ES2786569T3 - Derivados de pirimidina para el tratamiento de infecciones víricas - Google Patents
Derivados de pirimidina para el tratamiento de infecciones víricas Download PDFInfo
- Publication number
- ES2786569T3 ES2786569T3 ES17203628T ES17203628T ES2786569T3 ES 2786569 T3 ES2786569 T3 ES 2786569T3 ES 17203628 T ES17203628 T ES 17203628T ES 17203628 T ES17203628 T ES 17203628T ES 2786569 T3 ES2786569 T3 ES 2786569T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- mmol
- compound
- alkoxy
- reduced pressure
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 208000036142 Viral infection Diseases 0.000 title description 6
- 230000009385 viral infection Effects 0.000 title description 6
- 150000003230 pyrimidines Chemical class 0.000 title description 5
- 229940083082 pyrimidine derivative acting on arteriolar smooth muscle Drugs 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 95
- 150000003839 salts Chemical class 0.000 claims abstract description 24
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 23
- 125000003118 aryl group Chemical group 0.000 claims abstract description 21
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 17
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 16
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 16
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 14
- 239000001257 hydrogen Substances 0.000 claims abstract description 13
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 13
- 239000012453 solvate Substances 0.000 claims abstract description 12
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 11
- 150000002367 halogens Chemical group 0.000 claims abstract description 11
- 125000003710 aryl alkyl group Chemical group 0.000 claims abstract description 10
- 125000004446 heteroarylalkyl group Chemical group 0.000 claims abstract description 10
- 150000002825 nitriles Chemical class 0.000 claims abstract description 8
- 125000001424 substituent group Chemical group 0.000 claims abstract description 7
- 150000001408 amides Chemical class 0.000 claims abstract description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims abstract description 6
- 125000003342 alkenyl group Chemical group 0.000 claims abstract description 5
- 125000000304 alkynyl group Chemical group 0.000 claims abstract description 5
- 125000002618 bicyclic heterocycle group Chemical group 0.000 claims abstract description 5
- 150000001733 carboxylic acid esters Chemical class 0.000 claims abstract description 5
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims abstract description 5
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims abstract description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims abstract description 4
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims abstract description 3
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims abstract description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims abstract description 3
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims abstract 6
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract 6
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims abstract 5
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims abstract 4
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 claims abstract 4
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims abstract 3
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims abstract 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 9
- -1 hydroxy, amino Chemical group 0.000 claims description 9
- 239000008194 pharmaceutical composition Substances 0.000 claims description 9
- 239000003814 drug Substances 0.000 claims description 8
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 6
- 150000002148 esters Chemical class 0.000 claims description 5
- 239000000969 carrier Substances 0.000 claims description 3
- 239000003085 diluting agent Substances 0.000 claims description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 2
- 150000002431 hydrogen Chemical group 0.000 claims description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 claims description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 abstract description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 123
- OKKJLVBELUTLKV-UHFFFAOYSA-N methanol Substances OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 100
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 90
- 230000002829 reductive effect Effects 0.000 description 72
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 69
- 239000002904 solvent Substances 0.000 description 68
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 50
- 239000000203 mixture Substances 0.000 description 47
- 239000000543 intermediate Substances 0.000 description 46
- 238000000034 method Methods 0.000 description 46
- 239000011541 reaction mixture Substances 0.000 description 46
- 238000002360 preparation method Methods 0.000 description 39
- 239000000243 solution Substances 0.000 description 38
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 36
- 239000007787 solid Substances 0.000 description 35
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 34
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 32
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 31
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 28
- 238000003756 stirring Methods 0.000 description 28
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 24
- 238000006243 chemical reaction Methods 0.000 description 24
- 210000004027 cell Anatomy 0.000 description 21
- 239000012043 crude product Substances 0.000 description 21
- 239000000706 filtrate Substances 0.000 description 21
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 20
- 238000005160 1H NMR spectroscopy Methods 0.000 description 18
- 239000003921 oil Substances 0.000 description 17
- 239000012071 phase Substances 0.000 description 17
- 239000000377 silicon dioxide Substances 0.000 description 17
- 102000002689 Toll-like receptor Human genes 0.000 description 16
- 108020000411 Toll-like receptor Proteins 0.000 description 16
- 238000001914 filtration Methods 0.000 description 16
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 15
- 238000010992 reflux Methods 0.000 description 15
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 14
- 239000012044 organic layer Substances 0.000 description 13
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 13
- 239000000047 product Substances 0.000 description 13
- 238000010898 silica gel chromatography Methods 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- 241000711549 Hepacivirus C Species 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- 239000012267 brine Substances 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 10
- 238000003556 assay Methods 0.000 description 9
- 238000000746 purification Methods 0.000 description 9
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 8
- 238000005481 NMR spectroscopy Methods 0.000 description 8
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 8
- 229910000024 caesium carbonate Inorganic materials 0.000 description 8
- 229940125904 compound 1 Drugs 0.000 description 8
- 239000003208 petroleum Substances 0.000 description 8
- 239000002244 precipitate Substances 0.000 description 8
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- 239000003039 volatile agent Substances 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 7
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 7
- 238000011534 incubation Methods 0.000 description 7
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 7
- 235000019341 magnesium sulphate Nutrition 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- 101000669402 Homo sapiens Toll-like receptor 7 Proteins 0.000 description 6
- 101000800483 Homo sapiens Toll-like receptor 8 Proteins 0.000 description 6
- 102000014150 Interferons Human genes 0.000 description 6
- 108010050904 Interferons Proteins 0.000 description 6
- 108060001084 Luciferase Proteins 0.000 description 6
- 239000005089 Luciferase Substances 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 230000004913 activation Effects 0.000 description 6
- 239000012298 atmosphere Substances 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- STIAPHVBRDNOAJ-UHFFFAOYSA-N carbamimidoylazanium;carbonate Chemical compound NC(N)=N.NC(N)=N.OC(O)=O STIAPHVBRDNOAJ-UHFFFAOYSA-N 0.000 description 6
- 239000002552 dosage form Substances 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 229940079322 interferon Drugs 0.000 description 6
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 5
- 241000282412 Homo Species 0.000 description 5
- 102100033110 Toll-like receptor 8 Human genes 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 230000010076 replication Effects 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 5
- 239000003643 water by type Substances 0.000 description 5
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- 0 COC(COCCOCc1ccccc1)=* Chemical compound COC(COCCOCc1ccccc1)=* 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- 102100039390 Toll-like receptor 7 Human genes 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 125000000753 cycloalkyl group Chemical group 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- 150000003840 hydrochlorides Chemical class 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 239000005695 Ammonium acetate Substances 0.000 description 3
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 3
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 3
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 235000019257 ammonium acetate Nutrition 0.000 description 3
- 229940043376 ammonium acetate Drugs 0.000 description 3
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 3
- 239000001099 ammonium carbonate Substances 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 239000000411 inducer Substances 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000012299 nitrogen atmosphere Substances 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 3
- 239000013612 plasmid Substances 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- 235000011152 sodium sulphate Nutrition 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- VCUXVXLUOHDHKK-UHFFFAOYSA-N 2-(2-aminopyrimidin-4-yl)-4-(2-chloro-4-methoxyphenyl)-1,3-thiazole-5-carboxamide Chemical compound ClC1=CC(OC)=CC=C1C1=C(C(N)=O)SC(C=2N=C(N)N=CC=2)=N1 VCUXVXLUOHDHKK-UHFFFAOYSA-N 0.000 description 2
- ZISWRXJZUKDIOO-UHFFFAOYSA-N 3-(3,4-dimethoxyphenyl)propan-1-ol Chemical compound COC1=CC=C(CCCO)C=C1OC ZISWRXJZUKDIOO-UHFFFAOYSA-N 0.000 description 2
- LHHKQWQTBCTDQM-UHFFFAOYSA-N 3-(3,4-dimethoxyphenyl)propanoic acid Chemical compound COC1=CC=C(CCC(O)=O)C=C1OC LHHKQWQTBCTDQM-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- OXAYXWLBMCQAJC-UHFFFAOYSA-N COc(cnc(N)n1)c1Cl Chemical compound COc(cnc(N)n1)c1Cl OXAYXWLBMCQAJC-UHFFFAOYSA-N 0.000 description 2
- 241000710188 Encephalomyocarditis virus Species 0.000 description 2
- 229920001917 Ficoll Polymers 0.000 description 2
- 108090000331 Firefly luciferases Proteins 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 239000012979 RPMI medium Substances 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 150000001350 alkyl halides Chemical class 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 230000000840 anti-viral effect Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 239000006143 cell culture medium Substances 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 230000001143 conditioned effect Effects 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- ZANNOFHADGWOLI-UHFFFAOYSA-N ethyl 2-hydroxyacetate Chemical compound CCOC(=O)CO ZANNOFHADGWOLI-UHFFFAOYSA-N 0.000 description 2
- XWRLQRLQUKZEEU-UHFFFAOYSA-N ethyl(hydroxy)silicon Chemical class CC[Si]O XWRLQRLQUKZEEU-UHFFFAOYSA-N 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 102000045715 human TLR7 Human genes 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- TVZNFYXAPXOARC-LURJTMIESA-N methyl (2s)-2-aminohexanoate Chemical compound CCCC[C@H](N)C(=O)OC TVZNFYXAPXOARC-LURJTMIESA-N 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 244000052769 pathogen Species 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 125000006413 ring segment Chemical group 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- NESLWCLHZZISNB-UHFFFAOYSA-M sodium phenolate Chemical compound [Na+].[O-]C1=CC=CC=C1 NESLWCLHZZISNB-UHFFFAOYSA-M 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 229960005486 vaccine Drugs 0.000 description 2
- ULAXUFGARZZKTK-YFKPBYRVSA-N (2s)-2-aminopentan-1-ol Chemical compound CCC[C@H](N)CO ULAXUFGARZZKTK-YFKPBYRVSA-N 0.000 description 1
- STPKWKPURVSAJF-LJEWAXOPSA-N (4r,5r)-5-[4-[[4-(1-aza-4-azoniabicyclo[2.2.2]octan-4-ylmethyl)phenyl]methoxy]phenyl]-3,3-dibutyl-7-(dimethylamino)-1,1-dioxo-4,5-dihydro-2h-1$l^{6}-benzothiepin-4-ol Chemical compound O[C@H]1C(CCCC)(CCCC)CS(=O)(=O)C2=CC=C(N(C)C)C=C2[C@H]1C(C=C1)=CC=C1OCC(C=C1)=CC=C1C[N+]1(CC2)CCN2CC1 STPKWKPURVSAJF-LJEWAXOPSA-N 0.000 description 1
- CPHLODVMQBMDNC-UHFFFAOYSA-N 1-(3-bromopropyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCCBr)C=C1 CPHLODVMQBMDNC-UHFFFAOYSA-N 0.000 description 1
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 1
- ZTHHRSBDBPCCMZ-UHFFFAOYSA-N 2,4-dichloro-5-methoxypyrimidine Chemical compound COC1=CN=C(Cl)N=C1Cl ZTHHRSBDBPCCMZ-UHFFFAOYSA-N 0.000 description 1
- YYRIKJFWBIEEDH-UHFFFAOYSA-N 2-(chloromethyl)-3,4-dimethoxypyridine;hydrochloride Chemical compound [Cl-].COC1=CC=[NH+]C(CCl)=C1OC YYRIKJFWBIEEDH-UHFFFAOYSA-N 0.000 description 1
- QWQSXGCANXFBCW-UHFFFAOYSA-N 2-amino-5-methoxy-1h-pyrimidin-6-one Chemical compound COC1=CN=C(N)NC1=O QWQSXGCANXFBCW-UHFFFAOYSA-N 0.000 description 1
- UNCQVRBWJWWJBF-UHFFFAOYSA-N 2-chloropyrimidine Chemical compound ClC1=NC=CC=N1 UNCQVRBWJWWJBF-UHFFFAOYSA-N 0.000 description 1
- HXDLWJWIAHWIKI-UHFFFAOYSA-N 2-hydroxyethyl acetate Chemical compound CC(=O)OCCO HXDLWJWIAHWIKI-UHFFFAOYSA-N 0.000 description 1
- HJBWJAPEBGSQPR-GQCTYLIASA-N 3,4-dimethoxycinnamic acid Chemical compound COC1=CC=C(\C=C\C(O)=O)C=C1OC HJBWJAPEBGSQPR-GQCTYLIASA-N 0.000 description 1
- DFRAKBCRUYUFNT-UHFFFAOYSA-N 3,8-dicyclohexyl-2,4,7,9-tetrahydro-[1,3]oxazino[5,6-h][1,3]benzoxazine Chemical compound C1CCCCC1N1CC(C=CC2=C3OCN(C2)C2CCCCC2)=C3OC1 DFRAKBCRUYUFNT-UHFFFAOYSA-N 0.000 description 1
- PXUYIYBCHHFVRG-UHFFFAOYSA-N 3-(3,4-dimethoxyphenyl)propyl methanesulfonate Chemical compound COC1=CC=C(CCCOS(C)(=O)=O)C=C1OC PXUYIYBCHHFVRG-UHFFFAOYSA-N 0.000 description 1
- JVVRCYWZTJLJSG-UHFFFAOYSA-N 4-dimethylaminophenol Chemical compound CN(C)C1=CC=C(O)C=C1 JVVRCYWZTJLJSG-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-dimethylaminopyridine Substances CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- GCNTZFIIOFTKIY-UHFFFAOYSA-N 4-hydroxypyridine Chemical compound OC1=CC=NC=C1 GCNTZFIIOFTKIY-UHFFFAOYSA-N 0.000 description 1
- SGEZKPNUNBVVLB-UHFFFAOYSA-N 5-(chloromethyl)-1,3-dimethylpyrazole Chemical compound CC=1C=C(CCl)N(C)N=1 SGEZKPNUNBVVLB-UHFFFAOYSA-N 0.000 description 1
- VKLKXFOZNHEBSW-UHFFFAOYSA-N 5-[[3-[(4-morpholin-4-ylbenzoyl)amino]phenyl]methoxy]pyridine-3-carboxamide Chemical compound O1CCN(CC1)C1=CC=C(C(=O)NC=2C=C(COC=3C=NC=C(C(=O)N)C=3)C=CC=2)C=C1 VKLKXFOZNHEBSW-UHFFFAOYSA-N 0.000 description 1
- GDUANFXPOZTYKS-UHFFFAOYSA-N 6-bromo-8-[(2,6-difluoro-4-methoxybenzoyl)amino]-4-oxochromene-2-carboxylic acid Chemical compound FC1=CC(OC)=CC(F)=C1C(=O)NC1=CC(Br)=CC2=C1OC(C(O)=O)=CC2=O GDUANFXPOZTYKS-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical class NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- CSJHICKJXRBPMF-HTLJXXAVSA-N CC(C)[C@@H](C(CCO)Nc(nc(N)nc1)c1OC)S Chemical compound CC(C)[C@@H](C(CCO)Nc(nc(N)nc1)c1OC)S CSJHICKJXRBPMF-HTLJXXAVSA-N 0.000 description 1
- QMVSBIUUYCRVJD-UHFFFAOYSA-N CC(NC(N)N=C1Cl)=C1OCc1ccccc1 Chemical compound CC(NC(N)N=C1Cl)=C1OCc1ccccc1 QMVSBIUUYCRVJD-UHFFFAOYSA-N 0.000 description 1
- KNFKQDPQYVMZIH-MLWJPKLSSA-N CCC(C)[C@H](CCO)N Chemical compound CCC(C)[C@H](CCO)N KNFKQDPQYVMZIH-MLWJPKLSSA-N 0.000 description 1
- GRCQAQGTLSELSK-GKAPJAKFSA-N CCC(C)[C@H](CCO)Nc1nc(N)ncc1OC Chemical compound CCC(C)[C@H](CCO)Nc1nc(N)ncc1OC GRCQAQGTLSELSK-GKAPJAKFSA-N 0.000 description 1
- MCJACENWCYIRFQ-UHFFFAOYSA-N CCCCNC(C(OCCCc(cc1OC)ccc1OC)=CN1)=NC1N=C Chemical compound CCCCNC(C(OCCCc(cc1OC)ccc1OC)=CN1)=NC1N=C MCJACENWCYIRFQ-UHFFFAOYSA-N 0.000 description 1
- NDXNPFNOMLJCJP-UHFFFAOYSA-N CCCCNc(nc(N)nc1)c1OCc1cccc(CO)c1 Chemical compound CCCCNc(nc(N)nc1)c1OCc1cccc(CO)c1 NDXNPFNOMLJCJP-UHFFFAOYSA-N 0.000 description 1
- CVPXLLWTDKCHGJ-UHFFFAOYSA-N CCCCNc1nc(NC(OC(C)(C)C)=O)ncc1OCc1cccc(CO)c1 Chemical compound CCCCNc1nc(NC(OC(C)(C)C)=O)ncc1OCc1cccc(CO)c1 CVPXLLWTDKCHGJ-UHFFFAOYSA-N 0.000 description 1
- IFOVMKVRTFAQOT-MRVPVSSYSA-N CCCC[C@@H](CC=C)N[S-]=O Chemical compound CCCC[C@@H](CC=C)N[S-]=O IFOVMKVRTFAQOT-MRVPVSSYSA-N 0.000 description 1
- QCJPDFURJLAZCH-VIFPVBQESA-N CCCC[C@@H](CO)Nc(nc(NC(C)=O)nc1)c1O Chemical compound CCCC[C@@H](CO)Nc(nc(NC(C)=O)nc1)c1O QCJPDFURJLAZCH-VIFPVBQESA-N 0.000 description 1
- SCJNYBYSTCRPAO-LXBQGUBHSA-N CN(C)C\C=C\C(=O)NC1=CC=C(N=C1)C(=O)N[C@@]1(C)CCC[C@H](C1)NC1=NC(C2=CNC3=CC=CC=C23)=C(Cl)C=N1 Chemical compound CN(C)C\C=C\C(=O)NC1=CC=C(N=C1)C(=O)N[C@@]1(C)CCC[C@H](C1)NC1=NC(C2=CNC3=CC=CC=C23)=C(Cl)C=N1 SCJNYBYSTCRPAO-LXBQGUBHSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- DNZWXXANFRPKDV-UHFFFAOYSA-N Cc1nc(N)nc(Cl)c1Oc1ccccc1 Chemical compound Cc1nc(N)nc(Cl)c1Oc1ccccc1 DNZWXXANFRPKDV-UHFFFAOYSA-N 0.000 description 1
- WIHUNEIESRQDTE-UHFFFAOYSA-N Cc1nc(N)nc(O)c1Oc1ccccc1 Chemical compound Cc1nc(N)nc(O)c1Oc1ccccc1 WIHUNEIESRQDTE-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 238000003512 Claisen condensation reaction Methods 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- HJBWJAPEBGSQPR-UHFFFAOYSA-N DMCA Natural products COC1=CC=C(C=CC(O)=O)C=C1OC HJBWJAPEBGSQPR-UHFFFAOYSA-N 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 101000852870 Homo sapiens Interferon alpha/beta receptor 1 Proteins 0.000 description 1
- 101000763579 Homo sapiens Toll-like receptor 1 Proteins 0.000 description 1
- 102100036714 Interferon alpha/beta receptor 1 Human genes 0.000 description 1
- 108010025815 Kanamycin Kinase Proteins 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 108010006444 Leucine-Rich Repeat Proteins Proteins 0.000 description 1
- 101100481581 Mus musculus Tlr13 gene Proteins 0.000 description 1
- JJSFFUAPEGTXSM-UHFFFAOYSA-N NC(N=C1O)=C[IH]C=C1OCCCOCc1ccccc1 Chemical compound NC(N=C1O)=C[IH]C=C1OCCCOCc1ccccc1 JJSFFUAPEGTXSM-UHFFFAOYSA-N 0.000 description 1
- FXXPDGSSPYTMKH-UHFFFAOYSA-N Nc(nc1)nc(O)c1OCc1ccccc1 Chemical compound Nc(nc1)nc(O)c1OCc1ccccc1 FXXPDGSSPYTMKH-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- 108700008625 Reporter Genes Proteins 0.000 description 1
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 229940124613 TLR 7/8 agonist Drugs 0.000 description 1
- 241001441723 Takifugu Species 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- 102100027010 Toll-like receptor 1 Human genes 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 108091023045 Untranslated Region Proteins 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000008484 agonism Effects 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- HEDRZPFGACZZDS-UHFFFAOYSA-N chloroform Substances ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 1
- GGRHYQCXXYLUTL-UHFFFAOYSA-N chloromethyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OCCl GGRHYQCXXYLUTL-UHFFFAOYSA-N 0.000 description 1
- 208000020403 chronic hepatitis C virus infection Diseases 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 239000003636 conditioned culture medium Substances 0.000 description 1
- 230000003750 conditioning effect Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- JZCCFEFSEZPSOG-UHFFFAOYSA-L copper(II) sulfate pentahydrate Chemical compound O.O.O.O.O.[Cu+2].[O-]S([O-])(=O)=O JZCCFEFSEZPSOG-UHFFFAOYSA-L 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 150000004292 cyclic ethers Chemical class 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- RDULEYWUGKOCMR-UHFFFAOYSA-N ethyl 2-chloro-3-oxobutanoate Chemical compound CCOC(=O)C(Cl)C(C)=O RDULEYWUGKOCMR-UHFFFAOYSA-N 0.000 description 1
- WUDNUHPRLBTKOJ-UHFFFAOYSA-N ethyl isocyanate Chemical compound CCN=C=O WUDNUHPRLBTKOJ-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- 102000045720 human TLR8 Human genes 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000015788 innate immune response Effects 0.000 description 1
- 210000005007 innate immune system Anatomy 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000004901 leucine-rich repeat Anatomy 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- DAZSWUUAFHBCGE-KRWDZBQOSA-N n-[(2s)-3-methyl-1-oxo-1-pyrrolidin-1-ylbutan-2-yl]-3-phenylpropanamide Chemical compound N([C@@H](C(C)C)C(=O)N1CCCC1)C(=O)CCC1=CC=CC=C1 DAZSWUUAFHBCGE-KRWDZBQOSA-N 0.000 description 1
- NTPKDNQDGWXUIY-UHFFFAOYSA-N n-hexyl-1h-1,2,4-triazole-5-carboxamide Chemical compound CCCCCCNC(=O)C1=NC=NN1 NTPKDNQDGWXUIY-UHFFFAOYSA-N 0.000 description 1
- PVWOIHVRPOBWPI-UHFFFAOYSA-N n-propyl iodide Chemical compound CCCI PVWOIHVRPOBWPI-UHFFFAOYSA-N 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 239000003961 penetration enhancing agent Substances 0.000 description 1
- HGBOYTHUEUWSSQ-UHFFFAOYSA-N pentanal Chemical compound CCCCC=O HGBOYTHUEUWSSQ-UHFFFAOYSA-N 0.000 description 1
- 229940083251 peripheral vasodilators purine derivative Drugs 0.000 description 1
- 210000001539 phagocyte Anatomy 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 210000005134 plasmacytoid dendritic cell Anatomy 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 125000000561 purinyl group Chemical class N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- STIKETVNLGXQCS-UHFFFAOYSA-N pyridazin-3-ylmethanol Chemical compound OCC1=CC=CN=N1 STIKETVNLGXQCS-UHFFFAOYSA-N 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000005494 pyridonyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229960000329 ribavirin Drugs 0.000 description 1
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 description 1
- 239000004544 spot-on Substances 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 239000012096 transfection reagent Substances 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 239000012646 vaccine adjuvant Substances 0.000 description 1
- 229940124931 vaccine adjuvant Drugs 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Un compuesto de fórmula (I) **(Ver fórmula)** o su sal, tautómero(s), solvato o polimorfos farmacéuticamente aceptable, en donde R1 es hidrógeno, alquilo C1-4, ciclopropilo, alcoxi C1-6, halógeno, hidroxilo o trifluorometilo, R2 es alquilo C1-8, alcoxi (C1-4)-alquilo (C1-4), cicloalquilo C3-7, heterociclo C4-7, heterociclo bicíclico, arilalquilo, heteroarilo o heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido con uno o más sustituyentes independientemente seleccionados de halógeno, hidroxilo, amino, alquilo C1-6, di-alquilamino (C1-6), alquilamino C1-6, alquilo C1-6, alcoxi C1-6, cicloalquilo C3-6, ácido carboxílico, éster carboxílico, amida carboxílica, heterociclo, arilo, alquenilo, alquinilo, arilalquilo, heteroarilo, heteroarilalquilo, nitrilo; y R3 es alquilo C4-8, alcoxi C4-8, alquenilo C2-6 o alquinilo C2-6, cada uno de los cuales está opcionalmente sustituido con uno o más sustituyentes independientemente seleccionados de halógeno, hidroxilo, amino, alquilo C1-3, alcoxi C1-3, cicloalquilo C3-6 y nitrilo.
Description
DESCRIPCIÓN
Derivados de pirimidina para el tratamiento de infecciones víricas
La presente invención se refiere a derivados de pirimidina, procedimientos para su preparación, composiciones farmacéuticas y su uso en el tratamiento de infecciones víricas, como VHB o VHC.
La presente invención se refiere al uso de derivados de pirimidina en el tratamiento de infecciones víricas, trastornos inmunitarios o inflamatorios, en los cuales está implicada la modulación o el agonismo de receptores de tipo toll (TLR). Los receptores de tipo toll son proteínas transmembrana primarias caracterizadas por un dominio rico en leucina extracelular y una extensión citoplásmica que contiene una región conservada. El sistema inmunitario innato puede reconocer patrones moleculares asociados con patógenos mediante estos TLR expresados en la superficie celular de ciertos tipos de células inmunitarias. El reconocimiento de patógenos extraños activa la producción de citocinas y el aumento de la regulación de moléculas co-estimuladores en fagocitos. Esto conduce a la modulación de la conducta de las células T.
Se ha estimado que la mayoría de las especies mamíferas tienen entre diez y quince tipos de receptores de tipo toll. Se han identificado trece TLR (llamados TLR1 a TLR13) en seres humanos y ratones, y se han hallado formas equivalentes de muchos de estos en otras especies mamíferas. No obstante, los equivalentes de ciertos TLR hallados en seres humanos no están presentes en todos los mamíferos. Por ejemplo, un gen que codifica una proteína análoga a TLR10 en seres humanos está presente en ratones, pero parece haber sido dañado en algún punto en el pasado por un retrovirus. Por otra parte, los ratones expresan los TLR 11, 12 y 13, ninguno de los cuales está representado en seres humanos. Otros mamíferos pueden expresar TLR que no se hallan en seres humanos. Otras especies no mamíferas pueden tener TLR distintos de los de mamíferos, como lo demuestra el TLR14, que se encuentra en el pez globo Takifugu. Esto puede complicar el proceso de usar animales experimentales como modelos de inmunidad innata humana.
Para revisiones detalladas de receptores de tipo toll, ver los siguientes artículos científicos. Hoffmann, J.A., Nature, 426, pág. 33-38, 2003; Akira, S., Takeda, K., y Kaisho, T., Annual Rev. Immunology, 21, pág. 335-376, 2003; Ulevitch, R. J., Nature Reviews: Immunology, 4, pág. 512-520, 2004.
Los compuestos que indican actividad en receptores de tipo toll se han descrito previamente, como los derivados de purina en el documento WO 2006/117670, derivados de adenina en los documentos WO 98/01448 y WO 99/28321, y pirimidinas en el documento WO 2009/067081.
No obstante, existe una fuerte necesidad de nuevos moduladores de los receptores de tipo toll que tengan selectividad preferida, mayor potencia, mayor estabilidad metabólica y un mejor perfil de seguridad que los compuestos de la técnica anterior.
En el tratamiento de determinadas infecciones víricas se pueden administrar inyecciones regulares de interferón (IFNa), como en el caso del virus de hepatitis C (VHC), (Fried et. al. Peginterferon-alfa plus ribavirin for chronic hepatitis C virus infection, N Engl J Med 2002; 347: 975-82). Los inductores de IFN de moléculas pequeñas disponibles por vía oral ofrecen ventajas potenciales de inmunogenicidad reducida y conveniencia de administración. Por lo tanto, los nuevos inductores de IFN son una nueva clase de fármacos potencialmente eficaz para tratar infecciones víricas. Para un ejemplo en la bibliografía de un inductor de IFN de moléculas pequeñas que tiene efecto antivírico, véase De Clercq, E.; Descamps, J.; De Somer, P. Science 1978, 200, 563-565.
IFNa es también administrado en combinación con otros fármacos en el tratamiento de ciertos tipos de cáncer (Eur. J. Cancer 46, 2849-57, y Cancer Res. 1992, 52, 1056). Los agonistas de TLR 7/8 son también de interés como adyuvantes de vacunas debido a su capacidad de inducir una respuesta de Th1 pronunciada (Hum. Vaccines 2010, 6, 1-14; Hum. Vaccines 2009, 5, 381-394).
De acuerdo con la presente invención, se da a conocer un compuesto de fórmula (I)

o una de sus sales, tautómeros, solvatos o polimorfos farmacéuticamente aceptables, en donde
R1 es hidrógeno, alquilo C1-4, ciclopropilo, alcoxi C1-6, halógeno, hidroxilo o trifluorometilo,
R2 es alquilo C1-8, alcoxi (C1-4)-alquilo (C1-4), cicloalquilo C3-7, heterociclo C4-7, heterociclo bicíclico, arilalquilo, heteroarilo, o heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido con uno o más sustituyentes independientemente seleccionados entre halógeno, hidroxilo, amino, alquilo C1-6, di-alquilamino (C1-6), alquilamino
Ci -6, alquilo Ci -6, alcoxi Ci -6, cicloalquilo C3-6, ácido carboxílico, éster carboxílico, amida carboxílica, heterociclo, arilo, alquenilo, alquinilo, arilalquilo, heteroarilo, heteroarilalquilo, nitrilo; y
R3 es alquilo C4-8, alcoxi C4-8, alquenilo C2-6 o alquinilo C2-6, cada uno de los cuales está opcionalmente sustituido con uno o más sustituyentes independientemente seleccionados de halógeno, hidroxilo, amino, alquilo C1-3, alcoxi C1-3, cicloalquilo C3-6 y nitrilo:
En una primera realización, la presente invención proporciona compuestos de fórmula (I) en donde R3 es butilo o pentilo y en donde R2 y Ri son como se ha especificado antes.
En una realización adicional, la invención se refiere a compuestos de fórmula (I) en donde R3 es alquilo C4-8 sustituido con hidroxilo, y en donde R2 y R1 son como se han especificado antes.
Otra realización se refiere a compuestos de fórmula (I) en donde R3 , cuando es alquilo C4-8 sustituido con hidroxilo, es uno de los siguientes

Además, la presente invención también proporciona compuestos de fórmula (I), en donde R1 es hidrógeno o -CH3 y en donde R2 y R3 son como se ha especificado antes.
En otra realización de la presente invención se dan a conocer compuestos de fórmula (I) en donde R2 es arilalquilo o heteroarilalquilo, sustituido con alquilo C1-3, hidroxilo, alcoxi, nitrilo, heterociclo o éster y en donde R1 y R3 son como se mencionó anteriormente.
Otra realización de la presente invención se refiere a compuestos de fórmula (I) en donde R2 es alquilo C1-3 sustituido con arilo, heterociclo o heteroarilo que está además sustituido con alquilo C1-3, alcoxi, éster carboxílico o amida carboxílica y en donde R1 y R3 son como se definió anteriormente
Asimismo, la invención se refiere a compuestos de fórmula (I) en donde R2 es uno de los siguientes ejemplos que pueden además sustituirse con alquilo C1-3, hidroxilo, alcoxi, nitrilo, heterociclo o éster.

Los compuestos preferidos de acuerdo con la invención son:

Los compuestos de fórmula (I) y su sal, tautómero(s), solvato o polimorfos farmacéuticamente aceptables tienen actividad como productos farmacéuticos, en particular como moduladores de receptores de tipo toll (especialmente TLR7 y/o TLR8).
En otro aspecto, la presente invención da a conocer una composición farmacéutica que comprende un compuesto de fórmula (I) o su sal, solvato o polimorfos farmacéuticamente aceptable junto con uno o más excipientes, diluyentes o vehículos farmacéuticamente aceptables.
Asimismo, un compuesto de fórmula (I) o su sal, solvato o polimorfos farmacéuticamente aceptable de acuerdo con la presente invención, o una composición farmacéutica que comprende dicho compuesto de fórmula (I) o su sal, solvato o polimorfos farmacéuticamente aceptables, se puede usar como medicamento.
El compuesto de fórmula (I) o su sal, solvato o polimorfos farmacéuticamente aceptable, o dicha composición farmacéutica que comprende dicho compuesto de fórmula (I) o su sal, solvato o polimorfos farmacéuticamente aceptable se puede usar en el tratamiento de un trastorno o enfermedad en donde está implicada la modulación de TLR7 y/o TLR8.
El término "alquilo" se refiere a un hidrocarburo alifático saturado de cadena lineal o de cadena ramificada que contiene el número especificado de átomos de carbono.
El término "halógeno" se refiere a flúor, cloro, bromo o yodo.
El término "alquenilo" se refiere a un alquilo como se definió anteriormente, que consiste en por lo menos dos átomos de carbono y por lo menos un doble enlace carbono-carbono.
El término "alquinilo" se refiere a un alquilo como se definió anteriormente, que consiste en por lo menos dos átomos de carbono y por lo menos un triple enlace carbono-carbono.
El término "cicloalquilo" se refiere a un anillo carbocíclico que contiene el número especificado de átomos de carbono.
El término "heteroarilo" significa una estructura de anillo aromático según se define para el término "arilo" que comprende por lo menos 1 heteroátomo seleccionado entre N, O y S, en particular entre N y O.
El término "arilo" significa una estructura de anillo aromático que opcionalmente comprende uno o dos heteroátomos seleccionados entre N, O y S, en particular entre N y O. Dicha estructura de anillo aromático puede tener 4, 5, 6 o 7 átomos del anillo. En particular, dicha estructura de anillo aromático puede tener 5 o 6 átomos del anillo.
El término "heterociclo bicíclico" significa una estructura de anillo aromático, según se define para el término "arilo" comprendida por dos anillos aromáticos condensados. Cada anillo está opcionalmente comprendido por heteroátomos seleccionados entre N, O y S, en particular entre N y O.
El término arilalquilo" significa una estructura de anillo aromático como se define para el término "arilo" opcionalmente sustituida con un grupo alquilo.
El término "heteroarilalquilo" significa una estructura de anillo aromático según se define para el término "heteroarilo" opcionalmente sustituida con un grupo alquilo.
El término "alcoxi" se refiere a un grupo alquilo (cadena de carbono e hidrógeno) singular unido a oxígeno como por ejemplo un grupo metoxi o un grupo etoxi.
Heterociclo se refiere a moléculas que están saturadas o parcialmente saturadas e incluyen óxido de etilo, tetrahidrofurano, dioxano u otros éteres cíclicos. Los heterociclos que contienen nitrógeno incluyen, por ejemplo, azetidina, morfolina, piperidina, piperazina, pirrolidina y similares. Otros heterociclos incluyen, por ejemplo, tiomorfolina, dioxolinilo y sulfonas cíclicas.
Los grupos heteroarilo son grupos heterocíclicos que son aromáticos por naturaleza. Estos son heteroátomos monocíclicos, bicíclicos o policíclicos que contienen uno o más heteroátomos seleccionados entre N, O S. Los grupos heteroarilo pueden ser, por ejemplo, imidazolilo, isoxazolilo, furilo, oxazolilo, pirrolilo, piridonilo, piridilo, piridazinilo o pirazinilo.
Las sales farmacéuticamente aceptables de los compuestos de fórmula (I) incluyen las sales de adición con ácido y sus sales de base. Las sales de adición con ácido adecuadas se forman a partir de ácidos que forman sales no tóxicas. Las sales de base adecuadas se forman a partir de bases que forman sales no tóxicas.
Los compuestos de la invención pueden también existir en formas no solvatadas y solvatadas. El término "solvato" se usa en la presente invención para describir un complejo molecular que comprende el compuesto de la invención y una o más moléculas disolventes farmacéuticamente aceptables, por ejemplo, etanol.
El término "polimorfismo" se refiere a la capacidad del compuesto de la invención de existir en más de una forma o estructura cristalina.
Los compuestos de la presente invención se pueden administrar como productos cristalinos o amorfos. Se pueden obtener por ejemplo como tapones sólidos, polvos o películas por métodos tales como precipitación, cristalización, liofilización, secado por atomización o secado evaporativo. Se pueden administrar solos o combinados con uno o más de otros compuestos de la invención o en combinación con uno o más de otros fármacos. En general, se administrarán como una formulación en asociación con uno o más excipientes farmacéuticamente aceptables. El
término "excipiente" se usa en la presente invención para describir cualquier ingrediente distinto del compuesto(s) de la invención. La elección del excipiente depende en gran medida de factores tales como el modo de administración particular, el efecto del excipiente sobre la solubilidad y la estabilidad, y la naturaleza de la forma farmacéutica.
Los compuestos de la presente invención o cualquiera de sus subgrupos se pueden formular en diversas formas farmacéuticas para propósitos de administración. Como composiciones apropiadas se pueden citar todas las composiciones usualmente empleadas para fármacos de administración sistémica. Para preparar las composiciones farmacéuticas de la presente invención, se combina una cantidad eficaz del compuesto particular, opcionalmente en la forma de sal de adición, como el ingrediente activo en una mezcla íntima con un vehículo farmacéuticamente aceptable, en donde el vehículo puede adoptar una gran variedad de formas dependiendo de la forma de preparación deseada para administración. Estas composiciones farmacéuticas son convenientemente en forma de dosis unitaria adecuada, por ejemplo, para administración oral, rectal o percutánea. Por ejemplo, para preparar las composiciones en una presentación oral, se puede emplear cualquier medio farmacéutico habitual tal como, por ejemplo, agua, glicoles, aceites, alcoholes y similares en el caso de preparaciones líquidas orales tales como suspensiones, jarabes, elixires, emulsiones y disoluciones; o vehículos sólidos tales como almidones, azúcares, caolín, diluyentes, lubricantes, aglutinantes, disgregantes y similares en el caso de polvos, pastillas, cápsulas y comprimidos. Debido a su facilidad de administración, los comprimidos y las cápsulas representan las formas farmacéuticas unitarias orales más ventajosas, en cuyo caso se emplean obviamente los vehículos farmacéuticos sólidos. Además, se incluyen las preparaciones en forma sólida que se pueden convertir, justo antes de usar, en formas líquidas. En las composiciones adecuadas para administración percutánea, el vehículo opcionalmente comprende un agente potenciador de penetración y/o un agente humectante adecuado, opcionalmente combinado con aditivos adecuados de cualquier naturaleza en proporciones menores, en donde los aditivos no introducen un efecto perjudicial importante sobre la piel. Dichos aditivos pueden facilitar la administración a la piel y/o pueden ser útiles para preparar las composiciones deseadas. Estas composiciones se pueden administrar de diversas formas, p. ej., como un parche transdérmico, como un spot-on, como un ungüento. Los compuestos de la presente invención pueden además administrarse por inhalación o insuflación a través de métodos y formulaciones empleados en la técnica para administrar de esta manera. Por lo tanto, en general, los compuestos de la presente invención se pueden administrar a los pulmones en la forma de una disolución, una suspensión o un polvo seco.
Es especialmente ventajoso formular las composiciones farmacéuticas anteriormente mencionadas en forma farmacéutica unitaria para facilidad de administración y uniformidad de la dosis. La forma farmacéutica unitaria, tal como se usa en la presente memoria, se refiere a unidades físicamente discretas adecuadas como dosis unitarias, en donde cada unidad contiene una cantidad predeterminada de ingrediente activo calculada para producir el efecto terapéutico deseado en asociación con el vehículo farmacéutico requerido. Los ejemplos de dichas formas farmacéuticas unitarias son comprimidos (incluidos comprimidos ranurados o recubiertos), cápsulas, pastillas, bolsitas de polvo, obleas, supositorios, disoluciones inyectables o suspensiones y similares, y sus múltiples segregados.
Los expertos en el tratamiento de enfermedades infecciosas serán capaces de determinar la cantidad eficaz a partir de los resultados de los ensayos presentados en lo sucesivo. En general, se contempla que una cantidad diaria eficaz sería entre 0,01 mg/kg y 50 mg/kg de peso corporal, más preferiblemente entre 0,1 mg/kg y 10 mg/kg de peso corporal. Puede ser adecuado administrar la dosis requerida como dos, tres, cuatro o más sub-dosis en intervalos apropiados a lo largo del día. Dichas sub-dosis se pueden formular como formas farmacéuticas unitarias, por ejemplo, que contienen entre 1 y 1000 mg, y en particular entre 5 y 200 mg de ingrediente activo por forma farmacéutica unitaria.
La dosis y la frecuencia exactas de administración dependen del compuesto particular de fórmula (I) utilizado, la afección particular que se esté tratando, la gravedad de la afección que se esté tratando, la edad, el peso y el estado físico general del paciente particular, así como también otros medicamentos que el individuo pueda estar tomando, como conocen los expertos en la técnica. Asimismo, es obvio que la cantidad eficaz puede disminuirse o aumentarse dependiendo de la respuesta del sujeto tratado y/o dependiendo de la evaluación del médico que prescribe los compuestos de la presente invención. Los intervalos de cantidades eficaces anteriormente mencionados son por consiguiente solamente pautas y no tienen como fin limitar el alcance o el uso de la invención en grado alguno.
Preparación de compuestos
Los compuestos de fórmula (I), en donde R1 es un átomo de hidrógeno, se preparan de acuerdo con el esquema 1.

(i) Reacción de un alcohol heterocíclico con un éster halogenado y una base adecuada, por ejemplo, carbonato de potasio, carbonato de cesio o hidruro de sodio. El ejemplo se muestra en el esquema 2a.
(ii) Reacción de un alcohol, o hidroxi-éster, por ejemplo acetato de 2-hidroxi-etilo, con un haluro de alquilo usando una base apropiada, por ejemplo hidruro de sodio. El ejemplo se muestra en el esquema 2b.
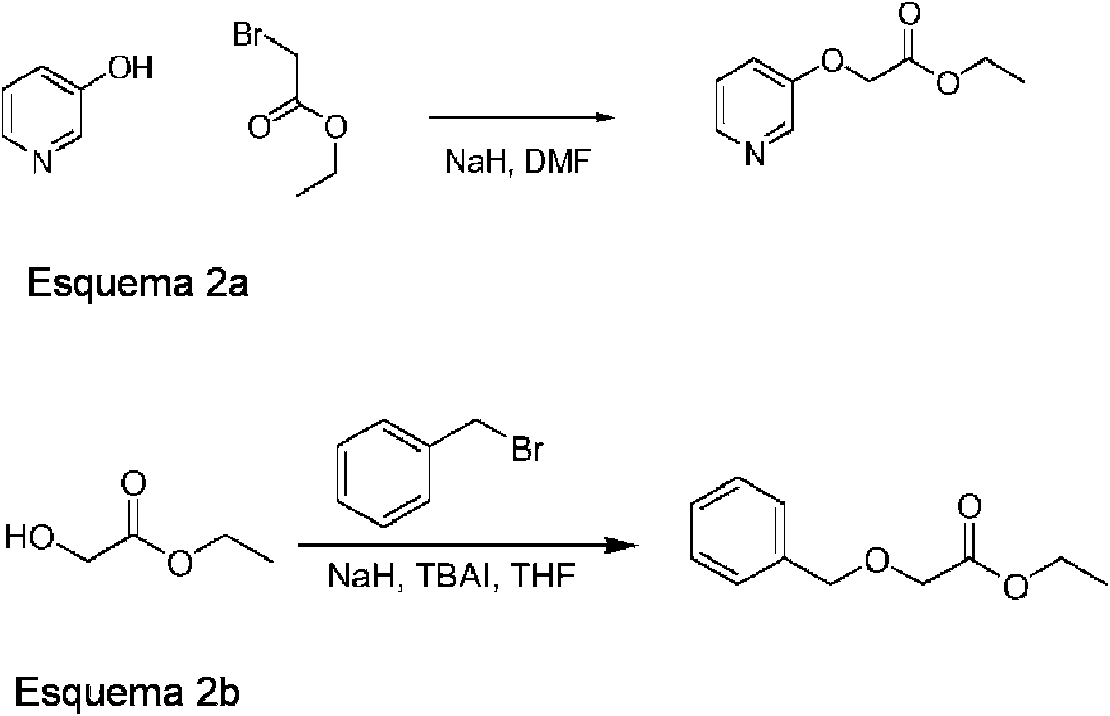
Los compuestos de fórmula (I), cuando R1 es alquilo, cicloalquilo, trifluorometilo o alcoxi y donde R2 es arilo o heteroarilo, se preparan como en el esquema 3 a continuación. El betacetoéster (E) se puede clorar usando, por ejemplo, cloruro de tionilo para proporcionar el 2-cloro-beta-cetoéster intermedio (F). El fenol o alcohol heteroaromático (R2OH) se combina con una relación equimolar de hidróxido de sodio acuoso. Los disolventes luego se eliminan a presión reducida para proporcionar la sal de fenol o alcohol heteroaromático de R2. Esta sal se combina con el 2-cloro-p-cetoéster intermedio (F) para dar el intermedio G de acuerdo con el procedimiento de la bibliografía. El intermedio G se combina entonces, con o sin base, con carbonato de guanidina en un disolvente apropiado, por ejemplo, etanol. Luego se somete a reacción el intermedio H con oxicloruro de fósforo para formar cloropirimidina intermedia (J). Los productos se forman entonces como consecuencia del calentamiento (J) en presencia de exceso de amina y opcionalmente exceso de base orgánica, por ejemplo trietilamina, a temperatura elevada. Este es un esquema general que emplea métodos conocidos para el experto, véase, por ejemplo, Organic Syntheses volumen 33, pág. 43 (1953).

Los compuestos de fórmula (I), cuando Ri es alquilo, cicloalquilo, trifluorometilo o alcoxi y donde R2 es aromático o alifático, se pueden preparar de acuerdo con el esquema 4. Este esquema de reacción comienza con una reacción de Claisen cruzada donde un cloruro de acilo reacciona con el éster intermedio A (se muestra en el esquema 1) para formar los intermedios (G) como en el esquema 3. A partir del intermedio G, el esquema de reacción sigue la misma ruta que los productos del esquema 3. Este es un esquema general que emplea métodos conocidos por el experto en la técnica, véase, por ejemplo, The Journal of American Chemical Society volumen 127, página 2854 (2005). Sección experimental.
Síntesis del intermedio A-1.

A una mezcla de glicolato de etilo [623-50-7] (250,00 g, 2,40 mol), NaH (105,65 g, 2,64 mol), yoduro de tetrabutilamonio (TBAI) (88,70 g, 240,14 mmol) en Th F anhidro (2 l) se le añadió bromuro de bencilo (451,80 g, 2,64 mol) gota a gota a 0°C. La mezcla resultante se agitó a 25°C durante 16 horas. La mezcla de reacción se inactivó con cloruro de amonio acuoso saturado (1 L), y la capa acuosa se extrajo con acetato de etilo (3 x 1 L). Las capas
orgánicas combinadas se lavaron con salmuera (1 L), se secaron sobre sulfato de magnesio, los sólidos se eliminaron por filtración, y los disolventes del filtrado se concentraron a presión reducida. El residuo se purificó por cromatografía en columna de gel de sílice (éter de petróleo: acetato de etilo = 6:1) para obtener el intermedio A-1 (200 g).
1H RMN (CDCl3400 MHz) 5 ppm 7,37-7,27 (m, 5H); 4,62 (s, 2H), 4,24-4,19 (q, J = 6,8 Hz, 2H); 4,07 (s, 2H); 1,29 1,25 (t, J = 6,8 Hz, 3H).
Procedimiento para la preparación del intermedio B-1.

A una suspensión agitada de NaH (45,30 g, 1,13 mol) en THF anhidro (1,2 L) se le añadió formiato de etilo (114,42 g, 1,54 mol). La suspensión se enfrió en un baño de hielo y luego se añadió gota a gota el compuesto A-1 (200 g, 1,03 mol) en THF anhidro (300 ml) con un embudo de adición. La mezcla blanca se agitó a 0°C hasta temperatura ambiente durante 5 horas. Durante este periodo, la reacción fue exotérmica y se tornó amarilla. En un matraz separado, se trató carbonato de guanidina [593-85-1] (111,31 g, 0,618 mol) con disolución de etóxido sódico, recién preparada por adición cautelosa de Na (28,41 g, 1,24 mol) a etanol anhidro (750 ml) a temperatura ambiente. La suspensión blanquecina obtenida después de agitar durante 1 hora se añadió a la disolución amarilla anteriormente preparada. La mezcla de reacción amarilla pálida resultante se calentó hasta reflujo durante 15 horas. El disolvente se eliminó y el residuo bruto se disolvió en agua (1,5 L). La mezcla se ajustó hasta pH=5 con ácido acético. El sólido se recogió, se lavó abundantemente con agua y etanol para dar el intermedio B-1 (160 g).
1H RMN (400 MHz, DMSO-ofe) 5 ppm 4,90 (s, 2 H), 6,33 (s a, 2 H), 7,25 (s, 1 H), 7,29 - 7,42 (m, 5 H), 11,21 (s a, 1 H) Procedimiento para la preparación del intermedio C-1.

Una suspensión del intermedio B-1 (160 g, 0,74 mol) en POCh (900 ml) se calentó hasta 100°C bajo N2 agitando durante 5 horas. La mezcla de reacción se enfrió hasta temperatura ambiente. Se eliminó el exceso de POCh a presión reducida, se vertió el residuo oleoso en NaHCO3 sat. acuoso frío (2 L) que se agitó durante 30 minutos. La mezcla se extrajo con acetato de etilo (3 x 1,5 L). Las capas orgánicas combinadas se separaron y lavaron con salmuera (1 L), se secaron sobre sulfato de sodio, los sólidos se eliminaron por filtración y los disolventes del filtrado se concentraron para dar el intermedio C-1 (70 g) en la forma de un sólido amarillo. El producto se usó en la etapa siguiente sin purificación adicional.
Procedimiento para la preparación del compuesto 1

A una suspensión de C-1 (70,00 g, 297,03 mmol) en etanol (1,4 L) se añadió n-butilamina (217,24 g, 2,97 mol) y trietilamina (60,11 g, 594,05 mmol). La mezcla de reacción se calentó a reflujo durante 16 horas. La mezcla de reacción se enfrió a temperatura ambiente y los disolventes se separaron a presión reducida. El residuo se purificó por cromatografía en gel de sílice ultrarrápida usando un gradiente de éter de petróleo a acetato de etilo para obtener el compuesto 1 (26 g) en forma de un sólido amarillo.
1H RMN (400 MHz, METANOL-C4) 5 ppm 0,96 (t, J=7,3 Hz, 3 H), 1,32 - 1,43 (m, 2 H), 1,52 - 1,61 (m, 2 H), 3,38 (t, J=7,2 Hz, 2 H), 5,01 (s, 2 H), 7,28 (s, 1 H), 7,31 - 7,46 (m, 5 H)

Preparación de intermedio D-1.
En un matraz de fondo redondo de 100 ml equipado con una barra agitadora magnética se puso el compuesto 1 (1 g, 3,67 mmol) en ácido acético (40 ml). La disolución amarilla se dejó agitar a reflujo durante 15 horas. Se eliminaron los disolventes a presión reducida. El producto puro se purificó por cromatografía en gel de sílice usando un gradiente de heptano a acetato de etilo. Se recogieron las mejores fracciones y los disolventes se eliminaron a presión reducida para dar un sólido blanco, D-1.
LC-MS: Anal. Calc. para C19H24N4O3 : 356,19; encontrado 357[M+H]+
1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 0,94 (t, J=7,4 Hz, 3 H), 1,31 - 1,45 (m, 2 H), 1,50 - 1,67 (m, 2 H), 2,31 (s, 6 H), 3,44 (m, J=6,0 Hz, 2 H), 5,12 (s, 2 H), 5,41 - 5,52 (m, 1 H), 7,43 (m, J=1,5 Hz, 5 H), 7,79 (s, 1 H) Preparación de intermedio D-2.

Método A. En un matraz Erlenmeyer de 250 ml equipado con una barra agitadora magnética se puso D-1 (1 g), y etanol (100 ml). Se pasa un chorro de nitrógeno por el matraz, seguido de la adición de Pd sobre carbono al 10% (100 mg). El matraz se cerró herméticamente y se eliminó la atmósfera y se sustituyó por hidrógeno. La reacción se dejó agitar a temperatura ambiente durante 15 horas. La mezcla heterogénea se filtró a través de Celite empaquetada y los disolventes del filtrado se separaron a presión reducida para dar D-2 con rendimiento cuantitativo.
Método B. Una disolución 0,1 M del material de partida en metanol se trató en e1H-cube, equipado con un cartucho de Pd/C al 10%, a 0,5 ml/min y 30 bar de presión de hidrógeno. El análisis por LC-m S mostró la conversión completa. Los disolventes se separaron a presión reducida. El producto bruto se purificó por cromatografía en gel de sílice usando un gradiente de diclorometano a metanol en diclorometano al 10%. Se mezclaron las mejores fracciones; los disolventes se eliminaron a presión reducida para dar un sólido blanco, D-2.
LC-MS: Anal. Calc. para C12H18N4O3 : 266,14; encontrado 267[M+H]+
1H RMN (400 MHz, DMSO-de) 5 ppm 0,87 (t, J=7,4 Hz, 3 H), 1,28 (dd, J=14,9, 7,4 Hz, 2 H), 1,49 (t, J=7,2 Hz, 2 H), 2,15 (s, 6 H), 3,20 - 3,37 (m, 2 H), 7,02 - 7,12 (m, 1 H), 7,58 (s, 1 H), 10,27 (s ancho, 1 H)
Preparación de intermedio D-3.

En un matraz de fondo redondo de 100 ml se puso el compuesto 1 (1 g, 3,67 mmol), dicarbonato de di-terc-butilo (7,5 g) y acetonitrilo (50 ml). La disolución amarilla se agitó a reflujo durante 16 horas. Los disolventes se eliminaron a presión reducida. El residuo se purificó por cromatografía en sílice usando una columna de sílice de 80 g preempaquetada y un gradiente de heptano a acetato de etilo con recolección automática a 254 nm. Las mejores fracciones se combinaron para dar un aceite amarillo, D-3.
LC-MS: Anal. Calc. para C25H36N4O5: 472,269; encontrado 473[M+H]+
1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 0,94 (t, J=7,4 Hz, 3 H), 1,33 - 1,42 (m, 2 H), 1,46 (s, 18 H), 1,50 - 1,65 (m, 2 H), 3,35 - 3,51 (m, 2 H), 5,09 (s, 2 H), 5,31 - 5,38 (m, 1 H), 7,36 - 7,48 (m, 5 H), 7,75 (s, 1 H)
Preparación de intermedio D-4.
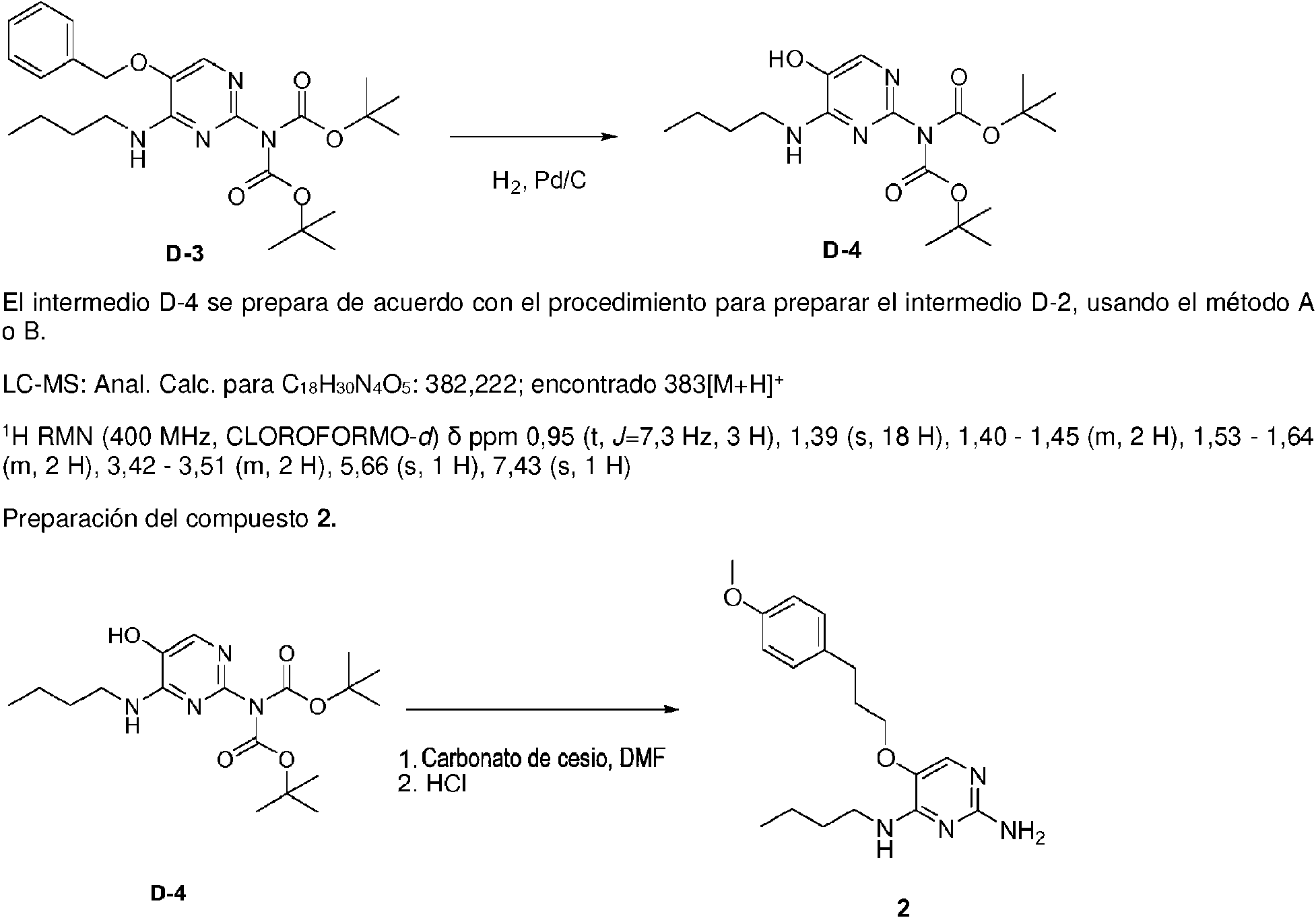
En un vial de 30 ml se puso el intermedio D-4 (200 mg, 0,52 mmol), DMF (5 ml), 1-(3-bromopropil)-4-metoxibenceno (130 mg, 0,57 mmol), y carbonato de cesio (508 mg, 1,56 mmol). La reacción se dejó agitar durante 15 horas a temperatura ambiente. Los sólidos se separaron por filtración. Los disolventes del filtrado se eliminaron a presión reducida y el producto bruto se reconstituyó en metanol y se le añadió HCl (6 M en isopropanol) y la reacción se dejó agitar 15 horas a temperatura ambiente. Los disolventes se eliminaron a presión reducida y el producto bruto se purificó por separación en fase inversa para dar el compuesto 2 como la base libre.
Preparación de intermedio G-1.

A una disolución agitada de A-1 (60 g, 309 mmol, 1 eq) y 1-metilimidazol (30,4 g, 370 mmol, 1,2 eq) en CH2Cl2 (1 L) se añadió cloruro de acetilo (24,3 g, 309 mmol, 1 eq) a -45°C en atmósfera de N2. Después de agitar durante 20 min, se añadieron TiCl4 (210 g, 1,08 mol, 3,5 eq) y tributilamina (230 g, 1,24 mol, 4 eq) a la mezcla a -45°C en atmósfera
de N2 , y se continúa agitando durante 50 minutos a -45°C en atmósfera de N2. Después de completarse, se añadieron agua y acetato de etilo. La capa orgánica se separó y la capa acuosa se extrajo con acetato de etilo dos veces. La capa orgánica se lavó con salmuera y se secó sobre sulfato de sodio. Los sólidos se separaron por filtración y los disolventes del filtrado se eliminaron a presión reducida. El producto bruto se purificó por cromatografía en columna de sílice usando un gradiente de heptano a acetato de etilo para dar un aceite amarillo pálido, G-1.
1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 1,30 (t, J=7,2 Hz, 3 H), 2,28 (s, 3 H), 4,27 (q, J=7,2 Hz, 2 H), 4,41 (s, 1 H), 4,58 (d, J=11,8 Hz, 1 H), 4,75 (d, J=11,8 Hz, 1 H), 7,32 - 7,43 (m, 5 H)
Preparación de intermedio H-1.

En un vial de microondas de 20 ml se puso el intermedio G-1 (500 mg, 2,12 mmol), etanol (5 ml), y carbonato de guanidina (200 mg, 2,22 mmol). El vial se cerró herméticamente y se dejó reaccionar a 120°C con agitación durante 4 horas. Los disolventes se separaron a presión reducida. Se añadió agua (25 ml). La mezcla se llevó a pH=5 por adición cuidadosa de ácido acético. El precipitado se aisló por filtración para dar un sólido blanco, H-1.
1H RMN (400 MHz, CLOROFORMO-a) 5 ppm 1,88 (s, 3 H), 4,85 (s, 2 H), 6,38 (s ancho, 2 H), 7,24 - 7,49 (m, 5 H), 11,16 (s, 1 H)
Preparación de intermedio G-2.

Etapa 1. Se preparó fenolato de sodio por evaporación de porciones equimolares de fenol e hidróxido de sodio en un matraz de fondo redondo de 1 litro en el rotavapor. Se usa tolueno en la separación azeotrópica de agua.
Etapa 2. El fenolato de sodio (116 g, 1 mol) preparado en la etapa 1 y tolueno (1 L) se pusieron en un matraz de tres bocas de 2 L equipado con agitador mecánico, embudo de adición y refrigerante de reflujo con tubo de secado. La suspensión se calentó a reflujo, después se añadió a-cloroacetoacetato de etilo (165 g, 1 mol) con agitación a través del embudo de adición donde la reacción se continúa calentando a reflujo durante 4 horas. La suspensión marrón claro se enfría a temperatura ambiente, se extrae con agua (2 x 500 ml), y se seca (sulfato magnésico anhidro). Los sólidos se separan por filtración y los disolventes del filtrado se separan a presión reducida. El producto bruto se usa en la siguiente etapa sin purificación.
Preparación de intermedio H-2.
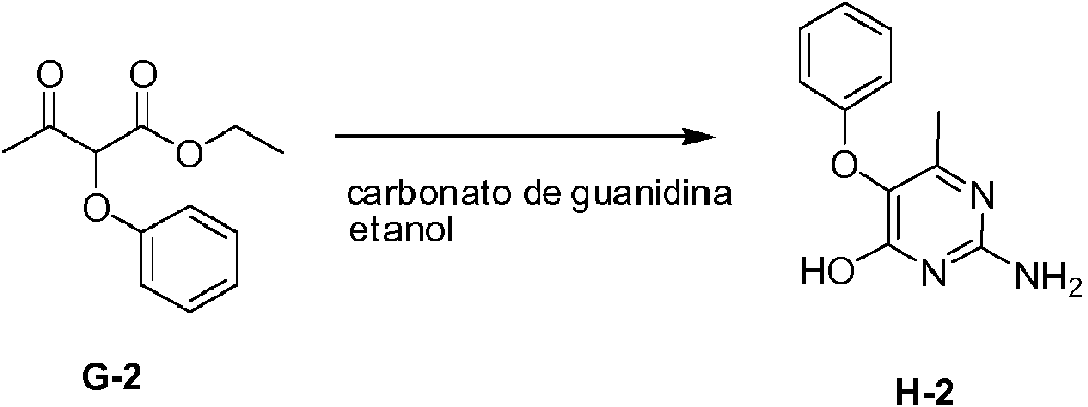
En un matraz de fondo redondo de 100 ml equipado con una barra agitadora magnética y refrigerante de reflujo se añadió el intermedio G-2 (1 g, 4,5 mmol), etanol (50 ml), y carbonato de guanidina [593-85-1](203 mg, 2,25 mmol). La mezcla de reacción se llevó a reflujo durante 15 horas. El disolvente se separó a presión reducida. Se añadió agua (25 ml). La mezcla se llevó a pH=5 por adición cuidadosa de ácido acético. El precipitado se aisló por filtración para dar un sólido blanco, H-2. Este se usa sin más purificación en la siguiente etapa.
Preparación de intermedio J-1.

En un matraz de fondo redondo de 50 ml equipado con una barra agitadora magnética y refrigerante de reflujo, se añadió el intermedio H-2 (500 mg, 2,3 mmol) y POCh (20 ml). La suspensión se calentó a reflujo con agitación durante 6 horas. Los disolventes se separaron a presión reducida para dar un aceite marrón bruto, J-1. No se hizo más purificación. El compuesto se usó como estaba en la siguiente etapa.
Preparación del compuesto 3.

En un tubo herméticamente cerrado de 50 ml con una barra agitadora magnética se puso el intermedio J-1 (150 mg, 0,64 mmol), n-butilamina (70 mg, 0,96 mmol), alúmina básica (100 mg), y dioxano (10 ml). El tubo se cerró herméticamente, se puso en un baño de aceite a 120°C, y la reacción se calentó con agitación durante 15 horas. El recipiente se enfrió a temperatura ambiente y se retiró con cuidado la tapa. El contenido se vertió en un matraz de fondo redondo donde los disolventes se separaron a presión reducida. El producto bruto se purificó por cromatografía en columna de gel de sílice usando un gradiente de diclorometano a metanol en diclorometano al 5%. Las mejores fracciones se combinaron, y los disolventes se separaron a presión reducida para dar el compuesto 3.
LC-MS: Anal. Calc. para C15H20N4O: 272,16; encontrado 273 [M+H]+
1H RMN (300 MHz, CLOROFORMO-d) 5 ppm 0,80 (t, J=7,3 Hz, 3 H), 1,20 (dq, J=15,0, 7,3 Hz, 2 H), 1,33 - 1,47 (m, 2 H), 1,98 (s, 3 H), 3,20 - 3,34 (m, 2 H), 4,74 (s ancho, 2 H), 4,79 (s ancho, 1 H), 6,78 - 6,84 (m, 2 H), 6,91 - 7,01 (m, 1 H), 7,18 - 7,28 (m, 2 H)
Preparación del compuesto 4

Etapa 1.
En un vial de microondas de 20 ml se añadió 2,4-dicloro-5-metoxi-pirimidina (300 mg, 1,68 mmol) disponible en el mercado, etanol (5 ml) y n-butilamina (0,166 ml, 1,68 mmol). El vial se cierra herméticamente y después se calienta en el microondas durante 10 minutos a 120°C. El análisis de LC-MS muestra conversión completa. Los disolventes se separaron a presión reducida. El producto bruto se usa como está en la etapa 2.
Etapa 2.
El compuesto de la etapa 1 se puso en un recipiente con presión de 20 ml con amoniaco acuoso (10 ml) y se le añadió bicarbonato amónico (200 mg, 2,6 mmol) y CuO (24 mg, 0,17 mmol, 0,1eq). El recipiente se cerró herméticamente y la mezcla se calentó a 120°C con agitación durante 24 horas. La mezcla de reacción se extrajo 3
veces con 5 ml de diclorometano:metanol 9:1 y los productos volátiles se separaron a presión reducida. El compuesto se filtró a través de sílice eluyendo con diclorometano:metanol 9:1 y los compuestos volátiles se separaron a presión reducida. El residuo se purificó por cromatografía de fase inversa.
LC/MS: Anal. Calc. para C9H16N4O: 196,13; encontrado 197[M+H]+
1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 0,97 (t, J=7,3 Hz, 3 H), 1,35 - 1,48 (m, 2 H), 1,56 - 1,68 (m, 2 H), 3,44 - 3,52 (m, 2 H), 3,80 (s, 3 H), 5,86 (s, 1 H), 5,97 (s, 2 H), 7,07 - 7,14 (m, 1 H)
Preparación del compuesto 5.
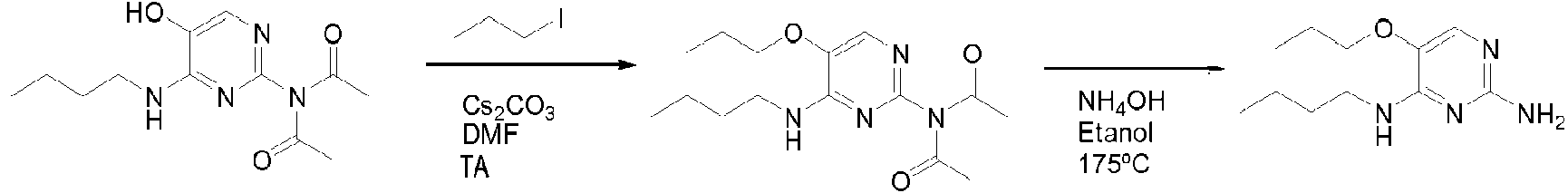
D-2 5
Etapa 1.
En un tubo de ensayo de 16x100 se puso el intermedio D-2 (180 mg, 0,66 mmol), DMF (5 ml), yoduro de propilo (111 mg, 0,656 mmol), y carbonato de cesio (320 mg, 0,98 mmol). La reacción se dejó agitar a temperatura ambiente durante 15 horas. Los sólidos se separaron por filtración y los disolventes del filtrado se separaron a presión reducida. El producto bruto se purificó por cromatografía en gel de sílice usando un gradiente de diclorometano a metanol en diclorometano al 10%. Las mejores fracciones se combinaron, los disolventes se separaron a presión reducida para dar un sólido blanco.
Etapa 2.
En un vial de microondas de 10 ml se puso el sólido blanco anterior (100 mg), hidróxido de amonio (1 ml) y etanol (1 ml). El vial se cerró herméticamente y se calentó con agitación a 175°C durante 10 minutos. El análisis de LC-MS mostró conversión completa al producto. Los disolventes se separaron a presión reducida. El producto bruto se purificó por cromatografía de gel de sílice usando un gradiente de diclorometano a metanol en diclorometano al 10%. Las mejores fracciones se combinaron, los disolventes se separaron a presión reducida para dar un aceite incoloro. La adición de un equivalente de HCl (usando HCl en isopropanol de 5 a 6 N) da un sólido blanco, 5.
LC/MS: Anal. Calc. para C11H20N4O: 224,16; encontrado 225[M+H]+
1H RMN (400 MHz, DMSO-afe) 5 ppm 0,90 (t, J=7,3 Hz, 3 H), 0,98 (t, J=7,4 Hz, 3 H), 1,20 - 1,35 (m, 2 H), 1,54 (t, J=7,2 Hz, 2 H), 1,69 - 1,75 (m, 2 H), 3,40 (d, J=7,0 Hz, 2 H), 3,87 (t, J=6,5 Hz, 2 H), 7,39 (d, J=5,5 Hz, 1 H), 7,46 (s ancho, 2 H), 8,28 - 8,37 (m, 1 H)
Esquema sintético para la preparación de AA-9

A una disolución de valeraldehído (43 g, 500 mmol) en THF (1 L) se le añadió AA-2 (200 g, 532 mmol) y la mezcla de reacción se agito durante 16 horas a temperatura ambiente. Los disolventes se evaporaron y el residuo se diluyó en éter de petróleo y se filtró. Los disolventes del filtrado se eliminaron a presión reducida y el residuo se purificó por cromatografía en sílice, usando un gradiente de éter de petróleo a acetato de etilo en éter de petróleo al 3% para dar AA-3 (90 g) en la forma de un aceite incoloro.
1H RMN (400 MHz, CDCla): 5 ppm 6,81-6,77 (m, 1H), 5,68-5,64 (td, J=1,2Hz, 15,6 Hz, 1H), 2,11-2,09 (m, 2H), 1,406 (s, 9H), 1,38-1,26(m, 4H), 0,85-0,81 (t, J=7,2Hz, 3H).
Síntesis del compuesto AA-5

Se añadió n-butil-litio (290 ml, 725 mmol, 1,5 eq.) a una disolución agitada de AA-4 (165 g, 781 mmol) en THF (800 ml) a -78°C. La mezcla de reacción se agitó durante 30 minutos, luego se añadió AA-3 (90 g, 488,4 mmol) en THF
(400 ml) y la reacción se agitó durante 2 horas a -78°C. La mezcla se inactivó con disolución saturada acuosa de NH4CI y se calentó hasta temperatura ambiente. El producto se repartió entre acetato de etilo y agua. La fase orgánica se lavó con salmuera, se secó y se evaporó. El residuo se purificó por cromatografía en columna eluyendo con acetato de etilo en éter de petróleo al 5% para dar un aceite incoloro, AA-5 (132 g).
1H RMN (400 MHz, CDCh ): 5 ppm 7,36-7,16 (m, 10H), 3,75-3,70 (m, 2H), 3,43-3,39 (d, J=15,2Hz, 1H), 3,33-3,15 (m, 1H), 1,86-1,80 (m, 2H), 1,47-1,37 (m, 2H), 1,32 (s, 9H), 1,26-1,17 (m, 7H), 0,83-0,79 (t, J=7,2Hz, 3H).
Síntesis de AA-6

Se disolvió AA-5 (130 g, 328 mmol) en THF (1,5 L) y se añadió LAH (20 g, 526 mmol) a 0°C en pequeñas porciones. La mezcla resultante se agitó a la misma temperatura durante 2 horas y luego se dejó calentar hasta temperatura ambiente. La mezcla se inactivó con una disolución saturada acuosa de NH4CL El producto se repartió entre acetato de etilo y agua. La fase orgánica se lavó con salmuera, se secó y se evaporó. Las capas orgánicas combinadas se secaron sobre sulfato de sodio, los sólidos se eliminaron por filtración y se concentraron para dar AA-6 bruto (100 g), que se usó en la etapa siguiente sin purificación adicional.
1H RMN (400 MHz, CDCh ): 5 ppm 7,33-7,14 (m, 10H), 3,91-3,86 (m, 1H), 3,80-3,77 (d, J=13,6Hz, 1H), 3,63-3,60 (d, J=13,6Hz, 1 H), 3,43-3,42 (m, 1 H), 3,15-3,10 (m, 1H), 2,70-2,63 (m, 2H), 1,65-1,28 (m, 10H), 0,89-0,81 (m, 3H). Síntesis de AA-9

Una disolución de AA-6 (38 g, 116,75 mmol) y 10% Pd/C en metanol (200 ml) se hidrogenó bajo 50 PSI de hidrógeno a 50°C durante 24 horas. La mezcla de reacción se filtró y el disolvente se evaporó para dar el producto bruto AA-7 (17 g).
El producto bruto se disolvió en diclorometano (200 ml), se añadieron trietilamina (26,17 g, 259,1 mmol) y dicarbonato de di-terc-butilo (84,7 g, 194,4 mmol) a 0°C. La mezcla resultante se agitó a temperatura ambiente durante 16 horas. La mezcla se repartió entre diclorometano y agua. La fase orgánica se lavó con salmuera, se secó y se evaporó. El residuo se purificó por cromatografía de gel de sílice eluyendo con 20% acetato de etilo en éter de petróleo al 20% para dar AA-8 (13 g) en la forma de un aceite incoloro.
1H RMN (400 MHz, CDCh ): 5 ppm 4,08-4,03 (a, 1H), 3,68 (m, 1H), 3,58-3,55 (m, 2H), 3,20-2,90 (a, 1H), 1,80-1,73 (m, 1 H), 1,42-1,17 (m, 15 H), 0,85-0,82 (t, J=6,8 Hz, 3H).
Se disolvió AA-8 (42 g, 0,182 mol) en dioxano (200 ml) y se añadió dioxano/HCl (4 M, 200 ml) a 0°C. La mezcla resultante se agitó a temperatura ambiente durante 2 h. El disolvente se evaporó para dar el producto bruto. Se
añadió una mezcla de diclorometano/éter de petróleo (50 ml, 1:1, v/v) al producto bruto y se decantó el líquido sobrenadante. Este procedimiento se repitió dos veces para obtener un aceite, AA-9 (26,6 g).
1H RMN (400 MHz, DMSO-ofe): 5 ppm 8,04 (s, 3H), 3,60-3,49 (m, 2H), 3,16-3,15 (m, 1H), 1,71-1,67 (m, 2H), 1,60-1,55(m, 2H), 1,33-1,26 (m, 4H), 0,90-0,87 (t, J=6,8Hz, 3H).
Preparación de AA-10

Etapa 1. Se disolvió ácido 3,4-dimetoxicinámico (5 g, 24 mmol) en THF (100 ml). Se añadió níquel Raney a esta disolución en una atmósfera de N2. La mezcla de reacción se expuso a una atmósfera de hidrógeno y se agitó 15 horas a temperatura ambiente. La mezcla de reacción se filtró sobre un cartucho empaquetado con tierra de diatomeas y el disolvente del filtrado se eliminó a presión reducida. El residuo se usó como estaba en la siguiente etapa.
LC-MS: Anal. Calc. para C11 H14O4 : 210,09; encontrado 209[M-H]

Etapa 2. Se disolvió ácido 3-(3,4-dimetoxifenil)propanoico en THF (100 ml). Se añadió complejo de boranodimetilsulfóxido (2 M en éter dietílico, 20 ml, 40 mmol). La mezcla de reacción se agitó durante la noche a temperatura ambiente. Se añadió metanol lentamente para inactivar la mezcla de reacción, después se añadió sílice y los productos volátiles se separaron a presión reducida. El residuo se purificó en sílice usando un gradiente de heptano a acetato de etilo, dando el producto en forma de un aceite. Este se usó como estaba en la siguiente etapa. LC-MS: Anal. Calc. para C11 H16O3 : 196,11; encontrado 195[M-H]

Etapa 3. Se disolvieron 3-(3,4-dimetoxifenil)propan-1-ol (3,8 g, 19,5 mmol) y trietilamina (3,8 ml, 27,3 mmol) en acetonitrilo (15 ml) y después se añadió cloruro de metanosulfonilo (1,5 ml, 19,5 mmol). La mezcla de reacción se agitó durante la noche a temperatura ambiente. Los compuestos volátiles se separaron a presión reducida y el residuo se purificó por cromatografía en gel de sílice usando un gradiente de heptano a acetato de etilo dando el producto en forma de un aceite transparente.
1H RMN (400 MHz, DMSO-ofe) 5 ppm 1,91 - 2,01 (m, 2 H), 2,58 - 2,64 (m, 2 H), 3,17 (s, 3 H), 3,72 (s, 3 H), 3,75 (s, 3 H), 4,19 (t, J=6,4 Hz, 2 H), 6,71 - 6,76 (m, 1 H), 6,81 - 6,89 (m, 2 H)

Etapa 4. Una disolución de D-4 (400 mg, 1 mmol), carbonato de cesio (511 mg, 1,6 mmol) y metanosulfonato de 3-(3,4-dimetoxifenil)propilo (430 mg, 1,6 mmol) en acetona (50 ml) se calentó a 50°C durante 15 horas. La mezcla de reacción se puso en una centrífuga y el líquido sobrenadante se decantó y después se evaporó hasta sequedad. El residuo se purificó por cromatografía en columna de sílice usando un gradiente de heptano a acetato de etilo. Las fracciones que contenían el producto se combinaron y los disolventes se eliminaron a presión reducida para dar D-5.
LC-MS: Anal. Calc. para C29H44N4O7: 560,2; encontrado 561 [M+H]+

Etapa 5. El compuesto protegido con boc se disolvió en diclorometano (5 ml) y se añadió HCl 6 M en isopropanol (3 ml). La mezcla de reacción se agitó 15 horas a temperatura ambiente (5 ml) y se formó un precipitado, el compuesto 74 se aisló por filtración y después se secó en el horno con vacío durante 15 horas.
Preparación del compuesto 75

Etapa 1. Se preparó el intermedio B-2 de acuerdo con el método descrito para la preparación del intermedio B-1. Etapa 2. A una disolución de B-2 (1 g, 3,62 mmol) y DBU (5,4 ml, 36 mmol) en acetonitrilo (20 ml) se le añadió BOP (2,08 g, 4,71 mmol) y la mezcla de reacción se tornó transparente y se agitó durante 15 minutos a temperatura ambiente. Se añadió AA-9 (910 mg, 5,43 mmol) y la mezcla de reacción se agitó durante 2 días a 50°C. Los volátiles se eliminaron a presión reducida y el residuo se purificó sobre sílice usando un gradiente de diclorometano a metanol en diclorometano. Las mejores fracciones se combinaron y los disolventes se eliminaron a presión reducida. El bruto se reconstituyó en diclorometano (2 ml), luego se añadió HCl en éter dietílico para formar la sal de HCl. El precipitado se aisló por filtración y se secó en la estufa de vacío para proporcionar el compuesto 75.
Preparación de 76

Etapa 1. Se disolvieron C-1 (2 g, 8,49 mmol), L-norvalinol (1,75 g, 17 mmol) y diisopropiletilamina (5,85 ml, 34 mmol) en acetonitrilo (200 ml) en un recipiente a presión recubierto con teflón de 500 ml y se calentó hasta 130°C durante 15 horas. La mezcla se dejó enfriar hasta temperatura ambiente, los volátiles se eliminaron a presión reducida y el bruto se purificó por cromatografía en columna de gel de sílice usando un gradiente de diclorometano a metanol en diclorometano al 10%. Las mejores fracciones se combinaron y los disolventes se eliminaron a presión reducida para proporcionar el intermedio D-6.
LC-MS: Anal. calc. para C16H22N4O2: 302,17; encontrado 303 [M+H]+
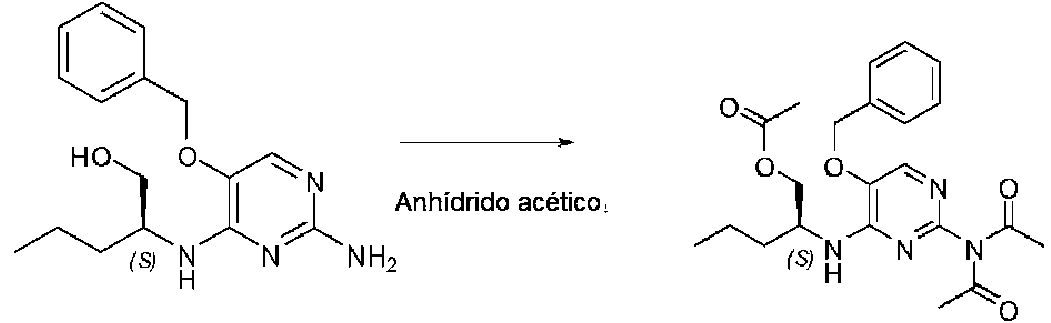
Etapa 2. Se calentó D-6 (2 g, 6,61 mmol) a reflujo en anhídrido acético (100 ml) en un matraz con fondo redondo de 250 ml durante 4 horas. Los volátiles se eliminaron a presión reducida y el residuo se purificó por cromatografía en columna de gel de sílice usando un gradiente de heptano a acetato de etilo produciendo un aceite amarillo, D-7. LC-MS: Anal. Calc. para C22H28N4O5: 428,21; observado 429 [M+H]+

Etapa 3. Se preparó D-8 de acuerdo con el método para preparar el intermedio D-2.
LC-MS: Anal. Calc. para C15H22N4O5 : 338.,16; observado 339 [M+H]+

Etapa 4. El intermedio D-9 se preparó de acuerdo con el método descrito en el ejemplo 75 a partir del intermedio D-4.
LC-MS: Anal. Calc. para C15H22N4O5 : 338,16; observado 339 [M+H]+
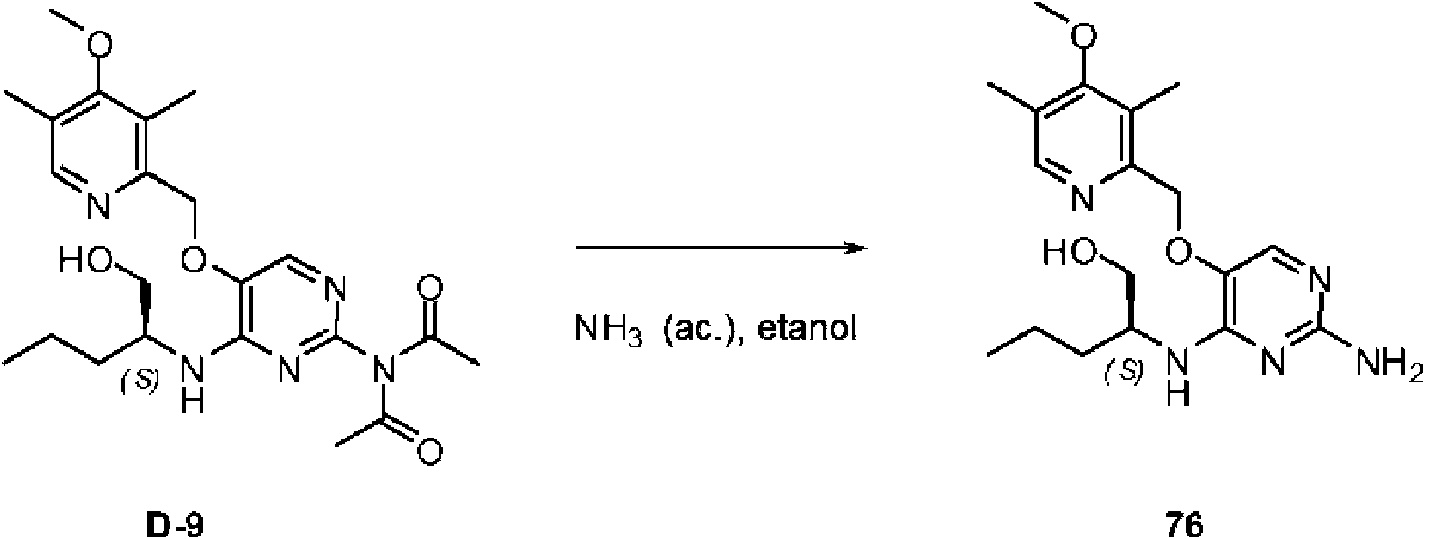
Etapa 5. La desprotección de D-9 se efectuó de acuerdo con el método descrito en la etapa 2 del compuesto 5 para dar 76.
Preparación del compuesto 77

Etapa 1. D-10 se preparó a partir de D-4 de acuerdo con el método para preparar el ejemplo 5, purificación por columna en sílice con gradiente de heptano a acetato de etilo.
LC-MS: Anal. Calc. para C27H38N4O7 : 530,27; observado 531 [M+H]+
1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 0,93 (t, J=7,3 Hz, 3 H), 1,37 (dd, J=14,9, 7,4 Hz, 2 H), 1,53 - 1,62 (m, 2 H), 3,40 - 3,50 (m, 2 H), 3,92 - 3,95 (m, 3 H), 5,13 (s, 2 H), 5,33 (s, 1 H), 7,46 - 7,52 (m, 1 H), 7,56 - 7,62 (m, 1 H), 7,73 (s, 1 H), 8,05 (dt, J=7,7, 1,4 Hz, 1 H), 8,09 (d, J=1,5 Hz, 1 H)

Etapa 2. D-10 (2,14 g, 3,91 mmol) se disolvió en THF anhidro (250 ml). Se añadió gota a gota hidruro de litio y aluminio (1 M en THF, 5,87 ml, 5,87 mmol) y la mezcla de reacción se agitó durante 3 horas a temperatura ambiente. Se añadió gota a gota NH4Cl (sat., ac.) a la mezcla de reacción y las sales precipitadas se separaron por filtración y se lavaron con THF. El filtrado se evaporó hasta sequedad y el producto bruto D-11 se usó como estaba en la siguiente etapa.
LC-MS: Anal. Calc. para C21H30N4O4 : 402,23; observado 403 [M+H]+

Etapa 3. D-11 (1,57 g, 3,91 mmol) se disolvió en diclorometano (20 ml) y se le añadió HCl (6 M en isopropanol, 50 ml). La mezcla de reacción se agitó durante 16 horas a temperatura ambiente. Los compuestos volátiles se separaron a presión reducida y el compuesto bruto se purificó por columna en sílice usando un gradiente de diclorometano a diclorometano en metanol al 10% dando el compuesto 77 en forma de un aceite que solidificó al reposar.

Etapa 1. Una disolución de D-4 (0,5 g, 1,31 mmol), 3-piridazinilmetanol (158 mg, 1,44 mmol) y trifenilfosfina (377 mg, 1,44 mmol) en THF anhidro (4 ml) se enfrió a 0°C y se añadió gota a gota una disolución de DIAD (0,28 ml, 1,44 mmol) a 0°C. Después de la adición, la mezcla de reacción se agitó durante 3 horas a temperatura ambiente. El disolvente se inactivó con agua (10 ml), se agitó durante 10 minutos y los compuestos volátiles se separaron a presión reducida. La capa de agua se extrajo con diclorometano, las capas orgánicas se combinaron y el disolvente se separó a presión reducida. El compuesto bruto se purificó por cromatografía en columna de gel de sílice usando gradiente de heptano a acetato de etilo. Las mejores fracciones se combinaron, los disolventes se separaron a presión reducida para dar D-12.
LC-MS: Anal. Calc. para C23H34N6O5: 474,26; observado 475 [M+H]+

Etapa 2. D-11 (620 mg, 1,31 mmol) se disolvió en diclorometano (10 ml) y se le añadió HCl (6 M en isopropanol, 10 ml). La mezcla de reacción se agitó durante 15 horas a temperatura ambiente. Los compuestos volátiles se separaron a presión reducida y el residuo se purificó por cromatografía de fase inversa para dar el compuesto 78.
Preparación de 79

Etapa 1. En un matraz de 500 ml se calentó una mezcla de B-1 (30 g, 138 mmol) y ácido sulfúrico (3 ml) en anhídrido acético (300 ml) a 90°C durante 3 horas. La reacción se enfrió hasta temperatura ambiente y el precipitado se aisló por filtración, se lavó con éter diisopropílico y se secó al vacío a 50°C para obtener un sólido blanco, B-5.

Etapa 2. En un reactor Multimax de 400 ml se agitó una mezcla de B-5 (21,8 g, 84 mmol) en acetonitrilo (244 ml) a 30°C bajo una corriente moderada de nitrógeno. Se añadió cloruro de fosforilo (18,14 ml, 195 mmol) gota a gota en un periodo de 5 minutos. Después de la adición, la mezcla de reacción se calentó hasta 45°C y la mezcla se agitó durante 15 minutos, luego se añadió lentamente DIPEA (33 ml, 195 mmol) en un periodo de 1,5 horas. La reacción se agitó a 45°C hasta completarse (monitoreada por LC-MS). Se calentó una disolución de etanoato de sodio (65 g) en agua (732 ml) en un matraz de 2 L hasta 35°C y la mezcla de reacción se repartió en esta disolución en un periodo de 5 minutos. La temperatura se mantuvo entre 35-40°C con un baño de enfriamiento externo. La mezcla se dejó alcanzar temperatura ambiente y se siguió agitando por durante 1 hora. El precipitado se aisló por filtración, se lavó con agua y se secó al vacío a 50°C para obtener C-2 en la forma de un sólido.
LC-MS: Anal. Calc. para C13H12ClN3O2: 277,06; observado 278 [M+H]+
1H RMN (400 MHz, DMSO-ds) 5 ppm 2,11 (s, 3 H), 5,31 (s, 2 H), 7,33 - 7,39 (m, 1 H), 7,43 (t, J=7,2 Hz, 2 H), 7,46 -7,51 (m, 2 H), 8,59 (s, 1 H), 10,65 (s, 1 H)

Etapa 3. Una disolución del intermedio C-2 (5,9 g, 21,2 mmol), (2S)-2-aminohexanoato de metilo (5,79 g, 31,9 mmol) y trietilamina (14,8 ml, 106 mmol) en acetonitrilo (100 ml) se calentó a reflujo durante 4 días. La mezcla de reacción se enfrió hasta temperatura ambiente y el disolvente se eliminó a presión reducida. El residuo se disolvió en diclorometano y se lavó con salmuera. La capa orgánica se secó (sulfato de magnesio), luego se purificó directamente mediante una columna de sílice usando un gradiente de diclorometano a metanol en diclorometano. Las mejores fracciones se combinaron y los disolventes se eliminaron a presión reducida para dar D-13.
LC-MS: Anal. Calc. para C20H26N4O4: 386,20; observado 387 [M+H]+
Etapa 2. Se disolvió D-13 (3,7 g, 9,57 mmol) en THF anhidro (100 ml). Se añadió gota a gota hidruro de aluminio y litio (1 M en THF, 9,6 ml, 9,6 mmol) y la mezcla de reacción se agitó por 3 horas a temperatura ambiente. Se añadió NH4Cl (sat., ac.) gota a gota a la mezcla de reacción y las sales precipitadas se eliminaron por filtración y se lavaron con THF. El filtrado se evaporó a sequedad y el residuo se purificó por cromatografía en columna de gel de sílice usando un gradiente de diclorometano a metanol en diclorometano al 10%. Las mejores fracciones se combinaron y los disolventes se eliminaron a presión reducida para dar D-14.
LC-MS: Anal. Calc. para C19H26N4O3: 358,20; observado 359 [M+H]+

Etapa 3. Se preparó D-15 de acuerdo con el método descrito para el intermedio D-2. Se usó sin purificación en la etapa siguiente.
LC-MS: Anal. Calc. para C12H20N4O3 : 268,15; observado 269 [M+H]+

Etapa 4. Una mezcla de D-15 (210 mg, 0,78 mmol) y carbonato de cesio (765 mg, 2,35 mmol) en DMF (25 ml) se calentó hasta 60°C con agitación y luego se añadió gota a gota una disolución de 5-(clorometil)-1,3-dimetil-1H-pirazol (113 mg, 0,78 mmol) en DMF (10 ml). La mezcla de reacción se agitó durante 1 hora a 60°C. Los sólidos se eliminaron por filtración y el disolvente se eliminó a presión reducida. Se usó D-16 bruto como tal en la siguiente etapa.
LC-MS: Anal. Calc. para C18H28N6O3: 376,22; observado 377 [M+H]+

Etapa 5. En un tubo de vidrio de 30 ml se dispusieron D-16 (295 mg, 0,78 mmol) y NaOCH3 (30% en metanol, 2 ml) y metanol (20 ml), y la mezcla se agitó a 60°C durante la noche. La mezcla de reacción se purificó por cromatografía de líquidos de fase inversa (Sunfire Prep C18 OBD 10mm, 30 x 150 mm. Fase móvil disolución de NH4OAc al 0,25% en agua, metanol) para proporcionar 79 como la base libre.
Preparación de 80

Etapa 1. Se preparó el intermedio D-17 de acuerdo con el método utilizado para D-16 por alquilación de D-15.
LC-MS: Anal. Calc. para C19H24N6O3: 384,19; observado 385 [M+H]+

Etapa 2. En un tubo de vidrio de 30 ml se disolvieron D-17 (301 mg, 0,78 mmol) y NaOCH3 (30% en metanol, 2 ml) en metanol (20 ml) y se agitó a 60°C durante la noche. Se añadieron 10 ml de agua a la mezcla de reacción y se agitó durante 2 horas a 60°C. La mezcla de reacción se purificó por cromatografía de líquidos de fase inversa (Sunfire Prep C18 OBD 10 mm, 30 x 150 mm. Fase móvil disolución de NH4OAc al 0,25% en agua, metanol) proporcionando 80 en la forma de un polvo.
Preparación de 81

Se calentó una disolución del intermedio C-2 (2 g, 7,2 mmol), AA-9 (3,02 g, 18 mmol) y trietilamina (5 ml, 36 mmol) en acetonitrilo (75 ml) a reflujo durante 6 horas. La mezcla de reacción se enfrió y el disolvente se eliminó a presión reducida. El residuo se disolvió en diclorometano y se lavó con salmuera. La capa orgánica se cargó en un cartucho de sílice y se aplicó un gradiente de diclorometano a metanol en diclorometano al 10%. Las fracciones que contenían el producto se evaporaron a sequedad proporcionando un polvo blanco, D-18.
LC-MS: Anal. Calc. para C20H28N4O3 : 372,22; observado 373 [M+H]+
1H RMN (400 MHz, DMSO-afe) 5 ppm 0,77 - 0,92 (m, 3 H) 1,15 - 1,36 (m, 4 H) 1,42 - 1,72 (m, 4 H) 2,12 (s, 3 H) 3,35 -3,42 (m, 2 H) 4,11 - 4,24 (m, 1 H) 4,35 - 4,52 (m, 1 H) 6,42 (d, J=8,80 Hz, 1 H) 7,42 (s, 1 H) 9,63 (s a, 1 H)

Se preparó D-19 a partir de D-18 de acuerdo con el método empleado para el intermedio D-2.
LC-MS: Anal. Calc. para C13H22N4O3 : 282,1; observado 283 [M+H]+

Se preparó D-20 a partir de D-19 de acuerdo con el método para preparar D-17.
LC-MS: Anal. Calc. para C19H30N6O3: 390,24; observado 391 [M+H]+

Se preparó 81 a partir de D-20 de acuerdo con el método para preparar el compuesto 79.
Preparación de 82
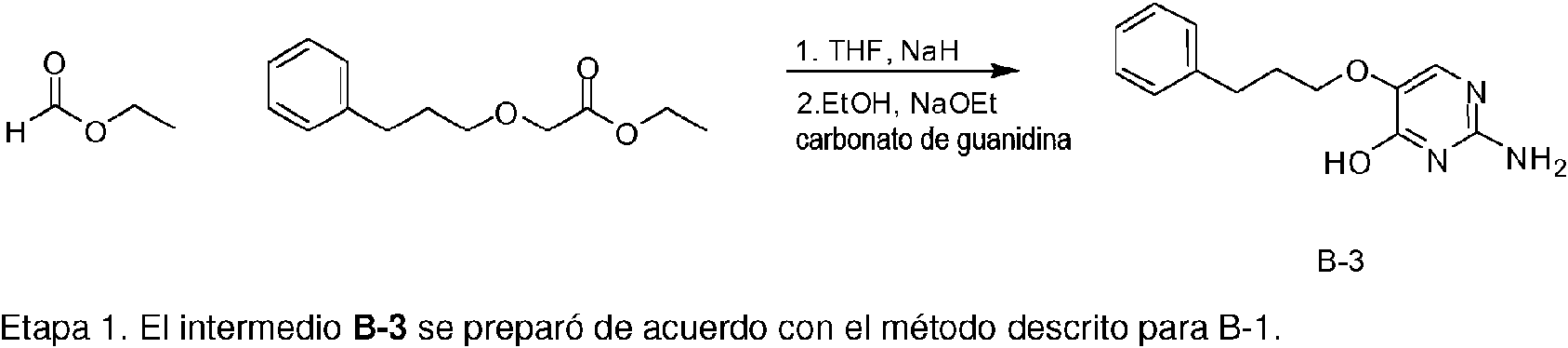
LC-MS: Anal. Calc. para C13H15N3O2 : 245,12; observado 246 [M+H]+
H RMN (400 MHz, DMSO-afe) ó ppm 1,79 - 1,93 (m, 2 H), 2,66 (t, J=7,8 Hz, 2 H), 3,76 (t, J=6,4 Hz, 2 H), 6,54 (s ancho, 2 H), 7,11 - 7,21 (m, 3 H), 7,22 - 7,29 (m, 3 H), 11,46 (s ancho, 1 H)
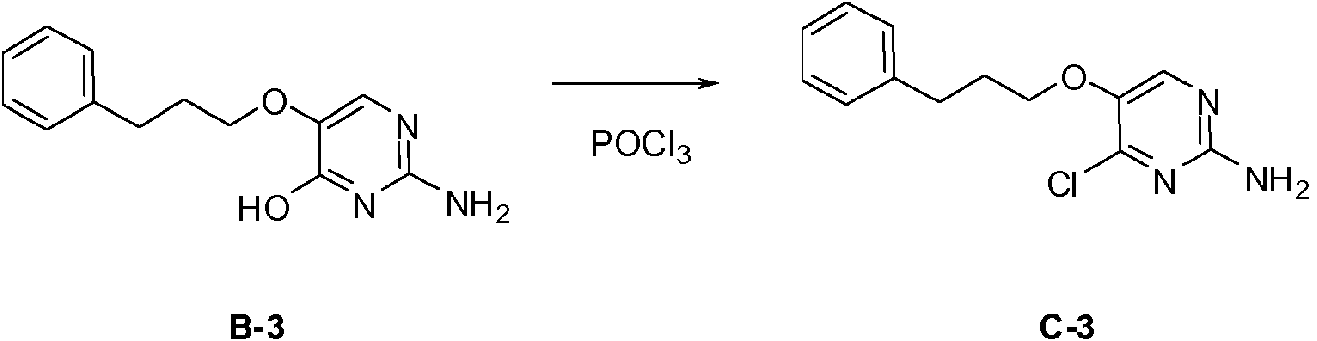
Etapa 2. En un matraz de fondo redondo de 250 ml se calentó una mezcla de B-3 (15 g, 61,15 mmol) en POCI3 (150 ml) a reflujo y se agitó durante 2 horas. La reacción se dejó enfriar y el disolvente se separó a presión reducida. La fracción residual se trituró con éter diisopropílico. El precipitado formado se aisló por filtración, se lavó con éter diisopropílico y se secó a vacío a 50°C para obtener un sólido, C-3, usado como estaba en la siguiente etapa.
LC-MS: Anal. Calc. para C13H14ClN3O: 263,08; observado [M+H]+

Etapa 3. En un tubo de 20 ml se puso C-3 (0,45 g, 1,05 mmol), éster metílico del ácido L-2-aminohexanoico HCl (0,48 g, 2,62 mmol), DIPEA (1,18 ml, 6,82 mmol), y acetonitrilo (5 ml). El tubo se cerró herméticamente y se calentó en un microondas durante 1,5 horas a 120°C. La reacción se dejó enfriar y el disolvente se separó a presión reducida.
La mezcla bruta se purificó por Prep HPLC en (RP Vydac Denali C18 - 10 pm, 250 g, 5 cm). Fase móvil (disolución de NH4OAc en agua al 0,25%, metanol), las fracciones deseadas se recogieron y se evaporaron hasta sequedad. La fracción residual se disolvió en una mezcla de diclorometano/metanol y se vertió sobre un cartucho de extracción de fase sólida modificado con ácido (SCX). El producto se liberó usando NH37 N en metanol. La disolución recogida se concentró a presión reducida para obtener el sólido deseado, 82.
Preparación de 83

Etapa 1. El intermedio B-4 se preparó de acuerdo con el método para preparar B-1.
LC-MS: Anal. Calc. para C14H17N3O3 : 275,13; observado 276 [M+H]+
1H RMN (400 MHz, DMSO-afe) 5 ppm 3,63 (dd, J=5,4, 3,9 Hz, 2 H), 3,95 (dd, J=5,4, 3,6 Hz, 2 H), 4,50 (s, 2 H), 6,33 (s ancho, 2 H), 7,22 - 7,29 (m, 2 H), 7,30 - 7,36 (m, 4 H), 10,71 - 11,58 (m, 1 H)

Etapa 2. En un matraz de fondo redondo de 250 ml se pusieron B-4 (10 g, 38,27 mmol) y POCh (75 ml). La mezcla se calentó a reflujo y se agitó durante 5 horas. Se dejó que la mezcla de reacción alcanzara temperatura ambiente y
se agitó durante 15 horas. El disolvente se separó a presión reducida. El compuesto C-4 bruto se usó como estaba en la siguiente etapa.
LC-MS: Anal. Calc. para C12H12GN3O2 : 265,06; observado 266 [M+H]+

Etapa 3. En tubos de 50 ml se pusieron C-4 (10 g, 35,75 mmol), n-butilamina (10,6 ml, 107,25 mmol) y DIPEA (30,8 ml, 178,75 mmol) en acetonitrilo (40 ml). La mezcla se calentó a 120°C con irradiación de microondas durante 3 horas. Las mezclas de reacción combinadas se concentraron a presión reducida y el aceite residual se disolvió en diclorometano y se lavó con HCl 1 N y agua. La capa orgánica se secó (sulfato de magnesio), los sólidos se separaron por filtración y el disolvente del filtrado se separó a presión reducida para obtener una espuma rojomarrón, 83.
Preparación de 84

Etapa 1. En un matraz de fondo redondo de 500 ml se pusieron el compuesto 83 (13,5 g, 25,6 mmol), Boc-anhídrido (27,94 g, 128 mmol) y acetonitrilo (150 ml). La disolución amarilla se agitó a reflujo durante 16 horas. El disolvente se separó a presión reducida. La fracción residual se disolvió en diclorometano y se lavó con una disolución acuosa saturada de NaHCO3 y agua. La capa orgánica se secó (sulfato magnésico), los sólidos se separaron por filtración, y los disolventes del filtrado se separaron a presión reducida para obtener un aceite, D-20.
LC-MS: Anal. Calc. para C22H32N4O4 : 416,24; observado 417 [M+H]+

Etapa 2. En un Erlenmeyer de 1 L se suspendió Pd/C al 10% (4 g) en metanol (350 ml) con corriente de N2 gaseoso, después se añadió D-20 (14,3 g, 34,33 mmol). La mezcla se agitó a 50°C en una atmósfera de hidrógeno hasta que se absorbió 1 equivalente de hidrógeno. El catalizador se separó por filtración sobre decalita empaquetada. El disolvente del filtrado se separó a presión reducida para obtener un aceite, D-21. El residuo se usó como estaba en la siguiente etapa.
LC-MS: Anal. Calc. para C15H26N4O4: 326,20; observado 327 [M+H]+
Etapa 3. En un matraz de fondo redondo de 1 L se agitaron D-21 (8,7 g, 26,66 mmol) y trietilamina (7,41 ml, 53,31 mmol) en acetonitrilo (300 ml) a temperatura ambiente y se añadió cloruro de metanosulfonilo (3,1 ml, 40 mmol). Después de la adición, la mezcla de reacción se agitó durante 1,5 horas a temperatura ambiente. El disolvente se separó a presión reducida. El producto bruto se disolvió en acetato de etilo y se lavó con disolución acuosa saturada de NaHCO3. Las capas orgánicas se combinaron, se secaron (sulfato magnésico), los sólidos se separaron por filtración y el disolvente del filtrado se evaporó hasta sequedad para obtener D-22 en forma de un aceite.
LC-MS: Anal. Calc. para C16H28N4O6S: 404,17; observado 405 [M+H]+

Etapa 4. En un tubo de vidrio de 30 ml se puso una mezcla de 4-hidroxipiridina (94 mg, 0,99 mmol) y Cs2CO3 (0,8 g, 2,47 mmol) en acetonitrilo (10 ml). El vial se cerró herméticamente y se agitó a temperatura ambiente durante 1 hora. Se añadió D-22 (400 mg, 0,99 mmol) en forma de una disolución en acetonitrilo (10 ml) a la mezcla de reacción y se agitó durante 18 horas adicionales a temperatura ambiente. Se añadió carbonato de cesio (320 mg, 1 mmol) y la mezcla se agitó durante 1 día a temperatura ambiente. El disolvente se separó a presión reducida y el producto bruto se trató con una mezcla de diclorometano/metanol 95/5 y se agitó durante 1 h, después se filtró sobre 2 g de sílice empaquetada. El filtrado se concentró a presión reducida y D-23 se usó como estaba en la siguiente etapa.
LC-MS: Anal. Calc. para C20H29N5O4 : 403,22; observado 404 [M+H]+

Etapa 5. D-23 se desprotegió para proporcionar el compuesto 84 usando el método aplicado para desproteger el compuesto 78.

Etapa 1. En un matraz de fondo redondo de 150 ml equipado con una barra agitadora magnética se pusieron el compuesto D-4 (0,35 g, 5,23 mmol) y carbonato de cesio (0,89 g, 2,75 mmol) en acetonitrilo (20 ml). La mezcla se agitó a temperatura ambiente durante 30 minutos. Se añadió una disolución del haluro de alquilo (0,19 g, 1 mmol) en acetonitrilo (5 ml) y la mezcla de reacción se agitó durante 1 día a temperatura ambiente. La reacción se completó y se separaron las sales por filtración. El filtrado se concentró a presión reducida y el producto bruto se purificó por cromatografía en columna de sílice usando un gradiente de heptano a acetato de etilo para dar el intermedio D-24.
LC-MS: Anal. Calc. para C24H37N7 O7 : 535,28; observado 536 [M+H]+

Etapa 2. En un matraz Erlenmeyer de 100 ml se suspendió Pt/C, al 5% (100 mg) en tiofeno (0,25 ml) y metanol (20 ml) bajo una capa de nitrógeno gaseoso, después se añadió D-24 (130 mg, 0,24 mmol). La mezcla de reacción se agitó a 50°C bajo una atmósfera de hidrógeno. El catalizador se eliminó por filtración sobre decalita empaquetada. Los disolventes del filtrado se separaron a presión reducida para obtener D-25 en forma de un aceite, que se usó como estaba en la siguiente etapa.
LC-MS: Anal. Calc. para C24H39N7 O5 : 505,30; observado 506 [M+H]+

Etapa 3. El intermedio D-25 se desprotege para dar el compuesto 85 de acuerdo con el método usado para preparar el compuesto 78.
Preparación de 86

Etapa 1. En un matraz de fondo redondo de 100 ml se puso azida sódica (6,85 g, 103,76 mmol) en agua (12,5 ml) después se añadió pivalato de clorometilo (10,6 g, 70,38 mmol) y se agitó enérgicamente a 90°C durante 16 horas. Se dejó que la mezcla de reacción se enfriara a temperatura ambiente y se añadió diclorometano (20 ml). La capa orgánica se separó, se secó sobre sulfato sódico anhidro, los sólidos se separaron por filtración y el disolvente del filtrado se separó a presión reducida para obtener A-2 en forma de un aceite.
LC-MS: Anal. Calc. para C6H11N3O2 : 157,09; observado 158 [M+H]+

Etapa 2. En un tubo de 25 ml se pusieron D-26 (100 mg, 0,238 mmol), A-2 (37,9 mg, 0,238 mmol), í-butanol (2,5 ml) y agua (2,5 ml). El tubo se cerró herméticamente y la mezcla se agitó a temperatura ambiente. Se añadieron sulfato de cobre(II) pentahidrato (3 mg, 0,012 mmol) y sal de sodio del ácido L-ascórbico (15,5 mg, 0,079 mmol). La mezcla de reacción se agitó durante 18 horas a temperatura ambiente, después se añadió agua (2,5 ml). El precipitado se aisló por filtración, se lavó con agua y se secó a vacío a 60°C para obtener un polvo blanco, D-27.
LC-MS: Anal. Calc. para C27H43N7 O7 : 577,32; observado 578 [M+H]+

Etapa 3. En un matraz de fondo redondo de 100 ml, una mezcla de D-27 (0,1 g, 0,17 mmol) en HCl (5 ml 6 M en isopropanol) y diclorometano (5 ml) se agitó a temperatura ambiente durante 16 horas. La reacción se calentó a 65°C y se agitó durante 16 horas adicionales. El disolvente se separó a presión reducida.
El producto bruto se purificó por cromatografía líquida de fase inversa (RP Vydac Denali C18 - 10 pm, 250 g, 5 cm). Fase móvil (disolución de NH4 HCO3 en agua al 0,25%, metanol), las fracciones deseadas se recogieron, se evaporaron, se disolvieron en metanol y se trataron con HCl 2 M en éter. El sólido se aisló por filtración para dar el compuesto 86 en forma de la sal de HCl.
Preparación de 87

Etapa 1. En un matraz de 100 ml con fondo redondo se dispuso una disolución de C-2 (500 mg, 1,8 mmol), AA-10 (692 mg, 4,5 mmol) y trietilamina (0.,75 ml, 5,4 mmol) en acetonitrilo (30 ml). La mezcla se calentó hasta 80°C durante 16 horas con agitación. La reacción se dejó enfriar y el disolvente se eliminó a presión reducida. El producto bruto se disolvió en diclorometano y se lavó con salmuera. La capa orgánica se secó (sulfato de magnesio), los sólidos se eliminaron por filtración y el disolvente del filtrado se eliminó para obtener un aceite, D-28.
LC-MS: Anal. Calc. para C19H26N4O3 : 358,20; observado 359 [M+H]+
1H RMN (360 MHz, DMSO-ofe) 5 ppm 0,85 (t, J=7,32 Hz, 3 H) 1,19 - 1,37 (m, 2 H) 1,38 - 1,53 (m, 1 H) 1,53 - 1,75 (m, 3 H) 2,13 (s, 3 H) 3,38 - 3,48 (m, 2 H) 4,19 - 4,31 (m, 1 H) 5,16 (s, 2 H) 6,69 (d, J=9,15 Hz, 1 H) 7,29 - 7,41 (m, 3 H) 7,45 - 7,53 (m, 2 H) 7,66 (s, 1 H) 9,77 (s, 1 H)

Etapa 2. Se preparó D-29 de acuerdo con el método utilizado para preparar D-21. Se añadió THF para incrementar la solubilidad de D-29.
LC-MS: Anal. Calc. para C12H20N4O3: 268,15; observado 269 [M+H]+

Etapa 3. En un matraz de 250 ml con fondo redondo se agitó una mezcla de D-29 (5 g, 18,6 mmol) y carbonato de cesio (18,2 g, 55,9 mmol) en DMF (80 ml) a temperatura ambiente durante 30 minutos. La mezcla se calentó hasta 60°C y se añadió gota a gota una disolución de hidrocloruro de 2-clorometil-3,4-dimetoxi-piridina (3,97 g, 17,7 mmol) en DMF (60 ml) gota a gota. La mezcla de reacción se agitó durante 2 horas a 60°C. La reacción se dejó enfriar y las sales se eliminaron por filtración. La mezcla de reacción se concentró a presión reducida y se usó D-30 como tal en la etapa siguiente.
LC-MS: Anal. Calc. para C20H29N5O5 : 419,22; observado 420 [M+H]+
1H RMN (400 MHz, DMSO-ds ) 5 ppm 0,83 (t, J=7,4 Hz, 3 H), 1,18 - 1,32 (m, 2 H), 1,41 - 1,71 (m, 4 H), 2,14 (s, 3 H), 3,34 - 3,40 (m, 2 H), 3,78 (s, 3 H), 3,91 (s, 3 H), 4,17 - 4,29 (m, 1 H), 4,41 (t, J=5,3 Hz, 1 H), 5,09 (s, 2 H), 6,79 (d, J=8,8 Hz, 1 H), 7,15 (d, J=5,7 Hz, 1 H), 7,75 (s, 1 H), 8,24 (d, J=5,5 Hz, 1 H), 9,75 (s, 1 H)

Etapa 4. Se preparó 87 de acuerdo con el mismo método utilizado para preparar 79 a partir del intermedio D-16. Se purificó 87 por cromatografía de fase inversa (Hyperprep C18 HS BDS. Fase móvil (Gradiente de 90% bicarbonato de amonio en agua 0,25%, 10% de acetonitrilo a 0% de bicarbonato de amonio en agua 0,25%, 100% de acetonitrilo). Las mejores fracciones se combinaron, los disolventes se eliminaron a presión reducida, se reconstituyeron en metanol y se trataron con HCl 2 M en éter y luego se concentraron a presión reducida para obtener un sólido blanco, la sal de HCl de 87.

El aislamiento de la sal de HCl de 87 mediante cromatografía de líquidos de fase inversa condujo al aislamiento concomitante de 88 con bajo rendimiento. Las mejores fracciones se combinaron, y los disolventes se eliminaron a presión reducida para dar un sólido blanco, 88.
Preparación de 89

Etapa 1. En un matraz de fondo redondo de 100 ml se pusieron AA-8 (2 g, 8,65 mmol), diclorometano (6 ml), isocianato de etilo (1,6 ml, 10,38 mmol) y DMAP (21 mg, 0,173 mmol). La mezcla de reacción se dejó agitar durante 16 horas a temperatura ambiente. El disolvente se separó a presión reducida y el compuesto AA-12 se usó en la siguiente etapa sin más purificación.
LC-MS: Anal. Calc. para C15H30N2O4 : 302,22; obtenido 303 [M+H]+

Etapa 2. En un matraz de fondo redondo de 100 ml se pusieron AA-12 bruto (2,61 g, 8,65 mmol) y diclorometano (30 ml). A esta disolución se añadió HCl (20 ml, 4 M en dioxano). La reacción se dejó agitar 3 horas a temperatura ambiente.
LC-MS: Anal. Calc. para C10H22N2O2 : 202,17; observado 203 [M+H]+

Etapa 3. En un matraz de fondo redondo de 100 ml equipado con una barra agitadora magnética se pusieron 2-amino-4-hidroxi-5-metoxi-pirimidina (500 mg, 3,54 mmol), DMF anhidra (30 ml), AA-13 (1,27 g, 5,31 mmol), DBU (2,12 ml, 14,17 mmol), y BOP (1,96 g, 4,43 mmol). La mezcla de reacción se dejó agitar a temperatura ambiente durante 30 minutos y después a 50°C durante 16 horas. El disolvente se separó a presión reducida y el residuo se repartió entre salmuera y acetato de etilo. Las capas orgánicas se combinaron, se secaron (sulfato magnésico), los sólidos se separaron por filtración y los disolventes del filtrado se separaron a presión reducida. El producto bruto se purificó por cromatografía de líquidos en fase inversa (RP Vydac Denali C18 - 10 gm, 250 g, 5 cm. Fase móvil
disolución de NH4 HCO3 al 0,25% en agua, metanol), las mejores fracciones se combinaron, los disolventes se separaron a presión reducida para dar 89.
Preparación de 264

Etapa 1. AA-14 se preparó de acuerdo con el procedimiento para preparar AA-10, usando el aldehído de partida adecuado.
LC-MS: Anal. Calc. para C7 H17NO: 131,13; observado 132 [M+H]+
1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 0,81 - 0,89 (m, 6 H), 1,15 - 1,25 (m, 2 H), 1,33 - 1,47 (m, 1 H), 1,54 -1,69 (m, 2 H), 2,71 (s ancho, 3 H), 2,88 - 2,98 (m, 1 H), 3,69 - 3,80 (m, 2 H)

Etapa 2. C-5 se preparó de acuerdo con el método usado para preparar C-2 a partir del material de partida disponible. El producto bruto se usó son más purificación.
LC-MS: Anal. Calc. para C5H6ClN3O: 159,02; observado 160 [M+H]+

Etapa 3. C-5 se combinó con AA-14 de acuerdo con el método usado para preparar el compuesto 1, excepto que se usó acetonitrilo como disolvente, para dar el compuesto 264.
Preparación de 278

Etapa 1. AA-15 se preparó de acuerdo con el procedimiento para preparar AA-10, usando el aldehído de partida adecuado.
LC-MS: Anal. Calc. para C7 H17NO: 131,13; observado 132 [M+H]+
1H RMN (400 MHz, DMSO-afe) 5 ppm 0,81 - 0,89 (m, 6 H), 1,05 - 1,20 (m, 1 H), 1,27 - 1,40 (m, 1 H), 1,43 - 1,77 (m, 3 H), 3,05 - 3,19 (m, 1 H), 3,44 - 3,57 (m, 2 H), 4,82 (s ancho, 1 H), 7,94 (d, J=18,6 Hz, 2 H)

Etapa 2. C-5 se combinó con AA-15 de acuerdo con el método usado para preparar el compuesto 1, excepto que se usó acetonitrilo como disolvente, para dar el compuesto 278.
Preparación de 295

Etapa 1. AA-16 se preparó de acuerdo con los procedimientos indicados en Chem. Rev., 2010, Vol. 110, No. 6, 3600-3740.
LC-MS: Anal. Calc. para C8H17N: 127,14; encontrado 128 [M+H]+
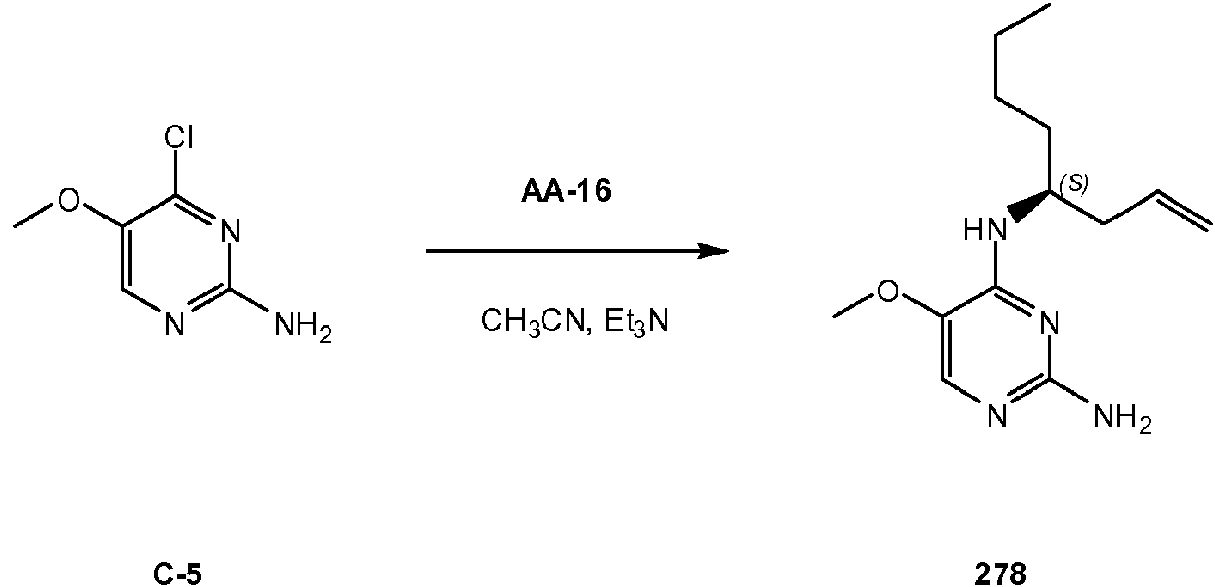
Etapa 2. C-5 se combinó con AA-16 de acuerdo con el método usado para preparar el compuesto 1, excepto que se usó acetonitrilo como disolvente, para dar el compuesto 295.
Preparación de 304
Etapa 1. AA-17 se preparó de acuerdo con los procedimientos indicados en Chem. Rev., 2010, Vol. 110, No. 6, 3600-3740.
LC-MS: Anal. Calc. para C8H19NO: 145,15; observado 146 [M+H]+

Etapa 2. C-5 se combinó con AA-17 de acuerdo con el método usado para preparar el compuesto 1, excepto que se usó acetonitrilo como un disolvente, para dar el compuesto 304.
Tabla I: Compuestos de fórmula (I).






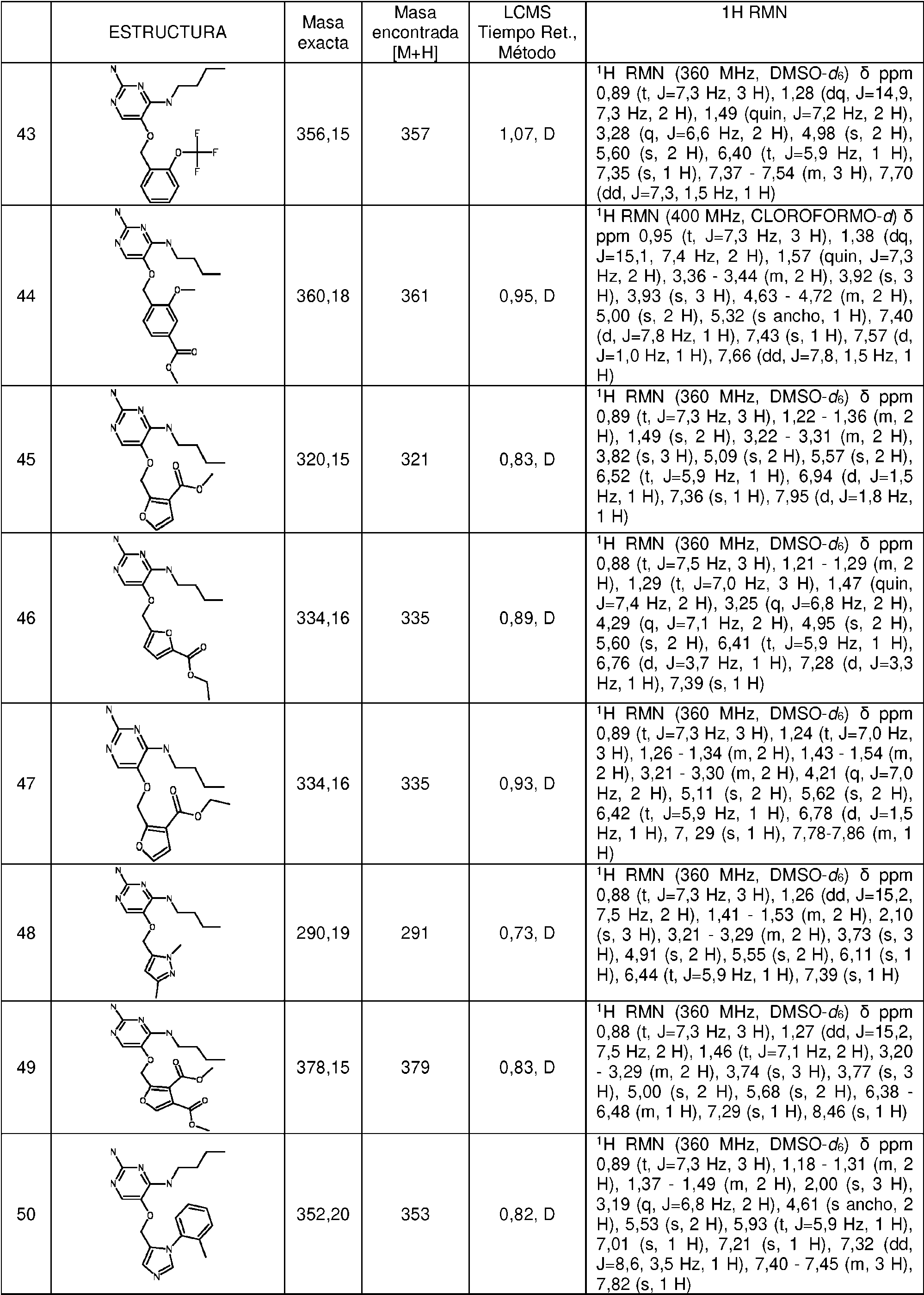












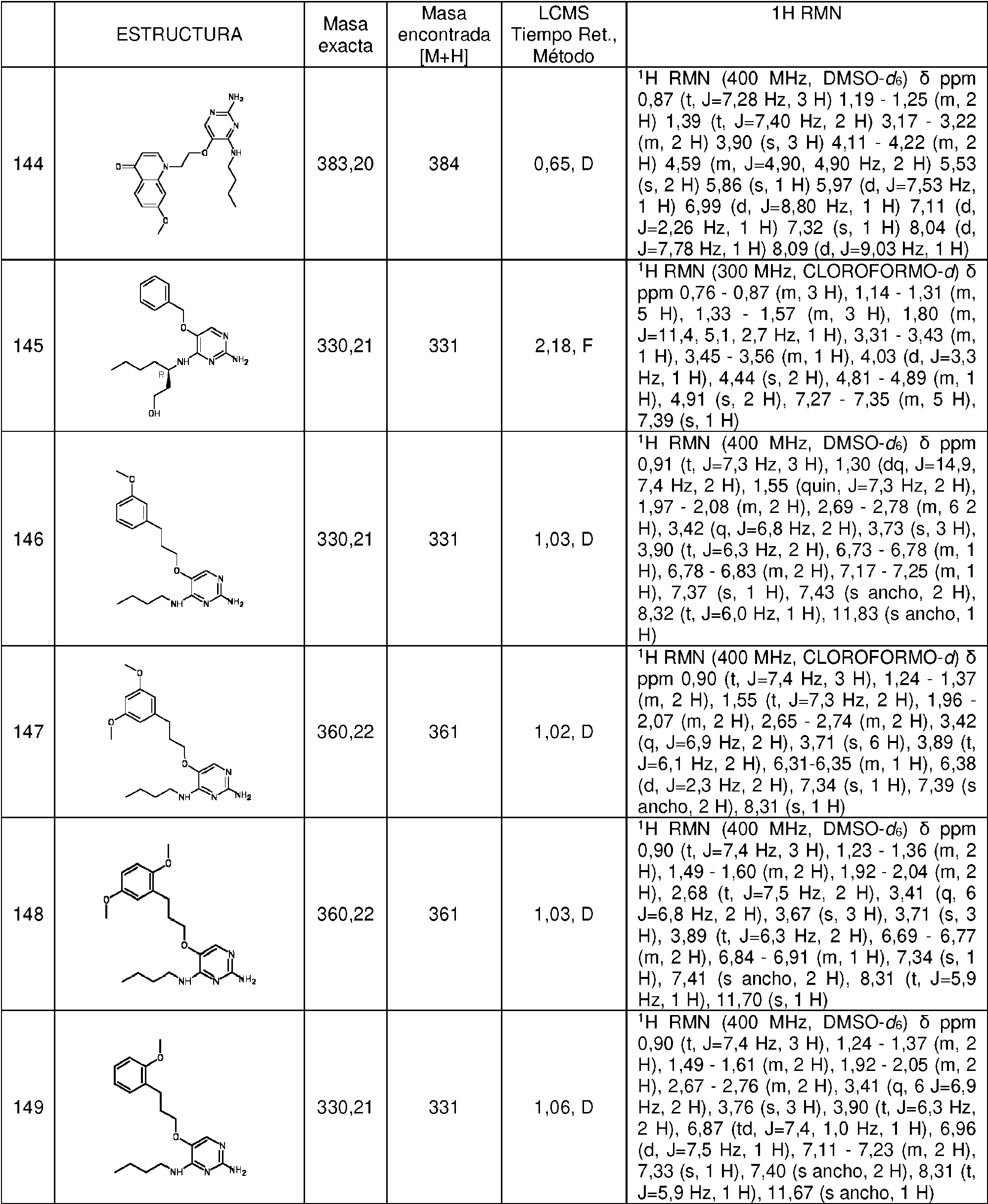


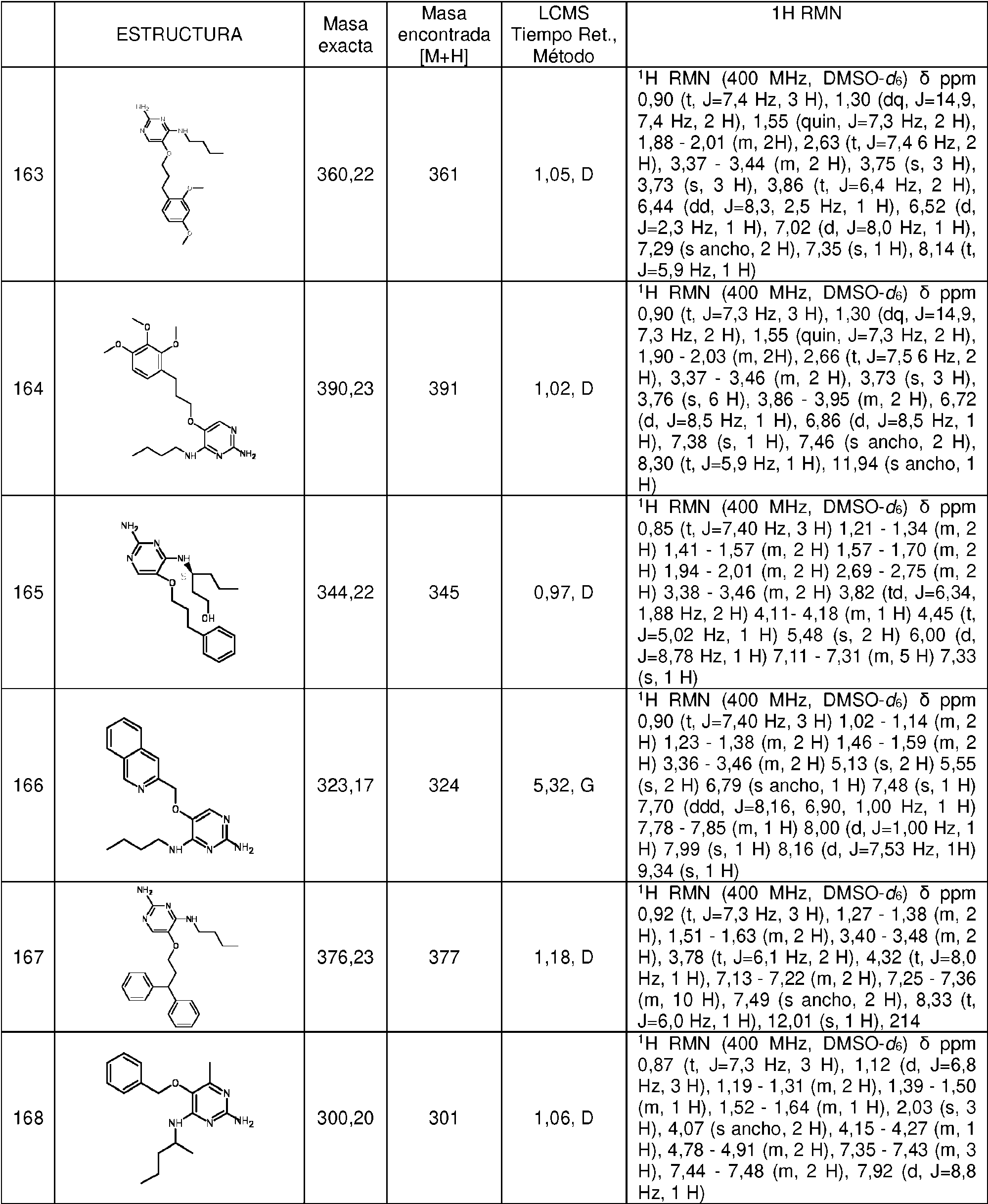






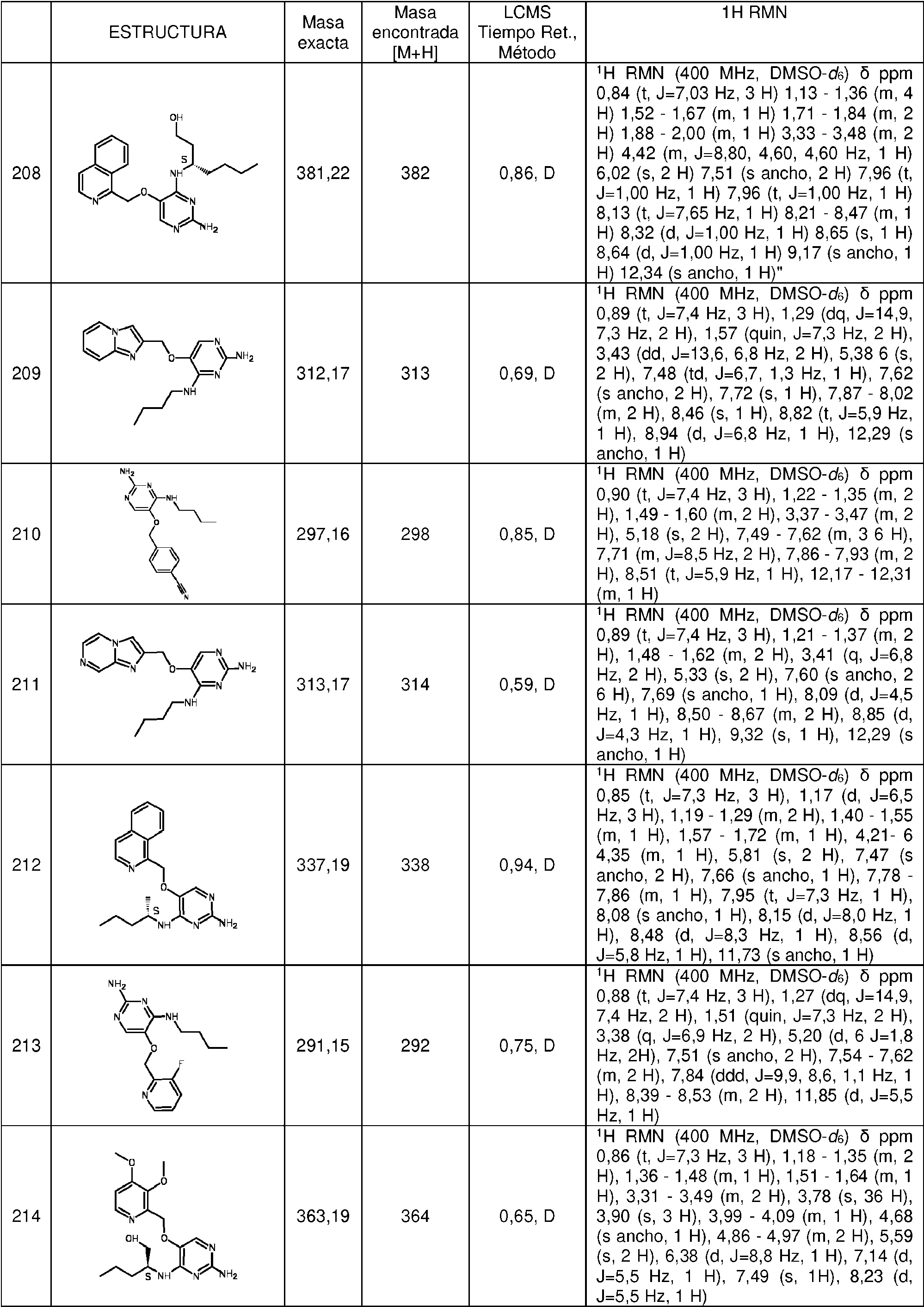
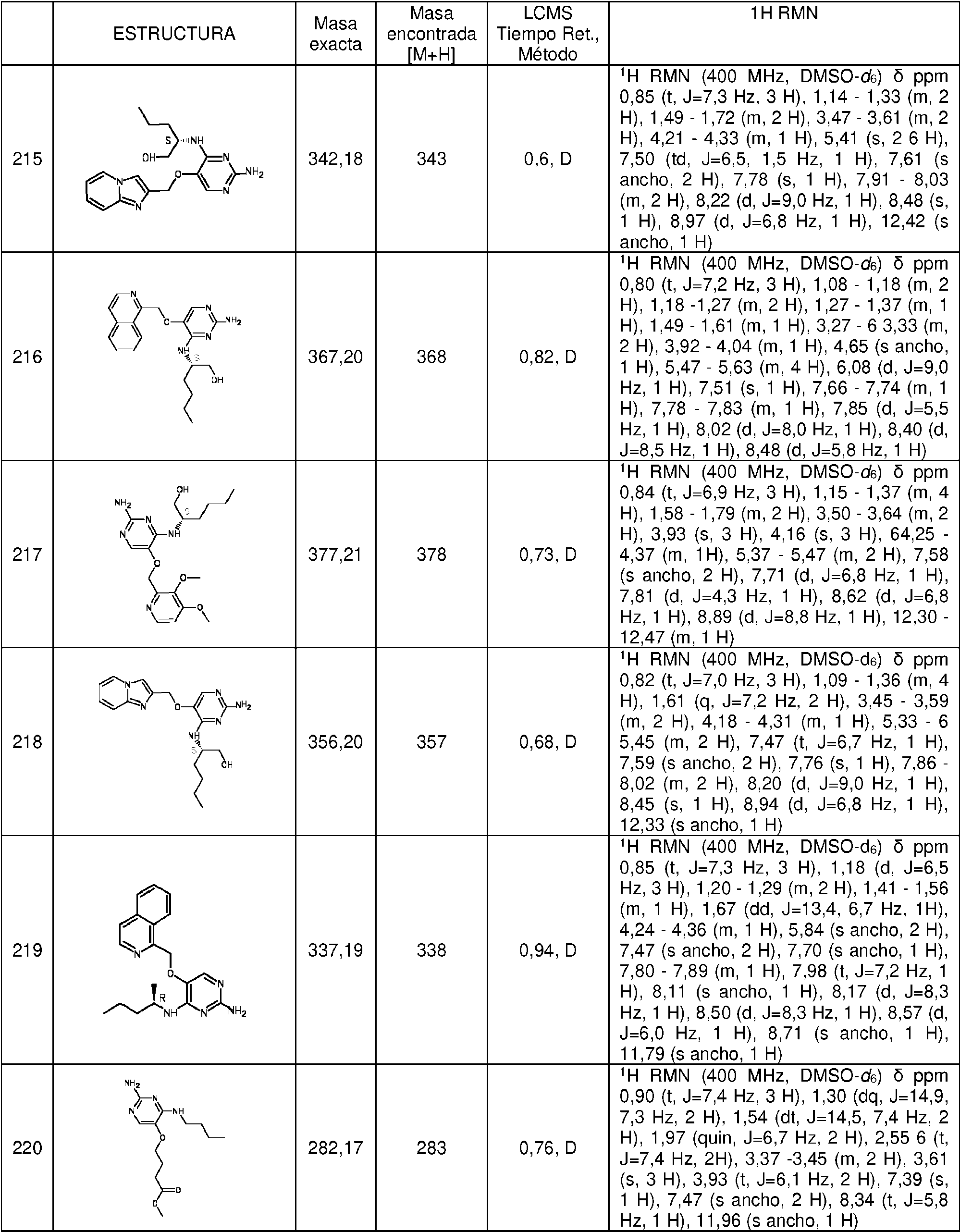












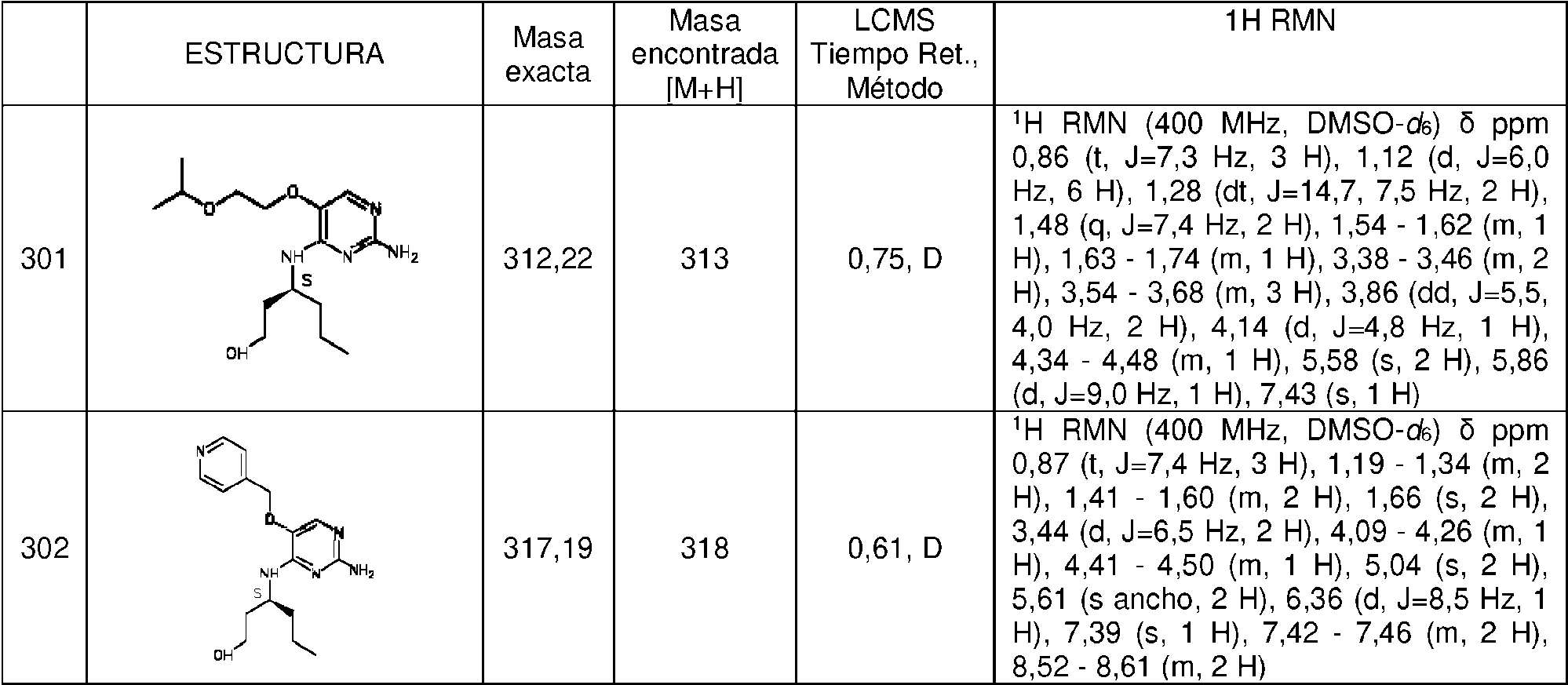
Métodos analíticos.
Todos los compuestos se caracterizaron por LC-MS. Se emplearon los siguientes métodos de LC-MS:
Método A. Waters Aquity UPLC equipado con un detector de PDA (210-400 nm) y un equipo Waters SQD con una fuente de iones de modo dual ES+/-. La columna utilizada fue una Halo C18, 2,7 pm, 2,1 x 50 mm, calentada hasta 50°C. Se utilizó un gradiente de ácido fórmico al 95% acuoso (0,1%)/5% de acetonitrilo hasta 100% de acetonitrilo durante 1,5 minutos, mantenido durante 0,6 minutos, luego retorna a ácido fórmico al 100% acuoso (0,1%) durante 0,5 minutos. El caudal fue 0,6 ml/min.
Método B.


Método C.

Método D. La UPLC (cromatografía de líquidos de ultra rendimiento) de fase inversa se llevó a cabo en una columna híbrida con puente de etilsiloxano/sílice (BEH) C18 (1,7 gm, 2,1 x 50 mm; Waters Acquity) con un caudal de 0,8 ml/min. Se utilizaron dos fases móviles (acetato de amonio 10 mM en H2Ü/acetonitrilo 95/5; fase móvil B: acetonitrilo) para ejecutar una condición de gradiente de 95% de A y 5% de B a 5% de A y 95% de B en 1,3 minutos durante 0,7 minutos. Se usó un volumen de inyección de 0,75 gl. El voltaje del cono fue 30 V para el modo de ionización positiva y 30 V para el modo de ionización negativa.
Método E. Usando una columna Phenomenex Kinetex (XB-C1850 x 4,6 mm I.D. 2,6u) mantenida a 35°C. Detección MS: modo de ionización positiva API-ES, intervalo de masas 100-1200. Detección p Da (A=190-400 nm). Se usó el siguiente gradiente con una inyección de 2 gl:

Método F. Usando YMC ODS-AQ C-18;50 x 4,6 mm, ID = 3 gm que se mantuvo a 35°C. Detección MS: API-ES modo ionización positiva, intervalo de masas 100-1400. Detección PDA (A=190-400 nm). Se empleó el siguiente gradiente con una inyección de 2 gl:

Método G. Sistema Alliance HT 2790 (Waters) que comprende una bomba cuaternaria con desgaseador, un automuestreador, un horno de columna (programada a 40°C). El caudal de la columna se separó a un espectrómetro de MS. El detector de MS se configuró con una fuente de ionización por electropulverización. El voltaje de la aguja capilar fue 3 kV y la temperatura de la fuente se mantuvo a 140°C. Se usó nitrógeno como gas nebulizador. Columna Xterra MS C18 (3,5 gm, 4,6 x 100 mm) con un caudal de 1,6 ml/min. Se emplearon tres fases móviles (fase móvil A: 95% de acetato de amonio 25 mM 5% de acetonitrilo; fase móvil B: acetonitrilo; fase móvil C: metanol) para ejecutar una condición de gradiente de 100% de A a 50% de B y 50% de C en 6,5 minutos, a 100% de B en 0,5 minuto, 100% de B durante 1 minuto y reequilibrado con 100% de A durante 1,5 minutos. Se utilizó un volumen de inyección de 10 gl.
Método H. Se llevó a cabo UPLC (cromatografía de líquidos de ultra rendimiento) de fase inversa en una columna híbrida con puente de etilsiloxano/sílice (BEH) C18 (1,7 gm, 2,1 x 50 mm; Waters Acquity) con un caudal de 0,8 ml/min. Se utilizaron dos fases móviles (fase móvil A: acetato de amonio 10 mM en H2O/acetonitrilo 95/5; fase móvil B: acetonitrilo) para ejecutar una condición de gradiente de 95% de A y 5% de B a 5% de A y 95% de B en 1,3 minutos y se mantuvo durante 0,2 minutos. Se empleó un volumen de inyección de 0,5 gl. El voltaje del cono fue 10 V para el modo de ionización positiva y 20 V para el modo de ionización negativa.
Actividad biológica de los compuestos de fórmula (I)
Descripción de ensayos biológicos
Evaluación de la actividad de TLR7 y TLR8
Se evaluó la capacidad de los compuestos de activar TLR7 y/o TLR8 humano en un ensayo indicador celular que utiliza células HEK293 transfectadas transitoriamente con un vector de expresión de TLR7 o TLR8 y un constructo indicador de NFkB-Iuc. En un caso el constructor de expresión de TLR expresa la respectiva secuencia de tipo salvaje o una secuencia mutante que comprende una eliminación en la segunda repetición rica en leucina del TLR. Dichas proteínas de TLR mutantes han demostrado previamente ser más susceptibles a la activación de agonistas (US 7498409).
En síntesis, se desarrollaron células HEK293 en medio de cultivo (DMEM enriquecido con 10% FCS y glutamina 2 mM). Para la transfección de células en placas de 10 cm, las células se desprendieron con Tripsina-EDTA, se transfectaron con una mezcla de CMV-TLR7 o plásmido TLR8 (750 ng), plásmido NFkB-Iuc (375 ng) y un reactivo de transfección, y se incubaron durante 48 horas a 37°C en una atmósfera de 5% de CO2 humidificada. Las células transfectadas se desprendieron luego con Tripsina-EDTA, se lavaron con PBS y se resuspendieron en medio hasta una densidad de 1,67 x 105 células/ml. Treinta microlitros de células se dispensaron luego en cada pocilio de placas de 384 pocilios, en donde ya estaban presentes 10 pl del compuesto en 4% de DMSO. Después de la incubación de 6 horas a 37°C, 5% de CO2, se determinó la actividad de la luciferasa añadiendo 15 pl de sustrato Steady Lite Plus (Perkin Elmer) a cada pocilio y la lectura se llevó a cabo en un lector de imágenes de microplacas ViewLux ultraHTS (Perkin Elmer). Las curvas de dosis y respuesta se generaron a partir de las mediciones cuadruplicadas. Los valores de las concentraciones eficaces más bajas (LEC), definidos como la concentración que induce un efecto que está al menos dos veces por encima de la desviación estándar del ensayo, se determinaron para cada compuesto.
La toxicidad del compuesto se determinó en paralelo usando una serie de diluciones similares del compuesto con 30 pl por pocilio de células transfectadas con el constructo CMV-TLR7 solo (1,67 x 105 células/ml), en placas de 384 pocilios. La viabilidad de las células se midió después de 6 horas de incubación a 37°C, 5% de CO2 añadiendo 15 pl de ATP lite (Perkin Elmer) por pocilio y leyendo en un lector de imágenes de microplacas ViewLux ultraHTS (Perkin Elmer). Los datos se describieron como CC50.
Supresión de la replicación del replicón del VHC
La activación de TLR7 humano resulta en la producción robusta de interferón por las células dendríticas plasmacitoides presentes en sangre humana. El potencial de los compuestos de inducir interferón se evaluó mirando la actividad antivírica en el sistema de replicón del VHC tras la incubación con medio acondicionado de células mononucleares de sangre periférica (PBMC). El ensayo del replicón del VHC se basa en un constructo de expresión bicistrónico, como lo describen Lohmann et al. (Science (1999) 285: 110-113; Journal of Virology (2003) 77: 3007-15 3019) con las modificaciones descritas por Krieger et al. (Journal of Virology (2001) 75: 4614-4624). El ensayo utilizó la línea celular establemente transfectada Huh-7 luc/neo que aloja un ARN que codifica un constructo de expresión bicistrónico que comprende las regiones NS3-NS5B de tipo salvaje del VHC de tipo 1b que se traducen de un sitio interno de entrada al ribosoma (IRES) del virus de encefalomiocarditis (EMCV), precedido por un gen indicador (luciérnaga-luciferasa) y un gen marcador seleccionable (neoR, neomicina fosfotransferasa). El constructo es flanqueado por NTR 5' y 3' (regiones no traducidas) de VHC de tipo 1b. El cultivo continuado de las células del replicón en presencia de G418 (neoR) depende de la replicación del ARN de VHC. Las células del replicón establemente transfectadas que replican el ARN del VHC de manera autónoma y en altos niveles, que codifican, entre otros, luciferasa, se usaron para obtener un perfil del medio de cultivo celular acondicionado.
En síntesis, se prepararon PBMC de capas leucocíticas de por lo menos dos donantes usando un protocolo de centrifugación Ficoll estándar. Las PBMC aisladas se resuspendieron en medio RPMI enriquecido con suero AB humano al 10% y se dispensaron 2 x 105 células/pocillo en placas de 384 pocilios que contenían los compuestos (70 pl de volumen total). Después de incubar durante la noche, se transfirieron 10 pl de sobrenadante a placas de 384 pocilios que contenían 2,2 x 103 células del replicón/pocillo en 30 pl (dispuestas en placas el día anterior). Después de 24 horas de incubación, se midió la replicación ensayando la actividad de la luciferasa usando 40 pl/pocillo de sustrato Steady Lite Plus (Perkin Elmer) y se midió con el generador de imágenes de microplacas ViewLux ultraHTS (Perkin Elmer). La actividad inhibidora de cada compuesto en Huh7-luc/células neo se describió como valores CE50, definidos como la concentración de compuesto aplicada a las PBMC que resulta en una reducción de 50% de actividad de luciferasa, que a su vez indica el grado de replicación del ARN del replicón en la transferencia de una cantidad definida de medio de cultivo de PBMC. Se usó interferón a-2a recombinante (Roferon-A) como compuesto control estándar.
Actividad biológica de los compuestos de fórmula (I). Todos los compuestos demostraron CC50 de >24 uM en el ensayo de HEK 293 TOX anteriormente descrito.
Activación de elementos promotores de ISRE
El potencial de los compuestos de inducir IFN-I se evaluó también midiendo la activación de elementos sensibles a la estimulación con interferón (ISRE) por medio acondicionado de PBMC. El elemento ISRE de la secuencia GAAACTGAAACT es altamente sensible al factor de transcripción STAT1-STAT2-IRF9, activado tras la unión de IFN-I a su receptor IFNAR (Clontech, PT3372-5W). El plásmido pISRE-Luc de Clontech (ref. 631913) contiene 5 copias de este elemento ISRE, seguido por luciferasa de luciérnaga ORF. Se estableció una línea celular HEK293
establemente transfectada con pISRE-Luc (HEK-ISREluc) para obtener un perfil del medio de cultivo celular de PBMC acondicionado.
En síntesis, se prepararon PBMC de capas leucocíticas de por lo menos dos donantes usando un protocolo de centrifugación Ficoll estándar. Las PBMC aisladas se resuspendieron en medio RPMI enriquecido con 10% suero AB humano y 2 x 105 células/pocillo se dispensaron en placas de 384 pocillos que contenían los compuestos (70 pl volumen total). Después de incubar durante toda la noche, se transfirieron 10 pl de sobrenadante a placas de 384 pocillos que contenían 5 x 103 células HEK-ISREluc/pocillo en 30 pl (dispuestas en placas el día anterior). Después de 24 horas de incubación, se midió la activación de los elementos ISRE ensayando la actividad de la luciferasa con el uso de 40 pl/pocillo de sustrato Steady Lite Plus (Perkin Elmer) y se midió con un generador de imágenes de microplacas ViewLux ultraHTS (Perkin Elmer). La actividad estimuladora de cada compuesto en las células HEK-ISREluc se describió como el valor LEC, definido como la concentración de compuesto aplicada a las PBMC que resulta en actividad de luciferasa por lo menos dos veces por encima de la desviación estándar del ensayo. El valor LEC a su vez indica el grado de activación de ISRE en la transferencia de una cantidad definida de medio de cultivo de PBMC. Se usó interferón recombinante a-2a (Roferon-A) como compuesto control estándar.
Para un compuesto determinado, el valor LEC obtenido a partir de este ensayo estuvo dentro del mismo intervalo que los valores CE50 obtenidos a partir de la "supresión del ensayo de replicación del VHC". Por lo tanto, es posible comparar el potencial de los compuestos para inducir IFN-I por PBMC, que se mide con cualquiera de los 2 ensayos.
Tabla II Actividad biológica de los compuestos.


















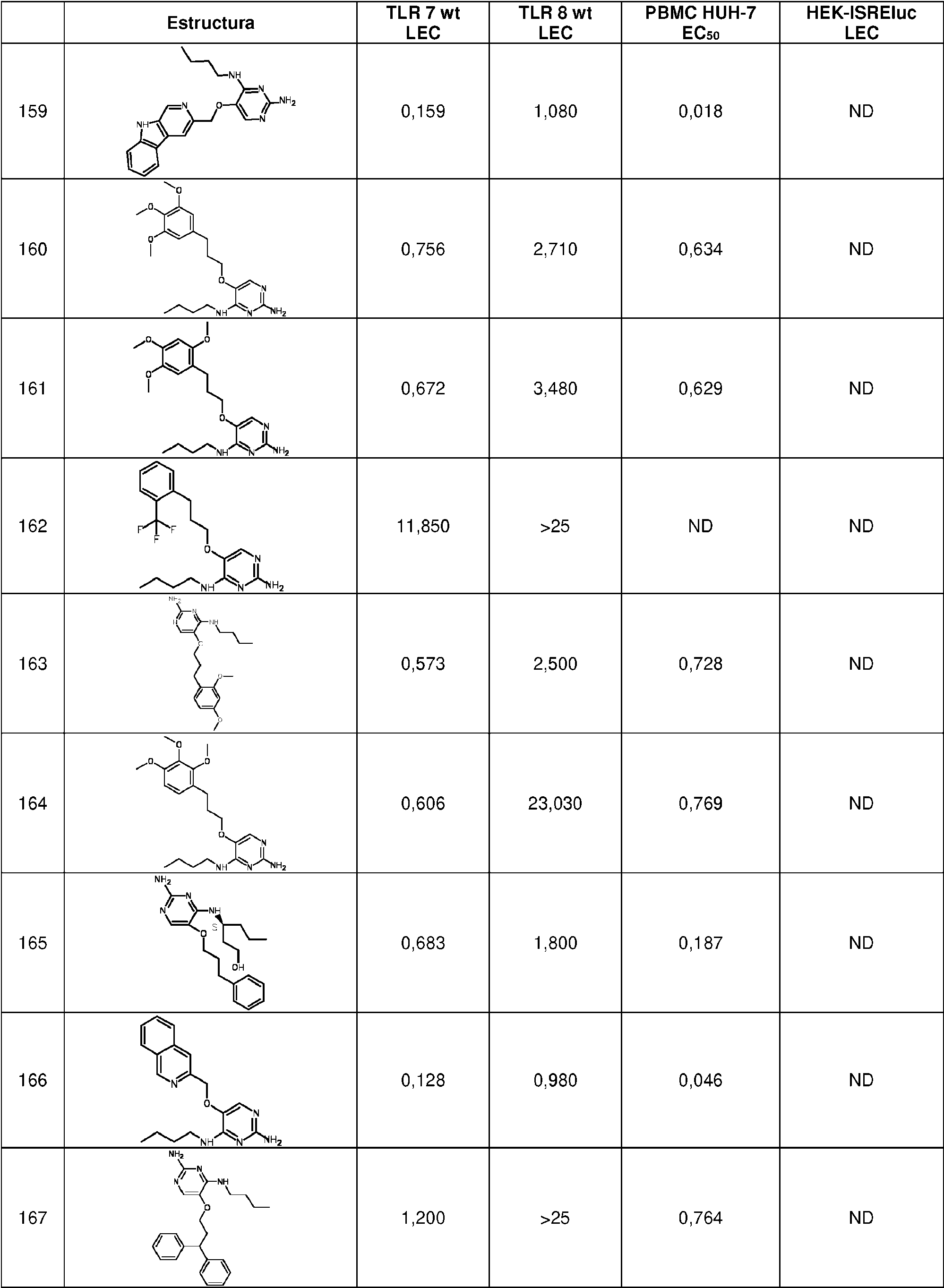















ND = No determinado.
Claims (11)
1. Un compuesto de fórmula (I)

o su sal, tautómero(s), solvato o polimorfos farmacéuticamente aceptable, en donde
R1 es hidrógeno, alquilo C1-4, ciclopropilo, alcoxi C1-6, halógeno, hidroxilo o trifluorometilo,
R2 es alquilo C1-8, alcoxi (C1-4)-alquilo (C1-4), cicloalquilo C3-7, heterociclo C4-7, heterociclo bicíclico, arilalquilo, heteroarilo o heteroarilalquilo, cada uno de los cuales está opcionalmente sustituido con uno o más sustituyentes independientemente seleccionados de halógeno, hidroxilo, amino, alquilo C1-6, di-alquilamino (C1-6), alquilamino C1-6, alquilo C1-6, alcoxi C1-6, cicloalquilo C3-6, ácido carboxílico, éster carboxílico, amida carboxílica, heterociclo, arilo, alquenilo, alquinilo, arilalquilo, heteroarilo, heteroarilalquilo, nitrilo; y
R3 es alquilo C4-8, alcoxi C4-8, alquenilo C2-6 o alquinilo C2-6, cada uno de los cuales está opcionalmente sustituido con uno o más sustituyentes independientemente seleccionados de halógeno, hidroxilo, amino, alquilo C1-3, alcoxi C1-3, cicloalquilo C3-6 y nitrilo.
2. El compuesto de la reivindicación 1, en donde R2 es alquilo C1-8, opcionalmente sustituido con uno o más sustituyentes independientemente seleccionados de halógeno, hidroxilo, amino, alquilo C1-6, di-alquilamino (C1-6), alquilamino C1-6, alquilo C1-6, alcoxi C1-6, cicloalquilo C3-6, ácido carboxílico, éster carboxílico, amida carboxílica, heterociclo, arilo, alquenilo, alquinilo, arilalquilo, heteroarilo, heteroarilalquilo y nitrilo.
3. El compuesto de la reivindicación 2, que se selecciona entre los compuestos
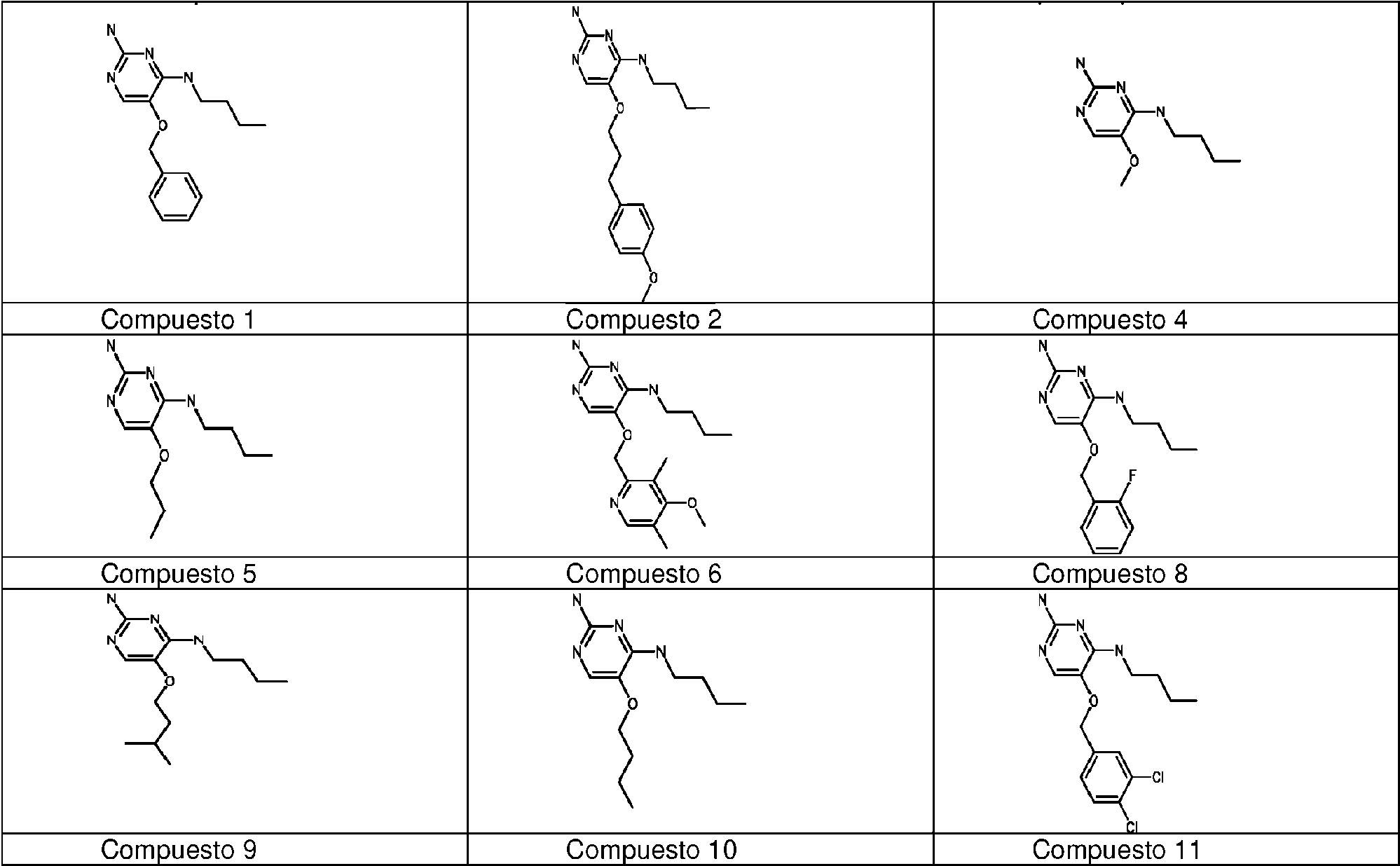













4. Un compuesto de fórmula (I) según la reivindicación 1, en donde R3 es butilo o pentilo y en donde R2 y Ri son como se ha especificado antes.
5. Un compuesto de fórmula (I) según la reivindicación 1, en donde R3 es alquilo C4-8 sustituido con hidroxilo, y en donde R2 y Ri son como se han especificado antes.
6. El compuesto de la reivindicación 5, en donde R2 es alquilo C1-8, alcoxi (Ci-4)-alquilo (C1-4), cicloalquilo C3-7, heterociclo C4-7, heterociclo bicíclico, arilalquilo, heteroarilo o heteroarilaquilo.
7. El compuesto de la reivindicación 6, que se selecciona entre los compuestos



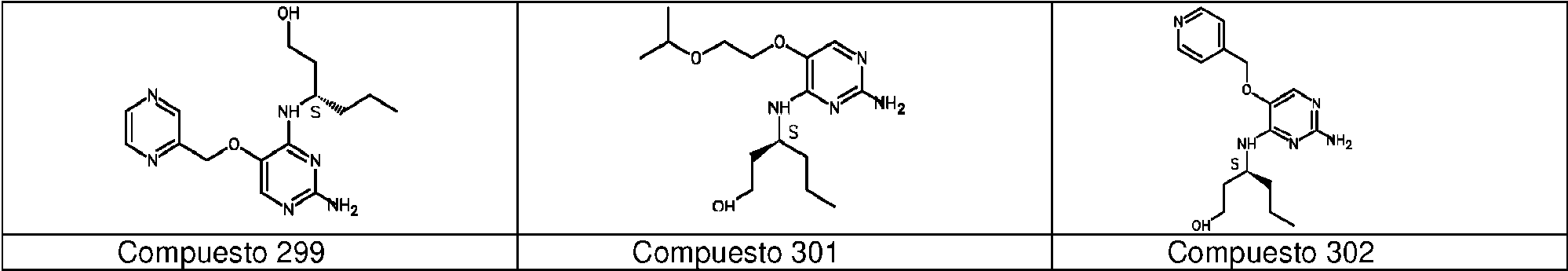
8. Un compuesto de fórmula (I) según la reivindicación 1, en donde Ri es hidrógeno o -CH3 y en donde R2 y R3 son como se han especificado antes.
9. Un compuesto de fórmula (I) según la reivindicación 1, en donde R2 es alquilo C1-3 sustituido con arilo, heterociclo o heteroarilo que está además sustituido con alquilo C1-3, alcoxi C1-6, éster carboxílico o amida carboxílica y en donde R1 y R3 son como se han especificado antes.
10. Una composición farmacéutica que comprende un compuesto de fórmula (I) o una de sus sales, tautómero(s), solvatos o polimorfos farmacéuticamente aceptables según una cualquiera de las reivindicaciones 1-9, junto con uno o más excipientes, diluyentes o vehículos farmacéuticamente aceptables.
11. Un compuesto de fórmula (I) o una de sus sales, tautómero(s), solvatos o polimorfos farmacéuticamente aceptables según una cualquiera de las reivindicaciones 1-9, o una composición farmacéutica según la reivindicación 10 para usar como un medicamento.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11161595 | 2011-04-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2786569T3 true ES2786569T3 (es) | 2020-10-13 |
Family
ID=43978015
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17203628T Active ES2786569T3 (es) | 2011-04-08 | 2012-04-10 | Derivados de pirimidina para el tratamiento de infecciones víricas |
| ES12712666.2T Active ES2691745T3 (es) | 2011-04-08 | 2012-04-10 | Derivados de pirimidina para el tratamiento de infecciones víricas |
| ES19192173T Active ES2887303T3 (es) | 2011-04-08 | 2012-04-10 | Derivados de pirimidina para el tratamiento de infecciones víricas |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES12712666.2T Active ES2691745T3 (es) | 2011-04-08 | 2012-04-10 | Derivados de pirimidina para el tratamiento de infecciones víricas |
| ES19192173T Active ES2887303T3 (es) | 2011-04-08 | 2012-04-10 | Derivados de pirimidina para el tratamiento de infecciones víricas |
Country Status (31)
| Country | Link |
|---|---|
| US (5) | US9422250B2 (es) |
| EP (3) | EP2694484B1 (es) |
| JP (1) | JP6046694B2 (es) |
| KR (3) | KR102024766B1 (es) |
| CN (1) | CN103608335B (es) |
| AU (4) | AU2012238564A1 (es) |
| BR (3) | BR122017025423B1 (es) |
| CA (1) | CA2832685C (es) |
| CL (1) | CL2013002877A1 (es) |
| CY (3) | CY1121363T1 (es) |
| DK (3) | DK2694484T3 (es) |
| EA (1) | EA027792B1 (es) |
| ES (3) | ES2786569T3 (es) |
| HR (3) | HRP20181464T1 (es) |
| HU (3) | HUE055286T2 (es) |
| IL (1) | IL228317A (es) |
| LT (3) | LT2694484T (es) |
| ME (1) | ME03782B (es) |
| MX (1) | MX349588B (es) |
| MY (1) | MY170941A (es) |
| PH (1) | PH12013502033B1 (es) |
| PL (3) | PL2694484T3 (es) |
| PT (3) | PT3330257T (es) |
| RS (3) | RS60191B1 (es) |
| SG (1) | SG194131A1 (es) |
| SI (3) | SI3330257T1 (es) |
| SM (3) | SMT201800527T1 (es) |
| TR (1) | TR201815062T4 (es) |
| UA (1) | UA113956C2 (es) |
| WO (1) | WO2012136834A1 (es) |
| ZA (1) | ZA201307472B (es) |
Families Citing this family (194)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EA027792B1 (ru) | 2011-04-08 | 2017-09-29 | Янссен Сайенсиз Айрлэнд Юси | Производные пиримидина для лечения вирусных инфекций |
| CN104066733A (zh) | 2011-11-09 | 2014-09-24 | 爱尔兰詹森研发公司 | 用于治疗病毒感染的嘌呤衍生物 |
| JP5977837B2 (ja) | 2011-12-21 | 2016-08-24 | ノヴィラ・セラピューティクス・インコーポレイテッド | B型肝炎抗ウイルス剤 |
| SG11201408542VA (en) | 2012-07-13 | 2015-02-27 | Janssen Sciences Ireland Uc | Macrocyclic purines for the treatment of viral infections |
| MX373711B (es) | 2012-08-28 | 2020-05-08 | Janssen Sciences Ireland Uc | Sulfamoilarilamidas y su uso como medicamentos para el tratamiento de la hepatitis b. |
| KR102252649B1 (ko) * | 2012-10-05 | 2021-05-17 | 얀센 사이언시즈 아일랜드 언리미티드 컴퍼니 | 바이러스 감염 및 추가 질환의 치료를 위한 아실아미노피리미딘 유도체 |
| KR102280595B1 (ko) | 2012-10-10 | 2021-07-22 | 얀센 사이언시즈 아일랜드 언리미티드 컴퍼니 | 바이러스 감염 및 다른 질환 치료를 위한 피롤로[3,2-d]피리미딘 유도체 |
| CN105051018B (zh) | 2012-11-16 | 2019-09-20 | 爱尔兰詹森科学公司 | 用于治疗病毒性感染的杂环的经取代的2-氨基-喹唑啉衍生物 |
| CN105189468B (zh) | 2013-02-21 | 2018-10-30 | 爱尔兰詹森科学公司 | 用于治疗病毒性感染的2-氨基嘧啶衍生物 |
| DK2961732T3 (en) | 2013-02-28 | 2017-07-10 | Janssen Sciences Ireland Uc | SULFAMOYLARYLAMIDS AND USE THEREOF AS MEDICINES TO TREAT HEPATITIS B |
| US8993771B2 (en) | 2013-03-12 | 2015-03-31 | Novira Therapeutics, Inc. | Hepatitis B antiviral agents |
| MX366481B (es) | 2013-03-29 | 2019-07-09 | Janssen Sciences Ireland Uc | Deaza-purinonas macrociclicas para el tratamiento de infecciones virales. |
| CN105102451B (zh) | 2013-04-03 | 2018-09-18 | 爱尔兰詹森科学公司 | N-苯基-氨甲酰衍生物及其作为药物用于治疗乙型肝炎的用途 |
| JO3603B1 (ar) | 2013-05-17 | 2020-07-05 | Janssen Sciences Ireland Uc | مشتقات سلفامويل بيرولاميد واستخدامها كادوية لمعالجة التهاب الكبد نوع بي |
| DK3004074T3 (da) * | 2013-05-24 | 2018-01-29 | Janssen Sciences Ireland Uc | Pyridonderivater til behandling af virusinfektioner og yderligere sygdomme |
| NO3030563T3 (es) | 2013-06-27 | 2018-01-06 | ||
| KR102244937B1 (ko) | 2013-07-25 | 2021-04-27 | 얀센 사이언시즈 아일랜드 언리미티드 컴퍼니 | 글리옥사미드 치환된 피롤아미드 유도체 및 b형 간염 치료용 의약으로서의 이의 용도 |
| MX368625B (es) * | 2013-07-30 | 2019-10-08 | Janssen Sciences Ireland Uc | Derivados de tieno[3,2-d]pirimidinas para el tratamiento de infecciones virales. |
| DK3060547T3 (en) | 2013-10-23 | 2018-01-15 | Janssen Sciences Ireland Uc | CARBOXAMIDE DERIVATIVES AND USE THEREOF AS MEDICINES FOR TREATMENT OF HEPATITS B |
| US10392349B2 (en) | 2014-01-16 | 2019-08-27 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis B infections |
| US9169212B2 (en) | 2014-01-16 | 2015-10-27 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis B infections |
| US9181288B2 (en) | 2014-01-16 | 2015-11-10 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis B infections |
| EP3102225B1 (en) | 2014-02-05 | 2020-03-25 | Novira Therapeutics Inc. | Combination therapy for treatment of hbv infections |
| US11078193B2 (en) | 2014-02-06 | 2021-08-03 | Janssen Sciences Ireland Uc | Sulphamoylpyrrolamide derivatives and the use thereof as medicaments for the treatment of hepatitis B |
| US9400280B2 (en) | 2014-03-27 | 2016-07-26 | Novira Therapeutics, Inc. | Piperidine derivatives and methods of treating hepatitis B infections |
| JP2017522334A (ja) * | 2014-07-24 | 2017-08-10 | バイエル・クロップサイエンス・アクチェンゲゼルシャフト | 殺真菌性ピラゾール誘導体 |
| KR102394917B1 (ko) * | 2015-03-04 | 2022-05-09 | 길리애드 사이언시즈, 인코포레이티드 | 톨 유사 수용체 조정제 화합물 |
| US9884831B2 (en) | 2015-03-19 | 2018-02-06 | Novira Therapeutics, Inc. | Azocane and azonane derivatives and methods of treating hepatitis B infections |
| US10875876B2 (en) | 2015-07-02 | 2020-12-29 | Janssen Sciences Ireland Uc | Cyclized sulfamoylarylamide derivatives and the use thereof as medicaments for the treatment of hepatitis B |
| JP2018525412A (ja) | 2015-08-26 | 2018-09-06 | ギリアード サイエンシーズ, インコーポレイテッド | 重水素化トール様受容体調節因子 |
| WO2017048727A1 (en) | 2015-09-15 | 2017-03-23 | Gilead Sciences, Inc. | Modulators of toll-like recptors for the treatment of hiv |
| TW201718496A (zh) | 2015-09-29 | 2017-06-01 | 諾維拉治療公司 | B型肝炎抗病毒劑之晶型 |
| MA43298A (fr) | 2015-11-27 | 2021-04-14 | Janssen Sciences Ireland Unlimited Co | Derives heterocycliques d'indole pour des infections par le virus de la influenza |
| CR20180321A (es) | 2015-12-15 | 2018-08-21 | Gilead Sciences Inc | Anticuerpos neutralizadores del virus de inmunodeficiencia humana |
| KR20180100375A (ko) * | 2016-01-20 | 2018-09-10 | 얀센 사이언시즈 아일랜드 유씨 | 인플루엔자 바이러스 감염에서 사용하기 위한 아릴 치환 피리미딘 |
| CN109640980A (zh) | 2016-04-15 | 2019-04-16 | 诺维拉治疗公司 | 包含壳体装配抑制剂的组合和方法 |
| MX2018014377A (es) | 2016-05-27 | 2019-03-14 | Gilead Sciences Inc | Metodos para tratar infecciones por virus de hepatitis b usando inhibidores de proteina no estructural 5a (ns5a), proteina no estructural 5b (ns5b) o proteina no estructural 3 (ns3). |
| BR102017010009A2 (pt) | 2016-05-27 | 2017-12-12 | Gilead Sciences, Inc. | Compounds for the treatment of hepatitis b virus infection |
| CA3027471A1 (en) | 2016-07-01 | 2018-01-04 | Janssen Sciences Ireland Unlimited Company | Dihydropyranopyrimidines for the treatment of viral infections |
| JOP20190024A1 (ar) | 2016-08-26 | 2019-02-19 | Gilead Sciences Inc | مركبات بيروليزين بها استبدال واستخداماتها |
| PT3507276T (pt) | 2016-09-02 | 2022-01-11 | Gilead Sciences Inc | Compostos moduladores do recetor de tipo toll |
| WO2018045150A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | 4,6-diamino-pyrido[3,2-d]pyrimidine derivaties as toll like receptor modulators |
| CN109790154B (zh) * | 2016-09-29 | 2023-06-23 | 爱尔兰詹森科学公司 | 用于治疗病毒感染和另外的疾病的嘧啶前药 |
| MA46535A (fr) | 2016-10-14 | 2019-08-21 | Prec Biosciences Inc | Méganucléases modifiées spécifiques de séquences de reconnaissance dans le génome du virus de l'hépatite b |
| CA3045517A1 (en) * | 2016-12-05 | 2018-06-14 | Apros Therapeutics, Inc. | Pyrimidine compounds containing acidic groups |
| TWI714820B (zh) | 2017-01-31 | 2021-01-01 | 美商基利科學股份有限公司 | 替諾福韋艾拉酚胺(tenofovir alafenamide)之晶型 |
| JOP20180008A1 (ar) | 2017-02-02 | 2019-01-30 | Gilead Sciences Inc | مركبات لعلاج إصابة بعدوى فيروس الالتهاب الكبدي b |
| FI3603619T3 (fi) | 2017-03-29 | 2025-09-25 | Sumitomo Pharma Co Ltd | Rokoteadjuvanttiformulaatio |
| JOP20180040A1 (ar) | 2017-04-20 | 2019-01-30 | Gilead Sciences Inc | مثبطات pd-1/pd-l1 |
| AR112412A1 (es) | 2017-08-17 | 2019-10-23 | Gilead Sciences Inc | Formas de sal de colina de un inhibidor de la cápside del vih |
| TWI687415B (zh) | 2017-08-17 | 2020-03-11 | 美商基利科學股份有限公司 | Hiv蛋白質膜抑制劑之固體形式 |
| JP6934562B2 (ja) | 2017-08-22 | 2021-09-15 | ギリアード サイエンシーズ, インコーポレイテッド | 治療用複素環式化合物 |
| WO2019077104A1 (en) * | 2017-10-20 | 2019-04-25 | Sabic Global Technologies B.V. | NOVEL SYNTHESIS PROCESS FOR THE PREPARATION OF DIBENZOATE COMPOUNDS, SUCH AS 4- [BENZOYL (METHYL) AMINO] PENTANE-2-YL DIBENZOATE |
| WO2019084060A1 (en) | 2017-10-24 | 2019-05-02 | Silverback Therapeutics, Inc. | CONJUGATES AND METHODS OF USE FOR THE SELECTIVE DELIVERY OF IMMUNOMODULATORY AGENTS |
| WO2019084271A1 (en) * | 2017-10-25 | 2019-05-02 | Children's Medical Center Corporation | PAPD5 INHIBITORS AND METHODS OF USE |
| WO2019118884A1 (en) | 2017-12-15 | 2019-06-20 | Silverback Therapeutics, Inc. | Antibody construct-drug conjugate for the treatment of hepatitis |
| AU2018392213B2 (en) | 2017-12-20 | 2021-03-04 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3' cyclic dinucleotides with phosphonate bond activating the STING adaptor protein |
| WO2019123339A1 (en) | 2017-12-20 | 2019-06-27 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3' cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| US12226480B2 (en) | 2017-12-21 | 2025-02-18 | Sumitomo Pharma Co., Ltd. | Combination drug including TLR7 agonist |
| SI3752501T1 (sl) | 2018-02-13 | 2023-08-31 | Gilead Sciences, Inc. | Inhibitorji pd-1/pd-l1 |
| PL3752495T3 (pl) | 2018-02-15 | 2024-01-15 | Gilead Sciences, Inc. | Pochodne pirydynowe i ich zastosowanie do leczenia infekcji hiv |
| CA3175384A1 (en) | 2018-02-16 | 2019-08-22 | Gilead Sciences, Inc. | Methods and intermediates for preparing therapeutic compounds useful in the treatment of retroviridae viral infection |
| JP7050165B2 (ja) | 2018-02-26 | 2022-04-07 | ギリアード サイエンシーズ, インコーポレイテッド | Hbv複製阻害剤としての置換ピロリジン化合物 |
| TW201945003A (zh) | 2018-03-01 | 2019-12-01 | 愛爾蘭商健生科學愛爾蘭無限公司 | 2,4-二胺基喹唑啉衍生物及其醫學用途 |
| JP2021515769A (ja) | 2018-03-14 | 2021-06-24 | ヤンセン・サイエンシズ・アイルランド・アンリミテッド・カンパニー | カプシド集合調節剤の投薬レジメン |
| EP3774883A1 (en) | 2018-04-05 | 2021-02-17 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis b virus protein x |
| TWI833744B (zh) | 2018-04-06 | 2024-03-01 | 捷克科學院有機化學與生物化學研究所 | 3'3'-環二核苷酸 |
| TWI818007B (zh) | 2018-04-06 | 2023-10-11 | 捷克科學院有機化學與生物化學研究所 | 2'3'-環二核苷酸 |
| TW202005654A (zh) | 2018-04-06 | 2020-02-01 | 捷克科學院有機化學與生物化學研究所 | 2,2,─環二核苷酸 |
| TW201945388A (zh) | 2018-04-12 | 2019-12-01 | 美商精密生物科學公司 | 對b型肝炎病毒基因體中之識別序列具有特異性之最佳化之經工程化巨核酸酶 |
| CA3093130C (en) | 2018-04-19 | 2023-10-17 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| TW202014193A (zh) | 2018-05-03 | 2020-04-16 | 捷克科學院有機化學與生物化學研究所 | 包含碳環核苷酸之2’3’-環二核苷酸 |
| US11168130B2 (en) | 2018-07-03 | 2021-11-09 | Gilead Sciences, Inc. | Antibodies that target HIV GP120 and methods of use |
| EP3818052A1 (en) | 2018-07-06 | 2021-05-12 | Gilead Sciences, Inc. | Therapeutic heterocyclic compounds |
| WO2020010223A1 (en) | 2018-07-06 | 2020-01-09 | Gilead Sciences, Inc. | Therapeutic heterocyclic compounds |
| SI3820572T1 (sl) | 2018-07-13 | 2023-12-29 | Gilead Sciences, Inc. | Inhibitorji pd-1/pd-l1 |
| CN120053445A (zh) | 2018-07-16 | 2025-05-30 | 吉利德科学公司 | 用于治疗hiv的衣壳抑制剂 |
| PH12021550175A1 (en) | 2018-07-23 | 2022-07-11 | Japan As Represented By Director General Of Nat Institute Of Infectious Diseases | Composition containing influenza vaccine |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| CN112673007A (zh) * | 2018-09-11 | 2021-04-16 | 豪夫迈·罗氏有限公司 | 用于治疗自身免疫性疾病的吡唑并吡啶胺化合物 |
| JP2022500414A (ja) | 2018-09-12 | 2022-01-04 | シルバーバック セラピューティックス インコーポレイテッド | 免疫刺激性コンジュゲートにより疾患を処置する方法および組成物 |
| EP3860717A1 (en) | 2018-10-03 | 2021-08-11 | Gilead Sciences, Inc. | Imidozopyrimidine derivatives |
| JP7158577B2 (ja) | 2018-10-24 | 2022-10-21 | ギリアード サイエンシーズ, インコーポレイテッド | Pd-1/pd-l1阻害剤 |
| TWI721624B (zh) | 2018-10-31 | 2021-03-11 | 美商基利科學股份有限公司 | 經取代之6-氮雜苯并咪唑化合物 |
| CA3117556A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| MX2021009496A (es) | 2019-02-08 | 2021-09-08 | Progeneer Inc | Complejo de colesterol-agonista del receptor 7 u 8 tipo larga distancia, y uso del mismo. |
| MA55020A (fr) | 2019-02-22 | 2021-12-29 | Janssen Sciences Ireland Unlimited Co | Dérivés d'amide utiles dans le traitement d'une infection par le virus de l'hépatite b ou de maladies induites par le virus de l'hépatite b |
| WO2020176505A1 (en) | 2019-02-25 | 2020-09-03 | Gilead Sciences, Inc. | Protein kinase c agonists |
| WO2020176510A1 (en) | 2019-02-25 | 2020-09-03 | Gilead Sciences, Inc. | Protein kinase c agonists |
| EP3935065A1 (en) | 2019-03-07 | 2022-01-12 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020178770A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| JP7350871B2 (ja) | 2019-03-07 | 2023-09-26 | インスティチュート オブ オーガニック ケミストリー アンド バイオケミストリー エーエスシーアール,ヴイ.ヴイ.アイ. | 2’3’-環状ジヌクレオチドおよびそのプロドラッグ |
| HUE059677T2 (hu) | 2019-03-22 | 2022-12-28 | Gilead Sciences Inc | Áthidalt triciklusos karbamoilpiridon-vegyületek és ezek gyógyszerészeti alkalmazása |
| TW202210480A (zh) | 2019-04-17 | 2022-03-16 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| TWI751517B (zh) | 2019-04-17 | 2022-01-01 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| CN113993848A (zh) | 2019-04-24 | 2022-01-28 | 儿童医学中心公司 | Papd5抑制剂及其使用方法 |
| AR119732A1 (es) | 2019-05-06 | 2022-01-05 | Janssen Sciences Ireland Unlimited Co | Derivados de amida útiles en el tratamiento de la infección por vhb o de enfermedades inducidas por vhb |
| TWI762925B (zh) | 2019-05-21 | 2022-05-01 | 美商基利科學股份有限公司 | 鑑別對使用gp120 v3聚醣導向之抗體的治療敏感之hiv病患的方法 |
| EP3972695A1 (en) | 2019-05-23 | 2022-03-30 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| WO2020255042A1 (en) | 2019-06-18 | 2020-12-24 | Janssen Sciences Ireland Unlimited Company | Combination of hepatitis b virus (hbv) vaccines and a pyrimidine derivative |
| WO2020255035A1 (en) | 2019-06-18 | 2020-12-24 | Janssen Sciences Ireland Unlimited Company | Combination of hepatitis b virus (hbv) vaccines and pyrimidine derivatives |
| CA3140708A1 (en) | 2019-06-18 | 2020-12-24 | Helen Horton | Combination of hepatitis b virus (hbv) vaccines and pyridopyrimidine derivatives |
| US20220241402A1 (en) | 2019-06-18 | 2022-08-04 | Janssen Sciences Ireland Unlimited Company | Combination of hepatitis b virus (hbv) vaccines and quinazoline derivatives |
| US20220249685A1 (en) | 2019-06-19 | 2022-08-11 | Silverback Therapeutics, Inc. | Anti-mesothelin antibodies and immunoconjugates thereof |
| CN114245807B (zh) | 2019-06-25 | 2025-05-02 | 吉利德科学公司 | Flt3l-fc融合蛋白和使用方法 |
| TWI879779B (zh) | 2019-06-28 | 2025-04-11 | 美商基利科學股份有限公司 | 類鐸受體調節劑化合物的製備方法 |
| CN112174823B (zh) * | 2019-07-01 | 2023-12-01 | 南京富润凯德生物医药有限公司 | 一种合成2,2-二甲基-3-氧杂环丁酮的中间体及其制备方法和应用 |
| KR20220047277A (ko) | 2019-07-16 | 2022-04-15 | 길리애드 사이언시즈, 인코포레이티드 | Hiv 백신, 및 이의 제조 및 사용 방법 |
| US20220257619A1 (en) | 2019-07-18 | 2022-08-18 | Gilead Sciences, Inc. | Long-acting formulations of tenofovir alafenamide |
| EP4017476A1 (en) | 2019-08-19 | 2022-06-29 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| MY208114A (en) | 2019-09-30 | 2025-04-16 | Gilead Sciences Inc | Hbv vaccines and methods treating hbv |
| WO2021067644A1 (en) | 2019-10-01 | 2021-04-08 | Silverback Therapeutics, Inc. | Combination therapy with immune stimulatory conjugates |
| US11795223B2 (en) | 2019-10-18 | 2023-10-24 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| NZ786589A (en) | 2019-10-31 | 2025-03-28 | Forty Seven Llc | Anti-cd47 and anti-cd20 based treatment of blood cancer |
| TWI778443B (zh) | 2019-11-12 | 2022-09-21 | 美商基利科學股份有限公司 | Mcl1抑制劑 |
| EP4643882A2 (en) | 2019-11-26 | 2025-11-05 | Gilead Sciences, Inc. | Capsid inhibitors for the prevention of hiv |
| CN116057068A (zh) | 2019-12-06 | 2023-05-02 | 精密生物科学公司 | 对乙型肝炎病毒基因组中的识别序列具有特异性的优化的工程化大范围核酸酶 |
| MX2022007930A (es) | 2019-12-24 | 2022-08-08 | Carna Biosciences Inc | Compuestos moduladores de diacilglicerol quinasa. |
| TWI832035B (zh) | 2020-02-14 | 2024-02-11 | 美商基利科學股份有限公司 | 結合ccr8之抗體及融合蛋白及其用途 |
| US11179473B2 (en) | 2020-02-21 | 2021-11-23 | Silverback Therapeutics, Inc. | Nectin-4 antibody conjugates and uses thereof |
| JP2023516372A (ja) | 2020-03-02 | 2023-04-19 | プロジェニア インコーポレイテッド | 病原菌外壁成分基盤の生病原体模倣ナノ粒子及びその製造方法 |
| WO2021188959A1 (en) | 2020-03-20 | 2021-09-23 | Gilead Sciences, Inc. | Prodrugs of 4'-c-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using the same |
| EP4143194A1 (en) | 2020-05-01 | 2023-03-08 | Gilead Sciences, Inc. | Cd73 inhibiting 2,4-dioxopyrimidine compounds |
| EP4153181A1 (en) | 2020-05-21 | 2023-03-29 | Gilead Sciences, Inc. | Pharmaceutical compositions comprising bictegravir |
| WO2021262990A1 (en) | 2020-06-25 | 2021-12-30 | Gilead Sciences, Inc. | Capsid inhibitors for the treatment of hiv |
| CN116209678A (zh) | 2020-07-01 | 2023-06-02 | 安尔士制药公司 | 抗asgr1抗体缀合物及其用途 |
| EP4194008A4 (en) | 2020-08-04 | 2025-01-08 | Progeneer Inc. | Kinetically acting adjuvant ensemble |
| JP7690222B2 (ja) | 2020-08-04 | 2025-06-10 | プロジェニア インコーポレイテッド | 動力学的制御が可能なアジュバントを含むmRNAワクチン |
| JP2023536954A (ja) | 2020-08-04 | 2023-08-30 | プロジェニア インコーポレイテッド | 活性化部位が一時的に不活性化したトール様受容体7または8作用薬と機能性薬物の結合体およびその用途 |
| BR112023002164A2 (pt) | 2020-08-07 | 2023-03-14 | Gilead Sciences Inc | Profármacos de análogos de nucleotídeos de fosfonamida e seu uso farmacêutico |
| TWI815194B (zh) | 2020-10-22 | 2023-09-11 | 美商基利科學股份有限公司 | 介白素2-Fc融合蛋白及使用方法 |
| CA3195799A1 (en) | 2020-11-11 | 2022-05-19 | Stephen R. Martin | Methods of identifying hiv patients sensitive to therapy with gp120 cd4 binding site-directed antibodies |
| TW202304524A (zh) | 2021-04-10 | 2023-02-01 | 美商普方生物製藥美國公司 | Folr1結合劑、其結合物及使用方法 |
| TW202302145A (zh) | 2021-04-14 | 2023-01-16 | 美商基利科學股份有限公司 | CD47/SIRPα結合及NEDD8活化酶E1調節次單元之共抑制以用於治療癌症 |
| AU2022262644A1 (en) | 2021-04-23 | 2023-11-09 | Genmab A/S | Anti-cd70 antibodies, conjugates thereof and methods of using the same |
| TW202348237A (zh) | 2021-05-13 | 2023-12-16 | 美商基利科學股份有限公司 | TLR8調節化合物及抗HBV siRNA療法之組合 |
| TW202313094A (zh) | 2021-05-18 | 2023-04-01 | 美商基利科學股份有限公司 | 使用FLT3L—Fc融合蛋白之方法 |
| JP7686091B2 (ja) | 2021-06-23 | 2025-05-30 | ギリアード サイエンシーズ, インコーポレイテッド | ジアシルグリセロールキナーゼ調節化合物 |
| CA3222277A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| EP4359389A1 (en) | 2021-06-23 | 2024-05-01 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| AU2022298639C1 (en) | 2021-06-23 | 2025-07-17 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| TW202320857A (zh) | 2021-07-06 | 2023-06-01 | 美商普方生物製藥美國公司 | 連接子、藥物連接子及其結合物及其使用方法 |
| CA3234909A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| JP2024541979A (ja) | 2021-10-29 | 2024-11-13 | ギリアード サイエンシーズ, インコーポレイテッド | Cd73化合物 |
| EP4440702B1 (en) | 2021-12-03 | 2025-05-21 | Gilead Sciences, Inc. | Therapeutic compounds for hiv virus infection |
| JP7736929B2 (ja) | 2021-12-03 | 2025-09-09 | ギリアード サイエンシーズ, インコーポレイテッド | Hivウイルス感染症のための治療化合物 |
| US12084467B2 (en) | 2021-12-03 | 2024-09-10 | Gilead Sciences, Inc. | Therapeutic compounds for HIV virus infection |
| US20230220106A1 (en) | 2021-12-08 | 2023-07-13 | Dragonfly Therapeutics, Inc. | Antibodies targeting 5t4 and uses thereof |
| WO2023107956A1 (en) | 2021-12-08 | 2023-06-15 | Dragonfly Therapeutics, Inc. | Proteins binding nkg2d, cd16 and 5t4 |
| CN118488946A (zh) | 2021-12-22 | 2024-08-13 | 吉利德科学公司 | Ikaros锌指家族降解剂及其用途 |
| KR20240125012A (ko) | 2021-12-22 | 2024-08-19 | 길리애드 사이언시즈, 인코포레이티드 | 이카로스 아연 핑거 패밀리 분해제 및 이의 용도 |
| TW202340168A (zh) | 2022-01-28 | 2023-10-16 | 美商基利科學股份有限公司 | Parp7抑制劑 |
| DK4245756T3 (da) | 2022-03-17 | 2024-10-21 | Gilead Sciences Inc | Ikaros zinkfinger-familiens nedbrydere og anvendelse heraf |
| US20230355796A1 (en) | 2022-03-24 | 2023-11-09 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| TWI876305B (zh) | 2022-04-05 | 2025-03-11 | 美商基利科學股份有限公司 | 用於治療結腸直腸癌之組合療法 |
| TWI843506B (zh) | 2022-04-06 | 2024-05-21 | 美商基利科學股份有限公司 | 橋聯三環胺甲醯基吡啶酮化合物及其用途 |
| IL316058A (en) | 2022-04-21 | 2024-11-01 | Gilead Sciences Inc | Compounds modulate KRAS G12D |
| CR20240570A (es) | 2022-07-01 | 2025-03-03 | Gilead Sciences Inc | Compuestos de cd73 |
| WO2024006982A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Therapeutic compounds useful for the prophylactic or therapeutic treatment of an hiv virus infection |
| AU2023307100A1 (en) | 2022-07-12 | 2025-01-02 | Gilead Sciences, Inc. | Hiv immunogenic polypeptides and vaccines and uses thereof |
| CN116120282B (zh) * | 2022-08-04 | 2024-02-20 | 苏州系统医学研究所 | 具有ev71和/或cva16病毒抑制活性的化合物及其应用 |
| US20240083984A1 (en) | 2022-08-26 | 2024-03-14 | Gilead Sciences, Inc. | Dosing and scheduling regimen for broadly neutralizing antibodies |
| WO2024064668A1 (en) | 2022-09-21 | 2024-03-28 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPα DISRUPTION ANTICANCER COMBINATION THERAPY |
| US20240226130A1 (en) | 2022-10-04 | 2024-07-11 | Gilead Sciences, Inc. | 4'-thionucleoside analogues and their pharmaceutical use |
| KR20250122479A (ko) | 2022-12-22 | 2025-08-13 | 길리애드 사이언시즈, 인코포레이티드 | Prmt5 억제제 및 이의 용도 |
| US20240383922A1 (en) | 2023-04-11 | 2024-11-21 | Gilead Sciences, Inc. | KRAS Modulating Compounds |
| AR132464A1 (es) | 2023-04-19 | 2025-07-02 | Gilead Sciences Inc | Régimen de dosificación de inhibidor de la cápside |
| AU2024259556A1 (en) | 2023-04-21 | 2025-10-23 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2024249573A1 (en) | 2023-05-31 | 2024-12-05 | Gilead Sciences, Inc. | Solid forms of compounds useful in the treatment of hiv |
| TW202448483A (zh) | 2023-05-31 | 2024-12-16 | 美商基利科學股份有限公司 | 用於hiv之治療性化合物 |
| WO2025006720A1 (en) | 2023-06-30 | 2025-01-02 | Gilead Sciences, Inc. | Kras modulating compounds |
| WO2025024663A1 (en) | 2023-07-26 | 2025-01-30 | Gilead Sciences, Inc. | Parp7 inhibitors |
| US20250100998A1 (en) | 2023-07-26 | 2025-03-27 | Gilead Sciences, Inc. | Parp7 inhibitors |
| WO2025029247A1 (en) | 2023-07-28 | 2025-02-06 | Gilead Sciences, Inc. | Weekly regimen of lenacapavir for the treatment and prevention of hiv |
| WO2025042394A1 (en) | 2023-08-23 | 2025-02-27 | Gilead Sciences, Inc. | Dosing regimen of hiv capsid inhibitor |
| US20250109147A1 (en) | 2023-09-08 | 2025-04-03 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2025054347A1 (en) | 2023-09-08 | 2025-03-13 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2025072406A1 (en) | 2023-09-26 | 2025-04-03 | Profoundbio Us Co. | Ptk7 binding agents, conjugates thereof and methods of using the same |
| TW202530226A (zh) | 2023-10-11 | 2025-08-01 | 美商基利科學股份有限公司 | 橋聯三環胺甲醯基吡啶酮化合物及其用途 |
| TW202515549A (zh) | 2023-10-11 | 2025-04-16 | 美商基利科學股份有限公司 | 橋聯三環胺甲醯基吡啶酮化合物及其用途 |
| WO2025080863A1 (en) | 2023-10-11 | 2025-04-17 | Gilead Sciences, Inc. | Bridged tricyclic carbamoylpyridone compounds and uses thereof |
| US20250154172A1 (en) | 2023-11-03 | 2025-05-15 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| US20250230168A1 (en) | 2023-12-22 | 2025-07-17 | Gilead Sciences, Inc. | Azaspiro wrn inhibitors |
| WO2025137245A1 (en) | 2023-12-22 | 2025-06-26 | Gilead Sciences, Inc. | Solid forms of hiv integrase inhibitors |
| US20250296992A1 (en) | 2024-01-10 | 2025-09-25 | Genmab A/S | Slitrk6 binding agents, conjugates thereof and methods of using the same |
| WO2025181219A1 (en) | 2024-02-29 | 2025-09-04 | Genmab A/S | Egfr and c-met bispecific binding agents, conjugates thereof and methods of using the same |
| US20250333424A1 (en) | 2024-03-01 | 2025-10-30 | Gilead Sciences, Inc. | Antiviral compounds |
| WO2025184447A1 (en) | 2024-03-01 | 2025-09-04 | Gilead Sciences, Inc. | Pharmaceutical compositions comprising hiv integrase inhibitors |
| US20250289822A1 (en) | 2024-03-01 | 2025-09-18 | Gilead Sciences, Inc. | Solid forms of hiv integrase inhibitors |
| WO2025240246A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| WO2025240244A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies comprising bulevirtide and lonafarnib for use in the treatment of hepatitis d virus infection |
| US20250345389A1 (en) | 2024-05-13 | 2025-11-13 | Gilead Sciences, Inc. | Combination therapies |
| WO2025240242A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| WO2025245003A1 (en) | 2024-05-21 | 2025-11-27 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
Family Cites Families (90)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2610889B2 (ja) | 1987-09-03 | 1997-05-14 | 日本臓器製薬株式会社 | 新規架橋アデニン誘導体 |
| ATE283855T1 (de) | 1996-07-03 | 2004-12-15 | Sumitomo Pharma | Neue purinderivate |
| DE69736711T2 (de) | 1996-08-28 | 2007-09-20 | Pfizer Inc. | Substituierte 6,5-heterobicyclische-derivate |
| AU733316B2 (en) | 1996-10-04 | 2001-05-10 | Kyorin Pharmaceutical Co. Ltd. | Pyrazolopyridylpyridazinone derivatives and process for preparing the same |
| AR012634A1 (es) | 1997-05-02 | 2000-11-08 | Sugen Inc | Compuesto basado en quinazolina, composicion famaceutica que lo comprende, metodo para sintetizarlo, su uso, metodos de modulacion de la funcion deserina/treonina proteinaquinasa con dicho compuesto y metodo in vitro para identificar compuestos que modulan dicha funcion |
| US6339089B2 (en) | 1997-08-13 | 2002-01-15 | Fujirebio Inc. | Pyrimidine nucleus-containing compound and a medicament containing the same for a blood oxygen partial pressure amelioration, and a method for preparing the same |
| CA2311742C (en) | 1997-11-28 | 2009-06-16 | Sumitomo Pharmaceuticals Co., Ltd. | 6-amino-9-benzyl-8-hydroxypurine derivatives |
| TW572758B (en) | 1997-12-22 | 2004-01-21 | Sumitomo Pharma | Type 2 helper T cell-selective immune response inhibitors comprising purine derivatives |
| US6187777B1 (en) | 1998-02-06 | 2001-02-13 | Amgen Inc. | Compounds and methods which modulate feeding behavior and related diseases |
| KR20010041015A (ko) | 1998-02-17 | 2001-05-15 | 윌리엄 제이. 리플린 | 항바이러스성 피리미딘 유도체 |
| US6110929A (en) | 1998-07-28 | 2000-08-29 | 3M Innovative Properties Company | Oxazolo, thiazolo and selenazolo [4,5-c]-quinolin-4-amines and analogs thereof |
| JP4315300B2 (ja) | 1998-08-10 | 2009-08-19 | 大日本住友製薬株式会社 | 新規なキナゾリン誘導体 |
| JP4342007B2 (ja) | 1998-08-10 | 2009-10-14 | 大日本住友製薬株式会社 | キナゾリン誘導体 |
| DE69917469T2 (de) | 1998-08-27 | 2005-05-12 | Sumitomo Pharmaceuticals Co., Ltd. | Pyrimidin derivate |
| US6583148B1 (en) * | 1999-04-08 | 2003-06-24 | Krenitsky Pharmaceuticals, Inc. | Neurotrophic substituted pyrimidines |
| CA2323008C (en) | 1999-10-11 | 2005-07-12 | Pfizer Inc. | Pharmaceutically active compounds |
| WO2002088079A2 (en) | 2001-05-01 | 2002-11-07 | Bristol-Myers Squibb Company | Dual inhibitors of pde 7 and pde 4 |
| AU2002364211A1 (en) | 2001-12-21 | 2003-07-15 | Bayer Pharmaceuticals Corporation | Thienopyrimidine derivative compounds as inhibitors of prolylpeptidase, inducers of apoptosis and cancer treatment agents |
| TW200407143A (en) | 2002-05-21 | 2004-05-16 | Bristol Myers Squibb Co | Pyrrolotriazinone compounds and their use to treat diseases |
| US7091232B2 (en) | 2002-05-21 | 2006-08-15 | Allergan, Inc. | 4-(substituted cycloalkylmethyl) imidazole-2-thiones, 4-(substituted cycloalkenylmethyl) imidazole-2-thiones, 4-(substituted cycloalkylmethyl) imidazol-2-ones and 4-(substituted cycloalkenylmethyl) imidazol-2-ones and related compounds |
| AU2003242252A1 (en) | 2002-06-07 | 2003-12-22 | Kyowa Hakko Kogyo Co., Ltd. | Bicyclic pyrimidine derivatives |
| NZ539064A (en) | 2002-09-27 | 2007-09-28 | Dainippon Sumitomo Pharma Co | Novel adenine compound and use thereof |
| US8455458B2 (en) | 2002-10-16 | 2013-06-04 | Arthrodynamic Technologies, Animal Health Division, Inc. | Composition and method for treating connective tissue damage |
| EA200600069A1 (ru) | 2003-06-20 | 2006-08-25 | Коли Фармасьютикал Гмбх | Низкомолекулярные антагонисты toll-подобных рецепторов (tlr) |
| WO2005025583A2 (en) | 2003-09-05 | 2005-03-24 | Anadys Pharmaceuticals, Inc. | Tlr7 ligands for the treatment of hepatitis c |
| EP1728792A4 (en) | 2004-03-26 | 2010-12-15 | Dainippon Sumitomo Pharma Co | 8-OXOADENINE COMPOUND |
| TWI392678B (zh) | 2004-03-26 | 2013-04-11 | Dainippon Sumitomo Pharma Co | 9-取代-8-氧基腺嘌呤化合物 |
| WO2007084413A2 (en) | 2004-07-14 | 2007-07-26 | Ptc Therapeutics, Inc. | Methods for treating hepatitis c |
| CA2575002C (en) | 2004-08-10 | 2015-01-20 | Janssen Pharmaceutica N.V. | Hiv inhibiting 1,2,4-triazin-6-one derivatives |
| MX2007005408A (es) | 2004-11-09 | 2007-05-16 | Hoffmann La Roche | Compuestos de aminoquinazolinas. |
| US7498409B2 (en) | 2005-03-24 | 2009-03-03 | Schering Corporation | Screening assay for TLR7, TLR8 and TLR9 agonists and antagonists |
| BRPI0611435A2 (pt) | 2005-05-04 | 2010-09-08 | Pfizer Ltd | derivados de 2-amido-6-amino-8-oxopurina, composições farmacêuticas, uso e processo de preparo dos mesmos |
| TW200716631A (en) | 2005-05-12 | 2007-05-01 | Tibotec Pharm Ltd | Pyrido[2,3-d]pyrimidines useful as HCV inhibitors, and methods for the preparation thereof |
| US7994360B2 (en) | 2005-05-16 | 2011-08-09 | Xtl Biopharmaceuticals Ltd. | Benzofuran compounds |
| TWI404537B (zh) | 2005-08-19 | 2013-08-11 | Array Biopharma Inc | 作為類鐸受體(toll-like receptor)調節劑之8-經取代苯并氮雜呯 |
| JP4850911B2 (ja) | 2005-09-01 | 2012-01-11 | エフ.ホフマン−ラ ロシュ アーゲー | P2x3およびp2x2/3モジュレーターとしてのジアミノピリミジン |
| EP1939198A4 (en) | 2005-09-22 | 2012-02-15 | Dainippon Sumitomo Pharma Co | NEW ADENINE CONNECTION |
| WO2007056208A2 (en) | 2005-11-02 | 2007-05-18 | Cytovia, Inc. | N-arylalkyl-thienopyrimidin-4-amines and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| EP1970373A1 (en) | 2005-12-02 | 2008-09-17 | Mitsubishi Tanabe Pharma Corporation | Alicyclic heterocyclic compound |
| MX2008010611A (es) | 2006-02-17 | 2008-11-12 | Pfizer Ltd | Derivados de 3-desazapurina como moduladores de receptores similares a toll. |
| WO2008009077A2 (en) | 2006-07-20 | 2008-01-24 | Gilead Sciences, Inc. | 4,6-dl- and 2,4,6-trisubstituted quinazoline derivatives and pharmaceutical compositions useful for treating viral infections |
| WO2008009078A2 (en) | 2006-07-20 | 2008-01-24 | Gilead Sciences, Inc. | 4,6-dl- and 2,4,6-trisubstituted quinazoline derivatives useful for treating viral infections |
| EP2114950B1 (en) | 2006-12-07 | 2016-03-09 | Genentech, Inc. | Phosphoinositide 3-kinase inhibitor compounds and methods of use |
| JP2010513450A (ja) | 2006-12-20 | 2010-04-30 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | 抗ウイルス性インドール |
| EA019151B1 (ru) | 2007-02-07 | 2014-01-30 | Дзе Регентс Оф Дзе Юниверсити Оф Калифорния | Конъюгаты синтетических агонистов tlr и их применение |
| JP2008222557A (ja) | 2007-03-08 | 2008-09-25 | Kotobuki Seiyaku Kk | ピロロ[3,2−d]ピリミジン誘導体及びこれを有効成分とする医薬組成物 |
| WO2008114008A1 (en) | 2007-03-19 | 2008-09-25 | Astrazeneca Ab | 9-substituted-8-oxo-adenine compounds as toll-like receptor (tlr7 ) modulators |
| AR065784A1 (es) | 2007-03-20 | 2009-07-01 | Dainippon Sumitomo Pharma Co | Derivados de 8-oxo adenina,medicamentos que los contienen y usos como agentes terapeuticos para enfermedades alergicas, antivirales o antibacterianas. |
| EP2138497A4 (en) | 2007-03-20 | 2012-01-04 | Dainippon Sumitomo Pharma Co | NEW ADENINE CONNECTION |
| US9150556B2 (en) | 2007-05-22 | 2015-10-06 | Boehringer Ingelheim International Gmbh | Benzimidazolone chymase inhibitors |
| ES2541434T3 (es) | 2007-06-29 | 2015-07-20 | Gilead Sciences, Inc. | Derivados de purina y su uso como moduladores del receptor de tipo Toll 7 |
| TW200922569A (en) | 2007-08-10 | 2009-06-01 | Genelabs Tech Inc | Certain nitrogen containing bicyclic chemical entities for treating viral infections |
| US8440681B2 (en) | 2007-08-28 | 2013-05-14 | Irm Llc | 2-biphenylamino-4-aminopyrimidine derivatives as kinase inhibitors |
| WO2009030998A1 (en) | 2007-09-05 | 2009-03-12 | Coley Pharmaceutical Group, Inc. | Pyrimidine compounds as toll-like receptor (tlr) agonists |
| PE20091236A1 (es) | 2007-11-22 | 2009-09-16 | Astrazeneca Ab | Derivados de pirimidina como immunomoduladores de tlr7 |
| AU2008339917B2 (en) | 2007-12-24 | 2013-02-07 | Tibotec Pharmaceuticals | Macrocyclic indoles as hepatitis C virus inhibitors |
| EP2259788A4 (en) | 2008-02-07 | 2011-03-16 | Univ California | TREATMENT OF BLADDER DISEASES WITH A TLR7 ACTIVATOR |
| US8461107B2 (en) | 2008-04-28 | 2013-06-11 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8946239B2 (en) | 2008-07-10 | 2015-02-03 | Duquesne University Of The Holy Spirit | Substituted pyrrolo, -furano, and cyclopentylpyrimidines having antimitotic and/or antitumor activity and methods of use thereof |
| UY31982A (es) | 2008-07-16 | 2010-02-26 | Boehringer Ingelheim Int | Derivados de 1,2-dihidropiridin-3-carboxamidas n-sustituidas |
| DK2313111T3 (da) | 2008-08-01 | 2013-12-02 | Ventirx Pharmaceuticals Inc | Toll-lignende receptoragonistformuleringer og anvendelse deraf |
| CA2760766A1 (en) | 2009-05-21 | 2010-11-25 | Nicholas James Bennett | Novel pyrimidine derivatives and their use in the treatment of cancer and further diseases |
| TWI468402B (zh) | 2009-07-31 | 2015-01-11 | 必治妥美雅史谷比公司 | 降低β-類澱粉生成之化合物 |
| US8637525B2 (en) | 2009-07-31 | 2014-01-28 | Bristol-Myers Squibb Company | Compounds for the reduction of beta-amyloid production |
| WO2011049988A2 (en) | 2009-10-20 | 2011-04-28 | Eiger Biopharmaceuticals, Inc. | Indazoles to treat flaviviridae virus infection |
| US8507507B2 (en) | 2009-10-22 | 2013-08-13 | Gilead Sciences, Inc. | Modulators of toll-like receptors |
| KR101094446B1 (ko) | 2009-11-19 | 2011-12-15 | 한국과학기술연구원 | 단백질 키나아제 저해활성을 가지는 2,4,7-치환된 티에노[3,2-d]피리미딘 화합물 |
| JP2013032290A (ja) | 2009-11-20 | 2013-02-14 | Dainippon Sumitomo Pharma Co Ltd | 新規縮合ピリミジン誘導体 |
| DE102010040233A1 (de) | 2010-09-03 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Bicyclische Aza-Heterocyclen und ihre Verwendung |
| BR112013007678A2 (pt) | 2010-10-01 | 2017-07-04 | Ventirx Pharmaceuticals Inc | método para tratamento de doenças alérgicas |
| WO2012067269A1 (en) | 2010-11-19 | 2012-05-24 | Dainippon Sumitomo Pharma Co., Ltd. | Aminoalkoxyphenyl compounds and their use in the treatment of disease |
| WO2012066335A1 (en) | 2010-11-19 | 2012-05-24 | Astrazeneca Ab | Phenol compounds als toll -like receptor 7 agonists |
| EA027792B1 (ru) * | 2011-04-08 | 2017-09-29 | Янссен Сайенсиз Айрлэнд Юси | Производные пиримидина для лечения вирусных инфекций |
| HRP20180447T1 (hr) | 2011-05-18 | 2018-04-20 | Janssen Sciences Ireland Uc | Derivati kinazolina za liječenje virusnih infekcija i drugih bolesti |
| CN104066733A (zh) | 2011-11-09 | 2014-09-24 | 爱尔兰詹森研发公司 | 用于治疗病毒感染的嘌呤衍生物 |
| EA033907B1 (ru) | 2012-02-08 | 2019-12-09 | Янссен Сайенсиз Айрленд Юси | Производные пиперидино-пиримидина, фармацевтическая композиция и их применение |
| SMT201800608T1 (it) | 2012-04-24 | 2019-01-11 | Vertex Pharma | Inibitori di dna-pk |
| SG11201408542VA (en) | 2012-07-13 | 2015-02-27 | Janssen Sciences Ireland Uc | Macrocyclic purines for the treatment of viral infections |
| EP2882721B1 (en) | 2012-08-10 | 2018-12-05 | Janssen Sciences Ireland Unlimited Company | Alkylpyrimidine derivatives for the treatment of viral infections and further diseases |
| EP2712866A1 (en) | 2012-10-01 | 2014-04-02 | Centre National de la Recherche Scientifique (CNRS) | 1,2,4-triazine derivatives for the treatment of viral infections |
| KR102252649B1 (ko) | 2012-10-05 | 2021-05-17 | 얀센 사이언시즈 아일랜드 언리미티드 컴퍼니 | 바이러스 감염 및 추가 질환의 치료를 위한 아실아미노피리미딘 유도체 |
| KR102280595B1 (ko) | 2012-10-10 | 2021-07-22 | 얀센 사이언시즈 아일랜드 언리미티드 컴퍼니 | 바이러스 감염 및 다른 질환 치료를 위한 피롤로[3,2-d]피리미딘 유도체 |
| CN105051018B (zh) | 2012-11-16 | 2019-09-20 | 爱尔兰詹森科学公司 | 用于治疗病毒性感染的杂环的经取代的2-氨基-喹唑啉衍生物 |
| CN105189468B (zh) | 2013-02-21 | 2018-10-30 | 爱尔兰詹森科学公司 | 用于治疗病毒性感染的2-氨基嘧啶衍生物 |
| MX366481B (es) | 2013-03-29 | 2019-07-09 | Janssen Sciences Ireland Uc | Deaza-purinonas macrociclicas para el tratamiento de infecciones virales. |
| DK3004074T3 (da) | 2013-05-24 | 2018-01-29 | Janssen Sciences Ireland Uc | Pyridonderivater til behandling af virusinfektioner og yderligere sygdomme |
| NO3030563T3 (es) | 2013-06-27 | 2018-01-06 | ||
| MX368625B (es) | 2013-07-30 | 2019-10-08 | Janssen Sciences Ireland Uc | Derivados de tieno[3,2-d]pirimidinas para el tratamiento de infecciones virales. |
| US9701661B2 (en) | 2014-07-11 | 2017-07-11 | Northwestern University | 2-imidazolyl-pyrimidine scaffolds as potent and selective inhibitors of neuronal nitric oxide synthase |
| CA3027471A1 (en) | 2016-07-01 | 2018-01-04 | Janssen Sciences Ireland Unlimited Company | Dihydropyranopyrimidines for the treatment of viral infections |
-
2012
- 2012-04-10 EA EA201391495A patent/EA027792B1/ru unknown
- 2012-04-10 US US14/110,054 patent/US9422250B2/en active Active
- 2012-04-10 BR BR122017025423-5A patent/BR122017025423B1/pt active IP Right Grant
- 2012-04-10 PT PT172036287T patent/PT3330257T/pt unknown
- 2012-04-10 PT PT12712666T patent/PT2694484T/pt unknown
- 2012-04-10 PL PL12712666T patent/PL2694484T3/pl unknown
- 2012-04-10 RS RS20200465A patent/RS60191B1/sr unknown
- 2012-04-10 PH PH12013502033A patent/PH12013502033B1/en unknown
- 2012-04-10 KR KR1020197003300A patent/KR102024766B1/ko not_active Expired - Fee Related
- 2012-04-10 MX MX2013011686A patent/MX349588B/es active IP Right Grant
- 2012-04-10 EP EP12712666.2A patent/EP2694484B1/en active Active
- 2012-04-10 DK DK12712666.2T patent/DK2694484T3/en active
- 2012-04-10 TR TR2018/15062T patent/TR201815062T4/tr unknown
- 2012-04-10 CN CN201280017291.4A patent/CN103608335B/zh not_active Expired - Fee Related
- 2012-04-10 SI SI201231762T patent/SI3330257T1/sl unknown
- 2012-04-10 PL PL19192173T patent/PL3590928T3/pl unknown
- 2012-04-10 AU AU2012238564A patent/AU2012238564A1/en not_active Abandoned
- 2012-04-10 SM SM20180527T patent/SMT201800527T1/it unknown
- 2012-04-10 BR BR122019023564-3A patent/BR122019023564B1/pt active IP Right Grant
- 2012-04-10 KR KR1020197027271A patent/KR102058946B1/ko not_active Expired - Fee Related
- 2012-04-10 HU HUE19192173A patent/HUE055286T2/hu unknown
- 2012-04-10 HR HRP20181464TT patent/HRP20181464T1/hr unknown
- 2012-04-10 MY MYPI2013003675A patent/MY170941A/en unknown
- 2012-04-10 LT LTEP12712666.2T patent/LT2694484T/lt unknown
- 2012-04-10 EP EP17203628.7A patent/EP3330257B1/en active Active
- 2012-04-10 DK DK17203628.7T patent/DK3330257T3/da active
- 2012-04-10 SM SM20200196T patent/SMT202000196T1/it unknown
- 2012-04-10 SM SM20210483T patent/SMT202100483T1/it unknown
- 2012-04-10 UA UAA201311375A patent/UA113956C2/uk unknown
- 2012-04-10 DK DK19192173.3T patent/DK3590928T3/da active
- 2012-04-10 ES ES17203628T patent/ES2786569T3/es active Active
- 2012-04-10 ES ES12712666.2T patent/ES2691745T3/es active Active
- 2012-04-10 PL PL17203628T patent/PL3330257T3/pl unknown
- 2012-04-10 KR KR1020137027547A patent/KR101946499B1/ko not_active Expired - Fee Related
- 2012-04-10 WO PCT/EP2012/056388 patent/WO2012136834A1/en not_active Ceased
- 2012-04-10 HU HUE17203628A patent/HUE048561T2/hu unknown
- 2012-04-10 EP EP19192173.3A patent/EP3590928B1/en active Active
- 2012-04-10 RS RS20181198A patent/RS57758B1/sr unknown
- 2012-04-10 SI SI201231938T patent/SI3590928T1/sl unknown
- 2012-04-10 SI SI201231386T patent/SI2694484T1/sl unknown
- 2012-04-10 CA CA2832685A patent/CA2832685C/en active Active
- 2012-04-10 ES ES19192173T patent/ES2887303T3/es active Active
- 2012-04-10 SG SG2013074976A patent/SG194131A1/en unknown
- 2012-04-10 ME MEP-2020-84A patent/ME03782B/me unknown
- 2012-04-10 BR BR112013025987-6A patent/BR112013025987B1/pt not_active IP Right Cessation
- 2012-04-10 RS RS20211049A patent/RS62254B1/sr unknown
- 2012-04-10 LT LTEP19192173.3T patent/LT3590928T/lt unknown
- 2012-04-10 LT LTEP17203628.7T patent/LT3330257T/lt unknown
- 2012-04-10 HU HUE12712666A patent/HUE040565T2/hu unknown
- 2012-04-10 JP JP2014503170A patent/JP6046694B2/ja not_active Expired - Fee Related
- 2012-04-10 PT PT191921733T patent/PT3590928T/pt unknown
-
2013
- 2013-09-09 IL IL228317A patent/IL228317A/en active IP Right Grant
- 2013-10-07 ZA ZA2013/07472A patent/ZA201307472B/en unknown
- 2013-10-07 CL CL2013002877A patent/CL2013002877A1/es unknown
-
2016
- 2016-07-13 US US15/209,637 patent/US10272085B2/en active Active
- 2016-11-09 AU AU2016256732A patent/AU2016256732B2/en not_active Ceased
-
2018
- 2018-01-10 US US15/867,041 patent/US10420767B2/en active Active
- 2018-09-24 AU AU2018236703A patent/AU2018236703B2/en not_active Ceased
- 2018-10-18 CY CY20181101071T patent/CY1121363T1/el unknown
-
2019
- 2019-08-02 US US16/530,385 patent/US10780089B2/en active Active
-
2020
- 2020-04-02 HR HRP20200538TT patent/HRP20200538T1/hr unknown
- 2020-06-09 CY CY20201100513T patent/CY1123324T1/el unknown
- 2020-09-04 US US17/012,286 patent/US11541050B2/en active Active
- 2020-12-07 AU AU2020281180A patent/AU2020281180A1/en not_active Abandoned
-
2021
- 2021-07-29 HR HRP20211224TT patent/HRP20211224T1/hr unknown
- 2021-08-31 CY CY20211100767T patent/CY1124469T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2786569T3 (es) | Derivados de pirimidina para el tratamiento de infecciones víricas | |
| ES2716811T3 (es) | Derivados piperidinopirimidínicos para el tratamiento de infecciones víricas | |
| ES2670513T3 (es) | Derivados pirrolo[3,2-d]pirimidínicos para el tratamiento de infecciones víricas y otras enfermedades | |
| ES2663601T3 (es) | Derivados de quinazolina para el tratamiento de infecciones víricas y otras enfermedades | |
| ES2690082T3 (es) | Derivados de purina para el tratamiento de infecciones virales | |
| ES2645950T3 (es) | Derivados de pirrolo[3,2-d]pirimidina para el tratamiento de infecciones víricas y otras enfermedades | |
| HK1256490B (en) | Pyrimidine derivatives for the treatment of viral infections | |
| HK1192547B (en) | Pyrimidine derivatives for the treatment of viral infections | |
| HK1192547A (en) | Pyrimidine derivatives for the treatment of viral infections |