CN1681844A - Has化的多肽,特别是has化的促红细胞生成素 - Google Patents
Has化的多肽,特别是has化的促红细胞生成素 Download PDFInfo
- Publication number
- CN1681844A CN1681844A CNA038214644A CN03821464A CN1681844A CN 1681844 A CN1681844 A CN 1681844A CN A038214644 A CNA038214644 A CN A038214644A CN 03821464 A CN03821464 A CN 03821464A CN 1681844 A CN1681844 A CN 1681844A
- Authority
- CN
- China
- Prior art keywords
- epo
- polypeptide
- carbohydrate
- halfcystine
- coupling
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images



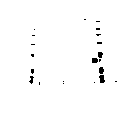









Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B31/00—Preparation of derivatives of starch
- C08B31/08—Ethers
- C08B31/12—Ethers having alkyl or cycloalkyl radicals substituted by heteroatoms, e.g. hydroxyalkyl or carboxyalkyl starch
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B31/00—Preparation of derivatives of starch
- C08B31/08—Ethers
- C08B31/10—Alkyl or cycloalkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/61—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule the organic macromolecular compound being a polysaccharide or a derivative thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0021—Intradermal administration, e.g. through microneedle arrays, needleless injectors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/475—Growth factors; Growth regulators
- C07K14/505—Erythropoietin [EPO]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/53—Colony-stimulating factor [CSF]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/54—Interleukins [IL]
- C07K14/5412—IL-6
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/54—Interleukins [IL]
- C07K14/55—IL-2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/555—Interferons [IFN]
- C07K14/56—IFN-alpha
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/52—Cytokines; Lymphokines; Interferons
- C07K14/555—Interferons [IFN]
- C07K14/565—IFN-beta
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K17/00—Carrier-bound or immobilised peptides; Preparation thereof
- C07K17/02—Peptides being immobilised on, or in, an organic carrier
- C07K17/10—Peptides being immobilised on, or in, an organic carrier the carrier being a carbohydrate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B31/00—Preparation of derivatives of starch
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B31/00—Preparation of derivatives of starch
- C08B31/003—Crosslinking of starch
- C08B31/006—Crosslinking of derivatives of starch
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B31/00—Preparation of derivatives of starch
- C08B31/08—Ethers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B31/00—Preparation of derivatives of starch
- C08B31/18—Oxidised starch
- C08B31/185—Derivatives of oxidised starch, e.g. crosslinked oxidised starch
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08H—DERIVATIVES OF NATURAL MACROMOLECULAR COMPOUNDS
- C08H1/00—Macromolecular products derived from proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Abstract
本发明涉及含有一个或多个HAS分子的羟烷基淀粉(HAS)-多肽偶联物(HAS-多肽),其中每个HAS通过碳水化合物部分或硫醚与多肽偶联,本发明还涉及该偶联物的生产方法。在一个优选实施方案中,该多肽是促红细胞生成素(EPO)。
Description
本发明涉及与羟烷基淀粉(HAS)特别是与羟乙基淀粉偶联的多肽,特别是促红细胞生成素。
为了获得一种特定的生理效果而对循环系统应用多肽特别是酶或细胞因子,是现代医学中一种众所周知的方法。
促红细胞生成素(EPO)是红系祖细胞成熟为红细胞所必需的一种糖蛋白激素。成人在肾脏中产生EPO。EPO是调节循环中红细胞水平所必需的。以低组织氧水平为标志的疾病引起EPO生物合成增加,随后刺激红细胞生成。例如,在慢性肾衰竭中,肾功能的丧失一般导致EPO的生物合成减少,伴有红细胞减少。
促红细胞生成素是一种分子量约为34,000Da的酸性糖蛋白激素。人促红细胞生成素是一种166个氨基酸的多肽,其作为一种单体天然存在(Lin等人,1985,PNAS 82,7580-7584,EP 148 605 B2,EP411 678 B2)。例如,美国专利4,703,008描述了对编码促红细胞生成素的基因的鉴定、克隆和表达。例如,美国专利4,667,016描述了从支持含有重组促红细胞生成素质粒的哺乳动物细胞生长的细胞培养基中纯化重组促红细胞生成素。
在本技术领域中公认,EPO的体内生物活性主要取决于唾液酸与EPO结合的程度(参见,例如,EP 428 267 B1)。理论上,14个唾液酸分子能够与一个EPO分子在与N-和O-糖基化位点连接的碳水化合物侧链末端结合。获得高度唾液酸化的EPO制品需要非常复杂的纯化步骤。
关于促红细胞生成素的更详细的信息,见Krantz,Erythropoietin,1991,Blood,77(3):419-34(Review),和Cerami,Beyond erythropoiesis:novel applications forrecombinant human erythropoietin,,2001,Semin Hematol.,(3Supp17):33-9(Review)。
多肽和酶应用的一个众所周知的问题是这些蛋白质通常不能显示令人满意的稳定性。具体而言,促红细胞生成素的血浆半衰期相对较短(Spivak和Hogans,1989,Blood 73,90;McMahon等人,1990,Blood 76,1718)。这意味着治疗性血浆水平快速降低,必须进行重复静脉内施用。此外,在某些情况下,还观察到针对这些肽的免疫应答。
通常认为,当多肽与聚合分子偶联时,多肽的稳定性能够提高且针对这些多肽的免疫应答降低。WO 94/28024公开了用聚乙二醇(PEG)修饰的生理活性多肽显示免疫原性和抗原性降低,在血流中的循环远长于未偶联的蛋白质,即清除所需时间更长。
然而,PEG-药物偶联物也有几个缺点,例如,它们不具有能够被体内降解途径组件识别的天然结构。因此,除了PEG-偶联物之外,还制备了其它偶联物和蛋白质聚合物。交联不同蛋白质和大分子(如聚合酶)的多种方法在文献中已有描述(参见,例如,Wong,《蛋白质偶联和交联的化学》(Chemistry of protein conjugation andcross-linking),1993,CRCS,Inc.)。
羟乙基淀粉(HES)是天然存在的支链淀粉衍生物,可被体内α-淀粉酶降解。本领域已经了解HES-蛋白质偶联物的制备(参见,例如,DE 26 16 086或DE 26 46 854中的HES-血红蛋白偶联物)。
DE 26 46 854公开了血红蛋白与HES的偶联方法。在这些方法中,HES与高碘酸钠反应,产生与血红蛋白连接的二醛。与此不同,DE 2616 086公开的血红蛋白与HES的偶联方法中,交联剂(例如bromocyane)首先与HES结合,随后血红蛋白与中间产物连接。
HES是碳水化合物聚合物支链淀粉的取代衍生物,在玉米淀粉中以浓度高达95%重量存在。HES显示有利的生物学性质,可以用作血容量替代剂,在临床血液稀释治疗中使用(Sommermeyer等人,1987,Krankenhauspharmazie,8(8),271-278;Weidler等人,1991,Arzneim.-Forschung/Drug Res.,41,494-498)。
支链淀粉由葡萄糖部分(moiety)组成,其中主链中存在α-1,4-糖苷键,在分支位点处有α-1,6-糖苷键。该分子的物理化学性质主要取决于糖苷键的类型。由于α-1,4-糖苷键具有切口,产生每圈大约有6个葡萄糖单体的螺旋结构。
该聚合物的物理化学以及生物化学性质能够通过取代改变。通过碱羟乙基化作用可以引入羟乙基。通过改变反应条件,能够研究未取代葡萄糖单体中各羟基对羟乙基化的不同反应性。由于这一事实,技术人员能够有限程度地影响取代模式。
因此,HES的特征主要在于分子量分布和取代程度。有两种方式可以描述取代程度:
1.以取代葡萄糖单体相对于所有葡萄糖部分的比例描述取代程度(DS)。
2.以描述每葡萄糖部分的羟乙基数的“摩尔取代”(MS)来描述取代程度。
HES溶液作以多分散组合物存在,每个分子在聚合程度、分支位点数量和型式以及取代型式等方面彼此不同。因此HES是不同分子量化合物的混合物。因此,利用统计学方法,根据平均分子量确定具体的HES溶液。在本文中,Mn被计算为取决于分子数的算术平均值。此外,重量平均值Mw代表取决于HES质量的单位。
本领域中公开的HES-药物偶联物具有HES不能与药物位点特异性偶联的缺点。因此,这种偶联产生含有多种成分的非常不均一的产物,由于在偶联步骤中三维结构被破坏,这些成分可能没有活性。
总之,仍然需要稳定性和/或生物活性提高的进一步改进的多肽。对于促红细胞生成素尤其如此,必须将具有含高度唾液酸因此具有高活性的同种型从唾液酸含量低的同种型中纯化(见EP 428 267 B1)。因此,如果可以采用能够产生高活性多肽而不需要复杂纯化的生产方法,将是非常有利的。遗憾的是,在细菌或昆虫细胞中生产多肽通常较困难,因为通常产生的多肽不具有正确折叠和天然构象,并且缺乏适当的糖基化。
因此,本发明的一个目的是提供体内生物活性高、能够以低成本容易生产的多肽衍生物,特别是促红细胞生成素衍生物。此外,本发明的另外一个目的是提供一种生产多肽衍生物的方法,其易于进行,并且产生高生物活性的产物。本发明的另外一个目的是提供含有高生物活性的多肽衍生物的药物组合物。
根据本发明的一个方面,该问题用含有一个或多个HAS分子的羟烷基淀粉(HAS)-促红细胞生成素(EPO)-偶联物(HAS-EPO)来解决,其中每个HAS与EPO通过
a)碳水化合物部分;或
b)硫醚偶联。
本发明的HAS-EPO具有以下优点:与偶联前的促红细胞生成素相比,生物稳定性提高。此外,它也显示比标准BRP EPO更高的生物活性。这主要是基于以下事实:HAS-EPO很少甚至不被肝脏和肾脏的清除系统识别,因此在循环系统中长时间存在。此外,由于HAS以位点特异性的方式连接,因此EPO体内生物活性因HAS与EPO偶联而被破坏的危险被最小化。
本发明的HAS-EPO主要含有两种成分,即促红细胞生成素(EPO)多肽和与之连接的羟烷基淀粉(HAS)。
EPO可以是任何人来源的(参见,例如,Inoue,Wada,Takeuchi,1994,An improved method for the purification of humanerythropoietin with high in vivo activity from the urine ofanemic patients,Biol Pharm Bull.17(2),180-4;Miyake,Kung,Goldwasser,1977,Purification of human erythropoietin,J BiolChem.,252(15),5558-64)或者另外一种哺乳动物来源的,可以从天然存在的来源(如人肾脏、人胚胎肝脏,或动物优选猴肾)中通过纯化获得。此外,表述“促红细胞生成素”或“EPO”也包括具有红细胞生成活性的EPO变体,其中一个或多个氨基酸(例如1-25个,优选1-10个,更优选1-5个,最优选1个或2个)已经被置换为另外的氨基酸(参见,例如EP 640 619 B1)。红细胞生成活性的测量在本领域中有描述(关于体外活性的测量参见例如Fibi等人,1991,Blood,77,1203 ff;Kitamura等人,1989,J.Cell Phys.,140,323-334;关于EPO体内活性的测量参见Ph.Eur.2001,911-917;Ph.Eur.2000,1316Erythropoietini solutio concentrata,780-785;欧洲药典(1996/2000);欧洲药典,1996,Erythropoietin concentratedsolution,Pharmaeuropa.,8,371-377;Fibi,Hermentin,Pauly,Lauffer,Zettlmeissl.,1995,N-and O-glycosylation muteins ofrecombinant human erythropoietin secreted from BHK-21 cells,Blood,85(5),1229-36;(在第1、2、3天向雌性NMRI小鼠注射EPO和修饰的EPO型(等量蛋白质50ng/小鼠),在第4天采集血样,测定网织红细胞))。测量EPO活性试验的其它出版物包括Barbone,Aparicio,Anderson,Natarajan,Ritchie,1994,Reticulocytesmeasurements as a bioassay for erythropoeitin,J.Pharm.Biomed.Anal.,12(4),515-22;Bowen,Culligan,Beguin,Kendall,Vlllis,1994,Estimation of effective and total erythropoiesis inmyelodysplasia using serum transferring receptor anderythropoietin concentrations,with automated reticulocyteparameters,Leukemi,8(1),151-5;Delorme,Lorenzini,Giffin,Martin,Jacobsen,Boone,Elliott,1992,Role of glycosylationon the secretion and biological activity of erythropoietin,Biochemistry,31(41),9871-6;Higuchi,Oh-eda,Kuboniwa,Tomonoh,Shimonaka,Ochi,1992;Role of sugar chains in theexpression of the biological activity of human erythropoietin,J.Biol.Chem.,267(11),7703-9;Yamaguchi,Akai,Kawanishi,Ueda,Masuda,Sasaki,1991,Effects of site-directed removalof N-glycosylation sites in human erythropoietin on itsproduction and biological properties,J.Biol.Chem.,266(30),20434-9;Takeuchi,Inoue,Strickland,Kubota,Wada,Shimizu,Hoshi,Kozutsumi,Takasaki,Kobata,1989,Relationship betweensugar chain structure and biological activity of recombinanthuman erythropoietin produced in Chinese hamster ovary cells,Proc.Natl.Acad.Sci.USA,85(20),7819-22;Kurtz,Eckardt,1989,Assay methods for erythropoietin,Nephron.,51(1),11-4(German);Zucali,Sulkowski,1985,Purification of humanurinary erythropoietin on controlled-pore glass and silicicacid,Exp.Hematol.,13(3),833-7;Krystal,1983,Physical andbiological characterization of erythroblast enhancingfactor(EEF),a late acting erythropoeticstimulator in serumdistinct from erythropoietin,Exp.Hematol.,11(1),18-31。
优选地,EPO通过重组产生。这包括在真核或原核细胞中产生,优选地在哺乳动物、昆虫、酵母、细菌细胞中或者在适于重组生产EPO的其它任何细胞类型中产生。此外,EPO也可以在转基因动物中(例如在体液中,如奶、血液等)、在转基因鸟特别是家禽优选在鸡的蛋中,或者在转基因植物中表达。
多肽的重组产生为本领域公知。通常包括用一种适当的表达载体转染宿主细胞,在能够生产多肽的条件下培养宿主细胞,以及从宿主细胞中纯化多肽。关于详细信息,参见例如Krystal,Pankratz,Farber,Smart,1986,Purification of human erythropoietin tohomogeneity by a rapid five-step procedure,Blood,67(1),71-9;Quelle,Caslake,Burkert,Wojchowski,1989,High-levelexpression and purification of a recombinant humanerythropoietin produced using a baculovirus vector,Blood,74(2),652-7;EP 640 619 B1和EP 668 351 B1。
在一个优选实施方案中,EPO具有人EPO的氨基酸序列(见EP 148605 B2)。
EPO可以含有通过N-和/或O-连接的糖基化作用与EPO连接的一个或多个碳水化合物侧链(优选1-4个,优选4个),即EPO被糖基化。当在真核细胞中生产EPO时,多肽通常在翻译后被糖基化。因此,在哺乳动物特别是在人、昆虫或酵母细胞内的生物合成过程中,碳水化合物侧链可以与EPO连接。糖基化EPO的结构和性质在本领域中已经广泛研究(参见EP 428 267 B1;EP 640 619 B1;Rush,Derby,Smith,Merry,Rogers,Rohde,Katta,1995,Microheterogeneity oferythropoietin carbohydrate structure,Anal Chem.,67(8),1442-52;Takeuchi,Kobata,1991,Structures and functionalroles of the sugar chains of human erythropoietins,Glycobiology,1(4),337-4 6(Review)。
HAS可以与EPO直接偶联,或者也可以通过一个接头分子偶联。接头分子的性质取决于HAS与EPO连接的方式。表1及下文描述了可能的接头官能团。有几种接头可以作为商品获得(例如,来自Pierce,可获自Perbio Science Deutschland GmbH,Bonn,德国))。表2描述了一些合适的接头。接头的性质及其用途在下文关于HES-EPO生产方法的部分中详细描述。
根据本发明HAS-EPO偶联物的一个优选实施方案,HAS通过一个碳水化合物部分与EPO偶联。
在本发明上下文中,术语“碳水化合物部分”指羟基醛或羟基酮,以及它们的化学修饰(见Rmpp Chemielexikon,ThiemeVerlagStuttgart,德国,第9版1990,第9卷,2281-2285,以及此处引用的文献)。此外它还指天然存在的碳水化合物部分(如葡萄糖、半乳糖、甘露糖、唾液酸等)的衍生物。该术语也包括环结构已经打开并被化学氧化的天然存在碳水化合物部分。
碳水化合物部分可以与EPO多肽骨架直接连接。优选地,碳水化合物部分是碳水化合物侧链的一部分。在这种情况下,HAS连接的碳水化合物部分与EPO多肽骨架之间还可能存在其它碳水化合物部分。更优选地,碳水化合物部分是碳水化合物侧链的末端部分。
在一个更优选的实施方案中,HAS与碳水化合物侧链的半乳糖残基偶联,优选与碳水化合物侧链的末端半乳糖残基偶联。通过除去末端唾液酸,随后氧化,该半乳糖残基能够用于偶联(见下文)。
在另外一个更优选的实施方案中,HAS与碳水化合物侧链的唾液酸残基偶联,优选与碳水化合物侧链的末端唾液酸残基偶联。
此外,HAS也可以通过硫醚与EPO偶联。如下文详述的,S原子可以来自于附于EPO上的任何天然或非天然存在的SH基团。
在一个优选实施方案中,S原子可以来源于被引入HES氧化碳水化合物部分(优选作为EPO碳水化合物侧链一部分的氧化碳水化合物部分)中的SH基团(见下文)。
优选地,硫醚中的S原子来源于天然存在的半胱氨酸或者添加的半胱氨酸。更优选EPO具有人EPO的氨基酸序列,天然存在的半胱氨酸是半胱氨酸29和/或33。在一个更优选的实施方案中,HAS与半胱氨酸29偶联,半胱氨酸33被置换为另外一种氨基酸。此外,HAS也可以与半胱氨酸33偶联,而半胱氨酸29被置换为另外一种氨基酸。
在本发明中,术语“添加的半胱氨酸”是指多肽(优选地EPO)含有在野生型多肽中不存在的半胱氨酸残基。
在本发明该方面,半胱氨酸可以是在EPO的N端或C端添加的额外氨基酸。
此外,也可以通过将天然存在的氨基酸置换为半胱氨酸来添加半胱氨酸。适当的方法在本领域中公知(见上文)。在本发明该方面的上下文中,EPO优选是人EPO,置换的氨基酸残基优选是丝氨酸126。
HAS-EPO的第二种组分是羟烷基淀粉(HAS)。
在本发明中,术语“羟烷基淀粉”指被羟烷基取代的淀粉衍生物。此处,烷基可以被取代。优选地,羟烷基含有2-10个碳原子,更优选2-4个碳原子。因此,“羟烷基淀粉”优选包括羟乙基淀粉、羟丙基淀粉和羟丁基淀粉,其中羟乙基淀粉和羟丙基淀粉是优选的。
HAS的羟烷基至少含有一个OH基团。
表述“羟烷基淀粉”也包括以下衍生物,其中烷基是单取代或多取代的。对此,只要HAS保持水溶性,优选烷基被卤素特别是氟、或者被芳基取代。此外,羟烷基的末端羟基也可以酯化或者醚化。另外,羟烷基淀粉的烷基也可以是线性的或分支的。
此外,也可以使用线性或分支的取代或未取代的链烯基代替烷基。
对于本发明的所有实施方案,羟乙基淀粉(HES)是最优选的。
在本发明中,羟乙基淀粉的平均分子量(重量平均值)可以是1-300kDa,其中5-100kDa的平均分子量是更优选的。羟乙基淀粉可以进一步显示相对于羟乙基的0.1-0.8的摩尔取代程度和2-20的C2∶C6取代比。
HAS-EPO的每个EPO分子可以含有1-12个,优选1-9、1-6或1-3个,最优选1-4个HAS分子。可以在产物水解及所得单糖衍生化后,利用GC-MS,通过定量碳水化合物组成分析确定每个EPO分子的HAS分子数(见Chaplin和Kennedy(编著),1986,《碳水化合物分析:一种实用方法》(Carbohydrate Analysis:apractical approach),IRL Press Practical approach series(ISBN 0-947946-44-3),具体见第1章,单糖,第1-36页;第2章,寡糖,第37-53页,第3章,中性多糖,第55-96页)。
本发明的HAS-EPO偶联物可以显示与重组天然EPO基本相同的体外生物活性,因为体外生物活性只取决于EPO受体的结合亲和力。测定体外生物活性的方法在本领域公知(见上文)。
此外,HAS-EPO也显示比用作偶联初始材料的EPO(未偶联的EPO)更高的体内活性。测定体内生物活性的方法在本领域公知(见上文)。此外,实施例9和10也给出了测定体内和体外EPO活性的测定法。
如果将未偶联的EPO的体内活性设为100%,则HAS-EPO偶联物可能显示出110-500%、优选300-400%,或者110%-300%、优选、110%-200%、更优选110%-180%,或者110-150%、最优选110%-140%的体内活性。
与Amgen的高度唾液酸化EPO相比(见EP 428 267 B1),如果将高度唾液酸化EPO的体内活性设为100%,则HAS-EPO显示优选至少50%、更优选至少70%、更优选至少85%,或者至少95%、至少150%、至少200%或者至少300%的高度唾液酸化EPO的体内活性。最优选地,它显示至少95%的高度唾液酸化EPO的体内活性。
本发明HAS-EPO偶联物的高体内生物活性主要是基于以下事实:该HAS-EPO偶联物在循环中保持的时间比未偶联的EPO更长,因为它较少被肝脏清除系统识别,并且由于分子量较高,肾清除减少。测定EPO在体内循环半衰期的方法在本领域公知(Sytkowski,Lunn,Davis,Feldman,Siekman,1998,Human erythropoietin dimmers withmarkedly enhanced in vivo activity,Proc.Natl.Acad.Sci.USA,95(3),1184-8)。
因此,本发明的一个明显优点是:提供可以比目前作为商品获得的EPO制品以更低频率施用的HAS-EPO。标准EPO制品必须至少连续施用3天,而本发明HAS-EPO偶联物优选每周施用两次,更优选每周一次。
下文公开的涉及EPO或HAS特性的关于本发明生产HAS-EPO方法的所有实施方案,也适用于本发明的HAS-EPO偶联物。
羟烷基淀粉是淀粉的一种醚衍生物。除了所述醚衍生物以外,在本发明中也能够使用其它淀粉衍生物。例如,可以使用含有酯化羟基的衍生物。这些衍生物可以是,例如,具有2-12个碳原子的未取代单羧酸或二羧酸的衍生物,或者其取代衍生物的衍生物。特别有用的是具有2-6个碳原子的未取代单羧酸的衍生物,特别是乙酸的衍生物。对此,乙酰基淀粉、丁基淀粉或丙基淀粉是优选的。
此外,含有2-6个碳原子的未取代二羧酸的衍生物也是优选的。
对于二羧酸的衍生物,二羧酸的第二个羧基也被酯化是有利的。此外,二羧酸的单烷基酯衍生物在本发明中也是适用的。
对于取代的单羧酸或二羧酸,取代基优选可以与上述用于取代烷基残基的取代基相同。
淀粉的酯化技术在本领域公知(参见,例如Klemm D.等人,《综合纤维素化学》(Comprehensive Cellulose Chemistry)第二卷,1998,Whiley-VCH,Weinheim,纽约,具体见第4.4章,纤维素的酯化(ISBN 3-527-29489-9)。
另一方面,本发明涉及生产羟烷基淀粉(HAS)-促红细胞生成素(EPO)偶联物(HAS-EPO)的方法,其包括下列步骤:
a)提供能够与修饰的HAS反应的EPO,
b)提供能够与步骤a)中EPO反应的修饰的HAS,和
c)使步骤a)的EPO与步骤b)的HAS反应,从而产生一种含有一个或多个HAS分子的HAS-EPO,其中每个HAS都与EPO通过
i)碳水化合物部分;或
ii)硫醚偶联。
本发明的方法具有产生显示高生物活性的HAS-EPO偶联物的优点。此外,本发明的方法也具有能够以低成本生产有效的EPO衍生物的优点,因为该方法不包括导致低终产率的复杂且耗时的纯化步骤,例如,不需要纯化掉已知显示低体内生物活性或者无体内生物活性的低唾液酸化的EPO型。具体而言,实施例20证实,用极少修饰步骤生产的一种HES-EPO显示3倍于标准BRP-EPO的活性。
因此,在本发明方法的第一个步骤中,提供一种能够与修饰的HAS反应的EPO。
本发明所用的术语“提供”解释为在各步骤后,可以获得一种具有所需特性的分子(在步骤a)中为EPO,在步骤b)中为HAS)。
对于步骤a),包括从天然来源中纯化EPO,以及在宿主细胞或生物中重组生产,以及必要时对这样获得的EPO进行修饰。
任何关于作为本发明初始材料的EPO的情况,同样适用于作为本发明HAS-EPO偶联物一部分的促红细胞生成素。对此,以上公开的优选实施方案也适用于本发明方法。
因此,在一个优选实施方案中,EPO具有人EPO的氨基酸序列。
优选EPO重组产生。包括在真核或原核细胞中产生,优选在哺乳动物、昆虫、酵母、细菌细胞中或者在适于重组生产EPO的其它任何细胞类型中产生。此外,EPO也可以在转基因动物中(例如在体液中,如奶、血液等)、在转基因鸟类特别是家禽优选在鸡的蛋中,或者在转基因植物中表达。
多肽的重组产生在本领域中公知。通常包括用一种适当的表达载体转染宿主细胞,在能够生产多肽的条件下培养宿主细胞,以及从宿主细胞中纯化多肽(Krystal,Pankratz,Farber,Smart,1986,Purification of human erythropoietin to homogeneity by a rapidfive-step procedure,Blood,67(1),71-9;Quelle,Caslake,Burkert,Wojchowski,1989,High-level expression andpurification of a recombinant human erythropoietin producedusing a baculoviru vector,Blood,74(2),652-7;EP 640 619 B1和EP 668 351 B1)。
EPO可以含有通过N-和/或O-连接的糖基化作用与EPO连接的一个或多个碳水化合物侧链,即EPO被糖基化。当在真核细胞中生产EPO时,多肽通常在翻译后糖基化。因此,在哺乳动物,特别是在人、昆虫或酵母细胞内的生物合成过程中,碳水化合物侧链可以与EPO连接,这些细胞可以是转基因动物的细胞(见上文),或从动物中提取的或者仍然保留在动物中的细胞。
这些碳水化合物侧链可以在适当细胞中表达后进行化学或酶修饰,例如通过除去或添加一个或多个碳水化合物部分(参见,例如,Dittmar,Conradt,Hauser,Hofer,Lindenmaier,1989,蛋白质设计进展(Advances in Protein design);Bloecker,Collins,Schmidt和Schomburg编著,GBF-专著(GBF-Monographs)12,231-246,VCHPublishers,Weinheim,New York,Cambridge)。
本发明方法的目的是提供一种HAS-EPO,其含有一个或多个HAS分子,其中该HAS通过碳水化合物部分(i)或者通过硫醚(ii)与EPO偶联。因此,步骤a)提供的EPO应当具有以下特性:能够通过碳水化合物部分和/或通过硫醚偶联。因此,在步骤a)之后,EPO优选地可以含有
(1)至少一个直接或者通过一个接头分子与巯基团或者碳水化合物部分连接的反应性基团,其能够与HES或修饰的HES反应,
(2)至少一个修饰的HAS能够与之偶联的碳水化合物部分,和/或
(3)至少一个游离SH基。
对于上述可能性(1),步骤a)的EPO优选地可以通过将适当的接头分子与EPO的SH基或碳水化合物部分偶联获得。实施例4中的2.1提供了这种修饰的EPO的一个实例。重要的是确保接头分子的添加不损害EPO。而这为本领域技术人员所公知。
对于上述可能性(2),在一个优选实施方案中,修饰的HAS通过碳水化合物部分与EPO偶联。
碳水化合物部分可以与EPO多肽骨架直接连接。优选地,碳水化合物部分是碳水化合物侧链的一部分。在这种情况下,与HAS连接的碳水化合物部分与EPO多肽骨架之间还可能存在其它碳水化合物部分。更优选地,碳水化合物部分是碳水化合物侧链的末端部分。
因此,在一个优选实施方案中,修饰的HAS与碳水化合物链连接(通过一个接头,或者不通过接头,见下文),后者与EPO的N-和/或O-糖基化位点连接。
然而,本发明也包括,EPO含有与修饰的HAS偶联的其它碳水化合物部分。本领域公知通过酶或者通过遗传工程连接碳水化合物部分与多肽的技术,以及随后在适当细胞中的表达(Berger,Greber,Mosbach,1986,Galactosyltransferase-dependent sialylation ofcomplex and endo-Nacetylglucosaminidase H-treated coreN-glycans in vitro,FEBS Lett.,203(1),64-8;Dittmar,Conradt,Hauser,Hofer,Lindenmaier,1989,《蛋白质设计进展》;Bloecker,Collins,Schmidt和Schomburg编著,GBF-专著,12,231-246,VCHPubishers,Weinheim,New York,Cambridge)。
在本发明方法的一个优选实施方案中,为了能够与修饰的HAS反应而氧化碳水化合物部分。这种氧化能够以化学或酶学方法进行。
化学氧化多肽碳水化合物部分的方法在本领域公知,包括用高碘酸盐处理(Chamow等人,1992,J.Biol.Chem.,267,15916-15922)。
通过化学氧化,原则上能够氧化位于末端或者不位于末端的任何碳水化合物部分。然而,通过选择温和的条件(1mM高碘酸盐,0℃,不同于严格条件:10mM高碘酸盐,室温1小时),可以优选地氧化碳水化合物侧链的末端碳水化合物部分,例如唾液酸或半乳糖。
此外,碳水化合物部分也可以酶氧化。用于氧化各碳水化合物部分的酶为本领域公知,例如,对于半乳糖的酶是半乳糖氧化酶。
如果准备氧化末端半乳糖部分,如果在能够连接唾液酸与碳水化合物链的细胞例如哺乳动物细胞中,或者在遗传修饰后能够连接唾液酸与碳水化合物链的细胞中生产EPO,最终必须(部分或完全)除去末端唾液酸。去除唾液酸的化学或酶法在本领域中公知(Chaplin和Kennedy(编著),1996,《碳水化合物分析:一种实用方法》(Carbohydrate Analysis:a practical approach),具体见第5章Montreuill,糖蛋白,第175-177页;IRL Press Practicalapproach series(ISBN0-94794 6-4 4-3))。
本发明还包括,在步骤a)中将要与修饰的HAS连接的碳水化合物部分与EPO连接。如果希望连接半乳糖,可以利用半乳糖转移酶实现。这些方法在本领域中公知(Berger,Greber,Mosbach,1986,Galactosyltransferase-dependent sialylation of complex andendo-N-acetylglucoaminidase H-treated core N-glycans in vitro,FEBS Lett.,203(1),64-8)。
在一个最优选的实施方案中,在步骤a)中,必要时,优选部分或完全除去(酶和/或化学)末端唾液酸之后,通过氧化EPO的一个或多个碳水化合物侧链的至少一个末端糖单位(优选半乳糖)来修饰EPO(见上文)。
因此,优选地,修饰的HAS与碳水化合物链氧化的末端糖单位偶联,优选与半乳糖偶联。
此外,修饰的HAS优选地也可以与末端唾液酸偶联,后者优选地在本发明的方法的步骤a)中氧化。
在一个进一步优选的实施方案中(见上述第(3)点),EPO含有至少一个游离SH基。
根据一个优选实施方案,该SH基可以与一个优选氧化的碳水化合物部分连接,例如通过使用羟胺衍生物,例如2-(氨氧基)乙硫醇盐酸盐(Bauer L.等人,1965,J.Org.Chem.,30,949),或者通过使用酰肼衍生物,例如巯基乙酸酰肼(Whitesides等人,1977,J.Org.Chem.,42,332)。偶联这些分子与EPO氧化碳水化合物部分的方法可以与实施例方案8和9所述类似。
在一个进一步优选的实施方案,游离SH基是天然存在的半胱氨酸或添加的半胱氨酸的一部分。
哺乳动物EPO含有几个通常形成二硫键的半胱氨酸。然而,通过将至少一个半胱氨酸置换为另外一种氨基酸(例如通过重组方法),可以获得具有至少一个含游离SH基的天然存在半胱氨酸的EPO。氨基酸置换方法在本领域中公知(Elliott,Lorenzini,Chang,Barzilay,Delorme,1997,Mapping of the active site of recombinant humanerythropoietin,Blood,89(2),493-502;Boissel,Lee,Presnell,Cohen,Bunn,1993,Erythropoietin structure-functionrelationships.Mutant proteins that test a model of tertiarystructure,J Biol Chem.,268(21),15983-93))。
优选地,EPO具有人EPO的氨基酸序列,天然存在的半胱氨酸是半胱氨酸29和/或33。
因此,在一个优选实施方案中,半胱氨酸33被置换为另外一种氨基酸,在步骤c)中,修饰的HAS与半胱氨酸29偶联。
在一个进一步优选的实施方案中,半胱氨酸29被置换为另一种氨基酸,在步骤c)中,修饰的HAS与半胱氨酸33偶联。
在本发明中,术语“添加的半胱氨酸”是指多肽(优选EPO)含有一个在野生型多肽中不存在的半胱氨酸残基。这能够如下实现:在多肽的N端或C端添加(例如通过重组方法)一个半胱氨酸残基,或者将一个天然存在的氨基酸置换(例如通过重组方法)为半胱氨酸。本领域技术人员公知这些方法(见上文)。
优选地,通过将一个天然存在的氨基酸置换为一个半胱氨酸来添加半胱氨酸。
在一个优选实施方案中,EPO是人EPO,被置换的氨基酸残基是丝氨酸126。
优选地,修饰的HAS在步骤c)中与添加的半胱氨酸偶联。
在本发明方法的步骤b)中,提供能够与步骤a)的EPO反应的修饰的HAS。
对此,HAS优选地可以在其还原性末端被修饰。这样有化学反应易于控制且技术人员能够确保HAS的哪个基团在反应过程中被修饰的优点。由于只将一个基团引入HAS中,能够防止不同EPO分子通过多功能HAS分子交联和其它副反应。
因此,修饰的HAS能够与下列成分反应:
(1)至少一个直接或者通过接头分子与EPO的巯基团或碳水化合物部分连接的基团,
(2)至少一个碳水化合物部分,优选其被氧化,和/或
(3)至少一个游离SH基。
对于上述第(1)点,HAS的修饰取决于与EPO连接的基团。其机制在本领域中公知。实施例4中的2.1给出了一个实例。
对于上述第(2)点和第(3)点,本领域公知几种修饰HAS的方法。这些方法的基本原理是,为了能够与碳水化合物部分或SH基反应而修饰HAS的反应性基团,或者一个接头分子与HAS偶联,后者含有能够与碳水化合物部分或SH基反应的反应性基团。
对于第(2)点,修饰的HAS能够与氧化的碳水化合物部分反应,优选与末端糖残基、更优选与半乳糖或者与末端唾液酸反应。
已知有几种方法可以修饰HAS,使其能够与氧化的优选末端糖残基反应。如上所述,可以在HES-链的还原性末端区域选择性地引入这种修饰。在这种情况下,在第一步中,醛基被氧化为内酯。这些修饰包括但不限于:直接或者通过一个接头向HAS上添加酰肼、氨基(以及羟氨基)、氨基脲或硫醇功能基团。这些技术在实施例2-4中进一步详细描述。此外,机制本身在本领域中公知(参见,例如DE19628705A1;Hpoe等人,1981,Carbohydrate Res.,91,39;Fissekis等人,1960,Journal of Medicinal and Pharmaceutical Chemistry,2,47;Frie,1998,毕业论文,Fachhochschule Hamburg,DE)。
在本发明中,优选添加酰肼或羟氨基官能团。在这种情况下,优选地通过在pH 5.5下进行本发明方法步骤c)的反应,确保修饰的HAS与EPO的氧化的碳水化合物部分选择性反应,而没有赖氨酸侧链与氧化的糖残基形成亚胺从而引起分子间或分子内EPO交联。
对于第(3)点,也公知几种用于修饰HAS的方法,使其能够与游离SH基反应。优选地,在HES-链的还原性末端区域选择性地引入这种修饰。这些方法包括但不限于向HAS添加马来酰亚胺、二硫醚或卤代乙酰胺官能团。这些技术在实施例2-4中进一步详细描述。
关于这些技术的进一步的细节,可见Chamov等人,1992,J.Biol.Chem.,267,15916;Thorpe等人,1984,Eur.J.Biochem.,140,63;Greenfield等人,1990,Cancer Research,50,6600,以及实施例2中的1.3引用的文献。
表1列出了其它一些可能的官能团,提供了对可能的接头分子的系统综述。此外,机制本身在本领域公知。
可以在本发明中使用的几种接头分子在本领域中公知,或者可以作为商品获得(例如来自Pierce,可以获自Perbio ScienceDeutschland GmbH,Bonn,德国)。表2列出了实例。
在本发明方法的步骤c)中,步骤a)的EPO与步骤b)的HAS反应,从而产生含一个或多个HAS分子的HAS-EPO,其中HAS通过碳水化合物部分或者通过硫醚与EPO偶联。
原则上,使EPO与修饰HAS反应的详细方法取决于EPO和/或HAS各自的修饰,为本领域公知(参见,例如,Rose,1994,J.Am.Chem.Soc.,116,30,O′Shannessay和Wichek,1990,AnalyticalBiochemistry,191,1;Thorpe等人,1984,Eur.J.Biochem.,140,63;Chamov等人,1992,J.Bioh.Chem.267,15916)。
关于本发明举例说明的方法,实施例2-4,特别是实施例4给出了详细描述。
步骤c)可以在至少含有10%重量H2O的反应介质中进行。
在本发明方法的这一优选实施方案中,反应介质含有至少10%重量的水,优选至少50%、更优选至少80%,例如90%或者可达100%。据此可以计算有机溶剂的含量。因此,该反应在水相中进行。优选的反应介质是水。
本发明方法这一实施方案的一个优点是,不必使用在毒理学上危险的溶剂,因此在生产过程后不必除去这些溶剂来避免溶剂的污染。此外,对于残余的毒理学上危险的溶剂,不必进行额外的质量控制。优选使用毒理学上不危险的溶剂作为有机溶剂,如乙醇或丙二醇。
本发明方法的另外一个优点是,避免了有机溶剂诱导的不可逆的或可逆的结构改变。因此,根据本发明方法获得的多肽不同于在有机溶剂(如DMSO)中制备的多肽。
此外,也意外地发现,HAS与药物在水溶液中的偶联最小化或者避免了副反应。因此,本发明方法的这个实施方案产生高纯度的改良产物。
在本发明中,术语“羟烷基淀粉”指被羟烷基取代的淀粉衍生物。其中,烷基可以被取代。优选羟烷基含有2-10个碳原子,更优选2-4个碳原子。因此“羟烷基淀粉”优选地包括羟乙基淀粉、羟丙基淀粉和羟丁基淀粉,其中羟乙基淀粉和羟丙基淀粉是优选的。
HAS的羟烷基含有至少一个OH基。
对于本发明的所有实施方案,羟乙基淀粉(HES)是最优选的。
表述“羟烷基淀粉”也包括以下衍生物,其中烷基是单取代或多取代的。对此,只要HAS保持水溶性,优选烷基可以被卤素特别是氟、或者芳基取代。此外,羟烷基的末端羟基也可以酯化或者醚化。另外,羟烷基淀粉的烷基也可以是线性的或分支的。
此外,除了烷基,也可以使用线性或分支的取代或未取代的烯基。
在本发明中,羟乙基淀粉的平均分子量可以是1-300kDa,其中5-100kDa的平均分子量是更优选的。羟乙基淀粉可以进一步显示相对于羟乙基的0.1-0.8的摩尔取代程度,和2-20的C2∶C6取代比。
通过本发明方法生产的HAS-EPO能够被如下纯化和表征:
HAS-EPO的分离可以用已知的纯化天然和重组EPO的方法进行(例如大小排阻层析、离子交换层析、RP-HPLC、羟基磷灰石层析、疏水作用层析、实施例20.8所述的方法或其组合)。
HAS与EPO多肽的共价连接能够在修饰蛋白质水解后通过碳水化合物组成分析证实(EPO的三个N-糖基化位点上存在的羟乙基葡萄糖与甘露糖之比)。
EPO的N-连接寡糖处的HAS修饰能够如下证实:除去HAS修饰的N-聚糖,利用SDS-PAGE+/-Western Blotting观察预知的向较高迁移率的转变。
EPO在半胱氨酸残基处的HAS修饰能够根据以下情况证实:通过RP-HPLC和MALDI/TOF-MS不能在HAS修饰产物的蛋白水解片段中检测到相应的蛋白水解Cys-肽(Zhou等人,1998,Application ofcapillary electrophoresis,liquid chromatogfaphy,electrospray-mass spectrometry and matrix-assistedlaserdesorption/ionization-time of flight-mass spectrometryto the characterization of recombinant human erythropoietin.Electrophoresis,19(13),2348-55)。在蛋白水解消化Cys-修饰的EPO后,分离含有HAS的级分能够通过常规氨基酸组成分析证实该级分中含有相应的肽。
以上公开的涉及EPO或HAS特性的关于本发明HAS-EPO的所有实施方案,也适用于本发明制备HAS-EPO的方法。
本发明还涉及一种可通过本发明方法获得的HAS-EPO。优选地,该HAS-EPO具有上述限定本发明HAS-EPO的特征。
本发明还涉及在治疗人或动物体的方法中使用的本发明HAS-EPO。
此外,本发明还涉及含有本发明HAS-EPO的药物组合物。在一个优选实施方案中,该药物组合物还含有至少一种在促红细胞生成素治疗中使用的药学可接受稀释剂、佐剂和/或载体。
优选地,该药物组合物用于治疗贫血性疾病或造血功能障碍疾病或与之有关的疾病。
如此处所用的“治疗有效量”是指对于指定病症和施用方案提供治疗效果的量。促红细胞生成素同种型优选地通过肠胃外途径施用。选择的具体途径取决于治疗的疾病。促红细胞生成素同工型优选作为含有适当载体(如人血清白蛋白)、适当稀释剂(如缓冲液)和/或适当佐剂的制剂的一部分施用。需要的剂量是足以提高患者血细胞比容的量,根据治疗疾病的严重程度、施用方法等而不同。
优选地,本发明的组合物治疗的目的是将血液中血红蛋白值提高至6.8mmol/l以上。为此,可以施用药物组合物,使得血红蛋白值每周提高0.6mmol/l-1.6mmol/l。如果血红蛋白值超过8.7mmol/l,优选地应当中断治疗,直到血红蛋白值低于8.1mmol/l。
本发明组合物优选地在适于皮下或静脉内或肠胃外注射的制剂中使用。为此,适当的赋形剂和载体是,例如:磷酸二氢钠、磷酸氢二钠、氯酸钠、聚山梨醇酯80(polysorbate 80)、HAS和注射用水。该组合物可以每周施用三次,优选每周两次,更优选每周一次,最优选每两周一次。
优选地,该药物组合物以0.01-10μg/kg患者体重的量施用,更优选0.1-5μg/kg,0.1-1μg/kg或0.2-0.9μg/kg,最优选0.3-0.7μg/kg,最优选0.4-0.6μg/kg体重。
优选地,每次剂量通常施用10ug-200μg,优选15μg-100μg。
本发明还涉及本发明HAS-EPO在制备治疗贫血疾病或造血功能障碍疾病或与之有关疾病的药物中的应用。
根据本发明的另外一个方面,用含有一个或多个羟烷基淀粉(HAS)分子的HAS-多肽偶联物(HAS-多肽)解决了问题,其中每个HAS与多肽通过
a)碳水化合物部分;或
b)硫醚偶联。
本发明HAS-多肽具有以下优点:与偶联前的多肽相比,其显示生物稳定性提高。这主要是由于以下事实:HAS-多肽很少或者不被肝脏和肾脏的清除系统识别,因此在循环系统中长时间存在。此外,由于HAS被位点特异性地连接,因HAS与多肽偶联而破坏该多肽体内生物活性的危险被最小化。
本发明HAS-多肽主要含有两种成分,即多肽和与之连接的羟烷基淀粉(HAS)。
该多肽可以是任何人或动物来源的。在一个优选实施例中,该多肽是人来源的。
该多肽可以是细胞因子特别是促红细胞生成素,抗凝血酶(AT)如AT III,白介素特别是白介素-2、IFN-β、IFN-α、G-CSF、CSF、白介素-6和治疗抗体。
根据一个优选实施方案,该多肽是抗凝血酶(AT),优选是ATIII(Levy JH,Weisinger A,Ziomek CA,Echelard Y,RecombinantAntithrombin:Production and Role in Cardiovascular Disorder,Seminars in Thrombosis and Hemostasis 27,4(2001)405-416;Edmunds T,Van Patten SM,Pollock J,HansonE,Bernasconi R,Higgins E,Manavalan P,Ziomek C,MeadeH,McPherson J,Cole ES,Transgenically Produced Human Antithrombin:Structural andFunctional Comparison to Human Plasma-Derived Antithrombin,Blood 91,12(1998)4661-4671;Minnema MC,Chang ACK,Jansen PM,Lubbers YTP,Pratt BM,Whittaker BG,Taylor FB,Hack CE,Friedman B,Recombinant human antithrombin III improvessurvival and attenuates inflammatory responses in baboonslethally challenged with Escherichia coli,Blood 95,4(2000)1117-1123;Van Patten SM,Hanson EH,Bernasconi R,Zhang K,Manavaln P,Cole ES,McPherson JM,Edmunds T,Oxidation ofMethionine Residues in An tithrombin,J.Biol.Chemistry 274,15(1999)10268-10276)。
根据另外一个优选实施方案,该多肽是人IFN-β,特别是IFN-β1a(参见Avonex,REBIF)和IFN-β1b(参见BETASERON)。
另外一种优选的多肽是人G-CSF(粒细胞集落刺激因子)。参见,例如,Nagata等人,The chromosomal gene structure and two mRNAsfor human granulocyte colony-stimulating fator,EMBO J.5:575-581,1986;Souza等人,Recombinant human granulocytecolony-stimulating factor:effects on normal and leukemicmyeloid cells,Science 232(1986)61-65;Herman等人,Characterization,formulation,and stability ofNeupogen(Filgrastim),a recombinant human granulocyte-colonystimulating factor,见《蛋白质药物的配制、表征和稳定性》(Formulalion,characterization,and stability of proteindrugs),Rodney Pearlman和Y.John Wang编著,Plenum Press,NewYork,1996,303-328。
关于促红细胞生成素,以上公开的所有实施方案在此也适用。
优选地,该多肽通过重组产生。包括在真核或原核细胞中产生,优选在哺乳动物、昆虫、酵母、细菌细胞中,或者在适于重组生产多肽的其它任何细胞类型中产生。此外,多肽也可以在转基因动物中(例如在体液中,如奶、血液等)、在转基因鸟特别是家禽优选在鸡的蛋中,或者在转基因植物中表达。
多肽的重组产生在本领域中公知。通常包括用一种适当的表达载体转染宿主细胞,在能够产生多肽的条件下培养宿主细胞,以及从宿主细胞中纯化多肽。关于详细信息,参见,例如:Krystal,Pankratz,Farber,Smart,1986,Purification of human erythropoietin tohomogeneity by a rapid five-step procedure,Blood,67(1),71-9;Quelle,Caslake,Burkert,Wojchowski,1989,High-levelexpression and purification of a recombinant humanerythropoietin produced using a baculovirus vector,Blood,74(2),652-7;EP 640 619 B1和EP 668 351 B1。
多肽可以含有通过N-和/或O-连接的糖基化作用与该多肽连接的一个或多个碳水化合物侧链,即该多肽被糖基化。当在真核细胞中生产多肽时,该多肽通常在翻译后被糖基化。因此,在哺乳动物特别是人、昆虫或酵母细胞中生物合成过程中,碳水化合物侧链可以与该多肽连接。
HAS可以直接与多肽偶联,或者可以通过接头分子偶联。接头分子的性质取决于HAS与多肽连接的方式。有几种接头可以作为商品获得(例如,来自Pierce,见上文)。接头的性质及其用途在以下关于HES-多肽生产方法的部分中详细描述。
根据本发明HAS-多肽偶联物的一个优选实施方案,HAS通过碳水化合物部分与多肽偶联。优选地,当多肽是抗凝血酶优选ATIII时,同样适用。
在本发明中,术语“碳水化合物部分”指羟基醛或羟基酮,以及它们的化学修饰(见Rmpp Chemielexikon,Thieme Verlag Stuttgart,德国,第9版,9,2281-2285,和此处引用的文献)。此外,它也指天然存在的碳水化合物部分(如葡萄糖、半乳糖、甘露糖、唾液酸等)的衍生物。该术语也包括环结构已经打开并被化学氧化的天然存在碳水化合物部分。
碳水化合物部分可以直接与多肽骨架连接。优选地,碳水化合物部分是碳水化合物侧链的一部分。在这种情况下,HAS连接的碳水化合物部分与多肽骨架之间还可能存在其它碳水化合物部分。更优选地,该碳水化合物部分是碳水化合物侧链的末端部分。
在一个更优选的实施方案中,HAS与碳水化合物侧链的半乳糖残基偶联,优选地与碳水化合物侧链的末端半乳糖残基偶联。通过除去末端唾液酸,随后氧化,该半乳糖残基可以用于偶联(见下文)。
在一个更优选的实施方案中,HAS与碳水化合物侧链的唾液酸残基偶联,优选与碳水化合物侧链的末端唾液酸残基偶联。
此外,HAS也可以通过硫醚与多肽偶联。如下文详述的,S原子可以来自于附于多肽上的任何天然或非天然存在的SH基团。
在一个优选实施方案中,S原子可以来源于被引入HES氧化碳水化合物部分(优选作为多肽碳水化合物侧链一部分的氧化碳水化合物部分)中的SH基团(见下文)。
优选地,硫醚中的S原子来源于天然存在的半胱氨酸或者来源于添加的半胱氨酸。
在本发明的上下文中,术语“添加的半胱氨酸”指多肽含有在野生型多肽中不存在的半胱氨酸残基。
在本发明该方面中,半胱氨酸可以是在多肽的N端或C端添加的额外氨基酸。
此外,也可以通过将天然存在的氨基酸置换为半胱氨酸来添加半胱氨酸。
HAS-多肽的第二种组分是HAS。
在本发明中,术语“羟烷基淀粉”指被羟烷基取代的淀粉衍生物。其中,烷基可以被取代。优选地,羟烷基含有2-10个碳原子,更优选2-4个碳原子。因此,“羟烷基淀粉”优选包括羟乙基淀粉、羟丙基淀粉和羟丁基淀粉,其中羟乙基淀粉和羟丙基淀粉是优选的。
HAS的羟烷基至少含有一个OH基。
“羟烷基淀粉”也包括以下衍生物,其中烷基是单取代或多取代的。对此,只要HAS保持水溶性,优选烷基被卤素特别是氟、或者芳基取代。此外,羟烷基的末端羟基也可以酯化或者醚化。另外,羟烷基淀粉的烷基也可以是线性的或分支的。
此外,也可以使用线性或分支的取代或未取代的链烯基代替烷基。
对于本发明的所有实施方案,羟乙基淀粉(HES)是最优选的。
在本发明中,羟乙基淀粉的平均分子量(重量平均值)可以是1-300kDa,其中5-100kDa的平均分子量更优选。羟乙基淀粉可以进一步显示相对于羟乙基的0.1-0.8的摩尔取代程度和2-20的C2∶C6取代比。
HAS-多肽每个多肽分子可以含有1-12个,优选1-9、1-6或1-3个,最优选1-4个HAS分子。每个多肽分子的HAS分子数能够在产物水解及所得单糖衍生化后,利用GC-MS,通过定量碳水化合物组成分析来确定(Chaplin和Kennedy(编著),1986,《碳水化合物分析:一种实用方法》(Carbohydrate Analysis:a practical approach),第1章,单糖,第1-36页;第2章,寡糖,第37-53页,第3章,中性多糖,第55-96页;IRL Press Practical approach series(ISBN0-947946-44-3))。
以下公开的涉及多肽或HAS性质的本发明生产HAS-多肽的方法的所有实施方案也适用于本发明HAS-多肽。此外,以上公开的通常涉及肽或HAS的关于HAS-EPO或其制备的所有实施方案也适用于本发明的HAS-多肽。
羟烷基淀粉是淀粉的一种醚衍生物。除了所述醚衍生物以外,在本发明中也能够使用其它淀粉衍生物。例如,可以使用含有酯化羟基的衍生物。这些衍生物可以是,例如,具有2-12个碳原子的未取代单羧酸或二羧酸的衍生物,或其取代衍生物的衍生物。特别有用的是具有2-6个碳原子的未取代单羧酸的衍生物,特别是乙酸的衍生物。在上下文中,乙酰基淀粉、丁基淀粉或丙基淀粉是优选的。
此外,具有2-6个碳原子的未取代二羧酸的衍生物也是优选的。
对于二羧酸的衍生物,二羧酸的第二个羧基也被酯化是有利的。此外,二羧酸的单烷基酯衍生物在本发明中也是适用的。
对于取代的单羧酸或二羧酸,取代基优选可以与上述用于取代烷基残基的取代基相同。
淀粉的酯化技术在本领域公知(参见,例如,Klemm D.等人,《综合纤维素化学》(Comprehensive Cellulose Chemistry)第2卷,1998,Whiley-VCH,Weinheim,纽约,具体见第4.4章,纤维素的酯化(ISBN3-527-29489-9)。
另一方面,本发明涉及生产羟烷基淀粉(HAS)-多肽偶联物(HAS-多肽)的方法,其包括下列步骤:
a)提供能够与修饰的HAS反应的多肽,
b)提供能够与步骤a)中多肽反应的修饰的HAS,和
c)使步骤a)的多肽与步骤b)的HAS反应,从而产生一种含有一个或多个HAS分子的HAS-多肽,其中每个HAS都与该多肽通过
i)碳水化合物部分;或
ii)硫醚偶联。
本发明的方法具有生产显示高生物活性的HAS-多肽偶联物的优点。此外,本发明的方法也具有能够以低成本生产有效的多肽衍生物的优点,因为该方法不包括导致低终产量的复杂且耗时的纯化步骤。
因此,在本发明的方法的第一个步骤中,提供能够与修饰的HAS反应的一种多肽。
本发明所用的术语“提供”解释为在各步骤后,可以获得一种具有所需特性的分子(在步骤a)中为多肽,在步骤b)中为HAS)。
对于步骤a),包括从天然来源中纯化多肽,以及在宿主细胞或生物中重组生产,以及必要时对这样获得的多肽进行修饰。
任何关于作为本发明初始材料多肽的情况,同样适用于作为本发明HAS-多肽偶联物一部分的促红细胞生成素。对此,以上公开的优选实施方案也适用于本发明方法。
优选多肽是重组产生的。包括在真核或原核细胞中产生,优选地在哺乳动物、昆虫、酵母、细菌细胞中或者在适于重组生产多肽的其它任何细胞类型中产生。此外,多肽也可以在转基因动物中(例如在体液中,如奶、血液等)、在转基因鸟特别是家禽优选在鸡的蛋中,或者在转基因植物中表达。
多肽的重组产生在本领域中公知。通常包括用一种适当的表达载体转染宿主细胞,在能够生产多肽的条件下培养宿主细胞,以及从宿主细胞中纯化多肽(Krystal,Pankratz,Farber,Smart,1986,Purification of human erythropoietin to homogeneity by a rapidfive-step procedure,Blood,67(1),71-9;Quelle,Caslake,Burkert,Wojchowski,1989,High-level expression andpurification of a recombinant human erythropoietin producedusing a baculovirus vector,Blood,74(2),652-7;EP 640 619 B1和EP 668 351 B1)。
多肽可以含有通过N-和/或O-连接的糖基化作用与该多肽连接的一个或多个碳水化合物侧链,即该多肽被糖基化。当在真核细胞中生产多肽时,多肽通常在翻译后糖基化。因此,在哺乳动物特别是在人、昆虫或酵母细胞中生物合成过程中,碳水化合物侧链可以与多肽连接,这些细胞可以是转基因动物的细胞(见上文)。
这些碳水化合物侧链可以在适当细胞中表达后化学或酶修饰,例如通过除去或添加一个或多个碳水化合物部分(参见,例如,Dittmar,Conradt,Hauser,Hofer,Lindenmaier,1989,蛋白质设计进展;Bloecker,Collins,Schmidt和Schomburg编著,GBF-专著,12,231-246,VCH Publishers,Weinheim,New York,Cambridge)。
本发明的方法的目的是提供一种含有一个或多个HAS分子的HAS-多肽,其中该HAS通过碳水化合物部分(i)或者通过硫醚(ii)与多肽偶联。因此,步骤a)提供的多肽应当具有以下特性:能够通过碳水化合物部分和/或通过硫醚偶联。因此,在步骤a)之后,多肽优选地可以含有
(1)至少一个直接或者通过接头分子与巯基团或者碳水化合物部分连接的反应性基团,能够与HES或修饰的HES反应,
(2)至少一个能够与修饰的HAS偶联的碳水化合物部分,和/或
(3)至少一个游离SH基。
对于上述可能性(1),步骤a)的多肽优选地可以通过将适当的接头分子与多肽的SH基或碳水化合物部分偶联获得。实施例4中的2.1提供了这种修饰的多肽的一个实例。重要的是确保接头分子的添加不损害多肽。而这为本领域技术人员所公知。
对于上述可能性(2),在一个优选实施方案中,修饰的HAS通过碳水化合物部分与多肽偶联。
碳水化合物部分可以与多肽骨架直接连接。优选地,碳水化合物部分是碳水化合物侧链的一部分。在这种情况下,与HAS连接的碳水化合物部分与多肽骨架之间还可能存在其它碳水化合物部分。更优选地,碳水化合物部分是碳水化合物侧链的末端部分。
因此,在一个优选实施方案中,修饰的HAS与碳水化合物链连接(通过一个接头,或者不通过接头,见下文),后者与多肽的N-和/或O-糖基化位点连接。
然而,本发明也包括,多肽含有与修饰的HAS偶联的其它碳水化合物部分。本领域公知通过酶或者通过遗传工程连接碳水化合物部分与多肽的技术,以及随后在适当细胞中的表达(Berger,Greber,Mosbach,1986,Galactosyltransferase-dependent sialylation ofcomplex and endo-Nacetylglucosaminidase H-treated coreN-glycans invitro,FEBS Lett.,203(1),64-8;Dittmar,Conradt,Hauser,Hofer,Lindenmaier,1989,《蛋白质设计进展》;Bloecker,Collins,Schmidt和Schomburg编著,GBF-专著,12,231-246,VCHPub lishers,Weinheim,New York,Cambridge)。
在本发明方法的一个优选实施方案中,为了能够与修饰的HAS反应而氧化碳水化合物部分。这种氧化能够以化学或酶学方法进行。
化学氧化多肽碳水化合物部分的方法在本领域公知,包括用高碘酸盐处理(Chamow等人,1992,J.Biol.Chem.,267,15916-15922)。
通过化学氧化,原则上能够氧化位于末端或者不位于末端的任何碳水化合物部分。然而,通过选择温和的条件(1mM高碘酸盐,0℃,不同于严格条件:10mM高碘酸盐,室温1小时),可以优选地氧化碳水化合物侧链的末端碳水化合物部分,例如唾液酸或半乳糖。
此外,碳水化合物部分也可以酶氧化。用于氧化各碳水化合物部分的酶在本领域公知,例如,对于半乳糖的酶是半乳糖氧化酶。
如果准备氧化末端半乳糖部分,如果在能够连接唾液酸与碳水化合物链的细胞例如在哺乳动物细胞中,或者在遗传修饰后能够连接唾液酸与碳水化合物链的细胞中生产多肽,最终必须(部分或完全)除去末端唾液酸。去除唾液酸的化学或酶法在本领域中公知(Chaplin和Kennedy(编著),1996,《碳水化合物分析:一种实用方法》,具体见第5章Montreuill,糖蛋白,第175-177页;IRL Press Practicalapproach series(ISBN 0-947946-44-3))。
然而,本发明也包括在步骤a)中将要与修饰的HAS连接的碳水化合物部分与多肽连接。如果希望连接半乳糖,可以利用半乳糖转移酶实现。这些方法在本领域中公知(Berger,Greber,Mosbach,1986,Galactosyltransferase-dependent sialylation of complex andendo-N-acetylglucosaminidase H-treated core N-glycans in vitro,FEBS Lett.,203(1),64-8)。
在一个最优选的实施方案中,在步骤a)中,必要时,优选在部分或完全(酶和/或化学)除去末端唾液酸之后,通过氧化多肽的一个或多个碳水化合物侧链的至少一个末端糖单位(优选半乳糖)来修饰多肽(见上文)。
因此,优选修饰的HAS与碳水化合物链被氧化的末端糖单位偶联,优选与半乳糖偶联。
在一个进一步优选的实施方案中(见上文第(3)点),多肽含有至少一个游离SH基。
根据一个优选实施方案,游离SH基是天然存在的半胱氨酸或添加的半胱氨酸的一部分。
氨基酸置换方法在本领域中公知(Elliott,Lorenzini,Chang,Barzilay,Delorme,1997,Mapping of the active site ofrecombinant human erythropoietin,Blood,89(2),493-502;Boissel,Lee,Presnell,Cohen,Bunn,1993,Erythropoietinstructure-function relationships.Mutant proteins that test amodel of tertiary structure,J Biol Chem.,268(21),15983-93))。
在本发明中,术语“添加的半胱氨酸”是指多肽含有一个在野生型多肽中不存在的半胱氨酸残基。这能够如下实现:在多肽的N端或C端添加(例如通过重组方法)一个半胱氨酸残基,或者将一个天然存在的氨基酸置换(例如通过重组方法)为半胱氨酸。本领域技术人员公知这些方法(见上文)。
优选地,通过将一个天然存在的氨基酸置换为一个半胱氨酸来添加半胱氨酸。
优选地,修饰的HAS在步骤c)中与添加的半胱氨酸偶联。
在本发明的方法的步骤b)中,提供能够与步骤a)的多肽反应的修饰的HAS。
对此,HAS优选地可以在其还原性末端被修饰。这样有化学反应易于控制且技术人员能够确保HAS的哪个基团在反应过程中被修饰的优点。由于只将一个基团引入HAS中,能够防止不同多肽分子通过多功能HAS分子交联和其它副反应。
因此,修饰的HAS能够与下列成分反应:
(1)至少一个直接或者通过接头分子与多肽的巯基团或碳水化合物部分连接的基团,
(2)至少一个碳水化合物部分,优选其被氧化,和/或
(3)至少一个游离SH基。
对于上述第(1)点,HAS的修饰取决于与多肽连接的基团。其机制在本领域中公知。实施例4中的2.1给出了一个实例。
对于上述第(2)点和第(3)点,本领域公知几种修饰HAS的方法。这些方法的基本原理是,为了能够与碳水化合物部分或SH基反应而修饰HAS的反应性基团,或者一个接头分子与HAS偶联,后者含有能够与碳水化合物部分或SH基反应的反应性基团。
对于第(2)点,修饰的HAS能够与氧化的碳水化合物部分反应,优选与末端糖残基、更优选与半乳糖或者与末端唾液酸反应。
公知几种方法可以修饰HAS,使其能够与氧化的优选末端糖残基反应。如上所述,可以在HES-链的还原性末端区域选择性地引入这种修饰。在这种情况下,在第一步中,醛基被氧化为内酯。这些修饰包括但不限于:直接或者通过接头向HAS上添加酰肼、氨基(以及羟氨基)、氨基脲或硫醇官能基团。这些技术在实施例2-4中进一步详细描述。此外,机制本身在本领域中公知(参见,例如DE 196 28 705 A1;Hpoe等人,1981,Carbohydrate Res.,91,39;Fissekis等人,1960,Journal of Medicinal and Pharmaceutical Chemistry,2,47;Frie,1998,毕业论文,Fachhochschule Hamburg,DE)。
在本发明中,优选添加酰肼或羟氨基官能团。在这种情况下,优选地通过在pH 5.5下进行本发明方法步骤c)的反应,确保修饰的HAS与多肽的氧化的碳水化合物部分选择性反应,而没有赖氨酸侧链与氧化的糖残基形成亚胺从而引起分子间或分子内多肽交联。
对于第(3)点,也公知几种用于修饰HAS的方法,使其能够与游离SH基反应。优选地,在HES-链的还原性末端区域选择性地引入这种修饰。这些方法包括但不限于向HAS添加马来酰亚胺、二硫醚或卤代乙酰胺官能团。这些技术在实施例2-4中进一步详细描述。
关于这些技术的进一步的细节,可见Chamov等人,1992,J.Biol.Chem.,267,15916;Thorpe等人,1984,Eur.J.Biochem.,140,63;Greenfield等人,1990,Cancer Research,50,6600,以及实施例2中的1.3引用的文献。
表1列出了其它一些可能的官能团,提供了对可能的接头分子的系统综述。此外,机制本身在本领域公知。
可以在本发明中使用的几种接头分子在本领域中公知,或者可以作为商品获得(例如来自Pierce,可以获自Perbio ScienceDeutschland GmbH,Bonn,德国)。
在本发明的方法的步骤c)中,步骤a)的多肽与步骤b)的HAS反应,从而产生含有一个或多个HAS分子的HAS-多肽,其中HAS通过碳水化合物部分或者通过硫醚与该多肽偶联。
原则上,使多肽与修饰HAS反应的详细方法取决于多肽和/或HAS各自的修饰,为本领域中公知(参见,例如Rose,1994,J.Am.Chem.Soc.,116,30,O′Shannessay 和 Wichek,1990,AnalyticalBiochemistry,191,1;Thorpe等人,1984,Eur.J.Bio-chem.,140,63;Chamov等人,1992,J.Biol.Chem.267,15916)。
对于本发明举例说明的方法,实施例2-4,特别是实施例4中给出了详细描述。
步骤c)可以在至少含有10%重量H2O的反应介质中进行。
在本发明方法的这一优选实施方案中,反应介质含有至少10%重量的水,优选至少50%,更优选至少80%,例如90%或者可达100%。据此可以计算有机溶剂的含量。因此,该反应在水相中进行。优选的反应介质是水。
本发明方法的这一实施方案的一个优点是,不必使用在毒理学上危险的溶剂,因此在生产过程后不必除去这些溶剂来避免溶剂的污染。此外,对于残余的毒理学上危险的溶剂,不必进行额外的质量控制。优选使用毒理学上不危险的溶剂作为有机溶剂,如乙醇或丙二醇。
本发明的方法的另外一个优点是,避免了有机溶剂诱导的不可逆的或可逆的结构改变。因此,根据本发明方法获得的多肽不同于在有机溶剂(如DMSO)中制备的多肽。
此外,也意外地发现,HAS与药物在水溶液中的偶联最小化或者避免了副反应。因此,本发明方法的这个实施方案产生高纯度的改良产物。
在本发明中,术语“羟烷基淀粉”是指被羟烷基取代的淀粉衍生物。其中,烷基可以被取代。优选羟烷基含有2-10个碳原子,更优选2-4个碳原子。因此“羟烷基淀粉”优选地包括羟乙基淀粉、羟丙基淀粉和羟丁基淀粉,其中羟乙基淀粉和羟丙基淀粉是优选的。
HAS的羟烷基含有至少一个OH基。
对于本发明的所有实施方案,羟乙基淀粉(HES)是最优选的。
术语“羟烷基淀粉”也包括以下衍生物,其中烷基是单取代或多取代的。对此,只要HAS保持水溶性,优选烷基可以被卤素特别是氟、或者芳基取代。此外,羟烷基的末端羟基也可以酯化或者醚化。另外,羟烷基淀粉的烷基也可以是线性的或分支的。
此外,也可以使用线性或分支的取代或未取代的链烯基代替烷基。
在本发明中,羟乙基淀粉的平均分子量可以是1-300kDa,其中5-100kDa的平均分子量是更优选的。羟乙基淀粉能够进一步显示相对于羟乙基的0.1-0.8的摩尔取代程度和2-20的C2∶C6取代比。
通过本发明的方法生产的HAS-多肽能够被如下纯化和表征:
HAS-多肽的分离可以用已知的纯化天然和重组多肽的方法进行(例如大小排阻层析、离子交换层析、RP-HPLC、羟基磷灰石层析、疏水相互作用层析、实施例20.8所述的方法或其组合)。
HAS与多肽的共价连接能够在修饰的蛋白质水解后通过碳水化合物组成分析证实。
在多肽的N-连接寡糖处的HAS修饰能够如下证实:除去HAS修饰的N-聚糖,利用SDS-PAGE+/-Western Blotting观察预知的向较高迁移率的转变。
多肽在半胱氨酸残基处的HAS修饰能够根据以下情况证实:在HAS修饰产物的蛋白水解片段中,通过RP-HPLC和MALDI/TOF-MS不能检测到相应的蛋白水解Cys-肽(Zhou等人,1998,Application ofcapillary electrophoresis,liquid chromatography,electrospray-mass spectrometry and matrix-assistedlaserdesorption/ionization-time of flight-mass spectrometryto the characterization of recombinant human erythropoietin.Electrophoresis,19(13),2348-55)。在蛋白水解消化Cys-修饰的多肽后,分离含有HAS的级分能够通过常规氨基酸组成分析证实该级分中含有相应的肽。
以上公开的涉及多肽或HAS性质的关于本发明HAS-多肽的所有实施方案,也适用于本发明生产HAS-多肽偶联物的方法。此外,以上公开的涉及一般性肽或HAS的关于HAS-EPO或其制备的所有实施方案,也适用于本发明生产HAS-多肽偶联物的方法。
本发明还涉及一种可通过本发明方法获得的HAS-多肽。优选地,该HAS-多肽具有上述用于限定本发明HAS-EPO的特征。
根据本发明的一个优选实施方案,使用的HAS具有下列通式(I)
其中,R1、R2和R3分别是氢,或者是直链或支链羟烷基。本发明所用的术语“羟烷基淀粉”不只限于末端碳水化合物部分含有如通式(I)所示的羟烷基R1、R2和/或R3的化合物(为了简短起见),也指如下化合物:在末端碳水化合物部分和/或淀粉分子的其余部分HAS’中任何处存在的至少一个羟基被羟烷基R1、R2或R3取代。其中,烷基可以是直链或支链烷基,它可以被适当的取代。优选地,羟烷基含有1-10个碳原子,更优选1-6个碳原子,更优选1-4个碳原子,更优选2-4个碳原子。因此,“羟烷基淀粉”优选包括羟乙基淀粉、羟丙基淀粉和羟丁基淀粉,其中羟乙基淀粉和羟丙基淀粉是特别优选的,尤其优选羟乙基淀粉。
HAS(优选HES)可以与交联化合物反应,该交联剂可以与HAS(优选HES)反应,并且可与多肽(如上述多肽)反应。
HAS与交联化合物之间的反应可以在HAS的还原性末端或在HAS的氧化的还原性末端发生。因此,具有通式(I)结构的HAS

和/或,如果还原性末端被氧化,则具有通式(IIa)
和/或通式(IIb)结构的HAS

可以反应。
如果通式(I)的HAS与交联化合物反应,则反应优选在水介质中发生。如果通式(IIa)和/或(IIb)的HAS与交联化合物反应,则反应优选在非水介质中发生,例如极性非质子溶剂或溶剂混合物如DMSO,和/或DMF。
如果通过含有HAS和交联化合物的HAS衍生物与多肽的氧化的碳水化合物部分反应产生本发明HAS-多肽偶联物,则该交联化合物优选地是下述化合物
H2N-NH2或
或


如果通过含有HAS和至少一种交联化合物的HAS衍生物与多肽的巯基反应产生本发明HAS-多肽偶联物,则优选地,HAS在任选氧化的还原性末端与第一种交联化合物反应,该交联化合物优选是下述化合物
H2N-NH2或
或

并且产生的HAS衍生物与第二种交联化合物反应,第二种交联化合物能够与该HAS衍生物和多肽的巯基反应。例如,如果HAS衍生物含有结构-NH-作为与第二种交联化合物反应的官能团,则如上详述的,含有官能团F1和F2的以下类型的第二种交联化合物尤其是优选的:
| 化合物类型(L) | F1 | F2 |
| C | 碘烷基 | N-琥珀酰亚胺酯 |
| D | 溴烷基 | N-琥珀酰亚胺酯 |
| E | 马来酰亚胺 | N-琥珀酰亚胺酯 |
| F | 吡啶基二硫基 | N-琥珀酰亚胺酯 |
| G | 乙烯基砜 | N-琥珀酰亚胺酯 |
第一种交联化合物的特别优选的例子是



化合物
和

是特别优选的,下列第二种交联化合物是优选的,
和
化合物
是特别优选的。
根据各自的反应条件、使用的溶剂或溶剂混合物和/或在水介质中与HAS反应的化合物R′-NH-R″的残基R′和/或R″,通过上述方法获得的羟烷基淀粉衍生物可以具有下列结构(IIIa):

因此,本发明也涉及如上所述具有通式(IIIa)结构的羟烷基淀粉衍生物。
例如,如果R′是氢,则通过上述方法获得的羟烷基淀粉衍生物也可能具有下列结构(IIIa)或(IIIb),其中(IIIa)和(IIIb)可以在反应混合物中同时存在,并具有一定的平衡分布:

因此,本发明也涉及如上所述具有根据通式(IIIb)结构的羟烷基淀粉衍生物。
而且,本发明也涉及以上述通式(IIIa)和(IIIb)结构的混合物存在的羟烷基淀粉衍生物。
根据反应条件和/或用于反应的化合物R′-NH-R″的化学性质,通式(IIIa)的化合物可以在平伏位置或直立位置含有N原子,也可能存在具有一定平衡分布的两种型式的混合物。
根据反应条件和/或用于反应的化合物R′-NH-R″的化学性质,通式(IIIb)化合物可能含有E或Z构象的C-N双键,也可能存在具有一定平衡分布的两种型式的混合物。
在某些情况下,可能希望稳定通式(IIIa)的化合物,特别是在水溶液中生产并使用通式(IIIa)的化合物时。作为稳定方法,特别优选酰化通式(IIIa)的化合物,特别是当R′是氢时。可以使用产生通式(IV
a)的羟烷基淀粉衍生物的任何适当试剂作为酰化剂。

根据本发明的特别优选的实施方案,作为酰化剂一部分的残基Ra是甲基。优选使用羧酸酐、羧酸酰卤和羧酸活化的酯作为酰化剂。
因此,本发明也涉及可以通过上述方法获得的羟烷基淀粉衍生物,其中该衍生物具有通式(IVa)的结构。
酰化在0-30℃下进行,优选地在2-20℃下进行,特别优选地在4-10下进行。
在其它一些情况下,可能希望稳定通式(IIIb)的化合物,特别是当在水溶液中生产和/或使用通式(IIIb)的化合物时。作为稳定方法,还原通式(IIIb)的化合物是特别优选的,特别是当R′是氢时。可以使用产生通式(IVb)的羟烷基淀粉衍生物的任何适当试剂作为还原剂。
根据本发明的特别优选的实施方案,使用硼氢化物如NaCNBH3或NaBH4,作为还原试剂。
因此,本发明也涉及可以通过上述方法获得的羟烷基淀粉衍生物,其中该衍生物具有通式(IVb)的结构。
还原在4-100℃下进行,优选地在10-90℃下进行,特别优选地在25-80℃下进行。
本发明还涉及化合物(IIIa)和(IIIb)、(IVa)和(IVb)、(IIIa)和(VIa)、(IIIa)和(IVb)、(IIIb)和(IVa)、(IIIb)和(IVb)、(IIIa)和(IIIb)和(IVa)、(IIIa)和(IIIb)和(IVb)、(IVa)和(IVb)和(IIIa),以及(IVa)和(IVb)和(IIIb)的混合物,其中(IIIa)和/或(IVa)可以独立地以N原子位于平伏或直立位置的构象存在,和/或(IIIb)可以以E或Z构象的C-N双键形式存在。
本发明还涉及在治疗人或动物体的方法中使用的本发明的HAS-多肽。
此外,本发明还涉及含有本发明HAS-多肽的药物组合物。在一个优选实施方案中,该药物组合物还含有至少一种在促红细胞生成素治疗中使用的药学可接受的稀释剂、佐剂和/或载体。
本发明还涉及本发明HAS-多肽在制备治疗贫血性疾病或造血功能障碍性疾病或与之相关疾病的药物中的应用。
本发明通过下列附图、表和实施例进一步说明,它们绝非旨在限制本发明的范围。
附图简述
图1
图1显示两种HES-EPO偶联物的SDS-PAGE分析
mw:分子量标准
1道:根据实施例方案8产生的HES-EPO:EPO与酰肼基-HES 12KDL偶联
2道:根据实施例方案9产生的HES-EPO:EPO与羟胺基HES 12KDK偶联
C:对照(未偶联的EPO);上面的一条带代表EPO二聚体
图2
图2通过显示多肽N-糖苷酶对HAS修饰的EPO型的消化,证明HES与碳水化合物侧链的碳水化合物部分偶联
1道:根据实施例方案8产生的HES-EPO,N-糖苷酶消化
2道:根据实施例方案9产生的HES-EPO,N-糖苷酶消化
3道:BRP EPO标准
4道:BRP EPO标准,N-糖苷酶消化
mw:分子量标准(Bio-Rad SDS-PAGE Standards Low range,目录号161-0305,Bio-Rad Laboratories,Hercules,CA,USA)
图3
图3显示根据实施例17.1产生的HES-EPO偶联物的SDS-PAGE分析。
A道:蛋白质分子量标准Roti-Mark PRESTAINED(Carl RothGmbH+Co,Karlsruhe,D);蛋白质标准的分子量(kD)从上到下依次为:245,123,77,42,30,25.4和17。
B道:根据实施例17.1偶联后的粗制品。
C道:EPO初始材料
图4
图4显示根据实施例17.3产生的HES-EPO偶联物的SDS-PAGE分析。
A道:根据实施例17.3偶联后的粗制品。
B道:EPO初始材料
C道:蛋白质分子量标准Roti-Mark PRESTAINED(Carl RothGmbH+Co,Karlsruhe,D);蛋白质标准的分子量(kD)从上到下依次为:245,123,77,42,30,25.4和17。
图5
图5显示根据实施例17.4和17.5产生的HES-EPO偶联物的SDS-PAGE分析。
A道:蛋白质分子量标准Roti-Mark PRESTAINED(Carl RothGmbH+Co,Karlsruhe,D);蛋白质标准的分子量(kD)从上到下依次为:245,123,77,42,30,25.4和17。
B道:根据实施例17.4偶联后的粗制品。
C道:根据实施例17.5偶联后的粗制品。
D道:EPO初始材料。
图6
图6显示根据实施例19.1和19.4产生的HES-EPO偶联物的SDS-PAGE分析。
A道:蛋白质分子量标准Roti-Mark PRESTAINED(Carl RothGmbH+Co,Karlsruhe,D);蛋白质标准的分子量(kD)从上到下依次为:245,123,77,42,30,25.4和17。
B道:根据实施例19.4偶联后的粗制品。
C道:根据实施例19.1偶联后的粗制品。
D道:EPO初始材料。
图7
图7显示根据实施例19.2、19.3、19.5和19.6产生的HES-EPO偶联物的SDS-PAGE分析。
A道:蛋白质分子量标准Roti-Mark PRESTAINED(Carl RothGmbH+Co,Karlsruhe,D);蛋白质标准的分子量(kD)从上到下依次为:245,123,77,42,30,25.4和17。
B道:根据实施例19.6偶联后的粗制品,基于实施例13.3b)。
C道:根据实施例19.5偶联后的粗制品,基于实施例13.1b)。
D道:根据实施例19.6偶联后的粗制品,基于实施例13.3a)。
E道:根据实施例19.5偶联后的粗制品,基于实施例13.1a)。
F道:根据实施例19.2偶联后的粗制品。
G道:根据实施例19.3偶联后的粗制品。
K道:EPO初始材料。
图8
图8显示根据实施例19.7、19.8、19.9、19.10、19.11和19.12产生的HES-EPO偶联物的SDS-PAGE分析。
A道:蛋白质分子量标准Roti-Mark PRESTAINED(Carl RothGmbH+Co,Karlsruhe,D);蛋白质标准的分子量(kD)从上到下依次为:245,123,77,42,30,25.4和17。
B道:根据实施例19.11偶联后的粗制品。
C道:根据实施例19.10偶联后的粗制品。
D道:根据实施例19.7偶联后的粗制品。
E道:根据实施例19.8偶联后的粗制品。
F道:根据实施例19.12偶联后的粗制品。
G道:EPO初始材料。
K道:根据实施例19.9偶联后的粗制品。
图9
EPO-GT-1的SDS-PAGE分析,其中弱酸处理5分钟=2道;10分钟=3道;60分钟=4道,未处理的EPO=1道;显示除去N-聚糖后EPO的迁移率变化(+N-糖苷酶(PNGase))。
图10
从未处理的EPO中以及在弱酸水解条件下温育5分钟、10分钟、60分钟的EPO中所分离的寡糖的HPAEC-PAD(高效阴离子交换色谱-积分脉冲安培检测法)图。罗马数字I-V表示洗脱位置,I=二唾液酸化的二分支结构,II=三唾液酸化的三分支结构(两种异构体),III=四唾液酸化的四分支结构+2个N-乙酰基乳糖胺重复,IV=四唾液酸化的四分支结构+1个N-乙酰基乳糖胺重复;V=四唾液酸化的四分支结构+不含N-乙酰基乳糖胺重复。不含及含有1-4个唾液酸的寡糖结构的洗脱区用括号表示。
图11
去唾液酸化后N-连接寡糖的HPAEC-PAD;显示了N-乙酰神经氨酸的洗脱位置;数字1-9表示标准寡糖的洗脱位置;1=二分支;2=三分支(2-4种异构体),3=三分支(2-6种异构体),4=四分支;5=三分支+1个重复;6=四分支+1个重复;7=三分支+2个重复;8=四分支+2个重复;9=四分支+3个重复。
图12
温和处理的和未处理的EPO在唾液酸残基经受高碘酸盐氧化后的SDS-PAGE分析。1=高碘酸盐氧化,未酸处理;2=高碘酸盐氧化5分钟,酸处理;3=高碘酸盐氧化和酸处理10分钟;4=高碘酸盐氧化,未酸处理;5=未高碘酸盐氧化且未酸处理的BRP EPO标准。
图13
从未处理的EPO中以及从在弱酸水解条件下温育5分钟和10分钟随后高碘酸盐处理的EPO中分离的天然寡糖的HPAEC-PAD模式。不含及含有1-4个唾液酸的寡糖结构的洗脱区用括号1-5表示。
图14
EPO-GT-1-A的HES修饰时间过程的SDS-PAGE分析:20μg等份EPO-GT-1-A与羟胺修饰的HES衍生物X反应30分钟、2、4、17小时。1道=30分钟反应时间;2道=2小时反应时间;3道=4小时反应时间;4道=17小时反应时间;5道=没有HES修饰的EPO-GT-1-A。左图显示在羟胺修饰的HES衍生物(流速:1ml.min-1)X存在下,随着温育时间延长,EPO-GT-1-A的迁移率改变:1道=30分钟反应时间;2道=2小时反应时间;3道=4小时反应时间;4道=17小时反应时间;5道=HES修饰的EPO-GT-1-A。右图显示相同样品用N-糖苷酶处理后的分析。
图15
HES-EPO偶联物的Q-Sepharose级分的SDS-PAGE分析。各1%的穿流液(flow-through)以及在高盐浓度下洗脱的1%级分用Speed Vac浓缩器浓缩,在样品缓冲液中加样到凝胶上。EPO蛋白用考马斯蓝染色。A=样品I;B=样品II;C=样品III;K=对照EPO-GT-1;A1、B1、C1和K1表示穿流液部分;A2、B2、C2和K2表示用高盐浓度洗脱的级分。
图16a
HES修饰的EPO样品A2(见图15)、对照EPO样品K2和EPO-GT-1-A的SDS-PAGE分析。为了除去N-连接的寡糖,在N-糖苷酶存在下消化EPO制品。所有EPO样品都显示迁移率向缺乏或含有O-聚糖的低分子量型转变。对于HES-修饰的EPO样品A2,在去-N-糖基化后观察到O-糖基化与非糖基化蛋白带的比值较低,在30KDa左右检测到一条弥散的蛋白带,这可能代表O-聚糖残基的唾液酸处的HES-修饰(见星号标记的箭头)。
图16b
未处理的或者在N-糖苷酶存在下消化除去N-连接寡糖(见图16a)的HES-修饰EPO样品A2(见图15)、对照EPO样品K2和EPO-GT-1A在温和水解后的SDS-PAGE分析。在N-糖苷酶处理之前的高分子量型A2和处理之后的高分子量型A(见有箭头及无箭头的括号)在样品酸处理后都消失。用于比较的BRP EPO标准没有进行弱酸处理。
图17
从HES修饰的样品A、EPO-GT-1-A以及与未修饰HES温育的对照EPO样品(K)中释放的N-连接寡糖物质的HPAEC-PAD分析。罗马数字I-V表示洗脱位置,I=二唾液酸化的二分支结构,II=三唾液酸化的三分支结构(两种异构体),III=四唾液酸化的四分支结构+2个N-乙酰基乳糖胺重复,IV=四唾液酸化的四分支结构+1个N-乙酰基乳糖胺重复;V=四唾液酸化的四分支结构+不含N-乙酰基乳糖胺重复;括号表示二、三、四唾液酸化的N-聚糖的洗脱区,如图10和图13的图例所述。
图18
从HES修饰的样品A、EPO-GT-1-A以及与未修饰HES温育的对照EPO样品(K)中释放的N-连接寡糖物质的HPAEC-PAD分析。显示了标准寡糖混合物的保留时间:数字1-9表示标准寡糖的洗脱位置:1=二分支;2=三分支(2-4种异构体),3=三分支(2-6种异构体),4=四分支;5=三分支+1个重复;6=四分支+1个重复;7=三分支+2个重复;8=四分支+2个重复;9=四分支+3个重复。
图19-25
图19-25表示从HES修饰的EPO和对照EPO制品中分离的酶释放的和化学去唾液酸化的N-聚糖的MALDI/TOF质谱。在m/z1809.7、2174.8、2539.9、2905.0和3270.1处的主要信号([M+Na]+)对应于二到四分支的不含、含有一个或两个N-乙酰基乳糖胺重复的复杂型N-聚糖结构,并且伴有由于对MS分析的样品去唾液酸化所使用的酸水解条件引起岩藻糖或半乳糖损失所致的弱信号。
图19
MALDI/TOF谱:HES-修饰的EPO A2的去唾液酸化寡糖。
图20
MALDI/TOF谱:EPO GT-1-A的去唾液酸化寡糖。
图21
MALDI/TOF谱:EPO K2的去唾液酸化寡糖。
图22
MALDI/TOF谱:EPO-GT-1的去唾液酸化寡糖。
图23
MALDI/TOF谱:酸水解5分钟EPO-GT-1的去唾液酸化寡糖。
图24
MALDI/TOF谱:酸水解10分钟EPO-GT-1的去唾液酸化寡糖。
图25
MALDI/TOF谱:酸水解60分钟EPO-GT-1的去唾液酸化寡糖。
实施例
实施例1
重组EPO的生产
A)在哺乳动物细胞中的生产
重组EPO如下在CHO细胞中生产
将携带人EPO cDNA的质粒克隆到真核表达载体中(pCR3,在下文中称为pCREPO)。如述利用标准方法进行定点诱变(Grabenhorst,Nimtz,Costa等人,1998,In vivo specificity of human alpha1,3/4-fucosyltrans ferases III-VII in the biosynthesis ofLewis(x)and sialyl Lewis(x)motifs on complex-type N-glycans-Coexpression studies from BHK-21 cells together with humanbeta-trace protein,J.Biol.Chem.,273(47),30985-30994)。
稳定表达人EPO或其氨基酸变体(例如Cys-29→Ser/Ala,或Cys-33→Ser/Ala,Ser-126→Ala等)的CHO细胞用磷酸钙沉淀法产生,如述(Grabenhorst等人)用硫酸G418筛选。转染三天后,将细胞1∶5传代培养,在含有10%FBS和1.5g/l硫酸G418的DMEM中筛选。
采用这种筛选操作,通常有100-500个克隆存活,在选择培养基中再繁殖2-3周。然后通过Western blot和IEF/Western Blot分析汇合生长之单层的细胞培养上清液的EPO表达水平。
EPO由稳定的亚克隆在旋转培养瓶中或者在21灌注反应器(perfusion reactors)中产生。按照发表的方案,含有不同数量NeuAc(例如2-8、4-10、8-12个NeuAc残基)的EPO的不同糖型(glycoform)用如下所述多种层析法的组合分离。
文献:
Grabenhorst,Conradt,1999,The cytoplasmic,transmembrane,and stem regions of glycosyltransferases specify their in vivofunctional sublocalization and stability in the Golgi.J BiolChem.,274(51),36107-16;Grabenhorst,Schlenke,Pohl,Nimtz,Conradt,1999,Genetic engineering of recombinantglycoproteins and the glycosylationpathway in mammalian hostcells,Glycoconj J.,16(2),81-97;Mueller,Schlenke,Nimtz,Conradt,Hauser,1999,Recombinant glycoprotein productqualiry in proliferation-controlled BHK-21 cells,Biotechnology and bioengineering,65(5),529-536;Schlenke,Grabenhorst,Nimtz,Conradt,1999,Construction andcharacterization of stably transfected BHK-21 cells withhuman-type sialylation characteristic,Cytotechnology,30(1-3),17-25。
B)在昆虫细胞中的生产
如文献所述,用含有受多角体蛋白启动子控制的人EPOcDNA的重组杆状病毒载体转染细胞后,由昆虫细胞系SF9和SF 21产生重组人EPO。
在无血清培养基中生长的细胞以2×106或2×107个细胞/mL的细胞密度转染,每天测定细胞培养上清液中的EPO滴度。EPO通过Bluesepharose层析、Q-Sepharose上的离子交换层析、最后通过C4相上的RP-HPLC纯化。
产物的纯度通过SDS-PAGE和N末端测序证实。详细的碳水化合物结构分析(N-和O-糖基化)按照发表的操作进行。
文献:
Grabenhorst,Hofer,Nimtz,Jager,Conradt,1993,Biosynthesis and secretion of human in terleukin 2 glycoproteinvariants from baculovirus-infecrted Sf21 cells.Charaterization of polypeptides and posttranslationalmodifications,Eur J Biochem.,215(1),189-97;Quelle,Caslake,Burkert,Wojchowski,1989,High-level expression andpurification of a recombinant human erythropoietin producedusing a baculovirus vector,Blood,74(2),652-7.
实施例2
反应性HES衍生物的形成
1.SH-反应性HES
1.1EMCH与氧代-HES12KD反应,形成SH-反应性HES 12KD B
B

将0.144g(0.012mmol)氧代-HES12KD(Fresenius GermanPatent DE 196 28 705 A1)溶解于0.3mL无水二甲亚砜(DMSO)中,在氮气下逐滴加至34mg(0.15mmol)EMCH(Perbio Science,Deutschland GmbH,Bonn,德国)在1.5mL DMSO中的混合物中。在60℃下搅拌19小时后,将反应混合物加至16mL乙醇与丙酮的1∶1混合物中。离心收集沉淀,再溶解于3mL DMSO中,如上所述再次沉淀。通过离心及真空干燥获得SH-反应性HES 12KD B。与巯基-EPO的偶联反应在实施例3的2.2中描述。
备选方案:
在该反应中可以使用具有被间隔区隔开的酰肼和马来酰亚胺官能基的所有交联剂。表2列出了可从Perbio Science,DeutschlandGmbH,Bonn,Germany获得的这类分子的另外一些实例;用“A”标记。此外,也可以使用具有活化的二硫醚功能团而不是马来酰亚胺的另外一类交联剂。
1.2HES糖基胺的卤代乙酰胺衍生物
a)糖基胺的形成1
将1mg HES12KD样品溶解于3mL饱和碳酸氢铵中。然后另外加入固体碳酸氢铵,以使溶液在30℃120小时的温育过程中保持饱和。氨基-HES12KD C通过直接冻干反应混合物脱盐。
1Manger,Wong,Rademacher,Dwek,1992,Biochemistry,31,10733-10740;Manger,Rademacher,Dwek,1992,Biochemistry,31,10724-10732
b)用氯乙酸酐酰化糖基胺C
将1mg氨基-HES12KD C样品溶解于1mL 1M碳酸氢钠中,在冰上冷却。加入固体氯乙酸酐晶体(~5mg),使反应混合物升温至室温。监测pH,如果pH下降到7.0以下则另外加入碱。在室温下2小时后,加入第二份碱和酸酐。6小时后产物氯乙酰胺-HES D1(X=Cl)通过混合床Amberlite MB-3(H)(OH)离子交换树脂脱盐。
c)用溴乙酸酐酰化糖基胺2
溴乙酸酐如Thomas所述制备3。将1mg氨基-HES12KD C样品溶解于0.1mL干DMF中,在冰上冷却,加入5mg溴乙酸酐。使反应混合物缓慢升温至室温,搅拌溶液3小时。在-20℃下将反应混合物加至1mL乙醇与丙酮的1∶1混合物中。离心收集沉淀,再溶解于0.1mLDMF中,如上所述再次沉淀。通过离心及真空干燥获得溴乙酰胺-HESD2(X=Br)。与巯基-EPO的偶联反应在实施例3的1.2中描述。
2Black,Kiss,Tull,Withers,1993,Carbohydr.Res.,250,195
3Thomas,1977,Methodes Enymol.,46,362
d)相应的碘衍生物D3(X=I)如合成D2所述合成。使用碘乙酸N-琥珀酰亚胺酯代替溴乙酸酐,所有步骤都避光进行。

备选方案:
对于氨基的酰化,可以使用卤素、酸的其它活化形式,例如
--溴化物或-氯化物
-酯,例如N-羟基琥珀酰亚胺酯,与取代酚的酯(对硝基酚、五氟酚、三氯酚等)
此外,也可以使用含有被间隔区隔开的反应性氨基和卤代乙酰基的所有交联剂。一个例子是SBAP。该分子及其它一些分子可获自Perbio Science Deutschland GmbH,Bonn,德国。它们在表2中用“D”标记。关于不需要分离卤代乙酰胺-HES衍生物就可以连接氨基-HES与巯基-EPO的交联剂,见实施例3中1.2的解释。
1.3氨基-HES E的卤代乙酰胺衍生物1
a)1,4-二氨基丁烷与氧代-HES12KD反应,产生氨基-HES12KDE4
4S.Frie,Diplomarbeit,Fachhochschule Hamburg,1998
将1.44g(0.12mmol)氧代-HES12KD溶解于3mL无水二甲亚砜(DMSO)中,在氮气下逐滴加至1.51mL(15mmol)1,4-二氨基丁烷在15mL DMSO中的混合物中。在40℃下搅拌19小时后,将反应混合物加至160mL乙醇与丙酮的1∶1混合物中。离心收集沉淀氨基-HES12KDE,再溶解于40mL水中,对水透析4天(SnakeSkin透析管,3.5KD截留分子量(cut off),Perbio Science Deutschland GmbH,Bonn,德国),冻干。
b)氯乙酰胺-HES12KD F1按上述1.3中制备氯乙酰胺-HES12KDD1的所述方法制备。
c)溴乙酰胺-HES12KD F2(X=Br)按上述1.3中制备溴乙酰胺-HES12KD D2的所述方法制备。与巯基-EPO的偶联反应在实施例3的1.2中描述。
d)相应的碘衍生物F3(X=I)在与巯基-EPO反应之前不分离。实验在实施例3的1.1中描述。
备选方案:
参见上文1.2。
2.CHO-反应性HES
2.1酰肼-HES
a)肼与氧代-HES12KD的反应
将1.44g(0.12mmol)氧代-HES12KD溶解于3mL无水二甲亚砜(DMSO)中,在氮气下逐滴加至0.47mL(15mmol)肼在15mLDMSO中的混合物中。在40℃下搅拌19小时后,将反应混合物加至160mL乙醇与丙酮的1∶1混合物中。离心收集沉淀产物J,再溶解于40mL水中,用0.5%(v/v)三乙胺水溶液透析2天,用水透析2天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,德国),冻干。与氧化的Glyco-EPO的偶联反应在实施例4的2.2中描述。
b)己二酸二酰肼与氧代-HES12KD的反应
在65℃下将1.74g(15mmol)己二酸二酰肼溶解于20mL无水二甲亚砜(DMSO)中,在氮气下逐滴加入溶解于3mL无水DMSO中的
1.44g(0.12mmol)氧代-HES12KD。在60℃下搅拌68小时后,将反应混合物加至200mL水中。含有L的溶液用0.5%(v/v)三乙胺水溶液透析2天,用水透析2天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,德国),冻干。与氧化的Glyco-EPO的偶联反应在实施例4的2.2中描述。
备选方案:
此外,也可以使用2个酰肼基被任一间隔区分隔的衍生物。
3.另外的氨基-HES12KD衍生物I和H1
D或F的氨解如下分开进行:将各1mg卤代乙酰胺样品溶解于0.1mL饱和碳酸铵中。然后加入另外的固体碳酸铵,以使溶液在30℃120小时的温育过程中保持饱和。在-20℃下将反应混合物加至1mL乙醇与丙酮的1∶1混合物中。离心收集沉淀物,再溶解于0.05mL水中,如上所述再次沉淀。通过离心及真空干燥获得产物氨基HESH或I。与氧化的Glyco-EPO的偶联反应在实施例4的4.1中描述。
4.羟胺修饰的HES12KD K
O-[2-(2-氨氧基-乙氧基)-乙基]-羟胺如Boturyn等人所述用可以购得的材料通过两步合成5。将1.44g(0.12mmol)氧代-HES12KD溶解于3mL无水二甲亚砜(DMSO)中,在氮气下逐滴加至2.04g(15mmol)O-[2-(2-氨氧基-乙氧基)-乙基]-羟胺在15mLDMSO中的混合物中。在65℃下搅拌48小时后,将反应混合物加至160mL乙醇与丙酮的1∶1混合物中。离心收集沉淀产物K,再溶解于40mL水中,用水透析4天(SnakeSkin透析管,3.5KD截留分子量,PerbioScienceDeutschland GmbH,Bonn,德国),冻干。与氧化的Glyco-EPO的偶联反应在实施例4的3.1中描述。
备选方案:
此外,也可以使用两个羟胺基团被任何间隔区隔开的衍生物。
5Boturyn,Boudali,Constant,Defrancq,Lhomme,1997,Tetrahedron,53,5485
5.巯基-HES12KD
5.1向氧代-HES12KD中添加

将1.44g(0.12mmol)氧代-HES12KD溶解于3mL无水二甲亚砜(DMSO)中,在氮气下逐滴加至1.16g(15mmol)巯乙胺在15mLDMSO中的混合物中。在40℃下搅拌24小时后,将反应混合物加至160mL乙醇与丙酮的1∶1混合物中。离心收集沉淀产物M,再溶解于40mL水中,用0.5%(v/v)三乙胺水溶液透析2天,用水透析2天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,德国),冻干。与氧化的Glyco-EPO的偶联反应在实施例4的2.1中描述。
备选方案:
也可以使用氨基和硫功能基被任何分隔区隔开的衍生物。此外,衍生物中的氨基也可以被置换为肼、酰肼或羟胺基团。硫功能基可以以例如二硫醚或三苯甲基衍生物的形式受到保护。但在这种情况下,在偶联之前必须进行额外的脱保护步骤,该步骤将释放一种类似于M的成分。
5.2氨基HES12KD E、H或I的修饰
a)用SATA/SATP修饰
将1.44g(0.12mmol)氨基-HES12KD E、H或I溶解于3mL无水二甲亚砜(DMSO)中,在氮气下逐滴加至139mg(0.6mmol)SATA在5mL DMSO中的混合物中。在室温下搅拌24小时后,将反应混合物加至160mL乙醇与丙酮的1∶1混合物中。离心收集沉淀产物N,再溶解于40mL水中,用水透析2天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,德国),冻干。
脱保护在含有25mM EDTA和0.5M羟胺的50mM磷酸钠缓冲液pH7.5中进行,室温下2小时,通过用含有1mM EDTA的0.1M乙酸钠缓冲液pH5.5透析来纯化产物O。在脱保护反应之后立即进行实施例4的2.1中描述的偶联反应。
b)用SPDP修饰
将1.44g(0.12mmol)氨基-HES12KD E、H或I溶解于3mL无水二甲亚砜(DMSO)中,在氮气下逐滴加至187mg(0.6mmol)SPDP在5mL DMSO中的混合物中。在室温下搅拌24小时后,将反应混合物加至160mL乙醇与丙酮的1∶1混合物中。离心收集沉淀产物P,再溶解于40mL水中,用水透析2天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,德国),冻干。
脱保护在每0.5mL含有12mg二硫苏糖醇(DTT)的100mM乙酸钠缓冲液(含100mM氯化钠)pH4.5中进行,室温下30分钟,通过用含有1mM EDTA的0.1M乙酸钠缓冲液pH5.5透析来纯化产物Q。在脱保护反应之后立即进行实施例4的2.1中描述的偶联反应。
备选方案:
对于氨基向游离形式或受保护的硫醇基的转化,可以使用几种试剂。在修饰后,可以分离产物。此外,关于交联剂的使用,它们可以直接用于偶联反应,优选在纯化步骤之后使用。受保护形式的巯基-HES衍生物的合成可以用于分离和贮存巯基-HES衍生物。为此,可以使用类似于SATA的所有衍生物,它们含有被任何间隔区隔开的活性酯官能团和硫酯官能团。表2中的SATP(用“H”标记)是该类的另外一个成员。与SPDP类似的衍生物可能含有被任何间隔区隔开的活性酯官能团和二硫醚官能团。表2中可见这些类的其它一些成员,用“F”标记。其它类似的衍生物可能含有被任何间隔区隔开的活性酯官能团和作为三苯甲基衍生物而受到保护的硫醇官能团。
实施例3
与巯基-EPO的偶联反应
1.巯基-EPO与卤代乙酰胺修饰的SH-反应性HES的反应
1.1实施例方案1
使用含有NHS-活性酯和碘乙酰胺基团的交联剂例如SIA偶联巯基-EPO与氨基-HES12KD(E、H或I)6
6Cumber,Forrester,Foxwell,Ross,Thorpe,1985,MethodsEnrymol.,112,207
材料
A.硼酸盐缓冲液。组成为50 mM硼酸钠,pH8.3,5mM EDTA。
B.PBS,磷酸盐缓冲盐水:10mM磷酸钠,150mM NaCl,pH7.4。
C.氨基-HES12KD E、H或I。在硼酸盐缓冲液中以1mg/mL制备。
D.交联剂贮存液:14mg SIA溶解于1mL DMSO中。
E.D-SaltTM葡聚糖脱盐柱,2×5mL柱床体积(Perbio ScienceDeutschland GmbH,Bonn,德国)
F.Coomassie蛋白质检测试剂(Perbio Science DeutschlandGmbH,Bonn,德国)
G.巯基EPO溶液:硼酸盐缓冲液中的5mg/mL巯基EPO1
H.微量浓缩器:Microcon YM-3(amicon,Milipore GmbH,Eschborn,德国)
方法
将100μL SIA溶液加至400μL氨基HES12KD E溶液中,在室温下搅拌使之反应0.5小时。用微量浓缩器以14000×g离心样品60分钟除去过量的交联剂。离心后,用硼酸盐缓冲液使样品达到其初始体积,该步骤重复两次以上。将所得溶液加至1mL巯基EPO溶液中,反应混合物在室温下温育16小时。在温育结束时,通过加入半胱氨酸至终浓度为10mM来终止过量碘乙酰胺的反应性。将反应混合物上样到用PBS缓冲液平衡的脱盐柱上,用Coomassie蛋白质检测试剂监测级分中的蛋白质含量。合并含蛋白质偶联物的所有级分,用水透析过夜后通过冻干获得偶联物。
备选方案:
在该反应中,可以使用含有被间隔区隔开的琥珀酰亚胺或硫代琥珀酰亚胺官能团和碘乙酰胺官能团的所有交联剂。表2中可见其它一些实例。它们用“C”标记,可获自Perbio Science Deutschland GmbH,Bonn,德国。
1.2实施例方案2
巯基EPO 1与SH反应性HES12KD溴乙酰胺D2、F2或碘乙酰胺D3的偶联7
材料
A.磷酸盐缓冲液:组成为100mM磷酸钠,pH6.1,5mM EDTA。
B.PBS,磷酸盐缓冲盐水:10mM磷酸钠,150mM NaCl,pH7.4。
C.SH反应性HES12KD溴乙酰胺D2。在磷酸盐缓冲盐水中以10mg/mL制备。
D.D-SaltTM葡聚糖脱盐柱,2×5mL柱床体积(Perbio ScienceDeutschland GmbH,Bonn,德国)
E.Coomassie蛋白质测定试剂(Perbio Science DeutschlandGmbH,Bonn,德国)
F.巯基EPO溶液:磷酸盐缓冲盐水中的5mg/mL巯基EPO1
方法
1mL SH反应性HES12KD溴乙酰胺D2溶液与1mL巯基-EPO溶液混合,反应混合物在室温下温育48小时。在温育结束时,通过加入半胱氨酸至终浓度10mM终止过量溴乙酰胺的反应性。将反应混合物上样到用PBS缓冲液平衡的脱盐柱上。用Coomassie蛋白质检测试剂监测级分的蛋白质含量,合并含有蛋白质偶联物的所有级分,在用水透析过夜后通过冻干获得偶联物。
7de Valasco,Merkus,Anderton,Verheul,Lizzio,Van der Zee,van Eden,Hoffmann,Verhoef,Snippe,1995,Infect.Immun.,63,961
备选方案:
代替SH反应性HES12KD-溴乙酰胺D2的分离,氨基HES12KD(E,H,I)可以通过琥珀酰亚胺官能团和溴乙酰胺官能团用交联剂连接(见上文1.1)。SBAP是这类交联剂的一个成员,在表2中可见,用“D”标记。
2.巯基-EPO与马来酰亚胺修饰的SH反应性HES的反应
2.1实施例方案3
用含有酰肼和马来酰亚胺官能团的交联剂例如M2C2H来偶联巯基EPO与HES12KD
材料
A.M2C2H贮存液:DMSO中的10mg/mL M2C2H,新鲜制备
B.HES12KD:0.1M乙酸钠缓冲液pH5.5中的10mg/mL
C.巯基EPO溶液:磷酸盐/NaCl缓冲液中的5mg/mL巯基EPO
D.磷酸盐/NaCl:0.1M磷酸钠,50mM NaCl,pH7.0
E.微量浓缩器:Microcon YM-3(amicon,Milipore GmbH,Eschborn,德国)
F.凝胶过滤柱:例如,SephadexG-200(1.5×45cm)
G.Coomassie蛋白质检测试剂(Perbio Science DeutschlandGmbH,Bonn,德国)
H.PBS,磷酸盐缓冲盐水:10mM磷酸钠,150mM NaCl,pH7.4。
方法
将M2C2H溶液加至400μL HES12KD溶液中至终浓度为1mM,使之在室温搅拌下反应2小时。用微量浓缩器以14000×g离心样品60分钟除去过量的交联剂。离心后,用磷酸盐/NaCl缓冲液使样品达到其初始体积,该步骤重复两次以上。向M2C2H修饰的HES12KD中加入0.5mL巯基EPO溶液,反应混合物在室温下温育2小时。在温育结束时,通过加入半胱氨酸至终浓度为10mM终止过量马来酰亚胺的反应性。将反应混合物上样到用PBS缓冲液平衡的ScphadexG-200(1.5×45cm)上,以1mL为单位收集级分。用Coomassie蛋白质检测试剂监测级分的蛋白质含量。合并含有蛋白质偶联物的所有级分,在用水透析过夜后通过冻干获得偶联物。
操作注意事项
腙加合物在极端pH下略不稳定。对于可能涉及低pH下处理的应用,我们通过用PBS缓冲液中的30mM氰基硼氢化钠处理,将腙还原为肼。对于大多数应用,该额外步骤是不必要的。
2.2实施例方案4
巯基EPO与马来酰亚氨基-HES12KD B的偶联
材料
A.马来酰亚氨基-HES12KD B:0.1M乙酸钠缓冲液pH5.5中的10mg/mL
B.巯基EPO溶液:磷酸盐/NaCl缓冲液中的5mg/mL巯基EPO
C.磷酸盐/NaCl:0.1M磷酸钠,50mM NaCl,pH7.0
D.凝胶过滤柱:例如,SephadexG-200(1.5×45cm)
E.Coomassie蛋白质检测试剂(Perbio Science DeutschlandGmbH,Bonn,德国)
F.PBS,磷酸盐缓冲盐水:10mM磷酸钠,150mM NaCl,pH7.4。
方法
1mL SH反应性HES12KD B溶液与1mL巯基EPO1溶液混合,在室温下温育2小时。在温育结束时,通过加入半胱氨酸至终浓度10mM终止过量马来酰亚胺的反应性。将反应混合物上样到用PBS缓冲液平衡的SephadexG-200(1.5×45cm)上,以1mL为单位收集级分。用Coomassie蛋白质检测试剂监测级分的蛋白质含量。合并含有蛋白质偶联物的所有级分,在用水透析过夜后通过冻干获得偶联物。
2.3实施例方案12
使用含有NHS-活性酯和马来酰亚胺官能团的交联剂例如SMCC来偶联巯基EPO与氨基HES12KD(E,H,I)
材料
A.微量浓缩器:Microcon YM-10(amicon,Milipore GmbH,Eschborn,德国)
B.PBS,磷酸盐缓冲盐水:10mM磷酸钠,150mM NaCl,pH7.4
C.氨基HES12KD E、H或I。在PBS缓冲液中以10mg/mL制备。
D.SMCC溶液:1mg SMCC溶解于50μLDMSO中
E.D-SaltTM葡聚糖脱盐柱,2×5mL柱床体积(Perbio ScienceDeutschland GmbH,Bonn,德国)
F.Coomassie蛋白质检测试剂(Perbio Science DeutschlandGmbH,Bonn,德国)
G.巯基EPO
1溶液:PBS缓冲液中的5mg/mL巯基EPO
方法
向50μL SMCC溶液中加入400μl氨基HES12KD E溶液,使反应混合物在室温搅拌下反应80分钟,在46℃下反应10分钟。通过使用微量浓缩器以14000×g离心样品60分钟除去过量的交联剂。用PBS缓冲液使体积达到450μL,该步骤重复两次以上。最后一次离心后,向所得溶液中加入PBS至450μL,加至1mL巯基EPO溶液中,反应混合物在室温下温育16小时。在温育结束时,通过加入半胱氨酸至终浓度10mM终止过量马来酰亚胺的反应性。将反应混合物上样到用PBS缓冲液平衡的脱盐柱上。用Coomassie蛋白质检测试剂监测级分的蛋白质含量,合并含有蛋白质偶联物的所有级分,用水透析过夜后通过冻干获得偶联物。
备选方案:
在该反应中,可以使用含有被间隔区隔开的琥珀酰亚胺或硫代琥珀酰亚胺官能团和马来酰亚胺官能团的所有交联剂。表2中可见这类分子的其它一些例子,用“E”标记,可以获自Perbio ScienceDeutschland GmbH,Bonn,德国。还有另外一类交联剂,它们含有活化的二硫醚官能团而不是马来酰亚胺官能团。这些交联剂也可以用于偶联。而该偶联物的二硫键在还原条件下可以被切割。这一类的成员在表2中用“F”标记。第三类交联剂利用乙烯砜官能团代替马来酰亚胺官能团作为SH反应性基团。这一类的一个成员是“SVSB”,在表2中用“G”标记。
实施例4
与氧化的EPO的偶联反应
1.Glyco-EPO的氧化
1.1用偏高碘酸钠氧化Glyco-EPO:实施例方案5
材料
A.Glyco-EPO溶液:乙酸盐缓冲液中的10mg/mL Glyco-EPO
B.偏高碘酸钠溶液:乙酸盐缓冲液中的10mM或100mM高碘酸钠,新鲜制备。暗处保存。使用这些溶液时,氧化混合物中高碘酸钠的终浓度分别为1mM或10mM。
C.乙酸盐缓冲液:0.1M乙酸钠缓冲液,pH5.5
D.甘油
E.微量浓缩器:Microcon YM-3(amicon,Milipore GmbH,Eschborn,德国)
方法
所有步骤都避光进行。
向1mL冷Glyco-EPO溶液中加入0.1mL冷偏高碘酸钠溶液,氧化反应避光进行1小时。如果待氧化的Glyco-EPO含有唾液酸残基,则氧化条件是1mM高碘酸钠,0℃。否则,使用室温下10mM高碘酸钠的条件。为了终止氧化,加入甘油至终浓度为15mM,并在0℃下温育5分钟。用微量浓缩器以14000×g离心产物60分钟,除去过量的试剂和副产物。离心后,用下一修饰步骤所用的缓冲液例如乙酸盐缓冲液,使样品达到其初始体积。该步骤重复两次以上。
1.2Glyco-EPO的酶氧化:实施例方案6
EPO的酶氧化在别处已经有描述(Chamow等人,1992,J.Biol.Chem.,267,15916-15922)。
2.与肼/酰肼衍生物的偶联
2.1实施例方案7
用含有酰肼和马来酰亚胺官能团的交联剂例如M2C2H(PerbioScience,Deutschland GmbH,Bonn,德国)来偶联氧化的Glyco-EPO与巯基-HES12KD M、O或Q。
材料
A.M2C2H贮存液:DMSO中的10mg/mL M2C2H,新鲜制备
B.来自6.1.1的氧化的Glyco-EPO溶液:乙酸盐缓冲液中的5mg/mL Glyco-EPO
C.巯基-HES12KD M、O或Q:磷酸盐/NaCl缓冲液中的10mg/mL
D.乙酸盐缓冲液:0.1M乙酸钠缓冲液,pH5.5
E.磷酸盐/NaCl:0.1M磷酸钠,50mM NaCl,pH7.0
F.微量浓缩器:Microcon YM-3(amicon,Milipore GmbH,Eschborn,德国)
G.凝胶过滤柱:例如,SephadexG-200(1.5×45cm)
H.Coomassie蛋白质检测试剂(Perbio Science DeutschlandGmbH,Bonn,德国)
I.PBS,磷酸盐缓冲盐水:10mM磷酸钠,150mM NaCl,pH7.4
方法
向1mL氧化的Glyco-EPO中加入M2C2H贮存液至终浓度为1mM,使之在室温搅拌下反应2小时。通过使用微量浓缩器以14000×g离心样品60分钟,除去过量的交联剂。离心后,用磷酸盐/NaCl缓冲液使样品达到其初始体积,该步骤重复两次以上。向M2C2H修饰的Glyco-EPO中加入1mL巯基-HES12KD M、O或Q溶液,反应混合物在室温下温育16小时。在温育结束时,通过加入半胱氨酸终止过量马来酰亚胺的反应性。将反应混合物上样到用PBS缓冲液平衡的SephadexG-200(1.5×45cm)上,以1mL为单位收集级分。用Coomassie蛋白质检测试剂监测级分的蛋白质含量,合并含有蛋白质偶联物的所有级分,用水透析过夜后通过冻干获得偶联物。
操作注意事项
腙加合物在极端pH下略不稳定。对于可能涉及低pH下处理的应用,我们用PBS缓冲液中的30mM氰基硼氢化钠处理,将腙还原为肼。对于大多数应用,该额外步骤是不必要的。
2.2实施例方案8
氧化的Glyco-EPO与酰肼基-HES12KD L或J直接偶联
材料
A.来自6.1.1的氧化的Glyco-EPO溶液:乙酸盐缓冲液中的5mg/mL Glyco-EPO
B.酰肼基-HES12KD L或J:乙酸盐缓冲液中10mg/mL
C.乙酸盐缓冲液:0.1M乙酸钠缓冲液,pH5.5
D.凝胶过滤柱:例如,SephadexG-200(1.5×45cm)
E.Coomassie蛋白质检测试剂(Perbio Science DeutschlandGmbH,Bonn,德国)
F.PBS,磷酸盐缓冲盐水:10mM磷酸钠,150mM NaCl,pH7.4
方法
1mL酰肼基-HES12KD L或J溶液与1mL氧化的Glyco-EPO溶液混合,反应混合物在搅拌下室温温育16小时。将反应混合物上样到用PBS缓冲液平衡的SephadexG-200(1.5×45cm)上,以1mL为单位收集级分。用Coomassie蛋白质检测试剂监测级分的蛋白质含量,合并含有蛋白质偶联物的所有级分,用水透析过夜后通过冻干获得偶联物。偶联结果在图24中显示。观察到的分子位移证明偶联是成功的。拖尾(smear)是由于HES的不均一性引起的。图25证明HES与碳水化合物侧链的碳水化合物部分偶联。
操作注意事项
腙加合物在极端pH下略不稳定。对于可能涉及低pH下处理的应用,我们通过用PBS缓冲液中的30mM氰基硼氢化钠处理,将腙还原为肼。对于大多数应用,该额外步骤是不必要的。
3.与羟胺衍生物的偶联8
3.1实施例方案9
氧化的Glyco-EPO与羟氨基-HES12KD K的偶联
材料
A.来自6.1.1的氧化的Glyco-EPO溶液:乙酸盐缓冲液中的5mg/mL Glyco-EPO
B.羟氨基-HES12KD K:乙酸盐缓冲液中的10mg/mL
C.乙酸盐缓冲液:0.1M乙酸钠缓冲液,pH5.5
D.凝胶过滤柱:例如,SephadexG-200(1.5×45cm)
E.Coomassie蛋白质检测试剂(Perbio Science DeutschlandGmbH,Bonn,德国)
F.PBS,磷酸盐缓冲盐水:10mM磷酸钠,150mM NaCl,pH7.4
8Rose,1994,Am.Chem.Soc.,116,30
方法
1mL羟氨基-HES12KD K溶液与1mL氧化的Glyco-EPO溶液混合,反应混合物在搅拌下室温温育16小时。将反应混合物上样到用PBS缓冲液平衡的SephadexG-200(1.5×45cm)上,以1mL为单位收集级分。用Coomassie蛋白质检测试剂监测级分的蛋白质含量,合并含有蛋白质偶联物的所有级分,用水透析过夜后通过冻干获得偶联物。偶联结果在图24中显示。在2道观察到的分子位移证明偶联是成功的。拖尾(smear)是由于HES的不均一性引起的。图25证明HES与碳水化合物侧链的碳水化合物部分偶联。
实施例5
半乳糖氧化酶处理的EPO N-聚糖的鉴定
在过氧化氢酶存在下,在0.05M磷酸钠缓冲液pH7.0中,重组EPO或部分去唾液酸化的EPO型(有限弱酸水解产生的)与半乳糖氧化酶在37℃温育30分钟-4小时。通过取出50μg等份EPO,随后用多肽N-聚糖酶处理蛋白质,监测反应的进展。
在除去唾液酸之前及之后,如(Grabenhorst等人,1999,Nimtz等人,1993/1994;Schlenke等人,1999)所述,对释放的N-连接寡糖(通过SDS-PAGE检测去-N-糖基化多肽进行监测)进行HPAEC-PAD作图。根据HPAEC-PAD中观察到的典型位移,定量各EPO寡糖中氧化的半乳糖残基,也通过寡糖混合物的MALDI/TOF MS证实定量。
实施例6
HAS修饰的EPO的鉴定
使用例如Ultrogel AcA 44/54或类似的凝胶过滤介质,通过凝胶过滤实现HAS修饰的EPO型与未反应的EPO和HAS前体分子的分离。此外也可以如下除去未反应的HAS:在含有与Affigel(BioRad)偶联的单克隆抗体的4mL柱上免疫亲和分离EPO,随后通过凝胶过滤分离未修饰的EPO(例如使用能够分离分子量在20kDa-200kDa之间的球蛋白的介质)。
通过SDS-PAGE分析(使用12.5%或10%丙稀酰胺凝胶),通过考马斯亮蓝染色凝胶后检测高于未修饰EPO的分子量,鉴定HAS修饰的EPO。使用抗重组人EPO的多克隆抗体对样品进行Western Blot分析,也能鉴定HAS修饰的EPO多肽的较高分子量。
EPO型的N-聚糖修饰通过用多肽N-聚糖酶(重组N-糖苷酶,来自德国Roche,使用25单位/mg EPO蛋白,37℃16小时)从EPO蛋白上成功去除来证明;SDS-PAGE分析可见EPO蛋白向N-糖苷酶处理的未修饰EPO之迁移位置典型位移约20KDa。
通过SDS-PAGE检测与未反应的去-N-糖基化EPO相比去-N-糖基化产物的迁移位置,证实单去唾液酸化并经半乳糖氧化酶处理的EPOO-聚糖在Ser 126处的修饰。必要时,在SDS-PAGE分析之前通过在C8-相上的RP-HPLC分级修饰的EPO。通过O-聚糖的β-去除以及使用抗重组人EPO多克隆抗体的Western blot检测EPO的去-O-糖基化形式,来分析EPO的HAS O-聚糖修饰。
实施例7
EPO和修饰EPO型的定量
用如欧洲药典(2000,Erythropoietini solutio concentrata,1316,780-785)所述的UV测量对EPO定量,并且与国际BRP参照EPO标准比较。此外,也可以根据使用RP-C4-柱的RP-HPLC分析以及在254nm处的吸光度(用20、40、80、120μg BRP标准EPO参照制品进行校准),测定EPO浓度。
实施例8
HES修饰的重组人EPO的体外生物活性
使用如Krystal[Krystal,1984,Exp.Heamatol.,11,649-660]描述的促红细胞生成素生物活性分析来检测纯化的HES修饰的EPO的活性。
通过盐酸苯肼处理在NMRI小鼠中诱导贫血,如[Fibi等人,1991,Blood,77,1203ff.]所述收集并使用脾细胞。EPO的稀释液在96孔微量滴定板中与3×105细胞/孔温育。在潮湿空气(5%CO2)下37℃温育24小时后,细胞用每孔1μCi 3H-胸苷标记4小时。通过液体闪烁计数测量掺入的放射性。国际参照EPO标准(BRP-标准)用于比较。
此外,也可以使用EPO敏感细胞系TF-1(Kitamura等人,[J.cellPhys.,140.323-334]),通过体外检测来测量EPO的生物活性。洗去指数生长的细胞中的生长因子,在连续稀释的EPO存在下再温育48小时。使用Mosmann[Mosman,1983,J.Immunol.Methods,65,55-63]所述的MTT还原检测来估计细胞的增殖。
实施例9
EPO和HAS-修饰的EPO型的体内活性测定
在红细胞正常的小鼠中进行体内活性测定,方法是在动物接受预定剂量的EPO或修饰EPO型4天后测量网织红细胞的增加。使用BRPEPO标准进行测定,该标准在红细胞增多小鼠测定中用WHO EPO标准校准。EPO样品用含有1mg/ml牛血清白蛋白(Sigma)的磷酸盐缓冲盐水稀释。
每只动物皮下注射0.5ml EPO在Dulbecco′s缓冲盐溶液中的检测溶液(相当于100、80、40或20IU/ml BRP标准EPO的EPO蛋白量)。注射4天后采集血样,网织红细胞用吖啶橙染色;在采集血样后5小时内用流式细胞仪计数总共30,000个血细胞,对网织红细胞定量(参见欧洲药典,2000,Erythropoietini solutio concentrata,1316,780-785和欧洲药典(1996/2000,附件2002))。
实施例10
体内半衰期测定
给兔静脉内注射指定量的未修饰或HAS-修饰的EPO型。在指定时间采集血样,制备血清。血清促红细胞生成素水平通过体外生物测定或者通过EPO-特异性商品化ELISA测定。
实施例11
体内药代动力学
小鼠:每只动物皮下接受300IU EPO/kg。处理7天后测量每只动物的血细胞比容。在修饰EPO处理的所有动物中有9只观察到血细胞比容显著增加,由于未处理的EPO的半衰期相对较短,这是一个预料之中的结果。修饰EPO处理组的血细胞比容的平均变化与未处理EPO组和对照组显著不同。
兔:兔用相当于200或者多达800ng/kg体重的单次剂量未修饰或HAS-修饰的EPO处理。2、6、16、24、48小时后用测定血浆浓度的商品化EPO-特异性ELISA分析血样。测定平均血浆EPO浓度,如(Zettlmissl等人,1989,J.Biol.Chem.,264,21153-21159)所述,根据ELISA值计算平均初始半衰期(α-相)和终末半衰期(β-相)。
文献:
Sytkowski,Lunn,Risinger和Davis,1999,An ErythropoietinFusion Protein Comprised of Identical Repeating DomainsExhititis Enhanced Biological Properites,J.Biol.Chem.,274,24773-24778。
实施例12
HES-修饰的重组人IL-2的体外生物活性评估
通过在Ultrogel AcA 54上凝胶过滤来回收修饰的IL2。无菌过滤相应级分的等份,使用依赖IL2的鼠CTLL-2细胞系测定IL2的生物活性[Gillis,Ferm,On和Smith,1978,J.Immunol.,120,2027-2032]。活性与国际参照IL2标准制品相关联。
实施例13
通过未氧化的还原性末端的还原胺化形成羟乙基淀粉衍生物
实施例13.1羟乙基淀粉与1,3-二氨基-2-羟基丙烷的反应
a)向200mg羟乙基淀粉(HES18/0.4(MW=18,000D,DS=0.4))在5ml水中的溶液中加入0.83mmol 1,3-二氨基-2-羟基丙烷和50mg氰基硼氢化钠NaCNBH3。获得的混合物在80℃下温育17小时。将反应混合物加至160mL丙酮与乙醇的1∶1冷混合物中。离心收集沉淀,用水透析4天(SnakeSkin透析管,3.5KD截留分子量,Perbio ScienceDeutschland GmbH,Bonn,D),冻干。
b)向200mg羟乙基淀粉的溶液中加入0.83mmol 1,3-二氨基-2-羟基丙烷和50mg氰基硼氢化钠NaCNBH3产生的混合物,也可以在25℃下温育3天。
实施例13.2羟乙基淀粉与1,2-二羟基-3-氨基丙烷的反应
a)向200mg羟乙基淀粉(HES18/0.4(MW=18,000D,DS=0.4))在5ml水中的溶液中加入0.83mmol 1,2-二羟基-3-氨基丙烷和50mg氰基硼氢化钠NaCNBH3。获得的混合物在80℃下温育17小时。将反应混合物加至160mL丙酮与乙醇的1∶1(v/v)冷混合物中。离心收集沉淀,用水透析4天(SnakeSkin透析管,3.5KD截留分子量,PerbioScience Deutschland GmbH,Bonn,D),冻干。
如G.Avigad,Anal.Biochem.134(1983)449-504所述,用高碘酸盐氧化切割反应产物中的1,2-二醇产生醛,通过对醛定量间接证实1,2-二羟基-3-氨基丙烷与HES的反应。
b)向200mg羟乙基淀粉的溶液中加入0.83mmol 1,2-二羟基-3-氨基丙烷和50mg氰基硼氢化钠NaCNBH3产生的混合物,也可以在25℃下温育3天。
实施例13.3羟乙基淀粉与1,4-二氨基丁烷的反应

a)向200mg羟乙基淀粉(HES18/0.4(MW=18,000D,DS=0.4))在5ml水中的溶液中加入0.83mmol 1,4-二氨基丁烷和50mg氰基硼氢化钠NaCNBH3。获得的混合物在80℃下温育17小时。将反应混合物加至160mL丙酮与乙醇的1∶1(v/v)冷混合物中。离心收集沉淀,用水透析4天(SnakeSkin透析管,3.5KD截留分子量,Perbio ScienceDeutschland GmbH,Bonn,D),冻干。
b)向200mg羟乙基淀粉的溶液中加入0.83mmol 1,4-二氨基丁烷和50mg氰基硼氢化钠NaCNBH3产生的混合物,也可以在25℃下温育3天。
实施例13.4羟乙基淀粉与1-巯基-2-氨基乙烷的反应
a)向200mg羟乙基淀粉(HES18/0.4(MW=18,000D,DS=0.4))在5ml水中的溶液中加入0.83mmol 1-巯基-2-氨基乙烷和50mg氰基硼氢化钠NaCNBH3。获得的混合物在80℃下温育17小时。将反应混合物加至160mL丙酮与乙醇的1∶1(v/v)冷混合物中。离心收集沉淀,用水透析4天(snakeSkin透析管,3.5KD截留分子量,PerbioScience Deutschland GmbH,Bonn,D),冻干。
b)向200mg羟乙基淀粉的溶液中加入0.83mmol 1-巯基-2-氨基乙烷和50mg氰基硼氢化钠NaCNBH3产生的混合物,也可以在25℃下温育3天。
实施例14:
通过与未氧化的还原性末端偶联形成羟乙基淀粉衍生物
实施例14.1:羟乙基淀粉与碳酰肼的反应

将0.96g HES18/0.4(MW=18,000D,DS=0.4)溶解于8ml 0.1M乙酸钠水性缓冲液pH5.2中,加入8mmol碳酰肼(Sigma Aldrich,Taufkirchen,D)。在25℃下搅拌18小时后,将反应混合物加至160mL丙酮与乙醇的1∶1(v/v)冷混合物中。离心收集沉淀产物,再溶解于40ml水中,用水透析4天(SnakeSkin透析管,3.5 KD截留分子量,Perbio Science Deutschland GmbH,Bonn,D),冻干。
实施例14.2:羟乙基淀粉与己二酸二酰肼的反应
将0.96g HES18/0.4(MW=18,000D,DS=0.4)溶解于8ml 0.1M乙酸钠水性缓冲液pH5.2中,加入8mmol己二酸二酰肼(LancasterSynthesis,Frankfurt/Main,D)。在25℃下搅拌18小时后,将反应混合物加至160mL丙酮与乙醇的1∶1(v/v)冷混合物中。离心收集沉淀产物,再溶解于40ml水中,用水透析3天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,D),冻干。
实施例14.3:羟乙基淀粉与1,4-亚苯基-二-3-氨基硫脲的反应
将0.96g HES18/0.4(MW=18,000D,DS=0.4)溶解于8ml 0.1M乙酸钠水性缓冲液pH5.2中,加入8mmol 1,4-亚苯基-二-3-氨基硫脲(Lancaster Synthesis,Frankfurt/Main,D)。在25℃下搅拌18小时后,向反应混合物中加入8ml水,悬液以4,500rpm离心15分钟。将澄清的上清液倾析并随后加至160mL丙酮与乙醇的1∶1(v/v)冷混合物中。离心收集沉淀产物,再溶解于40ml水中,以4,500rpm离心15分钟。澄清的上清液用水透析3天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,D),冻干。
实施例14.4:羟乙基淀粉与O-[2-(2-氨氧基-乙氧基)-乙基]-羟胺的反应

O-[2-(2-氨氧基-乙氧基)-乙基]-羟胺如Boturyn等人Tetrahedron 53(1997)5485-5492所述用可购得的材料通过两步合成。
将0.96g HES18/0.4(MW=18,000D,DS=0.4)溶解于8ml0.1M乙酸钠水性缓冲液pH5.2中,加入8mmol O-[2-(2-氨氧基-乙氧基)-乙基]-羟胺。在25℃下搅拌18小时后,将反应混合物加至160mL丙酮与乙醇的1∶1(v/v)冷混合物中。离心收集沉淀产物,再溶解于40ml水中,用水透析3天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,D),冻干。
实施例15
通过与氧化的还原性末端反应形成羟乙基淀粉衍生物
实施例15.1羟乙基淀粉与碳酰肼的反应

将0.12mmol氧代-HES 10/0.4(MW=10,000D,DS=0.4,根据DE196 28 705 A1制备)溶解于3ml无水二甲亚砜(DMSO)中,在氮气下逐滴加至15mmol碳酰肼(Sigma Aldrich,Taufkirchen,D)在15mlDMSO中的混合物中。在65℃下搅拌88小时后,将反应混合物加至160mL丙酮与乙醇的1∶1(v/v)冷混合物中。离心收集沉淀,用水透析4天(SnakeSkin透析管,3.5KD截留分子量,PerbioscienceDeutschland GmbH,Bonn,D),冻干。
实施例15.2羟乙基淀粉与1,4-亚苯基-二-3-氨基硫脲的反应

将0.12mmol氧代-HES 10/0.4(MW=10,000 D,DS=0.4,根据DE196 28 705 A1制备)溶解于3ml无水二甲亚砜(DMSO)中,在氮气下逐滴加至15mmol 1,4-亚苯基-二-3-氨基硫脲(Lancaster Synthesis,Frankfurt/Main,D)在15ml DMSO中的混合物中。在65℃下搅拌88小时后,将反应混合物加至160mL丙酮与乙醇的1∶1(v/v)冷混合物中。离心收集沉淀,用水透析4天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,D),冻干。
实施例15.3羟乙基淀粉与肼的反应
H2N-NH2
将1.44g(0.12mmol)氧代-HES 10/0.4(MW=10,000D,DS=0.4,根据DE 196 287 05 A1制备)溶解于3ml无水二甲亚砜(DMSO)中,在氮气下逐滴加至0.47ml(15mmol)肼在15ml DMSO中的混合物中。在40℃下搅拌19小时后,将反应混合物加至160mL乙醇与丙酮的1∶1(v/v)混合物中。离心收集沉淀产物,再溶解于40mL水中,用0.5%(v/v)三乙胺水溶液透析2天,用水透析2天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,德国),冻干。
实施例15.4羟乙基淀粉与羟胺的反应
O-[2-(2-氨氧基-乙氧基)-乙基]-羟胺如Boturyn等人所述用可购得的材料通过两步合成(Boturyn,Boudali,Constant,Defrancq,Lhomme,1997,Tetrahedron,53,5485)。
将1.44g(0.12mmol)氧代-HES 10/0.4(MW=10,000D,DS=0.4,根据DE 196 287 05 A1制备)溶解于3ml无水二甲亚砜(DMSO)中,在氮气下逐滴加至2.04g(15mmol)O-[2-(2-氨氧基-乙氧基)-乙基]-羟胺在15ml DMSO中的混合物中。在65℃下搅拌48小时后,将反应混合物加至160mL乙醇与丙酮的1∶1(v/v)混合物中。离心收集沉淀产物,再溶解于40mL水中,用水透析4天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,德国),冻干。
实施例15.5羟乙基淀粉与己二酸二酰肼的反应
在65℃下将1.74g(15mmol)己二酸二酰肼溶解于20ml无水二甲亚砜(DMSO)中,在氮气下逐滴加入溶解于3ml无水二甲亚砜中的1.44g(0.12mmol)氧代-HES 10/0.4(MW=10,000D,DS=0.4,根据DE 196 28 705 A1制备)。在60℃下搅拌68小时后,将反应混合物加至200mL水中。含有反应产物的溶液用0.5%(v/v)三乙胺水溶液透析2天,用水透析2天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,德国),冻干。
实施例15.6羟乙基淀粉与1,4-二氨基丁烷的反应
将1.44g(0.12mmol)氧代-HES 10/0.4(MW=10,000D,DS=0.4,根据DE 196 287 05 A1制备)溶解于3ml无水二甲亚砜(DMSO)中,在氮气下逐滴加至1.51ml(15mmol)1,4-二氨基丁烷在15ml DMSO中的混合物中。在40℃下搅拌19小时后,将反应混合物加至160mL乙醇与丙酮的1∶1(v/v)混合物中。离心收集沉淀氨基-HES10KD/0.4,再溶解于40ml水中,用水透析4天(SnakeSkin透析管,3.5KD截留分子量,Perbio Science Deutschland GmbH,Bonn,德国),冻干。
实施例16促红细胞生成素的氧化
氧化的促红细胞生成素如实施例20所述生产。使用实施例20.11(c)所述的EPO-GT-1-A作为氧化的促红细胞生成素(未进行酸水解,用温和的高碘酸盐氧化处理的EPO-GT-1)。
实施例17:
羟乙基淀粉衍生物与实施例4氧化的促红细胞生成素的偶联
实施例17.1氧化的促红细胞生成素与实施例14.1反应产物的反应
用5M乙酸钠缓冲液pH5.2将含氧化EPO(1.055μg/μl)的20mMPBS缓冲液调节为pH5.3。向19μl EPO溶液中加入根据实施例14.1产生的18μl HES衍生物溶液(MW18kD;18.7μg/μl,在0.1M乙酸钠缓冲液pH5.2中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图3中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例17.2氧化的促红细胞生成素与实施例14.3反应产物的反应
用5M乙酸钠缓冲液pH5.2将含氧化EPO(1.055μg/μl)的20mMPBS缓冲液调节为pH5.3。向19μlEPO溶液中加入根据实施例14.3产生的18μl HES衍生物溶液(MW 18kD;18.7μg/μl,在0.1M乙酸钠缓冲液pH5.2中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。
实施例17.3氧化的促红细胞生成素与实施例14.4反应产物的反应
用5M乙酸钠缓冲液pH5.2将含氧化EPO(1.055μg/μl)的20mMPBS缓冲液调节为pH5.3。向19μl EPO溶液中加入根据实施例14.4产生的18μl HES衍生物溶液(MW 18kD;18.7μg/μl,在0.1M乙酸钠缓冲液pH5.2中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图4中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例17.4氧化的促红细胞生成素与实施例15.1反应产物的反应
用5M乙酸钠缓冲液pH5.2将含氧化EPO(1.055μg/μl)的20mMPBS缓冲液调节为pH5.3。向19μl EPO溶液中加入根据实施例15.1产生的18μl HES衍生物溶液(MW 10kD;18.7μg/μl,在0.1M乙酸钠缓冲液pH5.2中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图5中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例17.5氧化的促红细胞生成素与实施例15.2反应产物的反应
用5M乙酸钠缓冲液pH5.2将含氧化EPO(1.055μg/μl)的20mMPBS缓冲液调节为pH5.3。向19μl EPO溶液中加入根据实施例15.1产生的18μl HES衍生物溶液(MW 10kD;18.7μg/μl,在0.1M乙酸钠缓冲液pH5.2中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10% Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品 。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图5中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例18通过还原促红细胞生成素形成巯基-EPO
241.5μg促红细胞生成素(EPO-GT-1,见实施例20)在500μl 0.1M硼酸钠缓冲液,5mM EDTA,10mM DTT(Lancaster,Morcambe,UK),pH8.3中37℃温育1小时。使用VIVASPIN0.5ml浓缩器,10KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤除去DTT,随后用硼酸盐缓冲液洗涤3次,用磷酸盐缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)洗涤2次。
实施例19:
用交联化合物偶联羟乙基淀粉衍生物与巯基促红细胞生成素
在下列每个实施例中,都使用N-(α-马来酰亚胺基乙酰氧基)琥珀酰亚胺酯(AMAS)作为交联化合物。
实施例19.1巯基促红细胞生成素与实施例14.1的反应产物和交联化合物的反应
向根据实施例14.1产生并且溶解于200μl 0.1 M磷酸钠缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufkirchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去剩余的AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图6中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例19.2巯基促红细胞生成素与实施例14.2的反应产物和交联化合物的反应
向根据实施例14.2产生并且溶解于200μl 0.1M磷酸钠缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufkirchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去剩余的AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图7中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例19.3巯基促红细胞生成素与实施例14.3的反应产物和交联化合物的反应
向根据实施例14.3产生并且溶解于200μl 0.1M磷酸钠缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufkirchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去剩余的AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图7中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例19.4巯基促红细胞生成素与实施例14.4的反应产物和交联化合物的反应
向根据实施例14.4产生并且溶解于200μl 0.1M磷酸钠缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufkirchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去剩余的AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图6中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例19.5巯基促红细胞生成素与实施例13.1的反应产物和交联化合物的反应
向在80℃温育17小时以及25℃温育3天的根据实施例13.1产生并且溶解于200μl 0.1M磷酸钠缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufkirchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去剩余的AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图7中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例19.6巯基促红细胞生成素与实施例13.3的反应产物和交联化合物的反应
向在80℃温育17小时以及25℃温育3天的根据实施例13.3产生并且溶解于200μl 0.1M磷酸钠缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)中的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufkirchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去剩余的AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图7中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例19.7巯基促红细胞生成素与实施例15.1的反应产物和交联化合物的反应
向根据实施例15.1产生并且溶解于200μl磷酸钠缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufkirchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图8中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例19.8巯基促红细胞生成素与实施例15.2的反应产物和交联化合物的反应
向根据实施例15.2产生并且溶解于200μl磷酸盐缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufkirchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图8中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例19.9巯基促红细胞生成素与实施例15.3的反应产物和交联化合物的反应
向根据实施例15.3产生并且溶解于200μl磷酸盐缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufklrchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图8中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用的HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例19.10巯基促红细胞生成素与实施例15.4的反应产物和交联化合物的反应
向根据实施例15.4产生并且溶解于200μl磷酸盐缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)中的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufkirchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图8中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例19.11巯基促红细胞生成素与实施例15.5的反应产物和交联化合物的反应
向根据实施例15.5产生并且溶解于200μl磷酸盐缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)中的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufkirchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图8中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例19.12巯基促红细胞生成素与实施例15.6的反应产物和交联化合物的反应
向根据实施例15.6产生并且溶解于200μl磷酸盐缓冲液(0.1M,9.15M NaCl,50mM EDTA,pH7.2)中的50nmol HES衍生物中加入10μl 2.5μmol AMAS(Sigma Aldrich,Taufkirchen,D)在DMSO中的溶液。将澄清溶液在25℃下温育80分钟,在40℃下温育20分钟。使用VIVASPIN 0.5ml浓缩器,5KD MWCO(VIVASCIENCE,Hannover,德国),以13,000rpm离心过滤,并用磷酸盐缓冲液洗涤4次各30分钟,除去AMAS。
向剩余溶液中加入15μg根据实施例18产生的巯基EPO(1μg/μl,在磷酸盐缓冲液中),混合物在25℃下温育16小时。冻干后,使用NuPAGE 10%Bis-Tris Gels/MOPS缓冲液(Invitrogen,Carlsbad,CA,USA),如Invitrogen说明书所述,通过SDS-PAGE分析粗制品。凝胶用Roti-考马斯蓝染色试剂(Roth,Karlsruhe,D)染色过夜。
实验结果在图8中显示。蛋白带向较高分子量的迁移表明偶联成功。带宽增加是由于所用HES衍生物的分子量分布和与该蛋白质连接的HES衍生物的数量。
实施例20HES-EPO偶联物的制备性生产
概述
HES-EPO偶联物如下合成:将HES衍生物(平均分子量为18,000道尔顿;羟乙基取代程度为0.4)与重组人EPO的寡糖链上部分(温和的高碘酸盐)氧化的唾液酸残基偶联。根据碳水化合物结构分析,引入的修饰不影响核心寡糖链的结构完整性,因为经弱酸处理的HES-修饰聚糖的MALDI/TOF-MS显示完整的中性N-乙酰基乳糖胺型链,它与未修饰EPO产物中所见的链没有差别。获得的结果表明,对于预先没有部分去除唾液酸而进行修饰的EPO制品,每个EPO分子至少连接3个修饰的HES残基。缺少前述蛋白质中约50%唾液酸残基的EPO变体在SDS-PAGE中显示类似的高表观分子量迁移率(60-110 KDa比BRPEPO标准的40KDa)。在室温和pH 3-10下的标准离子交换层析条件下,HES修饰的EPO稳定。
在红细胞正常的小鼠系中进行EPO生物检测,根据欧洲药典用紫外线吸收值测定蛋白质,RP-HPLC测定EPO蛋白质并经BRP EPO标准制品校准,在该检测中,HES修饰的EPO具有比国际BRP EPO参照标准高2.5-3.0倍的比活性(IU/mg)。
实施例20.1材料与方法
(a)通过N-糖苷酶消化释放N-连接的寡糖
样品与25单位(根据德国Roche Diagnostics的厂商说明书)重组PNGase F在37℃下温育过夜。根据蛋白质在SDS-PAGE中的比迁移率位移来监测完全消化。通过加入3倍体积的冷100%乙醇,并在-20℃下放置至少2小时,从多肽中分离释放的N-聚糖(Schroeter S等人,1999)。在4℃下以13000rpm离心10分钟除去沉淀的蛋白质。然后用500μl冰冷的75%乙醇再洗涤沉淀两次。合并上清液中的寡糖在真空离心机(Speed Vac浓缩器,Savant Instruments Inc.,USA)中干燥。聚糖样品在使用前用Hypercarb柱(25mg或100mg HyperCarb)如下脱盐:柱用500μl溶于0.1%TFA中的80%乙腈(v/v)洗涤3次,随后用500μl水洗涤3次。样品用水稀释至终体积300μl-600μl,之后上柱,然后用水充分洗涤柱子。寡糖用1.2ml(25mg柱;对于100mg柱为1.8ml)含有0.1%三氟乙酸(v/v)的25%乙腈水溶液洗脱。洗脱的寡糖用2M NH4OH中和,在Speed Vac浓缩器中干燥。在某些情况下,通过将总(糖)蛋白样品<100μg的消化混合物吸附到100mgHypercarb柱上,对N-糖苷酶释放的寡糖进行脱盐。
(b)通过基质辅助激光解吸/电离飞行时间质谱法(MALDI/TOF/TOF-MS)对寡糖的分析
使用Bruker ULTRAFLEX飞行时间(TOF/TOF)仪器:在阳离子及阴离子模式中使用2,5-二羟基苯甲酸作为紫外线吸收材料,在两种模式中均使用反射器(reflectron),分析天然去唾液酸化的寡糖。对于MS-MS分析,选择母离子进行激光诱导的解离(LID),获得的碎片离子用仪器的第二TOF阶段(LIFT)分离。浓度约为1-10pmol·μl-1的1μl样品溶液与等量的各自的基质混合。将该混合物点样到不锈钢靶上,分析前在室温下干燥。
实施例20.2重组人EPO(EPO-GT-1)的制备和鉴定
如(Mueller PP等人,1999,Dorner AJ等人,1984)所述由重组CHO细胞表达EPO,根据欧洲药典所述的方法鉴定这些制品(Ph.Eur.4,Monography 01/2002:1316:Erythropoietin concentratedsolution)。终产物的唾液酸含量为每nMol蛋白质12nMol(+/-1.5nMol)。N-连接寡糖的结构如(Nimtz等人,1999,Grabenhorst,1999)所述通过HPAEC-PAD和MALDI/TOF-MS测定。获得的EPO制品含有二、三、四唾液酸化的寡糖(分别为2-12%、15-28%和60-80%,少量存在硫酸化和五唾液酸化链)。EPO制品的总糖基化特征类似于国际BRPEPO标准制品。
重组EPO的等电聚焦模型与国际BRP参照EPO标准制品相当,表明是相应的同种型。25%的EPO蛋白在多肽链的Ser126处缺乏O-糖基化。
实施例20.3部分去唾液酸化的EPO形式的制备
EPO GT-1蛋白(2.84mg/ml)在20mM磷酸钠缓冲液pH7.0中加热到80℃,然后每1ml EPO溶液加入100μl 1N H2SO4;分别继续温育5分钟、10分钟、60分钟,产生不同唾液酸化程度的EPO制品。用多肽N-糖苷酶释放寡糖后对含有0-4个唾液酸的寡糖进行定量,通过用Hypercarb柱(25mg HyperSep Hypercarb;Thermo-Hypersil-Keystone,UK)脱盐进行N-连接链的分离。加入1N NaOH中和EPO制品,在液氮中冷冻,贮存于-20℃,直到下一步使用。
实施例20.4唾液酸化EPO形式的高碘酸盐氧化
向溶解于3.5ml 20mM磷酸钠缓冲液pH7.0中的10mg未处理或弱酸处理的EPO中加入1.5ml 0.1M乙酸钠缓冲液pH5.5,将混合物在冰浴中冷却到0℃;加入500μl10mM高碘酸钠,反应混合物在0℃下避光保持60分钟。加入10μl甘油,继续避光温育10分钟。使用VIVASPIN浓缩器(10,000 MWCO,PES Vivasciernce AG,Hannover,德国),根据厂商推荐,在配备固定角转子的实验室离心机中以3000rpm脱盐,从试剂中分离部分氧化的EPO形式。在液氮中冷冻后,EPO制品以4ml的终体积贮存于-20℃。
对100μg等份的部分氧化EPO制品进行N-糖苷酶处理,寡糖如上所述用Hypercarb柱分离。寡糖通过弱酸处理去唾液酸化,用HPAEC-PAD分析,它们的保留时间与如(Nimtz等人,1990和1993)所述的可信标准寡糖相当。
实施例20.5用二硫赤藓糖醇还原EPO二硫醚
在30mM二硫赤藓糖醇(DTT)存在下,将5mg EPO-GT-1在5ml 0.1M Tris/HCl缓冲液pH 8.1中,37℃温育60分钟;使用Vivaspin浓缩器在4℃下除去DTT,更换缓冲液4个循环。最终的还原EPO制品冷冻于液氮中,在50mM乙酸钠缓冲液pH5.5中贮存于-20℃。
实施例20.6 EPO蛋白测定
EPO蛋白的定量测定如下进行:根据欧洲药典(欧洲药典4,专题01/2002:1316:促红细胞生成素浓缩溶液),在1cm径长的比色杯中测定280nm处的紫外线吸光度。另外,也可以使用RP-C4柱(VydacProtein C4,目录号214TP5410,Grace Vydac,Ca,US),通过RP-HPLC方法对EPO定量;HPLC方法用促红细胞生成素BRP1参照标准校准(欧洲药典,Conseil de l′Europe B.P.907-F67029,Strasbourg Cedex1)。
实施例20.7用半乳糖氧化酶氧化去唾液酸化的EPO
在16μl过氧化氢酶(6214单位/200ml)和80μl半乳糖氧化酶(2250单位/ml,来自Dactylium dendroides(Sigma-Aldrich,Steinheim,德国)存在下,将4.485mg完全去唾液酸化的EPO在20mM磷酸钠缓冲液pH6.8中,37℃温育过夜;在温育开始4小时和8小时后分两次加入20μl半乳糖氧化酶。
实施例20.8用于生物检测的EPO样品的制备
从高碘酸盐或半乳糖氧化酶氧化的EPO蛋白制品与活化的HES温育反应中纯化EPO
EPO样品的纯化(除去未反应的HES衍生物)在室温下进行。EPO温育混合物(约5mg EPO蛋白)用缓冲液A(20mM N-吗啉丙磺酸[MOPS/NaOH],在双蒸水中,pH8.0)1∶10稀释,以0.5ml/min的流速上样到用10倍柱体积(CV)缓冲液A平衡的含有3ml Q-SepharoseHP(Pharmacia编号17-1014-03,批号220211)的柱上。用6-8倍柱体积的缓冲液A洗柱(流速=0.8ml/min),使用缓冲液B(20mM吗啉乙磺酸[MES/NaOH,0.5M NaCl,在双蒸水中,pH6.5)以0.5ml/min的流速进行洗脱。根据280nm处的紫外线吸光度检测EPO,洗脱到约6ml中。柱用3倍柱体积的缓冲液C(20mM MES,1.5M NaCl,在H2O中,调节为pH6.5)再生,用10倍柱体积的缓冲液A再平衡(流速=0.7ml/min)。
用Vivaspin浓缩器和磷酸盐缓冲盐水(PBS)对Q-Sepharose步骤获得的EPO洗脱物进行缓冲液更换,每个样品进行3个离心循环;样品用PBS调节为2ml,贮存于-20℃。
因为在所采用条件下基础EPO型不结合Q-Sepharose并发现与未反应的HES衍生物一起出现在穿流液中,因此从Q-Sepharose洗脱物中只获得<25%的部分唾液酸化、随后温和高碘酸盐氧化的经HES修饰的EPO型。
实施例20.9脉冲安培检测的高pH阴离子交换层析(HPAEC-PAD)
使用配有CarboPac PA1柱(0.4×25cm)的Dionex BioLC系统(Dionex,USA),通过高pH阴离子交换(HPAE)层析以及脉冲安培检测器(PAD),分析纯化的天然及去唾液酸化的寡糖(Schroter等人,1999;Nimtz等人,1999)。检测器电势(E)和脉冲持续时间(T)为:E1:+50mV,T1:480ms;E2:+500mV,T2:120ms;E3:-500mV,T3:60ms,输出范围为500-1500nA。然后将寡糖注射到用100%溶剂A平衡的CarboPac PAl柱上。对于去唾液酸化的寡糖,在40分钟内使用溶剂B的线性梯度(0-20%),随后在5分钟内将溶剂B从20%线性提高到100%,进行洗脱(流速:1ml·min-1)。溶剂A是溶于双蒸水的0.2MNaOH,溶剂B由溶于溶剂A的0.6M NaOAc组成。对于天然寡糖,柱子用100%溶剂C(0.1M NaOH,溶于双蒸水)平衡,在48分钟内使用线性梯度(0-35%)溶剂D,随后在10分钟内将溶剂D从35%线性提高到100%,进行洗脱(流速:1ml·min-1)。溶剂D由溶于溶剂C的0.6M NaAc组成。
实施例20.10通过GC-MS进行N-聚糖、HES-修饰的N-聚糖和EPO蛋白的单糖组成分析
在甲醇分解、N-再乙酰化和三甲基硅烷化后,通过GC/MS分析相应的甲基糖苷[Chaplin,M.F.(1982)一种快速、灵敏的碳水化合物分析方法.Anal.Biochem.123,336-341]来分析单糖。这些分析用Finnigan GCQ离子阱质谱仪(Finnigan MAT corp.,San Jose,CA)进行,其以配备一个30m DB5毛细管柱的阳离子EI模式运行。温度程序:80℃恒温2min,然后每分钟升高10度,至300℃。
根据保留时间和特有的断裂模式鉴定单糖。未校正的电子峰积分的结果用于定量。由于正位异构化和/或存在呋喃型和吡喃型而产生一个以上峰的单糖,通过加和所有主峰来定量。0.5μg肌醇用作内部标准化合物。
实施例20.11结果
实施例20.11(a)弱酸处理的(部分去唾液酸化的)EPO-GT-1的N-聚糖的鉴定
如图9所示,在与N-糖苷酶温育释放N-连接寡糖之前及之后,通过SDS-PAGE分析经弱酸处理5、10或60分钟的EPO-GT-1制品。对N-连接寡糖进行HPAEC-PAD寡糖作图(图10)。未处理的EPO-GT-1含有>90%的具有3或4个唾液酸残基的N-连接寡糖,而在弱酸存在下温育5分钟后,<40%的碳水化合物链含有3或4个唾液酸残基。去唾液酸化N-聚糖的HPAEC-PAD显示,检测的未处理EPO-GT-1的中性寡糖比例在酸处理5、10或60分钟的制品中保持稳定。去唾液酸化聚糖的MALDI/TOF-MS显示,在弱酸处理蛋白质后存在<90%的近端岩藻糖。
实施例20.11(b)高碘酸盐处理的EPO-GT-1的鉴定
对于预先酸处理5分钟和10分钟或者不予处理的温和高碘酸盐处理的EPO型,在图12中比较它们的SDS-PAGE迁移率。用于高碘酸盐氧化唾液酸的条件不改变EPO制品的SDS-PAGE图(比较图9)。唾液酸的氧化在HPAEC-PAD分析中导致寡糖向较早洗脱时间迁移(比较图10和13)。
实施例20.11(c)HES-修饰的EPO衍生物的鉴定
(aa)用根据实施例14.4产生的羟胺修饰的HES衍生物X对EPO-GT-1-A进行HES修饰的时程(time course)
将400μg羟胺修饰的HES衍生物X加入溶于20μl 0.5M NaOAc缓冲液pH5.5中的20μg EPO-GT-1-A(温和高碘酸盐氧化的EPO,在温和高碘酸盐氧化之前未经酸水解)中,分别在30分钟、2、4、17小时后,通过将样品冷冻于液氮中,终止反应。随后将样品贮存于-20℃,直到进一步分析。
加入SDS-PAGE样品缓冲液,将样品加热到90℃,加样到SDS-凝胶上。如图14所示,温育时间延长导致向较高分子量蛋白质的迁移增加。在羟胺修饰的HES衍生物X存在下温育17小时后,检测到一条弥散的考马斯蓝染色的蛋白带,根据分子量标准的位置,它在60-11KDa之间的区域迁移(见图14的左部分)。用N-糖苷酶处理后,大多数蛋白质向去-N-糖基化EPO的位置迁移(见图14,右侧凝胶;箭头A表示N-糖苷酶的迁移位置;箭头B表示去-N-糖基化EPO的位置;推定在28KDa与36KDa分子量标准之间区域内所见的弥散蛋白带代表HES在分子的O-糖基化位点修饰的EPO型。关于N-糖苷酶的特异性,我们由该结果得出结论,实际上HES-修饰发生在EPO蛋白的聚糖之高碘酸盐氧化的唾液酸残基处。
(bb)HES-EPO偶联物的鉴定
HES-EPO偶联物I(来源于温和高碘酸盐氧化后的EPO-GT-1,即来源于EPO-GT-1-A)、II(来源于酸水解5分钟且温和高碘酸盐氧化的EPO-GT-1)、III(来源于酸水解10分钟且温和高碘酸盐氧化的EPO-GT-1)如前所述合成。包括在相同缓冲液条件下的对照温育(K),其中包括未修饰的EPO-GT-1并加入等量的未修饰HES。为了随后进行的HES-EPO衍生物的生物化学分析,进一步纯化这些温育混合物。
HES-EPO偶联物I、II和III以及对照温育K的温育物如“材料与方法”(实施例20.8)所述进行Q-Sepharose纯化步骤,以除去过量未反应HES试剂,后者预计出现在离子交换柱的穿流液中。由于在预先酸处理的样品II和III中含有大量的基础EPO型,我们预计在穿流液中会含有来自这些温育物的相当量的修饰的EPO产物。如图15所示,几乎所有来源于样品I的EPO都被Q-Sepharose柱保留,而样品III和II中只有约20-30%被回收到用高盐浓度洗脱的级分中。在SDS-PAGE中,与对照EPO相比,在穿流液和用高盐洗脱的级分中,与HES衍生物X温育产生的所有蛋白质物质都具有更高的表观分子量。
为了更详细地鉴定HES-修饰的EPO样品A和K(见图13),我们与高碘酸盐氧化形式EPO-GT-1-A进行比较。样品进行N-糖苷酶处理,如图16a和16b所示,N-聚糖的释放在标准EPO制品的O-糖基化和非糖基化EPO型位置处产生两条低分子量带。对于样品A,检测到另一条在28KDa分子量标准位置处迁移的带,提示在该EPO变体的O-聚糖处发生HES-修饰(参见实施例20.11(c)(aa))。在温和水解样品后,这条带(以及高度HES-修饰的高分子量形式的N-糖基化EPO,见图16a和16b)消失,这与在促红细胞生成素的高碘酸盐氧化的唾液酸残基处发生HES修饰的看法一致。
使用能够从寡糖上完全除去唾液酸残基(以及唾液酸连接的HES衍生物)的条件,水解N-糖苷酶温育的混合物等份;中和后,将混合物吸附到小Hypercarb柱上进行脱盐。用水充分洗涤该柱,随后用含有0.1%三氟乙酸的40%乙腈水溶液洗脱结合的中性寡糖。对产生的寡糖进行MALDI/TOF-MS。来自样品A、EPO-GT-1-A和样品K的去唾液酸化寡糖级分的波谱显示复杂型寡糖的相同质量数:m/z=1810Da(二分支)、2175=三分支、2540=四分支、2906=四分支+1个N-乙酰基乳糖胺重复、3271=四分支+2个N-乙酰基乳糖胺重复;检测到对应于缺乏岩藻糖(-146)和半乳糖(-162)的弱信号,这可归因于去除唾液酸所使用的酸水解条件(见MALDI-图19、20、21)。
在一个平行实验中,将N-糖苷酶消化混合物吸附到1ml RP-C18柱上(寡糖预先没有用酸水解),用含有0.1%TFA的5%乙腈水溶液进行洗脱;在这些条件下,EPO蛋白完全保留在RP-材料上,用含有0.1%TFA的5%乙腈水溶液从柱上洗下寡糖。用含有0.1%TFA的70%乙腈水溶液洗脱去-N-糖基化的EPO蛋白。中和N-糖苷酶处理的样品A、EPOGT-1-A和样品K经过RP-C18步骤后获得的寡糖级分,如前所述用Hypercarb柱脱盐。在能够从聚糖上定量除去唾液酸的条件下进行弱酸处理之前(见图17)及之后(见图18),对分离的寡糖进行HPAEC-PAD作图。
从HES修饰的样品A中所得天然物质的HPAEC-PAD图只显示可以忽略的寡糖信号,而EPOGT-1-A-衍生的寡糖显示与图13所示样品(称为温和高碘酸盐处理后的EPO-GT-1样品)相同的聚糖HPAEC-PAD图。由对照EPO样品(K)获得的寡糖洗脱图产生预期的图案(图10中的比较图)。为了比较,包括了用于比较和参照标准的国际BRP-EPO标准的天然寡糖谱。
在弱酸水解后,所有寡糖制品都显示相同的中性寡糖结构的洗脱图(见图18),该结构具有二、三、四分支复杂型碳水化合物链的定性和定量组成,如在作为本研究初始材料的EPO制品的方法部分所述。该结果证实,EPO样品的HES-修饰导致HES衍生物的共价连接,该衍生物可以用N-糖苷酶从EPO-蛋白质上分离下来,使用碳水化合物去唾液酸化的弱酸处理条件可以将其从N-聚糖上去除(见图16a+b),因此该衍生物是酸不稳定的。
(cc)通过GC-MS对HES-EPO和HES-EPO N-聚糖的单糖组成分析
为了进一步证实EPO在分子N-聚糖处的HES-修饰,用N-糖苷酶消化EPO样品,EPO蛋白被吸附到RP-C18柱上,而寡糖物质如上所述洗掉。如表3所示,只在半胱氨酸残基处进行了HES修饰的EPO蛋白中和EPO样品A2之寡糖级分中检测到了葡萄糖和羟乙基化葡萄糖衍生物。
实施例20.11(d)HES-修饰的EPO的体内生物活性检测
按照欧洲药典所述方法在红细胞正常的小鼠系中进行EPO生物检测;进行EPO检测的实验室使用国际BRP EPO参照标准制品。对于HES-修饰的EPO A2制品,测得比活性平均值为每mg EPO蛋白294600单位,表明与同样进行活性测定的国际BRP EPO参照标准制品相比,比活性约高3倍。
研究结果总结于表4中。
实施例13-20的参考文献:
Nimtz M,Noll G,Paques EP,Conradt HS.
Carbohydrate structures of a human tissue plasminogenactivator expressed in recombinant Chinese hams ter ovary cells.
FEBS Lett.1990 Oct.1;271(1-2):14-8
Dorner AJ,Wasley LC,Kaufman RJ.
Increased synthesis of secreted proteins inducesexpression of glucose-regulated proteins in butyrate-treatedChinese hamster ovary cells.
J Biol Chem.1989 Dec 5;264(34):20602-7
Mueller PP,Schlenke P,Nimtz M,Conradt HS,Hauser H
Recombinant glycoprotein quality inproliferation-controlled BHK-21cells.
Biotechnol Bioeng.1999 Dec 5;65(5):529-36
Nimtz M,Martin W,Wray V,Kloppel KD,Augustin J,ConradtHS.
Structures of sialylated oligosaccharides of humanerythropoietine xpressed in recobminant BHK-21 cells.
Eur J Biochem.1993 Apr.1;213(1):39-56
Hermentin P,Witzel R,Vliegenthart JF,Kamerling JP,NimtzM.Conradt HS.
A strategy for the mapping of N-glycans by high-phanion-exchange chromatography with pulsed amperometricdetection.
Anal Biochem.1992 Jun;203 (2):281-9
Schroter S,Derr P,Conradt HS,NimtzM,Hale G,KirchhoffC.
Male specific modification of human CD52.
J Biol Chem.1999 Oct.15;274(42):29862-73
表1
| 接头类型 | 官能团1:与多肽、特别是EPO反应 | 官能团2:与HES反应 |
| A | 酰肼(醛反应性) | 马来酰亚胺(SH反应性) |
| B | 酰肼(醛反应性) | 吡啶基二硫(SH反应性) |
| C | 碘烷基(SH反应性) | N-琥珀酰亚胺酯(胺反应性) |
| D | 溴烷基(SH反应性) | N-琥珀酰亚胺酯(胺反应性) |
| E | 马来酰亚胺(SH反应性) | N-琥珀酰亚胺酯(胺反应性) |
| F | 吡啶基二硫(SH反应性) | N-琥珀酰亚胺酯(胺反应性) |
| G | 乙烯基砜(SH反应性) | N-琥珀酰亚胺酯(胺反应性) |
表2


表3
来自HES修饰的EPO和对照样品之聚糖的单糖组成分析
| **单糖 | I.来自A2的聚糖 | II.来自EPO-GT-1A的聚糖 | III.来自K2的聚糖 | III.来自A2的聚糖 | IV.来自EPO-GT-1A的聚糖 | V.来自K2的聚糖 | VI.半胱氨酸修饰的EPO蛋白* |
| 岩藻糖 | 1935 | 3924 | 2602 | 2246 | 4461 | 2601 | 2181 |
| 甘露糖 | 6028 | 11020 | 9198 | 6379 | 11668 | 6117 | 6260 |
| 半乳糖 | 8886 | 19935 | 14427 | 10570 | 16911 | 11555 | 10386 |
| 葡萄糖 | 17968 | --- | --- | 21193 | 痕量 | 痕量 | 33021 |
| GlcNAc | 7839 | 21310 | 14440 | 11360 | 15953 | 10503 | 10498 |
| GlcHe1 | 5583 | --- | --- | 5926 | --- | --- | 14857 |
| GlcHe2 | 1380 | --- | --- | 1552 | --- | --- | 3775 |
| NeuNAc | 5461 | 822 | 4504 | 3895 | 4871 | 13562 | 13003 |
| 肌醇 | 1230 | 2310 | 620 | 2050 | 1320 | 1134 | 1087 |
| *对等量的Cys-HES-修饰的EPO蛋白进行组成分析;如上所述用Q-Sepharose柱通过层析从HES-温育混合物中分离EPO蛋白,利用Vivaspin 5分离装置离心脱盐。**通过全三甲基甲硅烷基化甲基糖苷的单次GC,进行单糖测定;给出了峰的电子积分值,这些数值未对衍化过程和每种化合物回收过程中的损失进行校正。 | |||||||
表4
| 样品号 | 样品描述 | EPO样品的计算比活性(根据A280nm和RP-HPLC测定) |
| 850247 | 1.HES-修饰的EPO A2 | 344000U/mg |
| 850248 | 2.EPO-GT-1-A | 82268U/mg |
| 850249 | 3.对照EPO K2 | 121410U/mg |
| 850250 | 4.BRP EPO标准 | 86702U/mg |
| 850251 | 1.用4倍体积的PBS稀释 | 309129U/mg |
| 850252 | 2.用4倍体积的PBS稀释 | 94500U/mg |
| 850253 | 3.用4倍体积的PBS稀释 | 114100U/mg |
| 850254 | 4.用4倍体积的PBS稀释 | 81200U/mg |
| 850255 | 1.用4倍体积的PBS稀释 | 230720U/mg |
Claims (85)
1.一种含有一个或多个HAS分子的羟烷基淀粉(HAS)-促红细胞生成素(EPO)偶联物(HAS-EPO),其中每个HAS都与EPO通过
a)碳水化合物部分;或
b)硫醚偶联。
2.权利要求1的HAS-EPO,其中EPO具有人EPO的氨基酸序列。
3.权利要求1或2的HAS-EPO,其中EPO含有通过N-和/或O-连接糖基化作用与EPO连接的一个或多个碳水化合物侧链。
4.权利要求3的HAS-EPO,其中在哺乳动物特别是人、昆虫或酵母细胞内的生产过程中,所述碳水化合物侧链已与EPO连接。
5.权利要求1-4中任一项的HAS-EPO,其中HAS通过接头分子与EPO偶联。
6.权利要求3-5中任一项的HAS-EPO,其中HAS通过碳水化合物部分与EPO偶联,该碳水化合物部分是碳水化合物侧链的一部分并优选是被氧化的。
7.权利要求6的HAS-EPO,其中HAS与碳水化合物侧链的半乳糖或唾液酸残基偶联。
8.权利要求1-7中任一项的HAS-EPO,其中硫醚中的S原子来源于天然存在的半胱氨酸或者添加的半胱氨酸。
9.权利要求8的HAS-EPO,其中EPO具有人EPO的氨基酸序列,天然存在的半胱氨酸是半胱氨酸29和/或33。
10.权利要求9的HAS-EPO,其中HAS与半胱氨酸29偶联,半胱氨酸33被置换为另一种氨基酸。
11.权利要求9的HAS-EPO,其中HAS与半胱氨酸33偶联,半胱氨酸29被置换为另一种氨基酸。
12.权利要求8-11中任一项的HAS-EPO,其中通过将天然存在的氨基酸置换为半胱氨酸来添加半胱氨酸。
13.权利要求12的HAS-EPO,其中EPO是人EPO,被置换的氨基酸残基是丝氨酸126。
14.权利要求1-13中任一项的HAS-EPO,其中每个EPO分子具有1-12个、优选、1-6个或1-3个、最优选1-4个HAS分子。
15.权利要求1-14中任一项的HAS-EPO,其中HAS选自羟乙基淀粉、羟丙基淀粉和羟丁基淀粉。
16.权利要求15的HAS-EPO,其中HAS是羟乙基淀粉(HES)。
17.权利要求16的HAS-EPO,其中HES的分子量为1-300kDa,优选5-100kDa。
18.权利要求16或17的HAS-EPO,其中HES显示相对于羟乙基的0.1-0.8的摩尔取代度和2-20的C2∶C6取代比。
19.一种生产羟烷基淀粉(HAS)-促红细胞生成素(EPO)偶联物(HAS-EPO)的方法,包括下列步骤:
a)提供能够与修饰的HAS反应的EPO,
b)提供能够与步骤a)中EPO反应的修饰的HAS,和
c)使步骤a)的EPO与步骤b)的HAS反应,从而产生含有一个或多个HAS分子的HAS-EPO,其中每个HAS都与EPO通过
i)碳水化合物部分;或
ii)硫醚偶联。
20.权利要求19的方法,其中EPO具有人EPO的氨基酸序列。
21.权利要求19或20的方法,其中EPO是重组产生的。
22.权利要求19-21中任一项的方法,其中EPO含有通过N-和/或O-连接糖基化作用与EPO连接的一个或多个碳水化合物侧链。
23.权利要求22的方法,其中在哺乳动物特别是人、昆虫或酵母细胞内的生产过程中,所述碳水化合物侧链已与EPO连接。
24.权利要求22或23的方法,其中HAS通过碳水化合物部分与EPO偶联,并且该碳水化合物部分是碳水化合物侧链的一部分。
25.权利要求24的方法,其中在步骤a)中,通过氧化EPO的一个或多个碳水化合物侧链的至少一个碳水化合物部分、优选至少一个末端糖单元、更优选半乳糖来修饰EPO。
26.权利要求25的方法,其中在部分或完全(酶和/或化学)除去末端唾液酸后氧化末端糖单元。
27.权利要求25或26的方法,其中在步骤c)中,修饰的HAS与氧化的末端糖单元偶联。
28.权利要求19-27中任一项的方法,其中EPO含有至少一个游离SH基。
29.权利要求28的方法,其中游离SH基是天然存在的半胱氨酸或添加的半胱氨酸的一部分。
30.权利要求29的方法,其中EPO具有人EPO的氨基酸序列,天然存在的半胱氨酸是半胱氨酸29和/或33。
31.权利要求30的方法,其中半胱氨酸33被置换为另一种氨基酸,在步骤c)中修饰的HAS与半胱氨酸29偶联。
32.权利要求30的方法,其中半胱氨酸29被置换为另一种氨基酸,在步骤c)中修饰的HAS与半胱氨酸33偶联。
33.权利要求29-32中任一项的方法,其中通过将天然存在的氨基酸置换为半胱氨酸来添加半胱氨酸。
34.权利要求33的方法,其中EPO是人EPO,被置换的氨基酸残基是丝氨酸126。
35.权利要求33或34的方法,其中在步骤c)中,修饰的HAS与添加的半胱氨酸偶联。
36.权利要求19-35中任一项的方法,其中修饰HAS,使得如果HAS与氧化的碳水化合物部分偶联,则其含有游离的酰肼、羟胺、硫醇或氨基脲官能团,或者如果HAS与SH基偶联,则其含有游离的马来酰亚胺、二硫醚或卤代乙酰胺官能团。
37.权利要求19-36中任一项的方法,其中步骤c)在至少含有10%重量H2O的反应介质中进行。
38.权利要求19-37中任一项的方法,其中HAS通过接头分子与EPO偶联。
39.权利要求19-38中任一项的方法,其中HAS是羟乙基淀粉、羟丙基淀粉或羟丁基淀粉,优选羟乙基淀粉(HES)。
40.权利要求39的方法,其中HES具有如权利要求17或18中任一项所限定的性质。
41.通过权利要求19-40中任一项的方法获得的HAS-EPO。
42.权利要求41的HAS-EPO,其具有权利要求1-18中任一项所限定的特征。
43.用于人或动物体治疗方法的根据权利要求1-18、41或42中任一项的HAS-EPO在。
44.一种药物组合物,其含有根据权利要求1-18、41或42中任一项的HAS-EPO。
45.权利要求44的药物组合物,进一步含有至少一种药学可接受的载体。
46.根据权利要求1-18、41或42中任一项的HAS-EPO在制备治疗贫血性疾病或造血功能障碍性疾病的药物中的应用。
47.含有一个或多个HAS分子的羟烷基淀粉(HAS)-多肽偶联物(HAS-多肽),其中每个HAS都与多肽通过
a)碳水化合物部分;或
b)硫醚偶联。
48.权利要求47的HAS-多肽,其中多肽是人源性的。
49.权利要求47或48的HAS-多肽,其中多肽选自促红细胞生成素、白介素特别是白介素-2、IFN-β、IFN-α、CSF、白介素6和治疗性抗体。
50.权利要求47-49中任一项的HAS-多肽,其中多肽含有通过N-和/或O-连接糖基化作用与多肽连接的一个或多个碳水化合物侧链。
51.权利要求50的HAS-多肽,其中在哺乳动物特别是人、昆虫或酵母细胞内的生产过程中,所述碳水化合物侧链已与多肽连接。
52.权利要求47-51中任一项的HAS-多肽,其中HAS通过接头分子与多肽偶联。
53.权利要求49-52中任一项的HAS-多肽,其中HAS通过碳水化合物部分与多肽偶联,该碳水化合物部分是碳水化合物侧链的一部分,并优选是被氧化的。
54.权利要求53的HAS-多肽,其中HAS与碳水化合物侧链的半乳糖残基偶联。
55.权利要求47-54中任一项的HAS-多肽,其中硫醚中的S原子来源于天然存在的半胱氨酸或者添加的半胱氨酸。
56.权利要求55的HAS-多肽,其中通过将天然存在的氨基酸置换为半胱氨酸来添加半胱氨酸。
57.权利要求47-56中任一项的HAS-多肽,其中每个多肽分子具有1-12个、优选1-6或1-3个、最优选1-4个HAS分子。
58.权利要求47-57中任一项的HAS-多肽,其中HAS选自羟乙基淀粉、羟丙基淀粉和羟丁基淀粉。
59.权利要求58的HAS-多肽,其中HAS是羟乙基淀粉(HES)。
60.权利要求59的HAS-多肽,其中HES的分子量为1-300kDa,优选5-100kDa。
61.权利要求59或60中任一项的HAS-多肽,其中HES显示相对于羟乙基的0.1-0.8的摩尔取代程度和2-20的C2∶C6取代比。
62.一种生产羟烷基淀粉(HAS)-多肽偶联物(HAS-多肽)的方法,包括下列步骤:
a)提供能够与修饰的HAS反应的多肽,
b)提供能够与步骤a)中多肽反应的修饰的HAS,和
c)使步骤a)的多肽与步骤b)的HAS反应,从而产生含有一个或多个HAS分子的HAS-多肽,其中每个HAS都与多肽通过
i)碳水化合物部分;或
ii)硫醚偶联。
63.权利要求62的方法,其中多肽是人源性的。
64.权利要求62或63的方法,其中多肽选自促红细胞生成素、白介素特别是白介素-2、IFN-β、IFN-α、CSF、白介素6和治疗性抗体。
65.权利要求62-64中任一项的方法,其中多肽是重组产生的。
66.权利要求62-65中任一项的方法,其中多肽含有通过N-和/或O-连接糖基化作用与多肽连接的一个或多个碳水化合物侧链。
67.权利要求66的方法,其中在哺乳动物特别是人、昆虫或酵母细胞内的生产过程中所述碳水化合物侧链已与多肽连接。
68.权利要求66或67的方法,其中HAS通过碳水化合物部分与多肽偶联,该碳水化合物部分是碳水化合物侧链的一部分。
69.权利要求68的方法,其中在步骤a)中,通过氧化多肽的一个或多个碳水化合物侧链的至少一个碳水化合物部分、优选至少一个末端糖单元、更优选半乳糖来修饰该多肽。
70.权利要求69的方法,其中在部分或完全(酶和/或化学)除去末端唾液酸后氧化末端糖单元。
71.权利要求69或70的方法,其中在步骤c)中,修饰的HAS与氧化的末端糖单元偶联。
72.权利要求62-71中任一项的方法,其中多肽含有至少一个游离SH基。
73.权利要求72的方法,其中游离SH基是天然存在的半胱氨酸或添加的半胱氨酸的一部分。
74.权利要求62-73中任一项的方法,其中通过将天然存在的氨基酸置换为半胱氨酸来添加半胱氨酸。
75.权利要求73或74中任一项的方法,其中在步骤c)中,修饰的HAS与添加的半胱氨酸偶联。
76.权利要求62-75中任一项的方法,其中修饰HAS,使得如果HAS与氧化的碳水化合物部分偶联则含有游离的酰肼、羟胺、硫醇或氨基脲官能团,或者如果HAS与SH基偶联则含有游离的马来酰亚胺、二硫醚或卤代乙酰胺官能团。
77.权利要求62-76中任一项的方法,其中步骤c)在至少含有10%重量H2O的反应介质中进行。
78.权利要求62-78中任一项的方法,其中HAS通过接头分子与多肽偶联。
79.权利要求62-78中任一项的方法,其中HAS是羟乙基淀粉、羟丙基淀粉或羟丁基淀粉,优选羟乙基淀粉(HES)。
80.权利要求79的方法,其中HES具有如权利要求60或61中任一项所限定的性质。
81.可通过权利要求62-80中任一项的方法获得的HAS-多肽。
82.权利要求41的HAS-多肽,其具有权利要求47-61中任一项所限定的特征。
83.在人或动物体治疗方法中使用的根据权利要求47-61、81或82中任一项的HAS-多肽。
84.一种药物组合物,其含有根据权利要求47-61、81或82中任一项的HAS-多肽。
85.权利要求84的药物组合物,进一步含有至少一种药学可接受的载体。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US40978102P | 2002-09-11 | 2002-09-11 | |
| US60/409,781 | 2002-09-11 | ||
| EP02020425.1 | 2002-09-11 | ||
| EP02020425A EP1400533A1 (en) | 2002-09-11 | 2002-09-11 | HASylated polypeptides, especially HASylated erythropoietin |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN200910133235.7A Division CN101580547B (zh) | 2002-09-11 | 2003-08-08 | Has化的多肽,特别是has化的促红细胞生成素 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1681844A true CN1681844A (zh) | 2005-10-12 |
Family
ID=31995520
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA038214644A Pending CN1681844A (zh) | 2002-09-11 | 2003-08-08 | Has化的多肽,特别是has化的促红细胞生成素 |
| CNB038214652A Expired - Fee Related CN100348618C (zh) | 2002-09-11 | 2003-08-08 | 生产羟烷基淀粉衍生物的方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB038214652A Expired - Fee Related CN100348618C (zh) | 2002-09-11 | 2003-08-08 | 生产羟烷基淀粉衍生物的方法 |
Country Status (25)
| Country | Link |
|---|---|
| US (5) | US7815893B2 (zh) |
| EP (8) | EP1398327B1 (zh) |
| JP (4) | JP2006516534A (zh) |
| KR (3) | KR101174510B1 (zh) |
| CN (2) | CN1681844A (zh) |
| AR (3) | AR041097A1 (zh) |
| AT (3) | ATE323723T1 (zh) |
| AU (6) | AU2003260393B2 (zh) |
| BR (3) | BR0314107A (zh) |
| CA (2) | CA2495242A1 (zh) |
| CY (1) | CY1109813T1 (zh) |
| DE (7) | DE60323192D1 (zh) |
| DK (3) | DK1398327T3 (zh) |
| ES (4) | ES2213507T3 (zh) |
| HK (4) | HK1136226A1 (zh) |
| IL (5) | IL166506A0 (zh) |
| MX (4) | MXPA05002594A (zh) |
| NO (2) | NO20051419L (zh) |
| PL (3) | PL216545B1 (zh) |
| PT (3) | PT1398327E (zh) |
| RU (3) | RU2328505C2 (zh) |
| SI (2) | SI1398327T1 (zh) |
| TW (1) | TW200418875A (zh) |
| WO (3) | WO2004024777A1 (zh) |
| ZA (1) | ZA200600651B (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108219019A (zh) * | 2018-02-08 | 2018-06-29 | 华中科技大学 | 一种巯基化羟乙基淀粉及其修饰的纳米材料和制备方法 |
| CN108403641A (zh) * | 2018-02-08 | 2018-08-17 | 华中科技大学 | 一种载药纳米材料及其制备方法 |
| CN108503718A (zh) * | 2018-02-08 | 2018-09-07 | 华中科技大学 | 一种羟烷基淀粉偶联物及其制备方法和应用 |
| CN111157736A (zh) * | 2018-11-08 | 2020-05-15 | 中国科学院大连化学物理研究所 | 一种基于化学酶促的人血清o糖基化鉴定方法 |
Families Citing this family (152)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10112825A1 (de) * | 2001-03-16 | 2002-10-02 | Fresenius Kabi De Gmbh | HESylierung von Wirkstoffen in wässriger Lösung |
| DE10209822A1 (de) | 2002-03-06 | 2003-09-25 | Biotechnologie Ges Mittelhesse | Kopplung niedermolekularer Substanzen an ein modifiziertes Polysaccharid |
| DE10209821A1 (de) | 2002-03-06 | 2003-09-25 | Biotechnologie Ges Mittelhesse | Kopplung von Proteinen an ein modifiziertes Polysaccharid |
| BR0314107A (pt) | 2002-09-11 | 2005-07-19 | Fresenius Kabi De Gmbh | Método de produção de derivados de hidroxialquil amido |
| DE10242076A1 (de) * | 2002-09-11 | 2004-03-25 | Fresenius Kabi Deutschland Gmbh | HAS-Allergen-Konjugate |
| DE02020425T1 (de) * | 2002-09-11 | 2004-07-15 | Fresenius Kabi Deutschland Gmbh | Hasylierte Polypeptide, besonders hasyliertes Erythropoietin |
| DE60323756D1 (de) | 2002-10-08 | 2008-11-06 | Fresenius Kabi De Gmbh | Pharmazeutisch aktive oligosaccharid-conjugate |
| DE10256558A1 (de) * | 2002-12-04 | 2004-09-16 | Supramol Parenteral Colloids Gmbh | Ester von Polysaccharid Aldonsäuren, Verfahren zu ihrer Herstellung und Verwendung zur Kopplung an pharmazeutische Wirkstoffe |
| EP1653991A2 (en) * | 2003-08-08 | 2006-05-10 | Fresenius Kabi Deutschland GmbH | Conjugates of a polymer and a protein linked by an oxime linking group |
| SG145746A1 (en) * | 2003-08-08 | 2008-09-29 | Fresenius Kabi De Gmbh | Conjugates of hydroxyalkyl starch and g-csf |
| CA2534418A1 (en) * | 2003-08-08 | 2005-02-17 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyalkyl starch and g-csf |
| WO2005014655A2 (en) * | 2003-08-08 | 2005-02-17 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyalkyl starch and a protein |
| US8754031B2 (en) | 2004-03-08 | 2014-06-17 | Oncolix, Inc. | Use of prolactin receptor antagonists in combination with an agent that inactivates the HER2/neu signaling pathway |
| WO2005092928A1 (en) * | 2004-03-11 | 2005-10-06 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyalkyl starch and a protein, prepared by reductive amination |
| WO2005092391A2 (en) * | 2004-03-11 | 2005-10-06 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyalkyl starch and a protein |
| WO2005092369A2 (en) * | 2004-03-11 | 2005-10-06 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyethyl starch and erythropoietin |
| US7589063B2 (en) | 2004-12-14 | 2009-09-15 | Aplagen Gmbh | Molecules which promote hematopoiesis |
| WO2006094826A2 (en) * | 2005-03-11 | 2006-09-14 | Fresenius Kabi Deutschland Gmbh | Method for coupling enzymatically activated glycoconjugates to a hydroxyalkyl starch |
| AU2006222187A1 (en) * | 2005-03-11 | 2006-09-14 | Fresenius Kabi Deutschland Gmbh | Production of bioactive glycoproteins from inactive starting material by conjugation with hydroxyalkylstarch |
| EP1762250A1 (en) * | 2005-09-12 | 2007-03-14 | Fresenius Kabi Deutschland GmbH | Conjugates of hydroxyalkyl starch and an active substance, prepared by chemical ligation via thiazolidine |
| US8470963B2 (en) | 2006-06-30 | 2013-06-25 | Sku Asset Management Gmbh | Multifunctional compounds for pharmaceutical purposes |
| US8394921B2 (en) * | 2006-07-25 | 2013-03-12 | Lipoxen Technologies Limited | N-terminal derivatisation of proteins with polysaccharides |
| JP2010510794A (ja) | 2006-11-28 | 2010-04-08 | ハナル ファーマシューティカル カンパニー リミテッド | 修飾型エリスロポエチンポリペプチド及びこの治療用用途 |
| BRPI0720282B8 (pt) * | 2006-12-15 | 2021-05-25 | Baxalta GmbH | construção proteica, composição farmacêutica, e, kit |
| SI2457919T1 (sl) | 2007-01-18 | 2019-10-30 | Genzyme Corp | Oligosaharidi, ki vsebujejo skupino aminooksi in njihovi konjugati |
| EP2205280B1 (en) | 2007-09-27 | 2019-09-04 | Amgen Inc. | Pharmaceutical formulations |
| WO2009064838A1 (en) | 2007-11-15 | 2009-05-22 | Amgen, Inc. | Aqueous formulation of erythropoiesis stimulating protein stablised by antioxidants for parenteral administration |
| EP2070951A1 (en) * | 2007-12-14 | 2009-06-17 | Fresenius Kabi Deutschland GmbH | Method for producing a hydroxyalkyl starch derivatives with two linkers |
| EP2070950A1 (en) * | 2007-12-14 | 2009-06-17 | Fresenius Kabi Deutschland GmbH | Hydroxyalkyl starch derivatives and process for their preparation |
| CN104888263B (zh) | 2008-01-14 | 2018-11-30 | 北京环球利康科技有限公司 | 生物相容性止血、防粘连、促愈合、外科封闭的变性淀粉材料 |
| AU2009206306B2 (en) | 2008-01-25 | 2013-06-06 | Amgen Inc. | Ferroportin antibodies and methods of use |
| EP2310423A1 (en) * | 2008-04-28 | 2011-04-20 | SurModics, Inc. | Poly- (1 4)glucopyranose-based matrices with hydrazide crosslinking |
| EP2816059A1 (en) | 2008-05-01 | 2014-12-24 | Amgen, Inc | Anti-hepcidin antibodies and methods of use |
| EP2166085A1 (en) | 2008-07-16 | 2010-03-24 | Suomen Punainen Risti Veripalvelu | Divalent modified cells |
| US10138283B2 (en) | 2008-07-23 | 2018-11-27 | Ambrx, Inc. | Modified bovine G-CSF polypeptides and their uses |
| EP2350118B1 (en) * | 2008-09-19 | 2016-03-30 | Nektar Therapeutics | Carbohydrate-based drug delivery polymers and conjugates thereof |
| EP3693014A1 (en) | 2008-11-13 | 2020-08-12 | The General Hospital Corporation | Methods and compositions for regulating iron homeostasis by modulation bmp-6 |
| BRPI0920896B8 (pt) | 2008-12-06 | 2021-05-25 | Braun Melsungen Ag | composto de fórmula geral (i) (t-z)n-p (i), formulação farmacêutica e processo para preparar um composto de fórmula geral (i) |
| US8648046B2 (en) | 2009-02-26 | 2014-02-11 | Oncolix, Inc. | Compositions and methods for visualizing and eliminating cancer stem cells |
| US8754035B2 (en) | 2009-02-26 | 2014-06-17 | Oncolix, Inc. | Compositions and methods for visualizing and eliminating cancer stem cells |
| CN102365099A (zh) | 2009-03-31 | 2012-02-29 | B·布朗·梅尔松根有限公司 | 胺化多糖粘合制品 |
| ES2597954T3 (es) | 2009-07-27 | 2017-01-24 | Baxalta GmbH | Conjugados de proteína de la coagulación sanguínea |
| RU2744370C2 (ru) * | 2009-07-27 | 2021-03-05 | Баксалта Инкорпорейтед | Конъюгаты белков свертывания крови |
| US8642737B2 (en) | 2010-07-26 | 2014-02-04 | Baxter International Inc. | Nucleophilic catalysts for oxime linkage |
| US8809501B2 (en) | 2009-07-27 | 2014-08-19 | Baxter International Inc. | Nucleophilic catalysts for oxime linkage |
| EP2459226B1 (en) * | 2009-07-27 | 2016-06-29 | Lipoxen Technologies Limited | Glycopolysialylation of proteins other than blood coagulation proteins |
| US9662271B2 (en) | 2009-10-23 | 2017-05-30 | Amgen Inc. | Vial adapter and system |
| BR112012015597A2 (pt) | 2009-12-21 | 2017-01-31 | Ambrx Inc | peptídeos de somatotropina suínos modificados e seus usos |
| MX349301B (es) | 2009-12-21 | 2017-07-21 | Ambrx Inc | Polipéptidos de somatotropina bovina modificados y sus usos. |
| WO2011098095A1 (en) | 2010-02-09 | 2011-08-18 | Aplagen Gmbh | Peptides binding the tpo receptor |
| EA032537B1 (ru) | 2010-06-07 | 2019-06-28 | Эмджен Инк. | Способ работы устройства для доставки лекарственного средства |
| WO2012004004A1 (en) | 2010-07-09 | 2012-01-12 | Fresenius Kabi Deutschland Gmbh | Nitric oxide delivering hydroxyalkyl starch derivatives |
| WO2012004007A1 (en) * | 2010-07-09 | 2012-01-12 | Fresenius Kabi Deutschland Gmbh | Conjugates comprising hydroxyalkyl starch and a cytotoxic agent and process for their preparation |
| EP2590677A1 (en) * | 2010-07-09 | 2013-05-15 | Fresenius Kabi Deutschland GmbH | Conjugates comprising hydroxyalkyl starch and a cytotoxic agent and process for their preparation |
| WO2012004009A1 (en) * | 2010-07-09 | 2012-01-12 | Fresenius Kabi Deutschland Gmbh | Conjugates comprising hydroxyalkyl starch and a cytotoxic agent and process for their preparation |
| KR101969601B1 (ko) | 2010-07-30 | 2019-04-17 | 박스알타 인코퍼레이티드 | 옥심 결합용 친핵성 촉매 |
| AR083006A1 (es) | 2010-09-23 | 2013-01-23 | Lilly Co Eli | Formulaciones para el factor estimulante de colonias de granulocitos (g-csf) bovino y variantes de las mismas |
| ES2800983T3 (es) | 2010-12-22 | 2021-01-07 | Baxalta GmbH | Materiales y métodos para conjugar un derivado de ácido graso soluble en agua con una proteína |
| WO2012135315A1 (en) | 2011-03-31 | 2012-10-04 | Amgen Inc. | Vial adapter and system |
| LT2699293T (lt) | 2011-04-20 | 2019-04-25 | Amgen Inc. | Automatinio purškimo prietaisas |
| WO2012166622A1 (en) | 2011-05-27 | 2012-12-06 | Baxter International Inc. | Therapeutic proteins with increased half-life and methods of preparing same |
| WO2012170396A1 (en) * | 2011-06-07 | 2012-12-13 | Philadelphia Health & Education Corporation | Spla2 monitoring strip |
| WO2013021056A1 (en) | 2011-08-10 | 2013-02-14 | Ludwig-Maximilians-Universität München | Method for the controlled intracellular delivery of nucleic acids |
| EP3335747B1 (en) | 2011-10-14 | 2021-04-07 | Amgen Inc. | Injector and method of assembly |
| WO2013113503A1 (en) | 2012-01-31 | 2013-08-08 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyalkyl starch and an oligonucleotide |
| AU2013204754C1 (en) | 2012-05-16 | 2018-10-11 | Takeda Pharmaceutical Company Limited | Nucleophilic Catalysts for Oxime Linkage |
| ES2780395T3 (es) | 2012-11-21 | 2020-08-25 | Amgen Inc | Dispositivo de administración de fármacos |
| CN104936613B (zh) * | 2012-11-30 | 2018-05-22 | 株式会社糖锁工学研究所 | 糖链加成连接子、含有糖链加成连接子与生理活性物质的化合物或其盐、以及其制造方法 |
| US10492990B2 (en) | 2013-03-15 | 2019-12-03 | Amgen Inc. | Drug cassette, autoinjector, and autoinjector system |
| MX359794B (es) | 2013-03-15 | 2018-10-10 | Intrinsic Lifesciences Llc | Anticuerpos anti-hepcidina y usos de los mismos. |
| JP6336564B2 (ja) | 2013-03-15 | 2018-06-06 | アムゲン・インコーポレーテッド | 薬物カセット、自動注入器、および自動注入器システム |
| WO2014147174A1 (en) * | 2013-03-20 | 2014-09-25 | Fresenius Kabi Deutschland Gmbh | Process for the preparation of thiol functionalized hydroxyalkyl starch derivatives |
| CA2907471A1 (en) * | 2013-03-20 | 2014-09-25 | Fresenius Kabi Deutschland Gmbh | Hydroxyalkyl starch derivatives as reactants for coupling to thiol groups |
| CA2907371A1 (en) * | 2013-03-20 | 2014-09-25 | Fresenius Kabi Deutschland Gmbh | Hydroxyalkyl starch derivatives as reactants for coupling to thiol groups |
| CN113559363B (zh) | 2013-03-22 | 2023-10-31 | 美国安进公司 | 注射器及装配方法 |
| JP6520712B2 (ja) | 2013-09-24 | 2019-05-29 | 味の素株式会社 | 糖アミノ酸およびその用途 |
| JP7051293B2 (ja) | 2013-10-24 | 2022-04-11 | アムジエン・インコーポレーテツド | 温度感知制御を伴う薬剤送達システム |
| US11097055B2 (en) | 2013-10-24 | 2021-08-24 | Amgen Inc. | Injector and method of assembly |
| WO2015119906A1 (en) | 2014-02-05 | 2015-08-13 | Amgen Inc. | Drug delivery system with electromagnetic field generator |
| US20150225315A1 (en) * | 2014-02-10 | 2015-08-13 | Honeywell International Inc. | Reactor design for liquid phase fluorination |
| EP3114138B1 (en) | 2014-03-05 | 2021-11-17 | Pfizer Inc. | Improved muteins of clotting factor viii |
| SG11201609219QA (en) | 2014-05-07 | 2016-12-29 | Amgen Inc | Autoinjector with shock reducing elements |
| KR102506249B1 (ko) | 2014-06-03 | 2023-03-03 | 암겐 인코포레이티드 | 약물 전달 시스템 및 사용 방법 |
| CN104072608A (zh) * | 2014-06-23 | 2014-10-01 | 山东齐都药业有限公司 | 含羧基羟乙基淀粉衍生物修饰的牛血清白蛋白偶联物及其制备方法 |
| WO2016049036A1 (en) | 2014-09-22 | 2016-03-31 | Intrinsic Lifesciences Llc | Humanized anti-hepcidin antibodies and uses thereof |
| CA2957960C (en) | 2014-10-14 | 2023-08-22 | Amgen, Inc. | Drug injection device with visual and audible indicators |
| JP6716566B2 (ja) | 2014-12-19 | 2020-07-01 | アムジエン・インコーポレーテツド | 近接センサ付き薬物送達装置 |
| EP3233159B1 (en) | 2014-12-19 | 2020-03-04 | Amgen Inc. | Drug delivery device with live button or user interface field |
| ES2748750T3 (es) | 2015-02-17 | 2020-03-17 | Amgen Inc | Dispositivo de administración de fármacos con sujeción asistida por vacío y/o realimentación |
| ES2905870T3 (es) | 2015-02-27 | 2022-04-12 | Amgen Inc | Dispositivo de suministro de fármacos que tiene un mecanismo de protección de aguja con un umbral de resistencia ajustable al movimiento de la protección de aguja |
| WO2017039786A1 (en) | 2015-09-02 | 2017-03-09 | Amgen Inc. | Syringe assembly adapter for a syringe |
| US11351308B2 (en) | 2015-12-09 | 2022-06-07 | Amgen Inc. | Auto-injector with signaling cap |
| US11154661B2 (en) | 2016-01-06 | 2021-10-26 | Amgen Inc. | Auto-injector with signaling electronics |
| EP3721922B1 (en) | 2016-03-15 | 2022-05-04 | Amgen Inc. | Reducing probability of glass breakage in drug delivery devices |
| WO2017189089A1 (en) | 2016-04-29 | 2017-11-02 | Amgen Inc. | Drug delivery device with messaging label |
| WO2017192287A1 (en) | 2016-05-02 | 2017-11-09 | Amgen Inc. | Syringe adapter and guide for filling an on-body injector |
| AU2017263558B2 (en) | 2016-05-13 | 2022-12-22 | Amgen Inc. | Vial sleeve assembly |
| US11238150B2 (en) | 2016-05-16 | 2022-02-01 | Amgen Inc. | Data encryption in medical devices with limited computational capability |
| CN109152817A (zh) | 2016-05-20 | 2019-01-04 | 瑞士奥克特珐玛公司 | 具有改进的药代动力学的糖基化vwf融合蛋白 |
| GB201609083D0 (en) | 2016-05-24 | 2016-07-06 | Syntab Therapeutics Gmbh | Synthetic compound |
| US11541176B2 (en) | 2016-06-03 | 2023-01-03 | Amgen Inc. | Impact testing apparatuses and methods for drug delivery devices |
| WO2018004842A1 (en) | 2016-07-01 | 2018-01-04 | Amgen Inc. | Drug delivery device having minimized risk of component fracture upon impact events |
| US20190328965A1 (en) | 2016-08-17 | 2019-10-31 | Amgen Inc. | Drug delivery device with placement detection |
| US20200261643A1 (en) | 2016-10-25 | 2020-08-20 | Amgen Inc. | On-body injector |
| AU2018210301A1 (en) | 2017-01-17 | 2019-08-01 | Amgen Inc. | Injection devices and related methods of use and assembly |
| JP7064501B2 (ja) | 2017-02-17 | 2022-05-10 | アムジエン・インコーポレーテツド | 無菌流体流路を備える薬物送達デバイスおよび関連する組立方法 |
| AU2018221351B2 (en) | 2017-02-17 | 2023-02-23 | Amgen Inc. | Insertion mechanism for drug delivery device |
| CA3050927A1 (en) | 2017-03-06 | 2018-09-13 | Brian Stonecipher | Drug delivery device with activation prevention feature |
| IL268478B2 (en) | 2017-03-07 | 2023-10-01 | Amgen Inc | Inserting a needle using super pressure |
| JP2020509837A (ja) | 2017-03-09 | 2020-04-02 | アムジエン・インコーポレーテツド | 薬剤送達装置のための挿入機構 |
| CN110446499A (zh) | 2017-03-20 | 2019-11-12 | 豪夫迈·罗氏有限公司 | 一种体外糖基工程化红细胞生成刺激蛋白的方法 |
| CN110446512B (zh) | 2017-03-28 | 2022-03-18 | 美国安进公司 | 柱塞杆和注射器组件系统以及方法 |
| US11590294B2 (en) | 2017-06-08 | 2023-02-28 | Amgen Inc. | Syringe assembly for a drug delivery device and method of assembly |
| EP3634546A1 (en) | 2017-06-08 | 2020-04-15 | Amgen Inc. | Torque driven drug delivery device |
| MX2019015472A (es) | 2017-06-22 | 2020-02-19 | Amgen Inc | Reduccion del impacto/choque de la activacion del mecanismo. |
| WO2018237225A1 (en) | 2017-06-23 | 2018-12-27 | Amgen Inc. | ELECTRONIC DRUG DELIVERY DEVICE COMPRISING A CAP ACTIVATED BY A SWITCH ASSEMBLY |
| EP3651832B1 (en) | 2017-07-14 | 2023-12-13 | Amgen Inc. | Needle insertion-retraction system having dual torsion spring system |
| MA49626A (fr) | 2017-07-21 | 2020-05-27 | Amgen Inc | Élément d'étanchéité perméable aux gaz pour récipient à médicament et procédés d'assemblage |
| WO2019022951A1 (en) | 2017-07-25 | 2019-01-31 | Amgen Inc. | DRUG DELIVERY DEVICE WITH GEAR MODULE AND ASSEMBLY METHOD THEREOF |
| WO2019022950A1 (en) | 2017-07-25 | 2019-01-31 | Amgen Inc. | DRUG DELIVERY DEVICE WITH CONTAINER ACCESS SYSTEM AND ASSEMBLY METHOD THEREOF |
| EP3664863A2 (en) | 2017-08-09 | 2020-06-17 | Amgen Inc. | Hydraulic-pneumatic pressurized chamber drug delivery system |
| MA49897A (fr) | 2017-08-18 | 2020-06-24 | Amgen Inc | Injecteur sur-corps avec patch adhésif stérile |
| US11103636B2 (en) | 2017-08-22 | 2021-08-31 | Amgen Inc. | Needle insertion mechanism for drug delivery device |
| WO2019070472A1 (en) | 2017-10-04 | 2019-04-11 | Amgen Inc. | FLOW ADAPTER FOR MEDICATION DELIVERY DEVICE |
| CN111132711B (zh) | 2017-10-06 | 2022-07-01 | 安进公司 | 带有联锁组件的药物递送装置及相关组装方法 |
| EP3694578A1 (en) | 2017-10-09 | 2020-08-19 | Amgen Inc. | Drug delivery device with drive assembly and related method of assembly |
| WO2019090079A1 (en) | 2017-11-03 | 2019-05-09 | Amgen Inc. | System and approaches for sterilizing a drug delivery device |
| WO2019090303A1 (en) | 2017-11-06 | 2019-05-09 | Amgen Inc. | Fill-finish assemblies and related methods |
| WO2019089178A1 (en) | 2017-11-06 | 2019-05-09 | Amgen Inc. | Drug delivery device with placement and flow sensing |
| CA3079665A1 (en) | 2017-11-10 | 2019-05-16 | Amgen Inc. | Plungers for drug delivery devices |
| MX2020005066A (es) | 2017-11-16 | 2020-08-20 | Amgen Inc | Autoinyector con deteccion de detencion y punto final. |
| WO2019099324A1 (en) | 2017-11-16 | 2019-05-23 | Amgen Inc. | Door latch mechanism for drug delivery device |
| WO2019212377A1 (ru) * | 2018-05-04 | 2019-11-07 | Farber Boris Slavinovich | Фармацевтическая композиция на основе полимиксина для лечения инфекционных заболеваний |
| US10835685B2 (en) | 2018-05-30 | 2020-11-17 | Amgen Inc. | Thermal spring release mechanism for a drug delivery device |
| US11083840B2 (en) | 2018-06-01 | 2021-08-10 | Amgen Inc. | Modular fluid path assemblies for drug delivery devices |
| CA3103682A1 (en) | 2018-07-24 | 2020-01-30 | Amgen Inc. | Delivery devices for administering drugs |
| EP3826701A1 (en) | 2018-07-24 | 2021-06-02 | Amgen Inc. | Delivery devices for administering drugs |
| WO2020023220A1 (en) | 2018-07-24 | 2020-01-30 | Amgen Inc. | Hybrid drug delivery devices with tacky skin attachment portion and related method of preparation |
| US20210228815A1 (en) | 2018-07-24 | 2021-07-29 | Amgen Inc. | Hybrid drug delivery devices with grip portion |
| EP3829692A1 (en) | 2018-07-31 | 2021-06-09 | Amgen Inc. | Fluid path assembly for a drug delivery device |
| MA53724A (fr) | 2018-09-24 | 2021-12-29 | Amgen Inc | Systèmes et procédés de dosage interventionnel |
| AU2019350660A1 (en) | 2018-09-28 | 2021-03-18 | Amgen Inc. | Muscle wire escapement activation assembly for a drug delivery device |
| AR116679A1 (es) | 2018-10-02 | 2021-06-02 | Amgen Inc | Sistemas de inyección para la administración de fármacos con transmisión de fuerza interna |
| EP3860686A1 (en) | 2018-10-05 | 2021-08-11 | Amgen Inc. | Drug delivery device having dose indicator |
| SG11202103800RA (en) | 2018-10-15 | 2021-05-28 | Amgen Inc | Drug delivery device having damping mechanism |
| CA3109988A1 (en) | 2018-10-15 | 2020-04-23 | Amgen Inc. | Platform assembly process for drug delivery device |
| EP3873566A1 (en) | 2018-11-01 | 2021-09-08 | Amgen Inc. | Drug delivery devices with partial drug delivery member retraction |
| MA54057A (fr) | 2018-11-01 | 2022-02-09 | Amgen Inc | Dispositifs d'administration de médicament à rétraction partielle d'élément d'administration de médicament |
| WO2020092056A1 (en) | 2018-11-01 | 2020-05-07 | Amgen Inc. | Drug delivery devices with partial needle retraction |
| US20220160972A1 (en) | 2019-04-24 | 2022-05-26 | Amgen Inc. | Syringe sterilization verification assemblies and methods |
| CA3148261A1 (en) | 2019-08-23 | 2021-03-04 | Amgen Inc. | Drug delivery device with configurable needle shield engagement components and related methods |
| WO2022246055A1 (en) | 2021-05-21 | 2022-11-24 | Amgen Inc. | Method of optimizing a filling recipe for a drug container |
Family Cites Families (157)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE279486C (zh) | ||||
| US3191291A (en) | 1959-01-21 | 1965-06-29 | Continental Can Co | Art of producing very thin steel and like sheets in wide strips |
| CH397115A (de) | 1960-10-04 | 1965-08-15 | Hoechst Ag | Verfahren zur Herstellung wasserunlöslicher Farbstoffe |
| GB1385403A (en) | 1971-07-14 | 1975-02-26 | Unilever Ltd | Process for preparing oxidised carbohydrates |
| GB1419080A (en) | 1972-12-29 | 1975-12-24 | Cheminova As | Chemical compounds having juvenile hormone activity |
| US4179337A (en) | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| US4125492A (en) | 1974-05-31 | 1978-11-14 | Pedro Cuatrecasas | Affinity chromatography of vibrio cholerae enterotoxin-ganglioside polysaccharide and the biological effects of ganglioside-containing soluble polymers |
| US4001401A (en) | 1975-02-02 | 1977-01-04 | Alza Corporation | Blood substitute and blood plasma expander comprising polyhemoglobin |
| US4001200A (en) | 1975-02-27 | 1977-01-04 | Alza Corporation | Novel polymerized, cross-linked, stromal-free hemoglobin |
| US4061736A (en) | 1975-02-02 | 1977-12-06 | Alza Corporation | Pharmaceutically acceptable intramolecularly cross-linked, stromal-free hemoglobin |
| US4053590A (en) | 1975-02-27 | 1977-10-11 | Alza Corporation | Compositions of matter comprising macromolecular hemoglobin |
| CA1055932A (en) | 1975-10-22 | 1979-06-05 | Hematech Inc. | Blood substitute based on hemoglobin |
| DE2616086C2 (de) * | 1976-04-13 | 1986-04-03 | Dr. Eduard Fresenius, Chemisch-pharmazeutische Industrie KG, 6380 Bad Homburg | Substanz zur Verwendung in einem kolloidalen Blutvolumenersatzmittel aus Hydroxyethylstärke und Hämoglobin |
| GB1578348A (en) | 1976-08-17 | 1980-11-05 | Pharmacia Ab | Products and a method for the therapeutic suppression of reaginic antibodies responsible for common allergic |
| FR2378094A2 (fr) | 1977-01-24 | 1978-08-18 | Inst Nat Sante Rech Med | Nouveaux reactifs biologiques constitues par des supports solides sur lesquels sont couples des composes organiques comportant un residu glucidique |
| EP0019403B1 (en) * | 1979-05-10 | 1985-07-31 | American Hospital Supply Corporation | Hydroxyalkyl-starch drug carrier |
| DE3029307A1 (de) * | 1980-08-01 | 1982-03-04 | Dr. Eduard Fresenius, Chemisch-pharmazeutische Industrie KG, 6380 Bad Homburg | Haemoglobin enthaltendes blutersatzmittel |
| US4454161A (en) | 1981-02-07 | 1984-06-12 | Kabushiki Kaisha Hayashibara Seibutsu Kagaku Kenkyujo | Process for the production of branching enzyme, and a method for improving the qualities of food products therewith |
| JPS57206622A (en) | 1981-06-10 | 1982-12-18 | Ajinomoto Co Inc | Blood substitute |
| IE56026B1 (en) | 1982-10-19 | 1991-03-27 | Cetus Corp | Cysteine-depleted muteins of biologically active proteins |
| EP0127839B1 (en) | 1983-05-27 | 1992-07-15 | THE TEXAS A&M UNIVERSITY SYSTEM | Method for producing a recombinant baculovirus expression vector |
| IE57931B1 (en) | 1983-10-11 | 1993-05-19 | Fidia Spa | Hyaluronic acid fractions having pharmaceutical activity,methods for preparation thereof,and pharmaceutical compositions containing the same |
| NZ210501A (en) | 1983-12-13 | 1991-08-27 | Kirin Amgen Inc | Erythropoietin produced by procaryotic or eucaryotic expression of an exogenous dna sequence |
| US4703008A (en) | 1983-12-13 | 1987-10-27 | Kiren-Amgen, Inc. | DNA sequences encoding erythropoietin |
| US4496689A (en) | 1983-12-27 | 1985-01-29 | Miles Laboratories, Inc. | Covalently attached complex of alpha-1-proteinase inhibitor with a water soluble polymer |
| US4952496A (en) | 1984-03-30 | 1990-08-28 | Associated Universities, Inc. | Cloning and expression of the gene for bacteriophage T7 RNA polymerase |
| IL77081A (en) | 1984-12-04 | 1999-10-28 | Genetics Inst | AND sequence encoding human erythropoietin, a process for its preparation and a pharmacological preparation of human erythropoietin |
| DE3501616A1 (de) | 1985-01-17 | 1986-07-17 | Schering AG, 1000 Berlin und 4709 Bergkamen | Verfahren zur herstellung von hydroxylamin-derivaten |
| US4667016A (en) | 1985-06-20 | 1987-05-19 | Kirin-Amgen, Inc. | Erythropoietin purification |
| US4766106A (en) | 1985-06-26 | 1988-08-23 | Cetus Corporation | Solubilization of proteins for pharmaceutical compositions using polymer conjugation |
| US4863964A (en) | 1985-07-02 | 1989-09-05 | Biomedical Frontiers, Inc. | Method for the stabilization of deferoxamine to chelate free ions in physiological fluid |
| US5217998A (en) * | 1985-07-02 | 1993-06-08 | Biomedical Frontiers, Inc. | Composition for the stabilization of deferoxamine to chelate free ions in physiological fluid |
| GB8610551D0 (en) | 1986-04-30 | 1986-06-04 | Hoffmann La Roche | Polypeptide & protein derivatives |
| IT1203814B (it) | 1986-06-30 | 1989-02-23 | Fidia Farmaceutici | Esteri dell'acido alginico |
| FR2600894B1 (fr) | 1986-07-02 | 1989-01-13 | Centre Nat Rech Scient | Conjugues macromoleculaires d'hemoglobine, leur procede de preparation et leurs applications |
| US5214132A (en) | 1986-12-23 | 1993-05-25 | Kyowa Hakko Kogyo Co., Ltd. | Polypeptide derivatives of human granulocyte colony stimulating factor |
| US5362853A (en) | 1986-12-23 | 1994-11-08 | Kyowa Hakko Kogyo Co., Ltd. | Polypeptide derivatives of human granulocyte colony stimulating factor |
| JP2594123B2 (ja) | 1987-09-12 | 1997-03-26 | 株式会社林原生物化学研究所 | 減感作剤 |
| EP0307827A3 (en) | 1987-09-15 | 1989-12-27 | Kuraray Co., Ltd. | Novel macromolecular complexes, process for producing same and medicinal use of such complexes |
| IL84252A (en) | 1987-10-23 | 1994-02-27 | Yissum Res Dev Co | Phospholipase inhibiting compositions |
| US4904584A (en) * | 1987-12-23 | 1990-02-27 | Genetics Institute, Inc. | Site-specific homogeneous modification of polypeptides |
| US4847325A (en) * | 1988-01-20 | 1989-07-11 | Cetus Corporation | Conjugation of polymer to colony stimulating factor-1 |
| DK110188D0 (da) | 1988-03-02 | 1988-03-02 | Claus Selch Larsen | High molecular weight prodrug derivatives of antiinflammatory drugs |
| FR2630329B1 (fr) | 1988-04-20 | 1991-07-05 | Merieux Inst | Conjugues macromoleculaires d'hemoglobine, leur procede de preparation et leurs applications |
| IT1219942B (it) | 1988-05-13 | 1990-05-24 | Fidia Farmaceutici | Esteri polisaccaridici |
| US4900780A (en) | 1988-05-25 | 1990-02-13 | Masonic Medical Research Laboratory | Acellular resuscitative fluid |
| US4925677A (en) | 1988-08-31 | 1990-05-15 | Theratech, Inc. | Biodegradable hydrogel matrices for the controlled release of pharmacologically active agents |
| US5420105A (en) | 1988-09-23 | 1995-05-30 | Gustavson; Linda M. | Polymeric carriers for non-covalent drug conjugation |
| US5218092A (en) | 1988-09-29 | 1993-06-08 | Kyowa Hakko Kogyo Co., Ltd. | Modified granulocyte-colony stimulating factor polypeptide with added carbohydrate chains |
| DE3836600A1 (de) | 1988-10-27 | 1990-05-03 | Wolff Walsrode Ag | Kohlensaeureester von polysacchariden und verfahren zu ihrer herstellung |
| DE68925966T2 (de) | 1988-12-22 | 1996-08-29 | Kirin Amgen Inc | Chemisch modifizierte granulocytenkolonie erregender faktor |
| US6261800B1 (en) * | 1989-05-05 | 2001-07-17 | Genentech, Inc. | Luteinizing hormone/choriogonadotropin (LH/CG) receptor |
| DE3919729C3 (de) | 1989-06-16 | 1997-06-19 | Fresenius Ag | Hydroxyethylstärke als Plasmaexpander, Verfahren zu ihrer Herstellung und Verwendung als kolloidales Plasmaersatzmittel |
| JP2896580B2 (ja) | 1989-08-25 | 1999-05-31 | チッソ株式会社 | アミロース―リゾチームハイブリッドと活性化糖およびその製造法 |
| JP2838800B2 (ja) | 1989-09-02 | 1998-12-16 | 株式会社林原生物化学研究所 | 減感作剤 |
| JP2983629B2 (ja) | 1989-10-13 | 1999-11-29 | キリン―アムジエン・インコーポレイテツド | エリスロポエチンイソフォーム |
| JP2975632B2 (ja) | 1990-03-30 | 1999-11-10 | 生化学工業株式会社 | グリコサミノグリカン修飾プロテイン |
| US5169784A (en) | 1990-09-17 | 1992-12-08 | The Texas A & M University System | Baculovirus dual promoter expression vector |
| DK130991D0 (da) | 1991-07-04 | 1991-07-04 | Immunodex K S | Polymere konjugater |
| US5281698A (en) * | 1991-07-23 | 1994-01-25 | Cetus Oncology Corporation | Preparation of an activated polymer ester for protein conjugation |
| DE4130807A1 (de) * | 1991-09-17 | 1993-03-18 | Wolff Walsrode Ag | Verfahren zur herstellung von polysaccharidcarbonaten |
| US6172208B1 (en) | 1992-07-06 | 2001-01-09 | Genzyme Corporation | Oligonucleotides modified with conjugate groups |
| AU5098193A (en) * | 1992-09-01 | 1994-03-29 | Berlex Laboratories, Inc. | Glycolation of glycosylated macromolecules |
| GB2270920B (en) | 1992-09-25 | 1997-04-02 | Univ Keele | Alginate-bioactive agent conjugates |
| NO934477L (no) * | 1992-12-09 | 1994-06-10 | Ortho Pharma Corp | PEG hydrazon- og PEG oksim-bindingdannende reagenser og proteinderivater derav |
| NZ250375A (en) | 1992-12-09 | 1995-07-26 | Ortho Pharma Corp | Peg hydrazone and peg oxime linkage forming reagents and protein derivatives |
| CA2110543A1 (en) | 1992-12-09 | 1994-06-10 | David E. Wright | Peg hydrazone and peg oxime linkage forming reagents and protein derivatives thereof |
| EP0601417A3 (de) | 1992-12-11 | 1998-07-01 | Hoechst Aktiengesellschaft | Physiologisch verträglicher und physiologisch abbaubarer, Kohlenhydratrezeptorblocker auf Polymerbasis, ein Verfahren zu seiner Herstellung und seine Verwendung |
| US5581476A (en) | 1993-01-28 | 1996-12-03 | Amgen Inc. | Computer-based methods and articles of manufacture for preparing G-CSF analogs |
| DE69401109T2 (de) | 1993-03-16 | 1997-05-28 | Hemosol Inc | Selektive vernetzung von hemoglobin mittels oxidierten, ringgeöffneten sacchariden |
| WO1994028024A1 (en) * | 1993-06-01 | 1994-12-08 | Enzon, Inc. | Carbohydrate-modified polymer conjugates with erythropoietic activity |
| IL192290A0 (en) | 1993-08-17 | 2008-12-29 | Kirin Amgen Inc | Erythropoietin analogs |
| US5840900A (en) | 1993-10-20 | 1998-11-24 | Enzon, Inc. | High molecular weight polymer-based prodrugs |
| JPH07188291A (ja) | 1993-12-27 | 1995-07-25 | Hayashibara Biochem Lab Inc | 蛋白質とその製造方法並びに用途 |
| US5876980A (en) * | 1995-04-11 | 1999-03-02 | Cytel Corporation | Enzymatic synthesis of oligosaccharides |
| US6214331B1 (en) | 1995-06-06 | 2001-04-10 | C. R. Bard, Inc. | Process for the preparation of aqueous dispersions of particles of water-soluble polymers and the particles obtained |
| US5736533A (en) | 1995-06-07 | 1998-04-07 | Neose Technologies, Inc. | Bacterial inhibition with an oligosaccharide compound |
| WO1996040662A2 (en) * | 1995-06-07 | 1996-12-19 | Cellpro, Incorporated | Aminooxy-containing linker compounds and their application in conjugates |
| WO1997021452A2 (en) | 1995-12-14 | 1997-06-19 | Advanced Magnetics, Inc. | Macromolecular prodrugs of nucleotide analogs |
| US5723589A (en) | 1995-12-21 | 1998-03-03 | Icn Pharmaceuticals | Carbohydrate conjugated bio-active compounds |
| NZ332234A (en) | 1996-03-12 | 2000-06-23 | Pg Txl Company Lp | Water soluble paclitaxel prodrugs formed by conjugating paclitaxel or docetaxel with a polyglutamic acid polymer and use for treating cancer |
| US5696152A (en) | 1996-05-07 | 1997-12-09 | Wisconsin Alumni Research Foundation | Taxol composition for use as organ preservation and cardioplegic agents |
| TW517067B (en) * | 1996-05-31 | 2003-01-11 | Hoffmann La Roche | Interferon conjugates |
| DE19628705A1 (de) * | 1996-07-08 | 1998-01-15 | Fresenius Ag | Neue Sauerstoff-Transport-Mittel, diese enthaltende Hämoglobin-Hydroxyethylstärke-Konjugate, Verfahren zu deren Herstellung, sowie deren Verwendung als Blutersatzstoffe |
| US5770645A (en) | 1996-08-02 | 1998-06-23 | Duke University Medical Center | Polymers for delivering nitric oxide in vivo |
| US5851984A (en) | 1996-08-16 | 1998-12-22 | Genentech, Inc. | Method of enhancing proliferation or differentiation of hematopoietic stem cells using Wnt polypeptides |
| US6011008A (en) * | 1997-01-08 | 2000-01-04 | Yissum Research Developement Company Of The Hebrew University Of Jerusalem | Conjugates of biologically active substances |
| US5952347A (en) | 1997-03-13 | 1999-09-14 | Merck & Co., Inc. | Quinoline leukotriene antagonists |
| US6299881B1 (en) * | 1997-03-24 | 2001-10-09 | Henry M. Jackson Foundation For The Advancement Of Military Medicine | Uronium salts for activating hydroxyls, carboxyls, and polysaccharides, and conjugate vaccines, immunogens, and other useful immunological reagents produced using uronium salts |
| US5990237A (en) | 1997-05-21 | 1999-11-23 | Shearwater Polymers, Inc. | Poly(ethylene glycol) aldehyde hydrates and related polymers and applications in modifying amines |
| DE69825288D1 (de) | 1997-08-07 | 2004-09-02 | Univ Utah | Prodrugs und konjugate von selenolenthaltenden verbindungen und ihre verwendung |
| US5847110A (en) | 1997-08-15 | 1998-12-08 | Biomedical Frontiers, Inc. | Method of reducing a schiff base |
| US6875594B2 (en) | 1997-11-13 | 2005-04-05 | The Rockefeller University | Methods of ligating expressed proteins |
| US6624142B2 (en) | 1997-12-30 | 2003-09-23 | Enzon, Inc. | Trimethyl lock based tetrapartate prodrugs |
| DE19808079A1 (de) | 1998-02-20 | 1999-08-26 | Schering Ag | Hydroxyethylstärke-Konjugate, Verfahren zu ihrer Herstellung und diese enthaltende pharmazeutische Mittel |
| US6596135B1 (en) | 1998-03-05 | 2003-07-22 | Asahi Glass Company, Limited | Sputtering target, transparent conductive film, and method for producing the same |
| CA2233725A1 (en) | 1998-03-31 | 1999-09-30 | Hemosol Inc. | Hemoglobin-hydroxyethyl starch complexes |
| US6153655A (en) | 1998-04-17 | 2000-11-28 | Enzon, Inc. | Terminally-branched polymeric linkers and polymeric conjugates containing the same |
| US6660843B1 (en) | 1998-10-23 | 2003-12-09 | Amgen Inc. | Modified peptides as therapeutic agents |
| US6261594B1 (en) | 1998-11-25 | 2001-07-17 | The University Of Akron | Chitosan-based nitric oxide donor compositions |
| EP1035137A1 (en) * | 1999-03-12 | 2000-09-13 | Pasteur Merieux Serums Et Vaccins | Method for the reductive amination of polysaccharides |
| CZ299516B6 (cs) | 1999-07-02 | 2008-08-20 | F. Hoffmann-La Roche Ag | Konjugát erythropoetinového glykoproteinu, zpusobjeho výroby a použití a farmaceutická kompozice sjeho obsahem |
| US7279176B1 (en) | 1999-09-02 | 2007-10-09 | Rice University | Nitric oxide-producing hydrogel materials |
| US20020065410A1 (en) | 1999-12-02 | 2002-05-30 | Antrim Richard L. | Branched starches and branched starch hydrolyzates |
| US6555660B2 (en) * | 2000-01-10 | 2003-04-29 | Maxygen Holdings Ltd. | G-CSF conjugates |
| US6749865B2 (en) | 2000-02-15 | 2004-06-15 | Genzyme Corporation | Modification of biopolymers for improved drug delivery |
| US6586398B1 (en) | 2000-04-07 | 2003-07-01 | Amgen, Inc. | Chemically modified novel erythropoietin stimulating protein compositions and methods |
| JP2001294601A (ja) | 2000-04-11 | 2001-10-23 | Akita Prefecture | 高度分岐澱粉と該高度分岐澱粉の製造方法 |
| JP2002003398A (ja) | 2000-04-17 | 2002-01-09 | Ltt Institute Co Ltd | 徐放製剤、その製造法及びワクチン |
| DE10023051B4 (de) * | 2000-05-11 | 2004-02-19 | Roche Diagnostics Gmbh | Verfahren zur Herstellung von Fluoresceinisothiocyanat-Sinistrin, dessen Verwendung und Fluoresceinisothiocyanat-Sinistrin enthaltende diagnostische Zubereitung |
| FR2811967B1 (fr) | 2000-07-24 | 2002-12-13 | Cebal | Tube muni d'une tete de fixation pour divers bouchages et les divers bouchages munis de moyens de fixation sur ledit tube |
| DE10041541A1 (de) | 2000-08-24 | 2002-03-14 | Michael Duchene | Rekombinante Allergene aus der Motte Plodia interpunctella |
| US6417347B1 (en) | 2000-08-24 | 2002-07-09 | Scimed Life Systems, Inc. | High yield S-nitrosylation process |
| RU2003109746A (ru) | 2000-09-08 | 2005-01-27 | Грифон Терапьютикс, Инк. (Us) | Синтетические белки, стимулирующие эритропоэз |
| DE10105921A1 (de) | 2001-02-09 | 2002-08-14 | Braun Melsungen Ag | An Kolloide gebundene Arzneiwirkstoffe |
| DE10112825A1 (de) | 2001-03-16 | 2002-10-02 | Fresenius Kabi De Gmbh | HESylierung von Wirkstoffen in wässriger Lösung |
| DE10126158A1 (de) | 2001-05-30 | 2002-12-12 | Novira Chem Gmbh | Eine Methode zur Synthese von Gemischen einfach aktivierter und nicht aktivierter Polyoxyalkylene zur Modifizierung von Proteinen |
| DE10129369C1 (de) | 2001-06-21 | 2003-03-06 | Fresenius Kabi De Gmbh | Wasserlösliches, einen Aminozucker aufweisendes Antibiotikum in Form eines Pol ysaccharid-Konjugats |
| DE10135694A1 (de) | 2001-07-21 | 2003-02-06 | Supramol Parenteral Colloids | Amphiphile Stärke-und Hydroxyethylstärke-Konjugate |
| US7125843B2 (en) * | 2001-10-19 | 2006-10-24 | Neose Technologies, Inc. | Glycoconjugates including more than one peptide |
| US7179617B2 (en) | 2001-10-10 | 2007-02-20 | Neose Technologies, Inc. | Factor IX: remolding and glycoconjugation of Factor IX |
| DK2431377T3 (en) | 2001-10-26 | 2017-08-14 | Noxxon Pharma Ag | Modified L-nucleic acids |
| US6375846B1 (en) | 2001-11-01 | 2002-04-23 | Harry Wellington Jarrett | Cyanogen bromide-activation of hydroxyls on silica for high pressure affinity chromatography |
| DE10155098A1 (de) | 2001-11-09 | 2003-05-22 | Supramol Parenteral Colloids | Mittel zur Prävention von mykotischen Kontaminationen bei der Zell- und Gewebekultur bakteriellen, pflanzlichen, animalischen und humanen Ursprungs, bestehend aus Polyen-Makrolid-Konjugaten mit Polysacchariden |
| US6916962B2 (en) | 2001-12-11 | 2005-07-12 | Sun Bio, Inc. | Monofunctional polyethylene glycol aldehydes |
| DE10207072A1 (de) * | 2002-02-20 | 2003-08-28 | Supramol Parenteral Colloids | Stärkederivate, ihre Konjugate und Verfahren zur Herstellung derselben |
| DE10209821A1 (de) | 2002-03-06 | 2003-09-25 | Biotechnologie Ges Mittelhesse | Kopplung von Proteinen an ein modifiziertes Polysaccharid |
| DE10209822A1 (de) | 2002-03-06 | 2003-09-25 | Biotechnologie Ges Mittelhesse | Kopplung niedermolekularer Substanzen an ein modifiziertes Polysaccharid |
| JP4272537B2 (ja) | 2002-03-13 | 2009-06-03 | 北京鍵▲凱▼科技有限公司 | Y型分鎖親水性ポリマー誘導体、それらの調製方法、前記誘導体および薬剤分子の結合生成物、ならびに前記結合生成物を含む医薬組成物 |
| DE10217994A1 (de) | 2002-04-23 | 2003-11-06 | Supramol Parenteral Colloids | Konjugate von hyperverzweigten Polysacchariden |
| AU2003249692B2 (en) | 2002-06-03 | 2008-07-31 | The Institute For Systems Biology | Methods for quantitative proteome analysis of glycoproteins |
| US20040101546A1 (en) | 2002-11-26 | 2004-05-27 | Gorman Anne Jessica | Hemostatic wound dressing containing aldehyde-modified polysaccharide and hemostatic agents |
| UA80838C2 (en) | 2002-09-09 | 2007-11-12 | Water-soluble polymer alkanals | |
| EP1591467A1 (en) | 2002-09-09 | 2005-11-02 | Nektar Therapeutics Al, Corporation | Conjugate between a polyethylene glycol having a terminal alkanal group and a human growth hormone |
| BR0314107A (pt) | 2002-09-11 | 2005-07-19 | Fresenius Kabi De Gmbh | Método de produção de derivados de hidroxialquil amido |
| DE10242076A1 (de) | 2002-09-11 | 2004-03-25 | Fresenius Kabi Deutschland Gmbh | HAS-Allergen-Konjugate |
| DE02020425T1 (de) | 2002-09-11 | 2004-07-15 | Fresenius Kabi Deutschland Gmbh | Hasylierte Polypeptide, besonders hasyliertes Erythropoietin |
| DE60323756D1 (de) | 2002-10-08 | 2008-11-06 | Fresenius Kabi De Gmbh | Pharmazeutisch aktive oligosaccharid-conjugate |
| DE10254745A1 (de) | 2002-11-23 | 2004-06-03 | Supramol Parenteral Colloids Gmbh | Imidazolide von Polysaccharid Aldonsäuren, Verfahren zu ihrer Herstellung und Verwendung zur Kopplung an pharmazeutische Wirkstoffe |
| DE10256558A1 (de) | 2002-12-04 | 2004-09-16 | Supramol Parenteral Colloids Gmbh | Ester von Polysaccharid Aldonsäuren, Verfahren zu ihrer Herstellung und Verwendung zur Kopplung an pharmazeutische Wirkstoffe |
| WO2005014655A2 (en) | 2003-08-08 | 2005-02-17 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyalkyl starch and a protein |
| EP1653991A2 (en) | 2003-08-08 | 2006-05-10 | Fresenius Kabi Deutschland GmbH | Conjugates of a polymer and a protein linked by an oxime linking group |
| CA2534418A1 (en) | 2003-08-08 | 2005-02-17 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyalkyl starch and g-csf |
| SG145746A1 (en) | 2003-08-08 | 2008-09-29 | Fresenius Kabi De Gmbh | Conjugates of hydroxyalkyl starch and g-csf |
| AU2005209303A1 (en) | 2004-01-29 | 2005-08-11 | Biosynexus, Inc. | Use of amino-oxy functional groups in the preparation of vaccines conjugates |
| DE102004009783A1 (de) | 2004-02-28 | 2005-09-15 | Supramol Parenteral Colloids Gmbh | Hyperverzweigte Stärkefraktion, Verfahren zu ihrer Herstellung und ihre Konjugate mit pharmazeutischen Wirkstoffen |
| WO2005092928A1 (en) | 2004-03-11 | 2005-10-06 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyalkyl starch and a protein, prepared by reductive amination |
| WO2005092369A2 (en) | 2004-03-11 | 2005-10-06 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyethyl starch and erythropoietin |
| WO2005092391A2 (en) | 2004-03-11 | 2005-10-06 | Fresenius Kabi Deutschland Gmbh | Conjugates of hydroxyalkyl starch and a protein |
| WO2005112954A1 (en) | 2004-05-14 | 2005-12-01 | William Marsh Rice University | Nitric oxide releasing compositions and associated methods |
| AU2006222187A1 (en) | 2005-03-11 | 2006-09-14 | Fresenius Kabi Deutschland Gmbh | Production of bioactive glycoproteins from inactive starting material by conjugation with hydroxyalkylstarch |
| EP1762250A1 (en) | 2005-09-12 | 2007-03-14 | Fresenius Kabi Deutschland GmbH | Conjugates of hydroxyalkyl starch and an active substance, prepared by chemical ligation via thiazolidine |
| AU2006309212B2 (en) | 2005-10-31 | 2011-09-15 | Government Of The United States Of America, Represented By The Secretary Department Of Health And Human Services | Polysaccharide-derived nitric oxide-releasing carbon-bound diazeniumdiolates |
| JP2009093397A (ja) | 2007-10-09 | 2009-04-30 | Panasonic Corp | タッチパネル及びこれを用いた入力装置 |
| EP2070950A1 (en) | 2007-12-14 | 2009-06-17 | Fresenius Kabi Deutschland GmbH | Hydroxyalkyl starch derivatives and process for their preparation |
| EP2070951A1 (en) | 2007-12-14 | 2009-06-17 | Fresenius Kabi Deutschland GmbH | Method for producing a hydroxyalkyl starch derivatives with two linkers |
| EP2349347A2 (en) | 2008-10-07 | 2011-08-03 | Rexahn Pharmaceuticals, Inc. | Hpma - docetaxel or gemcitabine conjugates and uses therefore |
-
2003
- 2003-08-08 BR BR0314107-1A patent/BR0314107A/pt not_active Application Discontinuation
- 2003-08-08 PL PL374969A patent/PL216545B1/pl not_active IP Right Cessation
- 2003-08-08 WO PCT/EP2003/008859 patent/WO2004024777A1/en active Application Filing
- 2003-08-08 MX MXPA05002594A patent/MXPA05002594A/es active IP Right Grant
- 2003-08-08 CN CNA038214644A patent/CN1681844A/zh active Pending
- 2003-08-08 BR BR0314106-3A patent/BR0314106A/pt not_active IP Right Cessation
- 2003-08-08 CA CA002495242A patent/CA2495242A1/en not_active Abandoned
- 2003-08-08 RU RU2005110423/04A patent/RU2328505C2/ru not_active IP Right Cessation
- 2003-08-08 PL PL374490A patent/PL217085B1/pl unknown
- 2003-08-08 AU AU2003260393A patent/AU2003260393B2/en not_active Ceased
- 2003-08-08 IL IL16650603A patent/IL166506A0/xx unknown
- 2003-08-08 RU RU2005110412/04A patent/RU2326891C2/ru not_active IP Right Cessation
- 2003-08-08 CN CNB038214652A patent/CN100348618C/zh not_active Expired - Fee Related
- 2003-08-08 BR BR0314227-2A patent/BR0314227A/pt not_active Application Discontinuation
- 2003-08-08 KR KR1020057003928A patent/KR101174510B1/ko not_active IP Right Cessation
- 2003-08-08 WO PCT/EP2003/008858 patent/WO2004024761A1/en active Application Filing
- 2003-08-08 PL PL03375037A patent/PL375037A1/xx unknown
- 2003-08-08 RU RU2005110415/04A patent/RU2329274C2/ru not_active IP Right Cessation
- 2003-08-08 MX MXPA05002593A patent/MXPA05002593A/es active IP Right Grant
- 2003-08-08 JP JP2004535075A patent/JP2006516534A/ja not_active Withdrawn
- 2003-08-08 KR KR1020057004002A patent/KR101045401B1/ko not_active IP Right Cessation
- 2003-08-08 MX MXPA05002591A patent/MXPA05002591A/es active IP Right Grant
- 2003-08-08 WO PCT/EP2003/008829 patent/WO2004024776A1/en active Application Filing
- 2003-08-08 KR KR1020057003902A patent/KR101045422B1/ko not_active IP Right Cessation
- 2003-08-08 AU AU2003253399A patent/AU2003253399B2/en not_active Ceased
- 2003-08-08 CA CA2496317A patent/CA2496317C/en not_active Expired - Fee Related
- 2003-08-08 AU AU2003255406A patent/AU2003255406B2/en not_active Ceased
- 2003-08-29 AR ARP030103141A patent/AR041097A1/es active IP Right Grant
- 2003-09-10 TW TW092124954A patent/TW200418875A/zh unknown
- 2003-09-11 ES ES03020425T patent/ES2213507T3/es not_active Expired - Lifetime
- 2003-09-11 EP EP03020424A patent/EP1398327B1/en not_active Expired - Lifetime
- 2003-09-11 DE DE60323192T patent/DE60323192D1/de not_active Expired - Lifetime
- 2003-09-11 EP EP09012453A patent/EP2143736B1/en not_active Expired - Lifetime
- 2003-09-11 DK DK03020424T patent/DK1398327T3/da active
- 2003-09-11 EP EP10011836.3A patent/EP2272865A3/en not_active Withdrawn
- 2003-09-11 EP EP03020423A patent/EP1398322B1/en not_active Expired - Lifetime
- 2003-09-11 AT AT03020423T patent/ATE323723T1/de active
- 2003-09-11 AT AT03020425T patent/ATE449112T1/de active
- 2003-09-11 DE DE0001398322T patent/DE03020423T1/de active Pending
- 2003-09-11 DE DE60330098T patent/DE60330098D1/de not_active Expired - Lifetime
- 2003-09-11 EP EP09014352A patent/EP2154160A1/en not_active Withdrawn
- 2003-09-11 PT PT03020424T patent/PT1398327E/pt unknown
- 2003-09-11 DE DE60304640T patent/DE60304640T2/de not_active Expired - Lifetime
- 2003-09-11 EP EP08012908A patent/EP2017287A3/en not_active Withdrawn
- 2003-09-11 DE DE0001398328T patent/DE03020425T1/de active Pending
- 2003-09-11 EP EP03020425A patent/EP1398328B1/en not_active Expired - Lifetime
- 2003-09-11 ES ES03020423T patent/ES2213505T3/es not_active Expired - Lifetime
- 2003-09-11 DE DE0001398327T patent/DE03020424T1/de active Pending
- 2003-09-11 PT PT03020423T patent/PT1398322E/pt unknown
- 2003-09-11 EP EP10011835A patent/EP2316850A3/en not_active Withdrawn
- 2003-09-11 AT AT03020424T patent/ATE406387T1/de active
- 2003-09-11 PT PT03020425T patent/PT1398328E/pt unknown
- 2003-09-11 ES ES09012453T patent/ES2399006T3/es not_active Expired - Lifetime
- 2003-09-11 SI SI200331431T patent/SI1398327T1/sl unknown
- 2003-09-11 SI SI200331733T patent/SI1398328T1/sl unknown
- 2003-09-11 ES ES03020424T patent/ES2213506T3/es not_active Expired - Lifetime
- 2003-09-11 DK DK03020425.9T patent/DK1398328T3/da active
- 2003-09-11 DE DE20321836U patent/DE20321836U1/de not_active Expired - Lifetime
- 2003-09-11 DK DK03020423T patent/DK1398322T3/da active
-
2004
- 2004-08-06 ZA ZA200600651A patent/ZA200600651B/xx unknown
- 2004-08-09 AR ARP040102850A patent/AR045234A1/es active IP Right Grant
- 2004-08-09 AR ARP040102851A patent/AR045235A1/es active IP Right Grant
- 2004-08-18 HK HK10103783.3A patent/HK1136226A1/xx not_active IP Right Cessation
- 2004-08-18 HK HK04106171A patent/HK1063475A1/xx not_active IP Right Cessation
- 2004-08-18 HK HK04106174.1A patent/HK1063478A1/xx not_active IP Right Cessation
- 2004-08-18 HK HK04106173A patent/HK1063477A1/xx not_active IP Right Cessation
-
2005
- 2005-01-26 IL IL166506A patent/IL166506A/en not_active IP Right Cessation
- 2005-02-16 IL IL166931A patent/IL166931A/en not_active IP Right Cessation
- 2005-02-16 IL IL166930A patent/IL166930A/en not_active IP Right Cessation
- 2005-03-11 US US11/077,906 patent/US7815893B2/en not_active Expired - Fee Related
- 2005-03-11 US US11/078,098 patent/US20050238723A1/en not_active Abandoned
- 2005-03-11 US US11/078,582 patent/US8618266B2/en not_active Expired - Fee Related
- 2005-03-17 NO NO20051419A patent/NO20051419L/no active IP Right Review Request
- 2005-03-17 NO NO20051427A patent/NO20051427L/no not_active Application Discontinuation
-
2009
- 2009-07-30 IL IL200150A patent/IL200150A0/en unknown
- 2009-11-20 AU AU2009238372A patent/AU2009238372B2/en not_active Ceased
-
2010
- 2010-02-16 CY CY20101100153T patent/CY1109813T1/el unknown
- 2010-06-04 MX MX2010006175A patent/MX2010006175A/es active IP Right Grant
- 2010-06-28 US US12/824,618 patent/US20120046240A9/en not_active Abandoned
- 2010-07-06 JP JP2010153807A patent/JP5455821B2/ja not_active Expired - Fee Related
- 2010-07-06 JP JP2010153808A patent/JP5275293B2/ja not_active Expired - Fee Related
- 2010-08-12 US US12/855,381 patent/US8475765B2/en not_active Expired - Fee Related
- 2010-11-30 JP JP2010266646A patent/JP2011089125A/ja active Pending
-
2011
- 2011-01-14 AU AU2011200219A patent/AU2011200219B2/en not_active Ceased
- 2011-01-14 AU AU2011200158A patent/AU2011200158B2/en not_active Ceased
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108219019A (zh) * | 2018-02-08 | 2018-06-29 | 华中科技大学 | 一种巯基化羟乙基淀粉及其修饰的纳米材料和制备方法 |
| CN108403641A (zh) * | 2018-02-08 | 2018-08-17 | 华中科技大学 | 一种载药纳米材料及其制备方法 |
| CN108503718A (zh) * | 2018-02-08 | 2018-09-07 | 华中科技大学 | 一种羟烷基淀粉偶联物及其制备方法和应用 |
| CN108503718B (zh) * | 2018-02-08 | 2020-03-20 | 华中科技大学 | 一种羟烷基淀粉偶联物及其制备方法和应用 |
| CN108219019B (zh) * | 2018-02-08 | 2020-03-20 | 华中科技大学 | 一种巯基化羟乙基淀粉及其修饰的纳米材料和制备方法 |
| CN108403641B (zh) * | 2018-02-08 | 2020-03-20 | 华中科技大学 | 一种载药纳米材料及其制备方法 |
| CN111157736A (zh) * | 2018-11-08 | 2020-05-15 | 中国科学院大连化学物理研究所 | 一种基于化学酶促的人血清o糖基化鉴定方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1681844A (zh) | Has化的多肽,特别是has化的促红细胞生成素 | |
| CN1708514A (zh) | 羟烷基淀粉衍生物 | |
| CN1946742A (zh) | 通过还原氨基化制备的羟烷基淀粉和蛋白质的偶联物 | |
| CN1933857A (zh) | 羟烷基淀粉和蛋白质的偶联物 | |
| EP1681303B1 (en) | HASylated polypeptides, especially HASylated erythropoietin | |
| CN1832762A (zh) | 羟烷基淀粉与g-csf的偶联物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |