DE69918074T2 - Makrolid-formulierung mit verzögerter wirkstoffabgabe - Google Patents
Makrolid-formulierung mit verzögerter wirkstoffabgabe Download PDFInfo
- Publication number
- DE69918074T2 DE69918074T2 DE69918074T DE69918074T DE69918074T2 DE 69918074 T2 DE69918074 T2 DE 69918074T2 DE 69918074 T DE69918074 T DE 69918074T DE 69918074 T DE69918074 T DE 69918074T DE 69918074 T2 DE69918074 T2 DE 69918074T2
- Authority
- DE
- Germany
- Prior art keywords
- group
- sustained
- release formulation
- formulation according
- solid dispersion
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 239000000203 mixture Substances 0.000 title claims description 238
- 238000009472 formulation Methods 0.000 title claims description 166
- 230000003111 delayed effect Effects 0.000 title claims description 21
- 239000013543 active substance Substances 0.000 title description 3
- -1 fatty acid ester Chemical class 0.000 claims description 95
- 150000001875 compounds Chemical class 0.000 claims description 75
- 239000003120 macrolide antibiotic agent Substances 0.000 claims description 64
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 claims description 63
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 claims description 61
- 239000008101 lactose Substances 0.000 claims description 61
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 claims description 61
- 238000013268 sustained release Methods 0.000 claims description 39
- 239000012730 sustained-release form Substances 0.000 claims description 39
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 38
- 238000000034 method Methods 0.000 claims description 36
- 239000007962 solid dispersion Substances 0.000 claims description 36
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 claims description 33
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 claims description 32
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 29
- 125000000217 alkyl group Chemical group 0.000 claims description 25
- 235000014113 dietary fatty acids Nutrition 0.000 claims description 24
- 239000000194 fatty acid Substances 0.000 claims description 24
- 229930195729 fatty acid Natural products 0.000 claims description 24
- 239000002245 particle Substances 0.000 claims description 24
- 239000001856 Ethyl cellulose Substances 0.000 claims description 20
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical group CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 claims description 20
- 235000019325 ethyl cellulose Nutrition 0.000 claims description 20
- 229920001249 ethyl cellulose Polymers 0.000 claims description 20
- 239000000843 powder Substances 0.000 claims description 18
- 239000000243 solution Substances 0.000 claims description 18
- 239000002775 capsule Substances 0.000 claims description 16
- 229930006000 Sucrose Natural products 0.000 claims description 15
- 125000003545 alkoxy group Chemical group 0.000 claims description 15
- 125000004043 oxo group Chemical group O=* 0.000 claims description 15
- 239000005720 sucrose Substances 0.000 claims description 15
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical group CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 claims description 12
- 238000007922 dissolution test Methods 0.000 claims description 12
- 229960001967 tacrolimus Drugs 0.000 claims description 12
- 239000012085 test solution Substances 0.000 claims description 12
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 claims description 11
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 claims description 11
- 235000019700 dicalcium phosphate Nutrition 0.000 claims description 11
- 239000001863 hydroxypropyl cellulose Substances 0.000 claims description 11
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 claims description 11
- 125000001424 substituent group Chemical group 0.000 claims description 11
- 229920003176 water-insoluble polymer Polymers 0.000 claims description 10
- 125000004432 carbon atom Chemical group C* 0.000 claims description 9
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 claims description 9
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical group OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 claims description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 9
- 125000003118 aryl group Chemical group 0.000 claims description 8
- 229920000223 polyglycerol Polymers 0.000 claims description 8
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 claims description 7
- 229910052760 oxygen Inorganic materials 0.000 claims description 7
- 239000002904 solvent Substances 0.000 claims description 7
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 6
- SVSARCCKBMZNMR-UHFFFAOYSA-N [1-[2-[methyl-[2-[4-(oxoazaniumylmethylidene)pyridin-1-yl]ethyl]amino]ethyl]pyridin-4-ylidene]methyl-oxoazanium;dichloride Chemical compound [Cl-].[Cl-].C1=CC(=C[NH+]=O)C=CN1CCN(C)CCN1C=CC(=C[NH+]=O)C=C1 SVSARCCKBMZNMR-UHFFFAOYSA-N 0.000 claims description 6
- 125000003342 alkenyl group Chemical group 0.000 claims description 6
- 239000011230 binding agent Substances 0.000 claims description 6
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 claims description 6
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 claims description 6
- 239000001301 oxygen Substances 0.000 claims description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 5
- 125000005553 heteroaryloxy group Chemical group 0.000 claims description 5
- 150000003839 salts Chemical class 0.000 claims description 5
- 229920003169 water-soluble polymer Polymers 0.000 claims description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 4
- 239000001257 hydrogen Substances 0.000 claims description 4
- 239000000314 lubricant Substances 0.000 claims description 4
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 claims description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 3
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 claims description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 3
- 229910052794 bromium Inorganic materials 0.000 claims description 3
- 239000000460 chlorine Substances 0.000 claims description 3
- 229910052801 chlorine Inorganic materials 0.000 claims description 3
- 229920001577 copolymer Polymers 0.000 claims description 3
- 125000004122 cyclic group Chemical group 0.000 claims description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000003700 epoxy group Chemical group 0.000 claims description 3
- 239000008187 granular material Substances 0.000 claims description 3
- 125000000623 heterocyclic group Chemical group 0.000 claims description 3
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 claims description 3
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 3
- 239000003826 tablet Substances 0.000 claims description 3
- 125000005842 heteroatom Chemical group 0.000 claims description 2
- 229920006395 saturated elastomer Polymers 0.000 claims description 2
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 claims description 2
- 229910052799 carbon Inorganic materials 0.000 claims 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen(.) Chemical compound [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims 1
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 34
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 33
- 238000012360 testing method Methods 0.000 description 23
- 239000007903 gelatin capsule Substances 0.000 description 21
- 235000019359 magnesium stearate Nutrition 0.000 description 17
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 16
- 239000007787 solid Substances 0.000 description 16
- 238000004090 dissolution Methods 0.000 description 15
- 201000010099 disease Diseases 0.000 description 14
- 239000002585 base Substances 0.000 description 13
- 210000004369 blood Anatomy 0.000 description 13
- 239000008280 blood Substances 0.000 description 13
- 125000002252 acyl group Chemical group 0.000 description 12
- JYKSTGLAIMQDRA-UHFFFAOYSA-N tetraglycerol Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO JYKSTGLAIMQDRA-UHFFFAOYSA-N 0.000 description 12
- 229940079593 drug Drugs 0.000 description 9
- 239000003814 drug Substances 0.000 description 9
- 229920000642 polymer Polymers 0.000 description 9
- 208000006673 asthma Diseases 0.000 description 8
- 239000001993 wax Substances 0.000 description 8
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 229920003144 amino alkyl methacrylate copolymer Polymers 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 238000005549 size reduction Methods 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 125000005115 alkyl carbamoyl group Chemical group 0.000 description 5
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 5
- WOKDXPHSIQRTJF-UHFFFAOYSA-N 3-[3-[3-[3-[3-[3-[3-[3-[3-(2,3-dihydroxypropoxy)-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]-2-hydroxypropoxy]propane-1,2-diol Chemical compound OCC(O)COCC(O)COCC(O)COCC(O)COCC(O)COCC(O)COCC(O)COCC(O)COCC(O)COCC(O)CO WOKDXPHSIQRTJF-UHFFFAOYSA-N 0.000 description 4
- 208000023275 Autoimmune disease Diseases 0.000 description 4
- 201000004624 Dermatitis Diseases 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- 125000001931 aliphatic group Chemical group 0.000 description 4
- 125000005103 alkyl silyl group Chemical group 0.000 description 4
- ZDQSOHOQTUFQEM-PKUCKEGBSA-N ascomycin Chemical compound C/C([C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@]2(O)O[C@@H]([C@H](C[C@H]2C)OC)[C@@H](OC)C[C@@H](C)C\C(C)=C/[C@H](C(C[C@H](O)[C@H]1C)=O)CC)=C\[C@@H]1CC[C@@H](O)[C@H](OC)C1 ZDQSOHOQTUFQEM-PKUCKEGBSA-N 0.000 description 4
- 208000006454 hepatitis Diseases 0.000 description 4
- 206010023332 keratitis Diseases 0.000 description 4
- 210000004185 liver Anatomy 0.000 description 4
- 210000000056 organ Anatomy 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 238000011282 treatment Methods 0.000 description 4
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 210000002216 heart Anatomy 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 231100000283 hepatitis Toxicity 0.000 description 3
- 230000001506 immunosuppresive effect Effects 0.000 description 3
- 230000000968 intestinal effect Effects 0.000 description 3
- 230000000302 ischemic effect Effects 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 229920000609 methyl cellulose Polymers 0.000 description 3
- 239000001923 methylcellulose Substances 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 239000004570 mortar (masonry) Substances 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000008194 pharmaceutical composition Substances 0.000 description 3
- 239000000825 pharmaceutical preparation Substances 0.000 description 3
- 229940127557 pharmaceutical product Drugs 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 208000017520 skin disease Diseases 0.000 description 3
- 230000004936 stimulating effect Effects 0.000 description 3
- 238000002054 transplantation Methods 0.000 description 3
- 125000002861 (C1-C4) alkanoyl group Chemical group 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- 201000004384 Alopecia Diseases 0.000 description 2
- 208000032467 Aplastic anaemia Diseases 0.000 description 2
- 206010010744 Conjunctivitis allergic Diseases 0.000 description 2
- 208000003556 Dry Eye Syndromes Diseases 0.000 description 2
- 206010013774 Dry eye Diseases 0.000 description 2
- 206010060742 Endocrine ophthalmopathy Diseases 0.000 description 2
- 229920003151 Eudragit® RL polymer Polymers 0.000 description 2
- 229920003152 Eudragit® RS polymer Polymers 0.000 description 2
- 208000027761 Hepatic autoimmune disease Diseases 0.000 description 2
- 206010019663 Hepatic failure Diseases 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 2
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 2
- 241000282567 Macaca fascicularis Species 0.000 description 2
- 206010029240 Neuritis Diseases 0.000 description 2
- NYEWWQDBWFZZJS-UHFFFAOYSA-N OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCCCCCCCCCC(O)=O Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCCCCCCCCCC(O)=O NYEWWQDBWFZZJS-UHFFFAOYSA-N 0.000 description 2
- MMQZBEXYFLXHEN-UHFFFAOYSA-N OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCCCCCCCCCCCC(O)=O Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCCCCCCCCCCCC(O)=O MMQZBEXYFLXHEN-UHFFFAOYSA-N 0.000 description 2
- 206010034277 Pemphigoid Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 241000187747 Streptomyces Species 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 2
- YKTSYUJCYHOUJP-UHFFFAOYSA-N [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] Chemical compound [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] YKTSYUJCYHOUJP-UHFFFAOYSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 125000005042 acyloxymethyl group Chemical group 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- 208000002205 allergic conjunctivitis Diseases 0.000 description 2
- 231100000360 alopecia Toxicity 0.000 description 2
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- ZDQSOHOQTUFQEM-XCXYXIJFSA-N ascomycin Natural products CC[C@H]1C=C(C)C[C@@H](C)C[C@@H](OC)[C@H]2O[C@@](O)([C@@H](C)C[C@H]2OC)C(=O)C(=O)N3CCCC[C@@H]3C(=O)O[C@H]([C@H](C)[C@@H](O)CC1=O)C(=C[C@@H]4CC[C@@H](O)[C@H](C4)OC)C ZDQSOHOQTUFQEM-XCXYXIJFSA-N 0.000 description 2
- 208000010668 atopic eczema Diseases 0.000 description 2
- 230000001363 autoimmune Effects 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 229940084030 carboxymethylcellulose calcium Drugs 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 208000019425 cirrhosis of liver Diseases 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 2
- 150000005690 diesters Chemical class 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- UKMSUNONTOPOIO-UHFFFAOYSA-N docosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCC(O)=O UKMSUNONTOPOIO-UHFFFAOYSA-N 0.000 description 2
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 2
- FSXVSUSRJXIJHB-UHFFFAOYSA-M ethyl prop-2-enoate;methyl 2-methylprop-2-enoate;trimethyl-[2-(2-methylprop-2-enoyloxy)ethyl]azanium;chloride Chemical compound [Cl-].CCOC(=O)C=C.COC(=O)C(C)=C.CC(=C)C(=O)OCC[N+](C)(C)C FSXVSUSRJXIJHB-UHFFFAOYSA-M 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000019634 flavors Nutrition 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 208000024908 graft versus host disease Diseases 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 2
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 2
- VKOBVWXKNCXXDE-UHFFFAOYSA-N icosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCC(O)=O VKOBVWXKNCXXDE-UHFFFAOYSA-N 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 208000023589 ischemic disease Diseases 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 208000017169 kidney disease Diseases 0.000 description 2
- 208000019423 liver disease Diseases 0.000 description 2
- 208000007903 liver failure Diseases 0.000 description 2
- 231100000835 liver failure Toxicity 0.000 description 2
- 229940031703 low substituted hydroxypropyl cellulose Drugs 0.000 description 2
- 229940041033 macrolides Drugs 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 2
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 150000004682 monohydrates Chemical class 0.000 description 2
- DNKKLDKIFMDAPT-UHFFFAOYSA-N n,n-dimethylmethanamine;2-methylprop-2-enoic acid Chemical compound CN(C)C.CC(=C)C(O)=O.CC(=C)C(O)=O DNKKLDKIFMDAPT-UHFFFAOYSA-N 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- KMNJKLJBUHNCQM-UHFFFAOYSA-N octadecanoic acid propane-1,2,3-triol Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O KMNJKLJBUHNCQM-UHFFFAOYSA-N 0.000 description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 201000000306 sarcoidosis Diseases 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- OMDQUFIYNPYJFM-XKDAHURESA-N (2r,3r,4s,5r,6s)-2-(hydroxymethyl)-6-[[(2r,3s,4r,5s,6r)-4,5,6-trihydroxy-3-[(2s,3s,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]methoxy]oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O[C@H]2[C@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@H](O)[C@H](O)O1 OMDQUFIYNPYJFM-XKDAHURESA-N 0.000 description 1
- OYHQOLUKZRVURQ-NTGFUMLPSA-N (9Z,12Z)-9,10,12,13-tetratritiooctadeca-9,12-dienoic acid Chemical compound C(CCCCCCC\C(=C(/C\C(=C(/CCCCC)\[3H])\[3H])\[3H])\[3H])(=O)O OYHQOLUKZRVURQ-NTGFUMLPSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- UQRONKZLYKUEMO-UHFFFAOYSA-N 4-methyl-1-(2,4,6-trimethylphenyl)pent-4-en-2-one Chemical group CC(=C)CC(=O)Cc1c(C)cc(C)cc1C UQRONKZLYKUEMO-UHFFFAOYSA-N 0.000 description 1
- CKOZVEHVVHCMGD-UHFFFAOYSA-N 5-[(4-fluorophenyl)methyl]-n,n-dimethyltetrazole-1-carboxamide Chemical compound CN(C)C(=O)N1N=NN=C1CC1=CC=C(F)C=C1 CKOZVEHVVHCMGD-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 208000002874 Acne Vulgaris Diseases 0.000 description 1
- 208000009304 Acute Kidney Injury Diseases 0.000 description 1
- 208000007788 Acute Liver Failure Diseases 0.000 description 1
- 206010000804 Acute hepatic failure Diseases 0.000 description 1
- 208000026872 Addison Disease Diseases 0.000 description 1
- 201000010000 Agranulocytosis Diseases 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 208000037259 Amyloid Plaque Diseases 0.000 description 1
- 206010002065 Anaemia megaloblastic Diseases 0.000 description 1
- 206010002660 Anoxia Diseases 0.000 description 1
- 241000976983 Anoxia Species 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- 241001225321 Aspergillus fumigatus Species 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 208000037157 Azotemia Diseases 0.000 description 1
- 208000023328 Basedow disease Diseases 0.000 description 1
- 208000009137 Behcet syndrome Diseases 0.000 description 1
- 235000021357 Behenic acid Nutrition 0.000 description 1
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 1
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 208000019838 Blood disease Diseases 0.000 description 1
- 208000020084 Bone disease Diseases 0.000 description 1
- 101100476924 Caenorhabditis elegans sdc-1 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 239000005632 Capric acid (CAS 334-48-5) Substances 0.000 description 1
- 239000005635 Caprylic acid (CAS 124-07-2) Substances 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- 208000002177 Cataract Diseases 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 206010008088 Cerebral artery embolism Diseases 0.000 description 1
- 206010008111 Cerebral haemorrhage Diseases 0.000 description 1
- 206010008132 Cerebral thrombosis Diseases 0.000 description 1
- 206010008609 Cholangitis sclerosing Diseases 0.000 description 1
- 206010008805 Chromosomal abnormalities Diseases 0.000 description 1
- 208000031404 Chromosome Aberrations Diseases 0.000 description 1
- 206010008909 Chronic Hepatitis Diseases 0.000 description 1
- SBUKLPSBNFWJCU-UHFFFAOYSA-N ClIBr Chemical compound ClIBr SBUKLPSBNFWJCU-UHFFFAOYSA-N 0.000 description 1
- 206010009657 Clostridium difficile colitis Diseases 0.000 description 1
- 208000015943 Coeliac disease Diseases 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 208000027932 Collagen disease Diseases 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- 206010011831 Cytomegalovirus infection Diseases 0.000 description 1
- 206010012438 Dermatitis atopic Diseases 0.000 description 1
- 206010012442 Dermatitis contact Diseases 0.000 description 1
- 206010048768 Dermatosis Diseases 0.000 description 1
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 201000010374 Down Syndrome Diseases 0.000 description 1
- 206010014561 Emphysema Diseases 0.000 description 1
- 208000017701 Endocrine disease Diseases 0.000 description 1
- 208000004232 Enteritis Diseases 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 206010014950 Eosinophilia Diseases 0.000 description 1
- 206010014954 Eosinophilic fasciitis Diseases 0.000 description 1
- 206010014989 Epidermolysis bullosa Diseases 0.000 description 1
- 206010015150 Erythema Diseases 0.000 description 1
- 206010015218 Erythema multiforme Diseases 0.000 description 1
- 229920003148 Eudragit® E polymer Polymers 0.000 description 1
- 229920003134 Eudragit® polymer Polymers 0.000 description 1
- 101710103508 FK506-binding protein Proteins 0.000 description 1
- 101710104425 FK506-binding protein 2 Proteins 0.000 description 1
- 101710104423 FK506-binding protein 3 Proteins 0.000 description 1
- 101710104333 FK506-binding protein 4 Proteins 0.000 description 1
- 101710104342 FK506-binding protein 5 Proteins 0.000 description 1
- 101710149710 FKBP-type 16 kDa peptidyl-prolyl cis-trans isomerase Proteins 0.000 description 1
- 101710121306 FKBP-type 22 kDa peptidyl-prolyl cis-trans isomerase Proteins 0.000 description 1
- 101710180800 FKBP-type peptidyl-prolyl cis-trans isomerase FkpA Proteins 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 241000223221 Fusarium oxysporum Species 0.000 description 1
- 229920000926 Galactomannan Polymers 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- 208000024869 Goodpasture syndrome Diseases 0.000 description 1
- 208000009329 Graft vs Host Disease Diseases 0.000 description 1
- 206010018691 Granuloma Diseases 0.000 description 1
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 206010019196 Head injury Diseases 0.000 description 1
- 208000010496 Heart Arrest Diseases 0.000 description 1
- 208000035186 Hemolytic Autoimmune Anemia Diseases 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 206010019799 Hepatitis viral Diseases 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010020850 Hyperthyroidism Diseases 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- 206010021074 Hypoplastic anaemia Diseases 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 1
- 201000003838 Idiopathic interstitial pneumonia Diseases 0.000 description 1
- 206010021245 Idiopathic thrombocytopenic purpura Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010021929 Infertility male Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 201000001429 Intracranial Thrombosis Diseases 0.000 description 1
- 206010022941 Iridocyclitis Diseases 0.000 description 1
- 208000032382 Ischaemic stroke Diseases 0.000 description 1
- 208000002260 Keloid Diseases 0.000 description 1
- 206010023330 Keloid scar Diseases 0.000 description 1
- 208000009319 Keratoconjunctivitis Sicca Diseases 0.000 description 1
- 239000005639 Lauric acid Substances 0.000 description 1
- 206010024380 Leukoderma Diseases 0.000 description 1
- GWNVDXQDILPJIG-SHSCPDMUSA-N Leukotriene C4 Natural products CCCCCC=C/CC=C/C=C/C=C/C(SCC(NC(=O)CCC(N)C(=O)O)C(=O)NCC(=O)O)C(O)CCCC(=O)O GWNVDXQDILPJIG-SHSCPDMUSA-N 0.000 description 1
- 101710104030 Long-type peptidyl-prolyl cis-trans isomerase Proteins 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 208000007466 Male Infertility Diseases 0.000 description 1
- 208000000682 Megaloblastic Anemia Diseases 0.000 description 1
- 208000027530 Meniere disease Diseases 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 1
- 206010028116 Mucosal inflammation Diseases 0.000 description 1
- NIPNSKYNPDTRPC-UHFFFAOYSA-N N-[2-oxo-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 NIPNSKYNPDTRPC-UHFFFAOYSA-N 0.000 description 1
- 206010028851 Necrosis Diseases 0.000 description 1
- 206010051606 Necrotising colitis Diseases 0.000 description 1
- 206010029164 Nephrotic syndrome Diseases 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 208000027771 Obstructive airways disease Diseases 0.000 description 1
- 206010030111 Oedema mucosal Diseases 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 101710114693 Outer membrane protein MIP Proteins 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- QHZLMUACJMDIAE-UHFFFAOYSA-N Palmitic acid monoglyceride Natural products CCCCCCCCCCCCCCCC(=O)OCC(O)CO QHZLMUACJMDIAE-UHFFFAOYSA-N 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 201000011152 Pemphigus Diseases 0.000 description 1
- 241000721454 Pemphigus Species 0.000 description 1
- 101710116692 Peptidyl-prolyl cis-trans isomerase Proteins 0.000 description 1
- 101710111764 Peptidyl-prolyl cis-trans isomerase FKBP10 Proteins 0.000 description 1
- 101710111749 Peptidyl-prolyl cis-trans isomerase FKBP11 Proteins 0.000 description 1
- 101710111747 Peptidyl-prolyl cis-trans isomerase FKBP12 Proteins 0.000 description 1
- 101710111757 Peptidyl-prolyl cis-trans isomerase FKBP14 Proteins 0.000 description 1
- 101710111682 Peptidyl-prolyl cis-trans isomerase FKBP1A Proteins 0.000 description 1
- 101710111689 Peptidyl-prolyl cis-trans isomerase FKBP1B Proteins 0.000 description 1
- 101710147154 Peptidyl-prolyl cis-trans isomerase FKBP2 Proteins 0.000 description 1
- 101710147149 Peptidyl-prolyl cis-trans isomerase FKBP3 Proteins 0.000 description 1
- 101710147152 Peptidyl-prolyl cis-trans isomerase FKBP4 Proteins 0.000 description 1
- 101710147150 Peptidyl-prolyl cis-trans isomerase FKBP5 Proteins 0.000 description 1
- 101710147138 Peptidyl-prolyl cis-trans isomerase FKBP7 Proteins 0.000 description 1
- 101710147137 Peptidyl-prolyl cis-trans isomerase FKBP8 Proteins 0.000 description 1
- 101710147136 Peptidyl-prolyl cis-trans isomerase FKBP9 Proteins 0.000 description 1
- 102100038809 Peptidyl-prolyl cis-trans isomerase FKBP9 Human genes 0.000 description 1
- 101710174853 Peptidyl-prolyl cis-trans isomerase Mip Proteins 0.000 description 1
- 101710200991 Peptidyl-prolyl cis-trans isomerase, rhodopsin-specific isozyme Proteins 0.000 description 1
- 101710092145 Peptidyl-prolyl cis-trans isomerase-like 1 Proteins 0.000 description 1
- 101710092146 Peptidyl-prolyl cis-trans isomerase-like 2 Proteins 0.000 description 1
- 101710092148 Peptidyl-prolyl cis-trans isomerase-like 3 Proteins 0.000 description 1
- 101710092149 Peptidyl-prolyl cis-trans isomerase-like 4 Proteins 0.000 description 1
- 102000009658 Peptidylprolyl Isomerase Human genes 0.000 description 1
- 108010020062 Peptidylprolyl Isomerase Proteins 0.000 description 1
- 208000031845 Pernicious anaemia Diseases 0.000 description 1
- 206010034972 Photosensitivity reaction Diseases 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 206010065159 Polychondritis Diseases 0.000 description 1
- 229920002675 Polyoxyl Polymers 0.000 description 1
- 206010060932 Postoperative adhesion Diseases 0.000 description 1
- 208000002500 Primary Ovarian Insufficiency Diseases 0.000 description 1
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 1
- 101710113444 Probable parvulin-type peptidyl-prolyl cis-trans isomerase Proteins 0.000 description 1
- 101710090737 Probable peptidyl-prolyl cis-trans isomerase Proteins 0.000 description 1
- 206010036774 Proctitis Diseases 0.000 description 1
- 208000003100 Pseudomembranous Enterocolitis Diseases 0.000 description 1
- 206010037128 Pseudomembranous colitis Diseases 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 101710133309 Putative peptidyl-prolyl cis-trans isomerase Proteins 0.000 description 1
- 208000006311 Pyoderma Diseases 0.000 description 1
- 206010037779 Radiculopathy Diseases 0.000 description 1
- 208000033626 Renal failure acute Diseases 0.000 description 1
- 208000002200 Respiratory Hypersensitivity Diseases 0.000 description 1
- 208000007014 Retinitis pigmentosa Diseases 0.000 description 1
- 206010039705 Scleritis Diseases 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- 208000009359 Sezary Syndrome Diseases 0.000 description 1
- 208000021388 Sezary disease Diseases 0.000 description 1
- 101710124237 Short-type peptidyl-prolyl cis-trans isomerase Proteins 0.000 description 1
- 208000031709 Skin Manifestations Diseases 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 208000007107 Stomach Ulcer Diseases 0.000 description 1
- 241000187391 Streptomyces hygroscopicus Species 0.000 description 1
- 241001647839 Streptomyces tsukubensis Species 0.000 description 1
- 208000032851 Subarachnoid Hemorrhage Diseases 0.000 description 1
- 206010042742 Sympathetic ophthalmia Diseases 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 208000001106 Takayasu Arteritis Diseases 0.000 description 1
- 208000031981 Thrombocytopenic Idiopathic Purpura Diseases 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 241000223238 Trichophyton Species 0.000 description 1
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical class CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 1
- 206010044688 Trisomy 21 Diseases 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 208000024780 Urticaria Diseases 0.000 description 1
- 206010046851 Uveitis Diseases 0.000 description 1
- 208000001445 Uveomeningoencephalitic Syndrome Diseases 0.000 description 1
- 208000024248 Vascular System injury Diseases 0.000 description 1
- 208000035868 Vascular inflammations Diseases 0.000 description 1
- 208000012339 Vascular injury Diseases 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- 208000034705 Vogt-Koyanagi-Harada syndrome Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 206010000496 acne Diseases 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 231100000836 acute liver failure Toxicity 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 238000003915 air pollution Methods 0.000 description 1
- 230000010085 airway hyperresponsiveness Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000005107 alkyl diaryl silyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 201000009961 allergic asthma Diseases 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 208000004631 alopecia areata Diseases 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 238000013103 analytical ultracentrifugation Methods 0.000 description 1
- 238000002399 angioplasty Methods 0.000 description 1
- 230000007953 anoxia Effects 0.000 description 1
- 201000004612 anterior uveitis Diseases 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000000078 anti-malarial effect Effects 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000003435 aroyl group Chemical group 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 229940091771 aspergillus fumigatus Drugs 0.000 description 1
- 208000024998 atopic conjunctivitis Diseases 0.000 description 1
- 201000008937 atopic dermatitis Diseases 0.000 description 1
- 201000005000 autoimmune gastritis Diseases 0.000 description 1
- 201000003710 autoimmune thrombocytopenic purpura Diseases 0.000 description 1
- 229940116226 behenic acid Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 238000010322 bone marrow transplantation Methods 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 206010006451 bronchitis Diseases 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- NEDGUIRITORSKL-UHFFFAOYSA-N butyl 2-methylprop-2-enoate;2-(dimethylamino)ethyl 2-methylprop-2-enoate;methyl 2-methylprop-2-enoate Chemical compound COC(=O)C(C)=C.CCCCOC(=O)C(C)=C.CN(C)CCOC(=O)C(C)=C NEDGUIRITORSKL-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 235000001465 calcium Nutrition 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 239000007963 capsule composition Substances 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- 125000001721 carboxyacetyl group Chemical group 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 229920003123 carboxymethyl cellulose sodium Polymers 0.000 description 1
- 229940063834 carboxymethylcellulose sodium Drugs 0.000 description 1
- 231100000357 carcinogen Toxicity 0.000 description 1
- 239000003183 carcinogenic agent Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 239000004568 cement Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 208000026106 cerebrovascular disease Diseases 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 208000020832 chronic kidney disease Diseases 0.000 description 1
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 description 1
- 208000022831 chronic renal failure syndrome Diseases 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 206010009887 colitis Diseases 0.000 description 1
- 238000000748 compression moulding Methods 0.000 description 1
- 208000010247 contact dermatitis Diseases 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000002592 cumenyl group Chemical group C1(=C(C=CC=C1)*)C(C)C 0.000 description 1
- 208000004921 cutaneous lupus erythematosus Diseases 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 208000033679 diabetic kidney disease Diseases 0.000 description 1
- GPLRAVKSCUXZTP-UHFFFAOYSA-N diglycerol Chemical compound OCC(O)COCC(O)CO GPLRAVKSCUXZTP-UHFFFAOYSA-N 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 238000003912 environmental pollution Methods 0.000 description 1
- 201000001564 eosinophilic gastroenteritis Diseases 0.000 description 1
- 231100000321 erythema Toxicity 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- HHFAWKCIHAUFRX-UHFFFAOYSA-N ethoxide Chemical compound CC[O-] HHFAWKCIHAUFRX-UHFFFAOYSA-N 0.000 description 1
- 125000006351 ethylthiomethyl group Chemical group [H]C([H])([H])C([H])([H])SC([H])([H])* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 210000003414 extremity Anatomy 0.000 description 1
- 208000024711 extrinsic asthma Diseases 0.000 description 1
- 208000030533 eye disease Diseases 0.000 description 1
- 208000011318 facial edema Diseases 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000000855 fermentation Methods 0.000 description 1
- 230000004151 fermentation Effects 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 210000000245 forearm Anatomy 0.000 description 1
- 229940085942 formulation r Drugs 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 235000011389 fruit/vegetable juice Nutrition 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 201000005917 gastric ulcer Diseases 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 208000007565 gingivitis Diseases 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 208000018706 hematopoietic system disease Diseases 0.000 description 1
- 230000002949 hemolytic effect Effects 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 206010019692 hepatic necrosis Diseases 0.000 description 1
- 208000002672 hepatitis B Diseases 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- VZJYKFZAQPEYRF-UHFFFAOYSA-N hexadecanoic acid propane-1,2,3-triol Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCC(O)=O VZJYKFZAQPEYRF-UHFFFAOYSA-N 0.000 description 1
- AUORDTBEEXPMOT-UHFFFAOYSA-N hexadecanoic acid propane-1,2,3-triol Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCC(O)=O AUORDTBEEXPMOT-UHFFFAOYSA-N 0.000 description 1
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 1
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 230000003463 hyperproliferative effect Effects 0.000 description 1
- 201000009939 hypertensive encephalopathy Diseases 0.000 description 1
- 210000000835 hypertrophic cicatrix Anatomy 0.000 description 1
- 206010021198 ichthyosis Diseases 0.000 description 1
- 201000002597 ichthyosis vulgaris Diseases 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 229940099472 immunoglobulin a Drugs 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000004968 inflammatory condition Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 208000030603 inherited susceptibility to asthma Diseases 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 208000028774 intestinal disease Diseases 0.000 description 1
- 208000020658 intracerebral hemorrhage Diseases 0.000 description 1
- 201000010849 intracranial embolism Diseases 0.000 description 1
- 201000010659 intrinsic asthma Diseases 0.000 description 1
- 208000023569 ischemic bowel disease Diseases 0.000 description 1
- 125000006352 iso-propylthiomethyl group Chemical group [H]C([H])([H])C([H])(SC([H])([H])*)C([H])([H])[H] 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 210000001117 keloid Anatomy 0.000 description 1
- 201000010666 keratoconjunctivitis Diseases 0.000 description 1
- 238000004898 kneading Methods 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 201000002364 leukopenia Diseases 0.000 description 1
- 231100001022 leukopenia Toxicity 0.000 description 1
- VNYSSYRCGWBHLG-AMOLWHMGSA-N leukotriene B4 Chemical compound CCCCC\C=C/C[C@@H](O)\C=C\C=C\C=C/[C@@H](O)CCCC(O)=O VNYSSYRCGWBHLG-AMOLWHMGSA-N 0.000 description 1
- GWNVDXQDILPJIG-NXOLIXFESA-N leukotriene C4 Chemical compound CCCCC\C=C/C\C=C/C=C/C=C/[C@H]([C@@H](O)CCCC(O)=O)SC[C@@H](C(=O)NCC(O)=O)NC(=O)CC[C@H](N)C(O)=O GWNVDXQDILPJIG-NXOLIXFESA-N 0.000 description 1
- 201000011486 lichen planus Diseases 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- 231100000149 liver necrosis Toxicity 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 208000008585 mastocytosis Diseases 0.000 description 1
- 231100001016 megaloblastic anemia Toxicity 0.000 description 1
- IQSHMXAZFHORGY-UHFFFAOYSA-N methyl prop-2-enoate;2-methylprop-2-enoic acid Chemical compound COC(=O)C=C.CC(=C)C(O)=O IQSHMXAZFHORGY-UHFFFAOYSA-N 0.000 description 1
- 244000000010 microbial pathogen Species 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 201000006938 muscular dystrophy Diseases 0.000 description 1
- 206010028417 myasthenia gravis Diseases 0.000 description 1
- 201000005962 mycosis fungoides Diseases 0.000 description 1
- 210000003098 myoblast Anatomy 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- RIWRFSMVIUAEBX-UHFFFAOYSA-N n-methyl-1-phenylmethanamine Chemical class CNCC1=CC=CC=C1 RIWRFSMVIUAEBX-UHFFFAOYSA-N 0.000 description 1
- 125000001038 naphthoyl group Chemical group C1(=CC=CC2=CC=CC=C12)C(=O)* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 208000004995 necrotizing enterocolitis Diseases 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 201000008383 nephritis Diseases 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- CFZGEMKIQUVTCC-UHFFFAOYSA-N octacos-18-ene-2,3,10,16-tetrone Chemical compound CCCCCCCCCC=CCC(=O)CCCCCC(=O)CCCCCCC(=O)C(C)=O CFZGEMKIQUVTCC-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- BMANBDGYGRWCMJ-UHFFFAOYSA-N octadecanoic acid;propane-1,2,3-triol Chemical compound OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.OCC(O)CO.CCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O BMANBDGYGRWCMJ-UHFFFAOYSA-N 0.000 description 1
- 229960002446 octanoic acid Drugs 0.000 description 1
- 208000017945 ocular siderosis Diseases 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 235000021313 oleic acid Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 201000005737 orchitis Diseases 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 201000001976 pemphigus vulgaris Diseases 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 201000006195 perinatal necrotizing enterocolitis Diseases 0.000 description 1
- 208000028169 periodontal disease Diseases 0.000 description 1
- 201000001245 periodontitis Diseases 0.000 description 1
- 210000004261 periodontium Anatomy 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 208000015385 phacoanaphylactic uveitis Diseases 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 1
- 230000036211 photosensitivity Effects 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- KASDHRXLYQOAKZ-OLHLVPFQSA-N pimecrolimus Chemical compound C/C([C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@]2(O)O[C@@H]([C@H](C[C@H]2C)OC)[C@@H](OC)C[C@@H](C)C\C(C)=C/[C@H](C(C[C@H](O)[C@H]1C)=O)CC)=C\[C@@H]1CC[C@H](Cl)[C@H](OC)C1 KASDHRXLYQOAKZ-OLHLVPFQSA-N 0.000 description 1
- 210000002381 plasma Anatomy 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 201000006292 polyarteritis nodosa Diseases 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 208000005987 polymyositis Diseases 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 206010036601 premature menopause Diseases 0.000 description 1
- 201000000742 primary sclerosing cholangitis Diseases 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000003014 reinforcing effect Effects 0.000 description 1
- 238000002271 resection Methods 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 206010039083 rhinitis Diseases 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- 208000010157 sclerosing cholangitis Diseases 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229940023144 sodium glycolate Drugs 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- TUNFSRHWOTWDNC-HKGQFRNVSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCC[14C](O)=O TUNFSRHWOTWDNC-HKGQFRNVSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 238000002076 thermal analysis method Methods 0.000 description 1
- 230000001732 thrombotic effect Effects 0.000 description 1
- KJAMZCVTJDTESW-UHFFFAOYSA-N tiracizine Chemical compound C1CC2=CC=CC=C2N(C(=O)CN(C)C)C2=CC(NC(=O)OCC)=CC=C21 KJAMZCVTJDTESW-UHFFFAOYSA-N 0.000 description 1
- YXFVVABEGXRONW-UHFFFAOYSA-N toluene Substances CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 1
- 125000005147 toluenesulfonyl group Chemical group C=1(C(=CC=CC1)S(=O)(=O)*)C 0.000 description 1
- 125000005425 toluyl group Chemical group 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 230000008359 toxicosis Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 210000003437 trachea Anatomy 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 125000002306 tributylsilyl group Chemical group C(CCC)[Si](CCCC)(CCCC)* 0.000 description 1
- 150000005691 triesters Chemical class 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- PVNIQBQSYATKKL-UHFFFAOYSA-N tripalmitin Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCC PVNIQBQSYATKKL-UHFFFAOYSA-N 0.000 description 1
- JEJAMASKDTUEBZ-UHFFFAOYSA-N tris(1,1,3-tribromo-2,2-dimethylpropyl) phosphate Chemical compound BrCC(C)(C)C(Br)(Br)OP(=O)(OC(Br)(Br)C(C)(C)CBr)OC(Br)(Br)C(C)(C)CBr JEJAMASKDTUEBZ-UHFFFAOYSA-N 0.000 description 1
- 231100000397 ulcer Toxicity 0.000 description 1
- 208000009852 uremia Diseases 0.000 description 1
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 201000005539 vernal conjunctivitis Diseases 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 201000001862 viral hepatitis Diseases 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
- 125000005023 xylyl group Chemical group 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/453—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/436—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having oxygen as a ring hetero atom, e.g. rapamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/143—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/145—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/146—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
- A61K9/1623—Sugars or sugar alcohols, e.g. lactose; Derivatives thereof; Homeopathic globules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1635—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1641—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1641—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poloxamers
- A61K9/1647—Polyesters, e.g. poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Biophysics (AREA)
- Immunology (AREA)
- Oncology (AREA)
- Inorganic Chemistry (AREA)
- Communicable Diseases (AREA)
- Transplantation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Saccharide Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Pyrane Compounds (AREA)
Description
- Die vorliegende Erfindung bezieht sich auf eine Formulierung zur Verwendung auf einem medizinischen Gebiet, die eine Makrolidverbindung enthält und die mit der Eigenschaft einer außerordentlich guten, verzögerten Freisetzung ausgestattet ist.
- Eine orale Formulierung von einer der Makrolidverbindungen, nämlich Tacrolimus mit einer nützlichen immunosuppressiven Aktivität, wurde als eine Feststoffdispersions-zusammensetzung hergestellt, die eine schnelle Freisetzungseigenschaft unter Verwendung von Polymeren, wie Hydroxypropylmethylcellulose, und einem Lösungsvermittler (siehe zum Beispiel
EP 0 240 773 ) besitzt. Aufgrund der Gegenwart des Lösungsvermittlers ist diese Formulierung eine Formulierung mit schneller Freisetzung. Aufgrund der hohen Absorbierbarkeit wird sie auf dem klinischen Gebiet hoch eingeschätzt. Alternativ wurde in der klinischen Praxis das Auftreten einer oralen Tacrolimus-Formulierung mit einer ausreichenden langen Wirkung und einer ausgezeichneten oralen Absorbierbarkeit erwartet. Für einen Fachmann ist jedoch der Stand der Technik, dass die Absorbierbarkeit eines pharmazeutisch aktiven Wirkstoffes, der oral in der Form einer Formulierung mit verzögerter Freisetzung gegeben wird, allgemein reduziert ist, und/oder dass eine nicht vernachlässigbare Veränderung der Absorbierbarkeit beobachtet wird. Die Erfinder der vorliegenden Erfindung haben eine Reihe von Untersuchungen durchgeführt. Die Erfinder haben Formulierungen mit verzögerter Freisetzung von Makrolidverbindungen erfunden, wobei ein Vertreter davon Tacrolimus ist. Die Formulierungen sind dadurch gekennzeichnet, dass die Makrolidverbindung ausgezeichnet oral absorbiert wird und/oder dass die Variation ihrer Absorbierbarkeit unterdrückt ist. - Die vorliegende Erfindung bezieht sich auf eine Makrolid-Formulierung mit verzögerter Freisetzung, wobei das Lösen der Makrolidverbindung unter verzögerter Freisetzung erfolgt.
- Es ist eine Aufgabe der Erfindung eine Makrolid-Formulierung mit verzögerter Freisetzung bereitzustellen, worin die Zeit (T 63,2%), die für das Lösen von 63,2% der maximalen Menge der Makrolidverbindung notwendig ist, 0,7 bis 15 Stunden beträgt, wie gemessen gemäß der japanischen Pharmacopoeia, 13. Ausgabe, Lösungstest, Nr. 2 (Paddle-Verfahren, 50 Umdrehungen pro Minute) unter Verwendung einer Testlösung, die eine wässrige 0,005% Hydroxypropylcelluloselösung ist, welche auf einen pH von 4,5 eingestellt wurde.
- Es ist eine weitere Aufgabe der Erfindung eine Feststoffdispersionszusammensetzung einer Makrolidverbindung bereitzustellen, die für die oben erwähnte Formulierung mit verzögerter Freisetzung verwendbar ist, worin die Makrolidverbindung in einem amorphen Zustand auf Feststoffbasis vorliegt.
- Der T 63,2%-Wert, der gemäß der Erfindung mit dem Lösungstest bestimmt wird, kann aus der Freisetzungskurve bestimmt werden, die durch das Auftragen von Testdaten auf Koordinatenpapier erstellt wird. Das Freisetzungsprofil eines Arzneimittels kann jedoch im Allgemeinen durch Anpassung der Daten des Lösungstests auf ein Freisetzungsmodell analysiert werden und solch ein Verfahren kann auch bei der Berechnung des T 63,2%-Wertes benutzt werden. Das Modell für die Anpassung, welches verwendet werden kann, umfasst das Modell erster Ordnung oder ein lineares Modell, ein Modell nullter Ordnung, ein Kubikwurzelmodell, etc., wie dies beschrieben ist in Yamaoka, K. & Yagahara, Y.; Introduction to Pharmacokinetics wich a Microcomputer, Nankodo, S. 138. Als ein Modell, mit dem alle Arten von Freisetzungsmustern mit der höchsten Gültigkeit gezeigt werden können, ist die Weibull-Funktion bekannt, die in dem obigen Buch beschrieben ist sowie in L. J. Leeson & J. T. Carstensen (Hrsg.): Release of Pharmaceutical Products (American Pharmaceutical Society) (Chizin Shokan), S. 192–195.
- Die Weibull-Funktion ist eine Funktion, mit der die Auflösungsrate (%) mit der Zeit (T) durch die folgende Gleichung ausgedrückt werden kann;
- Um die Daten des Lösungstests auf die Weibull-Funktion anzupassen und die entsprechenden Parameter zu berechnen, wird die nicht-lineare Methode der kleinsten Quadrate verwendet, die beschrieben ist in Yamaoka, K. & Yagahara, Y.; Introduction to Pharmacokinetics with a Microcomputer, Nankodo, S. 40, wie oben erwähnt. Die Parameter werden zu einem Zeitpunkt bestimmt, wo die Summe der Quadrate der Differenzen zwischen den Werten, welche durch die obige Gleichung berechnet werden, und den gemessenen Werten zu jedem Zeitpunkt minimal ist. Die Auflösungskurve, welche unter Verwendung dieser Parameter mit der obigen Gleichung berechnet wird, ist die Kurve, welche am genauesten die gemessenen Werte repräsentiert.
- Die Bedeutung von jedem Parameter der Weibull-Funktion wird nun erläutert.
- Dmax (maximale Auflösungsrate) ist die maximale Auflösungsrate bei unendlicher Zeit, wie oben erwähnt, wobei im Allgemeinen der Wert von Dmax vorzugsweise so nah wie möglich an 100 (%) liegt.
- m (Skalenparameter) ist ein Parameter, der die Auflösungsgeschwindigkeit eines pharmazeutischen Produktes darstellt. Je kleiner der Wert von m ist, desto größer ist die Auflösungsgeschwindigkeit und in ähnlicher Weise, je größer der Wert von m ist, desto niedriger ist die Auflösungsgeschwindigkeit.
- n (Formparameter) ist ein Parameter, der die Form einer Auflösungskurve darstellt. Wenn der Wert von n 1 ist, kann die Weibull-Funktion als Auflösungsrate (%) = Dmax × (1 – exp[–(T – Ti)/m]) geschrieben werden. Da dies äquivalent zu einer Kinetik erster Ordnung ist, ist die Auflösungskurve linear. Wenn der Wert von n kleiner als 1 ist, bildet die Auflösungskurve ein Plateau. Wenn der Wert von n größer als 1 ist, herrscht eine sigmoide Auflösungskurve vor.
- Ti (Positionsparameter) ist ein Parameter, der die Verzögerungsphase bis zum Start der Auflösung darstellt.
- Die Makrolid-Formulierung mit verzögerter Freisetzung gemäß der vorliegenden Erfindung kann auch mittels dieser Weibull-Funktion charakterisiert werden. Die aufgabengemäße Formulierung mit verzögerter Freisetzung kann somit realisiert werden, indem Dmax (maximale Auflösungsrate) auf 80% oder mehr gesetzt wird, vorzugsweise auf 90% oder mehr, wobei 95% oder mehr noch stärker bevorzugt sind, m (Skalenparameter) auf 0,7–20 gesetzt wird, vorzugsweise 1–12, wobei 1,5–8 mehr bevorzugt ist, n (Formparameter) auf 0,2–5 gesetzt wird, vorzugsweise auf 0,3–3, wobei 0,5–1,5 mehr bevorzugt ist, und Ti (Positionsparameter) auf 0–12 gesetzt wird, vorzugsweise auf 0–8, wobei 0–4 mehr bevorzugt ist.
- Der Wert, der durch die Substitution der Parameterwerte von m und n von der obigen Weibull-Funktion in dem Ausdruck m1/n gefunden wird, stellt die Zeit dar, in der 63,2% der maximalen Auflösungsmenge des Wirkstoffes von der Formulierung (T 63,2%) freigesetzt wird. Dies bedeutet T 63,2% (Stunde) = m1/n. Die Freisetzungscharakteristik der Formulierung mit verzögerter Freisetzung gemäß der Erfindung kann mit dem Lösungstest, Verfahren 2 (Paddle-Verfahren, 50 Umdrehungen pro Minute) von JP XIII ausgewertet werden, wobei eine Testlösung verwendet wird, die eine wässrige, 0,005% Lösung von Hydroxypropylcellulose ist, die auf einen pH von 4,5 eingestellt ist. Bei der Makrolid-Formulierung mit verzögerter Freisetzung gemäß der vorliegenden Erfindung beträgt die Zeit (T 63,2%), in der 63,2% der maximalen Menge der zu lösenden Makrolidverbindung von der Formulierung freigesetzt wird, 0,7–15 Stunden. In der Vergangenheit wurde eine schnell freisetzende Formulierung mit einer Makrolidverbindung bereits hergestellt, wobei aber bisher noch keine Formulierungen mit verzögerter Freisetzung hergestellt wurden, worin T 63,2% 0,7–15 Stunden beträgt und die für die klinische Praxis nützlich wären. Die vorliegende Erfindung schafft dies zum ersten Mal. Wenn der T 63,2%-Wert kürzer als 0,7 Stunden ist, wird die Wirksamkeit der Makrolidverbindung nach oraler Verabreichung nicht ausreichend verzögert werden. Wenn die Formulierung einen T 63,2%-Wert von mehr als 15 Stunden hat, wird die Freisetzung des Wirkstoffes so verzögert werden, dass der Wirkstoff aus dem Körper eliminiert wird, bevor eine effektive Blutkonzentration erreicht ist. Dies wäre für eine Formulierung gemäß der Erfindung nicht geeignet. Wenn T 63,2% 1,0–12 Stunden beträgt, kann eine günstigere verzögerte Freisetzung erzielt werden. Mehr bevorzugt ist ein T 63,2%-Wert von 1,3–8,2 Stunden, wobei am meisten bevorzugt eine Formulierung mit verzögerter Freisetzung ist, wo der T 63,2%-Wert 2–5 Stunden beträgt.
- Der Ausdruck „Makrolidverbindung" zur Verwendung gemäß der Erfindung ist ein generischer Name von Verbindungen mit 12 Mitgliedern oder mehr, die zu den Lactonen mit einem großen Ring gehören. Zahlreiche Makrolidverbindungen, die von Mikroorganismen der Art Streptomyces, wie Rapamycin, Tacrolimus (FK506) und Ascomycin, sowie die Analoga und die Derivate davon sind von dem Ausdruck Makrolidverbindung umfasst.
- Die Makrolidverbindung nach der Erfindung ist eine tricyclische Verbindung mit der folgenden Formel (I).worin die benachbarten Paare von R1 und R2, R3 und R4 und R5 und
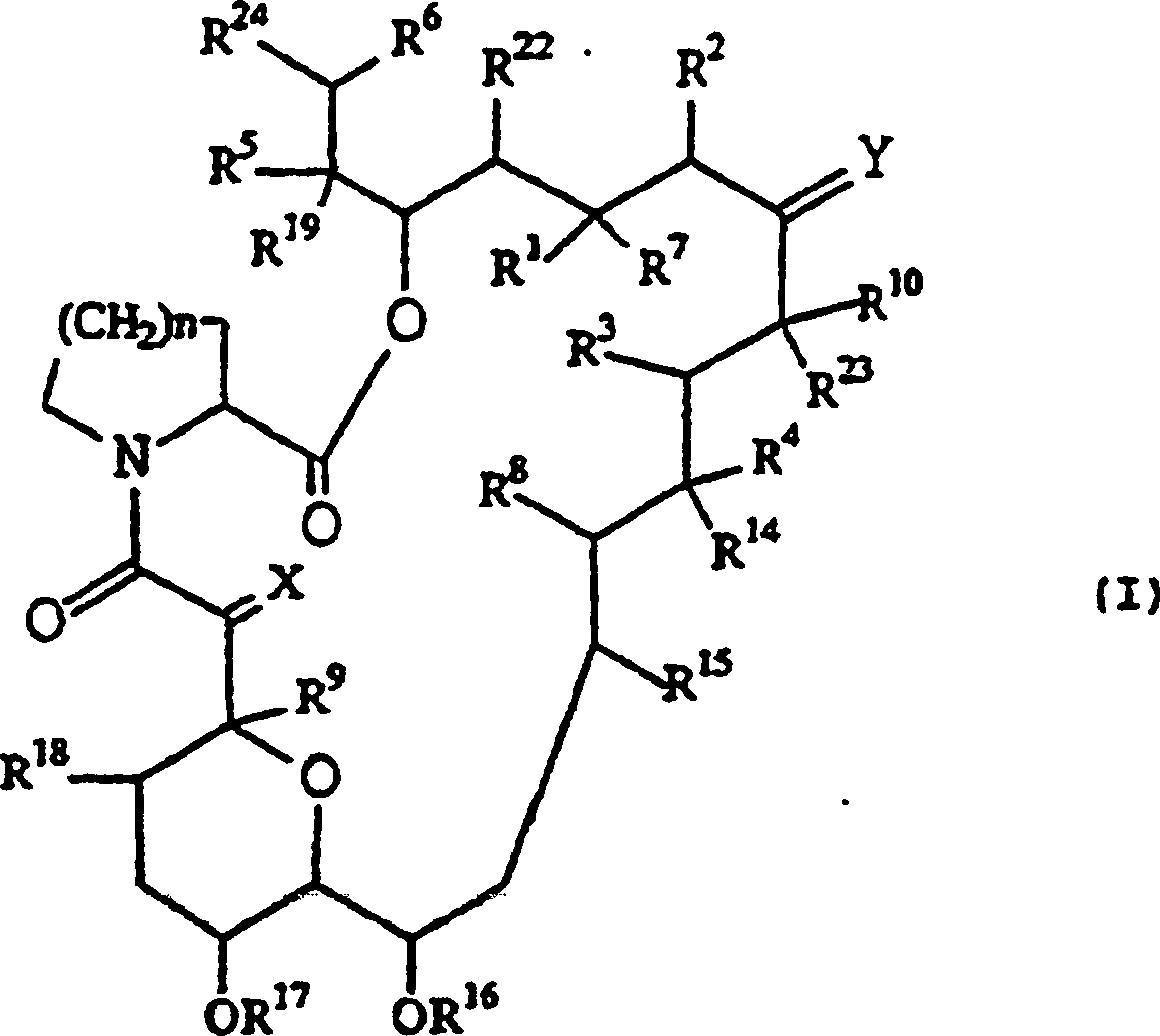
R6 jeweils unabhängig voneinander - (a) zwei benachbarte Wasserstoffatome sind, wobei aber R2 auch eine Alkylgruppe sein kann, oder
- (b) eine andere Bindung bilden können, die zwischen den Kohlenstoffatomen, mit denen sie verbunden sind, ausgebildet ist;
- R24 kann eine Cyclo(C5-7) alkylgruppe sein, die durch die folgenden Gruppen dargestellt ist,
- (a) eine 3,4-Dioxocyclohexylgruppe;
- (b) eine 3-R20-4-R21-Cyclohexylgruppe, bei der R20 Hydroxy, eine Alkoxygruppe, eine Oxogruppe oder eine -OCH2OCH2CH2OCH3-Gruppe ist, und R21 ist Hydroxy, -OCN, eine Alkoxygruppe, ein Heteroaryloxy, das mit geeigneten Substituenten substituiert sein kann, eine -OCH2OCH2CH2OCH3-Gruppe, eine geschützte Hydroxygruppe, Chlor, Brom, Iod, Aminooxalyloxy, eine Azidogruppe, p-Tolyloxythiocarbonyloxy oder R25R26CHCOO-, wobei R25 ein optional geschütztes Hydroxy oder geschütztes Amino ist und R26 ist Wasserstoff oder Methyl, oder R20 und R21 bilden zusammen ein Sauerstoffatom in einem Epoxidring; oder
- (c) eine Cyclopentylgruppe, die substituiert ist mit Methoxymethyl, optional geschütztes Hydroxymethyl, Acyloxymethyl (worin die Acylgruppe optional eine Dimethylaminogruppe, die quarternisiert sein kann, oder eine Carboxygruppe enthält, die verestert sein kann), eine oder mehrere Amino- und/oder Hydroxygruppen, die geschützt sein können, oder Aminooxalyloxymethyl. Ein bevorzugtes Beispiel ist eine 2-Formylcyclopentylgruppe.
- Die in der obigen, allgemeinen Formel (I) benutzten Definitionen sowie die spezifischen und bevorzugten Beispiele dafür werden nun erläutert und im Detail festgelegt werden.
- Der Ausdruck „nieder" bedeutet, soweit nichts anderes angezeigt ist, eine Gruppe mit 1 bis 6 Kohlenstoffatomen.
- Bevorzugte Beispiele für die „Alkylgruppen" und eine Alkylkomponente der „Alkoxygruppe" umfassen einen geraden oder verzweigten, aliphatischen Kohlenwasserstoffrest, zum Beispiel eine niedere Alkylgruppe, wie Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, Pentyl, Neopentyl und Hexyl.
- Bevorzugte Beispiele für die „Alkenylgruppen" umfassen einen geraden oder verzweigten, aliphatischen Kohlenwasserstoffrest mit einer Doppelbindung, zum Beispiel eine niedere Alkenylgruppe, wie Vinyl, Propenyl (z. B. Allylgruppe), Butenyl, Methylpropenyl, Pentenyl und Hexenyl.
- Bevorzugte Beispiele für die „Arylgruppen" umfassen Phenyl, Tolyl, Xylyl, Cumenyl, Mesityl und Naphthyl.
- Bevorzugte Schutzgruppen in den „geschützten Hydroxygruppen" und in dem geschützten Amino sind 1-(niederes Alkylthio)-(niedere) alkylgruppe, wie eine niedere Alkylthiomethylgruppe (z. B. Methylthiomethyl, Ethylthiomethyl, Propylthiomethyl, Isopropylthiomethyl, Butylthiomethyl, Isobutylthiomethyl, Hexylthiomethyl, etc.), wobei eine C1-C4 Alkylthiomethylgruppe mehr bevorzugt ist und eine Methylthiomethylgruppe am bevorzugtesten ist; eine trisubstituierte Silylgruppe, wie ein Tri (niederes) alkylsilyl (z. B. Trimethylsilyl, Triethylsilyl, Tributylsilyl, tert-Butyldimethylsilyl, tri-tert-Butylsilyl, etc.) oder niederes Alkyldiarylsilyl (z. B. Methyldiphenylsilyl, Ethyldiphenylsilyl, Propyldiphenylsilyl, tert-Butyldiphenylsilyl, etc.), wobei eine Tri (C1-C4) alkylsilylgruppe und eine C1-C4 Alkyldiphenylsilylgruppe mehr bevorzugt sind, wobei eine tert-Butyldimethylsilylgruppe und eine tert-Butyldiphenylsilylgruppe am meisten bevorzugt sind, und eine Acylgruppe, wie eine aliphatische, aromatische Acylgruppe oder eine aliphatische Acylgruppe, die mit einer aromatischen Gruppe substituiert ist, die von einer Carbonsäure, Sulfonsäure oder Carbamidsäure abstammen.
- Beispiele für die aliphatischen Acylgruppen umfassen eine niedere Alkanoylgruppe, die optional einen oder mehrere, geeignete Substituenten hat, wie Carboxy, z. B. Formyl, Acetyl, Propionyl, Butyryl, Isobutyryl, Valeryl, Isovaleryl, Pivaloyl, Hexanoyl, Carboxyacetyl, Carboxypropionyl, Carboxybutyryl, Carboxyhexanoyl, etc.; eine Cyclo (niedere) alkoxy (niedere) Alkanoylgruppe, die optional einen oder mehrere, geeignete Substituenten hat, wie niederes Alkyl, z. B. Cyclopropyloxyacetyl, Cyclobutyloxypropionyl, Cycloheptyloxybutyryl, Menthyloxyacetyl, Menthyloxypropionyl, Menthyloxybutyryl, Menthyloxypentanoyl, Menthyloxyhexanoyl, etc.; eine Camphorsulfonylgruppe oder eine niedere Alkylcarbamoylgruppe, die einen oder mehrere, geeignete Substituenten hat, wie Carboxy oder geschütztes Carboxy, z. B. Carboxy (niederes) alkylcarbamoylgruppe (z. B. Carboxymethylcarbamoyl, Carboxyethylcarbamoyl, Carboxypropylcarbamoyl, Carboxybutylcarbamoyl, Carboxypentylcarbamoyl, Carboxyhexylcarbamoyl, etc.), Tri (niederes) alkylsilyl (niederes) alkoxycarbonyl (niedere) alkylcarbamoylgruppe (z. B. Trimethylsilylmethoxycarbonylethylcarbamoyl, Trimethylsilylethoxycarbonylpropyl-carbamoyl, Triethylsilylethoxycarbonylpropylcarbamoyl, tert-Butyldimethylsilylethoxycarbonylpropylcarbamoyl, Trimethylsilylpropoxycarbonylbutylcarbamoyl, etc.) und so weiter.
- Beispiele für die aromatischen Acylgruppen umfassen eine Aroylgruppe, die optional einen oder mehrere, geeignete Substituenten hat, wie Nitro, z. B. Benzoyl, Toluoyl, Xyloyl, Naphthoyl, Nitrobenzoyl, Dinitrobenzoyl, Nitronaphthoyl, etc.; und eine Arensulfonylgruppe, die optional einen oder mehrere, geeignete Substituenten hat, z. B. Benzensulfonyl, Toluolsulfonyl, Xylensulfonyl, Naphthalinsulfonyl, Fluorbenzensulfonyl, Chlorbenzensulfonyl, Brombenzensulfonyl, Iodbenzensulfonyl, etc.
- Beispiele für die aliphatischen Acylgruppen, die mit einer aromatischen Gruppe substituiert sind, umfassen eine Ar (niedere) Alkanoylgruppe, die optional einen oder mehrere geeignete Substituenten hat, wie niederes Alkoxy oder Trihalo(niederes)alkyl, z. B. Phenylacetyl, Phenylpropionyl, Phenylbutyryl, 2-Trifluormethyl-2-methoxy-2-phenylacetyl, 2-Ethyl-2-trifluormethyl-2-phenylacetyl, 2-Trifluormethyl-2-propoxy-2-phenylacetyl, etc.
- Mehr bevorzugte Acylgruppen unter den oben genannten Acylgruppen sind eine C1-C4 Alkanoylgruppe, die optional Carboxy hat, Cyclo (C5-C6) alkoxy (C1-C4) Alkanoylgruppe mit zwei (C1-C4) Alkylen an der Cycloalkylkomponente, Camphorsulfonylgruppe, Carboxy (C1-C4) alkylcarbamoylgruppe, Tri (C1-C4) alkylsilyl (C1-C4) alkoxycarbonyl (C1-C4) Alkylcarbamoylgruppe, Benzoylgruppe, die optional eine oder zwei Nitrogruppen hat, Benzensulfonylgruppe mit Halogen, oder eine Phenyl(C1-C4)alkonoylgruppe mit C1-C4 Alkoxy und Trihalo(C1-C4)alkylgruppe. Unter diesen sind am meisten bevorzugt Acetyl, Carboxypropionyl, Menthyloxyacetyl, Camphorsulfonyl, Benzoyl, Nitrobenzoyl, Dinitrobenzoyl, Iodbenzensulfonyl und 2-Trifluormethyl-2-methoxy-2-phenylacetyl.
- Bevorzugte Beispiele für den "5- oder 6-gliedrigen, Stickstoff-, Schwefel- und/oder Sauerstoff- haltigen, heterocyclischen Ring" umfassen eine Pyrrolylgruppe und eine Tetrahydrofurylgruppe.
- "Ein Heteroaryl, das mit geeigneten Substituenten substituiert sein kann" -Komponente von „Heteroaryloxy, das mit geeigneten Substituenten substituiert sein kann" kann eine Komponente sein, die dargestellt ist für R1 der Verbindung der Formel von EP-A-532,088, wobei der Vorzug 1-Hydroxyethylindol-5-yl gegeben wird. Diese Veröffentlichung ist hiermit durch Verweis eingeführt.
- Die tricyclischen Verbindungen (I) und die pharmazeutisch akzeptablen Salze davon zur Verwendung gemäß der Erfindung sind hinsichtlich ihrer guten immunosuppressiven Aktivität, ihrer antimikrobiellen Aktivität und anderen pharmakologischen Aktivitäten gut bekannt und als solche von Wert für die Behandlung oder die Verhinderung von Abstoßungsreaktionen bei der Transplantation von Organen oder Geweben, Graft-versus-host-Krankheit, Autoimmunerkrankungen und Infektionserkrankungen [EP-A-0184162, EP-A-0323042, EP-A-423714, EP-A-427680, EP-A-465426, EP-A-480623, EP-A-532088, EP-A-532089, EP-A-569337, EP-A-626385, WO 89/05303, WO 93/05058, WO 96/31514, WO 91/13889, WO 91/19495, WO 93/5059, etc.].
- Insbesondere sind die Verbindungen, die bezeichnet sind als FR900506 (= FK506), FR900520 (ascomycin), FR900523 und FR900525, Produkte, die von Mikroorganismen der Art Streptomyces hergestellt werden, wie Streptomyces tsukubaensis Nr. 9993 [hinterlegt bei National Institute of Bioscience and Human Technology Agency of Industrial Science and Technology (zuvor Fementation Research Institute Agency of Industrial Science and Technology), 1–3, Higashi 1-chome, Tsukubashi, Ibaraki, Japan, Datum der Hinterlegung 5. Oktober 1984, Zugangsnummer FERM BP-927] oder Streptomyces hygroscopicus subsp. Yakushimaensis Nr. 7238 [hinterlegt bei National Institute of Bioscience and Human Technology Agency of Industrial Science and Technology (zuvor Fermentation Research Institute Agency of Industrial Science and Technology), 1–3, Higashi 1-chome, Tsukubashi, Ibaraki, Japan, Datum der Hinterlegung 12. Januar 1985, Zugangsnummer FERM BP-928] [EP-A-0184162]. FK506 (allgemeiner Name: Tacrolimus) mit der folgenden chemischen Struktur ist insbesondere eine repräsentative Verbindung.
-

- Chemischer Name: 17-Allyl-1,14-dihydroxy-12-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylvinyl]-23,25-dimethoxy-13,19,21,27-tetramethyl-11,28-dioxa-4-azatricyclo[22.3.1.04,9]octacos-18-en-2,3,10,16-tetraon.
- Die bevorzugten Beispiele für die tricyclischen Verbindungen (I) sind solche, worin jedes der benachbarten Paare von R3 und R4 oder von R5 und R6 unabhängig voneinander eine andere Bindung bilden, die zwischen den Kohlenstoffatomen, mit denen sie verbunden sind, ausgebildet ist;
R8 und R23 unabhängig voneinander ein Wasserstoffatom sind;
R9 eine Hydroxygruppe ist;
R10 eine Methylgruppe, eine Ethylgruppe, eine Propylgruppe oder eine Allylgruppe ist;
X (ein Wasserstoffatom und ein Wasserstoffatom) oder eine Oxogruppe ist;
Y eine Oxogruppe ist;
jedes von R14, R15, R16, R17, R18, R19 und R22 eine Methylgruppe ist;
R24 eine 3-R20-4-R21-Cyclohexylgruppe ist, worin R20 ein Hydroxy, eine Alkoxygruppe, eine Oxogruppe oder eine -OCH2OCH2CH2OCH3-Gruppe ist, und R23 ein Hydroxy, -OCN, eine Alkoxygruppe, ein Heteroaryloxy, das mit geeigneten Substituenten substituiert sein kann, eine -OCH2OCH2CH2OCH3-Gruppe, eine geschützte Hydroxygruppe, Chlor, Brom, Iod, Aminooxalyloxy, eine Azidogruppe, p-Tolyloxythiocarbonyloxy oder R25R26CHCOO- ist, worin R25 ein optional geschütztes Hydroxy oder ein geschütztes Amino ist, und R26 Wasserstoff oder Methyl ist, oder R20 und R21 zusammen ein Sauerstoffatom in einem Epoxidring bilden; und
n ist eine ganze Zahl von 1 oder 2. - Die am meisten bevorzugtesten, tricyclischen Verbindungen (I) sind zusätzlich zu FK506, Ascomycinderivate, wie ein halogeniertes Ascomycin (z. B. 33-epi-Chlor-33-desoxyascomycin), das in
EP 427,680 - Die tricyclischen Verbindungen (I) haben eine ähnliche Grundstruktur, das heißt, die tricyclische Makrolid-Struktur, und besitzen wenigstens eine ähnliche biologische Eigenschaft (z. B. die immunosuppressive Aktivität).
- Die tricyclischen Verbindungen (I) können in Salzform vorliegen, was bekannte, nicht-toxische und pharmazeutisch akzeptable Salze umfasst, wie ein Salz mit anorganischen oder organischen Basen, insbesondere ein Alkalimetallsalz, wie ein Natriumsalz und Kaliumsalz, ein Erdalkalimetallsalz, wie ein Calciumsalz und ein Magnesiumsalz, ein Ammoniumsalz und ein Aminsalz, wie ein Triethylaminsalz und ein N-Benzyl-N-methylaminsalz.
- Hinsichtlich der gemäß der vorliegenden Erfindung verwendeten Makrolidverbindung sei angemerkt, dass diese als Konformationsisomere und ein oder mehrere Stereoisomere, wie optische und geometrische Isomere, aufgrund des oder der asymmetrischen Kohlenstoffatome und Doppelbindungen vorliegen kann. Solche Konformationsisomere und Isomere sind auch vom Umfang der Makrolidverbindung gemäß der vorliegenden Erfindung umfasst. Die Makrolidverbindungen können auch als Solvate vorliegen, die ebenfalls vom Umfang der vorliegenden Erfindung umfasst sind. Bevorzugte Solvate sein ein Hydrat und ein Ethanolat.
- Die Formulierung mit verzögerter Freisetzung gemäß der vorliegenden Erfindung ist eine Formulierung, die eine Feststoffdispersionszusammensetzung umfasst, worin die Makrolidverbindung in einem amorphen Zustand auf Feststoffbasis vorliegt, die einen T 63,2%-Wert von 0,7 bis 15 Stunden zeigt. Die Gegenwart oder die Abwesenheit eines Beugungspeaks, der mit Röntgenkristallographie, thermischer Analyse usw. nachgewiesen wird, zeigt an, ob oder ob nicht eine Makrolidverbindung in dem amorphen Zustand auf einer Feststoffbasis in der Feststoffdispersionszusammensetzung vorhanden ist.
- Die Feststoffbasis nach der Erfindung ist eine wasserunlösliche, pharmazeutisch akzeptable Grundlage, die zur Zurückhaltung der Makrolidverbindung in einem amorphen Zustand in der Lage ist und die bei Umgebungstemperatur in einem festen Zustand vorliegt. Diese Grundlage wird ausgewählt aus Wachs und wasserunlöslichen Polymeren.
- Spezifische Beispiele für das Wachs umfassen Glycerinmonostearat und Saccharose-Fettsäureester (zum Beispiel Mono-, Di- oder Triester der Saccharose mit mittleren bis hohen Fettsäuren mit 8 bis 20 Kohlenstoffatomen, z. B. Caprylsäure, Caprinsäure, Laurinsäure, Myristinsäure, Palmitinsäure, Stearinsäure, Arachinsäure, Behensäure, Ölsäure, Linolsäure, etc.). Zusätzliche Beispiele für das Wachs umfassen Polyglycerin-Fettsäureester. Jeder Polyglycerin-Fettsäureester, einschließlich Monoester, Diester oder Polyester von Polyglycerin mit Fettsäuren, ist zufriedenstellend. Spezifische Beispiele für die Polyglycerin- Fettsäureester umfassen Tetraglycerinhexabehenat, Decaglycerinmonocaprylat, Triglycerindicaprylat, Triglycerindidecanoat, Tetraglycerinmonolaurat, Hexaglycerinmonolaurat, Decaglycerinmonolaurat, Tetraglycerinmonooleat, Hexaglycerinmonooleat, Decaglycerinmonooleat, Triglycerindioleat, Tetraglycerindioleat, Decaglycerinsesquioleat, Tetraglycerinpentaoleat, Hexaglycerinpentaoleat, Decaglycerindecaoleat, Heptaglycerinmonolinoleat, Triglycerindilinoleat, Tetraglycerindilinoleat, Hexaglycerindilinoleat, Diglycerinmonostearat, Tetraglycerinmonostearat, Hexaglycerinmonostearat, Decaglycerinmonostearat, Tetraglycerintristearat, Hexaglycerintristearat, Hexaglycerinsesquistearat, Tetraglycerinpentastearat, Hexaglycerinpentastearat, Decaglycerindecastearat, Tetraglycerinmonopalmitat, Hexaglycerinmonopalmitat, Decaglycerinmonopalmitat, Tetraglycerintripalmitat, Hexaglycerintripalmitat, Hexaglycerinsesquipalmitat, Tetraglycerinpentapalmitat, Hexaglycerinpentapalmitat und Decaglycerindecapalmitat. Bevorzugte Polyglycerin-Festtsäureester sind zum Beispiel Tetraglycerinhexabehenat (z. B. Poem J-46B unter einem Handelsnamen, hergestellt von RikenVitamin Co., Ltd.), Tetraglycerinpentastearat (z. B. PS-310 unter einem Handelsnamen, hergestellt von Sakamoto Yakuhin Kogyo Co., Ltd.), Tetraglycerinmonostearat (z. B. MS-310 unter einem Handelsnamen, hergestellt von Sakamoto Yakuhin Kogyo Co., Ltd.), Hexaglycerinpentastearat (PS-500 unter einem Handelsnamen, hergestellt von Sakamoto Yakuhin Kogyo Co., Ltd.), Hexaglycerinsesquistearat (SS-500 unter einem Handelsnamen, hergestellt von Sakamoto Yakuhin Kogyo Co., Ltd.), Decaglycerinmonostearat und eine Mischung davon. Mehr bevorzugte Wachse sind Glycerinmonostearat und Nieder-HLB Saccharose-Festtsäureester (z. B. F-50, F-20- F-10, etc., hergestellt von Daiichi Kogyo Seiyaku, Co., Ltd.).
- Das Gewichtsverhältnis von der Makrolidverbindung und Wachs beträgt vorzugsweise 1 : 10 bis 1 : 100, mehr bevorzugt sind 1 : 40 bis 1 : 60, wenn das Wachs zum Beispiel Glycerinmonostearat ist. Das Gewichtsverhältnis ist vorzugsweise 1 : 0,2 bis 1 : 20, mehr bevorzugt sind 1 : 0,5 bis 1 : 5, wenn das Wachs zum Beispiel ein Saccharose-Fettsäureester ist, Das Gewichtsverhältnis beträgt vorzugsweise 1 : 0,1 bis 1 : 100, wobei 1 : 0,5 bis 1 : 50 mehr bevorzugt ist, wenn das Wachs ein Polyglycerin-Fettsäureester ist.
- Bevorzugte wasserunlösliche Polymere umfassen zum Beispiel Ethylcellulose, Methacrylat-Copolymere (z. B. Eudragits, wie Eudragit E, R, S, RS, LD, etc.). Falls das wasserunlösliche Polymer Ethylcellulose ist, kann eine pharmazeutisch akzeptable Ethylcellulose gemäß der vorliegenden Erfindung verwendet werden. Jedoch beträgt die bevorzugte Viskosität 3 bis 110 cps, mehr bevorzugt sind 6 bis 49 cps, wobei 9 bis 11 cps am meisten bevorzugt ist, wenn die Viskosität einer 5% Ethylcellulose-Toluol/Ethanol (80/20)-Lösung mit einem Viskositätstest gemessen wird, der in USP 23, NF18, beschrieben ist. Bevorzugt ist zum Beispiel ETHOCELL (Viskosität: 10) (Marke, Dow Chemical (USA)).
- Das Gewichtsverhältnis der Makrolidverbindung und des wasserunlöslichen Polymers beträgt vorzugsweise 1 : 0,01 bis 1 : 10, mehr bevorzugt ist 1 : 0,1 bis 1 : 5, wobei 1 : 0,1 bis 1 : 1 am meisten bevorzugt ist, wenn das wasserunlösliche Polymer Ethylcellulose ist. Ein Gewichtsverhältnis von 1 : 0,5 bis 1 : 5 ist am bevorzugtesten, wenn das wasserunlösliche Polymer ein Methacrylat-Copolymer ist.
- Die Formulierung mit verzögerter Freisetzung gemäß der Erfindung kann ferner eine wasserlösliche Basis enthalten und diese zugesetzte Basis ist vorzugsweise eines der folgenden wasserlöslichen Polymere:
- Polyvinylpyrrolidon (PVP), Cellulosepolymer (Hydroxypropylmethylcellulose (HPMC), Hydroxypropylmethylcellulose-Phthalat, Methylcellulose (MC), Carboxymethylcellulose-Natrium (CMC-Na), Hydroxyethylcellulose, Hydroxypropylcellulose (HPC), etc.), Pektin, Cyclodextrine, Galactomannan, Polyethylenglycol (PEG) mit einem durchschnittlichen Molekulargewicht von 4.000 oder mehr, Gelatine, etc.
- Für die Verwendung werden die wasserlöslichen Polymere individuell oder in einer Mischung von zwei oder mehr zugesetzt. Eine mehr bevorzugte, wasserlösliche Basis ist Cellulosepolymer oder PVP. Die am meisten bevorzugteste, wasserlösliche Basis ist HPMC, PVP oder eine Kombination davon. Insbesondere kann HPMC mit einer geringen Viskosität einen Effekt mit der wünschenswerten verzögerten Freisetzung ausüben. Eine wässrige 2% Lösung von diesem Typ von HPMC hat eine Viskosität von 1 bis 4.000 cps, vorzugsweise 1 bis 50 cps, mehr bevorzugt von 1 bis 15 cps, gemäß Messung bei 20°C mit einem Viskometer des Brookfield-Typs, wobei insbesondere HPMC 2910 mit einer Viskosität von 3 cps bevorzugt ist (TC-5E, EW, Shin-estu Chemical Co., Ltd.).
- Das Gewichtsverhältnis der Makrolidverbindung zu dieser wasserlöslichen Basis beträgt vorzugsweise 1 : 0,05 bis 1 : 2, mehr bevorzugt 1 : 0,1 bis 1 : 1, wobei 1 : 0,2 bis 1 : 0,4 am meisten bevorzugt ist.
- Bei der Herstellung der Feststoffdispersionszusammensetzung gemäß der vorliegenden Erfindung kann die obige, wasserunlösliche Basis allein oder in Kombination mit der wasserlöslichen Basis verwendet werden. Da die wasserunlösliche Basis als die essentielle Feststoffbasis nach der Erfindung festgelegt ist, kann ein geeignetes Lösungsprofil der Feststoffdispersionszusammensetzung erreicht werden, indem eine geeignete Menge der wasserlöslichen Basis zugemischt wird, wie ein wasserlösliches Polymer (z. B. HPMC). Falls gewünscht, können auch andere Komponenten als die oben beschriebene Feststoffbasis zugesetzt werden, um eine Feststoffdispersionszusammensetzung herzustellen, wie geeignete Trägerstoffe, (Lactose, etc.), Bindemittel, Färbemittel, Süßstoffe, Aromen, Verdünnungsmittel, Antioxidationsmittel (Vitamin E, etc.) und Gleitmittel (z. B. synthetisches Aluminiumsilicat, Magnesiumstearat, Calciumhydrogenphosphat, Calciumstearat, Talk, etc.).
- In Abhängigkeit von dem Typ der Feststoffbasis ist die Auflösungsrate der Makrolidverbindung aus der Feststoffdispersionszusammensetzung manchmal etwas langsam oder die anfängliche Auflösungsrate davon muss manchmal etwas angehoben werden. In diesem Fall kann die Auflösungsrate der Makrolidverbindung aus der Feststoffdispersionszusammensetzung eingestellt werden, indem der Feststoffdispersionszusammensetzung geeignete Lösungsvermittler (z. B. Cross-Carmelose-Natrium (CC-Na), Carboxymethylcellulose-Calcium (CM-Ca), gering substituierte Hydroxypropylcellulose (L-HPC), Stärke-Natriumglycolat, mikrofeine Kristallcellulose, Cross-Povidon, etc.) oder geeignete oberflächenaktive Mittel (z. B. gehärtetes Polyoxyethylen-Castoröl, Polyoxylstearat 40, Polysorbat 80, Natriumlaurylsulfat, Saccharose-Fettsäureester (HLB ist mehr als 10), etc.) zugesetzt werden.
- Die Partikelgröße der Feststoffdispersionszusammensetzung, worin die Makrolidverbindung in einem amorphen Zustand in einer Feststoffbasis vorliegt, ist gleich oder kleiner als 500 μm. Die Zusammensetzung hat insbesondere eine Partikelgröße, welche durch ein 350 μm, mehr bevorzugt sind 250 μm, Sieb hindurchtreten kann.
- Die Feststoffdispersionszusammensetzung der Makrolidverbindung, die in der Formulierung mit verzögerter Freisetzung gemäß der vorliegenden Erfindung vorliegt, kann durch die Verfahren hergestellt werden, die beschrieben sind in
EP 0 240 773 und WO 91/19495 und ähnliches. Die Verfahren werden im folgenden beschrieben. - Die Makrolidverbindung wird in einem organischen Lösungsmittel (z. B. Ethanol, Dichlormethan oder einer wässrigen Mischung davon, etc.) gelöst und anschließend wird eine geeignete Menge einer Feststoffbasis zugesetzt. Die erzielte Mischung wird ausreichend gelöst oder suspendiert oder sie wird quellen gelassen. Dann wird die Mischung ausreichend zusammengeknetet. Nach Entfernung des organischen Lösungsmittels von der Mischung wird der Rest getrocknet und zermahlen und wird dann einer Größenreduktion unterworfen, wodurch eine Feststoffdispersionszusammensetzung hergestellt werden kann, worin die Makrolidverbindung in einem amorphen Zustand in der Feststoffbasis vorliegt. Während des Knetverfahrens können, falls notwendig, Gleitmittel, wie Calciumhydrogenphosphat, Trägerstoffe, wie Lactose, und ähnliches der Mischung zugesetzt werden.
- Die Formulierung mit verzögerter Freisetzung, die eine Makrolidverbindung gemäß der Erfindung umfasst, kann hergestellt werden unter Verwendung eines fein aufgeteilten Pulvers der Makrolidverbindung. Die Kontrolle der Partikelgröße der Makrolidverbindung kann mittels einer Mühlenvorrichtung durchgeführt werden, was ein Routineverfahren in der pharmazeutischen Industrie ist, wie eine Stiftmühle, Hammermühle, Strahlmühle und eine trockene oder nasse Kugelmühle, um einige Beispiele zu nennen. Das feine Pulver der Makrolidverbindung sollte eine Verteilung des Partikeldurchmessers innerhalb des Bereiches von 0,1–50 μm haben, vorzugsweise von 0,2–20 μm, wobei 0,5–10 μm mehr bevorzugt sind, und/oder einen durchschnittlichen Partikeldurchmesser von 0,2–20 μm, vorzugsweise 0,5–10 μm, wobei 1–5 μm mehr bevorzugt sind.
- Die Feststoffdispersionszusammensetzung der Makrolidverbindung, welche durch die obigen Verfahren hergestellt wird, kann als solche als eine Formulierung mit verzögerter Freisetzung verwendet werden. Unter Berücksichtigung der Handhabbarkeit als Formulierung, der Dispergierbarkeit in Wasser und der Dispergierbarkeit nach oraler Dosierung wird die Zusammensetzung jedoch mehr bevorzugt als eine Formulierung mit verzögerter Freisetzung hergestellt in Form eines Pulvers, feinen Pulvers, Granulats, Tablette oder Kapsel, wobei Routineformulierungsverfahren angewandt werden (z. B. Formpressen).
- Falls gewünscht, kann die Formulierung mit verzögerter Freisetzung durch Mischen der Feststoffdispersionszusammensetzung der Makrolidverbindungen mit zum Beispiel Verdünnungsmittel oder Gleitmittel (wie Saccharose, Lactose, Stärke, kristalline Cellulose, synthetisches Aluminiumsilicat, Magnesiumstearat, Calciumstearat, Calciumhydrogen-phosphat und Talk) und/oder mit Färbemittel, Süßungsmittel, Geschmackstoffen und Lösungsvermittler für Routineanwendungen hergestellt werden. Die erzielte Mischung wird dann gründlich miteinander vermischt, um eine Formulierung mit verzögerter Freisetzung zu erzielen. Die Formulierung mit verzögerter Freisetzung oder die Feststoffdispersions-zusammensetzung der Makrolidverbindung nach der vorliegenden Erfindung kann vorläufig in Wasser oder Saft dispergiert werden, um sie als eine Flüssigformulierung oral aufzunehmen.
- Die effektive Dosis der Makrolidverbindung variiert in Abhängigkeit von dem Typ der Verbindung, dem Alter des Patienten, seiner Krankheit, der Heftigkeit der Erkrankung sowie von anderen Faktoren. Im Allgemeinen wird der Wirkstoff mit einer Dosis von etwa 0,001 bis 1.000 mg verwendet, vorzugsweise mit 0,01 bis 500 mg, wobei 0,1 bis 100 mg pro Tag der therapeutischen Behandlung einer Krankheit am meisten bevorzugt ist. Im Allgemeinen beträgt die durchschnittliche Einzeldosis etwa 0,01 mg, 0,1 mg, 0,5 mg, 1 mg, 5 mg, 10 mg, 50 mg, 100 mg, 250 mg und 500 mg.
- Nach oraler Verabreichung setzt die Makrolidformulierung mit verzögerter Freisetzung gemäß der vorliegenden Erfindung in charakteristischer Weise die Makrolidverbindung verzögert frei, so dass die pharmazeutische Aktivität über einen langen Zeitraum erhalten bleibt. Gemäß der Erfindung kann die Frequenz der Verabreichung der pharmakologisch aktiven Makrolidverbindung reduziert werden. Insbesondere ist es möglich, eine ein Makrolid enthaltende, pharmazeutische Formulierung bereitzustellen, die nur einmal am Tag verabreicht werden kann. Es ist ferner nun möglich, eine pharmazeutische Zusammensetzung bereitzustellen, die frei ist von dem Risiko von unerwünschten Nebenwirkungen, die durch vorrübergehend sehr hohe Konzentrationen hervorgerufen werden. Dies stellt die Expression der pharmakologischen Wirksamkeit in ausreichender Weise über einen verlängerten Zeitraum sicher.
- Die Formulierung mit verzögerter Freisetzung gemäß der vorliegenden Erfindung ist nützlich für die Behandlung und/oder Vorbeugung der folgenden Krankheiten und gesundheitlichen Zustände aufgrund der pharmakologischen Aktivitäten, welche diese tricyclischen Makrolidverbindungen (I) besitzen.
- Abstoßungsreaktionen durch Transplantation von Organen oder Geweben, wie Herz, Niere, Leber, Knochenmark, Haut, Hornhaut, Lunge, Pankreas, Dünndarm, Extremität, Muskel, Nerven, Zwischenwirbelscheibe, Trachea, Myoblast, Knorpel, etc.; Graft-versus-Host-Reaktionen nach Knochenmarkstransplantation;
Autoimmunerkrankungen, wie rheumatische Arthritis, systemische Lupus erythematosus, Hashimoto's Thyroiditis, multiple Sklerose, Myasthenia gravis, Diabetes Typ I, etc.; und Infektionen, die durch pathogene Mikroorganismen hervorgerufen werden (z. B. Aspergillus fumigatus, Fusarium oxysporum, Trichophyton asteroides, etc.);
entzündliche oder hyperproliferierende Hauterkrankungen oder Hautmanifestationen von immunologisch-vermittelten Krankheiten (z. B. Psoriasis, atopische Dermatitis, Kontaktdermatitis, Ekzematoidreaktion, Dermatitits seborrhoeica, Lichen planus, Pemphigus, Blasen-Pemphigoid, Epidermolysis bullosa, Urtikaria, allergisches Gesichtsödem, Vasculitis, Erythem, dermale Eosinophilie, Lupus erythematosus, Akne und Alopecia areata);
Autoimmunkrankheiten des Auge (z. B. Keratoconjunktivitis, vernale Konjunktivitis, Uveitis, assoziiert mit der Behcet-Krankheit, Keratitis, Herpeskeratitis, konische Keratitis, konische Epitheldystrophie, Keratoleukom, okulare Premphigus, Mooren's Ulcus, Skleritis, Graves' Ophthalmopathie, Vogt-Koyanagi-Harada-Syndrom, Keratoconjunktivitis sicca (trockenes Auge), Phlyktäne, Iridozyclitis, Sarkoidose, endokrine Ophthalmopathie, etc.);
reversible, obstruktive Luftwegeerkrankungen [Asthma (z. B. Bronchialasthma, allergisches Asthma, Intrinsic-Asthma, Extrinsic-Asthma und Staubasthma), insbesondere chronisches oder hartnäckiges Asthma (z. B. spätes Asthma und Luftweg-Hyperresponsivität) Bronchitis, etc.];
Schleimhaut- oder Gefäßentzündungen (z. B. Magengeschwüre, ischämische oder thrombotische Gefäßverletzung, ischämische Darmkrankheiten, Enteritis, nekrotisierende Enterokolitis, Darmschädigungen, verbunden mit Hitzeverbrennungen, Leukotrien B4-vermittelte Krankheiten);
Darmentzündungen/Allergien (z. B. zöliakable Krankheiten, Proktitis, eosinophile Gastroenteritis, Mastocytose, Crohn's Krankheit und Colitis ulcerosa);
Nahrungsmittel bedingte, allergische Krankheiten mit symptomatischen Manifestationen entfernt vom Gastrointestinaltrakt (z. B. Migräne, Rhinitis und Ekzem);
Nierenerkrankungen (z. B. Intestitialnephritis, Goodpasture-Syndrom, hämolytisches Urämiesyndrom und diabetische Nephropathie);
Nervenkrankheiten (z. B. multiple Myositis, Guillain-Barre-Syndrom, Morbus Meniere, multiple Neuritis, Solitärneuritis, Hirnschlag, Alzheimer-Krankheit, Parkinson-Krankheit, amyotrophe laterale Sclerosis (ALS) und Radikulopathie);
cerebrale ischämische Krankheit (z. B. Kopfverletzung, Blutung im Gehirn (z. B. Subarachnoidblutung, intracerebrale Blutung, cerebrale Thrombose, cerebrale Embolie, Herzstillstand, Schlaganfall, transienter ischämischer Anfall (TIA), hypertensive Encephalopathie, cerebraler Infarkt);
endokrine Erkrankungen (z. B. Hyperthyroidism und Basedow's Krankheit);
Blutkrankheiten (z. B. reine rot Blutzelloplasie, aplastische Anämie, hypoplastische Anämie, idiopathische thrombozytopenische Purpura, hämolythische Autoimmunanämie, Agranulocytose, perniziöse Anämie, megaloblastische Anämie und Anerythroplasie);
Knochenkrankheiten (z. B. Osteoporose);
Atemwegserkrankungen (z. B. Sarkoidose, Lungenfibrose und idiopathische interstitiale Pneumonie);
Hautkrankheiten (z. B. Dermatomyositis, Leukoderma vulgaris, Ichthyosis vulgaris, Lichtempfindlichkeit und T-Zell-Hautlymphom);
Kreislauferkrankungen (z. B. Arteriosklerose, Atherosklerose, Aortitis-Syndrom, Polyarteritis nodosa und Myocardose);
Kollagenkrankheiten (z. B. Sclerodermie, Wegener's Granuloma und Sjorgren's Syndrom);
Adipositas;
eosinophile Fasziitis;
Paradontose (z. B. Schädigungen des Zahnfleisches, des Periodontiums, des Alveolarknochens oder Substantia Ossea Dentis);
nephrotisches Syndrom (z. B. Glomerulonephritis);
männliches Alopezie, Alopezie senile;
muskuläre Dystrophie;
Pyoderma und Sezary-Syndrom;
mit Chromosomenanomalien verbundene Krankheiten (z. B. Down's Syndrom);
Addisonsche Krankheit;
durch Aktivsauerstoff vermittelte Erkrankungen [z. B. Organverletzung (z. B. ischämische Kreislaufstörungen der Organe (z. B. Herz, Leber, Niere, Verdauungstrakt, etc.), verbunden mit Aufbewahrung, Transplantation oder ischämische Krankheiten (z. B. Thrombose, Herzinfarkt, etc.));
Darmerkrankungen (z. B. Endotoxinschock, pseudomembranöse Colitis und Colitis, die durch Arzneimittel oder Strahlung induziert wird);
Nierenkrankheiten (z. B. ischämische, akute renale Insuffizienz, chronisches Nierenversagen);
Lungenkrankheiten (z. B. Toxikose, hervorgerufen durch Lungensauerstoff oder Arzneimittel (z. B. Paracort, Bleomycin, etc.), Lungenkrebs und Lungenemphysum);
Augenkrankheiten (z. B. Katarakt, Eisenspeicherkrankheit (Siderosis bulbi), Retinitis pigmentosa, senile Plaques, Glaskörpervernarbung, Augenverätzung durch Laugen);
Dermatitis (z. B. Erythema multiforme, lineare Immunoglobulin A-Blasendermatose, Zementdermatitis);
und andere Krankheiten (z. B. Gingivitis, Periodontitis, Sepsis, Pancreatitis und Krankheiten, die durch Umweltverschmutzung hervorgerufen werden (z. B. Luftverschmutzung), Alterung, Carcinogen, Metastasen von Karcinomen und Hypobaropathie);
Krankheiten, die durch Histaminfreisetzung oder Leukotrien C4-Freisetzung hervorgerufen werden;
Restenose der Herzarterien nach Angioplastik und Behinderung von postoperativen Verwachsungen;
Autoimmunkrankheiten und entzündliche Zustände (z. B. primäres Mukosaödem, autoimmune atrophische Autoimmungastritis, vorzeitige Menopause, männliche Sterilität, juveniler Diabetes mellitus, Pemphigus vulgaris, Pemphigoid, Ophthalmia sympathica, Linsen induzierte Uveitis, idiopathische Leukopenie, aktive chronische Hepatitis, idiopathische Zirrhose, Lupus erythematodes discoides, Autoimmunorchitis, Arthritis (z. B. Arthritis deformans) oder Polychondritis);
HIV-Virusinfektion, Aids;
allergische Conjunctivitis;
hypertrophe Cicatrix und Keloid aufgrund von Trauma, Verbrennung oder Operation. - Zusätzlich haben die tricyclischen Makrolide eine Leber stimulierende Aktivität und/oder Aktivitäten zur Stimulierung der Hypertrophie und Hyperplasie von Hepatozyten. Somit ist die pharmazeutische Zusammensetzung gemäß der vorliegenden Erfindung nützlich für die Zunahme der Wirkung einer Therapie und/oder Prophylaxe von Leberkrankheiten [z. B. immunogenen Erkrankungen (z. B. chronische Autoimmunleberkrankheiten, wie Autoimmunleberkrankheiten, primäre biliäre Zirrhose oder sklerosierende Cholangitis), partielle Leberresektion, akute Lebernekrose (z. B. Nekrose, die hervorgerufen wird durch Toxine, virale Hepatitis, Schock oder Anoxie), Hepatitis B, Hepatitis des Nicht-A und Nicht-B Typs, Leberzirrhose und Leberversagen (z. B. plötzliche Hepatitis, spät einsetzende Hepatitis und akuter Schub eines Leberversagens (akutes Leberversagen bei chronischen Lebererkrankungen))].
- Die vorliegende Zusammensetzung ist ferner nützlich für die Zunahme der Wirkung für die Verhinderung und/oder Behandlung von verschiedenen Krankheiten aufgrund der nützlichen pharmakologischen Aktivität dieser tricyclischen Makrolide, wie Verstärkungsaktivität einer chemotherapeutischen Wirkung, Aktivität einer Cytomegalovirusinfektion, antiinflammatorische Aktivität, Hemmaktivität gegen Peptidyl-Prolyl-Isomerase oder Rotamase, Antimalariaaktivität, Antitumoraktivität usw.
- Die Erfindung stellt ferner ein Verfahren für einen Auflösungstest für eine Festformulierung bereit, die eine Makrolidverbindung umfasst, wobei der Test eine Testlösung verwendet, die eine geeignete Menge eines Cellulosepolymers enthält. Im Allgemeinen wird der Auflösungstest zum testen der Freisetzungseigenschaft eines medizinisch aktiven Stoffes, der in einer Feststoffformulierung gelöst vorliegt, gemäß „Dissolution Test", Verfahren 2 (Paddle-Verfahren, 50 Upm), JP XIII oder „Dissolution Test" gemäß USP 23, NF18 oder der europäischen Pharmacopoeia (3. Ausgabe) durchgeführt. Bei der Durchführung eines Auflösungstests für eine Formulierung, die nur eine geringe Menge einer Makrolidverbindung enthält, kann die Freisetzung der Makrolidverbindung, basierend auf deren intrinsischem Gehalt, selbst nach mehreren Stunden 100% nicht erreichen. Wenn die Menge der Makrolidverbindung klein ist, liegt dies daran, dass die Adsorption der Makrolidverbindung an den Oberflächen des Testgefäßes, Filter, etc. eine zunehmende Bedeutung haben wird. Nach solchen Untersuchungen haben die Erfinder gefunden, dass durch die Zugabe einer geeigneten Menge eines Cellulosepolymers (wie HPMC, Hydroxypropylcellulose-Phthalat, MC, CMC-Na, Hydroxyethylcellulose, Hydroxypropyl-cellulose (HPC) und so weiter) zu der Testlösung und, falls notwendig, durch Zugabe von Phosphorsäure oder ähnlichem zu der Testlösung, um diese auf einen pH von nicht größer als 7 zu bringen, um einen nachteiligen Effekt eines zunehmenden Anstiegs des pH-Wertes auf die Stabilität der Makrolidverbindung zu vermeiden, der Einfluss der Adsorption der Makrolidverbindung auf die Oberflächen des Testgerätes gehemmt werden kann, um eine Wiedergewinnungsrate von nahezu 100 zu erzielen. Ein bevorzugtes Cellulosepolymer ist Hyderoxypropylcellulose oder ein Äquivalent davon. Die bevorzugte Viskosität davon ist so, dass die Lösung eine Viskosität von 75–150 cps zeigt, wenn 5,0 g in 95 ml Wasser gelöst wird und der Schaum, falls notwendig, nach der Zentrifugation entfernt wird, wobei die Viskosität der Lösung mit einem Drehviscometer bei 25 ± 0,1°C gemessen wird. Eine entsprechende Hydroxypropylcellulose mit einem durchschnittlichen Molekulargewicht von etwa 100.000 ist zum Beispiel die Hydroxypropylcellulose, die von Aldrich verfügbar ist.
- Die „geeignete Menge" des Cellulosepolymers, welches der Testlösung hinzuzufügen ist, beträgt 0,001–0,1%, vorzugsweise 0,002–0,01%, wobei 0,005% am meisten bevorzugt ist, wobei dies auf der Gesamtmenge der Testlösung basiert.
- „Dissolution Test", Verfahren 2 (Paddle-Verfahren), JP XIII und „Dissolution Test", gemäß USP 23, NF 18 oder in der europäischen Pharmacopoeia (3. Ausgabe) sind gut bekannte Verfahren zur Testung der Freisetzungskinetik von Wirkstoffen aus einem pharmazeutischen Festprodukt. Dies sind Auflösungstests unter Verwendung eines spezifischen Gefäßes, Paddles und anderer Hardware, wobei die Quantität der Testlösung, die Temperatur der Testlösung, die Drehgeschwindigkeit und andere Bedingungen kontrolliert werden. Falls notwendig, wird der Test mit einer Testlösung durchgeführt, die auf einen geeigneten pH eingestellt ist. Gemäß der vorliegenden Erfindung beträgt der pH vorzugsweise nicht mehr als 7. Bei der vorliegenden Erfindung bedeutet "Lösungstest, Verfahren 2 (Paddle-Verfahren, 50 Upm), JP XIII" einen Lösungstest, Verfahren 2 (Paddle-Verfahren) JP XIII", der mit 50 Umdrehungen pro Minute gerührt wird. Die entsprechenden Beschreibungen in JP XIII, USP 23 (NF 18) und in der europäischen Pharmacopoeia (3. Ausgabe) sind durch Verweis auf diese Publikationen hiermit eingeführt.
- Die Erfindung wird im folgenden anhand von Beispielen weiter beschrieben. Bei den folgenden Beispielen wird FK506 als Monohydrat beigemischt, wenn die Zusammensetzungen FK506 enthalten, obwohl die Menge von FK506 als Gewicht von FK506 und nicht als das Monohydrat-Gewicht ausgedrückt ist. Beispiel 1 (Referenz)
FK506 1,0 mg HPMC 2910 1,0 mg gesamt 2,0 mg - FK506 wurde in Ethanol gelöst und HPMC 2910 wurde der erzielten Lösung zugesetzt, um FK506 in einem ausreichenden Maße quellen zu lassen. Dann wurde die Mischung zusammengeknetet. Die erzielte, geknetete Mischung wurde zu einer rostfreien Schale überführt, im Vakuum getrocknet und mit einer Kaffeemühle zermahlen. Anschließend wurde das erzielte Pulver einer Größenreduktion unterworfen, wobei die folgenden Verfahren angewandt wurden, um eine Feststoffdispersionszusammensetzung herzustellen (im folgenden bezeichnet als SDC) 1-1) bis 1-6).
- (1) Das gemahlene Pulver wurde durch ein 250 μm Sieb gegeben und die auf dem Sieb verbliebene Fraktion wurde als SDC 1-1)(> 250 μm) bezeichnet.
- (2) Die durch das Sieb nach Verfahren (1) hindurchgetretene Fraktion wurde durch ein 1 80 μm Sieb passiert und die auf dem Sieb verbliebene Fraktion wurde als SDC 1-2) (180–250 μm) bezeichnet.
- (3) Die durch das Sieb bei dem Verfahren (2) hindurchgetretene Fraktion wurde durch ein 150 μm Sieb passiert und die auf dem Sieb verbliebene Fraktion wurde als SDC 1-3)(150–180 μm) bezeichnet.
- (4) Die durch das Sieb bei dem Verfahren (3) hindurchgetretene Fraktion wurde durch ein 106 μm Sieb passiert und die auf dem Sieb verbliebene Fraktion wurde als SDC 1-4)(106–150 μm) bezeichnet.
- (5) Die durch das Sieb nach dem Verfahren (4) hindurchgetretene Fraktion wurde durch ein 75 μm Sieb passiert und die auf dem Sieb verbliebene Fraktion wurde als SDC 1-5)(75–106 μm) bezeichnet.
- (6) Die durch das Sieb bei dem Verfahren (5) hindurchgetretene Fraktion wurde als SDC 1-6)(< 75 μm) bezeichnet.
- Beispiel 2 (Referenz)
- SDC 1-2), die im Beispiel 1 erzielt wurde, wurde ausreichend mit Lactose (58,0 mg) gemischt und die erzielte Mischung wurde eingekapselt, um eine Kapsel herzustellen.
- Beispiel 3 (Referenz)
- In einer ähnlichen Weise wie beim Beispiel 1 wurde ein gemahlenes Pulver der folgenden SDC mit Partikelgröße von 180 bis 250 μm hergestellt.
-
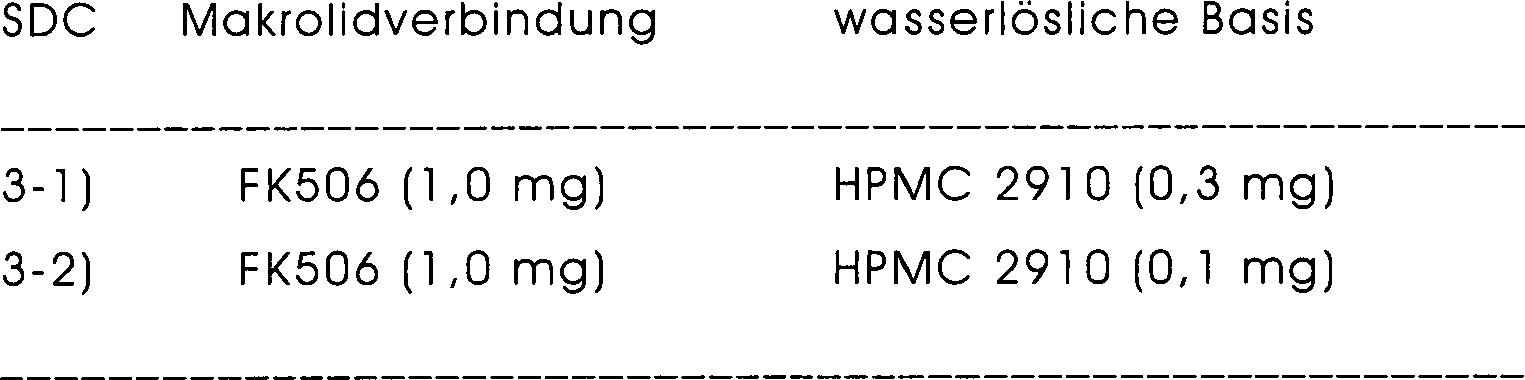
- Ferner wurde SDC 3-1) ausreichend mit Lactose (58,7 mg) gemischt und die erzielte Mischung wurde eingekapselt, um eine Kapsel 3-1) herzustellen. SDC 3-2) wurde ausreichend mit Lactose (58,9 mg) gemischt und die erzielte Mischung wurde eingekapselt, um die Kapsel 3-2) herzustellen.
- Beispiel 4 (Referenz)
- In einer ähnlichen Weise wie für SDC 1-2) nach Beispiel 1 wurden die folgenden SDCs hergestellt.
-

- In einer ähnlichen Weise wie bei Beispiel 2 wurden Lactose (mit einer geeigneten Menge) und Magnesiumstearat (0,6 mg) den entsprechenden SDCs zugesetzt, um Kapseln herzustellen von jeweils 60,0 mg.
- Beispiel 5 (Referenz)
- In einer ähnlichen Weise wie bei der SDC 1-2) von Beispiel 1 wurde eine SDC unter Verwendung von FK506 (1,0 mg) und HPMC 291 0 (1,0 mg) hergestellt. In einer ähnlichen Weise wie nach Beispiel 2 wurden dann die folgenden Additive der SDC zugesetzt, um die Kapseln 5-1) bis 5-4) von jeweils 60,0 mg herzustellen.Beispiel 6 (Referenz)

FK506 1,0 g HPMC 2910 0,3 g gesamt 1,3 g - FK506 wurde in Ethanol gelöst und der erzielten Mischung wurde HPMC 2910 zugesetzt, um eine ausreichende Quellung zu erzielen. Dann wurde die Mischung zusammengeknetet. Die erzielte, geknetete Substanz wurde auf eine rostfreie Schale überführt, im Vakuum getrocknet und mit einer Kaffeemühle zermahlen. Das erzielte Pulver wurde einer Größenreduktion unterworfen, um die SDCs 6-1) bis 6-6) herzustellen, wobei die folgenden Verfahren angewandt wurden.
- (1) Das gemahlene Pulver wurde durch ein 250 μm Sieb passiert und die auf dem Sieb verbliebene Fraktion wurde als SDC 6-1)(> 250 μm) bezeichnet.
- (2) Die durch das Sieb nach Verfahren (1) hindurchgetretene Fraktion wurde durch ein 180 μm Sieb passiert und die auf dem Sieb verbliebene Fraktion wurde bezeichnet als SDC 6-2)(180–250 μm).
- (3) Die auf dem Sieb nach Verfahren (2) hindurchgetretene Fraktion wurde durch ein 150 μm Sieb passiert und die auf dem Sieb verbliebene Fraktion wurde bezeichnet als SDC 6-3)(150–180 μm).
- (4) Die durch das Sieb nach Verfahren (3) hindurchgetretene Fraktion wurde durch ein 106 μm Sieb passiert und die auf dem Sieb verbliebene Fraktion wurde bezeichnet als SDC 6-4)(106–150 μm).
- (5) Die durch das Sieb nach dem Verfahren (4) hindurchgetretene Fraktion wurde durch ein 75 μm Sieb passiert und die auf dem Sieb verbliebene Fraktion wurde bezeichnet als SDC 6-5)(75–106 μm).
- (6) Die durch das Sieb nach Verfahren (5) hindurchgetretene Fraktion wurde bezeichnet als SDC 6-6).
- Beispiel 7 (Referenz)
- SDC 6-4)(1,3 mg), die im Beispiel 6 erzielt wurde, wurde gründlich mit Lactose (58,1 mg) und Magnesiumstearat (0,6 mg) gemischt und die erzielte Mischung wurde in Kapseln gefüllt, die als Kapsel 7 definiert wurden.
- Beispiel 8 (Referenz)
- In einer ähnlichen Weise wie nach Beispiel 1 wurden die folgenden SDCs mit Partikelgrößen von 180–250 μm hergestellt.
-

- In einer ähnlichen Weise wie nach Beispiel 7 wurde jede Kapsel hergestellt, indem Lactose (58,1 mg) und Magnesiumstearat (0,6 mg) zugesetzt wurden. Beispiel 9 (Referenz) SDC 9
FK506 10 g HPMC 2910 3 g Calciumhydrogenphosphat 3 g gesamt 16 g SDC 9 16 g Lactose qs Magnesiumstearat 7 g gesamt 700 g - FK506 wurde in Ethanol gelöst, HPMC 2910 wurde zugesetzt und mit der erzielten Lösung gründlich gemischt und anschließend wurde Calciumhydrogenphosphat hinzugefügt. Nach Trocknung im Vakuum über Nacht wurde die erzielte Mischung einer Größenreduktion unterworfen, wobei eine Geschwindigkeitsmühle und ein Walzengranulator verwendet wurden. Das erzielte Pulver wurde mit einem Sieb von 212 μm gesiebt und die das Sieb passierende Fraktion wurde bezeichnet als SDC 9. SDC 9 wurde mit Lactose und Magnesiumstearat gemischt, um die Formulierung 9 herzustellen. Die Formulierung 9 wurde zu 350 mg in die Nr. 1 Kapsel und mit 70 mg in die Nr. 5 Gelatinekapsel gefüllt, die als die Formulierungen A beziehungsweise B definiert wurden. Beispiel 10 (Referenz) SDC 10
FK506 10 g HPMC 2910 3 g Lactose 3 g gesamt 16 g SDC 10 16 g Lactose qs Magnesiumstearat 7 g gesamt 700 g - In einer ähnlichen Weise wie nach Beispiel 9 wurde die SDC 10 und die Formulierung 10 hergestellt. Beispiel 11 (Referenz) SDC 11
FK506 10 g HPMC 2910 3 g Calciumhydrogenphosghat 3 g gesamt 16 g SDC 11 16 g Lactose qs Magnesiumstearat 7 g gesamt 700 g - FK506 wurde in Ethanol gelöst, HPMC 2910 wurde zugesetzt und ausreichend mit der erzielten Lösung gemischt, wobei anschließend Calciumhydrogenphosphat hinzugefügt wurde. Nachdem die erzielte Mischung über Nacht im Vakuum getrocknet wurde, wurde die Mischung einer Größenreduktion unterworfen, wobei eine Geschwindigkeitsmühle und ein Walzengranulator verwendet wurden. Das erzielte Pulver wurde mit einem Sieb von 250 μm und einem Sieb von 180 μm gesiebt. Die Fraktion von 180–250 μm wurde als SDC 11 bezeichnet. SDC 11, Lactose und Magnesiumstearat wurden miteinander gemischt, um die Formulierung 11 herzustellen. Die Formulierung 11 wurde zu 350 mg in die Nr. 1 Kapsel und zu 70 mg in die Nr. 5 Gelatinekapsel eingefüllt, die als die Formulierungen C und D bezeichnet wurden. Beispiel 12 SDC 12
FK506 2 g Glycerinmonostearat 98 g HPMC 2910 20 g gesamt 120 g SDC 12 120 g Magnesiumstearat 1,2 g gesamt 121,2 g - Glycerinmonostearat wurde erhitzt, bei 80°C geschmolzen und unter Rühren wurde FK506 zugesetzt, um das FK506 zu lösen. Zu der erzielten Mischung wurde HPMC 2910 unter ausreichender Vermischung zugesetzt und die erzielte Mischung wurde dann auf eine Schale überführt und wurde für eine spontane Abkühlung stehen gelassen. Die durch das Abkühlen erzielte Festsubstanz wurde mit einer Kaffeemühle zermahlen und dann mit einem Sieb von 500 μm gesiebt. Die durch das Sieb hindurchtretende Fraktion wurde als SDC 12 bezeichnet. SDC 12 wurde mit Magnesiumstearat gemischt, um die Formulierung herzustellen, die dann mit 60,6 mg in die Nr. 5 Kapsel eingefüllt wurde. Die erzielte Kapsel wurde als Formulierung E bezeichnet. Beispiel 13 SDC 13
FK506 2 g Aminoalkyl-Methacrylat-Copolymer 6 g Calciumhydrogenphosphat 2 g gesamt 10 g SDC 13 10 g Lactose 130 g gesamt 140 g - FK506 und Aminoalkyl-Methacrylat-Copolymer wurden in Ethanol gelöst und anschließend wurde Calciumhydrogenphosphat zugesetzt und die erzielte Mischung wurde ausreichend miteinander vermischt. Die Mischung wurde im Vakuum über Nacht getrocknet, in einem Mörser zermahlen und mit Sieben von 150 μm und 106 μm gesiebt, um eine Fraktion von 106–150 μm als SDC 13 zu erzielen. SDC 13 wurde mit Lactose gemischt und als Formulierung 13 hergestellt. Diese wurde dann mit 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung F herzustellen. Beispiel 14 SDC 14
FK506 2 g Aminoalkyl-Methacrylat-Copolymer (Eudragit RL) 4,6 g Aminoalkyl-Methacrylat-Copolymer (Eudragit RS) 1,4 g Calciumhydrogenphosphat 2 q gesamt 10 g SDC 14 10 g Lactose 130 g gesamt 140 g - In einer ähnlichen Weise wie nach Beispiel 13 wurde die SDC 14 mit Partikelgrößen von 106–150 μm und die Formulierung 14 hergestellt.
- Dann wurde die Formulierung 14 mit 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung G herzustellen. Beispiel 15 SDC 15
FK506 2 g Aminoalkyl-Methacrylat-Copolymer (Eudragit RL) 3 g Aminoalkyl-Methacrylat-Copolymer (Eudragit RS) 3 g Calciumhvdrogenphosphat 2 g gesamt 10 g SDC 15 10 g Lactose 130 g gesamt 140 g - In einer ähnlichen Weise wie nach Beispiel 1 3 wurde SDC 1 5 mit Partikelgrößen von 106–150 μm und die Formulierung 15 hergestellt. Die Formulierung 15 wurde mit 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung H herzustellen. Beispiel 16 SDC 16
FK506 2 g Ethylcellulose 0,4 g Lactose 6 g gesamt 8,4 g SDC 16 8,4 g Lactose 131,6 g gesamt 140 g - FK506 und Ethylcellulose wurden in Ethanol gelöst und anschließend wurde Lactose zugesetzt. Die erzielte Mischung wurde in einem ausreichenden Maße gemischt. Die Mischung wurde im Vakuum über Nacht getrocknet, in einem Mörser zermahlen und mit Sieben von 150 μm und 106 μm gesiebt, um eine Fraktion von 106–150 μm als SDC 16 herzustellen. SDC 16 wurde mit Lactose gemischt und als Formulierung 16 hergestellt und dann wurden 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung I herzustellen. Beispiel 17 SDC 17
FK506 2 g Ethylcellulose 1 g Lactose 6 g gesamt 9 g SDC 17 9 g Lactose 131 g gesamt 140 g - In einer ähnlichen Weise wie nach Beispiel 16 wurde die SDC 17 mit Partikelgrößen von 106–150 μm und die Formulierung 17 hergestellt. Die Formulierung 17 wurde mit 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung J herzustellen. Beispiel 18 SDC 18
FK506 2 g Ethylcellulose 0,4 g Hydroxypropylmethylcellulose 0,6 g Lactose 6 g gesamt 9 g SDC 18 9 g Lactose 131 g gesamt 140 g - In einer ähnlichen Weise wie nach Beispiel 16 wurde SDC 18 mit Partikelgrößen von 106–150 μm und die Formulierung 18 hergestellt. Die Formulierung 18 wurde mit 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung K herzustellen. Beispiel 19 SDC 19
FK506 2 g Ethylcellulose 0,6 g HPMC 2910 0,6 g Lactose 6 g gesamt 9,2 g SDC 19 9,2 g Lactose 130,8 g gesamt 140 g - In einer ähnlichen Weise wie nach Beispiel 16 wurde SDC 19 mit Partikelgrößen von 106–150 μm und die Formulierung 19 hergestellt. Die Formulierung 19 wurde mit 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung L herzustellen Beispiel 20 SDC 20
FK506 10 g Ethylcellulose 3 g HPMC 2910 3 g Lactose 50 g gesamt 66 g SDC 20 66 g Lactose qs Magnesiumstearat 7 g gesamt 700 g - FK506 wurde in Ethanol gelöst und Ethylcellulose wurde hinzugesetzt und gelöst. HPMC 2910 und Lactose wurden der erzielten Lösung zugesetzt und ausreichend gemischt. Nachdem die Mischung über Nacht im Vakuum getrocknet wurde, wurde die erzielte Mischung einer Größenreduktion unter Verwendung einer Geschwindigkeitsmühle und einem Walzengranulator unterworfen. Das erzielte Pulver wurde mit einem Sieb von 250 μm gesiebt. Die Fraktion, welche das Sieb passierte, wurde als SDC 20 bezeichnet. SDC 20, Lactose und Magnesiumstearat wurden miteinander gemischt, um die Formulierung 20 herzustellen. Die Formulierung 20 wurde zu 350 mg in die Nr. 1 Kapsel abgefüllt und zu 70 mg in die Nr. 5 Gelatinekapsel, die als Formulierungen M beziehungsweise N bezeichnet wurden. Beispiel 21 SDC 21
FK506 10 g Ethylcellulose 3 g HPMC 2910 3 g Lactose 20 g gesamt 36 g SDC 21 36 g Lactose qs Magnesiumstearat 7 g gesamt 700 g - In einer ähnlichen Weise wie nach Bespiel 20 wurde eine Fraktion, welche das Sieb 212 μm passierte, als SDC 21 bezeichnet und die Formulierung 21 wurde hergestellt. Die Formulierung 21 wurde dann zu 350 mg in die Nr. 1 Gelatinekapsel und zu 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierungen O beziehungsweise P zu erzielen. Beispiel 22 SDC 22
F K506 1 g Saccharose-Fettsäureester (HLB = 6) (DK Ester F-50) 1 g gesamt 2 g SDC 22 2 g Lactose 68 g gesamt 70 g - FK506 wurde in Ethanol/Aceton (1/1) gelöst. Nach Erhitzung der Lösung auf 75°C wurde ein Saccharose-Fettsäureester zugesetzt, gelöst und dann auf Raumtemperatur abgekühlt. Die Mischung wurde über Nacht im Vakuum getrocknet, in einem Mörser zermahlen und mit Sieben von 150 μm und 106 μm gesiebt, um eine Fraktion von 106–150 μm als SDC 22 herzustellen. SDC 22 wurde mit Lactose gemischt und als Formulierung 22 hergestellt, die dann mit 70 mg in die Nr. 5 Gelatinekapsel abgefüllt wurde, um die Formulierung Q herzustellen. Beispiel 23 SDC 23
FK506 1 g Saccharose-Fettsäureester (HLB = 6) (DK Ester F-50) 0,75 g Saccharose-Fettsäureester (HLB = 2) (DK Ester F-20W) 0,25 g gesamt 2 g SDC 23 2 g Lactose 68 g gesamt 70 g - In einer ähnlichen Weise wie nach Beispiel 22 wurde SDC 23 mit Partikelgrößen von 106–150 μm sowie die Formulierung 23 hergestellt. Die Formulierung 23 wurde dann mit 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung R herzustellen. Beispiel 24 SDC 24
FK506 1 g Saccharose-Fettsäureester (HLB = 1) (DK Ester F-10) 1 g Lactose 1 g gesamt 3 g SDC 24 3 g Lactose 67 g gesamt 70 g - In einer ähnlichen Weise wie nach Beispiel 22 wurde SDC 24 mit Partikelgrößen von 106–150 μm sowie die Formulierung 24 hergestellt. Die Formulierung 24 wurde dann zu 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung S herzustellen. Beispiel 25 SDC 25
FK506 1 g Saccharose-Fettsäureester (HLB = 1) (DK Ester F-10) 1 g Lactose 3 g gesamt 5 g SDC 25 5 g Lactose 65 g gesamt 70 g - In einer ähnlichen Weise wie nach Beispiel 22 wurde SDC 25 mit Partikelgrößen von 106–150 μm und die Formulierung 25 hergestellt, Die Formulierung 25 wurde zu 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung T herzustellen. Beispiel 26 SDC 26
FK506 1 g Saccharose-Fettsäureester (HLB = 1) (DK Ester F-10) 1 g Lactose 5 g gesamt 7 g SDC 26 7 g Lactose 63 g gesamt 70 g - In einer ähnlichen Weise wie nach Beispiel 22 wurde SDC 26 mit Partikelgrößen von 106–150 μm und die Formulierung 26 hergestellt. Die Formulierung 26 wurde dann zu 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung U herzustellen. Beispiel 27 SDC 27
FK506 1 g Tetraglycerin-Trifettsäureester 30 g Lactose 15 g gesamt 46 g SDC 27 46 g Lactose 24 g gesamt 70 g - Ein Tetraglycerin-Trifettsäureester, der durch Erhitzen bei 80°C geschmolzen war, wurde mit zugesetztem FK506 gelöst. Lactose wurde zugesetzt, gemischt und dann auf einer Schale abkühlen gelassen. Die erzielte Festsubstanz wurde mit einer Kaffeemühle zermahlen und mit Sieben von 150 μm und 1 06 μm gesiebt, um eine Fraktion von 106–150 μm als SDC 27 herzustellen. SDC 27 wurde mit Lactose gemischt und als Formulierung 27 hergestellt. Die Formulierung 27 wurde dann zu 70 mg in die Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung V herzustellen. Beispiel 28 SDC 28
FK506 1 g Tetraglycerin-Trifettsäureester 30 g Polysolbat 0,3 g gesamt 31,3 g SDC 28 31,3 g Lactose 38,7 g gesamt 70 g - In einer ähnlichen Weise wie nach Beispiel 27 wurde SDC 28 mit Partikelgrößen von 106–150 μm hergestellt. Ferner wurde die Formulierung 28 hergestellt, die dann zu 70 mg in die Nr. 5 Gelatinekapsel abgefüllt wurde, um die Formulierung W herzustellen. Beispiel 29 SDC 29
FK506 1 g Tetraglycerin-Trifettsäureester 1 g Lactose 3 g gesamt 5 g SDC 29 5 g Lactose 65 g gesamt 70 g - Ethanol wurde einem Tetraglycerin-Trifettsäureester zugesetzt. Die erzielte Mischung wurde durch Erhitzen auf 40°C geschmolzen und FK506 wurde zugesetzt und geschmolzen unter Rühren. Lactose wurde zugesetzt, gemischt und dann in einer Schale abkühlen gelassen. Die erzielte, feste Substanz wurde mit einer Kaffeemühle gemahlen, im Vakuum über Nacht getrocknet und mit Sieben von 150 μm und 106 μm gesiebt, um eine Fraktion von 106–150 μm als SDC 29 herzustellen. SDC 29 wurde mit Lactose gemischt und als Formulierung 29 hergestellt. Dann wurde die Formulierung 29 zu 70 mg in Nr. 5 Gelatinekapsel abgefüllt, um die Formulierung X herzustellen. Beispiel 30 (Referenz) Formulierung 30
FK506 feines Pulver 0,5 g Lactose 29,2 g Magnesiumstearat 0,3 g gesamt 30 g - Kristalle von FK506 wurden mit einer Strahlenmühle zermahlen und dann mit Lactose und Magnesiumstearat gemischt, um die Formulierung 30 herzustellen. Dann wurde die Formulierung 30 mit 60 mg in Nr. 5 Gelatinekapseln abgefüllt, um die Formulierung Z herzustellen. Der Bereich der Partikelgröße des feinen Pulvers von FK506, das mit der Strahlenmühle zermahlen worden war, betrug 1–10 μm und die durchschnittliche Partikelgröße war etwa 3 μm.
- Beispiel 31 (Referenz)
- Lösungstest
- Testprobe
-
- (1) Die Formulierungen A und C wurden gemäß den oben erwähnten Beispielen hergestellt.
- (2) Kontrollformulierung (Formulierung mit schneller Freisetzung), die eine 1 mg Kapselformulierung war, welche die folgenden Bestandteile umfasste. Sie wurde in einer ähnlichen Weise wie die Beispiele 1 und 2 von WO 91/19495 hergestellt, indem die Bestandteile (e) und (f) mit der Feststoffdispersionszusammensetzung gemischt wurden, welche die folgenden Bestandteile (a) bis (d) enthielt. Die Formulierung wurde dann verkapselt.
-

- Testverfahren
- Gemäß der japanischen Pharmacopoeia, 13. Ausgabe, Lösungstest, Nr. 2 (Paddle-Verfahren, 50 Upm) wurde ein Test durchgeführt unter Verwendung einer 0,005% wässrigen Hydroxypropylcellulose-Lösung, die auf einen pH von 4,5 eingestellt worden war. Die erzielten Daten sind im folgenden dargestellt.

Zeit (Std.) Kontrolle (%) 0 0,0 0,17 30,1 0,5 68,4 1 92,8 2 100,1 - Beispiel 32
- In einer ähnlichen Weise wie nach Beispiel 31 wurde der Lösungstest durchgeführt. Die verschiedenen Parameter in der Weibull-Funktion und die Werte von T 63,2% wurden durch Berechnung erzielt.
- Ergebnis

-

- Beispiel 33 (Referenz)
- Orale Absorbierfähigkeit
- Testprobe
-
- (1) Die Formulierungen B und D wurden wie in den oben erwähnten Beispielen hergestellt.
- (2) Kontrollformulierung (die gleiche Kontrollformulierung wie im Beispiel 31).
- Testverfahren
- Die Testproben wurden oral 6 cynomologen Affen (1 mg/Affe als FK506-Dosis) gegeben, um die FK506 Blutkonzentration nach der Verabreichung zu bestimmen. 17 Stunden vor der Verabreichung wurde das Futter von dem Futtertisch für die cynomologen Affen, die ein Körpergewicht von etwa 6 kg hatten, entfernt. Dann hungerten die Tiere bis 12 Stunden nach der Verabreichung. Wasser wurde ad libitum vor Beginn des Tests durch die Verabreichung der Testproben und anschließend zugeführt. Bei der Dosierung wurde Wasser (20 ml) simultan den Tieren gegeben. Zu vorbestimmten Zeitpunkten nach der Dosierung wurde 1 ml Blut aus der Vorderarmvene unter Verwendung einer sterilen Spritze entnommen und in ein Plastikgefäß gegeben, das Heparin enthielt. Die Probe wurde bei –80°C bis zum Beginn der Bestimmung der Arzneimittelkonzentration aufbewahrt. Die Gesamtblutkonzentration von FK506 wurde mit einem FK506-spezifischen Enzymimmunoassay (EIA) bestimmt, der bekannt ist aus JP-A-1-92659. Die Veröffentlichung davon wird hiermit zitiert und ist Teil der vorliegenden Beschreibung.
- Durchschnittswert
-

- Die maximale Blutkonzentration (Cmax) wurde definiert als die maximale Menge des Arzneimittels im Gesamtblut. Tmax ist die Zeit, die für das Erreichen der maximalen Blutkonzentration notwendig ist. MRT wird als die durchschnittliche Retentionszeit definiert. Der Bereich unter der Blutkonzentration-Zeitkurve (AUC) wurde mit dem Trapezoidverfahren berechnet. Als Indikator für die Variation der oralen Absorbierfähigkeit wurde ferner CV (Standardabweichung/Durchschnitt in %) berechnet.
- Testergebnisse

- Beispiel 34
- In ähnlicher Weise wie nach Beispiel 33 wurde die orale Absorbierfähigkeit der verschiedenen Formulierungen gemäß der vorliegenden Erfindung bestimmt.
- Ergebnisse
-
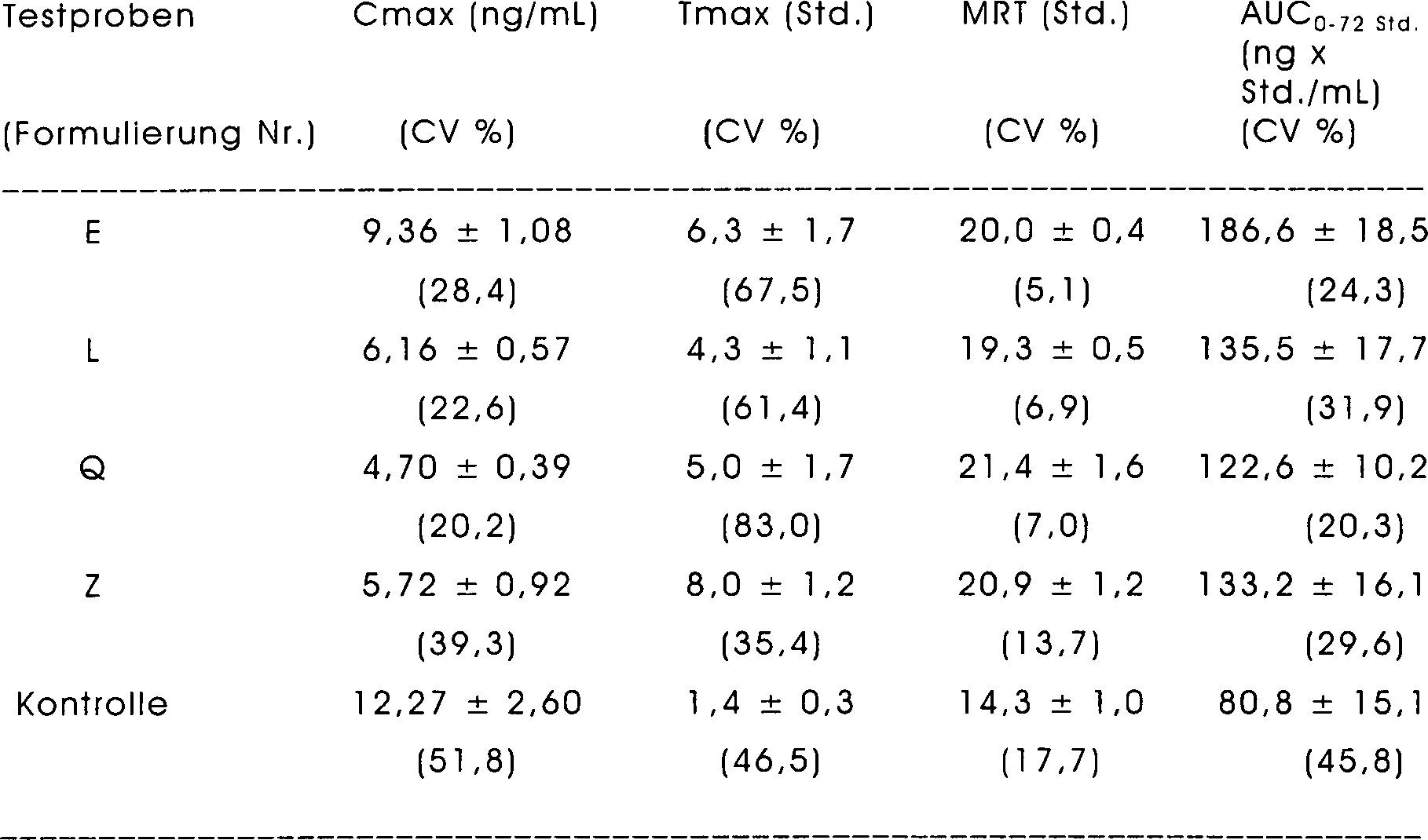
- Die obigen Ergebnisse zeigen, dass die für die obigen Experimente angewandten Formulierungen nach einer oralen Verabreichung eine kleinere Cmax und ausreichend verlängerte Tmax und MRT besitzen als die Formulierung mit schneller Freisetzung (Kontrolle). Im Vergleich mit der Formulierung mit einer schnellen Freisetzung waren die AUC der obigen Formulierungen fast die gleichen oder größer. Die obigen Formulierungen mit verzögerter Freisetzung zeigten bei den einzelnen Testtieren geringere Variationen von Cmax und/oder AUC im Vergleich mit der Formulierung mit schneller Freisetzung.
- Gemäß der Erfindung der vorliegenden Anmeldung kann eine geringe Variation in den Testtieren hinsichtlich der maximalen Blutkonzentration oder des Bereiches unter der Blutkonzentration-Zeitkurve der Makrolidverbindung nach oraler Dosierung bestimmt werden im Vergleich zu einer Formulierung mit schneller Freisetzung. Dafür wird ein Indikator für die Variation der Blutabsorbierfähigkeit der Makrolidverbindung verwendet, nämlich die Standardabweichung/Durchschnitt (CV in %) der maximalen Blutkonzentration oder des Bereiches unter der Blutkonzentration-Zeitkurve. Der Ausdruck „geringe Variation" bedeutet einen kleinen CV-Wert davon und dieser Ausdruck bedeutet insbesondere, dass der CV-Wert kleiner ist als der Wert der oben beschriebenen Formulierung mit schneller Freisetzung.
- Die Veröffentlichungen der Patente, Patentanmeldungen und der Referenzen, die hier in der Anmeldung erwähnt sind, ist von der hier vorliegenden Beschreibung mit umfasst.
R8 und R9 sind unabhängig voneinander ein Wasserstoffatom oder eine Hydroxygruppe;
R10 ist ein Wasserstoffatom, eine Alkylgruppe, eine Alkylgruppe, die mit einer oder mehreren Hydroxygruppen substituiert ist, eine Alkenylgruppe, eine Alkenylgruppe, die mit einer oder mehreren Hydroxygruppen substituiert ist, oder eine Alkylgruppe, die mit einer Oxogruppe substituiert ist;
X ist eine Oxogruppe, (ein Wasserstoffatom und eine Hydroxygruppe), (ein Wasserstoffatom und ein Wasserstoffatom) oder eine Gruppe, die dargestellt ist durch die Formel -CH2O -;
Y ist eine Oxogruppe, (ein Wasserstoffatom und eine Hydroxygruppe), (ein Wasserstoffatom und ein Wasserstoffatom) oder eine Gruppe, die dargestellt ist durch die Formeln N-NR11R12 oder N-OR13;
R11 und R12 sind unabhängig voneinander ein Wasserstoffatom, eine Alkylgruppe, eine Arylgruppe oder eine Tosylgruppe;
R13, R14, R15, R16, R17, R18, R19, R22 und R23 sind unabhängig voneinander ein Wasserstoffatom oder eine Alkylgruppe;
R24 ist ein optional substituiertes Ringsystem, das ein oder mehrere Heteroatome enthalten kann;
n ist eine ganze Zahl von 1 oder 2; und zusätzlich zu den obigen Definitionen können Y, R10 und R23, zusammen mit dem Kohlenstoffatomen, mit denen sie verbunden sind, einen gesättigten oder ungesättigten, 5- oder 6-gliedrigen, Stickstoff, Schwefel und/oder Sauerstoff enthaltenden, heterocyclischen Ring darstellen, der optional substituiert ist mit einer oder mehreren Gruppen, die aus der Gruppe ausgewählt werden, die besteht aus einem Alkyl, einem Hydroxy, einem Alkoxy, einem Benzyl, einer Gruppe der Formel -CH2Se(C6H5) und einem Alkyl, das mit einer oder mehreren Hydroxygruppen substituiert ist.
Claims (29)
- Makrolid-Formulierung mit verzögerter Freisetzung, wobei die Zeit (T 63,2%), die für das Lösen von 63,2% der maximalen Menge der zu lösenden Makrolidverbindung erforderlich ist, 0,7 bis 15 Stunden beträgt gemäß Messung nach der japanischen Pharmacopoeia, 13. Ausgabe, Lösungstest, Nr. 2 (Paddle-Verfahren, 50 Upm) unter Verwendung einer Testlösung, die eine auf einen pH von 4,5 eingestellte, wässrige, 0,005 Hydroxypropylcellulose-Lösung ist, und wobei die Makrolidverbindung eine tricyclische Verbindung, die durch die allgemeine Formel (I) dargestellt ist, und ein pharmazeutisch akzeptables Salz davon ist,wobei jedes der benachbarten Paare von R1 und R2, R3 und R4 sowie R5 und R6 unabhängig voneinander (a) zwei benachbarte Wasserstoffatome sind, wobei aber R2 auch eine Alkylgruppe sein kann, oder (b) eine andere Bindung bilden können, die zwischen den Kohlenstoffatomen ausgebildet ist, mit denen sie verbunden sind; R7 ein Wasserstoffatom, eine Hydroxygruppe, eine geschützte Hydroxygruppe, eine Alkoxygruppe oder eine Oxogruppe zusammen mit R1 ist; R8 und R9 unabhängig voneinander ein Wasserstoffatom oder eine Hydroxygruppe sind; R10 ein Wasserstoffatom, eine Alkylgruppe, eine Alkylgruppe, die mit einer oder mehreren Hydroxygruppen substituiert ist, eine Alkenylgruppe, eine Alkenylgruppe, die mit einer oder mehreren Hydroxygruppen substituiert ist, oder eine Alkylgruppe ist, die mit einer Oxogruppe substituiert ist; X eine Oxogruppe, (ein Wasserstoffatom und eine Hydroxygruppe), (ein Wasserstoffatom und ein Wasserstoffatom), oder eine Gruppe ist, die durch die Formel -CH2O- dargestellt ist; Y eine Oxogruppe, (ein Wasserstoffatom und eine Hydroxygruppe), (ein Wasserstoffatom und ein Wasserstoffatom), oder eine Gruppe ist, die durch die Formeln N-NR11R12 oder N-OR13 dargestellt ist; R11 und R12 unabhängig voneinander ein Wasserstoffatom, eine Alkylgruppe, eine Arylgruppe oder eine Tosylgruppe sind; R13, R14, R15, R16, R17, R18, R19, R22 und R23 unabhängig voneinander ein Wasserstoffatom oder eine Alkylgruppe sind; R24 ein optional substituiertes Ringsystem ist, das ein oder mehrere Heteroatome enthalten kann; n eine ganze Zahl von 1 oder 2 ist; und zusätzlich zu den obigen Definitionen können Y, R10 und R23 zusammen mit dem Kohlenstoffatom, mit dem sie verbunden sind, einen gesättigten oder ungesättigten, 5- oder 6-gliedrigen, Stickstoff-, Schwefel- und/oder Sauerstoff-enthaltenden, heterocyclischen Ring darstellen, der optional mit einer oder mehreren Gruppen substituiert ist, die aus der Gruppe ausgewählt sind, die besteht aus einem Alkyl, einem Hydroxy, einem Alkoxy, einem Benzyl, einer Gruppe der Formel -CH2Se(C6H5) und einem Alkyl, das mit einer oder mehreren Hydroxygruppen substituiert ist; wobei die Formulierung eine Feststoffdispersionszusammensetzung umfasst, worin die Makrolidverbindung (I) in einem amorphen Zustand in einer wasserunlöslichen Basis vorliegt, wobei die wasserunlösliche Basis, welche in der Feststoffdispersionszusammensetzung enthalten ist, aus einem wasserunlöslichen Polymer oder Wachs ausgewählt ist.
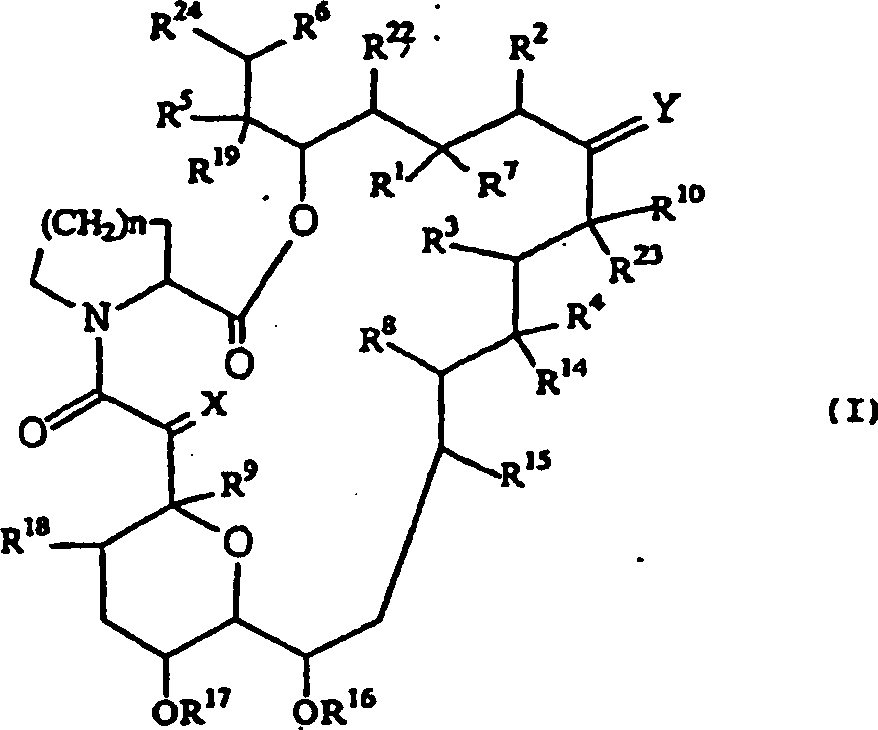
- Formulierung mit verzögerter Freisetzung nach Anspruch 1, worin die wasserunlösliche Basis ein wasserunlösliches Polymer ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 1, worin die Feststoffdispersionszusammensetzung dadurch gekennzeichnet ist, dass (1) Lactose oder Calciumhydrogenphosphat als Bindemittel und/oder Gleitmittel enthalten ist, (2) ein Lösungsvermittler nicht enthalten ist, und (3) die Partikelgröße der Feststoffdispersionszusammensetzung gleich oder kleiner als 350 μm ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 2, worin die Feststoffdispersionszusammensetzung dadurch gekennzeichnet ist, dass das wasserunlösliche Polymer in einer Gewichtsmenge von 0,1–5 zu der Verbindung (I)(1,0) vorliegt.
- Formulierung mit verzögerter Freisetzung nach Anspruch 2, worin das wasserunlösliche Polymer Ethylcellulose oder ein Methacrylat-Copolymer ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 5, worin das wasserunlösliche Polymer Ethylcellulose ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 2, worin ein wasserlösliches Polymer mit dem wasserunlöslichen Polymer gemischt ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 7, worin das wasserlösliche Polymer Hydroxypropylmethylcellulose ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 8, worin die Feststoffdispersionszusammensetzung dadurch gekennzeichnet ist, dass (1) die Makrolidverbindung (I) in einem amorphen Zustand in einer Mischung von Ethylcellulose und Hydroxypropylmethylcellulose vorliegt, (2) Lactose als Bindemittel enthalten ist, (3) die Partikelgröße der Feststoffdispersionszusammensetzung gleich oder kleiner als 250 μm ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 9, worin der Gewichtsanteil der Verbindung (I) zu Hydroxypropylmethylcellulose 1 bis 0,2–0,4 ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 1, worin die wasserunlösliche Basis Wachs ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 11, worin das Wachs Glycerinmonostearat, ein Polyglycerin-Fettsäureester oder ein Saccharose-Fettsäureester ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 11 oder 12, worin die Feststoffdispersionszusammensetzung dadurch gekennzeichnet ist, dass (1) Lactose oder Calciumhydrogenphosphat als ein Bindemittel enthalten ist, (2) Lösungsvermittler nicht enthalten sind, und (3) die Partikelgröße der Feststoffdispersionszusammensetzung gleich oder kleiner als 350 μm ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 12, die eine Feststoffdispersionszusammensetzung umfasst, welche durch die Verbindung (I) gekennzeichnet ist, die in einem amorphen Zustand in dem Saccharose-Fettsäureester vorliegt, der mit einem Gewichtsanteil von 0,2–20 zu der Verbindung (I)(1,0) vorhanden ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 12, die eine Feststoffdispersionszusammensetzung umfasst, welche durch die Verbindung (I) gekennzeichnet ist, die in einem amorphen Zustand in dem Glycerinmonostearat vorliegt, das mit einem Gewichtsanteil von 10–100 zu der Verbindung (I)(1,0) vorhanden ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 12, die eine Feststoffdispersionszusammensetzung umfasst, welche durch die Verbindung (I) gekennzeichnet ist, die in einem amorphen Zustand in dem Polyglycerin-Fettsäureester vorliegt, der mit einem Gewichtsanteil von 0,1–100 zu der Verbindung (I)(1,0) vorhanden ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 1, worin die Verbindungen (I) die sind, worin jedes der benachbarten Paare von R3 und R4 oder R5 und R6 unabhängig voneinander eine andere Bindung bilden, die zwischen den Kohlenstoffatomen ausgebildet sind, mit denen sie verbunden sind; R8 und R32 unabhängig voneinander ein Wasserstoffatom sind; R9 eine Hydroxygruppe ist; R10 eine Methylgruppe, eine Ethylgruppe, eine Propylgruppe oder eine Allylgruppe ist; X ein (Wasserstoffatom und ein Wasserstoffatom) oder eine Oxogruppe ist; Y eine Oxogruppe ist; jedes von R14, R15, R16, R17, R18, R19 und R22 eine Methylgruppe ist; R24 eine 3-R20-4-R21-Cyclohexylgruppe ist, worin R20 ein Hydroxy, eine Alkoxygruppe, eine Oxogruppe oder eine -OCH2OCH2CH2OCH3-Gruppe ist, und R21 Hydroxy, -OCN, eine Alkoxygruppe, ein Heteroaryloxy, das mit geeigneten Substituenten substituiert sein kann, eine -OCH2OCH2CH2OCH3-Gruppe, eine geschützte Hydroxygruppe, Chlor, Brom, Iod, Aminooxalyloxy, eine Azidogruppe, p-Tolyloxythiocarbonyloxy oder R25R26CHCOO- ist, worin R25 ein optional geschütztes Hydroxy oder ein geschütztes Amino ist, und R26 Wasserstoff oder Methyl ist, oder R20 und R21 in einem Epoxidring zusammen ein Sauerstoffatom bilden; und n eine ganze Zahl von 1 oder 2 ist.
- Formulierung mit verzögerter Freisetzung nach einem der Ansprüche 1 bis 17, worin die Verbindung (I) Tacrolimus oder das Hydrat davon ist.
- Formulierung mit verzögerter Freisetzung nach einem der Ansprüche 1 bis 18, die in Form eines Pulvers, feinen Pulvers, Granulats, Tablette oder einer Kapsel vorliegt.
- Formulierung mit verzögerter Freisetzung nach Anspruch 1, worin die Zeit (T 63,2%) 1,0 bis 12 Stunden beträgt.
- Formulierung mit verzögerter Freisetzung nach Anspruch 1, worin die Zeit (T 63,2%) 1,3 bis 8,2 Stunden beträgt.
- Formulierung mit verzögerter Freisetzung nach Anspruch 1, worin die Zeit (T 63,2%) 2 bis 5 Stunden beträgt.
- Formulierung mit verzögerter Freisetzung, die eine Feststoffdispersionszusammensetzung umfasst, welche dadurch gekennzeichnet ist, dass (1) Tacrolimus oder das Hydrat davon in einem amorphen Zustand in einer Mischung von Ethylcellulose und Hydroxypropylmethylcellulose vorhanden ist, wobei der Gewichtsanteil der Ethylcellulose und der Hydroxypropylmethylcellulose 0,1 bis 5 beziehungsweise 0,2 bis 0,4 zu Tacrolimus oder dem Hydrat davon (1,0) beträgt, (2) Lactose als ein Bindemittel enthalten ist, (3) die Partikelgröße der Feststoffdispersionszusammensetzung gleich oder kleiner als 250 μm ist.
- Formulierung mit verzögerter Freisetzung nach Anspruch 23, worin der Gewichtsanteil der Ethylcellulose 0,1–1 zu Tacrolimus (1,0) beträgt.
- Formulierung mit verzögerter Freisetzung nach Anspruch 23, worin der Gewichtsanteil der Lactose 2, 3 oder 5 zu Tacrolimus (1,0) beträgt.
- Formulierung mit verzögerter Freisetzung nach Anspruch 23, worin in der Feststoffdispersionszusammensetzung keine Lösungsvermittler enthalten sind.
- Formulierung mit verzögerter Freisetzung nach Anspruch 23, worin die Partikelgröße der Feststoffdispersionszusammensetzung gleich oder kleiner als 212 μm ist.
- Formulierung mit verzögerter Freisetzung, die eine Feststoffdispersionszusammensetzung umfasst, welche dadurch gekennzeichnet ist, dass (1) Tacrolimus oder das Hydrat davon in einem amorphen Zustand in einer Mischung von Ethylcellulose und Hydroxypropylmethylcellulose vorhanden ist, wobei der Gewichtsanteil der Ethylcellulose und der Hydroxypropylmethylcellulose jeweils 0,3 zu Tacrolimus oder einem Hydrat davon (1,0) beträgt, (2) Lactose als ein Bindemittel enthalten ist, (3) die Partikelgröße der Feststoffdispersionszusammensetzung gleich oder kleiner als 212 μm ist.
- Formulierung mit verzögerter Freisetzung nach einem der Ansprüche 23 bis 28, die in Form eines Pulvers, feinen Pulvers, Granulats, Tablette oder einer Kapsel vorliegt.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP7903998 | 1998-03-26 | ||
| JP7903998 | 1998-03-26 | ||
| JP18296398 | 1998-06-29 | ||
| JP18296398 | 1998-06-29 | ||
| PCT/JP1999/001499 WO1999049863A1 (en) | 1998-03-26 | 1999-03-25 | Sustained release preparations |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69918074D1 DE69918074D1 (de) | 2004-07-22 |
| DE69918074T2 true DE69918074T2 (de) | 2004-11-04 |
Family
ID=26420115
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69918074T Expired - Lifetime DE69918074T2 (de) | 1998-03-26 | 1999-03-25 | Makrolid-formulierung mit verzögerter wirkstoffabgabe |
| DE69942286T Expired - Lifetime DE69942286D1 (de) | 1998-03-26 | 1999-03-25 | Retardpräparat mit Makroliden wie Tacrolimus |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69942286T Expired - Lifetime DE69942286D1 (de) | 1998-03-26 | 1999-03-25 | Retardpräparat mit Makroliden wie Tacrolimus |
Country Status (31)
| Country | Link |
|---|---|
| US (5) | US6440458B1 (de) |
| EP (3) | EP1064942B9 (de) |
| JP (4) | JP3714970B2 (de) |
| KR (3) | KR100498765B1 (de) |
| CN (1) | CN1229111C (de) |
| AR (1) | AR023299A1 (de) |
| AT (3) | ATE269075T1 (de) |
| AU (1) | AU749623B2 (de) |
| BR (1) | BRPI9909201B8 (de) |
| CA (1) | CA2322516C (de) |
| CZ (1) | CZ300548B6 (de) |
| DE (2) | DE69918074T2 (de) |
| DK (3) | DK1421939T5 (de) |
| ES (3) | ES2343248T3 (de) |
| HR (1) | HRP20000707B1 (de) |
| HU (1) | HU230889B1 (de) |
| ID (1) | ID27825A (de) |
| IL (1) | IL138466A (de) |
| ME (2) | ME00189B (de) |
| NO (1) | NO330578B1 (de) |
| NZ (1) | NZ507211A (de) |
| PL (1) | PL193244B1 (de) |
| PT (3) | PT1421939E (de) |
| RS (1) | RS50164B (de) |
| RU (1) | RU2214244C9 (de) |
| SI (1) | SI1064942T1 (de) |
| SK (1) | SK286887B6 (de) |
| TR (1) | TR200002771T2 (de) |
| TW (2) | TW570814B (de) |
| WO (1) | WO1999049863A1 (de) |
| ZA (1) | ZA200004963B (de) |
Families Citing this family (105)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030129215A1 (en) * | 1998-09-24 | 2003-07-10 | T-Ram, Inc. | Medical devices containing rapamycin analogs |
| US7399480B2 (en) * | 1997-09-26 | 2008-07-15 | Abbott Laboratories | Methods of administering tetrazole-containing rapamycin analogs with other therapeutic substances using medical devices |
| US6890546B2 (en) | 1998-09-24 | 2005-05-10 | Abbott Laboratories | Medical devices containing rapamycin analogs |
| ME00189B (me) * | 1998-03-26 | 2011-02-10 | Astellas Pharma Inc | Preparati sa neprekidnim oslobađanjem |
| US7455853B2 (en) * | 1998-09-24 | 2008-11-25 | Abbott Cardiovascular Systems Inc. | Medical devices containing rapamycin analogs |
| US6855172B2 (en) | 1998-10-13 | 2005-02-15 | Dry, Inc. | Dry-cleaning article, composition and methods |
| US7807211B2 (en) | 1999-09-03 | 2010-10-05 | Advanced Cardiovascular Systems, Inc. | Thermal treatment of an implantable medical device |
| US6790228B2 (en) | 1999-12-23 | 2004-09-14 | Advanced Cardiovascular Systems, Inc. | Coating for implantable devices and a method of forming the same |
| US20070032853A1 (en) | 2002-03-27 | 2007-02-08 | Hossainy Syed F | 40-O-(2-hydroxy)ethyl-rapamycin coated stent |
| CN1212831C (zh) * | 2000-03-08 | 2005-08-03 | Awd.药品股份有限两合公司 | 药物制剂 |
| JP5698423B2 (ja) | 2000-06-27 | 2015-04-08 | ベクトゥラ・リミテッド | 医薬組成物で使用するための粒子の製造法 |
| US20040018228A1 (en) * | 2000-11-06 | 2004-01-29 | Afmedica, Inc. | Compositions and methods for reducing scar tissue formation |
| MX368013B (es) | 2001-02-19 | 2019-09-13 | Novartis Ag | Tratamiento de cáncer. |
| US6645946B1 (en) | 2001-03-27 | 2003-11-11 | Pro-Pharmaceuticals, Inc. | Delivery of a therapeutic agent in a formulation for reduced toxicity |
| US7012068B2 (en) | 2001-03-27 | 2006-03-14 | Pro-Pharmaceuticals, Inc. | Co-administration of a polysaccharide with a chemotherapeutic agent for the treatment of cancer |
| GB0108498D0 (en) * | 2001-04-04 | 2001-05-23 | Novartis Ag | Organic Compounds |
| BR0209474A (pt) * | 2001-05-09 | 2006-02-07 | Novartis Ag | Métodos para imunomodulação seletiva |
| AUPR529701A0 (en) * | 2001-05-28 | 2001-06-21 | Fujisawa Pharmaceutical Co., Ltd. | Pharmaceutical composition |
| US20080145402A1 (en) * | 2001-09-10 | 2008-06-19 | Abbott Cardiovascular Systems Inc. | Medical Devices Containing Rapamycin Analogs |
| GB0123400D0 (en) | 2001-09-28 | 2001-11-21 | Novartis Ag | Organic compounds |
| EP1443910A1 (de) * | 2001-11-02 | 2004-08-11 | Wockhardt Limited | Zubereitungen mit kontrollierter freigabe von makrolidantibiotika |
| JP2003327536A (ja) * | 2002-03-07 | 2003-11-19 | Kitasato Inst:The | ヒト免疫不全症候群ウイルスの感染、増殖抑制剤 |
| US20060171984A1 (en) * | 2002-09-06 | 2006-08-03 | Cromack Keith R | Device having hydration inhibitor |
| EP1638549A4 (de) | 2003-03-10 | 2011-06-15 | Optimer Pharmaceuticals Inc | Neue antibakterielle mittel |
| US20040185009A1 (en) * | 2003-03-19 | 2004-09-23 | Dexcel Pharma Technologies Ltd. | Composition and device for treating periodontal diseases |
| GB0307867D0 (en) * | 2003-04-04 | 2003-05-14 | Novartis Ag | Pharmaceutical composition |
| GB0307866D0 (en) * | 2003-04-04 | 2003-05-14 | Novartis Ag | Pharmaceutical composition |
| EP1638529B1 (de) * | 2003-06-16 | 2016-08-10 | ANDRX Pharmaceuticals, LLC. | Orale zusammensetzung mit verlängerter freisetzung |
| US20050118344A1 (en) | 2003-12-01 | 2005-06-02 | Pacetti Stephen D. | Temperature controlled crimping |
| EP1641437A4 (de) * | 2003-07-09 | 2009-06-03 | Chong Kun Dang Pharm Corp | Feste dispersion von tacrolimus |
| WO2005007110A2 (en) * | 2003-07-11 | 2005-01-27 | Pro-Pharmaceuticals, Inc. | Compositions and methods for hydrophobic drug delivery |
| US8591946B2 (en) * | 2003-08-29 | 2013-11-26 | Veloxis Pharmaceuticals A/S | Modified release compositions comprising tacrolimus |
| WO2005020993A1 (en) | 2003-08-29 | 2005-03-10 | Lifecycle Pharma A/S | Modified release compositions comprising tacrolimus |
| US20050053664A1 (en) * | 2003-09-08 | 2005-03-10 | Eliezer Zomer | Co-administration of a polysaccharide with a chemotherapeutic agent for the treatment of cancer |
| EP1675576A1 (de) * | 2003-10-03 | 2006-07-05 | LifeCycle Pharma A/S | Verfahren zur herstellung von pharmazeutischen zusammensetzungen mit modifizierter freisetzung |
| US7285544B2 (en) * | 2003-11-18 | 2007-10-23 | Bernstein Eric F | Use of nitroxides in treating skin disease |
| KR100485877B1 (ko) * | 2003-12-30 | 2005-04-28 | 종근당바이오 주식회사 | 타크롤리무스를 생산하는 미생물 및 이를 이용한타크롤리무스의 대량 생산방법 |
| JP2007516951A (ja) * | 2003-12-30 | 2007-06-28 | アステラス製薬株式会社 | 気道閉塞症の治療または予防のためのマクロライドの使用 |
| EP1718146A2 (de) * | 2004-02-13 | 2006-11-08 | Pro-Pharmaceuticals, Inc. | Zusammensetzungen und verfahren zur behandlung von akne und candida |
| US20080286251A1 (en) * | 2004-08-02 | 2008-11-20 | Propharmaceuticals, Inc. | Compositions and Methods for the Enhancement of Chemotherapy with Microbial Cytotoxins |
| WO2006021443A2 (en) * | 2004-08-26 | 2006-03-02 | Novartis Ag | Composition comprising an at1 receptor blocker and a macrolide t-cell immunomodulator |
| MX2007003731A (es) * | 2004-09-29 | 2007-08-14 | Johnson & Johnson | Formas de dosis farmaceuticas de compuestos similares a rapamicina amorfos estables. |
| CA2584430A1 (en) * | 2004-12-01 | 2006-06-08 | Teva Gyogyszergyar Zartkoruen Mukodo Reszvenytarsasag | Methods for preparing pimecrolimus |
| JP2007527434A (ja) * | 2005-01-05 | 2007-09-27 | テバ ジョジセルジャール ザ−トケルエン ムケド レ−スベニュタ−ルシャシャ−グ | 非晶質タクロリマス及びその調製 |
| KR100678824B1 (ko) * | 2005-02-04 | 2007-02-05 | 한미약품 주식회사 | 용해성이 증가된 무정형 타크로리무스 고체분산체 및 이를포함하는 약제학적 조성물 |
| MX2007011494A (es) * | 2005-03-17 | 2007-12-06 | Elan Pharma Int Ltd | Composiciones inyectables de compuestos inmunosupresores nanoparticulados. |
| KR101411100B1 (ko) * | 2006-01-23 | 2014-07-08 | 이슘 리서치 디벨롭먼트 컴퍼니 오브 더 히브루 유니버시티 오브 예루살렘, 엘티디. | 친지성 약물을 함유하는 나노캡슐을 포함하는 미소구체 |
| HUP0600097A3 (en) * | 2006-02-08 | 2008-07-28 | Richter Gedeon Nyrt | Pharmaceutical compositions comprising tacrolimus and process for their preparation |
| JP2007308479A (ja) | 2006-04-20 | 2007-11-29 | Shin Etsu Chem Co Ltd | 固体分散体製剤 |
| EP2067475A4 (de) * | 2006-09-26 | 2010-12-15 | Astellas Pharma Inc | Tacrolimus-zubereitung mit verzögerter freisetzung |
| EP1938804A1 (de) * | 2006-12-22 | 2008-07-02 | Novartis AG | Pharmazeutische Zubereitung enthaltend Neurokinin Antagonist |
| WO2008081829A1 (ja) * | 2006-12-27 | 2008-07-10 | Astellas Pharma Inc. | 難水溶性薬物の溶解性維持用アミノアルキルメタアクリレートコポリマーe |
| CA2673863A1 (en) * | 2006-12-28 | 2008-07-10 | Limerick Biopharma, Inc. | Methods and compositions for therapeutic treatment |
| US20090011018A1 (en) * | 2006-12-28 | 2009-01-08 | Astellas Pharma Inc., | Sustained release formulation for tacrolimus |
| US7521523B2 (en) | 2006-12-28 | 2009-04-21 | Eastman Chemical Company | Oxygen-scavenging polyester compositions useful in packaging |
| JP2010515753A (ja) | 2007-01-10 | 2010-05-13 | ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム | 肺内送達用免疫抑制剤組成物の送達の増強 |
| MX2009010556A (es) | 2007-03-29 | 2009-10-22 | Panacea Biotec Ltd | Formas de dosificacion modificada de tacrolimus. |
| ITMI20070720A1 (it) * | 2007-04-06 | 2008-10-07 | Monteresearch Srl | Composizioni orali contenenti tacrolimus in forma amorfa |
| US8703204B2 (en) | 2007-05-03 | 2014-04-22 | Bend Research, Inc. | Nanoparticles comprising a cholesteryl ester transfer protein inhibitor and anon-ionizable polymer |
| US8309129B2 (en) | 2007-05-03 | 2012-11-13 | Bend Research, Inc. | Nanoparticles comprising a drug, ethylcellulose, and a bile salt |
| HUE033011T2 (hu) | 2007-05-30 | 2017-11-28 | Veloxis Pharmaceuticals As | Takrolimuszt tartalmazó napi egyszeri orális dózisforma |
| US12083103B2 (en) | 2007-05-30 | 2024-09-10 | Veloxis Pharmaceuticals, Inc. | Tacrolimus for improved treatment of transplant patients |
| EP2162120B1 (de) | 2007-06-04 | 2016-05-04 | Bend Research, Inc | Nanopartikel mit nicht ionisierbarem zellulosepolymer und amphipilem nicht ionisierbarem blockcopolymer |
| WO2008149230A2 (en) | 2007-06-04 | 2008-12-11 | Pfizer Products Inc. | Nanoparticles comprising drug, a non-ionizable cellulosic polymer and tocopheryl polyethylene glycol succinate |
| JP2011500512A (ja) * | 2007-07-31 | 2011-01-06 | リメリック バイオファーマ,インコーポレイティド | リン酸化ピロン類似体および方法 |
| DK2200588T3 (da) * | 2007-09-25 | 2019-07-01 | Solubest Ltd | Sammensætninger, som omfatter lipofile aktive forbindelser, og fremgangsmåde til fremstilling deraf |
| EP2214484A4 (de) | 2007-10-25 | 2013-01-02 | Cempra Pharmaceuticals Inc | Verfahren zur herstellung von antibakteriellen makrolid-wirkstoffen |
| TW200932240A (en) | 2007-10-25 | 2009-08-01 | Astellas Pharma Inc | Pharmaceutical composition containing lipophilic substance which inhibits IL-2 production |
| KR20090044269A (ko) * | 2007-10-31 | 2009-05-07 | (주)아모레퍼시픽 | 서방성 나노입자 및 이를 함유하는 화장료 조성물 |
| WO2009073216A1 (en) | 2007-12-06 | 2009-06-11 | Bend Research, Inc. | Nanoparticles comprising a non-ionizable polymer and an amine-functionalized methacrylate copolymer |
| EP2231169B1 (de) | 2007-12-06 | 2016-05-04 | Bend Research, Inc. | Pharmazeutische zusammensetzungen mit nanopartikeln und resuspensionsmaterial |
| CL2008000374A1 (es) * | 2008-02-05 | 2008-04-04 | Igloo Zone Chile S A | Composicion farmaceutica que comprende un polvo para suspension oral de tacrolimus o una de sus sales, hidratos o solvatos y excipientes farmaceuticamente aceptables; procedimiento de preparacion de dicha composicion farmaceutica; y uso para la preve |
| US8222272B2 (en) * | 2008-04-11 | 2012-07-17 | Roxane Laboratories, Inc. | Pharmaceutical formulation and process comprising a solid dispersion of macrolide (tacrolimus) |
| US12403095B2 (en) | 2008-05-30 | 2025-09-02 | Veloxis Pharmaceuticals, Inc. | Stabilized tacrolimus composition |
| JP2011524413A (ja) * | 2008-06-17 | 2011-09-01 | バイオチカ テクノロジー リミテッド | 新規化合物、及びこれらの製造のための方法 |
| JP5711135B2 (ja) | 2008-10-24 | 2015-04-30 | センプラ ファーマシューティカルズ,インコーポレイテッド | 胃腸疾患を治療するための方法 |
| WO2010099508A1 (en) | 2009-02-26 | 2010-09-02 | Theraquest Biosciences, Inc. | Extended release oral pharmaceutical compositions of 3-hydroxy-n-methylmorphinan and method of use |
| US9937194B1 (en) | 2009-06-12 | 2018-04-10 | Cempra Pharmaceuticals, Inc. | Compounds and methods for treating inflammatory diseases |
| ES2608285T3 (es) | 2009-09-10 | 2017-04-07 | Cempra Pharmaceuticals, Inc. | Procedimientos para el tratamiento de paludismo, tuberculosis y enfermedades por MAC |
| TWI510238B (zh) | 2010-02-17 | 2015-12-01 | Lifecycle Pharma As | 穩定化他克莫司(tacrolimus)組合物 |
| WO2011105070A1 (ja) | 2010-02-25 | 2011-09-01 | パナソニック株式会社 | 需給制御装置、需給制御方法、およびプログラム |
| JP5823484B2 (ja) | 2010-07-14 | 2015-11-25 | 千寿製薬株式会社 | α―ケトアミド誘導体固体分散体 |
| WO2012026896A1 (en) * | 2010-08-25 | 2012-03-01 | Les Laboratoires Medis Sa | Surface modified micronized tacrolimus crystalline particles and pharmaceutical compositions thereof |
| EP2613630A4 (de) | 2010-09-10 | 2014-01-15 | Cempra Pharmaceuticals Inc | Wasserstoffbindung zur formung von fluorketoliden zur behandlung von krankheiten |
| JP2014521745A (ja) * | 2011-08-16 | 2014-08-28 | メルク・シャープ・アンド・ドーム・コーポレーション | 安定した非晶質分散体を調製するための無機マトリックスと有機ポリマーの組み合わせの使用 |
| ES2663744T3 (es) * | 2011-10-06 | 2018-04-16 | Novartis Ag | Composiciones farmacéuticas que comprenden 40-O-(2-hidroxi)etil-rapamicina |
| NZ700182A (en) | 2012-03-27 | 2017-02-24 | Cempra Pharmaceuticals Inc | Parenteral formulations for administering macrolide antibiotics |
| EP2871188B1 (de) * | 2012-07-06 | 2017-04-19 | Godo Shusei Co., Ltd. | Verfahren zur trennung einer cyclischen makrolidverbindung |
| AU2014239959A1 (en) | 2013-03-14 | 2015-10-01 | Cempra Pharmaceuticals, Inc. | Methods for treating respiratory diseases and formulations therefor |
| CN105188712A (zh) | 2013-03-15 | 2015-12-23 | 森普拉制药公司 | 用于制备大环内酯抗菌剂的收敛方法 |
| US9168246B2 (en) | 2013-06-27 | 2015-10-27 | Veloxis Pharmaceutical A/S | Regimen for suppressing organ rejection |
| CN103432099A (zh) * | 2013-08-13 | 2013-12-11 | 江苏正大清江制药有限公司 | 一种他克莫司缓释胶囊及其制备方法 |
| WO2016078481A1 (zh) | 2014-11-21 | 2016-05-26 | 杭州领业医药科技有限公司 | 一种含他克莫司的药物组合物及其制备方法 |
| CN104473907A (zh) * | 2014-12-25 | 2015-04-01 | 北京华禧联合科技发展有限公司 | 一种他克莫司的口服缓释制剂 |
| CA2986359A1 (en) | 2015-05-20 | 2016-11-24 | Novartis Ag | Pharmaceutical combination of everolimus with dactolisib |
| CN104840446B (zh) * | 2015-05-27 | 2017-08-11 | 福建科瑞药业有限公司 | 一种他克莫司药物组合物及其制备方法 |
| SMT202600033T1 (it) | 2016-04-15 | 2026-03-09 | Bioatla Inc | Anticorpi anti-axl, frammenti di anticorpo e loro immunoconiugati e loro utilizzi |
| EP3509572A4 (de) * | 2016-09-08 | 2020-05-13 | Synergistic Therapeutics, LLC | Topische haarwachstumsformulierung |
| CN110114070A (zh) | 2016-11-23 | 2019-08-09 | 诺华公司 | 使用依维莫司(everolimus)、达托里昔布(dactolisib)或二者增强免疫反应的方法 |
| US10596165B2 (en) | 2018-02-12 | 2020-03-24 | resTORbio, Inc. | Combination therapies |
| KR102081176B1 (ko) * | 2018-06-22 | 2020-02-25 | 주식회사 종근당 | 타크로리무스를 포함하는 서방형 약제학적 제제 |
| US20220062305A1 (en) * | 2018-12-18 | 2022-03-03 | DDP Specialty Electronic Materials US, Inc. | Sustained release composition comprising an ethylcellulose |
| EP3934677A4 (de) | 2019-03-13 | 2022-11-16 | University of Virginia Patent Foundation | Zusammensetzungen und verfahren zur förderung der insellebensfähigkeit und zur steigerung der insulinsekretion |
| GR1009790B (el) | 2019-03-20 | 2020-08-03 | Φαρματεν Α.Β.Ε.Ε. | Σκευασμα παρατεταμενης αποδεσμευσης που περιλαμβανει τακρολιμους |
| JP7681303B2 (ja) * | 2021-09-03 | 2025-05-22 | 国立研究開発法人農業・食品産業技術総合研究機構 | 養分供給量推定方法、養分供給体の利用方法、プログラム、および推定システム |
Family Cites Families (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL32929A (en) | 1968-09-03 | 1973-08-29 | Banker G | Controlled-release pharmaceutical compositions containing a polymer for chemisorbing the active drug |
| US4127647A (en) * | 1975-04-08 | 1978-11-28 | Meiji Seika Kaisha, Ltd. | Process for preparation of stable amorphous macrolide antibiotic solids |
| US4389393A (en) | 1982-03-26 | 1983-06-21 | Forest Laboratories, Inc. | Sustained release therapeutic compositions based on high molecular weight hydroxypropylmethylcellulose |
| US4894366A (en) | 1984-12-03 | 1990-01-16 | Fujisawa Pharmaceutical Company, Ltd. | Tricyclo compounds, a process for their production and a pharmaceutical composition containing the same |
| JPS62208103A (ja) | 1986-03-07 | 1987-09-12 | Yokogawa Hewlett Packard Ltd | ロボツト運動制御システム |
| GB8608080D0 (en) * | 1986-04-02 | 1986-05-08 | Fujisawa Pharmaceutical Co | Solid dispersion composition |
| JPS63150220A (ja) * | 1986-12-15 | 1988-06-22 | Dainippon Pharmaceut Co Ltd | 経口用固形製剤の製造方法 |
| US4968508A (en) * | 1987-02-27 | 1990-11-06 | Eli Lilly And Company | Sustained release matrix |
| JP2822389B2 (ja) | 1987-06-05 | 1998-11-11 | 藤沢薬品工業株式会社 | 抗fr−900506物質抗体、および高感度酵素免疫測定法 |
| WO1989005304A1 (en) | 1987-12-09 | 1989-06-15 | Fisons Plc | Macrocyclic compounds |
| DE3742525C2 (de) | 1987-12-11 | 1998-02-19 | Hertz Inst Heinrich | Verfahren zur Herstellung von Metallalkylverbindungen und deren Verwendung |
| FR2624732B1 (fr) * | 1987-12-21 | 1991-02-15 | Synthelabo | Formulation pharmaceutique a liberation prolongee |
| JPH02232814A (ja) | 1989-03-07 | 1990-09-14 | Fuji Photo Film Co Ltd | 磁気記録媒体 |
| DK0406791T3 (da) | 1989-07-05 | 1995-03-27 | Fujisawa Pharmaceutical Co | Vandigt flydende præparat til ekstern anvendelse |
| KR0159766B1 (ko) | 1989-10-16 | 1998-12-01 | 후지사와 토모키치로 | 양모제 조성물 |
| DK0427680T3 (da) | 1989-11-09 | 1995-12-18 | Sandoz Ltd | Heteroatom-holdige cykliske forbindelser |
| JPH03232814A (ja) * | 1990-02-08 | 1991-10-16 | Shin Etsu Chem Co Ltd | 徐放性錠剤の製造方法 |
| EP0594600A1 (de) | 1990-03-13 | 1994-05-04 | FISONS plc | Immunosuppressive makrozyklische verbindungen |
| US5643901A (en) | 1990-06-11 | 1997-07-01 | Fujisawa Pharmaceutical Co., Ltd. | Medicament for treating idiopathic thrombocytopenic purpura |
| JPH05507915A (ja) | 1990-06-11 | 1993-11-11 | 藤沢薬品工業株式会社 | 特発性血小板減少性紫斑病およびバセドウ病治療薬製造のためのfk506等のマクロライド化合物の用途 |
| MY110418A (en) | 1990-07-02 | 1998-05-30 | Novartis Ag | Heteroatoms-containing tricyclic compounds. |
| EP0480623A1 (de) | 1990-10-11 | 1992-04-15 | Merck & Co. Inc. | Halomakrolide und Derivate mit immunosuppressiver Wirkung |
| US5208241A (en) | 1991-09-09 | 1993-05-04 | Merck & Co., Inc. | N-heteroaryl, n-alkylheteroaryl, n-alkenylheteroaryl and n-alkynylheteroarylmacrolides having immunosuppressive activity |
| US5252732A (en) | 1991-09-09 | 1993-10-12 | Merck & Co., Inc. | D-heteroaryl, O-alkylheteroaryl, O-alkenylheteroaryl and O-alkynylheteroarylmacrolides having immunosuppressive activity |
| US5247076A (en) | 1991-09-09 | 1993-09-21 | Merck & Co., Inc. | Imidazolidyl macrolides having immunosuppressive activity |
| JP3128320B2 (ja) | 1992-03-28 | 2001-01-29 | 株式会社常盤電機 | 不燃性シート及びその製造方法 |
| GB9208492D0 (en) * | 1992-04-16 | 1992-06-03 | Glaxo Spa | Heterocyclic compounds |
| HUT66531A (en) | 1992-05-07 | 1994-12-28 | Sandoz Ag | Heteroatoms-containing tricyclic compounds, pharmaceutical prepns contg. them and process for producing them |
| GB9209882D0 (en) * | 1992-05-07 | 1992-06-24 | Glaxo Lab Sa | Compositions |
| JP2707023B2 (ja) * | 1992-07-01 | 1998-01-28 | 株式会社大塚製薬工場 | 経口吸収用製剤 |
| GB9221220D0 (en) * | 1992-10-09 | 1992-11-25 | Sandoz Ag | Organic componds |
| AU686115B2 (en) | 1992-11-02 | 1998-02-05 | Fujisawa Pharmaceutical Co., Ltd. | Imidazo (I,2-a) pyridine derivatives as bradykinin antagonists, pharmaceuticals and processes for their preparation |
| US5601844A (en) * | 1992-11-18 | 1997-02-11 | Fujisawa Pharmaceutical Co., Ltd. | Sustained release medicinal preparation |
| MY110603A (en) | 1993-05-27 | 1998-08-29 | Novartis Ag | Tetrahydropyran derivatives |
| US6204243B1 (en) * | 1993-09-01 | 2001-03-20 | Novatis Ag | Pharmaceutical preparations for the targeted treatment of crohn's disease and ulcerative colitis |
| DE4329503A1 (de) | 1993-09-01 | 1995-03-02 | Galenik Labor Freiburg Gmbh | Pharmazeutische Präparate zur gezielten Behandlung von Morbus Crohn und Colitis Ulcerosa |
| JP3232814B2 (ja) | 1993-09-22 | 2001-11-26 | ジェイエスアール株式会社 | 感熱記録媒体用インキ組成物 |
| HUT77530A (hu) * | 1994-05-06 | 1998-05-28 | Pfizer Inc. | Azitromicint tartalmazó, szabályozott hatóanyag-felszabadítású dózisformák |
| AR004480A1 (es) | 1995-04-06 | 1998-12-16 | Amico Derin C D | Compuestos de ascomicina que poseen actividad antiinflamatoria, pro cedimiento para prepararlos, uso de dichos compuestos para preparar agentesfarmaceuticos y composiciones farmaceuticas que los incluyen |
| FR2736550B1 (fr) * | 1995-07-14 | 1998-07-24 | Sandoz Sa | Composition pharmaceutique sous la forme d'une dispersion solide comprenant un macrolide et un vehicule |
| CA2229718A1 (en) * | 1995-08-24 | 1997-03-06 | David J. Mathre | Process for the preparation of imidazolyl macrolide immunosuppressants |
| US5705190A (en) * | 1995-12-19 | 1998-01-06 | Abbott Laboratories | Controlled release formulation for poorly soluble basic drugs |
| TW426516B (en) | 1996-12-06 | 2001-03-21 | Fujisawa Pharmaceutical Co | An oral pharmaceutical composition in solid dispersion containing water-insoluble tricyclic compounds |
| ME00189B (me) * | 1998-03-26 | 2011-02-10 | Astellas Pharma Inc | Preparati sa neprekidnim oslobađanjem |
-
1999
- 1999-03-25 ME MEP-2008-300A patent/ME00189B/me unknown
- 1999-03-25 HR HR20000707A patent/HRP20000707B1/xx not_active IP Right Cessation
- 1999-03-25 NZ NZ507211A patent/NZ507211A/en not_active IP Right Cessation
- 1999-03-25 PT PT04002277T patent/PT1421939E/pt unknown
- 1999-03-25 RS YUP-580/00A patent/RS50164B/sr unknown
- 1999-03-25 IL IL13846699A patent/IL138466A/xx not_active IP Right Cessation
- 1999-03-25 PT PT99909332T patent/PT1064942E/pt unknown
- 1999-03-25 KR KR10-2003-7016882A patent/KR100498765B1/ko not_active Expired - Lifetime
- 1999-03-25 KR KR10-2000-7010438A patent/KR100440553B1/ko not_active Expired - Lifetime
- 1999-03-25 SK SK1439-2000A patent/SK286887B6/sk not_active IP Right Cessation
- 1999-03-25 ID IDW20002175A patent/ID27825A/id unknown
- 1999-03-25 SI SI9930622T patent/SI1064942T1/xx unknown
- 1999-03-25 ME MEP-300/08A patent/MEP30008A/xx unknown
- 1999-03-25 CN CNB998064157A patent/CN1229111C/zh not_active Expired - Lifetime
- 1999-03-25 EP EP99909332A patent/EP1064942B9/de not_active Expired - Lifetime
- 1999-03-25 DK DK04002277.4T patent/DK1421939T5/da active
- 1999-03-25 ES ES04002277T patent/ES2343248T3/es not_active Expired - Lifetime
- 1999-03-25 EP EP10152774A patent/EP2198858B1/de not_active Expired - Lifetime
- 1999-03-25 TW TW088104712A patent/TW570814B/zh not_active IP Right Cessation
- 1999-03-25 RU RU2000126836A patent/RU2214244C9/ru active Protection Beyond IP Right Term
- 1999-03-25 JP JP54564199A patent/JP3714970B2/ja not_active Expired - Lifetime
- 1999-03-25 CA CA2322516A patent/CA2322516C/en not_active Expired - Lifetime
- 1999-03-25 AT AT99909332T patent/ATE269075T1/de active
- 1999-03-25 CZ CZ20003549A patent/CZ300548B6/cs not_active IP Right Cessation
- 1999-03-25 AU AU28563/99A patent/AU749623B2/en not_active Expired
- 1999-03-25 WO PCT/JP1999/001499 patent/WO1999049863A1/ja not_active Ceased
- 1999-03-25 BR BRPI9909201A patent/BRPI9909201B8/pt not_active IP Right Cessation
- 1999-03-25 DK DK10152774.5T patent/DK2198858T3/da active
- 1999-03-25 ES ES99909332T patent/ES2219000T3/es not_active Expired - Lifetime
- 1999-03-25 PL PL343096A patent/PL193244B1/pl unknown
- 1999-03-25 TR TR2000/02771T patent/TR200002771T2/xx unknown
- 1999-03-25 DE DE69918074T patent/DE69918074T2/de not_active Expired - Lifetime
- 1999-03-25 AT AT04002277T patent/ATE464900T1/de active
- 1999-03-25 AT AT10152774T patent/ATE514419T1/de active
- 1999-03-25 HU HU0101237A patent/HU230889B1/hu unknown
- 1999-03-25 TW TW092115044A patent/TWI235068B/zh not_active IP Right Cessation
- 1999-03-25 DE DE69942286T patent/DE69942286D1/de not_active Expired - Lifetime
- 1999-03-25 DK DK99909332T patent/DK1064942T3/da active
- 1999-03-25 KR KR10-2003-7005713A patent/KR100505464B1/ko not_active Expired - Lifetime
- 1999-03-25 ES ES10152774T patent/ES2367294T3/es not_active Expired - Lifetime
- 1999-03-25 US US09/403,787 patent/US6440458B1/en not_active Expired - Lifetime
- 1999-03-25 PT PT10152774T patent/PT2198858E/pt unknown
- 1999-03-25 EP EP04002277A patent/EP1421939B9/de not_active Revoked
- 1999-03-26 AR ARP990101353A patent/AR023299A1/es active IP Right Grant
-
2000
- 2000-09-18 ZA ZA200004963A patent/ZA200004963B/en unknown
- 2000-09-25 NO NO20004773A patent/NO330578B1/no not_active IP Right Cessation
-
2001
- 2001-10-17 US US09/978,025 patent/US6576259B2/en not_active Expired - Lifetime
-
2003
- 2003-04-14 US US10/412,281 patent/US6884433B2/en not_active Expired - Lifetime
-
2004
- 2004-08-18 JP JP2004237946A patent/JP3992031B2/ja not_active Expired - Lifetime
- 2004-08-18 JP JP2004237947A patent/JP4622382B2/ja not_active Expired - Lifetime
-
2005
- 2005-02-17 US US11/059,439 patent/US20050169993A1/en not_active Abandoned
-
2008
- 2008-07-17 JP JP2008186587A patent/JP4992845B2/ja not_active Expired - Lifetime
- 2008-11-05 US US12/265,108 patent/US8551522B2/en not_active Expired - Fee Related
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69918074T2 (de) | Makrolid-formulierung mit verzögerter wirkstoffabgabe | |
| CA2274485C (en) | Medicinal composition | |
| RS58000B1 (sr) | Derivati naftiridina kao antagonisti alfa v beta 6 integrina za lečenje npr. fibrotičnih bolesti | |
| JPWO1998024418A1 (ja) | 医薬組成物 | |
| JPWO1999049863A1 (ja) | 徐放性製剤 | |
| MXPA00009227A (en) | Sustained release preparations |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8327 | Change in the person/name/address of the patent owner |
Owner name: ASTELLAS PHARMA INC., TOKIO/TOKYO, JP |