WO2007004620A1 - 硬化性組成物及びそれを用いる部材の仮固定方法 - Google Patents
硬化性組成物及びそれを用いる部材の仮固定方法 Download PDFInfo
- Publication number
- WO2007004620A1 WO2007004620A1 PCT/JP2006/313247 JP2006313247W WO2007004620A1 WO 2007004620 A1 WO2007004620 A1 WO 2007004620A1 JP 2006313247 W JP2006313247 W JP 2006313247W WO 2007004620 A1 WO2007004620 A1 WO 2007004620A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- curable composition
- mass
- parts
- meth
- composition according
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J4/00—Adhesives based on organic non-macromolecular compounds having at least one polymerisable carbon-to-carbon unsaturated bond ; adhesives, based on monomers of macromolecular compounds of groups C09J183/00 - C09J183/16
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/04—Acids; Metal salts or ammonium salts thereof
- C08F220/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/67—Unsaturated compounds having active hydrogen
- C08G18/671—Unsaturated compounds having only one group containing active hydrogen
- C08G18/672—Esters of acrylic or alkyl acrylic acid having only one group containing active hydrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/14—Peroxides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
- C08L33/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, which oxygen atoms are present only as part of the carboxyl radical
- C08L33/10—Homopolymers or copolymers of methacrylic acid esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
- C08L33/14—Homopolymers or copolymers of esters of esters containing halogen, nitrogen, sulfur, or oxygen atoms in addition to the carboxy oxygen
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J133/00—Adhesives based on homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Adhesives based on derivatives of such polymers
- C09J133/04—Homopolymers or copolymers of esters
- C09J133/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, the oxygen atom being present only as part of the carboxyl radical
- C09J133/10—Homopolymers or copolymers of methacrylic acid esters
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J133/00—Adhesives based on homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Adhesives based on derivatives of such polymers
- C09J133/04—Homopolymers or copolymers of esters
- C09J133/14—Homopolymers or copolymers of esters of esters containing halogen, nitrogen, sulfur or oxygen atoms in addition to the carboxy oxygen
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J175/00—Adhesives based on polyureas or polyurethanes; Adhesives based on derivatives of such polymers
- C09J175/04—Polyurethanes
- C09J175/14—Polyurethanes having carbon-to-carbon unsaturated bonds
- C09J175/16—Polyurethanes having carbon-to-carbon unsaturated bonds having terminal carbon-to-carbon unsaturated bonds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/22—Mixtures comprising a continuous polymer matrix in which are dispersed crosslinked particles of another polymer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2666/00—Composition of polymers characterized by a further compound in the blend, being organic macromolecular compounds, natural resins, waxes or and bituminous materials, non-macromolecular organic substances, inorganic substances or characterized by their function in the composition
- C08L2666/02—Organic macromolecular compounds, natural resins, waxes or and bituminous materials
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
- C08L33/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, which oxygen atoms are present only as part of the carboxyl radical
- C08L33/10—Homopolymers or copolymers of methacrylic acid esters
- C08L33/12—Homopolymers or copolymers of methyl methacrylate
Definitions
- the present invention relates to a temporary fixing method when various members are cared, and to a curable composition and an adhesive suitable for the method. More specifically, the present invention relates to a method of temporarily fixing the optical member when processing the optical member, a curable adhesive suitable for this method, and a curable composition that can be easily peeled off.
- Double-sided tapes and hot-melt adhesives are used as temporary fixing adhesives for optical lenses, prisms, arrays, silicon wafers, semiconductor mounting components, etc., and these adhesives are joined or laminated.
- the adhesive is removed to produce a processed part.
- semiconductor mounting parts these parts are fixed to the substrate with double-sided tape, then the desired parts are cut, and the double-sided tape is irradiated with ultraviolet rays to remove the component strength. I do.
- a hot melt adhesive after joining the members, the adhesive is infiltrated into the gap by heating, and then the desired part is cut and the adhesive is peeled off in an organic solvent.
- the material of the material to be processed is limited to materials that transmit light such as ultraviolet rays, and materials that do not transmit light such as ceramic materials and colored plastic materials are applicable. There is a problem that you can not. Furthermore, since heat curable adhesives such as epoxy resin often require a heating device, the curing time becomes long, and there are problems of further labor saving, energy saving and shortening of working time (Patent Document 1). , 2, 3).
- Patent Document 1 JP-A-6-116534.
- Patent Document 2 JP-A-11 71553.
- Patent Document 3 Japanese Patent Application Laid-Open No. 2001-226641.
- the present invention has a gist characterized by the following.
- a curable composition (A): a polyfunctional (meth) acrylate, (B): a monofunctional (meth) acrylate, (C): a photopolymerization initiator, and (D): a polar solvent.
- a curable composition (A): a polyfunctional (meth) acrylate, (B): a monofunctional (meth) acrylate, (C): a photopolymerization initiator, and (D): a polar solvent.
- (D) is at least one selected from the group power consisting of water, methanol, ethanol, isopropyl alcohol, and n-butanol.
- (G) is an N-alkyl (meth) acrylamide derivative, a nitrogen-containing cyclic monomer, and a beryl group-containing amino acid group power of at least one selected polymer, and Z or N-alkyl (meta 6.
- N-alkyl (meth) acrylamide derivative strength The curable composition according to 6 or 7 above, which is N-isopropylacrylamide.
- (G) is a copolymer composed of N-isopropylacrylamide, diacetone acrylamide, and acrylic acid, and a copolymer composed of Z or N-isopropylacrylamide, acrylamide, and acrylic acid. Sex composition.
- (H) is at least one selected from the group consisting of crosslinked polymethyl methacrylate particles, crosslinked polystyrene particles, and spherical silica particles.
- the total of (A) and (B) is 100 parts by mass, (C) is 0.1-20 parts by mass, (D) is 0.1-20 parts by mass, (G) is 0.001 ⁇ : L0 10.
- the curable composition according to any one of 5 to 9 above, which is contained by mass part.
- An adhesive comprising the curable composition according to any one of 1 to 17 above.
- the temperature at which the member is temporarily fixed using the curable composition according to 14 or 15 above is , — Temporary fixing method for members, which is 10 ° C to 40 ° C.
- a member is temporarily fixed using the curable composition according to any one of 1 to 17, and after the temporarily fixed member is processed, the caulked member is heated to 30 to 90 ° C.
- the surface roughness (RMax) of the surface with respect to the curable composition of the substrate is 10 ⁇ m to 50 ⁇ m, and 10 m than the surface roughness (RMax) of the surface with respect to the curable composition of the member 24.
- the curable composition of the present invention has a room temperature curable property or a photocurable property because of its composition. For example, it is cured by visible light or ultraviolet light. For this reason, compared with the conventional hot melt adhesive, remarkable effects are obtained in terms of labor saving, energy saving, and work shortening. In addition, since the cured body can exhibit high adhesive strength without being affected by cutting water used during processing, a member excellent in dimensional accuracy that is less likely to be displaced during processing of the member can be easily obtained. .
- the resulting cured body can be easily recovered by contacting with hot water of 30 to 90 ° C. to reduce the adhesive strength and the bonding force between the members or between the member and the jig. Therefore, it is not necessary to use an organic solvent that is expensive, strongly ignitable, or generates gas that is harmful to the human body, compared with conventional adhesives.
- the cured product swells upon contact with hot water at 30 to 90 ° C. and can be recovered from the member in the form of a film, so that the workability is excellent.
- the method for temporarily fixing the member of the present invention uses a curable composition that decreases the adhesive strength by contacting with hot water of 30 to 90 ° C.
- the member can be easily recovered simply by contacting it.
- it is not necessary to use an organic solvent that is expensive, highly ignitable, or generates a gas harmful to the human body, as compared with the case of conventional adhesives.
- the cured product of the curable composition is surely peeled off as a member, and as a result, the processing efficiency of the member can be extremely high.
- the temperature of the hot water has no technical meaning to set the upper limit, but is preferably 30 to 90 ° C because it is easy to work.
- FIG. 1-1 shows the measurement results of the film thickness distribution of the composition according to Example 1 17.
- a polyfunctional (meth) acrylate which is used in the present invention is a polyfunctional (meth) acrylate polymer in which two or more (meth) acrylates are formed at the terminal or side chain of the oligomer / polymer.
- Z polymers and monomers having two or more (meth) acryloyl groups can be used.
- the polyfunctional (meth) acrylate oligomer Z polymer 1,2-polybutadiene-terminated urethane (meth) acrylate (eg, TE-2000, TEA-1000 manufactured by Nippon Soda Co., Ltd.), the hydrogenated product (eg, Nippon Soda Co., Ltd.
- TEAI-1000 1, 4 Polybutadiene-terminated urethane (meth) acrylate (for example, BAC-45 made by Osaka Organic Chemical Co., Ltd.), Polyisoprene-terminated (meth) acrylate, Polyester urethane (meth) acrylate Polyether-based urethane (meth) acrylate, polyester (meth) acrylate, bis A type epoxy (meth) acrylate (for example, Osaka Organic Chemical Co., Ltd. Biscoat # 540, Showa High Polymer Co., Ltd. Biscoat VR-77), etc. Can be mentioned.
- the bifunctional (meth) acrylate monomer includes 1,3 butylene glycol di (meth) acrylate, 1, 4 butanediol di (meth) acrylate, 1, 6 hexadiol di (meth) acrylate, 1 , 9-nonanediol di (meth) acrylate, neopentyl glycol di (meth) acrylate, dicyclopentadi (meth) acrylate, 2-ethyl 2-butyl-propanediol (meth) acrylate, modified with neopentyl glycol Trimethylol propandi (meth) acrylate, stearic acid-modified pentaerythritol diatalylate, polypropylene glycol di (meth) acrylate, 2, 2 Bis (4- (meth) attadidiethoxyphenol) propane, 2, 2 Bis (4- (meth) ataryloxypropoxyphenyl) propane,
- the trifunctional (meth) acrylate monomer is trimethylolpropane tri (meth) atelier And tris [(meth) atariloykischetil] isocyanurate.
- the tetra- or higher functional (meth) acrylate monomer includes dimethylolpropane tetra (meth) acrylate, pentaerythritol tetra (meth) acrylate, pentaerythritol etoxytetra (meth) acrylate, dipentaerythritol penta (meth) ate Rate, dipentaerythritol hexa (meth) acrylate, etc.
- (A) is more preferably hydrophobic.
- the water-soluble case is not preferable because the cured product of the composition swells during cutting, which may cause displacement and poor processing accuracy. Even if it is hydrophilic, it can be used as long as the cured product of the curable composition is not greatly swollen or partially dissolved by water.
- the addition amount of (A) is preferably 1 to 50 parts by mass in a total of 100 parts by mass of (A) and (B). In particular, 5 to 30 parts by mass is preferable. If the amount is 1 part by mass or more, the property that the cured product peels off from the adherend when the cured product of the curable composition is immersed in warm water (hereinafter simply referred to as “peelability”) is sufficiently promoted. In addition, it can be ensured that the cured product of the curable composition is peeled off into a film. Moreover, if it is 50 mass parts or less, there is no possibility that initial adhesiveness will fall.
- Monofunctional (meth) acrylate monomers include methyl (meth) acrylate, ethyl (meth) acrylate, propyl (meth) acrylate, butyl (meth) acrylate, 2-ethyl Xylyl (meth) acrylate, iso-octyl (meth) acrylate, isodecyl (meth) acrylate, lauryl (meth) acrylate, stearyl (meth) acrylate, phenol (meth) acrylate, cyclohexyl (meth) Atalylate, dicyclopental (meth) atarylate, dicyclopentaure (meth) atarylate, dicyclopente-mouth kichetyl (meth) atarylate, isobutyl (meth) atarylate, methoxylated cyclodecatriene (meth) Atalylate, 2-hydroxychetyl (meth) atarylate, 2-hydroxychety
- ( ⁇ ) is water-soluble, which is more preferably hydrophobic, as in ( ⁇ ) above, misalignment is caused by swelling of the cured product of the curable composition during cutting. This is not preferable because the raising accuracy may be inferior. Further, even if it is hydrophilic, it can be used if the cured product of the curable composition does not swell or partially dissolve with water.
- the addition amount of ( ⁇ ) is preferably 5 to 95 parts by mass in 100 parts by mass of ( ⁇ ) and ( ⁇ ) in total. In particular, 10 to 80 parts by mass is preferable. If it is 5 parts by mass or more, there is a fear that the initial adhesiveness will be lowered. If it is 95 parts by mass or less, the peelability can be secured, and the cured product of the curable composition will be peeled in a film form.
- the addition amount of (C) is preferably 0.1 to 20 parts by mass with respect to 100 parts by mass in total of (i) and (ii). More preferably, it is 3-20 mass parts. If the amount is 1 part by mass or more, the effect of promoting the curing can be surely obtained, and if it is 20 parts by mass or less, a sufficient curing rate can be obtained. More preferably, by adding 3 parts by mass or more of (C) as a form, it becomes possible to cure without depending on the amount of light irradiation, and further, the degree of cross-linking of the cured product of the curable composition is increased, resulting in misalignment during cutting, etc. It is more preferable in that it does not cause odor and the peelability is improved.
- organic peroxides include, for example, tertiary butyl peroxide, cumene hydride peroxide, diisopropylbenzene hydride, paramentane hydride, 2 , 5 Dimethylhexane
- ketone peroxides methyl ethyl ketone peroxide, cyclohexanone peroxide, 3, 3, 5-trimethylcyclohexanone peroxide, methylcyclohexanone peroxide, methyl acetoacetate peroxide and A cetyl acetone peroxide etc. are mentioned.
- acetyl chloride isobutyl peroxide, otanoyl peroxide, decanol peroxide, lauryl peroxide, 3, 3, 5-trimethylhexanoyl peroxide, succinic acid
- examples include peroxide, benzoyl peroxide, 2,4-dichlorobenzoyl peroxide, and methanol oil peroxide.
- One or more of these organic peroxides can be used.
- the addition amount of (E) is preferably 0.5 to: LO parts by mass with respect to a total of 100 parts by mass of (A) and (B). More preferably, it is 1-8 mass parts. 0.5 If it is 5 parts by mass or more, the curability can be surely obtained, and if it is 10 parts by mass or less, the adhesiveness and storage stability are not significantly reduced, and the skin irritation is reduced. So preferred.
- the organic peroxide decomposition accelerator is an organic acid metal salt, an organic metal, or the like when a hydrated peroxide or a ketone peroxide is used as the organic peroxide.
- Chelates e.g., cobalt naphthenate, copper naphthenate, manganese naphthenate, cobalt otatenate, copper oxalate, manganese oxalate, copper acetylacetonate, titanium acetylacetonate, manganese acetylethylacetonate, chromium acetylethylacetate Nate, iron acetylacetonate, vanadyl-rucetylacetonate, cobalt acetylacetonate and the like can be used.
- thiourea derivatives such as thiol urea, dibutyl thiourea, ethylene thiourea, tetramethyl thiourea, mercaptobenzoimidazole, benzoyl thiourea, etc. are used. can do.
- organic peroxide such as disilver oxides such as peroxybenzoyl
- organic peroxide decomposition accelerator amines such as N, N-dimethyl-p-toluidine, N, N-jetyl-p-toluidine, N, N-di (2-hydroxyethyl) ⁇ -toluidine, N, N-diisopropanol-p-toluidine, triethylamine, tripropylamine, ethyljetanolamine N, N dimethylaniline, ethylene diamine, triethanolamine, aldehyde monoamine condensation reaction products, and the like can be used.
- organic peroxide such as disilver oxides such as peroxybenzoyl
- organic peroxide decomposition accelerator amines such as N, N-dimethyl-p-toluidine, N, N-jetyl-p-toluidine, N, N-di (2-hydroxyethyl) ⁇ -toluidine, N,
- One or more of these organic peroxide decomposition accelerators can be used.
- the amount of (F) used is preferably 0.1 to: LO parts by mass with respect to a total of 100 parts by mass of (A) and (B). More preferably, it is 0.5 to 8 parts by mass. 0. If it is 1 part by mass or more, it is preferable because the curability is surely obtained, and if it is 10 parts by mass or less, the adhesiveness and storage stability are not significantly lowered. /.
- the (D) polar solvent used in the present invention has a boiling point of preferably 50 to 130 ° C, particularly preferably 60 to 120 ° C.
- a polar solvent having a boiling point within the above range is selected, it is preferable because the cured product after curing can be brought into contact with warm water to exhibit a phenomenon in which the adhesive strength is lowered.
- a polar solvent include water, alcohol, ketone, and ester. According to the inventor's investigation results, water and alcohol are preferably selected.
- Examples of the alcohol include methanol, ethanol, n-propanol, isopropanol, n-butanol, isobutanol, sec-butanol, tert-butanol, n-amyl alcohol, isoamyl alcohol, 2-ethylbutyl alcohol and the like. .
- methanol, ethanol, n-propanol, isopropanol, n -butanol, isobutanol, sec-butanol and tert-butanol having a boiling point of preferably 120 ° C. or less are preferred.
- methanol, ethanol, isopropanol, and n-butanol are more preferred.
- the addition amount of (D) is preferably 0.5 to 20 parts by mass, and more preferably 3 to 20 parts by mass with respect to 100 parts by mass of the total of (A) and (B). 0.5 If it is 5 parts by mass If the amount is 20 parts by mass or less, the cured product of the curable composition that does not have a risk of lowering the initial adhesiveness peels into a film.
- the temperature-responsive polymer is a homopolymer that also has one monomer force and a copolymer that also has Z or two or more monomer forces.
- This): temperature-responsive polymer can be dispersed and swollen in (D): polar solvent to form a mixture, and the mixture can be used together with (A) and (B).
- the temperature-responsive polymer of (G) shrinks at a certain temperature or higher, discharges water, and absorbs water at a temperature lower than this temperature, that is, the lower critical eutectic temperature (from water solubility). Represents a polymer having a temperature that changes to water insoluble). This polymer has been put to practical use as a water absorbent for water, a soil modifier, a drug release base for drug delivery systems, and a cell culture sheet.
- (G) examples include methyl cellulose, hydroxypropyl methyl cellulose, hydroxypropyl cellulose, poly (meth) acrylic acid, polyethylene polyacetate saponified product, polyethylene oxide, polybutymethyl ether, poly Temperature responsive polymers such as (N-alkyl (meth) acrylamide), poly (N-butylpyrrolidone), poly (ethyloxazoline), poly (hydroxypropyl attalylate), and Z or their polyfunctional vinyls.
- -Copolymers that are three-dimensionally crosslinked with a crosslinking agent such as a monomer.
- N-alkyl (meth) A copolymer comprising one or more monomers and a vinyl monomer, which are selected from the group power of acrylamide derivatives, nitrogen-containing cyclic monomers, and vinyl group-containing amino acids is preferred.
- the bull monomer represents a vinyl monomer copolymerizable with an N-alkyl (meth) acrylamide derivative, a nitrogen-containing cyclic monomer, a vinyl group-containing amino acid, or the like.
- N-alkyl (meth) acrylamide derivatives include N-n-propyl (meth) acrylic And N-isopropylacrylamide, N-cyclopropylacrylamide, Nn-ethylacrylamide and the like.
- nitrogen-containing cyclic monomer include N chloropyrrolidine, N acryl piperazine, N acrylomorpholine, 1,4 dimethyl piperazine and the like.
- amino acid containing a bur group examples include N-acryloyl L pyrroline, N acryloyl-L-pyrroline methyl ester, N-methacryloyl L-succinic methyl ester, N-methacloyl L-isoleucine methyl ester, N-methacryloyl-L-glutamic acid methyl ester, N-methacryloyl-L vanillin methyl ester, and the like.
- Examples of copolymerizable butyl monomers include (meth) acrylamide, diacetone attalylamide, (meth) acrylate, (meth) acryl-tolyl, styrene, and butyl acetate; N, N-methylenebis Crosslinkable monomers such as (meth) acrylamide, ethylene glycol di (meth) acrylate, polyethylene glycol di (meth) acrylate, propylene glycol di (meth) acrylate, glycerin tri (meth) acrylate; acrylic acid, methacryl Examples thereof include monocarboxylic acids such as acids; and bull monomers such as dicarboxylic acids such as maleic acid, phthalic acid and itaconic acid.
- (G) includes, among the above, one or more polymers selected from the group consisting of N alkyl (meth) acrylamide derivatives, nitrogen-containing cyclic monomers and vinyl group-containing amino acids, and Z or N- An alkyl (meth) acrylamide derivative, a nitrogen-containing cyclic monomer, and a group power that also has a bull group-containing amino acid power are selected.
- polymers selected from the group consisting of N alkyl (meth) acrylamide derivatives, nitrogen-containing cyclic monomers and vinyl group-containing amino acids, and Z or N- An alkyl (meth) acrylamide derivative, a nitrogen-containing cyclic monomer, and a group power that also has a bull group-containing amino acid power are selected.
- the copolymers consisting of one or more monomers and a bull monomer N-isopropylacrylamide polymer and Z Or the copolymer which consists of a bull monomer is preferable from a temperature-responsive viewpoint.
- a copolymer of an N-alkyl (meth) acrylamide derivative and a butyl monomer having a carboxylic acid group and a butyl monomer copolymerizable therewith is more preferable.
- a method for obtaining a temperature-responsive polymer a method of copolymerizing with a crosslinkable monomer when the monomer exhibiting the temperature responsiveness is superposed, or a temperature response obtained after polymerization.
- a method of irradiating a crosslinkable polymer with an electron beam or the like is not limited to these methods.
- the addition amount of (G) is preferably 0.001 to 10 parts by mass, particularly preferably 0.01 to 1 part by mass with respect to 100 parts by mass of the total amount of (A) and (B). It is. If it is at least 001 parts by mass, releasability can be ensured, and if it is at most 10 parts by mass, there is no risk of lowering the initial adhesiveness.
- the cured product of the curable composition will be peeled into a film.
- (G) As a method for adding (G), there is a method in which (D) is preliminarily dispersed and swollen at room temperature or lower, and the dispersed swollen product is added to a composition containing (A), (B) and (C).
- a method for adding (G) there is a method in which (D) is preliminarily dispersed and swollen at room temperature or lower, and the dispersed swollen product is added to a composition containing (A), (B) and (C).
- the blending ratio of (G): (D) is adjusted at room temperature or lower so that the blending ratio of mass ratio is 1: 0.1 to 1: 1000, and a mixture is obtained.
- a method of adding a composition comprising (B) and (C) is preferred,
- the (H): particulate material used in the present invention is one that does not dissolve in the above (A) to (G).
- organic particles or inorganic particles that are generally used can be used.
- organic particles include polyethylene particles, polypolypropylene particles, bridged polymethyl methacrylate particles, and crosslinked polystyrene particles.
- inorganic particles include ceramic particles such as glass, silica, alumina, and titanium.
- (H) is preferably spherical in view of improving processing accuracy, that is, controlling the film thickness of the adhesive.
- the long and short diameter ratio of the granular material is in the range of 0.8 to 1.
- the organic particles include cross-linked polymethyl methacrylate particles and cross-linked polystyrene particles obtained as monodisperse particles by a known emulsion polymerization method of a methyl methacrylate monomer, a styrene monomer and a cross-linkable monomer.
- the inorganic particles include spherical silica. These particles are preferable because the film thickness of the composition after curing is uniform due to variation in the particle size with less deformation.
- (H) preferably has an average particle diameter of 1 to 300 ⁇ m. In particular, 10 to 200 ⁇ m is preferable. If it is 1 ⁇ m or more, peelability can be secured, and if it is 300 ⁇ m or less, the processing accuracy does not decrease.
- the particle size distribution is preferably as narrow as possible.
- the addition amount of (H) is preferably 0.1 to 20 parts by mass, particularly 0.1 to part by mass with respect to 100 parts by mass of the total of (A) and (B).
- the content is 1 part by mass or more, the thickness of the cured body is almost constant, and when the content is 20 parts by mass or less, there is no fear that the initial adhesiveness is lowered.
- a small amount of a polymerization inhibitor can be used to improve the storage stability.
- polymerization inhibitors include methylno, idroquinone, hydride quinone, 2,2-methylene bis (4-methyl-6 tertiary butyl phenol), catechol nore, hydroquinone monomethylol ether, mono tertiary butyl hydroquinone, 2, 5 diter.
- the amount of these polymerization inhibitors used is preferably 0.001 to 3 parts by mass, more preferably 0.01 to 2 parts by mass with respect to 100 parts by mass in total of (A) and (B). 0. Storage stability is ensured at 001 parts by mass or more, good adhesion is obtained at 3 parts by mass or less, and it does not become uncured.
- the curable composition of the present invention includes various elastomers such as acrylic rubber, urethane rubber, acrylonitrile-butadiene-styrene rubber, inorganic filler, and the like that are generally used within the range not impairing the object of the present invention.
- Additives such as solvents, extenders, reinforcing materials, plasticizers, thickeners, dyes, pigments, flame retardants, silane coupling agents, and surfactants may be used.
- the curable composition can be used as a one-component type or a two-component type.
- (A), (B), (F) and (D) are mixed in advance and (E) is added when used, or (A), (B), ( E) and (D) are premixed and used when (F) And the like.
- mix (A) and (B) into two components add (E) to one solution and add (F) to the other.
- (D) can be added to both liquids or one of them, and the two liquids can be mixed when actually used.
- the temporary fixing method of the present invention is a method in which a member is bonded using a curable composition that is brought into contact with hot water of 30 to 90 ° C to reduce adhesive strength, and the curable composition is cured, Temporarily fix. Thereafter, the temporarily fixed member is processed, and the cured adhesive is removed by immersing the processed member in warm water.
- various members such as optical members without using an organic solvent can be processed with high accuracy.
- the cured body of the curable composition when the cured body of the curable composition is removed, the cured body swells in contact with hot water of 30 to 90 ° C, and forms a film-like member. By making it possible to recover from the waste, excellent workability can be obtained.
- the temperature of the hot water is 30 to 90 ° C, more preferably 40 to 90 ° C.
- the cured adhesive body swells in a short time and the residual strain stress generated when the curable composition is cured is released. Therefore, the adhesive strength is reduced, and in addition, the vapor pressure of (D) acts as a peeling force between the member and the cured product of the curable composition, and the adhesive cured product can be removed in the form of an adherend.
- the method of contacting the cured body with warm water is recommended because the method of immersing the entire joined body in warm water is simple.
- a member made of a material capable of transmitting ultraviolet rays is preferred when used as an ultraviolet ray curable adhesive, in which the material of the member used for temporary fixing is not particularly limited.
- examples of such a material include a crystal member, a glass member, and a plastic member. Therefore, the temporary fixing method of the present invention can be applied to the processing of crystal resonators, glass lenses, plastic lenses, and optical disks.
- silica, alumina, carbonic acid represented by epoxy resin for sealing etc.
- examples include reinforced plastics filled with inorganic fillers such as calcium and pigment, substrate materials using them, glass reinforced plastics (FRP), ceramics, stainless steel, aluminum, metals such as ferrite, and opaque materials such as silicon.
- Temporary fixing of a member using the curable composition of the present invention may be performed as follows.
- a suitable amount of adhesive is applied to the adhesive surface of one member to be fixed or the supporting substrate, and then the other member is overlapped, or temporarily fixed in advance. After a large number of members are laminated, the adhesive is applied by, for example, infiltrating the adhesive into the gap, and then the member is irradiated with visible light or ultraviolet light to cure the photocurable adhesive. The members are temporarily fixed.
- the temporarily fixed member is then processed into a desired shape by cutting, polishing ij, polishing, drilling, and the like, and the member is immersed in water, preferably warm water, thereby hardening the adhesive. Can be peeled off from the member.
- a base material is used when the member is temporarily bonded and fixed, but it is preferable that the cured body remains on the base material when the cured body is removed.
- the base material is made of a material that gives an adhesive strength higher than the adhesive strength between the adhesive and the member to be processed, or even the same material has different surface properties. As a result, the above situation is achieved. Select what you can do.
- the surface roughness (RMax) of the surface with respect to the curable composition of the substrate is 10 ⁇ m to 50 ⁇ m. Particularly preferred is 15 to 45 / ⁇ ⁇ .
- the thickness of the surface is preferably 10 ⁇ m or more, particularly preferably 15 to 50 ⁇ m larger than the surface roughness (RMax) of the surface of the member with respect to the curable composition. If the surface roughness of the surface of the member to be processed with respect to the curable composition is 10 ⁇ m or more, sufficient adhesive strength can be obtained after the curable composition is cured, and 50 m or less. Then, when the cured body is brought into contact with warm water, the cured body can be peeled off within a practical time.
- the cured body is made warm.
- the member-side force is surely peeled off, the cured body is temporarily kept on the substrate side, and the cured material of the substrate force-curable composition can be reliably peeled off after a further time has elapsed. .
- the effect can be obtained. .
- GR-600 average particle size 25 ⁇ m cross-linked polymethyl methacrylate particles (art pearl GR-600 manufactured by Negami Kogyo Co., Ltd.)
- GM-5047 average particle size 10 ⁇ m cross-linked polybutyl methacrylate particles (Ganz Pearl GM-1005-10 manufactured by Ganz Kasei)
- SGP-150C average particle size 55 ⁇ m cross-linked polystyrene particles (Kemisnow SGP-150C, manufactured by Sokeni Sogakusha)
- SGP-140C average particle size 42 ⁇ m cross-linked polystyrene particles (Kemisnow SGP-140C, manufactured by Sokeni Sogakusha)
- SGP-100C Cross-linked polystyrene particles with an average particle size of 25 ⁇ m (Chemisnow SGP-100C, manufactured by Sokeni Sogakusha)
- 2100JPD Average particle size 147 ⁇ m polyethylene particles (Hi-Zex2100JPD manufactured by Mitsui Chemicals, Inc. )
- PE—130 Average particle size 12.5 ⁇ m polyethylene particles (Caliant Japan CAERI DUST PE-130)
- PP-6071 Average particle size 20 ⁇ m polypropylene particles (CAERI DUST PP-6071 manufactured by Clariant Japan)
- LS -L100 Average particle size 10 Spherical silica particles (LS-L 100 manufactured by Tokama)
- MTEGMA Methoxytetraethylene glycol monometatalylate (Shin Nakamura ⁇ Gakusha Co., Ltd. ⁇ Ester M—90G)
- Trimethylolpropane tritalylate (KAYARAD ⁇ ⁇ manufactured by Nippon Kayaku Co., Ltd.)
- TE-2000 (1, 2-polybutadiene terminated urethane metatalylate, hereinafter abbreviated as “TE-2000”), 20 parts by mass, dicyclotentar tertialite Rate (KAYARAD R-684 manufactured by Nippon Kayaku Co., Ltd., hereinafter abbreviated as “R-684”) 15 parts by mass
- B 2- (1,2-cyclohexacarboxyimido as monofunctional (meth) atalylate
- Ethyl attalylate (Aronix M-140 manufactured by Toa Gosei Co., Ltd., hereinafter abbreviated as “M-140”) 40 parts by mass, phenol ethylene oxide 2 mol modified attalylatenicks M-101A (hereinafter abbreviated as “M-101A”) 25 parts by mass 100 parts by mass in total
- C Benzyldimethyl ketal (hereinafter abbreviated as “BD
- the major / minor diameter ratio of (H) used was determined by the following evaluation method. The results are shown in Table 1-1. Using the obtained curable composition, the tensile shear bond strength was measured and the peel test was conducted by the following evaluation method. The results are shown in Table 1-2.
- the ratio of major axis to minor axis As an index to indicate the sphericity of (H), an image analyzer (Nippon API-TASS) was used to measure the particle image that was close-up 20,000 times with a scanning electron microscope (“JS M-T200” manufactured by JEOL Ltd.). The average of the ratio of the major axis to the minor axis of 100 particles arbitrarily selected was obtained.
- Tensile shear bond strength Measured according to JIS K 6850. Specifically, a Pyrex (heat-resistant glass, registered trademark, the same applies hereinafter) glass plate (25 mm X 25 mm X thickness 2. Omm) was used as the adherend, and the bonding site was 8 mm in diameter. Two Pyrex glass plates were bonded together with the curable composition. After that, it was cured with a curing device using an electrodeless discharge lamp (manufactured by Fusion) under the condition of an integrated light quantity of 2000 mjZcm 2 at a wavelength of 365 nm to prepare a tensile shear bond strength test piece. The prepared specimens were measured for tensile shear bond strength at a tensile rate of lOmmZmin using a universal testing machine in an environment of a temperature of 23 ° C and a humidity of 50%.
- a Pyrex (heat-resistant glass, registered trademark, the same applies hereinafter) glass plate 25 mm X 25 mm
- Peel test The same conditions as above except that the curable composition was applied to the above-mentioned Noirex glass plate and bonded to a blue plate glass plate (150 mm X 150 mm X thickness 1.7 mm) as a support.
- the curable composition prepared in 1 was cured to prepare a peel test specimen.
- the obtained specimen was immersed in warm water (80 ° C), the time for the Pyrex glass plate to peel was measured, and the peeled state was also observed.
- a curable composition was prepared in the same manner as in Example 1 except that the raw materials of the types shown in Tables 1-2 and 1-3 were used in the compositions shown in Tables 1-2 and 1-3.
- the cured product of the obtained curable composition was subjected to measurement of tensile shear adhesive strength and peel test in the same manner as in Example 1-1. The results are shown in Tables 12 and 13.
- Rigid film The cured product is peeled off from the glass surface without any adhesive residue.
- a curable composition was prepared in the same manner as in Example 1-1 except that the raw materials shown in Table 14 were used in the composition shown in Table 14.
- the cured body of the obtained curable composition was subjected to measurement of tensile shear adhesive strength and peeling test in the same manner as in Example 1-1. The results are shown in Table 1-4.
- Example 1 The curable composition of 1 is applied to a 200 mm square polyethylene terephthalate (hereinafter abbreviated as PET) film, and a 150 mm X 150 mm X 2 mm Pyrex glass plate is laminated, so that the pressure is 32 kgZcm 2 A constant load was applied to the glass plate with a weight for 10 minutes, and then adhesive and cured in the same manner as in Example 1-1. Thereafter, by peeling off the PET film, Fig 1-1 n the results of the measurement of the film thickness of 16 divided any 16 forces plants curable composition cured on a glass plate of 0.99 mm square at a micrometer Shown in As a result, almost constant The fact that the film thickness was obtained proved powerful.
- PET polyethylene terephthalate
- Example 1-1 a 150 mm X 150 mm X 2 mm Pyrex glass plate and the blue plate glass plate used in Example 1-1 were used as dummy glass plates using the curable yarn composition of Example 1-1.
- the adhesive was cured. Only the Pyrex glass plate portion of this adhesion test specimen was cut into 10 mm squares using a dicing machine. The Pyrex glass plate did not fall off during cutting, indicating good workability.
- the adhesion test specimen in which only the Nolex glass plate was cut, was immersed in warm water at 80 ° C, it peeled off after 60 minutes.
- Hot melt adhesive (Nikkasei Energy Adfix A) was heated to 90 ° C and dissolved to bond the 150 mm X 150 mm X 2 mm Pyrex glass plate to the blue glass plate used in Example 11 I let you. Only the Pyrex glass plate portion of this adhesion test specimen was cut into 10 mm squares using a dicing machine. The Pyrex glass plate did not fall off during cutting, indicating good workability. The test piece was immersed in an N-methylbidolylone solution for 1 day, and the cut test piece was collected, and 10 pieces of the cut test pieces peeled off in the same manner as in Example 1-18 were taken out. Each piece of the surface temporarily fixed with a hot-melt adhesive was observed using an optical microscope, the maximum width of the portion where the glass plate was missing was measured, and the average value and standard deviation were obtained. The results are shown in Table 1-5.
- a Pyrex glass plate of 150 mm X 150 mm X 2 mm was bonded using UV curable PET adhesive tape. Only the Nolex glass plate part of this adhesion test specimen was cut into 10 mm squares using a dicing machine. The adhesive strength was reduced by irradiating the adhesive tape portion of the test piece with ultraviolet light, and the cut test piece was collected. Ten pieces of the cut specimens from which the cut specimens were peeled in the same manner as in Example 1-18 were taken out, and each piece on the back surface (the surface temporarily fixed with adhesive tape) of the cut specimens was taken using an optical microscope. The maximum width of the portion where the glass plate was missing was measured, and the average value and standard deviation were obtained. The results are shown in Table 15.
- TE-2000 (1, 2-polybutadiene terminated urethane metatalylate, hereinafter abbreviated as “TE-2000”), 20 parts by mass, dicyclotentar diatalylate (KAYARAD R-684 manufactured by Nippon Kayaku Co., Ltd., hereinafter abbreviated as “R-684”) 15 parts by mass
- R-684 dicyclotentar diatalylate
- B 2- (1,2-cyclohexacarboxyimide) as monofunctional (meth) acrylate
- Ethyl acrylate (Aronix M-140 manufactured by Toa Gosei Co., Ltd., hereinafter abbreviated as “M-140”) 40 parts by mass, 2 mol of phenol ethylene oxide M-101A ”) 25 parts by mass Total 100 parts by mass
- E Tamennoide mouth peroxide (Nippon Yushi Co., Ltd.
- CHP Park Mill H-80
- IPA Isopropyl alcohol as polar solvent
- H Cross-linked with an average particle size of 50 / zm as a particulate material
- Polymethyl methacrylate particles Negami Kogyo Art Pearl GR-200 or less “abbreviated as” GR-200 ”
- MDP 2,2-methylenebis (4-methyl-6 tertiary butylphenol) as a polymerization inhibitor
- a curable composition was prepared. Using the obtained curable composition, the tensile shear bond strength was measured and the peel test was performed by the following evaluation method. The results are shown in Table 2-1.
- Curing time After obtaining the curable composition, the time until it became a hardened body was measured under the condition of a temperature of 23 ° C.
- Tensile shear bond strength Measured according to JIS K 6850. Specifically, an iron test piece (SPCC, 100 X 25 X 1.6 mm) was used as the material to be bonded, and the bonded part was 25 mm wide x 12.5 mm wide. An iron specimen was adhered. Thereafter, it was cured for 1 day under the condition of a temperature of 23 ° C. to prepare a tensile shear bond strength test piece. The prepared specimens were measured for tensile shear adhesive strength at a tensile speed of lOmmZmin using a universal testing machine.
- Peel test The same conditions as above except that the curable composition was applied to the above-mentioned Neurex glass plate and bonded to a blue plate glass plate (150 mm X 150 mm X thickness 1.7 mm) as a support.
- the curable composition prepared in 1 was cured to prepare a peel test specimen.
- the obtained specimen was immersed in warm water (80 ° C), the time for the Pyrex glass plate to peel was measured, and the peeled state was also observed.
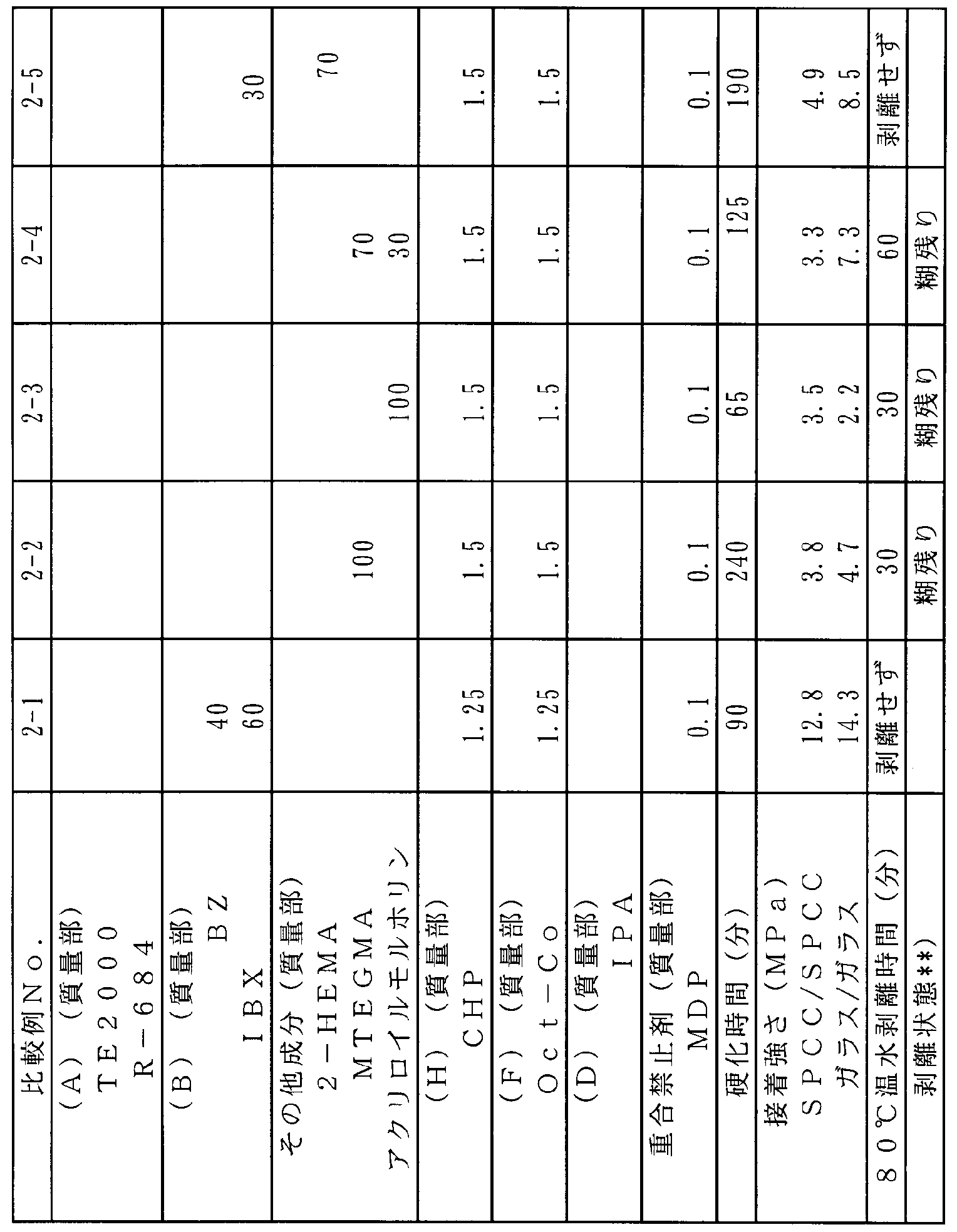
- a curable composition was prepared in the same manner as in Example 2-1, except that the raw materials of the type shown in Table 2-1 were used in the composition shown in Table 2-1.
- the obtained curable composition was subjected to measurement of tensile shear bond strength and peeling test in the same manner as in Example 2-1. The results are shown in Table 2-1.
- Example 2-2 except that the raw materials of the type shown in Table 2-2 were used in the composition shown in Table 2-2. 1 A curable composition was prepared in the same manner. About the obtained curable composition, Example 2 ZZI
- Example 2-1 Using the curable composition prepared in Example 1, except that the ambient temperature for curing was changed to -10 ° C, 0 ° C, 10 ° C, and 40 ° C, in the same manner as in Example 2-1. The curing time, tensile shear bond strength measurement and peel test were performed. The results are shown in Table 2-3. The result is It cures regardless of temperature and also exhibits releasability.
- Example 2-1 Using the curable composition prepared in Example 2-1 as a dummy glass plate, the Pyrex glass plate of 150 mm X 150 mm X 2 mm and the blue plate glass plate used in Example 2-1 were used in the same manner as in Example 2-1.
- the adhesive was cured. Only the Pyrex glass plate part of this adhesion test specimen was cut into 10 mm squares using a dicing machine. The Pyrex glass plate did not fall off during cutting, indicating good workability. Adhesion with only the Pyrex glass plate cut The test specimen was immersed in warm water at 80 ° C and peeled off after 60 minutes.
- G Thermogel R-60 (manufactured by Kojin Co., Ltd., N-isopropylacrylamide Z acrylic acid copolymer, hereinafter abbreviated as "R-60”) as a temperature-responsive polymer, 1 part by mass
- D As a polar solvent, isopropyl alcohol (hereinafter abbreviated as “IPA”) Dissolved and mixed in 50 parts by mass with stirring at 23 ° C. for 24 hours to a concentration of 0.5% (gZg) (G) Got.
- IPA isopropyl alcohol
- TE-2000 (1, 2-polybutadiene terminated urethane metatalylate, hereinafter abbreviated as “TE-2000”), 20 parts by mass, dicyclotentar diatalylate (KAYARAD R-684, manufactured by Nippon Kayaku Co., Ltd., hereinafter abbreviated as “R-684”) 15 parts by mass
- B 2- (1,2-cyclohexacarboxyimide) as monofunctional (meth) acrylate Ethyl attalylate (TO-1429 manufactured by Toa Gosei Co., Ltd., hereinafter abbreviated as “TO-1429”) 40 parts by mass, 2 mol of phenol ethylene oxide (Abbreviated as “101 ⁇ ”) 25 parts by mass total 100 parts by mass
- Example 3-1 was used except that the dispersion swollen material (1) prepared in Example 3-1 was used and the raw materials of the type shown in Table 3-1 were used in the composition shown in Table 3-1 A curable composition was prepared. The obtained curable composition was subjected to measurement of tensile shear adhesive strength and a peel test in the same manner as in Example 3-1. The results are shown in Table 3-1.
- a curable composition was prepared in the same manner as in Example 3-1, except that the raw materials of the type shown in Table 3-2 were used in the composition shown in Table 3-2.
- the obtained curable composition was subjected to measurement of tensile shear bond strength and peel test in the same manner as in Example 3-1. The results are shown in Table 3-2.
- Example 3-1 Using the curable composition of Example 3-1, a Pyrex glass plate of 150mm x 150mm x 2mm and the blue plate glass plate used in Example 3-1 were bonded in the same manner as in Example 3-1. Cured. Only the Pyrex glass plate portion of this adhesion test specimen was cut into 10 mm squares using a dicing machine. The Pyrex glass plate did not fall off during cutting, indicating good workability. The test specimen, which had been cut only on the Pyrex glass plate, was immersed in warm water at 80 ° C and peeled off after 60 minutes.
- TE-2000 (1, 2-polybutadiene terminated urethane metatalylate, hereinafter abbreviated as “TE-2000”), 20 parts by mass, dicyclotentar diatalylate (KAYARAD R-684, manufactured by Nippon Kayaku Co., Ltd., hereinafter abbreviated as “R-684”) 15 parts by mass
- B 2- (1,2-cyclohexacarboxyimide) as monofunctional (meth) acrylate Ethyl attalylate (TO-1429 manufactured by Toa Gosei Co., Ltd., hereinafter abbreviated as “TO-1429”) 40 parts by mass, 2 mol of phenol ethylene oxide (Abbreviated as “101 ⁇ ”) 25 parts by mass total 100 parts by mass
- Peel test Evaluation was performed in the same manner as described in Example 1-1.
- the adhesive coating surfaces of the Pyrex glass plate and the blue plate glass plate were previously blasted to have a surface roughness (RMax) of 1 m.
- the surface roughness (RMax) was measured with a stylus type surface roughness measuring instrument based on the surface roughness measuring method of JIS B 0651.
- a curable composition was prepared in the same manner as in Example 4-1, except that the raw materials of the types shown in Tables 4-1 and 4-2 were used in the compositions shown in Tables 4-1 and 42.
- the obtained curable composition was subjected to measurement of tensile shear adhesive strength and peeling test in the same manner as in Example 4-1. The results are shown in Tables 41 and 42.
- a curable composition was prepared in the same manner as in Example 41 except that the raw materials of the type shown in Table 43 were used in the composition shown in Table 43.
- the obtained curable composition was subjected to measurement of tensile shear bond strength and peeling test in the same manner as in Example 4-1. The results are shown in Table 43.
- Substrate The cured product remains on the substrate glass. ⁇ 0113 Using the compositions of Example 4-1 and Comparative Example 1-5, respectively, the accumulated light quantity at a wavelength of 365 nm was 500, 1000, 2000, 4000 mjZcm 2 using a curing device (made by Fusion) using an electrodeless discharge lamp. Then, the curable composition was cured to prepare a peel test specimen and a tensile shear bond strength test piece. Otherwise, the measurement of the tensile shear bond strength and the peel test were performed in the same manner as in Example 4-1. The results are shown in Table 45.
- Example 4-7 Using the curable compositions of Examples 4-1 and 4-7, a peel test specimen was prepared in the same manner as in Example 4-1, and the temperature of the hot water was 40 ° C, 50 ° C, 60, 70 ° C. A peel test was conducted instead of That Table 4 _ 6
- Example 4-1 Using the curable composition of Example 4-1, a Pyrex glass plate of 150 mm X 150 mm X 2 mm and the blue plate glass plate used in Example 4-1 were used as a dummy glass plate. I let you. Only the Pyrex glass plate portion of this adhesion test specimen was cut into 10 mm squares using a dicing machine. The Pyrex glass plate did not fall off during cutting, indicating good workability. When the adhesion test specimen, in which only the Nolex glass plate was cut, was immersed in warm water at 80 ° C, it peeled off after 60 minutes.
- the curable composition of Example 4-1 swelled even when immersed in water at 25 ° C, as compared to the curable composition using hydrophilic (meth) acrylate as in the comparative example. Because it is low, it is less susceptible to cutting water used during machining.
- the curable composition of the present invention has a room temperature curable property and a light curable property because of its composition, and is also cured by visible light or ultraviolet light. Since the cured body can exhibit high adhesion strength without being affected by cutting water or the like, it is possible to easily obtain a member excellent in dimensional accuracy that is less likely to be displaced during processing of the member. Furthermore, the adhesive strength is lowered by contact with hot water, and the bonding force between the members or between the member and the jig is reduced. Therefore, the member can be easily collected. That is, it is extremely useful industrially as an adhesive for temporarily fixing optical lenses, prisms, arrays, silicon wafers, semiconductor mounting parts, and the like.
- the method for temporarily fixing the member of the present invention uses the above-mentioned characteristic curable composition! It is easy to work because it can be recovered from the parts.
- the Japanese patent application 2005- 194752 filed on July 4, 2005, the Japanese patent application 2005-224101 filed on August 2, 2005, and the application filed August 22, 2005 The entire contents of the specification, claims, drawings and abstract of Japanese Patent Application 2005-239987 and Japanese Patent Application 2005-278984 filed on September 27, 2005 are incorporated herein by reference. The disclosure of this specification is incorporated.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Adhesives Or Adhesive Processes (AREA)
- Polymerisation Methods In General (AREA)
- Macromonomer-Based Addition Polymer (AREA)
Abstract
Description

















Claims
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/916,123 US8313604B2 (en) | 2005-07-04 | 2006-07-03 | Curable composition and temporary fixation method of member using it |
| EP06780751A EP1900761B1 (en) | 2005-07-04 | 2006-07-03 | Curable composition and method for temporal fixation of structural member using the same |
| CN2006800242184A CN101213225B (zh) | 2005-07-04 | 2006-07-03 | 固化性组合物以及使用该组合物的构件的暂时固定方法 |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005-194752 | 2005-07-04 | ||
| JP2005194752A JP4916681B2 (ja) | 2005-07-04 | 2005-07-04 | 仮固定方法用光硬化性接着剤及びそれを用いる部材の仮固定方法 |
| JP2005224101A JP5164316B2 (ja) | 2005-08-02 | 2005-08-02 | 接着剤及びそれを用いる部材の仮固定方法 |
| JP2005-224101 | 2005-08-02 | ||
| JP2005-239987 | 2005-08-22 | ||
| JP2005239987A JP5002139B2 (ja) | 2005-08-22 | 2005-08-22 | 組成物及びそれを用いる部材の仮固定方法 |
| JP2005278984 | 2005-09-27 | ||
| JP2005-278984 | 2005-09-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007004620A1 true WO2007004620A1 (ja) | 2007-01-11 |
Family
ID=37604481
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/313247 WO2007004620A1 (ja) | 2005-07-04 | 2006-07-03 | 硬化性組成物及びそれを用いる部材の仮固定方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8313604B2 (ja) |
| EP (2) | EP2383303A1 (ja) |
| KR (1) | KR100995257B1 (ja) |
| MY (1) | MY154745A (ja) |
| SG (1) | SG163531A1 (ja) |
| TW (1) | TWI383031B (ja) |
| WO (1) | WO2007004620A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007016141A (ja) * | 2005-07-08 | 2007-01-25 | Denki Kagaku Kogyo Kk | 組成物及びそれを用いる部材の仮固定方法 |
| JP2008214593A (ja) * | 2007-03-07 | 2008-09-18 | Denki Kagaku Kogyo Kk | 樹脂組成物及びそれを用いる被加工部材の仮固定方法と表面保護方法 |
| EP2050799A1 (en) * | 2006-08-10 | 2009-04-22 | Denki Kagaku Kogyo Kabushiki Kaisha | Adhesive composition and method for temporarily fixing member by using the same |
| WO2010010900A1 (ja) * | 2008-07-22 | 2010-01-28 | 電気化学工業株式会社 | 部材の仮固定・剥離方法及びそれに好適な仮固定用接着剤 |
| WO2013039226A1 (ja) * | 2011-09-14 | 2013-03-21 | 電気化学工業株式会社 | 組成物及びそれを用いた部材の仮固定方法 |
| WO2014128991A1 (ja) * | 2013-02-20 | 2014-08-28 | 株式会社オートネットワーク技術研究所 | 光硬化性材料、及びその硬化物 |
| WO2014192941A1 (ja) * | 2013-05-30 | 2014-12-04 | 電気化学工業株式会社 | 硬質基板積層体および硬質基板積層体の製造方法 |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080289750A1 (en) | 2006-01-13 | 2008-11-27 | Denki Kagaku Kogyo Kabushiki Kaisha | Curable Resin Composition, Surface Protection Method, Temporary Fixation Method, and Separation Method |
| US8492454B2 (en) | 2009-10-05 | 2013-07-23 | Creative Nail Design, Inc. | Removable color layer for artificial nail coatings and methods therefore |
| US8541482B2 (en) | 2009-10-05 | 2013-09-24 | Creative Nail Design, Inc. | Removable multilayer nail coating system and methods therefore |
| US8263677B2 (en) * | 2009-09-08 | 2012-09-11 | Creative Nail Design, Inc. | Removable color gel basecoat for artificial nail coatings and methods therefore |
| WO2011046120A1 (ja) * | 2009-10-14 | 2011-04-21 | 電気化学工業株式会社 | 樹脂組成物及び接着剤 |
| US9227385B2 (en) * | 2010-11-19 | 2016-01-05 | Denka Company Limited | Method for processing transluscent rigid substrate laminate and method for manufacturing plate shaped product |
| JP5831887B2 (ja) | 2010-11-30 | 2015-12-09 | デンカ株式会社 | 透光性硬質基板積層体の加工方法及びこれを使用した板状製品の製造方法 |
| US9416300B2 (en) | 2011-01-16 | 2016-08-16 | Simpson Strong-Tie Company, Inc. | Low temperature curable adhesive compositions |
| CN103501755A (zh) | 2011-03-07 | 2014-01-08 | 创意指甲设计公司 | 用于可uv固化的化妆品指甲涂层的组合物和方法 |
| CN102898958B (zh) * | 2011-07-25 | 2016-11-02 | 汉高股份有限公司 | 一种粘合剂组合物 |
| EP3357518B1 (en) | 2011-10-03 | 2020-12-02 | Hyalex Orthopaedics, Inc. | Polymeric adhesive for anchoring compliant materials to another surface |
| US10532020B2 (en) | 2012-08-22 | 2020-01-14 | Revlon Consumer Products Corporation | Nail coatings having enhanced adhesion |
| JP2014070191A (ja) * | 2012-09-28 | 2014-04-21 | Fujifilm Corp | 半導体装置製造用仮接着剤、並びに、それを用いた接着性支持体、及び、半導体装置の製造方法。 |
| JP5909460B2 (ja) * | 2012-09-28 | 2016-04-26 | 富士フイルム株式会社 | 半導体装置製造用仮接着剤、並びに、それを用いた接着性支持体、及び、半導体装置の製造方法。 |
| WO2014111292A1 (en) | 2013-01-18 | 2014-07-24 | Basf Se | Acrylic dispersion-based coating compositions |
| US9713585B2 (en) | 2013-04-22 | 2017-07-25 | Creative Nail Design, Inc. | Nail coatings having enhanced adhesion |
| CN105555899B (zh) | 2013-08-26 | 2019-07-16 | 国立研究开发法人科学技术振兴机构 | 一种粘合剂 |
| KR101891729B1 (ko) * | 2015-01-20 | 2018-08-24 | 주식회사 엘지화학 | 경화성 조성물 |
| CN109415473B (zh) * | 2016-07-01 | 2021-12-10 | 电化株式会社 | 组合物 |
| KR102494902B1 (ko) * | 2017-02-02 | 2023-02-01 | 쇼와덴코머티리얼즈가부시끼가이샤 | 전자 부품의 제조 방법, 가보호용 수지 조성물 및 가보호용 수지 필름 |
| US20200324018A1 (en) * | 2017-10-23 | 2020-10-15 | Endologix, Inc. | Endoluminal device and polymer |
| US11739172B2 (en) | 2020-12-17 | 2023-08-29 | 3M Innovative Properties Company | Composition including monomer with a carboxylic acid group, monomer with a hydroxyl group, and crosslinker and related articles and methods |
| CN113789129A (zh) * | 2021-07-27 | 2021-12-14 | 佳化化学科技发展(上海)有限公司 | 一种水凝胶型临时粘合剂及其制备方法和应用 |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0423875A (ja) * | 1990-05-19 | 1992-01-28 | Three Bond Co Ltd | 仮固定用接着剤組成物 |
| US5240989A (en) | 1987-12-11 | 1993-08-31 | Avery Dennison Corporation | Removable pressure-sensitive adhesive compositions comprising acrylic based emulsion polymers |
| JPH06116534A (ja) | 1992-10-05 | 1994-04-26 | Three Bond Co Ltd | 接着剤組成物 |
| JPH07228639A (ja) * | 1994-02-18 | 1995-08-29 | Nippon Kayaku Co Ltd | 温度応答型ハイドロゲル |
| JPH08277313A (ja) * | 1995-02-09 | 1996-10-22 | Denki Kagaku Kogyo Kk | 常温硬化型アクリル系土木建築用補修材及びそれを使用した補修方法 |
| JPH1171553A (ja) | 1997-08-28 | 1999-03-16 | Toagosei Co Ltd | 仮固定用光硬化性組成物及び物品の製造方法 |
| JP2001226641A (ja) | 2000-02-17 | 2001-08-21 | Aader:Kk | 仮固定用接着剤組成物及びその使用方法 |
| JP2002121230A (ja) * | 2000-10-19 | 2002-04-23 | National Institute Of Advanced Industrial & Technology | 高温低粘度低温高粘度型感温性高分子材料 |
| JP2002201229A (ja) * | 2000-11-02 | 2002-07-19 | Mitsubishi Chemicals Corp | 光硬化性樹脂組成物、低複屈折光学部材及びその製造方法 |
| JP2002322214A (ja) * | 2001-04-24 | 2002-11-08 | Central Glass Co Ltd | 反応性高分子化合物 |
| US6734249B1 (en) | 2000-06-14 | 2004-05-11 | Texas Research International, Inc. | Two-part adhesive with (poly)(meth)acrylate in part A and N,N-disubstituted aromatic amine and di(meth)acrylate in part B |
| JP2005170920A (ja) * | 2003-12-10 | 2005-06-30 | Shiyoufuu:Kk | 新規重合開始剤を含む歯科用組成物 |
| EP1860128A1 (en) | 2005-03-18 | 2007-11-28 | Denki Kagaku Kogyo Kabushiki Kaisha | Adherent composition and method of temporarily fixing member therewith |
Family Cites Families (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3745653A (en) * | 1971-12-06 | 1973-07-17 | M Cohl | Method of orthodontia |
| US3928113A (en) * | 1973-06-14 | 1975-12-23 | Clairol Inc | Method for coating human nails |
| JPS521958B2 (ja) * | 1973-08-15 | 1977-01-19 | ||
| US4575539A (en) * | 1985-06-03 | 1986-03-11 | E. R. Squibb & Sons, Inc. | Drug delivery systems including novel interpenetrating polymer networks and method |
| JPH01207371A (ja) | 1988-02-15 | 1989-08-21 | Seiko Epson Corp | 小型防水装置用接着剤 |
| JP3158731B2 (ja) | 1992-10-05 | 2001-04-23 | 株式会社スリーボンド | 接着剤組成物 |
| JP3107471B2 (ja) | 1992-12-17 | 2000-11-06 | 三井化学株式会社 | 密着性に優れた光硬化型水性樹脂組成物 |
| JPH06346040A (ja) * | 1993-06-08 | 1994-12-20 | Dainippon Ink & Chem Inc | 着色された感圧接着剤層およびそれを使用した感圧接着テープまたはシート |
| JP2865534B2 (ja) | 1993-09-22 | 1999-03-08 | 積水化学工業株式会社 | 接着性テープおよび粘着性テープ |
| JPH07157531A (ja) | 1993-12-09 | 1995-06-20 | Daicel Chem Ind Ltd | 水系ウレタンアクリレートの製造方法 |
| JPH07330835A (ja) | 1994-06-10 | 1995-12-19 | Toagosei Co Ltd | 低粘度速硬化性(メタ)アクリレート組成物 |
| JP3514601B2 (ja) | 1996-03-06 | 2004-03-31 | 電気化学工業株式会社 | 含浸用組成物、並びに、レジンコンクリート用プライマー組成物、その塗布方法及びそれを使用した複合躯体 |
| EP0802455B1 (en) * | 1996-04-15 | 2002-10-16 | Teijin Seiki Co., Ltd. | Use of a photocurable resin composition for the production of a stereolithographed object |
| JPH10130309A (ja) | 1996-10-25 | 1998-05-19 | Toagosei Co Ltd | 仮固定用光硬化性組成物及び物品の製造方法 |
| JP3634592B2 (ja) | 1997-01-08 | 2005-03-30 | セイコーエプソン株式会社 | 接着固定物、電子機器および時計 |
| JP3694153B2 (ja) | 1997-07-23 | 2005-09-14 | 尾池工業株式会社 | 防眩性ハードコートフィルム |
| JP2973991B2 (ja) | 1997-12-02 | 1999-11-08 | 株式会社スリーボンド | 小物品の加工方法 |
| JPH11180814A (ja) | 1997-12-24 | 1999-07-06 | Gc:Kk | 歯質接着剤セット |
| AU756369B2 (en) * | 1998-02-19 | 2003-01-09 | Britesmile Development, Inc. | Antimicrobial denture adhesive composition |
| JPH11279242A (ja) | 1998-03-27 | 1999-10-12 | Arakawa Chem Ind Co Ltd | 水性活性エネルギー線硬化性樹脂組成物およびその製造方法 |
| US6265061B1 (en) | 1998-05-04 | 2001-07-24 | 3M Innovative Properties Company | Retroflective articles including a cured ceramer composite coating having abrasion and stain resistant characteristics |
| JP2000038547A (ja) | 1998-07-24 | 2000-02-08 | Mitsubishi Rayon Co Ltd | 光硬化型接着剤組成物およびそれを用いた光学部材 |
| US6326417B1 (en) * | 1999-10-21 | 2001-12-04 | Jeneric/Pentron Incorporated | Anti-microbial dental compositions and method |
| JP2001172336A (ja) | 1999-12-16 | 2001-06-26 | Toagosei Co Ltd | 活性エネルギー線硬化型樹脂組成物 |
| JP4253977B2 (ja) | 1999-12-27 | 2009-04-15 | 東亞合成株式会社 | 活性エネルギー線硬化型組成物 |
| JP4011811B2 (ja) * | 2000-01-14 | 2007-11-21 | Jsr株式会社 | 光硬化性樹脂組成物及び光学部材 |
| EP1299770A2 (en) * | 2000-06-09 | 2003-04-09 | Dsm N.V. | Resin composition and three-dimensional object |
| JP2002060442A (ja) | 2000-08-18 | 2002-02-26 | Nippon Synthetic Chem Ind Co Ltd:The | 紫外線硬化型樹脂組成物及びその用途 |
| DE60143796D1 (de) * | 2000-11-02 | 2011-02-17 | Kuraray Co | Füllungen und dentalmaterialkomposite diese füllungen enthaltend. |
| JP2002173516A (ja) | 2000-12-05 | 2002-06-21 | Mitsubishi Chemicals Corp | 活性エネルギー線硬化型水性エマルジョン組成物 |
| JP2002338900A (ja) | 2001-05-11 | 2002-11-27 | Shin Etsu Polymer Co Ltd | 紫外線硬化型接着剤、接着方法及びそれから製造される成形品 |
| JP2002348534A (ja) | 2001-05-25 | 2002-12-04 | Nippon Arc Co Ltd | ハードコート組成物およびハードコート製品 |
| JP2003128714A (ja) | 2001-10-26 | 2003-05-08 | Arakawa Chem Ind Co Ltd | アクリル系部分重合体組成物の製造方法、当該製造方法により得られるアクリル系部分重合体組成物および当該組成物を紫外線重合させてなる粘着剤組成物 |
| JP2003155455A (ja) | 2001-11-19 | 2003-05-30 | Nippon Synthetic Chem Ind Co Ltd:The | 活性エネルギー線硬化型粘着剤組成物 |
| JP4326747B2 (ja) | 2002-04-23 | 2009-09-09 | 株式会社 アーデル | 仮固定用接着剤を用いる加工方法、及び仮固定用接着剤の剥離方法 |
| JP4404183B2 (ja) | 2002-10-23 | 2010-01-27 | 日本合成化学工業株式会社 | 活性エネルギー線硬化型粘着剤組成物 |
| JP4266623B2 (ja) | 2002-11-29 | 2009-05-20 | リンテック株式会社 | ハードコートフィルム |
| US7223826B2 (en) * | 2003-01-30 | 2007-05-29 | 3M Innovative Properties Company | Amide-functional polymers, compositions, and methods |
| EP1607437B2 (en) * | 2003-03-25 | 2016-08-24 | Sekisui Plastics Co., Ltd. | Expandable resin beads of styrene-modified, linear low-density polyethylene, process for production thereof, pre-expanded beads, and foams |
| US20040228998A1 (en) | 2003-05-12 | 2004-11-18 | Haas Hans E. | Curable film preform compositions |
| WO2005017487A2 (en) * | 2003-06-09 | 2005-02-24 | The Regents Of The University Of California | Matrix for maldi analysis based on porous polymer monoliths |
| US7214726B2 (en) | 2003-07-17 | 2007-05-08 | Kerr Corporation | Methods of using two-part self-adhering dental compositions |
| US7244793B2 (en) * | 2003-09-26 | 2007-07-17 | Illinois Tool Works Inc. | Adhesive compositions |
| JP2005224101A (ja) | 2003-09-29 | 2005-08-25 | Fujishiro:Kk | 食肉加工用筒状ケーシング |
| DE102004061116A1 (de) * | 2003-12-18 | 2005-07-28 | Tesa Ag | Haftklebemasse |
| CN100412128C (zh) * | 2003-12-22 | 2008-08-20 | 电气化学工业株式会社 | 固化性树脂组合物 |
| JP4396277B2 (ja) | 2004-01-06 | 2010-01-13 | 株式会社Ihi | 親子シールド機 |
| JP2005239987A (ja) | 2004-02-24 | 2005-09-08 | Kazuhiko Ishihara | 生体分子を固定化する反応性ポリマー、その製造方法及び用途 |
| JP2005278984A (ja) | 2004-03-30 | 2005-10-13 | Kunihiko Sakiyama | 手桶 |
-
2006
- 2006-07-03 US US11/916,123 patent/US8313604B2/en active Active
- 2006-07-03 EP EP20110004200 patent/EP2383303A1/en not_active Withdrawn
- 2006-07-03 WO PCT/JP2006/313247 patent/WO2007004620A1/ja active Application Filing
- 2006-07-03 SG SG201004661-3A patent/SG163531A1/en unknown
- 2006-07-03 KR KR1020087000044A patent/KR100995257B1/ko active IP Right Grant
- 2006-07-03 EP EP06780751A patent/EP1900761B1/en not_active Expired - Fee Related
- 2006-07-04 TW TW095124376A patent/TWI383031B/zh active
- 2006-07-04 MY MYPI20063178A patent/MY154745A/en unknown
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5240989A (en) | 1987-12-11 | 1993-08-31 | Avery Dennison Corporation | Removable pressure-sensitive adhesive compositions comprising acrylic based emulsion polymers |
| JPH0423875A (ja) * | 1990-05-19 | 1992-01-28 | Three Bond Co Ltd | 仮固定用接着剤組成物 |
| JPH06116534A (ja) | 1992-10-05 | 1994-04-26 | Three Bond Co Ltd | 接着剤組成物 |
| JPH07228639A (ja) * | 1994-02-18 | 1995-08-29 | Nippon Kayaku Co Ltd | 温度応答型ハイドロゲル |
| JPH08277313A (ja) * | 1995-02-09 | 1996-10-22 | Denki Kagaku Kogyo Kk | 常温硬化型アクリル系土木建築用補修材及びそれを使用した補修方法 |
| JPH1171553A (ja) | 1997-08-28 | 1999-03-16 | Toagosei Co Ltd | 仮固定用光硬化性組成物及び物品の製造方法 |
| JP2001226641A (ja) | 2000-02-17 | 2001-08-21 | Aader:Kk | 仮固定用接着剤組成物及びその使用方法 |
| US6734249B1 (en) | 2000-06-14 | 2004-05-11 | Texas Research International, Inc. | Two-part adhesive with (poly)(meth)acrylate in part A and N,N-disubstituted aromatic amine and di(meth)acrylate in part B |
| JP2002121230A (ja) * | 2000-10-19 | 2002-04-23 | National Institute Of Advanced Industrial & Technology | 高温低粘度低温高粘度型感温性高分子材料 |
| JP2002201229A (ja) * | 2000-11-02 | 2002-07-19 | Mitsubishi Chemicals Corp | 光硬化性樹脂組成物、低複屈折光学部材及びその製造方法 |
| JP2002322214A (ja) * | 2001-04-24 | 2002-11-08 | Central Glass Co Ltd | 反応性高分子化合物 |
| JP2005170920A (ja) * | 2003-12-10 | 2005-06-30 | Shiyoufuu:Kk | 新規重合開始剤を含む歯科用組成物 |
| EP1860128A1 (en) | 2005-03-18 | 2007-11-28 | Denki Kagaku Kogyo Kabushiki Kaisha | Adherent composition and method of temporarily fixing member therewith |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1900761A4 |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007016141A (ja) * | 2005-07-08 | 2007-01-25 | Denki Kagaku Kogyo Kk | 組成物及びそれを用いる部材の仮固定方法 |
| EP2050799A1 (en) * | 2006-08-10 | 2009-04-22 | Denki Kagaku Kogyo Kabushiki Kaisha | Adhesive composition and method for temporarily fixing member by using the same |
| EP2050799B1 (en) * | 2006-08-10 | 2014-06-18 | Denki Kagaku Kogyo Kabushiki Kaisha | Adhesive composition and method for temporarily fixing member by using the same |
| JP2008214593A (ja) * | 2007-03-07 | 2008-09-18 | Denki Kagaku Kogyo Kk | 樹脂組成物及びそれを用いる被加工部材の仮固定方法と表面保護方法 |
| WO2010010900A1 (ja) * | 2008-07-22 | 2010-01-28 | 電気化学工業株式会社 | 部材の仮固定・剥離方法及びそれに好適な仮固定用接着剤 |
| JP5675355B2 (ja) * | 2008-07-22 | 2015-02-25 | 電気化学工業株式会社 | 部材の仮固定・剥離方法及びそれに好適な仮固定用接着剤 |
| WO2013039226A1 (ja) * | 2011-09-14 | 2013-03-21 | 電気化学工業株式会社 | 組成物及びそれを用いた部材の仮固定方法 |
| JPWO2013039226A1 (ja) * | 2011-09-14 | 2015-03-26 | 電気化学工業株式会社 | 組成物及びそれを用いた部材の仮固定方法 |
| US9718996B2 (en) | 2011-09-14 | 2017-08-01 | Denka Company Limited | Composition and method for temporarily fixing member using same |
| WO2014128991A1 (ja) * | 2013-02-20 | 2014-08-28 | 株式会社オートネットワーク技術研究所 | 光硬化性材料、及びその硬化物 |
| JP2014159522A (ja) * | 2013-02-20 | 2014-09-04 | Auto Network Gijutsu Kenkyusho:Kk | 光硬化性材料、及びその硬化物 |
| WO2014192941A1 (ja) * | 2013-05-30 | 2014-12-04 | 電気化学工業株式会社 | 硬質基板積層体および硬質基板積層体の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1900761A4 (en) | 2009-07-15 |
| KR100995257B1 (ko) | 2010-11-19 |
| TWI383031B (zh) | 2013-01-21 |
| EP2383303A1 (en) | 2011-11-02 |
| TW200714686A (en) | 2007-04-16 |
| US20100012263A1 (en) | 2010-01-21 |
| EP1900761B1 (en) | 2012-06-13 |
| SG163531A1 (en) | 2010-08-30 |
| EP1900761A1 (en) | 2008-03-19 |
| US8313604B2 (en) | 2012-11-20 |
| KR20080031721A (ko) | 2008-04-10 |
| MY154745A (en) | 2015-07-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007004620A1 (ja) | 硬化性組成物及びそれを用いる部材の仮固定方法 | |
| JP4916681B2 (ja) | 仮固定方法用光硬化性接着剤及びそれを用いる部材の仮固定方法 | |
| WO2008018252A1 (fr) | Composition adhésive et procédé pour fixer temporairement un élément à l'aide de cette dernière | |
| JP5675355B2 (ja) | 部材の仮固定・剥離方法及びそれに好適な仮固定用接着剤 | |
| JP5563268B2 (ja) | (メタ)アクリル系樹脂組成物、接着剤組成物及び仮固定・剥離方法 | |
| JP5016224B2 (ja) | 組成物及びそれを用いた部材の仮固定方法 | |
| WO2006100788A1 (ja) | 接着性の組成物及びそれを用いる部材の仮固定方法 | |
| JP5674332B2 (ja) | 接着剤組成物及びそれを用いた部材の仮固定方法 | |
| JP5002139B2 (ja) | 組成物及びそれを用いる部材の仮固定方法 | |
| JP4459859B2 (ja) | 組成物及びそれを用いる部材の仮固定方法 | |
| JP2001503811A (ja) | 熱硬化可能な感圧接着剤 | |
| JP2007169560A (ja) | 組成物及びそれを用いた部材の仮固定方法 | |
| JP5350056B2 (ja) | 光硬化型易解体性接着剤及びこれを用いた解体方法 | |
| JP4932300B2 (ja) | 仮固定用組成物及びそれを用いた部材の仮固定方法 | |
| JP5164316B2 (ja) | 接着剤及びそれを用いる部材の仮固定方法 | |
| JPH11140388A (ja) | 硬化型粘接着シート | |
| JP5132045B2 (ja) | 組成物及びそれを用いた部材の仮固定方法 | |
| JP4749751B2 (ja) | 部材の仮固定方法 | |
| JP5408836B2 (ja) | 組成物及びそれを用いる部材の仮固定剥離方法 | |
| JPH11140406A (ja) | 粘着剤組成物 | |
| JP3043292B2 (ja) | エネルギー重合性組成物及び硬化型粘接着シート | |
| JP2005023114A (ja) | 感圧型両面接着テープ又はシート | |
| KR100476798B1 (ko) | 열경화형감압성접착제와이의접착시트류 | |
| KR101848038B1 (ko) | 광 후경화형 필름의 제조 방법 | |
| JP2006052358A (ja) | 回路基板保護用粘着シートおよびその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680024218.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006780751 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12007502504 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11916123 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087000044 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |