WO2012165638A1 - トナー - Google Patents
トナー Download PDFInfo
- Publication number
- WO2012165638A1 WO2012165638A1 PCT/JP2012/064334 JP2012064334W WO2012165638A1 WO 2012165638 A1 WO2012165638 A1 WO 2012165638A1 JP 2012064334 W JP2012064334 W JP 2012064334W WO 2012165638 A1 WO2012165638 A1 WO 2012165638A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- toner
- wax
- mass
- temperature
- parts
- Prior art date
Links
Images


Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/087—Binders for toner particles
- G03G9/08742—Binders for toner particles comprising macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
- G03G9/08764—Polyureas; Polyurethanes
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/0802—Preparation methods
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/0802—Preparation methods
- G03G9/0804—Preparation methods whereby the components are brought together in a liquid dispersing medium
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/0821—Developers with toner particles characterised by physical parameters
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/087—Binders for toner particles
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/087—Binders for toner particles
- G03G9/08742—Binders for toner particles comprising macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
- G03G9/08755—Polyesters
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/087—Binders for toner particles
- G03G9/08775—Natural macromolecular compounds or derivatives thereof
- G03G9/08782—Waxes
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/087—Binders for toner particles
- G03G9/08784—Macromolecular material not specially provided for in a single one of groups G03G9/08702 - G03G9/08775
- G03G9/08788—Block polymers
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/087—Binders for toner particles
- G03G9/08784—Macromolecular material not specially provided for in a single one of groups G03G9/08702 - G03G9/08775
- G03G9/08795—Macromolecular material not specially provided for in a single one of groups G03G9/08702 - G03G9/08775 characterised by their chemical properties, e.g. acidity, molecular weight, sensitivity to reactants
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/087—Binders for toner particles
- G03G9/08784—Macromolecular material not specially provided for in a single one of groups G03G9/08702 - G03G9/08775
- G03G9/08797—Macromolecular material not specially provided for in a single one of groups G03G9/08702 - G03G9/08775 characterised by their physical properties, e.g. viscosity, solubility, melting temperature, softening temperature, glass transition temperature
Definitions
- the present invention relates to a toner used in an image forming method using an electrophotographic method, an electrostatic recording method, and a toner jet method recording method.
- Patent Document 2 shows that fixing by low temperature heating is possible by using a block copolymer obtained by esterification of a crystalline polyester block and an amorphous polyester block.
- Patent Document 3 discloses a toner having improved heat storage stability and hot offset resistance using a urea-modified polyester obtained by modifying a crystalline polyester segment and an amorphous polyester segment with an amino crosslinking agent.
- Patent Document 4 a solution obtained by dissolving a resin composed of a crystalline part and an amorphous part containing an aliphatic polyester (that is, a crystalline polyester) as an essential component in an organic solvent is dissolved in carbon dioxide in a liquid or supercritical state.
- a toner obtained by dispersing to form resin particles containing the resin and an organic solvent, and then removing the organic solvent and carbon dioxide.
- the present invention has been made in view of the above problems, and is a toner containing a resin having a crystalline part excellent in sharp melt properties that is advantageous for low-temperature fixability.
- An object of the present invention is to provide a toner having a wide fixing width and high heat-resistant storage stability.
- Another object of the present invention is to prevent wax exudation while using a wax with improved dispersibility, to suppress a decrease in crystallinity due to the compatibility between the crystalline part and the wax, and to improve heat resistant storage stability.
- the toner of the present invention is a toner having toner particles containing a binder resin mainly composed of polyester, a colorant and a wax, and the binder resin can take a portion having a crystal structure and a crystal structure.
- the endothermic amount of the maximum endothermic peak is 30 J / g or more and 100 J / g or less, and the wax is a tri- or more functional ester wax.
- the present invention although it is a toner containing a resin having a crystalline part excellent in sharp melt properties, which is advantageous for low temperature fixability, it has a wide fixing range from a low temperature part to a high temperature part, and the heat resistant storage of the toner Toner with high properties can be provided.
- wax with improved dispersibility as wax, it prevents the exudation of wax, suppresses the decrease in crystallinity due to the compatibility between the crystalline part and the wax, and improves the heat resistant storage stability. I can do it.
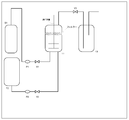
- FIG. 1 is a schematic view of a toner manufacturing apparatus.
- FIG. 2 shows a time chart in a toner heat cycle test.
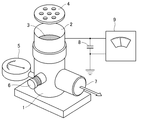
- FIG. 3 shows a schematic view of an apparatus for measuring the triboelectric charge amount.
- FIG. 4 shows a schematic diagram of a viscoelasticity measuring device (rheometer).
- the toner of the present invention is a toner having toner particles containing a binder resin mainly composed of polyester, a colorant and a wax, and the binder resin can take a portion having a crystal structure and a crystal structure.
- a peak polymer having a maximum endothermic peak derived from the binder resin which is obtained from differential scanning calorimetry (DSC) measurement of the toner, containing a block polymer in which no site is bonded,
- the endothermic amount of the maximum endothermic peak is 30 J / g or more and 100 J / g or less
- the wax is a tri- or more functional ester wax.
- the peak temperature (Tp) of the maximum endothermic peak derived from the binder resin, determined from the differential scanning calorimetry (DSC) measurement of the toner is 50 ° C. or higher and 80 ° C. or lower.
- the peak temperature (Tp) of the maximum endothermic peak derived from the binder resin is an aggregate of “sites that can take a crystal structure”, which is a constituent element of the block polymer contained in the binder resin used in the present invention (hereinafter referred to as the It can be controlled by changing the peak temperature (Tp) of the maximum endothermic peak measured by the differential scanning calorific value (DSC) of the crystalline part. Specifically, it can be controlled by the monomer composition and crystallinity of the portion that can take a crystal structure. By setting the peak temperature (Tp) to 50 ° C. or more and 80 ° C. or less, it becomes possible to design a toner satisfying heat-resistant storage stability and low-temperature fixability.
- the lower limit of the peak temperature (Tp) is preferably 55 ° C or higher, and the upper limit of the peak temperature (Tp) is preferably 70 ° C or lower.
- the endothermic amount ( ⁇ H) of the maximum endothermic peak derived from the binder resin, obtained from the differential scanning calorimetry (DSC) measurement of the toner is 30 J / g or more and 100 J / g or less.
- the ⁇ H reflects the ratio of the crystalline portion present in the toner in a state where the crystallinity is maintained in the entire binder resin. That is, even when many crystalline parts are present in the toner, ⁇ H is small if the crystallinity is impaired.
- the ratio of the crystalline part existing in a state where the crystallinity is maintained in the toner is appropriate, and good low-temperature fixability can be obtained.
- the ⁇ H is smaller than 30 J / g
- the ratio of aggregates of “sites that cannot take a crystalline structure” in the block polymer (hereinafter also referred to as amorphous parts) is relatively increased.
- the glass transition point (Tg) derived from the amorphous part is more greatly affected than the sharp melt property of the crystalline part. Therefore, it becomes difficult to show good low-temperature fixability.
- the ⁇ H is larger than 100 J / g, the ratio of the crystalline portion is increased, the dispersion of the colorant in the toner is easily inhibited, and the image density may be lowered.
- the preferable range of ⁇ H is 35 J / g or more and 90 J / g or less.
- the ⁇ H can be adjusted by changing the content of a portion capable of forming a crystal structure, and can be controlled within the above range by subjecting the toner particles to an annealing treatment described later.
- ester waxes are excellent in dispersibility in toner and are effective in preventing cold offset when fixing is performed under low temperature conditions.
- ester waxes have a structure similar to that of crystalline resins (for example, crystalline polyesters) and are therefore easily compatible with crystalline polyesters. When they are compatible, they enter the crystal of the crystalline polyester and easily break the crystal structure of the crystalline polyester. For this reason, the crystallinity is lowered and the heat resistance tends to be insufficient.
- the toner of the present invention preferably has a number average molecular weight (Mn) of 8,000 or more and 30,000 or less, and a weight average molecular weight in gel permeation chromatography (GPC) measurement of tetrahydrofuran (THF) soluble content.
- Mn number average molecular weight
- GPC gel permeation chromatography
- Mw is preferably 15,000 or more and 60,000 or less. By being in this range, it is possible to impart appropriate viscoelasticity to the toner.
- a more preferable range of Mn is 10,000 or more and 25,000 or less, and a more preferable range of Mw is 25,000 or more and 50,000 or less.
- Mw / Mn is preferably 6 or less.
- a more preferable range of Mw / Mn is 3 or less.
- the toner of the present invention is a toner having toner particles containing a polyester-based binder resin, a colorant, and a wax, and the binder resin cannot take a crystalline structure and a crystalline structure.
- a block polymer with bonded sites containing a polyester-based binder resin, a colorant, and a wax, and the binder resin cannot take a crystalline structure and a crystalline structure.
- “based on polyester” means that the polyester portion occupies 50% by mass or more based on the total amount of the binder resin.
- part contained in the said block polymer is also contained in the above-mentioned polyester site
- the block polymer is a polymer in which polymers are covalently bonded within one molecule.
- the portion capable of taking the crystal structure is a portion that regularly arranges and expresses crystallinity when a large number of itself are assembled, and means a crystalline polymer chain.
- the crystalline polymer chain is preferably a crystalline polyester.
- the part which cannot take the said crystal structure is a part which does not arrange regularly even if it collects itself, but becomes amorphous, and means an amorphous polymer chain.
- the block polymer is, for example, an AB type diblock polymer having a crystalline polymer chain (A) and an amorphous polymer chain (B), an ABA type triblock polymer, a BAB type triblock polymer, an ABAB... Type multiblock. Any form of polymer may be used.
- the control of viscoelasticity of the amorphous part where the urethane bond is an aggregate of the amorphous polymer chain especially the viscosity at high temperature. It is effective to raise
- the crystalline part which is an aggregate of the part (crystalline polymer chain) capable of taking the crystal structure of the block polymer is a crystalline polyester obtained by reacting an aliphatic dicarboxylic acid and an aliphatic diol.
- the crystalline part will be described by taking crystalline polyester as an example, but the present invention is not limited thereto.
- the crystalline polyester preferably uses an aliphatic diol having 4 to 20 carbon atoms and a polyvalent carboxylic acid as at least raw materials.
- the aliphatic diol is preferably linear. It is preferable that it is a straight-chain type because the crystallinity of polyester is easily increased.
- Examples of the aliphatic diol include, but are not limited to, the following. In some cases, it is also possible to use a mixture. 1,4-butanediol, 1,5-pentanediol, 1,6-hexanediol, 1,7-heptanediol, 1,8-octanediol, 1,9-nonanediol, 1,10-decanediol, , 11-undecanediol, 1,12-dodecanediol, 1,13-tridecanediol, 1,14-tetradecanediol, 1,18-octadecanediol, 1,20-eicosanediol.
- 1,4-butanediol, 1,5-pentanediol, and 1,6-hexanediol are preferable from the viewpoint of melting point.
- An aliphatic diol having a double bond can also be used. Examples of the aliphatic diol having a double bond include the following. 2-butene-1,4-diol, 3-hexene-1,6-diol, 4-octene-1,8-diol.
- the acid component used for the preparation of the crystalline polyester will be described.
- the acid component used for the preparation of the crystalline polyester is preferably a polyvalent carboxylic acid.
- an aromatic dicarboxylic acid and an aliphatic dicarboxylic acid are preferable.
- an aliphatic dicarboxylic acid is more preferable, and a linear dicarboxylic acid is more preferable from the viewpoint of crystallinity.
- Examples of the aliphatic dicarboxylic acid include, but are not limited to, the following. In some cases, it is also possible to use a mixture.
- Succinic acid malonic acid, succinic acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid, sebacic acid, 1,9-nonanedicarboxylic acid, 1,10-decanedicarboxylic acid, 1,11-undecanedicarboxylic acid, 1,12-dodecanedicarboxylic acid, 1,13-tridecanedicarboxylic acid, 1,14-tetradecanedicarboxylic acid, 1,16-hexadecanedicarboxylic acid, 1,18-octadecanedicarboxylic acid. Or its lower alkyl ester or acid anhydride.
- sebacic acid adipic acid, 1,10-decanedicarboxylic acid, or a lower alkyl ester or acid anhydride thereof is preferable.
- aromatic dicarboxylic acid include the following. Terephthalic acid, isophthalic acid, 2,6-naphthalenedicarboxylic acid, 4,4′-biphenyldicarboxylic acid. Of these, terephthalic acid is preferable because it is easily available and a polymer having a low melting point is easily formed. A dicarboxylic acid having a double bond can also be used.
- a dicarboxylic acid having a double bond can be suitably used in order to prevent high temperature offset at the time of fixing, because the entire resin can be crosslinked using the double bond.
- dicarboxylic acids include, but are not limited to, fumaric acid, maleic acid, 3-hexenedioic acid, and 3-octenedioic acid. These lower alkyl esters or acid anhydrides may also be mentioned. Among these, fumaric acid or maleic acid is preferable in terms of cost.
- the method for producing the crystalline polyester is not particularly limited, and can be produced by a general polyester polymerization method in which an acid component and an alcohol component are reacted. For example, direct polycondensation and transesterification can be performed using different types of monomers.
- the production of the crystalline polyester is preferably carried out at a polymerization temperature of 180 ° C. or higher and 230 ° C. or lower, and the reaction system can be reacted while reducing the pressure in the reaction system and removing water and alcohol generated during condensation as necessary. preferable.
- the monomer is not dissolved or compatible at the reaction temperature, it is preferable to dissolve it by adding a high boiling point solvent as a solubilizing agent. In the polycondensation reaction, the dissolution auxiliary solvent is distilled off.
- the monomer having poor compatibility and the monomer and the acid or alcohol to be polycondensed are condensed in advance and then polycondensed together with the main component.
- the catalyst that can be used in the production of the crystalline polyester include the following. Titanium catalyst such as titanium tetraethoxide, titanium tetrapropoxide, titanium tetraisopropoxide, titanium tetrabutoxide; tin catalyst such as dibutyltin dichloride, dibutyltin oxide, diphenyltin oxide.
- the crystalline polyester is preferably alcohol-terminated.
- the molar ratio of the acid component to the alcohol component is preferably 1.02 or more and 1.20 or less.
- the crystalline polyester preferably has a number average molecular weight (Mn) of 2,000 or more and 20,000 or less, and a weight average molecular weight in gel permeation chromatography (GPC) measurement of tetrahydrofuran (THF) soluble matter.
- Mn number average molecular weight
- GPC gel permeation chromatography
- Mw is preferably 4,000 or more and 100,000 or less.
- a more preferable range of Mn is 3,000 or more and 15,000 or less, and a more preferable range of Mw is 6,000 or more and 80,000 or less.
- the peak temperature (Tp) of the maximum endothermic peak measured by a differential scanning calorimeter (DSC) is preferably 50 ° C. or higher and 85 ° C. or lower, and more preferably 55 ° C. or higher and 80 ° C. or lower.
- the amorphous part which is an aggregate of the part (non-crystalline polymer chain) that cannot take the crystal structure of the block polymer is polyester resin, polyurethane resin, polyurea resin, polyamide resin, polystyrene resin, styrene acrylic.
- the polymer are preferably polyurethanes, but polyurethanes obtained by reacting diols with diisocyanates are preferred.
- polyurethane as an amorphous part will be described.
- the polyurethane is a reaction product of a diol and a substance containing a diisocyanate group, and those having various functions can be obtained by adjusting the diol and the diisocyanate.
- diisocyanate examples include the following. C6-C20 aromatic diisocyanate, C2-C18 aliphatic diisocyanate, C4-C15 alicyclic diisocyanate, and diisocyanates thereof (excluding carbon in NCO group, the same applies hereinafter) Modified products (urethane groups, carbodiimide groups, allophanate groups, urea groups, burette groups, uretdione groups, uretoimine groups, isocyanurate groups, oxazolidone group-containing modified products, hereinafter also referred to as modified diisocyanates), and two or more of these Mixture of. Examples of the aliphatic diisocyanate include the following.
- alicyclic diisocyanate include the following. Isophorone diisocyanate (IPDI), dicyclohexylmethane-4,4′-diisocyanate, cyclohexylene diisocyanate, methylcyclohexylene diisocyanate.
- aromatic diisocyanate include the following. m- and / or p-xylylene diisocyanate (XDI), ⁇ , ⁇ , ⁇ ′, ⁇ ′-tetramethylxylylene diisocyanate.
- the said polyurethane resin can also use a trifunctional or more than trifunctional isocyanate compound. Moreover, the following are mentioned as diol which can be used for the said urethane resin.
- Alkylene glycol (ethylene glycol, 1,2-propylene glycol, 1,3-propylene glycol); alkylene ether glycol (polyethylene glycol, polypropylene glycol); alicyclic diol (1,4-cyclohexanedimethanol); bisphenols (bisphenol) A); an alkylene oxide (ethylene oxide, propylene oxide) adduct of the alicyclic diol; a polyester diol.
- the alkyl part of the alkylene ether glycol may be linear or branched. In the present invention, an alkylene glycol having a branched structure can also be preferably used.
- the glass transition temperature of the amorphous part is preferably 50 ° C. or higher and 130 ° C. or lower, more preferably 70 ° C. or higher and 130 ° C. or lower. By being in this range, the elasticity in the fixing region is easily maintained.
- the block polymer can be prepared by separately preparing a portion that can take a crystal structure and a portion that cannot take a crystal structure, and bonding them together (two-stage method). It is possible to use a method (one-step method) in which raw materials at a site that can be obtained and a site that cannot take a crystal structure are simultaneously charged and prepared at one time.
- the block polymer in the present invention can be selected from various methods in consideration of the reactivity of each terminal functional group to be a block polymer. When using a binder, various binders can be used.
- a dehydration reaction or an addition reaction can be performed using a polyvalent carboxylic acid, a polyhydric alcohol, a polyvalent isocyanate, a polyfunctional epoxy, or a polyacid anhydride.
- the alcohol terminal of the crystalline polyester and the polyurethane It can be prepared by subjecting an isocyanate terminal to a urethanization reaction.
- the synthesis can also be performed by mixing a crystalline polyester having an alcohol terminal and a diol and diisocyanate constituting polyurethane and heating.
- the block polymer preferably has a urethane bond concentration of 1.00 mmol / g or more and 3.20 mmol / g or less.
- the urethane bond concentration is 1.40 mmol / g or more and 2.60 mmol / g or less.
- the urethane bond concentration of the block polymer can be controlled by adjusting the amount of diisocyanate used at this time, for example, when a urethane structure is introduced as a site that cannot take a crystal structure.
- the block polymer has a storage elastic modulus G ′ (at a temperature (Tp + 25) (° C.) 25 ° C. higher than the peak temperature Tp of the maximum endothermic peak derived from the binder resin, which is determined by differential scanning calorimetry (DSC) measurement.
- Tp + 25 ° C.) is preferably 1.0 ⁇ 10 3 Pa or more and 1.0 ⁇ 10 5 Pa or less, more preferably 2.0 ⁇ 10 3 Pa or more and 7.0 ⁇ 10 4 Pa or less.
- the said block polymer contains 50 mass% or more and 85 mass% or less of the site
- the content of the part capable of taking a crystal structure in the block polymer is 50% by mass or more, the sharp melt property of the crystalline part, which is an aggregate of the parts that can take the crystal structure, is easily expressed effectively. More preferably, it is 60 mass% or more and 85 mass% or less.
- the content of the portion of the block polymer that cannot take the crystal structure is preferably 10% by mass or more.
- the elasticity of the amorphous portion that is an aggregate of the portions that cannot take the crystal structure after the sharp melt is improved. More preferably, it is 15 mass% or more, More preferably, it is 15 mass% or more and less than 50 mass%, Most preferably, it is 15 mass% or more and 40 mass% or less.
- the block polymer preferably has a number average molecular weight (Mn) of 8,000 or more and 30,000 or less in gel permeation chromatography (GPC) measurement of tetrahydrofuran (THF) soluble matter, and a weight average molecular weight ( Mw) is preferably 15,000 or more and 60,000 or less.
- a more preferable range of Mn is 10,000 or more and 25,000 or less, and a more preferable range of Mw is 25,000 or more and 50,000 or less. Moreover, it is preferable that Mw / Mn is 6 or less. A more preferable range of Mw / Mn is 3 or less.
- the peak temperature (Tp) of the maximum endothermic peak measured with a differential scanning calorimeter (DSC) is preferably 50 ° C. or higher and 80 ° C. or lower, and more preferably 55 ° C. or higher and 70 ° C. or lower.
- the binder resin in the present invention may contain other known resins known as binder resins for toners in addition to the block polymer. In that case, it is preferable that the block polymer contains 50 mass% or more and 85 mass% or less of a portion capable of forming a crystal structure with respect to the total amount of the binder resin.
- the wax used in the present invention is an ester wax having an ester bond in the wax molecule.
- the ester wax used in the present invention is a tri- or higher functional ester wax, preferably a tetra-functional or higher functional ester wax, more preferably a hexa-functional or higher functional ester wax.
- the tri- or higher functional ester wax can be obtained by, for example, condensation of a tri- or higher functional acid and a long-chain linear saturated alcohol, or synthesis of a tri- or higher-functional alcohol and a long-chain linear saturated fatty acid.
- Examples of the trifunctional or higher functional alcohol that can be used in the present invention include, but are not limited to, the following. In some cases, it is also possible to use a mixture.
- Glycerin trimethylolpropane, erythritol, pentaerythritol, sorbitol.
- polyglycerin such as diglycerin condensed with glycerin, triglycerin, tetraglycerin, hexaglycerin and decaglycerin, condensed trimethylolpropane with ditrimethylolpropane, tristrimethylolpropane and pentaerythritol. And dipentaerythritol and trispentaerythritol.
- a structure having a branched structure is preferable, pentaerythritol or dipentaerythritol is more preferable, and dipentaerythritol is particularly preferable.
- the long-chain linear saturated fatty acid that can be used in the present invention is represented by the general formula C n H 2n + 1 COOH, and those in which n is 5 or more and 28 or less are preferably used.
- n is 5 or more and 28 or less are preferably used.
- the present invention is not limited thereto. In some cases, it is also possible to use a mixture.
- myristic acid, palmitic acid, stearic acid, and behenic acid are preferable from the viewpoint of the melting point of the wax.
- the trifunctional or higher functional acid that can be used in the present invention include, but are not limited to, the following. In some cases, these acids may be used in combination. Trimellitic acid, butanetetracarboxylic acid.
- the long-chain linear saturated alcohol that can be used in the present invention is represented by C n H 2n + 1 OH, and those in which n is 5 or more and 28 or less are preferably used.
- n is 5 or more and 28 or less are preferably used.
- the present invention is not limited thereto.
- myristyl alcohol, palmityl alcohol, stearyl alcohol, and behenyl alcohol are preferable from the viewpoint of the melting point of the wax.
- the peak temperature of the maximum endothermic peak in the differential scanning calorimetry (DSC) measurement of the wax is preferably 65 ° C. or higher, more preferably 65 ° C. or higher and 85 ° C. or lower, and further preferably 65 ° C. or higher and 80 ° C. or lower. It is below °C. Since the peak temperature of the maximum endothermic peak of the wax is within the above range, the wax melts properly at the time of fixing while maintaining heat-resistant storage stability, so that better low-temperature fixability and offset resistance can be obtained. it can.
- the saponification value of the wax is preferably 160 mgKOH / g or more, more preferably 160 mgKOH / g or more and 230 mgKOH / g or less.
- the saponification value is 160 mgKOH / g or more
- the dispersibility of the wax in the toner becomes better.
- the molecular weight of the wax is preferably 1500 or more and 2200 or less, more preferably 1600 or more and 2000 or less.
- the molecular weight of the wax is within the above range, it is easy to maintain fluidity after heat-resistant storage in a heat cycle environment in which the temperature is raised and lowered repeatedly. In addition, the wax easily oozes out during fixing, and the offset resistance at low temperatures can be further improved.
- the wax content is preferably 2.0 parts by mass or more and 8.0 parts by mass or less with respect to 100 parts by mass of the binder resin.
- the content of the wax is within the above range, it is difficult for the wax to ooze out during storage of the toner, and the anti-offset property can be further improved by obtaining a good release effect during fixing.
- the toner of the present invention requires a colorant in order to impart coloring power.
- the colorants conventionally used in toners can be used, but preferred colorants include the following organic pigments, organic dyes, inorganic pigments, carbon black as a black colorant, and magnetic powder. .
- Examples of the colorant for yellow include the following. Condensed azo compounds, isoindolinone compounds, anthraquinone compounds, azo metal complexes, methine compounds, allylamide compounds.
- C.I. I. Pigment Yellow 12, 13, 14, 15, 17, 62, 74, 83, 93, 94, 95, 109, 110, 111, 128, 129, 147, 155, 168, 180 are preferably used.
- magenta colorant examples include the following. Condensed azo compounds, diketopyrrolopyrrole compounds, anthraquinones, quinacridone compounds, basic dye lake compounds, naphthol compounds, benzimidazolone compounds, thioindigo compounds, perylene compounds. Specifically, C.I. I. Pigment Red 2, 3, 5, 6, 7, 23, 48: 2, 48: 3, 48: 4, 57: 1, 81: 1, 122, 144, 146, 166, 169, 177, 184, 185, 202, 206, 220, 221, and 254 are preferably used.
- the colorant for cyan examples include the following. Copper phthalocyanine compounds and derivatives thereof, anthraquinone compounds, basic dye lake compounds.
- C.I. I. Pigment Blue 1, 7, 15, 15: 1, 15: 2, 15: 3, 15: 4, 60, 62, 66 are preferably used.
- the colorant used in the toner of the present invention is selected from the viewpoints of hue angle, saturation, brightness, light resistance, OHP transparency, and dispersibility in the toner.
- the colorant is preferably used by adding 1 part by weight or more and 20 parts by weight or less to 100 parts by weight of the binder resin.
- carbon black is used as the black colorant, it is preferable to add 1 to 20 parts by mass with respect to 100 parts by mass of the binder resin.
- the addition amount is 40 to 150 mass parts with respect to 100 mass parts of binder resin.
- a charge control agent can be mixed with toner particles and used as necessary. Further, it may be added when the toner particles are produced. By blending the charge control agent, the charge characteristics can be stabilized and the optimum triboelectric charge amount can be controlled according to the development system.
- the charge control agent a known one can be used, and a charge control agent that has a high charging speed and can stably maintain a constant charge amount is particularly preferable. Examples of the charge control agent that control the toner to be negatively charged include the following.
- Organic metal compounds and chelate compounds are effective, and monoazo metal compounds, acetylacetone metal compounds, aromatic oxycarboxylic acids, aromatic dicarboxylic acids, oxycarboxylic acids, and dicarboxylic acid-based metal compounds.
- the toner of the present invention may contain these charge control agents alone or in combination of two or more.
- the blending amount of the charge control agent is preferably 0.01 parts by mass or more and 20 parts by mass or less, more preferably 0.5 parts by mass or more and 10 parts by mass or less with respect to 100 parts by mass of the binder resin.
- One measure for realizing the properties of the toner of the present invention is to produce the toner in an unheated state.
- a toner manufactured in an unheated state once passes a temperature higher than the melting point of a crystalline resin such as a crystalline polyester or a resin having a crystalline polyester portion contained in the toner at the time of toner production. It means nothing. However, heating during the production of the crystalline resin is not considered. Crystalline polyester tends to lose its crystallinity when heated above its melting point. Therefore, if the toner is produced without heating, a toner containing a crystalline polyester that maintains the crystallinity can be obtained without destroying the crystallinity of the crystalline polyester contained in the toner.
- Examples of the non-heated toner production method include the following dissolution suspension method, but the present invention is not limited thereto.
- toner particles used in the toner of the present invention (i) a step of obtaining a solution or dispersion in which a binder resin, a colorant and a wax are dissolved or dispersed in an organic solvent, (Ii) a step of dispersing the dissolved material or dispersion in a dispersion medium having carbon dioxide in a supercritical state or a liquid state in which resin fine particles are dispersed, and (iii) removing the organic solvent from the dispersion.
- a toner manufactured through a step of forming toner particles is included.
- high-pressure carbon dioxide can be used as the dispersion medium as described above. That is, after the granulation is performed by dispersing the binder resin solution used for the toner in high-pressure carbon dioxide, the organic solvent contained in the granulated particles is extracted and removed into the carbon dioxide phase. In this method, carbon dioxide is separated by releasing the pressure to obtain toner particles.
- the high pressure carbon dioxide preferably used in the present invention is liquid or supercritical carbon dioxide.
- the dispersion medium is preferably mainly composed of high-pressure carbon dioxide (50% by mass or more).
- the dispersion medium may contain an organic solvent as another component. In this case, it is preferable that carbon dioxide and the organic solvent form a homogeneous phase.
- toner particles that uses liquid or supercritical carbon dioxide as a dispersion medium, which is suitable for obtaining the toner particles of the present invention, will be described as an example.
- a method for producing toner particles that uses liquid or supercritical carbon dioxide as a dispersion medium which is suitable for obtaining the toner particles of the present invention, will be described as an example.
- a binder resin, colorant, wax, and other additives as required in an organic solvent that can dissolve the binder resin, and disperse it as a homogenizer, ball mill, colloid mill, or ultrasonic disperser. Dissolve or disperse uniformly with a machine.
- the organic solvent used in the present invention those capable of dissolving the binder resin are preferable, and ketone solvents such as acetone and methyl ethyl ketone are preferable.
- acetone can be preferably used.
- the binder resin preferably has an acetone insoluble content of 1.0% by mass or less. When the acetone insoluble content exceeds 1.0% by mass, the viscosity at the time of toner preparation becomes high, and the toner particle size tends to be large or the particle size distribution tends to be wide. More preferably, it is 0.5 mass% or less.
- the thus obtained solution or dispersion (hereinafter simply referred to as a binder resin solution) is dispersed in liquid or supercritical carbon dioxide to form oil droplets.
- a binder resin solution it is preferable to disperse the dispersant in the liquid or supercritical carbon dioxide as the dispersion medium.
- the dispersant may be any of an inorganic fine particle dispersant, an organic fine particle dispersant, and a mixture thereof, and may be used alone or in combination of two or more depending on the purpose.
- the inorganic fine particle dispersant include inorganic fine particles such as silica, alumina, zinc oxide, titania, and calcium oxide.
- organic fine particle dispersant examples include vinyl resin, urethane resin, epoxy resin, ester resin, polyamide, polyimide, silicone resin, fluorine resin, phenol resin, melamine resin, benzoguanamine resin, urea resin, aniline resin, and ionomer resin.
- Organic particulates such as polycarbonate, cellulose and mixtures thereof.
- carbon dioxide dissolves in the resin, plasticizes the resin, and lowers the glass transition temperature. It is easy to wake up. Therefore, it is preferable to use a resin having crystallinity as the organic resin fine particles, and when an amorphous resin is used, it is preferable to introduce a crosslinked structure.
- grain with the crystalline resin may be sufficient.
- the said dispersing agent may be used as it is, what improved the surface by various processes may be used in order to improve the adsorptivity to the said oil-droplet surface at the time of granulation. Specific examples include surface treatment with a coupling agent such as silane, titanate, and aluminate, surface treatment with various surfactants, and coating with a polymer. Since the dispersant adsorbed on the surface of the oil droplet remains as it is after the toner particles are formed, when resin fine particles are used as the dispersant, toner particles whose surfaces are coated with the resin fine particles can be formed.
- the resin fine particles preferably have a volume average particle size of 30 nm or more and 300 nm or less, and more preferably 50 nm or more and 100 nm or less.
- the blended amount of the resin fine particles is preferably 3.0 parts by mass or more and 15.0 parts by mass or less with respect to 100 parts by mass of the solid content in the binder resin solution used for forming the oil droplets.
- the oil droplets can be adjusted as appropriate according to the stability of the oil droplets and the desired particle size. In the present invention, any method may be used as a method of dispersing the dispersant in a liquid or supercritical carbon dioxide.
- the dispersant and liquid or supercritical carbon dioxide are charged in a container and directly dispersed by stirring or ultrasonic irradiation.
- a dispersion liquid in which the dispersant is dispersed in an organic solvent is introduced into a container charged with liquid or supercritical carbon dioxide using a high-pressure pump.
- any method may be used as a method of dispersing the binder resin solution in liquid or supercritical carbon dioxide.
- the binder resin solution is introduced into a container containing a liquid in which the dispersant is dispersed or carbon dioxide in a supercritical state using a high-pressure pump.
- a liquid in which the dispersant is dispersed or carbon dioxide in a supercritical state may be introduced into a container charged with the binder resin solution.
- the dispersion medium of carbon dioxide in the liquid or supercritical state is preferably a single phase.
- the temperature and pressure of the dispersion medium, and the amount of the binder resin solution with respect to the liquid or supercritical carbon dioxide are preferably adjusted within a range in which carbon dioxide and the organic solvent can form a homogeneous phase.
- the temperature and pressure of the dispersion medium attention must be paid to granulation properties (ease of oil droplet formation) and solubility of the constituent components in the binder resin solution in the dispersion medium.
- the binder resin and wax in the binder resin solution may be dissolved in the dispersion medium depending on temperature conditions and pressure conditions.
- the temperature of the dispersion medium is preferably in the temperature range of 10 ° C. or higher and lower than the melting point of the crystalline polyester.
- the pressure in the container forming the dispersion medium is preferably 1 MPa or more and 20 MPa or less, more preferably 2 MPa or more and 15 MPa or less.
- the pressure in this invention shows the total pressure, when components other than a carbon dioxide are contained in a dispersion medium.
- the proportion of carbon dioxide in the dispersion medium in the present invention is preferably 70% by mass or more, more preferably 80% by mass or more, and further preferably 90% by mass or more.
- liquid or supercritical carbon dioxide is further mixed with the dispersion medium in which oil droplets are dispersed, and the remaining organic solvent is extracted into a carbon dioxide phase, and carbon dioxide containing the organic solvent is removed. Further, it is performed by substituting with carbon dioxide in a liquid or supercritical state.
- a higher pressure liquid or supercritical carbon dioxide may be added to the dispersion medium. May also be added to low pressure liquid or supercritical carbon dioxide.
- the toner particles to be formed are performed while being supplemented by a filter.
- the container is decompressed in order to recover the obtained toner particles.
- the organic solvent dissolved in the toner may condense and the toner particles may be redissolved or the toner particles may coalesce. Therefore, the substitution with carbon dioxide in the liquid or supercritical state must be performed until the organic solvent is completely removed.
- the amount of liquid to be circulated or carbon dioxide in a supercritical state is preferably 1 to 100 times, more preferably 1 to 50 times, most preferably 1 or more times the volume of the dispersion medium. 30 times or less.
- the container When extracting toner particles from a liquid containing toner particles dispersed or a dispersion containing carbon dioxide in a supercritical state, the container may be depressurized to room temperature and normal pressure at once, but the container is pressure controlled independently. The pressure may be reduced stepwise by providing multiple stages. The decompression speed is preferably set within a range where the toner particles do not foam.
- the organic solvent, liquid, or supercritical carbon dioxide used in the present invention can be recycled.
- the toner of the present invention preferably undergoes a heat treatment step under a temperature condition lower than the melting point of the crystalline polyester. In the present invention, this heat treatment is hereinafter referred to as annealing treatment.
- crystallinity of a crystalline resin increases when an annealing treatment is performed.
- the principle is considered as follows. That is, when annealing is performed on a crystalline material, the molecular mobility of the polymer chain is increased to some extent by the heat, so that the polymer chain is reoriented to a more stable structure, that is, a regular crystal structure. Thus, crystallization occurs.
- the polymer chain When treated at a temperature equal to or higher than the melting point of the crystalline material, the polymer chain obtains an energy higher than that required for reorientation, so recrystallization does not occur.
- the annealing temperature is determined according to the peak temperature of the endothermic peak derived from the crystalline polyester after measuring the differential scanning calorimetry (DSC) of the toner particles obtained in advance. That's fine. Specifically, it is preferable to perform the heat treatment at a temperature obtained by subtracting 15 ° C. from a peak temperature obtained when DSC measurement is performed at a temperature increase rate of 10.0 ° C./min.
- the annealing treatment may be performed at any stage as long as it is after the toner particle forming step.
- the annealing treatment time can be appropriately adjusted depending on the ratio and type of crystalline polyester in the toner and the crystalline state, but it is usually preferable to perform the annealing treatment in the range of 1 hour or more and 50 hours or less. More preferably, it is the range of 2 hours or more and 24 hours or less.
- the annealing rate may change due to the compatibility with the crystalline polyester. When the compatibility between the crystalline polyester and the wax is small, the crystallization speed of the crystalline polyester is likely to be high, and it is effective to use a wax with suppressed compatibility in terms of production.
- the toner of the present invention preferably contains toner particles and inorganic fine powder as an external additive.
- the inorganic fine powder include fine powder such as silica fine powder, titanium oxide fine powder, alumina fine powder, and double oxide fine powder thereof.
- silica fine powder and titanium oxide fine powder are preferable.
- the silica fine powder include dry silica or fumed silica produced by vapor phase oxidation of silicon halide, and wet silica produced from water glass.
- the inorganic fine powder dry silica having less silanol groups on the surface and inside the silica fine powder and less Na 2 O and SO 3 2 ⁇ is preferable.
- the dry silica may be a composite fine powder of silica and another metal oxide produced by using a metal halogen compound such as aluminum chloride or titanium chloride together with a silicon halogen compound in the production process.
- the inorganic fine powder is preferably externally added to the toner particles in order to improve the fluidity of the toner and make the chargeability of the toner uniform. By hydrophobizing the inorganic fine powder, it is possible to adjust the charge amount of the toner, improve the environmental stability, and improve the characteristics in a high-humidity environment. More preferably, it is used.
- treatment agents for the hydrophobic treatment of inorganic fine powder unmodified silicone varnish, various modified silicone varnishes, unmodified silicone oil, various modified silicone oils, silane compounds, silane coupling agents, other organosilicon compounds, organotitanium Compounds.
- these treatment agents may be used alone or in combination.
- inorganic fine powder treated with silicone oil is preferable. More preferably, the inorganic fine powder is hydrophobized with a coupling agent, or at the same time or after the treatment, the hydrophobized inorganic fine powder treated with silicone oil maintains a high toner charge amount even in a high humidity environment, and is selectively developed. It is good in reducing the property.
- the amount of the inorganic fine powder added is preferably 0.1 parts by mass or more and 4.0 parts by mass or less, more preferably 0.2 parts by mass or more and 3.5 parts by mass with respect to 100 parts by mass of the toner particles. It is as follows.
- the toner of the present invention has a weight average particle diameter (D4) of preferably 3.0 ⁇ m or more and 8.0 ⁇ m or less, and more preferably 5.0 ⁇ m or more and 7.0 ⁇ m or less.
- the use of a toner having such a weight average particle diameter (D4) is preferable from the viewpoint of sufficiently satisfying the dot reproducibility while improving the handleability.
- the ratio D4 / D1 of the weight average particle diameter (D4) to the number average particle diameter (D1) of the toner of the present invention is preferably 1.25 or less. More preferably, it is 1.20 or less.
- Tp Maximum Endothermic Peak Peak Temperature
- ⁇ H Maximum Endothermic Peak Endothermic Peak
- DSC Differential Scanning Calorimetry
- the temperature correction of the device detection unit uses the melting points of indium and zinc, and the correction of heat uses the heat of fusion of indium. Specifically, about 5 mg of a sample is precisely weighed, placed in a silver pan, and measured once. A silver empty pan is used as a reference. When a toner is used as a sample, the maximum endothermic peak (maximum endothermic peak derived from the binder resin) overlaps with the endothermic peak of a resin other than wax and binder resin (for example, a shell phase resin in a toner having a core-shell structure).
- the endothermic amount of the maximum endothermic peak obtained is treated as it is as the endothermic amount of the maximum endothermic peak derived from the binder resin.
- the endothermic amount derived from the resin other than the wax and the binder resin is calculated. It is necessary to subtract from the endothermic amount of the maximum endothermic peak obtained.
- the endothermic amount derived from the resin other than the wax and the binder resin is subtracted from the endothermic amount of the obtained maximum endothermic peak to obtain the endothermic amount of the maximum endothermic peak derived from the binder resin.
- the DSC measurement of a single wax is separately performed to obtain the endothermic characteristics.
- the content of wax in the toner is determined.
- the measurement of the content of the wax in the toner is not particularly limited, but can be performed by, for example, peak separation in DSC measurement or known structural analysis.
- the endothermic amount resulting from the wax is calculated from the wax content in the toner, and this amount is subtracted from the endothermic amount of the maximum endothermic peak obtained above.
- the compatibility ratio of the binder resin determined in advance is the endothermic amount obtained for the binder resin and wax mixed at a ratio of 100: 6 (weight of binder resin: weight of wax). Calculated from the value obtained by dividing the endothermic amount by the theoretical endothermic amount calculated from the endothermic amount of the wax alone.
- the endothermic amount derived from the resin other than the binder resin is also determined by the same method as that for the wax.
- the compatibility is the endothermic amount obtained for the binder resin and the binder resin other than the binder resin (mass of the binder resin: mass of the resin other than the binder resin) at a ratio of 100: 6.
- the value is calculated from a value obtained by dividing the endothermic amount of the binder resin obtained in advance and the theoretical endothermic amount calculated from the endothermic amount of the resin other than the binder resin. Further, in the measurement, in order to obtain an endothermic amount per 1 g of the binder resin, it is necessary to remove the mass of components other than the binder resin from the mass of the sample.
- the content of components other than the binder resin can be measured by a known analysis means.
- the incineration residual ash content in the toner is obtained by the following procedure. About 2 g of toner is placed in a pre-weighed 30 ml magnetic crucible. Place the crucible in an electric furnace, heat at about 900 ° C.
- the maximum endothermic peak means a peak where the endothermic amount is maximum when there are a plurality of peaks. Further, the endothermic amount ( ⁇ H) of the maximum endothermic peak is obtained from the peak area by calculation using analysis software attached to the apparatus.
- the temperature rising rate is 10 ° C./min between a measurement temperature range of 30 to 180 ° C.
- the measurement was performed.
- the temperature is once raised to 180 ° C., subsequently lowered to 30 ° C., and then the temperature is raised again.
- the temperature showing the maximum endothermic peak of the DSC curve in the second temperature raising process was defined as the melting point of the wax.
- the maximum endothermic peak means a peak having the largest endothermic amount when a plurality of peaks are present.
- Tg glass transition temperature
- DSC Q1000 manufactured by TA Instruments
- Measurement start temperature 25 °C -Measurement end temperature: 130 ° C
- a new measurement sample was prepared. The temperature was raised only once, and the “Reversing Heat Flow” was plotted on the vertical axis to obtain a DSC curve, and the onset value was Tg.
- the weight average particle diameter (D4) and number average particle diameter (D1) of the toner are calculated as follows.
- a precise particle size distribution measuring device “Coulter Counter Multisizer 3” (registered trademark, manufactured by Beckman Coulter, Inc.) using a pore electrical resistance method equipped with a 100 ⁇ m aperture tube is used.
- the attached dedicated software “Beckman Coulter Multisizer 3 Version 3.51” (manufactured by Beckman Coulter, Inc.) is used. The measurement is performed with 25,000 effective measurement channels.
- the electrolytic aqueous solution used for the measurement special grade sodium chloride is dissolved in ion-exchanged water so as to have a concentration of about 1% by mass, for example, “ISOTON II” (manufactured by Beckman Coulter) can be used.
- ISOTON II manufactured by Beckman Coulter
- the dedicated software is set as follows. On the “Change Standard Measurement Method (SOM)” screen of the dedicated software, set the total count in the control mode to 50000 particles, set the number of measurements once, and set the Kd value to “standard particles 10.0 ⁇ m” (Beckman Coulter Set the value obtained using By pressing the “Threshold / Noise Level Measurement Button”, the threshold and noise level are automatically set.
- the current is set to 1600 ⁇ A
- the gain is set to 2
- the electrolyte is set to ISOTON II
- the “aperture tube flush after measurement” is checked.
- the bin interval is set to logarithmic particle size, the particle size bin to 256 particle size bin, and the particle size range from 2 ⁇ m to 60 ⁇ m.
- the specific measurement method is as follows. (1) About 200 ml of the electrolytic solution is placed in a glass 250 ml round bottom beaker exclusively for Multisizer 3, set on a sample stand, and the stirrer rod is stirred counterclockwise at 24 rpm.
- the height position of a beaker is adjusted so that the resonance state of the liquid level of the electrolyte solution in a beaker may become the maximum.
- (5) In a state where the electrolytic aqueous solution in the beaker of (4) is irradiated with ultrasonic waves, about 10 mg of toner is added to the electrolytic aqueous solution little by little and dispersed. Then, the ultrasonic dispersion process is continued for another 60 seconds. In the ultrasonic dispersion, the temperature of the water tank is appropriately adjusted so as to be 10 ° C. or higher and 40 ° C. or lower.
- the electrolytic solution (5) in which the toner is dispersed is dropped using a pipette, and the measurement concentration is adjusted to about 5%. . Then, measurement is performed until the number of measured particles reaches 50,000.
- the measurement data is analyzed with the dedicated software attached to the apparatus, and the weight average particle diameter (D4) and the number average particle diameter (D1) are calculated.
- the “average diameter” on the “analysis / volume statistics (arithmetic average)” screen when the graph / volume% is set in the dedicated software is the weight average particle size (D4).
- the “average diameter” on the “analysis / number statistics (arithmetic average)” screen is the number average particle diameter (D1).
- sample Preparation of measurement sample Resin (sample) and THF are mixed at a concentration of 5 mg / ml, left at room temperature for 5 to 6 hours, and then shaken sufficiently so that THF and the sample are combined. Mix well until no more. Furthermore, it left still at room temperature for 12 hours or more. At this time, the time from the start of mixing the sample and THF to the end of standing was set to 24 hours or longer. After that, a sample processed filter (pore size 0.45 to 0.5 ⁇ m, Myssho Disc H-25-2 [manufactured by Tosoh Corporation]) is used as a GPC sample.
- a sample processed filter pore size 0.45 to 0.5 ⁇ m, Myssho Disc H-25-2 [manufactured by Tosoh Corporation]
- ⁇ Measuring method of the ratio of the part which can take a crystal structure The proportion (% by mass) of the block polymer capable of taking a crystal structure is measured by 1 H-NMR under the following conditions.
- Measuring apparatus FT NMR apparatus JNM-EX400 (manufactured by JEOL Ltd.) Measurement frequency: 400MHz Pulse condition: 5.0 ⁇ s Frequency range: 10500Hz Integration count: 64 times Measurement temperature: 30 ° C
- a peak independent of the peaks attributed to the other constituent elements is selected from the peaks attributed to the constituent elements of the portion that can take the crystal structure, and the integration of this peak is selected. to calculate the value S 1.
- an integrated value S 2 is calculated for this peak.
- the ratio (mol%) of the site capable of taking the crystal structure is converted to mass% by the molecular weight of each component.
- volume average particle size of resin fine particles in resin fine particle dispersion and volume average particle diameter of wax particles in wax dispersion is 0.001 ⁇ m to 10 ⁇ m in particle size using a Microtrac particle size distribution analyzer HRA (X-100) (manufactured by Nikkiso Co., Ltd.). Measurement is performed with a range setting, and the volume average particle size ( ⁇ m or nm) is measured.
- ⁇ Method for measuring urethane bond concentration of resin The measurement of the urethane bond concentration of the resin is performed using 1H-NMR. 1H-NMR measurement is performed under the following conditions. Measuring apparatus: FT NMR apparatus JNM-EX400 (manufactured by JEOL Ltd.) Measurement frequency: 400MHz Pulse condition: 5.0 ⁇ s Frequency range: 10500Hz Integration count: 64 times Measurement temperature: 30 ° C Sample: 50 mg of a measurement sample is put into a sample tube having an inner diameter of 5 mm, CDCl 3 is added as a solvent, and this is dissolved in a constant temperature bath at 40 ° C. to prepare.
- the molar ratio of the constituents is obtained by determining the hydrogen amount ratio of the constituent units of the resin used.
- concentration of the structural unit constituting the urethane bond per gram is determined from the determined mol ratio and molecular weight, and this is defined as the concentration [mmol / g] of the urethane bond.
- ⁇ Method for measuring storage modulus G ′ of block polymer> The storage elastic modulus G ′ of the block polymer is measured using a viscoelasticity measuring apparatus (rheometer) ARES (manufactured by Rheometrics Scientific). The outline of the measurement is described in ARES operation manuals 902-30004 (August 1997 version) and 902-00153 (July 1993 version) published by Rheometrics Scientific, Inc., and is as follows.
- ⁇ Measurement jig torsion rectangular Measurement sample: A rectangular parallelepiped sample having a width of about 12 mm, a height of about 20 mm, and a thickness of about 2.5 mm is prepared from the block polymer using a pressure molding machine (maintaining 15 kN for 1 minute at room temperature).
- the press molding machine uses a 100 kN press NT-100H manufactured by NPa Systems. After the jig and sample are left at room temperature (23 ° C.) for 1 hour, the sample is attached to the jig. See FIG. As shown in FIG. 4, the measurement unit is fixed so that the width is about 12 mm, the thickness is about 2.5 mm, and the height is 10 mm.
- Measurement temperature Temperature is increased from 30 ° C to 150 ° C at a rate of 2 ° C per minute.
- Measurement interval Viscoelasticity data is measured every 30 seconds, that is, every 1 ° C.
- RSI Orchestrator operating on Microsoft Windows 2000 (registered trademark) Data is transferred through the interface to (control, data collection and analysis software) (manufactured by Rheometrics Scientific).
- the storage elastic modulus (G ′ (Tp + 25) of the block polymer at a temperature 25 ° C. higher than the peak temperature Tp of the maximum endothermic peak derived from the binder resin, which is obtained from the scanning scanning calorimetry (DSC) measurement of the toner. ) Value is read.
- Crystalline polyester 1 was synthesize
- Table 1 shows the physical properties of the crystalline polyester 1.
- the crystalline polyester 5 was synthesized in the same manner except that the raw material charge was changed as follows. Table 1 shows the physical properties of the crystalline polyester 5.
- the crystalline polyester 7 was synthesized in the same manner except that the raw material charge was changed as follows.
- Table 1 shows the physical properties of the crystalline polyester 7.
- the crystalline polyester 8 was synthesized in the same manner except that the raw materials were changed as follows.
- Table 1 shows the physical properties of the crystalline polyester 8.
- Block polymer resin solutions 1 to 12 To a beaker equipped with a stirrer, 100.0 parts by mass of acetone and 100.0 parts by mass of block polymer 1 were added, and stirring was continued until the solution was completely dissolved at a temperature of 40 ° C. to prepare a block polymer resin solution 1. Similarly, block polymer resin solutions 2 to 12 were prepared.
- Neogen RK Neogen RK
- 5.0 parts by mass-Ion-exchanged water 180.0 parts by mass
- the above components are mixed and heated to 100 ° C.
- dispersion treatment is performed with a pressure discharge type gorin homogenizer for 1 hour, and a crystalline polyester resin dispersion having a volume average particle size of 180 nm and a solid content of 38.3% by mass is dispersed. Liquid 1 was obtained.
- amorphous resin dispersion 1 115.0 parts by mass-Ionic surfactant Neogen RK (Daiichi Kogyo Seiyaku) 5.0 parts by mass-Ion-exchanged water 180.0 parts by mass
- Neogen RK Neogen RK
- amorphous resin 1 115.0 parts by mass-Ionic surfactant Neogen RK (Daiichi Kogyo Seiyaku) 5.0 parts by mass-Ion-exchanged water 180.0 parts by mass
- the above components are mixed and heated to 100 ° C. Then, after sufficiently dispersing with IKA Ultra Turrax T50, dispersion treatment is performed with a pressure discharge type gorin homogenizer for 1 hour, an amorphous resin having a volume average particle size of 200 nm and a solid content of 38.3 mass%. Dispersion 1 was obtained.
- wax dispersion-1 was put into a heat-resistant container together with 20 parts by mass of 1 mm glass beads, and dispersed for 3 hours with a paint shaker (manufactured by Toyo Seiki) to obtain wax dispersion-1.
- the wax particle size in the wax dispersion-1 was 0.15 ⁇ m in terms of volume average particle size.
- the properties of the obtained wax dispersion-1 and the wax used (WAX-1) are shown in Table 3.
- ⁇ Preparation of wax dispersions 2 to 12> Preparation of Wax Dispersion-1 except that the waxes (WAX-2 to 12) shown in Table 3 were used instead of the dipentaerythritol palmitate wax (WAX-1) used in Wax Dispersion-1.
- wax dispersions 2 to 12 were prepared. Table 3 shows the properties of the obtained wax dispersions-2 to 12 and the WAX-2 to 12 used.
- ⁇ Preparation of Wax Dispersion-13> ⁇ Dipentaerythritol behenate wax (WAX-2) 30.0 parts by mass / nitrile group-containing styrene acrylic resin (styrene 60 parts by mass, n-butyl acrylate 30 parts by mass, acrylonitrile 10 parts by mass, peak molecular weight 8500) 15.0 parts by mass, cationic surfactant Neogen RK (Daiichi Kogyo Seiyaku) 5.0 parts by mass, ion-exchanged water 200.0 parts by mass
- the above is mixed and heated to 95 ° C. After sufficiently dispersing at T50, the mixture was dispersed with a pressure discharge type gorin homogenizer to obtain a wax dispersion 13 having a volume average particle size of 0.20 ⁇ m and a solid content of 20
- Pigment Blue 15 3 45.0 parts by mass, ionic surfactant Neogen RK (Daiichi Kogyo Seiyaku) 5.0 parts by mass, ion-exchanged water 200.0 parts by mass, glass beads (1 mm) 250.0 parts by mass
- Neogen RK Diichi Kogyo Seiyaku
- ion-exchanged water 200.0 parts by mass
- glass beads (1 mm) 250.0 parts by mass
- the material was put into a heat-resistant glass container, dispersed for 5 hours with a paint shaker, glass beads were removed with a nylon mesh, and a colorant dispersion 2 was obtained.
- a silane coupling agent 3- (2-aminoethylaminopropyl) trimethyl
- a mixed solvent coating solution of methyl ethyl ketone and toluene was prepared so that the coating resin content was 2.5 parts by mass with respect to the carrier core (solution concentration: 10% by mass).
- the coating solution was applied to the surface of the magnetic resin particles by volatilizing the solvent at 70 ° C. while continuously applying shear stress.
- the resin-coated magnetic carrier particles were heat-treated with stirring at 100 ° C.
- Example 1> Manufacturing process of toner particles (before treatment) 1)
- the valves V1 and V2 and the pressure adjustment valve V3 are closed, and the resin fine particle dispersion 1 (in the pressure resistant granulation tank T1 having a filter and a stirring mechanism for capturing toner particles is provided.
- Table 4 “resin fine particle-1” was charged, and the internal temperature was adjusted to 30 ° C.
- the valve V1 was opened, carbon dioxide (purity 99.99%) was introduced into the pressure vessel T1 from the cylinder B1 using the pump P1, and the valve V1 was closed when the internal pressure reached 5 MPa.
- the block polymer resin solution 1, the wax dispersion 1, the colorant dispersion 1, and acetone were charged into the resin solution tank T2, and the internal temperature was adjusted to 30 ° C.
- the valve V2 is opened and the contents of the resin solution tank T2 are introduced into the granulation tank T1 using the pump P2 while stirring the inside of the granulation tank T1 at 2000 rpm. Valve V2 was closed. After the introduction, the internal pressure of the granulation tank T1 was 8 MPa.
- the preparation amount (mass ratio) of various materials is as follows.
- Block polymer resin solution 1 175.0 parts by mass Wax dispersion 1 31.3 parts by mass (as solids, WAX-1 is 5 parts by mass, nitrile group-containing styrene acrylic resin ("Dispersant-1" in Table 4) And 2.5 parts by mass) -Colorant dispersion 1 12.5 parts by mass-Acetone 31.2 parts by mass-Resin fine particle dispersion 1 25.0 parts by mass-Carbon dioxide 280.0 parts by mass From the temperature (30 ° C.) and the pressure (8 MPa), the density of carbon dioxide is calculated from the equation of state described in the literature (Journal of Physical and Chemical Reference data, vol. 25, P. 1509 to 1596), and granulated in this Calculation was performed by multiplying the volume of the tank T1.
- the introduction of carbon dioxide into the granulation tank T1 was stopped when it reached 5 times the mass of carbon dioxide initially introduced into the granulation tank T1. At this point, the operation of replacing carbon dioxide containing an organic solvent with carbon dioxide not containing an organic solvent was completed. Further, the pressure regulating valve V3 was opened little by little, and the internal pressure of the granulation tank T1 was reduced to atmospheric pressure, whereby the toner particles (before treatment) 1 captured by the filter were collected. The obtained toner particles (before treatment) 1 were subjected to DSC measurement, and the peak temperature (Tp) of the maximum endothermic peak derived from the binder resin was determined to be 58 ° C.
- the annealing treatment was performed using a constant temperature dryer (Satake Chemical 41-S5). The internal temperature of the constant temperature dryer was adjusted to 50 ° C. The toner particles (before treatment) 1 were spread evenly in a stainless steel vat, placed in the constant temperature dryer and allowed to stand for 2 hours, and then taken out. Thus, annealed toner particles (after treatment) 1 were obtained.
- Toner 1 preparation of Toner 1 Then, (after treatment) the toner particles with respect to 100.0 parts by weight of 1, anatase-type titanium oxide fine powder (BET specific surface area 80 m 2 / g, number average particle diameter (D1) 15 nm, isobutyl trimethoxysilane 12 mass % Treatment) First, 0.9 part by mass was externally added using a Henschel mixer FM-10B (manufactured by Mitsui Miike Chemical Co., Ltd.).
- the toner subjected to the heat cycle test was passed through a sieve of 200 mesh (aperture 75 ⁇ m) and allowed to stand for 1 day in a normal temperature and humidity environment (temperature 23 ° C./humidity 60%) to obtain a sample toner.
- 1.0 g and 19.0 g of toner and carrier Japanese Image Society Standard Carrier Spherical Carrier N-01 with a ferrite core surface-treated are placed in a plastic bottle with a lid, respectively, and left in the measurement environment for one day.
- the suction machine 1 (at least the part in contact with the measurement container 2) is suctioned from the suction port 7 and the air volume control valve 6 is adjusted so that the pressure of the vacuum gauge 5 is 250 mmAq.
- suction is performed for 2 minutes to remove the toner by suction.
- the potential of the electrometer 9 at this time is set to V (volt).
- 8 is a capacitor, and the capacity is C (mF).
- W2 (g) The triboelectric charge amount (mC / kg) of this sample is calculated as follows.
- Sample triboelectric charge (mC / kg) C ⁇ V / (W1-W2) (Evaluation criteria for charge retention)
- D The difference between the charge amount of the sample toner and the charge amount of the standard toner is 20% or more.
- the low-temperature fixability of the toner was evaluated by two methods, a fixing start temperature due to peelability and a fixing start temperature due to cold offset property.
- a two-component developer 1 was prepared by mixing 8.0 parts by mass of toner 1 and 92.0 parts by mass of carrier A produced as described above.
- the above-described two-component developer 1 and a color laser copying machine CLC5000 (manufactured by Canon Inc.) were used.
- the development contrast of the copying machine is adjusted so that the toner loading on paper is 1.2 mg / cm 2 , and a “solid” unfixed image with a front margin of 5 mm, a width of 100 mm, and a length of 28 mm is obtained in the single color mode. It was created in a normal temperature and humidity environment (23 ° C./60% RH).
- thick paper A4 paper (“Prober bond paper”: 105 g / m 2 , manufactured by Fox River) was used.
- the fixing device of LBP5900 manufactured by Canon Inc.
- the rotation speed of the fixing device was changed to 270 mm / s and the pressure in the nip: 120 kPa.
- each of the above-mentioned “solid” unfixed images was increased while increasing the fixing temperature by 10 ° C. in the range of 80 ° C. to 180 ° C. in a normal temperature and humidity environment (23 ° C./60% RH). A fixed image at temperature was obtained.
- the image area of the obtained fixed image is covered with a soft thin paper (trade name “Dasper”, manufactured by Ozu Sangyo Co., Ltd.), and the image area is rubbed five times while applying a load of 4.9 kPa from the thin paper. did.
- the image density before and after rubbing was measured, and the reduction rate ⁇ D (%) of the image density due to peeling was calculated by the following formula.
- the temperature when this ⁇ D (%) was less than 10% was defined as the fixing start temperature due to peelability, and evaluation was performed according to the following evaluation criteria.
- the image density was measured with a color reflection densitometer (Color reflection densitometer X-Rite 404A: manufacturer X-Rite).
- ⁇ D (%) ⁇ (Image density before rubbing ⁇ Image density after rubbing) / Image density before rubbing ⁇ ⁇ 100 (Evaluation criteria)
- the higher of the fixing start temperature due to the above-mentioned peelability and the fixing start temperature due to the cold offset property is set as the fixing start temperature, and the difference between the fixing start temperature and the high temperature fixing temperature (high temperature fixing temperature ⁇ fixing start temperature) is set as the fixing region. Judgment was made.
- amorphous resin dispersion 1 13.0 parts by mass of the amorphous resin dispersion 1, was gradually added thereto. Thereafter, the pH of the system was adjusted to 5.4 with a 0.5 mol / L sodium hydroxide aqueous solution, and then the stainless steel flask was sealed and heated to 96 ° C. while continuing to stir using a magnetic seal for 5 hours. Retained. After completion of the reaction, the mixture was cooled, filtered, sufficiently washed with ion exchange water, and then subjected to solid-liquid separation by Nutsche suction filtration. This was further redispersed in 3 L of ion exchange water at 40 ° C., and stirred and washed at 300 rpm for 15 minutes.
- Examples 2 to 24 The same procedure as in Example 1 was conducted except that the block polymer resin solution and the wax dispersion were selected so that the block polymer and wax in the production process of toner particles (before treatment) 1 would become the block polymer and wax shown in Table 4. Toners 8 to 30 were obtained. Toner preparation conditions and toner characteristics are shown in Tables 4 and 5, and evaluation results are shown in Table 6. In the toners 9, 11, 17 and 20 according to Examples 3, 5, 11 and 14, the endothermic peak of the wax overlapped the maximum endothermic peak derived from the binder resin in the endothermic curve of the toner.
- each value was obtained as the endothermic amount of the maximum endothermic peak derived from the binder resin by subtracting the endothermic amount of the wax from the endothermic amount of the maximum endothermic peak.
- the endothermic peaks of the phase resins overlapped. Therefore, each value was determined by subtracting the endothermic amount of the shell phase resin from the endothermic amount of the maximum endothermic peak as the endothermic amount of the maximum endothermic peak derived from the binder resin.
- the maximum endothermic peak in the endothermic curve of the toner was directly used as the maximum endothermic peak derived from the binder resin.
- Tp of the toner particles (after treatment) represents “Tp derived from the binder resin of the toner”, and the endothermic amount of the maximum endothermic peak derived from the binder resin of the toner particles (after treatment) is “toner of the toner”. It represents the “endothermic amount of the maximum endothermic peak derived from the binder resin”.
Landscapes
- Physics & Mathematics (AREA)
- General Physics & Mathematics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Developing Agents For Electrophotography (AREA)
Abstract
Description
従来、より低温での定着を可能とするためには結着樹脂をよりシャープメルトにする手法が効果的な方法の一つとして知られている。この点において結晶性ポリエステル樹脂を用いたトナーが紹介されている。結晶性ポリエステルは、分子鎖が配列することにより、明確なガラス転移を示さず、結晶融点まで軟化しにくいという特性を持つため、耐熱保存性と低温定着性を両立できる材料として検討が行われている。
しかしながら、トナーの結着樹脂に結晶性ポリエステルを単独で用いる場合、シャープメルト性は有するものの、高温での弾性がなく、ホットオフセット、紙上への染み込みのための光沢度の低下が発生し、定着の温度幅が狭くなってしまうことがある。そのため、プリンターでの低温環境下での連続画像形成では、オフセットや光沢ムラが発生しやすくなり、安定した画像を得ることが出来なかった。
そのため、結晶性ポリエステルの添加量を下げ、結晶性ポリエステルと非晶性ポリエステルとを混合して用いたトナーが提案されている。
特許文献1では、結晶性ポリエステルと非晶性ポリエステルを含有したカプセル型のトナーで、融点+20℃での貯蔵弾性率及び損失弾性率を制御することにより、定着のラチチュードの向上を図っている。
また、非晶性ポリエステルに結晶性ポリエステルを少量添加した場合、非晶性ポリエステルの粘度を変えることにより、高温での粘度を調整でき、ホットオフセットを抑えることが出来る。しかしながら、この場合、結晶性ポリエステルのシャープメルト性を十分に示すことができず、低温定着性に対する効果が十分に発揮できなかった。
このような問題を解決するため、結晶性ポリエステルと非晶性ポリエステルとをブロック化した結着樹脂を用いたトナーが提案なされている。
特許文献2では、結晶性ポリエステルブロック及び非結晶性ポリエステルブロックのエステル化によりにより得られたブロック共重合体を用いることにより、低温度加熱による定着が可能であることが示されている。
特許文献3では、結晶性ポリエステルのセグメントと無定形ポリエステルのセグメントがアミノ架橋剤によって変性されてなるウレア変性ポリエステルにより、耐熱保存性及び耐ホットオフセット性を改良したトナーが示されている。
特許文献4では、脂肪族ポリエステル(すなわち結晶性ポリエステル)を必須成分とする結晶部と非晶部から構成される樹脂を有機溶剤に溶解させた溶液を、液状又は超臨界状態の二酸化炭素中に分散させることによって、前記樹脂と有機溶剤を含有する樹脂粒子を形成し、次いで前記有機溶剤及び二酸化炭素を除去することによって得られるトナーが提案されている。
しかしながら、このようなブロックポリマーを含有したトナーを用いる場合においても、低温定着性に関し、特にトナー中のワックス分散性が十分でないと、コールドオフセットが発生し、十分な効果が得られない場合があった。また、結晶性ポリエステルの結晶化が不十分な場合、耐熱保存性が十分でなかったり、用いるワックスによっては、ワックスの染み出しや、結晶性ポリエステルとワックスとの相溶による結晶性の低下により、耐熱保存性の低下を引き起こしたりした。特に、昇温と、降温が繰り返えされるような環境下に長期間放置した場合に耐熱保存性の劣化現象が発生しやすかった。そのため更に改良したトナーが望まれている。
本発明者らは、上述した結晶性ポリエステルを用いたトナーの種々の問題点について検討を重ねた結果、特定の構造を有するワックスとの組み合わせにおいて、これらの問題を解決できることを見出し本発明に至った。
本発明のトナーは、ポリエステルを主成分にする結着樹脂、着色剤及びワックスを含有するトナー粒子を有するトナーであって、前記結着樹脂は、結晶構造をとりうる部位と結晶構造をとりえない部位を結合したブロックポリマーを含有し、前記トナーの示差走査熱量(DSC)測定から求められる、前記結着樹脂に由来する最大吸熱ピークのピーク温度が50℃以上、80℃以下であり、前記最大吸熱ピークの吸熱量が30J/g以上、100J/g以下であり、前記ワックスは、3官能以上のエステルワックスであることを特徴とする。
本発明においては、トナーの示差走査熱量(DSC)測定から求められる、結着樹脂に由来する最大吸熱ピークのピーク温度(Tp)が、50℃以上、80℃以下である。当該結着樹脂に由来する最大吸熱ピークのピーク温度(Tp)は、本発明に用いられる結着樹脂に含まれるブロックポリマーの構成要素である「結晶構造をとりうる部位」の集合体(以下、結晶性部位ともいう)の示差走査熱量(DSC)により測定される最大吸熱ピークのピーク温度(Tp)を変更することにより制御することが出来る。具体的には結晶構造をとりうる部位のモノマー組成及び結晶化度により制御することが可能である。上記ピーク温度(Tp)を50℃以上、80℃以下にすることにより、耐熱保存性と低温定着性を満足するトナーを設計することが可能になる。上記ピーク温度(Tp)の下限は好ましくは55℃以上であり、ピーク温度(Tp)の上限は好ましくは70℃以下である。
本発明においては、トナーの示差走査熱量(DSC)測定から求められる、結着樹脂に由来する最大吸熱ピークの吸熱量(ΔH)が30J/g以上、100J/g以下である。ここで、当該ΔHは、トナー中において結晶性が維持された状態で存在している結晶性部位の結着樹脂全体における割合を反映する。すなわち、トナー中に結晶性部位を多く存在させた場合であっても、結晶性が損なわれている場合は、ΔHは小さくなる。従って、ΔHが上記範囲にあるようなトナーは、トナー中において結晶性を維持した状態で存在する結晶性部位の割合が適正であり、良好な低温定着性が得られる。上記ΔHが30J/gよりも小さいと、相対的にブロックポリマー中の「結晶性構造をとりえない部位」の集合体(以下、非晶性部位ともいう)の割合が大きくなり、その結果、結晶性部位のシャープメルト性よりも非晶性部位に由来するガラス転移点(Tg)の影響をより大きく受けるようになる。そのため、良好な低温定着性を示すことが困難となる。一方、上記ΔHが100J/gよりも大きいと、結晶性部位の割合が大きくなり、トナー中の着色剤の分散を阻害しやすくなり、画像濃度の低下が起こることがある。上記ΔHの好ましい範囲は、35J/g以上、90J/g以下である。
上記ΔHは、結晶構造をとりうる部位の含有量を変更することで調整することができ、更にトナー粒子に対して後述するアニール処理を施すことにより上記範囲に制御することが可能である。
この理由は以下の通り推察される。エステル系ワックスはトナー中における分散性に優れ、低温度条件で定着を行う場合のコールドオフセットの防止に対し有効である。ところがエステル系ワックスは結晶性樹脂(例えば、結晶性ポリエステル)と類似構造を有するため結晶性ポリエステルと相溶しやすい。相溶した場合、結晶性ポリエステルの結晶内部に入り、結晶性ポリエステルの結晶構造を崩しやすい。そのため、結晶性が低下し、耐熱性が不十分になりやすかった。特に、昇温と降温が繰り返されるような環境下にトナーを長期間放置した場合に耐熱保存性が不十分となる現象が発生しやすかった。また、耐熱保存性を満足する場合においても、ワックスの染み出しによる帯電の維持性が問題になる場合があった。
本発明のように3官能以上のエステルワックスを用いた場合、当該ワックスは分岐構造を有するため、直鎖構造をもつ結晶性ポリエステルと相溶し難くなり、結晶性ポリエステルの結晶構造を維持しやすくなる。本発明においては、さらに分岐構造の多い4官能以上のエステルワックスを用いることが好ましく、6官能以上のエステルワックスを用いることがより好ましい。
ここで「ポリエステルを主成分にする」とは、結着樹脂の総量に対し、ポリエステル部位が50質量%以上占めることを意味する。また、上記ブロックポリマーに含有されるポリエステル部位も前述のポリエステル部位に含まれる。
また、上記ブロックポリマーとは、一分子内でポリマー同士が共有結合にて結ばれたポリマーである。上記結晶構造をとりうる部位とは、それ自体が多数集合すると、規則的に配列し結晶性を発現する部位であり、結晶性ポリマー鎖を意味する。本発明において結晶性ポリマー鎖は、結晶性ポリエステルであることが好ましい。また、上記結晶構造をとりえない部位とは、それ自体が集合しても規則的に配列せず、非晶性となる部位であり、非晶性ポリマー鎖を意味する。
上記ブロックポリマーは、例えば結晶性ポリマー鎖(A)と非晶性ポリマー鎖(B)とのAB型ジブロックポリマー、ABA型トリブロックポリマー、BAB型トリブロックポリマー、ABAB・・・・型マルチブロックポリマーの、いずれの形態であってもよい。また、上記ブロックポリマーの結晶性ポリマー鎖と非晶性ポリマー鎖との結合形態としては、ウレタン結合が非晶性ポリマー鎖の集合体である非晶性部位の粘弾性の制御、とりわけ高温における粘性を上げるために有効である。
以下、上記結晶性部位について、結晶性ポリエステルを例に挙げて説明するが、これに限定されるわけではない。
結晶性ポリエステルは、炭素数4以上20以下の脂肪族ジオールおよび多価カルボン酸を少なくとも原料として用いることが好ましい。
さらに、前記脂肪族ジオールは直鎖型であることが好ましい。直鎖型であることで、ポリエステルの結晶性を上げやすく好ましい。
上記脂肪族ジオールとしては、例えば以下のものを挙げることが出来るが、これに限定されるものではない。また、場合によっては混合して用いることも可能である。1,4-ブタンジオール、1,5-ペンタンジオール、1,6-ヘキサンジオール、1,7-ヘプタンジオール、1,8-オクタンジオール、1,9-ノナンジオール、1,10-デカンジオール、1,11-ウンデカンジオール、1,12-ドデカンジオール、1,13-トリデカンジオール、1,14-テトラデカンジオール、1,18-オクタデカンジオール、1,20-エイコサンジオール。これらのうち、融点の観点から、1,4-ブタンジオール、1,5-ペンタンジオール、1,6-ヘキサンジオールが好ましい。
また、二重結合を持つ脂肪族ジオールを用いることもできる。前記二重結合を持つ脂肪族ジオールとしては、例えば以下のものを挙げることができる。2-ブテン-1,4-ジオール、3-ヘキセン-1,6-ジオール、4-オクテン-1,8-ジオール。
次に、結晶性ポリエステルの調製に用いられる酸成分について述べる。結晶性ポリエステルの調製に用いられる酸成分は、多価カルボン酸が好ましい。多価カルボン酸としては、芳香族ジカルボン酸および脂肪族ジカルボン酸が好ましく、中でも脂肪族ジカルボン酸がより好ましく、結晶性の観点から、特に直鎖型のジカルボン酸が更に好ましい。
脂肪族ジカルボン酸としては、例えば以下のものを挙げることができるが、これに限定されるものではない。また、場合によっては混合して用いることも可能である。蓚酸、マロン酸、琥珀酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、1,9-ノナンジカルボン酸、1,10-デカンジカルボン酸、1,11-ウンデカンジカルボン酸、1,12-ドデカンジカルボン酸、1,13-トリデカンジカルボン酸、1,14-テトラデカンジカルボン酸、1,16-ヘキサデカンジカルボン酸、1,18-オクタデカンジカルボン酸。あるいはその低級アルキルエステルや酸無水物。これらのうち、セバシン酸、アジピン酸、1,10-デカンジカルボン酸、または、その低級アルキルエステルまたは酸無水物が好ましい。
芳香族ジカルボン酸としては、例えば以下のものを挙げることができる。テレフタル酸、イソフタル酸、2,6-ナフタレンジカルボン酸、4,4’-ビフェニルジカルボン酸。
これらのうち、テレフタル酸が入手の容易性、及び、低融点のポリマーを形成しやすいという点で好ましい。
また、二重結合を有するジカルボン酸を用いることもできる。二重結合を有するジカルボン酸は、その二重結合を利用して樹脂全体を架橋させ得る点で、定着時の高温オフセットを防ぐために好適に用いることができる。このようなジカルボン酸としては、例えば、フマル酸、マレイン酸、3-ヘキセンジオイック酸、3-オクテンジオイック酸が挙げられるが、これらに限定されるわけではない。また、これらの低級アルキルエステルまたは酸無水物も挙げられる。これらの中でも、コストの点で、フマル酸またはマレイン酸が好ましい。
前記結晶性ポリエステルの製造方法としては、特に制限はなく、酸成分とアルコール成分とを反応させる一般的なポリエステル重合法で製造することができ、例えば直接重縮合、エステル交換法を、モノマーの種類によって使い分けて製造する。
前記結晶性ポリエステルの製造は、重合温度180℃以上230℃以下の間で行うことが好ましく、必要に応じて反応系内を減圧にし、縮合時に発生する水やアルコールを除去しながら反応させることが好ましい。モノマーが、反応温度下で溶解または相溶しない場合は、高沸点の溶剤を溶解補助剤として加え溶解させることが好ましい。重縮合反応においては、溶解補助溶剤を留去しながら行う。共重合反応において相溶性の悪いモノマーが存在する場合は、あらかじめ相溶性の悪いモノマーとそのモノマーと重縮合予定の酸またはアルコールとを縮合させておいてから主成分とともに重縮合させることが好ましい。
前記結晶性ポリエステルの製造時に使用可能な触媒としては、例えば以下を挙げることができる。チタンテトラエトキシド、チタンテトラプロポキシド、チタンテトライソプロポキシド、チタンテトラブトキシドの如きチタン触媒;ジブチルスズジクロライド、ジブチルスズオキシド、ジフェニルスズオキシドの如きスズ触媒。
前記結晶性ポリエステルはアルコール末端であることが前記ブロックポリマーを調製する上で好ましい。そのため、前記結晶性ポリエステルの調製では酸成分とアルコール成分のモル比(アルコール成分/カルボン酸成分)は1.02以上1.20以下であることが好ましい。上記結晶性ポリエステルは、テトラヒドロフラン(THF)可溶分のゲルパーミエーションクロマトグラフィー(GPC)測定において、数平均分子量(Mn)が2,000以上、20,000以下であることが好ましく、重量平均分子量(Mw)が4,000以上、100,000以下であることが好ましい。Mnのより好ましい範囲は、3,000以上、15,000以下であり、Mwのより好ましい範囲は、6,000以上、80,000以下である。また、Mw/Mnは6以下であることが好ましい。Mw/Mnのより好ましい範囲は3以下である。さらに、示差走査熱量計(DSC)により測定される最大吸熱ピークのピーク温度(Tp)が50℃以上、85℃以下であることが好ましく、55℃以上、80℃以下であることがより好ましい。
以下、非晶性部位としてのポリウレタンについて述べる。前記ポリウレタンはジオールとジイソシアネート基を含有する物質との反応物であり、ジオール、ジイソシアネートの調整により、各種機能性をもつものを得ることができる。
前記ジイソシネートとしては以下のものが挙げられる。
炭素数(NCO基中の炭素を除く、以下同様)6以上20以下の芳香族ジイソシアネート、炭素数2以上18以下の脂肪族ジイソシアネート、炭素数4以上15以下の脂環式ジイソシアネート、及びこれらのジイソシアネートの変性物(ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物。以下、変性ジイソシアネートともいう)、並びにこれらの2種以上の混合物。
前記脂肪族ジイソシアネートとしては、以下のものが挙げられる。エチレンジイソシアネート、テトラメチレンジイソシアネート、ヘキサメチレンジイソシアネート(HDI)、ドデカメチレンジイソシアネート。
前記脂環式ジイソシアネートとしては、以下のものが挙げられる。イソホロンジイソシアネート(IPDI)、ジシクロヘキシルメタン-4,4’-ジイソシアネート、シクロヘキシレンジイソシアネート、メチルシクロヘキシレンジイソシアネート。
前記芳香族ジイソシアネートとしては、例えば以下のものが挙げられる。m-及び/またはp-キシリレンジイソシアネート(XDI)、α,α,α’,α’-テトラメチルキシリレンジイソシアネート。
これらのうちで好ましいものは炭素数6以上15以下の芳香族ジイソシアネート、炭素数4以上12以下の脂肪族ジイソシアネート及び炭素数4以上15以下の脂環式ジイソシアネートであり、特に好ましいものはHDI、IPDI及びXDIである。
また、前記ポリウレタン樹脂は、前記したジイソシアネートに加えて、3官能以上のイソシアネート化合物を用いることもできる。
また、前記ウレタン樹脂に用いることのできるジオールとしては、以下のものが挙げられる。
アルキレングリコール(エチレングリコール、1,2-プロピレングリコール、1,3-プロピレングリコール);アルキレンエーテルグリコール(ポリエチレングリコール、ポリプロピレングリコール);脂環式ジオール(1,4-シクロヘキサンジメタノール);ビスフェノール類(ビスフェノールA);前記脂環式ジオールのアルキレンオキサイド(エチレンオキサイド、プロピレンオキサイド)付加物;ポリエステルジオール。
前記アルキレンエーテルグリコールのアルキル部分は直鎖状であっても、分岐していてもよい。本発明においては分岐構造のアルキレングリコールも好ましく用いることができる。
前記非晶性部位のガラス転移温度は、50℃以上130℃以下であることが好ましく、より好ましくは70℃以上130℃以下である。この範囲であることで、定着領域における弾性が維持されやすい。
結合剤を使う場合は、種々の結合剤が使用できる。多価カルボン酸、多価アルコール、多価イソシアネート、多官能エポキシ、多酸無水物を用いて、脱水反応や付加反応を行うことができる。
また、結晶構造をとりうる部位が結晶性ポリエステルで、結晶構造をとりえない部位がポリウレタン樹脂であるブロックポリマーの場合は、各部位を別々に調製した後、結晶性ポリエステルのアルコール末端とポリウレタンのイソシアネート末端とをウレタン化反応させることにより調製できる。また、アルコール末端を持つ結晶性ポリエステルおよびポリウレタンを構成するジオール、ジイソシアネートを混合し、加熱することでも合成が可能である。この場合、前記ジオールおよびジイソシアネートの濃度が高い反応初期は、これらが選択的に反応してポリウレタンを形成し、ある程度分子量が大きくなった後に、ポリウレタンのイソシアネート末端と結晶性ポリエステルのアルコール末端とのウレタン化が起こる。
上記ブロックポリマーの効果を有効に発現するためには、可能な限り結晶性ポリエステルのホモポリマーや非晶性ポリマーのホモポリマーがトナー中に存在しないほうが好ましい。すなわち、ブロック化率が高いことが好ましい。
上記ブロックポリマーは、ウレタン結合濃度が1.00mmol/g以上、3.20mmol/g以下であることが好ましい。
ウレタン結合濃度が1.00mmol/g以上、3.20mmol/gにすることにより、結晶構造をとりうる部位を多く含むようなブロックポリマーであっても高温での粘性を更に高いレベルで維持することができ、良好な光沢度を高温領域まで維持させることが可能である。より好ましくはウレタン結合濃度が1.40mmol/g以上、2.60mmol/g以下である。なお、ブロックポリマーのウレタン結合濃度は、例えば結晶構造をとりえない部位としてウレタン構造を導入する場合、この時に用いるジイソシアネートの添加量を調整することによって制御することができる。
また、上記ブロックポリマーは、示差走査熱量(DSC)測定から求められる、上記結着樹脂に由来する最大吸熱ピークのピーク温度Tpより25℃高い温度(Tp+25)(℃)における貯蔵弾性率G’(Tp+25℃)が1.0×103Pa以上、1.0×105Pa以下であることが好ましく、より好ましくは2.0×103Pa以上、7.0×104Pa以下である。この要件を満たすことによって高温領域での定着性及び光沢度を更に向上させることが可能である。
また、上記ブロックポリマーは、結晶構造をとりうる部位を、50質量%以上、85質量%以下含有することが好ましい。ブロックポリマーにおける結晶構造をとりうる部位の含有量が50質量%以上であることで、結晶構造をとりうる部位の集合体である結晶性部位のシャープメルト性が有効に発現されやすくなる。より好ましくは、60質量%以上85質量%以下である。
一方、ブロックポリマーにおける上記結晶構造をとりえない部位の含有量は、10質量%以上であることが好ましい。結晶構造をとりえない部位の含有量が10質量%以上であることで、シャープメルト後の結晶構造をとりえない部位の集合体である非晶性部位の弾性の維持が良好になる。より好ましくは、15質量%以上であり、更に好ましくは15質量%以上、50質量%未満であり、特に好ましくは15質量%以上、40質量%以下である。
上記ブロックポリマーは、テトラヒドロフラン(THF)可溶分のゲルパーミエーションクロマトグラフィー(GPC)測定において、数平均分子量(Mn)が8,000以上、30,000以下であることが好ましく、重量平均分子量(Mw)が15,000以上、60,000以下であることが好ましい。Mnのより好ましい範囲は、10,000以上、25,000以下であり、Mwのより好ましい範囲は、25,000以上、50,000以下である。また、Mw/Mnは6以下であることが好ましい。Mw/Mnのより好ましい範囲は3以下である。さらに、示差走査熱量計(DSC)で測定された最大吸熱ピークのピーク温度(Tp)が50℃以上、80℃以下であることが好ましく、55℃以上、70℃以下であることがより好ましい。
本発明における結着樹脂としては、上記ブロックポリマーに加えて、トナー用の結着樹脂として知られているその他の公知の樹脂を含有させてもよい。その場合、当該ブロックポリマーは、結晶構造をとりうる部位を、結着樹脂全量に対し、50質量%以上、85質量%以下含有することが好ましい。
3官能以上のエステルワックスは、例えば3官能以上の酸と長鎖直鎖飽和アルコールの縮合、または、3官能以上のアルコールと長鎖直鎖飽和脂肪酸の合成によって得られる。
本発明にて使用可能な3官能以上のアルコールとしては以下を挙げることが出来るが、これに限定されるものではない。また、場合によっては混合して用いることも可能である。グリセリン、トリメチロールプロパン、エリスリトール、ペンタエリスリトール、ソルビトール。また、これらの縮合物として、グリセリンの縮合したジグリセリン、トリグリセリン、テトラグリセリン、ヘキサグリセリン及びデカグリセリンの如きポリグリセリン、トリメチロールプロパンの縮合したジトリメチロールプロパン、トリストリメチロールプロパン及びペンタエリスリトールの縮合したジペンタエリスリトール及びトリスペンタエリスリトールが挙げられる。これらのうち、分岐構造をもつ構造が好ましく、ペンタエルスリトール又はジペンタエリスリトールがより好ましく、特にジペンタエリスリトールが好ましい。
本発明にて使用可能な長鎖直鎖飽和脂肪酸は、一般式CnH2n+1COOHで表され、nが5以上28以下のものが好ましく用いられる。
例えば以下のものを挙げることが出来るが、これに限定されるものではない。また、場合によっては混合して用いることも可能である。カプロン酸、カプリル酸、オクチル酸、ノニル酸、デカン酸、ドデカン酸、ラウリン酸、トリデカン酸、ミリスチン酸、パルミチン酸、ステアリン酸、ベヘン酸。その中でも、ワックスの融点の面からミリスチン酸、パルミチン酸、ステアリン酸、ベヘン酸が好ましい。
本発明にて使用可能な3官能以上の酸としては以下のものを挙げることが出来るが、これに限定されるものではない。また、場合によってはこれらの酸を混合して用いることも可能である。トリメリット酸、ブタンテトラカルボン酸。
本発明にて使用可能な長鎖直鎖飽和アルコールはCnH2n+1OHで表され、nが5以上28以下のものが好ましく用いられる。
例えば以下のものを挙げることが出来るが、これに限定されるものではない。また、場合によっては混合して用いることも可能である。カプリルアルコール、ラウリルアルコール、ミリスチルアルコール、パルミチルアルコール、ステアリルアルコール、ベヘニルアルコール。その中でも、ワックスの融点の面からミリスチルアルコール、パルミチルアルコール、ステアリルアルコール、ベヘニルアルコールが好ましい。
本発明において、ワックスのけん化価は、160mgKOH/g以上であることが好ましく、より好ましくは160mgKOH/g以上230mgKOH/g以下である。けん化価が160mgKOH/g以上の場合は、トナー中へのワックスの分散性がより良好になる。
本発明において、ワックスの分子量は、1500以上2200以下であることが好ましく、より好ましくは1600以上2000以下である。ワックスの分子量が上記の範囲内であることにより、昇温、降温を繰り返すヒートサイクル環境下で耐熱保存した後の流動性を維持しやすい。また、定着時にワックスが染み出しやすく、低温での耐オフセット性を更に向上させることができる。
本発明において、ワックスの含有量は、結着樹脂100質量部に対し2.0質量部以上8.0質量部以下が好ましい。ワックスの含有量が上記の範囲内であることにより、トナー保存時のワックスの染み出しが生じにくく、定着時には離型効果を良好に得ることで耐オフセット性を更に向上させることができる。
イエロー用着色剤としては、以下のものが挙げられる。縮合アゾ化合物、イソインドリノン化合物、アンスラキノン化合物、アゾ金属錯体、メチン化合物、アリルアミド化合物。具体的には、C.I.ピグメントイエロー12、13、14、15、17、62、74、83、93、94、95、109、110、111、128、129、147、155、168、180が好適に用いられる。
マゼンタ用着色剤としては、以下のものが挙げられる。縮合アゾ化合物、ジケトピロロピロール化合物、アンスラキノン、キナクリドン化合物、塩基染料レーキ化合物、ナフトール化合物、ベンズイミダゾロン化合物、チオインジゴ化合物、ペリレン化合物。具体的には、C.I.ピグメントレッド2、3、5、6、7、23、48:2、48:3、48:4、57:1、81:1、122、144、146、166、169、177、184、185、202、206、220、221、254が好適に用いられる。
シアン用着色剤としては、以下のものが挙げられる。銅フタロシアニン化合物およびその誘導体、アンスラキノン化合物、塩基染料レーキ化合物。具体的には、C.I.ピグメントブルー1、7、15、15:1、15:2、15:3、15:4、60、62、66が好適に用いられる。
本発明のトナーに用いられる着色剤は、色相角、彩度、明度、耐光性、OHP透明性、トナー中での分散性の観点から選択される。該着色剤は、結着樹脂100質量部に対し、1質量部以上20質量部以下添加して用いられることが好ましい。
黒色着色剤としてカーボンブラックを用いる場合も、結着樹脂100質量部に対し、1質量部以上20質量部以下添加して用いることが好ましい。また、黒色着色剤として磁性粉体を用いる場合、その添加量は結着樹脂100質量部に対し、40質量部以上150質量部以下であることが好ましい。
荷電制御剤としては、公知のものが利用でき、特に帯電スピードが速く、かつ、一定の帯電量を安定して維持できる荷電制御剤が好ましい。
荷電制御剤として、トナーを負荷電性に制御するものとしては、以下のものが挙げられる。有機金属化合物、キレート化合物が有効であり、モノアゾ金属化合物、アセチルアセトン金属化合物、芳香族オキシカルボン酸、芳香族ダイカルボン酸、オキシカルボン酸及びダイカルボン酸系の金属化合物。
本発明のトナーは、これら荷電制御剤を単独で或いは2種類以上組み合わせて含有させてもよい。
荷電制御剤の配合量は、結着樹脂100質量部に対して0.01質量部以上20質量部以下であることが好ましく、より好ましくは0.5質量部以上10質量部以下である。
結晶性ポリエステルは、融点以上の加熱を行うと、結晶性が崩れやすい。従って、非加熱にてトナーの製造を行えば、トナーに含有される結晶性ポリエステルの結晶性を崩すことなく、結晶性を維持した結晶性ポリエステルを含有するトナーが得られる。
非加熱でのトナー製法としては、例えば、下記に示す溶解懸濁法が挙げられるが、本発明はこれに限定されるわけではない。具体的には、本発明のトナーに用いられるトナー粒子の一形態として、(i)結着樹脂、着色剤及びワックスを、有機溶媒中に溶解または分散させた溶解物または分散物を得る工程、(ii)溶解物または分散物を、樹脂微粒子を分散させた超臨界状態または液体状態の二酸化炭素を有する分散媒体中に分散させて分散体を得る工程、(iii)分散体から有機溶媒を除去することによってトナー粒子を形成する工程を経て製造されたものが含まれる。
上記結晶性ポリエステルを含有するトナーの製造においては、上記のように分散媒体として高圧状態の二酸化炭素を用いることができる。すなわち、トナーに使用する結着樹脂の溶解液を高圧状態の二酸化炭素中に分散させて造粒を行い、造粒後の粒子に含まれる有機溶媒を二酸化炭素の相に抽出して除去した後、圧力を開放することによって二酸化炭素を分離し、トナー粒子として得る方法である。本発明において好適に用いられる高圧状態の二酸化炭素とは、液体あるいは超臨界状態の二酸化炭素である。なお、分散媒体は高圧状態の二酸化炭素が主成分(50質量%以上)であることが好ましい。
ここで、液体の二酸化炭素とは、二酸化炭素の相図上における三重点(温度=-57℃、圧力=0.5MPa)と臨界点(温度=31℃、圧力=7.4MPa)を通る気液境界線、臨界温度の等温線、および固液境界線に囲まれた部分の温度、圧力条件にある二酸化炭素を表す。また、超臨界状態の二酸化炭素とは、上記二酸化炭素の臨界点以上の温度、圧力条件にある二酸化炭素を表す。
本発明において、分散媒体中には他の成分として有機溶媒が含まれていてもよい。この場合、二酸化炭素と有機溶媒とが均一相を形成することが好ましい。
この方法によれば、高圧下で造粒が行われるため、結晶性ポリエステルの結晶性を維持しやすいばかりでなく、より高めることも可能である点で特に好適である。
以下に、本発明のトナー粒子を得る上で好適な、液体あるいは超臨界状態の二酸化炭素を分散媒体として用いるトナー粒子の製造法を例示して説明する。
まず、結着樹脂を溶解することのできる有機溶媒中に、結着樹脂、着色剤、ワックスおよび必要に応じて他の添加物を加え、ホモジナイザー、ボールミル、コロイドミル、超音波分散機の如き分散機によって均一に溶解または分散させる。
本発明で用いる有機溶媒としては、結着樹脂を溶解できるものが好ましく、アセトン及びメチルエチルケトンの如きケトン系溶媒が好ましい。その中でも、アセトンを好ましく用いることができる。
前記結着樹脂はアセトン不溶分が1.0質量%以下であることが好ましい。アセトン不溶分が1.0質量%を超える場合、トナー作製時の粘度が高くなり、トナー粒度が大きくなったり、粒度分布が広くなりやすい。より好ましくは、0.5質量%以下である。
次に、こうして得られた溶解液あるいは分散液(以下、単に結着樹脂溶解液という)を、液体あるいは超臨界状態の二酸化炭素中に分散させて油滴を形成する。
このとき、分散媒体としての液体あるいは超臨界状態の二酸化炭素中には、分散剤を分散させておくことが好ましい。分散剤としては、無機微粒子分散剤、有機微粒子分散剤、それらの混合物のいずれでもよく、目的に応じて単独で用いても2種以上を併用してもよい。
上記無機微粒子分散剤としては、例えば、シリカ、アルミナ、酸化亜鉛、チタニア、酸化カルシウムの如き無機微粒子が挙げられる。
上記有機微粒子分散剤としては、例えば、ビニル樹脂、ウレタン樹脂、エポキシ樹脂、エステル樹脂、ポリアミド、ポリイミド、シリコーン樹脂、フッ素樹脂、フェノール樹脂、メラミン樹脂、ベンゾグアナミン系樹脂、ユリア樹脂、アニリン樹脂、アイオノマー樹脂、ポリカーボネート、セルロースおよびこれらの混合物の如き有機微粒子が挙げられる。
非晶性樹脂からなる有機樹脂微粒子を分散剤として使用すると、二酸化炭素が前記樹脂中に溶解して樹脂を可塑化させ、ガラス転移温度を低下させるため、造粒の際に粒子同士が凝集を起こしやすくなる。したがって、有機樹脂微粒子としては結晶性を有する樹脂を使用することが好ましく、非晶性樹脂を用いる場合は、架橋構造を導入することが好ましい。また非晶性樹脂粒子を結晶性樹脂で被覆した微粒子であってもよい。
上記分散剤は、そのまま用いてもよいが、造粒時における上記油滴表面への吸着性を向上させるため、各種処理によって表面改質したものを用いてもよい。具体的には、シラン系、チタネート系、アルミネート系の如きカップリング剤による表面処理や、各種界面活性剤による表面処理、ポリマーによるコーティング処理が挙げられる。
油滴の表面に吸着した分散剤は、トナー粒子形成後もそのまま残留するため、分散剤として樹脂微粒子を用いた場合には、樹脂微粒子で表面が被覆されたトナー粒子を形成することができる。
上記樹脂微粒子の粒径は、体積平均粒径で30nm以上、300nm以下であることが好ましく、より好ましくは、50nm以上、100nm以下である。樹脂微粒子の粒径を上記の範囲内にすることによって、造粒時の油滴の安定性がより向上させることができる。
また、上記樹脂微粒子の配合量は、油滴の形成に使用する上記結着樹脂溶解液中の固形分100質量部に対して3.0質量部以上15.0質量部以下であることが好ましく、油滴の安定性や所望する粒径に合わせて適宜調整することができる。
本発明において、上記分散剤を液体あるいは超臨界状態の二酸化炭素中に分散させる方法は、如何なる方法を用いてもよい。具体例としては、上記分散剤と液体あるいは超臨界状態の二酸化炭素を容器内に仕込み、撹拌や超音波照射により直接分散させる方法が挙げられる。また、液体あるいは超臨界状態の二酸化炭素を仕込んだ容器に、上記分散剤を有機溶媒に分散させた分散液を、高圧ポンプを用いて導入する方法が挙げられる。
また、本発明において、上記結着樹脂溶解液を液体あるいは超臨界状態の二酸化炭素中に分散させる方法は、如何なる方法を用いてもよい。具体例としては、上記分散剤を分散させた状態の液体あるいは超臨界状態の二酸化炭素を入れた容器に、上記結着樹脂溶解液を、高圧ポンプを用いて導入する方法が挙げられる。また、上記結着樹脂溶解液を仕込んだ容器に、上記分散剤を分散させた状態の液体あるいは超臨界状態の二酸化炭素を導入してもよい。
本発明において、上記液体あるいは超臨界状態の二酸化炭素による分散媒体は、単一相であることが好ましい。上記結着樹脂溶解液を液体あるいは超臨界状態の二酸化炭素中に分散させて造粒を行う場合、油滴中の有機溶媒の一部は分散体中に移行する。このとき、二酸化炭素の相と有機溶媒の相が分離した状態で存在することは、油滴の安定性が損なわれる原因となり好ましくない。したがって、上記分散媒体の温度や圧力、液体あるいは超臨界状態の二酸化炭素に対する上記結着樹脂溶解液の量は、二酸化炭素と有機溶媒とが均一相を形成し得る範囲内に調整することが好ましい。
また、上記分散媒体の温度および圧力については、造粒性(油滴形成のし易さ)や上記結着樹脂溶解液中の構成成分の上記分散媒体への溶解性にも注意が必要である。例えば、上記結着樹脂溶解液中の結着樹脂やワックスは、温度条件や圧力条件によっては、上記分散媒体に溶解することがある。通常、低温、低圧になるほど上記成分の分散媒体への溶解性は抑制されるが、形成した油滴が凝集・合一を起こし易くなり、造粒性は低下する。一方、高温、高圧になるほど造粒性は向上するものの、上記成分が上記分散媒体に溶解し易くなる傾向を示す。
さらに、上記分散媒体の温度については、結晶性ポリエステル成分の結晶性が損なわれないよう、結晶性ポリエステルの融点よりも低い温度でなければならない。
したがって、トナー粒子の製造において、上記分散媒体の温度は10℃以上、結晶性ポリエステルの融点未満の温度範囲であることが好ましい。
また、上記分散媒体を形成する容器内の圧力は、1MPa以上、20MPa以下であることが好ましく、2MPa以上、15MPa以下であることがより好ましい。尚、本発明における圧力とは、分散媒体中に二酸化炭素以外の成分が含まれる場合には、その全圧を示す。
また、本発明における分散媒体中に占める二酸化炭素の割合は、70質量%以上であることが好ましく、80質量%以上であることがより好ましく、90質量%以上であることがさらに好ましい。
こうして造粒が完了した後、油滴中に残留している有機溶媒を、液体あるいは超臨界状態の二酸化炭素による分散媒体を介して除去する。具体的には、油滴が分散された上記分散媒体にさらに液体あるいは超臨界状態の二酸化炭素を混合して、残留する有機溶媒を二酸化炭素の相に抽出し、この有機溶媒を含む二酸化炭素を、さらに液体あるいは超臨界状態の二酸化炭素で置換することによって行う。
上記分散媒体と上記液体あるいは超臨界状態の二酸化炭素の混合は、上記分散媒体に、これよりも高圧の液体あるいは超臨界状態の二酸化炭素を加えてもよく、また、上記分散媒体を、これよりも低圧の液体あるいは超臨界状態の二酸化炭素中に加えてもよい。
そして、有機溶媒を含む二酸化炭素をさらに液体あるいは超臨界状態の二酸化炭素で置換する方法としては、容器内の圧力を一定に保ちつつ、液体あるいは超臨界状態の二酸化炭素を流通させる方法が挙げられる。このとき、形成されるトナー粒子は、フィルターで補足しながら行う。
上記液体あるいは超臨界状態の二酸化炭素による置換が十分でなく、分散媒体中に有機溶媒が残留した状態であると、得られたトナー粒子を回収するために容器を減圧する際、上記分散媒体中に溶解した有機溶媒が凝縮してトナー粒子が再溶解したり、トナー粒子同士が合一したりするといった不具合が生じる場合がある。したがって、上記液体あるいは超臨界状態の二酸化炭素による置換は、有機溶媒が完全に除去されるまで行う必要がある。流通させる液体あるいは超臨界状態の二酸化炭素の量は、上記分散媒体の体積に対して1倍以上、100倍以下が好ましく、さらに好ましくは1倍以上、50倍以下、最も好ましくは1倍以上、30倍以下である。
容器を減圧し、トナー粒子が分散した液体あるいは超臨界状態の二酸化炭素を含む分散体からトナー粒子を取り出す際は、一気に常温、常圧まで減圧してもよいが、独立に圧力制御された容器を多段に設けることによって段階的に減圧してもよい。減圧速度は、トナー粒子が発泡しない範囲で設定することが好ましい。
尚、本発明において使用する有機溶媒や、液体あるいは超臨界状態の二酸化炭素は、リサイクルすることが可能である。
更に本発明のトナーは、結晶性ポリエステルの融点よりも低い温度条件にて加熱処理する工程を経ることが好ましい。本発明では、以後、この熱処理をアニール処理と称する。一般に、結晶性樹脂は、アニール処理を施すことによって結晶性が高まることが知られている。その原理は以下のように考えられている。すなわち、結晶性材料にアニール処理を行うと、その熱によって高分子鎖の分子運動性がある程度高くなるために、高分子鎖がより安定な構造、すなわち規則的な結晶構造へと再配向することで、結晶化が起こるというものである。結晶性材料の融点以上の温度で処理した場合には、高分子鎖は再配向に必要なエネルギーよりも高いエネルギーを得ることになるため、再結晶化は起こらない。
したがって、本発明におけるアニール処理は、トナー中の結晶性ポリエステルの分子運動を可能な限り活発化させるため、結晶性ポリエステルの融点に対して、限られた温度範囲内で行うことが重要である。
本発明において、アニール処理の温度は、予め得られたトナー粒子の示差走査熱量(DSC)測定を行い、結晶性ポリエステルに由来する吸熱ピークのピーク温度を求めた後、このピーク温度に応じて決めればよい。具体的には、昇温速度10.0℃/minの条件でDSC測定したときに求められるピーク温度から15℃差し引いた温度以上、5℃差し引いた温度以下で熱処理を行うことが好ましい。より好ましくは、上記ピーク温度から10℃差し引いた温度以上、5℃差し引いた温度以下の温度範囲である。
本発明において、アニール処理は、トナー粒子の形成工程後であれば、どの段階で行ってもよい。
また、アニール処理時間は、トナー中の結晶性ポリエステルの割合や種類、結晶状態によって適宜調整可能であるが、通常は1時間以上、50時間以下の範囲で行うことが好ましい。より好ましくは、2時間以上、24時間以下の範囲である。
ワックスを含有するトナーを用いる場合、結晶性ポリエステルとの相溶で、アニール速度が変わる場合がある。結晶性ポリエステルとワックスの相溶が小さい場合、結晶性ポリエステルの結晶化速度は速くなりやすく、製造面において相溶を抑えたワックスを用いることは有効である。
シリカ微粉体としては、ケイ素ハロゲン化物の蒸気相酸化により生成された乾式シリカ又はヒュームドシリカ、及び水ガラスから製造される湿式シリカが挙げられる。無機微粉体としては、表面及びシリカ微粉体の内部にあるシラノール基が少なく、またNa2O、SO3 2-の少ない乾式シリカの方が好ましい。また乾式シリカは、製造工程において、塩化アルミニウム、塩化チタン他の如き金属ハロゲン化合物をケイ素ハロゲン化合物と共に用いることによって製造された、シリカと他の金属酸化物の複合微粉体であっても良い。無機微粉体は、トナーの流動性の改良及びトナーの帯電性の均一化のためにトナー粒子に外添されることが好ましい。無機微粉体を疎水化処理することによって、トナーの帯電量の調整、環境安定性の向上、高湿環境下での特性の向上を達成することができるので、疎水化処理された無機微粉体を用いることがより好ましい。
無機微粉体の疎水化処理の処理剤としては、未変性のシリコーンワニス、各種変性シリコーンワニス、未変性のシリコーンオイル、各種変性シリコーンオイル、シラン化合物、シランカップリング剤、その他有機ケイ素化合物、有機チタン化合物が挙げられる。これらの処理剤は単独で或いは併用して用いられても良い。
その中でも、シリコーンオイルにより処理された無機微粉体が好ましい。より好ましくは、無機微粉体をカップリング剤で疎水化処理すると同時或いは処理した後に、シリコーンオイル処理された疎水化処理無機微粉体が高湿環境下でもトナーの帯電量を高く維持し、選択現像性を低減する上でよい。
上記無機微粉体の添加量は、トナー粒子100質量部に対して、0.1質量部以上4.0質量部以下であることが好ましく、より好ましくは0.2質量部以上3.5質量部以下である。
本発明のトナーの重量平均粒径(D4)は、3.0μm以上、8.0μm以下であることが好ましく、より好ましくは、5.0μm以上、7.0μm以下である。このような重量平均粒径(D4)のトナーを用いることは、ハンドリング性を良好にしつつ、ドットの再現性を十分に満足する上で好ましい。
更に、本発明のトナーの重量平均粒径(D4)と個数平均粒径(D1)の比D4/D1は1.25以下であることが好ましい。より好ましくは1.20以下である。
<トナー等の示差走査熱量(DSC)測定における最大吸熱ピークのピーク温度(Tp)及び最大吸熱ピークの吸熱量(ΔH)の測定方法>
本発明におけるトナー等(トナー、結晶性ポリエステル、ブロックポリマーを含む)の最大吸熱ピークのピーク温度(Tp)は、DSC Q1000(TA Instruments社製)を使用して以下の条件にて測定を行う。
昇温速度:10℃/min
測定開始温度:20℃
測定終了温度:180℃
装置検出部の温度補正はインジウムと亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いる。
具体的には、試料約5mgを精秤し、銀製のパンの中に入れ、一回測定を行う。リファレンスとしては銀製の空パンを用いる。
トナーを試料とする場合において、最大吸熱ピーク(結着樹脂由来の最大吸熱ピーク)がワックス及び結着樹脂以外の樹脂(例えば、コアシェル構造を有するトナーにおけるシェル相の樹脂)の吸熱ピークと重なっていない場合には、得られた最大吸熱ピークの吸熱量をそのまま結着樹脂に由来する最大吸熱ピークの吸熱量として扱う。一方、トナーを試料とする場合において、ワックス及び結着樹脂以外の樹脂の吸熱ピークが結着樹脂の最大吸熱ピークと重なっている場合は、ワックス及び結着樹脂以外の樹脂に由来する吸熱量を、得られた最大吸熱ピークの吸熱量から差し引く必要がある。
以下の方法により、ワックス及び結着樹脂以外の樹脂に由来する吸熱量を、得られた最大吸熱ピークの吸熱量から差し引き、結着樹脂に由来する最大吸熱ピークの吸熱量を得る。
まず、別途ワックス単体のDSC測定を行い、吸熱特性を求める。次いでトナー中のワックスの含有量を求める。トナー中のワックスの含有量の測定は、特に制限されないが、例えば、DSC測定におけるピーク分離や、公知の構造解析によって行うことができる。その後、トナー中のワックス含有量からワックスに起因する吸熱量を算出し、上記得られた最大吸熱ピークの吸熱量からこの分を差し引けばよい。ワックスが結着樹脂と相溶しやすい場合には、前記ワックスの含有量に相溶率を乗じた上でワックスに起因する吸熱量を算出して差し引いておく必要がある。相溶率は、結着樹脂とワックスとを(結着樹脂の質量:ワックスの質量)で100:6の比率で混合したものについて求めた吸熱量を、予め求めておいた前記結着樹脂の吸熱量とワックス単体の吸熱量から算出される理論吸熱量で除した値から算出する。
なお、結着樹脂以外の樹脂に由来する吸熱量についても前記のワックスと同様の方法によって求める。ここで、相溶率は、結着樹脂と結着樹脂以外とを(結着樹脂の質量:結着樹脂以外の樹脂の質量)で100:6の比率で混合したものについて求めた吸熱量を、予め求めておいた前記結着樹脂の吸熱量と結着樹脂以外の樹脂の吸熱量から算出される理論吸熱量で除した値から算出する。
また、測定においては、結着樹脂1g当りの吸熱量とするために、試料の質量から結着樹脂以外の成分の質量を除く必要がある。
結着樹脂以外の成分の含有量は、公知の分析手段によって測定することができる。分析が困難な場合には、トナーの焼却残灰分量を求め、それにワックス等の焼却される結着樹脂以外の成分の量を加えた量を結着樹脂以外の成分の含有量と見なして、トナーの質量から差し引くことによって求めることができる。
トナー中の焼却残灰分は以下の手順で求める。予め秤量した30mlの磁性るつぼに約2gのトナーを入れる。るつぼを電気炉に入れ、約900℃で約3時間加熱し、電気炉中で放冷し、常温下でデシケーター中に1時間以上放冷し、焼却残灰分を含むるつぼの質量を秤量し、るつぼの質量を差し引くことにより焼却残灰分を算出する。
なお、最大吸熱ピークとは、ピークが複数あった場合に、吸熱量が最大となるピークのことを意味する。また、最大吸熱ピークの吸熱量(ΔH)はピークの面積から装置付属の解析ソフトを用いて計算により求める。
<ワックスの示差走査熱量(DSC)測定における最大吸熱ピークのピーク温度(ワックスの融点)の測定方法>
ワックスの示差走査熱量(DSC)測定における最大吸熱ピークのピーク温度(ワックスの融点)は、示差走査熱量計であるDSC Q1000(TA Instruments社製)を用い、ASTM D3418-82に準じて測定した。
装置検出部の温度補正はインジウムと亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いた。
具体的には、試料約2mgを精秤、アルミニウム製のパンの中に入れ、リファレンスとして空のアルミニウム製のパンを用い、測定温度範囲30乃至180℃の間で、昇温速度10℃/minで測定を行った。尚、測定においては、一度180℃まで昇温させ、続いて30℃まで降温し、その後に再度昇温を行う。この2度目の昇温過程におけるDSC曲線の最大の吸熱ピークを示す温度をワックスの融点とした。このとき、上記最大吸熱ピークとは、ピークが複数存在する場合には、最も吸熱量の大きいピークをいう。
Tgの測定方法は、DSC Q1000(TA Instruments社製)を用いて以下の条件にて測定を行う。
《測定条件》
・モジュレーションモード
・昇温速度:0.5℃/分
・モジュレーション温度振幅:±1.0℃/分
・測定開始温度:25℃
・測定終了温度:130℃
昇温速度を変えるときは、新しい測定サンプルを用意した。昇温は1度のみ行い、「Reversing Heat Frow」を縦軸にとることでDSCカーブを得、オンセット値をTgとした。
トナーの重量平均粒径(D4)および個数平均粒径(D1)は、以下のようにして算出する。測定装置としては、100μmのアパーチャーチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer 3」(登録商標、ベックマン・コールター社製)を用いる。測定条件の設定及び測定データの解析は、付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター社製)を用いる。尚、測定は実効測定チャンネル数2万5千チャンネルで行う。
測定に使用する電解水溶液は、特級塩化ナトリウムをイオン交換水に溶解して濃度が約1質量%となるようにしたもの、例えば、「ISOTON II」(ベックマン・コールター社製)が使用できる。
尚、測定、解析を行う前に、以下のように前記専用ソフトの設定を行う。
前記専用ソフトの「標準測定方法(SOM)を変更」画面において、コントロールモードの総カウント数を50000粒子に設定し、測定回数を1回、Kd値は「標準粒子10.0μm」(ベックマン・コールター社製)を用いて得られた値を設定する。「閾値/ノイズレベルの測定ボタン」を押すことで、閾値とノイズレベルを自動設定する。また、カレントを1600μAに、ゲインを2に、電解液をISOTON IIに設定し、「測定後のアパーチャーチューブのフラッシュ」にチェックを入れる。
前記専用ソフトの「パルスから粒径への変換設定」画面において、ビン間隔を対数粒径に、粒径ビンを256粒径ビンに、粒径範囲を2μmから60μmまでに設定する。
具体的な測定法は以下の通りである。
(1)Multisizer 3専用のガラス製250ml丸底ビーカーに前記電解水溶液約200mlを入れ、サンプルスタンドにセットし、スターラーロッドの撹拌を反時計回りで24回転/秒にて行う。そして、専用ソフトの「アパーチャーのフラッシュ」機能により、アパーチャーチューブ内の汚れと気泡を除去しておく。
(2)ガラス製の100ml平底ビーカーに前記電解水溶液約30mlを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で約3質量倍に希釈した希釈液を約0.3ml加える。
(3)発振周波数50kHzの発振器2個を、位相を180度ずらした状態で内蔵し、電気的出力120Wの超音波分散器「Ultrasonic Dispersion System Tetora150」(日科機バイオス社製)を準備する。超音波分散器の水槽内に約3.3lのイオン交換水を入れ、この水槽中にコンタミノンNを約2ml添加する。
(4)前記(2)のビーカーを前記超音波分散器のビーカー固定穴にセットし、超音波分散器を作動させる。そして、ビーカー内の電解水溶液の液面の共振状態が最大となるようにビーカーの高さ位置を調整する。
(5)前記(4)のビーカー内の電解水溶液に超音波を照射した状態で、トナー約10mgを少量ずつ前記電解水溶液に添加し、分散させる。そして、さらに60秒間超音波分散処理を継続する。尚、超音波分散にあたっては、水槽の水温が10℃以上40℃以下となる様に適宜調節する。
(6)サンプルスタンド内に設置した前記(1)の丸底ビーカーに、ピペットを用いてトナーを分散した前記(5)の電解水溶液を滴下し、測定濃度が約5%となるように調整する。そして、測定粒子数が50000個になるまで測定を行う。
(7)測定データを装置付属の前記専用ソフトにて解析を行い、重量平均粒径(D4)および個数平均粒径(D1)を算出する。尚、前記専用ソフトでグラフ/体積%と設定したときの、「分析/体積統計値(算術平均)」画面の「平均径」が重量平均粒径(D4)であり、前記専用ソフトでグラフ/個数%と設定したときの、「分析/個数統計値(算術平均)」画面の「平均径」が個数平均粒径(D1)である。
樹脂(ブロックポリマーを含む)のゲルパーミエーションクロマトグラフィー(GPC)による分子量分布、数平均分子量(Mn)及び重量平均分子量(Mw)は、樹脂のテトラヒドロフラン(THF)可溶分を、THFを溶媒としたGPC(ゲルパーミエーションクロマトグラフィー)により測定する。測定条件は以下の通りである。
(1)測定試料の作製
樹脂(試料)とTHFとを5mg/mlの濃度で混合し、室温にて5乃至6時間放置した後、充分に振とうし、THFと試料を試料の合一体がなくなるまで良く混ぜた。更に、室温にて12時間以上静置した。この時、試料とTHFの混合開始時点から、静置終了の時点までの時間が24時間以上となる様にした。
その後、サンプル処理フィルター(ポアサイズ0.45乃至0.5μm、マイショリディスクH-25-2[東ソー社製])を通過させたものをGPCの試料とする。
(2)試料の測定
40℃のヒートチャンバー中でカラムを安定化させ、この温度に於けるカラムに、溶媒としてTHFを毎分1mlの流速で流し、試料濃度を5mg/mlに調整した樹脂のTHF試料溶液を200μl注入して測定する。
試料の分子量測定にあたっては、試料の有する分子量分布を数種の単分散ポリスチレン標準試料により作製された検量線の対数値とカウント数との関係から算出する。
検量線作成用の標準ポリスチレン試料としては、Pressure ChemicalCo.製或いは東洋ソーダ工業社製の、分子量が6×102、2.1×103、4×103、1.75×104、5.1×104、1.1×105、3.9×105、8.6×105、2×106、4.48×106のものを用いる。又、検出器にはRI(屈折率)検出器を用いる。
尚、カラムとしては、1×103乃至2×106の分子量領域を適確に測定する為に、市販のポリスチレンゲルカラムを下記のように複数組合せて用いた。本発明における、GPCの測定条件は以下の通りである。
[GPC測定条件]
装置:LC-GPC 150C(ウォーターズ社製)
カラム:KF801、802、803、804、805、806、807(ショウデックス製)の7連
カラム温度:40℃
移動相:THF(テトラヒドロフラン)
ブロックポリマーにおける結晶構造をとりうる部位の割合(質量%)の測定は、1H-NMRにより以下の条件にて行う。
測定装置 :FT NMR装置 JNM-EX400(日本電子社製)
測定周波数:400MHz
パルス条件:5.0μs
周波数範囲:10500Hz
積算回数 :64回
測定温度 :30℃
試料 :ブロックポリマー50mgを内径5mmのサンプルチューブに入れ、溶媒として重クロロホルム(CDCl3)を添加し、これを40℃の恒温槽内で溶解させて調製する。得られた1H-NMRチャートより、結晶構造をとりうる部位の構成要素に帰属されるピークの中から、他の構成要素に帰属されるピークとは独立したピークを選択し、このピークの積分値S1を算出する。同様に、非晶性部位の構成要素に帰属されるピークの中から、他の構成要素に帰属されるピークとは独立したピークを選択しこのピークの積分値S2を算出する。結晶構造をとりうる部位の割合は、上記積分値S1および積分値S2を用いて、以下のようにして求める。尚、n1、n2は着眼したピークが帰属される構成要素における水素の数である。
結晶構造をとりうる部位の割合(モル%)=
{(S1/n1)/((S1/n1)+(S2/n2))}×100
上記結晶構造をとりうる部位の割合(モル%)を各成分の分子量により質量%に換算する。
基本操作はJIS K-0070に準ずる。
(1)試料を1乃至3g精秤し、重さをW(g)とする。
(2)300(ml)の三角フラスコに試料を入れ、0.5mol/lのKOHのエタノール溶液25mlを加える。
(3)三角フラスコに空気冷却器を取り付け、ときどき内容物を振り混ぜながら30分間水浴、砂浴又は熱板で穏やかに加熱して反応させる。加熱するときは還流するエタノールの環が空気冷却器の上端に達しないように加熱温度を調節する。
(4)反応が終わった後、直ちに冷却し、内容物が寒天状に固まらないうちに空気冷却器の上から少量の水、又はキシレン/エタノール(1/3)混合溶液を吹き付けてその内壁を洗浄した後、空気冷却器を外す。
(5)0.5mol/lの塩酸を用いて、電位差滴定装置を用いて滴定する(例えば、京都電子株式会社製の電位差滴定装置AT-400(win workstation)とABP-410電動ビューレットを用いての自動滴定が利用できる。)。
(6)この時の塩酸の使用量をS(ml)とし、同時にブランクを測定し、この時の塩酸の使用量をB(ml)とする。
(7)次式によりけん化価を計算する。fは塩酸のファクターである。
けん化価(mgKOH/g)={(B-S)×f×28.05}/W
樹脂微粒子分散液中の樹脂微粒子およびワックス分散液中のワックス粒子の体積平均粒径はマイクロトラック粒度分布測定装置HRA(X-100)(日機装社製)を用い、粒径0.001μm乃至10μmのレンジ設定で測定を行い、体積平均粒径(μm又はnm)として測定する。
樹脂のウレタン結合濃度の測定は、1H-NMRを用いて行う。
1H-NMRの測定は、以下の条件にて行う。
測定装置 :FT NMR装置 JNM-EX400(日本電子社製)
測定周波数:400MHz
パルス条件:5.0μs
周波数範囲:10500Hz
積算回数 :64回
測定温度 :30℃
試料 :測定試料50mgを内径5mmのサンプルチューブに入れ、溶媒としてCDCl3を添加し、これを40℃の恒温槽内で溶解させて調製する。
得られた1H-NMRチャートより、用いている樹脂の構成単位の水素量比をもとめ、構成物のmol比を求める。
求めた、mol比と、分子量から1gあたりのウレタン結合を構成する構成単位の濃度を求め、これをウレタン結合の濃度〔mmol/g〕とする。
ブロックポリマーの貯蔵弾性率G’は、粘弾性測定装置(レオメーター)ARES(Rheometrics Scientific社製)を用いて測定を行う。測定の概略は、Rheometrics Scientific社製発行のARES操作マニュアル902-30004(1997年8月版)、902-00153(1993年7月版)に記載されているが、以下の通りである。
・測定治具:torsion rectangular
・測定試料:ブロックポリマーを、加圧成型機を用い幅約12mm、高さ約20mm、厚み約2.5mmの直方体型試料を作製する(常温で1分間15kNを維持する)。加圧成型機はNPaシステム社製100kNプレスNT-100Hを用いる。
治具及びサンプルを常温(23℃)に1時間放置した後、治具にサンプルを取り付ける。図4参照。図4のように、測定部の幅約12mm、厚さ約2.5mm、高さ10mmになるように固定する。測定開始温度30.00℃まで10分間かけて温度調節した後、下記設定で測定を行う。
・測定周波数 :6.28ラジアン/秒
・測定歪みの設定:初期値を0.1%に設定し、自動測定モードにて測定を行う
・試料の伸長補正:自動測定モードにて調整
・測定温度 :30℃から150℃まで毎分2℃の割合で昇温する
・測定間隔 :30秒おき、すなわち1℃おきに粘弾性データを測定する
Microsoft社製Windows2000(登録商標)上で動作するRSI Orchesrator(制御、データ収集および解析ソフト)(Rheometrics Scientific社製)へ、インターフェースを通じてデータ転送する。
このうち、上記トナーの示査走査熱量(DSC)測定から求められる、結着樹脂に由来する最大吸熱ピークのピーク温度Tpより、25℃高い温度におけるブロックポリマーの貯蔵弾性率(G’(Tp+25))の値を読み取る。
<結晶性ポリエステル1の合成>
加熱乾燥した二口フラスコに、窒素を導入しながら以下の原料を仕込んだ。
・セバシン酸 136.2質量部
・1,4-ブタンジオール 63.8質量部
・酸化ジブチルスズ 0.1質量部
減圧操作により系内を窒素置換した後、180℃にて6時間撹拌を行った。その後、撹拌を続けながら減圧下にて230℃まで徐々に昇温し、更に2時間保持した。粘稠な状態となったところで空冷し、反応を停止させることで、結晶性ポリエステル1を合成した。結晶性ポリエステル1の物性を表1に示す。
<結晶性ポリエステル2の合成>
結晶性ポリエステル1の合成において、原料の仕込みを以下のように変えた以外は全て同様にして、結晶性ポリエステル2を合成した。結晶性ポリエステル2の物性を表1に示す。
・セバシン酸 75.9質量部
・アジピン酸 53.9質量部
・1,4-ブタンジオール 70.2質量部
・酸化ジブチルスズ 0.1質量部
<結晶性ポリエステル3の合成>
結晶性ポリエステル1の合成において、原料の仕込みを以下のように変えた以外は全て同様にして、結晶性ポリエステル3を合成した。結晶性ポリエステル3の物性を表1に示す。
・ドデカン二酸 116.5質量部
・1,10-デカンジオール 83.5質量部
・酸化ジブチルスズ 0.1質量部
<結晶性ポリエステル4の合成>
結晶性ポリエステル1の合成において、原料の仕込みを以下のように変えた以外は全て同様にして、結晶性ポリエステル4を合成した。結晶性ポリエステル4の物性を表1に示す。
・セバシン酸 105.0質量部
・アジピン酸 28.0質量部
・1,4-ブタンジオール 67.0質量部
・酸化ジブチルスズ 0.1質量部
<結晶性ポリエステル5の合成>
結晶性ポリエステル1の合成において、原料の仕込みを以下のように変えた以外は全て同様にして、結晶性ポリエステル5を合成した。結晶性ポリエステル5の物性を表1に示す。
・オクタデカン二酸 152.9質量部
・1,4-ブタンジオール 47.1質量部
・酸化ジブチルスズ 0.1質量部
<結晶性ポリエステル6の合成>
結晶性ポリエステル1の合成において、原料の仕込みを以下のように変えた以外は全て同様にして、結晶性ポリエステル6を合成した。結晶性ポリエステル6の物性を表1に示す。
・セバシン酸 111.7質量部
・アジピン酸 21.9質量部
・1,4-ブタンジオール 66.4質量部
・酸化ジブチルスズ 0.1質量部
<結晶性ポリエステル7の合成>
結晶性ポリエステル1の合成において、原料の仕込みを以下のように変えた以外は全て同様にして、結晶性ポリエステル7を合成した。結晶性ポリエステル7の物性を表1に示す。
・テトラデカン二酸 134.0質量部
・1,6-ヘキサンジオール 66.0質量部
・酸化ジブチルスズ 0.1質量部
<結晶性ポリエステル8の合成>
結晶性ポリエステル1の合成において、原料の仕込みを以下のように変えた以外は全て同様にして、結晶性ポリエステル8を合成した。結晶性ポリエステル8の物性を表1に示す。
・セバシン酸 137.5質量部
・1,4-ブタンジオール 62.5質量部
・酸化ジブチルスズ 0.1質量部
・結晶性ポリエステル1 210.0質量部
・キシリレンジイソシアネート(XDI) 56.0質量部
・シクロヘキサンジメタノール(CHDM) 34.0質量部
・テトラヒドロフラン(THF) 300.0質量部
撹拌装置および温度計を備えた反応容器中に、窒素置換をしながら上記を仕込んだ。50℃まで加熱し、15時間かけてウレタン化反応を施した。その後、ターシャリーブチルアルコール3.0質量部を添加し、イソシアネート末端を修飾した。溶媒であるTHFを留去し、ブロックポリマー1を得た。得られたブロックポリマーの物性を表2に示す。
<ブロックポリマー2乃至12の合成>
ブロックポリマー1の合成において、表2に示す材料、配合量に変更することによりブロックポリマー2乃至12を得た。得られたブロックポリマー2乃至12の物性を表2に示す。
撹拌装置のついたビーカーに、アセトンを100.0質量部、ブロックポリマー1を100.0質量部投入し、温度40℃で完全に溶解するまで撹拌を続け、ブロックポリマー樹脂溶液1を調製した。同様にして、ブロックポリマー樹脂溶液2乃至12を調製した。
・キシリレンジイソシアネート(XDI) 117.0質量部
・シクロヘキサンジメタノール(CHDM) 83.0質量部
・アセトン 200.0質量部
撹拌装置および温度計を備えた反応容器中に、窒素置換をしながら上記を仕込んだ。50℃まで加熱し、15時間かけてウレタン化反応を施した。その後、ターシャリーブチルアルコール3.0質量部を添加し、イソシアネート末端を修飾した。溶媒であるアセトンを留去し、非晶性樹脂1を得た。得られた非晶性樹脂1はMnが4,400、Mwが20,000であった。
・結晶性ポリエステル8 115.0質量部
・イオン性界面活性剤ネオゲンRK(第一工業製薬) 5.0質量部
・イオン交換水 180.0質量部
以上の各成分を混合し100℃に加熱して、IKA社製ウルトラタラックスT50にて十分に分散後、圧力吐出型ゴーリンホモジナイザーで分散処理を1時間行い、体積平均粒径が180nm、固形分量が38.3質量%の結晶性ポリエステル樹脂分散液1を得た。
・非晶性樹脂1 115.0質量部
・イオン性界面活性剤ネオゲンRK(第一工業製薬) 5.0質量部
・イオン交換水 180.0質量部
以上の各成分を混合し100℃に加熱して、IKA社製ウルトラタラックスT50にて十分に分散後、圧力吐出型ゴーリンホモジナイザーで分散処理を1時間行い、体積平均粒径が200nm、固形分量が38.3質量%の非晶性樹脂分散液1を得た。
滴下漏斗を備え、加熱乾燥した二口フラスコに、ノルマルヘキサン870質量部を仕込んだ。別のビーカーに、ノルマルヘキサン42質量部、ベヘニルアクリレート(炭素数22個の直鎖アルキル基を有するアルコールのアクリレート)52質量部、アゾビスメトキシジメチルバレロニトリル0.3質量部を仕込み、20℃にて攪拌、混合して単量体溶液を調製し、滴下漏斗に導入した。反応容器を窒素置換した後、密閉下、40℃にて1時間かけて単量体溶液を滴下した。滴下終了から3時間攪拌を続け、アゾビスメトキシジメチルバレロニトリル0.3質量部およびノルマルヘキサン42質量部の混合物を再度滴下し、40℃にて3時間攪拌を行った。その後、室温まで冷却し,体積平均粒径200nm、固形分量20質量%の樹脂微粒子分散液1を得た。なお、この樹脂微粒子分散液中の樹脂微粒子の最大吸熱ピークのピーク温度は66℃であった。
・ジペンタエリスリトールパルチミン酸エステルワックス(WAX-1)
16質量部
・ニトリル基含有スチレンアクリル樹脂(スチレン60質量部、n-ブチルアクリレート30質量部、アクリロニトリル10質量部、ピーク分子量8500)
8質量部
・アセトン 76質量部
上記を撹拌羽根突きのガラスビーカー(IWAKIガラス製)に投入し、系内を50℃に加熱することでワックスをアセトンに溶解させた。ついで、系内を50rpmで緩やかに撹拌しながら徐々に冷却し、3時間かけて25℃にまで冷却させ乳白色の液体を得た。この溶液を1mmのガラスビーズ20質量部とともに耐熱性の容器に投入し、ペイントシェーカー(東洋精機製)にて3時間の分散を行い、ワックス分散液-1を得た。
上記ワックス分散液-1中のワックス粒径は、体積平均粒径で0.15μmであった。得られたワックス分散液-1、および使用したワックス(WAX-1)の特性を表3に示す。
<ワックス分散液-2乃至12の調製>
ワックス分散液-1で用いたジペンタエリスリトールパルチミン酸エステルワックス(WAX-1)の代わりに、表3に示すワックス(WAX-2乃至12)を用いたこと以外はワックス分散液-1の調製と同様にしてワックス分散液-2乃至12を調製した。得られたワックス分散液-2乃至12、および使用したWAX-2乃至12の特性を表3に示す。
<ワックス分散液の調製-13>
・ジペンタエリスリトールベヘン酸エステルワックス(WAX-2)
30.0質量部
・ニトリル基含有スチレンアクリル樹脂(スチレン60質量部、n-ブチルアクリレート30質量部、アクリロニトリル10質量部、ピーク分子量8500)
15.0質量部
・カチオン性界面活性剤ネオゲンRK(第一工業製薬) 5.0質量部
・イオン交換水 200.0質量部
以上を混合し95℃に加熱して、IKA社製ウルトラタラックスT50にて十分に分散後、圧力吐出型ゴーリンホモジナイザーで分散処理し、体積平均粒径が0.20μm、固形分量が20.0質量%のワックス分散液13を得た。
・C.I.ピグメントブルー15:3 100.0質量部
・アセトン 150.0質量部
・ガラスビーズ(1mm) 200.0質量部
上記材料を耐熱性のガラス容器に投入し、ペイントシェーカーにて5時間分散を行い、ナイロンメッシュでガラスビーズを取り除き、着色剤分散液1を得た。
<着色剤分散液2の調製>
・C.I.ピグメントブルー15:3 45.0質量部
・イオン性界面活性剤ネオゲンRK(第一工業製薬) 5.0質量部
・イオン交換水 200.0質量部
・ガラスビーズ(1mm) 250.0質量部
上記材料を耐熱性のガラス容器に投入し、ペイントシェーカーにて5時間分散を行い、ナイロンメッシュにてガラスビーズを取り除き、着色剤分散液2を得た。
個数平均粒径0.25μmのマグネタイト粉と、個数平均粒径0.60μmのヘマタイト粉に対して、夫々4.0質量%のシラン系カップリング剤(3-(2-アミノエチルアミノプロピル)トリメトキシシラン)を加え、容器内で、100℃以上で高速混合撹拌し、それぞれの微粒子を親油化処理した。
・フェノール 10質量部
・ホルムアルデヒド溶液(ホルムアルデヒド40質量%、メタノール10質量%、水50質量%) 6質量部
・親油化処理したマグネタイト 63質量部
・親油化処理したヘマタイト 21質量部
上記材料と、28%アンモニア水5質量部、水10質量部をフラスコに入れ、攪拌、混合しながら30分間で85℃まで昇温・保持し、3時間重合反応させて硬化させた。その後、30℃まで冷却し、更に水を添加した後、上澄み液を除去し、沈殿物を水洗した後、風乾した。次いで、これを減圧下(5mmHg以下)、60℃で乾燥して、磁性体が分散された状態の球状の磁性樹脂粒子を得た。
コート樹脂として、メチルメタクリレートとパーフルオロアルキル基(CF3-(CF2)m- :m=7)を有するメチルメタクリレートの共重合体(共重合比8:1、重量平均分子量45,000)を用いた。該コート樹脂100質量部に、個数平均粒径290nmのメラミン粒子を10質量部、比抵抗1×10-2Ω・cmで個数平均粒径30nmのカーボン粒子を6質量部加え、超音波分散機で30分間分散させた。更に、コート樹脂分がキャリアコアに対し、2.5質量部となるようにメチルエチルケトン及びトルエンの混合溶媒コート溶液を作製した(溶液濃度10質量%)。
このコート溶液を、剪断応力を連続して加えながら溶媒を70℃で揮発させて、上記磁性樹脂粒子表面への樹脂コートを行った。この樹脂コートされた磁性キャリア粒子を100℃で2時間撹拌しながら熱処理し、冷却、解砕した後、200メッシュの篩で分級して個数平均粒径33μm、真比重3.53g/cm3、見かけ比重1.84g/cm3、磁化の強さ42Am2/kgのキャリアAを得た。
(トナー粒子(処理前)1の製造工程)
図1の実験装置において、まず、バルブV1、V2、および圧力調整バルブV3を閉じ、トナー粒子を捕捉するためのフィルターと撹拌機構とを備えた耐圧の造粒タンクT1に樹脂微粒子分散液1(表4では「樹脂微粒子-1」と記載)を仕込み、内部温度を30℃に調整した。次に、バルブV1を開き、ボンベB1からポンプP1を用いて二酸化炭素(純度99.99%)を耐圧容器T1に導入し、内部圧力が5MPaに到達したところでバルブV1を閉じた。
一方、樹脂溶解液タンクT2にブロックポリマー樹脂溶液1、ワックス分散液1、着色剤分散液1、アセトンを仕込み、内部温度を30℃に調整した。
次に、バルブV2を開き、造粒タンクT1の内部を2000rpmで撹拌しながら、ポンプP2を用いて樹脂溶解液タンクT2の内容物を造粒タンクT1内に導入し、すべて導入を終えたところでバルブV2を閉じた。
導入後の、造粒タンクT1の内部圧力は8MPaとなった。
尚、各種材料の仕込み量(質量比)は、次の通りである。
・ブロックポリマー樹脂溶液1 175.0質量部
・ワックス分散液1 31.3質量部
(固形分として、WAX-1が5質量部、ニトリル基含有スチレンアクリル樹脂(表4では「分散剤-1」と記載)が2.5質量部)
・着色剤分散液1 12.5質量部
・アセトン 31.2質量部
・樹脂微粒子分散液1 25.0質量部
・二酸化炭素 280.0質量部
尚、導入した二酸化炭素の質量は、二酸化炭素の温度(30℃)、および圧力(8MPa)から、二酸化炭素の密度を文献(Journal of Physical andChemical Reference data、vol.25、P.1509~1596)に記載の状態式より算出し、これに造粒タンクT1の体積を乗じることにより算出した。
樹脂溶解液タンクT2の内容物の造粒タンクT1への導入を終えた後、さらに、2000rpmで3分間撹拌して造粒を行った。
次に、バルブV1を開き、ボンベB1からポンプP1を用いて二酸化炭素を造粒タンクT1内に導入した。この際、圧力調整バルブV3を10MPaに設定し、造粒タンクT1の内部圧力を10MPaに保持しながら、さらに二酸化炭素を流通させた。この操作により、造粒後の液滴中から抽出された有機溶媒(主にアセトン)を含む二酸化炭素を、溶剤回収タンクT3に排出し、有機溶媒と二酸化炭素を分離した。
造粒タンクT1内への二酸化炭素の導入は、最初に造粒タンクT1に導入した二酸化炭素質量の5倍量に到達した時点で停止した。この時点で、有機溶媒を含む二酸化炭素を、有機溶媒を含まない二酸化炭素で置換する操作は完了した。
さらに、圧力調整バルブV3を少しずつ開き、造粒タンクT1の内部圧力を大気圧まで減圧することで、フィルターに捕捉されているトナー粒子(処理前)1を回収した。得られたトナー粒子(処理前)1のDSC測定を行い、結着樹脂に由来する最大吸熱ピークのピーク温度(Tp)を求めたところ、58℃であった。
(アニール処理工程)
アニール処理は、恒温乾燥器(佐竹化学製41-S5)を用いて行った。恒温乾燥器の内部温度を50℃に調整した。
上記トナー粒子(処理前)1を、ステンレス製バットに均等になるように広げて入れ、これを前記恒温乾燥器に入れて2時間静置した後、取り出した。こうして、アニール処理されたトナー粒子(処理後)1を得た。
(トナー1の調製)
次に、上記トナー粒子(処理後)1の100.0質量部に対し、アナターゼ型酸化チタン微粉末(BET比表面積80m2/g、個数平均粒径(D1)15nm、イソブチルトリメトキシシラン12質量%処理)0.9質量部をまずヘンシェルミキサー FM-10B(三井三池化工機(株)製)により外添した。その後、さらにオイル処理シリカ微粒子(BET比表面積95m2/g、シリコーンオイル15質量%処理)1.2質量部、ゾルゲルシリカ微粒子(BET比表面積24m2/g、個数平均粒径(D1)110nm)1.5質量部を同じヘンシェルミキサーにて混合し、トナー1を得た。得られたトナー1(処理後)のDSC測定を行い、結着樹脂に由来する最大吸熱ピークのピーク温度(Tp)を求めたところ、61℃であった。トナー1の作製条件、及び、トナー1の特性を表4及び5に示す。また、以下に示す手順に従って行った評価の結果を表6に示す。
約10gのトナー1を100mlのポリカップに入れ、50℃で3日及び53℃で3日放置した後、目視で評価した。
(評価基準)
A:まったく凝集物は確認されず、初期とほぼ同様の状態。
B:若干、凝集気味であるが、ポリカップを軽く5回振る程度で崩れる状態であり、特に問題とならない。
C:凝集気味であるが、指でほぐすと簡単にほぐれる状態であり、実使用に耐えうる。
D:凝集が激しく発生。
E:固形化しており、使用できない。
約10gのトナー1を100mlのポリカップに入れ、50℃で1日放置後、50℃と53℃の間を1℃/時間の速度で変化させ、3日の間12サイクル行った後トナーを取り出し凝集を確認した。ヒートサイクル試験のタイムチャートを図2に示す。
(耐熱保存性の評価基準)
A:まったく凝集物は確認されず、初期とほぼ同様の状態。
B:若干、凝集気味であるが、ポリカップを軽く5回振る程度で崩れる状態であり、特に問題とならない。
C:凝集気味であるが、指でほぐすと簡単にほぐれる状態であり、実使用に耐えうる。
D:凝集が激しく発生。
E:固形化しており、使用できない。
<ヒートサイクル試験後の帯電維持性の評価>
上記ヒートサイクル試験を行っていないトナーを常温常湿環境下(温度23℃/湿度60%)に1日放置し、標準トナーとして用意した。上記ヒートサイクル試験を行ったトナーを200メッシュ(目開き75μm)の篩にかけ、常温常湿環境下(温度23℃/湿度60%)に1日放置し、サンプルトナーとした。
トナー及びキャリア(日本画像学会標準キャリア フェライトコアを表面処理した球形キャリア N-01)を蓋付きのプラスチックボトルにそれぞれ、1.0g、19.0g入れ、測定環境に1日放置する。トナーとキャリアとを入れたプラスチックボトルを振盪器(YS-LD、(株)ヤヨイ製)にセットし、1秒間に4往復のスピードで1分間振とうし、トナーとキャリアからなる現像剤を帯電させる。
次に、図3に示す摩擦帯電量を測定する装置において摩擦帯電量を測定する。図3において、底に500メッシュ(目開き25μm)のスクリーン3のある金属製の測定容器2に、前述した現像剤約0.5~1.5gを入れ、金属製のフタ4をする。この時の測定容器2全体の質量を秤りW1(g)とする。次に吸引機1(測定容器2と接する部分は少なくとも絶縁体)において、吸引口7から吸引し風量調節弁6を調整して真空計5の圧力を250mmAqとする。この状態で2分間吸引を行い、トナーを吸引除去する。この時の電位計9の電位をV(ボルト)とする。ここで、8はコンデンサーであり容量をC(mF)とする。また、吸引後の測定容器全体の質量を秤りW2(g)とする。この試料の摩擦帯電量(mC/kg)は下式の如く算出される。
試料の摩擦帯電量(mC/kg)=C×V/(W1-W2)
(帯電維持性の評価基準)
A:サンプルトナーの帯電量と標準トナーの帯電量との差が5%未満。
B:サンプルトナーの帯電量と標準トナーの帯電量との差が5%以上10%未満。
C:サンプルトナーの帯電量と標準トナーの帯電量との差が10%以上20%未満。
D:サンプルトナーの帯電量と標準トナーの帯電量との差が20%以上。
E:サンプルトナーが凝集、固形化しており、帯電評価できない。
当該評価は、トナー粒子を構成するコアの低分子量成分やワックスの染み出しの状態を評価するものである。
トナーの低温定着性は、剥離性による定着開始温度とコールドオフセット性による定着開始温度の2種類の方法で評価した
<剥離性による定着開始温度の評価>
上記トナー1を8.0質量部と上記のように製造されたキャリアA 92.0質量部を混合してなる二成分現像剤1を調製した。
評価には上記二成分現像剤1、カラーレーザー複写機CLC5000(キヤノン社製)を用いた。紙上のトナー載り量を1.2mg/cm2になるように上記複写機の現像コントラストを調整し、単色モードで、先端余白5mm、幅100mm、長さ28mmの、「べた」の未定着画像を常温常湿度環境下(23℃/60%RH)で作成した。紙は、厚紙A4用紙(「プローバーボンド紙」:105g/m2、フォックスリバー社製)を用いた。
次に、LBP5900(キヤノン社製)の定着器を手動で定着温度設定が可能となるように改造し、定着器の回転速度を270mm/s、ニップ内圧力:120kPaに変更した。該改造定着器を用い、常温常湿度環境下(23℃/60%RH)で、80℃から180℃の範囲で10℃ずつ定着温度を上昇させながら、上記「べた」の未定着画像の各温度における定着画像を得た。
得られた定着画像の画像領域に、柔和な薄紙(商品名「ダスパー」、小津産業社製)を被せ、該薄紙の上から4.9kPaの荷重をかけつつ5往復、該画像領域を摺擦した。摺擦前と摺擦後の画像濃度をそれぞれ測定して、下記式により剥離による画像濃度の低下率ΔD(%)を算出した。このΔD(%)が10%未満のときの温度を剥離性による定着開始温度とし、以下のような評価基準で評価した。
尚、画像濃度はカラー反射濃度計(Color reflection densitometer X-Rite 404A:製造元 X-Rite社製)で測定した。
(式):ΔD(%)={(摺擦前の画像濃度-摺擦後の画像濃度)/摺擦前の画像濃度}×100
(評価基準)
A:定着開始温度が100℃以下
B:定着開始温度が110℃
C:定着開始温度が120℃
D:定着開始温度が130℃
E:定着開始温度が140℃以上
尚、本発明においてはCランクまでを良好な低温定着性と判断した。
<コールドオフセット(C.O.)性による定着開始温度の評価>
上記剥離性による定着開始温度の評価で得られた定着画像を用いて、コールドオフセットの評価を行った。評価は、「べた」画像の周方向末端から定着ベルト1周分後方の白地になる部位で濃度変化を確認した。測定は東京電色技術センター製DENSITOMETER TC-6DSを用い、反射率(%)を、測定し、濃度の値とした。濃度が0.5%変化したところをコールドオフセット発生点とし、コールドオフセットが発生しなかった最低温度をコールドオフセット性による定着開始温度とした。
(評価基準)
A:定着開始温度が100℃以下
B:定着開始温度が110℃
C:定着開始温度が120℃
D:定着開始温度が130℃
E:定着開始温度が140℃以上
尚、本発明においてはCランクまでを良好なコールドオフセット性と判断した。
上記低温定着性の評価より、紙を普通紙A4用紙(「オフィスプランナー」:64g/m2、キヤノン製)に変更して定着性の評価を行った。定着後の画像より、目視にて定着器2周目に、前周期の高温オフセットトナーが見られた点を高温オフセット開始温度と判断し、高温オフセット開始温度より低い温度の最高温度を高温定着温度と判断した。なお、180℃まで高温オフセットが発生しなかったものに関しては、180℃を高温定着温度とした。
上記剥離性による定着開始温度と、コールドオフセット性による定着開始温度の高い方を定着開始温度とし、定着開始温度と高温定着温度の差(高温定着温度-定着開始温度)を定着領域とし、以下の判断を行った。
(評価基準)
A:定着領域が70℃以上
B:定着領域が60℃
C:定着領域が50℃
D:定着領域が40℃
E:定着領域が30℃以下
(トナー粒子(処理前)2の製造工程)
・結晶性ポリエステル樹脂分散液1 159.7質量部
・非晶性樹脂分散液1 68.6質量部
・着色剤分散液2 27.8質量部
・ワックス分散液13 41.7質量部
・ポリ塩化アルミニウム 0.41質量部
以上の各成分を丸型ステンレス製フラスコ中に入れ、ウルトラタラックスT50で十分に混合・分散した。次いで、これにポリ塩化アルミニウム0.36質量部を加え、ウルトラタラックスT50で分散操作を継続した。加熱用オイルバスでフラスコを撹拌しながら47℃まで加熱し、この温度で60分間保持した後、ここに非晶性樹脂分散液1、13.0質量部を緩やかに追加した。その後、0.5mol/Lの水酸化ナトリウム水溶液で系内のpHを5.4にした後、ステンレス製フラスコを密閉し、磁力シールを用いて撹拌を継続しながら96℃まで加熱し、5時間保持した。
反応終了後、冷却し、濾過、イオン交換水で十分に洗浄した後、ヌッチェ式吸引濾過により固液分離を施した。これを更に40℃のイオン交換水3Lに再分散し、300rpmで15分間撹拌・洗浄した。これを更に5回繰り返し、濾液のpHが7.0になったところで、ヌッチェ式吸引濾過によりNo.5Aろ紙を用いて固液分離を行った。次いで真空乾燥を12時間継続し、トナー粒子(処理前)2を得た。得られたトナー粒子2のDSC測定での結着樹脂に由来する最大吸熱ピークのピーク温度は58℃であった。
(アニール処理工程)
アニール処理は、恒温乾燥器(佐竹化学製41-S5)を用いて行った。恒温乾燥器の内部温度を50℃に調整した。
上記トナー粒子(処理前)2を、ステンレス製バットに均等になるように広げて入れ、これを前記恒温乾燥器に入れて2時間静置した後、取り出した。こうして、アニール処理されたトナー粒子(処理後)2を得た。
(トナー2の調製)
次に、上記トナー粒子(処理後)2について、実施例1のトナーの調製1と同様の操作を行い、トナー2を得た。得られたトナー2(処理後)のDSC測定を行い、結着樹脂に由来する最大吸熱ピークのピーク温度(Tp)を求めたところ、61℃であった。トナー2の作製条件、及び、トナー2の特性を表4及び5に示す。また、実施例1と同様の方法にて評価した結果を表6に示す。
トナー粒子(処理前)1の製造工程におけるブロックポリマーおよびワックスを表4に示すように、ブロックポリマー樹脂溶液およびワックス分散液を選択する以外は、実施例1と同様に行い、トナー3乃至7を得た。トナーの作製条件、及び、トナーの特性を表4及び5に、評価結果を表6に示す。
トナー粒子(処理前)1の製造工程におけるブロックポリマーおよびワックスを表4に示すブロックポリマーおよびワックスになるように、ブロックポリマー樹脂溶液およびワックス分散液を選択する以外は、実施例1と同様に行いトナー8乃至30を得た。トナーの作製条件、及び、トナーの特性を表4及び5に、評価結果を表6に示す。
なお、実施例3、5、11及び14に係るトナー9、11、17及び20では、トナーの吸熱量曲線において、結着樹脂に由来する最大吸熱ピークにワックスの吸熱ピークが重なっていた。そのため、最大吸熱ピークの吸熱量からワックスの吸熱量を差し引いたものを結着樹脂に由来する最大吸熱ピークの吸熱量として各値を求めた。また、実施例に係るトナー1、2、5、11~15、17~28及び30では、トナーの吸熱量曲線において、結着樹脂に由来する最大吸熱ピークにトナーの樹脂微粒子によって形成されたシェル相の樹脂の吸熱ピークが重なっていた。そのため、最大吸熱ピークの吸熱量からシェル相の樹脂の吸熱量を差し引いたものを結着樹脂に由来する最大吸熱ピークの吸熱量として各値を求めた。
これら以外の例においては、トナーの吸熱量曲線における最大吸熱ピークをそのまま結着樹脂に由来する最大吸熱ピークとして各値を求めた。





表5中、トナー粒子(処理後)のTpは「トナーの結着樹脂に由来するTp」を表し、トナー粒子(処理後)の結着樹脂に由来する最大吸熱ピークの吸熱量は「トナーの結着樹脂に由来する最大吸熱ピークの吸熱量」を表す。

Claims (10)
- ポリエステルを主成分にする結着樹脂、着色剤及びワックスを含有するトナー粒子を有するトナーであって、
前記結着樹脂は、結晶構造をとりうる部位と結晶構造をとりえない部位を結合したブロックポリマーを含有し、
前記トナーの示差走査熱量(DSC)測定から求められる、前記結着樹脂に由来する最大吸熱ピークのピーク温度が50℃以上、80℃以下であり、前記最大吸熱ピークの吸熱量が30J/g以上、100J/g以下であり、
前記ワックスは、3官能以上のエステルワックスであることを特徴とするトナー。 - 前記ワックスの示差走査熱量(DSC)測定における最大吸熱ピークのピーク温度は65℃以上であり、けん化価が160mgKOH/g以上であることを特徴とする請求項1に記載のトナー。
- 前記ワックスは、分子量が1500以上、2200以下であることを特徴とする請求項1または2に記載のトナー。
- 前記ワックスは、6官能以上のエステルワックスであることを特徴とする請求項1乃至3のいずれか1項に記載のトナー。
- 前記ワックスは、ジペンタエリスリトールと長鎖直鎖飽和脂肪酸とをエステル結合させて得られるエステルワックスであることを特徴とする請求項1乃至4のいずれか1項に記載のトナー。
- 前記ワックスの含有量は、結着樹脂100質量部に対し2.0質量部以上8.0質量部以下であることを特徴とする請求項1乃至5のいずれか1項に記載のトナー。
- 前記ブロックポリマーの結晶構造をとりうる部位は、脂肪族ジカルボン酸と脂肪族ジオールを反応して得られる結晶性ポリエステルであることを特徴とする請求項1乃至6のいずれか1項に記載のトナー。
- 前記ブロックポリマーの結晶性構造をとりえない部位は、ジオールとジイソシアネートを反応して得られるポリウレタンであることを特徴とする請求項1乃至7のいずれか1項に記載のトナー。
- 前記ブロックポリマーは、結晶構造をとりうる部位を前記結着樹脂の全量に対し50質量%以上、85質量%以下含有することを特徴とする請求項1乃至8のいずれか1項に記載のトナー。
- 前記トナー粒子は、
(i)前記結着樹脂、前記着色剤及び前記ワックスを、有機溶媒中に溶解または分散させた溶解物または分散物を得る工程、
(ii)該溶解物または該分散物を、樹脂微粒子を分散させた超臨界状態または液体状態の二酸化炭素を有する分散媒体中に分散させて分散体を得る工程、
(iii)該分散体から該有機溶媒を除去することによってトナー粒子を形成する工程
を経て製造されたものであることを特徴とする請求項1乃至9のいずれか1項に記載のトナー。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020137034333A KR101533704B1 (ko) | 2011-06-03 | 2012-06-01 | 토너 |
| EP12791997.5A EP2717100B1 (en) | 2011-06-03 | 2012-06-01 | Toner |
| CN201280026872.4A CN103562799B (zh) | 2011-06-03 | 2012-06-01 | 调色剂 |
| US13/741,356 US8785101B2 (en) | 2011-06-03 | 2013-01-14 | Toner |
| US14/250,328 US9625844B2 (en) | 2011-06-03 | 2014-04-10 | Toner |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011125760 | 2011-06-03 | ||
| JP2011-125760 | 2011-06-03 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/741,356 Continuation US8785101B2 (en) | 2011-06-03 | 2013-01-14 | Toner |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012165638A1 true WO2012165638A1 (ja) | 2012-12-06 |
Family
ID=47259486
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/064334 WO2012165638A1 (ja) | 2011-06-03 | 2012-06-01 | トナー |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US8785101B2 (ja) |
| EP (1) | EP2717100B1 (ja) |
| JP (1) | JP5274692B2 (ja) |
| KR (1) | KR101533704B1 (ja) |
| CN (1) | CN103562799B (ja) |
| WO (1) | WO2012165638A1 (ja) |
Families Citing this family (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103562799B (zh) * | 2011-06-03 | 2016-08-31 | 佳能株式会社 | 调色剂 |
| JP5709065B2 (ja) | 2011-10-17 | 2015-04-30 | 株式会社リコー | トナー、該トナーを用いた現像剤、画像形成装置 |
| JP5892089B2 (ja) * | 2013-03-07 | 2016-03-23 | 株式会社リコー | 電子写真画像形成用トナー、画像形成方法、画像形成装置及びプロセスカートリッジ |
| JP6079325B2 (ja) * | 2013-03-14 | 2017-02-15 | 株式会社リコー | トナー |
| JP6157200B2 (ja) * | 2013-04-30 | 2017-07-05 | キヤノン株式会社 | トナー |
| JP6331599B2 (ja) * | 2013-09-17 | 2018-05-30 | 株式会社リコー | 電子写真用トナー |
| CN104678724B (zh) * | 2013-11-29 | 2018-10-09 | 佳能株式会社 | 调色剂 |
| US9575424B2 (en) * | 2014-03-12 | 2017-02-21 | Canon Kabushiki Kaisha | Method of producing a toner particle |
| JP6335582B2 (ja) | 2014-03-28 | 2018-05-30 | キヤノン株式会社 | トナー |
| US9857713B2 (en) | 2014-12-26 | 2018-01-02 | Canon Kabushiki Kaisha | Resin particle and method of producing the resin particle, and toner and method of producing the toner |
| JP6727837B2 (ja) | 2015-03-25 | 2020-07-22 | キヤノン株式会社 | トナー及びトナーの製造方法 |
| US9823595B2 (en) | 2015-06-30 | 2017-11-21 | Canon Kabushiki Kaisha | Toner |
| US9798256B2 (en) | 2015-06-30 | 2017-10-24 | Canon Kabushiki Kaisha | Method of producing toner |
| JP2017083822A (ja) | 2015-10-29 | 2017-05-18 | キヤノン株式会社 | トナーの製造方法および樹脂粒子の製造方法 |
| JP6565050B2 (ja) * | 2016-02-22 | 2019-08-28 | 花王株式会社 | 電子写真用トナーの製造方法 |
| JP6833341B2 (ja) * | 2016-04-19 | 2021-02-24 | キヤノン株式会社 | トナーおよびトナーの製造方法 |
| JP7062373B2 (ja) * | 2016-04-19 | 2022-05-06 | キヤノン株式会社 | トナー |
| JP6798691B2 (ja) * | 2016-11-29 | 2020-12-09 | 花王株式会社 | 電子写真用トナー |
| US10409180B2 (en) | 2017-02-13 | 2019-09-10 | Canon Kabushiki Kaisha | Resin fine particles, method of producing resin fine particles, method of producing resin particles, and method of producing toner |
| JP6919279B2 (ja) * | 2017-03-31 | 2021-08-18 | 日油株式会社 | トナー用ワックス組成物 |
| JP2019012220A (ja) * | 2017-06-30 | 2019-01-24 | 花王株式会社 | 液体現像剤の製造方法 |
| US10545420B2 (en) | 2017-07-04 | 2020-01-28 | Canon Kabushiki Kaisha | Magnetic toner and image-forming method |
| JP7066439B2 (ja) | 2018-02-14 | 2022-05-13 | キヤノン株式会社 | トナー用外添剤、トナー用外添剤の製造方法及びトナー |
| US10768540B2 (en) | 2018-02-14 | 2020-09-08 | Canon Kabushiki Kaisha | External additive, method for manufacturing external additive, and toner |
| CN110597029A (zh) | 2018-06-13 | 2019-12-20 | 佳能株式会社 | 调色剂和调色剂的制造方法 |
| EP3582020B1 (en) | 2018-06-13 | 2023-08-30 | Canon Kabushiki Kaisha | Toner |
| US10877388B2 (en) | 2018-06-13 | 2020-12-29 | Canon Kabushiki Kaisha | Toner |
| CN110597028A (zh) | 2018-06-13 | 2019-12-20 | 佳能株式会社 | 磁性调色剂和磁性调色剂的制造方法 |
| EP3582017B1 (en) | 2018-06-13 | 2023-04-26 | Canon Kabushiki Kaisha | Toner and method for producing toner |
| JP7267706B2 (ja) | 2018-10-02 | 2023-05-02 | キヤノン株式会社 | 磁性トナー |
| JP7267705B2 (ja) | 2018-10-02 | 2023-05-02 | キヤノン株式会社 | 磁性トナー |
| JP7207981B2 (ja) | 2018-12-10 | 2023-01-18 | キヤノン株式会社 | トナー及びトナーの製造方法 |
| JP2020095083A (ja) | 2018-12-10 | 2020-06-18 | キヤノン株式会社 | トナー |
| JP7224885B2 (ja) | 2018-12-10 | 2023-02-20 | キヤノン株式会社 | トナー |
| JP7292965B2 (ja) | 2019-05-13 | 2023-06-19 | キヤノン株式会社 | トナーおよびトナーの製造方法 |
| JP7341718B2 (ja) | 2019-05-13 | 2023-09-11 | キヤノン株式会社 | トナー |
| JP7313930B2 (ja) | 2019-06-27 | 2023-07-25 | キヤノン株式会社 | トナー |
| JP7313931B2 (ja) | 2019-06-27 | 2023-07-25 | キヤノン株式会社 | トナー |
| JP2021036316A (ja) | 2019-08-21 | 2021-03-04 | キヤノン株式会社 | トナー |
| JP7463086B2 (ja) | 2019-12-12 | 2024-04-08 | キヤノン株式会社 | トナー |
| JP7475887B2 (ja) | 2020-02-14 | 2024-04-30 | キヤノン株式会社 | 磁性トナーの製造方法 |
| JP7483428B2 (ja) | 2020-03-16 | 2024-05-15 | キヤノン株式会社 | トナー |
| US20220187725A1 (en) * | 2020-12-15 | 2022-06-16 | Canon Kabushiki Kaisha | Toner and method for producing toner |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0315079A (ja) * | 1989-06-13 | 1991-01-23 | Sanyo Chem Ind Ltd | 荷電制御剤 |
| JPH1195482A (ja) * | 1997-09-17 | 1999-04-09 | Sanyo Chem Ind Ltd | 電子写真用トナーバインダー |
| JP2002287405A (ja) * | 2001-03-27 | 2002-10-03 | Konica Corp | 静電荷像現像用トナー及び画像形成方法 |
| JP2003302785A (ja) * | 2002-04-12 | 2003-10-24 | Fujitsu Ltd | 電子写真用トナー及び電子写真用フラッシュ定着装置 |
| JP2004191927A (ja) | 2002-11-29 | 2004-07-08 | Fuji Xerox Co Ltd | 静電荷像現像用トナー、その製造方法、並びに、これを用いた静電荷像現像剤及び画像形成方法 |
| JP2005003883A (ja) * | 2003-06-11 | 2005-01-06 | Sharp Corp | 現像剤及びその製造方法 |
| JP2005024863A (ja) * | 2003-07-01 | 2005-01-27 | Seiko Epson Corp | トナーの製造方法およびトナー |
| WO2005013012A1 (ja) * | 2003-07-09 | 2005-02-10 | Matsushita Electric Industrial Co., Ltd. | トナー、トナーの製造方法、二成分現像剤及び画像形成装置 |
| JP2005215253A (ja) * | 2004-01-29 | 2005-08-11 | Konica Minolta Business Technologies Inc | 電子写真画像形成方法 |
| JP2006003513A (ja) * | 2004-06-16 | 2006-01-05 | Ricoh Co Ltd | トナー、その製造方法、該トナーを用いた現像剤、トナー入り容器、画像形成方法およびプロセスカートリッジ |
| JP2006276293A (ja) * | 2005-03-28 | 2006-10-12 | Nippon Zeon Co Ltd | 静電荷像現像用トナー |
| JP2007114635A (ja) | 2005-10-24 | 2007-05-10 | Fuji Xerox Co Ltd | 画像形成方法及び静電荷像現像用トナーの製造方法 |
| JP2008052192A (ja) | 2006-08-28 | 2008-03-06 | Konica Minolta Business Technologies Inc | トナー |
| JP2009168915A (ja) * | 2008-01-11 | 2009-07-30 | Canon Inc | トナー用樹脂組成物、トナー及び二成分現像剤 |
| JP2010168529A (ja) | 2008-05-23 | 2010-08-05 | Sanyo Chem Ind Ltd | 樹脂粒子およびその製造方法 |
Family Cites Families (71)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0421416B1 (en) | 1989-10-05 | 1998-08-26 | Canon Kabushiki Kaisha | Heat fixable toner and heat fixing method |
| SG48073A1 (en) | 1990-01-19 | 1998-04-17 | Canon Kk | Electrostatic image developing toner and fixing method |
| US5300386A (en) | 1991-03-22 | 1994-04-05 | Canon Kabushiki Kaisha | Developer for developing electrostatic image, image forming method and heat fixing method |
| JP3262378B2 (ja) | 1991-08-29 | 2002-03-04 | キヤノン株式会社 | 静電荷像現像用カラートナー |
| JP2899177B2 (ja) | 1991-09-19 | 1999-06-02 | キヤノン株式会社 | 静電荷像現像用トナー及び静電荷像現像用二成分系現像剤 |
| US5354640A (en) | 1991-09-25 | 1994-10-11 | Canon Kabushiki Kaisha | Toner for developing electrostatic image |
| EP0578093B1 (en) | 1992-06-29 | 2000-11-29 | Canon Kabushiki Kaisha | Image forming method and heat fixing method |
| US5529873A (en) | 1993-04-20 | 1996-06-25 | Canon Kabushiki Kaisha | Toner for developing electrostatic images and process for producing toner |
| EP0651292B1 (en) | 1993-10-20 | 1998-07-15 | Canon Kabushiki Kaisha | Toner for developing electrostatic images, and process for its production |
| JP3028276B2 (ja) | 1993-10-29 | 2000-04-04 | キヤノン株式会社 | 静電荷像現像用カラートナー,その製造方法及びカラー画像形成方法 |
| US6002895A (en) | 1994-05-13 | 1999-12-14 | Canon Kabushiki Kaisha | Process cartridge |
| DE69511328T2 (de) | 1994-05-13 | 2000-03-30 | Canon Kk | Toner zur Entwicklung elektrostatischer Bilder, Prozesskassette und Bilderzeugungsverfahren |
| EP0725318B1 (en) | 1995-02-01 | 1999-07-28 | Canon Kabushiki Kaisha | Developer for developing an electrostatic image and image forming method |
| US5972553A (en) | 1995-10-30 | 1999-10-26 | Canon Kabushiki Kaisha | Toner for developing electrostatic image, process-cartridge and image forming method |
| US5712073A (en) | 1996-01-10 | 1998-01-27 | Canon Kabushiki Kaisha | Toner for developing electrostatic image, apparatus unit and image forming method |
| JP3347646B2 (ja) | 1996-07-31 | 2002-11-20 | キヤノン株式会社 | 静電荷潜像現像用磁性黒色トナー及びマルチカラー又はフルカラー画像形成方法 |
| DE69926685T2 (de) | 1998-05-13 | 2006-01-19 | Canon K.K. | Toner und Bildherstellungsverfahren |
| US6300024B1 (en) | 1999-06-30 | 2001-10-09 | Canon Kabushiki Kaisha | Toner, two-component type developer, heat fixing method, image forming method and apparatus unit |
| KR100402219B1 (ko) | 1999-10-06 | 2003-10-22 | 캐논 가부시끼가이샤 | 토너, 토너 제조 방법, 화상 형성 방법 및 장치 유니트 |
| US6485875B1 (en) | 1999-10-26 | 2002-11-26 | Canon Kabushiki Kaisha | Toner and resin composition for the toner |
| JP4387613B2 (ja) | 2000-07-10 | 2009-12-16 | キヤノン株式会社 | マゼンタトナー |
| US6875549B2 (en) | 2001-04-10 | 2005-04-05 | Canon Kabushiki Kaisha | Dry toner, toner production process, image forming method and process cartridge |
| WO2002084408A1 (en) * | 2001-04-11 | 2002-10-24 | Sekisui Chemical Co., Ltd. | Resin composition for toner and toner |
| EP1760536A3 (en) | 2001-12-28 | 2007-03-14 | Canon Kabushiki Kaisha | Image-forming method having at least two speed modes |
| EP1329774B1 (en) | 2002-01-18 | 2006-12-20 | Canon Kabushiki Kaisha | Color toner, and full-color image-forming method |
| US6881527B2 (en) | 2002-03-26 | 2005-04-19 | Canon Kabushiki Kaisha | Toner, and process cartridge |
| EP1398673A3 (en) | 2002-09-12 | 2005-08-31 | Canon Kabushiki Kaisha | Developer |
| EP1403723B1 (en) | 2002-09-27 | 2013-02-20 | Canon Kabushiki Kaisha | Toner |
| EP1406129B8 (en) | 2002-10-02 | 2012-05-23 | Canon Kabushiki Kaisha | Silica fine particle, toner, two-component developer and image forming method |
| JP4290015B2 (ja) | 2003-01-10 | 2009-07-01 | キヤノン株式会社 | カラートナー及び画像形成装置 |
| CN1550919B (zh) | 2003-05-14 | 2010-04-28 | 佳能株式会社 | 磁性载体及双组分显影剂 |
| EP1502933B1 (en) | 2003-07-30 | 2010-09-08 | Canon Kabushiki Kaisha | Toner comprising hydrophobic inorganic fine particles |
| JP2005062797A (ja) | 2003-07-30 | 2005-03-10 | Canon Inc | 磁性トナー |
| US7288354B2 (en) | 2003-08-01 | 2007-10-30 | Canon Kabushiki Kaisha | Toner |
| EP1505448B1 (en) | 2003-08-01 | 2015-03-04 | Canon Kabushiki Kaisha | Toner |
| US7452649B2 (en) | 2003-09-12 | 2008-11-18 | Canon Kabushiki Kaisha | Magnetic toner, and image forming method |
| US7514190B2 (en) | 2004-01-29 | 2009-04-07 | Konica Minolta Business Technologies, Inc. | Method for forming an electrophotographic image |
| US7943281B2 (en) | 2005-04-15 | 2011-05-17 | Canon Kabushiki Kaisha | Black toner |
| DE602006003681D1 (de) | 2005-04-22 | 2009-01-02 | Canon Kk | Toner |
| KR100960198B1 (ko) | 2005-04-22 | 2010-05-27 | 캐논 가부시끼가이샤 | 자성 토너 |
| US7678524B2 (en) | 2005-05-19 | 2010-03-16 | Canon Kabushiki Kaisha | Magnetic toner |
| JP4544095B2 (ja) * | 2005-08-24 | 2010-09-15 | 富士ゼロックス株式会社 | 電子写真用トナー、電子写真用トナーの製造方法、電子写真用現像剤並びに画像形成方法 |
| CN102608885A (zh) | 2005-11-11 | 2012-07-25 | 佳能株式会社 | 调色剂用树脂和调色剂 |
| US20070117945A1 (en) | 2005-11-11 | 2007-05-24 | Canon Kabushiki Kaisha | Novel polymer, charge control agent, and toner for developing electrostatic latent images |
| US8110329B2 (en) | 2005-11-11 | 2012-02-07 | Canon Kabushiki Kaisha | Charge controlling agent and toner |
| US7935771B2 (en) | 2005-11-11 | 2011-05-03 | Canon Kabushiki Kaisha | Polymer having sulfonic acid group or sulfonic acid ester group and amide group, and toner for developing electrostatic latent image having the polymer |
| KR101014991B1 (ko) | 2006-06-08 | 2011-02-16 | 캐논 가부시끼가이샤 | 토너 |
| EP2063322B1 (en) | 2006-10-11 | 2015-12-30 | Canon Kabushiki Kaisha | Toner |
| JP4468482B2 (ja) | 2007-03-12 | 2010-05-26 | キヤノン株式会社 | 重合トナーの製造方法、トナー用結着樹脂の製造方法及びトナー |
| CN101681134B (zh) | 2007-05-21 | 2012-09-19 | 佳能株式会社 | 生产聚合调色剂的方法、聚合调色剂、生产调色剂用粘结剂树脂的方法和调色剂用粘结剂树脂 |
| JP5158081B2 (ja) * | 2007-05-29 | 2013-03-06 | 日本ゼオン株式会社 | 静電荷像現像用正帯電性トナー |
| WO2008156117A1 (ja) | 2007-06-19 | 2008-12-24 | Canon Kabushiki Kaisha | カラートナー |
| JP5159239B2 (ja) | 2007-10-15 | 2013-03-06 | キヤノン株式会社 | トナー |
| JP4537496B2 (ja) | 2007-12-27 | 2010-09-01 | キヤノン株式会社 | トナー |
| KR101261106B1 (ko) | 2008-02-25 | 2013-05-06 | 캐논 가부시끼가이샤 | 토너 |
| CN101960390A (zh) | 2008-02-25 | 2011-01-26 | 佳能株式会社 | 调色剂 |
| JP5153864B2 (ja) | 2008-03-10 | 2013-02-27 | キヤノン株式会社 | トナー |
| JP4590486B2 (ja) | 2008-05-16 | 2010-12-01 | キヤノン株式会社 | 疎水性無機微粒子及びトナー |
| WO2009142010A1 (ja) | 2008-05-23 | 2009-11-26 | 三洋化成工業株式会社 | 樹脂粒子およびその製造方法 |
| JP5164715B2 (ja) | 2008-07-25 | 2013-03-21 | キヤノン株式会社 | トナー |
| EP2309334A4 (en) | 2008-07-31 | 2013-05-01 | Canon Kk | TONER CYAN |
| JP4565053B2 (ja) | 2009-02-27 | 2010-10-20 | キヤノン株式会社 | マゼンタトナー |
| JP4565054B2 (ja) | 2009-02-27 | 2010-10-20 | キヤノン株式会社 | 黒トナー |
| WO2010098226A1 (en) | 2009-02-27 | 2010-09-02 | Canon Kabushiki Kaisha | Yellow toner |
| US8652725B2 (en) | 2009-12-04 | 2014-02-18 | Canon Kabushiki Kaisha | Toner |
| KR101402566B1 (ko) | 2010-05-31 | 2014-05-30 | 캐논 가부시끼가이샤 | 자성 토너 |
| US8877417B2 (en) * | 2010-07-22 | 2014-11-04 | Canon Kabushiki Kaisha | Toner |
| JP5836888B2 (ja) | 2011-06-03 | 2015-12-24 | キヤノン株式会社 | トナー |
| WO2012165636A1 (ja) | 2011-06-03 | 2012-12-06 | キヤノン株式会社 | トナー |
| CN103562799B (zh) * | 2011-06-03 | 2016-08-31 | 佳能株式会社 | 调色剂 |
| JP6053336B2 (ja) | 2011-06-03 | 2016-12-27 | キヤノン株式会社 | トナー及びトナーの製造方法 |
-
2012
- 2012-06-01 CN CN201280026872.4A patent/CN103562799B/zh active Active
- 2012-06-01 WO PCT/JP2012/064334 patent/WO2012165638A1/ja active Application Filing
- 2012-06-01 KR KR1020137034333A patent/KR101533704B1/ko not_active IP Right Cessation
- 2012-06-01 JP JP2012126582A patent/JP5274692B2/ja active Active
- 2012-06-01 EP EP12791997.5A patent/EP2717100B1/en active Active
-
2013
- 2013-01-14 US US13/741,356 patent/US8785101B2/en active Active
-
2014
- 2014-04-10 US US14/250,328 patent/US9625844B2/en not_active Expired - Fee Related
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0315079A (ja) * | 1989-06-13 | 1991-01-23 | Sanyo Chem Ind Ltd | 荷電制御剤 |
| JPH1195482A (ja) * | 1997-09-17 | 1999-04-09 | Sanyo Chem Ind Ltd | 電子写真用トナーバインダー |
| JP2002287405A (ja) * | 2001-03-27 | 2002-10-03 | Konica Corp | 静電荷像現像用トナー及び画像形成方法 |
| JP2003302785A (ja) * | 2002-04-12 | 2003-10-24 | Fujitsu Ltd | 電子写真用トナー及び電子写真用フラッシュ定着装置 |
| JP2004191927A (ja) | 2002-11-29 | 2004-07-08 | Fuji Xerox Co Ltd | 静電荷像現像用トナー、その製造方法、並びに、これを用いた静電荷像現像剤及び画像形成方法 |
| JP2005003883A (ja) * | 2003-06-11 | 2005-01-06 | Sharp Corp | 現像剤及びその製造方法 |
| JP2005024863A (ja) * | 2003-07-01 | 2005-01-27 | Seiko Epson Corp | トナーの製造方法およびトナー |
| WO2005013012A1 (ja) * | 2003-07-09 | 2005-02-10 | Matsushita Electric Industrial Co., Ltd. | トナー、トナーの製造方法、二成分現像剤及び画像形成装置 |
| JP2005215253A (ja) * | 2004-01-29 | 2005-08-11 | Konica Minolta Business Technologies Inc | 電子写真画像形成方法 |
| JP2006003513A (ja) * | 2004-06-16 | 2006-01-05 | Ricoh Co Ltd | トナー、その製造方法、該トナーを用いた現像剤、トナー入り容器、画像形成方法およびプロセスカートリッジ |
| JP2006276293A (ja) * | 2005-03-28 | 2006-10-12 | Nippon Zeon Co Ltd | 静電荷像現像用トナー |
| JP2007114635A (ja) | 2005-10-24 | 2007-05-10 | Fuji Xerox Co Ltd | 画像形成方法及び静電荷像現像用トナーの製造方法 |
| JP2008052192A (ja) | 2006-08-28 | 2008-03-06 | Konica Minolta Business Technologies Inc | トナー |
| JP2009168915A (ja) * | 2008-01-11 | 2009-07-30 | Canon Inc | トナー用樹脂組成物、トナー及び二成分現像剤 |
| JP2010168529A (ja) | 2008-05-23 | 2010-08-05 | Sanyo Chem Ind Ltd | 樹脂粒子およびその製造方法 |
Non-Patent Citations (3)
| Title |
|---|
| "902-00153", July 1993, RHEOMETRICS SCIENTIFIC |
| "ARES operating manuals 902-30004", August 1997 |
| JOURNAL OF PHYSICAL AND CHEMICAL REFERENCE DATA, vol. 25, pages 1509 - 1596 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN103562799A (zh) | 2014-02-05 |
| US8785101B2 (en) | 2014-07-22 |
| EP2717100A4 (en) | 2015-03-04 |
| KR101533704B1 (ko) | 2015-07-03 |
| KR20140016396A (ko) | 2014-02-07 |
| CN103562799B (zh) | 2016-08-31 |
| JP2013011882A (ja) | 2013-01-17 |
| JP5274692B2 (ja) | 2013-08-28 |
| US20140220486A1 (en) | 2014-08-07 |
| EP2717100A1 (en) | 2014-04-09 |
| US9625844B2 (en) | 2017-04-18 |
| US20130130168A1 (en) | 2013-05-23 |
| EP2717100B1 (en) | 2017-09-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5274692B2 (ja) | トナー | |
| JP5812737B2 (ja) | トナー | |
| JP5558952B2 (ja) | トナー | |
| JP4929411B2 (ja) | トナー | |
| JP6000660B2 (ja) | トナーおよび該トナーの製造方法 | |
| JP5734123B2 (ja) | トナー | |
| JP5582956B2 (ja) | トナー | |
| JP6335582B2 (ja) | トナー | |
| JP5836888B2 (ja) | トナー | |
| JP6000850B2 (ja) | トナー及びトナーの製造方法 | |
| JP5289614B2 (ja) | トナー | |
| JP2012027212A5 (ja) | ||
| JP5541717B2 (ja) | トナーの製造方法 | |
| JP2012042508A5 (ja) | ||
| JP6033062B2 (ja) | トナー | |
| JP2014032232A (ja) | トナー | |
| JP6004815B2 (ja) | トナー | |
| JP6157200B2 (ja) | トナー | |
| JP6272105B2 (ja) | トナーの製造方法 | |
| JP5773765B2 (ja) | トナー |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12791997 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012791997 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012791997 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20137034333 Country of ref document: KR Kind code of ref document: A |