JP5159239B2 - トナー - Google Patents
トナー Download PDFInfo
- Publication number
- JP5159239B2 JP5159239B2 JP2007267662A JP2007267662A JP5159239B2 JP 5159239 B2 JP5159239 B2 JP 5159239B2 JP 2007267662 A JP2007267662 A JP 2007267662A JP 2007267662 A JP2007267662 A JP 2007267662A JP 5159239 B2 JP5159239 B2 JP 5159239B2
- Authority
- JP
- Japan
- Prior art keywords
- toner
- resin
- mass
- parts
- less
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images


Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/093—Encapsulated toner particles
- G03G9/09307—Encapsulated toner particles specified by the shell material
- G03G9/09314—Macromolecular compounds
- G03G9/09328—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/0827—Developers with toner particles characterised by their shape, e.g. degree of sphericity
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/083—Magnetic toner particles
- G03G9/0835—Magnetic parameters of the magnetic components
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/093—Encapsulated toner particles
- G03G9/09307—Encapsulated toner particles specified by the shell material
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03G—ELECTROGRAPHY; ELECTROPHOTOGRAPHY; MAGNETOGRAPHY
- G03G9/00—Developers
- G03G9/08—Developers with toner particles
- G03G9/093—Encapsulated toner particles
- G03G9/09392—Preparation thereof
Landscapes
- Physics & Mathematics (AREA)
- General Physics & Mathematics (AREA)
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Developing Agents For Electrophotography (AREA)
Description
ポリエステル樹脂、イソシアネート基を有する低分子化合物、およびその他の成分を酢酸エチルに溶解及び分散して油相を調製し、水中で液滴を調製する。これにより、液滴界面でイソシアネート基を有する化合物を界面重合させることで、ポリウレタンもしくはポリウレアを最外殻としたカプセルトナー粒子を調製する。
特許文献6には、ポリウレタン樹脂(a)からなる皮膜状の1層以上のシェル層(P)
と樹脂(b)からなる1層のコア層(Q)とで構成されるコア・シェル型のトナー粒子が提案されている。
このコア・シェル型のトナー粒子においては、コア部分を低粘度にし、耐熱性保存性に劣る性質を、シェル部分の耐熱保存性で補う構成をとる。この場合、シェル部分はやや熱
的に固いものを用いるために、高度に架橋したり、高い分子量にしたりするなどの工夫が必要であるため低温定着性を阻害してしまう傾向にある。
しかしながら、溶解懸濁法を用いた磁性トナーには様様な問題が生じやすかった。一つは、磁性体の分散が不十分であると、脱離した磁性体が多く発生し、トナーの抵抗を下げやすい。その結果、トナー帯電量が下がり、現像不良、転写不良等が発生しやすくなったり、剤汚染等を引き起こしやすかった。また、離型剤の添加量を大きくした場合、トナー粒子表面に離型剤がでやすくなり、流動性不良による、画像品位の低下しやすくなった。
前記表面層(B)は樹脂(b)を含有し、前記樹脂(b)は、ポリエステル樹脂(b1)、ビニル樹脂(b2)またはウレタン樹脂(b3)から選ばれる樹脂を含有する樹脂であり、
前記樹脂(a)のガラス転移温度Tg(a)と前記樹脂(b)のガラス転移温度Tg(b)が下記式(1)の関係を満たし、
Tg(a)<Tg(b)・・・(1)
前記樹脂(b)はスルホン酸基を有し、前記樹脂(b)のスルホン酸基価が1mgKOH/g以上25mgKOH/g以下であり、
前記トナーの79.6kA/mの外部磁場における磁化(σt)が、12Am2/kg以上30Am2/kg以下であり、
前記トナーの平均円形度が、0.960以上1.000以下であることを特徴とする。
特にトナー母粒子(A)にポリエステルを主成分にする樹脂(a)を用いることにより、トナーのシャープメルト性を向上させた一方で、磁性体及びワックスの分散性を制御することが出来た。
また、表面層(B)でカプセル型構造を有することにより、磁性体の表面露出を減らし、帯電性に優れるトナーを提供することが可能となり、トナー飛散、かぶりといった黒トナーで抱える問題を解決できた。
更に、本発明の好ましい形態によれば、トナーの形状制御、表面性制御も可能である。従って、帯電性、現像性、転写性、クリーニング性といった電子写真特性に求められる特性を満足できるトナーを提供することが可能となった。
Tg(a)<Tg(b)・・・(1)
トナーの79.6kA/mの外部磁場における磁化(σt)が、12Am2/kg以上30Am2/kg以下であり、トナーの平均円形度が、0.960以上1.000以下であることを特徴とする。
本発明のトナーは、ポリエステルを主成分とする樹脂(a)、磁性体及びワックスを少なくとも含有するトナー母粒子(A)の表面に、表面層(B)を有するカプセル型の構造(カプセル構造)を有している。
上記カプセル構造をとらない場合、例えば、ワックスを含有するトナーでは、トナー表面にワックスが析出することにより、トナーが凝集しやすくなり、現像領域での攪拌不良、クリーナーでのつまりを引き起こしやすい。また、磁性体がトナー表面に出ることで、トナー表面の抵抗値が下がり帯電量の低下を引き起こしやすい。帯電の低下は、現像領域だけでなく、感光体への電荷注入や転写時の剥離放電によるトナー帯電量の変化も発生しやすい。
(b1)、ビニル樹脂(b2)またはウレタン樹脂(b3)から選ばれる樹脂を含有する樹脂である。
Tg(a)<Tg(b) ・・・・・・ (1)
即ち、カプセル型トナーの、Tg(b)をTg(a)より大きくすることにより、トナーの熱特性を低温での低粘度を実現したまま、耐熱性を維持できるトナーが達成できる。
ここで、Tg(a)は35℃以上、65℃以下が好ましく、更に、40℃以上60℃以下がより好ましい。Tg(b)の好ましい範囲は後述する。
上記トナーの磁化(σt)は、磁性体の添加量、用いる磁性体の磁化等を調節することで上記範囲を満たすことが可能である。
そのため、以下[1]〜[3]の方法を用いることにより高画質に対応できるトナーの提供が可能である。
[1]ポリエステルを主成分にする樹脂(a)と磁性体を十分に予備混合し、磁性体の分散性を高めておくこと。
[2]表面層(B)に用いる樹脂(b)の極性を上げ、トナー粒子中に磁性体を密閉する。
[3]磁性体を疎水化処理し水相への親和力を下げる。
磁性体の分散性を上げるため本発明では以下の手法を実施することが好ましい。
1)湿式分散(メディア分散)
2)乾式混練
更に、磁性体の分散性を上げるため、以下の手法が好ましい。
3)乾式混練品の湿式分散
4)乾式混練作製時の溶媒添加
5)乾式混練作製時のワックス添加
これら手法は、単独又は組み合わせて行うことも出来る。
また、各種材料の予備分散後、油相の調製時の混合過程において、分散過程が不十分になりやすい。特に、本発明において、磁性体の分散不良は性能低下に顕著な傾向が現れる
。本発明では、通常の機械式撹拌翼での分散だけでなく、超音波による微分散工程あるいは油相混合液のメディア分散を入れることで、磁性体のトナー粒子への分散を改善することが出来る。
表面層(B)に用いる樹脂(b)に極性の高い官能基を導入する。例えは、カルボキシル基、スルホン酸基を樹脂(b)に導入する。更には、ウレタン結合を有するポリウレタンを主鎖に用い、上記官能基を導入することも有効である。
LogRt>14−σt/25・・・(2)
これは、上記磁性体の遊離、表面存在量を減少させ、カプセル型のトナーとして、樹脂でトナー母粒子の表面を覆うとともに、磁性体の分散を良くしたためと考えられる。
また、本発明のトナーは下記式(3)の関係を満足することがより好ましい。
LogRt>15−σt/25・・・(3)
更に、本発明のトナーは下記式(4)の関係を満足することが好ましい。
LogRt>15−σt/40・・・(4)
上記トナーの体積抵抗率とトナーの磁化の関係は、磁性体の分散を上げること、及びコア・シェル構造を形成することで上記範囲を満たすことが可能である。
より好ましくは0.010以下である。一方、上記誘電損失(tanδ)は、周波数100000Hzにおいて、0.004以上であることが好ましい。
トナーの誘電損失が0.015より大きい場合、帯電量が低く、飛び散り飛散、現像性の低下を引き起こすだけでなく、現像、転写時等にバイアス等の電気的影響を受けやすくなり、帯電が不安定になりやすい。
上記トナーの誘電損失(tanδ)は、磁性体の分散状態を制御すること、すなわち、磁性体の分散方法で調節することで上記範囲を満たすことが可能である。特に油相調製時に超音波分散をかけることにより、出力、照射時間等を調整し分散状態の制御が可能である。
0μm以上であることが好ましく、より好ましくは0.20μm以上である。
上記トナー粒子の断面拡大写真における、磁性体の個数平均分散径は、油相調製時の超音波分散時の出力、照射時間等を調節することで上記範囲を満たすことが可能である。
トナーの重量平均粒子径が4.0μmより小さいと、特に長時間の使用後などにおいて
トナーがチャージアップし、濃度が低下するなどの問題を生じやすい。また、トナーの重
量平均粒子径が9.0μmよりも大きい場合には、ライン画像等を出力する場合に於いて飛び散りやボタ落ちを招き易くなり、細線再現性に劣ることがある。
また、トナーの重量平均粒子径(D4)は、樹脂(b)の添加量、油相や分散液の配合量を制御することで上記範囲に調整することが可能である。
更に、本発明トナーにおいて、トナーを水分散体中で超音波処理した後における、トナーの0.6μm以上2.0μm以下の粒子が5.0個数%以下である事が好ましい。特に、高速機等の現像器中でシェアがかかる場合、トナー割れや、シェル剥がれといった問題が発生しやすくなり、上記問題の原因となる。より好ましくは、2.0個数%以下である。
上記トナーの微粉量は、乳化時の撹拌強度や、乳化後の脱溶剤時の撹拌羽の回転速度等を調節することで上記範囲を満たすことが可能である。
4/D1は1.00以上であることが好ましい。
本発明に用いられるトナー母粒子(A)は、ポリエステルを主成分とする樹脂(a)、磁性体及びワックスを少なくとも含有する。従って、必要に応じて上記以外に、他の添加剤を含んでもよい。
上記脂肪族ジオールは、好ましくは炭素数が2〜8であり、より好ましくは炭素数が2〜6である。
上記炭素数2〜8の脂肪族ジオールとしては、エチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、ネオペンチルグリコール、1,4−ブテンジオール、1,7−ヘプタンジオール、1,8−オクタンジオールのジオール、グリセリン、ペンタエリスリトール、トリメチロールプロパンの3価以上の多価アルコールが挙げられる。これらの中では、α,ω−直鎖アルカンジオール好ましく、1,4−ブタンジオール及び1,6−ヘキサンジオールがより好ましい。更に耐久性の観点から、脂肪族ジオールの含有量はポリエステルを構成するアルコール成分中、30〜100モル%であることが好ましく、より好ましくは50〜100モル%である。
フタル酸、イソフタル酸、テレフタル酸、トリメリット酸、ピロメリット酸等の芳香族多価カルボン酸、フマル酸、マレイン酸、アジピン酸、コハク酸、ドデセニルコハク酸、
オクテニルコハク酸等の炭素数1〜20のアルキル基又は炭素数2〜20のアルケニル基で置換されたコハク酸等の脂肪族多価カルボン酸、それらの酸の無水物及びそれらの酸のアルキル(炭素数1〜8)エステル等。
また、原料モノマー中には、定着性の観点から、3価以上の多価モノマー、即ち3価以上の多価アルコール及び/又は3価以上の多価カルボン酸化合物が含有されていてもよい。
分子量が1000以下の割合が10.0%より多い場合には、比較的熱的に不安定である低分子量成分が部材を汚染してしまう場合がある。
ために、以下のような調製方法を好適に用いることができる。
ましい。樹脂(a)と磁性体とワックスとを溶解乃至分散した溶液を、水系媒体中で懸濁させる前に、水系媒体と接触させたまま放置する方法を用いることで効率的に低分子量成分を除去することができる。
上記結晶性ポリエステルは、2価以上の多価アルコールからなるアルコール成分と、2価以上の多価カルボン酸化合物からなるカルボン酸成分とを含有した単量体を用いて得られる。その中でも、炭素数が2〜6、好ましくは4〜6の脂肪族ジオールを60モル%以上含有したアルコール成分と炭素数が2〜8、好ましくは4〜6、より好ましくは4の脂肪族ジカルボン酸化合物を60モル%以上含有したカルボン酸成分を縮重合させて得られた樹脂が好ましい。
エチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、ネオペンチルグリコール、1,4−ブテンジオール。これらの中でも、1,4−ブタンジオール、1,6−ヘキサンジオールが好ましい。
プロパン、ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン等のビスフェノールAのアルキレン(炭素数2〜3)オキサイド(平均付加モル数1〜10)付加物等の2価の芳香族アルコールやグリセリン、ペンタエリスリトール、トリメチロールプロパン等の3価以上のアルコール。
低分子量ポリエチレン、低分子量ポリプロピレン、低分子量オレフィン共重合体、マイクロクリスタリンワックス、パラフィンワックス、フィッシャートロプシュワックスの如き脂肪族炭化水素系ワックス;酸化ポリエチレンワックスの如き脂肪族炭化水素系ワックスの酸化物;脂肪族炭化水素系エステルワックスの如き脂肪酸エステルを主成分とするワックス;及び脱酸カルナバワックスの如き脂肪酸エステルを一部又は全部を脱酸化したもの;ベヘニン酸モノグリセリドの如き脂肪酸と多価アルコールの部分エステル化物;植物性油脂を水素添加することによって得られるヒドロキシル基を有するメチルエステル化合物。
本発明において特に好ましく用いられるワックスは、溶解懸濁法において、ワックス分散液の作製のしやすさ、作製したトナー中への取り込まれやすさ、定着時におけるトナーからの染み出し性、離型性から、エステルワックスが好ましい。
更に、エステルワックスが磁性体のトナー中への分散助剤として働き、凝集物、遊離物を減らすことに有利に働いているものと思われる。
質量%、より好ましくは5.0〜15.0質量%である。5.0質量%より少ないと、トナーの離型性を保てなくなり、20.0質量%より多い場合は、トナー表面にワックスが露
出し易くなり、耐熱保存性の低下を招く恐れがある。
一方、最大吸熱ピークが90℃より高いと、定着時に適切にワックスが溶融せず低温定着性や耐オフセット性に劣る場合がある。
本発明に用いられる磁性体の構成及び製造法の一例について説明する。
本発明に用いられる磁性体は、例えば、下記方法で製造される。第一鉄塩水溶液に所定量のZnの金属塩及びケイ酸塩等を添加した後に、鉄成分に対して当量以上の水酸化ナトリウムの如きアルカリを加え、水酸化第一鉄を含む水溶液を調製する。調製した水溶液のpHをpH7以上(好ましくはpH8乃至10)に維持しながら空気を吹き込み、水溶液
を70℃以上に加温しながら水酸化第一鉄の酸化反応をおこない、磁性体の芯となる種晶をまず生成する。
ることで磁性体の分散性を向上させることが可能となり、色味に優れるトナーを得ることが出来る。
上記磁性体の個数平均粒径及び変動係数は、磁性体作製時の温度、処理時間等を調節することで上記範囲を満たすことが可能である。
特に、赤味を帯びる磁性体を用いた場合、青やシアン系の着色剤を添加して用いることは有効である。
ニグロシン系染料、トリフェニルメタン系染料、含金アゾ錯体染料、モリブデン酸キレート顔料、ローダミン系染料、アルコキシ系アミン、4級アンモニウム塩(フッ素変性4級アンモニウム塩を含む)、アルキルアミド、燐の単体又は化合物、タングステンの単体又は化合物、フッ素系活性剤、サリチル酸金属塩及びサリチル酸誘導体の金属塩。
コピーチャージPSY VP2038、トリフェニルメタン誘導体のコピーブルーPR、第四級アンモニウム塩のコピーチャージ NEG VP2036、コピーチャージ NX
VP434(以上、ヘキスト社製)、LRA−901、ホウ素錯体であるLR−147(日本カーリット社製)、銅フタロシアニン、ペリレン、キナクリドン、アゾ系顔料、その他スルホン酸基、カルボキシル基及び四級アンモニウム塩等の官能基を有する高分子系の化合物。
上記表面層(B)は樹脂(b)を含有する。樹脂(b)としては、ポリエステル樹脂(b1)、ビニル樹脂(b2)、ウレタン樹脂(b3)から選ばれる樹脂を含有する樹脂である。樹脂(b)としては、上記樹脂の2種以上を併用しても差し支えない。
表面層(B)の粘度を下げるためには、ポリエステルを構成要素にもつポリエステル樹脂(b1)、ウレタン系樹脂(b3)が好ましい。また、溶剤に対する適度の親和性を示し、水分散性、粘度の調整、粒径の揃えやすさから、樹脂(b)はウレタン結合により形成された化合物であるウレタン系樹脂(b3)を含有することが特に好ましい。
本発明で用いられる樹脂(b)のガラス転移温度Tg(b)は樹脂(a)のガラス転移温度Tg(a)より大きい。そのため樹脂(b)のガラス転移温度Tg(b)を所定の値にするために、モノマー種、分子量、分岐構造をコントロールして、用いることが好ましい。Tg(b)は50℃以上、100℃以下が好ましく、更に、55℃以上90℃以下がより好ましい。これにより、耐熱保存性を満足し、定着阻害を抑えられたトナーを得ることができる。
但し、樹脂(a)と同じ組成を用いる場合、用いる溶剤に溶けやすく、造粒工程や、シェル構成時にトナー粒子を維持しにくくなる。そのため、樹脂(a)に対し、極性の高いモノマーを導入することが好ましい。
ポリエステル樹脂(b1)はスルホン酸基を有することが好ましい。ポリエステル樹脂(b1)のスルホン酸基価が1mgKOH/g以上25mgKOH/g以下であることが好ましい。より好ましくは、10mgKOH/g以上25mgKOH/g以下である。
(1)ビニル系炭化水素:
(1−1)脂肪族ビニル系炭化水素:アルケン類、例えばエチレン、プロピレン、ブテン、イソブチレン、ペンテン、ヘプテン、ジイソブチレン、オクテン、ドデセン、オクタデセン、前記以外のα−オレフィン等;アルカジエン類、例えばブタジエン、イソプレン、1,4−ペンタジエン、1,6−ヘキサジエン、1,7−オクタジエン。
(1−2)脂環式ビニル系炭化水素:モノ−もしくはジ−シクロアルケンおよびアルカジエン類、例えばシクロヘキセン、(ジ)シクロペンタジエン、ビニルシクロヘキセン、エチリデンビシクロヘプテン等;テルペン類、例えばピネン、リモネン、インデン。
(1−3)芳香族ビニル系炭化水素:スチレンおよびそのハイドロカルビル(アルキル、シクロアルキル、アラルキルおよび/またはアルケニル)置換体、例えばα−メチルスチレン、ビニルトルエン、2,4−ジメチルスチレン、エチルスチレン、イソプロピルスチレン、ブチルスチレン、フェニルスチレン、シクロヘキシルスチレン、ベンジルスチレン、クロチルベンゼン、ジビニルベンゼン、ジビニルトルエン、ジビニルキシレン、トリビニルベンゼン;およびビニルナフタレン。
炭素数3〜30の不飽和モノカルボン酸、不飽和ジカルボン酸ならびにその無水物およびそのモノアルキル(炭素数1〜24)エステル、例えば(メタ)アクリル酸、(無水)マレイン酸、マレイン酸モノアルキルエステル、フマル酸、フマル酸モノアルキルエステル、クロトン酸、イタコン酸、イタコン酸モノアルキルエステル、イタコン酸グリコールモノエーテル、シトラコン酸、シトラコン酸モノアルキルエステル、桂皮酸等のカルボキシル基含有ビニル系モノマー。
炭素数2〜14のアルケンスルホン酸、例えばビニルスルホン酸、(メタ)アリルスルホン酸、メチルビニルスルホン酸、スチレンスルホン酸;およびその炭素数2〜24のアルキル誘導体、例えばα−メチルスチレンスルホン酸等;スルホ(ヒドロキシ)アルキル−(メタ)アクリレートもしくは(メタ)アクリルアミド、例えば、スルホプロピル(メタ)アクリレート、2−ヒドロキシ−3−(メタ)アクリロキシプロピルスルホン酸、2−(メタ)アクリロイルアミノ−2,2−ジメチルエタンスルホン酸、2−(メタ)アクリロイルオキシエタンスルホン酸、3−(メタ)アクリロイルオキシ−2−ヒドロキシプロパンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸、3−(メタ)アクリルアミド−2−ヒドロキシプロパンスルホン酸、アルキル(炭素数3〜18)アリルスルホコハク酸、ポリ(n=2〜30)オキシアルキレン(エチレン、プロピレン、ブチレン:単独、ランダム、ブロックでもよい)モノ(メタ)アクリレートの硫酸エステル[ポリ(n=5〜15)オキシプロピレンモノメタクリレート硫酸エステル等]、ポリオキシエチレン多環フェニルエーテル硫酸エステル、および下記一般式(1−1)〜(1−3)で示される硫酸エステルもしくはスルホン酸基含有モノマー;ならびそれらの塩。

炭素数(NCO基中の炭素を除く、以下同様)6〜20の芳香族ジイソシアネート、炭素数2〜18の脂肪族ジイソシアネート、炭素数4〜15の脂環式ジイソシアネート、炭素数8〜15の芳香族炭化水素ジイソシアネート、及びこれらのジイソシアネートの変性物(ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物。以下、変性ジイソシアネートともいう)、並びにこれらの2種以上の混合物。
1,3−フェニレンジイソシアネート、1,4−フェニレンジイソシアネート、1,5−ナフチレンジイソシアネート、2,4−トリレンジイソシアネート、2,6−トリレンジイソシアネート(TDI)、粗製TDI、2,4’−ジフェニルメタンジイソシアネート、4,4’−ジフェニルメタンジイソシアネート(MDI)、粗製MDI[粗製ジアミノフェニルメタン〔ホルムアルデヒドと芳香族アミン(アニリン)又はその混合物との縮合生成物〕]。
エチレンジイソシアネート、テトラメチレンジイソシアネート、ヘキサメチレンジイソシアネート(HDI)、ドデカメチレンジイソシアネート、1,6,11−ウンデカントリイソシアネート、2,2,4−トリメチルヘキサメチレンジイソシアネート、リジンジイソシアネート、2,6−ジイソシアナトメチルカプロエート、ビス(2−イソシアナトエチル)フマレート、ビス(2−イソシアナトエチル)カーボネート、2−イソシアナトエチル−2,6−ジイソシアナトヘキサノエート。
イソホロンジイソシアネート(IPDI)、ジシクロヘキシルメタン−4,4’−ジイソシアネート(水添MDI)、シクロヘキシレンジイソシアネート、メチルシクロヘキシレンジイソシアネート(水添TDI)、ビス(2−イソシアナトエチル)−4−シクロヘキセン−1,2−ジカルボキシレート、2,5−ノルボルナンジイソシアネート、2,6
−ノルボルナンジイソシアネート。
m−キシリレンジイソシアネート、p−キシリレンジイソシアネート(XDI)、α,α,α’,α’−テトラメチルキシリレンジイソシアネート(TMXDI)。
変性MDI(ウレタン変性MDI、カルボジイミド変性MDI、トリヒドロカルビルホスフェート変性MDI)、ウレタン変性TDI等のイソシアネートの変性物及びこれらの2種以上の混合物[例えば変性MDIとウレタン変性TDI(イソシアネート含有プレポリマー)との併用]が挙げられる。
これらのうちで好ましいものは6〜15の芳香族ジイソシアネート、炭素数4〜12の脂肪族ジイソシアネート、及び炭素数4〜15の脂環式ジイソシアネートであり、特に好ましいものはTDI、MDI、HDI、水添MDI、及びIPDIである。
アルキレングリコール(エチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、1,6−ヘキサンジオール、オクタンジオール、デカンジオール、ドデカンジオール、テトラデカンジオール、ネオペンチルグリコール、2,2−ジエチル−1,3−プロパンジオール);
アルキレンエーテルグリコール(ジエチレングリコール、トリエチレングリコール、ジプロピレングリコール、ポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレンエーテルグリコール);
脂環式ジオール(1,4-シクロヘキサンジメタノール、水素添加ビスフェノールAなど);
ビスフェノール類(ビスフェノールA、ビスフェノールF、ビスフェノールSなど);
上記脂環式ジオールのアルキレンオキサイド(エチレンオキサイド、プロピレンオキサイド、ブチレンオキサイドなど)付加物;
上記ビスフェノール類のアルキレンオキサイド(エチレンオキサイド、プロピレンオキサイド、ブチレンオキサイドなど)付加物;
その他、ポリラクトンジオール(ポリε−カプロラクトンジオールなど)、ポリブタジエンジオール。
上記したアルキレンエーテルグリコールのアルキル部分は直鎖状であっても、分岐していてもよい。本発明においては分岐構造のアルキレングリコールも好ましく用いることができる。
ジアミノエタン、ジアミノプロパン、ジアミノブタン、ジアミノヘキサン、ピペラジン、2,5−ジメチルピペラジン、アミノ−3−アミノメチル−3,5,5−トリメチルシクロヘキサン(イソホロンジアミン、IPDA)、4,4′−ジアミノジシクロヘキシルメタン、1,4−ジアミノシクロヘキサン、アミノエチルエタノールアミン、ヒドラジン、ヒドラジン水和物などのジアミン。
トリエチルアミン、ジエチレントリアミンおよび1,8−ジアミノ−4−アミノメチルオクタンなどのトリアミン。
酢酸エチル、キシレン、ヘキサン等の炭化水素系溶媒、塩化メチレン、クロロホルム、ジクロルエタン等のハロゲン化炭化水素系溶媒、酢酸メチル、酢酸エチル、酢酸ブチル、酢酸イソプロピル等のエステル系溶媒、ジエチルエーテル等のエーテル系溶媒、アセトン、メチルエチルケトン、ジイソブチルケトン、シクロヘキサノン、メチルシクロヘキサン等のケトン系溶媒、メタノール、エタノール、ブタノール等のアルコール系溶媒。
トナー粒子は、樹脂(b)を含有する樹脂微粒子を分散させた水系媒体中(以下、水相ともいう)に、少なくとも、ポリエステルを主成分とする樹脂(a)、磁性体及びワックスを有機媒体中で溶解又は分散させて得られた溶解物又は分散物(以下、油相ともいう)を分散させ、得られた分散液から溶媒を除去し乾燥することによって得られることが好ましい。
上記の系においては、樹脂微粒子が上記溶解物又は分散物(油相)を上記水相に懸濁する際の分散剤としても機能する系である。上記方法でトナー粒子を調製することにより、トナー表面への凝集工程などを必要とせず、簡便にカプセル型のトナー粒子を調製することができる。
酢酸エチル、キシレン、ヘキサン等の炭化水素系溶媒、塩化メチレン、クロロホルム、ジクロルエタン等のハロゲン化炭化水素系溶媒、酢酸メチル、酢酸エチル、酢酸ブチル、酢酸イソプロピル等のエステル系溶媒、ジエチルエーテル等のエーテル系溶媒、アセトン、メチルエチルケトン、ジイソブチルケトン、シクロヘキサノン、メチルシクロヘキサン等のケトン系溶媒。
上記油相は、これら、樹脂分散液、ワックス分散液、磁性体分散液、及び有機媒体を所望量配合し、上記各成分を該有機媒体中に分散させることで調製することが出来る。
本発明において、磁性体の分散性を通常以上に上げるために以下の手法を用いた。
(1)湿式分散(メディア分散)
磁性体を分散用メディア存在下で溶媒に分散する方法である。例えば、磁性体、樹脂、
その他添加剤と上記有機溶媒を混合し、分散用メディア存在下で分散機を用いて、混合物を分散する。用いた分散用メディアは回収し磁性体分散液を得る。上記分散機としては、例えば、アトライター(三井三池工機(株))を使用する。上記分散用メディアとしては、アルミナ、ジルコニア、ガラス及び鉄のビーズが挙げられるが、メディア汚染が極めて少ないジルコニアビーズが好ましい。その際のビーズ径は、2mm〜5mmが分散性に優れており好ましい。
(2)乾式混練
樹脂、磁性体、その他添加剤を、ニーダー、ロール式の分散器で溶融混錬し(乾式)、得られた樹脂と磁性体の溶融混練物を粉砕後、上記有機溶媒に溶解させることにより、磁性体分散液を得る。
(3)乾式溶融混練物の湿式分散
上記乾式で得られた樹脂と磁性体の溶融混練物を用いて作製された磁性体分散液を、上記分散用メディア及び分散機を用いて更に湿式分散する。
(4)乾式溶融混練物作製時の溶媒添加
上記乾式溶融混練物の作製時に、溶媒を添加する。溶融混練時の温度は、樹脂のガラス転移温度(Tg)以上、溶媒の沸点以下が好ましい。用いる溶媒は、樹脂を溶解できるものが好ましく、上記油相に用いられる溶媒が好ましい。
(5)乾式溶融混練物作製時のワックス添加
上記乾式溶融混練物の作製時に、ワックスを添加する。溶融混練時の温度は、樹脂のガラス転移温度(Tg)以上、溶媒の沸点以下が好ましい。用いるワックスは、上記油相に溶解するワックスを用いてもいいが、他の比較的高融点のワックスを用いてもよい。
(6)樹脂に磁性体との親和性の高い樹脂を用いる。
上記乾式溶融混練物の作製に用いる樹脂に、磁性体との親和性の高い樹脂を用いる。例えば、ポリエステルを主成分とする樹脂(a)に、少なくとも2種の樹脂(a1)、(a2)を用い、一方の樹脂(a2)で、磁性体を分散する。ここで、樹脂(a1)には少なくとも脂肪族ジオールより合成される樹脂を用い、樹脂(a2)には結晶性ポリエステルまたは、少なくとも芳香族ジオールより合成される樹脂を用いる。
アクリル酸、メタクリル酸、α−シアノアクリル酸、α−シアノメタクリル酸、イタコン酸、クロトン酸、フマール酸、マレイン酸または無水マレイン酸などの酸類;
アクリル酸β−ヒドロキシエチル、メタクリル酸β−ヒドロキシエチル、アクリル酸β−ヒドロキシプロビル、メタクリル酸β−ヒドロキシプロピル、アクリル酸γ−ヒドロキシプロピル、メタクリル酸γ−ヒドロキシプロピル、アクリル酸3−クロロ2−ヒドロキシプロビル、メタクリル酸3−クロロ−2−ヒドロキシプロピル、ジエチレングリコールモノアクリル酸エステル、ジエチレングリコールモノメタクリル酸エステル、グリセリンモノアクリル酸エステル、グリセリンモノメタクリル酸エステル、N−メチロールアクリルアミド、N−メチロールメタクリルアミド等の水酸基を含有する(メタ)アクリル系単量体;
ビニルアルコール、又はビニルメチルエーテル、ビニルエチルエーテル、ビニルプロピルエーテル等のビニルアルコールとのエ一テル類;
酢酸ビニル、プロピオン酸ビニル、酪酸ビニル等のビニルアルコールとカルボキシル基を含有する化合物のエステル類;
アクリルアミド、メタクリルアミド、ジアセトンアクリルアミド或いはこれらのメチロール化合物;アクリル酸クロライド、メタクリル酸クロライド等の酸クロライド類;ビニルピリジン、ビニルピロリドン、ビニルイミダゾール、エチレンイミン等の窒素原子、又はその複素環を有するもの等のホモポリマー又は共重合体;
ポリオキシエチレン、ポリオキシプロピレン、ポリオキシエチレンアルキルアミン、ポリオキシプロピレンアルキルアミン、ポリオキシエチレンアルキルアミド、ポリオキシプロピレンアルキルアミド、ポリオキシエチレンノニルフェニルエーテル、ポリオキシエチレンラウリルフェニルエーテル、ポリオキシエチレンステアリルフェニルエステル、ポリオキシエチレンノニルフェニルエステル等のポリオキシエチレン類;
メチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース等のセルロース類。
、高剪断力で有機媒体を微細に分散して形成された油滴の周囲を分散安定剤が囲み、油滴同士が再凝集するのを防ぎ、安定化させる。
例えば、ウルトラタラックス(IKA社製)、ポリトロン(キネマティカ社製)、TKオートホモミキサー(特殊機化工業(株)製)、エバラマイルダー(荏原製作所(株)製)、TKホモミックラインフロー(特殊機化工業(株)製)、コロイドミル(神鋼パンテック社製)、スラッシャー、トリゴナル湿式微粉砕機(三井三池化工機(株)製)、キャビトロン(ユーロテック社製)、ファインフローミル(太平洋機工(株)製)等の連続式乳化機、クレアミックス(エムテクニック社製)、フィルミックス(特殊機化工業(株)製)のバッチ式、若しくは連続両用乳化機が挙げられる。
シリカ、アルミナ、酸化チタン、チタン酸バリウム、チタン酸マグネシウム、チタン酸カルシウム、チタン酸ストロンチウム、酸化亜鉛、酸化スズ、ケイ砂、クレー、雲母、ケイ灰石、ケイソウ土、酸化クロム、酸化セリウム、ベンガラ、三酸化アンチモン、酸化マグネシウム、酸化ジルコニウム、硫酸バリウム、炭酸バリウム、炭酸カルシウム、炭化ケイ素、窒化ケイ素。
上記ポリマー微粒子は、比較的粒度分布が狭く、体積平均粒径が0.01から1μmのものが好ましい。
<樹脂の軟化点(Tm)の測定方法>
樹脂の軟化点(Tm)は、定荷重押出し式細管式レオメーターであるフローテスターにより測定した。
即ち、樹脂の軟化点(Tm)は、島津製作所製の高架式フローテスターCFT500C型を用い、下記条件にて測定した。得られたデータに基づき、フローテスターカーブを作製した(図1(a)および(b)に表示)。該図より樹脂の軟化点(Tm)を求めた。
図1中、Tfb:流出開始温度を樹脂の軟化点(Tm)とした。
(測定条件)
荷重 :10kgf/cm2 (9.807×105 Pa)
昇温速度:4.0℃/min
ダイ口径:1.0mm
ダイ長さ:1.0mm
ワックスの融点は、ワックスを、示差走査熱量計(DSC)「Q1000」(TA Instruments社製)を用い、ASTM D3418−82に準じて測定した。
装置検出部の温度補正はインジウムと亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いた。
具体的には、試料約10mgを精秤し、アルミニウム製のパンの中に入れ、リファレンスとして空のアルミニウム製のパンを用い、測定温度範囲30〜200℃の間で、昇温速度10℃/minで測定を行った。尚、測定においては、一度200℃まで昇温させ、続いて30℃まで降温し、その後に再度昇温を行う。この2度目の昇温過程で、温度30〜200℃の範囲におけるDSC曲線の最大の吸熱ピークを示す温度をワックスの融点とした。上記最大吸熱ピークとは、ピークが複数存在する場合には、最も吸熱量の大きいピークをいう。
樹脂のガラス転移温度(Tg)は、示差走査熱量計(DSC)「Q1000」(TA
Instruments社製)を用い、ASTM D3418−82に準じて測定した。装置検出部の温度補正はインジウムと亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いた。
具体的には、試料約10mgを精秤し、アルミニウム製のパンの中に入れ、リファレンスとして空のアルミニウム製のパンを用い、測定温度範囲30〜200℃の間で、昇温速度10℃/minで測定を行った。 この昇温過程で、温度30℃〜100℃の範囲にお
いて比熱変化が得られた。このときの比熱変化が出る前と出た後のベースラインの中間点の線と示差熱曲線との交点を、結着樹脂のガラス転移温度(Tg)とした。
本発明の磁性体ののBET比表面積の測定は次の様にして行った。
BET比表面積は、湯浅アイオニクス(株)製、全自動ガス吸着量測定装置(オートソープ1)を使用し、吸着ガスに窒素を用い、BET多点法により求めた。サンプルの前処理としては、50℃で10時間の脱気を行った。
トナーの重量平均粒径(D4)および数平均粒径(D1)は、100μmのアパーチャーチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer 3」(登録商標、ベックマン・コールター社製)と、測定条件設定及び測定データ解析をするための付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター社製)を用いて、実効測定チャンネル数2万5千チャンネルで測定し、測定データの解析を行ない、算出した。
測定に使用する電解水溶液は、特級塩化ナトリウムをイオン交換水に溶解して濃度が約1質量%となるようにしたもの、例えば、「ISOTON II」(ベックマン・コールター社製)が使用できる。
尚、測定、解析を行なう前に、以下のように専用ソフトの設定を行なった。
専用ソフトの「標準測定方法(SOM)を変更画面」において、コントロールモードの総カウント数を50000粒子に設定し、測定回数を1回、Kd値は「標準粒子10.0μm」(ベックマン・コールター社製)を用いて得られた値を設定した。閾値/ノイズレ
ベルの測定ボタンを押すことで、閾値とノイズレベルを自動設定した。また、カレントを1600μAに、ゲインを2に、電解液をISOTON IIに設定し、測定後のアパーチャーチューブのフラッシュにチェックを入れた。
専用ソフトの「パルスから粒径への変換設定画面」において、ビン間隔を対数粒径に、粒径ビンを256粒径ビンに、粒径範囲を2μmから60μmまでに設定した。
具体的な測定法は以下の通りである。
(1)Multisizer 3専用のガラス製250ml丸底ビーカーに前記電解水溶液約200mlを入れ、サンプルスタンドにセットし、スターラーロッドの撹拌を反時計回りで24回転/秒にて行った。そして、解析ソフトの「アパーチャーのフラッシュ」機能により、アパーチャーチューブ内の汚れと気泡を除去しておいた。
(2)ガラス製の100ml平底ビーカーに前記電解水溶液約30mlを入れ、この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で3質量倍に希釈した希釈液を約0.3ml加えた。
(3)発振周波数50kHzの発振器2個を、位相を180度ずらした状態で内蔵し、電気的出力120Wの超音波分散器「Ultrasonic Dispension System Tetora150」(日科機バイオス社製)の水槽内に所定量のイオン交換水を入れ、この水槽中に前記コンタミノンNを約2ml添加した。
(4)前記(2)のビーカーを前記超音波分散器のビーカー固定穴にセットし、超音波分散器を作動させた。そして、ビーカー内の電解水溶液の液面の共振状態が最大となるようにビーカーの高さ位置を調整した。
(5)前記(4)のビーカー内の電解水溶液に超音波を照射した状態で、トナー約10mgを少量ずつ前記電解水溶液に添加し、分散させた。そして、さらに60秒間超音波分散処理を継続した。尚、超音波分散にあたっては、水槽の水温が10℃以上40℃以下となる様に適宜調節した。
(6)サンプルスタンド内に設置した前記(1)の丸底ビーカーに、ピペットを用いてトナーを分散した前記(5)の電解質水溶液を滴下し、測定濃度が約5%となるように調整した。そして、測定粒子数が50000個になるまで測定を行った。
(7)測定データを装置付属の前記専用ソフトにて解析を行ない、重量平均粒径(D4)および数平均粒径(D1)を算出した。尚、専用ソフトでグラフ/体積%と設定したときの、分析/体積統計値(算術平均)画面の「平均径」が重量平均粒径(D4)であり、専用ソフトでグラフ/個数%と設定したときの、分析/個数統計値(算術平均)画面の「平均径」が数平均粒径(D1)である。
トナーの平均円形度は、フロー式粒子像分析装置「FPIA−3000」(シスメックス社製)を用い、校正作業時の測定及び解析条件で測定した。
具体的な測定方法としては、イオン交換水20mlに、分散剤として界面活性剤、好ましくはドデシルベンゼンスルホン酸ナトリウム塩を適量加えた後、測定試料0.02gを加え、発振周波数50kHz、電気的出力150Wの卓上型の超音波洗浄器分散機(例えば「VS−150」(ヴェルヴォクリーア社製))を用いて2分間分散処理を行い、測定用の分散液とした。その際、分散液の温度が10℃以上40℃以下となる様に適宜冷却した。
測定には、標準対物レンズ(10倍)を搭載した前記フロー式粒子像分析装置を用い、シース液にはパーティクルシース「PSE−900A」(シスメックス社製)を使用した。前記手順に従い調製した分散液を前記フロー式粒子像分析装置に導入し、HPF測定モードで、トータルカウントモードにて3000個のトナー粒子を計測して、粒子解析時の2値化閾値を85%とし、解析粒子径を円相当径2.00μm以上、200.00μm以下に限定し、トナー粒子の平均円形度を求めた。
測定にあたっては、測定開始前に標準ラテックス粒子(例えばDuke Scient
ific社製の「5100A」をイオン交換水で希釈)を用いて自動焦点調整を行う。その後、測定開始から2時間毎に焦点調整を実施することが好ましい。
なお、本願実施例では、シスメックス社による校正作業が行われた、シスメックス社が発行する校正証明書の発行を受けたフロー式粒子像分析装置を使用し、解析粒子径を円相当径2.00μm以上、200.00μm以下に限定した以外は、校正証明を受けた時の測定及び解析条件で測定を行った。
一方、トナーの微分量は、解析粒子径を0.60μm以上、200.00μm以下の範
囲で、平均円形度の測定と同様に測定し、0.60μm以上、2.00μm以下の個数頻度を求め、0.60μm以上、200.00μm以下の全範囲に対する割合を求めた。これ
を、トナーの微粉量とした。
上記トナーの微分量を求めた測定用分散液を更に、発振周波数50kHz、電気的出力150Wの卓上型の超音波洗浄器分散機(「VS−150」(ヴェルヴォクリーア社製))を用いて30分間分散処理を行い、測定用の分散液とした。
この分散液を、上記トナーの微粉量測定と同様にして、測定を行い0.60μm以上、
2.00μm以下の個数頻度を求め、0.60μm以上、200.00μm以下の全範囲に対する割合を求めた。
樹脂微粒子、及びワックス分散液中のワックス粒子の粒子径は、マイクロトラック粒度分布測定装置HRA(X−100)(日機装社製)を用い、0.001μm〜10μmのレンジ設定で測定を行い、個数平均粒子径(μm又はnm)として測定した。なお、希釈溶媒として樹脂微粒子には水、ワックス粒子には酢酸エチルを選択した。
樹脂のゲルパーミエーションクロマトグラフ(GPC)による分子量分布、ピーク分子量、及び数平均分子量は、樹脂のテトラヒドロフラン(THF)可溶分を、THFを溶媒としたGPC(ゲルパーメイションクロマトグラフィ)により測定した。測定条件は以下の通りである。
(1)測定試料の作製
樹脂(試料)とTHFとを約0.5〜5mg/ml(例えば約5mg/ml)の濃度で混合し、室温にて数時間(例えば5〜6時間)放置した後、充分に振とうし、THFと試料を試料の合一体がなくなるまで良く混ぜた。更に、室温にて12時間以上(例えば24時間)静置した。この時、試料とTHFの混合開始時点から、静置終了の時点までの時間が24時間以上となる様にした。
その後、サンプル処理フィルタ(ポアサイズ0.45〜0.5μm、マイショリディスクH−25−2[東ソー社製]、エキクロディスク25CR[ゲルマン サイエンスジャパン社製]が好ましく利用出来る)を通過させたものをGPCの試料とした。
(2)試料の測定
40℃のヒートチャンバー中でカラムを安定化させ、この温度に於けるカラムに、溶媒としてTHFを毎分1mlの流速で流し、試料濃度を0.5〜5mg/mlに調整した樹脂のTHF試料溶液を50〜200μl注入して測定した。
試料の分子量測定にあたっては、試料の有する分子量分布を数種の単分散ポリスチレン標準試料により作製された検量線の対数値とカウント数との関係から算出した。
検量線作成用の標準ポリスチレン試料としては、Pressure Chemical
Co.製或いは東洋ソーダ工業社製の、分子量が6×102、2.1×103、4×103、1.75×104、5.1×104、1.1×105、3.9×105、8.6×105、2×106、4.48×106のものを用いた。又、検出器にはRI(屈折率)
検出器を用いた。
尚、カラムとしては、1×103〜2×106の分子量領域を適確に測定する為に、市販のポリスチレンゲルカラムを下記のように複数組合せて用いた。本発明に於ける、GPCの測定条件は以下の通りである。
[GPC測定条件]
装 置 :LC−GPC 150C(ウォーターズ社製)
カラム :KF801,802,803,804,805,806,807(ショウデックス製)の7連
カラム温度 :40℃
移動相 :THF(テトラヒドロフラン)
トナーの誘電損率ε”/誘電率ε’で示される誘電損失(tanδ)は、4284AプレシジョンLCRメータ(ヒューレット・パッカード社製)を用いて、1000Hz及び1MHzの周波数で校正後、周波数100000Hzにおける複素誘電率の測定値より誘電損失正接(tanδ=ε”/ε’)を算出した。
即ち、トナーを1.0g秤量し、19600kPa(200kgf/cm2)の荷重を1分間かけて成形し、直径25mm、厚さ2mm以下(好ましくは0.5mm以上1.5mm以下)の円盤状の測定試料を調製した。この測定試料を直径25mmの誘電率測定治具(電極)を装着したARES(レオメトリック・サイエンティフィック・エフ・イー社製)に装着し、室温で、1000Hzから1MHzの周波数の範囲で測定試料の複素誘電率を測定し、誘電損失正接(tanδ=ε”/ε’)を算出した。周波数100000Hzにおける値を、トナーの誘電損率ε”/誘電率ε’で示される誘電損失(tanδ)とした。
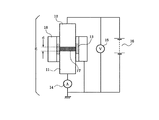
トナーの体積抵抗率(Rt:Ω・cm)は、図2に示した測定装置を用いて行った。
即ち、抵抗測定セルEに、トナーを充填し、該トナーに接するように下部電極11及び上部電極12を配し、これらの電極間に電圧を印加し、そのときに流れる電流を測定することによって体積抵抗率を求めた。測定条件は、以下の通りである。
充填トナーと電極との接触面積:S=約2.3cm2
厚み :d=約0.5mm
上部電極12の荷重 :180g
印加電圧 :500V
クライオミクロト―ム(Reichert社製 ULTRACUT N FC4E)装置に水溶性樹脂に分散したトナー粒子を入れた。液体窒素により該装置を−80℃まで冷却し、トナー粒子が分散された水溶性樹脂を凍結した。凍結された水溶性樹脂を、ガラスナイフにより切削面形状が約0.1ミリ幅、約0.2ミリ長になるようにトリミングした。次にダイヤモンドナイフを用いて、水溶性樹脂を含むトナーの超薄切片(厚み設定:70nm)を作製し、まつげプローブを用いてTEM観察用グリッドメッシュ上に移動した。水溶性樹脂を含むトナー粒子の超薄切片を室温に戻した後、水溶性樹脂を純水に溶解させて透過型電子顕微鏡(TEM)の観察試料とした。該試料は、透過型電子顕微鏡H−7500(日立製作所製)を用い、加速電圧100kVにて観察し、トナー粒子の断面の拡大写真を撮影した。トナー粒子の断面は任意に選んだ。また、拡大写真の倍率は10000倍とした。
上記写真撮影により得られた画像は、インターフェースを介して、600dpiで読み取り、画像解析装置Win ROOF Version5.0(マイクロソフト社製−三
谷商事)に導入し、2値の画像データに変換した。そのうち、磁性体についてのみ無作為に解析を行うこととし、サンプリング数が100回まで測定を繰り返し、磁性体の凝集径を求め、その個数平均をトナー粒子中に存在する磁性体の個数平均分散径とした。
磁性体及びトナーの磁化の強さは、磁気特性と質量とから求めた。磁性体及びトナーの磁気特性は、「振動資料型磁力計VSM−3S−15」(東英工業(株)製)を用いて測定した。
測定方法としては、円筒状のプラスチック容器に十分密になるように磁性体またはトナーを充填し、一方で1.00キロエルステッド(79.6kA/m)の外部磁場を作り、
この状態で前記容器に充填した磁性体またはトナーの磁化モーメントを測定した。
次に、前記容器に充填した磁性体またはトナーの実際の質量を測定して、磁性体またはトナーの磁化の強さ(Am2/kg)を求めた。
また、最大印加磁場を1.00キロエルステッド(79.6kA/m)とした際のヒス
テリシスループを描くことにより、残留磁化(σr)を求めた。
磁性体の個数平均粒径(D50)及び標準偏差σは、電子顕微鏡観察で撮影した粒子画像(任意に350個)を、統計解析(グラフテック株式会社製デジタイザKD4620)を用いて計測し、算出した。
また、磁性体の粒径の変動係数は、上記個数平均径D50(μm)と上記標準偏差σ(μm)とから下記式に従って算出した。粒度分布の値が小さくなるほど、粒度分布に優れていることを表している。
(式)磁性体の粒径の変動係数=σ/D50×100(%)
磁性体のかさ密度は、パウダーテスタPT−R(ホソカワミクロン社製)を用い、
該機器の操作マニュアルに従い、測定した。
目開き500μmの篩を用いて、振幅を1mmで振動させながら、ちょうど10mlとなるまで磁性体を補給しつつ、金属性カップを振幅18mmにて上下往復180回タッピングさせ、タッピング後の磁性体量から、かさ密度(g/cm3)を計算した。
固形分20質量%の樹脂微粒子分散液を、塩酸、又は水酸化ナトリウムで、中性(pH=7.0±0.1)にした後、塩酸を滴定しながら、分散液のpH及びゼータ電位を測定する。pHが2.0以上3.0以下の範囲において、ゼータ電位が負の値から、正の値に変わることを観測する。この範囲でゼータ電位が0になる点を求め、要した塩酸のモル数を求める。同じモル数の水酸化カリウムの質量を求める。一方で、樹脂微粒子分散液の固形分の質量を求めることにより、単位質量あたりのスルホン酸基価の値とした。
なお、pHが3.0以上の値でゼータ電位が負の値から、正の値に変わる場合、スルホン酸基価は0mgKOH/gとする。
酸価は試料1gに含まれる酸を中和するために必要な水酸化カリウムのmg数である。結着樹脂の酸価はJIS K 0070−1966に準じて測定されるが、具体的には、以下の手順に従って測定する。
(1)試薬の準備
フェノールフタレイン1.0gをエチルアルコール(95vol%)90mlに溶かし、イオン交換水を加えて100mlとし、「フェノールフタレイン溶液」を得る。
特級水酸化カリウム7gを5mlの水に溶かし、エチルアルコール(95vol%)を
加えて1lとする。炭酸ガスに触れないように、耐アルカリ性の容器に入れて3日間放置後、ろ過して、「水酸化カリウム溶液」を得る。得られた水酸化カリウム溶液は、耐アルカリ性の容器に保管する。標定はJIS K 0070−1996に準じて行う。
(2)操作
(A)本試験
粉砕した結着樹脂の試料2.0gを200mlの三角フラスコに精秤し、トルエン/エタノール(2:1)の混合溶液100mlを加え、5時間かけて溶解する。次いで、指示薬として前記フェノールフタレイン溶液を数滴加え、前記水酸化カリウム溶液を用いて滴定する。尚、滴定の終点は、指示薬の薄い紅色が約30秒間続いたときとする。
(B)空試験
試料を用いない(すなわちトルエン/エタノール(2:1)の混合溶液のみとする)以外は、上記操作と同様の滴定を行う。
(3)得られた結果を下記式に代入して、酸価を算出する。
A=[(B−C)×f×5.61]/S
ここで、A:酸価(mgKOH/g)、B:空試験の水酸化カリウム溶液の添加量(ml)、C:本試験の水酸化カリウム溶液の添加量(ml)、f:水酸化カリウム溶液のファクター、S:試料(g)である。
・プロピレングリコール、エチレングリコール、ブタンジオールの40:50:10モル混合物とテレフタル酸、イソフタル酸の等モル混合物から得られた、数平均分子量約2000のポリエステルジオール 120質量部
・ジメチロールプロパン酸 94質量部
・3−(2,3−ジヒドロキシプロポキシ)−1−プロパンスルホン酸
8質量部
・イソホロンジイソシアネート 120質量部
上記原材料をアセトン60質量部中に溶解し、67℃1時間反応させた。
ついで、イソホロンジイソシアネート271質量部を添加し、更に67℃で30分反応させ冷却した。
上記反応物に更に100質量部のアセトンを追加した後、トリエチルアミン80質量部を投入し攪拌した。
上記アセトン溶液をイオン交換水1000質量部に500rpmで攪拌しながら滴下し、微粒子分散液を調製した。
ついで10%アンモニア水100質量部にトリエチルアミン50質量部を溶解させた水溶液を投入し、50℃、8時間反応させることで伸長反応を行った。更に、イオン交換水を固形分20質量%になるまで添加し樹脂微粒子分散液−1を得た。特性を表1に示す。
温度計、撹拌機を備えたオ−トクレ−ブ中に、
ジメチルテレフタレ−ト 116質量部、
ジメチルイソフタレ−ト 66質量部、
5−ナトリウムスルホイソフタレ−トメチルエステル 30質量部、
無水トリメリット酸 5質量部、
プロピレングリコ−ル 150質量部、
テトラブトキシチタネ−ト 0.1質量部、
を仕込み200℃で120分間加熱してエステル交換反応を行った。ついで反応系を220℃まで昇温し、系の圧力1〜10mmHgとして60分間反応を続け、ポリエステル樹脂を得た。該ポリエステル樹脂40質量部、メチルエチルケトン15質量部、テトラヒドロフラン10質量部を80℃にて溶解した後、80℃の水60質量部を攪拌しながら添加し、減圧にて溶剤を除去し、イオン交換水を添加することにより、固形分20質量%である樹脂微粒子分散液−2を得た。特性を表1に示す。
冷却管、窒素導入管および攪拌機のついた反応容器中に、下記を投入した。
・スチレン 300質量部
・n−ブチルアクリレート 110質量部
・アクリル酸 10質量部
・スチレンスルホン酸ナトリウム 30質量部
・2−ブタノン(溶媒) 50質量部
重合開始剤として2,2’−アゾビス(2,4−ジメチルバレロニトリル)8質量部を上記組成物に溶解し、重合性単量体組成物を調製した。60℃で8時間、重合性単量体組成物を重合した後、150℃まで昇温させ、減圧下で脱溶剤し、反応容器から取り出した。反応物を室温まで冷却した後、粉砕、粒子化し、線形ビニル樹脂を得た。該樹脂100質量部と、トルエン400質量部とを混合し、80℃まで加温し、樹脂を溶解し、樹脂溶解液を得た。
次に、イオン交換水360質量とドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液(「エレミノールMON−7」、三洋化成工業製)40質量部とを混合し、上記樹脂溶解液を加え混合攪拌し乳白色の液体を得た。減圧にてトルエンを除去し、イオン交換水を添加することにより、固形分20質量%である樹脂微粒子分散液−3を得た。特性を表1に示す。
・プロピレングリコール、エチレングリコール、ブタンジオールの40:50:10モル混合物とテレフタル酸、イソフタル酸の等モル混合物から得られた、数平均分子量約2000のポリエステルジオール 100質量部
・プロピレングリコール 16質量部
・ジメチロールプロパン酸 94質量部
・N,N−ビス(2−ヒドロキシエチル)−2−アミノエタンスルホン酸ナトリウム
8質量部
・トリレンジイソシアネート 30質量部
上記原材料をアセトン60質量部に溶解し、67℃で1時間反応させた。
更に、イソホロンジイソシアネート271質量部(1.2モル)を添加し、更に67℃で30分反応させ冷却した。
上記反応物に更に100質量部のアセトンを追加した後、トリエチルアミン80質量部(0.8モル)を投入し攪拌した。
上記アセトン溶液をイオン交換水1000質量部に500rpmで攪拌しながら滴下し、微粒子分散液を調製した。
ついで10%アンモニア水100質量部にトリエチルアミン50質量部を溶解させた水溶液を投入し、50℃で8時間反応させることで伸長反応を行った。更に、イオン交換水を固形分20質量%になるまで添加し、樹脂微粒子分散液−4を得た。特性を表1に示す。
・プロピレングリコール、エチレングリコール、ブタンジオールの40:50:10モル
混合物とテレフタル酸、イソフタル酸の等モル混合物から得られた、数平均分子量約2000のポリエステルジオール 120質量部
・プロピレングリコール 8質量部
・ジメチロールプロパン酸 94質量部
・3−(2,3−ジヒドロキシプロポキシ)−1−プロパンスルホン酸
8質量部
・イソホロンジイソシアネート 39質量部
上記原材料をアセトン60質量部中に溶解し、67℃で1時間反応させた。
ついで、イソホロンジイソシアネート271質量部を添加し、更に67℃で30分反応させ冷却した。
上記アセトン溶液をイオン交換水1000質量部に500rpmで攪拌しながら滴下し、微粒子分散液を調製した。
上記反応物に更に100質量部のアセトンを追加した後、トリエチルアミン80質量部を投入し攪拌した。
ついで10%アンモニア水100質量部にトリエチルアミン50質量部を溶解させた水溶液を投入し、50℃で8時間反応させることで伸長反応を行った。更に、イオン交換水を固形分20質量%になるまで添加し、樹脂微粒子分散液−5を得た。特性を表1に示す。
・プロピレングリコール、エチレングリコール、ブタンジオールの40:50:10モル混合物とテレフタル酸、イソフタル酸の等モル混合物から得られた、数平均分子量約2000のポリエステルジオール 120質量部
・プロピレングリコール 8質量部
・ジメチロールプロパン酸 94質量部
・3−(2,3−ジヒドロキシプロポキシ)−1−プロパンスルホン酸
8質量部
・イソホロンジイソシアネート 39質量部
上記原材料をアセトン60質量部中に溶解し、67℃で1時間反応させた。
ついで、イソホロンジイソシアネート150質量部を添加し、更に65℃で20分反応させ冷却した。
上記アセトン溶液をイオン交換水1000質量部に500rpmで攪拌しながら滴下し、微粒子分散液を調製した。
上記反応物に更に100質量部のアセトンを追加した後、トリエチルアミン80質量部を投入し攪拌した。
ついで10%アンモニア水100質量部にトリエチルアミン50質量部を溶解させた水溶液を投入し、50℃で8時間反応させることで伸長反応を行った。更に、イオン交換水を固形分20質量%になるまで添加し、樹脂微粒子分散液−6を得た。特性を表1に示す。
冷却管、窒素導入管および攪拌機のついた反応容器中に、下記を投入した。
・1,4−ブタンジオール 928質量部
・テレフタル酸ジメチルエステル 776質量部
・1,6−ヘキサン二酸 292質量部
・テトラブトキシチタネート(縮合触媒) 3質量部
160℃で窒素気流下、生成するメタノールを留去しながら8時間反応させた。ついで210℃まで徐々に昇温させながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに20mmHgの減圧下にて1時間反応させた。ついで160℃まで冷却し、無水トリメリット酸173質量部および1,3−プロパン二酸125質量部を加え、常圧密閉下2時間反応後、200℃常圧で反応させ、軟化点が170℃になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕、粒子化し、非線形ポリエステル樹脂であるポリエステル−1を得た。ポリエステル−1のTgは53℃、酸価は25mgKOH/gであった。
・ポリオキシプロピレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
30質量部
・ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
33質量部
・テレフタル酸 21質量部
・無水トリメリット酸 1質量部
・フマル酸 3質量部
・ドデセニルコハク酸 12質量部
・酸化ジブチル錫 0.1質量部
をガラス製4リットルの4つ口フラスコに入れ、温度計、撹拌棒、コンデンサー及び窒素導入管を取りつけマントルヒーター内においた。窒素雰囲気下で、215℃で5時間反応させ、ポリエステル−2を得た。ポリエステル−2のTgは62℃、酸価は6mgKOH/gであった。
冷却管、窒素導入管および攪拌機のついた反応容器中に、下記を投入した。
・1,2−プロパンジオール 799質量部
・テレフタル酸ジメチルエステル 815質量部
・1,5−ペンタン二酸 238質量部
・テトラブトキシチタネート(縮合触媒) 3質量部
180℃で窒素気流下、生成するメタノールを留去しながら8時間反応させた。ついで230℃まで徐々に昇温させながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに20mmHgの減圧下にて1時間反応させた。ついで180℃まで冷却し、無水トリメリット酸173質量部を加え、常圧密閉下2時間反応後、220℃常圧で反応させ、軟化点が180℃になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕、粒子化し、非線形ポリエステル樹脂であるポリエステル−3を得た。ポリエステル−3のTgは62℃、酸価は2mgKOH/gであった。
冷却管、窒素導入管および攪拌機のついた反応容器中に、下記を投入した。
・1,3−ブタンジオール 1036質量部
・テレフタル酸ジメチルエステル 892質量部
・1,6−ヘキサン二酸 205質量部
・テトラブトキシチタネート(縮合触媒) 3質量部
180℃で窒素気流下、生成するメタノールを留去しながら8時間反応させた。ついで230℃まで徐々に昇温させながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに20mmHgの減圧下にて反応させ、軟化点が150℃になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕、粒子化し、線形ポリエステル樹脂であるポリエステル−4を得た。ポリエステル−4のTgは38℃、酸価は15mgKOH/gであった。
冷却管、窒素導入管および攪拌機のついた反応容器中に、下記を投入した。
・1,2プロパンジオール 858質量部
・テレフタル酸ジメチルエステル 873質量部
・1,6−ヘキサン二酸 219質量部
・テトラブトキシチタネート(縮合触媒) 3質量部
180℃で窒素気流下、生成するメタノールを留去しながら8時間反応させた。ついで230℃まで徐々に昇温させながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに20mmHgの減圧下にて反応させ、軟化点が150℃になった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕、粒子化し、線形ポリエステル樹脂であるポリエステル−5を得た。ポリエステル−5のTgは44℃、酸価は13mgKOH/gであった。
攪拌羽つきの密閉性容器に酢酸エチルを投入し、100rpmで攪拌しているところに、上記ポリエステル−1〜5を入れ室温で3日攪拌することでポリエステル樹脂溶液−1〜5を調製した。樹脂含有量(質量%)は表2に示す。
・カルナウバワックス(融点81℃) 20質量部
・酢酸エチル 80質量部
上記を攪拌羽根突きのガラスビーカー(IWAKIガラス製)に投入し、系内を70℃に加熱することでカルナウバワックスを酢酸エチルに溶解させた。
ついで、系内を50rpmで緩やかに攪拌しながら徐々に冷却し、3時間かけて25℃にまで冷却させ乳白色の液体を得た。
この溶液を1mmのガラスビーズ20質量部とともに耐熱性の容器に投入し、ペイントシェーカー(東洋精機製)にて3時間の分散を行い、ワックス分散液−1を得た。
上記ワックス分散液−1中のワックス粒子径をマイクロトラック粒度分布測定装置HRA(X−100)(日機装社製)にて測定したところ、個数平均粒子径で0.15μmであった。特性を表3に示す。
・ステアリン酸ステアリル(融点67℃) 16質量部
・ニトリル基含有スチレンアクリル樹脂(スチレン65質量部、n−ブチルアクリレート35質量部、アクリロニトリル10質量部、ピーク分子量8500)8質量部
・酢酸エチル 76質量部
上記を攪拌羽根突きのガラスビーカー(IWAKIガラス製)内に投入し、系内を65℃に加熱することでステアリン酸ステアリルを酢酸エチルに溶解させた。
ついで、ワックス分散液−1と同様操作を行い、ワックス分散液−2を得た。上記ワックス分散液−2中のワックス粒子径をマイクロトラック粒度分布測定装置HRA(X−100)(日機装社製)にて測定したところ、個数平均粒子径で0.12μmであった。特性を表4に示す。
・トリメチロールプロパントリベヘネート(融点58℃) 16質量部
・ニトリル基含有スチレンアクリル樹脂(スチレン65質量部、n−ブチルアクリレート35質量部、アクリロニトリル10質量部、ピーク分子量8500)8質量部
・酢酸エチル 76質量部
上記を攪拌羽根突きのガラスビーカー(IWAKIガラス製)内に投入し、系内を60℃に加熱することでトリメチロールプロパントリベヘネートを酢酸エチルに溶解させた。
ついで、ワックス分散液−1と同様操作を行い、ワックス分散液−3を得た。上記ワックス分散液−3中のワックス粒子径をマイクロトラック粒度分布測定装置HRA(X−100)(日機装社製)にて測定したところ、個数平均粒子径で0.18μmであった。特性を表4に示す。
・酢酸エチル 100質量部
・ポリエステル−1 50質量部
・マグネタイト−1 100質量部
(個数平均粒子径 0.22μm、比表面積 9.6m2/g、変動係数 44%、磁化 68.4Am2/kg)
・ガラスビーズ(1mm) 100質量部
上記原材料を耐熱性のガラス容器に投入し、ペイントシェーカー(東洋精機製)にて5時間分散を行い、ナイロンメッシュでガラスビーズを取り除き、磁性体分散液−1を得た。
・ポリエステル−2 50質量部
・マグネタイト−2(物性は表4参照) 100質量部
上記の原材料をニーダー型ミキサーに仕込み、混合しながら非加圧下で昇温した。130℃まで昇温し、約10分間加熱溶融混練を行ない、マグネタイトを樹脂に分散させた。その後、冷却しながら混練を続け、80℃まで冷却し、50質量部の酢酸エチルを徐々に加えた。酢酸エチルを添加後、系を75℃に固定し、30分混練した後、冷却し、混練物を得た。次いで、上記混錬物を、ハンマーを用いて粗粉砕後、固形分濃度が、60質量%になるように、酢酸エチルと混ぜた後、ディスパーを用いて、8000rpmで10分間撹拌し、磁性体分散液−2を得た。
・マグネタイト−3(物性は表4参照) 250質量部
・酢酸エチル 250質量部
・ガラスビーズ(1mm) 300質量部
上記原材料を耐熱性のガラス容器に投入し、ペイントシェーカー(東洋精機製)にて5時間分散を行い、ナイロンメッシュでガラスビーズを取り除き、磁性体分散液−3を得た。
・ポリエステル−4 50質量部
・マグネタイト−4(物性は表4参照) 100質量部
上記の原材料をニーダー型ミキサーに仕込み、混合しながら非加圧下で昇温した。13
0℃まで昇温し、約60分間加熱溶融混練を行ない、マグネタイトを樹脂に分散させた。その後、冷却し、混練物を得た。次いで、上記混錬物を、ハンマーを用いて粗粉砕後、固形分濃度が、60質量%になるように、酢酸エチルと混ぜた後、ディスパーを用いて、8000rpmで10分間撹拌し、磁性体分散液−4を得た。
・ポリエステル−5 50質量部
・マグネタイト−5(物性は表4参照) 100質量部
上記の原材料をニーダー型ミキサーに仕込み、混合しながら非加圧下で昇温した。130℃まで昇温し、約60分間加熱溶融混練を行ない、マグネタイトを樹脂に分散させた。その後、冷却し、混練物を得た。次いで、上記混錬物を、ハンマーを用いて粗粉砕し粗粉砕物を得た。
・上記粗粉砕物 150質量部
・酢酸エチル 100質量部
・ガラスビーズ(1mm) 100質量部
上記原材料を耐熱性のガラス容器に投入し、ペイントシェーカー(東洋精機製)にて5時間分散を行い、ナイロンメッシュでガラスビーズを取り除き、磁性体分散液−5を得た。
(油相の調製)
・ワックス分散液−1 50質量部
・磁性体分散液−1 75質量部
・ポリエステル樹脂溶液−1 90質量部
・トリエチルアミン 0.5質量部
・酢酸エチル 34.5質量部
上記溶液を容器内に投入し、ホモディスパー(特殊機化工業(株)社製)で、1500rpmで10分間攪拌・分散した。更に、上記溶液を常温下で超音波分散器により30分間分散させることにより油相1を調製した。
容器に下記を投入し、TKホモミクサー(特殊機化社製)にて5000rpmで1分攪拌し、水相を調製した。
・イオン交換水 255質量部
・樹脂微粒子分散液−1 25質量部
(トナー母粒子100質量部に対して、樹脂微粒子5質量部仕込み)
・ドデシルジフェニルエーテルジスルホン酸ナトリウムの50%水溶液
(エレミノールMON−7、三洋化成工業製) 25質量部
・酢酸エチル 30質量部
上記水相中に油相を投入し、TKホモミクサーで回転数を8000rpmまでの条件で、3分間攪拌を続け、油相1を懸濁させた。
ついで、容器に攪拌羽をセットし、200rpmで攪拌しながら系内を50℃に昇温し、かつ500mmHgに減圧した状態で5時間かけて脱溶剤を行い、トナー粒子の水分散液を得た。
ついで、上記のトナー粒子の水分散液をろ過し、イオン交換水500質量部にリスラリーした後、系内を攪拌しつつ、系内がpH4になるまで塩酸を加えて、5分間攪拌した。上記スラリーを再度ろ過し、イオン交換水200質量部添加し5分間攪拌する操作を3回繰り返すことで、系内に残存したトリエチルアミンを除去し、トナー粒子のろ過ケーキを得た。
上記ろ過ケーキを温風乾燥機にて45℃で3日間乾燥し、目開き75μmメッシュでふるい、トナー粒子1を得た。
次に、上記トナー粒子1の100質量部に対し、平均径20nmの疎水性シリカ0.7質量部と、平均径120nmのチタン酸ストロンチウム3.0質量部をヘンシェルミキサ
ー(三井三池化工機(株)製)FM−10Bにて混合し、トナー1を得た。
トナーの成分組成比を表5に、トナーの特性を表6に示す。
得られたトナーの評価方法について説明する。画像評価には市販のキヤノン製白黒複写機(商品名:IR3570)を用いた。トナーの画像評価の結果を表7に示す。
かぶりの評価は、上記耐久試験中、1000枚終了時点で、現像バイアスの交流成分の振幅を1.8kVに設定し、べた白を2枚プリントし、2枚目のかぶりを以下の方法により測定した。
反射濃度計(リフレクトメーター:モデル TC−6DS:東京電色社製)を用いて画像形成前後の転写材を測定し、画像形成後の反射濃度最悪値をDs、画像形成前の転写材の反射平均濃度をDrとし、Ds−Drを求め、これをかぶり量として評価した。数値の少ない方が、かぶりが少ないことを示す。かぶりの評価基準を以下に示す。
(評価基準)
A:1.0未満。
B:1.0以上2.0未満。
C:2.0以上3.5未満。
D:3.5以上。
細線再現性の評価は、上記耐久試験中、1000枚、10000枚終了時点で行った。
まず、潜像のライン幅が85μmになるようにレーザー露光して、厚紙(105g/m2)にプリントした定着画像を測定用サンプルとした。測定装置として、ルーゼックス450粒子アナライザー(株式会社ニレコ)を用いて、拡大したモニター画像から、インジケーターを用いて線幅の測定を行った。このとき、線幅の測定位置はトナーの細線画像の幅方向に凹凸があるため、凹凸の平均的線幅をもって測定点とした。細線再現性の評価は、線幅測定値の、潜像線幅(85μm)に対する比(線幅比)を算出することによって評価した。細線再現性の評価基準を以下に示す。
(評価基準)
線幅測定値の、潜像線幅に対する比(線幅比)が、
A:1.08未満である。
B:1.08以上、1.12未満である。
C:1.12以上、1.18未満である。
D:1.18以上である。
1000枚後に細線再現性に引き続き転写効率を測定した。細線再現性を測定した設定条件でベタ画像を出力し、転写紙上に転写した画像と、感光体上の転写残の画像濃度を、濃度計(X−rite 500Series:X−rite社)で測定した。画像濃度から、載り量を換算し転写紙上への転写効率を求めた。
(評価基準)
A:トナーの転写効率が95%以上である。
B:トナーの転写効率が93%以上である。
C:トナーの転写効率が90%以上である。
D:トナーの転写効率が90%未満である。
画像濃度は、以下の手順で評価した。即ち、上記試験機を用い、常温常湿度環境下(23℃/60%RH)において、キヤノンリサイクルペーパーEN−100紙(キヤノン社)上に、ベタ画像でトナー乗り量が0.35mg/cm2になるように調整し、定着後の画像を準備した。
該画像を、X−rite社製反射濃度計500 Series Spectrodensitemeterを用いて評価した。なお、黒色である本トナーについてはVisualでの値を濃度の値とする。
上記試験機を用い、常温常湿度環境下(23℃/60%RH)において、紙上のトナー載り量を0.35mg/cm2になるよう現像コントラストを調整し、先端余白5mm、幅100mm、長さ280mmのべたの未定着画像を作成した。紙としては、厚紙A4用紙(「プローバーボンド紙」:105g/m2、フォックスリバー社製)を用いた。
上記試験機の定着器を改造し、定着ユニットは手動で定着温度が設定できるようにした状態で、常温常湿度環境下(23℃/60%RH)に於いて80℃から200℃の範囲で順に10℃ずつ上げ定着試験を行った。
得られた定着画像の画像領域に、柔和な薄紙(例えば、商品名「ダスパー」、小津産業社製)の上から4.9KPaの荷重をかけつつ5往復摺擦し、摺擦前と摺擦後の画像濃度をそれぞれ測定して、下記式により画像濃度の低下率ΔD(%)を算出した。このΔD(%)が10%未満のときの温度を定着開始温度とし、低温定着性の基準とした。
尚、画像濃度はX−Rite社製カラー反射濃度計(Color reflection densitometer X−Rite 404A)で測定した。
(式): ΔD(%)=(摺擦前の画像濃度−摺擦後の画像濃度)×100/摺擦前の画
像濃度
(評価基準)
A:定着開始温度が120℃以下
B:120℃<定着開始温度≦140℃
C:140℃<定着開始温度≦160℃
D:160℃<定着開始温度
なお、本発明においてはBランクまでを良好な低温定着性と判断した。
帯電性(トリボ)の評価は、トナーの摩擦帯電量を用いて評価した。
以下にトナーの摩擦帯電量の測定方法について説明する。
まず、所定のキャリア(日本画像学会標準キャリア フェライトコアを表面処理した球形キャリア N-01)とトナーとを蓋付きのプラスチックボトルに入れ、振盪器(YS−
LD、(株)ヤヨイ製)で、1秒間に4往復のスピードで1分間振とうし、トナーとキャリアからなる現像剤を帯電させる。次に、図3に示す摩擦帯電量を測定する装置において摩擦帯電量を測定する。図3において、底に500メッシュのスクリーン3のある金属製の測定容器2に、前述した現像剤約0.5〜1.5gを入れ、金属製のフタ4をする。この時の測定容器2全体の質量を秤りW1(g)とする。次に吸引機1(測定容器2と接する部分は少なくとも絶縁体)において、吸引口7から吸引し風量調節弁6を調整して真空計5の圧力を250mmAqとする。この状態で2分間吸引を行い、トナーを吸引除去する。この時の電位計9の電位をV(ボルト)とする。ここで、8はコンデンサーであり容量をC(mF)とする。また、吸引後の測定容器全体の質量を秤りW2(g)とする。この試料の摩擦帯電量(mC/kg)は下式の如く算出される。
試料の摩擦帯電量(mC/kg)=C×V/(W1−W2)
約10gのトナーを100mlのポリカップに入れ、50℃で3日放置した後、目視で評価した。
(評価基準)
A:凝集物は見られない。
B:凝集物は見られるが容易に崩れる。
C:凝集物をつかむことができ容易に崩れない。
実施例1において、以下に示す(水相の調製)、(乳化及び脱溶剤工程)及び(洗浄〜乾燥工程)を用いた以外は、実施例1と同様にしてトナー2を得た。トナーの成分組成比を表5に、トナーの特性を表6に、トナーの画像評価の結果を表7に示す。
(水相の調製)
[無機系水系分散媒体の調製]
イオン交換水709質量部に0.1mol/L Na3PO4水溶液451質量部を投入し60℃に加温した後、TKホモミクサー(特殊機化工業製)で12,000 rpmにて攪拌し、1.0mol/L CaCl2水溶液67.7質量部を徐々に添加してCa3(PO4)2を含む無機系水系分散媒体を得た。
・上記無機系水系分散媒体 200質量部
・ドデシルジフェニルエーテルジスルホン酸ナトリウムの50%水溶液
(エレミノールMON−7、三洋化成工業製) 4質量部
・酢酸エチル 16質量部
上記をビーカーに投入し、TKホモミクサーにて5000rpmで1分攪拌し、水相を
調製した。
(乳化及び脱溶剤工程)
上記水相中に油相を投入し、TKホモミクサーの回転数を8000rpmまでの条件で、3分間攪拌を続け、上記油相1を懸濁させた。
ついで、ビーカーに攪拌羽をセットし、200rpmで攪拌しながら系内を50℃に昇温し、ドラフトチャンバー内で10時間かけて脱溶剤を行い、トナー粒子の水分散液を得た。
(洗浄〜乾燥工程)
上記トナー水分散液をろ過し、イオン交換水500質量部に投入しリスラリーとした後、系内を攪拌しつつ、系内がpH 1.5になるまで塩酸を加えてCa3(PO4)2を溶解し、さらに5分間攪拌した。
上記スラリーを再度ろ過し、イオン交換水200質量部添加し、5分間攪拌する操作を3回繰り返すことで、系内に残存したトリエチルアミンを除去し、トナー粒子のろ過ケーキを得た。上記ろ過ケーキを温風乾燥機にて45℃で3日間乾燥し、目開き75μmメッシュでふるい、トナー粒子2を得た。
実施例1で用いた水相の代わりに、以下に示す水相を用いた以外は、実施例1と同様にしてトナー3を得た。トナーの成分組成比を表5に、トナーの特性を表6に、トナーの画像評価の結果を表7に示す。
(水相の調製)
容器に下記を投入し、TKホモミクサー(特殊機化社製)にて5000rpmで1分攪拌し、水相を調製した。
・イオン交換水 255質量部
・樹脂微粒子分散液−6 25質量部
(トナー粒子100質量部に対して、樹脂微粒子5質量部仕込み)
・ドデシルジフェニルエーテルジスルホン酸ナトリウムの50%水溶液
(エレミノールMON−7、三洋化成工業製) 25質量部
・酢酸エチル 30質量部
実施例1で用いた油相中の、磁性体分散液−1、ポリエステル樹脂溶液−1の添加量を表5に示すように変更した以外は、実施例1と同様にしてトナー4、5を得た。トナーの成分組成比を表5に、トナーの特性を表6に、トナーの画像評価の結果を表7に示す。
実施例1において、(乳化及び脱溶剤工程)を下記に記載したように変更した以外は、実施例と同様にしてトナー6を得た。トナーの成分組成比を表5に、トナーの特性を表6に、トナーの画像評価の結果を表7に示す。
(乳化及び脱溶剤工程)
水相中に油相を投入し、TKホモミクサーで回転数を8000rpmまでの条件で、3分間攪拌を続け、油相1を懸濁した。
容器に攪拌羽をセットし、200rpmで攪拌しながら系内を25℃に維持し、かつ200mmHgに減圧した状態で5時間かけて脱溶剤を行い、トナー粒子の水分散液を得た。
(油相の調製)
・ワックス分散液−1 50 質量部
・磁性体分散液−2 112.5質量部
・ポリエステル樹脂溶液−2 45 質量部
・トリエチルアミン 0.5質量部
・酢酸エチル 42 質量部
上記溶液を容器内に投入し、ホモディスパー(特殊機化工業(株)社製)で、1500rpmで10分間攪拌・分散した。更に、上記溶液にガラスビーズ100質量部を加え、ペイントシェーカー(東洋精機製)にて1時間分散を行い、ナイロンメッシュでガラスビ
ーズを取り除き、油相7を調製した。
容器に下記を投入し、TKホモミクサー(特殊機化社製)にて5000rpmで1分攪拌し、水相を調製した。
・イオン交換水 245質量部
・樹脂微粒子分散液−4 35質量部
(トナー母粒子100質量部に対して、樹脂微粒子7質量部仕込み)
・ドデシルジフェニルエーテルジスルホン酸ナトリウムの50%水溶液
(エレミノールMON−7、三洋化成工業製) 25質量部
・酢酸エチル 30質量部
上記水相中に油相7を投入し、TKホモミクサーで回転数を8000rpmまでの条件で、3分間攪拌を続け、油相7を懸濁させた。
ついで、容器に攪拌羽をセットし、200rpmで攪拌しながら系内を50℃に昇温し、かつ500mmHgに減圧した状態で5時間かけて脱溶剤を行い、トナー粒子の水分散液を得た。
ついで、上記のトナー粒子の水分散液をろ過し、イオン交換水500質量部にリスラリーした後、系内を攪拌しつつ、系内がpH4になるまで塩酸を加えて、5分間攪拌した。上記スラリーを再度ろ過し、イオン交換水200質量部添加し5分間攪拌する操作を3回繰り返すことで、系内に残存したトリエチルアミンを除去し、トナー粒子のろ過ケーキを得た。
上記ろ過ケーキを温風乾燥機にて45℃で3日間乾燥し、目開き75μmメッシュでふるい、トナー粒子7を得た。
次に、上記トナー粒子7の100質量部に対し、平均径20nmの疎水性シリカ0.7質量部と、平均径120nmのチタン酸ストロンチウム3.0質量部をヘンシェルミキサ
ー(三井三池化工機(株)製)FM−10Bにて混合し、トナー7を得た。
トナーの成分組成比を表5に、トナーの特性を表6に、トナーの画像評価の結果を表7に示す。
参考例1で用いた油相中の、磁性体分散液−2、ポリエステル樹脂溶液−2の添加量、及び水相中の樹脂微粒子分散液−4の添加量を、表5に示すように変更した以外は、参考例1と同様にしてトナー8を得た。トナーの成分組成比を表5に、トナーの特性を表6に、トナーの画像評価の結果を表7に示す。
(油相の調製)
・ワックス分散液−2 62.5質量部
・磁性体分散液−3 70.0質量部
・ポリエステル樹脂溶液−3 100.0質量部
・トリエチルアミン 0.5質量部
・酢酸エチル 17.0質量部
上記溶液を容器内に投入し、ホモディスパー(特殊機化工業(株)社製)で、1500rpmで10分間攪拌・分散した。更に、上記溶液を常温下で超音波分散器により30分間分散させることにより油相9を調製した。
容器に下記を投入し、TKホモミクサー(特殊機化社製)にて5000rpmで1分攪拌し、水相を調製した。
・イオン交換水 215.0質量部
・樹脂微粒子分散液−2 65.0質量部
(トナー母粒子100質量部に対して、樹脂微粒子13.0質量部仕込み)
・ドデシルジフェニルエーテルジスルホン酸ナトリウムの50%水溶液
(エレミノールMON−7、三洋化成工業製) 25.0質量部
・酢酸エチル 30.0質量部
(油相の調製)
・ワックス分散液−3 62.5質量部
・磁性体分散液−4 62.5質量部
・ポリエステル樹脂溶液−4 95質量部
・トリエチルアミン 0.5質量部
・酢酸エチル 29.5質量部
上記溶液を容器内に投入し、ホモディスパー(特殊機化工業(株)社製)で、1500rpmで10分間攪拌・分散した。更に、上記溶液を常温下で超音波分散器により30分間分散させることにより油相10を調製した。
容器に下記を投入し、TKホモミクサー(特殊機化社製)にて5000rpmで1分攪拌し、水相を調製した。
・イオン交換水 265質量部
・樹脂微粒子分散液−3 15質量部
(トナー母粒子100質量部に対して、樹脂微粒子 3質量部仕込み)
・ドデシルジフェニルエーテルジスルホン酸ナトリウムの50%水溶液
(エレミノールMON−7、三洋化成工業製) 25質量部
・酢酸エチル 30質量部
成分組成比を表5に、トナーの特性を表6に、トナーの画像評価の結果を表7に示す。
(油相の調製)
・ワックス分散液−1 50.0質量部
・磁性体分散液−5 100.0質量部
・ポリエステル樹脂溶液−5 60.0質量部
・トリエチルアミン 0.5質量部
・酢酸エチル 39.5質量部
上記溶液を容器内に投入し、ホモディスパー(特殊機化工業(株)社製)で、1500rpmで10分間攪拌・分散した。更に、上記溶液を常温下で超音波分散器により30分間分散させることにより油相11を調製した。
容器に下記を投入し、TKホモミクサー(特殊機化社製)にて5000rpmで1分攪拌し、水相を調製した。
・イオン交換水 267.5質量部
・樹脂微粒子分散液−5 12.5質量部
(トナー母粒子100質量部に対して、樹脂微粒子 2.5質量部仕込み)
・ドデシルジフェニルエーテルジスルホン酸ナトリウムの50%水溶液
(エレミノールMON−7、三洋化成工業製) 25質量部
・酢酸エチル 30質量部
(油相の調製)
・ワックス分散液−1 50.0質量部
・磁性体分散液−2 100.0質量部
・ポリエステル樹脂溶液−2 60.0質量部
・トリエチルアミン 0.5質量部
・酢酸エチル 39.5質量部
上記溶液を容器内に投入し、ホモディスパー(特殊機化工業(株)社製)で、1500rpmで10分間攪拌・分散し、油相12を調製した。
容器に下記を投入し、TKホモミクサー(特殊機化社製)にて5000rpmで1分攪拌し、水相を調製した。
・イオン交換水 267.5質量部
・樹脂微粒子分散液4 12.5質量部
(トナー母粒子100質量部に対して、樹脂微粒子 2.5質量部仕込み)
・ドデシルジフェニルエーテルジスルホン酸ナトリウムの50%水溶液
(エレミノールMON−7、三洋化成工業製) 25質量部
・酢酸エチル 30質量部
2 金属製の測定容器
3 500メッシュのスクリーン
4 金属製のフタ
5 真空計
6 風量調節弁
7 吸引口
8 コンデンサー
9 電位計
11 下部電極
12 上部電極
13 絶縁物
14 電流計
15 電圧計
16 定電圧装置
17 キャリア
18 ガイドリング
d 試料厚み
E 抵抗測定セル
Claims (9)
- ポリエステルを主成分とする樹脂(a)、磁性体及びワックスを少なくとも含有するトナー母粒子(A)の表面に、表面層(B)を有するカプセル型のトナー粒子を含有するトナーであって、
前記表面層(B)は樹脂(b)を含有し、前記樹脂(b)は、ポリエステル樹脂(b1)、ビニル樹脂(b2)またはウレタン樹脂(b3)から選ばれる樹脂を含有する樹脂であり、
前記樹脂(a)のガラス転移温度Tg(a)と前記樹脂(b)のガラス転移温度Tg(b)が下記式(1)の関係を満たし、
Tg(a)<Tg(b)・・・(1)
前記樹脂(b)はスルホン酸基を有し、前記樹脂(b)のスルホン酸基価が1mgKOH/g以上25mgKOH/g以下であり、
前記トナーの79.6kA/mの外部磁場における磁化(σt)が、12Am2/kg以上30Am2/kg以下であり、
前記トナーの平均円形度が、0.960以上1.000以下であることを特徴とするトナー。 - 前記トナーの、誘電損率ε”/誘電率ε’で示される誘電損失(tanδ)が、周波数100000Hzにおいて、0.015以下であることを特徴とする請求項1に記載のトナー。
- 前記トナーの重量平均粒子径(D4)が4.0μm以上9.0μm以下であり、前記トナーの0.6μm以上2.0μm以下の粒子が5.0個数%以下であることを特徴とする請求項1または2に記載のトナー。
- 前記トナーの超音波処理後における0.6μm以上2.0μm以下の粒子が5.0個数%以下であることを特徴とする請求項1乃至3のいずれかに記載のトナー。
- 前記トナーの超音波処理後における0.6μm以上2.0μm以下の粒子が2.0個数
%以下であることを特徴とする請求項1乃至4のいずれかに記載のトナー。 - 前記表面層(B)は、前記トナー母粒子(A)に対し、2.0質量%以上15.0質量%以下であることを特徴とする請求項1乃至5のいずれかに記載のトナー。
- 前記ワックスは、エステルワックスであることを特徴とする請求項1乃至6のいずれかに記載のトナー。
- 前記樹脂(b)はウレタン系樹脂(b3)を含有することを特徴とする請求項1乃至7のいずれかに記載のトナー。
- 前記トナーの断面拡大写真における、前記磁性体の個数平均分散径が、0.10μm以上0.50μm以下であることを特徴とする請求項1乃至8のいずれかに記載のトナー。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007267662A JP5159239B2 (ja) | 2007-10-15 | 2007-10-15 | トナー |
| EP08840250.8A EP2204699B1 (en) | 2007-10-15 | 2008-10-10 | Toner |
| CN200880111646XA CN101828150B (zh) | 2007-10-15 | 2008-10-10 | 调色剂 |
| PCT/JP2008/068443 WO2009051072A1 (ja) | 2007-10-15 | 2008-10-10 | トナー |
| KR1020107010068A KR101176283B1 (ko) | 2007-10-15 | 2008-10-10 | 토너 |
| US12/395,792 US7794908B2 (en) | 2007-10-15 | 2009-03-02 | Toner |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007267662A JP5159239B2 (ja) | 2007-10-15 | 2007-10-15 | トナー |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2009098257A JP2009098257A (ja) | 2009-05-07 |
| JP2009098257A5 JP2009098257A5 (ja) | 2010-12-02 |
| JP5159239B2 true JP5159239B2 (ja) | 2013-03-06 |
Family
ID=40567339
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007267662A Expired - Fee Related JP5159239B2 (ja) | 2007-10-15 | 2007-10-15 | トナー |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7794908B2 (ja) |
| EP (1) | EP2204699B1 (ja) |
| JP (1) | JP5159239B2 (ja) |
| KR (1) | KR101176283B1 (ja) |
| CN (1) | CN101828150B (ja) |
| WO (1) | WO2009051072A1 (ja) |
Families Citing this family (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2161624B1 (en) * | 2007-06-19 | 2013-09-11 | Canon Kabushiki Kaisha | Color toner |
| JP5153864B2 (ja) * | 2008-03-10 | 2013-02-27 | キヤノン株式会社 | トナー |
| JP5414188B2 (ja) * | 2008-03-19 | 2014-02-12 | キヤノン株式会社 | 画像形成方法 |
| JP5253506B2 (ja) * | 2008-07-31 | 2013-07-31 | キヤノン株式会社 | シアントナー |
| JP4565053B2 (ja) | 2009-02-27 | 2010-10-20 | キヤノン株式会社 | マゼンタトナー |
| JP4565054B2 (ja) * | 2009-02-27 | 2010-10-20 | キヤノン株式会社 | 黒トナー |
| CN102334073B (zh) * | 2009-02-27 | 2013-12-04 | 佳能株式会社 | 黄色调色剂 |
| JP5494922B2 (ja) * | 2009-06-10 | 2014-05-21 | 株式会社リコー | トナー、現像剤、トナー入り容器、プロセスカートリッジ、画像形成方法及び画像形成装置 |
| US20110033794A1 (en) * | 2009-08-05 | 2011-02-10 | Naohiro Watanabe | Toner, method for producing the same, and process cartridge |
| KR20130113507A (ko) * | 2010-12-28 | 2013-10-15 | 캐논 가부시끼가이샤 | 토너 |
| US20120270146A1 (en) * | 2011-04-20 | 2012-10-25 | Xerox Corporation | Magnetic toner compositions |
| JP5743959B2 (ja) | 2011-06-03 | 2015-07-01 | キヤノン株式会社 | トナー |
| JP6000660B2 (ja) | 2011-06-03 | 2016-10-05 | キヤノン株式会社 | トナーおよび該トナーの製造方法 |
| CN103562799B (zh) | 2011-06-03 | 2016-08-31 | 佳能株式会社 | 调色剂 |
| KR101600160B1 (ko) | 2011-06-03 | 2016-03-04 | 캐논 가부시끼가이샤 | 토너 |
| JP5748592B2 (ja) * | 2011-07-26 | 2015-07-15 | キヤノン株式会社 | 磁性トナー |
| JP6399804B2 (ja) * | 2013-06-24 | 2018-10-03 | キヤノン株式会社 | トナー |
| KR102171245B1 (ko) * | 2013-12-31 | 2020-10-28 | 롯데정밀화학 주식회사 | 토너 및 그의 제조방법 |
| CN105767158B (zh) * | 2014-07-21 | 2019-08-23 | 上海惠昌化工厂 | 一种水果保鲜果蜡及其制备方法 |
| US9470993B2 (en) * | 2014-08-07 | 2016-10-18 | Canon Kabushiki Kaisha | Magnetic toner |
| US9857713B2 (en) | 2014-12-26 | 2018-01-02 | Canon Kabushiki Kaisha | Resin particle and method of producing the resin particle, and toner and method of producing the toner |
| US9798256B2 (en) | 2015-06-30 | 2017-10-24 | Canon Kabushiki Kaisha | Method of producing toner |
| US9823595B2 (en) | 2015-06-30 | 2017-11-21 | Canon Kabushiki Kaisha | Toner |
| JP6047224B1 (ja) * | 2015-12-25 | 2016-12-21 | 出光興産株式会社 | 鉱油系基油、潤滑油組成物、内燃機関、及び内燃機関の潤滑方法 |
| EP3395931B1 (en) | 2015-12-25 | 2023-05-31 | Idemitsu Kosan Co.,Ltd. | Mineral base oil, lubricant composition, internal combustion engine, lubricating method of internal combustion engine |
| US10295921B2 (en) * | 2016-12-21 | 2019-05-21 | Canon Kabushiki Kaisha | Toner |
| US10545420B2 (en) * | 2017-07-04 | 2020-01-28 | Canon Kabushiki Kaisha | Magnetic toner and image-forming method |
| US20190227447A1 (en) * | 2018-01-19 | 2019-07-25 | Static Control Components, Inc. | Magnetic Spherical Toner |
| JP7500245B2 (ja) * | 2019-04-25 | 2024-06-17 | 日鉄鉱業株式会社 | コバルトフェライト粒子の製造方法とそれにより製造されたコバルトフェライト粒子 |
| CN115838568B (zh) * | 2022-12-31 | 2023-11-28 | 上海纳米技术及应用国家工程研究中心有限公司 | 一种透明隔热浆料及其制备方法和应用 |
Family Cites Families (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5754950A (en) * | 1980-09-20 | 1982-04-01 | Canon Inc | Microcapsule toner |
| JPS61120161A (ja) * | 1984-11-15 | 1986-06-07 | Konishiroku Photo Ind Co Ltd | マイクロカプセル型トナ− |
| JPS62150262A (ja) * | 1985-12-24 | 1987-07-04 | Fuji Photo Film Co Ltd | カプセルトナ− |
| JP2827697B2 (ja) | 1992-04-22 | 1998-11-25 | 富士ゼロックス株式会社 | 電子写真用トナー組成物および画像形成方法 |
| JPH06148924A (ja) * | 1992-10-30 | 1994-05-27 | Kao Corp | 現像方法 |
| JP3237385B2 (ja) * | 1994-03-16 | 2001-12-10 | 富士ゼロックス株式会社 | 磁性トナー |
| US5620826A (en) | 1995-01-30 | 1997-04-15 | Agfa-Gevaert, N.V. | Polymer suspension method for producing toner particles |
| JPH08286423A (ja) | 1995-04-11 | 1996-11-01 | Minolta Co Ltd | 磁性現像剤 |
| JP3798204B2 (ja) * | 1998-12-18 | 2006-07-19 | 三井化学株式会社 | 静電荷像現像用トナー |
| JP3455523B2 (ja) | 2000-02-16 | 2003-10-14 | 三洋化成工業株式会社 | 粒径が均一である樹脂粒子およびその製造方法 |
| US6638674B2 (en) * | 2000-07-28 | 2003-10-28 | Canon Kabushiki Kaisha | Magnetic toner |
| JP4086487B2 (ja) | 2001-07-30 | 2008-05-14 | キヤノン株式会社 | 磁性トナー及び画像形成装置 |
| JP3826029B2 (ja) * | 2001-12-27 | 2006-09-27 | キヤノン株式会社 | 磁性トナー |
| JP2003295512A (ja) * | 2002-04-03 | 2003-10-15 | Canon Inc | 磁性トナーの製造方法 |
| JP4164289B2 (ja) * | 2002-05-16 | 2008-10-15 | キヤノン株式会社 | トナー、現像カートリッジ及び画像形成方法 |
| JP4072385B2 (ja) * | 2002-06-21 | 2008-04-09 | キヤノン株式会社 | 画像形成方法、トナーおよび二成分現像剤 |
| EP1439429B1 (en) | 2003-01-20 | 2013-03-13 | Ricoh Company, Ltd. | Toner and developer |
| JP4234022B2 (ja) * | 2003-01-20 | 2009-03-04 | 株式会社リコー | トナー、現像剤、現像装置、及び画像形成装置 |
| JP4066346B2 (ja) | 2003-01-21 | 2008-03-26 | 株式会社リコー | 静電荷像現像用トナー及びプロセスカートリッジ |
| JP4049688B2 (ja) | 2003-03-07 | 2008-02-20 | 株式会社リコー | 静電荷像現像用トナー、現像剤及び画像形成装置 |
| EP1455237B1 (en) * | 2003-03-07 | 2011-05-25 | Canon Kabushiki Kaisha | Toner and two-component developer |
| EP1477864B1 (en) * | 2003-05-14 | 2008-01-02 | Canon Kabushiki Kaisha | Magnetic carrier and two-component developer |
| WO2005073287A1 (ja) | 2004-01-30 | 2005-08-11 | Sanyo Chemical Industries, Ltd. | 樹脂分散体及び樹脂粒子 |
| JP2006154026A (ja) * | 2004-11-26 | 2006-06-15 | Toyo Ink Mfg Co Ltd | トナー用荷電制御剤とその製造方法及びそれを用いたトナー |
| JP2006195079A (ja) * | 2005-01-12 | 2006-07-27 | Canon Inc | 二成分現像方法 |
| US7214463B2 (en) * | 2005-01-27 | 2007-05-08 | Xerox Corporation | Toner processes |
| JP4625386B2 (ja) * | 2005-03-11 | 2011-02-02 | 株式会社リコー | 静電荷像現像用トナー及びその製造方法 |
| US7901857B2 (en) * | 2005-03-15 | 2011-03-08 | Fuji Xerox Co., Ltd. | Electrostatic latent image developing toner, production method thereof, electrostatic latent image developer, and image forming method |
| US7396628B2 (en) | 2005-03-15 | 2008-07-08 | Fuji Xerox Co., Ltd. | Toner for electrostatic charge image developing, developer for electrostatic charge image developing, and image forming apparatus |
| JP2006276062A (ja) | 2005-03-25 | 2006-10-12 | Fuji Xerox Co Ltd | 電子写真用二成分現像剤 |
| JP4628961B2 (ja) * | 2006-01-05 | 2011-02-09 | 株式会社リコー | 静電荷現像用トナー及び製造方法 |
| JP4660402B2 (ja) * | 2006-03-16 | 2011-03-30 | 株式会社リコー | 静電荷像現像用非磁性トナー |
-
2007
- 2007-10-15 JP JP2007267662A patent/JP5159239B2/ja not_active Expired - Fee Related
-
2008
- 2008-10-10 EP EP08840250.8A patent/EP2204699B1/en not_active Not-in-force
- 2008-10-10 WO PCT/JP2008/068443 patent/WO2009051072A1/ja not_active Ceased
- 2008-10-10 KR KR1020107010068A patent/KR101176283B1/ko not_active Expired - Fee Related
- 2008-10-10 CN CN200880111646XA patent/CN101828150B/zh not_active Expired - Fee Related
-
2009
- 2009-03-02 US US12/395,792 patent/US7794908B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009098257A (ja) | 2009-05-07 |
| EP2204699A1 (en) | 2010-07-07 |
| KR20100065202A (ko) | 2010-06-15 |
| CN101828150B (zh) | 2012-07-04 |
| EP2204699A4 (en) | 2013-03-13 |
| KR101176283B1 (ko) | 2012-08-22 |
| US20090170021A1 (en) | 2009-07-02 |
| CN101828150A (zh) | 2010-09-08 |
| EP2204699B1 (en) | 2017-03-01 |
| US7794908B2 (en) | 2010-09-14 |
| WO2009051072A1 (ja) | 2009-04-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5159239B2 (ja) | トナー | |
| JP5153864B2 (ja) | トナー | |
| JP4565054B2 (ja) | 黒トナー | |
| JP5253506B2 (ja) | シアントナー | |
| JP4565052B2 (ja) | イエロートナー | |
| JP4565053B2 (ja) | マゼンタトナー | |
| JP5570124B2 (ja) | トナー | |
| JPWO2008156117A1 (ja) | カラートナー | |
| JP5078506B2 (ja) | トナー | |
| JP5455475B2 (ja) | トナー | |
| JP2012048014A (ja) | トナー | |
| JP5495532B2 (ja) | トナー | |
| JP5300243B2 (ja) | トナー | |
| JP2009053501A (ja) | トナー | |
| JP5371588B2 (ja) | トナーの製造方法 | |
| JP2009015212A (ja) | トナー | |
| JP5697774B2 (ja) | トナー |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101015 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20101015 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120703 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120831 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121113 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121211 |
|
| R151 | Written notification of patent or utility model registration |
Ref document number: 5159239 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R151 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151221 Year of fee payment: 3 |
|
| S802 | Written request for registration of partial abandonment of right |
Free format text: JAPANESE INTERMEDIATE CODE: R311802 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| LAPS | Cancellation because of no payment of annual fees |