DE69738468T2 - Substituierte pyrrolopyrimidine und verfahren zu ihrer herstellung - Google Patents
Substituierte pyrrolopyrimidine und verfahren zu ihrer herstellung Download PDFInfo
- Publication number
- DE69738468T2 DE69738468T2 DE69738468T DE69738468T DE69738468T2 DE 69738468 T2 DE69738468 T2 DE 69738468T2 DE 69738468 T DE69738468 T DE 69738468T DE 69738468 T DE69738468 T DE 69738468T DE 69738468 T2 DE69738468 T2 DE 69738468T2
- Authority
- DE
- Germany
- Prior art keywords
- formula
- pyrrolo
- pyrimidine
- chloroanilino
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 238000000034 method Methods 0.000 title claims description 48
- 238000004519 manufacturing process Methods 0.000 title description 4
- 150000004944 pyrrolopyrimidines Chemical class 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 203
- JJTNLWSCFYERCK-UHFFFAOYSA-N 7h-pyrrolo[2,3-d]pyrimidine Chemical class N1=CN=C2NC=CC2=C1 JJTNLWSCFYERCK-UHFFFAOYSA-N 0.000 claims abstract description 5
- -1 phenylureido Chemical group 0.000 claims description 227
- 125000006239 protecting group Chemical group 0.000 claims description 76
- 125000000217 alkyl group Chemical group 0.000 claims description 61
- 125000001424 substituent group Chemical group 0.000 claims description 54
- 238000006243 chemical reaction Methods 0.000 claims description 50
- 150000003839 salts Chemical class 0.000 claims description 48
- 125000000524 functional group Chemical group 0.000 claims description 32
- 238000002360 preparation method Methods 0.000 claims description 31
- 239000000460 chlorine Substances 0.000 claims description 29
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 25
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 24
- 239000001257 hydrogen Substances 0.000 claims description 20
- 229910052739 hydrogen Inorganic materials 0.000 claims description 20
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 20
- 206010028980 Neoplasm Diseases 0.000 claims description 15
- 239000008194 pharmaceutical composition Substances 0.000 claims description 15
- 125000004432 carbon atom Chemical group C* 0.000 claims description 14
- 125000003545 alkoxy group Chemical group 0.000 claims description 13
- 229910052801 chlorine Inorganic materials 0.000 claims description 13
- 125000004299 tetrazol-5-yl group Chemical group [H]N1N=NC(*)=N1 0.000 claims description 13
- 125000003282 alkyl amino group Chemical group 0.000 claims description 12
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 10
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 10
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 9
- 230000008569 process Effects 0.000 claims description 9
- 125000004343 1-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C([H])([H])[H] 0.000 claims description 8
- 241001465754 Metazoa Species 0.000 claims description 7
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 claims description 7
- 150000001412 amines Chemical class 0.000 claims description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 7
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 7
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 claims description 7
- BYYLPECJKOEHBV-UHFFFAOYSA-N 4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidine-6-carbaldehyde Chemical compound ClC1=CC=CC(NC=2C=3C=C(C=O)NC=3N=CN=2)=C1 BYYLPECJKOEHBV-UHFFFAOYSA-N 0.000 claims description 6
- 229910052783 alkali metal Inorganic materials 0.000 claims description 6
- 125000005036 alkoxyphenyl group Chemical group 0.000 claims description 6
- 150000001448 anilines Chemical class 0.000 claims description 6
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 6
- 229910052794 bromium Chemical group 0.000 claims description 6
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 6
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 claims description 6
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 claims description 6
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 5
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 claims description 5
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 claims description 5
- 125000000972 4,5-dimethylthiazol-2-yl group Chemical group [H]C([H])([H])C1=C(N=C(*)S1)C([H])([H])[H] 0.000 claims description 4
- BNMRACHWXQYRHO-UHFFFAOYSA-N 4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidine-6-carbothioamide Chemical compound N1=CN=C2NC(C(=S)N)=CC2=C1NC1=CC=CC(Cl)=C1 BNMRACHWXQYRHO-UHFFFAOYSA-N 0.000 claims description 4
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 4
- 239000012024 dehydrating agents Substances 0.000 claims description 4
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 claims description 4
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 claims description 4
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 4
- YYELWCZQXDCXCI-UHFFFAOYSA-N 1-[3-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-3-ethylurea Chemical compound CCNC(=O)NC1=CC=CC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=C1 YYELWCZQXDCXCI-UHFFFAOYSA-N 0.000 claims description 3
- VAKYVBRASQJLDI-UHFFFAOYSA-N 2-methylpropyl n-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]carbamate Chemical compound C1=CC(NC(=O)OCC(C)C)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 VAKYVBRASQJLDI-UHFFFAOYSA-N 0.000 claims description 3
- 125000000590 4-methylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 claims description 3
- NIFHYNGZYDVDBU-UHFFFAOYSA-N 6-[4-(benzylamino)phenyl]-n-(3-chlorophenyl)-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C=2C=CC(NCC=3C=CC=CC=3)=CC=2)=C1 NIFHYNGZYDVDBU-UHFFFAOYSA-N 0.000 claims description 3
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 claims description 3
- 241000282412 Homo Species 0.000 claims description 3
- OGEBYZFAAZVIHT-UHFFFAOYSA-N N-(3-chlorophenyl)-6-(2H-tetrazol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C=2NN=NN=2)=C1 OGEBYZFAAZVIHT-UHFFFAOYSA-N 0.000 claims description 3
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 claims description 3
- 125000002777 acetyl group Chemical class [H]C([H])([H])C(*)=O 0.000 claims description 3
- 150000001351 alkyl iodides Chemical class 0.000 claims description 3
- 125000005281 alkyl ureido group Chemical group 0.000 claims description 3
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 claims description 3
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- NUXWAOSDMYECDX-UHFFFAOYSA-N ethyl n-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]carbamate Chemical compound C1=CC(NC(=O)OCC)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 NUXWAOSDMYECDX-UHFFFAOYSA-N 0.000 claims description 3
- 150000002443 hydroxylamines Chemical class 0.000 claims description 3
- 239000012948 isocyanate Substances 0.000 claims description 3
- BRBPDQKNOPJHDS-UHFFFAOYSA-N n'-[3-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-n,n-diethylmethanimidamide Chemical compound CCN(CC)C=NC1=CC=CC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=C1 BRBPDQKNOPJHDS-UHFFFAOYSA-N 0.000 claims description 3
- HYSPQBYMQIZTMH-UHFFFAOYSA-N n'-[3-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-n,n-dimethylmethanimidamide Chemical compound CN(C)C=NC1=CC=CC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=C1 HYSPQBYMQIZTMH-UHFFFAOYSA-N 0.000 claims description 3
- PRVIBBCNUUBOSX-UHFFFAOYSA-N n-(3-chlorophenyl)-6-(1,3-thiazol-2-yl)-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C=2SC=CN=2)=C1 PRVIBBCNUUBOSX-UHFFFAOYSA-N 0.000 claims description 3
- OKQBRJKNDDAIKW-UHFFFAOYSA-N n-(3-chlorophenyl)-6-[4-(morpholin-4-ylmethylideneamino)phenyl]-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C=2C=CC(=CC=2)N=CN2CCOCC2)=C1 OKQBRJKNDDAIKW-UHFFFAOYSA-N 0.000 claims description 3
- GBCGNNNFIRWGCW-UHFFFAOYSA-N n-(3-chlorophenyl)-6-[4-[(4-methylpiperazin-1-yl)methylideneamino]phenyl]-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound C1CN(C)CCN1C=NC1=CC=C(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)C=C1 GBCGNNNFIRWGCW-UHFFFAOYSA-N 0.000 claims description 3
- ROLDUDKLNYYONL-UHFFFAOYSA-N n-[3-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]ethanesulfonamide Chemical compound CCS(=O)(=O)NC1=CC=CC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=C1 ROLDUDKLNYYONL-UHFFFAOYSA-N 0.000 claims description 3
- RBEFWIDYHAIBSL-UHFFFAOYSA-N n-[3-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]methanesulfonamide Chemical compound CS(=O)(=O)NC1=CC=CC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=C1 RBEFWIDYHAIBSL-UHFFFAOYSA-N 0.000 claims description 3
- PKNAMUVGIGLHAY-UHFFFAOYSA-N n-[3-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]propane-2-sulfonamide Chemical compound CC(C)S(=O)(=O)NC1=CC=CC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=C1 PKNAMUVGIGLHAY-UHFFFAOYSA-N 0.000 claims description 3
- WVICNPJYDFMIGL-OAHLLOKOSA-N n-[3-[4-[[(1r)-1-phenylethyl]amino]-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]ethanesulfonamide Chemical compound CCS(=O)(=O)NC1=CC=CC(C=2NC3=NC=NC(N[C@H](C)C=4C=CC=CC=4)=C3C=2)=C1 WVICNPJYDFMIGL-OAHLLOKOSA-N 0.000 claims description 3
- KQWSURRAHQPGAF-CQSZACIVSA-N n-[3-[4-[[(1r)-1-phenylethyl]amino]-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]methanesulfonamide Chemical compound N([C@H](C)C=1C=CC=CC=1)C(C=1C=2)=NC=NC=1NC=2C1=CC=CC(NS(C)(=O)=O)=C1 KQWSURRAHQPGAF-CQSZACIVSA-N 0.000 claims description 3
- DZRXPIWDNCNPOM-MRXNPFEDSA-N n-[3-[4-[[(1r)-1-phenylethyl]amino]-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]propane-2-sulfonamide Chemical compound CC(C)S(=O)(=O)NC1=CC=CC(C=2NC3=NC=NC(N[C@H](C)C=4C=CC=CC=4)=C3C=2)=C1 DZRXPIWDNCNPOM-MRXNPFEDSA-N 0.000 claims description 3
- WLMINMLKCZPIDN-UHFFFAOYSA-N n-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]benzenesulfonamide Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C=2C=CC(NS(=O)(=O)C=3C=CC=CC=3)=CC=2)=C1 WLMINMLKCZPIDN-UHFFFAOYSA-N 0.000 claims description 3
- RJHPCDDFUQAQPU-UHFFFAOYSA-N n-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]ethanesulfonamide Chemical compound C1=CC(NS(=O)(=O)CC)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 RJHPCDDFUQAQPU-UHFFFAOYSA-N 0.000 claims description 3
- PKPOERZOLNFQEC-UHFFFAOYSA-N n-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]furan-2-carboxamide Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C=2C=CC(NC(=O)C=3OC=CC=3)=CC=2)=C1 PKPOERZOLNFQEC-UHFFFAOYSA-N 0.000 claims description 3
- WBVNHFVJQUXGDJ-UHFFFAOYSA-N n-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]methanesulfonamide Chemical compound C1=CC(NS(=O)(=O)C)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 WBVNHFVJQUXGDJ-UHFFFAOYSA-N 0.000 claims description 3
- NDIAGRBKTSAOQM-UHFFFAOYSA-N n-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]propane-2-sulfonamide Chemical compound C1=CC(NS(=O)(=O)C(C)C)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 NDIAGRBKTSAOQM-UHFFFAOYSA-N 0.000 claims description 3
- KFHQCHGEIQBNKA-UHFFFAOYSA-N n-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]thiophene-2-carboxamide Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C=2C=CC(NC(=O)C=3SC=CC=3)=CC=2)=C1 KFHQCHGEIQBNKA-UHFFFAOYSA-N 0.000 claims description 3
- ZXEAZQRLYXDOIU-OAHLLOKOSA-N n-[4-[4-[[(1r)-1-phenylethyl]amino]-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]ethanesulfonamide Chemical compound C1=CC(NS(=O)(=O)CC)=CC=C1C(NC1=NC=N2)=CC1=C2N[C@H](C)C1=CC=CC=C1 ZXEAZQRLYXDOIU-OAHLLOKOSA-N 0.000 claims description 3
- KJPHJVDDQOMOSH-MRXNPFEDSA-N n-[4-[4-[[(1r)-1-phenylethyl]amino]-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]propane-2-sulfonamide Chemical compound C1=CC(NS(=O)(=O)C(C)C)=CC=C1C(NC1=NC=N2)=CC1=C2N[C@H](C)C1=CC=CC=C1 KJPHJVDDQOMOSH-MRXNPFEDSA-N 0.000 claims description 3
- MKLSHESXKCZSML-UHFFFAOYSA-N propan-2-yl n-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]carbamate Chemical compound C1=CC(NC(=O)OC(C)C)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 MKLSHESXKCZSML-UHFFFAOYSA-N 0.000 claims description 3
- LDUVGYTUBRSPKL-UBKPWBPPSA-N (NE)-N-[[4-(3-chloroanilino)-7H-pyrrolo[2,3-d]pyrimidin-6-yl]methylidene]hydroxylamine Chemical compound N1=CN=C2NC(/C=N/O)=CC2=C1NC1=CC=CC(Cl)=C1 LDUVGYTUBRSPKL-UBKPWBPPSA-N 0.000 claims description 2
- LDUVGYTUBRSPKL-FMQZQXMHSA-N (NZ)-N-[[4-(3-chloroanilino)-7H-pyrrolo[2,3-d]pyrimidin-6-yl]methylidene]hydroxylamine Chemical compound N1=CN=C2NC(\C=N/O)=CC2=C1NC1=CC=CC(Cl)=C1 LDUVGYTUBRSPKL-FMQZQXMHSA-N 0.000 claims description 2
- TWNRMTYWTBYQQS-UHFFFAOYSA-N 1-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-3-ethylurea Chemical compound C1=CC(NC(=O)NCC)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 TWNRMTYWTBYQQS-UHFFFAOYSA-N 0.000 claims description 2
- UCRKSQGCZVGBGX-UHFFFAOYSA-N 1-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-3-methylthiourea Chemical compound C1=CC(NC(=S)NC)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 UCRKSQGCZVGBGX-UHFFFAOYSA-N 0.000 claims description 2
- JEWOIKNJTXJTGH-UHFFFAOYSA-N 4-(4-anilino-7h-pyrrolo[2,3-d]pyrimidin-6-yl)-2-methoxyphenol;hydrochloride Chemical compound Cl.C1=C(O)C(OC)=CC(C=2NC3=NC=NC(NC=4C=CC=CC=4)=C3C=2)=C1 JEWOIKNJTXJTGH-UHFFFAOYSA-N 0.000 claims description 2
- WXZNMFZVZMNGIO-UHFFFAOYSA-N 4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]-2-methoxyphenol;hydrochloride Chemical compound Cl.C1=C(O)C(OC)=CC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=C1 WXZNMFZVZMNGIO-UHFFFAOYSA-N 0.000 claims description 2
- BCZCVGXQOLPACI-UHFFFAOYSA-N N-(3-chlorophenyl)-6-(2-methyltetrazol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound CN1N=NC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=N1 BCZCVGXQOLPACI-UHFFFAOYSA-N 0.000 claims description 2
- ZRSLTYRSYOZHIR-UHFFFAOYSA-N N-(3-chlorophenyl)-6-(4-ethyl-1,3-thiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound CCC1=CSC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=N1 ZRSLTYRSYOZHIR-UHFFFAOYSA-N 0.000 claims description 2
- KJCWFUJEFVWZSE-UHFFFAOYSA-N [4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]-morpholin-4-ylmethanone Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C(=O)N2CCOCC2)=C1 KJCWFUJEFVWZSE-UHFFFAOYSA-N 0.000 claims description 2
- SWTAWOJNKFZMMT-UHFFFAOYSA-N [4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-morpholin-4-ylmethanone Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C=2C=CC(=CC=2)C(=O)N2CCOCC2)=C1 SWTAWOJNKFZMMT-UHFFFAOYSA-N 0.000 claims description 2
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 claims description 2
- 150000002431 hydrogen Chemical class 0.000 claims description 2
- 150000002513 isocyanates Chemical class 0.000 claims description 2
- 150000002540 isothiocyanates Chemical class 0.000 claims description 2
- YBZDNHCFYNZPHI-UHFFFAOYSA-N n'-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-n,n-dimethylmethanimidamide Chemical compound C1=CC(N=CN(C)C)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 YBZDNHCFYNZPHI-UHFFFAOYSA-N 0.000 claims description 2
- JJJQHTCJNMSSJV-UHFFFAOYSA-N pyrrolo[2,3-d]pyrimidin-4-one Chemical class O=C1N=CN=C2N=CC=C12 JJJQHTCJNMSSJV-UHFFFAOYSA-N 0.000 claims description 2
- 150000004943 pyrrolo[2,3-d]pyrimidines Chemical class 0.000 claims description 2
- 150000003512 tertiary amines Chemical class 0.000 claims description 2
- 230000001225 therapeutic effect Effects 0.000 claims description 2
- UTIXVEICQYJJRY-UHFFFAOYSA-N 1-[3-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-3-methylthiourea Chemical compound CNC(=S)NC1=CC=CC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=C1 UTIXVEICQYJJRY-UHFFFAOYSA-N 0.000 claims 1
- YPCURGZEPMFYLX-OAHLLOKOSA-N 1-ethyl-3-[4-[4-[[(1r)-1-phenylethyl]amino]-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]urea Chemical compound C1=CC(NC(=O)NCC)=CC=C1C(NC1=NC=N2)=CC1=C2N[C@H](C)C1=CC=CC=C1 YPCURGZEPMFYLX-OAHLLOKOSA-N 0.000 claims 1
- IKELJGAIXVSXJM-CQSZACIVSA-N 1-methyl-3-[4-[4-[[(1r)-1-phenylethyl]amino]-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]thiourea Chemical compound C1=CC(NC(=S)NC)=CC=C1C(NC1=NC=N2)=CC1=C2N[C@H](C)C1=CC=CC=C1 IKELJGAIXVSXJM-CQSZACIVSA-N 0.000 claims 1
- QOPHSBUZTVLVBJ-UHFFFAOYSA-N N-(3-chlorophenyl)-6-(1-methyltetrazol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound CN1N=NN=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 QOPHSBUZTVLVBJ-UHFFFAOYSA-N 0.000 claims 1
- GAABGZUDSKAVGH-UHFFFAOYSA-N N-(3-chlorophenyl)-6-(4,5-dimethyl-1,3-thiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound S1C(C)=C(C)N=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 GAABGZUDSKAVGH-UHFFFAOYSA-N 0.000 claims 1
- BPEIBTXCKPKWLE-UHFFFAOYSA-N N-(3-chlorophenyl)-6-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]-7H-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound C1=CC(OC)=CC=C1C1=CSC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=N1 BPEIBTXCKPKWLE-UHFFFAOYSA-N 0.000 claims 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims 1
- 238000002512 chemotherapy Methods 0.000 claims 1
- SDSLGPIDULPJQG-MRXNPFEDSA-N n,n-dimethyl-n'-[3-[4-[[(1r)-1-phenylethyl]amino]-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]methanimidamide Chemical compound N([C@H](C)C=1C=CC=CC=1)C(C=1C=2)=NC=NC=1NC=2C1=CC=CC(N=CN(C)C)=C1 SDSLGPIDULPJQG-MRXNPFEDSA-N 0.000 claims 1
- UNUHIYOQLRIQNQ-MRXNPFEDSA-N n,n-dimethyl-n'-[4-[4-[[(1r)-1-phenylethyl]amino]-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]methanimidamide Chemical compound N([C@H](C)C=1C=CC=CC=1)C(C=1C=2)=NC=NC=1NC=2C1=CC=C(N=CN(C)C)C=C1 UNUHIYOQLRIQNQ-MRXNPFEDSA-N 0.000 claims 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 abstract description 13
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 abstract description 13
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 abstract description 10
- 230000000694 effects Effects 0.000 abstract description 9
- 108091000080 Phosphotransferase Proteins 0.000 abstract description 8
- 102000020233 phosphotransferase Human genes 0.000 abstract description 8
- 239000002246 antineoplastic agent Substances 0.000 abstract 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 114
- 150000003254 radicals Chemical class 0.000 description 80
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 75
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 61
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 57
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 56
- 239000000203 mixture Substances 0.000 description 53
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 48
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 45
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 44
- 235000002639 sodium chloride Nutrition 0.000 description 43
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 32
- 239000000243 solution Substances 0.000 description 32
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 27
- 239000011541 reaction mixture Substances 0.000 description 23
- 239000007858 starting material Substances 0.000 description 23
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 21
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 21
- 238000009835 boiling Methods 0.000 description 21
- 239000002904 solvent Substances 0.000 description 21
- 238000004128 high performance liquid chromatography Methods 0.000 description 20
- 239000000725 suspension Substances 0.000 description 19
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 17
- 239000004480 active ingredient Substances 0.000 description 17
- 210000004027 cell Anatomy 0.000 description 17
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 239000002253 acid Substances 0.000 description 15
- 239000013078 crystal Substances 0.000 description 15
- 229910052757 nitrogen Inorganic materials 0.000 description 15
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 14
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 14
- 229910052736 halogen Inorganic materials 0.000 description 14
- 150000002367 halogens Chemical class 0.000 description 14
- 230000005764 inhibitory process Effects 0.000 description 14
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 14
- 239000002775 capsule Substances 0.000 description 13
- 235000014113 dietary fatty acids Nutrition 0.000 description 13
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 13
- 239000000194 fatty acid Substances 0.000 description 13
- 229930195729 fatty acid Natural products 0.000 description 13
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- 102000001253 Protein Kinase Human genes 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 108060006633 protein kinase Proteins 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 229920001223 polyethylene glycol Chemical class 0.000 description 11
- 239000000741 silica gel Substances 0.000 description 11
- 229910002027 silica gel Inorganic materials 0.000 description 11
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 10
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 10
- 125000003277 amino group Chemical group 0.000 description 10
- 125000003118 aryl group Chemical group 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 9
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 9
- 239000013543 active substance Substances 0.000 description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- 238000001914 filtration Methods 0.000 description 9
- SPEUIVXLLWOEMJ-UHFFFAOYSA-N 1,1-dimethoxyethane Chemical compound COC(C)OC SPEUIVXLLWOEMJ-UHFFFAOYSA-N 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 8
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 8
- 239000003112 inhibitor Substances 0.000 description 8
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 8
- 239000003921 oil Substances 0.000 description 8
- 235000019198 oils Nutrition 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 8
- 238000006722 reduction reaction Methods 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 238000005406 washing Methods 0.000 description 8
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 7
- UZDFIMXOVIIKQO-UHFFFAOYSA-N 6-(4-aminophenyl)-n-(3-chlorophenyl)-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound C1=CC(N)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 UZDFIMXOVIIKQO-UHFFFAOYSA-N 0.000 description 7
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 7
- 102400001368 Epidermal growth factor Human genes 0.000 description 7
- 101800003838 Epidermal growth factor Proteins 0.000 description 7
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 7
- 239000002202 Polyethylene glycol Chemical class 0.000 description 7
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 7
- 125000005115 alkyl carbamoyl group Chemical group 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 7
- 201000010099 disease Diseases 0.000 description 7
- 229940116977 epidermal growth factor Drugs 0.000 description 7
- 235000019253 formic acid Nutrition 0.000 description 7
- 239000012442 inert solvent Substances 0.000 description 7
- 239000000543 intermediate Substances 0.000 description 7
- 239000012074 organic phase Substances 0.000 description 7
- 230000009467 reduction Effects 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 102000001301 EGF receptor Human genes 0.000 description 6
- 108060006698 EGF receptor Proteins 0.000 description 6
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 6
- LGDSHSYDSCRFAB-UHFFFAOYSA-N Methyl isothiocyanate Chemical compound CN=C=S LGDSHSYDSCRFAB-UHFFFAOYSA-N 0.000 description 6
- 239000007868 Raney catalyst Substances 0.000 description 6
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 6
- 229910000564 Raney nickel Inorganic materials 0.000 description 6
- 239000008346 aqueous phase Substances 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 125000001589 carboacyl group Chemical group 0.000 description 6
- 239000003054 catalyst Substances 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 238000002425 crystallisation Methods 0.000 description 6
- 230000008025 crystallization Effects 0.000 description 6
- 239000003995 emulsifying agent Substances 0.000 description 6
- 238000000605 extraction Methods 0.000 description 6
- 229920000159 gelatin Polymers 0.000 description 6
- 235000019322 gelatine Nutrition 0.000 description 6
- 235000011187 glycerol Nutrition 0.000 description 6
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 6
- 229930195733 hydrocarbon Natural products 0.000 description 6
- 150000002430 hydrocarbons Chemical class 0.000 description 6
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 239000002674 ointment Substances 0.000 description 6
- 229920006395 saturated elastomer Polymers 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 6
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 5
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 150000007513 acids Chemical class 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 238000006136 alcoholysis reaction Methods 0.000 description 5
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 5
- 239000007795 chemical reaction product Substances 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 239000006071 cream Substances 0.000 description 5
- JLYIDLKBGFXUTE-UHFFFAOYSA-N ethyl 4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidine-6-carboxylate Chemical compound N1=CN=C2NC(C(=O)OCC)=CC2=C1NC1=CC=CC(Cl)=C1 JLYIDLKBGFXUTE-UHFFFAOYSA-N 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 238000003797 solvolysis reaction Methods 0.000 description 5
- 239000000600 sorbitol Substances 0.000 description 5
- 150000005846 sugar alcohols Polymers 0.000 description 5
- 239000000454 talc Substances 0.000 description 5
- 229910052623 talc Inorganic materials 0.000 description 5
- 235000012222 talc Nutrition 0.000 description 5
- 238000004809 thin layer chromatography Methods 0.000 description 5
- HRRSTGSFDNCRBK-UHFFFAOYSA-N 4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidine-6-carboxamide Chemical compound N1=CN=C2NC(C(=O)N)=CC2=C1NC1=CC=CC(Cl)=C1 HRRSTGSFDNCRBK-UHFFFAOYSA-N 0.000 description 4
- KFNHYCUETQTMIC-UHFFFAOYSA-N 6-(3-aminophenyl)-n-(3-chlorophenyl)-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound NC1=CC=CC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=C1 KFNHYCUETQTMIC-UHFFFAOYSA-N 0.000 description 4
- OVWSZWFGFGIFNR-CYBMUJFWSA-N 6-(3-aminophenyl)-n-[(1r)-1-phenylethyl]-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound N([C@H](C)C=1C=CC=CC=1)C(C=1C=2)=NC=NC=1NC=2C1=CC=CC(N)=C1 OVWSZWFGFGIFNR-CYBMUJFWSA-N 0.000 description 4
- SRSAKUNDUJJZAN-CYBMUJFWSA-N 6-(4-aminophenyl)-n-[(1r)-1-phenylethyl]-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound N([C@H](C)C=1C=CC=CC=1)C(C=1C=2)=NC=NC=1NC=2C1=CC=C(N)C=C1 SRSAKUNDUJJZAN-CYBMUJFWSA-N 0.000 description 4
- 239000005711 Benzoic acid Substances 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 239000005662 Paraffin oil Substances 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 229910004298 SiO 2 Inorganic materials 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- 125000004423 acyloxy group Chemical group 0.000 description 4
- 125000005236 alkanoylamino group Chemical group 0.000 description 4
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- 235000010233 benzoic acid Nutrition 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 239000001768 carboxy methyl cellulose Substances 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 238000004090 dissolution Methods 0.000 description 4
- 239000008298 dragée Substances 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 230000008020 evaporation Effects 0.000 description 4
- 150000002191 fatty alcohols Chemical class 0.000 description 4
- 125000005456 glyceride group Chemical group 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 4
- 229910052740 iodine Chemical group 0.000 description 4
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 4
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 4
- WQEPLUUGTLDZJY-UHFFFAOYSA-N pentadecanoic acid Chemical compound CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 4
- 235000019271 petrolatum Nutrition 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- 238000001953 recrystallisation Methods 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 239000007940 sugar coated tablet Substances 0.000 description 4
- DLYUQMMRRRQYAE-UHFFFAOYSA-N tetraphosphorus decaoxide Chemical compound O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 4
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 4
- 230000004614 tumor growth Effects 0.000 description 4
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 3
- PNPCRKVUWYDDST-UHFFFAOYSA-N 3-chloroaniline Chemical compound NC1=CC=CC(Cl)=C1 PNPCRKVUWYDDST-UHFFFAOYSA-N 0.000 description 3
- YOAQRPAYMXTKQI-UHFFFAOYSA-N 4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid Chemical compound N1=CN=C2NC(C(=O)O)=CC2=C1NC1=CC=CC(Cl)=C1 YOAQRPAYMXTKQI-UHFFFAOYSA-N 0.000 description 3
- UIDMSVGNGNXGET-UHFFFAOYSA-N 4-chloro-6-(4-nitrophenyl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound C1=CC([N+](=O)[O-])=CC=C1C1=CC2=C(Cl)N=CN=C2N1 UIDMSVGNGNXGET-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 3
- 102000054300 EC 2.7.11.- Human genes 0.000 description 3
- 108700035490 EC 2.7.11.- Proteins 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 239000001828 Gelatine Substances 0.000 description 3
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 3
- 239000004166 Lanolin Substances 0.000 description 3
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 3
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 3
- 108090000315 Protein Kinase C Proteins 0.000 description 3
- 102000003923 Protein Kinase C Human genes 0.000 description 3
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 3
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 235000021355 Stearic acid Nutrition 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 150000001335 aliphatic alkanes Chemical class 0.000 description 3
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 229960000541 cetyl alcohol Drugs 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 238000011049 filling Methods 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 150000002334 glycols Chemical class 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 210000004962 mammalian cell Anatomy 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 239000012299 nitrogen atmosphere Substances 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 3
- 229920000136 polysorbate Polymers 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 239000001632 sodium acetate Substances 0.000 description 3
- 235000017281 sodium acetate Nutrition 0.000 description 3
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 3
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 3
- 239000007901 soft capsule Substances 0.000 description 3
- 239000008117 stearic acid Substances 0.000 description 3
- 229960004274 stearic acid Drugs 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- RQEUFEKYXDPUSK-SSDOTTSWSA-N (1R)-1-phenylethanamine Chemical compound C[C@@H](N)C1=CC=CC=C1 RQEUFEKYXDPUSK-SSDOTTSWSA-N 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- RXYPXQSKLGGKOL-UHFFFAOYSA-N 1,4-dimethylpiperazine Chemical compound CN1CCN(C)CC1 RXYPXQSKLGGKOL-UHFFFAOYSA-N 0.000 description 2
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- OIQOAYVCKAHSEJ-UHFFFAOYSA-N 2-[2,3-bis(2-hydroxyethoxy)propoxy]ethanol;hexadecanoic acid;octadecanoic acid Chemical compound OCCOCC(OCCO)COCCO.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O OIQOAYVCKAHSEJ-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- CYMAZKIEYPVYOH-UHFFFAOYSA-N 2-methoxy-4-[4-(3-methylanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenol Chemical compound C1=C(O)C(OC)=CC(C=2NC3=NC=NC(NC=4C=C(C)C=CC=4)=C3C=2)=C1 CYMAZKIEYPVYOH-UHFFFAOYSA-N 0.000 description 2
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 2
- SJSOFNCYXJUNBT-UHFFFAOYSA-N 3,4,5-trimethoxybenzoic acid Chemical compound COC1=CC(C(O)=O)=CC(OC)=C1OC SJSOFNCYXJUNBT-UHFFFAOYSA-N 0.000 description 2
- DKRKYQJAGJAZRT-UHFFFAOYSA-N 4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidine-6-carbonitrile Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C#N)=C1 DKRKYQJAGJAZRT-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- IDLAFTRDGALWDS-UHFFFAOYSA-N 4-chloro-6-(3-nitrophenyl)-7h-pyrrolo[2,3-d]pyrimidine Chemical compound [O-][N+](=O)C1=CC=CC(C=2NC3=NC=NC(Cl)=C3C=2)=C1 IDLAFTRDGALWDS-UHFFFAOYSA-N 0.000 description 2
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 2
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 2
- 125000004195 4-methylpiperazin-1-yl group Chemical group [H]C([H])([H])N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 2
- VGFSMURRDQKEKB-UHFFFAOYSA-N 6-(3-methoxy-4-phenylmethoxyphenyl)-n-(3-methylphenyl)-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound COC1=CC(C=2NC3=NC=NC(NC=4C=C(C)C=CC=4)=C3C=2)=CC=C1OCC1=CC=CC=C1 VGFSMURRDQKEKB-UHFFFAOYSA-N 0.000 description 2
- AWFOZCHHTUUXAI-UHFFFAOYSA-N 6-(4-nitrophenyl)-1,7-dihydropyrrolo[2,3-d]pyrimidin-4-one Chemical compound C=1C=2C(O)=NC=NC=2NC=1C1=CC=C([N+]([O-])=O)C=C1 AWFOZCHHTUUXAI-UHFFFAOYSA-N 0.000 description 2
- QOLIVYYSALQXGF-LJQANCHMSA-N 6-[3-(benzylamino)phenyl]-n-[(1r)-1-phenylethyl]-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound N([C@H](C)C=1C=CC=CC=1)C(C=1C=2)=NC=NC=1NC=2C(C=1)=CC=CC=1NCC1=CC=CC=C1 QOLIVYYSALQXGF-LJQANCHMSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- DPUOLQHDNGRHBS-UHFFFAOYSA-N Brassidinsaeure Natural products CCCCCCCCC=CCCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-UHFFFAOYSA-N 0.000 description 2
- 102000029330 CSK Tyrosine-Protein Kinase Human genes 0.000 description 2
- 108010069682 CSK Tyrosine-Protein Kinase Proteins 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 201000009030 Carcinoma Diseases 0.000 description 2
- DZMUFMFMZLAPEB-UHFFFAOYSA-N ClC=1C=C(NC=2C3=C(N=CN2)NC(=C3)C3=CC=C(C=C3)N=CN(CC)CC)C=CC1 Chemical compound ClC=1C=C(NC=2C3=C(N=CN2)NC(=C3)C3=CC=C(C=C3)N=CN(CC)CC)C=CC1 DZMUFMFMZLAPEB-UHFFFAOYSA-N 0.000 description 2
- AVVCEXDWGOYMHB-UHFFFAOYSA-N ClC=1C=C(NC=2C3=C(N=CN=2)NC(=C3)C2=CC(=CC=C2)NC(=O)NC2=CC=CC=C2)C=CC=1 Chemical compound ClC=1C=C(NC=2C3=C(N=CN=2)NC(=C3)C2=CC(=CC=C2)NC(=O)NC2=CC=CC=C2)C=CC=1 AVVCEXDWGOYMHB-UHFFFAOYSA-N 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 2
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 2
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 238000005684 Liebig rearrangement reaction Methods 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 2
- OKJIRPAQVSHGFK-UHFFFAOYSA-N N-acetylglycine Chemical compound CC(=O)NCC(O)=O OKJIRPAQVSHGFK-UHFFFAOYSA-N 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 235000021314 Palmitic acid Nutrition 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 102000011653 Platelet-Derived Growth Factor Receptors Human genes 0.000 description 2
- 108010071563 Proto-Oncogene Proteins c-fos Proteins 0.000 description 2
- 102000007568 Proto-Oncogene Proteins c-fos Human genes 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 108091005682 Receptor kinases Proteins 0.000 description 2
- 102000038012 SFKs Human genes 0.000 description 2
- 108091008118 SFKs Proteins 0.000 description 2
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- UPDHEPFPHMXRKP-UHFFFAOYSA-N [4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]methanol Chemical compound N1=CN=C2NC(CO)=CC2=C1NC1=CC=CC(Cl)=C1 UPDHEPFPHMXRKP-UHFFFAOYSA-N 0.000 description 2
- XQAXGZLFSSPBMK-UHFFFAOYSA-M [7-(dimethylamino)phenothiazin-3-ylidene]-dimethylazanium;chloride;trihydrate Chemical compound O.O.O.[Cl-].C1=CC(=[N+](C)C)C=C2SC3=CC(N(C)C)=CC=C3N=C21 XQAXGZLFSSPBMK-UHFFFAOYSA-M 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 150000001241 acetals Chemical class 0.000 description 2
- 125000004442 acylamino group Chemical group 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 2
- 150000005840 aryl radicals Chemical class 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 239000004359 castor oil Substances 0.000 description 2
- 235000019438 castor oil Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 150000004292 cyclic ethers Chemical class 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- UKJLNMAFNRKWGR-UHFFFAOYSA-N cyclohexatrienamine Chemical group NC1=CC=C=C[CH]1 UKJLNMAFNRKWGR-UHFFFAOYSA-N 0.000 description 2
- YEWDOWIBJZTOCM-UHFFFAOYSA-N diethyl 5-amino-1h-pyrrole-2,4-dicarboxylate Chemical compound CCOC(=O)C1=CC(C(=O)OCC)=C(N)N1 YEWDOWIBJZTOCM-UHFFFAOYSA-N 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- 108010037444 diisopropylglutathione ester Proteins 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- UKMSUNONTOPOIO-UHFFFAOYSA-N docosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCC(O)=O UKMSUNONTOPOIO-UHFFFAOYSA-N 0.000 description 2
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- ZQPPMHVWECSIRJ-MDZDMXLPSA-N elaidic acid Chemical compound CCCCCCCC\C=C\CCCCCCCC(O)=O ZQPPMHVWECSIRJ-MDZDMXLPSA-N 0.000 description 2
- 239000002702 enteric coating Substances 0.000 description 2
- 238000009505 enteric coating Methods 0.000 description 2
- 210000002919 epithelial cell Anatomy 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- HAXXOPWEDLHULE-UHFFFAOYSA-N ethyl 2-amino-5-(4-nitrophenyl)-1h-pyrrole-3-carboxylate Chemical compound N1C(N)=C(C(=O)OCC)C=C1C1=CC=C([N+]([O-])=O)C=C1 HAXXOPWEDLHULE-UHFFFAOYSA-N 0.000 description 2
- BMVGJDTURCUCNP-UHFFFAOYSA-N ethyl 4-chloro-7h-pyrrolo[2,3-d]pyrimidine-6-carboxylate Chemical compound N1=CN=C2NC(C(=O)OCC)=CC2=C1Cl BMVGJDTURCUCNP-UHFFFAOYSA-N 0.000 description 2
- RSJUPDUDZRGVRW-UHFFFAOYSA-N ethyl 4-oxo-1,7-dihydropyrrolo[2,3-d]pyrimidine-6-carboxylate Chemical compound N1C=NC(=O)C2=C1NC(C(=O)OCC)=C2 RSJUPDUDZRGVRW-UHFFFAOYSA-N 0.000 description 2
- WUDNUHPRLBTKOJ-UHFFFAOYSA-N ethyl isocyanate Chemical compound CCN=C=O WUDNUHPRLBTKOJ-UHFFFAOYSA-N 0.000 description 2
- 230000007717 exclusion Effects 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 238000005469 granulation Methods 0.000 description 2
- 230000003179 granulation Effects 0.000 description 2
- 150000008282 halocarbons Chemical class 0.000 description 2
- KEMQGTRYUADPNZ-UHFFFAOYSA-N heptadecanoic acid Chemical compound CCCCCCCCCCCCCCCCC(O)=O KEMQGTRYUADPNZ-UHFFFAOYSA-N 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 239000003906 humectant Substances 0.000 description 2
- 238000007327 hydrogenolysis reaction Methods 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 239000011630 iodine Chemical group 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- TWBYWOBDOCUKOW-UHFFFAOYSA-N isonicotinic acid Chemical compound OC(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-N 0.000 description 2
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 2
- 150000003951 lactams Chemical class 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 235000019388 lanolin Nutrition 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- CUONGYYJJVDODC-UHFFFAOYSA-N malononitrile Chemical compound N#CCC#N CUONGYYJJVDODC-UHFFFAOYSA-N 0.000 description 2
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 229960000907 methylthioninium chloride Drugs 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 239000012452 mother liquor Substances 0.000 description 2
- LALUOAHCWIEXII-UHFFFAOYSA-N n'-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-n,n,2-trimethylpropanimidamide Chemical compound C1=CC(N=C(C(C)C)N(C)C)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 LALUOAHCWIEXII-UHFFFAOYSA-N 0.000 description 2
- LIPUGMMVKJZMRG-UHFFFAOYSA-N n-(3-chlorophenyl)-6-(3-methoxy-4-phenylmethoxyphenyl)-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound COC1=CC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=CC=C1OCC1=CC=CC=C1 LIPUGMMVKJZMRG-UHFFFAOYSA-N 0.000 description 2
- MIZGMBWXPIRGEE-UHFFFAOYSA-N n-(3-chlorophenyl)-6-[3-(morpholin-4-ylmethylideneamino)phenyl]-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C=2C=C(C=CC=2)N=CN2CCOCC2)=C1 MIZGMBWXPIRGEE-UHFFFAOYSA-N 0.000 description 2
- GOQYKNQRPGWPLP-UHFFFAOYSA-N n-heptadecyl alcohol Natural products CCCCCCCCCCCCCCCCCO GOQYKNQRPGWPLP-UHFFFAOYSA-N 0.000 description 2
- XTEGVFVZDVNBPF-UHFFFAOYSA-N naphthalene-1,5-disulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1S(O)(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-N 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 239000002304 perfume Substances 0.000 description 2
- 210000001428 peripheral nervous system Anatomy 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 230000026267 regulation of growth Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 229910052717 sulfur Chemical group 0.000 description 2
- 239000011593 sulfur Chemical group 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical class [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- SZHOJFHSIKHZHA-UHFFFAOYSA-N tridecanoic acid Chemical compound CCCCCCCCCCCCC(O)=O SZHOJFHSIKHZHA-UHFFFAOYSA-N 0.000 description 2
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 2
- 230000001228 trophic effect Effects 0.000 description 2
- 229940100445 wheat starch Drugs 0.000 description 2
- 238000010626 work up procedure Methods 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 0 *C(C(*)=O)N* Chemical compound *C(C(*)=O)N* 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- CCXQVBSQUQCEEO-UHFFFAOYSA-N 1-bromobutan-2-one Chemical compound CCC(=O)CBr CCXQVBSQUQCEEO-UHFFFAOYSA-N 0.000 description 1
- GEGLCBTXYBXOJA-UHFFFAOYSA-N 1-methoxyethanol Chemical compound COC(C)O GEGLCBTXYBXOJA-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- WPSSRLMJMYUENZ-UHFFFAOYSA-N 2-(4-aminophenyl)-n-(1-phenylethyl)-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound C=1C=CC=CC=1C(C)NC(C=1C=CNC=1N=1)=NC=1C1=CC=C(N)C=C1 WPSSRLMJMYUENZ-UHFFFAOYSA-N 0.000 description 1
- BVEJOGZHTREYKM-UHFFFAOYSA-N 2-(4-nitrophenyl)-N-(1-phenylethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound C=1C=CC=CC=1C(C)NC(C=1C=CNC=1N=1)=NC=1C1=CC=C([N+]([O-])=O)C=C1 BVEJOGZHTREYKM-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- XQJAHBHCLXUGEP-UHFFFAOYSA-N 2-bromo-1-(4-methoxyphenyl)ethanone Chemical compound COC1=CC=C(C(=O)CBr)C=C1 XQJAHBHCLXUGEP-UHFFFAOYSA-N 0.000 description 1
- MBUPVGIGAMCMBT-UHFFFAOYSA-N 2-bromo-1-(4-nitrophenyl)ethanone Chemical compound [O-][N+](=O)C1=CC=C(C(=O)CBr)C=C1 MBUPVGIGAMCMBT-UHFFFAOYSA-N 0.000 description 1
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 1
- PKRSYEPBQPFNRB-UHFFFAOYSA-N 2-phenoxybenzoic acid Chemical compound OC(=O)C1=CC=CC=C1OC1=CC=CC=C1 PKRSYEPBQPFNRB-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- IWPZKOJSYQZABD-UHFFFAOYSA-N 3,4,5-trimethoxybenzoic acid Natural products COC1=CC(OC)=CC(C(O)=O)=C1 IWPZKOJSYQZABD-UHFFFAOYSA-N 0.000 description 1
- LJGHYPLBDBRCRZ-UHFFFAOYSA-N 3-(3-aminophenyl)sulfonylaniline Chemical group NC1=CC=CC(S(=O)(=O)C=2C=C(N)C=CC=2)=C1 LJGHYPLBDBRCRZ-UHFFFAOYSA-N 0.000 description 1
- QHJJSLUZWHFHTK-UHFFFAOYSA-N 3-amino-3-azaniumylidenepropanoate Chemical compound NC(=N)CC(O)=O QHJJSLUZWHFHTK-UHFFFAOYSA-N 0.000 description 1
- JDQDSEVNMTYMOC-UHFFFAOYSA-N 3-methylbenzenesulfonic acid Chemical compound CC1=CC=CC(S(O)(=O)=O)=C1 JDQDSEVNMTYMOC-UHFFFAOYSA-N 0.000 description 1
- OSJPPGNTCRNQQC-UHFFFAOYSA-N 3-phosphoglyceric acid Chemical compound OC(=O)C(O)COP(O)(O)=O OSJPPGNTCRNQQC-UHFFFAOYSA-N 0.000 description 1
- 125000001999 4-Methoxybenzoyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C(*)=O 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 1
- 125000000242 4-chlorobenzoyl group Chemical group ClC1=CC=C(C(=O)*)C=C1 0.000 description 1
- YGSQUEYKJVADFW-CYBMUJFWSA-N 6-(3-nitrophenyl)-n-[(1r)-1-phenylethyl]-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound N([C@H](C)C=1C=CC=CC=1)C(C=1C=2)=NC=NC=1NC=2C1=CC=CC([N+]([O-])=O)=C1 YGSQUEYKJVADFW-CYBMUJFWSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- WDJHALXBUFZDSR-UHFFFAOYSA-N Acetoacetic acid Natural products CC(=O)CC(O)=O WDJHALXBUFZDSR-UHFFFAOYSA-N 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical class [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 235000021357 Behenic acid Nutrition 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 238000006418 Brown reaction Methods 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical class [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 229910052684 Cerium Inorganic materials 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- 244000060011 Cocos nucifera Species 0.000 description 1
- 235000013162 Cocos nucifera Nutrition 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- AEMOLEFTQBMNLQ-YMDCURPLSA-N D-galactopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-YMDCURPLSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- URXZXNYJPAJJOQ-UHFFFAOYSA-N Erucic acid Natural products CCCCCCC=CCCCCCCCCCCCC(O)=O URXZXNYJPAJJOQ-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical class C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- 108091008794 FGF receptors Proteins 0.000 description 1
- 102000044168 Fibroblast Growth Factor Receptor Human genes 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- DHCLVCXQIBBOPH-UHFFFAOYSA-N Glycerol 2-phosphate Chemical compound OCC(CO)OP(O)(O)=O DHCLVCXQIBBOPH-UHFFFAOYSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical class C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 1
- 102100026720 Interferon beta Human genes 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- OWIKHYCFFJSOEH-UHFFFAOYSA-N Isocyanic acid Chemical compound N=C=O OWIKHYCFFJSOEH-UHFFFAOYSA-N 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- 150000000996 L-ascorbic acids Chemical class 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 239000005639 Lauric acid Substances 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical class [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 1
- ZKGNPQKYVKXMGJ-UHFFFAOYSA-N N,N-dimethylacetamide Chemical compound CN(C)C(C)=O.CN(C)C(C)=O ZKGNPQKYVKXMGJ-UHFFFAOYSA-N 0.000 description 1
- SVYKKECYCPFKGB-UHFFFAOYSA-N N,N-dimethylcyclohexylamine Chemical compound CN(C)C1CCCCC1 SVYKKECYCPFKGB-UHFFFAOYSA-N 0.000 description 1
- DXLYRVFZCHMUAX-UHFFFAOYSA-N N-(3-chlorophenyl)-2-(3-nitrophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound [O-][N+](=O)C1=CC=CC(C=2N=C3NC=CC3=C(NC=3C=C(Cl)C=CC=3)N=2)=C1 DXLYRVFZCHMUAX-UHFFFAOYSA-N 0.000 description 1
- FSZWTIFZYUHKJN-UHFFFAOYSA-N N-(3-chlorophenyl)-6-(methoxyiminomethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound N1=CN=C2NC(C=NOC)=CC2=C1NC1=CC=CC(Cl)=C1 FSZWTIFZYUHKJN-UHFFFAOYSA-N 0.000 description 1
- OTCCIMWXFLJLIA-UHFFFAOYSA-N N-acetyl-DL-aspartic acid Natural products CC(=O)NC(C(O)=O)CC(O)=O OTCCIMWXFLJLIA-UHFFFAOYSA-N 0.000 description 1
- OTCCIMWXFLJLIA-BYPYZUCNSA-N N-acetyl-L-aspartic acid Chemical compound CC(=O)N[C@H](C(O)=O)CC(O)=O OTCCIMWXFLJLIA-BYPYZUCNSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 208000003788 Neoplasm Micrometastasis Diseases 0.000 description 1
- BZQFBWGGLXLEPQ-UHFFFAOYSA-N O-phosphoryl-L-serine Natural products OC(=O)C(N)COP(O)(O)=O BZQFBWGGLXLEPQ-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 108091008606 PDGF receptors Proteins 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical class [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 108010077895 Sarcosine Proteins 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 102000013275 Somatomedins Human genes 0.000 description 1
- NWGKJDSIEKMTRX-AAZCQSIUSA-N Sorbitan monooleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O NWGKJDSIEKMTRX-AAZCQSIUSA-N 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 1
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- NLPUTBDZNNXHCO-CGHBYZBKSA-N Val(5)-angiotensin II Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CC=C(O)C=C1 NLPUTBDZNNXHCO-CGHBYZBKSA-N 0.000 description 1
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- FGUZFFWTBWJBIL-XWVZOOPGSA-N [(1r)-1-[(2s,3r,4s)-3,4-dihydroxyoxolan-2-yl]-2-hydroxyethyl] 16-methylheptadecanoate Chemical compound CC(C)CCCCCCCCCCCCCCC(=O)O[C@H](CO)[C@H]1OC[C@H](O)[C@H]1O FGUZFFWTBWJBIL-XWVZOOPGSA-N 0.000 description 1
- SMEGJBVQLJJKKX-HOTMZDKISA-N [(2R,3S,4S,5R,6R)-5-acetyloxy-3,4,6-trihydroxyoxan-2-yl]methyl acetate Chemical compound CC(=O)OC[C@@H]1[C@H]([C@@H]([C@H]([C@@H](O1)O)OC(=O)C)O)O SMEGJBVQLJJKKX-HOTMZDKISA-N 0.000 description 1
- LNPXDMGETBINDL-UHFFFAOYSA-N [4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]-(4-methylpiperazin-1-yl)methanone Chemical compound C1CN(C)CCN1C(=O)C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 LNPXDMGETBINDL-UHFFFAOYSA-N 0.000 description 1
- FXJLDAPRIWSWJJ-UHFFFAOYSA-N [4-[4-(3-chloroanilino)-7H-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]methanol Chemical compound ClC=1C=C(NC=2C3=C(N=CN=2)NC(=C3)C2=CC=C(C=C2)CO)C=CC=1 FXJLDAPRIWSWJJ-UHFFFAOYSA-N 0.000 description 1
- RLCKNAUVJCZGAP-UHFFFAOYSA-N [4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]cyclohexa-2,4-dien-1-ylidene]methanone Chemical compound ClC1=CC=CC(NC=2C=3C=C(NC=3N=CN=2)C=2C=CC(CC=2)=C=O)=C1 RLCKNAUVJCZGAP-UHFFFAOYSA-N 0.000 description 1
- GRKDKLKIGVYWFI-UHFFFAOYSA-N [4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-(4-methylpiperazin-1-yl)methanone;1-methylpiperazine Chemical compound CN1CCNCC1.C1CN(C)CCN1C(=O)C1=CC=C(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)C=C1 GRKDKLKIGVYWFI-UHFFFAOYSA-N 0.000 description 1
- CIUQDSCDWFSTQR-UHFFFAOYSA-N [C]1=CC=CC=C1 Chemical compound [C]1=CC=CC=C1 CIUQDSCDWFSTQR-UHFFFAOYSA-N 0.000 description 1
- ZKHQWZAMYRWXGA-KNYAHOBESA-N [[(2r,3s,4r,5r)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] dihydroxyphosphoryl hydrogen phosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)O[32P](O)(O)=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KNYAHOBESA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 1
- 229940081735 acetylcellulose Drugs 0.000 description 1
- 229960004308 acetylcysteine Drugs 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- JIMXXGFJRDUSRO-UHFFFAOYSA-N adamantane-1-carboxylic acid Chemical compound C1C(C2)CC3CC2CC1(C(=O)O)C3 JIMXXGFJRDUSRO-UHFFFAOYSA-N 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910052784 alkaline earth metal Chemical class 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- OENHQHLEOONYIE-UKMVMLAPSA-N all-trans beta-carotene Natural products CC=1CCCC(C)(C)C=1/C=C/C(/C)=C/C=C/C(/C)=C/C=C/C=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C OENHQHLEOONYIE-UKMVMLAPSA-N 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- DTOSIQBPPRVQHS-PDBXOOCHSA-N alpha-linolenic acid Chemical compound CC\C=C/C\C=C/C\C=C/CCCCCCCC(O)=O DTOSIQBPPRVQHS-PDBXOOCHSA-N 0.000 description 1
- 235000020661 alpha-linolenic acid Nutrition 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229960004909 aminosalicylic acid Drugs 0.000 description 1
- 125000004682 aminothiocarbonyl group Chemical group NC(=S)* 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229940046836 anti-estrogen Drugs 0.000 description 1
- 230000001833 anti-estrogenic effect Effects 0.000 description 1
- 235000021342 arachidonic acid Nutrition 0.000 description 1
- 229940114079 arachidonic acid Drugs 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 239000003886 aromatase inhibitor Substances 0.000 description 1
- 229940046844 aromatase inhibitors Drugs 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 239000003849 aromatic solvent Substances 0.000 description 1
- 125000003435 aroyl group Chemical group 0.000 description 1
- 125000005101 aryl methoxy carbonyl group Chemical group 0.000 description 1
- 125000005110 aryl thio group Chemical group 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 229940116226 behenic acid Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000011648 beta-carotene Substances 0.000 description 1
- 235000013734 beta-carotene Nutrition 0.000 description 1
- TUPZEYHYWIEDIH-WAIFQNFQSA-N beta-carotene Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CCCC1(C)C)C=CC=C(/C)C=CC2=CCCCC2(C)C TUPZEYHYWIEDIH-WAIFQNFQSA-N 0.000 description 1
- 229960002747 betacarotene Drugs 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- 239000011575 calcium Chemical class 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 235000001465 calcium Nutrition 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 125000003739 carbamimidoyl group Chemical group C(N)(=N)* 0.000 description 1
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- ZMIGMASIKSOYAM-UHFFFAOYSA-N cerium Chemical compound [Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce] ZMIGMASIKSOYAM-UHFFFAOYSA-N 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- HNEGQIOMVPPMNR-IHWYPQMZSA-N citraconic acid Chemical compound OC(=O)C(/C)=C\C(O)=O HNEGQIOMVPPMNR-IHWYPQMZSA-N 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000007859 condensation product Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229950006137 dexfosfoserine Drugs 0.000 description 1
- WOWBFOBYOAGEEA-UHFFFAOYSA-N diafenthiuron Chemical compound CC(C)C1=C(NC(=S)NC(C)(C)C)C(C(C)C)=CC(OC=2C=CC=CC=2)=C1 WOWBFOBYOAGEEA-UHFFFAOYSA-N 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- FFYPMLJYZAEMQB-UHFFFAOYSA-N diethyl pyrocarbonate Chemical compound CCOC(=O)OC(=O)OCC FFYPMLJYZAEMQB-UHFFFAOYSA-N 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- GRWZHXKQBITJKP-UHFFFAOYSA-L dithionite(2-) Chemical compound [O-]S(=O)S([O-])=O GRWZHXKQBITJKP-UHFFFAOYSA-L 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- LQZZUXJYWNFBMV-UHFFFAOYSA-N dodecan-1-ol Chemical compound CCCCCCCCCCCCO LQZZUXJYWNFBMV-UHFFFAOYSA-N 0.000 description 1
- LLRANSBEYQZKFY-UHFFFAOYSA-N dodecanoic acid;propane-1,2-diol Chemical compound CC(O)CO.CCCCCCCCCCCC(O)=O LLRANSBEYQZKFY-UHFFFAOYSA-N 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000002451 electron ionisation mass spectrometry Methods 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 208000037828 epithelial carcinoma Diseases 0.000 description 1
- DPUOLQHDNGRHBS-KTKRTIGZSA-N erucic acid Chemical compound CCCCCCCC\C=C/CCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-KTKRTIGZSA-N 0.000 description 1
- 239000000328 estrogen antagonist Substances 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- ZKQFHRVKCYFVCN-UHFFFAOYSA-N ethoxyethane;hexane Chemical compound CCOCC.CCCCCC ZKQFHRVKCYFVCN-UHFFFAOYSA-N 0.000 description 1
- FPIQZBQZKBKLEI-UHFFFAOYSA-N ethyl 1-[[2-chloroethyl(nitroso)carbamoyl]amino]cyclohexane-1-carboxylate Chemical compound ClCCN(N=O)C(=O)NC1(C(=O)OCC)CCCCC1 FPIQZBQZKBKLEI-UHFFFAOYSA-N 0.000 description 1
- HSDKTLKBDJXJQU-UHFFFAOYSA-N ethyl 3-amino-3-iminopropanoate Chemical compound CCOC(=O)CC(N)=N HSDKTLKBDJXJQU-UHFFFAOYSA-N 0.000 description 1
- VICYTAYPKBLQFB-UHFFFAOYSA-N ethyl 3-bromo-2-oxopropanoate Chemical compound CCOC(=O)C(=O)CBr VICYTAYPKBLQFB-UHFFFAOYSA-N 0.000 description 1
- FLBNYSVQCAAWOC-UHFFFAOYSA-N ethyl 4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]benzoate Chemical compound C1=CC(C(=O)OCC)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 FLBNYSVQCAAWOC-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- SIVVHUQWDOGLJN-UHFFFAOYSA-N ethylsulfamic acid Chemical group CCNS(O)(=O)=O SIVVHUQWDOGLJN-UHFFFAOYSA-N 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 150000002194 fatty esters Chemical class 0.000 description 1
- 239000010685 fatty oil Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 238000005227 gel permeation chromatography Methods 0.000 description 1
- 230000008570 general process Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 244000144993 groups of animals Species 0.000 description 1
- 239000003630 growth substance Substances 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 1
- UKJFVOWPUXSBOM-UHFFFAOYSA-N hexane;oxolane Chemical compound C1CCOC1.CCCCCC UKJFVOWPUXSBOM-UHFFFAOYSA-N 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- XNXVOSBNFZWHBV-UHFFFAOYSA-N hydron;o-methylhydroxylamine;chloride Chemical compound Cl.CON XNXVOSBNFZWHBV-UHFFFAOYSA-N 0.000 description 1
- 150000002440 hydroxy compounds Chemical class 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- WTDHULULXKLSOZ-UHFFFAOYSA-N hydroxylamine hydrochloride Substances Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 1
- WCYJQVALWQMJGE-UHFFFAOYSA-M hydroxylammonium chloride Chemical compound [Cl-].O[NH3+] WCYJQVALWQMJGE-UHFFFAOYSA-M 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 1
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000003978 infusion fluid Substances 0.000 description 1
- 238000011081 inoculation Methods 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000003049 inorganic solvent Substances 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- XUGNVMKQXJXZCD-UHFFFAOYSA-N isopropyl palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC(C)C XUGNVMKQXJXZCD-UHFFFAOYSA-N 0.000 description 1
- GRHBQAYDJPGGLF-UHFFFAOYSA-N isothiocyanic acid Chemical compound N=C=S GRHBQAYDJPGGLF-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 229960004488 linolenic acid Drugs 0.000 description 1
- KQQKGWQCNNTQJW-UHFFFAOYSA-N linolenic acid Natural products CC=CCCC=CCC=CCCCCCCCC(O)=O KQQKGWQCNNTQJW-UHFFFAOYSA-N 0.000 description 1
- 235000014666 liquid concentrate Nutrition 0.000 description 1
- 150000004668 long chain fatty acids Chemical class 0.000 description 1
- 239000011777 magnesium Chemical class 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000001055 magnesium Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Chemical class 0.000 description 1
- 229910044991 metal oxide Inorganic materials 0.000 description 1
- 150000004706 metal oxides Chemical class 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 1
- ROZJWSLFOMITNR-UHFFFAOYSA-N methyl n-[4-[4-(3-chloroanilino)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]carbamate Chemical compound C1=CC(NC(=O)OC)=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 ROZJWSLFOMITNR-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-N methyl sulfate Chemical compound COS(O)(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-N 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 229960002900 methylcellulose Drugs 0.000 description 1
- 230000003641 microbiacidal effect Effects 0.000 description 1
- 229940124561 microbicide Drugs 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- 125000006518 morpholino carbonyl group Chemical group [H]C1([H])OC([H])([H])C([H])([H])N(C(*)=O)C1([H])[H] 0.000 description 1
- 125000001064 morpholinomethyl group Chemical group [H]C([H])(*)N1C([H])([H])C([H])([H])OC([H])([H])C1([H])[H] 0.000 description 1
- RCOAJPOHJGOPPC-UHFFFAOYSA-N n-(3-chlorophenyl)-6-(3-nitrophenyl)-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound [O-][N+](=O)C1=CC=CC(C=2NC3=NC=NC(NC=4C=C(Cl)C=CC=4)=C3C=2)=C1 RCOAJPOHJGOPPC-UHFFFAOYSA-N 0.000 description 1
- DHSPAWKUZSBXHS-UHFFFAOYSA-N n-(3-chlorophenyl)-6-(4-nitrophenyl)-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound C1=CC([N+](=O)[O-])=CC=C1C(NC1=NC=N2)=CC1=C2NC1=CC=CC(Cl)=C1 DHSPAWKUZSBXHS-UHFFFAOYSA-N 0.000 description 1
- OHTCWCLASYCDQT-UHFFFAOYSA-N n-(3-chlorophenyl)-6-(morpholin-4-ylmethyl)-7h-pyrrolo[2,3-d]pyrimidin-4-amine Chemical compound ClC1=CC=CC(NC=2C=3C=C(CN4CCOCC4)NC=3N=CN=2)=C1 OHTCWCLASYCDQT-UHFFFAOYSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- PSKGKFLVQJOKPI-CQSZACIVSA-N n-[4-[4-[[(1r)-1-phenylethyl]amino]-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]methanesulfonamide Chemical compound N([C@H](C)C=1C=CC=CC=1)C(C=1C=2)=NC=NC=1NC=2C1=CC=C(NS(C)(=O)=O)C=C1 PSKGKFLVQJOKPI-CQSZACIVSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 230000017095 negative regulation of cell growth Effects 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- BDJRBEYXGGNYIS-UHFFFAOYSA-N nonanedioic acid Chemical compound OC(=O)CCCCCCCC(O)=O BDJRBEYXGGNYIS-UHFFFAOYSA-N 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 238000011580 nude mouse model Methods 0.000 description 1
- YTJSFYQNRXLOIC-UHFFFAOYSA-N octadecylsilane Chemical class CCCCCCCCCCCCCCCCCC[SiH3] YTJSFYQNRXLOIC-UHFFFAOYSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 239000003791 organic solvent mixture Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 125000003232 p-nitrobenzoyl group Chemical group [N+](=O)([O-])C1=CC=C(C(=O)*)C=C1 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 238000004810 partition chromatography Methods 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical class OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- DGTNSSLYPYDJGL-UHFFFAOYSA-N phenyl isocyanate Chemical compound O=C=NC1=CC=CC=C1 DGTNSSLYPYDJGL-UHFFFAOYSA-N 0.000 description 1
- 150000003017 phosphorus Chemical class 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- BZQFBWGGLXLEPQ-REOHCLBHSA-N phosphoserine Chemical compound OC(=O)[C@@H](N)COP(O)(O)=O BZQFBWGGLXLEPQ-REOHCLBHSA-N 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920000223 polyglycerol Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000137 polyphosphoric acid Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 239000011591 potassium Chemical class 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 235000007686 potassium Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229940116317 potato starch Drugs 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- HLIBNTOXKQCYMV-UHFFFAOYSA-N propylsulfamic acid Chemical compound CCCNS(O)(=O)=O HLIBNTOXKQCYMV-UHFFFAOYSA-N 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 239000001944 prunus armeniaca kernel oil Substances 0.000 description 1
- DXVAQZJPPDWTNY-UHFFFAOYSA-N pyrrolo[3,2-d]pyrimidin-2-one Chemical compound O=C1N=CC2=NC=CC2=N1 DXVAQZJPPDWTNY-UHFFFAOYSA-N 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 108091008597 receptor serine/threonine kinases Proteins 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 229940100486 rice starch Drugs 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 235000003441 saturated fatty acids Nutrition 0.000 description 1
- 150000004671 saturated fatty acids Chemical class 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000008054 signal transmission Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 235000015424 sodium Nutrition 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 229940080236 sodium cetyl sulfate Drugs 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- JVBXVOWTABLYPX-UHFFFAOYSA-L sodium dithionite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])=O JVBXVOWTABLYPX-UHFFFAOYSA-L 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- RZWQDAUIUBVCDD-UHFFFAOYSA-M sodium;benzenethiolate Chemical compound [Na+].[S-]C1=CC=CC=C1 RZWQDAUIUBVCDD-UHFFFAOYSA-M 0.000 description 1
- GGHPAKFFUZUEKL-UHFFFAOYSA-M sodium;hexadecyl sulfate Chemical compound [Na+].CCCCCCCCCCCCCCCCOS([O-])(=O)=O GGHPAKFFUZUEKL-UHFFFAOYSA-M 0.000 description 1
- NWZBFJYXRGSRGD-UHFFFAOYSA-M sodium;octadecyl sulfate Chemical compound [Na+].CCCCCCCCCCCCCCCCCCOS([O-])(=O)=O NWZBFJYXRGSRGD-UHFFFAOYSA-M 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 229940057429 sorbitan isostearate Drugs 0.000 description 1
- 229950004959 sorbitan oleate Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- TUNFSRHWOTWDNC-HKGQFRNVSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCC[14C](O)=O TUNFSRHWOTWDNC-HKGQFRNVSA-N 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 1
- DPUOLQHDNGRHBS-MDZDMXLPSA-N trans-Brassidic acid Chemical compound CCCCCCCC\C=C\CCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-MDZDMXLPSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 229940078499 tricalcium phosphate Drugs 0.000 description 1
- 235000019731 tricalcium phosphate Nutrition 0.000 description 1
- 229910000391 tricalcium phosphate Inorganic materials 0.000 description 1
- ILWRPSCZWQJDMK-UHFFFAOYSA-N triethylazanium;chloride Chemical compound Cl.CCN(CC)CC ILWRPSCZWQJDMK-UHFFFAOYSA-N 0.000 description 1
- 150000004072 triols Chemical class 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 238000010518 undesired secondary reaction Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 150000003738 xylenes Chemical class 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- OENHQHLEOONYIE-JLTXGRSLSA-N β-Carotene Chemical compound CC=1CCCC(C)(C)C=1\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C OENHQHLEOONYIE-JLTXGRSLSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
- Die Erfindung bezieht sich auf 7H-Pyrrolo[2,3-d]pyrimidinderivate und auf Verfahren und neue Zwischenprodukte zu deren Herstellung, auf pharmazeutische Formulierungen, die solche Derivate enthalten, und auf die Verwendung dieser Derivate als Arzneimittel.
- Demnach betrifft die Erfindung 7H-Pyrrolo[2,3-d]pyrimidinderivate der Formel Iworin
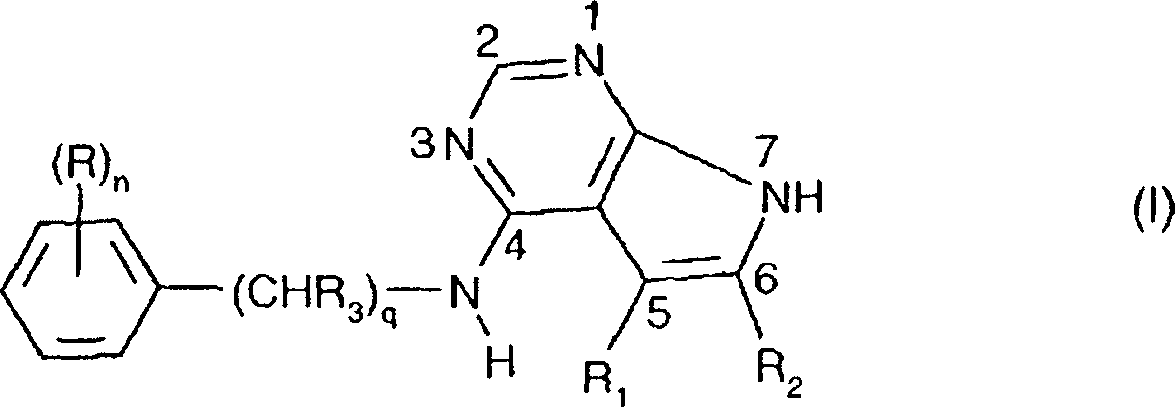
n für 0 oder 1 steht,
q für 0 oder 1 steht,
R für Chlor steht,
R1 für Wasserstoff steht, und
R2 steht für - a) einen Rest der Formel II worin u für 1 steht und R4 für N3-Niederalkylureido, N3-Phenylureido, N3-Niederalkylthioureido, Niederalkoxycarbonylamino, Benzyloxycarbonylamino, Morpholin-4-carbonyl, Piperazin-1-carbonyl, 4-Niederalkylpiperazin-1-carbonyl, Niederalkylsulfonylamino, Benzolsulfonylamino, Toluolsulfonylamino, Furan-2-carbonylamino, Thiophen-2-carbonylamino, Benzylamino, Hydroxymethyl oder einen Rest der Formel -N=C(R5)-R6 steht, worin R5 für Wasserstoff oder Niederalkyl steht und R6 für Diniederalkylamino, Piperidino, 4-Niederalkylpiperazino oder Morpholino steht, oder

- b) einen Rest der Formel III worin R7 für Niederalkoxy steht und R8 für Hydroxyl oder Benzyloxy steht, oder
- c) Piperazin-1-carbonyl, 4-Niederalkylpiperazin-1-carbonyl, Morpholin-4-carbonyl, Thiocarbamoyl, Thiazol-2-yl, 4-(4-Methoxyphenyl)-thiazol-2-yl, 4-Ethylthiazol-2-yl, 4,5-Dimethylthiazol-2-yl, Tetrazol-5-yl, 2-Methyltetrazol-5-yl oder 1-Methyltetrazol-5-yl, oder
- d) einen Rest der Formel -CH=N-OR9, worin R9 für Wasserstoff oder Niederalkyl steht und worin R3 für Wasserstoff oder Niederalkyl steht, oder ein pharmazeutisch akzeptables Salz hiervon, worin das Präfix Nieder für einen Rest mit bis zu und einschließlich maximal 7 Kohlenstoffatomen steht.
- Unter dem Präfix nieder wird hierin und im Folgenden ein Rest von bis zu und einschließlich maximal 7 Kohlenstoffatomen verstanden, besonders mit bis zu und einschließlich maximal 4 Kohlenstoffatomen und besonders mit ein oder zwei Kohlenstoffatomen.
- Vorzugsweise steht n für 0 oder besonders für 1. Gibt es nur einen Substituenten R, dann befindet sich dieser Substituent vorzugsweise in Stellung 3 am Phenylring. Sind zwei Substituenten R vorhanden, dann befinden sich diese Substituenten vorzugsweise in den Stellungen 3 und 4.
- Das Halogen R ist Brom oder Iod oder vorzugsweise Fluor oder Chlor. Steht n für 1, dann ist R vorzugsweise Chlor.
- Niederalkyl ist beispielsweise Methyl.
- Niederalkanoyloxy ist beispielsweise Acetoxy.
- Niederalkoxy ist beispielsweise Methoxy.
- Niederalkanoyl ist beispielsweise Acetyl.
- Niederalkoxycarbonyl ist beispielsweise Methoxycarbonyl.
- N-Niederalkylcarbamoyl ist beispielsweise N-Methylcarbamoyl, N-(n-Butyl)-carbamoyl oder N-(3-Methylbut-1-yl)-carbamoyl.
- N,N-Diniederalkylcarbamoyl ist beispielsweise N,N-Dimethylcarbamoyl.
- Niederalkanoylamino ist beispielsweise Acetylamino.
- Niederalkylamino ist beispielsweise Methylamino.
- N,N-Diniederalkylamino ist beispielsweise Dimethylamino.
- Niederalkoxycarbonylmethoxy ist beispielsweise Methoxycarbonylmethoxy.
- Der Rest R1 ist vorzugsweise Wasserstoff.
- Das Symbol u ist vorzugsweise 1, wobei sich in diesem Fall der Rest R4 vorzugsweise in Stellung 3 oder 4 des Phenylrings befindet.
- Amidino ist ein Rest der Formel -C(=NH)-NH2.
- Guanidino ist ein Rest der Formel -NH-C(=NH)-NH2.
- Ureido ist ein Rest der Formel -NH-C(=O)-NH2.
- N3-Niederalkylureido ist ein Rest der Formel -NH-C(=O)-NH-Niederalkyl, vorzugsweise N3-Ethylureido.
- N3,N3-Diniederalkylureido ist ein Rest der Formel -NH-C(=O)-N-(Niederalkyl)2.
- N3-Phenylureido ist ein Rest der Formel -NH-C(=O)-NH-Phenyl.
- N3,N3-Diphenylureido ist ein Rest der Formel -NH-C(=O)-N-(Phenyl)2.
- Thioureido ist ein Rest der Formel -NH-C(=S)-NH2.
- N3-Niederalkylthioureido ist ein Rest der Formel -NH-C(=S)-NH-Niederalkyl, vorzugsweise N3-Methylthioureido.
- N3,N3-Diniederalkylthioureido ist ein Rest der Formel -NH-C(=S)-N-(Niederalkyl)2.
- Niederalkoxycarbonylamino ist beispielsweise Methoxycarbonylamino, Ethoxycarbonylamino, Isopropyloxycarbonylamino oder 2-Methylpropyloxycarbonylamino.
- Morpholin-4-carbonyl wird auch als Morpholinocarbonyl bezeichnet.
- 4-Niederalkylpiperazin-1-carbonyl ist vorzugsweise 4-Methylpiperazin-1-carbonyl.
- Niederalkylsulfonylamino ist vorzugsweise Methylsulfonylamino, Ethylsulfonylamino oder Isopropylsulfonylamino.
- Der Rest der Formel -N=C(R5)-R6, worin R5 für Wasserstoff steht und R6 für Diniederalkylamino steht, wird als Diniederalkylaminomethylenamino bezeichnet. Entsprechende Reste, worin R6 für Piperidino, 4-Niederalkylpiperazino oder Morpholino steht, werden als R6-Methylenaminoreste bezeichnet, worin R6 wie oben definiert ist, beispielsweise als Piperidinomethylamino.
- R5 ist vorzugsweise Wasserstoff.
- Diniederalkylamino R6 ist vorzugsweise Dimethylamino oder Diethylamino.
- 4-Niederalkylpiperazino ist 4-Niederalkylpiperazin-1-yl, vorzugsweise 4-Methylpiperazin-1-yl.
- Morpholino ist 4-Morpholinyl.
- Niederalkoxy R7 ist vorzugsweise Methoxy.
- Aminoniederalkyl R1 oder R2, dessen Aminogruppe substituiert ist durch ein oder zwei der Reste Hydroxyniederalkyl, Aminoniederalkyl, Carboxyniederalkyl, Niederalkoxycarbonylniederalkyl, Benzyloxycarbonylniederalkyl oder Benzyl, worin der Phenylrest unsubstituiert oder substituiert ist durch Halogen, Niederalkyl, Hydroxymethyl, Aminomethyl, Hydroxyl, Niederalkanoyloxy, Niederalkoxy, Carboxyl, Niederalkanoyl, Benzoyl, Niederalkoxycarbonyl, Carbamoyl, N-Niederalkylcarbamoyl, N,N-Diniederalkylcarbamoyl, Cyano, Amino, Niederalkanoylamino, Niederalkylamino, N,N-Diniederalkylamino oder Trifluormethyl, wobei diese Reste entsprechend substituiert sind durch Aminomethyl.
- Aminoniederalkyl R1 oder R2, worin die Aminogruppe substituiert ist durch ein oder zwei Hydroxyniederalkylreste, ist vorzugsweise beispielsweise ein Rest der Formel -CH2-NH(CH2-CH2)2.
- Aminoniederalkyl R1 oder R2, worin die Aminogruppe substituiert ist durch ein oder zwei Benzylreste, worin der Phenylrest unsubstituiert oder substituiert ist durch Halogen, Niederalkyl, Hydroxymethyl, Aminomethyl, Hydroxyl, Niederalkanoyloxy, Niederalkoxy, Carboxyl, Niederalkanoyl, Benzoyl, Niederalkoxycarbonyl, Carbamoyl, N-Niederalkylcarbamoyl, N,N-Diniederalkylcarbamoyl, Cyano, Amino, Niederalkanoylamino, Niederalkylamino, N,N-Diniederalkylamino oder Trifluormethyl, ist vorzugsweise beispielsweise ein Rest der Formel -CH2-NH-CH2-C6H4-OCH3, wie besonders 4-Methoxyphenylmethylaminomethyl oder 4-Hydroxyphenylmethylaminomethyl.
- Thiocarbamoyl ist der Rest der Formel -C(=S)-NH2 und wird auch als Aminothiocarbonyl bezeichnet.
- Ein heterocyclischer Rest R1 oder R2, der über ein Ringkohlenstoffatom gebunden ist und fünf Ringglieder und 1 bis 4 Ringheteroatome aufweist, die ausgewählt sind aus Sauerstoff, Stickstoff und Schwefel, ist unsubstituiert oder substituiert, und beispielsweise Pyrrolyl, Thienyl, Furyl oder vorzugsweise Tetrazol-5-yl, das unsubstituiert oder substituiert ist durch Niederalkyl, oder Thiazol-2-yl, das unsubstituiert oder substituiert ist durch Niederalkoxyphenyl, wie beispielsweise Thiazol-2-yl, 4-(4-Methoxyphenyl)-thiazol-2-yl, 4-Ethylthiazol-2-yl oder 4,5-Dimethylthiazol-2-yl.
- Tetrazol-5-yl ist 1H-Tetrazol-5-yl oder das tautomere 2H-Tetrazol-5-yl oder ein Gemisch dieser beiden tautomeren Formen.
- Tetrazol-5-yl, das durch Niederalkyl substituiert ist, ist N1-Niederalkyltetrazol-5-yl oder N2-Niederalkyltetrazol-5-yl, besonders 1-Methyltetrazol-5-yl oder 2-Methyltetrazol-5-yl.
- Ein heterocyclischer Rest, der kein Piperazinyl ist und der fünf oder sechs Ringglieder enthält und 1 bis 4 Ringheteroatome, die ausgewählt sind aus Sauerstoff, Stickstoff und Schwefel, ist ein solcher unsubstituierter oder substituierter Rest, beispielsweise Pyrrolyl, Thienyl, Furyl oder Tetrazol-5-yl, das unsubstituiert oder substituiert ist durch Niederalkyl, Thiazol-2-yl, das unsubstituiert oder substituiert ist durch Niederalkoxyphenyl, oder Morpholino oder 4-Niederalkylpiperazin-1-yl.
- Niederalkyl R1 oder R2, das durch einen solchen heterocyclischen Rest substituiert ist, ist vorzugsweise geeignet substituiertes Methyl, vorzugsweise 4-Methylpiperazin-1-ylmethyl oder Morpholinomethyl.
- Der Rest -CH=N-OR9R1 oder R2 kann die trans-Konfiguration oder die cis-Konfiguration haben.
- Phenyl R1 oder R2, das substituiert ist durch Halogen, Niederalkyl, Trifluormethyl oder Niederalkoxy, ist beispielsweise 4-Methoxyphenyl. R1 oder R2 können aber nur Phenyl sein, das so substituiert ist, wenn in der Formel I das Symbol q für 1 steht.
- R3 ist vorzugsweise Methyl.
- Salze von Verbindungen der Formel I sind besonders Säureadditionssalze mit organischen oder anorganischen Säuren, besonders die pharmazeutisch akzeptablen und nicht toxischen Salze. Geeignete anorganische Salze sind beispielsweise Salze von Kohlensäure, vorzugsweise in der Form von Carbonaten oder Hydrogencarbonaten, Halogenwasserstoffsäuren, wie Chlorwasserstoffsäure, Schwefelsäure oder Phosphorsäure. Geeignete organische Säuren sind beispielsweise Carbonsäuren, Phosphonsäuren, Schwefelsäuren oder Sulfaminsäuren, wie beispielsweise Essigsäure, Propionsäure, Octansäure, Decansäure, Dodecansäure, Glycolsäure, Milchsäure, 2-Hydroxybuttersäure, Gluconsäure, Glucosemonocarbonsäure, Fumarsäure, Bernsteinsäure, Adipinsäure, Pimelinsäure, Suberinsäure, Azelainsäure, Apfelsäure, Weinsäure, Citronensäure, Glucarsäure, Galactarsäure, Aminosäuren, wie Glutaminsäure, Asparaginsäure, N-Methylglycin, Acetylaminoessigsäure, N-Acetylasparaginsäure oder N-Acetylcystein, Pyruvasäure, Acetoessigsäure, Phosphoserin, 2- oder 3-Glycerophosphorsäure, Glucose-6-phosphorsäre, Glucose-1-phosphorsäure, Fructose-1,6-bisphosphorsäre, Maleinsäure, Hydroxymaleinsäure, Methylmaleinsäure, Cyclohexancarbonsäure, Adamantancarbonsäure, Benzoesäure, Salicylsäure, 1- oder 3-Hydroxynaphthyl-2-carbonsäure, 3,4,5-Trimethoxybenzoesäure, 2-Phenoxybenzoesäure, 2-Acetoxybenzoesäure, 4-Aminosalicylsäure, Phthalsäure, Phenylessigsäure, Mandelsäure, Zimtsäure, Nicotinsäure, Isonicotinsäure, Glucuronsäure, Galacturonsäure, Methansulfonsäure, Ethansulfonsäure, 2-Hydroxyethansulfonsäure, Ethan-1,2-disulfonsäure, Benzolsulfonsäure, 2-Naphthalinsulfonsäure, 1,5-Naphthalindisulfonsäure, 2-, 3- oder 4-Methylbenzolsulfonsäure, Methylschwefelsäure, Ethylschwefelsäure, Dodecylschwefelsäure, N-Cyclohexylsulfaminsäure, N-Methyl-, N-Ethyl- oder N-Propylsulfaminsäure oder andere organische Protonensäuren, wie Ascorbinsäuren.
- Verbindungen der Formel I, die wenigstens eine saure Gruppe enthalten, wie beispielsweise eine freie Carboxylgruppe, sind auch zur Bildung innerer Salze, Metallsalze oder Ammoniumsalze befähigt, wie Salze von Alkalimetallen oder Erdalkalimetallen, beispielsweise Salze von Natrium, Kalium, Magnesium oder Calcium, Salze von Ammonium mit Ammoniak oder geeigneten organischen Aminen, wie tertiä ren Monoaminen, beispielsweise Triethylamin oder Tri-(2-hydroxyethyl)-amin, oder heterocyclische Basen, beispielsweise N-Ethylpiperidin oder N,N'-Dimethylpiperazin.
- Zu Zwecken einer Isolierung oder Reinigung können auch Salze verwendet werden, die nicht pharmazeutisch geeignet sind, beispielsweise Picrate oder Perchlorate. Für therapeutische Zwecke werden aber nur Salze verwendet, die – bei den geeigneten Dosen – pharmazeutisch akzeptabel und nicht toxisch sind, und diese Salze sind daher bevorzugt.
- Im Hinblick auf die nahe Verwandtschaft zwischen den neuen Verbindungen in freier Form oder in Form ihrer Salze unter Einschluss der Salze, die als Zwischenprodukte verwendet werden können, beispielsweise zur Reinigung oder Identifikation der neuen Verbindungen, werden hierin die freien Verbindungen verstanden, wie die entsprechenden Salze, sofern diese geeignet und zweckdienlich sind.
- Die Verbindungen der Formel I verfügen über wertvolle pharmazeutisch brauchbare Eigenschaften. Sie entwickeln besonders spezifische Inhibitoraktivitäten, die von pharmakologischem Interesse sind. Sie sind primär wirksam als Protein-Tyrosin-Kinase Inhibitoren und/oder – weiterhin – auch als Inhibitoren für Protein-Serin/Threonin-Kinasen. Sie verfügen beispielsweise über eine starke Inhibition der Tyrosin-Kinase Aktivität des Rezeptors für den epidermalen Wachstumsfaktor (EGF) und von c-erbB2-Kinase. Diese Rezeptor-spezifischen Enzymaktivitäten spielen eine Schlüsselrolle bei einer Signaltransmission in einer großen Anzahl an Säugerzellen unter Einschluss menschlicher Zellen, besonders Epithelzellen, Zellen des Immunsystems und Zellen des zentralen und peripheren Nervensystems. So ist beispielsweise in verschiedenen Zellarten eine EGF-induzierte Aktivierung der Rezeptor-assoziierten Protein-Tyrosin-Kinase (EGF-R-PTK) eine Voraussetzung für eine Zellteilung und somit für die Proliferation der Zellpopulation. Gleiches gilt analog auch für anderen Proteinkinasen, wie sie oben und im Folgenden erwähnt sind.
- Zusätzlich zu oder anstatt einer Inhibition einer EGF-Rezeptor-Protein-Tyrosin-Kinase inhibieren die Verbindungen der Formel I auch in unterschiedlichen Ausmaßen andere Proteintyrosinkinasen, die bei einer Signaltransmission involviert sind, welche durch trophische Faktoren mediiert werden, wie beispielsweise abl Kinase, besonders v-abl Kinase, Kinasen aus der Familie der src Kinasen, besonders der c-src Kinase, Ick, fyn, andere Kinasen aus der EGF Familie, beispielsweise c-erbB2 Kinase (HER-2), c-erbB3 Kinase, c-erbB4 Kinase, Mitglieder der Familie der PDGF Rezeptor-Protein-Tyrosin-Kinasen, beispielsweise von PDGF-Rezeptor-Kinase, CSF-1 Rezeptor-Kinase, Kit-Rezeptor-Kinase, VEGF-Rezeptor-Kinase und FGF-Rezeptor-Kinase, der Rezeptor-Kinase des Insulin-ähnlichen Wachstumsfaktors (IGF-1 Kinase) und der Serin/Threonin-Kinasen, beispielsweise von Protein-Kinase C oder cdc Kinasen, die alle einen Teil in einer Wachstumsregulation und Transformation in Säugerzellen unter Einschluss menschlicher Zellen spielen.
- Die Inhibition von EGF-Rezeptor-spezifischer Protein-Tyrosin-Kinase (EGF-R-PTK) kann unter Verwendung bekannter Verfahren gezeigt werden, beispielsweise unter Verwendung der rekombinanten intrazellularen Domain des EGF-Rezeptors (EGF-R ICD, wozu beispielsweise hingewiesen wird auf E. McGlynn et al., Europ. J. Biochem. 207, Seiten 265 bis 275 (1992)). Im Vergleich zur Kontrolle ohne Inhibitor inhibieren die Verbindungen der Formel I die Enzymaktivität um 50% (IC50-Wert) beispielsweise in einer Konzentration von 0,0005 bis 1 μM, besonders von 0,001 bis 0,1 μM.
- Die Wirkung der Verbindungen der Formel I auf eine EGF-stimulierte zellulare Tyrosin-Phosphorylierung im EGF-Rezeptor kann bestimmt werden bei der humanen A431 Epithelkarzinomzelllinie mittels eines ELISAs, wie dies beispielsweise beschrieben ist von U. Trinks et al. in J. Med. Chem. 37:7, Seiten 1015 bis 1027 (1994). Bei diesem Test (EGFR-ELISA) zeigen die Verbindungen der Formel I einen IC50-Wert von etwa 0,001 bis 1 μM.
- Eine Stimulation mit quieszenten BALB/c3T3 Zellen mit EGF ergibt eine rasche Induktion der Expression von c-fos mRNA. Eine Vorbehandlung der Zellen mit einer Verbindung der Formel I vor der Stimulation mit EGF inhibiert eine c-fos Expression bei einem IC50-Wert von etwa 0,001 bis 0,1 μM. Dieses Testverfahren wird ebenfalls von U. Trinks et al. in J. Med. Chem. 37:7, Seiten 1015 bis 1027 (1994) beschrieben.
- Auch im mikromolaren Bereich zeigen die Verbindungen der Formel I eine Inhibition, beispielsweise eine Inhibition des Zellwachstums der EGF-abhängigen Zelllinien, beispielsweise der Kartinocytenzelllinie einer epidermoiden BALB/c-Maus, wozu beispielsweise hingewiesen wird auf B. A. Weissmann und S. A. Aaronson, Cell 32, Seite 599 (1983), oder der A431 Zelllinie, welche beide anerkanntermaßen als Standardquellen von EGF-abhängigen Epithelzellen brauchbar sind, wozu hingewiesen wird auf G. Carpenter und J. Zendegni in J. Anal. Biochem. 153, Seiten 279 bis 282 (1985). Bei einer bekannten Testmethode, wozu beispielsweise hingewiesen wird auf Meyer et al. in Int. J. Cancer 43, Seite 851 (1989), wird die inhibitorische Wirksamkeit der Verbindungen der Formel I kurz wie folgt bestimmt.
- BALB/MK-Zellen (10000/Loch einer Mikrotiterplatte) werden in Mikrotiterplatten mit 96 Löchern übertragen. Die Testverbindungen werden (gelöst in DMSO) in einer Reihe an Konzentrationen (Verdünnungsreihen) derart zugegeben, dass die Endkonzentration an DMSO nicht über 1% (Vol./Vol.) hinausgeht. Nach Zusatz werden die Platten 3 Tage inkubiert, wobei während dieser Zeit die Kontrollkulturen ohne eine Testverbindung wenigstens 3 Zellteilzyklen erfahren können. Das Wachstum der MK Zellen wird durch eine Anfärbung mit Methylenblau wie folgt bestimmt: Nach der Inkubation werden die Zellen mit Glutaraldehyd fixiert, mit Wasser gewaschen und mit 0,05% Methylenblau angefärbt. Nach einer Waschstufe wird die Anfärbung mit 3% HCl eluiert und die optische Dichte pro Loch der Mikrotiterplatte mittels eines Geräts Titertek Multiscan bei 665 nm gemessen. Die IC50-Werte werden durch ein computerunterstütztes System unter Anwendung der folgenden Formel bestimmt:
- Der IC50-Wert dieser Experimente ist angegeben als die Konzentration der jeweiligen Testverbindung, die eine Zellzählung ergibt, die über 50% niedriger ist als die unter Verwendung der Kontrolle ohne Inhibitor erhaltene Zellzählung. Dabei entwickeln die Verbindungen der Formel I eine Inhibitoraktivität im mikromolaren Bereich, beispielsweise einen IC50-Wert von etwa 0,1 bis 1 μM.
- Die Verbindungen der Formel I zeigen auch eine Inhibition des Wachstums von Tumorzellen in vivo, was beispielsweise durch folgenden Test belegt werden kann. Dieser Test beruht auf einer Inhibition des Wachstums des humanen Epidermoidkarzinoms A431 (ATCC Nr. CRL 1555, American Type Culture Collection, Rockville, Maryland, USA, wozu hingewiesen wird auf J. B. Santon et al., Cancer Research 46, Seiten 4701 bis 4705 (1986) und auf S. Ozawa et al., Int. J. Cancer 40, Seiten 706 bis 710 (1987), das in eine weibliche BALG/c Nacktmaus (Bomholtgard, Dänemark) transplantiert wird. Dieses Karzinom weist ein Wachstum auf, das mit dem Ausmaß der Expression des EGF-Rezeptors korreliert. Bei diesem Experiment werden Tumoren, die ein Volumen von etwa 1 cm3 haben und in vivo gezüchtet worden sind, chirurgisch unter sterilen Bedingungen aus den Experimentaltieren entfernt. Anschließend werden die Tumoren zerkleinert und in 10 Volumina (Gewicht pro Volumen) einer Phosphat-gepufferten Kochsalzlösung suspendiert. Sodann wird diese Suspension subkutan in die linke Flanke der Tiere injiziert (0,2 ml/Maus in einer Phosphat-gepufferten Kochsalzlösung). Alternativ können 1 × 106 Zellen aus einer in vitro Kultur in 0,2 ml Phosphat-gepufferter Kochsalzlösung injiziert werden. Eine Behandlung mit Testverbindungen der Formel I wird 5 oder 7 Tage nach Transplantation gestartet, sobald die Tumoren einen Durchmesser von 4 bis 5 mm erreicht haben. Der jeweilige Wirkstoff wird einmal täglich während 15 aufeinanderfolgender Tage verabreicht, und zwar in unterschiedlichen Dosen für unterschiedliche Tiergruppen. Das Tumorwachstum wird durch Messung des Durchmessers der Tumoren längs der drei Achsen gemessen, die senkrecht zueinander stehen. Die Tumorvolumina werden unter Anwendung der bekannten Formel p × L × D2/6 berechnet, wozu hingewiesen wird auf B. D. Evans et al. in Brit. J. Cancer 45, Seiten 466 bis 468 (1982). Die Ergebnisse werden in Prozentwerten des Verhältnisses von Behandlung zu Kontrolle angegeben (T/C × 100 = T/C%). Bei einer Dosis von 3 bis 50 mg/kg Wirkstoff wird eine distinkte Inhibition des Tumorwachstums gefunden, beispielsweise bei T/C% Werten von weniger als 10, was für eine starke Inhibition des Tumorwachstums spricht.
- Genauso wie oder anstelle einer Inhibition der EGF-Rezeptor-Protein-Tyrosin-Kinase zeigen die Verbindungen der Formel I auch eine Hemmung bei anderen Protein-Tyrosin-Kinasen, die in einer Signaltransmission involviert sind, welche durch trophische Faktoren mediiert wird, wie beispielsweise bei abl-Kinase, besonders bei v-abl-Kinase (IC50-Werte von beispielsweise 0,01 bis 5 μM), bei Kinasen aus der Familie der src-Kinasen, wie besonders von c-src-Kinase (IC50-Werte von beispielsweise 0,1 bis 10 μM) und bei c-erbB2-Kinase (HER-2) und bei Serin/Threonin-Kinasen, beispielsweise bei Protein-Kinase C, wobei alle diese Kinasen in der Wachstumsregulierung und Transformation in Säugerzellen unter Einschluss menschlicher Zellen involviert sind.
- Die oben erwähnte Inhibition von v-abl-Tyrosin-Kinase wird unter Anwendung der Verfahren bestimmt, wie sie beispielsweise beschrieben sind von N. Lydon et al., Oncogene Research 5, Seiten 161 bis 173 (1990) und von J. F. Geissler et al., Cancer Research 52, Seiten 4492 bis 4498 (1992). Bei diesen Verfahren werden [Val5]-Angiotensin II und [γ32P]-ATP als Substrate verwendet.
- Die Inhibition von c-erbB2-Tyrosin-Kinase (HER-2) kann beispielsweise bestimmt werden in Analogie zum für EGF-R-TPK verwendeten Verfahren, wozu hingewiesen wird auf C. House et al., Europ. J. Biochem. 140, Seiten 363 bis 367 (1984). Die c-erbB2-Kinase kann isoliert und bezüglich ihrer Aktivität bestimmt werden unter Anwendung an sich bekannter Protokolle, beispielsweise entsprechend T. Akiyama et al., Science 232, Seite 1644 (1986).
- Die Verbindungen der Formel I, die die Tyrosin-Kinase-Aktivität des Rezeptors für den epidermalen Wachstumsfaktor (EGF) und weitere der oben erwähnten anderen Protein-Tyrosin-Kinasen hemmen, eignen sich daher beispielsweise für die Behandlung benigner oder maligner Tumoren. Sie sind brauchbar zur Bewirkung einer Tumorregression und Verhinderung der Bildung von Tumormetastasen und des Wachstums von Mikrometastasen. Sie können besonders im Fall einer epidermalen Hyperproliferation (Psoriasis) für die Behandlung von Neoplasien von epithelialem Charakter, beispielsweise von Mammakarzinomen, und von Leukämien verwendet werden. Darüber hinaus lassen sich die Verbindungen der Formel I und besonders die neuen Verbindungen hiervon auch verwenden für die Behandlung solcher Störungen des Immunsystems, bei denen mehrere oder besonders individuelle Protein-Tyrosin-Kinasen und/oder weiterhin auch Protein-Serin/Threonin-Kinasen involviert sind. Weiter können diese Verbindungen der Formel I auch verwendet werden für die Behandlung von Störungen des zentralen oder peripheren Nervensystems, bei denen eine Signaltransmission durch mehrere oder besonders eine einzelne Protein-Tyrosin-Kinase und/oder weiterhin Protein-Serin/Threonin-Kinasen involviert sind.
- Allgemein bezieht sich die vorliegende Erfindung daher auch auf die Verwendung der Verbindungen der Formel I für die Inhibition der erwähnten Protein-Kinasen.
- Dabei können die erfindungsgemäßen Verbindungen sowohl allein als auch in Kombination mit anderen pharmakologisch wirksamen Verbindungen angewandt werden, beispielsweise zusammen mit Inhibitoren der Enzyme einer Polyaminsynthese, Inhibitoren für Protein-Kinase C, Inhibitoren anderer Tyrosin-Kinasen, Cytokine, negative Wachstumsregulatoren, beispielsweise TGF-β oder IFN-β, Aromatase-Inhibitoren, Antiöstrogenen und/oder cytostatischen Mitteln angewandt werden.
- Bei den im Folgenden erwähnten bevorzugten Gegenständen der Erfindung können allgemeine Definitionen durch speziellere Definitionen, wie sie zu Beginn angegeben worden sind, ersetzt werden, sofern dies geeignet und zweckdienlich ist.
- Am meisten bevorzugt sind die Verbindungen der Formel I, welche in den Beispielen beschrieben sind, und deren pharmazeutisch akzeptable Salze.
- Die Verbindungen der Formel I und ihre Salze werden nach an sich bekannten Verfahren hergestellt, nämlich durch
- a) Umsetzung eines Pyrrolo[2,3-d]pyrimidinderivats
der Formel IV worin X eine geeignete Abgangsgruppe ist, Z für Wasserstoff oder 1-Arylniederalkyl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei in den Resten R1 und R2 vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, mit einem Anilinderivat der Formel V
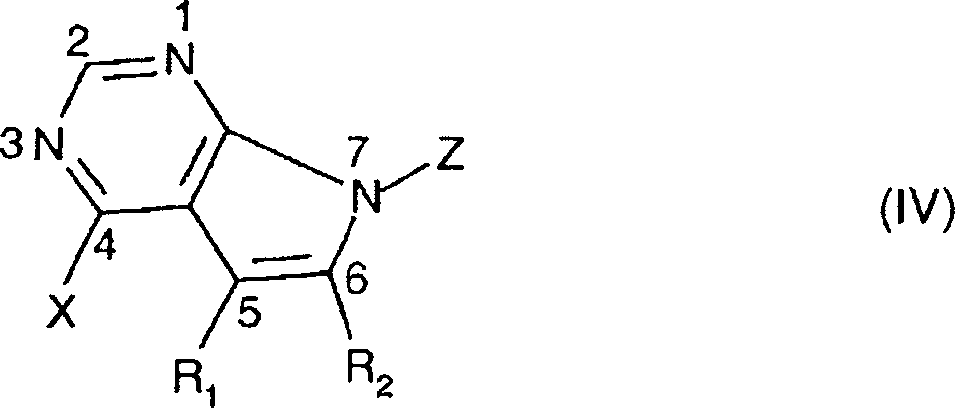 worin R, R3, n und q wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und Entfernung der vorhandenen Schutzgruppen und, falls vorhanden, auch des 1-Arylniederalkylrests Z, oder
worin R, R3, n und q wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und Entfernung der vorhandenen Schutzgruppen und, falls vorhanden, auch des 1-Arylniederalkylrests Z, oder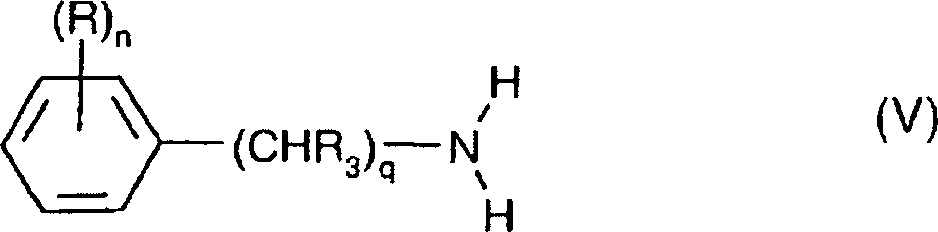
- b) Umsetzung eines Pyrrolo[2,3-d]pyrimidin-4-on-derivats der
Formel VI worin Z' für 1-Arylniederalkyl steht und R1 und R2 wie oben für die Verbindungen der Formel I definiert sind, wobei in den Resten R1 und R2 vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, in Gegenwart eines Dehydratationsmittels und eines tertiären Amins, mit einem Phenylamin der obigen Formel V und Entfernung der vorhandenen Schutzgruppen, oder
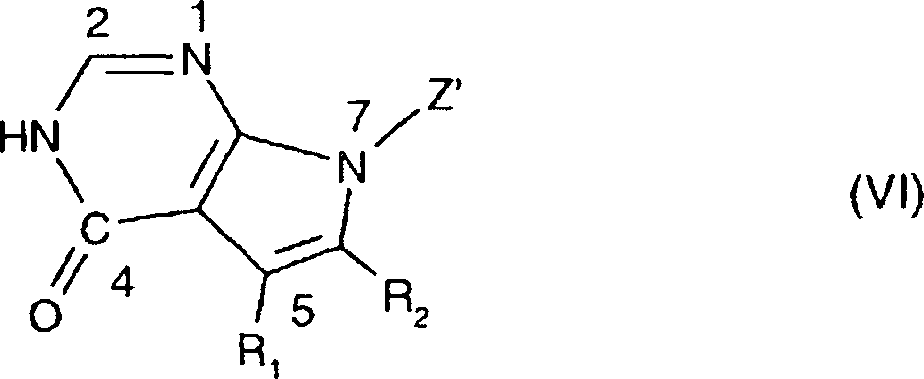
- e) zur Herstellung einer Verbindung der Formel I, worin einer
der Reste R1 und R2 ein
Rest der Formel -CH=N-OR9 ist, worin R9 für
Wasserstoff oder Niederalkyl steht, und die anderen Substituenten
wie oben für
die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung
der Formel I, worin einer der Reste R1 und
R2 für
Formyl steht und die anderen Substituenten wie oben für die Verbindungen
der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale
Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind,
mit einem Hydroxylaminderivat der Formel VIII
- f) zur Herstellung einer Verbindung der Formel I, worin einer
der Reste R1 und R2 für Piperidin-1-carbonyl, Piperazin-1-carbonyl,
4-Niederalkylpiperazin-1-carbonyl oder Morpholin-4-carbonyl steht,
und der andere dieser Substituenten wie oben für die Verbindung der Formel
I definiert ist, Umsetzung einer Verbindung der Formel I, worin
einer der Reste R1 und R2 für Carboxyl
steht und die anderen Substituenten wie oben für die Verbindungen der Formel
I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen
erforderlichenfalls durch Schutzgruppen geschützt sind, oder einem reaktiven
Carbonsäurederivat
einer solchen Verbindung, mit einem Amin der Formel VII
- g) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Thiocarbamoyl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Aminocarbonyl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei freie funktionale Gruppen, die im Rest R vorhanden sind, erforderlichenfalls durch Schutzgruppen geschützt werden, mit einem Lawesson-Reagenz, und anschließende Entfernung der vorhandenen Schutzgruppen, oder
- h) zur Herstellung einer Verbindung der Formel I, worin einer
der Reste R1 und R2 für R13-Thiazol-2-yl steht, worin R13 jeweils
für Ethyl
oder Niederalkoxyphenyl steht, und die anderen Substituenten wie
oben für
die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung
der Formel I, worin einer der Reste R1 und
R2 für
Thiocarbamoyl steht, und die anderen Substituenten wie oben für die Verbindungen
der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale
Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind,
mit einer Verbindung der Formel IX worin X für eine Abgangsgruppe steht und R13 jeweils für Ethyl oder Niederalkoxyphenyl steht, wobei im Rest R13 vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und anschließende Entfernung der vorhandenen Schutzgruppen, oder

- i) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Tetrazol-5-yl steht, und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Cyano steht, und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, mit einem geeigneten Alkalimetallazid, und anschließende Entfernung der vorhandenen Schutzgruppen, oder
- j) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für 2-Niederalkyltetrazol-5-yl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Tetrazol-5-yl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, mit dem geeigneten Niederalkyliodid, und anschließende Entfernung der vorhandenen Schutzgruppen, oder
- k) zur Herstellung einer Verbindung der Formel I, worin einer
der Reste R1 und R2 für einen
Rest der Formel II steht worin wenigstens einer der Reste R4 für Niederalkylsulfonylamino, Benzolsulfonylamino oder Toluolsulfonylamino steht, und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch Schutzgruppen geschützt sind, mit einer Verbindung der Formel X

- l) zur Herstellung einer Verbindung der Formel I, worin einer
der Reste R1 und R2 für einen
Rest der Formel II steht worin wenigstens ein Rest R4 für einen Rest der Formel -N=C(R5)-R6 steht, worin R5 für Wasserstoff steht und R6 wie oben für die Verbindungen der Formel I definiert ist, und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch Schutzgruppen geschützt sind, mit einem Acetal der Formel XI
 worin R15 und R16 jeweils individuell für Niederalkyl stehen oder zusammen für Pentan-1,5-diyl, 3-N-Niederalkyl-3-azapentan-1,5-diyl oder 3-Oxapentan-1,5-diyl stehen, und R17 für Niederalkyl steht, und anschließende Entfernung der vorhandenen Schutzgruppen, oder
worin R15 und R16 jeweils individuell für Niederalkyl stehen oder zusammen für Pentan-1,5-diyl, 3-N-Niederalkyl-3-azapentan-1,5-diyl oder 3-Oxapentan-1,5-diyl stehen, und R17 für Niederalkyl steht, und anschließende Entfernung der vorhandenen Schutzgruppen, oder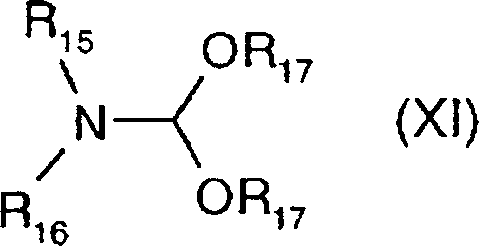
- m) zur Herstellung einer Verbindung der Formel I, worin einer
der Reste R1 und R2 für einen
Rest der Formel II steht worin wenigstens ein Rest R4 für N3-Niederalkylureido oder N3-Phenylureido steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch Schutzgruppen geschützt sind, mit einem Isocyanat der Formel XII
- n) zur Herstellung einer Verbindung der Formel I, worin einer
der Reste R1 und R2 für einen
Rest der Formel II steht worin wenigstens ein Rest R4 für N3-Niederalkylthioureido oder N3-Phenylthioureido steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch Schutzgruppen geschützt sind, mit Isothiocyanat der Formel XIII

- o) zur Herstellung einer Verbindung der Formel I, worin einer
der Reste R1 und R2 für einen
Rest der Formel II steht, worin wenigstens ein Rest R4 für Niederalkoxycarbonylamino oder Benzyloxycarbonylamino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch Schutzgruppen geschützt sind, mit einem Chlorameisensäureester der Formel XIV

- Detaillierte Beschreibung der einzelnen Verfahrensstufen
- Die obigen Verfahren werden im Folgenden im Detail beschrieben, wozu beispielsweise hingewiesen wird auf
DE 30 36 390 A und A. Jorgensen et al., J. Heterocycl. Chem. 22, 859 (1985). In der nun folgenden präziseren Beschreibung sind, sofern nichts anderes gesagt ist, die Reste R, R1 und R2 und der Index n wie für die Verbindungen der Formel I definiert. - Allgemeine Punkte
- Die Endprodukte der Formel I können Substituenten enthalten, die auch als Schutzgruppen in Ausgangsmaterialien für die Herstellung anderer Endprodukte der Formel I verwendet werden können. Sofern im vorliegenden Kontext nichts anderes gesagt ist, bezieht sich die Bezeichnung Schutzgruppe, wie sie in diesem Text verwendet wird, auf die Bezeichnung nur einer leicht entfernbaren Gruppe, die kein Bestandteil des besonderen gewünschten Endprodukts der Formel I ist.
- Verfahren a)
- In der Verbindung der Formel IV ist eine geeignete Abgangsgruppe X vorzugsweise Halogen, wie Brom, Iod oder insbesondere Chlor. Ein 1-Arylniederalkyl Z ist vorzugsweise 1-Phenylniederalkyl, besonders 1-Phenylethyl oder insbesondere Benzyl.
- Die in den Resten R1 und R2 vorhandenen funktionalen Gruppen, die erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, sind besonders Amino oder Niederalkylamino.
- Schutzgruppen und ihre Einführung und Entfernung werden beispielsweise beschrieben in Protective Groups in Organic Chemistry, Plenum Press, London, New York, 1973, und in Methoden der Organischen Chemie, Houben-Weyl, 4. Auflage, Band 15/1, Georg-Thieme-Verlag, Stuttgart, 1974, und in Theodors W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1981. Ein Charakteristikum der Schutzgruppen ist, dass sie leicht entfernt werden können, nämlich ohne dass unerwünschte Sekundärreaktionen stattfinden, beispielsweise durch Solvolyse, Reduktion, Photolyse oder alternativ unter physiologischen Bedingungen.
- Eine Aminogruppe kann beispielsweise in Form einer leicht abspaltbaren Acylamino-, Arylmethylamino-, veretherten Mercaptoamino- oder 2-Acylniederalk-1-enylaminogruppe vorhanden sein.
- In einer entsprechenden Acylaminogruppe bedeutet Acyl beispielsweise den Acylrest einer organischen Carbonsäure mit beispielsweise bis zu 18 Kohlenstoffatomen, besonders einer unsubstituierten oder substituierten Carbonsäure, beispielsweise einer durch Halogen oder Aryl substituierten Alkancarbonsäure oder einer unsubstituierten oder substituierten, beispielsweise durch Halogen, Niederalkoxy oder Nitro substituierten Benzoesäure, oder eines Carbonsäurehemiesters. Solche Acylgruppen sind beispielsweise Niederalkanoyl, wie Formyl, Acetyl oder Propionyl, Halogenniederalkanoyl, wie 2-Halogenacetyl, besonders 2-Chlor-, 2-Brom-, 2-Iod-, 2,2,2-Trifluor- oder 2,2,2-Trichloracetyl, das unsubstituiert oder substituiert sein kann, beispielsweise durch Halogen, Niederalkoxy oder Nitro, Benzoyl, beispielsweise Benzoyl, 4-Chlorbenzoyl, 4-Methoxybenzoyl oder 4-Nitrobenzoyl, oder durch Niederalkoxycarbonyl, das in der Position 1 des Niederalkylrests verzweigt sein kann oder in den Positionen 1 und 2 geeignet substituiert sein kann, besonders tert-Niederalkoxycarbonyl, beispielsweise tert-Butoxycarbonyl, Arylmethoxycarbonyl mit ein oder zwei Arylresten, die vorzugsweise Phenyl sind, das unsubstituiert oder monosubstituiert oder polysubstituiert sein kann beispielsweise durch Niederalkyl, besonders tert-Niederalkyl, wie tert-Butyl, Niederalkoxy, wie Methoxy, Hydroxyl, Halogen, beispielsweise Chlor, und/oder durch Nitro, wie unsubstituiertes oder substituiertes Benzyloxycarbonyl, beispielsweise 4-Nitrobenzyloxycarbonyl, oder substituiertes Diphenylmethoxycarbonyl, beispielsweise Benzhydryloxycarbonyl oder Di-(4-methoxyphenyl)-methoxycarbonyl, Aroylmethoxycarbonyl, worin die Aroylgruppe vorzugsweise Benzoyl ist, das unsubstituiert oder substituiert sein kann, beispielsweise durch Halogen, wie Brom, beispielsweise Phenacyloxycarbonyl, 2-Halogenniederalkoxycarbonyl, beispielsweise 2,2,2-Trichlorethoxycarbonyl, 2-Bromethoxycarbonyl oder 2-Iodethoxycarbonyl, oder in Stellung 2 trisubstituiertes Silylethoxycarbonyl, worin die Substituenten jeweils unabhängig voneinander unsubstituiert oder substituiert sind beispielsweise durch Niederalkyl, Niederalkoxy, Aryl, Halogen oder Nitro, einen aliphatischen, araliphatischen, cycloaliphatischen oder aromatischen Kohlenwasserstoffrest mit bis zu 15 Kohlenstoffatomen, wie entsprechend unsubstituiertes oder substituiertes Niederalkyl, Phenylniederalkyl, Cycloalkyl oder Phenyl, beispielsweise 2-Triniederalkylsilylethoxycarbonyl, wie 2-Trimethylsilylethoxycarbonyl oder 2-(Di-n-butylmethylsilyl)-ethoxycarbonyl oder 2-Triarylsilylethoxycarbonyl, wie 2-Triphenylsilylethoxycarbonyl.
- In einer Arylmethylaminogruppe, die eine monosubstituierte, disubstituierte oder besonders eine trisubstituierte Arylmethylaminogruppe ist, sind die Arylreste besonders unsubstituierte oder substituierte Phenylreste. Solche Gruppen sind beispielsweise Benzylamino, Diphenylmethylamino und besonders Tritylamino.
- Eine veretherte Mercaptogruppe in einer Aminogruppe, die durch solche Reste geschützt ist, ist besonders Arylthio oder Arylniederalkylthio, worin Aryl besonders für Phenyl steht, das unsubstituiert oder substituiert ist, beispielsweise durch Niederalkyl, wie Methyl oder tert-Butyl, Niederalkoxy, wie Methoxy, Halogen, wie Chlor, und/oder durch Nitro. Eine entsprechende Aminoschutzgruppe ist beispielsweise 4-Nitrophenylthio.
- In einem 2-Acylniederalkyl-1-en-1-ylrest, der als eine Aminoschutzgruppe verwendet werden kann, ist Acyl beispielsweise der entsprechende Rest einer Niederalkancarbonsäure, einer Benzoesäure, die unsubstituiert oder substituiert ist, beispielsweise durch Niederalkyl, wie Methyl oder tert-Butyl, Niederalkoxy, wie Methoxy, Halogen, wie Chlor, und/oder durch Nitro, oder besonders ein Carbonsäurehemiester, wie ein Carbonsäureniederalkylhemiester. Entsprechende Schutzgruppen sind besonders 1-Niederalkanoylprop-1-en-2-yl, wie 1-Acetylprop-1-en-2-yl, oder 1-Niederalkoxycarbonylprop-1-en-2-yl, beispielsweise 1-Ethoxycarbonylprop-1-en-2-yl.
- Bevorzugte Aminoschutzgruppen sind Acylreste von Carbonsäurehemiestern, besonders tert-Butyloxycarbonyl, Benzyloxycarbonyl, das unsubstituiert oder beispielsweise wie angegeben substituiert ist, beispielsweise 4-Nitrobenzyloxycarbonyl, oder Diphenylmethoxycarbonyl, oder 2-Halogenniederalkoxycarbonyl, wie 2,2,2-Trichlorethoxycarbonyl oder auch Trityl oder Formyl.
- Die Umsetzung zwischen dem Derivat der Formel IV und dem Anilinderivat der Formel V findet statt in geeigneten inerten polaren Lösemitteln, besonders Alkoholen, beispielsweise Niederalkanolen, wie Methanol, Propanol, Isopropanol und vor allem Ethanol oder n-Butanol. In einigen Fällen ist der Zusatz eines Lösevermittlers von Vorteil, wie von 1,3-Dimethyl-3,4,5,6-tetrahydro-2-(1H)-pyrimidinon (DMPU). Die Umsetzung findet bei erhöhten Temperaturen statt, beispielsweise bei Temperaturen im Bereich von 70 bis 150°C, vorzugsweise unter Rückflussbedingungen.
- Steht Z in der Verbindung der Formel IV für 1-Arylniederalkyl, dann wird dieser Rest vom erhaltenen Vorläufer der Verbindung der Formel I (mit Z anstelle des Wasserstoffatoms am Stickstoffatom) entfernt. Dies wird beispielsweise bewirkt durch eine Behandlung mit Protonsäuren, wie Chlorwasserstoffsäure, Phosphorsäuren oder Polyphosphorsäure, bei bevorzugten Temperaturen von 20°C bis 150°C und geeignetenfalls in Gegenwart von Wasser, was besonders das bevorzugte Verfahren ist, wenn Z für 1-Phenylethyl steht, oder vorzugsweise durch Behandlung mit Lewis-Säuren, besonders AlCl3, in einem aromatischen Lösemittel, besonders in Benzol und/oder Toluol, bei erhöhter Temperatur, besonders unter Rückflusstemperatur, wobei dies die speziell bevorzugte Variante ist, wenn Z für Benzyl steht, wozu auch auf die analogen Verfahren in Chem. Pharm. Bull. 39(5), Seite 1152 (1991) hingewiesen wird.
- Die Entfernung der Schutzgruppen, die keine Bestandteile des gewünschten Endprodukts der Formel I sind, wird in bekannter Weise durchgeführt, beispielsweise durch Solvolyse, besonders Hydrolyse, Alkoholyse oder Acidolyse, oder durch Reduktion, besonders durch Hydrogenolyse oder chemische Reduktion geeignetenfalls stufenweise oder simultan.
- Eine geschützte Aminogruppe wird in an sich bekannter Weise freigesetzt und der Art der Schutzgruppen entsprechend unter Anwendung verschiedener Methoden, vorzugsweise durch Solvolyse oder Reduktion. Die Gruppen 2-Halogenniederalkoxycarbonylamino (geeignetenfalls nach einer Um wandlung einer 2-Bromniederalkoxycarbonylaminogruppe in eine 2-Iodniederalkoxycarbonylaminogruppe), Aroylmethoxycarbonylamino oder 4-Nitrobenzyloxycarbonylamino können gespalten werden beispielsweise durch Behandlung mit einem geeigneten chemischen Reduktionsmittel, wie Zink, in Gegenwart einer geeigneten Carbonsäure, wie wässriger Essigsäure. Aroylmethoxycarbonylamino kann auch durch Behandlung mit einem nukleophilen Mittel, vorzugsweise mit einem Salz bildenden Mittel, gespalten werden, wie mit Natriumthiophenolat, während 4-Nitrobenzyloxycarbonylamino auch durch Behandlung mit einem Alkalimetalldithionit, beispielsweise mit Natriumdithionit, gespalten werden kann. Unsubstituiertes oder substituiertes Diphenylmethoxycarbonylamino, tert-Niederalkoxycarbonylamino oder 2-trisubstituiertes Silylethoxycarbonylamino kann durch Behandlung mit einer geeigneten Säure gespalten werden, beispielsweise mit Ameisensäure oder Trifluoressigsäure. Unsubstituiertes oder substituiertes Benzyloxycarbonylamino kann beispielsweise durch Hydrogenolyse gespalten werden, wie durch Behandlung mit Wasserstoff in Gegenwart eines geeigneten Hydrierkatalysators, wie eines Palladiumkatalysators. Unsubstituiertes oder substituiertes Triarylmethylamino oder Formylamino kann beispielsweise gespalten werden durch Behandlung mit einer Säure, wie einer Mineralsäure, beispielsweise mit Chlorwasserstoffsäure, oder mit einer organischen Säure, wie beispielsweise Ameisensäure, Essigsäure oder Trifluoressigsäure, geeignetenfalls in Anwesenheit von Wasser. Eine Aminogruppe, die durch eine organische Silylgruppe geschützt ist, kann freigesetzt werden beispielsweise durch Hydrolyse oder Alkoholyse. Eine Aminogruppe, die durch 2-Halogenacetyl geschützt ist, beispielsweise 2-Chloracetyl, kann freigesetzt werden durch Behandlung mit Thioharnstoff in Gegenwart einer Base oder mit einem Thiolatsalz, wie einem Alkalimetallthiolat, von Thioharnstoff, und anschließende Solvolyse, wie eine Alkoholyse oder Hydrolyse, des erhaltenen Kondensationsprodukts. Eine Aminogruppe, die durch ein 2-substituiertes Silylethoxycarbonyl geschützt ist, kann in die freie Aminogruppe auch durch Behandlung mit einem Salz von Fluorwasserstoffsäure umgewandelt werden, das Fluoridanionen ergibt.
- Verfahren b)
- Z' steht für 1-Arylniederalkyl in einer Verbindung der Formel VI, besonders 1-Phenylethyl und auch Benzyl.
- Die Verbindung der Formel VI befindet sich in einem tautomeren Gleichgewicht (Lactam/Lactim-Form), wobei die Lactam-Form (Formel VI) wahrscheinlich dominiert. Die Formel VI dient zur Darstellung der beiden möglichen Gleichgewichtsformen.
- Die Lactim-Form hat die Formel VIaworin die angegebenen Reste wie für Verbindungen der Formel VI definiert sind.
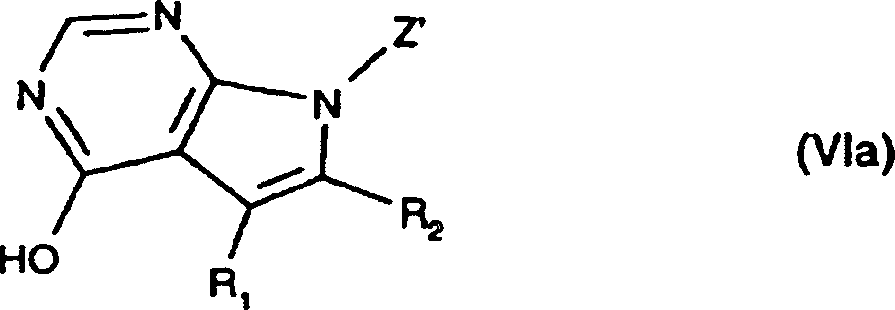
- Die vorliegende Erfindung bezieht sich auch auf die neuen Verbindungen der Formeln VI und VIa.
- Das verwendete Dehydrationsmittel ist besonders ein starkes chemisches Dehydrationsmittel, vor allem Phosphorpentoxid (P4O10).
- Ein geeignetes tertiäres Amin ist primär Ammoniak, der durch drei Reste substituiert ist, die unabhängig voneinander ausgewählt sind aus Alkyl, besonders Niederalkyl, wie Methyl oder Ethyl, und Cycloalkyl mit 3 bis 7 Kohlenstoffatomen, besonders Cyclohexyl, beispielsweise N,N-Dimethyl-N-cyclohexylamin, N-Ethyl-N,N-diisopropylamin oder Triethylamin, oder auch Pyridin, N-Methylmorpholin oder 4-Dimethylaminopyridin.
- Die Umsetzung zwischen dem Pyrrolopyrimidinon der Formel VI und dem Anilinderivat der Formel V findet bei erhöhter Temperatur statt, beispielsweise bei 200 bis 250°C.
- Verfahren e)
- Diese Umsetzung wird vorzugsweise in einem geeigneten inerten Lösemittel, beispielsweise einem geeigneten Alkohol, besonders Methanol, bei Temperaturen von etwa +10°C bis +100°C, vorzugsweise bei Siedetemperatur des Reaktionsgemisches durchgeführt. In diesem Fall wird das Hydroxylaminderivat der Formel VIII vorzugsweise als ein Salz eingesetzt und durch Zugabe einer wässrigen Lösung von Natriumacetat zum Reaktionsgemisch in die freie Base umgewandelt.
- Verfahren f)
- Ein reaktives Carbonsäurederivat einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Carboxyl steht und die anderen Substituenten wie oben für Verbindungen der Formel I definiert sind, ist beispielsweise ein geeigneter Ester, wie besonders ein Ethylester.
- Wo möglich, beispielsweise wenn Morpholin das Amin der Formel VII ist, kann das Amin der Formel VII auch direkt als ein Lösemittel verwendet werden. In anderen Fällen wird ein geeignetes inertes Lösemittel, beispielsweise Dimethylformamid, zusammen mit TPTU (O-(1,2-Dihydro-2-oxo-1-pyridyl)-N,N,N',N'-tetramethyluroniumtetrafluorborat) verwendet. Diese Umsetzung wird vorzugsweise bei Temperaturen von etwa +10°C bis +150°C und vorzugsweise bei Raumtemperatur bis 100°C durchgeführt.
- Verfahren g)
- Ein Lawesson-Reagenz ist 2,4-Di-[4-methoxyphenyl]-1,3-dithia-2,4-diphosphetan-2,4-disulfid und ist im Handel unter anderem von Sigma, Fluka und dergleichen erhältlich. Diese Umsetzung wird vorzugsweise in einem geeigneten inerten Lösemittel durchgeführt, beispielsweise einem geeigneten Ether, wie besonders einem cyclischen Ether, beispielsweise Tetrahydrofuran, bei Temperaturen von etwa 50°C bis 150°C und vorzugsweise bei Siedetemperatur des Reaktionsgemisches.
- Verfahren h)
- Hier ist R13 vorzugsweise Niederalkyl, besonders Ethyl, oder Phenyl, das unsubstituiert oder substituiert ist durch Halogen, Niederalkyl, Hydroxymethyl, Aminomethyl, Hydroxyl, Niederalkanoyloxy, Niederalkoxy, Carboxyl, Niederalkanoyl, Benzoyl, Niederalkoxycarbonyl, Carbamoyl, N-Niederalkylcarbamoyl, N,N-Diniederalkylcarbamoyl, Cyano, Amino, Niederalkanoylamino, Niederalkylamino, N,N-Diniederalkylamino oder Trifluormethyl, wobei R13 besonders für 4-Methoxyphenyl steht.
- Diese Umsetzung wird vorzugsweise in einem geeigneten inerten Lösemittel, beispielsweise einem geeigneten Alkohol, besonders Methanol, oder einem geeigneten Ether, beispielsweise Dioxan, bei Temperaturen von etwa +20°C bis +180°C und vorzugsweise bei Siedetemperatur des Reaktionsgemisches durchgeführt.
- Verfahren i)
- Diese Umsetzung wird vorzugsweise in einem geeigneten inerten Lösemittel, beispielsweise einem geeigneten Alkohol, wie besonders 1-Methoxyethanol, in Gegenwart von Lithiumchlorid, beispielsweise einer 1½fachen Menge der molaren Menge an Lithiumchlorid, bei Temperaturen von etwa +20°C bis +180°C und vorzugsweise bei Siedetemperatur des Reaktionsgemisches durchgeführt.
- Verfahren j)
- Diese Umsetzung wird vorzugsweise in einem geeigneten inerten Lösemittel, beispielsweise in Dimethylformamid, in Gegenwart von Natriumhydrogencarbonat, bei Temperaturen von etwa 0°C bis +180°C und vorzugsweise bei Raumtemperatur durchgeführt. In diesem Fall wird das Niederalkyliodid, wie besonders Methyliodid, vorzugsweise als eine Lösung zugegeben, beispielsweise eine 1 M Lösung, in einem geeigneten Ether, wie Dioxan.
- Verfahren k)
- Hierin steht X vorzugsweise für Chlor.
- Die Umsetzung wird vorzugsweise in einem geeigneten inerten und wasserfreien Lösemittel, beispielsweise Dimethylacetamid, bei Temperaturen von –30°C bis +70°C und vorzugsweise bei 0°C durchgeführt.
- Verfahren l)
- Hierin steht R17 vorzugsweise für Methyl.
- Diese Umsetzung wird vorzugsweise in einem geeigneten inerten und wasserfreien Lösemittel, beispielsweise einem Ether, wie Tetrahydrofuran, in Gegenwart von Triethylamin bei Temperaturen von etwa –10°C bis +70°C und vorzugsweise bei Raumtemperatur durchgeführt.
- Verfahren m)
- Diese Umsetzung wird vorzugsweise in einem geeigneten inerten und wasserfreien Lösemittel, beispielsweise einem Ether, wie Tetrahydrofuran, zweckmäßig in Gegenwart von Dimethylacetamid, bei Temperaturen von etwa 0°C bis +150°C und vorzugsweise bei Siedetemperatur des Reaktionsgemisches durchgeführt.
- Verfahren n)
- Diese Umsetzung wird vorzugsweise in einem geeigneten inerten und wasserfreien Lösemittel, beispielsweise einem Ether, wie Tetrahydrofuran, zweckmäßig in Gegenwart von Dimethylacetamid, bei Temperaturen von etwa 0°C bis +150°C und vorzugsweise bei der Siedetemperatur des Reaktionsgemisches durchgeführt.
- Verfahren o)
- Diese Umsetzung wird vorzugsweise in einem geeigneten inerten und wasserfreien Lösemittel, beispielsweise einem Ether, wie Dioxan, zweckmäßig in Gegenwart von 2,6-Lutidin, bei Temperaturen von etwa –30°C bis +150°C und vorzugsweise bei Raumtemperatur durchgeführt.
- Ausgangsmaterialien
- Die Ausgangsmaterialien der Formel IV sind neu, so dass sich die Erfindung auch darauf bezieht. Sie können in Analogie zu Verfahren hergestellt werden, wie sie beschrieben werden in
DE 28 18 676 A undDE 30 36 390 A . - Das Ausgangsmaterial der Formel IV, worin X für Chlor steht, wird erhalten beispielsweise aus einer zur Formel IV analogen Verbindung, worin X für Hydroxyl steht (siehe die Verbindungen der Formel VIa), durch Umsetzung mit Phosphoroxychlorid (Phosphorylchlorid, P(=O)Cl3), unter Ausschluss von Feuchtigkeit bei Rückflusstemperatur. Gewünschtenfalls kann die weitere Umsetzung des so erhaltenen Ausgangsmaterials der Formel IV, worin X für Chlor steht, mit einem Anilinderivat der Formel V im gleichen Reaktionsgefäß umgesetzt werden, nämlich in einer Eintopfreaktion. Zu diesem Zweck wird nach Beendigung der Umsetzung mit Phosphoroxychlorid das Reaktionsgemisch zur Trockne eingedampft, der Rückstand mit einem geeigneten Lösemittel, wie n-Butanol, suspendiert, und die Suspension dann mit dem Anilinderivat der Formel V weiter umgesetzt.
- Eine zur Formel IV, worin X für Hydroxyl steht, analoge Verbindung lässt sich beispielsweise aus einer Verbindung der Formel XV erhaltenworin die Symbole wie oben definiert sind, durch Umsetzung mit Ameisensäure, die vorzugsweise in einem Überschuss im Verhältnis zur Verbindung der Formel XV verwendet wird, beispielsweise in einem 10 bis 30 molaren Überschuss, geeignetenfalls in Gegenwart inerter Lösemittel, wie Dimethylformamid, bei erhöhter Temperatur, beispielsweise bei Temperaturen von 80°C bis zur Siedetemperatur.

- Alternativ wird eine Verbindung erhalten, die zur Formel IV analog ist, worin X für Hydroxyl steht und die anderen Symbole wie oben definiert sind, beispielsweise aus einer Verbindung der Formel XVIworin R19 für Niederalkyl steht, besonders für Ethyl, und die anderen Symbole wie oben definiert sind, durch Umsetzung mit einem großen Überschuss an Formamid in einem Gemisch von wasserfreiem Dimethylformamid und Ameisensäure. Die Umsetzung wird bei erhöhter Temperatur, beispielsweise von 100°C bis 150°C und vorzugsweise unter einem Schutzgas durchgeführt.
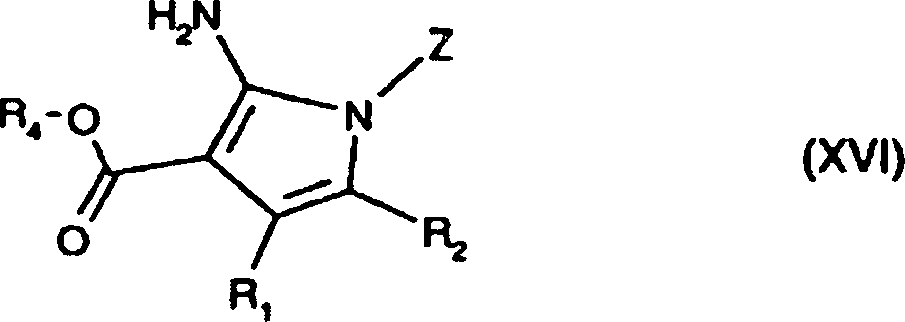
- Die vorliegende Erfindung bezieht sich auch auf die neuen Ausgangsmaterialien der Formeln XV und XVI.
- Die 1-(Z')-2-Amino-3-cyanopyrrole der Formel XV, die als Zwischenprodukte verwendet werden, können in guten Ausbeuten hergestellt werden nach an sich bekannten Verfahren und sind bereits publiziert worden, wozu beispielsweise hingewiesen wird auf H. J. Roth und K. Eger, Arch. Pharmaz. 308, Seite 179 (1975). Zu diesem Zweck wird beispielsweise eine Verbindung der Formel XVIIzuerst mit einem Amin der Formel Z'-NH2 unter Bildung einer Verbindung der Formel XVIII umgesetzt
 die dann durch Umsetzung mit Malonsäurenitril der Formel CH2(CN)2 zum gewünschten Zwischenprodukt umgewandelt wird. Im Einzelnen wird diese Umsetzung mit dem Amin Z'-NH2 unter üblichen Kondensationsbedingungen durchgeführt, beispielsweise in Gegenwart einer katalytischen Menge einer starken Säure, beispielsweise Chlorwasserstoffsäure oder p-Toluolsulfonsäure, bei erhöhter Temperatur, vorzugsweise Siedetemperatur, in einem geeigneten Lösemittel, beispielsweise Benzol oder Toluol, unter Abtrennung von Wasser, wodurch das entsprechende α-Aminoketon der Formel XVIII erhalten wird. Dieses wird nicht isoliert, sondern sofort mit Malonsäurenitril unter Erhitzung und weiterer Abtrennung von Wasser kondensiert, was erforderlichenfalls durch Zusatz einer geringen Menge einer Base, wie Piperidin, geschieht, wodurch eine Verbindung der Formel XV erhalten wird.
die dann durch Umsetzung mit Malonsäurenitril der Formel CH2(CN)2 zum gewünschten Zwischenprodukt umgewandelt wird. Im Einzelnen wird diese Umsetzung mit dem Amin Z'-NH2 unter üblichen Kondensationsbedingungen durchgeführt, beispielsweise in Gegenwart einer katalytischen Menge einer starken Säure, beispielsweise Chlorwasserstoffsäure oder p-Toluolsulfonsäure, bei erhöhter Temperatur, vorzugsweise Siedetemperatur, in einem geeigneten Lösemittel, beispielsweise Benzol oder Toluol, unter Abtrennung von Wasser, wodurch das entsprechende α-Aminoketon der Formel XVIII erhalten wird. Dieses wird nicht isoliert, sondern sofort mit Malonsäurenitril unter Erhitzung und weiterer Abtrennung von Wasser kondensiert, was erforderlichenfalls durch Zusatz einer geringen Menge einer Base, wie Piperidin, geschieht, wodurch eine Verbindung der Formel XV erhalten wird.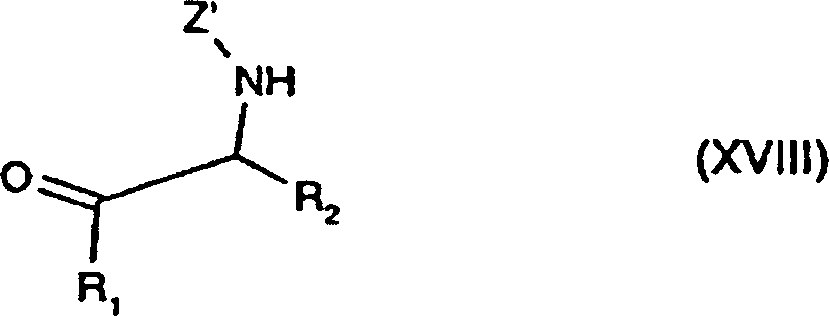
- Die Verbindungen der Formel XVI, die als Zwischenprodukte verwendet werden, werden beispielsweise erhalten durch Umsetzung eines 2-Amidinoessigsäureniederalkylesters der Formel XIXworin R19 wie oben definiert ist, mit einem 2-X-1-R2-Ethan-1-onderivat der Formel XX
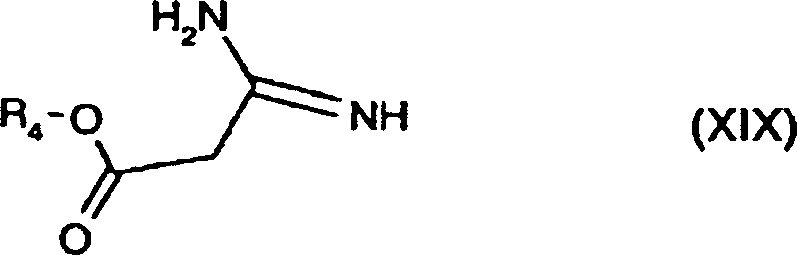 worin die angegebenen Symbole wie oben definiert sind. Die Abgangsgruppe X ist vorzugsweise Brom. Der 2-Amidinoessigsäureniederalkylester der Formel XIX wird aus seinem Säureadditionssalz, wie besonders Hydrochlorid, freigesetzt, bevor mit der Umsetzung mit äquimolaren Mengen einer Base, besonders Natriumethoxid, unter Eiskühlung begonnen wird. Diese Umsetzung wird in einem geeigneten Lösemittel durchgeführt, wie speziell einem Niederalkanol, vorzugsweise Ethanol, bei bevorzugten Temperaturen von 0°C bis 50°C, besonders bei Raumtemperatur.
worin die angegebenen Symbole wie oben definiert sind. Die Abgangsgruppe X ist vorzugsweise Brom. Der 2-Amidinoessigsäureniederalkylester der Formel XIX wird aus seinem Säureadditionssalz, wie besonders Hydrochlorid, freigesetzt, bevor mit der Umsetzung mit äquimolaren Mengen einer Base, besonders Natriumethoxid, unter Eiskühlung begonnen wird. Diese Umsetzung wird in einem geeigneten Lösemittel durchgeführt, wie speziell einem Niederalkanol, vorzugsweise Ethanol, bei bevorzugten Temperaturen von 0°C bis 50°C, besonders bei Raumtemperatur.
- Ausgangsverbindungen der Formel I, worin einer der Reste R1 und R2 für Formyl, Carboxyl, Niederalkoxycarbonyl, Cyano, Carbamoyl, Thiocarbamoyl, Aminophenyl oder Tetrazol-5-yl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene funktionale Gruppen erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, werden erhalten, wie dies exemplarisch im Beispielsteil beschrieben ist. Ein Durchschnittsfachmann kann die speziellen beispielhaften Reaktionen ohne größere Anstrengung zu Verbindungen umwandeln, die andere Reste R und R1 oder R2 haben als bei den speziellen Beispielen, solange die im Rest R vorhandenen freien funktionalen Gruppen leicht entfernbare Schutzgruppen sind, was bedeutet, dass diese die gewünschte Reaktion nicht stören. In vielen Fällen, besonders wenn R beispielsweise für Halogen steht, ist kein Schutz erforderlich.
- Allgemeine Verfahrensbedingungen
- Freie Verbindungen der Formel I mit Salz bildenden Eigenschaften, die wie dem Verfahren entsprechend erhältlich sind, können in an sich bekannter Weise in ihre Salze überführt werden, beispielsweise durch Behandlung mit Säuren oder geeigneten Derivaten hiervon, beispielsweise durch Zusatz der geeigneten Säure zur Verbindung der Formel I, die in einem geeigneten Lösemittel gelöst ist, beispielsweise einem Ether, wie einem cyclischen Ether, besonders Dioxan und vor allem Tetrahydrofuran.
- Isomerengemische, die erfindungsgemäß erhältlich sind, können in an sich bekannter Weise in die einzelnen Isomeren aufgetrennt werden. Racemate können beispielsweise aufgetrennt werden durch Bildung von Salzen mit optisch reinen Salz bildenden Reagenzien und Auftrennung des so erhältlichen Gemisches an Diastereomeren beispielsweise durch eine fraktionierte Kristallisation.
- Die oben beschriebenen Umsetzungen können unter an sich bekannten Reaktionsbedingungen durchgeführt werden in Abwesenheit oder gewöhnlich in Anwesenheit von Lösemitteln oder Verdünnungsmitteln, vorzugsweise solchen, die gegenüber den verwendeten Reagenzien inert und Lösemittel hierfür sind, in Abwesenheit oder Anwesenheit von Katalysatoren, Kondensationsmitteln, beispielsweise Phosphorpentoxid, oder Neutralisationsmitteln, beispielsweise Basen, speziell Stickstoffbasen, wie Triethylaminhydrochlorid, in Abhängigkeit von der Art der Reaktion und/oder der Reaktanten, bei einer reduzierten, normalen oder erhöhten Temperatur, beispielsweise einer Temperatur im Bereich von etwa –80°C bis etwa 200°C, vorzugsweise von etwa –20°C bis etwa 150°C, beispielsweise beim Siedepunkt des verwendeten Lösemittels, unter atmosphärischem Druck oder in einem geschlossenen Behältnis, geeignetenfalls unter Druck und/oder in einer inerten Atmosphäre, beispielsweise unter einer Stickstoffatmosphäre.
- Die angegebenen speziellen Reaktionsbedingungen sind jeweils bevorzugt.
- Lösemittel und Verdünnungsmittel sind beispielsweise Wasser, Alkohole, beispielsweise Niederalkylhydroxide, wie Methanol, Ethanol, Propanol oder besonders Butanol, Diole, wie Ethylenglycol, Triole, wie Glycerin, oder Arylalkohole, wie Phenol, Säureamide, beispielsweise Carbonsäureamide, wie Dimethylformamid, Dimethylacetamid oder 1,3-Dimethyl-3,4,5,6-tetrahydro-2-(1H)-pyrimidinon (DMPU), Carbonsäuren, besonders Ameisensäure oder Essigsäure, Amide anorganischer Säuren, wie Hexa methylphosphorsäuretriamid, Ether, beispielsweise cyclische Ether, wie Tetrahydrofuran oder Dioxan, oder acyclische Ether, wie Diethylether oder Ethylenglycoldimethylether, Halogenkohlenwasserstoffe, wie Halogenniederalkane, beispielsweise Methylenchlorid oder Chloroform, Ketone, wie Aceton, Nitrile, wie Acetonitril, Säureanhydride, wie Essigsäureanhydrid, Ester, wie Ethylacetat, Bisalkansulfine, wie Dimethylsulfoxid, Stickstoff enthaltende heterocyclische Verbindungen, wie Pyridin, Kohlenwasserstoffe, beispielsweise Niederalkane, wie Heptan, oder aromatische Verbindungen, wie Benzol, Toluol oder Xylole, oder Gemische dieser Lösemittel, wobei die für die obigen Umsetzungen jeweils geeigneten Lösemittel ohne weiteres ausgewählt werden können.
- Zur Aufarbeitung der erhaltenen Verbindung der Formel I oder ihrer Salze werden herkömmliche Verfahren angewandt, beispielsweise eine Solvolyse mit einem Überschuss an Reagenzien, eine Umkristallisation, eine Chromatographie, beispielsweise eine Verteilungschromatographie, eine Ionenchromatographie oder Gelchromatographie, eine Verteilung zwischen anorganischen und organischen Lösemittelphasen, eine einzelne oder multiple Extraktion, besonders nach einer Ansäuerung oder Erhöhung der Basizität oder des Salzgehalts, eine Trocknung über hygroskopische Salze, eine Digestion, Filtration, Waschung, Auflösung, Verdampfung, erforderlichenfalls unter Vakuum oder Hochvakuum, Destillation, Kristallisation, beispielsweise von in einer Ölform erhaltenen Verbindungen oder aus der Mutterlauge, wobei eine Animpfung mit einem Kristall des Endprodukts ebenfalls möglich ist, oder durch eine Kombination von zwei oder mehr der erwähnten Aufarbeitungsstufen, die auch wiederholt angewandt werden können, und dergleichen.
- Ausgangsmaterialien und Zwischenprodukte können auch in reiner Form, beispielsweise nach einer Aufarbeitung, wie dies gerade erwähnt ist, in partiell gereinigter Form oder alternativ beispielsweise direkt als ein Rohprodukt verwendet werden.
- Die Verbindungen und ihre Salze können auch in Form von Hydraten erhalten werden oder ihre Kristalle können beispielsweise das für eine Kristallisation verwendete Lösemittel enthalten. Die vorliegende Erfindung bezieht sich auch auf diese Hydrate oder Solvate der Verbindungen der Formel I und auf die beschriebenen Ausgangsmaterialien, die ebenfalls zur Erfindung gehören.
- Im Hinblick auf die enge Verwandtschaft zwischen den Verbindungen der Formel I in freier Form oder in Form ihrer Salze werden oben und im Folgenden unter den freien Verbindungen und ihren Salzen auch die entsprechenden Salze und freien Verbindungen verstanden, sofern dies geeignet und zweckdienlich ist, mit der Maßgabe, dass diese Verbindungen Salz bildende Gruppen enthalten. Gleiches gilt auch für die Hydrate und Solvate.
- Beim erfindungsgemäßen Verfahren werden vorzugsweise solche Ausgangsmaterialien verwendet, die zu den neuen Verbindungen der Formel I führen, wie dies zu Beginn beschrieben worden ist, da diese besonders wertvoll sind.
- Die Erfindung bezieht sich auch auf die Formen des Verfahrens, bei denen eine Verbindung, die an irgendeiner Stufe des Verfahrens als ein Zwischenprodukt erhalten werden kann, das als ein Ausgangsmaterial verwendet wird, und wobei die restlichen Vefahrensstufen durchgeführt werden oder bei denen unter den Reaktionsbedingungen ein Ausgangsmaterial gebildet wird oder in der Form eines Derivats, beispielsweise eines Salzes hiervon, verwendet wird.
- Pharmazeutische Zusammensetzungen, ihre Herstellung und die erfindungsgemäße Verwendung der Verbindungen der Formel I und von Zusammensetzungen, die diese Verbindungen als Wirkstoff umfassen
- Die vorliegende Erfindung bezieht sich auch auf pharmazeutische Zusammensetzungen, die ein oder mehr Verbindungen der Formel I als Wirkstoff enthalten und die besonders zur Behandlung von Krankheiten der am Beginn erwähnten Art verwendet werden. Zusammensetzungen für eine enterale Verabreichung, wie eine nasale, buccale, rektale oder besonders eine orale Verabreichung, und für eine parenterale Verabreichung, wie eine intravenöse, intramuskulare oder subkutane Verabreichung an Warmblüter, speziell an Menschen, sind besonders bevorzugt. Diese Zusammensetzungen enthalten den Wirkstoff als solchen oder vorzugsweise zusammen mit einem pharmazeutisch akzeptablen Träger. Die Dosis des Wirkstoff ist abhängig von der zu behandelnden Krankheit und der Spezies, dem Alter, dem Gewicht und dem individuellen Zustand, den individuellen pharmakokinetischen Daten, der zu behandelnden Krankheit und auch von der Art der Verabreichung.
- Weiter bezieht sich die Erfindung auf pharmazeutische Zusammensetzungen zur Verwendung bei einem Verfahren für die Behandlung des menschlichen und tierischen Körpers, auf ein Verfahren zu deren Herstellung, speziell auf Zusammensetzungen für die Behandlung von Tumoren, und auf ein Verfahren für die Behandlung neoplastischer Krankheiten, besonders der oben erwähnten Krankheiten.
- Bevorzugt ist eine pharmazeutische Zusammensetzung, die sich für eine Verabreichung an einen Warmblüter eignet, speziell einen Menschen, der an einer Krankheit leidet, die auf eine Inhibition einer Protein-Kinase responsiv ist, beispielsweise Psoriasis oder ein Tumor, wobei diese Zusammensetzung eine Verbindung der Formel I oder ein Salz hiervon, falls Salz bildende Gruppen vorhanden sind, in einer Menge umfasst, die für die Inhibition der Protein-Kinase wirksam ist, zusammen mit wenigstens einem pharmazeutisch akzeptablen Träger.
- Die pharmazeutischen Zusammensetzungen enthalten etwa 1% bis etwa 95% Wirkstoff, wobei Verabreichungsformen in Form einer einzelnen Dosierungsform vorzugsweise etwa 20% bis etwa 90% Wirkstoff enthalten, und Verabreichungsformen, die keine einzelnen Dosierungsformen sind, vorzugsweise etwa 5% bis etwa 20% Wirkstoff enthalten. Die Einheitsdosierungsformen sind beispielsweise mit Zucker beschichtete Tabletten, Tabletten, Ampullen, Fläschchen, Suppositorien oder Kapseln. Andere Verabreichungsformen sind beispielsweise Salben, Cremes, Pasten, Schäume, Tinkturen, Lippenstifte, Tropfen, Sprays, Dispersionen und dergleichen. Beispiele sind Kapseln, die etwa 0,05 g bis etwa 1,0 g Wirkstoff enthalten.
- Die erfindungsgemäßen pharmazeutischen Zusammensetzungen werden in an sich bekannter Weise hergestellt, beispielsweise durch herkömmliche Techniken zur Vermischung, Granulation, Zuckerbeschichtung, Auflösung oder Lyophilisation.
- Vorzugsweise werden Lösungen des Wirkstoffs verwendet, und auch Suspensionen oder Dispersionen, besonders isotonische wässrige Lösungen, Dispersionen und Suspensionen, die beispielsweise im Fall lyophilisierter Zusammensetzungen den Wirkstoff als solchen oder zusammen mit einem Träger, beispielsweise Mannit, enthalten und vor ihrer Verwendung zubereitet werden können. Die pharmazeutischen Zusammensetzungen können sterilisiert sein und/oder Exzipientien enthalten, beispielsweise Konservierungsmittel, Stabilisatoren, Netzmittel und/oder Emulgiermittel, Lösevermittler, Salze zur Regulierung des osmotischen Drucks und/oder Puffer, und werden in an sich bekannter Weise hergestellt, beispielsweise durch herkömmliche Verfahren zur Auflösung und Lyophilisation. Die erwähnten Lösungen oder Suspensionen können auch Substanzen zur Erhöhung der Viskosität enthalten, wie Natriumcarboxymethylcellulose, Carboxymethylcellulose, Dextran, Polyvinylpyrrolidon oder Gelatine.
- Suspensionen in Öl enthalten als die Ölkomponente pflanzliche, synthetische oder halbsynthetische Öle, wie sie für Injektionszwecke üblich sind. Zu erwähnen sind dabei speziell flüssige Fettsäureester, die als die Säurekomponente eine langkettige Fettsäure mit 8 bis 22 Kohlenstoffatomen, speziell mit 12 bis 22 Kohlenstoffatomen, enthält, beispielsweise Laurinsäure, Tridecylsäure, Myristinsäure, Pentadecylsäure, Palmitinsäure, Margarinsäure, Stearinsäure, Arachidonsäure oder Behensäure, oder entsprechende ungesättigte Säuren, beispielsweise Ölsäure, Elaidinsäure, Erucasäure, Brassidinsäure oder Linolensäure, gewünschtenfalls unter Zusatz von Antioxidantien, beispielsweise Vitamin E,β-Carotin oder 3,5-Di-tert-butyl-4-hydroxytoluol. Die Alkoholkomponente dieser Fettsäureester hat maximal 6 Kohlenstoffatome und ist ein monohydrischer oder polyhydrischer Alkohol, beispielsweise ein monohydrischer, dihydrischer oder trihydrischer Alkohol, beispielsweise Methanol, Ethanol, Propanol, Butanol oder Pentanol oder ein Isomer hiervon, aber speziell ein Glycol oder Glycerin. In diesem Zusammenhang sind daher folgende Beispiele von Fettsäureestern zu erwähnen: Ethyloleat, Isopropylmyristat, Isopropylpalmitat, Labrafil M 2375 (ein Polyoxyethylenglycerintrioleat von Gattefossé, Paris), Labrafil M 1944 CS (ungesättigte polyglycolierte Glyceride, die durch Alkoholyse von Aprikosenkernöl hergestellt werden und aus Glyceriden und Polyethylenglycolestern bestehen, Gattefossé, Frankreich), Labrasol (gesättigte polyglycolierte Glyceride, die durch Alkoholyse von TCM hergestellt werden und Glyceride und Polyethylenglycolester enthalten, Gattefossé, Frankreich) und/oder Miglyol 812 (ein Triglycerid von gesättigten Fettsäuren mit einer Kettenlänge von C8 bis C12, Hüls AG, Deutschland), wobei hierfür aber besonders Pflanzenöle verwendet werden, wie Baumwollsaatöl, Mandelöl, Olivenöl, Rizinusöl, Sesamöl, Sojabohnenöl und vor allem Erdnussöl.
- Injektionslösungen werden in bekannter Weise unter sterilen Bedingungen hergestellt, wobei gleiches auch für eine Abfüllung der Zusammensetzungen beispielsweise in Ampullen oder Fläschchen und beispielsweise eine Versiegelung der jeweiligen Behältnisse gilt.
- Pharmazeutische Zusammensetzungen für eine orale Verabreichung können beispielsweise erhalten werden durch Kombination des Wirkstoffs mit ein oder mehr festen Trägern, geeignetenfalls durch Granulation eines erhaltenen Gemisches, und gewünschtenfalls durch entsprechende Verarbeitung des Gemisches oder der Granulate, geeignetenfalls durch Zugabe weiterer Exzipientien, unter Bildung von Tabletten oder mit Zucker beschichteten Tablettenkernen.
- Geeignete Träger sind besonders Füllstoffe, wie Zucker, beispielsweise Lactose, Saccharose, Mannit oder Sorbit, Cellulosepräparationen und/oder Calciumphosphate, beispielsweise Tricalciumphos phat oder Calciumhydrogenphosphat, und auch Bindemittel, wie Stärken, beispielsweise Maisstärke, Weizenstärke, Reisstärke oder Kartoffelstärke, Methylcellulose, Hydroxypropylmethylcellulose, Natriumcarboxymethylcellulose und/oder Polyvinylpyrrolidon, und/oder gewünschtenfalls Zerfallhilfsmittel, wie die oben erwähnten Stärken, auch Carboxymethylstärke, vernetztes Polyvinylpyrrolidon, Alginsäure oder Salze hiervon, wie Natriumalginat. Weitere Adjunkte sind hauptsächlich Fließkonditionierer und Schmiermittel, beispielsweise Kieselsäure, Talk, Stearinsäure oder Salze hiervon, wie Magnesiumstearat oder Calciumstearat, und/oder Polyethylenglycol oder Derivate hiervon.
- Mit Zucker beschichtete Tablettenkerne können mit geeigneten und optional enterischen Überzügen versehen werden, unter anderem mit konzentrierten Zuckerlösungen, die Gummi arabicum, Talk, Polyvinylpyrrolidon, Polyethylenglycol und/oder Titandioxid enthalten, oder mit Beschichtungslösungen in geeigneten organischen Lösemitteln oder Lösemittelgemischen, oder für die Herstellung enterischer Überzüge Lösungen geeigneter Cellulosepräparationen, wie Acetylcellulosephthalat oder Hydroxypropylmethylcellulosephthalat. Den Tabletten oder den mit Zucker überzogenen Tabletten können auch Farbstoffe oder Pigmente beigegeben werden, und zwar beispielsweise für Identifikationszwecke oder zur Kenntlichmachung unterschiedlicher Wirkstoffdosierungen.
- Zu oral verabreichbaren pharmazeutischen Zusammensetzungen gehören auch trocken gefüllte Kapseln, die Gelatine umfassen, oder auch weiche versiegelte Kapseln, die Gelatine und einen Weichmacher umfassen, wie Glycerin oder Sorbit. Die trocken gefüllten Kapseln können den Wirkstoff in Form von Granulaten enthalten, beispielsweise als ein Gemisch mit Füllstoffen, wie Maisstärke, Bindemitteln und/oder Gleitmitteln, wie Talkum oder Magnesiumstearat, und geeignetenfalls mit Stabilisatoren. In Weichkapseln ist der Wirkstoff vorzugsweise gelöst oder suspendiert in geeigneten flüssigen Exzipientien, wie Fettölen, Paraffinöl oder flüssigen Polyethylenglycolen oder Fettsäureestern von Ethylen oder Propylenglycol, die auch mit Stabilisatoren und Tensiden versetzt sein können, beispielsweise mit üblichen Polyethylensorbitanfettsäureestern.
- Andere orale Verabreichungsformen sind beispielsweise in herkömmlicher Weise hergestellte Sirupe, die den Wirkstoff beispielsweise in suspendierter Form und in einer Konzentration von etwa 5% bis 20%, vorzugsweise etwa 10%, oder in einer ähnlichen Konzentration enthalten, die für eine geeignete Einzeldosis sorgt, beispielsweise bei Verabreichung in Mengen von 5 oder 10 ml. Ebenfalls geeignet sind beispielsweise pulverisierte oder flüssige Konzentrate zur Herstellung von Shakes, beispielsweise in Milch. Solche Konzentrate können auch in Einzeldosismengen abgepackt sein.
- Geeignete rektal verabreichbare pharmazeutische Zusammensetzungen sind beispielsweise Suppositorien, die aus einer Kombination des Wirkstoffs und einer Suppositoriengrundlage bestehen. Geeignete Suppositoriengrundlagen sind beispielsweise natürliche oder synthetische Triglyceride, Paraffinkohlenwasserstoffe, Polyethylenglycole oder höhere Alkanole.
- Für eine parenterale Verabreichung gibt es hauptsächlich geeignete wässrige Lösungen des Wirkstoffs in einer in Wasser löslichen Form, beispielsweise in Form eines in Wasser löslichen Salzes, oder als Injektionssuspension, die Substanzen zur Erhöhung der Viskosität enthalten, beispielsweise Natriumcarboxymethylcellulose, Sorbit und/oder Dextran, und gewünschtenfalls auch Stabilisatoren. Der Wirkstoff, der zweckdienlich auch Adjunkts enthält, kann auch in Form eines Lyophilisats vorliegen und vor einer parenteralen Verabreichung durch Zugabe geeigneter Lösemittel in Lösung gebracht werden.
- Lösungen, die beispielsweise für eine parenterale Verabreichung verwendet werden können, können auch als Infusionslösungen verwendet werden.
- Bevorzugte Konservierungsmittel sind beispielsweise Antioxidantien, wie Ascorbinsäure, oder Mikrobiozide, wie Sorbinsäure oder Benzoesäure.
- Salben sind Emulsionen von Öl-in-Wasser, die bis zu 70%, bevorzugt aber 20% bis 50%, Wasser oder eine wässrige Phase enthalten. Geeignet als Fettphase sind hauptsächlich Kohlenwasserstoffe, beispielsweise Petroleumgel, Paraffinöl oder Hartparaffine, die zur Verbesserung des Wasserbindevermögens vorzugsweise geeignete Hydroxyverbindungen enthalten, wie Fettalkohole oder Fettester, beispielsweise Cetylalkohol, oder Wollwachsalkohole, wie Wollwachs. Emulgatoren sind geeignetenfalls lipophile Substanzen, wie Sorbitanfettsäureester (Spans), beispielsweise Sorbitanoleat und/oder Sorbitanisostearat. Additive für die wässrige Phase sind beispielsweise Befeuchtungsmittel, wie Polyalkohole, beispielsweise Glycerin, Propylenglycol, Sorbit und/oder Polyethylenglycol, und auch Konservierungsmittel und Parfüme.
- Fettsalben sind wasserfrei und enthalten als Grundlage speziell Kohlenwasserstoffe, beispielsweise Paraffin, Petroleumgel oder Paraffinöl, und auch natürliche oder teilsynthetische Fette, beispielsweise Kokosnussfettsäuretriglycerid oder vorzugsweise gehärtete Öle, beispielsweise hydriertes Ernussöl oder Rizinusöl, ferner Fettsäurepartialester von Glycerin, beispielsweise Glycerinmonostearat und Glycerindistearat, und weiter beispielsweise die Absorption von Wasser erhöhenden Fettalkohole, Emulgatoren und/oder Additive, wie dies im Zusammenhang mit den Salben erwähnt ist.
- Cremes sind Öl-in-Wasser-Emulsionen, die mehr als 50% Wasser enthalten. Als Ölbase werden primär verwendet Fettalkohole, beispielsweise Laurylalkohol, Cetylalkohol oder Stearylalkohol, Fettsäuren, beispielsweise Palmitinsäure oder Stearinsäure, flüssige bis feste Waches, beispielsweise Isopropylmyristat, Wollwachs oder Bienenwachs, und/oder Kohlenwasserstoffe, beispielsweise Petroleumgel (Petrolatum) oder Paraffinöl. Geeignete Emulgatoren sind Substanzen mit vorwiegend hydrophilen Anteilen, wie geeignete nicht ionische Emulgatoren, beispielsweise Fettsäureester von Polyalkoholen oder Ethylenoxidaddukten hiervon, wie Polyglycerinfettsäureester oder Polyoxyethylensorbitanfettsäureester (Tweens), und auch Polyoxyethylenfettalkoholether oder Fettsäureester, oder geeignete ionische Emulgatoren, wie Alkalimetallsalze von Fettalkoholsulfaten, beispielsweise Natriumlaurylsulfat, Natriumcetylsulfat oder Natriumstearylsulfat, die herkömmlich in Anwesenheit von Fettalkoholen verwendet werden, beispielsweise Cetylalkohol oder Stearylalkohol. Additive für die wässrige Phase sind unter anderem Mittel zur Reduktion einer Austrocknung der Cremes, beispielsweise Polyalkohole, wie Glycerin, Sorbit, Propylenglycol und/oder Polyethylenglycole, und auch Konservierungsmittel und Parfüme.
- Pasten sind Cremes oder Salben mit Sekretions-absorbierenden Pulverbestandteilen, wie Metalloxiden, beispielsweise mit Titanoxid oder Zinkoxid, oder auch Talk und/oder Aluminiumsilicate, durch die sich Feuchtigkeit oder vorhandene Sekretionen binden lassen.
- Schäume werden verabreicht aus Druckbehältnissen und sind flüssige Öl-in-Wasser-Emulsionen in Aerosolform, wobei als Treibgase Halogenkohlenwasserstoffe, wie Chlorfluorniederalkane, beispielsweise Dichlordifluormethan und Dichlortetrafluorethan, oder vorzugsweise nicht halogenierte gasförmige Kohlenwasserstoffe, Luft, N2O oder Kohlendioxid. Als Ölphase werden unter anderem solche Phasen verwendet, wie die oben im Fall von Salben oder Cremes verwendet werden und auch die oben in diesem Zusammenhang erwähnten Additive.
- Tinkturen und Lösungen haben gewöhnlich eine wässrige-ethanolische Grundlage, der unter anderem auch Polyalkohole, wie Glycerin, Glycole und/oder Polyethylenglycol als Befeuchtungsmittel zur Reduktion einer Verdampfung zugesetzt werden, und Fetthaltesubstanzen, wie Fettsäureester mit niedermolekularen Polyethylenglycolen, wie lipophile Substanzen, die im wässrigen Gemisch löslich sind, als ein Ersatz für die Fettsubstanzen, die aus der Haut durch das Ethanol und, falls vorhanden, durch Adjunkte und Additive extrahiert werden.
- Zur Erfindung gehört auch ein Verfahren oder eine Methode zur Behandlung der oben erwähnten pathologischen Zustände, besonders der Zustände, die responsiv sind für eine Inhibition von Protein-Kinasen. Die Verbindungen der Formel I können als solche oder in Form pharmazeutischer Zusammensetzungen hiervon prophylaktisch oder therapeutisch vorzugsweise in einer Menge, die gegen die erwähnten Krankheiten wirksam ist, an Warmblüter, beispielsweise einen behandlungsbedürftigen Menschen, verabreicht werden, wobei die Verbindungen speziell in Form pharmazeutischer Zusammensetzungen angewandt werden. Im Fall eines Körpergewichts von etwa 70 kg wird eine Tagesdosis von etwa 0,1 g bis etwa 5 g, vorzugsweise von etwa 0,5 g bis etwa 2 g, einer erfindungsgemäßen Verbindung verabreicht.
- Die folgenden Beispiele dienen zur weiteren Illustration der Erfindung.
- Sofern nichts anderes gesagt ist, ist das Verhältnis von Lösemitteln zueinander in Volumenteilen angegeben.
- Die in den Beispielen verwendeten Kurzformen und Abkürzungen haben die folgenden Bedeutungen:
- Elutionsmittel (Gradienten)
- HPLC Gradienten:
-
- Grad20 20% → 100% a) in b) während 20 min.
- Eluens a): Acetonitril + 0,05% TFA, Eluens b): Wasser + 0,05% TFA. Säule (250 × 4,6 mm), die mit dem Umkehrphasenmaterial C18-Nucleosil® bepackt ist (5 μm mittlere Teilchengröße, Silicagel, das mit Octadecylsilanen kovalent derivatisiert ist, Macherey & Nagel, Düren, Deutschland).
- Detektion durch UV Absorption bei 254 nm. Die Retentionszeiten (tRet) sind in min angegeben.
- Flussrate: 1 ml/min.
- Abkürzungen
-
-
- abs.
- absolut (wasserfrei)
- TLC-Rf
- Rf entsprechend einer Dünnschichtchromatographie
- DEPC
- Diethylpyrocarbonat
- DIPE
- Diisopropylether
- DMA
- Dimethylacetamid
- DMF
- Dimethylformamid
- DMPU
- 1,3-Dimethyl-3,4,5,6-tetrahydro-2-(1H)-pyrimidinon
- DMSO
- Dimethylsulfoxid
- EI-MS
- Massenspektroskopie durch Elektronenstoßionisation
- FAB-MS
- Massenspektroskopie durch Bombardment mit schnellen Atomen
- ges.
- gesättigt
- h
- Stunde(n)
- HPLC
- Hochdruckflüssigkeitschromatographie
- HV
- Hochvakuum
- min
- Minute(n)
- MS
- Massenspektroskopie
- RT
- Raumtemperatur
- RE
- Rotationsverdampfer
- Smp.
- Schmelzpunkt
- Salzlösung
- gesättigte Kochsalzlösung
- TFA
- Trifluoressigsäure
- THF
- Tetrahydrofuran
- TPTU
- O-(1,2-Dihydro-2-oxo-1-pyridyl)-N,N,N',N'-tetramethyluroniumtetrafluorborat
- In den NMR Spektraldaten verwendete Abkürzungen
- b
- breit
- d
- doppelt
- J
- Kopplungskonstante
- m
- Multiplett
- q
- Quartett
- s
- Singulett
- t
- Triplett
- Beispiel 1:
- 136,4 mg (0,50 mmol) 4-(3-Chloranilino)-6-formyl-7H-pyrrolo[2,3-d]pyrimidin und 67 μl N-Methylpiperazin in 5 ml Methanol, 15 ml DMPU und 63 μl Essigsäure werden bei 50°C in Gegenwart von 30 mg Raney-Nickel hydriert. Der Katalysator wird abfiltriert, das Filtrat eingedampft und der Rückstand in Ethylacetat und gesättigter NaHCO3 Lösung gelöst. Die sich abtrennende wässrige Phase wird zweimal mit Ethylacetat extrahiert. Die organischen Phasen werden mit gesättigter NaHCO3 Lösung und viermal mit Wasser und Kochsalzlösung gewaschen, über Na2SO4 getrocknet und eingedampft. Durch Säulenchromatographie (SiO2, CH2Cl2/Methanol = 7:2) und Rührung in Diethylether wird 4-(3-Chloranilino)-6-[(4-methylpiperazin-1-yl)-methyl]-7H-pyrrol[2,3-d]pyrimidin erhalten; HPLC: tRet(Grad20) = 7,4; TLC-Rf = 0,16 (CH2Cl2/Methanol = 7:3); FAB-MS: (M+H)+ = 357.
- Das Ausgangsmaterial wird wie folgt erhalten:
- Stufe 1.1:
- Bei 0 bis 5°C werden zu Beginn 56,0 g (0,43 mol) Ethyl-2-amidinoacetat [zur Herstellung siehe Liebigs Ann. Chem., 1561 (1981)] in 172 ml 1,3-Dimethyl-3,4,5,6-tetrahydro-2-(1H)-pyrimidinon einge führt. Nach tropfenweiser Zugabe von 56,0 ml (0,45 mol) von Ethylbrompyruvat während 30 min wird das Gemisch 3 h auf 60°C erwärmt. Die dunkelbraune Reaktionsmischung wird in 1 l Eiswasser gegossen, worauf sich eine Extraktion mit 1 l Ethylacetat und zweimal mit 0,5 l Ethylacetat jedes Mal anschließt. Die organischen Phasen werden dreimal mit 0,5 l Wasser und 0,5 l Kochsalzlösung gewaschen, getrocknet (Na2SO4) und eingedampft. Durch Säulenchromatographie (SiO2, Hexan/Ethylacetat [1:1]) und Umkristallisation aus Diethylether-Hexan wird 2-Amino-3,5-bis-(ethoxycarbonyl)-1H-pyrrol erhalten; Smp. 147 bis 149°C; MS: (M)+ = 226.
- Stufe 1.2:
- Unter Ausschluss von Luft wird ein Gemisch von 51,5 g (227 mmol) 2-Amino-3,5-bis-(ethoxycarbonyl)-1H-pyrrol, 455 mmol Formamid, 227 ml DMF und 113 ml Ameisensäure während 27 h bei 140°C gerührt. Die erhaltene gelbe Suspension wird auf 0 bis 5°C gekühlt. Durch anschließende Filtration und Waschung mit Isopropanol und Hexan wird 6-Ethoxycarbonyl-4-hydroxy-7H-pyrrolo[2,3-d]pyrimidin erhalten; 1H-NMR (DMSO-d6): 13-12 (2 HX), 7,99 und 7,11 (2s, 2H), 4,31 (q, J = 7, 2H), 1,32 (t, J = 7, 3H).
- Stufe 1.3:
- Unter einer N2 Atmosphäre werden 32,0 g (154 mmol) 6-Ethoxycarbonyl-4-hydroxy-7H-pyrrolo[2,3-d]pyrimidin in 308 ml (338 mmol) POCl3 bei RT suspendiert und unter Rührung auf 120°C erwärmt, wobei der Feststoff während dieser Zeit in Lösung geht. Nach Rührung bei 120°C während 3 h wird das überschüssige POCl3 abdestilliert (65°C Außentemperatur, 15 mbar). Sodann erfolgt eine Suspension des Rückstands in 50 ml eiskaltem Toluol, Filtration und Waschung mit Toluol, wodurch 4-Chlor-6-ethoxycarbonyl-7H-pyrrolo[2,3-d]pyrimidin erhalten wird; Smp. 219-221°C; 1H-NMR (DMSO-d6) 8,77 und 7,24 (2s, 2H), 4,39 (q, J = 7, 2H), 1,36 (t, J = 7, 3H). Weiteres Produkt kann aus dem verdampften Filtrat durch Rührung in Ethylacetat/Wasser erhalten werden.
- Stufe 1.4:
- Unter einer Argonatmosphäre werden 29,0 g (128 mmol) 4-Chlor-6-ethoxycarbonyl-7H-pyrrolo[2,3-d]pyrimidin und 18,0 ml (171 mmol) 3-Chloranilin in 430 ml n-Butanol während 3 h bei 100°C gerührt, wobei nach 1 h praktisch alles in Lösung gegangen ist und sich dann eine dicke Suspension bildet. Hierauf wird das Reaktionsgemisch mit 400 ml Isopropanol/Hexan (1:1) unter Abkühlung auf = 50°C versetzt, das Produkt abfiltriert und mit Isopropanol und Hexan gewaschen. Nach Rührung in Diethylether wird 4-(3-Chloranilino)-6-ethoxycarbonyl-7H-pyrrolo[2,3-d]pyrimidin erhalten; 1H-NMR (DMSO-d6) 13,0 und 10,53 (2 sb, 2HN), 8,48 (s, 1H), 8,13 (m, 1H), 7,78 (dm, J = 8, 1H), 7,76 (s, 1H), 7,45 (t, J = 8, 1H), 7,21 (dm, J = 8, 1H), 4,37 (q, J = 7, 2H), 1,37 (t, J = 7, 3H).
- Stufe 1.5:
- Unter einer N2 Atmosphäre werden 1,4 g (37 mmol) Lithiumaluminiumhydrid portionsweise zu 5,70 g (18 mmol) 4-(3-Chloranilino)-6-ethoxycarbonyl-7H-pyrrolo[2,3-d]pyrimidin gegeben. Nach einer Rührung bei 50°C während 2 h wird das Reaktionsgemisch tropfenweise mit 100 ml Wasser versetzt und dann durch Celite filtriert. Das Filtrat wird mit Wasser versetzt, und das Gemisch dreimal mit Ethylacetat extrahiert. Die organischen Phasen werden dreimal mit Wasser und Kochsalzlösung gewaschen, getrocknet (MgSO4) und eingedampft. Durch Umkristallisation aus Isopropanol wird 4-(3-Chloranilino)-6- hydroxymethyl-7H-pyrrolo[2,3-d]pyrimidin erhalten; HPLC: tRet(Grad20) = 8,2; TLC-Rf = 0,11 (CH2Cl2/Methanol [10:1]); MS: (M)+ = 274.
- Stufe 1.6:
- Unter Eiskühlung werden 1,9 g Mangandioxid (85%) zu einer Suspension von 715 mg (2,6 mmol) 4-(3-Chloranilino)-6-hydroxymethyl-7H-pyrrolo[2,3-d]pyrimidin in 170 ml Methylenchlorid gegeben, wobei das Gemisch 20 h bei RT gerührt wird. Hierauf wird das Reaktionsgemisch mit 20 ml 1,3-Dimethyl-3,4,5,6-tetrahydro-2-(1H)-pyrimidinon (DMPU) versetzt und 1 h gerührt, worauf es durch Hyflo filtriert wird. Der Filtrationsrückstand wird erneut gerührt in 50 ml Methylenchlorid/DMPU (1:1) und dann wiederum filtriert. Die beiden Filtrate werden vereinigt, eingedampft und in Ethylacetat/THF und Wasser aufgenommen. Die wässrigen Phasen werden zweimal mit Ethylacetat extrahiert, worauf die organischen Phasen viermal mit Wasser und Kochsalzlösung gewaschen, getrocknet (MgSO4) und bis auf ein Restvolumen von ≈ 20 ml verdampft werden. Durch anschließende Zugabe von Diethylether und Filtration wird 4-(3-Chloranilino)-6-formyl-7H-pyrrolo[2,3-d]pyrimidin erhalten; HPLC: tRet(Grad20) = 10,1; TLC-Rf = 0,24 (CH2Cl2/Methanol [10:1]).
- Beispiel 2:
- Ein Gemisch von 109 mg (0,40 mmol) 4-(3-Chloranilino)-6-formyl-7H-pyrrolo[2,3-d]pyrimidin (Stufe 1.6) und 70 μl (0,8 mmol) Morpholin in 6 ml Methanol, 2 ml DMPU und 50 μl Essigsäure wird 2 h auf 50°C erhitzt. Sodann wird das Gemisch mit 30 mg Raney-Nickel versetzt und bei 50°C hydriert. Der Katalysator wird abfiltriert, das Filtrat eingedampft und der Rückstand in Ethylacetat und gesättigter Na2CO3 Lösung gelöst. Die abgetrennte wässrige Phase wird zweimal mit Ethylacetat extrahiert, worauf die organischen Phasen mit ges. NaHCO3 Lösung gewaschen, zweimal mit Wasser und Kochsalzlösung gewaschen, getrocknet (MgSO4) und eingedampft wird. Der Rückstand wird in Methanol aufgenommen, worauf die Lösung mit Silicagel behandelt und getrocknet wird. Das Pulver wird auf eine Silicagelsäule gegeben und mit CH2Cl2/Methanol (10:1) eluiert. Durch Kristallisation aus Ethylacetat/Diethylether/Hexan wird 4-(3-Chloranilino)-6-[(morpholin-4-yl)-methyl]-7H-pyrrolo[2,3-d]pyrimidin erhalten; Smp. 244–246°C; HPLC: tRet(Grad20) = 7,4; TLC-Rf = 0,20 (CH2Cl2/Methanol 10:1); FAB-MS: (M+H)+ = 344.
- Beispiel 6:
- Ein Gemisch von 115 mg (1,66 mmol) Hydroxylammoniumchlorid und 139 mg (1,69 mmol) Natriumacetat in 2 ml Wasser wird zu 273 mg (1,00 mmol) 4-(3-Chloranilino)-6-formyl-7H-pyrrolo[2,3-d]pyrimidin (Stufe 1.6) in 3 ml Methanol gegeben. Die Suspension wird 3 h auf Siedetemperatur erhitzt, abgekühlt und filtriert, und der Rückstand wird mit Wasser/Isopropanol (1:1) gewaschen. Das Rohprodukt wird in etwa 80 ml heißem THF gelöst und mit Aktivkohle geklärt. Das Filtrat wird mit 10 ml Isopropanol versetzt und dann partial eingedampft. Die dabei gebildeten Kristalle werden abfiltriert und mit DIPE und Hexan gewaschen, worauf reines (E)-4-(3-Chloranilino)-7H-pyrrolo[2,3-d]pyrimidin-6-carbaldehydoxim erhalten wird; 1H-NMR (DMSO-d6): 12,16 und 11,35 (2s, 2HN), 9,60 (s, 1H), 8,35 (s, 1H), 8,19 (sb, 1H), 8,17 (s, 1H), 7,79 (db, J = 8, 1H), 7,35 (t, J = 8, 1H), 7,05 (db, J = 8, 1H), 7,03 (s, 1H); HPLC: tRet(Grad20) = 9,5; FAB-MS: (M+H)+ = 288.
- Nach Zugabe von Diethylether und Stehenlassen bei –20°C kristallisiert aus der eingedampften Mutterlauge (Z)-4-(3-Chloranilino)-7H-pyrrolo[2,3-d]pyrimidin-6-carbaldehydoxim als ein Gemisch mit ≈ 10% des (E) Isomers aus: 1H-NMR (DMSO-d6): 12,00 (s, 2HN), 9,71 (s, 1H), 8,39 (s, 1H), 8,24 (sb, 1H), 7,84 (db, J = 8, 1H), 7,67 (s, 1H), 7,52 (s, 1H), 7,35 (t, J = 8, 1H), 7,05 (db, J = 8, 1H); HPLC: tRet(Grad20) = 9,1; FAB-MS: (M+H)+ = 288.
- Beispiel 7:
- Ein Gemisch von 69,3 mg (0,83 mmol) O-Methylhydroxylaminhydrochlorid und 69,3 mg (0,845 mmol) Natriumacetat in 1 ml Wasser wird zu 163,3 mg (0,5 mmol) 4-(3-Chloranilino)-6-formyl-7H-pyrrolo[2,3-d]pyrimidin (Stufe 1.6) in 1 ml Methanol gegeben. Die Suspension wird 4 h auf Siedetemperatur erhitzt, abgekühlt und filtriert, worauf der Rückstand mit Wasser/Isopropanol und schließlich mit Diethylether gewaschen wird, wodurch 4-(3-Chloranilino)-7H-pyrrolo[2,3-d]pyrimidin-6-carbaldehyd-O-methyloxim erhalten wird; HPLC: tRet(Grad20) = 11,3; TLC-Rf = 0,31 (CH2Cl2/Methanol 10:1) FAB-MS: (M+H)+ = 302.
- Beispiel 8:
- Ein Gemisch von 168,5 mg (0,53 mmol) 4-(3-Chloranilino)-6-ethoxycarbonyl-7H-pyrrolo[2,3-d]pyrimidin (Stufe 1.4) in 3 ml Morpholin wird zuerst 5 Tage bei 50°C und dann 1 Tag bei 100°C gerührt. Das Reaktionsgemisch wird mit Silicagel versetzt und eingedampft, und der erhaltene Rückstand wird auf eine Silicagelsäule gegeben, die mit Ethanol/Methylenchlorid (1:15) eluiert wird. Durch Rührung des Reaktionsprodukts in Propanol wird 4-(3-Chloranilino)-6-(morpholin-4-ylcarbonyl)-7H-pyrrolo[2,3-d]pyrimidin erhalten; HPLC: tRet(Grad20) = 9,2; TLC-Rf = 0,56 (CH2Cl2/Methanol = 10:1); MS: (M)+ = 357.
- Beispiel 9:
- Es werden zuerst 98 mg (0,33 mmol) TPTU und dann 133 μl (1,2 mmol) N-Methylpiperazin in 1 ml DMF (→ Lösung) zu einer Suspension von 97,6 mg (0,30 mmol) 6-Carboxy-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin in 7 ml DMF gegeben. Nach 1 h erfolgt ein weiterer Zusatz von 30 mg TPTU, wobei das Gemisch über Nacht bei RT gerührt wird. Der nach Verdampfung des Reaktionsgemisches unter HV erhaltene Rückstand wird mit Ethanol/Methylenchlorid (1:2) und dann mit etwa 4 ml Ethanol/Wasser (3:1) behandelt. Das bei diesem Prozess auskristallisierte 4-(3-Chloranilino)-6-[(4-methylpiperazin-1-yl)carbonyl]-7H-pyrrolo[2,3-d]pyrimidin wird abfiltriert und mit Ethanol/Wasser (1:1) und dann mit Isopropanol/Hexan (1:1) gewaschen; Smp. 276–278°C; HPLC: tRet(Grad20) = 7,3; TLC-Rf = 0,21 (CH2Cl2/Ethanol = 2:1); MS: (M)+ = 370.
- Das Ausgangsmaterial wird wie folgt erhalten:
- Stufe 9.1:
- Eine Lösung von 25 mg (0,6 mmol) LiOH·H2O in 0,4 ml H2O wird tropfenweise zu einer Suspension von 95 mg (0,30 mmol) 4-(3-Chloranilino)-6-ethoxycarbonyl-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 1.4) in 0,7 ml Methanol gegeben, wobei das Gemisch 4,5 h auf Siedetemperatur erhitzt wird. Sodann wird das Gemisch in einem Eisbad abgekühlt und mit 0,6 ml 1 normaler HCl Lösung angesäuert. Durch an schließende Filtration und Waschung mit Wasser wird 6-Carboxy-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin erhalten; HPLC: tRet(Grad20) = 8,7; FAB-MS: (M+H)+ = 289.
- Beispiel 10:
- Ein Gemisch von 144 mg (0,50 mmol) 6-Aminocarbonyl-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin und 202 mg (0,5 mmol) Lawesson-Reagenz in 5 ml THF wird 17 h auf Siedetemperatur erhitzt und dann eingedampft. Durch Säulenchromatographie (SiO2, CH2Cl2/Ethanol = 15:1) des Rückstands und Rührung in Diethylether wird 4-(3-chloranilino)-6-(thiocarbamoyl)-7H-pyrrolo[2,3-d]pyrimidin erhalten; HPLC: tRet(Grad20) = 9,8; TLC-Rf = 0,44 (CH2Cl2/Methanol = 10:1); FAB-MS: (M+H)+ = 304; IR: (KBr) inter alia 1614s, 1566s, 1506m, 1474s, 1380m, 1294m, 1134m.
- Das Ausgangsmaterial wird wie folgt erhalten:
- Stufe 10.1:
- Ein Gemisch von 90 mg (0,285 mmol) 4-(3-Chloranilino)-6-ethoxycarbonyl-7H-pyrrolo[2,3-d]pyrimidin (Stufe 1.4) in 30 ml Methanol und = 5 g Ammoniak wird 48 h in einem Autoklav auf 120°C erhitzt. Das Reaktionsgemisch wird mit Silicagel behandelt, eingedampft und auf eine Silicagelsäule als ein Pulver gegeben und dann mit Methylenchlorid/Methanol/THF (210:35:10) eluiert. Durch Filtration mit Methanol durch eine Aluminiumoxidsäule (basisch) und Rührung in Ethylacetat wird 6-Aminocarbonyl-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin erhalten; HPLC: tRet(Grad20) = 8,1; TLC-Rf = 0,18 (CH2Cl2/Methanol [10:1]); hohe Auflösung MS: (M+H)+ = 288,0669 (berechnet 288,0652).
- Das Ausgangsmaterial kann alternativ und vorteilhaft auch wie folgt erhalten werden:
- Alternative Stufe 10.1:
- Ein Gemisch von 2,165 g (7,5 mmol) 6-Carboxy-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin in 60 ml THF und 10 ml DMPU wird 30 min unter Rückflusstemperatur gehalten und dann auf 0°C abgekühlt, wodurch eine feine Suspension erhalten wird. Sodann wird diese Suspension mit 824 μl (7,5 mmol) N-Methylmorpholin und hierauf mit 981 μl (7,5 mmol) Isobutylchlorformiat in 10 ml THF versetzt, worauf bei 0°C nach 1 h tropfenweise zuerst 824 μl (7,5 mmol) N-Methylmorpholin und dann 981 μl (7,5 mmol) Isobutylchlorformiat zugegeben werden. Das Gemisch wird 1 h gerührt und dann tropfenweise mit 70 ml einer gesättigten Lösung von Ammoniak in Dioxan versetzt. Nach 3 h wird das Gemisch unter Vakuum eingeengt. Der Rückstand wird in Wasser gegossen, und der Niederschlag wird abfiltriert, und dann mit Wasser und siedendem Isopropanol gewaschen, wodurch 6-Aminocarbonyl-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin erhalten wird. Aus dem Isopropanolfiltrat wird weiteres Produkt erhalten.
- Beispiel 11
- Ein Gemisch von 57,5 mg (0,19 mmol) 4-(3-Chloranilino)-6-(thiocarbamoyl)-7H-pyrrolo[2,3-d]pyrimidin (siehe Beispiel 10) in 3 ml Methanol und 57,3 mg (0,25 mmol) 4-Methoxyphenacylbromid wird 17 h auf Siedetemperatur erhitzt. Durch Abkühlung der fahlgelben Suspension, Filtration und anschließende Waschung mit Isopropanol/Diethylether wird 4-(3-Chloranilino)-6-[4-(4-methoxyphenyl)-thiazol-2- yl]-7H-pyrrolo[2,3-d]pyrimidin erhalten; HPLC: tRet(Grad20) = 15,4; TLC-Rf = 0,33 (CH2Cl2/Methanol = 10:1); FAB-MS: (M+H)+ = 434.
- Beispiel 12:
- Ein Gemisch von 57,5 mg (0,19 mmol) 4-(3-Chloranilino)-6-(thiocarbamoyl)-7H-pyrrolo[2,3-d]pyrimidin (siehe Beispiel 10) und 34 ml 1-Brom-2-butanon (Aldrich, Milwaukee/USA) in 3 ml Dioxan wird 2 h auf Siedetemperatur erhitzt. Sodann wird die abgekühlte Suspension filtriert und der Rückstand mit Isopropanol/Diethylether gewaschen, wodurch 4-(3-Chloranilino)-6-(4-ethylthiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin erhalten wird; HPLC: tRet(Grad20) = 13,6; FAB-MS: (M+H)+ = 356.
- Beispiel 13:
- 4-(3-Chloranlino)-6-(4,5-dimethylthiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin wird nach einem in diesem Text beschriebenen Verfahren erhalten.
- Beispiel 14:
- Ein Gemisch von 19 mg (0,45 mmol) Lithiumchlorid und 19,5 mg (0,30 mmol) Natriumazid wird zu einer Lösung von 80,9 mg (0,30 mmol) 4-(3-Chloranilino)-6-cyano-7H-pyrrolo[2,3-d]pyrimidin in 1 ml Methoxyethanol gegeben, und das Gemisch wird 6,5 h auf Siedetemperatur erhitzt. Nach Abkühlung wird das Reaktionsgemisch in Wasser/HCl (konz.) 10:1 gegeben und 30 min gerührt, worauf der Feststoff abfiltriert und mit Wasser gewaschen wird. Durch Auflösung der Kristalle in THF/Isopropanol, Teilverdampfung bis zur Kristallisation, Filtration und Waschung mit Diethylether wird 4-(3-Chloranilino)-6-(tetrazol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin erhalten; HPLC: tRet(Grad20) = 9,5; TLC-Rf = 0,45 (CH2Cl2/Methanol = 7:1); FAB-MS: (M+H)+ = 313.
- Das Ausgangsmaterial wird wie folgt erhalten:
- Stufe 14.1:
- Ein Gemisch von 1,048 g (3,6 mmol) 6-Aminocarbonyl-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 10.1) und 0,7 ml N,N-Dimethylacetamid wird mit 13 ml Phosphoroxychlorid versetzt. Nach Rührung bei RT während 1 h und bei 100°C während 4 h wird das Reaktionsgemisch in eine eiskalte gesättigte Lösung von NaHCO3 gegeben. Anschließend erfolgt eine Extraktion mit Ethylacetat (dreimal), Waschung der organischen Schichten mit einer gesättigten NaHCO3 Lösung sowie mit Wasser und einer gesättigten Natriumchloridlösung, Trocknung (Na2SO4) und Einengung führt zu einem Feststoff. Durch Säulenchromatographie (SiO2, Ethylacetat) und Rührung des Rohprodukts in Diethylether und Hexan wird 4-(3-Chloranilino)-6-cyano-7H-pyrrolo[2,3-d]pyrimidin erhalten; Smp. 284–287°C; TLC-Rf = 0,71 (CH2Cl2/Methanol [10:1]); HPLC: tRet(Grad20) = 11,8.
- Beispiel 15:
- Man gibt 50,4 mg (0,60 mmol) in NaHCO3 und bei 0 bis 5°C 600 μl (0,60 mmol) Methyliodid (1 M in Dioxan) zu 187,6 mg (0,60 mmol) 4-(3-Chloranilino)-6-(tetrazol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin (siehe Beispiel 14) in 12 ml DMF. Nach einer Rührung bei RT während 18 h wird das Gemisch mit Ethylacetat, etwas THF und Wasser verdünnt und die wässrige Phase abgetrennt, worauf sich eine erneute Extraktion mit Ethylacetat und etwas THF anschließt. Die organischen Phasen werden zweimal mit Wasser und Kochsalzlösung gewaschen, mit MgSO4 getrocknet und eingedampft. Durch Rührung des Rückstands in THF/Ethanol wird ein 2:1 Gemisch von 4-(3-Chloranilino)-6-(2-methyltetrazol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin und 4-(3-Chloranilino)-6-(1-methyltetrazol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin erhalten; HPLC: tRet(Grad20) = 10,5 (1 Teil) und 10,7 (2 Teile), 1H-NMR (DMSO-d6) inter alia 4,45 (s, 2-H3C-Tetrazol), 4,32 (s, 1-H3C-Tetrazol); FAB-MS: (M+H)+ = 327.
- Beispiel 16:
- Ein Gemisch von 0,1 g (0,304 mmol) (R)-6-(4-Aminophenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin und 0,026 ml (0,33 mmol) Methansulfochlorid in 1 ml absolutem Dimethylacetamid wird bei 0°C während 4 h so lange gerührt, bis sich im TLC kein Ausgangsmaterial mehr zeigt. Das Reaktionsgemisch wird dann in 10 ml Eiswasser gegossen. Hierauf erfolgt eine Extraktion mit Ethylacetat und mit 20 ml einer wässrigen NaHCO3 Lösung. Die organische Phase wird mit Wasser gewaschen, getrocknet und eingeengt, wodurch (R)-6-(4-Methylsulfonylaminophenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin auskristallisiert. Dieses Produkt wird mit Hexan gewaschen; Smp. 253–256°C; FAB-MS: (M+H)+ = 408.
- Das Ausgangsmaterial wird wie folgt erhalten:
- Stufe 16.1:
- In Analogie zur Stufe 19.4 werden durch Kochen von 6,0 g (220 mmol) 4-Chlor-6-(4-nitrophenyl)-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 19.3) mit 6,04 g (R)-(+)-1-Phenylethylamin in 120 ml n-Butanol (R)-6-(4-Nitrophenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin in Form rostbrauner Kristalle mit einem Smp. von > 250°C erhalten.
- Stufe 16.2:
- In Analogie zur Stufe 19.5 wird durch Reduktion von (R)-6-(4-Nitrophenyl-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin mit Raney-Nickel in THF/Methanol (R)-6-(4-Aminophenyl-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin erhalten; Smp. 234–235°C; MS: M+ = 329.
- Beispiel 17:
- In Analogie zum Beispiel 16 werden ausgehend von (R)-6-(4-Aminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 16.2) und dem geeigneten Alkylsulfochlorid die folgenden Verbindungen erhalten:
- a) (R)-6-(4-Ethylsulfonylaminophenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]-pyrimidin; Smp. 260–261°C; FAB-MS: (M+H)+ = 422, und
- b) (R)-6-(4-Isopropylsulfonylaminophenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]-pyrimidin; Smp. 262–263°C; FAB-MS: (M+H)+ = 436.
- Beispiel 18:
- In Analogie zum Beispiel 16 werden ausgehend von (R)-6-(3-Aminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin und dem geeigneten Alkylsulfochlorid die folgenden Verbindungen hergestellt:
- a) (R)-6-(3-Methylsulfonylaminophenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]-pyrimidin; Smp. 139–148°C (amorph); MS: (M+) = 407,
- b) (R)-6-(3-Ethylsulfonylaminophenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]-pyrimidin; Smp. 235–236°C; FAB-MS: (M+H)+ = 422, und
- c) (R)-6-(3-Isopropylsulfonylaminophenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]-pyrimidin; Smp. 257–258°C; FAB-MS: (M+H)+ = 436.
- Das Ausgangsmaterial wird wie folgt erhalten:
- Stufe 18.1:
- In Analogie zu dem in der Stufe 19.4 beschriebenen Verfahren werden durch Kochen von 20,0 g (700 mmol) 4-Chlor-6-(3-nitrophenyl)-7H-pyrrolo[2,3-d]pyrimidin (hergestellt in Analogie zu den Stufen 19.1 bis 19.3) mit 23,1 ml (168 mmol) (R)-Phenylethylamin in 23,1 ml n-Butanol (R)-6-(3-Nitrophenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin in Form rostbrauner Kristalle mit einem Smp. von > 250°C hergestellt; MS: M+ = 359.
- Stufe 18.2:
- In Analogie zur Stufe 19.5 wird durch Reduktion von (R)-6-(3-Nitrophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin mit Raney-Nickel in THF/Methanol (R)-6-(3-Aminophenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin hergestellt; Smp. 129–190°C (amorph); MS: M+ = 329.
- Beispiel 19:
- In Analogie zum Beispiel 16 werden aus 6-(4-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin und dem entsprechenden Alkylsulfochlorid die folgenden Verbindungen hergestellt:
- a) 6-(4-Methylsulfonylaminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin, Smp. > 300°C; FAB-MS: (M+H)+ = 414,
- b) 4-(3-Chloranilino)-6-(4-ethylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C; FAB-MS: (M+H)+ = 428,
- c) 4-(3-Chloranilino)-6-(4-isopropylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C; FAB-MS: (M+H)+ = 442 und
- d) 4-(3-Chloranilino)-6-(4-phenylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C; FAB-MS: (M+H)+ = 475.
- Das Ausgangsmaterial wird wie folgt erhalten:
- Stufe 19.1:
- In einen trockenen Dreihalskolben werden 75 ml absolutes Ethanol und 6,5 g (390 mmol) Ethyl-2-amidinoacetathydrochlorid [zur Herstellung siehe Liebigs Ann. Chem., 1895 (1977)], initial unter Argon eingeführt, wobei das Gemisch auf 0 bis 5°C gekühlt wird und mit 2,65 g (390 mmol) Natriumethoxid behandelt wird. Hierauf werden 5 g (195 mmol) 2-Brom-1-(4-nitrophenyl)-ethan-1-on zugesetzt, wobei man das Gemisch auf RT kommen lässt und weitere 48 h rührt. Sodann wird das Reaktionsgemisch zwischen Wasser und Ethylacetat verteilt. Die Ethylacetatphase wird dreimal mit Wasser und einmal mit ges. NaCl Lösung gewaschen, getrocknet und filtriert, und das Filtrat wird eingedampft. Der rotbraune Rückstand wird in Hexan suspendiert, wodurch 2-Amino-3-ethoxycarbonyl-5-(4-nitrophenyl)-pyrrol als ein Rohpro dukt (Reinheit 93%) ausfällt, das ohne weitere Reinigung für die nächste Stufe verwendet wird; MS: (M)+ = 275. Stufe 19.2:
- Ein Gemisch von 2,5 g (97 mmol) 2-Amino-3-ethoxycarbonyl-5-(4-nitrophenyl)-pyrrol, 19,4 ml Formamid, 9,7 ml DMF und 3,1 ml Ameisensäure wird bei 150°C zusammen für 22 h gerührt. Sodann wird das warme Reaktionsgemisch mit 1 ml Isopropanol versetzt. Das Reaktionsgemisch wird gekühlt, und das dabei ausgefallene Produkt wird abfiltriert. Dieses Produkt wird dann der Reihe nach dreimal mit 10 ml Ethanol jedes Mal, zweimal mit 10 ml Isopropanol jedes Mal und zweimal mit 10 ml Hexan jeweils gewaschen. Hierdurch wird 4-Hydroxy-6-(4-nitrophenyl)-7H-pyrrolo[2,3-d]pyrimidin in Form rostbrauner Kristalle erhalten, die für die nächste Stufe verwendet werden; MS: (M)+ = 256.
- Stufe 19.3:
- Durch Erhitzung von 4-Hydroxy-6-(4-nitrophenyl)-7H-pyrrolo[2,3-d]pyrimidin mit POCl3 wird 4-Chlor-6-(4-nitrophenyl)-7H-pyrrolo[2,3-d]pyrimidin (Reinheit 93%) hergestellt; Smp. > 280°C; FAB-MS: (M+H)+ = 275.
- Stufe 19.4:
- Durch Kochen von 0,25 g (0,91 mmol) 4-Chlor-6-(4-nitrophenyl)-7H-pyrrolo[2,3-d]pyrimidin mit 0,19 ml 3-Chloranilin in 5 ml n-Butanol wird 4-(3-Chloranilin)-6-(4-nitrophenyl)-7H-pyrrolo[2,3-d]pyrimidin in Form rostbrauner Kristalle erhalten; Smp. > 250°C; 1H-NMR (360 MHz, DMSO-d6): 12,95 (s, Pyrrol-NH), 10,3 (s, Anilin-NH), 8,45 (s, Pyrimidin-H), 8,24 (s, aromatisches H), 7,18-8,4 (7 aromatisches H + Pyrrol-5H); MS: (M)+ = 365.
- Stufe 19.5:
- Durch Hydrierung von 150 mg (0,41 mmol) 4-(3-Chloranilino)-6-(4-nitrophenyl)-7H-pyrrolo[2,3-d]pyrimidin mit 50 mg Raney-Nickel in 20 ml THF/Methanol bei RT und normalem Druck während 5 h fällt das gewünschte Produkt im Verlauf dieser Reaktion aus. Der Katalysator wird abfiltriert und der Filterrückstand mit warmer THF gewaschen. Das Filtrat wird zur Trockne eingedampft. Das Rohprodukt wird durch mehrmalige Digestion in Methanol und Ausfällung aus THF/Hexan gereinigt, wodurch 6-(4-Aminophenyl)-4-(3-chloranilin)-7H-pyrrolo[2,3-d]pyrimidin in Form fahlbeiger Kristalle erhalten wird; Smp. > 290°C; 1H-NMR (360 MHz, DSMO-d6); 12,05 (s, Pyrrol-NH), 9,38 (s, Anilin-NH), 8,31 (s, Pyrimidin-H), 8,24 (s, aromatisches H), 7,80 (d, aromatisches H), 7,53 (d, 2 aromatische H), 7,35 (t, aromatisches H), 7,05 (d, aromatisches H), 6,90 (s, Pyrrol-5H), 6,64 (d, 2 aromatische H), 5,35 (s, NH2); MS: (M)+ = 335.
- Beispiel 20:
- In Analogie zum Beispiel 16 werden ausgehend von 6-(3-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin und dem entsprechenden Alkylsulfochlorid die folgenden Verbindungen hergestellt:
- a) 4-(3-Chloranilino)-6-(3-methylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C; FAB-MS: (M+H)+ = 414,
- b) 4-(3-Chloranilino)-6-(3-ethylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. 228–230°C; FAB-MS: (M+H)+ = 428, und
- c) 4-(3-Chloranilino)-6-(3-isopropylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. 233°C; FAB-MS: (M+H)+ = 442.
- Das Ausgangsmaterial wird wie folgt erhalten:
- Stufe 20.1:
- In Analogie zur Stufe 19.4 wird durch Kochen von 2,0 g (7,28 mmol) 4-Chlor-6-(3-nitrophenyl)-7H-pyrrolo[2,3-d]pyrimidin mit 4,2 ml (40 mmol) 3-Chloranilin in 150 ml n-Butanol 6-(3-Nitrophenyl-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin in Form gelblicher Kristalle erhalten; Smp. > 250°C; MS: M+ = 365.
- Stufe 20.2:
- In Analogie zur Stufe 19.5 wird durch Reduktion von 6-(3-Nitrophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin mit Raney-Nickel in THF/Methanol 6-(3-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin erhalten; Smp. 293–295°C; MS: M+ = 336.
- Beispiel 21:
- Ein Gemisch von 0,2 g (0,56 mmol) 6-(4-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 19.5), 0,097 ml (0,67 mmol) N,N-Dimethylformamiddimethylacetal und 0,112 ml (0,73 mmol) Triethylamin wird bei RT 24 h in 10 ml THF so lange gerührt, bis das gesamte Ausgangsmaterial im TLC verschwunden ist. Sodann wird die Reaktionslösung unter Vakuum zur Trockne konzentriert und der Rückstand auf einer Silicagelsäule chromatographiert. Durch Kristallisation aus THF/Hexan oder Ethylacetat/Hexan wird 4-(3-Chloranilino)-6-(4-[dimethylaminomethylenamino]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin erhalten; Smp. > 300°C; MS: (M)+ = 390.
- Beispiel 22:
- In Analogie zum Beispiel 21 werden ausgehend von 6-(4-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 19.5) und dem entsprechenden N,N-Dialkylformamiddimethylacetal oder Heterocyclylaldehyddimethylacetal die folgenden Verbindungen hergestellt:
- a) 4-(3-Chloranilino)-6-(4-[diethylaminomethylenamino]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 310°C; FAB-MS: (M+H)+ = 419,
- b) 4-(3-Chloranilino)-6-(4-[piperidinomethylenaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C; MS: (M)+ = 430,
- c) 4-(3-Chloranilino)-6-(4-[morpholinomethylenamino]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 310°C; MS: (M)+ = 432, und
- d) 4-(3-Chloranilino)-6-{4-[(4-methylpiperazino)-methylenamino]-phenyl}-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 310°C; FAB-MS: (M+H)+ = 446.
- Beispiel 23:
- In Analogie zum Beispiel 21 werden ausgehend von 6-(3-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 20.2) und dem entsprechenden N,N-Dialkylformamiddimethylacetal oder Morpholinoaldehyddimethylacetal die folgenden Verbindungen hergestellt:
- a) 4-(3-Chloranilino)-6-(3-[dimethylaminomethylenamino]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. 266–268°C; MS: (M)+ = 390,
- b) 4-(3-Chloranilino)-6-(3-[diethylaminomethylenamino]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. 171–172°C; FAB-MS: (M+H)+ = 419, und
- c) 4-(3-Chloranilino)-6-(3-[morpholinomethylenamino]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. 284–286°C; FAB-MS: (M+H)+ = 433.
- Beispiel 24:
- In Analogie zum Beispiel 21 wird ausgehend von (R)-6-(4-Aminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 16.2) und N,N-Dimethylformamiddimethylacetal die folgende Verbindung hergestellt:
(R)-6-(4-[Dimethylaminomethylenamino]-phenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin;
Smp. > 300°C; MS: (M+H)+ = 385. - Beispiel 25:
- In Analogie zum Beispiel 21 wird ausgehend von (R)-6-(3-Aminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 18.2) und N,N-Dimethylformamiddimethylacetal die folgende Verbindung hergestellt:
(R)-6-(3-[Dimethylaminomethylenamino]-phenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin;
Smp. 267–269°C; FAB-MS: (M+H)+ = 385. - Beispiel 26:
- 738 mg (2,02 mmol) 6-(4-Carbonylphenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin werden bei RT während 3 h mit 3 Äquivalenten Morpholin und 3,5 Äquivalenten DEPC (Aldrich) in DMF gerührt. Die Reaktionslösung wird auf Wasser gegossen, worauf der Feststoff abfiltriert und mit Wasser und Methanol gewaschen wird. Das erhaltene Produkt wird über Silicagel chromatographiert und mit Chloroform/Methanol/Essigsäure/Wasser (850:130:15:5, Vol./Vol.) eluiert. Das so erhaltene Produkt wird in Diethylether digestiert, abfiltriert und getrocknet, wodurch 4-(3-Chloranilino)-6-(4-[morpholin-4-ylcarbonyl]phenyl)-7H-pyrrolo[2,3-d]pyrimidin in Form beiger Kristalle erhalten wird. Smp. > 250°C; FAB-MS: (M+H)+ = 434.
- Beispiel 27:
- In Analogie zum Beispiel 26 wird ausgehend von 3-(Chloranilino)-6-(4-carbonylphenyl)-7H-pyrrolo[2,3-d]pyrimidin und N-Methylpiperazin 4-(3-Chloranilino)-6-(4-[4-methylpiperazin-1-yl]-carbonylphenyl)-7H-pyrrolo[2,3-d]pyrimidin erhalten; Smp. 250°C; FAB-MS: (M+H)+ = 447.
- Beispiel 28:
- 300 mg (0,89 mmol) 6-(4-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 19.5) werden in 20 ml absolutem THF und 2 ml (3,56 mmol) DMA gelöst, worauf diese Lösung mit 0,29 ml Ethylisocyanat (Fluka) unter Ausschluss von Feuchtigkeit und so langes Erhitzen auf Rückflusstempe ratur behandelt wird, bis im TLC (24 h) kein Ausgangsmaterial mehr vorhanden ist. Das Reaktionsgemisch wird unter Vakuum zur Trockne eingedampft und das Rohprodukt über Silicagel chromatographiert, wobei die Elution unter Verwendung von Ethylacetat/Methanol erfolgt. Die das gewünschte Produkt enthaltenden Fraktionen werden vereinigt und zur Trockne eingedampft. Der Rückstand wird in wenig THF gelöst. Sodann wird die Zielverbindung durch Zugabe von n-Hexan ausgefällt/kristallisiert, wodurch Kristalle von 4-(3-Chloranilino)-6-(4-[N3-ethylureido]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin erhalten werden; Smp. > 290°C; FAB-MS: (M+H)+ = 407.
- Beispiel 29:
- In Analogie zum Beispiel 28 werden ausgehend von 6-(3-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 20.2) und dem entsprechenden Alkylisocyanat oder Phenylisocyanat die folgenden Verbindungen hergestellt:
- a) 4-(3-Chloranilino)-6-(3-[N3-ethylureido]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C; FAB-MS: (M+H)+ = 407, und
- b) 4-(3-Chloranilino)-6-(3-[N3-phenylureido]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C; FAB-MS: (M+H)+ = 455.
- Beispiel 30:
- In Analogie zum Beispiel 28 wird ausgehend von (R)-6-(4-Aminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 16...) und Ethylisocyanat (R)-6-(4-[N3-Ethylureido]phenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin hergestellt; Smp. 238–240°C; FAB-MS: (M+H)+ = 401.
- Beispiel 31:
- In Analogie zum Beispiel 28 wird ausgehend von (R)-6-(3-Aminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 18.2) und Ethylisocyanat (R)-6-(3-[N3-Ethylureido]-phenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrmidin hergestellt; Smp. 177–178°C; FAB-MS: (M+H)+ = 401.
- Beispiel 32:
- In Analogie zum im Beispiel 28 beschriebenen Verfahren werden 300 mg (0,89 mmol) 6-(4-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 19.5) in 20 ml absolutem THF und 2 ml DMA mit 0,27 ml (3,56 mmol) Methylisothiocyanat (EGA) umgesetzt, wodurch nach Umkristallisation aus THF-n-Hexan 4-(3-Chloranilino)-6-(4-[N3-methylthioureido]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin in Form farbloser Kristalle erhalten wird; Smp. 275–276°C; FAB-MS: (M+H)+ = 409.
- Beispiel 33:
- In Analogie zum Beispiel 32 wird ausgehend von 6-(3-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin und Methylisothiocyanat 4-(3-Chloranilino)-6-(3-[N3-methylthioureido]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin hergestellt; Smp. 198–200°C; FAB-MS: (M+H)+ = 409.
- Beispiel 34:
- In Analogie zum Beispiel 32 wird ausgehend von (R)-6-(4-Aminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 16.2) und Methylisothiocyanat (R)-6-(4-[N3-Methylthioureido]-phenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin hergestellt; Smp. 225–228°C; FAB-MS: (M+H)+ = 403.
- Beispiel 35:
- In Analogie zum Beispiel 32 wird ausgehend von (R)-6-(3-Aminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 18.2) und Methylisothiocyanat (R)-6-(3-[N3-Methylthioureido]-phenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin hergestellt; Smp. 188–190°C; FAB-MS: (M+H)+ = 403.
- Beispiel 36:
- 300 mg (0,89 mmol) 6-(4-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 19.5) werden in 2,7 ml absolutem Dioxan und 0,13 ml 2,6-Lutidin mit einer Lösung von 0,10 ml (0,98 mmol) Methylchlorformiat in 1,8 ml absolutem Dioxan unter Ausschluss von Feuchtigkeit so lange bei RT gerührt, bis im TLC kein Ausgangsmaterial mehr zu sehen ist. Sodann wird das Reaktionsgemisch zu 50 ml Wasser gegeben. Anschließend erfolgt eine Extraktion mit 200 ml Ethylacetat und 10 ml 5%iger wässriger Natrium hydrogencarbonatlösung. Die vereinigten Ethylacetatphasen werden dreimal mit Wasser gewaschen, getrocknet und unter Vakuum eingedampft. Der Rückstand wird auf Silicagel chromatographiert unter Elution mit Methylenchlorid und zunehmend höher werdenden Mengen Methanol. Sodann wird die Zielverbindung 4-(3-Chloranilino)-6-(4-methoxycarbonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin aus Methanol oder Aceton in Form leicht gelber Kristalle kristallisiert. Smp. > 300°C; FAB-MS: (M+H)+ = 394.
- Beispiel 37:
- In Analogie zum Beispiel 36 werden ausgehend von 6-(4-Aminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin (siehe Stufe 19.5) und dem entsprechenden Alkylchlorformiat die folgenden Verbindungen hergestellt:
- a) 4-(3-Chloranilino)-6-(4-ethoxycarbonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C; FAB-MS: (M+H)+ = 408,
- b) 4-(3-Chloranilino)-6-(4-isopropyloxycarbonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C; FAB-MS: (M+H)+ = 422, und
- c) 4-(3-Chloranilino)-6-{4-[(2-methylpropyloxy)-carbonylamino]-phenyl}-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 282–284°C; FAB-MS: (M+H)+ = 436.
- Beispiel 38:
- 3,53 g (9,0 mmol) 4-(3-Chloranilino)-6-(4-ethoxycarbonylphenyl)-7H-pyrrolo[2,3-d]pyrimidin werden in 150 ml THF suspendiert. Hierauf werden bei 54°C während 5 h insgesamt 0,856 g (21,6 mmol) Lithiumaluminiumhydrid zugeführt. Anschließend erfolgt eine Aufarbeitung durch eine aufeinanderfolgen de Zugabe von 1 ml Wasser, 2 ml 1 N Natriumhydroxidlösung und 1 ml Wasser und anschließend 10 g Natriumsulfat. Der Feststoff wird abfiltriert und das Filtrat eingeengt. Der erhaltene Rückstand wird mit Methanol und Diethylether gewaschen und dann getrocknet, wodurch 4-(3-Chloranilino)-6-(4-hydroxymethylphenyl)-7H-pyrrolo[2,3-d]pyrimidin in Form farbloser Kristalle erhalten wird; Smp. > 250°C; FAB-MS: (M+H)+ = 351.
- Beispiel 39:
- In Analogie zu dem in diesem Text beschriebenen Verfahren werden die folgenden Verbindungen erhalten:
- a) (R)-6-(3-Benzylaminophenyl)-4-[(1-phenylethyl)-amino]-7H-pyrrolo[2,3-d]pyrimidin; Smp. 253–255°C,
- b) 6-(4-Benzyloxy-3-methoxyphenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin; Smp. 237–238°C,
- c) 6-(4-Benzyloxy-3-methoxyphenyl)-4-(3-methylanilino)-7H-pyrrolo[2,3-d]pyrimidin; Smp. 231–234°C,
- d) 6-(4-Hydroxy-3-methoxyphenyl)-4-(3-methylanilino)-7H-pyrrolo[2,3-d]pyrimidin; Smp. 234–236°C,
- e) 4-Anilino-6-(4-hydroxy-3-methoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidinhydrochlorid; Smp. 242–246°C,
- f) 4-(3-Chloranilino)-6-(4-hydroxy-3-methoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidinhydrochlorid; Smp. > 250°C,
- g) 4-(3-Chloranilino)-6-(4-[fur-2-ylcarbonylamino]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C,
- h) 4-(3-Chloranilino)-6-(4-[thien-2-ylcarbonylamino]-phenyl)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C,
- i) 6-(4-Benzylaminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin; Smp. > 300°C,
- k) 4-(3-Chloranilino)-6-{4-[(1-dimethylamino-1-isopropylmethylen)-amino]-phenyl}-7H-pyrrolo[2,3-d]pyrimidin; Smp. 255–257°C, und
- l) 4-(3-Chloranilino)-6-(thiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin.
- Beispiel 40: Trocken gefüllte Kapseln
- 5000 Kapseln, die als Wirkstoff jeweils 0,25 g einer der Verbindungen der Formel I enthalten, wie sie in den vorhergehenden Beispielen beschrieben sind, werden wie folgt hergestellt:
Zusammensetzung Menge Wirkstoff 1250 g Talk 180 g Weizenstärke 120 g Magnesiumstearat 80 g Lactose 20 g - Herstellungsverfahren: Die erwähnten Substanzen werden pulverisiert und durch ein Sieb mit einer Siebgröße von 0,6 mm gedrückt. Jeweils Anteile von 0,33 g des Gemisches werden unter Verwendung einer Kapselfüllmaschine in Gelatinekapseln abgefüllt.
- Beispiel 41: Weichkapseln
- 5000 Weichgelatinekapseln, die als Wirkstoff jeweils 0,05 g einer der Verbindungen der Formel I enthalten, wie sie in den vorhergehenden Beispielen beschrieben sind, werden wie folgt hergestellt:
Zusammensetzung Menge Wirkstoff 250 g Lauroglykol 2 l - Herstellungsverfahren: Der pulverisierte Wirkstoff wird in Lauroglykol® (Propylenglycollaurat, Gattefossé S. A., Saint Priest, Frankreich) suspendiert und in einem Nasspulverisator auf eine Teilchengröße von etwa 1 bis 3 μm vermahlen. Anschließend werden Anteile von 0,419 g des Gemisches mit einer Kapselfüllmaschine in Weichgelatinekapseln abgefüllt.
- Beispiel 42: Weichkapseln
- 5000 Weichgelatinekapseln, die als Wirkstoff jeweils 0,05 g einer der Verbindungen der Formel I enthalten, wie sie in den vorhergehenden Beispielen erwähnt sind, werden wie folgt hergestellt:
Zusammensetzung Menge Wirkstoff 250 g PEG 400 1 l Tween 80 1 l - Herstellungsverfahren: Der pulverisierte Wirkstoff wird in PEG 400 (Polyethylenglycol mit einem Molekulargewicht Mr von etwa 380 bis etwa 420, Fluka, Schweiz) und Tween® 80 (Polyoxyethylensorbitanmonolaurat, Atlas Chem. Ind. Inc., USA, Hersteller Fluka, Schweiz) suspendiert und in einem Nasspulverisator auf eine Teilchengröße von etwa 1 bis 3 μm vermahlen. Sodann werden Anteile von 0,43 g des Gemisches unter Verwendung einer Kapselfüllmaschine in Weichgelatinekapseln abgefüllt.
worin das Präfix Nieder für einen Rest mit bis zu und einschließlich maximal 7 Kohlenstoffatomen steht.
Claims (7)
- 7H-Pyrrolo[2,3-d]pyrimidinderivate der Formel Iworin n für 0 oder 1 steht, q für 0 oder 1 steht, R für Chlor steht, R1 für Wasserstoff steht, und R2 steht für a) einen Rest der Formel II
 worin u für 1 steht und R4 für N3-Niederalkylureido, N3-Phenylureido, N3-Niederalkylthioureido, Niederalkoxycarbonylamino, Benzyloxycarbonylamino, Morpholin-4-carbonyl, Piperazin-1-carbonyl, 4-Niederalkylpiperazin-1-carbonyl, Niederalkylsulfonylamino, Benzolsulfonylamino, Toluolsulfonylamino, Furan-2-carbonylamino, Thiophen-2-carbonylamino, Benzylamino, Hydroxymethyl oder einen Rest der Formel -N=C(R5)-R6 steht, worin R5 für Wasserstoff oder Niederalkyl steht und R6 für Diniederalkylamino, Piperidino, 4-Niederalkylpiperazino oder Morpholino steht, oder b) einen Rest der Formel III
worin u für 1 steht und R4 für N3-Niederalkylureido, N3-Phenylureido, N3-Niederalkylthioureido, Niederalkoxycarbonylamino, Benzyloxycarbonylamino, Morpholin-4-carbonyl, Piperazin-1-carbonyl, 4-Niederalkylpiperazin-1-carbonyl, Niederalkylsulfonylamino, Benzolsulfonylamino, Toluolsulfonylamino, Furan-2-carbonylamino, Thiophen-2-carbonylamino, Benzylamino, Hydroxymethyl oder einen Rest der Formel -N=C(R5)-R6 steht, worin R5 für Wasserstoff oder Niederalkyl steht und R6 für Diniederalkylamino, Piperidino, 4-Niederalkylpiperazino oder Morpholino steht, oder b) einen Rest der Formel III worin R7 für Niederalkoxy steht und R8 für Hydroxyl oder Benzyloxy steht, oder c) Piperazin-1-carbonyl, 4-Niederalkylpiperazin-1-carbonyl, Morpholin-4-carbonyl, Thiocarbamoyl, Thiazol-2-yl, 4-(4-Methoxyphenyl)thiazol-2-yl, 4-Ethylthiazol-2-yl, 4,5-Dimethylthiazol-2-yl, Tetrazol-5-yl, 2-Methyltetrazol-5-yl oder 1-Methyltetrazol-5-yl, oder d) einen Rest der Formel -CH=N-OR9, worin R9 für Wasserstoff oder Niederalkyl steht und worin R3 für Wasserstoff oder Niederalkyl steht, oder ein pharmazeutisch akzeptables Salz hiervon, worin das Präfix Nieder für einen Rest mit bis zu und einschließlich maximal 7 Kohlenstoffatome steht.
worin R7 für Niederalkoxy steht und R8 für Hydroxyl oder Benzyloxy steht, oder c) Piperazin-1-carbonyl, 4-Niederalkylpiperazin-1-carbonyl, Morpholin-4-carbonyl, Thiocarbamoyl, Thiazol-2-yl, 4-(4-Methoxyphenyl)thiazol-2-yl, 4-Ethylthiazol-2-yl, 4,5-Dimethylthiazol-2-yl, Tetrazol-5-yl, 2-Methyltetrazol-5-yl oder 1-Methyltetrazol-5-yl, oder d) einen Rest der Formel -CH=N-OR9, worin R9 für Wasserstoff oder Niederalkyl steht und worin R3 für Wasserstoff oder Niederalkyl steht, oder ein pharmazeutisch akzeptables Salz hiervon, worin das Präfix Nieder für einen Rest mit bis zu und einschließlich maximal 7 Kohlenstoffatome steht.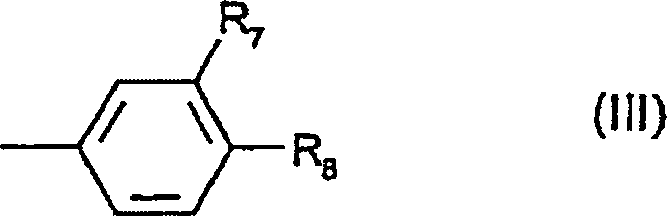
- Verbindung der Formel I nach Anspruch 1 oder ein pharmazeutisch akzeptables Salz hiervon, ausgewählt aus (E)-4-(3-Chloranilino)-7H-pyrrolo[2,3-d]pyrimidin-6-carbaldehydoxim, (Z)-4-(3-Chloranilino)-7H-pyrrolo[2,3-d]pyrimidin-6-carbaldehydoxim, 4-(3-Chloranilino)-7H-pyrrolo[2,3-d]pyrimidin-6-carbaldehyd-O-methyoxim, 4-(3-Chloranilino)-6-(morpholin-4-yl-carbonyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-[(4-methylpiperazin-1-yl)carbonyl]-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(thiocarbamoyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-[4-(4-methoxyphenyl)thiazol-2-yl]-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-ethylthiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4,5-dimethylthiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(tetrazol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(2-methyltetrazol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(1-methyltetrazol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin, (R)-6-(4-Methylsulfonylaminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d)pyrimidin, (R)-6-(4-Ethylsulfonylaminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]-pyrimidin, (R)-6-(4-Isopropylsulfonylaminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin, (R)-6-(3-Methylsulfonylaminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin, (R)-6-(3-Ethylsulfonylaminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin, (R)-6-(3-Isopropylsulfonylaminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin, 6-(4-Methylsulfonylaminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-ethylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-isopropylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-phenylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(3-methylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(3-ethylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(3-isopropylsulfonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-[dimethylaminomethylenamino]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-[diethylaminomethylenamino]phenyl-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-[piperidinomethylenaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-[morpholinomethylenamino]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-{4-[(4-methylpiperazino)methylenamino]phenyl}-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(3-[dimethylaminomethylenamino]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(3-[diethylaminomethylenamino]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(3-[morpholinomethylenamino)phenyl)-7H-pyrrolo[2,3-d]pyrimidin, (R)-6-(4-[Dimethylaminomethylenamino]phenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin, (R)-6-(3-[Dimethylaminomethylenamino]phenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-[morpholin-4-ylcarbonyl]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-[4-methylpiperazin-1-ylcarbonyl]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-[N3-ethylureido]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(3-[N3-ethylureido]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(3-[N3-phenylureidophenyl)-7H-pyrrolo[2,3-d]pyrimidin, (R)-6-(4-[N3-Ethylureido]phenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin, (R)-6-(3-[N3-Ethylureido]phenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-[N3-methylthioureido]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(3-[N3-methylthioureido]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, (R)-6-(4-[N3-Methylthioureido]phenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin, (R)-6-(3-[N3-Methylthioureido]phenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-methoxycarbonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-ethoxycarbonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-isopropyloxycarbonylaminophenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-{4-[(2-methylpropyloxy)carbonylamino]phenyl}-7H-pyrrolo[2,3-d]pyrimidin, (R)-6-(3-Benzylaminophenyl)-4-[(1-phenylethyl)amino]-7H-pyrrolo[2,3-d]pyrimidin, 6-(4-Benzyloxy-3-methoxyphenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin, 6-(4-Benzyloxy-3-methoxyphenyl)-4-(3-methylanilino)-7H-pyrrolo[2,3-d]pyrimidin, 6-(4-Hydroxy-3-methoxyphenyl)-4-(3-methylanilino)-7H-pyrrolo(2,3-d]pyrimidin, 4-Anilino-6-(4-hydroxy-3-methoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidinhydrochlorid, 4-(3-Chloranilino)-6-(4-hydroxy-3-methoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidinhydrochlorid, 4-(3-Chloranilino)-6-(4-[fur-2-ylcarbonylamino]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-(4-[thien-2-ylcarbonylamino]phenyl)-7H-pyrrolo[2,3-d]pyrimidin, 6-(4-Benzylaminophenyl)-4-(3-chloranilino)-7H-pyrrolo[2,3-d]pyrimidin, 4-(3-Chloranilino)-6-{4-[(1-dimethylamino-1-isopropylmethylen)amino}phenyl)-7H-pyrrolo[2,3-d]pyrimidin und 4-(3-Chloranilino)-6-(thiazol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin.
- Verbindung der Formel I nach einem der Ansprüche 1 oder 2 oder ein pharmazeutisch akzeptables Salz einer solchen Verbindung zur Verwendung bei einem Verfahren für die therapeutische Behandlung des menschlichen oder tierischen Körpers.
- Pharmazeutische Zusammensetzung, umfassend eine Verbindung der Formel I nach einem der Ansprüche 1 oder 2 oder ein pharmazeutisch akzeptables Salz einer solchen Verbindung zusammen mit einem pharmazeutischen Träger.
- Pharmazeutische Zusammensetzung für die Behandlung von Tumoren bei Warmblütern unter Einschluss des Menschen, umfassend in einer gegen Tumoren wirksamen Menge eine Verbindung der Formel I nach einem der Ansprüche 1 oder 2 oder ein pharmazeutisch akzeptables Salz einer solchen Verbindung zusammen mit einem pharmazeutischen Träger.
- Verwendung einer Verbindung der Formel I nach einem der Ansprüche 1 oder 2 oder eines pharmazeutisch akzeptablen Salzes einer solchen Verbindung zur Herstellung pharmazeutischer Zusammensetzungen für die Verwendung in der Chemotherapie von Tumoren.
- Verfahren zur Herstellung eines 7H-Pyrrolo[2,3-d]pyrimidinderivats der Formel Inach Anspruch 1, oder eines Salzes hiervon, durch a) Umsetzung eines Pyrrolo[2,3-d]pyrimidinderivats der Formel IV
 worin X eine geeignete Abgangsgruppe ist, Z für Wasserstoff oder 1-Arylniederalkyl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei in den Resten R1 und R2 vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, mit einem Anilinderivat der Formel V
worin X eine geeignete Abgangsgruppe ist, Z für Wasserstoff oder 1-Arylniederalkyl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei in den Resten R1 und R2 vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, mit einem Anilinderivat der Formel V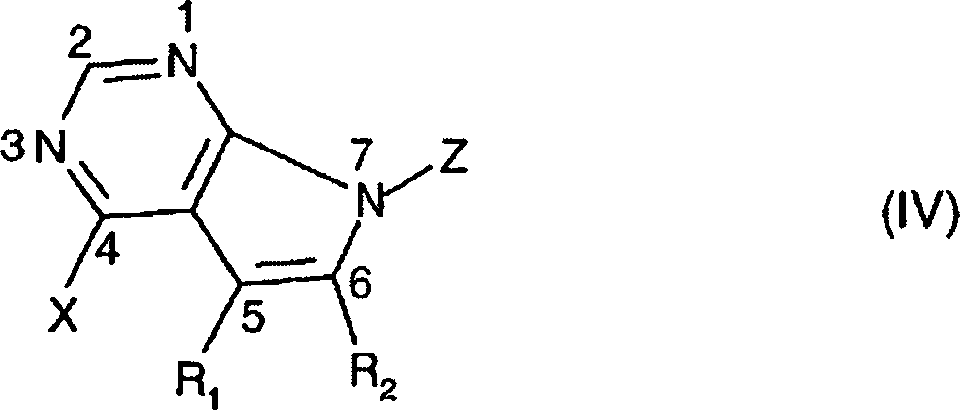 worin R, R3, n und q wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und Entfernung der vorhandenen Schutzgruppen und, falls vorhanden, auch des 1-Arylniederalkylrests Z, oder b) Umsetzung eines Pyrrolo[2,3-d]pyrimidin-4-on-derivats der Formel VI
worin R, R3, n und q wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und Entfernung der vorhandenen Schutzgruppen und, falls vorhanden, auch des 1-Arylniederalkylrests Z, oder b) Umsetzung eines Pyrrolo[2,3-d]pyrimidin-4-on-derivats der Formel VI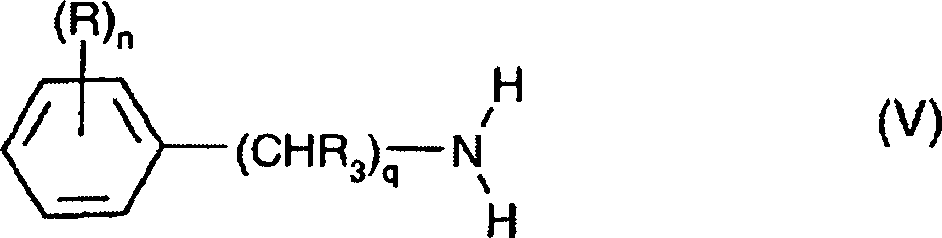 worin Z' für 1-Arylniederalkyl steht und R1 und R2 wie oben für die Verbindungen der Formel I definiert sind, wobei in den Resten R1 und R2 vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, in Gegenwart eines Dehydratationsmittels und eines tertiären Amins, mit einem Phenylamin der obigen Formel V und Entfernung der vorhandenen Schutzgruppen, oder e) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 ein Rest der Formel -CH=N-OR9 ist, worin R9 für Wasserstoff oder Niederalkyl steht, und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Formyl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, mit einem Hydroxylaminderivat der Formel VIII
worin Z' für 1-Arylniederalkyl steht und R1 und R2 wie oben für die Verbindungen der Formel I definiert sind, wobei in den Resten R1 und R2 vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, in Gegenwart eines Dehydratationsmittels und eines tertiären Amins, mit einem Phenylamin der obigen Formel V und Entfernung der vorhandenen Schutzgruppen, oder e) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 ein Rest der Formel -CH=N-OR9 ist, worin R9 für Wasserstoff oder Niederalkyl steht, und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Formyl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, mit einem Hydroxylaminderivat der Formel VIII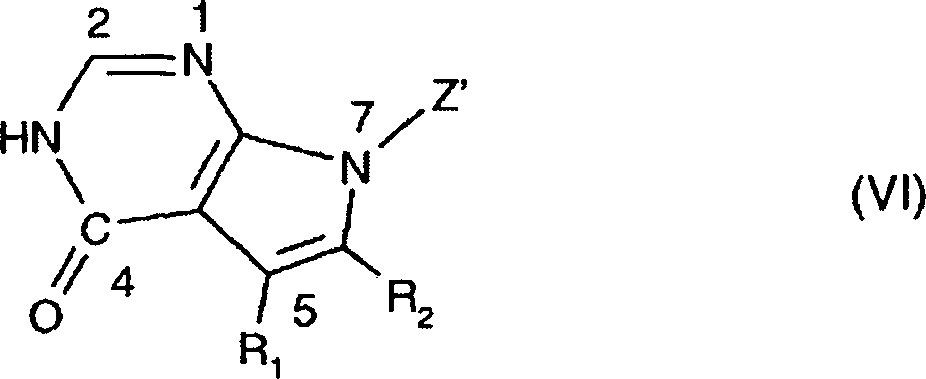 worin X für eine Abgangsgruppe steht und R13 jeweils für Ethyl oder Niederalkoxyphenyl steht, wobei im Rest R13 vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und anschließende Entfernung der vorhandenen Schutzgruppen, oder i) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Tetrazol-5-yl steht, und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Cyano steht, und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, mit einem geeigneten Alkalimetallazid, und anschließende Entfernung der vorhandenen Schutzgruppen, oder j) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für 2-Niederalkyltetrazol-5-yl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Tetrazol-5-yl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, mit dem geeigneten Niederalkyliodid, und anschließende Entfernung der vorhandenen Schutzgruppen, oder k) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht
worin X für eine Abgangsgruppe steht und R13 jeweils für Ethyl oder Niederalkoxyphenyl steht, wobei im Rest R13 vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und anschließende Entfernung der vorhandenen Schutzgruppen, oder i) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Tetrazol-5-yl steht, und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Cyano steht, und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, mit einem geeigneten Alkalimetallazid, und anschließende Entfernung der vorhandenen Schutzgruppen, oder j) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für 2-Niederalkyltetrazol-5-yl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für Tetrazol-5-yl steht und die anderen Substituenten wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, mit dem geeigneten Niederalkyliodid, und anschließende Entfernung der vorhandenen Schutzgruppen, oder k) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht worin wenigstens einer der Reste R4 für Niederalkylsulfonylamino, Benzolsulfonylamino oder Toluolsulfonylamino steht, und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, mit einer Verbindung der Formel X
worin wenigstens einer der Reste R4 für Niederalkylsulfonylamino, Benzolsulfonylamino oder Toluolsulfonylamino steht, und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, mit einer Verbindung der Formel X worin wenigstens ein Rest R4 für einen Rest der Formel -N=C(R5)-R6 steht, worin R5 für Wasserstoff steht und R6 wie oben für die Verbindungen der Formel I definiert ist, und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, mit einem Acetal der Formel XI
worin wenigstens ein Rest R4 für einen Rest der Formel -N=C(R5)-R6 steht, worin R5 für Wasserstoff steht und R6 wie oben für die Verbindungen der Formel I definiert ist, und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, mit einem Acetal der Formel XI worin R15 und R16 jeweils individuell für Niederalkyl stehen oder zusammen für Pentan-1,5-diyl, 3-N-Niederalkyl-3-azapentan-1,5-diyl oder 3-Oxapentan-1,5-diyl stehen, und R17 für Niederalkyl steht, und anschließende Entfernung der vorhandenen Schutzgruppen, oder m) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht
worin R15 und R16 jeweils individuell für Niederalkyl stehen oder zusammen für Pentan-1,5-diyl, 3-N-Niederalkyl-3-azapentan-1,5-diyl oder 3-Oxapentan-1,5-diyl stehen, und R17 für Niederalkyl steht, und anschließende Entfernung der vorhandenen Schutzgruppen, oder m) zur Herstellung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht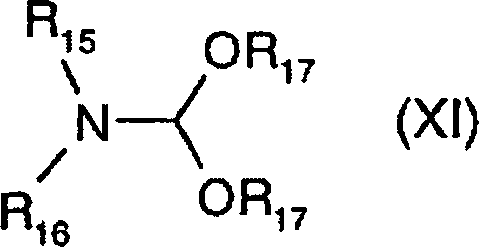 worin wenigstens ein Rest R4 für N3-Niederalkylureido oder N3-Phenylureido steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, mit einem Isocyanat der Formel XII
worin wenigstens ein Rest R4 für N3-Niederalkylureido oder N3-Phenylureido steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, mit einem Isocyanat der Formel XII worin wenigstens ein Rest R4 für N3-Niederalkylthioureido oder N3-Phenylthioureido steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, mit Isothiocyanat der Formel XIII
worin wenigstens ein Rest R4 für N3-Niederalkylthioureido oder N3-Phenylthioureido steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, mit Isothiocyanat der Formel XIII worin wenigstens ein Rest R4 für Niederalkoxycarbonylamino oder Benzyloxycarbonylamino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, mit einem Chlorameisensäureester der Formel XIV
worin wenigstens ein Rest R4 für Niederalkoxycarbonylamino oder Benzyloxycarbonylamino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, Umsetzung einer Verbindung der Formel I, worin einer der Reste R1 und R2 für einen Rest der Formel II steht, worin wenigstens ein Rest R4 für Amino steht und die anderen Substituenten und Symbole wie oben für die Verbindungen der Formel I definiert sind, wobei im Rest R vorhandene freie funktionale Gruppen erforderlichenfalls durch Schutzgruppen geschützt sind, und, falls vorhanden, die anderen Reste R4 erforderlichenfalls durch leicht entfernbare Schutzgruppen geschützt sind, mit einem Chlorameisensäureester der Formel XIV
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CH207196 | 1996-08-23 | ||
| CH207196 | 1996-08-23 | ||
| PCT/EP1997/004564 WO1998007726A1 (en) | 1996-08-23 | 1997-08-21 | Substituted pyrrolopyrimidines and processes for their preparation |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69738468D1 DE69738468D1 (de) | 2008-03-06 |
| DE69738468T2 true DE69738468T2 (de) | 2009-01-08 |
Family
ID=4225221
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69738468T Expired - Lifetime DE69738468T2 (de) | 1996-08-23 | 1997-08-21 | Substituierte pyrrolopyrimidine und verfahren zu ihrer herstellung |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US6180636B1 (de) |
| EP (1) | EP0938486B1 (de) |
| JP (1) | JP4242928B2 (de) |
| AT (1) | ATE384062T1 (de) |
| AU (1) | AU720429B2 (de) |
| CA (1) | CA2262421C (de) |
| DE (1) | DE69738468T2 (de) |
| DK (1) | DK0938486T3 (de) |
| ES (1) | ES2297864T3 (de) |
| PT (1) | PT938486E (de) |
| WO (1) | WO1998007726A1 (de) |
Families Citing this family (148)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6395733B1 (en) * | 1995-06-07 | 2002-05-28 | Pfizer Inc | Heterocyclic ring-fused pyrimidine derivatives |
| DE69720965T2 (de) | 1996-02-13 | 2004-02-05 | Astrazeneca Ab | Chinazolinderivate und deren verwendung als vegf hemmer |
| NZ331191A (en) | 1996-03-05 | 2000-03-27 | Zeneca Ltd | 4-anilinoquinazoline derivatives and pharmaceutical compositions thereof |
| US6251911B1 (en) | 1996-10-02 | 2001-06-26 | Novartis Ag | Pyrimidine derivatives and processes for the preparation thereof |
| US6187777B1 (en) | 1998-02-06 | 2001-02-13 | Amgen Inc. | Compounds and methods which modulate feeding behavior and related diseases |
| US6878716B1 (en) | 1998-06-02 | 2005-04-12 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A1 receptor and uses thereof |
| US6686366B1 (en) | 1998-06-02 | 2004-02-03 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A3 receptor and uses thereof |
| JP4611524B2 (ja) | 1998-06-02 | 2011-01-12 | オーエスアイ・ファーマスーティカルズ・インコーポレーテッド | ピロロ[2,3d]ピリミジン組成物およびその使用 |
| PA8474101A1 (es) | 1998-06-19 | 2000-09-29 | Pfizer Prod Inc | Compuestos de pirrolo [2,3-d] pirimidina |
| SK286640B6 (sk) * | 1998-06-19 | 2009-03-05 | Pfizer Products Inc. | Pyrol [2,3-d] pyrimidínová zlúčenina, jej použitie na výrobu liečiva, farmaceutická kompozícia s jej obsahom, použitie jej kombinácie s ďalšími činidlami a súpravy s jej obsahom na výrobu liečiva |
| UA71945C2 (en) * | 1999-01-27 | 2005-01-17 | Pfizer Prod Inc | Substituted bicyclic derivatives being used as anticancer agents |
| KR100881105B1 (ko) | 1999-11-05 | 2009-02-02 | 아스트라제네카 아베 | Vegf 억제제로서의 퀴나졸린 유도체 |
| US6680322B2 (en) | 1999-12-02 | 2004-01-20 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A1 receptors and uses thereof |
| US6664252B2 (en) | 1999-12-02 | 2003-12-16 | Osi Pharmaceuticals, Inc. | 4-aminopyrrolo[2,3-d]pyrimidine compounds specific to adenosine A2a receptor and uses thereof |
| US7160890B2 (en) | 1999-12-02 | 2007-01-09 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A3 receptor and uses thereof |
| DE60037345T2 (de) * | 1999-12-10 | 2008-11-13 | Pfizer Products Inc., Groton | Pyrrolo(2,3-d)pyrimidin-Verbindungen |
| GB0001930D0 (en) * | 2000-01-27 | 2000-03-22 | Novartis Ag | Organic compounds |
| US7201891B1 (en) * | 2000-05-19 | 2007-04-10 | Otsuka Pharmaceutical Co., Ltd. | Pharmaceutical preparation for the diagnosis of helicobacter pylori infection |
| JP2004503553A (ja) | 2000-06-14 | 2004-02-05 | ワーナー−ランバート・カンパニー、リミテッド、ライアビリティ、カンパニー | 6,5−縮合二環式複素環 |
| ATE423120T1 (de) * | 2000-06-26 | 2009-03-15 | Pfizer Prod Inc | Pyrroloä2,3-düpyrimidin verbindungen als immunosuppressive wirkstoffe |
| US6680324B2 (en) | 2000-12-01 | 2004-01-20 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A1 receptors and uses thereof |
| US6673802B2 (en) | 2000-12-01 | 2004-01-06 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A3 receptor and uses thereof |
| GB0031080D0 (en) * | 2000-12-20 | 2001-01-31 | Novartis Ag | Organic compounds |
| US7301023B2 (en) | 2001-05-31 | 2007-11-27 | Pfizer Inc. | Chiral salt resolution |
| GB0119249D0 (en) * | 2001-08-07 | 2001-10-03 | Novartis Ag | Organic compounds |
| US7323469B2 (en) | 2001-08-07 | 2008-01-29 | Novartis Ag | 7H-pyrrolo[2,3-d]pyrimidine derivatives |
| DK1450811T3 (da) | 2001-11-30 | 2010-02-15 | Osi Pharm Inc | Forbindelser specifikke af adenosin A1- og A3-receptorer og anvendelser heraf |
| PY0228255A (es) * | 2001-12-06 | 2004-06-01 | Pfizer Prod Inc | Compuestos cristalinos novedosos |
| US20030229067A1 (en) | 2001-12-20 | 2003-12-11 | Arlindo Castelhano | Pyrrolopyrimidine A2b selective antagonist compounds, their synthesis and use |
| CN1620294A (zh) | 2001-12-20 | 2005-05-25 | Osi药物公司 | 嘧啶a2b选择性拮抗剂化合物,它们的合成及用途 |
| DE60313339T2 (de) * | 2002-07-31 | 2008-01-03 | Critical Outcome Technologies, Inc. | Protein tyrosin kinase inhibitoren |
| US20040121407A1 (en) * | 2002-09-06 | 2004-06-24 | Elixir Pharmaceuticals, Inc. | Regulation of the growth hormone/IGF-1 axis |
| US7629347B2 (en) * | 2002-10-09 | 2009-12-08 | Critical Outcome Technologies, Inc. | Protein tyrosine kinase inhibitors |
| NZ539901A (en) * | 2002-11-26 | 2007-09-28 | Pfizer Prod Inc | Method of treatment of transplant rejection |
| GB0403606D0 (en) | 2004-02-18 | 2004-03-24 | Novartis Ag | Organic compounds |
| CA2569016C (en) * | 2004-06-02 | 2012-11-27 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compound |
| JP2008508358A (ja) * | 2004-08-02 | 2008-03-21 | オーエスアイ・ファーマスーティカルズ・インコーポレーテッド | アリール−アミノ置換ピロロピリミジンマルチキナーゼ阻害化合物 |
| EP2121681B1 (de) | 2007-01-11 | 2015-04-15 | Critical Outcome Technologies, Inc. | Verbindungen und verfahren zur behandlung von krebs |
| CA2710039C (en) * | 2007-12-26 | 2018-07-03 | Critical Outcome Technologies, Inc. | Semicarbazones, thiosemicarbazones and related compounds and methods for treatment of cancer |
| EP2247596A2 (de) | 2008-01-11 | 2010-11-10 | Natco Pharma Limited | Neuartige pyrazol-[3,4-d-]pyrimidin-derivate als wirkstoffe gegen krebs |
| EP2318406B1 (de) | 2008-07-17 | 2016-01-27 | Critical Outcome Technologies, Inc. | Thiosemicarbazonhemmerverbindungen und krebsbehandlungsverfahren |
| KR101335843B1 (ko) | 2008-08-20 | 2013-12-02 | 조에티스 엘엘씨 | 피롤로[2,3-d]피리미딘 화합물 |
| US8385364B2 (en) * | 2008-09-24 | 2013-02-26 | Nec Laboratories America, Inc. | Distributed message-passing based resource allocation in wireless systems |
| EP2349235A1 (de) | 2008-11-07 | 2011-08-03 | Triact Therapeutics, Inc. | Verwendung von catecholbutan-derivaten in der krebstherapie |
| WO2010099139A2 (en) | 2009-02-25 | 2010-09-02 | Osi Pharmaceuticals, Inc. | Combination anti-cancer therapy |
| JP2012519170A (ja) | 2009-02-26 | 2012-08-23 | オーエスアイ・ファーマシューティカルズ,エルエルシー | 生体内の腫瘍細胞のemtステータスをモニターするためのinsitu法 |
| WO2010099364A2 (en) | 2009-02-27 | 2010-09-02 | Osi Pharmaceuticals, Inc. | Methods for the identification of agents that inhibit mesenchymal-like tumor cells or their formation |
| WO2010099363A1 (en) | 2009-02-27 | 2010-09-02 | Osi Pharmaceuticals, Inc. | Methods for the identification of agents that inhibit mesenchymal-like tumor cells or their formation |
| WO2010099138A2 (en) | 2009-02-27 | 2010-09-02 | Osi Pharmaceuticals, Inc. | Methods for the identification of agents that inhibit mesenchymal-like tumor cells or their formation |
| EP2408479A1 (de) | 2009-03-18 | 2012-01-25 | OSI Pharmaceuticals, LLC | Kombinationskrebstherapie mit verabreichung eines egfr-hemmers und eines igf-1r-hemmers |
| IN2012DN01251A (de) * | 2009-07-28 | 2015-05-15 | Ube Industries | |
| CA2780875A1 (en) | 2009-11-13 | 2011-05-19 | Pangaea Biotech, S.L. | Molecular biomarkers for predicting response to tyrosine kinase inhibitors in lung cancer |
| AU2010339444A1 (en) | 2009-12-30 | 2012-07-19 | Arqule, Inc. | Substituted pyrrolo-aminopyrimidine compounds |
| EP2552915B1 (de) | 2010-04-01 | 2017-07-19 | Critical Outcome Technologies Inc. | Verbindungen zur behandlung von hiv |
| EP2468883A1 (de) | 2010-12-22 | 2012-06-27 | Pangaea Biotech S.L. | Molekulare Biomarker zur Vorhersage der Reaktion auf Tyrosinkinaseinhibitoren bei Lungenkrebs |
| US9134297B2 (en) | 2011-01-11 | 2015-09-15 | Icahn School Of Medicine At Mount Sinai | Method and compositions for treating cancer and related methods |
| EP2492688A1 (de) | 2011-02-23 | 2012-08-29 | Pangaea Biotech, S.A. | Molekulare Biomarker zur Vorhersage der Reaktion auf die Antitumorbehandlung bei Lungenkrebs |
| WO2012129145A1 (en) | 2011-03-18 | 2012-09-27 | OSI Pharmaceuticals, LLC | Nscle combination therapy |
| WO2012149014A1 (en) | 2011-04-25 | 2012-11-01 | OSI Pharmaceuticals, LLC | Use of emt gene signatures in cancer drug discovery, diagnostics, and treatment |
| DE102011111400A1 (de) * | 2011-08-23 | 2013-02-28 | Merck Patent Gmbh | Bicyclische heteroaromatische Verbindungen |
| EP2751285B2 (de) | 2011-08-31 | 2020-04-01 | Genentech, Inc. | Methode zur bestimmung der empfindlichkeit eines tumors für egfr kinase-inhibitoren |
| EP2761025A1 (de) | 2011-09-30 | 2014-08-06 | Genentech, Inc. | Diagnostische methylierungsmarker vom epithelialen oder mesenchymalen phenotyp und reaktion auf egfr-kinaseinhibitor bei tumoren oder tumorzellen |
| WO2013152252A1 (en) | 2012-04-06 | 2013-10-10 | OSI Pharmaceuticals, LLC | Combination anti-cancer therapy |
| WO2013190089A1 (en) | 2012-06-21 | 2013-12-27 | Pangaea Biotech, S.L. | Molecular biomarkers for predicting outcome in lung cancer |
| GB201223308D0 (en) | 2012-12-21 | 2013-02-06 | Univ Sunderland | Enzyme inhibitors |
| US9834575B2 (en) | 2013-02-26 | 2017-12-05 | Triact Therapeutics, Inc. | Cancer therapy |
| EP2976085A1 (de) | 2013-03-21 | 2016-01-27 | INSERM - Institut National de la Santé et de la Recherche Médicale | Verfahren und pharmazeutische zusammensetzung zur verwendung bei der behandlung von chronischen lebererkrankungen im zusammenhang mit einer niedrigen hepcidinexpression |
| WO2015035410A1 (en) | 2013-09-09 | 2015-03-12 | Triact Therapeutic, Inc. | Cancer therapy |
| WO2015156674A2 (en) | 2014-04-10 | 2015-10-15 | Stichting Het Nederlands Kanker Instituut | Method for treating cancer |
| WO2016105503A1 (en) | 2014-12-24 | 2016-06-30 | Genentech, Inc. | Therapeutic, diagnostic and prognostic methods for cancer of the bladder |
| US9630968B1 (en) | 2015-12-23 | 2017-04-25 | Arqule, Inc. | Tetrahydropyranyl amino-pyrrolopyrimidinone and methods of use thereof |
| TW201811795A (zh) | 2016-08-24 | 2018-04-01 | 美商亞闊股份有限公司 | 胺基-吡咯并嘧啶酮化合物及其用途 |
| CN110366550A (zh) | 2016-12-22 | 2019-10-22 | 美国安进公司 | 作为用于治疗肺癌、胰腺癌或结直肠癌的KRAS G12C抑制剂的苯并异噻唑、异噻唑并[3,4-b]吡啶、喹唑啉、酞嗪、吡啶并[2,3-d]哒嗪和吡啶并[2,3-d]嘧啶衍生物 |
| AU2018273356B2 (en) | 2017-05-22 | 2021-09-16 | Amgen Inc. | KRAS G12C inhibitors and methods of using the same |
| US11548867B2 (en) | 2017-07-19 | 2023-01-10 | Idea Ya Biosciences, Inc. | Amido compounds as AhR modulators |
| AU2018329920B2 (en) | 2017-09-08 | 2022-12-01 | Amgen Inc. | Inhibitors of KRAS G12C and methods of using the same |
| CA3098574A1 (en) | 2018-05-04 | 2019-11-07 | Amgen Inc. | Kras g12c inhibitors and methods of using the same |
| EP3788053B1 (de) | 2018-05-04 | 2024-07-10 | Amgen Inc. | Kras-g12c-inhibitoren und verfahren zu deren verwendung |
| MA52564A (fr) | 2018-05-10 | 2021-03-17 | Amgen Inc | Inhibiteurs de kras g12c pour le traitement du cancer |
| US11096939B2 (en) | 2018-06-01 | 2021-08-24 | Amgen Inc. | KRAS G12C inhibitors and methods of using the same |
| MX2020012204A (es) | 2018-06-11 | 2021-03-31 | Amgen Inc | Inhibidores de kras g12c para tratar el cáncer. |
| US11285156B2 (en) | 2018-06-12 | 2022-03-29 | Amgen Inc. | Substituted piperazines as KRAS G12C inhibitors |
| CN113194954A (zh) | 2018-10-04 | 2021-07-30 | 国家医疗保健研究所 | 用于治疗角皮病的egfr抑制剂 |
| JP7516029B2 (ja) | 2018-11-16 | 2024-07-16 | アムジエン・インコーポレーテツド | Kras g12c阻害剤化合物の重要な中間体の改良合成法 |
| JP7377679B2 (ja) | 2018-11-19 | 2023-11-10 | アムジエン・インコーポレーテツド | がん治療のためのkrasg12c阻害剤及び1種以上の薬学的に活性な追加の薬剤を含む併用療法 |
| AU2019384118B2 (en) | 2018-11-19 | 2025-06-12 | Amgen Inc. | KRAS G12C inhibitors and methods of using the same |
| MA54543A (fr) | 2018-12-20 | 2022-03-30 | Amgen Inc | Inhibiteurs de kif18a |
| AU2019401495B2 (en) | 2018-12-20 | 2025-06-26 | Amgen Inc. | Heteroaryl amides useful as KIF18A inhibitors |
| JP7686559B2 (ja) | 2018-12-20 | 2025-06-02 | アムジエン・インコーポレーテツド | Kif18a阻害剤 |
| JP2022513967A (ja) | 2018-12-20 | 2022-02-09 | アムジエン・インコーポレーテツド | Kif18a阻害剤として有用なヘテロアリールアミド |
| WO2020132384A1 (en) | 2018-12-21 | 2020-06-25 | Celgene Corporation | Thienopyridine inhibitors of ripk2 |
| NL2022471B1 (en) | 2019-01-29 | 2020-08-18 | Vationpharma B V | Solid state forms of oclacitinib |
| MX2021010323A (es) | 2019-03-01 | 2021-12-10 | Revolution Medicines Inc | Compuestos bicíclicos de heterociclilo y usos de este. |
| MX2021010319A (es) | 2019-03-01 | 2021-12-10 | Revolution Medicines Inc | Compuestos biciclicos de heteroarilo y usos de estos. |
| WO2020231990A1 (en) | 2019-05-13 | 2020-11-19 | Relay Therapeutics, Inc. | Fgfr inhibitors and methods of use thereof |
| EP3738593A1 (de) | 2019-05-14 | 2020-11-18 | Amgen, Inc | Dosierung von kras-inhibitor zur behandlung von krebserkrankungen |
| WO2020236947A1 (en) | 2019-05-21 | 2020-11-26 | Amgen Inc. | Solid state forms |
| EP4007753B1 (de) | 2019-08-02 | 2025-09-24 | Amgen Inc. | Kif18a-inhibitoren |
| EP4007756B1 (de) | 2019-08-02 | 2025-12-24 | Amgen Inc. | Kif18a-inhibitoren |
| EP4007752B1 (de) | 2019-08-02 | 2025-09-24 | Amgen Inc. | Kif18a-inhibitoren |
| EP4007638A1 (de) | 2019-08-02 | 2022-06-08 | Amgen Inc. | Pyridinderivate als kif18a-inhibitoren |
| EP4048671B1 (de) | 2019-10-24 | 2026-03-18 | Amgen Inc. | Pyridopyrimidinderivate als kras-g12c- und kras-g12d-inhibitoren zur behandlung von krebs |
| TW202132316A (zh) | 2019-11-04 | 2021-09-01 | 美商銳新醫藥公司 | Ras抑制劑 |
| AU2020377925A1 (en) | 2019-11-04 | 2022-05-05 | Revolution Medicines, Inc. | Ras inhibitors |
| IL322454A (en) | 2019-11-04 | 2025-09-01 | Revolution Medicines Inc | ras inhibitors |
| CA3156359A1 (en) | 2019-11-08 | 2021-05-14 | Adrian Liam Gill | Bicyclic heteroaryl compounds and uses thereof |
| JP7837865B2 (ja) | 2019-11-14 | 2026-03-31 | アムジエン・インコーポレーテツド | Kras g12c阻害剤化合物の改良合成法 |
| JP2023501528A (ja) | 2019-11-14 | 2023-01-18 | アムジエン・インコーポレーテツド | Kras g12c阻害剤化合物の改善された合成 |
| EP4065231A1 (de) | 2019-11-27 | 2022-10-05 | Revolution Medicines, Inc. | Kovalente ras-inhibitoren und verwendungen davon |
| WO2021142026A1 (en) | 2020-01-07 | 2021-07-15 | Revolution Medicines, Inc. | Shp2 inhibitor dosing and methods of treating cancer |
| IL299131A (en) | 2020-06-18 | 2023-02-01 | Revolution Medicines Inc | Methods for delaying, preventing and treating acquired resistance to RAS inhibitors |
| US11814388B1 (en) | 2020-08-28 | 2023-11-14 | Ferris State University | Substituted pyrrolo[2,3-d]pyrimidines and pyrazolo[3,4-d]pyrimidines as inhibitors for multi-resistant cancers |
| US20250195521A1 (en) | 2020-09-03 | 2025-06-19 | Revolution Medicines, Inc. | Use of sos1 inhibitors to treat malignancies with shp2 mutations |
| CA3194067A1 (en) | 2020-09-15 | 2022-03-24 | Revolution Medicines, Inc. | Ras inhibitors |
| TW202233625A (zh) | 2020-11-18 | 2022-09-01 | 美商傳達治療有限公司 | Fgfr抑制劑及其製造及使用方法 |
| JP7849366B2 (ja) | 2020-12-22 | 2026-04-21 | キル・レガー・セラピューティクス・インコーポレーテッド | Sos1阻害剤およびその使用 |
| JP7840066B2 (ja) | 2021-05-03 | 2026-04-03 | ジェービーケーラボ カンパニー リミテッド | 2,6-ジクロロ-4-(4-(4-ヒドロキシシクロヘキシルアミノ)-7h-ピロロ[2,3-d]ピリミジン-5-イル)フェノールを活性成分として含有する、がんを予防、緩和、または処置するための医薬組成物 |
| TW202309052A (zh) | 2021-05-05 | 2023-03-01 | 美商銳新醫藥公司 | Ras抑制劑 |
| KR20240017811A (ko) | 2021-05-05 | 2024-02-08 | 레볼루션 메디슨즈, 인크. | 암의 치료를 위한 ras 억제제 |
| JP2024516450A (ja) | 2021-05-05 | 2024-04-15 | レボリューション メディシンズ インコーポレイテッド | 共有結合性ras阻害剤及びその使用 |
| AR127308A1 (es) | 2021-10-08 | 2024-01-10 | Revolution Medicines Inc | Inhibidores ras |
| CN119212994A (zh) | 2021-12-17 | 2024-12-27 | 建新公司 | 作为shp2抑制剂的吡唑并吡嗪化合物 |
| EP4227307A1 (de) | 2022-02-11 | 2023-08-16 | Genzyme Corporation | Pyrazolopyrazinverbindungen als shp2-inhibitoren |
| KR20240156373A (ko) | 2022-03-07 | 2024-10-29 | 암젠 인크 | 4-메틸-2-프로판-2-일-피리딘-3-카르보니트릴의 제조 방법 |
| JP2025510572A (ja) | 2022-03-08 | 2025-04-15 | レボリューション メディシンズ インコーポレイテッド | 免疫不応性肺癌を治療するための方法 |
| WO2023240263A1 (en) | 2022-06-10 | 2023-12-14 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| AU2023358792A1 (en) | 2022-10-14 | 2025-04-17 | Black Diamond Therapeutics, Inc. | Methods of treating cancers using isoquinoline or 6-aza-quinoline derivatives |
| CN116332940B (zh) * | 2023-02-14 | 2024-09-13 | 广西民族大学 | 一种7-脱氮嘌呤衍生物及其制备方法和应用 |
| AU2024241633A1 (en) | 2023-03-30 | 2025-11-06 | Revolution Medicines, Inc. | Compositions for inducing ras gtp hydrolysis and uses thereof |
| AR132338A1 (es) | 2023-04-07 | 2025-06-18 | Revolution Medicines Inc | Inhibidores de ras |
| CR20250420A (es) | 2023-04-07 | 2025-11-20 | Revolution Medicines Inc | Inhibidores macrocíclicos de ras |
| CN121100123A (zh) | 2023-04-14 | 2025-12-09 | 锐新医药公司 | Ras抑制剂的结晶形式 |
| CN121464140A (zh) | 2023-04-14 | 2026-02-03 | 锐新医药公司 | Ras抑制剂的结晶形式、含有其的组合物及其使用方法 |
| TW202508595A (zh) | 2023-05-04 | 2025-03-01 | 美商銳新醫藥公司 | 用於ras相關疾病或病症之組合療法 |
| US20250049810A1 (en) | 2023-08-07 | 2025-02-13 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| AU2024360465A1 (en) | 2023-10-12 | 2026-04-09 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| TW202542151A (zh) | 2023-12-22 | 2025-11-01 | 美商銳格醫藥有限公司 | Sos1抑制劑及其用途 |
| WO2025171296A1 (en) | 2024-02-09 | 2025-08-14 | Revolution Medicines, Inc. | Ras inhibitors |
| TW202547461A (zh) | 2024-05-17 | 2025-12-16 | 美商銳新醫藥公司 | Ras抑制劑 |
| WO2025255438A1 (en) | 2024-06-07 | 2025-12-11 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| WO2025265060A1 (en) | 2024-06-21 | 2025-12-26 | Revolution Medicines, Inc. | Therapeutic compositions and methods for managing treatment-related effects |
| WO2026006747A1 (en) | 2024-06-28 | 2026-01-02 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026015801A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015825A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Use of ras inhibitor for treating pancreatic cancer |
| WO2026015790A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015796A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026050446A1 (en) | 2024-08-29 | 2026-03-05 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026072904A2 (en) | 2024-09-26 | 2026-04-02 | Revolution Medicines, Inc. | Compositions and methods for treating lung cancer |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3036930C2 (de) | 1980-09-30 | 1982-06-16 | Windmöller & Hölscher, 4540 Lengerich | Ungleichförmigkeitsgetriebe, vorzugsweise zum Antrieb von Messerwalzen |
| US5661148A (en) * | 1989-09-19 | 1997-08-26 | Teijin Limited | Pyrrolo[2,3-d]pyrimidine derivatives, process for producing the same and pharmaceutical preparation comprising the same as active ingredient |
| SG64322A1 (en) | 1991-05-10 | 1999-04-27 | Rhone Poulenc Rorer Int | Bis mono and bicyclic aryl and heteroaryl compounds which inhibit egf and/or pdgf receptor tyrosine kinase |
| IL112249A (en) | 1994-01-25 | 2001-11-25 | Warner Lambert Co | Pharmaceutical compositions containing di and tricyclic pyrimidine derivatives for inhibiting tyrosine kinases of the epidermal growth factor receptor family and some new such compounds |
| IL112248A0 (en) | 1994-01-25 | 1995-03-30 | Warner Lambert Co | Tricyclic heteroaromatic compounds and pharmaceutical compositions containing them |
| US5654307A (en) * | 1994-01-25 | 1997-08-05 | Warner-Lambert Company | Bicyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| DE59500788D1 (de) | 1994-05-03 | 1997-11-20 | Ciba Geigy Ag | Pyrrolopyrimidinderivate mit antiproliferativer Wirkung |
| US5869485A (en) | 1994-09-29 | 1999-02-09 | Novartis Finance Corp. | Pyrrolo 2,3-d!pyrimidines and their use |
| CA2214086C (en) | 1995-04-03 | 2008-07-29 | Novartis Ag | Pyrazole derivatives and processes for the preparation thereof |
| US6403599B1 (en) | 1995-11-08 | 2002-06-11 | Pfizer Inc | Corticotropin releasing factor antagonists |
| JP3290666B2 (ja) | 1995-06-07 | 2002-06-10 | ファイザー・インコーポレーテッド | 複素環式の縮合環ピリミジン誘導体 |
| SI9620103A (sl) | 1995-07-06 | 1998-10-31 | Novartis Ag | Pirolopirimidini in postopki za njihovo pripravo |
| AU1441497A (en) | 1996-01-23 | 1997-08-20 | Novartis Ag | Pyrrolopyrimidines and processes for their preparation |
| GB9604361D0 (en) | 1996-02-29 | 1996-05-01 | Pharmacia Spa | 4-Substituted pyrrolopyrimidine compounds as tyrosine kinase inhibitors |
| US6251911B1 (en) | 1996-10-02 | 2001-06-26 | Novartis Ag | Pyrimidine derivatives and processes for the preparation thereof |
| AU4779897A (en) | 1996-10-02 | 1998-04-24 | Novartis Ag | Fused pyrazole derivatives and processes for their preparation |
-
1997
- 1997-08-21 WO PCT/EP1997/004564 patent/WO1998007726A1/en not_active Ceased
- 1997-08-21 CA CA002262421A patent/CA2262421C/en not_active Expired - Fee Related
- 1997-08-21 JP JP51042598A patent/JP4242928B2/ja not_active Expired - Fee Related
- 1997-08-21 DK DK97940108T patent/DK0938486T3/da active
- 1997-08-21 AT AT97940108T patent/ATE384062T1/de not_active IP Right Cessation
- 1997-08-21 PT PT97940108T patent/PT938486E/pt unknown
- 1997-08-21 ES ES97940108T patent/ES2297864T3/es not_active Expired - Lifetime
- 1997-08-21 US US09/242,592 patent/US6180636B1/en not_active Expired - Fee Related
- 1997-08-21 DE DE69738468T patent/DE69738468T2/de not_active Expired - Lifetime
- 1997-08-21 AU AU42064/97A patent/AU720429B2/en not_active Ceased
- 1997-08-21 EP EP97940108A patent/EP0938486B1/de not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| DE69738468D1 (de) | 2008-03-06 |
| WO1998007726A1 (en) | 1998-02-26 |
| CA2262421A1 (en) | 1998-02-26 |
| JP2000516626A (ja) | 2000-12-12 |
| AU4206497A (en) | 1998-03-06 |
| DK0938486T3 (da) | 2008-07-07 |
| EP0938486A1 (de) | 1999-09-01 |
| ES2297864T3 (es) | 2008-05-01 |
| EP0938486B1 (de) | 2008-01-16 |
| AU720429B2 (en) | 2000-06-01 |
| US6180636B1 (en) | 2001-01-30 |
| JP4242928B2 (ja) | 2009-03-25 |
| ATE384062T1 (de) | 2008-02-15 |
| CA2262421C (en) | 2007-10-02 |
| PT938486E (pt) | 2008-03-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69738468T2 (de) | Substituierte pyrrolopyrimidine und verfahren zu ihrer herstellung | |
| DE69619114T2 (de) | Pyrolopyrimidine und verfahren zu ihrer herstellung | |
| DE69732780T2 (de) | Pyrimiderivate und verfahren zu ihrer herstellung | |
| DE69712745T2 (de) | Pyrrolopyrimidinen und verfahren zu deren herstellung | |
| EP0682027B1 (de) | Pyrrolopyrimidinderivate mit antiproliferativer Wirkung | |
| DE60030002T2 (de) | Für den adenosin-a1-a2a-und-a3-rezeptor spezifische verbindungen und verwendungen davon | |
| DE69111077T2 (de) | Chinazolinderivate um die antitumorwirkung zu verbessern. | |
| DE10057751A1 (de) | Neue Carbamat-substituierte Pyrazolopyridinderivate | |
| WO1998014451A1 (en) | Fused pyrazole derivative and process for its preparation | |
| DE60129174T2 (de) | Neue pleuromutilinderivate | |
| DE69729890T2 (de) | Heilmittel/vorbeugemittel für erkrankungen der atemwege | |
| US4916137A (en) | 5-(Substituted-amino)-8-(phenyl or substituted-phenyl)-3H,6H-1,4,5A,8A-tetraazaacenaphthylen-3-ones and treatment of neural behavior disorders | |
| DE68911951T2 (de) | 5-(Substituierte Amino)-8-(phenyl oder substituierte phenyl)-3H,6H-1,4,5a,8a-tetraazaacenaphthylen-3-one. | |
| AU707626C (en) | Pyrrolopyrimidines and processes for the preparation thereof | |
| AT395156B (de) | Neue 9-deazaguanine | |
| HK1008222B (en) | Pyrrolopyrimidines and processes for the preparation thereof | |
| DE10122895A1 (de) | Neue Lactam-substituierte Pyrazolopyridinderivate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8328 | Change in the person/name/address of the agent |
Representative=s name: KROHER, STROBEL RECHTS- UND PATENTANWAELTE, 80336 |
|
| 8328 | Change in the person/name/address of the agent |
Representative=s name: DR. SCHOEN & PARTNER, 80336 MUENCHEN |