgrund der Fortschritte
bei den landwirtschaftlichen Kulturmaßnahmen (Bewässerung, Düngung,
Pestizidbehandlungen) findet man bei angebauten Kulturpflanzen nicht
oft starken Stress. Daher ist das durch leichten Stress induzierte
geschwächte Wachstum häufig ein unerwünschtes
Merkmal in der Landwirtschaft. Leichter Stress ist der alltägliche
biotische und/oder abiotische Stress (Umweltstress), dem eine Pflanze
ausgesetzt ist. Abiotischer Stress kann durch Trockenheit oder Wasserüberschuss,
anaeroben Stress, Salzstress, chemische Toxizität, oxidativen
Stress und Hitze, Kälte oder Gefriertemperaturen verursacht
werden. Bei dem abiotischen Stress kann es sich um einen osmotischen
Stress handeln, der durch Wasserstress (insbesondere aufgrund von
Trockenheit), Salzstress, oxidativen Stress oder ionenbedingten
Stress verursacht wird. Biotischer Stress wird gewöhnlich
durch Pathogene, wie Bakterien, Viren, Pilze und Insekten verursacht.
-
Insbesondere
können die erfindungsgemäßen Verfahren
unter Nichtstressbedingungen oder unter leichten Trockenheitsbedingungen
durchgeführt werden, wodurch man Pflanzen mit einem im
Vergleich zu Kontrollpflanzen erhöhten Ertrag erhält.
Wie von Wang et al. (Planta (2003), 218: 1–14)
beschrieben, führt abiotischer Stress zu einer Reihe von
morphologischen, physiologischen, biochemischen und molekularen
Veränderungen, die das Pflanzenwachstum und die Produktivität
negativ beeinflussen. Trockenheit, Salinität, extreme Temperaturen
und oxidativer Stress stehen bekanntlich miteinander in Verbindung
und können über ähnliche Mechanismen
Wachstums- und Zellschäden induzieren. Rabbani
et al. (Plant Physiol. (2003), 133: 1755–1767)
beschreiben ein besonders hohes Ausmaß an gegenseitiger
Beeinflussung von Trockenheitsstress und durch hohe Salinität
verursachtem Stress. Trockenheit und/oder Versalzung äußern
sich beispielsweise in erster Linie als osmotischer Stress, was
zur Störung der Homöostase und der Ionenverteilung
in der Zelle führt. Oxidativer Stress, der häufig
Hoch- oder Niedertemperatur-, Salinitäts- oder Trockenheitsstress
begleitet, kann zur Denaturierung funktioneller und struktureller
Proteinen führen. Demzufolge aktivieren diese verschiedenen
Umweltstressfaktoren häufig ähnliche Signalleitungswege
der Zelle und Zellreaktionen, wie die Produktion von Stressproteinen,
die Aufwärtsregulation von Antioxidantien, die Anreicherung
kompatibler gelöster Stoffe und ein Einstellen des Wachstums.
Der Begriff ”Nichtstress”-Bedingungen bedeutet
im vorliegenden Zusammenhang diejenigen Umweltbedingungen, die ein
optimales Wachstum der Pflanzen ermöglichen. Der Fachmann
ist mit den normalen Bodenbedingungen und Klimabedingungen für
einen bestimmten Standort vertraut. Pflanzen mit optimalen Wachstumsbedingungen
(die unter Nicht-Stress-Bedingungen gezüchtet wurden) ergeben
gewöhnlich in steigender Reihenfolge der Präferenz
mindestens 90%, 87%, 85%, 83%, 80%, 77% oder 75% der durchschnittlichen
Produktion einer solchen Pflanze bei einer gegebenen Umgebung. Die durchschnittliche
Produktion kann auf der Basis der Ernte und/oder der Jahreszeit
berechnet werden. Dem Fachmann sind Produktionen einer Kulturpflanze
mit durchschnittlichem Ertrag geläufig.
-
Die
Durchführung der erfindungsgemäßen Verfahren
ergibt Pflanzen, die bei Wachstum unter Nichtstressbedingungen oder
unter leichten Trockenheitsbedingungen einen im Vergleich zu Kontrollpflanzen,
die unter vergleichbaren Bedingungen wachsen, erhöhten
Ertrag aufweisen. Gemäß der vorliegenden Erfindung wird
daher ein Verfahren zur Erhöhung des Ertrags von Pflanzen,
die unter Nichtstressbedingungen oder unter leichten Trockenheitsbedingungen
heranwachsen, bereitgestellt, wobei man bei dem Verfahren in einer
Pflanze die Expression einer Nukleinsäure, die ein SPL11-Polypeptid
kodiert, erhöht.
-
Die
Durchführung der erfindungsgemäßen Verfahren
ergibt Pflanzen, die unter Nährstoffmangelbedingungen gezüchtet
wurden, insbesondere unter Stickstoffmangelbedingungen, mit besserem
Ertrag im Vergleich zu unter vergleichbaren Bedingungen gezüchteten
Kontrollpflanzen. Daher wird erfindungsgemäß ein Verfahren
zur Steigerung des Ertrags bei Pflanzen bereitgestellt, die unter
Nährstoffmangelbedingungen gezüchtet wurden, bei
dem man die Expression einer Nukleinsäure, die ein SPL11-Polypeptid
kodiert, in einer Pflanze steigert. Nährstoffmangel kann
sich unter Anderem aus einem Fehlen von Nährstoffen, wie
Stickstoff, Phosphaten und anderen phosphorhaltigen Verbindungen,
Kalium, Calcium, Cadmium, Magnesium, Mangan, Eisen und Bor, ergeben.
-
Die
vorliegende Erfindung umfasst Pflanzen oder Teile davon (einschließlich
Samen), die durch die erfindungsgemäßen Verfahren
erhältlich sind. Die Pflanzen oder Teile davon umfassen
ein Nukleinsäuretransgen, das ein SPL11-Polypeptid wie
oben definiert, kodiert.
-
Die
Erfindung stellt auch Genkonstrukte und Vektoren bereit, um die
Einführung und/oder Expression von Nukleinsäuren,
die SPL11-Polypeptide kodieren, in Pflanzen zu erleichtern. Die
Genkonstrukte können in handelsübliche Vektoren
eingeführt werden, die für die Transformation
in Pflanzen und für die Expression des interessierenden
Gens in den transformierten Zellen geeignet sind. Die Erfindung
stellt auch die Verwendung eines wie im vorliegenden Text definierten
Genkonstrukts in den erfindungsgemäßen Verfahren
bereit.
-
Genauer
ausgedrückt stellt die vorliegende Erfindung ein Konstrukt
bereit, umfassend:
- (d) eine Nukleinsäure,
die ein SPL11-Polypeptid wie oben definiert kodiert;
- (e) eine oder mehrere Kontrollsequenzen, die die Expression
der Nukleinsäuresequenz gemäß (a) steuern kann
bzw. können; sowie gegebenenfalls
- (f) eine Transkriptionsterminationssequenz.
-
Vorzugsweise
ist die Nukleinsäure, die eine SPL11-Polypeptid kodiert,
wie oben definiert. Der Begriff ”Kontrollsequenz” und ”Terminationssequenz” sind
wie hier definiert.
-
Die
Pflanzen werden mit einem Vektor transformiert, der eine beliebige
der oben beschriebenen Nukleinsäuren umfasst. Der Fachmann
ist mit den genetischen Elementen, die in einem Vektor vorhanden
sein müssen, um Wirtszellen, die die interessierende Sequenz
enthalten, erfolgreich zu transformieren, zu selektieren und zu
vermehren, bestens vertraut. Die interessierende Sequenz ist funktionsfähig
mit einer oder mehreren Kontrollsequenzen (mindestens mit einem
Promotor) verbunden.
-
Jede
Art von Promotor, sowohl natürlich als auch synthetisch,
kann vorteilhaft für das Steuern der Expression der Nukleinsäuresequenz
verwendet werden. Ein konstitutiver Promotor ist besonders bei den
erfindungsgemäßen Verfahren geeignet. Für
die Definitionen der verschiedenen Promotortypen siehe Abschnitt ”Definitionen” im
vorliegenden Text.
-
Man
muss sich darüber im Klaren sein, dass die Anwendbarkeit
der vorliegenden Erfindung weder auf die SPL11-Polypeptid-kodierende
Nukleinsäure, die durch SEQ ID NO: 427 veranschaulicht
wird, noch auf die Expression einer SPL11-Polypeptid-kodierenden
Nukleinsäure, die von einem konstitutiven Promotor gesteuert
wird, beschränkt ist.
-
Der
samenspezifische Promotor ist vorzugsweise ein GOS2-Promotor, vorzugsweise
ein GOS2-Promotor aus Reis. Weiter bevorzugt ist der konstitutive
Promotor durch eine Nukleinsäuresequenz veranschaulicht,
die im wesentlichen SEQ ID NO: 476 ähnelt, am stärksten
bevorzugt ist der samenspezifische Promotor durch SEQ ID NO: 476
veranschaulicht. Im Abschnitt ”Defintionen” sind
weitere Beispiele für samenspezifische Promotoren beschrieben.
-
In
einer alternativen Ausführungsform wird ein samenspezifischer
Promotor verwendet. Der samenspezifische Promotor ist vorzugsweise
durch ABA (Abszisinsäure) induzierbar, er ist vorzugsweise
der WSI18-Promotor, vorzugsweise WSI18 aus Reis. Weiter bevorzugt
ist der konstitutive Promotor durch eine Nukleinsäuresequenz
dargestellt, die im Wesentlichen SEQ ID NO: 477 ähnelt.
Siehe im Abschnitt ”Definitionen” für
weitere Beispiele samenspezifischer Promotoren.
-
Es
sollte eindeutig sein, dass die Promotoren, die sich in den erfindungsgemäßen
Verfahren eignen, nicht auf diejenigen eingeschränkt sind,
die in den vorstehenden Ausführungsformen angegeben sind.
-
Gegebenenfalls
können eine oder mehrere Terminatorsequenzen in dem in
eine Pflanze eingebrachten Konstrukt verwendet werden. Zusätzliche
regulatorische Elemente können Transkriptions- und Translations-Enhancer
umfassen. Der Fachmann kennt Terminator- und Enhancer-Sequenzen,
die sich zur Durchführung der Erfindung eignen. Eine Intronsequenz
kann ebenfalls zu der 5'-untranslatierten Region (UTR) oder in der
kodierenden Sequenz hinzugefügt werden, damit die Menge
der reifen Botschaft gesteigert wird, die sich im Cytosol anreichert,
wie es im Abschnitt Definitionen beschrieben ist. Andere Kontrollsequenzen
(neben Promotor, Enhancer, Silencer, Intronsequenzen, 3'UTR- und/oder
5'UTR-Regionen) können Protein- und/oder RNA-stabilisierende
Elemente sein. Solche Sequenzen sind dem Fachmann bekannt oder können
leicht von ihm erhalten werden.
-
Die
erfindungsgemäßen Genkonstrukte können
weiterhin eine Replikationsursprungssequenz beinhalten, die zur
Aufrechterhaltung und/oder Replikation in einem bestimmten Zelltyp
erforderlich ist. Bei einem Beispiel muss ein Genkonstrukt in einer Bakterienzelle
als episomales genetisches Element aufrechterhalten werden (z. B.
Plasmid- oder Cosmidmolekül). Bevorzugte Replikationsursprünge
umfassen f1-ori und colE1, sind jedoch nicht darauf beschränkt.
-
Für
den Nachweis des erfolgreichen Transfers der Nukleinsäuresequenzen,
wie sie bei den erfindungsgemäßen Verfahren verwendet
wurden, und/oder für die Selektion der transgenen Pflanzen,
die diese Nukleinsäuren umfassen, werden vorteilhafterweise
Markergene (oder Reportergene) verwendet. Das Genkonstrukt kann
daher gegebenenfalls ein Selektionsmarkergen umfassen. Selektionsmarker
sind hier eingehender im Abschnitt ”Definitionen” beschrieben.
-
Es
ist bekannt, dass je nach dem verwendeten Expressionsvektor und
der verwendeten Transfektionstechnik bei der stabilen oder transienten
Integration von Nukleinsäuren in Pflanzenzellen nur ein
kleiner Teil der Zellen die Fremd-DNA aufnimmt und falls gewünscht
in ihr Genom integriert. Um diese Integranten zu identifizieren
und zu selektieren, wird üblicherweise ein Gen, das einen
Selektionsmarker (wie die oben beschriebenen Selektionsmarker) kodiert,
zusammen mit dem interessierenden Gen in die Wirtszellen eingeführt.
Diese Marker können zum Beispiel in Mutanten, in denen
diese Gene nicht funktionell sind, verwendet werden, und zwar zum
Beispiel durch Deletion nach herkömmlichen Verfahren. Weiterhin
können Nukleinsäuremoleküle, die einen
Selektionsmarker kodieren, in eine Wirtszelle auf demselben Vektor
eingeführt werden, der die Sequenz, die die erfindungsgemäßen
Polypeptide oder die Polypeptide, die bei den Verfahren der Erfindung verwendet
werden, umfasst, oder auch auf einen separaten Vektor. Zellen, die
mit der eingeführten Nukleinsäure stabil transfiziert
worden sind, können zum Beispiel durch Selektion identifiziert
werden (zum Beispiel Zellen, die den Selektionsmarker integriert
haben, überleben, während die anderen Zellen absterben).
Die Markergene können aus der transgenen Zelle entfernt
oder gespalten werden, sobald sie nicht mehr benötigt werden.
Techniken zur Entfernung des Markengens sind im Stand der Technik
bekannt, geeignete Techniken sind oben im Abschnitt Definitionen
beschrieben.
-
Die
Erfindung stellt auch ein Verfahren zur Produktion von transgenen
Pflanzen mit verbesserten Ertragsmerkmalen vorzugsweise verbesserten
Samenertragsmerkmalen gegenüber Kontrollpflanzen bereit,
wobei eine beliebige Nukleinsäure, die ein SPL11-Polypeptid
wie oben definiert, in eine(r) Pflanze eingeführt und exprimiert
wird.
-
Insbesondere
stellt die vorliegende Erfindung ein Verfahren zur Produktion von
transgenen Pflanzen mit verbesserten Ertragsmerkmalen, vorzugsweise
gesteigertem (Samen)ertrag bereit, wobei das Verfahren umfasst:
- (iii) Einführen und Exprimieren einer
SPL11-Polypeptid-kodierenden Nukleinsäure in eine(r) Pflanze
oder Pflanzenzelle unter der Kontrolle eines konstitutiven Pflanzenpromotors;
und
- (iv) Züchten der Pflanzenzelle unter Bedingungen, die
das Wachstum und die Entwicklung der Pflanze fördern.
-
Die
Nukleinsäure von (i) kann eine beliebige Nukleinsäure
sein, die ein SPL11-Polypeptid wie hier definiert kodieren kann.
-
Die
Nukleinsäure kann in eine Pflanzenzelle oder in die Pflanze
selbst direkt eingeführt werden (was das Einführen
in ein Gewebe, Organ oder einen beliebigen anderen Teil einer Pflanze
beinhaltet). Gemäß einem bevorzugten Merkmal der
vorliegenden Erfindung wird die Nukleinsäure vorzugsweise
mittels Transformation in eine Pflanze eingeführt. Der
Begriff ”Transformation” wird im vorliegenden
Text im Abschnitt ”Definitionen” genauer beschrieben.
-
Die
genetisch modifizierten Pflanzenzellen können nach allen
Verfahren, mit denen der Fachmann vertraut ist, regeneriert werden.
Geeignete Verfahren finden sich in den oben genannten Veröffentlichungen von
S. D. Kung und R. Wu, Potrykus oder Höfgen und Willmitzer.
-
Im
Allgemeinen werden nach der Transformation die Pflanzenzellen oder
Zellgruppen auf das Vorhandensein von einem oder mehreren Markern
selektiert, die von den mit dem interessierenden Gen gemeinsam transferierten,
in Pflanzen exprimierbaren Genen kodiert werden, wonach das transformierte
Material zu einer ganzen Pflanze regeneriert wird. Für
die Selektion von transformierten Pflanzen wird das bei der Transformation
erhaltene Pflanzenmaterial im Allgemeinen Selektionsbedingungen
unterworfen, so dass man transformierte Pflanzen von untransformierten
Pflanzen unterscheiden kann. So können zum Beispiel Samen,
die auf die oben beschriebene Art und Weise erhalten wurden, ausgepflanzt
werden und nach einer anfänglichen Wachstumsphase durch
Spritzen einer geeigneten Selektion unterworfen werden. Eine weitere
Möglichkeit besteht darin, dass man die Samen, gegebenenfalls
nach Sterilisieren, auf Agarplatten heranzieht, wobei man ein geeignetes
Selektionsmittel verwendet, so dass nur die transformierten Samen
zu Pflanzen heranwachsen können. Alternativ werden die
transformierten Pflanzen auf das Vorhandensein eines Selektionsmarkers
wie die oben beschriebenen gescreent.
-
Nach
dem DNA-Transfer und der Regeneration können mutmaßlich
transformierte Pflanzen auch zum Beispiel unter Verwendung der Southern-Analyse
auf das Vorhandensein des interessierenden Gens, die Kopienzahl
und/oder die Genomorganisation ausgewertet werden. Alternativ dazu
oder zusätzlich können die Expressionsniveaus
der neu eingeführten DNA mittels Northern- und/oder Western-Analyse überwacht
werden, wobei beide Techniken dem Durchschnittsfachmann gut bekannt
sind.
-
Die
erzeugten transformierten Pflanzen können mit unterschiedlichen
Mitteln vermehrt werden, wie durch klonale Vermehrung oder durch
klassische Züchtungstechniken. So kann zum Beispiel eine
transformierte Pflanze der ersten Generation (T1-Pflanze) geselbstet werden,
und homozygote Transformanten der zweiten Generation (T2-Pflanze)
können selektiert werden, und die T2-Pflanzen können
dann mit Hilfe von klassischen Züchtungstechniken weiter
vermehrt werden. Die erzeugten transformierten Organismen können in
verschiedener Form vorliegen. Sie können beispielsweise
Chimären von transformierten Zellen und untransformierten
Zellen sein; klonale Transformanten (z. B. alle Zellen wurden dahingehend
transformiert, dass sie die Expressionskassette enthalten); Pfropfmaterial
von transformiertem und untransformiertem Gewebe (z. B. bei Pflanzen
ein transformierter Wurzelstock, der auf ein untransformiertes Edelreis
gepfropft wurde).
-
Die
vorliegende Erfindung erstreckt sich eindeutig auf eine jede Pflanzenzelle
oder Pflanze, die nach einem der hier beschriebenen Verfahren erzeugt
wurde, und auf sämtliche Pflanzenteile und sämtliches
Vermehrungsmaterial davon. Die vorliegende Erfindung umfasst weiterhin
die Nachkommenschaft einer/eines primär transformierten
oder transfizierten Zelle, Gewebes, Organs oder ganzen Pflanze,
die/das nach einem der oben genannten Verfahren erzeugt wurde, wobei
die einzige Voraussetzung ist, dass die Nachkommenschaft dasselbe/dieselben
genotypische(n) und/oder phänotypische(n) Merkmal(e) aufweist,
wie es/sie von dem Elter bei den erfindungsgemäßen
Verfahren gezeigt wird.
-
Die
Erfindung beinhaltet auch Wirtszellen, die eine isolierte Nukleinsäure,
die ein wie oben definiertes SPL11-Polypeptid kodiert, enthalten.
Bevorzugte erfindungsgemäße Wirtszellen sind Pflanzenzellen.
Wirtspflanzen für die bei dem erfindungsgemäßen
Verfahren verwendeten Nukleinsäuren oder den Vektor, für
die Expressionskassette oder das Konstrukt oder den Vektor, sind
im Prinzip vorteilhafterweise alle Pflanzen, die die bei dem erfindungsgemäßen
Verfahren verwendeten Polypeptide synthetisieren können.
-
Die
erfindungsgemäßen Verfahren lassen sich vorteilhaft
auf jede beliebige Pflanze anwenden. Zu den Pflanzen, die bei den
erfindungsgemäßen Verfahren besonders geeignet
sind, gehören alle Pflanzen, die zu der Superfamilie Viridiplantae
gehören, insbesondere monokotyle und dikotyle Pflanzen,
einschließlich Futter- oder Weidepflanzenleguminosen, Zierpflanzen,
Nahrungsmittelkulturpflanzen, Bäume oder Sträucher.
Gemäß einer bevorzugten Ausführungsform
der vorliegenden Erfindung handelt es sich bei der Pflanze um eine
Kulturpflanze. Zu den Kulturpflanzen gehören zum Beispiel
Sojabohne, Sonnenblume, Canola-Raps, Luzerne, Raps, Baumwolle, Tomate,
Kartoffel und Tabak. Weiter bevorzugt handelt es sich bei der Pflanze
um eine monokotyle Pflanze. Zu den monokotylen Pflanzen gehört
zum Beispiel Zuckerrohr. Weiter bevorzugt handelt es sich bei der
Pflanze um ein Getreide. Zu den Getreiden gehören zum Beispiel
Reis, Mais, Weizen, Gerste, Hirse, Roggen, Triticale, Sorghum und
Hafer.
-
Die
Erfindung betrifft auch erntefähige Teile einer Pflanze,
wie zum Beispiel, jedoch nicht ausschließlich, Samen, Blätter,
Früchte, Blüten, Stängel, Rhizome, Knollen
und Zwiebeln. Die Erfindung betrifft weiterhin Produkte, die von
einem erntefähigen Teil einer solchen Pflanze abstammen,
vorzugsweise direkt abstammen, wie trockene Pellets oder Pulver, Öl,
Fett und Fettsäuren, Stärke oder Proteine.
-
Gemäß einem
bevorzugten Merkmal der Erfindung handelt es sich bei der modulierten
Expression um erhöhte Expression. Verfahren zur Erhöhung
der Expression von Nukleinsäuren oder Genen oder Genprodukten,
sind im Fachgebiet gut beschrieben; und Beispiele sind im Abschnitt
Definitionen gegeben.
-
Wie
vorstehend erwähnt, ist ein bevorzugtes Verfahren zum Modulieren
(vorzugsweise Erhöhen) der Expression einer Nukleinsäure,
die ein SPL11-Polypeptid kodiert, durch Einführen und Exprimieren
einer Nukleinsäure, die ein SPL11-Polypeptid kodiert, in
eine(r) Pflanze; die Wirkungen der Durchführung des Verfahrens,
d. h. das Erhöhen der Ertragsmerkmale, können
auch unter Verwendung anderer bekannter Techniken erzielt werden,
einschließlich, aber nicht beschränkt auf T-DNA-Activation-Tagging,
TILLING, homologe Rekombination. Eine Beschreibung dieser Techniken
findet sich im Abschnitt Definitionen.
-
Die
vorliegende Erfindung umfasst auch die Verwendung von Nukleinsäuren,
die SPL11-Polypeptide, wie hier beschrieben, kodieren, und die Verwendung
dieser SPL11-Polypeptide zur Verbesserung eines der oben genannten
Ertragsmerkmale in Pflanzen.
-
Hier
beschriebene Nukleinsäuren, die die SPL11-Polypeptide kodieren,
oder die SPL11-Polypeptide selbst können Verwendung in
Züchtungsprogrammen finden, in denen ein DNA-Marker identifiziert
wird, der mit einem für ein SPL11-Polpeptid kodierenden
Gen genetisch verknüpft sein kann. Die Nukleinsäuren/Gene oder
die SPL11-Polypeptide selbst können dazu verwendet werden,
einen molekularen Marker zu bestimmen. Dieser DNA- oder Protein-Marker
kann dann in Züchtungsprogrammen zur Selektion von Pflanzen
mit verbesserten Ertragsmerkmalen verwendet werden, wie hier unter
den erfindungsgemäßen Verfahren definiert.
-
Allelische
Varianten einer Nukleinsäure/eines Gens, die/das ein SPL11-Polypeptid
kodiert, können auch Verwendung in Marker-unterstützten
Züchtungsprogrammen finden. Bei solchen Züchtungsprogrammen ist
manchmal das Einführen von allelischer Variation durch
Mutagenbehandlung der Pflanzen erforderlich, wobei zum Beispiel
EMS-Mutagenese verwendet wird; alternativ kann das Programm von
einer Sammlung von allelischen Varianten so genannten ”natürlichen” Ursprungs
ausgehen, die ohne Absicht hervorgerufen wurden. Dann erfolgt die
Identifikation allelischer Varianten, zum Beispiel mittels PCR.
Darauf folgt ein Schritt zur Selektion besserer allelischer Varianten
der in Frage kommenden Sequenz, die bessere Erträge liefern.
Die Selektion wird gewöhnlich durchgeführt, indem
man die Wachstumsleistung von Pflanzen verfolgt, die verschiedene
allelische Varianten der in Frage kommenden Sequenz enthalten. Die
Wachstumsleistung kann im Gewächshaus oder auf dem Feld
verfolgt werden. Weitere Schritte sind gegebenenfalls u. a. das
Kreuzen der Pflanzen, in denen die bessere allelische Variante identifiziert
wurde, mit einer anderen Pflanze. Dies kann zum Beispiel zur Herstellung
einer Kombination interessanter phänotypischer Merkmale
verwendet werden.
-
SPL11-Polypeptide
kodierende Nukleinsäuren können auch als Sonden
zur genetischen und physikalischen Kartierung der Gene, von denen
sie ein Teil sind, und als Marker für Merkmale, die mit
diesen Genen verknüpft sind, verwendet werden. Diese Information
kann für die Pflanzenzüchtung nützlich
sein, um Linien mit gewünschten Phänotypen zu
entwickeln. Eine solche Verwendung der SPL11-Polypeptid-kodierenden
Nukleinsäuren benötigt nur eine Nukleinsäuresequenz
von mindestens 15 Nukleotiden Länge. Die SPL11-Polypeptid-kodierenden
Nukleinsäuren können als Restriktionsfragmentlängenpolymorphismus
(RFLP)-Marker verwendet werden. Southern-Blots (Sambrook
J, Fritsch EF and Maniatis T (1989) Molecular Cloning, A Laboratory
Manual) von restriktions-gespaltener genomischer Pflanzen-DNA
kann mit den SPL11-kodierenden Nukleinsäuren sondiert werden.
Die erhaltenen Bandenmuster können dann genetischen Analysen
mit Computerprogrammen unterworfen werden, wie MapMaker (Lander
et al. (1987) Genomics 1: 174–181), so dass man eine
genetische Karte erhält. Die Nukleinsäuren können
zudem zum Sondieren von Southern-Blots verwendet werden, die Restriktionsendonuklease
behandelte genomische DNAs von einem Satz von Individuen enthalten,
die die Eltern und die Nachfahren einer definierten genetischen
Kreuzung repräsentieren. Die Segregation der DNA-Polymorphismen
wird festgestellt und zur Berechnung der Position der SPL11-Polypeptidkodierenden
Nukleinsäure in der genetischen Karte verwendet, die mit
dieser Population erhalten wurde (Botstein et al. (1980)
Am. J. Hum. Genet. 32: 314–331).
-
Die
Herstellung und Verwendung von Sonden, die von Pflanzengenen stammen,
für die Verwendung bei der genetischen Kartierung ist in Bernatzky
und Tanksley (1986) Plant Mol. Biol. Reporter 4: 37–41 beschrieben.
Zahlreiche Veröffentlichungen beschreiben die genetische
Kartierung spezifischer cDNA-Klone, wobei die vorstehend erläuterte
Methodik oder Varianten davon verwendet werden. Für die
Kartierung können zum Beispiel F2-Inzuchtpopulationen,
Rückkreuzungspopulationen, zufallsgemäß gepaarte
Populationen, nahezu isogene Linien und andere Sätze von
Individuen verwendet werden. Diese Methoden sind dem Fachmann bestens
bekannt.
-
Die
Nukleinsäuresonden können auch zur physikalischen
Kartierung (d. h. zum Anordnen von Sequenzen in physikalischen Karten;
siehe Hoheisel et al. In: Non-mammalian Genomic Analysis:
A Practical Guide, Academic Press 1996, S. 319–346 und
darin genannte Literaturstellen) verwendet werden.
-
In
einer anderen Ausführungsform können die Nukleinsäuresonden
zur Kartierung durch direkte Fluoreszenz-in-situ-Hybridisierung
(FISH) (Trask (1991) Trends Genet. 7: 149–154)
verwendet werden. Zwar bevorzugen derzeitige FISH-Kartierungsverfahren
die Verwendung großer Klone (mehrere kb bis mehrere Hundert
kb; siehe Laan et al. (1995) Genome Res. 5: 13–20),
aber durch Verbesserungen in der Empfindlichkeit kann die FISH-Kartierung
auch unter Verwendung kürzerer Sonden durchgeführt
werden.
-
Eine
Reihe von Verfahren auf der Basis von Nukleinsäureamplifikation
für die genetische und physikalische Kartierung kann mit
Hilfe der Nukleinsäuren durchgeführt werden. Beispiele
sind u. a. die allelspezifische Amplifikation (Kazazian
(1989) J. Lab. Clin. Med 11: 95–96), Polymorphismus
PCR-amplifizierter Fragmente (CAPS; Sheffield et al. (1993)
Genomics 16: 325–332), allelspezifische Ligation
(Landegren et al. (1988) Science 241: 1077–1080),
Nukleotidverlängerungsreaktionen (Sokolov (1990)
Nucleic Acid Res. 18: 3671), Radiation-Hybrid-Kartierung
(Walter et al. (1997) Nat. Genet. 7: 22–28)
und Happy-Kartierung (Dear und Cook (1989) Nucleic Acid
Res. 17: 6795–6807). Bei diesen Verfahren wird
die Sequenz einer Nukleinsäure dazu verwendet, Primer-Paare
zu gestalten und zu erzeugen, die bei der Amplifikationsreaktion
oder Primerverlängerungsreaktionen verwendet werden. Die
Gestaltung dieser Primer ist dem Fachmann bekannt. Bei Verfahren, die
eine genetische Kartierung auf PCR-Basis einsetzen, kann es notwendig
sein, dass man DNA-Sequenz-Unterschiede zwischen den Eltern der
Kartierungskreuzung in dem Bereich identifiziert, welcher der vorliegenden
Nukleinsäuresequenz entspricht. Dies ist jedoch für
Kartierungsverfahren nicht allgemein erforderlich.
-
Die
erfindungsgemäßen Verfahren ergeben Pflanzen mit
verbesserten Ertragsmerkmalen, wie hier vorstehend beschrieben.
Diese Merkmale können auch mit anderen ökonomisch
vorteilhaften Merkmalen kombiniert werden, wie weiteren Ertragsmerkmalen,
Toleranz gegenüber anderen abiotischen und biotischen Stressbedingungen,
Merkmalen, die verschiedene architektonische Merkmale und/oder biochemische und/oder
physiologische Merkmale modifizieren.
-
Die
vorliegende Erfindung wird nun anhand der folgenden Punkte beschrieben:
- 1. Verfahren zur Verbesserung von Ertragsmerkmalen
in Pflanzen im Vergleich zu Kontrollpflanzen, bei dem in einer Pflanze
eine Nukleinsäure, die ein LBD-Polypeptid kodiert, moduliert
wird, wobei das LBD-Polypeptid eine DUF206-Domäne umfasst.
- 2. Verfahren nach Punkt 1, wobei das LBD-Polypeptid eines oder
mehrere der folgenden Motive umfasst:
(i) Motiv 1: MSCNGCRXLRKGCX
(SEQ ID NO: 5),
(ii) Motiv 2: QXXATXFXAKFXGR (SEQ ID NO: 6),
(iii)
Motiv 3: FXSLLXEAXG (SEQ ID NO: 7)
- 3. Verfahren nach Punkt 1 oder 2, wobei die modulierte Expression
durch Einführen und Exprimieren einer Nukleinsäure,
die ein LBD-Polypeptid kodiert in eine(r) Pflanze herbeigeführt
wird.
- 4. Verfahren nach einem vorhergehenden Punkt, wobei die Nukleinsäure,
die ein LBD-Polypeptid kodiert, eines der in Tabelle A1 aufgeführten
Proteine kodiert, oder ein Teil einer solchen Nukleinsäure
ist, oder eine Nukleinsäure, die mit einer solchen Nukleinsäure
hybridisieren kann.
- 5. Verfahren nach einem vorhergehenden Punkt, wobei die Nukleinsäuresequenz
ein Orthologon oder Paralogon von einem der in Tabelle A1 angegebenen
Proteine kodiert.
- 6. Verfahren nach einem vorhergehenden Punkt, wobei die verbesserten
Ertragsmerkmale einen gesteigerten Ertrag, vorzugsweise gesteigerte
Biomasse und/oder gesteigerten Samenertrag im Vergleich zu Kontrollpflanzen
umfasst.
- 7. Verfahren nach einem der Punkte 1 bis 6, wobei die verbesserten
Ertragsmerkmale unter Nichtstressbedingungen erhalten werden.
- 8. Verfahren nach einem der Punkte 1 bis 6, wobei die verbesserten
Ertragsmerkmale unter Stickstoffmangelbedingungen erhalten werden.
- 9. Verfahren nach einem der Punkte 3 bis 8, wobei die Nukleinsäure
funktionsfähig mit einem konstitutiven Promotor, vorzugsweise
einem GOS2-Promotor, am stärksten bevorzugt einem GOS2
Promotor aus Reis, verknüpft ist.
- 10. Verfahren nach einem vorhergehenden Punkt, wobei die Nukleinsäure,
die ein LBD-Polypeptid kodiert, aus einer Pflanze, vorzugsweise
aus einer dikotylen Pflanze, weiter bevorzugt aus der Familie Brassicaceae,
stärker bevorzugt aus der Gattung Arabidopsis, am stärksten
bevorzugt aus Arabidopsis thaliana, stammt.
- 11. Isoliertes Nukleinsäuremolekül, umfassend
eines der folgenden Merkmale:
(i) eine Nukleinsäure
nach SEQ ID NO: 69;
(ii) eine Nukleinsäure oder ein
Fragment davon, das zu einer der in (i) angegebenen SEQ ID NOs komplementär
ist;
(iii) eine Nukleinsäure, die ein LBD-Polypeptid
kodiert, das in steigender Reihenfolge der Präferenz mindestens
60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder
mehr Sequenzidentität zu SEQ ID NO: 70 aufweist;
(iv)
eine Nukleinsäure, die unter stringenten Bedingungen mit
einer der in (i), (ii) oder
(iii) oben abgegebenen Nukleinsäuren
hybridisieren kann.
- 12. Isoliertes Polypeptid, umfassend:
(i) eine Aminosäuresequenz,
das in steigender Reihenfolge der Präferenz mindestens
60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder
100% Sequenzidentität zu der in SEQ ID NO: 70 angegebenen
Aminosäuresequenz aufweist.
(ii) Derivate von einer
der in (i) angegebenen Aminosäuresequenzen.
- 13. Pflanze oder Teil davon, einschließlich Samen,
die durch ein Verfahren nach einem vorhergehenden Punkt erhältlich
sind, wobei die Pflanze oder das Teil davon eine rekombinante Nukleinsäure
umfasst, die ein LBD-Polypeptid kodiert.
- 14. Konstrukt, umfassend:
(i) eine Nukleinsäure,
die ein LBD-Polypeptid nach den Punkten 1, 2 oder 12 kodiert;
(ii)
eine oder mehrere Kontrollsequenzen, die die Expression der Nukleinsäuresequenz
von (a) steuern können; und gegebenenfalls
(iii) eine
Transkriptionsterminationssequenz.
- 15. Konstrukt nach Punkt 14, wobei eine der Kontrollsequenzen
ein konstitutiver Promotor, vorzugsweise ein GOS2-Promotor, am stärksten
bevorzugt ein GOS2-Promotor aus Reis, ist.
- 16. Verwendung eines Konstrukts nach Punkt 14 oder 15 in einem
Verfahren zur Herstellung von Pflanzen mit gesteigertem Ertrag,
vorzugsweise gesteigerter Biomasse und/oder gesteigertem Samenertrag
im Vergleich zu Kontrollpflanzen.
- 17. Pflanze, Pflanzenteil oder Pflanzenzelle, die mit einem
Konstrukt nach Punkt 14 oder 15 transformiert ist.
- 18. Verfahren zur Produktion einer transgenen Pflanze mit gesteigertem
Ertrag, vorzugsweise gesteigerter Biomasse und/oder gesteigertem
Samenertrag im Vergleich zu Kontrollpflanzen, umfassend:
(i)
Einführen und Exprimieren einer Nukleinsäure,
die ein LBD-Polypeptid nach Punkt 1, 2 oder 12 kodiert, in eine(r)
Pflanze; und
(ii) Züchten der Pflanzenzelle unter
Bedingungen, die das Wachstum und die Entwicklung der Pflanze fördern.
- 19. Transgene Pflanze mit gesteigertem Ertrag, vorzugsweise
gesteigerter Biomasse und/oder gesteigertem Samenertrag im Vergleich
zu Kontrollpflanzen, der sich aus einer gesteigerten Expression
einer Nukleinsäure, die ein LBD-Polypeptid nach Punkt 1,
2 oder 12 kodiert, ergibt, oder eine Pflanzenzelle, die aus der
transgenen Pflanze stammt.
- 20. Transgene Pflanze nach Punkt 13, 17 oder 19, oder eine daraus
stammende transgene Pflanzenzelle, wobei die Pflanze eine Kulturpflanze
oder eine Monokotyle oder ein Getreide, wie Reis, Mais, Weizen, Gerste,
Hirse, Roggen, Triticale, Sorghum und Hafer ist.
- 21. Erntefähige Teile einer Pflanze nach Punkt 19,
wobei die erntefähigen Teile vorzugsweise Sprossbiomasse
und/oder Samen sind.
- 22. Produkte, die aus einer Pflanze nach Punkt 19 und/oder aus
erntefähigen Teilen einer Pflanze nach Punkt 20 stammen.
- 23. Verwendung einer Nukleinsäure, die ein LBD-Polypeptid
kodiert, bei der Steigerung des Ertrags, vorzugsweise Steigerung
des Samenertrags und/oder Sprossbiomasse in Pflanzen im Vergleich
zu Kontrollpflanzen.
- 24. Verfahren zur Verbesserung der Ertragsmerkmale in Pflanzen
im Vergleich zu Kontrollpflanzen, bei dem in einer Pflanze eine
Nukleinsäure, die ein JMJC-Polypeptid kodiert, moduliert
wird, wobei das JMJC-Polypeptid eine JmjC-Domäne umfasst.
- 25. Verfahren nach Punkt 24, wobei die JmjC-Domäne
durch eine Sequenz veranschaulicht wird, die in steigender Präferenz
mindestens 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, oder
mehr Sequenzidentität aufweist zu:
(i) SEQ ID NO:
78; und/oder
(ii) einer der JmjC-Domänen, die sich
in den JMJC-Polypeptiden befinden, die durch SEQ ID NO: 84; SEQ ID
NO: 86; SEQ ID NO: 96; SEQ ID NO: 98; SEQ ID NO: 104; SEQ ID NO:
108; SEQ ID NO: 110; SEQ ID NO: 112; SEQ ID NO: 114; SEQ ID NO:
116; SEQ ID NO: 118; SEQ ID NO: 120; SEQ ID NO: 122; SEQ ID NO:
124; SEQ ID NO: 128; SEQ ID NO: 130; SEQ ID NO: 132; und SEQ ID
NO: 134 veranschaulicht werden, deren Aminosäurekoordinaten
in der Tabelle B4 angegeben sind.
- 26. Verfahren nach Punkt 24 und Punkt 25, wobei das JMJC-Polypeptid
ein Motiv umfasst, das in steigender Reihenfolge der Präferenz
mindestens 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% oder mehr
Sequenzidentität aufweist zu einer Sequenz von:
(i)
SEQ ID NO: 79,
(ii) SEQ ID NO: 80,
(iii) SEQ ID NO: 81,
(iv)
SEQ ID NO: 82;
- 27. Verfahren nach Punkt 24 oder 26, wobei die modulierte Expression
durch Einführen und Exprimieren einer Nukleinsäure,
die ein JMJC-Polypeptid kodiert, in eine(r) Pflanze herbeigeführt
wird.
- 28. Verfahren nach einem der Punkte 24 bis 27, wobei es sich
bei der modulierten Expression um eine erhöhte Expression
handelt.
- 29. Verfahren nach einem der Punkte 24 bis 28, wobei die Nukleinsäure,
die ein JMJC-Polypeptid kodiert, eines der in Tabelle B1 aufgeführten
Proteine kodiert, oder ein Teil einer solchen Nukleinsäure
oder eine Nukleinsäure ist, die mit einer solchen Nukleinsäure
hybridisieren kann.
- 30. Verfahren nach einem der Punkte 24 bis 29, wobei die Nukleinsäuresequenz
ein Orthologon oder Paralogon von einem der in Tabelle B1 angegebenen
Proteine kodiert.
- 31. Verfahren nach Punkt 30, wobei die Nukleinsäure
SEQ ID NO: 74 kodiert.
- 32. Verfahren nach einem der Punkte 24 to 31, wobei die verbesserten
Ertragsmerkmale einen gesteigerten Ertrag, vorzugsweise gesteigerten
Harvest-Index und/oder Samenertrag im Vergleich zu Kontrollpflanzen umfassen.
- 33. Verfahren nach einem der Punkte 24 bis 32, wobei die verbesserten
Ertragsmerkmale Jungpflanzenvitalität im Vergleich zu Kontrollpflanzen
umfassen.
- 34. Verfahren nach einem der Punkte 24 bis 33, wobei die verbesserten
Ertragsmerkmale unter Nichtstressbedingungen erhalten werden.
- 35. Verfahren nach einem der Punkte 24 bis 33, wobei die verbesserten
Ertragsmerkmale unter Wachstumsbedingungen eines leichten Trockenheitsstress
erhalten werden.
- 36. Verfahren nach einem der Punkte 24 bis 33, wobei die verbesserten
Ertragsmerkmale unter Stickstoffmangel-Wachstumsbedingungen erhalten
werden.
- 37. Verfahren nach einem der Punkte 27 bis 36, wobei die Nukleinsäure
funktionsfähig mit einem konstitutiven Promotor, vorzugsweise
einem GOS2-Promotor, am stärksten bevorzugt einem GOS2
Promotor aus Reis verknüpft ist.
- 38. Verfahren nach einem der Punkte 24 bis 37, wobei die Nukleinsäure,
die ein JMJC-Polypeptid kodiert, aus einer Pflanze, vorzugsweise
aus einer dikotylen Pflanze, weiter bevorzugt aus der Familie Brassicaceae,
stärker bevorzugt aus der Gattung Arabidopsis, am stärksten
bevorzugt aus Arabidopsis thaliana stammt.
- 39. Pflanze oder Teil davon, einschließlich Samen,
die durch ein Verfahren nach einem der Punkte 24 bis 38 erhältlich
sind, wobei die Pflanze oder das Teil davon eine rekombinante Nukleinsäure
umfasst, die JMJC-Polypeptid kodiert.
- 40. Konstrukt, umfassend:
(i) eine Nukleinsäure,
die ein JMJC-Polypeptid nach den Punkten 24 bis 26 kodiert;
(ii)
eine oder mehrere Kontrollsequenzen, die die Expression der Nukleinsäure
von (i) steuern können; und gegebenenfalls
(iii) eine
Transkriptionsterminationssequenz.
- 41. Konstrukt nach Punkt 40, wobei eine der Kontrollsequenzen
ein konstitutiver Promotor, vorzugsweise ein GOS2-Promotor, am stärksten
bevorzugt ein GOS2-Promotor aus Reis, ist.
- 42. Verwendung eines Konstrukts nach Punkt 40 oder 41 in einem
Verfahren zur Herstellung von Pflanzen mit verbesserten Ertragsmerkmalen,
insbesondere Jungpflanzenvitalität und/oder gesteigertem
Samenertrag im Vergleich zu Kontrollpflanzen.
- 43. Pflanze, Pflanzenteil oder Pflanzenzelle, die mit einem
Konstrukt nach Punkt 40 oder 41 transformiert ist.
- 44. Verfahren zur Herstellung einer transgenen Pflanze mit verbesserten
Ertragsmerkmalen, insbesondere Jungpflanzenvitalität und/oder
gesteigertem Samenertrag im Vergleich zu Kontrollpflanzen, umfassend:
(i)
Einführen und Exprimieren einer Nukleinsäure,
die ein JMJC-Polypeptid nach Punkt 24 bis 26 kodiert, in eine(r)
Pflanze; und
(ii) Züchten der Pflanzenzelle unter
Bedingungen, die das Wachstum und die Entwicklung der Pflanze fördern.
- 45. Transgene Pflanze mit gesteigertem Ertrag, insbesondere
gesteigerter Jungpflanzenvitalität beim Keimling und/oder
gesteigertem Samenertrag im Vergleich zu Kontrollpflanzen, der sich
aus der gesteigerten Expression einer Nukleinsäure ergibt,
die ein JMJC-Polypeptid nach Punkt 24 bis 26 kodiert, oder eine
transgene Pflanzenzelle, die aus der transgenen Pflanze stammt.
- 46. Transgene Pflanze nach Punkt 39, 43 oder 45, oder eine daraus
stammende transgene Pflanzenzelle, wobei die Pflanze eine Kulturpflanze
oder eine Monokotyle oder ein Getreide, wie Reis, Mais, Weizen, Gerste,
Hirse, Roggen, Triticale, Sorghum und Hafer ist.
- 47. Erntefähige Teile einer Pflanze nach Punkt 46,
wobei die erntefähigen Teile vorzugsweise Sprossbiomasse
und/oder Samen sind.
- 48. Produkte, die aus einer Pflanze nach Punkt 46 und/oder aus
erntefähigen Teilen einer Pflanze nach Punkt 47 stammen.
- 49. Verwendung einer Nukleinsäure, die ein JMJC-Polypeptid
kodiert, bei der Steigerung des Ertrags, insbesondere bei der Steigerung
des Samenertrags und/oder Sprossbiomasse in Pflanzen, im Vergleich
zu Kontrollpflanzen.
- 50. Isoliertes Nukleinsäuremolekül, umfassend
eines der folgenden Merkmale:
(i) eine Nukleinsäure
nach SEQ ID NO: 169;
(ii) eine Nukleinsäure oder ein
Fagment davon, das zu SEQ ID NO: 169 komplementär ist;
(iii)
eine Nukleinsäure, die ein JMJC-Polypeptid kodiert, das
in steigender Reihenfolge der Präferenz mindestens 70%,
75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
zu der in SEQ ID NO: 170 angegebenen Aminosäuresequenz
aufweist;
(iv) eine Nukleinsäure, die unter stringenten
Bedingungen an eine der in (i), (ii) oder (iii) oben angegebenen Nukleinsäuren
hybridisieren kann.
- 51. Isoliertes Polypeptidmolekül, umfassend:
(i)
eine Aminosäuresequenz, die in steigender Reihenfolge der
Präferenz mindestens 70%, 75%, 80%, 85%, 90%, 95%, 96%,
97%, 98%, 99% oder 100% oder mehr Sequenzidentität zu der
in SEQ ID NO: 170 angegebenen Aminosäuresequenz aufweist;
(ii)
Derivate von einer der in (i) angegebenen Aminosäuresequenzen.
- 52. Verfahren zur Verbesserung der Ertragsmerkmale in Pflanzen
im Vergleich zu Kontrollpflanzen, bei der man die Expression einer
Nukleinsäure, die eine Caseinkinase I (CKI) kodiert, in
einer Pflanze moduliert, wobei die CKI aus SEQ ID NO: 174 oder einem
Orthologon oder Paralogon davon ausgewählt ist.
- 53. Verfahren nach Punkt 52, wobei die modulierte Expression
durch Einführen und Exprimieren einer Nukleinsäure,
die das CKI-Polypeptid kodiert, in eine(r) Pflanze herbeigeführt
wird.
- 54. Verfahren nach Punkt 52 oder 53, wobei die Nukleinsäure,
die das CKI-Polypeptid kodiert, ein Abschnitt von SEQ ID NO: 173,
oder eine Nukleinsäure ist, die mit einer solchen Nukleinsäure
hybridisieren kann.
- 55. Verfahren nach einem der Punkte 52 bis 54, wobei die Nukleinsäuresequenz
SEQ ID NO: 174 kodiert.
- 56. Verfahren nach einem der Punkte 52 bis 55, wobei die verbesserten
Ertragsmerkmale eine gesteigerte Jungpflanzenvitalität
und/oder gesteigerten Ertrag, vorzugsweise gesteigerte Biomasse
und/oder gesteigerten Samenertrag im Vergleich zu Kontrollpflanzen
umfassen.
- 57. Verfahren nach einem der Punkte 52 bis 56, wobei die verbesserten
Ertragsmerkmale unter Nichtstressbedingungen erhalten werden.
- 58. Verfahren nach einem der Punkte 52 to 56, wobei die verbesserten
Ertragsmerkmale unter abiotischen Stressbedingungen erhalten werden.
- 59. Verfahren nach Punkt 58, wobei die abiotischen Stressbedingungen
ausgewählt sind aus einer oder mehreren Bedingungen von
Trockenheitsstress, Salzstressbedingungen, und Stickstoffmangelbedingungen.
- 60. Verfahren nach einem der Punkte 53 bis 59, wobei die Nukleinsäure
funktionsfähig mit einem samenspezifischen Promotor verknüpft
ist.
- 61. Verfahren nach einem der Punkte 53 bis 60, wobei der samenspezifische
Promotor ein WSI18-Promotor, vorzugsweise ein WSI18-Promotor aus
Reis, ist.
- 62. Verfahren nach einem der Punkte 52 bis 61, wobei die Nukleinsäure,
die ein CKI-Polypeptid kodiert, aus einer Pflanze stammt.
- 63. Verfahren nach einem der Punkte 52 bis 62, wobei die Ursprungspflanze
vorzugsweise eine dikotyle Pflanze ist, weiter bevorzugt aus der
Familie der Solanaceae, stärker bevorzugt aus der Gattung
Nicotiana tabacum.
- 64. Pflanze oder Teil davon, einschließlich Samen,
die durch ein Verfahren nach einem der Punkte 52 bis 63 erhältlich
sind, wobei die Pflanze oder das Teil davon eine rekombinante Nukleinsäure
umfasst, die ein CKI-Polypeptid kodiert.
- 65. Konstrukt, umfassend:
(i) eine Nukleinsäure,
die ein CKI-Polypeptid nach Punkt 52 kodiert;
(ii) eine oder
mehrere Kontrollsequenzen, die die Expression der Nukleinsäuresequenz
von (a) steuern können; und gegebenenfalls
(iii) eine
Transkriptionsterminationssequenz.
- 66. Konstrukt nach Punkt 65, wobei eine der Kontrollsequenzen
ein samenspezifischer Promotor, vorzugsweise ein WSI18-Promotor,
ist.
- 67. Konstrukt nach Punkt 65, wobei einer der samenspezifischen
Promotoren ein WSI18-Promotor, am stärksten bevorzugt ein
WSI18-Promotor aus Reis, ist.
- 68. Verwendung eines Konstrukts nach einem der Punkte 65 bis
67 in einem Verfahren zur Herstellung von Pflanzen mit verbesserten
Ertragsmerkmalen, insbesondere gesteigerter Jungpflanzenvitalität,
gesteigerter Biomasse und/oder gesteigertem Samenertrag im Vergleich
zu Kontrollpflanzen.
- 69. Pflanze, Pflanzenteil, oder Pflanzenzelle, die mit einem
Konstrukt nach einem der Punkte 65 bis 67 transformiert sind.
- 70. Verfahren zur Herstellung einer transgenen Pflanze mit einem
gesteigerten Ertrag, insbesondere gesteigerter Biomasse und/oder
gesteigertem Samenertrag im Vergleich zu Kontrollpflanzen, umfassend:
(i)
Einführen und Exprimieren einer Nukleinsäure,
die ein CKI-Polypeptid nach Punkt 52 kodiert, in eine(r) Pflanze;
and
(ii) Züchten der Pflanzenzelle unter Bedingungen
unter Bedingungen, die das Wachstum und die Entwicklung der Pflanze
fördern.
- 71. Transgene Pflanze mit verbesserten Ertragsmerkmalen, insbesondere
gesteigerter Jungpflanzenvitalität gesteigerter Biomasse
und/oder gesteigertem Samenertrag im Vergleich zu Kontrollpflanzen,
der sich aus einer gesteigerten Expression einer Nukleinsäure
ergibt, die ein CKI-Polypeptid nach Punkt 52 kodiert, oder eine
transgene Pflanzenzelle, die aus der transgenen Pflanze stammt.
- 72. Transgene Pflanze nach Punkt 64, 69 oder 71, oder eine davon
stammende transgene Pflanzenzelle, wobei die Pflanze eine Kulturpflanze
oder eine Monokotyle oder ein Getreide, wie Reis, Mais, Weizen, Gerste,
Hirse, Roggen, Triticale, Sorghum und Hafer ist.
- 73. Erntefähige Teile einer Pflanze nach Punkt 72,
wobei die erntefähigen Teile vorzugsweise Sprossbiomasse
und/oder Samen sind.
- 74. Produkte, die von einer Pflanze nach Punkt 72, und/oder
von erntefähigen Teilen einer Pflanze nach Punkt 73 stammen.
- 75. Verwendung einer Nukleinsäure, die ein CKI-Polypeptid
kodiert, zur Verbesserung von Ertragsmerkmalen, insbesondere zur
Steigerung von einem oder mehreren Merkmalen von Jungpflanzenvitalität,
Samenertrag und Sprossbiomasse in Pflanzen, im Vergleich zu Kontrollpflanzen.
- 76. Verfahren zur Verbesserung von Ertragsmerkmalen, vorzugsweise
zur Verbesserung von Samenertragsmerkmalen, in Pflanzen im Vergleich
zu Kontrollpflanzen, bei dem die Expression einer Nukleinsäuresequenz,
die ein Pflanzen-Homöodomänen-Finger-Homöodomänen-(plant
homeodomain fnger-homeodomain (PHDf-HD))-Polypeptid kodiert, in
einer Pflanze moduliert, vorzugsweise gesteigert wird, wobei das PHDf-HD-Polypeptid
umfasst: (i) eine Domäne mit mindestens 25%, 30%, 35%,
40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%,
99% oder mehr Aminosäuresequenzidentität zu einer Leucin-Zipper/Pflanzen-Homöodomänen-Finger(ZIP/PHDf)-Domäne,
wie sie durch SEQ ID NO: 233 veranschaulicht wird; und (ii) eine
Domäne mit mindestens 25%, 30%, 35%, 40%, 45%, 50%, 55%,
60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99% oder mehr Aminosäuresequenzidentität
zu einer Homöodomäne (HD) wie sie durch SEQ ID
NO: 234 veranschaulicht wird, und gegebenenfalls Selektieren auf
Pflanzen mit verbesserten Ertragsmerkmalen.
- 77. Verfahren nach Punkt 76, wobei das PHDf-HD-Polypeptid umfasst:
(i) eine PHD-Polypeptid-Domäne wie sie durch PFAM00628
veranschaulicht ist; und (ii) eine HD wie sie durch PFAM00046 veranschaulicht wird.
- 78. Verfahren nach Punkt 76 oder 77, wobei das PHDf-HD-Polypeptid
bei der Verwendung zur Konstruktion eines HD-Stammbaums, wie in 13 gezeigt, eher mit der PHDf-HD Gruppe von Polypeptiden,
umfassend die Polypeptidsequenz wie sie in SEQ ID NO: 180 veranschaulicht
ist, Cluster bildet, als mit einer anderen HD-Gruppe.
- 79. Verfahren nach einem der Punkte 76 bis 78, wobei das PHDf-HD-Polypeptid
in steigender Reihenfolge der Präferenz mindestens 30%,
35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%,
99% oder mehr Sequenzidentität zu dem PHDf-HD-Polypeptid
wie es durch SEQ ID NO: 180 veranschaulicht ist oder zu einer der
in Tabelle D1 hier angegebenen Polypeptid-Sequenzen aufweist.
- 80. Verfahren nach einem der Punkte 76 bis 79, wobei die Nukleinsäuresequenz,
die ein PHDf-HD-Polypeptid kodiert, durch eine der in Tabelle D1
angegebenen Nukleinsäuresequenz-SEQ ID NOs oder einen Abschnitt
davon, oder eine Sequenz, die mit eine der in Tabelle D1 angegebenen
Nukleinsäuresequenz SEQ ID NOs hybridisieren kann veranschaulicht
ist.
- 81. Verfahren nach einem der Punkte 76 bis 80, wobei die Nukleinsäuresequenz
ein Orthologon oder Paralogon von einer der in Tabelle D1 angegebenen
SEQ ID NOs kodiert.
- 82. Verfahren nach einem der Punkte 76 bis 81, wobei die modulierte,
vorzugsweise gesteigerte, Expression durch eine oder mehrere von
T-DNA activation tagging, TILLING, oder homologer Rekombination
herbeigeführt wird.
- 83. Verfahren nach einem der Punkte 76 bis 82, wobei die gesteigerte
Expression durch Einführen und Exprimieren einer Nukleinsäuresequenz,
die ein PHDf-HD-Polypeptid kodiert, in eine(r) Pflanze herbeigeführt wird.
- 84. Verfahren nach einem der Punkte 76 bis 83, wobei die Ertragsmerkmale
Samenertragsmerkmale sind, umfassend eines oder mehrere der folgenden
Merkmale: (i) erhöhte Anzahl Primärrispen; (ii)
gesteigertes Gesamtsamengewicht pro Pflanze; (iii) gesteigerte Anzahl
(gefüllter) Samen; (iv) gesteigertes TKG; oder (v) gesteigerter
Harvest-Index.
- 85. Verfahren nach einem der Punkte 76 bis 84, wobei die Nukleinsäuresequenz
funktionsfähig mit einem konstitutiven Promotor, vorzugsweise
einem konstitutiven Pflanzenpromotor, stärker bevorzugt
einem GOS2-Promotor, am stärksten bevorzugt einem GOS2-Promotor
aus Reis verknüpft ist.
- 86. Verfahren nach einem der Punkte 76 bis 85, wobei die Nukleinsäuresequenz,
die ein PHDf-HD-Polypeptid kodiert aus einer Pflanze, vorzugsweise
aus einer monokotylen Pflanze, weiter bevorzugt aus der Familie
Poacae, stärker bevorzugt aus der Gattung Oryza, am stärksten
bevorzugt aus Oryza sativa, stammt.
- 87. Pflanzen, Teile davon (einschließlich Samen) oder
Pflanzenzellen, die durch ein Verfahren nach einem der Punkte 76
bis 86 erhältlich sind, wobei die Pflanze, ein Teil oder
eine Zelle davon ein isoliertes Nukleinsäure-Transgen umfasst,
das ein PHDf-HD Polypeptid kodiert, und das funktionsfähig
mit einem konstitutiven Pflanzenpromotor verknüpft ist.
- 88. Konstrukt, umfassend:
(i) eine Nukleinsäuresequenz,
die ein PHDf-HD Polypeptid nach einem der Punkte 76 bis 81 kodiert;
(ii)
eine oder mehrere Kontrollsequenzen, die die Expression der Nukleinsäuresequenz
von (i) steuern können; und gegebenenfalls
(iii) eine
Transkriptionsterminationssequenz.
wobei mindestens eine der
Kontrollsequenzen ein konstitutiver Pflanzenpromotor, vorzugsweise
ein GOS2-Promotor, ist.
- 89. Verwendung eines Konstrukts nach Punkt 87 in einem Verfahren
zur Herstellung von Pflanzen mit verbesserten Ertragsmerkmalen im
Vergleich zu Kontrollpflanzen, wobei die verbesserten Ertragsmerkmale, vorzugsweise
verbesserten Samenertragsmerkmale ein oder mehrere der folgenden
Merkmale sind: (i) erhöhte Anzahl Primärrispen;
(ii) gesteigertes Gesamtsamengewicht pro Pflanze; (iii) gesteigerte
Anzahl (gefüllter) Samen; (iv) gesteigertes TKG; oder (v)
gesteigerter Harvest-Index.
- 90. Pflanze, Pflanzenteil oder Pflanzenzelle, die mit einem
Konstrukt nach Punkt 87 oder 88 transformiert ist.
- 91. Verfahren zur Produktion transgener Pflanzen mit verbesserten
Ertragsmerkmalen im Vergleich zu Kontrollpflanzen, umfassend:
(i)
Einführen und Exprimieren einer Nukleinsäuresequenz,
die ein PHDf-HD Polypeptid nach einem der Punkte 76 bis 81 kodiert,
in eine(r) Pflanze, eine(m) Pflanzenteil oder eine(r) Pflanzezelle
unter der Kontrolle eines konstitutiven Pflanzenpromotors; und
(ii)
Züchten der Pflanzenzelle unter Bedingungen, die das Wachstum
und die Entwicklung der Pflanze fördern.
- 92. Transgene Pflanze mit verbesserten Ertragsmerkmalen, vorzugsweise
verbesserten Samenertragsmerkmalen, im Vergleich zu Kontrollpflanzen,
der sich aus einer modulierten, vorzugsweise gesteigerten Expression
einer Nukleinsäuresequenz ergibt, die ein PHDf-HD Polypeptid
nach einem der Punkte 76 bis 81 kodiert, die funktionsfähig
mit einem konstitutiven Pflanzenpromotor verknüpft ist,
oder eine transgene Pflanzenzelle, die aus der transgenen Pflanze
stammt.
- 93. Transgene Pflanze nach Punkt 87, 90 oder 92, wobei die Pflanze
eine Kulturpflanze oder eine Monokotyle oder ein Getreide, wie Reis,
Mais, Weizen, Gerste, Hirse, Roggen, Triticale, Sorghum und Hafer
ist, oder eine transgene Pflanzenzelle, die aus der transgenen Pflanze
stammt.
- 94. Erntefähige Teile, umfassend eine Nukleinsäuresequenz,
die ein PHDf-HD-Polypeptid kodiert, einer Pflanze nach Punkt 93,
wobei die erntefähigen Teile vorzugsweise Samen sind.
- 95. Produkte, die von einer Pflanze nach Punkt 93 und/oder von
erntefähigen Teilen einer Pflanze nach Punkt 94 stammen.
- 96. Verwendung einer Nukleinsäuresequenz, die ein PHDf-HD-Polypeptid
nach einem der Punkte 76 bis 81 kodieren, bei der Verbesserung von
Ertragsmerkmalen bei Pflanzen, vorzugsweise Samenertragsmerkmalen,
umfassend eines oder mehrere der folgenden Merkmale: (i) gesteigerte
Anzahl Primärrispen; (ii) gesteigertes Gesamtsamengewicht
pro Pflanze; (iii) gesteigerte Anzahl (gefüllter) Samen;
(iv) gesteigertes TKG; oder (v) gesteigerter Harvest-Index.
- 97. Isoliertes Nukleinsäuremolekül, umfassend
eines der folgenden Merkmale:
(i) eine Nukleinsäure
nach SEQ ID NO: 242;
(ii) eine Nukleinsäure oder ein
Fragment davon, das zu SEQ ID NO: 242 komplementär ist;
(iii)
eine Nukleinsäure, die ein JMJC-Polypeptid kodiert, das
in steigender Reihenfolge der Präferenz mindesten 70%,
75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
zu der in SEQ ID NO: 243 angegebenen Aminosäuresequenz
aufweist;
(iv) eine Nukleinsäure, die unter stringenten
Bedingungen mit einer der in (i), (ii) oder
(iii) oben angegebenen
Nukleinsäuren hybridisieren kann.
- 98. Isoliertes Polypeptid-Molekül, umfassend:
(i)
eine Aminosäuresequenz, die in steigender Reihenfolge der
Präferenz mindestens 70%, 75%, 80%, 85%, 90%, 95%, 96%,
97%, 98%, 99% oder 100% oder mehr Sequenzidentität zu der
in SEQ ID NO: 243 angegebenen Aminosäuresequenz aufweist;
(ii)
Derivate von einer der in (i) angegebenen Aminosäuresequenzen.
- 99. Verfahren zur Verbesserung der Ertragsmerkmale in Pflanzen
im Vergleich zu Kontrollpflanzen, umfassend das Modulieren der Expression
einer Nukleinsäure, die ein bHLH11-ähnliches Polypeptid
kodiert, in einer Pflanze, wobei das bHLH-11-ähnliche Polypeptid
eine Helix-Loop-Helix Domäne umfasst.
- 100. Verfahren nach Punkt 99, wobei das bHLH11-ähnliche
Polypeptid eines oder mehrere der folgenden Motive umfasst:
(i)
Motiv 1 (SEQ ID NO: 246);
(ii) Motiv 2 (SEQ ID NO: 247);
(iii)
Motiv 3 (SEQ ID NO: 248);
(iv) Motiv 4 (SEQ ID NO: 249);
(v)
Motiv 5 (SEQ ID NO: 250);
(vi) Motiv 6 (SEQ ID NO: 251);
(vii)
Motiv 7 (SEQ ID NO: 252).
- 101. Verfahren nach Punkt 99 oder 100, wobei die modulierte
Expression durch Einführen und Exprimieren einer Nukleinsäure,
die ein bHLH11-ähnliches Polypeptid kodiert, in eine(r)
Pflanze herbeigeführt wird.
- 102. Verfahren nach einem der Punkte 99 bis 101, wobei die Nukleinsäure,
die ein bHLH11-ähnliches Polypeptid kodiert, eines der
in Tabelle E1 aufgeführten Proteine kodiert, oder ein Teil
einer solchen Nukleinsäure, oder eine Nukleinsäure
ist, die mit einer solchen Nukleinsäure hybridisieren kann.
- 103. Verfahren nach einem der Punkte 99 bis 102, wobei die Nukleinsäuresequenz
ein Orthologon oder Paralogon von einem der in Tabelle E1 angegebenen
Proteine kodiert.
- 104. Verfahren nach einem der Punkte 99 bis 103, wobei die verbesserten
Ertragsmerkmale einen gesteigerten Ertrag, vorzugsweise gesteigerten
Samenertrag, im Vergleich zu Kontrollpflanzen umfassen.
- 105. Verfahren nach einem der Punkte 99 bis 104, wobei die verbesserten
Ertragsmerkmale unter Nichtstressbedingungen erhalten werden.
- 106. Verfahren nach einem der Punkte 101 bis 105, wobei die
Nukleinsäure funktionsfähig mit einem konstitutiven
Promotor, vorzugsweise einem GOS2-Promotor, am stärksten
bevorzugt einem GOS2-Promotor aus Reis, verknüpft ist.
- 107. Verfahren nach einem der Punkte 99 bis 106, wobei die Nukleinsäure,
die ein bHLH11-ähnliches Polypeptid kodiert, aus einer
Pflanze, vorzugsweise eine monokotylen Pflanze, weiter bevorzugt
aus der Familie Poaceae, stärker bevorzugt aus der Gattung
Triticum, am stärksten bevorzugt aus Triticum aestivum stammt.
- 108. Pflanze oder Teil davon, einschließlich Samen,
erhältlich durch ein Verfahren nach einem der Punkte 99
bis 107, wobei die Pflanze oder das Teil davon eine rekombinante
Nukleinsäure umfasst, die ein bHLH11-ähnliches
Polypeptid kodiert.
- 109. Konstrukt, umfassend:
(i) eine Nukleinsäure,
die ein bHLH11-ähnliches Polypeptid nach den Punkten 99
oder 100 kodiert;
(ii) eine oder mehrere Kontrollsequenzen,
die die Expression der Nukleinsäuresequenz von (i) steuern
können; und gegebenenfalls
(iii) eine Transkriptionsterminationssequenz.
- 110. Konstrukt nach Punkt 109, wobei eine der Kontrollsequenzen
ein konstitutiver Promotor, vorzugsweise ein GOS2-Promotor, am stärksten
bevorzugt ein GOS2-Promotor aus Reis, ist.
- 111. Verwendung eines Konstrukts nach Punkt 109 oder 110 in
einem Verfahren zur Herstellung von Pflanzen mit gesteigertem Ertrag,
insbesondere gesteigertem Samenertrag im Vergleich zu Kontrollpflanzen.
- 112. Pflanze, Pflanzenteil oder Pflanzenzelle, die mit einem
Konstrukt nach Punkt 109 oder 110 transformiert sind.
- 113. Verfahren zur Herstellung einer transgenen Pflanze mit
gesteigertem Ertrag, insbesondere gesteigertem Samenertrag, im Vergleich
zu Kontrollpflanzen, umfassend:
(i) Einführen und
Exprimieren einer Nukleinsäure, die ein bHLH11-ähnliches
Polypeptid nach Punkt 99 oder 100 kodiert, in eine(r) Pflanze; und
(ii)
Züchten der Pflanzenzelle unter Bedingungen, die das Wachstum
und die Entwicklung der Pflanze fördern.
- 114. Transgene Pflanze mit gesteigertem Ertrag, insbesondere
gesteigertem Samenertrag im Vergleich zu Kontrollpflanzen, der sich
aus der modulierten Expression einer Nukleinsäure, die
ein bHLH11-ähnliches Polypeptid nach Punkt 99 oder 100
kodiert, ergibt, oder eine transgene Pflanzenzelle, die aus der
transgenen Pflanze stammt.
- 115. Transgene Pflanze nach Punkt 108, 112 oder 114, oder eine
daraus stammende transgene Pflanzenzelle, wobei die Pflanze eine
Kulturpflanze oder eine Monokotyle oder ein Getreide, wie Reis,
Mais, Weizen, Gerste, Hirse, Roggen, Triticale, Sorghum, Emmer,
Dinkel, Secale, Einkorn, Zwerghirse, Mohrenhirse and Hafer ist.
- 116. Erntefähige Teile einer Pflanze nach Punkt 115,
wobei die erntefähigen Teile vorzugsweise Samen sind.
- 117. Produkte, die aus einer Pflanze nach Punkt 115 und/oder
aus erntefähigen Teilen einer Pflanze nach Punkt 116 stammen.
- 118. Verwendung einer Nukleinsäure, die ein bHLH11-ähnliches
Polypeptid kodiert, bei der Steigerung des Ertrags, insbesondere
bei der Steigerung des Samenertrags in Pflanzen, im Vergleich zu
Kontrollpflanzen.
- 119. Verfahren zur Verbesserung der Ertragsmerkmale in Pflanzen
im Vergleich zu Kontrollpflanzen, umfassend die Modulation der Expression
einer Nukleinsäure, die ein ASR kodiert, wobei das ASR
durch SEQ ID NO: 397 oder einem Orthologon oder Paralogon veranschaulicht
wird, in einer Pflanze.
- 120. Verfahren nach Punkt 119, wobei die modulierte Expression
durch Einführen und Exprimieren einer Nukleinsäure,
die das ASR-Polypeptid kodiert, in eine(r) Pflanze herbeigeführt
wird.
- 121. Verfahren nach Punkt 119 oder 120, wobei die Nukleinsäure,
die das ASR-Polypeptid kodiert, ein Abschnitt von SEQ ID NO: 396
ist, oder eine Nukleinsäure, die mit einer solchen Nukleinsäure
hybridisieren kann.
- 122. Verfahren nach einem der Punkte 119 bis 121, wobei die
Nukleinsäuresequenz SEQ ID NO: 397 oder ein Orthologon
oder Paralogon davon kodiert.
- 123. Verfahren nach einem der Punkte 119 bis 122, wobei die
verbesserten Ertragsmerkmale einen gesteigerten Ertrag, vorzugsweise
gesteigerten Samenertrag im Vergleich zu Kontrollpflanzen umfassen.
- 124. Verfahren nach einem der Punkte 119 bis 123, wobei die
verbesserten Ertragsmerkmale unter Nichtstressbedingungen erhalten
werden.
- 125. Verfahren nach einem der Punkte 120 bis 124, wobei die
Nukleinsäure funktionsfähig mit einem konstitutiven
Promotor, vorzugsweise einem GOS2-Promotor, am stärksten
bevorzugt einem GOS2-Promotor aus Reis, verknüpft ist.
- 126. Verfahren nach einem der Punkte 119 bis 125, wobei die
Nukleinsäure, die ein ASR-Polypeptid kodiert, aus einer
Pflanze, vorzugsweise aus einer dikotylen Pflanze, weiter bevorzugt
aus der Familie Poaceae, stärker bevorzugt aus der Gattung
Oryza, am stärksten bevorzugt aus Oryza sativa stammt.
- 127. Pflanzen oder Teil davon, einschließlich Samen,
erhältlich durch ein Verfahren nach einem der Punkte 119
bis 126, wobei die Pflanze oder das Teil davon eine rekombinante
Nukleinsäure umfasst, die ein ASR-Polypeptid kodiert.
- 128. Konstrukt, umfassend:
(i) eine Nukleinsäure,
die ein ASR-Polypeptid nach Punkt 119 kodiert, oder eine von SEQ
ID NO: 401, 403, 405, 407, 409, 411, 413, 415 und 417;
(ii)
eine oder mehrere Kontrollsequenzen, die die Expression der Nukleinsäuresequenz
von (a) steuern können; und gegebenenfalls
(iii) ein
Transkriptionsterminationssequenz.
- 129. Konstrukt nach Punkt 128, wobei eine der Kontrollsequenzen
ein konstitutiver Promotor, vorzugsweise ein GOS2-Promotor, am stärksten
bevorzugt ein GOS2-Promotor aus Reis, ist.
- 130. Verwendung eines Konstrukts nach einem der Punkte 128 oder
129 in einem Verfahren zur Herstellung von Pflanzen mit verbesserten
Ertragsmerkmalen, insbesondere gesteigertem Samenertrag im Vergleich
zu Kontrollpflanzen.
- 131. Pflanze, Pflanzenteil oder Pflanzenzelle, die mit einem
Konstrukt nach einem der Punkte 128 oder 129 transformiert sind.
- 132. Verfahren zur Herstellung einer transgenen Pflanze mit
gesteigertem Ertrag, insbesondere gesteigerter Biomasse und/oder
gesteigertem Samenertrag im Vergleich zu Kontrollpflanzen, umfassend:
(i)
Einführen und Exprimieren einer Nukleinsäure,
die ein ASR-Polypeptid nach Punkt 119 kodiert, in eine(r) Pflanze;
und
(ii) Züchten der Pflanzenzelle unter Bedingungen,
die das Wachstum und die Entwicklung der Pflanze fördern.
- 133. Transgene Pflanze mit verbesserten Samenertragsmerkmalen,
insbesondere gesteigertem Samenertrag, im Vergleich zu Kontrollpflanzen,
der sich aus einer gesteigerten Expression einer Nukleinsäure,
die ein ASR-Polypeptid nach Punkt 119 kodiert, ergibt, oder eine
transgene Pflanzenzelle, die aus der transgenen Pflanze stammt.
- 134. Transgene Pflanze nach Punkt 127, 131 oder 133, oder eine
davon abgeleitete transgene Pflanzenzelle, wobei die Pflanze eine
Kulturpflanze oder eine Monokotyle oder ein Getreide, wie Reis,
Mais, Weizen, Gerste, Hirse, Roggen, Triticale, Sorghum und Hafer
ist.
- 135. Erntefähige Teile einer Pflanze nach Punkt 134,
wobei die erntefähigen Teile vorzugsweise Samen sind.
- 136. Produkte, die aus einer Pflanze nach Punkt 134 und/oder
aus erntefähigen Teilen einer Pflanze nach Punkt 135 stammen.
- 137. Verwendung einer Nukleinsäure, die ein ASR-Polypeptid
kodiert, bei der Verbesserung der Ertragsmerkmale, insbesondere
bei der Steigerung des Samenertrags bei Pflanzen im Vergleich zu
Kontrollpflanzen.
- 138. Isoliertes Nukleinsäuremolekül, umfassend
eines der folgenden Merkmale:
(i) eine Nukleinsäure
nach einer der Sequenzen von SEQ ID NO: 401, 403, 405, 407, 409,
411, 413, 415 und 417;
(ii) das Komplement einer Nukleinsäure,
dargestellt durch eine der Sequenzen von SEQ ID NO: 401, 403, 405,
407, 409, 411, 413, 415 und 417;
(iii) eine Nukleinsäure,
die ein ASR-Polypeptid kodiert, das in steigender Reihenfolge der
Präferenz mindestens 50%, 55%, 60%, 65%, 70%, 75%, 80%,
85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
zu der Aminosäuresequenz aufweist, die durch eine der Sequenzen
von SEQ ID NO: 402, 404, 406, 408, 410, 412, 414, 416 und 418 veranschaulicht
wird.
- 139. Isoliertes Polypeptid, umfassend:
(i) eine Aminosäuresequenz,
nach einer der Sequenzen von SEQ ID NO: 402, 404, 406, 408, 410,
412, 414, 416 und 418;
(ii) eine Aminosäuresequenz,
die in steigender Reihenfolge der Präferenz mindestens
50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%,
99% oder mehr Sequenzidentität zu der Aminosäuresequenz
aufweist, die durch eine der Sequenzen von SEQ ID NO: 402, 404,
406, 408, 410, 412, 414, 416 und 418 veranschaulicht wird.
(iii)
Derivate von einer der in (i) oder (ii) oben angegebenen Aminosäuresequenzen.
- 140. Verfahren zur Verbesserung von Ertragsmerkmalen in Pflanzen
im Vergleich zu Kontrollpflanzen, bei dem man in einer Pflanze die
Expression einer Nukleinsäure, die ein SPL11-Polypeptid
kodiert, moduliert, wobei das SPL11-Polypeptid eine SBP-Domäne
umfasst, die in steigender Reihenfolge der Präferenz mindestens
70%, 75%, 80%, 85%, 90%, 95%, 97% oder mehr Sequenzidentität
zu einer der Sequenzen von SEQ ID NO: 456 to SEQ ID NO: 468 und
SEQ ID NO: 478 aufweist.
- 141. Verfahren nach Punkt 140, wobei das SPL11-Polypeptid neben
der SBP-Domäne eine oder mehrere der folgenden konservierten
Motive umfasst:
(i) Motiv 1 nach SEQ ID NO: 469, wobei jede
konservative Aminosäuresubstitution und/oder 1 oder 2 nichtkonservative
Substitutionen erlaubt sind;
(ii) Motiv 2 nach SEQ ID NO: 470,
wobei jede Änderung erlaubt ist, vorausgesetzt dass mindestens
4 Aminosäuren eine polare Seitenkette, vorzugsweise Serin
oder Threonin, aufweisen, und vorausgesetzt dass sich die Domäne
am N-terminalen Ende der SBP-Domäne befindet;
(iii)
Motiv 3 nach SEQ ID: 471 wobei 1 oder 2 Mismatches erlaubt sind;
(iv)
Motiv 4 nach SEQ ID: 472 wobei 1, 2 oder 3 Mismatches erlaubt sind.
- 142. Verfahren nach Punkt 140 oder 141, wobei die modulierte
Expression durch Einführen und Exprimieren einer Nukleinsäure,
die ein SPL11-Polypeptid nach Punkt 140 oder 141 kodiert, in einer
Pflanze herbeigeführt wird.
- 143. Verfahren nach einem der Punkte 140 bis 142, wobei die
modulierte Expression eine gesteigerte Expression ist.
- 144. Verfahren nach einem der Punkte 140 bis 143, wobei die
Nukleinsäure, die ein SPL11-Polypeptid kodiert, eines der
in Tabelle G1 aufgeführten Proteine kodiert, oder ein Abschnitt
einer solchen Nukleinsäure, oder eine Nukleinsäure
ist, die mit einer solchen Nukleinsäure hybridisieren kann.
- 145. Verfahren nach einem der Punkte 140 bis 144, wobei die
Nukleinsäuresequenz ein Orthologon oder Paralogon von einem
der in Tabelle G1 angegebenen Proteine kodiert.
- 146. Verfahren nach Punkt 145, wobei die Nukleinsäure
SEQ ID NO: 428 kodiert.
- 147. Verfahren nach einem der Punkte 140 bis 146, wobei die
verbesserten Ertragsmerkmale einen gesteigerten Ertrag, vorzugsweise
gesteigertes Gesamtsamengewicht, Anzahl der gefüllten Samen,
Anzahl der Samen oder Blütchen pro Rispe, Tausendkorngewicht,
Samenfüllungsrate und/oder Harvest-Index im Vergleich zu
Kontrollpflanzen umfassen.
- 148. Verfahren nach einem der Punkte 140 bis 147, wobei die
verbesserten Ertragsmerkmale die Jungpflanzenvitalität
der Pflanze (des Keimlings) im Vergleich zu Kontrollpflanzen umfassen.
- 149. Verfahren nach einem der Punkte 140 bis 147, wobei die
verbesserten Ertragsmerkmale unter Nichtstressbedingungen erhalten
werden.
- 150. Verfahren nach einem der Punkte 140 bis 147, wobei die
verbesserten Ertragsmerkmale unter Wachstumsbedingungen eines leichten
Trockenheits-Stresses erhalten werden.
- 151. Verfahren nach einem der Punkte 140 bis 147, wobei die
verbesserten Ertragsmerkmale unter Stickstoffmangelbedingungen erhalten
werden.
- 152. Verfahren nach einem der Punkte 140 bis 151, wobei die
Nukleinsäure funktionsfähig mit einem konstitutiven
Promotor, vorzugsweise einem GOS2-Promotor, am stärksten
bevorzugt einem GOS2-Promotor aus Reis, verknüpft ist.
- 153. Verfahren nach einem der Punkte 140 bis 151, wobei die
Nukleinsäure funktionsfähig mit einem samenspezifischen
Promotor, vorzugsweise einem WSI18-Promotor, am stärksten
bevorzugt einem WSI18-Promotor aus Reis, verknüpft ist.
- 154. Verfahren nach einem der Punkte 140 bis 153, wobei die
Nukleinsäure, die ein SPL11-Polypeptid kodiert, aus einer
Pflanze, vorzugsweise aus einer dikotylen Pflanze, weiter bevorzugt
aus der Familie Brassicaceae, stärker bevorzugt aus der
Gattung Arabidopsis, am stärksten bevorzugt aus Arabidopsis
thaliana stammt.
- 155. Pflanze oder Teil davon, einschließlich Samen,
erhältlich durch ein Verfahren nach einem der Punkte 140
bis 154, wobei die Pflanze oder das Teil davon eine rekombinante
Nukleinsäure umfasst, die ein SPL11-Polypeptid kodiert.
- 156. Isoliertes Nukleinsäuremolekül, umfassend:
(i)
eine Nukleinsäure nach SEQ ID NO: 448;
(ii) das Komplement
einer Nukleinsäure nach SEQ ID NO: 448;
(iii) eine
Nukleinsäure, die ein SPL11-Polypeptid kodiert, das in
steigender Reihenfolge der Präferenz mindestens 70%, 75%,
80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
zu der Aminosäuresequenz nach SEQ ID NO: 449 aufweist,
und die in steigender Reihenfolge der Präferenz mindestens
70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
zu SEQ ID NO: 465: SYCQVEGCRTDLSSAKDYHRKHRVCEPHSKAPKVWAGLERRFCQQCSRFHG
LAEFDQKKKSCRRRLNDHNARRRKPQPEAL aufweist;
(iv) eine Nukleinsäure,
die unter stringenten Bedingungen mit SEQ ID NO: 448 hybridisiert.
- 157. Isoliertes Polypeptid, umfassend:
(i) eine Aminosäuresequenz
nach SEQ ID NO: 449;
(ii) eine Aminosäuresequenz,
die in steigender Reihenfolge der Präferenz mindestens
70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
zu der Aminosäuresequenz nach SEQ ID NO: 449 aufweist,
und die in steigender Reihenfolge der Präferenz mindestens
70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% oder mehr Sequenzidentität
zu SEQ ID NO: 465:
SYCQVEGCRTDLSSAKDYHRKHRVCEPHSKAPKVVVAGLERRFCQQCSRFHG
LAEFDQKKKSCRRRLNDHNARRRKPQPEAL aufweist.
(iii) Derivate von
einer der in (i) oder (ii) oben angegebenen Aminosäuresequenzen.
- 158. Konstrukt, umfassend:
(i) eine Nukleinsäure,
die ein SPL11-Polypeptid kodiert;
(ii) eine oder mehrere Kontrollsequenzen,
die die Expression der Nukleinsäuresequenz von (i) steuern
können; und gegebenenfalls
(iii) eine Transkriptionsterminationssequenz
- 159. Konstrukt nach Punkt 158, wobei die Nukleinsäure,
die ein SPL11-Polypeptid kodiert, eine Nukleinsäure nach
Punkt 156 ist.
- 160. Konstrukt nach Punkt 158 oder 159, wobei eine der Kontrollsequenzen
ein konstitutiver Promotor, vorzugsweise ein GOS2-Promotor, am stärksten
bevorzugt ein GOS2-Promotor aus Reis, ist.
- 161. Konstrukt nach Punkt 158 oder 159, wobei eine der Kontrollsequenzen
ein samenspezifischer Promotor, vorzugsweise ein WSI18-Promotor,
am stärksten bevorzugt ein WSI18-Promotor aus Reis, ist.
- 162. Verwendung eines Konstrukts nach den Punkten 158 bis 161
in einem Verfahren zur Herstellung von Pflanzen mit verbesserten
Ertragsmerkmalen, insbesondere gesteigertem Samenertrag im Vergleich
zu Kontrollpflanzen und/oder einer Pflanze mit Jungpflanzenvitalität
beim Keimling.
- 163. Pflanze, Pflanzenteil oder Pflanzenzelle, die mit einem
Konstrukt nach den Punkten 158 bis 161 transformiert sind.
- 164. Verfahren zur Herstellung einer transgenen Pflanze mit
verbesserten Ertragsmerkmalen, insbesondere gesteigertem Samenertrag
und/oder Jungpflanzenvitalität bei der Pflanze oder dem
Keimling im Vergleich zu Kontrollpflanzen, umfassend:
(i) Einführen
und Exprimieren einer Nukleinsäure, die ein SPL11-Polypeptid
kodiert, in eine(r) Pflanze; und
(ii) Züchten der
Pflanzenzelle unter Bedingungen, die das Wachstum und die Entwicklung
der Pflanze fördern.
- 165. Transgene Pflanze mit gesteigertem Ertrag, insbesondere
gesteigerter Jungpflanzenvitalität beim Keimling und/oder
gesteigertem Samenertrag, im Vergleich zu Kontrollpflanzen, der
sich aus einer gesteigerten Expression einer Nukleinsäure,
die ein SPL11-Polypeptid kodiert, ergibt, oder eine transgene Pflanzenzelle,
die aus der transgenen Pflanze stammt.
- 166. Transgene Pflanze nach Punkt 155, 163 oder 165, oder eine
daraus stammende transgene Pflanzenzelle, wobei die Pflanze eine
dikotyle Kulturpflanze, wie Sojabohne, Baumwolle oder Canola-Raps,
oder eine monokotyle Kulturpflanze oder ein Getreide, wie Reis,
Mais, Weizen, Gerste, Hirse, Roggen, Triticale, Sorghum und Hafer,
ist.
- 167. Erntefähige Teile einer Pflanze nach Punkt 166,
wobei die erntefähigen Teile vorzugsweise Sprossbiomasse,
Blüten und/oder Samen sind.
- 168. Produkte, die aus einer Pflanze nach Punkt 166 und/oder
aus erntefähigen Teilen einer Pflanze nach Punkt 167 stammen.
- 169. Verwendung einer Nukleinsäure, die ein SPL11-Polypeptid
kodiert, bei der Steigerung des Ertrags, insbesondere Samenertrags,
und/oder der Sprossbiomasse in Pflanzen im Vergleich zu Kontrollpflanzen.
-
Beschreibung der Figuren
-
Die
vorliegende Erfindung wird nun anhand der folgenden Figuren veranschaulicht.
Es zeigt:
-
I. LOB-Domäne umfassendes Protein
(LOB: Lateral Organ Boundaries, Laterale Organgrenzen)
-
1 die
Sequenz von SEQ ID NO: 2, wobei die DUF260 Domäne fett
gezeigt ist, die konservierten Motive 1, 2 and 3 unterstrichen sind
und die konservierten Cys-Reste (Motiv von SEQ ID NO: 9) kursiv
gezeigt ist.
-
2 ein
multiples Alignment von Sequenzen verschiedener LBD-Proteine, die
sich in den erfindungsgemäßen Verfahren eignen.
-
3 einen
Stammbaum der LBD-Proteine der Klasse II und Klasse I. Die Sequenz
von SEQ ID NO: 2 ist durch AtLBD37 veranschaulicht.
-
4 den
binären Vektor für die gesteigerte Expression
in Oryza sativa einer LBD-kodierenden Nukleinsäure unter
der Kontrolle eines GOS2-Promotors (pGOS2) aus Reis.
-
5 genaue
Beispiele für LBD-Sequenzen, die sich zur Durchführung
der erfindungsgemäßen Verfahren eignen.
-
II. JMJC(JUMONJI-C)-Polypeptid
-
6 die
Aminosäuresequenz und die Domänenstruktur von
SEQ ID NO: 74. Es sind konservierte Motive und Domänen
gezeigt. Die JmjC-Domäne ist fett gezeigt. Die konservierten
Motive SEQ ID NO: 79, SEQ ID NO: 80 und SEQ ID NO: 81 sind durch
einfach, doppelt bzw. dreifach gestrichelte Rechtecke gezeigt. Unterstrichen
sind die Aminosäurereste T (Theonin) und K (Lysin), die
gewöhnlich an der 2-Oxogutarat-Koordinierung beteiligt
sind. Ein groß geschriebenes H zeigt den Aminosäurerest
Histidin an, der das Eisenion koordiniert.
-
7 ein
multiples Alignment von JMJC-Proteinen. Der Ursprung des Proteins
ist durch die beiden ersten alphanumerischen Zeichen im Namen angegeben,
At: Arabidopsis thaliana, Pp: Populus trichocarpa, Os: Oryza sativa,
Ot: streococcus tauri, Ce: Caenorhabditis elegans, Hs: Homo sapiens.
Die Position der konservierten Aminosäurereste und der
Domänen, die denen der 6 entsprechen,
ist angegeben. Es ist eine Konsensussequenz angegeben.
-
Hochkonservierte
Aminosäuren in den Konsensussequenzen sind angegeben; Leerstellen
stehen für eine beliebige Aminosäure.
-
8 einen
Stammbaum der JMJC-Polypeptide. Der Pfeil zeigt SEQ ID NO: 74. Andere
JMJC-Polypeptide sind nach der Genbank-Zugangsnummer bezeichnet.
Gruppe I (G I) umfasst Proteine pflanzlichen Ursprungs; Gruppe II
(GII) umfasst Proteine nicht-pflanzlichen Ursprungs.
-
9 den
binären Vektor für eine gesteigerte Expression
in Oryza sativa von SEQ ID NO: 73 unter der Kontrolle eines GOS2-Promotors
(pGOS2) aus Reis.
-
10 genaue Beispiele für JMJC-Sequenzen,
die sich zur Durchführung der erfindungsgemäßen Verfahren
eignen.
-
III. Casein Kinase I
-
11 den binären Vektor für eine
gesteigerte Expression in Oryza sativa einer GRP-kodierenden Nukleinsäure
unter der Kontrolle eines WSI18-Promotors (pWSI18::GRP) aus Reis.
-
12 genaue Beispiele für GRP-Sequenzen,
die sich zur Durchführung der erfindungsgemäßen
Verfahren eignen.
-
IV. Pflanzen-Homöodomänen-Finger-Homöodomänen(Plant
homeodomain finger-homeodomain (PHDf-HD))-Polypeptid
-
13 ein Dendrogramm, konstruiert nach einem Alignment
sämtlicher Transkriptionsfaktoren, die zu der HD-Familie
gehören (heruntergeladen von der riceTFDB Datenbank, die
auf dem Server der Universität Potsdam gehostet wird) und
sämtlicher Polypeptid-Sequenzen von Tabelle D1 (wenn es
sich um Volllänge handelt), durchgeführt mit dem
Clustal-Algorithmus (1.83) des progressiven Alignments, unter Verwendung
von Default Werten. Die interessierende Gruppe, die zwei Reis-Paraloga
umfasst (SEQ ID NO: 180 oder Os02g05450.1, und SEQ ID NO: 202 oder
Os06g12400.1) wurde eingekreist. Jedes Polypeptid, das in dieser HD-Gruppe
liegt (nach einem neuen multiplen Alignment-Schritt wie vorstehend
beschrieben) wird zur Durchführung der hier beschriebenen
erfindungsgemäßen Verfahren als geeignet angesehen.
-
14 einen Cartoon eines PHDf-HD Polypeptids nach
SEQ ID NO: 180, das eines oder mehrere der folgenden Merkmale umfasst:
ein vorhergesagtes Kernlokalisierungssignal (NLS), einen Leucin-Zipper
(ZIP), einen PHD-Finger (PHDf, Pfam00628), einen sauren Abschnitt,
zwei basische Abschnitte, eine Homöodomäne (HD,
Pfam00046).
-
15 das Sequenzlogo für die Homöodomäne
(HD) der PHDf-HD-Polypeptide von Tabelle D1, wobei die Gesamthöhe
des Stapels die Sequenzkonservierung an dieser Position angibt,
wohingegen die Höhe der Symbole innerhalb des Stapels die
relative Häufigkeit jeder Aminosäure oder Nukleinsäure
an dieser Position angibt. Die HD nach SEQ ID NO: 234, die sich
auch in SEQ ID NO: 180 befindet, entspricht dem in dieser Figur
gezeigten Sequenzlogo.
-
16 die graphische Ausgabe des COILS-Algorithmus,
der zwei Coiled Coil-Domänen in der N-terminalen Hälfte
des Polypeptids nach SEQ ID NO: 180 vorhersagt. Die X-Achse veranschaulicht
die Aminosäurereste-Koordinaten, die Y-Achse die Wahrscheinlichkeit
(die von 0 bis 1 reicht), dass eine Coiled Coil-Domäne zugegen
ist, und die drei Linien, die drei untersuchten Fenster (14, 21,
28).
-
17 ein CLUSTAL W (1; 83) multiples Sequenzalignment
von PHDf-HD-Polypeptiden aus Tabelle D1 (wenn es sich um Volllänge
handelt), wobei eine Anzahl von Merkmalen identifiziert werden.
Vom N-Terminus zum C-Terminus der Polypeptide sind: (i) ein vorhergesagtes
Kernlokalisierungssignal (NLS); (ii) ein Leucin-Zipper (ZIP), mit
vier Heptaden (im Kasten, wobei gewöhnlich ein Leucin (mitunter
ein Isoleucin, ein Valin, oder ein Methionin) an jeder siebten Aminosäure
erscheint; (iii) ein PHD-Finger (PHDf), mit dem üblichen C4HC3
(4 Cysteine, 1 Histidin, 3 Cysteine) und einem charakteristischen
Cystein-Spacing; (iv) ein saurer Abschnitt (reich an sauren Aminosäuren
D und E); (v) saure Abschnitte (reich an basischen Aminosäuren
K und R); (vi) eine Homöodomäne (HD).
-
18 den binären Vektor für modulierte,
vorzugsweise gesteigerte Expression in Oryza sativa einer Nukleinsäuresequenz,
die ein PHDf-HD Polypeptid kodiert, unter der Kontrolle des GOS2-Promotors
(pGOS2) aus Reis.
-
19 genaue Beispiele für PHDf-HD Sequenzen,
die sich zur Durchführung der erfindungsgemäßen Verfahren
eignen.
-
V. bHLH11-ähnliches(basisches
Helix-Loop-Helix 11)-Protein
-
20 die Domänenstruktur von SEQ ID NO:
245 wobei die konservierten Motive durch Unterstreichen und ihre
Zahl angegeben ist. Die HLH-Domäne (Motiv 8) wie durch
SMART bestimmt ist fett unterstrichen gezeigt.
-
21 ein multiples Alignment verschiedener bHLH11-ähnlicher
Proteine. Ein Punkt zeigt konservierte Reste, ein Komma hochkonservierte
Reste, und ein Sternchen steht für perfekt konservierte
Reste. Der höchste Grad an Sequenzkonservierung findet
sich in der Region der bHLH Domäne. Der C-terminale Teil
von AT2G24260, der über die anderen Proteine hinausgeht,
ist weggelassen.
-
22 Kreis-Kladogramm ausgewählter bHLH-Proteine.
bHLH11-ähnliche Proteine und ein Arabidopsis-Protein, die
jeweils eine der anderen Klassen repräsentieren, die von
Heim 2003 definiert wurden, wurden verwendet. Das Alignment erfolgte
mit ”CLUSTALX”, und es wurde ein Dendrogramm berechnet.
Das Kreis-Kladogramm wurde mit einem Dendroskop gezeichnet (Huson
et al. BMC Bioinformatics 2007). Bootstrap-Ergebnisse für
100 Replikate sind für einige Hauptknoten angegeben, wobei
der Bootstrap-Wert im Kasten zeigt, dass die Gruppe der bHLH11-ähnlichen
Proteine klar von den anderen bHLH Proteinen abgegrenzt ist.
-
23 den binären Vektor zur gesteigerten
Expression in Oryza sativa einer bHLH11-ähnlich-kodierenden
Nukleinsäure unter der Kontrolle eines GOS2-Promotors (pGOS2)
aus Reis.
-
24 genaue Beispiele für bHLH11-ähnliche
Sequenzen, die sich zur Durchführung in den erfindungsgemäßen
Verfahren eignen.
-
VI. ASR (abszisinsäure-, stress-,
und reifeinduziertes) Protein
-
25 den binären Vektor für eine
gesteigerte Expression in Oryza sativa einer GRP-kodierenden Nukleinsäure
unter der Kontrolle eines GOS2-Promotors (pGOS2::GRP) aus Reis.
-
26 genaue Beispiele für GRP-Sequenzen,
die sich zur Durchführung der erfindungsgemäßen
Verfahren eignen.
-
VII. Squamosa-Promotor-Bindungsprotein-ähnlich
11 (SPL11)
-
27 die Aminosäuresequenz und die Domänenstruktur
von SEQ ID NO: 428. Konservierte Motive und Domänen sind
gezeigt. Die SBP-Domäne ist durch eine unterbrochene Linie
gezeigt; Motiv 1 ist fett gezeigt; Motiv 2 ist fett und unterstrichen
gezeigt; Motiv 3 ist in fetten Großbuchstaben und unterstrichen
und Motiv 4 ist fett und doppelt unterstrichen.
-
28 ein multiples Sequenzalignment von SPL11-Polypeptiden.
Die Position der konservierten Aminosäurereste und Domänen,
die denjenigen der 27 entsprechen, ist angegeben.
Eine Konsensussequenz, die SPL11 Polypeptide veranschaulicht, ist
angegeben. In der Konsensussequenz sind die hochkonservierten Aminosäuren
angegeben und Leerstellen dazwischen stehen für eine beliebige
Aminosäure.
-
29 einen Stammbaum der SPL11-Polypeptide. Der
Stammbaum entspricht demjenigen in Xie et al. Plant Physiology,
2006, Vol. 142, pp. 280–293, die hiermit vollinhaltlich
durch Bezugnahme aufgenommen ist. SPL11-Polypeptide bilden in der
gleichen Gruppe Cluster wie AtSPL11 (identisch zu SEQ ID NO: 2)
in der als Klass S3 bezeichneten Klasse.
-
30 eine Sequenzanalyse von miR156 Genen
aus (OsmiR156) Reis und ihren angezielten Sequenzen in SPL-Polypeptiden
(30A) aus Reis, sowie ein multiples Alignment von
SPL11-Nukleinsäuren (30B).
Die 30A zeigt ein Sequenz-Alignment
von reifen OsmiR156-Sequenzen mit komplementären Sequenzen
von OsSPL-Genen. Die konservierte Aminosäuresequenz, die
durch die Zielsequenzen kodiert wird, ist unten gezeigt. Die Punkte
zwischen miR156 und den angezielten OsSPL-Sequenzen zeigen Mismatches.
Die 4B zeigt ein multiples Alignment
von SPL11-Nukleinsäuren, wobei die hochkonservierten Nukleinsäurereste
in der Konsensussequenz angezeigt sind. Die Position der hochkonservierten
miR156-Zielstelle ist fett gegenüber der Konsensussequenz
angegeben. SEQ ID NO: 431, SEQ ID NO: 440 und SEQ ID NO: 454 veranschaulichen
SPL11-Nukleinsäuren, die miR156-unempfindlich sind, die
nicht die konservierte miR156 Zielstelle haben oder eine große
Divergenz in ihrer Position an dieser Stelle haben.
-
31 den binären Vektor für
eine gesteigerte Expression in Oryza sativa von SEQ ID NO: 427 unter der
Kontrolle eines GOS2-Promotors (pGOS2) aus Reis (31A) oder unter einem WSI 18-Promotor aus Reis
(31B).
-
32 genaue Beispiele für SPL11-Sequenzen,
die sich bei der Durchführung der erfindungsgemäßen
Verfahren eignen.
-
Beispiele
-
Die
vorliegende Erfindung wird nun anhand der folgenden Beispiele beschrieben,
die lediglich beispielhaft sind. Die folgenden Beispiele sollen
den Schutzbereich der Erfindung keinesfalls vollständig
definieren oder ansonsten einschränken.
-
Die
DNA-Manipulation: wenn nicht anders angegeben werden DNA-Rekombinationstechniken
gemäß Standard-Protokollen durchgeführt,
beschrieben in Sambrook (2001) Molecular Cloning: a laboratory
manual, 3rd Edition Cold Spring Harbor Laboratory Press, CSH, New
York, oder in Bd. 1 und 2 von Ausubel et al. (1994),
Current Protocols in Molecular Biology, Current Protocols. Standard-Materialien
und Verfahren zur molekularen Arbeit an der Pflanze sind in Plant
Molecular Biology Labfax (1993) von R. D. D. Croy, veröffentlicht von
BIOS Scientific Publications Ltd (UK) und Blackwell Scientific Publications
(UK) beschrieben.
-
I. LOB-Domäne umfassendes Protein
(LOB: Laterale Organ Grenzen)
-
Beispiel 1: Identifikation von Sequenzen,
die mit der in den erfindungsgemäßen Verfahren
verwendeten Nukleinsäuren verwandt sind
-
Unter
den Sequenzen in der Entrez Nucleotides Datenbank am National Center
for Biotechnology Information (NCBI) wurden unter Verwendung von
Datenbanksequenzabfragewerkzeugen, wie dem Basic Local Alignment
Tool (BLAST) (Altschul et al., (1990), J. Mol. Biol. 215:
403–410; und Altschul et al., (1997),
Nucleic Acids Res. 25: 3389–3402), (Volllängen-cDNA,
ESTs oder genomische) Sequenzen, die mit der in den erfindungsgemäßen
Verfahren verwendeten Nukleinsäuren verwandt sind, identifiziert.
Das Programm wurde zum Auffinden von Regionen mit lokaler Ähnlichkeit
zwischen Sequenzen verwendet, und zwar durch Vergleichen von Nukleinsäure-
oder Polypeptid-Sequenzen mit Sequenz-Datenbanken und durch Berechnen
der statistischen Signifikanz der Matches. Das durch die erfindungsgemäß verwendete
Nukleinsäure kodierte Polypeptid wurde beispielsweise für
den TBLASTN-Algorithmus verwendet, und zwar unter Ausschaltung der
Default-Einstellungen und des Filters für die Nichtmiteinbeziehung
von Sequenzen mit niedriger Komplexität. Der Output der
Analyse wurde mittels paarweisem Vergleich betrachtet und gemäß der
Wahrscheinlichkeits-Wertung (E-Wert) gereiht, wobei die Wertung
die Wahrscheinlichkeit, dass ein bestimmtes Alignment statistisch
vorkommt, widerspiegelt (je niedriger der E-Wert, desto signifikanter
der Hit). Zusätzlich zu den E-Werten wurden die Vergleiche
auch durch den Prozentsatz der Identität gewertet. Der
Prozentsatz der Identität bezieht sich auf die Anzahl identischer
Nukleotide (oder Aminosäuren) zwischen den zwei verglichenen
Aminosäure (oder Polypeptid)-Sequenzen über eine
bestimmte Länge.
-
Die
Tabelle A1 stellt eine Liste von Nukleinsäuresequenzen
bereit, die mit der in den erfindungsgemäßen Verfahren
verwendeten LBD Nukleinsäuresequenz verwandt sind Tabelle A1: Beispiele für LBD-Polypeptide:
zugefügt DNA Sequenzen von Reis, Mais, Weizen, Canola-Raps, Kartoffel,
Soja, Arabidopsis
| Ursprungspflanze* | Nukleinsäure
SEQ ID NO: | Protein
SEQ ID NO: |
| Arabidopsis
thaliana LBD-Protein | 1 | 2 |
| Os01g03890,
Oryza sativa | 59 | 11 |
| Os01g32770,
Oryza sativa | 60 | 12 |
| Os03g33090,
Oryza sativa | 61 | 13 |
| Os03g41330,
Oryza sativa | 62 | 14 |
| Os07g40000,
Oryza sativa | 63 | 15 |
| TC9404,
Nicotiana benthamiana | | 16 |
| TC227562,
Glycine max | | 17 |
| TC216138,
Glycine max | | 18 |
| TC147776,
Hordeum vulgare | | 19 |
| TC104758,
Sorghum bicolor | | 20 |
| TC18561,
Aquilegia sp. | | 21 |
| TC60668,
Vitis vinifera | | 22 |
| TC15459,
Lotus japonicus | | 23 |
| TC30552,
Gossypium hirsutum | | 24 |
| TC235711,
Triticum aestivum | 71 | 25 |
| TC133081,
Solanum tuberosum | | 26 |
| TC107091,
Medicago truncatula | | 27 |
| TC147808,
Hordeum vulgare | | 28 |
| TC55931,
Vitis vinifera | | 29 |
| TC162239,
Solanum tuberosum | | 30 |
| TC69225,
Pinus taeda | | 31 |
| TC67269,
Pinus taeda | | 32 |
| TC220806,
Glycine max | | 33 |
| TC270332,
Triticum aestivum | | 34 |
| TC18329,
Aquilegia sp. | | 35 |
| TC137193,
Solanum tuberosum | | 36 |
| TC133385,
Solarium tuberosum | | 37 |
| TC140088,
Solanum tuberosum | | 38 |
| TC14656,
Picea alba | | 39 |
| TC59178,
Pinus taeda | | 40 |
| TC67974,
Pinus taeda | | 41 |
| TC178827,
Lycopersicon esculentum | | 42 |
| Pt-III.589,
Populus tremuloides | | 43 |
| Pt-V.543,
Populus tremuloides | | 44 |
| Pt-XIV.94,
Populus tremuloides | | 45 |
| Pt-II105,
Populus tremuloides | | 46 |
| Pt-123.86,
Populus tremuloides | | 47 |
| Pt-X180,
Populus tremuloides | | 48 |
| Pt-XII.481,
Populus tremuloides | | 49 |
| DQ787782,
Caragana korshinskii | | 50 |
| AAP37970,
Brassica napus | | 51 |
| ABE82505,
Medicago truncatula | 72 | 52 |
| ABE78739,
Medicago truncatula | | 53 |
| Q9SN23,
Arabidopsis thaliana | 64 | 54 |
| Q9SZE8,
Arabidopsis thaliana | 65 | 55 |
| Q9ZW96,
Arabidopsis thaliana | 66 | 56 |
| Q9M886,
Arabidopsis thaliana | 67 | 57 |
| Q9CA30,
Arabidopsis thaliana | 68 | 58 |
| Ls_LBD,
Linum usitatissimum | 69 | 70 |
- *: GenBank oder SwissProt Datenbank Zugangsnummern
sind angegeben, wenn vorhanden, TC-Codes stammen von TIGR.
-
Beispiel 2: Alignment von LBD-Polypeptidsequenzen
-
Das
Alignment von Polypeptid-Sequenzen erfolgte mit dem AlignX Programm
von Vector NTI (Invitrogen), das auf dem bekannten Clustal W-Algorithmus
des progressiven Alignments beruht (Thompson et al. (1997)
Nucleic Acids Res 25: 4876–4882; Chenna et al. (2003).
Nucleic Acids Res 31: 3497–3500). Die Default-Werte
lauten: gap open penalty 10, gap extension penalty 0,1, und die
gewählte ”weight matrix” ist Blosum 62.
Um das Alignment weiter zu optimieren, wurden kleinere Veränderungen
von Hand durchgeführt. Die Sequenzkonservierung unter LBD-Polypeptiden
befand sich im Wesentlichen in der N-terminalen DUF260-Domäne
der Polypeptide, wobei die C-terminale Region gewöhnlich
hinsichtlich Sequenzlänge und Zusammensetzung variabler
ist. Das Alignment der LBD-Polypeptide ist in 2 gezeigt.
-
Mit
einem ”Neighbour joining”-Clustering-Algorithmus,
wie er von dem AlignX-Programm von Vektor NTI (Invitrogen) bereitgestellt
wird, wurde ein Stammbaum der LBD-Polypeptide (3)
konstruiert. Die Sequenzen der LBD-Proteine der Klasse I, die bei
der Konstruktion des Stammbaums verwendet wurden, sind öffentlich
zugänglich und sind mit ihren GenBank oder SwissProt-Zugangsnummern
gekennzeichnet.
-
Beispiel 3: Berechnung des Gesamtidentitätsprozentsatzes
zwischen Polypeptidsequenzen, die bei der Durchführung
der erfindungsgemäßen Verfahren geeignet sind
-
Die
Gesamtprozentsätze für Ähnlichkeit und
Identität zwischen Volllängenpolypeptidsequenzen,
die bei der Durchführung der erfindungsgemäßen
Verfahren geeignet sind, wurden unter Verwendung von einer der in
der Fachwelt verfügbaren Techniken, nämlich der
MatGAT (Matrix Global Alignment Tool) software (BMC Bioinformatics.
2003 4: 29. MatGAT: an application that generstes similarity/identity
matrices using Protein or DNA sequences. Campanella JJ, Bitincka
L, Smalley J; von Ledion Bitincka gehostete software) bestimmt.
Die MatGAT-Software erzeugt Ähnlichkeits-/Identitätsmatrizes
für DNA- oder Proteinsequenzen, ohne dass ein vorheriges
Alignment der Daten erforderlich ist. Das Programm führt
eine Reihe von paarweisen Alignments unter Verwendung des globalen
Alignment-Algorithmus von Myers und Miller durch (gap opening penalty
12, gap extension penalty 2), berechnet die Ähnlichkeit
und Identität unter Verwendung von z. B. Blosum 62 (für Polypeptide)
und platziert dann die Ergebnisse in einer Abstandsmatrix. Die Sequenzähnlichkeit
ist in der unteren Hälfte der Trennlinie und die Sequenzidentität
in der oberen Hälfte der diagonalen Trennlinie dargestellt.
-
Bei
den in dem Vergleich eingesetzten Parametern handelte es sich um:
| Scoring
matrix: | Blosum
62 |
| First
Gap: | 12 |
| Extending
gap: | 2 |
-
Die
Ergebnisse der Software-Analyse sind in Tabelle B für die
Gesamtähnlichkeit und -identität über die
volle Länge der Polypeptidsequenzen dargestellt. Der Identitätsprozentsatz
ist fett über der Diagonale und der Ähnlichkeitsprozentsatz
unter der Diagonale (normale Schrift) angegeben.
-
Der
Identitätsprozentsatz zwischen den LBD-Polypeptidsequenzen,
die bei der Durchführung der erfindungsgemäßen
Verfahren geeignet sind, kann nur 30,2% Aminosäureidentität
im Vergleich zu SEQ ID NO: 2 (veranschaulicht durch At-LBD37, Reihe
44) betragen. Der Identitätsprozentsatz ist wahrscheinlich
höher, wenn nur die Sequenzen der DUF206-Domäne
verglichen werden. Zur Identifikation der DUF260-Domäne (wie
in
1 beschrieben) in anderen LBD Proteinen, kann
das multiple Alignment von
2 verwendet
werden. Tabelle
A2: MatGAT-Ergebnis für globale Ähnlichkeit und
Identität über die volle Länge der LBD-Polypeptidsequenzen.










| | 47 | 48 | 49 |
| 1.
Os-Os01g03890 | 41,5 | 41,3 | 38,2 |
| 2.
Os-Os01g32770 | 41,5 | 44,8 | 39,0 |
| 3.
Os-Os03g33090 | 40,4 | 35,0 | 40,8 |
| 4.
Os-Os03g41330 | 38,2 | 33,7 | 36,0 |
| 5.
Os-Os07g40000 | 40,2 | 33,8 | 36,3 |
| 6.
Nb-TC9404 | 39,4 | 34,6 | 40,7 |
| 7.
Gm-TC227562 | 40,8 | 35,7 | 39,8 |
| 8.
Gm-TC216138 | 36,5 | 34,0 | 35,5 |
| 9.
Hv-TC147776 | 38,2 | 34,6 | 37,7 |
| 10.
Sb-TC104758 | 36,2 | 34,5 | 38,6 |
| 11.
Aq-TC18561 | 42,1 | 37,2 | 36,6 |
| 12.
Vv-TC60668 | 40,2 | 36,1 | 38,2 |
| 13.
Lj-TC15459 | 38,1 | 35,1 | 36,4 |
| 14.
Gh-TC30552 | 39,8 | 34,6 | 36,6 |
| 15.
Ta-TC235711 | 44,3 | 44,3 | 41,0 |
| 16.
St-TC133081 | 50,7 | 55,9 | 45,7 |
| 17.
Mt-TC107091 | 52,6 | 56,1 | 40,2 |
| 18.
Hv-TC147808 | 42,1 | 46,5 | 39,7 |
| 19.
Vv-TC55931 | 49,2 | 49,3 | 47,3 |
| 20.
St-TC162239 | 46,5 | 44,5 | 47,5 |
| 21.
Pta-TC69225 | 36,4 | 36,2 | 34,1 |
| 22.
Pta-TC67269 | 36,0 | 36,3 | 33,3 |
| 23.
Gm-TC220806 | 52,8 | 50,8 | 49,8 |
| 24.
Ta-TC270332 | 39,2 | 30,4 | 36,3 |
| 25.
Aq-TC18329 | 49,8 | 53,2 | 43,7 |
| 26.
St-TC137193 | 39,2 | 34,2 | 38,6 |
| 27.
St-TC133385 | 40,1 | 34,1 | 38,4 |
| 28.
St-TC140088 | 37,8 | 35,7 | 36,6 |
| 29.
Pa-TC14656 | 33,8 | 30,4 | 31,3 |
| 30.
Pta-TC59178 | 30,2 | 32,5 | 27,2 |
| 31.
Pta-TC67974 | 34,8 | 31,8 | 33,0 |
| 32.
Le-TC178827 | 39,0 | 34,6 | 38,5 |
| 33.
Pt-III.589 | 36,6 | 35,0 | 35,3 |
| 34.
Pt-V.543 | 38,2 | 35,4 | 39,2 |
| 35.
Pt-XIV.94 | 37,8 | 33,8 | 38,3 |
| 36.
Pt-II105 | 40,2 | 35,7 | 37,4 |
| 37.
Pt-123.86 | 53,8 | 59,1 | 43,6 |
| 38.
Pt-X180 | 46,8 | 41,5 | 49,6 |
| 39.
Pt-XII.481 | 38,5 | 37,6 | 36,4 |
| 40.
Ck-LOB-DQ787782 | 40,2 | 31,6 | 38,2 |
| 41.
Bn-AAP37970 | 55,5 | 83,5 | 42,1 |
| 42.
Mt-ABE82505 | 40,3 | 33,8 | 36,8 |
| 43.
Mt-ABE78739 | 53,3 | 51,6 | 43,8 |
| 44.
At-LBD37-Q9FN11 | 37,5 | 34,5 | 34,9 |
| 45.
At-LBD38-Q9SN23 | 40,5 | 33,5 | 34,8 |
| 46.
At-LBD39-Q9SZE8 | 38,3 | 35,0 | 37,1 |
| 47.
At-LBD40-Q9ZW96 | | 56,7 | 46,5 |
| 48.
At-LBD41-Q9M886 | 68,4 | | 42,6 |
| 49.
At-LBD42-Q9CA30 | 61,8 | 56,3 | |
-
Beispiel 4: Identifikation von Domänen
in Polypeptidsequenzen, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet sind
-
Bei
der Integrated Resource of Protein Families, Domains and Sites-(InterPro-)Datenbank
handelt es sich um eine integrierte Plattform für die häufig
verwendeten Signatur-Datenbanken für Suchen auf Text- und Sequenzbasis.
Die InterPro-Datenbank kombiniert diese Datenbanken, in denen unterschiedliche
Methodiken und verschiedene Mengen an biologischer Information über
gut charakterisierte Proteine verwendet werden, um Proteinsignaturen
abzuleiten. Zu den angeschlossenen Datenbanken zählen SWISS-PROT,
PROSITS, TrEMBL, PRINTS, ProDom und Pfam, Smart und TIGRFAMs. Pfam
ist eine große Sammlung multipler Sequenzalignments und
versteckter Markov-Modelle, die viele häufige Proteindomänen
und Familien abdeckt. Pfam wird auf dem Server des Sanger Institut
im Vereinigten Königreich gehostet. InterPro wird vom European Bioinformatics
Institute im Vereinigten Königreich gehostet.
-
Die
Ergebnisse des Interpro-Scans der Polypeptidsequenz gemäß SEQ
ID NO: 2 sind in Tabelle A3 angegeben. Tabelle A3: InterPro-Scan-Ergebnisse (Haupt-Zugangsnummern)
der Polypeptidsequenz gemäß SEQ ID NO: 2.
| Datenbank | Zugangsnummer | Zugangsname | Aminosäurekoordinaten auf
SEQ ID NO: 2 |
| PFAM | PF03195 | DUF260 | 2–107 |
| PROFILE | PS50891 | LOB | 1–107 |
-
Beispiel 5: Topologie-Vorhersage für
Polypeptidsequenzen, die sich für die Durchführung
der erfindungsgemäßen Verfahren eignen
-
TargetP
1.1 sagt die subzelluläre Lokalisierung eukaryotischer
Proteine voraus. Die Zuordnung der Lokalisierung beruht auf der
vorhergesagten Anwesenheit von einer der folgenden N-terminalen
Präsequenzen: Chloroplasten-Transitpeptid (cTP), mitochondriales
Targeting-Peptid (mTP) oder Signalpeptid (SP) für den sekretorischen
Weg. Die Bewertungen (Scores), auf denen die endgültige
Vorhersage beruht, sind keine echten Wahrscheinlichkeiten, und sie
tragen auch nicht notwendigerweise zu einer bei. Die Lokalisierung
mit dem höchsten Score ist jedoch gemäß TargetP
am wahrscheinlichsten und das Verhältnis zwischen den Scores
(die Verlässlichkeitsklasse) kann ein Anzeichen dafür
sein, wie sicher die Vorhersage ist. Die Verlässlichkeitsklasse (Reliability
Class, RC) reicht von 1 bis 5, wobei 1 die stärkste Vorhersage
angibt. TargetP wird auf dem Server der Technischen Universität
Dänemark gehalten.
-
Für
die Sequenzen, die den Vorhersagen zufolge eine N-terminale Präsequenz
enthalten, kann auch eine potentielle Spaltstelle vorhergesagt werden.
-
Es
wurde eine Anzahl Parameter ausgewählt, wie Organismusgruppe
(Nicht-Pflanze oder Pflanze), Sätze von Ausschlussgrenzen
(keine, vordefinierte Sätze von Ausschlussgrenzen oder
nutzerspezifische Sätze von Ausschlussgrenzen) und die
Berechnung der Vorhersage von Spaltstellen (Ja oder Nein).
-
Die
Ergebnisse der TargetP 1.1-Analyse der Polypeptidsequenz, wie sie
durch SEQ ID NO: 2 veranschaulicht wird, sind in Tabelle A4 dargestellt.
Es wurde die Organismusgruppe ”Pflanze” gewählt,
keine Ausschlussgrenzen definiert und die vorhergesagte Länge
des Transitpeptids abgefragt. Tabelle A4: TargetP 1.1-Analyse der Polypeptidsequenz
gemäß SEQ ID NO: 2
| Länge
(AA) | 250 |
| Chloroplasten-Transitpeptid | 0,026 |
| Mitochondriales
Transitpeptid | 0,401 |
| Signalpeptid
des sekretorischen Wegs | 0,019 |
| Anderes
subzelluläres Ziel | 0,573 |
| Vorhergesagte
Lokalisierung | / |
| Verlässlichkeitsklasse | 5 |
| Vorhergesagte
Länge des Transitpeptids | / |
-
Die
subzelluläre Lokalisierung der Polypeptidsequenz gemäß SEQ
ID NO: 2 kann das Cytoplasma oder der Kern sein, es wird kein Transitpeptid
vorhergesagt. SubLoc (Hua & Sun, Bioinformatics 17, 721–728, 2001)
sagt eine Kernlokalisierung voraus (Verlässlichkeitsindex:
2, Genauigkeit: 74%); diese Voraussage stimmt mit den Daten von Liu
et al (2005) überein.
-
Zur
Durchführung solcher Analysen können viele andere
Algorithmen verwendet werden, wie u. a.:
- • ChloroP
1.1, das auf dem Server der Technischen Univerität Dänemark
gehostet wird;
- • Protein Prowler Subcellular Localisation Predictor
Version 1.2, das auf dem Server des Instituts für Molekulare
Biowissenschaft, Universität Queensland, Brisbane, Australien,
gehostet wird;
- • PENCE Proteome Analyst PA-GOSUB 2.5, das auf dem
Server der Universität von Alberts, Edmonton, Alberts,
Kanada, gehostet wird;
- • TMHMM, das auf dem Server der Technischen Universität
Dänemark gehostet wird
-
Beispiel 6: Klonierung der LBD-Nukleinsäuresequenz,
die bei den erfindungsgemäßen Verfahren verwendet wird
-
Die
Nukleinsäuresequenz, die bei den erfindungsgemäßen
Verfahren werden wurde, wurde mittels PCR amplifiziert, wobei als
Matrize eine maßgeschneiderte cDNA-Bank aus Arabidopsis
thaliana-Keimlingen (in pCMV Sport 6.0; Invitrogen, Paisley, UK)
verwendet wurde. Die PCR wurde unter Verwendung von Hifi Taq DNA-Polymerase
unter Standardbedingungen mit 200 ng Matrize in 50 μl PCR-Mix
durchgeführt. Die verwendeten Primer waren prm009067 (SEQ
ID NO: 3; sense, Startcodon fettgedruckt):
5'-ggggacaagtttgtacaaaaaagcaggcttaaacaatgagctgcaatggttgc-3'
und prm009068 (SEQ ID NO: 4; revers, komplementär):
5'-ggggaccactttgtacaagaaagctgggtactaactctgagaaaaccgcc-3',
die
die AttB-Stellen für die Gateway-Rekombination enthalten.
Das amplifizierte PCR-Fragment wurde ebenfalls unter Verwendung
von Standardverfahren aufgereinigt. Anschließend wurde
der erste Schritt des Gateway-Verfahrens, die BP-Reaktion, durchgeführt,
bei der das PCR-Fragment in vivo mit dem Plasmid pDONR201 unter
Bildung eines – gemäß der Gateway-Teminologie – ”entry
clone”, pLBD, rekombinierte. Das Plasmid pDONR201 wurde
von Invitrogen als Bestandteil der Gateway®-Technologie
erworben.
-
Der ”entry
clone”, der die SEQ ID NO: 1 enthielt, wurde anschließend
in einer LR-Reaktion mit einem Zielvektor, der für die
Transformation von Oryza sativa verwendet wird, verwendet. Dieser
Vektor enthielt als funktionelle Elemente innerhalb der T-DNA-Grenzen
Folgendes: Einen pflanzlichen Selektionsmarker; eine screenbare
Markerexpressionskassette und eine Gateway-Kassette, die für
die LR-in-vivo-Rekombination mit der interessierenden Nukleinsäuresequenz,
die bereits in den ”entry clone” kloniert wurde,
vorgesehen ist. Ein GOS2-Promotor aus Reis (SEQ ID NO: 10) für
die spezifische Expression in Wurzeln befand sich stromaufwärts
von dieser Gateway-Kassette.
-
Nach
dem LR-Rekombinationsschritt wurde der erhaltene Expressionsvektor
pGOS2::LBD (4) nach im Stand der Technik
bekannten Verfahren in den Agrobacterium-Stamm LBA4044 transformiert.
-
Beispiel 7: Pflanzentransformation
-
Reistransformation
-
Agrobacterium,
das den Expressionsvektor enthielt, wurde zur Transformation von
Oryza sativa-Pflanzen verwendet. Reife trockene Samen der japonica-Reissorte
Nipponbare wurden entspelzt. Die Sterilisation erfolgte durch einminütiges
Inkubieren in 70%igem Ethanol und anschließend 30 Minuten
in 0,2% HgCl2, wonach 6 Mal je 15 Minuten
mit sterilem destilliertem Wasser gewaschen wurde. Anschließend
wurden die sterilen Samen auf einem Medium, das 2,4-D enthielt,
(Kallusinduktionsmedium) zur Keimung gebracht. Nach vierwöchiger
Inkubation im Dunkeln wurden embryogene, vom Scutellum stammende
Kalli herauspräpariert und mittels Subkultur auf dem gleichen
Medium vermehrt. Nach zwei Wochen wurden die Kalli mittels Subkultur auf
dem gleichen Medium für weitere 2 Wochen vervielfacht oder
vermehrt. Stückchen der embryogenen Kalli wurden 3 Tage
vor der Cokultivierung auf frischem Medium subkultiviert (um die
Zellteilungsaktivität zu fördern).
-
Für
die Cokultivierung verwendete man den Agrobacterium–Stamm
LBA4404, der den Expressionsvektor enthielt. Agrobacterium wurde
auf AB-Medium mit den entsprechenden Antibiotika überimpft
und für 3 Tage bei 28°C kultiviert. Anschließend
wurden die Bakterien gewonnen und bis zum Erreichen einer Dichte (OD600) von ungefähr 1 in flüssigem
Cokultivierungsmedium suspendiert. Anschließend wurde die
Suspension in eine Petrischale überführt und die
Kalli wurden 15 Minuten in der Suspension eingetaucht. Dann wurden
die Kallusgewebe auf einem Papierfilter trocken getupft, auf ein
verfestigtes Cokultivierungsmedium umgesetzt und für 3
Tage im Dunkeln bei 25°C inkubiert. Die cokultivierten
Kalli wurden 4 Wochen im Dunkeln bei 28°C in Gegenwart
eines Selektionsmittels auf 2,4-D-haltigem Medium herangezogen.
Während dieses Zeitraums entwickelten sich rasch wachsende
resistente Kallusinseln. Nach dem Umsetzen dieses Materials auf
ein Regenerationsmedium und Inkubation im Hellen wurde das embryogene
Potential freigesetzt und in den nächsten 4 bis 5 Wochen
entwickelten sich Sprosse. Die Sprosse wurden aus den Kalli herauspräpariert
und 2 bis 3 Wochen lang auf auxinhaltigem Medium inkubiert, von
dem sie in Erde umgesetzt wurden. Abgehärtete Sprosse wurden
im Gewächshaus unter hoher Feuchtigkeit und im Kurztag
herangezogen.
-
Pro
Konstrukt wurden ungefähr 35 unabhängige T0-Reistransformanten
erzeugt. Die Primärtransformanten wurden von einer Gewebekulturkammer
in ein Gewächshaus umgesetzt. Nach einer quantitativen PCR-Analyse
zur Überprüfung der Kopienzahl des T-DNA-Inserts
wurden für die Ernte von T1-Samen nur transgene Ein-Kopien-Pflanzen
zurückbehalten, die eine Toleranz gegen das Selektionsmittel
zeigten. Die Samen wurden dann 3 bis 5 Monate nach der Transplantation
geerntet. Das Verfahren ergab Einzel-Locus-Transformanten mit einer
Rate von über 50% (Aldemita und Hodges 1996, Chan
et al., 1993, Hiei et al., 1994).
-
Maistransformation
-
Die
Transformation von Mais (Zea mays) erfolgt durch eine Modifikation
des von Ishida et al. (1996) Nature Biotech 14 (6): 745–50 beschriebenen
Verfahrens. Die Transformation in Mais ist genotypabhängig
und nur spezifische Genotypen sind für eine Transformation
und Regeneration zugänglich. Die Inzuchtlinie A188 (University
of Minnesota) oder Hybride mit A188 als einem Elter sind gute Quellen
für Donormaterial für die Transformation, aber
es können auch andere Genotypen erfolgreich verwendet werden.
Die Ähren werden von Maispflanzen etwa 11 Tage nach der
Bestäubung (days after pollination, DAP) geerntet, wenn
die Länge des unreifen Embryos etwa 1 bis 1,2 mm beträgt.
Unreife Embryonen werden mit Agrobacterium tumefaciens cokultiviert,
das den Expressionsvektor enthält, und transgene Pflanzen
werden mittels Organogenese gewonnen. Herauspräparierte
Embryonen werden auf Kallusinduktionsmedium, dann auf Mais-Regenerationsmedium,
das das Selektionsmittel (zum Beispiel Imidazolinon, aber es können
verschiedene Selektionsmarker verwendet werden) enthält,
herangezogen. Die Petrischalen werden im Licht bei 25°C
für 2–3 Wochen oder, bis sich Sprosse entwickeln,
inkubiert. Die grünen Sprosse werden von jedem Embryo auf
Mais-Wurzelmedium überführt und bei 25°C
für 2–3 Wochen inkubiert, bis sich Wurzeln entwickeln.
Die bewurzelten Sprosse werden dann auf Boden im Gewächshaus
umgesetzt. T1-Samen werden von Pflanzen gewonnen, die eine Toleranz
gegen das Selektionsmittel aufweisen und eine Einzelkopie des T-DNA-Inserts
enthalten.
-
Weizentransformation
-
Die
Transformation von Weizen erfolgt mit dem von Ishida et
al. (1996) Nature Biotech 14 (6): 745–50 beschriebenen
Verfahren. Die Sorte Bobwhite (von CIMMYT, Mexico, erhältlich)
wird üblicherweise zur Transformation verwendet. Unreife
Embryonen werden mit Agrobacterium tumefaciens cokultiviert, das
den Expressionsvektor enthält, und transgene Pflanzen werden
mittels Organogenese gewonnen. Nach der Inkubation mit Agrobacterium
werden die Embryonen in vitro auf Kallusinduktionsmedium, dann auf
Regenerationsmedium, das das Selektionsmittel (zum Beispiel Imidazolinon,
aber es können verschiedene Selektionsmarker verwendet
werden) enthält, herangezogen. Die Petrischalen werden
im Licht bei 25°C für 2–3 Wochen oder,
bis sich Sprosse entwickeln, inkubiert. Die grünen Sprosse
werden von jedem Embryo auf Wurzelmedium überführt
und bei 25°C für 2–3 Wochen inkubiert,
bis sich Wurzeln entwickeln. Die bewurzelten Sprosse werden dann
auf Boden im Gewächshaus umgesetzt. T1-Samen werden von
Pflanzen gewonnen, die eine Toleranz gegen das Selektionsmittel
aufweisen und eine Einzelkopie des T-DNA-Inserts enthalten.
-
Sojabohnentransformation
-
Sojabohne
wird gemäß einer Modifikation des Verfahrens,
das im Texas A&M-Patent
US 5,164,310 beschrieben
ist, transformiert. Es sind mehrere im Handel erhältliche
Sojanbohnen-Sorten für die Transformation durch dieses
Verfahren zugänglich. Die (von der Illinois Seed Foundation
erhältliche) Sorte Jack wird üblicherweise für
die Transformation verwendet. Sojabohnensamen wird für
die In-vitro-Aussaat sterilisiert. Das Hypokotyl, die Keimwurzel
und ein Keimblatt werden von jungen, 7 Tage alten Keimlingen präpariert.
Das Epikotyl und das verbleibende Keimblatt werden weiter gezüchtet,
damit sich Achselknoten entwickeln. Diese Achselknoten werden herauspräpariert
und mit Agrobacterium tumefaciens, das den Expressionsvektor enthält,
inkubiert. Nach der Cokultivierungsbehandlung werden die Explantate
gewaschen und auf Selektionsmedien umgesetzt. Regenerierte Sprosse
werden herauspräpariert und auf Sprossverlängerungsmedium
umgesetzt. Sprosse mit nicht mehr als 1 cm Länge werden
auf Wurzelmedium umgesetzt, bis sich Wurzeln entwickeln. Die bewurzelten
Sprosse werden auf Boden im Gewächshaus umgesetzt. T1-Samen
werden von Pflanzen gewonnen, die eine Toleranz gegen das Selektionsmittel
aufweisen und eine Einzelkopie des T-DNA-Inserts enthalten.
-
Raps-/Canolatransformation
-
Keimblattpetiolen
und -hypokotyle von 5–6 Tage alten jungen Keimlingen werden
als Explantate für die Gewebekultur verwendet und gemäß Babic
et al. (1998, Plant Cell Rep 17: 183–188) transformiert.
Die im Handel erhältliche Sorte Westar (Agriculture Canada)
ist die für die Transformation eingesetzte Standardsorte, aber
es können auch andere Sorten verwendet werden. Canolasamen
werden für die in-vitro-Aussaat oberflächensterilisiert.
Die Kotyledonenpetiolenexplantate mit dem daran befindlichen Keimblatt
werden aus den In-vitro-Keimlingen herauspräpariert und
mit Agrobacterium (das den Expressionsvektor enthält) beimpft,
indem das abgeschnittene Ende des Petiolenexplantats in die Bakterienlösung
getaucht wird. Die Explantate werden dann für 2 Tage auf
MSBAP-3-Medium, das 3 mg/l BAP, 3% Saccharose, 0,7% Phytagar enthält,
bei 23°C, 16 Std. Licht gezüchtet. Nach zweitägiger
Cokultur mit Agrobacterium werden die Petiolenexplantate für 7
Tage auf MSBAP-3-Medium umgesetzt, das 3 mg/l BAP, Cefotaxim, Carbenicillin
oder Timentin (300 mg/l) enthält, und dann auf MSBAP-3-Medium
mit Cefotaxim, Carbenicillin oder Timentin und Selektionsmittel
bis zur Sprossregeneration gezüchtet. Wenn die Sprosse
eine Länge von 5–10 mm haben, werden sie abgeschnitten
und auf Sprossverlängerungsmedium (MSBAP-0,5 mit 0,5 mg/l
BAP) umgesetzt. Sprosse mit einer Länge von etwa 2 cm werden
für die Wurzelinduktion auf Wurzelmedium (MS0) umgesetzt.
Die bewurzelten Sprosse werden auf Boden im Gewächshaus
umgesetzt. T1-Samen werden von Pflanzen gewonnen, die eine Toleranz gegen
das Selektionsmittel aufweisen und eine Einzelkopie des T-DNA-Inserts
enthalten.
-
Alfalfatransformation
-
Ein
regenerierender Alfalfa-(Medicago sativa-)Klon wird unter Verwendung
des Verfahrens von (McKersie et al., 1999 Plant Physiol
119: 839–847) transformiert. Die Regeneration
und Transformation von Alfalfa ist genotypabhängig und
daher wird eine regenerierende Pflanze benötigt. Verfahren
zur Gewinnung regenerierender Pflanzen sind beschrieben. Diese können
zum Beispiel von der Sorte Rangelander (Agriculture Canada) oder
jeder anderen im Handel erhältlichen Alfalfa-Sorte wie
von Brown DCW und A Atanassov (1985. Plant Cell Tissue Organ
Culture 4: 111–112) beschrieben erhalten werden.
Alternativ hat man die Sorte RA3 (University of Wisconsin) für
die Verwendung bei der Gewebekultur ausgewählt (Walker
et al., 1978 Am J Bot 65: 654–659). Petiolenexplantate
werden mit einer Übernachtkultur von Agrobacterium tumefaciens C58C1
pMP90 (McKersie et al., 1999 Plant Physiol 119: 839–847)
oder LBA4404, die den Expressionsvektor enthalten, cokultiviert.
Die Explantate werden für 3 Tage im Dunkeln auf SH-Induktionsmedium,
das 288 mg/l Pro, 53 mg/l Thioprolin, 4,35 g/l K2SO4 und 100 μm Acetosyringinon enthält,
gezüchtet. Die Explantate werden in Murashige-Skoog-Medium
(Murashige and Skoog, 1962) der halben Stärke
gewaschen und auf demselben SH-Induktionsmedium ohne Acetosyringinon,
aber mit einem geeigneten Selektionsmittel und einem geeigneten
Antibiotikum für die Hemmung des Agrobacterium-Wachstums
herangezogen. Nach mehreren Wochen werden somatische Embryonen auf
BOi2Y-Entwicklungsmedium umgesetzt, das keine Wachstumsregulatoren,
keine Antibiotika und 50 g/l Saccharose enthält. Die somatischen
Embryonen lässt man anschließend auf Murashige-Skoog-Medium
der halben Stärke keimen. Bewurzelte Keimlinge werden in
Töpfe umgesetzt und im Gewächshaus herangezogen.
T1-Samen werden von Pflanzen gewonnen, die eine Toleranz gegen das
Selektionsmittel aufweisen und eine Einzelkopie des T-DNA-Inserts
enthalten.
-
Baumwolltransformation
-
Die
Transformation von Baumwolle (Gossypium hirsutum L.) erfolgt unter
Verwendung von Agrobacterium tumefaciens an Hypokotylenexplantaten.
Die handelsüblichen Sorten, wie Coker 130 oder Coker 312 (SeedCo,
Lubbock, TX) sind für die Transformation verwendete Standardsorten,
aber es können auch andere Sorten verwendet werden. Die
Samen werden oberflächensterilisiert und im Dunkeln gekeimt.
Hypokotylenexplantate von den gekeimten Keimlingen werden auf eine
Länge von etwa 1–1,5 Zentimeter geschnitten. Das Hypokotylenexplantat
wird in dem Agrobacterium tumefaciens-Impfgut, das den Expressionsvektor
enthält, für 5 Minuten eingetaucht und dann für
etwa 48 Stunden auf MS + 1,8 mg/l KNO3 +
2% Glucose bei 24°C im Dunkeln cokultiviert. Die Explantate
werden in das gleiche Medium, das geeignete bakterielle und pflanzliche
Selektionsmarker enthält (und mehrmals erneuert wird) umgesetzt,
bis embryogene Kalli beobachtet werden. Die Kalli werden dann getrennt
und subkultiviert, bis somatische Embryonen auftreten. Pflänzchen,
die von den somatischen Embryonen stammen, lässt man auf
Wurzelmedium reifen, bis sich Wurzeln entwickeln. Die bewurzelten
Sprosse werden auf Blumenerde im Gewächshaus umgesetzt.
T1-Samen werden von Pflanzen gewonnen, die eine Toleranz gegen das
Selektionsmittel aufweisen und eine Einzelkopie des T-DNA-Inserts
enthalten.
-
Beispiel 8: Vorgehen bei der phänotypischen
Auswertung
-
8.1 Aufbau der Auswertung
-
Es
wurden ungefähr 35 unabhängige T0-Reistransformanten
erzeugt. Die Primärtransformanten wurden für das
Heranziehen und Ernten von T1-Samen von einer Gewebekulturkammer
in ein Gewächshaus umgesetzt. Es wurden sechs Ereignisse
zurückbehalten, von denen die T1-Nachkommenschaft für
Vorliegen/Abwesenheit des Transgens im Verhältnis 3:1 aufspaltete.
Für jedes dieser Ereignisse wurden ungefähr 10 T1-Keimpflanzen,
die das Transgen enthielten (Hetero- und Homozygote), und ungefähr
10 T1-Keimpflanzen, denen das Transgen fehlte (Nullizygoten), durch
Beobachten der sichtbaren Markerexpression selektiert. Die transgenen
Pflanzen und die entsprechenden Nullizygoten wurden nebeneinander
in zufälliger Anordnung herangezogen. Die Gewächshausbedingungen
waren Kurztage (12 Stunden Licht), 28°C im Hellen und 22°C
im Dunkeln und eine relative Feuchtigkeit von 70%. Die unter Nichtstressbedingungen
gezogenen Pflanzen werden in regelmäßigen Abständen
mit Wasser versorgt, so dass gewährleistet ist, dass Wasser
und Nährstoffe nicht limitierend sind, damit die Bedürfnisse
der Pflanze hinsichtlich der Vollendung von Wachstum und Entwicklung
erfüllt werden.
-
Screening auf Effizienz der Stickstoffnutzung
-
Reispflanzen
aus T2-Samen wurden in Blumenerde und mit Ausnahme der Nährlösung
unter normalen Bedingungen gezüchtet. Die Töpfe
wurden von der Transplantierung bis zur Reifung mit einer spezifischen Nährlösung
gewässert, die einen reduzierten Stickstoff-(N-)Gehalt,
und zwar gewöhnlich 7 bis 8 Mal niedriger, aufwies. Die übrige
Anzucht (Pflanzenreifung, Samenernte) entsprach ansonsten derjenigen
von Pflanzen, die nicht unter abiotischem Stress gezüchtet
werden. Wachstums- und Ertragsparameter werden wie für
das Wachstum unter normalen Bedingungen beschrieben ausgewertet.
-
8.2 Statistische Analyse: F-Test
-
Als
statistisches Modell für die Gesamtauswertung der phänotypischen
Eigenschaften der Pflanze wurde eine zweifaktorielle ANOVA (Varianzanalyse)
verwendet. Mit allen Parametern, die bei allen Pflanzen von allen
Ereignissen, die mit dem erfindungsgemäßen Gen
transformiert worden waren, bestimmt wurden, wurde ein F-Test durchgeführt.
Der F-Test erfolgte zur Überprüfung im Hinblick
auf einen Effekt des Gens über alle Transformationsereignisse
und zur Feststellung eines Gesamteffekts des Gens, was auch als
globaler Geneffekt bezeichnet wird. Die Signifikanzschwelle für
einen echten globalen Geneffekt wurde bei dem F-Test auf dem 5%-Wahrscheinlichkeitsniveau
festgelegt. Ein signifikanter F-Test-Wert weist auf einen Geneffekt
hin, was bedeutet, dass mehr als nur das einfache Vorhandensein
oder die Lage des Gens die Unterschiede im Phänotyp verursacht.
-
38.3 Messparameter
-
Biomasseparameter-Messung
-
Vom
Sästadium bis zum Reifestadium wurden die Pflanzen mehrmals
in eine Digital-Imaging-Kammer gestellt. Zu jedem Zeitpunkt wurden
von jeder Pflanze aus mindestens 6 verschiedenen Winkeln Digitalbilder (2048×1536
Pixel, 16 Millionen Farben) aufgenommen. Die oberirdische Pflanzenfläche
(oder Blattbiomasse) wurde dadurch bestimmt, dass man die Gesamtanzahl
der Pixel auf den Digitalbildern von oberirdischen Pflanzenteilen
zählte, die sich vom Hintergrund unterschieden. Dieser
Wert wurde über die zu demselben Zeitpunkt aus unterschiedlichen
Winkeln aufgenommenen Bilder gemittelt und mittels Kalibrierung
in einen physikalischen Oberflächenwert, ausgedrückt
in Quadratmillimeter, umgewandelt. Experimente zeigen, dass die
auf diese Weise gemessene oberirdische Pflanzenfläche mit
der Biomasse der oberirdischen Pflanzenteile korreliert. Die oberirdische
Fläche ist diejenige Fläche, die zu dem Zeitpunkt
gemessen wurde, an dem die Pflanzen ihre maximale Blattbiomasse
erreicht hatten. Die Jungpflanzenvitalität wurde bestimmt,
indem die Gesamtzahl der Pixel von oberirdischen Pflanzenteilen,
die sich vom Hintergrund unterschieden, gezählt wurde.
Dieser Wert wurde über die zu demselben Zeitpunkt aus unterschiedlichen
Winkeln aufgenommenen Bilder gemittelt und mittels Kalibrierung
in einen physikalischen Oberflächenwert, ausgedrückt
in Quadratmillimeter, umgewandelt. Die nachstehend beschriebenen
Ergebnisse gelten für Pflanzen drei Wochen nach der Keimung.
-
Samenparameter-Messungen
-
Die
reifen Primärrispen wurden geerntet, gezählt,
eingetütet, mit einem Strichcode versehen und dann drei
Tage bei 37°C in einem Ofen getrocknet. Dann wurden die
Rispen gedroschen und alle Samen wurden gesammelt und gezählt.
Die gefüllten Spelzen wurden von den leeren Spelzen mittels
einer Luftblaseeinrichtung getrennt. Die leeren Spelzen wurden verworfen
und die restliche Fraktion wurde nochmals gezählt. Die gefüllten
Spelzen wurden auf einer Analysewaage gewogen. Die Anzahl gefüllter
Samen wurde dadurch bestimmt, dass man die Anzahl der gefüllten
Spelzen, die nach dem Abtrennungsschritt verblieben, auszählte. Der
Gesamtsamenertrag wurde dadurch gemessen, dass man alle von einer
Pflanze geernteten gefüllten Spelzen wog. Die Gesamtsamenzahl
pro Pflanze wurde dadurch gemessen, dass man die von einer Pflanze geerntete
Anzahl Spelzen auszählte. Das Tausendkorngewicht (TKG)
wird aus der Anzahl der gezählten gefüllten Samen
und ihrem Gesamtgewicht extrapoliert. Der Harvest Index (HI) ist
bei der vorliegenden Erfindung als das Verhältnis zwischen
dem Gesamtsamenertrag pro Pflanze und der oberirdischen Fläche
(mm2), multipliziert mit einem Faktor 106, definiert. Die Samenfüllungsrate
ist bei der vorliegenden Erfindung als das Verhältnis (ausgedrückt
als %) der Anzahl gefüllter Samen zu der Gesamtzahl Samen
(oder Blüten) definiert.
-
Beispiel 9: Ergebnisse der phänotypischen
Untersuchung der transgenen Pflanzen, die die SEQ ID NO: 1 umfassen
-
Die
Untersuchung transgener Reispflanzen, die eine LBD-Nukleinsäuresequenz
exprimierten, unter Nicht-Stress-Bedingungen zeigte, dass es zu
einem Anstieg von mehr als 5% bei der oberirdischen Biomasse (AreaMax),
dem Gesamtsamenertrag, der Anzahl gefüllter Samen, der
Füllungsrate, beim Harvest Index und von mehr als 3% beim
Tausendkorngewicht kam.
-
Die
Untersuchung transgener Reispflanzen, die eine LBD-Nukleinsäuresequenz
exprimierten, im Screening auf Effizienz der Stickstoffnutzung ergab
einen Anstieg von mehr als 5% für die Vitalität
beim Aufgehen (Jungpflanzenvitalität).
-
Beispiel 10: Ergebnisse der phänotypischen
Untersuchung der transgenen Pflanzen, die die SEQ ID NO: 71 umfassen
-
Die
in der SEQ ID NO: 71 enthaltene kodierende Region wurde unter der
Kontrolle des GOS2-Promotors aus Reis in einen Reis-Transformationsvektor
kloniert, wie in Beispiel 6 beschrieben. Transgene Reispflanzen,
die die kodierende Region der SEQ ID NO: 71 enthielten, wurden gemäß den
Verfahren von Beispiel 7 hergestellt. Die Pflanzen wurden nach dem
in Beispiel 8 beschriebenen Verfahren untersucht. SEQ ID NO: 71
kodiert das durch SEQ ID NO: 25 dargestellte LBD-Protein.
-
Die
Untersuchung transgener Reispflanzen, die die SEQ ID NO: 71 exprimierten,
unter Nicht-Stress-Bedingungen zeigte, dass es zu einem Anstieg
von mehr als 5% bei der oberirdischen Biomasse (AreaMax), der Anzahl
an Blüten pro Rispe, der Gesamtanzahl an Samen pro Pflanze
und von mehr als 3% beim Tausendkorngewicht kam.
-
Beispiel 11: Ergebnisse der phänotypischen
Untersuchung der transgenen Pflanzen, die die SEQ ID NO: 72 umfassen
-
Die
in der SEQ ID NO: 72 enthaltene kodierende Region wurde unter der
Kontrolle des GOS2-Promotors aus Reis in einen Reis-Transformationsvektor
kloniert, wie in Beispiel 6 beschrieben. Transgene Reispflanzen,
die die kodierende Region der SEQ ID NO: 72 enthielten, wurden gemäß den
Verfahren von Beispiel 7 hergestellt. Die Pflanzen wurden nach dem
in Beispiel 8 beschriebenen Verfahren untersucht. SEQ ID NO: 72
kodiert das durch SEQ ID NO: 52 dargestellte LBD-Protein.
-
Die
Untersuchung transgener Reispflanzen, die die SEQ ID NO: 72 exprimierten,
unter Nicht-Stress-Bedingungen zeigte, dass es zu einem Anstieg
von mehr als 5% bei der oberirdischen Biomasse (AreaMax), dem Samengewicht
pro Pflanze, der Anzahl gefüllter Samen, der Anzahl an
Blüten pro Rispe, der Gesamtanzahl an Samen pro Pflanze,
beim Harvest Index und von mehr als 3% beim Tausendkorngewicht kam.
-
Die
Untersuchung transgener Reispflanzen, die die SEQ ID NO: 72 exprimierten,
im Screening auf Effizienz der Stickstoffnutzung ergab einen Anstieg
von mehr als 5% für die oberirdische Biomasse (AreaMax).
-
II. JMJC-(JUMONJI-C-)Polypeptid
-
Beispiel 12: Identifikation von Sequenzen,
die mit der in den erfindungsgemäßen Verfahren
verwendeten JMJC-Nukleinsäuresequenz verwandt sind
-
Sequenzen
(Volllängen-cDNA, ESTs oder genomische), die mit der in
den erfindungsgemäßen Verfahren verwendeten Nukleinsäuresequenz
verwandt sind, wurden unter den Sequenzen in der Entrez Nucleotides Datenbank
am National Center for Biotechnology Information (NCBI) unter Verwendung
von Datenbanksequenzabfragewerkzeugen, wie dem Basic Local Alignment
Tool (BLAST) (Altschul et al. (1990), J. Mol. Biol. 215:
403–410; und Altschul et al., (1997),
Nucleic Acids Res. 25: 3389–3402), identifiziert.
Das Programm wird zum Auffinden von Regionen mit lokaler Ähnlichkeit
zwischen Sequenzen verwendet, und zwar durch Vergleichen von Nukleinsäure-
oder Polypeptidsequenzen mit Sequenz-Datenbanken und durch Berechnen
der statistischen Signifikanz der Übereinstimmungen. Das
Polypeptid, das durch die bei der vorliegenden Erfindung verwendete
Nukleinsäure kodiert wird, wurde zum Beispiel für
den TBLASTN-Algorithmus verwendet, und zwar unter Ausschaltung der
Default-Einstellungen und des Filters für die Nichtmiteinbeziehung
von Sequenzen mit niedriger Komplexität. Der Output der
Analyse wurde mittels paarweisem Vergleich betrachtet und anhand
der Wahrscheinlichkeits-Wertung (E-Wert) in eine Reihenfolge gebracht,
wobei die Wertung die Wahrscheinlichkeit widerspiegelt, dass ein
bestimmtes Alignment zufällig vorkommt (je niedriger der
E-Wert, desto signifikanter der Hit). Zusätzlich zu den
E-Werten wurden die Vergleiche auch anhand der Prozent Identität
gewertet. Prozent Identität bezieht sich auf die Anzahl
identischer Nukleotide (oder Aminosäuren) zwischen den zwei
verglichenen Aminosäure-(oder Polypeptid)-Sequenzen über
eine bestimmte Länge.
-
Die
Tabelle B1 stellt eine Liste von Nukleinsäuresequenzen
bereit, die mit der JMJC-Nukleinsäuresequenz, die in den
erfindungsgemäßen Verfahren verwendet wird, verwandt
sind. Polypeptide, an deren Zugang ein Punkt und eine Stelle angehängt
ist, stellen Spleißvarianten dar. Tabelle B1: Beispiele für JMJC-Polypeptide:
| Ursprungspflanze | Genbank-Zugangs(oder Locus-)nummer | Nukleinsäure
SEQ ID NO: | Protein
SEQ ID NO: |
| Arabidopsis
thaliana | AT3G20810 | 73 | 74 |
| Arabidopsis
thaliana | AT3G20810 | 83 | 84 |
| Arabidopsis
thaliana | AT5G19840 | 85 | 86 |
| Arabidopsis
thaliana | AT3G45880 | 87 | 88 |
| Medicago
truncatula | ABE92082 | 89 | 90 |
| Brachypodium
sylvaticum | CAJ26373 | 91 | 92 |
| Oryza
sativa | Os09g0483600 | 93 | 94 |
| Arabidopsis
thaliana | AT1G08620 | 95 | 96 |
| Arabidopsis
thaliana | AT1G09060 | 97 | 98 |
| Arabidopsis
thaliana | AT1G09060 | 99 | 100 |
| Arabidopsis
thaliana | AT1G09060 | 101 | 102 |
| Arabidopsis
thaliana | AT1G11950 | 103 | 104 |
| Arabidopsis
thaliana | AT1G30810 | 105 | 106 |
| Arabidopsis
thaliana | AT1G62310 | 107 | 108 |
| Arabidopsis
thaliana | AT1G63490 | 109 | 110 |
| Arabidopsis
thaliana | AT1G78280 | 111 | 112 |
| Arabidopsis
thaliana | AT2G34880 | 113 | 114 |
| Arabidopsis
thaliana | AT2G38950 | 115 | 116 |
| Arabidopsis
thaliana | AT3G07610 | 117 | 118 |
| Arabidopsis
thaliana | AT3G48430 | 119 | 120 |
| Arabidopsis
thaliana | AT4G00990 | 121 | 122 |
| Arabidopsis
thaliana | AT4G20400 | 123 | 124 |
| Arabidopsis
thaliana | AT4G20400 | 125 | 126 |
| Arabidopsis
thaliana | AT5G04240 | 127 | 128 |
| Arabidopsis
thaliana | AT5G06550 | 129 | 130 |
| Arabidopsis
thaliana | AT5G46910 | 131 | 132 |
| Arabidopsis
thaliana | AT5G63080 | 133 | 134 |
| Oryz
asativa | Os01g36630 | 135 | 136 |
| Oryz
asativa | Os01g67970 | 137 | 138 |
| Oryza
sativa | Os02g01940 | 139 | 140 |
| Oryza
sativa | Os02g58210 | 141 | 142 |
| Oryza
sativa | Os02g58210 | 143 | 144 |
| Oryza
sativa | Os03g05680 | 145 | 146 |
| Oryza
sativa | Os03g22540 | 147 | 148 |
| Oryza
sativa | Os03g27250 | 149 | 150 |
| Oryza
sativa | Os03g31594 | 151 | 152 |
| Oryza
sativa | Os03g31594 | 153 | 154 |
| Oryza
sativa | Os05g10770 | 155 | 156 |
| Oryza
sativa | Os05g23670 | 157 | 158 |
| Oryza
sativa | Os09g22540 | 159 | 160 |
| Oryza
sativa | Os10g42690 | 161 | 162 |
| Oryza
sativa | Os11g36450 | 163 | 164 |
| Oryza
sativa | Os12g18149 | 165 | 166 |
| Oryza
sativa | Os12g18150 | 167 | 168 |
| Glycine
max | Gm_JMJ_1 | 169 | 170 |
-
In
einigen Fällen sind verwandte Sequenzen vorläufig
zusammengebaut und durch Forschungsinstitutionen, wie The Institute
for Genomic Research (TIGR), öffentlich offenbart worden.
Die Eukaryotic Gene Orthologs (EGO) Datenbank kann zur Identifikation
solcher verwandten Sequenzen verwendet werden, und zwar entweder über
die Schlüsselwort-Suche oder unter Verwendung des BLAST-Algorithmus
mit der interessierenden Nukleinsäure- oder Polypeptidsequenz.
-
Beispiel 13: Alignment von JMJC-Polypeptisequenzen
-
Das
Alignment von Polypeptidsequenzen erfolgte mit dem AlignX-Programm
von dem Vector NTI (Invitrogen), das auf dem bekannten Clustal W-Algorithmus
für progressives Alignment beruht (Thompson et
al. (1997) Nucleic Acids Res 25: 4876–4882; Chenna
et al. (2003). Nucleic Acids Res 31: 3497–3500).
Die Default-Werte lauten: gap open penalty 10, gap extension penalty
0,1 und die gewählte ”weight matrix” ist
Blosum 62 (wenn Polypeptide einem Alignment unterworfen werden).
Um das Alignment weiter zu optimieren, können kleinere
Veränderungen von Hand durchgeführt wurden. Die
Sequenzkonservierung unter JMJC-Polypeptiden findet sich größtenteils
in der JmjC-Domäne der Polypeptide. Die Region, die den
durch SEQ ID NO: 79 und SEQ ID NO: 81 dargestellten Motiven entspricht,
ist stärker konserviert als diejenige von Motiv B. Eine
Konsensussequenz ist angegeben. Die Aminosäurereste in
der Konsensussequenz sind hochkonserviert. Das Alignment der JMJC-Polypeptide
ist in 7 gezeigt.
-
Beispiel 14: Berechnung der Gesamtprozent
an Identität zwischen JMJC-Polypeptidsequenzen, die für
die Durchführung der erfindungsgemäßen
Verfahren geeignet sind
-
Die
Gesamtprozent an Ähnlichkeit und Identität zwischen
Volllängenpolypeptidsequenzen, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet sind,
wurden unter Verwendung von einem der im Stand der Technik verfügbaren
Verfahren, nämlich der MatGAT (Matrix Global Alignment
Tool) Software (BMC Bioinformatics. 2003 4: 29. MatGAT:
an application that generstes similarity/identity matrices using
Protein or DNA sequences. Campanella JJ, Bitincka L, Smalley J;
von Ledion Bitincka gehostete Software), bestimmt. Die
MatGAT-Software erzeugt Ähnlichkeits-/Identitätsmatrizes
für DNA- oder Proteinsequenzen, ohne dass ein vorheriges
Alignment der Daten erforderlich ist. Das Programm führt
eine Reihe von paarweisen Alignments unter Verwendung des globalen
Alignment-Algorithmus von Myers und Miller durch (gap opening penalty
12, gap extension penalty 2), berechnet die Ähnlichkeit
und die Identität unter Verwendung von z. B. Blosum 62 (für
Polypeptide) und platziert dann die Ergebnisse in einer Abstandsmatrix.
Die Sequenzähnlichkeit ist in der unteren Hälfte
der Trennlinie und die Sequenzidentität in der oberen Hälfte
der diagonalen Trennlinie dargestellt.
-
Bei
den in dem Vergleich eingesetzten Parametern handelte es sich um:
| Scoring
matrix: | Blosum
62 |
| First
Gap: | 12 |
| Extending
gap: | 2 |
-
Die
Ergebnisse der Software-Analyse sind in Tabelle B2 für
die Gesamt-Ähnlichkeit und -Identität über die
volle Länge der Polypeptidsequenzen dargestellt. Die Prozent
Identität sind fettgedruckt über der Diagonalen
und die Prozent Ähnlichkeit sind (in normaler Schrift)
unter der Diagonalen angegeben.
-
Die
Prozent Identität zwischen den JMJC-Polypeptidsequenzen,
die für die Durchführung der erfindungsgemäßen
Verfahren geeignet sind, kann nur 15% Aminosäure-Identität
im Vergleich zu SEQ ID NO: 74 betragen. Tabelle B2: MatGAT-Ergebnisse für
Gesamt-Ähnlichkeit und -Identität über
die volle Länge der Polypeptidsequenzen.
| | | SEQ ID
NO: 74 | SEQ ID
NO: 84 | SEQ ID
NO: 86 | SEQ ID
NO: 90 | SEQ ID
NO: 92 | SEQ ID
NO: 94 | SEQ ID
NO: 136 |
| AT3G20810.1 | SEQ
ID NO: 74 | | 97,4 | 16,1 | 15,5 | 16,9 | 17,3 | 15,5 |
| AT3G20810.2 | SEQ
ID NO: 84 | 97,4 | | 15,5 | 14,5 | 17,5 | 17,1 | 14,9 |
| AT5G19840 | SEQ
ID NO: 86 | 30,3 | 30,7 | | 16,9 | 17 | 16,9 | 16,9 |
| ABE92082 | SEQ
ID NO: 90 | 28,2 | 27,7 | 28,5 | | 51,8 | 51,7 | 17,3 |
| CAJ26373 | SEQ
ID NO: 92 | 30,1 | 29,6 | 27,7 | 67,6 | | 82,5 | 12,4 |
| Os09g0483600 | SEQ
ID NO: 94 | 30,1 | 30,1 | 27,5 | 67,3 | 89,6 | | 16,1 |
| Os01g36630 | SEQ
ID NO: 136 | 33,7 | 31,2 | 32,9 | 34,2 | 29,1 | 29,6 | |
-
Beispiel 15: Identifikation von Domänen
in JMJC-Polypeptidsequenzen, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet sind
-
Bei
der Integrated Resource of Protein Families, Domains and Sites-(InterPro-)Datenbank
handelt es sich um eine integrierte Plattform für die häufig
verwendeten Signatur-Datenbanken für Suchen auf Text- und Sequenzbasis.
Die InterPro-Datenbank kombiniert diese Datenbanken, in denen unterschiedliche
Methodiken und verschiedene Mengen an biologischer Information über
gut charakterisierte Proteine verwendet werden, um Proteinsignaturen
abzuleiten. Zu den angeschlossenen Datenbanken zählen SWISS-PROT,
PROSITS, TrEMBL, PRINTS, ProDom und Pfam, Smart und TIGRFAMs. Pfam
ist eine große Sammlung multipler Sequenzalignments und
versteckter Markov-Modelle, die viele häufige Proteindomänen
und Familien abdeckt. Pfam wird auf dem Server des Sanger Institut
im Vereinigten Königreich gehostet. InterPro wird am European Bioinformatics
Institute im Vereinigten Königreich gehostet. Tabelle B3
zeigt die Einstellungen (Gathering cut off, trusted cut off und
noise cut off), wie in der Pfam-Datenbank beschrieben, die zur Erstellung
der HMMs_fs für die verschiedenen Domänen verwendet
wurden. Tabelle B3: HMMs_fs-Einstellungen.
| Domäne | Gathering
cutoff | Trusted
cutoff | Noise
cutoff |
| PF02373 | 16 | 16,1 | 15,9 |
| PF02375 | 15 | 16,8 | 13,8 |
| PF02928 | 25 | 44,4 | 21,1 |
| PF00646 | 17,7 | 17,7 | 17,6 |
| PF00096 | 22,5 | 22,5 | 22,4 |
| PF04967 | 15,4 | 15,4 | 3 |
-
Die
Ergebnisse des Pfam-Scans für beispielhafte JMJC-Polypeptide
pflanzlichen Ursprungs sind in Tabelle B4 dargestellt. Die Aminosäurekoordinaten
für jede Domäne in der Referenz-Sequenz sind in
den Spalten Start und Ende angegeben. Der E-Wert für das
Alignment ist ebenfalls angegeben. Die InterPro ID-Zugangsnummer
jeder identifizierten Domäne ist ebenfalls genannt. Tabelle
B4: Pfam-Scan-Ergebnisse (Haupt-Zugangsnummern) beispielhafter JMJC-Polypeptide
pflanzlichen Ursprungs.
-
Beispiel 16: Klonierung der JMJC-Nukleinsäuresequenz,
die bei den erfindungsgemäßen Verfahren verwendet wird
-
Die
Nukleinsäuresequenz, die bei den erfindungsgemäßen
Verfahren werden wurde, wurde mittels PCR amplifiziert, wobei als
Matrize eine maßgeschneiderte cDNA-Bank aus Arabidopsis
thaliana-Keimlingen (in pCMV Sport 6.0; Invitrogen, Paisley, UK)
verwendet wurde. Die PCR wurde unter Verwendung von Hifi Taq DNA-Polymerase
unter Standardbedingungen mit 200 ng Matrize in 50 μl PCR-Mix
durchgeführt. Es wurden ein stromaufwärts- bzw.
ein stromabwärts-Primer, die in SEQ ID NO: 75 und SEQ ID
NO: 76 dargestellt sind, für die Amplifikation der kodierenden
Region von JMJC gemäß SEQ ID NO: 73 mittels PCR
(Polymerasekettenreaktion) verwendet. Die Primer enthalten die AttB-Stellen
für die Gateway-Rekombination, wodurch die Klonierung des
amplifizierten PCR-DNA-Fragments in einen Gateway-Klonierungsvektor
erleichtert wird.
-
Das
amplifizierte PCR-Fragment wurde ebenfalls unter Verwendung von
Standardmethoden aufgereinigt. Anschließend wurde der erste
Schritt des Gateway-Verfahrens, die BP-Reaktion, durchgeführt,
bei der das PCR-Fragment in vivo mit dem Plasmid pDONR201 unter
Bildung eines – gemäß der Gateway-Teminologie – ”entry
clone”, pJMJC, rekombinierte. Das Plasmid pDONR201 wurde
von Invitrogen als Bestandteil der Gateway®-Technologie
erworben.
-
Der
die SEQ ID NO: 73 enthaltende ”entry clone” wurde
anschließend in einer LR-Reaktion mit einem Zielvektor,
der für die Transformation von Oryza sativa verwendet wird,
verwendet.
-
Dieser
Vektor enthielt als funktionelle Elemente innerhalb der T-DNA-Grenzen
Folgendes: Einen pflanzlichen Selektionsmarker; eine screenbare
Markerexpressionskassette und eine Gateway-Kassette, die für
die LR-in-vivo-Rekombination mit der interessierenden Nukleinsäuresequenz,
die bereits in den ”entry clone” kloniert wurde,
vorgesehen ist. Ein GOS2-Promotor aus Reis (SEQ ID NO: 77) für
die konstitutive Expression befand sich stromaufwärts von
dieser Gateway-Kassette.
-
Nach
dem LR-Rekombinationsschritt wurde der erhaltene Expressionsvektor
pGOS2::JMJC (9) nach im Stand der Technik
bekannten Verfahren in den Agrobacterium-Stamm LBA4044 transformiert.
-
Beispiel 17: Pflanzentransformation
-
Die
Transformation von Pflanzen wurde gemäß dem in
Beispiel 7 dargestellten Verfahren durchgeführt.
-
Beispiel 18: Verfahren bei der phänotypischen
Untersuchung
-
18.1 Aufbau der Auswertung
-
Es
wurden ungefähr 35 unabhängige T0-Reistransformanten
erzeugt. Die Primärtransformanten wurden für das
Heranziehen und Ernten von T1-Samen von einer Gewebekulturkammer
in ein Gewächshaus umgesetzt. Es wurden acht Ereignisse
zurückbehalten, von denen die T1-Nachkommenschaft für
Vorliegen/Abwesenheit des Transgens im Verhältnis 3:1 aufspaltete.
Für jedes dieser Ereignisse wurden ungefähr 10 T1-Keimpflanzen,
die das Transgen enthielten (Hetero- und Homozygote), und ungefähr
10 T1-Keimpflanzen, denen das Transgen fehlte (Nullizygoten), durch
Beobachten der sichtbaren Markerexpression selektiert. Die transgenen
Pflanzen und die entsprechenden Nullizygoten wurden nebeneinander
in zufälliger Anordnung herangezogen. Die Gewächshausbedingungen
waren Kurztage (12 Stunden Licht), 28°C im Hellen und 22°C
im Dunkeln und eine relative Feuchtigkeit von 70%. Die unter Nichtstressbedingungen
gezogenen Pflanzen werden in regelmäßigen Abständen
mit Wasser versorgt, so dass gewährleistet ist, dass Wasser
und Nährstoffe nicht limitierend sind, damit die Bedürfnisse
der Pflanze hinsichtlich der Vollendung von Wachstum und Entwicklung
erfüllt werden.
-
Vier
T1-Ereignisse wurden in der T2-Generation nach dem gleichen Untersuchungsverfahren
wie für die T1-Generation, jedoch mit mehr Individuen pro
Ereignis, weiter untersucht. Vom Sästadium bis zum Reifestadium
wurden die Pflanzen mehrmals in eine Digital-Imaging-Kammer gestellt.
Zu jedem Zeitpunkt wurden von jeder Pflanze aus mindestens 6 verschiedenen
Winkeln Digitalbilder (2048×1536 Pixel, 16 Millionen Farben)
aufgenommen.
-
Screening unter Trockenheitsbedingungen
-
Pflanzen
aus T2-Samen wurden unter Normalbedingungen in Blumenerde herangezogen,
bis sie das Stadium des Rispenschiebens erreichten. Anschließend
wurden sie in ein ”trockenes” Areal gestellt,
wo keine Bewässerung erfolgte. Um den Bodenwassergehalt
(BWG) zu verfolgen, wurden Feuchtigkeitssonden in statistisch ausgewählte
Töpfe eingeführt. Sank der BWG unter gewisse Schwellenwerte,
wurden die Pflanzen automatisch ständig erneut gegossen,
bis wieder ein Normalgehalt erreicht wurde. Dann wurden die Pflanzen wiederum
in Normalbedingungen umgestellt. Die übrige Anzucht (Pflanzenreife,
Samenernte) erfolgte wie bei den nicht unter abiotischen Stressbedingungen
herangezogenen Pflanzen. Wachstums- und Ertragsparameter werden
aufgezeichnet, wie für das Wachstum unter Normalbedingungen
beschrieben.
-
Screening auf Effizienz der Stickstoffnutzung
-
Reispflanzen
aus T2-Samen werden in Blumenerde und mit Ausnahme der Nährlösung
unter normalen Bedingungen gezüchtet. Die Töpfe
werden von der Transplantierung bis zur Reifung mit einer spezifischen Nährlösung
gewässert, die einen reduzierten Stickstoff-(N-)Gehalt,
und zwar gewöhnlich 7 bis 8 Mal niedriger, aufwies. Die übrige
Anzucht (Pflanzenreifung, Samenernte) entspricht ansonsten derjenigen
von Pflanzen, die nicht unter abiotischem Stress gezüchtet
werden. Wachstums- und Ertragsparameter werden wie für
das Wachstum unter normalen Bedingungen beschrieben ausgewertet.
-
18.2 Statistische Analyse: F-Test
-
Als
statistisches Modell für die Gesamtauswertung der phänotypischen
Eigenschaften der Pflanze wurde eine zweifaktorielle ANOVA (Varianzanalyse)
verwendet. Mit allen Parametern, die bei allen Pflanzen von allen
Ereignissen, die mit dem erfindungsgemäßen Gen
transformiert worden waren, bestimmt wurden, wurde ein F-Test durchgeführt.
Der F-Test erfolgte zur Überprüfung im Hinblick
auf einen Effekt des Gens über alle Transformationsereignisse
und zur Feststellung eines Gesamteffekts des Gens, was auch als
globaler Geneffekt bezeichnet wird. Die Signifikanzschwelle für
einen echten globalen Geneffekt wurde bei dem F-Test auf dem 5%-Wahrscheinlichkeitsniveau
festgelegt. Ein signifikanter F-Test-Wert weist auf einen Geneffekt
hin, was bedeutet, dass mehr als nur das einfache Vorhandensein
oder die Lage des Gens die Unterschiede im Phänotyp verursacht.
-
Da
zwei Experimente mit überlappenden Ereignissen durchgeführt
wurden, wurde eine kombinierte Analyse durchgeführt. Dies
ist dazu geeignet, die Übereinstimmung der Effekte über
die beiden Versuche zu überprüfen, und wenn dies
der Fall ist, Aussagen von beiden Versuchen zu vereinigen, um die
Verlässlichkeit der Schlussfolgerung zu erhöhen.
Bei dem verwendeten Verfahren handelte es sich um einen ”Mixed-Modell”-Ansatz,
der die mehrschichtige Struktur der Daten (d. h. Versuch – Ereignis – Segreganten)
berücksichtigt. Die p-Werte wurden dadurch erhalten, dass
man den Wahrscheinlichkeits-Quotienten-Test mit Chi-Quadrat-Verteilungen
verglich.
-
18.3 Messparameter
-
Biomasseparameter-Messung
-
Vom
Sästadium bis zum Reifestadium wurden die Pflanzen mehrmals
in eine Digital-Imaging-Kammer gestellt. Zu jedem Zeitpunkt wurden
von jeder Pflanze aus mindestens 6 verschiedenen Winkeln Digitalbilder (2048×1536
Pixel, 16 Millionen Farben) aufgenommen.
-
Die
oberirdische Pflanzenfläche (oder Blattbiomasse) wurde
dadurch bestimmt, dass man die Gesamtanzahl der Pixel auf den Digitalbildern
von oberirdischen Pflanzenteilen zählte, die sich vom Hintergrund unterschieden.
Dieser Wert wurde über die zu demselben Zeitpunkt aus unterschiedlichen
Winkeln aufgenommenen Bilder gemittelt und mittels Kalibrierung
in einen physikalischen Oberflächenwert, ausgedrückt
in Quadratmillimeter, umgewandelt. Versuche zeigen, dass die auf
diese Weise gemessene oberirdische Pflanzenfläche mit der
Biomasse der oberirdischen Pflanzenteile korreliert. Die oberirdische
Fläche ist diejenige Fläche, die zu dem Zeitpunkt
gemessen wurde, an dem die Pflanze ihre maximale Blattbiomasse erreicht
hatte. Die Jungpflanzenvitalität ist die oberirdische Fläche
der Pflanze (des Keimlings) drei Wochen nach der Keimung. Eine Erhöhung
der Wurzelbiomasse wird als Anstieg der Gesamtwurzelbiomasse (die
als maximale Biomasse der Wurzeln, die während der Lebenszeit
einer Pflanze beobachtet werden, gemessen wird) oder als ein Anstieg
des Wurzel/Spross-Index (der als das Verhältnis zwischen
Wurzelmasse und Sprossmasse im aktiven Wachstumszeitraum von Wurzel
und Spross gemessen wird) ausgedrückt.
-
Die
Jungpflanzenvitalität wurde bestimmt, indem die Gesamtzahl
der Pixel von oberirdischen Pflanzenteilen, die sich vom Hintergrund
unterschieden, gezählt wurde. Dieser Wert wurde über
die zu demselben Zeitpunkt aus unterschiedlichen Winkeln aufgenommenen
Bilder gemittelt und mittels Kalibrierung in einen physikalischen
Oberflächenwert, ausgedrückt in Quadratmillimeter,
umgewandelt. Die nachstehend beschriebenen Ergebnisse gelten für
Pflanzen drei Wochen nach der Keimung.
-
Samenparameter-Messungen
-
Die
reifen Primärrispen wurden geerntet, gezählt,
eingetütet, mit einem Strichcode versehen und dann drei
Tage bei 37°C in einem Ofen getrocknet. Dann wurden die
Rispen gedroschen und alle Samen wurden gesammelt und gezählt.
Die gefüllten Spelzen wurden von den leeren Spelzen mittels
einer Luftblaseeinrichtung getrennt. Die leeren Spelzen wurden verworfen
und die verbleibende Fraktion wurde nochmals gezählt. Die
gefüllten Spelzen wurden auf einer Analysewaage gewogen.
Die Anzahl gefüllter Samen wurde dadurch bestimmt, dass
man die Anzahl der gefüllten Spelzen, die nach dem Abtrennungsschritt
verblieben, auszählte. Der Gesamtsamenertrag wurde dadurch
gemessen, dass man alle von einer Pflanze geernteten gefüllten Spelzen
wog. Die Gesamtsamenzahl pro Pflanze wurde dadurch gemessen, dass
man die von einer Pflanze geerntete Anzahl Spelzen auszählte.
Das Tausendkorngewicht (TKG) wird aus der Anzahl der gezählten
gefüllten Samen und ihrem Gesamtgewicht extrapoliert. Der
Harvest Index (HI) ist bei der vorliegenden Erfindung als das Verhältnis
zwischen dem Gesamtsamenertrag und der oberirdischen Fläche
(mm2), multipliziert mit einem Faktor 106, definiert. Die Gesamtanzahl an Blüten
pro Rispe ist bei der vorliegenden Erfindung als das Verhältnis
zwischen der Gesamtanzahl an Samen und der Anzahl an reifen Primärrispen
definiert. Die Samenfüllungsrate ist bei der vorliegenden
Erfindung als der Anteil (ausgedrückt als %) der Anzahl
gefüllter Samen zu der Gesamtzahl Samen (oder Blüten)
definiert.
-
Beispiel 19: Ergebnisse der phänotypischen
Untersuchung der transgenen Pflanzen
-
Die
Ergebnisse der Untersuchung transgener Reispflanzen, die die JMJC-Nukleinsäure
gemäß SEQ ID NO: 73 exprimierten, unter Nicht-Stress-Bedingungen
sind nachstehend dargestellt. Es wurde ein Anstieg von mindestens
5% bei der Vitalität beim Aufgehen (Jungpflanzenvitalität),
beim Wurzel/Spross-Index, Gesamtsamenertrag, Harvest Index und von
mindestens 3% beim Tausendkorngewicht bei den transgenen Pflanzen
im Vergleich zu den Nullizygoten-Kontrollpflanzen beobachtet.
-
Die
Ergebnisse der Untersuchung transgener Reispflanzen, die SEQ ID
NO: 73 exprimierten, unter Trockenheitsstressbedingungen sind im
Folgenden dargestellt. Es wurde ein Anstieg von mindestens 5% beim Wurzel/Spross-Index,
Gesamtsamengewicht, der Anzahl an gefüllten Samen, der
Füllungsrate, beim Harvest Index und von mindestens 3%
beim Tausendkorngewicht bei den transgenen Pflanzen im Vergleich
zu den Nullizygoten-Kontrollpflanzen beobachtet.
-
III. Casein Kinase 1
-
Beispiel 20: Identifikation von Sequenzen,
die mit der in den erfindungsgemäßen Verfahren
verwendeten CKI-Nukleinsäuresequenz verwandt sind
-
Sequenzen
(Volllängen-cDNA, ESTs oder genomische), die mit der in
den erfindungsgemäßen Verfahren verwendeten Nukleinsäuresequenz
verwandt sind, werden unter den Sequenzen in der Entrez Nucleotides Datenbank
am National Center for Biotechnology Information (NCBI) unter Verwendung
von Datenbanksequenzabfragewerkzeugen, wie dem Basic Local Alignment
Tool (BLAST) (Altschul et al. (1990), J. Mol. Biol. 215:
403–410; und Altschul et al., (1997),
Nucleic Acids Res. 25: 3389–3402), identifiziert.
Das Programm wird zum Auffinden von Regionen mit lokaler Ähnlichkeit
zwischen Sequenzen verwendet, und zwar durch Vergleichen von Nukleinsäure-
oder Polypeptidsequenzen mit Sequenz-Datenbanken und durch Berechnen
der statistischen Signifikanz der Übereinstimmungen. Das
Polypeptid, das durch die bei der vorliegenden Erfindung verwendeten
Nukleinsäuren kodiert wird, wird zum Beispiel für
den TBLASTN-Algorithmus verwendet, und zwar unter Ausschaltung der
Default-Einstellungen und des Filters für die Nichtmiteinbeziehung
von Sequenzen mit niedriger Komplexität. Der Output der
Analyse wird mittels paarweisem Vergleich betrachtet und anhand
der Wahrscheinlichkeits-Wertung (E-Wert) in eine Reihenfolge gebracht,
wobei die Wertung die Wahrscheinlichkeit widerspiegelt, dass ein
bestimmtes Alignment zufällig vorkommt (je niedriger der
E-Wert, desto signifikanter der Hit). Zusätzlich zu den
E-Werten wurden die Vergleiche auch anhand der Prozent Identität
gewertet. Prozent Identität bezieht sich auf die Anzahl
identischer Nukleotide (oder Aminosäuren) zwischen den zwei
verglichenen Nukleinsäure-(oder Polypeptid-)sequenzen über
eine bestimmte Länge. In einigen Fällen können
die Default-Parameter angepasst werden, um die Stringenz der Suche
zu modifizieren. Zum Beispiel kann man den E-Wert erhöhen,
so dass weniger stringente Übereinstimmungen angezeigt
werden. Auf diese Weise lassen sich kurze fast genaue Übereinstimmungen
identifizieren.
-
In
einigen Fällen können verwandte Sequenzen vorläufig
zusammengebaut und öffentlich durch Forschungsinstitutionen,
wie The Institute for Genomic Research (TIGR), offenbart werden.
Die Eukaryotic Gene Orthologs (EGO) Datenbank kann zur Identifikation
solcher verwandten Sequenzen verwendet werden, und zwar entweder über
die Schlüsselwort-Suche oder unter Verwendung des BLAST-Algorithmus
mit der interessierenden Nukleinsäure- oder Polypeptidsequenz.
-
Beispiel 21: Alignment von GRP-Polypeptisequenzen
-
Das
Alignment von Polypeptidsequenzen erfolgt mit dem AlignX-Programm
aus dem Vector NTI-Paket (Invitrogen), das auf dem bekannten Clustal
W-Algorithmus für progressives Alignment beruht (Thompson
et al. (1997) Nucleic Acids Res 25: 4876–4882; Chenna
et al. (2003). Nucleic Acids Res 31: 3497–3500).
Die Default-Werte lauten: gap open penalty 10, gap extension penalty
0,1 und die gewählte ”weight matrix” ist
Blosum 62 (wenn Polypeptide einem Alignment unterworfen werden).
Um das Alignment weiter zu optimieren, können kleinere Veränderungen
von Hand durchgeführt werden.
-
Ein
Stammbaum von GRP-Polypeptiden wird unter Verwendung eines Neighbour-Joining-Cluster-Algorithmus
konstruiert, der in dem AlignX Programm von Vector NTI (Invitrogen)
bereitgestellt wird.
-
Beispiel 22: Berechnung der Gesamtprozent
an Identität zwischen Polypeptidsequenzen, die für
die Durchführung der erfindungsgemäßen
Verfahren geeignet sind
-
Die
Gesamtprozent an Ähnlichkeit und Identität zwischen
Volllängenpolypeptidsequenzen, die bei der Durchführung
der erfindungsgemäßen Verfahren geeignet sind,
werden unter Verwendung von einem der im Stand der Technik verfügbaren
Verfahren, nämlich der MatGAT (Matrix Global Alignment
Tool) Software (BMC Bioinformatics. 2003 4: 29. MatGAT:
an application that generstes similarity/identity matrices using
Protein or DNA sequences. Campanella JJ, Bitincka L, Smalley J;
von Ledion Bitincka gehostete Software), bestimmt. Die
MatGAT-Software erzeugt Ähnlichkeits-/Identitätsmatrizes
für DNA- oder Proteinsequenzen, ohne dass ein vorheriges
Alignment der Daten erforderlich ist. Das Programm führt
eine Reihe von paarweisen Alignments unter Verwendung des globalen
Alignment-Algorithmus von Myers und Miller durch (gap opening penalty
12, gap extension penalty 2), berechnet die Ähnlichkeit
und die Identität unter Verwendung von z. B. Blosum 62 (für
Polypeptide) und platziert dann die Ergebnisse in einer Abstandsmatrix.
Die Sequenzähnlichkeit ist in der unteren Hälfte
der Trennlinie und die Sequenzidentität in der oberen Hälfte
der diagonalen Trennlinie dargestellt.
-
Bei
den in dem Vergleich eingesetzten Parametern handelte es sich um:
| Scoring
matrix: | Blosum
62 |
| First
Gap: | 12 |
| Extending
gap: | 2 |
-
Eine
MATGAT-Tabelle für das lokale Alignment einer spezifischen
Domäne oder Daten zu % Identität/Ähnlichkeit
zwischen spezifischen Domänen können ebenfalls
erzeugt werden.
-
Beispiel 23: Identifikation von Domänen
in Polypeptidsequenzen, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet sind
-
Bei
der Integrated Resource of Protein Families, Domains and Sites-(InterPro-)Datenbank
handelt es sich um eine integrierte Plattform für die häufig
verwendeten Signatur-Datenbanken für Suchen auf Text- und Sequenzbasis.
Die InterPro-Datenbank kombiniert diese Datenbanken, in denen unterschiedliche
Methodiken und verschiedene Mengen an biologischer Information über
gut charakterisierte Proteine verwendet werden, um Proteinsignaturen
abzuleiten. Zu den angeschlossenen Datenbanken zählen SWISS-PROT,
PROSITS, TrEMBL, PRINTS, ProDom und Pfam, Smart und TIGRFAMs. Pfam
ist eine große Sammlung multipler Sequenzalignments und
versteckter Markov-Modelle, die viele häufige Proteindomänen
und Familien abdeckt. Pfam wird auf dem Server des Sanger Institut
im Vereinigten Königreich gehostet. Interpro wird am European Bioinformatics
Institute im Vereinigten Königreich gehostet.
-
Die
Proteinsequenzen, welche die GRP wiedergeben, werden als Abfrage
bei einer Suche in der InterPro-Datenbank verwendet.
-
Beispiel 24: Topologie-Vorhersage für
Polypeptidsequenzen, die sich für die Durchführung
der erfindungsgemäßen Verfahren eignen
-
TargetP
1.1 sagt die subzelluläre Lokalisierung eukaryotischer
Proteine voraus. Die Zuordnung der Lokalisierung beruht auf der
vorhergesagten Anwesenheit von einer der folgenden N-terminalen
Präsequenzen: Chloroplasten-Transitpeptid (cTP), mitochondriales
Targeting-Peptid (mTP) oder Signalpeptid (SP) für den sekretorischen
Weg. Die Scores, auf denen die endgültige Vorhersage beruht,
sind keine echten Wahrscheinlichkeiten, und sie tragen auch nicht
notwendigerweise zu einer bei. Die Lokalisierung mit dem höchsten
Score ist jedoch gemäß TargetP am wahrscheinlichsten
und das Verhältnis zwischen den Scores (die Verlässlichkeitsklasse)
kann ein Anzeichen dafür sein, wie sicher die Vorhersage
ist. Die Verlässlichkeitsklasse (RC) reicht von 1 bis 5,
wobei 1 die stärkste Vorhersage angibt. TargetP wird auf
dem Server der Technischen Universität Dänemark
gehalten.
-
Für
die Sequenzen, die den Vorhersagen zufolge eine N-terminale Präsequenz
enthalten, kann auch eine potentielle Spaltstelle vorhergesagt werden.
-
Es
wurde eine Anzahl an Parametern ausgewählt, wie Organismusgruppe
(Nicht-Pflanze oder Pflanze), Sätze von Ausschlussgrenzen
(keine, vordefinierte Sätze von Ausschlussgrenzen oder
nutzerspezifische Sätze von Ausschlussgrenzen) und die
Berechnung der Vorhersage von Spaltstellen (Ja oder Nein).
-
Die
Proteinsequenzen, welche die GRP darstellen, werden für
die Abfrage von TargetP 1.1 verwendet. Es wurde die Organismusgruppe ”Pflanze” gewählt,
keine Ausschlussgrenzen definiert und die vorhergesagte Länge
des Transitpeptids abgefragt.
-
Zur
Durchführung solcher Analysen können viele andere
Algorithmen verwendet werden, wie u. a.:
- • ChloroP
1.1, das auf dem Server der Technischen Univerität Dänemark
gehostet wird;
- • Protein Prowler Subcellular Localisation Predictor
Version 1.2, das auf dem Server des Instituts für Molekulare
Biowissenschaft, Universität Queensland, Brisbane, Australien,
gehostet wird;
- • PENCE Proteome Analyst PA-GOSUB 2.5, das auf dem
Server der Universität von Alberts, Edmonton, Alberts,
Kanada, gehostet wird;
- • TMHMM, das auf dem Server der Technischen Universität
Dänemark gehostet wird
-
Beispiel 25: Klonierung der Nukleinsäuresequenz,
die bei den erfindungsgemäßen Verfahren verwendet
wird
-
Klonierung von SEQ ID NO: 171:
-
Die
Nukleinsäuresequenz SEQ ID NO: 171, die bei den erfindungsgemäßen
Verfahren verwendet wurde, wurde mittels PCR amplifiziert, wobei
als Matrize eine maßgeschneiderte cDNA-Bank aus Arabidopsis
thaliana-Keimlingen (in pCMV Sport 6.0; Invitrogen, Paisley, UK)
verwendet wurde. Die PCR wurde unter Verwendung von Hifi Taq DNA-Polymerase
unter Standardbedingungen mit 200 ng Matrize in 50 μl PCR-Mix
durchgeführt. Die verwendeten Primer waren prm8667 (SEQ
ID NO: 177; sense, Startcodon fettgedruckt):
5'-ggggacaagtttgtacaaaaaagcaggcttaaacaatggactgcaacatggtatct-3'
und prm8668 (SEQ ID NO: 178; revers, komplementär):
5'-ggggaccactttgtacaagaaagctgggtcacattacttactcatctattttgg-3',
die
die AttB-Stellen für die Gateway-Rekombination enthalten.
Das amplifizierte PCR-Fragment wurde ebenfalls unter Verwendung
von Standardmethoden aufgereinigt. Anschließend wurde der
erste Schritt des Gateway-Verfahrens, die BP-Reaktion, durchgeführt,
bei der das PCR-Fragment in vivo mit dem Plasmid pDONR201 unter
Bildung eines – gemäß der Gateway-Teminologie – ”entry
clone” rekombinierte. Das Plasmid pDONR201 wurde von Invitrogen
als Bestandteil der Gateway®-Technologie
erworben.
-
Klonierung von SEQ ID NO: 173:
-
Ein
cDNA-AFLP-Experiment wurde an einer synchronisierten Tabak-BY2-Zellkultur
(Nicotiana tabacum L. cv. Bright Yellow-2) durchgeführt
und in BY2 exprimierte Sequenz-Tags, die mit dem Verlauf des Zellzyklus
moduliert wurden, wurden für die weitere Klonierung ausgewählt.
Die exprimierten Sequenz-Tags wurden für das Screening
einer Tabak-cDNA-Bank und zur Isolation einer interessierenden Volllängen-cDNA, nämlich
einer DNA, die SEQ ID NO: 173 kodierte, verwendet.
-
Eine
Suspension von kultivierten Tabak-BY2-Zellen (Nicotiana tabacum
L. cv. Bright Yellow-2) wurde synchronisiert, indem die Zellen mit
Aphidicolin wie folgt in der frühen S-Phase blockiert wurden.
Die Zellsuspension von Nicotiana tabacum L. cv. Bright Yellow 2
wurde gehalten, wie beschrieben (Nagata et al. Int. Rev. Cytol.
132, 1–30, 1992). Für die Synchronisation
wurde eine 7 Tage alte stationäre Kultur 10-fach in frischem Medium
verdünnt, das mit Aphidicolin (Sigma-Aldrich, St. Louis,
MO; 5 mg/l) angereichert war. Nach 24 Std. wurde die Blockierung
der Zellen durch mehrmaliges Waschen mit frischem Medium aufgehoben,
wonach sie das Durchlaufen des Zellzyklus wieder aufnahmen.
-
Gesamt-RNA
wurde unter Verwendung von LiCI-Fällung extrahiert und
die poly(A+)-RNA wurde aus 500 μg
Gesamt-RNA unter Verwendung von Oligotex-Säulen (Qiagen,
Hilden, Deutschland) gemäß der Anleitung des Herstellers
isoliert. Ausgehend von 1 μg poly(A+)-RNA
wurde der erste Strang der cDNA mittels reverser Transkription mit
einem biotinylierten oligo-dT25-Primer (Genset,
Paris, Frankreich) und Superscript II (Life Technologies, Gaithersburg,
MD) synthetisiert. Die Zweistrangsynthese erfolgte durch Strangverdrängung
mit Escherichia coli-Ligase (Life Technologies), DNA-Polymerase
I (USB, Cleveland, OH) und RNAse H (USB).
-
Es
wurden 500 ng doppelsträngige cDNA für die AFLP-Analyse,
wie beschrieben (Vos et al., Nucleic Acids Res. 23 (21)
4407–4414, 1995; Bachem et al., Plant
J. 9 (5) 745–53, 1996) mit Modifikationen, verwendet.
Die verwendeten Restriktionsenzyme waren BstYI und MseI (Biolabs)
und die Spaltung erfolgte in zwei getrennten Schritten. Nach der
ersten Restriktionsspaltung mit einem der Enzyme wurden die 3'-Ende-Fragmente
mithilfe ihres biotinylierten Schwanzes an Dyna-Kugeln (Dynal, Oslo,
Norwegen) eingefangen, wohingegen die übrigen Fragmente
abgewaschen wurden. Nach der Spaltung mit dem zweiten Enzym wurden
die freigesetzten Restriktionsfragmente gesammelt und als Matrizen
bei den anschließenden AFLP-Schritten eingesetzt. Für
Voramplifikationen wurden ein MseI-Primer ohne selektive Nukleotide
mit einem BstYI-Primer kombiniert, der entweder ein T oder ein C
als häufigstes 3'-Nukleotid enthielt. Die PCR-Bedingungen
waren wie beschrieben (Vos et al., 1995). Die erhaltenen
Amplifikationsgemische wurden 600-fach verdünnt und 5 μl wurden
für selektive Amplifikationen unter Verwendung eines P33-markierten BstYI-Primers und der Amplitaq-Gold-Polymerase
(Roche Diagnostics, Brüssel, Belgien) eingesetzt. Die Amplifikationsprodukte
wurden auf 5%igen Polyacrylamidgelen unter Verwendung des Sequigel-Systems
(Biorad) aufgetrennt. Die getrockneten Gele wurden mit Kodak Biomax-Filmen
exponiert und in einem PhosphorImager (Amersham Pharmacia Biotech,
Little Chalfont, UK) gescannt.
-
Banden,
die differentiell exprimierten Transkripten entsprachen, darunter
das (partielle) Transkript, das der SEQ ID NO: 173 entspricht, wurden
aus dem Gel isoliert und die eluierte DNA wurde unter den gleichen Bedingungen
wie bei der selektiven Amplifikation reamplifiziert. Sequenzinformation
wurde entweder durch direkte Sequenzierung des reamplifizierten
Produkts der Polymerasekettenreaktion mit dem selektiven BstYI-Primer
oder nach Klonierung der Fragmente in pGEM-T easy (Promega, Madison,
WI) und Sequenzierung von Einzelklonen erhalten. Die erhaltenen
Sequenzen wurden mit Nukleotid- und Proteinsequenzen in den öffentlich
zugänglichen Datenbanken mittels BLAST-Sequenz-Alignments
(Altschul et al., Nucleic Acids Res. 25 (17) 3389–3402
1997) verglichen. Wenn verfügbar, wurden die Tag-Sequenzen
gegen längere EST- oder isolierte cDNA-Sequenzen ausgetauscht,
um die Wahrscheinlichkeit zu erhöhen, dass eine signifikante Homologie
gefunden wurde. Der physikalische cDNA-Klon, der der SEQ ID NO 173
entspricht, wurde anschließend aus einer im Handel erhältlichen
Tabak-cDNA-Bank wie folgt isoliert:
Eine cDNA-Bank mit einer
durchschnittlichen Insertgröße von 1400 bp wurde
aus poly(A+)-RNA hergestellt, die aus sich
aktiv teilenden, nicht-synchronisierten BY2-Tabakzellen isoliert
worden war. Die Inserts dieser Bank waren in dem Vektor pCMVSPORT6.0
kloniert, der eine attB-Gateway-Kassette (Life Technologies) umfasst. Aus
dieser Bank wurden 46 000 Klone ausgewählt, in Arrays auf
Mikrotiterplatten mit 384 Vertiefungen angeordnet und anschließend
in Doppelproben auf Nylonfilter getupft. Die Klon-Arrays wurden
unter Verwendung von Pools von mehreren Hundert radioaktiv markierten
Tags (einschließlich des BY2-Tags, das der SEQ ID NO: 173
entspricht) als Sonden gescreent. Positive Klone wurden isoliert
(darunter der Klon, der der SEQ ID NO: 173 entspricht), sequenziert
und einem Alignment mit der Tag-Sequenz unterzogen. Wenn die Hybridisierung
mit dem Tag nicht gelang, wurde die Volllängen-cDNA, die
dem Tag entsprach, mittels PCR-Amplifikation selektiert: Tag-spezifische
Primer wurden unter Verwendung von öffentlich zugänglicher
Software gestaltet und in Kombination mit einem üblichen
Vektor-Primer für die Amplifikation partieller cDNA-Inserts
verwendet. Pools der DNA von 50 000, 100 000, 150 000 und 300 000
cDNA-Klonen wurden bei den PCR-Amplifikationen als Matrizen eingesetzt.
Die Amplifikationsprodukte wurden dann aus Agarosegelen isoliert,
kloniert sequenziert und ihre Sequenz wurde einem Alignment mit
der Tag-Sequenz unterzogen. Danach wurde die Volllängen-cDNA,
die der Nukleotidsequenz der SEQ ID NO 173 entsprach, aus dem pCMVsport6.0-Bankvektor über
eine LR-Reaktion in pDONR201, einen Gateway®-Donorvektor
(Invitrogen, Paisley, UK), kloniert, wodurch ein ”entry
clone” erhalten wurde.
-
Der
die SEQ ID NO: 171 oder SEQ ID NO: 173 enthaltende ”entry
clone” wurde anschließend in einer LR-Reaktion
mit einem Zielvektor, der für die Transformation von Oryza
sativa verwendet wird, verwendet. Dieser Vektor enthielt als funktionelle
Elemente innerhalb der T-DNA-Grenzen Folgendes: Einen pflanzlichen
Selektionsmarker; eine screenbare Markerexpressionskassette und
eine Gateway-Kassette, die für die LR-in-vivo-Rekombination
mit der interessierenden Nukleinsäuresequenz, die bereits
in den ”entry clone” kloniert wurde, vorgesehen
ist. Ein WSI18-Promotor aus Reis (SEQ ID NO: 175) für die
spezifische Expression in Samen befand sich stromaufwärts
von dieser Gateway-Kassette.
-
Nach
dem LR-Rekombinationsschritt wurde der erhaltene Expressionsvektor
pWSI18::GRP (11) nach im Stand der Technik
bekannten Verfahren in den Agrobacterium-Stamm LBA4044 transformiert.
-
In
einem alternativen Konstrukt wurde der GOS2-Promotor aus Reis (SEQ
ID NO: 176) verwendet, so dass der Expressionsvektor pGOS2::GRP
erhalten wurde.
-
Beispiel 26: Pflanzentransformation
-
Die
Transformation von Pflanzen wurde gemäß dem in
Beispiel 7 ausgeführten Verfahren durchgeführt.
-
Beispiel 27: Verfahren bei der phänotypischen
Auswertung
-
27.1 Aufbau der Auswertung
-
Es
wurden ungefähr 35 unabhängige T0-Reistransformanten
erzeugt. Die Primärtransformanten wurden für das
Heranziehen und Ernten von T1-Samen von einer Gewebekulturkammer
in ein Gewächshaus umgesetzt. Es wurden sechs Ereignisse
zurückbehalten, von denen die T1-Nachkommenschaft für
Vorliegen/Abwesenheit des Transgens im Verhältnis 3:1 aufspaltete.
Für jedes dieser Ereignisse wurden ungefähr 10 T1-Keimpflanzen,
die das Transgen enthielten (Hetero- und Homozygote), und ungefähr
10 T1-Keimpflanzen, denen das Transgen fehlte, (Nullizygoten) durch
Beobachten der sichtbaren Markerexpression selektiert. Die transgenen
Pflanzen und die entsprechenden Nullizygoten wurden nebeneinander
in zufälliger Anordnung herangezogen. Die Gewächshausbedingungen
waren Kurztage (12 Stunden Licht), 28°C im Hellen und 22°C
im Dunkeln und eine relative Feuchtigkeit von 70%. Die unter Nichtstressbedingungen
gezogenen Pflanzen werden in regelmäßigen Abständen
mit Wasser versorgt, so dass gewährleistet ist, dass Wasser
und Nährstoffe nicht limitierend sind, damit die Bedürfnisse
der Pflanze hinsichtlich der Vollendung von Wachstum und Entwicklung
erfüllt werden.
-
Vier
T1-Ereignisse wurden in der T2-Generation nach dem gleichen Untersuchungsverfahren
wie für die T1-Generation, jedoch mit mehr Individuen pro
Ereignis, weiter untersucht. Vom Sästadium bis zum Reifestadium
wurden die Pflanzen mehrmals in eine Digital-Imaging-Kammer gestellt.
Zu jedem Zeitpunkt wurden von jeder Pflanze aus mindestens 6 verschiedenen
Winkeln Digitalbilder (2048×1536 Pixel, 16 Millionen Farben)
aufgenommen.
-
Screening unter Trockenheitsbedingungen
-
Pflanzen
aus T2-Samen wurden unter Normalbedingungen in Blumenerde herangezogen,
bis sie das Stadium des Rispenschiebens erreichten. Anschließend
wurden sie in ein ”trockenes” Areal gestellt,
wo keine Bewässerung erfolgte. Um den Bodenwassergehalt
(BWG) zu verfolgen, wurden Feuchtigkeitssonden in statistisch ausgewählte
Töpfe eingeführt. Sank der BWG unter gewisse Schwellenwerte,
wurden die Pflanzen automatisch ständig erneut gegossen,
bis wieder ein Normalgehalt erreicht wurde. Dann wurden die Pflanzen wiederum
in Normalbedingungen umgestellt. Die übrige Anzucht (Pflanzenreife,
Samenernte) erfolgte wie bei den nicht unter abiotischen Stressbedingungen
herangezogenen Pflanzen. Wachstums- und Ertragsparameter werden
aufgezeichnet, wie für das Wachstum unter Normalbedingungen
beschrieben.
-
Screening auf Effizienz der Stickstoffnutzung
-
Reispflanzen
aus T2-Samen werden in Blumenerde und mit Ausnahme der Nährlösung
unter normalen Bedingungen gezüchtet. Die Töpfe
werden von der Transplantierung bis zur Reifung mit einer spezifischen Nährlösung
gewässert, die einen reduzierten Stickstoff-(N-)Gehalt,
und zwar gewöhnlich 7 bis 8 Mal niedriger, aufweist. Die übrige
Anzucht (Pflanzenreifung, Samenernte) entspricht ansonsten derjenigen
von Pflanzen, die nicht unter abiotischem Stress gezüchtet
werden. Wachstums- und Ertragsparameter werden wie für
das Wachstum unter normalen Bedingungen beschrieben ausgewertet.
-
27.2 Statistische Analyse: F-Test
-
Als
statistisches Modell für die Gesamtauswertung der phänotypischen
Eigenschaften der Pflanze wurde eine zweifaktorielle ANOVA (Varianzanalyse)
verwendet. Mit allen Parametern, die bei allen Pflanzen von allen
Ereignissen, die mit dem erfindungsgemäßen Gen
transformiert wurden, bestimmt wurden, wurde ein F-Test durchgeführt.
Der F-Test erfolgte zur Überprüfung im Hinblick
auf einen Effekt des Gens über alle Transformationsereignisse
und zur Feststellung eines Gesamteffekts des Gens, was auch als
globaler Geneffekt bezeichnet wird. Die Signifikanzschwelle für
einen echten globalen Geneffekt wurde bei dem F-Test auf dem 5%-Wahrscheinlichkeitsniveau
festgelegt. Ein signifikanter F-Test-Wert weist auf einen Geneffekt
hin, was bedeutet, dass mehr als nur das einfache Vorhandensein
oder die Lage des Gens die Unterschiede im Phänotyp verursacht.
-
Da
zwei Versuche mit überlappenden Ereignissen durchgeführt
wurden, wurde eine kombinierte Analyse durchgeführt. Dies
ist dazu geeignet, die Übereinstimmung der Effekte über
die beiden Versuche zu überprüfen, und wenn dies
der Fall ist, Befunde von beiden Versuchen zu vereinigen, um die
Verlässlichkeit der Schlussfolgerung zu erhöhen.
Bei dem verwendeten Verfahren handelte es sich um einen ”Mixed-Modell”-Ansatz,
der die mehrschichtige Struktur der Daten (d. h. Versuch – Ereignis – Segreganten)
berücksichtigt. Die p-Werte wurden dadurch erhalten, dass
der Wahrscheinlichkeits-Quotienten-Test mit Chi-Quadrat-Verteilungen
verglichen wurde.
-
27.3 Messparameter
-
Biomasseparameter-Messung
-
Vom
Sästadium bis zum Reifestadium wurden die Pflanzen mehrmals
in eine Digital-Imaging-Kammer gestellt. Zu jedem Zeitpunkt wurden
von jeder Pflanze aus mindestens 6 verschiedenen Winkeln Digitalbilder (2048×1536
Pixel, 16 Millionen Farben) aufgenommen. Die oberirdische Pflanzenfläche
(oder Blattbiomasse) wurde dadurch bestimmt, dass man die Gesamtanzahl
der Pixel auf den Digitalbildern von oberirdischen Pflanzenteilen
zählte, die sich vom Hintergrund unterschieden. Dieser
Wert wurde über die zu demselben Zeitpunkt aus unterschiedlichen
Winkeln aufgenommenen Bilder gemittelt und mittels Kalibrierung
in einen physikalischen Oberflächenwert, ausgedrückt
in Quadratmillimeter, umgewandelt. Versuche zeigen, dass die auf
diese Weise gemessene oberirdische Pflanzenfläche mit der
Biomasse der oberirdischen Pflanzenteile korreliert. Die oberirdische
Fläche ist diejenige Fläche, die zu dem Zeitpunkt
gemessen wurde, an dem die Pflanzen ihre maximale Blattbiomasse
erreicht hatten.
-
Die
Jungpflanzenvitalität ist die oberirdische Fläche
der Pflanze (des Keimlings) drei Wochen nach der Keimung. Die Jungpflanzenvitalität
wurde bestimmt, indem die Gesamtzahl der Pixel von oberirdischen
Pflanzenteilen, die sich vom Hintergrund unterschieden, gezählt
wurde. Dieser Wert wurde über die zu demselben Zeitpunkt
aus unterschiedlichen Winkeln aufgenommenen Bilder gemittelt und
mittels Kalibrierung in einen physikalischen Oberflächenwert,
ausgedrückt in Quadratmillimeter, umgewandelt. Die nachstehend
beschriebenen Ergebnisse gelten für Pflanzen drei Wochen
nach der Keimung.
-
Samenparameter-Messungen
-
Die
reifen Primärrispen wurden geerntet, gezählt,
eingetütet, mit einem Strichcode versehen und dann drei
Tage bei 37°C in einem Ofen getrocknet. Dann wurden die
Rispen gedroschen und alle Samen wurden gesammelt und gezählt.
Die gefüllten Spelzen wurden von den leeren Spelzen mittels
einer Luftblaseeinrichtung getrennt. Die leeren Spelzen wurden verworfen
und die restliche Fraktion wurde nochmals gezählt. Die gefüllten
Spelzen wurden auf einer Analysewaage gewogen. Die Anzahl gefüllter
Samen wurde dadurch bestimmt, dass man die Anzahl der gefüllten
Spelzen, die nach dem Abtrennungsschritt verblieben, auszählte. Der
Gesamtsamenertrag wurde dadurch gemessen, dass man alle von einer
Pflanze geernteten gefüllten Spelzen wog. Die Gesamtsamenzahl
pro Pflanze wurde dadurch gemessen, dass man die von einer Pflanze geerntete
Anzahl Spelzen auszählte. Das Tausendkorngewicht (TKG)
wird aus der Anzahl der gezählten gefüllten Samen
und ihrem Gesamtgewicht extrapoliert. Der Harvest Index (HI) ist
bei der vorliegenden Erfindung als das Verhältnis zwischen
dem Gesamtsamenertrag und der oberirdischen Fläche (mm2), multipliziert mit einem Faktor 106, definiert. Die Gesamtanzahl an Blüten
pro Rispe ist bei der vorliegenden Erfindung als das Verhältnis
zwischen der Gesamtanzahl an Samen und der Anzahl an reifen Primärrispen
definiert. Die Samenfüllungsrate ist bei der vorliegenden
Erfindung als der Anteil (ausgedrückt als %) der Anzahl
gefüllter Samen zu der Gesamtzahl Samen (oder Blüten)
definiert.
-
Beispiel 28: Ergebnisse der phänotypischen
Untersuchung der transgenen Pflanzen
-
Die
transgenen Reispflanzen, welche die durch SEQ ID NO: 171 dargestellte
GRP-Nukleinsäure unter der Kontrolle des WSI18-Promotors
exprimierten, zeigten einen Anstieg von mehr als 5% bei der Biomasse, dem
Gesamtgewicht der Samen, der Anzahl gefüllter Samen, der
Füllungsrate und beim Harvest Index, wenn sie unter Trockenheitsstressbedingungen
herangezogen wurden. Bei Anzucht unter Nichtstressbedingungen wurde
ein Anstieg von mehr als 5% bei der Biomasse, der Anzahl gefüllter
Samen, dem Gesamtgewicht der Samen und der Anzahl an ersten Rispen
beobachtet.
-
Bei
dem Konstrukt mit SEQ ID NO: 171 unter der Kontrolle des GOS2-Promotors
wurde bei den transgenen Pflanzen ein Anstieg bezüglich
der Jungpflanzenvitalität und der Blüten pro Rispe
beobachtet, wobei der Anstieg bei jedem dieser Parameter mehr als
5% betrug.
-
Transgene
Pflanzen, die die SEQ ID NO: 173 unter der Kontrolle des WSI18-Promotors
exprimierten und unter Trockenheitsstressbedingungen herangezogen
wurden, zeigten eine Zunahme beim Gesamtgewicht der Samen, der Anzahl
an gefüllten Samen, der Anzahl an Blüten pro Rispe,
beim Harvest Index und beim Tausendkorngewicht. Beim Tausendkorngewicht
betrug die beobachtetet Zunahme mindestens 3,5% und bei den anderen
Parametern mehr als 5%.
-
IV. Pflanzen-Homöodomäne-Finger-Homöodomäne-(PHDf-HD-)Polypeptid
-
Beispiel 29: Identifikation von Sequenzen,
die mit der in den erfindungsgemäßen Verfahren
verwendeten Nukleinsäuresequenz verwandt sind
-
Sequenzen
(Volllängen-cDNA, ESTs oder genomische), die mit der in
den erfindungsgemäßen Verfahren verwendeten Nukleinsäuresequenz
verwandt sind, wurden unter den Sequenzen in der Entrez Nucleotides Datenbank
am National Center for Biotechnology Information (NCBI) unter Verwendung
von Datenbanksequenzabfragewerkzeugen, wie dem Basic Local Alignment
Tool (BLAST) (Altschul et al. (1990), J. Mol. Biol. 215:
403–410; und Altschul et al., (1997),
Nucleic Acids Res. 25: 3389–3402), identifiziert.
Das Programm wird zum Auffinden von Regionen mit lokaler Ähnlichkeit
zwischen Sequenzen verwendet, und zwar durch Vergleichen von Nukleinsäure-
oder Polypeptidsequenzen mit Sequenz-Datenbanken und durch Berechnen
der statistischen Signifikanz der Übereinstimmungen. Das
Polypeptid, das durch die erfindungsgemäße Nukleinsäuresequenz
kodiert wird, wurde zum Beispiel für den TBLASTN-Algorithmus
verwendet, und zwar unter Ausschaltung der Default-Einstellungen
und des Filters für die Nichtmiteinbeziehung von Sequenzen
mit niedriger Komplexität. Der Output der Analyse wurde
mittels paarweisem Vergleich betrachtet und anhand der Wahrscheinlichkeits-Wertung
(E-Wert) in eine Reihenfolge gebracht, wobei die Wertung die Wahrscheinlichkeit
widerspiegelt, dass ein bestimmtes Alignment zufällig vorkommt
(je niedriger der E-Wert, desto signifikanter der Hit). Zusätzlich
zu den E-Werten wurden die Vergleiche auch anhand der Prozent Identität
gewertet. Prozent Identität bezieht sich auf die Anzahl
identischer Nukleotide (oder Aminosäuren) zwischen den
zwei verglichenen Nukleinsäure-(oder Polypeptid-)sequenzen über
eine bestimmte Länge. In einigen Fällen können
die Default-Parameter angepasst werden, um die Stringenz der Suche
zu modifizieren. Zum Beispiel kann man den E-Wert erhöhen,
so dass weniger stringente Übereinstimmungen angezeigt
werden. Auf diese Weise lassen sich kurze, fast genaue Übereinstimmungen
identifizieren.
-
Tabelle
D1 zeigt eine Auflistung von Nukleinsäuresequenzen, die
mit der bei den erfindungsgemäßen Verfahren verwendeten
PHDf-HD-Nukleinsäuresequenz verwandt sind. Tabelle
D1: Beispiele für PHDf-HD-Polypeptide:
-
Beispiel 30: Alignment von PHDf-HD-Polypeptidsequenzen
-
Die
Universität Potsdam, Deutschland, hat eine Datenbank von
Pflanzen-Transkriptionsfaktoren, einschließlich derjenigen
von Oryza sativa mit der Bezeichnung riceTFDB, erstellt. Sämtliche
Polypeptidsequenzen, die den zur HD-Familie gehörenden
Transkriptionsfaktoren entsprechen (bisher 120 identifizierte Genmodelle
(91 Loci)) wurden einschließlich der beiden bisher identifizierten
PHDf-HD-Polypeptide (Os02g05450.1 und Os06g12400.1) heruntergeladen.
Die Polypeptidsequenzen in Tabelle D1 der vorliegenden Anmeldung (bei
Volllänge, d. h. 21 Polypeptidsequenzen) wurden zu dem
Satz der HD-Familie hinzugefügt.
-
Das
Alignment aller Polypeptidsequenzen erfolgte mit dem Clustal Algorithmus
(1.83) für progressives Alignment unter Verwendung der
Default-Werte (Thompson et al. (1997) Nucleic Acids Res
25: 4876–4882; Chenna et al. (2003). Nucleic
Acids Res 31: 3497–3500). Anschließend
wurde ein Neighbour-Joining-Tree konstruiert, der in 13 der vorliegenden Anmeldung dargestellt ist.
Die interessierende Gruppe, welche die beiden Paraloge aus Reis
(Os02g05450.1 und Os06g12400.1) umfasst, ist eingekreist. Jedes
Polypeptid, das (nach einem Mehrfach-Alignment-Schritt, wie hier
vorstehend beschrieben) in diese HD-Gruppe fällt, wird
als geeignet für die Durchführung der erfindungsgemäßen
Verfahren, wie hier beschrieben, erachtet.
-
In
einem mehrfachen Sequenz-Alignment der Volllangen-PHDf-HD-Polypeptide
in Tabelle D1 kann eine Reihe von Merkmalen identifiziert werden,
die in 17 markiert sind. Vom N-Terminus
zum C-Terminus der Polypeptide findet man: (i) ein vorhergesagtes
Kern-Lokalisierungssignal (NLS); (ii) einen Leucin-Zipper (ZIP)
mit vier Heptaden (im Kasten, worin in der Regel ein Leucin (gelegentlich
ein Isoleucin, ein Valin oder ein Methionin) als jede siebte Aminosäure
auftritt); (iii) einen PHD-Finger (PHDf) mit dem typischen C4HC3
(vier Cysteine, ein Histidin, drei Cysteine) mit charakteristischem
Cystein-Abstand; (iv) eine saure Abfolge (die reich an den sauren
Aminosäuren D und E ist); (v) basische Abfolgen (die reich
an den basischen Aminosäuren K und R sind); (vi) eine Homöodomäne
(HD).
-
Beispiel 31: Berechnung der Gesamtprozent
an Identität zwischen Polypeptidsequenzen, die für
die Durchführung der erfindungsgemäßen
Verfahren geeignet sind
-
Die
Gesamtprozent an Ähnlichkeit und Identität zwischen
Volllängenpolypeptidsequenzen, die bei der Durchführung
der erfindungsgemäßen Verfahren geeignet sind,
wurden unter Verwendung von einem der im Stand der Technik verfügbaren
Verfahren, nämlich der MatGAT (Matrix Global Alignment
Tool) Software (BMC Bioinformatics. 2003 4: 29. MatGAT:
an application that generstes similarity/identity matrices using
Protein or DNA sequences. Campanella JJ, Bitincka L, Smalley J;
von Ledion Bitincka gehostete Software), bestimmt. Die
MatGAT-Software erzeugt Ähnlichkeits-/Identitätsmatrizes
für DNA- oder Proteinsequenzen, ohne dass ein vorheriges
Alignment der Daten erforderlich ist. Das Programm führt
eine Reihe von paarweisen Alignments unter Verwendung des globalen
Alignment-Algorithmus von Myers und Miller durch (gap opening penalty
12, gap extension penalty 2), berechnet die Ähnlichkeit
und die Identität unter Verwendung von z. B. Blosum 62 (für
Polypeptide) und platziert dann die Ergebnisse in einer Abstandsmatrix.
Die Sequenzähnlichkeit ist in der unteren Hälfte
der Trennlinie und die Sequenzidentität in der oberen Hälfte
der diagonalen Trennlinie dargestellt.
-
Bei
den in dem Vergleich eingesetzten Parametern handelte es sich um:
| Scoring
matrix: | Blosum
62 |
| First
Gap: | 12 |
| Extending
gap: | 2 |
-
Die
Ergebnisse der Software-Analyse sind in Tabelle D2 für
die Gesamt-Ähnlichkeit und -Identität über die
volle Länge der Polypeptidsequenzen dargestellt (wobei
die partiellen Polypeptidsequenzen ausgeschlossen wurden). Die Prozent
Identität sind oberhalb der Diagonalen und die Prozent Ähnlichkeit
sind unterhalb der Diagonalen angegeben.
-
Die
Prozent Identität zwischen den PHDf-HD-Polypeptidsequenzen,
die für die Durchführung der erfindungsgemäßen
Verfahren geeignet sind, kann nur 15% Aminosäure-Identität
im Vergleich zu SEQ ID NO: 180 betragen. Tabelle
D2: MatGAT-Ergebnisse für Gesamt-Ähnlichkeit und
-Identität über die volle Länge der Polypeptidsequenzen.
-
Die
Prozent Identität können erheblich erhöht
werden, wenn man die Identitätsberechnung zwischen der
konservierten ZIP/PHDf-Domäne (konservierte Leucin-Zipper/Pflanzen-Homöodomäne-Finger-Domäne, die
die Zip- und die PHDf-Domäne enthält) der SEQ
ID NO: 180 (wie durch SEQ ID NO: 233 dargestellt) und der konservierten
ZIP/PHDf-Domäne der Polypeptide, die für die Durchführung
der Erfindung geeignet sind, durchführt. Die konservierte
ZIP/PHDf der SEQ ID NO: 233 ist insgesamt 180 aufeinander folgende
Aminosäuren lang. Die Prozent Identität über
die konservierte ZIP/PHDf-Domäne zwischen den Polypeptidsequenzen, die
zur Durchführung der erfindungsgemäßen
Verfahren geeignet sind, reicht von 30% bis 75% Aminosäure-Identität,
wie aus Tabelle D3 zu entnehmen ist. Tabelle
D3: MatGAT-Ergebnisse für die Gesamt-Ähnlichkeit
und -Identität zwischen den Polypeptidsequenzen über
die konservierte ZIP/PHDf-Domäne.
-
Die
Prozent Identität können auch zwischen der konservierten
HD der SEQ ID NO: 180 (wie in SEQ ID NO: 234 dargestellt) und der
konservierten HD der Polypeptide, die für die Durchführung
der Erfindung geeignet sind, berechnet werden. Die Prozent Identität über
die konservierte HD zwischen den Polypeptidsequenzen, die zur Durchführung
der erfindungsgemäßen Verfahren geeignet sind,
reicht von 25% bis 70% Aminosäure-Identität, wie
aus Tabelle D4 zu entnehmen ist. Tabelle
D4: MatGAT-Ergebnisse für die Gesamt-Ähnlichkeit
und -Identität zwischen den Polypeptidsequenzen über
die konservierte HD.
-
Beispiel 32: Identifikation von Domänen
in Polypeptidsequenzen, die für die Durchführung
der erfindungsgemäßen Verfahren geeignet sind
-
Bei
der Integrated Resource of Protein Families, Domains and Sites-(InterPro-)Datenbank
handelt es sich um eine integrierte Plattform für die häufig
verwendeten Signatur-Datenbanken für Suchen auf Text- und Sequenzbasis.
Die InterPro-Datenbank kombiniert diese Datenbanken, in denen unterschiedliche
Methodiken und verschiedene Mengen an biologischer Information über
gut charakterisierte Proteine verwendet werden, um Proteinsignaturen
abzuleiten. Zu den angeschlossenen Datenbanken zählen SWISS-PROT,
PROSITS, TrEMBL, PRINTS, ProDom und Pfam, Smart und TIGRFAMs. Interpro
wird am European Bioinformatics Institute im Vereinigten Königreich
gehostet.
-
Die
Ergebnisse aus dem InterPro-Scan der Polypeptidsequenz gemäß SEQ
ID NO: 180 sind in Tabelle D5 dargestellt. Tabelle D5: InterPro-Scan-Ergebnisse für
die Polypeptidsequenz gemäß SEQ ID NO: 180
| InterPro-Zugangsnummer | Name
in der integrierten Datenbank | Zugangsnummer in
der integrierten Datenbank | Zugangsname
in der integrierten Datenbank | Aminosäure-koordinaten
auf SEQ ID NO: 180 |
| IPR0013556
Homeobox | ProDom | PD000010 | PRH_ARATH_P48785 | 439–497 |
| IPR0013556
Homeobox | PFAM | PF00046 | Homeobox | 439–495 |
| IPR0013556
Homeobox | SMART | SM00389 | HOX | 438–500 |
| IPR0013556
Homeobox | Profile | PS50071 | Homeobox_2 | 436–496 |
| IPR00195
Zinc-finger, PHDtype | PFAM | PF00628 | PHD | 197–251 |
| IPR00195
Zinc-finger, PHD-type | SMART | SM00249 | PHD | 197–249 |
| IPR00195
Zinc-finger, PHD-type | Profile | PS50016 | ZF_PHD_2 | 195–251 |
| IPR012287
Homeodomainrelated | Gene3D | G3DSA:1.10.10.60 | keine
Beschreibung | 436–501 |
| IPR013256
Chromatin SPT2 | SMART | SM00784 | keine
Beschreibung | 78–154 |
| Untegrated
IPR | PANTHER | PTHR19418 | Homeobox
protein | 397–414
431–590 |
-
Die
Ergebnisse des InterPro-Scans identifizieren deutlich die wesentlichen
Merkmale eines PHDf-HD-Polypeptids, d. h. einen PHDf-Zinkfinger
und eine Homöobox, wie zum Beispiel an den Pfam-Einträgen
PF00628 beziehungsweise PF00046 zu erkennen ist.
-
Bekanntlich
enthalten HDs kanonische Reste. Die HD von PHDf-HD-Polypeptiden
zeigt sogar an den Positionen, die unter den Homöodomänen
nahezu unveränderlich sind, große Sequenzunterschiede.
Das Sequenz-Logo der HD der PHDf-HD-Polypeptide in Tabelle D1 ist
in 15 dargestellt. Sequenz-Logos sind eine graphische
Darstellung eines Alignments mehrerer Aminosäure- oder
Nukleinsäuresequenzen. Jedes Logo besteht aus Stapeln von
Symbolen mit jeweils einem Stapel für jede Position in
der Sequenz. Die Gesamthöhe des Stapels zeigt die Sequenzkonservierung
an dieser Position an, wohingegen die Höhe der Symbole
in dem Stapel die relative Häufigkeit jeder Amino- oder
Nukleinsäure an dieser Position anzeigt. Im Allgemeinen
liefert ein Logo eine reichhaltigere und genauere Beschreibung zum
Beispiel einer Bindungsstelle als eine Konsensussequenz. Der Algorithmus (WebLogo),
der zum Erzeugen solcher Logos verwendet wird” ist auf
dem Server der Universität of California, Berkeley, verfügbar.
Die HD gemäß SEQ ID NO: 234, die in SEQ ID NO:
180 enthalten ist, stimmt mit dem in 15 dargestellten
Sequenz-Logo überein. Polypeptide, die für die
Durchführung der erfindungsgemäßen Verfahren
geeignet sind, umfassen eine HD, die das in 15 dargestellte Sequenz-Logo
umfasst.
-
Beispiel 33: Vorhersage von Sekundärstrukturmerkmalen
der Polypeptidsequenzen, die für die Durchführung der
erfindungsgemäßen Verfahren geeignet sind
-
Coiled
Coils enthalten gewöhnlich ein Muster aus sieben wiederholten
Aminosäuren, das man als Heptad-Wiederholung bezeichnet.
Coiled Coils sind wichtig für die Identifikation von Protein-Protein-Wechselwirkungen,
wie Oligomerisierung, entweder von identischen Proteinen, Proteinen
aus derselben Familie oder von nicht miteinander verwandten Proteinen.
Vor kurzem wurden große Fortschritte bei der Computer-Vorhersage
von Coiled Coils aus Sequenzdaten gemacht. Unter den ExPASy Proteomics-Werkzeugen
sind viele Algorithmen verfügbar, die dem Fachmann bekannt
sind. Eines unter ihnen, COILS, ist ein Programm, das eine Sequenz
mit einer Datenbank bekannter paralleler zweisträngiger
Coiled Coils vergleicht und einen Ähnlichkeitswert ableitet.
Durch Vergleichen dieses Werts mit der Verteilung der Bewertungen
in globulären und Coiled Coil-Proteinen berechnet das Programm
die Wahrscheinlichkeit, dass die Sequenz eine Coiled Coil-Konformation
annimmt.
-
Für
das PHDf-HD-Polypeptid gemäß SEQ ID NO: 180 werden
zwei N-terminale Coiled Coil-Domänen mit einer hohen Wahrscheinlichkeit
in allen drei untersuchten Fenstern (14, 21 and 28) vorhergesagt.
In Tabelle D6 sind die Koordinaten der Reste, die Reste, die drei
Fenster und die entsprechenden Wahrscheinlichkeitswerte dargestellt.
16 zeigt die graphische Ausgabe des COILS-Algorithmus
an dem Polypeptid gemäß SEQ ID NO: 180, wobei
die beiden vorhergesagten Coiled Coils in der N-terminalen Hälfte
des Polypeptids in allen drei Fenstern (durch die drei Linien dargestellt)
deutlich sichtbar sind. Tabelle D6: Numerische Ausgabe des COILS-Algorithmus
an dem Polypeptid gemäß SEQ ID NO: 180. Die Koordinaten
der Reste (#), die Reste, die drei Fenster und die entsprechenden
Wahrscheinlichkeitswerte sind dargestellt. Wahrscheinlichkeiten über
0,09 sind grau dargestellt.
| # | Rest | Fenster
= 14 | Wahr | Fenster
= 21 | Wahr | Fenster
= 28 | Wahr |
| 87 | P | d | 0,001 | e | 0,001 | E | 0,001 |
| 88 | T | e | 0,063 | f | 0,274 | F | 0,099 |
| 89 | R | f | 0,063 | g | 0,567 | G | 0,099 |
| 90 | R | g | 0,063 | a | 0,567 | A | 0,099 |
| 91 | K | b | 0,066 | b | 0,642 | B | 0,188 |
| 92 | H | c | 0,066 | c | 0,642 | C | 0,188 |
| 93 | K | d | 0,069 | d | 0,642 | D | 0,591 |
| 94 | Q | e | 0,178 | e | 0,642 | E | 0,672 |
| 95 | K | f | 0,335 | f | 0,642 | F | 0,672 |
| 96 | R | g | 0,335 | g | 0,642 | G | 0,772 |
| 97 | K | a | 0,335 | a | 0,642 | A | 0,772 |
| 98 | N | b | 0,347 | b | 0,642 | B | 0,772 |
| 99 | D | c | 0,347 | c | 0,642 | C | 0,772 |
| 100 | E | d | 0,347 | d | 0,642 | D | 0,772 |
| 101 | S | e | 0,347 | e | 0,642 | E | 0,808 |
| 102 | D | f | 0,347 | f | 0,642 | F | 0,808 |
| 103 | E | g | 0,347 | g | 0,642 | g | 0,808 |
| 104 | V | a | 0,347 | a | 0,642 | a | 0,808 |
| 105 | S | b | 0,347 | b | 0,642 | b | 0,808 |
| 106 | R | c | 0,347 | c | 0,642 | c | 0,808 |
| 107 | M | d | 0,347 | d | 0,642 | d | 0,808 |
| 108 | E | e | 0,347 | e | 0,642 | e | 0,808 |
| 109 | K | f | 0,347 | f | 0,642 | F | 0,808 |
| 110 | R | g | 0,347 | g | 0,642 | g | 0,808 |
| 111 | A | a | 0,347 | a | 0,642 | a | 0,808 |
| 112 | R | b | 0,307 | b | 0,514 | b | 0,808 |
| 113 | Y | c | 0,066 | c | 0,503 | c | 0,808 |
| 114 | L | d | 0,262 | d | 0,503 | d | 0,808 |
| 115 | L | e | 0,262 | e | 0,503 | e | 0,808 |
| 116 | I | f | 0,262 | f | 0,503 | F | 0,808 |
| 117 | K | g | 0,262 | g | 0,503 | g | 0,808 |
| 118 | I | a | 0,262 | a | 0,503 | a | 0,808 |
| 119 | K | b | 0,262 | b | 0,503 | b | 0,808 |
| 120 | Q | c | 0,262 | c | 0,503 | c | 0,808 |
| 121 | E | d | 0,262 | d | 0,503 | d | 0,808 |
| 122 | Q | e | 0,262 | e | 0,503 | e | 0,808 |
| 123 | N | f | 0,262 | f | 0,503 | F | 0,808 |
| 124 | L | g | 0,262 | g | 0,434 | g | 0,808 |
| 125 | L | a | 0,262 | a | 0,434 | a | 0,808 |
| 126 | D | b | 0,262 | b | 0,434 | b | 0,808 |
| 127 | A | c | 0,262 | c | 0,304 | c | 0,808 |
| 128 | Y | d | 0,145 | d | 0,304 | d | 0,808 |
| 129 | S | e | 0,145 | e | 0,044 | e | 0,808 |
| 130 | G | f | 0,119 | f | 0,009 | F | 0,367 |
| 131 | D | g | 0,078 | g | 0,007 | g | 0,136 |
| 132 | G | a | 0,002 | a | 0,001 | a | 0,005 |
| 133 | W | b | 0 | a | 0,001 | a | 0,001 |
| 134 | N | b | 0,001 | b | 0,045 | b | 0,057 |
| 135 | G | c | 0,001 | c | 0,045 | c | 0,057 |
| 136 | H | d | 0,003 | d | 0,07 | d | 0,219 |
| 137 | S | e | 0,008 | e | 0,132 | e | 0,231 |
| 138 | R | f | 0,025 | f | 0,357 | F | 0,231 |
| 139 | E | g | 0,025 | g | 0,357 | g | 0,231 |
| 140 | K | a | 0,025 | a | 0,357 | a | 0,231 |
| 141 | I | b | 0,028 | b | 0,357 | b | 0,231 |
| 142 | K | c | 0,07 | c | 0,357 | c | 0,462 |
| 143 | P | d | 0,07 | d | 0,357 | d | 0,462 |
| 144 | E | e | 0,998 | e | 0,997 | e | 0,974 |
| 145 | K | f | 0,998 | f | 0,997 | f | 0,974 |
| 146 | E | g | 0,998 | g | 0,997 | g | 0,974 |
| 147 | L | a | 0,998 | a | 0,997 | a | 0,974 |
| 148 | Q | b | 0,998 | b | 0,997 | b | 0,974 |
| 149 | R | c | 0,998 | c | 0,997 | c | 0,974 |
| 150 | A | d | 0,998 | d | 0,997 | d | 0,974 |
| 151 | K | e | 0,998 | e | 0,997 | e | 0,974 |
| 152 | K | f | 0,998 | f | 0,997 | f | 0,974 |
| 153 | Q | g | 0,998 | g | 0,997 | g | 0,974 |
| 154 | I | a | 0,998 | a | 0,997 | a | 0,974 |
| 155 | M | b | 0,998 | b | 0,997 | b | 0,974 |
| 156 | K | c | 0,998 | c | 0,997 | c | 0,974 |
| 157 | Y | d | 0,998 | d | 0,997 | d | 0,974 |
| 158 | K | e | 0,989 | e | 0,997 | e | 0,974 |
| 159 | I | f | 0,896 | f | 0,997 | f | 0,974 |
| 160 | A | g | 0,486 | g | 0,997 | g | 0,974 |
| 161 | I | a | 0,409 | a | 0,997 | a | 0,974 |
| 162 | R | b | 0,274 | b | 0,997 | b | 0,974 |
| 163 | D | c | 0,255 | c | 0,997 | c | 0,974 |
| 164 | V | d | 0,095 | d | 0,997 | d | 0,974 |
| 165 | I | e | 0,025 | e | 0,871 | e | 0,974 |
| 166 | H | f | 0,025 | f | 0,622 | f | 0,974 |
| 167 | Q | g | 0,025 | g | 0,496 | g | 0,974 |
| 168 | L | a | 0,025 | a | 0,496 | a | 0,974 |
| 169 | D | b | 0,025 | b | 0,468 | b | 0,974 |
| 170 | L | c | 0,005 | c | 0,111 | c | 0,974 |
| 171 | C | d | 0,002 | d | 0,02 | d | 0,974 |
| 172 | S | e | 0,001 | e | 0,007 | e | 0,719 |
| 173 | S | f | 0,001 | f | 0,003 | f | 0,482 |
| 174 | S | g | 0,001 | g | 0,001 | g | 0,066 |
| 175 | G | a | 0 | a | | a | 0,001 |
-
Ein
weiteres Coiled Coil wird in der C-terminalen Hälfte des
PHDf-HD-Polypeptids gemäß SEQ ID NO: 180 mit einer
kleineren Wahrscheinlichkeit als für die zwei Coiled Coil-Domänen
in der N-terminalen Hälfte des Polypeptids vorhergesagt.
-
Beispiel 34: Subzelluläre Lokalisierungsvorhersage
der Polypeptid-Sequenzen, die bei der Durchführung der erfindungsgemäßen
Verfahren geeignet sind.
-
LOCtree
ist ein Algorithmus, mit dem man die subzelluläre Lokalisierung
und die DNA-Bindungsneigung von Nichtmembranproteinen in nichtpflanzlichen
und pflanzlichen Eukaryoten sowie Prokaryoten vorhersagen kann.
LOCtree klassifiziert eukaryotische Tierproteine in eine von 5 Subzellklassen,
wohingegen pflanzliche Proteine in eine von 6 Klassen und prokaryotische
Proteine in eine von 3 Klassen unterteilt werden.
-
LOCtree
beschreibt sofern vorhanden auch Vorhersagen, die sich auf folgendes
gründen: 1) Kernlokalisierungssignale, die mit dem PredictNLS-Algorithmus
ermittelt werden, 2) eine Lokalisierung, die über die in
dem Protein befindlichen Prosite-Motiven und Pfam-Domänen
hergeleitet wird, und 3) die mit einem Protein einhergehenden SWISS-PROT- Schlüsselwörter.
Die Lokalisierung wird in den letzten beiden Fällen mit
dem LOCkey-Algorthmus auf Entropie-Basis hergeleitet. Die Software
wird an der Universität von Columbia, USA, gehostet. Vorhersage der subzellulären
Lokalisierung eines PHDf-HD-Polypeptids nach SEQ ID NO: 180 auf
Motiv- und Schlüsselwort-Basis mittels LOCkey:
| Vorhergesagte
Lokalisierung | Vertrauen | Alternative
Vorhersage | SWISS-PROT
Schlüsselwörter die für die Bestimmung
der Lokalisierung |
| Kern | 100 | - | verwendet
wurden Homöobox, DNA-Bindung, Transkriptionsregulation,
Kern-Protein, Transkription, Zinkfinger |
-
Die
Vorhersage eines Kernlokalisierungssignals (NLS) erfolgt beispielsweise
mit dem PredictNLS-Algorithmus. Der Algorithmus wird ebenfalls auf
dem Server an der Universität von Columbia, USA, gehostet.
In der nachstehenden Tabelle ist die Vorhersage des NLS auf dem
Polypeptid von SEQ ID NO: 180 mit dem PredictNLS-Algorithmus, gezeigt.
| Generalisiertes
Motiv ([KR]{3,5} zwischen 3- und 5-fach K oder R) | Hit
auf das Polypeptid von SEQ ID NO: 180 | Koordinaten
auf dem Polypeptid von SEQ ID NO: 180 |
| K[RK]{3,5}×{11,18}[RK]K×{2,3}K | KRRRGSDAATGKSATGPTRRKHKQK | 71–95 |
| [KR]{4}×{20,24}K{1,4}×K | KRRRGSDAATGKSATGPTRRKHKQKRK | 71–97 |
-
In
der nachstehend gezeigten Polypeptidsequenz (SEQ ID NO: 180), ist
die Position des vorhergesagten NLS fett und doppelt unterstrichen
gezeigt:
-
Zur
Durchführung solcher Analysen können viele andere
Algorithmen verwendet werden, wie u. a.:
- • ChloroP
1.1, das auf dem Server der Technischen Universität Dänemark
gehostet wird;
- • Protein Prowler Subcellular Localisation Predictor
Version 1.2, das auf dem Server des Instituts für Molekulare
Biowissenschaft, Universität Queensland, Brisbane, Australien,
gehostet wird;
- • PENCE Proteome Analyst PA-GOSUB 2.5, das auf dem
Server der Universität von Alberts, Edmonton, Alberts,
Canada gehostet wird;
- • TMHMM, das auf dem Server der Technischen Universität
Dänemark gehostet wird.
-
Beispiel 35: Klonierung der Nukleinsäuresequenz
nach SEQ ID NO: 179
-
Falls
nicht anders erwähnt werden die DNA-Rekombinationstechniken
nach den Standardprotokollen durchgeführt, die in den folgenden
Veröffentlichungen beschrieben sind: (Sambrook
(2001), Molecular Cloning: a laboratory manual, 3. Ausgabe, Cold
Spring Harbor Laboratory Press, CSH, New York) oder in Band
1 und 2 von Ausubel et al., (1994), Current Protocols in Molecular
Biology, Current Protocols. Standardmaterialien und -verfahren für
molekularbiologische Arbeiten an Pflanzen sind beschrieben in Plant
Molecular Biology Labfax (1993), von R. D. D. Croy, verlegt bei
BIOS Scientific Publications Ltd (UK) und Blackwell Scientific Publications
(UK).
-
Das
PHDf-HD-Gen aus Oryza sativa wurde mittels PCR unter Verwendung
einer Reis-cDNA-Bank als Matrize, die aus mRNA, extrahiert aus gemischten
Pflanzengeweben, synthetisiert wurde. Für die PCR-Amplifikation
wurden die Primer prm09687 (SEQ ID NO: 236; Sense: 5'-ggggacaagtttgtacaaaaaagcaggcttaaacaatgaataccccagaaaagaaaa-3')
und Primer prm09688 SEQ ID NO: 237; revers, komplementär: 5'-ggggaccactttgtacaagaaagctgggtgatgcaaggttaagatgcttt-3'),
die die AttB-Stellen für die Gateway-Rekombination beinhalten,
verwendet. Die PCR erfolgte mittels Hifi-Taq-DNA-Polymerase unter
Standard-Bedingungen. Ein PCR-Fragment mit der erwarteten Größe
(einschließlich AttB-Spaltstellen) wurde ebenfalls unter
Verwendung von Standardmethoden amplifiziert und aufgereinigt. Anschließend
wurde der erste Schritt des Gateway-Verfahrens, die BP-Reaktion,
durchgeführt, bei der das PCR-Fragment in vivo mit dem
Plasmid pDONR201 unter Bildung eines – gemäß Gateway-Teminologie – ”entry
clone” rekombinierte. Das Plasmid pDONR201 wurde käuflich
von Invitrogen als Bestandteil der Gateway® Technik
erworben.
-
Beispiel 36: Expressionsvektorkonstruktion
unter Verwendung der Nukleinsäuresequenz gemäß SEQ
ID NO: 179
-
Der
die SEQ ID NO: 179 enthaltende ”entry clone” wurde
anschließend in einer LR-Reaktion mit einem Zielvektor,
der für die Transformation von Oryza sativa verwendet wird,
verwendet. Dieser Vektor enthält als funktionelle Elemente
innerhalb der T-DNA-Grenzen Folgendes: Einen pflanzlichen Selektionsmarker;
eine screenbare Markerexpressionskassette, und eine Gateway-Kassette,
die für die LR-in-vivo-Rekombination mit der interessierenden
Nukleinsäuresequenz, die bereits in den ”entry
clone” kloniert wurde, vorgesehen ist. Ein GOS2-Promotor
(SEQ ID NO: 235) aus Reis zur konstitutiven Expression befand sich
stromaufwärts dieser Gateway-Kassette.
-
Nach
dem LR-Rekombinationsschritt wurde der erhaltene Expressionsvektor
pGOS2::PHDf-HD (18) nach Verfahren des Standes
der Technik in den Agrobacterium-Stamm LBA4044 transformiert.
-
Beispiel 37: Pflanzentransformation
-
Reistransformation
-
Agrobacterium,
das den Expressionsvektor enthielt, wurde unabhängig zur
Transformation von Oryza-sativa-Pflanzen verwendet. Reife trockene
Samen der japonica-Reissorte Nipponbare wurden entspelzt. Die Sterilisation
erfolgte durch einminütiges Inkubieren in 70%igem Ethanol
und anschließend 30 Minuten in 0,2% HgCl2,
wonach 6 mal je 15 Minuten mit sterilem destilliertem Wasser gewaschen
wurde. Anschließend wurden die sterilen Samen auf einem
Medium, das 2,4-D (Kallusinduktionsmedium) enthielt zur Keimung
gebracht. Nach vierwöchiger Inkubation im Dunkeln wurden
embryogene, von Scutellum stammende Kalli herauspräpariert
und auf dem gleichen Medium vermehrt. Nach zwei Wochen wurden die
Kalli durch Subkultur auf dem gleichen Medium für weitere
2 Wochen vervielfacht oder vermehrt. Embryogene Kallusstückchen
wurden auf frischem Medium 3 Tage vor der Cokultivierung subkultiviert
(um die Zellteilungsaktivität zu fördern).
-
Für
die Cokultivierung verwendete man unabhängig den Agrobacterium-Stamm
LBA4404, der jeden einzelnen Expressionsvektor enthielt. Agrobacterium
wurde auf AB-Medium mit den entsprechenden Antibiotika überimpft
und 3 Tage bei 28°C kultiviert. Anschließend wurden
die Bakterien gewonnen und bis zum Erreichen einer Dichte (OD600) von ungefähr 1 in flüssigem
Cokultivierungsmedium suspendiert. Anschließend wurde die
Suspension in eine Petrischale überführt, und
die Kalli wurden 15 Minuten in der Suspension eingetaucht. Dann
wurden die Kallusgewebe auf einem Papierfilter trocken getupft und
auf ein verfestigtes Cokultivierungsmedium umgesetzt und 3 Tage
im Dunkeln bei 25°C inkubiert. Die cokultivierten Kalli
wurden 4 Wochen im Dunkeln bei 28°C in Gegenwart eines
Selektionsmittels auf 2,4-D-haltigem Medium herangezogen. Während
dieses Zeitraums entwickelten sich rasch wachsende resistente Kallusinseln.
Nach dem Umsetzen dieses Materials auf ein Regenerationsmedium und
Inkubation im Hellen wurde das embryogene Potential freigesetzt,
und in den nächsten 4 bis 5 Wochen entwickelten sich Sprosse.
Die Sprosse wurden von den Kalli herauspräpariert und 2
bis 3 Wochen lang auf auxinhaltigem Medium inkubiert, von dem sie
in Erde umgesetzt wurden. Abgehärtete Sprosse wurden im
Gewächshaus unter hoher Feuchtigkeit und im Kurztag herangezogen.
-
Pro
Konstrukt wurden ungefähr 35 unabhängige T0-Reistransformanten
erzeugt. Die Primärtransformanten wurden von einer Gewebekulturkammer
in ein Gewächshaus umgesetzt. Nach einer quantitativen PCR-Analyse
zur Überprüfung der Kopienzahl des T-DNA-Inserts
wurden nur transgene Ein-Kopien-Pflanzen mit Toleranz für
die Selektionsmittel zurückbehalten, um T1-Samen zu ernten.
Die Samen wurden dann 3 bis 5 Monate nach dem Umsetzen geerntet.
Das Verfahren ergab Ein-Locus-Transformanten mit einer Rate von über
50% (Aldemita und Hodges 1996, Chan et al., 1993, Hiei et
al., 1994).
-
Beispiel 38: Verfahren bei der phänotypischen
Untersuchung
-
38.1 Aufbau der Untersuchung
-
Es
wurden ungefähr 35 unabhängige T0-Reistransformanten
erzeugt. Die Primärtransformanten wurden für das
Heranziehen und Ernten von T1-Samen von einer Gewebekulturkammer
in ein Gewächshaus umgestellt. Sechs Ereignisse, von denen
sich die T1-Nachkommenschaft im Verhältnis 3:1 für
Vorliegen/Abwesenheit des Transgens aufspaltete, wurden behalten.
Für jedes dieser Ereignisse wurden ungefähr 10 T1-Keimpflanzen,
die das Transgen enthielten (Hetero- und Homozygoten) und ungefähr
10 T1-Keimpflanzen, denen das Transgen fehlte (Nullizygoten) durch
Beobachten der visuellen Markerexpression selektiert. Die transgenen
Pflanzen und die entsprechenden Nullizygoten wurden nebeneinander
in zufälliger Anordnung herangezogen. Die Gewächshausbedingungen
waren kurze Tage (12 Stunden Licht), 28°C im Hellen und
22°C im Dunkeln, und eine relative Feuchtigkeit von 70%.
Die unter Nichtstressbedingungen gezogenen Pflanzen werden in regelmäßigen
Abständen mit Wasser versorgt, so dass gewährleistet
war, dass Wasser und Nährstoffe nicht limitierend waren,
damit die Bedürfnisse der Pflanze, zu wachsen und sich
zu entwickeln, erfüllt werden.
-
Vom
Sästadium bis zum Reifestadium wurden die Pflanzen mehrmals
in eine Digital-Imaging-Kammer gestellt. Zu jedem Zeitpunkt wurden
von jeder Pflanze aus mindestens 6 verschiedenen Winkeln Digitalbilder (2048×1536
Pixel, 16 Millionen Farben) aufgenommen.
-
38.2 Statistische Analyse: F-Test
-
Als
statistisches Modell für die Gesamtuntersuchung der phänotypischen
Eigenschaften der Pflanze wurde eine zweifaktorielle ANOVA (Varianzanalyse)
verwendet. Mit allen Parametern, die bei allen Pflanzen von allen
Ereignissen, die mit dem erfindungsgemäßen Gen
transformiert wurden, bestimmt wurden, wurde ein F-Test durchgeführt.
Der F-Test erfolgte als Überprüfung auf einen
Effekt des Gens über alle Transformations-Ereignisse und
zur Feststellung eines Gesamteffekts des Gens, der auch als globaler
Geneffekt bezeichnet wird. Die Signifikanzschwelle für
einen echten globalen Geneffekt wurde bei dem F-Test auf dem 5%-Wahrscheinlichkeitsniveau
festgelegt. Ein signifikanter F-Test-Wert weist auf einen Geneffekt
hin, was bedeutet, dass mehr als nur das einfache Vorhandensein
oder die Lage des Gens die Unterschiede im Phänotyp verursacht.
-
38.3 Messparameter
-
Biomasseparameter-Messung
-
Vom
Sästadium bis zum Reifestadium wurden die Pflanzen mehrmals
in eine Digital-Imaging-Kammer gestellt. Zu jedem Zeitpunkt wurden
von jeder Pflanze aus mindestens 6 verschiedenen Winkeln Digitalbilder (2048×1536
Pixel, 16 Millionen Farben) aufgenommen.
-
Die
oberirdische Pflanzenfläche (oder Blattbiomasse) wurde
dadurch bestimmt, dass man die Gesamtanzahl Pixel der Digitalbilder
der vom Hintergrund unterschiedlichen oberirdischen Pflanzenteile
zählte. Dieser Wert wurde über die am selben Zeitpunkt
aus unterschiedlichen Winkeln aufgenommenen Bilder gemittelt und
mittels Kalibrierung in einen physikalischen Oberflächenwert,
ausgedrückt in Quadratmillimeter, umgewandelt. Versuche zeigen,
dass die auf diese Weise bestimmte oberirdische Pflanzenfläche
mit der Biomasse der oberirdischen Pflanzenteile korreliert. Die
oberirdische Fläche ist diejenige Fläche, die
zu dem Zeitpunkt, an dem die Pflanze ihre maximale Blattbiomasse
erreicht hatte, bestimmt wurde. Die Jungpflanzenvitalität
ist die oberirdische Fläche der Pflanze (Keimpflanze) 3
Wochen nach der Keimung. Die Steigerung der Wurzelbiomasse wird
als Steigerung der Gesamtwurzelbiomasse (gemessen als maximale Wurzelbiomasse, die
während der Lebensdauer einer Pflanze beobachtet wurde)
oder als Steigerung des Wurzel/Spross-Index (gemessen als Verhältnis
zwischen Wurzelmasse und Sprossmasse im Zeitraum des aktiven Wachstums
von Wurzel und Spross) ausgedrückt.
-
Samenparameter-Messungen
-
Die
reifen Primärrispen wurden geerntet, gezählt,
eingetütet, mit einem Strichcode versehen und dann drei
Tage bei 37°C in einem Ofen getrocknet. Dann wurden die
Rispen gedroschen, und alle Samen wurden gesammelt und gezählt.
Die gefüllten Spelzen wurden von den leeren Spelzen mittels
einer Luftblaseeinrichtung getrennt. Die leeren Spelzen wurden verworfen,
und die restliche Fraktion wurde nochmals gezählt. Die gefüllten
Spelzen wurden auf einer Analysewaage gewogen. Die Anzahl gefüllter
Samen wurde dadurch bestimmt, dass man die Anzahl der gefüllten
Spelzen, die nach dem Abtrennungsschritt verblieben, auszählte. Das
Gesamtsamengewicht wurde dadurch bestimmt, dass man alle von einer
Pflanze geernteten gefüllten Spelzen wog. Die Gesamtsamenzahl
pro Pflanze wurde dadurch bestimmt, dass man die von einer Pflanze geerntete
Anzahl Spelzen auszählte. Das Tausendkorngewicht (TKG)
wird aus der Anzahl der gezählten gefüllten Samen
und ihres Gesamtgewichts extrapoliert. Der Harvest Index (HI) wird
in der vorliegenden Erfindung als das Verhältnis zwischen
dem Gesamtsamengewicht pro Pflanze und der oberirdischen Fläche
(mm2), multipliziert mit einem Faktor 106, definiert. Die Gesamtzahl Blüten
pro Rispe wird in der vorliegenden Erfindung als das Verhältnis
zwischen der Gesamtsamenzahl und der Anzahl der reifen Primärrispen
definiert. Die Samenfüllungsrate wird in der vorliegenden
Erfindung als das Verhältnis (als Prozent ausgedrückt)
der Anzahl gefüllter Samen zu der Gesamtzahl Samen (oder
Blüten) definiert.
-
Screening auf reduzierte Nährstoff-(Stickstoff-)Verfügbarkeit
-
Reispflanzen
aus T2-Samen wurden in Blumenerde und mit Ausnahme der Nährlösung
unter normalen Bedingungen gezüchtet. Die Töpfe
wurden von der Umsetzung bis zur Reifung mit einer spezifischen
Nährlösung gewässert, die einen reduzierten
Stickstoff (N)-Gehalt, und zwar gewöhnlich 7 bis 8mal niedriger,
aufwies. Die Anzucht (Pflanzenreifung, Samenernte) entsprach ansonsten
derjenigen von Pflanzen, die nicht unter abiotischem Stress gezüchtet
werden. Wachstums- und Ertragsparameter wurden wie für
das Wachstum unter normalen Bedingungen beschrieben ausgewertet.
-
Beispiel 39: Ergebnisse der phänotypischen
Untersuchung transgener Reispflanzen
-
Die
Ergebnisse der Untersuchung transgener Reispflanzen, die die Nukleinsäuresequenz,
die ein PHDf-HD Polypeptid nach SEQ ID NO: 180 kodiert, unter der
Kontrolle des GOS2-Promotors für eine konstitutive Expression
exprimieren, sind nachstehend aufgeführt.
-
Es
kam zu einem signifikanten Anstieg der Anzahl Rispen, des Gesamtsamenertrags
pro Pflanze, der Gesamtanzahl gefüllter Samen, der Gesamtanzahl
Samen, des Tausendkorngewichts (TKG) und des Harvest Index der transgenen
Pflanzen im Vergleich zu den entsprechenden Nullizygoten (Kontrollen),
wie in Tabelle D7 gezeigt. Tabelle D7: Ergebnisse der Untersuchung
transgener Reispflanzen, die die Nukleinsäuresequenz, die
ein PHDf-HD Polypeptid nach SEQ ID NO: 180 kodiert, unter der Kontrolle
des GOS2-Promotors für konstitutive Expression exprimieren.
| | Durchschn.
% Anstieg in 3 Ereignissen in der T1 Generation |
| Anzahl
der ersten Rispen | 22% |
| Gesamtsamenertrag
pro Pflanze | 30% |
| Gesamtanzahl
gefüllter Samen | 24% |
| Gesamtsamenanzahl | 24% |
| TKG | 4% |
| Harvest
Index | 17% |
-
Beispiel 40: Ergebnisse der phänotypischen
Untersuchung transgener Reispflanzen, die unter reduzierter Nährstoffverfügbarkeit
gewachsen waren.
-
Die
Ergebnisse der Untersuchung transgener Reispflanzen, die die Nukleinsäuresequenz,
die ein PHDf-HD Polypeptid nach SEQ ID NO: 180 kodiert, unter der
Kontrolle des GOS2-Promotors für eine konstitutive Expression
exprimieren, und die unter reduzierter Nährstoffverfügbarkeit
gewachsen waren, sind nachstehend aufgeführt.
-
Es
kam zu einem signifikanten Anstieg der Biomasse, beim Auftreten
von Jungpflanzenvitalität, des Gesamtsamenertrags pro Pflanze,
der Gesamtanzahl gefüllter Samen, der Gesamtanzahl Samen,
des Tausendkorngewichts (TKG) und des Harvest Index der transgenen
Pflanzen im Vergleich zu den entsprechenden Nullizygoten (Kontrollen),
wie in Tabelle D8 gezeigt. Tabelle D8: Ergebnisse der Untersuchung
transgener Reispflanzen, die die Nukleinsäuresequenz, die
ein PHDf-HD Polypeptid nach SEQ ID NO: 180 kodiert, unter der Kontrolle
des GOS2-Promotors für konstitutive Expression exprimieren,
und die unter reduzierter Nährstoffverfügbarkeit
gewachsen waren.
| | durchschn.
Gesamt % Anstieg (6 Ereignisse in der T2-Generation) |
| Biomasse | 8% |
| Auftreten
von Vitalität | 20% |
| Gesamtsamenertrag
pro Pflanze | 17% |
| Gesamtanzahl
gefüllter Samen | 12% |
| Samenfüllungsrate | 3% |
| Gesamtanzahl
Samen | 9% |
| TKG | 5% |
| Harvest
Index | 9% |
-
Beispiel 41: Beispiele für die
Transformation anderer Kulturpflanzen
-
Die
Transformation von Mais, Weizen, Sojabohne, Raps/Canola-Raps, Luzerne,
und Baumwolle erfolgte wie in Beispiel 7 beschrieben
-
Beispiel 42: Beispiele für Screenings
bei abiotischem Stress
-
Screening unter Trockenheitsbedingungen
-
Pflanzen
von einer ausgewählten Anzahl Ereignisse wurden unter Normalbedingungen
in Blumenerde herangezogen, bis sie das Stadium des Rispenschiebens
erreichten. Anschließend wurden sie in ein ”trockenes” Areal
gestellt, wo keine Bewässerung erfolgte. Um den Bodenwassergehalt
(BWG) zu verfolgen, wurden Feuchtigkeitssonden in statistisch ausgewählte
Töpfe eingeführt. Sank der BWG unter gewisse Schwellenwerte,
wurden die Pflanzen automatisch erneut gegossen, bis wiederum ein
Normalgehalt erreicht wurde. Dann wurden die Pflanzen wiederum in
Normalbedingungen umgestellt. Ansonsten wurde wie bei den nicht
unter abiotischen Stressbedingungen herangezogenen Pflanzen kultiviert
(Pflanzenreife, Samenernte). Wachstums- und Ertragsparameter werden
wie für das Wachstum unter Normalbedingungen beschrieben
aufgezeichnet.
-
Salzstress-Screening
-
Die
Pflanzen werden auf einem Substrat aus Kokosfasern und Argex (Verhältnis
3 zu 1) herangezogen. Während der ersten zwei Wochen nach
dem Umsetzen der Pflänzchen in das Gewächshaus
wird eine normale Nährlösung verwendet. Nach den
ersten zwei Wochen wird die Nährlösung mit 25
mM Salz (NaCl) versetzt, bis die Pflanzen geerntet werden. Anschließend
wurden die Wachstums- und Ertragsparameter bestimmt, wie für
Wachstum unter normalen Bedingungen eingehend beschrieben.
-
VIII. bHLH11-ähnliches (basisches
Helix-Loop-Helix 11) Protein
-
Beispiel 43: Identifikation von Sequenzen,
die mit der in den erfindungsgemäßen Verfahren
verwendeten bHLH11-ähnlichen Nukleinsäuresequenz
verwandt sind
-
Unter
den Sequenzen in der Entrez Nucleotides Datenbank am National Center
for Biotechnology Information (NCBI) wurden unter Verwendung von
Datenbanksequenzabfragewerkzeugen, wie dem Basic Local Alignment
Tool (BLAST) (
Altschul et al., (1990), J. Mol. Biol. 215:
403–410; und
Altschul et al., (1997),
Nucleic Acids Res. 25: 3389–3402), (Volllängen-cDNA,
ESTs oder genomische) Sequenzen, die mit der in den erfindungsgemäßen
Verfahren verwendeten Nukleinsäuresequenz verwandt sind,
identifiziert. Das Programm wurde zum Auffinden von Regionen mit
lokaler Ähnlichkeit zwischen Sequenzen verwendet, und zwar
durch Vergleichen von Nukleinsäure- oder Polypeptid-Sequenzen
mit Sequenz-Datenbanken und durch Berechnen der statistischen Signifikanz
der Matches. Das durch die erfindungsgemäß verwendete
Nukleinsäure kodierte Polypeptid wurde beispielsweise für
den TBLASTN-Algorithmus verwendet, und zwar unter Ausschaltung der
Default-Einstellungen und des Filters für die Nichtmiteinbeziehung
von Sequenzen mit niedriger Komplexität. Der Output der
Analyse wurde mittels paarweisem Vergleich betrachtet und gemäß der
Wahrscheinlichkeits-Wertung (E-Wert) gereiht, wobei die Wertung
die Wahrscheinlichkeit, dass ein bestimmtes Alignment statistisch vorkommt,
widerspiegelt (je niedriger der E-Wert, desto signifikanter der
Hit). Zusätzlich zu den E-Werten wurden die Vergleiche
auch durch die Prozent der Identität gewertet. Die Prozent
der Identität bezieht sich auf die Anzahl identischer Nukleotide
(oder Aminosäuren) zwischen den zwei verglichenen Nukleinsäure-(oder
Polypeptid)-Sequenzen über eine bestimmte Länge.
In einigen Fällen können die Default-Parameter
so eingestellt werden, dass die Stringenz der Suche angepasst wird.
Der E-Wert kann beispielsweise erhöht werden, dass weniger
stringente Matches gezeigt werden. Auf diese Weise können
nahezu exakte Matches identifiziert werden. Die Tabelle E1 stellt
eine Liste von Nukleinsäuresequenzen bereit, die mit der
in den erfindungsgemäßen Verfahren verwendeten
Nukleinsäuresequenz verwandt sind. Tabelle E1: Beispiele für bHLH11-ähnliche
Polypeptide:
| Ursprungspflanze | Nukleinsäure
SEQ ID NO: | Protein
SEQ ID NO: |
| Triticum
aestivum | 244 | 245 |
| Allium
cepa | 258 | 327 |
| Arabidopsis
thaliana | 259 | 328 |
| Arabidopsis
thaliana | 260 | 329 |
| Arabidopsis
thaliana | 261 | 330 |
| Arabidopsis
thaliana | 262 | 331 |
| Arabidopsis
thaliana | 263 | 332 |
| Aquilegia
vulgaris | 264 | 333 |
| Aquilegia
vulgaris | 265 | 334 |
| Brassica
napus | 266 | 335 |
| Citrus
clementina | 267 | 336 |
| Curcuma
longa | 268 | 337 |
| Citrus
paridisi hybrid | 269 | 338 |
| Citrus
sinensis | 270 | 339 |
| Eucalyptus
grandis | 271 | 340 |
| Eucalyptus
grandis | 272 | 341 |
| Gossypium
hirsutum | 273 | 342 |
| Gossypium
hirsutum | 274 | 343 |
| Gossypium
hirsutum | 275 | 344 |
| Gossypium
hirsutum | 276 | 345 |
| Gossypium
hirsutum | 277 | 346 |
| Gossypium
hirsutum | 278 | 347 |
| Glycine
max | 279 | 348 |
| Glycine
max | 280 | 349 |
| Glycine
max | 281 | 350 |
| Glycine
max | 282 | 351 |
| Gossypium
raimondii | 283 | 352 |
| Helianthus
petiolaris | 284 | 353 |
| Hordeum
vulgare | 285 | 354 |
| Lactuca
perennis | 286 | 355 |
| Nicotiana
benthamiana | 287 | 356 |
| Nicotiana
benthamiana | 288 | 357 |
| Nicotiana
benthamiana | 289 | 358 |
| Nicotiana
benthamiana | 290 | 359 |
| Nicotiana
tabacum | 291 | 360 |
| Oryza
sativa | 292 | 361 |
| Oryza
sativa | 293 | 362 |
| Oryza
sativa | 294 | 363 |
| Oryza
sativa | 295 | 364 |
| Oryza
sativa | 296 | 365 |
| Oryza
sativa | 297 | 366 |
| Oryza
sativa | 298 | 367 |
| Picea
abies | 299 | 368 |
| Populus
deltoides | 300 | 369 |
| Pinus
radiata | 301 | 370 |
| Picea
sitchensis | 302 | 371 |
| Pinus
taeda | 303 | 372 |
| Populus
trichocarpa | 304 | 373 |
| Populus
trichocarpa | 305 | 374 |
| Populus
trichocarpa | 306 | 375 |
| Populus
trichocarpa | 307 | 376 |
| Populus
trichocarpa | 308 | 377 |
| Poncirus
trifoliata | 309 | 378 |
| Ricinus
communis | 310 | 379 |
| Ricinus
communis | 311 | 380 |
| Sorghum
bicolor | 312 | 381 |
| Solanum
lycopersicum | 313 | 382 |
| Solanum
lycopersicum | 314 | 383 |
| Solanum
lycopersicum | 315 | 384 |
| Solanum
tuberosum | 316 | 385 |
| Solanum
tuberosum | 317 | 386 |
| Solanum
tuberosum | 318 | 387 |
| Solanum
tuberosum | 319 | 388 |
| Triticum
aestivum | 320 | 389 |
| Vitis
vinifera | 321 | 390 |
| Vitis
vinifera | 322 | 391 |
| Vitis
vinifera | 323 | 392 |
| Vitis
vinifera | 324 | 393 |
| Zea
mays | 325 | 394 |
| Zea
mays | 326 | 395 |
-
In
einigen Fällen wurden verwandte Sequenzen vorläufig
zusammengebaut und öffentlich durch Forschungsinstitutionen
offenbart, wie The Institute for Genomic Research (TIGR). Die Eukaryotic
Gene Orthologs (EGO) Datenbank kann zur Identifikation solcher verwandten
Sequenzen verwendet werden, und zwar entweder über die
Schlüsselwort-Suche oder durch den BLAST-Algorithmus mit
der interessierenden Nukleinsäure- oder Polypeptid-Sequenz.
-
Beispiel 44: Alignment von bHLH11-ähnlichen
Polypeptisequenzen
-
Das
Alignment von Polypeptid-Sequenzen erfolgte mit dem AlignX-Programm
von Vector NTI (Invitrogen), das auf dem bekannten Clustal W-Algorithmus
des progressiven Alignments beruht (Thompson et al. (1997)
Nucleic Acids Res 25: 4876–4882; Chenna
et al. (2003). Nucleic Acids Res 31: 3497–3500).
Die Default-Werte lauten: gap open penalty 10, gap extension penalty
0,1, und die gewählte ”weight matrix” ist
Blosum 62 (wenn Polypeptide einem Alignment unterworfen werden).
Um das Alignment weiter zu optimieren, wurden kleinere Veränderungen
von Hand durchgeführt. Die Sequenzkonservierung unter bHLH11-ähnlichen
Polypeptiden befand sich im Wesentlichen in der C-terminalen bHLH-Domäne
der Polypeptide, wobei die N-terminale Region gewöhnlich
hinsichtlich Sequenzlänge und Zusammensetzung variabler
ist. Das Alignment der bHLH11-ähnlichen Polypeptide ist
in 21 gezeigt.
-
Mit ”CLUSTALX” wurde
ein Stammbaum der bHLH11-ähnlichen Polypeptide (22) konstruiert und es wurde ein Dendrogramm berechnet.
Das Kreis-Kladogramm wurde mit einem Dendroskop gezeichnet (Huson
et al., 2007).
-
Beispiel 45: Berechnung der Gesamtidentitätsprozent
zwischen Polypeptidsequenzen, die bei der Durchführung
der erfindungsgemäßen Verfahren geeignet sind
-
Die
Gesamtprozent für Ähnlichkeit und Identität
zwischen Volllängenpolypeptidsequenzen, die bei der Durchführung
der erfindungsgemäßen Verfahren geeignet sind,
wurden unter Verwendung von einer der in der Fachwelt verfügbaren
Techniken, nämlich der MatGAT (Matrix Global Alignment
Tool) Software (BMC Bioinformatics. 2003 4: 29. MatGAT:
an application that generstes similarity/identity matrices using
Protein or DNA sequences. Campanella JJ, Bitincka L, Smalley J;
Ledion Bitincka gehostete software) bestimmt. Die MatGAT-Software
erzeugt Ähnlichkeits-/Identitätsmatrizes für
DNA- oder Proteinsequenzen, ohne dass ein vorheriges Alignment der
Daten erforderlich ist. Das Programm führt eine Reihe von
paarweisen Alignments unter Verwendung des globalen Alignment-Algorithmus
von Myers und Miller durch (gap opening penalty 12, gap extension
penalty 2), berechnet die Ähnlichkeit und Identität
unter Verwendung von z. B. Blosum 62 (für Polypeptide)
und platziert dann die Ergebnisse in einer Abstandsmatrix. Die Sequenzähnlichkeit
ist in der unteren Hälfte der Trennlinie und die Sequenzidentität
in der oberen Hälfte der diagonalen Trennlinie dargestellt.
-
Bei
den in dem Vergleich eingesetzten Parametern handelte es sich um:
| Scoring
matrix: | Blosum
62 |
| First
Gap: | 12 |
| Extending
gap: | 2 |
-
Die
Ergebnisse der Software-Analyse sind in Tabelle E2 für
die Gesamtähnlichkeit und -identität über die
volle Länge der Polypeptidsequenzen dargestellt. Die Identitätsprozent
sind über der Diagonale und die Ähnlichkeitsprozent
unter der Diagonale angegeben. SEQ ID NO: 245 ist als TabHLH11 dargestellt.
-
Die
Identitätsprozent zwischen den bHLH11-ähnlichen
Polypeptidsequenzen, die bei der Durchführung der erfindungsgemäßen
Verfahren geeignet sind, kann nur 20% Aminosäureidentität
im Vergleich zu SEQ ID NO: 245 betragen. Die Identitätsprozent
sind jedoch viel höher, wenn die HLH-Domänen verglichen
werden. (Tabelle E3).
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
Beispiel 46: Identifikation von Domänen
in Polypeptidsequenzen, die bei der Durchführung der erfindungsgemäßen
Verfahren geeignet sind
-
Bei
der Integrated Resource of Protein Families, Domains and Sites (InterPro)
Datenbank handelt es sich um eine integrierte Plattform für
die häufig verwendeten Signatur-Datenbanken für
Suchen auf Text- und Sequenzbasis. Die InterPro-Datenbank kombiniert
diese Datenbanken, in denen unterschiedliche Methodiken und verschiedene
Mengen an biologischer Information über gut charakterisierte
Proteine verwendet werden, um Proteinsignaturen abzuleiten. Zu den
angeschlossenen Datenbanken zählen SWISS-PROT, PROSITS, TrEMBL,
PRINTS, ProDom und Pfam, Smart und TIGRFAMs. Pfam ist eine große
Sammlung multipler Sequenzalignments und versteckter Markov-Modelle,
die viele gemeinsame Proteindomänen und Familien abdecken.
Pfam wird am Sanger Institute Server im Vereinigten Königreich
gehostet. InterPro wird von dem European Bioinformatics Institute
im Vereinigten Königreich gehostet.
-
Die
Ergebnisse des Interpro-Scans der Polypeptidsequenz nach SEQ ID
NO: 245 sind in Tabelle E4 angegeben. Tabelle E4: InterPro-Scan Ergebnisse (Haupt-Zugangsnummern)
der Polypeptidsequenz nach SEQ ID NO: 245.
| Datenbank | Zugangsnummer | Zugangsname | Aminosäurekoordinaten
auf SEQ ID NO 245; e-Wert |
| InterPro | IPR001092 | Basischer
Helix-Loop-Helix Dimerisierungsregion bHLH | |
| HMMPfam | PF00010.14 | Helix-Loop-Helix
DNA-Bindungsdomäne | T[127–176] 1.6e–06 |
| HMMSmart | SM00353 | keine
Beschreibung | T[132–181] 1.5e–09 |
| Profil-Scan | PS50888 | HLH | T[120–176] 12.451 |
| InterPro | IPR011598 | Helix-Loop-Helix
DNA-Bindung | |
| Superfamilie | SSF47459 | Helix-Loop-Helix
DNA-Bindungsdomäne | T[122–195] 1.8e–14 |
-
Beispiel 47: Topologie-Vorhersage der
Polypeptidsequenzen, die sich bei der Durchführung der
erfindungsgemäßen Verfahren eignen
-
TargetP
1.1 bestimmt die subzelluläre Lokalisierung eukaryotischer
Proteine. Die Zuordnung der Lokalisierung beruht auf der vorhergesagten
Anwesenheit von einer der N- terminalen Präsequenzen: Chloroplasten-Transitpeptid
(cTP), mitochondriales Targeting-Peptid (mTP) oder Signalpeptid
(SP) des sekretorischen Wegs. Die Werte, auf denen die endgültige
Vorhersage beruht, sind keine echten Wahrscheinlichkeiten, und sie
tragen auch nicht notwendigerweise zu einer bei. Die Lokalisierung
mit dem höchsten Wert ist gemäß TargetP
am wahrscheinlichsten, und das Verhältnis zwischen den
Werten (die Verlässlichkeitsklasse) kann ein Anzeichen
dafür sein, wie sicher die Vorhersage ist. Die Verlässlichkeitsklasse
(RC) reicht von 1 bis 5, wobei 1 die stärkste Vorhersage
angibt. TargetP wird auf dem Server der Technischen Universität
Dänemark gehalten.
-
Für
die Sequenzen, die den Vorhersagen entsprechend eine N-terminale
Präsequenz enthalten, kann auch eine potentielle Spaltstelle
vorhergesagt werden.
-
Es
wurde eine Anzahl Parameter ausgewählt, wie Organismus-Gruppe
(Nicht-Pflanze oder Pflanze), Ausschlussgrenz-Sätze für
(kein, vordefinierte Ausschlussgrenz-Sätze, oder verwenderspezifische
Ausschlussgrenz-Sätze), und die Berechnung der Vorhersage
der Spaltstellen (Ja oder Nein).
-
Die
Ergebnisse der TargetP 1.1-Analyse der Polypeptidsequenz, wie sie
durch SEQ ID NO: 245 veranschaulicht ist, sind in der Tabelle E5
veranschaulicht. Es wurde die Organismus-Gruppe ”Pflanze” gewählt, es
wurden keine Ausschlussgrenzen definiert, und die vorhergesagte
Länge des Transitpeptids abgefragt. Die subzelluläre
Lokalisierung der Polypeptidsequenz nach SEQ ID NO: 245 kann das
Cytoplasma oder der Kern sein, es wurde kein Transitpeptid vorhergesagt. Tabelle E5: TargetP 1.1-Analyse der Polypeptidsequenz
nach SEQ ID NO: 245
| Länge
(AA) | 281 |
| Chloroplasten-Transitpeptid | 0,094 |
| Mitochondriales
Transitpeptid | 0,166 |
| Signalpeptid
des sekretorischen Wegs | 0,034 |
| Anderes
subzelluläres Ziel | 0,855 |
| Vorhergesagte
Lokalisierung | / |
| Verlässlichkeitsklasse | 2 |
| Vorhergesagte
Länge des Transitpeptids | / |
-
Gemäß der
Analysen mit PLOC (Park und Kanehisa, Bioinformatics, 19,
1656–1663, 2003) oder mit PSORT (URL: psort.org),
hat das Protein den Vorhersagen zufolge eine Kernlokalisierung.
-
Zur
Durchführung solcher Analysen können viele andere
Algorithmen verwendet werden, wie u. a.:
- • ChloroP
1.1, das auf dem Server der Technischen Universität Dänemark
gehostet wird;
- • Protein Prowler Subcellular Localisation Predictor
Version 1.2, das auf dem Server des Instituts für Molekulare
Biowissenschaft, Universität Queensland, Brisbane, Australien,
gehostet wird;
- • PENCE Proteome Analyst PA-GOSUB 2.5, das auf dem
Server der Universität von Alberts, Edmonton, Alberts,
Canada gehostet wird;
- • TMHMM, das auf dem Server der Technischen Universität
Dänemark gehostet wird
-
Beispiel 48: Funktioneller Test für
das bHLH11-ähnliche Polypeptid
-
Ein
DNA-Bindungstest für AtbHLH6 ist in Dombrecht et
al. (2007) angegeben, dieser Ansatz kann für ein
beliebiges bHLH11-ähnliches Protein verwendet werden. Kurz
gesagt wurde ein 1000-bp Fragment der AtbHLH6 kodierenden Sequenz,
die die Codons 285 bis 623 umfasst, aus der genomischen DNA amplifiziert und
in den Nhel-BamHI-gespaltenen pTacLCELD6·His Vektor kloniert
(Xue, Plant J. 41, 638–649, 2005). Das resultierende
Konstrukt kodierte die letzten 338 Aminosäuren von AtbHLH6
(einschließlich der bHLH-Region) im Leseraster mit dem
Reporterprotein CelD und einem 6xHis-Tag. Die korrekte Amplifikation
und Klonierung wurden durch DNA-Sequenzierung verifiziert.
-
Die
Bestimmung der Konsensussequenz des MYC2-DNA-Bindungsmotivs und
die relative Bindungsaffinität dieser Stellen erfolgte
gemäß Xue (2005), wobei eine
Fusion eines DNA-Bindungsproteins (DBP) an 6·His-tagged
Cellulase D (CELD) sowohl als Mittel für eine Affinitätsreinigung
des DBP-DNA Komplexes bei der Auswahl der Bindungsstellen aus einem
Pool biotinylierter Oligonukleotide mit statistischer Sequenz als auch
als Reporter zur Messung der DNA-Bindungsaktivität dienen
kann.
-
Zur
Auswahl der Bindungsstellen mittels Cellulosepapier als Affinitätsmatrix
wurde DBD-CELD bei Raumtemperatur für 1 Std. mit 20 ng
eines Biotin-markierten Bio-RS-Oligo in 40 μl Bindungs-/Waschpuffer (oben
beschrieben) mit 1 mM EDTA, 0,25 μg μl–1 poly d(AC-TG), 1 mg ml–1 BSA und 10% Glycerin inkubiert. Bei
einer geeigneten Menge rohes DBP-CELD, das zur Selektion der Bindungsstelle
verwendet wurde, wurden 20 bis 30% relative Cellulose-Bindungseffizienz
(Prozent Cellulose-Aktivität, die nach dem Waschen an Cellullose
band) erzielt. DBP-CELD/Bio-RS-Oligo Gemische wurden in Mikrotiterplatten
mit 96 Vertiefungen überführt, welche Whatman
1 Filterpapier (4 mm2) enthielten, das mit
10 μl Blockierungslösung [Bindungs-/Waschpuffer
mit 0,5 μg μl–1 poly
d(AC-TG) und 10% Glycerin] vorgetränkt wurde. Nach der
Inkubation bei 0°C für 1 Std. unter leichtem Schütteln,
wurde das Cellulosepapier sechsmal mit Bindungs-/Waschpuffer mit
1 mM EDTA und 0,1 mg ml–1 BSA gemischt.
Die Verwendung von ausgiebigem Waschen bei der Auswahl der Zielstelle
beruhte auf Beobachtungen der relativ hohen Stabilität
des auf der festen Matrix immobilisierten DBP-Oligonukleotid-Komplexes.
DBP-CELD tragende Ziel-Bindungsstellen wurden bei 40°C
für 15 min mit 40 μl Cellulase-Elutionspuffer
[10 mM HEPES, pH-Wert 7, 50 mM KCl, 0,2 mM EDTA, 4 mM Cellobiose,
0,05 mg ml–1 BSA und 10 μg
ml–1 eines sense-sequenzspezifischen
Primer SP-S] eluiert. Das Eluat wurde zur PCR-Amplifikation ausgewählter
Oligonukleotide verwendet. Das PCR-Produkt (0,1 μl) ohne
Reinigung wurde in der nächsten Runde der Stellenselektion
verwendet.
-
Alternativ
wurde 6·His tagged DBP-CELD (10–25 μg
rohes Protein) in 60 μl PNT-Puffer (50 mM Natriumphosphat,
pH-Wert 8,0, 300 mM NaCl und 0,05% Tween 20) mit 10 mM Imidazol
und 350 μg Ni-NTA Magnetagaroseperlen (Qiagen) bei Raumtemperatur
für 45–60 min unter leichtem Schütteln
in einem Micro-Röhrchen-Mischer (Tomy Seiko Co., Tokyo,
Japan) inkubiert. Die Ni-NTA Magnetperlen wurden auf der Seite der Röhrchen
gesammelt, indem die Röhrchen in einem Magnet für
12 Röhrchen (Qiagen) untergebracht wurden. Ungebundene
Proteine wurden durch zweimaliges Waschen mit 150–200 μl
PNT mit 20 mM Imidazol und einmal mit 60 μl Bindungs-/Waschpuffer
mit 2 mM MgCl2, 1 mg ml–1 BSA
und 10% Glycerin entfernt. Die gewaschenen Perlen wurde- in 40 μl
of Bindungs-/Waschpuffer mit 2 mM MgCl2,
0,25 μg μl–1 poly
d(AC-TG), 1 mg ml–1 BSA, 0,0025%
Nonidet P-40, 10% Glycerin und biotin-markierten Oligonukleotiden
(50 ng Bio-RS-Oligo oder 1 μl PCR-Produkt, das aus vorher
ausgewählten Oligonukleotiden amplifiziert wurde) resuspendiert.
Die Suspension wurde bei Raumtemperatur für 1,5 Std. unter
leichtem Schütteln in dem Mikro-Röhrchen-Mischer inkubiert.
Nach viermaligem Waschen (3–4 min bei jedem Waschvorgang)
mit 150–200 μl Bindungs-/Waschpuffer mit 2 mM
MgCl2, 0,1 mg ml–1 BSA
und 0,0025% Nonidet P-40, wurden die Perlen, die das an der Zielstelle
gebundene BDP-CELD tragen, in 8 μl 5 mM Tris-Cl (pH-Wert
8,0)/0,5 mM EDTA mit 5 μg ml–1 eines
sense-sequenzspezifischen Primers (SP-S) resuspendiert, und die
Suspension wurde in ein sauberes Röhrchen überführt
und für die PCR-Amplifikation ausgewählter Oligonukleotide
verwendet. Das PCR-Produkt (1 μl) wurde ohne Reinigung
in der nächsten Runde der Stellenselektion verwendet.
-
Für
EMSA Tests wurden 6·His-tagged DBP-CELD Proteine mit Ni-NTA
Magnetagaroseperlen mit einem hochstringenten Bindungspuffer (50
mM Natriumphosphat, pH-Wert 8,0, 1 M NaCl, 10% Glycerin, 1% Tween
20 und 10 mM Imidazol) gereinigt, und der Rest des Verfahrens folgte
den Herstellangaben. Doppelsträngige synthetische Oligonukleotide
(30–45 fmol), markiert mit Digoxigenin am 3'-Ende, wurde
mit einem gereinigten DBP (30–75 ng) in 15 μl
Bindungspuffer [25 mM HEPES/KOH, pH-Wert 7,0, 50 mM KCl, 0,5 mM DTT,
2 mM MgCl2, 0,2 μg μl–1 poly d(AC-TG), 0,3 mg ml–1 BSA und 10% Glycerin] inkubiert.
Nach der Inkubation bei Raumtemperatur für 30 min, wurden
die DBP/DNA-Komplexe von freien Sonden auf einem 6% Polyacrylamidgel
in einem 40-mM Tris-Acetat-Puffer (pH-Wert 7,5) mit 5 mM Na-Acetat,
0,5 mM EDTA und 5% Glycerin getrennt. Die DBP/DNA-Komplexe und die
freien Sonden in den Gelen nach der Elektrophorese wurden auf eine
Hybond Nþ-Membran überführt. Alkalische
Phosphatase-konjugierter Anti-Digoxigenin-Antikörper und
ein chemilumineszierendes Substrat, CDP-Star (Roche Diagnostics)
wurde zum Nachweisen von Digoxigenin gemäß den
Herstellerangaben verwendet. Weitere Einzelheiten sind in Xue
(2005) angegeben.
-
Beispiel 49: Klonierung der in den erfindungsgemäßen
Verfahren verwendeten Nukleinsäuresequenz
-
Die
in den erfindungsgemäßen Verfahren verwendete
Nukleinsäuresequenz gemäß SEQ ID NO:
244 wurde durch PCR mit einer speziell angefertigten cDNA-Bank von
Triticum aestivum Keimlingen (in pCMV Sport 6.0; Invitrogen, Paisley,
UK) als Matrize amplifiziert. Die PCR wurde mit Hifi Taq DNA-Polymerase
in Standardbedingungen, mit 200 ng Matrize in einem 50 μl
PCR-Mix durchgeführt. Die verwendeten Primer waren prm009718
(SEQ ID NO: 254; Sense, Start-Codon in fett):
5'-ggggacaagtttgtacaaaaaagcaggcttaaacaatggcggggcanccg-3'
und prm009719 (SEQ ID NO: 255; revers, komplementär):
5'-ggggaccactttgtacaagaaagctgggtatgggtgttgcagctgctgtt-3',
die
die AttB-Stellen für die Gateway-Rekombination enthalten.
Das amplifizierte PCR-Fragment wurde ebenfalls mit Standardverfahren
gereinigt. Der erste Schritt des Gateway-Verfahrens, die BP-Reaktion,
wurde dann durchgeführt, bei der das PCR-Fragment in vivo
mit dem Plasmid pDONR201 unter Bildung eines pbHLH11-ähnlichen – gemäß Gateway-Teminologie – ”entry
clone” rekombinierte. Das Plasmid pDONR201 wurde käuflich
von Invitrogen als Bestandteil der Gateway® Technik
erworben.
-
Der
SEQ ID NO: 244 enthaltende ”entry clone” wurde
anschließend in einer LR-Reaktion mit einem Zielvektor,
der für die Transformation von Oryza sativa verwendet wird,
verwendet. Dieser Vektor enthält als funktionelle Elemente
innerhalb der T-DNA-Grenzen Folgendes: Einen pflanzlichen Selektionsmarker;
eine screenbare Markerexpressionskassette, und eine Gateway-Kassette,
die für die LR-in-vivo-Rekombination mit der interessierenden
Nukleinsäuresequenz, die bereits in den ”entry
clone” kloniert wurde, vorgesehen ist. Ein GOS2-Promotor
aus Reis (SEQ ID NO: 256) für die konstitutive Expression
befand sich stromaufwärts dieser Gateway-Kassette.
-
Nach
dem LR-Rekombinationsschritt wurde der erhaltene Expressionsvektor
pGOS2::bHLH11-ähnlich (23)
nach fachbekannten Methoden in den Agrobacterium-Stamm LBA4044 transformiert.
-
Beispiel 50: Pflanzentransformation
-
Die
Transformation der Pflanzen erfolgte gemäß dem
in Beispiel 7 aufgeführten Verfahren.
-
Beispiel 51: Verfahren bei der phänotypischen
Untersuchung
-
51.1 Aufbau der Untersuchung
-
Es
wurden ungefähr 35 unabhängige T0-Reistransformanten
erzeugt. Die Primärtransformanten wurden für das
Heranziehen und Ernten von T1-Samen von einer Gewebekulturkammer
in ein Gewächshaus umgestellt. Sechs Ereignisse, von denen
sich die T1-Nachkommenschaft im Verhältnis 3:1 für
Vorliegen/Abwesenheit des Transgens aufspaltete, wurden behalten.
Für jedes dieser Ereignisse wurden ungefähr 10 T1-Keimpflanzen,
die das Transgen enthielten (Hetero- und Homozygoten) und ungefähr
10 T1-Keimpflanzen, denen das Transgen fehlte (Nullizygoten) durch
Beobachten der visuellen Markerexpression selektiert. Die transgenen
Pflanzen und die entsprechenden Nullizygoten wurden nebeneinander
in zufälliger Anordnung herangezogen. Die Gewächshausbedingungen
waren kurze Tage (12 Stunden Licht), 28°C im Hellen und
22°C im Dunkeln, und eine relative Feuchtigkeit von 70%.
Die unter Nichtstressbedingungen gezogenen Pflanzen werden in regelmäßigen
Abständen mit Wasser versorgt, so dass gewährleistet
war, dass Wasser und Nährstoffe nicht limitierend waren,
damit die Bedürfnisse der Pflanze, zu wachsen und sich
zu entwickeln, erfüllt werden.
-
Vom
Sästadium bis zum Reifestadium wurden die Pflanzen mehrmals
in eine Digital-Imaging-Kammer gestellt. Zu jedem Zeitpunkt wurden
von jeder Pflanze aus mindestens 6 verschiedenen Winkeln Digitalbilder (2048×1536
Pixel, 16 Millionen Farben) aufgenommen.
-
Screening unter Trockenheitsbedingungen
-
Pflanzen
aus T2-Samen wurden unter Normalbedingungen in Blumenerde herangezogen,
bis sie das Stadium des Rispenschiebens erreichten. Anschließend
wurden sie in ein ”trockenes” Areal gestellt,
wo keine Bewässerung erfolgte. Um den Bodenwassergehalt
(BWG) zu verfolgen, wurden Feuchtigkeitssonden in statistisch ausgewählte
Töpfe eingeführt. Sank der BWG unter gewisse Schwellenwerte,
wurden die Pflanzen automatisch erneut gegossen, bis wiederum ein
Normalgehalt erreicht wurde. Dann wurden die Pflanzen wiederum in
Normalbedingungen umgestellt. Ansonsten wurde wie bei den nicht
unter abiotischen Stressbedingungen herangezogenen Pflanzen kultiviert
(Pflanzenreife, Samenernte). Wachstums- und Ertragparameter werden
wie für das Wachstum unter Normalbedingungen beschrieben
aufgezeichnet.
-
Effizienz-Screening der Stickstoff-Verwerfung
-
Reispflanzen
aus T2-Samen werden in Blumenerde unter Normalbedingungen mit Ausnahme
der Nährlösung herangezogen. Die Töpfe
werden vom Umsetzen bis zur Reife mit einer spezifischen Nährlösung mit
verringertem N-Stickstoff-Gehalt (N), üblicherweise zwischen
7 bis 8 mal weniger, gegossen. Ansonsten wird wie bei den nicht
unter abiotischen Stressbedingungen herangezogenen Pflanzen kultiviert
(Pflanzenreife, Samenernte). Wachstums- und Ertragsparameter werden
wie für das Wachstum unter Normalbedingungen aufgezeichnet
-
Salzstress-Screening
-
Die
Pflanzen werden auf einem Substrat aus Kokosfasern und Argex (Verhältnis
3 zu 1) herangezogen. Während der ersten zwei Wochen nach
dem Umsetzen der Pflänzchen in das Gewächshaus
wird eine normale Nährlösung verwendet. Nach den
ersten zwei Wochen wird die Nährlösung mit 25
mM Salz (NaCl) versetzt, bis die Pflanzen geerntet werden. Anschließend
werden die Wachstums- und Ertragsparameter bestimmt, wie für
Wachstum unter normalen Bedingungen eingehend beschrieben.
-
51.2 Statistische Analyse: F-Test
-
Als
statistisches Modell für die Gesamtuntersuchung der phänotypischen
Eigenschaften der Pflanze wurde eine zweifaktorielle ANOVA (Varianzanalyse)
verwendet. Mit allen Parametern, die bei allen Pflanzen von allen
Ereignissen, die mit dem erfindungsgemäßen Gen
transformiert wurden, bestimmt wurden, wurde ein F-Test durchgeführt.
Der F-Test erfolgte als Überprüfung auf einen
Effekt des Gens über alle Transformations-Ereignisse und
zur Feststellung eines Gesamteffekts des Gens, der auch als globaler
Geneffekt bezeichnet wird. Die Signifikanzschwelle für
einen echten globalen Geneffekt wurde bei dem F-Test auf dem 5%-Wahrscheinlichkeitsniveau
festgelegt. Ein signifikanter F-Test-Wert weist auf einen Geneffekt
hin, was bedeutet, dass mehr als nur das einfache Vorhandensein
oder die Lage des Gens die Unterschiede im Phänotyp verursacht.
-
51.3 Messparameter
-
Biomasseparameter-Messung
-
Vom
Sästadium bis zum Reifestadium wurden die Pflanzen mehrmals
in eine Digital-Imaging-Kammer gestellt. Zu jedem Zeitpunkt wurden
von jeder Pflanze aus mindestens 6 verschiedenen Winkeln Digitalbilder (2048×1536
Pixel, 16 Millionen Farben) aufgenommen.
-
Die
oberirdische Pflanzenfläche (oder Blattbiomasse) wurde
dadurch bestimmt, dass man die Gesamtanzahl Pixel der Digitalbilder
der vom Hintergrund unterschiedlichen oberirdischen Pflanzenteile
zählte. Dieser Wert wurde über die am selben Zeitpunkt
aus unterschiedlichen Winkeln aufgenommenen Bilder gemittelt und
mittels Kalibrierung in einen physikalischen Oberflächenwert,
ausgedrückt in Quadratmillimeter, umgewandelt. Versuche
zeigen, dass die auf diese Weise bestimmte oberirdische Pflanzenfläche
mit der Biomasse der oberirdischen Pflanzenteile korreliert. Die
oberirdische Fläche ist diejenige Fläche, die
zu dem Zeitpunkt, an dem die Pflanze ihre maximale Blattbiomasse
erreicht hatte, bestimmt wurde. Die Jungpflanzenvitalität
ist die oberirdische Fläche der Pflanze (Keimpflanze) 3
Wochen nach der Keimung. Die Steigerung der Wurzelbiomasse wird
als Steigerung der Gesamtwurzelbiomasse (gemessen als maximale Wurzelbiomasse, die
während der Lebensdauer einer Pflanze beobachtet wurde)
oder als Steigerung des Wurzel/Spross-Index (gemessen als Verhältnis
zwischen Wurzelmasse und Sprossmasse im Zeitraum des aktiven Wachstums
von Wurzel und Spross) ausgedrückt.
-
Für
die Bestimmung der Jungpflanzenvitalität wurde die Gesamtzahl
der Pixel aus überirdischen Pflanzenteilen vom Hintergrund
unterschieden. Dieser Wert wurde für die Bilder gemittelt,
die zum gleichen Zeitpunkt aus verschiedenen Winkeln aufgenommen
wurden, und wurde durch Kalibrierung in einen physikalischen Oberflächenwert
umgewandelt, der in mm2 ausgedrückt
wird. Die nachstehend beschriebenen Ergebnisse stammen von Pflanzen
drei Wochen nach der Keimung.
-
Samenparameter-Messungen
-
Die
reifen Primärrispen wurden geerntet, gezählt,
eingetütet, mit einem Strichcode versehen und dann drei
Tage bei 37°C in einem Ofen getrocknet. Dann wurden die
Rispen gedroschen, und alle Samen wurden gesammelt und gezählt.
Die gefüllten Spelzen wurden von den leeren Spelzen mittels
einer Luftblaseeinrichtung getrennt. Die leeren Spelzen wurden verworfen,
und die restliche Fraktion wurde nochmals gezählt. Die gefüllten
Spelzen wurden auf einer Analysewaage gewogen. Die Anzahl gefüllter
Samen wurde dadurch bestimmt, dass man die Anzahl der gefüllten
Spelzen, die nach dem Abtrennungsschritt verblieben, auszählte. Der
Gesamtsamenertrag wurde dadurch bestimmt, dass man alle von einer
Pflanze geernteten gefüllten Spelzen wog. Die Gesamtsamenzahl
pro Pflanze wurde dadurch bestimmt, dass man die von einer Pflanze
geerntete Anzahl Spelzen auszählte. Das Tausendkorngewicht
(TKG) wird aus der Anzahl der gezählten gefüllten Samen
und ihres Gesamtgewichts extrapoliert. Der Harvest Index (HI) wird
in der vorliegenden Erfindung als das Verhältnis zwischen
dem Gesamtsamengewicht und der oberirdischen Fläche (mm2), multipliziert mit einem Faktor 106, definiert. Die Gesamtzahl Blüten
pro Rispe wird in der vorliegenden Erfindung als das Verhältnis
zwischen der Gesamtsamenzahl und der Anzahl der reifen Primärrispen
definiert. Die Samenfüllungsrate wird in der vorliegenden
Erfindung als das Verhältnis (als Prozent ausgedrückt)
der Anzahl gefüllter Samen zu der Gesamtzahl Samen (oder
Blüten) definiert.
-
Beispiel 52: Ergebnisse der phänotypischen
Untersuchung transgener Pflanzen
-
Die
Ergebnisse der Untersuchung transgener Reispflanzen, die die bHLH11-ähnliche
Nukleinsäuresequenz unter Nichtstressbedingungen exprimieren,
sind nachstehend aufgeführt. Eine Steigerung von mehr als 5%
in mindestens 2 Linien wurde für den Gesamtsamenertrag,
die Anzahl gefüllter Samen, Füllungsrate, Harvest-Index,
Anzahl der ersten Rispen, und mindestens 3% für das Tausendkorngewicht
beobachtet. Die Daten der Gesamtsteigerung (gemessen über
alle untersuchten Linien) für jeden Parameter sind in der
Tabelle E6 angegeben: Tabelle E6:
| Parameter | Gesamtanstieg
(%) |
| Gesamtgewicht
der Samen (Gesamtsamenertrag) | 20 |
| Anzahl
gefüllter Samen | 20 |
| Füllungsrate | 18 |
| Harvest
Index | 21 |
-
VI. ASR (abszisinsäure-, stress-,
und reifungsinduziertes) Protein
-
Beispiel 53: Identifikation von Sequenzen,
die mit der in den erfindungsgemäßen Verfahren
verwendeten ASR Nukleinsäuresequenz verwandt sind
-
Unter
den Sequenzen in der Entrez Nucleotides Datenbank am National Center
for Biotechnology Information (NCBI) wurden unter Verwendung von
Datenbanksequenzabfragewerkzeugen, wie dem Basic Local Alignment
Tool (BLAST) (Altschul et al., (1990), J. Mol. Biol. 215:
403–410; und Altschul et al., (1997),
Nucleic Acids Res. 25: 3389–3402), (Volllängen-cDNA,
ESTs oder genomische) Sequenzen, die mit der in den erfindungsgemäßen
Verfahren verwendeten Nukleinsäuren verwandt sind, identifiziert.
Das Programm wurde zum Auffinden von Regionen mit lokaler Ähnlichkeit
zwischen Sequenzen verwendet, und zwar durch Vergleichen von Nukleinsäure-
oder Polypeptid-Sequenzen mit Sequenz-Datenbanken und durch Berechnen
der statistischen Signifikanz der Matches. Das durch die erfindungsgemäß verwendete
Nukleinsäure kodierte Polypeptid wurde beispielsweise für
den TBLASTN-Algorithmus verwendet, und zwar unter Ausschaltung der
Default-Einstellungen und des Filters für die Nichtmiteinbeziehung
von Sequenzen mit niedriger Komplexität. Der Output der
Analyse wurde mittels paarweisem Vergleich betrachtet und gemäß der
Wahrscheinlichkeits-Wertung (E-Wert) gereiht, wobei die Wertung
die Wahrscheinlichkeit, dass ein bestimmtes Alignment statistisch
vorkommt, widerspiegelt (je niedriger der E-Wert, desto signifikanter
der Hit). Zusätzlich zu den E-Werten wurden die Vergleiche
auch durch die Prozent der Identität gewertet. Die Prozent
der Identität bezieht sich auf die Anzahl identischer Nukleotide
(oder Aminosäuren) zwischen den zwei verglichenen Nukleinsäure-(oder
Polypeptid)-Sequenzen über eine bestimmte Länge.
-
Die
Tabelle F1 stellt eine Liste von Nukleinsäure- und Polypeptidsequenzen
bereit, die mit der in den erfindungsgemäßen Verfahren
verwendeten Nukleinsäuresequenz verwandt sind. Tabelle F1: Beispiele für ASR
Nukleinsäuresequenzen und Polypeptide.
| Name | Ursprungspflanze | Nukleinsäure
SEQ ID NO: | Protein
SEQ ID NO: |
| Orysa_ASR_1 | Oryza
sativa | 396 | 397 |
| Zeama_ASR_1 | Zea
mays | 401 | 402 |
| Zeama_ASR_2 | Zea
mays | 403 | 404 |
| Zeama_ASR_3 | Zea
mays | 405 | 406 |
| Zeama_ASR_4 | Zea
mays | 407 | 408 |
| Zeama_ASR_5 | Zea
mays | 409 | 410 |
| Zeama_ASR_6 | Zea
mays | 411 | 412 |
| Zeama_ASR_7 | Zea
mays | 413 | 414 |
| Zeama_ASR_8 | Zea
mays | 415 | 416 |
| Zeama_ASR_9 | Zea
mays | 417 | 418 |
| Lyces_ASR_1 | Lycopersicon
esculentum | 419 | 420 |
| Lyces_ASR_2 | Lilium
longiflorum | 421 | 422 |
| Sacof_ASR_1 | Saccharum
officinarum | 423 | 424 |
| Zeama_ASR_10 | Zea
mays | 425 | 426 |
-
Beispiel 54: Alignment von ASR-Polypeptidsequenzen
-
Das
Alignment von Polypeptid-Sequenzen erfolgte mit dem AlignX Programm
aus dem Vector NTI Package (Invitrogen), das auf dem bekannten Clustal
W-Algorithmus des progressiven Alignments beruht (Thompson
et al. (1997) Nucleic Acids Res 25: 4876–4882; Chenna
et al. (2003). Nucleic Acids Res 31: 3497–3500).
Die Default-Werte lauten: gap open penalty 10, gap extension penalty
0,1, und die gewählte ”weight matrix” ist
Blosum 62 (wenn Polypeptide einem Alignment unterworfen werden).
Um das Alignment weiter zu optimieren, wurden kleinere Veränderungen
von Hand durchgeführt.
-
Ein
Stammbaum der GRP-Polypeptide wird mit einem Neighbour joining clustering
Algorithmus wie er in dem AlignX-Programm von Vector NTI (Invitrogen)
bereitgestellt wird, konstruiert.
-
Beispiel 55: Berechnung der Gesamtidentitätsprozent
zwischen ASR-Polypeptidsequenzen, die bei der Durchführung
der erfindungsgemäßen Verfahren geeignet sind
-
Die
Gesamtprozent für Ähnlichkeit und Identität
zwischen Volllängenpolypeptidsequenzen, die bei der Durchführung
der erfindungsgemäßen Verfahren geeignet sind,
wurden unter Verwendung von einer der in der Fachwelt verfügbaren
Techniken, nämlich der MatGAT (Matrix Global Alignment
Tool) Software (BMC Bioinformatics. 2003 4: 29. MatGAT:
an application that generstes similarity/identity matrices using
Protein or DNA sequences. Campanella JJ, Bitincka L, Smalley J;
software hosted by Ledion Bitincka) bestimmt. Die MatGAT-Software
erzeugt Ähnlichkeits-/Identitätsmatrizes für
DNA- oder Proteinsequenzen, ohne dass ein vorheriges Alignment der
Daten erforderlich ist. Das Programm führt eine Reihe von
paarweisen Alignments unter Verwendung des globalen Alignment-Algorithmus
von Myers und Miller durch (gap opening penalty 12, gap extension
penalty 2), berechnet die Ähnlichkeit und Identität
unter Verwendung von z. B. Blosum 62 (für Polypeptide)
und platziert dann die Ergebnisse in einer Abstandsmatrix. Die Sequenzähnlichkeit
ist in der unteren Hälfte der Trennlinie und die Sequenzidentität
in der oberen Hälfte der diagonalen Trennlinie dargestellt.
-
Bei
den in dem Vergleich eingesetzten Parametern handelte es sich um:
| Scoring
matrix: | Blosum
62 |
| First
Gap: | 12 |
| Extending
gap: | 2 |
-
Eine
MATGAT-Tabelle für lokales Alignment einer spezifischen
Domäne oder die Daten bezüglich % Identität/Ähnlichkeit
zwischen spezifischen Domänen können ebenfalls
erzeugt werden.
-
Beispiel 56: Identifikation von Domänen
in ASR-Polypeptidsequenzen, die bei der Durchführung der
erfindungsgemäßen Verfahren geeignet sind
-
Bei
der Integrated Resource of Protein Families, Domains and Sites (InterPro)
Datenbank handelt es sich um eine integrierte Plattform für
die häufig verwendeten Signatur-Datenbanken für
Suchen auf Text- und Sequenzbasis. Die InterPro-Datenbank kombiniert
diese Datenbanken, in denen unterschiedliche Methodiken und verschiedene
Mengen an biologischer Information über gut charakterisierte
Proteine verwendet werden, um Proteinsignaturen abzuleiten. Zu den
angeschlossenen Datenbanken zählen SWISS-PROT, PROSITS, TrEMBL,
PRINTS, ProDom und Pfam, Smart und TIGRFAMs. Pfam ist eine große
Sammlung multipler Sequenzalignments und versteckter Markov-Modelle,
die viele gemeinsame Proteindomänen und Familien abdecken.
Pfam wird am Sanger Institute Server im Vereinigten Königreich
gehostet. InterPro wird von dem European Bioinformatics Institute
im Vereinigten Königreich gehostet.
-
Die
das GRP veranschaulichenden Proteinsequenzen werden als Abfrage
zum Durchsuchen der InterPro Datenbank verwendet.
-
Beispiel 57: Topologie-Vorhersage der
ASR-Polypeptidsequenzen, die sich bei der Durchführung
der erfindungsgemäßen Verfahren eignen
-
TargetP
1.1 bestimmt die subzelluläre Lokalisierung eukaryotischer
Proteine. Die Zuordnung der Lokalisierung beruht auf der vorhergesagten
Anwesenheit von einer der N-terminalen Präsequenzen: Chloroplasten-Transitpeptid
(cTP), mitochondriales Targeting-Peptid (mTP) oder Signalpeptid
(SP) des sekretorischen Wegs. Die Werte, auf denen die endgültige
Vorhersage beruht, sind keine echten Wahrscheinlichkeiten, und sie
tragen auch nicht notwendigerweise zu einer bei. Die Lokalisierung
mit dem höchsten Wert ist am wahrscheinlichsten gemäß TargetP,
und das Verhältnis zwischen den Werten (die Verlässlichkeitsklasse)
kann ein Anzeichen dafür sein, wie sicher die Vorhersage
ist. Die Verlässlichkeitsklasse (RC) reicht von 1 bis 5,
wobei 1 die stärkste Vorhersage angibt. TargetP wird auf
dem Server der Technischen Universität Dänemark
gehalten.
-
Für
die Sequenzen, die den Vorhersagen entsprechend eine N-terminale
Präsequenz enthalten, kann auch eine potentielle Spaltstelle
vorhergesagt werden.
-
Es
wurde eine Anzahl Parameter ausgewählt, wie Organismus-Gruppe
(Nicht-Pflanze oder Pflanze) Ausschlussgrenz-Sätze (kein,
vordefinierte Ausschlussgrenz-Sätze, oder verwenderspezifische
Ausschlussgrenz-Sätze), und die Berechnung der Vorhersage
der Spaltstellen (Ja oder Nein).
-
Die
das GRP veranschaulichenden Sequenzen werden zur Abfrage von TargetP
1.1 verwendet. Es wurde die Organismus-Gruppe ”Pflanze” gewählt,
es wurden keine Ausschlussgrenzen definiert, und die vorhergesagte
Länge des Transitpeptids abgefragt.
-
Zur
Durchführung solcher Analysen können viele andere
Algorithmen verwendet werden, wie u. a.:
- • ChloroP
1.1, das auf dem Server der Technischen Universität Dänemark
gehostet wird;
- • Protein Prowler Subcellular Localisation Predictor
Version 1.2, das auf dem Server des Instituts für Molekulare
Biowissenschaft, Universität Queensland, Brisbane, Australien,
gehostet wird;
- • PENCE Proteome Analyst PA-GOSUB 2.5, das auf dem
Server der Universität von Alberts, Edmonton, Alberts,
Canada gehostet wird;
- • TMHMM, das auf dem Server der Technischen Universität
Dänemark gehostet wird.
-
Beispiel 58: Klonierung einer in den erfindungsgemäßen
Verfahren verwendeten ASR-Nukleinsäure
-
Klonierung von SEQ ID NO: 396:
-
Die
Nukleinsäuresequenz SEQ ID NO: 396, die in den erfindungsgemäßen
Verfahren verwendet wurde, wurde durch PCR mit einer speziell angefertigten
cDNA-Bank aus Oryza sativa Keimlingen (in pCMV Sport 6.0; Invitrogen,
Paisley, UK) amplifiziert. Die PCR erfolgte mit Hifi Taq DNA-Polymerase
in Standard-Bedingungen, mit 200 ng Matrize in einem 50 μl
PCR-Mix. Die verwendeten Primer waren prm06442 (SEQ ID NO: 399; Sense,
Startcodon in fett):
5'-ggggacaagtttgtacaaaaaagcaggcttaaacaatggcggaggagaagca-3'
und prm06443 (SEQ ID NO: 400; revers, komplementär):
5'-ggggaccactttgtacaagaaagctgggtcggcgacgttgtgatga-3',
die
die AttB-Stellen für die Gateway-Rekombination enthalten.
Das amplifizierte PCR-Fragment wurde ebenfalls mit Standardverfahren
gereinigt. Der erste Schritt des Gateway-Verfahrens, die BP-Reaktion,
wurde dann durchgeführt, bei der das PCR-Fragment in vivo
mit dem Plasmid pDONR201 unter Bildung eines – gemäß Gateway-Teminologie – ”entry
clone” rekombinierte. Das Plasmid pDONR201 wurde käuflich
von Invitrogen als Bestandteil der Gateway® Technik
erworben.
-
Der
SEQ ID NO: 396 enthaltende ”entry clone” wurde
anschließend in einer LR-Reaktion mit einem Zielvektor,
der für die Transformation von Oryza sativa verwendet wird,
verwendet. Dieser Vektor enthält als funktionelle Elemente
innerhalb der T-DNA-Grenzen Folgendes: Einen pflanzlichen Selektionsmarker;
eine screenbare Markerexpressionskassette, und eine Gateway-Kassette,
die für die LR-in-vivo-Rekombination mit der interessierenden
Nukleinsäuresequenz, die bereits in den ”entry
clone” kloniert wurde, vorgesehen ist. Ein GOS2-Promotor
aus Reis (SEQ ID NO: 398) für die konstitutive Expression
befand sich stromaufwärts dieser Gateway-Kassette.
-
Nach
dem LR-Rekombinationsschritt wurde der erhaltene Expressionsvektor
pGOS2::GRP (25) in den Agrobacterium Stamm
LBA4044 gemäß den Verfahren des Standes der Technik
transformiert.
-
Beispiel 59: Pflanzentransformation
-
Die
Transformation der Pflanzen erfolgte gemäß dem
in Beispiel 7 beschriebenen Verfahren
-
Beispiel 60: Verfahren bei der phänotypischen
Untersuchung
-
60.1 Aufbau der Untersuchung
-
Es
wurden ungefähr 35 unabhängige T0-Reistransformanten
erzeugt. Die Primärtransformanten wurden für das
Heranziehen und Ernten von T1-Samen von einer Gewebekulturkammer
in ein Gewächshaus umgestellt. Sechs Ereignisse, von denen
sich die T1-Nachkommenschaft im Verhältnis 3:1 für
Vorliegen/Abwesenheit des Transgens aufspaltete, wurden behalten.
Für jedes dieser Ereignisse wurden ungefähr 10 T1-Keimpflanzen,
die das Transgen enthielten (Hetero- und Homozygoten) und ungefähr
10 T1-Keimpflanzen, denen das Transgen fehlte (Nullizygoten) durch
Beobachten der visuellen Markerexpression selektiert. Die transgenen
Pflanzen und die entsprechenden Nullizygoten wurden nebeneinander
in zufälliger Anordnung herangezogen. Die Gewächshausbedingungen
waren kurze Tage (12 Stunden Licht), 28°C im Hellen und
22°C im Dunkeln, und eine relative Feuchtigkeit von 70%.
-
Vier
T1-Ereignisse wurden weiterhin in der T2-Generation nach dem gleichen
Bewertungsverfahren wie für die T1-Generation, jedoch mit
mehr Individuen pro Ereignis bewertet.
-
Vom
Sästadium bis zum Reifestadium wurden die Pflanzen mehrmals
in eine Digital-Imaging-Kammer gestellt. Zu jedem Zeitpunkt wurden
von jeder Pflanze aus mindestens 6 verschiedenen Winkeln Digitalbilder (2048×1536
Pixel, 16 Millionen Farben) aufgenommen.
-
Screening unter Trockenheitsbedingungen
-
Pflanzen
aus T2-Samen wurden unter Normalbedingungen in Blumenerde herangezogen,
bis sie das Stadium des Rispenschiebens erreichten. Anschließend
wurden sie in ein ”trockenes” Areal gestellt,
wo keine Bewässerung erfolgte. Um den Bodenwassergehalt
(BWG) zu verfolgen, wurden Feuchtigkeitssonden in statistisch ausgewählte
Töpfe eingeführt. Sank der BWG unter gewisse Schwellenwerte,
wurden die Pflanzen automatisch erneut gegossen, bis wiederum ein
Normalgehalt erreicht wurde. Dann wurden die Pflanzen wiederum in
Normalbedingungen umgestellt. Ansonsten wurde wie bei den nicht
unter abiotischen Stressbedingungen herangezogenen Pflanzen kultiviert
(Pflanzenreife, Samenernte). Wachstums- und Ertragparameter werden
wie für das Wachstum unter Normalbedingungen beschrieben
aufgezeichnet. Pflanzen, die unter Nicht-Stressbedingungen gewachsen
waren, werden in regelmäßigen Abständen
mit Wasser versorgt, so dass das Wasser und die Nährstoffe
nicht limitierend waren, damit die Bedürfnisse der Pflanzen,
das Wachstum und die Entwicklung zu beenden erfüllt wurde.
-
Effizienz-Screening der Stickstoff-Verwertung
-
Reispflanzen
aus T2-Samen werden in Blumenerde unter Normalbedingungen mit Ausnahme
der Nährlösung herangezogen. Die Töpfe
werden vom Umsetzen bis zur Reife mit einer spezifischen Nährlösung mit
verringertem N-Stickstoff-Gehalt (N), üblicherweise zwischen
7 bis 8 mal weniger, gegossen. Ansonsten wird wie bei den nicht
unter abiotischen Stressbedingungen herangezogenen Pflanzen kultiviert
(Pflanzenreife, Samenernte). Wachstums- und Ertragsparameter werden
wie für das Wachstum unter Normalbedingungen aufgezeichnet.
-
60.2 Statistische Analyse: F-Test
-
Als
statistisches Modell für die Gesamtuntersuchung der phänotypischen
Eigenschaften der Pflanze wurde eine zweifaktorielle ANOVA (Varianzanalyse)
verwendet. Mit allen Parametern, die bei allen Pflanzen von allen
Ereignissen, die mit dem erfindungsgemäßen Gen
transformiert wurden, bestimmt wurden, wurde ein F-Test durchgeführt.
Der F-Test erfolgte als Überprüfung auf einen
Effekt des Gens über alle Transformations-Ereignisse und
zur Feststellung eines Gesamteffekts des Gens, der auch als globaler
Geneffekt bezeichnet wird. Die Signifikanzschwelle für
einen echten globalen Geneffekt wurde bei dem F-Test auf dem 5%-Wahrscheinlichkeitsniveau
festgelegt. Ein signifikanter F-Test-Wert weist auf einen Geneffekt
hin, was bedeutet, dass mehr als nur das einfache Vorhandensein
oder die Lage des Gens die Unterschiede im Phänotyp verursacht.
-
Da
zwei Versuche mit überlappenden Ereignissen durchgeführt
wurden, wurde eine kombinierte Analyse durchgeführt. Dies
ist geeignet, um die Übereinstimmung der Effekte über
die beiden Versuche zu überprüfen, und wenn dies
der Fall ist, Aussagen von beiden Versuchen zu vereinigen, um die
Verlässlichkeit der Schlussfolgerung zu erhöhen.
Bei dem verwendeten Verfahren handelte es sich um einen ”Mixed-Modell”-Ansatz,
der die mehrschichtige Struktur der Daten (d. h. Versuch – Ereignis – Segreganten)
berücksichtigt. Die p-Werte wurden dadurch erhalten, dass
man den Wahrscheinlichkeits-Quotienten-Test mit Chi-Quadrat-Verteilungen
vergleicht.
-
60.3 Messparameter
-
Biomasseparameter-Messung
-
Die
oberirdische Pflanzenfläche (oder Blattbiomasse) wurde
dadurch bestimmt, dass man die Gesamtanzahl Pixel der Digitalbilder
der vom Hintergrund unterschiedlichen oberirdischen Pflanzenteile
zählte. Dieser Wert wurde über die am selben Zeitpunkt
aus unterschiedlichen Winkeln aufgenommenen Bilder gemittelt und
mittels Kalibrierung in einen physikalischen Oberflächenwert,
ausgedrückt in Quadratmillimeter, umgewandelt. Versuche
zeigen, dass die auf diese Weise bestimmte oberirdische Pflanzenfläche
mit der Biomasse der oberirdischen Pflanzenteile korreliert. Die
oberirdische Fläche ist diejenige Fläche, die
zu dem Zeitpunkt, an dem die Pflanzen ihre maximale Blattbiomasse
erreicht hatten, bestimmt wurde.
-
Samenparameter-Messungen
-
Die
reifen Primärrispen wurden geerntet, gezählt,
eingetütet, mit einem Strichcode versehen und dann drei
Tage bei 37°C in einem Ofen getrocknet. Dann wurden die
Rispen gedroschen, und alle Samen wurden gesammelt und gezählt.
Die gefüllten Spelzen wurden von den leeren Spelzen mittels
einer Luftblaseeinrichtung getrennt. Die leeren Spelzen wurden verworfen,
und die restliche Fraktion wurde nochmals gezählt. Die gefüllten
Spelzen wurden auf einer Analysewaage gewogen. Die Anzahl gefüllter
Samen wurde dadurch bestimmt, dass man die Anzahl der gefüllten
Spelzen, die nach dem Abtrennungsschritt verblieben, auszählte. Der
Gesamtsamenertrag wurde dadurch bestimmt, dass man alle von einer
Pflanze geernteten gefüllten Spelzen wog. Die Gesamtsamenzahl
pro Pflanze wurde dadurch bestimmt, dass man die von einer Pflanze
geerntete Anzahl Spelzen auszählte. Das Tausendkorngewicht
(TKG) wird aus der Anzahl der gezählten gefüllten Samen
und ihres Gesamtgewichts extrapoliert. Der Harvest Index (HI) wird
in der vorliegenden Erfindung als das Verhältnis zwischen
dem Gesamtsamengewicht und der oberirdischen Fläche (mm2), multipliziert mit einem Faktor 106, definiert. Die Gesamtzahl Blüten
pro Rispe wird in der vorliegenden Erfindung als das Verhältnis
zwischen der Gesamtsamenzahl und der Anzahl der reifen Primärrispen
definiert. Die Samenfüllungsrate wird in der vorliegenden
Erfindung als das Verhältnis (als Prozent ausgedrückt)
der Anzahl gefüllter Samen zu der Gesamtzahl Samen (oder
Blüten) definiert.
-
Beispiel 61: Ergebnisse der phänotypischen
Untersuchung der transgenen Pflanzen
-
Die
transgenen Reispflanzen, die die GRP-Nukleinsäure nach
SEQ ID NO: 396 unter der Kontrolle des GOS2-Promotors exprimieren,
zeigten einen Anstieg von mehr als 5% für das Gesamtgewicht
der Samen, Anzahl der gefüllten Samen, und Harvest-Index,
wenn sie unter Nichtstress-Bedingungen gewachsen waren.
-
VII. Squamosa-Promotor-Bindungsprotein-ähnlich
11 (SPL11)
-
Beispiel 62: Identifikation von Sequenzen,
die mit der in den erfindungsgemäßen Verfahren
verwendeten SPL11-Nukleinsäuresequenz verwandt sind
-
Unter
den Sequenzen in der Entrez Nucleotides Datenbank am National Center
for Biotechnology Information (NCBI) wurden (Volllängen-cDNA,
ESTs oder genomische) Sequenzen, die mit der in den erfindungsgemäßen
Verfahren verwendeten Nukleinsäuresequenz verwandt sind,
identifiziert. Die Durchsuchung der Sequenzen erfolgte auch mit
anderen öffentlichen und geschützten Datenbanken.
Es wurden Datenbanksequenzabfragewerkzeuge verwendet, wie das Basic
Local Alignment Tool (BLAST) (Altschul et al., (1990), J. Mol.
Biol. 215: 403–410; und Altschul et al.,
(1997), Nucleic Acids Res. 25: 3389–3402). Das
Programm wurde zum Auffinden von Regionen mit lokaler Ähnlichkeit
zwischen Sequenzen verwendet, und zwar durch Vergleichen von Nukleinsäure-
oder Polypeptid-Sequenzen mit Sequenz-Datenbanken und durch Berechnen
der statistischen Signifikanz der Matches. Das durch die erfindungsgemäß verwendete
Nukleinsäure kodierte Polypeptid wurde beispielsweise für
den TBLASTN-Algorithmus verwendet, und zwar unter Ausschaltung der
Default-Einstellungen und des Filters für die Nichtmiteinbeziehung
von Sequenzen mit niedriger Komplexität. Der Output der
Analyse wurde mittels paarweisem Vergleich betrachtet und gemäß der
Wahrscheinlichkeits-Wertung (E-Wert) gereiht, wobei die Wertung
die Wahrscheinlichkeit, dass ein bestimmtes Alignment statistisch vorkommt,
widerspiegelt (je niedriger der E-Wert, desto signifikanter der
Hit). Zusätzlich zu den E-Werten wurden die Vergleiche
auch durch die Prozent der Identität gewertet. Die Prozent
der Identität bezieht sich auf die Anzahl identischer Nukleotide
(oder Aminosäuren) zwischen den zwei verglichenen Nukleinsäure-(oder
Polypeptid)-Sequenzen über eine bestimmte Länge.
In einigen Fällen können die Default-Parameter
so eingestellt werden, dass die Stringenz der Suche angepasst wird.
Der E-Wert kann beispielsweise erhöht werden, dass weniger
stringente Matches gezeigt werden. Auf diese Weise können
nahezu exakte Matches identifiziert werden.
-
Die
Tabelle G1 stellt eine Liste von Nukleinsäuresequenzen
bereit, die mit der in den erfindungsgemäßen Verfahren
verwendeten Nukleinsäuresequenz verwandt sind. Die Polypeptide
mit einer um einen Punkt und eine Ziffer verlängerten Zugangsnummer
veranschaulichen Spleißvarianten. Tabelle G1: Beispiele von SPL11 Nukleinsäuren
und Polypeptiden
| Name | Alias | Ursprungspflanze | Nukleinsäure SEQ
ID NO: | Protein
SEQ ID NO: |
| Arath_SPL11a | At1g27360 | Arabidopsis thaliana | 427 | 428 |
| Arath_SPL11aa | NM_202191
(Spleißvariante) | Arabidopsis thaliana | 429 | 428 |
| Arath_SPL11aaa | NM_001084135 | Arabidopsis thaliana | 430 | 428 |
| Arath_SPL11aaaa | SEQ
ID NO: 1 Variant MiR156 insensitiv | Synthetisch | 431 | 428 |
| Arath_SPL11b | At1g27370 | Arabidopsis thaliana | 432 | 433 |
| Arath_SPL11c | At5g43270 | Arabidopsis thaliana | 434 | 435 |
| Orysa_SPL11a | LOC_Os02g04680 | Oryza
sativa | 436 | 437 |
| Orysa_SPL11b | LOC_Os06g49010 | Oryza
sativa | 438 | 439 |
| Popth_SPL11a | Populus
SPL11a | Populus
Art | 440 | 441 |
| Popth_SPL11b | Populus
SPL11b | Populus
Art | 442 | 443 |
| Popth_SPL11c | Populus
SPL11c | Populus
Art | 444 | 445 |
| Brara_SPL11a | Br_AC189413 | Brassica
rapa | 446 | 447 |
| Zeama_SPL11a | ZM_60752693 | Zea
mays | 448 | 449 |
| Zeama_SPL11b | ZM_SPL11b | Zea
mays | 450 | 451 |
| Triae_SPL11a | Ta_SPi11a | Triticum
aestivum | 452 | 453 |
| Vitvi_SPL11a | Vv_SPL11a | Vitis
vinifera | 454 | 455 |
-
Beispiel 63: Alignment von SPL11-Polypeptidsequenzen
-
Das
Alignment von Polypeptid-Sequenzen erfolgte mit dem AlignX-Programm
von Vector NTI (Invitrogen), das auf dem bekannten Clustal W-Algorithmus
des progressiven Alignments beruht (Thompson et al. (1997)
Nucleic Acids Res 25: 4876–4882; Chenna
et al. (2003). Nucleic Acids Res 31: 3497–3500).
Die Default-Werte lauten: gap open penalty 10, gap extension penalty
0,1, und die gewählte ”weight matrix” ist
Blosum 62 (wenn Polypeptide einem Alignment unterworfen werden).
Um das Alignment weiter zu optimieren, wurden kleinere Veränderungen
von Hand durchgeführt. Eine Konsensussequenz, die die hochkonservierten
Aminosäuren an jeder Position umfasst, ist angegeben; Leerstellen
zwischen bestimmten Aminosäuren in der Konsensussequenz
veranschaulichen eine beliebige Aminosäure. Eine hohe Sequenzkonservierung
unter SPL11-Polypeptiden wird hauptsächlich entlang der
SBP-Domäne der Polypeptide beobachtet. Die Position der
anderen konservierten Sequenzmotive außerhalb der SBP-Domäne,
die hier durch Motiv 1 bis Motiv 4 (SEQ ID NO: 466 to SEQ ID NO:
472) dargestellt ist, ist über die Konsensussequenz angegeben.
-
Die 28 stellt ein Alignment repräsentativer
SPL11 Polypeptide bereit.
-
Beispiel 64: Berechnung der Gesamtidentitätsprozent
zwischen SPL11 Polypeptidsequenzen, die bei der Durchführung
der erfindungsgemäßen Verfahren geeignet sind
-
Die
Gesamtprozent für Ähnlichkeit und Identität
zwischen Volllängenpolypeptidsequenzen, die bei der Durchführung
der erfindungsgemäßen Verfahren geeignet sind,
wurden unter Verwendung von einer der in der Fachwelt verfügbaren
Techniken, nämlich der MatGAT (Matrix Global Alignment
Tool) Software (BMC Bioinformatics. 2003 4: 29. MatGAT:
an application that generstes similarity/identity matrices using
Protein or DNA sequences. Campanella JJ, Bitincka 1, Smalley J;
software hosted by Ledion Bitincka) bestimmt. Die MatGAT-Software
erzeugt Ähnlichkeits-/Identitätsmatrizes für
DNA- oder Proteinsequenzen, ohne dass ein vorheriges Alignment der
Daten erforderlich ist.
-
Das
Programm führt eine Reihe von paarweisen Alignments unter
Verwendung des globalen Alignment-Algorithmus von Myers und Miller
durch (gap opening penalty 12, gap extension penalty 2), berechnet die Ähnlichkeit
und Identität unter Verwendung von z. B. Blosum 62 (für
Polypeptide) und platziert dann die Ergebnisse in einer Abstandsmatrix.
Die Sequenzähnlichkeit ist in der unteren Hälfte
der Trennlinie und die Sequenzidentität in der oberen Hälfte
der diagonalen Trennlinie dargestellt.
-
Bei
den in dem Vergleich eingesetzten Parametern handelte es sich um:
| Scoring
matrix: | Blosum
62 |
| First
Gap: | 12 |
| Extending
gap: | 2 |
-
Die
Ergebnisse der Softwareanalyse sind in der Tabelle G2 für
die Gesamtähnlichkeit und -identität über
die volle Länge der Polypeptidsequenzen und in der Tabelle
G3 für die SBP-Domäne dargestellt. Die Identitätsprozent
sind über der Diagonale und die Ähnlichkeitsprozent
unter der Diagonale (normale Schriftsatz) angegeben.
-
Die
Identitätsprozent zwischen den SP11-Polypeptidsequenzen,
die bei der Durchführung der erfindungsgemäßen
Verfahren geeignet sind, kann nur 20% Aminosäureidentität
gegenüber SEQ ID NO: 428 ausmachen. Gewöhnlich
haben die SPL11 Polypeptide eine Gesamt (entlang der Sequenz des
gesamten Polypeptids) Sequenzidentität im Bereich von 40%.
-
Die
Identitätsprozent zwischen den SBP-Domänen in
den SPL11-Polypeptidsequenzen, die sich zur Durchführung
der erfindungsgemäßen Verfahren eignen, kann nur
30% Aminosäureidentität im Vergleich zu SEQ ID
NO: 456 (SBP-Domäne in Arath_SPL11a oder SEQ ID NO: 428)
betragen. Die SBP-Domänen in den in Table G1 angegebenen
SPL11 Polypeptidsequenzen haben eine Sequenzidentität im
Bereich von 31 bis 93,6%. Tabelle
G2: MatGAT-Ergebnisse für die Gesamtähnlichkeit
und -identität über die Volllänge der
Polypeptidsequenzen.
Tabelle
G3: MatGAT-Ergebnisse für die Gesamtähnlichkeit
und -identität über die SBP-Domäne der
Polypeptidsequenzen.
-
Beispiel 65: Identifikation von Domänen
in Polypeptidsequenzen, die bei der Durchführung der erfindungsgemäßen
Verfahren geeignet sind
-
Bei
der Integrated Resource of Protein Families, Domains and Sites (InterPro)
Datenbank handelt es sich um eine integrierte Plattform für
die häufig verwendeten Signatur-Datenbanken für
Suchen auf Text- und Sequenzbasis. Die InterPro-Datenbank kombiniert
diese Datenbanken, in denen unterschiedliche Methodiken und verschiedene
Mengen an biologischer Information über gut charakterisierte
Proteine verwendet werden, um Proteinsignaturen abzuleiten. Zu den
angeschlossenen Datenbanken zählen SWISS-PROT, PROSITS, TrEMBL,
PRINTS, ProDom und Pfam, Smart und TIGRFAMs. Pfam ist eine große
Sammlung multipler Sequenzalignments und versteckter Markov-Modelle,
die viele gemeinsame Proteindomänen und Familien abdecken.
Pfam wird am Sanger Institute Server im Vereinigten Königreich
gehostet. InterPro wird von dem European Bioinformatics Institute
im Vereinigten Königreich gehostet.
-
Die
Tabelle G4 zeigt die Einstellungen (Gathering cut off, trusted cut
off, und noise cut off), wie in der Pfam-Datenbank beschrieben,
die zum Aufbau der HMMs_fs für verschiedene SBP-Domänen
verwendet wurden. Tabelle G4: HMMs Aufbauinformation in
der Domänendatenbank Pfam Version 21.0
| HMMER
Aufbau information | | |
| | Pfam_Is
[Download HMM] | Pfam
fs [Download HMM] |
| Gathering
cut off | 25.0
25.0; | 25.0
25.0 |
| Trusted
cut off | 41.4
41.4; | 26.2
54.1 |
| Noise
cut off | 20.8
20.8; | 18.0
11.2 |
| Aufbauverfahren
von HMM | hmmbuild
-F HMM_Is SEED | hmmbuild
-f -F HMM_fs SEED |
| | hmmcalibrate
--seed 0 HMM_Is | hmmcalibrate
--seed 0 HMM_fs |
-
Die
Ergebnisse des Pfam-Scans für die repräsentativen
SPL11-Polypeptide pflanzlichen Ursprungs sind in der Tabelle G5
angegeben. Die Aminosäurekoordinaten für die SBP-Domäne
in der Referenzsequenz sind in der entsprechenden Spalte angegeben.
Der E-Wert des Alignments ist ebenfalls angegeben. Tabelle G5: Pfam-Scan-Ergebnisse für
die SBP-Domäne (Pfam Referenz PF03110) der repräsentativen SPL11-Polypeptide,
wie sie auf Pfam Version 21.0 erhalten wurden.
| Sequenzname | Aminosäurekoordinaten
der SBP Domäne | e-Wert
des Alignments |
| Arath_SPL11a | 174–252 | 1.10E–50 |
| Arath_SPL11b | 175–253 | 1.61E–47 |
| Arath_SPL11c | 168–246 | 1.90E–52 |
| Orysa_SPL11a | 181–259 | 5.10E–50 |
| Orysa_SPL11b | 179–257 | 3.90E–47 |
| Popth_SPL11a | 170–277 | 170–277 |
| Popth_SPL11b | 180–277 | 1.50E–41 |
| Popth_SPL11c | 171–249 | 7.80E–52 |
| Brara_SPL11a | 180–279 | 5.20E–44 |
| Zeama_SPL11a | 161–239 | 2.20E–51 |
| Zeama_SPL11b | 159–237 | 1.10E–48 |
| Triae_SPL11a | 184–262 | 2.90E–52 |
| Vitvi_SPL11a | 171–249 | 7.80E–52 |
-
Beispiel 66: Klonierung der in den erfindungsgemäßen
Verfahren verwendeten Nukleinsäuresequenz
-
Die
in den erfindungsgemäßen Verfahren verwendete
Nukleinsäuresequenz wurde durch PCR mit einer speziell
angefertigten cDNA-Bank von Arabidopsis thaliana Keimlingen (in
pCMV Sport 6.0; Invitrogen, Paisley, UK) als Matrize amplifiziert.
Die PCR wurde mit Hifi Taq DNA-Polymerase in Standardbedingungen, mit
200 ng Matrize in einem 50 μl PCR-Mix durchgeführt.
Ein stromaufwärts und ein stromabwärts gelegener Primer
nach SEQ ID NO: 473 bzw. SEQ ID NO: 474 wurden zur Amplifikation
des kodierenden Bereichs der SPL11 Nukleinsäure nach SEQ
ID NO: 427 durch PCR (Polymerasekettenreaktion) verwendet. Die Primer
umfassen die AttB-Stellen für Gateway-Rekombination zur
Erleichterung der Klonierung des amplifizierten PCR DNA-Fragments
in einen Gateway-Klonierungsvektor.
-
Das
amplifizierte PCR-Fragment wurde ebenfalls mit Standard-Verfahren
gereinigt. Der erste Schritt des Gateway-Verfahrens, die BP-Reaktion,
wurde dann durchgeführt, bei der das PCR-Fragment in vivo
mit dem Plasmid pDONR201 unter Bildung eines – gemäß Gateway-Teminologie – ”entry
clone” pSPL11 rekombinierte. Das Plasmid pDONR201 wurde
käuflich von Invitrogen als Bestandteil der Gateway® Technik erworben.
-
Der
SEQ ID NO: 427 enthaltende ”entry clone” wurde
anschließend in einer LR-Reaktion mit einem Zielvektor,
der für die Transformation von Oryza sativa verwendet wird,
verwendet. Dieser Vektor enthält als funktionelle Elemente
innerhalb der T-DNA-Grenzen Folgendes: Einen pflanzlichen Selektionsmarker;
eine screenbare Markerexpressionskassette, und eine Gateway-Kassette,
die für die LR-in-vivo-Rekombination mit der interessierenden
Nukleinsäuresequenz, die bereits in den ”entry
clone” kloniert wurde, vorgesehen ist. Ein GOS2-Promotor
aus Reis (SEQ ID NO: 476) für die konstitutive Expression
befand sich stromaufwärts dieser Gateway-Kassette in dem
Vektor pGOS2::SPL11. Die SPL11-kodierende Sequenz wurde auch in
einen Expressionsvektor kloniert, der einen WSI18-Promotor aus Reis
(SEQ ID NO: 477) für eine (ABA induzierbare) Expression
(pWSI18::SPL11) enthielt.
-
Nach
dem LR-Rekombinationsschritt wurden die erhaltenen Expressionsvektoren
pGOS2::SPL11 und pWSI18::SPL11 (31)
in den Agrobacterium Stamm LBA4044 nach fachbekannten Verfahren
transformiert.
-
Beispiel 67: Pflanzentransformation
-
Die
Transformation der Pflanzen erfolgte gemäß dem
in Beispiel 7 beschriebenen Verfahren.
-
Beispiel 68: Verfahren bei der phänotypischen
Untersuchung
-
60.1 Aufbau der Untersuchung
-
Es
wurden ungefähr 35 unabhängige T0-Reistransformanten
erzeugt. Die Primärtransformanten wurden für das
Heranziehen und Ernten von T1-Samen von einer Gewebekulturkammer
in ein Gewächshaus umgestellt. Sechs Ereignisse, von denen
sich die T1-Nachkommenschaft im Verhältnis 3:1 für
Vorliegen/Abwesenheit des Transgens aufspaltete, wurden behalten.
Für jedes dieser Ereignisse wurden ungefähr 10 T1-Keimpflanzen,
die das Transgen enthielten (Hetero- und Homozygoten) und ungefähr
10 T1-Keimpflanzen, denen das Transgen fehlte (Nullizygoten) durch
Beobachten der visuellen Markerexpression selektiert. Die transgenen
Pflanzen und die entsprechenden Nullizygoten wurden nebeneinander
in zufälliger Anordnung herangezogen. Die Gewächshausbedingungen
waren kurze Tage (12 Stunden Licht), 28°C im Hellen und
22°C im Dunkeln, und eine relative Feuchtigkeit von 70%.
Die unter Nichtstressbedingungen gezogenen Pflanzen werden in regelmäßigen
Abständen mit Wasser versorgt, so dass gewährleistet
war, dass Wasser und Nährstoffe nicht limitierend waren,
damit die Bedürfnisse der Pflanze, zu wachsen und sich
zu entwickeln, erfüllt werden.
-
Vier
T1-Ereignisse wurden weiterhin in der T2-Generation nach dem gleichen
Untersuchungsverfahren wie für die T1-Generation, jedoch
mit mehr Individuen pro Ereignis untersucht. Vom Sästadium
bis zum Reifestadium wurden die Pflanzen mehrmals in eine Digital-Imaging-Kammer
gestellt. Zu jedem Zeitpunkt wurden von jeder Pflanze aus mindestens
6 verschiedenen Winkeln Digitalbilder (2048×1536 Pixel,
16 Millionen Farben) aufgenommen.
-
Screening unter Trockenheitsbedingungen
-
Pflanzen
aus T2-Samen wurden unter Normalbedingungen in Blumenerde herangezogen,
bis sie das Stadium des Rispenschiebens erreichten. Anschließend
wurden sie in ein ”trockenes” Areal gestellt,
wo keine Bewässerung erfolgte. Um den Bodenwassergehalt
(BWG) zu verfolgen, wurden Feuchtigkeitssonden in statistisch ausgewählte
Töpfe eingeführt. Sank der BWG unter gewisse Schwellenwerte,
wurden die Pflanzen automatisch erneut gegossen, bis wiederum ein
Normalgehalt erreicht wurde. Dann wurden die Pflanzen wiederum in
Normalbedingungen umgestellt. Ansonsten wurde wie bei den nicht
unter abiotischen Stressbedingungen herangezogenen Pflanzen kultiviert
(Pflanzenreife, Samenernte). Wachstums- und Ertragparameter werden
wie für das Wachstum unter Normalbedingungen beschrieben
aufgezeichnet.
-
Effizienz-Screening der Stickstoff-Verwertung
-
Reispflanzen
aus T2-Samen werden in Blumenerde unter Normalbedingungen mit Ausnahme
der Nährlösung herangezogen. Die Töpfe
werden vom Umsetzen bis zur Reife mit einer spezifischen Nährlösung mit
verringertem N-Stickstoff-Gehalt (N), üblicherweise zwischen
7 bis 8 mal weniger, gegossen. Ansonsten wird wie bei den nicht
unter abiotischen Stressbedingungen herangezogenen Pflanzen kultiviert
(Pflanzenreife, Samenernte). Wachstums- und Ertragsparameter werden
wie für das Wachstum unter Normalbedingungen aufgezeichnet.
-
68.2 Statistische Analyse: F-Test
-
Als
statistisches Modell für die Gesamtuntersuchung der phänotypischen
Eigenschaften der Pflanze wurde eine zweifaktorielle ANOVA (Varianzanalyse)
verwendet. Mit allen Parametern, die bei allen Pflanzen von allen
Ereignissen, die mit dem erfindungsgemäßen Gen
transformiert wurden, bestimmt wurden, wurde ein F-Test durchgeführt.
Der F-Test erfolgte als Überprüfung auf einen
Effekt des Gens über alle Transformations-Ereignisse und
zur Feststellung eines Gesamteffekts des Gens, der auch als globaler
Geneffekt bezeichnet wird. Die Signifikanzschwelle für
einen echten globalen Geneffekt wurde bei dem F-Test auf dem 5%-Wahrscheinlichkeitsniveau
festgelegt. Ein signifikanter F-Test-Wert weist auf einen Geneffekt
hin, was bedeutet, dass mehr als nur das einfache Vorhandensein
oder die Lage des Gens die Unterschiede im Phänotyp verursacht.
-
Wenn
zwei Versuche mit überlappenden Ereignissen durchgeführt
wurden, wurde eine kombinierte Analyse durchgeführt. Dies
ist geeignet, um die Übereinstimmung der Effekte über
die beiden Versuche zu überprüfen, und wenn dies
der Fall ist, Aussagen von beiden Versuchen zu vereinigen, um die
Verlässlichkeit der Schlussfolgerung zu erhöhen.
Bei dem verwendeten Verfahren handelte es sich um einen ”Mixed-Modell”-Ansatz,
der die mehrschichtige Struktur der Daten (d. h. Versuch – Ereignis – Segreganten)
berücksichtigt. Die p-Werte wurden dadurch erhalten, dass
man den Wahrscheinlichkeits-Quotienten-Test mit Chi-Quadrat-Verteilungen
vergleicht.
-
68.3 Messparameter
-
Biomasseparameter-Messung
-
Vom
Sästadium bis zum Reifestadium wurden die Pflanzen mehrmals
in eine Digital-Imaging-Kammer gestellt. Zu jedem Zeitpunkt wurden
von jeder Pflanze aus mindestens 6 verschiedenen Winkeln Digitalbilder (2048×1536
Pixel, 16 Millionen Farben) aufgenommen. Die oberirdische Pflanzenfläche
(oder Blattbiomasse) wurde dadurch bestimmt, dass man die Gesamtanzahl
Pixel der Digitalbilder der vom Hintergrund unterschiedlichen oberirdischen
Pflanzenteile zählte. Dieser Wert wurde über die
am selben Zeitpunkt aus unterschiedlichen Winkeln aufgenommenen
Bilder gemittelt und mittels Kalibrierung in einen physikalischen
Oberflächenwert, ausgedrückt in Quadratmillimeter,
umgewandelt. Versuche zeigen, dass die auf diese Weise bestimmte oberirdische
Pflanzenfläche mit der Biomasse der oberirdischen Pflanzenteile
korreliert. Die oberirdische Fläche ist diejenige Fläche,
die zu dem Zeitpunkt, an dem die Pflanze ihre maximale Blattbiomasse
erreicht hatte, bestimmt wurde. Die Jungpflanzenvitalität
ist die oberirdische Fläche der Pflanze (Keimpflanze) 3
Wochen nach der Keimung. Die Steigerung der Wurzelbiomasse wird
als Steigerung der Gesamtwurzelbiomasse (gemessen als maximale Wurzelbiomasse,
die während der Lebensdauer einer Pflanze beobachtet wurde)
oder als Steigerung des Wurzel/Spross-Index (gemessen als Verhältnis
zwischen Wurzelmasse und Sprossmasse im Zeitraum des aktiven Wachstums
von Wurzel und Spross) ausgedrückt.
-
Für
die Bestimmung der Jungpflanzenvitalität wurde die Gesamtzahl
der Pixel aus überirdischen Pflanzenteilen vom Hintergrund
unterschieden. Dieser Wert wurde für die Bilder gemittelt,
die zum gleichen Zeitpunkt aus verschiedenen Winkeln aufgenommen
wurden, und wurde durch Kalibrierung in einen physikalischen Oberflächenwert
umgewandelt, der in mm2 ausgedrückt
wird. Die nachstehend beschriebenen Ergebnisse stammen von Pflanzen
drei Wochen nach der Keimung.
-
Samenparameter-Messungen
-
sDie
reifen Primärrispen wurden geerntet, gezählt,
eingetütet, mit einem Strichcode versehen und dann drei
Tage bei 37°C in einem Ofen getrocknet. Dann wurden die
Rispen gedroschen, und alle Samen wurden gesammelt und gezählt.
Die gefüllten Spelzen wurden von den leeren Spelzen mittels
einer Luftblaseeinrichtung getrennt. Die leeren Spelzen wurden verworfen,
und die restliche Fraktion wurde nochmals gezählt. Die
gefüllten Spelzen wurden auf einer Analysewaage gewogen.
Die Anzahl gefüllter Samen wurde dadurch bestimmt, dass
man die Anzahl der gefüllten Spelzen, die nach dem Abtrennungsschritt
verblieben, auszählte. Der Gesamtsamenertrag wurde dadurch
bestimmt, dass man alle von einer Pflanze geernteten gefüllten
Spelzen wog. Die Gesamtsamenzahl pro Pflanze wurde dadurch bestimmt,
dass man die von einer Pflanze geerntete Anzahl Spelzen auszählte.
Das Tausendkorngewicht (TKG) wird aus der Anzahl der gezählten
gefüllten Samen und ihres Gesamtgewichts extrapoliert.
Der Harvest Index (HI) wird in der vorliegenden Erfindung als das
Verhältnis zwischen dem Gesamtsamengewicht und der oberirdischen
Fläche (mm2), multipliziert mit
einem Faktor 106, definiert. Die Gesamtzahl
Blüten pro Rispe wird in der vorliegenden Erfindung als
das Verhältnis zwischen der Gesamtsamenzahl und der Anzahl
der reifen Primärrispen definiert. Die Samenfüllungsrate wird
in der vorliegenden Erfindung als das Verhältnis (als Prozent
ausgedrückt) der Anzahl gefüllter Samen zu der
Gesamtzahl Samen (oder Blüten) definiert.
-
Beispiel 69: Ergebnisse der phänotypischen
Untersuchung transgener Pflanzen
-
Die
Ergebnisse der Untersuchung der transgenen Reispflanzen, die eine
SP11-Nukleinsäure exprimieren, die dem Konstrukt pGOS2::SPL11
und pWSI18::SPL11 entsprechen, sind unabhängig davon, ob
sie unter Trockenheitsstress oder unter Nichtstressbedingungen erhalten
wurde, hier angegeben. Eine Steigerung von mehr als 5% wurde für
einen oder mehrere der folgenden Parameter erhalten, das Auftreten
von Jungpflanzenvitalität (Jungpflanzenvitalität
der Keimpflanze), den Gesamtsamenertrag (Samengewicht), Samenfüllrate
(Samenfüllungsrate), die Anzahl gefüllter Samen,
die Anzahl Blüten (Samen) pro Rispe, und Harvest-Index,
und mindestens 3% für das Tausendkorn (1000-Samen)-Gewicht
in den transgenen Pflanzen im Vergleich zu Kontroll-Nullizygoten-Pflanzen
in mindestens einer der Wachstumsbedingungen beobachtet.
-
Zusammenfassung
-
Pflanzen mit verbesserten
Ertragsmerkmalen und Verfahren zu ihrer Herstellung
-
Die
vorliegende Erfindung betrifft allgemein das Gebiet der Molekularbiologie
und befasst sich mit einem Verfahren zur Verbesserung von Ertragsmerkmalen
und/oder zur Verbesserung verschiedener Pflanzenwachstumseigenschaften,
bei dem man eine Nukleinsäure, die ein GRP (wachstumsregulierendes
Protein) kodiert, in einer Pflanze moduliert. Das GRP ist ausgewählt
aus einem Protein mit LOB-Domäne (LOB: Lateral Organ Boundaries,
Laterale Organgrenzen), hier abgekürzt als LBD-Polypeptid,
einem JMJC (JUMONJI-C)-Polypeptid, einem CKI (Casein Kinase I) Polypeptid,
einem bHLH11-ähnlichen (basische Helix-Loop-Helix 11) Protein,
einem Pflanzen-Homöodomänen-Finger-Homöodomänen(PHDf-HD)
Polypeptid, einem ASR (abszisinsäure-, stress-, und reifungsinduzierten)
Polypeptid und/oder einem Squamosa-Promotor-Bindungsprotein-ähnlichen
11 (SPL11) Transkriptionsfaktorpolypeptid. Die vorliegende Erfindung
betrifft auch Pflanzen mit einer modulierten Expression einer Nukleinsäure,
die ein GRP kodiert, wobei die Pflanzen verbesserte Wachstumseigenschaften
im Vergleich zu entsprechenden Wildtyp-Pflanzen oder anderen Kontrollpflanzen aufweisen.
Die Erfindung stellt auch neue GRP-Nukleinsäuren und GRP-Polypeptide,
sowie Konstrukte bereit, die sich in den erfindungsgemäßen
Verfahren eignen.
-
Es folgt ein
Sequenzprotokoll nach WIPO St. 25.
Dieses kann von der
amtlichen Veröffentlichungsplattform des DPMA heruntergeladen
werden.
-
ZITATE ENTHALTEN IN DER BESCHREIBUNG
-
Diese Liste
der vom Anmelder aufgeführten Dokumente wurde automatisiert
erzeugt und ist ausschließlich zur besseren Information
des Lesers aufgenommen. Die Liste ist nicht Bestandteil der deutschen
Patent- bzw. Gebrauchsmusteranmeldung. Das DPMA übernimmt
keinerlei Haftung für etwaige Fehler oder Auslassungen.
-
Zitierte Patentliteratur
-
- - US 20060218674 [0012]
- - US 7196245 [0031]
- - US 7154025 [0040]
- - EP 1033405 [0044]
- - US 5811238 [0069]
- - US 6395547 [0069]
- - WO 2004/070039 [0074, 0080, 0080, 0080, 0080, 0080, 0080, 0080, 0080, 0080, 0082]
- - WO 2004/065596 [0074]
- - US 4962028 [0074]
- - WO 01/14572 [0074]
- - WO 95/14098 [0074]
- - WO 94/12015 [0074]
- - US 5401836 [0079]
- - US 20050044585 [0079]
- - EP 99106056 [0080]
- - US 5565350 [0088, 0117]
- - WO 9322443 [0088]
- - WO 98/53083 [0095]
- - WO 99/53050 [0095]
- - US 4987071 [0104]
- - US 5116742 [0104]
- - WO 94/00012 [0104]
- - WO 95/03404 [0104]
- - WO 00/00619 [0104]
- - WO 97/13865 [0104]
- - WO 97/38116 [0104]
- - WO 98/36083 [0105]
- - WO 99/15682 [0105]
- - WO 00/15815 [0117]
- - EP 1198985 A1 [0120]
- - US 5164310 [0779]
-
Zitierte Nicht-Patentliteratur
-
- - Wang et al.,
Planta (2003), 218: 1–14 [0006]
- - Shuai et al., Plant Physiol. 129, 747–761, 2002 [0012]
- - Bortiri et al., Plant Cell 18, 574–587, 2006 [0012]
- - Liu et al., Plant J. 43, 47–56, 2005 [0012]
- - Inukai et al., Plant Cell 17, 1387–1396, 2005 [0012]
- - Evans et al., Plant Cell 19, 46–62, 2007 [0012]
- - Chalfun-Junior et al., Plant Mol. Biol. 57, 559–575,
2005 [0012]
- - Iwakawa et al., Plant Cell Physiol. 43, 467–478,
2002 [0012]
- - Yang et al. (Molecular Phylogenetics and Evolution 39, 248–262,
2006) [0012]
- - Iwakawa et al. (2005) [0012]
- - Shuai et al. (2002) [0012]
- - Toyoda et al. 2003 Dev Cell. 5 (1): 85–97. [0016]
- - Tsukada, Nature. 2006; 439 (7078): 811–6 [0017]
- - Linke et al. (2004) J. Biol. Chem., Vol. 279, 14391–14397 [0017]
- - Dann et al; Proc Natl Acad Sci U S A. 2002; 99 (24): 15351–15356 [0018]
- - Trewick et al. EMBO Rep. 2005 (4): 15–20 [0018]
- - Noh et al. Plant Cell. 2004. 16 (10): 2601–13 [0019]
- - Hoekstra MF et al., Science 253: Brockman JL et al., Proc.
Natl Acad Sci. USA 89: 9454–9458, 1992 [0022]
- - Dhillon N und Hoekstra MF, EMBO J 13: 2777–2788,
1994 [0022]
- - Gross and Anderson, 1998 Cell Signal 10: 699–711 [0022]
- - Craves und Roach, 1995, J Biol Chem 270: 21689–21694 [0022]
- - Klimczak und Cashmore AR, 1993. Biochem. J. 293: 283–288 [0023]
- - Liu et al. 2003, Plant Journal. 36, 189–202 [0023]
- - Lee et al. 2005 Plant Cell. 17 (10): 2817–2831 [0023]
- - Lee et al. 2005 [0023]
- - Liu et al. [0023]
- - Riechmann et al. (2000) Science 290: 2105–2109 [0026]
- - Gao et al. (2006) Bioinformatics 2006, 22 (10): 1286–7 [0026]
- - Richardt et al. (2007) Plant Phys 143 (4): 1452–1466 [0027]
- - Bellman & Werr
(1992) EMBO J 11: 3367–3374 [0028]
- - Schindler et al. (1993) Plant J 4: 137–150 [0028]
- - Halbach et al. (2000) Nucleic Acid Res 28 (18): 3542–3550 [0028]
- - Halbach et al. (2000) [0029]
- - Halbach et al. (2000) [0029]
- - Uberlacker et al. (1996) Plant Cell 8: 349–362 [0030]
- - Ito et al. (2004) Gene 331: 9–15 [0030]
- - Toledo-Ortiz et al., Plant Cell 15, 1749–1770, 2003 [0034]
- - Bailey et al., Plant Cell 15, 2497–2501, 2003 [0034]
- - Li et al Plant Physiol. 141, 1167–1184, 2006 [0034]
- - Li et al. (2006) [0034]
- - Vi et al., Plant Physiol. 138, 2087–2096 [0035]
- - Iusem et al 1993 Plant Physiol 102: 1353–1354 [0038]
- - Carrai et al. 2004 Trends Plant Sci 9: 57–59 [0038]
- - Frankel et al. 2006. Gene Pages 74–83 [0038]
- - Kalifa et al., 2004a Biochem J 381: 373–378 [0038]
- - Cakir et al., 2003 Plant Cell 15: 2165–2180 [0038]
- - Goldgur et al. Plant Physiol. 2007 Feb; 143 (2): 617–28 [0038]
- - Frankel et al. Plant Mol Biol. 2007 Mar; 63 (5): 719–30 [0039]
- - Kalifa et al. 2004 Biochem J. 2004 Jul 15; 381 (Pt 2): 373–8 [0039]
- - Rom et al. 2006. Biochimie. 88 (6): 621–8 [0039]
- - Goldgur et al. 2007. Plant Physiol. Feb; 143 (2): 617–28 [0039]
- - Yang et al. 2005. Plant Physiol 139: 836–846 [0040]
- - Klein et al. (1996) Mol Gen Genet 259: 7–16 [0042]
- - Cardon et al. (1999) Gene 237: 91–104 [0042]
- - Yamasaki et al. (2004) J Mol Biol 337: 49–63 [0042]
- - Rhoades et al. (2002) Cell 110: 513–520 [0042]
- - Riechmann et al. (2000) Science 290: 2105–2109 [0043]
- - Cardon et al. (1999) [0043]
- - Cardon et al. (1997) Plant J 12: 367–377 [0044]
- - Creighton (1984), Proteins. W. H. Freeman and Company (Hrsg.) [0054]
- - Terpe, Appl. Microbiol. Biotechnol. 60, 523–533,
2003 [0056]
- - Meinkoth und Wahl, Anal. Biochem., 138: 267–284,
1984 [0062]
- - Sambrook et al., (2001), Molecular Cloning: a laboratory manual,
3. Ausgabe, Cold Spring Harbor Laboratory Press, CSH, New York [0066]
- - Current Protocols in Molecular Biology, John Wiley & Sons, N. Y. (1989
und jährliche Neufassungen) [0066]
- - Foissac und Schiex (2005) BMC Bioinformatics 6: 25 [0067]
- - Castle et al., (2004), Science 304 (5674): 1151–4 [0069]
- - Neid et al., 1996 Genome Methods 6: 986–994 [0072]
- - McElroy et al., Plant Cell, 2: 163–171, 1990 [0074]
- - Odell et al., Nature, 313: 810–812, 1985 [0074]
- - Nilsson et al., Physiol. Plant. 100: 456–462, 1997 [0074]
- - Christensen et al., Plant Mol. Biol. 18: 675–689,
1992 [0074]
- - Buchholz et al., Plant Mol. Biol. 25 (5): 837–43,
1994 [0074]
- - Lepetit et al., Mol. Gen. Genet. 231: 276–285, 1992 [0074]
- - Wu et al., Plant Mol. Biol. 11: 641–649, 1988 [0074]
- - An et al., Plant J. 10 (1); 107–121, 1996 [0074]
- - Sanger et al., Plant. Mol. Biol., 14, 1990: 433–443 [0074]
- - Leisner (1988), Proc. Natl. Acad. Sci. USA 85 (5): 2553 [0074]
- - Jain et al., Crop Science, 39 (6), 1999: 1696 [0074]
- - Jain et al., Crop Science, 39 (6), 1999: 1696 [0074]
- - Shaw et al., (1984), Nucleic Acids Res. 12 (20): 7831–7846 [0074]
- - Gatz 1997, Annu. Rev. Plant Physiol. Plant Mol. Biol., 48:
89–108 [0077]
- - Plant Mol. Biol. 1995 Jan; 27 (2): 237–48 [0079]
- - Kovama et al., 2005; Mudge et al. (2002, Plant J. 31: 341) [0079]
- - Xiao et al., 2006 [0079]
- - Nitz et al. (2001) Plant Sci. 161 (2): 337–346 [0079]
- - Tingey et al., EMBO J. 6: 1, 1987 [0079]
- - Van der Zaal et al., Plant Mol. Biol. 16, 983, 1991 [0079]
- - Oppenheimer et al., Gene 63: 87, 1988 [0079]
- - Conkling et al., Plant Physiol. 93: 1203, 1990 [0079]
- - Suzuki et al., Plant Mol. Biol. 21: 109–119, 1993 [0079]
- - Baumberger et al., 2001, Genes & Dev. 15: 1128 [0079]
- - Lauter et al. (1996, PNAS 3: 8139) [0079]
- - Lauter et al. (1996, PNAS 3: 8139) [0079]
- - Liu et al., Plant Mol. Biol. 153: 386–395, 1991 [0079]
- - Downey et al. (2000, J. Biol. Chem. 275: 39420) [0079]
- - W Song (1997) PhD Thesis, North Carolina State University,
Raleigh, NC USA [0079]
- - Wang et al., 2002, Plant Sci. 163: 273 [0079]
- - Diener et al. (2001, Plant Cell 13: 1625) [0079]
- - Quesada et al. (1997, Plant Mol. Biol. 34: 265) [0079]
- - Qing Qu und Takaiwa (Plant Biotechnol. J. 2, 113–125,
2004) [0080]
- - Simon et al., Plant Mol. Biol. 5: 191, 1985 [0080]
- - Scofield et al., J. Biol. Chem. 262: 12202, 1987 [0080]
- - Baszczynski et al., Plant Mol. Biol. 14: 633, 1990 [0080]
- - Pearson et al., Plant Mol. Biol. 18: 235–245, 1992 [0080]
- - Ellis et al., Plant Mol. Biol. 10: 203–214, 1988 [0080]
- - Takaiwa et al., Mol. Gen. Genet. 208: 15–22, 1986 [0080]
- - Takaiwa et al., FEBS Letts. 221: 43–47, 1987 [0080]
- - Matzke et al., Plant Mol. Biol., 14 (3): 323–32,
1990 [0080]
- - Stalberg et al., Planta 199: 515–519, 1996 [0080]
- - Mol. Gen. Genet. 216: 81–90, 1989; NAR 17: 461–2,
1989 [0080]
- - Albani et al., Plant Cell, 9: 171–184, 1997 [0080]
- - EMBO J. 3: 1409–15, 1984 [0080]
- - Diaz et al., (1995), Moll Gen. Genet. 248 (5): 592–8 [0080]
- - Theor. Appl. Gen. 98: 1253–62, 1999 [0080]
- - Plant J. 4: 343–55, 1993 [0080]
- - Mol. Gen. Genet. 250: 750–60, 1996 [0080]
- - Mena et al., The Plant Journal, 116 (1): 53–62, 1998 [0080]
- - Vicente-Carbajosa et al., Plant J. 13: 629–640, 1998 [0080]
- - Wu et al., Plant Cell Physiology 39 (8) 885–889,
1998 [0080]
- - Wu et al., Plant Cell Physiology 39 (8) 885–889,
1998 [0080]
- - Sato et al., Proc. Natl. Acad. Sci. USA, 93: 8117–8122,
1996 [0080]
- - Nakase et al., Plant Mol. Biol. 33: 513–522, 1997 [0080]
- - Trans. Res. 6: 157–68, 1997 [0080]
- - Plant J. 12: 235–46, 1997 [0080]
- - DeRose et al., Plant Mol. Biol. 32: 1029–35, 1996 [0080]
- - Postma-Haarsma et al., Plant Mol. Biol. 39: 257–71,
1999 [0080]
- - Wu et al., J. Biochem. 123: 386, 1998 [0080]
- - Cummins et al., Plant Mol. Biol. 19: 873–876, 1992 [0080]
- - Lanahan et al., Plant Cell 4: 203–211, 1992 [0080]
- - Skriver et al., Proc. Natl. Acad. Sci., USA 88: 7266–7270,
1991 [0080]
- - Cejudo et al., Plant Mol. Biol. 20: 849–856, 1992 [0080]
- - Kalla et al., Plant J. 6: 849–60, 1994 [0080]
- - Leah et al., Plant J. 4: 579–89, 1994 [0080]
- - Selinger et al., Genetics 149; 1125–38, 1998 [0080]
- - Takaiwa et al., (1986), Mol. Gen. Genet. 208: 15–22 [0080]
- - Takaiwa et al., (1987), FERS Letts. 221: 43–47 [0080]
- - Matzke et al., (1990), Plant Mol. Biol. 14 (3): 323–32 [0080]
- - Colot et al., (1989), Mol. Gen. Genet. 216: 81–90 [0080]
- - Anderson et al., (1989), NAR 17: 461–2 [0080]
- - Albani et al., (1997), Plant Cell 9: 171–184 [0080]
- - Rafalski et al., (1984), EMBO 3: 1409–15 [0080]
- - Diaz et al., (1995), Mol. Gen. Genet. 248 (5): 592–8 [0080]
- - Cho et al., (1999), Theor. Appl. Genet. 98: 1253–62 [0080]
- - Muller et al., (1993), Plant J. 4: 343–55 [0080]
- - Sorenson et al., (1996), Mol. Gen. Genet. 250: 750–60 [0080]
- - Mena et al., (1998), Plant J. 116 (1): 53–62 [0080]
- - Onate et al., (1999), J. Biol. Chem. 274 (14): 9175–82 [0080]
- - Vicente-Carbajosa et al., (1998), Plant J. 13: 629–640 [0080]
- - Wu et al., (1998), Plant Cell Physiol. 39 (8) 885–889 [0080]
- - Wu et al., (1998), Plant Cell Physiol. 39 (8) 885–889 [0080]
- - Nakase et al., (1997), Plant Molec. Biol. 33: 513–522 [0080]
- - Russell et al., (1997), Trans. Res. 6: 157–68 [0080]
- - Opsahl-Ferstad et al., (1997), Plant J. 12: 235–46 [0080]
- - DeRose et al., (1996), Plant Mol. Biol. 32: 1029–35 [0080]
- - Sato et al., Proc. Natl. Acad. Sci. USA, 93: 8117–8122,
1996 [0080]
- - Postma-Haarsma et al., Plant Mol. Biol. 39: 257–71,
1999 [0080]
- - Lanahan et al., Plant Cell 4: 203–211, 1992 [0080]
- - Skriver et al., Proc. Natl. Acad. Sci. USA 88: 7266–7270,
1991 [0080]
- - Cejudo et al., Plant Mol. Biol. 20: 849–856, 1992 [0080]
- - Kalla et al., Plant J. 6: 849–60, 1994 [0080]
- - Leah et al., Plant J. 4: 579–89, 1994 [0080]
- - Selinger et al., Genetics 149; 1125–38, 1998 [0080]
- - Fukavama et al., 2001 [0082]
- - Kausch et al., 2001 [0082]
- - Liu et al., 2003 [0082]
- - Nomura et al., 2000 [0082]
- - Panguluri et al., 2005 [0082]
- - Sato et al. (1996) Proc. Natl. Acad. Sci. USA, 93: 8117–8122 [0083]
- - Wagner & Kohorn
(2001) Plant Cell 13 (2): 303–318 [0083]
- - Buchman and Berg (1988) Mol. Cell biol. 8: 4395–4405 [0090]
- - Callis et al. (1987) Genes Dev 1: 1183–1200 [0090]
- - The Maize Handbook, Chapter 116, Freeling and Walbot, Eds.,
Springer, N. Y. (1994) [0090]
- - Gaultier et al. (1987) Nucl Ac Res 15: 6625–6641 [0103]
- - Inoue et al. (1987) Nucl Ac Res 15, 6131–6148 [0103]
- - Inoue et al. (1987) FERS Lett. 215, 327–330 [0103]
- - Haselhoff and Gerlach (1988) Nature 334, 585–591 [0104]
- - Bartel and Szostak (1993) Science 261, 1411–1418 [0104]
- - Angell and Baulcombe ((1999) Plant J 20 (3): 357–62 [0105]
- - Helene, C., Anticancer Drug Res. 6, 569–84, 1991;
Helene et al., Ann. N. Y. Acad. Sci. 660, 27–36 1992 [0107]
- - Maher, L. J. Bioassays 14, 807–15, 1992 [0107]
- - Schwab et al., Dev. Cell 8, 517–527, 2005 [0111]
- - Schwab et al., Plant Cell 18, 1121–1133, 2006 [0111]
- - Tribble et al., J. Biol. Chem., 275, 2000: 22255–22267 [0116]
- - Velmurugan et al., J. Cell Biol., 149, 2000: 553–566 [0116]
- - Krens, F. A. et al., (1982), Nature 296, 72–74 [0120]
- - Negrutiu I et al., (1987), Plant Mol. Biol. 8: 363–373 [0120]
- - Shillito R. D. et al., (1985), Bio/Technol. 3, 1099–1102 [0120]
- - Crossway A et al., (1986), Mol. Gen. Genet. 202: 179–185 [0120]
- - Klein TM et al., (1987), Nature 327: 70 [0120]
- - Clough und Bent, Plant J. (1998), 16, 735–743 [0120]
- - Aldemita und Hodges (Planta 199: 612–617, 1996 [0120]
- - Chan et al., (Plant Mol. Biol. 22 (3): 491–506, 1993 [0120]
- - Hiei et al., (Plant J. 6 (2): 271–282, 1994 [0120]
- - Ishida et al., (Nat. Biotechnol. 14 (6): 745–50,
1996 [0120]
- - Frame et al. (Plant Physiol. 129 (1): 13–22, 2002 [0120]
- - B. Jenes et al., Techniques for Gene Transfer, in: Transgenic
Plants, Band 1, Engineering and Utilization, Hrsg. S. D. Kung und
R. Wu, Academic Press (1993), 128–143 [0120]
- - Potrykus Annu. Rev. Plant Physiol. Plant Molec. Biol. 42 (1991)
205–225 [0120]
- - Bevan et al., Nucl. Acids Res. 12 (1984) 8711 [0120]
- - Höfgen und Willmitzer in Nucl. Acid Res. (1988),
16, 9877 [0120]
- - F. F. White, Vectors for Gene Transfer in Higher Plants; in
Transgenic Plants, Band 1, Engineering and Utilization, Hrsg. S.
D. Kung und R. Wu, Academic Press, 1993, S. 15–38 [0120]
- - Feldman, KA und Marks MD (1987). Mol. Gen. Genet. 208: 274–289 [0121]
- - Feldmann K (1992). In: C Koncz, N-H Chua und J Shell, Hrsg.,
Methods in Arabidopsis Research. Word Scientific, Singapur, S. 274–289 [0121]
- - Chang (1994). Plant J. 5: 551–558; Katavic (1994).
Mol. Gen. Genet., 245: 363–370 [0121]
- - Bechthold, N (1993). C. R. Acad. Sci. Paris Life Sci., 316:
1194–1199 [0121]
- - Clough, SJ und Bent AF (1998), The Plant J. 16, 735–743 [0121]
- - Klaus et al., 2004, [Nature Biotechnology 22 (2), 225–229] [0121]
- - Bock (2001) Transgenic plastids in basic research and plant
biotechnology. J. Mol. Biol. 2001 Sept. 21; 312 (3): 425–38 [0121]
- - Maliga, P (2003), Progress towards commercialization of plastid
transformation technology. Trends Biotechnol. 21, 20–28 [0121]
- - Klaus et al., 2004, Nature Biotechnology 22 (2), 225–229 [0121]
- - Hayashi et al. Science (1992) 1350–1353 [0122]
- - Redei GP und Koncz C (1992), In Methods in Arabidopsis Research,
Koncz C, Chua NH, Schell J, Hrsg. Singapur, World Scientific Publishing
Co, S. 16–82 [0123]
- - Feldmann et al., (1994) In Meyerowitz EM, Somerville CR, Hrsg,
Arabidopsis. Cold Spring Harbor Laboratory Press, Cold Spring Harbor,
NY, S. 137–172 [0123]
- - Lightner J und Caspar T (1998), In J Martinez-Zapater, J Salinas,
Hrsg., Methods an Molecular Biology, Band 82. Humana Press, Totowa,
NJ, S. 91–104 [0123]
- - McCallum et al., (2000), Nat. Biotechnol. 18: 455–457;
in einem Übersichtsartikel von Stemple (2004), Nat. Rev.
Genet. 5 (2): 145–50 [0123]
- - Offringa et al. (1990) EMBO J 9 (10): 3077–84 [0124]
- - Terada et al. (2002) Nat Biotech 20 (10): 1030–4 [0124]
- - Iida and Terada (2004) Curr Opin Biotech 15 (2): 132–8 [0124]
- - Miller et al, Nature Biotechnol. 25, 778–785, 2007 [0124]
- - Yang et al., 2006 [0136]
- - Hasegawa et al., J. Mol. Evol. 22, 160–174, 1985 [0136]
- - Schultz et al., (1998), Proc. Natl. Acad. Sci. USA 95, 5857–5864 [0140]
- - Letunic et al., (2002), Nucleic Acids Res. 30, 242–244 [0140]
- - Mulder et al., (2003), Nucl. Acids Res. 31, 315–318 [0140]
- - Sucher und Bairoch (1994), A generalized Profile syntax for
biomolecular sequences motifs and its function in automatic sequence
interpretation. (In) ISMB-94; Proceedings 2nd International Conference
an Intelligent Systems for Molecular Biology. Altman R., Brutlag
D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61, AAAI
Press, Menlo Park; [0140]
- - Hulo et al., Nucl. Acids Res. 32: D134–D137, (2004) [0140]
- - Bateman et al., Nucleic Acids Research 30 (1): 276–280
(2002) [0140]
- - Gasteiger et al., ExPASy: the proteomics server for in-depth
Protein knowledge and analysis, Nucleic Acids Res. 31: 3784–3788
(2003) [0140]
- - Needleman und Wunsch ((1970), J. Mol. Biol. 48: 443–453 [0141]
- - Altschul et al., (1990), J. Mol. Biol. 215: 403–10 [0141]
- - Campanella et al., BMC Bioinformatics. 2003, Juli, 10; 4:
29. MatGAT: an application that generstes similarity/identity matrices
using Protein or DNA sequences. [0141]
- - Sambrook et al. (2001) Molecular Cloning: a laboratory manual,
3. Ausgabe Cold Spring Harbor Laboratory Press, CSH, New York [0142]
- - Ausubel et al., Current Protocols in Molecular Biology, Current
Protocols [0142]
- - Yang et al., 2006 [0153]
- - Hasegawa et al., J. Mol. Evol. 22, 160–174, 1985 [0153]
- - Yang et al., 2006 [0157]
- - Hasegawa et al., J. Mol. Evol. 22, 160–174, 1985 [0157]
- - Yang et al., 2006 [0160]
- - Hasegawa et al., J. Mol. Evol. 22, 160–174, 1985 [0160]
- - Yang et al., 2006 [0163]
- - Hasegawa et al., J. Mol. Evol. 22, 160–174, 1985 [0163]
- - Yang et al., 2006 [0166]
- - Hasegawa et al., J. Mol. Evol. 22, 160–174, 1985 [0166]
- - Current Protocols in Molecular Biology. Wiley Hrsg. [0167]
- - Wang et al. (Planta (2003), 218: 1–14 [0177]
- - Rabbani et al., (Plant Physiol. (2003), 133: 1755–1767 [0177]
- - S. D. Kung und R. Wu, Potrykus oder Höfgen und Willmitzer [0196]
- - Sambrook J, Fritsch EF und Maniatis T (1989) Molecular Cloning,
A Laboratory Manual [0210]
- - Lander et al. (1987) Genomics 1: 174–181 [0210]
- - Botstein et al. (1980) Am. J. Hum. Genet. 32: 314–331 [0210]
- - Bernatzky und Tanksley (1986) Plant Mol. Biol. Reporter 4:
37–41 [0211]
- - Hoheisel et al. In: Non-mammalian Genomic Analysis: A Practical
Guide, Academic Press 1996, S. 319–346 [0212]
- - Trask (1991) Trends Genet. 7: 149–154 [0213]
- - Laan et al. (1995) Genome Res. 5: 13–20 [0213]
- - Kazazian (1989) J. Lab. Clin. Med 11: 95–96 [0214]
- - CAPS; Sheffield et al. (1993) Genomics 16: 325–332 [0214]
- - Landegren et al. (1988) Science 241: 1077–1080 [0214]
- - Sokolov (1990) Nucleic Acid Res. 18: 3671 [0214]
- - Walter et al. (1997) Nat. Genet. 7: 22–28 [0214]
- - Dear und Cook (1989) Nucleic Acid Res. 17: 6795–6807 [0214]
- - Clissold et al. Trends Biochem Sci. 2001 Jan; 26 (1): 7–9 [0220]
- - Finn et al. Nucleic Acids Research (2006) Database Issue 34:
D247–D251 [0221]
- - Balciunas und Ronne. Trends Biochem Sci 2000; 25: 274–276 [0228]
- - Takeuchi et al. Dev Dyn. 2006 235 (9): 2449–59 [0231]
- - Schultz et al., (1998), Proc. Natl. Acad. Sci. USA 95, 5857–5864 [0232]
- - Letunic et al., (2002), Nucleic Acids Res. 30, 242–244 [0232]
- - Mulder et al., (2003), Nucl. Acids Res. 31, 315–318 [0232]
- - Sucher und Bairoch (1994), A generalized Profile syntax for
biomolecular sequences motives und its function in automatic sequence
interpretation. (In) ISMB-94; Proceedings 2nd International Conference
an Intelligent Systems for Molecular Biology. Altman R., Brutlag
D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61, AAAIPress,
Menlo Park [0232]
- - Hulo et al., Nucl. Acids Res. 32: D134–D137, (2004) [0232]
- - Bateman et al., Nucleic Acids Research 30 (1): 276–280
(2002) [0232]
- - Gasteiger et al., ExPASy: the proteomics server for in-depth
Protein knowledge and analysis, Nucleic Acids Res. 31: 3784–3788
(2003) [0232]
- - Needleman und Wunsch ((1970), J. Mol. Biol. 48: 443–453 [0233]
- - Altschul et al., (1990), J. Mol. Biol. 215: 403–10 [0233]
- - Campanella et al., BMC Bioinformatics. Juli 2003, 10; 4:29.
MatGAT: an application that generstes similarity/identity matrices
using Protein or DNA sequences. [0233]
- - Lavaissiere et al. J Clin Invest. 1996; 98 (6): 1313–23 [0234]
- - Linke et al. J Biol Chem. 2004; 279 (14): 14391–7 [0234]
- - Lee, et al. J. Biol. Chem. 278: 7558–7563; 2003 [0234]
- - Lando et al. Genes Dev. 16: 1466–1471; 2002 [0234]
- - Trewick et al. 2005 [0234]
- - Takeuchi et al. Dev Dyn. 2006 235 (9): 2449–59 [0242]
- - Takeuchi et al. Dev Dyn. 2006 235 (9): 2449–59 [0246]
- - Takeuchi et al. Dev Dyn. 2006 235 (9): 2449–59 [0249]
- - Takeuchi et al. Dev Dyn. 2006 235 (9): 2449–59 [0252]
- - Takeuchi et al. Dev Dyn. 2006 235 (9): 2449–59 [0258]
- - Current Protocols in Molecular Biology. Wiley Hrsg [0259]
- - Wang et al. (Planta (2003), 218: 1–14 [0269]
- - Rabbani et al. (Plant Physiol. (2003), 133: 1755–1767 [0269]
- - Sambrook J, Fritsch EF und Maniatis T (1989) Molecular Cloning,
A Laboratory Manual [0302]
- - Lander et al. (1987) Genomics 1: 174–181 [0302]
- - Botstein et al. (1980) Am. J. Hum. Genet. 32: 314–331 [0302]
- - Bernatzky und Tanksley (1986) Plant Mol. Biol. Reporter 4:
37–41 [0303]
- - Hoheisel et al. In: Non-mammalian Genomic Analysis: A Practical
Guide, Academic Press 1996, S. 319–346 [0304]
- - Trask (1991) Trends Genet. 7: 149–154 [0305]
- - Laan et al. (1995) Genome Res. 5: 13–20 [0305]
- - Kazazian (1989) J. Lab. Clin. Med 11: 95–96 [0306]
- - CAPS; Sheffield et al. (1993) Genomics 16: 325–332 [0306]
- - Landegren et al. (1988) Science 241: 1077–1080 [0306]
- - Sokolov (1990) Nucleic Acid Res. 18: 3671 [0306]
- - Walter et al. (1997) Nat. Genet. 7: 22–28 [0306]
- - Dear und Cook (1989) Nucleic Acid Res. 17: 6795–6807 [0306]
- - Schultz et al. (1998), Proc. Natl. Acad. Sci. USA 95, 5857–5864 [0312]
- - Letunic et al., (2002), Nucleic Acids Res. 30, 242–244 [0312]
- - Mulder et al., (2003), Nucl. Acids Res. 31, 315–318 [0312]
- - Sucher und Bairoch (1994), A generalized Profile syntax for
biomolecular sequences Motivs and its function in automatic sequence
interpretation. (In) ISMB-94; Proceedings 2nd International Conference
an Intelligent Systems for Molecular Biology. Altman R., Brutlag
D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61, AAAI
Press, Menlo Park [0312]
- - Hulo et al., Nucl. Acids Res. 32: D134–D137, (2004) [0312]
- - Bateman et al., Nucleic Acids Research 30 (1): 276–280
(2002) [0312]
- - Gasteiger et al., ExPASy: the Proteomics server for in-depth
Protein knowledge and analysis, Nucleic Acids Res. 31: 3784–3788
(2003) [0312]
- - Needleman und Wunsch ((1970), J. Mol. Biol. 48: 443–453) [0313]
- - Altschul et al., (1990), J. Mol. Biol. 215: 403–10) [0313]
- - Campanella et al., BMC Bioinformatics. 2003, Juli, 10; 4:
29. MatGAT: an application that generstes similarity/identity matrices
using Protein or DNA sequences. [0313]
- - Current Protocols in Molecular Biology. Wiley Hrsg. [0332]
- - Wang et al. (Planta (2003), 218: 1–14 [0342]
- - Rabbani et al. (Plant Physiol. (2003), 133: 1755–1767 [0342]
- - Sambrook J, Fritsch EF and Maniatis T (1989) Molecular Cloning,
A Laboratory Manual [0373]
- - Lander et al. (1987) Genomics 1: 174–181 [0373]
- - Botstein et al. (1980) Am. J. Hum. Genet. 32: 314–331 [0373]
- - Bernatzky und Tanksley (1986) Plant Mol. Biol. Reporter 4:
37–41 [0374]
- - Hoheisel et al. In: Non-mammalian Genomic Analysis: A Practical
Guide, Academic Press 1996, S. 319–346 [0375]
- - Trask (1991) Trends Genet. 7: 149–154 [0376]
- - Laan et al. (1995) Genome Res. 5: 13–20 [0376]
- - Kazazian (1989) J. Lab. Clin. Med 11: 95–96 [0377]
- - CAPS; Sheffield et al. (1993) Genomics 16: 325–332 [0377]
- - Landegren et al. (1988) Science 241: 1077–1080 [0377]
- - Sokolov (1990) Nucleic Acid Res. 18: 3671 [0377]
- - Walter et al. (1997) Nat. Genet. 7: 22–28 [0377]
- - Dear und Cook (1989) Nucleic Acid Res. 17: 6795–6807 [0377]
- - Schultz et al. (1998), Proc. Natl. Acad. Sci. USA 95, 5857–5864 [0386]
- - Letunic et al. (2002), Nucleic Acids Res. 30, 242–244 [0386]
- - Mulder et al. (2003), Nucl. Acids Res. 31, 315–318 [0386]
- - Sucher und Bairoch (1994), A generalized Profile syntax for
biomolecular sequences motifs and its function in automatic sequence
interpretation. (In) ISMB-94; Proceedings 2nd International Conference
an Intelligent Systems for Molecular Biology. Altman R., Brutlag
D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61 [0386]
- - AAAI Press, Menlo Park; Hulo et al., Nucl. Acids Res. 32:
D134–D137, (2004) [0386]
- - Bateman et al., Nucleic Acids Research 30 (1): 276–280
(2002) [0386]
- - Gasteiger et al., ExPASy: the proteomics server for in-depth
Protein knowledge and analysis, Nucleic Acids Res. 31: 3784–3788
(2003) [0386]
- - Needleman und Wunsch ((1970), J. Mol. Biol. 48: 443–453 [0387]
- - Altschul et al. (1990), J. Mol. Biol. 215: 403–10 [0387]
- - Campanella et al., BMC Bioinformatics. 2003, Juli, 10; 4:
29. MatGAT: an application that generstes similarity/identity matrices
using Protein or DNA sequences. [0387]
- - Gasteiger E et al. (2003) ExPASy: the proteomics server for
in-depth Protein knowledge and analysis. Nucleic Acids Res 31: 3784–3788 [0388]
- - Current Protocols in Molecular Biology, Bd. 1 und 2, Ausubel
et al. (1994) [0391]
- - Korfhage et al. (1994) Plant C 6: 695–708 [0391]
- - Schindler et al. (1993) Plant J 4 (1): 137–150 [0391]
- - Halbach et al. (2000) Nucleic Acid Res 28 (18): 3542–3550 [0391]
- - Viola & Gonzalez
(May 26, 2007) Biochemistry [0391]
- - Current Protocols in Molecular Biology. Wiley Eds. [0416]
- - Wang et al., (Planta (2003), 218: 1–14 [0427]
- - Rabbani et al. (Plant Physiol. (2003), 133: 1755–1767 [0427]
- - Frink et al. (1999), Proc. Natl. Acad. Sci. USA 96 (4): 1175–1180 [0430]
- - Sambrook J, Fritsch EF and Maniatis T (1989) Molecular Cloning,
A Laboratory Manual [0462]
- - Lander et al. (1987) Genomics 1: 174–181 [0462]
- - Botstein et al. (1980) Am. J. Hum. Genet. 32: 314–331 [0462]
- - Bernatzky und Tanksley (1986) Plant Mol. Biol. Reporter 4:
37–41 [0463]
- - Hoheisel et al. In: Non-mammalian Genomic Analysis: A Practical
Guide, Academic Press 1996, S. 319–346 [0464]
- - Trask (1991) Trends Genet. 7: 149–154 [0465]
- - Laan et al. (1995) Genome Res. 5: 13–20 [0465]
- - Kazazian (1989) J. Lab. Clin. Med 11: 95–96 [0466]
- - CAPS; Sheffield et al. (1993) Genomics 16: 325–332 [0466]
- - Landegren et al. (1988) Science 241: 1077–1080 [0466]
- - Sokolov (1990) Nucleic Acid Res. 18: 3671 [0466]
- - Walter et al. (1997) Nat. Genet. 7: 22–28 [0466]
- - Dear und Cook (1989) Nucleic Acid Res. 17: 6795–6807 [0466]
- - Li et al. (2006) [0473]
- - Schultz et al. (1998), Proc. Natl. Acad. Sci. USA 95, 5857–5864 [0474]
- - Letunic et al. (2002), Nucleic Acids Res. 30, 242–244 [0474]
- - Mulder et al. (2003), Nucl. Acids Res. 31, 315–318 [0474]
- - Sucher und Bairoch (1994), A generalized Profile syntax for
biomolecular sequences motifs and its function in automatic sequence
interpretation. (In) ISMB-94; Proceedings 2nd International Conference
an Intelligent Systems for Molecular Biology. Altman R., Brutlag
D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61, AAAI
Press, Menlo Park [0474]
- - Hulo et al., Nucl. Acids Res. 32: D134–D137, (2004) [0474]
- - Bateman et al., Nucleic Acids Research 30 (1): 276–280
(2002) [0474]
- - Gasteiger et al., ExPASy: the proteomics server for in-depth
Protein knowledge and analysis, Nucleic Acids Res. 31: 3784–3788
(2003) [0474]
- - Needleman und Wunsch ((1970), J. Mol. Biol. 48: 443–453 [0475]
- - Altschul et al., (1990), J. Mol. Biol. 215: 403–10 [0475]
- - Campanella et al., BMC Bioinformatics. 2003, Juli, 10;4: 29.
MatGAT: an application that generstes similarity/identity matrices
using Protein or DNA sequences. [0475]
- - Smith TF, Waterman MS (1981) J. Mol. Biol 147 (1); 195–7 [0475]
- - Li et al. (2006) [0484]
- - Li et al. (2006) [0488]
- - Li et al. (2006) [0491]
- - Li et al. (2006) [0494]
- - Li et al. (2006) [0497]
- - Current Protocols in Molecular Biology. Wiley Hrsg. [0498]
- - Wang et al. (Planta (2003), 218: 1–14 [0508]
- - Rabbani et al. (Plant Physiol. (2003), 133: 1755–1767 [0508]
- - Sambrook J, Fritsch EF und Maniatis T (1989) Molecular Cloning,
A Laboratory Manual [0540]
- - Lander et al. (1987) Genomics 1: 174–181 [0540]
- - Botstein et al. (1980) Am. J. Hum. Genet. 32: 314–331 [0540]
- - Bernatzky und Tanksley (1986) Plant Mol. Biol. Reporter 4:
37–41 [0541]
- - Hoheisel et al. In: Non-mammalian Genomic Analysis: A Practical
Guide, Academic Press 1996, S. 319–346 [0542]
- - Trask (1991) Trends Genet. 7: 149–154 [0543]
- - Laan et al. (1995) Genome Res. 5: 13–20 [0543]
- - Kazazian (1989) J. Lab. Clin. Med 11: 95–96 [0544]
- - CAPS; Sheffield et al. (1993) Genomics 16: 325–332 [0544]
- - Landegren et al. (1988) Science 241: 1077–1080 [0544]
- - Sokolov (1990) Nucleic Acid Res. 18: 3671 [0544]
- - Walter et al. (1997) Nat. Genet. 7: 22–28 [0544]
- - Dear und Cook (1989) Nucleic Acid Res. 17: 6795–6807 [0544]
- - Schultz et al., (1998), Proc. Natl. Acad. Sci. USA 95, 5857–5864 [0550]
- - Letunic et al., (2002), Nucleic Acids Res. 30, 242–244 [0550]
- - Mulder et al., (2003), Nucl. Acids Res. 31, 315–318 [0550]
- - Sucher und Bairoch (1994), A generalized Profile syntax for
biomolecular sequences motives and its function in automatic sequence
interpretation. (In) ISMB-94; Proceedings 2nd International Conference
an Intelligent Systems for Molecular Biology. Altman R., Brutlag
D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61, AAAIPress,
Menlo Park [0550]
- - Hulo et al., Nucl. Acids Res. 32: D134–D137, (2004) [0550]
- - Bateman et al., Nucleic Acids Research 30 (1): 276–280
(2002) [0550]
- - Gasteiger et al., ExPASy: the Proteomics server for in-depth
Protein knowledge and analysis, Nucleic Acids Res. 31: 3784–3788
(2003) [0550]
- - Needleman und Wunsch ((1970), J. Mol. Biol. 48: 443–453 [0551]
- - Altschul et al., (1990), J. Mob. Biol. 215: 403–10 [0551]
- - Campanella et al., BMC Bioinformatics. Juli 2003, 10; 4: 29.
MatGAT: an application that generstes similarity/identity matrices
using Protein or DNA sequences. [0551]
- - Wang et al. (Planta (2003), 218: 1–14 [0583]
- - Rabbani et al. (Plant Physiol. (2003), 133: 1755–1767 [0583]
- - Sambrook J, Fritsch EF und Maniatis T (1989) Molecular Cloning,
A Laboratory Manual [0614]
- - Lander et al. (1987) Genomics 1: 174–181 [0614]
- - Botstein et al. (1980) Am. J. Hum. Genet. 32: 314–331 [0614]
- - Bernatzky und Tanksley (1986) Plant Mol. Biol. Reporter 4:
37–41 [0615]
- - Hoheisel et al. In: Non-mammalian Genomic Analysis: A Practical
Guide, Academic Press 1996, S. 319–346 [0616]
- - Trask (1991) Trends Genet. 7: 149–154 [0617]
- - Laan et al. (1995) Genome Res. 5: 13–20 [0617]
- - Kazazian (1989) J. Lab. Clin. Med 11: 95–96 [0618]
- - CAPS; Sheffield et al. (1993) Genomics 16: 325–332 [0618]
- - Landegren et al. (1988) Science 241: 1077–1080 [0618]
- - Sokolov (1990) Nucleic Acid Res. 18: 3671 [0618]
- - Walter et al. (1997) Nat. Genet. 7: 22–28 [0618]
- - Dear und Cook (1989) Nucleic Acid Res. 17: 6795–6807 [0618]
- - Finn et al. Nucleic Acids Research (2006) Database Issue 34:
D247–D251 [0629]
- - Schultz et al., (1998), Proc. Natl. Acad. Sci. USA 95, 5857–5864 [0639]
- - Letunic et al., (2002), Nucleic Acids Res. 30, 242–244 [0639]
- - Mulder et al., (2003), Nucl. Acids Res. 31, 315–318 [0639]
- - Sucher und Bairoch (1994), A generalized Profile syntax for
biomolecular sequences motifs and its function in automatic sequence
interpretation. (In) ISMB-94; Proceedings 2nd International Conference
an Intelligent Systems for Molecular Biology. Altman R., Brutlag
D., Karp P., Lathrop R., Searls D., Hrsg., S. 53–61, AAAIPress,
Menlo Park [0639]
- - Hulo et al., Nucl. Acids Res. 32: D134–D137, (2004) [0639]
- - (Bateman et al., Nucleic Acids Research 30 (1): 276–280
(2002) [0639]
- - Gasteiger et al., ExPASy: the proteomics server for in-depth
Protein knowledge and analysis, Nucleic Acids Res. 31: 3784–3788
(2003) [0639]
- - Needleman und Wunsch ((1970), J. Mol. Biol. 48: 443–453 [0640]
- - Altschul et al., (1990), J. Mob. Biol. 215: 403–10 [0640]
- - Campanella et al., BMC Bioinformatics. Juli 2003, 10;4: 29.
MatGAT: an application that generstes similarity/identity matrices
using Protein or DNA sequences. [0640]
- - Klein at al. Mol Gen Genet. 1996, 15; 250 (1): 7–16 [0641]
- - Yamasaki et al. 2004 J Mol Biol. 2004, 12; 337 (1): 49–63 [0641]
- - Griffiths-Jones et al. 2006 Nucleic Acids Research, 2006,
Bd. 34, Database issue D140–D144 [0650]
- - Wang et al. (Planta (2003), 218: 1–14 [0678]
- - Rabbani et al. (Plant Physiol. (2003), 133: 1755–1767 [0678]
- - Sambrook J, Fritsch EF and Maniatis T (1989) Molecular Cloning,
A Laboratory Manual [0712]
- - Lander et al. (1987) Genomics 1: 174–181 [0712]
- - Botstein et al. (1980) Am. J. Hum. Genet. 32: 314–331 [0712]
- - Bernatzky und Tanksley (1986) Plant Mol. Biol. Reporter 4:
37–41 [0713]
- - Hoheisel et al. In: Non-mammalian Genomic Analysis: A Practical
Guide, Academic Press 1996, S. 319–346 [0714]
- - Trask (1991) Trends Genet. 7: 149–154 [0715]
- - Laan et al. (1995) Genome Res. 5: 13–20 [0715]
- - Kazazian (1989) J. Lab. Clin. Med 11: 95–96 [0716]
- - CAPS; Sheffield et al. (1993) Genomics 16: 325–332 [0716]
- - Landegren et al. (1988) Science 241: 1077–1080 [0716]
- - Sokolov (1990) Nucleic Acid Res. 18: 3671 [0716]
- - Walter et al. (1997) Nat. Genet. 7: 22–28 [0716]
- - Dear und Cook (1989) Nucleic Acid Res. 17: 6795–6807 [0716]
- - Huson et al. BMC Bioinformatics 2007 [0742]
- - Xie et al. Plant Physiology, 2006, Vol. 142, pp. 280–293 [0749]
- - Sambrook (2001) Molecular Cloning: a laboratory manual, 3rd
Edition Cold Spring Harbor Laboratory Press, CSH, New York [0754]
- - in Bd. 1 und 2 von Ausubel et al. (1994), Current Protocols
in Molecular Biology, Current Protocols. Standard-Materialien und
Verfahren zur molekularen Arbeit an der Pflanze sind in Plant Molecular
Biology Labfax (1993) von R. D. D. Croy, veröffentlicht
von BIOS Scientific Publications Ltd (UK) und Blackwell Scientific
Publications (UK) [0754]
- - Altschul et al., (1990), J. Mol. Biol. 215: 403–410 [0755]
- - Altschul et al., (1997), Nucleic Acids Res. 25: 3389–3402 [0755]
- - Thompson et al. (1997) Nucleic Acids Res 25: 4876–4882;
Chenna et al. (2003). Nucleic Acids Res 31: 3497–3500 [0757]
- - BMC Bioinformatics. 2003 4: 29. MatGAT: an application that
generstes similarity/identity matrices using Protein or DNA sequences.
Campanella JJ, Bitincka L, Smalley J; von Ledion Bitincka gehostete
software [0759]
- - Hua & Sun,
Bioinformatics 17, 721–728, 2001 [0769]
- - Liu et al (2005) [0769]
- - Aldemita und Hodges 1996, Chan et al., 1993, Hiei et al.,
1994 [0776]
- - Ishida et al. (1996) Nature Biotech 14 (6): 745–50 [0777]
- - Ishida et al. (1996) Nature Biotech 14 (6): 745–50 [0778]
- - Babic et al. (1998, Plant Cell Rep 17: 183–188 [0780]
- - McKersie et al., 1999 Plant Physiol 119: 839–847 [0781]
- - Brown DCW und A Atanassov (1985. Plant Cell Tissue Organ Culture
4: 111–112) [0781]
- - Walker et al., 1978 Am J Bot 65: 654–659 [0781]
- - McKersie et al., 1999 Plant Physiol 119: 839–847 [0781]
- - Murashige and Skoog, 1962 [0781]
- - Altschul et al. (1990), J. Mol. Biol. 215: 403–410 [0795]
- - Altschul et al., (1997), Nucleic Acids Res. 25: 3389–3402 [0795]
- - Thompson et al. (1997) Nucleic Acids Res 25: 4876–4882 [0798]
- - Chenna et al. (2003). Nucleic Acids Res 31: 3497–3500 [0798]
- - BMC Bioinformatics. 2003 4: 29. MatGAT: an application that
generstes similarity/identity matrices using Protein or DNA sequences.
Campanella JJ, Bitincka L, Smalley J; von Ledion Bitincka gehostete
Software [0799]
- - Altschul et al. (1990), J. Mol. Biol. 215: 403–410 [0823]
- - Altschul et al., (1997), Nucleic Acids Res. 25: 3389–3402 [0823]
- - Thompson et al. (1997) Nucleic Acids Res 25: 4876–4882 [0825]
- - Chenna et al. (2003). Nucleic Acids Res 31: 3497–3500 [0825]
- - BMC Bioinformatics. 2003 4: 29. MatGAT: an application that
generstes similarity/identity matrices using Protein or DNA sequences.
Campanella JJ, Bitincka L, Smalley J; von Ledion Bitincka gehostete
Software [0827]
- - Nagata et al. Int. Rev. Cytol. 132, 1–30, 1992 [0839]
- - Vos et al., Nucleic Acids Res. 23 (21) 4407–4414,
1995 [0841]
- - Bachem et al., Plant J. 9 (5) 745–53, 1996 [0841]
- - Vos et al., 1995 [0841]
- - Altschul et al., Nucleic Acids Res. 25 (17) 3389–3402
1997 [0842]
- - Altschul et al. (1990), J. Mol. Biol. 215: 403–410 [0859]
- - Altschul et al., (1997), Nucleic Acids Res. 25: 3389–3402 [0859]
- - Thompson et al. (1997) Nucleic Acids Res 25: 4876–4882 [0862]
- - Chenna et al. (2003). Nucleic Acids Res 31: 3497–3500 [0862]
- - BMC Bioinformatics. 2003 4: 29. MatGAT: an application that
generstes similarity/identity matrices using Protein or DNA sequences.
Campanella JJ, Bitincka L, Smalley J; von Ledion Bitincka gehostete
Software [0864]
- - Sambrook (2001), Molecular Cloning: a laboratory manual, 3.
Ausgabe, Cold Spring Harbor Laboratory Press, CSH, New York [0882]
- - Band 1 und 2 von Ausubel et al., (1994), Current Protocols
in Molecular Biology, Current Protocols. Standardmaterialien und
-verfahren für molekularbiologische Arbeiten an Pflanzen
sind beschrieben in Plant Molecular Biology Labfax (1993), von R.
D. D. Croy, verlegt bei BIOS Scientific Publications Ltd (UK) und Blackwell
Scientific Publications (UK) [0882]
- - Aldemita und Hodges 1996, Chan et al., 1993, Hiei et al.,
1994 [0888]
- - Altschul et al., (1990), J. Mol. Biol. 215: 403–410 [0903]
- - Altschul et al., (1997), Nucleic Acids Res. 25: 3389–3402 [0903]
- - Thompson et al. (1997) Nucleic Acids Res 25: 4876–4882 [0905]
- - Chenna et al. (2003). Nucleic Acids Res 31: 3497–3500 [0905]
- - Huson et al., 2007 [0906]
- - BMC Bioinformatics. 2003 4: 29. MatGAT: an application that
generstes similarity/identity matrices using Protein or DNA sequences.
Campanella JJ, Bitincka L, Smalley J; Ledion Bitincka gehostete
software [0907]
- - Park und Kanehisa, Bioinformatics, 19, 1656–1663,
2003 [0917]
- - psort.org [0917]
- - Dombrecht et al. (2007) [0919]
- - Xue, Plant J. 41, 638–649, 2005 [0919]
- - Xue (2005) [0920]
- - Xue (2005) [0923]
- - Altschul et al., (1990), J. Mol. Biol. 215: 403–410 [0939]
- - Altschul et al., (1997), Nucleic Acids Res. 25: 3389–3402 [0939]
- - Thompson et al. (1997) Nucleic Acids Res 25: 4876–4882 [0941]
- - Chenna et al. (2003). Nucleic Acids Res 31: 3497–3500 [0941]
- - BMC Bioinformatics. 2003 4: 29. MatGAT: an application that
generstes similarity/identity matrices using Protein or DNA sequences.
Campanella JJ, Bitincka L, Smalley J; software hosted by Ledion
Bitincka [0943]
- - Altschul et al., (1990), J. Mol. Biol. 215: 403–410 [0967]
- - Altschul et al., (1997), Nucleic Acids Res. 25: 3389–3402 [0967]
- - Thompson et al. (1997) Nucleic Acids Res 25: 4876–4882 [0969]
- - Chenna et al. (2003). Nucleic Acids Res 31: 3497–3500 [0969]
- - (BMC Bioinformatics. 2003 4: 29. MatGAT: an application that
generstes similarity/identity matrices using Protein or DNA sequences.
Campanella JJ, Bitincka 1, Smalley J; software hosted by Ledion
Bitincka [0971]
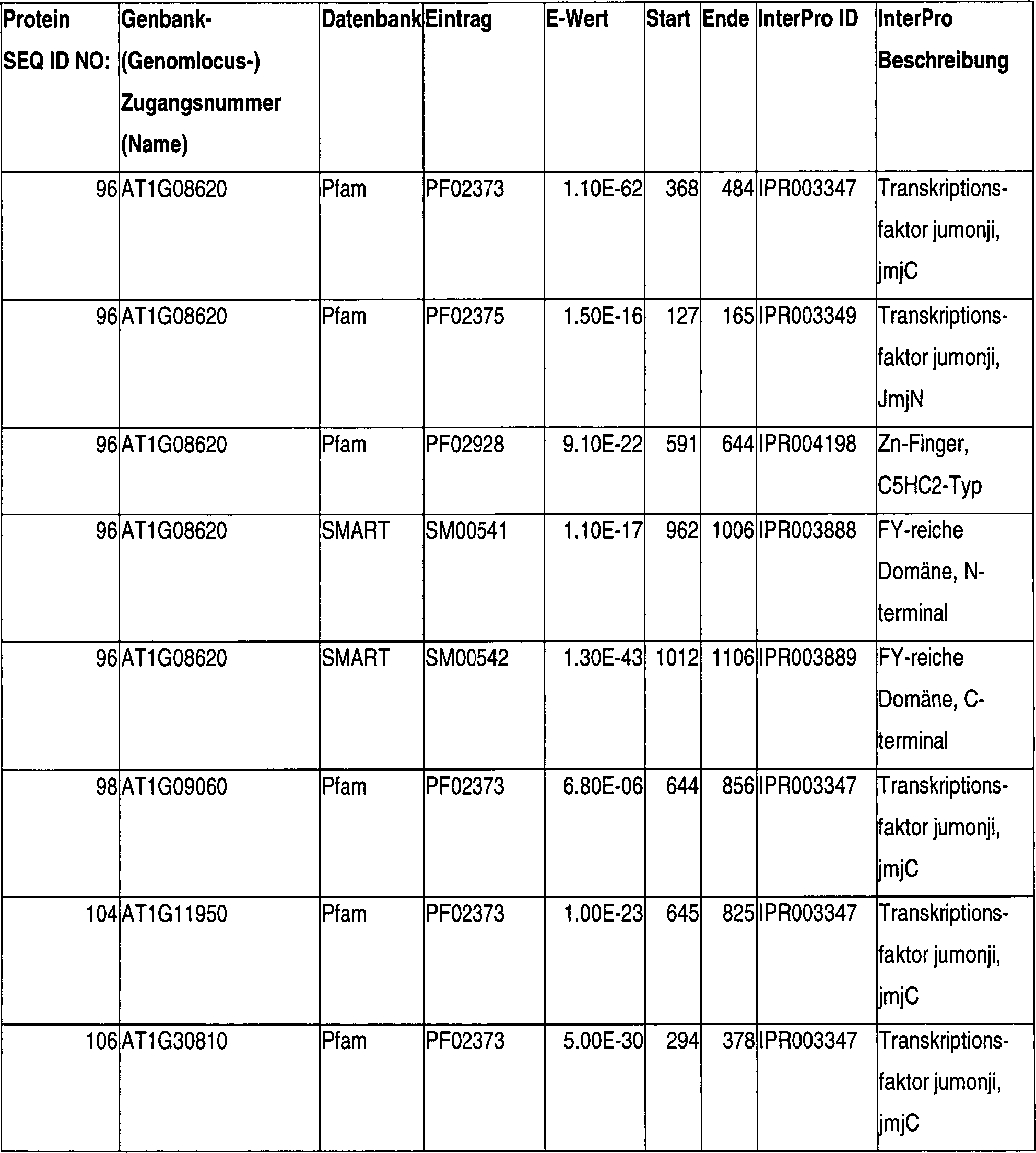






























 Tabelle G3: MatGAT-Ergebnisse für die Gesamtähnlichkeit und -identität über die SBP-Domäne der Polypeptidsequenzen.
Tabelle G3: MatGAT-Ergebnisse für die Gesamtähnlichkeit und -identität über die SBP-Domäne der Polypeptidsequenzen.
