DE69917469T2 - Pyrimidin derivate - Google Patents
Pyrimidin derivate Download PDFInfo
- Publication number
- DE69917469T2 DE69917469T2 DE69917469T DE69917469T DE69917469T2 DE 69917469 T2 DE69917469 T2 DE 69917469T2 DE 69917469 T DE69917469 T DE 69917469T DE 69917469 T DE69917469 T DE 69917469T DE 69917469 T2 DE69917469 T2 DE 69917469T2
- Authority
- DE
- Germany
- Prior art keywords
- alkyl
- radical
- branched
- straight
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 229940083082 pyrimidine derivative acting on arteriolar smooth muscle Drugs 0.000 title description 15
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical class C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 title 1
- 150000003839 salts Chemical class 0.000 claims abstract description 40
- 150000003230 pyrimidines Chemical class 0.000 claims abstract description 39
- 208000026935 allergic disease Diseases 0.000 claims abstract description 13
- -1 carboxy, carbamoyl Chemical group 0.000 claims description 61
- 230000028993 immune response Effects 0.000 claims description 36
- 125000000217 alkyl group Chemical group 0.000 claims description 32
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 24
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 24
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 23
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 18
- 125000001424 substituent group Chemical group 0.000 claims description 17
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 16
- 125000005843 halogen group Chemical group 0.000 claims description 15
- 210000002443 helper t lymphocyte Anatomy 0.000 claims description 14
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 13
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 claims description 13
- 230000001225 therapeutic effect Effects 0.000 claims description 13
- 125000002947 alkylene group Chemical group 0.000 claims description 12
- 230000003449 preventive effect Effects 0.000 claims description 12
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 12
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 12
- JWCVYQRPINPYQJ-UHFFFAOYSA-N thiepane Chemical compound C1CCCSCC1 JWCVYQRPINPYQJ-UHFFFAOYSA-N 0.000 claims description 12
- 208000006673 asthma Diseases 0.000 claims description 11
- 125000000623 heterocyclic group Chemical group 0.000 claims description 11
- YPWFISCTZQNZAU-UHFFFAOYSA-N Thiane Chemical compound C1CCSCC1 YPWFISCTZQNZAU-UHFFFAOYSA-N 0.000 claims description 9
- 239000003795 chemical substances by application Substances 0.000 claims description 9
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 9
- UHHKSVZZTYJVEG-UHFFFAOYSA-N oxepane Chemical compound C1CCCOCC1 UHHKSVZZTYJVEG-UHFFFAOYSA-N 0.000 claims description 8
- 239000004480 active ingredient Substances 0.000 claims description 7
- 125000002541 furyl group Chemical group 0.000 claims description 7
- 230000001965 increasing effect Effects 0.000 claims description 7
- 125000004076 pyridyl group Chemical group 0.000 claims description 7
- 125000006296 sulfonyl amino group Chemical group [H]N(*)S(*)(=O)=O 0.000 claims description 7
- RAOIDOHSFRTOEL-UHFFFAOYSA-N tetrahydrothiophene Chemical compound C1CCSC1 RAOIDOHSFRTOEL-UHFFFAOYSA-N 0.000 claims description 7
- 125000001544 thienyl group Chemical group 0.000 claims description 7
- 125000000304 alkynyl group Chemical group 0.000 claims description 6
- 208000010668 atopic eczema Diseases 0.000 claims description 6
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 claims description 5
- 125000003545 alkoxy group Chemical group 0.000 claims description 5
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 5
- 125000004432 carbon atom Chemical group C* 0.000 claims description 5
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 5
- 201000010099 disease Diseases 0.000 claims description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 5
- 239000001257 hydrogen Substances 0.000 claims description 5
- 229910052739 hydrogen Inorganic materials 0.000 claims description 5
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 4
- YCNYCBYHUAGZIZ-UHFFFAOYSA-N 7-oxabicyclo[2.2.1]hept-2-ene Chemical compound O1C2CCC1C=C2 YCNYCBYHUAGZIZ-UHFFFAOYSA-N 0.000 claims description 4
- YPWFNLSXQIGJCK-UHFFFAOYSA-N 7-oxabicyclo[2.2.1]heptane Chemical compound C1CC2CCC1O2 YPWFNLSXQIGJCK-UHFFFAOYSA-N 0.000 claims description 4
- HXJUTPCZVOIRIF-UHFFFAOYSA-N Tetrahydrothiophene-1,1-dioxide, Natural products O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 claims description 4
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 claims description 4
- 201000008937 atopic dermatitis Diseases 0.000 claims description 4
- 150000001924 cycloalkanes Chemical class 0.000 claims description 4
- 150000001925 cycloalkenes Chemical class 0.000 claims description 4
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 claims description 4
- ISXOBTBCNRIIQO-UHFFFAOYSA-N tetrahydrothiophene 1-oxide Chemical compound O=S1CCCC1 ISXOBTBCNRIIQO-UHFFFAOYSA-N 0.000 claims description 4
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 claims description 4
- LBMZLLXZMSMJPJ-UHFFFAOYSA-N thiepane 1-oxide Chemical compound O=S1CCCCCC1 LBMZLLXZMSMJPJ-UHFFFAOYSA-N 0.000 claims description 4
- XSROQCDVUIHRSI-UHFFFAOYSA-N thietane Chemical compound C1CSC1 XSROQCDVUIHRSI-UHFFFAOYSA-N 0.000 claims description 4
- PTBOOHAPESUXGV-UHFFFAOYSA-N 2-iodo-3-methylpyridine Chemical compound CC1=CC=CN=C1I PTBOOHAPESUXGV-UHFFFAOYSA-N 0.000 claims description 3
- 206010012434 Dermatitis allergic Diseases 0.000 claims description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- 125000002252 acyl group Chemical group 0.000 claims description 3
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 claims description 3
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 3
- 125000005842 heteroatom Chemical group 0.000 claims description 3
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 3
- 206010039085 Rhinitis allergic Diseases 0.000 claims description 2
- 125000003342 alkenyl group Chemical group 0.000 claims description 2
- 201000010105 allergic rhinitis Diseases 0.000 claims description 2
- 229910052736 halogen Inorganic materials 0.000 claims description 2
- 150000002367 halogens Chemical class 0.000 claims description 2
- 239000002955 immunomodulating agent Substances 0.000 claims description 2
- 229940121354 immunomodulator Drugs 0.000 claims description 2
- 230000002584 immunomodulator Effects 0.000 claims description 2
- 125000004043 oxo group Chemical group O=* 0.000 claims description 2
- 125000003258 trimethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])[*:1] 0.000 claims description 2
- 102000004127 Cytokines Human genes 0.000 abstract description 26
- 108090000695 Cytokines Proteins 0.000 abstract description 26
- 108090000978 Interleukin-4 Proteins 0.000 abstract description 19
- 108010002616 Interleukin-5 Proteins 0.000 abstract description 18
- 239000003814 drug Substances 0.000 abstract description 15
- 238000004519 manufacturing process Methods 0.000 abstract description 11
- 208000023275 Autoimmune disease Diseases 0.000 abstract description 4
- 230000002401 inhibitory effect Effects 0.000 abstract description 3
- 206010025135 lupus erythematosus Diseases 0.000 abstract description 2
- 230000009885 systemic effect Effects 0.000 abstract description 2
- 229940124597 therapeutic agent Drugs 0.000 abstract description 2
- 208000011580 syndromic disease Diseases 0.000 abstract 1
- 150000001875 compounds Chemical class 0.000 description 102
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 69
- 239000000203 mixture Substances 0.000 description 51
- 238000005160 1H NMR spectroscopy Methods 0.000 description 49
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 45
- 238000000034 method Methods 0.000 description 38
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 34
- 150000003254 radicals Chemical class 0.000 description 33
- 239000002904 solvent Substances 0.000 description 33
- 238000012360 testing method Methods 0.000 description 31
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 27
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 27
- 239000000243 solution Substances 0.000 description 27
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 26
- 238000006243 chemical reaction Methods 0.000 description 25
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 24
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 24
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 22
- 239000011541 reaction mixture Substances 0.000 description 22
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 21
- 102000004388 Interleukin-4 Human genes 0.000 description 18
- 229940028885 interleukin-4 Drugs 0.000 description 18
- 102000000743 Interleukin-5 Human genes 0.000 description 17
- 229940100602 interleukin-5 Drugs 0.000 description 17
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 16
- 239000012044 organic layer Substances 0.000 description 16
- 238000010898 silica gel chromatography Methods 0.000 description 16
- 229910052938 sodium sulfate Inorganic materials 0.000 description 16
- 235000011152 sodium sulphate Nutrition 0.000 description 16
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 15
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 15
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 15
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 13
- 108010074328 Interferon-gamma Proteins 0.000 description 13
- 230000015572 biosynthetic process Effects 0.000 description 13
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- 210000004027 cell Anatomy 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 12
- DPBLXKKOBLCELK-UHFFFAOYSA-N pentan-1-amine Chemical compound CCCCCN DPBLXKKOBLCELK-UHFFFAOYSA-N 0.000 description 12
- BTLNLOHFQGIGLV-UHFFFAOYSA-N 4-chloro-5,6,7,8-tetrahydroquinazolin-2-amine Chemical compound C1CCCC2=NC(N)=NC(Cl)=C21 BTLNLOHFQGIGLV-UHFFFAOYSA-N 0.000 description 11
- 229940079593 drug Drugs 0.000 description 11
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 10
- 102100037850 Interferon gamma Human genes 0.000 description 10
- 241000699666 Mus <mouse, genus> Species 0.000 description 10
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 10
- 206010070834 Sensitisation Diseases 0.000 description 10
- 210000001165 lymph node Anatomy 0.000 description 10
- 238000005259 measurement Methods 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- DNIAPMSPPWPWGF-UHFFFAOYSA-N propylene glycol Substances CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 10
- 230000008313 sensitization Effects 0.000 description 10
- 241000699670 Mus sp. Species 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 229940086542 triethylamine Drugs 0.000 description 9
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 8
- 230000004913 activation Effects 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 7
- 230000003287 optical effect Effects 0.000 description 7
- 150000001298 alcohols Chemical class 0.000 description 6
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 6
- 210000003979 eosinophil Anatomy 0.000 description 6
- 150000002170 ethers Chemical class 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 239000002609 medium Substances 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 5
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 5
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 235000011114 ammonium hydroxide Nutrition 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- STIAPHVBRDNOAJ-UHFFFAOYSA-N carbamimidoylazanium;carbonate Chemical compound NC(N)=N.NC(N)=N.OC(O)=O STIAPHVBRDNOAJ-UHFFFAOYSA-N 0.000 description 5
- 239000012442 inert solvent Substances 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 230000000638 stimulation Effects 0.000 description 5
- HJRJRUMKQCMYDL-UHFFFAOYSA-N 1-chloro-2,4,6-trinitrobenzene Chemical compound [O-][N+](=O)C1=CC([N+]([O-])=O)=C(Cl)C([N+]([O-])=O)=C1 HJRJRUMKQCMYDL-UHFFFAOYSA-N 0.000 description 4
- MUGVXCHGASIZAN-UHFFFAOYSA-N 2-amino-5,6,7,8-tetrahydro-1h-quinazolin-4-one Chemical compound C1CCCC2=C1C(=O)N=C(N)N2 MUGVXCHGASIZAN-UHFFFAOYSA-N 0.000 description 4
- VYCNDBQWMCGZAA-UHFFFAOYSA-N 2-amino-5-butyl-6-methyl-1h-pyrimidin-4-one Chemical compound CCCCC1=C(C)NC(N)=NC1=O VYCNDBQWMCGZAA-UHFFFAOYSA-N 0.000 description 4
- DKQJLZGVCBMQGR-UHFFFAOYSA-N 2-chloro-n-pentyl-6,7-dihydro-5h-cyclopenta[d]pyrimidin-4-amine Chemical compound CCCCCNC1=NC(Cl)=NC2=C1CCC2 DKQJLZGVCBMQGR-UHFFFAOYSA-N 0.000 description 4
- JTFIACKVVYYOAN-UHFFFAOYSA-N 3-(2-amino-4-chloro-6-methylpyrimidin-5-yl)propanenitrile Chemical compound CC1=NC(N)=NC(Cl)=C1CCC#N JTFIACKVVYYOAN-UHFFFAOYSA-N 0.000 description 4
- RGEBQSGYHZEFEV-UHFFFAOYSA-N 4-butyl-6-chloro-5-methylpyrimidin-2-amine Chemical compound CCCCC1=NC(N)=NC(Cl)=C1C RGEBQSGYHZEFEV-UHFFFAOYSA-N 0.000 description 4
- ZUORSWNFLYOARB-UHFFFAOYSA-N 4-chloro-5-(2-methoxyethyl)-6-methylpyrimidin-2-amine Chemical compound COCCC1=C(C)N=C(N)N=C1Cl ZUORSWNFLYOARB-UHFFFAOYSA-N 0.000 description 4
- MGZDAOPTTMOUKA-UHFFFAOYSA-N 4-chloro-7,8-dihydro-5h-pyrano[4,3-d]pyrimidin-2-amine Chemical compound C1OCCC2=NC(N)=NC(Cl)=C21 MGZDAOPTTMOUKA-UHFFFAOYSA-N 0.000 description 4
- OJSWAIOKPQDUQY-UHFFFAOYSA-N 5-butyl-4-chloro-6-methylpyrimidin-2-amine Chemical compound CCCCC1=C(C)N=C(N)N=C1Cl OJSWAIOKPQDUQY-UHFFFAOYSA-N 0.000 description 4
- 238000002965 ELISA Methods 0.000 description 4
- 108010002350 Interleukin-2 Proteins 0.000 description 4
- 102000000588 Interleukin-2 Human genes 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 4
- 230000000172 allergic effect Effects 0.000 description 4
- 239000000427 antigen Substances 0.000 description 4
- 108091007433 antigens Proteins 0.000 description 4
- 102000036639 antigens Human genes 0.000 description 4
- 229960002685 biotin Drugs 0.000 description 4
- 235000020958 biotin Nutrition 0.000 description 4
- 239000011616 biotin Substances 0.000 description 4
- 238000009835 boiling Methods 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 230000016396 cytokine production Effects 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 239000000945 filler Substances 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 229920000609 methyl cellulose Polymers 0.000 description 4
- 239000001923 methylcellulose Substances 0.000 description 4
- 235000010981 methylcellulose Nutrition 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 239000002953 phosphate buffered saline Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 150000003431 steroids Chemical class 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- GINSRDSEEGBTJO-UHFFFAOYSA-N thietane 1-oxide Chemical compound O=S1CCC1 GINSRDSEEGBTJO-UHFFFAOYSA-N 0.000 description 4
- 238000011200 topical administration Methods 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 3
- GDHHAOFBLGZCMI-UHFFFAOYSA-N 2,4-dichloro-6,7-dihydro-5h-cyclopenta[d]pyrimidine Chemical compound ClC1=NC(Cl)=NC2=C1CCC2 GDHHAOFBLGZCMI-UHFFFAOYSA-N 0.000 description 3
- VWNZPXHTSQBFKJ-UHFFFAOYSA-N 2-amino-1,5,7,8-tetrahydropyrano[4,3-d]pyrimidin-4-one Chemical compound C1OCCC2=NC(N)=NC(O)=C21 VWNZPXHTSQBFKJ-UHFFFAOYSA-N 0.000 description 3
- MGMJINGLVXWGAO-UHFFFAOYSA-N 2-amino-6-butyl-5-methyl-1h-pyrimidin-4-one Chemical compound CCCCC1=NC(N)=NC(O)=C1C MGMJINGLVXWGAO-UHFFFAOYSA-N 0.000 description 3
- JYVLIDXNZAXMDK-UHFFFAOYSA-N 2-pentanol Substances CCCC(C)O JYVLIDXNZAXMDK-UHFFFAOYSA-N 0.000 description 3
- NUGQOZJAWOOGSB-UHFFFAOYSA-N 5-benzyl-4-chloro-6-methylpyrimidin-2-amine Chemical compound CC1=NC(N)=NC(Cl)=C1CC1=CC=CC=C1 NUGQOZJAWOOGSB-UHFFFAOYSA-N 0.000 description 3
- 0 C[C@]1C[C@@](C)*C1 Chemical compound C[C@]1C[C@@](C)*C1 0.000 description 3
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- 206010020751 Hypersensitivity Diseases 0.000 description 3
- 102000008070 Interferon-gamma Human genes 0.000 description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- 108010058846 Ovalbumin Proteins 0.000 description 3
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 230000002159 abnormal effect Effects 0.000 description 3
- 208000037883 airway inflammation Diseases 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- LFYJSSARVMHQJB-QIXNEVBVSA-N bakuchiol Chemical compound CC(C)=CCC[C@@](C)(C=C)\C=C\C1=CC=C(O)C=C1 LFYJSSARVMHQJB-QIXNEVBVSA-N 0.000 description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 230000001684 chronic effect Effects 0.000 description 3
- 208000037976 chronic inflammation Diseases 0.000 description 3
- 230000006020 chronic inflammation Effects 0.000 description 3
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- UQLDLKMNUJERMK-UHFFFAOYSA-L di(octadecanoyloxy)lead Chemical compound [Pb+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O UQLDLKMNUJERMK-UHFFFAOYSA-L 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 230000008595 infiltration Effects 0.000 description 3
- 238000001764 infiltration Methods 0.000 description 3
- 229960003130 interferon gamma Drugs 0.000 description 3
- 239000000543 intermediate Substances 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 239000006210 lotion Substances 0.000 description 3
- 206010025482 malaise Diseases 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 229940092253 ovalbumin Drugs 0.000 description 3
- 238000012746 preparative thin layer chromatography Methods 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- BUGOPWGPQGYYGR-UHFFFAOYSA-N thiane 1,1-dioxide Chemical compound O=S1(=O)CCCCC1 BUGOPWGPQGYYGR-UHFFFAOYSA-N 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 2
- DDWDBNCLFQQEJS-UHFFFAOYSA-N 1-aminopentan-2-ol;hydrochloride Chemical compound Cl.CCCC(O)CN DDWDBNCLFQQEJS-UHFFFAOYSA-N 0.000 description 2
- RRQYJINTUHWNHW-UHFFFAOYSA-N 1-ethoxy-2-(2-ethoxyethoxy)ethane Chemical compound CCOCCOCCOCC RRQYJINTUHWNHW-UHFFFAOYSA-N 0.000 description 2
- KGHIDMFJEQPDPA-UHFFFAOYSA-N 2-amino-5-(2-methoxyethyl)-6-methyl-1h-pyrimidin-4-one Chemical compound COCCC1=C(C)N=C(N)N=C1O KGHIDMFJEQPDPA-UHFFFAOYSA-N 0.000 description 2
- HJFHZMPLTXXTPB-UHFFFAOYSA-N 2-amino-5-(2-phenylethyl)-1h-pyrimidin-6-one Chemical compound OC1=NC(N)=NC=C1CCC1=CC=CC=C1 HJFHZMPLTXXTPB-UHFFFAOYSA-N 0.000 description 2
- VRCGFYUFABMDSD-UHFFFAOYSA-N 2-amino-5-benzyl-6-methyl-1h-pyrimidin-4-one Chemical compound N1C(N)=NC(=O)C(CC=2C=CC=CC=2)=C1C VRCGFYUFABMDSD-UHFFFAOYSA-N 0.000 description 2
- BLHUMPJZJWQBRP-UHFFFAOYSA-N 2-amino-5-hexyl-6-methyl-1h-pyrimidin-4-one Chemical compound CCCCCCC1=C(C)NC(N)=NC1=O BLHUMPJZJWQBRP-UHFFFAOYSA-N 0.000 description 2
- XQFXFCTVDCZGMS-UHFFFAOYSA-N 2-chloro-5,6-dimethyl-n-pentylpyrimidin-4-amine Chemical compound CCCCCNC1=NC(Cl)=NC(C)=C1C XQFXFCTVDCZGMS-UHFFFAOYSA-N 0.000 description 2
- KOVRUHZFIUMYIY-UHFFFAOYSA-N 2-chloro-n-pentyl-5,6,7,8-tetrahydroquinazolin-4-amine Chemical compound C1CCCC2=C1N=C(Cl)N=C2NCCCCC KOVRUHZFIUMYIY-UHFFFAOYSA-N 0.000 description 2
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 2
- CPIBPTZOODLBPS-UHFFFAOYSA-N 3-(2-amino-6-methyl-4-oxo-1h-pyrimidin-5-yl)propanenitrile Chemical compound CC=1NC(N)=NC(=O)C=1CCC#N CPIBPTZOODLBPS-UHFFFAOYSA-N 0.000 description 2
- AZAPTJADIJNMGJ-UHFFFAOYSA-N 4-chloro-5-(2-phenylethyl)pyrimidin-2-amine Chemical compound ClC1=NC(N)=NC=C1CCC1=CC=CC=C1 AZAPTJADIJNMGJ-UHFFFAOYSA-N 0.000 description 2
- PSUQYEONRIYSPX-UHFFFAOYSA-N 4-chloro-5-hexyl-6-methylpyrimidin-2-amine Chemical compound CCCCCCC1=C(C)N=C(N)N=C1Cl PSUQYEONRIYSPX-UHFFFAOYSA-N 0.000 description 2
- QKOJJWMXCCQJKO-UHFFFAOYSA-N 5-benzyl-4-chloropyrimidin-2-amine Chemical compound ClC1=NC(N)=NC=C1CC1=CC=CC=C1 QKOJJWMXCCQJKO-UHFFFAOYSA-N 0.000 description 2
- DDHJPZZLNWYTES-UHFFFAOYSA-N 6-butyl-5-methyl-4-n-pentylpyrimidine-2,4-diamine Chemical compound CCCCCNC1=NC(N)=NC(CCCC)=C1C DDHJPZZLNWYTES-UHFFFAOYSA-N 0.000 description 2
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical class O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- 125000000041 C6-C10 aryl group Chemical group 0.000 description 2
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 2
- 229930105110 Cyclosporin A Natural products 0.000 description 2
- 108010036949 Cyclosporine Proteins 0.000 description 2
- 206010012438 Dermatitis atopic Diseases 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- 102000003814 Interleukin-10 Human genes 0.000 description 2
- 108090000174 Interleukin-10 Proteins 0.000 description 2
- 102000003816 Interleukin-13 Human genes 0.000 description 2
- 108090000176 Interleukin-13 Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 239000004166 Lanolin Substances 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- LOUPRKONTZGTKE-WZBLMQSHSA-N Quinine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-WZBLMQSHSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 230000029662 T-helper 1 type immune response Effects 0.000 description 2
- 241000700605 Viruses Species 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 230000009285 allergic inflammation Effects 0.000 description 2
- 208000030961 allergic reaction Diseases 0.000 description 2
- ZXVOCOLRQJZVBW-UHFFFAOYSA-N azane;ethanol Chemical compound N.CCO ZXVOCOLRQJZVBW-UHFFFAOYSA-N 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- KMPWYEUPVWOPIM-KODHJQJWSA-N cinchonidine Chemical compound C1=CC=C2C([C@H]([C@H]3[N@]4CC[C@H]([C@H](C4)C=C)C3)O)=CC=NC2=C1 KMPWYEUPVWOPIM-KODHJQJWSA-N 0.000 description 2
- KMPWYEUPVWOPIM-UHFFFAOYSA-N cinchonidine Natural products C1=CC=C2C(C(C3N4CCC(C(C4)C=C)C3)O)=CC=NC2=C1 KMPWYEUPVWOPIM-UHFFFAOYSA-N 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- LPIQUOYDBNQMRZ-UHFFFAOYSA-N cyclopentene Chemical compound C1CC=CC1 LPIQUOYDBNQMRZ-UHFFFAOYSA-N 0.000 description 2
- 229930182912 cyclosporin Natural products 0.000 description 2
- 229940019778 diethylene glycol diethyl ether Drugs 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 230000004727 humoral immunity Effects 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 229960003444 immunosuppressant agent Drugs 0.000 description 2
- 239000003018 immunosuppressive agent Substances 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 229940076144 interleukin-10 Drugs 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 235000019388 lanolin Nutrition 0.000 description 2
- 229940039717 lanolin Drugs 0.000 description 2
- 229940083747 low-ceiling diuretics xanthine derivative Drugs 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 2
- NHFFUAXLXREJPB-UHFFFAOYSA-N methyl 2-[(2-amino-5,6,7,8-tetrahydroquinazolin-4-yl)amino]hexanoate Chemical compound C1CCCC2=C1N=C(N)N=C2NC(CCCC)C(=O)OC NHFFUAXLXREJPB-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- LEHBURLTIWGHEM-UHFFFAOYSA-N pyridinium chlorochromate Chemical compound [O-][Cr](Cl)(=O)=O.C1=CC=[NH+]C=C1 LEHBURLTIWGHEM-UHFFFAOYSA-N 0.000 description 2
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000000021 stimulant Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- NNLBRYQGMOYARS-UHFFFAOYSA-N thiane 1-oxide Chemical compound O=S1CCCCC1 NNLBRYQGMOYARS-UHFFFAOYSA-N 0.000 description 2
- CFRVORMUGQWQNZ-UHFFFAOYSA-N thiepane 1,1-dioxide Chemical compound O=S1(=O)CCCCCC1 CFRVORMUGQWQNZ-UHFFFAOYSA-N 0.000 description 2
- FCFMKFHUNDYKEG-UHFFFAOYSA-N thietane 1,1-dioxide Chemical compound O=S1(=O)CCC1 FCFMKFHUNDYKEG-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000001960 triggered effect Effects 0.000 description 2
- 230000029069 type 2 immune response Effects 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- TXTWXQXDMWILOF-UHFFFAOYSA-N (2-ethoxy-2-oxoethyl)azanium;chloride Chemical compound [Cl-].CCOC(=O)C[NH3+] TXTWXQXDMWILOF-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- YMYLGVIBUGDMLT-QMMMGPOBSA-N (2s)-2-(phenylmethoxyamino)propanoic acid Chemical compound OC(=O)[C@H](C)NOCC1=CC=CC=C1 YMYLGVIBUGDMLT-QMMMGPOBSA-N 0.000 description 1
- 125000006701 (C1-C7) alkyl group Chemical group 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- UPLURGSDLNKCSU-UHFFFAOYSA-N 1,5,6,7-tetrahydrocyclopenta[d]pyrimidine-2,4-dione Chemical compound O=C1NC(=O)NC2=C1CCC2 UPLURGSDLNKCSU-UHFFFAOYSA-N 0.000 description 1
- ZDJXTZMWRPBODK-UHFFFAOYSA-N 1-[(2-amino-5-butyl-6-methylpyrimidin-4-yl)amino]pentan-2-ol Chemical compound CCCCC1=C(C)N=C(N)N=C1NCC(O)CCC ZDJXTZMWRPBODK-UHFFFAOYSA-N 0.000 description 1
- 125000006218 1-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- RQEUFEKYXDPUSK-UHFFFAOYSA-N 1-phenylethylamine Chemical compound CC(N)C1=CC=CC=C1 RQEUFEKYXDPUSK-UHFFFAOYSA-N 0.000 description 1
- PSTIJXHSQUNQST-UHFFFAOYSA-N 2-[(2-amino-5,6,7,8-tetrahydroquinazolin-4-yl)amino]hexanamide Chemical compound C1CCCC2=C1N=C(N)N=C2NC(CCCC)C(N)=O PSTIJXHSQUNQST-UHFFFAOYSA-N 0.000 description 1
- VWVRASTUFJRTHW-UHFFFAOYSA-N 2-[3-(azetidin-3-yloxy)-4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]pyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound O=C(CN1C=C(C(OC2CNC2)=N1)C1=CN=C(NC2CC3=C(C2)C=CC=C3)N=C1)N1CCC2=C(C1)N=NN2 VWVRASTUFJRTHW-UHFFFAOYSA-N 0.000 description 1
- XXZCIYUJYUESMD-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-3-(morpholin-4-ylmethyl)pyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C(=NN(C=1)CC(=O)N1CC2=C(CC1)NN=N2)CN1CCOCC1 XXZCIYUJYUESMD-UHFFFAOYSA-N 0.000 description 1
- FYELSNVLZVIGTI-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-5-ethylpyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C=NN(C=1CC)CC(=O)N1CC2=C(CC1)NN=N2 FYELSNVLZVIGTI-UHFFFAOYSA-N 0.000 description 1
- BPGIOCZAQDIBPI-UHFFFAOYSA-N 2-ethoxyethanamine Chemical compound CCOCCN BPGIOCZAQDIBPI-UHFFFAOYSA-N 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- SMVIKWIBYJGAIJ-UHFFFAOYSA-N 3-[2-amino-4-methyl-6-(pentylamino)pyrimidin-5-yl]propanenitrile Chemical compound CCCCCNC1=NC(N)=NC(C)=C1CCC#N SMVIKWIBYJGAIJ-UHFFFAOYSA-N 0.000 description 1
- WADQOGCINABPRT-UHFFFAOYSA-N 3-chloro-2-methylphenol Chemical compound CC1=C(O)C=CC=C1Cl WADQOGCINABPRT-UHFFFAOYSA-N 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- XZKIHKMTEMTJQX-UHFFFAOYSA-N 4-Nitrophenyl Phosphate Chemical compound OP(O)(=O)OC1=CC=C([N+]([O-])=O)C=C1 XZKIHKMTEMTJQX-UHFFFAOYSA-N 0.000 description 1
- VJVQURFWVIPIFM-UHFFFAOYSA-N 4-bromo-5,6,7,8-tetrahydroquinazolin-2-amine Chemical compound C1CCCC2=NC(N)=NC(Br)=C21 VJVQURFWVIPIFM-UHFFFAOYSA-N 0.000 description 1
- PYPBCCFJYPGJBA-UHFFFAOYSA-N 4-chloro-5-ethyl-6-methylpyrimidin-2-amine Chemical compound CCC1=C(C)N=C(N)N=C1Cl PYPBCCFJYPGJBA-UHFFFAOYSA-N 0.000 description 1
- ATSGNLDQRXTBHY-UHFFFAOYSA-N 4-n-butyl-5,6,7,8-tetrahydroquinazoline-2,4-diamine Chemical compound C1CCCC2=C1N=C(N)N=C2NCCCC ATSGNLDQRXTBHY-UHFFFAOYSA-N 0.000 description 1
- HADJIBFQXNGHFR-UHFFFAOYSA-N 4-n-hexyl-5,6,7,8-tetrahydroquinazoline-2,4-diamine Chemical compound C1CCCC2=C1N=C(N)N=C2NCCCCCC HADJIBFQXNGHFR-UHFFFAOYSA-N 0.000 description 1
- AZSGQOZVSDCWCU-UHFFFAOYSA-N 4-n-pentyl-5,6,7,8-tetrahydroquinazoline-2,4-diamine Chemical compound C1CCCC2=C1N=C(N)N=C2NCCCCC AZSGQOZVSDCWCU-UHFFFAOYSA-N 0.000 description 1
- GBUHVEFCTQINBK-UHFFFAOYSA-N 4-n-pentyl-5-(2-phenylethyl)pyrimidine-2,4-diamine Chemical compound CCCCCNC1=NC(N)=NC=C1CCC1=CC=CC=C1 GBUHVEFCTQINBK-UHFFFAOYSA-N 0.000 description 1
- LINDJJJZQPKKLO-UHFFFAOYSA-N 4-n-pentyl-6,7-dihydro-5h-cyclopenta[d]pyrimidine-2,4-diamine Chemical compound CCCCCNC1=NC(N)=NC2=C1CCC2 LINDJJJZQPKKLO-UHFFFAOYSA-N 0.000 description 1
- ATCQYBFSNUJNBX-UHFFFAOYSA-N 4-n-pentyl-7,8-dihydro-5h-pyrano[4,3-d]pyrimidine-2,4-diamine Chemical compound C1COCC2=C1N=C(N)N=C2NCCCCC ATCQYBFSNUJNBX-UHFFFAOYSA-N 0.000 description 1
- SEUGQUWUMKOJRA-UHFFFAOYSA-N 5,6-dimethyl-4-n-pentylpyrimidine-2,4-diamine Chemical compound CCCCCNC1=NC(N)=NC(C)=C1C SEUGQUWUMKOJRA-UHFFFAOYSA-N 0.000 description 1
- PAWSCFSCHHTUSD-UHFFFAOYSA-N 5-(2-methoxyethyl)-6-methyl-4-n-pentylpyrimidine-2,4-diamine Chemical compound CCCCCNC1=NC(N)=NC(C)=C1CCOC PAWSCFSCHHTUSD-UHFFFAOYSA-N 0.000 description 1
- HGEMUUFTVSAPOM-UHFFFAOYSA-N 5-benzyl-4-n-pentylpyrimidine-2,4-diamine Chemical compound CCCCCNC1=NC(N)=NC=C1CC1=CC=CC=C1 HGEMUUFTVSAPOM-UHFFFAOYSA-N 0.000 description 1
- JCZDGUXZPPWROA-UHFFFAOYSA-N 5-benzyl-6-methyl-4-n-pentylpyrimidine-2,4-diamine Chemical compound CCCCCNC1=NC(N)=NC(C)=C1CC1=CC=CC=C1 JCZDGUXZPPWROA-UHFFFAOYSA-N 0.000 description 1
- AEBBCRQCZPTMBF-UHFFFAOYSA-N 5-butyl-6-methyl-4-n-pentylpyrimidine-2,4-diamine Chemical compound CCCCCNC1=NC(N)=NC(C)=C1CCCC AEBBCRQCZPTMBF-UHFFFAOYSA-N 0.000 description 1
- LQSOVJUZPZOESH-UHFFFAOYSA-N 5-ethyl-6-methyl-4-n-pentylpyrimidine-2,4-diamine Chemical compound CCCCCNC1=NC(N)=NC(C)=C1CC LQSOVJUZPZOESH-UHFFFAOYSA-N 0.000 description 1
- SLSYMKZGXBFRJT-UHFFFAOYSA-N 5-hexyl-6-methyl-4-n-pentylpyrimidine-2,4-diamine Chemical compound CCCCCCC1=C(C)N=C(N)N=C1NCCCCC SLSYMKZGXBFRJT-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- GRELAOXZRDTXLF-UHFFFAOYSA-N CCOCCNc1c(CCCC2)c2nc(N)n1 Chemical compound CCOCCNc1c(CCCC2)c2nc(N)n1 GRELAOXZRDTXLF-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 235000001258 Cinchona calisaya Nutrition 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- PMPVIKIVABFJJI-UHFFFAOYSA-N Cyclobutane Chemical compound C1CCC1 PMPVIKIVABFJJI-UHFFFAOYSA-N 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- XDWQYMXQMNUWID-UHFFFAOYSA-N Ethyl 2-benzylacetoacetate Chemical compound CCOC(=O)C(C(C)=O)CC1=CC=CC=C1 XDWQYMXQMNUWID-UHFFFAOYSA-N 0.000 description 1
- GGFNXKFGVQQNRV-UHFFFAOYSA-N Ethyl 4-phenylbutanoate Chemical compound CCOC(=O)CCCC1=CC=CC=C1 GGFNXKFGVQQNRV-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 241001295925 Gegenes Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 101001044384 Mus musculus Interferon gamma Proteins 0.000 description 1
- 101001002703 Mus musculus Interleukin-4 Proteins 0.000 description 1
- XTUVJUMINZSXGF-UHFFFAOYSA-N N-methylcyclohexylamine Chemical compound CNC1CCCCC1 XTUVJUMINZSXGF-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical class CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- QMGVPVSNSZLJIA-UHFFFAOYSA-N Nux Vomica Natural products C1C2C3C4N(C=5C6=CC=CC=5)C(=O)CC3OCC=C2CN2C1C46CC2 QMGVPVSNSZLJIA-UHFFFAOYSA-N 0.000 description 1
- 208000001388 Opportunistic Infections Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 206010040007 Sense of oppression Diseases 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 230000024932 T cell mediated immunity Effects 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- GUGOEEXESWIERI-UHFFFAOYSA-N Terfenadine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GUGOEEXESWIERI-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108010000134 Vascular Cell Adhesion Molecule-1 Proteins 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001387 anti-histamine Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 239000000043 antiallergic agent Substances 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- VIQRCOQXIHFJND-UHFFFAOYSA-N bicyclo[2.2.2]oct-2-ene Chemical compound C1CC2CCC1C=C2 VIQRCOQXIHFJND-UHFFFAOYSA-N 0.000 description 1
- GPRLTFBKWDERLU-UHFFFAOYSA-N bicyclo[2.2.2]octane Chemical compound C1CC2CCC1CC2 GPRLTFBKWDERLU-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229940124630 bronchodilator Drugs 0.000 description 1
- 239000000168 bronchodilator agent Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 231100000315 carcinogenic Toxicity 0.000 description 1
- 206010061592 cardiac fibrillation Diseases 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 229940082500 cetostearyl alcohol Drugs 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 208000023819 chronic asthma Diseases 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 208000010247 contact dermatitis Diseases 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000004850 cyclobutylmethyl group Chemical group C1(CCC1)C* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- WJTCGQSWYFHTAC-UHFFFAOYSA-N cyclooctane Chemical compound C1CCCCCCC1 WJTCGQSWYFHTAC-UHFFFAOYSA-N 0.000 description 1
- 239000004914 cyclooctane Substances 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 125000005117 dialkylcarbamoyl group Chemical group 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 229910000397 disodium phosphate Inorganic materials 0.000 description 1
- 235000019800 disodium phosphate Nutrition 0.000 description 1
- 230000003028 elevating effect Effects 0.000 description 1
- JFQTXHMZBZRXET-UHFFFAOYSA-N ethyl 2-(2-cyanoethyl)-3-oxobutanoate Chemical compound CCOC(=O)C(C(C)=O)CCC#N JFQTXHMZBZRXET-UHFFFAOYSA-N 0.000 description 1
- ZVJPCEUTGFHQRQ-UHFFFAOYSA-N ethyl 2-(2-methoxyethyl)-3-oxobutanoate Chemical compound CCOC(=O)C(C(C)=O)CCOC ZVJPCEUTGFHQRQ-UHFFFAOYSA-N 0.000 description 1
- SMGRHCCILTYVRF-UHFFFAOYSA-N ethyl 2-[(2-amino-5,6,7,8-tetrahydroquinazolin-4-yl)amino]-3-hydroxypropanoate Chemical compound C1CCCC2=C1N=C(N)N=C2NC(CO)C(=O)OCC SMGRHCCILTYVRF-UHFFFAOYSA-N 0.000 description 1
- NBGPINNGASMYLQ-UHFFFAOYSA-N ethyl 2-[(2-amino-5,6,7,8-tetrahydroquinazolin-4-yl)amino]-4-methylpentanoate Chemical compound C1CCCC2=C1N=C(N)N=C2NC(CC(C)C)C(=O)OCC NBGPINNGASMYLQ-UHFFFAOYSA-N 0.000 description 1
- AJBUKHBSSSXWPR-UHFFFAOYSA-N ethyl 2-[(2-amino-5,6,7,8-tetrahydroquinazolin-4-yl)amino]acetate Chemical compound C1CCCC2=C1N=C(N)N=C2NCC(=O)OCC AJBUKHBSSSXWPR-UHFFFAOYSA-N 0.000 description 1
- NJPQFACDTRCYAD-UHFFFAOYSA-N ethyl 2-[(2-amino-5,6,7,8-tetrahydroquinazolin-4-yl)amino]propanoate Chemical compound C1CCCC2=C1N=C(N)N=C2NC(C)C(=O)OCC NJPQFACDTRCYAD-UHFFFAOYSA-N 0.000 description 1
- ZTOQBHVLCJERBS-UHFFFAOYSA-N ethyl 2-acetylhexanoate Chemical compound CCCCC(C(C)=O)C(=O)OCC ZTOQBHVLCJERBS-UHFFFAOYSA-N 0.000 description 1
- BRGRZPQESQKATK-UHFFFAOYSA-N ethyl 2-acetyloctanoate Chemical compound CCCCCCC(C(C)=O)C(=O)OCC BRGRZPQESQKATK-UHFFFAOYSA-N 0.000 description 1
- JCXLZWMDXJFOOI-UHFFFAOYSA-N ethyl 2-aminopropanoate;hydron;chloride Chemical compound [Cl-].CCOC(=O)C(C)[NH3+] JCXLZWMDXJFOOI-UHFFFAOYSA-N 0.000 description 1
- DGGOELSWSLCMNU-UHFFFAOYSA-N ethyl 2-methyl-3-oxoheptanoate Chemical compound CCCCC(=O)C(C)C(=O)OCC DGGOELSWSLCMNU-UHFFFAOYSA-N 0.000 description 1
- FGSGHBPKHFDJOP-UHFFFAOYSA-N ethyl 2-oxocyclohexane-1-carboxylate Chemical compound CCOC(=O)C1CCCCC1=O FGSGHBPKHFDJOP-UHFFFAOYSA-N 0.000 description 1
- ZUJSLBBFQOCMFB-UHFFFAOYSA-N ethyl 4-oxooxane-3-carboxylate Chemical compound CCOC(=O)C1COCCC1=O ZUJSLBBFQOCMFB-UHFFFAOYSA-N 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 230000002600 fibrillogenic effect Effects 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229960000789 guanidine hydrochloride Drugs 0.000 description 1
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- UBHWBODXJBSFLH-UHFFFAOYSA-N hexadecan-1-ol;octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO.CCCCCCCCCCCCCCCCCCO UBHWBODXJBSFLH-UHFFFAOYSA-N 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 235000014380 magnesium carbonate Nutrition 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- XNLICIUVMPYHGG-UHFFFAOYSA-N methyl n-propyl ketone Natural products CCCC(C)=O XNLICIUVMPYHGG-UHFFFAOYSA-N 0.000 description 1
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 150000002762 monocarboxylic acid derivatives Chemical class 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- ZSIAUFGUXNUGDI-UHFFFAOYSA-N n-hexyl alcohol Natural products CCCCCCO ZSIAUFGUXNUGDI-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- JFNLZVQOOSMTJK-UHFFFAOYSA-N norbornene Chemical compound C1C2CCC1C=C2 JFNLZVQOOSMTJK-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 229940039748 oxalate Drugs 0.000 description 1
- YNOGYQAEJGADFJ-UHFFFAOYSA-N oxolan-2-ylmethanamine Chemical compound NCC1CCCO1 YNOGYQAEJGADFJ-UHFFFAOYSA-N 0.000 description 1
- 229940056211 paraffin Drugs 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 230000024241 parasitism Effects 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 229940100684 pentylamine Drugs 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 235000020030 perry Nutrition 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- UXCDUFKZSUBXGM-UHFFFAOYSA-N phosphoric tribromide Chemical compound BrP(Br)(Br)=O UXCDUFKZSUBXGM-UHFFFAOYSA-N 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- ALDITMKAAPLVJK-UHFFFAOYSA-N prop-1-ene;hydrate Chemical group O.CC=C ALDITMKAAPLVJK-UHFFFAOYSA-N 0.000 description 1
- 229960004063 propylene glycol Drugs 0.000 description 1
- 125000004742 propyloxycarbonyl group Chemical group 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 229960001404 quinidine Drugs 0.000 description 1
- 229960000948 quinine Drugs 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 210000001533 respiratory mucosa Anatomy 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229920003051 synthetic elastomer Polymers 0.000 description 1
- 239000005061 synthetic rubber Substances 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- OULAJFUGPPVRBK-UHFFFAOYSA-N tetratriacontyl alcohol Natural products CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCO OULAJFUGPPVRBK-UHFFFAOYSA-N 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical class OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Immunology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Description
- Die vorliegende Erfindung betrifft Pyrimidinderivate und medizinische Verwendungen davon. Im Einzelnen betrifft die vorliegende Erfindung Pyrimidinderivate mit Wirksamkeiten zur Unterdrückung von Immunantworten der Typ 2 T-Helferzelle (Th2) und die Erhöhung von Immunantworten der Typ 1 T-Helferzelle (Th1) und therapeutische Verfahren für Immunkrankheiten unter Verwendung der Pyrimidinderivate und therapeutische Zusammensetzungen, die die Pyrimidinderivate enthalten.
- Als erstes wird von Mosmann et al. vorgeschlagen, dass Lymphocyten, genannt T-Helferzellen, die die zentrale Rolle bei Immunantworten spielen, in zwei Untergruppen klassifiziert werden. Sie klassifizierten T-Helferzellen (Th) von Mäusen in Th1 und Th2, abhängig von den Arten der gebildeten Cytokine (J. Immunol. 136, 2348–2357 (1986)).
- Als Cytokine des Th1-Typs werden Interleukin 2 (IL-2), Interferon γ (IFN-γ) usw. veranschaulicht. Als Cytokine des Th2-Typs werden Interleukin 4 (IL-4), Interleukin 5 (IL-5), Interleukin 10 (IL-10), Interleukin 13 (IL-13) usw. veranschaulicht.
- Heutzutage wird der Gedankengang der Klassifizierung in Th1/Th2 auf die Klassifizierung in Untergruppen von T-Helferzellen und auch in Bezug auf eine Reihe von Immunantworten im lebenden Körper unter dem Gesichtspunkt, welche Untergruppe von T-Helferzellen hauptsächlich teilnimmt, angewandt, die Immunantworten wurden als „Immunantworten auf Th1-Typ" bzw. „Immunantworten auf Th2-Typ" interpretiert.
- Immunantworten auf Th1-Typ werden hauptsächlich durch Cytokine bewirkt, wie Interleukin 2 (IL-2), Interferon γ (IFN-γ) usw., die durch aktivierte Th1 gebildet werden. So ist bekannt, dass Th1-Cytokine an durch Zellen vermittelter Immunität, wie dem Schutz hauptsächlich vor Infektionen durch Viren, Bakterien usw., durch Aktivieren von Makrophagen, natürlichen Killerzellen usw. oder durch weitere Aktivierung von Th1 über IL-12 usw., gebildet durch die aktivierten Makrophagen, beteiligt sind.
- Andererseits sind Immunantworten auf den Th2-Typ hauptsächlich durch Cytokine, wie IL-4, IL-5 usw., bewirkt, die durch aktivierte Th2 gebildet werden. So ist bekannt, dass Th2-Cytokine an der humoralen Immunität, wie der Bildung von Antikörpern (z. B. IgE-Klasse) aus B-Zellen, beteiligt sind.
- Da Th2 Cytokine, wie IL-4 oder IL-5, bilden, die eine allergische Reaktion, wie nachstehend erwähnt, betreffen, wird vorgeschlagen, dass Th2 die verantwortlichen Zellen für eine allergische Reaktion sind. Zum Beispiel induziert IL-4, ein typisches Cytokin des Th2-Typs, die Bildung von IgE-Antikörpern aus B-Zellen. IL-4 induziert auch die Expression des VCAM-1 Gens, ein wichtiges Molekül, das funktioniert, wenn Eosinophile an den vasculären Endothelzellen haften und in das Gewebe infiltrieren (Farumashia, 29, 1123–1128 (1993). Vor kurzem wurde IL-4 Aufmerksamkeit als einem eine Differenzierung induzierenden Faktor für Th2 geschenkt. IL-5, ein anderes Cytokin des Th2-Typs, induziert eine Differenzierung, Wanderung und Aktivierung von Eosinophilen. Eine allergische Entzündung ist dadurch gekennzeichnet, dass sie durch Infiltration, Aktivierung und Degranulierung von Eosinophilen ausgelöst wird, wie eine typische chronische Atemwegsentzündung bei Asthma. So wird IL-5 als Faktor angesehen, der eine allergische Entzündung induziert.
- Da Th2-Cytokine die vorstehenden Eigenschaften aufweisen, wird erkannt, dass Th2 sowohl allergische Reaktionen der „Reaktion des frühen Stadiums" durch IgE-Antikörper oder Mastzellen als auch „Reaktion des späten Stadiums" durch Eosinephile steuert, und daher sind Th2 die zentralen Zellen bei einer allergischen Entzündung. Und es wird angenommen, dass allergische Krankheiten durch die Überexpression von Immunantworten des Th2-Typs bewirkt werden. Diese Annahme wird auch durch die Feststellung des Vorhandenseins von Th2 oder der Bildung von Cytokinen des Th2-Typs, wie IL-4, IL-5 usw., bei dem erkrankten Teil bei allergischen Krankheiten, wie Luftweg oder Haut, unterstützt.
- Daher wird angenommen, dass es wichtig ist, die Immunantworten von Th2 zu unterdrücken, um sowohl die Reaktionen im frühen Stadium als auch späten Stadium zu unterdrücken oder die allergische entzündliche Reaktion zu hemmen, die durch die Infiltration und Aktivierung von Eosinophilen in dem Stadium der fundamentalen Quelle gekennzeichnet sind, und allgemeine allergische Krankheiten therapeutisch und vorbeugend zu behandeln. D. h. wenn ein Arzneistoff zum Unterdrücken der Immunantworten des Th2-Typs entwickelt wird, wird der Arzneistoff ein ein therapeutisches und vorbeugendes Mittel für allergische Krankheiten sein.
- Insbesondere bei ernstem chronischen Asthma oder atopischer Dermatitis unter den allergischen Krankheiten wird angenommen, dass die Reaktion im späten Stadium eine wichtige Rolle spielt. Jedoch basieren die heutzutage verwendeten antiallergischen Mittel hauptsächlich auf der Antihistamin-Wirksamkeit und hemmen nur die Reaktion im frühen Stadium und die klinische Wirkung davon ist nicht zufriedenstellend. Auch im Hinblick darauf war die Entwicklung eines Arzneistoffs erwünscht, der sowohl Reaktionen im frühen Stadium als auch späten Stadium durch Unterdrücken der Immunantworten von Th2 hemmt und therapeutisch und vorbeugend allgemeine allergische Krankheiten, wie vorstehend erwähnt, behandelt.
- Außerdem ist von Bronchodilatoren, die durch Xanthinderivate oder β-Stimulantien repräsentiert werden, die als Asthmamittel für lange Jahre verwendet wurden, bekannt, dass sie unterdrückende Wirksamkeit des Zusammenziehens des glatten Bronchienmuskels durch verschiedene Stimulation aufweisen. Jedoch sind diese nicht wirksam gegenüber chronischer Atemwegsentzündung, die eine grundlegende Ursache für Asthma ist. Zusätzlich sind Nebenwirkungen von Xanthinderivaten oder β-Stimulantien gegenüber Kreislauforganen besorgniserregend. In der neueren Asthmatherapie, wie defininitiv in der Richtlinie der WHO gezeigt, wird Asthma als chronische Entzündung des Atemwegs angesehen, und es wurde zu einer Hauptaufgabe gemacht, die chronische Entzündung des Atemwegs zu behandeln. Die chronische Entzündung des Atemwegs bei Asthma wird durch die Infiltration, Aktivierung und Degranulierung von Eosinophilen getriggert und weist ein pathologisches charakteristisches Merkmal auf, das eine Hypertrophie und Fibrillierung des Atemwegepithels ergibt. Gemäß der vorstehenden Richtlinie werden die einzigen Steroid-Inhalationsmittel, die gegenüber chronischer Atemwegsentzündung wirksam sind, jetzt als zuerst gewähltes Arzneimittel für Asthma mit mehr als mittlerem Grad positioniert.
- Als Ergebnis wurden Steroide häufig für ernstes Asthma und atopische Dermatitis verwendet, da sie als die einzig wirksamen Arzneistoffe angesehen werden. Jedoch wird es ein Problem, dass unter Verwendung solcher Steroide für lange Zeit verschiedene Nebenwirkungen (steroide Dermatitis, induzierte Infektionskrankheit, Dyscorticismus usw.) auftreten.
- Ebenfalls im Hinblick darauf war erwünscht, den Arzneistoff zu entwickeln, der Immunantworten auf Th2 seletiv unterdrückt und Reaktionen sowohl im frühen als auch späten Stadium hemmt oder eine allergische entzündliche Reaktion hemmt, die durch eine Infiltration und Aktivierung von Eosinophilen im Stadium der fundamentalen Quelle gekennzeichnet ist und therapeutisch und vorbeugend für allgemeine allergische Krankheiten wirksam ist.
- Außerdem scheint es, wenn geplant wird, therapeutische oder vorbeugende Arzneistoffe zu entwickeln, die weniger Nebenwirkungen aufweisen, dass die Arzneistoffe, die Immunantworten auf Th2 wie vorstehend erwähnt unterdrücken und Immunantworten auf Th1 gleichzeitig verstärken, als Arzneimittel stärker bevorzugt sind. Wie vorstehend erwähnt sind, da Th1 eine wichtige Rolle für den lebenden Körper spielt, d. h. Infektionsschutz gegen Viren und Bakterien hauptsächlich durch Produktion von IFN-γ, die Arnzeistoffe, die die Immunantworten auf Th2 unterdrücken und die Wirksamkeit von Th1 verstärken, sehr bevorzugt im Hinblick auf Nebenwirkungen. Zum Beispiel ist von Immunsuppressiva, z. B. Cyclosporin oder FK506, bekannt, dass sie in starkem Maße die Aktivierung von Th2 hemmen. Jedoch zeigen sowohl Cyclosporin als auch FK506 keine spezifische Unterdrückung gegenüber Immunantworten, d. h. sie hemmen nicht nur die Aktivierung von Th2, sondern hemmen auch stärker die Aktivierung von Th1. Daher waren ernste Nebenwirkungen, wie opportunistische Infektion oder Zunahme der carcinogenen Rate durch eine solche nicht spezifische Unterdrückung gegenüber Immunantworten ein Problem. Von anderen nicht spezifischen Immunsuppressiva wird angenommen, dass sie die gleichen Probleme aufweisen.
- Wie vorstehend erwähnt ist der Arzneistoff, der die Immunantwort auf Th1, wiedergegeben durch die Produktion von IFN-γ, verstärkt und die Immunantworten auf Th2, wiedergegeben durch die Produktion von IL-4 und IL-5, gleichzeitig unterdrückt, ein therapeutisches und vorbeugendes Mittel für allergische Krankheiten mit weniger Nebenwirkungen.
- Autoimmunkrankheiten in dem Zustand, dass die Bildung eines Antikörpers oder von humoraler Immunität abnorm erhöht ist, wie systemischer Lupus erythematosus, werden ebenfalls als im Zustand angesehen, dass Immunantworten auf Th2 abnorm verstärkt sind (Medical Immunology 15, 401 (1988)). Daher wird von dem Arzneistoff, der Immunantworten auf Th1 erhöht und Immunantworten von Th2 unterdrückt, erwartet, dass er ein therapeutisches Mittel für Autoimmunkrankheiten wird.
- Pyrimidinderivate mit allgemeiner antiviraler Wirksamkeit sind in der japanischen Patentveröffentlichung A-9-301958 und der japanischen Patentveröffentlichung A 8-134044 offenbart. Jedoch gibt es keinen Vorschlag für Pyrimidinderivate der vorliegenden Erfindung, die Immunantworten von Th1 erhöhen und Immunantworten von Th2 unterdrücken.
- WO 97/09325 offenbart Pyrimidinderivate mit immunschwächender Wirksamkeit usw. Jedoch sind diese Derivate strukturell verschieden zu den erfindungsgemäßen Pyrimidinderivaten.
- Zusammenfassung der Erfindung
- Unter solchen Umständen haben die in der vorliegenden Anmeldung genannten Erfinder verschiedene Verbindungen synthetisiert und sie auf die Wirkung auf Th1- und Th2-Immunantworten untersucht. Als Ergebnis wurde festgestellt, dass bestimmte Pyrimidinderivate die Th1-Immunantworten erhöhen und die Th2-Immunantworten unterdrücken, und daher das Gleichgewicht von Th1/Th2 in eine bevorzugte Richtung ändern.
- D. h. die vorliegende Erfindung betrifft:
- [1]
ein Pyrimidinderivat der Formel (1) oder ein Salz davon; wobei der Rest R1 durch eine Formel (2) wiedergegeben ist;
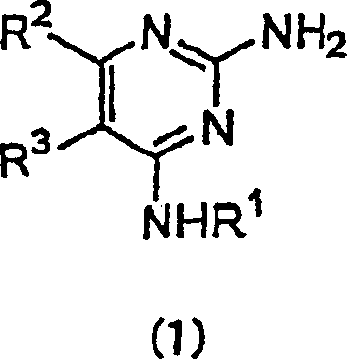 {in der Formel (2) ist der Ring A ein substituiertes oder unsubstituiertes C3-10-Cycloalkan, ein substituiertes oder unsubstituiertes C5-10-Cycloalken, ein substituiertes oder unsubstituiertes C7-10-Bicycloalkan oder ein heterocyclischer Ring, der ausgewählt ist aus Oxetan, Thietan (Trimethylensulfid), Thietan-1-oxid, Thiethan-1,1-Dioxid, Tetrahydrofuran, Tetrahydrothiophen, Tetrahydrothiophen-1-oxid, Tetrahydrothiophen-1,1-dioxid, Tetrahydro-4H-pyran, Thian (Pentamethylensulfid), Thian-1-oxid, Thian-1,1-dioxid, Oxepan (Hexamethylenoxid), Thiepan (Hexamethylensulfid), Thiepan-1-oxid, Thiepan-1,1-dioxid, 7-Oxabicyclo[2.2.1]heptan und 7-Oxabicyclo[2.2.1]hepta-5-en, oder wobei der heterocyclische Ring einen oder mehrere Substituenten, ausgewählt aus C1-3Alkyl, Hydroxy, C1-3-Alkoxycarbonyl, Carboxy, Carbamoyl und Oxo, aufweist, und die Substituenten an den benachbarten Kohlenstoffatomen am heterocyclischen Ring eine Tetramethylenbrücke bilden können, ist der Rest R4 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkenyl, C3-6-Cycloalkyl, C4-10-Cycloalkylalkyl, oder OR8 (wobei der Rest R8 ein geradkettiges oder verzweigtes C1-10-Alkyl, C3-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl oder C4-10-Cycloalkylalkyl ist)}, oder durch eine Formel (3) wiedergegeben ist;
{in der Formel (2) ist der Ring A ein substituiertes oder unsubstituiertes C3-10-Cycloalkan, ein substituiertes oder unsubstituiertes C5-10-Cycloalken, ein substituiertes oder unsubstituiertes C7-10-Bicycloalkan oder ein heterocyclischer Ring, der ausgewählt ist aus Oxetan, Thietan (Trimethylensulfid), Thietan-1-oxid, Thiethan-1,1-Dioxid, Tetrahydrofuran, Tetrahydrothiophen, Tetrahydrothiophen-1-oxid, Tetrahydrothiophen-1,1-dioxid, Tetrahydro-4H-pyran, Thian (Pentamethylensulfid), Thian-1-oxid, Thian-1,1-dioxid, Oxepan (Hexamethylenoxid), Thiepan (Hexamethylensulfid), Thiepan-1-oxid, Thiepan-1,1-dioxid, 7-Oxabicyclo[2.2.1]heptan und 7-Oxabicyclo[2.2.1]hepta-5-en, oder wobei der heterocyclische Ring einen oder mehrere Substituenten, ausgewählt aus C1-3Alkyl, Hydroxy, C1-3-Alkoxycarbonyl, Carboxy, Carbamoyl und Oxo, aufweist, und die Substituenten an den benachbarten Kohlenstoffatomen am heterocyclischen Ring eine Tetramethylenbrücke bilden können, ist der Rest R4 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkenyl, C3-6-Cycloalkyl, C4-10-Cycloalkylalkyl, oder OR8 (wobei der Rest R8 ein geradkettiges oder verzweigtes C1-10-Alkyl, C3-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl oder C4-10-Cycloalkylalkyl ist)}, oder durch eine Formel (3) wiedergegeben ist;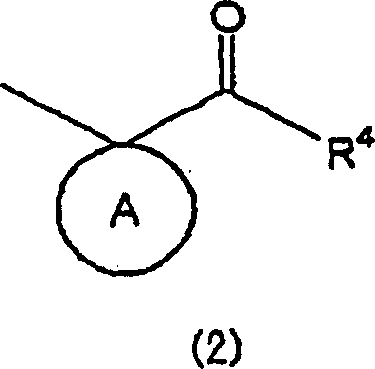 {in der Formel (3) ist der Rest R5 ein geradkettiges oder verzweigtes C1-10-Alkyl; C2-6-Alkenyl; C3-6-Alkinyl; ein geradkettiges oder verzweigtes C1-10-Alkyl, das mit Hydroxy, einem Halogenatom oder C1-4-Alkoxy substituiert ist; Phenyl; C3-8-Cycloalkyl; ein 5 bis 7 gliedriger gesättigter heterocyclischer Ring, der ein oder zwei Sauerstoffatome als Heteroatome enthält; oder C(=O)R9 (wobei der Rest R9 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl, C4-10-Cycloalkylalkyl oder OR10 ist (wobei der Rest R10 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl oder C4-10-Cycloalkylalkyl ist)), ist der Rest R6 ein Wasserstoffatom, ein geradkettiges oder verzweigtes C1-10-Alkyl, C6-10-Aryl, ein Halogenatom, C6-10-Aryl, das mit C1-4-Alkoxy oder C1-4-Alkyl, Carbamoyl, oder Hydroxymethyl substituiert ist, und ist der Rest R7 ein Wasserstoffatom oder ein geradkettiges oder verzweigtes C1-10-Alkyl}, ist der Rest R2 ein Wasserstoffatom oder ein geradkettiges oder verzweigtes C1-10-Alkyl, und ist der Rest R3 ein geradkettiges oder verzweigtes C1-10-Alkyl; C3-6-Cycloalkyl; ein geradkettiges oder verzweigtes C1-10-Alkyl, das mit C1-2-Alkylcarbamoyl, C2-4-Dialkylcarbamoyl, C1-4-Alkoxy, C1-4-Alkoxycarbonyl, C3-6-Cycloalkyl, Hydroxy, C1-4-Alkylcarbonyloxy, einem Halogenatom, Amino, C2-4-Acyl-substituiertem Amino, C1-4-Alkyl-substituiertem Sulfonylamino oder C1-5-Alkoxycarbonylamino substituiert ist; oder durch eine Formel (4) wiedergegeben ist;
{in der Formel (3) ist der Rest R5 ein geradkettiges oder verzweigtes C1-10-Alkyl; C2-6-Alkenyl; C3-6-Alkinyl; ein geradkettiges oder verzweigtes C1-10-Alkyl, das mit Hydroxy, einem Halogenatom oder C1-4-Alkoxy substituiert ist; Phenyl; C3-8-Cycloalkyl; ein 5 bis 7 gliedriger gesättigter heterocyclischer Ring, der ein oder zwei Sauerstoffatome als Heteroatome enthält; oder C(=O)R9 (wobei der Rest R9 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl, C4-10-Cycloalkylalkyl oder OR10 ist (wobei der Rest R10 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl oder C4-10-Cycloalkylalkyl ist)), ist der Rest R6 ein Wasserstoffatom, ein geradkettiges oder verzweigtes C1-10-Alkyl, C6-10-Aryl, ein Halogenatom, C6-10-Aryl, das mit C1-4-Alkoxy oder C1-4-Alkyl, Carbamoyl, oder Hydroxymethyl substituiert ist, und ist der Rest R7 ein Wasserstoffatom oder ein geradkettiges oder verzweigtes C1-10-Alkyl}, ist der Rest R2 ein Wasserstoffatom oder ein geradkettiges oder verzweigtes C1-10-Alkyl, und ist der Rest R3 ein geradkettiges oder verzweigtes C1-10-Alkyl; C3-6-Cycloalkyl; ein geradkettiges oder verzweigtes C1-10-Alkyl, das mit C1-2-Alkylcarbamoyl, C2-4-Dialkylcarbamoyl, C1-4-Alkoxy, C1-4-Alkoxycarbonyl, C3-6-Cycloalkyl, Hydroxy, C1-4-Alkylcarbonyloxy, einem Halogenatom, Amino, C2-4-Acyl-substituiertem Amino, C1-4-Alkyl-substituiertem Sulfonylamino oder C1-5-Alkoxycarbonylamino substituiert ist; oder durch eine Formel (4) wiedergegeben ist;
- [2] das Pyrimidinderivat oder sein Salz nach [1], wobei der Rest R3 ein geradkettiges oder verzweigtes C1-10-Alkyl; C3-6-Cycloalkyl; oder ein geradkettiges oder verzweigtes C1-10-Alkyl ist, das mit C1-2-Alkylcarbamoyl, C2-4-Dialkylcarbamoyl, C1-4-Alkoxy, C1-4-Alkoxycarbonyl, C3-6-Cycloalkyl, Hydroxy, C1-4-Alkylcarbonyloxy, einem Halogenatom, Amino, C2-4-Acyl-substituiertem Amino, C1-4-Alkyl-substituiertem Sulfonylamino oder C1-5-Alkoxycarbonylamino substituiert ist; oder die Reste R2 und R3 zusammen C3-5-Alkylen oder genanntes Alkylen bilden, wobei Methylen mit einem O-Atom ersetzt ist, mit der Maßgabe dass, wenn der Rest R5 Phenyl ist und die Reste R6 und R7 ein Wasserstoffatom sind, die Reste R2 und R3 zusammen C4-5-Alkylen oder C3-5-Alkylen bilden, wobei Methylen durch ein O-Atom ersetzt ist,
- [3] das Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach [1] oder [2], wobei die Reste R2 und R3 zusammen Trimethylen oder Tetramethylen sind, mit der Maßgabe dass, wenn der Rest R5 Phenyl ist und die Reste R6 und R7 ein Wasserstoffatom sind, die Reste R2 und R3 zusammen Tetramethylen sind,
- [4] das Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach [1] oder [2], wobei der Rest R3 ein geradkettiges oder verzweigtes C1-7-Alkyl ist,
- [5] das Pyrimidinderivat oder sein pharmazeutisch verträgliches
Salz nach [1], wobei der Rest R3 durch die Formel
(4) wiedergegeben ist;
- [6] das Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach [1] oder [5], wobei der Rest R11 der Formel (4) Pyridyl, Thienyl oder Furyl ist,
- [7] das Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach [1], [5] oder [6], wobei n der Formel (4) eine ganze Zahl von 2–4 ist,
- [8] das Pyrimidinderivat oder sein pharmazeutisch verträgliches
Salz nach einem von [1] bis [7], wobei der Rest R1 durch
die Formel (2) wiedergegeben ist; wobei der Ring A und R4 die gleiche vorstehend in [1] angegebene Bedeutung haben,
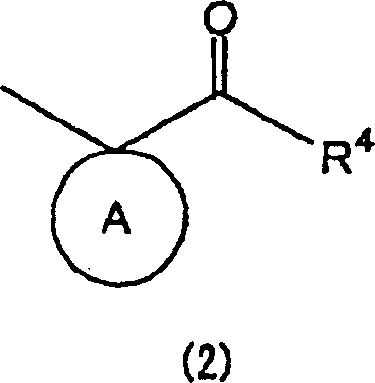
- [9] das Pyrimidinderivat oder sein pharmazeutisch verträgliches
Salz nach einem von [1] bis [7], wobei der Rest R1 durch
die Formel (3) wiedergegeben ist; wobei die Reste R5, R6 und R7, die gleiche vorstehend in [1] angegebene Bedeutung haben,
- [10] das Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach einem von [1] bis [7] oder [9], wobei der Rest R5 ein geradkettiges C2-4-Alkyl oder ein geradkettiges C2-4-Alkyl ist, das mit Hydroxy substituiert ist,
- [11] ein Immunmodulator, der die Immunantworten der Typ 2 T-Helferzelle unterdrückt und die Immunantworten der Typ 1 T-Helferzelle verstärkt, umfassend das Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach einem von [1] bis [10] als einen Wirkstoff,
- [12] ein therapeutisches oder vorbeugendes Mittel gegen Krankheiten in dem Stadium, in dem Immunantworten der T-Helferzelle von Typ 2 abnormal erhöht sind, umfassend das Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach einem von [1] bis [10] als einen Wirkstoff,
- [13] das therapeutische oder vorbeugende Mittel nach [12], wobei die Krankheit in dem Stadium, in dem die Immunantworten der Typ 2 T-Helferzelle abnormal erhöht sind, eine allergische Krankheit ist, und
- [14] das therapeutische oder vorbeugende Mittel nach [13], wobei die allergische Krankheit Asthma, allergische Rinitis oder allergische Dermatitis ist.
- DETAILLIERTE ERKLÄRUNG DER ERFINDUNG
- Die vorliegende Erfindung wird nachstehend im Einzelnen erklärt.
- Definition der Begriffe
- Die „Substituenten R1, R2 und R3" am Pyrimidinring der vorliegenden Erfindung werden wie folgt erklärt:
In Bezug auf den Rest R1
sind Beispiele von C3-10-Cycloalkan im Ring A Cyclopropan, Cyclobutan, Cyclopentan, Cyclohexan, Cycloheptan, Cyclooctan usw. Beispiele von C5-10-Cycloalken sind Cyclopenten, Cyclohexen usw. Beispiele von C7-10-Bicycloalkan sind Bicyclo[2.2.1]heptan, Bicyclo[2.2.1]hepta-5-en, Bicyclo[2.2.2]octan, Bicyclo[2.2.2]octa-5-en usw. Beispiele des heterocyclischen Rings sind Oxetan, Thietan (Trimethylensulfid), Thietan-1-oxid (Trimethylensulfoxid), Thietan-1,1-dioxid (Trimethylensulfon), Tetrahydrofuran, Tetrahydrothiophen, Tetrahydrothiophen-1-oxid, Tetrahydrothiophen-1,1-dioxid, Tetrahydro- 4H-pyran, Thian (Pentamethylensulfid), Thian-1,1-dioxid (Pentamethylensulfon), Thian-1-oxid (Pentamethylensulfoxid), Oxepan (Hexamethylenoxid), Thiepan (Hexamethylensulfid), Thiepan-1-oxid (Hexamethylensulfoxid), Thiepan-1,1-dioxid (Hexamethylensulfon), 7-Oxabicyclo[2.2.1]heptan, 7-Oxabicyclo[2.2.1]hepta-5-en usw. und
Beispiele der Substituenten von substituiertem Cycloalkan, substituiertem Cycloalken, substituiertem Bicycloalkan und substituiertem heterocyclischen Ring im Ring A sind C1-3-Alkyl, Hydroxy, C1-3-Alkoxycarbonyl, Carboxy, Carbamoyl usw., und die Substituenten an den benachbarten Kohlenstoffatomen können eine Tetramethylenbrücke bilden oder Kohlenstoffatom(e) im Ring können mit Carbonyl (C=O) substituiert sein. Der (die) Substituent(en) ist (sind) einer oder mehrere und gleich oder verschieden. Beispiele von C1-3-Alkyl sind Methyl, Ethyl, n-Propyl, 2-Propyl usw. Beispiele von C1-3-Alkoxycarbonyl sind Methoxycarbonyl, Ethoxycarbonyl, n-Propoxycarbonyl, 2-Propoxycarbonyl usw. - Beispiele von geradkettigem oder verzweigtem C1-10-Alkyl in den Resten R2, R3, R4, R5, R6, R7, R8, R9 und R10 sind Methyl, Ethyl, Propyl, 1-Methylethyl, Butyl, 1-Methylpropyl, 2-Methylpropyl, Pentyl, 1-Methylbutyl, 2-Methylbutyl, 3-Methylbutyl, 1-Ethylpropyl, Hexyl, 1-Methylpentyl, 2-Methylpentyl, 3-Methylpentyl, 4-Methylpentyl, 1-Ethylbutyl, 2-Ethylbutyl, Heptyl, Octyl, Nonyl, Decyl usw.
- Beispiele von C2-6-Alkenyl in den Resten R4, R5, R8, R9 und R10 sind Vinyl, Allyl, Butenyl, Pentenyl, Hexenyl usw.
- Beispiele von C3-6-Alkinyl in den Resten R4, R5, R8, R9 und R10 sind Propargyl, Butinyl, Pentinyl usw.
- Beispiele von C3-8-Cycloalkyl in Substituenten des geradkettigen oder verzweigten C1-10-Alkyls in den Resten R3, R4, R5, R8, R9 und R10 sind Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl, Cycloheptyl, Cyclooctyl usw.
- Beispiele von C4-10-Cycloalkylalkyl in den Resten R4, R8, R9 und R10 sind Cyclopropylmethyl, Cyclobutylmethyl, Cyclopentylethyl, Cyclohexylmethyl, Cyclohexylpropyl usw.
- Beispiele der Halogenatome in den Resten R3, R5 und R6 sind ein Fluoratom, Chloratom, Bromatom oder Jodatom.
- Beispiele von C1-4-Alkoxy in den Resten R3, R5 und R6 sind Methoxy, Ethoxy, Propoxy, Butoxy usw.
- Bevorzugte Beispiele von geradkettigem oder verzweigtem C1-10-Alkyl im Rest R3 sind ein geradkettiges oder verzweigtes C1-7-Alkyl, z. B. Methyl, Ethyl, Propyl, 1-Methylethyl, Butyl, 1-Methylpropyl, 2-Methylpropyl, Pentyl, 1-Methylbutyl, 2-Methylbutyl, 3-Methylbutyl, 1-Ethylpropyl, Hexyl, Heptyl usw.
- In Bezug auf die Substituenten des geradkettigen oder verzweigten C1-10-Alkyls im Rest R3 sind Beispiele von C1-2-Alkylcarbamoyl Methylcarbamoyl, Ethylcarbamoyl usw.; sind Beispiele von C2-4-Dialkylcarbamoyl Dimethylcarbamoyl, Methylethylcarbamoyl, Diethylcarbamoyl usw.; sind Beispiele von C1-4-Alkoxycarbonyl Methoxycarbonyl, Ethoxycarbonyl, Propoxycarbonyl, 2-Propoxycarbonyl usw.; sind Beispiele von C1-4-Alkylcarbonyloxy Acetoxy, Ethylcarbonyloxy, Propylcarbonyloxy usw., sind Beispiele von C2-4-Acyl-substituiertem Amino Acetylamino, Propanoylamino usw.; sind Beispiele von C1-4-Alkyl-substituiertem Sulfonylamino Methylsulfonylamino, Ethylsulfonylamino, Propylsulfonylamino, Butylsulfonylamino usw.; sind Beispiele von C1-5-Alkoxycarbonylamino Methoxycarbonylamino, Ethoxycarbonylamino, Propyloxycarbonylamino, Butoxycarbonylamino usw.
- R11 in R3 bedeutet Phenyl, Pyridyl, Thienyl oder Furyl und jedes davon kann mit einem oder mehreren Substituenten substituiert sein. Phenyl und Pyridyl sind bevorzugt, und Phenyl ist besonders bevorzugt. Die Substituenten sind Halogenatome, wie F, Cl, Br usw., Cyano, Carbamoyl, C1-4-Alkoxy, wie Methoxy, Ethoxy, Propoxy usw., C1-4-Alkyl, wie Methyl, Ethyl, Propyl, Butyl usw. n ist eine ganze Zahl von 0–4, mit der Maßgabe, dass n eine ganze Zahl von 1–4 ist, wenn R11 Phenyl ist. n ist vorzugsweise eine ganze Zahl von 0–2, stärker bevorzugt 1 oder 2.
- Beispiele eines 5- bis 7-gliedrigen gesättigten heterocyclischen Rings, der ein oder zwei Sauerstoffatome als Heteroatome enthält, in R5 sind Tetrahydrofuran, Oxan, 1,4-Dioxan, Oxepan usw.
- Bevorzugte Substituenten des geradkettigen oder verzweigten C1-10-Alkyls in R5 sind Hydroxy, die bevorzugte Zahl ist ein oder mehrere, und seine bevorzugte Position ist 1 oder 2 (Position 2 oder 3 bei Zählen von der Aminogruppe des Pyridinrings). Wenn der Substituent des geradkettigen oder verzweigten C1-10-Alkyls in R5 Hydroxy ist, ist seine Position vorzugsweise nicht die Endposition der Alkylkette.
- Beispiele von C6-10-Aryl in R6 sind Phenyl, Naphthyl usw.
- Bevorzugte Beispiele von geradkettigem oder verzweigtem C1-10-Alkyl in R9 sind ein geradkettiger oder verzweigter C2-4-Alkyl, z. B. Ethyl, Propyl, 1-Methylethyl, Butyl usw. Beispiele von C3-5-Alkylen, das R2 und R3 zusammen bilden, sind Trimethylen, Tetramethylen, Pentamethylen usw. Sie werden durch die folgenden Formeln (4), (5) und (6) wiedergegeben;
-

- Beispiele von C3-5-Alkylen, das R2 und R3 zusammen bilden, wobei Methylen mit einem O-Atom ersetzt ist, sind Oxybismethylen, Oxymethylenethylen, Oxybisethylen usw. Sie werden durch die folgenden Formeln (7), (8), (9), (10), (11) und (12) wiedergegeben;
-
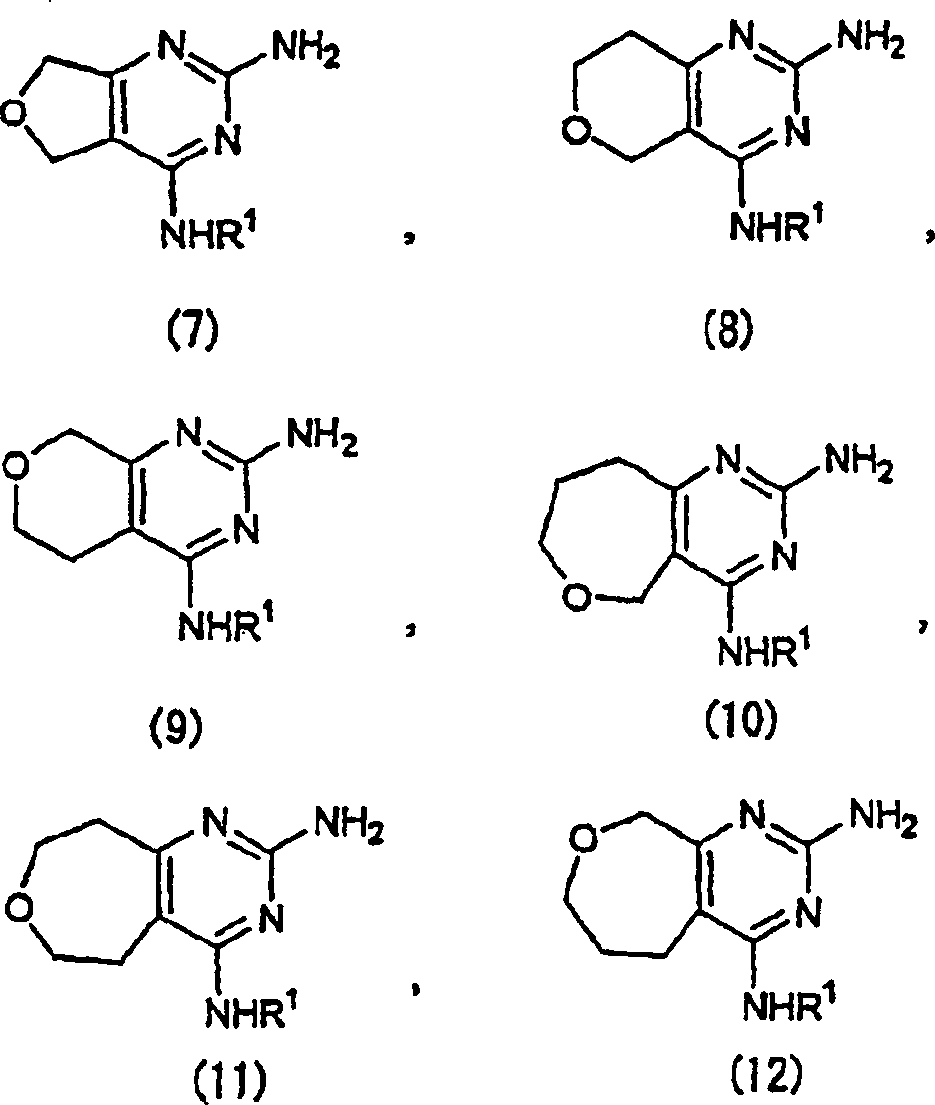
- Die Pyrimidinderivate der vorliegenden Erfindung, die Wirkstoffe als medizinische Arzneistoffe sind, werden zu pharmazeutisch verträglichen Salzen geformt. Als pharmazeutisch verträgliche Salze werden Säureadditionssalze und Basenadditionssalze veranschaulicht. Als Säureadditionssalze werden anorganische Säuresalze, wie Hydrochlorid, Hydrobromid, Sulfat oder Phosphat, oder organische Säuresalze, wie Citrat, Oxalat, Malat, Tartrat, Fumarat oder Maleat, veranschaulicht. Als Basenadditionssalze werden anorganische Basensalze, wie Natriumsalze oder Calciumsalze, oder organische Basensalze, wie Megluminsalz, Tris(hydroxymethyl)aminomethansalz, veranschaulicht. Die erfindungsgemäßen Pyrimidinderivate oder pharmazeutisch verträglichen Salze davon schließen auch Solvate, wie Hydrate usw., ein.
- Die erfindungsgemäßen Verbindungen der Formel (1) können mit folgendem Verfahren oder gemäß folgendem Verfahren hergestellt werden.wobei R1, R2 und R3 die gleiche vorstehend in Formel (1) angegebene Bedeutung haben.
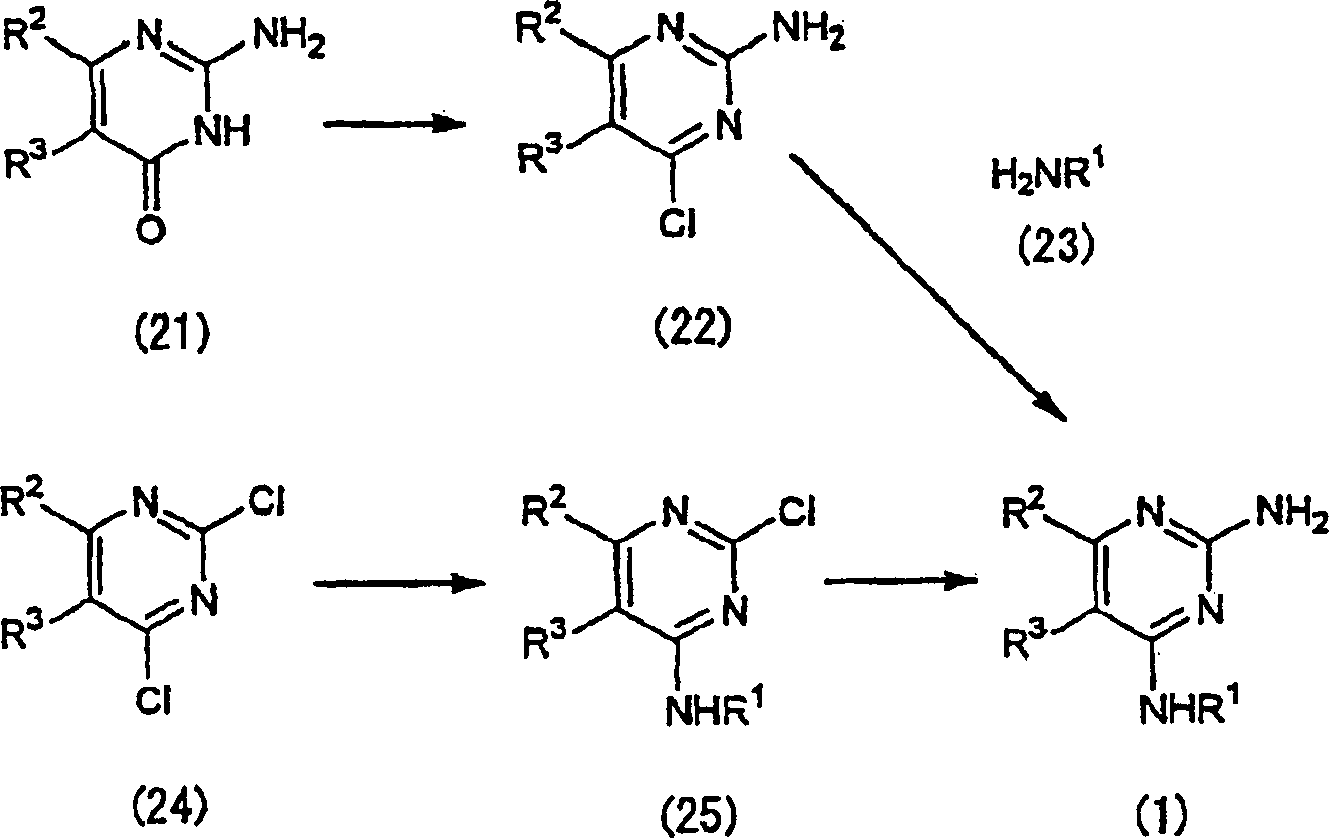
- Verfahren 1
- Die Verbindung (22) wird durch Umsetzung der Verbindung (21) mit Phosphoroxychlorid hergestellt. Die Reaktion kann, falls erforderlich, in Gegenwart eines Lösungsmittels durchgeführt werden. Als Lösungsmittel werden aromatische Kohlenwasserstoffe, wie Toluol oder Xylol, aufgeführt. Die Reaktion kann in Gegenwart eines Reaktionsbeschleunigers, wie N,N-Dimethylaminopyridin, durchgeführt werden. Die Reaktionstemperatur wird zwischen Raumtemperatur und Rückflußtemperatur des Lösungsmittels gewählt.
- Die erfindungsgemäße Verbindung (1) kann durch Umsetzung der Verbindung (22) mit der Verbindung (23) hergestellt werden. Als Lösungsmittel werden aromatische Kohlenwasserstoffe, wie Toluol oder Xylol, Ether, wie Tetrahydrofuran (THF) oder Dioxan, Alkohole, wie Ethanol, 2-Propanol oder Butanol, oder inerte Lösungsmittel, wie Dimethylformamid (DMF) oder Acetonitril, aufgeführt. Die Reaktion wird, falls erforderlich, in Gegenwart einer organischen Base, wie Triethylamin, oder einer anorganischen Base, wie Natriumcarbonat oder Kaliumcarbonat, durchgeführt. Die Reaktionstemperatur wird zwischen zum Beispiel Raumtemperatur und Rückflußtemperatur des Lösungsmittels gewählt.
- Verfahren 2
- Die Verbindung (25) kann durch Umsetzung der Verbindung (24) mit der Verbindung (23) hergestellt werden. Als Lösungsmittel werden aromatische Kohlenwasserstoffe, wie Toluol oder Xylol, Ether, wie Tetrahydrofuran (THF) oder Dioxan, Alkohole, wie Ethanol, 2- Propanol oder Butanol, oder inerte Lösungsmittel, wie Dimethylformamid (DMF) oder Acetonitril, aufgeführt. Die Reaktion kann, falls erforderlich, in Gegenwart einer organischen Base, wie Triethylamin, oder anorganischen Base, wie Natriumcarbonat oder Kaliumcarbonat, durchgeführt werden. Die Reaktionstemperatur wird zwischen zum Beispiel Raumtemperatur und Rückflußtemperatur des Lösungsmittels gewählt.
- Die erfindungsgemäße Verbindung (1) kann durch Umsetzung der Verbindung (25) mit Ammoniak in einem Lösungsmittel hergestellt werden. Als Lösungsmittel werden Alkohole, wie Methanol oder Ethanol, Ether, wie Dioxan oder Ethylenglycoldimethylether, aufgeführt. Die Reaktion wird in einem Autoklaven bei Raumtemperatur bis etwa 200°C durchgeführt.
- Die erfindungsgemäße Verbindung (1) kann auch durch Umsetzung der Verbindung (25) mit Natriumazid, gefolgt von Reduktion mit Triphenylphosphin, hergestellt werden. Die Umsetzung mit Natriumazid wird in einem inerten Lösungsmittel, wie DMF usw., durchgeführt. Die Reaktionstemperatur wird von etwa Raumtemperatur bis etwa dem Siedepunkt des Lösungsmittels gewählt. Die Reduktion durch Triphenylphosphin wird in einem Ether, wie THF usw., durchgeführt. Die Reaktionstemperatur wird von etwa Raumtemperatur bis etwa dem Siedepunkt des Lösungsmittels gewählt.
- Die erfindungsgemäßen Verbindungen der Formel (1) und Zwischenprodukte zu ihrer Herstellung können mit üblichen Verfahren, wie Säulenchromatographie, Umkristallisation usw., gereinigt werden. Als Lösungsmittel zur Umkristallisation werden Alkohole, wie Methanol, Ethanol oder 2-Propanol, Ether, wie Diethylether, Ester, wie Essigsäureethylester, aromatische Kohlenwasserstoffe, wie Toluol, Ketone, wie Aceton, Kohlenwasserstoffe, wie Hexan, oder ein Gemisch davon aufgeführt.
- Bei Durchführen der vorstehenden Umsetzungen können falls erforderlich Verfahren zum Schützen und zur Schutzgruppenabspaltung verwendet werden. Die Verfahren zum Schützen und zur Schutzgruppenabspaltung sind im Einzelnen in „Protecting Groups in Organic Synthesis" von T. W. Greene und P. G. M. Wuts (1991), JOHN WILEY & SONS INC. beschrieben.
- Die erfindungsgemäßen Pyrimidinderivate oder pharmazeutisch verträglichen Salze davon können Solvate, wie Hydrate, bilden, und daher schließt die vorliegende Erfindung auch die Solvate ein.
- Wenn die erfindungsgemäßen Verbindungen ein oder mehrere asymmetrische Kohlenstoffatom(e) aufweisen, existieren optische Isomere, und daher sind ein Gemisch davon und ein isoliertes optisches Isomer in die erfindungsgemäßen Verbindungen eingeschlossen. Zum Reinigen eines solchen optischen Isomers wird eine optische Trennung verwendet.
- Als optische Trennung können die erfindungsgemäßen Verbindungen oder ihre Zwischenprodukte Salze mit einer optisch aktiven Säure (z. B. Monocarbonsäure, wie Mandelsäure, N-Benzyloxyalanin oder Milchsäure, Dicarbonsäure, wie Weinsäure, o-Diisopropylidenweinsäure oder Äpfelsäure, oder Sulfonsäuren, wie Camphersulfonsäure, Bromcamphersulfonsäure) in einem inerten Lösungsmittel (z. B. Alkohole, wie Methanol, Ethanol oder 2-Propanol, Ether, wie Diethylether, Ester, wie Essigsäureethylester, aromatische Kohlenwasserstoffe, wie Toluol, Acetonitril, oder ein Gemisch davon) bilden.
- Wenn die erfindungsgemäßen Verbindungen oder Zwischenprodukte davon einen sauren Substituenten, wie eine Carboxygruppe usw., aufweisen, können sie auch Salze mit einem optisch aktiven Amin (z. B. einem organischen Amin, wie α-Phenethylamin, Chinin, Chinidin, Cinchonidin, Cinchonin, Strychnin usw.) bilden.
- Die Salze bildende Temperatur beträgt Raumtemperatur bis zum Siedepunkt des Lösungsmittels. Um die optische Reinheit der Verbindung zu erhöhen, wird die Temperatur vorzugsweise einmal auf eine Temperatur um den Siedepunkt des Lösungsmittels erhöht. Die Ausbeute kann, falls erforderlich, durch Abkühlen des Lösungsmittels vor Filtrieren eines ausgefällten Salzes erhöht werden. Die Menge einer optisch aktiven Säure oder eines Amins beträgt etwa 0,5–2,0 Äquimole zum Substrat, vorzugsweise etwa 1 Äquimol. Falls erforderlich werden die Kristalle in einem inerten Lösungsmittel (z. B. Alkohole, wie Methanol, Ethanol oder 2-Propanol, Ether, wie Diethylether, Ester, wie Essigsäureethylester, aromatische Kohlenwasserstoffe, wie Toluol, Acetonitril, oder ein Gemisch davon) umkristallisiert, um ein optisch aktives Salz mit in hohem Maß optischer Reinheit zu erhalten.
- Das erhaltene Salz wird falls erforderlich auf herkömmliche Weise mit einer Säure oder einer Base behandelt, wobei eine freie Verbindung erhalten wird.
- Die erfindungsgemäßen Pyrimidinderivate können oral oder parenteral verabreicht werden. Bei oraler Verabreichung wird die Verbindung in der üblichen Verabreichungsform verabreicht. Bei parenteraler Verabreichung kann die Verbindung in topischen Verabreichungsformen, Injektionen, transdermalen Aufbringungsformen oder nasalen Aufbringungsformen verabreicht werden. Präparate für orale oder rektale Verabreichung schließen zum Beispiel Kapseln, Tabletten, Pillen, Pulver, Caches, Suppositorien, Lösungen usw. ein. Injektionen schließen zum Beispiel sterilisierte Lösungen oder Emulsionen usw. ein. Topische Verabreichungspräparate schließen zum Beispiel Cremes, Salben, Lotionen, transdermale Präparate (übliche Pflaster, Matrizen) usw. ein.
- Die vorstehenden Präparate werden mit pharmazeutisch verträglichen Füllstoffen und Zusätzen mit dem üblichen Verfahren hergestellt. Pharmazeutisch verträgliche Füllstoffe und Zusätze schließen Träger, Bindemittel, Geschmacksstoffe, Puffermittel, Mittel zum Erhöhen der Viskosität, färbende Mittel, Stabilisationsmittel, Emulgatoren, Dispergiermittel, Suspendiermittel, Konservierungsstoffe usw. ein.
- Pharmazeutisch verträgliche Träger schließen zum Beispiel Magnesiumcarbonat, Magnesiumstearat, Talkum, Zucker, Lactose, Pectin, Dextrin, Stärke, Gelatine, Traganth, Methylcellulose, Natriumcarboxymethylcellulose, Wachs (geringerer Schmelzpunkt), Kakaobutter usw. ein. Kapseln können durch Einbringen der erfindungsgemäßen Verbindung mit pharmazeutisch verträglichen Trägern hergestellt werden. Die erfindungsgemäße Verbindung wird mit den pharmazeutisch verträglichen Füllstoffen gemischt und das Gemisch in Kapseln gegeben oder die Verbindung ohne Füllstoff in Kapseln gegeben. Caches können mit dem gleichen Verfahren wie die Kapseln hergestellt werden.
- Lösungen zur Injektion schließen zum Beispiel Lösungen, Suspensionen, Emulsionen usw., wie eine wässrige Lösung, Wasser-Propylenglycol-Lösung ein. Die Lösung kann Wasser enthalten und kann in Propylenglycol und/oder Propylenglycollösung hergestellt werden. Die für orale Verabreichung geeigneten Lösungen können durch Zugabe der erfindungsgemäßen Verbindung zu Wasser und falls erforderlich Zugabe eines färbenden Mittels, Geschmacksstoffs, Stabilisationsmittels, Süßstoffs, Mittels zum Löslichmachen, Mittels zum Erhöhen der Viskosität usw. hergestellt werden. Ebenfalls können die für orale Verabreichung geeigneten Lösungen durch Zugabe der erfindungsgemäßen Verbindung und eines Dispersionsmittels zu Wasser zum Bilden viskoser Lösungen hergestellt werden. Die Mittel zum Erhöhen der Viskosität schließen zum Beispiel natürlichen oder synthetischen Gummi, Harz, Methylcellulose, Natriumcarboxymethylcellulose oder bekannte Emulgatoren ein.
- Die Präparate für topische Verabreichung schließen zum Beispiel die vorstehend genannten Lösungen, Cremes, Aerosole, Sprays, Pulver, Lotionen, Salben usw. ein. Die Präparate für topische Verabreichung können durch Mischen der erfindungsgemäßen Verbindung, pharmazeutisch verträglichen Verdünnungsmitteln und herkömmlich verwendeten Trägern hergestellt werden. Cremes und Salben können zum Beispiel durch Mischen von wässrigen oder Ölgrundstoffen und Mitteln zum Erhöhen der Viskosität und/oder Geliermitteln hergestellt werden. Die Grundstoffe schließen zum Beispiel Wasser, flüssiges Paraffin, Pflanzenöl (Erdnußöl, Rhizinusöl) usw. ein. Die Mittel zum Erhöhen der Viskosität schließen zum Beispiel weiches Paraffin, Aluminiumstearat, Cetostearylalkohol, Propylenglycol, Polyethylenglycol, Lanolin, hydriertes Lanolin, Bienenwachs usw. ein. Die Lotionen können durch Mischen von wässrigen oder Ölgrundstoffen und einem oder mehreren pharmazeutisch verträglichen Stabilisationsmitteln, Suspendiermitteln, Emulgatoren, Dispergiermitteln, Mitteln zum Erhöhen der Viskosität, färbenden Mitteln, Geschmacksstoffen usw. hergestellt werden.
- Die Pulver werden mit pharmazeutisch verträglichen Pulvergrundstoffen hergestellt. Die Grundstoffe sind Talkum, Lactose, Stärke usw. Tropfen können mit wässrigen oder nicht wässrigen Grundstoffen und einem oder mehreren pharmazeutisch verträglichen Dispergiermitteln, Suspendiermitteln, Mitteln zum Löslichmachen usw. hergestellt werden.
- Die Präparate für topische Verabreichung können, falls erforderlich, Konservierungsmittel, wie Hydroxybenzoesäuremethylester, Hydroxybenzoesäurepropylester, Chlorcresol, Benzalkoniumchlorid, und antibakterielle Mittel enthalten.
- Flüssigkeiten für Spray, Pulver oder Tropfen, die die erfindungsgemäße Verbindung enthalten, können nasal verabreicht werden.
- Die Dosierung und Zahl der Verabreichungen variieren mit einer zu behandelnden Krankheit, dem Alter, Körpergewicht, dem Weg der Verabreichung. Bei oraler Verabreichung wird ein Wirkstoff einem Erwachsenen im Allgemeinen mit etwa 1–500 mg pro Tag, vorzugsweise etwa 5–100 mg, ein- oder mehrmals verabreicht. Bei Injektionen wird ein Wirkstoff im Allgemeinen mit etwa 0,1–300 mg pro Tag, vorzugsweise etwa 1–100 mg, ein- oder mehrmals verabreicht.
- Beispiele
- Die vorliegende Erfindung wird im Einzelnen durch die Beispiele, Bezugsbeispiele und Tests erklärt, aber die vorliegende Erfindung sollte nicht auf sie beschränkt sein.
- Beispiel 1 Ethyl-2-[(2-amino-5,6,7,8-tetrahydrochinazolin-4-yl)amino]acetat
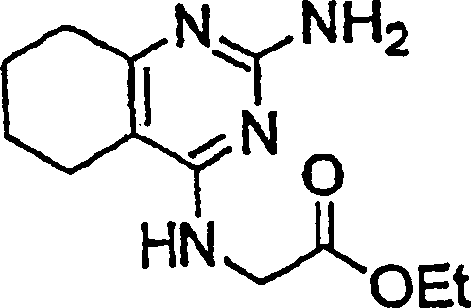
- Zu einem Gemisch von 4-Chlor-5,6,7,8-tetrahydrochinazolin-2-yl-amin (100 mg, 0,545 mmol), Triethylamin (221 mg, 2,18 mmol) und Butanol (3 ml) wurde Glycinethylester-Hydrochlorid (152 mg, 1,10 mmol) bei Raumtemperatur gegeben. Nachdem das Gemisch 4 Stunden bei 90°C gerührt worden war, wurde das Reaktionsgemisch in Wasser gegossen und mit Chloroform extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet, filtriert und das Lösungsmittel im Filtrat unter Vakuum entfernt. Der Rückstand wurde durch Kieselgelchromatographie (3% MeOH/CHCl3) gereinigt, wobei die gewünschte Verbindung (98,3 mg, 72,1%) erhalten wurde.
1H-NMR (CDCl3): δ 1,30 (3H, t, J = 7,0 Hz), 1.78 (4H, m), 2,30 (2H, m), 2,55 (2H, m), 4,20 (2H, m), 4,24 (2H, q, J = 7.0 Hz), 4,76 (2H, bs), 5,13 (1H, bs). - Beispiel 2 N-(2-Amino-5,6,7,8-tetrahydrochinazolin-4-yl)-N-(cyclohexylmethyl)amin

- Ein Gemisch von 4-Chlor-5,6,7,8-tetrahydrochinazolin-2-yl-amin (107 mg, 0,58 mmol), Triethylamin (221 mg, 2,18 mmol), Cyclohexylmethylamin (132 mg, 1,17 mmol) und n-Butanol (3 ml) wurde 4 Stunden bei 80–90°C umgesetzt. Gemäß der Nachbehandlung von Beispiel 1 wurde die gewünschte Verbindung erhalten (102 mg, 67,9%).
1H-NMR (CDCl3): δ 0,97 (2H, m), 1,22 (3H, m), 1,56 (1H, m), 1,76 (9H, m), 2,21 (2H, m), 2,55 (2H, m), 3,28 (2H, t, J = 6,8 Hz), 4,71 (1H, bt), 5,03 (2H, bs). - Beispiel 3 Ethyl-2-[(2-amino-5,6,7,8-tetrahydrochinazolin-4-yl)amino]-4-methylpentanoat
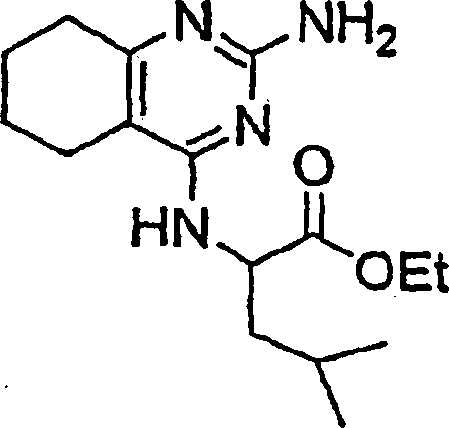
- Ein Gemisch von 4-Chlor-5,6,7,8-tetrahydrochinazolin-2-yl-amin (117 mg, 0,64 mmol), Triethylamin (259 mg, 2,56 mmol), d1-Leucinethylester-Hydrochlorid (250 mg, 1,28 mmol) und n-Butanol (2 ml) wurde 6 Stunden bei 80–90°C umgesetzt. Gemäß der Nachbehandlung von Beispiel 1 wurde die gewünschte Verbindung erhalten (104,3 mg, 72,1%).
1H-NMR (CDCl3): δ 0,92 (6H, m), 1,30 (3H, t, J = 7,1 Hz), 1,60–1,70 (3H, m), 1,79 (4H, m), 2,29 (2H, m), 2,54 (2H, m), 4,18 (2H, q, J = 7,1 Hz), 4,80 (1H, m), 4,88 (2H, bs), 4,90 (1H, bs). - Beispiel 4 N-(2-Amino-5,6,7,8-tetrahydrochinazolin-4-yl)-N-(2-ethoxyethyl)amin

- Zu einem Gemisch von 4-Chlor-5,6,7,8-tetrahydrochinazolin-2-yl-amin (100 mg, 0,545 mmol), Triethylamin (221 mg, 2,18 mmol) und Dimethylformamid (2 ml) wurde Ethoxyethylamin (98 mg, 1,10 mmol) bei Raumtemperatur gegeben. Nachdem das Gemisch 2,5 Stunden bei 90°C gerührt worden war, wurde das Reaktionsgemisch in Wasser gegossen und mit Chloroform extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet, filtriert und das Lösungsmittel im Filtrat unter Vakuum entfernt. Der Rückstand wurde durch präparative DC (10% MeOH/CHCl3) gereinigt, wobei die gewünschte Verbindung (41,7 mg, 32,4%) erhalten wurde.
1H-NMR (CDCl3): δ 1,22 (3H, t, J = 6,8 Hz), 1,80 (4H, m), 2,23 (2H, m), 2,59 (2H, m), 3,53 (2H, q, J = 6,8 Hz), 3,62 (4H, m), 5,17 (4H, bt), 5,30 (2H, bs). - Beispiel 5 N-(2-Amino-5,6,7,8-tetrahydrochinazolin-4-yl)-N-butylamin
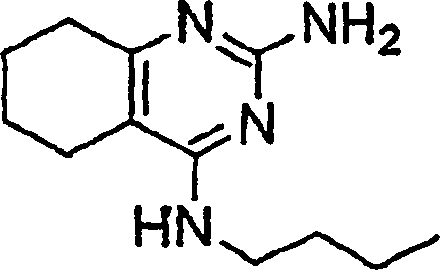
- Ein Gemisch von 4-Chlor-5,6,7,8-tetrahydrochinazolin-2-yl-amin (100 mg, 0,545 mmol) und Butylamin (2 ml) wurde 4 Stunden bei 90°C gerührt. Das Reaktionsgemisch wurde in Wasser gegossen und mit Chloroform extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet, filtriert und das Lösungsmittel im Filtrat unter Vakuum entfernt. Der Rückstand wurde durch Säulenchromatographie (10% MeOH/CHCl3) gereinigt, wobei die gewünschte Verbindung erhalten wurde (94,5 mg, 78,9%).
1H-NMR (CDCl3): δ 0,93 (3H, t, J = 7,0 Hz), 1,36 (2H, m), 1,63 (2H, m), 1,78 (4H, m), 2,31 (2H, m), 2,58 (2H, m), 3,47 (2H, q, J = 7,0 Hz), 6,00 (1H, bs), 6,03 (1H, t ähnlich), 7,34 (1H, bs). - Beispiel 6 N-(2-Amino-5,6,7,8-tetrahydrochinazolin-4-yl)-N-hexylamin
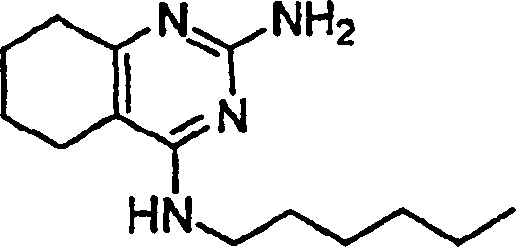
- Gemäß dem Verfahren von Beispiel 5 wurde die vorstehende Verbindung erhalten.
1H-NMR (CDCl3): δ 0,89 (3H, m), 1,32 (6H, m), 1,59 (2H, m), 1,81 (4H, m), 2,21 (2H, m), 2,62 (2H, m), 3,44 (2H, q, J = 7,0 Hz), 4,99 (1H, bs), 5,73 (2H, brs). - Beispiel 7 Ethyl-2-[(2-amino-5,6,7,8-tetrahydrochinazolin-4-yl)amino]propanoat
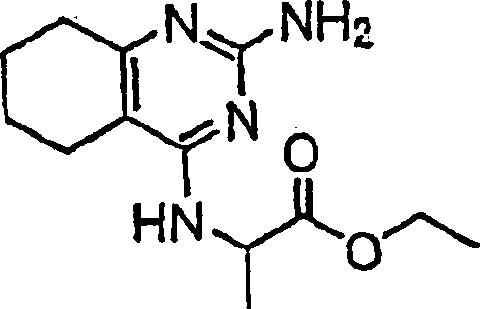
- Zu einem Gemisch von 4-Chlor-5,6,7,8-tetrahydrochinazolin-2-yl-amin (100 mg, 0,545 mmol), Triethylamin (221 mg, 2,18 mmol) und Dimethylformamid (4 ml) wurde 2-Aminopropionsäureethylester-Hydrochlorid (167 mg, 1,09 mmol) bei Raumtemperatur gegeben. Nachdem das Gemisch 2,5 Stunden bei 100°C gerührt worden war, wurde das Reaktionsgemisch in Wasser gegossen und mit Chloroform extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet, filtriert und das Lösungsmittel im Filtrat unter Vakuum entfernt. Der Rückstand wurde durch Kieselgelchromatographie (5% MeOH/CHCl3) gereinigt, wobei die gewünschte Verbindung (42,1 mg, 29,3%) erhalten wurde.
1H-NMR (300 MHz, CDCl3): δ 5,07 (brd, 1H, J = 6,8 Hz), 4,79–4,69 (3H, m), 4,21 (q, 2H, J = 7,1 Hz), 2,56–2,53 (2H, m), 2,32–2,25 (2H, m), 1,85–1,73 (4H, m), 1,47 (d, 3H, J = 7,1 Hz), 1,29 (t, 3H, J = 7,1 Hz). - Beispiel 8 Ethyl-2-[(2-amino-5,6,7,8-tetrahydrochinazolin-4-yl)amino]-3-hydroxypropanoat
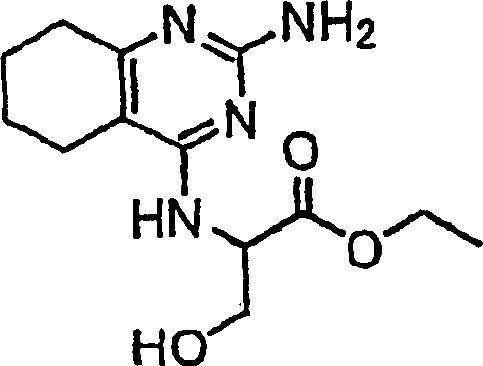
- Gemäß dem Verfahren von Beispiel 7 wurde die vorstehende Verbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ 5,63 (d, 1H, J = 6,2 Hz), 4,84–4,76 (3H, m), 4,26–4,20 (2H, m), 4,08 (dd, 1H, J = 11,0, 3,1 Hz), 3,94 (dd, 1H, J = 11,0, 1,9 Hz), 2,55–2,47 (2H, m), 2,32–2,25 (2H, m), 1,80–1,70 (4H, m), 1,31 (t, 3H, J = 7,1 Hz). - Beispiel 9 Methyl-2-[(2-amino-5,6,7,8-tetrahydrochinazolin-4-yl)amino]hexanoat

- Gemäß dem Verfahren von Beispiel 7 wurde die vorstehende Verbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ 4,94 (d, 1H, J = 7,7 Hz), 4,85–4,75 (1H, m), 4,74 (1H, brs), 3,74 (3H, s), 2,57–2,50 (2H, m), 2,30–2,50 (2H, m), 1,95–1,65 (6H, m), 1,40–1,25 (4H, m), 0,92–0,87 (3H, m). - Beispiel 10 2-[(2-Amino-5,6,7,8-tetrahydrochinazolin-4-yl)amino]hexan-1-ol

- Methyl-2-[(2-amino-5,6,7,8-tetrahydrochinazolin-2-yl)amino]hexanoat (122 mg, 0,417 mmol) wurde in THF (3 ml) gelöst. Zur Lösung wurde Lithiumaluminiumhydrid (15 mg, 0,417 mmol) bei 0°C gegeben und auf Raumtemperatur erwärmt. Das Reaktionsgemisch wurde, abgekühlt und THF (10 ml) zugetropft, gefolgt von Zutropfen von Wasser (1 ml). Dann wurde 1 M wässrige NaOH-Lösung zugegeben, bis sich ein Feststoff entwickelte. MgSO4 wurde zum Reaktionsgemisch gegeben und das Gemisch filtriert. Zum Filtrat wurden eine gesättigte wässrige Natriumhydrogencarbonatlösung und Chloroform gegeben und es extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet, filtriert und das Lösungsmittel im Filtrat unter Vakuum entfernt. Der Rückstand wurde durch präparative DC (15% MeOH/CHCl3) gereinigt, wobei die gewünschte Verbindung (27 mg, 24,5%) erhalten wurde.
1H-NMR (300 MHz, CDCl3): δ 5,46 (brs, 1H), 4,97 (d, 1H, J = 7,1 Hz), 4,50 (2H, brs), 4,20–4,10 (1H, m), 3,76 (dd, 1H, J = 11,0, 3,1 Hz), 3,62 (dd, 1H, J = 11,0, 6,6 Hz), 2,60–2,50 (2H, m), 2,35–2,15 (2H, m), 1,85–1,70 (4H, m), 1,70–1,45 (2H, m), 0,93–0,88 (3H, m). - Beispiel 11 1-[(2-Amino-5,6,7,8-tetrahydrochinazolin-4-yl)amino]pentan-2-ol

- Ein Gemisch von 4-Chlor-5,6,7,8-tetrahydrochinazolin-2-yl-amin (184 mg, 1 mmol), 2-Hydroxypentylamin-Hydrochlorid (140 mg, 1 mmol), Triethylamin (202 mg, 2 mmol) und DMF (1 ml) wurde 5 Stunden in einem Bad (Badtemperatur 90°C) erwärmt. Das Lösungsmittel im Filtrat wurde unter Vakuum entfernt und der Rückstand durch Kieselgelsäulenchromatographie (CHCl3 : MeOH : NH4OH wässr. = 100 : 10 : 0,4) gereinigt, wobei das Rohprodukt (210 mg) erhalten wurde. Zum Rohprodukt wurden wässrige Ammoniaklösung (5 ml) und Chloroform (30 ml) gegeben und es wurde extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung (20 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und das Lösungsmittel im Filtrat unter Vakuum entfernt, wobei die gewünschte Verbindung (128 mg, 51%) erhalten wurde.
1H-NMR (300 MHz, CDCl3): δ 4,93 (1H, brm), 4,62 (2H, brs), 3,75–3,85 (1H, m), 3,55–3,65 (1H, m), 3,33–3,44 (1H, m), 2,50–2,54 (2H, m), 2,20–2,22 (2H, m), 1,77–1,79 (4H, m), 1,38–1,54 (4H, m), 0,95 (3H, t, J = 7,3 Hz). - Beispiel 12 1-[(2-Amino-5,6,7,8-tetrahydrochinazolin-4-yl)amino]pentan-2-on
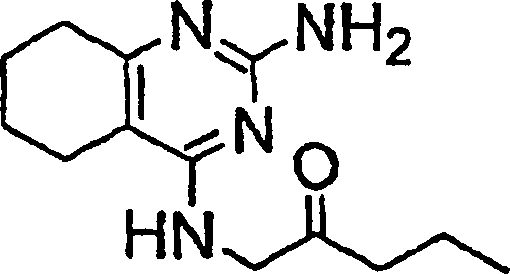
- Zu einer Lösung von 1-[2-Amino-5,6,7,8-tetrahydrochinazolin-4-yl)amino]pentan-2-ol (120 mg, 0,479 mmol) in Dichlormethan (20 ml) wurde Pyridiniumchlorchromat (517 mg, 23,97 mmol) gegeben und das Gemisch 3,5 Stunden gerührt. Kieselgel (10 g) wurde zum Reaktionsgemisch gegeben und es wurde filtriert. Das Kieselgel wurde mit 5% MeOH/CHCl3 gewaschen. Die Filtrate wurden gesammelt und das Lösungsmittel unter Vakuum entfernt. Der Rückstand wurde durch Kieselgelchromatographie (CHCl3 : MeOH : NH4OH wässr. = 100 : 5 : 0,4) gereinigt, wobei die gewünschte Verbindung (32 mg, 26%) erhalten wurde.
1H-NMR (300 MHz, CDCl3): δ 5,36 (1H, brs), 4,62 (2H, brs), 4,28 (2E, d, J = 4,0 Hz), 2,46–2,57 (4H, m), 2,30–2,32 (2H, m), 2,02 (1H, brm), 1,65–1,81 (6H, m), 0,96 (3H, t, J = 7,3 Hz). - Beispiel 13 N-(2-Amino-5,6,7,8-tetrahydrochinazolin-4-yl)-N-(tetrahydrofuran-2-yl-methyl)amin

- Ein Gemisch von 4-Chlor-5,6,7,8-tetrahydrochinazolin-2-yl-amin (184 mg, 1 mmol), Tetrahydrofurfurylamin (101 mg, 1 mmol) und Diethylenglycoldiethylether (1 ml) wurde 2 Stunden auf 100–110°C erwärmt gehalten. Das Reaktionsgemisch wurde mit Essigsäureethylester (50 ml) und gesättigtem wässrigem Natriumhydrogencarbonat (20 ml) extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet, filtriert und das Lösungsmittel im Filtrat unter Vakuum entfernt. Der Rückstand wurde durch Säulenchromatographie (CHCl3 : MeOH : NH4OH wässr. = 100 : 10 : 0,4) gereinigt, wobei die gewünschte Verbindung (80 mg, 32,3%) erhalten wurde.
1H-NMR (300 MHz, CDCl3): δ 4,98 (1H, brs), 4,87 (1H, brs), 4,01–4,11 (1H, m), 3,71–3,92 (3H, m), 3,29–3,38 (1H, m), 3,14 (1H, brm), 2,54–2,58 (2H, m), 2,22–2,24 (2H, m), 1,77–2,07 (8H, m). - Beispiel 14 N-(2-Amino-5-butyl-6-methylpyrimidin-4-yl)-N-pentylamin

- Ein Gemisch von 5-Butyl-4-chlor-6-methylpyrimidin-2-yl-amin (100 mg, 0,5 mmol) und Amylamin (2 ml) wurde 11 Stunden unter Rückfluß erhitzt. Das Reaktionsgemisch wurde abgekühlt und das Lösungsmittel unter Vakuum entfernt und der Rückstand durch Kieselgelsäulenchromatographie (MeOH : CHCl3 = 1 : 20) gereinigt, wobei die gewünschte Verbindung (98 mg, 78%) in Form eines Öls erhalten wurde.
1H-NMR (CDCl3): δ 0,93 (6H, m), 1,37 (8H, brm), 1,60 (2H, m), 2,30 (3H, s), 2,32 (2H, m), 3,44 (2H, q-ähnlich), 4,96 (1H, br), 5,59 (2H, br). - Beispiel 15 N-(2-Amino-5-hexyl-6-methylpyrimidin-4-yl)-N-pentylamin
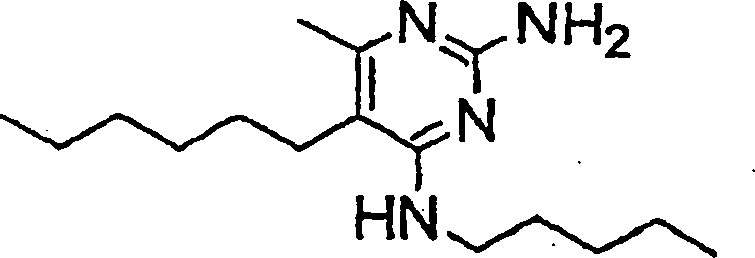
- Ein Gemisch von 4-Chlor-5-hexyl-6-methylpyrimidin-2-yl-amin (1,00 mg, 0,44 mmol) und Amylamin (2 ml) wurde 11 Stunden unter Rückfluß erhitzt. Das Reaktionsgemisch wurde abgekühlt und das Lösungsmittel unter Vakuum entfernt. Der Rückstand wurde durch Kieselgelsäulenchromatographie (MeOH : CHCl3 = 1 : 20) gereinigt, wobei die gewünschte Verbindung (107 mg, 87%) in Form eines Öls erhalten wurde.
1H-NMR (TMS/CDCl3): δ 0,91 (6H, m), 1,36 (12H, brm), 1,60 (2H, m), 2,29 (3H, s), 2,31 (2H, m), 3,43 (2H, q-ähnlich), 4,90 (1H, br), 5,50 (2H, br). - Beispiel 16 N-(2-Amino-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-yl)-N-pentylamin

- Ein Gemisch von 4-Chlor-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-2-yl-amin (29,3 mg, 0,158 mmol) und Amylamin (1,0 ml) wurde 2,5 Stunden unter Rückfluß erhitzt. Nach der Umsetzung wurde das Verfahren gemäß der Vorbehandlung von Beispiel 7 durchgeführt, wobei die gewünschte Verbindung (22,3 mg, 59%) erhalten wurde.
1H-NMR (300 MHz, CDCl3): δ 4,86 (2H, brs), 4,40 (2H, d, J = 1,1 Hz), 4,09 (1H, brs), 3,94 (2H, t, J = 5,6 Hz), 3,41 (2H, dt, J = 7,1, 5,4 Hz), 2,64 (2H, t, J = 5,6 Hz), 1,64–1,50 (2H, m), 1,42–1,25 (4H, m), 0,96–0,86 (3E, m). - Beispiel 17 N-(2-Amino-6-butyl-5-methylpyrimidin-4-yl)-N-pentylamin

- Ein Gemisch von 4-Butyl-6-chlor-5-methylpyrimidin-2-yl-amin (93,5 mg, 0,47 mmol) und Amylamin (1,5 ml) wurde 8 Stunden unter Rückfluß erhitzt. Nach der Umsetzung wurde das Verfahren gemäß der Vorbehandlung von Beispiel 7 durchgeführt, wobei die gewünschte Verbindung (50 mg, 42,7%) erhalten wurde.
1H-NMR (CDCl3): δ 0,93 (6H, tx 2), 1,37 (6H, m), 1,57 (4H, m), 1,91 (3H, s), 2,51 (2H, t, J = 7,6 Hz), 3,40 (2H, q, J = 7,3 Hz), 4,61 (1H, bs), 4,98 (2H, bs). - Beispiel 18 N-(2-Amino-5,6-dimethylpyrimidin-4-yl)-N-pentylamin

- Ein Gemisch von N-(2-Chlor-5,6-dimethylpyrimidin-4-yl)-N-pentylamin (131 mg, 0,575 mmol) und 5 M Ammoniak-Ethanol (40 ml) wurde 10 Stunden auf 170°C gehalten. Das Reaktionsgemisch wurde unter Vakuum konzentriert, durch präparative DC (20% MeOH/CHCl3) gereinigt, wobei die gewünschte Verbindung (4,2 mg, 3,5%) erhalten wurde.
1H-NMR (300 MHz, CDCl3): δ 5,17 (2H, brs), 4,56 (1H, brs), 3,82–3,44 (2H, m), 2,25 (3H, s), 1,90 (3H, s), 1,65–1,55 (2H, m), 1,37–1,32 (4H, m), 0,94–0,89 (3H, m). - Beispiel 19 N-(2-Amino-5,6,7,8-tetrahydrochinazolin-4-yl)-N-pentylamin
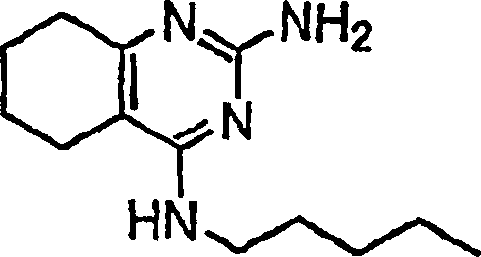
- Unter Verwendung von N-(2-Chlor-5,6,7,8-tetrahydrochinazolin-4-yl)-N-pentylamin als Ausgangssubstanz und gemäß dem Verfahren von Beispiel 18 wurde die gewünschte Verbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ 5,11 (2H, brs), 4,52 (1H, brs), 3,86–3,52 (2H, m), 2,57–2,54 (2H, m), 2,21–2,18 (2H, m), 1,83–1,75 (4H, m), 1,64–1,74 (2H, m), 1,40–1,30 (4H, m), 0,94–0.89 (3H, m). - Beispiel 20 N-(2-Amino-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)-N-pentylamin

- Unter Verwendung von 2-Chlor-N-pentyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amin als Ausgangssubstanz und gemäß dem Verfahren von Beispiel 18 wurde die gewünschte Verbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ 4,89 (2H, brs), 4,31 (1H, brs), 3,46–3,88 (2H, m), 2,75 (2H, t, J = 7,7 Hz), 2,55 (2H, t, J = 7,7 Hz), 2,07 (2H, tt, J = 7,7, 7,7 Hz), 1,64–1,54 (2H, m), 1,37–1,32 (4H, m), 0,94–0,89 (3H, m). - Beispiel 21 2-[(2-Amino-5,6,7,8-tetrahydrochinazolin-4-yl)amino]hexanamid
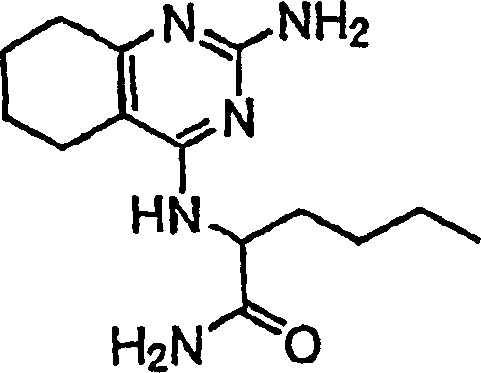
- Ein Gemisch von Methyl-2-[(2-amino-5,6,7,8-tetrahydrochinazolin-4-yl)amino]hexanoat (520 mg, 1,77 mmol) und 5 M Ammoniak-Ethanol (60 ml) wurde 24 Stunden auf 120°C gehalten. Das Reaktionsgemisch wurde unter Vakuum konzentriert, durch Kieselgelchromatographie (20% MeOH/CHCl3) gereinigt, wobei die gewünschte Verbindung (67,7 mg, 7,6%) erhalten wurde.
1H-NMR (300 MHz, DMSO-d6): δ 7,26 (1H, brs), 7,01 (1H, brs), 5,80 (1H, d, J = 7,9 Hz), 5,58 (2H, brs), 4,46 (1H, dt, J = 8,1, 7,9 Hz), 2,43–2,21 (4H, m), 1,85–1,56 (6H, m), 1,34–1,13 (4H, m), 0,92–0,77 (3H, m). - Beispiel 22 N-(2-Amino-5-(2-methoxyethyl)-6-methylpyrimidin-4-yl)-N-pentylamin

- Ein Gemisch von 4-Chlor-5-(2-methoxyethyl)-6-methylpyrimidin-2-yl-amin (150 mg, 0,74 mmol), Amylamin (0,86 ml) und Dioxan (1,5 ml) wurde 7 Stunden auf 90°C gehalten. Das Reaktionsgemisch wurde im Vakuum konzentriert und der Rückstand mit Chloroform und einer gesättigten wässrigen NaHCO3-Lösung) extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet, filtriert und das Filtrat unter Vakuum konzentriert. Der Rückstand wurde durch Säulenchromatographie (4% MeOH : CHCl3) gereinigt, wobei die gewünschte Verbindung (108 mg, 57,5%) erhalten wurde.
1H-NMR (CDCl3): δ 0,92 (3H, t, J = 6,6), 1,40–1,32 (4H, m), 1,57 (2H, m), 2,21 (3H, s), 2,62 (2H, t, J = 5,9), 3,31–3,38 (5H, m), 3,50 (2H, t, J = 5,9), 4,72 (2H, brs), 5,62 (1H, m). - Beispiel 23 3-[2-Amino-4-methyl-6-(pentylamino)pyrimidin-5-yl]propannitril

- Ein Gemisch von 3-(2-Amino-4-chlor-6-methylpyrimidin-5-yl)propannitril (500 mg, 2,54 mmol), Amylamin (2,94 ml) und Dioxan (5 ml) wurde 8,5 Stunden auf 90°C gehalten. Das Verfahren gemäß der Vorbehandlung von Beispiel 23 wurde durchgeführt, wobei die gewünschte Verbindung (346 mg, 55,0%) erhalten wurde.
1H-NMR (CDCl3): δ 0.92 (3H, t, J = 6,9), 1,35 (4H, m), 1,60 (2H, m), 2,25 (3H, s), 2,45 (2H, t, J = 7,9), 2,75 (2H, t, J = 7,9), 3,40 (2H, m), 4,45 (1H, m), 4,65 (2H, brs). - Beispiel 24 N-(2-Amino-5-ethyl-6-methylpyrimidin-4-yl)-N-pentylamin

- Ein Gemisch von 4-Chlor-5-ethyl-6-methylpyrimidin-2-yl-amin (400 mg, 33 mmol), Amylamin (1,35 ml) und Dioxan (5 ml) wurde 17 Stunden auf 95–100°C gehalten. Das Verfahren gemäß der Vorbehandlung von Beispiel 23 wurde durchgeführt, wobei die gewünschte Verbindung (301 mg, 58,1%) erhalten wurde.
1H-NMR (CDCl3): δ 0,91 (3H, t, J = 6,9), 1,06 (3H, t, J = 7,6), 1,23–1,43 (4H, m), 1,59 (2H, m), 2,22 (3H, s), 2,35 (2H, q, J = 7,6), 3,40 (2H, m), 4,50 (1H, m), 4,61 (2H, brs). - Beispiel 25
- Gemäß dem Verfahren von Beispiel 23 wurde die folgende Verbindung erhalten: 1-[(2-Amino-5-butyl-6-methylpyrimidin-4-yl)amino]pentan-2-ol1H-NMR (CDCl3): δ 0,94 (6H, t), 1,45 (8H, m), 2,34 (3H, s), 2,37 (2H, m), 3,31 (1H, m), 3,48 (1H, s), 3,76 (2H, m), 6,10 (1H, brs), 6,32 (2H, brs).
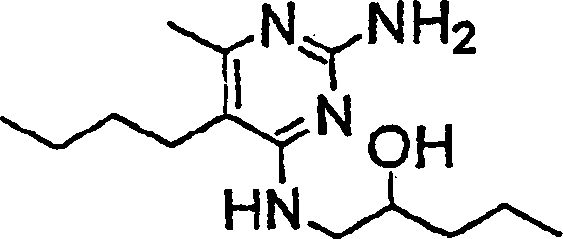
- Beispiel 26 N-(2-Amino-5-benzyl-6-methylpyrimidin-4-yl)-N-pentylamin
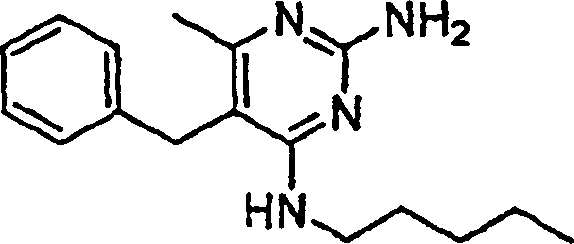
- Ein Gemisch von 5-Benzyl-4-chlor-6-methylpyrimidin-2-yl-amin (500 mg, 2,14 mmol), Amylamin (1,24 ml) und Dioxan (4 ml) wurde 19 Stunden auf 95–100°C gehalten. Das Reaktionsgemisch wurde unter Vakuum konzentriert und der Rückstand mit Chloroform und einer gesättigten wässrigen NaHCO3-Lösung extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet und unter Vakuum konzentriert. Der Rückstand wurde durch Kieselgelsäulenchromatographie (2% MeOH/CHCl3) gereinigt, wobei die gewünschte Verbindung (546 mg, 89,7%) erhalten wurde.
1H-NMR CDCl3): δ 0,81 (3H, t, J = 7,3), 1,05 (2H, m), 1,19 (2H, m), 1,35 (2H, m), 2,28 (3H, s), 3,27 (2H, m), 3,76 (2H, s), 4,30 (1H, m), 4,64 (2H, brs), 7,12–7,31 (5H, m), 0,94–0,89 (3H, m). - Beispiel 27 N-(2-Amino-5-benzylpyrimidin-4-yl)-N-pentylamin
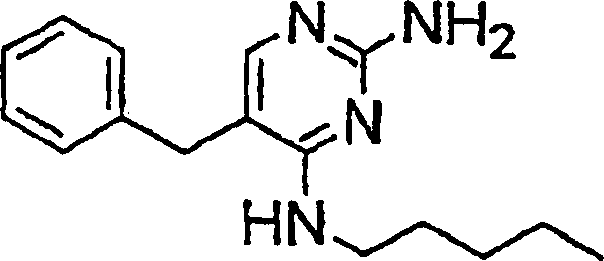
- Ein Gemisch von 5-Benzyl-4-chlorpyrimidin-2-yl-amin (350 mg, 0,74 mmol), Amylamin (0,74 ml) und Dioxan (4 ml) wurde 8 Stunden auf 90–100°C gehalten. Das Reaktionsgemisch wurde unter Vakuum konzentriert und der Rückstand mit Ether und einer gesättigten wässrigen NaHCO3-Lösung extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet, filtriert und konzentriert. Der Rückstand wurde durch Kieselgelsäulenchromatographie (MeOH : CHCl3 = 70 : 1) gereinigt, wobei die gewünschte Verbindung (355 mg, 82,2%) erhalten wurde.
1H-NMR (CDCl3): δ 0.82 (3H, t, J = 6,9), 1,04 (2H, m), 1,21 (2H, m), 1,35 (2H, m), 3,26 (2H, m), 3,66 (2H, s), 4,26 (1H, m), 4,64 (2H, brs), 7,16–7,33 (5H, m), 7,68 (1H, s). - Beispiel 28 N-(2-Amino-5-phenethylpyrimidin-4-yl)-N-pentylamin
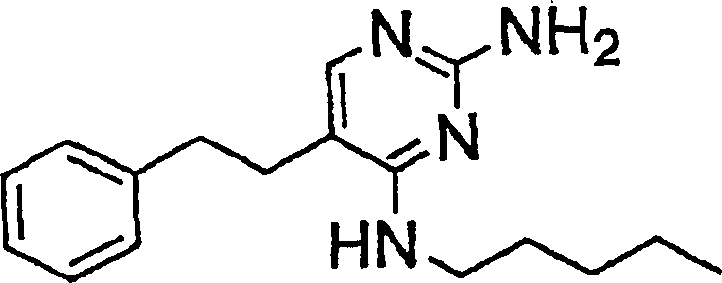
- Ein Gemisch von 4-Chlor-5-phenethylpyrimidin-2-yl-amin (234 mg, 1 mmol), Amylamin (0,58 ml) und Dioxan (2 ml) wurde 8,5 Stunden auf 95–100°C gehalten. Das Verfahren gemäß der Vorbehandlung von Beispiel 27 wurde durchgeführt, wobei die gewünschte Verbindung (227 mg, 79,7%) erhalten wurde.
1H-NMR (CDCl3): δ 0,91 (3H, t, J = 6,9), 1,25–1,42 (4H, m), 1,50 (2H, m), 2,55 (2H, t, J = 7,3), 2,84 (2H, t, J = 7,3), 3,31 (2H, m), 4,27 (1H, m), 4,60 (2H, brs), 7,15–7,33 (5H, m), 7,56 (1H, s). - Beispiel 29 N-(2-Amino-5-benzyl-6-methylpyrimidin-4-yl)-N-pentan-2-ol
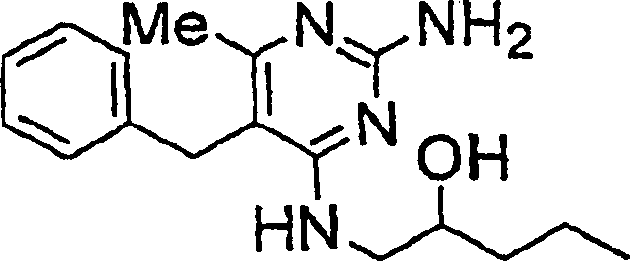
- Ein Gemisch von 5-Benzyl-4-chlor-6-methylpyrimidin-2-yl-amin (1,5 g, 6,42 mmol), 2-Hydroxypentylamin-Hydrochlorid (990 mg, 7,06 mmol), Triethylamin (1,4 g, 14,18 mmol) und Diethylenglycoldiethylether (5 ml) wurde 15 Stunden in einem Bad (Badtemperatur: 90–100°C) warmgehalten. Das Lösungsmittel wurde im Vakuum entfernt und der Rückstand durch Kieselgelsäulenchromatographie (CHCl3 : MeOH : NH4OH wässr. = 100 : 10 : 0,4) gereinigt, wobei die gewünschte Verbindung (800 mg, 41,5%) erhalten wurde.
1H-NMR (TMS/CDCl3): δ 0,86 (3H, t, J = 6,9 Hz), 1,18–1,40 (4H, m), 2,27 (3H, s), 3,17–3,27 (1H, m), 3,60–3,71 (1H, m), 3,78 (2H, d, J = 6,6 Hz), 4,76 (3H, br), 7,23 (2H, d, J = 6,9 Hz), 7,28–7,33 (3H, m). - Beispiel 30
- Die Verbindungen in der folgenden Tabelle können gemäß den Verfahren der vorstehenden Beispiele hergestellt werden.
-

-

-
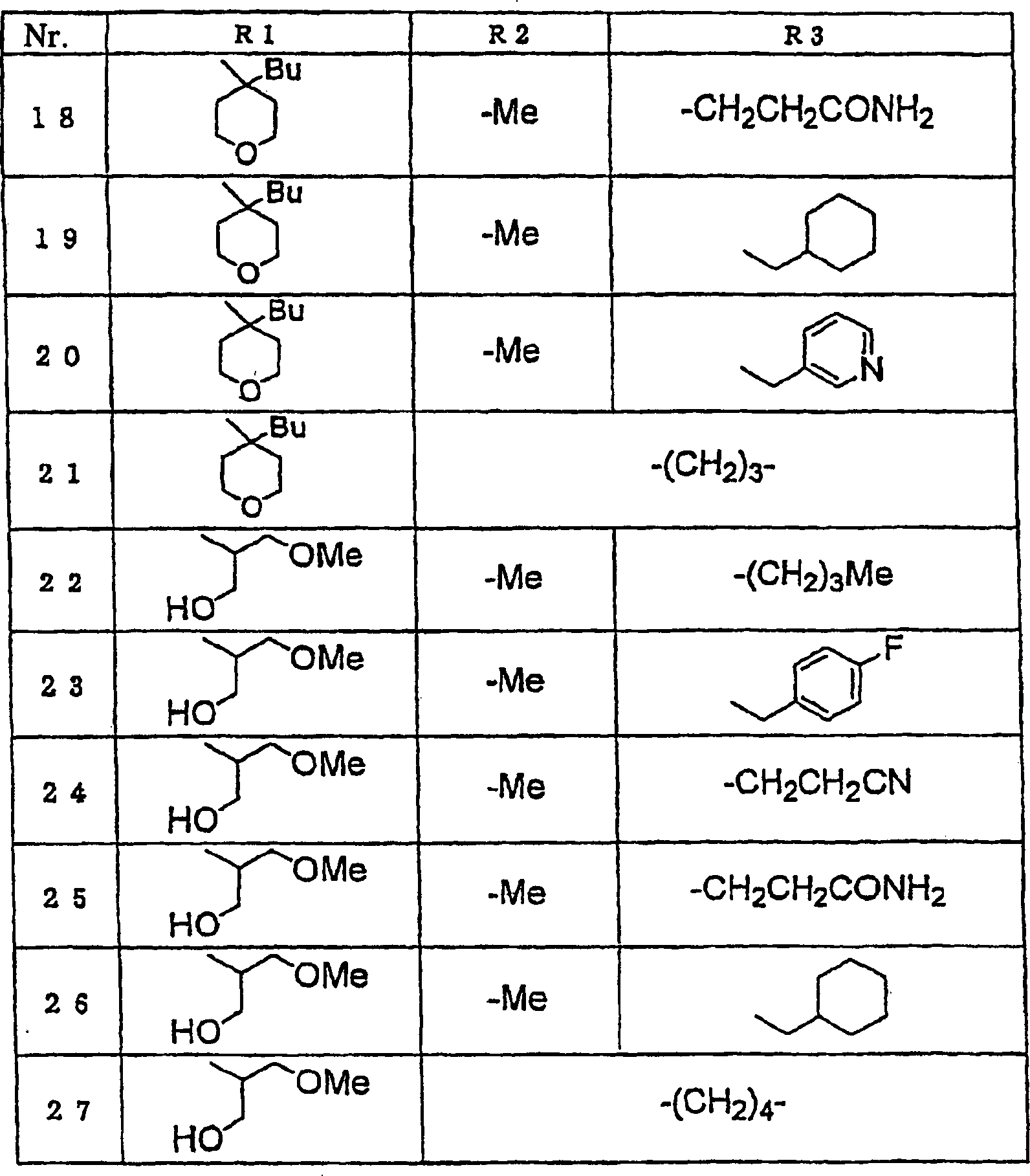
- Bezugsbeispiel 1 4-Chlor-5,6,7,8-tetrahydrochinazolin-2-ylamin (1-1) 2-Amino-5,6,7,8-tetrahydrochinazolin-4-ol

- Zu einer Lösung von Ethyl-2-oxocyclohexancarboxylat (41 g, 241 mmol) in Ethanol (200 ml) wurde Guanidincarbonat (26,0 g, 289 mmol) unter Rühren bei Raumtemperatur gegeben. Das Reaktionsgemisch wurde 1 Stunde unter Rückfluß erhitzt und dann auf Raumtemperatur abgekühlt. Die ausgefallenen Kristalle wurden filtriert und mit Wasser gewaschen, gefolgt von Methanol. Die Kristalle wurden im Vakuum getrocknet, wobei die gewünschte Verbindung (35,5 g, 89%) erhalten wurde.
1H-NMR (300 MHz, DMSO-d6): δ 10,64 (1H, brs), 6,18 (2H, brs), 2,35–2,25 (2H, m), 2,23–2,15 (2H, m), 1,70–1,54 (4H, m). - (1-2) 4-Chlor-5,6,7,8-tetrahydrochinazolin-2-ylamin

- Zu einer Suspension von 2-Amino-5,6,7,8-tetrahydrochinazolin-4-ol (20,0 g, 121 mmol) in Toluol (150 ml) wurde Phosphoroxychlorid (55,7 g, 363 mmol) bei 90°C getropft. Das Gemisch wurde 1 Stunde gerührt und das Lösungsmittel im Vakuum entfernt. Der Rückstand wurde in 28%ige wässrige Ammoniaklösung bei 0°C gegossen. Der Feststoff wurde filtriert und durch Kieselgelsäulenchromatographie (3% MeOH/CHCl3) gereinigt, wobei die gewünschte Verbindung (13,5 g, 60%) erhalten wurde.
1H-NMR (300 MHz, DMSO-d6): δ 6,69 (2H, brs), 2,60–2,52 (2H, m), 2,52–2,44 (2H, m), 1,76–1,66 (4H, m).
13C NMR (75 Hz, DMSO-d6): δ 168,4, 161,0, 160,1, 114,8, 31,8, 24,3, 22,1, 21,7. - Bezugsbeispiel 2 5-Butyl-4-chlor-6-methylpyrimidin-2-ylamin (2-1) 2-Amino-5-butyl-6-methylpyrimidin-4-ol

- Ein Gemisch von Ethyl-2-acetylhexanoat (5,59 g, 30 mmol), Guanidincarbonat (6,49 g, 30 mmol) und Ethanol (20 ml) wurde 11 Stunden unter Rückfluß erhitzt und dann eisgekühlt. Die ausgefallenen Kristalle wurden filtriert, mit Ethanol gewaschen und unter Vakuum getrocknet, wobei 2-Amino-5-butyl-6-methylpyrimidin-4-ol (2,59 g, 47%) erhalten wurde.
- (2-2) 5-Butyl-4-chlor-6-methylpyrimidin-2-ylamin
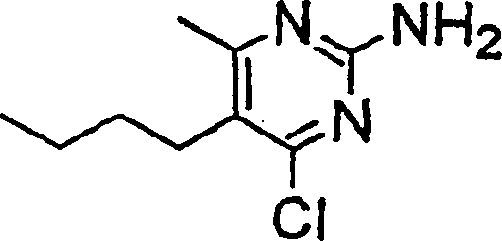
- 2-Amino-5-butyl-6-methylpyrimidin-4-ol (1,0 g, 5,52 mmol) und Phosphoroxychlorid (12 ml) wurden 3 Stunden unter Rückfluß erhitzt. Das Lösungsmittel wurde im Vakuum entfernt und der Rückstand durch Kieselgelsäulenchromatographie (n-Hexan : Essigsäureethylester = 2 : 1) gereinigt, wobei die gewünschte Verbindung (325 mg, 29%) erhalten wurde.
1H-NMR (CDCl3): δ 0,96 (3H, t, J = 7,1 Hz), 1,37–1,50 (4H, m), 2,38 (3H, s), 2,60 (2H, m), 5,01 (2H, brs). - Bezugsbeispiel 3 4-Chlor-5-hexyl-6-methylpyrimidin-2-ylamin
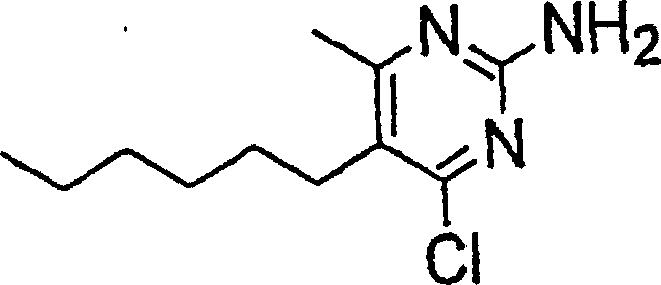
- Unter Verwendung von Ethyl-2-acetyloctanoat (6,43 g, 30 mmol) als Ausgangssubstanz und gemäß dem Verfahren von Bezugsbeispiel 2 wurde 2-Amino-5-hexyl-6-methylpyrimidin-4-ol (4,70 g, 74%) erhalten. Durch Umsetzung des erhaltenen 2-Amino-5-hexyl-6- methylpyrimidin-4-ols (1 g, 4,78 mmol) und Phosphoroxychlorid (12 ml) wurde die gewünschte Verbindung (196 mg, 18%) erhalten
1H-NMR (CDCl3): δ 0,90 (3H, t, J = 6,8 Hz), 1,31–1,52 (8H, m), 2,37 (3H, s), 2,59 (2H, m), 4,95 (2H, brs). - Bezugsbeispiel 4 4-Chlor-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-2-ylamin (4-1) 2-Amino-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-ol

- Unter Verwendung von Ethyl-4-oxotetrahydro-2H-pyran-3-carboxylat (600 mg, 3,49 mmol) als Ausgangssubstanz und gemäß dem Verfahren von Bezugsbeispiel 2 wurde 2-Amino-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-ol (230 mg, 39%) erhalten.
1H-NMR (300 MHz, DMSO-d6): δ 10,78 (1H, brs), 6,34 (2H, brs), 4,24 (2H, brs), 3,78 (2H, t, J = 5,5 Hz), 2,36 (2H, t). - (4-2) 4-Chlor-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-2-ylamin

- Durch Umsetzung von 2-Amino-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-ol (562 mg, 3,36 mmol) und Phosphoroxychlorid (3 ml) wurde die gewünschte Verbindung (136 mg, 22%) erhalten.
1H-NMR (300 MHz, CDCl3): δ 5,10 (1H, brs), 4,62 (2H, s), 3,99 (2H, t, J = 5,4 Hz), 2,78 (2H, t, J = 5,4 Hz). - Bezugsbeispiel 5 4-Butyl-6-chlor-5-methylpyrimidin-2-ylamin (5-1) 2-Amino-6-butyl-5-methylpyrimidin-4-ol

- Unter Verwendung von Ethyl-2-methyl-3-oxoheptanoat (1,06 g, 5,69 mmol) als Ausgangssubstanz und gemäß dem Verfahren von Bezugsbeispiel 2 wurde 2-Amino-6-butyl-5-methylpyrimidin-4-ol (420 mg) erhalten.
1H-NMR (DMSO-d6): δ 0,88 (3H, t, J = 7,3 Hz), 1,30 (2H, m), 1,49 (2H, m), 1,78 (3H, s), 2,32 (2H, t, J = 7,3 Hz), 6,18 (2H, bs), 10,69 (1H, bs). - (5-2) 4-Butyl-6-chlor-5-methylpyrimidin-2-ylamin
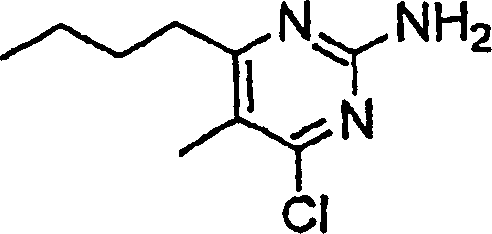
- Durch Umsetzung von 2-Amino-6-butyl-5-methylpyrimidin-4-ol (0,82 g, 4,52 mmol) und Phosphoroxychlorid (10 ml) wurde die gewünschte Verbindung (720 mg) erhalten.
1H-NMR (CDCl3): δ 0,93 (3H, t, J = 7,3 Hz), 1,40 (2H, m), 1,60 (2H, m), 2,20 (3H, s), 2,63 (2H, t, J = 7,3 Hz), 5,73 (2H, bs). - Bezugsbeispiel 6 4-Chlor-5-(2-methoxyethyl)-6-methylpyrimidin-2-ylamin (6-1) 2-Amino-5-(2-methoxyethyl)-6-methylpyrimidin-4-ol
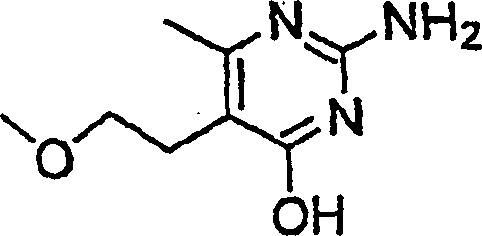
- Ein Gemisch von Ethyl-2-(2-methoxyethyl)-3-oxobutanoat (4 g, 21 mmol), Guanidincarbonat (2,27 g, 16,3 mmol) und Ethanol (16 ml) wurde 9 Stunden unter Rückfluß erhitzt. Nach Abkühlen wurde der Niederschlag filtriert und mit Wasser, Ethanol und Ether in der Reihenfolge gewaschen, wobei die gewünschte Verbindung (1,24 g, 31,9%) erhalten wurde.
1H-NMR (DMSO-d6): δ 2,06 (3H, s), 2,49–2,54 (4H (2H), m, überlappt mit DMSO), 3,22 (3H, s), 3,28 (2H, t, J = 7,3), 6,40 (2H, brs), 10.90 (1H, brs). - (6-2) 4-Chlor-5-(2-methoxyethyl)-6-methylpyrimidin-2-ylamin

- Ein Gemisch von 2-Amino-5-(2-methoxyethyl)-6-methylpyrimidin-4-ol (600 mg, 3,27 mmol) und Phosphoroxychlorid (6 ml) wurde 5,5 Stunden auf 90°C gehalten. Das Reaktionsgemisch wurde im Vakuum konzentriert. wurde zum Rückstand gegeben und eine wässrige Ammoniaklösung vorsichtig zugegeben. Das Gemisch wurde mit Chloroform extrahiert und die organische Schicht mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet und das Lösungsmittel konzentriert. Der Rückstand wurde durch Kieselgelsäulenchromatographie (Chloroform : Essigsäureethylester = 8 : 2) gereinigt, wobei die gewünschte Verbindung (200 mg, 30,3%) erhalten wurde.
1H-NMR (CDCl3): δ 2,42 (3H, s), 2,91 (2H, t, J = 7,3), 3,34 (3H, s), 3,51 (2H, t, J = 7,3), 5,03 (2H, brs). - Bezugsbeispiel 7 3-(2-Amino-4-chlor-6-methylpyrimidin-5-yl)propannitril (7-1) 3-(2-Amino-4-hydoxy-6-methylpyrimidin-5-yl)propannitril
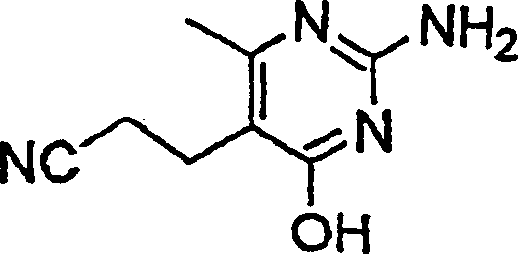
- Ein Gemisch von Ethyl-2-(2-cyanoethyl)-3-oxobutanoat (9 g, 49 mmol), Guanidincarbonat (5,30 g, 29,4 mmol) und Pyridin (49 ml) wurde 8 Stunden auf 100°C gehalten. Das Verfahren gemäß der Vorbehandlung von Bezugsbeispiel 6 wurde durchgeführt, wobei die gewünschte Verbindung (3,38 g, 38,6%) erhalten wurde.
1H-NMR (DMSO-d6): δ 2,11 (3H, s), 2,58 (4H, s), 6,44 (2H, brs), 10,91 (1H, brs). - (7-2) 3-(2-Amino-4-chlor-6-methylpyrimidin-5-yl)propannitril

- Ein Gemisch von 3-(2-Amino-4-hydroxy-6-methylpyrimidin-5-yl)propannitril (2 g, 11,2 mmol) und Phosphoroxychlorid (13 ml) wurde 5 Stunden auf 90°C gehalten. Das Verfahren gemäß der Vorbehandlung von Bezugsbeispiel 6 wurde durchgeführt, wobei die gewünschte Verbindung (1,06 g, 48%) erhalten wurde.
1H-NMR (CDCl3): δ 2,47 (3H, s), 2,61 (2H, t, J = 7,6), 3,02 (2H, t, J = 7,6), 5,11 (2H, brs). - Bezugsbeispiel 8 4-Brom-5,6,7,8-tetrahydrochinazolin-2-yl-amin
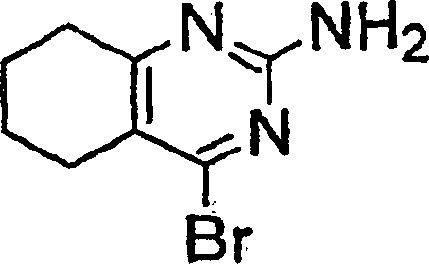
- Zu einer Suspension von 2-Amino-5,6,7,8-tetrahydrochinazolin-4-ol (1,65 g, 10 mmol) in Toluol (16,5 ml) wurde Phosphoroxybromid (3 g) gegeben und das Gemisch in einem Bad (Badtemperatur 90–100°C) 2 Stunden warmgehalten. Nach Bestätigen des Verschwindens der Ausgangssubstanzen wurde das Reaktionsgemisch in Eiswasser gegossen und mit Chloroform und einer gesättigten wässrigen NaHCO3-Lösung extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand wurde durch Kieselgelsäulenchromatographie (CHCl3) gereinigt, wobei die gewünschte Verbindung (1,7 g, 75%) erhalten wurde.
1H-NMR (300 MHz, CDCl3): δ 5,13 (2H, brs), 2,66 (2H, brm), 2,57 (2H, brm), 1,77–1.82 (4H, m). - Bezugsbeispiel 9 2-Chlor-N-pentyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amin (9-1) 2,4-Dichlor-6,7-dihydro-5H-cyclopenta[d]pyrimidin
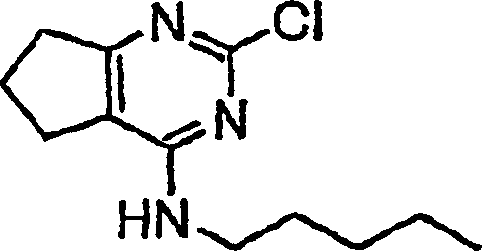
- 6,7-Dihydro-5H-cyclopenta[d]pyrimidin-2,4-diol (359 mg) und Phosphoroxychlorid (5 ml) wurden 3 Stunden unter Rückfluß erhitzt. Nach der Umsetzung wurde das Gemisch unter Vakuum konzentriert. Der Rückstand wurde in Wasser gegossen und mit Chloroform extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet und unter Vakuum konzentriert, wobei 2,4-Dichlor-6,7-dihydro-5H-cyclopenta[d]pyrimidin (410 mg) erhalten wurde.
- (9-2) 2-Chlor-N-pentyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-amin
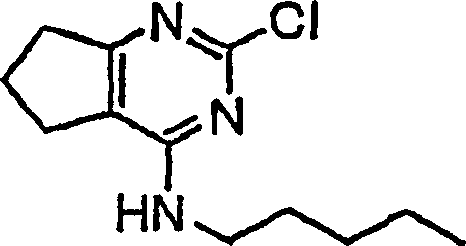
- Ein Gemisch von 2,4-Dichlor-6,7-dihydro-5H-cyclopenta[d]pyrimidin (410 mg) und Pentylamin (1 ml) wurde 8 Stunden bei Raumtemperatur gerührt. Das Reaktionsgemisch wurde in eine wässrige Ammoniumchloridlösung gegossen und die Lösung mit Chloroform extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet und unter Vakuum konzentriert, wobei die gewünschte Verbindung (296 mg, 65%) erhalten wurde.
1H-NMR (300 MHz, CDCl3): δ 4,56 (1H, brs), 3,52–3,46 (2H, m), 2,86 (2H, t, J = 7,5 Hz), 2,63 (2H, t, J = 7,5 Hz), 2,13 (2H, tt, J = 7,5, 7,5 Hz), 1,66–1,57 (2H, m), 1,40–1,33 (4H, m), 0,94–0,89 (3H, m). - Bezugsbeispiel 10
- N-(2-Chlor-5,6,7,8-tetrahydrochinazolin-4-yl)-N-pentylamin
- Die vorstehende Verbindung wurde gemäß dem Verfahren von Bezugsbeispiel 9 hergestellt.1H-NMR (300 MHz, CDCl3): δ 4,64 (1H, brs), 3,51–3,45 (2H, m), 2,68–2,65 (2H, m), 2,27–2,23 (2H, m), 1,90–1,75 (4H, m), 1,70–1,55 (2H, m), 1,45–1,30 (4H, m), 0,93–0,89 (3H, m).

- Bezugsbeispiel 11
- N-(2-Chlor-5,6-dimethylpyrimidin-4-yl)-N-pentylamin
- Die vorstehende Verbindung wurde gemäß dem Verfahren von Bezugsbeispiel 9 hergestellt.1H-NMR (300 MHz, CDCl3): δ 4,65 (1H, brs), 3,51–3,44 (2H, m), 2,34 (3H, s), 1,97 (3H, s), 1,70–1,55 (2H, m), 1,45–1,30 (4H, m), 0,94–0.89 (3H, m).
- Bezugsbeispiel 12 2-Amino-5-benzyl-6-methylpyrimidin-4-ol

- Ein Gemisch von Ethyl-2-benzyl-3-oxobutanoat (6 g, 27,2 mmol), Guanidincarbonat (2,94 g, 16,3 mmol) und Ethanol (20 ml) wurde 10 Stunden unter Rückfluß erhitzt. Nach Abkühlen wurde der Niederschlag filtriert und mit Wasser, Ethanol und Ether in der Reihenfolge gewaschen, wobei die gewünschte Verbindung (3,62 g, 61,7%) erhalten wurde.
1H-NMR DMSO-d6): δ 2,01 (3H, s), 3,64 (2H, s), 6,39 (3H, brs), 7,10–7,26 (5H, m), 10,89 (1H, brs). - Bezugsbeispiel 13 5-Benzyl-4-chlor-6-methylpyrimidin-2-ylamin
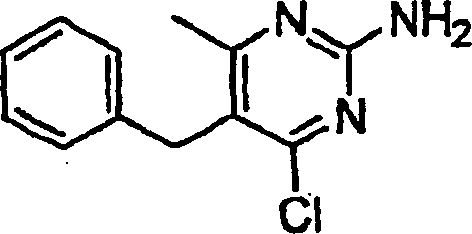
- Ein Gemisch von 2-Amino-5-benzyl-6-methylpyrimidin-4-ol (1,2 g, 5,57 mmol) und Phosphoroxychlorid (9 ml) wurde 6 Stunden auf 90°C erwärmt gehalten. Das Reaktionsgemisch wurde im Vakuum konzentriert. Eiswasser wurde zum Rückstand gegeben und wässriges Ammoniak vorsichtig zugegeben. Die Lösung wurde mit Chloroform extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet und im Vakuum konzentriert. Der Rückstand wurde durch Kieselgelsäulenchromatographie (Chloroform : Essigsäureethylester = 8 : 2) gereinigt, wobei die gewünschte Verbindung (700 mg, 53,7%) erhalten wurde.
1H-NMR (CDCl3): δ 2,30 (3H, s), 4,05 (2H, s), 5,07 (2H, brs), 7,10–7,31 (5H, m). - Bezugsbeispiel 14 2-Amino-5-phenethylpyrimidin-4-ol
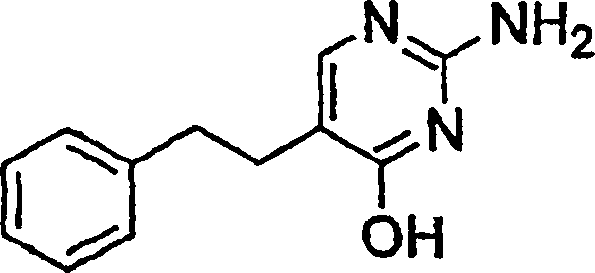
- Metallisches Natrium (966 mg, 42 mmol) wurde unter Stickstoffgas zu Ether (42 ml) gegeben. Dazu wurde ein Gemisch von 4-Phenylbuttersäureethylester (8 g, 42 mmol) und Ethylformiat (3,42 g, 42 mmol) unter Rühren bei Raumtemperatur während eines Zeitraums von 30 Minuten getropft. Das Gemisch wurde 10 Stunden gerührt, um eine Ketoesterverbindung herzustellen.
- Dann wurde Natriumethoxid (3,14 g, 46,2 mmol) unter Stickstoffgas zu Ethanol (42 ml) gegeben. Dazu wurde Guanidin-Hydrochlorid (4,41 g, 46,2 mmol) gegeben und das Gemisch 30 Minuten gerührt. Das Salz wurde abfiltriert und das Filtrat zur vorher hergestellten Ketoesterverbindung in Ether gegeben. Das Reaktionsgemisch wurde 6 Stunden auf 80–90°C gehalten. Nach der Umsetzung wurde das Lösungsmittel unter Vakuum entfernt. Eine 10%ige wässrige Zitronensäurelösung wurde zum Rückstand gegeben, um den pH-Wert auf 8 einzustellen. Essigsäureethylester wurde zum Gemisch gegeben und die erhaltene unlösliche Substanz filtriert, mit Ethanol und Ether gewaschen, wobei die gewünschte Verbindung (853 mg, 9,5%) erhalten wurde.
1H-NMR (DMSO-d6): δ 2,46 (2H, t, J = 7,3), 2,73 (2H, t, J = 7,3), 6,32 (2H, brs), 7,16–7,29 (5H, m), 10,88 (1R, brs). - Bezugsbeispiel 15 4-Chlor-5-phenethylpyrimidin-2-ylamin
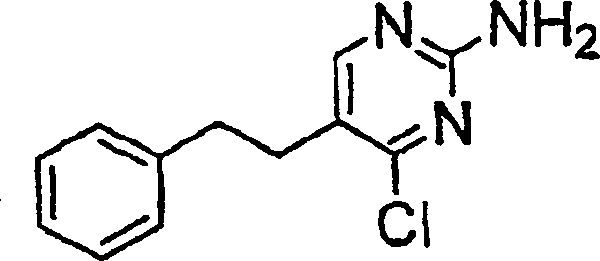
- Ein Gemisch von 2-Amino-5-phenethylpyrimidin-4-ol (600 mg, 2,79 mmol) und Phosphoroxychlorid (5 ml) wurde 6 Stunden auf 90°C erwärmt gehalten. Das Reaktionsgemisch wurde unter Vakuum kondensiert. Eiswasser wurde zum Rückstand gegeben und wässriges Ammoniak vorsichtig zugegeben. Die Lösung wurde mit Chloroform extrahiert. Die organische Schicht wurde mit gesättigter Salzlösung gewaschen, über Natriumsulfat getrocknet und im Vakuum konzentriert. Der Rückstand wurde durch Kieselgelsäulenchromatographie (Chloroform : Essigsäureethylester = 8 : 2) gereinigt, wobei die gewünschte Verbindung (265 mg, 40,7%) erhalten wurde.
1H-NMR (CDCl3): δ 2,87 (4H, s), 5,08 (2H, brs), 7,15–7,32 (5H, m), 7,90 (1H, s). - Bezugsbeispiel 16 5-Benzyl-4-chlorpyrimidin-2-ylamin
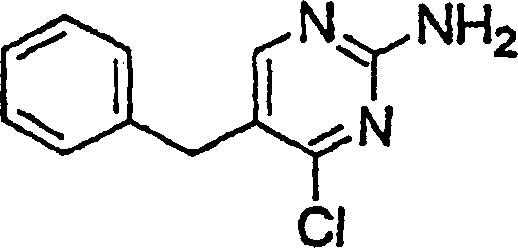
- Die vorstehende Verbindung wurde gemäß dem in J. Amer. Chem. Soc., 73, 3758–3762 (1951) beschriebenen Verfahren hergestellt.
- Test 1
- Wirksamkeit auf die Cytokinenbildung aus Mauslymphknotenzellen durch Verbindungen der Arbeitsbeispiele Experimentelles Verfahren
- 1) Tiere
- BALB/c Mäuse wurden von Japan Charlse River (Yokohama) erworben und 8 Wochen alte weibliche Mäuse wurden verwendet.
- 2) Kulturmedium
- D-MEM (hohe Glucosekonzentration)-Medium (Nikken Biomedical Research Lab. (Kyoto), Code Nr. CM4402), versetzt mit 20%igem durch Wärme inaktiviertem (56°C, 30 min) fötalem Rinderserum (charakterisiert, Code Nr. A-1115-L, HyClone Lab., Logan, Utah), 50 μM 2-Mercaptoethanol (Sigma, St. Louis, MO, Code Nr. M-6250), 100 Einheiten/ml Penicillin und 100 μg/ml Streptomycin (Penicillin-Streptomycin, Bibco-BRL, Code Nr. 15140-122) wurden für den Versuch verwendet.
- 3) Testverbindungen
- Jede Testverbindung, gelöst in DMSO (Nacalai Tesque (Kyoto) Code Nr. 11J) in einer Konzentration von 100 mM wurde auf die Endkonzentration mit dem Medium verdünnt.
- 4) Sensibilisierung und Präparierung der Lymphknotenzellen
- KLH (0,2 mg) wurde subcutan in den Mäusefuß mit Freund-vollständigem Hilfsmittel (Difco Lab., Detroit, Michigan, Code Nr. 3113-60-5) verabreicht. Acht Tage später wurde der Kniekehlen-Lymphknoten aufgenommen und seine Zellsuspension hergestellt.
- 5) Bildung der Cytokine durch Stimulation mit einem Antigen
- KLH (0,1 mg/ml) und die Testverbindung wurden zu Lymphknotenzellen (2,5 × 106 Zellen/ml) gegeben und das Gemisch bei 37°C unter 5% CO2 4 Tage inkubiert (Corning 25850, 0,15 ml/Mulde). Dann wurde die Menge an im Überstand gebildeten Cytokin mit gegenüber Cytokin spezifischen ELISA gemessen.
- Die Mengen an Interleukin 4 (IL-4) und Interleukin 5 (IL-5) als typisches Th2-Typ Cytokin und die Mengen an Interferon γ (IFN-γ) als typisches Th1-Typ Cytokin wurden gemessen.
- 6) Verfahren der Messung (ELISA)
- Die Menge an IL-1 wurde mit ELISA wie nachstehend aufgeführt gemessen. Ein Ratten-Antimaus-IL-4-Antikörper (Pharmingen, San Diego, CA, Code Nr. 18031D, 0,5 mg/ml) als primärer Antikörper wurde 250fach mit Hydrogencarbonat-Puffer verdünnt und in eine Platte mit 96 Mulden (Falcon 3912, Becton Dickinson and Company, Flanklin Lakes, NJ) (50 μ/Mulde) angeimpft und jede Mulde über Nacht bei 4°C bedeckt. Dann wurde die Platte mit PBS (–) Lösung (phosphatgepufferte Salzlösung ohne Calciumchlorid und Magnesiumchlorid) blockiert, die 3% BSA (200 μl/Mulde) enthielt. Nach dreimaligem Spülen der Platte mit PBS (–) Lösung, die 0,05% Polyoxyethylensorbitanmonolaurat (Tween 20TM, Nacalai Tesque (Kyoto) Code Nr. 281-51) (PBST) enthielt, wurde der Überstand des Kulturmediums zu den Mulden (50 μl/Mulde) gegeben und 4 Stunden bei Raumtemperatur inkubiert. Rekombinantes Maus-IL-4 (Pharmingen, Code Nr. 19231 W) wurde zur Erstellung einer Kalibrierungskurve verwendet.
- Nach dreimaligem Spülen der Platte mit PBST wurde ein mit Biotin markierter Ratten-Antimaus-IL-4-Antikörper (Pharmingen, Code Nr. 18042D, 0,5 mg/ml) als sekundärer Antikörper, der 500fach mit PBS (–) Lösung verdünnt war, die 0,1% BSA enthielt, in die Mulden (100 μl/Mulde) gegossen. Die Platte wurde eine Stunde bei Raumtemperatur inkubiert. Der an die Platte gebundene sekundäre Antikörper wurde mit Streptoabidin-Alkaliphosphatase (Kirkegaad & Perry Lab., Gaithersburg, MD, Code Nr. 15-30-00) (0,25 μg/ml, 100 μl/Mulde) nachgewiesen. Nach Inkubieren der Platte für eine Stunde bei 37°C und dreimaligem Spülen der Platte mit PBST wurde das Färben durch Zugabe von PNPP-Substrat (p-Nitrophenyldinatriumphosphat Substrat (Nacalai Tesque) (1 mg/ml, 100 μl/Mulde)) durchgeführt. Die Absorption bei 415 nm wurde mit einem Mikroplattenableser (MTP-120 Microplate reader, Corona Electric Co.) gemessen.
- Die Messung der Menge des IFN-γ wurde mit dem gleichen Verfahren wie vorstehend genannt unter Verwendung eines Ratten-Antimaus-IFN-γ-Antikörpers (Pharmingen, San Diego, CA, Code Nr. 18181D, 0,5 mg/ml) als primärer Antikörper und eines Ratten-Antimaus-IL-5-Antikörpers, markiert mit Biotin (Pharmingen, Code Nr. 18112D, 0,5 mg/ml) als sekundärer Antikörper durchgeführt. Rekombinantes Maus-IFN-γ (Pharmingen, Code Nr. 19301U) wurde zur Erstellung einer Kalibrierungskurve verwendet.
- Die Messung der Menge an IL-5 wurde mit dem gleichen Verfahren wie vorstehend genannt unter Verwendung eines Ratten-Antimaus-IL-5-Antikörpers (Pharmingen, San Diego, CA, Code Nr. 18051D, 0,5 mg/ml) als primärer Antikörper und eines Ratten Antimaus IL-5 Antikörpers, markiert mit Biotin (Pharmingen, Code Nr. 18062D, 0,5 mg/ml) als sekundärer Antikörper, durchgeführt. Der Test wurde dreimal durchgeführt und der Mittelwert berechnet.
- 7) Ergebnisse
- Die Verbindungen der Beispiele 10, 11, 14, 19 und 25 wurden als Testverbindungen in diesem Test verwendet.
- Von jeder Verbindung wurde bestätigt, dass sie die Bildung von IL-4 und IL-5 hemmt und die Bildung von IFN-γ erhöht.
- Test 2
- Die Wirksamkeit auf die Cytokinbildung aus Lymphknotenzellen der Maus durch die Verbindungen der Arbeitsbeispiele
- Experimentelles Verfahren
- Wie in Test 1 wurde jede in DMSO (Nacalai Tesque (Kyoto) Code Nr. 11J) mit einer Konzentration von 100 mM gelöste Testverbindung auf die Endkonzentration mit dem Medium verdünnt. Die Sensibilisierung und Präparierung von Lymphknotenzellen, Bildung von Cytokinen durch Stimulation mit einem Antigen und Messung der Mengen der Cytokine wurden mit dem gleichen Verfahren wie in Test 1 durchgeführt.
- Durch Messung des Hemmungsverhältnisses der Bildung von IL-4 bei verschiedenen Konzentrationen jeder Testverbindung und Verwendung einer Grafik, die die Konzentration der Verbindung und das Hemmungsverhältnis betrifft, wurde eine 50%ige Hemmungskonzentration (IC50) auf jede Testverbindung berechnet.
- Die Ergebnisse sind in Tabelle 1 gezeigt.
- Tabelle 1

- Test 3
- Wirksamkeit auf die Cytokinbildung aus Lymphknotenzellen der Maus durch die Verbindungen der Arbeitsbeispiele
- Experimentelles Verfahren und Ergebnisse
- Wie im Test 1 wurde jede in DMSO (Nacalai Tesque (Kyoto) Code Nr. 11J) mit einer Konzentration von 100 mM gelöste Testverbindung auf die Endkonzentration mit dem Medium verdünnt. Die Sensibilisierung und Herstellung der Lymphknotenzellen, Bildung von Cytokinen durch Stimulation mit einem Antigen und Messung der Mengen der Cytokine wurden mit dem gleichen Verfahren wie in Test 1 durchgeführt.
- Als Ergebnis wurde bestätigt, dass die Verbindungen der Beispiele 26, 27 und 28 die Bildung von IL-4 und IL-5 hemmen und die Bildung von IFN-γ erhöhen.
- Test 4
- Wirksamkeit auf Cytokinbildung aus Lymphknotenzellen der Maus durch die Verbindungen der Arbeitsbeispiele
- Experimentelles Verfahren und Ergebnisse
- Wie im Test 1 wurde jede in DMSO (Nacalai Tesque (Kyoto) Code Nr. 11J) mit einer Konzentration von 100 mM gelöste Testverbindung auf die Endkonzentration mit dem Medium verdünnt. Die Sensibilisierung und Herstellung der Lymphknotenzellen, Bildung von Cytokinen durch Stimulation mit einem Antigen und Messung der Mengen der Cytokine wurden mit dem gleichen Verfahren wie in Test 1 durchgeführt.
- Durch Messung des Hemmungsverhältnisses der Bildung von IL-4 bei verschiedenen Konzentrationen jeder Testverbindung und Verwendung einer Grafik, die die Verbindungskonzentration und das Hemmungsverhältnis betrifft, wurde die 50% Hemmungskonzentration (IC50) auf jede Testverbindung berechnet.
- Die Ergebnisse sind in Tabelle 2 gezeigt.
- Tabelle 2
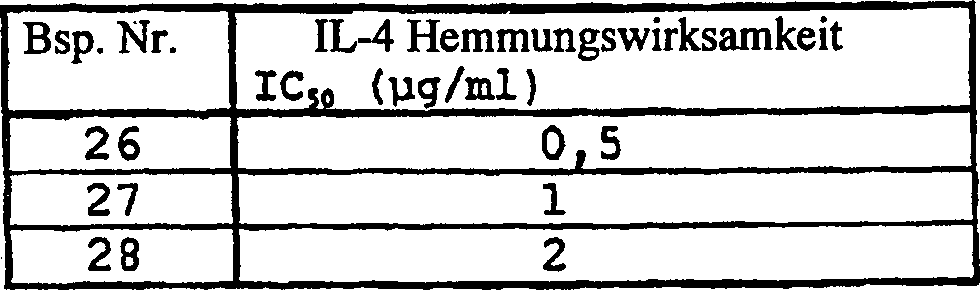
- Test 5
- Wirksamkeit auf die IgE-Bildung aus der Maus in vivo durch Verbindungen der Arbeitsbeispiele Experimentelle Verfahren
- 1) Tier
- BALB/c Mäuse (8 Wochen alte weibliche Mäuse) wurden von Japan Charles River (Yokohama) erworben und nach Vorfüttern für 9 Tage wurden die Mäuse verwendet.
- 2) Sensibilisierung mit Ovalbumin
- Physiologische Salzlösung, die Ovalbumin (Sigma Chemical Co., St Louis, Mo) (4 μg/ml) enthält, und Aluminiumhydroxid·Hilfsmittel (Alu-Gel-S; Serva Feinbiochemica GmbH & Co., Code Nr. 12261) wurden in der gleichen Menge gemischt und das Gemisch einer Maus intraperitoneal verabreicht.
- 3) Verabreichungsverfahren der Testverbindung
- Die Testverbindung wurde in Methylcellulose suspendiert und die Suspension eine Stunde vor der Sensibilisierung mit Ovalbumin und einmal am Tag für 12 Tage nach der Verabreichung verabreicht. Methylcellulose wurde als Kontrolle verwendet.
- 4) Entnahme von Blut und Herstellung von Serum
- Am 13. Tag nach der Sensibilisierung wurde Blut aus dem orbitalen Venenplexus unter Anästhesie mit einer mit Heparin behandelten Kapillare entnommen und zentrifugiert, um ein Serum herzustellen.
- 5) Messung von IgE im Blut
- Die Messung von IgE im Blut wurde mit ELISA durchgeführt. Unter Verwendung eines monoclonalen Ratten-Antimaus-IgE-Antikörpers (Yamasa soy sauce Co., Chiba, Code Nr. 7627) als primären Antikörper und eines mit Biotin markierten monoclonalen Ratten- Antimaus-IgE-Antikörpers (Yamasa soy sauce Co., Chiba, Code Nr. 7617) wurde die Messung der Menge an IgE mit dem gleichen Verfahren wie in Test 2 durchgeführt. Der Assay wurde unter 500fachem Verwenden des Serums durchgeführt. Die Menge an IgE im Blut wurde unter Verwendung einer Standardkurve von Maus-IgE (Yamasa soy sauce Co., Chiba, Code Nr. 7626) berechnet.
- 6) Statistische Behandlung
- Das Ergebnis wurde statistisch mit t-Kalibrierung oder Welch-Kalibrierung behandelt.
- Test 6
- Wirksamkeit gegen Kontaktüberempfindlichkeitsreaktion, die durch TNCB induziert wird
- Testverfahren
- BALB/c Mäuse (6–8 Wochen alte weibliche Mäuse) wurden von Japan Charlse River (Yokohama) erworben. Vor Verwendung ließ man die Mäuse eine Woche aklimatisieren.
- 2) Sensibilisierung
- Das Haar am Mäusebauch wurde geschnitten und darauf 7%iges 2,4,6-Trinitrochlorbenzol (TNCB) in Aceton (0,1 ml/Maus) zum Sensibilisieren verteilt.
- 3) Verfahren der Messung der Dicke der Aurikel
- Sechs Tage nach der Sensibilisierung wurde 1%ige TNCB-Lösung in Aceton auf beiden Seiten der linken Aurikel für die Induktion verteilt. Vierundzwanzig Stunden später wurde die Dicke der Aurikel gemessen.
- 4) Verabreichungsverfahren der Testverbindung
- Die durch Lösen einer Testverbindung (0,4 mg) in Aceton (20 μl) hergestellte Lösung wurde auf dem linken Aurikel 1–2 Stunden vor Sensibilisierung verteilt.
- Industrielle Anwendbarkeit
- Die Pyrimidinderivate oder ihre Salze der vorliegenden Erfindung zeigen die Wirksamkeiten, die Immunantworten auf Th1 erhöhen und die Immunantworten auf Th2 gleichzeitig unterdrücken und weiter die Immunantworten durch Ändern des Gleichgewichts von Th1 und Th2 steuern. Zum Beispiel erhöhen sie die Produktion von Cytokinen des Th1-Typs, wie IFN-γ usw., und hemmen die Produktion von Cytokinen des Th2-Typs, wie IL-4, IL-5 usw. Durch diese Wirksamkeiten können sie als therapeutische und vorbeugende Mittel für allergische Krankheiten, Parasitismus, Autoimmunkrankheiten, wie systemischer Lupus erythemathosus, infektiöse Viren- oder Bakterienkrankheiten, malignen Tumor und erworbenes Immundefizienzsyndrom (AIDS), verwendet werden.
Claims (14)
- Pyrimidinderivat der Formel (1) oder sein Salz;wobei der Rest R1 durch eine Formel (2) wiedergegeben ist;
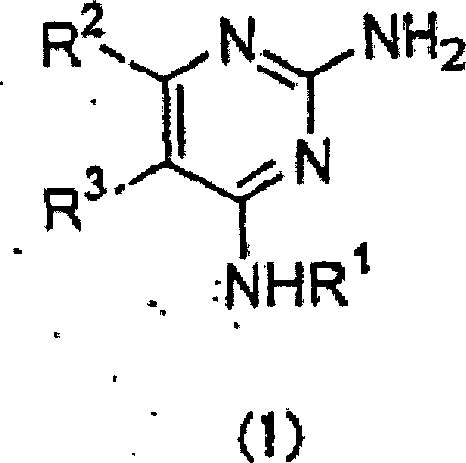 {in der Formel (2) ist der Ring A ein substituiertes oder unsubstituiertes C3-10-Cycloalkan, ein substituiertes oder unsubstituiertes C5-10-Cycloalken, ein substituiertes oder unsubstituiertes C7-10-Bicycloalkan, oder ein heterocyclischer Ring, der ausgewählt ist aus Oxetan, Thietan (Trimethylensulfid), Thiethan-1-oxid, Thiethan-1,1-Dioxid, Tetrahydrofuran, Tetrahydrothiophen, Tetrahydrothiophen-1-oxid, Tetrahydrothiophen-1,1-dioxid, Tetrahydro-4H-pyran, Thian (Pentamethylensulfid), Thian-1-oxid, Thian-1,1-dioxid, Oxepan (Hexamethylenoxid), Thiepan (Hexamethylensulfid), Thiepan-1-oxid, Thiepan-1,1-dioxid, 7-Oxabicyclo[2.2.1]heptan, und 7-Oxabicyclo[2.2.1]hepta-5-en, oder wobei der heterocyclische Ring einen oder mehrere Substituenten, ausgewählt aus C1-3-Alkyl, Hydroxy, C1-3-Alkoxycarbonyl, Carboxy, Carbamoyl und Oxo, aufweist, und die Substituenten mit den benachbarten Kohlenstoffatomen am heterocyclischen Ring eine Tetramethylenbrücke bilden können, ist der Rest R4 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl, C4-10-Cycloalkylalkyl, oder OR8 (wobei der Rest R8 ein geradkettiges oder verzweigtes C1-10-Alkyl, C3-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl oder C4-10-Cycloalkylalkyl ist)}, oder durch eine Formel (3) wiedergeben ist;
{in der Formel (2) ist der Ring A ein substituiertes oder unsubstituiertes C3-10-Cycloalkan, ein substituiertes oder unsubstituiertes C5-10-Cycloalken, ein substituiertes oder unsubstituiertes C7-10-Bicycloalkan, oder ein heterocyclischer Ring, der ausgewählt ist aus Oxetan, Thietan (Trimethylensulfid), Thiethan-1-oxid, Thiethan-1,1-Dioxid, Tetrahydrofuran, Tetrahydrothiophen, Tetrahydrothiophen-1-oxid, Tetrahydrothiophen-1,1-dioxid, Tetrahydro-4H-pyran, Thian (Pentamethylensulfid), Thian-1-oxid, Thian-1,1-dioxid, Oxepan (Hexamethylenoxid), Thiepan (Hexamethylensulfid), Thiepan-1-oxid, Thiepan-1,1-dioxid, 7-Oxabicyclo[2.2.1]heptan, und 7-Oxabicyclo[2.2.1]hepta-5-en, oder wobei der heterocyclische Ring einen oder mehrere Substituenten, ausgewählt aus C1-3-Alkyl, Hydroxy, C1-3-Alkoxycarbonyl, Carboxy, Carbamoyl und Oxo, aufweist, und die Substituenten mit den benachbarten Kohlenstoffatomen am heterocyclischen Ring eine Tetramethylenbrücke bilden können, ist der Rest R4 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl, C4-10-Cycloalkylalkyl, oder OR8 (wobei der Rest R8 ein geradkettiges oder verzweigtes C1-10-Alkyl, C3-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl oder C4-10-Cycloalkylalkyl ist)}, oder durch eine Formel (3) wiedergeben ist;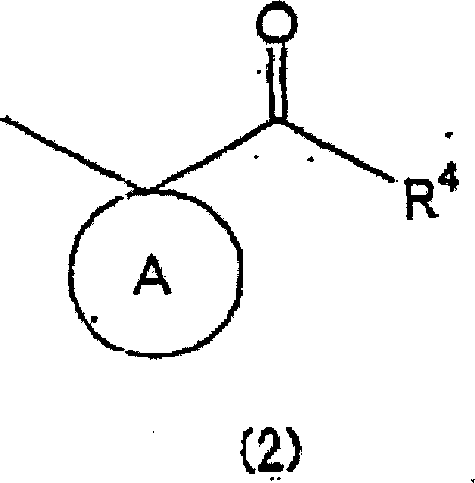 {in der Formel (3) ist der Rest R5 ein geradkettiges oder verzweigtes C1-10-Alkyl; C2-6-Alkenyl; C3-6-Alkinyl; ein geradkettiges oder verzweigtes C1-10-Alkyl, das mit Hydroxy, einem Halogenatom oder C1-4-Alkoxy substituiert ist; Phenyl; C3-8-Cycloalkyl; ein 5 bis 7 gliedriger gesättigter heterocyclischer Ring, der ein oder zwei Sauerstoffatome als Heteroatome enthält; oder C(=O)R9 (wobei der Rest R9 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl, C4-10-Cycloalkylalkyl oder OR10 ist (wobei der Rest R10 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl oder C4-10-Cycloalkylalkyl ist)), ist der Rest R6 ein Wasserstoffatom, ein geradkettiges oder verzweigtes C1-10-Alkyl, C6-10-Aryl, ein Halogenatom, C6-10-Aryl, das mit C1-4-Alkoxy oder C1-4-Alkyl, Carbamoyl, oder Hydroxymethyl substituiert ist, und ist der Rest R7 ein Wasserstoffatom oder ein geradkettiges oder verzweigtes C1-10-Alkyl}, ist der Rest R2 ein Wasserstoffatom, ein geradkettiges oder verzweigtes C1-10-Alkyl, und ist der Rest R3 ein geradkettiges oder verzweigtes C1-10-Alkyl; C3-6-Cycloalkyl; ein geradkettiges oder verzweigtes C1-10-Alkyl, das mit C1-2-Alkylcarbamoyl, C2-4-Dialkylcarbamoyl, C1-4-Alkoxy, C1-4-Alkoxycarbonyl, C3-6-Cycloalkyl, Hydroxy, C1-4-Alkylcarbonyloxy, einem Halogenatom, Amino, C2-4-Acyl-substituiertem Amino, C1-4-Alkyl-substituiertem Sulfonylamino oder C1-5-Alkoxycarbonylamino substituiert ist; oder durch eine Formel (4) wiedergegeben ist;
{in der Formel (3) ist der Rest R5 ein geradkettiges oder verzweigtes C1-10-Alkyl; C2-6-Alkenyl; C3-6-Alkinyl; ein geradkettiges oder verzweigtes C1-10-Alkyl, das mit Hydroxy, einem Halogenatom oder C1-4-Alkoxy substituiert ist; Phenyl; C3-8-Cycloalkyl; ein 5 bis 7 gliedriger gesättigter heterocyclischer Ring, der ein oder zwei Sauerstoffatome als Heteroatome enthält; oder C(=O)R9 (wobei der Rest R9 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl, C4-10-Cycloalkylalkyl oder OR10 ist (wobei der Rest R10 ein geradkettiges oder verzweigtes C1-10-Alkyl, C2-6-Alkenyl, C3-6-Alkinyl, C3-6-Cycloalkyl oder C4-10-Cycloalkylalkyl ist)), ist der Rest R6 ein Wasserstoffatom, ein geradkettiges oder verzweigtes C1-10-Alkyl, C6-10-Aryl, ein Halogenatom, C6-10-Aryl, das mit C1-4-Alkoxy oder C1-4-Alkyl, Carbamoyl, oder Hydroxymethyl substituiert ist, und ist der Rest R7 ein Wasserstoffatom oder ein geradkettiges oder verzweigtes C1-10-Alkyl}, ist der Rest R2 ein Wasserstoffatom, ein geradkettiges oder verzweigtes C1-10-Alkyl, und ist der Rest R3 ein geradkettiges oder verzweigtes C1-10-Alkyl; C3-6-Cycloalkyl; ein geradkettiges oder verzweigtes C1-10-Alkyl, das mit C1-2-Alkylcarbamoyl, C2-4-Dialkylcarbamoyl, C1-4-Alkoxy, C1-4-Alkoxycarbonyl, C3-6-Cycloalkyl, Hydroxy, C1-4-Alkylcarbonyloxy, einem Halogenatom, Amino, C2-4-Acyl-substituiertem Amino, C1-4-Alkyl-substituiertem Sulfonylamino oder C1-5-Alkoxycarbonylamino substituiert ist; oder durch eine Formel (4) wiedergegeben ist;
- Pyrimidinderivat oder sein Salz nach Anspruch 1, wobei der Rest R3 ein geradkettiges oder verzweigtes C1-10-Alkyl; C3-6-Cycloalkyl; oder ein geradkettiges oder verzweigtes C1-10-Alkyl ist, das mit C1-2-Alkylcarbamoyl, C2-4-Dialkylcarbamoyl, C1-4-Alkoxy, C1-4-Alkoxycarbonyl, C3-6-Cycloalkyl, Hydroxy, C1-4-Alkylcarbonyloxy, einem Halogenatom, Amino, C2-4-Acyl-substituiertem Amino, C1-4-Alkyl-substituiertem Sulfonylamino oder C1-5-Alkoxycarbonylamino substituiert ist; oder die Reste R2 und R3 zusammen C3-5-Alkylen oder Alkylen bilden, wobei Methylen mit einem O-Atom ersetzt ist, mit der Maßgabe dass, wenn der Rest R5 Phenyl ist und die Reste R6 und R7 ein Wasserstoffatom sind, die Reste R2 und R3 zusammen C4-5-Alkylen oder C3-5-Alkylen bilden, wobei Methylen durch ein O-Atom ersetzt ist.
- Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach Anspruch 1 oder 2, wobei die Reste R2 und R3 zusammen Trimethylen oder Tetramethylen sind, mit der Maßgabe dass, wenn der Rest R5 Phenyl ist und die Reste R6 und R7 ein Wasserstoffatom sind, die Reste R2 und R3 zusammen Tetramethylen sind.
- Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach Anspruch 1 oder 2, wobei der Rest R3 ein geradkettiges oder verzweigtes C1-7-Alkyl ist.
- Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach Anspruch 1, wobei der Rest R3 durch die Formel (4) wiedergegeben ist;
- Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach Anspruch 1 oder 5, wobei der Rest R11 der Formel (4) Pyridyl, Thienyl oder Furyl ist.
- Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach Anspruch 1, 5 oder 6, wobei n der Formel (4) eine ganze Zahl von 2–4 ist.
- Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach einem der Ansprüche 1 bis 7, wobei der Rest R1 durch die Formel (2) wiedergegeben ist;wobei der Ring A und der Rest R4 die gleiche vorstehend in Anspruch 1 angegebene Bedeutung haben.
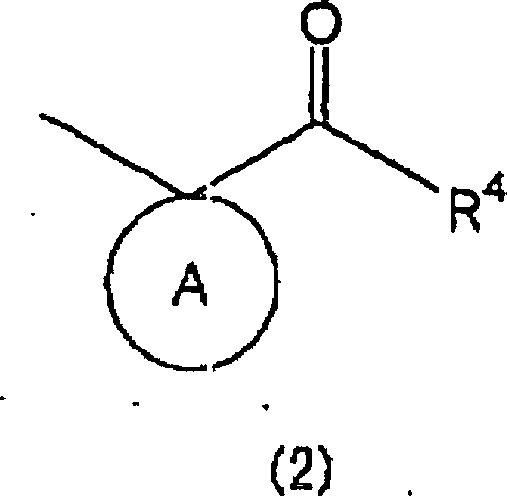
- Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach einem der Ansprüche 1 bis 7, wobei der Rest R1 durch die Formel (3) wiedergegeben ist;wobei die Reste R3, R6 und R7 die gleiche vorstehend in Anspruch 1 angegebene Bedeutung haben.

- Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach einem der Ansprüche 1 bis 7 oder 9, wobei der Rest R5 ein geradkettiges C2-4-Alkyl oder ein geradkettiges C2-4-Alkyl ist, das mit Hydroxy substituiert ist.
- Immunmodulator, der die Immunantworten der Typ 2 T-Helferzelle unterdrückt und die Immunantworten der Typ 1 T-Helferzelle erhöht, umfassend das Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach einem der Ansprüche 1 bis 10 als einen Wirkstoff.
- Therapeutisches oder vorbeugendes Mittel gegen Krankheiten in dem Stadium, in dem Immunantworten der T-Helferzelle vom Typ 2 abnormal erhöht sind, umfassend das Pyrimidinderivat oder sein pharmazeutisch verträgliches Salz nach einem der Ansprüche 1 bis 10 als einen Wirkstoff.
- Therapeutisches oder vorbeugendes Mittel nach Anspruch 12, wobei die Krankheit in dem Stadium, in dem die Immunantworten der Typ 2 T-Helferzelle abnormal erhöht sind, eine allergische Krankheit ist.
- Therapeutisches oder vorbeugendes Mittel nach Anspruch 13, wobei die allergische Krankheit Asthma, allergische Rinitis, oder allergische Dermatitis ist.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP24184298 | 1998-08-27 | ||
| JP24184298 | 1998-08-27 | ||
| JP25350698 | 1998-09-08 | ||
| JP25350698 | 1998-09-08 | ||
| PCT/JP1999/004505 WO2000012487A1 (en) | 1998-08-27 | 1999-08-20 | Pyrimidine derivatives |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69917469D1 DE69917469D1 (de) | 2004-06-24 |
| DE69917469T2 true DE69917469T2 (de) | 2005-05-12 |
Family
ID=26535473
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69917469T Expired - Lifetime DE69917469T2 (de) | 1998-08-27 | 1999-08-20 | Pyrimidin derivate |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US6458798B1 (de) |
| EP (1) | EP1110951B1 (de) |
| JP (1) | JP4497340B2 (de) |
| AT (1) | ATE267175T1 (de) |
| CA (1) | CA2341707C (de) |
| DE (1) | DE69917469T2 (de) |
| ES (1) | ES2221414T3 (de) |
| WO (1) | WO2000012487A1 (de) |
Families Citing this family (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7772432B2 (en) | 1991-09-19 | 2010-08-10 | Astrazeneca Ab | Amidobenzamide derivatives which are useful as cytokine inhibitors |
| WO2000020402A1 (en) | 1998-10-01 | 2000-04-13 | Astrazeneca Ab | Chemical compounds |
| BR0009083B1 (pt) | 1999-03-17 | 2011-11-01 | derivado de amida compreendendo um núcleo de quinazolinona, processo para a preparação de um derivado de amida, composição farmacêutica, e, uso de um derivado de amida. | |
| US7037916B2 (en) | 1999-07-15 | 2006-05-02 | Pharmacopeia Drug Discovery, Inc. | Pyrimidine derivatives as IL-8 receptor antagonists |
| GB9924092D0 (en) | 1999-10-13 | 1999-12-15 | Zeneca Ltd | Pyrimidine derivatives |
| GB0324790D0 (en) | 2003-10-24 | 2003-11-26 | Astrazeneca Ab | Amide derivatives |
| US8012964B2 (en) | 2004-03-26 | 2011-09-06 | Dainippon Sumitomo Pharma Co., Ltd. | 9-substituted 8-oxoadenine compound |
| WO2006006616A1 (ja) * | 2004-07-13 | 2006-01-19 | Taiho Pharmaceutical Co., Ltd. | トシル酸スプラタスト結晶の均一性の評価方法並びに均一な結晶及びその製造方法 |
| TW200801003A (en) * | 2005-09-16 | 2008-01-01 | Astrazeneca Ab | Novel compounds |
| WO2008004948A1 (en) | 2006-07-05 | 2008-01-10 | Astrazeneca Ab | 8-oxoadenine derivatives acting as modulators of tlr7 |
| CA2656535C (en) * | 2006-07-14 | 2011-08-23 | Amgen Inc. | Alkyne-substituted pyridone compounds and methods of use |
| TW200831105A (en) | 2006-12-14 | 2008-08-01 | Astrazeneca Ab | Novel compounds |
| JP5480637B2 (ja) * | 2007-03-19 | 2014-04-23 | アストラゼネカ・アクチエボラーグ | Toll様受容体(tlr7)モジュレーターとしての9−置換−8−オキソ−アデニン化合物 |
| WO2008114006A1 (en) | 2007-03-19 | 2008-09-25 | Astrazeneca Ab | 9-substituted-8-oxo-adenine compounds as toll-like receptor (tlr7) modulators |
| TW200902018A (en) * | 2007-03-20 | 2009-01-16 | Dainippon Sumitomo Pharma Co | Novel adenine compound |
| WO2008114819A1 (ja) * | 2007-03-20 | 2008-09-25 | Dainippon Sumitomo Pharma Co., Ltd. | 新規アデニン化合物 |
| KR20100016289A (ko) | 2007-05-08 | 2010-02-12 | 아스트라제네카 아베 | 면역-조정 특성의 이미다조퀴놀린 |
| PE20091236A1 (es) * | 2007-11-22 | 2009-09-16 | Astrazeneca Ab | Derivados de pirimidina como immunomoduladores de tlr7 |
| PE20091156A1 (es) | 2007-12-17 | 2009-09-03 | Astrazeneca Ab | Sales de (3-{[[3-(6-amino-2-butoxi-8-oxo-7,8-dihidro-9h-purin-9-il)propil](3-morfolin-4-ilpropil)amino]metil}fenil)acetato de metilo |
| WO2010022121A1 (en) * | 2008-08-20 | 2010-02-25 | Schering Corporation | Substituted pyridine and pyrimidine derivatives and their use in treating viral infections |
| WO2010064705A1 (ja) * | 2008-12-05 | 2010-06-10 | 大日本住友製薬株式会社 | H4受容体アンタゴニスト作用を有する新規7位置換ジヒドロピラノピリミジン誘導体 |
| CA2760766A1 (en) * | 2009-05-21 | 2010-11-25 | Nicholas James Bennett | Novel pyrimidine derivatives and their use in the treatment of cancer and further diseases |
| GB0908772D0 (en) * | 2009-05-21 | 2009-07-01 | Astrazeneca Ab | New salts 756 |
| JP2013512859A (ja) * | 2009-12-03 | 2013-04-18 | 大日本住友製薬株式会社 | トール様受容体(tlr)を介して作用するイミダゾキノリン |
| JP5978225B2 (ja) | 2010-12-16 | 2016-08-24 | 大日本住友製薬株式会社 | 治療に有用なイミダゾ[4,5−c]キノリン−1−イル誘導体 |
| US8895570B2 (en) | 2010-12-17 | 2014-11-25 | Astrazeneca Ab | Purine derivatives |
| PH12013502033A1 (en) | 2011-04-08 | 2014-01-06 | Janssen Sciences Ireland Uc | Pyrimidine derivatives for the treatment of viral infections |
| EP2776439B1 (de) | 2011-11-09 | 2018-07-04 | Janssen Sciences Ireland UC | Purinderivate zur behandlung von virusinfektionen |
| DK2812331T3 (en) * | 2012-02-08 | 2019-04-08 | Janssen Sciences Ireland Unlimited Co | PIPERIDINOPYRIMIDINE DERIVATIVES FOR TREATING VIRUS INFECTIONS |
| AU2013261267B2 (en) | 2012-05-18 | 2017-06-29 | Sumitomo Pharma Co., Ltd. | Carboxylic acid compounds |
| WO2014009509A1 (en) | 2012-07-13 | 2014-01-16 | Janssen R&D Ireland | Macrocyclic purines for the treatment of viral infections |
| MX369417B (es) * | 2012-08-10 | 2019-11-07 | Janssen Sciences Ireland Uc | Derivados de alquilpirimidina para el tratamiento de infecciones víricas y otras enfermedades. |
| NZ705360A (en) * | 2012-10-05 | 2018-11-30 | Janssen Sciences Ireland Uc | Acylaminopyrimidine derivatives for the treatment of viral infections and further diseases |
| US9499549B2 (en) | 2012-10-10 | 2016-11-22 | Janssen Sciences Ireland Uc | Pyrrolo[3,2-]pyrimidine derivatives for the treatment of viral infections and other diseases |
| UA118341C2 (uk) | 2012-11-16 | 2019-01-10 | ЯНССЕН САЙЄНСІЗ АЙРЛЕНД ЮСі | Гетероциклічні заміщені похідні 2-амінохіназоліну для лікування вірусних інфекцій |
| UA118751C2 (uk) | 2013-02-21 | 2019-03-11 | ЯНССЕН САЙЄНСІЗ АЙРЛЕНД ЮСі | Похідні 2-амінопіримідину для лікування вірусних інфекцій |
| BR112015024411B1 (pt) | 2013-03-29 | 2022-02-22 | Janssen Sciences Ireland Uc | Desaza-purinonas macrocíclicas para o tratamento de infeções virais e composição farmacêutica que os compreende |
| AU2014270418B2 (en) | 2013-05-24 | 2017-11-30 | Janssen Sciences Ireland Uc | Pyridone derivatives for the treatment of viral infections and further diseases |
| NO3030563T3 (de) | 2013-06-27 | 2018-01-06 | ||
| CA2913691C (en) | 2013-07-30 | 2022-01-25 | Janssen Sciences Ireland Uc | Thieno[3,2-d]pyrimidines derivatives for the treatment of viral infections |
| TWI718188B (zh) | 2015-10-07 | 2021-02-11 | 日商大日本住友製藥股份有限公司 | 嘧啶化合物 |
| CN109476675B (zh) * | 2016-07-01 | 2022-12-09 | 爱尔兰詹森科学公司 | 用于治疗病毒性感染的二氢吡喃并嘧啶衍生物 |
| AU2017335205B2 (en) | 2016-09-29 | 2021-11-04 | Janssen Sciences Ireland Unlimited Company | Pyrimidine prodrugs for the treatment of viral infections and further diseases |
| US10662161B2 (en) * | 2017-01-30 | 2020-05-26 | Regents Of The University Of Minnesota | Pyrimidines and uses thereof |
| FI3603619T3 (fi) | 2017-03-29 | 2025-09-25 | Sumitomo Pharma Co Ltd | Rokoteadjuvanttiformulaatio |
| RU2020124007A (ru) | 2017-12-21 | 2022-01-21 | Сумитомо Дайниппон Фарма Ко., Лтд. | Комбинированный препарат, включающий агонист tlr7 |
| TW202415645A (zh) | 2018-03-01 | 2024-04-16 | 愛爾蘭商健生科學愛爾蘭無限公司 | 2,4-二胺基喹唑啉衍生物及其醫學用途 |
| IL280332B2 (en) | 2018-07-23 | 2025-05-01 | Japan As Represented By Director General Of Nat Institute Of Infectious Diseases | A preparation containing influenza vaccine |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3185691A (en) * | 1965-05-25 | Gycloocteno-pyrimroines | ||
| GB152883A (en) | 1919-11-26 | 1920-10-28 | Carl Conrad Albin Perlich | Improvements in means for controlling the supply of gas to gas cooking apparatus |
| US3034954A (en) * | 1960-03-23 | 1962-05-15 | Monsanto Chemicals | Virus inhibitors |
| DE1470336A1 (de) * | 1962-07-04 | 1969-03-20 | Thomae Gmbh Dr K | Neue Dihydrothieno-[3,4-d]-pyrimidine und Verfahren zu ihrer Herstellung |
| GB1152883A (en) * | 1967-01-25 | 1969-05-21 | Ucb Sa | 4,5-Polymethylene-Pyrimidine Derivatives |
| JP2762430B2 (ja) | 1991-01-18 | 1998-06-04 | 宇部興産株式会社 | アラルキルアミノピリミジン類の製法 |
| WO1997009325A1 (en) * | 1995-09-01 | 1997-03-13 | Signal Pharmaceuticals, Inc. | Pyrimidine carboxylates and related compounds and methods for treating inflammatory conditions |
-
1999
- 1999-08-20 ES ES99938567T patent/ES2221414T3/es not_active Expired - Lifetime
- 1999-08-20 DE DE69917469T patent/DE69917469T2/de not_active Expired - Lifetime
- 1999-08-20 CA CA2341707A patent/CA2341707C/en not_active Expired - Fee Related
- 1999-08-20 JP JP2000567517A patent/JP4497340B2/ja not_active Expired - Fee Related
- 1999-08-20 EP EP99938567A patent/EP1110951B1/de not_active Expired - Lifetime
- 1999-08-20 WO PCT/JP1999/004505 patent/WO2000012487A1/ja not_active Ceased
- 1999-08-20 US US09/763,728 patent/US6458798B1/en not_active Expired - Fee Related
- 1999-08-20 AT AT99938567T patent/ATE267175T1/de not_active IP Right Cessation
-
2002
- 2002-08-19 US US10/222,922 patent/US6951866B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| US6951866B2 (en) | 2005-10-04 |
| US6458798B1 (en) | 2002-10-01 |
| EP1110951A1 (de) | 2001-06-27 |
| WO2000012487A1 (en) | 2000-03-09 |
| ES2221414T3 (es) | 2004-12-16 |
| EP1110951A4 (de) | 2002-01-02 |
| JP4497340B2 (ja) | 2010-07-07 |
| CA2341707A1 (en) | 2000-03-09 |
| EP1110951B1 (de) | 2004-05-19 |
| US20030105323A1 (en) | 2003-06-05 |
| CA2341707C (en) | 2010-02-16 |
| DE69917469D1 (de) | 2004-06-24 |
| ATE267175T1 (de) | 2004-06-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69917469T2 (de) | Pyrimidin derivate | |
| JPWO2000012487A1 (ja) | ピリミジン誘導体 | |
| JP4890723B2 (ja) | TNFαインヒビターとして有用なクマリン誘導体 | |
| DE69930773T2 (de) | Bicyclische heteroaromatische Verbindungen als Protein-Tyrosinekinase-Inhibitoren | |
| DE69906554T2 (de) | P38 inhibitoren | |
| DE69130683T2 (de) | PYRAZOLO[1,5-a]PYRIMIDINDERIVATE UND SIE ENTHALTENDE ANTIINFLAMMATORISCHE MITTEL | |
| DE60110749T2 (de) | Pyridinderivative als angiogenese- und/oder vegf-rezeptor-tyrosinkinase-inhibitoren | |
| DE69531623T2 (de) | Neue benzoxazole | |
| DE69329574T2 (de) | Pharmazeutisch wirksame bicyclisch heterocyclische amine | |
| DE69934805T2 (de) | Substituierte imidazole mit cytokin-inhibierender wirkung | |
| DE60120138T2 (de) | Amid-verbindungen und deren verwendung | |
| DE69120581T2 (de) | 1,8-Naphthyridin-2-on-Derivate | |
| DE602004001397T2 (de) | Spirosubstituierte pyrrolopyrimidine | |
| JP4342007B2 (ja) | キナゾリン誘導体 | |
| DE10013126A1 (de) | Arzneimittel gegen virale Erkrankungen | |
| DE69511240T2 (de) | Derivate des hydroxamsäure als metalloproteinaseinhibitoren | |
| DE69813605T2 (de) | Benzofuranderivate | |
| JPH08253466A (ja) | N−(3−ベンゾフラニル)ウレア−誘導体 | |
| DE3331808A1 (de) | Carbostyrilderivate, verfahren zu deren herstellung und arzneimittel, welche diese enthalten | |
| EP0114270B1 (de) | Acylderivate von 1,4:3,6-Dianhydro-hexiten, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| DE60222465T2 (de) | Pyrazolopyridin-derivate als antiherpesmittel | |
| DE69817810T2 (de) | Cyanoguanidine als zellproliferation inhibitoren | |
| WO2001068639A1 (de) | Dihydropyrimidin-5-carbonsäureester und ihre verwendung als arzneimittel gegen virale erkrankungen | |
| DE69128254T2 (de) | Antihypertensive benzopyran-derivate | |
| DE69630799T2 (de) | Pyrimidinderivate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8327 | Change in the person/name/address of the patent owner |
Owner name: DAINIPPON SUMITOMO PHARMA CO., LTD., OSAKA, JP |