JP6316784B2 - ペプチドの改造および複合糖質化 - Google Patents
ペプチドの改造および複合糖質化 Download PDFInfo
- Publication number
- JP6316784B2 JP6316784B2 JP2015184218A JP2015184218A JP6316784B2 JP 6316784 B2 JP6316784 B2 JP 6316784B2 JP 2015184218 A JP2015184218 A JP 2015184218A JP 2015184218 A JP2015184218 A JP 2015184218A JP 6316784 B2 JP6316784 B2 JP 6316784B2
- Authority
- JP
- Japan
- Prior art keywords
- peptide
- glycan
- intended
- remodeling
- expressed
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 108090000765 processed proteins & peptides Proteins 0.000 title claims description 1605
- 238000007634 remodeling Methods 0.000 title description 1099
- 238000000034 method Methods 0.000 claims description 592
- 235000000346 sugar Nutrition 0.000 claims description 36
- 229920003169 water-soluble polymer Polymers 0.000 claims description 22
- 229920001223 polyethylene glycol Polymers 0.000 claims description 21
- 150000001720 carbohydrates Chemical group 0.000 claims description 18
- 102100022641 Coagulation factor IX Human genes 0.000 claims description 17
- 108010076282 Factor IX Proteins 0.000 claims description 17
- 229960004222 factor ix Drugs 0.000 claims description 17
- 125000003147 glycosyl group Chemical group 0.000 claims description 17
- OVRNDRQMDRJTHS-UHFFFAOYSA-N N-acelyl-D-glucosamine Natural products CC(=O)NC1C(O)OC(CO)C(O)C1O OVRNDRQMDRJTHS-UHFFFAOYSA-N 0.000 claims description 13
- MBLBDJOUHNCFQT-LXGUWJNJSA-N N-acetylglucosamine Natural products CC(=O)N[C@@H](C=O)[C@@H](O)[C@H](O)[C@H](O)CO MBLBDJOUHNCFQT-LXGUWJNJSA-N 0.000 claims description 13
- 108700023372 Glycosyltransferases Proteins 0.000 claims description 11
- SQVRNKJHWKZAKO-UHFFFAOYSA-N beta-N-Acetyl-D-neuraminic acid Natural products CC(=O)NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO SQVRNKJHWKZAKO-UHFFFAOYSA-N 0.000 claims description 8
- SQVRNKJHWKZAKO-OQPLDHBCSA-N sialic acid Chemical compound CC(=O)N[C@@H]1[C@@H](O)C[C@@](O)(C(O)=O)OC1[C@H](O)[C@H](O)CO SQVRNKJHWKZAKO-OQPLDHBCSA-N 0.000 claims description 7
- 125000005629 sialic acid group Chemical group 0.000 claims description 7
- 238000000338 in vitro Methods 0.000 claims description 6
- 239000000758 substrate Substances 0.000 claims description 6
- 108010019236 Fucosyltransferases Proteins 0.000 claims description 5
- 102000006471 Fucosyltransferases Human genes 0.000 claims description 5
- 102000003838 Sialyltransferases Human genes 0.000 claims description 5
- 108090000141 Sialyltransferases Proteins 0.000 claims description 5
- 102000051366 Glycosyltransferases Human genes 0.000 claims description 4
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 claims description 4
- 108060003306 Galactosyltransferase Proteins 0.000 claims description 3
- 102000030902 Galactosyltransferase Human genes 0.000 claims description 3
- 239000002773 nucleotide Substances 0.000 claims description 3
- 125000003277 amino group Chemical group 0.000 claims description 2
- 229930182830 galactose Natural products 0.000 claims description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 2
- 239000008194 pharmaceutical composition Substances 0.000 claims description 2
- 125000005647 linker group Chemical group 0.000 claims 6
- 125000003275 alpha amino acid group Chemical group 0.000 claims 2
- 125000003729 nucleotide group Chemical group 0.000 claims 2
- 108010055629 Glucosyltransferases Proteins 0.000 claims 1
- 102000000340 Glucosyltransferases Human genes 0.000 claims 1
- 108010087568 Mannosyltransferases Proteins 0.000 claims 1
- 102000006722 Mannosyltransferases Human genes 0.000 claims 1
- OVRNDRQMDRJTHS-CBQIKETKSA-N N-Acetyl-D-Galactosamine Chemical group CC(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-CBQIKETKSA-N 0.000 claims 1
- OVRNDRQMDRJTHS-FMDGEEDCSA-N N-acetyl-beta-D-glucosamine Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-FMDGEEDCSA-N 0.000 claims 1
- 108010066816 Polypeptide N-acetylgalactosaminyltransferase Proteins 0.000 claims 1
- 108010090473 UDP-N-acetylglucosamine-peptide beta-N-acetylglucosaminyltransferase Proteins 0.000 claims 1
- 239000000203 mixture Substances 0.000 claims 1
- 229950006780 n-acetylglucosamine Drugs 0.000 claims 1
- 108010091748 peptide A Proteins 0.000 claims 1
- 150000003839 salts Chemical class 0.000 claims 1
- 150000004676 glycans Chemical group 0.000 description 1681
- 210000004027 cell Anatomy 0.000 description 553
- 230000008569 process Effects 0.000 description 540
- 238000010586 diagram Methods 0.000 description 284
- 108010047761 Interferon-alpha Proteins 0.000 description 217
- 102000006992 Interferon-alpha Human genes 0.000 description 186
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 161
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 161
- 229960000187 tissue plasminogen activator Drugs 0.000 description 161
- 125000000539 amino acid group Chemical group 0.000 description 146
- 102000004196 processed proteins & peptides Human genes 0.000 description 124
- 108010029485 Protein Isoforms Proteins 0.000 description 93
- 102000001708 Protein Isoforms Human genes 0.000 description 93
- 108010063954 Mucins Proteins 0.000 description 62
- 102000015728 Mucins Human genes 0.000 description 62
- 230000004927 fusion Effects 0.000 description 62
- 108090000467 Interferon-beta Proteins 0.000 description 58
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 44
- 108010074328 Interferon-gamma Proteins 0.000 description 42
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 39
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 39
- 229960005356 urokinase Drugs 0.000 description 39
- 108010017544 Glucosylceramidase Proteins 0.000 description 34
- 102000004547 Glucosylceramidase Human genes 0.000 description 34
- 108090000394 Erythropoietin Proteins 0.000 description 30
- 108010079345 Follicle Stimulating Hormone Proteins 0.000 description 30
- 102000012673 Follicle Stimulating Hormone Human genes 0.000 description 30
- 230000027455 binding Effects 0.000 description 28
- 241000282414 Homo sapiens Species 0.000 description 27
- 230000004988 N-glycosylation Effects 0.000 description 27
- 102000016911 Deoxyribonucleases Human genes 0.000 description 24
- 108010053770 Deoxyribonucleases Proteins 0.000 description 24
- 108010002350 Interleukin-2 Proteins 0.000 description 23
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 22
- 102000003951 Erythropoietin Human genes 0.000 description 20
- 102000003996 Interferon-beta Human genes 0.000 description 20
- 229940105423 erythropoietin Drugs 0.000 description 20
- 229940028334 follicle stimulating hormone Drugs 0.000 description 20
- 229960001388 interferon-beta Drugs 0.000 description 20
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 20
- 102000005744 Glycoside Hydrolases Human genes 0.000 description 18
- 108010031186 Glycoside Hydrolases Proteins 0.000 description 18
- 102000000588 Interleukin-2 Human genes 0.000 description 16
- 102000015395 alpha 1-Antitrypsin Human genes 0.000 description 16
- 108010050122 alpha 1-Antitrypsin Proteins 0.000 description 16
- 235000014633 carbohydrates Nutrition 0.000 description 16
- 239000002753 trypsin inhibitor Substances 0.000 description 15
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 14
- 102000008070 Interferon-gamma Human genes 0.000 description 14
- OVRNDRQMDRJTHS-RTRLPJTCSA-N N-acetyl-D-glucosamine Chemical group CC(=O)N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-RTRLPJTCSA-N 0.000 description 14
- 229960003130 interferon gamma Drugs 0.000 description 14
- 108010054218 Factor VIII Proteins 0.000 description 13
- 102000001690 Factor VIII Human genes 0.000 description 13
- 108010015899 Glycopeptides Proteins 0.000 description 13
- 102000002068 Glycopeptides Human genes 0.000 description 13
- 229940088598 enzyme Drugs 0.000 description 13
- 229960000301 factor viii Drugs 0.000 description 13
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 12
- 239000002202 Polyethylene glycol Substances 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 12
- 150000002482 oligosaccharides Chemical class 0.000 description 12
- 230000001225 therapeutic effect Effects 0.000 description 12
- 102000004190 Enzymes Human genes 0.000 description 11
- 108090000790 Enzymes Proteins 0.000 description 11
- 238000003786 synthesis reaction Methods 0.000 description 11
- 125000000837 carbohydrate group Chemical group 0.000 description 10
- 230000002255 enzymatic effect Effects 0.000 description 9
- 102100023804 Coagulation factor VII Human genes 0.000 description 8
- 108010023321 Factor VII Proteins 0.000 description 8
- 108010054265 Factor VIIa Proteins 0.000 description 8
- 229940012413 factor vii Drugs 0.000 description 8
- 229940012414 factor viia Drugs 0.000 description 8
- 229920001542 oligosaccharide Polymers 0.000 description 8
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 7
- 230000013595 glycosylation Effects 0.000 description 7
- 238000006206 glycosylation reaction Methods 0.000 description 7
- 102000045442 glycosyltransferase activity proteins Human genes 0.000 description 7
- 108700014210 glycosyltransferase activity proteins Proteins 0.000 description 7
- 210000003714 granulocyte Anatomy 0.000 description 7
- SHZGCJCMOBCMKK-DHVFOXMCSA-N L-fucopyranose Chemical group C[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@@H]1O SHZGCJCMOBCMKK-DHVFOXMCSA-N 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 description 5
- PNNNRSAQSRJVSB-SLPGGIOYSA-N Fucose Natural products C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C=O PNNNRSAQSRJVSB-SLPGGIOYSA-N 0.000 description 5
- 239000003814 drug Substances 0.000 description 5
- 125000000311 mannosyl group Chemical group C1([C@@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 5
- -1 nucleotide sugars Chemical class 0.000 description 5
- 150000008163 sugars Chemical class 0.000 description 5
- 238000012546 transfer Methods 0.000 description 5
- DQJCDTNMLBYVAY-ZXXIYAEKSA-N (2S,5R,10R,13R)-16-{[(2R,3S,4R,5R)-3-{[(2S,3R,4R,5S,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}-5-(ethylamino)-6-hydroxy-2-(hydroxymethyl)oxan-4-yl]oxy}-5-(4-aminobutyl)-10-carbamoyl-2,13-dimethyl-4,7,12,15-tetraoxo-3,6,11,14-tetraazaheptadecan-1-oic acid Chemical compound NCCCC[C@H](C(=O)N[C@@H](C)C(O)=O)NC(=O)CC[C@H](C(N)=O)NC(=O)[C@@H](C)NC(=O)C(C)O[C@@H]1[C@@H](NCC)C(O)O[C@H](CO)[C@H]1O[C@H]1[C@H](NC(C)=O)[C@@H](O)[C@H](O)[C@@H](CO)O1 DQJCDTNMLBYVAY-ZXXIYAEKSA-N 0.000 description 4
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 4
- 102000003886 Glycoproteins Human genes 0.000 description 4
- 108090000288 Glycoproteins Proteins 0.000 description 4
- 102000035195 Peptidases Human genes 0.000 description 4
- 108091005804 Peptidases Proteins 0.000 description 4
- 229940024606 amino acid Drugs 0.000 description 4
- 235000001014 amino acid Nutrition 0.000 description 4
- 150000001413 amino acids Chemical class 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 239000000562 conjugate Substances 0.000 description 4
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 4
- 229930182470 glycoside Natural products 0.000 description 4
- 238000002715 modification method Methods 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 241000196324 Embryophyta Species 0.000 description 3
- 241000238631 Hexapoda Species 0.000 description 3
- SQVRNKJHWKZAKO-LUWBGTNYSA-N N-acetylneuraminic acid Chemical compound CC(=O)N[C@@H]1[C@@H](O)CC(O)(C(O)=O)O[C@H]1[C@H](O)[C@H](O)CO SQVRNKJHWKZAKO-LUWBGTNYSA-N 0.000 description 3
- 108010071384 Peptide T Proteins 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- 125000000613 asparagine group Chemical group N[C@@H](CC(N)=O)C(=O)* 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 150000002338 glycosides Chemical class 0.000 description 3
- 229920001427 mPEG Polymers 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 229940060155 neuac Drugs 0.000 description 3
- CERZMXAJYMMUDR-UHFFFAOYSA-N neuraminic acid Natural products NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO CERZMXAJYMMUDR-UHFFFAOYSA-N 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 2
- 102100032487 Beta-mannosidase Human genes 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 2
- PNIWLNAGKUGXDO-UHFFFAOYSA-N Lactosamine Natural products OC1C(N)C(O)OC(CO)C1OC1C(O)C(O)C(O)C(CO)O1 PNIWLNAGKUGXDO-UHFFFAOYSA-N 0.000 description 2
- 108010046068 N-Acetyllactosamine Synthase Proteins 0.000 description 2
- 102100023315 N-acetyllactosaminide beta-1,6-N-acetylglucosaminyl-transferase Human genes 0.000 description 2
- 108010056664 N-acetyllactosaminide beta-1,6-N-acetylglucosaminyltransferase Proteins 0.000 description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 239000000370 acceptor Substances 0.000 description 2
- NIGUVXFURDGQKZ-UQTBNESHSA-N alpha-Neup5Ac-(2->3)-beta-D-Galp-(1->4)-[alpha-L-Fucp-(1->3)]-beta-D-GlcpNAc Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]1[C@H](O[C@H]2[C@@H]([C@@H](O[C@]3(O[C@H]([C@H](NC(C)=O)[C@@H](O)C3)[C@H](O)[C@H](O)CO)C(O)=O)[C@@H](O)[C@@H](CO)O2)O)[C@@H](CO)O[C@@H](O)[C@@H]1NC(C)=O NIGUVXFURDGQKZ-UQTBNESHSA-N 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 2
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 2
- 108010055059 beta-Mannosidase Proteins 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 230000004087 circulation Effects 0.000 description 2
- 230000021615 conjugation Effects 0.000 description 2
- IERHLVCPSMICTF-XVFCMESISA-N cytidine 5'-monophosphate Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(O)=O)O1 IERHLVCPSMICTF-XVFCMESISA-N 0.000 description 2
- 238000001212 derivatisation Methods 0.000 description 2
- 239000010432 diamond Substances 0.000 description 2
- 229910003460 diamond Inorganic materials 0.000 description 2
- 230000005847 immunogenicity Effects 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- DOVBXGDYENZJBJ-ONMPCKGSSA-N lactosamine Chemical compound O=C[C@H](N)[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O DOVBXGDYENZJBJ-ONMPCKGSSA-N 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 230000000269 nucleophilic effect Effects 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 230000017854 proteolysis Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 108010066476 ribonuclease B Proteins 0.000 description 2
- 235000004400 serine Nutrition 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 238000009966 trimming Methods 0.000 description 2
- JLJZRAUDCHWQPS-NYONGVCJSA-N 4-oxo-n-[(3s,4r,5s,6r)-2,4,5-trihydroxy-6-(hydroxymethyl)oxan-3-yl]pentanamide Chemical compound CC(=O)CCC(=O)N[C@@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O JLJZRAUDCHWQPS-NYONGVCJSA-N 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- TXCIAUNLDRJGJZ-BILDWYJOSA-N CMP-N-acetyl-beta-neuraminic acid Chemical compound O1[C@@H]([C@H](O)[C@H](O)CO)[C@H](NC(=O)C)[C@@H](O)C[C@]1(C(O)=O)OP(O)(=O)OC[C@@H]1[C@@H](O)[C@@H](O)[C@H](N2C(N=C(N)C=C2)=O)O1 TXCIAUNLDRJGJZ-BILDWYJOSA-N 0.000 description 1
- 102100035149 Cytosolic endo-beta-N-acetylglucosaminidase Human genes 0.000 description 1
- 108010089072 Dolichyl-diphosphooligosaccharide-protein glycotransferase Proteins 0.000 description 1
- 108700034637 EC 3.2.-.- Proteins 0.000 description 1
- 101710144190 Endo-beta-N-acetylglucosaminidase Proteins 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- LQEBEXMHBLQMDB-UHFFFAOYSA-N GDP-L-fucose Natural products OC1C(O)C(O)C(C)OC1OP(O)(=O)OP(O)(=O)OCC1C(O)C(O)C(N2C3=C(C(N=C(N)N3)=O)N=C2)O1 LQEBEXMHBLQMDB-UHFFFAOYSA-N 0.000 description 1
- 108010015133 Galactose oxidase Proteins 0.000 description 1
- 208000015872 Gaucher disease Diseases 0.000 description 1
- 229920001503 Glucan Polymers 0.000 description 1
- 101000766306 Homo sapiens Serotransferrin Proteins 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 108010035766 P-Selectin Proteins 0.000 description 1
- 102100023472 P-selectin Human genes 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108010009736 Protein Hydrolysates Proteins 0.000 description 1
- 101000895926 Streptomyces plicatus Endo-beta-N-acetylglucosaminidase H Proteins 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 235000016127 added sugars Nutrition 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 108010047754 beta-Glucosidase Proteins 0.000 description 1
- 102000006995 beta-Glucosidase Human genes 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 229940049197 cerezyme Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 235000021310 complex sugar Nutrition 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- IERHLVCPSMICTF-UHFFFAOYSA-N cytidine monophosphate Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(COP(O)(O)=O)O1 IERHLVCPSMICTF-UHFFFAOYSA-N 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000000386 donor Substances 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- WUSMNKZFOXUXJK-UHFFFAOYSA-N ethene;oxolane-2,5-dione Chemical compound C=C.O=C1CCC(=O)O1 WUSMNKZFOXUXJK-UHFFFAOYSA-N 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 238000001215 fluorescent labelling Methods 0.000 description 1
- 150000008267 fucoses Chemical class 0.000 description 1
- 230000033581 fucosylation Effects 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 108010001671 galactoside 3-fucosyltransferase Proteins 0.000 description 1
- 239000000348 glycosyl donor Substances 0.000 description 1
- 102000035122 glycosylated proteins Human genes 0.000 description 1
- 108091005608 glycosylated proteins Proteins 0.000 description 1
- 229930004094 glycosylphosphatidylinositol Natural products 0.000 description 1
- 210000002288 golgi apparatus Anatomy 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 108010039650 imiglucerase Proteins 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000009851 immunogenic response Effects 0.000 description 1
- 229940051026 immunotoxin Drugs 0.000 description 1
- 239000002596 immunotoxin Substances 0.000 description 1
- 230000002637 immunotoxin Effects 0.000 description 1
- 231100000608 immunotoxin Toxicity 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 230000010189 intracellular transport Effects 0.000 description 1
- 150000002576 ketones Chemical group 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 108091005601 modified peptides Proteins 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 239000006225 natural substrate Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000813 peptide hormone Substances 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 230000006461 physiological response Effects 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- XNSAINXGIQZQOO-SRVKXCTJSA-N protirelin Chemical compound NC(=O)[C@@H]1CCCN1C(=O)[C@@H](NC(=O)[C@H]1NC(=O)CC1)CC1=CN=CN1 XNSAINXGIQZQOO-SRVKXCTJSA-N 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 125000006853 reporter group Chemical group 0.000 description 1
- 108010038196 saccharide-binding proteins Proteins 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 230000009450 sialylation Effects 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 150000004044 tetrasaccharides Chemical class 0.000 description 1
- 125000000341 threoninyl group Chemical group [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 238000011282 treatment Methods 0.000 description 1
- 150000004043 trisaccharides Chemical class 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
Images


































































































































































































































































































































































































































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
- C12N9/6421—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue from mammals
- C12N9/6424—Serine endopeptidases (3.4.21)
- C12N9/6437—Coagulation factor VIIa (3.4.21.21)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/549—Sugars, nucleosides, nucleotides or nucleic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6845—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a cytokine, e.g. growth factors, VEGF, TNF, a lymphokine or an interferon
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6855—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from breast cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6863—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from stomach or intestines cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6867—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from a cell of a blood cancer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/10—Drugs for disorders of the endocrine system of the posterior pituitary hormones, e.g. oxytocin, ADH
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/006—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length of peptides containing derivatised side chain amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/107—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides
- C07K1/1072—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides by covalent attachment of residues or functional groups
- C07K1/1077—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides by covalent attachment of residues or functional groups by covalent attachment of residues other than amino acids or peptide residues, e.g. sugars, polyols, fatty acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/13—Labelling of peptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/475—Growth factors; Growth regulators
- C07K14/505—Erythropoietin [EPO]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/53—Colony-stimulating factor [CSF]
- C07K14/535—Granulocyte CSF; Granulocyte-macrophage CSF
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/54—Interleukins [IL]
- C07K14/55—IL-2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/555—Interferons [IFN]
- C07K14/56—IFN-alpha
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/555—Interferons [IFN]
- C07K14/565—IFN-beta
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/555—Interferons [IFN]
- C07K14/57—IFN-gamma
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/59—Follicle-stimulating hormone [FSH]; Chorionic gonadotropins, e.g.hCG [human chorionic gonadotropin]; Luteinising hormone [LH]; Thyroid-stimulating hormone [TSH]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/61—Growth hormone [GH], i.e. somatotropin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/62—Insulins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/715—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons
- C07K14/7151—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons for tumor necrosis factor [TNF], for lymphotoxin [LT]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/745—Blood coagulation or fibrinolysis factors
- C07K14/755—Factors VIII, e.g. factor VIII C (AHF), factor VIII Ag (VWF)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/81—Protease inhibitors
- C07K14/8107—Endopeptidase (E.C. 3.4.21-99) inhibitors
- C07K14/811—Serine protease (E.C. 3.4.21) inhibitors
- C07K14/8121—Serpins
- C07K14/8125—Alpha-1-antitrypsin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K9/00—Peptides having up to 20 amino acids, containing saccharide radicals and having a fully defined sequence; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases RNAses, DNAses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/24—Hydrolases (3) acting on glycosyl compounds (3.2)
- C12N9/2402—Hydrolases (3) acting on glycosyl compounds (3.2) hydrolysing O- and S- glycosyl compounds (3.2.1)
- C12N9/2405—Glucanases
- C12N9/2434—Glucanases acting on beta-1,4-glucosidic bonds
- C12N9/2445—Beta-glucosidase (3.2.1.21)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
- C12N9/6421—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue from mammals
- C12N9/6424—Serine endopeptidases (3.4.21)
- C12N9/644—Coagulation factor IXa (3.4.21.22)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
- C12N9/6421—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue from mammals
- C12N9/6424—Serine endopeptidases (3.4.21)
- C12N9/6456—Plasminogen activators
- C12N9/6459—Plasminogen activators t-plasminogen activator (3.4.21.68), i.e. tPA
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
- C12N9/6421—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue from mammals
- C12N9/6424—Serine endopeptidases (3.4.21)
- C12N9/6456—Plasminogen activators
- C12N9/6462—Plasminogen activators u-Plasminogen activator (3.4.21.73), i.e. urokinase
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y302/00—Hydrolases acting on glycosyl compounds, i.e. glycosylases (3.2)
- C12Y302/01—Glycosidases, i.e. enzymes hydrolysing O- and S-glycosyl compounds (3.2.1)
- C12Y302/01021—Beta-glucosidase (3.2.1.21)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21021—Coagulation factor VIIa (3.4.21.21)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21022—Coagulation factor IXa (3.4.21.22)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21069—Protein C activated (3.4.21.69)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21073—Serine endopeptidases (3.4.21) u-Plasminogen activator (3.4.21.73), i.e. urokinase
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Zoology (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Wood Science & Technology (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Gastroenterology & Hepatology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Engineering & Computer Science (AREA)
- Toxicology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Cell Biology (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Endocrinology (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Virology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Communicable Diseases (AREA)
- Analytical Chemistry (AREA)
- Reproductive Health (AREA)
- Rheumatology (AREA)
Description
本発明はペプチドに結合した特別なグルカン構造を有するペプチドの多数の改造方法を含む。特別なグリカン構造を本明細書に記載するが、本発明は任意の1つの特定のグリカン構造に限定されると解釈されるべきではない。加えて本明細書には特別なペプチドを記載するが、本発明は記載するペプチドの性質に限定されるべきではなく、むしろ任意のすべての適当なペプチドおよびその変形物を包含するべきである。

AAはペプチドの末端または内部アミノ酸残基であり;
X1−X2は該AAに共有結合したサッカリドであり、ここで
X1は第1グリコシル残基であり;そして
X2はX1に共有結合した第2グリコシル残基であり、ここでX1およびX2は単糖およびオリゴ糖残基から選択される、
を有するペプチドの無細胞のインビトロ改造方法を提供する。この方法は:
(a)X2またはその糖サブユニットを該ペプチドから除去し、これにより短縮化グリカンを形成し;そして
(b)短縮化グリカンを、少なくとも1つのグリコシルトランスフェラーゼおよび少なくとも1つのグリコシル供与体と、少なくとも1つのグリコシル供与体を短縮化グリカンへ転移させるために適する条件下で接触させ、これにより該ペプチドを改造する、
ことを含んでなる。
(c)X1を除去し、これによりAAを露出し;そして
(d)AAを、少なくとも1つのグリコシルトランスフェラーゼおよび少なくとも1つのグリコシル供与体と、少なくとも1つのグリコシル供与体をAAへ転移させるために適する条件下で接触させ、これにより該ペプチドを改造する、
ことを含んでなる。
(e)工程(b)の前に、翻訳後修飾中にサッカリドに付加した基を除去する、
ことを含んでなる。
ZはO、S、NHおよびクロスリンカーから選択される員である、
を有する。
X3、X4、X5、X6、X7およびX17は、単糖またはオリゴ糖残基から独立して選択され;そして
a、b、c、d、eおよびxは、整数0、1および2から独立して選択されるが、ただしa、b、c、d、eおよびxから選択される少なくとも1つの員は1または2である、を有するペプチドの無細胞のインビトロ改造方法である。この方法は;
(a)少なくとも1つのX3、X4、X5、X6、X7またはX17、またはその糖サブユニットをペプチドから除去し、これにより短縮化グリカンを形成し;そして
(b)短縮化グリカンを、少なくとも1つのグリコシルトランスフェラーゼおよび少なくとも1つのグリコシル供与体と、少なくとも1つのグリコシル供与体を該短縮化グリカンヘ転移させるために適する条件下で接触させ、これにより該ペプチドを改造する、
ことを含んでなる。
式中、
X8は単−およびオリゴ−糖から選択されるグリコシル部分であり;
yは0および1から選択される整数であり;そして
zは1から20の間の整数であり、ここで
Zは3以上である時、(マンノース)zは直鎖および分岐構造から選択される、
から成る群から選択される。
r、sおよびtは、0および1から独立して選択される整数である、
を有するグリカンを含んでなるペプチドの無細胞のインビトロ改造方法である。この方法は:
(a)ペプチドを、少なくとも1つのグリコシルトランスフェラーゼおよび少なくとも1つのグリコシル供与体と、少なくとも1つのグリコシル供与体をグリカンへ転移させるために適する条件下で接触させ、これにより該ペプチドを改造する、
ことを含んでなる。
X9およびX10は、単糖またはオリゴ糖残基から独立して選択され;そして
m、nおよびfは0および1から選択される整数である、
を有する。
X11およびX12は、独立して選択されるグリコシル部分であり;そして
rおよびxは0および1から独立して選択される整数である、
を有する。
X13、X14およびX15は、独立して選択されるグリコシル残基であり;そして
g、h、i、j、kおよびpは整数0および1から独立して選択されるが、ただし少なくとも1つのg、h、i、j、kおよびpは1である、
を有する。
X16は
から選択される員である、
を有する請求項1に記載の方法。
AAはペプチドの末端または内部アミノ酸残基であり;
X1は、単糖およびオリゴ糖残基から選択される、AAに共有結合したグリコシル残基であり;そして
uは0および1から選択される整数である、
を有するペプチドの無細胞のインビトロ改造方法である。この方法は:ペプチドを、少なくとも1つのグリコシルトランスフェラーゼおよび少なくとも1つのグリコシル供与体と、少なくとも1つのグリコシル供与体を該短縮化グリカンへ転移させるために適する条件下で接触させ、ここで該グリコシル供与体は修飾基を含んでなり、これにより該ペプチドを改造することを含んでなる。
第1の完全なグリコシル連結基を介してペプチドの第1残基に共有結合した第1修飾基、および
第2の完全なグリコシル連結基を介してペプチドの第2残基に結合した第2グリコシル連結基、
を含んでなる。
ここで
リンカー部分は、第1ペプチドおよびリンカー部分の間に挿入され、そして両方を共有結合した第1の完全なグリコシル連結基を介して第1ペプチドに結合し、そして
リンカー部分は、第2ペプチドおよ該リンカー部分の間に挿入され、そして両方に共有結合した第2の完全なグリコシル連結基を介して該第2ペプチドに結合している、
との間に共有結合物を形成する方法を提供する。この方法は:
(a)第1ペプチドを、第1の完全なグリコシル連結基の前駆体および第2の完全なグリコシル連結基の前駆体を含んでなるリンカー部分前駆体の誘導体と接触させ;
(b)(a)からの混合物を、第1グリコシル連結基の前駆体が基質であるグリコシルトランスフェラーゼと、第1の完全なグリコシル連結基の前駆体を第1の完全なグリコシル連結基に転換するために十分な条件下で接触させ、これによりリンカー部分前駆体と第1ペプチドとの間に第1結合物を形成し;
(c)第1結合物を、第2ペプチドおよび第2の完全なグリコシル基の前駆体が基質であるグリコシルトランスフェラーゼと、第2の完全なグリコシル連結基の前駆体を第2のグリコシル連結基に転換するために十分な条件下で接触させ、これによりリンカー部分と第1グリコシル化または非グリコシル化ペプチドとの間、および第2グリコシル化または非−グリコシル化ペプチドとの間に該結合物を形成する、
ことを含んでなる。
ここで
リンカー部分は第1ペプチドに共有結合し、そして
リンカー部分は、第2ペプチドとリンカー部分の間に挿入され、そして両方に共有結合した完全なグリコシル連結基を介して第2ペプチドに結合している、
との間に共有結合物を形成する方法を提供する。この方法は:
(a)第1ペプチドを、
第1ペプチド上の残基に相補的な反応性の反応性官能基、および完全なグリコシル連結基の前駆体、
を含んでなるリンカー部分の活性化誘導体と、反応性官能基と残基との間に共有結合を形成するために十分な条件下で接触させ、これにより第1結合物を形成し;そして
(b)第1結合物を、第2ペプチドおよび完全なグリコシル連結基の前駆体が基質であるグリコシルトランスフェラーゼと、完全なグリコシル連結基の前駆体を完全なグリコシル連結基に転換するために十分な条件下で接触させ、これにより第1グリコシル化または非グリコシル化ペプチドと、第2グリコシル化または非−グリコシル化ペプチドとの間にリンカー部分により連結された結合物を形成する、
ことを含んでなる。
MSは修飾基に共有結合した糖を含んでなる修飾された糖であり;
Nuはヌクレオシドであり;そして
bは0から2の整数である、
を有する化合物である。
X、Y、Z、AおよびBは、S、OおよびNHから独立して選択される員であり;
R21、R22、R23、R24およびR25は、Hおよびポリマーから独立して選択される員であり;
R26は、H、OHおよびポリマーから選択される員であり;
R27はCOO−およびNa+から選択される員であり;
Nuはヌクレオシドであり;そして
aは1から3の整数である、
を有する化合物を含む。
AAはペプチドの末端または内部アミノ酸残基である、
を有するペプチドの無細胞のインビトロ改造方法を提供する。この方法は;
該ペプチドを、少なくとも1つのグリコシルトランスフェラーゼおよび少なくとも1つのグリコシル供与体と、少なくとも1つのグリコシル供与体を該アミノ酸残基へ転移させるために適する条件下で接触させ、ここでグリコシル供与体は修飾基を含んでなり、これにより該ペプチドを改造する、
ことを含んでなる。
a、b、cおよびeは、0および1から独立して選択される員であり;
dは0であり;そして
Rは修飾基、マンノースまたはオリゴマンノースである、
を有するグリコシル残基を含んでなる。この方法は
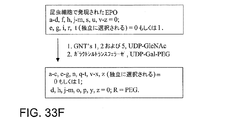
(a)G−CSFペプチドを、グリコシルトランスフェラーゼおよび該修飾基に共有結合した該グリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基の形成に適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、G−CSFペプチドをシアリダーゼと、G−CSFペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の前に、G−CSFペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースをG−CSFペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と部分との間に結合物を形成することを含んでなる。
(e)工程(a)の前に、G−CSFペプチドをN−アセチルガラクトサミントランスフェラーゼおよびGalNAc供与体と、GalNAcをG−CSFペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(f)工程(a)の前に、G−CSFペプチドを、合成的に作用するエンド−N−アセチルガラクトサミニダーゼおよびGalNAc供与体と、GalNAc該G−CSFペプチドへ転移させるために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、o、p、q、r、s、t、u、aa、bb、cc、ddおよびeeは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の整数から独立して選択される員であり;
j、k、lおよびmは0から20の整数から独立して選択される員であり;
v、w、x、yおよびzは0であり;そして
Rは修飾基、マンノースまたはオリゴマンノースであり、
R’はH、グリコシル残基、修飾基または複合糖質である、
から選択される式を有するグリコシル残基を含んでなる。
(a)糖ペプチドを、グリコシルトランスフェラーゼ、合成的に作用するエンド−アセチルガラクトサミニダーゼおよびトランス−シアリダーゼから選択される員、および修飾基に共有結合した該グリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、該完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、糖ペプチドをシアリダーゼと、糖ペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と部分との間に結合物を形成することを含んでなる。
(d)工程(a)の前に、糖ペプチドをグリコシダーゼおよびシアリダーゼの組み合わせと接触させることを含んでなる。
(e)工程(a)の前に、糖ペプチドをエンドグリカナーゼと、糖ペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(f)工程(a)の前に、糖ペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcを糖ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(g)工程(a)の前に、糖ペプチドを、ガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを生成物へ転移させるために適する条件下で接触させることを含んでなる。
(h)工程(b)の前に、糖ペプチドをエンドグリカナーゼと、糖ペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(i)工程(a)の前に、糖ペプチドをマンノシダーゼと、糖ペプチドからマンノースを除去するために適する条件下で接触させることを含んでなる。
(j)工程(a)の生成物をシアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
a、b、c、d、i、p、q、r、s、tおよびuは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の間の整数から独立して選択される員であり;
j、k、lおよびmは0から100の間の整数から独立して選択される員であり;
v、w、xおよびyは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はH、またはグリコシル、修飾基または複合糖質基である、
を有するグリコシル残基を含んでなる。この方法は
(a)インターフェロンベータペプチドを、グリコシルトランスフェラーゼおよびトランス−シアリダーゼから選択される員および修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、インターフェロンベータペプチドをシアリダーゼと、インターフェロンベータペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)からの生成物を、修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と部分との間に結合物を形成することを含んでなる。
(d)工程(a)の前に、上記インターフェロンベータペプチドをグリコシダーゼおよびシアリダーゼの組み合わせと接触させることを含んでなる。
(e)工程(a)の前に、インターフェロンベータペプチドをエンドグリカナーゼと、インターフェロンベータペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(f)工程(a)の前に、インターフェロンベータペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcをインターフェロンベータペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(g)工程(a)の前に、インターフェロンベータペプチドを、ガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを生成物へ転移させるために適する条件下で接触させることを含んでなる。
(h)工程(b)の前に、インターフェロンベータペプチドをエンドグリカナーゼと、インターフェロンベータペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(i)工程(a)の前に、インターフェロンベータペプチドをマンノシダーゼと、インターフェロンベータペプチドからマンノースを除去するために適する条件下で接触させることを含んでなる。
(j)工程(a)の生成物をシアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
a、b、c、d、i、o、p、q、r、s、tおよびuは、0および1から独立して選択される員であり;
e、f、g、hおよびnは0から6の整数から独立して選択される員であり;
j、k、lおよびmは0から20の整数から独立して選択される員であり;
v、w、xおよびyは0であり;そして
Rは修飾基、マンノース、オリゴマンノース、シアリルルイスx(SialylLewisx)またはシアリルルイスa(SialylLewisa)である、
から選択される員である式を有するグリコシル残基を含んでなる。
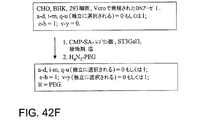
(a)第VIIa因子ペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合した該グリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、第VIIa因子ペプチドをシアリダーゼと、第VIIa因子ペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の前に、第VIIa因子ペプチドをガラクトシダーゼと、第VIIa因子ペプチドからガラクトースを除去するためにに適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、第VIIa因子ペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを第VIIa因子ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(e)工程(a)の生成物をシアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、o、p、q、r、s、t、u、bb、cc、dd、ee、ffおよびggは、0および1から独立して選択される員であり;
e、f、g、hおよびaaは0から6の整数から独立して選択される員であり;
j、k、lおよびmは0から20の整数から独立して選択される員であり;
v、w、x、yおよびzは0であり;
Rは修飾基、マンノースまたはオリゴマンノースである、
から選択される員である式を有するグリコシル残基を含んでなる。この方法は
(a)第IX因子ペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、該完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、第IX因子ペプチドをシアリダーゼと、第IX因子ペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(d)工程(b)からの生成物を、ガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを生成物へ転移させるために適する条件下で接触させることを含んでなる。
(e)工程(d)からの生成物を、ST3Gal3およびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と部分との間に結合物を形成することを含んでなる。
a、b、c、d、i、q、r、s、tおよびuは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の間の整数から独立して選択される員であり;
j、k、lおよびmは0から100の間の整数から独立して選択される員であり;
v、w、xおよびyは0であり;そして
Rは修飾基、マンノースまたはオリゴマンノースである、
を有するグリコシル残基を含んでなる。この方法は:
(a)FSHペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合した該グリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、該完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、FSHペプチドをシアリダーゼと、FSHペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の生成物を、シアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、該FSHペプチドをガラクトシダーゼと、FSHペプチドからガラクトースを除去するために適する条件下で接触させることを含んでなる。
(e)工程(a)の前に、FSHペプチドをグリコシダーゼおよびシアリダーゼの組み合わせと接触させることを含んでなる。
(f)工程(a)の前に、FSHペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースをFSHペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)からの生成物を、修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と部分との間に結合物を形成する。
(e)工程(b)の前に、FSHペプチドをエンドグリカナーゼと、FSHペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(f)工程(a)の前に、FSHペプチドをN−アセチルアセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcをFSHペプチドへ転移させるために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、o、p、q、r、s、tおよびuは、0および1から独立して選択される員であり;
e、f、gおよびhは0から4の間の整数から独立して選択される員であり;
j、k、lおよびmは0から20の間の整数から独立して選択される員であり;
v、w、x、yおよびzは0であり;そして
Rは修飾基、マンノースまたはオリゴマンノースである、から選択される員である式を有するグリコシル残基を含んでなる。この方法は:
(a)EPOペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、該完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、EPOペプチドをシアリダーゼと、EPOペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の生成物を、シアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、EPOペプチドを合成的に作用するガラクトシダーゼと、EPOペプチドにガラクトースを付加するために適する条件下で接触させることを含んでなる。
(e)工程(a)の前に、EPOペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースをEPOペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(f)工程(e)からの生成物をST3Gal3およびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(g)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と部分との間に結合物を形成することを含んでなる。
(h)工程(a)の前に、EPOペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcをEPOペプチドへ転移させるために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、o、p、q、r、s、t、u、aa、bbおよびccは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の間の整数から独立して選択される員であり;
j、k、lおよびmは0から100の間の整数から独立して選択される員であり;
v、w、xおよびyは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はHまたはグリコシル残基または修飾基または複合糖質である、
から選択される式を有するグリコシル残基を含んでなる。この方法は:
(a)GM−CSFペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合した該グリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、GM−CSFペプチドをシアリダーゼと、GM−CSFペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と該部分との間に結合物を形成することを含んでなる。
(d)工程(a)の前に、GM−CSFペプチドをグリコシダーゼおよびシアリダーゼの組み合わせと接触させることを含んでなる。
(e)工程(a)の前に、GM−CSFペプチドをエンドグリカナーゼと、、GM−CSFペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(f)工程(a)の前に、GM−CSFペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcをGM−CSFペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(g)工程(a)の前に、GM−CSFペプチドをマンノシダーゼと、GM−CSFペプチドからマンノース残基を開裂するために適する条件下で接触させることを含んでなる。
(h)工程(a)の前に、GM−CSFペプチドをST3Gal3およびシアル酸供与体と、シアル酸を生成物に転移させるために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、p、q、r、s、tおよびuは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の間の整数から独立して選択される員であり;
j、k、lおよびmは0から100の間の整数から独立して選択される員であり;
v、w、xおよびyは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はH、またはグリコシル残基、複合糖質または修飾基である、
を有するグリコシル残基を含んでなる。
(a)インターフェロンガンマペプチドを、グリコシルトランスフェラーゼおよび合成的に作用するガラクトシダーゼから選択される員および修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、インターフェロンガンマペプチドをシアリダーゼと、インターフェロンガンマペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と部分との間に結合物を形成することを含んでなる。
(d)工程(a)の前に、インターフェロンガンマペプチドをグリコシダーゼおよびシアリダーゼの組み合わせと接触させることを含んでなる。
(e)工程(a)の前に、インターフェロンガンマペプチドをエンドグリカナーゼと、インターフェロンガンマペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(f)工程(a)の前に、インターフェロンガンマペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcをインターフェロンガンマペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(g)工程(a)の前に、インターフェロンガンマペプチドを、ガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを生成物へ転移させるために適する条件下で接触させることを含んでなる。
(h)工程(a)の生成物を、シアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、p、q、r、s、tおよびuは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の間の整数から独立して選択される員であり;
j、k、lおよびmは0から100の間の整数から独立して選択される員であり;
v、w、xおよびyは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はH、またはグリコシル残基、複合糖質または修飾基である、
を有するグリコシル残基を含んでなる。
(a)A−1−PIペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、A−1−PIペプチドをシアリダーゼと、A−1−PIペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)からの生成物を修飾基に反応する部分と接触させ、こうして完全なグリコシル連結基と部分との間に結合物を形成することを含んでなる。
(d)工程(a)の前に、A−1−PIペチドをグリコシダーゼおよびシアリダーゼの組み合わせ物と接触させることを含んでなる。
(e)工程(a)の前に、A−1−PIペプチドをエンドグリカナーゼと、A−1−PIペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(f)工程(a)の前に、A−1−PIペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcをA−1−PIペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(g)工程(a)の前に、A−1−PIペプチドをマンノシダーゼと、A−1−PIペプチドからマンノースを除去するために適する条件下で接触させることを含んでなる。
(h)工程(a)の前に、A−1−PIペプチドをマンノシダーゼ、キシロシダーゼ、ヘキソサミニダーゼおよびその組み合わせから選択される員と、A−1−PIペプチドからグリコシル残基を除去するために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、p、q、r、s、tおよびuは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の間の整数から独立して選択される員であり;
j、k、lおよびmは0から100の間の整数から独立して選択される員であり;
v、w、xおよびyは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はH、またはグリコシル残基、複合糖質または修飾基である、
を有するグリコシル残基を含んでなる。
(a)ベータグルコシダーゼペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、ベータグルコシダーゼペプチドをシアリダーゼと、ベータグルコシダーゼペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と部分との間に結合物を形成することを含んでなる。
(d)工程(a)の前に、ベータグルコシダーゼペプチドをグリコシダーゼおよびシアリダーゼの組み合わせ物と接触させることを含んでなる。
(e)工程(a)の前に、ベータグルコシダーゼペプチドをエンドグリカナーゼと、ベータグルコシダーゼペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(f)工程(a)の前に、ベータグルコシダーゼペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcを該ベータグルコシダーゼペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(g)工程(a)の前に、ベータグルコシダーゼペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを生成物へ転移させるために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、o、p、q、r、s、t、u、v、w、xおよびyは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の整数から独立して選択される員であり;
j、k、lおよびmは0から100の整数から独立して選択される員であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;
R’はH、またはグリコシル残基、複合糖質または修飾基であり;そして
R”はグリコシル基、複合糖質または修飾基である、
を含んでなるグリコシルサブユニットを有する。この方法は:
(a)TPAペプチドを、グリコシルトランスフェラーゼおよび合成的に作用するグリコシダーゼから選択される員および修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、TPAペプチドをシアリダーゼと、TPAペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の生成物をシアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、TPAペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースをTPAペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(e)工程(a)の前に、上記TPAペプチドをグリルコシダーゼおよびシアリダーゼの組み合わせと接触させることを含んでなる。
(f)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と部分との間に結合物を形成することを含んでなる。
(g)工程(a)の前に、上PAペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcをTPAプチドへ転移させるために適する条件下で接触させることを含んでなる。
(h)工程(a)の前に、TPAペプチドをエンドグリカナーゼと、TPAペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(i)工程(a)の前に、TPAペプチドをマンノシダーゼ、キシロシダーゼ、ヘキソサミニダーゼおよびそれらの組み合わせから選択される員と、TPAペプチドからグリコシル残基を除去するために適する条件下で接触させることを含んでなる。
a、b、cおよびeは0および1から独立して選択される員であり;
dは0であり;そして
Rは修飾基である、
を有するグリコシル残基を含んでなる。この方法は:
(a)IL−2ペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、IL−2ペプチドをシアリダーゼと、IL−2ペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の前に、IL−2ペプチドを合成的に作用するエンド−N−アセチルガラクトサミニダーゼと、GalNAcをIL−2ペプチドに付加するために適する条件下で接触させることを含んでなる。
(d)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と該部分との間に結合物を形成することを含んでなる。
(e)工程(a)の前に、IL−2ペプチドをN−アセチルガラクトサミントランスフェラーゼおよびGalNAc供与体と、GalNAcをIL−2ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(f)工程(a)の前に、IL−2ペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースをIL−2ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、o、p、q、r、s、t、u、aa、ccおよびddは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の間の整数から独立して選択される員であり;
j、k、lおよびmは0から20の間の整数から独立して選択される員であり;
v、w、x、yおよびzは0であり;そして
Rは修飾基、マンノースまたはオリゴマンノースであり;
R’はH、グリコシル残基、修飾基および複合糖質から選択される員である、
から選択される員である式を有するグリコシル残基を含んでなる。この方法は:
(a)該糖ペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、糖ペプチドをシアリダーゼと、糖ペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の生成物をシアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、糖ペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを糖ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(e)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と該部分との間に結合物を形成することを含んでなる。
(f)工程(a)の前に、糖ペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcを糖ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(g)工程(a)の前に、糖ペプチドをエンドグリカナーゼと、糖ペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(h)工程(a)の前に、糖ペプチドをST3Gal3およびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(i)工程(a)の前に、糖ペプチドをマンノシダーゼと、糖ペプチドからマンノースを除去するために適する条件下で接触させることを含んでなる。
a、b、c、d、i、j、k、l、m、q、r、s、t、u、w、wwおよびzは0および1から独立して選択される員であり;
e、f、gおよびhは0から4の間の整数から独立して選択される員であり;
n、v、xおよびyは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はH、グリコシル残基、修飾基および複合糖質から選択される員である、
を有するグリコシル残基を含んでなる。この方法は:
(a)該糖ペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合した該グリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、糖ペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを糖ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(c)工程(a)の前に、糖ペプチドをエンドグリカナーゼと、糖ペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、p、q、r、s、tおよびuは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の間の整数から独立して選択される員であり;
j、k、lおよびmは0から100の間の整数から独立して選択される員であり;
v、w、xおよびyは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はH、グリコシル残基、複合糖質または修飾基である、
を有するグリコシル残基を含んでなる。
(a)該ウロキナーゼペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、ウロキナーゼペプチドをシアリダーゼと、ウロキナーゼペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の生成物をシアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、ウロキナーゼペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースをウロキナーゼペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(e)工程(a)の前に、記ウロキナーゼペプチドをグリコシダーゼおよびシアリダーゼの組み合わせと接触させることを含んでなる。
(f)工程(a)からの生成物を修飾基に反応する部分と接触させ、これによりの完全なグリコシル連結基と該部分との間に結合物を形成することを含んでなる。
(g)工程(a)の前に、ウロキナーゼペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcをウロキナーゼペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(h)工程(a)の前に、ウロキナーゼペプチドをエンドグリカナーゼと、ウロキナーゼペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
a、b、c、d、i、j、k、l、m、r、s、t、u、z、aa、bb、ccおよびeeは、0および1から独立して選択される員であり;
e、f、gおよびhは0から4の整数から独立して選択される員であり;
n、v、w、x、yおよびddは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はH、グリコシル残基、修飾基および複合糖質から選択される員である、
から選択される員である式を有するグリコシル残基を含んでなる。この方法は:
(a)糖ペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合した該グリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、糖ペプチドをシアリダーゼと、糖ペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の生成物をシアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、糖ペプチドを合成的に作用するガラクトシダーゼと、ガラクトシダーゼを糖ペプチドに付加するために適する条件下で接触させることを含んでなる。
(e)工程(a)の前に、糖ペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを糖ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(f)工程(a)からの生成物を、ST3Gal3およびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(g)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と該部分との間に結合物を形成することを含んでなる。
(h)工程(a)の前に、糖ペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcを糖ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(i)工程(a)の前に、糖ペプチドをエンドグリカナーゼと、糖ペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
a、b、c、d、i、j、k、l、q、r、s、t、uおよびzは、0および1から独立して選択される員であり;
e、f、gおよびhは0から4の間の整数から独立して選択される員であり;
n、v、w、xおよびyは0であり;
mは0〜20であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’は水素およびグリコシル残基および修飾基から選択される員である、
を有するグリコシル残基を含んでなる。この方法は:
(a)キメラ抗−HER2抗体ペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、キメラ抗−HER2抗体ペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースをキメラ抗−HER2抗体ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(c)工程(a)の前に、キメラ抗−HER2抗体ペプチドをエンドグリカナーゼと、キメラ抗−HER2抗体ペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
a、b、c、d、i、j、k、l、m、p、q、r、s、t、uおよびzは、0および1から独立して選択される員であり;
e、f、gおよびhは0から4の整数から独立して選択される員であり;
n、v、w、xおよびyは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はHおよびグリコシル残基、複合糖質および修飾基から選択される員である、
を有するグリコシル残基を含んでなる。この方法は:
(a)抗−RSV Fペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、抗−RSV Fペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを抗−RSV Fペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(c)工程(b)の前に、抗−RSV Fペプチドをエンドグリカナーゼと、抗−RSV
Fペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
a、b、c、d、i、j、k、l、m、q、r、s、t、uおよびzは、0および1から独立して選択される整数であり;
e、f、gおよびhは0から4の整数から独立して選択され;
n、v、w、xおよびyは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はH、グリコシル残基、複合糖質または修飾基から選択される員である、
を含んでなるグリコシルサブユニットを有する。この方法は:
(a)抗−CD20抗体ペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、抗−CD20抗体ペプチドをガラクトシルトランスフェラーゼおよびガラクトシル供与体と、ガラクトシル供与体を抗−CD20抗体ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(c)工程(b)の前に、抗−CD20抗体ペプチドをエンドグリカナーゼと、抗−CD20抗体ペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、抗−CD20抗体ペプチドをマンノシダーゼと、抗−CD20抗体ペプチドからマンノースを除去するために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、p、q、r、s、tおよびuは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の間の整数から独立して選択される員であり;
j、k、lおよびmは0から100の間の整数から独立して選択される員であり;
v、w、xおよびyは0であり;そして
Rはポリマー、複合糖質、マンノース、オリゴマンノースおよび修飾基から選択される員である、
を有するグリコシル残基を含んでなり、該方法が
(a)組換えDNaseペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、組換えDNaseペプチドをシアリダーゼと、組換えDNaseペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の生成物をシアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、組換えDNaseペプチドを、ガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを組換えDNaseペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(e)工程(a)の前に、組換えDNaseペプチドをグリコシダーゼおよびシアリダーゼの組み合わせと接触させることを含んでなる。
(f)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と部分との間に結合物を形成することを含んでなる。
(g)工程(a)の前に、組換えDNaseペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcを該組換えDNaseペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(h)工程(a)の前に、組換えDNaseペプチドをエンドグリカナーゼと、組換えDNaseペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
a、b、c、d、i、n、o、p、q、r、s、t、uおよびzは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の間の整数から独立して選択される員であり;
j、k、lおよびmは0から20の間の整数から独立して選択される員であり;
n、v、w、xおよびyは0であり;そして
Rは修飾基、マンノースまたはオリゴマンノースであり;
R’は複合糖質または修飾基である、
を有するグリコシル残基を含んでなる。この方法は:
(a)抗−TNFアルファペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、抗−TNFアルファペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを抗−TNFアルファペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(c)工程(a)の前に、抗−TNFアルファペプチドをエンドグリカナーゼと、抗−TNFアルファペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
a、b、c、d、i、j、k、l、m、r、s、t、u、z、aa、bb、ccおよびeeは、0および1から独立して選択される員であり;
e、f、gおよびhは0から4の間の整数から独立して選択される員であり;
dd、n、v、w、xおよびyは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はH、グリコシル残基、修飾基および複合糖質から選択される員である、
から選択される員である、式を有するグリコシル残基を含んでなる。この方法は:
(a)糖ペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合した該グリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、糖ぺプチドをシアリダーゼと、糖ペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の生成物を、シアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、糖プチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcを糖ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(e)工程(a)の前に、糖ペプチドをエンドグリカナーゼと、糖ペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
aa、bb、a、b、c、d、i、n、q、r、s、tおよびuは、0および1から独立して選択される員であり;
e、f、gおよびhは0から6の間の整数から独立して選択される員であり;
o、p、j、k、lおよびmは0から100の間の整数から独立して選択される員であり;
cc、v、w、xおよびyは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はH、またはグリコシル残基、複合糖質、または修飾基である、
から選択される員である式を有するグリコシル残基を含んでなる。
(a)HBsAgペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、HBsAgペプチドをシアリダーゼと、HBsAgペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の生成物をシアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、HBsAgペプチドをガラクトシダーゼと、HBsAgペプチドからグリコシル残基を開裂するために適する条件下で接触させることを含んでなる。
(e)工程(a)の前に、HBsAgペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースをHBsAgペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(f)工程(d)の生成物を、ST3Gal3およびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(g)工程(a)からの生成物を修飾基に反応する部分と接触させ、これにより完全なグリコシル連結基と部分との間に結合物を形成することを含んでなる。
(h)工程(a)の前に、HBsAgペプチドをN−アセチルグルコサミントランスフェラーゼおよびGlcNAc供与体と、GlcNAcをHBsAgペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(i)工程(a)の前に、HBsAgペプチドをマンノシダーゼと、HBsAgペプチドからマンノースを開裂するために適する条件下で接触させることを含んでなる。
(j)工程(a)の前に、HBsAgペプチドをエンドグリカナーゼと、HBsAgペプチドからグリコシル基を開裂するために適する条件下で接触させることを含んでなる。
a、b、c、d、i、j、k、l、m、r、s、t、u、z、aa、bb、ccおよびeeは、0および1から独立して選択される員であり;
e、f、gおよびhは0から4の間の整数から独立して選択される員であり;
n、v、w、x、yおよびddは0であり;
Rは修飾基、マンノースまたはオリゴマンノースであり;そして
R’はH、グリコシル残基、修飾基および複合糖質から選択される員である、
から選択される員である式を有するグリコシル残基を含んでなる。この方法は:
(a)糖ペプチドを、グリコシルトランスフェラーゼおよび修飾基に共有結合したグリコシルトランスフェラーゼの基質であるグリコシル部分を含んでなる修飾されたグリコシル供与体と、完全なグリコシル連結基が形成するために適する条件下で接触させることを含んでなる。
(b)工程(a)の前に、糖ペプチドをシアリダーゼと、糖ペプチドからシアル酸を除去するために適する条件下で接触させることを含んでなる。
(c)工程(a)の前に、糖ペプチドをエンドグリカナーゼと、糖ペプチドからグリコシル部分を開裂するために適する条件下で接触させることを含んでなる。
(c)工程(a)の前に、糖ペプチドをガラクトシルトランスフェラーゼおよびガラクトース供与体と、ガラクトースを糖ペプチドへ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)の生成物をシアリルトランスフェラーゼおよびシアル酸供与体と、シアル酸を生成物へ転移させるために適する条件下で接触させることを含んでなる。
(d)工程(a)の前に、糖ペプチドをガラクトシダーゼと、糖ペプチドからグリコシル残基を開裂するために適する条件下で接触させることを含んでなる。
本発明は、特別にカスタマイズされた、または所望のグリコシル化パターンを有する糖ペプチド分子を提供する様式で、ペプチド分子に、またはペプチド分子から糖を無細胞でインビトロ付加および/または削除するための方法および組成物を含み、ここで糖ペプチドは工業的規模で生産される。本発明の好適な態様では、そのように生産された糖ペプチドは酵素反応を介してペプチドに付加された修飾糖が糖ペプチドに結合している。本発明の重要な特徴は、任意の細胞型により生産されるペプチドを取り、そしてペプチド上にコアグリカン構造を生成し、その後にグリカン構造をインビトロで改造して、哺乳動物の治療的使用に適するグリコシル化パターンを有する糖ペプチドを生成するように改造することである。より具体的には、結合分子がペプチドに有利な特性を付与するように、本発明に従い修飾された糖分子またはそれに結合した他の化合物を有する糖ペプチド分子を調製することが可能である。本発明により、結合分子のペプチドへの酵素に基づく付加は位置選択性および立体選択性の利点を有するので、結合分子はペプチドに酵素的に付加される。したがって本明細書に提供する方法および組成物を使用することにより、ペプチドを改造して好ましくはペプチドに結合した修飾された糖を有する望ましいグリカン構造をペプチドに付与することが可能である。また本発明の方法および組成物を使用することにより、工業的規模で所望する、かつまたは修飾されたグリカン構造を有するペプチド分子を生成することも可能であり、これにより初めて当該技術分野に改善された治療用ペプチドを効率的に生産するための現実的解決を提供する。
他に定義しない限り、本明細書で使用するすべての技術でおよび科学的用語は、一般に本発明が属する分野で当業者が普通に理解している意味と同じ意味を有する。一般に細胞培養、分子遺伝学、有機化学、および核酸化学およびハイブリダイゼーションで本明細書で使用する命名法および研究手順は、当該技術分野において周知なものであり、そして普通に使用されている。核酸およびペプチド合成については標準的な技術を使用する。これらの技法および手順は当該技術分野の常法および様々な一般的参考書(例えばSambrook et al.,1989,モレキュラークローニング:ア ラボラトリーマニュアル(Molecular Cloning:A Laboratory Manual)、第2版、コールドスプリングハーバーラボラトリー出版、コールドスプリングハーバー、ニューヨーク州)に従い一般に行われ、これらはこの文献全体にわたって提供されている。本明細書で使用する命名法および以下に記載する分析化学および有機合成で使用する研究手順は周知で、しかも当該技術分野では普通に使用されているものである。標準技法およびそれらの変法を、化学合成および化学分析に使用する。
al.、1989、抗体:ア ラボラトリーマニュアル(Antibodies:A Laboratory Manual)、コールドスプリングハーバー、ニューヨーク;Houston et al.,1988,Proc.Natl.Acad.Sci.USA85:5879−5883;Bird et al.,1988,Science242:423−426)。
et al.,編集、1999、CSHL出版を参照にされたい。
al.,(1986)J.Biol.Chem.261:11550−11557;Kanamori et al.,J.Biol.Chem.265:21811−21819(1990))。また含まれるのは9−O−C1−C6アシル−Neu5Ac様の9−O−ラクチル−Neu5Acまたは9−O−アセチル−Neu5Ac、9−デオキシ−9−フルオロ−Neu5Acおよび9−アジド−9−デオキシ−Neu5Acのような9−置換シアル酸である。シアル酸ファミリーの総説については、例えばVarki,Glycobiology,2:25−40(1992);シアル酸:化学、代謝および機能(Sialic Acid:Chemistry,Metabolism and Function)、R.Schauer、編集(スプリンガーファーラーグ、ニューヨーク(1992))を参照にされたい。シアリル化手順におけるシアル酸化合物の合成および使用は、1992年、10月1日に公開された国際公開第92/16640号パンフレットに開示されている。
グリシン、アラニン;
バリン、イソロイシン、ロイシン;
アスパラギン酸、グルタミン酸;
アスパラギン、グルタミン;
セリン、トレオニン;
リシン、アルギニン;
フェニルアラニン、チロシン。
PHARMACOLOGICAL ASPECTS)」で、Feeney et al.、アメリカ化学学会、ワシントン、D.C.、1982、第370〜387頁;Kasina et al.,Bioconjugate Chem.,9:108−117(1988);Song et al.,Bioconjugate Chem.,8:249−255(1997)を参照にされたい。
I.グリカン鎖の改造するための方法
本発明は、好ましくは修飾された糖をペプチドに付加することを含む特別にカスタマイズされた、または所望のグリコシル化パターンを有するペプチド分子を提供するような様式で、糖ペプチド分子へ、またはそれから糖をインビトロで付加および/または削除するための方法および組成物を含む。したがって本発明の重要な特徴は、任意の細胞型により生産されるペプチドを取り、そしてペプチド上にコアグリカン構造を生成し、その後にグリカン構造をインビトロで改造して哺乳動物における治療的使用に適するグリコシル化パターンを有するペプチドを生成することである。
1つの観点では、本発明は商業的または製薬学的に興味深いほとんどのペプチドが本明細書でトリマンノシルコア(trimannosyl core)と呼ぶ共通する5個の糖構造を含んでなり、これはペプチド鎖上の配列Asn−X−Ser/ThrでアスパラギンにN−結合しているという事実を利用する。基本的なトリマンノシルコアは本質的にペプチドに結合した2個のN−アセチルグルコサミン(GlcNAc)残基および3個のマンノース(Man)残基からなり、すなわちこれは場合によりフコース残基を含むことができる場合を除き、このような5個の糖残基を含んでなり、そしてさらなる糖は含まない。第1GlcNAcはアスパラギンのアミド基に結合し、そして第2のGlcNAcはβ1,4−結合を介して第1に結合している。マンノース残基はβ1,4−結合を介して第2のGlcNAcに結合し、そして2個のマンノース残基がそれぞれα1,3およびα1,6結合を介してこのマンノースに結合している。トリマンノシルコア構造の図解的説明は図2、左側に示す。ほとんどのペプチド上のグリカン構造はトリマンノシルコアに加えて他の糖を含んでなるのが通例であるが、トリマンノシルコア構造は哺乳動物のペプチド上のN−結合グリカンの本質的特徴を表す。
本発明の基本的なトリマンノシルコアまたはMan3GlcNAc4糖ペプチドは、必要ならばペプチド精製の分野で周知な技術を使用して単離し、そして精製することができる。適当な技術にはクロマトグラフィー技法、等電点電気泳動技法、超遠心技法等を含む。そのような任意の技法を使用して、本発明の組成物を調製することができ、ここで本発明の糖ペプチドは通常、細胞培養基内に見いだされる他のペプチドおよび他の成分から単離されている。精製の程度は例えば他のペプチドに関して90%または95%、あるいは一層高い、例えば98%となり得る。例えばDeutscher et al.(編集、1990、ペプチド精製に対する手引き(Guide to Peptide Purification)、Harcourt Brace Jovanovich、サンディエゴ)を参照にされたい。
N−結合糖ペプチドが細胞により生産される時、本明細書のいたるところで記載するように、これは他の糖部分をそれに付加する前に共通の、一般的には基本的なトリマンノシルコアまたはMan3GlcNAc4構造に減少されなければならないグリカン構造の異種の混合物を含んでなるかもしれない。どの糖を特定のグリカン構造から除去するべきであるかを正確に決定するために、1次グリカン構造を同定することが必要となる場合がある。グリカンの1次構造を決定するための技術は当該技術分野では周知であり、そして例えばMontreuil、「糖ペプチドの構造および生合成(Structure and Biosynthesis of Glycopeptides)」、医学的応用における多糖(Polysaccharides in Medicinal Applications)、第273〜327頁、1996、Severian Damitriu編集、マルセルデッカー、ニューヨーク州、に詳細に記載されている。したがって糖生物学の分野の当業者には、細胞により生産されたペプチド群を単離し、そしてそれに結合しているグリカンの構造(1つまたは複数)を決定することは単純な事柄である。例えば(i)加水分解、アセトリシス、ヒドラジン分解のような化学的開裂により、または亜硝酸のデアミネーションによりいずれかでグリコシド結合を分解し(splitting);(ii)メチル化を完成し、続いて加水分解またはメタノリシスを行い、そして部分的にメチル化された単糖のガス−液体クロマトグラフィーおよび質量分析を行い;そして(iii)エキソグリコシダーゼを使用して単糖間のアノマー結合を決定し、これはまた順次分解により1次グリカン構造の洞察も提供するための効率的な方法を利用することができる。特に質量分析および核磁気共鳴(NMR)分光法の技術、特に高磁場NMRはグリカン1次構造を決定するために成功裏に使用されてきた。
N−結合グリコシル化の改造は、式1:
a、b、c、d、eおよびxは(独立して選択された)0、1または2であるが、ただしa、b、c、d、eおよびxから選択される少なくとも1つの員は1または2である、を参照にして最もよく説明される。
X3およびX5=|−GlcNAc−Gal−SA;
aおよびc=1;
d=0または1;
b、eおよびx=0
を有するように改造される。
X3およびX5は|−GlcNAc−Gal−SA;
aおよびc=1;
X4がGlcNAcであり;
b=1;
d=0または1;
eおよびx=0
を有するように改造される。
X3およびX5は
a、c、d=1;
b、eおよびx=0;
X6=フコース
を有するように改造される。
X3およびX5は|−GlcNAc−Gal−SA−R;
aおよびc=1または2;
d=0または1;
b、d、eおよびx=0;
ここでR=結合基
を有するように改造される。
X3およびX5は|−GlcNAc−Gal−(SA)であり;
aおよびc=1;
b、d、eおよびx=0
である。
X3およびX5は|−GlcNAcであり;
aおよびc=0または1;
b=0;
X6はフコースであり;
d=0、1または2;そして
eおよびx=0
である。
セルビシエ(Saccharomyces cerevisiae)のような酵母で発現させる。N−結合グリカンの共通部位を持つペプチドがS.セルビシエ(cerevisiae)細胞から発現され、そして分泌される時、N−結合グリカンは図5の左に表す構造を有するだろう。N−結合グリカンは常にトリマンノシルコアを有し、これはしばしばマンノースまたは関連する多糖を用いて1000残基まで合成される。式1の用語において、
X3およびX5=|−Man−Man−(Man)0−1000;
aおよびc=1または2;
b、d、eおよびx=0
である。
この工程は図2、3および4で具体的に説明する。
O−グリコシル化は種々の単糖がO−グリコシド結合でヒドロキシアミノ酸へ結合していることが特徴である。O−グリコシル化は動物および植物界で広く行き渡った翻訳後修飾である。タンパク質にO−結合したグリカンの構造的複雑さは、N−結合グリカンのそれをはるかに越える。新たに翻訳されたペプチドのセリンまたはトレオニン残基は、ペプチドのGalNAcトランスフェラーゼによりゴルジのシスからトランス区画で修飾されるようになる。O−グリコシル化の部位はグリコシルトランスフェラーゼの配列特異性により決定されだけでなく、異なる基質部位間の競合およびグリカンを形成する原因となる他のグリコシルトランスフェラーゼとの競合により媒介される後成的調節によっても決定される。
O−結合グリコシル化の改造は、式2を参照にして最もよく具体的に説明される:
X2は|−SA;または|−SA−SAであり;
fおよびn=0または1;
X10はSAであり;
m=0
を有するように改造される。
X2は|−SAであり; X10はFucまたは|−GalNAc(Fuc)−Gal−SAであり;
fおよびn=1;
m=0
を有するように改造される。
X2は|−SA−Rであり;
f=1;
nおよびm=0
ここでRは結合基である
を有するように改造される。
X2=|−SA;
f=1;
mおよびn=0
である構造を有することが多い。
X2=H;
f=0または1;
nおよびm=0
である構造を有する。
|−AA−Man−Man1−2
を有する。Gemmill and Trimble(1999,Biochim.Biophys.Acta 1426:227−237)を参照にされたい。このようなO−結合グリカンをヒトでの使用に改造するために、グリカンをアミノ酸レベルで開裂し、そしてここから再構築することが好ましい。
本発明はグリコシル化または非グリコシル化ペプチドの結合物の調製法を提供する。本発明の結合物はペプチドと水溶性ポリマー、治療部分、診断部分、標的部分等のような多様な種との間で形成される。また提供されるのはリンカーアーム(linker arm)を介して一緒に連結された2以上のペプチドを含む結合物、すなわち多機能性結合物(multifunctional conjugate)である。本発明の多機能性結合物は、同じペプチドの2以上のコピー、または異なる構造および/または特性を持つ多様なペプチドの集合を含むことができる。
第1の観点では、本発明はペプチドと選択部分との間に結合物を提供する。ペプチドと選択部分との間の連結には、ペプチドと選択部分の間に挿入された完全なグリコシル連結基を含む。本明細書で検討するように、選択部分は、サッカリド単位に結合することができる本質的には任意の種であり、ペプチドに修飾された糖を付ける適切なトランスフェラーゼ酵素により認識される「修飾された糖」を生じる。修飾された糖のサッカリド成分は、ペプチドと選択部分との間に挿入された時、「完全なグリコシル連結基」となる。このグリコシル連結基は、選択部分を用いた修飾後に適切なトランスフェラーゼの基質となる単−またはオリゴ糖から形成される。
に相当する。「作用物質(agent)」は治療薬、生物活性剤、検出可能な標識、水溶性部分等である。この「作用物質」はペプチド、例えば酵素、抗体、抗原等であることができる。リンカーは任意の広い配列の上記連結基であることができる。あるいはリンカーは単結合または「ゼロ次リンカー(zero order linker)」でもよい。ペプチドの同一性には限界がない。ペプチドの例を図1に提供する。
et al.,Biotechnol.Appl.Biochem.,12:119−128(1990));N−ヒドロキシスクシンイミド誘導化活性エステル(Buckmann et al.,Makromol.Chem.,182:1379−1384(1981);Joppich et al.,Makromol.Chem.,180:1381−1384(1979);Abuchowski et al.,Cancer
Biochem.Biophys.,7:175−186(1984);Katreet al.,Proc.Natl.Acad.Sci.U.S.A.,84:1487−1491(1987);Kitamura et al.,Cancer Res.,51:4310−4315(1991);Boccu et al.,Z.Naturforsch.,38C:94−99(1983);カーボネート(Zalipsky et
al.,ポリ(エチレングリコール)化学:生物工学的および生物医学的応用(POLY(ETHYLENE GLYCOL)CHEMISTRY:BIOTECHNICAL
AND BIOMEDICAL APPLICATIONS)、Harris編集、プレナム出版、ニューヨーク、1992、第347−370頁;Zalipsky et al.,Biotechnol.Appl.Biochem.,15:100−114(1992);Veronese et al.,Appl.Biochem.Biotech.,11:141−152(1985));イミダゾリルホルメート(Beauchamp et al.,Anal.Biochem.,131:25−33(1983);Berger et al.,Blood,71:1641−1647(1988));4−ジチオピリジン(Woghiren et al.,Bioconjugate Chem.,4:314−318(1993));イソシアネート(Byun et al.,ASAIO Journal,M649−M−653(1992))およびエポキシド(Noishiki et al.へ発効された米国特許第4,806,595号明細書(1989)を参照にされたい。他の連結基にはアミノ基と活性化PEGとの間のウレタン結合を含む。Veronese,et al.,Appl.Biochem.Biotechnol.,11:141−152(1985)を参照にされたい。
修飾されたグリコシル供与体種(「修飾された糖」)は好ましくは、修飾された糖ヌクレオチド、活性化された修飾された糖およびヌクレオチドでもなく活性化されてもいない単なるサッカリドである修飾された糖から選択される。任意の所望する炭水化物構造が本発明の方法を使用してペプチドに加えられる。典型的には構造は単糖であるが、本発明は修飾された単糖の糖の使用に限定されず;オリゴ糖および多糖も有用である。
a)水溶性ポリマー
選択したペプチドの親水性は、アミン−、エステル−、ヒドロキシル−およびポリヒドロキシル−含有分子のような極性分子との結合により強化される。代表例には限定するわけではないがポリリシン、ポリエチレンイミン、ポリ(エチレングリコール)およびポリ(プロピレングリコール)を含む。好適な水溶性ポリマーは本質的に非−蛍光性であるか、あるいはアッセイで蛍光マーカーとして使用するためには不適当であるような最少量の蛍光を発する。天然に存在する糖ではないポリマーを使用してもよい。さらに他の物質(例えばポリ(エチレングリコール)、ポリ(プロピレン グリコール)、ポリ(アスパルテート)、生体分子、治療部分、診断部分等)の共有結合により修飾されている外は天然に存在する糖の使用も意図している。別の例示態様では、治療用の糖部分をリンカーアームに結合し、そして糖−リンカーアームを続いて本発明の方法を介してペプチドに結合する。
AND CROSSLINKING)、CRC出版、ボカラトン;G.T.Hermanson et al.,(1993)、固定化アフィニティーリガンド技術(IMMOBILIZED AFFINITY LIGAND TECHNIQUES)、アカデミック出版、ニューヨーク;Dunn,R.L.,et al.,編集、ポリマー性薬剤および薬剤送達系(POLYMERIC DRUGS AND DELIVERY SYSTEMS)、ACSシンポジウムシリーズ、第469巻、アメリカ化学学会、ワシントン、D.C.1991)を参照にされたい。
X、Y、W、U(独立して選択される)=O、S、NH、N−R’;
R’、R”’(独立して選択される)=アルキル、ベンジル、アリール、アルキルアリール、ピリジル、置換アリール、アリールアルキル、アシルアリール;
n=1〜2000;
m、q、p(独立して選択される)=0〜20
o=0〜20
Z=HO、NH2、ハロゲン、S−R”’、活性化エステル、
V=HO、NH2、ハロゲン、S−R”’、活性化エステル、活性化アミド、−糖−ヌクレオチド、タンパク質。
X、Y、W、A、B(独立して選択される)=O、S、NH、N−R’(CH2)l;
n、p(独立して選択される)=1〜2000;
m、q、o(独立して選択される)=0〜20
Z=HO、NH2、ハロゲン、S−R”’、活性化エステル、
V=HO、NH2、ハロゲン、S−R”’、活性化エステル、活性化アミド、−糖−ヌクレオチド、タンパク質。
et al.J.Clin.Invest.89:1643−1651(1992);Pyatak et al.,Res.Commun.Chem.Pathol Pharmacol.29:113−127(1980))。インターロイキン−2のPEG化は、インビボで抗腫瘍効力を上昇させることが報告され(Katre et al.,Proc.Natl.Acad.Sci.USA.84:1487−1491(1987))、そしてモノクローナル抗体A7に由来するF(ab’)2のPEG化は、その腫瘍への局在化を向上させた(Kitamura et al.,Biochem.Biophys.Res.Commun.28:1387−1394(1990))。
Inc.)、ランカスター、ペンシルベニア州)に見出すことができる。
本発明の結合物は1以上の水−不溶性ポリマーを含んでもよい。本発明のこの態様は、治療用ペプチドを制御された様式で送達する賦形剤としての結合物の使用により具体的に説明される。ポリマー性の薬剤送達系は当該技術分野では既知である。例えばDunn et al.、ポリマー性薬剤および薬剤送達系(POLYMERIC DRUG AND DRUG DELIVERY SYSTEMS)、ACSシンポジウムシリーズ、第469巻、アメリカ化学学会、ワシントン、D.C.1991を参照にされたい。当業者は実質的に任意の既知の薬剤送達系を本発明の結合物に応用できると考えるだろう。
c)生体分子
別の好適な態様では、修飾された糖は生体分子を持つ。さらに好適な態様では、生体分子は機能的タンパク質、酵素、抗原、抗体、核酸(例えばシングルヌクレオチドまたはヌクレオシド、オリゴヌクレオチド、ポリヌクレオチドおよび1−およびより多い鎖の核酸)、レクチン、レセプターまたはそれらの組み合わせである。
Morgan,Cellular and Molecular Neurobiology 20:77−95(2000)。トランスフェリン−トランスフェリンレセプター相互作用に基づく送達系が、ペプチド、タンパク質およびリポソームを脳に効率的に送達するために確立された。例えばペプチドは脳によるより多い取り込みを達成するために、トランスフェリンレセプターに対して向けられたMabにカップリングすることができる、Moos and Morgan、同上。同様に、トランスフェリンレセプターに向けられたMAbにカップリングした時、血液脳関門全域の塩基性繊維芽細胞増殖因子(bFGF)の輸送が強化される。Song et al.,The Journal of
Pharmacology and Experimental Therapeutics 301:605−610(2002);Wu et al.,Journal of Drug Targeting 10:239−245(2002)。さらに化学療法剤、ドキソルビシンのC6グリオーマへの効果的輸送のためのリポソーム送達系が報告され、ここでトランスフェリンはリポソームPEG鎖の遠位末端に結合された。Eavarone et al.,J.Biomed.Mater.Res.51:10−14(2000)。多数の米国特許もトランスフェリン−トランスフェリンレセプター相互作用に基づき血液−脳関門をバイパスする送達法に関する。例えば米国特許第5,154,924号;同第5,182,107号;同第5,527,527号;同第5,833,988号;同第6,015,555号明細書を参照にされたい。
別の好適な態様では、修飾された糖は治療部分を含む。当業者は治療部分と生体分子の範疇の間で重複する部分があると考えるだろう;多くの生体分子が治療特性または潜在性を有する。
本発明の結合物を形成するために有用な修飾された糖を本明細書で検討する。この検討は説明の簡明さのために水溶性ポリマーで修飾された糖の調製に焦点をあてる。特にこの検討はポリ(エチレングリコール)部分を含む修飾された糖の調製に焦点をあてる。当業者は本明細書に記載する方法が修飾された糖の調製に広く応用可能であり、したがってこの検討は本発明の範囲を限定すると解釈すべきでないと考えるだろう。
(a)カルボキシル基およびそれらの種々の誘導体、限定するわけではないが、N−ヒドロキシスクシンイミドエステル、N−ヒドロキシベンゾトリアゾールエステル、酸ハライド、アシルイミダゾール、チオエステル、p−ニトロフェニルエステル、アルキル、アルケニル、アルキニルおよび芳香族エステルを含む;
(b)例えばエステル、エーテル、アルデヒド等に転換できるヒドロキシル基;
(c)ハロアルキル基、ここでハライドは例えばアミン、カルボキシレートアニオン、チオールアニオン、カルバニオン、またはアルコキシドイオンのような求核性基に後で置き換わることができ、これによりハロゲン原子の官能基で新たな基の共有結合をもたらす;(d)ディールス−アルダー反応に参加することができるジエノフィル基、例えばマレイミド基;
(e)後の誘導化が例えばイミン、ヒドラゾン、セミカルバゾンまたはオキシムのようなカルボニル誘導体の形成を介して、あるいはグリニャール付加またはアルキルリチウム付加のようなメカニズムを介して可能となるようなアルデヒドまたはケトン基;
(f)例えばスルホンアミドを形成するために、アミンとの後の反応のためのスルホニルハライド基;(g)例えばジスルフィドに転換されるか、またはアルキルおよびアシルハライドと反応することができるチオール基;
(h)例えばアシル化、アルキル化または酸化され得るアミンまたはスルフヒドリル基;(i)例えば付加環化、アシル化、マイケル付加等を受けることができるアルケン;および
(j)例えばアミンおよびヒドロキシル化合物と反応することができるエポキシド。
上記の方法により生成されたヌクレオチド糖および誘導体は、精製せずに使用することができる。しかし通常、生成物を回収することが好ましい。薄または厚層(thick layer)クロマトグラフィー、カラムクロマトグラフィー、イオン交換クロマトグラフィー、または膜濾過のような標準的な周知技法を使用することができる。膜濾過を使用すること、より好ましくは逆浸透膜を使用することが好ましく、または回収のためにはこれから、そして本明細書に引用する技術文献で検討するように1以上のカラムクロマトグラフィー技術を使用することが好適である。例えば膜濾過(ここで膜は約3000〜約10,000の分子量カットオフを有する)を使用して、10,000Da未満の分子量を有する試薬についてタンパク質を取り出すために使用することができる。次に膜濾過または逆浸透を使用して塩を除去し、かつ/または生成物サッカリドを精製するために使用することができる(例えば国際公開第98/15581号パンフレットを参照にされたい)。ナノフィルター(nanofilter)膜は1種の逆浸透膜であり、これは使用する膜に依存して1価の塩を通すが、多価の塩および約100から約2,000ダルトンより大きい非電荷溶質は保持する。このように典型的な応用では、本発明の方法により調製されるサッカリドは膜内に保持され、そして混入する塩は通過する。
本発明の方法で使用するための修飾された糖の調製には、修飾基の糖残基への結合、そしてグリコシルトランスフェラーゼの基質となる安定な付加物の形成を含む。このように修飾基を糖へ結合するために架橋剤を使用することが好ましいことが多い。修飾基を炭水化物部分に結合するために使用することができる二官能性化合物の例には、限定するわけではないが二官能性ポリ(エチレングリコール)、ポリアミド、ポリエーテル、ポリエステル等を含む。炭水化物を他の分子に連結するための一般的取り組みは、技術文献にて知られている。例えば、Lee et al.,Biochemistry 28:1856(1989);Bhatia et al.,Anal.Biochem.178:408(1989);Janda et al.,J.Am.Chem.Soc.112:8886(1990)およびBednarski et al.、国際公開第92/18135号パンフレットを参照にされたい。以下の考察は反応基が新生の修飾された糖の糖部分上で良性(benign)として処置される。この考察に焦点をあてるのは具体的説明の明瞭性のためである。当業者はこの考察が修飾基上の反応基にも関連すると考えるだろう。
a.アミノ−反応基
1つの好適な態様では、クロス−リンカー上のこの部位はアミノ−反応基である。アミノ−反応基の有用な非限定的例には、N−ヒドロキシスクシンイミド(NHS)エステル、イミドエステル、イソシアネート、アシルハライド、アリールアジド、p−ニトロフェニルエステル、アルデヒドおよびスルホニルクロライドを含む。
別の好適な態様では、部位はスルフヒドリル−反応性基である。スルフヒドリル−反応基の有用な非限定的例には、マレイミド、アルキルハライド、ピリジルジスルフィドおよびチオフタルイミドを含む。
別の態様では、水および有機溶媒に可溶性のカルボジイミドをカルボキシル−反応試薬として使用する。これらの化合物は遊離のカルボキシル基と反応してプソイドウレアを形成し、次にこれは利用可能なアミンにカップリングしてアミド結合を形成することができる。カルボジイミドを用いてカルボキシル基を修飾する手順は、当該技術分野では周知である(Yamada et al.,Biochemistry 20:4836−4842,1981を参照にされたい)。
部位−特異的な反応性部分に加えて、本発明は糖を修飾基に連結するための非特異的な反応性基も企図する。
et al.,J.Org.Chem.55:3640−3647,1990)。
a.第一アミンと反応性のホモ二官能性クロスリンカー
アミン−反応性クロス−リンカーの合成、特性および応用は、技術文献で工業的使用のために記載されている(架橋の手順および試薬の総説については上記を参照にされたい)。多くの試薬が利用可能である(例えば、ピアスケミカル(Pierce Chemical)社、ロックフォード、イリノイ州;シグマケミカル社、セントルイス、モンタナ州;モレキュラープローブス社、ユージーン、オレゴン州)。
そのような試薬の合成、特性および応用は、技術文献に記載されている(架橋の手順および試薬の総説については上記を参照にされたい)。多くの試薬が市販されている(例えば、ピアスケミカル社、ロックフォード、イリノイ州;シグマケミカル社、セントルイス、モンタナ州;モレキュラープローブス社、ユージーン、オレゴン州)。
そのような試薬の合成、特性および応用は、技術文献に記載されている(架橋の手順および試薬の総説については上記を参照にされたい)。数種の試薬が市販されている(例えば、ピアスケミカル社、ロックフォード、イリノイ州;シグマケミカル社、セントルイス、モンタナ州;モレキュラープローブス社、ユージーン、オレゴン州)。
a.ピリジルジスルフィド部分を持つアミノ−反応性ヘテロ二官能性試薬
そのような試薬の合成、特性および応用は、技術文献に記載されている(架橋結合の手順および試薬の総説については上記を参照にされたい)。多くの試薬が市販されている(例えば、ピアスケミカル社、ロックフォード、イリノイ州;シグマケミカル社、セントルイス、モンタナ州;モレキュラープローブス社、ユージーン、オレゴン州)。
そのような試薬の合成、特性および応用は、技術文献に記載されている。マレイミド部分およびアミノ−反応性NHSエステルを持つヘテロ二官能性試薬の好適な非限定的例には、スクシンイミジルマレイミジルアセテート(AMAS)、スクシンイミジル3−マレイミジルプロピオネート(BMPS)、N−γ−マレイミドブチリルオキシスクシンイミドエステル(GMBS)N−γ−マレイミドブチリルオキシスルホスクシンイミドエステル(スルホ−GMBS)スクシンイミジル6−マレイミジルヘキサノエート(EMCS)、スクシンイミジル3−マレイミジルベンゾエート(SMB)、m−マレイミドベンゾイル−N−ヒドロキシスクシンイミドエステル(MBS)、m−マレイミドベンゾイル−N−ヒドロキシスルホスクシンイミドエステル(スルホ−MBS)、スクシンイミジル4−(N−マレイミドメチル)−シクロヘキサン−1−カルボキシレート(SMCC)、スルホスクシンイミジル 4−(N−マレイミドメチル)シクロヘキサン−1−カルボキシレート(スルホ−SMCC)、スクシンイミジル 4−(p−マレイミドフェニル)ブチレート(SMPB)およびスルホスクシンイミジル 4−(p−マレイミドフェニル)ブチレート(スルホ−SMPB)を含む。
そのような試薬の合成、特性および応用は、技術文献に記載されている。アルキルハライド部分およびアミノ−反応性NHSエステルを持つヘテロ二官能性試薬の好適な非限定的例には、N−スクシンイミジル−(4−ヨードアセチル)アミノベンゾエート(SIAB)、スルホスクシンイミジル−(4−ヨードアセチル)アミノベンゾエート(スルホ−SIAB)、スクシンイミジル−6−(ヨードアセチル)アミノヘキサノエート(SIAX)、スクシンイミジル−6−(6−((ヨードアセチル)−アミノ)ヘキサノイルアミノ)ヘキサノエート(SIAXX)、スクシンイミジル−6−(((4−ヨードアセチル)−アミノ)−メチル)−シクロヘキサン−1−カルボニル)アミノヘキサノエート(SIACX)およびスクシンイミジル−4((ヨードアセチル)−アミノ)−メチルシクロヘキサン−1−カルボキシレート(SIAC)を含む。
さらなる態様では、リンカー基には開裂して糖残基から修飾基を放出することができる基が提供される。多くの開裂性基が当該技術分野では知られている。例えばJung et al.,Biochem.Biophys.Acta 761:152−162(1983);Joshi et al.,J.Biol.Chem.265:14518−14525(1990);Zarling et al.,J.Immunol.124:913−920(1980);Bouizar et al.,Eur.J.Biochem.155:141−147(1986);Park et al.,J.Biol.Chem.261:205−210(1986);Browning et al.,Immunol.143:1859−1867(1989)を参照にされたい。さらに広範な開裂性の二官能性(ホモ−およびヘテロ−二官能性の両方)リンカー基がピアスのような供給元から市販されている。
修飾された糖は、結合を媒介する適切な酵素を使用してグリコシル化または非グリコシル化ペプチドに結合される。好ましくは修飾された供与体の糖(1つまたは複数)、酵素(1つまたは複数)および受容体ペプチド(1つまたは複数)の濃度は、受容体が消費されるまでグリコシル化が進行するように選択される。以下に検討する考察は、シアリルトランスルフェラーゼの内容について説明すると同時に、一般に他のグリコシルトランスフェラーゼ反応に応用することできる。
例示態様では、ペプチドを少なくとも1つのマンノース−6−ホスフェート部分で誘導化する。マンノース−6−ホスフェート部分はペプチドを細胞のリソソームに標的し、そして例えばリソソーム蓄積疾患(lysosomal storage disease)の治療に治療用タンパク質をリソソームに標的するために有用である。
Duve,Fed.Proc.23:1045(1964))。その時から、酵素代替治療が様々なリソソーム蓄積疾患を処置するために有益になり得るという種々の研究が提案された。最高の成功は、胎盤から(Ceredase(商標))またはより最近では組換え的(Cerezyme(商標))に調製した外因性酵素(β−グルコセレブロシダーゼ)で処置したI型ゴーシェ病を持つ個体で示された。酵素代替はファブリー病ならびに他のリソソーム蓄積疾患を処置するためにも有益であることが示唆された。例えばDawson et al.,Ped.Res.7(8):684−690(1973)(インビトロ)およびMapes et al.,Science 169:987(1970)(インビボ)を参照にされたい。酵素代替治療の臨床試験は、正常血漿(Mapes et al.,Science 169:987−989(1970))、胎盤から精製したα−ガラクトシダーゼA(Brady et al.,N.Eng.J.Med.279:1163(1973));または脾臓または血漿から精製したα−ガラクトシダーゼA(Desnick et al.,Proc.Natl.Acad.Sci.USA76:5326−5330(1979))の潅流を使用したファブリー患者について報告され、そしてファブリー病の直接的酵素代替の生物化学的効力が証明された。これらの研究は反復した酵素代替により病理学的な糖脂質蓄積を排除し、または有意に減少するための可能性を示す。例えば1つの実験では(Desnick et al.,同上)、精製された酵素の静脈内注入で、蓄積される脂質物質、グロボトリアシルセラミドの血漿レベルに一過性の減少がもたらされた。
al.,Proc.Natl.Acad.Sci.USA93:7917−7922(1996);Novo,F.J.,Gene Therapy.4:488−492(1997);Ohshima et al.,Proc.Natl.Acad.Sci.USA94:2540−2544(1997);およびSugimoto et al.,Human Gene Therapy 6:905−916(1995)を参照にされたい。マンノース−6−ホスフェートは治療用ペプチドがリソソームを標的とすることを媒介し、本発明は十分量の生物学的に活性なリソソームペプチドを欠損細胞に送達するための組成物および方法を提供する。
1つの態様では、本発明は基本的なトリマンノシルコアを主要なグリカン構造としてペプチドに結合して有する1つのペプチドの多数のコピーを含んでなる組成物を提供する。好適な態様では、ペプチドは治療用分子である。ペプチドの天然な形態は複雑なN−結合グリカンを含んでなることができ、あるいは高マンノースグリカンでよい。ペプチドは哺乳動物のペプチドでよく、そして好ましくはヒトのペプチドである。幾つかの態様では、ペプチドは免疫グロブリン、エリスロポエチン、組織型アクチベーターペプチドおよびその他からなる群から選択される(図1を参照にされたい)。
I、II、IVおよびVとを接触させることにより加える。TPAはさらにガラクトース供与体およびガラクトシルトランスフェラーゼを使用してガラクトシル化される。次いでPEGは、ST3Gal3により触媒されるシアル化により、そしてPEG部分で誘導化されたシアル酸の供与体を使用して分子に結合させる。図38Cでは、TPAが昆虫または真菌細胞で発現する。修飾には、適切なN−アセチルグルコサミンの供与体およびGnT Iおよび/またはIIを使用したN−アセチルグルコサミンの付加:ガラクトース供与体およびガラクトシルトランスフェラーゼを使用したガラクトシル化:およびST3Gal3およびPEGで誘導化されたシアル酸の供与体を使用したシアリル化によるPEGの結合工程を含む。図38Dでは、TPAが酵母で発現し、そして続いてエンドグリカナーゼで処理されてサッカリド鎖を戻し改作する。ポリペプチドは、供与体からTPAへのPEG−ガラクトースの転移を触媒するガラクトシルトランスフェラーゼの作用を介してさらにPEG化される。図38Eでは、TPAが昆虫または酵母細胞で発現する。ついでポリペプチドをα−およびβ−マンノシダーゼで処理して末端マンノシル残基を戻し改作する。さらにPEG部分は、ガラクトシルトランスフェラーゼにより媒介される適当な供与体からTPAへのPEG−ガラクトースの転移を介して分子へ結合させる。図38Fは昆虫または酵母系から得たTPAの異なる修飾法を提供する:ポリペプチドは、N−アセチルグルコサミンの供与体およびGnT Iおよび/またはIIを使用したN−アセチルグルコサミンの付加、続いてガラクトシルトランスフェラーゼおよびPEG化ガラクトースの供与体を使用したガラクトシル化により改造される。図38Gは昆虫または酵母細胞で発現したTPAの別の改造スキームを提供する。末端N−アセチルグルコサミンは、N−アセチルグルコサミンの供与体およびGnT Iおよび/またはIIを使用して加える。加水分解的様式というよりむしろ合成的に作用するように修飾されたガラクトシダーゼを利用して、適切な供与体からN−アセチルグルコサミン残基へPEG化ガラクトースを加える。図38Iでは、哺乳動物系で発現したTPAを最初にシアリダーゼおよびガラクトシダーゼで処理してシアル酸およびガラクトース残基を戻し改作する。ポリペプチドはさらに、レブリン酸で修飾したシアル酸供与体で適切な末端残基をキャッピングし、反応性ケトンをシアル酸供与体に付加することにより修飾する。ペプチドのグリコシル残基への付加後、ケトンをヒドラジン−またはアミン−PEGのような部分で誘導化する。図38Jでは、哺乳動物系で発現したTPAをこのスキームに従い改造する:最初にポリペプチドをα−およびβ−マンノシダーゼで処理して末端マンノシル残基を戻し改作する:次いでシアル酸残基をシアル酸供与体およびST3Gal3を使用して末端ガラクトシル残基に結合させる:さらにTPAは、ガラクトシルトランスフェラーゼにより触媒される供与体からN−アセチルグルコサミニル残基へのPEG化ガラクトースの転移を介してPEG化される。図38KではTPAが植物細胞で発現する。この例の修飾手順は以下の通りである:TPAを最初にヘキソサミニダーゼ、マンノシダーゼおよびキシロシダーゼで処理してそのグリコシル基を戻し改作する;次いでPEG化N−アセチルグルコサミンを、適当な供与体およびN−アセチルグルコサミントランスフェラーゼを使用してTPAに加える。図38Mでは、哺乳動物細胞で発現したTPA突然変異体(TNK TPA)を改造する。末端シアル酸残基を最初にシアリダーゼを使用して戻し改作する;次いでポリペプチドがPEG化されるようにST3Gal3を使用してPEG化シアル酸を供与体からTNK TPAへ転移する。図38Nでは、哺乳動物系で発現したTNK TPAを最初にシアリダーゼで処理して末端シアル酸残基を戻し改作する。次いでタンパク質を、供与体としてCMP−SA−PEGおよびST3Gal3を使用してPEG化し、そしてさらにシアル酸供与体およびST3Gal3を使用してシアリル化する。図38Oでは、NSO細胞が発現したTNK TPAを最初にシアリダーゼおよびα−ガラクトシダーゼで処理して末端シアル酸およびガラクトース残基を戻し改作する。ついでTNK TPAは、ガラクトース供与体およびガラクトシルトランスフェラーゼを使用してガラクトシル化される。この改造スキームの最終工程は、PEG部分で誘導化されたシアル酸を、シアリルトランスフェラーゼまたはST3Gal3を使用して供与体からTNK TPAへ転移することである。図38QではTNK TPAが哺乳動物系で発現し、そして最初にシアリダーゼで処理されて末端シアル酸残基が戻し改作される。次いでタンパク質はST3Gal3およびPEG化シアル酸の供与体を使用してシアリル化される。次いでタンパク質はシアル酸供与体およびST3Gal3を使用してシアリル化される。図38Rでは哺乳動物系で発現したTNK TPAを、レブリン酸で修飾したシアル酸供与体で適切な末端残基をキャッピングし、反応性ケトンをシアル酸供与体に付加することにより修飾する。ペプチドのグリコシル残基への付加後、ケトンをヒドラジン−またはアミン−PEGのような部分で誘導化する。図38Sでは哺乳動物細胞で発現したTNK TPAを異なる方法を介して修飾する:ポリペプチドは、シアル酸供与体およびα2,8−シアリルトランスフェラーゼを使用したシアル酸の付加により改造する。図38Uでは、昆虫細胞で発現したTNK TPAを、適当な供与体および1以上のGnT I、II、IVおよびVを使用してN−アセチルグルコサミンの付加により改造する。このタンパク質はさらに、PEG化ガラクトースの供与体およびガラクトシルトランスフェラーゼを使用したPEG部分の付加により修飾される。図38Vでは、TNK TPAは酵母で発現する。ポリペプチドを最初にエンドグリカナーゼで処理してそのグリコシル鎖を戻し改作し、そして次いでPEGで誘導化したガラクトース供与体およびガラクトシルトランスフェラーゼを使用してPEG化する。図38WではTNK TPAが哺乳動物系で生産される。ポリペプチドを最初に、ST3Gal3およびリンカーを介して反応性ガラクトースで誘導化したシアル酸の供与体と接触させて、ポリペプチドをリンカーおよびシアル酸残基を介して反応性ガラクトースに結合させる。次いでポリペプチドをガラクトシルトランスフェラーゼおよびCHOで生産された抗−TNF IGキメラと接触させて、これによりキメラはガラクトース残基を介して連結するようになる。
本発明は、ペプチド上のグリカン鎖(1つまたは複数)の部位が天然なペプチドグリカン鎖から改変されたペプチドの使用を意図する。典型的にはN−結合グリカン鎖はアスパラギン残基で1次ペプチド構造に連結され、ここでアスパラギン残基は小胞体(ER)内の膜−結合グリコシルトランスフェラーゼにより認識されるアミノ酸配列内にある。典型的には1次ペプチド構造上の認識部位は配列アスパラギン−X−セリン/トレオニでンあり、ここでXはプロリンおよびアスパラギン酸を除く任意のアミノ酸であることができる。この認識部位は典型的なものであるが、本発明はさらに、N−結合鎖が天然なまたは組換えグリコシルトランスフェラーゼを使用して加えられた他の認識部位にN−結合グリカン鎖を有するペプチドを包含する。
ペプチドにO−結合グリコシル化部位を付加することは、ペプチドの始めの1次アミノ酸配列に比べてペプチドの1次アミノ酸配列が1以上のさらなるO−結合グリコシル化部位を含むように、ペプチドの1次アミノ酸配列を改変させることにより都合よく行う。ペプチドにO−結合グリコシル化部位を加えることは、OH基が接近可能となり、そしてO−結合グリコシル化に利用できるようになるように、ペプチドの配列内に−OH基、好ましくはセリンまたはトレオニン残基を含んでなる1以上のアミノ酸種をペプチドに包含させることにより行うこともできる。ペプチド内のN−結合グリコシル化部位の改変の検討と同様に、ペプチドの1次アミノ酸配列は好ましくはヌクレオチドレベルで改変する。ペプチドをコードするDNA配列中の特別なヌクレオチドは、所望のアミノ酸が配列にコードされるように改変することができる。DNA中の突然変異(1つまたは複数)は好ましくは、上記のホスホロアミダイト法のDNA合成および部位指向的突然変異誘発法の技術のような当該技術分野で既知の方法を使用して作成される。
本発明に有用なペプチドの1次構造は細胞に基づく発現系で最も効率的に生成され得るが、ペプチドが合成的に生成できることも本発明の範囲内である。ペプチドの化学合成は当該技術分野では周知であり、そして限定するわけてはないが段階的な固相合成、および溶液中または固相上でのフラグメント縮合を含む。古典的な段階的固相合成には、所望のペプチド鎖のカルボキシ−末端アミノ酸に対応するアミノ酸を固体支持体に共有結合し、そしてペプチド鎖を、活性化カルボキシル基を有する活性化アミノ酸誘導体の段階的カップリングにより、アミノ末端に向けて延長することを含む。完全に保護された固相結合ペプチド鎖の集成体が完成した後、ペプチド−固相の共有結合を適当な化学により開裂し、そして保護基を除去して生成物であるペプチドを得る。R.Merrifield、固相ペプチド合成:テトラペプチドの合成(Solid Phase Peptide Synthesis:The Synthesis of Tetrapeptide)、J.Am.Chem.Soc.,85:2149−2154(1963)を参照にされたい。より長いペプチド鎖ほど、高純度の十分に規定された生成物を得ることが難しくなる。複雑な混合物の生産のために、段階的な固相合成法にはサイズに限界がある。一般に100の連続するアミノ酸残基またはそれ以上の十分に規定されたペプチドが、段階的な固相合成を介して日常的に調製されることはない。
当業者はペプチドがN−結合および/またはO−結合グリカンを付加される上に、翻訳後修飾も受け得ると考えるだろう。グリコシル化以外の翻訳後修飾を有するペプチドは、ペプチドの所望する生物学的活性または機能を保持または改善するかぎり、本発明でペプチドとして使用できることを意図している。そのような翻訳後修飾は通常、インビボで起こる天然な修飾、またはペプチドにインビトロで行われる操作された修飾であることができる。意図する既知の修飾には限定するわけではないがアセチル化、アシル化、ADP−リボシル化、アミド化、フラビンの共有結合、ヘム部分の共有結合、ヌクレオチドまたはヌクレオチド誘導体の共有結合、脂質または脂質誘導体の共有結合、ホスファチジルイノシトールの共有結合、架橋結合、環化、ジスルフィド結合の形成、脱メチル化、共有架橋結合の形成、システインの形成、ピログルタメートの形成、ホルミル化、ガンマカルボキシル化、グリコシル化、GPIアンカーの形成、ヒドロキシル化、ヨード化、メチル化、ミリストイル化、酸化、タンパク質分解プロセッシング、リン酸化、プレニル化、ラセミ化、セレノイル化、硫黄化、アルギニレーションのようなトランスファー−RNAが媒介するアミノ酸のペプチドへの付加、およびユビキチン化を含む。多くのこれらの修飾を行うために使用され得る酵素は当該技術分野では周知であり、そして中でもベーリンガーマンハイム(Boehringer Mannheim)(インディアナポリス、イリノイ州)およびシグマケミカル社(セントルイス、モンタナ州)のような会社から市販されている。
et al.(Ann.N.Y.Acad.Sci.663:48−62(1992))によるような多くの詳細な総説が入手可能である。
本発明に有用なペプチドは融合ペプチドを含んでなることができる。融合ペプチドは2つのペプチドの生物学的および/または機能的特性が1つのペプチド分子内に組み合わされることが望まれる場合、特に有利である。そのような融合ペプチドは、治療用および工業的応用の新規かつ有用な分子を作成するために、天然には見いだされない生物学的活性および機能の組み合わせを提示することができる。問題の生物学的活性には限定するわけではないが酵素活性、レセプターおよび/またはリガンド活性、免疫原性モチーフおよび構造的ドメインを含む。
本発明で使用するペプチドは天然なペプチドの変異体でよく、ここで天然なペプチドのフラグメントが完全長の天然なペプチドの代わりに使用される。さらにプレ−プロ−およびプレ−ペプチドを企図する。変異体ペプチドは天然なペプチドよりもサイズが小さくてもよく、そしてより大きなペプチドの1以上のドメインを含んでなってもよい。特別なペプチドドメインの選択は、ペプチド中の特定のドメインの生物学的活性が望まれるが、ペプチドの他のドメインの生物学的活性は望まない時に有利となり得る。またペプチドの所望する治療的効果を強化することができるペプチドの短縮化および内部削除も含む。そのような任意な形のペプチドは、ペプチドの所望する活性が保存される限り、本発明に有用となることを意図している。
本発明のペプチドは天然なペプチドの1次配列に由来するものでよく、または当業者に知られている多くの手段を使用して工作することができる。そのように工作したペプチドは、天然なペプチドに比べて強化された、または新規な特性から設計され、かつ/または選択されることができる。例えばペプチドは、上昇した酵素反応速度、上昇したまたは低下した基質もしくはリガンドへの結合親和性、上昇または低下したレセプターへの結合親和性、基質、リガンド、レセプターまたは他の結合パートナーに対する改変された特異性、上昇または低下したインビトロおよび/またはインビボの安定性、あるいは動物において上昇または低下した免疫原性を有するように工作することができる。
1.合理的な設計の突然変異
本発明の方法に有用なペプチドは、所望する生物学的活性または機能を強化し、ペプチドの望ましくない特性を縮小し、かつ/またはペプチドに新規活性または機能を加えるために突然変異させることができる。「合理的なペプチド設計」はそのような改変されたペプチドを生成するために使用することができる。いったんペプチドのアミノ酸配列および構造を知り、そして所望する突然変異を計画すれば、突然変異は突然変異させることを望むアミノ酸残基をコードする対応する核酸コドンに対して最も都合良く作成することができる。当業者は一般的(universal)な遺伝子コード、および選択した発現系でのコドン優先性の知識に基づき、どのように核酸配列を改変すべきかを容易に決定することができる。コドン中の突然変異は翻訳中にペプチドに重合化されるアミノ酸残基を変化させるために作ることができる。あるいはコドンは対応するコードされるアミノ酸残基は同じであるが、コドンの選択が所望のペプチド発現系により適するように突然変異させることができる。例えばシス−残基を別のアミノ酸に置き換えて成熟ペプチドからジスルフィド結合を除去してもよく、触媒ドメインを突然変異させて生物学的活性を改変してもよく、そして一般にペプチドのアイソフォームを工作することができる。そのような突然変異はとりわけ点突然変異、削除、挿入および短縮であることができる。
al.,1999,Peptide Eng.12:635−638)への導入により向上した。さらに疎水的接触の導入が3−イソプロピルマレートデヒドロゲナーゼ(Akanuma et al.,1999,Eur.J.Biochem.260:499−504)およびシュードモナス種(Pseudomonas)種から得たギ酸デヒドロゲナーゼ(Rojkova et al.,1999,FEBS Lett.445:183−188)を安定化した。これら突然変異の安定化効果の背後にあるメカニズムは、一般に多くのペプチドに応用することができる。これらのおよび同様の突然変異は、本発明の方法で改造されるペプチドに関して有用になると予想する。
本発明の方法に有用な新規ペプチドは、核酸のコード配列にランダムな突然変異を導入する技術を使用して生成することができる。次いで核酸は望ましい発現系で発現され、そして生成したペプチドを問題の特性について評価する。ランダムな突然変異をDNA配列に導入するための技術は当該技術分野では周知であり、そしてPCR突然変異誘発法、飽和(saturation)突然変異誘発法および縮重オリゴヌクレオチド法を含む。Sambrook and Russell(2001、モレキュラークローニング、ア ラボラトリーアプローチ、コールドスプリングハーバー出版、コールドスプリングハーバー、ニューヨーク州)およびAusubel et al.(2002、分子生物学の現在の手法(Current Protocols in Molecular Biology)、ジョン ウィリー&サンズ、ニューヨーク州)を参照にされたい。
本発明の方法で有用なペプチドは、「方向付けされる進化」の技術を使用して生成することもできる。ペプチドの構造の知識を必要とする部位指向的突然変異誘発技術とは対照的に、今ではペプチドの構造的特徴の知識無しで改善された特性を持つペプチドが得られる突然変異のライブラリーを作成する方法が存在する。これらの方法は一般に「方向付けされる進化」の技術として知られ、そして問題のペプチドをコードする核酸配列を循環的(recursive round)な突然変異、スクリーニングそして増幅に供する点でこれまでのランダムな変異誘導手順とは異なる。
本発明の方法に有用な新規ペプチドは、遺伝子−シャフリング、モチーフ−シャフリング、エキソン−シャフリングおよび/またはコドン−シャフリング(集合的に「DNAシャフリング」と呼ぶ)を使用して生成することもできる。DNAシャフリング技術は本発明に有用なペプチドの活性をモジュレートするために使用することができ、そして改変した活性を有するペプチドを生成するために使用することができる。一般に米国特許第5,605,793号;同第5,811,238号;同第5,830,721号;同第5,834,252号;および同第5,837,458号明細書、およびStemmer et
al.(1994,Nature 370(6488):389−391);Crameri et al.(1998,Nature 391(6664):288−291);Zhang et al.(1997,Proc.Natl.Acad.Sci.USA 94(9):4504−4509);Stemmer et al.(1994,Proc.Natl.Acad.Sci USA 91(22):10747−10751),Patten et al.(1997,Curr.Opinion Biotechnol.8:724−33);Harayama,(1998,Trends Biotechnol.16(2):76−82);Hansson,et al.,(1999,J.Mol.Biol.287:265−76);and Lorenzo and Blasco(1998,Biotec hniques 24(2):308−13)
を参照にされたい(これら各々は、引用により本明細書に編入する)。
「進化」の各循環後、突然変異した遺伝子により発現された所望のペプチドを問題の特性についてスクリーニングする。次いで「候補」遺伝子を増幅し、そして次回のDNAシャフリング用にプールする。使用するスクリーニング手順は、「進化」したペプチドおよび問題の特性に高度に依存する。中でもとりわけペプチド安定性、生物学的活性、抗原性のような特性は、当該技術分野で周知な手順を使用して選択することができる。本発明の方法に有用な好適ペプチドの生物学的活性に関する個々のアッセイは、本明細書のいたるところに記載する。
当業者には突然変異および選択の上記技術は、本発明の方法に有用な最高に可能なペプチド分子を生成するために、互いに、そしてさらなる手順と組み合わせることができると考えるだろう。すなわち本発明はペプチドの生成に関して任意の1つの方法に限定されることはなく、そして本明細書に記載するありとあらゆる方法論を包含すると解釈すべきである。例えば点突然変異を核酸配列に導入する手順を最初に行い、続いて循環的DNAシャフリング、選択および増幅を行うことができる。初期の点突然変異の導入を使用して、それを欠く遺伝子群に多様性を導入し、次回のDNAのシャフリングおよびスクリーニングにより有利な点突然変異を選択し、そして組み合わせることができる。
A.グリコシダーゼ
グリコシダーゼは受容体分子として水を使用し、そしてそのままで典型的なグリコシド−加水分解酵素であるグリコシルトランスフェラーゼである。グリコシダーゼは、反応混合物の熱力学または動力学を制御することによりインビトロでグリコシド結合を形成するために使用することができる。変更された反応条件でも、グリコシダーゼ反応はうまく作用することが難しく、そしてグリコシダーゼは可逆的なトランスグリコシラーゼ反応および競合する加水分解反応の結果、低い合成収量を与える傾向がある。
グリコシルトランスフェラーゼは活性化糖(供与体NDP−糖)のタンパク質、糖ペプチド、脂質または糖脂質への、あるいは成長しているオリゴ糖の非還元末端への付加を段階的様式で触媒する。N−結合糖ペプチドは、トランスフェラーゼおよび脂質−結合オリゴ糖供与体Dol−PP−NAG2Glc3Man9をenブロック転移で、続いてコアの改作を介して合成される。この場合、「コア」サッカリドの性質は後の結合とは幾分異なる。大変多数のグリコシルトランスフェラーゼが当該技術分野で知られている。
of glycosyltransferases and related genes)、スプリンガー、東京、を参照にされたい。
幾つかの態様では、本発明の方法で使用するグリコシルトランスフェラーゼは、フコシルトランスフェラーゼである。フコシルトランスフェラーゼは当業者に知られている。フコシルトランスフェラーゼの例には、L−フコースをGDP−フコースから受容体の糖のヒドロキシ部位へ転移する酵素を含む。非−ヌクレオチド糖から受容体へ転移するフコシルトランスフェラーゼも本発明で使用される。
別の態様群では、グリコシルトランスフェラーゼはガラクトシルトランスフェラーゼである。ガラクトシルトランスフェラーゼの例にはα(1,3)ガラクトシルトランスフェラーゼ(E.C.No.2.4.1.151、Dabkowski et al.,Transplant Proc.25:2921(1993)およびYamamoto et al.,Nature345:229−233(1990)を参照にされたい)、ウシ(GenBank j04989、Joziasse et al.,J.Biol.Chem.264:14290−14297(1989))、マウス(GenBank m26925;Larsen et al.,Proc.Nat’l.Acad.Sci.USA 86:8227−8231(1989)、ブタ(GenBank L36152;Strahan et al.,Immunogenetics 41:101−105(1995)を含む。別の適当なα1,3ガラクトシルトランスフェラーゼは、血液のB抗原群の合成に関与するものである(E.C.2.4.1.37、Yamamoto
et al.,J.Biol.Chem.265:1146−1151(1990)(ヒト))。
シアリルトランスフェラーゼは、組換え細胞および本発明の反応混合物に有用な別の種類のグリコシルトランスフェラーゼである。本発明の使用に適するシアリルトランスフェラーゼの例には、ST3GalIII(例えばラットまたはヒトST3GalIII)、ST3GalIV、ST3GalI、ST6GalI、ST3GalV、ST6GalII、ST6GalNAcI、ST6GalNAcIIおよびST6GalNAcIIIを含む(本明細書で使用するシアリルトランスフェラーゼの命名法は、Tsuji et al.,Glycobiology,6:v−xiv(1996)に記載されている)。α(2,3)シアリルトランスフェラーゼ(EC2.4.99.6)と呼ぶα(2,3)シアリルトランスフェラーゼの例は、シアル酸をGalβ1→3Glc二糖またはグリコシドの非還元末端のGalへ転移する。Van den Eijnden et al.,J.Biol.Chem.256:3159(1981)、Weinstein et
al.,J.Biol.Chem.257:13845(1982)およびWen et al.,J.Biol.Chem.267:21011(1992)を参照にされたい。別の例のα2,3シアリルトランスフェラーゼ(EC2.4.99.4)は、シアル酸を二糖またはグリコシドの非還元末端のGalへ転移する。Rearick et al.,J.Biol.Chem.254:4444(1979)およびGillespie et al.,J.Biol.Chem.267:21004(1992)を参照にされたい。さらなる酵素の例にはGal−β−1,4−GlcNAcα−2,6−シアリルトランスフェラーゼを含む(Kurosawa et al.Eur.J.Biochem.219:375−381(1994)を参照にされたい)。
当業者は他のグリコシルトランスフェラーゼを、シアリルトランスフェラーゼについて詳細に記載したように同様なトランスフェラーゼサイクルで置き換え得ることができると理解するだろう。特にグリコシルトランスフェラーゼは例えばグルコシルトランスフェラーゼ、例えばAlg8(Stagljov et al.,Proc.Natl.Acad.Sci.USA 91:5977(1994))またはAlg5(Heesen et al.,Eur.J.Biochem.224:71(1994))であることもできる。
et al.、Critical Reviews in Microbiology23(3):139−180(1996))。そのような酵素には限定するわけではないが、β1,6ガラクトシルトランスフェラーゼおよびβ1,3ガラクトシルトランスフェラーゼを含む大腸菌(E.coli)およびネズミチフス菌(Salmonella typhimurium)のような種のrfaオペロンのタンパク質(例えばEMBL寄託番号M80599およびM86935(大腸菌(E.coli));EMBL寄託番号S56361(ネズミチフス菌(S.typhimurium)を参照にされたい)、グルコシルトランスフェラーゼ(スイス−プロット寄託番号P25740(大腸菌(E.coli))、β1,2−グルコシルトランスフェラーゼ(rfaJ)(スイス−プロット寄託番号P27129(大腸菌(E.coli))およびスイス−プロット寄託番号P19817(ネズミチフス菌(S.typhimurium))、およびβ1,2−N−アセチルグルコサミニルトランスフェラーゼ(rfaK)(EMBL寄託番号U00039(大腸菌(E.coli))を含む。アミノ酸配列が既知の他のグリコシルトランスフェラーゼには、肺炎桿菌(Klebsiella pneumoniae)、大腸菌(E.coli)、ネズミチフス菌(Salmonella typhimurium)、サルモネラ エンテリカ(Salmonella enterica)、腸炎エルシニア(Yersinia enterocolitica)、マイコバクテリウム レプロサム(Mycobacterium leprosum)のような微生物で特徴付けられたrfaBのようなオペロン、および緑膿菌(Pseudomonas aeruginosa)のrh1オペロンにコードされるものを含む。
本発明は、例えばヘパリン、硫酸ヘパラン、カラゲニン(carragenen)のような硫黄化多糖および関連化合物を含む硫黄化した分子を含むペプチドの生産法を提供する。適当なスルホトランスフェラーゼには例えば、コンドロイチン−6−スルホトランスフェラーゼ(Fukuta et al.,J.Biol.Chem.270:18575−18580(1995)により記載されたニワトリcDNA;GenBank寄託番号D49915)、グルコサミノグリカンN−アセチルグルコサミンN−デアセチラーゼ/N−スルホトランスフェラーゼ1(Dixon et al.,Genomics 26:239−241(1995);UL18918)、およびグルコサミノグリカンN−アセチルグルコサミンN−デアセチラーゼ/N−スルホトランスフェラーゼ2(Orellana et al.,J.Biol.Chem.269:2270−2276(1994)およびEriksson et al.,J.Biol.Chem.269:10438−10443(1994)に記載されたマウスcDNA;GenBank寄託番号U2304に記載されたヒトcDNA)を含む。
別の態様では、本発明の方法で採用する酵素は細胞−結合グリコシルトランスフェラーゼである。多くの可溶性グリコシルトランスフェラーゼが知られているが(例えば米国特許第5,032,519号明細書を参照にされたい)、グリコシルトランスフェラーゼは細胞に付随する時は一般に膜に結合している状態である。これまでに研究された多くの膜に結合酵素は内在性のタンパク質であると考えられ;すなわちそれらは超音波により膜から放出されず、そして可溶化に界面活性剤を要する。表面のグリコシルトランスフェラーゼは脊椎動物および無脊椎動物の細胞上の表面で同定され、そしてこれら表面のトランスフェラーゼが生理学的条件下で触媒活性を維持していることも認識された。しかし細胞表面のグリコシルトランスフェラーゼのより認識されている機能は細胞間の認識である(Roth,1990,超細胞現象に対する分子的取り組み(Molecular Approaches to Supracellular Phenomena)。
別の例示態様では、本発明の方法は所望の糖ペプチド結合物の合成に関与する1より多くの酵素活性を有する融合ペプチドを利用する。融合ペプチドは例えば、アクセサリー酵素の触媒的に活性なドメインに連結されたグリコシルトランスフェラーゼの触媒的に活性なドメインからなることができる。アクセサリー酵素の触媒ドメインは、例えばグリコシルトランスフェラーゼの供与体であるヌクレオチド糖の形成における工程を触媒するか、あるいはグリコシルトランスフェラーゼサイクルに関与する反応を触媒することができる。例えばグリコシルトランスフェラーゼをコードするポリヌクレオチドはフレイム内で、ヌクレオチド糖合成に関与する酵素をコードするポリヌクレオチドに結合され得る。生成した融合ペプチドは次いでヌクレオチド糖合成を触媒するだけでなく、アクセプター分子への糖部分の転移も触媒する。融合ペプチドは1つの発現可能なヌクレオチド配列に連結された2以上のサイクル酵素であることができる。別の態様では融合ペプチドは2以上のグリコシルトランスフェラーゼの触媒的に活性なドメインを含む。例えば米国特許5,641,668第号明細書を参照にされたい。本発明の修飾された糖ペプチドは、様々な適切な融合ペプチドを利用して容易に設計し、そして製造することができる(例えば国際公開第99/31224号パンフレットとして1999年6月24日に公開されたPCT特許出願PCT/CA98/01180を参照にされたい)。
細胞に結合した酵素に加えて、本発明は固体および/または可溶性支持体上に固定化された酵素の使用も提供する。例示態様では、本発明の方法に従い完全なグリコシルリンカーを介してPEGに結合されたグリコシルトランスフェラーゼを提供する。PEG−リンカー−酵素結合物は、場合により固体支持体に結合される。本発明の方法において固体に支持された酵素の使用は、反応混合物の処理および反応生成物の精製を簡単にし、そしてまた酵素の容易な回収を可能とする。このグリコシルトランスフェラーゼ結合物は本発明の方法に利用する。他の酵素および支持体の組み合わせも当業者には明らかであろう。
本発明の方法で使用するグリコシルトランスフェラーゼ、シアリルトランスフェラーゼ、スルホトランスフェラーゼおよび任意の他の酵素の新規形態は、これまでに記載した任意の方法ならびに当該技術分野で周知の他の方法を使用して作成することができる。特に興味深いのは、改変された受容体特異性および/または供与体特異性を持つトランスフェラーゼである。また興味深いのは、中でもより高い転換速度およびより高い安定性を持つ酵素である。
Biology 11:593−600)。グリコシルトランスフェラーゼはそれらの構造に基づき2つのファミリーに分類される:GT−AおよびGT−B。GT−Aファミリーのグリコシルトランスフェラーゼは2つの似ていないドメインを含んでなり、1つはヌクレオチド結合に関与し、そしてもう1つは受容体結合に関与する。このように1つの遺伝子に由来するドメインをコードするDNA配列を、第2の遺伝子に由来するドメインとフレイム内で都合よく融合して、新たな受容体/供与体特異性を持つタンパク質をコードする新たな遺伝子を作成することができる。そのようなドメインの交換にはさらに炭水化物モジュールおよび他のアクセサリードメインを含むことができる。
A.糖ペプチドを生産するための細胞
グリコシルトランスフェラーゼの作用はペプチドのグリコシル化に重要であり、すなわち所定の細胞型での1組のグリコシルトランスフェラーゼの発現における差異は、その細胞で生産される所定のペプチドのグリコシル化パターンに影響を及ぼす。宿主細胞に依存的なペプチドのグリコシル化の総説に関しては、Kabata and Takasaki、「細胞表面の炭水化物の構造および生合成(Structure and Biosynthesis of Cell Surface Carbohydrates)」、細胞表面の炭水化物および細胞生育(Cell Surface Carbohydrates and Cell Development)で、1991、第1〜24頁、編集.Minoru Fukuda、CRC出版、ボカ ラトン、フロリダ州を参照にされたい。
酵母で生産されたペプチドはグリコシル化されており、そしてそこに存在するグリカン構造は主に高マンノース構造である。N−グリカンの場合では、酵母で生産されたグリカン構造は、9以上ものマンノース残基を含むことができ、この残基はそれに結合したさらなる糖を含んでも、含まなくてもよい。酵母細胞により生産されるペプチド上のこの型のグリカンの例を、図5の左側に示す。マンノース残基の番号およびそれに結合したさらなる糖の型および複雑さに関係なく、酵母細胞で生産されたペプチドの成分としてN−グリカンは、図5に示すトリマンノシルコア構造を含んでなる。酵母細胞で生産されたペプチド上のグリカン構造が高マンノース構造である時、当業者が利用可能なマンノシダーゼ酵素を使用してグリカンのトリマンノシルコアを含んでなる残基を除き、分子からすべてのマンノース残基をインビトロで除去することは単純なことであり、これにより結合した基本的なトリマンノシルコア構造を有するペプチドを生成する。ここで今、当該技術分野で利用可能な技術を使用し、そして本開示から着手すれば、インビトロで酵素的にさらなる糖部分を基本的なトリマンノシルコア構造に加え、所望のグリカン構造をトリマンノシルコアに結合して有するペプチドを生成することは簡単である。同様に酵母細胞により生産されたペプチドが、結合した他の複雑な糖に加えて高マンノース構造を含んでなる時、余分なマンノース残基を含めさらなる糖のすべてを酵素的に開裂して基本的なトリマンノシルコア構造に到達することは簡単な事柄である。いったん基本的なトリマンノシルコアが生成されれば、所望のグリコシル化を有するペプチドの生成は本明細書に提供する指針に従い可能となる。
サッカロミセス(Saccharomyces)では、ペプチドを生産するために使用する適当な酵母ベクターにはYRp7(Struhl et al.,Proc.Natl.Acad.Sci.USA76:1035−1039,1978)、YEp13(Broach et al.,Gene 8:121−133,1979)、POTベクター(Kawasaki et al.,米国特許第4,931,373号明細書、これは引用により本明細書に編入する)、pJDB249およびpJDB219(Beggs,Nature 275:104−108,1978)およびそれらの誘導体を含む。酵母での使用に好適なプロモーターは、酵母の解糖遺伝子の発現に関するプロモーター(Hitzeman et al.,J.Biol.Chem.255:12073−12080,1980;Alber and Kawasaki,J.Mol.Appl.Genet.1:419−434,1982;Kawasaki,米国特許第4,599,311号明細書)、またはアルコールデヒドロゲナーゼ遺伝子(Young et al.、化学品のための微生物の遺伝子工学(Genetic Engineering of Microorganisms for Chemicals)、Hollaender
et al.,(編集)、第355頁、プレナム出版、ニューヨーク、1982;Ammerer,Meth.Enzymol.101:192−201,1983)、およびADH2−4cプロモーター(Russell et al.,Nature 304:652−654,1983;Irani and Kilgore,米国特許出願第07/784,653号明細書、カナダ国特許第1,304,020号明細書および欧州特許第284 044号明細書、これらは引用により本明細書に編入する)を含む。発現単位は転写ターミネーターも含むことができる。好適な転写ターミネーターはTPI1ターミネーター(Alber and Kawasaki,同上)である。
組換えペプチドの生産のための宿主としてピヒア メタノリカ(Pichia methanolica)の使用は、PCT出願、国際公開第97/17450号、同第97/17451号、同第98/02536号および同第98/02565号パンフレットに開示されている。P.メタノリカ(methanolica)の形質転換に使用するDNA分子は一般に、形質転換前に好ましくは直線化される二本鎖の環状プラスミドとして調製される。P.メタノリカ(methanolica)でのペプチド生産には、プラスミド中のプロモーターおよびターミネーターは、P.メタノリカ(methanolica)のアルコール利用遺伝子(AUG1またはAUG2)のようなP.メタノリカ(methanolica)遺伝子のものであることが好ましい。他の有用なプロモーターにはジヒドロキシアセトンシンターゼ(DHAS)、ギ酸デヒドロゲナーゼ(FMD)およびカタラーゼ(CAT)遺伝子のもの、ならびに米国特許第5,252,726号明細書に開示されているものを含む。DNAの宿主染色体への組み込みを容易にするために、宿主DNA配列により両末端を挟まれたプラスミドの全発現セグメントを有することが好ましい。ピヒア メタノリカ(Pichia methanolica)での使用に好適な選択マーカーは、ホスホリボシル−5−アミノイミダゾールカルボキシラーゼ(AIRC;EC4.1.1.21)をコードするP.メタノリカ(methanolica)ADE2遺伝子であり、これはade2宿主細胞がアデニンの不存在下で成長することを可能とする。メタノールの使用を最少にすることが望まれる大規模な工業的方法には、両方のメタノール利用遺伝子(AUG1およびAUG2)が削除されている宿主細胞が好ましい。分泌するペプチドの生産には、液胞プロテアーゼ遺伝子(PEP4およびPRB1)を欠く宿主細胞が好適である。目的のペプチドをコードするDNAを含むプラスミドを、P.メタノリカ(methanolica)細胞に導入することを容易にするために、電気穿孔を使用する。指数的に減少する、2.5〜4.5kV/cm、好ましくは約3.75kV/cmの場の強さを有するパルス電場、および1〜40ミリ秒、最も好ましくは約20ミリ秒の時間定数(t)を使用して、電気穿孔によりP.メタノリカ(methanolica)細胞を形質転換することが好ましい。抗体フラグメントの大規模生産のためのピヒア パストリス(Pichia pastoris)の使用に関する総説は、Fischer et al.(1999,Biotechnol.Appl Biochem.30(Pt2):117−120)を参照にされたい。
アスペルギルス(Aspergillus)種でペプチドを発現させる方法は当該技術分野では周知であり、それらには限定するわけではないが引用により全部、本明細書に編入するCarrez et al.,1990,Gene 94:147−154;Contreras,1991,Bio/Technology 9:378−381;Yelton et al.,1984,Proc.Natl.Acad.Sci.USA 81:1470−1474;Tilburn et al.,1983,Gene 26:205−221;Kelly and.Hynes,1985,EMBO J.4:475−479;Ballance et al.,1983,Biochem.Biophys.Res.Comm.112:284−289;Buxton et al.,1985,Gene 37:207−214,and U.S.Pat.No.4,935,349に記載されているものを含む。アスペルギルス(Aspergillus)で有用なプロモーターの例は、米国特許第5,252,726号明細書に見いだされる。ペプチドの発現に有用なアスペルギルス(Aspergillus)の株は、米国特許第4,935,349号明細書に見いだされる。外因性ペプチドの工業的生産は、アスペルギルス ニガー(Aspergillus niger)およびアスペルギルス オリーゼ(Aspergillus oryzae)についてノボエンザイム(Novoenzymes)から入手可能である。
トリコデルマ(Trichoderma)は所望のペプチドを発現させるために、他の種の組換え宿主細胞に優るある一定の利点を有する。この微生物は大量に成長させることが容易であり、そしてグリコシル化する能力を有し、そして組換え哺乳動物ペプチドを培地に高い収量で効率よく分泌し、ペプチドの単離を比較的容易とする。さらに発現したペプチド上のグリコシル化パターンは、他の系で発現されるものよりもヒトペプチドに類似している。しかしこれらの細胞から発現したペプチド上のグリカン構造にも差異が存在する。例えば末端シアル酸残基は、グリカン構造の末端のこれらの部分の存在が哺乳動物の血流からペプチドの排除を遅らせるので、哺乳動物系におけるペプチドの治療的機能に対して重要である。シアリル化分子が生物学的半減期を上昇させる背後にあるメカニズムは、レクチンによるそれらの認識の低下にあると考えられる(Drickamer,1988,J.Biol.Chem.263:9557−9560)。しかし一般に真菌細胞は末端シアル酸残基をペプチド上のグリカンに加えず、故に真菌細胞で合成されたペプチドはシアル酸が除かれている。本発明に従い、この欠如は本明細書のいたるところに詳細に記載する本発明のインビトログリカン改造方法を使用して補修する(remedied)ことができる。
クルイベロミセス(Kluyveromyces)属に属する酵母は、組換えペプチドの生産に宿主微生物として使用されてきた。この属の酵母により生産されるペプチドは特にキモシン(欧州特許第96430号明細書)、タウマチン(欧州特許第96910号明細書)、アルブミン、インターロイキン−1β、TPA、TPA、TIMP(欧州特許第361991号明細書)および治療的機能を有するアルブミン誘導体(欧州特許第413622号明細書)である。クルイベロミセス(Kluyveromyces)属の中で特に興味深い種にはK.ラクティス(lactis)を含む。
真菌の属であるクリソポリウム(Chrysoporium)は最近、外来組換えペプチドの発現に使用された。外来ペプチドを発現するために当業者がクリソポリウム(Chrysoporium)を使用することができる手順の記載は、国際公開第00/20555号明細書に見いだされる(これは引用により全部、本明細書に編入する)。発現系に特に適する種は限定するわけではないが、C.ボトリオイデス(botryoides)、C.カルミカエリー(carmichaelii)、C.クラッシツニカタム(crassitunicatum)、C.ヨーローパ(europae)、C.エボルセアンヌイ(evolceannui)、F.ファスチジウム(fastidium)、C.フィリホルメ(filiforme)、C.ゲロギエ(gerogiae)、C.グロビフェラム(globiferum)、C.グロビフェラム 変種 アーチクラタム(globiferum var.articulatum)、C.グロビフェラム 変種 ニベウム(globiferum var.niveum)、C.ヒルンド(hirundo)、C.ヒスパニカム(hispanicum)、C.ホルミー(holmii)、C.インジカム(indicum)、C.イノプス(inops)、C.ケラチノフィラム(keratinophilum)、C.クレイセリー(kreiselii)、C.クズロビアナム(kuzurovianum)、C.リグノラム(lignorum)、C.ロバタム(lobatum)、C.ラックノヴェンセ(lucknowense)、C.ラックノヴェンセ(lucknowense)Garg27K、C.メディウム(medium)、C.メディウム 変種 スピッセスセンス(medium var.spissescens)、C.メフィチカム(mephiticum)、C.メルダリウム(merdarium)、C.メルダリウム 変種 ロゼウム(merdarium var.roseum)、C.マイナー(minor)、C.パンニコーラ(pannicola)、C.パルバム(parvum)、C.パルバム 変種 クレセンス(parvum var.crescens)、C.ピロサム(pilosum)、C.ペオドメルデリウム(peodomerderium)、C.ピリホルミス(pyriformis)、C.クィーンズランディカム(queenslandicum)、C.シグレリ(sigleri)、C.スルフレウム(sulfureum)、C.シンクロナム(synchronum)、C.トロピカム(tropicum)、C.ウンダラタム(undulatum)、C.バレレナレンス(vallenarense)、C.ベスペルチリウム(vespertilium)およびC.ゾナタム(zonatum)を含む。
シュワニオマイセス(Schwanniomyces)を形質転換する方法は欧州特許第394538号明細書に開示されている。アクレモニウム クリソゲナム(Acremonium chrysogenum)を形質転換する方法は、米国特許第5,162,228号明細書に開示されている。ニューロスポーラ(Neurospora)を形質転換する方法は、米国特許第4,486,533号明細書に開示されている。またシゾサッカロミセス ポンベ(Schizosaccharomyces pombe)に特異的な発現系も知られている(欧州特許第385391号明細書)。分裂酵母シゾサッカロミセス ポンベ(Schizosaccharomyces pombe)でペプチドを発現させるための一般的方法は、Giga−Hama and Kumagai(1997、分裂酵母:シゾサッカロミセス ポンベでの外来遺伝子発現(Foreign gene expression in fission yeast:Schizosaccharomyces pombe)、スプリンガー、ベルリン)に見いだすことができる。
上記のように、哺乳動物細胞は典型的にはトリマンノシルコアに結合したさらなる糖の数および配列について変動するN−グリカン構造のヘテロロガスな混合物を生産する。典型的には哺乳動物細胞は図4右側に示すような複雑なグリカン構造を有するペプチドを生産する。本発明の方法を使用して、最初に1次グリカン構造を同定し、次にグリカン構造を改造するためにはどの糖が除去されるなけれければならないかを決定することにより、哺乳動物細胞で生産されたペプチドをインビトロで改造して、所望のグリコシル化を有するペプチドを生成することができる。本明細書で検討するように、除去される糖によりどの開裂酵素が使用されるかが決定され、すなわ改造方法の正確な工程は最初の基質として使用される1次グリカン構造に依存して変動するだろう。哺乳動物細胞で一般に生産されるグリカン構造を改造するための実例スキームは図3に示す。哺乳動物細胞でのN−グリカン生合成経路は十分に特徴付けられた(Moremen,1994,Glycobiology4:113−125を参照にされたい)。グリカン合成に必要な酵素の多くが同定され、そしてチャイニーズハムスター卵巣(CHO)細胞系Lec23(アルファ−グルコシダーゼI欠損)およびLec18(新規GlcNAc−TVIII)を含め、この酵素的経路を欠く突然変異体細胞系が単離された。これらの突然変異体細胞により生産されるペプチドのグリコシル化パターンは、正常CHO細胞に対して改変されている。本明細書で検討するように、これらおよび他の突然変異体細胞におけるグリコシル化の欠損は、複雑なグリカン構造を欠くペプチドを生産する目的に活かすことができる。例えばLec23細胞により生産されたペプチドはシアル酸残基を欠き、すなわち基本的なトリマンノシルコアまたはMan3GlcNac4にグリカン構造を減少させるために必要な酵素的操作が少ない。このようにこれらの細胞で生産されるペプチドはグリカン改造用の好適な基質として役立つ。当業者は既知の方法、例えばStanley et al.,1990,Somatic Cell Mol.Genet.,16:211−223に記載されている方法に基づき、他のグリコシル化−欠損細胞系を単離し、そして同定することができる。同定された、または未だ同定されていないグリコシル化−欠損細胞系の使用は、本明細書に記載する改造方法のための好適なペプチド基質を生成する目的で本発明に含まれる。
and van der Eb,1973;Corsaro and Pearson,1981,Somatic Cell Genetics 7:603),ジエチルアミノエチル(DEAE)−デキストラン トランスフェクション(Fujita et al.,1986;Lopata et al.,1984;Selden et al.,1986,),電気窄孔(Neumann et al.,1982,;Potter,1988,;Potter et al.,1984,;Wong and Neuman,1982),カチオン性脂質試薬のトランスフェクション(Elroy−Stein and Moss,1990;Feigner et al.,1987;Rose et al.,1991;Whitt et al.,1990;Hawley−Nelson et al.,1993,Focus 15:73;Ciccarone et al.,1993,Focus 15:80),レトロウイルス(Cepko et al.,1984;Miller and Baltimore,1986;Pear et al.,1993;Austin and Cepko,1990;Bodine et al.,1991;Fekete and Cepko,1993;Lemischka et al.,1986;Turner et al.,1990;Williams et al.,1984;Miller and Rosman,1989,BioTechniques 7:980−90;Wang and Finer ,1996,Nature Med.2:714−6),ポリブレン(Chaney et al,1986;Kawai and Nishizawa,1984),マイクロインジェクション(Capecchi,1980),およびプロトプラスト融合(Rassoulzadegan et al.,1982;Sandri−Goldin et
al.,1981;Schaffer,1980)を含む。一般に形質転換技術に関しては、Sambrook et al.(2001、モレキュラークローニング:ア ラボラトリーマニュアル、コールドスプリングハーバーラボラトリー、ニューヨーク)およびAusubel et al.(2002、分子生物学の現在のプロトコール、ジョン
ウィリー&サンズ、ニューヨーク)を参照にされたい。
昆虫細胞、そして特に培養した昆虫細胞は、シアリル化されることが稀で、そして通常は結合したさらなるフコース残基を持つかまたは持たないかもしれないマンノース残基を含んでなるN−結合グリカン構造を有するペプチドを発現する。培養した昆虫細胞で生産されるペプチド上に存在するグリカン構造の型の例は、図7およびそれらのマンノースグリカンに示す。
Manual)、オックスフォード大学出版)、およびKing and Possee(1992、バキュロウイルス発現系:ア ラボラトリーガイド(Baculovirus Expression System:A Laboratory Guide)、チャップマン&ホール(Chapman&Hall))を含む。さらにLucklow(1993,Curr.Opin.Biotechnol.4:564−572)およびMiller(1993,Curr.Opin.Genet.Dev.3:97−101)のような報告もある。
ペプチド生産体としての植物細胞は、別口の話題を提示する。植物により生産されるN−結合グリカンはトリマンノシルコア構造を含んでなり、この五糖骨格は図6に示すように数個の異なるさらなる糖を含んでなることができる。例えば例として、トリマンノシルコア構造はβ1,2結合キシロース残基およびα1,3結合フコース残基に置き換えられる。さらに植物細胞はMan5GlcNAc2構造も生産することができる。植物細胞中で生産されたペプチドはしばしば、グリカン構造上のコアα1,3フコースおよびキシロースの存在の結果として高度に抗原性であり、そして末端シアル酸残基の不存在から哺乳動物に導入された時、血流から迅速に排除される。したがってこれらのペプチドは本明細書で提供する方法を使用して改造されない限り、それらは一般には哺乳動物の治療薬としては適さないと考えられる。植物細胞中で発現した幾つかのモノクローナル抗体がマウスで非−免疫原性であることが判明したが、これはグリカン鎖がこれらの抗体中のFc領域に埋め込まれたので免疫原性ではなかったらしい(Chargelegue et al.,2000,Transgenic Res.9(3):187−194)。
et al.,1999;Hansen and Chilton,1999;Shilito,1999),プロトプラストの電気窄孔(Fromm et al.,1985,Ou−Lee et al.,1986;Rhodes et al.,1988;Saunders et al.,1989;Trick et al.,1997),ポリエチレン グリコール処理(Shiltio,1999;Trick et al.,1997),植物内 マイクロインジェクション(Leduc et al.,1996;Zhou et al.,1983),種子 インビビッション(Trick et
al.,1997),レーザー ビーム(1996),およびシリコン カーバイド ウイスカ(Thompson et al.,1995;米国特許出願第20020100077号明細書、引用
により全部、本明細書に編入する)を含む。
unnuus),ギニア アブラヤシ(Elaeis guineeis),アメリカホドイモ(ラッカセイ,Arachis hypogaea;Deng et al.,2001,Cell.Res.11:156−160),ココヤシ(Cocus nucifera),キャスター(Ricinus communis),ベニバナ(Carthamus tinctorius),カラシナ(Brassica spp.and Sinapis alba),コリアンダー,(Coriandrum sativum),カボチャ(Cucurbita maxima;Spencer and Snow,2001,Heredity 86(Pt 6):694−702),亜麻子/亜麻(Linum usitatissimum;Lamblin et al.,2001,Physiol Plant 112:223−232),ブラジル ナッツ(Bertholletia excelsa),ホホバ(Simmondsia chinensis),トウモロコシ(Zea mays;Hood et al.,1999,Adv.Exp.Med.Biol.464:127−147;Hood et al.,1997,Mol.Breed.3:291−306;Petolino et al.,2000,Transgenic Research 9:1−9),アルファルファ(Khoudi et al.,1999,Biotechnol.Bioeng.64:135−143),タバコ(Nicotiana tabacum;Wright et al.,Transgenic Res.10:177−181;Frigerio et al.,2000,Plant Physiol.123:1483−1493;Cramer et al.,1996,Ann.New York Acad.Sci.792:62−8−71;Cabanes−Macheteau et al.,1999,Glycobiology 9:365−372;Ruggiero et al.,2000,FEBS Lett.469:132−1 36),キャノーラ(Bai et al.,2001,Biotechnol.Prog.17:168− 174;Zhang et al.,2000,J.Anim.Sci.78:2868−2878)),ジャガイモ(Tacket et al.,1998,J.Infect.Dis.182:302−305;Richter et al.,2000,Nat.Biotechnol.18:1167−1171;Chong et al.,2000,Transgenic Res.9:71−78),アルファルファ(Wingdorovitz et al.,1999,Virology 255:347−353),エンドウ(Pisum sativum;Perrin et al.,2000,Mol.Breed.6:345−352),コメ(Oryza sativa;Stoger
et al.,2000,Plant Mol.Biol.42:583−590),ワタ(Gossypium hirsutum;Kornyeyev et al.,2001,Physiol Plant 113:323−331),オオムギ(Hordeum vulgare;Petersen et al.,2002,Plant Mol Biol 49:45−58);コムギ(Triticum spp.;Pellegrineschi et al.,2002,Genome 45:421−430)およびインゲン(Vicia spp.;Saalbach et al.,1994,Mol Gen Genet 242:226−236)を含む。
and practice)、エルセビア サイエンス出版、アムステルダム)、Islam(1996、植物組織培養(Plant tissue culture)、オックスフォード&IBH出版社、ニューデリー、インド)、Dodds and Roberts(1995、植物組織培養における実験(Experiments in plant tissue culture)、ニューヨーク;ケンブリッジ大学出版、ケンブリッジ、イギリス)、Bhojwani(植物組織培養:応用および限界(Plant tissue culture:applications and limitations)、エルセビア、アムステルダム、1990)、Trigiano and Gray(2000、植物組織培養の概念および実験実習(Plant tissue culture concepts and laboratory exercises)、CRC出版、ボカ ラトン、フロリダ州)およびLindsey(1991、植物組織培養マニュアル:基礎および応用(Plant tissue culture manual:fundamentals and applications)、クルヴァー
アカデミック(Kluwer Academic)、ボストン)を含む。
動物(例えば哺乳動物)の受精卵への組換えDNAの導入は、トランスジェニック動物技術で標準的な多数の技術を使用して行うことができる。例えばHogan et al.,マウス胚の操作;ア ラボラトリーマニュアル(Manipulating the
Mouse Embryo:A Laboratory Manual)、コールドスプリングハーバーラボラトリー出版、コールドスプリングハーバー、ニューヨーク州、1986;および米国特許第5,811,634号明細書(これは引用により全部、本明細書に編入する)を参照にされたい。最も普通には組換えDNAは前核マイクロインジェクションにより胚に導入されるGordon et al.,1980,PNAS 77:7380−7384;Gordon and Ruddle,1981,Science
214:1244−1246;Brinster et al.,1981,Cell
27:223−231;Costantini and Lacy,1981,Nature 294:92−94)。マイクロインジェクションは広範な様々な種に応用可能な利点を有する。着床前の胚もレトロウイルスで形質転換することができる(Jaenisch and Mintz,1974,Proc.Natl.Acad.Sci.U.S.A.71:1250−1254;Jaenisch et al.,1976,Hamatol Bluttransfus.19:341−356;Stuhlmann et al.,1984,Proc.Natl.Acad.Sci.U.S.A.81:7151−7155)。レトロウイルスが媒介する形質転換は1コピーの組換え核酸を細胞へ加える利点があるが、高度なモザイク現象を生じる。最も最近では、胚性幹細胞が媒介する技法が使用され(Gossler et al.,1986,Proc.Natl.Acad.Sci.USA.,83:9065−9069)、全染色体セグメントの転移(Lavitrano et al.,1989,Cell 57:717−723)およびインビトロ受精と組み合わせた配偶子トランスフェクション(Lavitrano
et al.,1989,Cell 57:717−723)も使用された。これらの技法を開示した研究手順に関する数冊の本が出版された:Cid−Arregui and Garcia−Caaranca(1998,マイクロインジェクションおよびトランスジェネシス:方法およびプロトコール(Microinjection and Transgenesis:Strategies and Protocols)、スプリンガー、ベルリン)、Clarke(2002、トランスジェネシス技法:原理およびプロトコール(Transgenesis Techniques:Principles and Protocols)、ヒューマナ出版、トトワ、ニュージャージー州)およびPinkert(1994、トランスジェニック動物技法:ア ラボラトリーハンドブック(Transgenic Animal Technology:A Laboratory Handbook)、アカデミック出版、サンディエゴ)。
細菌で組換え的に発現したペプチドは一般にグリコシル化されていない。しかしペプチドをグリコシル化することができる細菌系は明らかになりつつあり、したがって将来はグリコシル化された組換えペプチドを細菌で生産することができるかもしれない。
Genomic Analysis:A Practical Guide)、アカデミック出版、サンディエゴ)を参照にされたい。
本開示から、細胞により生産される出発原料が均一なほど、より効率的に所望のグリコシル化を有する大量のペプチドがインビトロで生成されることは明白である。すなわち本明細書で開示するインビトロの酵素的反応の出発原料として、均一にグリコシル化されたペプチドを生産するための宿主細胞の遺伝子工学は、ヘテロロガスな組のグリカン構造を結合して有するペプチド出発原料を使用することよりも有意な利点を提供する。本発明で使用するために好適な1つのペプチド出発原料は、基本的なトリマンノシルコア構造のみからなる1次グリカン分子を有するペプチドである。別の好適な出発原料はMan3GlcNAc4である。改造方法の後、好適なペプチドは所望のグリコシル化、すなわち改善された臨床的効力を有する最大量のペプチドを生じるようになる。しかし他のグリカン出発原料も、例えば高マンノースグリカンは一連のマンノシダーゼを使用してインビトロで基本的なトリマンノシルコア構造に容易に減少されるので、本明細書に記載する方法での使用に適する。本明細書のいたるところに記載するように、基本的なトリマンノシルコア構造またはMan3GlcNAc4が生成されるようにすべての過剰な糖部分が開裂可能であるならば、他のグリカン出発原料も使用することができる。このように本発明のペプチドを生産するために、遺伝的に工作した細胞を使用する目的は、出来る限り均一なグリカン構造を結合して有するペプチドを生成することであり、ここでグリカン構造はインビトロで改造されて所望のグリコシル化を有するペプチドを生成することができる。これによりこのようなペプチドの生産コストに劇的な減少をもたらす。この方法を使用して生産された糖ペプチドは、大部分が同じN−結合グリカン構造を有するので、生産後修飾プロトコールはバッチ毎の一貫性がより高い最終産物を生成するために標準化し、そして至適化することができる。結果として、最終的に完成された−鎖(chain)生成物は、現在入手可能なものよりも異質性が低くなり得る。この生成物は従来技術の生成物よりも改善された生物学的半減期および生物学活性を有する。あるいは所望により本発明は、例えば異なる糖部分の付加をもたらす反応条件を選択することにより、限定され、そして特異的な異質性を導入するために使用することができる。
et al.,(In vitrogen Conference,March,1999,abstract),Hollister and Jarvis,(2001,Glycobiology 11(1):1−9),and Palacpac et al.,(1999,PNAS USA 96:4697),Jarvis et al.,(1998.Curr.Opin.Biotechnol.9:528−533),Gerngross(米国特許出願20020137134第号明細書)を参照にされたい、これらすべてが昆虫または植物細胞をグリコシルトランスフェラーゼ遺伝子でトランスフェクションすることにより、昆虫または植物発現系を「哺乳動物化する(mammalianize)」ための技術を開示する。
修飾された糖タンパク質が細胞内で生産され、または分泌される場合、第1工程として特定の屑、宿主細胞、分解したフラグメントを、例えば遠心または超遠心により除去し;場合によりタンパク質は市販されているタンパク質濃縮フィルターを用いて濃縮することができ、続いてペプチド変異体はイムノアフィニティークロマトグラフィー、イオン交換カラム分画(例えばジエチルアミノエチル(DEAE)またはカルボキシメチルもしくはスルホプロピル基を含むマトリックス)、Blue−Sepharose、CM Blue−Sepharose、MONO−Q、MONO−S、レンチルレクチン−Sepharose、WGA−Sepharose、ConA−Sepharose、Ether Toyopearl、Butyl Toyopearl、Phenyl ToyopearlまたはプロテインA Sepharoseでのカラムクロマトグラフィー、SDS−PAGE−クロマトグラフィー、シリカクロマトグラフィー、クロマトフォーカシング、逆相HPLC(RP−HPLC)、ゲル濾過(例えばSephadex分子篩またはサイズ−排除クロマトグラフィー)、ペプチドを選択的に結合するカラムでのクロマトグラフィー、およびエタノール、pHまたは硫酸アンモニウム沈殿、膜濾過および種々の技法から選択される1以上の工程により他の不純物から分離することができる。
本発明は種々のペプチドおよびタンパク質、および同様な分子またはそれらのフラグメントをコードする単離された核酸を含む。そのようなペプチドには限定するわけではないが、ヒト顆粒球コロニー刺激因子(G−CSF)、ヒトインターフェロンアルファ(IFN−アルファ)、ヒトインターフェロンベータ(IFN−ベータ)、ヒト第VII因子(第VII因子)、ヒト第IX因子(第IX因子)、ヒト濾胞刺激ホルモン(FSH)、ヒトエリトロポエチン(EPO)、ヒト顆粒球/マクロファージコロニー刺激因子(GM−CSF)、ヒトインターフェロンガンマ(IFN−ガンマ)、ヒト−アルファ−1プロテアーゼインヒビター(アルファ−1アンチトリプシンまたはアルファ−1−トリプシンインヒビター、A−1−P1としても知られている)、グルコセレブロシダーゼ、ヒト組織型アクチベーター(TPA)、ヒトインターロイキン−2(IL−2)、ヒト第VIII因子(第VIII因子)、ヒトIgG免疫グロブリンFc部分に融合した75kDa腫瘍壊死因子レセプター(ENBREL(商標)またはETANERCEPT(商標)(キメラTNFR)として商業的に知られている)、ヒトウロキナーゼ(ウロキナーゼ)、糖タンパク質IIb/IIIaおよびビトロネクチンアルファvベータ3レセプターに特異的に結合するヒト/マウスキメラモノクローナル抗体のFabフラグメント(REOPRO(商標)またはABCIXIMAB(キメラ抗−糖タンパク質IIb/IIIa)として商業的に知られている)、ヒトHER2に特異的に結合するマウス/ヒトキメラモノクローナル抗体(HERCEPTIN(商標)(キメラ抗−HER2)として商業的に知られている)、呼吸合胞体ウイルスのA抗原部位またはFタンパク質に特異的に結合するヒト/マウスキメラ抗体(SYNAGIS(商標)またはPALIVIZUMAB(キメラ抗−RSV)として商業的に知られている)、ヒトB−細胞上のCD20に特異的に結合するキメラヒト/マウスモノクローナル抗体(RITUXAN(商標)またはRITUXAMAB(商標)(キメラ抗−CD20)として商業的に知られている)、ヒト組換えDNase(DNase)、ヒト腫瘍壊死因子に特異的に結合するキメラヒト/マウスモノクローナル抗体(REMICADE(商標)またはINFLIXIMAB(キメラ抗−TNF)として商業的に知られている)、ヒトインスリン、B型肝炎ウイルスの表面抗原(adwサブタイプ;HBsAg)、およびヒト成長ホルモン(HGH)等を含む。
本発明はG−CSF上のグリカン構造を修飾する方法を包含する。G−CSFは活性化T−細胞、マクロファージ、内皮細胞およびストロマの繊維芽細胞により生産されるサイトカインとして当該技術分野では周知である。G−CSFは主に骨髄に作用して炎症性白血球の生産を増加し、そしてさらに内分泌ホルモンとして機能して炎症機能中に消費される好中球の補充を開始する。またG−CSFは化学療法後の骨髄代用に臨床的応用を有する。
本発明はさらに、IFNアルファおよびIFNベータの改造および修飾法を包含する。IFNアルファは重量が約18kDaの約20個のペプチドのファミリーの一部である。IFNアルファおよびIFNベータは集合的にI型インターフェロンとして知られるが、同じ細胞レセプターに結合し、そして類似の応答を誘導する。I型IFNは中でも、ウイルス複製を阻害し、NK細胞の溶解能力を上げ、MHC分子の発現を調節し、そして細胞増殖を阻害する。I型IFNはウイルス感染、特に肝炎ウイルスの治療として、および多発性硬化症の治療として使用されてきた。
さらに本発明は第VII因子の改造および修飾法を包含する。血液凝固経路は、多くの出来事(event)を含んでなる複雑な反応である。この経路の中間の出来事が第VII因子であり、これは組織因子およびカルシウムイオンの存在下で第X因子をXa因子に転換することにより(第VIIa因子への活性化で)、血液凝固の外因系経路に参加するプロ酵素である。次に第Xa因子は、第Va因子、カルシウムイオンおよびリン脂質の存在下でプロトロンビンをトロンビンに転換する。第X因子から第Xa因子への活性化は、内因系および外因系の血液凝固経路の両方により分けられる(shared)出来事であり、したがって第VIIa因子は第VIII因子の欠損またはインヒビターを有する患者の処置に使用することができる。また第VIIa因子が内因系の経路にも参加できることを示唆する証拠もあるので、血液凝固における第VII因子の役割の突出および重要性が増している。
本発明はさらに第IX因子を改造および/または修飾するための方法を包含する。上記のように第IX因子は血液凝固カスケードで活性で(vital)ある。体内での第IX因子の欠損は、血友病(B型)の型の特徴である。この疾患の処置は通常、ヒト血漿タンパク質の第IX因子濃縮物の静脈内輸血に限られている。しかし時間および経費の現実的な欠点に加えて、血液濃縮物の輸血にはウイルス性肝炎、後天的免疫不全症候群の伝播、または受容者に対する血栓塞栓性疾患の危険性を含む。
and Thrombosis)、オックスフォード、ブラックウェル、サイエンティフィック、第614頁)に記載されているような1段階の活性化部分トロンボプラスチン時間アッセイのような周知方法を使用して行うことができる。簡単に説明すると、本発明の方法により開発された第IX分子の生物学的活性をアッセイするために、このアッセイは等容量の活性化部分トロンボプラスチン試薬、B型血友病患者から単離した第IX因子欠損血漿を用いて、当該技術分野で周知の滅菌瀉血技法、および標準として正常プール血清またはサンプルを使用して行うことができる。このアッセイでは1単位の活性を、1ミリリットルの正常プール血清に存在する量と定める。さらに正常に対して第IX因子欠損患者に由来する血漿の凝固時間を下げる第IX因子の能力に基づく生物学的活性に関するアッセイが、例えばProctor and Rapaport(1961,Amer.J.Clin.Path.36:212)に記載されているように行うことができる。
本発明はさらFSHを改造および/または修飾する方法を含む。ヒトの生殖機能は部分的には、共通する92アミノ酸糖タンパク質アルファサブユニットを有するが、ホルモン−特異的ベータサブユニットが異なるヘテロ二量体のヒト糖タンパク質ホルモンのファミリーにより制御されている。このファミリーには濾胞刺激ホルモン(FSH)、黄体形成ホルモン(LH)、チロトロピンまたは甲状腺−刺激ホルモン(TSH)およびヒト絨毛性性腺刺激ホルモン(hCG)を含む。ヒトFSHおよびLHは治療的に使用されて、ヒト女性において生殖に関する様々な代謝的な観点を調節する。例えば尿から部分精製されたFSHは、無排卵症候群または黄体期欠損の無排卵女性の小胞成熟を刺激するために臨床的に使用されている。黄体形成ホルモン(LH)およびFSHは、インビトロの受精のために卵胞の生育の刺激に組み合わせて使用されている。生殖サイクルにおけるFSHの役割は十分によく知られているので治療的使用が可能であるが、部分的には天然な供給源からの調製物の異質性および不純さから困難に遭遇した。
本発明はさらEPOを改造および/または修飾する方法を含んでなる。EPOは約34kDaの酸性糖タンパク質であり、そして3つの天然な形態で存在することができる:アルファ、ベータおよびアシアロ。アルファおよびベータ形は炭水化物成分がわずかに異なるが、同じ能力、生物学的活性および分子量を有する。アシアロ形はアルファまたはベータ形であり、末端シアル酸が除去されている。EPOは身体が健康な状態である時は血漿中に大変低濃度で存在し、ここで組織は既存の数の赤血球から十分な酸素を受容する。この正常濃度は通常は加齢を通して失われる赤血球細胞の代替を刺激するために十分である。循環中のエリトロポエチンの量は、循環中の血液細胞による酸素輸送が低下する時の低酸素の条件下で増加する。低酸素は出血による大量の血液損失、照射に過剰暴露された赤血球細胞の破壊、高山病または長期の意識不明による酸素取り込みの低下、または様々な状態の貧血によるより引き起こされ得る。したがってEPOは、照射療法後の赤血球細胞の補充、貧血および他の生命を脅かす状態に有用な化合物である。
本発明はGM−CSFを修飾する方法を包含する。GM−CSFは活性化T−細胞、マクロファージ、内皮細胞およびストロマの繊維芽細胞により生産されるサイトカインとして当該技術分野では周知である。GM−CSFは主に骨髄に作用して炎症性白血球の生産を増加し、そしてさらに内分泌ホルモンとして機能して炎症機能中に消費される好中球の補充を開始する。さらにGM−CSFはマクロファージ活性化因子であり、そしてランゲルハンス細胞の樹状細胞への分化を促進する。G−CSFと同様に、GM−CSFは化学療法後の骨髄代用にも臨床的応用を有する。
IFN−ガンマを修飾および/または改造する方法を包含することは、本発明の目的である。IFN−ガンマはII型インターフェロンとしても知られているが、IFNアルファおよびIFNベータとは対照的に、約21〜24kDaの2つのサブユニットを含んでなるホモ二量体糖タンパク質である。サイズの変動は、当該技術分野で知られている種々の発現系で組換え的に再生した時、通常は複製しない可変性のグリコシル化パターンによるものである。IFN−ガンマはマクロファージの有力なアクチベーターであり、MHCクラスI分子の発現、そして程度は低いがMHCクラスII分子刺激物を上昇させる。さらにIFN−ガンマはT−細胞の分化およびB−細胞中でのアイソタイプスイッチを促進する。IFN−ガンマはまた好中球、NK細胞、および食細胞が媒介する排除を導く抗体応答の刺激物としてもよく知られている。IFN−ガンマは、結核症およびリーシュマニアのような細胞内病原体による感染と関連して使用される処置として、および種々の癌および他の新形成のような、顕著な特徴として異常な細胞増殖を伴う状態に有用な抗−増殖性治療剤としても提案された。
本発明はさらに、アルファ−アンチトリプシンとしても知られているアルファ−プロテアーゼインヒビター(A−I−PI、α−1−アンチトリプシンまたはα−1−トリプシンインヒビター)の改造方法を含む。A−I−PIは53kDaの分子量を有する糖タンパク質である。A−I−PIは内因性のセリンプロテアーゼによる組織破壊の制御に役割を有し、そして血漿中で最も有名なセリンプロテアーゼインヒビターである。特にA−I−PIは好中球エラスターゼを含む種々のエラスターゼを阻害する。エラスターゼは組織を破壊するプロテアーゼであり、そしてその活性が肺組織で調節されない時、特に問題を生じる。このプロテアーゼは外来タンパク質を分解することにより機能する。しかしAPIがエラスターゼ活性を調節するために十分な量で存在しない時、エラスターゼは肺組織を分解する。早晩、この平衡失調が慢性の肺組織傷害および気腫をもたらす。実際に、A−1−PIの遺伝的欠損は、肺気腫の未成熟な発生を伴うことが示された。A−1−PIの補充はこの状態の気腫の治療に成功裏に使用された。さらにA−1−PIの欠損は嚢胞性繊維症および関節炎のような他の疾患の悪化にも関与し、この場合、白血球は感染と戦うために肺または関節に移動する。
本明細書に記載する本発明はさらに、グルコセレブロシダーゼを修飾する方法を含む。グルコセレブロシダーゼは糖脂質であるグルコセレブロシドをグルコースおよびセラミドへ加水分解することを触媒するリソソームの糖タンパク質酵素である。グルコセレブロシダーゼの変異体は、Cerezyme(商標)およびCeredase(商標)として市販されており、ゴーシェ病の処置に治療薬として認可されている。Ceredase(商標)は完全なN−結合構造を有するグルコセレブロシダーゼの胎盤に由来する形態である。Cerezyme(商標)は497アミノ酸長で、CHO細胞で発現されたグルコセレブロシダーゼの組換え変異体である。Cerezymeの4N−結合グリカンはトリマンノースコアで終結するように修飾された。
本発明はさらに組織−型アクチベーター(TPA)の改造方法も包含する。TPAはプラスミノーゲンを活性化して、フィブリン(血栓のタンパク質物質の主成分)を溶解するプラスミンを形成する。TPA調製物は、心筋梗塞および脳梗塞を引き起こす血栓症の血栓溶解処置において血栓に対して大変高い選択性を有する血栓溶解剤として開発された。
本発明はさらにIL−2を改造および修飾する方法を包含する。IL−2はTリンパ球の主要な増殖因子であり、そして細胞傷害性CD8T細胞およびNK細胞を刺激することにより液性および細胞性免疫応答を上昇させる。したがってIL−2は腫瘍およびウイルス感染に対する防御メカニズムにおいて重要である。IL−2は転移性メラノーマおよび腎腺癌に対する治療にも使用され、そして臨床試験で多くの形態の癌に使用されてきた。さらにIL−2は、CD4カウントの有意な上昇を導くHIVに感染した患者にも使用されてきた。
本発明はさらに第VIII因子の改造および修飾法を包含する。第VIIおよび第IX因子について前に記載したように、第VIII因子は血液凝固経路の重要な成分である。ヒト第VIII因子(抗血友病因子;FVIII:C)は2つのペプチド(80kDaの軽鎖分子量、およびグリコシル化状態に依存して90から220kDaまで可変の重鎖分子量)からなるヒト血漿タンパク質である。これは凝固経路の必須なコファクターであり、そして第X因子がその活性状態(第Xa)因子に転換するために必要である。第VIII因子は、2050aaペプチドの二量体であるフォン ビルブラント(von Willibrand)因子(aka FVIII:RP)との非共有複合体として血漿中を循環する(米国特許第6,307,032号明細書を参照にされたい)。正常の20%未満の第VIII因子の血液濃度は、血友病Aと呼ばれる出血性障害を引き起こす。1%未満の第VIII因子の血液レべルは、重篤な出血障害を生じ、最も共通する症状である天然な関節出血を伴う。
本発明はウロキナーゼを改造および/または修飾する方法も含む。ウロキナーゼはプラスミノーゲンをプラスミンに活性化するセリンプロテアーゼである。このタンパク質は内皮および腎臓を含む種々の組織中で合成され、そして尿中に微量分泌される。精製されたウロキナーゼは2つの活性形態、高分子量形(HUK:約50kDa)および低分子量形(LUK:約30kDa)で存在する。LUKはHUKから最初の135アミノ酸を放出するリシン35以降のタンパク質分解により、HUKから誘導されることが示された。HUKまたはLUKは、プロウロキナーゼと呼ばれる1本鎖前駆体の、リシン158とイソロイシン159との間のタンパク質分解的加水分解によりタンパク質分解的に活性な形に転換されて、二本鎖の活性化形(これはHUKまたはLUKをいずれかに対応し続ける)を生じるはずであるという従来の見識が維持された。この加水分解的切断から生じるタンパク質分解的に活性なウロキナーゼ種は、1つのジスルフィド結合により一緒に保持される2つのアミノ酸鎖を含む。活性化切断により形成された2つの鎖をAまたはA1鎖(それぞれHUKまたはLUK)と呼び、そしてB鎖は分子のプロテアーゼドメインを含んでなる。 ウロキナーゼは効果的な血栓溶解剤であることが示されてきた。しかしこれは天然には微量でしか生産されないので、効果的な投薬用量のための酵素のコストは高い。ウロキナーゼは組換え細胞培養で生産され、そしてウロキナーゼをコードするDNAは適当なベクターおよび宿主微生物と一緒に知られている。ウロキナーゼを含んでなる現在の組成物およびウロキナーゼを組換え的に生産する方法はグリコシル化パターンが欠如した産物により阻まれ、そしてウロキナーゼの活性化を取り巻く複雑なタンパク質分解的開裂を考慮すると、この異型のグリコシル化は効果が低く、そして能力が低い生成物を導く。
本発明はさらに組換えヒトDNaseを改造および/または修飾する方法を包含する。ヒトDNaseIは治療薬として試験され、そしてインビトロで嚢胞性線維症の粘液の粘度を低下させることが示された。化膿した粘液は約10〜13mg/mlのDNAを含むと測定され、イオン性ポリマーが気道流体の流動学的特性に影響を及ぼすと予想された。したがってウシ膵臓DNaseI(DNAを分解する酵素)が粘液溶解剤として何年も前に試験されたが、抗原性および/または混入プロテアーゼにより誘導される副作用から臨床的実施には入らなかった。組換えヒトDNaseは現在、嚢胞性線維症のような疾患の症状を緩和するために治療薬として使用されている。
本発明はさらにインスリンの改造方法を含む。インスリンは、血中グルコースレベルを調節するために膵臓のベータ小島細胞がインスリンを生産しないI型糖尿病に最も効果的な処置であることは周知である。糖尿病の系統および非制御血中グルコースには、循環器系および足の問題、および失明、挙げることはしないが糖尿病の悪化からもたらされるか、または悪化のいずれかである他の様々な合併症を含む。
本発明はさらにB型肝炎ワクチン(HbsAgまたはB型肝炎sAg)で使用する抗原を改造する方法を含んでなる。HBsAgはB型肝炎S−タンパク質の組換え的に生産される表面抗原であり、そしてB型肝炎ウイルス(とりわけ肝硬変および癌腫を含む肝臓疾患を引き起こす危険なウイルスを増やし、そして世界中で年間に百万人以上の死をもたらす)に対する免疫応答を誘導するために使用される。現在HBsAgワクチンは防御および中和免疫応答を誘導するために、6カ月間の間隔で3回投与される。
本発明はさらにヒト成長ホルモン(HGH)の改造方法を包含する。ヒト下垂体で分泌されるHGHのアイソフォームは、191個のアミノ酸からなり、そして約21,500の分子量を有する。胎盤で作られるHGHのアイソフォームはグリコシル化形である。HGHは、とりわけ一次成長(linear growth)(体細胞発生)、哺乳期、マクロファージの活性化およびインスリン−様および糖尿病原性効果を含む正常なヒトの成長および発生の調節に大いに関与している。
本発明はさらに、キメラTNFR、キメラ抗−糖タンパク質IIb/IIIa、キメラ抗−HER2、キメラ抗−RSV、キメラ抗−CD20およびキメラ抗−TNFを含む種々のキメラ抗体調製物を改造する方法を含んでなる。キメラ抗体調製物は、IgG抗体に由来するヒトのFc部分および抗原に特異的なモノクローナル抗体に由来する可変領域を含んでなる。他の調製物はレセプター、例えばヒトIgGFc部分に融合した75kDaのTNFレセプターを含んでなる。これらの分子はさらに、ヒトおよびマウスに由来する軽および重鎖を含んでなるFabフラグメントを含む。キメラTNFRは、慢性関節リウマチのような炎症性疾患の処置に有用である。キメラ抗−糖タンパク質IIb/IIIaは、心臓異常、血液凝固および血小板機能障害の処置に有用である。キメラ抗−HER2は胸部癌の処理として有用であり、キメラ抗−RSVは呼吸合胞体ウイルスの処理に有用であり、キメラ抗−CD20は非−ホジキンリンパ腫の処置に有用であり、そしてキメラ抗−TNFはクローン病の処置に有用である。
本明細書で使用する用語「抗体」は、抗原上の特異的なエピトープに特異的な結合することができる免疫グロブリン分子を称する。抗体は天然な供給源に由来するか、または組換え源に由来する完全な免疫グロブリンであることができ、そして完全な免疫グロブリンの免疫反応性部分であることができる。抗体は典型的には免疫グロブリン分子の四量体である。本発明の抗体は、例えばポリクローナル抗体、モノクローナル抗体、Fv、FabおよびF(ab)2を含む種々の形態、ならびに一本鎖抗体およびヒト化抗体で存在することができる(Harlow et al.,1999,抗体を使用して(Using Antibodies):ア ラボラトリーマニュアル、コールドスプリングハーバーラボラトリー出版、ニューヨーク州;Harlow et al.,1989,抗体:ア ラボラトリーマニュアル、コールドスプリングハーバー、ニューヨーク;Houston
et al.,1988,Proc.Natl.Acad.Sci.USA85:5879−5883;Bird et al.,1988,Science242:423−426)。
ペプチドの1クラス、すなわち免疫グロブリンの特異的なグリコシル化は、これらペプチドの生物学的活性に特に重要な効果を有する。本発明はIgGクラスの免疫グロブリンのみに限定されると解釈すべきではなく、抗体のIgA、IgEおよびIgMクラスの免疫グロブリンも含むと解釈すべきである。
al.,1995,Biotechnology13:592−596)。
al.,1998,Immunological Rev.163:59−76)。バイセクティングGlcNAcがそのように生産された免疫グロブリン分子の群全体に存在することを確実とするために、分子の混合物を以下のエキソグリコシダーゼと連続的に、または混合物で処理することができる:ノイラミニダーゼ、β−ガラクトシダーゼ、β−グルコサミニダーゼ、α−フコシダーゼ。生成したトリマンノシルコアは次いで上記のようにグリコシルトランスフェラーゼを使用して改造することができる。
75kDaのヒトTNFレセプターのヌクレオチドおよびアミノ酸配列を、本明細書でそれぞれ配列番号31および配列番号32として説明する(それぞれ図71Aおよび71B)。キメラ抗−HER2の軽および重可変領域のアミノ酸配列を、それぞれ配列番号35および配列番号36として説明する(それぞれ図72Aおよび72B)。キメラ抗−RSVの軽および重可変領域のアミノ酸配列を、それぞれ配列番号38および配列番号37として説明する(それぞれ図73Aおよび73B)。抗−TNFの非−ヒト可変領域のアミノ酸配列を、本明細書中でそれぞれ配列番号41および配列番号42として説明する(それぞれ図74Aおよび74B)。ヒトIgGのFc部分のヌクレオチドおよびアミノ酸配列を、配列番号49および配列番号50として説明する(それぞれ図75Aおよび75B)。
マウス抗−糖タンパク質IIb/IIIa抗体の可変領域のアミノ酸配列を、配列番号52(マウス成熟可変軽鎖、図76)および配列番号54(マウス成熟可変重鎖、図77)で説明する。これらのマウス配列は、米国特許第5,777.085号明細書に見いだされる手順に従い、ヒトIgGアミノ酸配列の配列番号51(ヒト成熟可変軽鎖、図78)、配列番号53(ヒト成熟可変重鎖、図79)、配列番号55(ヒト軽鎖、図80)および配列番号56(ヒト重鎖、図81)と組み合わせて、キメラヒト化マウス抗−糖タンパク質IIb/IIIa抗体を作成することができる。他の抗−糖タンパク質IIb/IIIaヒト化抗体は、米国特許第5,877,006号明細書に見いだされる。抗−糖タンパク質IIb/IIIa MAb 7E3を発現する細胞系は、ATCC(マナッサス、バージニア州)から寄託番号HB−8832で商業的に得ることができる。
キメラ抗−CD20抗体の核酸およびアミノ酸配列は、配列番号59(マウス可変領域軽鎖の核酸配列、図82A)、配列番号60(マウス可変領域軽鎖のアミノ酸配列、図82B)、配列番号61(マウス可変領域重鎖の核酸配列、図83A)および配列番号62(マウス可変領域重鎖のアミノ酸配列、図83B)に説明する。マウス抗体をヒト化するために、ヒトIgG重および軽定常ドメインを含むTCAE8(配列番号57、図84A−84E)を都合よく使用することができる。上記マウス可変領域コードDNAを、米国特許第5,736,137号明細書に与えられた使用説明に従いTCAE8ベクターにクローニングすることにより、哺乳動物細胞系で形質転換した時、キメラ抗−CD20抗体を発現するベクターが作成される(配列番号58、図85A−85E)。他のヒト化抗−CD20抗体は、米国特許第6,120,767号明細書に見いだされる。抗−CD20MAb C273を発現する細胞系は、ATCC(マナッサス、バージニア州)から寄託番号HB−9303で商業的に得ることができる。
別の観点では、本発明は製薬学的組成物を提供する。製薬学的組成物には、製薬学的に許容され得る希釈剤および天然には存在しない、水溶性ポリマー、治療部分または生体分子とグリコシル化または非グリコシル化ペプチドとの間の共有結合物を含む。ポリマー、治療部分または生体分子は、ペプチドとポリマー、治療部分または生体分子との間に挿入され、そして両方を共有結合する完全なグリコシル連結基を介してペプチドに結合される。
本発明をこれから以下の実施例を参照にして記載する。これらの実施例は具体的説明の目的のみに提供し、そして本発明がこれらの実施例に限定されるべきでは全くなく、むしろ本明細書に提供する教示の結果として明らかになるありとあらゆる変更を包含すると解釈すべきである。
この実施例で提示するこの実験に使用する材料および方法をこれから記載する。
この実施例は、シアリルルイスX部分を用いたTP10の調製および強化された生物学的活性の分析を説明する。
この実施例では、数種の組換えペプチドのシアリル化形態の調製を説明する。
2遊離放射性標識前駆体からゲル濾過により分離した後に、14C−NeuAcの取り込みにより測定された取り込まれたNeuAc。
3%Rxnは理論的な最大値として末端Gal含量に基づく反応の%完成を示す。
4アンチトロンビンIII。
5α1アンチトリプシン。
この実施例は、N−結合シアリルルイスX抗原を持つ組織組織プラスミノーゲンアクチベーター(TPA)の調製を説明する。
、FTV、FTIIIを含み;ならびに微生物トランスフェラーゼも使用することができる。
この実施例は、高マンノースグリカンからの戻し改作によりトリマンノースコアを持つ組織プラスミノーゲンアクチベータの調製を説明する。
この実施例は、GlcNAc残基のトリマンノシルコア上への付加を説明する。
100mM MES pH6.5、または100mM Tris pH7.5
5mM UDP−GlcNAc
20mM MnCl2
100mU/ml GlcNAcT−I
100mU/ml GlcNAcT−II
1mg/ml EPO(精製され、Sf9細胞で発現した、
プロテインサイエンスから購入)
この実施例はハイブリッドまたは複雑なN−グリカンを持つウシ膵臓RNaseの調製について説明する。RNaseの高マンノースN−結合グリカンを酵素的に消化し、そしてハイブリッドN−結合グリカンを作成するために仕上げる(elaborated)。さらにRNaseの高マンノースN−結合グリカンを酵素的に消化して、複合N−結合グリカンを作成するために仕上げる。
この実施例は、昆虫細胞が発現したEPOからPEG化された2アンテナ状EPO、および3アンテナ状のシアリル化されたEPOの調製を説明する。
8.CMP−SA−PEGの調製
この実施例はCMP−SA−PEGの調製を説明する。
1726.4,1770.3,1814.4,1858.2,1881.5,1903.5,1947.3。
この実施例は本発明の結合物の集成体を説明する。濾胞刺激ホルモン(FSH)は脱シアリル化し、そしてCMP−(シアル酸)−PEGを結合した。
この実施例は本発明の結合物の集成体を説明する。ジシアリル化FSHにCMP−(シアル酸)−PEGを結合した。
この実施例は非−PEG化FSHと比べて、本発明の方法に従い調製したグリコPEG化濾胞刺激ホルモン(FSH)の薬物動態学的特性の試験を説明する。
この実施例は培養したセルトリー(Sertori)細胞に基づく濾胞刺激ホルモン(FSH)活性に関するバイオアッセイを説明する。このアッセイは複合糖質化を含むグリカン改造後のFSHの生物活性を測定するために有用である。
この実施例はアシアロトランスフェリンの調製およびPEG−CMP−シアル酸でのそのシアリル化を説明する。
この実施例はCHO細胞で作られた組換え第VIIa因子のPEG化を説明する。
この実施例はアシアロ第IX因子の調製およびPEG−CMP−シアル酸を用いたそのシアリル化を説明する。
この実施例は、事前のシアリダーゼ処理無しのペプチドのシアリル−グリコPEG化の調製を説明する。ここで第IX因子は例示ペプチドである。
この実施例は、シアリル−グリコPEG化ペプチドのシアル酸キャッピングの手順を説明する。ここで第IX因子は例示ペプチドである。
この実施例は哺乳動物または昆虫系で発現したPEG化ペプチドの調製を説明する。
CaCl2を加えて含む)を、0.2U/mLの固定化シアリダーゼ(ビブリオ コレラ:Vibrio cholerae由来、カルビオケム)と32℃で24時間インキューベーションすることを含む。微生物の成長は、滅菌濾過または0.02%のアジ化ナトリウムを包含することにより止めることができる。次いで樹脂を遠心または濾過により取り出し、次いで洗浄してトラップされたペプチドを回収する。この時点で、EDTAを溶液に加えて樹脂から浸出したシアリダーゼは阻害することができる。
この実施例は昆虫細胞が発現したEPOからPEG化2アンテナ状EPOの調製法を説明する。
アシアロ−インターフェロンαの調製。CHO細胞から生産されたインターフェロンαを2.5mg/mLで50mM Tris 50mM Tris−HCl pH7.4、0.15M NaCl、5mM CaCl2に溶解し、そして500μLにCentricon Plus20遠心フィルター中で濃縮する。この溶液を300mU/mLのノイラミニダーゼII(ビブリオ コレラ:Vibrio cholerae)と16時間、32℃インキューベーションする。反応を監視するために、反応の小アリコートを適当なバッファーで希釈し、そしてIEFゲルを行う。次いで反応混合物を、前洗浄したN−(p−アミノフェニル)オキサミン酸−アガロース結合体(800μL/mL反応容量)に加え、そして洗浄したビーズを24時間、4℃で穏やかに回転する。混合物を10,000rpmで遠心し、そして上清を集める。ビーズをTris−EDTAバッファーで3回洗浄し、0.4mLのTris−EDTAバッファーで1回、そして0.2mLのTris−EDTAバッファーで1回洗浄し、すべての上清をプールする。上清は4℃で50mM Tris−HCl、pH7.4、1M NaCl、0.05%NaN3に対して透析し、そして50mM Tris−HCl、pH7.4、1M NaCl、0.05%NaN3に対してさらに2回透析する。次いで透析した溶液はCentricon Plus20遠心フィルターを使用して濃縮し、そして−20℃で保存する。IEFゲルに関する条件は、インビトロゲンにより提供される手順および試薬に従い泳動する。天然な、および脱シアリル化G−CSFのサンプルを水に対して透析し、そしてMALDI−TOF MSにより分析する。
CMP−シアル酸−PEGおよび0.1U/mLのST3Gal1と32℃で2日間インキューベーションする。シアル酸−PEGの取り込みを監視するために、反応の小アリコートにCMP−SA−PEG−蛍光リガンドを加え;ペプチドへ取り込まれた標識は、PBSバッファー(pH7.1)を使用してトーソー ハースG3000SW分析カラムでのゲル濾過により遊離標識から分離する。ペプチドへの蛍光標識の取り込みは、イン−ラインの蛍光検出器を使用して定量する。2日後、反応混合物はPBSバッファー(pH7.1)を使用してトーソー ハースG3000SW調製カラムを使用して精製し、そしてUV吸収に基づき画分を集める。反応の生成物は、インビトロゲンにより供給される手順および試薬に従いSDS−PAGEおよびIEF分析を使用して分析する。天然な、および脱シアリル化インターフェロン−アルファのサンプルを水に対して透析し、そしてMALDI−TOF MSにより分析する。
Tris−HCl、0.15M NaCl、0.05%NaN3、pH7.2に溶解する。この溶液を1mM CMP−シアル酸−PEGおよび0.1U/mLのST6GlcNAcIまたはIIと32℃で2日間インキューベーションする。シアル酸−PEGの取り込みを監視するために、反応の小アリコートにCMP−SA−PEG−蛍光リガンドを加え;ペプチドへの取り込まれた標識は、PBSバッファー(pH7.1)を使用してトーソー ハースG3000SW分析カラムでのゲル濾過により遊離標識から分離する。ペプチドへの蛍光標識の取り込みは、イン−ラインの蛍光検出器を使用して定量する。2日後、反応混合物はPBSバッファー(pH7.1)を使用してトーソー ハースG3000SW調製カラムを使用して精製し、そしてUV吸収に基づき画分を集める。反応の生成物は、インビトロゲンにより供給される手順および試薬に従いSDS−PAGEおよびIEF分析を使用して分析する。天然な、およびPEG化インターフェロン−アルファのサンプルを水に対して透析し、そしてMALDI−TOF MSにより分析する。
アシアロ−顆粒球−コロニー刺激因子(G−CSF)の調製。CHO細胞で生産されたG−CSFを2.5mg/mLで50mM Tris 50mM Tris−HCl、pH7.4、0.15M NaCl、5mM CaCl2に溶解し、そして500μLにCentricon Plus20遠心フィルター中で濃縮する。この溶液を300mU/mLのノイラミニダーゼII(ビブリオ コレラ:Vibrio cholerae)と16時間、32℃でインキューベーションする。反応を監視するために、反応の小アリコートを適当なバッファーで希釈し、そしてIEFゲルを行う。次いで反応混合物は前洗浄したN−(p−アミノフェニル)オキサミン酸−アガロース結合体(800μL/mL反応容量)に加え、そして洗浄したビーズを24時間、4℃で穏やかに回転する。混合物を10,000rpmで遠心し、そして上清を集める。ビーズをTris−EDTAバッファーで3回洗浄し、0.4mLのTris−EDTAバッファーで1回、そして0.2mLのTris−EDTAバッファーで1回洗浄し、すべての上清をプールする。上清は4℃で50mM Tris−HCl、pH7.4、1M NaCl、0.05%NaN3に対して透析し、そして50mM Tris−HCl、pH7.4、1M NaCl、0.05%NaN3に対してさらに2回透析する。次いで透析した溶液はCentricon
Plus20遠心フィルターを使用して濃縮し、そして−20℃で保存する。IEFゲルに関する条件は、インビトロゲンにより提供される手順および試薬に従い泳動する。天然な、および脱シアリル化G−CSFのサンプルを水に対して透析し、そしてMALDI−TOF MSにより分析する。
MSにより分析する。
O−結合EPO−SA−PEG(10kDa)の調製。元々CHO細胞で生産されたアシアロ−EPOを2.5mg/mLで、50mM Tris−HCl、0.15M NaCl、0.05%NaN3、pH7.2に溶解する。この溶液を5mM CMP−SAおよび0.1U/mL ST3Gal3と32℃で2日間インキューベーションする。シアル酸のN−結合グリカンへの取り込みを監視するために、反応の小アリコートにCMP−SA−14Cを加え;ペプチドは、メタノール、水を使用してトーソー ハースG3000SW分析カラムでのゲル濾過により分離し、そして生成物は放射能検出器を使用して検出する。反応が完了した時、溶液はCentricon−20フィルターを使用して濃縮する。残る溶液はCMP−SAがもはや検出できなくなるまで0.05M Tris(pH7.2)、0.15M NaCl、0.05%NaN3とバッファーと交換して7.2mLの最終容量とする。次いで残りを0.05M Tris(pH7.2)、0.15M
NaCl、0.05%NaN3に2.5mg/mLタンパク質で再懸濁する。溶液はO−結合部位をグルコシル化するために、1mMのCMP−SA−PEG(10KDa)およびST3Gal1と32℃で2日間インキューベーションする。シアル酸−PEGの取り込みを監視するために反応の小アリコートは、トーソー ハースTSK−gel−3000分析カラムを使用したゲル濾過により分離し、PBSpH7.0で溶出し、そしてUV検出により分析する。反応が完了した時、反応混合物はPBSバッファー(pH7.0)を使用してトーソー ハースTSK−gel−3000調製カラムを使用して精製し、UV吸収に基づき画分を集める。反応の生成物は、インビトロゲンにより供給される手順および試薬に従いSDS−PAGEおよびIEF分析を使用して分析する。サンプルを水に対して透析し、そしてMALDI−TOF MSにより分析する。
この実施例は、抗体分子のO−結合グリカンをPEG化するための手順を説明する。ここでEnbrel(商標)は例として使用するが、当業者はこの手順が多くの抗体分子に使用できると考えるだろう。
この実施例は、PEG分子をFc領域グリカンに導入することにより組換え抗体分子をグリコPEG化するための手順を説明する。ここでRemicade(商標)、TNF−R:IgGFc領域融合タンパク質は例示ペプチドである。
ハースTSK−Gel−3000調製カラムを使用して精製し、そしてUV吸収に基づき画分を集める。生成物を含む画分を合わせ、濃縮し、バッファー交換し、そして凍結乾燥する。反応の生成物は、インビトロゲンにより供給される手順および試薬に従いSDS−PAGEおよびIEF分析を使用して分析する。サンプルを水に対して透析し、そしてMALDI−TOF MSにより分析する。
この実施例は、酵母で発現されたTPAのようなペプチド上のPEG化GlcNAc−ASN構造の調製を説明する。
この実施例は、PEG化されたGlcNAc−Asn構造を持つ組織アクチベーターの調製を説明する。
27.CMP−SA−レブリネートの合成
この実施例はCMP−SA−レブリネートの合成手順を説明する。
[M+1] 277.9。
定値([M−1]−),363.97。
この実施例はマンノース−6−ホスフェートをCHO細胞で生産されたグルコセレブロシダーゼのようなペプチドに複合糖質化する手順を説明する。
この実施例は、ミトラマイシンのような低分子を哺乳動物細胞で生産された抗体分子のFc領域グリカンへ複合糖質化するため手順を説明する。ここで抗体Herceptin(商標)を使用するが、当業者はこの方法が多くの他の抗体に使用できると考えるだろう。
ハース TSK−Gel−3000調製カラムを使用して精製し、そしてUV吸収に基づき画分を集める。生成物を含む画分を合わせ、濃縮し、バッファー交換し、そして凍結乾燥する。反応の生成物は、インビトロゲンにより供給される手順および試薬に従いSDS−PAGEおよびIEF分析を使用して分析する。サンプルを水に対して透析し、そしてMALDI−TOF MSにより分析する。
この実施例は、ゲルダナマイシンのような低分子をCHO細胞で生産されたRituxan(商標)のような抗体のFc領域グリカンへ複合糖質化するための手順を説明する。ここで抗体Rituxan(商標)を使用するが、当業者はこの方法が多くの他の抗体に使用できると考えるだろう。
ハース TSK−Gel−3000調製カラムを使用して精製し、そしてUV吸収に基づき画分を集める。生成物を含む画分を合わせ、濃縮し、バッファー交換し、そして凍結乾燥する。反応の生成物は、インビトロゲンにより供給される手順および試薬に従いSDS−PAGEおよびIEF分析を使用して分析する。サンプルを水に対して透析し、そしてMALDI−TOF MSにより分析する。
31.トランスフェリン−GDNF
この実施例はタンパク質の複合糖質化、そして特にトランスフェリンをGDNFに複合糖質化する手順について説明する。トランスフェリン−SA−リンカー−Gal−UDP−はトランスフェリンから調製する。ガラクトース残基をGNDFグリカンから除去し、そしてトランスフェリン−SA−リンカー−Gal−UDPをGNDFグリカンにガラクトシルトランスフェラーゼを使用して結合する。
ハースG3000SW分析カラムでのゲル濾過により遊離標識から分離する。放射能標識のペプチドへの取り込みは、イン−ライン放射能検出器を使用して定量する。
この実施例はタンパク質の複合糖質化、そして特にトランスフェリンをグルコセレブロシダーゼに複合糖質化する手順について説明する。GlcNAc−ASN構造をグルコセレブロシダーゼ上に構築し、そしてトランスフェリン−SA−リンカー−Gal−UDPはGNDF GlcNAc−ASN構造にガラクトシルトランスフェラーゼを使用して結合する。
Tris−HCl、0.15M NaCl、5mM MnCl2、0.05%NaN3、pH7.2に溶解する。この溶液を2.5mg/mLのGlcNAc−グルコセレブロシダーゼおよび0.1U/mのガラクトシルトランスフェラーゼと32℃で2日間インキューベーションする。グルコセレブロシダーゼの取り込みを監視するために、ペプチドは、PBSバッファー(pH7.1)を使用してトーソー ハースG3000SW分析カラムでのゲル濾過により分離し、そしてU吸収により生成物を検出する。反応混合物はPBSバッファー(pH7.1)を使用したトーソー ハースG3000SW調製カラムを使用して精製し、UV吸収に基づき画分を集める。反応の生成物は、インビトロゲンにより供給される手順および試薬に従いSDS−PAGEおよびIEF分析を使用して分析する。サンプルを水に対して透析し、そしてMALDI−TOF MSにより分析する。
この実施例はタンパク質のO−結合グリカンへの複合糖質化、そして特にトランスフェリンをEPOに複合糖質化する手順について説明する。シアル酸残基はEPOのO−結合グリカンから除去し、そしてEPO−SA−リンカー−SA−CMPを調製する。EPO−SA−リンカー−SA−CMPをアシアロトランスフェリンにST3Gal3で複合糖質化する。
NaCl、0.05%NaN3、pH7.2に溶解する。この溶液を2.5mg/mLのアシアロ−トランスフェリンおよび0.1U/mLのST3Gal3と32℃で2日間インキューベーションする。トランスフェリンの取り込みを監視するために、ペプチドはPBSバッファー(pH7.1)を使用してトーソー ハースG3000SW分析カラムでのゲル濾過により分離し、そして生成物をUV吸収により検出する。反応が完了した時、溶液を5mMのCMP−SAおよび0.1U/mLのST3Gal3(未反応のトランスフェリングリカンをキャップするために)と32℃で2日間インキューベーションする。反応混合物はPBSバッファー(pH7.1)を使用してトーソー ハースG3000SW調製カラムを使用して精製し、そして画分をUV吸収に基づき集める。反応の生成物は、インビトロゲンにより供給される手順および試薬に従いSDS−PAGEおよびIEF分析を使用して分析する。サンプルを水に対して透析し、そしてMALDI−TOF MSにより分析する。
この実施例はペプチドの複合糖質化の手順、そして特にEPO−SA−リンカー−SA−GDNFの調製について説明する。
Claims (10)
- ファクターIXであるペプチドと水溶性ポリマーとの共有結合物であって、
前記水溶性ポリマーがポリ(エチレングリコール)であり、かつ、前記水溶性ポリマーがインタクトなグリコシル連結基を介して前記ペプチドに結合している、共有結合物。 - 前記ポリ(エチレングリコール)が直鎖ポリ(エチレングリコール)および分岐ポリ(エチレングリコール)から選択されるメンバーである、請求項1に記載の共有結合物。
- 前記インタクトなグリコシル連結基が炭水化物部分、アミノ酸部分、及びそれらの組み合わせから選択されるメンバーに結合している、請求項1又は2に記載の共有結合物。
- 前記インタクトなグリコシル連結基がO−またはN−結合グリカンおよびそれらの組み
合わせから選択されるメンバーである炭水化物部分に結合している、請求項3に記載の共
有結合物。 - 前記インタクトなグリコシル連結基が前記アミノ酸部分のヒドロキシルもしくはアミノ基またはそれらの組み合わせに結合している、請求項3に記載の共有結合物。
- 前記インタクトなグリコシル連結基がシアル酸、ガラクトース、N−アセチルグルコサミンまたはN−アセチルガラクトサミン残基を含む、請求項3に記載の共有結合物。
- 前記水溶性ポリマーがシアル酸残基に、前記シアル酸残基の5位及び9位から選択される位置で結合している、請求項1に記載の共有結合物。
- 請求項1〜7のいずれか一項に記載の共有結合物および製薬学的に許容可能な塩を含有する製薬学的組成物。
- ファクターIXであるペプチドと水溶性ポリマーとの共有結合物を形成する、細胞フリーな、インビトロの方法であって、
前記水溶性ポリマーがポリ(エチレングリコール)であり、かつ、インタクトなグリコシル連結基を介して前記ペプチドに共有結合し、
ファクターIXである前記ペプチドと、少なくとも1つの前記水溶性ポリマーに共有結合するヌクレオチド糖および少なくとも1つの前記ヌクレオチド糖が基質であるグリコシルトランスフェラーゼを含む混合物と、を接触させ、それにより前記ペプチドの前記共有結合物を形成する、方法。 - 前記グリコシルトランスフェラーゼが、シアリルトランスフェラーゼ、ガラクトシルトランスフェラーゼ、グルコシルトランスフェラーゼ、GalNAcトランスフェラーゼ、GlcNAcトランスフェラーゼ、フコシルトランスフェラーゼおよびマンノシルトランスフェラーゼからなる群から選択される、請求項9に記載の方法。
Applications Claiming Priority (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US32852301P | 2001-10-10 | 2001-10-10 | |
| US60/328,523 | 2001-10-10 | ||
| US34469201P | 2001-10-19 | 2001-10-19 | |
| US60/344,692 | 2001-10-19 | ||
| US33423301P | 2001-11-28 | 2001-11-28 | |
| US33430101P | 2001-11-28 | 2001-11-28 | |
| US60/334,233 | 2001-11-28 | ||
| US60/334,301 | 2001-11-28 | ||
| US38729202P | 2002-06-07 | 2002-06-07 | |
| US60/387,292 | 2002-06-07 | ||
| US39177702P | 2002-06-25 | 2002-06-25 | |
| US60/391,777 | 2002-06-25 | ||
| US39659402P | 2002-07-17 | 2002-07-17 | |
| US60/396,594 | 2002-07-17 | ||
| US40424902P | 2002-08-16 | 2002-08-16 | |
| US60/404,249 | 2002-08-16 | ||
| US40752702P | 2002-08-28 | 2002-08-28 | |
| US60/407,527 | 2002-08-28 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014013799A Division JP2014087370A (ja) | 2001-10-10 | 2014-01-28 | ペプチドの改造および複合糖質化 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2016037503A JP2016037503A (ja) | 2016-03-22 |
| JP6316784B2 true JP6316784B2 (ja) | 2018-04-25 |
Family
ID=27578810
Family Applications (8)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003534446A Expired - Lifetime JP5232352B2 (ja) | 2001-10-10 | 2002-10-09 | ペプチドの改造および複合糖質化 |
| JP2008297209A Expired - Lifetime JP5376912B2 (ja) | 2001-10-10 | 2008-11-20 | ペプチドの改造および複合糖質化 |
| JP2010024516A Expired - Lifetime JP5258809B2 (ja) | 2001-10-10 | 2010-02-05 | ペプチドの改造および複合糖質化 |
| JP2010217733A Expired - Fee Related JP5739632B2 (ja) | 2001-10-10 | 2010-09-28 | ペプチドの改造および複合糖質化 |
| JP2012106130A Pending JP2012143250A (ja) | 2001-10-10 | 2012-05-07 | ペプチドの改造および複合糖質化 |
| JP2014013799A Ceased JP2014087370A (ja) | 2001-10-10 | 2014-01-28 | ペプチドの改造および複合糖質化 |
| JP2015184218A Expired - Lifetime JP6316784B2 (ja) | 2001-10-10 | 2015-09-17 | ペプチドの改造および複合糖質化 |
| JP2016118316A Expired - Lifetime JP6321724B2 (ja) | 2001-10-10 | 2016-06-14 | ペプチドの改造および複合糖質化 |
Family Applications Before (6)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003534446A Expired - Lifetime JP5232352B2 (ja) | 2001-10-10 | 2002-10-09 | ペプチドの改造および複合糖質化 |
| JP2008297209A Expired - Lifetime JP5376912B2 (ja) | 2001-10-10 | 2008-11-20 | ペプチドの改造および複合糖質化 |
| JP2010024516A Expired - Lifetime JP5258809B2 (ja) | 2001-10-10 | 2010-02-05 | ペプチドの改造および複合糖質化 |
| JP2010217733A Expired - Fee Related JP5739632B2 (ja) | 2001-10-10 | 2010-09-28 | ペプチドの改造および複合糖質化 |
| JP2012106130A Pending JP2012143250A (ja) | 2001-10-10 | 2012-05-07 | ペプチドの改造および複合糖質化 |
| JP2014013799A Ceased JP2014087370A (ja) | 2001-10-10 | 2014-01-28 | ペプチドの改造および複合糖質化 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016118316A Expired - Lifetime JP6321724B2 (ja) | 2001-10-10 | 2016-06-14 | ペプチドの改造および複合糖質化 |
Country Status (20)
| Country | Link |
|---|---|
| US (3) | US7416858B2 (ja) |
| EP (7) | EP2279754B1 (ja) |
| JP (8) | JP5232352B2 (ja) |
| CN (3) | CN101724075B (ja) |
| AU (2) | AU2002360264B2 (ja) |
| BE (1) | BE2014C004I2 (ja) |
| BR (1) | BRPI0213207B1 (ja) |
| CA (1) | CA2462930C (ja) |
| DK (2) | DK1578771T3 (ja) |
| ES (9) | ES2564688T3 (ja) |
| FR (1) | FR14C0007I2 (ja) |
| HK (4) | HK1079797A1 (ja) |
| IL (3) | IL161251A0 (ja) |
| LU (1) | LU92359I2 (ja) |
| MX (1) | MXPA04003333A (ja) |
| NL (1) | NL300912I2 (ja) |
| NZ (1) | NZ532027A (ja) |
| PT (2) | PT2279755E (ja) |
| SG (2) | SG159381A1 (ja) |
| WO (1) | WO2003031464A2 (ja) |
Families Citing this family (312)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5581476A (en) * | 1993-01-28 | 1996-12-03 | Amgen Inc. | Computer-based methods and articles of manufacture for preparing G-CSF analogs |
| US5545553A (en) * | 1994-09-26 | 1996-08-13 | The Rockefeller University | Glycosyltransferases for biosynthesis of oligosaccharides, and genes encoding them |
| DE60030323T2 (de) | 1999-12-24 | 2007-10-11 | Genentech, Inc., South San Francisco | Verfahren und verbindungen zur verlängerung der halbwertzeiten bei der ausscheidung von biowirksamen verbindungen |
| EP2236152B1 (en) * | 2000-04-12 | 2014-06-04 | Novozymes Biopharma DK A/S | Albumin fusion proteins |
| US7625756B2 (en) | 2000-06-28 | 2009-12-01 | GycoFi, Inc. | Expression of class 2 mannosidase and class III mannosidase in lower eukaryotic cells |
| US7449308B2 (en) | 2000-06-28 | 2008-11-11 | Glycofi, Inc. | Combinatorial DNA library for producing modified N-glycans in lower eukaryotes |
| US7598055B2 (en) | 2000-06-28 | 2009-10-06 | Glycofi, Inc. | N-acetylglucosaminyltransferase III expression in lower eukaryotes |
| US7863020B2 (en) | 2000-06-28 | 2011-01-04 | Glycofi, Inc. | Production of sialylated N-glycans in lower eukaryotes |
| US7723296B2 (en) | 2001-01-18 | 2010-05-25 | Genzyme Corporation | Methods for introducing mannose-6-phosphate and other oligosaccharides onto glycoproteins and its application thereof |
| US7507413B2 (en) * | 2001-04-12 | 2009-03-24 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| US7112410B1 (en) | 2001-08-29 | 2006-09-26 | Human Genome Sciences, Inc. | Human tumor necrosis factor TR21 and methods based thereon |
| AU2004236174B2 (en) * | 2001-10-10 | 2011-06-02 | Novo Nordisk A/S | Glycopegylation methods and proteins/peptides produced by the methods |
| US7399613B2 (en) * | 2001-10-10 | 2008-07-15 | Neose Technologies, Inc. | Sialic acid nucleotide sugars |
| US7173003B2 (en) | 2001-10-10 | 2007-02-06 | Neose Technologies, Inc. | Granulocyte colony stimulating factor: remodeling and glycoconjugation of G-CSF |
| US7214660B2 (en) * | 2001-10-10 | 2007-05-08 | Neose Technologies, Inc. | Erythropoietin: remodeling and glycoconjugation of erythropoietin |
| US7795210B2 (en) * | 2001-10-10 | 2010-09-14 | Novo Nordisk A/S | Protein remodeling methods and proteins/peptides produced by the methods |
| US8008252B2 (en) | 2001-10-10 | 2011-08-30 | Novo Nordisk A/S | Factor VII: remodeling and glycoconjugation of Factor VII |
| US7179617B2 (en) * | 2001-10-10 | 2007-02-20 | Neose Technologies, Inc. | Factor IX: remolding and glycoconjugation of Factor IX |
| US7157277B2 (en) | 2001-11-28 | 2007-01-02 | Neose Technologies, Inc. | Factor VIII remodeling and glycoconjugation of Factor VIII |
| MXPA04003333A (es) * | 2001-10-10 | 2006-02-22 | Neose Technologies Inc | Remodelado y glicoconjugacion de peptidos. |
| US7439043B2 (en) * | 2001-10-10 | 2008-10-21 | Neose Technologies, Inc. | Galactosyl nucleotide sugars |
| US7696163B2 (en) | 2001-10-10 | 2010-04-13 | Novo Nordisk A/S | Erythropoietin: remodeling and glycoconjugation of erythropoietin |
| DK1440157T3 (da) * | 2001-10-29 | 2012-05-07 | Crucell Holland Bv | Methods and means for producing proteins with predetermined post-translational modifications |
| US7473680B2 (en) * | 2001-11-28 | 2009-01-06 | Neose Technologies, Inc. | Remodeling and glycoconjugation of peptides |
| EP1463751B1 (en) * | 2001-12-21 | 2013-05-22 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| CA2472937C (en) | 2002-01-11 | 2014-06-17 | Biomarin Pharmaceutical, Inc. | Use of p97 as an enzyme delivery system for the delivery of therapeutic lysosomal enzymes |
| DE10209821A1 (de) | 2002-03-06 | 2003-09-25 | Biotechnologie Ges Mittelhesse | Kopplung von Proteinen an ein modifiziertes Polysaccharid |
| DE60336555D1 (de) | 2002-06-21 | 2011-05-12 | Novo Nordisk Healthcare Ag | Pegylierte glykoformen von faktor vii |
| CA2494223C (en) * | 2002-08-01 | 2012-10-02 | National Research Council Of Canada | Campylobacter glycans and glycopeptides |
| EP1534324B1 (en) | 2002-08-20 | 2019-01-16 | Glykos Finland Oy | Tumor specific oligosaccharide epitopes and use thereof |
| CA2498319A1 (en) * | 2002-09-09 | 2004-03-18 | Nautilus Biotech | Rational evolution of cytokines for higher stability, the cytokines and encoding nucleic acid molecules |
| EP1558747B1 (en) | 2002-10-16 | 2009-10-14 | The Scripps Research Institute | Glycoprotein synthesis |
| US7241282B2 (en) * | 2002-11-08 | 2007-07-10 | The Brigham And Women's Hospital, Inc. | Compositions and methods for prolonging survival of platelets |
| US7332299B2 (en) | 2003-02-20 | 2008-02-19 | Glycofi, Inc. | Endomannosidases in the modification of glycoproteins in eukaryotes |
| ES2420581T3 (es) * | 2003-03-14 | 2013-08-26 | Biogenerix Gmbh | Polímeros solubles en agua ramificados y sus conjugados |
| WO2006127896A2 (en) * | 2005-05-25 | 2006-11-30 | Neose Technologies, Inc. | Glycopegylated factor ix |
| WO2004091499A2 (en) * | 2003-04-09 | 2004-10-28 | Neose Technologies, Inc. | Intracellular formation of peptide conjugates |
| CA2520970A1 (en) * | 2003-04-09 | 2004-10-28 | Robert G. Schaub | Hemophilia treatment by inhalation of coagulation factors |
| US8791070B2 (en) | 2003-04-09 | 2014-07-29 | Novo Nordisk A/S | Glycopegylated factor IX |
| CA2524936A1 (en) | 2003-05-09 | 2004-12-02 | Neose Technologies, Inc. | Compositions and methods for the preparation of human growth hormone glycosylation mutants |
| WO2005012484A2 (en) | 2003-07-25 | 2005-02-10 | Neose Technologies, Inc. | Antibody-toxin conjugates |
| WO2005014655A2 (en) | 2003-08-08 | 2005-02-17 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyalkyl starch and a protein |
| US20090226397A1 (en) * | 2003-10-24 | 2009-09-10 | Nora Therapeutics, Inc. | Compositions and methods for reducing the likelihood of implantation failure or miscarriage in recipients of artificial insemination |
| US8338373B2 (en) * | 2003-10-24 | 2012-12-25 | Nora Therapeutics, Inc. | Method for reducing the risk of spontaneous abortion in a human female subject |
| EP2198875A1 (en) * | 2003-10-24 | 2010-06-23 | Nora, LLC | A method for reducing the likelihood of preterm labour in a subject in need thereof |
| US7507573B2 (en) * | 2003-11-14 | 2009-03-24 | Vib, Vzw | Modification of protein glycosylation in methylotrophic yeast |
| WO2005051429A2 (en) * | 2003-11-19 | 2005-06-09 | Government Of The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Targeted conjugates with a modified saccharide linker |
| WO2005051327A2 (en) * | 2003-11-24 | 2005-06-09 | Neose Technologies, Inc. | Glycopegylated erythropoietin |
| US20080305992A1 (en) | 2003-11-24 | 2008-12-11 | Neose Technologies, Inc. | Glycopegylated erythropoietin |
| US8633157B2 (en) * | 2003-11-24 | 2014-01-21 | Novo Nordisk A/S | Glycopegylated erythropoietin |
| WO2005054275A2 (en) | 2003-12-01 | 2005-06-16 | Novo Nordisk Health Care Ag | Nanofiltration of factor vii solutions to remove virus |
| US20080318850A1 (en) * | 2003-12-03 | 2008-12-25 | Neose Technologies, Inc. | Glycopegylated Factor Ix |
| US20060040856A1 (en) | 2003-12-03 | 2006-02-23 | Neose Technologies, Inc. | Glycopegylated factor IX |
| US7956032B2 (en) | 2003-12-03 | 2011-06-07 | Novo Nordisk A/S | Glycopegylated granulocyte colony stimulating factor |
| CA2549409C (en) * | 2003-12-03 | 2013-10-29 | Neose Technologies, Inc. | Glycopegylated granulocyte colony stimulating factor |
| CA2549413A1 (en) * | 2003-12-03 | 2005-06-23 | Neose Technologies, Inc. | Glycopegylated factor ix |
| ATE507822T1 (de) | 2003-12-19 | 2011-05-15 | Novo Nordisk Healthcare Ag | Stabilisierte zusammensetzungen von faktor-vii- polypeptiden |
| EP1765853B1 (en) * | 2004-01-08 | 2015-10-28 | ratiopharm GmbH | O-linked glycosylation of G-CSF peptides |
| NZ548308A (en) * | 2004-01-26 | 2010-11-26 | Biogenerix Ag | Branched polymeric sugars and nucleotides thereof |
| EP1720892B1 (en) * | 2004-01-26 | 2013-07-24 | BioGeneriX AG | Branched polymer-modified sugars and nucleotides |
| BRPI0507159A (pt) * | 2004-02-02 | 2007-06-26 | Ambrx Inc | polipeptìdeos em feixe de quatro hélices humanos modificados e seus usos |
| CA2554089C (en) * | 2004-02-09 | 2013-10-22 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| TW200539855A (en) * | 2004-03-15 | 2005-12-16 | Wyeth Corp | Calicheamicin conjugates |
| CA2560289A1 (en) * | 2004-03-23 | 2005-10-13 | Amgen Inc. | Chemically modified protein compositions and methods |
| DE602005019038D1 (de) | 2004-05-04 | 2010-03-11 | Novo Nordisk Healthcare Ag | O-verknüpfte glykoformen von faktor vii und verfahren zu deren herstellung |
| US20050287639A1 (en) | 2004-05-17 | 2005-12-29 | California Institute Of Technology | Methods of incorporating amino acid analogs into proteins |
| US7579444B2 (en) * | 2004-06-30 | 2009-08-25 | Nektar Therapeutics Al, Corporation | Polymer-factor IX moiety conjugates |
| AU2014280936B2 (en) * | 2004-06-30 | 2016-12-15 | Nektar Therapeutics | Polymer-factor ix moiety conjugates |
| WO2006010143A2 (en) * | 2004-07-13 | 2006-01-26 | Neose Technologies, Inc. | Branched peg remodeling and glycosylation of glucagon-like peptide-1 [glp-1] |
| WO2006020372A2 (en) * | 2004-07-23 | 2006-02-23 | Neose Technologies, Inc. | Enzymatic modification of glycopeptides |
| US20120225044A9 (en) * | 2004-09-07 | 2012-09-06 | Zymequest, Inc. | Compositions and methods for prolonging survival of platelets |
| AU2005282449B2 (en) * | 2004-09-07 | 2011-07-14 | Velico Medical, Inc. | Apparatus for prolonging survival of platelets |
| US8268967B2 (en) | 2004-09-10 | 2012-09-18 | Novo Nordisk A/S | Glycopegylated interferon α |
| WO2006041641A2 (en) * | 2004-10-05 | 2006-04-20 | Genentech, Inc. | Therapeutic agents with decreased toxicity |
| JP5123663B2 (ja) * | 2004-10-15 | 2013-01-23 | ベリコ メディカル インコーポレイティッド | 血小板の生存延長のための組成物および方法 |
| US20080064059A1 (en) | 2004-10-20 | 2008-03-13 | Scripps Research Institute | In Vivo Site-Specific Incorporation of N-Acetyl-Galactosamine Amino Acids in Eubacteria |
| CA2585758C (en) | 2004-10-29 | 2017-08-01 | Neose Technologies, Inc. | Remodeling and glycopegylation of fibroblast growth factor (fgf) |
| WO2006066258A2 (en) * | 2004-12-17 | 2006-06-22 | Neose Technologies, Inc. | Lipoconjugation of peptides |
| PL1831242T3 (pl) | 2004-12-23 | 2013-04-30 | Novo Nordisk Healthcare Ag | Zmniejszanie zawartości zanieczyszczeń białkowych w kompozycjach zawierających zależne od witaminy K białko będące przedmiotem zainteresowania |
| WO2006074279A1 (en) * | 2005-01-06 | 2006-07-13 | Neose Technologies, Inc. | Glycoconjugation using saccharyl fragments |
| ES2449195T3 (es) | 2005-01-10 | 2014-03-18 | Ratiopharm Gmbh | Factor estimulante de colonias de granulocitos glicopegilado |
| WO2006079155A1 (en) * | 2005-01-25 | 2006-08-03 | Apollo Life Sciences Limited | Molecules and chimeric molecules thereof |
| JP2008529505A (ja) * | 2005-02-15 | 2008-08-07 | アポロ ライフ サイエンシズ リミテッド | 分子およびそのキメラ分子 |
| JP2008532966A (ja) * | 2005-03-11 | 2008-08-21 | フレゼニウス・カビ・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | 不活性出発物質からの生理活性糖タンパク質の生成 |
| WO2006102652A2 (en) * | 2005-03-24 | 2006-09-28 | Neose Technologies, Inc. | Expression of soluble, active eukaryotic glycosyltransferases in prokaryotic organisms |
| EP1866427A4 (en) * | 2005-03-30 | 2010-09-01 | Novo Nordisk As | METHOD OF MANUFACTURING PEPTIDES PRODUCED IN INSECT CELL LINES |
| EP1876441B1 (en) * | 2005-03-31 | 2010-10-06 | The Noguchi Institute | Analysis method for biological sample and screening method for disease marker |
| EP1871795A4 (en) | 2005-04-08 | 2010-03-31 | Biogenerix Ag | COMPOSITIONS AND METHOD FOR PRODUCING GLYCOSYLATION MUTANTS OF A PROTEASE-RESISTANT HUMAN GROWTH HORMONE |
| US20060246523A1 (en) | 2005-04-28 | 2006-11-02 | Christopher Bieniarz | Antibody conjugates |
| US20110003744A1 (en) * | 2005-05-25 | 2011-01-06 | Novo Nordisk A/S | Glycopegylated Erythropoietin Formulations |
| WO2006127910A2 (en) * | 2005-05-25 | 2006-11-30 | Neose Technologies, Inc. | Glycopegylated erythropoietin formulations |
| JP5335422B2 (ja) | 2005-06-17 | 2013-11-06 | ノボ ノルディスク ヘルス ケア アクチェンゲゼルシャフト | 少なくとも1つの非天然のシステインを含んでいる操作されたタンパク質の選択的な還元および誘導体化 |
| US7696318B2 (en) | 2005-07-13 | 2010-04-13 | Novo Nordisk Health Care Ag | Host cell protein knock-out cells for production of therapeutic proteins |
| EP1937306B1 (en) * | 2005-08-19 | 2016-02-17 | Janssen Biotech, Inc. | Proteolysis resistant antibody preparations |
| US20070105755A1 (en) * | 2005-10-26 | 2007-05-10 | Neose Technologies, Inc. | One pot desialylation and glycopegylation of therapeutic peptides |
| US20090305967A1 (en) * | 2005-08-19 | 2009-12-10 | Novo Nordisk A/S | Glycopegylated factor vii and factor viia |
| JP5894722B2 (ja) | 2005-09-01 | 2016-03-30 | ノボ ノルディスク ヘルス ケア アーゲー | 第vii因子ポリペプチドの疎水性相互作用クロマトグラフィ精製 |
| US20090055942A1 (en) | 2005-09-14 | 2009-02-26 | Novo Nordisk Healthcare A/G | Human Coagulation Factor VII Polypeptides |
| AU2006304856A1 (en) * | 2005-10-21 | 2007-04-26 | Synageva Biopharma Corp. | Glycolated and glycosylated poultry derived therapeutic proteins |
| WO2007056191A2 (en) * | 2005-11-03 | 2007-05-18 | Neose Technologies, Inc. | Nucleotide sugar purification using membranes |
| US8247530B2 (en) * | 2005-11-08 | 2012-08-21 | Palatin Technologies, Inc. | N-alkylated cyclic peptide melanocortin agonists |
| US20070117153A1 (en) | 2005-11-23 | 2007-05-24 | Christopher Bieniarz | Molecular conjugate |
| EP1816201A1 (en) * | 2006-02-06 | 2007-08-08 | CSL Behring GmbH | Modified coagulation factor VIIa with extended half-life |
| DK1991273T4 (da) | 2006-02-10 | 2022-02-07 | Life Technologies Corp | Mærkning og påvisning af post-translationelt modificerede proteiner |
| CA2647632C (en) | 2006-03-27 | 2017-06-27 | University Of Maryland Biotechnology Institute | Glycoprotein synthesis and remodeling by enzymatic transglycosylation |
| US7985839B2 (en) * | 2006-03-31 | 2011-07-26 | Baxter International Inc. | Factor VIII polymer conjugates |
| US7645860B2 (en) * | 2006-03-31 | 2010-01-12 | Baxter Healthcare S.A. | Factor VIII polymer conjugates |
| KR20080108147A (ko) | 2006-03-31 | 2008-12-11 | 백스터 인터내셔널 인코포레이티드 | 페질화된 인자 viii |
| US7982010B2 (en) * | 2006-03-31 | 2011-07-19 | Baxter International Inc. | Factor VIII polymer conjugates |
| JP2009533347A (ja) | 2006-04-07 | 2009-09-17 | ネクター セラピューティックス エイエル,コーポレイション | 抗TNFα抗体の複合体 |
| US8198045B2 (en) | 2006-04-19 | 2012-06-12 | Biogenerix Ag | Expression of O-glycosylated therapeutic proteins in prokaryotic microorganisms |
| WO2007130453A2 (en) | 2006-05-02 | 2007-11-15 | Allozyne, Inc. | Non-natural amino acid substituted polypeptides |
| AU2007258609B2 (en) * | 2006-06-07 | 2013-01-24 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| US20100173323A1 (en) * | 2006-06-09 | 2010-07-08 | University Of Maryland, Baltimore | Glycosylation engineered antibody therapy |
| AR078117A1 (es) | 2006-06-20 | 2011-10-19 | Protech Pharma S A | Una muteina recombinante del interferon alfa humano glicosilado, un gen que codifica para dicha muteina, un metodo de produccion de dicho gen, un metodo para obtener una celula eucariota productora de dicha muteina, un metodo para producir dicha muteina, un procedimiento para purificar dicha muteina |
| US9187532B2 (en) | 2006-07-21 | 2015-11-17 | Novo Nordisk A/S | Glycosylation of peptides via O-linked glycosylation sequences |
| EP2630972B1 (en) | 2006-07-25 | 2017-08-30 | Lipoxen Technologies Limited | N-terminal polysialylation |
| WO2008057683A2 (en) * | 2006-10-03 | 2008-05-15 | Novo Nordisk A/S | Methods for the purification of polypeptide conjugates |
| PT2068907T (pt) * | 2006-10-04 | 2018-01-15 | Novo Nordisk As | Açúcares e glicopéptidos peguilados ligados a glicerol |
| WO2008073620A2 (en) * | 2006-11-02 | 2008-06-19 | Neose Technologies, Inc. | Manufacturing process for the production of polypeptides expressed in insect cell-lines |
| KR101248252B1 (ko) | 2006-11-28 | 2013-03-27 | 한올바이오파마주식회사 | 변형된 에리스로포이에틴 폴리펩티드와 이의 치료용 용도 |
| EP3323430A1 (en) | 2006-12-15 | 2018-05-23 | Baxalta GmbH | Factor viia-(poly)sialic acid conjugate having prolonged in vivo half-life |
| LT2457920T (lt) | 2007-01-18 | 2018-02-12 | Genzyme Corporation | Oligosacharidai, apimantys aminooksigrupę, ir jų konjugatai |
| WO2008089339A2 (en) * | 2007-01-18 | 2008-07-24 | Genzyme Corporation | Oligosaccharide conjugates for cellular targeting |
| US7960139B2 (en) | 2007-03-23 | 2011-06-14 | Academia Sinica | Alkynyl sugar analogs for the labeling and visualization of glycoconjugates in cells |
| PL2068909T3 (pl) * | 2007-03-30 | 2012-09-28 | Ambrx Inc | Modyfikowane polipeptydy fgf-21 i ich zastosowanie |
| PL2144923T3 (pl) | 2007-04-03 | 2013-12-31 | Biogenerix Ag | Sposoby leczenia z użyciem glikopegilowanego G-CSF |
| WO2008124706A2 (en) | 2007-04-06 | 2008-10-16 | Arizona Board Of Regents Acting For And On Behalf Of Arizona State University | Devices and methods for target molecule characterization |
| US20090053167A1 (en) * | 2007-05-14 | 2009-02-26 | Neose Technologies, Inc. | C-, S- and N-glycosylation of peptides |
| EP2170919B8 (en) * | 2007-06-12 | 2016-01-20 | ratiopharm GmbH | Improved process for the production of nucleotide sugars |
| US20100268055A1 (en) * | 2007-07-19 | 2010-10-21 | Arizona Board of Regents, a body corporate acting for and on behalf of Arizona State University | Self-Anchoring MEMS Intrafascicular Neural Electrode |
| KR20100058541A (ko) | 2007-08-15 | 2010-06-03 | 아뮤닉스 인코포레이티드 | 생물학적 활성 폴리펩티드의 특성을 변경하기 위한 조성물 및 방법 |
| CN104689333B (zh) | 2007-08-27 | 2018-05-29 | 拉蒂奥法姆有限责任公司 | G-csf 缀合物的液体制剂 |
| US8207112B2 (en) | 2007-08-29 | 2012-06-26 | Biogenerix Ag | Liquid formulation of G-CSF conjugate |
| KR20100095441A (ko) * | 2007-11-09 | 2010-08-30 | 백스터 인터내셔널 인코포레이티드 | 변형된 재조합 인자 ⅷ 및 폰 빌레브란트 인자 및 사용 방법 |
| KR101343485B1 (ko) * | 2008-01-07 | 2013-12-20 | 시나게바 바이오파르마, 코포레이션 | 조류에서의 당화 |
| US20100286067A1 (en) * | 2008-01-08 | 2010-11-11 | Biogenerix Ag | Glycoconjugation of polypeptides using oligosaccharyltransferases |
| MX2010009154A (es) * | 2008-02-27 | 2010-09-09 | Novo Nordisk As | Moleculas conjugadas del factor viii. |
| US8961757B2 (en) | 2008-03-18 | 2015-02-24 | Arizona Board Of Regents, A Body Corporate Of The State Of Arizona Acting For And On Behalf Of Arizona State University | Nanopore and carbon nanotube based DNA sequencer |
| WO2009117522A2 (en) | 2008-03-18 | 2009-09-24 | Reinhart, Kevin | Nanopore and carbon nanotube based dna sequencer and a serial recognition sequencer |
| CA2728708A1 (en) * | 2008-06-25 | 2009-12-30 | Bayer Healthcare Llc | Factor viii muteins with reduced immunogenicity |
| WO2010009271A2 (en) | 2008-07-15 | 2010-01-21 | Academia Sinica | Glycan arrays on ptfe-like aluminum coated glass slides and related methods |
| WO2010042514A1 (en) | 2008-10-06 | 2010-04-15 | Arizona Board Of Regents | Nanopore and carbon nanotube based dna sequencer and a serial recognition elements |
| EP2465542B1 (en) | 2008-12-16 | 2015-01-21 | Genzyme Corporation | Oligosaccharide-protein conjugates |
| CA2750262C (en) | 2009-01-28 | 2016-08-09 | Smartcells, Inc. | Conjugate based systems for controlled drug delivery |
| CN108530543B (zh) | 2009-02-03 | 2023-06-23 | 阿穆尼克斯制药公司 | 延伸重组多肽和包含该延伸重组多肽的组合物 |
| US9238878B2 (en) | 2009-02-17 | 2016-01-19 | Redwood Bioscience, Inc. | Aldehyde-tagged protein-based drug carriers and methods of use |
| EP2410846B1 (en) | 2009-03-25 | 2016-09-07 | Seneb Biosciences, Inc. | Glycolipids as treatment for disease |
| ES2427627T3 (es) | 2009-04-06 | 2013-10-31 | Novo Nordisk A/S | Entrega dirigida de proteínas de Factor VIII a plaquetas |
| JP6091892B2 (ja) * | 2009-06-16 | 2017-03-08 | グライセラ・リミテッド | グリコシル化に関連する物質および方法 |
| AU2010281453C1 (en) * | 2009-07-27 | 2019-08-15 | Takeda Pharmaceutical Company Limited | Blood coagulation protein conjugates |
| US8809501B2 (en) | 2009-07-27 | 2014-08-19 | Baxter International Inc. | Nucleophilic catalysts for oxime linkage |
| EP2459226B1 (en) | 2009-07-27 | 2016-06-29 | Lipoxen Technologies Limited | Glycopolysialylation of proteins other than blood coagulation proteins |
| US8642737B2 (en) | 2010-07-26 | 2014-02-04 | Baxter International Inc. | Nucleophilic catalysts for oxime linkage |
| SG10201401194VA (en) | 2009-07-27 | 2014-07-30 | Lipoxen Technologies Ltd | Glycopolysialylation of non-blood coagulation proteins |
| WO2011018515A1 (en) | 2009-08-14 | 2011-02-17 | Novo Nordisk Health Care Ag | Method of purifying pegylated proteins |
| JP2013502458A (ja) | 2009-08-24 | 2013-01-24 | アムニクス オペレーティング インコーポレイテッド | 凝固第vii因子組成物ならびにそれを製造および使用する方法 |
| GB0915403D0 (en) * | 2009-09-04 | 2009-10-07 | London School Hygiene & Tropical Medicine | Protein glycosylation |
| CA2777226A1 (en) | 2009-10-12 | 2011-04-21 | Pfizer Inc. | Cancer treatment |
| EP2504349B1 (en) | 2009-11-24 | 2020-02-26 | Novo Nordisk Health Care AG | Method of purifying pegylated proteins |
| EP2507267B1 (en) * | 2009-12-02 | 2016-09-14 | Acceleron Pharma, Inc. | Compositions and methods for increasing serum half-life of fc fusion proteins |
| US10087236B2 (en) * | 2009-12-02 | 2018-10-02 | Academia Sinica | Methods for modifying human antibodies by glycan engineering |
| US11377485B2 (en) | 2009-12-02 | 2022-07-05 | Academia Sinica | Methods for modifying human antibodies by glycan engineering |
| CN105153313A (zh) | 2010-02-16 | 2015-12-16 | 诺沃—诺迪斯克有限公司 | 因子viii融合蛋白 |
| CN105524164A (zh) | 2010-02-16 | 2016-04-27 | 诺沃—诺迪斯克有限公司 | 具有降低的vwf结合的因子viii分子 |
| RU2573909C2 (ru) | 2010-03-04 | 2016-01-27 | Пфенекс Инк. | Способ получения растворимого рекомбинантного белка интерферона без денатурирования |
| EP2552949B1 (en) | 2010-04-01 | 2016-08-17 | Pfenex Inc. | Methods for g-csf production in a pseudomonas host cell |
| WO2011130332A1 (en) | 2010-04-12 | 2011-10-20 | Academia Sinica | Glycan arrays for high throughput screening of viruses |
| US8962345B2 (en) * | 2010-05-21 | 2015-02-24 | The United States Of America As Represented By The Secretary Of Commerce | Method of characterizing glycans attached to glycoproteins |
| TW201212938A (en) | 2010-06-30 | 2012-04-01 | Novo Nordisk As | Antibodies that are capable of specifically binding tissue factor pathway inhibitor |
| EA037095B1 (ru) | 2010-07-09 | 2021-02-05 | Биовератив Терапьютикс Инк. | Способ лечения гемофилии в и эпизодов кровотечения |
| US20130183280A1 (en) | 2010-07-15 | 2013-07-18 | Novo Nordisk A/S | Stabilized factor viii variants |
| WO2012015692A2 (en) * | 2010-07-28 | 2012-02-02 | Smartcells, Inc. | Recombinantly expressed insulin polypeptides and uses thereof |
| WO2012013823A2 (en) | 2010-07-30 | 2012-02-02 | Glycode | A yeast artificial chromosome carrying the mammalian glycosylation pathway |
| PT2415779E (pt) | 2010-08-02 | 2015-05-13 | Ratiopharm Gmbh | Processo de produção e purificação de uma sialiltransferase solúvel activa |
| WO2012032181A2 (en) | 2010-09-10 | 2012-03-15 | Allozyne, Inc | Novel antibody derivatives |
| EP3466968A1 (en) | 2010-09-15 | 2019-04-10 | Stichting Sanquin Bloedvoorziening | Factor viii variants having a decreased cellular uptake |
| JP2013542188A (ja) | 2010-09-22 | 2013-11-21 | ノヴォ ノルディスク アー/エス | 治療用第viii因子抗体 |
| EP2635607B1 (en) | 2010-11-05 | 2019-09-04 | Zymeworks Inc. | Stable heterodimeric antibody design with mutations in the fc domain |
| KR20130125789A (ko) | 2010-12-16 | 2013-11-19 | 노보 노르디스크 에이/에스 | 수성 인자ⅷ 용액 |
| AU2011349574B9 (en) | 2010-12-22 | 2017-06-15 | Takeda Pharmaceutical Company Limited | Materials and methods for conjugating a water soluble fatty acid derivative to a protein |
| AU2012204241A1 (en) * | 2011-01-06 | 2013-06-06 | The Johns Hopkins University | Method of production of recombinant glycoproteins with increased circulatory half-life in mammalian cells |
| BR112013017980A2 (pt) | 2011-01-14 | 2017-06-27 | Redwood Bioscience Inc | polipeptídeos de imunoglobulina marcado com aldeído e método de uso dos mesmos |
| CA2827732A1 (en) * | 2011-02-25 | 2012-08-30 | Merck Sharp & Dohme Corp. | Yeast strain for the production of proteins with modified o-glycosylation |
| JP6215056B2 (ja) | 2011-03-01 | 2017-10-18 | ノヴォ ノルディスク アー/エス | 拮抗性dr3リガンド |
| WO2012117091A1 (en) | 2011-03-02 | 2012-09-07 | Novo Nordisk A/S | Coagulation factor-targeting to tlt-1 on activated platelets |
| US9499605B2 (en) | 2011-03-03 | 2016-11-22 | Zymeworks Inc. | Multivalent heteromultimer scaffold design and constructs |
| EP2895592A1 (en) | 2011-03-23 | 2015-07-22 | Société Industrielle Limousine d'Application Biologique (SILAB) | A yeast recombinant cell capable of producing gdp-fucose |
| EP3536708A1 (en) | 2011-04-19 | 2019-09-11 | Pfizer Inc | Combinations of anti-4-1bb antibodies and adcc-inducing antibodies for the treatment of cancer |
| JP6148228B2 (ja) | 2011-05-18 | 2017-06-14 | ユーメデリス ファーマシューティカルズ,インク. | 改善されたペプチド製剤 |
| KR20230023059A (ko) * | 2011-05-18 | 2023-02-16 | 메더리스 다이어비티즈, 엘엘씨 | 인슐린 저항성에 대한 개선된 펩티드 제약 |
| WO2012164034A1 (en) | 2011-05-31 | 2012-12-06 | Probiogen Ag | Methods for preparation of fucose-linked site specific conjugates of proteins with toxins, adjuvants, detection labels and pharmacokinetic half life extenders |
| US8883982B2 (en) | 2011-06-08 | 2014-11-11 | Acceleron Pharma, Inc. | Compositions and methods for increasing serum half-life |
| LT2717898T (lt) | 2011-06-10 | 2019-03-25 | Bioverativ Therapeutics Inc. | Pro-koagulianto junginiai ir jų gavimo būdai |
| EP2717917B1 (en) | 2011-07-05 | 2016-04-20 | biOasis Technologies Inc | P97-antibody conjugates |
| AU2012294673B2 (en) | 2011-08-05 | 2015-11-26 | Bioasis Technologies Inc. | p97 fragments with transfer activity |
| MY167234A (en) | 2011-09-23 | 2018-08-14 | Novo Nordisk As | Novel glucagon analogues |
| CA2850469C (en) * | 2011-10-01 | 2020-07-07 | Glytech, Inc. | Glycosylated polypeptide and pharmaceutical composition containing same |
| KR101456174B1 (ko) * | 2011-10-18 | 2014-11-03 | 한국생명공학연구원 | 용해도가 증가된 비―퀴논 젤다나마이신 당전이 유도체 또는 이의 약학적으로 허용가능한 염, 이의 제조방법 및 이의 열충격 단백질(Hsp90) 에이티피아제(ATPase) 활성 저해용 약학적 조성물 |
| WO2013058582A2 (ko) * | 2011-10-18 | 2013-04-25 | 한국생명공학연구원 | 당전이 효소를 이용한 안사마이신 배당체의 제조방법 |
| US9034829B1 (en) * | 2011-10-27 | 2015-05-19 | Northwestern University | pH-sensitive polymer-drug conjugates for targeted delivery of therapeutics |
| CN109897103B (zh) | 2011-11-04 | 2024-05-17 | 酵活英属哥伦比亚有限公司 | 在Fc结构域中具有突变的稳定异源二聚的抗体设计 |
| HUE043537T2 (hu) | 2012-02-15 | 2019-08-28 | Bioverativ Therapeutics Inc | Rekombináns VIII faktor fehérjék |
| KR102097263B1 (ko) | 2012-02-15 | 2020-04-06 | 바이오버라티브 테라퓨틱스 인크. | Viii 인자 조성물 및 이를 제조하고 사용하는 방법 |
| GB2501611B (en) | 2012-02-21 | 2015-03-04 | Cytonics Corp | Alpha-2-macroglobulin compositions and therapeutic uses |
| US10130714B2 (en) | 2012-04-14 | 2018-11-20 | Academia Sinica | Enhanced anti-influenza agents conjugated with anti-inflammatory activity |
| JP2015519313A (ja) | 2012-04-24 | 2015-07-09 | ノヴォ ノルディスク アー/エス | 血友病の治療に適する医薬組成物 |
| WO2013083858A1 (en) | 2012-04-24 | 2013-06-13 | Novo Nordisk A/S | Compounds suitable for treatment of haemophilia |
| WO2014004586A1 (en) | 2012-06-25 | 2014-01-03 | Zymeworks Inc. | Process and methods for efficient manufacturing of highly pure asymmetric antibodies in mammalian cells |
| CN102786030B (zh) * | 2012-07-06 | 2014-03-12 | 江苏大学 | 一种通过溶剂处理制备多肽纳米薄膜的方法 |
| WO2014012082A2 (en) | 2012-07-13 | 2014-01-16 | Zymeworks Inc. | Multivalent heteromultimer scaffold design an constructs |
| CA3140358A1 (en) | 2012-07-31 | 2014-02-06 | Bioasis Technologies, Inc. | Dephosphorylated lysosomal storage disease proteins and methods of use thereof |
| JP6302909B2 (ja) | 2012-08-18 | 2018-03-28 | アカデミア シニカAcademia Sinica | シアリダーゼの同定および画像化のための細胞透過性プローブ |
| JP6321658B2 (ja) | 2012-09-24 | 2018-05-09 | メディミューン リミテッド | アミノ酸誘導体 |
| WO2014059144A1 (en) | 2012-10-10 | 2014-04-17 | Arizona Board Of Regents Acting For And On Behalf Of Arizona State University | Systems and devices for molecule sensing and method of manufacturing thereof |
| CN104717973A (zh) | 2012-10-10 | 2015-06-17 | 诺和诺德保健Ag(股份有限公司) | 因子vii多肽的液体药物组合物 |
| JP2015533152A (ja) | 2012-10-15 | 2015-11-19 | ノヴォ・ノルディスク・ヘルス・ケア・アーゲー | 第vii因子コンジュゲート |
| EP2906693A1 (en) | 2012-10-15 | 2015-08-19 | Novo Nordisk Health Care AG | Coagulation factor vii polypeptides |
| EP2922876B1 (en) | 2012-11-20 | 2018-10-10 | Mederis Diabetes, LLC | Improved peptide pharmaceuticals for insulin resistance |
| CA3157899A1 (en) | 2012-11-20 | 2014-05-30 | Mederis Diabetes, Llc | Improved peptide pharmaceuticals |
| US9914785B2 (en) | 2012-11-28 | 2018-03-13 | Zymeworks Inc. | Engineered immunoglobulin heavy chain-light chain pairs and uses thereof |
| WO2014088836A1 (en) | 2012-12-03 | 2014-06-12 | Merck Sharp & Dohme Corp. | O-glycosylated carboxy terminal portion (ctp) peptide-based insulin and insulin analogues |
| CN103044509B (zh) * | 2013-01-05 | 2015-04-01 | 宁辉 | γ-胞嘧啶核苷-5’-三磷酸二钠结晶化合物、其制备方法及其药物组合物 |
| EP4063389A3 (en) * | 2013-03-11 | 2022-12-28 | Genzyme Corporation | Site-specific antibody-drug conjugation through glycoengineering |
| BR112015022416A2 (pt) | 2013-03-13 | 2017-10-24 | Bioasis Technologies Inc | fragmentos de p97 e seus usos |
| WO2014140240A1 (en) | 2013-03-15 | 2014-09-18 | Novo Nordisk A/S | Antibodies capable of specifically binding two epitopes on tissue factor pathway inhibitor |
| CN105307672B (zh) | 2013-04-18 | 2021-01-05 | 诺和诺德股份有限公司 | 用于医学用途的稳定、延长的glp-1/胰高血糖素受体共激动剂 |
| SI2991683T1 (sl) * | 2013-05-02 | 2020-01-31 | Glykos Finland Oy | Konjugati glikoproteina ali glikana s toksično obremenitvijo |
| AU2014262566A1 (en) | 2013-05-08 | 2015-11-12 | Zymeworks Inc. | Bispecific HER2 and HER3 antigen binding constructs |
| EP3013365B1 (en) | 2013-06-26 | 2019-06-05 | Academia Sinica | Rm2 antigens and use thereof |
| EP3013347B1 (en) | 2013-06-27 | 2019-12-11 | Academia Sinica | Glycan conjugates and use thereof |
| US10548953B2 (en) | 2013-08-14 | 2020-02-04 | Bioverativ Therapeutics Inc. | Factor VIII-XTEN fusions and uses thereof |
| WO2015031654A2 (en) | 2013-08-28 | 2015-03-05 | Cytonics Corporation | Systems, compositions, and methods for transplantation and treating conditions |
| KR102298172B1 (ko) | 2013-09-06 | 2021-09-06 | 아카데미아 시니카 | 변화된 글리코실 그룹을 갖는 당지질을 사용한 인간 iNKT 세포 활성화 |
| RU2676307C2 (ru) | 2013-10-04 | 2018-12-27 | Мерк Шарп И Доум Корп. | Чувствительные к глюкозе конъюгаты инсулина |
| EP3058083B1 (en) | 2013-10-14 | 2018-04-11 | SynAffix B.V. | Modified glycoprotein, protein-conjugate and process for the preparation thereof |
| EP3057618B1 (en) * | 2013-10-14 | 2022-12-14 | SynAffix B.V. | Glycoengineered antibody, antibody-conjugate and methods for their preparation |
| CN105637088A (zh) | 2013-10-15 | 2016-06-01 | 诺和诺德保健股份有限公司 | 凝固因子vii多肽 |
| WO2015057622A1 (en) | 2013-10-16 | 2015-04-23 | Momenta Pharmaceuticals, Inc. | Sialylated glycoproteins |
| US10150818B2 (en) | 2014-01-16 | 2018-12-11 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| WO2015109180A2 (en) | 2014-01-16 | 2015-07-23 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US20170009266A1 (en) * | 2014-01-24 | 2017-01-12 | Synaffix B.V. | Process for the attachment of a galnac moiety comprising a (hetero)aryl group to a glcnac moiety, and product obtained thereby |
| WO2015112016A1 (en) | 2014-01-24 | 2015-07-30 | Synaffix B.V. | Process for the cycloaddition of a halogenated 1,3-dipole compound with a (hetero)cycloalkyne |
| US11168085B2 (en) | 2014-01-24 | 2021-11-09 | Synaffix B.V. | Process for the cycloaddition of a hetero(aryl) 1,3-dipole compound with a (hetero)cycloalkyne |
| AU2015210612B2 (en) | 2014-02-03 | 2020-04-09 | Bioasis Technologies Inc. | P97 fusion proteins |
| AR099340A1 (es) * | 2014-02-12 | 2016-07-13 | Novo Nordisk As | Conjugados del factor de coagulación ix |
| WO2015126729A1 (en) | 2014-02-19 | 2015-08-27 | Bioasis Technologies, Inc. | P97-ids fusion proteins |
| US10188739B2 (en) | 2014-02-27 | 2019-01-29 | Xenetic Biosciences, Inc. | Compositions and methods for administering insulin or insulin-like protein to the brain |
| TWI687428B (zh) | 2014-03-27 | 2020-03-11 | 中央研究院 | 反應性標記化合物及其用途 |
| JP6750148B2 (ja) * | 2014-04-25 | 2020-09-02 | 公益財団法人野口研究所 | 糖鎖切断抗体の製造方法及び均一糖鎖抗体 |
| US10058619B2 (en) | 2014-05-01 | 2018-08-28 | Bioasis Technologies, Inc. | P97-polynucleotide conjugates |
| TWI654202B (zh) | 2014-05-27 | 2019-03-21 | 中央研究院 | 增進抗體功效之通用糖型之組合物及方法 |
| EP4116329A1 (en) | 2014-05-27 | 2023-01-11 | Academia Sinica | Anti-her2 glycoantibodies and uses thereof |
| TWI670078B (zh) | 2014-05-27 | 2019-09-01 | 中央研究院 | 抗cd20醣抗體及其用途 |
| US10118969B2 (en) | 2014-05-27 | 2018-11-06 | Academia Sinica | Compositions and methods relating to universal glycoforms for enhanced antibody efficacy |
| CN106714829A (zh) | 2014-05-28 | 2017-05-24 | 中央研究院 | 抗TNF‑α醣抗体及其用途 |
| EP4397374A3 (en) | 2014-05-28 | 2024-10-16 | Mederis Diabetes, LLC | Improved peptide pharmaceuticals for insulin resistance |
| WO2015185640A1 (en) | 2014-06-04 | 2015-12-10 | Novo Nordisk A/S | Glp-1/glucagon receptor co-agonists for medical use |
| AU2015282627B2 (en) | 2014-06-30 | 2020-04-02 | Glykos Finland Oy | Saccharide derivative of a toxic payload and antibody conjugates thereof |
| JP6355842B2 (ja) * | 2014-07-10 | 2018-07-11 | アカデミア シニカAcademia Sinica | マルチドラッグ送達システム及びその使用 |
| EP3177317B1 (en) | 2014-08-04 | 2020-03-18 | CSL Limited | Factor viii formulation |
| CN107001404B (zh) | 2014-09-08 | 2021-06-29 | 中央研究院 | 使用醣脂激活人类iNKT细胞 |
| PL3204425T3 (pl) * | 2014-10-09 | 2021-03-08 | Genzyme Corporation | Glikomodyfikowane koniugaty przeciwciał z lekami |
| US10889631B2 (en) | 2014-11-20 | 2021-01-12 | Cytonics Corporation | Therapeutic variant alpha-2-macroglobulin compositions |
| WO2016091268A2 (en) | 2014-12-12 | 2016-06-16 | University Of Copenhagen | N-glycosylation |
| CA2972757A1 (en) | 2014-12-31 | 2016-07-07 | Arthur M. Krieg | Combination tumor immunotherapy |
| US9975965B2 (en) | 2015-01-16 | 2018-05-22 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US10495645B2 (en) | 2015-01-16 | 2019-12-03 | Academia Sinica | Cancer markers and methods of use thereof |
| KR102691114B1 (ko) | 2015-01-24 | 2024-08-01 | 아카데미아 시니카 | 신규한 글리칸 콘주게이트 및 이를 사용하는 방법 |
| SG11201706659WA (en) | 2015-03-06 | 2017-09-28 | Csl Behring Recombinant Facility Ag | Modified von willebrand factor having improved half-life |
| EA201890423A1 (ru) | 2015-08-03 | 2018-07-31 | Биовератив Терапьютикс Инк. | Слитые белки фактора ix, способы их получения и применения |
| AU2017205268B2 (en) | 2016-01-08 | 2022-02-17 | Ascendis Pharma Growth Disorders A/S | CNP prodrugs with large carrier moieties |
| IL293979B2 (en) | 2016-01-08 | 2024-08-01 | Ascendis Pharma Growth Disorders As | Controlled-release CNP agonists with increased NEP stability |
| AU2017205694B2 (en) | 2016-01-08 | 2022-05-26 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with low NPR-C binding |
| SMT202200466T1 (it) | 2016-01-08 | 2023-01-13 | Ascendis Pharma Growth Disorders As | Profarmaci di cnp con legame a supporto in corrispondenza del raggruppamento ciclico |
| IL259658B2 (en) * | 2016-01-08 | 2024-06-01 | Ascendis Pharma Growth Disorders As | Controlled-release CNP agonists with reduced side effects |
| CA3007982C (en) | 2016-01-08 | 2023-12-19 | Ascendis Pharma Growth Disorders A/S | Controlled-release cnp agonists with low initial npr-b activity |
| EP3426693A4 (en) | 2016-03-08 | 2019-11-13 | Academia Sinica | PROCESS FOR MODULAR SYNTHESIS OF N-GLYCANES AND ARRANGEMENTS THEREOF |
| MA45473A (fr) * | 2016-04-04 | 2019-02-13 | Shire Human Genetic Therapies | Inhibiteur de c1 estérase conjugué et ses utilisations |
| CA3019398A1 (en) | 2016-04-26 | 2017-11-02 | R.P. Scherer Technologies, Llc | Antibody conjugates and methods of making and using the same |
| DK3464318T3 (da) | 2016-06-02 | 2021-06-28 | Abbvie Inc | Glucocorticoidreceptoragonist og immunkonjugater deraf |
| US10538592B2 (en) | 2016-08-22 | 2020-01-21 | Cho Pharma, Inc. | Antibodies, binding fragments, and methods of use |
| CA3037448A1 (en) | 2016-09-29 | 2018-04-05 | Ascendis Pharma Growth Disorders A/S | Combination therapy with controlled-release cnp agonists |
| IL266972B2 (en) | 2016-12-02 | 2024-04-01 | Bioverativ Therapeutics Inc | Methods for the treatment of hemophilic arthritis with the help of chimeric blood coagulation factors |
| JP6852397B2 (ja) * | 2016-12-28 | 2021-03-31 | 株式会社島津製作所 | 分析用試料の調製方法および分析方法 |
| KR102294517B1 (ko) * | 2016-12-29 | 2021-08-27 | 재단법인 생물기술개발중심 | 당단백질-약물 컨주게이트를 제조하는 방법 |
| TW201837051A (zh) | 2017-02-08 | 2018-10-16 | 美商必治妥美雅史谷比公司 | 包含藥物動力學增強劑之經修飾之鬆弛素(relaxin)多肽及其用途 |
| HUE060983T2 (hu) | 2017-02-17 | 2023-05-28 | Denali Therapeutics Inc | Mesterségesen elõállított transferrin receptort kötõ polipeptidek |
| JP2020511499A (ja) | 2017-03-20 | 2020-04-16 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | 赤血球生成刺激タンパク質のインビトロでの糖鎖改変のための方法 |
| MA49578A (fr) | 2017-07-11 | 2021-04-07 | Synthorx Inc | Incorporation de nucléotides non naturels et procédés associés |
| EP3661955A4 (en) | 2017-08-03 | 2021-05-26 | Synthorx, Inc. | CYTOKINE CONJUGATES FOR THE TREATMENT OF AUTOIMMUNE DISEASES |
| EP3678687B1 (en) | 2017-09-04 | 2022-08-17 | 89Bio Ltd. | Mutant fgf-21 peptide conjugates and uses thereof |
| EP3713592A4 (en) | 2017-11-21 | 2022-01-12 | The Board of Trustees of the Leland Stanford Junior University | INTERLEUKIN-2 PARTIAL AGONISTS |
| AU2018374634A1 (en) | 2017-12-01 | 2020-05-28 | Abbvie Inc. | Glucocorticoid receptor agonist and immunoconjugates thereof |
| MX2020006999A (es) | 2018-01-03 | 2020-12-10 | Mederis Diabetes Llc | Farmaceuticos de peptidos mejorados para el tratamiento de nash y otros trastornos. |
| JP7511478B2 (ja) | 2018-04-09 | 2024-07-05 | チェックメイト ファーマシューティカルズ | ウイルス様粒子中へのオリゴヌクレオチドのパッケージ化 |
| CN112512555A (zh) | 2018-05-18 | 2021-03-16 | 比奥维拉迪维治疗股份有限公司 | 治疗血友病a的方法 |
| EP3796941A4 (en) * | 2018-05-30 | 2022-12-28 | Purdue Research Foundation | Targeting anabolic drugs for accelerated fracture repair |
| CN113301925A (zh) * | 2018-12-19 | 2021-08-24 | 小利兰·斯坦福大学理事会 | 用于溶酶体靶向的双官能分子以及相关的组合物和方法 |
| CA3127689A1 (en) | 2019-02-06 | 2020-08-13 | Synthorx, Inc. | Il-2 conjugates and methods of use thereof |
| JP2022538357A (ja) | 2019-07-04 | 2022-09-01 | ツェー・エス・エル・ベーリング・レングナウ・アクチエンゲゼルシャフト | 第VIII凝固因子のin vitroでの安定性を向上させるための切断型フォン・ヴィレブランド因子(VWF) |
| US20220348637A1 (en) | 2019-11-11 | 2022-11-03 | CSL Behring Lengnau AG | Polypeptides for inducing tolerance to factor viii |
| PE20230036A1 (es) | 2019-12-23 | 2023-01-10 | Denali Therapeutics Inc | Variantes de progranulina |
| IL294659A (en) | 2020-01-14 | 2022-09-01 | Synthekine Inc | Biased il2 muteins methods and compositions |
| KR20230042524A (ko) * | 2020-08-07 | 2023-03-28 | 주식회사 유틸렉스 | 항-her2 / 항-4-1bb 이중특이적 항체 및 이의 용도 |
| WO2022079211A1 (en) * | 2020-10-16 | 2022-04-21 | Adc Therapeutics Sa | Glycoconjugates |
| JP2023554215A (ja) * | 2020-10-16 | 2023-12-27 | ユニバーシティー オブ ジョージア リサーチ ファウンデーション,インコーポレイテッド | 糖共役体 |
| CN117202924A (zh) | 2020-12-07 | 2023-12-08 | 斯皮特弗尔制药有限责任公司 | 使用glp-1r和gcgr平衡激动剂降低血糖和/或体重的治疗方案和方法 |
| EP4267195A1 (en) * | 2020-12-24 | 2023-11-01 | Synaffix B.V. | Glycan-conjugated antibodies binding to fc-gamma receptor |
| CN113429443B (zh) * | 2021-07-08 | 2023-07-25 | 江南大学 | 一种双唾液酸-甘露寡糖复合物及其合成方法 |
| IL313686A (en) * | 2021-12-20 | 2024-08-01 | 89Bio Inc | Chemical synthesis of cytidine-5'-monophospho-N-glycyl-sialic acid |
| TW202442259A (zh) * | 2022-12-29 | 2024-11-01 | 嘉正生物科技股份有限公司 | 醣蛋白修飾方法 |
Family Cites Families (261)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3691016A (en) | 1970-04-17 | 1972-09-12 | Monsanto Co | Process for the preparation of insoluble enzymes |
| CA1023287A (en) | 1972-12-08 | 1977-12-27 | Boehringer Mannheim G.M.B.H. | Process for the preparation of carrier-bound proteins |
| GB1479268A (en) | 1973-07-05 | 1977-07-13 | Beecham Group Ltd | Pharmaceutical compositions |
| US4179337A (en) | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| DE2433883C2 (de) * | 1973-07-20 | 1986-03-27 | Research Corp., New York, N.Y. | Verwendung von physiologisch aktiven Polypeptiden |
| CH596313A5 (ja) | 1975-05-30 | 1978-03-15 | Battelle Memorial Institute | |
| US4385260A (en) * | 1975-09-09 | 1983-05-24 | Beckman Instruments, Inc. | Bargraph display |
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4342832A (en) | 1979-07-05 | 1982-08-03 | Genentech, Inc. | Method of constructing a replicable cloning vehicle having quasi-synthetic genes |
| IL63270A0 (en) | 1980-08-05 | 1981-10-30 | Univ Leland Stanford Junior | Eukaryotic autonomously replicating segment |
| US4414147A (en) | 1981-04-17 | 1983-11-08 | Massachusetts Institute Of Technology | Methods of decreasing the hydrophobicity of fibroblast and other interferons |
| JPS57206622A (en) | 1981-06-10 | 1982-12-18 | Ajinomoto Co Inc | Blood substitute |
| US4579821A (en) | 1981-11-23 | 1986-04-01 | University Patents, Inc. | Control of DNA sequence transcription |
| US4656134A (en) | 1982-01-11 | 1987-04-07 | Board Of Trustees Of Leland Stanford Jr. University | Gene amplification in eukaryotic cells |
| US4975276A (en) | 1982-01-15 | 1990-12-04 | Cetus Corporation | Interferon-alpha 54 |
| US4748233A (en) | 1982-03-23 | 1988-05-31 | Bristol-Myers Company | Alpha-interferon Gx-1 |
| US4695543A (en) | 1982-03-23 | 1987-09-22 | Bristol-Myers Company | Alpha Interferon GX-1 |
| US6936694B1 (en) | 1982-05-06 | 2005-08-30 | Intermune, Inc. | Manufacture and expression of large structural genes |
| DE3381090D1 (de) | 1982-05-19 | 1990-02-15 | Gist Brocades Nv | Klonierungssystem fuer kluyveromyce spezies. |
| US4486533A (en) | 1982-09-02 | 1984-12-04 | St. Louis University | Filamentous fungi functional replicating extrachromosomal element |
| US4522811A (en) | 1982-07-08 | 1985-06-11 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US4599311A (en) | 1982-08-13 | 1986-07-08 | Kawasaki Glenn H | Glycolytic promotersfor regulated protein expression: protease inhibitor |
| US5151511A (en) | 1982-09-16 | 1992-09-29 | Amgen Inc. | DNA encoding avian growth hormones |
| US4737462A (en) | 1982-10-19 | 1988-04-12 | Cetus Corporation | Structural genes, plasmids and transformed cells for producing cysteine depleted muteins of interferon-β |
| US4588585A (en) | 1982-10-19 | 1986-05-13 | Cetus Corporation | Human recombinant cysteine depleted interferon-β muteins |
| US4966843A (en) | 1982-11-01 | 1990-10-30 | Cetus Corporation | Expression of interferon genes in Chinese hamster ovary cells |
| US4438253A (en) | 1982-11-12 | 1984-03-20 | American Cyanamid Company | Poly(glycolic acid)/poly(alkylene glycol) block copolymers and method of manufacturing the same |
| US4501728A (en) | 1983-01-06 | 1985-02-26 | Technology Unlimited, Inc. | Masking of liposomes from RES recognition |
| US4603044A (en) | 1983-01-06 | 1986-07-29 | Technology Unlimited, Inc. | Hepatocyte Directed Vesicle delivery system |
| US4713339A (en) | 1983-01-19 | 1987-12-15 | Genentech, Inc. | Polycistronic expression vector construction |
| US4518584A (en) | 1983-04-15 | 1985-05-21 | Cetus Corporation | Human recombinant interleukin-2 muteins |
| US4745051A (en) | 1983-05-27 | 1988-05-17 | The Texas A&M University System | Method for producing a recombinant baculovirus expression vector |
| DK168016B1 (da) | 1983-12-26 | 1994-01-17 | Kyowa Hakko Kogyo Kk | Derivat af humant interferon-gamma-polypeptid |
| US4496689A (en) | 1983-12-27 | 1985-01-29 | Miles Laboratories, Inc. | Covalently attached complex of alpha-1-proteinase inhibitor with a water soluble polymer |
| DE3572982D1 (en) | 1984-03-06 | 1989-10-19 | Takeda Chemical Industries Ltd | Chemically modified lymphokine and production thereof |
| IL71691A (en) | 1984-04-27 | 1991-04-15 | Yeda Res & Dev | Production of interferon-ypsilon |
| US4879236A (en) | 1984-05-16 | 1989-11-07 | The Texas A&M University System | Method for producing a recombinant baculovirus expression vector |
| US4931373A (en) | 1984-05-25 | 1990-06-05 | Zymogenetics, Inc. | Stable DNA constructs for expression of α-1 antitrypsin |
| US4761371A (en) | 1985-02-12 | 1988-08-02 | Genentech, Inc. | Insulin receptor |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| GR860984B (en) | 1985-04-17 | 1986-08-18 | Zymogenetics Inc | Expression of factor vii and ix activities in mammalian cells |
| DE3684546D1 (de) | 1985-04-22 | 1992-04-30 | Genetics Inst | Herstellung mit hoher leistung des aktivfaktors ix. |
| WO1986006358A1 (en) | 1985-04-30 | 1986-11-06 | Büro Patent Ag | Installation and method for the automatic feeding of filled cans and the automatic evacuation of empty cans in a spinning machine |
| JP2524586B2 (ja) | 1985-06-26 | 1996-08-14 | シタス コーポレイション | ポリマ−接合を利用する医薬組成物用蛋白質の可溶化 |
| JPS6238172A (ja) | 1985-08-12 | 1987-02-19 | 株式会社 高研 | 抗血栓性医用材料の製造方法 |
| US4810643A (en) | 1985-08-23 | 1989-03-07 | Kirin- Amgen Inc. | Production of pluripotent granulocyte colony-stimulating factor |
| US6004548A (en) | 1985-08-23 | 1999-12-21 | Amgen, Inc. | Analogs of pluripotent granulocyte colony-stimulating factor |
| JPS62236497A (ja) | 1985-09-17 | 1987-10-16 | Chugai Pharmaceut Co Ltd | 顆粒球コロニー刺激因子活性を有する糖蛋白質の製造方法 |
| WO1987002060A1 (en) | 1985-10-03 | 1987-04-09 | Biogen N.V. | Human granulocyte-macrophage colony stimulating factor-like polypeptides and processes for producing them in high yields in microbial cells |
| IL80529A0 (en) | 1985-11-14 | 1987-02-27 | Daiichi Seiyaku Co | Method of producing peptides |
| DE3712985A1 (de) | 1987-04-16 | 1988-11-03 | Hoechst Ag | Bifunktionelle proteine |
| US4935349A (en) | 1986-01-17 | 1990-06-19 | Zymogenetics, Inc. | Expression of higher eucaryotic genes in aspergillus |
| EP0272253A4 (en) * | 1986-03-07 | 1990-02-05 | Massachusetts Inst Technology | METHOD FOR IMPROVING GLYCOPROTE INSTABILITY. |
| US4925796A (en) * | 1986-03-07 | 1990-05-15 | Massachusetts Institute Of Technology | Method for enhancing glycoprotein stability |
| IT1203758B (it) | 1986-03-27 | 1989-02-23 | Univ Roma | Vettori di clonazione e di espressione di geni eterologhi in lieviti e lieviti trasformati con tali vettori |
| GB8610600D0 (en) | 1986-04-30 | 1986-06-04 | Novo Industri As | Transformation of trichoderma |
| US4902505A (en) | 1986-07-30 | 1990-02-20 | Alkermes | Chimeric peptides for neuropeptide delivery through the blood-brain barrier |
| US5075109A (en) | 1986-10-24 | 1991-12-24 | Southern Research Institute | Method of potentiating an immune response |
| US4894330A (en) | 1986-12-23 | 1990-01-16 | Cetus Corporation | Purification of recombinant beta-interferon incorporating RP-HPLC |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US4857505A (en) * | 1987-03-09 | 1989-08-15 | American Cyanamid Company | Sustained release compositions for parenteral administration and their use |
| DK175243B1 (da) | 1987-03-23 | 2004-07-19 | Zymogenetics Inc | Ekspressionsvektor, der er i stand til at styre ekspression af heterologe gener eller cDNA i gær, gærværtscelle samt fremgangsmåde til öget produktion af proteiner i gærværtsceller |
| IL82834A (en) | 1987-06-09 | 1990-11-05 | Yissum Res Dev Co | Biodegradable polymeric materials based on polyether glycols,processes for the preparation thereof and surgical artiicles made therefrom |
| ATE97446T1 (de) | 1987-07-28 | 1993-12-15 | Gist Brocades Nv | Kluyveromyces als wirtsstamm. |
| US4897268A (en) | 1987-08-03 | 1990-01-30 | Southern Research Institute | Drug delivery system and method of making the same |
| US5252726A (en) | 1987-09-04 | 1993-10-12 | Novo Nordisk A/S | Promoters for use in aspergillus |
| GB8725529D0 (en) | 1987-10-30 | 1987-12-02 | Delta Biotechnology Ltd | Polypeptides |
| US5153265A (en) | 1988-01-20 | 1992-10-06 | Cetus Corporation | Conjugation of polymer to colony stimulating factor-1 |
| US4847325A (en) * | 1988-01-20 | 1989-07-11 | Cetus Corporation | Conjugation of polymer to colony stimulating factor-1 |
| GB8810808D0 (en) | 1988-05-06 | 1988-06-08 | Wellcome Found | Vectors |
| JP2511494B2 (ja) | 1988-05-12 | 1996-06-26 | 善治 松浦 | 日本脳炎ウイルス表面抗原蛋白質の製造法 |
| FR2631974B1 (fr) | 1988-05-31 | 1992-12-11 | Agronomique Inst Nat Rech | Baculovirus modifie, son procede de preparation et son application en tant que vecteur d'expression de genes |
| FR2649991B2 (fr) | 1988-08-05 | 1994-03-04 | Rhone Poulenc Sante | Utilisation de derives stables du plasmide pkd1 pour l'expression et la secretion de proteines heterologues dans les levures du genre kluyveromyces |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| US5162215A (en) | 1988-09-22 | 1992-11-10 | Amgen Inc. | Method of gene transfer into chickens and other avian species |
| US5534617A (en) | 1988-10-28 | 1996-07-09 | Genentech, Inc. | Human growth hormone variants having greater affinity for human growth hormone receptor at site 1 |
| FR2638643B1 (fr) | 1988-11-09 | 1991-04-12 | Transgene Sa | Sequence d'adn codant pour le facteur ix humain ou une proteine analogue, vecteur d'expression, cellules transformees, procede de preparation du facteur ix et produits obtenus correspondants |
| US5047335A (en) | 1988-12-21 | 1991-09-10 | The Regents Of The University Of Calif. | Process for controlling intracellular glycosylation of proteins |
| US6166183A (en) * | 1992-11-30 | 2000-12-26 | Kirin-Amgen, Inc. | Chemically-modified G-CSF |
| ATE176924T1 (de) | 1988-12-23 | 1999-03-15 | Genentech Inc | Verfahren zur herstellung von menschlicher dnase |
| US5162228A (en) | 1988-12-28 | 1992-11-10 | Takeda Chemical Industries, Ltd. | Gylceraldehyde-3-phosphate dehydrogenase gene and promoter |
| US5096815A (en) | 1989-01-06 | 1992-03-17 | Protein Engineering Corporation | Generation and selection of novel dna-binding proteins and polypeptides |
| US5198346A (en) | 1989-01-06 | 1993-03-30 | Protein Engineering Corp. | Generation and selection of novel DNA-binding proteins and polypeptides |
| US4957773A (en) | 1989-02-13 | 1990-09-18 | Syracuse University | Deposition of boron-containing films from decaborane |
| US5475090A (en) | 1989-02-21 | 1995-12-12 | Boyce Thompson Institute For Plant Research | Gene coded for a polypeptide which enhances virus infection of host insects |
| US5194376A (en) | 1989-02-28 | 1993-03-16 | University Of Ottawa | Baculovirus expression system capable of producing foreign gene proteins at high levels |
| DE3906540A1 (de) | 1989-03-02 | 1990-09-13 | Behringwerke Ag | Expressionsvektoren zur synthese von proteinen in der spalthefe schizosaccharomyces pombe |
| US5179023A (en) | 1989-03-24 | 1993-01-12 | Research Corporation Technologies, Inc. | Recombinant α-galactosidase a therapy for Fabry disease |
| US5122614A (en) | 1989-04-19 | 1992-06-16 | Enzon, Inc. | Active carbonates of polyalkylene oxides for modification of polypeptides |
| AU5526690A (en) | 1989-04-19 | 1990-11-29 | Enzon, Inc. | Active carbonates of polyalkylene oxides for modification of polypeptides |
| US5324844A (en) | 1989-04-19 | 1994-06-28 | Enzon, Inc. | Active carbonates of polyalkylene oxides for modification of polypeptides |
| DE68927344T2 (de) | 1989-04-28 | 1997-02-20 | Rhein Biotech Proz & Prod Gmbh | Hefezellen der Gattung-Schwanniomyces |
| US5766883A (en) | 1989-04-29 | 1998-06-16 | Delta Biotechnology Limited | Polypeptides |
| US5244805A (en) | 1989-05-17 | 1993-09-14 | University Of Georgia Research Foundation, Inc. | Baculovirus expression vectors |
| US5162222A (en) | 1989-07-07 | 1992-11-10 | Guarino Linda A | Use of baculovirus early promoters for expression of foreign genes in stably transformed insect cells or recombinant baculoviruses |
| US5179007A (en) | 1989-07-07 | 1993-01-12 | The Texas A & M University System | Method and vector for the purification of foreign proteins |
| US5077214A (en) | 1989-07-07 | 1991-12-31 | The Texas A&M University System | Use of baculovirus early promoters for expression of foreign genes in stably transformed insect cells |
| FR2650598B1 (fr) | 1989-08-03 | 1994-06-03 | Rhone Poulenc Sante | Derives de l'albumine a fonction therapeutique |
| US5155037A (en) | 1989-08-04 | 1992-10-13 | The Texas A&M University System | Insect signal sequences useful to improve the efficiency of processing and secretion of foreign genes in insect systems |
| US5023328A (en) | 1989-08-04 | 1991-06-11 | The Texas A&M University System | Lepidopteran AKH signal sequence |
| US5154924A (en) | 1989-09-07 | 1992-10-13 | Alkermes, Inc. | Transferrin receptor specific antibody-neuropharmaceutical agent conjugates |
| US5527527A (en) | 1989-09-07 | 1996-06-18 | Alkermes, Inc. | Transferrin receptor specific antibody-neuropharmaceutical agent conjugates |
| US5182107A (en) | 1989-09-07 | 1993-01-26 | Alkermes, Inc. | Transferrin receptor specific antibody-neuropharmaceutical or diagnostic agent conjugates |
| US5672683A (en) | 1989-09-07 | 1997-09-30 | Alkermes, Inc. | Transferrin neuropharmaceutical agent fusion protein |
| US5977307A (en) | 1989-09-07 | 1999-11-02 | Alkermes, Inc. | Transferrin receptor specific ligand-neuropharmaceutical agent fusion proteins |
| US5032519A (en) | 1989-10-24 | 1991-07-16 | The Regents Of The Univ. Of California | Method for producing secretable glycosyltransferases and other Golgi processing enzymes |
| US5312808A (en) | 1989-11-22 | 1994-05-17 | Enzon, Inc. | Fractionation of polyalkylene oxide-conjugated hemoglobin solutions |
| DE4009630C2 (de) * | 1990-03-26 | 1995-09-28 | Reinhard Prof Dr Dr Brossmer | CMP-aktivierte fluoreszierende Sialinsäuren sowie Verfahren zu ihrer Herstellung |
| JP2975632B2 (ja) * | 1990-03-30 | 1999-11-10 | 生化学工業株式会社 | グリコサミノグリカン修飾プロテイン |
| US5606031A (en) | 1990-04-06 | 1997-02-25 | Lile; Jack | Production and purification of biologically active recombinant neurotrophic protein in bacteria |
| GB9107846D0 (en) | 1990-04-30 | 1991-05-29 | Ici Plc | Polypeptides |
| US5951972A (en) * | 1990-05-04 | 1999-09-14 | American Cyanamid Company | Stabilization of somatotropins and other proteins by modification of cysteine residues |
| US5399345A (en) | 1990-05-08 | 1995-03-21 | Boehringer Mannheim, Gmbh | Muteins of the granulocyte colony stimulating factor |
| US5219564A (en) | 1990-07-06 | 1993-06-15 | Enzon, Inc. | Poly(alkylene oxide) amino acid copolymers and drug carriers and charged copolymers based thereon |
| FR2664905B1 (fr) | 1990-07-18 | 1994-08-12 | Agronomique Inst Nat Rech | Baculovirus modifie, son procede d'obtention, et vecteurs d'expression obtenus a partir dudit baculovirus. |
| US5169784A (en) | 1990-09-17 | 1992-12-08 | The Texas A & M University System | Baculovirus dual promoter expression vector |
| US5529914A (en) | 1990-10-15 | 1996-06-25 | The Board Of Regents The Univeristy Of Texas System | Gels for encapsulation of biological materials |
| US5410016A (en) | 1990-10-15 | 1995-04-25 | Board Of Regents, The University Of Texas System | Photopolymerizable biodegradable hydrogels as tissue contacting materials and controlled-release carriers |
| GB9023111D0 (en) | 1990-10-24 | 1990-12-05 | Wellcome Found | Expression system |
| US5401650A (en) | 1990-10-24 | 1995-03-28 | The Mount Sinai School Of Medicine Of The City University Of New York | Cloning and expression of biologically active α-galactosidase A |
| CA2073511A1 (en) * | 1990-11-14 | 1992-05-29 | Matthew R. Callstrom | Conjugates of poly(vinylsaccharide) with proteins for the stabilization of proteins |
| US5420027A (en) | 1991-01-10 | 1995-05-30 | Board Of Regents, The University Of Texas System | Methods and compositions for the expression of biologically active fusion proteins comprising a eukaryotic cytochrome P450 fused to a reductase in bacteria |
| US5650554A (en) | 1991-02-22 | 1997-07-22 | Sembiosys Genetics Inc. | Oil-body proteins as carriers of high-value peptides in plants |
| US6288304B1 (en) | 1991-02-22 | 2001-09-11 | Sembiosys Genetics Inc. | Expression of somatotropin in plant seeds |
| US5948682A (en) | 1991-02-22 | 1999-09-07 | Sembiosys Genetics Inc. | Preparation of heterologous proteins on oil bodies |
| US5278299A (en) * | 1991-03-18 | 1994-01-11 | Scripps Clinic And Research Foundation | Method and composition for synthesizing sialylated glycosyl compounds |
| EP0576592B1 (en) | 1991-03-18 | 2000-05-31 | The Scripps Research Institute | Oligosaccharide enzyme substrates and inhibitors: methods and compositions |
| US5212075A (en) | 1991-04-15 | 1993-05-18 | The Regents Of The University Of California | Compositions and methods for introducing effectors to pathogens and cells |
| US5472858A (en) | 1991-06-04 | 1995-12-05 | Wisconsin Alumni Research Foundation | Production of recombinant proteins in insect larvae |
| US5374655A (en) | 1991-06-10 | 1994-12-20 | Alberta Research Council | Methods for the synthesis of monofucosylated oligosaccharides terminating in di-N-acetyllactosaminyl structures |
| US5352670A (en) * | 1991-06-10 | 1994-10-04 | Alberta Research Council | Methods for the enzymatic synthesis of alpha-sialylated oligosaccharide glycosides |
| KR950014915B1 (ko) * | 1991-06-19 | 1995-12-18 | 주식회사녹십자 | 탈시알로당단백-포함화합물 |
| AU2147192A (en) * | 1991-06-28 | 1993-01-25 | Genentech Inc. | Method of stimulating immune response using growth hormone |
| US5352570A (en) | 1991-06-28 | 1994-10-04 | Eastman Kodak Company | Method and photographic material and process comprising a benzotriazole compound |
| US5633146A (en) | 1991-07-02 | 1997-05-27 | Rhone-Poulenc Rorer S.A. | Method for producing recombinant proteins and host cells used therein |
| US5281698A (en) | 1991-07-23 | 1994-01-25 | Cetus Oncology Corporation | Preparation of an activated polymer ester for protein conjugation |
| DK8892D0 (da) | 1992-01-23 | 1992-01-23 | Symbicom Ab | Humant proteingen |
| IT1260468B (it) | 1992-01-29 | 1996-04-09 | Metodo per mantenere l'attivita' di enzimi proteolitici modificati con polietilenglicole | |
| US5965106A (en) | 1992-03-04 | 1999-10-12 | Perimmune Holdings, Inc. | In vivo binding pair pretargeting |
| DE69332538T2 (de) | 1992-04-02 | 2003-11-06 | Sembiosys Genetics, Inc. | Cis elemente des ölkörperproteins als regulationische signale |
| US6037452A (en) | 1992-04-10 | 2000-03-14 | Alpha Therapeutic Corporation | Poly(alkylene oxide)-Factor VIII or Factor IX conjugate |
| US5516657A (en) | 1992-05-11 | 1996-05-14 | Cambridge Biotech Corporation | Baculovirus vectors for expression of secretory and membrane-bound proteins |
| US5614184A (en) * | 1992-07-28 | 1997-03-25 | New England Deaconess Hospital | Recombinant human erythropoietin mutants and therapeutic methods employing them |
| CA2141673A1 (en) * | 1992-08-07 | 1994-02-17 | Graham P. Allaway | Non-peptidyl moiety-conjugated cd4-gamma2 and cd4-igg2 immunoconjugates, and uses thereof |
| AU5006993A (en) | 1992-08-21 | 1994-03-15 | Enzon, Inc. | Novel attachment of polyalkylene oxides to bio-effecting substances |
| AU5098193A (en) * | 1992-09-01 | 1994-03-29 | Berlex Laboratories, Inc. | Glycolation of glycosylated macromolecules |
| US5348886A (en) | 1992-09-04 | 1994-09-20 | Monsanto Company | Method of producing recombinant eukaryotic viruses in bacteria |
| WO1994012646A1 (en) | 1992-11-27 | 1994-06-09 | Ciba-Geigy Ag | Proteins having glycosyltransferase activity |
| US6210671B1 (en) | 1992-12-01 | 2001-04-03 | Protein Design Labs, Inc. | Humanized antibodies reactive with L-selectin |
| NZ250375A (en) * | 1992-12-09 | 1995-07-26 | Ortho Pharma Corp | Peg hydrazone and peg oxime linkage forming reagents and protein derivatives |
| AU6029594A (en) | 1993-01-15 | 1994-08-15 | Enzon, Inc. | Factor viii - polymeric conjugates |
| US5349001A (en) | 1993-01-19 | 1994-09-20 | Enzon, Inc. | Cyclic imide thione activated polyalkylene oxides |
| US5321095A (en) | 1993-02-02 | 1994-06-14 | Enzon, Inc. | Azlactone activated polyalkylene oxides |
| US5202413A (en) | 1993-02-16 | 1993-04-13 | E. I. Du Pont De Nemours And Company | Alternating (ABA)N polylactide block copolymers |
| US5374541A (en) | 1993-05-04 | 1994-12-20 | The Scripps Research Institute | Combined use of β-galactosidase and sialyltransferase coupled with in situ regeneration of CMP-sialic acid for one pot synthesis of oligosaccharides |
| US6001364A (en) | 1993-05-05 | 1999-12-14 | Gryphon Sciences | Hetero-polyoxime compounds and their preparation by parallel assembly |
| US6174530B1 (en) | 1993-05-05 | 2001-01-16 | Gryphon Sciences | Homogeneous polyoxime compositions and their preparation by parallel assembly |
| AU7097094A (en) * | 1993-06-01 | 1994-12-20 | Enzon, Inc. | Carbohydrate-modified polymer conjugates with erythropoietic activity |
| US5621039A (en) * | 1993-06-08 | 1997-04-15 | Hallahan; Terrence W. | Factor IX- polymeric conjugates |
| JPH09503905A (ja) * | 1993-07-15 | 1997-04-22 | ネオゼ ファーマシューティカルス | 糖組成物の合成方法 |
| DE4325317C2 (de) * | 1993-07-29 | 1998-05-20 | Univ Dresden Tech | Verfahren zur radioaktiven Markierung von Immunglobulinen |
| US5446090A (en) | 1993-11-12 | 1995-08-29 | Shearwater Polymers, Inc. | Isolatable, water soluble, and hydrolytically stable active sulfones of poly(ethylene glycol) and related polymers for modification of surfaces and molecules |
| US5827690A (en) | 1993-12-20 | 1998-10-27 | Genzyme Transgenics Corporatiion | Transgenic production of antibodies in milk |
| WO1995017515A1 (en) | 1993-12-23 | 1995-06-29 | University Technologies International Inc. | Methods of expressing proteins in insect cells and methods of killing insects |
| US5597709A (en) | 1994-01-27 | 1997-01-28 | Human Genome Sciences, Inc. | Human growth hormone splice variants hGHV-2(88) and hGHV-3(53) |
| US5369017A (en) * | 1994-02-04 | 1994-11-29 | The Scripps Research Institute | Process for solid phase glycopeptide synthesis |
| US6117679A (en) | 1994-02-17 | 2000-09-12 | Maxygen, Inc. | Methods for generating polynucleotides having desired characteristics by iterative selection and recombination |
| US5837458A (en) | 1994-02-17 | 1998-11-17 | Maxygen, Inc. | Methods and compositions for cellular and metabolic engineering |
| US5605793A (en) | 1994-02-17 | 1997-02-25 | Affymax Technologies N.V. | Methods for in vitro recombination |
| US6165793A (en) | 1996-03-25 | 2000-12-26 | Maxygen, Inc. | Methods for generating polynucleotides having desired characteristics by iterative selection and recombination |
| US5834252A (en) | 1995-04-18 | 1998-11-10 | Glaxo Group Limited | End-complementary polymerase reaction |
| JP3090586B2 (ja) | 1994-03-15 | 2000-09-25 | 片倉工業株式会社 | システインプロテアーゼ遺伝子欠損バキュロウイルスおよびその製造法並びにこれを利用する有用タンパク質の製造法 |
| US5545723A (en) | 1994-03-15 | 1996-08-13 | Biogen Inc. | Muteins of IFN-β |
| US5432059A (en) | 1994-04-01 | 1995-07-11 | Specialty Laboratories, Inc. | Assay for glycosylation deficiency disorders |
| GB9408717D0 (en) | 1994-05-03 | 1994-06-22 | Biotech & Biolog Scien Res | DNA sequences |
| DE69526395D1 (de) | 1994-08-13 | 2002-05-23 | Roche Diagnostics Gmbh | Verwendung von Interferon-gamma zur Vermeidung der Proliferation und Differenzierung der primitiven Hämapoietischen Vorläuferzellen |
| US5871986A (en) | 1994-09-23 | 1999-02-16 | The General Hospital Corporation | Use of a baculovirus to express and exogenous gene in a mammalian cell |
| US5545553A (en) | 1994-09-26 | 1996-08-13 | The Rockefeller University | Glycosyltransferases for biosynthesis of oligosaccharides, and genes encoding them |
| EP0785276A4 (en) * | 1994-09-29 | 2002-01-09 | Ajinomoto Kk | MODIFICATION OF A PEPTIDE AND A PROTEIN |
| US5521299A (en) | 1994-11-22 | 1996-05-28 | National Science Council | Oligonucleotides for detection of baculovirus infection |
| US5834251A (en) * | 1994-12-30 | 1998-11-10 | Alko Group Ltd. | Methods of modifying carbohydrate moieties |
| US5932462A (en) | 1995-01-10 | 1999-08-03 | Shearwater Polymers, Inc. | Multiarmed, monofunctional, polymer for coupling to molecules and surfaces |
| CA2211743A1 (en) | 1995-02-21 | 1996-08-29 | Cantab Pharmaceuticals Research Limited | Viral preparations, vectors, immunogens, and vaccines |
| US5843705A (en) | 1995-02-21 | 1998-12-01 | Genzyme Transgenic Corporation | Transgenically produced antithrombin III |
| GB9506249D0 (en) | 1995-03-27 | 1995-05-17 | Karobio Ab | Media for insect cell cultures |
| US5876980A (en) * | 1995-04-11 | 1999-03-02 | Cytel Corporation | Enzymatic synthesis of oligosaccharides |
| US5922577A (en) | 1995-04-11 | 1999-07-13 | Cytel Corporation | Enzymatic synthesis of glycosidic linkages |
| US6030815A (en) * | 1995-04-11 | 2000-02-29 | Neose Technologies, Inc. | Enzymatic synthesis of oligosaccharides |
| US5728554A (en) | 1995-04-11 | 1998-03-17 | Cytel Corporation | Enzymatic synthesis of glycosidic linkages |
| EP0833663A4 (en) * | 1995-05-15 | 1999-04-07 | Constantin A Bona | CARBOHYDRATE MEDIATED COUPLING OF PEPTIDES TO IMMUNOGLOBULINES |
| US6015555A (en) | 1995-05-19 | 2000-01-18 | Alkermes, Inc. | Transferrin receptor specific antibody-neuropharmaceutical or diagnostic agent conjugates |
| AU6255096A (en) | 1995-06-07 | 1996-12-30 | Mount Sinai School Of Medicine Of The City University Of New York, The | Pegylated modified proteins |
| US5672662A (en) | 1995-07-07 | 1997-09-30 | Shearwater Polymers, Inc. | Poly(ethylene glycol) and related polymers monosubstituted with propionic or butanoic acids and functional derivatives thereof for biotechnical applications |
| US5811634A (en) | 1995-09-12 | 1998-09-22 | Thomas G. O'Brien | Transgenic mammal encoding ornithine decarboxylase |
| ATE301196T1 (de) * | 1995-09-21 | 2005-08-15 | Genentech Inc | Varianten des menschlichen wachstumshormons |
| SE9503380D0 (sv) * | 1995-09-29 | 1995-09-29 | Pharmacia Ab | Protein derivatives |
| US5767379A (en) | 1995-11-06 | 1998-06-16 | John Howard | Commercial production of avidin in plants |
| WO1997017451A2 (en) | 1995-11-09 | 1997-05-15 | Zymogenetics, Inc. | Production of gad65 in methylotrophic yeast |
| US6171820B1 (en) | 1995-12-07 | 2001-01-09 | Diversa Corporation | Saturation mutagenesis in directed evolution |
| US6361974B1 (en) | 1995-12-07 | 2002-03-26 | Diversa Corporation | Exonuclease-mediated nucleic acid reassembly in directed evolution |
| US5965408A (en) | 1996-07-09 | 1999-10-12 | Diversa Corporation | Method of DNA reassembly by interrupting synthesis |
| US5716812A (en) | 1995-12-12 | 1998-02-10 | The University Of British Columbia | Methods and compositions for synthesis of oligosaccharides, and the products formed thereby |
| US5728580A (en) | 1996-02-20 | 1998-03-17 | Cornell Research Foundation, Inc. | Methods and culture media for inducing single cell suspension in insect cell lines |
| US6096548A (en) | 1996-03-25 | 2000-08-01 | Maxygen, Inc. | Method for directing evolution of a virus |
| US5734024A (en) | 1996-04-19 | 1998-03-31 | Boris Y. Zaslavsky | Method for determining the biological activity of recombinant human growth hormone |
| US5750383A (en) | 1996-05-14 | 1998-05-12 | Boyce Thompson Institute For Plant Research, Inc. | Baculovirus cloning system |
| AU708572B2 (en) | 1996-07-17 | 1999-08-05 | Zymogenetics Inc. | Preparation of (pichia methanolica) auxotrophic mutants |
| JP2002515746A (ja) | 1996-07-17 | 2002-05-28 | ザイモジェネティクス,インコーポレイティド | ピヒア・メタノリカの形質転換 |
| US5682823A (en) | 1996-08-23 | 1997-11-04 | Bethlehem Steel Corporation | Removable insulated cover and method for transporting hot oversized steel ingots |
| US20020064546A1 (en) | 1996-09-13 | 2002-05-30 | J. Milton Harris | Degradable poly(ethylene glycol) hydrogels with controlled half-life and precursors therefor |
| JP2001502005A (ja) | 1996-10-10 | 2001-02-13 | サイテル コーポレイション | 限外濾過、逆浸透及びナノ濾過を利用する炭水化物の精製 |
| US5856452A (en) | 1996-12-16 | 1999-01-05 | Sembiosys Genetics Inc. | Oil bodies and associated proteins as affinity matrices |
| BR9606270A (pt) | 1996-12-18 | 1998-09-22 | Univ Minas Gerais | Processo para a produção da proteína do interferon beta-cis humano recombinante e proteína de interferon beta-cis humano recombinante |
| DE69823046T2 (de) | 1997-01-16 | 2005-03-31 | Neose Technologies, Inc. | Praktische in vitro sialylierung von rekombinanten glykpproteinen |
| US5945314A (en) | 1997-03-31 | 1999-08-31 | Abbott Laboratories | Process for synthesizing oligosaccharides |
| US6183738B1 (en) * | 1997-05-12 | 2001-02-06 | Phoenix Pharamacologics, Inc. | Modified arginine deiminase |
| US7585645B2 (en) | 1997-05-27 | 2009-09-08 | Sembiosys Genetics Inc. | Thioredoxin and thioredoxin reductase containing oil body based products |
| ES2282788T3 (es) | 1997-06-13 | 2007-10-16 | Amylin Pharmaceuticals, Inc. | Ligamiento quimico nativo en fase solida de peptidos no protegidos o protegidos en cisteinas n-terminales en disolucion acuosa. |
| US6210736B1 (en) | 1997-06-17 | 2001-04-03 | Genzyme Transgenics Corporation | Transgenically produced prolactin |
| AU757627B2 (en) | 1997-06-24 | 2003-02-27 | Genentech Inc. | Methods and compositions for galactosylated glycoproteins |
| WO1999000150A2 (en) | 1997-06-27 | 1999-01-07 | Regents Of The University Of California | Drug targeting of a peptide radiopharmaceutical through the primate blood-brain barrier in vivo with a monoclonal antibody to the human insulin receptor |
| US6090584A (en) | 1997-08-21 | 2000-07-18 | University Technologies International Inc. | Baculovirus artificial chromosomes and methods of use |
| GB9722604D0 (en) * | 1997-10-28 | 1997-12-24 | Cancer Res Campaign Tech | Heparin-binding growth factor derivatives |
| CA2307166A1 (en) | 1997-10-31 | 1999-05-14 | Genentech, Inc. | Methods and compositions comprising glycoprotein glycoforms |
| US6168937B1 (en) | 1997-12-08 | 2001-01-02 | Alan D. Elbein | Purified β1,2-xylosyltransferase and uses thereof |
| US7244601B2 (en) | 1997-12-15 | 2007-07-17 | National Research Council Of Canada | Fusion proteins for use in enzymatic synthesis of oligosaccharides |
| CA2316834C (en) | 1998-01-07 | 2006-01-03 | Shearwater Polymers, Inc. | Degradable heterobifunctional poly(ethylene glycol) acrylates and gels and conjugates derived therefrom |
| AR014491A1 (es) | 1998-01-29 | 2001-02-28 | Dow Agrosciences Llc | Metodo para obtener plantas transgenicas fertiles de gossypium hirsutum. |
| US6362254B2 (en) | 1998-03-12 | 2002-03-26 | Shearwater Corporation | Poly(ethylene glycol) derivatives with proximal reactive groups |
| US6689604B1 (en) | 1998-03-20 | 2004-02-10 | National Research Council Of Canada | Lipopolysaccharide α-2,3 sialyltransferase of Campylobacter jejuni and its uses |
| PT2180007E (pt) | 1998-04-20 | 2013-11-25 | Roche Glycart Ag | Engenharia de glicosilação de anticorpos para melhorar a citotoxicidade celular dependente de anticorpos |
| KR100618495B1 (ko) | 1998-10-06 | 2006-08-31 | 마크 아론 에말파브 | 섬유상의 진균성 숙주 영역에서의 형질전환 시스템:크리소스포륨속에서 |
| US6245539B1 (en) | 1998-10-06 | 2001-06-12 | Board Of Trustees Operating Michigan State University | Human asparaginyl-tRNA synthetase DNA |
| CN101062419B (zh) * | 1998-10-16 | 2016-06-29 | 生物基因Ma公司 | 干扰素-β-1a的聚合物缀合物及其使用 |
| US6374295B2 (en) * | 1998-10-29 | 2002-04-16 | Nortel Networks Limited | Active server management |
| DE19852729A1 (de) * | 1998-11-16 | 2000-05-18 | Werner Reutter | Rekombinante Glycoproteine, Verfahren zu ihrer Herstellung, sie enthaltende Arzneimittel und ihre Verwendung |
| JP2002530087A (ja) * | 1998-11-18 | 2002-09-17 | ネオーズ テクノロジーズ, インコーポレイテッド | オリゴ糖の安価な産生 |
| DE69942671D1 (de) | 1998-12-01 | 2010-09-23 | Facet Biotech Corp | Humanisierte antikoerper gegen gamma-interferon |
| WO2000044785A1 (en) * | 1999-01-29 | 2000-08-03 | F. Hoffmann-La Roche Ag | Gcsf conjugates |
| US6365410B1 (en) | 1999-05-19 | 2002-04-02 | Genencor International, Inc. | Directed evolution of microorganisms |
| PE20010288A1 (es) * | 1999-07-02 | 2001-03-07 | Hoffmann La Roche | Derivados de eritropoyetina |
| AU6357900A (en) * | 1999-07-20 | 2001-02-05 | Amgen, Inc. | Hyaluronic acid-protein conjugates, pharmaceutical compositions and related methods |
| US6348558B1 (en) | 1999-12-10 | 2002-02-19 | Shearwater Corporation | Hydrolytically degradable polymers and hydrogels made therefrom |
| AU781729B2 (en) | 1999-12-22 | 2005-06-09 | Nektar Therapeutics | Method for the preparation of 1-benzotriazolyl carbonate esters of poly(ethylene glycol) |
| IL150314A0 (en) | 1999-12-30 | 2002-12-01 | Maxygen Aps | Improved lysosomal enzymes and lysosomal enzyme activators |
| CA2395713C (en) * | 2000-01-10 | 2012-04-10 | Torben Lauesgaard Nissen | G-csf conjugates |
| US6423488B1 (en) | 2000-01-15 | 2002-07-23 | Avigenics, Inc | High throughput screening assay for detecting a DNA sequence |
| AU2001232337A1 (en) | 2000-02-18 | 2001-08-27 | Kanagawa Academy Of Science And Technology | Pharmaceutical composition, reagent and method for intracerebral delivery of pharmaceutically active ingredient or labeling substance |
| NZ523476A (en) | 2000-06-28 | 2004-04-30 | Glycofi Inc | Methods for humanizing glycosylation of recombinant glycoproteins expressed in lower eukaryotes |
| WO2002002597A2 (en) | 2000-06-30 | 2002-01-10 | Maxygen Aps | Peptide extended glycosylated polypeptides |
| AU2001273348A1 (en) | 2000-07-10 | 2002-01-21 | Diosynth Rtp, Inc. | Purification of human troponin i |
| AU2001283740A1 (en) | 2000-08-17 | 2002-02-25 | University Of British Columbia | Chemotherapeutic agents conjugated to p97 and their methods of use in treating neurological tumours |
| WO2002013873A2 (en) | 2000-08-17 | 2002-02-21 | Synapse Technologies, Inc. | P97-active agent conjugates and their methods of use |
| JP2004534523A (ja) | 2001-02-27 | 2004-11-18 | マキシゲン・エイピーエス | 新規なインターフェロンβ様分子 |
| MXPA04003333A (es) * | 2001-10-10 | 2006-02-22 | Neose Technologies Inc | Remodelado y glicoconjugacion de peptidos. |
| JP3894776B2 (ja) | 2001-11-09 | 2007-03-22 | 富士通メディアデバイス株式会社 | 吐出装置および吐出方法 |
| ATE503498T1 (de) * | 2002-06-21 | 2011-04-15 | Novo Nordisk Healthcare Ag | Pegylierte glykoformen von faktor vii |
| WO2004091499A2 (en) * | 2003-04-09 | 2004-10-28 | Neose Technologies, Inc. | Intracellular formation of peptide conjugates |
| JP4251399B2 (ja) * | 2004-05-21 | 2009-04-08 | 独立行政法人産業技術総合研究所 | O結合型糖鎖が付加されたペプチドのスクリーニング方法 |
-
2002
- 2002-10-09 MX MXPA04003333A patent/MXPA04003333A/es active IP Right Grant
- 2002-10-09 ES ES09151346.5T patent/ES2564688T3/es not_active Expired - Lifetime
- 2002-10-09 ES ES10012794.3T patent/ES2538342T3/es not_active Expired - Lifetime
- 2002-10-09 SG SG200605529-7A patent/SG159381A1/en unknown
- 2002-10-09 NZ NZ532027A patent/NZ532027A/en not_active IP Right Cessation
- 2002-10-09 ES ES10012798T patent/ES2561985T3/es not_active Expired - Lifetime
- 2002-10-09 EP EP10012938.6A patent/EP2279754B1/en not_active Expired - Lifetime
- 2002-10-09 ES ES02795509T patent/ES2411007T3/es not_active Expired - Lifetime
- 2002-10-09 EP EP10012536.8A patent/EP2279753B1/en not_active Expired - Lifetime
- 2002-10-09 EP EP10012939.4A patent/EP2279755B1/en not_active Expired - Lifetime
- 2002-10-09 PT PT100129394T patent/PT2279755E/pt unknown
- 2002-10-09 EP EP10012795A patent/EP2292271A3/en not_active Withdrawn
- 2002-10-09 ES ES10012938.6T patent/ES2516041T3/es not_active Expired - Lifetime
- 2002-10-09 PT PT100125368T patent/PT2279753E/pt unknown
- 2002-10-09 CA CA2462930A patent/CA2462930C/en not_active Expired - Lifetime
- 2002-10-09 JP JP2003534446A patent/JP5232352B2/ja not_active Expired - Lifetime
- 2002-10-09 DK DK02795509.5T patent/DK1578771T3/da active
- 2002-10-09 WO PCT/US2002/032263 patent/WO2003031464A2/en active Application Filing
- 2002-10-09 CN CN200910145812.4A patent/CN101724075B/zh not_active Expired - Lifetime
- 2002-10-09 IL IL16125102A patent/IL161251A0/xx unknown
- 2002-10-09 CN CN2011100354568A patent/CN102180944A/zh active Pending
- 2002-10-09 US US10/492,261 patent/US7416858B2/en not_active Expired - Lifetime
- 2002-10-09 EP EP09000818.6A patent/EP2042196B1/en not_active Expired - Lifetime
- 2002-10-09 EP EP02795509A patent/EP1578771B1/en not_active Expired - Lifetime
- 2002-10-09 EP EP09151346.5A patent/EP2080525B1/en not_active Expired - Lifetime
- 2002-10-09 ES ES10012939.4T patent/ES2466024T3/es not_active Expired - Lifetime
- 2002-10-09 ES ES10012536.8T patent/ES2556338T3/es not_active Expired - Lifetime
- 2002-10-09 ES ES09000818.6T patent/ES2606840T3/es not_active Expired - Lifetime
- 2002-10-09 CN CN02820073XA patent/CN1635901B/zh not_active Expired - Lifetime
- 2002-10-09 AU AU2002360264A patent/AU2002360264B2/en not_active Expired
- 2002-10-09 DK DK10184886.9T patent/DK2322229T3/en active
- 2002-10-09 BR BRPI0213207-9A patent/BRPI0213207B1/pt not_active IP Right Cessation
- 2002-10-09 ES ES10184886T patent/ES2619371T3/es not_active Expired - Lifetime
- 2002-10-09 SG SG2006055305A patent/SG177002A1/en unknown
- 2002-11-05 US US10/287,994 patent/US7138371B2/en active Active
-
2004
- 2004-04-01 IL IL161251A patent/IL161251A/en active IP Right Grant
-
2005
- 2005-12-23 HK HK05111922.5A patent/HK1079797A1/xx not_active IP Right Cessation
- 2005-12-23 HK HK11107561.1A patent/HK1149513A1/xx not_active IP Right Cessation
-
2006
- 2006-01-04 HK HK06100136.9A patent/HK1080090B/zh not_active IP Right Cessation
- 2006-04-12 US US11/404,266 patent/US7276475B2/en not_active Expired - Lifetime
-
2008
- 2008-11-20 JP JP2008297209A patent/JP5376912B2/ja not_active Expired - Lifetime
-
2009
- 2009-06-16 AU AU2009202405A patent/AU2009202405B2/en not_active Expired
-
2010
- 2010-02-05 JP JP2010024516A patent/JP5258809B2/ja not_active Expired - Lifetime
- 2010-09-28 JP JP2010217733A patent/JP5739632B2/ja not_active Expired - Fee Related
- 2010-12-23 IL IL210257A patent/IL210257A/en not_active IP Right Cessation
-
2011
- 2011-08-18 HK HK11108713.6A patent/HK1154498A1/xx not_active IP Right Cessation
-
2012
- 2012-05-07 JP JP2012106130A patent/JP2012143250A/ja active Pending
-
2014
- 2014-01-23 LU LU92359C patent/LU92359I2/fr unknown
- 2014-01-24 BE BE2014C004C patent/BE2014C004I2/fr unknown
- 2014-01-24 FR FR14C0007C patent/FR14C0007I2/fr active Active
- 2014-01-28 JP JP2014013799A patent/JP2014087370A/ja not_active Ceased
-
2015
- 2015-09-17 JP JP2015184218A patent/JP6316784B2/ja not_active Expired - Lifetime
-
2016
- 2016-06-14 JP JP2016118316A patent/JP6321724B2/ja not_active Expired - Lifetime
-
2017
- 2017-11-21 NL NL300912C patent/NL300912I2/nl unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6321724B2 (ja) | ペプチドの改造および複合糖質化 | |
| JP4674702B2 (ja) | グリコペギレ−ション法およびその方法により生成されたタンパク質/ペプチド | |
| JP5413548B2 (ja) | エリスロポエチン:エリスロポエチンの改造および複合糖質化 | |
| US7473680B2 (en) | Remodeling and glycoconjugation of peptides | |
| AU2002360264A1 (en) | Remodeling and glycoconjugation of peptides | |
| EP2305314B1 (en) | Remodelling and glycoconjugation of antibodies |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160920 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20161219 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170220 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20170802 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171201 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20180130 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20180326 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20180328 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6316784 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |