JP5769965B2 - ヒト胚性幹細胞の分化 - Google Patents
ヒト胚性幹細胞の分化 Download PDFInfo
- Publication number
- JP5769965B2 JP5769965B2 JP2010520194A JP2010520194A JP5769965B2 JP 5769965 B2 JP5769965 B2 JP 5769965B2 JP 2010520194 A JP2010520194 A JP 2010520194A JP 2010520194 A JP2010520194 A JP 2010520194A JP 5769965 B2 JP5769965 B2 JP 5769965B2
- Authority
- JP
- Japan
- Prior art keywords
- cells
- days
- treated
- activin
- expression
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 210000001671 embryonic stem cell Anatomy 0.000 title claims description 201
- 230000004069 differentiation Effects 0.000 title description 112
- 210000004027 cell Anatomy 0.000 claims description 690
- 210000001900 endoderm Anatomy 0.000 claims description 334
- 108010023082 activin A Proteins 0.000 claims description 280
- 238000000034 method Methods 0.000 claims description 168
- 210000001778 pluripotent stem cell Anatomy 0.000 claims description 122
- 239000003550 marker Substances 0.000 claims description 106
- 230000009996 pancreatic endocrine effect Effects 0.000 claims description 82
- 108010011459 Exenatide Proteins 0.000 claims description 75
- JUFFVKRROAPVBI-PVOYSMBESA-N chembl1210015 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N[C@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@]3(O[C@@H](C[C@H](O)[C@H](O)CO)[C@H](NC(C)=O)[C@@H](O)C3)C(O)=O)O2)O)[C@@H](CO)O1)NC(C)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 JUFFVKRROAPVBI-PVOYSMBESA-N 0.000 claims description 75
- 229960001519 exenatide Drugs 0.000 claims description 75
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 claims description 64
- 229930002330 retinoic acid Natural products 0.000 claims description 60
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 59
- 239000008103 glucose Substances 0.000 claims description 58
- 229960001727 tretinoin Drugs 0.000 claims description 56
- 238000012258 culturing Methods 0.000 claims description 47
- 102000018233 Fibroblast Growth Factor Human genes 0.000 claims description 41
- 108050007372 Fibroblast Growth Factor Proteins 0.000 claims description 41
- 229940125373 Gamma-Secretase Inhibitor Drugs 0.000 claims description 35
- 229940126864 fibroblast growth factor Drugs 0.000 claims description 35
- 239000003540 gamma secretase inhibitor Substances 0.000 claims description 35
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 claims description 10
- 210000001161 mammalian embryo Anatomy 0.000 claims description 8
- 229960003966 nicotinamide Drugs 0.000 claims description 5
- 235000005152 nicotinamide Nutrition 0.000 claims description 5
- 239000011570 nicotinamide Substances 0.000 claims description 5
- 238000004519 manufacturing process Methods 0.000 claims description 2
- 210000005260 human cell Anatomy 0.000 claims 30
- 101000898034 Homo sapiens Hepatocyte growth factor Proteins 0.000 claims 12
- 101001076408 Homo sapiens Interleukin-6 Proteins 0.000 claims 12
- 101000868152 Homo sapiens Son of sevenless homolog 1 Proteins 0.000 claims 12
- 230000014509 gene expression Effects 0.000 description 259
- 239000002609 medium Substances 0.000 description 183
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 94
- 108010082117 matrigel Proteins 0.000 description 88
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 83
- 239000012091 fetal bovine serum Substances 0.000 description 83
- 210000002966 serum Anatomy 0.000 description 82
- 102100031650 C-X-C chemokine receptor type 4 Human genes 0.000 description 77
- 101000922348 Homo sapiens C-X-C chemokine receptor type 4 Proteins 0.000 description 77
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 75
- 239000003112 inhibitor Substances 0.000 description 71
- 238000011282 treatment Methods 0.000 description 71
- 210000001519 tissue Anatomy 0.000 description 63
- 108090000623 proteins and genes Proteins 0.000 description 54
- 102000004877 Insulin Human genes 0.000 description 47
- 108090001061 Insulin Proteins 0.000 description 47
- 229940125396 insulin Drugs 0.000 description 47
- 102000013814 Wnt Human genes 0.000 description 44
- 108050003627 Wnt Proteins 0.000 description 44
- 108010087745 Hepatocyte Nuclear Factor 3-beta Proteins 0.000 description 42
- -1 Mix11 Proteins 0.000 description 41
- 239000003102 growth factor Substances 0.000 description 40
- 230000015572 biosynthetic process Effects 0.000 description 39
- 238000003753 real-time PCR Methods 0.000 description 39
- 239000000758 substrate Substances 0.000 description 39
- 101100310648 Mus musculus Sox17 gene Proteins 0.000 description 38
- 238000010790 dilution Methods 0.000 description 36
- 239000012895 dilution Substances 0.000 description 36
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 36
- 239000003446 ligand Substances 0.000 description 36
- 210000000130 stem cell Anatomy 0.000 description 35
- 241000699666 Mus <mouse, genus> Species 0.000 description 33
- 210000004039 endoderm cell Anatomy 0.000 description 33
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 32
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 32
- 108090000100 Hepatocyte Growth Factor Proteins 0.000 description 31
- 102000003745 Hepatocyte Growth Factor Human genes 0.000 description 31
- SAQUSDSPQYQNBG-UHFFFAOYSA-N chembl355496 Chemical compound N1C2=CC=CC=C2C(N=O)=C1C1=C(O)NC2=CC(Br)=CC=C21 SAQUSDSPQYQNBG-UHFFFAOYSA-N 0.000 description 31
- 102000009094 Hepatocyte Nuclear Factor 3-beta Human genes 0.000 description 30
- 230000000694 effects Effects 0.000 description 29
- 101000819074 Homo sapiens Transcription factor GATA-4 Proteins 0.000 description 27
- 101710183548 Pyridoxal 5'-phosphate synthase subunit PdxS Proteins 0.000 description 27
- 102100021380 Transcription factor GATA-4 Human genes 0.000 description 27
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 26
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 26
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 25
- 210000002744 extracellular matrix Anatomy 0.000 description 25
- 230000001965 increasing effect Effects 0.000 description 25
- 210000003890 endocrine cell Anatomy 0.000 description 24
- 210000002950 fibroblast Anatomy 0.000 description 24
- 230000012010 growth Effects 0.000 description 23
- 102000052547 Wnt-1 Human genes 0.000 description 21
- 102000004169 proteins and genes Human genes 0.000 description 21
- 102100035423 POU domain, class 5, transcription factor 1 Human genes 0.000 description 20
- 150000001875 compounds Chemical class 0.000 description 19
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 18
- 238000004113 cell culture Methods 0.000 description 18
- 101710126211 POU domain, class 5, transcription factor 1 Proteins 0.000 description 17
- 108010049955 Bone Morphogenetic Protein 4 Proteins 0.000 description 16
- 102100024505 Bone morphogenetic protein 4 Human genes 0.000 description 16
- 238000004458 analytical method Methods 0.000 description 16
- 108020004999 messenger RNA Proteins 0.000 description 16
- 102100028412 Fibroblast growth factor 10 Human genes 0.000 description 15
- 108090000031 Hedgehog Proteins Proteins 0.000 description 15
- 102000003693 Hedgehog Proteins Human genes 0.000 description 15
- 102000000905 Cadherin Human genes 0.000 description 14
- 108050007957 Cadherin Proteins 0.000 description 14
- 239000000523 sample Substances 0.000 description 14
- 101500016415 Lophius americanus Glucagon-like peptide 1 Proteins 0.000 description 13
- 108050000588 Neurogenic differentiation factor 1 Proteins 0.000 description 13
- 102100032063 Neurogenic differentiation factor 1 Human genes 0.000 description 13
- 239000003636 conditioned culture medium Substances 0.000 description 13
- 230000002829 reductive effect Effects 0.000 description 13
- 102100028072 Fibroblast growth factor 4 Human genes 0.000 description 12
- 102100029284 Hepatocyte nuclear factor 3-beta Human genes 0.000 description 12
- 101100043062 Mus musculus Sox7 gene Proteins 0.000 description 12
- 230000005913 Notch signaling pathway Effects 0.000 description 12
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 11
- 102000052651 Pancreatic hormone Human genes 0.000 description 11
- 108700020987 Wnt-1 Proteins 0.000 description 11
- 239000003795 chemical substances by application Substances 0.000 description 11
- TWSALRJGPBVBQU-PKQQPRCHSA-N glucagon-like peptide 2 Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(O)=O)[C@@H](C)CC)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)CC)C1=CC=CC=C1 TWSALRJGPBVBQU-PKQQPRCHSA-N 0.000 description 11
- 210000003494 hepatocyte Anatomy 0.000 description 11
- 239000004025 pancreas hormone Substances 0.000 description 11
- 229940032957 pancreatic hormone Drugs 0.000 description 11
- MFBOGIVSZKQAPD-UHFFFAOYSA-M sodium butyrate Chemical compound [Na+].CCCC([O-])=O MFBOGIVSZKQAPD-UHFFFAOYSA-M 0.000 description 11
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 10
- 108010059616 Activins Proteins 0.000 description 10
- OHCQJHSOBUTRHG-KGGHGJDLSA-N FORSKOLIN Chemical compound O=C([C@@]12O)C[C@](C)(C=C)O[C@]1(C)[C@@H](OC(=O)C)[C@@H](O)[C@@H]1[C@]2(C)[C@@H](O)CCC1(C)C OHCQJHSOBUTRHG-KGGHGJDLSA-N 0.000 description 10
- 108090000385 Fibroblast growth factor 7 Proteins 0.000 description 10
- 102000003972 Fibroblast growth factor 7 Human genes 0.000 description 10
- 108700014808 Homeobox Protein Nkx-2.2 Proteins 0.000 description 10
- 101000781955 Homo sapiens Proto-oncogene Wnt-1 Proteins 0.000 description 10
- 102100026818 Inhibin beta E chain Human genes 0.000 description 10
- 101000610097 Mesocricetus auratus Pancreatic beta cell growth factor Proteins 0.000 description 10
- 102100041030 Pancreas/duodenum homeobox protein 1 Human genes 0.000 description 10
- 101710144033 Pancreas/duodenum homeobox protein 1 Proteins 0.000 description 10
- 101800001268 Pancreatic hormone Proteins 0.000 description 10
- 241000288906 Primates Species 0.000 description 10
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 10
- 101150109862 WNT-5A gene Proteins 0.000 description 10
- 108700020483 Wnt-5a Proteins 0.000 description 10
- 102000043366 Wnt-5a Human genes 0.000 description 10
- 239000000488 activin Substances 0.000 description 10
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 10
- 101100156752 Caenorhabditis elegans cwn-1 gene Proteins 0.000 description 9
- 108090001047 Fibroblast growth factor 10 Proteins 0.000 description 9
- 108090000381 Fibroblast growth factor 4 Proteins 0.000 description 9
- 108060003199 Glucagon Proteins 0.000 description 9
- 102000044880 Wnt3A Human genes 0.000 description 9
- 108700013515 Wnt3A Proteins 0.000 description 9
- 230000010261 cell growth Effects 0.000 description 9
- 238000010899 nucleation Methods 0.000 description 9
- 210000001811 primitive streak Anatomy 0.000 description 9
- 230000001737 promoting effect Effects 0.000 description 9
- 238000003757 reverse transcription PCR Methods 0.000 description 9
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 9
- VOUAQYXWVJDEQY-QENPJCQMSA-N 33017-11-7 Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)NCC(=O)NCC(=O)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)CCC1 VOUAQYXWVJDEQY-QENPJCQMSA-N 0.000 description 8
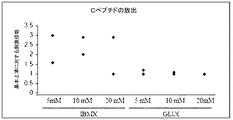
- 108010075254 C-Peptide Proteins 0.000 description 8
- 102000051325 Glucagon Human genes 0.000 description 8
- 102000040945 Transcription factor Human genes 0.000 description 8
- 108091023040 Transcription factor Proteins 0.000 description 8
- 239000007640 basal medium Substances 0.000 description 8
- 206010012601 diabetes mellitus Diseases 0.000 description 8
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 8
- 229960004666 glucagon Drugs 0.000 description 8
- 238000000338 in vitro Methods 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 230000003248 secreting effect Effects 0.000 description 8
- 230000003827 upregulation Effects 0.000 description 8
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 7
- 241000283690 Bos taurus Species 0.000 description 7
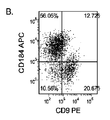
- 102100037904 CD9 antigen Human genes 0.000 description 7
- 102100037362 Fibronectin Human genes 0.000 description 7
- 108010067306 Fibronectins Proteins 0.000 description 7
- 108010090254 Growth Differentiation Factor 5 Proteins 0.000 description 7
- 102100035379 Growth/differentiation factor 5 Human genes 0.000 description 7
- 101000738354 Homo sapiens CD9 antigen Proteins 0.000 description 7
- 101000855004 Homo sapiens Protein Wnt-7a Proteins 0.000 description 7
- 101500016432 Lophius americanus Glucagon-like peptide 2 Proteins 0.000 description 7
- 108050000637 N-cadherin Proteins 0.000 description 7
- 102000003982 Parathyroid hormone Human genes 0.000 description 7
- 108090000445 Parathyroid hormone Proteins 0.000 description 7
- 102100020729 Protein Wnt-7a Human genes 0.000 description 7
- 102000013275 Somatomedins Human genes 0.000 description 7
- 102000043168 TGF-beta family Human genes 0.000 description 7
- 108091085018 TGF-beta family Proteins 0.000 description 7
- 102000046299 Transforming Growth Factor beta1 Human genes 0.000 description 7
- 101800002279 Transforming growth factor beta-1 Proteins 0.000 description 7
- DFPAKSUCGFBDDF-ZQBYOMGUSA-N [14c]-nicotinamide Chemical compound N[14C](=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-ZQBYOMGUSA-N 0.000 description 7
- 238000001514 detection method Methods 0.000 description 7
- 230000002124 endocrine Effects 0.000 description 7
- 235000020776 essential amino acid Nutrition 0.000 description 7
- 239000003797 essential amino acid Substances 0.000 description 7
- WDHRPWOAMDJICD-FOAQWNCLSA-N n-[2-[(3'r,3'as,6's,6as,6bs,7'ar,9r,11as,11br)-3',6',10,11b-tetramethyl-3-oxospiro[1,2,4,6,6a,6b,7,8,11,11a-decahydrobenzo[a]fluorene-9,2'-3,3a,5,6,7,7a-hexahydrofuro[3,2-b]pyridine]-4'-yl]ethyl]-6-(3-phenylpropanoylamino)hexanamide Chemical compound C([C@@H](C)C[C@@H]1[C@@H]2[C@H]([C@]3(C(=C4C[C@@H]5[C@@]6(C)CCC(=O)CC6=CC[C@H]5[C@@H]4CC3)C)O1)C)N2CCNC(=O)CCCCCNC(=O)CCC1=CC=CC=C1 WDHRPWOAMDJICD-FOAQWNCLSA-N 0.000 description 7
- 102000045246 noggin Human genes 0.000 description 7
- 108700007229 noggin Proteins 0.000 description 7
- 210000000496 pancreas Anatomy 0.000 description 7
- 239000000199 parathyroid hormone Substances 0.000 description 7
- 229960001319 parathyroid hormone Drugs 0.000 description 7
- 239000008177 pharmaceutical agent Substances 0.000 description 7
- 210000004623 platelet-rich plasma Anatomy 0.000 description 7
- 230000001105 regulatory effect Effects 0.000 description 7
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 7
- 101800001382 Betacellulin Proteins 0.000 description 6
- 102400001242 Betacellulin Human genes 0.000 description 6
- 102100024506 Bone morphogenetic protein 2 Human genes 0.000 description 6
- DWJXYEABWRJFSP-XOBRGWDASA-N DAPT Chemical compound N([C@@H](C)C(=O)N[C@H](C(=O)OC(C)(C)C)C=1C=CC=CC=1)C(=O)CC1=CC(F)=CC(F)=C1 DWJXYEABWRJFSP-XOBRGWDASA-N 0.000 description 6
- 101710201246 Eomesodermin Proteins 0.000 description 6
- 102100030751 Eomesodermin homolog Human genes 0.000 description 6
- 101800000221 Glucagon-like peptide 2 Proteins 0.000 description 6
- 101000917237 Homo sapiens Fibroblast growth factor 10 Proteins 0.000 description 6
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 6
- 101100013973 Mus musculus Gata4 gene Proteins 0.000 description 6
- 102100038553 Neurogenin-3 Human genes 0.000 description 6
- 101710096141 Neurogenin-3 Proteins 0.000 description 6
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 6
- 102000005157 Somatostatin Human genes 0.000 description 6
- 108010056088 Somatostatin Proteins 0.000 description 6
- FOIVPCKZDPCJJY-JQIJEIRASA-N arotinoid acid Chemical compound C=1C=C(C(CCC2(C)C)(C)C)C2=CC=1C(/C)=C/C1=CC=C(C(O)=O)C=C1 FOIVPCKZDPCJJY-JQIJEIRASA-N 0.000 description 6
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 6
- 238000011977 dual antiplatelet therapy Methods 0.000 description 6
- 210000004602 germ cell Anatomy 0.000 description 6
- 238000003364 immunohistochemistry Methods 0.000 description 6
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 6
- 229960000553 somatostatin Drugs 0.000 description 6
- WWUZIQQURGPMPG-UHFFFAOYSA-N (-)-D-erythro-Sphingosine Natural products CCCCCCCCCCCCCC=CC(O)C(N)CO WWUZIQQURGPMPG-UHFFFAOYSA-N 0.000 description 5
- 108010039627 Aprotinin Proteins 0.000 description 5
- 108010049974 Bone Morphogenetic Protein 6 Proteins 0.000 description 5
- 108010049870 Bone Morphogenetic Protein 7 Proteins 0.000 description 5
- 102100022525 Bone morphogenetic protein 6 Human genes 0.000 description 5
- 102100022544 Bone morphogenetic protein 7 Human genes 0.000 description 5
- 102000024905 CD99 Human genes 0.000 description 5
- 108060001253 CD99 Proteins 0.000 description 5
- 108060005980 Collagenase Proteins 0.000 description 5
- 102000029816 Collagenase Human genes 0.000 description 5
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 5
- QASFUMOKHFSJGL-LAFRSMQTSA-N Cyclopamine Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H](CC2=C3C)[C@@H]1[C@@H]2CC[C@@]13O[C@@H]2C[C@H](C)CN[C@H]2[C@H]1C QASFUMOKHFSJGL-LAFRSMQTSA-N 0.000 description 5
- 102000004127 Cytokines Human genes 0.000 description 5
- 108090000695 Cytokines Proteins 0.000 description 5
- SUZLHDUTVMZSEV-UHFFFAOYSA-N Deoxycoleonol Natural products C12C(=O)CC(C)(C=C)OC2(C)C(OC(=O)C)C(O)C2C1(C)C(O)CCC2(C)C SUZLHDUTVMZSEV-UHFFFAOYSA-N 0.000 description 5
- 101800000224 Glucagon-like peptide 1 Proteins 0.000 description 5
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 5
- 102400000326 Glucagon-like peptide 2 Human genes 0.000 description 5
- 101000578254 Homo sapiens Homeobox protein Nkx-6.1 Proteins 0.000 description 5
- 102100024392 Insulin gene enhancer protein ISL-1 Human genes 0.000 description 5
- 108090000978 Interleukin-4 Proteins 0.000 description 5
- 101100351017 Mus musculus Pax4 gene Proteins 0.000 description 5
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 5
- 229940122315 Notch pathway inhibitor Drugs 0.000 description 5
- 102100040918 Pro-glucagon Human genes 0.000 description 5
- 229940079156 Proteasome inhibitor Drugs 0.000 description 5
- 108010071390 Serum Albumin Proteins 0.000 description 5
- 102000007562 Serum Albumin Human genes 0.000 description 5
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 5
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 5
- RTKIYFITIVXBLE-UHFFFAOYSA-N Trichostatin A Natural products ONC(=O)C=CC(C)=CC(C)C(=O)C1=CC=C(N(C)C)C=C1 RTKIYFITIVXBLE-UHFFFAOYSA-N 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 229930013930 alkaloid Natural products 0.000 description 5
- 229960004405 aprotinin Drugs 0.000 description 5
- 230000024245 cell differentiation Effects 0.000 description 5
- OHCQJHSOBUTRHG-UHFFFAOYSA-N colforsin Natural products OC12C(=O)CC(C)(C=C)OC1(C)C(OC(=O)C)C(O)C1C2(C)C(O)CCC1(C)C OHCQJHSOBUTRHG-UHFFFAOYSA-N 0.000 description 5
- 229960002424 collagenase Drugs 0.000 description 5
- 229910052802 copper Inorganic materials 0.000 description 5
- 239000010949 copper Substances 0.000 description 5
- 210000004748 cultured cell Anatomy 0.000 description 5
- QASFUMOKHFSJGL-UHFFFAOYSA-N cyclopamine Natural products C1C=C2CC(O)CCC2(C)C(CC2=C3C)C1C2CCC13OC2CC(C)CNC2C1C QASFUMOKHFSJGL-UHFFFAOYSA-N 0.000 description 5
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 5
- 210000002308 embryonic cell Anatomy 0.000 description 5
- 229940031098 ethanolamine Drugs 0.000 description 5
- 108010043649 gastrin I Proteins 0.000 description 5
- 229960000890 hydrocortisone Drugs 0.000 description 5
- 230000001939 inductive effect Effects 0.000 description 5
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 210000003716 mesoderm Anatomy 0.000 description 5
- 230000014511 neuron projection development Effects 0.000 description 5
- 102000039446 nucleic acids Human genes 0.000 description 5
- 108020004707 nucleic acids Proteins 0.000 description 5
- 150000007523 nucleic acids Chemical class 0.000 description 5
- 230000001817 pituitary effect Effects 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 239000000186 progesterone Substances 0.000 description 5
- 229960003387 progesterone Drugs 0.000 description 5
- 230000035755 proliferation Effects 0.000 description 5
- 239000003207 proteasome inhibitor Substances 0.000 description 5
- 230000008410 smoothened signaling pathway Effects 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- WWUZIQQURGPMPG-KRWOKUGFSA-N sphingosine Chemical compound CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](N)CO WWUZIQQURGPMPG-KRWOKUGFSA-N 0.000 description 5
- 230000003637 steroidlike Effects 0.000 description 5
- RTKIYFITIVXBLE-QEQCGCAPSA-N trichostatin A Chemical compound ONC(=O)/C=C/C(/C)=C/[C@@H](C)C(=O)C1=CC=C(N(C)C)C=C1 RTKIYFITIVXBLE-QEQCGCAPSA-N 0.000 description 5
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 5
- 229960000604 valproic acid Drugs 0.000 description 5
- OAFQSZJQTKEEPK-UHFFFAOYSA-N 2-phenyl-1H-indole-3,4-dicarboximidamide dihydrochloride Chemical compound Cl.Cl.N1C2=CC=CC(C(N)=N)=C2C(C(=N)N)=C1C1=CC=CC=C1 OAFQSZJQTKEEPK-UHFFFAOYSA-N 0.000 description 4
- ZDEJZKULWCZIHL-UHFFFAOYSA-N 3-[1-(3-hydroxypropyl)pyrrolo[2,3-b]pyridin-3-yl]-4-pyrazin-2-ylpyrrole-2,5-dione Chemical compound C12=CC=CN=C2N(CCCO)C=C1C(C(NC1=O)=O)=C1C1=CN=CC=N1 ZDEJZKULWCZIHL-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 108010049931 Bone Morphogenetic Protein 2 Proteins 0.000 description 4
- 108010069241 Connexin 43 Proteins 0.000 description 4
- 102000001045 Connexin 43 Human genes 0.000 description 4
- 102100030074 Dickkopf-related protein 1 Human genes 0.000 description 4
- 108010003471 Fetal Proteins Proteins 0.000 description 4
- 102000004641 Fetal Proteins Human genes 0.000 description 4
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 4
- 102100039290 Gap junction gamma-1 protein Human genes 0.000 description 4
- 108010086527 Hepatocyte Nuclear Factor 6 Proteins 0.000 description 4
- 102100029087 Hepatocyte nuclear factor 6 Human genes 0.000 description 4
- 101000864646 Homo sapiens Dickkopf-related protein 1 Proteins 0.000 description 4
- 101001027128 Homo sapiens Fibronectin Proteins 0.000 description 4
- 229930182816 L-glutamine Natural products 0.000 description 4
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 description 4
- 102000007354 PAX6 Transcription Factor Human genes 0.000 description 4
- 101150081664 PAX6 gene Proteins 0.000 description 4
- 102000018886 Pancreatic Polypeptide Human genes 0.000 description 4
- 101150075928 Pax4 gene Proteins 0.000 description 4
- 101000983124 Sus scrofa Pancreatic prohormone precursor Proteins 0.000 description 4
- 210000002459 blastocyst Anatomy 0.000 description 4
- 230000032823 cell division Effects 0.000 description 4
- 239000002299 complementary DNA Substances 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 210000002257 embryonic structure Anatomy 0.000 description 4
- 230000001605 fetal effect Effects 0.000 description 4
- 108010069764 helospectin I Proteins 0.000 description 4
- HTMVMVKJOPFRMK-OYZAELBCSA-N helospectin i Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=C(O)C=C1 HTMVMVKJOPFRMK-OYZAELBCSA-N 0.000 description 4
- 229940088597 hormone Drugs 0.000 description 4
- 239000005556 hormone Substances 0.000 description 4
- 238000003365 immunocytochemistry Methods 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 230000006698 induction Effects 0.000 description 4
- 238000012423 maintenance Methods 0.000 description 4
- 239000011159 matrix material Substances 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 238000010186 staining Methods 0.000 description 4
- 239000013589 supplement Substances 0.000 description 4
- 238000002054 transplantation Methods 0.000 description 4
- 230000009278 visceral effect Effects 0.000 description 4
- HFDKKNHCYWNNNQ-YOGANYHLSA-N 75976-10-2 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)N)C(C)C)[C@@H](C)O)C1=CC=C(O)C=C1 HFDKKNHCYWNNNQ-YOGANYHLSA-N 0.000 description 3
- 108010088751 Albumins Proteins 0.000 description 3
- 102000009027 Albumins Human genes 0.000 description 3
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 3
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 3
- 239000005711 Benzoic acid Substances 0.000 description 3
- 102100036008 CD48 antigen Human genes 0.000 description 3
- 102100037986 Dickkopf-related protein 4 Human genes 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 102100035308 Fibroblast growth factor 17 Human genes 0.000 description 3
- 102100037680 Fibroblast growth factor 8 Human genes 0.000 description 3
- 102000002254 Glycogen Synthase Kinase 3 Human genes 0.000 description 3
- 108010014905 Glycogen Synthase Kinase 3 Proteins 0.000 description 3
- 108700031316 Goosecoid Proteins 0.000 description 3
- 102000050057 Goosecoid Human genes 0.000 description 3
- 102000014015 Growth Differentiation Factors Human genes 0.000 description 3
- 108010050777 Growth Differentiation Factors Proteins 0.000 description 3
- 102000012046 Hepatocyte Nuclear Factor 1-beta Human genes 0.000 description 3
- 108010061414 Hepatocyte Nuclear Factor 1-beta Proteins 0.000 description 3
- 102100028098 Homeobox protein Nkx-6.1 Human genes 0.000 description 3
- 102100030634 Homeobox protein OTX2 Human genes 0.000 description 3
- 102000009331 Homeodomain Proteins Human genes 0.000 description 3
- 108010048671 Homeodomain Proteins Proteins 0.000 description 3
- 101000716130 Homo sapiens CD48 antigen Proteins 0.000 description 3
- 101000951340 Homo sapiens Dickkopf-related protein 4 Proteins 0.000 description 3
- 101000878124 Homo sapiens Fibroblast growth factor 17 Proteins 0.000 description 3
- 101001052035 Homo sapiens Fibroblast growth factor 2 Proteins 0.000 description 3
- 101001060274 Homo sapiens Fibroblast growth factor 4 Proteins 0.000 description 3
- 101001027382 Homo sapiens Fibroblast growth factor 8 Proteins 0.000 description 3
- 101000584400 Homo sapiens Homeobox protein OTX2 Proteins 0.000 description 3
- 101001094700 Homo sapiens POU domain, class 5, transcription factor 1 Proteins 0.000 description 3
- 101000713275 Homo sapiens Solute carrier family 22 member 3 Proteins 0.000 description 3
- 101000976622 Homo sapiens Zinc finger protein 42 homolog Proteins 0.000 description 3
- 108010090306 Member 2 Subfamily G ATP Binding Cassette Transporter Proteins 0.000 description 3
- 102000013013 Member 2 Subfamily G ATP Binding Cassette Transporter Human genes 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 238000000636 Northern blotting Methods 0.000 description 3
- 238000010222 PCR analysis Methods 0.000 description 3
- 229930040373 Paraformaldehyde Natural products 0.000 description 3
- 108010004729 Phycoerythrin Proteins 0.000 description 3
- 238000011530 RNeasy Mini Kit Methods 0.000 description 3
- 108091006299 SLC2A2 Proteins 0.000 description 3
- 102100023537 Solute carrier family 2, facilitated glucose transporter member 2 Human genes 0.000 description 3
- 102100023550 Zinc finger protein 42 homolog Human genes 0.000 description 3
- 108010004469 allophycocyanin Proteins 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- 239000000427 antigen Substances 0.000 description 3
- 108091007433 antigens Proteins 0.000 description 3
- 102000036639 antigens Human genes 0.000 description 3
- 210000002469 basement membrane Anatomy 0.000 description 3
- 235000010233 benzoic acid Nutrition 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 108010015426 connexin 45 Proteins 0.000 description 3
- 238000007405 data analysis Methods 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 231100000673 dose–response relationship Toxicity 0.000 description 3
- 238000010195 expression analysis Methods 0.000 description 3
- 238000000684 flow cytometry Methods 0.000 description 3
- 230000007045 gastrulation Effects 0.000 description 3
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 3
- 238000003018 immunoassay Methods 0.000 description 3
- 238000002991 immunohistochemical analysis Methods 0.000 description 3
- 238000007901 in situ hybridization Methods 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 210000004153 islets of langerhan Anatomy 0.000 description 3
- 229960004857 mitomycin Drugs 0.000 description 3
- 238000010606 normalization Methods 0.000 description 3
- 210000004940 nucleus Anatomy 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 229920002866 paraformaldehyde Polymers 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- LFKDJXLFVYVEFG-UHFFFAOYSA-N tert-butyl carbamate Chemical compound CC(C)(C)OC(N)=O LFKDJXLFVYVEFG-UHFFFAOYSA-N 0.000 description 3
- 238000001262 western blot Methods 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- 108010083123 CDX2 Transcription Factor Proteins 0.000 description 2
- 102000006277 CDX2 Transcription Factor Human genes 0.000 description 2
- 101100257359 Caenorhabditis elegans sox-2 gene Proteins 0.000 description 2
- 241000202252 Cerberus Species 0.000 description 2
- 108050009340 Endothelin Proteins 0.000 description 2
- 102000002045 Endothelin Human genes 0.000 description 2
- 102400001368 Epidermal growth factor Human genes 0.000 description 2
- 101800003838 Epidermal growth factor Proteins 0.000 description 2
- 238000001134 F-test Methods 0.000 description 2
- 108090000386 Fibroblast Growth Factor 1 Proteins 0.000 description 2
- 102100031706 Fibroblast growth factor 1 Human genes 0.000 description 2
- 108090000368 Fibroblast growth factor 8 Proteins 0.000 description 2
- 240000008168 Ficus benjamina Species 0.000 description 2
- 241000287828 Gallus gallus Species 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 101800001586 Ghrelin Proteins 0.000 description 2
- 102400000442 Ghrelin-28 Human genes 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- 101150092640 HES1 gene Proteins 0.000 description 2
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 2
- 108700005087 Homeobox Genes Proteins 0.000 description 2
- 102100028096 Homeobox protein Nkx-6.2 Human genes 0.000 description 2
- 101000762366 Homo sapiens Bone morphogenetic protein 2 Proteins 0.000 description 2
- 101000578258 Homo sapiens Homeobox protein Nkx-6.2 Proteins 0.000 description 2
- 101001053263 Homo sapiens Insulin gene enhancer protein ISL-1 Proteins 0.000 description 2
- 101000971533 Homo sapiens Killer cell lectin-like receptor subfamily G member 1 Proteins 0.000 description 2
- 101000576323 Homo sapiens Motor neuron and pancreas homeobox protein 1 Proteins 0.000 description 2
- 101000979205 Homo sapiens Transcription factor MafA Proteins 0.000 description 2
- 101000652324 Homo sapiens Transcription factor SOX-17 Proteins 0.000 description 2
- 101000642523 Homo sapiens Transcription factor SOX-7 Proteins 0.000 description 2
- 102000004375 Insulin-like growth factor-binding protein 1 Human genes 0.000 description 2
- 108090000957 Insulin-like growth factor-binding protein 1 Proteins 0.000 description 2
- 108010085895 Laminin Proteins 0.000 description 2
- 102000007547 Laminin Human genes 0.000 description 2
- 102000004058 Leukemia inhibitory factor Human genes 0.000 description 2
- 108090000581 Leukemia inhibitory factor Proteins 0.000 description 2
- 102000043136 MAP kinase family Human genes 0.000 description 2
- 108091054455 MAP kinase family Proteins 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 102100025170 Motor neuron and pancreas homeobox protein 1 Human genes 0.000 description 2
- 101100284799 Mus musculus Hesx1 gene Proteins 0.000 description 2
- 101100350576 Mus musculus Otx1 gene Proteins 0.000 description 2
- 101100257363 Mus musculus Sox2 gene Proteins 0.000 description 2
- 101100369076 Mus musculus Tdgf1 gene Proteins 0.000 description 2
- 101100519293 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pdx-1 gene Proteins 0.000 description 2
- 108010070047 Notch Receptors Proteins 0.000 description 2
- 102000005650 Notch Receptors Human genes 0.000 description 2
- 102100039277 Pleiotrophin Human genes 0.000 description 2
- 239000013614 RNA sample Substances 0.000 description 2
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 2
- 239000012891 Ringer solution Substances 0.000 description 2
- 101150086694 SLC22A3 gene Proteins 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 102100030243 Transcription factor SOX-17 Human genes 0.000 description 2
- 102100036730 Transcription factor SOX-7 Human genes 0.000 description 2
- 102000004338 Transferrin Human genes 0.000 description 2
- 108090000901 Transferrin Proteins 0.000 description 2
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 2
- 101001029301 Xenopus tropicalis Forkhead box protein D3 Proteins 0.000 description 2
- 230000001464 adherent effect Effects 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 238000000540 analysis of variance Methods 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 150000003857 carboxamides Chemical class 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000002738 chelating agent Substances 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 230000003831 deregulation Effects 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 210000003981 ectoderm Anatomy 0.000 description 2
- 210000002242 embryoid body Anatomy 0.000 description 2
- ZUBDGKVDJUIMQQ-UBFCDGJISA-N endothelin-1 Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)NC(=O)[C@H]1NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](C(C)C)NC(=O)[C@H]2CSSC[C@@H](C(N[C@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N2)=O)NC(=O)[C@@H](CO)NC(=O)[C@H](N)CSSC1)C1=CNC=N1 ZUBDGKVDJUIMQQ-UBFCDGJISA-N 0.000 description 2
- 229940116977 epidermal growth factor Drugs 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 229940029303 fibroblast growth factor-1 Drugs 0.000 description 2
- 210000003953 foreskin Anatomy 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- GNKDKYIHGQKHHM-RJKLHVOGSA-N ghrelin Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)CN)COC(=O)CCCCCCC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C1=CC=CC=C1 GNKDKYIHGQKHHM-RJKLHVOGSA-N 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 238000009396 hybridization Methods 0.000 description 2
- 238000010191 image analysis Methods 0.000 description 2
- 238000003125 immunofluorescent labeling Methods 0.000 description 2
- 238000012744 immunostaining Methods 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 108010090448 insulin gene enhancer binding protein Isl-1 Proteins 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 210000000936 intestine Anatomy 0.000 description 2
- OGQSCIYDJSNCMY-UHFFFAOYSA-H iron(3+);methyl-dioxido-oxo-$l^{5}-arsane Chemical compound [Fe+3].[Fe+3].C[As]([O-])([O-])=O.C[As]([O-])([O-])=O.C[As]([O-])([O-])=O OGQSCIYDJSNCMY-UHFFFAOYSA-H 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 238000010208 microarray analysis Methods 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- 230000005305 organ development Effects 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 230000035935 pregnancy Effects 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000008093 supporting effect Effects 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 2
- 230000036962 time dependent Effects 0.000 description 2
- 239000012581 transferrin Substances 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 210000002438 upper gastrointestinal tract Anatomy 0.000 description 2
- 210000003556 vascular endothelial cell Anatomy 0.000 description 2
- 230000035899 viability Effects 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- LAQPKDLYOBZWBT-NYLDSJSYSA-N (2s,4s,5r,6r)-5-acetamido-2-{[(2s,3r,4s,5s,6r)-2-{[(2r,3r,4r,5r)-5-acetamido-1,2-dihydroxy-6-oxo-4-{[(2s,3s,4r,5s,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxy}hexan-3-yl]oxy}-3,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy}-4-hydroxy-6-[(1r,2r)-1,2,3-trihydrox Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]([C@@H](NC(C)=O)C=O)[C@@H]([C@H](O)CO)O[C@H]1[C@H](O)[C@@H](O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@H](O)CO)C(O)=O)[C@@H](O)[C@@H](CO)O1 LAQPKDLYOBZWBT-NYLDSJSYSA-N 0.000 description 1
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 1
- FHYNZKLNCPUNEU-UHFFFAOYSA-N 4-[(3,4-dihydroxyphenyl)methyl]-3-[(4-hydroxyphenyl)methyl]oxolan-2-one Chemical compound C1=CC(O)=CC=C1CC1C(=O)OCC1CC1=CC=C(O)C(O)=C1 FHYNZKLNCPUNEU-UHFFFAOYSA-N 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- 239000004382 Amylase Substances 0.000 description 1
- 102000013142 Amylases Human genes 0.000 description 1
- 108010065511 Amylases Proteins 0.000 description 1
- 102100022987 Angiogenin Human genes 0.000 description 1
- 108060000903 Beta-catenin Proteins 0.000 description 1
- 102000015735 Beta-catenin Human genes 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 102000007350 Bone Morphogenetic Proteins Human genes 0.000 description 1
- 108010007726 Bone Morphogenetic Proteins Proteins 0.000 description 1
- 101150096994 Cdx1 gene Proteins 0.000 description 1
- 102100025745 Cerberus Human genes 0.000 description 1
- 101710010675 Cerberus Proteins 0.000 description 1
- 208000031404 Chromosome Aberrations Diseases 0.000 description 1
- 206010010099 Combined immunodeficiency Diseases 0.000 description 1
- 102100027907 Cytoplasmic tyrosine-protein kinase BMX Human genes 0.000 description 1
- 102000000541 Defensins Human genes 0.000 description 1
- 108010002069 Defensins Proteins 0.000 description 1
- 101100373143 Drosophila melanogaster Wnt5 gene Proteins 0.000 description 1
- 102000012804 EPCAM Human genes 0.000 description 1
- 101150084967 EPCAM gene Proteins 0.000 description 1
- 102100033167 Elastin Human genes 0.000 description 1
- 108010014258 Elastin Proteins 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 102100028071 Fibroblast growth factor 7 Human genes 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 101710178004 Gap junction gamma-1 protein Proteins 0.000 description 1
- 102000030595 Glucokinase Human genes 0.000 description 1
- 108010021582 Glucokinase Proteins 0.000 description 1
- 101150036261 HNF1B gene Proteins 0.000 description 1
- 229920002971 Heparan sulfate Polymers 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101001060261 Homo sapiens Fibroblast growth factor 7 Proteins 0.000 description 1
- 101001066129 Homo sapiens Glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 1
- 101000911772 Homo sapiens Hsc70-interacting protein Proteins 0.000 description 1
- 101001069749 Homo sapiens Prospero homeobox protein 1 Proteins 0.000 description 1
- 101000655352 Homo sapiens Telomerase reverse transcriptase Proteins 0.000 description 1
- 101000819088 Homo sapiens Transcription factor GATA-6 Proteins 0.000 description 1
- 102100027037 Hsc70-interacting protein Human genes 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 101150070110 Isl1 gene Proteins 0.000 description 1
- 102100021457 Killer cell lectin-like receptor subfamily G member 1 Human genes 0.000 description 1
- 108010052014 Liberase Proteins 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- 101000607573 Mus musculus Unknown protein from 2D-PAGE of fibroblasts Proteins 0.000 description 1
- 101000976618 Mus musculus Zinc finger protein 42 Proteins 0.000 description 1
- 108010069196 Neural Cell Adhesion Molecules Proteins 0.000 description 1
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 1
- 102100037369 Nidogen-1 Human genes 0.000 description 1
- 101150041192 Otx1 gene Proteins 0.000 description 1
- 101150111723 PDX1 gene Proteins 0.000 description 1
- 108091007960 PI3Ks Proteins 0.000 description 1
- 108700020479 Pancreatic hormone Proteins 0.000 description 1
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 1
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 102100033880 Prospero homeobox protein 1 Human genes 0.000 description 1
- 101710150336 Protein Rex Proteins 0.000 description 1
- 108010067787 Proteoglycans Proteins 0.000 description 1
- 102000016611 Proteoglycans Human genes 0.000 description 1
- 108010014608 Proto-Oncogene Proteins c-kit Proteins 0.000 description 1
- 102000016971 Proto-Oncogene Proteins c-kit Human genes 0.000 description 1
- 238000001190 Q-PCR Methods 0.000 description 1
- 238000002123 RNA extraction Methods 0.000 description 1
- 238000010240 RT-PCR analysis Methods 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- ZSJLQEPLLKMAKR-UHFFFAOYSA-N Streptozotocin Natural products O=NN(C)C(=O)NC1C(O)OC(CO)C(O)C1O ZSJLQEPLLKMAKR-UHFFFAOYSA-N 0.000 description 1
- 102100021669 Stromal cell-derived factor 1 Human genes 0.000 description 1
- 101710088580 Stromal cell-derived factor 1 Proteins 0.000 description 1
- 102000004874 Synaptophysin Human genes 0.000 description 1
- 108090001076 Synaptophysin Proteins 0.000 description 1
- 101150057140 TACSTD1 gene Proteins 0.000 description 1
- 108010008125 Tenascin Proteins 0.000 description 1
- 102000007000 Tenascin Human genes 0.000 description 1
- 206010043276 Teratoma Diseases 0.000 description 1
- 108010077673 Tetraspanin 29 Proteins 0.000 description 1
- 102000010431 Tetraspanin 29 Human genes 0.000 description 1
- 108090000190 Thrombin Proteins 0.000 description 1
- 102100021382 Transcription factor GATA-6 Human genes 0.000 description 1
- 108010009583 Transforming Growth Factors Proteins 0.000 description 1
- 102000009618 Transforming Growth Factors Human genes 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 102100035140 Vitronectin Human genes 0.000 description 1
- 108010031318 Vitronectin Proteins 0.000 description 1
- 102000052549 Wnt-3 Human genes 0.000 description 1
- 108700020985 Wnt-3 Proteins 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 102000013529 alpha-Fetoproteins Human genes 0.000 description 1
- 108010026331 alpha-Fetoproteins Proteins 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 235000019418 amylase Nutrition 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 108010072788 angiogenin Proteins 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 230000002424 anti-apoptotic effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 210000001172 blastoderm Anatomy 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 229940112869 bone morphogenetic protein Drugs 0.000 description 1
- 238000010804 cDNA synthesis Methods 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 108060001132 cathelicidin Proteins 0.000 description 1
- 102000014509 cathelicidin Human genes 0.000 description 1
- POIUWJQBRNEFGX-XAMSXPGMSA-N cathelicidin Chemical compound C([C@@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(O)=O)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CC(C)C)C1=CC=CC=C1 POIUWJQBRNEFGX-XAMSXPGMSA-N 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000011712 cell development Effects 0.000 description 1
- 230000033026 cell fate determination Effects 0.000 description 1
- 238000003779 cell fixation method Methods 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 230000003399 chemotactic effect Effects 0.000 description 1
- 210000001612 chondrocyte Anatomy 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000001143 conditioned effect Effects 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 230000002559 cytogenic effect Effects 0.000 description 1
- 230000032459 dedifferentiation Effects 0.000 description 1
- 229920006237 degradable polymer Polymers 0.000 description 1
- 238000004925 denaturation Methods 0.000 description 1
- 230000036425 denaturation Effects 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 108010007093 dispase Proteins 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 230000002183 duodenal effect Effects 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 210000000750 endocrine system Anatomy 0.000 description 1
- 230000020619 endoderm development Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 210000001842 enterocyte Anatomy 0.000 description 1
- 210000000646 extraembryonic cell Anatomy 0.000 description 1
- 210000003754 fetus Anatomy 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 108010074605 gamma-Globulins Proteins 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 101150003286 gata4 gene Proteins 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 102000006602 glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 1
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 1
- 210000002149 gonad Anatomy 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 239000012145 high-salt buffer Substances 0.000 description 1
- 238000010562 histological examination Methods 0.000 description 1
- 230000005745 host immune response Effects 0.000 description 1
- 102000047486 human GAPDH Human genes 0.000 description 1
- 210000003917 human chromosome Anatomy 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 210000002660 insulin-secreting cell Anatomy 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000031864 metaphase Effects 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 230000000921 morphogenic effect Effects 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- YAEMHJKFIIIULI-UHFFFAOYSA-N n-(4-methoxybenzyl)-n'-(5-nitro-1,3-thiazol-2-yl)urea Chemical compound C1=CC(OC)=CC=C1CNC(=O)NC1=NC=C([N+]([O-])=O)S1 YAEMHJKFIIIULI-UHFFFAOYSA-N 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 108010008217 nidogen Proteins 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 230000015031 pancreas development Effects 0.000 description 1
- 210000004303 peritoneum Anatomy 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 239000004633 polyglycolic acid Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 210000003240 portal vein Anatomy 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 238000003498 protein array Methods 0.000 description 1
- 239000012474 protein marker Substances 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 150000004728 pyruvic acid derivatives Chemical class 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 238000007634 remodeling Methods 0.000 description 1
- 238000009256 replacement therapy Methods 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 108090000064 retinoic acid receptors Proteins 0.000 description 1
- 102000003702 retinoic acid receptors Human genes 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 239000012679 serum free medium Substances 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 102000035025 signaling receptors Human genes 0.000 description 1
- 108091005475 signaling receptors Proteins 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000009331 sowing Methods 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 230000009469 supplementation Effects 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229960004072 thrombin Drugs 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 230000008467 tissue growth Effects 0.000 description 1
- 230000017423 tissue regeneration Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 230000008189 vertebrate development Effects 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 230000036266 weeks of gestation Effects 0.000 description 1
- 101150068520 wnt3a gene Proteins 0.000 description 1
Images
























































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0676—Pancreatic cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/37—Digestive system
- A61K35/39—Pancreas; Islets of Langerhans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/12—Antidiuretics, e.g. drugs for diabetes insipidus
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/30—Organic components
- C12N2500/34—Sugars
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/115—Basic fibroblast growth factor (bFGF, FGF-2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/12—Hepatocyte growth factor [HGF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/155—Bone morphogenic proteins [BMP]; Osteogenins; Osteogenic factor; Bone inducing factor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/16—Activin; Inhibin; Mullerian inhibiting substance
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/21—Chemokines, e.g. MIP-1, MIP-2, RANTES, MCP, PF-4
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/30—Hormones
- C12N2501/335—Glucagon; Glucagon-like peptide [GLP]; Exendin
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/30—Hormones
- C12N2501/38—Hormones with nuclear receptors
- C12N2501/385—Hormones with nuclear receptors of the family of the retinoic acid recptor, e.g. RAR, RXR; Peroxisome proliferator-activated receptor [PPAR]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/40—Regulators of development
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/40—Regulators of development
- C12N2501/415—Wnt; Frizzeled
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/40—Regulators of development
- C12N2501/42—Notch; Delta; Jagged; Serrate
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/70—Enzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/999—Small molecules not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/02—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from embryonic cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2533/00—Supports or coatings for cell culture, characterised by material
- C12N2533/90—Substrates of biological origin, e.g. extracellular matrix, decellularised tissue
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biomedical Technology (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- General Health & Medical Sciences (AREA)
- Cell Biology (AREA)
- Diabetes (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Hematology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Physiology (AREA)
- Gastroenterology & Hepatology (AREA)
- Nutrition Science (AREA)
- Developmental Biology & Embryology (AREA)
- Immunology (AREA)
- Virology (AREA)
- Epidemiology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Obesity (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Materials For Medical Uses (AREA)
Description
本発明は、多能性幹細胞の分化を促進するための方法を提供する。詳細には、本発明は膵臓内胚葉、膵臓ホルモン発現細胞及び膵臓ホルモン分泌細胞を形成するための改良された方法を提供する。本発明は更に、フィーダー細胞層を使用することなく多能性幹細胞の分化を促進する方法を提供する。
1型糖尿病の細胞置換療法における進歩及び移植可能なランゲルハンス島の不足のため、生着に適したインスリン産生細胞即ち、β細胞の供給源の開発に注目が集まっている。1つの方法として、例えば、胚性幹細胞のような多能性幹細胞から機能性のβ細胞を生成することがある。
〔発明が解決しようとする課題〕
したがって、膵臓内分泌細胞、膵臓ホルモン発現細胞、又は膵臓ホルモン分泌細胞への分化能を維持する一方で、今日の臨床的要求に応えるように拡張することが可能な多能性細胞系を樹立するための条件を開発することが依然、大きく求められている。本発明者等は、ヒト胚性幹細胞を膵臓内分泌細胞に分化させる効率を高めることを目的として代替的な手法を取ったものである。
一実施形態において、本発明は、多能性幹細胞を分化させるための方法において、
a.多能性幹細胞を培養する工程と、
b.前記多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させる工程と、を含む、方法を提供する。
a.血清の非存在下、アクチビンAを含む培地中で多能性幹細胞を培養し、次いで細胞をアクチビンA及び血清と培養し、次いで細胞をアクチビンA及び異なる濃度の血清と培養するか、
b.血清の非存在下、アクチビンAを含む培地中で多能性幹細胞を培養し、次いで細胞をアクチビンA及び別の濃度の血清と培養するか、
c.血清の非存在下、アクチビンA及びWntリガンドを含む培地中で多能性幹細胞を培養し、次いでWntリガンドを除去して細胞をアクチビンA及び血清と培養するか、
d.多能性幹細胞を、細胞外基質でコーティングした組織培養基質上で培養し、この多能性幹細胞をアクチビンA及びWntリガンドと培養するか、
e.多能性幹細胞を、細胞外基質でコーティングした組織培養基質上で培養し、次いでこの多能性幹細胞を、血清を含む第1の培地中でアクチビンA及びWntリガンドと培養し、次いでこの多能性幹細胞を、血清を含む第2の培地中でアクチビンAと培養するか、
f.多能性幹細胞を、細胞外基質でコーティングした組織培養基質上で培養し、次いでこの多能性幹細胞を、血清を含む第1の培地中でアクチビンA及びWntリガンドと培養し、次いでこの多能性幹細胞を、異なる濃度の血清を含む第2の培地中でアクチビンA及びWntリガンドと培養する。
a.胚体内胚葉系統に特徴的なマーカーを発現する細胞を、繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤で処理し、次いで繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤を含む培地を取り除いた後、レチノイン酸、繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤を含む培地中で細胞を培養するか、
b.胚体内胚葉系統に特徴的なマーカーを発現する細胞を、レチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理するか、
c.胚体内胚葉系統に特徴的なマーカーを発現する細胞をレチノイン酸で処理し、次いでレチノイン酸を除去した後、細胞を少なくとも1種類の繊維芽細胞増殖因子で処理する。
a.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、DAPT及びエキセンジン4を含む培地中で培養し、次いでDAPT及びエキセンジン4を含む培地を除去した後、細胞をエキセンジン1、IGF−1及びHGFを含む培地中で培養するか、
b.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、エキセンジン4を含む培地中で培養し、次いでエキセンジン4を含む培地を除去した後、細胞をエキセンジン1、IGF−1及びHGFを含む培地中で培養するか、
c.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、DAPT及びエキセンジン4を含む培地中で培養するか、
d.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、エキセンジン4を含む培地中で培養するか、
e.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、ノッチシグナル伝達経路を阻害する因子で処理するか、
f.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMのグルコース及びエキセンジン4を含む培地中で培養する。
a.多能性幹細胞を培養する工程と、
b.前記多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、β細胞系統の細胞に分化させる工程と、
e.前記β細胞系統の細胞を前記患者に移植する工程と、を含む、方法を提供する。
幹細胞とは、1個の細胞レベルで自己再生し、かつ、自己再生性の前駆細胞、非自己再生性の前駆細胞、及び最終分化細胞のような前駆細胞を生ずるように分化する能力によって定義される未分化細胞のことである。幹細胞はまた、複数の胚細胞層(内胚葉、中胚葉及び外胚葉)から異なる細胞系の機能性細胞へとインビトロで分化する能力、並びに、移植後に複数の胚細胞層の組織を生ずる能力、及び胚盤胞への注入後にすべての組織とまではいかないにしてもほぼ大部分の組織に分化する能力によっても特徴付けられる。
多能性幹細胞の特徴付け
多能性幹細胞は、発生段階特異的胚性抗原(SSEA)3及び4の1つ以上、及びTra−1−60及びTra−1−81と呼ばれる抗体を用いて検出可能なマーカーを発現しうる(Thomson et al., Science 282:1145, 1998)。インビトロで多能性幹細胞を分化させると、SSEA−4、Tra−1−60、及びTra−1−81の発現が消失し(存在する場合)、SSEA−1の発現が増大する。未分化の多能性幹細胞は通常アルカリホスファターゼ活性を有し、これは、細胞を4%パラホルムアルデヒドで固定し、次いで製造業者(ベクターラボラトリーズ、カリフォルニア州バーリンゲーム所在)によって述べられるようにVectorRedを基質として現像することによって検出することができる。未分化の多能性幹細胞は、RT−PCRによって検出されるOct−4及びTERTを通常更に発現している。
使用が可能な多能性幹細胞の種類としては、妊娠期間中の任意の時期(必ずしもではないが、通常は妊娠約10〜12週よりも前)に採取した前胚性組織(例えば、胚盤胞等)、胚性組織、胎児組織等の、妊娠後に形成される組織に由来する多能性幹細胞の株化細胞系が含まれる。非限定的な例としては、例えば、ヒト胚性幹細胞系H1、H7及びH9(WiCell)等のヒト胚性幹細胞又はヒト胚性生殖細胞の株化細胞系がある。更に、こうした細胞の初期の株化又は安定化の際に本開示の組成物を使用することも考えられるが、その場合は、供給源となる細胞は供給源組織から直接採取される1次多能性細胞である。フィーダー細胞の非存在下で既に培養された多能性幹細胞集団から得られる細胞も好適である。例えば、BG01v(ブレサジェン社(BresaGen)、ジョージア州アテネ所在)等の変異型ヒト胚性幹細胞系も好適である。
一実施形態では、通常、多能性幹細胞を、様々な点で多能性幹細胞を支持するフィーダー細胞の層上で培養する。あるいは、多能性幹細胞を、フィーダー細胞を基本的に含まないにも関わらず、細胞を大きく分化させることなく多能性幹細胞の増殖を支持する培養システム中で培養する。分化をともなわない無フィーダー細胞培養中での多能性幹細胞の増殖は、別の細胞種と予め培養することによって調整した培地を用いることで支持される。また、分化をともなわない無フィーダー細胞培養中での多能性幹細胞の増殖は、合成培地を用いることによっても支持される。
本発明における使用に適した多能性幹細胞としては例えば、胚性幹細胞系H9(NIHコード:WA09)、ヒト胚性幹細胞系H1(NIHコード:WA01)、ヒト胚性幹細胞系H7(NIHコード:WA07)、及びヒト胚性幹細胞系SA002(セラーティス社(Cellartis)、スウェーデン)が挙げられる。多能性細胞に特徴的な以下のマ−カー、即ち、ABCG2、クリプト(cripto)、CD9、FoxD3、コネキシン43、コネキシン45、Oct4、Sox2、ナノグ(Nanog)、hTERT、UTF−1、ZFP42、SSEA−3、SSEA−4、Tra1−60、Tra1−81の内の少なくとも1つを発現する細胞も本発明における使用に適している。
多能性幹細胞を、当該技術分野のいかなる方法、又は本発明で提案されるいかなる方法によって胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させてもよい。
a.細胞外基質でコーティングされた組織培養基質上に多能性幹細胞を播種する工程と、
b.前記多能性幹細胞を、アクチビンA及びWntリガンドと培養する工程と、を含む、方法を提供する。
本発明の一態様では、多能性細胞を、細胞外基質でコーティングされた組織培養基質上で培養して分化させる。細胞外基質は、マウスの肉腫細胞から抽出された可溶化基底膜の調製物(BDバイオサイエンス社(BD Biosciences)よりMATRIGELの商品名で販売されている)であってよい。また、細胞外基質は、増殖因子を低減させたMATRIGELであってもよい。また、細胞外基質はフィブロネクチンであってもよい。代替的な一実施形態では、多能性幹細胞を、ヒト血清でコーティングされた組織培養基質上で培養して分化させる。
1個の培地を使用する場合、培地は、例えば(国際特許出願公開第2006020919号に開示されるような)インスリン及びIGF等の特定の因子を、多能性幹細胞が胚体内胚葉に分化できるように充分に低い濃度で含有しなければならない。これは血清濃度を低下させるか、あるいはインスリン及びIGFを含まない合成培地を使用することによって実現することが可能である。合成培地の例はワイルズ(Wiles)等によって開示されている(Wiles et al. Exp Cell Res. 1999 Feb 25; 247(1): 241-8)。
多能性幹細胞の胚体内胚葉系統細胞への分化は、2個の培地を使用して多能性幹細胞をアクチビンA及びWntリガンドと培養することによって実現することが可能である。即ち、多能性幹細胞の分化は以下のようにして実現することが可能である。
a.細胞外基質でコーティングされた組織培養基質上に多能性幹細胞を播種し、
b.前記多能性幹細胞を第1の培地中でアクチビンA及びWntリガンドと培養し、
c.前記多能性幹細胞を第2の培地中でアクチビンAと培養する。
胚体内胚葉系統に特徴的なマーカーを発現する細胞が形成されたことは、特定のプロトコールを行う前後にマーカーの存在について試験を行うことによって判定することができる。多能性幹細胞は通常、こうしたマーカーを発現しない。したがって、多能性細胞の分化は、細胞がマーカーを発現し始めることで検出される。
胚体内胚葉系統に特徴的なマーカーを発現する細胞を、当該技術分野におけるいずれかの方法又は本発明において提案されるいずれかの方法によって膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させることができる。
a.胚体内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.胚体内胚葉系統に特徴的なマーカーを発現する前記細胞をレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理する工程と、を含む、方法を提供する。
a.胚体内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.胚体内胚葉系統に特徴的なマーカーを発現する前記細胞をレチノイン酸で処理する工程と、
c.レチノイン酸を取り除き、その後、細胞を少なくとも1種類の繊維芽細胞増殖因子で処理する工程と、を含む、方法を提供する。
膵臓内胚葉系統に特徴的なマーカーは当業者にはよく知られたものであり、膵臓内胚葉系統に特徴的な更なるマーカーも次々に特定されている。これらのマーカーを用いて、本発明に基づいて処理を行った細胞が、膵臓内胚葉系統に特徴的な性質を獲得するように分化したことを確認することが可能である。膵臓内胚葉系統に特異的なマーカーとしては、例えば、Hlxb9、PTF−1a、PDX−1、HNF−6、HNF−1β等、1以上の転写因子の発現が挙げられる。
膵臓内胚葉系統に特徴的なマーカーを発現する細胞は、当該技術分野におけるいずれかの方法又は本発明において開示されるいずれかの方法によって膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させることができる。
a.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.ノッチシグナル伝達経路を阻害する因子で前記細胞を処理する工程と、を含む、方法を提供する。
a.膵臓内胚葉系統に特徴的なマーカーを発現する細胞を培養する工程と、
b.グルコースを約10mM〜約20mMの濃度で含む培地中で、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させることが可能な因子で前記細胞を処理する工程と、を含む、方法を提供する。
膵臓内分泌系統の細胞に特徴的なマーカーは当業者にはよく知られたものであり、膵臓内分泌系統に特徴的な更なるマーカーも次々に特定されている。これらのマーカーを用いて、本発明に基づいて処理を行った細胞が膵臓内分泌系統に特徴的な性質を獲得するように分化したことを確認することが可能である。膵臓内分泌系統に特異的なマーカーとしては、例えば、NGN−3、ニューロD(NeuroD)、Islet−1等、1以上の転写因子の発現が挙げられる。
一態様において、本発明は、1型糖尿病を罹患しているかあるいは発症するリスクを有する患者を治療するための方法を提供する。この方法では、多能性幹細胞を培養し、その多能性幹細胞をインビトロでβ細胞系統に分化させ、そのβ細胞系統の細胞を患者に移植することを含む。
ヒト胚性幹細胞の培養
ヒト胚性幹細胞系H1、H7及びH9をウィセル・リサーチ・インスティテュート社(WiCell Research Institute, Inc.)(ウイスコンシン州マディソン所在)より入手し、当該供給元機関によって与えられる指示に従って培養した。簡単に述べると、20%ノックアウト血清リプレースメント、100nM MEM非必須アミノ酸、0.5mM βメルカプトエタノール、2mM L−グルタミンと4ng/mlヒト塩基性繊維芽細胞増殖因子(bFGF)(すべてインビトロジェン/ギブコ社(Invitrogen/GIBCO)より入手)を添加したDMEM/F12(インビトロジェン/ギブコ社(Invitrogen/GIBCO))からなるES細胞培地中、マウス胚繊維芽(MEF)フィーダー細胞上で細胞を培養した。MEF細胞はE13〜13.5のマウス胚から誘導されたものをチャールズリバー社(Charles River)より購入した。MEF細胞は、10%FBS(ハイクローン社(Hyclone))、2mMグルタミン、及び100mM MEM非必須アミノ酸を添加したDMEM培地で増殖させた。サブコンフルエンスに達したMEF細胞培養を、10μg/mlマイトマイシンC(シグマ社(Sigma)ミズーリ州セントルイス所在)で3時間処理して細胞分裂を停止させた後、トリプシン処理し、0.1%ウシゼラチンコートディッシュに2×104/cm2で播いた。継代数2〜4からのMEF細胞をフィーダー層として使用した。MEF細胞のフィーダー層に播いたヒト胚性幹細胞を、加湿した組織培養インキュベーター内で、5%CO2雰囲気中37℃で培養した。コンフルエンスに達した時点(播種後約5〜7日後)で、ヒト胚性幹細胞を1mg/ml IV型コラゲナーゼ(インビトロジェン/ギブコ社(Invitrogen/GIBCO))で5〜10分間処理した後、5mlピペットを用いて表面から静かに掻き取った。細胞を900rpmで5分間遠心し、得られたペレットを再懸濁して新鮮な培地に1:3〜1:4の細胞比で再び播いた。
胚体内胚葉細胞の形成
胚体内胚葉のマーカーの発現に対するアクチビンAの影響を調べた。アクチビンA(100ng/ml)をマウス胚繊維芽細胞上で培養したヒト胚性幹細胞の集団に添加した。細胞をアクチビンAの存在下で継続的に培養し、示した時間において採取した。胚体内胚葉マーカーの発現レベルをPCR(図1)、FACS(表IIに結果をまとめる)、及び免疫組織化学法(図2)によって調べた。
膵臓内胚葉細胞の形成
ヒト胚性幹細胞の膵臓内胚葉への分化を誘導することが知られている増殖因子を細胞培養に添加した。具体的には、膵臓内胚葉の形成を誘導することが知られているアクチビンA、bFGF、及びレチノイン酸を細胞培養に添加した。
膵臓内分泌細胞の形成
ヒト胚性幹細胞の培養を実施例1に述べた方法に従って3〜4日間未分化条件に維持した。細胞が50〜60%コンフルエンスに達した後、100ng/mlのアクチビンAを含む、FBSを含まないDMEM/F12に培地を換え、この培地中で細胞を1日間培養した。1日間の培養の後、培地を除去し、100ng/mlのアクチビンAを加えた0.5%FBSを含む培地と交換し、細胞を1日間培養した。この2回目の1日の培養の後、培地を除去し、100ng/mlのアクチビンAを加えた2%FBSを含む培地と交換し、細胞を1日間培養した。この時間間隔後、実施例2に概略を述べたように細胞をレチノイン酸とFGFの組み合わせで6日間処理し、その後、培地を除去し、10μMのγ−セクレターゼ阻害剤L−685,458を含む、2%FBSを含んだDMEM/F12からなる培地と交換し、3日間培養した。細胞を収穫し、mRNAの試料を収集して分析に供した。アクチビンA単独で5日間処理した細胞からなるコントロール培養も含めた。
Nkx2.2を発現する膵臓内分泌細胞の形成
実施例2に概略を述べた方法に従って得られた胚体内胚葉細胞を以下のように処理した。即ち、50ng/mlアクチビンA、50ng/ml塩基性FGF及び1μMのレチノイン酸を加えた2%FBS含有DMEM/F12からなる基本培地中で細胞を3〜5日間培養した。1μMのレチノイン酸を単独で又はbFGFとともに含む基本培地中で細胞を更に3〜5日間引き続き培養した。方向付けされた細胞の分化の評価を助けるため、このプロセスに沿った異なる時点で細胞からRNA試料を採取した。更に、この分化プロトコールの全体を通じて培地及び各因子を定期的に除去して補充した。アクチビンAの添加により、アクチビンAを加えない試料と比較してNkx2.2の発現は約35倍に増大した。培養の最初の3日間にアクチビンAで処理した試料では、Pdx1の発現はアクチビンAを含まない試料と同等のレベルに維持された(図11)。以上を考え合わせると、これらのデータは、膵臓内分泌マーカーであるNkx2.2の発現は、レチノイン酸及びbFGFによる処理の最初の3日間にアクチビンAを添加することによって更に促進されることを示唆するものである。
膵臓内胚葉細胞の培養における継代及び増殖
本実施例では、本願でヒト胚性幹細胞より誘導された膵臓内胚葉細胞を細胞培養中で維持し、更に分化させることなく継代することが可能であることを実証する。膵臓内胚葉細胞を低血清DMEM/F12中、100ng/mlのアクチビンAの存在下で分化させた。低血清DMEM/F12は、1日目には0%(v/v)のウシ胎児血清(FBS)、2日目には0.5%(v/v)のFBS、その後は毎日2%(v/v)のFBSを含有するものを使用した。4日間分化させた後、細胞を2%(v/v)FBS、1μMのレチノイン酸、及び50ng/mlのbFGFを含む低血清DMEM/F12中で、全体で6日間更に培養した。6日間の分化の後、細胞を、2%(v/v)FBSを含む低血清DMEM/F12中、50ng/mlのFGF10の存在下で、全体で6日間、培養中で維持した。6日間の培養期間中、膵臓内胚葉細胞を2回継代した。細胞集団の倍加時間はこの6日間の培養期間で約36〜48時間であった。培養の0、3及び6日目において、Q−PCRを用いて膵臓内胚葉を示すマーカー遺伝子の発現を測定した。図12は、50ng/mlのFGF10の存在下で増殖させた細胞が、誘導後の6日間の培養期間中、膵臓内胚葉マーカーであるPdx1の発現を維持したことを示している。
ヒト胚性幹細胞からの肝細胞の誘導
ヒト胚性幹細胞の培養を実施例1で述べた方法に従って未分化培養条件に2〜3日間維持した。細胞が70〜80%コンフルエンスに達した後、100ng/mlのアクチビンAを含む2%FBS含有DMEM/F12に培地を換え、アクチビンAの存在下で細胞を7日間培養した。7日間のアクチビンA処理の後、細胞を図13に示す各条件で5日間処理した。この後、細胞を採取し、mRNAの試料を収集して分析に供した。
H9ヒト胚性幹細胞系の特徴付け
未分化のES細胞が発現する幾つかのマーカーの発現を評価することにより、H9細胞の性質を経時的に監視した(Carpenter et al., 2001; Reubinoff et al., 2000; Thomson et al., 1998a)。H9細胞では、段階特異的胚抗原の交互発現が見られた(表III)。H9細胞は、いずれも未分化のヒト胚性幹細胞に特徴的なSSEA−3、SSEA−4、Tra−1−60、Tra−1−81、AP及びCD9抗原に対して強力な免疫応答性を示す。
蛍光活性化細胞選別(FACS)分析
接着した細胞をTrypLE(商標)Express溶液(インビトロジェン社(Invitrogen)カリフォルニア州所在)と5分間インキュベートすることによって培養皿から剥がした。剥がした細胞をヒト胚性幹細胞培地に再懸濁し、遠心して回収した後、洗浄し、この細胞を2%BSA、0.05%アジ化ナトリウムを含むPBSからなる染色緩衝液(シグマ社(Sigma)ミズーリ州)に再懸濁した。必要に応じて、0.1%γグロブリン(シグマ社(Sigma))溶液を用いて15分間、細胞のFc受容体をブロックした。一定分量(約105個の細胞)を表Iに示されるようなフィコエリスリン(PE)若しくはアロフィコシアニン(APC)を結合したモノクローナル抗体(106個の細胞毎に5μLの抗体)、又は非結合一次抗体とインキュベートした。コントロールには、適当なアイソタイプ一致抗体、非染色細胞、及び二次結合抗体のみで染色した細胞を含めた。抗体とのインキュベートをすべて、4℃で30分間行い、その後、細胞を染色緩衝液で洗った。非結合一次抗体で染色した試料を、PE又はAPC結合標識二次抗体と更に30分間、4℃でインキュベートした。使用した二次抗体の一覧については表Iを参照されたい。洗った細胞はペレットとし、染色緩衝液に再懸濁した。細胞表面の分子をFACS Array(BDバイオサイエンス社(BD Biosciences))装置を使用し、少なくとも10,000のイベントを収集して特定した。
免疫細胞化学法
0.1%Matrigel(BD社)でコーティングした培養皿に播種した細胞を室温で20分間、4%パラホルムアルデヒドで固定した。固定した細胞を室温で1時間、PBS/0.1%BSA/10%正常ニワトリ血清/0.5%TritonX−100でブロックした後、PBS/0.1%BSA/10%正常ニワトリ血清中、4℃で一次抗体と一晩インキュベートした。一次抗体及びその実用希釈率を表IBに示す。PBS/0.1%BSA中で3回洗った後、PBS中に1:100の希釈率で希釈した蛍光二次抗体と細胞を室温で1時間インキュベートして結合させた。コントロール試料として、一次抗体を省略するか、一次抗体と同じ濃度の、対応する一致したネガティブコントロールとしての免疫グロブリンで一次抗体を置き換えた反応を含めた。染色した試料はすすいだ。ジアミジノ−2−フェニルインドール二塩酸塩(DAPI)を含むPROLONG(登録商標)(インビトロジェン社(Invitrogen)カリフォルニア州所在)を、核を対比染色し、蛍光退色防止剤として機能するように各試料に1滴ずつ加えた。ニコンコンフォーカルEclipseC−1倒立顕微鏡(ニコン社、日本)及び10〜60倍の対物レンズを使用して画像を得た。
未分化細胞のPCR分析
RNAの抽出、精製及びcDNAの合成:エタノール含有高塩濃度緩衝液の存在下でシリカゲル膜(Rneasy Mini Kit、キアゲン社(Qiagen)カリフォルニア州所在)に結合させた後、洗浄して夾雑物を除去することによりRNA試料を精製した。TURBO DNA−Free Kit(アンビオン社(Ambion, Inc.))を使用してRNAを更に精製し、高品質RNAを水で溶出した。収率及び純度を分光光度計のA260及びA280の指示値によって評価した。ABI(ABI、カリフォルニア州所在)製高容量cDNA Archive Kitを使用して精製したRNAからcDNAコピーを作製した。
核型の分析
H9細胞の核型を、標準的なGバンディング核型分析法によって調べた。全部で100個の中期染色体展開像を評価した(アプライドジェネティクスラボラトリー社(Applied Genetics Laboratories, Inc.))。分析した100個の細胞で染色体異常は見られなかった。細胞遺伝子分析により、常染色体の数は正常であり、染色体数(modal chromosome number)は46本であることが示された。図15は、ヒト胚性幹細胞系H9から得られる典型的な核型を示したものである。
細胞外基質でコーティングした組織培養基質上でのヒト胚性幹細胞の培養
ヒト胚性幹細胞系H1、H7及びH9をウィセル・リサーチ・インスティテュート社(WiCell Research Institute, Inc.)(ウイスコンシン州マディソン所在)より入手し、当該供給元機関によって与えられる指示に従って培養した。簡単に述べると、20%ノックアウト血清リプレースメント、100nM MEM非必須アミノ酸、0.5mM βメルカプトエタノール、2mM L−グルタミンと4ng/mlヒト塩基性繊維芽細胞増殖因子(bFGF)を添加したDMEM/F12(インビトロジェン/ギブコ社(Invitrogen/GIBCO))からなるES細胞培地中、マウス胚繊維芽(MEF)フィーダー細胞上で細胞を培養した。MEF細胞はE13〜13.5のマウス胚から誘導されたものをチャールズリバー社(Charles River)より購入した。MEF細胞は、10%FBS(ハイクローン社(Hyclone))、2mMグルタミン、及び100mM MEM非必須アミノ酸を添加したDMEM培地で増殖させた。サブコンフルエンスに達したMEF細胞培養を、10μg/mlマイトマイシンC(シグマ社(Sigma)ミズーリ州セントルイス所在)で3時間処理して細胞分裂を停止させた後、トリプシン処理し、0.1%ウシゼラチンコートディッシュに2×104/cm2で播いた。継代数2〜4からのMEF細胞をフィーダー層として使用した。MEF細胞のフィーダー層に播いたヒト胚性幹細胞を、加湿した組織培養インキュベーター内で、5%CO2雰囲気中37℃で培養した。コンフルエンスに達した時点(播種後約5〜7日後)で、ヒト胚性幹細胞を1mg/ml IV型コラゲナーゼ(インビトロジェン/ギブコ社(Invitrogen/GIBCO))で5〜10分間処理した後、5mlのガラス製ピペットを用いて表面から静かに掻き取った。細胞を900rpmで5分間遠心し、得られたペレットを再懸濁して希釈率が1:30の増殖因子低減MATRIGEL(商標)(BDバイオサイエンス社(BD Biosciences))でコーティングしたプレート上に1:3〜1:4の細胞比で再播種した。この後、細胞を、8ng/mlのbFGF及びコラゲナーゼを添加したMEF馴化培地中で培養し、MATRIGELでコーティングしたプレート上で少なくとも5代にわたって継代した。MATRIGEL(商標)上で培養した細胞を、IV型コラゲナーゼ(インビトロジェン/ギブコ社(Invitrogen/GIBCO))、ディスパーゼ(BDバイオサイエンス社(BD Biosciences))、又はリベラーゼ酵素(ロシュ社(Roche)、インディアナ州)を用いて日常的に継代した。
細胞外基質でコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
Nature Biotechnology 23,1534〜1541(Dec 2005)に既に記載されているようにして胚性幹細胞を胚体内胚葉に分化させた。簡単に述べると、約60〜70%コンフルエンスのH9培養を、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。H9細胞は希釈率1:30〜1:10の増殖因子低減MATRIGELでコーティングしたプレート上又は希釈率1:30〜1:10の通常のMATRIGEL上で培養した。各プレートは室温で1時間、MATRIGELでコーティングした。
胚体内胚葉形成後のヒト胚性幹細胞における遺伝子発現の変化のマイクロアレイによる分析
RNeasy mini kit(キアゲン社(Qiagen))を使用して以下のヒト胚性幹細胞の培養から全RNAを単離した。即ち、MATRIGELコーティングしたプレート上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P83細胞、及び、MEF細胞上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地で更に3日間処理したH9P44細胞。各群のコントロールには、MATRIGELでコーティングした培養皿に播種し、MEF馴化培地中で培養した細胞、又はMEF上に播種し、ES培地中で培養した細胞を含めた。
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
Nature Biotechnology 23,1534〜1541(Dec 2005)に既に記載されているようにして胚性幹細胞を胚体内胚葉に分化させた。簡単に述べると、約60〜70%コンフルエンスに達した増殖因子低減MATRIGEL(商標)(希釈率1:30)培養上に播いたH9、H7又はH1細胞を、0.5%FBS及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。以下のすべての実施例において、特に断らないかぎり、この処理レジメンを胚体内胚葉(DE)プロトコールと称する。
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−Wntリガンドの役割
H7P44及びH9P46胚性幹細胞を、MATRIGEL(商標)(希釈度1:10)でコーティングした培養皿上で培養し、0.5%FBS及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。培養の一部のものには、20ng/mlのWnt−3a(カタログNo.1324−WN−002、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、20ng/mlのWnt−5a(カタログNo.654−WN−010、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、25ng/mlのWnt−7a(カタログNo.3008−WN−025、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、又は25ng/mlのWnt−5b(カタログNo.3006−WN−025、R&Dシステムズ社(R&D Systems)ミネソタ州所在)を5日間の処理期間の全体を通じて加えた。図24は、高濃度の(A)AA、又は(B)AA+20ng/mlのWnt−3aの存在下でのH9P46胚体内胚葉培養の位相差顕微鏡画像を示したものである。図25は、MATRIGEL(商標)(希釈率1:30)上で培養し、DEプロトコール+Wnt−3a(図25、パネルb及びd)、及び−Wnt−3a(図25、パネルa及びc)に供したH7P44及びH9P46細胞系について、FACSにより調べた5日目のCXCR4の発現を示したものである。Wnt−3aがDE培養中に存在する場合、低血清及び高濃度のAAで処理したDE培養と比較して、CXCR4(CD184)の著明な発現が見られた。図26は、低血清+AA±Wntリガンドで処理した、A)H7及びB)H9の培養についてリアルタイムPCRのデータを示したものである。いずれのH細胞系においても、Wnt−3aを添加することにより、胚体内胚葉マーカーの顕著なアップレギュレーションが認められた。これに対し、Wnt−5a、Wnt−5b及びWnt−7aは、胚体内胚葉マーカーの発現にほとんど影響を及ぼさなかった。
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−Wnt−3aの有効量
H9P46胚性幹細胞を、MATRIGEL(商標)(希釈度1:10)でコーティングした培養皿上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)及び10〜50ng/mlのWnt−3a(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS、100ng/mlアクチビンA(AA)、及び10〜50ng/mlのWnt−3aを添加したDMEM/F12培地で更に3日間処理した。コントロール培養はWnt−3aで処理しなかった。図27において、パネルa)はWnt−3aの非存在下、b)10ng/mlのWnt−3a、c)20ng/mlのWnt−3a、及びd)50ng/mlのWnt−3aの存在下における5日目のFACSによるCXCR4の発現を示したものである。Wnt−3aの非存在下ではCXCR4の発現は極めて低かった。これに対し、10〜50ng/mlのWnt−3aを添加することにより、CXCR4陽性細胞の数が顕著に増加した。更に、10ng/mlのWnt−3aの添加は、50ng/mlのWnt−3aの添加と同様に効果的であった。リアルタイムPCRの結果によってもこの知見が確認された(図28、パネルa)。
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−GSK−3B阻害剤の影響
Wnt−3aの影響がWnt経路を介したものであることを確認するため、GSK−3阻害剤を使用してβカテニン等のWntの下流の標的分子を活性化した。H9P46〜P48胚性幹細胞をMATRIGEL(商標)でコーティングした培養皿(希釈率1:10)上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)、及び10〜1000nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)カリフォルニア州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS、100ng/mlアクチビンA(AA)、及び0〜1000nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)カリフォルニア州所在)を添加したDMEM/F12培地で更に3日間処理した。コントロール培養は、低血清及び高用量のアクチビンA±Wnt−3aで処理した。図29において、パネルaはWnt−3aもGSK−3B阻害剤も加えない場合、b)+20ng/mlのWnt−3a、c)+1000ng/mlのGSK−3B阻害剤IX、d)+500ng/mlのGSK−3B阻害剤IX、e)+100ng/mlのGSK−3B阻害剤IX、f)+10ng/mlのGSK−3B阻害剤IX、g)+100ng/mlのGSK−3B阻害剤IXを1〜2日目、及びh)+10ng/mlのGSK−3B阻害剤IXを1〜2日目に加えた場合の5日目のFACSによるCXCR4の発現を示したものである。
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−GSK−3B阻害剤又はWnt−3aの存在下におけるアクチビンAの有効量
H9P49及びH1P46胚性幹細胞をMATRIGEL(商標)でコーティングした培養皿(希釈率1:10)上で培養し、0.5%FBS、10〜100ng/mlアクチビンA(AA)、及び100nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)又は20ng/mlのWnt−3aを添加したDMEM/F12培地に2日間曝露した後、2%FBS、10〜100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。コントロール培養は、低血清及び100ng/mlのアクチビンAで処理した。図31は、a)10ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、b)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3Aを最初の2日間、c)100ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、d)10ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、e)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3Aを最初の2日間、並びに、f)10ng/mlのアクチビンA及び20ng/mlのWnt−3Aを最初の2日間加えた場合の、5日目のH9P49及びH1P46におけるFACSによるCXCR4の発現を示したものである。図31のパネルa〜dはH9P49細胞のものであり、パネルE〜FはH1P46細胞のものである。図32は、10、50、又は100ng/mlのアクチビンA及び20ng/mlのWnt−3aで処理したH9P49細胞における胚体内胚葉マーカーの遺伝子発現を示したものであり、パネルaはAFP、Bry、CXCR4、GSC、HNF−3B、及びPOU5F(Oct−4)の発現を示し、パネルbはSOX−17及びGATA4の発現を示す。50ng/mlのAA+20ng/mlのWnt−3A又は100nMのGSK−3B阻害剤IXを使用することによって胚体内胚葉マーカーが著明に発現するようである。アクチビンAを低用量で使用すると胚体外内胚葉が形成される。
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−Wnt−3aとGSK−3B阻害剤の組み合わせ
H9P53胚性幹細胞をMATRIGEL(商標)でコーティングした培養皿(希釈率1:30)上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)、及び100nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)を添加し、更に20ng/mlのWnt−3aを加える(+)か、加えない(−)DMEM/F12培地に2日間曝露した後、2%FBS、10〜100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。これと並行して、H9P53培地を、25ng/mlのBMP−4(カタログNo.314−BP−010、R&Dシステムズ社(R&D Systems)ミネソタ州所在)±20ng/mlのWnt−3A±100ng/mlのアクチビンAで処理した。コントロール培養は、低血清及び100ng/mlのアクチビンAで処理した。図33は、a)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間及び25ng/mlのBMP−4を3〜5日目、b)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3aを最初の2日間、c)100ng/mlのアクチビンAを5日間全体、及び100nMのGSK−3B阻害剤IXを最初の2日間、d)20ng/mlのWnt−3a及び25ng/mlのBMP−4を5日間全体、e)100ng/mlのアクチビンAを5日間全体、及び20ng/mlのWnt−3a及び100nMのGSK−3B阻害剤IXを最初の2日間、並びに、f)100ng/mlのアクチビンA及び25ng/mlのBMP−4を5日間全体にわたって加えた場合の、5日目のFACSによるCXCR4の発現を示したものである。図34は、10又は100ng/mlのアクチビンA及び20ng/mlのWnt−3a又は100nMのGSK−3B阻害剤で処理した、継代数46のヒト胚性幹細胞系H1の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現を示す。パネル(a):AFP、Bry、CXCR4、GSC、及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17、HNF−3B及びGATA4の発現。結果は非処理細胞に対する増加倍率として表わす。図35は、50又は100ng/mlのアクチビンA及び10又は100nMのGSK−3B阻害剤で処理した、継代数49のヒト胚性幹細胞系H9の培養についてリアルタイムPCRで調べた胚体内胚葉マーカーの遺伝子発現を示す。パネル(a):AFP、Bry、CXCR4、GSC、HNF−3B及びPOU5F(Oct−4)の発現、並びに、パネル(b):SOX−17及びGATA4の発現。結果は非処理細胞に対する増加倍率として表わす。図36は、アクチビンA、Wnt−3a、GSK−3阻害剤、及びBMP−4の組み合わせで処理したH9P53培養における胚体内胚葉マーカーの遺伝子発現を示す。A)AFP、Bry、CXCR4、GSC、HNF−3B及びSOX7の発現、並びに、B)SOX−17、HNF−3B及びGATA4の発現。DEプロトコールにBMP−4を加えることにより、中胚葉マーカーであるBRYの形成が誘導されるようであるが、Wnt−3AとGSK−4B阻害剤の組み合わせでは、アクチビンAの存在下で各物質を添加した場合と比較して、胚体内胚葉マーカーの顕著なアップレギュレーションは認められなかった。
MEF上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−低血清中でのWnt−3a、アクチビンA、Wnt−5a、BMP−2、BMP−4、BMP−6、BMP−7、IL−4及びSDF−1の組み合わせ
H9P44細胞を、マイトマイシン処理したマウス胚繊維芽細胞(MEF)で予めコーティングした6穴プレート上に播種した。20%ノックアウト血清リプレースメント、100nM MEM非必須アミノ酸、0.5mM βメルカプトエタノール、2mM L−グルタミン(すべてインビトロジェン/ギブコ社(Invitrogen/GIBCO)より入手)、及び8ng/mlヒト塩基性繊維芽細胞増殖因子(bFGF)(R&Dシステムズ社(R&D Systems))を添加したDMEM/F12(インビトロジェン/ギブコ社(Invitrogen/GIBCO))からなるES細胞培地中で70〜80%コンフルエンスに達するまで細胞を培養した。
0日目:0%のFBSをDMEM/F12に加えた。
1日目:0.5%のFBSをDMEM/F12に加えた。
2日目:2%のFBSをDMEM/F12に加えた。
3日目:細胞を採取してFACS分析及びRT−PCRに供した。
1.コントロール:増殖因子を添加しなかった。
2.アクチビンA(100ng/ml)
3.アクチビンA(100ng/ml)+Wnt−3a(10ng/ml)+Wnt5a(10ng/ml)
4.アクチビンA(100ng/ml)+Wnt−3a(10ng/ml)+Wnt5a(10ng/ml)+BMP2(100ng/ml)
5.アクチビンA(100ng/ml)+BMP−4(100ng/ml)
6.アクチビンA(100ng/ml)+BMP−6(100ng/ml)
7.アクチビンA(100ng/ml)+BMP−7(100ng/ml)
8.アクチビンA(100ng/ml)+BMP−4(100ng/ml)+BMP−6(100ng/ml)+BMP−7(100ng/ml)
9.IL−4(10ng/ml)
10.SDF1a(20ng/ml)
11.アクチビンA(100ng/ml)+IL−4(10ng/ml)+SDF1a(20ng/ml)
12.BMP2(100ng/ml)+BMP−4(100ng.ml)+BMP−6(100ng/ml)+BMP−7(100ng/ml)
13.アクチビンA(100ng/ml)BMP−2(100ng/ml)+BMP−4(100ng.ml)+BMP−6(100ng/ml)+BMP−7(100ng/ml)
14.アクチビンA(100ng/ml)+IL−4(10ng/ml)
15.アクチビンA(100ng/ml)+(SDF1a(20ng/ml)
16.アクチビンA(100ng/ml)+Wnt−3a(10ng/ml)+Wnt−5a(10ng/ml)+Wnt−7a(10ng/ml)
17.アクチビンA(100ng/ml)+IL−4(10ng/ml)+SDF1a(20ng/ml)+BMP−4(100ng/ml)
DEプロトコール処理の3日目に細胞を採取した。分析に際しては、処理細胞の一定分量をRT−PCR用のRNAの調製に使用し、残りの細胞をFACS分析に使用した。CXCR4の出現頻度(%)を図37に示す。上記の増殖因子の添加により、CXCR4の発現は、低血清中での100ng/mlのAAによる処理以上には促進されなかった。
MATRIGEL又はヒトフィブロネクチンでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
異なる濃度のMATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
細胞外基質でコーティングした組織培養基質上で培養した後、MEF上で培養したヒト胚性幹細胞の胚体内胚葉への分化−Wnt−3aの役割
少なくとも5代にわたってMATRIGEL(商標)上で培養したヒト胚性幹細胞系H9から得た細胞をES培地中でMEFフィーダー上に播いた。細胞が約60〜70%コンフルエンスに達した時点で、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露し、その後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。更なる処理群として、20ng/mlのWnt−3aで5日間すべて、及び10〜100ng/mlのアクチビンAで処理した群を置いた。
細胞外基質でコーティングした組織培養基質上で培養されたヒト胚性幹細胞の、Wnt阻害剤DKK−1による処理後の胚体内胚葉への分化
Wnt−3aの添加によって分化の増大がもたらされるか否かを調べるため、Wnt−3シグナル伝達の阻害剤を培養に加えた。約60〜70%コンフルエンスのH9培養を、0.5%FBS、20ng/mlのWnt3a、100ng/mlのDikkopf−1(DKK−1)及び100ng/mlのアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlのアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。H9細胞は希釈率1:30の増殖因子低減MATRIGELでコーティングしたプレート上で培養した。各プレートはMATRIGELで1時間、室温でコーティングした。
MATRIGELでコーティングした組織培養基質上で培養し、低血清及びアクチビンA±Wnt−3aで分化させたH9胚性幹細胞におけるDEマーカーの免疫蛍光染色
5日目のH9細胞のDE培養を、実施例10に従ってSOX−17、HNF−3B、GATA−4、N−カドヘリン及びE−カドヘリンについて染色した。すべての核をDAPIで対比染色した。20ng/mlのWnt−3aにより、Wnt−3aの非存在下で分化させた培養と比較して大幅に多数の核がSOX−17、HNF−3β及びGATA−4について陽性に染色された。更に、Wnt−3aの添加により、E−カドヘリンの発現が著しく消失し、N−カドヘリンの発現が昂進した(図43、パネルa及び図43、パネルb)。
MEF又はMATRIGEL上での胚体内胚葉形成後の胚性幹細胞における遺伝子発現の変化のマイクロアレイによる分析
RNeasy mini kit(キアゲン社(Qiagen))を使用して以下の胚性幹細胞の培養から全RNAを単離した。即ち、A)MATRIGEL(商標)コーティングしたプレート(希釈率1:30)上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P33細胞、B)MEF細胞上で培養し、0.5%FBS及び100ng/mlアクチビンAを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P44細胞、及びC)MATRIGEL(商標名)コーティングしたプレート(希釈率1:30)上で培養し、0.5%FBS及び100ng/mlアクチビンA及び20ng/mlのWnt−3aを添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理したH9P48細胞。各群のコントロールには、MATRIGELコーティングした培養皿に播種し、MEF馴化培地中で培養した細胞、又はMEF上に播種し、ES培地中で培養した細胞を含めた。すべての群で3つの生物学的複製物を用い、各生物学的複製物を2個の別々の遺伝子チップ上で繰り返し用いた。
MATRIGELでコーティングした組織培養基質上で培養されたSA002 ES細胞系の胚体内胚葉への分化
8ng/mlのbFGFを添加したMEF−CM中、MATRIGELコーティングしたプレート(希釈率1:30)上で少なくとも3代にわたって予め培養したSA002 P38細胞(セラーティス社(Cellartis)スウェーデン)を、0.5%FBS、及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)±20ng/mlのWnt−3a又は100nMのGSK−3BIX阻害剤を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。リアルタイムPCRの結果を図44のパネルa及びbに示す。H1、H7及びH9細胞系と同様、SA002細胞系も、DEマーカーの顕著な発現にはWnt−3Aの添加をやはり必要とした。図45にCXCR4の発現を示す。a)AAで処理、b)AA+Wnt−3a、c)AA+GSK−3B阻害剤。
ヒト血清でコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化
継代数55のヒト胚性幹細胞系H1の培養をヒト血清(シグマ社(Sigma)No.H1388、ミズーリ州所在)でコーティングしたプレート上で増殖及び分化させた。0.5mlのヒト血清を組織培養処理した6穴培養皿の各ウェルに加え、室温で1時間インキュベートし、ヒト胚性幹細胞を加える前に吸引した。細胞が80%コンフルエンスに達した後、以下のように処理した。即ち、10ng/mlのマウス組み換えWnt3a(R&D)又は100nMのGSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)及び100ng/mlのアクチビンA(R&D)を含む0.5%FBS中で2日間処理した。この後、2%FBS及び100ng/mlアクチビンAで3日間処理した。この後、培養をリアルタイムPCRで分析した(図46、パネルa及びb)。アクチビンAのみで処理した細胞と比較して、アクチビンA+GSK−3B阻害剤又はWnt−3Aで処理した細胞では胚体内胚葉マーカーの著明な発現が認められた。これらの所見は、MATRIGEL(商標)又はヒトフィブロネクチンでコーティングしたプレート上で培養したヒト胚性幹細胞における我々の発見に対応したものである。
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化−異なるGSK−3B阻害剤の評価
ヒト胚性幹細胞からの胚体内胚葉(DE)の形成における市販の多くのGSK−3B阻害剤の有効性を評価した。以下のGSK−3B阻害剤を100nMで評価した。即ち、GSK−3B阻害剤VIII(カタログNo.361549、カルバイオケム社(Calbiochem)、カリフォルニア州所在)、GSK−3B阻害剤IX(カタログNo.361550、カルバイオケム社(Calbiochem)、カリフォルニア州所在)、GSK−3B阻害剤XI(カタログNo.361553、カルバイオケム社(Calbiochem)、カリフォルニア州所在)、GSK−3B阻害剤XII(カタログNo.361554、カルバイオケム社(Calbiochem)、カリフォルニア州所在)。H1P54 ES細胞を、MATRIGEL(商標名)(希釈度1:30)でコーティングした培養皿上で培養し、0.5%FBS、100ng/mlアクチビンA(AA)±各種GSK−3B阻害剤を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。コントロール培養を低血清及び高用量のAAで処理した。図47のパネルa及びbは、5日目における胚体内胚葉マーカーの遺伝子発現を示したものである。GSK−3B阻害剤IX及びXIは、GSK−3B阻害剤VIII及びXIIと比較してDEの形成を誘導するうえでいずれも有効であった。
無フィーダー条件下で培養されたヒト胚性幹細胞による膵臓内胚葉の形成−レチノイン酸類似体の評価
H9P49胚性幹細胞を、MATRIGEL(商標)(希釈度1:30)でコーティングした培養皿上で培養し、0.5%FBS、20ng/mlのWnt−3a(カタログNo.1324−WN−002、R&Dシステムズ社(R&D Systems)ミネソタ州所在)、及び100ng/mlアクチビンA(R&Dシステムズ社(R&D Systems)ミネソタ州所在)を添加したDMEM/F12培地に2日間曝露した後、2%FBS及び100ng/mlアクチビンA(AA)を添加したDMEM/F12培地で更に3日間処理した。5日目に細胞を回収してFACS及びリアルタイムPCRによる評価を行った。上記の実施例において示したように、このプロトコールによりCXCR4及びSOX−17のような胚体内胚葉マーカーが顕著にアップレギュレートされた。得られた5日目の胚体内胚葉細胞を以下の培地条件に曝露して膵臓内胚葉の形成を誘導した。即ち、2%FBS及び1μMのオールトランスレチノイン酸(RA)(カタログNo.R2625、シグマ社(Sigma)、ミズーリ州所在)、又は0.1〜10μMのAM−580(4−[(5,6,7,8−テトラヒドロ−5,5,8,8−テトラメチル−2−ナフタレニル)カルボキサミド]安息香酸、カタログNo.A8843、シグマ社(Sigma)、ミズーリ州所在)、又は0.1〜1μMのTTNPB(4−[(E)−2−(5,6,7,8−テトラヒドロ−5,5,8,8−テトラメチル−2−ナフタレニル)−1−プロペニル]安息香酸(アロチノイド酸)、カタログNo.T3757、シグマ社(Sigma)、ミズーリ州所在)を添加したDMEM/F12培地中で3日間培養した。AM−580及びTTNPBは、レチノイン酸受容体に対する親和性を有するレチノイン酸類似体である。RA処理の後、2%FBS及び20〜50ng/mlのbFGF(カタログNo.F0291、シグマ社(Sigma)、ミズーリ州所在)を添加したDMEM/F12培地中で更に3日間処理した。細胞を収穫し、mRNAの試料を採取して分析に供した。
ヒト胚性幹細胞におけるサイトカイン発現に対するWnt−3a処理の影響
Wnt−3aによる処理がサイトカイン発現に与える影響についてタンパク質アレイを用いて分析を行った。ヒト胚性幹細胞系H9の細胞を実施例15で述べた方法に従って培養した。継代数54において、0.5%FBSを含むDMEM/F12中、100ng/mlのアクチビンA±10ng/mlのWnt−3aの存在下で細胞を2日間分化させた。この後、100ng/mlのアクチビンA及び2%FBSを含むDMEM/F12中で細胞を更に3日間培養した。5日目の終わりに、CXCR4の発現を各処理群についてFACSにより調べた。アクチビンAのみで処理した細胞ではCXCR4を発現した細胞は1%に過ぎなかった。アクチビンA及びWnt3aで処理した細胞では、73%の細胞がCXCR4の発現について陽性であった。
MATRIGELでコーティングした組織培養基質上で培養されたヒト胚性幹細胞の胚体内胚葉への分化:Wnt1の役割
H1P55 ES細胞を、MATRIGEL(商標)(希釈度1:30)でコーティングした培養皿上で培養し、0.5%FBS及び100ng/mlのアクチビンA±10〜20ng/mlのWnt−1(ペプロテック社(PeproTech)、ニュージャージー州所在、カタログNo.120−17)を添加したDMEM/F12培地に2日間曝露した後、2%FBS、100ng/mlアクチビンA(AA)±10又は20ng/mlのWnt−1を添加したDMEM/F12培地で更に3日間処理した。以下のWnt1+AAの組み合わせについて試験を行った。
膵臓内分泌分化に対するグルコースの影響
膵臓内胚葉細胞の膵臓内分泌細胞への分化の効率は、例えば基本培地の選択、又はグルコースの濃度等の多くの因子に依存する。胚性幹細胞から誘導された膵臓内胚葉細胞の膵臓内分泌細胞への分化に対するグルコース濃度の影響を調べた。
(1) 膵臓内分泌系統に特徴的なマーカーを発現する細胞を生成するための方法において、膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMの濃度のグルコースを添加した培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させること、を含む、方法。
(2) 前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、多能性幹細胞から分化させられる、実施態様1に記載の方法。
(3) 前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞が、胚体内胚葉系統に特徴的なマーカーを発現する細胞から誘導される、実施態様1に記載の方法。
(4) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞が、多能性幹細胞から誘導される、実施態様1に記載の方法。
(5) 前記多能性幹細胞が、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に更に分化させられる前に胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させられる、実施態様2に記載の方法。
(6) 前記グルコースが約10mMの濃度で使用される、実施態様1に記載の方法。
(7) 前記グルコースが約20mMの濃度で使用される、実施態様1に記載の方法。
(8) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約30日間処理する、実施態様1に記載の方法。
(9) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約20日間処理する、実施態様1に記載の方法。
(10) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約10日間処理する、実施態様1に記載の方法。
(12) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約4日間処理する、実施態様1に記載の方法。
(13) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間処理する、実施態様1に記載の方法。
(14) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様6に記載の方法。
(15) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様7に記載の方法。
(16) 多能性幹細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させるための方法において、
a.前記多能性幹細胞を培養する工程と、
b.前記多能性細胞をアクチビンAで処理することによって、前記多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、少なくとも1種類の繊維芽細胞増殖因子、又はレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理することによって、前記胚体内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内胚葉系統に特徴的なマーカーを発現する細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、約10mM〜約20mMの濃度のグルコースを添加した培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現する細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させる工程と、を含む、方法。
(17) 前記グルコースが約10mMの濃度で使用される、実施態様16に記載の方法。
(18) 前記グルコースが約20mMの濃度で使用される、実施態様16に記載の方法。
(19) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約30日間処理する、実施態様16に記載の方法。
(20) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間〜約20日間処理する、実施態様16に記載の方法。
(22) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約10日間処理する、実施態様16に記載の方法。
(23) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約4日間処理する、実施態様16に記載の方法。
(24) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって約2日間処理する、実施態様16に記載の方法。
(25) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様21に記載の方法。
(26) 前記膵臓内分泌系統に特徴的なマーカーを発現する細胞を、グルコースによって約2日間〜約30日間処理する、実施態様22に記載の方法。
(27) 糖尿病を有する患者に実施態様1に記載の細胞を移植することによって前記患者を治療するための方法。
(28) 糖尿病を有する患者に実施態様16に記載の細胞を移植することによって前記患者を治療するための方法。
(29) 前記多能性幹細胞が胚性幹細胞である、実施態様16に記載の方法。
(30) 前記胚性幹細胞がヒト胚性幹細胞である、実施態様29に記載の方法。
Claims (26)
- 膵臓内分泌系統に特徴的なマーカーを発現するヒトの細胞を生成するための方法において、膵臓内胚葉系統に特徴的なマーカーを発現するヒトの細胞を、グルコースを添加した、ニコチンアミドを含まない培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって2〜30日の間処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現するヒトの細胞を、膵臓内分泌系統に特徴的なマーカーを発現する細胞に分化させること、を含み、前記グルコースの濃度は、10mM〜20mMの間で調整する、方法。
- 請求項1に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現するヒトの細胞が、ヒトの多能性幹細胞から分化させられる、方法。
- 請求項1に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞が、前記胚体内胚葉系統に特徴的なマーカーを発現する細胞から誘導される、方法。
- 請求項1に記載の方法において、前記膵臓内分泌系統に特徴的なマーカーを発現する前記ヒトの細胞が、ヒトの多能性幹細胞から誘導される、方法。
- 請求項2に記載の方法において、前記ヒトの多能性幹細胞が、前記膵臓内胚葉系統に特徴的なマーカーを発現するヒトの細胞に更に分化させられる前に胚体内胚葉系統に特徴的なマーカーを発現するヒトの細胞に分化させられる、方法。
- 請求項1に記載の方法において、前記グルコースが10mMの濃度で使用される、方法。
- 請求項1に記載の方法において、前記グルコースが20mMの濃度で使用される、方法。
- 請求項1に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって2日間〜20日間処理する、方法。
- 請求項1に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって2日間〜10日間処理する、方法。
- 請求項1に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって10日間処理する、方法。
- 請求項1に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって4日間処理する、方法。
- 請求項1に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって2日間処理する、方法。
- 請求項6に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、グルコースによって2日間〜30日間処理する、方法。
- 請求項7に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、グルコースによって2日間〜30日間処理する、方法。
- ヒトの多能性幹細胞を、膵臓内分泌系統に特徴的なマーカーを発現するヒトの細胞に分化させるための方法において、
a.前記ヒトの多能性幹細胞を培養する工程と、
b.前記ヒトの多能性細胞をアクチビンAで処理することによって、前記ヒトの多能性幹細胞を、胚体内胚葉系統に特徴的なマーカーを発現するヒトの細胞に分化させる工程と、
c.前記胚体内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、少なくとも1種類の繊維芽細胞増殖因子、又はレチノイン酸及び少なくとも1種類の繊維芽細胞増殖因子で処理することによって、前記胚体内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、膵臓内胚葉系統に特徴的なマーカーを発現するヒトの細胞に分化させる工程と、
d.前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、グルコースを添加した、ニコチンアミドを含まない培地中で、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって2〜30日の間処理することによって、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、膵臓内分泌系統に特徴的なマーカーを発現するヒトの細胞に分化させる工程と、を含み、前記グルコースの濃度は、10mM〜20mMの間で調整する、
方法。 - 請求項15に記載の方法において、前記グルコースが10mMの濃度で使用される、方法。
- 請求項15に記載の方法において、前記グルコースが20mMの濃度で使用される、方法。
- 請求項15に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって2日間〜20日間処理する、方法。
- 請求項15に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって2日間〜10日間処理する、方法。
- 請求項15に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって10日間処理する、方法。
- 請求項15に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって4日間処理する、方法。
- 請求項15に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、γセクレターゼ阻害剤、又はエキセンジン4、又はエキセンジン4及びHGFによって2日間処理する、方法。
- 請求項19に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、グルコースによって2日間〜30日間処理する、方法。
- 請求項20に記載の方法において、前記膵臓内胚葉系統に特徴的なマーカーを発現する前記ヒトの細胞を、グルコースによって2日間〜30日間処理する、方法。
- 請求項15に記載の方法において、前記ヒトの多能性幹細胞がヒトの胚性幹細胞である、方法。
- 請求項25に記載の方法において、前記ヒト胚性幹細胞が、H1及びH9からなる群の細胞系から誘導される、方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US95317807P | 2007-07-31 | 2007-07-31 | |
| US60/953,178 | 2007-07-31 | ||
| PCT/US2008/071782 WO2009018453A1 (en) | 2007-07-31 | 2008-07-31 | Differentiation of human embryonic stem cells |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015094014A Division JP2015144615A (ja) | 2007-07-31 | 2015-05-01 | ヒト胚性幹細胞の分化 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010535036A JP2010535036A (ja) | 2010-11-18 |
| JP5769965B2 true JP5769965B2 (ja) | 2015-08-26 |
Family
ID=40084239
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010520194A Active JP5769965B2 (ja) | 2007-07-31 | 2008-07-31 | ヒト胚性幹細胞の分化 |
| JP2015094014A Pending JP2015144615A (ja) | 2007-07-31 | 2015-05-01 | ヒト胚性幹細胞の分化 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015094014A Pending JP2015144615A (ja) | 2007-07-31 | 2015-05-01 | ヒト胚性幹細胞の分化 |
Country Status (11)
| Country | Link |
|---|---|
| US (4) | US9096832B2 (ja) |
| EP (2) | EP2610336A1 (ja) |
| JP (2) | JP5769965B2 (ja) |
| KR (1) | KR101617243B1 (ja) |
| CN (1) | CN101952415B (ja) |
| BR (1) | BRPI0814894A2 (ja) |
| CA (3) | CA2695225C (ja) |
| DK (1) | DK2185693T3 (ja) |
| MX (1) | MX2010001335A (ja) |
| RU (1) | RU2473685C2 (ja) |
| WO (1) | WO2009018453A1 (ja) |
Families Citing this family (78)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8017395B2 (en) | 2004-12-17 | 2011-09-13 | Lifescan, Inc. | Seeding cells on porous supports |
| US9074189B2 (en) | 2005-06-08 | 2015-07-07 | Janssen Biotech, Inc. | Cellular therapy for ocular degeneration |
| US8741643B2 (en) | 2006-04-28 | 2014-06-03 | Lifescan, Inc. | Differentiation of pluripotent stem cells to definitive endoderm lineage |
| US9080145B2 (en) | 2007-07-01 | 2015-07-14 | Lifescan Corporation | Single pluripotent stem cell culture |
| KR101617243B1 (ko) | 2007-07-31 | 2016-05-02 | 라이프스캔, 인코포레이티드 | 인간 배아 줄기 세포의 분화 |
| EP2229434B1 (en) | 2007-11-27 | 2011-09-07 | Lifescan, Inc. | Differentiation of human embryonic stem cells |
| CN102046779A (zh) | 2008-02-21 | 2011-05-04 | 森托科尔奥索生物科技公司 | 用于细胞粘附、培养和分离的方法、表面改性培养板和组合物 |
| US7939322B2 (en) * | 2008-04-24 | 2011-05-10 | Centocor Ortho Biotech Inc. | Cells expressing pluripotency markers and expressing markers characteristic of the definitive endoderm |
| US8623648B2 (en) | 2008-04-24 | 2014-01-07 | Janssen Biotech, Inc. | Treatment of pluripotent cells |
| PL2310492T3 (pl) | 2008-06-30 | 2015-12-31 | Janssen Biotech Inc | Różnocowanie pluripotencjalnych komórek macierzystych |
| AU2009308967C1 (en) | 2008-10-31 | 2017-04-20 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells to the pancreatic endocrine lineage |
| KR102025158B1 (ko) | 2008-10-31 | 2019-09-25 | 얀센 바이오테크 인코포레이티드 | 인간 배아 줄기 세포의 췌장 내분비 계통으로의 분화 |
| AU2009316583B2 (en) | 2008-11-20 | 2016-04-21 | Janssen Biotech, Inc. | Methods and compositions for cell attachment and cultivation on planar substrates |
| MX356756B (es) | 2008-11-20 | 2018-06-11 | Centocor Ortho Biotech Inc | Células madre pluripotentes en microportadores. |
| RU2576003C2 (ru) | 2008-12-03 | 2016-02-27 | Интернэшнл Стем Селл Корпорейшн | Способ получения дифференцированных клеток из стволовых клеток |
| EP2233566A1 (en) * | 2009-03-17 | 2010-09-29 | Vrije Universiteit Brussel | Generation of pancreatic progenitor cells |
| SG177416A1 (en) | 2009-07-20 | 2012-02-28 | Janssen Biotech Inc | Differentiation of human embryonic stem cells |
| AU2010276440B2 (en) | 2009-07-20 | 2014-07-03 | Janssen Biotech Inc. | Differentiation of human embryonic stem cells |
| EP2456862A4 (en) | 2009-07-20 | 2013-02-27 | Janssen Biotech Inc | DIFFERENTIATION OF HUMAN EMBRYONIC STEM CELLS |
| US8754890B2 (en) * | 2009-07-31 | 2014-06-17 | Oracle International Corporation | Graphical interface with data presence indicators |
| SG10201407102QA (en) * | 2009-10-30 | 2014-11-27 | Univ North Carolina | Multipotent stem cells from the extrahepatic billary tree and methods of isolating same |
| WO2011064549A2 (en) * | 2009-11-27 | 2011-06-03 | Stem Cells For Safer Medicines Limited | Composition and method for differentiation of human embryonic stem cells |
| CN102712902B (zh) * | 2009-12-23 | 2019-01-08 | 詹森生物科技公司 | 人胚胎干细胞的分化 |
| CN102741395B (zh) | 2009-12-23 | 2016-03-16 | 詹森生物科技公司 | 人胚胎干细胞的分化 |
| US8932853B2 (en) * | 2009-12-29 | 2015-01-13 | Takeda Pharmaceutical Company Limited | Method for manufacturing pancreatic-hormone-producing cells |
| RU2702198C2 (ru) | 2010-03-01 | 2019-10-04 | Янссен Байотек, Инк. | Способы очистки клеток, производных от плюрипотентных стволовых клеток |
| RU2587634C2 (ru) | 2010-05-12 | 2016-06-20 | Янссен Байотек, Инк. | Дифференцирование эмбриональных стволовых клеток человека |
| RU2576000C2 (ru) * | 2010-08-09 | 2016-02-27 | Такеда Фармасьютикал Компани Лимитед | Способ получения клеток, продуцирующих панкреатические гормоны |
| AU2015213422A1 (en) * | 2010-08-12 | 2015-09-10 | Janssen Biotech, Inc. | Treatment of diabetes with pancreatic endocrine precursor cells |
| JP2013533319A (ja) * | 2010-08-12 | 2013-08-22 | ヤンセン バイオテツク,インコーポレーテツド | 膵内分泌腺前駆体細胞による糖尿病の治療 |
| BR112013004614A2 (pt) | 2010-08-31 | 2024-01-16 | Janssen Biotech Inc | Diferenciação de células-tronco pluripotentes |
| KR101851956B1 (ko) | 2010-08-31 | 2018-04-25 | 얀센 바이오테크 인코포레이티드 | 인간 배아 줄기 세포의 분화 |
| WO2012030539A2 (en) | 2010-08-31 | 2012-03-08 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells |
| WO2012056997A1 (ja) | 2010-10-28 | 2012-05-03 | 国立大学法人熊本大学 | 多能性幹細胞の分化誘導効率を改善するための方法及び培地 |
| KR101259074B1 (ko) * | 2010-11-23 | 2013-04-29 | 서울대학교산학협력단 | 분비신호펩타이드가 연결된 엑센딘-4를 도입한 췌장소도세포 및 이를 이용한 당뇨병 치료방법 |
| US20140234963A1 (en) * | 2011-06-21 | 2014-08-21 | Novo Nordisk A/S | Efficient induction of definitive endoderm from pluripotent stem cells |
| US20140242038A1 (en) * | 2011-10-11 | 2014-08-28 | The Trustees Of Columbia University In The City Of New York | Method for generating beta cells |
| WO2013062140A1 (en) * | 2011-10-28 | 2013-05-02 | Kyoto University | Method for efficiently inducing differentiation of pluripotent stem cells into hepatic lineage cells |
| HUE041929T2 (hu) | 2011-12-06 | 2019-06-28 | Astellas Inst For Regenerative Medicine | Eljárás szaruhártya endotél sejtek irányított differenciálására |
| KR102203056B1 (ko) * | 2011-12-22 | 2021-01-14 | 얀센 바이오테크 인코포레이티드 | 인간 배아 줄기 세포의 단일 인슐린 호르몬 양성 세포로의 분화 |
| BR112014019328A8 (pt) * | 2012-02-17 | 2017-07-11 | The Schepens Eye Res Institute | Perfil de fenótipo de células progenitoras retinais humanas |
| SG11201405052RA (en) | 2012-03-07 | 2014-10-30 | Janssen Biotech Inc | Defined media for expansion and maintenance of pluripotent stem cells |
| EP2847319A4 (en) * | 2012-05-07 | 2015-12-16 | Janssen Biotech Inc | DIFFERENTIATION OF HUMAN EMBRYONAL STEM CELLS IN THE PANCREAS ENDODERM |
| CN108034633B (zh) | 2012-06-08 | 2022-08-02 | 詹森生物科技公司 | 人胚胎干细胞向胰腺内分泌细胞的分化 |
| US20150247123A1 (en) * | 2012-09-03 | 2015-09-03 | Novo Nordisk A/S | Generation of pancreatic endoderm from Pluripotent Stem cells using small molecules |
| CN103710302B (zh) * | 2012-09-28 | 2016-10-19 | 中国医学科学院基础医学研究所 | 制备胰腺干细胞的方法及其专用培养基组合 |
| US10370644B2 (en) | 2012-12-31 | 2019-08-06 | Janssen Biotech, Inc. | Method for making human pluripotent suspension cultures and cells derived therefrom |
| BR112015015714A2 (pt) | 2012-12-31 | 2017-07-11 | Janssen Biotech Inc | suspensão e aglomeração de células pluripotentes humanas para diferenciação em célu-las endócrinas pancreáticas |
| AU2013368224B2 (en) | 2012-12-31 | 2018-09-27 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells into pancreatic endocrine cells using HB9 regulators |
| EP2938724B1 (en) | 2012-12-31 | 2020-10-28 | Janssen Biotech, Inc. | Culturing of human embryonic stem cells at the air-liquid interface for differentiation into pancreatic endocrine cells |
| CN103194424A (zh) * | 2013-03-28 | 2013-07-10 | 于涛 | 一种诱导胚胎干细胞为胰腺组织样细胞的方法 |
| MX2015017103A (es) | 2013-06-11 | 2016-11-07 | Harvard College | Celulas beta derivadas de células madre ( sc-beta ) y composiciones y métodos para generarlas. |
| EP3143127B1 (en) | 2014-05-16 | 2021-07-14 | Janssen Biotech, Inc. | Use of small molecules to enhance mafa expression in pancreatic endocrine cells |
| EP3147356B1 (en) * | 2014-05-20 | 2024-03-13 | Tokyo Institute of Technology | Method for inducing differentiation of insulin-producing cells |
| EP3234110B1 (en) | 2014-12-18 | 2024-02-28 | President and Fellows of Harvard College | METHODS FOR GENERATING STEM CELL-DERIVED ß CELLS AND USES THEREOF |
| WO2016100930A1 (en) | 2014-12-18 | 2016-06-23 | President And Fellows Of Harvard College | Methods for generating stem cell-derived b cells and methods of use thereof |
| WO2016100898A1 (en) | 2014-12-18 | 2016-06-23 | President And Fellows Of Harvard College | Serum-free in vitro directed differentiation protocol for generating stem cell-derived b cells and uses thereof |
| CN104694462B (zh) * | 2015-03-17 | 2018-02-13 | 奥思达干细胞有限公司 | 一种胚胎干细胞定向诱导分化为肝细胞的方法 |
| CN107787363B (zh) | 2015-04-24 | 2021-06-25 | 哥本哈根大学 | 真正的胰腺祖细胞的分离 |
| CN106554936B (zh) * | 2015-09-30 | 2020-05-05 | 海门雨霖细胞科技有限责任公司 | 诱导人干细胞向肝细胞定向分化的新方法 |
| MA45479A (fr) | 2016-04-14 | 2019-02-20 | Janssen Biotech Inc | Différenciation de cellules souches pluripotentes en cellules de l'endoderme de l'intestin moyen |
| US10767164B2 (en) | 2017-03-30 | 2020-09-08 | The Research Foundation For The State University Of New York | Microenvironments for self-assembly of islet organoids from stem cells differentiation |
| CA3061915A1 (en) * | 2017-06-27 | 2019-01-03 | Trustees Of Boston University | Methods and compositions related to differentiated lung cells |
| US10391156B2 (en) | 2017-07-12 | 2019-08-27 | Viacyte, Inc. | University donor cells and related methods |
| CA2983845C (en) | 2017-10-26 | 2024-01-30 | University Of Copenhagen | Generation of glucose-responsive beta cells |
| IL305391B2 (en) | 2017-11-15 | 2024-09-01 | Vertex Pharma | Preparations for the production of islet cells and methods of use |
| US10426424B2 (en) | 2017-11-21 | 2019-10-01 | General Electric Company | System and method for generating and performing imaging protocol simulations |
| EP3833365A4 (en) | 2018-08-10 | 2022-05-11 | Vertex Pharmaceuticals Incorporated | ISLE DIFFERENTIATION DERIVED FROM STEM CELLS |
| US10724052B2 (en) | 2018-09-07 | 2020-07-28 | Crispr Therapeutics Ag | Universal donor cells |
| CN114401752B (zh) | 2019-05-31 | 2023-04-04 | W.L.戈尔及同仁股份有限公司 | 具有受控氧扩散距离的细胞封装装置 |
| AU2020282355B2 (en) | 2019-05-31 | 2023-11-02 | Viacyte, Inc. | A biocompatible membrane composite |
| CA3139590C (en) | 2019-05-31 | 2024-01-23 | W. L. Gore & Associates, Inc. | A biocompatible membrane composite |
| EP3975926A1 (en) | 2019-05-31 | 2022-04-06 | W.L. Gore & Associates, Inc. | A biocompatible membrane composite |
| KR20220058579A (ko) | 2019-09-05 | 2022-05-09 | 크리스퍼 테라퓨틱스 아게 | 보편적 공여자 세포 |
| CA3150235A1 (en) | 2019-09-05 | 2021-03-11 | Alireza Rezania | UNIVERSAL DONOR CELLS |
| JP2023537327A (ja) | 2020-07-31 | 2023-08-31 | バーテックス ファーマシューティカルズ インコーポレイテッド | 膵内分泌細胞の分化 |
| AU2021414617A1 (en) | 2020-12-31 | 2023-08-10 | Crispr Therapeutics Ag | Universal donor cells |
| IL311636A (en) | 2021-11-01 | 2024-05-01 | Vertex Pharma | Differentiation of stem cell-derived pancreatic islets |
Family Cites Families (274)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3209652A (en) | 1961-03-30 | 1965-10-05 | Burgsmueller Karl | Thread whirling method |
| AT326803B (de) | 1968-08-26 | 1975-12-29 | Binder Fa G | Maschenware sowie verfahren zur herstellung derselben |
| US3935067A (en) | 1974-11-22 | 1976-01-27 | Wyo-Ben Products, Inc. | Inorganic support for culture media |
| CA1201400A (en) | 1982-04-16 | 1986-03-04 | Joel L. Williams | Chemically specific surfaces for influencing cell activity during culture |
| US4499802A (en) | 1982-09-29 | 1985-02-19 | Container Graphics Corporation | Rotary cutting die with scrap ejection |
| US4537773A (en) | 1983-12-05 | 1985-08-27 | E. I. Du Pont De Nemours And Company | α-Aminoboronic acid derivatives |
| DE3401610A1 (de) * | 1984-01-18 | 1985-07-18 | Siemens AG, 1000 Berlin und 8000 München | Integrierte halbleiterschaltung mit einem ringoszillator |
| US4557264A (en) | 1984-04-09 | 1985-12-10 | Ethicon Inc. | Surgical filament from polypropylene blended with polyethylene |
| US5089396A (en) | 1985-10-03 | 1992-02-18 | Genentech, Inc. | Nucleic acid encoding β chain prodomains of inhibin and method for synthesizing polypeptides using such nucleic acid |
| US5215893A (en) | 1985-10-03 | 1993-06-01 | Genentech, Inc. | Nucleic acid encoding the ba chain prodomains of inhibin and method for synthesizing polypeptides using such nucleic acid |
| US4737578A (en) | 1986-02-10 | 1988-04-12 | The Salk Institute For Biological Studies | Human inhibin |
| US5863531A (en) | 1986-04-18 | 1999-01-26 | Advanced Tissue Sciences, Inc. | In vitro preparation of tubular tissue structures by stromal cell culture on a three-dimensional framework |
| US5759830A (en) | 1986-11-20 | 1998-06-02 | Massachusetts Institute Of Technology | Three-dimensional fibrous scaffold containing attached cells for producing vascularized tissue in vivo |
| CA1340581C (en) | 1986-11-20 | 1999-06-08 | Joseph P. Vacanti | Chimeric neomorphogenesis of organs by controlled cellular implantation using artificial matrices |
| US5567612A (en) | 1986-11-20 | 1996-10-22 | Massachusetts Institute Of Technology | Genitourinary cell-matrix structure for implantation into a human and a method of making |
| NZ229354A (en) | 1988-07-01 | 1990-09-26 | Becton Dickinson Co | Treating polymer surfaces with a gas plasma and then applying a layer of endothelial cells to the surface |
| EP0363125A3 (en) | 1988-10-03 | 1990-08-16 | Hana Biologics Inc. | Proliferated pancreatic endocrine cell product and process |
| SU1767433A1 (ru) | 1989-11-27 | 1992-10-07 | Пермский государственный медицинский институт | Способ определени инсулинорезистентности имунного генеза у больных сахарным диабетом I типа |
| US5837539A (en) | 1990-11-16 | 1998-11-17 | Osiris Therapeutics, Inc. | Monoclonal antibodies for human mesenchymal stem cells |
| DK0628639T3 (da) | 1991-04-25 | 2000-01-24 | Chugai Pharmaceutical Co Ltd | Rekonstitueret humant antistof mod human interleukin-6-receptor |
| US5449383A (en) | 1992-03-18 | 1995-09-12 | Chatelier; Ronald C. | Cell growth substrates |
| GB9206861D0 (en) | 1992-03-28 | 1992-05-13 | Univ Manchester | Wound healing and treatment of fibrotic disorders |
| CA2114282A1 (en) | 1993-01-28 | 1994-07-29 | Lothar Schilder | Multi-layered implant |
| JP3525221B2 (ja) | 1993-02-17 | 2004-05-10 | 味の素株式会社 | 免疫抑制剤 |
| JP2813467B2 (ja) | 1993-04-08 | 1998-10-22 | ヒューマン・セル・カルチャーズ・インコーポレーテッド | 細胞培養法および培地 |
| US5523226A (en) * | 1993-05-14 | 1996-06-04 | Biotechnology Research And Development Corp. | Transgenic swine compositions and methods |
| GB9310557D0 (en) | 1993-05-21 | 1993-07-07 | Smithkline Beecham Plc | Novel process and apparatus |
| TW257671B (ja) | 1993-11-19 | 1995-09-21 | Ciba Geigy | |
| US6001647A (en) | 1994-04-28 | 1999-12-14 | Ixion Biotechnology, Inc. | In vitro growth of functional islets of Langerhans and in vivo uses thereof |
| US5834308A (en) | 1994-04-28 | 1998-11-10 | University Of Florida Research Foundation, Inc. | In vitro growth of functional islets of Langerhans |
| US6703017B1 (en) | 1994-04-28 | 2004-03-09 | Ixion Biotechnology, Inc. | Reversal of insulin-dependent diabetes by islet-producing stem cells, islet progenitor cells and islet-like structures |
| US5840832A (en) | 1994-10-21 | 1998-11-24 | The Johns Hopkins University | Transcription factor regulating MHC expression, CDNA and genomic clones encoding same and retroviral expression constructs thereof |
| US6083903A (en) | 1994-10-28 | 2000-07-04 | Leukosite, Inc. | Boronic ester and acid compounds, synthesis and uses |
| JP4079461B2 (ja) | 1994-12-29 | 2008-04-23 | 中外製薬株式会社 | Il−6アンタゴニストを含んでなる抗腫瘍剤の作用増強剤 |
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US5718922A (en) | 1995-05-31 | 1998-02-17 | Schepens Eye Research Institute, Inc. | Intravitreal microsphere drug delivery and method of preparation |
| US5908782A (en) | 1995-06-05 | 1999-06-01 | Osiris Therapeutics, Inc. | Chemically defined medium for human mesenchymal stem cells |
| US5681561A (en) | 1995-06-07 | 1997-10-28 | Life Medical Sciences, Inc. | Compositions and methods for improving autologous fat grafting |
| JP2001508302A (ja) | 1997-01-10 | 2001-06-26 | ライフ テクノロジーズ,インコーポレイテッド | 胚性幹細胞血清置換 |
| UA65572C2 (en) | 1997-04-24 | 2004-04-15 | Ortho Mcneil Pharm Inc | Substituted imidazoles, intermediate compounds for the preparation thereof, a method for the preparation of substituted imidazoles and a method for the treatment of inflammatory diseases |
| DK1028737T3 (da) | 1997-07-03 | 2007-08-13 | Osiris Therapeutics Inc | Humane mesenchymale stamceller fra perifert blod |
| US6670127B2 (en) | 1997-09-16 | 2003-12-30 | Egea Biosciences, Inc. | Method for assembly of a polynucleotide encoding a target polypeptide |
| WO1999014318A1 (en) | 1997-09-16 | 1999-03-25 | Board Of Regents, The University Of Texas System | Method for the complete chemical synthesis and assembly of genes and genomes |
| JP3880795B2 (ja) | 1997-10-23 | 2007-02-14 | ジェロン・コーポレーション | フィーダー細胞を含まない培養物中で、霊長類由来始原幹細胞を増殖させるための方法 |
| AR014195A1 (es) | 1997-12-29 | 2001-02-07 | Ortho Mcneil Pharm Inc | Compuestos de trifenilpropanamida utiles para el tratamiento de procesos inflamatorios, composiciones anti-inflamatorias que los comprenden, ymetodos para prepararlos |
| US6328960B1 (en) | 1998-03-18 | 2001-12-11 | Osiris Therapeutics, Inc. | Mesenchymal stem cells for prevention and treatment of immune responses in transplantation |
| CZ302070B6 (cs) * | 1998-04-21 | 2010-09-29 | Micromet Ag | Jednoretezcový multifunkcní polypeptid, polynukleotid, vektor obsahující tento polynukleotid, bunka transfekovaná tímto polynukleotidem, prostredek obsahující tento polypeptid, polynukleotid nebo vektor a jejich použití a zpusob identifikace aktiváto |
| MY132496A (en) | 1998-05-11 | 2007-10-31 | Vertex Pharma | Inhibitors of p38 |
| US6413773B1 (en) | 1998-06-01 | 2002-07-02 | The Regents Of The University Of California | Phosphatidylinositol 3-kinase inhibitors as stimulators of endocrine differentiation |
| US6667176B1 (en) | 2000-01-11 | 2003-12-23 | Geron Corporation | cDNA libraries reflecting gene expression during growth and differentiation of human pluripotent stem cells |
| US7410798B2 (en) | 2001-01-10 | 2008-08-12 | Geron Corporation | Culture system for rapid expansion of human embryonic stem cells |
| US6610540B1 (en) | 1998-11-18 | 2003-08-26 | California Institute Of Technology | Low oxygen culturing of central nervous system progenitor cells |
| US6413556B1 (en) | 1999-01-08 | 2002-07-02 | Sky High, Llc | Aqueous anti-apoptotic compositions |
| EP1144597A2 (en) | 1999-01-21 | 2001-10-17 | Vitro Diagnostics, Inc. | Immortalized cell lines and methods of making the same |
| US6800460B1 (en) | 1999-03-11 | 2004-10-05 | Schering Corporation | Mammalian cytokine complexes |
| US6815203B1 (en) | 1999-06-23 | 2004-11-09 | Joslin Diabetes Center, Inc. | Methods of making pancreatic islet cells |
| US6333029B1 (en) | 1999-06-30 | 2001-12-25 | Ethicon, Inc. | Porous tissue scaffoldings for the repair of regeneration of tissue |
| US6306424B1 (en) | 1999-06-30 | 2001-10-23 | Ethicon, Inc. | Foam composite for the repair or regeneration of tissue |
| US6333298B1 (en) * | 1999-07-16 | 2001-12-25 | Infineum International Limited | Molybdenum-free low volatility lubricating oil composition |
| EP1224259A4 (en) | 1999-09-27 | 2005-04-27 | Univ Florida | INVERSION OF INSULIN DEPENDENT DIABETES BY ISOLATED STEM CELLS, PROGENITOR ISLANDIC CELLS, AND INSULAR TYPE STRUCTURES |
| US6685936B2 (en) | 1999-10-12 | 2004-02-03 | Osiris Therapeutics, Inc. | Suppressor cells induced by culture with mesenchymal stem cells for treatment of immune responses in transplantation |
| US20030082155A1 (en) * | 1999-12-06 | 2003-05-01 | Habener Joel F. | Stem cells of the islets of langerhans and their use in treating diabetes mellitus |
| US6753153B2 (en) | 1999-12-13 | 2004-06-22 | The Scripps Research Institute | Markers for identification and isolation of pancreatic islet α and β progenitors |
| US7005252B1 (en) | 2000-03-09 | 2006-02-28 | Wisconsin Alumni Research Foundation | Serum free cultivation of primate embryonic stem cells |
| US7439064B2 (en) | 2000-03-09 | 2008-10-21 | Wicell Research Institute, Inc. | Cultivation of human embryonic stem cells in the absence of feeder cells or without conditioned medium |
| US6436704B1 (en) | 2000-04-10 | 2002-08-20 | Raven Biotechnologies, Inc. | Human pancreatic epithelial progenitor cells and methods of isolation and use thereof |
| US6458589B1 (en) | 2000-04-27 | 2002-10-01 | Geron Corporation | Hepatocyte lineage cells derived from pluripotent stem cells |
| KR100947577B1 (ko) | 2000-06-26 | 2010-03-15 | 엔씨 메디컬 리서치 가부시키가이샤 | 신경계 세포로 분화할 수 있는 세포를 포함하는 세포분획 |
| DK1333833T3 (da) | 2000-10-23 | 2011-12-12 | Glaxosmithkline Llc | Ny trisubstitueret 8H-pyridol[2,3-d]pyrimidin-7-on-forbindelse til behandling af CSBP/RK/p38-kinnasemedierede sygdomme |
| ES2263681T3 (es) | 2000-12-08 | 2006-12-16 | Ortho-Mcneil Pharmaceutical, Inc. | Compuestos de pirrolina indazolil-substituidos como inhibidores de la kinasa. |
| AU2737102A (en) | 2000-12-08 | 2002-06-18 | Ortho Mcneil Pharm Inc | Macroheterocylic compounds useful as kinase inhibitors |
| US6599323B2 (en) | 2000-12-21 | 2003-07-29 | Ethicon, Inc. | Reinforced tissue implants and methods of manufacture and use |
| CA2435826A1 (en) | 2001-01-24 | 2002-08-01 | The Government Of The United States Of America | Differentiation of stem cells to pancreatic endocrine cells |
| CA2435124A1 (en) | 2001-01-25 | 2002-08-01 | Millennium Pharmaceuticals, Inc. | Formulation of boronic acid compounds |
| US6656488B2 (en) | 2001-04-11 | 2003-12-02 | Ethicon Endo-Surgery, Inc. | Bioabsorbable bag containing bioabsorbable materials of different bioabsorption rates for tissue engineering |
| EP1379626A2 (en) | 2001-04-19 | 2004-01-14 | DeveloGen Aktiengesellschaft für entwicklungsbiologische Forschung | A method for differentiating stem cells into insulin-producing cells |
| WO2002088335A1 (fr) | 2001-04-24 | 2002-11-07 | Ajinomoto Co., Inc. | Cellules souches et procede d'extraction de ces cellules |
| CA2447015A1 (en) | 2001-05-15 | 2002-11-21 | Rappaport Family Institute For Research In The Medical Sciences | Insulin producing cells derived from human embryonic stem cells |
| US6626950B2 (en) | 2001-06-28 | 2003-09-30 | Ethicon, Inc. | Composite scaffold with post anchor for the repair and regeneration of tissue |
| KR100418195B1 (ko) | 2001-07-05 | 2004-02-11 | 주식회사 우리기술 | 전력케이블의 다중절연진단장치 및 그 방법 |
| GB0117583D0 (en) | 2001-07-19 | 2001-09-12 | Astrazeneca Ab | Novel compounds |
| WO2003014313A2 (en) | 2001-08-06 | 2003-02-20 | Bresagen, Ltd. | Alternative compositions and methods for the culture of stem cells |
| US6617152B2 (en) | 2001-09-04 | 2003-09-09 | Corning Inc | Method for creating a cell growth surface on a polymeric substrate |
| EP1298201A1 (en) | 2001-09-27 | 2003-04-02 | Cardion AG | Process for the production of cells exhibiting an islet-beta-cell-like state |
| US20030138951A1 (en) | 2001-10-18 | 2003-07-24 | Li Yin | Conversion of liver stem and progenitor cells to pancreatic functional cells |
| US6420237B1 (en) * | 2001-10-22 | 2002-07-16 | Macronix International Co. Ltd. | Method of manufacturing twin bit cell flash memory device |
| JP4330995B2 (ja) | 2001-11-15 | 2009-09-16 | チルドレンズ メディカル センター コーポレーション | 絨毛膜絨毛、羊水、および胎盤からの胎児性幹細胞を単離、増殖、および分化させる方法、ならびにその治療的使用方法 |
| WO2003050249A2 (en) | 2001-12-07 | 2003-06-19 | Geron Corporation | Islet cells from human embryonic stem cells |
| KR100930139B1 (ko) | 2001-12-07 | 2009-12-07 | 사이토리 테라퓨틱스, 인크. | 처리된 리포애스퍼레이트 세포로 환자를 치료하기 위한시스템 및 방법 |
| WO2003054169A1 (en) | 2001-12-21 | 2003-07-03 | Thromb-X Nv | Compositions for the in vitro derivation and culture of embryonic stem (es) cell lines with germline transmission capability |
| EP1461421A2 (en) | 2001-12-28 | 2004-09-29 | Cellartis AB | A method for the establishment of a pluripotent human blastocyst-derived stem cell line |
| US20030162290A1 (en) | 2002-01-25 | 2003-08-28 | Kazutomo Inoue | Method for inducing differentiation of embryonic stem cells into functioning cells |
| US20030180268A1 (en) | 2002-02-05 | 2003-09-25 | Anthony Atala | Tissue engineered construct for supplementing or replacing a damaged organ |
| JPWO2003087349A1 (ja) | 2002-04-17 | 2005-08-18 | 大塚製薬株式会社 | 間葉系細胞から膵β細胞を形成する方法 |
| US20040161419A1 (en) | 2002-04-19 | 2004-08-19 | Strom Stephen C. | Placental stem cells and uses thereof |
| GB0210539D0 (en) | 2002-05-08 | 2002-06-19 | Univ Edinburgh | Control of es cell self renewal and lineage specification, and medium therefor |
| EP1506192B1 (en) | 2002-05-08 | 2008-02-27 | Janssen Pharmaceutica N.V. | Substituted pyrroline kinase inhibitors |
| US20060003446A1 (en) | 2002-05-17 | 2006-01-05 | Gordon Keller | Mesoderm and definitive endoderm cell populations |
| BR0311360A (pt) | 2002-05-28 | 2006-06-06 | Becton Dickinson Co | métodos para expansão e transdiferenciação in vitro de células acinares pancreáticas humanas em células produtoras de insulina |
| JP2005531609A (ja) | 2002-06-05 | 2005-10-20 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | キナーゼ阻害剤としてのシスインドリル−マレイミド誘導体 |
| GB0212976D0 (en) | 2002-06-06 | 2002-07-17 | Tonejet Corp Pty Ltd | Ejection method and apparatus |
| CN1171991C (zh) | 2002-07-08 | 2004-10-20 | 徐如祥 | 人神经干细胞的培养方法 |
| US6877147B2 (en) | 2002-07-22 | 2005-04-05 | Broadcom Corporation | Technique to assess timing delay by use of layout quality analyzer comparison |
| US7838290B2 (en) | 2002-07-25 | 2010-11-23 | The Scripps Research Institute | Hematopoietic stem cells and methods of treatment of neovascular eye diseases therewith |
| CA2494040A1 (en) | 2002-07-29 | 2004-02-05 | Es Cell International Pte Ltd. | Multi-step method for the differentiation of insulin positive, glucose |
| AU2003262628A1 (en) * | 2002-08-14 | 2004-03-03 | University Of Florida | Bone marrow cell differentiation |
| AU2003268534A1 (en) | 2002-09-06 | 2004-03-29 | Amcyte Inc. | Cd56 positive human adult pancreatic endocrine progenitor cells |
| US9969977B2 (en) | 2002-09-20 | 2018-05-15 | Garnet Biotherapeutics | Cell populations which co-express CD49c and CD90 |
| US20040062753A1 (en) | 2002-09-27 | 2004-04-01 | Alireza Rezania | Composite scaffolds seeded with mammalian cells |
| AU2003285172A1 (en) | 2002-11-08 | 2004-06-03 | The Johns Hopkins University | Human embryonic stem cell cultures, and compositions and methods for growing same |
| US7144999B2 (en) | 2002-11-23 | 2006-12-05 | Isis Pharmaceuticals, Inc. | Modulation of hypoxia-inducible factor 1 alpha expression |
| WO2004050827A2 (en) | 2002-12-05 | 2004-06-17 | Technion Research & Development Foundation Ltd. | Cultured human pancreatic islets, and uses thereof |
| JP4613069B2 (ja) | 2002-12-16 | 2011-01-12 | テクニオン リサーチ アンド ディベロップメント ファウンデーション リミテッド | 支持細胞非含有、異種非含有のヒト胚性幹細胞の調製方法およびこれらを使用して調製された幹細胞培養物 |
| US20050118148A1 (en) | 2002-12-20 | 2005-06-02 | Roland Stein | Compositions and methods related to mammalian Maf-A |
| RU2359671C2 (ru) | 2003-01-29 | 2009-06-27 | Такеда Фармасьютикал Компани Лимитед | Способ получения препарата с покрытием |
| EP1588708A4 (en) | 2003-01-29 | 2006-03-01 | Takeda Pharmaceutical | METHOD FOR PRODUCING COATED PREPARATION |
| WO2004073633A2 (en) | 2003-02-14 | 2004-09-02 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and compositions for modulating the development of stem cells |
| US20070154981A1 (en) | 2003-02-14 | 2007-07-05 | The Board Of Trustees Of The Leland Stanford Junior University | Insulin-producing cells derived from stem cells |
| CA2520861A1 (en) | 2003-03-27 | 2004-10-14 | Ixion Biotechnology, Inc. | Method for transdifferentiation of non-pancreatic stem cells to the pancreatic pathway |
| WO2004090110A2 (en) | 2003-03-31 | 2004-10-21 | Bresagen Inc. | Compositions and methods for the control, differentiation and/or manipulation of pluripotent cells through a gamma-secretase signaling pathway |
| US20090203141A1 (en) | 2003-05-15 | 2009-08-13 | Shi-Lung Lin | Generation of tumor-free embryonic stem-like pluripotent cells using inducible recombinant RNA agents |
| PL1641914T3 (pl) | 2003-06-27 | 2017-01-31 | DePuy Synthes Products, Inc. | Komórki pochodzące z poporodowej tkanki łożyska oraz sposoby uzyskiwania i zastosowania tych komórek |
| IL161903A0 (en) | 2003-07-17 | 2005-11-20 | Gamida Cell Ltd | Ex vivo progenitor and stem cell expansion for usein the treatment of disease of endodermally- deri ved organs |
| ITRM20030395A1 (it) | 2003-08-12 | 2005-02-13 | Istituto Naz Per Le Malattie Infettive Lazz | Terreno di coltura per il mantenimento, la proliferazione e il differenziamento di cellule di mammifero. |
| US20050042595A1 (en) | 2003-08-14 | 2005-02-24 | Martin Haas | Banking of multipotent amniotic fetal stem cells |
| US7157275B2 (en) | 2003-08-15 | 2007-01-02 | Becton, Dickinson And Company | Peptides for enhanced cell attachment and growth |
| US20060205072A1 (en) | 2003-08-27 | 2006-09-14 | Nobuko Uchida | Enriched pancreatic stem cell and progenitor cell populations, and methods for identifying, isolating and enriching for such populations |
| EP1696899A1 (en) | 2003-12-17 | 2006-09-06 | Allergan, Inc. | Methods for treating retinoid responsive disorders using selective inhibitors of cyp26a and cyp26b |
| US20060030042A1 (en) | 2003-12-19 | 2006-02-09 | Ali Brivanlou | Maintenance of embryonic stem cells by the GSK-3 inhibitor 6-bromoindirubin-3'-oxime |
| US7625753B2 (en) | 2003-12-23 | 2009-12-01 | Cythera, Inc. | Expansion of definitive endoderm cells |
| CN109628371B (zh) | 2003-12-23 | 2021-02-19 | 维亚希特公司 | 定形内胚层 |
| US7541185B2 (en) * | 2003-12-23 | 2009-06-02 | Cythera, Inc. | Methods for identifying factors for differentiating definitive endoderm |
| US20050266554A1 (en) | 2004-04-27 | 2005-12-01 | D Amour Kevin A | PDX1 expressing endoderm |
| CN103898045B (zh) | 2003-12-23 | 2019-02-01 | 维亚希特公司 | 定形内胚层 |
| TWI334443B (en) | 2003-12-31 | 2010-12-11 | Ind Tech Res Inst | Method of single cell culture of undifferentiated human embryonic stem cells |
| WO2005065354A2 (en) | 2003-12-31 | 2005-07-21 | The Burnham Institute | Defined media for pluripotent stem cell culture |
| US20070027063A1 (en) | 2004-01-12 | 2007-02-01 | Mannkind Corporation | Method of preserving the function of insulin-producing cells |
| WO2005071066A1 (en) | 2004-01-23 | 2005-08-04 | Board Of Regents, The University Of Texas System | Methods and compositions for preparing pancreatic insulin secreting cells |
| US7794704B2 (en) * | 2004-01-23 | 2010-09-14 | Advanced Cell Technology, Inc. | Methods for producing enriched populations of human retinal pigment epithelium cells for treatment of retinal degeneration |
| US20070298453A1 (en) | 2004-02-12 | 2007-12-27 | University Of Newcastle Upon Tyne | Stem Cells |
| WO2005080598A1 (ja) | 2004-02-19 | 2005-09-01 | Dainippon Sumitomo Pharma Co., Ltd. | 体細胞核初期化物質のスクリーニング方法 |
| US20060281174A1 (en) | 2004-03-09 | 2006-12-14 | Gang Xu | Methods for generating insulin-producing cells |
| EP1730261A4 (en) | 2004-03-10 | 2007-11-28 | Univ California | COMPOSITIONS AND METHODS FOR GROWING EMBRYONIC STEM CELLS |
| CN1934245B (zh) | 2004-03-23 | 2012-07-04 | 第一三共株式会社 | 多能干细胞的增殖方法 |
| WO2005097980A2 (en) | 2004-03-26 | 2005-10-20 | Geron Corporation | New protocols for making hepatocytes from embryonic stem cells |
| AU2005230832B2 (en) | 2004-04-01 | 2010-11-11 | Wisconsin Alumni Research Foundation | Differentiation of stem cells to endoderm and pancreatic lineage |
| DK2377922T3 (da) | 2004-04-27 | 2020-05-04 | Viacyte Inc | PDX1-eksprimerende endoderm |
| CN102925406B (zh) | 2004-07-09 | 2019-11-22 | 维亚希特公司 | 鉴定用于分化定型内胚层的因子的方法 |
| WO2006020919A2 (en) | 2004-08-13 | 2006-02-23 | University Of Georgia Research Foundation, Inc. | Compositions and methods for self-renewal and differentiation in human embryonic stem cells |
| US20080268533A1 (en) | 2004-08-25 | 2008-10-30 | University Of Georgia Research Foundation, Inc. | Methods and Compositions Utilizing Myc and Gsk3Beta to Manipulate the Pluripotency of Embryonic Stem Cells |
| DE102004043256B4 (de) | 2004-09-07 | 2013-09-19 | Rheinische Friedrich-Wilhelms-Universität Bonn | Skalierbarer Prozess zur Kultivierung undifferenzierter Stammzellen in Suspension |
| JP5420837B2 (ja) | 2004-09-08 | 2014-02-19 | ウィスコンシン アラムニ リサーチ ファンデーション | 胚幹細胞の培地及び培養 |
| KR20070058584A (ko) | 2004-09-08 | 2007-06-08 | 위스콘신 얼럼나이 리서어치 화운데이션 | 인간 배아 줄기 세포의 배양 |
| US20060275899A1 (en) | 2004-12-30 | 2006-12-07 | Stemlifeline, Inc. | Methods and compositions relating to embryonic stem cell lines |
| WO2006079854A1 (en) | 2005-01-28 | 2006-08-03 | Novathera Ltd | Methods for embryonic stem cell culture |
| JP2008528038A (ja) * | 2005-01-31 | 2008-07-31 | エス セル インターナショナル ピーティーイー リミテッド | 胚性幹細胞の指示された分化及びその利用 |
| WO2006088867A2 (en) | 2005-02-15 | 2006-08-24 | Medistem Laboratories, Incorporated | Method for expansion of stem cells |
| ES2627419T3 (es) | 2005-03-04 | 2017-07-28 | Lifescan, Inc. | Células estromales adultas derivadas del páncreas |
| GB0505970D0 (en) | 2005-03-23 | 2005-04-27 | Univ Edinburgh | Culture medium containing kinase inhibitor, and uses thereof |
| CN103356703B (zh) | 2005-03-31 | 2016-04-06 | 斯丹姆涅恩有限公司 | 制备用以治疗慢性感染伤口的药物的方法 |
| CN100425694C (zh) * | 2005-04-15 | 2008-10-15 | 北京大学 | 诱导胚胎干细胞向胰腺细胞分化的方法 |
| WO2006113470A2 (en) | 2005-04-15 | 2006-10-26 | Geron Corporation | Cancer treatment by combined inhibition of proteasome and telomerase activities |
| US20080208351A1 (en) | 2005-04-26 | 2008-08-28 | Aarhus Universitet | Biocompatible Material for Surgical Implants and Cell Guiding Tissue Culture Surfaces |
| JP2008541707A (ja) * | 2005-05-24 | 2008-11-27 | ノボザイムス アクティーゼルスカブ | ペニシリウム・カプスラタムアラビノフラノシダーゼ |
| JP5092124B2 (ja) | 2005-05-24 | 2012-12-05 | 国立大学法人 熊本大学 | Es細胞の分化誘導方法 |
| AU2006202209B2 (en) | 2005-05-27 | 2011-04-14 | Lifescan, Inc. | Amniotic fluid derived cells |
| US20100234400A1 (en) | 2005-06-10 | 2010-09-16 | Irm Llc | Compounds that maintain pluripotency of embryonic stem cells |
| WO2006138433A2 (en) | 2005-06-14 | 2006-12-28 | The Regents Of The University Of California | Induction of cell differentiation by class i bhlh polypeptides |
| EP1931764A1 (en) | 2005-06-21 | 2008-06-18 | GE Healthcare Bio-Sciences AB | Method for cell culture |
| CN101233226B (zh) * | 2005-06-22 | 2017-08-11 | 阿斯特利亚斯生物治疗股份公司 | 人胚胎干细胞的悬浮培养物 |
| NZ564179A (en) | 2005-06-30 | 2010-09-30 | Janssen Pharmaceutica Nv | Cyclic anilino - pyridinotriazines as GSK-3 inhibitors |
| US20080194021A1 (en) | 2005-07-29 | 2008-08-14 | Mays Robert W | Use of a Gsk-3 Inhibitor to Maintain Potency of Culture Cells |
| US20090087907A1 (en) | 2005-07-29 | 2009-04-02 | Alice Pebay | Compositions and Methods for Growth of Pluripotent Cells |
| WO2007025234A2 (en) | 2005-08-26 | 2007-03-01 | The Trustees Of Columbia University In The City Of New York | Generation of pancreatic endocrine cells from primary duct cell cultures and methods of use for treatment of diabetes |
| EP3354723B1 (en) | 2005-08-29 | 2023-12-13 | Technion Research & Development Foundation Ltd. | Media for culturing stem cells |
| KR20080056182A (ko) | 2005-09-02 | 2008-06-20 | 에이전시 포 사이언스, 테크놀로지 앤드 리서치 | 간엽 줄기 세포의 유도 방법 |
| WO2007030469A2 (en) * | 2005-09-06 | 2007-03-15 | University Of Pittsburgh - Of The Commonwealth System Of Higher Education | Transplantable cell growth niche and related compositions and methods |
| GB2444686B (en) | 2005-09-12 | 2010-08-25 | Es Cell Int Pte Ltd | Differentiation of pluripotent stem cells using p38 MAPK inhibitors or prostaglandins |
| EP1941032A2 (en) | 2005-10-14 | 2008-07-09 | Regents Of The University Of Minnesota | Differentiation of non-embryonic stem cells to cells having a pancreatic phenotype |
| US7732202B2 (en) | 2005-10-21 | 2010-06-08 | International Stem Cell Corporation | Oxygen tension for the parthenogenic activation of human oocytes for the production of human embryonic stem cells |
| EP1957636B1 (en) | 2005-10-27 | 2018-07-04 | Viacyte, Inc. | Pdx1-expressing dorsal and ventral foregut endoderm |
| EP3418297B1 (en) | 2005-12-13 | 2023-04-05 | Kyoto University | Nuclear reprogramming factor |
| WO2007082963A1 (es) | 2006-01-18 | 2007-07-26 | Fundación Instituto Valenciano De Infertilidad | Líneas de células madre embrionarias humanas y métodos para usar las mismas |
| SG170021A1 (en) | 2006-02-23 | 2011-04-29 | Novocell Inc | Compositions and methods useful for culturing differentiable cells |
| US7695965B2 (en) | 2006-03-02 | 2010-04-13 | Cythera, Inc. | Methods of producing pancreatic hormones |
| EP2650359B1 (en) | 2006-03-02 | 2022-05-04 | Viacyte, Inc. | Endocrine precursor cells, pancreatic hormone-expressing cells and methods of production |
| GB0615327D0 (en) | 2006-03-30 | 2006-09-13 | Univ Edinburgh | Culture medium containing kinase inhibitors and uses thereof |
| US8741643B2 (en) | 2006-04-28 | 2014-06-03 | Lifescan, Inc. | Differentiation of pluripotent stem cells to definitive endoderm lineage |
| EP4438720A2 (en) | 2006-04-28 | 2024-10-02 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells |
| US8685730B2 (en) | 2006-05-02 | 2014-04-01 | Wisconsin Alumni Research Foundation | Methods and devices for differentiating pluripotent stem cells into cells of the pancreatic lineage |
| US20070259423A1 (en) | 2006-05-02 | 2007-11-08 | Jon Odorico | Method of differentiating stem cells into cells of the endoderm and pancreatic lineage |
| US9598673B2 (en) | 2006-05-19 | 2017-03-21 | Creative Medical Health | Treatment of disc degenerative disease |
| US7964402B2 (en) | 2006-05-25 | 2011-06-21 | Sanford-Burnham Medical Research Institute | Methods for culture and production of single cell populations of human embryonic stem cells |
| WO2007143193A1 (en) | 2006-06-02 | 2007-12-13 | University Of Georgia Research Foundation, Inc. | Pancreatic and liver endoderm cells and tissue by differentiation of definitive endoderm cells obtained from human embryonic stems |
| CN101541953A (zh) | 2006-06-02 | 2009-09-23 | 佐治亚大学研究基金会 | 通过从人胚胎干细胞获得的定形内胚层细胞的分化得到胰和肝内胚层细胞及组织 |
| US8415153B2 (en) | 2006-06-19 | 2013-04-09 | Geron Corporation | Differentiation and enrichment of islet-like cells from human pluripotent stem cells |
| CN100494359C (zh) | 2006-06-23 | 2009-06-03 | 中日友好医院 | 神经干细胞三维立体培养体外扩增的方法 |
| WO2008036447A2 (en) | 2006-06-26 | 2008-03-27 | Lifescan, Inc. | Pluripotent stem cell culture |
| US20080003676A1 (en) | 2006-06-26 | 2008-01-03 | Millipore Corporation | Growth of embryonic stem cells |
| WO2008004990A2 (en) | 2006-07-06 | 2008-01-10 | Es Cell International Pte Ltd | Method for stem cell culture and cells derived therefrom |
| WO2008013664A2 (en) | 2006-07-26 | 2008-01-31 | Cythera, Inc. | Methods of producing pancreatic hormones |
| EP2733203B1 (en) | 2006-08-02 | 2018-10-10 | Technion Research & Development Foundation Ltd. | Methods of expanding embryonic stem cells in a suspension culture |
| KR101331510B1 (ko) | 2006-08-30 | 2013-11-20 | 재단법인서울대학교산학협력재단 | 저농도의 포도당을 함유하는 인간 배아줄기세포용 배지조성물 및 이를 이용한 인간 배아 줄기세포로부터 인슐린생산 세포 또는 세포괴로 분화시키는 방법, 그리고그로부터 유도된 인슐린 생산 세포 또는 세포괴 |
| JP2008099662A (ja) | 2006-09-22 | 2008-05-01 | Institute Of Physical & Chemical Research | 幹細胞の培養方法 |
| WO2008039521A2 (en) | 2006-09-26 | 2008-04-03 | Nmt Medical, Inc. | Method for modifying a medical implant surface for promoting tissue growth |
| BRPI0718310A2 (pt) | 2006-10-17 | 2013-11-19 | Stiefel Laboratories | Metabólitos de talarozol |
| WO2008048647A1 (en) | 2006-10-17 | 2008-04-24 | Cythera, Inc. | Modulation of the phosphatidylinositol-3-kinase pathway in the differentiation of human embryonic stem cells |
| CA2666789C (en) | 2006-10-18 | 2016-11-22 | Yong Zhao | Embryonic-like stem cells derived from adult human peripheral blood and methods of use |
| EP2088190A4 (en) | 2006-11-09 | 2011-01-05 | Japan Government | METHOD FOR THE CULTURE AND PASSAGE OF PRIMATE EMBRYONIC STRAIN CELL, AND METHOD FOR INDUCING DIFFERENTIATION OF EMBRYONIC STEM CELL |
| WO2008086005A1 (en) | 2007-01-09 | 2008-07-17 | University Of South Florida | Compositions including triciribine and bortezomib and derivatives thereof and methods of use thereof |
| WO2008094597A2 (en) | 2007-01-30 | 2008-08-07 | University Of Georgia Research Foundation, Inc. | Early mesoderm cells, a stable population of mesendoderm cells that has utility for generation of endoderm and mesoderm lineages and multipotent migratory cells (mmc) |
| GB0703188D0 (en) | 2007-02-19 | 2007-03-28 | Roger Land Building | Large scale production of stem cells |
| US20090053182A1 (en) | 2007-05-25 | 2009-02-26 | Medistem Laboratories, Inc. | Endometrial stem cells and methods of making and using same |
| JP5991796B2 (ja) | 2007-06-29 | 2016-09-14 | セルラー ダイナミクス インターナショナル, インコーポレイテッド | 胚性幹細胞培養のための自動化された方法および装置 |
| BRPI0814425A2 (pt) | 2007-07-18 | 2014-10-21 | Lifescan Inc | Diferenciação de células-tronco embrionárias humanas |
| ES2665434T3 (es) | 2007-07-31 | 2018-04-25 | Lifescan, Inc. | Diferenciación de células madre pluripotentes usando células alimentadoras humanas |
| KR101617243B1 (ko) | 2007-07-31 | 2016-05-02 | 라이프스캔, 인코포레이티드 | 인간 배아 줄기 세포의 분화 |
| KR101544498B1 (ko) | 2007-08-24 | 2015-08-17 | 스티칭 허트 네덜란드 칸커 인스티튜트 | 종양성 질환의 치료를 위한 조성물 |
| WO2009061442A1 (en) | 2007-11-06 | 2009-05-14 | Children's Medical Center Corporation | Method to produce induced pluripotent stem (ips) cells form non-embryonic human cells |
| EP2229434B1 (en) | 2007-11-27 | 2011-09-07 | Lifescan, Inc. | Differentiation of human embryonic stem cells |
| SG154367A1 (en) | 2008-01-31 | 2009-08-28 | Es Cell Int Pte Ltd | Method of differentiating stem cells |
| WO2009096049A1 (ja) | 2008-02-01 | 2009-08-06 | Kyoto University | 人工多能性幹細胞由来分化細胞 |
| EP2250252A2 (en) | 2008-02-11 | 2010-11-17 | Cambridge Enterprise Limited | Improved reprogramming of mammalian cells, and the cells obtained |
| CN102046779A (zh) | 2008-02-21 | 2011-05-04 | 森托科尔奥索生物科技公司 | 用于细胞粘附、培养和分离的方法、表面改性培养板和组合物 |
| JPWO2009110215A1 (ja) | 2008-03-03 | 2011-07-14 | 独立行政法人科学技術振興機構 | 繊毛細胞の分化誘導方法 |
| SG188918A1 (ja) | 2008-03-17 | 2013-04-30 | Agency Science Tech & Res | |
| RU2359030C1 (ru) | 2008-03-19 | 2009-06-20 | Общество С Ограниченной Ответственностью "Лаборатория Клеточных Технологий" | Способ получения эндотелиальных клеток из эмбриональных стволовых клеток человека (варианты) |
| WO2009131568A1 (en) | 2008-04-21 | 2009-10-29 | Cythera, Inc. | Methods for purifying endoderm and pancreatic endoderm cells derived from human embryonic stem cells |
| US8338170B2 (en) | 2008-04-21 | 2012-12-25 | Viacyte, Inc. | Methods for purifying endoderm and pancreatic endoderm cells derived from human embryonic stem cells |
| WO2009132083A2 (en) | 2008-04-22 | 2009-10-29 | President And Fellows Of Harvard College | Compositions and methods for promoting the generation of pdx1+ pancreatic cells |
| US8623648B2 (en) | 2008-04-24 | 2014-01-07 | Janssen Biotech, Inc. | Treatment of pluripotent cells |
| US7939322B2 (en) | 2008-04-24 | 2011-05-10 | Centocor Ortho Biotech Inc. | Cells expressing pluripotency markers and expressing markers characteristic of the definitive endoderm |
| EP2993226B1 (en) | 2008-06-03 | 2020-12-16 | Viacyte, Inc. | Growth factors for production of definitive endoderm |
| US20090298178A1 (en) | 2008-06-03 | 2009-12-03 | D Amour Kevin Allen | Growth factors for production of definitive endoderm |
| PL2310492T3 (pl) | 2008-06-30 | 2015-12-31 | Janssen Biotech Inc | Różnocowanie pluripotencjalnych komórek macierzystych |
| DE102008032236A1 (de) | 2008-06-30 | 2010-04-01 | Eberhard-Karls-Universität Tübingen | Isolierung und/oder Identifizierung von Stammzellen mit adipozytärem, chondrozytärem und pankreatischem Differenzierungspotential |
| US20100028307A1 (en) | 2008-07-31 | 2010-02-04 | O'neil John J | Pluripotent stem cell differentiation |
| US9683215B2 (en) | 2008-08-22 | 2017-06-20 | President And Fellows Of Harvard College | Methods of reprogramming cells |
| AU2009308967C1 (en) | 2008-10-31 | 2017-04-20 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells to the pancreatic endocrine lineage |
| KR102025158B1 (ko) | 2008-10-31 | 2019-09-25 | 얀센 바이오테크 인코포레이티드 | 인간 배아 줄기 세포의 췌장 내분비 계통으로의 분화 |
| US8008075B2 (en) | 2008-11-04 | 2011-08-30 | Viacyte, Inc. | Stem cell aggregate suspension compositions and methods of differentiation thereof |
| CN102361970B (zh) | 2008-11-04 | 2018-03-27 | 韦尔赛特公司 | 干细胞聚集体悬浮组合物及其分化方法 |
| EP4176888A1 (en) | 2008-11-14 | 2023-05-10 | ViaCyte, Inc. | Encapsulation of pancreatic cells derived from human pluripotent stem cells |
| MX356756B (es) | 2008-11-20 | 2018-06-11 | Centocor Ortho Biotech Inc | Células madre pluripotentes en microportadores. |
| WO2010063848A1 (en) | 2008-12-05 | 2010-06-10 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and medium for neural differentiation of pluripotent cells |
| EP2456862A4 (en) | 2009-07-20 | 2013-02-27 | Janssen Biotech Inc | DIFFERENTIATION OF HUMAN EMBRYONIC STEM CELLS |
| SG177416A1 (en) | 2009-07-20 | 2012-02-28 | Janssen Biotech Inc | Differentiation of human embryonic stem cells |
| CN107058389A (zh) | 2009-10-29 | 2017-08-18 | 詹森生物科技公司 | 多能干细胞 |
| FI20096288A0 (fi) | 2009-12-04 | 2009-12-04 | Kristiina Rajala | Formulations and methods for culturing stem cells |
| CN102741395B (zh) | 2009-12-23 | 2016-03-16 | 詹森生物科技公司 | 人胚胎干细胞的分化 |
| CN102712902B (zh) | 2009-12-23 | 2019-01-08 | 詹森生物科技公司 | 人胚胎干细胞的分化 |
| US8932853B2 (en) | 2009-12-29 | 2015-01-13 | Takeda Pharmaceutical Company Limited | Method for manufacturing pancreatic-hormone-producing cells |
| CN102858958A (zh) | 2010-02-03 | 2013-01-02 | 日本国立癌症研究中心 | 诱导肝干细胞及其制造方法、以及该细胞的用途 |
| CA2791846A1 (en) | 2010-03-02 | 2011-09-09 | National University Of Singapore | Culture additives to boost stem cell proliferation and differentiation response |
| JP5909482B2 (ja) | 2010-03-31 | 2016-04-26 | ザ スクリプス リサーチ インスティテュート | 細胞の再プログラム |
| EP2563908B1 (en) | 2010-04-25 | 2019-01-09 | Icahn School of Medicine at Mount Sinai | Generation of anterior foregut endoderm from pluripotent cells |
| RU2587634C2 (ru) | 2010-05-12 | 2016-06-20 | Янссен Байотек, Инк. | Дифференцирование эмбриональных стволовых клеток человека |
| US9085757B2 (en) | 2010-06-17 | 2015-07-21 | Regents Of The University Of Minnesota | Production of insulin producing cells |
| CA2807418C (en) | 2010-08-05 | 2017-04-04 | Wisconsin Alumni Research Foundation | Simplified basic media for human pluripotent cell culture |
| RU2576000C2 (ru) | 2010-08-09 | 2016-02-27 | Такеда Фармасьютикал Компани Лимитед | Способ получения клеток, продуцирующих панкреатические гормоны |
| WO2012030539A2 (en) | 2010-08-31 | 2012-03-08 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells |
| BR112013004614A2 (pt) | 2010-08-31 | 2024-01-16 | Janssen Biotech Inc | Diferenciação de células-tronco pluripotentes |
| MY177150A (en) | 2011-02-28 | 2020-09-08 | Stempeutics Res Malaysia Sdn Bhd | Isolation and expansion of adult stem cells, their therapeutic composition and uses thereof |
| US20130274184A1 (en) | 2011-10-11 | 2013-10-17 | The Trustees Of Columbia University In The City Of New York | Er stress relievers in beta cell protection |
| EP2766474B1 (en) | 2011-10-14 | 2020-10-07 | Children's Medical Center Corporation | Inhibition and enhancement of reprogramming by chromatin modifying enzymes |
| KR102203056B1 (ko) | 2011-12-22 | 2021-01-14 | 얀센 바이오테크 인코포레이티드 | 인간 배아 줄기 세포의 단일 인슐린 호르몬 양성 세포로의 분화 |
| US10519422B2 (en) | 2012-02-29 | 2019-12-31 | Riken | Method of producing human retinal pigment epithelial cells |
| CN108034633B (zh) | 2012-06-08 | 2022-08-02 | 詹森生物科技公司 | 人胚胎干细胞向胰腺内分泌细胞的分化 |
| US20150247123A1 (en) | 2012-09-03 | 2015-09-03 | Novo Nordisk A/S | Generation of pancreatic endoderm from Pluripotent Stem cells using small molecules |
| EP2938724B1 (en) | 2012-12-31 | 2020-10-28 | Janssen Biotech, Inc. | Culturing of human embryonic stem cells at the air-liquid interface for differentiation into pancreatic endocrine cells |
| AU2013368224B2 (en) | 2012-12-31 | 2018-09-27 | Janssen Biotech, Inc. | Differentiation of human embryonic stem cells into pancreatic endocrine cells using HB9 regulators |
| US8859286B2 (en) | 2013-03-14 | 2014-10-14 | Viacyte, Inc. | In vitro differentiation of pluripotent stem cells to pancreatic endoderm cells (PEC) and endocrine cells |
| EP2970892A1 (en) | 2013-03-15 | 2016-01-20 | The Jackson Laboratory | Isolation of non-embryonic stem cells and uses thereof |
| MX2015017103A (es) | 2013-06-11 | 2016-11-07 | Harvard College | Celulas beta derivadas de células madre ( sc-beta ) y composiciones y métodos para generarlas. |
-
2008
- 2008-07-31 KR KR1020107004455A patent/KR101617243B1/ko active IP Right Grant
- 2008-07-31 BR BRPI0814894-5A2A patent/BRPI0814894A2/pt not_active IP Right Cessation
- 2008-07-31 RU RU2010107224/10A patent/RU2473685C2/ru active
- 2008-07-31 CA CA2695225A patent/CA2695225C/en active Active
- 2008-07-31 CN CN200880109580.0A patent/CN101952415B/zh active Active
- 2008-07-31 JP JP2010520194A patent/JP5769965B2/ja active Active
- 2008-07-31 US US12/183,656 patent/US9096832B2/en active Active
- 2008-07-31 WO PCT/US2008/071782 patent/WO2009018453A1/en active Application Filing
- 2008-07-31 DK DK08782571.7T patent/DK2185693T3/da active
- 2008-07-31 CA CA3207103A patent/CA3207103A1/en active Pending
- 2008-07-31 EP EP12186995.2A patent/EP2610336A1/en not_active Withdrawn
- 2008-07-31 EP EP08782571.7A patent/EP2185693B1/en active Active
- 2008-07-31 MX MX2010001335A patent/MX2010001335A/es active IP Right Grant
- 2008-07-31 CA CA3114827A patent/CA3114827C/en active Active
-
2015
- 2015-05-01 JP JP2015094014A patent/JP2015144615A/ja active Pending
- 2015-06-26 US US14/751,416 patent/US9744195B2/en active Active
-
2017
- 2017-07-26 US US15/660,885 patent/US10456424B2/en active Active
-
2019
- 2019-09-25 US US16/582,279 patent/US11890304B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN101952415A (zh) | 2011-01-19 |
| CN101952415B (zh) | 2017-06-27 |
| EP2185693A1 (en) | 2010-05-19 |
| US20170354690A1 (en) | 2017-12-14 |
| WO2009018453A1 (en) | 2009-02-05 |
| US10456424B2 (en) | 2019-10-29 |
| RU2473685C2 (ru) | 2013-01-27 |
| MX2010001335A (es) | 2010-06-02 |
| US20150290249A1 (en) | 2015-10-15 |
| JP2015144615A (ja) | 2015-08-13 |
| US11890304B2 (en) | 2024-02-06 |
| RU2010107224A (ru) | 2011-09-10 |
| CA3207103A1 (en) | 2009-02-05 |
| CA2695225A1 (en) | 2009-02-05 |
| CA3114827C (en) | 2023-09-05 |
| US20200030383A1 (en) | 2020-01-30 |
| EP2185693B1 (en) | 2019-07-03 |
| US9096832B2 (en) | 2015-08-04 |
| DK2185693T3 (da) | 2019-09-23 |
| JP2010535036A (ja) | 2010-11-18 |
| KR101617243B1 (ko) | 2016-05-02 |
| CA2695225C (en) | 2021-06-01 |
| EP2610336A1 (en) | 2013-07-03 |
| CA3114827A1 (en) | 2009-02-05 |
| US20100015100A1 (en) | 2010-01-21 |
| BRPI0814894A2 (pt) | 2014-10-21 |
| KR20100065307A (ko) | 2010-06-16 |
| US9744195B2 (en) | 2017-08-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11890304B2 (en) | Pancreatic endocrine cells and methods thereof | |
| US20230287353A1 (en) | Differentiation of human embryonic stem cells | |
| JP5738591B2 (ja) | ヒト胚幹細胞の分化 | |
| JP6453277B2 (ja) | ヒトフィーダー細胞を用いた多能性幹細胞の分化 | |
| JP5882226B2 (ja) | ヒト胚性幹細胞の分化 | |
| KR102025158B1 (ko) | 인간 배아 줄기 세포의 췌장 내분비 계통으로의 분화 | |
| JP6392496B2 (ja) | ヒト胚性幹細胞の分化 | |
| EP3527658A1 (en) | Differentiation of human embryonic stem cells | |
| JP2013536686A (ja) | ヒト胚性幹細胞の分化 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110614 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20111208 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130514 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130808 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130815 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130913 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130924 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20131011 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20131021 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140527 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20140827 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140903 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140925 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20150106 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20150501 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20150512 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20150602 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20150624 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5769965 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |