DE69918552T2 - Pyrrolo(2,3-d)pyrimidin-verbindungen - Google Patents
Pyrrolo(2,3-d)pyrimidin-verbindungen Download PDFInfo
- Publication number
- DE69918552T2 DE69918552T2 DE69918552T DE69918552T DE69918552T2 DE 69918552 T2 DE69918552 T2 DE 69918552T2 DE 69918552 T DE69918552 T DE 69918552T DE 69918552 T DE69918552 T DE 69918552T DE 69918552 T2 DE69918552 T2 DE 69918552T2
- Authority
- DE
- Germany
- Prior art keywords
- alkyl
- aryl
- pyrrolo
- alkoxy
- heteroaryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 150000004943 pyrrolo[2,3-d]pyrimidines Chemical class 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims description 94
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 76
- -1 morpholino, thiomorpholino, piperidino, pyrrolidino, piperidyl Chemical group 0.000 claims description 56
- 150000003839 salts Chemical class 0.000 claims description 34
- 108010019421 Janus Kinase 3 Proteins 0.000 claims description 33
- 102000006500 Janus Kinase 3 Human genes 0.000 claims description 33
- 125000001072 heteroaryl group Chemical group 0.000 claims description 32
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims description 28
- 229910052739 hydrogen Inorganic materials 0.000 claims description 24
- 239000001257 hydrogen Substances 0.000 claims description 24
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 18
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 claims description 16
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 15
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 14
- 241000124008 Mammalia Species 0.000 claims description 14
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 13
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 13
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 12
- 239000001301 oxygen Substances 0.000 claims description 12
- 229910052760 oxygen Inorganic materials 0.000 claims description 12
- 125000000217 alkyl group Chemical group 0.000 claims description 11
- 238000002360 preparation method Methods 0.000 claims description 11
- 206010028980 Neoplasm Diseases 0.000 claims description 10
- 208000006673 asthma Diseases 0.000 claims description 10
- 201000011510 cancer Diseases 0.000 claims description 10
- 239000003795 chemical substances by application Substances 0.000 claims description 10
- 239000003814 drug Substances 0.000 claims description 10
- 229910052736 halogen Inorganic materials 0.000 claims description 10
- 150000002367 halogens Chemical class 0.000 claims description 10
- 150000002431 hydrogen Chemical class 0.000 claims description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 10
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 10
- 125000006719 (C6-C10) aryl (C1-C6) alkyl group Chemical group 0.000 claims description 9
- 208000024827 Alzheimer disease Diseases 0.000 claims description 9
- 208000023275 Autoimmune disease Diseases 0.000 claims description 9
- 206010009900 Colitis ulcerative Diseases 0.000 claims description 9
- 208000011231 Crohn disease Diseases 0.000 claims description 9
- 206010012438 Dermatitis atopic Diseases 0.000 claims description 9
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 claims description 9
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 claims description 9
- 201000004681 Psoriasis Diseases 0.000 claims description 9
- 208000024799 Thyroid disease Diseases 0.000 claims description 9
- 206010052779 Transplant rejections Diseases 0.000 claims description 9
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 9
- 201000006704 Ulcerative Colitis Diseases 0.000 claims description 9
- 201000008937 atopic dermatitis Diseases 0.000 claims description 9
- 206010012601 diabetes mellitus Diseases 0.000 claims description 9
- 208000032839 leukemia Diseases 0.000 claims description 9
- 206010025135 lupus erythematosus Diseases 0.000 claims description 9
- 201000006417 multiple sclerosis Diseases 0.000 claims description 9
- 210000000056 organ Anatomy 0.000 claims description 9
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 claims description 8
- 230000001363 autoimmune Effects 0.000 claims description 8
- 125000004432 carbon atom Chemical group C* 0.000 claims description 8
- LTVOKYUPTHZZQH-UHFFFAOYSA-N difluoromethane Chemical group F[C]F LTVOKYUPTHZZQH-UHFFFAOYSA-N 0.000 claims description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 8
- 230000002265 prevention Effects 0.000 claims description 8
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 7
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 7
- 125000002252 acyl group Chemical group 0.000 claims description 7
- 229940121363 anti-inflammatory agent Drugs 0.000 claims description 7
- 239000002260 anti-inflammatory agent Substances 0.000 claims description 7
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 7
- 208000035475 disorder Diseases 0.000 claims description 6
- 230000002401 inhibitory effect Effects 0.000 claims description 6
- 239000008194 pharmaceutical composition Substances 0.000 claims description 6
- 125000005936 piperidyl group Chemical group 0.000 claims description 6
- 229910052717 sulfur Inorganic materials 0.000 claims description 6
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 6
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims description 5
- 125000003545 alkoxy group Chemical group 0.000 claims description 5
- 125000000266 alpha-aminoacyl group Chemical group 0.000 claims description 5
- 229910052805 deuterium Inorganic materials 0.000 claims description 5
- 229940079593 drug Drugs 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 210000000987 immune system Anatomy 0.000 claims description 5
- 229910052757 nitrogen Inorganic materials 0.000 claims description 5
- 125000004738 (C1-C6) alkyl sulfinyl group Chemical group 0.000 claims description 4
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 claims description 4
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims description 4
- ZYUZIQCTDGCTPB-UHFFFAOYSA-N 5-chloro-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C=12C(Cl)=CNC2=NC=NC=1N1CCCCC1 ZYUZIQCTDGCTPB-UHFFFAOYSA-N 0.000 claims description 4
- CIPYWMYUPREAMJ-UHFFFAOYSA-N 5-ethynyl-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C=12C(C#C)=CNC2=NC=NC=1N1CCCCC1 CIPYWMYUPREAMJ-UHFFFAOYSA-N 0.000 claims description 4
- 125000004442 acylamino group Chemical group 0.000 claims description 4
- 125000005256 alkoxyacyl group Chemical group 0.000 claims description 4
- 125000006620 amino-(C1-C6) alkyl group Chemical group 0.000 claims description 4
- 125000003118 aryl group Chemical group 0.000 claims description 4
- 125000005135 aryl sulfinyl group Chemical group 0.000 claims description 4
- 125000004391 aryl sulfonyl group Chemical group 0.000 claims description 4
- 125000005110 aryl thio group Chemical group 0.000 claims description 4
- 125000000000 cycloalkoxy group Chemical group 0.000 claims description 4
- 229910052721 tungsten Inorganic materials 0.000 claims description 4
- 229910052727 yttrium Inorganic materials 0.000 claims description 4
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 claims description 3
- BHEXESBBAOCUEW-UHFFFAOYSA-N 4-(3,3-dimethylpiperidin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1C(C)(C)CCCN1C1=NC=NC2=C1C=CN2 BHEXESBBAOCUEW-UHFFFAOYSA-N 0.000 claims description 3
- 125000004193 piperazinyl group Chemical group 0.000 claims description 3
- 125000003386 piperidinyl group Chemical group 0.000 claims description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 3
- 125000006528 (C2-C6) alkyl group Chemical group 0.000 claims description 2
- 125000006763 (C3-C9) cycloalkyl group Chemical group 0.000 claims description 2
- KPPVNWGJXFMGAM-UUILKARUSA-N (e)-2-methyl-1-(6-methyl-3,4-dihydro-2h-quinolin-1-yl)but-2-en-1-one Chemical compound CC1=CC=C2N(C(=O)C(/C)=C/C)CCCC2=C1 KPPVNWGJXFMGAM-UUILKARUSA-N 0.000 claims description 2
- LERUYXUJSAVVIZ-UHFFFAOYSA-N 2-[1-[5-(3-methylphenyl)-7h-pyrrolo[2,3-d]pyrimidin-4-yl]piperidin-4-yl]ethanol Chemical compound CC1=CC=CC(C=2C3=C(N4CCC(CCO)CC4)N=CN=C3NC=2)=C1 LERUYXUJSAVVIZ-UHFFFAOYSA-N 0.000 claims description 2
- UZFMOKQJFYMBGY-UHFFFAOYSA-N 4-hydroxy-TEMPO Chemical compound CC1(C)CC(O)CC(C)(C)N1[O] UZFMOKQJFYMBGY-UHFFFAOYSA-N 0.000 claims description 2
- WOJIDULOACFYJL-UHFFFAOYSA-N 4-piperidin-1-yl-5-(3-propan-2-ylphenyl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CC(C)C1=CC=CC(C=2C3=C(N4CCCCC4)N=CN=C3NC=2)=C1 WOJIDULOACFYJL-UHFFFAOYSA-N 0.000 claims description 2
- YAJAWZHVCVQZIW-UHFFFAOYSA-N 4-piperidin-1-yl-5-(trifluoromethyl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C=12C(C(F)(F)F)=CNC2=NC=NC=1N1CCCCC1 YAJAWZHVCVQZIW-UHFFFAOYSA-N 0.000 claims description 2
- GUWZKQIDHRZALH-UHFFFAOYSA-N 5-(1-methylimidazol-4-yl)-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CN1C=NC(C=2C3=C(N4CCCCC4)N=CN=C3NC=2)=C1 GUWZKQIDHRZALH-UHFFFAOYSA-N 0.000 claims description 2
- XMDULOSPOORHTJ-UHFFFAOYSA-N 5-(2-methylpyridin-4-yl)-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1=NC(C)=CC(C=2C3=C(N4CCCCC4)N=CN=C3NC=2)=C1 XMDULOSPOORHTJ-UHFFFAOYSA-N 0.000 claims description 2
- LWQNDUSDVLVELT-UHFFFAOYSA-N 5-(3-methylimidazol-4-yl)-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CN1C=NC=C1C1=CNC2=NC=NC(N3CCCCC3)=C12 LWQNDUSDVLVELT-UHFFFAOYSA-N 0.000 claims description 2
- WZFLNSVJMWZIGG-UHFFFAOYSA-N 5-(3-methylphenyl)-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CC1=CC=CC(C=2C3=C(N4CCCCC4)N=CN=C3NC=2)=C1 WZFLNSVJMWZIGG-UHFFFAOYSA-N 0.000 claims description 2
- LCTMPEQAHUTSGO-UHFFFAOYSA-N 5-fluoro-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C=12C(F)=CNC2=NC=NC=1N1CCCCC1 LCTMPEQAHUTSGO-UHFFFAOYSA-N 0.000 claims description 2
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 2
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 2
- 125000004423 acyloxy group Chemical group 0.000 claims description 2
- 125000001769 aryl amino group Chemical group 0.000 claims description 2
- 125000004657 aryl sulfonyl amino group Chemical group 0.000 claims description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 2
- 230000005764 inhibitory process Effects 0.000 claims description 2
- 125000002757 morpholinyl group Chemical group 0.000 claims description 2
- SDCGYRLQORKUBU-UHFFFAOYSA-N n',n'-dimethyl-n-[[3-(4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl]methyl]ethane-1,2-diamine Chemical compound CN(C)CCNCC1=CC=CC(C=2C3=C(N4CCCCC4)N=CN=C3NC=2)=C1 SDCGYRLQORKUBU-UHFFFAOYSA-N 0.000 claims description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 claims description 2
- 125000002112 pyrrolidino group Chemical group [*]N1C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 2
- 125000006296 sulfonyl amino group Chemical group [H]N(*)S(*)(=O)=O 0.000 claims description 2
- 239000011593 sulfur Substances 0.000 claims description 2
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 2
- 125000006624 (C1-C6) alkoxycarbonylamino group Chemical group 0.000 claims 1
- 125000004845 (C1-C6) alkylsulfonylamino group Chemical group 0.000 claims 1
- 125000004093 cyano group Chemical group *C#N 0.000 claims 1
- 238000000034 method Methods 0.000 description 54
- 238000002844 melting Methods 0.000 description 51
- 230000008018 melting Effects 0.000 description 51
- 239000000203 mixture Substances 0.000 description 38
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 34
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 34
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 33
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 33
- 239000000243 solution Substances 0.000 description 33
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 30
- 238000006243 chemical reaction Methods 0.000 description 26
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 22
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- 239000011541 reaction mixture Substances 0.000 description 17
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 17
- 239000000047 product Substances 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- 210000004027 cell Anatomy 0.000 description 13
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 11
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 10
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 10
- 239000002585 base Substances 0.000 description 10
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 9
- 108010036949 Cyclosporine Proteins 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- BPTCCCTWWAUJRK-UHFFFAOYSA-N 4-chloro-7h-pyrrolo[2,3-d]pyrimidine Chemical compound ClC1=NC=NC2=C1C=CN2 BPTCCCTWWAUJRK-UHFFFAOYSA-N 0.000 description 6
- 102000004388 Interleukin-4 Human genes 0.000 description 6
- 108090000978 Interleukin-4 Proteins 0.000 description 6
- 230000003197 catalytic effect Effects 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- 235000019441 ethanol Nutrition 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 6
- 229930105110 Cyclosporin A Natural products 0.000 description 5
- 210000001744 T-lymphocyte Anatomy 0.000 description 5
- 239000000443 aerosol Substances 0.000 description 5
- 239000012267 brine Substances 0.000 description 5
- 239000000460 chlorine Substances 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 231100000252 nontoxic Toxicity 0.000 description 5
- 230000003000 nontoxic effect Effects 0.000 description 5
- 229910052763 palladium Inorganic materials 0.000 description 5
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 5
- 238000010898 silica gel chromatography Methods 0.000 description 5
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 4
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 4
- XTUVJUMINZSXGF-UHFFFAOYSA-N N-methylcyclohexylamine Chemical compound CNC1CCCCC1 XTUVJUMINZSXGF-UHFFFAOYSA-N 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 4
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 4
- 229940077388 benzenesulfonate Drugs 0.000 description 4
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- 229960001265 ciclosporin Drugs 0.000 description 4
- DIOQZVSQGTUSAI-UHFFFAOYSA-N decane Chemical compound CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 239000003018 immunosuppressive agent Substances 0.000 description 4
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 239000012312 sodium hydride Substances 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- POTIYWUALSJREP-UHFFFAOYSA-N 1,2,3,4,4a,5,6,7,8,8a-decahydroquinoline Chemical compound N1CCCC2CCCCC21 POTIYWUALSJREP-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 206010062016 Immunosuppression Diseases 0.000 description 3
- 108060001084 Luciferase Proteins 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 150000001447 alkali salts Chemical class 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 150000001768 cations Chemical class 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 239000012458 free base Substances 0.000 description 3
- 230000001506 immunosuppresive effect Effects 0.000 description 3
- 229960003444 immunosuppressant agent Drugs 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 229920002554 vinyl polymer Polymers 0.000 description 3
- 239000011592 zinc chloride Substances 0.000 description 3
- 235000005074 zinc chloride Nutrition 0.000 description 3
- 0 *c1c(*)[n]c2c1c(*)ncn2 Chemical compound *c1c(*)[n]c2c1c(*)ncn2 0.000 description 2
- NUPWQTAPUBVIPU-UHFFFAOYSA-N 1-(5-chloro-7h-pyrrolo[2,3-d]pyrimidin-4-yl)-3,4,4a,5,6,7,8,8a-octahydro-2h-quinoline Chemical compound C1CCC2CCCCC2N1C1=C2C(Cl)=CNC2=NC=N1 NUPWQTAPUBVIPU-UHFFFAOYSA-N 0.000 description 2
- NNWUEBIEOFQMSS-UHFFFAOYSA-N 2-Methylpiperidine Chemical compound CC1CCCCN1 NNWUEBIEOFQMSS-UHFFFAOYSA-N 0.000 description 2
- QBBKKFZGCDJDQK-UHFFFAOYSA-N 2-ethylpiperidine Chemical compound CCC1CCCCN1 QBBKKFZGCDJDQK-UHFFFAOYSA-N 0.000 description 2
- CDODDZJCEADUQQ-UHFFFAOYSA-N 3,3-dimethylpiperidine Chemical compound CC1(C)CCCNC1 CDODDZJCEADUQQ-UHFFFAOYSA-N 0.000 description 2
- PRRYQINUAIQTFJ-UHFFFAOYSA-N 5-phenyl-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CCCCN1C1=NC=NC2=C1C(C=1C=CC=CC=1)=CN2 PRRYQINUAIQTFJ-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- 229930182816 L-glutamine Natural products 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- PAMIQIKDUOTOBW-UHFFFAOYSA-N N-methylcyclohexylamine Natural products CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 2
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 2
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- 108010011005 STAT6 Transcription Factor Proteins 0.000 description 2
- 102100023980 Signal transducer and activator of transcription 6 Human genes 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 229960001138 acetylsalicylic acid Drugs 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 150000001350 alkyl halides Chemical class 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 230000003110 anti-inflammatory effect Effects 0.000 description 2
- 239000000010 aprotic solvent Substances 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 150000001503 aryl iodides Chemical class 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 229940092117 atgam Drugs 0.000 description 2
- 229960002170 azathioprine Drugs 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 229940107810 cellcept Drugs 0.000 description 2
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- MGNZXYYWBUKAII-UHFFFAOYSA-N cyclohexa-1,3-diene Chemical compound C1CC=CC=C1 MGNZXYYWBUKAII-UHFFFAOYSA-N 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 229960002806 daclizumab Drugs 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 229910001882 dioxygen Inorganic materials 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 210000004602 germ cell Anatomy 0.000 description 2
- 229960002706 gusperimus Drugs 0.000 description 2
- 229960001680 ibuprofen Drugs 0.000 description 2
- 229940073062 imuran Drugs 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 125000002346 iodo group Chemical group I* 0.000 description 2
- GHXZPUGJZVBLGC-UHFFFAOYSA-N iodoethene Chemical compound IC=C GHXZPUGJZVBLGC-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 2
- 229960000681 leflunomide Drugs 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 235000010755 mineral Nutrition 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 229940014456 mycophenolate Drugs 0.000 description 2
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 2
- IDINUJSAMVOPCM-INIZCTEOSA-N n-[(1s)-2-[4-(3-aminopropylamino)butylamino]-1-hydroxy-2-oxoethyl]-7-(diaminomethylideneamino)heptanamide Chemical compound NCCCNCCCCNC(=O)[C@H](O)NC(=O)CCCCCCN=C(N)N IDINUJSAMVOPCM-INIZCTEOSA-N 0.000 description 2
- XUOJALWUMHTTIM-UHFFFAOYSA-N n-cyclohexyl-n,6-dimethyl-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2NC(C)=CC=2C=1N(C)C1CCCCC1 XUOJALWUMHTTIM-UHFFFAOYSA-N 0.000 description 2
- DMNNVMWOBHRGQW-UHFFFAOYSA-N n-cyclohexyl-n-methyl-6-phenyl-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2NC(C=3C=CC=CC=3)=CC=2C=1N(C)C1CCCCC1 DMNNVMWOBHRGQW-UHFFFAOYSA-N 0.000 description 2
- UCOZXARHJOLLFO-UHFFFAOYSA-N n-cyclohexyl-n-methyl-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound N=1C=NC=2NC=CC=2C=1N(C)C1CCCCC1 UCOZXARHJOLLFO-UHFFFAOYSA-N 0.000 description 2
- 229960002009 naproxen Drugs 0.000 description 2
- CMWTZPSULFXXJA-VIFPVBQESA-M naproxen(1-) Chemical compound C1=C([C@H](C)C([O-])=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-M 0.000 description 2
- 239000006199 nebulizer Substances 0.000 description 2
- 229940063121 neoral Drugs 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- 229960002702 piroxicam Drugs 0.000 description 2
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 229960005205 prednisolone Drugs 0.000 description 2
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 229940063122 sandimmune Drugs 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 230000008054 signal transmission Effects 0.000 description 2
- 229960002930 sirolimus Drugs 0.000 description 2
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- XJLSEXAGTJCILF-RXMQYKEDSA-N (R)-nipecotic acid zwitterion Chemical compound OC(=O)[C@@H]1CCCNC1 XJLSEXAGTJCILF-RXMQYKEDSA-N 0.000 description 1
- 125000004502 1,2,3-oxadiazolyl group Chemical group 0.000 description 1
- 125000004511 1,2,3-thiadiazolyl group Chemical group 0.000 description 1
- 125000004529 1,2,3-triazinyl group Chemical group N1=NN=C(C=C1)* 0.000 description 1
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000004530 1,2,4-triazinyl group Chemical group N1=NC(=NC=C1)* 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- 125000003363 1,3,5-triazinyl group Chemical group N1=C(N=CN=C1)* 0.000 description 1
- XHKRDGMEADPJIH-UHFFFAOYSA-N 1-(6-phenyl-7h-pyrrolo[2,3-d]pyrimidin-4-yl)-3,4,4a,5,6,7,8,8a-octahydro-2h-quinoline Chemical compound C1CCCC2C1CCCN2C(C=1C=2)=NC=NC=1NC=2C1=CC=CC=C1 XHKRDGMEADPJIH-UHFFFAOYSA-N 0.000 description 1
- UJTWZEIHVBHEQZ-UHFFFAOYSA-N 1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-ol Chemical compound C1C(O)CCCN1C1=NC=NC2=C1C=CN2 UJTWZEIHVBHEQZ-UHFFFAOYSA-N 0.000 description 1
- CASUPQYZIYNRRS-UHFFFAOYSA-N 1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-3-carboxamide Chemical compound C1C(C(=O)N)CCCN1C1=NC=NC2=C1C=CN2 CASUPQYZIYNRRS-UHFFFAOYSA-N 0.000 description 1
- WWNAKDRFVWZIMY-UHFFFAOYSA-N 1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-3-carboxylic acid Chemical compound C1C(C(=O)O)CCCN1C1=NC=NC2=C1C=CN2 WWNAKDRFVWZIMY-UHFFFAOYSA-N 0.000 description 1
- FLFIJNXDDGGTBM-UHFFFAOYSA-N 1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-3-ol Chemical compound C1C(O)CCN1C1=NC=NC2=C1C=CN2 FLFIJNXDDGGTBM-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- HTQWGIHCFPWKAS-UHFFFAOYSA-N 1-phenyl-1,3,8-triazaspiro[4.5]decan-4-one Chemical compound C1CNCCC11C(=O)NCN1C1=CC=CC=C1 HTQWGIHCFPWKAS-UHFFFAOYSA-N 0.000 description 1
- FXBLDFWUGCHSBP-UHFFFAOYSA-N 1-phenyl-8-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1,3,8-triazaspiro[4.5]decan-4-one Chemical compound C1CN(C=2C=3C=CNC=3N=CN=2)CCC11C(=O)NCN1C1=CC=CC=C1 FXBLDFWUGCHSBP-UHFFFAOYSA-N 0.000 description 1
- GSPAVZIEDMALNO-UHFFFAOYSA-N 1-piperidin-2-ylethanol Chemical compound CC(O)C1CCCCN1 GSPAVZIEDMALNO-UHFFFAOYSA-N 0.000 description 1
- LDSZALOVNNCTFJ-UHFFFAOYSA-N 1-piperidin-3-ylpropan-2-ol Chemical compound CC(O)CC1CCCNC1 LDSZALOVNNCTFJ-UHFFFAOYSA-N 0.000 description 1
- NDJKRLGXVKYIGQ-UHFFFAOYSA-N 1-piperidin-4-ylethanol Chemical compound CC(O)C1CCNCC1 NDJKRLGXVKYIGQ-UHFFFAOYSA-N 0.000 description 1
- GHQBJLLYEHMIQT-UHFFFAOYSA-N 1-piperidin-4-ylpropan-2-ol Chemical compound CC(O)CC1CCNCC1 GHQBJLLYEHMIQT-UHFFFAOYSA-N 0.000 description 1
- VTDIWMPYBAVEDY-UHFFFAOYSA-N 1-propylpiperidine Chemical compound CCCN1CCCCC1 VTDIWMPYBAVEDY-UHFFFAOYSA-N 0.000 description 1
- YNOPDCKXNICFQS-UHFFFAOYSA-N 1-pyrrolidin-2-ylpropan-2-ol Chemical compound CC(O)CC1CCCN1 YNOPDCKXNICFQS-UHFFFAOYSA-N 0.000 description 1
- PDELQDSYLBLPQO-UHFFFAOYSA-N 2,3,3a,4,5,6,7,7a-octahydro-1h-indole Chemical compound C1CCCC2NCCC21 PDELQDSYLBLPQO-UHFFFAOYSA-N 0.000 description 1
- ZEBFPAXSQXIPNF-UHFFFAOYSA-N 2,5-dimethylpyrrolidine Chemical compound CC1CCC(C)N1 ZEBFPAXSQXIPNF-UHFFFAOYSA-N 0.000 description 1
- LDFGITIRBMZIRF-UHFFFAOYSA-N 2,6-dimethyl-4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)morpholine Chemical compound C1C(C)OC(C)CN1C1=NC=NC2=C1C=CN2 LDFGITIRBMZIRF-UHFFFAOYSA-N 0.000 description 1
- SDGKUVSVPIIUCF-UHFFFAOYSA-N 2,6-dimethylpiperidine Chemical compound CC1CCCC(C)N1 SDGKUVSVPIIUCF-UHFFFAOYSA-N 0.000 description 1
- DKMYDCOSDPNPCZ-UHFFFAOYSA-N 2-[1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-2-yl]ethanol Chemical compound OCCC1CCCCN1C1=NC=NC2=C1C=CN2 DKMYDCOSDPNPCZ-UHFFFAOYSA-N 0.000 description 1
- RQMRQUPOSGXLGO-UHFFFAOYSA-N 2-[1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-yl]propan-2-ol Chemical compound C1C(C(C)(O)C)CCCN1C1=NC=NC2=C1C=CN2 RQMRQUPOSGXLGO-UHFFFAOYSA-N 0.000 description 1
- AIPOYWXBMBROCL-UHFFFAOYSA-N 2-[1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-yl]ethanol Chemical compound C1CC(CCO)CCN1C1=NC=NC2=C1C=CN2 AIPOYWXBMBROCL-UHFFFAOYSA-N 0.000 description 1
- AUVVHKWZFJUNCQ-UHFFFAOYSA-N 2-[1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-yl]propan-2-ol Chemical compound C1CC(C(C)(O)C)CCN1C1=NC=NC2=C1C=CN2 AUVVHKWZFJUNCQ-UHFFFAOYSA-N 0.000 description 1
- XCKSFBNJMCSRHO-UHFFFAOYSA-N 2-[1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-2-yl]propan-2-ol Chemical compound CC(C)(O)C1CCCN1C1=NC=NC2=C1C=CN2 XCKSFBNJMCSRHO-UHFFFAOYSA-N 0.000 description 1
- OAYWAWQHKXLYCZ-UHFFFAOYSA-N 2-[3-(4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl]propan-2-ol Chemical compound CC(C)(O)C1=CC=CC(C=2C3=C(N4CCCCC4)N=CN=C3NC=2)=C1 OAYWAWQHKXLYCZ-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- ADNQOPWHBFPHKI-UHFFFAOYSA-N 2-piperidin-3-ylpropanamide Chemical compound NC(=O)C(C)C1CCCNC1 ADNQOPWHBFPHKI-UHFFFAOYSA-N 0.000 description 1
- SYAXBEMNIXCDBQ-UHFFFAOYSA-N 2-piperidin-3-ylpropanoic acid Chemical compound OC(=O)C(C)C1CCCNC1 SYAXBEMNIXCDBQ-UHFFFAOYSA-N 0.000 description 1
- XEWJXCNCHWVTPP-UHFFFAOYSA-N 2-pyrrolidin-3-ylacetamide Chemical compound NC(=O)CC1CCNC1 XEWJXCNCHWVTPP-UHFFFAOYSA-N 0.000 description 1
- IDWRJRPUIXRFRX-UHFFFAOYSA-N 3,5-dimethylpiperidine Chemical compound CC1CNCC(C)C1 IDWRJRPUIXRFRX-UHFFFAOYSA-N 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-N 3-Hydroxy-2-naphthoate Chemical compound C1=CC=C2C=C(O)C(C(=O)O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-N 0.000 description 1
- ADFCTPSWZLLXQI-UHFFFAOYSA-N 3-[1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-yl]propanoic acid Chemical compound C1C(CCC(=O)O)CCCN1C1=NC=NC2=C1C=CN2 ADFCTPSWZLLXQI-UHFFFAOYSA-N 0.000 description 1
- ORIMVPIFXCBEQV-UHFFFAOYSA-N 3-[1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-yl]-1h-benzimidazol-2-one Chemical compound O=C1NC2=CC=CC=C2N1C(CC1)CCN1C1=NC=NC2=C1C=CN2 ORIMVPIFXCBEQV-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- AEERELMAGMCGLN-UHFFFAOYSA-N 3-methyl-1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-3-ol Chemical compound C1C(C)(O)CCN1C1=NC=NC2=C1C=CN2 AEERELMAGMCGLN-UHFFFAOYSA-N 0.000 description 1
- UFACSEIVZXTBMF-UHFFFAOYSA-N 3-methyl-8-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-8-azabicyclo[3.2.1]octan-3-ol Chemical compound C1C(C)(O)CC2CCC1N2C1=NC=NC2=C1C=CN2 UFACSEIVZXTBMF-UHFFFAOYSA-N 0.000 description 1
- AJPSDOLVFRJEID-UHFFFAOYSA-N 3-methyl-8-azabicyclo[3.2.1]octan-3-ol Chemical compound C1C(C)(O)CC2CCC1N2 AJPSDOLVFRJEID-UHFFFAOYSA-N 0.000 description 1
- JEGMWWXJUXDNJN-UHFFFAOYSA-N 3-methylpiperidine Chemical compound CC1CCCNC1 JEGMWWXJUXDNJN-UHFFFAOYSA-N 0.000 description 1
- ILBDVRCFTYQLOE-UHFFFAOYSA-N 3-methylpyrrolidin-3-ol Chemical compound CC1(O)CCNC1 ILBDVRCFTYQLOE-UHFFFAOYSA-N 0.000 description 1
- PGALEJNMXOZBGY-UHFFFAOYSA-N 3-piperidin-4-yl-1,2-dihydrobenzimidazole Chemical compound C1NC2=CC=CC=C2N1C1CCNCC1 PGALEJNMXOZBGY-UHFFFAOYSA-N 0.000 description 1
- VFTPONHUNNOSKG-UHFFFAOYSA-N 4,5-dichloro-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1=NC(Cl)=C2C(Cl)=CNC2=N1 VFTPONHUNNOSKG-UHFFFAOYSA-N 0.000 description 1
- SASXLRNIYRKHAE-UHFFFAOYSA-N 4-(2,3,3a,4,5,6,7,7a-octahydroindol-1-yl)-5-chloro-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CC2CCCCC2N1C1=C2C(Cl)=CNC2=NC=N1 SASXLRNIYRKHAE-UHFFFAOYSA-N 0.000 description 1
- OBTMEDIKWQTUGY-UHFFFAOYSA-N 4-(2,5-dimethylpyrrolidin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CC1CCC(C)N1C1=NC=NC2=C1C=CN2 OBTMEDIKWQTUGY-UHFFFAOYSA-N 0.000 description 1
- DKSODRCCGCVCKJ-UHFFFAOYSA-N 4-(2,6-dimethylpiperidin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CC1CCCC(C)N1C1=NC=NC2=C1C=CN2 DKSODRCCGCVCKJ-UHFFFAOYSA-N 0.000 description 1
- SNMPCOQLKHXPES-UHFFFAOYSA-N 4-(2-ethylpiperidin-1-yl)-6-phenyl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CCC1CCCCN1C1=NC=NC2=C1C=C(C=1C=CC=CC=1)N2 SNMPCOQLKHXPES-UHFFFAOYSA-N 0.000 description 1
- CNUYLOUPQNSQOW-UHFFFAOYSA-N 4-(2-ethylpiperidin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CCC1CCCCN1C1=NC=NC2=C1C=CN2 CNUYLOUPQNSQOW-UHFFFAOYSA-N 0.000 description 1
- TUDFWDRQFZNJSM-UHFFFAOYSA-N 4-(2-methylpiperidin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CC1CCCCN1C1=NC=NC2=C1C=CN2 TUDFWDRQFZNJSM-UHFFFAOYSA-N 0.000 description 1
- HYSBRCJBAUATFT-UHFFFAOYSA-N 4-(2-propylpiperidin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CCCC1CCCCN1C1=NC=NC2=C1C=CN2 HYSBRCJBAUATFT-UHFFFAOYSA-N 0.000 description 1
- JMYNKIANGIPQHN-UHFFFAOYSA-N 4-(3,5-dimethylpiperidin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1C(C)CC(C)CN1C1=NC=NC2=C1C=CN2 JMYNKIANGIPQHN-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- IJQQAHLJKAUYAK-UHFFFAOYSA-N 4-(3-methylpiperidin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1C(C)CCCN1C1=NC=NC2=C1C=CN2 IJQQAHLJKAUYAK-UHFFFAOYSA-N 0.000 description 1
- HASRFXGIJALRRB-UHFFFAOYSA-N 4-(3-phenylpropyl)piperidine Chemical compound C=1C=CC=CC=1CCCC1CCNCC1 HASRFXGIJALRRB-UHFFFAOYSA-N 0.000 description 1
- FWFRNYDQDLYPRQ-UHFFFAOYSA-N 4-(4-benzylpiperidin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CN(C=2C=3C=CNC=3N=CN=2)CCC1CC1=CC=CC=C1 FWFRNYDQDLYPRQ-UHFFFAOYSA-N 0.000 description 1
- XPRRJFARPURYNF-UHFFFAOYSA-N 4-(4-methylpiperazin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CN(C)CCN1C1=NC=NC2=C1C=CN2 XPRRJFARPURYNF-UHFFFAOYSA-N 0.000 description 1
- GMUPYKQZMBLHEB-UHFFFAOYSA-N 4-(4-phenylpiperidin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CN(C=2C=3C=CNC=3N=CN=2)CCC1C1=CC=CC=C1 GMUPYKQZMBLHEB-UHFFFAOYSA-N 0.000 description 1
- GIAZHSRKWYDANE-UHFFFAOYSA-N 4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)morpholine Chemical compound C1COCCN1C1=NC=NC2=C1C=CN2 GIAZHSRKWYDANE-UHFFFAOYSA-N 0.000 description 1
- TYFOAZRGZASNFR-UHFFFAOYSA-N 4-(azepan-1-yl)-5-(3-methylphenyl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CC1=CC=CC(C=2C3=C(N4CCCCCC4)N=CN=C3NC=2)=C1 TYFOAZRGZASNFR-UHFFFAOYSA-N 0.000 description 1
- SVUIUARUAACCHA-UHFFFAOYSA-N 4-(azepan-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CCCCCN1C1=NC=NC2=C1C=CN2 SVUIUARUAACCHA-UHFFFAOYSA-N 0.000 description 1
- UBVHGEPXSKEAII-UHFFFAOYSA-N 4-(azocan-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CCCCCCN1C1=NC=NC2=C1C=CN2 UBVHGEPXSKEAII-UHFFFAOYSA-N 0.000 description 1
- RDRQUUWCJTYHCT-UHFFFAOYSA-N 4-(trifluoromethyl)piperidine Chemical compound FC(F)(F)C1CCNCC1 RDRQUUWCJTYHCT-UHFFFAOYSA-N 0.000 description 1
- JPPGNVXNDFPPGJ-UHFFFAOYSA-N 4-[2-(methoxymethyl)pyrrolidin-1-yl]-7h-pyrrolo[2,3-d]pyrimidine Chemical compound COCC1CCCN1C1=NC=NC2=C1C=CN2 JPPGNVXNDFPPGJ-UHFFFAOYSA-N 0.000 description 1
- JZDDREBDUSIFQF-UHFFFAOYSA-N 4-[4-(3-phenylpropyl)piperidin-1-yl]-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CN(C=2C=3C=CNC=3N=CN=2)CCC1CCCC1=CC=CC=C1 JZDDREBDUSIFQF-UHFFFAOYSA-N 0.000 description 1
- AKUHOBWUZYSCMB-UHFFFAOYSA-N 4-[4-(trifluoromethyl)piperidin-1-yl]-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CC(C(F)(F)F)CCN1C1=NC=NC2=C1C=CN2 AKUHOBWUZYSCMB-UHFFFAOYSA-N 0.000 description 1
- ABGXADJDTPFFSZ-UHFFFAOYSA-N 4-benzylpiperidine Chemical compound C=1C=CC=CC=1CC1CCNCC1 ABGXADJDTPFFSZ-UHFFFAOYSA-N 0.000 description 1
- CBWBJFJMNBPWAL-UHFFFAOYSA-N 4-chloro-5-iodo-7h-pyrrolo[2,3-d]pyrimidine Chemical compound ClC1=NC=NC2=C1C(I)=CN2 CBWBJFJMNBPWAL-UHFFFAOYSA-N 0.000 description 1
- FTYXRILYMGQZIW-UHFFFAOYSA-N 4-chloro-5-nitro-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1=NC(Cl)=C2C([N+](=O)[O-])=CNC2=N1 FTYXRILYMGQZIW-UHFFFAOYSA-N 0.000 description 1
- PGQJBJDESGAJSO-UHFFFAOYSA-N 4-chloro-6-methyl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound N1=CN=C2NC(C)=CC2=C1Cl PGQJBJDESGAJSO-UHFFFAOYSA-N 0.000 description 1
- AGLAPJLLGQYKAH-UHFFFAOYSA-N 4-chloro-6-phenyl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C=1C=2C(Cl)=NC=NC=2NC=1C1=CC=CC=C1 AGLAPJLLGQYKAH-UHFFFAOYSA-N 0.000 description 1
- KAUCHHKKEDFVMC-UHFFFAOYSA-N 4-chloro-7h-pyrrolo[2,3-d]pyrimidin-5-amine Chemical compound N1C=NC(Cl)=C2C(N)=CN=C21 KAUCHHKKEDFVMC-UHFFFAOYSA-N 0.000 description 1
- NEWCOPCSNFUQQY-UHFFFAOYSA-N 4-chloro-7h-pyrrolo[2,3-d]pyrimidine-5-carbonitrile Chemical compound ClC1=NC=NC2=C1C(C#N)=CN2 NEWCOPCSNFUQQY-UHFFFAOYSA-N 0.000 description 1
- ZSLNHSFKLKLHDR-UHFFFAOYSA-N 4-methyl-1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-ol Chemical compound C1CC(C)(O)CCN1C1=NC=NC2=C1C=CN2 ZSLNHSFKLKLHDR-UHFFFAOYSA-N 0.000 description 1
- CXBLQEIODBBSQD-UHFFFAOYSA-N 4-methylpiperidin-4-ol Chemical compound CC1(O)CCNCC1 CXBLQEIODBBSQD-UHFFFAOYSA-N 0.000 description 1
- XDQJMAHHTKXBHX-UHFFFAOYSA-N 4-phenyl-1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-ol Chemical compound C1CN(C=2C=3C=CNC=3N=CN=2)CCC1(O)C1=CC=CC=C1 XDQJMAHHTKXBHX-UHFFFAOYSA-N 0.000 description 1
- KQKFQBTWXOGINC-UHFFFAOYSA-N 4-phenylpiperidin-4-ol Chemical compound C=1C=CC=CC=1C1(O)CCNCC1 KQKFQBTWXOGINC-UHFFFAOYSA-N 0.000 description 1
- UTBULQCHEUWJNV-UHFFFAOYSA-N 4-phenylpiperidine Chemical compound C1CNCCC1C1=CC=CC=C1 UTBULQCHEUWJNV-UHFFFAOYSA-N 0.000 description 1
- LZSXQBNNRGZPES-UHFFFAOYSA-N 4-piperazin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CNCCN1C1=NC=NC2=C1C=CN2 LZSXQBNNRGZPES-UHFFFAOYSA-N 0.000 description 1
- AJJYZGJMURHTAU-UHFFFAOYSA-N 4-piperidin-1-yl-5-[3-(trifluoromethyl)phenyl]-7h-pyrrolo[2,3-d]pyrimidine Chemical compound FC(F)(F)C1=CC=CC(C=2C3=C(N4CCCCC4)N=CN=C3NC=2)=C1 AJJYZGJMURHTAU-UHFFFAOYSA-N 0.000 description 1
- ZYZDSWNXHGSTEX-UHFFFAOYSA-N 4-piperidin-1-yl-6-thiophen-2-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CCCCN1C1=NC=NC2=C1C=C(C=1SC=CC=1)N2 ZYZDSWNXHGSTEX-UHFFFAOYSA-N 0.000 description 1
- PSOQOVKARRFKTL-UHFFFAOYSA-N 4-piperidin-1-yl-6-thiophen-3-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CCCCN1C1=NC=NC2=C1C=C(C1=CSC=C1)N2 PSOQOVKARRFKTL-UHFFFAOYSA-N 0.000 description 1
- VXAXIOIDQGMFIP-UHFFFAOYSA-N 4-pyrazol-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1=CC=NN1C1=NC=NC2=C1C=CN2 VXAXIOIDQGMFIP-UHFFFAOYSA-N 0.000 description 1
- XSBMIQAAQVEHAL-UHFFFAOYSA-N 5-(2-methylphenyl)-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CC1=CC=CC=C1C1=CNC2=NC=NC(N3CCCCC3)=C12 XSBMIQAAQVEHAL-UHFFFAOYSA-N 0.000 description 1
- DMFICJGMHXJAJG-UHFFFAOYSA-N 5-(3-chloro-4-fluorophenyl)-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1=C(Cl)C(F)=CC=C1C1=CNC2=NC=NC(N3CCCCC3)=C12 DMFICJGMHXJAJG-UHFFFAOYSA-N 0.000 description 1
- QSJSVHCOJILSCM-UHFFFAOYSA-N 5-(3-chlorophenyl)-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound ClC1=CC=CC(C=2C3=C(N4CCCCC4)N=CN=C3NC=2)=C1 QSJSVHCOJILSCM-UHFFFAOYSA-N 0.000 description 1
- FMRFFGIVGWQSGX-UHFFFAOYSA-N 5-(3-methylphenyl)-4-(2-methylpiperidin-1-yl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound CC1CCCCN1C1=NC=NC2=C1C(C=1C=C(C)C=CC=1)=CN2 FMRFFGIVGWQSGX-UHFFFAOYSA-N 0.000 description 1
- SJNRYKGHYLIZBF-UHFFFAOYSA-N 5-(4-chlorophenyl)-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1=CC(Cl)=CC=C1C1=CNC2=NC=NC(N3CCCCC3)=C12 SJNRYKGHYLIZBF-UHFFFAOYSA-N 0.000 description 1
- ZFWLSMFAONKYKE-UHFFFAOYSA-N 5-(4-fluorophenyl)-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1=CC(F)=CC=C1C1=CNC2=NC=NC(N3CCCCC3)=C12 ZFWLSMFAONKYKE-UHFFFAOYSA-N 0.000 description 1
- ILMKPRHMPOWHTR-UHFFFAOYSA-N 5-(4-methoxyphenyl)-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1=CC(OC)=CC=C1C1=CNC2=NC=NC(N3CCCCC3)=C12 ILMKPRHMPOWHTR-UHFFFAOYSA-N 0.000 description 1
- QMSQNQGNQFIGNJ-UHFFFAOYSA-N 5-(4-methylphenyl)-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1=CC(C)=CC=C1C1=CNC2=NC=NC(N3CCCCC3)=C12 QMSQNQGNQFIGNJ-UHFFFAOYSA-N 0.000 description 1
- SPJFFPVWQITRTN-UHFFFAOYSA-N 5-[3,5-bis(trifluoromethyl)phenyl]-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C=2C3=C(N4CCCCC4)N=CN=C3NC=2)=C1 SPJFFPVWQITRTN-UHFFFAOYSA-N 0.000 description 1
- OXLMTRZWMHIZBY-UHFFFAOYSA-N 5-bromo-4-chloro-7h-pyrrolo[2,3-d]pyrimidine Chemical compound ClC1=NC=NC2=C1C(Br)=CN2 OXLMTRZWMHIZBY-UHFFFAOYSA-N 0.000 description 1
- MVOUQQZCPLFWBE-UHFFFAOYSA-N 6-methyl-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound N1=CN=C2NC(C)=CC2=C1N1CCCCC1 MVOUQQZCPLFWBE-UHFFFAOYSA-N 0.000 description 1
- YVBAVSHYNWGPEU-UHFFFAOYSA-N 6-phenyl-4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1CCCCN1C1=NC=NC2=C1C=C(C=1C=CC=CC=1)N2 YVBAVSHYNWGPEU-UHFFFAOYSA-N 0.000 description 1
- CDJBDMNUBKPNQF-UHFFFAOYSA-N 7-(benzenesulfonyl)-4-chloro-5-iodopyrrolo[2,3-d]pyrimidine Chemical compound C1=C(I)C=2C(Cl)=NC=NC=2N1S(=O)(=O)C1=CC=CC=C1 CDJBDMNUBKPNQF-UHFFFAOYSA-N 0.000 description 1
- QSIDTDGOKALJNU-UHFFFAOYSA-N 7-(benzenesulfonyl)-4-chloro-6-methylpyrrolo[2,3-d]pyrimidine Chemical compound CC1=CC2=C(Cl)N=CN=C2N1S(=O)(=O)C1=CC=CC=C1 QSIDTDGOKALJNU-UHFFFAOYSA-N 0.000 description 1
- RBTRNZJHABCLOO-UHFFFAOYSA-N 7-(benzenesulfonyl)-4-chloro-6-phenylpyrrolo[2,3-d]pyrimidine Chemical compound C=1C=2C(Cl)=NC=NC=2N(S(=O)(=O)C=2C=CC=CC=2)C=1C1=CC=CC=C1 RBTRNZJHABCLOO-UHFFFAOYSA-N 0.000 description 1
- HPWMGRLPNPQCQX-UHFFFAOYSA-N 7-(benzenesulfonyl)-4-chloropyrrolo[2,3-d]pyrimidine Chemical compound C1=CC=2C(Cl)=NC=NC=2N1S(=O)(=O)C1=CC=CC=C1 HPWMGRLPNPQCQX-UHFFFAOYSA-N 0.000 description 1
- IXXOMMSCNWTOPX-UHFFFAOYSA-N 7-(benzenesulfonyl)-5-bromo-4-chloropyrrolo[2,3-d]pyrimidine Chemical compound C1=C(Br)C=2C(Cl)=NC=NC=2N1S(=O)(=O)C1=CC=CC=C1 IXXOMMSCNWTOPX-UHFFFAOYSA-N 0.000 description 1
- ODSVXRSHXWHNNL-UHFFFAOYSA-N 7-(benzenesulfonyl)-5-bromo-4-piperidin-1-ylpyrrolo[2,3-d]pyrimidine Chemical compound C=12C(Br)=CN(S(=O)(=O)C=3C=CC=CC=3)C2=NC=NC=1N1CCCCC1 ODSVXRSHXWHNNL-UHFFFAOYSA-N 0.000 description 1
- DTTNUXRTLDFOQH-UHFFFAOYSA-N 7-(benzenesulfonyl)-5-iodo-4-piperidin-1-ylpyrrolo[2,3-d]pyrimidine Chemical compound C=12C(I)=CN(S(=O)(=O)C=3C=CC=CC=3)C2=NC=NC=1N1CCCCC1 DTTNUXRTLDFOQH-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- QKYQEWXQVQGDJR-UHFFFAOYSA-N 9-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1,2,3,4,4a,9a-hexahydrocarbazole Chemical compound C1CCCC(C2=CC=CC=C22)C1N2C1=NC=NC2=C1C=CN2 QKYQEWXQVQGDJR-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 208000002874 Acne Vulgaris Diseases 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- DSLZVSRJTYRBFB-LLEIAEIESA-N D-glucaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O DSLZVSRJTYRBFB-LLEIAEIESA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 101000934996 Homo sapiens Tyrosine-protein kinase JAK3 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 108090000172 Interleukin-15 Proteins 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 102000010787 Interleukin-4 Receptors Human genes 0.000 description 1
- 108010038486 Interleukin-4 Receptors Proteins 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 108010002335 Interleukin-9 Proteins 0.000 description 1
- 102000008986 Janus Human genes 0.000 description 1
- 108050000950 Janus Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241001024304 Mino Species 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 108010020346 Polyglutamic Acid Proteins 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 208000012827 T-B+ severe combined immunodeficiency due to gamma chain deficiency Diseases 0.000 description 1
- 208000037913 T-cell disorder Diseases 0.000 description 1
- 229910021627 Tin(IV) chloride Inorganic materials 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 208000023940 X-Linked Combined Immunodeficiency disease Diseases 0.000 description 1
- 201000007146 X-linked severe combined immunodeficiency Diseases 0.000 description 1
- FXSPEZBKQFGEDH-UHFFFAOYSA-N [1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-2-yl]methanol Chemical compound OCC1CCCCN1C1=NC=NC2=C1C=CN2 FXSPEZBKQFGEDH-UHFFFAOYSA-N 0.000 description 1
- JKLUKVHATSDXCO-UHFFFAOYSA-N [1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-yl]methanol Chemical compound C1C(CO)CCCN1C1=NC=NC2=C1C=CN2 JKLUKVHATSDXCO-UHFFFAOYSA-N 0.000 description 1
- VOENIPBLZDNSJO-UHFFFAOYSA-N [1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-2-yl]methanol Chemical compound OCC1CCCN1C1=NC=NC2=C1C=CN2 VOENIPBLZDNSJO-UHFFFAOYSA-N 0.000 description 1
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 1
- 206010000496 acne Diseases 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 150000001543 aryl boronic acids Chemical class 0.000 description 1
- YCOXTKKNXUZSKD-UHFFFAOYSA-N as-o-xylenol Natural products CC1=CC=C(O)C=C1C YCOXTKKNXUZSKD-UHFFFAOYSA-N 0.000 description 1
- ZSIQJIWKELUFRJ-UHFFFAOYSA-N azepane Chemical compound C1CCCNCC1 ZSIQJIWKELUFRJ-UHFFFAOYSA-N 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- LPCWKMYWISGVSK-UHFFFAOYSA-N bicyclo[3.2.1]octane Chemical compound C1C2CCC1CCC2 LPCWKMYWISGVSK-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M bisulphate group Chemical group S([O-])(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 229960004424 carbon dioxide Drugs 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 238000000451 chemical ionisation Methods 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cis-cyclohexene Natural products C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229920001577 copolymer Chemical group 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 230000000994 depressogenic effect Effects 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000012738 dissolution medium Substances 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000007824 enzymatic assay Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- JFUUMQWWSYKTEG-UHFFFAOYSA-N ethyl 1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxylate Chemical compound C1CC(C(=O)OCC)CCN1C1=NC=NC2=C1C=CN2 JFUUMQWWSYKTEG-UHFFFAOYSA-N 0.000 description 1
- KYHCFMIDWXXQSQ-UHFFFAOYSA-N ethyl 3-[1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-yl]propanoate Chemical compound C1C(CCC(=O)OCC)CCCN1C1=NC=NC2=C1C=CN2 KYHCFMIDWXXQSQ-UHFFFAOYSA-N 0.000 description 1
- 239000002024 ethyl acetate extract Substances 0.000 description 1
- RUJPPJYDHHAEEK-UHFFFAOYSA-N ethyl piperidine-4-carboxylate Chemical compound CCOC(=O)C1CCNCC1 RUJPPJYDHHAEEK-UHFFFAOYSA-N 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 238000000249 far-infrared magnetic resonance spectroscopy Methods 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical compound [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 230000009395 genetic defect Effects 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 102000049912 human JAK3 Human genes 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000007327 hydrogenolysis reaction Methods 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 230000003463 hyperproliferative effect Effects 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000005965 immune activity Effects 0.000 description 1
- 229940125721 immunosuppressive agent Drugs 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- SNHMUERNLJLMHN-UHFFFAOYSA-N iodobenzene Chemical compound IC1=CC=CC=C1 SNHMUERNLJLMHN-UHFFFAOYSA-N 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 238000000021 kinase assay Methods 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- OVEHNNQXLPJPPL-UHFFFAOYSA-N lithium;n-propan-2-ylpropan-2-amine Chemical compound [Li].CC(C)NC(C)C OVEHNNQXLPJPPL-UHFFFAOYSA-N 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 238000003670 luciferase enzyme activity assay Methods 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- HHQJWDKIRXRTLS-UHFFFAOYSA-N n'-bromobutanediamide Chemical compound NC(=O)CCC(=O)NBr HHQJWDKIRXRTLS-UHFFFAOYSA-N 0.000 description 1
- UONJJEHOXJJRJO-UHFFFAOYSA-N n,n-diethyl-1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-3-carboxamide Chemical compound C1C(C(=O)N(CC)CC)CCCN1C1=NC=NC2=C1C=CN2 UONJJEHOXJJRJO-UHFFFAOYSA-N 0.000 description 1
- ZXQKYQVJDRTTLZ-UHFFFAOYSA-N n,n-diethylpiperidine-3-carboxamide Chemical compound CCN(CC)C(=O)C1CCCNC1 ZXQKYQVJDRTTLZ-UHFFFAOYSA-N 0.000 description 1
- AVAWMINJNRAQFS-UHFFFAOYSA-N n,n-dimethylpyrrolidin-3-amine Chemical compound CN(C)C1CCNC1 AVAWMINJNRAQFS-UHFFFAOYSA-N 0.000 description 1
- QEDHDCLYRONBGE-UHFFFAOYSA-N n-[1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-3-yl]acetamide Chemical compound C1C(NC(=O)C)CCN1C1=NC=NC2=C1C=CN2 QEDHDCLYRONBGE-UHFFFAOYSA-N 0.000 description 1
- FVSXUDJYTCKUNR-UHFFFAOYSA-N n-ethyl-2-pyrrolidin-3-ylacetamide Chemical compound CCNC(=O)CC1CCNC1 FVSXUDJYTCKUNR-UHFFFAOYSA-N 0.000 description 1
- FRIKTXJBUKTPOB-UHFFFAOYSA-N n-ethyl-n-[1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-3-yl]acetamide Chemical compound C1C(N(CC)C(C)=O)CCN1C1=NC=NC2=C1C=CN2 FRIKTXJBUKTPOB-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- BVOCPVIXARZNQN-UHFFFAOYSA-N nipecotamide Chemical compound NC(=O)C1CCCNC1 BVOCPVIXARZNQN-UHFFFAOYSA-N 0.000 description 1
- XJLSEXAGTJCILF-UHFFFAOYSA-N nipecotic acid Chemical compound OC(=O)C1CCCNC1 XJLSEXAGTJCILF-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 125000005593 norbornanyl group Chemical group 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 229940100688 oral solution Drugs 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 235000010603 pastilles Nutrition 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- DCWXELXMIBXGTH-UHFFFAOYSA-N phosphotyrosine Chemical compound OC(=O)C(N)CC1=CC=C(OP(O)(O)=O)C=C1 DCWXELXMIBXGTH-UHFFFAOYSA-N 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- PRAYXGYYVXRDDW-UHFFFAOYSA-N piperidin-2-ylmethanol Chemical compound OCC1CCCCN1 PRAYXGYYVXRDDW-UHFFFAOYSA-N 0.000 description 1
- BIWOSRSKDCZIFM-UHFFFAOYSA-N piperidin-3-ol Chemical compound OC1CCCNC1 BIWOSRSKDCZIFM-UHFFFAOYSA-N 0.000 description 1
- VUNPWIPIOOMCPT-UHFFFAOYSA-N piperidin-3-ylmethanol Chemical compound OCC1CCCNC1 VUNPWIPIOOMCPT-UHFFFAOYSA-N 0.000 description 1
- 239000003880 polar aprotic solvent Substances 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920002643 polyglutamic acid Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- HVVNJUAVDAZWCB-UHFFFAOYSA-N prolinol Chemical compound OCC1CCCN1 HVVNJUAVDAZWCB-UHFFFAOYSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical class CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 229940083082 pyrimidine derivative acting on arteriolar smooth muscle Drugs 0.000 description 1
- XVIAPHVAGFEFFN-UHFFFAOYSA-N pyrimidine-5-carbonitrile Chemical compound N#CC1=CN=CN=C1 XVIAPHVAGFEFFN-UHFFFAOYSA-N 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- JHHZLHWJQPUNKB-UHFFFAOYSA-N pyrrolidin-3-ol Chemical compound OC1CCNC1 JHHZLHWJQPUNKB-UHFFFAOYSA-N 0.000 description 1
- 150000004944 pyrrolopyrimidines Chemical class 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000008279 sol Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 125000003011 styrenyl group Chemical class [H]\C(*)=C(/[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000005504 styryl group Chemical group 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229960001967 tacrolimus Drugs 0.000 description 1
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000005306 thianaphthenyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 229940062627 tribasic potassium phosphate Drugs 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- FWSPXZXVNVQHIF-UHFFFAOYSA-N triethyl(ethynyl)silane Chemical group CC[Si](CC)(CC)C#C FWSPXZXVNVQHIF-UHFFFAOYSA-N 0.000 description 1
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- LSGOVYNHVSXFFJ-UHFFFAOYSA-N vanadate(3-) Chemical compound [O-][V]([O-])([O-])=O LSGOVYNHVSXFFJ-UHFFFAOYSA-N 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
- A61P5/16—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4 for decreasing, blocking or antagonising the activity of the thyroid hormones
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Immunology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Psychiatry (AREA)
- Pulmonology (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Emergency Medicine (AREA)
- Obesity (AREA)
- Transplantation (AREA)
- Hospice & Palliative Care (AREA)
- Oncology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Description
- Hintergrund der Erfindung
- Die vorliegende Erfindung betrifft Pyrrolo[2,3-d]pyrimidin-Verbindungen, die Inhibitoren von Proteintyrosinkinasen wie dem Enzym Janus-Kinase 3 (im weiteren auch als JAK3 bezeichnet) sind und die als solche therapeutische Nützlichkeit bzw. Anwendbarkeit als Immunsupressiva für Organtransplantate, Lupus, multiple Sklerose, rheumatoide Arthritis, Psoriasis, Typ I-Diabetes und Komplikationen von Diabetes, Krebs, Asthma, atopische Dermatitis, Autoimmun-Thyroid-Störungen, Colitis ulcerosa, Crohn'sche Krankheit, Alzheimer-Krankheit, Leukämie und andere Indikationen sind, bei denen eine Immunsuppression wünschenswert wäre.
- Diese Erfindung betrifft auch die Verwendung solcher Verbindungen zur Herstellung eines Medikaments für die Behandlung der vorgenannten Indikationen bei Säugetieren, insbesondere Menschen, und dafür nützliche pharmazeutische Zusammensetzungen.
- JAK3 ist ein Mitglied der Janus-Familie von Proteinthyrosinkinasen. Obwohl die anderen Mitglieder dieser Familie im Wesentlichen durch sämtliche Gewebe exprimiert werden, ist die JAK3-Expression auf hämatopoetische Zellen beschränkt. Dies ist in Übereinstimmung mit seiner wesentlichen Funktion in der Signalübertragung durch die Rezeptoren für IL-2, IL-4, IL-7, IL-9 und IL-15 durch nicht kovalente Verbindung von JAK3 mit der Gammakette, die diesen vielkettigen Rezeptoren gemein ist. XSCID-Patientenpopulationen sind mit stark reduzierten JAK3-Protein-Spiegeln identifiziert worden oder mit genetischen Defekten der gemeinsamen Gammakette, was nahe legt, dass die Immunsuppression von der Blockierung der Signalübertragung durch den JAK3-Pfad herrührt. Tierstudien legen nahe, dass JAK3 nicht nur eine kritische Rolle bei der B- und T-Lymphozytenreifung spielt, sondern dass JAK3 in konstitutiver Form erforderlich ist, um die T-Zellenfunktion aufrecht zu erhalten. Die Modulation der Immunaktivität durch diesen neuen Mechanismus kann sich in der Behandlung von proliferativen T-Zellen-Störungen, wie Transplantatabstoßung und Autoimmunerkrankungen, nützlich erweisen.
- Die WO-A-97/27199 beschreibt Pyrrolopyrimidinverbindungen, die Inhibitoren von Proteinkinasen sind und zum Beispiel Antitumoraktivität aufweisen. Die WO-A-96/40142 und WO-A-98/23613 beschreiben heterozyklische ringverknüpfte Pyrimidinderivate, die nützlich in der Behandlung von hyperproliferativen Krankheiten, wie zum Beispiel Krebs und Akne, bei Säugetieren sind.
- Zusammenfassung der Erfindung
- Die vorliegende Erfindung betrifft eine Verbindung der Formeloder das pharmazeutisch annehmbare Salz davon, wobei
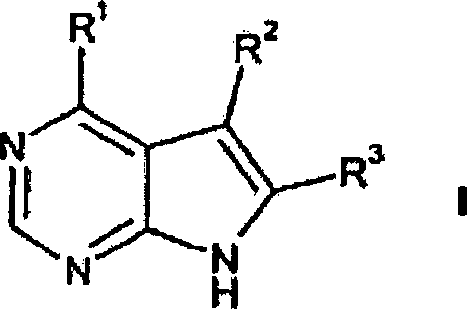
R1 eine Gruppe der Formelist, worin die gestrichelte Linie fakultative Doppelbindungen darstellt;
m 0, 1, 2 oder 3 ist;
n 0, 1, 2 oder 3 ist;
X, B und D jeweils unabhängig voneinander Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8 sind;
A und E jeweils CR7R8 sind; und
R6 ausgewählt ist aus der Gruppe, die besteht aus Wasserstoff, (C1-C6)Alkyl, Trifluormethyl, Trifluormethyl(C1-C6) alkyl, (C1-C6)Alkyl(difluormethylen), (C1-C3)Alkyl(difluormethylen)(C1-C3)alkyl, (C1-C6)Alkoxy(C1-C6)acyl, (C1-C6)Alkylamino(C1-C6)acyl, ((C1-C6)Alkyl)2amino(C1-C6)acyl, (C6-C10)Aryl, (C5-C9)Heteroaryl, (C6-C10)Aryl(C1-C6)alkyl, (C5-C9)Heteroaryl(C1-C6)alkyl, (C6-C10)Aryl(C6-C10)aryl, (C6-C10)Aryl(C6-C10)aryl(C1-C6)alkyl, (C3-C6)Cycloalkyl, (C3-C6)Cycloalkyl (C1-C6)alkyl, Hydroxy(C2-C6)alkyl, (C1-C6)Acyloxy(C2-C6)alkyl, (C1-C6)Alkoxy(C2-C6)alkyl, Piperazinyl(C1-C6)alkyl, (C1-C6)Acylamino(C1-C6)alkyl, (C6-C10)Aryl(C1-C6)alkoxy(C1-C6)alkyl, (C5-C9)Heteroaryl(C1-C6)alkoxy(C1-C6)alkyl, (C1-C6)Alkylthio(C1-C6)alkyl, (C6-C10)Arylthio(C1-C6)alkyl, (C1-C6)Alkylsulfinyl(C1-C6)alkyl, (C6-C10)Arylsulfinyl(C1-C6)alkyl, (C1-C6)Alkylsulfonyl(C1-C6)alkyl, (C6-C10)Arylsulfonyl(C1-C6)alkyl, Amino(C1-C6)alkyl, (C1-C6)Alkylamino(C1-C6)alkyl, ((C1-C6)Alkyl)2amino(C1-C6)alkyl, R13CO(C1-C6)Alkyl, worin R13 für R20O oder R20R21N steht, worin R20 und R21 jeweils unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, (C1-C6)Alkyl, (C6-C10)Aryl(C1-C6)alkyl oder (C5-C9)Heteroaryl(C1-C6)alkyl; und R19 (C2-C6)Alkyl, worin R19 für (C1-C6)Acylpiperazino, (C6-C10)-Arylpiperazino, (C5-C9)Heteroarylpiperazino, (C1-C6)Alkylipiperazino, (C6-C10)Aryl(C1-C6)alkylpiperazino, (C5-C9)Heteroaryl(C1-C6)alkylpiperazino, Morpholino, Thiomorpholino, Piperidino, Pyrrolidino, Piperidyl, (C1-C6)Alkylpiperidyl, (C6-C10)Arylpiperidyl, (C5-C9)Heteroarylpiperidyl, (C6-C10)Aryl(C1-C6)alkylpiperidyl, (C5-C9)Heteroaryl(C1-C6)alkylpiperidyl, (C1- C6)Alkoxyacyl, (C1-C6)Alkylaminoaryl, ((C1-C6)Alkyl)2aminoacyl oder (C1-C6)Acylpiperidyl steht;
R7 und R8 jeweils unabhängig ausgewählt sind aus der Gruppe, bestehend aus Wasserstoff, Deuterium, (C1-C6)Alkyl, Amino, Hydroxy, (C1-C6)Alkoxy, (C1-C6)Alkylamino, ((C1-C6)Alkyl)amino, (C1-C6)Acylamino, (C1-C6)Acyl(C1-C6)alkylamino, Carboxy, (C1-C6)Alkoxyacyl, (C1-C6)Alkylaminoacyl, ((C1-C6)Alkyl)2aminoacyl, Aminoacyl, Trifluormethyl, Trifluormethyl(C1-C6)alkyl, (C1-C6)Alkyl(difluormethylen), (C1-C3)Alkyl(difluormethylen)(C1-C3)alkyl, (C6-C10)Aryl, (C5-C9)Heteroaryl, (C6-C10)Aryl(C1-C6)alkyl, (C5-C9)Heteroaryl(C1-C6)alkyl, (C6-C10)Aryl(C6-C10)aryl, (C6-C10)Aryl(C6-C10)aryl(C1-C6)alkyl, (C3-C6)Cycloalkyl, (C3-C6)Cycloalkyl(C1-C6)alkyl, Hydroxy(C1-C6)alkyl, (C1-C6)Acyloxy(C1-C6)alkyl, (C1-C6)Alkoxy(C1-C6)alkyl, Piperazinyl(C1-C6)alkyl, (C1-C6)Acylamino(C1-C6)alkyl, Piperidyl, (C1-C6)Alkylpiperidyl, (C6-C10)Aryl(C1-C6)alkoxy(C1-C6)alkyl, (C5-C9)Heteroaryl(C1-C6)alkoxy(C1-C6)alkyl, (C1-C6)Alkylthio(C1-C6)alkyl, (C6-C10)Arylthio(C1-C6)alkyl, (C1-C6)Alkylsulfinyl (C1-C6)alkyl, (C6-C10)Arylsulfinyl(C1-C6)alkyl, (C1-C6)Alkylsulfonyl(C1-C6)alkyl, (C6-C10)Arylsulfonyl(C1-C6)alkyl, Amino (C1-C6)alkyl, (C1-C6)Alkylamino(C1-C6)alkyl, ((C1-C6)Alkyl)2amino(C1-C6)alkyl, R13CO(C1-C6)Alkyl oder R13CO(C3-C10)Cyloalkyl, worin R13 für R20O oder R20R21N steht, worin R20 und R21 jeweils unabhängig voneinander aus der Gruppe, bestehend aus Wasserstoff, (C1-C6)Alkyl, (C6-C10)Aryl(C1-C6)alkyl oder (C5-C9)Heteroaryl(C1-C6)alkyl, ausgewählt sind; R14, R14(C1-C6)Alkyl oder R14(C3-C10)Cycloalkyl, worin R14 (C1-C6)Acylpiperazino, (C6-C10)Arylpiperazino, (C5-C9)Heteroarylpiperazino, (C1-C6)Alkylpiperazino, (C6-C10)Aryl(C1-C6)alkylpiperazino, (C5-C9)Heteroaryl(C1-C6)alkylpiperazino, Morpholino, Thiomorpholino, Piperidino, Pyrrolidino, Piperidyl, (C1-C6)Alkylpiperidyl, (C6-C10)Arylpiperidyl, (C5-C9)Heteroarylpiperidyl, (C6-C10)Aryl (C1-C6)alkylpiperidyl, (C5-C9)Heteroaryl(C1-C6)alkylpiperidyl oder (C1-C6)Acylpiperidyl ist; oder einer Gruppe der Formelworin p 0, 1, 2 oder 3 ist; und
Z Hydroxy, (C1-C6)Alkoxy oder NR1R2 ist, worin R1 und R2 jeweils unabhängig voneinander ausgewählt sind aus der Gruppe, bestehend aus Wasserstoff, (C1-C6)Alkyl, Piperidyl, (C1-C6)Alkylpiperidyl, (C6-C10)Arylpiperidyl, (C5-C9)Heteroarylpiperidyl, (C6-C10)Aryl(C1-C6)alkylpiperidyl, (C5-C9)Heteroaryl(C1-C6)alkylpiperidyl, (C1-C6)Acylpiperidyl, (C6-C10)Aryl, (C5-C9)Heteroaryl, (C6-C10)Aryl(C1-C6)alkyl, (C5-C9)Heteroaryl(C1-C6)alkyl, (C6-C10)Aryl(C6-C10)aryl, (C6-C10)Aryl(C6-C10)aryl(C1-C6)alkyl, (C3-C6)Cycloalkyl, (C3-C6)Cycloalkyl(C1-C6)alkyl, R5(C1-C6)Alkyl, (C1-C6)Alkyl(CHR5)(C1-C6)alkyl, worin R5 Hydroxy, (C1-C6)Acyloxy, (C1-C6)Alkoxy, Piperazino, (C1-C6)Acylamino, (C1-C6)Alkylthio, (C6-C10)Arylthio, (C1-C6)Alkylsulfinyl, (C6-C10)Arylsulfinyl, (C1-C6)Alkylsulfoxyl, (C6-C10)Arylsulfoxyl, Amino, (C1-C6)Alkylamino, ((C1-C6)Alkyl)2amino, (C1-C6)Acylpiperazino, (C1-C6)Alkylpiperazino, (C6-C10)Aryl(C1-C6)alkylpiperazino, (C5-C9)Heteroaryl(C1-C6)alkylpiperazino, Morpholino, Thiomorpholino, Piperidino oder Pyrrolidino ist; R6 (C1-C6)Alkyl, (C1-C6)Alkyl-(CHR6)(C1-C6)alkyl, worin R6 Piperidyl, (C1-C6)Alkylpiperidyl, (C6-C10)Arylpiperidyl, (C6-C10)Aryl(C1-C6)alkylpiperidyl, (C5-C9)Heteroarylpiperidyl oder (C5-C9)Heteroaryl(C1-C6)alkylpiperidyl ist;
oder wenn n mindestens 1 ist, D und E oder D und X jeweils CR7R8 sind, die benachbarten R7-Gruppen zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, Gruppen der Formelnbilden, wobei die gestrichelten Linien fakultative Doppelbindungen darstellen;
a 0, 1 oder 2 ist;
m, A, B und X wie oben definiert sind; und
G, J, L und M jeweils unabhängig voneinander Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8, worin R6, R7 und R8 wie oben definiert sind, sind;
oder wenn n 1 ist, D und E jeweils CR7R8 sind und m 1 ist, A und B jeweils CR7R8 sind, die jeweiligen benachbarten R7-Gruppen zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, eine Gruppe der folgenden Formel bildenworin die gestrichelte Bindung fakultative Doppelbindungen darstellt;
a, G, J, L und M wie oben definiert sind;
r 0 oder 1 ist;
c 0, 1 oder 2 ist; und
R, W, Y und S jeweils unabhängig voneinander Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8, worin R6, R7 oder R8 wie oben definiert sind; sind;
R2 und R3 jeweils unabhängig voneinander ausgewählt sind aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Amino, Halogen, Hydroxy, Nitro, Carboxy, (C2-C6)Alkenyl, (C2-C6)Alkinyl, Trifluormethyl, Trifluormethoxy, (C1-C6)Alkyl, (C1-C6)Alkoxy, worin die Alkyl- oder Alkoxygruppen gegebenenfalls durch eine bis drei Gruppen, ausgewählt aus Halogen, Hydroxy, Carboxy, Amino(C1-C6)alkylthio, (C1-C6)Alkylamino, ((C1-C6)Alkyl)2amino, (C5-C9)Heteroaryl, (C2-C9)Heterocycloalkyl, (C3-C9)Cycloalkyl oder (C6-C10)Aryl substituiert sind; oder R2 und R3 jeweils unabhängig voneinander sind: (C3-C10)Cycloalkyl, (C3-C10)Cycloalkoxy, (C1-C6)Alkylamino, ((C1-C6)Alkyl)2amino, (C6-C10)Arylamino, (C1-C6)Alkylthio, (C6-C10)Arylthio, (C1-C6)Alkylsulfinyl, (C6-C10)Arylsulfinyl, (C1-C6)Alkylsulfonyl, (C6-C10)Arylsulfonyl, (C1-C6)Acyl, (C1-C6)Alkoxy-CO-NH-, (C1-C6)Alkylamino-CO-, (C5-C9)Heteroaryl, (C2-C9)Heterocycloalkyl oder (C6-C10)Aryl, wobei die Heteroaryl-, Heterocycloalkyl- und Arylgruppen gegebenenfalls substituiert sind durch ein bis drei Halogene, (C1-C6)Alkyl, (C1-C6)Alkyl-CO-NH-, (C1-C6)Alkoxy-CO-NH-, (C1-C6)Alkyl-CO-NH- (C1-C6)alkyl, (C1-C6)Alkoxy-CO-NH-(C1-C6)alkyl, (C1-C6)Alkoxy-CO-NH-(C1-C6)alkoxy, Carboxy, Carboxy(C1-C6)alkyl, Carboxy (C1-C6)alkoxy, Benzyloxycarbonyl(C1-C6)alkoxy, (C1-C6)Alkoxycarbonyl(C1-C6)alkoxy, (C6-C10)Aryl, Amino, Amino(C1-C6)alkyl, (C1-C6)Alkoxycarbonylamino, (C6-C10)Aryl(C1-C6)alkoxycarbonylamino, (C1-C6)Alkylamino, ((C1-C6)Alkyl)2amino, (C1-C6)Alkylamino(C1-C6)alkyl, ((C1-C6)Alkyl)2amino(C1-C6)alkyl, Hydroxy, (C1-C6)Alkoxy, Carboxy, Carboxy(C1-C6)alkyl, (C1-C6)Alkoxycarbonyl, (C1-C6)Alkoxycarbonyl(C1-C6)alkyl, (C1-C6)Alkoxy-CO-NH-, (C1-C6)Alkyl-CO-NH-, Cyano, (C5-C9)Heterocycloalkyl, Amino-CO-NH-, (C1-C6)Alkylamino-CO-NH-, ((C1-C6)Alkyl)2amino-CO-NH-, (C6-C10)Arylamino-CO-NH-, (C5-C9)Heteroarylamino-CO-NH-, (C1-C6)Alkylamino-CO-NH-(C1-C6)alkyl, ((C1-C6)Alkyl)2amino-CO-NH-(C1-C6)alkyl, (C6-C10)Arylamino-CO-NH-(C1-C6)alkyl, (C5-C9)Heteroarylamino-CO-NH-(C1-C6)alkyl, (C1-C6)Alkylsulfonyl, (C1-C6)Sulfonylamino, (C1-C6)Alkylsulfonylamino(C1-C6)alkyl, (C6- C10)Arylsulfonyl, (C6-C10)Arylsulfonylamino, (C6-C10)Arylsulfonylamino(C1-C6)alkyl, (C1-C6)Alkylsulfonylamino, (C1-C6)Alkylsulfonylamino(C1-C6)alkyl, (C5-C9)Heteroaryl oder (C2-C9)Heterocycloalkyl;
mit der Maßgabe, dass, wenn A, B oder X in der Formel V oder VI als NR6 oder CR7R8 definiert ist, R2 und/oder R3 Halogen sein muss/müssen;
mit der Maßgabe, dass, wenn R2 und R3 jeweils unabhängig voneinander Wasserstoff oder (C1-C6)Alkyl sind, R1 nicht unsubstituiertes Piperidinyl sein kann;
mit der Maßgabe, dass, wenn R2 und R3 jeweils Wasserstoff sind, R1 nicht unsubstituiertes Morpholinyl oder Pyrrolidinyl sein kann;
mit der Maßgabe, dass, wenn R2 und R3 jeweils Wasserstoff sind, R1 nicht Piperazinyl sein kann; und
mit der Maßgabe, dass die Gruppen der Formeln IV, V, VI oder XIII nicht zwei oder mehr Sauerstoff, Schwefel oder Kombinationen davon in benachbarten Positionen enthalten. - Die vorliegende Erfindung betrifft auch pharmazeutisch annehmbare Säureadditionssalze der Verbindungen der Formel I. Die Säuren, die verwendet werden, um die pharmazeutisch annehmbaren Säureadditionssalze der vorher erwähnten erfindungsgemäßen basischen Verbindungen herzustellen, sind solche, die nichttoxische Säureadditionssalze bilden, d. h. Salze, die pharmakologisch annehmbare Anionen enthalten, wie das Hydrochlorid, Hydrobromid, Hydroiodid, Nitrat, Sulfat, Bisulfat, Phosphat, Säurephosphat, Acetat, Lactat, Citrat, Säurecitrat, Tartrat, Bitartrat, Succinat, Maleat, Fumarat, Gluconat, Saccharat, Benzoat, Methansulfonat, Ethansulfonat, Benzolsulfonat, p-Toluolsulfonat und Pamoat [d. h. 1,1'-Methylen-bis-(2-hydroxy-3-naphthoat)]-Salze.
- Die Erfindung betrifft auch basische Additionssalze der Formel I. Die chemischen Basen, die als Reagenzien verwendet werden können, um die pharmazeutisch annehmbaren Basensalze solcher Verbindungen der Formel I herzustellen, die ihrer Natur nach sauer sind, sind solche, die nicht toxische Basensalze mit solchen Verbindungen bilden. Solche nicht toxischen Basensalze schließen solche ein, die von solchen pharmakologisch annehmbaren Kationen, wie Alkalimetallkationen (zum Beispiel Kalium und Natrium) und Erdalkalimetallkationen (zum Beispiel Calcium und Magnesium), abgeleitet sind, Ammonium oder wasserlösliche Aminadditionssalze, wie N-Methylglucamin(meglumin), und die niederen Alkanolammonium- und andere Basensalze pharmazeutisch annehmbarer organischer Amine, sind aber nicht darauf beschränkt.
- Der Begriff „Alkyl" schließt, so wie er hier verwendet wird, sofern nichts anderes angegeben ist, gesättigte einwertige Kohlenwasserstoffradikale mit geradkettigen, verzweigtkettigen oder zyklischen Gruppen oder Kombinationen davon ein.
- Der Begriff „Alkoxy" schließt, so wie er hier verwendet wird, O-Alkylgruppen ein, worin „Alkyl" wie oben definiert ist.
- Der Begriff „Halogen" schließt, so wie er hier verwendet wird, wenn nichts anderes angegeben ist, Fluor, Chlor, Brom oder Iod ein.
- Die erfindungsgemäßen Verbindungen können Doppelbindungen enthalten. Wenn solche Doppelbindungen vorliegen, können die erfindungsgemäßen Verbindungen in cis- und trans-Konfigurationen und als Gemische davon vorliegen.
- Soweit nichts anderes angegeben ist, können die hier erwähnten Alkyl- und Alkenylgruppen sowie die Alkylreste anderer hier erwähnten Gruppen (zum Beispiel Alkoxy) linear oder verzweigt sein und sie können auch zyklisch (zum Beispiel Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl oder Cyclohep tyl) sein oder linear oder verzweigt sein und zyklische Reste enthalten. Soweit nichts anderes angegeben ist, schließt Halogen Fluor, Chlor, Brom und Iod ein.
- (C3-C10) Cycloalkyl bezieht sich, wenn es hier verwendet wird, auf Cycloalkylgruppen, die null bis zwei Unsättigungsgrade enthalten, wie Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclopentenyl, Cyclohexyl, Cyclohexenyl, 1,3-Cyclohexadien, Cycloheptyl, Cycloheptenyl, Bicyclo[3.2.1]octan, Norbornanyl etc.
- (C2-C9)Heterocycloalkyl bezieht sich, wenn es hier verwendet wird, auf Pyrrolidinyl, Tetrahydrofuranyl, Dihydrofuranyl, Tetrahydropyranyl, Pyranyl, Thiopyranyl, Aziridinyl, Oxiranyl, Methylendioxyl, Chromenyl, Isoxazolidinyl, 1,3-Oxazolidin-3-yl, Isothiazolidinyl, 1,3-Thiazolidin-3-yl, 1,2-Pyrazolidin-2-yl, 1,3-Pyrazolidin-1-yl, Piperidinyl, Thio- morpholinyl, 1,2-Tetrahydrothiazin-2-yl, 1,3-Tetrahydrothiazin-1-yl, Tetrahydroazepinyl, Piperazinyl, Chromanyl etc. Der Fachmann wird kennen, dass die Verknüpfung der genannten (C2-C9)Heterocycloalkyl-Ringe über ein Kohlenstoffatom oder ein sp3-hybridisiertes Stickstoffheteroatom erfolgt.
- (C2-C9)Heteroaryl bezieht sich, wenn es hier verwendet wird, auf Furyl, Thienyl, Thiazolyl, Pyrazolyl, Isothiazolyl, Oxazolyl, Isoxazolyl, Pyrrolyl, Triazolyl, Tetrazolyl, Imidazolyl, 1,3,5-Oxadiazolyl, 1,2,4-Oxadiazolyl, 1,2,3-Oxadiazolyl, 1,3,5-Thiadiazolyl, 1,2,3-Thiadiazolyl, 1,2,4-Thiadiazolyl, Pyridyl, Pyrimidyl, Pyrazinyl, Pyridazinyl, 1,2,4-Triazinyl, 1,2,3-Triazinyl, 1,3,5-Triazinyl, Pyrazolo[3,4-b]pyridinyl, Cinnolinyl, Pteridinyl, Purinyl, 6,7-Dihydro-5H-[1]pyridinyl, Benzo[b]thiophenyl, 5,6,7,8-Tetrahydrochinolin-3-yl, Benzoxazolyl, Benzothiazolyl, Benzisothiazolyl, Benzisoxazolyl, Benzimidazolyl, Thianaphthenyl, Isothianaphthenyl, Benzofuranyl, Isobenzofuranyl, Isoindolyl, Indolyl, Indolizinyl, Indazolyl, Isochinolyl, Chinolyl, Phthalazinyl, Chinoxalinyl, Chinazolinyl, Benzoxazinyl etc. Es versteht sich für den Fachmann, dass die Verknüpfung der (C2-C9)Heterocycloalkyl-Ringe über ein Kohlenstoffatom oder ein sp3-hybridisiertes Stickstoffheteroatom erfolgt.
- (C6-C10)Aryl bezieht sich, wenn es hier verwendet wird, auf Phenyl oder Naphthyl.
- Verbindungen der Formel (I) können in pharmazeutisch annehmbarer Form entweder einzeln oder in Verbindung mit einem oder mehreren zusätzlichen Mitteln, die ein Säugetierimmunsystem modulieren, oder zusammen mit antiinflammatorischen Mitteln verabreicht werden. Diese Mittel schließen Cyclosporin A (zum Beispiel Sandimmune® oder Neoral®), Rapamycin, FK-506 (Tacrolimus), Leflunomid, Deoxyspergualin, Mycophenolat (zum Beispiel Cellcept®), Azathioprin (zum Beispiel Imuran®), Daclizumab (zum Beispiel Zenapax®), OKT3 (zum Beispiel Orthoclone®), AtGam, Aspirin, Acetaminophen, Ibuprofen, Naproxen, Piroxicam und antiinflammatorische Steroide (zum Beispiel Prednisolon oder Dexamethason) ein. Diese Mittel können als Teil derselben oder als separate Dosierungsformen über denselben oder einen verschiedenen Verabreichungsweg und mit derselben oder einer verschiedenen Verabreichungshäufigkeit gemäß üblicher pharmazeutischer Praxis verabreicht werden.
- Die erfindungsgemäßen Verbindungen schließen sämtliche Konfigurationsisomeren (zum Beispiel cis- und trans-Isomere) und sämtliche optischen Isomeren der Verbindungen der Formel I (zum Beispiel Enantiomere und Diastereomere) sowie racemische, diastereomere und andere Gemische solcher Isomeren ein. Die Erfindung schließt auch sämtliche Rotamere der Verbindungen der Formel I sowie scelemische Gemische ein.
- Bevorzugte Verbindungen der Formel I schließen solche ein, worin R1 eine Gruppe der Formelist, worin die gestrichelte Linie fakultative Doppelbindungen darstellt;

m 0, 1, 2 oder 3 ist;
n 0, 1, 2 oder 3 ist;
X, B und D jeweils unabhängig voneinander Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8 sind;
A und E jeweils unabhängig voneinander CR7R8 oder NR6 sind;
oder wenn n 1 ist, D und E jeweils CR7R8 sind und m 1 ist, A und B jeweils CR7R8 sind, die jeweiligen benachbarten R7-Gruppen zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, eine Gruppe der folgenden Formel bilden können:worin die gestrichelte Bindung fakultative Doppelbindungen darstellt;
a, G, J, L und M wie oben definiert sind;
r 0 oder 1 ist;
c 0, 1 oder 2 ist; und
R, W, Y und S jeweils unabhängig Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8, worin R6, R7 und R8 wie oben definiert sind, sind. - Weitere bevorzugte Verbindungen der Formel I schließen solche ein, worin R2 und R3 jeweils unabhängig voneinander aus der Gruppe, bestehend aus Wasserstoff, (C1-C6)Alkyl, (C1-C6)Alkoxy, (C3-C10)Cycloalkyl, (C3-C10)Cycloalkoxy, (C2-C9)Heterocycloalkyl, (C5-C9)Heteroaryl oder (C6-C10)Aryl ausgewählt sind.
- Spezielle bevorzugte Verbindungen der Formel I schließen die folgenden ein:
5-Fluor-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin;
4-Piperidin-1-yl-5-trifluormethyl-7H-pyrrolo[2,3-d]pyrimidin;
N,N-Dimethyl-N'-[3-(4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin-5-yl)benzyl]ethan-1,2-diamin;
2-[1-(5-m-Tolyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-yl]ethanol;
5-(3-Isopropylphenyl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin;
5-(3-Methyl-3H-imidazol-4-yl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin;
5-(1-Methyl-1H-imidazol-4-yl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin;
5-(2-Methylpyridin-4-yl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin;
5-Chlor-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin;
5-Ethinyl-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin;
4-Piperidin-1-yl-5-m-tolyl-7H-pyrrolo[2,3-d]pyrimidin; und
4-(3,3-Dimethylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin. - Die vorliegende Erfindung betrifft auch eine pharmazeutische Zusammensetzung zur (a) Behandlung oder Prävention einer Störung oder eines Zustandes, ausgewählt aus Organtransplantat abstoßung, Lupus, multipler Sklerose, rheumatoider Arthritis, Psoriasis, Typ I-Diabetes und Komplikationen, die aus Diabetes resultieren, Krebs, Asthma, atopischer Dermatitis, Autoimmun-Schilddrüsen-Störungen, Colitis ulcerosa, Crohn-Krankheit, Alzheimer-Krankheit, Leukämie und anderen Autoimmunkrankheiten, oder (b) Inhibierung von Proteintyrosinkinasen oder Janus-Kinase 3 (JAK3) in einem Säuger, einschließlich eines Menschen, die eine Menge einer Verbindung der Formel I oder eines pharmazeutisch annehmbaren Salzes davon, die bei solchen Störungen oder Zuständen wirksam ist, und einen pharmazeutisch annehmbaren Träger umfasst.
- Die vorliegende Erfindung betrifft auch eine pharmazeutische Zusammensetzung zur (a) Behandlung oder Prävention einer Störung oder eines Zustandes, ausgewählt aus Organtransplantatabstoßung, Lupus, multipler Sklerose, rheumatoider Arthritis, Psoriasis, Typ-I-Diabetes und Komplikationen, die aus Diabetes resultieren, Krebs, Asthma, atopischer Dermatitis, Autoimmun-Schilddrüsen-Störungen, Colitis ulcerosa, Crohn-Krankheit, Alzheimer-Krankheit, Leukämie und anderen Autoimmunkrankheiten, oder (b) Inhibierung von Proteintyrosinkinasen oder Janus-Kinase 3 (JAK3) in einem Säuger, einschließlich eines Menschen, die eine Menge einer Verbindung der Formel I oder eines pharmazeutisch annehmbaren Salzes allein oder in Kombination mit T-Zellen-immunsuppressiven Mitteln oder mit Mitteln gegen Entzündungen, die bei solchen Störungen oder Zuständen wirksam sind, und einen pharmazeutisch annehmbaren Träger umfasst.
- Die vorliegende Erfindung betrifft auch die Verwendung einer wirksamen Menge einer Verbindung der Formel I oder eines pharmazeutisch annehmbaren Salzes davon zur Herstellung eines Medikaments für die Inhibierung von Proteintyrosinkinasen oder Janus-Kinase 3 (JAK3) in einem Säuger, einschließlich eines Menschen.
- Die vorliegende Erfindung betrifft auch die Verwendung einer wirksamen Menge einer Verbindung der Formel I oder eines pharmazeutisch annehmbaren Salzes davon zur Herstellung eines Medikaments für die Behandlung oder Prävention einer Störung oder eines Zustandes, ausgewählt aus Organtransplantatabstossung, Lupus, multipler Sklerose, rheumatoider Arthritis, Psoriasis, Typ-I-Diabetes und Komplikationen, die aus Diabetes resultieren, Krebs, Asthma, atopischer Dermatitis, Autoimmun-Schilddrüsen-Störungen, Colitis ulcerosa, Crohn-Krankheit, Alzheimer-Krankheit, Leukämie und anderen Autoimmunkrankheiten, in einem Säuger, einschließlich eines Menschen.
- Die vorliegende Erfindung betrifft auch die Verwendung einer wirksamen Menge einer Verbindung der Formel I oder eines pharmazeutisch annehmbaren Salzes davon, allein oder in Kombination mit T-Zellen-immunsuppressiven Mitteln oder mit Mitteln gegen Entzündungen, zur Herstellung eines Medikaments zur Inhibierung von Proteintyrosinkinasen oder Janus-Kinase 3 (JAK3) in einem Säuger, einschließlich eines Menschen.
- Die vorliegende Erfindung betrifft auch die Verwendung einer wirksamen Menge einer Verbindung der Formel I oder eines pharmazeutisch annehmbaren Salzes davon, allein oder in Kombination mit T-Zellen-immunsuppressiven Mitteln oder mit Mitteln gegen Entzündungen zur Herstellung eines Medikaments für die Behandlung oder Prävention einer Störung oder eines Zustandes, ausgewählt aus Organtransplantatabstossung, Lupus, multipler Sklerose, rheumatoider Arthritis, Psoriasis, Typ-I-Diabetes und Komplikationen, die aus Diabetes resultieren, Krebs, Asthma, atopischer Dermatitis, Autoimmun-Schilddrüsen-Störungen, Colitis ulcerosa, Crohn-Krankheit, Alzheimer-Krankheit, Leukämie und anderen Autoimmunkrankheiten in einem Säuger, einschließlich eines Menschen.
- Detaillierte Beschreibung der Erfindung
- Die folgenden Reaktionsschemata illustrieren die Herstellung der erfindungsgemäßen Verbindungen. Soweit nichts anderes angegeben ist, sind R1, R2, R3 und R9 in den folgenden Reaktionsschemata und in der folgenden Erörterung wie oben definiert.
- SCHEMA 1
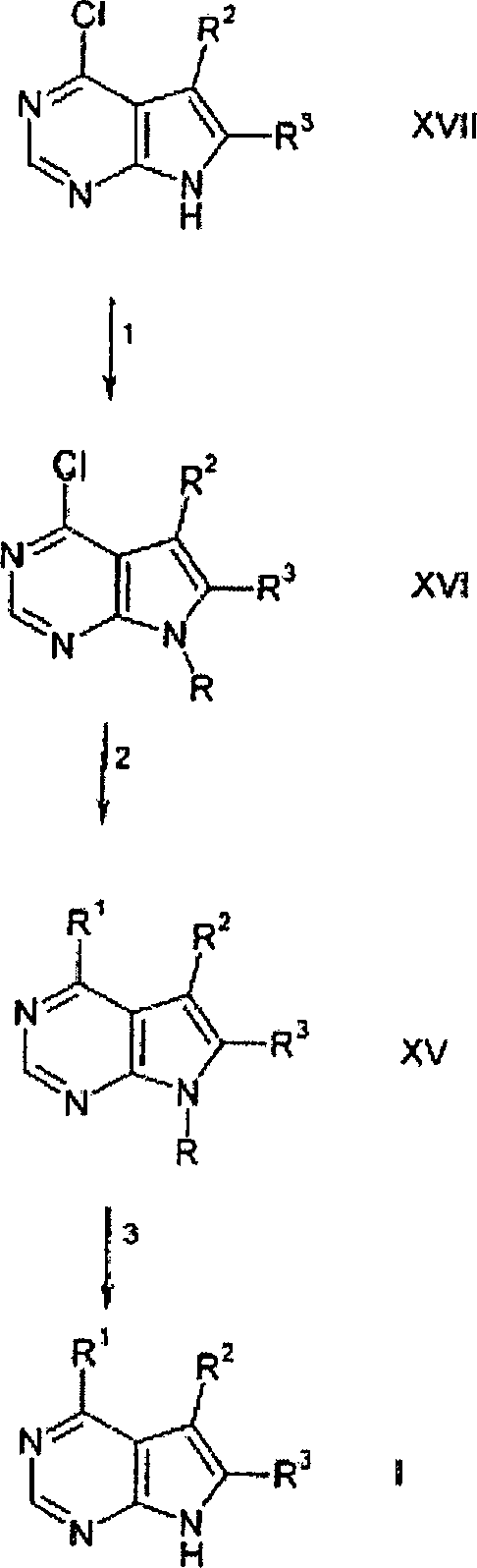
- SCHEMA 2
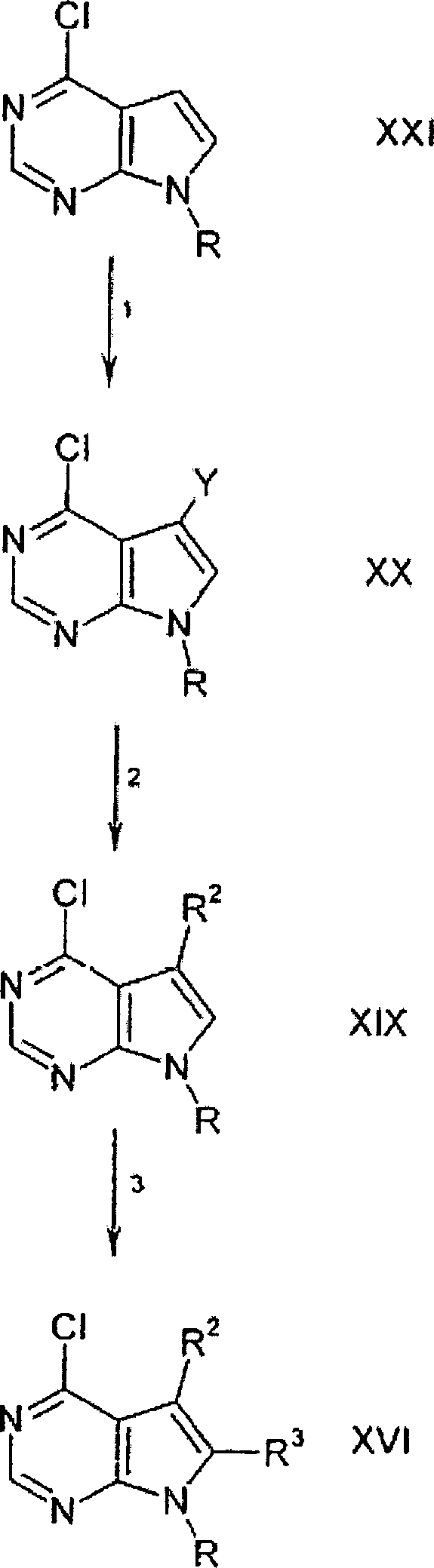
- SCHEMA 3

- SCHEMA 4
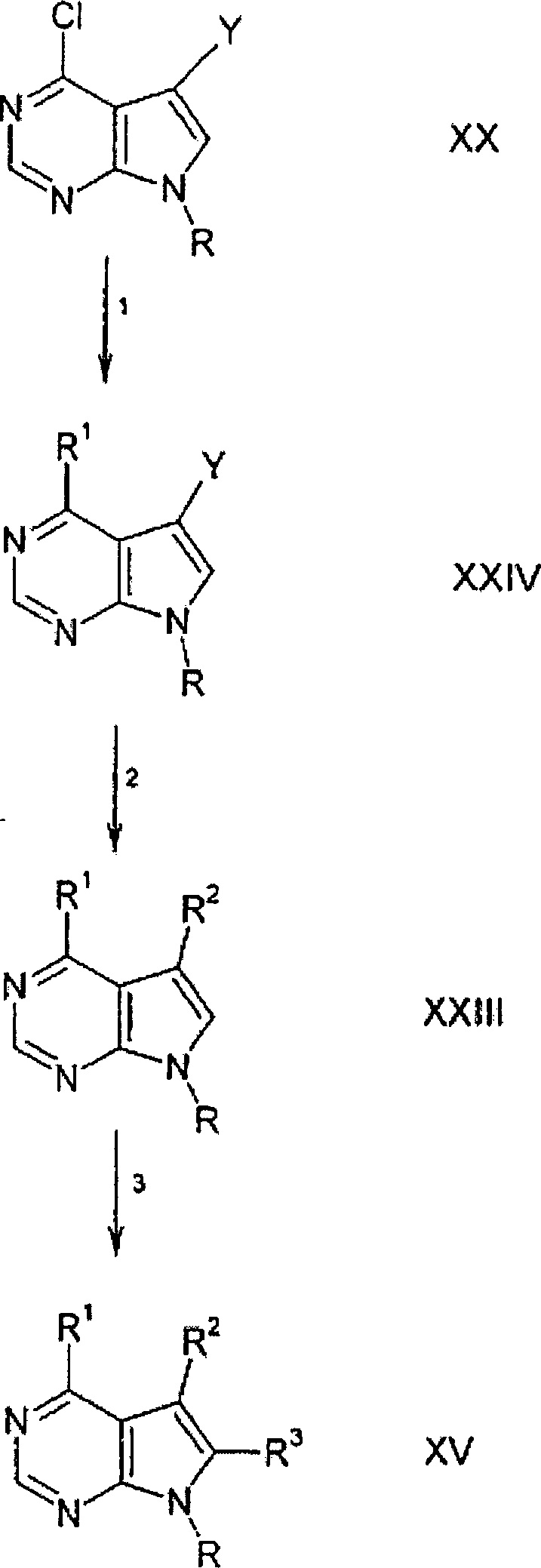
- In Reaktion 1 von Schema 1 wird die 4-Chlorpyrrolo[2,3-d]pyrimidin-Verbindung der Formel XVII zu der entsprechenden Verbindung der Formel XVI, worin R für Benzolsulfonyl oder Benzyl steht, durch Behandlung von XVII mit Benzolsulfonylchlorid, Benzylchlorid oder Benzylbromid in Gegenwart einer Base, wie Natriumhydrid oder Kaliumcarbonat, und eines polaren aprotischen Lösungsmittels, wie Dimethylformamid oder Tetrahydrofuran, umgewandelt. Das Reaktionsgemisch wird bei einer Temperatur zwischen etwa 0°C bis etwa 70°C, vorzugsweise etwa 30°C, über einen Zeitraum zwischen etwa einer Stunde und etwa drei Stunden, vorzugsweise etwa zwei Stunden, gerührt.
- In Reaktion 2 von Schema 1 wird die 4-Chlorpyrrolo[2,3-d]pyrimidin-Verbindung der Formel XVI zu der entsprechenden 4-Aminopyrrolo[2,3-d]pyrimidin-Verbindung der Formel XV durch Kupplung von XVI mit einer Verbindung der Formel R1H umgewandelt. Die Reaktion wird in einem alkoholischen Lösungsmittel, wie tert-Butanol, Methanol oder Ethanol, oder anderen hochsiedenden organischen Lösungsmitteln, wie Dimethylformamid, 1,4-Dioxan oder 1,2-Dichlorethan, bei einer Temperatur zwischen etwa 60°C bis etwa 120°C, vorzugsweise etwa 80°C durchgeführt. Typische Reaktionszeiten liegen zwischen etwa zwei Stunden bis etwa 48 Stunden, vorzugsweise etwa 16 Stunden.
- In Reaktion 3 von Schema 1 wird die Schutzgruppe von der Verbindung der Formel XV, worin R für Benzolsulfonyl steht, zur Herstellung der entsprechenden Verbindung der Formel I abgespalten, indem XV mit einer Alkalibase, wie Natriumhydroxid oder Kaliumhydroxid, in einem alkoholischen Lösungsmittel, wie Methanol oder Ethanol, oder gemischten Lösungsmitteln, wie Alkohol/Tetrahydrofuran oder Alkohol/Wasser, behandelt wird. Die Reaktion wird bei Raumtemperatur über einen Zeitraum zwischen etwa 15 Minuten bis etwa eine Stunde, vorzugsweise 30 Minuten durchgeführt. Die Entfernung der Schutzgruppe von der Verbindung der Formel XV, worin R Benzyl ist, wird durch Behandlung von XV mit Natrium in Ammoniak bei einer Temperatur von etwa –78°C über einen Zeitraum zwischen etwa 15 Minuten bis etwa eine Stunde durchgeführt.
- In Reaktion 1 von Schema 2 wird die 4-Chlorpyrrolo[2,3-d]pyrimidin-Verbindung der Formel XXI, worin R Wasserstoff oder Benzolsulfonat ist, zu der 4-Chlor-5-halopyrrolo[2,3-d]pyrimidin-Verbindung der Formel XX, worin Y für Chlor, Brom oder Iod steht, durch Umsetzung von XXI mit N-Chlorsuccinimid, N-Bromsuccinimid oder N-Iodsuccinimid umgewandelt. Das Reaktionsgemisch wird in Chloroform über einen Zeitraum zwischen etwa einer Stunde bis etwa drei Stunden, vorzugsweise etwa einer Stunde unter Rückfluss erhitzt. Alternativ wird in Reaktion 1 von Schema 2 4-Chlorpyrrolo[2,3-d]pyrimidin der Formel XXI, worin R für Wasserstoff steht, zu dem entsprechenden 4-Chlor-5-Nitropyrrolo[2,3-d]pyrimidin der Formel XX, worin Y Nitro bedeutet, durch Umsetzung von XXI mit Salpetersäure in Schwefelsäure bei einer Temperatur zwischen etwa –10°C bis etwa 10°C, vorzugsweise etwa 0°C, über einen Zeitraum zwischen etwa fünf Minuten bis etwa 15 Minuten, vorzugsweise etwa 10 Minuten, umgewandelt. Die Verbindung der Formel XXI, worin Y Nitro ist, wird zu dem entsprechenden 4-Chlor-5-aminopyrrolo[2,3-d]pyrimidin der Formel XX, worin Y für Amino steht, durch Umsetzen von XXI unter einer Reihe von Bedingungen, die dem Fachmann bekannt sind, wie Palladium-Hydrogenolyse oder Zinn(IV)chlorid und Salzsäure, umgewandelt.
- In Reaktion 2 von Schema 2 wird die 4-Chlor-5-halopyrrolo[2,3-d]pyrimidin-Verbindung der Formel XX, worin R für Wasserstoff steht, zu der entsprechenden Verbindung der Formel XIX, worin R2 (C1-C6)Alkyl oder Benzyl ist, durch Behandlung von XX mit N-Butyllithium bei einer Temperatur von etwa –78°C und Umsetzung der Dianionzwischenstufe, die so hergestellt worden ist, mit einem Alkylhalogenid oder Benzylhalogenid bei einer Temperatur zwischen etwa –78°C bis Raumtemperatur, vorzugsweise Raumtemperatur, umgewandelt. Alternativ wird das so hergestellte bzw. gebildete Dianion mit molekularem Sauerstoff unter Bildung der entsprechenden 4-Chlor-5-hydroxypyrrolo[2,3-d]pyrimidin-Verbindung der Formel XIX, worin R2 Hydroxy ist, umgesetzt. Die Verbindung der Formel XX, worin Y für Brom oder Iod steht und R Benzolsulfonat bedeutet, wird zu der Verbindung der Formel XIX, worin R2 (C6-C12)Aryl oder Vinyl ist, durch Behandlung von XX mit N-Butyllithium bei einer Temperatur von etwa –78°C und anschließender Zugabe von Zinkchlorid bei einer Temperatur von etwa –78°C umgewandelt. Die so hergestellte entsprechende Organozinkzwischenstufe wird anschließend mit Aryliodid oder Vinyliodid in Gegenwart einer katalytischen Menge Palladiums umgesetzt. Das Reaktionsgemisch wird bei einer Temperatur zwischen etwa 50°C bis etwa 80°C, vorzugsweise etwa 70°C, über einen Zeitraum zwischen etwa einer Stunde bis etwa drei Stunden, vorzugsweise etwa eine Stunde, gerührt.
- In Reaktion 3 von Schema 2 wird die Verbindung der Formel XIX zu der entsprechenden Verbindung der Formel XVI durch Behandlung von XIX mit N-Butyllithium, Lithiumdiisopropylamin oder Natriumhydrid bei einer Temperatur von etwa –78°C in Gegenwart eines polaren aprotischen Lösungsmittels, wie Tetrahydrofuran, umgewandelt. Die so hergestellte anionische Zwischenstufe wird weiter mit (a) einem Alkylhalogenid oder Benzylhalogenid bei einer Temperatur zwischen etwa –78°C bis Raumtemperatur, vorzugsweise –78°C, wenn R3 für Alkyl oder Benzyl steht, umgesetzt, (b) einem Aldehyd oder Keton bei einer Temperatur zwischen etwa –78°C bis Raumtemperatur, vorzugsweise –78°C, wenn R3 für Alkoxy steht und (c) Zinkchlorid bei einer Temperatur zwischen etwa –78°C bis Raumtemperatur, vorzugsweise –78°C, und die so gebildete entsprechende Organozinkzwischenstufe wird anschließend mit Aryliodid oder Vinyliodid in Gegenwart einer katalytischen Menge Palladiums umgesetzt. Das resultierende Reaktionsgemisch wird bei einer Temperatur zwischen etwa 50°C bis etwa 80°C, vorzugsweise etwa 70°C, über einen Zeitraum zwischen etwa einer Stunde bis etwa drei Stunden, vorzugsweise etwa eine Stunde, gerührt. Alternativ wird das so gebildete Anion mit molekularem Sauerstoff umgesetzt, um die entsprechende 4-Chlor-6-hydroxypyrrolo[2,3-d]pyrimidin-Verbindung der Formel XVI, worin R3 Hydroxy ist, herzustellen.
- In Reaktion 1 von Schema 3 wird die 4-Chlorpyrrolo[2,3-d]pyrimidin-Verbindung der Formel XXI zu der entsprechenden Verbindung der Formel XXII gemäß den oben in Reaktion 3 von Schema 2 beschriebenen Verfahren umgewandelt.
- In Reaktion 2 von Schema 3 wird die Verbindung der Formel XXII zu der entsprechenden Verbindung der Formel XVI gemäß den oben in Reaktionen 1 und 2 von Schema 3 beschriebenen Verfahren umgewandelt.
- In Reaktion 1 von Schema 4 wird die 4-Chlorpyrrolo[2,3-d]pyrimidin-Verbindung der Formel XX zu der entsprechenden 4-Aminopyrrolo[2,3-d]pyrimidin-Verbindung der Formel XXIV gemäß dem oben in Reaktion 2 beschriebenen Verfahren umgewandelt.
- In Reaktion 2 von Schema 4 wird die 4-Amino-5-halopyrrolo[2,3-d]pyrimidin-Verbindung der Formel XXIV, worin R Benzolsulfonat ist und Z für Brom oder Iod steht, zu der entsprechenden Verbindung der Formel XXIII durch Umsetzung von XXIV mit (a) Arylboronsäure, wenn R2 für Aryl steht, in einem aprotischen Lösungsmittel, wie Tetrahydrofuran oder Dioxan, in Gegenwart einer katalytischen Menge Palladium(0) bei einer Temperatur zwischen etwa 50°C bis etwa 100°C, vorzugsweise etwa 70°C, über einen Zeitraum zwischen etwa zwei Stunden bis etwa 48 Stunden, vorzugsweise etwa 12 Stunden, (b) Alkinen, wenn R2 für Alkinyl steht, in Gegenwart einer katalytischen Menge von Kupfer(I)iodid und Palladium(0) und einem polaren Lösungsmittel wie Dimethylformamid bei Raumtemperatur über einen Zeitraum zwischen etwa einer Stunde und etwa fünf Stunden, vorzugsweise etwa drei Stunden und (c) Alkenen oder Styrolen, wenn R2 für Vinyl oder Styryl steht, in Gegenwart einer katalytischen Menge Palladium in Dimethylformamid, Dioxan oder Tetrahydrofuran bei einer Temperatur zwischen etwa 80°C bis etwa 100°C, vorzugsweise etwa 100°C, über einen Zeitraum zwischen etwa zwei Stunden bis etwa 48 Stunden, vorzugsweise etwa 48 Stunden, umgewandelt.
- In Reaktion 3 von Schema 4 wird die Verbindung der Formel XXIII zu der entsprechenden Verbindung der Formel XV gemäß dem oben in Reaktion 3 von Schema 2 beschriebenen Verfahren umgewandelt.
- Die erfindungsgemäßen Verbindungen, die ihrer Natur nach basisch sind, können eine große Vielzahl verschiedener Salze mit verschiedenen anorganischen und organischen Säuren bilden. Obwohl solche Salze für die Verabreichung an Tieren pharmazeutisch annehmbar sein müssen, ist es in der Praxis häufig zweckmäßig, die erfindungsgemäße Verbindung aus dem Reaktionsgemisch als ein pharmazeutisch nicht annehmbares Salz zu isolieren und das letztere einfach zu der freien Base durch Behandlung mit einem alkalischen Reagens zurückzuverwandeln und die letztgenannte freie Base in ein pharmazeutisch annehmbares Säureadditionssalz umzuwandeln. Die Säureadditionssalze der erfindungsgemäßen basischen Verbindungen werden in einfacher Weise durch Behandeln der basischen Verbindung mit einer im wesentlichen äquivalenten Menge der gewählten Mineral- oder organischen Säure in einem wässrigen Lösungsmedium oder in einem geeigneten organischen Lösungsmittel, wie Methanol oder Ethanol, hergestellt. Bei vorsichtigem Eindampfen des Lösungsmittels wird das gewünschte feste Salz leicht erhalten. Das gewünschte Säuresalz kann auch aus einer Lösung der freien Base in einem organischen Lösungsmittel durch Zugabe einer geeigneten Mineral- oder organischen Säure zu der Lösung ausgefällt werden.
- Die erfindungsgemäßen Verbindungen, die ihrer Natur nach sauer sind, können mit zahlreichen pharmakologisch annehmbaren Kationen basische Salze bilden. Beispiele solcher Salze schließen die Alkalimetall- oder Erdalkalimetallsalze und insbesondere die Natrium- und Kaliumsalze ein. Diese Salze werden sämtlich durch übliche Verfahren hergestellt. Die chemischen Basen, die als Reagenzien verwendet werden, um die pharmazeutisch annehmbaren erfindungsgemäßen basischen Salze herzustellen, sind solche, die nichttoxische basische Salze mit den erfindungsgemäßen sauren Verbindungen bilden. Solche nichttoxischen basischen Salze schließen solche ein, die von solchen pharmakologisch annehmbaren Kationen, wie Natrium, Kalium, Calcium und Magnesium usw., abgeleitet sind. Diese Salze können in einfacher Weise durch Behandlung der entsprechenden sauren Verbindungen mit einer wässrigen Lösung, die die gewünschten pharmakologisch annehmbaren Kationen enthält, und anschließendem Eindampfen der resultierenden Lösung zur Trockne, vorzugsweise unter vermindertem Druck, hergestellt werden. Alternativ können sie auch durch Mischen von Niedrigalkanol-Lösungen der sauren Verbindungen und des gewünschten Alkalimetallalkoxids und anschließendem Eindampfen der resultierenden Lösung zur Trockene in derselben Weise wie zuvor hergestellt werden. In beiden Fällen werden vorzugsweise stöchiometrische Mengen der Reagenzien verwendet, um die Vollständigkeit der Reaktion und maximale Ausbeuten an dem gewünschten Endprodukt sicherzustellen.
- Die erfindungsgemäßen Zusammensetzungen können auf herkömmliche Weise unter Verwendung von einem oder mehreren pharmazeutisch annehmbaren Trägern formuliert werden. So können die aktiven erfindungsgemäßen Verbindungen für die orale, bukka le, intranasale, parenterale (zum Beispiel intravenöse, intramuskuläre oder subkutane) oder rektale Verabreichung oder in einer für die Verabreichung durch Inhalation oder Insufflation geeignete Form formuliert werden. Die aktiven erfindungsgemäßen Verbindungen können auch für die verzögerte Freisetzung formuliert werden.
- Für die orale Verabreichung können die pharmazeutischen Zusammensetzungen die Form von beispielsweise Tabletten oder Kapseln annehmen, die durch herkömmliche Mittel mit pharmazeutisch annehmbaren Exzipienzien wie Bindemitteln (zum Beispiel vorgelatinierter Maisstärke, Polyvinylpyrrolidon oder Hydroxypropylmethylcellulose); Füllstoffen (zum Beispiel Lactose, mikrokristalline Cellulose oder Calciumphosphat); Gleitmitteln (zum Beispiel Magnesiumstearat, Talk oder Siliciumdioxid); Sprengmitteln (zum Beispiel Kartoffelstärke oder Natriumstärkeglycolat); oder Netzmitteln (zum Beispiel Natriumlaurylsulfat) hergestellt werden. Die Tabletten können nach im Stand der Technik gut bekannten Verfahren beschichtet werden. Flüssige Zubereitungen für die orale Verabreichung können die Form von beispielsweise Lösungen, Sirupen oder Suspensionen annehmen oder sie können als ein Trockenprodukt für die Zubereitung mit Wasser oder einem anderen geeigneten Vehikel vor der Verwendung dargereicht werden. Solche flüssige Zubereitungen können durch herkömmliche Mittel mit pharmazeutisch annehmbaren Zusätzen, wie Suspendiermittel (zum Beispiel Sorbitsirup, Methylcellulose oder hydrierten Speisefetten); Emulgatoren (zum Beispiel Lecithin oder Akazienöl); nichtwässrigen Vehikeln (zum Beispiel Mandelöl, Ölester oder Ethylalkohol); und Konservierungsstoffen (zum Beispiel Methyl- oder Propyl-p-hydroxybenzoaten oder Sorbinsäure), hergestellt werden.
- Für die bukkale Verabreichung kann die Zusammensetzung die Form von Tabletten oder Pastillen, die auf übliche Weise formuliert werden, annehmen.
- Die erfindungsgemäßen aktiven Verbindungen können für die parenterale Verabreichung durch Injektion, einschließlich der Verwendung konventioneller Katheter-Techniken oder Infusion, formuliert werden. Formulierungen für die Injektion können in Einheitsdosisform, zum Beispiel in Ampullen oder in Behältern mit Mehrfachdosis, zusammen mit einem zugesetzten Konservierungsmittel dargereicht werden. Die Zusammensetzungen können in der Form von Suspensionen, Lösungen oder Emulsionen in ölartigen oder wässrigen Vehikeln vorliegen und können Formulierungshilfsmittel wie Suspensions-, Stabilisierungs- und/oder Dispergiermittel enthalten. Alternativ kann der Wirkstoff in Pulverform für die Zubereitung mit einem geeigneten Vehikel, zum Beispiel sterilem pyrogenfreien Wasser, vor der Verwendung vorliegen.
- Die erfindungsgemäßen aktiven Verbindungen können auch in Form von rektalen Zusammensetzungen, wie Suppositorien oder Retentionsklestiren, zum Beispiel mit üblichen Grundlagen für Suppositorien, wie Kakaobutter oder anderen Glyceriden, formuliert werden.
- Für die intranasale Verabreichung oder die Verabreichung durch Inhalation werden die erfindungsgemäßen aktiven Verbindungen zweckmäßigerweise in der Form einer Lösung oder einer Suspension aus einem Pumpspraybehälter, der durch den Patienten bedient, d. h. gedrückt oder gepumpt wird, oder als Aerosolspray-Darreichungsform aus einem unter Druck stehenden Behälter oder Vernebler unter Verwendung eines geeigneten Treibmittels, wie Dichlordifluormethan, Trichlorfluormethan, Dichlortetrafluorethan, Kohlendioxid oder einem anderen geeigneten Gas, dargereicht. Im Fall eines unter Druck stehen den Aerosols kann die Druckeinheit bestimmt werden, indem ein Ventil vorgesehen wird, das eine abgemessene bzw. bestimmte Menge abgibt. Der unter Druck stehende Behälter oder der Vernebler können eine Lösung oder Suspension der aktiven Verbindungen enthalten. Kapseln und Patronen (die zum Beispiel aus Gelatine hergestellt sind) für die Verwendung in einem Inhalator oder Insufflator können so formuliert werden, dass sie ein Pulvergemisch einer erfindungsgemäßen Verbindung und einer geeigneten Pulvergrundlage wie Lactose oder Stärke enthalten.
- Eine empfohlene Dosis der aktiven erfindungsgemäßen Verbindungen für die orale, parenterale oder bukkale Verabreichung beträgt für die Behandlung der oben erwähnten Krankheitszustände (zum Beispiel Asthma) bei einem durchschnittlichen Erwachsenen 0,1 bis 1000 mg des Wirkstoffs pro Einheitsdosis, die beispielsweise ein- bis viermal pro Tag verabreicht werden könnte.
- Aerosolformulierungen zur Behandlung der oben genannten Krankheitszustände (zum Beispiel rheumatoider Arthritis) werden vorzugsweise für den durchschnittlichen Erwachsenen so eingestellt, dass jede abgemessene Dosis oder jeder „Hub" des Aerosols 20 μg bis 1000 μg der erfindungsgemäßen Verbindungμ enthält. Die tägliche Gesamtdosis an Aerosol liegt innerhalb des Bereichs von 0,1 mg bis 1000 mg. Die Verabreichung kann mehrmals am Tag, zum Beispiel 2-, 3-, 4-, oder 8-mal erfolgen, wobei beispielsweise jedes Mal 1, 2 oder 3 Dosen verabreicht werden.
- Eine Verbindung der Formel (I) wird in einer pharmazeutisch annehmbaren Form entweder einzeln oder in Kombination mit einem oder mehreren zusätzlichen Mitteln, die ein Säugetierimmunsystem modulieren, oder mit antiinflammatorischen Mitteln, Mitteln, die Cyclosporin A (zum Beispiel Sandimmun® oder Neo ral®), Rapamycin, FK-506 (Tacrolimus), Leflunomid, Deoxyspergualin, Mycophenolat (zum Beispiel Cellcept®), Azathioprin (zum Beispiel Imuran®), Daclizumab (zum Beispiel Zenapax®), OKT3 (zum Beispiel Orthocolon®), AtGam, Aspirin, Acctaminophen, Ibuprofen, Naproxen, Piroxicam und antiinflammatorische Steroide (zum Beispiel Prednisolon oder Dexamethanson) einschließen, aber nicht darauf beschränkt sind, verabreicht und solche Mittel können als ein Teil derselben oder separater Dosierungsformen über denselben oder einen verschiedenen Verabreichungsweg und mit derselben oder einer verschiedenen Häufigkeit gemäß üblicher pharmazeutischer Praxis verabreicht werden.
- FK506 (Tacrolimus) wird oral in einer Menge von 0,10 bis 0,15 mg/kg Körpergewicht alle 12 Stunden innerhalb der ersten 48 Stunden postoperativ verabreicht. Die Dosis wird über den Minimumbereich der Tacrolimus-Spiegel im Serum überwacht.
- Cyclosporin A (eine oral oder intravernöse Sandimmun-Formulierung oder Neoral®, eine orale Lösung oder Kapseln) wird oral mit einer Menge von 5 mg/kg Köpergewicht alle 12 Stunden innerhalb von 48 Stunden postoperativ verabreicht. Die Dosis wird über den Minimumbereich der Cyclosporin-A-Spiegel im Blut überwacht.
- Die Wirkstoffe können für die verzögerte Freisetzung nach dem Fachmann an sich bekannten Verfahren formuliert werden. Beispiele für solche Formulierungen finden sich in den US-Patenten 3,538,214, 4,060,598, 4,173,626, 3,119,742 und 3,492,397.
- Die Fähigkeit der Verbindungen der Formel I oder ihrer pharmazeutisch annehmbarer Salze, die Janus-Kinase 3 zu inhibieren und folglich ihre Wirksamkeit zur Behandlung der durch die Janus-Kinase 3 gekennzeichneten Störungen oder Zustände zu demonstrieren, wird durch die folgenden in-vitro-Assay-Tests gezeigt.
- Biologischer Assay
- JAK3 (JH1 : GST) Enzymatischer Assay
- Der JAK3-Kinase-Assay verwendet ein Protein, das in mit Baculovirus infizierten SF9-Zellen exprimiert wurde (ein Fusionsprotein aus GST und der katalytischen Domäne von menschlichem JAK3), welches durch Affinitätschromatographie auf Glutadion-Sepharose gereinigt wurde. Das Substrat für die Reaktion ist poly-Glutaminsäure-Tyrosin (PGT (4 : 1), Sigma-Katalog Nr. P0275), welches auf Nunc-Maxi-Sorp-Platten in einer Menge von 100 μg/ml über Nacht bei 37°C aufgetragen wurde. Am Morgen nach dem Aufbringen werden die Platten dreimal gewaschen und JAK3 wird in die Vertiefungen gegeben, die 100 μl Kinasepuffer (50 mM HEPES, pH 7,3, 125 mM NaCl, 24 mM MgCl2) + 0,2 μM ATP + 1 mM Na-Orthovanadat) enthielten. Die Reaktion läuft 30 Minuten lang bei Raumtemperatur ab und die Platten werden noch dreimal gewaschen. Die Menge bzw. Konzentration an phosphoryliertem Tyrosin in einer bestimmten Vertiefung wird durch einen Standard-ELISA-Assay unter Verwendung eines Antiphosphotyrosin-Antikörpers (ICN PY20, Kat. Nr. 69-151-1) quantifiziert.
- DND39/IL-4 Zellulärer Assay für JAK3-Kinaseinhibitoren
- Der DND39/IL-4-Assay ist angelegt, um Inhibitoren der JAK3-Kinaseaktivität zu identifizieren, welche die besten Kandidaten für die Immunsuppression und/oder Allergie wären. Dieser Assay verwendet eine B-Zelllinie, genannt DND39, welche das Luciferasegen durch den Keimlinien-IgE-Promoter stabil in eines der Chromosomen integriert treiben ließ. Wenn diese Zellen mit IL-4 stimuliert werden, phosphoryliert die Kinase JAK3, die mit dem IL-4-Rezeptor assoziiert ist, den Signal überträger STAT6. STAT6 bindet anschließend an den Keimlinien-IgE-Promoter und beginnt die Transkription des Luciferasegens. Die Luciferase wird in einem Lysat dieser Zellen unter Verwendung des Promega-Luciferase-Assayreagenssystems bestimmt.
- Anmerkung: Die DND39-Zellen werden in RPMI 1640 kultiviert, das mit 10% durch Wärme inaktiviertem FCS, 2 mM L-Glutamin und 100 Einheiten/ml Pen./Strep. ergänzt wurde. Die Zellen werden in einer Menge von 1 × 105 bis 1 × 106 Zellen/ml gehalten. Freitags auf 1 × 105 geteilt beträgt ihre Anzahl montags etwa 1 × 106. Anschließend werden sie unter der Woche im Verhältnis 1 : 2 geteilt und 200 ml in einem Kolben wie erforderlich aufbewahrt.
- 3 × 105 DND39-Zellen werden in 100 μl RPMI 1640 plattiert, das mit 1% durch Wärme inaktiviertem FCS, 2 mM L-Glutamin und 100 Einheiten/ml Pen/Step in einer Vee-Tüpfelplatte mit 96 Vertiefungen (Nunc) ergänzt wurde. Die Verbindungen werden aufeinander folgend 1 : 2 in DMSO verdünnt, wobei man bei 4 mM bis 1,9 μM beginnt. Auf einer Polypropylen-Platte mit 96 Vertiefungen werden die Pipettenspitzen nach jeder Verdünnung gewechselt. Anschließend werden 5 μl jeder Verdünnung zu 500 μl an RPMI/1% Serum in einem Gestell mit 96 Röhrchen gegeben. 125 μl der verdünnten Lösungen der Verbindungen werden zu den Zellen gegeben und eine Stunde lang bei 37°C und 5% CO2 inkubiert. Nach einer Stunde werden 25 μl von 25 ng/ml IL-4 den Zellen zugesetzt und vermischt. Die Endkonzentration an IL-4 beträgt 2,5 ng/ml und die Endkonzentration der Verbindung reicht von 20 μM bis 156 nM. Die Zellen werden anschließend über Nacht 16 bis 18 Stunden lang inkubiert. Die Platte wird anschließend in einer Tischzentrifuge fünf Minuten lang bei 2500–3000 U/min zentrifugiert. Der Kulturüberstand wird vorsichtig durch Ansaugen mit einer achtfachen Spitze entfernt. 100 μl PBS mit Calcium und Magnesium werden den pelletierten Zellen zugesetzt. Die Zellen werden in dem PBS wieder suspendiert und auf eine weiße Packard-OptiPlate transferiert. 100 μl Packards LucLite-Reagenz werden in die Vertiefungen der OptiPlate gegeben.
- Die folgenden Beispiele illustrieren die Herstellung der erfindungsgemäßen Verbindungen, sie ist aber nicht auf Einzelheiten davon beschränkt. Die Schmelzpunkte sind unkorrigiert. Die NMR-Daten sind in „Parts per Million" (δ) angegeben und beziehen sich auf das Deuterium-Lock-Signal aus dem Lösungsmittel der Probe (Deuterochloroform, sofern nichts anderes angegeben). Handelsübliche Reagenzien wurden ohne weitere Reinigung verwendet. THF bedeutet Tetrahydrofuran. DMF bedeutet N,N-Dimethylformamid. Niedrig auflösende Massenspektren (LRMS) wurden entweder auf einem Hewlett Packard 5989®-Spektrometer unter Verwendung chemischer Ionisation (Ammonium) oder auf einer Fisons- (oder MicroMass-) Plattform mit chemischer Ionisation bei Atmosphärendruck (APCI) aufgenommen, wobei ein 50/50-Gemisch aus Acetonitril/Wasser mit 0,1% Ameisensäure als Ionisierungsmittel verwendet wird. Raum- oder Umgebungstemperatur bedeutet 20–25°C.
- BEISPEIL 1
- Cyclohexylmethyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amin
- METHODE A
- Cyclohexylmethyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amin
- Ein Gemisch von 200 mg (1,30 mmol) von 4-Chlor-7H-pyrrolo[2,3-d]pyrimidin (hergestellt nach dem Verfahren von Davoll, J. Am. Chem. Soc., (1960), 82, 131), dem Produkt aus Methode A (589 mg/5,21 mmol) und 3 ml tert-Butanol wurde in einem verschlossen Rohr 24 Stunden lang bei 100°C gerührt. Das Reaktionsgemisch wurde in Wasser gegossen, mit 1 N Salzsäure auf pH 1 angesäuert, zweimal mit Diethylether (Ether) gewaschen und mit 1 N Natriumhydroxid (NaOH) auf pH 14 gebracht. Der resultierende Niederschlag wurde abfiltriert und im Vakuum getrocknet, wodurch 263 mg (88%) der Titelverbindung erhalten wurden, Schmelzpunkt 177–180°C. 1H NMR (400 MHz, CDCl3): δ 1,11–1,22 (m, 1H), 1,43–1,63 (m, 4H), 1,73 (br d, 1H, J = 13,3 Hz), 1,83–1,90 (m, 4H), 3,23 (s, 3H), 4,69 (br, 1H), 6,53 (d, 1H, J = 3,5 Hz), 7,03 (d, 1H, J = 3,5 Hz), 8,30 (s, 1H), 10,6 (br, 1H), LMRS: 231 (M + 1).
- Die Titelverbindungen von Beispielen 2 bis 51 wurden durch eine Methode hergestellt, die der in Beispiel 1 beschriebenen analog ist.
- BEISPIEL 2
- 9-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)-2,3,4,4a,9,9a-hexahydro-1H-carbazol
- BEISPIEL 3
- 4-(2,6-Dimethylmorpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 2,6-Dimethylmorpholin. LRMS: 233,3.
- BEISPIEL 4
- 4-Morpholin-4-yl-7H-pyrrolo[2,3-d]pyrimidin
- 4-Morpholin. LRMS: 205.
- BEISPIEL 5
- 4-(2,5-Dimethylpyrrolidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 2,5-Dimethylpyrrolidin. Schmelzpunkt: 227–229°C; LRMS: 216,3.
- BEISPIEL 6
- 4-(4-Benzylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 4-Benzylpiperidin. Schmelzpunkt: 188–190°C; LRMS: 292,4.
- BEISPIEL 7
- 4-Phenyl-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-ol
- 4-Phenylpiperidin-4-ol. Schmelzpunkt: 201–202°C; LRMS: 294,4.
- BEISPIEL 8
- 1-[1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-yl]-1,3-dihydro-benzoimidazol-2-on
- Piperidin-4-yl-1,3-dihydrobenzimidazol. Schmelzpunkt: 182–184°C; LRMS: 334,4.
- BEISPIEL 9
- 1-Phenyl-8-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1,3,8-triazaspiro[4,5]decan-4-on
- 1-Phenyl-1,3,8-triazaspiro[4,5]decan-4-on. Schmelzpunkt: 232–234°C.
- BEISPIEL 10
- 4-(3-Methylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 3-Methylpiperidin. Schmelzpunkt: 176–178°C; LRMS: 217,1.
- BEISPIEL 11
- 4-(3,5-Dimethylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 3,5-Dimethylpiperidin. Schmelzpunkt: 258–260°C; LRMS: 231.
- BEISPIEL 12
- 4-(2-Methylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 2-Methylpiperidin. Schmelzpunkt: 144–146°C; LRMS: 217,1.
- BEISPIEL 13
- 4-(2-Ethylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 2-Ethylpiperidin. Schmelzpunkt: 112–114°C; LRMS: 231.
- BEISPIEL 14
- [1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-2-yl]methanol
- Piperidin-2-ylmethanol. Schmelzpunkt: 135–136°C; LRMS: 232,9.
- BEISPIEL 15
- 1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-carbonsäurediethylamid
- Piperidin-3-carbonsäurediethylamid. LRMS: 302,1.
- BEISPIEL 16
- 2-[1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-2-yl]ethanol
- Piperidin-2-ylethanol. Schmelzpunkt: 139–140°C.
- BEISPIEL 17
- 4-Azocan-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Azapan. Schmelzpunkt: 225–226°C; LRMS: 231,3.
- BEISPIEL 18
- 1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-carbonsäure amid
- Piperidin-3-carbonsäureamid. Schmelzpunkt: 283–285°C.
- BEISPIEL 19
- Dimethyl-[1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-3-yl]amin
- Dimethylpyrrolidin-3-ylamin. Schmelzpunkt: 210–212°C; LRMS: 232,2.
- BEISPIEL 20
- N-Ethyl-N-[1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-3-yl]acetamid
- N-Ethylpyrrolidin-3-ylacetamid. Schmelzpunkt: 197–199°C; LRMS: 274,3.
- BEISPIEL 21
- 4-(2-Methoxymethylpyrrolidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 2-Methoxymethylpyrrolidin. Schmelzpunkt: 134–135°C; LRMS: 233,2.
- BEISPIEL 22
- [1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-2-yl]methanol
- Pyrrolidin-2-ylmethanol. Schmelzpunkt: 188–189°C; LRMS: 219,3.
- BEISPIEL 23
- N-[1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-3-yl]acetamid
- Pyrrolidin-3-ylacetamid. Schmelzpunkt: 260–261°C; LRMS: 246,3.
- BEISPIEL 24
- 4-(2-Propylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- Propylpiperidin. Schmelzpunkt: 106–107°C; LRMS: 245,3.
- BEISPIEL 25
- 4-(4-Methylpiperazin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 4-Methylpiperazin. Schmelzpunkt: 141–142°C.
- BEISPIEL 26
- 4-Piperazin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Piperazin. Schmelzpunkt: 164–166°C.
- BEISPIEL 27
- 4-Azepan-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Azapan. Schmelzpunkt: 210°C; LRMS: 217,3.
- BEISPIEL 28
- 1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-3-ol
- Pyrrolidin-3-ol. Schmelzpunkt: 220–225°C; LRMS: 205,2.
- BEISPIEL 29
- [1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-yl]methanol
- Piperidin-3-ylmethanol. Schmelzpunkt: 161,5–163,5°C; LRMS: 234,3.
- BEISPIEL 30
- 1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-carbonsäureethylester
- Piperidin-4-carbonsäureethylester. Schmelzpunkt: 139–141°C; LRMS: 275,3.
- BEISPIEL 31
- 1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-carbonsäureethylester
- Piperidin-3-carbonsäureethylester. Schmelzpunkt: 139,5–141,5°C; LRMS: 275,3.
- BEISPIEL 32
- 2-[1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-yl]ethanol
- Piperidin-4-ylethanol. Schmelzpunkt: 129–131°C; LRMS: 265,3.
- BEISPIEL 33
- 4-(4-Phenylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 4-Phenylpiperidin. Schmelzpunkt: 195°C; LRMS: 279.
- BEISPIEL 34
- 4-(4-Trifluormethylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 4-Trifluormethylpiperidin. Schmelzpunkt: 198°C; LRMS: 271.
- BEISPIEL 35
- 4-[4-(3-Phenylpropyl)piperidin-1-yl]-7H-pyrrolo[2,3-d]pyrimidin
- 4-(3-Phenylpropyl)piperidin. Schmelzpunkt: 134°C; LRMS: 321.
- BEISPIEL 36
- 4-(3,3-Dimethylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 3,3-Dimethylpiperidin. Schmelzpunkt: 204°C; LRMS: 231.
- BEISPIEL 37
- 1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-carbonsäure
- Piperidin-3-carbonsäure. Schmelzpunkt: 159–160°C; LRMS: 307,3.
- BEISPIEL 38
- 1-Methyl-10-oxa-4-azatricyclo[5.2.1.0%2,6&]decan
- 1-Methyl-4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-10-oxa-4-azatricyclo[5.2.1]decan. Schmelzpunkt: 251–252°C; LRMS: 271,3.
- BEISPIEL 39
- 1-(5-Chlor-7H-pyrrolo[2,3-d]pyrimidin-4-yl)decahydrochinolin
- Decahydrochinolin. Schmelzpunkt: 190–192°C; LRMS: 291,8.
- BEISPIEL 40
- 3-[1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-yl]propionsäureethylester
- Piperidin-3-ylpropionsäureethylester. Schmelzpunkt: 101–103°C; LRMS: 303,4.
- BEISPIEL 41
- 3-[1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-yl]propionsäure
- Piperidin-3-ylpropionsäure. Schmelzpunkt: 217–219°C; LRMS: 275,3.
- BEISPIEL 42
- 1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-ol
- Piperidin-3-ol. Schmelzpunkt: 152–154°C; LRMS: 219,3.
- BEISPIEL 43
- 3-[1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-yl]propionamid
- Piperidin-3-ylpropionamid. Schmelzpunkt: 212–214°C; LRMS: 274,3.
- BEISPIEL 44
- 4-(2,6-Dimethylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- 2,6-Dimethylpiperidin. LRMS: 231.
- BEISPIEL 45
- 2-[1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3-yl]propan-2-ol
- Piperidin-3-ylpropan-2-ol: Schmelzpunkt: 182,8–183,6°C; LMRS: 261.
- BEISPIEL 46
- 2-[1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-yl]propan-2-ol
- Piperidin-4-ylpropan-2-ol. Schmelzpunkt: 170,1–171,3°C; LRMS: 261.
- BEISPIEL 47
- 4-Methyl-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-ol
- 4-Methylpiperidin-4-ol. Schmelzpunkt: 163,8–165,1°C; LRMS: 233,1.
- BEISPIEL 48
- 3-Methyl-8-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-8-azabicyclo[3.2.1]octan-3-ol
- 3-Methyl-8-azabicyclo[3.2.1]octan-3-ol. Schmelzpunkt: 142,1–143,8°C; LRMS: 259,1.
- BEISPIEL 49
- 2-[1-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-2-yl]propan-2-ol
- Pyrrolidin-2-ylpropan-2-ol. Schmelzpunkt: 173°C (Zers.); LRMS: 247,1.
- BEISPIEL 50
- 3-Methyl-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrrolidin-3-ol
- 3-Methylpyrrolidin-3-ol. LRMS: 219.
- BEISPIEL 51
- 4-Pyrazol-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Pyrazol. LRMS: 186,2.
- BEISPIEL 52
- Cyclohexylmethyl-(6-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl) amin
- Cyclohexylmethylamin.
- METHODE B
- 7-Benzolsulfonyl-4-chlor-7H-pyrrolo[2,3-d]pyrimidin
- In einem mit einer Flamme getrockneten Kolben wurden unter Stickstoff 780 mg 60%iges Natriumhydrid (19,5 mmol) in Mineralöl 30 ml Dimethylformamid (DMF) zugesetzt und das resultierende Gemisch auf 0°C abgekühlt. Eine Lösung von 2,0 g (13,0 mmol) 4-Chlor-7H-pyrrolo[2,3-d]pyrimidin in 10 ml DMF wurde langsam über einen Zeitraum von 5 Minuten zugesetzt. Das Reaktionsgemisch wurde 10 Minuten lang gerührt, wonach die Wasserstoffbildung (H2) aufhörte. Benzolsulfonylchlorid (1,7 ml/13,0 mmol) wurde zugesetzt, das Reaktionsgemisch auf Raumtemperatur erwärmt und eine Stunde lang gerührt. Wasser wurde zugesetzt, der resultierende Niederschlag abfiltriert und im Vakuum getrocknet, wodurch 3,4 g (89%) der Titelverbindung als kristalliner Feststoff erhalten wurden, Schmelzpunkt 163–167°C.
- METHODE C
- 7-Benzolsulfonyl-4-chlor-6-phenyl-7H-pyrrolo[2,3-d]pyrimidin
- In einem mit einer Flamme getrockneten Kolben wurden unter Stickstoff 0,53 ml (3,79 mmol) Diisopropylamin in 5 ml Tetrahydrofuran (THF) aufgelöst und die Lösung auf –78°C abgekühlt. n-Butyllithium (3,75 mmol als eine 2,5 M-Lösung in Hexan) wurde zugegeben und das resultierende Gemisch auf 0°C unter kontinuierlichem Rühren über einen Zeitraum von 10 Minuten abgekühlt. Das Reaktionsgemisch wurde wiederum auf –78°C abgekühlt und zu diesem Gemisch wurde eine Lösung von 1,0 g (3,40 mmol) des Produkts von Methode B in 10 ml THF über einen Zeitraum von 10 Minuten zugesetzt. Das Reaktionsgemisch wurde eine Stunde lang bei –78°C gerührt, wonach 8,2 ml (4,10 mmol) einer 0,5 M-Lösung Zinkchlorid in THF zugesetzt wurden. Anschließend wurde das Reaktionsgemisch auf Raumtemperatur erwärmt und eine Stunde lang gerührt. Iodbenzol (0,46 ml/4,11 mmol) und eine Suspension von 197 mg Tetrakis(triphenylphosphin)palladium in 2 ml THF wurden zugesetzt. Das resultierende Gemisch wurde unter Rückfluss drei Stunden lang gerührt, auf Raumtemperatur abgekühlt und zwischen Dichlormethan und Wasser verteilt. Die wässrige Phase wurde mit 1 N HCl angesäuert und zweimal mit Dichlormethan extrahiert. Die Dichlormethanphasen wurden vereinigt mit 1 N HCl und Kochsalzlösung gewaschen, über Magnesiumsulfat (MgSO4) getrocknet, filtriert und im Vakuum konzentriert, wodurch die Titelverbindung erhalten wurde. LRMS: 370, 372 (M + 2).
- METHODE D
- 4-Chlor-6-phenyl-7H-pyrrolo[2,3-d]pyrimidin
- Das Produkt von Methode C wurde in 10 ml THF aufgelöst und diese Lösung wurde mit 5,0 ml Methanol und 1,0 g NaOH versetzt. Das Reaktionsgemisch wurde 15 Minuten lang gerührt, im Vakuum konzentriert und zwischen einer gesättigten wässrigen Ammoniumchlorid-(NH4Cl)-Lösung und Ethylacetat verteilt. Die resultierende wässrige Schicht wurde zweimal mit Ethylacetat extrahiert. Die Ethylacetatschichten wurden vereinigt, mit Kochsalzlösung gewaschen, über MgSO4 getrocknet, filtriert und im Vakuum konzentriert. Das Rohprodukt wurde mittels Kieselgelchromatographie (1 : 5 Ethylacetat/Hexan) gereinigt, wodurch 0,59 g (76%) der Titelverbindung als blassgelber Feststoff erhalten wurden, Schmelzpunkt: 145°C (Zers.). LRMS: 230, 232 (M + 2).
- METHODE E
- Cyclohexylmethyl-(6-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)amin
- Das Produkt von Methode D (50 mg/0,218 mmol) wurde mit 0,12 ml N-Methylcyclohexylamin (0,920 mmol) wie in Methode beschrieben umgesetzt. Das Reaktionsgemisch wurde im Vakuum konzentriert, Methanol wurde zugesetzt und der resultierende Niederschlag filtriert, wodurch 7 mg (10%) der Titelverbindung als gelber Feststoff erhalten wurden. 1H NMR (400 MHz, CDCl3) δ 1,18–1,25 (m, 1H), 1,47–1,66 (m, 4H), 1,75–1,90 (m, 5H), 3,30 (s, 3H), 4,74 (br, 1H), 6,79 (s, 1H), 7,32–7,36 (m, 1H), 7,47–7,51 (m, 2H), 7,77 (d, 2H, J = 7,9 Hz), 8,33 (s, 1H). LMRS: 307 (M + 1).
- Die Titelverbindungen der Beispiele 53–58 wurden nach einer Methode hergestellt, die der in Beispiel 52 beschriebenen analog ist.
- BEISPIEL 53
- 1-(6-Phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)decahydrochinolin
- Decahydrochinolin. LRMS: 333,4.
- BEISPIEL 54
- 4-(2-Ethylpiperidin-1-yl)-6-phenyl-7H-pyrrolo[2,3-d]pyrimidin
- 2-Ethylpiperidin. LRMS: 307,4.
- BEISPIEL 55
- 4-(3,3-Dimethylpiperidin-1-yl)-6-phenyl-78-pyrrolo[2,3-d]pyrimidin
- 3,3-Dimethylpiperidin. LRMS: 307,4.
- BEISPIEL 56
- 6-Phenyl-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 279,4.
- BEISPIEL 57
- 4-Piperidin-1-yl-6-thiophen-3-yl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 285,4.
- BEISPIEL 58
- 4-Piperidin-1-yl-6-thiophen-2-yl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 285,4.
- BEISPIEL 59
- Cyclohexylmethyl-(6-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)amin
- Cyclohexylmethylamin.
- METHODE F
- 7-Benzolsulfonyl-4-chlor-6-methyl-7H-pyrrolo[2,3-d]pyrimidin
- In einem mit einer Flamme getrockneten Kolben wurden unter N2 0,57 ml (4,07 mmol) Diisopropylamin und 5,0 ml trockenes THF vorgelegt. Die Lösung wurde auf –78°C abgekühlt und 1,63 ml (4,08 mmol) einer 2,5 M-Lösung von n-Butyllithium in Hexan zugesetzt. Das resultierende Gemisch wurde auf 0°C erwärmt und 10 Minuten lang gerührt. Nach Abkühlen des Gemischs auf nochmals –78°C wurde eine Lösung von 1,0 g (3,40 mmol) des Rohprodukts von Methode C in 10 ml trockenem THF über einen Zeitraum von 10 Minuten zugegeben. Das resultierende Gemisch wurde eine Stunde lang gerührt und danach wurden 0,28 ml (4,50 mmol) Iodmethan zugegeben. Das Reaktionsgemisch wurde zwei Stunden lang gerührt, mit einer gesättigten NH4Cl-Lösung gequencht und auf Raumtemperatur erwärmt. Das Gemisch wurde 5 Minuten lang gerührt, mit Wasser verdünnt und dreimal mit Ethylacetat extrahiert. Die vereinigten Extrakte wurden mit Kochsalzlösung gewaschen, über MgSO4 getrocknet, filtriert und im Vakuum eingedampft, wodurch die Titelverbindung erhalten wurde. LRMS: 308, 310 (M + 2).
- METHODE G
- 4-Chlor-6-methyl-7H-pyrrolo[2,3-d]pyrimidin
- Das Produkt von Methode F wurde wie in Methode E beschrieben entschützt. Das Rohprodukt wurde durch Verreibung mit Hexan und Dichlormethan gereinigt, wodurch 250 mg (44%) der Titelverbindung als gelber Feststoff erhalten wurden. Schmelzpunkt: 205°C (Zers.), LRMS 168, 170 (M + 2).
- METHODE H
- Cyclohexylmethyl-(6-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)amin
- Das Produkt von Methode G (50 mg/0,298 mmol) wurde mit 100 mg (0,883 mmol) N-Methylcyclohexylamin wie in Methode A beschrieben umgesetzt. Das Reaktionsgemisch wurde wie in Methode A aufgearbeitet mit der Ausnahme, dass Ethylacetat anstelle von Ether verwendet wurde. Die Titelverbindung (42 mg, 58% Ausbeute) wurde als weißer Feststoff erhalten. Schmelzpunkt 221°C (Zers.). 1H NMR (400 MHz, CDCl3) δ: 1,15–1,25 (m, 1H), 1,43–1,62 (m, 4H), 1,73 (br s, 1H, J = 13,7 Hz), 1,82–1,90 (m, 4H), 2,41 (d, 3H, J = 0,8 Hz), 3,21 (s, 3H) 4,63 (br s, 1H), 6,20 (s, 1H), 8,22 (s, 1H), 10,1 (br s, 1H). LRMS: 245 (M + 1).
- Die Titelverbindung von Beispiel 60 wurde durch eine Methode hergestellt, die der in Beispiel 59 beschriebenen analog ist.
- BEISPIEL 60
- 6-Methyl-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 217,3.
- BEISPIEL 61
- 5-Chlor-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- METHODE I
- 4,5-Dichlor-7H-pyrrolo[2,3-d]pyrimidin
- 4-Chlor-7H-pyrrolo[2,3-d]pyrimidin (154 mg, 1,0 mmol) wurden in 6,0 ml trockenem Dichlormethan in einem mit einer Flamme getrockneten Kolben suspendiert und diesem Gemisch wurde N-Chlorsuccinimid (147 mg, 1,1 mmol) in einer Portion zugegeben. Das resultierende Gemisch wurde bei Raumtemperatur 18 Stunden lang gerührt, danach wurde das Lösungsmittel unter reduziertem Druck abgezogen. Der Rückstand wurde mit Wasser verrieben und durch Filtration isoliert, wodurch 137 mg (72%) der Titelverbindung als grauer Feststoff erhalten wurde, Schmelzpunkt: 224–227°C (dec). LRMS: 188 (M + 1).
- METHODE J
- 5-Chlor-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Das Produkt von Methode I (57 mg, 0,3 mmol) wurde in 3,0 ml tert-Butanol suspendiert und diese Lösung wurde mit Piperidin (90 μl, 0,9 mmol) versetzt und das resultierende System eine Stunde lang refluxiert. Das Reaktionsgemisch wurde auf Raumtemperatur abgekühlt und mit Wasser (4,0 ml) versetzt. Die Lösung wurde mit 1 N HCl auf pH 1 eingestellt und anschließend mit Ether gewaschen. Die wässrige Phase wurde abgetrennt und mit 2 N NaOH auf pH 12 eingestellt. Anschließend wurde die Lösung mit 2 × 15 ml Dichlormethan extrahiert und die vereinigten organischen Phasen mit Wasser und anschließend Kochsalzlösung gewaschen und über MgSO4 getrocknet. Das Verdampfen des Lösungsmittels lieferte 45 mg eines gelben Feststoffs, der durch Kieselgelchromatographie (3 : 1 Ethylacetat/Hexan) gereinigt wurde, wodurch 23 mg (32%) der Titelverbindung als schwachgelber Feststoff erhalten wurden. Schmelzpunkt 170–172°C. 1H NMR (400 MHz, CDCl3) δ 1,67–1,74 (m, 6H), 3,65–3,67 (m, 4H), 7,10 (s, 1H), 8,31 (s, 1H). LRMS: 237 (M + 1).
- Die Titelverbindungen der Beispiele 62–63 wurden durch eine Methode hergestellt, die der in Beispiel 61 beschriebenen analog ist.
- BEISPIEL 62
- 5-Chlor-4-(octahydroindol-1-yl)-7H-pyrrolo[2,3-d]pyrimidin
- Octahydroindol. Schmelzpunkt: 193°C; LRMS: 277,8.
- BEISPIEL 63
- 1-(5-Chlor-7H-pyrrolo[2,3-d]pyrimidin-4-yl)decahydrochinolin
- Decahydrochinolin. Schmelzpunkt: 190–192°C; LRMS: 291,8.
- BEISPIEL 64
- 5-Phenyl-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- METHODE K
- 5-Brom-4-chlor-7H-pyrrolo[2,3-d]pyrimidin
- Zu einer gerührten Lösung von 4-Chlor-7H-pyrrolo[2,3-d]pyrimidin (30 g/0,02 mol), aufgelöst in 75 ml Chloroform, wurden 3,5 g (0,02 mol) N-Bromsuccinamid zugesetzt und das resultierende Gemisch eine Stunde lang unter Rückfluss erhitzt. Nach Abkühlen auf Raumtemperatur wurde der Niederschlag durch Filtration abgetrennt und unter reduziertem Druck getrocknet, wodurch 4,1 g (89%) der Titelverbindung er halten wurden. 1H NMR (400 MHz) (CDCl3) δ: 7,93 (d, 1H, J = 2,8 Hz). 8,60 (s, 1H).
- METHODE L
- 7-Benzolsulfonyl-5-brom-4-chlor-7H-pyrrolo[2,3-d]pyrimidin
- Zu einer Aufschlämmung des Produkts von Methode K (4,1 g/0,018 mol) in DMF (15 ml), das auf 0°C abgekühlt worden war, wurden 1,0 g (0,025 mol) 60% Natriumhydrid in Mineralöl gegeben und das resultierende Gemisch 15 Minuten lang bei 0°C gerührt. Benzolsulfonylchlorid (3,2 g/0,018 mol) wurde zugegeben, das Reaktionsgemisch auf Raumtemperatur erwärmt und zwei Stunden lang gerührt. Anschließend wurde Wasser (15 ml) zugesetzt und der resultierende Feststoff durch Filtration abgetrennt und anschließend im Vakuum getrocknet, wodurch 5,9 g (89%) der Titelverbindung erhalten wurden.
- METHODE M
- 7-Benzolsulfonyl-5-brom-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Ein Gemisch aus 2,0 g (5,37 mmol) des Produkts von Methode L und 1,1 g (13,4 mmol) Pipieridin in 10 ml tert-Butanol wurde zwei Stunden lang unter Rühren bei 60°C gehalten. Nach Abkühlen auf Raumtemperatur wurde das Reaktionsgemisch zwischen Dichlormethan (25 ml) und Wasser (25 ml) verteilt. Die Dichlormethan-Phase wurde über Natriumsulfat (Na2SO4) getrocknet und im Vakuum zur Trockne konzentriert, wodurch 2,2 g (97%) der Titelverbindung erhalten wurden. 1H NMR (400 MHz) (CDCl3) δ: 1,63–1,72 (m, 6H), 3,54–3,57 (m, 4H), 7,53 (t, 2H, J = 2,0 Hz), 7,60 (s, 1H), 7,61 (t, 1H, J = 2,0 Hz), 8,17–8,20 (m, 2H), 8,43 (s, 1H). LRMS: 422,7, 420,7 (M + 1).
- METHODE N
- 5-Phenyl-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Zu einer gerührten Lösung des Produkts von Methode M (100 mg/ 0,237 mmol) in 1,0 ml Dioxan wurden 32 mg (0,261 mmol) Phenylboronsäure und 75 mg (0,356 mmol) von dreibasischem Kaliumphosphat gegeben und danach 7 mg (0,006 mmol) Tetrakis(triphenylphosphin)palladium. Das resultierende Gemisch wurde mit Stickstoff entgast und 48 Stunden bei 100°C gerührt. Nach Abkühlen auf Raumtemperatur wurden 1,0 ml Methanol zugesetzt und anschließend 50 mg NaOH und das neue Gemisch eine Stunde lang bei Raumtemperatur gerührt. Das resultierende Gemisch wurde anschließend zwischen Dichlormethan und Wasser verteilt, die Dichlormethan-Phase über MgSO4 getrocknet und im Vakuum zur Trockne konzentriert. Das Rohprodukt wurde mittels Kieselgelchromatographie (2 : 1 Ethylacetat/Hexan) gereinigt, wodurch 13 mg (20%) der Titelverbindung erhalten wurden. 1H NMR (400 MHz) (CDCl3) δ: 1,33–1,34 (m, 4H), 1,43–1,44 (m, 2H), 3,26–3,28 (m, 4H), 7,12 (s, 1H), 7,27 (t, 1H, J = 7,2 Hz), 7,38 (t, 2H, J = 8,0 Hz), 7,45 (d, 2H, J = 0,8 Hz), 8,42 (s, 1H). LRMS: 279,2 (M + 1).
- Die Titelverbindungen der Beispiele 65–77 wurden durch eine Methode hergestellt, die der in Beispiel 64 beschriebenen analog ist.
- BEISPIEL 65
- 5-(3-Chlor-4-fluorphenyl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 331,8.
- BEISPIEL 66
- 5-(4-Fluorphenyl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 297.
- BEISPIEL 67
- 5-(4-Chlorphenyl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 313.
- BEISPIEL 68
- 5-(3,5-Bistrifluormethylphenyl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 415,4.
- BEISPIEL 69
- 4-Piperidin-1-yl-5-o-tolyl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 293,4.
- BEISPIEL 70
- 4-Piperidin-1-yl-5-p-tolyl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 293,4.
- BEISPIEL 71
- 5-(4-Methoxyphenyl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 309,4.
- BEISPIEL 72
- 4-Piperidin-1-yl-5-(3-trifluormethylphenyl)-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 347,4.
- BEISPIEL 73
- 5-(3-Chlorphenyl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Piperidin. LRMS: 427,8.
- BEISPIEL 74
- 3-(4-Piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin-5-yl)benzoesäureethylester
- Piperidin. LRMS: 465,4.
- BEISPIEL 75
- 2-[3-(4-Piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl]propan-2-ol
- Piperidin. LRMS: 451,4.
- BEISPIEL 76
- 4-(2-Methylpiperidin-1-yl)-5-m-tolyl-7H-pyrrolo[2,3-d]pyrimidin
- 2-Methylpiperidin. LRMS: 307,2.
- BEISPIEL 77
- 4-Azepan-1-yl-5-m-tolyl-7H-pyrrolo[2,3-d]pyrimidin
- Azepan. LRMS: 307,2.
- BEISPIEL 78
- METHODE O
- 4-Piperidin-1-yl-78-pyrrolo[2,3-d]pyrimidin-5-carbonitril
- Zu einer gerührten Lösung von 4-Chlor-7H-pyrrolo[2,3-d]pyrimidin-5-carbonitril (54 mg/0,3 mmol) (hergestellt durch die Methode von Townsend, et al., J. Am. Chem. Soc., 1969, 91, 2102), das in 3,0 ml tert-Butanol suspendiert war, wurde Piperidin (59 μl/0,60 mmol) zugesetzt. Das resultierende Gemisch wurde anschließend 2,5 Stunden lang unter Rückfluss gekocht und nach Abkühlen auf Raumtemperatur in einen Scheidetrichter überführt und mit Ether (20 ml) verdünnt. Die Lösung wurde mit 2 × 10 ml 1 N HCl extrahiert, die vereinigten wässrigen Schichten auf pH 7 mit 2 N Kaliumhydroxid-(KOH)-Lösung eingestellt, wodurch ein Niederschlag gebildet wurde, welcher durch Filtration gesammelt, mit Wasser gewaschen und unter reduziertem Druck getrocknet wurde, wodurch 29 mg (42%) der Titelverbindung als farbloser Feststoff erhalten wurden. Schmelzpunkt 209–211°C; 1H NMR (400 MHz) (Aceton-d6) δ: 1,72–1,74 (m, 6H), 3,72–3,79 (m, 4H), 8,12 (s, 1H), 8,29 (s, 1H). LRMS: 228 (M + 1).
- BEISPIEL 79
- 5-Ethinyl-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- METHODE P
- 4-Chlor-5-iod-7H-pyrrolo[2,3-d]pyrimidin
- Zu einer gerührten Lösung von 4-Chlor-7H-pyrrolo[2,3-d]pyrimidin (30 g/0,02 mol), aufgelöst in 80 ml Chloroform, wurden 4,5 g (0,02 mol) N-Iodsuccinimid gegeben und das resultierende Gemisch eine Stunde lang unter Rückfluss erhitzt. Nach Abkühlen auf Raumtemperatur wurde der Niederschlag durch Filtration abgetrennt und unter reduziertem Druck getrocknet, wodurch 4,6 g (82%) der Titelverbindung erhalten wurden.
- METHODE Q
- 7-Benzolsulfonyl-4-chlor-5-iod-7H-pyrrolo[2,3-d]pyrimidin
- Die Titelverbindung wurde wie vorher in Methode L beschrieben hergestellt, indem das Produkt von Methode O verwendet wurde, wodurch 5,4 g (80%) des Materials erhalten wurden. LRMS: 419,6 (M + 1), 279,7.
- METHODE R
- 7-Benzolsulfonyl-5-iod-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Die Titelverbindung wurde durch das in Methode M beschriebene Verfahren hergestellt, wobei das Produkt von Methode O verwendet wurde, um die Titelverbindung herzustellen. LRMS: 469 (M + 1), 329,1.
- METHODE S
- 7-Benzolsulfonyl-4-piperidin-1-yl-5-triethylsilanylethinyl-7H-pyrrolo[2,3-d]pyrimidin
- In einem mit einer Flamme getrockneten Kolben wurden unter Stickstoff 211 mg (0,5 mmol) des Produkts aus Methode R, 19 mg (0,1 mmol) Kupfer(I)iodid und 58 mg (0,05 mmol) Tetrakis(triphenylphosphin)palladium vorgelegt. Dieses Gemisch wurde anschließend mit 0,14 ml (1,0 mmol) Triethylamin und 0,27 ml (1,5 mmol) Triethylsilylacetylen als Lösung in 1,5 ml trockenem DMF versetzt. Das resultierende Gemisch wurde drei Stunden lang bei Raumtemperatur gerührt und danach wurden 5,0 ml Wasser zugesetzt und das Gemisch mit Ethylacetat extrahiert. Der Ethylacetatextrakt wurde über MgSO4 getrocknet und im Vakuum konzentriert. Das resultierende Rohprodukt wurde anschließend durch Kieselgelchromatographie (7 : 1 Hexan/Ethylacetat) gereinigt, wodurch 194 mg (89%) der Titelverbindung erhalten wurden. LRMS: 481 (M + 1), 341.
- METHODE T
- 5-Ethinyl-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin
- Eine gerührte Lösung des Produkts von Methode S (194 mg/0,40 mmol) aufgelöst in 2,0 ml trockenem THF wurde tropfenweise mit 0,4 ml (0,4 mmol) einer 1 M-Lösung Tetrabutylammoniumfluorid in THF versetzt. Das resultierende Gemisch wurde bei Raumtemperatur 10 Minuten lang gerührt, anschließend in eine methanolische Lösung (3,0 ml), die 1 g KOH enthielt, transferiert, das neue Gemisch 15 Minuten lang bei Raumtemperatur gerührt und im Vakuum konzentriert. Der Rückstand wurde zwischen Wasser und Ethylacetat verteilt, die Ethylacetatschicht mit Wasser und Kochsalzlösung gewaschen, über MgSO4 getrocknet und im Vakuum bis zur Trockne konzentriert. Das Rohprodukt wurde mittels Kieselgelchromatographie (2 : 1 Ethylacetat/Hexan) gereinigt, wodurch 72 mg (64%) der Titelverbindung als weißer kristalliner Feststoff erhalten wurden. Schmelzpunkt 179–181°C. 1H NMR (400 MHz) (CDCl3) δ: 1,72 (br s, 6H), 3,20 (s, 1H), 3,82–3,83 (m, 4H), 7,47 (s, 1H), 8,35 (s, 1H). LRMS: 227 (M + 1).
Claims (11)
- Verbindung der Formeloder das pharmazeutisch annehmbare Salz davon, wobei R1 eine Gruppe der Formel
 ist, worin die gestrichelte Linie fakultative Doppelbindungen darstellt; m 0, 1, 2 oder 3 ist; n 0, 1, 2 oder 3 ist; X, B und D jeweils unabhängig voneinander Sauerstoff, S(O)d, worin d 0,1 oder 2 ist, NR6 oder CR7R8 sind; A und E jeweils CR7R8 sind; und R6 ausgewählt ist aus der Gruppe, die besteht aus Wasserstoff, (C1-C6)Alkyl, Trifluormethyl, Trifluormethyl(C1-C6)alkyl, (C1-C6)Alkyl(difluormethylen), (C1-C3)Alkyl(difluormethylen)(C1-C3)alkyl, (C1-C6)Alkoxy(C1-C6)acyl, (C1-C6)Alkylamino(C1-C6)acyl, ((C1-C6)Alkyl)2amino(C1-C6) acyl), (C6-C10)Aryl, (C5-C9)Heteroaryl, (C6-C10)Aryl(C1-C6)alkyl, (C5-C9)Heteroaryl(C1-C6)alkyl, (C6-C10)Aryl(C6-C10)aryl, (C6-C10)Aryl(C6-C10)aryl(C1-C6)alkyl, (C3-C6)Cycloalkyl, (C3-C6)Cycloalkyl(C1-C6)alkyl, Hydroxy(C2-C6)alkyl, (C1-C6)-Acyloxy(C2-C6)alkyl, (C1-C6)Alkoxy(C2-C6)alkyl, Piperazinyl-(C1-C6)alkyl, (C1-C6)Acylamino(C1-C6)alkyl, (C6-C10)Aryl-(C1-C6)alkoxy(C1-C6)alkyl, (C5-C9)Heteroaryl(C1-C6)alkoxy(C1-C6)-alkyl, (C1-C6)Alkylthio(C1-C6)alkyl, (C6-C10)Arylthio(C1-C6)alkyl, (C1-C6)Alkylsulfinyl(C1-C6)alkyl, (C6-C10)Arylsulfinyl-(C1-C6)alkyl, (C1-C6)Alkylsulfonyl(C1-C6)alkyl, (C6-C10)Arylsulfonyl(C1-C6)alkyl, Amino(C1-C6)alkyl, (C1-C6)Alkylamino-(C1-C6)alkyl, ((C1-C6)Alkyl)2amino(C1-C6)alkyl, R13CO(C1-C6)-Alkyl, worin R13 für R20O oder R20R21N steht, worin R20 und R21 j eweils unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, (C1-C6)Alkyl, (C6-C10)Aryl(C1-C6)alkyl oder (C5-C9)Heteroaryl (C1-C6)alkyl; und R14(C2-C6)Alkyl, worin R14 für (C1-C6)Acylpiperazino, (C6-C10)Arylpiperazino, (C5-C9)Heteroarylpiperazino, (C1-C6)Alkylipiperazino, (C6-C10)Aryl(C1-C6)alkylpiperazino, (C5-C9)Heteroaryl(C1-C6)-alkylpiperazino, Morpholino, Thiomorpholino, Piperidino, Pyrrolidino, Piperidyl, (C1-C5)Alkylpiperidyl, (C6-C10)-Arylpiperidyl, (C5-C9)Heteroarylpiperidyl, (C6-C10)Aryl-(C1-C6)alkylpiperidyl, (C5-C9)Heteroaryl(C1-C6)alkylpiperidyl, (C1-C6)Alkoxyacyl, (C1-C5)Alkylaminoaryl, ((C1-C6)-Alkyl)2aminoacyl oder (C1-C6)Acylpiperidyl steht; R7 und R8 jeweils unabhängig ausgewählt sind aus der Gruppe, bestehend aus Wasserstoff, Deuterium, (C1-C6)Alkyl, Amino, Hydroxy, (C1-C6)Alkoxy, (C1-C6)Alkylamino, ((C1-C6)-Alkyl)amino, (C1-C6)Acylamino, (C1-C6)Acyl(C1-C6)alkylamino, Carboxy, (C1-C6)Alkoxyacyl, (C1-C6)Alkylaminoacyl, ((C1-C6)-Alkyl)2aminoacyl, Aminoacyl, Trifluormethyl, Trifluormethyl(C1-C6)alkyl, (C1-C6)Alkyl(difluormethylen), (C1-C3)-Alkyl(difluormethylen)(C1-C3)alkyl, (C6-C10)Aryl, (C5-C9)-Heteroaryl, (C6-C10)Aryl(C1-C6)alkyl, (C5-C9)Heteroaryl-(C1-C6)alkyl, (C6-C10)Aryl(C6-C10)aryl, (C6-C10)Aryl(C6-C10)-aryl(C1-C6)alkyl, (C3-C6)Cycloalkyl, (C3- C6)Cycloalkyl-(C1-C6)alkyl, Hydroxy(C1-C6)alkyl, (C1-C6)Acyloxy(C1-C6)alkyl, Alkoxy(C1-C6)alkyl, (C1-C6)Acylamino-(C1-C6)alkyl, Piperidyl, (C1-C6)Alkylpiperidyl, (C6-C10)Aryl-(C1-C6)alkoxy(C1-C6)alkyl, (C5-C9) Heteroaryl (C1-C6)alkoxy-(C1-C6)alkyl, (C1-C6)Alkylthio(C1-C6)alkyl, (C6-C10)Arylthio-(C1-C6)alkyl, (C1-C6)Alkylsulfinyl(C1-C6)alkyl, (C6-C10)Aryl-sulfinyl(C1-C6)alkyl, (C1-C6)Alkylsulfonyl(C1-C6)alkyl, (C6-C10)Arylsulfonyl(C1-C6)alkyl, Amino(C1-C6)alkyl, (C1-C6)-Alkylamino(C1-C6)alkyl, ((C1-C6)Alkyl)2amino(C1-C6)alkyl, R13CO(C1-C6)Alkyl oder R13CO(C3-C10)Cyloalkyl, worin R13 für R20O oder R20R21N steht, worin R20 und R21 unabhängig voneinander aus der Gruppe, bestehend aus Wasserstoff, (C1-C6)-Alkyl, (C6-C10)Aryl(C1-C6)alkyl oder (C5-C9)Heteroaryl(C1-C6)-alkyl ausgewählt sind; R14, R14(C1-C6)Alkyl oder R14(C3-C10)-Cycloalkyl, worin R14 (C1-C6)Acylpiperazino, (C6-C10)Aryl-piperazino, (C5-C9)Heteroarylpiperazino, (C1-C6)Alkylpiperazino, (C6-C10)Aryl(C1-C6)alkylpi- perazino, (C5-C9)Heteroaryl(C1-C6)alkylpiperazino, Morpholino, Thiomorpholino, Piperidino, Pyrrolidino, Piperidyl, (C1-C6)Alkylpiperidyl, (C6-C10)Arylpiperidyl, (C5-C9)Heteroarylpiperidyl, (C6-C10)-Aryl(C1-C6)alkylpiperidyl, (C5-C9)Heteroaryl(C1-C6)alkyl-piperidyl oder (C1-C6)Acylpiperidyl ist; oder einer Gruppe der Formel
ist, worin die gestrichelte Linie fakultative Doppelbindungen darstellt; m 0, 1, 2 oder 3 ist; n 0, 1, 2 oder 3 ist; X, B und D jeweils unabhängig voneinander Sauerstoff, S(O)d, worin d 0,1 oder 2 ist, NR6 oder CR7R8 sind; A und E jeweils CR7R8 sind; und R6 ausgewählt ist aus der Gruppe, die besteht aus Wasserstoff, (C1-C6)Alkyl, Trifluormethyl, Trifluormethyl(C1-C6)alkyl, (C1-C6)Alkyl(difluormethylen), (C1-C3)Alkyl(difluormethylen)(C1-C3)alkyl, (C1-C6)Alkoxy(C1-C6)acyl, (C1-C6)Alkylamino(C1-C6)acyl, ((C1-C6)Alkyl)2amino(C1-C6) acyl), (C6-C10)Aryl, (C5-C9)Heteroaryl, (C6-C10)Aryl(C1-C6)alkyl, (C5-C9)Heteroaryl(C1-C6)alkyl, (C6-C10)Aryl(C6-C10)aryl, (C6-C10)Aryl(C6-C10)aryl(C1-C6)alkyl, (C3-C6)Cycloalkyl, (C3-C6)Cycloalkyl(C1-C6)alkyl, Hydroxy(C2-C6)alkyl, (C1-C6)-Acyloxy(C2-C6)alkyl, (C1-C6)Alkoxy(C2-C6)alkyl, Piperazinyl-(C1-C6)alkyl, (C1-C6)Acylamino(C1-C6)alkyl, (C6-C10)Aryl-(C1-C6)alkoxy(C1-C6)alkyl, (C5-C9)Heteroaryl(C1-C6)alkoxy(C1-C6)-alkyl, (C1-C6)Alkylthio(C1-C6)alkyl, (C6-C10)Arylthio(C1-C6)alkyl, (C1-C6)Alkylsulfinyl(C1-C6)alkyl, (C6-C10)Arylsulfinyl-(C1-C6)alkyl, (C1-C6)Alkylsulfonyl(C1-C6)alkyl, (C6-C10)Arylsulfonyl(C1-C6)alkyl, Amino(C1-C6)alkyl, (C1-C6)Alkylamino-(C1-C6)alkyl, ((C1-C6)Alkyl)2amino(C1-C6)alkyl, R13CO(C1-C6)-Alkyl, worin R13 für R20O oder R20R21N steht, worin R20 und R21 j eweils unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, (C1-C6)Alkyl, (C6-C10)Aryl(C1-C6)alkyl oder (C5-C9)Heteroaryl (C1-C6)alkyl; und R14(C2-C6)Alkyl, worin R14 für (C1-C6)Acylpiperazino, (C6-C10)Arylpiperazino, (C5-C9)Heteroarylpiperazino, (C1-C6)Alkylipiperazino, (C6-C10)Aryl(C1-C6)alkylpiperazino, (C5-C9)Heteroaryl(C1-C6)-alkylpiperazino, Morpholino, Thiomorpholino, Piperidino, Pyrrolidino, Piperidyl, (C1-C5)Alkylpiperidyl, (C6-C10)-Arylpiperidyl, (C5-C9)Heteroarylpiperidyl, (C6-C10)Aryl-(C1-C6)alkylpiperidyl, (C5-C9)Heteroaryl(C1-C6)alkylpiperidyl, (C1-C6)Alkoxyacyl, (C1-C5)Alkylaminoaryl, ((C1-C6)-Alkyl)2aminoacyl oder (C1-C6)Acylpiperidyl steht; R7 und R8 jeweils unabhängig ausgewählt sind aus der Gruppe, bestehend aus Wasserstoff, Deuterium, (C1-C6)Alkyl, Amino, Hydroxy, (C1-C6)Alkoxy, (C1-C6)Alkylamino, ((C1-C6)-Alkyl)amino, (C1-C6)Acylamino, (C1-C6)Acyl(C1-C6)alkylamino, Carboxy, (C1-C6)Alkoxyacyl, (C1-C6)Alkylaminoacyl, ((C1-C6)-Alkyl)2aminoacyl, Aminoacyl, Trifluormethyl, Trifluormethyl(C1-C6)alkyl, (C1-C6)Alkyl(difluormethylen), (C1-C3)-Alkyl(difluormethylen)(C1-C3)alkyl, (C6-C10)Aryl, (C5-C9)-Heteroaryl, (C6-C10)Aryl(C1-C6)alkyl, (C5-C9)Heteroaryl-(C1-C6)alkyl, (C6-C10)Aryl(C6-C10)aryl, (C6-C10)Aryl(C6-C10)-aryl(C1-C6)alkyl, (C3-C6)Cycloalkyl, (C3- C6)Cycloalkyl-(C1-C6)alkyl, Hydroxy(C1-C6)alkyl, (C1-C6)Acyloxy(C1-C6)alkyl, Alkoxy(C1-C6)alkyl, (C1-C6)Acylamino-(C1-C6)alkyl, Piperidyl, (C1-C6)Alkylpiperidyl, (C6-C10)Aryl-(C1-C6)alkoxy(C1-C6)alkyl, (C5-C9) Heteroaryl (C1-C6)alkoxy-(C1-C6)alkyl, (C1-C6)Alkylthio(C1-C6)alkyl, (C6-C10)Arylthio-(C1-C6)alkyl, (C1-C6)Alkylsulfinyl(C1-C6)alkyl, (C6-C10)Aryl-sulfinyl(C1-C6)alkyl, (C1-C6)Alkylsulfonyl(C1-C6)alkyl, (C6-C10)Arylsulfonyl(C1-C6)alkyl, Amino(C1-C6)alkyl, (C1-C6)-Alkylamino(C1-C6)alkyl, ((C1-C6)Alkyl)2amino(C1-C6)alkyl, R13CO(C1-C6)Alkyl oder R13CO(C3-C10)Cyloalkyl, worin R13 für R20O oder R20R21N steht, worin R20 und R21 unabhängig voneinander aus der Gruppe, bestehend aus Wasserstoff, (C1-C6)-Alkyl, (C6-C10)Aryl(C1-C6)alkyl oder (C5-C9)Heteroaryl(C1-C6)-alkyl ausgewählt sind; R14, R14(C1-C6)Alkyl oder R14(C3-C10)-Cycloalkyl, worin R14 (C1-C6)Acylpiperazino, (C6-C10)Aryl-piperazino, (C5-C9)Heteroarylpiperazino, (C1-C6)Alkylpiperazino, (C6-C10)Aryl(C1-C6)alkylpi- perazino, (C5-C9)Heteroaryl(C1-C6)alkylpiperazino, Morpholino, Thiomorpholino, Piperidino, Pyrrolidino, Piperidyl, (C1-C6)Alkylpiperidyl, (C6-C10)Arylpiperidyl, (C5-C9)Heteroarylpiperidyl, (C6-C10)-Aryl(C1-C6)alkylpiperidyl, (C5-C9)Heteroaryl(C1-C6)alkyl-piperidyl oder (C1-C6)Acylpiperidyl ist; oder einer Gruppe der Formel worin p 0, 1, 2 oder 3 ist; und Z Hydroxy, (C1-C6)Alkoxy oder NR1R2 ist, worin R1 und R2 jeweils unabhängig voneinander ausgewählt sind aus der Gruppe, bestehend aus Wasserstoff, (C1-C6)Alkyl, Piperidyl, (C1-C6)Alkylpiperidyl, (C6-C10)Arylpiperidyl, (C5-C9)Hetero-arylpiperidyl, (C6-C10)Aryl(C1-C6)alkylpiperidyl, (C5-C9)-Heteroaryl(C1-C6)alkylpiperidyl, (C1-C6)Acylpiperidyl, (C6-C10)Aryl, (C5-C9)Heteroaryl, (C6-C10)Aryl(C1-C6)alkyl, (C5-C9)Heteroaryl(C1-C6)alkyl, (C6-C10)Aryl(C6-C10)aryl, (C6-C10)Aryl(C6-C10)aryl(C1-C6)alkyl, (C3-C6)Cycloalkyl, (C3-C6)Cycloalkyl(C1-C5)alkyl, R5(C1-C6)Alkyl, (C1-C6)Alkyl(CHR5)(C1-C6)alkyl, worin R5 Hydroxy, (C1-C6)Acyloxy, (C1-C6)-Alkoxy, Piperazino, (C1-C6)Acylamino, (C1-C6)Alkylthio, (C6-C10)Arylthio, (C1-C6)Alkylsulfinyl, (C6-C10)Arylsulfinyl, (C1-C6)Alkylsulfoxyl, (C6-C10)Arylsulfoxyl, Amino, (C1-C6)-Alkylamino, ((C1-C6)Alkyl)2amino, (C1-C6)Acylpiperazino, (C1-C6)Alkylpiperazino, (C6-C10)Aryl(C1-C6)alkylpiperazino, (C5-C9)Heteroaryl(C1-C6)alkylpiperazino, Morpholino, Thiomorpholino, Piperidino oder Pyrrolidino ist; R6(C1-Cs)Alkyl, (C1-C5)Alkyl-(CHR6)(C1-C6)alkyl, worin R6 Piperidyl, (C1-C6)-Alkylpiperidyl, (C6-C10)Arylpiperidyl, (C6-C10)Aryl(C1-C6)-alkylpiperidyl, (C5-C9)Heteroarylpiperidyl oder (C5-C9)Heteroaryl(C1-C6)-alkylpiperidyl ist; oder wenn n mindestens 1 ist, D und E oder D und X jeweils CR7R8 sind, die benachbarten R7-Gruppen zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, Gruppen der Formeln
worin p 0, 1, 2 oder 3 ist; und Z Hydroxy, (C1-C6)Alkoxy oder NR1R2 ist, worin R1 und R2 jeweils unabhängig voneinander ausgewählt sind aus der Gruppe, bestehend aus Wasserstoff, (C1-C6)Alkyl, Piperidyl, (C1-C6)Alkylpiperidyl, (C6-C10)Arylpiperidyl, (C5-C9)Hetero-arylpiperidyl, (C6-C10)Aryl(C1-C6)alkylpiperidyl, (C5-C9)-Heteroaryl(C1-C6)alkylpiperidyl, (C1-C6)Acylpiperidyl, (C6-C10)Aryl, (C5-C9)Heteroaryl, (C6-C10)Aryl(C1-C6)alkyl, (C5-C9)Heteroaryl(C1-C6)alkyl, (C6-C10)Aryl(C6-C10)aryl, (C6-C10)Aryl(C6-C10)aryl(C1-C6)alkyl, (C3-C6)Cycloalkyl, (C3-C6)Cycloalkyl(C1-C5)alkyl, R5(C1-C6)Alkyl, (C1-C6)Alkyl(CHR5)(C1-C6)alkyl, worin R5 Hydroxy, (C1-C6)Acyloxy, (C1-C6)-Alkoxy, Piperazino, (C1-C6)Acylamino, (C1-C6)Alkylthio, (C6-C10)Arylthio, (C1-C6)Alkylsulfinyl, (C6-C10)Arylsulfinyl, (C1-C6)Alkylsulfoxyl, (C6-C10)Arylsulfoxyl, Amino, (C1-C6)-Alkylamino, ((C1-C6)Alkyl)2amino, (C1-C6)Acylpiperazino, (C1-C6)Alkylpiperazino, (C6-C10)Aryl(C1-C6)alkylpiperazino, (C5-C9)Heteroaryl(C1-C6)alkylpiperazino, Morpholino, Thiomorpholino, Piperidino oder Pyrrolidino ist; R6(C1-Cs)Alkyl, (C1-C5)Alkyl-(CHR6)(C1-C6)alkyl, worin R6 Piperidyl, (C1-C6)-Alkylpiperidyl, (C6-C10)Arylpiperidyl, (C6-C10)Aryl(C1-C6)-alkylpiperidyl, (C5-C9)Heteroarylpiperidyl oder (C5-C9)Heteroaryl(C1-C6)-alkylpiperidyl ist; oder wenn n mindestens 1 ist, D und E oder D und X jeweils CR7R8 sind, die benachbarten R7-Gruppen zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, Gruppen der Formeln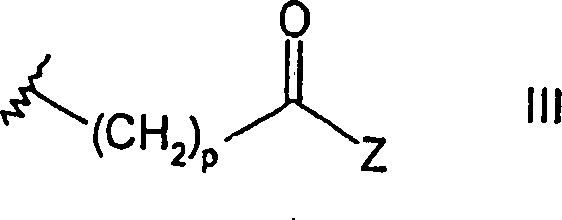 bilden, wobei die gestrichelten Linien fakultative Doppelbindungen darstellen; a 0, 1 oder 2 ist; m, A, B und X wie oben definiert sind; und G, J, L und M jeweils unabhängig voneinander Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8, worin R6, R7 und R8 wie oben definiert sind, sind; oder wenn n 1 ist, D und E jeweils CR7R8 sind und m 1 ist, A und B jeweils CR7R8 sind, die jeweiligen benachbarten R7-Gruppen zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, eine Gruppe der folgenden Formel bilden:
bilden, wobei die gestrichelten Linien fakultative Doppelbindungen darstellen; a 0, 1 oder 2 ist; m, A, B und X wie oben definiert sind; und G, J, L und M jeweils unabhängig voneinander Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8, worin R6, R7 und R8 wie oben definiert sind, sind; oder wenn n 1 ist, D und E jeweils CR7R8 sind und m 1 ist, A und B jeweils CR7R8 sind, die jeweiligen benachbarten R7-Gruppen zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, eine Gruppe der folgenden Formel bilden: worin die gestrichelte Bindung fakultative Doppelbindungen darstellt; a, G, J, L und M wie oben definiert sind; r 0 oder 1 ist; c 0, 1 oder 2 ist; und R, W, Y und S jeweils unabhängig voneinander Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8, worin R6, R7 oder R8 wie oben definiert sind; sind; R2 und R3 jeweils unabhängig voneinander ausgewählt sind aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Amino, Halogen, Hydroxy, Nitro, Carboxy, (C2-C6)Alkenyl, (C2-C6)Alkinyl, Trifluormethyl, Trifluormethoxy, (C1-C6)-Alkyl, (C1-C6)Alkoxy, worin die Alkyl- oder Alkoxygruppen gegebenenfalls durch eine oder drei Gruppen, ausgewählt aus Halogen, Hydroxy, Carboxy, Amino(C1-C6)alkylthio, (C1-C6)Alkylamino, ((C1-C6)Alkyl)2amino, (C5-C9)Heteroaryl, (C2-C9)Heterocycloalkyl, (C3-C9)Cycloalkyl oder (C6-C10)Aryl substituiert sind; oder R2 und R3 jeweils unabhängig voneinander sind: (C3-C10)Cycloalkyl, (C3-C10)Cycloalkoxy, (C1-C6)Alkylamino, ((C1-C6)Alkyl)2amino, (C6-C10)Arylamino, (C1-C6)Alkylthio, (C6-C10)Arylthio, (C1-C6)Alkylsulfinyl, (C6-C10)Arylsulfinyl, (C1-C6)Alkylsulfonyl, (C6-C10)Arylsulfonyl, (C1-C6)-Acyl, (C1-C6)Alkoxy-CO-NH-, (C1-C6)Alkylamino-CO-, (C5-C9)-Heteroaryl, (C2-C9)Heterocycloalkyl oder (C6-C10)Aryl, wobei die Heteroaryl-, Heterocycloalkyl- und Arylgruppen gegebenenfalls substituiert sind durch ein bis drei Halogene, (C1-C6)Alkyl, (C1-C6)Alkyl-CO-NH-, (C1-C6)Alkoxy-CO-NH-, (C1-C6)Alkyl-CO-NH-(C1-C6)alkyl, (C1-C6)Alkoxy-CO-NH-(C1-C6)-alkyl, (C1-C6)Alkoxy-CO-NH-(C1-C6)alkoxy, Carboxy, Carboxy(C1-C6)alkyl, Carboxy(C1-C6)alkoxy, Benzyloxycarbonyl-(C1-C6)Alkoxy, (C1-C6)Alkoxycarbonyl(C1-C6)alkoxy, (C6-C10)-Rryl, Amino, Amino(C1-C6)alkyl, (C1-C6)Alkoxycarbonylamino, (C6-C10)Aryl(C1-C6)alkoxycarbonylamino, (C1-C6)Alkylamino, ((C1-C6)alkyl)2amino, (C1-C6)Alkylamino(C1-C6)alkyl, ((C1-C6)-Alkyl)2amino(C1-C6)alkyl, Hydroxy, (C1-C6)Alkoxy, Carboxy, Carboxy(C1-C6)alkyl, (C1-C6)Alkoxycarbonyl, (C1-C6)Alkoxy-carbonyl(C1-C6)alkyl, (C1-C6)Alkoxy-CO-NH-, (C1-C6)Alkyl-CO-NH-, Cyano, (C5-C9)Heterocycloalkyl, Amino-CO-NH-, (C1-C6)-Alkylamino-CO-NH-, ((C1-C6)Alkyl)2amino-CO-NH-, (C6-C10)Aryl-amino-CO-NH-, (C5-C9)Heteroarylamino-CO-NH-, (C1-C6)Alkyl-amino-CO-NH-(C1-C6)alkyl, (C1-C6)Alkyl)2amino-CO-NH-(C1-C6)-alkyl, (C6-C10)Arylamino-CO-NH-(C1-C6)alkyl, (C5-C9)Hetero-arylamino-CO-NH-(C1-C6)alkyl, (C1-C6)Alkylsulfonyl, (C1-C6)-Sulfonylamino, (C1-C6)Alkylsulfonylamino(C1-C6)alkyl, (C6-C10)Arylsulfonyl, (C6-C10)Arylsulfonylamino, (C6-C10)Aryl-sulfonylamino(C1-C6)alkyl, (C1-C6)Alkylsulfonylamino, (C1-C6)Alkylsulfonylamino(C1-C6)alkyl, (C5-C9) Heteroaryl oder (C2-C9)Heterocycloalkyl; mit der Maßgabe, dass, wenn A, B oder X in der Formel V oder VI als NR6 oder CR7R8 definiert ist, R2 und/oder R3 Halogen sein muss/müssen; mit der Maßgabe, dass, wenn R2 und R3 j eweils unabhängig voneinander Wasserstoff oder (C1-C6)Alkyl sind, R1 nicht unsubstituiertes Piperidinyl sein kann; mit der Maßgabe, dass, wenn R2 und R3 jeweils Wasserstoff sind, R1 nicht unsubstituiertes Morpholinyl oder Pyrrolidinyl sein kann; mit der Maßgabe, dass, wenn R2 und R3 jeweils Wasserstoff sind, R1 nicht Piperazinyl sein kann; und mit der Maßgabe, dass die Gruppen der Formeln IV, V, VI oder XIII nicht zwei oder mehr Sauerstoff, Schwefel oder Kombinationen davon in benachbarten Positionen enthalten.
worin die gestrichelte Bindung fakultative Doppelbindungen darstellt; a, G, J, L und M wie oben definiert sind; r 0 oder 1 ist; c 0, 1 oder 2 ist; und R, W, Y und S jeweils unabhängig voneinander Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8, worin R6, R7 oder R8 wie oben definiert sind; sind; R2 und R3 jeweils unabhängig voneinander ausgewählt sind aus der Gruppe, bestehend aus Wasserstoff, Deuterium, Amino, Halogen, Hydroxy, Nitro, Carboxy, (C2-C6)Alkenyl, (C2-C6)Alkinyl, Trifluormethyl, Trifluormethoxy, (C1-C6)-Alkyl, (C1-C6)Alkoxy, worin die Alkyl- oder Alkoxygruppen gegebenenfalls durch eine oder drei Gruppen, ausgewählt aus Halogen, Hydroxy, Carboxy, Amino(C1-C6)alkylthio, (C1-C6)Alkylamino, ((C1-C6)Alkyl)2amino, (C5-C9)Heteroaryl, (C2-C9)Heterocycloalkyl, (C3-C9)Cycloalkyl oder (C6-C10)Aryl substituiert sind; oder R2 und R3 jeweils unabhängig voneinander sind: (C3-C10)Cycloalkyl, (C3-C10)Cycloalkoxy, (C1-C6)Alkylamino, ((C1-C6)Alkyl)2amino, (C6-C10)Arylamino, (C1-C6)Alkylthio, (C6-C10)Arylthio, (C1-C6)Alkylsulfinyl, (C6-C10)Arylsulfinyl, (C1-C6)Alkylsulfonyl, (C6-C10)Arylsulfonyl, (C1-C6)-Acyl, (C1-C6)Alkoxy-CO-NH-, (C1-C6)Alkylamino-CO-, (C5-C9)-Heteroaryl, (C2-C9)Heterocycloalkyl oder (C6-C10)Aryl, wobei die Heteroaryl-, Heterocycloalkyl- und Arylgruppen gegebenenfalls substituiert sind durch ein bis drei Halogene, (C1-C6)Alkyl, (C1-C6)Alkyl-CO-NH-, (C1-C6)Alkoxy-CO-NH-, (C1-C6)Alkyl-CO-NH-(C1-C6)alkyl, (C1-C6)Alkoxy-CO-NH-(C1-C6)-alkyl, (C1-C6)Alkoxy-CO-NH-(C1-C6)alkoxy, Carboxy, Carboxy(C1-C6)alkyl, Carboxy(C1-C6)alkoxy, Benzyloxycarbonyl-(C1-C6)Alkoxy, (C1-C6)Alkoxycarbonyl(C1-C6)alkoxy, (C6-C10)-Rryl, Amino, Amino(C1-C6)alkyl, (C1-C6)Alkoxycarbonylamino, (C6-C10)Aryl(C1-C6)alkoxycarbonylamino, (C1-C6)Alkylamino, ((C1-C6)alkyl)2amino, (C1-C6)Alkylamino(C1-C6)alkyl, ((C1-C6)-Alkyl)2amino(C1-C6)alkyl, Hydroxy, (C1-C6)Alkoxy, Carboxy, Carboxy(C1-C6)alkyl, (C1-C6)Alkoxycarbonyl, (C1-C6)Alkoxy-carbonyl(C1-C6)alkyl, (C1-C6)Alkoxy-CO-NH-, (C1-C6)Alkyl-CO-NH-, Cyano, (C5-C9)Heterocycloalkyl, Amino-CO-NH-, (C1-C6)-Alkylamino-CO-NH-, ((C1-C6)Alkyl)2amino-CO-NH-, (C6-C10)Aryl-amino-CO-NH-, (C5-C9)Heteroarylamino-CO-NH-, (C1-C6)Alkyl-amino-CO-NH-(C1-C6)alkyl, (C1-C6)Alkyl)2amino-CO-NH-(C1-C6)-alkyl, (C6-C10)Arylamino-CO-NH-(C1-C6)alkyl, (C5-C9)Hetero-arylamino-CO-NH-(C1-C6)alkyl, (C1-C6)Alkylsulfonyl, (C1-C6)-Sulfonylamino, (C1-C6)Alkylsulfonylamino(C1-C6)alkyl, (C6-C10)Arylsulfonyl, (C6-C10)Arylsulfonylamino, (C6-C10)Aryl-sulfonylamino(C1-C6)alkyl, (C1-C6)Alkylsulfonylamino, (C1-C6)Alkylsulfonylamino(C1-C6)alkyl, (C5-C9) Heteroaryl oder (C2-C9)Heterocycloalkyl; mit der Maßgabe, dass, wenn A, B oder X in der Formel V oder VI als NR6 oder CR7R8 definiert ist, R2 und/oder R3 Halogen sein muss/müssen; mit der Maßgabe, dass, wenn R2 und R3 j eweils unabhängig voneinander Wasserstoff oder (C1-C6)Alkyl sind, R1 nicht unsubstituiertes Piperidinyl sein kann; mit der Maßgabe, dass, wenn R2 und R3 jeweils Wasserstoff sind, R1 nicht unsubstituiertes Morpholinyl oder Pyrrolidinyl sein kann; mit der Maßgabe, dass, wenn R2 und R3 jeweils Wasserstoff sind, R1 nicht Piperazinyl sein kann; und mit der Maßgabe, dass die Gruppen der Formeln IV, V, VI oder XIII nicht zwei oder mehr Sauerstoff, Schwefel oder Kombinationen davon in benachbarten Positionen enthalten.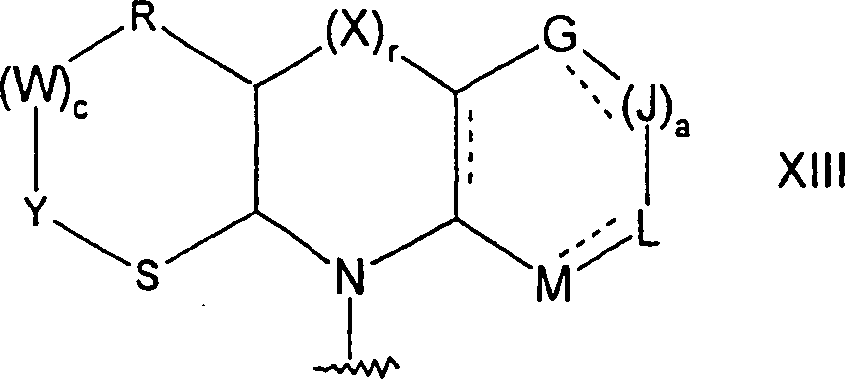
- Verbindung nach Anspruch 1, wobei R1 eine Gruppe der Formelist, worin die gestrichelte Linie fakultative Doppelbindungen darstellt; m 0, 1, 2 oder 3 ist; n 0, 1, 2 oder 3 ist; X, B und D jeweils unabhängig voneinander Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8 sind; A und E jeweils unabhängig voneinander CR7R8 oder NR6 sind; oder wenn n 1 ist, D und E jeweils CR7R8 sind und m 1 ist, A und B jeweils CR7R8 sind, die jeweiligen benachbarten R7-Gruppen zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, eine Gruppe der folgenden Formel bilden können:
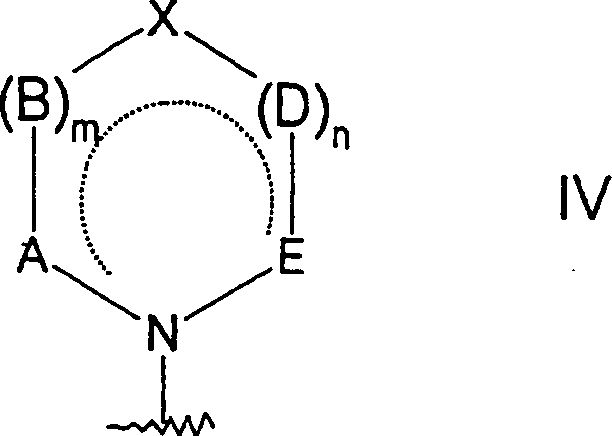 worin die gestrichelte Bindung fakultative Doppelbindungen darstellt; a, G, J, L und M wie oben definiert sind; r 0 oder 1 ist; c 0, 1 oder 2 ist; und R, W, Y und S jeweils unabhängig Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8, worin R6, R7 und R8 wie oben definiert sind, sind.
worin die gestrichelte Bindung fakultative Doppelbindungen darstellt; a, G, J, L und M wie oben definiert sind; r 0 oder 1 ist; c 0, 1 oder 2 ist; und R, W, Y und S jeweils unabhängig Sauerstoff, S(O)d, worin d 0, 1 oder 2 ist, NR6 oder CR7R8, worin R6, R7 und R8 wie oben definiert sind, sind.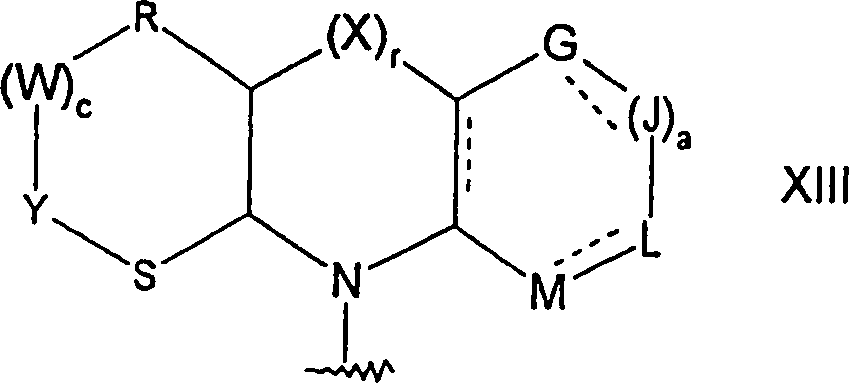
- Verbindung nach Anspruch 1 oder Anspruch 2, wobei R2 und R3 jeweils unabhängig voneinander aus der Gruppe, bestehend aus Wasserstoff, (C1-C6)Alkyl, (C1-C6)Alkoxy, (C3-C10)Cycloalkyl, (C3-C10)Cycloalkoxy, (C2-C9)Heterocycloalkyl, (C5-C9)Heteroaryl oder (C6-C10)Aryl ausgewählt sind.
- Verbindung nach einem der Ansprüche 1 bis 3, wobei die Verbindung aus der Gruppe, bestehend aus: 5-Fluor-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin; 4-Piperidin-1-yl-5-trifluormethyl-7H-pyrrolo[2,3-d]pyrimidin; N,N-Dimethyl-N'-[3-(4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin-5-yl)benzyl]ethan-1,2-diamin; 2-[1-(5-m-Tolyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-yl]ethanol; 5-(3-Isopropylphenyl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin; 5-(3-Methyl-3H-imidazol-4-yl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin; 5-(1-Methyl-1H-imidazol-4-yl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin; 5-(2-Methylpyridin-4-yl)-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin; 5-Chlor-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin; 5-Ethinyl-4-piperidin-1-yl-7H-pyrrolo[2,3-d]pyrimidin; 4-Piperidin-1-yl-5-m-tolyl-7H-pyrrolo[2,3-d]pyrimidin; und 4-(3,3-Dimethylpiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin ausgewählt ist.
- Verbindung nach einem der Ansprüche 1 bis 4 zur Verwendung als Medikament.
- Pharmazeutische Zusammensetzung zur (a) Behandlung oder Prävention einer Störung oder eines Zustandes, ausgewählt aus Organtransplantatabstossung, Lupus, multipler Sklerose, rheumatoider Arthritis, Psoriasis, Typ I-Diabetes und Komplikationen, die aus Diabetes resultieren, Krebs, Asthma, atopischer Dermatitis, Autoimmun-Schild-drüsen-Störungen, Colitis ulcerosa, Crohn-Krankheit, Alzheimer-Krankheit, Leukämie und anderen Autoimmunkrankheiten, oder (b) Inhibierung von Proteintyrosinkinasen oder Janus-Kinase 3 (JAK3) in einem Säuger, einschließlich eines Menschen, die eine Menge einer Verbindung nach einem der Ansprüche 1 bis 4 oder eines pharmazeutisch annehmbaren Salzes davon, die bei solchen Störungen oder Zuständen wirksam ist, und einen pharmazeutisch annehmbaren Träger umfasst.
- Pharmazeutische Zusammensetzung zur (a) Behandlung oder Prävention einer Störung oder eines Zustandes, ausgewählt aus Organtransplantatabstossung, Lupus, multipler Sklerose, rheumatoider Arthritis, Psoriasis, Typ I-Diabetes und Komplikationen, die aus Diabetes resultieren, Krebs, Asthma, atopischer Dermatitis, Autoimmun-Schild-drüsen-Störungen, Colitis ulcerosa, Crohn-Krankheit, Alzheimer-Krankheit, Leukämie und anderen Autoimmunkrankheiten, oder (b) Inhibierung von Proteintyrosinkinasen oder Janus-Kinase 3 (JAK3) in einem Säuger, einschließlich eines Menschen, die eine Menge einer Verbindung nach einem der Ansprüche 1 bis 4 oder eines pharmazeutisch annehmbaren Salzes allein oder in Kombination mit einem oder mehreren zusätzlichen Agentien, die ein Säugerimmunsystem modulieren, oder mit Mitteln gegen Entzündungen, die bei solchen Störungen oder Zuständen wirk sam ist, und einen pharmazeutisch annehmbaren Träger umfasst.
- Verwendung einer wirksamen Menge einer Verbindung nach einem der Ansprüche 1 bis 4 und eines pharmazeutisch annehmbaren Salzes davon zur Herstellung eines Medikaments für die Inhibierung von Proteintyrosinkinasen oder Janus-Kinase 3 (JAK3) in einem Säuger, einschließlich eines Menschen.
- Verwendung einer wirksamen Menge einer Verbindung nach einem der Ansprüche 1 bis 4 oder eines pharmazeutisch annehmbaren Salzes davon zur Herstellung eines Medikaments für die Behandlung oder Prävention einer Störung oder eines Zustandes, ausgewählt aus Organtransplantatabstossung, Lupus, multipler Sklerose, rheumatoider Arthritis, Psoriasis, Typ I-Diabetes und Komplikationen, die aus Diabetes resultieren, Krebs, Asthma, atopischer Dermatitis, Autoimmun-Schilddrüsen-Störungen, Colitis ulcerosa, Crohn-Krankheit, Alzheimer-Krankheit, Leukämie und anderen Autoimmunkrankheiten, in einem Säuger, einschließlich eines Menschen.
- Verwendung einer wirksamen Menge einer Verbindung nach einem der Ansprüche 1 bis 4 oder eines pharmazeutisch annehmbaren Salzes davon, allein oder in Kombination mit einem oder mehreren zusätzlichen Agentien, die ein Säugerimmunsystem modulieren, oder mit Mitteln gegen Entzündungen, zur Herstellung eines Medikaments zur Inhibierung von Proteintyrosinkinasen oder Janus-Kinase 3 (JAK3) in einem Säuger, einschließlich eines Menschen.
- Verwendung einer wirksamen Menge einer Verbindung nach einem der Ansprüche 1 bis 4 oder eines pharmazeutisch annehmbaren Salzes davon, allein oder in Kombination mit einem oder mehreren zusätzlichen Agentien, die ein Säugerimmunsystem modulieren, oder mit Mitteln gegen Ent zündungen zur Herstellung eines Medikaments für die Behandlung oder Prävention einer Störung oder eines Zustandes, ausgewählt aus Organtransplantatabstossung, Lupus, multipler Sklerose, rheumatoider Arthritis, Psoriasis, Typ I-Diabetes und Komplikationen, die aus Diabetes resultieren, Krebs, Asthma, atopischer Dermatitis, Autoimmun-Schild-drüsen-Störungen, Colitis ulcerosa, Crohn-Krankheit, Alzheimer-Krankheit, Leukämie und anderen Autoimmunkrankheiten in einem Säuger, einschließlich eines Menschen.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US8988698P | 1998-06-19 | 1998-06-19 | |
| US89886P | 1998-06-19 | ||
| PCT/IB1999/001110 WO1999065909A1 (en) | 1998-06-19 | 1999-06-14 | PYRROLO[2,3-d]PYRIMIDINE COMPOUNDS |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69918552D1 DE69918552D1 (de) | 2004-08-12 |
| DE69918552T2 true DE69918552T2 (de) | 2004-11-04 |
Family
ID=22220084
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69918552T Expired - Lifetime DE69918552T2 (de) | 1998-06-19 | 1999-06-14 | Pyrrolo(2,3-d)pyrimidin-verbindungen |
Country Status (46)
Families Citing this family (169)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PA8474101A1 (es) | 1998-06-19 | 2000-09-29 | Pfizer Prod Inc | Compuestos de pirrolo [2,3-d] pirimidina |
| EP1065207A1 (de) * | 1999-07-02 | 2001-01-03 | Aventis Pharma Deutschland GmbH | Naphthyridin derivate, Verfahren zu ihrer Herstellung, ihre Anwendung und sie enthaltende pharmazeutische Zusammenfassungen |
| MXPA02003436A (es) | 1999-10-07 | 2002-08-20 | Amgen Inc | Inhibidores de triazina cinasa. |
| DE19948417A1 (de) * | 1999-10-07 | 2001-04-19 | Morphochem Ag | Imidazol-Derivate und ihre Verwendung als Arzneimittel |
| DE60037345T2 (de) | 1999-12-10 | 2008-11-13 | Pfizer Products Inc., Groton | Pyrrolo(2,3-d)pyrimidin-Verbindungen |
| EP1782800B1 (de) * | 2000-01-24 | 2013-12-18 | Genzyme Corporation | Verfahren zur Detektion von JAK3 Inhibitoren |
| EP1250137B1 (de) * | 2000-01-24 | 2007-08-15 | Genzyme Corporation | Inhibitoren des jak/stat-weges und deren verwendung zur behandlung von allgemeiner primärer osteoarthritis |
| MY145722A (en) * | 2000-04-27 | 2012-03-30 | Abbott Lab | Diazabicyclic central nervous system active agents |
| ATE423120T1 (de) * | 2000-06-26 | 2009-03-15 | Pfizer Prod Inc | Pyrroloä2,3-düpyrimidin verbindungen als immunosuppressive wirkstoffe |
| US6881737B2 (en) | 2001-04-11 | 2005-04-19 | Amgen Inc. | Substituted triazinyl acrylamide derivatives and methods of use |
| US6864255B2 (en) | 2001-04-11 | 2005-03-08 | Amgen Inc. | Substituted triazinyl amide derivatives and methods of use |
| US7301023B2 (en) | 2001-05-31 | 2007-11-27 | Pfizer Inc. | Chiral salt resolution |
| GB0115393D0 (en) * | 2001-06-23 | 2001-08-15 | Aventis Pharma Ltd | Chemical compounds |
| AP2003002929A0 (en) * | 2001-06-23 | 2003-12-31 | Aventis Pharma Inc | Pyrrolopyrimidines as protein kinase inhibitors |
| US7323469B2 (en) | 2001-08-07 | 2008-01-29 | Novartis Ag | 7H-pyrrolo[2,3-d]pyrimidine derivatives |
| GB0119249D0 (en) | 2001-08-07 | 2001-10-03 | Novartis Ag | Organic compounds |
| PY0228255A (es) | 2001-12-06 | 2004-06-01 | Pfizer Prod Inc | Compuestos cristalinos novedosos |
| US6642381B2 (en) | 2001-12-27 | 2003-11-04 | Hoffman-La Roche Inc. | Pyrimido[5,4-e][1,2,4]triazine-5,7-diamine compounds as protein tyrosine phosphatase inhibitors |
| DK1474425T3 (da) | 2002-01-07 | 2006-09-25 | Eisai Co Ltd | Deazapuriner og anvendelser deraf |
| GB0202679D0 (en) * | 2002-02-05 | 2002-03-20 | Glaxo Group Ltd | Novel compounds |
| EP1388541A1 (de) | 2002-08-09 | 2004-02-11 | Centre National De La Recherche Scientifique (Cnrs) | Pyrrolopyrazine als Kinasehemmer |
| NZ539901A (en) | 2002-11-26 | 2007-09-28 | Pfizer Prod Inc | Method of treatment of transplant rejection |
| US7253166B2 (en) * | 2003-04-22 | 2007-08-07 | Irm Llc | 6-phenyl-7H-pyrrolo[2,3-d]pyrimidine compounds that induce neuronal differentiation in embryonic stem cells |
| CA2543116A1 (en) * | 2003-10-27 | 2005-05-19 | Genelabs Technologies, Inc. | Methods for preparing 7-(2'-substituted-.szlig.-d-ribofuranosyl)-4-(nr2r3)-5-(substituted ethyn-1-yl)-pyrrolo[2,3-d]pyrimidine derivatives |
| US20050239806A1 (en) * | 2004-01-13 | 2005-10-27 | Ambit Biosciences Corporation | Pyrrolopyrimidine derivatives and analogs and their use in the treatment and prevention of diseases |
| GB0403606D0 (en) | 2004-02-18 | 2004-03-24 | Novartis Ag | Organic compounds |
| WO2005117909A2 (en) * | 2004-04-23 | 2005-12-15 | Exelixis, Inc. | Kinase modulators and methods of use |
| WO2006115509A2 (en) | 2004-06-24 | 2006-11-02 | Novartis Vaccines And Diagnostics Inc. | Small molecule immunopotentiators and assays for their detection |
| MXPA06015237A (es) | 2004-06-29 | 2007-12-10 | Amgen Inc | Pirrolo[2-3-d]pirimidinas que modulan la actividad de ack1 y lck. |
| JP2008508358A (ja) | 2004-08-02 | 2008-03-21 | オーエスアイ・ファーマスーティカルズ・インコーポレーテッド | アリール−アミノ置換ピロロピリミジンマルチキナーゼ阻害化合物 |
| UY29177A1 (es) | 2004-10-25 | 2006-05-31 | Astex Therapeutics Ltd | Derivados sustituidos de purina, purinona y deazapurina, composiciones que los contienen métodos para su preparación y sus usos |
| MY179032A (en) | 2004-10-25 | 2020-10-26 | Cancer Research Tech Ltd | Ortho-condensed pyridine and pyrimidine derivatives (e.g.purines) as protein kinase inhibitors |
| AR054416A1 (es) | 2004-12-22 | 2007-06-27 | Incyte Corp | Pirrolo [2,3-b]piridin-4-il-aminas y pirrolo [2,3-b]pirimidin-4-il-aminas como inhibidores de las quinasas janus. composiciones farmaceuticas. |
| FR2880626B1 (fr) * | 2005-01-13 | 2008-04-18 | Aventis Pharma Sa | Derives de la purine, compositions les contenant et utilisation |
| WO2006091450A1 (en) * | 2005-02-18 | 2006-08-31 | Lexicon Genetics Incorporated | 4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine compounds |
| US8183248B2 (en) | 2005-05-13 | 2012-05-22 | Irm Llc | Substituted pyrrolo[2,3-d]pyrimidines and compositions as protein kinase inhibitors |
| CN102127078A (zh) | 2005-07-14 | 2011-07-20 | 安斯泰来制药株式会社 | Janus激酶3的杂环类抑制剂 |
| WO2007007919A2 (en) | 2005-07-14 | 2007-01-18 | Astellas Pharma Inc. | Heterocyclic janus kinase 3 inhibitors |
| CA2621261C (en) | 2005-09-22 | 2014-05-20 | Incyte Corporation | Azepine inhibitors of janus kinases |
| ZA200802685B (en) | 2005-09-30 | 2009-10-28 | Vertex Pharma | Deazapurines useful as inhibitors of janus kinases |
| SG10202003901UA (en) * | 2005-12-13 | 2020-05-28 | Incyte Holdings Corp | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| AU2015201850B2 (en) * | 2005-12-13 | 2017-03-02 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| EA200801968A1 (ru) * | 2006-03-11 | 2009-02-27 | Вернэлис (Р&Д) Лтд. | Пирролопиримидиновые производные, используемые в качестве ингибиторов hsp90 |
| SG170828A1 (en) | 2006-04-05 | 2011-05-30 | Vertex Pharmaceuticals Inc Us | Deazapurines useful as inhibitors of janus kinases |
| DK3421471T3 (da) | 2006-04-25 | 2021-06-14 | Astex Therapeutics Ltd | Purin- og deazapurinderivater som farmaceutiske forbindelser |
| GB0608176D0 (en) * | 2006-04-25 | 2006-06-07 | Astex Therapeutics Ltd | Pharmaceutical Compounds |
| WO2008012635A2 (en) * | 2006-07-26 | 2008-01-31 | Pfizer Products Inc. | Amine derivatives useful as anticancer agents |
| US20080161320A1 (en) * | 2006-09-11 | 2008-07-03 | Xiong Cai | Fused bicyclic pyrimidines as ptk inhibitors containing a zinc binding moiety |
| WO2008033747A2 (en) * | 2006-09-11 | 2008-03-20 | Curis, Inc. | Multi-functional small molecules as anti-proliferative agents |
| WO2008075007A1 (en) * | 2006-12-21 | 2008-06-26 | Cancer Research Technology Limited | Morpholino-substituted bicycloheteroaryl compounds and their use as anti cancer agents |
| AR064416A1 (es) * | 2006-12-21 | 2009-04-01 | Cancer Rec Tech Ltd | Derivados de purina, piridina y pirimidina condensadas con heterociclos, moduladores de pka y/o pkb, composiciones farmaceuticas que los contienen, y usos para el tratamiento de enfermedades hiperproliferativas. |
| CA2673038C (en) | 2006-12-22 | 2015-12-15 | Incyte Corporation | Substituted tricyclic heteroaryl compounds as janus kinase inhibitors |
| AR064879A1 (es) | 2007-01-12 | 2009-04-29 | Astellas Pharma Inc | Compuesto de piridina condensado |
| CN101679440A (zh) * | 2007-04-02 | 2010-03-24 | 帕劳制药股份有限公司 | 作为jak3抑制剂的吡咯并嘧啶衍生物 |
| UA99284C2 (ru) | 2007-05-11 | 2012-08-10 | Елі Ліллі Енд Компані | ИНГИБИТОРЫ р70 S6-КИНАЗЫ |
| KR20150036210A (ko) | 2007-06-13 | 2015-04-07 | 인사이트 코포레이션 | 야누스 키나제 억제제(R)―3―(4―(7H―피롤로[2,3-d]피리미딘―4―일)―1H―피라졸―1―일)―3―사이클로펜틸프로판니트릴의 염 |
| CL2008001709A1 (es) | 2007-06-13 | 2008-11-03 | Incyte Corp | Compuestos derivados de pirrolo [2,3-b]pirimidina, moduladores de quinasas jak; composicion farmaceutica; y uso en el tratamiento de enfermedades tales como cancer, psoriasis, artritis reumatoide, entre otras. |
| GB0713602D0 (en) * | 2007-07-12 | 2007-08-22 | Syngenta Participations Ag | Chemical compounds |
| AU2008281849B2 (en) * | 2007-07-27 | 2013-11-28 | Janssen Pharmaceutica Nv | Pyrrolopyrimidines |
| DK2188289T3 (en) | 2007-08-08 | 2016-01-25 | Lexicon Pharmaceuticals Inc | (7H-Pyrrolo [2,3-D] pyrimidin-4-yl) -piperazines as kinase inhibitors for the treatment of cancer and inflammation |
| WO2009030871A1 (en) * | 2007-09-07 | 2009-03-12 | Vernalis R & D Ltd | Pyrrolopyrimidine derivatives having hsp90 inhibitory activity |
| PL2201012T3 (pl) | 2007-10-11 | 2014-11-28 | Astrazeneca Ab | Pochodne pirolo[2,3-d]pirymidyny jako inhibitory kinazy białkowej b |
| UA104849C2 (uk) | 2007-11-16 | 2014-03-25 | Інсайт Корпорейшн | 4-піразоліл-n-арилпіримідин-2-аміни і 4-піразоліл-n-гетероарилпіримідин-2-аміни як інгібітори кіназ janus |
| HUE029767T2 (en) | 2008-03-11 | 2017-04-28 | Incyte Holdings Corp | JAK inhibitor azetidine and cyclobutane derivatives |
| AU2009239500B2 (en) | 2008-04-21 | 2014-01-30 | Lexicon Pharmaceuticals, Inc. | LIMK2 inhibitors, compositions comprising them, and methods of their use |
| EP2274288A2 (de) | 2008-04-24 | 2011-01-19 | Incyte Corporation | Makrocyclische verbindungen und ihre verwendung als kinaseinhibitoren |
| KR101335843B1 (ko) | 2008-08-20 | 2013-12-02 | 조에티스 엘엘씨 | 피롤로[2,3-d]피리미딘 화합물 |
| US8385364B2 (en) * | 2008-09-24 | 2013-02-26 | Nec Laboratories America, Inc. | Distributed message-passing based resource allocation in wireless systems |
| CL2009001884A1 (es) * | 2008-10-02 | 2010-05-14 | Incyte Holdings Corp | Uso de 3-ciclopentil-3-[4-(7h-pirrolo[2,3-d]pirimidin-4-il)-1h-pirazol-1-il)propanonitrilo, inhibidor de janus quinasa, y uso de una composición que lo comprende para el tratamiento del ojo seco. |
| TWI720517B (zh) | 2009-01-15 | 2021-03-01 | 美商英塞特公司 | 製造jak抑制劑之方法及相關中間化合物 |
| DE102009005193A1 (de) * | 2009-01-20 | 2010-07-22 | Merck Patent Gmbh | Neue heterocyclische Verbindungen als MetAP-2 Inhibitoren |
| WO2010085597A1 (en) | 2009-01-23 | 2010-07-29 | Incyte Corporation | Macrocyclic compounds and their use as kinase inhibitors |
| WO2010093808A1 (en) | 2009-02-11 | 2010-08-19 | Reaction Biology Corp. | Selective kinase inhibitors |
| AU2010239396B2 (en) | 2009-04-20 | 2016-06-23 | Auspex Pharmaceuticals, Llc | Piperidine inhibitors of Janus Kinase 3 |
| AR076794A1 (es) * | 2009-05-22 | 2011-07-06 | Incyte Corp | Derivados de n-(hetero)aril-pirrolidina de pirazol-4-il-pirrolo [2,3-d]pirimidinas y pirrol-3-il-pirrolo [2,3-d ]pirimidinas como inhibidores de la quinasa janus y composiciones farmaceuticas que los contienen |
| SI2432472T1 (sl) * | 2009-05-22 | 2019-11-29 | Incyte Holdings Corp | 3-(4-(7H-pirolo(2,3-d)pirimidin-4-il)-1H-pirazol-1-il)oktan- ali heptan-nitril kot inhibitorji JAK |
| WO2011003418A1 (en) | 2009-07-08 | 2011-01-13 | Leo Pharma A/S | Heterocyclic compounds as jak receptor and protein tyrosine kinase inhibitors |
| TWI466885B (zh) | 2009-07-31 | 2015-01-01 | Japan Tobacco Inc | 含氮螺環化合物及其醫藥用途 |
| TW201111385A (en) * | 2009-08-27 | 2011-04-01 | Biocryst Pharm Inc | Heterocyclic compounds as janus kinase inhibitors |
| TW201113285A (en) | 2009-09-01 | 2011-04-16 | Incyte Corp | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| NZ598985A (en) | 2009-09-04 | 2013-07-26 | Biogen Idec Inc | Bruton's tyrosine kinase inhibitors |
| CN102984941B (zh) * | 2009-09-04 | 2016-08-17 | 密执安大学评议会 | 用于治疗白血病的组合物和方法 |
| EA021478B1 (ru) | 2009-10-09 | 2015-06-30 | Инсайт Корпорейшн | ГИДРОКСИЛЬНЫЕ, КЕТО И ГЛЮКУРОНИДНЫЕ ПРОИЗВОДНЫЕ 3-(4-(7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)-1Н-ПИРАЗОЛ-1-ИЛ)-3-ЦИКЛОПЕНТИЛПРОПАННИТРИЛА |
| ES2426407T3 (es) | 2009-10-15 | 2013-10-23 | Pfizer Inc. | Compuestos de pirrolo[2,3-d]pirimidina |
| US9074143B2 (en) * | 2009-12-11 | 2015-07-07 | Uop Llc | Process for producing hydrocarbon fuel |
| WO2011075334A1 (en) | 2009-12-18 | 2011-06-23 | Pfizer Inc. | Pyrrolo[2,3-d]pyrimidine compounds |
| MX2012009074A (es) | 2010-02-05 | 2012-08-23 | Pfizer | Compuestos de urea pirrolo [2, 3-d] pirimidina como inhibidores de janus quinasa. |
| JP5858434B2 (ja) * | 2010-02-18 | 2016-02-10 | インサイト・ホールディングス・コーポレイションIncyte Holdings Corporation | Janusキナーゼ阻害薬としてのシクロブタンおよびメチルシクロブタン誘導体 |
| EP3354652B1 (de) | 2010-03-10 | 2020-05-06 | Incyte Holdings Corporation | Piperidin-4-yl azetidin-derivate als jak1-inhibitoren |
| PE20130216A1 (es) | 2010-05-21 | 2013-02-27 | Incyte Corp | Formulacion topica para un inhibidor de jak |
| US9586961B2 (en) | 2010-07-09 | 2017-03-07 | Leo Pharma A/S | Homopiperazine derivatives as protein tyrosine kinase inhibitors and pharmaceutical use thereof |
| WO2012022045A1 (en) * | 2010-08-20 | 2012-02-23 | Hutchison Medipharma Limited | Pyrrolopyrimidine compounds and uses thereof |
| KR101541086B1 (ko) | 2010-08-20 | 2015-08-03 | 허치슨 메디파르마 리미티드 | 피롤로피리미딘 화합물 및 그 용도 |
| CN102372717B (zh) * | 2010-08-20 | 2014-06-18 | 和记黄埔医药(上海)有限公司 | 吡咯并嘧啶类化合物及其用途 |
| TWI401258B (zh) * | 2010-09-08 | 2013-07-11 | Hutchison Medipharma Ltd | 吡咯並嘧啶類化合物及其用途 |
| PE20140146A1 (es) | 2010-11-19 | 2014-02-06 | Incyte Corp | Derivados de pirrolopiridina y pirrolopirimidina sustituidos con ciclobutilo como inhibidores de jak |
| US9034884B2 (en) | 2010-11-19 | 2015-05-19 | Incyte Corporation | Heterocyclic-substituted pyrrolopyridines and pyrrolopyrimidines as JAK inhibitors |
| ES2617339T3 (es) | 2010-12-16 | 2017-06-16 | Calchan Limited | Derivados de pirrolopirimidina inhibidores de ASK1 |
| WO2012088682A1 (en) * | 2010-12-29 | 2012-07-05 | Shanghai Fochon Pharmaceutical Co Ltd. | 2-(3-aminopiperidin-1-yl)-[1,2,4]triazolo[1,5-c]pyrimidine-5,7(3h,6h)-dione derivates as dipeptidyl peptidase iv(dpp-iv) inhibitors |
| US9233964B2 (en) | 2011-01-07 | 2016-01-12 | Leo Pharma A/S | Sulfamide piperazine derivatives as protein tyrosine kinase inhibitors and pharmaceutical use therof |
| DE102011008352A1 (de) * | 2011-01-12 | 2012-07-12 | Merck Patent Gmbh | 5-([1,2,3]Triazol-4-yl)-7H-pyrrolo-[2,3-d]pyrimidinderivate |
| WO2012106522A2 (en) * | 2011-02-04 | 2012-08-09 | Duquesne University Of The Holy Spirit | Bicyclic and tricyclic pyrimidine tyrosine kinase inhibitors with antitubulin activity and methods of treating a patient |
| ES2547916T3 (es) | 2011-02-18 | 2015-10-09 | Novartis Pharma Ag | Terapia de combinación de inhibidores de mTOR/JAK |
| CN103547580B (zh) | 2011-03-22 | 2016-12-07 | 阿迪维纳斯疗法有限公司 | 取代的稠合三环化合物、其组合物及医药用途 |
| EA027506B1 (ru) | 2011-04-01 | 2017-08-31 | Астразенека Аб | Способ лечения рака молочной железы |
| CN103797010B (zh) | 2011-06-20 | 2016-02-24 | 因塞特控股公司 | 作为jak抑制剂的氮杂环丁烷基苯基、吡啶基或吡嗪基甲酰胺衍生物 |
| TWI574965B (zh) * | 2011-06-29 | 2017-03-21 | 默沙東藥廠 | 二肽基肽酶-iv抑制劑之新穎結晶型 |
| ES2548414T3 (es) | 2011-07-08 | 2015-10-16 | Novartis Ag | Novedosos derivados de pirrolo pirimidina |
| EP2741747A1 (de) | 2011-08-10 | 2014-06-18 | Novartis Pharma AG | Jak-/p13k-mtor-kombinationstherapie |
| TW201313721A (zh) | 2011-08-18 | 2013-04-01 | Incyte Corp | 作為jak抑制劑之環己基氮雜環丁烷衍生物 |
| UA111854C2 (uk) | 2011-09-07 | 2016-06-24 | Інсайт Холдінгс Корпорейшн | Способи і проміжні сполуки для отримання інгібіторів jak |
| HRP20191982T4 (hr) | 2011-11-30 | 2023-01-06 | Astrazeneca Ab | Kombinacijsko liječenje raka |
| ES3018133T3 (en) | 2011-11-30 | 2025-05-14 | Univ Emory | Jak inhibitors for use in the prevention or treatment of a viral disease caused by a coronaviridae |
| EP2788000B1 (de) * | 2011-12-06 | 2018-05-30 | Merck Sharp & Dohme Corp. | Pyrrolopyrimidine als januskinaseinhibitoren |
| AU2013204533B2 (en) | 2012-04-17 | 2017-02-02 | Astrazeneca Ab | Crystalline forms |
| TW201406761A (zh) | 2012-05-18 | 2014-02-16 | Incyte Corp | 做爲jak抑制劑之哌啶基環丁基取代之吡咯并吡啶及吡咯并嘧啶衍生物 |
| AR091273A1 (es) | 2012-06-08 | 2015-01-21 | Biogen Idec Inc | Inhibidores de pirimidinil tirosina quinasa |
| EP2867236B1 (de) * | 2012-06-29 | 2017-06-14 | Pfizer Inc | Neue 4-(substituierte amino)-7h-pyrrolo[2,3-d]pyrimidine als lrrk2 hemmer |
| EP2892534B8 (de) * | 2012-09-06 | 2021-09-15 | Plexxikon Inc. | Verbindungen und verfahren zur kinasemodulation und indikationen dafür |
| US9593115B2 (en) | 2012-09-21 | 2017-03-14 | Advinus Therapeutics Ltd. | Substituted fused tricyclic compounds, compositions, and medicinal applications thereof |
| KR20150060839A (ko) * | 2012-10-26 | 2015-06-03 | 에프. 호프만-라 로슈 아게 | 브루톤 티로신 키나아제의 억제제 |
| CN102936251A (zh) * | 2012-11-05 | 2013-02-20 | 上海毕得医药科技有限公司 | 一种吡咯并[2,3-d]嘧啶衍生物的制备方法 |
| PH12020551186B1 (en) | 2012-11-15 | 2024-03-20 | Incyte Holdings Corp | Sustained-release dosage forms of ruxolitinib |
| GEP201606600B (en) | 2013-02-22 | 2017-01-10 | Pfizer | Pyrrolo [2, 3 -d]pyrimidine derivatives as inhibitors of janus- related kinases (jak) |
| RS62867B1 (sr) | 2013-03-06 | 2022-02-28 | Incyte Holdings Corp | Postupci i intermedijeri za dobijanje inhibitora jak |
| WO2014139388A1 (en) * | 2013-03-14 | 2014-09-18 | Merck Sharp & Dohme Corp. | Novel indole derivatives useful as anti-diabetic agents |
| SMT202000315T1 (it) | 2013-08-07 | 2020-07-08 | Incyte Corp | Forme di dosaggio a rilascio prolungato per un inibitore di jak1 |
| SI3077395T1 (en) * | 2013-12-05 | 2018-03-30 | Pfizer Inc. | Pyrrolo(2,3-d)pyrimidinyl, pyrrolo(2,3-b)pyrazinyl and pyrrolo(2,3-d)pyridinyl acrylamides |
| EP3083618B1 (de) | 2013-12-17 | 2018-02-21 | Pfizer Inc | Neuartige 3,4-disubstituierte-1h-pyrrolo[2,3-b]pyridine und 4,5-disubstituierte 7h-pyrrolo[2,3-c]pyridazine als lrrk2-hemmer |
| US9498467B2 (en) | 2014-05-30 | 2016-11-22 | Incyte Corporation | Treatment of chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML) by inhibitors of JAK1 |
| WO2016024185A1 (en) | 2014-08-12 | 2016-02-18 | Pfizer Inc. | Pyrrolo[2,3-d]pyrimidine derivatives useful for inhibiting janus kinase |
| USRE49687E1 (en) | 2014-09-09 | 2023-10-10 | The Regents Of The University Of Michigan | Thienopyrimidine and thienopyridine compounds and methods of use thereof |
| CN105524067A (zh) * | 2014-09-28 | 2016-04-27 | 江苏柯菲平医药股份有限公司 | 4-取代吡咯并[2,3-d]嘧啶化合物及其用途 |
| EP3288943B1 (de) | 2015-05-01 | 2022-09-28 | Pfizer Inc. | Pyrrolo[2,3-b]pyrazinyl acrylamide und epoxide davon als hemmstoffe von janus kinase |
| TWI703150B (zh) | 2015-06-04 | 2020-09-01 | 美商庫拉腫瘤技術股份有限公司 | 用於抑制menin及mll蛋白之交互作用的方法及組合物 |
| HK1246593A1 (zh) | 2015-06-04 | 2018-09-14 | Kura Oncology, Inc. | 用於抑制menin蛋白与mll蛋白的相互作用的方法及组合物 |
| EP3330271B1 (de) | 2015-07-31 | 2022-09-07 | Taiho Pharmaceutical Co., Ltd. | Pyrrolo [ 2,3-d ] pyrimidinverbindung oder salz davon |
| KR101771219B1 (ko) * | 2015-08-21 | 2017-09-05 | 양지화학 주식회사 | 야누스 키나제 1 선택적 억제제 및 그 의약 용도 |
| RU2722149C1 (ru) | 2015-09-14 | 2020-05-27 | Пфайзер Инк. | Новые производные имидазо[4,5-c] хинолинов и имидазо[4,5-c][1,5] нафтиридинов в качестве ингибиторов LRRK2 |
| US10045981B2 (en) | 2015-11-24 | 2018-08-14 | Jakpharm, Llc | Selective kinase inhibitors |
| CN107098908B (zh) * | 2016-02-23 | 2021-01-08 | 欣凯医药科技(上海)有限公司 | 一种吡咯并嘧啶类化合物的制备方法和应用 |
| PL3429591T3 (pl) | 2016-03-16 | 2023-07-17 | Kura Oncology, Inc. | Podstawione pochodne tieno[2,3-d]pirymidyny jako inhibitory meniny-mll i metody ich zastosowania |
| PH12018501955B1 (en) | 2016-03-16 | 2024-01-24 | Kura Oncology Inc | Bridged bicyclic inhibitors of menin-mll and methods of use |
| CN107513067A (zh) | 2016-06-16 | 2017-12-26 | 北京赛林泰医药技术有限公司 | 含有取代环戊基的吡咯并嘧啶化合物 |
| CN107513069A (zh) * | 2016-06-16 | 2017-12-26 | 正大天晴药业集团股份有限公司 | 手性吡咯并嘧啶化合物的制备方法 |
| JP7214632B2 (ja) | 2016-07-21 | 2023-01-30 | バイオジェン エムエー インク. | ブルトン型チロシンキナーゼ阻害剤のコハク酸塩形態および組成物 |
| US10316038B2 (en) | 2017-01-25 | 2019-06-11 | Aclaris Therapeutics, Inc. | Pyrrolopyrimidine ITK inhibitors for treating inflammation and cancer |
| RU2761626C2 (ru) * | 2017-02-03 | 2021-12-13 | Лео Фарма А/С | ПРОИЗВОДНЫЕ 5-(7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)-5-АЗАСПИРО[2.5]ОКТАН-8-КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ НОВЫХ ИНГИБИТОРОВ JAK-КИНАЗЫ |
| WO2018175746A1 (en) | 2017-03-24 | 2018-09-27 | Kura Oncology, Inc. | Methods for treating hematological malignancies and ewing's sarcoma |
| CN108794480A (zh) * | 2017-04-28 | 2018-11-13 | 天津药物研究院有限公司 | 吡咯并嘧啶类化合物、其制备方法和用途 |
| WO2018226976A1 (en) | 2017-06-08 | 2018-12-13 | Kura Oncology, Inc. | Methods and compositions for inhibiting the interaction of menin with mll proteins |
| EP3684361A4 (de) | 2017-09-20 | 2021-09-08 | Kura Oncology, Inc. | Substituierte inhibitoren von menin-mll und verfahren zur verwendung |
| WO2019090143A1 (en) | 2017-11-03 | 2019-05-09 | Aclaris Therapeutics, Inc. | Pyrazolyl pyrrolo[2,3-b]pyrimidine-5-carboxylate analogs and methods of making the same |
| KR102821964B1 (ko) | 2017-11-03 | 2025-06-19 | 어클라리스 쎄라퓨틱스, 인코포레이티드 | 치환된 피롤로피리미딘 jak 억제제 및 이의 제조 및 사용 방법 |
| WO2019113487A1 (en) | 2017-12-08 | 2019-06-13 | Incyte Corporation | Low dose combination therapy for treatment of myeloproliferative neoplasms |
| SG11202007164UA (en) | 2018-01-30 | 2020-08-28 | Incyte Corp | Processes for preparing (1 -(3-fluoro-2-(trifluoromethyl)isonicotinyl)piperidine-4-one) |
| SMT202400306T1 (it) | 2018-03-30 | 2024-09-16 | Incyte Corp | Trattamento dell'idrosadenite suppurativa utilizzando inibitori di jak. |
| CN119080779A (zh) | 2018-08-10 | 2024-12-06 | 阿克拉瑞斯治疗股份有限公司 | 吡咯并嘧啶itk抑制剂 |
| GB201818750D0 (en) * | 2018-11-16 | 2019-01-02 | Institute Of Cancer Res Royal Cancer Hospital | Lox inhibitors |
| CN109394768B (zh) * | 2018-12-10 | 2019-08-23 | 牡丹江医学院 | 一种治疗湿疹的药物及其制备方法 |
| NL2022471B1 (en) | 2019-01-29 | 2020-08-18 | Vationpharma B V | Solid state forms of oclacitinib |
| WO2020198583A1 (en) | 2019-03-27 | 2020-10-01 | Insilico Medicine Ip Limited | Bicyclic jak inhibitors and uses thereof |
| CA3138544A1 (en) | 2019-05-02 | 2020-11-05 | Aclaris Therapeutics, Inc. | Substituted pyrrolopyridines as jak inhibitors |
| WO2020232247A1 (en) | 2019-05-14 | 2020-11-19 | Provention Bio, Inc. | Methods and compositions for preventing type 1 diabetes |
| JP7717065B2 (ja) | 2019-11-22 | 2025-08-01 | インサイト コーポレーション | Alk2阻害剤及びjak2阻害剤を含む併用療法 |
| US11833155B2 (en) | 2020-06-03 | 2023-12-05 | Incyte Corporation | Combination therapy for treatment of myeloproliferative neoplasms |
| IL298999A (en) | 2020-06-11 | 2023-02-01 | Provention Bio Inc | Methods and compositions for the prevention of type 1 diabetes |
| JP2024520444A (ja) | 2021-05-24 | 2024-05-24 | プロヴェンション・バイオ・インコーポレイテッド | 1型糖尿病を治療するための方法 |
| US20250051337A1 (en) * | 2021-09-28 | 2025-02-13 | Sanford Burnham Prebys Medical Discovery Institute | Inhibitors of serine/threonine protein kinase stk3 or stk4 and uses thereof |
Family Cites Families (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3037980A (en) * | 1955-08-18 | 1962-06-05 | Burroughs Wellcome Co | Pyrrolopyrimidine vasodilators and method of making them |
| GB915303A (en) | 1958-03-13 | 1963-01-09 | Wellcome Found | Pyrrolo[2,3-d]pyrimidine derivatives and the manufacture thereof |
| NO169490C (no) | 1988-03-24 | 1992-07-01 | Takeda Chemical Industries Ltd | Analogifremgangsmaate for fremstilling av terapeutisk aktive pyrrolopyrimidinderivater |
| JP2983254B2 (ja) * | 1989-06-14 | 1999-11-29 | 武田薬品工業株式会社 | ピロロ〔2,3―d〕ピリミジン誘導体の製造法およびその中間体 |
| ES2150941T3 (es) * | 1992-04-03 | 2000-12-16 | Upjohn Co | Aminas biciclicas-heterociclicas eficaces farmaceuticamente. |
| US6136595A (en) | 1993-07-29 | 2000-10-24 | St. Jude Children's Research Hospital | Jak kinases and regulations of cytokine signal transduction |
| US5389509A (en) | 1993-10-04 | 1995-02-14 | Eastman Kodak Company | Ultrathin high chloride tabular grain emulsions |
| IL112249A (en) | 1994-01-25 | 2001-11-25 | Warner Lambert Co | Pharmaceutical compositions containing di and tricyclic pyrimidine derivatives for inhibiting tyrosine kinases of the epidermal growth factor receptor family and some new such compounds |
| DE59500788D1 (de) | 1994-05-03 | 1997-11-20 | Ciba Geigy Ag | Pyrrolopyrimidinderivate mit antiproliferativer Wirkung |
| JP3290666B2 (ja) * | 1995-06-07 | 2002-06-10 | ファイザー・インコーポレーテッド | 複素環式の縮合環ピリミジン誘導体 |
| JP4010563B2 (ja) | 1995-07-05 | 2007-11-21 | イー・アイ・デユポン・ドウ・ヌムール・アンド・カンパニー | 殺菌・殺カビ性のピリミジノン類 |
| SI9620103A (sl) * | 1995-07-06 | 1998-10-31 | Novartis Ag | Pirolopirimidini in postopki za njihovo pripravo |
| AR004010A1 (es) | 1995-10-11 | 1998-09-30 | Glaxo Group Ltd | Compuestos heterociclicos |
| BR9607089A (pt) | 1995-11-14 | 1997-11-11 | Pharmacia & Upjohn Spa | Compostos de pirimidina condensada biciclica composição farmacéutica utilização e produtos que contém os mesmos e processo para a preparação desses compostos |
| AU1441497A (en) * | 1996-01-23 | 1997-08-20 | Novartis Ag | Pyrrolopyrimidines and processes for their preparation |
| CH690773A5 (de) | 1996-02-01 | 2001-01-15 | Novartis Ag | Pyrrolo(2,3-d)pyrimide und ihre Verwendung. |
| GB9604361D0 (en) * | 1996-02-29 | 1996-05-01 | Pharmacia Spa | 4-Substituted pyrrolopyrimidine compounds as tyrosine kinase inhibitors |
| AU1794697A (en) | 1996-03-06 | 1997-09-22 | Novartis Ag | 7-alkyl-pyrrolo{2,3-d}pyrimidines |
| AU3176297A (en) | 1996-06-25 | 1998-01-14 | Novartis Ag | Substituted 7-amino-pyrrolo{3,2-d}pyrimidines and the use thereof |
| EA199900021A1 (ru) | 1996-07-13 | 1999-08-26 | Глаксо, Груп Лимитед | Бициклические гетероароматические соединения в качестве ингибиторов протеинтирозинкиназы |
| HRP970371A2 (en) | 1996-07-13 | 1998-08-31 | Kathryn Jane Smith | Heterocyclic compounds |
| WO1998007726A1 (en) * | 1996-08-23 | 1998-02-26 | Novartis Ag | Substituted pyrrolopyrimidines and processes for their preparation |
| WO1998023613A1 (en) * | 1996-11-27 | 1998-06-04 | Pfizer Inc. | Fused bicyclic pyrimidine derivatives |
| WO1998033798A2 (en) | 1997-02-05 | 1998-08-06 | Warner Lambert Company | Pyrido[2,3-d]pyrimidines and 4-amino-pyrimidines as inhibitors of cell proliferation |
| JP2001520748A (ja) | 1997-03-24 | 2001-10-30 | ファルマシア・アンド・アップジョン・カンパニー | Jak2/サイトカイン受容体結合の阻害剤を同定する方法 |
| AU3378799A (en) | 1998-04-02 | 1999-10-25 | Neurogen Corporation | Aminoalkyl substituted pyrrolo(2,3-b)pyridine and pyrrolo(2,3-d)pyrimidine derivatives: modulators of crf1 receptors |
| IL139641A0 (en) | 1998-05-28 | 2002-02-10 | Parker Hughes Inst | Quinazolines for treating brain tumor |
| PA8474101A1 (es) * | 1998-06-19 | 2000-09-29 | Pfizer Prod Inc | Compuestos de pirrolo [2,3-d] pirimidina |
| SK286640B6 (sk) | 1998-06-19 | 2009-03-05 | Pfizer Products Inc. | Pyrol [2,3-d] pyrimidínová zlúčenina, jej použitie na výrobu liečiva, farmaceutická kompozícia s jej obsahom, použitie jej kombinácie s ďalšími činidlami a súpravy s jej obsahom na výrobu liečiva |
| WO2000000202A1 (en) | 1998-06-30 | 2000-01-06 | Parker Hughes Institute | Method for inhibiting c-jun expression using jak-3 inhibitors |
| KR20010089171A (ko) | 1998-08-21 | 2001-09-29 | 추후제출 | 퀴나졸린 유도체 |
| ID29028A (id) | 1998-09-18 | 2001-07-26 | Basf Ag | Pirolopirimidina sebagai penghambat protein kinase |
| US6080747A (en) | 1999-03-05 | 2000-06-27 | Hughes Institute | JAK-3 inhibitors for treating allergic disorders |
| US6664252B2 (en) * | 1999-12-02 | 2003-12-16 | Osi Pharmaceuticals, Inc. | 4-aminopyrrolo[2,3-d]pyrimidine compounds specific to adenosine A2a receptor and uses thereof |
| DE60037345T2 (de) | 1999-12-10 | 2008-11-13 | Pfizer Products Inc., Groton | Pyrrolo(2,3-d)pyrimidin-Verbindungen |
| ATE423120T1 (de) * | 2000-06-26 | 2009-03-15 | Pfizer Prod Inc | Pyrroloä2,3-düpyrimidin verbindungen als immunosuppressive wirkstoffe |
| US6673802B2 (en) * | 2000-12-01 | 2004-01-06 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A3 receptor and uses thereof |
| US6680324B2 (en) * | 2000-12-01 | 2004-01-20 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A1 receptors and uses thereof |
| US7301023B2 (en) | 2001-05-31 | 2007-11-27 | Pfizer Inc. | Chiral salt resolution |
| PY0228255A (es) | 2001-12-06 | 2004-06-01 | Pfizer Prod Inc | Compuestos cristalinos novedosos |
| KR20040006555A (ko) * | 2002-07-12 | 2004-01-24 | 삼성전자주식회사 | 액정 표시 장치 |
-
1999
- 1999-05-26 PA PA19998474101A patent/PA8474101A1/es unknown
- 1999-05-27 HN HN1999000083A patent/HN1999000083A/es unknown
- 1999-06-14 DK DK99923800T patent/DK1087971T3/da active
- 1999-06-14 PL PL345118A patent/PL198639B1/pl unknown
- 1999-06-14 AU AU40545/99A patent/AU758427B2/en not_active Ceased
- 1999-06-14 TR TR2000/03720T patent/TR200003720T2/xx unknown
- 1999-06-14 YU YU79700A patent/YU79700A/sh unknown
- 1999-06-14 ES ES99923800T patent/ES2223172T3/es not_active Expired - Lifetime
- 1999-06-14 SI SI9930604T patent/SI1087971T1/xx unknown
- 1999-06-14 AT AT99923800T patent/ATE270673T1/de not_active IP Right Cessation
- 1999-06-14 EP EP99923800A patent/EP1087971B1/de not_active Expired - Lifetime
- 1999-06-14 SK SK1899-2000A patent/SK286685B6/sk not_active IP Right Cessation
- 1999-06-14 KR KR10-2000-7014403A patent/KR100452054B1/ko not_active Expired - Fee Related
- 1999-06-14 BR BR9912171-9A patent/BR9912171A/pt not_active Application Discontinuation
- 1999-06-14 CN CN99807519A patent/CN1125070C/zh not_active Expired - Fee Related
- 1999-06-14 PT PT99923800T patent/PT1087971E/pt unknown
- 1999-06-14 UA UA2000127301A patent/UA63013C2/uk unknown
- 1999-06-14 CZ CZ20004726A patent/CZ20004726A3/cs unknown
- 1999-06-14 DE DE69918552T patent/DE69918552T2/de not_active Expired - Lifetime
- 1999-06-14 JP JP2000554734A patent/JP3497823B2/ja not_active Expired - Fee Related
- 1999-06-14 NZ NZ508034A patent/NZ508034A/en unknown
- 1999-06-14 HU HU0103472A patent/HUP0103472A3/hu unknown
- 1999-06-14 TW TW088109933A patent/TW542834B/zh not_active IP Right Cessation
- 1999-06-14 ID IDW20002642A patent/ID27595A/id unknown
- 1999-06-14 EA EA200001188A patent/EA006034B1/ru not_active IP Right Cessation
- 1999-06-14 OA OA1200000348A patent/OA11571A/en unknown
- 1999-06-14 WO PCT/IB1999/001110 patent/WO1999065909A1/en not_active Ceased
- 1999-06-14 EA EA200500614A patent/EA010377B1/ru not_active IP Right Cessation
- 1999-06-14 CA CA002335186A patent/CA2335186C/en not_active Expired - Fee Related
- 1999-06-14 IL IL13959899A patent/IL139598A0/xx active IP Right Grant
- 1999-06-14 HR HR20000886A patent/HRP20000886B1/xx not_active IP Right Cessation
- 1999-06-16 TN TNTNSN99125A patent/TNSN99125A1/fr unknown
- 1999-06-16 EG EG72599A patent/EG23758A/xx active
- 1999-06-16 MA MA25629A patent/MA26653A1/fr unknown
- 1999-06-16 PE PE1999000530A patent/PE20000639A1/es not_active Application Discontinuation
- 1999-06-17 ZA ZA9904003A patent/ZA994003B/xx unknown
- 1999-06-17 AP APAP/P/1999/001583A patent/AP1157A/en active
- 1999-06-17 AR ARP990102915A patent/AR016498A1/es active IP Right Grant
- 1999-06-17 US US09/335,030 patent/US6635762B1/en not_active Expired - Fee Related
- 1999-06-17 MY MYPI99002503A patent/MY125802A/en unknown
- 1999-06-18 CO CO99038335A patent/CO5320601A1/es not_active Application Discontinuation
- 1999-06-18 GT GT199900091A patent/GT199900091A/es unknown
- 1999-06-28 SA SA99200285A patent/SA99200285B1/ar unknown
-
2000
- 2000-11-09 IL IL139598A patent/IL139598A/en not_active IP Right Cessation
- 2000-11-21 IS IS5721A patent/IS2395B/is unknown
- 2000-11-21 IS IS5722A patent/IS2461B/is unknown
- 2000-12-18 NO NO20006454A patent/NO318786B1/no not_active IP Right Cessation
-
2001
- 2001-01-08 BG BG105122A patent/BG65063B1/bg unknown
-
2003
- 2003-08-13 US US10/640,079 patent/US7569569B2/en not_active Expired - Fee Related
-
2005
- 2005-01-13 NO NO20050201A patent/NO20050201L/no unknown
-
2007
- 2007-07-02 GE GEAP200710158A patent/GEP20084336B/en unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69918552T2 (de) | Pyrrolo(2,3-d)pyrimidin-verbindungen | |
| DE69916833T2 (de) | Pyrrolo(2,3-d)pyrimidin-verbindungen | |
| DE60118917T2 (de) | PYRROLO[2,3-d]PYRIMIDIN-VERBINDUNGEN ALS IMMUNOSUPPRESSIVE WIRKSTOFFE ' | |
| DE60037345T2 (de) | Pyrrolo(2,3-d)pyrimidin-Verbindungen | |
| US7494992B2 (en) | Antiparasitic terpene alkaloids | |
| DE60216115T2 (de) | BENZIMIDAZO 4,5-föISOCHINOLINON-DERIVATE | |
| DE60209251T2 (de) | Adenosine a2a receptor antagonisten | |
| DE4116582A1 (de) | Bicyclische 1-aza-cycloalkane | |
| DE60026848T2 (de) | C-6 ringsubstituierte pyridoä1,2-aübenzimidazolderivate zur behandlung von störungen des zentralen nervensystems | |
| MXPA00012853A (en) | PYRROLO[2,3-d]PYRIMIDINE COMPOUNDS | |
| DE10309927A1 (de) | Neue substituierte Imidazo-pyridinone und Imidazo-pyridazinone, ihre Herstellung und ihre Verwendung als Arzneimittel |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition |