CN1144097C - 色调剂、双组分显影剂和成像方法 - Google Patents
色调剂、双组分显影剂和成像方法 Download PDFInfo
- Publication number
- CN1144097C CN1144097C CNB98103585XA CN98103585A CN1144097C CN 1144097 C CN1144097 C CN 1144097C CN B98103585X A CNB98103585X A CN B98103585XA CN 98103585 A CN98103585 A CN 98103585A CN 1144097 C CN1144097 C CN 1144097C
- Authority
- CN
- China
- Prior art keywords
- particle
- toner
- fine powder
- inorganic
- image
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images


Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G15/00—Apparatus for electrographic processes using a charge pattern
- G03G15/06—Apparatus for electrographic processes using a charge pattern for developing
- G03G15/08—Apparatus for electrographic processes using a charge pattern for developing using a solid developer, e.g. powder developer
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/0827—Developers with toner particles characterised by their shape, e.g. degree of sphericity
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/0819—Developers with toner particles characterised by the dimensions of the particles
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/097—Plasticisers; Charge controlling agents
- G03G9/09708—Inorganic compounds
Abstract
本发明公开了一种具有色调剂粒子和外部添加剂的色调剂,这种色调剂具有:(a)以流动式粒子图像分析仪测得的粒子成圆率分布中平均成圆率为0.920-0.995,所含成圆率小于0.950的粒子数量为2%~40%;(b)以Coulter法测得重均粒子直径为2.0~9.0μm;这种外部添加剂具有在色调剂粒子上的至少(i)一种以初级粒子或二级粒子的状态存在的、平均粒子长度为10mμm~400mμm和形状因子SF-1为100~130的无机细粉(A)和(ii)一种由多个粒子结合形成的、形状因子SF-1大于150的非球形无机细粉(B)。本发明还公开了一种采用这种色调剂的双组分显影剂以及成像方法。
Description
本发明涉及一种用在利用静电照相、静电记录、磁记录和喷墨记录或其它类似方法的记录方法中的色调剂。更具体地说,本发明涉及一种适于使带静电的图像显影的色调剂,这种色调剂用于复印机、打印机和传真机,在这些机器中色调剂图像是预先形成在静电潜像承载元件上,然后将色调剂图像转印到转印介质以形成图像。本发明还涉及使用该色调剂的一种双组分显影剂和一种成像方法。
在显影剂携带元件表面上携带一种干法显影剂作为潜像显影剂,并将该显影剂转送和提供到携带有静电潜像的潜像承载元件表面附近,同时给潜像承载元件和显影剂携带元件提供一交流电场,由该显影剂的色调剂使该静电潜像显影,以使静电潜像可见的方法是常规公知的。
显影剂携带元件在以下的介绍中常常称为“显影套筒”,因为显影套筒通常被广泛地用作显影剂携带元件。潜像承载元件(光敏元件)在以下的介绍中也被称为“感光鼓”,因为感光鼓通常被广泛地用作潜像承载元件。
作为上述的显影方法,通常公知的是所谓的磁刷显影法。该方法中,利用,例如,一种由双组分(载体粒子和色调剂粒子)组成的显影剂(双组分显影剂),内部提供一磁体的显影套筒表面形成磁刷,由此而形成的磁刷与正对着显影套筒放置的感光鼓摩擦或者接近,在它们之间保持一微小显影间隙,同时不断地给显影套筒和感光鼓(在S-D之间)提供一交流电场以反复地引起色调剂粒子从显影套筒侧转移至感光鼓侧,反之亦然,以实现显影(参见,例如,日本专利申请公开No.55-32060和No.59-165082)。
在这种使用双组分显影剂的磁刷显影法中,将色调剂粒子与载体粒子混合在一起使色调剂粒子摩擦充电。由于载体粒子具有比色调剂粒子更高的比重,混合时色调剂粒子与载体粒子之间的摩擦使色调剂粒子经受了高机械应变,以致在重复显影操作过程中色调剂的损耗趋向加速。
一旦色调剂的损耗发生,则会具体引起这样的现象:由于长时间使用的结果,固定影像的浓度发生改变、粘附在非影像区域的色调剂粒子引起所谓的“图象模糊”现象以及瞬间图像的可再现性很差。
在静电照相过程中,在形成于感光鼓上的色调剂图像被转印到转印介质之后,没有被转印到转印介质的残存在感光鼓上的色调剂被清洁步骤中的清洁装置从感光鼓表面除去并收集起来。这种清洁装置可以是刮板清洁,毛刷清洁或辊式清洁。
然而,当感光鼓上的色调剂通过清洁装置被除去和收集时,从设备方面讲,由于要提供这样一种清洁装置,设备必须制造得更大一些。在试图使设备变得更紧凑的情况下,这是一个难题。因此,希望有一种不需清洁装置的成像设备。
从生态学的观点看,在有效利用色调剂的意义上,一种不会产生废色调剂的无清洁器系统或者可重复利用色调剂系统是期待已久的。
这种技术称之为显影清洁技术,在这种技术中,转印后残存在感光鼓上的色调剂(转印残留色调剂)在显影时收集在显影装置里,收集的色调剂又被用于显影中。
作为这种称为“显影清洁”(或“无清洁器”)系统的技术,例如,日本专利公报No.5-69427公开了感光鼓每旋转一圈形成一个图像,以使转印残留色调剂在同一图像上不会出现任何的影响。日本专利申请公开No.64-20587、No.2-259784、No.4-50886和No.5-165378公开了一种系统,在此系统中通过一种驱除元件把转印残留色调剂分散或驱除,使之变为非图象,以使即使同一个感光鼓的表面对一图像使用好几遍时,转印残留色调剂也几乎不会出现在图像上。
日本专利申请公开No.5-2287公开了一种系统,在此系统中,感光鼓周围色调剂荷电量的比例是确定的,以使任何由转印残留色调剂引起的正记忆或负记忆不可能出现在图像上。然而,该申请并没有公开怎样去控制色调剂荷电量的具体结构。
在日本专利申请公开No.59-133573、No.62-203182、No.63-133179、No.2-302772、No.4-155361、No.5-2289、No.5-53482和No.5-61383中公开了涉及无清洁器系统的技术,其中对于成影像曝光,建议使用具有高强度的光进行曝光或者使用一种能够透射具有曝光波长的光的色调剂。然而,仅仅采用更高曝光强度可能会在潜像本身的点形成过程中产生模糊,以致引起不充分的隔离点可再现性,在图像质量上导致图像的分辨率低,尤其是,图片类图像缺乏层次。
对于采用能够透射具有曝光波长的光的色调剂的方法来说,光的透射当然对已处理得很光滑且没有粒子界面的固定色调剂具有很强的影响。然而,根据网点曝光的机理,这种方法的效果不大,因为这种方法更主要地是强调光在色调剂粒子表面的散射,而不是色调剂自身的着色。此外,色调剂着色剂必须选自一个较窄的范围,同时当想要形成全色时,至少需要具有不同波长的三种类型的曝光装置。这种方法与简化设备背道而弛,而尽量简化设备正是显影清洁的特征之一。
有一种成像方法采用了接触充电系统,此系统中通过一种接触充电元件以注入电荷的方式使待充电的元件感光鼓初次充电,任何由于充电元件的污染(废色调剂)所致的故障充电会产生不完善的图像,并引起运行方面的故障。因而,为使多页复印成为可能,抑制因充电元件污染导致故障充电所带来的影响已经成为一个迫切的需要。
接触充电系统用于使用无清洁器或显影清洁系统的成像系统中的实例可在日本专利申请公开No.4-234063和No.6-230652中找到,其中公开了一种成像方法,该方法是在反曝光同时显影系统中同时完成从感光鼓除去转印残留色调剂的清洁过程。
然而,在这些出版物中的建议均适用于充电电压和显影外加偏压是在低电场下形成的这样一种成像方法。在较高电场充电-显影外加偏压下成像(这种成像方法通常被广泛地应用于静电照相设备当中)时,可能会产生漏电,导致图像不佳例如出现行线和斑点。
又一种建议的方法是,粘附在充电元件上的色调剂在没有图像形成时即被移到感光鼓上,以致于可以防止任何由转印残留色调剂的粘连引起的不良效果。然而,这项建议没有谈及任何关于在显影步骤中被转移到感光鼓上的色调剂回收率改进方面的事情,也没有谈及因在显影步骤中收集色调剂对显影可能产生的任何影响。
此外,如果在显影时对转印残留色调剂的清洁效果不够充分,就可能会产生正重影问题,这是因为后来的色调剂会参与在已经有转印残留色调剂存在的感光鼓上的显影,因而在感光鼓上形成的图像可能具有比周围更高的浓度,而且,如果这些转印残留色调剂数量太大,由于色调剂不可能被彻底收集在显影部件中,图像上就会产生正记忆。这些问题的根本解决办法仍没有找到。
当感光鼓重复在一页转印介质上使用时,也就是当相应于感光鼓一圆周的长度小于转印介质移动方向的长度时,由转印残留介质引起的光屏蔽问题尤为突出。由于充电、曝光和显影必须在转印残留色调剂存在于感光鼓上的状态下进行,在存在转印残留色调剂的感光鼓表面部分的电压不可能完全降低,使得显影对比度不足,这种情况在反向显影过程中表现在图像上即为负重影,其密度比周围的密度低。经过静电转印步骤的感光鼓所带电荷的极性与整个色调剂电荷的极性相反,在这种情况下,由于重复使用导致感光鼓中电荷注入性能降低,充电元件中未能控制带有正常电荷极性的转印残留色调剂在成像时可能从充电元件中泄漏,遮断曝光光线,以致潜像出现无序,并且得不到任何理想的电压,从而在图像上产生一个负记忆。上述问题可能会继续发生,仍在寻找解决这些问题的根本解决方案。
近几年来,使用上述静电照相方法的输出设备例如复印机和激光束打印机成本越来越低,在数字技术方面也取得了进展。相应地,通过利用许多图像信息形成更忠实于原像的高质量图像,也就成为必需。尤其是当图像例如打印的照片、目录和地图被复制时,需要在整个细节上非常精细地和如实地复制,不会产生碎线图像和虚线图像。
在这样的技术趋势中,未来的色调剂应具有如此性能,即在显影、转印和定影过程中,色调剂较少引起色调剂在潜像周围的散射,色调剂本身维持高充电性能,同时显影后的色调剂可以以几乎100%的转印效率被转印到转印介质。
为提高静电照相方法中的图像质量,下列方法是可行的:(i)一种方法,其中使潜像承载元件上的潜像与显影剂的耳状物摩擦,同时保持显影剂携带元件上显影剂的耳状物密集地增长;(ii)一种方法,其中提供一种偏压电场,使其穿过显影剂携带元件和潜像承载元件,从而使色调剂能容易地流动;(iii)一种方法,其中使显影装置自身在装置内部的搅拌性能更好,从而永久地保持高带电能力;以及(iv)一种方法,其中使潜像的点子大小本身变得更加细微以提高分辨率。
这些涉及到显影的方法是非常有效的,并且为获得高质量图像的部分重要技术。然而,考虑到在图像质量上的更多改进,显影剂自身的性能被认为具有极大影响。
尤其在全色图像的成像过程中,单色色调剂被多次用于显影和转印,所以在潜像区域形成了多层色调剂,其中由于这些层接近最外层而趋于具有一较低的电压,在某些情况下导致在最低层和最上层之间出现色调剂显影性能的差别。
更进一步地说,由于在热熔处理后颜色混合不佳,不但得不到一幅真实的颜色可再现性,而且常常还会引起缺陷例如转印性能的降低和色调剂在非潜像电压区的散射。
从工艺因素的观点看,如上所述,在改进图像质量方面色调剂性能被认为具有极大影响。为了改进图像质量,迄今提出了各种各样的显影剂。例如,日本专利申请公开No.51-3244公开了一种非磁性色调剂,其中通过控制色调剂的粒子大小分布以改进图像质量。这种色调剂主要由粒子直径为8~12μm的色调剂粒子组成,这些粒子是较粗的。根据本发明者所做的研究,对具有这样粒子直径的色调剂来说,在密集状态下飞到潜像上是困难的。而且,具有下述特征的色调剂,即粒子直径是5μm或更小的粒子数量不超过30%且粒子直径是20μm或更大的粒子数量不超过5%,由于其粒子大小分布的广度,趋向导致一种低均匀度。在使用包括较粗色调剂粒子和具有宽范围粒子大小分布的色调剂时,为形成清晰的图像,在如上所述的多层构造中,每层的色调剂粒子必须被厚厚地叠加覆盖,以便于色调剂粒子之间的任何空隙都被填满,提高表观图像浓度。这样就带来一个问题,即为获得一指定的图像浓度,需要增加色调剂的消耗量。
日本专利申请公开No.58-129437公开了一种非磁性色调剂,平均粒子直径为6~10μm,其中直径为5~8μm的粒子占最大多数。可是,这种色调剂中粒子直径等于或小于5μm的色调剂粒子含量仅为15%,会形成清晰度低的图像。
本发明者研究发现粒子直径等于或小于5μm的色调剂粒子有助于潜像微点的清晰复制,且具有把色调剂浓浓地施于整个潜像之上的主要功能。尤其是,由于电力线的集中,在感光鼓上静电潜像边缘的电场强度比在其内侧的更高,聚集在那部分的色调剂粒子的质量会影响图像品质的清晰度。本发明者所做的研究揭示控制粒子直径等于或小于5μm的色调剂粒子的含量对提高辉亮部分的层次是有效的。
然而,粒子直径等于或小于5μm的色调剂粒子对潜像承载元件表面具有很强的粘附作用,以致于以清洁方式除去转印残留色调剂很困难。此外,由于连续印刷,结果导致一些低电阻物质例如纸屑或臭氧化物和色调剂粘附在感光鼓上。
为了刮去粘附的这些低电阻物质和色调剂,日本专利申请公开No.60-32060和No.60-136752公开了一种方案,建议添加一种无机细粉作为研磨剂,此细粉以氮吸附方法测得BET比表面积为0.5~30m2/g。这对防止色调剂粘附非常有效,但是获得期望的研磨效果很困难,除非显影剂的充电稳定性有改进。因此,这不足以获得稳定的清洁。
日本专利申请公开No.61-188546、No.63-289559和No.7-261446也公开了一种有关色调剂的建议,此建议是在色调剂中添加混入两种或三种无机细粒子。然而,这种建议主要目标在于达到研磨效果以获得流动性和除掉粘附在感光鼓上的物质,并没有取得使色调剂转印性能有极大提高的效果。采用同种类型的无机细粒子(例如,二氧化硅)不仅使色调剂的流动性提供效应变得不稳定,而且使色调剂的供电性也变得不稳定,乃至可能引起色调剂散射和图象模糊现象。此外,这个建议仅涉及到无机细粒子的平均粒子直径,关于粒子大小分布仍是不清楚的。因此,仍然有引起色调剂粘附在感光鼓上的可能。
为获得更好的图像质量,日本专利申请公开No.2-222966公开了组合使用二氧化硅和氧化铝细粒子。然而,这种二氧化硅细粒子具有过大的BET比表面积,以致于很难使其在色调剂粒子之间作为一种间隔物而获得任何显著效应。
本发明的一个目的是提供一种色调剂,该色调剂能够形成具有优良图像浓度稳定性和微小图像再现性的无模糊图像,同时即使在长期使用中也不会产生色调剂损耗,本发明还提供了一种利用这种色调剂的双组分显影剂和成像方法。
本发明的另一目的是提供一种能以几乎100%的转印效率转印到转印介质的色调剂和一种采用这种色调剂的双组分显影剂和成像方法。
本发明的另一目的是提供一种色调剂,它不会引起因长期使用所带来的色调剂损耗,也不会引起显影剂携带元件的表面损耗以及潜像承载元件的表面损耗和磨损,尤其是可抑制色调剂粘附在感光鼓表面上,本发明还提供了一种利用这种色调剂的双组分显影剂和成像方法。
本发明进一步的目的是提供一种利用具有优越充电性能的充电元件的成像方法。
本发明另一进一步的目的是提供一种基本上不使用清洁装置和具有优越运转性能的成像方法。
本发明进一步的目的是提供一种能简化成像设备本身的成像方法。
本发明另一进一步的目的是提供一种成像方法,该方法使用一种具有间隔粒子和优越电荷提供性能的色调剂,还提供一种能与这种色调剂共同保持良好充电性能的充电元件。
为达到上述目的,本发明提供了一种含有色调剂粒子和外部添加剂的色调剂,这种色调剂具有:
(a)以流动式粒子图像分析仪测得的粒子成圆率分布中平均成圆率为0.920-0.995,所含成圆率小于0.950的粒子数量为2%~40%;和
(b)以Coulter法测得重均粒子直径为2.0~9.0μm;和
外部添加剂具有在色调剂粒子上的至少(i)一种以初级粒子或二级粒子的状态存在的、平均粒子长度为10mμm~400mμm和形状因子SF-1为100~130的无机细粉(A)和(ii)一种由多个粒子结合形成的、形状因子SF-1大于150的非球形无机细粉(B)。
本发明也提供了一种双组分显影剂,它含有一种至少具有色调剂粒子和外部添加剂的色调剂,和一种载体,其中:
色调剂具有:
(a)以流动式粒子图像分析仪测得的粒子成园分布中平均成圆率为0.920-0.995,所含成圆率小于0.950的粒子数量为2%~40%;和
(b)以Coulter法测得重均粒子直径为2.0~9.0μm;和
外部添加剂具有在色调剂粒子上的至少(i)一种以初级粒子或二级粒子的状态存在的、平均粒子长度为10mμm~400 mμm和形状因子SF-1为100~130的无机细粉(A)和(ii)一种由多个粒子结合形成的、形状因子SF-1大于150的非球形无机细粉(B)。
本发明也提供了一种包括以下步骤的成像方法:
(I)给待载静电潜像的潜像承载元件静电充电;
(II)在充电后的潜像承载元件上形成静电潜像;
(III)利用色调剂使潜像承载元件上的静电潜像显影,形成色调剂像;和
(IV)将潜像承载元件上形成的色调剂图像转印到转印介质;
其中,
色调剂包括色调剂粒子和外部添加剂;以及
色调剂具有:
(a)以流动式粒子图像分析仪测得的粒子成圆率分布中平均成圆率为0.920-0.995,所含成圆率小于0.950的粒子数量为2%~40%;和
(b)以Coulter法测得重均粒子直径为2.0~9.0μm;和
外部添加剂具有在色调剂粒子上的至少(i)一种以初级粒子或二级粒子的状态存在的、平均粒子长度为10mμm~400 mμm和形状因子SF-1为100~130的无机细粉(A)和(ii)一种由多个粒子结合形成的、形状因子SF-1大于150的非球形无机细粉(B)。
图1是说明一个优选的可实施本发明成像方法的成像设备实施例的简图。
图2是另一可实施本发明成像方法的成像设备实施例简图。
图3是另一可实施本发明成像方法的成像设备实施例简图。
图4是另一可实施本发明成像方法的成像设备实施例简图。
图5是另一可实施本发明成像方法的成像设备实施例简图。
图6是一个用于描述本发明成像方法的优选成像设备的简图。
图7说明用于实施例1的交流电场。
图8说明测量摩擦电量的装置。
图9说明测量体积电阻率的装置。
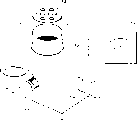
图10图示说明非球形无机细粉(B)的粒子形状。
本发明可提供一种具有优良图像浓度稳定性和微小图像再现性并能形成无模糊图像的色调剂,该色调剂即使在长期使用中也不会产生色调剂损耗。
色调剂损耗的原因有三点:色调剂粒子凸起变成细粒时的破裂;成为埋于色调剂粒子表面中的外部添加剂;以及成为充电性能不均匀的色调剂粒子。
本发明中,使用了具有特定形态和成圆率分布,并含有至少两种不同形状和粒子直径的外部添加剂细粒的色调剂粒子,这样,即使在长期使用中,也能形成具有优良图像浓度稳定性和微小图像再现性的无模糊图像,而且不会引起色调剂损耗。
本发明的实施方案在下面将被详细介绍。
本发明的色调剂具有以流动式粒子图像分析仪测得的平均成圆率0.920~0.995,优选0.950~0.995,更优选0.960~0.995。这里的流动式粒子图像分析仪指一种能统计分析被照相的粒子图像的仪器。平均成圆率是以一种根据下述成圆率确定的成圆率的算术平均计算的。
在上述的表达式中,粒子投影图像的圆周长意指由连接一种二进制代码粒子图像的边缘点形成的轮廓线长度。对应圆周长意指一种具有与二进制代码粒子图像一样的面积的圆的周长。
如果色调剂具有小于0.920的平均成圆率,则外部添加剂趋于定位在色调剂粒子表面,同时趋于导致一种不稳定的图像浓度。如果色调剂的平均成圆率大于0.995,外部添加剂难于保留在色调剂粒子表面,导致不稳定充电,趋于产生图象模糊现象。
色调剂含有成圆率小于0.950的粒子数量为2~40%,优选是3~30%。
如果色调剂含有的成圆率小于0.950的粒子数量小于2%,则色调剂趋于紧密堆集,导致不稳定充电,产生图象模糊现象。如果成圆率小于0.950的粒子数量大于40%,则色调剂的流动性降低,趋于引起图像劣化,例如细线再现性低下。
本发明中,制备具有上述特定平均成圆率和平均成圆率分布的色调剂的方法优选是一种热水浴法,该方法中把以研磨法(后面将要描述)制备的色调剂粒子分散在水中并加热;或者是一种热处理方法,该方法中使色调剂粒子通过热空气蒸汽,或一种机械冲击法,该方法中通过施加一种机械能处理粒子。在本发明中,从防止聚集作用和生产量的观点看,优选机械冲击法,尤其优选一种热机械冲击法,在这种方法中,在色调剂粒子的玻璃化转变温度Tg左右(Tg±10℃)进行处理。更优选是在色调剂粒子玻璃化转变温度Tg加减5℃的温度范围内处理。这对于在色调剂粒子表面降低半径至少10nm的孔是尤其有效的,以致于外部添加剂粒子可对提高转印效率有作用。
作为一种以上述研磨法制备色调剂粒子的方法,可以通过下述步骤制备色调剂粒子:利用一种混合设备如Henschel混合机或介质分散机均匀地分散构成材料,例如一种粘合剂树脂和一种着色剂以及任选的一种隔离剂和一种电荷控制剂,将它们制成混合物,然后以一种捏和加压捏和器或挤压机捏和混合物得到一种捏和产物,冷却这种捏和产物,然后以粉碎机如锤磨机粉碎,以机械法或通过使粉碎的产物以射流方式撞击靶体而精细地研磨所得粉碎产物,得到所需直径的色调剂粒子,把所得研磨产物进一步分级以使其粒子大小分布窄,最终得到色调剂粒子。
本发明中,除按上述研磨法将制备的色调剂粒子处理成球形的方法外,制备具有上述特定平均成圆率和特定成圆率分布色调剂的方法可以优选使用以下方法:日本专利公报No.56-13945中公开的方法,这种方法是以盘或多射流喷嘴将熔融捏和产物在空气中雾化制备球形色调剂粒子;日本专利公报No.36-10231和日本专利申请公开No.59-53856、No.59-61842公开的方法,在这些方法中,用悬浮聚合法制备聚合色调剂粒子;分散聚合方法,该方法中使用一种能够溶解可聚合单体和能少量溶解所得聚合物的含水有机溶剂制备聚合色调剂粒子;乳液聚合方法,典型的是无皂聚合,该方法是在水溶性极性聚合引发剂存在下使可聚合单体聚合制备色调剂粒子。
本发明中,悬浮聚合法是优选的,因为制备的色调剂粒子具有窄的粒子大小分布,而且能够将一种蜡作为隔离剂大量地混合到色调剂粒子中。种子聚合法也可优选用于本发明中,该方法是使单体进一步被吸附到获得的聚合色调剂粒子上,其后加入聚合引发剂实现聚合作用。
在本发明的色调剂中,如果具有聚合产生的色调剂粒子,这些色调剂粒子可由下述生产过程制备:将一种含有可聚合单体和加入其中的隔离剂(含有一种低软化点物质)、一种着色剂、一种电荷控制剂、一种聚合引发剂和其它添加剂的单体组合物通过均化器或一种超声分散器均匀溶解或分散,并用一种常用搅拌器或一种分散机如均相混合机或均化器将其分散在含有一种分散稳定剂的水相中。优选在控制搅拌速度和时间的同时进行造粒,以使单体组合物小滴具有所需的色调剂粒子大小。造粒后,搅拌保持一定的程度,这种程度下粒子状态不变,且分散稳定剂的作用能够防止粒子沉积。可以在40℃或40℃以上,通常是50~90℃的聚合温度下进行聚合反应。
这里,可通过选择分散稳定剂的类型和数量、搅拌强度、水相的pH和聚合温度来控制成圆率分布。
本发明中,色调剂粒子对应圆直径的成圆率分布是以下面的方法测得的,使用FPIA-1000流动式粒子图像分析仪,Toa Iyoudenshi K.K.制造。
为了进行测量,加入重量百分比为0.1~0.5%的表面活性剂(优选CONTAMINON,商标名;购自Wako Pure Chemical Industries,Ltd.)到离子交换水中制备溶液,这种离子交换水已经通过过滤器滤去细粉,因而在10-3cm3体积的水中在测量范围内(例如,对应圆直径为0.6μm至159.21μm以下)只含有20个或更少的粒子。将约0.02g测量样品加入到约10ml这样的溶液(20℃)中并均匀分散,即制备成样品分散体。以K.K.SMT制造的UH-50超声分散机(振动器:直径5mm的钛合金片)分散,分散时间至少为5分钟,同时适当地冷却分散介质,防止温度超过40℃。使用上述流动式粒子图像分析仪,测量对应圆直径为0.6μm至159.21μm以下的粒子的粒子大小分布和成圆率分布。
Toa Iyoudenshi K.K.出版的此测量仪器的操作手册FPIA-1000目录中和日本专利申请公开No.8-136439中概括描述了这种测量方法,如下所示:
样品分散体经过一种扁平透明导流池(厚度:200μm左右)的通道(沿流动方向)。将一种频闪放电管和一种CCD(电荷耦合器件)照像机安装在相对于导流池彼此正对的位置,以形成一能够穿过导流池厚度的光路。在样品分散体流动过程中,以间隔1/30秒的频闪光照射分散体,得到流经此池的粒子图像,从而以具有平行于导流池的特定范围的二维图像摄取每个粒子的照片。由每个粒子二维图像的面积计算出具有同样面积的圆的直径作为对应圆直径。用与每个粒子二维图像相同面积的圆的圆周长除以每个粒子二维图像的圆周长,就计算出每个粒子的成圆率。
通过把0.06μm到400μm的范围分成226个通道(一倍频程分为30通道)可以得到如下表1所示结果(相对百分频率和累积百分频率)。在实际测量中,在对应圆直径为0.6μm至159.21μm以下的范围内测量粒子。
在下表1中,每个粒子直径范围内的上限数字并不包括该数字本身,也就是表示“小于”的意思。
表1
粒子直径范围
(μm) (μm) (μm) (μm)
0.60-0.61 1.12-1.16 2.12-2.18 4.00-4.12
0.61-6.63 1.16-1.19 2.18-2.25 4.12-4.24
0.63-0.65 1.19-1.23 2.25-2.31 4.24-4.26
0.65-0.67 1.23-1.26 2.31-2.38 4.36-4.49
0.67-0.69 1.26-1.30 2.38-2.45 4.49-4.62
0.69-0.71 1.30-1.34 2.45-2.52 4.62-4.76
0.71-0.73 1.34-1.38 2.52-2.60 4.76-4.90
0.73-0.75 1.38-1.42 2.60-2.67 4.90-5.04
0.75-0.77 1.42-1.46 2.67-2.75 5.04-5.19
0.77-0.80 1.46-1.50 2.75-2.83 5.19-5.34
0.80-0.82 1.50-1.55 2.83-2.91 5.34-5.49
0.82-0.84 1.55-1.59 2.91-3.00 5.49-5.65
0.84-0.87 1.59-1.64 3.00-3.09 5.65-5.82
0.87-0.89 1.64-1.69 3.09-3.18 5.82-5.99
0.89-0.92 1.69-1.73 3.18-3.27 5.99-6.16
0.92-0.95 1.73-1.79 3.27-3.37 6.16-6.34
0.95-0.97 1.79-1.84 3.37-3.46 6.34-6.53
0.97-1.00 1.84-1.89 3.46-3.57 6.53-6.72
1.00-1.03 1.89-1.95 3.57-3.67 6.72-6.92
1.03-1.06 1.95-2.00 3.67-3.78 6.92-7.12
1.06-1.09 2.00-2.06 3.78-3.89 7.12-7.33
1.09-1.12 2.06-2.12 3.89-4.00 7.33-7.54
表1(续)
粒子直径范围
(μm) (μm) (μm) (μm)
7.54-7.76 14.20-14.62 26.75-27.53 50.37-51.84
7.76-7.99 14.62-15.04 27.53-28.33 51.84-53.36
7.99-8.22 15.04-15.48 28.33-29.16 53.36-54.91
8.22-8.46 15.48-15.93 29.16-30.01 54.91-56.52
8.46-8.71 15.93-16.40 30.01-30.89 56.52-58.17
8.71-8.96 16.40-16.88 30.89-31.79 58.17-59.86
8.96-9.22 16.88-17.37 31.79-32.72 59.86-61.61
9.22-9.49 17.37-17.88 32.72-33.67 61.61-63.41
9.49-9.77 17.88-18.40 33.67-34.65 63.41-65.26
9.77-10.05 18.40-18.94 34.65-35.67 65.26-67.16
10.05-10.35 18.94-19.49 35.67-36.71 67.16-69.12
10.35-10.65 19.49-20.06 36.71-37.78 69.12-71.14
10.65-10.96 20.06-20.65 37.78-38.88 71.14-73.22
10.96-11.28 20.65-21.25 38.88-40.02 73.22-75.36
11.28-11.61 21.25-21.87 40.02-41.18 75.36-77.56
11.61-11.95 21.87-22.51 41.18-42.39 77.56-79.82
11.95-12.30 22.51-23.16 42.39-43.62 79.82-82.15
12.30-12.66 23.16-23.84 43.62-44.90 82.15-84.55
12.66-13.03 23.84-24.54 44.90-46.21 84.55-87.01
13.03-13.41 24.54-25.25 46.21-47.56 87.01-89.55
13.41-13.80 25.25-25.99 47.56-48.94 89.55-92.17
13.80-14.20 25.99-26.75 48.94-50.37 92.17-94.86
表1(续)
粒子直径范围
(μm) (μm) (μm)
94.86-97.63 178.63-183.84 336.37-346.19
97.63-100.48 183.84-189.21 346.19-356.29
100.48-103.41 189.21-194.73 356.29-366.69
103.41-106.43 194.73-200.41 366.69-377.40
106.43-109.53 200.41-206.26 377.40-388.41
109.53-112.73 206.26-212.28 388.41-400.00
112.73-116.02 212.28-218.48
116.02-119.41 218.48-224.86
119.41-122.89 224.86-231.42
122.89-126.48 231.42-238.17
126.48-130.17 238.17-245.12
130.17-133.97 245.12-252.28
133.97-137.88 252.28-259.64
137.88-141.90 259.64-267.22
141.90-146.05 267.22-275.02
146.05-150.31 275.02-283.05
150.31-154.70 283.05-291.31
154.70-159.21 291.31-299.81
159.21-163.86 299.81-308.56
163.86-168.64 308.56-317.56
168.64-173.56 317.56-326.83
173.56-178.63 326.83-336.37
为了改进实际应用中成膜阻力和转印显影性能,本发明色调剂中色调剂粒子所具有的形状因子SF-1优选为100到150,更优选为100到130。
含有具有上述形状因子的色调剂粒子的色调剂不仅对于精确地复制微小潜像点以提高图像质量是必要的,而且该色调剂还能承受显影组件内的高机械应力从而减少对显影剂的损耗。而且,这种色调剂能良好地保证在高速复印时的转印显影性能。
当载体粒子的形状因子SF-1超过150时,这些粒子的球形度逐渐变小以致变成无定形。因此这种色调剂粒子会带来一些麻烦,使得难以达到均匀的充电性能,并可能破坏这些粒子的流动性。另外,色调剂粒子本身之间以及色调剂粒子与电荷提供元件(例如载体粒子)之间的摩擦力很大,以致于色调剂粒子破裂,变为细粒,从而使图像变得模糊,也导致了图像精密度降低。
在本发明中,用来表示形状因子的SF-1是这样得到的一个值:利用FE-SEM(S-800,一种由Hitachi公司制造的场致发射扫描电子显微镜)对粒子图像中100个粒子进行随机取样,将它们的图像信息通过某一接口传入一台图像分析仪(LUZEX-III;由Nikore公司制造)进行分析,并根据以下表达式计算数据。将得到的值定义为形状因子SF-1。
SF-1=(MXLNG)2/面积×π/4×100
这里,MXLNG代表图像中色调剂粒子的绝对最大长度,面积代表一个色调剂粒子的投影面积。
色调剂粒子的形状因子SF-1是色调剂粒子在FE-SEM中放大10000倍时进行测量的。
本发明的色调剂含有这种色调剂粒子和外部添加剂。外部添加剂含有在色调剂粒子上的至少一种以初级粒子或二级粒子状态存在的无机细粉(A)和由多个粒子合并形成的非球形无机细粉(B),从而使这种色调剂具有明显的摩擦电荷分布,其流动性得到了改进,并且可以减少由于运转引起的损耗。
更为特别的是,这种无机细粉(A)在色调剂粒子表面发生适当的移动,从而可以使这种色调剂粒子表面的充电一致,使色调剂中的电荷数量分布窄,并可以提高色调剂的流动性。非球形无机细粉(B)用作色调剂粒子的隔离物,用来抑制色调剂粒子被无机细粉(A)包埋。
通常,表面较规则和近似球形的色调剂粒子减少了逸出,这种逸出中,外加到色调剂粒子表面的外部添加剂在色调剂粒子与赋予色调剂以摩擦电荷的成分(例如:载体粒子)接触时会发生滑脱,这使外部添加剂趋于包埋在色调剂粒子表面,从而趋于导致色调剂的损耗。
本发明的色调剂是一种几乎为球形的色调剂,它的平均成圆率为0.920到0.995,并且如上所述含有以数量计为2到40%的成圆率小于0.950的粒子。但是,由于它含有无机细粉(A)和非球形无机细粉(B)作为这种色调剂粒子的外部添加剂,所以能有效地防止这种无机细粉(A)包埋在色调剂粒子表面。
在色调剂粒子上无机细粉(A)的平均粒子长度可为10mμm到400mμm,优选为15mμm到200mμm,更优选为15mμm到100mμm,其在色调剂粒子上的形状因子SF-1为100到130,优选为100到125。
如果无机细粉(A)的平均粒子长度小于10mμm,则即使它与非球形无机细粉(B)的粒子联合使用也还是往往包埋在色调剂粒子表面中,使色调剂损耗,不利地使色调剂浓度控制稳定性降低。如果无机细粉(A)的的平均粒子长度大于400mμm,则难以使色调剂获得良好的流动性,势必使色调剂充电不一致,从而势必导致色调剂发生散射并使图像变得模糊。
如果无机细粉(A)的形状因子SF-1大于130,则无机细粉(A)难以在色调剂粒子表面移动,势必导致色调剂的流动性降低。
色调剂粒子上的无机细粉(A)的形状因子SF-1是在FE-SEM中被放大100,000倍测量的。
无机细粉(A)的粒子长度/宽度比优选为小于或等于1.5,更优选为小于或等于1.3,以使无机细粉(A)能够容易地在色调剂粒子表面移动并改进色调剂的流动性。
根据BET法由氮吸附测得无机细粉(A)的比表面积(BET比表面积)优选为60到230m2/g,更优选为70到180m2/g,以使色调剂具有良好的充电特性和流动性,从而能够实现高图像质量和高图像浓度。
如果无机细粉(A)的BET比表面积小于60m2/g,则色调剂流动性小,势必形成细线再现性差的图像。如果无机细粉(A)的BET比表面积大于230m2/g,则色调剂的充电特性不稳定,导致色调剂发生散射的问题,特别是当色调剂长时间处于高湿度的环境中时。
本发明在色调剂粒子上所用的非球形无机细粉(B)的形状因子SF-1大于150,优选为大于190,更优选为大于200,以抑制无机细粉(A)包埋在色调剂粒子表面中。
如果非球形无机细粉(B)的形状因子SF-1小于等于150,则非球形无机细粉(B)本身趋于包埋在色调剂粒子表面中,从而不能很有效地抑制无机细粉(A)包埋在色调剂粒子表面中。
色调剂粒子上非球形无机细粉(B)的形状因子SF-1是在FE-SEM中被放到100,000倍来测量的。
在色调剂粒子上的非球形无机细粉(B)具有的长度/宽度比优选为大于或等于1.7,更优选为大于或等于2.0,进一步优选为大于或等于3.0,以高度有效地抑制无机细粉(A)包埋在色调剂粒子表面中。
优选非球形无机细粉(B)具有的粒子的平均长度大于无机细粉(A)的平均长度,优选为至少20mμm,更优选为至少40mμm,以抑制无机细粉(A)包埋在色调剂粒子表面中。
色调剂粒子上的非球形无机细粉(B)所具有的平均粒子长度优选为120到600mμm,更优选为130到500mμm。
如果非球形无机细粉(B)的平均粒子长度小于120mμm,则它所具有的抑制无机细粉(A)包埋在色调剂粒子表面中的隔离物作用减小,因而色调剂的显影转印性能低,导致图像浓度的降低。如果非球形无机细粉(B)的平均粒子长度大于600mμm,则可以获得上述的隔离物作用,但非球形无机细粉(B)会从色调剂粒子表面逸出,从而势必导致感光鼓上出现刮擦的痕迹。
在本发明中,根据电子显微镜放大的色调剂图像观察,色调剂粒子表面上存在的无机细粉(A)数量优选为平均每0.5×0.5μm单位面积上含有至少5个粒子,更优选为至少7个粒子,进一步优选为至少10个粒子,色调剂粒子表面上存在的非球形无机细粉(B)数量优选为平均每1.0×1.0μm单位面积上含有1到30个粒子,更优选为1到25个粒子,进一步优选为5到25个粒子。色调剂粒子表面上存在的无机细粉(A)的粒子数量指的是初级粒子和二级粒子的总数。
如果色调剂粒子表面上存在的无机细粉(A)的粒子数小于上述平均5个,则色调剂流动性不足,从而势必导致图像浓度的降低。
如果色调剂粒子表面上存在的非球形无机细粉(B)的粒子在上述面积中的平均个数小于1个,则非球形无机细粉(B)不能继续发挥隔离物的作用。如果粒子数大于30,则粉末(B)势必从色调剂粒子表面逸出,从而势必产生感光鼓上出现刮擦痕迹的问题。
外部添加剂的平均粒子长度,其粒子的长度/宽度之比以及色调剂粒子表面的外部添加剂的粒子数通过以下方法进行测定。
利用FE-SEM(S-800,Hitachi公司制造)将色调剂粒子表面放大100,000倍后拍摄得到放大的图像,利用该放大的图像测定无机细粉(A)的相关数值。
首先,色调剂粒子上的无机细粉(A)的平均长度是这样测定的,测定10个视野范围内可在放大的图像上看到存在于色调剂粒子上的每个无机细粉(A)的粒子长度,将这些粒子长度的平均值作为平均长度。类似地,每个无机细粉(A)的粒子宽度的平均值和长度/宽度比也可被测定。这里,粒子的长度对应于两条平行线之间的距离,该距离是在无机细粉(A)粒子轮廓上所画的一系列平行切线之间的最大距离,粒子的宽度对应于一系列这种平行切线之间的最小距离。
色调剂粒子表面上无机细粉(A)的粒子数量是这样测定的,在放大的图像上的10个视野中计算每0.5μm×0.5μm(在放大100,000倍图像下为50mm×50mm)单位面积下色调剂粒子表面的无机细粉(A)的粒子数量,计算它的平均值。当计算无机细粉(A)的粒子数量时,计数在对应于放大图像中心处0.5μm×0.5μm面积中以初级粒子或二级粒子状态存在的无机细粉(A)粒子数。
利用FE-SEM(S-800,Hitachi公司制造)将色调剂粒子表面放大30,000倍拍摄得到放大的图像,利用该放大的图像测量非球形无机细粉(B)的相关数值。
首先,非球形无机细粉(B)的平均长度是这样测定的,测量放大图像上10个视野范围内每个非球形无机细粉(B)的粒子长度,将这些粒子长度的平均值作为平均长度直径。类似地,每个非球形无机细粉(B)的粒子宽度的平均值和长度/宽度比也可被测定。这里,粒子的长度对应于两条平行线之间的距离,该距离是在非球形无机细粉(B)粒子轮廓上所画的一系列平行切线之间的最大距离,粒子的宽度对应于一系列这种平行切线之间的最小距离。
色调剂粒子表面上非球形无机细粉(B)的粒子数量是这样测定的,在放大的图像上的10个视野范围内计算每1.0μm×1.0μm(在放大30,000倍图像下为300mm×300mm)单位面积下色调剂粒子表面上非球形无机细粉(B)的粒子数量,计算它的平均值。当计算非球形无机细粉(B)的粒子数量时,它是计算存在于对应于放大图像中心处1.0μm×1.0μm的面积中的非球形无机细粉(B)。
为了将由电子显微镜放大的图像中的无机细粉(A)和非球形无机细粉(B)区分开来,采用一种方法可以独立检测到无机细粉(A)和非球形无机细粉(B),在这种方法中,当这些无机细粉的组成有差异时,这些无机细粉粒子存在的位置在FE-SEM上确定,并用X射线微量分析仪只检测特定的成分。或者,当这些无机细粉的粒子形状有明显差别时,可以依据在由电子显微镜放大的图像上粒子形状的差别来进行判断。两种方法都可以使用。
根据BET氮吸附法测得非球形无机细粉(B)的比表面积(BET比表面积)优选为20到90m2/g,更优选为25到80m2/g,以使粉末(B)易于均匀地分散于色调剂粒子表面,也可以使该非球形无机细粉(B)长时间地发挥作为隔离物的作用。
如果非球形无机细粉(B)的BET比表面积小于20m2/g,粉末(B)势必从感光鼓上的色调剂中逸出,从而在感光鼓上形成刮擦的痕迹。如果它具有的BET比表面积大于90m2/g,则粉末(B)在感光鼓上作为隔离物的作用减小,势必降低转印性能,尤其是在低湿度的环境里。
无机细粉(A)和非球形无机细粉(B)的BET比表面积可以利用Autosorb I(由Quantach Rome公司制造的比表面积测量仪)通过以下方法测定。
在一个小室中放入大约0.1g称好的试样,在40℃温度下让其在小于或等于1.0×10-3mmHg的真空度下至少脱气12小时。然后,让试样在被液氮冷却的状态下吸附氮气,接着用多测试点法测定比表面积的值。
本发明所用的色调剂的外部添加剂可以是任何材料,只要它在色调剂粒子表面的分散状态能得到满足,例如,它可以包括象氧化铝,二氧化钛,二氧化硅,氧化锆和氧化镁之类的氧化物,以及碳化硅、氮化硅、氮化硼,氮化铝,碳酸镁和有机硅化合物。
其中,氧化铝,二氧化钛,氧化锆,氧化镁或它们的被二氧化硅处理过的细粒子,以及氮化硅作为无机细粉(A)是优选的,因为它们不受温度和湿度的影响而且使色调剂的充电稳定。更优选为氧化铝细粒子、二氧化钛细粒子或者它们的被二氧化硅处理过的细粒子,以用来改进色调剂的流动性。
对于如何制成这种细粒子没有特别的限制,可使用的一种方法是,在气相中对卤化物或醇盐进行氧化,另一种方法为在水存在下水解形成。优选在足够低而不使初级粒子聚集的温度下进行焙烧。
在本发明中,在低温时焙烧得到的不定形或锥型钛白粉和不定形或γ氧化铝由于易于使之以球形和初级粒子形式单分散,因此是优选的。
优选可将无机细粉(A)进一步作疏水处理,以使色调剂的电荷数量较少依赖于诸如温度和湿度等环境因素,并且阻止粉末(A)从色调剂粒子表面逸出。这种疏水处理所用的试剂可包括偶合试剂,如硅烷偶合试剂,钛偶合试剂和铝偶合试剂,以及油,如硅油,氟油和各种改性油。
在上述疏水处理试剂中,偶合试剂更为优选,因为它们与残余基团或无机细粉上吸附的水发生反应,使无机细粉(A)获得均匀的处理,从而使色调剂充电稳定,并具有流动性。
因此,作为本发明所用的无机细粉(A),在水解硅烷偶合试剂的同时进行过表面处理的细氧化铝粒子或者细二氧化钛粒子是非常有效的,因为它们使充电稳定并能带来流动性。
经过疏水处理的无机细粉(A)的疏水性优选为20到80%,更优选为40到80%。
如果无机细粉(A)的疏水性小于20%,则当色调剂较长时间处于高湿度环境中时,电荷将会大量减少,这样硬件上需要一个用于加速充电作用的机构,导致设备的复杂。如果它的疏水性大于80%,则难以控制无机细粉自身的充电,势必造成在低湿度环境下色调剂的过度充电。
经过疏水处理的无机细粉(A)在波长等于400nm时,它具有的透光率优选为40%或更大。
更为具体的是,即使本发明所用的无机细粉(A)具有小的初级粒子直径,当它实际掺入色调剂中时,这种无机细粉(A)并不一定以初级粒子的形式分散,有时它以二级粒子的形式分散。因而,不管初级粒子直径多小,如果这种作为二级粒子的粒子具有较大的有效直径,本发明的效果将会减弱。但是,在400nm(在可见区域最短的波长)下有较高的透光率的无机细粉(A)相应地具有较小的二级粒子直径。因此,可以预见这种无机细粉(A)在提供流动性能和在OHP(高架投影仪)中投影图像的清晰方面具有良好效果。
选用400nm的原因在于它是紫外与可见光边界区域的波长,而且人们认为光能够通过直径不大于光波长的1/2的粒子。从这一点来说,任何在大于400nm波长下的透光率当然要更高,但这没有什么意义。
在本发明中,作为一种对无机细粉(A)进行疏水处理的方法,优选这样一种方法,其中在水的存在下对无机细粉(A)进行表面处理,同时用机械方法将它们分散,从而在水解偶合试剂时形成初级粒子。这种处理使粒子本身不易发生聚集,并且使这些粒子之间静电排斥,因此,这种无机细粉(A)可基本以初级粒子的状态进行表面处理。
由于在水的存在下在水解偶合试剂的同时处理粒子表面时采用机械力以使无机细粉(A)被分散成初级粒子,因此没有必要使用诸如氯化硅烷或硅氮烷之类可能会产生气体的偶合试剂。而且,也有可能使用因为在气相中粒子发生聚集而一直不能用的高度粘稠的偶合试剂,从而粒子能被极有效地疏水化。
上述的偶合试剂可以含有硅烷偶合试剂和钛偶合试剂中的任意一种。下式所示的硅烷偶合试剂是尤为优选的:
RmSiYn
其中R是烷氧基;m为1到3的整数,Y为烷基或含有乙烯基的烃基,缩水甘油氧基(glycidoxyl)或甲基丙烯酸基,n为1到3的整数;而且该偶合试剂可包括例如乙烯基三甲氧基硅烷,乙烯基三乙氧基硅烷,γ-甲基丙烯酰基氧丙基三甲氧基硅烷,乙烯基三乙酰氧基硅烷,甲基三甲氧基硅烷,甲基三乙氧基硅烷,异丁基三甲氧基硅烷,二甲基二甲氧基硅烷,二甲基二乙氧基硅烷,三甲基甲氧基硅烷,羟丙基三甲氧基硅烷,苯基三甲氧基硅烷,正十六烷基三甲氧基硅烷和正十八烷基三甲氧基硅烷。
这种偶合试剂可更优选为以CaH2a+1-Si(OCbH2b+1)3表示的那些,其中a为4到12而b为1到3。
这里,如果在分子式中a小于4,则处理更容易进行,但却不能得到满意的疏水性。如果a大于12,则可以获得满意的疏水性,但更容易发生粒子的聚集,这提供流动性的性能降低。
如果b大于3,则反应活性降低,导致粒子的疏水性不足。因此,在上述分子式中a的值应为4到12,优选为4到8,b的值应为1到3,优选为1到2。
用这种处理试剂对无机细粉(A)进行处理,其用量为100重量份的无机细粉(A)使用1到50重量份,优选3到40重量份,以使处理均匀进行而不造成任何聚集,而无机细粉(A)的疏水性可为20到98%,优选为30到90%,更优选为40到80%。
在本发明中,非球形无机细粉(B)优选选自二氧化硅,氧化铝,二氧化钛或它们的双氧化物的细粉,以改进充电稳定性、显影性能,流动性和储存稳定性。特别优选二氧化硅细粉末,因为可以通过原材料和氧化条件(如氧化温度)将初级粒子的聚集任意地控制到某种程度。例如,二氧化硅细粉末包括所谓的干法二氧化硅或煅制二氧化硅(通过在气相氧化硅的卤化物或烷氧化物得到)和所谓的湿法二氧化硅(由烷氧化物或水玻璃制得),二者都可以使用。优选干法二氧化硅,因为在它的表面和内部具有较少的硅烷醇基,不会遗留反应残余物(如Na2O和SO3 2-)。在干法二氧化硅中,在其生产步骤中还可以与卤化硅一起使用其他金属卤化物,如氯化铝或者氯化钛,以获得二氧化硅与其他金属氧化物的复合粉末。细二氧化硅粉末也包括这些物质。
关于粒子的形状,这些粒子可能不是非球形粒子(如棒状粒子或块状粒子),但可能是具有如图10所示的凹凸不平的部分或凹痕的非球形粒子。上述情况是优选的,因为无机细粉(A)可以免于在色调剂粒子表面被包埋,同时可以防止显影剂的密集,因此这种显影剂的松密度只发生很小变化。
这种非球形无机氧化物细粒子优选通过以下方法得到。
以二氧化硅细粉末为例,将一种硅卤化物在气相中氧化形成二氧化硅细粉末,让这种二氧化硅细粉末接受疏水化处理而得到非球形二氧化硅细粉。特别是当在气相中氧化时,优选在高到足以使二氧化硅初级粒子发生聚集的温度下进行焙烧。
特别优选通过以下方法得到的非球形无机细粉(B),将由初级粒子相互聚集而成的聚集粒子分类,挑出其中相对粗的粒子,调整粒子的大小分布以满足它们在色调剂粒子表面存在状态中的平均长度条件。
在本发明中,以100重量份色调剂计,该色调剂含有0.1-2.0重量份的无机细粉(A),以使色调剂的电荷数量稳定,从提供流动性而言,该无机细粉(A)的含量优选为0.2到2.0重量份,从改善定影性能而言,更优选为0.2到1.5重量份,为了使显影剂的松密度稳定,非球形无机细粉(B)的量为0.3到3.0重量份,从避免感光鼓产生刮痕而言,优选0.3到2.5重量份,从高湿度状态下的储存稳定性而言,更优选为0.3到2.0重量份,从OHP透明度而言,进一步优选为0.3到1.5重量份。
如果色调剂中含有的无机细粉(A)量少于0.1重量份,则色调剂的流动性不足,势必导致图像浓度的降低。如果它的数量超过20重量份,则色调剂势必充电不稳定,特别当较长时间处于高湿度环境中时,从而势必使色调剂发生散射。
如果色调剂中含有的非球形无机细粉(B)的数量少于0.3重量份,则不能有效地防止无机细粉(A)在色调剂粒子中被包埋。如果它的数量大于3.0重量份,势必使感光鼓上发生擦痕,从而产生错误的图像。
本发明中,对于外加入由聚合反应得到的聚合色调剂粒子中的外部添加剂,一个优选的具体例子是至少用氧化铝细粒子作为无机细粉(A),用二氧化硅细粒子作为非球形无机细粉(B)。
外加的氧化铝细粒子在粒子大小分布中含有的粒子直径至少为平均粒子直径的2倍的粒子数优选为0到5%,而外加的细二氧化硅粒子在组成聚集粒子的粒子的粒子大小分布中含有的粒子直径为平均初级粒子直径的2-3倍的粒子数优选为5到15%。
本发明的外部添加剂的特征是,氧化铝细粒子具有很窄的粒子大小分布,而组成细二氧化硅粒子的聚集粒子的粒子具有相对宽的粒子大小分布。氧化铝细粒子具有高度的流动性提供能力,以及具有能极大地影响色调剂的充电性以大大降低与湿度极为相关的环境之间的充电差别性的作用。
本发明者发现,除了聚合色调剂粒子的形状因子和外部添加剂的粒径比(长度/宽度比)之外,使氧化铝细粒子具有窄的粒子大小分布也能使充电高度稳定,并且能保证由色调剂粒子之间摩擦产生的色调剂粒子表面电荷的均匀性。本发明者同时还发现,作为本发明的最显著效果,通过使氧化铝细粒子具有窄的粒子大小分布,可以得到一种高水平的转印性能。这些效果,如同后面将要描述的,与组成二氧化硅细粒子的聚集粒子的粒子的粒子大小分布有关,有助于使得这些粒子在色调剂粒子之间有效地发挥隔离物粒子的作用,因为氧化铝细粒子由均匀的粒子组成,并且粒子直径小。因此,可以认为将这些粒子外加于色调剂粒子表面之后也不易于形成聚集粒子。如果氧化铝细粒子的数量分布不在上述的范围内,则它们势必形成聚集粒子或者聚集后难以得到本发明所需的效果。
此外,使组成二氧化硅细粒子的聚集粒子的粒子具有相对宽的粒子大小分布。这样,它们被认为具有充分的电荷提供性能而与色调剂的粒子大小分布无关。在给色调剂提供电荷的能力方面,二氧化硅细粒子比氧化铝细粒子强。因此,前者能够分散地提供对所有粒子都一致的电荷,而与不但具有小粒子而且还具有相对较大的粒子的色调剂粒子无关,同时还能获得在氧化铝细粒子中得到的隔离物效应。至于它们的粒子大小分布,如果这种分布在上述范围的下限之外,则二氧化硅细粒子趋于粘附在感光鼓表面,而它们所粘附的区域作为中心而导致色调剂成膜。如果这种分布在上述范围的上限之外,则可能导致色调剂的流动性大大受损,并且在长时间地重复地复印操作时使显影剂损耗。同样根据这些事实,本发明者发现二氧化硅细粒子能使色调剂均匀地被充电,并保持色调剂的流动性,因为该色调剂具有一个宽的粒子大小分布。
本发明所用的氧化铝细粒子和二氧化硅细粒子所具有的BET比表面积对于氧化铝细粒子来说优选为60到150m2/g,对于二氧化硅细粒子来说优选为20到70m2/g。如果两种粒子的BET比表面积值在上述范围之外,则得不到上述所需的粒子直径并导致图像质量受到影响。
氧化铝细粒子优选是以下述方法获得的一种氧化铝细粉为母体材料制得的氧化铝细粒子,该方法是在1,000到1,200℃的温度范围内热分解碳酸铝铵氢氧化物,随后在一种溶液中进行疏水化处理。
氧化铝细粉母体原料优选为日本专利申请公开号61-146794公开的γ氧化铝,或者是在较低温度下焙烧的无定形氧化铝。
这种氧化铝细粉可优选通过在例如氧气气氛中于1000到1200℃温度范围内焙烧碳酸铝铵氢氧化物(用式NH4AlO(OH)HCO3或NH4AlCO3(OH)2表示)得到。更为具体的是,优选氧化铝细粉是在下述化学反应之后得到的氧化铝细粉。
这里,选择1000到1200℃作为焙烧的温度范围,这是因为在该温度范围内能得到本发明所希望的粒子直径。
如果焙烧温度高于1200℃,则形成的氧化铝细粉中α氧化铝的比例突然增大。当然,这种粉末在结构上增长使得具有大的初级粒子直径和小的BET比表面积。此外,粉末粒子以更大的强度相互聚集,在处理步骤中必须提供大的能量来分散母体材料。变成这种状态的粉末不管处理步骤如何优化,都不再会成为具有较少聚集粒子的细粉。
如果焙烧的温度小于1000℃,则这种粉末的粒子直径会小于期望的大小,并且使其发挥不了足够的隔离物的作用,也难以实现高转印性能。
可以根据表面改性的目的来选择本发明中所用的氧化铝细粒子的表面疏水处理试剂,例如,为了控制充电性能,也可以是为了在高湿度环境中充电的稳定性和反应性。例如,可以使用硅烷类有机化合物,如烷氧基硅烷,硅氧烷,硅烷和硅油,这些物质在反应和处理温度下本身不会热分解。
如上述所优选的那些试剂,可以使用偶合试剂烷基烷氧硅烷,这种试剂具有挥发性,并且同时具有疏水基和富有反应活性的结合基。
为了计算氧化铝细粒的平均初级粒子直径和组成二氧化硅细粒的聚集粒子的粒子的平均初级粒子直径,将粒子分散于环氧树脂中以包埋于其中,然后切成薄的切片,利用一种透射电子显微镜(TEM)(放大10,000到100,000倍)获得如此分散的粒子的照相图像。在这种照相图像上,随机取样20到50个粒子。然后,对于球形粒子来说,将它们的直径作为粒子直径,而对于扁平的粒子,将它们的长度作为粒子直径。求得这些粒子直径的算术平均值以计算平均初级粒子直径。
在本发明中,一种优选的具体方案是,除了具有以上述方式组成的无机细粉(A)和非球形无机细粉(B)之外,还加入了无机或有机的近似球形的粒子,这种粒子具有大于或等于50mμm的初级粒子直径(且优选具有小于50m2/g的比表面积),以改进转印性能和/或清洁性能。例如,可以优选使用球形二氧化硅粒子,球形聚甲基倍半硅氧烷(Polymethylsilses-quioxane)粒子或者球形树脂粒子。
在本发明的色调剂中,还可以使用少量的其他添加剂粒子,只要这些粒子基本上不会对色调剂起到相反的作用。这些其他的添加剂粒子可以包括润滑粉(例如聚四氟乙烯粉,硬脂酸锌粉和聚偏二氟乙烯粉);研磨剂(例如氧化铈粉,碳化硅粉和钛酸锶粉);抗结块剂(例如氧化钛粉和氧化铝粉);导电性提供试剂(例如炭黑粉,氧化锌粉和氧化锡粉);显影性改进剂(例如反极性有机细粒子和反极性无机细粒子)。
在本发明中,为了真实地显影精细的潜像点以提高图像的质量,色调剂优选为具有小的粒子直径。具体地说,这种色调剂的重均粒子直径以用Coulter计数器测定为2.0μm到9.0μm,优选为4.0μm到8.0μm。这种色调剂也可以优选为具有小于或等于35%的数量分布变异系数,更优选为5到30%。
如果色调剂的重均粒子直径小于2μm,则它的转印效率低到在感光鼓上出现大量转印残留色调剂,不仅使图像不均匀,而且在感光鼓上发生熔融-粘着。如果色调剂的重均粒子直径大于9μm,则趋于导致图像质量的降低(例如,在特征线条图像周围出现黑点),也趋于造成色调剂在各种元件上发生熔融-粘着。
如果色调剂的数量分布变异系数大于35%,则趋于使色调剂充电不均匀,从而造成图像模糊不清。
本发明色调剂粒子大小分布是用TA-II Coulter计数器测定的。可使用Coulter Multisizer(Coulter电子仪器公司制造)。用一级氯化钠制备1%的NaCl水溶液作为一种电解质溶液。例如,可以使用ISOTON R-II(商品名,Coulter科学日本公司制造)。测定是这样进行的:往100到150ml的上述电解质水溶液中加入0.1到5ml的表面活性剂作为分散剂,优选为烷基苯磺酸盐,再加入2到20mg的待试样品。样品悬浮于电解质溶液中,利用超声波分散仪对该溶液进行约1到3分钟的分散处理。将一台用以输出数量分布和体积分布的接口(由Nikkaki K.K.公司制造)与一台个人计算机PC9801(NEC.公司制造)相连接。通过利用上述的测量装置(采用100μm的孔径)测量色调剂粒子的体积和数量,计算出直径为大于或等于2.00μm的色调剂粒子的数量分布和体积分布。
接着,由体积分布确定基于重量的重均粒子直径(D4)(以每个通道的中间值作为每个通道的代表值),并确定数量分布变异系数,作为本发明的各种数值。
数量分布变异系数是根据下式计算出来的。
变异系数(%)=(数量分布的标准偏差/数均粒子直径)×100
所用的孔道有13个,它们的大小分别为2.00μm至小于2.52μm,2.52μm至小于3.17μm,3.17μm至小于4.00μm,4.00μm至小于5.04μm,5.04μm至小于6.35μm,6.35μm至小于8.00μm,8.00μm至小于10.08μm,10.08μm至小于12.70μm,12.70μm至16.00μm,16.00μm至小于20.20μm,20.20μm至25.40μm,25.40μm至小于32.00μm,以及32.00μm至小于40.30μm。
本发明的色调剂粒子至少含有一种粘结剂树脂和一种着色剂。
本发明所用的粘结剂树脂可以包括苯乙烯及其衍生物的均聚物如聚苯乙烯和聚乙烯基甲苯;苯乙烯的共聚物,如苯乙烯-丙烯共聚物,苯乙烯-乙烯基甲苯共聚物,苯乙烯-乙烯基萘共聚物,苯乙烯-丙烯酸甲酯共聚物,苯乙烯-丙烯酸乙酯的共聚物,苯乙烯-丙烯酸丁酯的共聚物,苯乙烯-丙烯酸辛酯的共聚物,苯乙烯-丙烯酸二甲氨基乙酯的共聚物,苯乙烯-甲基丙烯酸甲酯的共聚物,苯乙烯-甲基丙烯酸乙酯的共聚物,苯乙烯-甲基丙烯酸丁酯的共聚物,苯乙烯-甲基丙烯酸二甲氨基乙酯的共聚物,苯乙烯-甲基乙烯基醚的共聚物,苯乙烯-乙基乙烯基醚的共聚物,苯乙烯-甲基乙烯基酮的共聚物,苯乙烯-丁二烯的共聚物,苯乙烯-异戊二烯的共聚物,苯乙烯-马来酸的共聚物和苯乙烯-马来酸酯共聚物;聚丙烯或甲基丙烯酸树脂如聚甲基丙烯酸酯,聚甲基丙烯酸甲酯,聚甲基丙烯酸丁酯,聚丙烯酸酯和聚丙烯酸甲酯;聚乙酸乙烯酯;聚乙烯;聚丙烯;聚乙烯醇缩丁醛;聚酯树脂;松香;改性松香;萜烯树脂;酚树酯;脂族或脂环族烃的树脂;芳香族石油树脂;石蜡和巴西棕榈蜡。上述任何一种可以单独使用或混合使用。
在本发明色调剂粒子中,可选择性地使用一种低软化点的称为蜡的物质。
本发明所用的低软化点物质可以包括聚亚甲基蜡(如石蜡),聚烯烃蜡,微晶蜡和费-托合成过程中得到的蜡,酰胺蜡,高级脂肪酸,长链醇,酯蜡,矿脂,巴西棕榈蜡,酮,硬化蓖麻油,植物蜡,动物蜡,矿物蜡,以及它们的衍生物,如接枝化合物或嵌段化合物。优选的是已从中除去低分子量的成分并在DSC吸热曲线上具有明显的最大吸热峰的那些。
优选的蜡是含15到100个碳原子的直链烷基醇,直链脂肪酸,直链酰胺,直链酯或褐煤酸衍生物。也优选已从中除去杂质如液体脂肪酸的任何蜡。
更为优选的蜡是低分子量的烯属烃聚合物,这些聚合物是通过在高压条件下自由基聚合而得到或在齐格勒催化剂或其他催化剂的存在下低压时聚合而得到;通过热分解高分子量的烯属烃聚合物得到的烯属烃聚合物;通过分离和纯化在烯属烃发生聚合时以副产物形成的低分子量烯属烃聚合物得到的那些物质;以及通过下述方法获得的聚亚甲基蜡,该方法是对一种烃聚合物的蒸馏残留物的特定组分进行分馏提取,这种烃聚合物是通过Arge法从由一氧化碳和氢气组成的合成气获得,或通过氢化蒸馏残留物得到的合成烃。在这些蜡中可以加入一些抗氧剂。
在本发明中,这种蜡可以是一种酯蜡,这种酯蜡主要成分为由具有15到45个碳原子的长链烷基醇和具有15到45个碳原子的长链烷基羧酸反应得到的酯化化合物。特别优选这种物质是考虑到在使用高架投影仪时形成高透明度的投影图像和良好的全色投影图像。
在本发明中作为隔离剂组分的低软化点物质优选具有重均分子量(Mw)为300到3,000,更优选重均分子量(Mw)为500到2500和重均分子重量/数均分子量(Mw/Mn)不超过3.0,更优选为1.0到2.0。
如果低软化点物质的Mw小于300,则色调剂具有低抗粘性。如果低软化点物质的Mw大于3,000,则会显示出它的可结晶性,导致低透明性。如果低软化点物质的Mw/Mn大于3.0,则色调剂流动性低,势必使图像浓度不均匀,也势必会造成充电元件的污染。
本发明所用的隔离剂在根据ASTM D3418-8通过DSC(差示扫描量热法)所测得的吸热曲线上,优选在温度为40℃到120℃范围内具有一个吸热的主峰,更优选为在40℃到90℃范围内,进一步优选为在40℃到85℃范围内具有一个吸热的主峰。如果它在40℃以下出现吸热主峰,则低软化点物质的自粘力弱,导致不期望的高温抗透印(anti-offset)特性降低。如果它的吸热主峰位于120℃以上,不理想的是色调剂具有较高的定影温度,特别是当通过聚合反应产生色调剂粒子时,如果吸热主峰温度很高,则这种低软化点物质可能会在形成粒子的过程中发生沉积,不良地扰乱了该悬浮体系。
本发明采用DSC测量法,例如选用了Perkin Elmer公司制造的DSC-7。在这种仪器的检测部分的温度是根据铟和锌的熔点来进行纠正的,热量是用铟的熔化热进行纠正的。将样品放于由铝制成的面板上,设置一个空的面板用作对比,使测量在温度为20℃到200℃,升温速率为10℃/分的条件下进行。
在本发明中,以色调剂粒子的重量计,优选色调剂粒子含有1到30%(重量)的低软化点物质,更优选5到30%(重量)。如果色调剂粒子所含的低软化点物质少于1%(重量),则导致色调剂的抗透印性降低。低软化点物质的含量若大于30%(重量),则通过聚合反应得到的色调剂粒子在成粒时发生相互聚集,趋于使粒子大小分布较宽。
已知的电荷控制剂都可以在本发明中使用。对于彩色色调剂来说,特别优选使用无色,使色调剂充电速度较高并且能够稳定地保留恒定的电荷数的电荷控制剂。当采用由聚合反应得到的色调剂粒子时,特别优选使用既不会有聚合抑制作用,又不溶于含水分散介质中的电荷控制剂。
作为负电荷控制剂,这种电荷控制剂包括水杨酸金属化合物,萘甲酸金属化合物,二元羧酸金属化合物,在侧链上带有磺酸或羧酸的聚合物类型的化合物,硼化合物,尿素化合物,硅化合物,carixarene,可以使用上述任何一种物质。作为正电荷控制剂,可以包括季铵盐,侧链上带有这种季铵盐的聚合类型的化合物,胍化合物和咪唑化合物,可以使用上述任何一种物质。
以100重量份的粘合树脂为基准计,电荷控制剂的用量优选为0.5到10重量份。但在本发明中,并非必须加入电荷控制剂。当采用双组份显影时,可以利用载体摩擦充电。当采用单组份显影(非磁性的单组分刮板涂覆显影)时,可利用调节色调剂层厚度用的刮板部件或携带色调剂用的套筒部件摩擦充电。因此,色调剂粒子不一定必须含有电荷控制剂。
本发明所用的粘合剂树脂可以包括苯乙烯及其衍生物的均聚物如聚苯乙烯,聚对氯苯乙烯和聚乙烯基甲苯;苯乙烯的共聚物,如苯乙烯-对氯苯乙烯共聚物,苯乙烯-乙烯基甲苯共聚物,苯乙烯-乙烯基萘的共聚物,苯乙烯-丙烯酸酯共聚物,苯乙烯-甲基丙烯酸酯共聚物,苯乙烯-α-氯甲基丙烯酸甲酯共聚物,苯乙烯-丙烯腈共聚物,苯乙烯-甲基乙烯基醚共聚物,苯乙烯-乙基乙烯基醚共聚物,苯乙烯-甲基乙烯基酮共聚物,苯乙烯-丁二烯共聚物,苯乙烯-异戊二烯共聚物和苯乙烯-丙烯腈-茚共聚物;聚氯乙烯;酚树脂;天然树脂改性酚树酯;天然树脂改性马来酸树脂;丙烯酸树脂;甲基丙烯酸树脂;聚乙酸乙烯酯;硅氧烷树脂;聚酯树脂;聚脲;聚酰胺树脂;呋喃树脂;环氧树脂;二甲苯树脂;聚乙烯醇缩丁醛;萜烯树脂;香豆酮(cumarone)茚树脂;石油树脂。同样,优选的粘合剂树脂为交联的苯乙烯树脂。
作为在苯乙烯共聚物中能与苯乙烯单体发生共聚的共聚用单体,乙烯基单体可以单独使用或者以两种或两种以上的单体联合使用。乙烯基单体可包括具有双键的一元羧酸和它们的衍生物,例如,丙烯酸,丙烯酸甲酯,丙烯酸乙酯,丙烯酸丁酯,丙烯酸十二烷基酯,丙烯酸辛酯,丙烯酸2-乙基己基酯,丙烯酸苯基酯,甲基丙烯酸,甲基丙烯酸甲酯,甲基丙烯酸乙酯,甲基丙烯酸丁酯,甲基丙烯酸辛酯,丙烯腈,甲基丙烯腈和丙烯酰胺;具有一个双键的二元羧酸及其衍生物,例如,马来酸,马来酸丁酯,马来酸甲酯和马来酸二甲酯;乙烯基酯,例如,氯乙烯,乙酸乙烯基酯和苯甲酸乙烯酯;乙烯属烯烃,例如,乙烯,丙烯和丁烯;乙烯基酮,如甲基乙烯基酮和己基乙烯基酮;乙烯基醚,如甲基乙烯基醚,乙基乙烯基醚和异丁基乙烯基醚。
在本发明中,可使用至少具有两根可聚合的双键的化合物作为交联剂。例如,它们包括芳族二乙烯基化合物,如二乙烯基苯和二乙烯基萘;具有两根双键的羧酸酯,如二丙烯酸乙二醇酯,二甲基丙烯酸乙二醇酯和二甲基丙烯酸1,3-丁二醇酯;二乙烯基化合物,如二乙烯基苯胺,二乙烯基醚,二乙烯硫醚和二乙烯基砜;以及至少具有三个乙烯基的化合物。它们之中任何一个都可以单独或混合使用。
除了上述苯乙烯共聚物之外,特别优选还进一步加入一种极性树脂,例如苯乙烯-丙烯酸或-甲基丙烯酸的共聚物,苯乙烯-马来酸的共聚物或饱和聚酯树脂。
用于压力定影的色调剂的粘合剂树脂可包括低分子量的聚乙烯,低分子量的聚丙烯,乙烯-乙酸乙烯酯的共聚物,乙烯-丙烯酸酯的共聚物,高级脂肪酸,聚酰胺树脂和聚酯树脂。它们之中任何一个都可以单独或混合使用。特别是当色调剂粒子是由聚合反应产生时,优选使用那些既不会有聚合反应抑制作用也不溶于含水分散介质中的树脂。
作为本发明使用的着色剂,可以使用炭黑,磁性物质,和用以下所示的黄色,品红色和青色着色剂调成黑色的着色剂作为黑色着色剂。
就黄色着色剂而言,可以使用缩合的偶氮化合物,异二氢吲哚酮(isoindolinone)化合物,蒽醌化合物,偶氮金属配合物,次甲基化合物和烯丙基酰胺化合物代表的化合物。具体而言,优选使用C.I.颜料黄12,13,14,15,17,62,74,83,93,94,95,97,109,110,111,120,127,128,129,147,168,174,176,180,181和191。
就品红色着色剂而言,可使用缩合的偶氮化合物,二酮基焦吡咯(diketopyropyyrole)化合物,蒽醌合物,奎吖啶酮(quinacridone)化合物,碱性染料色淀化合物,萘酚化合物,苯并咪唑啉酮化合物,硫靛化合物和苝化合物。具体说,特别优选使用C.I.颜料红2,3,5,6,7,23,48:2,48:3,48:4,57:1,81:1,144,146,166,169,177,184,185,202,206,220,221和254。
就青色着色剂而言,可以使用酞菁铜化合物及其衍生物,蒽醌化合物和碱性染料色淀化合物。具体说,特别优选使用C.I.颜料蓝1,7,15,15:1,15:2,15:3,15:4,60,62和66。
这些着色剂中的任何一种都可以单独或混合使用,也可以在固体溶液状态下使用。
本发明所用的着色剂的选择应考虑色调角度,色饱和度,亮度,耐候性,在OHP胶片上的透明度和在色调剂粒子中的分散性。每100重量份的粘合剂树脂中这种着色剂的用量为1到20重量份。
当使用磁性材料作为黑色着色剂,与其他着色剂不同的是,每100重量份的粘合剂树脂中这种着色剂的加入量为40到150重量份。
在本发明中,如果使用部分或全部由聚合形成的聚合色调剂粒子,则本发明的效果更好。特别是,当色调剂粒子的表面部分是通过聚合反应形成时,色调剂粒子在分散介质中以色调剂前体(单体组合物)粒子形式存在,并且它们的必要部分是通过聚合反应形成的。这样便能得到具有相当光滑表面特性的粒子。
在本发明中,色调剂粒子可能具有核/壳结构,其中其壳是由聚合反应合成的聚合物组成,而核是由低软化点物质组成。这种粒子是优选的,因为这种色调剂的定影性能得到改进而不会破坏它的抗粘性,而且还可以容易地将色调剂粒子中的残留单体除去。
更为具体的是,与不具有核的本体聚合色调剂粒子比较而言,如果只将壳部分进行聚合,则在聚合步骤之后所进行的后处理过程中,清除残留单体变得更为容易。
在本发明中,特别优选的是使悬浮聚合反应在常压或减压状态下进行,这样能相对容易地得到具有窄粒子大小分布和重均粒子直径为2.0到9.0μm或对于更高的图像质量为3.0到8.0μm的色调剂细粒子,因为这样可以容易形成将低软化点物质蜡包封在色调剂粒子中的核/壳结构。作为一种包封低软化点物质的具体方法,将含水介质中可聚合单体组合物中的主要单体的极性设定为小于低软化点物质的极性,且优选向可聚合单体组合物中少量加入具有高极性的树脂或单体,借此可以得到具有核/壳结构的色调剂粒子,在这种结构中,由低软化点物质组成的核表面被由壳树脂组成的壳所包围。色调剂粒子的粒子大小分布和粒子直径可以用如下两种方法来控制:一种方法是改变微溶于水的无机盐或具有保护性胶体作用的分散剂的类型或用量,另一种方法是改变机械设备条件,如搅拌条件,例如转子的周边速度,搅拌叶片的形状和通过次数以及反应器的形状,或者在含水介质中固体物质的浓度。
作为一种证实色调剂粒子具有核/壳结构的具体方法,将这种色调剂粒子在室温下充分分散于固化环氧树脂,接着在40℃的环境中进行2天的固化处理,将得到的固化产物用四氧化三钌(任选与四氧化三锇混合)进行染色,然后用具有一个钻石刀片的切片机将样品切成薄片,用透射电子显微镜(TEM)观察色调剂粒子的截面形状。在本发明中,优选使用四氧化三钌染色法,以能利用组成核的低软化点物质和组成壳的树脂之间的结晶性差异形成材料之间的对比。
在本发明中,当色调剂粒子采用聚合法制备时,用于合成粘合剂树脂的可聚合单体可以包括苯乙烯类单体,如苯乙烯,邻-或间-或对-甲基苯乙烯,和间-或对-乙基苯乙烯;丙烯酸或甲基丙烯酸酯单体,如丙烯酸甲酯或甲基丙烯酸甲酯,丙烯酸乙酯或甲基丙烯酸乙酯,丙烯酸丙酯或甲基丙烯酸丙酯,丙烯酸丁酯或甲基丙烯酸丁酯,丙烯酸辛酯或甲基丙烯酸辛酯,丙烯酸十二烷基酯或甲基丙烯酸十二烷基酯,丙烯酸十八烷基酯或甲基丙烯酸十八烷基酯,丙烯酸二十二烷基酯或甲基丙烯酸二十二烷基酯,丙烯酸2-乙基己基酯或甲基丙烯酸2-乙基己基酯,丙烯酸二甲基氨基乙酯或甲基丙烯酸二甲基氨基乙酯;丙烯酸二乙基氨基乙酯或甲基丙烯酸二乙基氨基乙酯;烯单体,如丁二烯,异戊二烯,环己烯,丙烯腈或甲基丙烯腈和丙烯酰胺;上述中的任何一种都可以优选使用。
任何一种可聚合单体可单独使用或者常常以适当的单体混合物的形式使用,混合后的理论玻璃化转变温度(Tg,如出版物《聚合物手册》(POLYMER HANDBOOK)第2版,第139-192页(John Wiley&Sons,Inc.)中所描述)为40到80℃。如果理论玻璃化转变温度低于40℃,则色调剂的储存稳定性和显影剂的运转稳定性方面就会发生问题。另一方面,如果理论玻璃化转变温度大于80℃,则色调剂的定影点就会变高。特别是如果将彩色色调剂用于形成全色图像,则在定影时各种颜色的色调剂的混色性能不足,导致彩色再现性不足,也可能使OHP图像的透明度严重降低。因此,从高图像质量考虑,这样的温度不是优选的。
在本发明中,组成壳的壳树脂的树脂组分具有的数均分子量(Mn)优选为5,000到1,000,000,更优选为6,000到500,000,重均分子量(Mw)与数均分子量(Mn)之比(Mw/Mn)优选为2到100,更优选为3到70。
如果壳树脂的数均分子量(Mn)小于5,000,则低软化点物质势必会逸出到粒子表面,趋于导致色调剂的抗粘性降低。
如果壳树脂的重均分子量(Mw)大于1,000,000,则低温定影性能受到影响。
如果重均分子量(Mw)/数均分子量(Mn)(Mw/Mn)小于2,则既难以得到低温定影性能,也难以得到抗粘性。如果重均分子量(Mw)/数均分子量(Mn)(Mw/Mn)大于100,则色调剂由于透明度低而使彩色OHP图像质量低劣。
壳树脂的树脂组分的分子量是通过GPC(凝胶渗透色谱法)来测定的。一种采用GPC测定的具体方法是,将色调剂预先通过索格利特提取器用甲苯溶剂提取20小时,然后使甲苯通过旋转蒸发仪蒸发,接着加入一种能溶解低软化点物质但不溶解壳树脂的有机溶剂(例如,氯仿),将色调剂彻底地清洗。然后,将这种溶液溶于THF(四氢呋喃),随后用孔径为0.3μm的耐溶剂性膜滤器过滤得到样品。样品的分子量用150C检测器(Waters公司制造)测定。作为柱构成,将可由Showa Denko K.K公司得到的A-801,A-802,A-803,A-804,A-805,A-806和A-807连接起来,用标准聚苯乙烯树脂的校正曲线测定分子量分布。
当生产具有核/壳结构的色调剂粒子时,除壳树脂以外,该壳中优选还加入极性树脂以使核的低软化点物质更好地被壳所包封。本发明所用极性树脂优选为苯乙烯与丙烯酸或甲基丙烯酸的共聚物,马来酸共聚物,饱和聚酯树脂和环氧树脂。特别优选的极性树脂其分子中不含有任何可能与可聚合的单体发生反应的不饱和基团。当使用不含有这种不饱和基团的极性树脂时,不会与组成壳树脂的单体发生交联反应。之所以优选这种树脂,是因为特别是在用作全色色调剂时,壳树脂不会显示出太大的分子量而且不会降低四种彩色色调剂的色彩混合。
在本发明中,具有核/壳结构的色调剂粒子表面还可进一步形成最外层的壳树脂层。
这种最外层壳树脂层优选为设置其玻璃化转变温度大于成壳的壳树脂的玻璃化转变温度,以提高抗粘性,也可优选为具有一种不致于影响定影性能的交联度。最外层壳树脂层优选进一步掺入极性树脂或电荷控制剂,以提高充电性能。
对于如何提供最外层壳树脂层没有特别的限定。例如,这种树脂层可以通过以下1)到3)之中一种方法来提供。
1)一种方法是,在聚合反应后半期或完成后,将通过溶解或分散可聚合单体,极性树脂,电荷控制剂和交联剂(在必要时)得到的单体组合物加入到反应系统中,并被聚合粒子吸附,然后加入聚合引发剂以进行聚合反应。
2)一种方法是,将通过使含有可聚合单体,极性树脂,电荷控制剂和交联剂(在必要时)的可聚合单体组合物发生聚合反应合成的乳液聚合粒子或无皂聚合粒子加入到反应系统中,使之附在聚合粒子的表面,随后可以任选地通过加热使之固定。
3)一种方法是,将通过使含有可聚合单体,极性树脂,电荷控制剂和交联剂(在必要时)的可聚合单体组合物发生聚合反应合成的乳液聚合粒子或无皂聚合粒子通过干燥方法机械地使之固定在色调剂粒子的表面。
当通过聚合反应产生本发明中的色调剂粒子时,聚合引发剂可包括,如偶氮类型的聚合引发剂,如2,2’-偶氮二-(2,4-二甲基戊腈),2,2’-偶氮二异丁腈,1,1’-偶氮二-(环己烷-1-腈),2,2’-偶氮二-4-甲氧基-2,4-二甲基戊腈和偶氮二异丁腈;过氧化物类型的聚合引发剂,如过氧化苯甲酰,甲基乙基酮过氧化物,过碳酸二异丙基酯,氢过氧化枯烯,2,4-二氯苯甲酰基过氧化物和过氧化月桂酰。这种聚合引发剂的加入量通常为0.5到20%(重量)(基于可聚合单体的重量),其加入量还可以根据本发明所需要的聚合程度加以变化。聚合引发剂类型在这些聚合方法中变化较小,可以单独或以混合物的形式使用,并参考其10-小时的半衰期温度。
为了使用更少量的引发剂能长时间保持高聚合物生长反应,从而能使作为链转移试剂的引发剂加入量更小,本发明的色调剂可采用以下方法得到,例如,往反应系统中加入一种在分子量为2,000到5,000之间区域有一尖峰的聚合物,在反应系统中能保证很少有分子量为2,000到5,000的聚合物形成。在造粒之前,将这种聚合物以合适的量加入到单体组合物中。
在本发明中,为了控制聚合度,还可进一步加入已知的交联剂,链转移试剂和聚合反应抑制剂。
在本发明中,当通过悬浮聚合反应制得色调剂粒子时,任何一种无机化合物和有机化合物都可以用作分散剂。这些无机化合物可包括磷酸三钙,磷酸镁,磷酸铝,磷酸锌,碳酸钙,碳酸镁,氢氧化钙,氢氧化镁,氢氧化铝,硅酸钙,硫酸钙,硫酸钡,膨润土,二氧化硅,氧化铝,磁性材料和铁酸盐。这些有机化合物可包括,如聚乙烯醇,明胶,甲基纤维素,甲基羟丙基纤维素,乙基纤维素,羧甲基纤维素钠盐和淀粉。这些分散剂在水相中分散。其中任何一种分散剂的用量以100重量份的可聚合单体为基准计为0.2到10.0重量份。
商业上可得到的分散剂都可以直接用作本发明的分散剂。不过,为了获得具有小而均匀粒子大小的分散粒子,可在高速搅拌下于分散介质中形成无机化合物的细粒子。例如,就磷酸三钙而言,将磷酸钠水溶液和氯化钙溶液在高速搅拌的条件混合得到一种优选用于悬浮聚合反应的细粒子分散剂。在这些分散剂中,可以联合使用0.001到0.1重量份的表面活性剂。具体讲,可使用商业上可得到的非离子、阴离子或阳离子表面活性剂。例如,优选使用十二烷基硫酸钠,十四烷基硫酸钠,十五烷基硫酸钠,辛基硫酸钠,油酸钠,月桂酸钠,硬脂酸钾和油酸钙。
当采用聚合反应制备色调剂粒子时,其具体可采用以下方法制备。将一种加入低软化点物质隔离剂的含有可聚合单体的单体组合物,着色剂,电荷控制剂,聚合引发剂和其它添加剂用混合机械(如均化器或超声分散器)预先均匀溶解或分散,再用已知的搅拌机,均相混合机或均化器将它们分散在含有分散稳定剂的水相中。进行造粒,同时控制搅拌速度和搅拌时间以使形成的单体组合物小滴具有所需的色调剂粒子大小。造粒之后,进行搅拌,搅拌程度应能通过分散稳定剂的作用保持粒子状态,使粒子不会发生沉淀。这种聚合反应进行时可将聚合温度设置为40℃或更高,优选为50℃到90℃。在聚合反应的后半期,可升高温度,也可在反应的后半期或反应结束后从反应系统中除去部分含水介质,从而除去了未反应的可聚合单体和副产物。反应结束后,通过洗涤、过滤和干燥得到色调剂粒子。在这种悬浮聚合反应中,通常将水作为分散介质,以100重量份的单体组合物为基准计,其用量优选为300到3,000重量份。
本发明的色调剂既可以用作单组分显影剂也可以用作双组分显影剂。就双组分显影剂而言,这种色调剂与称为载体的磁性显影粒子(下文也称“载体粒子”)混合。
这种载体具有的重均粒子直径为15到60μm,优选为20到45μm,其中粒子直径小于22μm的载体粒子含量不超过20%,优选为0.05到15%,更优选为0.1到12%,而粒子直径小于16μm的载体粒子数量为不超过3%,优选为不大于2%,更优选为不大于1%。
粒子直径大于62μm的载体粒子的粗粉末与图像的清晰度密切相关,其数量需为0.2到10%。
如果载体具有的重均粒子直径小于15μm,则载体的流动性低而不能与色调剂很好地混合,势必造成图像模糊。如果载体具有的重均粒子直径大于60μm,则载体保持色调剂的能力低,势必造成色调剂的分散。具有更细粉末的载体往往引起载体的粘附,而具有更粗粉末的载体往往降低图像浓度。
本发明所用的载体粒子可以包括,例如,磁性金属粒子,如表面氧化或未氧化的铁,镍,铜,锌,钴,锰,铬和稀土元素;合金和其氧化物;铁氧体;以及其中分散有磁性粉末的树脂载体。
为了使载体粒子表面光滑并提高球形度,优选使用(i) 以下式(I)表示的铁氧体载体,或(ii)通过悬浮聚合反应制得的含磁铁矿的聚合树脂载体。为了使这种载体粒子具有高电阻而不会扰乱潜像电压,特别优选这种含磁铁矿的聚合树脂载体。
式(I)
(Fe2O3)x(A)y(B)z
其中A代表MgO,Ag2O或它们的混合物;B代表Li2O,MnO,CaO,SrO,Al2O3,SiO2或这些物质之中任何一种的混合物;x,y和z各自代表重量比并满足以下条件:
0.2≤x≤0.95;
0.005≤y≤0.3;
0<z≤0.795;和
x+y+z≤1。
这种聚合树脂载体优选可含有Fe3O4磁铁矿,此外还含有Fe2O3,Al2O3,SiO2,CaO,SrO,MgO,MnO或这些物质之中任何一种的混合物。Fe3O4的量优选为全部氧化物重量的0.2到0.8。
如果在式(I)表示的铁氧体载体中x小于0.2,而聚合树脂载体中Fe3O4的量小于0.2,则这种载体具有低磁性,趋于造成载体的分散或者在感光鼓表面形成刮痕。如果x大于0.95或Fe3O4的量大于0.8,则载体的电阻降低,以致于载体粒子的表面必须覆盖大量的树脂,从而势必造成不希望发生的载体粒子聚集。
在铁氧体载体中,如果y小于0.005,则难于得到合适的磁性,如果y大于0.3,则载体粒子表面在某些情况下不能变得均一和圆整,引起堆积密度的很大变化,感应检测和精度降低。同样,如果z等于0,也就是,不含有组分B,将难以得到具有窄粒子大小分布的粒子,而且载体的超细粉末可能严重地造成感光鼓表面的刮痕,或者严重造成焙烧时粒子的聚集从而难以形成载体。如果z大于0.795,则载体的磁性降低从而严重导致载体的分散。
关于式(I)中的B,在Li2O,MnO,CaO,SrO,Al2O3和SiO2中,因为在使用高电压时载体的电阻变化较小,优选MnO,CaO,SiO2和Al2O3,由于能更好地适应所提供的色调剂,更优选为MnO和CaO。
关于聚合树脂载体,其粒子形状易于制成球形,根据其制备方法可以得到这种载体的窄粒子大小分布,因此,既使被制成更小粒子直径的粒子,它比铁氧体载体也更能抑制载体与感光鼓的粘附。而且,前者由于堆积密度的变化较小而比后者为优选。
本发明优选使用的载体是含有磁性粉末的磁性粉末分散型树脂载体,例如铁粉末,铁氧体粉末或氧化铁粉末已分散于该树脂中。更为优选的是通过聚合反应制得的含磁铁矿的聚合树脂载体,因为这种载体的压紧程度变化较小,特别优选含有非磁性金属氧化物和磁铁矿的聚合树脂载体。
这种非磁性金属氧化物优选为Fe2O3,Al2O3,SiO2,CaO,SrO,MnO或这些物质中任何一种的混合物。磁铁矿的量优选占全部氧化物重量的20到80%(重量)。
可选择性地对上述的磁铁矿进行处理使之具有亲脂性。当进行这种处理时,为了改进它的疏水性,可预先用二氧化硅,氧化铝或二氧化钛对它进行表面处理,并随后进行亲脂处理。
类似地,也可以优选地对非磁性金属氧化物进行处理使之亲脂。
其中待分散磁性粉末的树脂可以包括苯乙烯-丙烯酸酯或苯乙烯-甲基丙烯酸酯共聚物,聚酯树脂,环氧树脂,苯乙烯-丁二烯共聚物,酰胺树脂和密胺树脂。
特别地,这种树脂还可优选含有酚树脂。当它含有酚树脂时,它具有优异的耐热性和耐溶剂性,当这种粒子的表面被树脂包覆时,能达到满意的包覆效果。
本发明中所用的载体优选为由聚合反应产生的载体,这同样是为了实现均一的显影剂转送性能。
优选的载体粒子是其中磁性材料细粒子与固化酚醛树脂基质结合在一起的那些。这种载体粒子可由下述方法制得。
在碱性催化剂、磁性粉末和悬浮稳定剂的存在下,使酚和醛在含水介质中发生反应。
这里所用的酚可包括苯酚和带有酚羟基的化合物,例如,烷基苯酚,如间甲酚,对叔丁基苯酚,邻丙基苯酚,间苯二酚和双酚-A,卤代苯酚,其中苯环或其烷基已部分或全部被一个或多个氯或溴原子取代。具体而言,最优选苯酚。当以除苯酚之外的化合物作为酚类使用时,粒子的形成比较困难,或者即使能形成粒子其形状也为无定形。因此,考虑到粒子的形状,最优选为苯酚。
所用的醛可以包括以甲醛水溶液或多聚甲醛形式存在的甲醛以及糠醛。特别优选甲醛。醛与酚的摩尔比优选为1到2,特别优选为1.1到1.6。
关于所用碱性催化剂,可以使用制备常规可熔酚醛树脂所用的碱性催化剂。例如,它可以包括氨水和烷基胺,如六亚甲基四胺,二甲胺,二乙基三胺和聚乙烯亚胺。这些碱性催化剂中任何一种与酚的摩尔比优选为0.02到0.3。
当酚与醛在碱性催化剂的存在下发生反应时,与其同时存在的磁性粉末可以包括前述的磁性粉末。优选所用的磁性粉末量为酚重量的0.5至200倍。同样,考虑到粒子的饱和磁化强度值和粒子的强度,更优选用量为酚重量的4到100倍。
磁性粉末优选粒子直径为0.01到10μm,考虑到在含水介质中细粒子的分散度和待形成的载体粒子的强度,更优选为0.05到5μm。
悬浮稳定剂可以包括亲水有机化合物,如羧甲基纤维素和聚乙烯醇,氟化合物,如氟化钙和基本上水不溶的无机盐,如硫酸钙。
使用悬浮稳定剂时,其加入量优选以酚重量为基准为0.2到10%(重量),更优选为0.5到3.5%(重量)。
在这种制备方法中,反应是在含水介质中进行的。例如,这里所加入水的量优选为能够达到使载体中固体含量浓度为30到95%(重量),更优选为60到90%(重量)。
可以在升温速率为0.5到1.5℃/分钟的条件下逐渐升高温度来进行这种反应,优选升温速率为0.8到1.2℃/分钟,进行搅拌,控制反应温度在70到90℃,更优选为83到87℃,反应进行的时间为60到150分钟,更优选为80到110分钟。在这种反应中,使固化反应同时进行以生成固化酚树脂基质。
这种反应和固化过程完成后,将得到的反应产物冷却到40℃或40℃以下,以得到由磁性粉末粒子均匀地分散于固化酚树脂基质中形成的球形粒子的水分散体。
接着,采用常规的方法如过滤或离心方法将这种水分散体进行固液分离,随后进行洗涤和干燥。这样,便得到其中磁性粉末分散于酚树脂基质中的载体粒子。
上述方法可以连续进行也可以分批进行。在通常情况下,采用分批处理方式。
为了控制载体粒子的充电、电阻等,优选使用一种涂料涂布在载体粒子的表面。可根据用于色调剂的材料的不同选用不同的涂布在载体粒子表面的涂料。例如,它可以包括氨基丙烯酸酯或氨基甲基丙烯酸酯树脂,丙烯酸或甲基丙烯酸树脂,这些树脂中的任何一种与苯乙烯树脂的共聚物,丙烯酸或甲基丙烯酸树脂与氟树脂的聚合物,硅氧烷树脂,聚酯树脂,氟树脂,聚四氟乙烯,一氯三氟乙烯聚合物和聚偏二氟乙烯。特别优选硅氧烷树脂,氟树脂和由丙烯酸或甲基丙烯酸树脂与氟树脂组成的混合物或共聚物,因为这样能使载体粒子长时间保持高充电性能。这些涂料的涂布重量可以适当地选定以获得满意的载体的电荷提供性能且通常为载体粒子总重的0.1到30%(重量),优选为0.3到20%(重量)。
可以采用以下方法中任何一种在磁性载体核粒子表面形成树脂涂层:一种方法是将树脂组合物溶于适当的溶剂中,将磁性载体核粒子浸入所形成的溶液中,然后进行去溶剂化,干燥和高温焙烤;另一种方法是将磁性载体核粒子悬浮在流化体系中,用溶有上述树脂组合物的溶液进行喷涂,然后进行干燥和高温焙烤;还有一种方法是将磁性载体核粒子与树脂组合物的粉末或水乳液混和。
本发明优选的一种方法是使用一种混合溶剂,该混合溶剂通过在100重量份含有至少5%(重量),优选至少20%(重量)极性溶剂如酮或醇的溶剂中加入0.1到5重量份,优选为0.3到3重量份的水制备。优选这种方法是因为反应性硅氧烷树脂能够稳固地粘在磁性载体核粒子上。如果水的含量少于0.1重量份,则这种反应性硅氧烷树脂的水解反应不能很好地进行,从而在磁性载体核粒子上难以得到一种薄而均匀的涂层。如果水的含量大于5重量份,则难以控制这种反应,反而造成低涂层强度。
在本发明中,当载体与色调剂混合用于制备双组分显影剂时,如果它们按下述比例混合则能取得良好的效果,这种比例是在双组分类型显影剂中色调剂的浓度为1到15%(重量),优选3到12%(重量),更优选5到10%(重量)。如果色调剂的浓度小于1%(重量),则图像浓度势必降低。如果色调剂浓度大于15%(重量),则图像变得模糊,机器内部发生散射,从而缩短了双组分显影剂的运转寿命。
本发明的成像方法如以下所述。
本发明的成像方法包括(I)充电步骤,对用于带有静电潜像的潜像承载元件进行静电充电,(II)潜像形成步骤,即在这种充电的潜像承载元件上形成静电潜像,(III)显影步骤,用色调剂对潜像承载元件上的静电潜像进行显影,以形成色调剂图像,以及(IV)转印步骤,将潜像承载元件上形成的色调剂图像转印到转印介质上。作为这种色调剂,使用上述的色调剂。
在充电步骤中,非接触充电元件,如电晕充电装置和接触式充电元件,如刮板,辊或电刷,都可用作充电元件;前者是一种不与潜像承载元件表面接触而对它进行充电的元件,而后者是一种与潜像承载元件表面接触而对它进行充电的元件。优选使用接触式充电元件,因为在充电时产生较少的臭氧。
接触式充电元件中,优选导电刷,如纤维刷或磁刷,因为它与潜像承载元件的表面可以有很多接触点,这样与刮板和辊之类的元件(它们的光滑表面与潜像承载元件的表面相接触)相比,能使充电均匀。
优选用作组成纤维刷的纤维聚集体的物质包括含有超细成纤维共轭纤维(fiber-generation conjugate fibers)的聚集体;含有用酸,碱或有机溶剂化学处理的纤维聚集体;起绒的纤维缠绕材料;以及静电植绒材料。
电刷充电的基本充电原理被认为,充电元件的导电充电层与感光鼓表面的电荷注入层发生接触,导致电荷从导电充电层注入电荷注入层。因此,对接触式充电元件的性能要求是,它能够提供具有充足密度的电荷注入层表面和适于电荷转移的合适的电阻。
这样,通过以下方法能够与电荷注入层更频繁接触并实现均匀充分的充电:一种方法为采用超细成纤维共轭纤维使纤维密度增大,另一种方法是用化学蚀刻法处理纤维使纤维数目变大,还有一种方法是给表面提供挠性纤维端,它是利用通过使纤维缠绕材料起绒制备的元件或利用静电植绒材料而完成的。也就是说,本发明优选使用这样形成的电刷,即该电刷具有更高的纤维密度、更多的接触点数量,并使纤维端能与电荷注入层发生接触。
由超细成纤维共轭纤维组成的聚集体优选为其中超细纤维已通过物理或化学方法产生的那些。这种起绒的纤维缠绕材料优选为其中纤维缠绕材料由超细成纤维共轭纤维形成的那些。这种超细成纤维共轭纤维更优选为由物理或化学方法生成并使之起绒。
这种静电植绒材料优选为其中其组成纤维已经用酸、碱或有机溶剂化学处理的那些。作为该静电植绒材料的另一种优选形式,它可以具有一种其中其构成纤维是超细成纤维共轭纤维的形式,其超细纤维是通过物理或化学方法产生的。
磁刷可由作为磁性粒子承载元件的磁辊或内部具有一个磁辊的导电套筒组成,其表面上已经通过磁力结合有磁性粒子。
磁性粒子优选具有5到100μm的平均粒子直径。平均粒子直径小于5μm的磁性粒子往往使磁刷粘在感光鼓上。平均粒子直径大于100μm的磁性粒子则不能使套管上的磁刷磁耳(ears)密度升高,趋于造成对电荷注入层的电荷注入性能不足。磁性粒子更优选为具有10到80μm的平均粒子直径。如果采用粒子直径在这个范围之内的磁性粒子,则感光鼓上的转印残留色调剂能被更为有效地刮去,并能更有效地静电聚集到磁刷上而且并暂时附在磁刷上以更准确地控制色调剂的充电。磁性粒子可进一步优选为具有10到50μm的平均粒子直径。
磁性粒子的平均粒子直径可通过激光衍射粒子大小分布测定仪器HEROS(商品名,由日本Denshi K.K.公司制造)来测定,在这种测定仪器里,可将直径为0.05μm到200μm的粒子通过32-对数分开来测定直径,用它们的50%平均粒子直径作为磁性粒子的平均粒子直径。
将具有这种粒子直径的磁性粒子用于接触式充电元件可以与感光鼓表面具有大量的接触点,并有利于给感光鼓赋予更为均匀的充电电势。而且,当磁刷旋转时,直接与感光鼓发生接触的磁性粒子彼此相互取代,这样所带来的另一个优点是可以极大地减少任何由于磁性粒子表面的污染导致的电荷注入性能降低。
磁性粒子优选具有体积电阻率为1×104Ωcm到1×109Ωcm,更优选为1×107Ωcm到1×109Ωcm。当体积电阻系数小于1×104Ωcm时,磁性粒子趋于附着在潜像承载元件上。当体积电阻系数大于1×109Ωcm时,则磁性粒子向潜像承载元件提供摩擦电荷的能力趋于降低,特别是在低湿度环境下,会造成充电不足。
带有磁性粒子的承载元件和感光鼓优选设定为在它们之间有一个0.2到2mm范围的间隙,更优选为0.3到2.0mm,进一步优选为0.3到1.0mm,最优选为0.3到0.7mm。如果它们之间的间隙设置小于0.2mm,则磁性粒子不能容易地通过间隙,从而磁性粒子不能平稳地在承载元件上输送,进而导致不良充电,或者磁性粒子过度停滞在辊隙处而使其粘在感光鼓上,而且一定的应用电压会引起承载元件的导电部分和感光鼓之间的漏电,从而损坏感光鼓。大于2mm的间隙不是优选的,因为它使感光鼓和磁性粒子之间难以形成宽辊隙。
利用静电转移到磁刷上的转印残留色调剂在给定的时间因施加交流电压而被送到感光鼓表面。被送出并留在感光鼓表面的转印残留色调剂在感光鼓的旋转方向上移动,并开始朝向显影套管(显影剂携带元件),在转印残留色调剂被显影套管刮去(该显影套管以相反的方向转动,其上被施加一个偏压电场)的位点,即被收集到显影装置中,并再次被用作显影色调剂。
在那种情况下,保持在色调剂粒子上的外部添加剂粒子的表现是与接触式充电元件中的色调剂粒子发生分离,并在色调剂被送走后保持在那里。作为本发明者进行广泛研究的结果,他们发现在吸附到接触式充电元件上的转印残留色调剂被送出后进行充电时,磁刷中存在的外部添加剂粒子与感光鼓表面发生接触和摩擦,而这对于清除沉积物,如臭氧产物、纸屑和其他任何沉积物是非常有效的。他们还发现这样一个优点,即当磁刷与感光鼓表面发生接触和摩擦时,外部添加剂粒子起到了隔离物的作用,而这使感光鼓表面受到刮擦的程度减小,从而延长了感光鼓的寿命。
充电磁刷既可沿常规的方向移动,也可以与其接触部分的感光鼓表面移动的相反方向进行移动。为了使转印残留色调剂能良好被吸附到磁刷上,这种磁刷优选按相反方向移动。
充电磁性粒子保持在磁刷的充电磁性粒子承载元件上的数量优选为50到500mg/cm2,更优选为100到300mg/cm2,这样能获得一种稳定的充电性能。
关于在接触式充电元件上施加的充电偏压,可以只施加DC成分,但也可以少量施加AC成分以求图像质量的提高。关于AC成分,其可以依充电过程的速度而变,优选为具有约100Hz到10kHz的频率,且施加的AC成分的峰-峰电压优选为约1,000V或1,000V以下。如果它高于1,000V,因为感光鼓上的电势是相对于施加的电压得到的,则在某些情况下,潜像表面可能会由于电势的原因而波动,引起图象模糊不清或浓度降低。在采用放电的方法中,AC成分可依充电过程的速度而变化,具有的频率优选为大约100Hz到10kHz,而所施加的AC成分优选具有的峰-峰电压为约1000伏或更高,该电压优选至少是放电起始电压的两倍。这样设置是为了对磁刷和感光鼓表面获得足够的校平作用。作为AC组分的波形,可以使用正弦波、矩形波和锯齿波。
过量的充电磁性粒子可以保持在充电装置中并在其中循环。
为了通过磁性使磁耳浮起,并使所产生的磁刷与光敏元件接触而进行充电,作为磁性粒子使用的材料可以包括合金或含有具有铁磁性的元素的化合物,例如铁、钴和镍,以及已经通过氧化或还原对其电阻率进行调节的铁氧体,如通过组成调节的铁氧体和通过氢还原处理的Zn-Cu铁氧体、Mn-Mg铁氧体和Li-Mg铁氧体。为了把该铁氧体的电阻率设定在如前所述所施加电场之下的上述范围内,也可以通过调节金属的组成来达到该电阻率。二价铁以外的金属的增加一般导致电阻率的降低和趋向于引起电阻率的突然下降。
本发明中所用的磁性粒子的摩擦电优选具有与感光鼓电荷极性相同的极性。如前所述,由于摩擦电而引起的感光鼓电势的下降将促进磁性粒子向感光鼓移动,这就使将磁性粒子保留在接触充电元件上的条件更为苛刻。通过涂覆该磁性粒子表面来提供表面层就很容易控制磁性粒子摩擦电的极性。
用于本发明的具有表面层的磁性粒子是其表面用涂覆材料如一种沉积膜、导电树脂膜或分散有导电颜料的树脂膜涂覆的粒子,或者是用反应性化合物处理过表面的粒子。每个磁性粒子不必完全被表面层覆盖,只要能够达到本发明的效果,可以部分覆盖磁性粒子。换句话说,表面层可以不连续地形成。
从生产率和成本的角度看,优选使用一种分散有导电颜料的树脂膜覆盖的磁性粒子。
从控制电阻率的电场依赖性来看,优选也使用一种由高电阻率的粘合剂树脂和分散于其中的传导电子的导电颜料组成的树脂膜覆盖该磁性粒子。
当然,这样覆盖的磁性粒子必须具有在上述范围内的电阻率。从扩大对高电场一边电阻率突然下降的耐受范围和取决于感光鼓上划痕大小和深度可能发生的裂缝图像(leak images)的耐受范围来看,母体磁性粒子优选具有在上所范围内的电阻率。
作为用来覆盖磁性粒子的粘合剂树脂,它可以包括苯乙烯类如苯乙烯和氯代苯乙烯的均聚物或共聚物;单烯烃类如乙烯、丙烯、丁烯和异丁烯;乙烯基酯如乙酸乙烯基酯、丙酸乙烯基酯、苯甲酸乙烯基酯和乙酸乙烯基酯;α-亚甲基脂族一元羧酸酯如丙烯酸甲酯、丙烯酸乙酯、丙烯酸丁酯、丙烯酸十二烷基酯、丙烯酸辛酯、丙烯酸苯酯、甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸丁酯和甲基丙烯酸十二烷基酯;乙烯基醚类如甲基乙烯基醚、乙基乙烯基醚和丁基乙烯基醚;以及乙烯基酮类如甲基乙烯基酮、己基乙烯基酮和异丙烯基乙烯基酮。从导电细粒子的分散性、作为覆盖层的成膜特性和生产率来看,作为一种特别典型的粘合剂树脂,有聚苯乙烯、苯乙烯-丙烯酸烷基酯共聚物、苯乙烯-丙烯腈共聚物、苯乙烯-丁二烯共聚物、苯乙烯-马来酸酐共聚物、聚乙烯和聚丙烯。它还可以包括聚碳酸酯、酚树脂、聚酯类、聚氨酯类、环氧树脂类、聚烯烃类、含氟树脂、硅氧烷树脂和聚酰胺类。特别是从预防色调剂污染的角度来说,更优选含有一种临界表面张力小的树脂,如聚烯烃树脂、含氟树脂和硅氧烷树脂。
另外,从保持高电场一边电阻率突然下降的宽耐受性和防止由感光鼓上划痕所引起的裂缝图像的出现这两个方面来看,涂覆在磁性粒子上的树脂优选是具有高电压电阻的含氟树脂或硅氧烷树脂。
含氟树脂可以包括如,溶剂可溶的共聚物类,这些共聚物是通过把氟乙烯、1,1-二氟乙烯、三氟乙烯、三氟一氯乙烯、二氯二氟乙烯、四氟乙烯或六氟丙烯与其他单体共聚得到的。
硅氧烷树脂可以包括如KR271、KR282、KR311、KR255、KR255和KR155(直链硅氧烷清漆)、KR211、KR212、KR216、KR213、KR217和KR9218(改良的硅氧烷清漆)、SA-4、KR206和KR5206(硅氧烷醇酸清漆)、ES1001、ES1001N、ES1002T和ES1004(硅氧烷环氧清漆)、KR9706(硅氧烷丙烯酸清漆)、KR5203和KR5221(硅氧烷聚酯清漆),所有这些都可以从Shin-Etsu硅氧烷有限公司得到;以及SR2100、SR2101、SR2107、SR2110、SR2108、SR2109、SR2400、SR2410、SR2411、SH805、SH806A和SH840,这些可以从Toray硅氧烷有限公司得到。
当该磁性粒子是用一种反应性化合物处理表面时,优选一种偶合反应产物,但该化合物不局限于此。
下列将描述用于本发明的潜像承载元件(感光鼓)的一个优选实施方案的例子。
它基本上包含一个导电基底和从功能上可分离为电荷产生层和电荷输送层的一个光敏层。
作为导电基底来说,可以使用圆柱形元件或一种带,它们由如下材料制成:金属如铝或不锈钢,合金如铝合金或氧化铟-氧化锡合金、具有由任何这些金属和合金形成的涂层的塑料,用导电粒子浸渍的纸或塑料或含有导电聚合物的塑料。
在导电基底上,提供一种胶层以便改善光敏层的粘附性,改善涂层特性,保护基底,覆盖基底上的缺陷,改进电荷从基底上注入的性能,并且保护光敏层不被电击穿。用于形成胶层的材料有聚乙烯醇、聚-N-乙烯基咪唑、聚氧乙烯、乙基纤维素、甲基纤维素、硝基纤维素、乙烯-丙烯酸共聚物、聚乙烯醇缩丁醛、酚树脂、酪蛋白、聚酰胺、共聚物尼龙、胶液、明胶、聚氨酯或氧化铝。该胶层的厚度一般大约是0.1到10μm,优选从0.1到3μm。
用一种由产生电荷的物质分散到适当粘合剂中制得的液体涂覆或者真空沉积产生电荷的物质来形成电荷产生层。产生电荷的物质包括偶氮颜料、酞菁颜料、靛蓝颜料、苝颜料、多环醌颜料、squarilium染料、吡喃鎓盐、硫代吡喃鎓盐、三苯甲烷染料和无机物质如硒和非晶形硅。该粘合剂树脂可以从极宽的粘合剂树脂范围中选择,这些粘合剂树脂包括如聚碳酸酯树脂、聚酯树脂、聚乙烯醇缩丁醛树脂、聚苯乙烯树脂、丙烯酸树脂、甲基丙烯酸树脂、酚树脂、硅氧烷树脂、环氧树脂和乙酸乙烯基酯树脂。电荷产生层中所含的粘合剂树脂含量可以不超过80%(重量),优选0到40%(重量)。电荷产生层的厚度优选是5μm或更小,最好是0.05到2μm。
电荷输送层具有在电场存在下接受来自电荷产生层中的电荷载流子并输送它们的功能。该电荷输送层可以通过涂覆一种将电荷输送物质任选与一种粘合剂树脂一起溶解在一种溶剂中制得的溶液而形成,并且一般优选层厚度为5到40μm。该电荷输送物质包括在主链或侧链中具有一种结构如亚联苯基、蒽、芘或菲的多环芳族化合物;含氮的环状化合物如吲哚、咔唑、噁二唑和吡唑啉;腙化合物;苯乙烯基化合物;以及无机化合物如硒、硒-碲、非晶形硅和硫化镉。
用来把这类电荷输送物质分散于其中的粘合剂树脂可以包括绝缘树脂如聚碳酸酯树脂、聚酯树脂、聚甲基丙烯酸酯、聚苯乙烯树脂、丙烯酸树脂和聚酰胺树脂,以及有机光电导性聚合物如聚-N-乙烯基咔唑和聚乙烯基蒽。
用于本发明的感光鼓(潜像承载元件)优选具有一个离基底最远的电荷注入层,即表面层。这种电荷注入层可以有从1×108Ω·cm到1×1015Ω·cm的体积电阻率,为的是达到一种满意的充电性能和较少的模糊图像。特别是从图像的模糊性角度看,它优选是1×1010Ω·cm到1×1015Ω·cm,考虑到环境变化等因素,它优选是1×1010Ω·cm到1×1013Ω·cm。如果它低于1×108Ω·cm,产生的电荷在高湿度环境下不能保持在表面上,从而势必引起模糊图像。如果它高于1×1015Ω·cm,充电元件的电荷注入不足,并且不能很好地保持这些电荷,从而容易引起错误充电。感光鼓表面提供的这种功能层具有在光照射下保持从充电元件注入的电荷的功能,并且也具有使电荷释放到感光鼓支承体上从而使剩余的电势下降的功能。
使用上述充电元件和上述感光鼓的本发明构造使得电荷启动电压Vth较小和感光鼓的充电电压几乎为施加到充电元件上的电压的90%或更多。
例如,当把绝对值为100到2,000V的DC电压施加到以1,000mm/分钟或更低的处理速度运转的充电元件上时,能够把具有本发明电荷注入层的静电照相感光鼓的充电电压控制为所加电压的80%或更多或90%或更多。另一方面,当施加700V的DC电压时,通过常规放电达到的感光鼓的充电电压大约是200V,这只是所加电压的大约30%。
这种电荷注入层是一种由金属沉积膜组成的无机层或者是通过把导电细粒子分散到电荷注入层粘合剂树脂中形成的分散有导电细粒子的树脂层。该沉积膜能够通过真空沉积形成,并且分散有导电细粒子的树脂层能够通过用一种适当的涂覆工艺如浸涂、喷涂、辊涂或束流涂覆形成。该层也可以通过把一种绝缘粘合剂树脂和一种具有透光特性和高离子传导性的树脂混合或共聚合形成,或者可以只由具有中等电阻和光电导性的树脂形成。
在分散有导电细粒子的树脂层中,以该电荷注入层粘合剂树脂的重量为基准计,导电细粒子优选以2到250%(重量)的量加入,并更优选以2到190%(重量)的量加入。如果导电细粒子以低于2%(重量)的量加入,就很难达到所需的体积电阻率。如果以大于250%(重量)的量加入导电细粒子,该层的薄膜强度会较低,并且电荷注入层容易被刮去,造成感光鼓的短寿命,而且它们的电阻率也较低,由于潜像电压的流动而容易引起错误的图像。
电荷注入层的粘合剂树脂可以包括聚酯、聚碳酸酯、丙烯酸树脂、环氧树脂和酚树脂,以及这些树脂的固化剂,它们可以单独使用或两个或多个组合使用。当把大量导电细粒子分散时,优选把该导电细粒子分散在反应性单体或反应性低聚物中,并且把得到的分散体涂到感光鼓表面,之后用光或热固化。当光敏层92是由非晶形硅形成时,该电荷注入层可以优选是由SiC形成的。
作为分散在电荷注入层93的电荷注入层粘合剂树脂中的导电细粒子的例子有金属或金属氧化物的细粒子。它们优选是金属氧化物的超细粒子,这些金属氧化物如氧化锌、二氧化钛、氧化锡、氧化锑、氧化铟、氧化铋、氧化锡涂覆的二氧化钛、锡涂覆的氧化铟、锑涂覆的氧化锡和氧化锆。可以单独使用这些中的任何一种或可以是两种或多种合用。
当把粒子分散到电荷注入层时,一般粒子必需具有小于入射光的波长的直径,为的是防止入射光由分散的粒子引起散射。作为分散在本发明表面层(电荷注入层)中的导电细粒子,它优选具有0.5μm或更小的粒子直径。
在本发明中,电荷注入层优选含有润滑剂粒子。原因是可降低充电时感光鼓和充电元件之间的摩擦,并且可以增大充电间隙使得充电性能得到改善。作为润滑剂粒子,特别优选使用临界表面张力较低的含氟树脂、硅氧烷树脂或聚烯烃树脂。更优选使用四氟乙烯树脂(PTFE)。在这种情况下,以粘合剂树脂的重量为基数,润滑剂粒子可以2到50%(重量),优选5到40%(重量)的量加入。如果它们少于2%(重量),润滑剂粒子的量将不足,由此不能足以改善导电性能,如果它们多于50%(重量),图像的分辨率和感光鼓的敏感度会大大降低。
本发明的电荷注入层可优选具有0.1到10μm的层厚,特别优选有1到7μm的层厚。如果层厚小于0.1μm,该层将失去它对细划痕的耐久性,并由于发生错误的注入导致错误的图像。如果层厚大于10μm,注入的电荷会扩散,容易引起图像的紊乱。
在本发明中,可以将含氟树脂的细粒用于潜像承载元件中。该含氟的树脂的细粒子是由一种或多种选自于下列的物质组成:聚四氟乙烯、聚一氯三氟乙烯、聚偏二氟乙烯、聚二氯二氟乙烯、四氟乙烯-全氟烷基乙烯基醚的共聚物、四氟乙烯-六氟丙烯共聚物、四氟乙烯-乙烯共聚物和四氟乙烯-六氟丙烯-全氟烷基乙烯基醚共聚物。可以直接使用商业上可得到的含氟树脂细粒子。可以使用分子量为3,000到5,000,000的那些,并且它们的粒子直径为0.01到10μm,并优选0.05到2.0μm。
在许多情况下,分别把上述含氟树脂细粒子、电荷产生物质和电荷输送物质分散和掺入一种具有成膜特性的粘合剂树脂中,以分别形成保护层和光敏层。这类粘合剂树脂包括聚酯、聚氨酯、聚丙烯酸酯、聚乙烯、聚苯乙烯、聚碳酸酯、聚酰胺、聚丙烯、聚酰亚胺、酚树脂、丙烯酸树脂、硅氧烷树脂、环氧树脂、尿素树脂、烯丙基树脂、醇酸树脂、聚酰胺-酰亚胺、尼龙、聚砜类、聚烯丙醚、聚缩醛和丁醛树脂。
潜像承载元件的导电基底可以由金属如铁、铜、金、银、铝、锌、钛、铅、镍、锡、锑或铟或它们的合金,这些金属中任何一个的氧化物、碳或导电聚合物制成的。它可为鼓形如圆柱形、带形或片形。上述导电材料可以直接模塑,可以是以涂覆材料的形式使用,可以是真空沉积,或者可以通过蚀刻法或等离子体处理来加工。
在本发明中,使用具有中等电阻的接触充电元件将电荷注入到具有中等电阻的表面电阻的感光鼓的表面部分。优选不把电荷注入到光敏元件表面材料所具有的陷阱能级,但是把电荷提供给电荷注入层的导电细粒子,该电荷注入层是由导电细粒子分散于其中的透光性绝缘粘合剂形成的。
具体地说,本发明基于这样的理论,即从接触充电元件提供电荷到微小的电容器,每个电容器用电荷输送层作介电层,以金属基底和电荷注入层中的导电细粒子作为两个电极。在这种情况下,导电细粒子在电学上互相独立,并形成一种微小的漂浮电极。因此,宏观来看,光敏元件表面似乎被充至均匀的电势,但实际上处于这样一种条件,即微小的和无数的充电的导电细粒子涂覆在光敏元件表面。所以,甚至当用激光进行成影像曝光时,也能保持静电潜像,这是由于单个的导电细粒子在电性上独立于另外的导电细粒子的缘故。
这样,甚至少量的用于替代存在于常规光敏元件表面的陷阱能级的导电细粒子也能够提高电荷注入性能和电荷的保持性。
这里,用下列方法测定电荷注入层的体积电阻率:在其表面上已真空沉积有导电膜的聚对苯二甲酸乙二醇酯(PET)膜上形成电荷注入层。用一种体积电阻率测量仪(4140B pAMATER,由Hullet Packard公司生产)在23℃/65%RH的环境用100V电压测定它的电阻率。
在潜像形成步骤中,作为成影像曝光的方法,可以使用如激光和LEDs的已知方式。
在显影步骤中,作为静电潜像的显影方法,可以使用单组分显影或双组分显影;前者是使用只由色调剂组成的单组分显影剂的方法而后者是使用由色调剂和载体组成的双组分显影剂的方法。
当使用含有磁性材料的磁性色调剂作单组分显影剂时,可行的方法是使用在显影套筒中的磁体输送该磁性色调剂并使其充电。当用一种含有非磁性材料的非磁性色调剂作单组分显影剂时,可行的方法是通过刮板和毛刷在显影套筒上使非磁性色调剂强迫摩擦充电,从而使该色调剂被吸引到显影套筒上而被输送。
下面将描述使用上述双组分显影剂的双组分显影法。
双组分显影法包括,在由潜像承载元件和与其对置的显影剂携带元件所限定的显影区内,在显影剂携带元件上循环输送由色调剂和载体组成的双组分显影剂,用显影剂携带元件上运载的双组分显影剂的色调剂使潜像承载元件上的潜像显影。
在显影套筒中的磁体辊能影响载体的磁性,并大大影响显影剂的显影性和输送性。
在本发明的成像方法中,例如,在显影套筒(显影剂携带元件)中的磁体辊是静止的,而只有显影套筒旋转,由此把双组分显影剂循环输送到显影套筒上,并用双组分显影剂使潜像承载元件表面上的静电潜像显影。
在本发明的成像方法中,当(1)磁体辊是由排斥极组成,(2)显影区的磁通量密度为500到1,200高斯以及(3)显影载体的饱和磁化强度为20到50Am2/g时,复印能够获得很好的图像均匀度和层次再现性。
在本发明成像方法中,优选在显影区施加显影偏压下由双组分显影剂的色调剂使静电潜像显影。
下面将详细描述特别优选的显影偏压。
在本发明的成像方法中,为了在由潜像承载元件和显影剂携带元件所限定的显影区形成显影电场,优选把图7所示的具有不连续AC成分的显影电压施加到显影剂携带元件上,由此,使用显影剂携带元件上所运载的双组分显影剂的色调剂,将潜像承载元件上的潜像显影。具体来说,这种显影电压由把色调剂从潜像承载元件引向显影区中的显影剂携带元件的第一电压、引向潜像承载元件的第二电压和介于第一电压和第二电压之间的第三电压组成。这样,在潜像承载元件和显影剂携带元件之间就形成了显影电场。
另外,使介于第一电压和第二电压的第三电压施加到显影剂携带元件的时间(T2),即AC成分停止的时间,比把色调剂从潜像承载元件引向显影剂携带元件的第一电压和把色调剂从显影剂携带元件引向潜像承载元件的第二电压施加到显影剂携带元件的总时间(T1),即AC成分运行的时间,更长。因为色调剂能够在潜像承载元件上重排,从而使图像能够真正再现潜像,所以这是特别优选的。
实际上,在显影区中潜像承载元件和显影剂携带元件之间,把色调剂从潜像承载元件引向显影剂携带元件的电场和把色调剂从显影剂携带元件引向潜像承载元件的电场可以至少形成一次,并且之后在潜像承载元件的图像区形成一定时间的把色调剂从显影剂携带元件引向潜像承载元件的电场,和在潜像承载元件的非图像区形成一定时间的把色调剂从潜像承载元件引向显影剂携带元件的电场,用显影剂携带元件上所运载的双组分显影剂的色调剂将潜像承载元件上的潜像显影,其中使在潜像承载元件的图像区形成把色调剂从显影剂携带元件引向潜像承载元件的电场和在潜像承载元件的非图像区形成把色调剂从潜像承载元件引向显影剂携带元件的电场的时间(T2)优选长于形成把色调剂从潜像承载元件引向显影剂携带元件的电场和把色调剂从显影剂携带元件引向潜像承载元件的电场的总时间(T1)。
当在显影电场存在下使潜像显影时,载体粘着更难发生,在显影方法中周期性地使电场交替消失,即进行显影的同时形成上述特殊的显影电场,即,一种交替电场。原因还不清楚,但推测如下:
在常规连续的正弦或矩形波中,为了试图获得较高的图形浓度而使电场强度较高时,色调剂和载体在潜像承载元件和显影剂携带元件之间结合而互换,从而载体强烈地与潜像承载元件摩擦,引起载体粘着。当细粉载体增加时这种现象更明显地发生。
但是,当用一个脉冲施加本发明所述的特定显影电场时,色调剂或载体会在显影剂携带元件和潜像承载元件之间非全程地来回运动。因此,当潜像承载元件的表面电势和显影偏压直流成分的电势之间的电位差Vcont低于零,即,Vcont<0时,Vcont以这样一种方式起作用,即它使载体从显影剂携带元件飞走。但是,通过控制载体的磁性和磁体辊显影区的磁通量密度能够防止该载体的粘着。在Vcont>0时,磁场力和Vcont以这样一种方式起作用,即它把载体吸到显影剂携带元件一侧从而没有载体粘着发生。
如前所述,显影套筒中的磁体辊会影响载体的磁性,并且大大地影响显影剂的显影性能和输送性能。
在本发明中,在其中有磁体辊的显影套筒上,可以循环和运送由含有磁性粒子的载体和一种绝缘彩色色调剂组成的双组分显影剂,同时磁体辊被设置为静止而只使显影套管旋转,用双组分显影剂使潜像承载元件表面上的静电潜像显影。在该例子中,当(1)磁体辊是由排斥极组成,(2)在显影区的磁通量密度为500到1,200高斯以及(3)载体的饱和磁化强度为20到70Am2/g时,彩色复印能够得到很好的图像均匀性和层次再现性。
如果载体具有大于70Am2/g的饱和磁化强度(相对于施加的磁场为3,000奥斯特),在显影时,在与感光鼓(潜像承载元件)上形成的静电潜像相对的显影套筒上的载体和色调剂之外,会形成紧密的刷状磁耳,从而引起色彩层次和网点阶调再现性降低。如果它具有小于20Am2/g的饱和磁化强度,对色调剂和载体来说很难在显影套管上充分携带,从而容易引起载体粘合或色调剂散射的问题。
在转印步骤中,可以使用电晕充电组件、转印辊或转印带作为转印工具。而且,当把转印步骤后存在于感光鼓上的转印残留色调剂通过感光鼓表面运送到显影部件,从而被收集和再利用时,不改变感光鼓充电偏压也能进行。但是,在实际应用中,可以认为在转印纸卡纸或连续复印具有高图像区百分比的图像时,把过量的色调剂混合到色调剂充电组件中。
在这种情况下,在静电照相设备的运行过程中,能够利用感光鼓上没有形成图像的面积(即无图像区)把色调剂从充电组件转移到显影组件。这种无图像区指的是在向前旋转时、向后旋转时和转印纸张之间的区域存在的区域。在这种情况下,也优选把充电偏压改变成能够使色调剂很容易地从充电组件转移到感光鼓上的充电偏压。施加这种能使色调剂很容易地从充电组件出来的偏压的方法可以是使AC成分的峰-峰电压稍微变小或用DC成分替代的方法,或者是把峰-峰电压设定为相等和改变波形使AC的有效值变低的方法。
在转印步骤中,作为转印介质,(i)可以使用记录纸(一种记录介质)从而使在潜像承载元件上所形成的色调剂图像直接转印到这种记录介质上,并且(ii)也可以使用一种中介转印元件,从而使在潜像承载元件上所形成的色调剂图像首先转送到中介转印元件上,随后再将转印到中介转印元件上的色调剂图像转印到记录介质上。
本发明的色调剂具有优良的释放性能和优异的转印性能,并因此在上述成像方法中优选使用它,所述的成像方法是把在潜像承载元件上所形成的色调剂图像通过中介转印元件转印到记录介质上。
在把在潜像承载元件上或中介转印元件上所形成的色调剂图像转印到记录介质上的成像方法中,可以优选使用这样一种方法,即把在潜像承载元件上或中介转印元件上使用许多色调剂形成的多色调剂图像一次全部地转印到记录介质上。
本发明的色调剂具有优异的无凝结特性和均匀充电性能。因此,它能真实再现细小的潜像,并能优美地显出数字潜像。特别对全色图像,它能实现辉亮区的优异再现性和精细色差的再现,并且能形成全色图像,这些图像充满了物质感,并且光滑、鲜亮和有图画感。所以,也能漂亮地得到图表图像和线条字符图像,该色调剂优选用于数字全色复印机或打印机中。
下面将参考图2描述上述成像方法,其中把多色调剂通过中介转印元件一次转印到记录介质上。
通过充电辊2与感光鼓3接触旋转,使得作为潜像承载元件的感光鼓3的表面具有表面电势,并通过曝光设备1形成静电潜像。通过第一显影组件4、第二显影组件5、第三显影组件6和第四显影组件7把静电潜像连续显影,来形成相应的色调剂图像。把这样形成的每种色彩的色调剂图像多次转印到中介转印元件11上,形成一种多色调剂图像。
作为中介转印元件11,使用一种鼓状元件,其中可以使用一种在其周围安置一种固定元件的元件,或者可以使用一种包含基底和一种设于其上的导电性供应元件的元件,导电性供应元件如一种弹性层(如丁腈橡胶),其中已充分分散有炭黑、氧化锌、氧化锡、碳化硅或二氧化钛。也可以使用带状中介转印元件。该中介转印元件优选由硬度为10到50度(JIS K-6310)的弹性层组成,或者在转印带的情况中,由带有一种在转印区具有这种硬度的弹性层的支撑元件组成,在转印区把色调剂图像转印到转印介质(记录介质)上。
为了把色调剂图像从感光鼓3转印到中介转印元件11上,从电源13向中介转印元件11的磁心金属9施加一个偏压,从而形成转印电流,并转印色调剂图像。可以使用从固定元件或带背后的电晕放电或辊式充电。
转印充电组件114把中介转印元件11上的多色调剂图像一次全部地转印到记录介质S上。作为转印充电组件,可以使用利用转印辊或转印带的电晕充电组件或接触静电转印设备。
在定影步骤中,通过热和/或压力的帮助,将通过上述任何一种方法转印到记录介质上的色调剂图像定影在记录介质上。
在本发明中,对于在转印步骤中没被转印的潜像承载元件上的转印残留色调剂,可通过下述任一方法收集:(i)一种显影前清洁系统,该系统中使一种清洁元件与潜像承载元件的表面接触以除去和收集转印残留色调剂,和(ii)一种显影即时清洁系统,其中在显影时显影组件同时收集转印残留色调剂。为了使整个成像设备小型化和使潜像承载元件具有较长的寿命,优选显影即时清洁系统。
在显影即时清洁系统中,按潜像承载元件表面移动方向,依次按顺序设置显影区、转印区和充电区,而且该系统没有任何用于除去潜像承载元件表面上存在的转印残留色调剂的清洁元件,如果不是这种清洁系统,在转印区和充电区之间以及在充电区和显影区之间需要提供这种与潜像承载元件表面接触的清洁元件。
通过给出一个反转显影的例子来描述使用显影即时清洁系统的成像方法,该例子中,把色调剂的电荷极性设置得与潜像承载元件的静电潜像的电荷极性一致来实现显影。当使用可充负电的感光鼓和可充负电的色调剂时,在转印步骤中借助正极性的转印元件把可见的图像转印到转印介质,转印残留色调剂的电荷极性随着转印介质的类型(厚度、电阻和介电常数的差异)和图像区的不同而从正变到负。但是,即使转印残留色调剂的极性在转印步骤中已与感光鼓表面的极性一起变为正性,用来使可充负电的光敏元件充电的负极性充电元件能够均匀地调节电荷极性为负性。因此,当使用反转显影作显影方法时,即使在显影时均匀带负电的色调剂粒子存在于感光鼓表面,保持负电的转印残留色调剂保留在待显影的色调剂的亮区电势区。在不应被色调剂显影的色调剂黑区电势区,色调剂被吸附到与显影电场相关的显影剂携带元件,而不保留在负极性的感光鼓上。
图1图示说明成像设备,它能实现本发明的成像方法。
成像设备的主体是并排提供的第一成像装置Pa、第二成像装置Pb、第三成像装置Pc和第四成像装置Pd,通过潜像形成、显影和转印的工艺在转印介质上形成各个不同色彩的图像。
在成像设备中并排提供的各个成像装置都是按如下所述构成的,以第一成像装置为例。
第一成像装置Pa有直径为30mm的静电照相感光鼓61a作为潜像承载元件。该感光鼓61a以箭头a的方向旋转。参考号62a表示作充电设备的初级充电组件,并使用了一个磁刷充电组件,它包括直径16mm的套筒,该套管上运载的磁性粒子与感光鼓61a接触。参考号67a表示一种作为潜像形成设备的曝光设施,用于在感光鼓61a上形成一种静电潜像,该感光鼓61a表面已经通过初级充电组件62a进行均匀充电。参考号63a表示一种作为显影设备的显影组件,它带有一种彩色色调剂,用于使感光鼓61a上的静电潜像显影,形成彩色色调剂图像。参考号64a表示作为转印设备的转印刮板,用于把在感光鼓61a表面上形成的彩色色调剂图像转印到由带状转印介质携带元件68运送的转印介质表面。该转印刮板64a与转印介质携带元件68的背面接触,并能施加转印偏压。
在此第一成像装置Pa中,用初级充电组件62a使感光鼓61a的光敏元件首次均匀充电,由此通过曝光设备67a在光敏元件上形成静电潜像。用彩色色调剂通过显影组件63a把静电潜像显影。在第一转印区(光敏元件和转印介质之间相互接触的部位)通过施加转印偏压把这样显影形成的色调剂图像转印到转印介质表面上,该偏压来自与携带和输送转印介质的带状转印介质携带元件68背部接触的转印刮板64a。
该第一成像装置Pa没有任何用于从感光鼓表面除去转印残留色调剂的清洁元件,而一般的其他设备是要在与感光鼓表面接触的转印区和充电区之间以及充电区和显影区之间提供这种清洁元件的。本发明是使用了显影即时清洁系统,该系统中显影组件收集感光鼓上的转印残留色调剂,并在显影的同时清洁它的表面。
在本成像设备中,并排提供的第二成像装置Pb、第三成像装置Pc和第四成像装置Pd的构造与第一成像装置Pa相同,只是在显影组件上装有不同的彩色色调剂。例如,黄色色调剂用于第一成像装置Pa,品红色调剂用于第二成像装置Pb,青色色调剂用于第三成像装置Pc和黑色色调剂用于第四成像装置Pd,并且在各成像装置的转印区连续将转各彩色色调剂转印到转印介质上。在这个过程中,在定位的同时将各彩色色调剂于转印介质一次移动的过程中重叠在相同转印介质上。完成转印后,利用一种分离充电组件69把转印介质与转印介质携带元件68的表面分开,然后通过输送设备如输送带送到定影组件70,在此通过一次定影就形成了最终的全色图像。
定影组件70具有相匹配的直径40mm的定影辊71和直径30mm的压力辊72。定影辊71具有加热装置75和76。参考号73表示用于除去定影辊上的任何污点的网。
使转印到转印介质上的未定影彩色色调剂图像通过定影辊71和压力辊72之间的压力接触区,通过热和压力的作用把彩色色调剂图像定影到转印介质上。
在图1所示的设备中,转印介质携带元件68是一个循环的带状元件。通过传动辊80在箭头e的方向使带状元件运动。参考号79表示转印带清洁设备;81,传动带随动辊;82,传动带电荷消除器。参考号83表示一对相对抗的辊,用于将存在于转印介质盒中的转印介质运送到转印介质携带元件68上。
作为转印设备,可以用与转印介质携带元件的背面相接触的接触转印设备替代与转印介质携带元件的背面接触的转印刮板,该接触转印设备能够直接施加偏压,这种接触转印设备的例子为一种滚筒型转印辊。
上述接触转印设备也可以用一种非接触的转印设备替代,该非接触转印设备通过施加来自与常用的转印介质携带元件的背部不接触的电晕充电组件的转印偏压来进行转印。
但是,从能够控制施加偏压时产生的臭氧量的益处考虑,更优选使用接触转印设备。
将参考图3描述成像方法,其中在多个成像部件上分别形成不同色彩的色调剂图像,并将它们转印到相同的转移介质上同时将这些图像连续叠加在一起。
在这种方法中,排列形成第一、第二、第三和第四成像部分29a、29b、29c和29d,该成像部分分别具有其专用的潜像承载元件,即感光鼓19a、19b、19c和9d。
感光鼓19a到19d的周围分别提供潜像形成装置23a、23b、23c和23d,显影装置17a、17b、17c和17d,转印放电装置24a、24b、24c和24d以及清洁装置18a、18b、18c和18d。
在这种结构下,首先,例如,在第一成像部分29a的感光鼓19a上,通过潜像形成装置23a形成一种黄色成分的彩色潜像。用显影装置17a中具有黄色色调剂的显影剂把该潜像转化成可见图像(色调剂图像),并通过转印装置24a把该色调剂图像转印到转印介质S(记录介质)上。
当如上所述把黄色色调剂图像转印到转印介质S上时,在第二成像部分29b中,在感光鼓19b上形成了品红成分的彩色潜像,并随后使用具有品红色调剂的显影剂,在显影装置17b中将该彩色潜像转化成一种可见图像(一种色调剂图像)。当把在第一成像部分29a中已完成转印的转印介质S输送到转印装置24b上时,重叠这种可见图像(品红色调剂图像),并转印到转移介质S的预置位置上。
随后,以如上描述的相同方式,分别在第三和第四成像部分29c和29d上形成青色和黑色的色调剂图像,重叠青色和黑色的色调剂图像并转印到相同转印介质S上。一旦完成了这种成像步骤,就把转印介质S输送到定影部分22,在那儿把转印介质S上的色调剂图像定影。这样,在转印介质S上就获得了多色图像。分别用清洁装置18a、18b、18c和18d清洁已完成转印的感光鼓19a、19b、19c和19d来除去剩余的色调剂,并为随后将要进行的下一个潜像形成作准备。
在上述成像设备中,用输送带25运送记录介质即转印介质S。如图3中所看到的,把转印介质S从右边运送到左边,并且在运送过程中,分别通过各个成像部分29a、29b、29c和29d的转印装置24a、24b、24c和24d。
在该成像方法中,作为输送转印介质的输送装置,从容易工作和耐久性来看,使用由Tetoron纤维组成的网构成的输送带和由聚对苯二甲酸乙二醇酯树脂、聚亚胺树脂或尿素树脂制成的薄介电纸构成的输送带。
在转印介质S通过了第四成像部分29d之后,把AC电压施加到电荷清除器20,在其上转印介质S放电,从带68中分离出来,之后将其送到定影组件22,将色调剂图像定影,并且最后通过纸出口26送出。
在这种成像方法中,成像部分分别提供有独立的潜像承载元件,并且转印介质可以被带型输送装置连续送到各个潜像承载元件的转印区。
另一方面,在这种成像方法中,可以提供对各个成像部分通用的潜像承载元件,并且由鼓型输送装置把转印介质重复地送到潜像承载元件的转印区,从而获取各种色彩的色调剂图像。
但是,由于该转印带具有高体积电阻率,如同在彩色成像设备中,当转印重复几次的同时输送带继续增加电荷量。所以,除非在每次转印时连续地使转印电流变高,否则不能维持均匀的转印。
本发明的色调剂具有如此优异的转印性能,以致于在类似转印电流下,即使在每次重复转印时充电装置的充电已经增加,都能够使每次转印的色调剂转印性能达到均匀,从而能够获得高水平的优质图像。
参考图4将进一步描述按照另一实施方案形成全色图像的成像方法。
用具有第一彩色色调剂和载体的双组分显影剂使通过适当装置在感光鼓33上形成的静电潜像可见,该显影剂存放在作为显影装置的显影组件36中,该显影组件连接到沿箭头方向旋转的旋转显影单元39上。用转印充电组件44把这样形成在感光鼓33上的彩色色调剂图像(第一色彩)转印到被抓纸器47保持在转印鼓48上的转印介质(一种记录介质S)上。
在转印充电组件44中,使用电晕充电组件或接触转印充电组件。在转印充电组件44中使用电晕充电组件时,施加-10kV到+10kV的电压,并把转印电流设置为-500μA到+500μA。在转印鼓48的外面提供一种保持元件。这种保持元件是由膜状介电纸如聚偏二氟乙烯树脂膜或聚对苯二甲酸乙二醇酯膜组成。例如,使用100μm到200μm厚和1012到1014Ω·cm体积电阻率的纸。
接下来,对第二色彩,使旋转显影单元旋转直到显影组件35面对感光鼓33。然后,用存在于显影组件35中的具有第二彩色色调剂和载体的双组分显影剂把第二色彩的潜像显影,重叠这样形成的彩色色调剂图像,并转印到相同的转印介质上,如上述的记录介质S。
对第三和第四色彩重复相似的操作。这样,使转印鼓48旋转一定时间,同时保持把转印介质即记录介质S被抓在其上,从而将相应给定色彩的色调剂图像多次转印到记录介质上。优选按第一色彩、第二色彩、第三色彩和第四色彩的顺序依次使静电转印的转印电流变大,从而使转印后残留在感光鼓上的色调剂可以更少。
同时,由于转印图像会模糊,所以不优选高的转印电流。但是,因为本发明的色调剂具有良好的转印性能,待多次转印的第二、第三和第四彩色图像能够被准确转印。所以,清楚地形成了每种色彩的图像,并能够获得色调鲜明的多色图像。在全色图像中,也能获得具有良好色彩重现性的漂亮图像。而且,由于不再必需使转印电流这么高,所以在转印步骤中也很少出现图像模糊。当从转印鼓48分离出记录介质S时,用分离充电组件45消除电荷,在这里如果转印电流较大的话,记录介质S可以被牢牢静电吸引到转印鼓上,并且转印介质不能被分离下来,除非在分离时电流变得更大。如果变得更大,由于这种电流有相反于转印电流的极性,色调剂图像就模糊,或者色调剂从转印介质扩散而污染成像设备的内部。由于本发明的色调剂可以很容易地转印,转印介质能够很容易地分离而不使分离电流变大,从而避免了在分离时图像的模糊和色调剂的扩散。所以,本发明的色调剂特别是在具有多次转印步骤形成多色图像或全色图像的成像方法中优选使用。
用分离充电组件45从转印鼓48上分离其上已完成多次转印的记录介质S。然后借助热压辊定影组件3定影保留在上面的色调剂图像,该热压辊组件具有用硅油浸渍的网,并在定影时加色混合,这就形成复印的全色图像。
按照供应信号,从提供给各种彩色色调剂的装料斗中,通过色调剂输送线路,将待供给显影组件34-37中的供应色调剂按预定量输送到位于旋转显影单元中心的色调剂供应圆筒,在此把色调剂供给各个相应显影组件。
参考图5,以全色成像设备为例,描述多重显影一次转印的方法。
借助显影组件104、105、106和107,通过使用色调剂的连续显影步骤,使通过充电组件102和利用激光的曝光装置101在感光鼓103上形成的静电潜像显影。在显影步骤中,优选使用非接触显影。在非接触显影中,在显影组件中形成的显影剂层不会在感光鼓103的表面上摩擦,因此在第二次和随后进行的显影步骤中进行显影时不会使在前一步骤形成的图像模糊。关于显影的的顺序,在多色显影时,优选首先用一种不是黑色且亮度和色品较高的色彩使潜像显影。在全色显影时,显影顺序优选是黄色,然后是品红或者青色,之后是品红或者青色中的剩余者,最后为黑色。
用转印充电组件109,将在感光鼓103上重叠形成的多色图像或全色图像的色调剂图像转印到一种转印介质,即记录介质S上。在转印步骤中,优选使用静电转印,即使用电晕放电转印或接触转印。在前者电晕放电转印方法中,产生电晕放电的转印充电组件109与色调剂图像相对而置,在它们之间插入转印介质即记录介质S,并使电晕放电作用在记录介质的背部,来静电转印色调剂图像。在后者的接触转印方法中,使转印辊或转印带与感光鼓103接触,然后在转印色调剂图像的同时向该辊施加偏压,或者通过从该带背部静电充电,在它们之间插入转印介质即记录介质S。通过这种静电转印,一次将保留在感光鼓103上的多色色调剂图像转印到转印介质即记录介质S上。由于在这种一次转印系统中转印的色调剂是大量的,转印后会残留大量的色调剂,容易引起不均匀的转印,在全色图像中容易引起色彩的不均匀。
但是,本发明的色调剂在转印性能上是这样的优良以致于能够匀整地形成任何多色图像的彩色图像。在全色图像中,能够获得较好色彩再现性的漂亮图像。而且,由于即使在低电流下该色调剂也能够优质转印,因此能够抑制模糊图像的出现。再者,由于容易分离记录介质,在分离时也能抑制任何色调剂的扩散。另外,因为较好的释放性,在接触转印设备中也能获得好的转印性能。所以,本发明的色调剂也优选用于具有多图像一次转印步骤的成像方法中。
从感光鼓103上分离已经一次转印有多色色调剂图像的记录介质S,然后借助热辊定影组件112定影,最后在其上形成多色图像。
如图1到5所示成像设备中的显影组件可以使用图6中所示的双组分显影组件,该设备利用本发明的双组分显影剂进行显影。
如图6所示,用来使形成在感光鼓1(作为潜像承载元件)上的静电潜像显影的显影组件133具有显影容器126,其内部通过分隔壁127分隔成显影室(第一室)R1和搅拌器室(第二室)R2。在搅拌器室R2的上部,在分隔壁127的另一侧上形成色调剂储藏室R3。把显影剂129放于显影室R1中和搅拌室R2中,把补充色调剂(非磁性色调剂)128放于色调剂储藏室R3中。在色调剂储藏室R3上提供一个供料口130,从而通过该供料口130卸下相应消耗量的补充色调剂128,并供入搅拌器室R2。
在显影室R1内部安装一个输送叶轮123。当旋转输送叶轮123时,以显影套筒121的纵向输送显影室R1内的显影剂129。同样,在搅拌器室R2中安装一个输送叶轮124并且当旋转输送叶轮124时,以显影套筒121的纵向输送已从供料口130卸入搅拌器室R2中的色调剂。
显影剂129是一种包含非磁性色调剂129a和一种磁性载体129b的双组分显影剂。
在显影容器126相邻感光鼓120的部位设置一个开口,显影套筒121从开口处伸出,在显影套筒121和感光鼓120之间形成一个间隙。给由非磁性材料制成的显影套筒121提供一种在显影时施加偏压的偏压施加设备(在图中没有显示)。
固定在显影套筒121内部的作为磁场产生装置的磁体辊,即磁体122,有一个显影磁极N,在其下游侧的磁极S,以及磁极N、S和S,用于运送显影剂129。在显影套筒121中以这样一种方式提供磁体122,即显影磁极S面对感光鼓120。显影磁极S在显影区附近产生了一个磁场,显影区被定义在显影套筒121和感光鼓120之间,在此磁场形成了一个磁刷。
在显影套筒121下面,提供一个由非磁性材料如铝或SUS316不锈钢制成的非磁性刮板125,来调整在显影套筒121上显影剂129的层厚。在作为调整元件的非磁性刮板125的末端和显影套筒121的表面之间距离为300到1,000μm,并优选400到900μm。如果这个距离小于300μm,在它们之间可以捕获磁性载体,容易使显影剂层不匀,并且也不能在套筒上施加进行优良显影所需的显影剂,产生的问题是只能获得低浓度和很不均匀的图像。为了预防由于显影剂中包括不合格的粒子而造成的不均匀涂层(称之为刮板阻塞),这个距离优选400μm或更大。如果它大于1,000μm,涂覆在显影套筒121上的显影剂的量增加到不需要调整显影剂的层厚,造成下列问题,大量磁性载体粒子粘着到感光鼓120上,以及显影剂的循环和非磁性刮板125对显影剂的控制变得对显影剂的调整无效,容易引起图像模糊,这是由于缺乏色调剂的摩擦电的缘故。
即使显影套筒121在箭头方向旋转时,这层磁性载体粒子在随着根据磁力和重力所施加的粘合力和作用在套筒121运送的输送力之间的平衡进一步从套筒表面离开时移动较慢。当然一些粒子由于重力的作用会掉下来。
所以,可以适当选择安排磁极N和N的位置以及磁性载体粒子的流动性和磁性能,使得当接近套筒时把磁性载体粒子层送向磁极N,形成一个移动层。随着磁性载体粒子的移动,在显影套筒121旋转的同时把显影剂运送到显影区,供显影使用。
在图6所示的设备中,使感光鼓120初级充电的充电装置是一个磁刷充电组件,由内部有磁体辊的非磁性导电套筒131通过磁力将磁性粒子132粘着其上。
如上所述,本发明的色调剂具有特定成圆率分布和特定的重均粒子直径。而且,色调剂粒子上的色调剂外部添加剂为具有特定平均粒子长度和特定形状因子的无机细粉(A)和通过聚结粒子形成的具有特定形状因子的非球状无机细粉(B)。本发明的色调剂能够使较细小的潜像点以高图像质量真实地再现,并能经得起显影组件内部的任何机械应力,从而抑制色调剂的变质。
下面显示本发明的实施例。本发明决不受这些实施例的限定。在下面,“份”指的是“重量份”。
实施例1
在710份离子交换水中加入450份0.1M Na3PO4的水溶液,之后加热到60℃,然后用Clear混合机(由M Technic K.K.生产)以12,000rpm搅拌。向所得到的混合物中一点一点地加入68份1.0M CaCl2水溶液,得到含有磷酸钙化合物的含水介质。
(单体)
苯乙烯 165份
丙烯酸正丁酯 35份
(色料)
C.I.颜料兰15:3 15份
用球磨机把上述材料研细,之后加入下列材料。用加热到60℃的TK-型均匀混合机(由Tokushu Kika Kogyo有限公司生产)将所得到的混合物以12,000rpm均匀溶解和分散。接着,溶解10份聚合引发剂2,2’-偶氮双(2,4-二甲基戊腈)得到可聚合的单体组合物。
(电荷控制剂)
水杨酸金属化合物 3份
(极性树脂)
饱和的聚酯树脂 10份
(隔离剂)
酯蜡(熔点70℃) 50份
把上述可聚合的单体组合物加入上述含水介质中,之后在氮气下于60℃用Clear混合机以12,000rpm搅拌10分钟,来粒化可聚合的单体组合物。然后,把得到的粒化产物转移到反应容器中,并用一个浆状搅拌叶片搅拌,其间温度升高到80℃,进行聚合10小时。完成聚合后,减压蒸发掉剩余的单体,冷却反应体系,然后向其中加入盐酸来溶解磷酸钙,再过滤,用水洗涤,然后干燥得到着色的悬浮液粒子(色调剂粒子),其重均粒子直径为6.1μm,具有窄的粒子大小分布。
向如此得到的100份色调剂粒子中,外加入1.0份锐钛矿型疏水二氧化钛细粉(1)(体积电阻率:7×109Ω·cm),该细粉已经用10份异丁基三甲氧基硅烷在含水介质中处理,其BET比表面积为100m2/g,再外加入1.0份BET比表面积为43m2/g的非球形二氧化硅细粉(1),得到悬浮聚合青色色调剂1。
上述的二氧化硅细粉(1)是按下述方法获得的产品,即用10份六甲基二硅氮烷对100份商业上购买的二氧化硅细粒AEROSIL#50(Nippon Aerosil C0.,Ltd提供)进行表面处理,然后用空气分级器分级,收集相对粗糙的粒子,以控制粒子大小分布。在透射电镜(TEM)得到的100,000倍放大照片和扫描电镜(SEM)得到的30,000倍放大照片上,可以证实二氧化硅细粉(1)是由许多平均粒子直径为40mμm的初级粒子聚结形成的粒子。
悬浮聚合青色色调剂1中色调剂粒子上的二氧化钛细粉(1)具有形状因子SF-1为120,而其上的二氧化硅细粉(1)的形状因子SF-1为195。
用扫描电镜得到的悬浮聚合青色色调剂1的100,000倍放大照片中,可以证实二氧化钛细粉(1)的平均长度为50mμm,长/宽比为1.1,其数量为每0.5μm×0.5μm单位面积中有25个粒子。用扫描电镜得到的悬浮聚合青色色调剂1的30,000倍放大照片中,可以证实二氧化硅细粉(1)的平均长度为168mμm,长/宽比为2.8,其数量为每1μm×1μm单位面积中有17个粒子。放大照片中证实的二氧化硅细粉(1)的粒子形状如图10所示。
用Coulter粒子计数器可测出悬浮聚合青色色调剂1的重均粒子直径为6.1μm,用流动型粒子图像分析仪测出其成圆率分布中平均成圆率为0.983,并且其中11%(数量)的色调剂粒子的成圆率小于0.95。
将上述悬浮聚合青色色调剂1与后面的显影载体I混合成色调剂浓度为8%,制成双组分青色显影剂(1)(表观密度:1.45;紧密度:12%)
双组分青色显影剂(1)的表观密度和紧密度的值由下面的测试方法测定。
表观密度测定:
使用粉末测定仪,用75μm目的筛子在震动幅度为1nm的条件下振动,当粒子通过时测定表观密度A。
紧密度的测定:
使用粉末测定仪,测定180次上下反转后的堆密度P,用于计算双组分显影剂的紧密度。
紧密度=(P-A)/P×100(%)
其中A代表双组分显影剂的表观密度,P代表堆密度。
显影载体I的制备
苯酚/甲醛(50∶50)单体混合并分散在含水介质中。然后基于单体的重量,将经钛酸异丙氧基三异硬脂基酯(isopropoxytriisostearoyltitanate)表面处理后的600份0.25μm磁铁矿粒子和400份0.6μm的赤铁矿粒子均匀分散,加入适量的氨后将单体聚合,得到包含有球形磁性树脂载体核的磁性颗粒(平均粒子直径:33μm;饱和磁化强度:38Am2/kg)。
将20份甲苯,20份丁醇,20份水和40份冰加入四颈烧瓶中,然后边搅拌边加入40份由15摩尔CH3SiCl3、10摩尔(CH3)2SiCl2和一种催化剂组成的混合物,进一步搅拌30分钟后,在60℃下进行缩合反应1小时。然后,将硅氧烷用水充分清洗,溶解于甲苯/甲基乙基酮/丁醇的混合溶剂中得到含10%固体的硅氧烷清漆。
以100份硅氧烷固体为基数,向如此得到的硅氧烷清漆中,同时加入2.0份离子交换水,2.0份式(1)所示的固化剂,1.0份式(2)所示的氨基硅烷偶联剂和5.0份式(3)所示的硅烷偶联剂,制成载体涂层溶液I。

(CH3)2N-C3H6-Si-(OCH3)3 (2)
n-C3H7-Si-(OCH3)3 (3)
用涂覆仪器(SPIRACOATER,Okada Seiko K.K.制造),将由此得到的载体涂层溶液I涂在前述的载体核上,1重量份树脂涂层涂覆100份核,可得到涂覆载体I(显影载体I)。
用下面的方法可测出这种显影载体I的体积电阻率为4×1013Ω·cm,矫顽力为55奥斯特。
体积电阻率的测定:
使用如图9所示的电池测定体积电阻率。更确切的说,电池A与样品143封装在一起,使下层的电极141和上层电极142与封装的样品143接触,在电极上外加1,000V DC的电压,用电表测定即时电流以确定体积电阻率。标注144表示一种绝缘材料。测定条件为:封装样品与电池的接触面积S为2cm2,厚度d为3mm,上层电极载荷15kg。
磁性能测定:
使用BHU-60型磁化测量设备(Riken Sokutei Co.制造)。称量待测样品1.0g,封装在直径7mm高10mm的电池中,并将电池放入上述测量设备中。逐步增加外加磁场至最大值1,000奥斯特进行测量。然后降低外加磁场,最后在记录纸上得到一条样品的滞后曲线。由此可以测定饱和磁化强度,剩磁强度和矫顽力。
将双组分显影剂(1)加入图1所示的成像设备中的第一成像单元Pa的显影组件63a中,将悬浮聚合青色色调剂1加入色调剂料斗65a中。使用色斑浓度检测工具(未示出)控制显影组件63a中双组分显影剂(1)的色调剂浓度以维持在7%到9%之间。通过色调剂供给元件66a从色调剂料斗65a向显影组件63a中补充悬浮聚合青色色调剂1,在23℃/65%RH,30℃/80%RH和20℃/10%RH的环境中连续复印30,000张青色单色纸。
成像设备上的第一成像单元Pa由后面作为感光鼓61a的感光元件No.1和作为初级充电组件62a的磁刷充电组件No.1组成。磁刷充电组件以与感光鼓61a表面移动方向相反的方向以120%的速度旋转。首先将感光鼓61a充电至-700V,同时施加通过在-700V DC电流上叠加1kHz和1.2kVpp的AC电压而形成的充电偏压。另外,第一成像单元Pa没有用于清除和收集存在于感光鼓61a表面上的转印残留色调剂的清洗元件,而其他装置中在与感光鼓61a表面接触的转印区和充电区之间以及充电区和显影区之间却提供了这种清洗部件,这种单元的构造是具有显影清除系统,其中在进行显影时,利用双组分显影剂的磁刷清除和收集转印步骤完成后感光鼓61a表面上的转印残留色调剂。在显影组件63a中显影时,显影对比设为250V,防模糊反转对比为-150V,以在显影套管上施加如图7所示的不连续交流电压的同时进行显影。
-感光元件No.1-
感光元件No.1是OPC感光元件,使用了一种用于充负电的有机光电导材料。在直径为30mm的铝圆柱上从第一到第五层形成了下面5层功能层。
第一层是大约20μm厚的分散有导电粒子的树脂层。用于使铝圆柱上的任何缺陷变得光滑水平,也可防止由于激光曝光光线反射形成的斑纹。
第二层是正电荷注入防止层(胶层),是大约1μm厚的中等电阻层,其功能是防止从铝基底注入的正电荷对由于充电而在感光元件表面产生的负电荷的抵消。用6-66-610-12尼龙和甲氧基甲基化的尼龙调整这一层的电阻率为约106Ω·cm。
第三层为电荷产生层,厚度大约0.3μm,由二偶氮颜料分散在树脂中形成,暴露于激光下可产生正负电荷对。
第四层是电荷输送层,由腙粒子分散在聚碳酸酯树脂中形成,是p型半导体。因此,感光元件表面因充电形成的负电荷不能通过这一层,只有电荷产生层形成的正电荷可输送到感光元件表面。
第五层是电荷注入层。由可光固化的丙烯酸树脂形成,其中分散有SnO2超细粒子,并且为了延长充电元件与感光元件的接触时间以进行均匀充电,还分散有粒子直径大约为0.25μm的四氟乙烯树脂粒子。具体地说,以树脂重量为基准,分散有160%(重量)的粒子直径大约为0.03μm的无氧型低电阻SnO2粒子和30%(重量)的四氟乙烯树脂粒子和1.2%(重量)的分散剂。
由此得到的感光元件1表面层的体积电阻率与电荷输送层这一层的电阻率5×1015Ω·cm相比要低,为6×1011Ω·cm。
磁刷充电组件No.1
将5份MgO,8份MnO,4份SrO和83份Fe2O3均制成细粒子,然后加入水混合进行粒化,接下来在1300℃下焙烧,调整粒子大小得到平均粒子直径为28μm的铁氧体载体核(饱和磁化强度:63Am2/kg,矫顽力:55奥斯特)。
将上面的载体核用10份混合于99份己烷和1份水形成的混合溶剂中的钛酸异丙氧基三异硬脂基酯进行表面处理,处理量为0.1份,得到磁性粒子a。
用与测定显影载体I的体积电阻率相同的方法测出这种磁性粒子的体积电阻率为3×107Ω·cm,加热时的重量损失为0.1份。
磁刷充电装置No.1由一磁体辊放入其内侧的导电非磁性套筒和将上述磁性粒子通过磁性粘合在其表面上形成的磁刷组成,充电时磁体辊固定,导电非磁性套筒可旋转。
在上述30,000张连续复印实验中,对开始阶段图像上实体的均匀性,30,000张后的模糊性,开始阶段与运行30,000张后的图像浓度的差别显示出的运行性能,开始阶段与运行30,000张后的转印性能进行评价。根据色调剂在低湿度环境(20℃/10%RH)和高湿度环境(30℃/80%RH)之间摩擦电荷数量的不同对色调剂的环境稳定性进行了评价。
评价结果列于表3。图像浓度稳定,模糊性和转印性能方面没有问题,得到了很好的结果。
实体均匀性:
供复制用的原像是5个直径为20mm的圆点,用反射密度计RD918(Macbeth Co.制造)测得图像浓度为1.5。复制后,用反射密度计RD918测量图像区域的图像浓度以确定测量的最大值与最小值之间的差。
图像浓度:
供复制用的原像是直径为20mm的圆,用反射密度计RD918(Macbeth Co.制造)测得图像浓度为1.5。复制后,用反射密度计RD918测定图形区域的图像浓度。
模糊量:
用成像前测定的纸上10个点的反射密度平均值(Dr)减去成像后在非图像区域(白色背景)上测定的10点的反射密度最差值(Ds),得到的差值(Dr-Ds)表示为模糊量。
用TC-6DS型反射计(Tokyo Denshoku Co.,Ltd.制造)测量反射密度。模糊量小于等于2%为优质图像,基本没有模糊;模糊量大于5%的为粗糙图像,有明显的模糊。
转印性能:
在感光鼓上使实心图像显影,在转印过程中停止机器运行。用Mylar带收集感光鼓上的色调剂,该带固定在转印纸的白色背景区域。转印纸上的色调剂也用Mylar带固定。按以下方式计算转印性能(转印效率):转印性能(%)=(转印纸上的Macbeth密度/鼓上的Macbeth密度)×100
色调剂摩擦电荷的数量:
色调剂摩擦电荷的数量用下面的方式进行测量,如图8所示,使用了一个测量摩擦电荷数量的装置。
首先,将用于测量的色调剂和磁性粒子以1∶19的比例混合(放入50到100ml聚乙烯瓶容器中并手动震荡约10到40秒)而制备的大约0.5到1.5g混合物放入金属测量容器52中,容器底部有500目的筛子53,将容器用金属板54盖起来。此时测出测量容器52的总重量,记为W1(g)。然后,在一个抽气装置51中(至少与测量容器52接触的部分应用绝缘材料制造),将空气从抽气口57抽出,用空气流量控制阀56控制真空表55的压力读数为250mmAq。这时优选充分抽气大约2分钟,通过抽气移去色调剂。这一阶段用电位表59测得的电压记为V(伏)。在图8中,标注58代表一电容器,其电容记为C(mF)。完成抽气后测出测量容器的总重量,记为W2(g)。摩擦电荷数量Q(mC/kg)用下面的表达式计算:
色调剂的摩擦电荷数量:
(mC/kg)=(C×V)/(W1-W2)
(测量条件:低湿度为20℃/10%RH,高湿度为30℃/80%RH)
用构成双组分显影剂的载体与色调剂一起作为测量中所用的磁性粒子。
实施例2
悬浮聚合青色色调剂2的物理性能见表2,制备方法与实施例1相同,不同的是用BET比表面积为40m2/g的二氧化硅细粉(2)代替了二氧化硅细粉(1),二氧化硅细粉(2)由许多平均粒子直径为60mμm的初级粒子聚结形成的的聚结粒子组成。
按照与实施例1相同的方法使用上述的悬浮聚合青色色调剂2制备双组分显影剂(2)(表观密度:1.49,紧密度:13%)。性能评价方法也与实施例1相同。
评价结果列于表3。尽管在30,000次运行后转印性能有一点低,但仍得到了较好的结果。
对比例1
聚酯树脂由下述组分缩合而成
丙氧基化双酚,富马酸和1,2,4-苯三酸 100份
酞菁颜料 4份
二-叔丁基水杨酸的铝化合物 4份
低分子量聚丙烯 4份
将上述材料用Henshel混合器预混合,然后用双螺杆挤出机型捏和机进行熔融捏和。冷却后,用锤磨器将捏和产物粉碎形成直径约为1到2mm的粗糙粒子,接着用空气喷射系统的精细研磨器进行精细研磨。将得到的精细研磨产物进行分级得到蓝色粉末(色调剂粒子),重均粒子直径为6.0μm。然后用与实施例2相同的方法外加入二氧化钛细粉(1)和二氧化硅细粉(2)得到粉碎青色色调剂3,其物理性能见表2。
将上述球形处理的青色色调剂3用与实施例1相同的方法制成双组分显影剂(3)(表观密度:1.37;紧密度:21%)。性能评价方法也与
实施例1相同。
评价结果列于表3。在转印性能、模糊性和图像浓度方面均不能得到令人满意的结果。
实施例3
聚酯树脂由下述组分缩合而成
丙氧基化双酚,富马酸和1,2,4-苯三酸 100份
酞菁颜料 4份
二-叔丁基水杨酸的铝化合物 4份
低分子量聚丙烯 4份
将上述材料用Henshel混合器预混合,然后用双螺杆挤出机型捏和机进行熔融捏和。冷却后,用锤磨器将捏和产物粉碎形成直径约为1到2mm的粗糙粒子,接着用空气喷射系统的精细研磨器进行精细研磨。将得到的精细研磨产物进行进一步分级,然后用机械冲击的方法进行球化处理。这样可得到蓝色粉末(色调剂粒子),重均粒子直径为6.0μm。然后用与实施例2相同的方法外加入二氧化钛细粉(1)和二氧化硅细粉(2)得到球形处理的青色色调剂4,物理性能见表2。
用与实施例1相同的方法将上述球形处理的青色色调剂4制成双组分显影剂(4)(表观密度:1.41;紧密度:19%)。性能评价方法也与实施例1相同。
评价结果列于表3。尽管在30,000次运行后转印性能有一点低,仍得到了较好的结果。
实施例4
聚酯树脂由下述组分缩合而成
丙氧基化双酚,富马酸和1,2,4-苯三酸100份
酞菁颜料 4份
二-叔丁基水杨酸的铝化合物 4份
低分子量聚丙烯 4份
将上述材料用Henshel混合器预混合,然后用双螺杆挤出机型捏和机进行熔融捏和。冷却后,用锤磨器将捏和产物粉碎形成直径约为1到2mm的粗糙粒子,接着用空气喷射系统的精细研磨器进行精细研磨。将得到的精细研磨产物进行进一步分级,然后用热空气进行球化处理。这样可得到蓝色粉末(色调剂粒子),重均粒子直径为6.0μm。然后用与实施例2相同的方法外加入二氧化钛细粉(1)和二氧化硅细粉(2)得到球形处理的青色色调剂5,物理性能见表2。
用与实施例1相同的方法将上述球形处理的青色色调剂5制成双组分显影剂(5)(表观密度:1.43;紧密度:17%)。性能评价方法也与
实施例1相同。
评价结果列于表3。尽管环境稳定性有一点低,仍得到了较好的结果。
对比例2
聚酯树脂由下述组分缩合而成
丙氧基化双酚,富马酸和1,2,4-苯三酸 100份
酞菁颜料 4份
二-叔丁基水杨酸的铝化合物 4份
低分子量聚丙烯 4份
将上述材料用Henshel混合器预混合,然后用双螺杆挤出机型捏合机进行熔融捏和。冷却后,用锤磨器将捏和产物粉碎形成直径约为1到2mm的粗糙粒子,接着用空气喷射系统的精细研磨器进行精细研磨。将得到的精细研磨产物进行进一步分级,然后用热水浴进行球化处理。这样可得到蓝色粉末(色调剂粒子),重均粒子直径为6.0μm。然后用与实施例2相同的方法外加入二氧化钛细粉(1)和二氧化硅细粉(2)得到球形处理的青色色调剂6,物理性能见表2。
用与实施例1相同的方法将上述粉碎青色色调剂6制成双组分显影剂(6)(表观密度:1.89;紧密度:9%)。性能评价方法也与实施例1相同。
评价结果列于表3。图像模糊程度和图像浓度均不理想。
对比例3
悬浮聚合青色色调剂7的物理性能见表2,制备方法与实施例1相同,不同的是没有使用二氧化硅细粉(1),只是以100份色调剂粒子为基准外加入了2份二氧化钛细粉(1)。
按与实施例1相同的方法用上述的悬浮聚合青色色调剂7制成双组分显影剂(7)(表观密度:1.47,紧密度:13%)。性能评价方法也与
实施例1相同。
评价结果列于表3。转印性能与图像浓度均不理想。
对比例4
色调剂粒子制备方法与实施例1相同,不同的是通过加入0.1MNa3PO4水溶液和1.0M CaCl2水溶液制备磷酸钙化合物,同时保持Clear混合器的转速为6,000rpm。这样得到了粒子大小分布较宽的重均粒子直径为7.1μm的彩色悬浮粒子。将此粒子进行分级得到粒子大小分布较窄的重均粒子直径为6.5μm的彩色悬浮粒子(色调剂粒子)。用与实施例2相同的方法外加入二氧化钛细粉(1)和二氧化硅细粉(2)得到悬浮聚合青色色调剂8,物理性能见表2。
按与实施例1相同的方法用上述的悬浮聚合青色色调剂8制成双组分显影剂(8)(表观密度:1.40,紧密度:21%)。性能评价方法也与
实施例1相同。
评价结果列于表3。结果与对比例1得到的结果相似。这可以认为是色调剂的成圆率分布大体相同的原因,尽管色调剂制备方法不同。
实施例5
悬浮聚合青色色调剂粒子9的物理性能见表2,制备方法与实施例2相同,不同的是用锐钛矿型二氧化钛细粉(2)(体积电阻率2×1010Ω·cm;BET比表面积:92m2/g)代替了二氧化钛细粉(1),二氧化钛细粉(2)在Henschel混合器中用10份50厘泊的二甲基硅油进行过处理,处理方法为干处理。
按与实施例1相同的方法用上述的悬浮聚合青色色调剂9制成双组分显影剂(9)(表观密度:1.43,紧密度:14%)。性能评价方法也与
实施例1相同。
评价结果列于表3。与实施例2相比,实心图像浓度稍不均匀,可能是由于二氧化钛细粉的形状因子SF-1更小,但得到的结果较好。
对比例5
悬浮聚合青色色调剂粒子10物理性能见表2,制备方法与实施例1相同,不同的是用二氧化硅细粉(3)代替了二氧化硅细粉(1)。二氧化硅细粉(3)BET比表面积为26m2/g,并用10份六甲基二硅氮烷和10份50厘泊的二甲基硅油进行了处理,其由许多平均粒子直径为70mμm的初级粒子聚结形成的聚结粒子组成。
按与实施例1相同的方法用上述的悬浮聚合青色色调剂10制成双组分显影剂(10)(表观密度:1.40,紧密度:21%)。性能评价方法也与实施例1相同。
评价结果列于表3。与实施例1相比,图像浓度和模糊性均不理想,可能是由于二氧化硅细粉的形状因子SF-1更小。
实施例6
悬浮聚合青色色调剂粒子11物理性能见表2,制备方法与实施例1相同,不同的是外部添加剂的量变为:二氧化钛细粉(1)0.02份,二氧化硅细粉(1)1.0份。
按与实施例1相同的方法用上述的悬浮聚合青色色调剂11制成双组分显影剂(11)(表观密度:1.40,紧密度:22%)。性能评价方法也与实施例1相同。
评价结果列于表3。环境稳定性,图像模糊性和图像浓度均在较低的水平,但在这一水平上的实际应用没有问题。
实施例7
悬浮聚合青色色调剂粒子12物理性能见表2,制备方法与实施例1相同,不同的是外部添加剂的量变为:二氧化钛细粉(1)1.0份,二氧化硅细份(1)2.0份。
按与实施例1相同的方法用上述的悬浮聚合青色色调剂12制成双组分显影剂(12)(表观密度:1.49,紧密度:13%)。性能评价方法也与实施例1相同。
评价结果列于表3。环境稳定性与图像模糊性有一点低,但得到的结果较好。
实施例8
悬浮聚合青色色调剂粒子13物理性能见表2,制备方法与实施例1相同,不同的是用二氧化硅细粉(4)代替了二氧化硅细粉(1),二氧化硅细粉(4)是通过改变二氧化硅细粉(1)的分级条件以控制粒子大小分布而收集到的相对细小的粒子。
按与实施例1相同的方法用上述的悬浮聚合青色色调剂13制成双组分显影剂(13)(表观密度:1.52,紧密度:17%)。性能评价方法也与实施例1相同。
评价结果列于表3。图像有一点模糊,但得到的结果较好。
实施例9
悬浮聚合青色色调剂粒子14物理性能见表2,制备方法与实施例1相同,不同的是用二氧化硅细粉(5)代替了二氧化硅细粉(1),二氧化硅细粉(5)是通过改变二氧化硅细粉(1)的分级条件并重复进行几次分级以控制粒子大小分布从而能够收集到的更粗糙的粒子。
按与实施例1相同的方法用上述的悬浮聚合青色色调剂14制成双组分显影剂(14)(表观密度:1.41,紧密度:12%)。性能评价方法也与实施例1相同。
评价结果列于表3。实心图像浓度有一点低,转印性能也有一点低,但得到的结果较好。
对比例6
悬浮聚合青色色调剂粒子15物理性能见表2,制备方法与实施例1相同,不同的是没有用二氧化钛细粉(1),而只是以100份色调剂粒子为基准外加入了2份二氧化硅细粉(1)。
按与实施例1相同的方法将上述的悬浮聚合青色色调剂15制成双组分显影剂(15)(表观密度:1.41,紧密度:12%)。性能评价方法也与实施例1相同。
评价结果列于表3。模糊性、图像浓度和环境稳定性均不理想。
实施例10
双组分显影剂(16)(表观密度:1.88,紧密度:11%)的制备方法与实施例1相同,不同的是用下面的显影载体II代替了显影载体I。性能评价方法也与实施例1相同。出现了轻微的图像模糊,但得到的结果较好。
这可能是由于显影载体变成了铁氧体,同时由于重力的作用,补充色调剂的混合性能有点低。
显影载体II的制备
8份MgO,5份MnO和87份Fe203均制成粒子直径不超过0.1μm的细粒子,然后加入水均匀混合。将得到的混合物用喷雾干燥的方法粒化,使其平均粒子直径为35μm。接下来在1200℃下焙烧,去掉粗糙和细小的粉末得到铁氧体载体核。将由此得到的铁氧体载体核代替制备显影载体I中所用的含有磁性粒子的球形磁性树脂载体核,并用与制备显影载体I相同的方法进行表面涂覆。这样得到的显影载体II体积电阻率为2×1012Ω·cm,饱和磁化强度为37Am2/kg,矫顽力为5奥斯特。
实施例11
双组分显影剂(17)(表观密度:1.51,紧密度:14%)制备方法与实施例1相同,不同的是用下面的显影载体III代替了显影载体I。性能评价方法也与实施例1相同。结果在第30,000张时实心图像均匀性有一点低,但在这一水平的实际应用没有问题。这可能是由于显影载体有如此高的磁性以致于对显影区的色调剂有点破坏作用,影响了显影性能。
显影载体III的制备
显影载体III的制备方法与显影载体I相同,不同的是使用的磁铁矿粒子数量从600份变成了100份。
这样得到的显影载体III体积电阻率为8×1011Ω·cm,饱和磁化强度为65Am2/kg,矫顽力为78奥斯特。
实施例12
重复实施例2,只是显影套筒与感光鼓的旋转方向相同。结果是实心图像浓度有点不均匀,但得到的结果较好。
这可能是由于显影套筒旋转方向的改变使得难以平衡显影后显影剂的脱离和新鲜显影剂的表面涂覆,导致色调剂浓度的控制有一点不稳定。
实施例13
用与实施例1中悬浮聚合青色色调剂1相同的制备方法制备悬浮聚合黄色色调剂16,悬浮聚合品红色色调剂17和悬浮聚合黑色色调剂18,不同的是采用了C.I.颜料黄93,喹吖啶酮颜料和碳黑分别代替了C.I.颜料兰15:3。
用与实施例2相同的方法分别用上述悬浮聚合黄色色调剂16,悬浮聚合品红色色调剂17和悬浮聚合黑色色调剂18制成双组分黄色显影剂(18),双组分品红色显影剂(19)和双组分黑色显影剂(20)。
将由上面三种彩色双组分显影剂和实施例1中所用的双组分显影剂(1)组成的四彩色双组分显影剂用于如图1所示的成像设备中,按黄色,品红色,青色,黑色的颜色顺序构成色调剂图像,不使用任何清洗单元。将色调剂图像连续多次转印至一转印介质即记录介质上形成全色图像,这样连续转印30,000张。图像浓度只有很小的改变,得到了没有任何模糊的较好结果。
合成实施例1
苯乙烯 125份
甲基丙烯酸甲酯 35份
丙烯酸正丁酯 40份
酞菁铜颜料 14份
二-叔丁基水杨酸铝化合物 3份
饱和聚酯(酸值:10;峰值分子量:9100) 10份
酯蜡(Mw:450;Mn:400;Mw/Mn:1.13;
熔点:68℃;粘度:6.1mPa·s;维克斯硬度:
1.2;SP值:8.3) 40份
按上面配方配制的原料加热到60℃,然后用TK型均匀混合机(Tokushu Kika Kogyo C0.,Ltd.制造)在10,000rpm的条件下均匀分散溶解。在得到的混合物中溶入10份2,2′-偶氮双(2,4-二甲基戊腈)聚合引发剂。这样制备一种可聚合单体组合物。
另外,在710g离子交换水中加入450份0.1M Na3PO4水溶液,然后加热至60℃,用TK-型均匀混合机(Tokushu Kika Kogyo Co.,Ltd.制造)在1,300rpm的条件下进行搅拌。在得到的混合物中慢慢加入68份1.0M的CaCl2水溶液,制得含Ca3(PO4)2的含水介质。
将上述可聚合单体组合物加入上述的含水介质中,再加入2份聚乙烯,然后在Clear混合器中,以12,000rpm的转速,在氮气环境中于60℃下搅拌20分钟,将可聚合单体组合物粒化。接着升温至80℃,用桨叶搅拌器搅拌含水介质进行聚合反应8小时。
聚合完成后,冷却反应系统,然后加入盐酸以溶解磷酸钙,过滤后用水洗,接着干燥得到聚合粒子(聚合色调剂粒子)A。聚合色调剂粒子A的形状因子SF-1为115。
合成实施例2
苯乙烯 170份
丙烯酸2-乙基己基酯 30份
喹吖啶酮颜料 15份
二-叔丁基水杨酸铬化合物 3份
饱和聚酯(酸值:10;峰值分子量:9100) 10份
酯蜡(Mw:450;Mn:400;Mw/Mn:1.25;
熔点:70℃;粘度:6.5mPa·s;维克斯硬度:
1.1;SP值:8.6) 40份
用与合成实施例1相同的方法处理上面的材料制得可聚合单体组合物,然后加入合成实施例1中制得的含水介质中,重复后续的步骤得到聚合粒子(聚合色调剂粒子)B。
合成实施例3
苯乙烯 170份
丙烯酸2-乙基己基酯 30份
炭黑 15份
二-叔丁基水杨酸铬化合物 3份
饱和聚酯(酸值:10;峰值分子量:9100) 10份
酯蜡(Mw:450;Mn:400;Mw/Mn:1.25;
熔点:70℃;粘度:6.5mPa·s;维克斯硬度:
1.1;SP值:8.6) 40份
用与合成实施例1相同的方法处理上面的材料制得可聚合单体组合物,然后加入到合成实施例1中制得的含水介质中,重复后面的步骤得到聚合粒子(聚合色调剂粒子)C。
合成实施例4
苯乙烯 170份
丙烯酸正丁酯 30份
C.I.颜料黄93 15份
二-叔丁基水杨酸铬化合物 3份
饱和聚酯(酸值:10;峰值分子量:9100) 10份
二酯蜡(Mw:480;Mn:410;Mw/Mn:1.17;
熔点:73℃;粘度:10.5mPa·s;
维克斯硬度:1.0;SP值:9.1) 30份
用与合成实施例1相同的方法处理上面的材料制得可聚合单体组合物,然后加入到合成实施例1中制得的含水介质中,接着在Clear混合器中,以12,000rpm的转速,氮气环境下,60℃下搅拌20分钟将可聚合单体组合物粒化。然后升温至80℃,用桨叶搅拌器搅拌含水介质进行聚合反应10小时。
聚合完成后,冷却反应系统,然后加入盐酸以溶解磷酸钙,过滤后用水洗,接着干燥得到聚合粒子(聚合色调剂粒子)D。
合成实施例5
苯乙烯 170份
丙烯酸正丁酯 30份
喹吖啶酮颜料 15份
二-叔丁基水杨酸铬化合物 3份
饱和聚酯(酸值:10;峰值分子量:9100) 10份
石蜡(Mw:3390;Mn:2254;Mw/Mn:1.50;
熔点:72℃;粘度:6.3mPa·s;
维克斯硬度:6.8;SP值:8.7) 30份
用与合成实施例1相同的方法处理上面的材料制得可聚合单体组合物,然后加入到合成实施例1中制得的含水介质中,重复后面的步骤得到聚合粒子(聚合色调剂粒子)E。
合成实施例6
苯乙烯 170份
丙烯酸2-乙基己基酯 30份
炭黑 15份
单偶氮铁配合物 3份
饱和聚酯(酸值:10;峰值分子量:9100) 10份
石蜡(Mw:570;Mn:380;Mw/Mn:1.50;
熔点:69℃;粘度:6.8mPa·s;
维克斯硬度:0.7;SP值:8.3) 30份
用与合成实施例1相同的方法处理上面的材料制得可聚合单体组合物,然后加入到合成实施例1中制得的含水介质中,重复后面的步骤得到聚合粒子(聚合色调剂粒子)F。
合成实施例7
用与合成实施例1相同的方法制备可聚合单体组合物,并获得聚合粒子(聚合色调剂粒子)G,不同的是没有用极性树脂饱和聚酯。
合成实施例8
聚酯树脂 100份
酞菁铜颜料 4份
二-叔丁基水杨酸铝化合物 5份
石蜡(Mw:3390;Mn:2254;
Mw/Mn:1.50;熔点:72℃;粘度:
6.3mPa·s;维克斯硬度:6.8;SP值:8.7) 5份
用Henshel混合器预混合上述材料,然后用双螺杆挤出型捏和机进行熔融捏和。冷却后,用锤磨器将捏和产物粉碎形成直径约为1到2mm的粗糙粒子,接着用空气喷射系统的精细研磨器进行精细研磨。将得到的精细研磨产物进行进一步分级得到粉碎的色调剂粒子H。
前述合成实施例1到8中的聚合色调剂粒子A到G和粉碎的色调剂粒子H的形状因子SF-1的值见表4。
实施例14
向100份合成实施例1得到的聚合色调剂粒子A中,外加入已经用15份异丁基三甲氧基硅烷处理且BET比表面积为145m2/g的氧化铝细粉(A)1份和BET比表面积为68m2/g的非球形二氧化硅细粉(A)1份,可得到重均粒子直径为6.8μm的悬浮聚合色调剂(A)。
上述二氧化硅细粉(A)的制备方法是,用10份六甲基二硅氮烷对100份商业上获得的二氧化硅细粒AEROSIL#50(Nippon Aerosil Co.,Ltd提供)进行表面处理,然后用空气分级器分级,收集相对粗糙的粒子以控制粒子大小的分布。在透射电镜(TEM)得到的100,000倍放大照片和扫描电镜(SEM)得到的30,000倍放大照片上,可以证实二氧化硅细粉(A)是由许多平均粒子直径为38mμm的初级粒子聚结形成的聚结粒子。
悬浮聚合色调剂(A)的色调剂粒子上存在的氧化铝细粉(A)的形状因子SF-1为118,而其上的二氧化硅细粉(A)的形状因子SF-1为155。
用扫描电镜得到的悬浮聚合色调剂(A)的100,000倍放大照片中,可以证实氧化铝细粉(A)的平均长度为10mμm,长/宽比为1.1,在单位面积0.5μm×0.5μm中有至少90个粒子。用扫描电镜得到的悬浮聚合色调剂(A)的30,000倍放大照片中,可以证实二氧化硅细粉(A)的平均长度为150mμm,长/宽比为1.9,在单位面积1μm×1μm中有19个粒子。
将上述悬浮聚合色调剂(A)和铁氧体涂覆的载体(通过在Mg-Mn铁氧体核粒子表面上涂一层0.5μm厚的硅氧烷树脂得到,重均粒子直径为35μm)以重量比7∶100混合制得双组分显影剂(A)。
将上述双组分显影剂(A)用于一种改进的数字复印机(GP-55,Canon制造)作为电子照相装置的显影组件中,这种数字复印机进行了改进以使其可使用如图6所示的双组分显影组件和磁刷充电组件,当施用叠加由如图7所示的不连续交变电压而形成的显影偏压时,通过双组分显影剂(A)对300dpi的二进制静电潜像进行显影从而成像。
在这种电子照相装置中,磁刷充电组件中的磁性粒子是由Cu-Zn-铁氧体组成的,其平均粒子直径为25μm,其组成可表示为(Fe2O3)2.3∶(CuO)1∶(ZnO)1,磁性粒子通过磁力固定在内部有一磁体辊的非磁性套筒上形成磁刷。磁刷与感光鼓表面接触,感光鼓表面施加-700VDC和1kHz/1.2kVpp AC的充电偏压进行初级充电。
在磁刷充电组件中,如果保持磁刷固定,由于磁刷本身没有复位的物理力,磁刷因感光鼓弯曲或偏心运动而偏移时将使磁刷和感光鼓之间的缝隙变得难以维持,从而造成充电失败。因此优选使用总是清洁的磁刷表面。所以在此实施例中,磁刷以两倍于感光鼓圆周速度的速度旋转,其运动方向与感光鼓表面运动方向相反。
在23℃/65%RH环境中成像,连续进行50,000张运行测试。对开始阶段图像上的实心均匀性,运行50,000张后的图像模糊性,开始阶段与运行50,000张后的图像浓度差别表现出的运行性能,开始阶段与运行50,000张后的转印性能和色调剂在低湿度环境(20℃/10%RH)与高湿度环境(30℃/80%RH)之间摩擦电荷数量的不同表现出的环境稳定性进行评价。
悬浮聚合色调剂(A)的物理性能列于表4,性能评价结果见表5。
对比实施例7
双组分显影剂(B)制备方法与实施例14相同,不同的是用重均粒子直径为6.5μm的粉碎色调剂(B)代替了悬浮聚合色调剂(A),其中,如表4所示,将1.0份BET比表面积为72m2/g的硅氧烷-处理后的氧化铝细粉(B)和1.0份BET比表面积为66m2/g的二氧化硅细粉(B)外加入100份合成实施例8制备的粉碎色调剂粒子H中。性能评价方法也与实施例14相同。
粉碎色调剂(B)的物理性能列于表4,性能评价结果见表5。
实施例15
双组分显影剂(C)制备方法与实施例14相同,不同的是用重均粒子直径为6.6μm的悬浮聚合色调剂(C)代替了悬浮聚合色调剂(A),其中,如表4所示,向100份合成实施例2制备的聚合色调剂粒子B中外加入1.0份BET比表面积为120m2/g的烷基烷氧基硅烷处理后的氧化铝细粉(C)和1.0份BET比表面积为68m2/g的二氧化硅细粉(C)。性能评价方法也与实施例14相同。
悬浮聚合色调剂(C)的物理性能列于表4,性能评价结果见表5。
实施例16
双组分显影剂(D)制备方法与实施例14相同,不同的是用重均粒子直径为6.6μm的悬浮聚合色调剂(D)代替了悬浮聚合色调剂(A)。其中,如表4所示,向100份合成实施例3制备的聚合色调剂粒子C中外加入1.0份BET比表面积为140m2/g的烷基烷氧基硅烷处理后的氧化铝细粉(D)和1.0份BET比表面积为22m2/g的二氧化硅细粉(D)。性能评价方法也与实施例14相同。
悬浮聚合色调剂(D)的物理性能列于表4,性能评价结果见表5。
实施例17
双组分显影剂(E)制备方法与实施例14相同,不同的是用重均粒子直径为7.1μm的悬浮聚合色调剂(E)代替了悬浮聚合色调剂(A)。其中,如表4所示,向100份合成实施例4制备的聚合色调剂粒子D中外加入1.0份BET比表面积为66m2/g的硅油处理后的氧化铝细粉(E)和1.0份BET比表面积为23m2/g的二氧化硅细粉(E)。性能评价方法也与实施例14相同。
悬浮聚合青色色调剂(E)的物理性能列于表4,性能评价结果见表5。
实施例18
双组分显影剂(F)制备方法与实施例14相同,不同的是用重均粒子直径为6.8μm的悬浮聚合色调剂(F)代替了悬浮聚合色调剂(A)。其中,如表4所示,向100份合成实施例4制备的聚合色调剂粒子D中外加入1.0份BET比表面积为68m2/g的硅油处理后的氧化铝细粉(F)和1.0份BET比表面积为71m2/g的二氧化硅细粉(F)。性能评价方法也与实施例14相同。
悬浮聚合色调剂(F)的物理性能列于表4,性能评价结果见表5。
对比实施例8
双组分显影剂(G)制备方法与实施例14相同,不同的是用重均粒子直径为7.2μm的悬浮聚合色调剂(G)代替了悬浮聚合色调剂(A)。其中,如表4所示,向100份合成实施例3制备的聚合色调剂粒子C中外加入1.0份BET比表面积为210m2/g的烷基烷氧基硅烷处理后的氧化铝细粉(G)和1.0份BET比表面积为25m2/g的二氧化硅细粉(G)。性能评价方法也与实施例14相同。
悬浮聚合色调剂(G)的物理性能列于表4,性能评价结果见表5。
比较实施例9
双组分显影剂(H)制备方法与实施例14相同,不同的是用重均粒子直径为7.0μm的悬浮聚合色调剂(H)代替了悬浮聚合色调剂(A)。其中,如表4所示,向100份合成实施例3制备的聚合色调剂粒子C中外加入1.0份BET比表面积为147m2/g的烷基烷氧基硅烷处理后的氧化铝细粉(H)和1.0份BET比表面积为13m2/g的二氧化硅细粉(H)。性能评价方法也与实施例14相同。
悬浮聚合色调剂(H)的物理性能列于表4,性能评价结果见表5。
比较实施例10
双组分显影剂(I)制备方法与实施例14相同,不同的是用重均粒子直径为6.1μm的悬浮聚合色调剂(I)代替了悬浮聚合色调剂(A)。其中,如表4所示,向100份合成实施例2制备的聚合色调剂粒子B中外加入1.5份BET比表面积为151m2/g的二氧化硅细粉(I)。性能评价方法也与实施例14相同。
悬浮聚合色调剂(I)的物理性能列于表4,性能评价结果见表5。
比较实施例11
双组分显影剂(J)制备方法与实施例14相同,不同的是用重均粒子直径为6.1μm的悬浮聚合色调剂(J)代替了悬浮聚合色调剂(A)。其中,如表4所示,向100份合成实施例2制备的聚合色调剂粒子B中外加入1.5份BET比表面积为150m2/g的硅油处理后的氧化铝细粉(I)。性能评价方法也与实施例14相同。
悬浮聚合色调剂(J)的物理性能列于表4,性能评价结果见表5。
实施例19
双组分显影剂(K)制备方法与实施例14相同,不同的是用重均粒子直径为6.7μm的悬浮聚合色调剂(K)代替了悬浮聚合色调剂(A)。其中,如表4所示,向100份合成实施例5制备的聚合色调剂粒子E中外加入1.0份BET比表面积为122m2/g的硅氧烷处理后的氧化铝细粉(J)和1.0份BET比表面积为22m2/g的二氧化硅细粉(J)。性能评价方法也与实施例14相同。
悬浮聚合色调剂(K)的物理性能列于表4,性能评价结果见表5。
实施例20
双组分显影剂(L)制备方法与实施例14相同,不同的是用重均粒子直径为6.4μm的悬浮聚合色调剂(L)代替了悬浮聚合色调剂(A)。其中,如表4所示,向100份合成实施例7制备的聚合色调剂粒子G中外加入1.0份BET比表面积为145m2/g的烷基烷氧基硅烷处理后的氧化铝细粉(A)和1.0份BET比表面积为68m2/g的二氧化硅细粉(A)。性能评价方法也与实施例14相同。
悬浮聚合色调剂(L)的物理性能列于表4,性能评价结果见表5。
实施例21
双组分显影剂(M)制备方法与实施例14相同,不同的是用重均粒子直径为6.4μm的悬浮聚合色调剂(M)代替了悬浮聚合色调剂(A)。其中,如表4所示,向100份合成实施例6制备的聚合色调剂粒子F中外加入1.0份BET比表面积为74m2/g的未经过疏水处理的氧化铝细粉(K)和1.0份BET比表面积为67m2/g的二氧化硅细粉(K)。性能评价方法也与实施例14相同。
悬浮聚合色调剂(M)的物理性能列于表4,性能评价结果见表5。
实施例22
将实施例15制备的含悬浮聚合色调剂(C)的双组分显影剂(C)用于如图4所示的成像设备中的显影组件36,连续成像50,000张品红单色图像。性能评价方法也与实施例14相同。
性能评价结果见表6。
实施例23
将实施例16制备的含悬浮聚合色调剂(D)的双组分显影剂(D)用于如图5所示的成像设备中的显影组件107,连续成像50,000张黑色单色图像。性能评价方法也与实施例14相同。
性能评价结果见表6。
实施例24
将实施例17制备的含悬浮聚合色调剂(E)的双组分显影剂(E)用于如图3所示的成像设备中的显影组件29d,连续成像50,000张黄色单色图像。性能评价方法也与实施例14相同。
性能评价结果见表6。
实施例25
将实施例18制备的含悬浮聚合色调剂(F)的双组分显影剂(F)用于如图4所示的成像设备中的显影组件34,连续成像50,000张黄色单色图像。性能评价方法也与实施例14相同。
性能评价结果见表6。
对比实施例12
将比较实施例8制备的含悬浮聚合色调剂(G)的双组分显影剂(G)用于如图4所示的成像设备中的显影组件37,连续成像50,000张黑色单色图像。性能评价方法也与实施例14相同。
性能评价结果见表6。
比较实施例13
将比较实施例10制备的含悬浮聚合色调剂(I)的双组分显影剂(I)用于如图5所示的成像设备中的显影组件105,连续成像50,000张品红单色图像。性能评价方法也与实施例14相同。
性能评价结果见表6。
对比实施例14
将比较实施例11制备的含悬浮聚合色调剂(J)的双组分显影剂(J)用于如图3所示的成像设备中的显影组件17b,连续成像50,000张品红单色图像。性能评价方法也与实施例14相同。
性能评价结果见表6。
实施例26
将实施例19制备的含悬浮聚合色调剂(K)的双组分显影剂(K)用于如图4所示的成像设备中的显影组件36,连续成像50,000张品红单色图像。性能评价方法也与实施例14相同。
性能评价结果见表6。
实施例27
将实施例20制备的含悬浮聚合色调剂(L)的双组分显影剂(L)用于如图3所示的成像设备中的显影组件17c,连续成像50,000张青色单色图像。性能评价方法也与实施例14相同。
性能评价结果见表6。
实施例28
评价方法与实施例14相同,不同的是用平均粒子直径为150μm的磁性粒子代替了实施例14使用的磁刷充电组件中的磁性粒子。与实施例14相比,其结果是形成的实心图像均匀度稍微有一点低。
实施例29
使用合成实施例1制备的悬浮聚合色调剂粒子A,合成实施例2制备的悬浮聚合色调剂粒子B,合成实施例3制备的悬浮聚合色调剂粒子C和合成实施例4制备的悬浮聚合色调剂粒子D,向100份上述悬浮聚合色调剂粒子A至D中的每一种当中外加入如表4所示的1.0份用硅油处理后BET比表面积为66m2/g的氧化铝细粉(E)和1.0份BET比表面积为23m2/g的二氧化硅细粉(E),可分别制得悬浮聚合青色色调剂(N),悬浮聚合品红色调剂(O),悬浮聚合黑色色调剂(P)和悬浮聚合黄色色调剂(Q)。
将上述四种彩色色调剂按重量比7∶100的比例各自与实施例14中使用的铁氧体涂覆的载体混合可分别制得双组分显影剂(N)到(Q)。将这些双组分显影剂分别用于如图2所示的成像设备中的显影组件4到7,用这种方法按照黄色,品红,青色和黑色的颜色顺序将潜像显影。这样可形成单色和全色图像。
在全色图像的形成方面,多色调剂层形成的图像表现出颜色充分的混色性,色度更好及图像质量高。在单色图像的形成方面,以与实施例14相同的方法进行评价,评价结果见表7,得到的结果较好。
表2(待续)
| 色调剂 | |||
| 色调剂号 | 重均颗粒直径(μm) | 成圆率分布 | |
| 平均成圆率 | 成圆率小于0.950的粒子含量(数量%) | ||
| 实施例:1悬浮聚合青色色调剂12悬浮聚合青色色调剂2 | 6.16.1 | 0.9830.983 | 1111 |
| 比较实施例:1粉碎青色色调剂3 | 6.0 | 0.913 | 42 |
| 实施例:3球形处理后的青色色调剂44球形处理后的青色色调剂5 | 6.06.0 | 0.9250.953 | 3121 |
| 比较实施例:2球形处理后的青色色调剂63悬浮聚合青色色调剂74悬浮聚合青色色调剂8 | 6.06.16.5 | 0.9960.9840.927 | 1.51143 |
| 实施例:5悬浮聚合青色色调剂9 | 6.1 | 0.983 | 12 |
| 比较实施例:5悬浮聚合青色色调剂10 | 6.1 | 0.983 | 12 |
| 实施例:6悬浮聚合青色色调剂117悬浮聚合青色色调剂128悬浮聚合青色色调剂139悬浮聚合青色色调剂14 | 6.16.16.16.1 | 0.9830.9830.9830.983 | 11111111 |
| 比较实施例:6悬浮聚合青色色调剂15 | 6.1 | 0.983 | 11 |
表2
| 外部添加剂 | |||||||||||||
| 无机细粉(A) | 无机精细粉(B) | ||||||||||||
| 类型 | 含量(pbw) | BET比表面积(m2/g) | 外部添加剂物理性能* | 类型 | 含量(pbw) | BET比表面积(m2/g) | 外部添加剂物理性能* | ||||||
| 形状因子SF-1 | L/B | 平均长度mμm) | (N) | 形状因子SF-1 | L/B | 平均长度(mμm) | (N’) | ||||||
| 实施例:1FTP(1)2FTP(1) | 1.01.0 | 100100 | 120120 | 1.11.1 | 5050 | 7575 | 实施例:1FSP(1)2FSP(2) | 1.01.0 | 4340 | 195160 | 2.82.1 | 178160 | 1715 |
| 比较实施例:1FTP(1) | 1.0 | 100 | 120 | 1.1 | 50 | 72 | 比较实施例:1FSP(2) | 1.0 | 40 | 160 | 2.1 | 160 | 13 |
| 实施例:3FTP(1)4FTP(1) | 1.01.0 | 100100 | 120120 | 1.11.1 | 5050 | 7073 | 实施例:3FSP(2)4FSP(2) | 1.01.0 | 4040 | 160160 | 2.12.1 | 160160 | 1415 |
| 比较实施例:2FTP(1)3FTP(1)4FTP(1) | 1.02.01.0 | 100100100 | 120120120 | 1.11.11.1 | 505050 | 7513874 | 比较实施例:2FSP(2)3-4FSP(2) | 1.0-1.0 | 40-40 | 160-160 | 2.12.1 | 160-160 | 16-15 |
| 实施例:5FTP(2) | 1.0 | 92 | 128 | 1.3 | 50 | 68 | 实施例:5FSP(2) | 1.0 | 40 | 160 | 2.1 | 160 | 14 |
| 比较实施例:5FTP(1) | 1.0 | 100 | 121 | 1.2 | 50 | 71 | 比较实施例:5FSP(3) | 1.0 | 26 | 136 | 1.5 | 205 | 9 |
| 实施例:6FTP(1)7FTP(1)8FTP(1)9FTP(1) | 0.021.01.01.0 | 100100100100 | 120120120120 | 1.21.21.21.2 | 50505050 | 4747574 | 实施例:6FSP(2)7FSP(2)8FSP(4)9FSP(5) | 1.02.01.01.0 | 40403745 | 160160143205 | 2.12.81.93.1 | 160180115650 | 15352112 |
| 比较实施例:6--- | - | - | - | - | - | - | 比较实施例:6FSP(1) | 2.0 | 43 | 195 | 2.8 | 178 | 34 |
FTP:二氧化钛细粉; FSP:二氧化硅细粉;
L/B:长度/宽度比; *:在色调剂FEM图片中存在于色调剂粒子上;
(N):每0.5×0.5面积中的粒子数; (N’):每1.0×1.0面积中的粒子数
表3
| 色调剂号 | 初始阶段的实象均匀度 | 运行性能图像浓度 | (1) | 图像模糊程度(运行30,000张后)(%) | 转印性能 | |||
| (a)初始阶段 | (b)运行30,000张后 | (a)-(b)差值 | 色调剂摩擦电荷数量在L/L-H/H之间的差(Δ)(mC/kg) | 初始阶段(%) | 运行30,000张后(%) | |||
| 实施例:1悬浮聚合青色色调剂12悬浮聚合青色色调剂2 | 0.010.01 | 1.451.47 | 1.471.45 | 0.050.05 | 3.84.0 | 0.20.2 | 98.898.5 | 98.598.0 |
| 比较实施例:1粉碎青色色调剂3 | 0.05 | 1.48 | 1.35 | 0.18 | 8.3 | 1.5 | 96.1 | 94.2 |
| 实施例:3球形处理后的青色色调剂44球形处理后的青色色调剂5 | 0.030.02 | 1.451.43 | 1.401.41 | 0.090.07 | 4.55.2 | 0.20.2 | 98.298.6 | 97.198.3 |
| 比较实施例:2球形处理后的青色色调剂63悬浮聚合青色色调剂74悬浮聚合青色色调剂8 | 0.070.050.04 | 1.411.431.46 | 1.311.331.35 | 0.210.150.14 | 6.54.75.3 | 1.81.31.5 | 99.196.696.0 | 95.294.194.3 |
| 实施例:5悬浮聚合青色色调剂9 | 0.03 | 1.46 | 1.43 | 0.06 | 4.3 | 0.3 | 98.7 | 97.9 |
| 比较实施例:5悬浮聚合青色色调剂10 | 0.05 | 1.42 | 1.31 | 0.15 | 4.8 | 1.4 | 98.0 | 95.2 |
| 实施例:6悬浮聚合青色色调剂117悬浮聚合青色色调剂128悬浮聚合青色色调剂139悬浮聚合青色色调剂14 | 0.030.020.020.04 | 1.451.441.471.41 | 1.401.411.401.40 | 0.080.060.090.05 | 5.84.74.14.5 | 0.50.30.50.4 | 98.298.998.597.8 | 97.098.698.197.5 |
| 比较实施例:6悬浮聚合青色色调剂15 | 0.05 | 1.41 | 1.30 | 0.15 | 8.5 | 1.6 | 96.1 | 95.0 |
(1):环境稳定性
L/L:低温/低湿度环境;H/H:高温/高湿度环境
表4(待续)
| 色调剂 | ||||
| 色调剂号 | 重均粒子直径(μm) | 形状因子SF-1 | 成圆率分布 | |
| 平均成圆率 | 成圆率小于0.950的粒子含量(数量%) | |||
| 实施例:14悬浮聚合色调剂A | 6.8 | 115 | 0.985 | 9 |
| 比较实施例:7粉碎色调剂B | 6.5 | 155 | 0.918 | 44 |
| 实施例:15悬浮聚合色调剂C16悬浮聚合色调剂D17悬浮聚合色调剂E18悬浮聚合色调剂F | 6.66.67.16.8 | 140103118109 | 0.9620.9900.9800.987 | 2561610 |
| 比较实施例:8悬浮聚合色调剂G9悬浮聚合色调剂H10悬浮聚合色调剂I11悬浮聚合色调剂J | 7.29.56.16.6 | 103111103106 | 0.9880.9860.9900.985 | 101069 |
| 实施例:19悬浮聚合色调剂K20悬浮聚合色调剂L21悬浮聚合色调剂M | 6.76.46.4 | 110132119 | 0.9840.9470.976 | 153423 |
表4(待续)
| 外部添加剂 | ||||||||
| 无机细粉(A) | ||||||||
| 类型 | 含量(pbw) | BET比表面积(m2/g) | (a)初级粒子的平均初级粒子直径(mμm) | 至少为(a)的2倍的数量百分数 | 外部添加剂物理性能* | |||
| 形状因子SF-1 | L/B | 平均长度(mμm) | (N) | |||||
| 实施例:14氧化铝细粉(A) | 1.0 | 145 | 10 | 0 | 118 | 1.1 | 15 | 190 |
| 比较实施例:7氧化铝细粉(B) | 1.0 | 72 | 18 | 0 | 120 | 1.2 | 30 | 143 |
| 实施例:15氧化铝细粉(C)16氧化铝细粉(D)17氧化铝细粉(E)18氧化铝细粉(F) | 1.01.01.01.0 | 1201406668 | 15131918 | 0.300.500.400.40 | 123120125124 | 1.21.11.31.3 | 28253536 | 1151299095 |
| 比较实施例:8氧化铝细粉(G)9氧化铝细粉(H)10---11氧化铝细粉(I) | 1.01.0-1.5 | 210147-150 | 320-11 | 00.20-0 | 120119-118 | 1.11.1-1.1 | 845-15 | >200180->200 |
| 实施例:19氧化铝细粉(J)20氧化铝细粉(A)21氧化铝细粉(K) | 1.01.01.0 | 12214574 | 141017 | 0.0300 | 119118120 | 1.11.11.2 | 281531 | 155185140 |
*:在色调剂的FEM图片中出现在色调剂粒子上; L/B:长度/宽度比;
(N):在每0.5×0.5面积中出现的粒子数
表4
| 外部添加剂 | ||||||||
| 无机细粉(B) | ||||||||
| 类型 | 含量(pbw) | BET比表面积(m2/g) | (b):组成聚集粒子的初级粒子的平均初级粒子直径(mμm) | 至少2至3倍于(b)的粒子的数量百分数 | 在色调剂FEM照片中存在于色调剂粒子上的外部添加剂物理性能 | |||
| 形状因子SF-1 | L/B | 平均长度(mμm) | (N’) | |||||
| 实施例:14二氧化硅细粉(A) | 1.0 | 68 | 25 | 8.00 | 185 | 1.9 | 150 | 19 |
| 比较实施例:7二氧化硅细粉(B) | 1.0 | 66 | 27 | 6.40 | 180 | 2.0 | 145 | 16 |
| 实施例:15二氧化硅细粉(C)16二氧化硅细粉(D)17二氧化硅细粉(E)18二氧化硅细粉(F) | 1.01.01.01.0 | 68222371 | 25333425 | 7.406.109.302.50 | 165198205160 | 1.92.12.21.7 | 145195200140 | 179917 |
| 比较实施例:8二氧化硅精粉(G)9二氧化硅精粉(H)10二氧化硅细粉(I)11--- | 1.01.01.5- | 2513151- | 322510- | 9.108.208.10- | 205240135- | 2.02.31.6- | 19041070- | 14535- |
| 实施例:19二氧化硅精细粉(J)20二氧化硅精细粉(A)21二氧化硅精细粉(K) | 1.01.01.0 | 226867 | 322523 | 11.108.007.50 | 190185175 | 2.01.91.8 | 175150140 | 131820 |
L/B:长度/宽度比; (N’):每1.0×1.0面积中的粒子数
表5
| 色调剂号 | 初始阶段实像均匀度 | 运行性能图像浓度 | (1) | 图像模糊程度(运行50,000张后)(%) | 转印性能 | |||
| (a)初始阶段 | (b)运行50,000张后 | (a)-(b)差值 | 色调剂摩擦电荷数量在L/L-H/H之间的差(Δ)(mC/kg) | 初始阶段(%) | 运行50000张后(%) | |||
| 实施例:14悬浮聚合色调剂A | 0.02 | 1.46 | 1.43 | 0.05 | 3.0 | 0.1 | 98.9 | 98.0 |
| 比较实施例:7粉碎色调剂B | 0.06 | 1.45 | 1.32 | 0.15 | 11.3 | 1.5 | 95.8 | 93.2 |
| 实施例:15悬浮聚合色调剂C16悬浮聚合色调剂D17悬浮聚合色调剂E18悬浮聚合色调剂F | 0.030.030.020.02 | 1.461.451.451.45 | 1.401.441.401.39 | 0.070.040.070.06 | 9.07.59.58.5 | 0.30.30.20.3 | 97.299.098.598.4 | 96.198.297.997.5 |
| 比较实施例:8悬浮聚合色调剂G9悬浮聚合色调剂H10悬浮聚合色调剂I11悬浮聚合色调剂J | 0.030.050.080.03 | 1.441.401.411.48 | 1.301.281.251.25 | 0.160.150.180.25 | 12.36.810.311.7 | 1.41.71.81.1 | 97.398.295.198.0 | 94.096.993.394.9 |
| 实施例:19悬浮聚合色调剂K20悬浮聚合色调剂L21悬浮聚合色调剂M | 0.030.040.03 | 1.451.411.45 | 1.381.371.38 | 0.070.070.07 | 9.48.85.8 | 0.40.40.4 | 98.397.097.2 | 97.496.096.3 |
(1):环境稳定性;
L/L:低温/低湿度环境;H/H:高温/高湿度环境
表6
| 色调剂号 | 成像装置 | 初始阶段实像均匀度 | 运行性能图像浓度 | (1) | 图像模糊程度(运行50,000张后)(%) | 转印性能 | |||
| (a)初始阶段 | (b)运行50,000张后 | (a)-(b)差值 | 色调剂摩擦电荷数量在L/L-H/H之间的差(Δ)(mC/kg) | 初始阶段(%) | 运行50,000张后(%) | ||||
| 实施例:22悬浮聚合色调剂C23悬浮聚合色调剂D24悬浮聚合色调剂E25悬浮聚合色调剂F | 图4图5图3图4 | AABB | 1.701.651.671.58 | 1.611.591.511.49 | 0.090.060.160.09 | 9.37.89.68.5 | 0.20.30.20.3 | 98.396.595.895.6 | 96.795.693.594.2 |
| 比较实施例:12悬浮聚合色调剂G13悬浮聚合色调剂I14悬浮聚合色调剂J | 图4图5图3 | DAA | 1.671.721.69 | 1.481.511.63 | 0.190.210.06 | 10.615.610.2 | 1.61.71.2 | 89.295.288.7 | 85.194.882.1 |
| 实施例:26悬浮聚合色调剂K27悬浮聚合色调剂L | 图4图3 | BA | 1.561.64 | 1.471.52 | 0.090.12 | 9.58.8 | 0.40.4 | 95.496.3 | 94.695.1 |
(1):环境稳定性;L/L:低温/低湿度环境;H/H:高温/高湿度环境
表7
| 色调剂号 | 成像装置 | 初始阶段实像均匀度 | 运行性能 | (1) | 图像模糊程度 | 转印性能 | |||
| 图像浓度 | 色调剂摩擦电荷数量在L/L-H/H之间的差(Δ)(mC/kg) | (运行50,000张后)(%) | 初始阶段(%) | 运行50,000张后(%) | |||||
| (a)初始阶段 | (b)运行50,000张后 | (a)-(b)差值 | |||||||
| 实施例:29悬浮聚合色调剂N悬浮聚合色调剂O悬浮聚合色调剂P悬浮聚合色调剂Q | 图2图2图2图2 | AABB | 1.681.721.611.66 | 1.551.631.551.59 | 0.130.090.060.07 | 7.66.87.28.3 | 0.20.30.30.3 | 97.296.495.295.8 | 95.395.694.895.7 |
(1):环境稳定性;L/L:低温/低湿度环境;H/H:高温/高湿度环境
Claims (157)
1.一种色调剂,包括色调剂粒子和外部添加剂;所述的色调剂具有:
(a)以流动式粒子图像分析仪测得的粒子成圆率分布中平均成圆率为0.920-0.995,所含成圆率小于0.950的粒子数量为2%-40%;和
(b)以Coulter法测得重均粒子直径为2.0-9.0μm;和
所述外部添加剂具有在色调剂粒子上的(i)一种以初级粒子或二级粒子的状态存在的、平均粒子长度为10mμm-400mμm和形状因子SF-1为100-130的无机细粉(A)和(ii)一种由多个粒子结合形成的、形状因子SF-1大于150的非球形无机细粉(B)。
2.根据权利要求1的色调剂,其中色调剂的平均成圆率为0.950-0.995。
3.根据权利要求1的色调剂,其中色调剂的平均成圆率为0.960-0.995。
4.根据权利要求1的色调剂,其中成圆率小于0.950的粒子数量为3%-30%。
5.根据权利要求1的色调剂,其形状因子SF-1为100-150。
6.根据权利要求1的色调剂,其形状因子SF-1为100-130。
7.根据权利要求1的色调剂,其中色调剂粒子上的所述无机细粉(A)的平均粒子长度范围为15mμm-200mμm。
8.根据权利要求1的色调剂,其中色调剂粒子上的所述无机细粉(A)的平均粒子长度范围为15mμm-100mμm。
9.根据权利要求1的色调剂,其中色调剂粒子上的所述非球形无机细粉(B)的平均粒子长度范围为120mμm-600mμm。
10.根据权利要求1的色调剂,其中色调剂粒子上的所述非球形无机细粉(B)的平均粒子长度范围为130mμm-500mμm。
11.根据权利要求1的色调剂,其中色调剂粒子上的所述非球形无机细粉(B)的平均粒子长度比色调剂粒子上的所述无机细粉(A)的平均粒子长度长。
12.根据权利要求1的色调剂,其中色调剂粒子上的所述非球形无机细粉(B)的平均粒子长度比色调剂粒子上的所述无机细粉(A)的平均粒子长度至少长20mμm。
13.根据权利要求1的色调剂,其中色调剂粒子上的所述非球形无机细粉(B)的平均粒子长度比色调剂粒子上的所述无机细粉(A)的平均粒子长度至少长40mμm。
14.根据权利要求1的色调剂,其中色调剂粒子上的所述无机细粉(A)的平均粒子长度范围为15-100mμm;色调剂粒子上的所述非球形无机细粉(B)的平均粒子长度为120-600mμm。
15.根据权利要求1的色调剂,其中所述的无机细粉(A)的比表面积按照BET法以氮吸附测得为60m2/g-230m2/g。
16.根据权利要求1的色调剂,其中所述的无机细粉(A)的比表面积按照BET法以氮吸附测得为70m2/g-180m2/g。
17.根据权利要求1的色调剂,其中所述的非球形无机细粉(B)的比表面积按照BET法以氮吸附测得为20m2/g-90m2/g。
18.根据权利要求1的色调剂,其中所述的非球形无机细粉(B)的比表面积按照BET法以氮吸附测得为25m2/g-80m2/g。
19.根据权利要求1的色调剂,其中色调剂粒子上的所述无机细粉(A)的形状因子SF-1为100-125。
20.根据权利要求1的色调剂,其中色调剂粒子上的所述非球形无机细粉(B)的形状因子SF-1大于190。
21.根据权利要求1的色调剂,其中色调剂粒子上的所述非球形无机细粉(B)的形状因子SF-1大于200。
22.根据权利要求1的色调剂,其中在电子显微镜放大的色调剂照片上观察,所述的无机细粉(A)和所述的非球形无机细粉(B)在色调剂粒子表面上存在的数量分别为平均每0.5μm×0.5μm单位面积5-90个粒子和平均每1.0μm×1.0μm单位面积1-30个粒子。
23.根据权利要求1的色调剂,其中所述的色调剂是在以流动式粒子图像分析仪测得的成圆率分布中具有0.950-0.995的平均成圆率且所含成圆率小于0.950的粒子数量为2%-40%的色调剂;
所述的外部添加剂是一种在色调剂粒子上具有如下成分的外部添加剂:(i)一种以初级粒子或二级粒子状态存在的、平均粒子长度为15mμm-100mμm和形状因子SF-1为100-130的无机细粉(A)和(ii)一种由多个粒子结合形成的、平均成圆率为120mμm-600mμm和形状因子SF-1大于150的非球形无机细粉(B);和
在电子显微镜放大的色调剂照片上观察,所述的无机细粉(A)和所述的非球形无机细粉(B)在色调剂粒子表面上存在的数量分别为平均每0.5μm×0.5μm单位面积5-90个粒子和平均每1.0μm×1.0μm单位面积1-30个粒子。
24.根据权利要求1的色调剂,其中包括的所述无机细粉(A)按照100重量份色调剂计为0.1-2.0重量份。
25.根据权利要求1的色调剂,其中包括的所述非球形无机细粉(B)按照100重量份色调剂计为0.3-3.0重量份。
26.根据权利要求1的色调剂,其中所述的无机细粉(A)具有的细粒子选自氧化铝细粒子、二氧化钛细粒子、氧化锆细粒子、氧化镁细粒子,经二氧化硅处理的这些细粒子中的任一种和氮化硅细粒子。
27.根据权利要求1的色调剂,其中所述的无机细粉(A)具有的细粒子选自氧化铝细粒子、二氧化钛细粒子,以及经二氧化硅处理的这些细粒子中的任一种。
28.根据权利要求1的色调剂,其中所述的非球形无机细粉(B)具有的细粒子选自二氧化硅细粒子、氧化铝细粒子、二氧化钛细粒子以及上述任一种的双氧化物细粒子。
29.根据权利要求1的色调剂,其中所述的非球形无机细粉(B)具有二氧化硅细粒子。
30.根据权利要求1的色调剂,其中所述的无机细粉(A)具有的细粒子选自氧化铝细粒子、二氧化钛细粒子以及经二氧化硅处理的上述任一种细粒子,所述的非球形无机细粉(B)具有二氧化硅细粒子。
31.根据权利要求1的色调剂,其中所述的无机细粉(A)具有氧化铝细粒子,所述的非球形无机细粉(B)具有二氧化硅细粒子。
32.根据权利要求31的色调剂,其中所述的氧化铝细粒子具有这样的粒子大小分布,即直径是平均粒子直径至少两倍的粒子在数量上占0%-5%;所述的非球形无机细粉(B)具有这样的粒子大小分布,即直径是平均粒子直径的两倍到三倍的粒子在数量上占5%-15%。
33.根据权利要求31的色调剂,其中所述的氧化铝细粒子具有的比表面积按照BET法以氮吸附测得为60m2/g-150m2/g,所述的非球形无机细粉(B)的比表面积按照BET法以氮吸附测得为20m2/g-70m2/g。
34.根据权利要求31的色调剂,其中所述的氧化铝细粒子已经疏水性处理。
35.根据权利要求1的色调剂,其中所述的色调剂粒子含有一种粘合剂树脂和一种着色剂。
36.根据权利要求1的色调剂,其中所述的色调剂粒子含有一种粘合剂树脂、一种着色剂和一种隔离剂。
37.根据权利要求1的色调剂,其中所述的色调剂粒子含有一种粘合剂树脂、一种着色剂、一种隔离剂和一种电荷控制剂。
38.根据权利要求1的色调剂,其中所述隔离剂的重均分子量为300-3000。
39.根据权利要求1的色调剂,其中所述的色调剂粒子是由聚合方法制得的粒子,此方法是在一种聚合引发剂存在的条件下,使含有一种可聚合单体和一种着色剂的可聚合单体组合物在一种液体介质中聚合。
40.根据权利要求1的色调剂,其中所述的色调剂粒子是由悬浮聚合法制得的粒子,此方法是在一种聚合引发剂存在的条件下,使含有一种可聚合单体和一种着色剂的可聚合单体组合物在一种含水介质中聚合。
41.根据权利要求1的色调剂,其中所述的色调剂粒子是由悬浮聚合法制得的粒子,此方法是在一种聚合引发剂存在的条件下,使含有一种可聚合单体,一种着色剂和一种用做隔离剂的蜡的可聚合单体组合物在一种含水介质中聚合。
42.根据权利要求1的色调剂,其中所述的色调剂粒子是通过处理成球形制得的粒子,通过粉碎法制得的粒子,此粉碎法包括熔融捏和一种含有一种粘合剂树脂和着色剂的混合物以得到一种捏和产物以及粉碎该捏和产物的步骤。
43.一种双组分显影剂,包含一种具有色调剂粒子和外部添加剂的色调剂和一种载体,其中:
所述的色调剂具有:
(a)以流动式粒子图像分析仪测得的粒子成圆率分布中平均成圆率为0.920-0.995,所含成圆率小于0.950的粒子数量为2%-40%;和
(b)以Coulter法测得重均粒子直径为2.0-9.0μm;和
所述外部添加剂具有在色调剂粒子上的(i)一种以初级粒子或二级粒子的状态存在的,平均粒子长度为10mμm-400mμm和形状因子SF-1为100-130的无机细粉(A)和(ii)一种由多个粒子结合形成的、形状因子SF-1大于150的非球形无机细粉(B)。
44.根据权利要求43的双组分显影剂,其中所述色调剂的平均成圆率为0.950-0.995。
45.根据权利要求43的双组分显影剂,其中所述色调剂的平均成圆率为0.960-0.995。
46.根据权利要求43的双组分显影剂,其中成圆率小于0.950的粒子数量为3%-30%。
47.根据权利要求43的双组分显影剂,其中所述色调剂的形状因子SF-1为100-150。
48.根据权利要求43的双组分显影剂,其中所述色调剂的形状因子SF-1为100-130。
49.根据权利要求43的双组分显影剂,其中所述的无机细粉(A)在色调剂粒子上的平均粒子长度范围为15-200mμm。
50.根据权利要求43的双组分显影剂,其中所述的无机细粉(A)在色调剂粒子上的平均粒子长度范围为15-100mμm。
51.根据权利要求43的双组分显影剂,其中所述的非球形无机细粉(B)在色调剂粒子上的平均粒子长度范围为120-600mμm。
52.根据权利要求43的双组分显影剂,其中所述的非球形无机细粉(B)在色调剂粒子上的平均粒子长度范围为130-500mμm。
53.根据权利要求43的双组分显影剂,其中所述的非球形无机细粉(B)在色调剂粒子上的平均粒子长度大于色调剂粒子上的所述无机细粉(A)的平均粒子长度。
54.根据权利要求43的双组分显影剂,其中所述的非球形无机细粉(B)在色调剂粒子上的平均粒子长度比色调剂粒子上的所述无机细粉(A)的平均粒子长度长20-140mμm。
55.根据权利要求43的双组分显影剂,其中所述的无机细粉(A)在色调剂粒子上的平均粒子长度范围为15-100mμm;所述的非球形无机细粉(B)在色调剂粒子上的平均粒子长度为120-600mμm。
56.根据权利要求43的双组分显影剂,其中所述的无机细粉(A)的比表面积按照BET法以氮吸附测得为60m2/g-230m2/g。
57.根据权利要求43的双组分显影剂,其中所述的无机细粉(A)比表面积按照BET法以氮吸附测得为70m2/g-180m2/g。
58.根据权利要求43的双组分显影剂,其中所述的非球形无机细粉(B)比表面积按照BET法以氮吸附测得为20m2/g-90m2/g。
59.根据权利要求43的双组分显影剂,其中所述的非球形无机细粉(B)比表面积按照BET法以氮吸附测得为25m2/g-80m2/g。
60.根据权利要求43的双组分显影剂,其中所述的无机细粉(A)在色调剂粒子上的形状因子SF-1为100-125。
61.根据权利要求43的双组分显影剂,其中所述的非球形无机细粉(B)在色调剂粒子上的形状因子SF-1大于190。
62.根据权利要求43的双组分显影剂,其中所述的非球形无机细粉(B)在色调剂粒子上的形状因子SF-1大于200。
63.根据权利要求43的双组分显影剂,其中在电子显微镜放大的色调剂照片上观察,所述的无机细粉(A)和所述的非球形无机细粉(B)在色调剂粒子表面上存在的数量分别为平均每0.5μm×0.5μm单位面积5-90个粒子和平均每1.0μm×1.0μm单位面积1-30个粒子。
64.根据权利要求43的双组分显影剂,其中所述的色调剂是在以流动式粒子图像分析仪测得的成圆率分布中具有0.950-0.995的平均成圆率且所含成圆率小于0.950的粒子数量为2%-40%的色调剂;
所述的外部添加剂是一种在色调剂粒子上具有如下成分的外部添加剂:(i)一种以初级粒子或二级粒子状态存在的、平均粒子长度为15mμm-100mμm和形状因子SF-1为100-130的无机细粉(A)和(ii)一种由多个粒子结合形成的、平均成圆率为120mμm-600mμm和形状因子SF-1大于150的非球形无机细粉(B);和
在电子显微镜放大的色调剂照片上观察,所述的无机细粉(A)和所述的非球形无机细粉(B)在色调剂粒子表面上存在的数量分别为平均每0.5μm×0.5μm单位面积5-90个粒子和平均每1.0μm×1.0μm单位面积1-30个粒子。
65.根据权利要求43的双组分显影剂,其中所述的色调剂含有的所述无机细粉(A)的量以100重量份色调剂计为0.1-2.0重量份。
66.根据权利要求43的双组分显影剂,其中所述的色调剂含有的所述非球形无机细粉(B)的量以100重量份色调剂计为0.3-3.0重量份。
67.根据权利要求43的双组分显影剂,其中所述的无机细粉(A)具有的细粒子选自氧化铝细粒子、二氧化钛细粒子、氧化锆细粒子、氧化镁细粒子,经二氧化硅处理的这些细粒子中的任一种和氮化硅细粒子。
68.根据权利要求43的双组分显影剂,其中所述的无机细粉(A)具有的细粒子选自氧化铝细粒子、二氧化钛细粒子以及经二氧化硅处理的这些细粒子中的任一种。
69.根据权利要求43的双组分显影剂,其中所述的非球形无机细粉(B)具有的细粒子选自二氧化硅细粒子、氧化铝细粒子、二氧化钛细粒子和这些细粒子中任何一种的双氧化物细粒子。
70.根据权利要求43的双组分显影剂,其中所述的非球形无机细粉(B)具有二氧化硅细粒子。
71.根据权利要求43的双组分显影剂,其中所述的无机细粉(A)具有的细粒子选自氧化铝细粒子、二氧化钛细粒子以及经二氧化硅处理的这些细粒子中的任一种,和所述的非球形无机细粉(B)具有二氧化硅细粒子。
72.根据权利要求43的双组分显影剂,其中所述的无机细粉(A)具有氧化铝细粒子,所述的非球形无机细粉(B)具有二氧化硅细粒子。
73.根据权利要求72的双组分显影剂,其中所述的氧化铝细粒子具有这样的粒子大小分布,即直径是平均粒子直径至少两倍的粒子在数量上占0%-5%;所述的非球形无机细粉(B)具有这样的粒子大小分布,即直径是平均粒子直径的两倍到三倍的粒子在数量上占5%-15%。
74.根据权利要求72的双组分显影剂,其中所述的氧化铝细粒子具有的比表面积按照BET法以氮吸附测得为60m2/g-150m2/g,所述的非球形无机细粉(B)的比表面积按照BET法以氮吸附测得为20m2/g-70m2/g。
75.根据权利要求72的双组分显影剂,其中所述的氧化铝细粒子已经疏水性处理。
76.根据权利要求43的双组分显影剂,其中所述的色调剂粒子包括一种粘合剂树脂和一种着色剂。
77.根据权利要求43的双组分显影剂,其中所述的色调剂粒子包括一种粘合剂树脂、一种着色剂和一种隔离剂。
78.根据权利要求43的双组分显影剂,其中所述的色调剂粒子包括一种粘合剂树脂、一种着色剂、一种隔离剂和一种电荷控制剂。
79.根据权利要求43的双组分显影剂,其中所述隔离剂的重均分子量为300-3000。
80.根据权利要求43的双组分显影剂,其中所述的色调剂粒子是由聚合方法制得的粒子,此方法是将含有一种可聚合单体和一种着色剂的可聚合单体组合物在一种聚合引发剂存在的条件下在一种液体介质中聚合。
81.根据权利要求43的双组分显影剂,其中所述的色调剂粒子是由悬浮聚合法制得的粒子,此方法是将含有一种可聚合单体和一种着色剂的可聚合单体组合物在一种聚合引发剂存在的条件下在一种含水介质中聚合。
82.根据权利要求43的双组分显影剂,其中所述的色调剂粒子是由悬浮聚合法制得的粒子,此方法是将含有一种可聚合单体、一种着色剂和一种用做隔离剂的蜡的可聚合单体组合物在一种聚合引发剂存在的条件下在一种含水介质中聚合。
83.根据权利要求43的双组分显影剂,其中所述的色调剂粒子是通过处理成球形制得的粒子,通过粉碎法制得的粒子,此粉碎法包括熔融捏和一种含有一种粘合剂树脂和着色剂的混合物以得到一种捏和产物以及粉碎该捏和产物的步骤。
84.根据权利要求43的双组分显影剂,其具有的表观密度为1.2-2.0g/cm3。
85.根据权利要求43的双组分显影剂,其具有的表观密度为1.2-1.8g/cm3。
86.根据权利要求43的双组分显影剂,其具有的紧密度为5%-19%。
87.根据权利要求43的双组分显影剂,其具有的紧密度为5%-15%。
88.根据权利要求43的双组分显影剂,其中所述的载体含有一种磁性树脂载体,该磁性树脂载体含有一种树脂和一种磁性金属氧化物。
89.根据权利要求88的双组分显影剂,其中所述的磁性树脂载体包括一种树脂、一种磁性粉末和一种非磁性金属氧化物。
90.根据权利要求88的双组分显影剂,其中所述的磁性树脂载体是一种由聚合方法制得的载体。
91.根据权利要求88的双组分显影剂,其中所述的磁性树脂载体含有一种酚树脂作为粘合剂。
92.根据权利要求43的双组分显影剂,其中所述的载体具有的重均粒子直径为15μm-60μm。
93.根据权利要求43的双组分显影剂,其中所述的载体具有的重均粒子直径为20μm-45μm。
94.一种成像方法,包括:
(I)充电步骤,即给待载静电潜像的潜像承载元件进行静电充电;
(II)潜像形成步骤,即在上述充电之后的潜像承载元件上形成静电潜像;
(III)显影步骤,即采用色调剂将潜像承载元件上的静电潜像显影,形成彩色色调剂图像;和
(IV)转印步骤,将潜像承载元件上形成的色调剂图像转印到转印介质;
其中;
所述的色调剂含有色调剂粒子和外部添加剂;以及
所述的色调剂具有:
(a)以流动式粒子图像分析仪测得的粒子成圆率分布中平均成圆率为0.920-0.995,所含成圆率小于0.950的粒子数量为2%-40%;和
(b)以Coulter法测得重均粒子直径为2.0-9.0μm;和
所述外部添加剂具有在色调剂粒子上的(i)一种以初级粒子或二级粒子的状态存在的、平均粒子长度为10mμm-400mμm和形状因子SF-1为100-130的无机细粉(A)和(ii)一种由多个粒子结合形成的、形状因子SF-1大于150的非球形无机细粉(B)。
95.根据权利要求94的成像方法,其中所述的色调剂的平均成圆率为0.950-0.995。
96.根据权利要求94的成像方法,其中所述的色调剂的平均成圆率为0.960-0.995。
97.根据权利要求94的成像方法,其中成圆率小于0.950的粒子数量为3%-30%。
98.根据权利要求94的成像方法,其中所述的色调剂的形状因子SF-1为100-150。
99.根据权利要求94的成像方法,其中所述的色调剂的形状因子SF-1为100-130。
100.根据权利要求94的成像方法,其中所述的无机细粉(A)的初级或二级粒子在色调剂粒子上的平均粒子长度为15mμm-200mμm。
101.根据权利要求94的成像方法,其中所述的无机细粉(A)在色调剂粒子上的平均粒子长度为15mμm-100mμm。
102.根据权利要求94的成像方法,其中所述的非球形无机细粉(B)在色调剂粒子上的平均粒子长度为120mμm-600mμm。
103.根据权利要求94的成像方法,其中所述的非球形无机细粉(B)在色调剂粒子上的平均粒子长度为130mμm-500mμm。
104.根据权利要求94的成像方法,其中所述的非球形无机细粉(B)在色调剂粒子上的平均粒子长度大于色调剂粒子上所述无机细粉(A)的平均粒子长度。
105.根据权利要求94的成像方法,其中所述的非球形无机细粉(B)在色调剂粒子上的平均粒子长度比色调剂粒子上所述无机细粉(A)的平均粒子长度长20-140mμm。
106.根据权利要求94的成像方法,其中所述的非球形无机细粉(B)在色调剂粒子上的平均粒子长度比色调剂粒子上所述无机细粉(A)的平均粒子长度长40mμm。
107.根据权利要求94的成像方法,其中所述的无机细粉(A)在色调剂粒子上的平均粒子长度范围为15-100mμm;所述的非球形无机细粉(B)在色调剂粒子上的平均粒子长度为120-600mμm。
108.根据权利要求94的成像方法,其中所述的无机细粉(A)的比表面积按照BET法以氮吸附测得为60m2/g-230m2/g。
109.根据权利要求94的成像方法,其中所述的无机细粉(A)的比表面积按照BET法以氮吸附测得为70m2/g-180m2/g。
110.根据权利要求94的成像方法,其中所述的非球形无机细粉(B)的比表面积按照BET法以氮吸附测得为20m2/g-90m2/g。
111.根据权利要求94的成像方法,其中所述的非球形无机细粉(B)的比表面积按照BET法以氮吸附测得为25m2/g-80m2/g。
112.根据权利要求94的成像方法,其中所述的无机细粉(A)在色调剂粒子上的形状因子SF-1为100-125。
113.根据权利要求94的成像方法,其中所述的非球形无机细粉(B)在色调剂粒子上的形状因子SF-1大于190。
114.根据权利要求94的成像方法,其中所述的非球形无机细粉(B)在色调剂粒子上的形状因子SF-1大于200。
115.根据权利要求94的成像方法,其中所述的无机细粉(A)和所述的非球形无机细粉(B)在电子显微镜放大的色调剂照片上观察,在色调剂粒子表面上存在的数量分别为平均每0.5μm×0.5μm单位面积有5-90个粒子和平均每1.0μm×1.0μm单位面积1-30个粒子。
116.根据权利要求94的成像方法,其中所述的色调剂是在以流动式粒子图像分析仪测得的成圆率分布中具有0.950-0.995的平均成圆率且所含成圆率小于0.950的粒子数量为2%-40%的色调剂;
所述的外部添加剂是一种在色调剂粒子上具有如下成分的外部添加剂:(i)一种以初级粒子或二级粒子状态存在的、平均粒子长度为15mμm-100mμm和形状因子SF-1为100-130的无机细粉(A)和(ii)一种由多个粒子结合形成的、平均成圆率为120mμm-600mμm和形状因子SF-1大于150的非球形无机细粉(B);和
在电子显微镜放大的色调剂照片上观察,所述的无机细粉(A)和所述的非球形无机细粉(B)在色调剂粒子表面上存在的数量分别为平均每0.5μm×0.5μm单位面积5-90个粒子和平均每1.0μm×1.0μm单位面积1-30个粒子。
117.根据权利要求94的成像方法,其中所述的色调剂含有的所述无机细粉(A)的量按照100重量份色调剂计为0.1-2.0重量份。
118.根据权利要求94的成像方法,其中所述的色调剂含有的所述非球形无机细粉(B)的量按照100重量份色调剂计为0.3-3.0重量份。
119.根据权利要求94的成像方法,其中所述的无机细粉(A)具有的细粒子选自氧化铝细粒子、二氧化钛细粒子、氧化锆细粒子、氧化镁细粒子,经二氧化硅处理的这些细粒子中的任一种和氮化硅细粒子。
120.根据权利要求94的成像方法,其中所述的无机细粉(A)具有的细粒子选自氧化铝细粒子、二氧化钛细粒子以及经二氧化硅处理的上述细粒子中的任一种。
121.根据权利要求94的成像方法,其中所述的非球形无机细粉(B)具有的细粒子选自二氧化硅细粒子、氧化铝细粒子、二氧化钛细粒子以及上述细粒子中任一种的双氧化物细粒子。
122.根据权利要求94的成像方法,其中所述的非球形无机细粉(B)具有二氧化硅细粒子。
123.根据权利要求94的成像方法,其中所述的无机细粉(A)具有的细粒子选自氧化铝细粒子、二氧化钛细粒子以及经二氧化硅处理的上述细粒子中的任一种;所述的非球形无机细粉(B)具有二氧化硅细粒子。
124.根据权利要求94的成像方法,其中所述的无机细粉(A)具有氧化铝细粒子,所述的非球形无机细粉(B)具有二氧化硅细粒子。
125.根据权利要求124的成像方法,其中所述的氧化铝细粒子具有这样的粒子大小分布,即直径是平均粒子直径至少两倍的粒子在数量上占0%-5%;所述的非球形无机细粉(B)具有这样的粒子大小分布,即直径是平均粒子直径的两倍到三倍的粒子在数量上占5%-15%。
126.根据权利要求124的成像方法,其中所述的氧化铝细粒子具有的比表面积按照BET法以氮吸附测得为60m2/g-150m2/g,所述的非球形无机细粉(B)的比表面积按照BET法以氮吸附测得为20m2/g-70m2/g。
127.根据权利要求124的成像方法,其中所述的氧化铝细粒子已经疏水性处理。
128.根据权利要求94的成像方法,其中所述的色调剂粒子包括一种粘合剂树脂和一种着色剂。
129.根据权利要求94的成像方法,其中所述的色调剂粒子包括一种粘合剂树脂、一种着色剂和一种隔离剂。
130.根据权利要求94的成像方法,其中所述的色调剂粒子包括一种粘合剂树脂、一种着色剂、一种隔离剂和一种电荷控制剂。
131.根据权利要求94的成像方法,其中所述隔离剂的重均分子量为300-3000。
132.根据权利要求94的成像方法,其中所述的色调剂粒子是由聚合方法制得的粒子,此方法是将含有一种可聚合单体和一种着色剂的可聚合单体组合物在一种聚合引发剂存在的条件下在一种液体介质中聚合。
133.根据权利要求94的成像方法,其中所述的色调剂粒子是由悬浮聚合法制得的粒子,此方法是将含有一种可聚合单体和一种着色剂的可聚合单体组合物在一种聚合引发剂存在的条件下在一种含水介质中聚合。
134.根据权利要求94的成像方法,其中所述的色调剂粒子是由悬浮聚合法制得的粒子,此方法是将含有一种可聚合单体、一种着色剂和一种用做隔离剂的蜡的可聚合单体组合物在一种聚合引发剂存在的条件下在一种含水介质中聚合。
135.根据权利要求94的成像方法,其中所述的色调剂粒子是通过处理成球形制得的粒子,通过粉碎法制得的粒子,此粉碎法包括熔融捏和一种含有一种粘合剂树脂和着色剂的混合物以得到一种捏和产物以及粉碎该捏和产物的步骤。
136.根据权利要求94的成像方法,其中所述的显影步骤是利用含有所述色调剂和载体的双组分显影剂以及以双组分显影剂的所述色调剂显影潜像承载元件上的静电潜像的显影步骤。
137.根据权利要求136的成像方法,其中所述的双组分显影剂具有的表观密度为1.2-2.0g/cm3。
138.根据权利要求136的成像方法,其中所述的双组分显影剂具有的表观密度为1.2-1.8g/cm3。
139.根据权利要求136的成像方法,其中所述的双组分显影剂具有的紧密度为5%-19%。
140.根据权利要求136的成像方法,其中所述的双组分显影剂具有的紧密度为5%-15%。
141.根据权利要求136的成像方法,其中所述的载体含有一种磁性树脂载体,该磁性树脂载体含有一种树脂和一种磁性金属氧化物。
142.根据权利要求136的成像方法,其中所述的磁性树脂载体含有一种树脂、一种磁性粉末和一种非磁性金属氧化物。
143.根据权利要求136的成像方法,其中所述的磁性树脂载体是一种由聚合生产的载体。
144.根据权利要求136的成像方法,其中所述的磁性树脂载体含有酚树脂作为粘合剂。
145.根据权利要求136的成像方法,其中所述载体的重均粒子直径为15μm-60μm。
146.根据权利要求136的成像方法,其中所述载体的重均粒子直径为20μm-45μm。
147.根据权利要求94的成像方法,其中所述的转印介质是一种记录介质,其中将潜像承载元件上形成的色调剂图像直接转印到记录介质上,再将转印到记录介质的色调剂图像定影在记录介质上。
148.根据权利要求94的成像方法,其中所述的转印介质包含一种中间转印元件和一种记录介质,在潜像承载元件上形成的色调剂图像最初转印到这种中间转印元件上,然后将转印到中间转印元件上的色调剂图像又转印到记录介质,将转印到记录介质上的图像定影在记录介质上。
149.根据权利要求94的成像方法,其中所述的步骤I-IV是包括如下的步骤:
(i)充电步骤,即给待载静电潜像的潜像承载元件进行静电充电;
(ii)潜像形成步骤,即在上述充电之后的潜像承载元件上形成静电潜像;
(iii)显影步骤,采用彩色色调剂将潜像承载元件上的静电潜像显影,形成彩色色调剂图像;所述的彩色色调剂选自青色色调剂、品红色色调剂和黄色色调剂;和
(iv)转印步骤,将潜像承载元件上形成的彩色色调剂图像转印到转印介质;
采用具有不同颜色的彩色色调剂依次进行所述的步骤(i)-(iv)两次,在转印介质上形成多色色调剂图像;
其中:
青色色调剂包含i)含有一种粘合剂树脂和一种青色着色剂的青色色调剂粒子,和ii)所述的外部添加剂;
品红色色调剂包含i)含有一种粘合剂树脂和一种品红色着色剂的品红色调剂粒子和ii)所述的外部添加剂;
黄色色调剂包含i)含有一种粘合剂树脂和一种黄色着色剂的黄色色调剂粒子和ii)所述的外部添加剂。
150.根据权利要求149的成像方法,其中,采用包括所述青色色调剂、所述品红色色调剂、所述黄色色调剂和另外的黑色色调剂的四种彩色色调剂,以这些具有不同颜色的彩色色调剂按照所述的步骤(i)-(iv)连续执行四次,在转印介质上形成四色的彩色色调剂图像;
所述的黑色色调剂含有i)含有一种粘合剂树脂和一种黑色着色剂的黑色调剂粒子和ii)所述的外部添加剂。
151.根据权利要求149的成像方法,其中所述的转印介质是一种记录介质,其中将潜像承载元件上形成的色调剂图像直接转印到记录介质上,再将转印到记录介质的色调剂图像定影在记录介质上。
152.根据权利要求149的成像方法,其中所述的转印介质包含一种中间转印元件和一种记录介质,在潜像承载元件上形成的色调剂图像最初转印到这种中间转印元件上,然后将转印到中间转印元件上的色调剂图像又转印到记录介质,将转印到记录介质上的图像定影在记录介质上。
153.根据权利要求94的成像方法,其还包括一个在所述的转印步骤之后收集残留在潜像承载元件表面的色调剂的清洁步骤。
154.根据权利要求153的成像方法,其中所述的清洁步骤采用一种显影前清洁系统,在这种系统中,以一种与潜像承载元件表面接触的清洁元件清洁潜像承载元件的表面。
155.根据权利要求154的成像方法,其中所述显影前清洁系统中的清洁步骤是在转印步骤之后和充电步骤之前执行。
156.根据权利要求153的成像方法,其中:
所述转印步骤中的转印区、所述充电步骤中的充电区和所述显影步骤中的显影区的位置顺序以潜像承载元件表面的移动方向排列为转印区、充电区和显影区,而和潜像承载元件表面接触的任一用以除去潜像承载元件表面残留的色调剂的清洁元件不存在于转印区和充电区两者之间及充电区和显影区两者之间;和
所述清洁步骤采用一种显影时清洁系统,在这种系统中,储存有所述色调剂的显影组件使存在于潜像承载元件上的静电潜像显影,同时,显影组件又收集残留在潜像承载元件表面上的色调剂以清洁潜像承载元件表面。
157.根据权利要求156的成像方法,其中所述的潜像承载元件包括一种静电照相的感光元件。
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP160792/97 | 1997-06-18 | ||
| JP16079297 | 1997-06-18 | ||
| JP160792/1997 | 1997-06-18 | ||
| JP274049/97 | 1997-10-07 | ||
| JP27404997 | 1997-10-07 | ||
| JP274049/1997 | 1997-10-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1204783A CN1204783A (zh) | 1999-01-13 |
| CN1144097C true CN1144097C (zh) | 2004-03-31 |
Family
ID=26487188
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB98103585XA Expired - Lifetime CN1144097C (zh) | 1997-06-18 | 1998-06-18 | 色调剂、双组分显影剂和成像方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US6077635A (zh) |
| EP (1) | EP0886187B1 (zh) |
| KR (1) | KR100282952B1 (zh) |
| CN (1) | CN1144097C (zh) |
| DE (1) | DE69818912T2 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7803508B2 (en) | 2006-03-17 | 2010-09-28 | Ricoh Company, Ltd. | Toner as well as developer and image forming method using the same |
Families Citing this family (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0967527B1 (en) | 1998-06-24 | 2004-11-03 | Canon Kabushiki Kaisha | Toner and image forming method |
| EP0982636B1 (en) * | 1998-08-27 | 2005-12-14 | Ricoh Company, Ltd. | Toner for use in electrophotography, image formation method using the toner, method of producing the toner, and apparatus for producing the toner |
| JP3337994B2 (ja) | 1999-01-26 | 2002-10-28 | キヤノン株式会社 | 画像形成装置 |
| JP2000267357A (ja) * | 1999-03-16 | 2000-09-29 | Fuji Xerox Co Ltd | 静電潜像現像用トナー及び画像形成方法 |
| JP2000284560A (ja) * | 1999-03-31 | 2000-10-13 | Canon Inc | 画像形成装置 |
| WO2000060417A1 (fr) * | 1999-04-06 | 2000-10-12 | Fujitsu Limited | Toner pour electrophotographie, et procede de formation d'images |
| JP2000310885A (ja) * | 1999-04-27 | 2000-11-07 | Minolta Co Ltd | トナージェット用トナー |
| JP2001034011A (ja) * | 1999-05-17 | 2001-02-09 | Minolta Co Ltd | トナージェット用トナー |
| US6447969B1 (en) * | 1999-06-02 | 2002-09-10 | Canon Kabushiki Kaisha | Toner and image forming method |
| EP1109069B1 (en) * | 1999-12-15 | 2005-07-27 | Mitsubishi Chemical Corporation | Toner for the development of electrostatic image and method for producing the same |
| US7169526B2 (en) * | 1999-12-16 | 2007-01-30 | Mitsubishi Chemical Corporation | Toner for the development of electrostatic image and the production process thereof |
| JP4298114B2 (ja) * | 2000-02-21 | 2009-07-15 | キヤノン株式会社 | 現像剤並びに該現像剤を用いた画像形成方法及びプロセスカートリッジ |
| DE60111436T2 (de) * | 2000-02-21 | 2006-05-11 | Canon K.K. | Entwickler, Bildherstellungsverfahren und Prozesskartusche |
| JP2001312091A (ja) * | 2000-02-25 | 2001-11-09 | Canon Inc | 画像形成装置 |
| US6656654B2 (en) * | 2000-03-31 | 2003-12-02 | Ricoh Company, Ltd. | Toner and two-component developer, container therefor, and image forming apparatus |
| US20020039698A1 (en) * | 2000-07-17 | 2002-04-04 | Ricoh Company, Ltd. | Electrophotographic image formation method |
| US6589701B2 (en) * | 2000-07-28 | 2003-07-08 | Canon Kabushiki Kaisha | Dry toner, image forming method and process cartridge |
| US6638674B2 (en) | 2000-07-28 | 2003-10-28 | Canon Kabushiki Kaisha | Magnetic toner |
| KR20020018931A (ko) * | 2000-09-04 | 2002-03-09 | 오쿠무라 고조 | 정전하상 현상용 토너 및 그 제조 방법 |
| US6790572B2 (en) * | 2000-11-08 | 2004-09-14 | Ricoh Company Limited | Electrophotographic photoreceptor, and image forming method and apparatus using the photoreceptor |
| EP1239334B1 (en) * | 2001-03-08 | 2011-05-11 | Ricoh Company, Ltd. | Toner composition |
| JP2003005434A (ja) * | 2001-04-18 | 2003-01-08 | Oki Data Corp | トナー、トナーカートリッジ及び画像形成装置 |
| EP1273977B1 (en) * | 2001-07-03 | 2008-03-26 | Ricoh Company, Ltd. | Dry toner and method of preparing same |
| JP3997065B2 (ja) | 2001-08-20 | 2007-10-24 | キヤノン株式会社 | プロセスカートリッジ及び画像形成装置 |
| JP4004022B2 (ja) * | 2001-11-26 | 2007-11-07 | 株式会社リコー | 現像装置及び画像形成装置 |
| US20050011409A1 (en) * | 2001-12-25 | 2005-01-20 | Yasuhide Isobe | Inorganic oxide |
| JP4242097B2 (ja) * | 2002-01-08 | 2009-03-18 | シャープ株式会社 | 画像形成装置 |
| AU2003219323A1 (en) * | 2002-04-10 | 2003-10-27 | Avecia Limited | Chemically produced toner and process therefor |
| EP1535951B1 (en) * | 2002-06-17 | 2012-08-08 | Sanyo Chemical Industries, Ltd. | Resin particle and method for preparation thereof |
| JP4003877B2 (ja) * | 2002-08-22 | 2007-11-07 | 株式会社リコー | 静電荷像現像用トナー、現像剤、画像形成方法および画像形成装置 |
| CN1318923C (zh) * | 2002-09-20 | 2007-05-30 | 株式会社理光 | 显影剂限制部件,显影装置,处理卡盒及图像形成装置 |
| EP1406129B8 (en) * | 2002-10-02 | 2012-05-23 | Canon Kabushiki Kaisha | Silica fine particle, toner, two-component developer and image forming method |
| EP1437627B1 (en) * | 2003-01-09 | 2012-08-22 | Ricoh Company, Ltd. | Toner feeder and elelctrophotographic image forming apparatus using the toner feeder and toner |
| JP4290015B2 (ja) * | 2003-01-10 | 2009-07-01 | キヤノン株式会社 | カラートナー及び画像形成装置 |
| EP1455237B1 (en) * | 2003-03-07 | 2011-05-25 | Canon Kabushiki Kaisha | Toner and two-component developer |
| JP2005053620A (ja) * | 2003-08-01 | 2005-03-03 | Fuji Photo Film Co Ltd | シート体搬送用ガイド構造 |
| JP2005070276A (ja) * | 2003-08-22 | 2005-03-17 | Ricoh Co Ltd | 画像形成装置、プロセスカートリッジ及びこれらに用いるトナー |
| JP4270557B2 (ja) * | 2004-04-20 | 2009-06-03 | 花王株式会社 | トナーの製造方法 |
| EP1628171B1 (en) | 2004-04-27 | 2017-02-01 | Canon Kabushiki Kaisha | Developing method for an image forming apparatus and developing device using the same |
| US8064796B2 (en) * | 2006-03-30 | 2011-11-22 | Mitsubishi Chemical Corporation | Image forming apparatus |
| JP2007322916A (ja) | 2006-06-02 | 2007-12-13 | Ricoh Co Ltd | 現像剤供給装置、現像剤容器、現像剤、及び、画像形成装置 |
| CA2630933C (en) * | 2006-09-04 | 2012-08-28 | Ricoh Company, Ltd. | Electrostatic image developing toner, two-component developer, image forming method and process cartridge |
| JP5081538B2 (ja) * | 2006-12-05 | 2012-11-28 | 花王株式会社 | 電子写真用トナーの製造方法。 |
| JP4461169B2 (ja) * | 2007-12-06 | 2010-05-12 | シャープ株式会社 | カラートナー、現像剤、現像装置、および画像形成装置 |
| JP5473725B2 (ja) * | 2009-04-15 | 2014-04-16 | キヤノン株式会社 | 磁性トナー |
| US20110033797A1 (en) * | 2009-08-10 | 2011-02-10 | Kyocera Mita Corporation | Image formation apparatus |
| JP5640801B2 (ja) | 2010-02-24 | 2014-12-17 | 三菱化学株式会社 | 画像形成装置および電子写真カートリッジ |
| US8841056B2 (en) | 2010-03-31 | 2014-09-23 | Canon Kabushiki Kaisha | Toner and process for producing toner |
| US8426094B2 (en) | 2010-05-31 | 2013-04-23 | Canon Kabushiki Kaisha | Magnetic toner |
| US8614044B2 (en) | 2010-06-16 | 2013-12-24 | Canon Kabushiki Kaisha | Toner |
| KR101445048B1 (ko) | 2010-09-16 | 2014-09-26 | 캐논 가부시끼가이샤 | 토너 |
| WO2012046827A1 (en) | 2010-10-04 | 2012-04-12 | Canon Kabushiki Kaisha | Toner |
| EP2625568B1 (en) | 2010-10-04 | 2018-01-10 | Canon Kabushiki Kaisha | Toner |
| JP5644464B2 (ja) | 2010-12-15 | 2014-12-24 | 富士ゼロックス株式会社 | 静電荷像現像用トナー、静電荷像現像剤、トナーカートリッジ、プロセスカートリッジ及び画像形成装置 |
| JP2012189960A (ja) | 2011-03-14 | 2012-10-04 | Fuji Xerox Co Ltd | 静電荷像現像用トナー、静電荷像現像剤、トナーカートリッジ、プロセスカートリッジ、画像形成装置、及び、画像形成方法 |
| JP5712824B2 (ja) * | 2011-07-06 | 2015-05-07 | 富士ゼロックス株式会社 | シリカ粒子及びその製造方法 |
| JP5879931B2 (ja) | 2011-10-26 | 2016-03-08 | 富士ゼロックス株式会社 | 静電荷像現像用トナー、静電荷像現像剤、トナーカートリッジ、プロセスカートリッジ、画像形成装置及び画像形成方法 |
| JP2013137508A (ja) * | 2011-11-30 | 2013-07-11 | Konica Minolta Inc | 静電荷像現像用トナーおよび画像形成方法 |
| JP5868165B2 (ja) | 2011-12-27 | 2016-02-24 | キヤノン株式会社 | 現像装置及び現像方法 |
| JP5966464B2 (ja) * | 2012-03-14 | 2016-08-10 | 株式会社リコー | トナー、二成分現像剤、及び画像形成装置 |
| JP2013190647A (ja) * | 2012-03-14 | 2013-09-26 | Ricoh Co Ltd | トナー、二成分現像剤、及び画像形成装置 |
| JP2015011331A (ja) * | 2013-07-02 | 2015-01-19 | 富士ゼロックス株式会社 | 画像形成装置、及びプロセスカートリッジ |
| JP6487730B2 (ja) | 2014-03-20 | 2019-03-20 | キヤノン株式会社 | トナーおよび二成分現像剤 |
| JP6632249B2 (ja) | 2014-08-26 | 2020-01-22 | キヤノン株式会社 | 磁性キャリア及び二成分系現像剤 |
| JP6418992B2 (ja) | 2015-03-13 | 2018-11-07 | キヤノン株式会社 | 磁性キャリアおよびその製造方法 |
| DE112016001562B4 (de) | 2015-03-31 | 2021-12-09 | Canon Kabushiki Kaisha | Magnetischer träger |
| JP6584225B2 (ja) | 2015-08-25 | 2019-10-02 | キヤノン株式会社 | 磁性キャリア、二成分系現像剤、補給用現像剤、及び画像形成方法 |
| DE102016200324A1 (de) * | 2016-01-14 | 2017-07-20 | MTU Aero Engines AG | Verfahren zum Ermitteln einer Konzentration wenigstens eines Werkstoffs in einem Pulver für ein additives Herstellverfahren |
| JP6403816B2 (ja) | 2016-02-08 | 2018-10-10 | キヤノン株式会社 | 磁性キャリア、二成分系現像剤、補給用現像剤、及び画像形成方法 |
| CN108602672B (zh) * | 2016-02-29 | 2022-06-03 | 富士胶片株式会社 | 组合物、组合物的制造方法、固化膜、滤色器、遮光膜、固体摄像元件及图像显示装置 |
| JP6776570B2 (ja) * | 2016-03-22 | 2020-10-28 | 富士ゼロックス株式会社 | 静電荷像現像用トナー、静電荷像現像剤、トナーカートリッジ、プロセスカートリッジ、画像形成装置及び画像形成方法 |
| US10409188B2 (en) | 2017-02-10 | 2019-09-10 | Canon Kabushiki Kaisha | Magnetic carrier, two-component developer, replenishing developer, and image forming method |
| US10451985B2 (en) | 2017-02-28 | 2019-10-22 | Canon Kabushiki Kaisha | Toner |
| JP7069788B2 (ja) * | 2017-03-17 | 2022-05-18 | 株式会社リコー | トナーおよびその製造方法、画像形成方法、画像形成装置並びにプロセスカートリッジ |
| JP6938345B2 (ja) | 2017-11-17 | 2021-09-22 | キヤノン株式会社 | トナー |
| US10520884B2 (en) * | 2017-12-15 | 2019-12-31 | Canon Kabushiki Kaisha | Cartridge and image forming apparatus |
| JP7293010B2 (ja) | 2018-08-08 | 2023-06-19 | キヤノン株式会社 | 磁性キャリア、二成分系現像剤、補給用現像剤、及び画像形成方法 |
| JP7293009B2 (ja) | 2018-08-08 | 2023-06-19 | キヤノン株式会社 | 磁性キャリア、二成分系現像剤、補給用現像剤、及び画像形成方法 |
| JP7171314B2 (ja) | 2018-08-28 | 2022-11-15 | キヤノン株式会社 | トナー |
| JP7130518B2 (ja) | 2018-09-28 | 2022-09-05 | キヤノン株式会社 | 磁性キャリア、二成分系現像剤、補給用現像剤、及び画像形成方法 |
| US11249410B2 (en) | 2018-12-12 | 2022-02-15 | Canon Kabushiki Kaisha | Toner |
| JP7443048B2 (ja) * | 2018-12-28 | 2024-03-05 | キヤノン株式会社 | トナー |
Family Cites Families (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1043149A (en) * | 1974-05-30 | 1978-11-28 | Lewis O. Jones | Classified toner materials, developer mixture and imaging system |
| JPS5532060A (en) * | 1978-08-29 | 1980-03-06 | Canon Inc | Method and apparatus for electrophotographic developing |
| JPS58129437A (ja) * | 1982-01-29 | 1983-08-02 | Konishiroku Photo Ind Co Ltd | 静電荷像現像剤 |
| JPS5953856A (ja) * | 1982-09-21 | 1984-03-28 | Canon Inc | トナ−の製造方法 |
| JPS5961842A (ja) * | 1982-09-30 | 1984-04-09 | Canon Inc | 磁性トナ−の製造方法 |
| JPH0677166B2 (ja) * | 1983-01-20 | 1994-09-28 | 株式会社東芝 | 画像形成装置 |
| US4496644A (en) * | 1983-02-28 | 1985-01-29 | Eastman Kodak Company | Electric field adjustment for magnetic brushes |
| JPS6032060A (ja) * | 1983-08-03 | 1985-02-19 | Canon Inc | 磁性トナー |
| JPS60136752A (ja) * | 1983-12-26 | 1985-07-20 | Canon Inc | 画像形成方法 |
| JPS61146794A (ja) * | 1984-12-20 | 1986-07-04 | Toshiba Ceramics Co Ltd | 単結晶アルミナの製造方法 |
| JPS61188546A (ja) * | 1985-02-16 | 1986-08-22 | Konishiroku Photo Ind Co Ltd | 静電荷像現像用トナ− |
| JPS62203182A (ja) * | 1986-03-04 | 1987-09-07 | Toshiba Corp | 画像形成装置 |
| JPS63133179A (ja) * | 1986-11-26 | 1988-06-04 | Toshiba Corp | 記録装置 |
| JPH0786696B2 (ja) * | 1987-05-22 | 1995-09-20 | コニカ株式会社 | 静電像現像用トナ− |
| JP2637104B2 (ja) * | 1987-07-16 | 1997-08-06 | 株式会社東芝 | 画像形成装置 |
| JP2567018B2 (ja) * | 1988-02-26 | 1996-12-25 | 三田工業株式会社 | 静電荷像現像用トナーの製造方法 |
| US4904558A (en) * | 1988-03-08 | 1990-02-27 | Canon Kabushiki Kaisha | Magnetic, two-component developer containing fluidity improver and image forming method |
| JPH0222966A (ja) * | 1988-07-12 | 1990-01-25 | Sony Corp | クランプ回路 |
| JPH0259784A (ja) * | 1988-08-25 | 1990-02-28 | Sony Corp | 出席管理装置 |
| JPH02302772A (ja) * | 1989-05-18 | 1990-12-14 | Koichi Kinoshita | 電子写真プリンターのプリンティング方法 |
| US5240803A (en) * | 1989-08-29 | 1993-08-31 | Mita Industrial Co., Ltd. | Toner for developing statically charged images and process for preparation thereof |
| JPH0450886A (ja) * | 1990-06-15 | 1992-02-19 | Toshiba Corp | 記録装置 |
| JPH04155361A (ja) * | 1990-10-18 | 1992-05-28 | Konica Corp | 画像形成装置 |
| JPH04234063A (ja) * | 1990-12-28 | 1992-08-21 | Kyocera Corp | 画像形成方法 |
| JP3074037B2 (ja) * | 1991-06-25 | 2000-08-07 | 株式会社東芝 | 画像形成方法 |
| JPH0750337B2 (ja) * | 1991-06-25 | 1995-05-31 | 村田機械株式会社 | クリーナレス画像形成方法 |
| JPH053482A (ja) * | 1991-06-26 | 1993-01-08 | Nec Corp | ポーリング・セレクテイング方式 |
| JPH0561383A (ja) * | 1991-08-30 | 1993-03-12 | Murata Mach Ltd | クリーナレス画像形成方法 |
| JP3015825B2 (ja) * | 1991-09-13 | 2000-03-06 | 太平洋セメント株式会社 | コンクリート製品用型枠 |
| JP2629509B2 (ja) * | 1991-12-17 | 1997-07-09 | 村田機械株式会社 | クリーナレス画像形成装置 |
| JP3177644B2 (ja) * | 1993-02-02 | 2001-06-18 | 京セラ株式会社 | 画像形成用感光体の帯電方法および画像形成方法 |
| JPH072319A (ja) * | 1993-06-16 | 1995-01-06 | Sekisui Chem Co Ltd | 収納装置 |
| JP3016535B2 (ja) * | 1993-11-26 | 2000-03-06 | キヤノン株式会社 | 画像形成装置 |
| JP3066943B2 (ja) * | 1993-11-29 | 2000-07-17 | キヤノン株式会社 | 画像形成方法 |
| JPH07261446A (ja) * | 1994-03-17 | 1995-10-13 | Sharp Corp | 静電荷現像用トナー |
| EP1059567B1 (en) * | 1995-02-10 | 2003-05-21 | Canon Kabushiki Kaisha | Image forming apparatus comprising developing means provided with a black toner with specific sphericity , use of this black toner in an imaging process and toner kit |
| US5712072A (en) * | 1995-02-28 | 1998-01-27 | Canon Kabusbiki Kaisha | Toner for developing electrostatic image |
-
1998
- 1998-06-18 CN CNB98103585XA patent/CN1144097C/zh not_active Expired - Lifetime
- 1998-06-18 EP EP98304829A patent/EP0886187B1/en not_active Expired - Lifetime
- 1998-06-18 DE DE69818912T patent/DE69818912T2/de not_active Expired - Lifetime
- 1998-06-18 KR KR1019980022878A patent/KR100282952B1/ko not_active IP Right Cessation
- 1998-06-18 US US09/099,527 patent/US6077635A/en not_active Expired - Lifetime
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7803508B2 (en) | 2006-03-17 | 2010-09-28 | Ricoh Company, Ltd. | Toner as well as developer and image forming method using the same |
| CN101271287B (zh) * | 2006-03-17 | 2011-10-26 | 株式会社理光 | 调色剂、使用该调色剂的显影剂以及图像形成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0886187A2 (en) | 1998-12-23 |
| CN1204783A (zh) | 1999-01-13 |
| EP0886187A3 (en) | 1999-02-17 |
| DE69818912D1 (de) | 2003-11-20 |
| KR19990007092A (ko) | 1999-01-25 |
| KR100282952B1 (ko) | 2001-03-02 |
| DE69818912T2 (de) | 2004-08-19 |
| EP0886187B1 (en) | 2003-10-15 |
| US6077635A (en) | 2000-06-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1144097C (zh) | 色调剂、双组分显影剂和成像方法 | |
| CN1187656C (zh) | 磁性色调剂以及利用其的成像方法 | |
| CN1181403C (zh) | 磁性调色剂及其生产方法和成象方法,设备和处理盒 | |
| CN1231818C (zh) | 调色剂和成像方法 | |
| CN1105332C (zh) | 磁性涂层载体和双组分型显影剂及显影方法 | |
| CN1086233C (zh) | 磁性调色剂、磁性显影剂、设备装置、影象形成设备和传真设备 | |
| CN1181401C (zh) | 磁性调色剂 | |
| CN1204461C (zh) | 显影装置、处理卡盒和图像形成方法 | |
| CN1289973C (zh) | 调色剂 | |
| CN1324408C (zh) | 调色剂 | |
| CN1217240C (zh) | 图像形成方法,补给用墨粉及其制造方法,含载体墨粉匣 | |
| CN1237723A (zh) | 色粉,双组分显影剂,成象方法及成象设备 | |
| CN1174288C (zh) | 用于显影静电图像的色调剂、成像方法以及操作盒 | |
| CN1384401A (zh) | 补充显影剂和显影方法 | |
| CN1495550A (zh) | 对静电图像进行显影的显影剂,成像装置及成像方法 | |
| CN1673878A (zh) | 调色剂、使用其的显影设备 | |
| CN1752855A (zh) | 调色剂 | |
| CN1831665A (zh) | 显影装置和配有其的操作盒 | |
| CN1755529A (zh) | 磁性单组分调色剂及图像形成方法 | |
| CN1577117A (zh) | 调色剂 | |
| CN1301447C (zh) | 图像形成方法 | |
| CN1179247C (zh) | 色调剂和成像方法 | |
| CN1169026C (zh) | 电摄影装置,成像方法和处理盒 | |
| CN1873553A (zh) | 成像设备 | |
| CN1154019C (zh) | 用于静电影像显影的调色剂、成像方法、显影装置和工艺盒总成 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CX01 | Expiry of patent term |
Granted publication date: 20040331 |
|
| CX01 | Expiry of patent term |