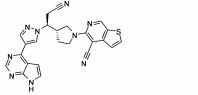
CAMPO TÉCNICO
A presente invenção refere-se aos derivados de N-(hetero)aril- pirrolidina de fórmula I, bem como suas composições e métodos de uso, que são inibidores de JAK, tal como inibidores de JAK1 seletiva, úteis no tratamento de doenças associadas com JAK incluindo, por exemplo, distúrbios inflamatórios e autoimunes, bem como câncer.
ANTECEDENTES
Proteínas quinases (PKs) são um grupo de enzimas que regulam diversos processos biológicos importante incluindo crescimento, sobrevivência e diferenciação celular, formação de órgão e morfogênese, neovascularização, regeneração e reparo de tecido, entre outros. Proteínas quinases exercem suas funções fisiológicas através da catalisação da fosforilação de proteínas (ou substratos) e desse modo modulam as atividades celulares dos substratos em vários contextos biológicos. Além das várias funções em tecidos/órgãos normais, muitas proteínas quinases também desempenham papéis mais especializados em um hospedeiro de doenças humanas incluindo câncer. Um subgrupo de proteínas quinases (também referidas como proteínas quinases oncogênicas), quando desregulado, pode causar a formação e desenvolvimento de tumor, e, além disso, contribui para a manutenção e progressão de tumor. Até aqui, as proteínas quinases oncogênicas representam um dos maiores grupos e mais atrativos de proteínas alvos para intervenção de câncer e desenvolvimento de fármaco.
A família de Janus quinase (JAK) desempenha um papel na re-gulação dependente de citocina de proliferação e função de células envolvidas em resposta imune. Atualmente, existem quarto membros de família JAK mamífera: JAK1 (também conhecida como Janus Quinase-1), JAK2 (também conhecida como Janus Quinase-2), JAK3 (também conhecida como Janus Quinase, leucócito; JAKL; L-JAK e Janus quinase-3) e TYK2 (também conhecida como proteína tirosina quinase 2). As proteínas JAK variam em tamanho de 120 a 140 kDa e compreendem sete domínios de homologia de JAK (JH) conservados; um destes é um domínio de quinase catalítica funcional, e outro é um domínio de pseudoquinase potencialmente exercendo 5 uma função reguladora e/ou funcionando como um sítio de acoplamento para STATs.
O bloqueio de transdução de sinal no nível das JAK quinases sustenta a promessa de desenvolvimento de tratamento para doenças infla-matórias, doenças autoimunes, doenças mieloproliferativas, e cânceres hu- 10 manos, para citar alguns. A inibição das JAK quinases é também pretendida ter benefícios terapêuticos em pacientes que estão sofrendo de distúrbios imunes de pele tais como psoríase, e senbilidade da pele. Consequentemente, os inibidores de Janus quinases ou quinases relacionadas são amplamente pesquisados e diversas publicações reportam as classes eficazes de 15 compostos. Por exemplo, certos inibidores de JAK, incluindo pirrolopiridina e pirrolopirimidinas, são reportados na Publicação de Pedido dos Estados Unidos n° 2007/0135461, depositada em 12 de Dezembro de 2006.
Desse modo, agentes novos ou melhorados que inibem quinases tal como Janus quinases são continuamente necessários para o desen- 20 volvimento de produtos farmacêuticos eficazes para tratar câncer e outras doenças. As composições e métodos descritos aqui são direcionados a estas necessidades e outras finalidades.
SUMÁRIO
A presente invenção fornece, entre outras coisas, compostos de 25 fórmula I:

ou sais farmaceuticamente aceitáveis ou N-óxidos dos mesmos; em que: X é ciano ou halogênio; Y é CH ou N; Z é hidrogênio, C1-4 alquila, C1-4 alquila fluorada, ou flúor; Ar é C6-14 arila, C1-14 heteroarila, C7-14 cicloalquilarila fundida, C6-14 heterocicloalquilarila fundida, C2-14 cicloalquil-heterocicloarila fundi- 5 da, ou C2-14 heterocicloalquil-heteroarila fundida, cada das quais é opcionalmente substituída por 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, C2-6 alquinila, C3-14 10 cicloalquila, C3-14 cicloalquil-C1 -4-alquila, C2-14 heterocicloalquila, C2-14 heterocicloalquil-C1-4-alquila, C6-14 arila, C6-14 aril-C 1 -4-alquila, C1-13 heteroarila, C1-13 heteroaril-C1-4-alquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)NReRf, -C(=O)Rb, -C(=O)ORb, -C(=O)NReRf, -OC(=O)Rb, -OC(=O)NReRf, -NReRf, -NRcC(=O)Rd, -NRcC(=O)ORd, 15 -NRcC(=O)NRd, -NRcS(=O)2Rd, e -NRbS(=O)2NReRf; em que as referidas C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, e C2-6 alquinila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos R1a independentemente selecionados; e em que as referidas C3-14 cicloalquila, C3-14 cicloalquíl-C1- 4-alquila, C2-14 heterocicloalquila, C2-14 heterocicloalquil-C1 -4-alquila, C620 14 arila, C6-14 aril-C 1-4-alquila, C1-13 heteroarila, e C1-13 heteroaril-C1-4- alquila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos R2a independentemente selecionados; cada R1a é independentemente selecionado de halogênio, cia-no, nitro, hidroxíla, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1-4 alquilsul- 25 finila, C1-4 alquilsulfonila, amino, C1-4 alquilamino, did-4-alquilamino, C1-4 alquilcarbonila, carbóxi, C1-4 alcoxicarbonila, C1-4-alquilcarbonilamino, did- 4-alquilcarbonilamino, C1-4-alcoxicarbonilamino, C1-4-alcoxicarbonil-(C1-4 alquil)amino, carbamila, C1-4 alquilcarbamila, e did-4-alquilcarbamila; cada R2a é independentemente selecionado de halogênio, cia- 30 no, nitro, hidroxila, C1-4 alquila, C1-4 haloalquila, C2-4 alquenila, C2-4 alqui-nila, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1-4 alquilsulfinila, C1-4 alquilsulfonila, amino, C1-4 alquilamino, did-4-alquilamino, C1-4 alquilcar- bonila, carbóxi, C1-4 alcoxicarbonila, C1-4-alquilcarbonilamino, did-4-alquil- carbonilamino, CI-4-alcoxicarbonilamino, C1-4-alcoxicarbonil-(C1^t al- quil)amino, carbamila, C1-4 alquilcarbamila, e did-4-alquilcarbamila; cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente seleciona- do de horas, C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, C2-6 alquinila, C3-7 cicloalquila, C3-7 cicloalquil-C 1 -4-alquila, C2-7 heterocicloalquila, C2-7 heterocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila; em que as referidas C1-6 alquila, C1-6 haloal-quila, C2-6 alquenila, e C2-6 alquinila são cada qual opcionalmente substitu- idas por 1, 2, 3, ou 4 grupos Rx independentemente selecionados; e em que as referidas C3-7 cicloalquila, C3-7 cicloalquil-C1 -4-alquila, C2-7 heteroci-cloalquila, C2-7 heterocicloalquil-C1-4-alquila, fenila, fenil-C1 -4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos Ry independentemente selecionados; ou qualquer Rc e Rd, juntamente com a porção a qual eles são ligados, podem formar um anel heterocicloalquila de 3, 4, 5, 6, ou 7 membros, em que o referido anel heterocicloalquila é opcionalmente substituído com 1,2, 3, ou 4 grupos independentemente selecionados de hidroxila, C1-4 alquila, C1-4 haloalquila, C1-4 alcóxi, C1-4 haloalcóxi, amino, C1-4 alquila- mino, e did-4-alquilamino; ou qualquer Re e Rf, juntamente com o átomo de nitrogênio ao qual eles são ligados, podem formar um anel heterocicloalquila ou anel hete-roarila de 3, 4, 5, 6, ou 7 membros, em que o referido anel heterocicloalquila ou heteroarila é opcionalmente substituído com 1, 2, 3, ou 4 grupos inde- pendentemente selecionados de hidroxila, C1-4 alquila, C1-4 haloalquila, C1-4 alcóxi, C1-4 haloalcóxi, amino, C1-4 alquilamino, e did-4-alquilamino; cada Rx é independentemente selecionado de hidroxila, C1-4 alcóxi, C1-4 haloalcóxi, amino, C1-4 alquilamino, e did-4-alquilamino; e cada Ry é independentemente selecionado de hidroxila, halo- 30 gen, ciano, nitro, C1-4 alquila, C1-4 haloalquila, C1-4 alcóxi, C1-4 haloalcóxi, amino, C1-4 alquilamino, e did-4-alquilamino; contanto que a valência de cada átomo nas porções opcional mente substituídas não seja excedida.
A presente invenção também fornece composições farmacêuti-cas, que compreendem um composto de fórmula I como descrito aqui, ou um sal farmaceuticamente aceitável ou N-óxido do mesmo, e um veículo far- 5 maceuticamente aceitável.
A presente invenção também fornece métodos de modulação de uma atividade de JAK1 que compreende contatar JAK1 com um composto de fórmula I como descrito aqui, ou um sal farmaceuticamente aceitável ou N-óxido do mesmo.
A presente invenção também fornece métodos de tratamento de uma doença autoimune, um câncer, um distúrbio mieloproliferativo, uma do-ença inflamatória, ou rejeição ao transplante de órgão em um paciente em necessidade dos mesmos, que compreendem administrar ao referido paciente uma quantidade terapeuticamente eficaz de um composto de fórmula I como descrito aqui, ou um sal farmaceuticamente aceitável ou N-óxido do mesmo.
A presente invenção também fornece compostos de fórmula I, ou sais farmaceuticamente aceitáveis ou N-óxidos dos mesmos, como descrito aqui para uso em métodos de tratamento de doenças autoimunes, cân- 20 cer, distúrbios mieloproliferativos, doenças inflamatórias, ou rejeição ao transplante de órgão.
A presente invenção também fornece compostos de fórmula I como descrito aqui, ou sais farmaceuticamente aceitáveis ou N-óxidos dos mesmos, para uso em métodos de modulação de uma JAK1.
A presente invenção também fornece usos de compostos de fórmula I como descrito aqui, ou sais farmaceuticamente aceitáveis ou N- óxidos dos mesmos, para a preparação de medicaments para uso em métodos de modulação de uma JAK1.
Este Pedido reivindica o benefício de Pedidos de Patente Provi- sória dos Estados Unidos n° de série 61/180.622, depositado em 22 de Maio de 2009 e n° de série 61/225.092, depositado em 13 de Julho de 2009 ambos os quais são incorporados aqui por referência em sua totalidade.
Os detalhes de uma ou mais modalidades da invenção são men-cionados nos desenhos acompanhantes e na descrição abaixo. Outras ca-racterísticas, objetivos, e vantagens da invenção serão evidentes a partir da descrição e desenhos, e a partir das reivindicações.
DESCRIÇÃO DETALHADA
Em vários lugares na presente especificação, substituintes de compostos da invenção são descritos em grupos ou em faixas. Especificamente entende-se que a invenção inclui cada e toda subcombinação individual dos membros de tais grupos e faixas. Por exemplo, o termo "C1-6 alqui- 10 la" destina-se especificamente a descrever individualmente metila, etila, C3 alquila, C4 alquila, C5 alquila, e C6 alquila.
Entende-se também que os compostos da invenção são estáveis. Como usado aqui "estável" refere-se a um composto que é suficientemente robusto para sobreviver ao isolamento em um grau útil de uma mistu- 15 ra reacional, e preferivelmente capaz de formulação em um agente terapêutico eficaz.
Aprecia-se também que certas características da invenção, que são, para clareza, descritas no contexto de modalidades separadas, possam também ser fornecidas em combinação em uma única modalidade. Contrari- 20 amente, várias características da invenção que são, para brevidade, descritas no contexto de uma única modalidade, podem também ser fornecidas separadamente ou em qualquer subcombinação adequada.
O termo "membro n" onde n é um número inteiro tipicamente descreve o número de átomos de formação de anel em uma porção onde o 25 número de átomos de formação de anel é n. Por exemplo, piperidinila é um exemplo de um anel heterocicloalquila de 6 membros e 1,2,3,4-tetraidro- naftaleno é um exemplo de um grupo cicloalquila de 10 membros.
Para os compostos da invenção em que uma variável aparece mais do que uma vez, cada variável pode ser uma porção diferente indepen- 30 dentemente selecionada do grupo que define a variável. Por exemplo, onde uma estrutura é descrita tendo dois grupos R que são simultaneamente presents no mesmo composto, os dois grupos R podem representar diferentes porções independentemente selecionadas do grupo definido por R. Em outro exemplo, quando um substituinte opcionalmente múltiplo é designado na forma:

em seguida entende-se que o substituinte R pode ocorrer diversas vezes no anel, e R pode ser uma porção diferente em cada ocorrência. Entende-se que cada grupo R pode ser substituir qualquer átomo de hidrogênio ligado a um átomo de anel, incluindo um ou ambos os átomos de hidrogênio (CH2)n. Além disso, no exemplo acima, deve a variável Q ser definida incluir hidro-génios, tal como quando Q é referido ser CH2, NH, etc., qualquer substituinte flutuante tal como R no exemplo acima, pode substituir um hidrogênio da variável Q bem como um hidrogênio em qualquer outro componente não va-riável do anel.
Para compostos da invenção em que uma variável aparece mais do que uma vez, cada variável pode ser uma porção diferente independen-temente selecionada do grupo que define a variável. Por exemplo, onde uma estrutura é descrita tendo dois grupos R que são simultaneamente presentes no mesmo composto, os dois grupos R podem representar diferentes porções independentemente selecionadas do grupo definido por R.
Como usada aqui, a frase "opcionalmente substituído" significa não substituído ou substituído. Como usado aqui, o termo "substituído" signi-fica que um átomo de hidrogênio é removido e substituído por um substituin-te. Como usada aqui, a frase "substituído com oxo" significa que dois átomos de hidrogênio são removidos de um átomo de cabono e substituídos por uma ligação de oxigênio por uma ligação dupla ao átomo de carbono. En- tende-se que a substituição em um determinado átomo seja limitada por valência.
Como usado aqui, o termo "Cn-m alquila", empregado sozinho ou em combinação com outros termos, refere-se a um grupo hidrocarboneto saturado que pode ser de cadeia linear ou ramificada, tendo átomos de car-bono de n a m. Em algumas modalidades, o grupo alquila contém 1 a 12, 1 a 8, 1 a 6, ou 1 a 4 átomos de carbono. Exemplos de porções alquila incluem, porém não estão limitados a grupos químicos tais como metila, etila, n-pro- pila, isopropila, n-butila, terc-butila, isobutila, sec-butila, 2-metil-1-butíla, n- pentila, 3-pentila, n-hexila, 1,2,2-trimetilpropila, n-heptila, n-octila, e similares.
Como usado aqui, o termo "Cn-m alquileno", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de ligação de alquila divalente tendo átomos de carbono de n a m. Em algumas modalida-des, a porção alquileno contém 2 a 6, ou 2 a 4 átomos de carbono. Exemplos de grupos alquileno incluem, porém não estão limitados a etan-1,2-di- ila, propan-1,3-di-ila, propan-1,2-di-ila, butan-1,4-di-ila, butan-1,3-di-ila, bu- tan-1,2-di-ila, 2-metil-propan-1,3-di-ila, e similares.
Como usado aqui, "Cn-m alquenila", empregado sozinho ou em combinação com outros termos, refere-se a um grupo alquila tendo uma ou mais ligações de carbono-carbono duplas e átomos de carbono de n a m. Em algumas modalidades, a porção alquenila contém 2 a 6, ou 2 a 4 átomos de carbono. Exemplo grupos alquenila incluem, porém não estão limitados à etenila, n-propenila, isopropenila, n-butenila, sec-butenila, e similares.
Como usado aqui, "Cn-m alquenileno", empregado sozinho ou em combinação com outros termos, refere-se a um grupo alquenila divalen- te. Em algumas modalidades, a porção alquenileno contém 2 a 6, ou 2 a 4 átomos de carbono.
Como usado aqui, "Cn-m alquinila", empregado sozinho ou em combinação com outros termos, refere-se a um grupo alquila tendo uma ou mais ligações de carbono-carbono triplas e átomos de carbono de n a m. Exemplo grupos alquinila incluem, porém não estão limitados à etinila, pro- pin-1-ila, propin-2-ila, e similares. Em algumas modalidades, a porção alqui-nila contém 2 a 6 ou 2 a 4 átomos de carbono.
Como usado aqui, "Cn-m alquinileno", empregado sozinho ou em combinação com outros termos, refere-se a um grupo alquinila divalente. Em algumas modalidades, a porção alquinileno contém 2 a 6, ou 2 a 4 átomos de carbono.
Como usado aqui, o termo "Cn-m alcóxi", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -O- alquila, em que o grupo alquila tem átomos de carbono de n a m. Exemplo de grupos alcóxi metóxi, etóxi, propóxi (por exemplo, n-propóxi e isopropóxi), t-butóxi, e similares. Em algumas modalidades, o grupo alquila tem 1 a 6 ou 1 a 4 átomos de carbono.
Como usado aqui, o termo "carbóxi", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -C(=O)OH.
Como usado aqui, o termo "Cn-M-alcoxicarbonil-(Cn-m al- quil)amino", empregado sozinho ou em combinação com outros termos, refe- re-se a um grupo de fórmula -N(alquil)-C(=O)O(alquil), em que cada alquila independentemente tem átomos de carbono de n a m.
Como usado aqui, o termo "Cn-M-alcoxicarbonilamino", empre-gado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -NH-C(=O)O(alquil), em que a alquila tem átomos de carbono de n a m.
Como usado aqui, o termo "Cn-m alcoxicarbonila’1, empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fór-mula -C(=O)O-alquila, em que o grupo alquila tem átomos de carbono de n a m. Em algumas modalidades, o grupo alquila tem 1 a 6 ou 1 a 4 átomos de carbono.
Como usado aqui, o termo "Cn-m alquilcarbonila", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -C(=O)-alquila, em que o grupo alquila tem átomos de carbono de n a m. Em algumas modalidades, o grupo alquila tem 1 a 6 ou 1 a 4 átomos de carbono.
Como usado aqui, o termo "Cn-M-alquilcarbonilamino", empre-gado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -NHC(=O)-alquila, em que o grupo alquila tem átomos de carbono de n a m. Em algumas modalidades, o grupo alquila tem 1 a 6 ou 1 a 4 átomos de carbono.
Como usado aqui, o termo "dicn-M-alquilcarbonilamino", empre-gado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -N(alquil)C(=O)-alquila, em que cada alquila group independen-temente tem átomos de carbono de n a m. Em algumas modalidades, cada grupo alquila independentemente tem 1 a 6 ou 1 a 4 átomos de carbono.
Como usado aqui, o termo "amino" refere-se a um grupo de fór- 5 mula -NH2.
Como usado aqui, o termo "Cn-m alquilamino", empregado sozi-nho ou em combinação com outros termos, refere-se a um grupo de fórmula -NH(alquil), em que o grupo alquila tem átomos de carbono de n a m.
Como usado aqui, o termo "dicn-M-alquilamino", empregado so- 10 zinho ou em combinação com outros termos, refere-se a um grupo de fórmula -N(alquil)2, em que cada grupo alquila tem independentemente átomos de carbono de n a m. Em algumas modalidades, cada grupo alquila independentemente tem 1 a 6 ou 1 a 4 átomos de carbono.
Como usado aqui, o termo "carbamila", empregado sozinho ou 15 em combinação com outros termos, refere-se a um grupo de fórmula - C(=O)NH2.
Como usado aqui, o termo "Cn-m alquilcarbamila", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -C(=O)-NH(alquil), em que o grupo alquila tem átomos de carbono de n 20 a m. Em algumas modalidades, o grupo alquila tem 1 a 6 ou 1 a 4 átomos de carbono.
Como usado aqui, o termo "dicn-M-alquilcarbamila", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -C(=O)-N(alquil)2, em que cada grupo alquila independentemente tem 25 átomos de carbono de n a m. Em algumas modalidades, cada grupo alquila independentemente tem 1 a 6 ou 1 a 4 átomos de carbono.
Como usado aqui, o termo "Cn-M-alquiltio", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -S- alquila, em que a alquila tem átomos de carbono de n a m. Em algumas mo- 30 dalidades, o grupo alquila tem 1 a 6 ou 1 a 4 átomos de carbono.
Como usado aqui, o termo "Cn-M-alquilsulfinila", empregado so-zinho ou em combinação com outros termos, refere-se a um grupo de fórmu- la -S(=O)-alquila, em que a alquila tem átomos de carbono de n a m. Em algumas modalidades, o grupo alquila tem 1 a 6 ou 1 a 4 átomos de carbono.
Como usado aqui, o termo "Cn-M-alquilsulfonila", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -S(=O)2-alquila, em que a alquila tem átomos de carbono de n a m. Em algumas modalidades, o grupo alquila tem 1 a 6 ou 1 a 4 átomos de car-bono.
Como usado aqui, o termo "carbonila", empregado sozinho ou em combinação com outros termos, refere-se a um grupo -C(=O)-, que é uma porção de um carbono divalente também ligada a um átomo de oxigênio com uma ligação dupla.
Como usado aqui, o termo "ciano", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -CN, em que os átomos de carbono e nitrogênio são ligados uns aos outros por uma ligação tripla.
Como usado aqui, os termos "halo" e "halogênio", empregado sozinho ou em combinação com outros termos, refereM-se a flúor, cloro, bromo, e iodo.
Como usado aqui, "Cn-m haloalcóxi", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -O-ha- loalquila, em que o grupo haloalquila tem átomos de carbono de n a m. Em algumas modalidades, o grupo alquila tem 1 a 6 ou 1 a 4 átomos de carbono. Um grupo haloalcóxi exemplo é -OCF3.
Como usado aqui, o termo "Cn-m haloalquila", empregado sozi-nho ou em combinação com outros termos, refere-se a um grupo alquila tendo de átomos de carbono de n a m e um átomo de halogênio a 2x+1 átomos de halogênio que podem ser iguais ou diferentes, onde "x" é o número de átomos de carbono em um grupo alquila. Em algumas modalidades, os áto-mos de halogênio são átomos de flúor. Em algumas modalidades, o grupo alquila tem 1 a 6 ou 1 a 4 átomos de carbono. Um exemplo de um grupo ha-loalquila é -CF3.
Como usado aqui, o termo "Cn-m alquila fluorado", empregado sozinho ou em combinação com outros termos, refere-se a um Cn-m haloalquila em que os átomos de halogênio são selecionados de flúor. Em algumas modalidades, Cn-m haloalquila fluorado é fluormetila, difluorometila, ou 5 trifluorometila.
Como usado aqui, o termo "Cn-m cicloalquila", empregado sozi-nho ou em combinação com outros termos, refere-se a uma porção hidro- carboneto cíclico não aromático, que pode opcionalmente conter um ou mais grupos alquenileno como parte da estrutura de anel, e que tem átomos de 10 carbono de membro de anel n a m. Grupos cicloalquila podem incluir sistemas de anel mono- ou policiclicos (por exemplo, tendo 2, 3, ou 4 anéis fundidos, em ponte, ou espiros). Também incluídas na definição de cicloalquila são porções que têm um ou mais anéis aromáticos fundidos (isto é, tendo uma ligação em comum com) ao anel cicloalquila, por exemplo, derivados de 15 benzo de ciclopentano, ciclopenteno, ciclo-hexano, e similares. O termo "cicloalquila" também inclui grupos cicloalquila cabeça de ponte e grupos espi- rocicloalquila. Como usado aqui, "grupos cicloalquila cabeça de ponte" refere-se às porções hidrocarboneto cíclico não aromático contendo pelo menos um carbono cabeça de ponte, tal como adamantan-1-ila. Como usado aqui, 20 "grupos espirocicloalquila" refere-se às porções hidrocarboneto não aromático contendo pelo menos dois anéis fundidos em um único átomo de carbono, tal como espiro[2,5]octano e similares. Em algumas modalidades, o grupo cicloalquila tem 3 a 14 membros de anel, 3 a 10 membros de anel, ou 3 a 7 membros de anel. Em algumas modalidades, o grupo cicloalquila é mono- 25 cíclico ou bicíclico. Em algumas modalidades, o grupo cicloalquila é monocí- clico. Em algumas modalidades, o grupo cicloalquila é um grupo C3-7 ciclo-alquila monocíclico. Um ou mais átomos de carbono de formação de anel de um grupo cicloalquila pode ser oxidado para formar ligações de carbonila. Exemplos de grupos cicloalquila incluem ciclopropila, ciclobutila, ciclopentila, 30 ciclo-hexila, cicloeptila, ciclopentenila, ciclo-hexenila, ciclo-hexadienila, ciclo- eptatrienila, norbornila, norpinila, norcarnila, adamantila, e similares. Em algumas modalidades, o grupo cicloalquila é adamantan-1-ila.
Como usado aqui, o termo "Cn-m cicloalquil-Co-p alquila", em-pregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -alquileno-cicloalquila, em que a porção cicloalquila tem átomos de carbono de n a m e a porção alquileno tem átomos de carbono do a 5 p. Em algumas modalidades, a porção alquileno tem 1 a 4, 1 a 3, 1 a 2, ou 1 átomo(s) de carbono. Em algumas modalidades, a porção alquileno é meti- leno. Em algumas modalidades, a porção cicloalquila tem 3 a 14 membros de anel, 3 a 10 membros de anel, ou 3 a 7 membros de anel. Em algumas modalidades, o grupo cicloalquila é monocíclico ou bícíclico. Em algumas 10 modalidades, a porção cicloalquila é monocíclica. Em algumas modalidades, a porção cicloalquila é um grupo C3-7 cicloalquila monocíclico.
Como usado aqui, o termo "Cn-m heterocicloalquila", "anel Cn-m heterocicloalquila", ou "grupo Cn-m heterocicloalquila", empregado sozinho ou em combinação com outros termos, refere-se ao anel não aromático ou 15 sistema de anel, que pode opcionalmente conter um ou mais grupos alqueni- leno como parte da estrutura de anel, que tem pelo menos um membro de anel heteroátomo independentemente selecionado de nitrogênio, enxofre e oxigênio, e que tem átomos de carbono de membro de anel de n a m. Grupos heterocicloalquila podem incluir sistemas de anel mono- ou policíclicos 20 (por exemplo, tendo 2, 3 ou 4 anéis fundidos, em ponte ou espiro). Em algumas modalidades, o grupo heterocicloalquila é um grupo monocíclico ou bicíclico tendo 1, 2, 3, ou 4 heteroátomos independentemente selecionados de nitrogênio, enxofre e oxigênio. Estão também incluídas na definição de heterocicloalquila porções que têm um ou mais anéis aromáticos fundidos 25 (isto é, tendo uma ligação em comum com) ao anel não aromático, por e- xemplo, 1,2,3,4-tetraidro-quinolina e similares. Grupos heterocicloalquila podem também incluir grupos heterocicloalquila cabeça de ponte e espiroete- rocicloalquila. Como usado aqui, "grupo heterocicloalquila cabeça de ponte" refere-se a uma porção heterocicloalquila contendo pelo menos um átomo 30 cabeça de ponte, tal como azadamantan-1-ila e similares. Como usado aqui, "grupo espiroeterocicloalquila" refere-se a uma porção heterocicloalquila contendo pelo menos dois anéis fundidos em um única átomo, tal como [1,4-dioxa-8-aza-espiro[4,5]decan-N-il] e similares. Em algumas modalidades, o grupo heterocicloalquila tem 3 a 20 átomos de formação de anel, 3 a 10 á- tomos de formação de anel, ou cerca de 3 a 8 átomos de formação de anel. Os átomos de carbono ou heteroátomos nos anel(s) do grupo heterocicloal- 5 quila podem ser oxidados para formar um grupo carbonila, ou sulfonila (ou outra ligação oxidada) ou um átomo de nitrogênio pode ser quartenizado. Em algumas modalidades, a porção de heterocicloalquila é um grupo C2-7 heterocicloalquila monociclico.
Como usado aqui, o termo "Cn-m heterocicloalquil-Co-p alquila", 10 empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -alquileno-heterocicloalquila, em que a porção heterocicloalquila tem átomos de carbono de n a m e a porção alquileno tem átomos de carbono o a p. Em algumas modalidades, a porção alquileno tem 1 a 4, 1 a 3, 1 a 2, ou 1 átomo(s) de carbono. Em algumas modalidades, a porção al- 15 quileno é metileno. Em algumas modalidades, a porção heterocicloalquila tem 3 a 14 membros de anel, 3 a 10 membros de anel, ou 3 a 7 membros de anel. Em algumas modalidades, o grupo heterocicloalquila é monociclico ou bicíclico. Em algumas modalidades, a porção heterocicloalquila é monocicli- ca. Em algumas modalidades, a porção heterocicloalquila é um grupo C2-7 20 heterocicloalquila monocíclica.
Como usado aqui, o termo "Cn-m arila", empregado sozinho ou em combinação com outros termos, refere-se à porção hidrocarboneto aromático monociclico ou policíclico (por exemplo, tendo 2, 3 ou 4 anéis fundidos) tendo átomos de carbono de membro de anel de n a m, tal como, po- 25 rém não limitados à fenila, 1-naftila, 2-naftila, antracenila, fenantrenila, e similares. Em algumas modalidades, grupos arila têm de 6 a 14 átomos de carbono, cerca de 6 a 10 átomos de carbono, ou cerca de 6 átomos de carbono. Em algumas modalidades, o grupo arila é um grupo monociclico ou bi- ciclico.
Como usado aqui, o termo "Cn-m aril-Co-p-alquila", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fór-mula -alquileno-arila, em que a porção arila tem átomos de carbono de mem- bro de anel de n a m e a porção alquileno tem átomos de carbono do a p. Em algumas modalidades, a porção alquileno tern 1 a 4, 1 a 3, 1 a 2, ou 1 átomo(s) de carbono. Em algumas modalidades, a porção alquileno é meti- leno. Em algumas modalidades, a porção arila é fenila. Em algumas modalidades, o grupo arila é um grupo monocíclico ou bicíclico. Em algumas modalidades, a arilalquila é benzila.
Como usado aqui, o termo "Cn-m heteroarila", "anel Cn-m hete-roarila", ou "grupos Cn-m heteroarila", empregado sozinho ou em combinação com outros termos, refere-se a uma porção hidrocarboneto monocíclico ou policiclico (por exemplo, tendo 2, 3 ou 4 anéis fundidos), tendo um ou mais membros de anel de heteroátomo independentemente selecionados de nitrogênio, enxofre e oxigênio e tendo átomos de carbono de membro de anel de n a m. Em algumas modalidades, o grupo heteroarila é um grupo monocíclico ou bicíclico tendo 1, 2, 3, ou 4 heteroátomos independentemente selecionados de nitrogênio, enxofre e oxigênio. Grupos heteroarila exemplo incluem, porém não estão limitados à pirrolila, azolila, oxazolila, tiazolila, imidazolila, furila, tienila, quinolinila, isoquinolinila, indolila, benzotienila, ben- zofuranila, benzisoxazolila, imidazo[1,2-b]tiazolila ou similares. Os átomos de carbono ou heteroátomos no anel(s) do grupo heteroarila podem ser oxidados para formar um grupo carbonila, ou sulfonila (ou outra ligação oxidada) ou um átomo de nitrogênio pode ser quaternizado, contanto que a natureza aromática do anel seja preservada. Em algumas modalidades, o grupo hete-roarila tem 5 a 10 átomos de carbono.
Como usado aqui, o termo "Cn-m heteroaril-Co-p-alquila", em-pregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -alquileno-heteroarila, em que a porção heteroarila tem átomos de carbono de membro de anel de n a m e a porção alquileno tem átomos de carbono do a p. Em algumas modalidades, a porção alquileno tem 1 a 4, 1 a 3, 1 a 2, ou 1 átomo(s) de carbono. Em algumas modalidades, a porção alquileno é metileno. Em algumas modalidades, a porção heteroarila é um grupo monocíclico ou bicíclico tendo 1,2, 3, ou 4 heteroátomos independentemente selecionado de nitrogênio, enxofre e oxigênio. Em algumas mo dalidades, a porção heteroarila tem 5 a 10 átomos de carbono.
Como usado aqui, o termo "Cn-m heterocicloalquil-heteroarila fundida" refere-se a uma porção tendo um grupo heteroarila fundido a um grupo heterocicloalquila, com átomos de carbono de membro de anel de n a m na porção, em que a porção é ligada ao átomo de nitrogênio do anel pirro- lidina de fórmula I através do grupo heteroarila. Em algumas modalidades, a Cn-m heterocicloalquil-heteroarila fundida tem 2 a 14, 3 a 14, 4a 14, 5 a 14, 5 a 12, 5 a 10, ou 6 a 9 átomos de carbono. Em algumas modalidades, a Cn- m heterocicloalquil-heteroarila fundida é 2,3-di-hidrotieno[2,3-b]pÍridin-6-ila, S-oxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3- b]piridin-6-ila, 2,3-di-hidrofuro[2,3-b]piridin-6-ila, 2,3-di-hidrotieno[3,2-c]piri- din-6-ila, S-oxo-2,3-di-hidrotieno[3,2-c]piridin-6-ila, S,S-dioxo-2,3-di-hidro- tieno[3,2-c]piridin-6-ila, ou similares.
Como usado aqui, o termo "Cn-m cicloalquil-heteroarila fundida" refere-se a uma porção tendo um grupo heteroarila fundida a um grupo ci-cloalquila, com átomos de carbono de membro de anel de n a m na porção, em que a porção é ligada ao átomo de nitrogênio do anel pirrolidina de fórmula I através do grupo heteroarila. Em algumas modalidades, a Cn-m cicloalquil-heteroarila fundida tem de 2 a 14, 3 a 14, 4 a 14, 5 a 14, 5 a 12, ou 5 a 10 átomos de carbono. Em algumas modalidades, a Cn-m cicloalquil-hete- roarila fundida é 6,7-di-hidro-5H-ciclopenta[b]piridin-2-ila ou similares.
Como usado aqui, o termo "Cn-m cicloalquilarila fundida" refere- se a uma porção tendo um grupo arila fundido a um grupo cicloalquila, com átomos de carbono de membro de anel de n a m na porção, em que a porção é ligada ao átomo de nitrogênio do anel pirrolidina de fórmula I através do grupo arila. Em algumas modalidades, a Cn-m cicloalquilarila fundida tem 6 a 16, 6 a 15, 6 a 14, 6 a 13, 6 a 12, ou 6 a 10 átomos de carbono.
Como usado aqui, o termo "Cn-m heterocicloalquilarila fundida" refere-se a uma porção tendo um grupo arila fundido a um grupo heteroci-cloalquila, com átomos de carbono de membro de anel de n a m na porção, em que a porção é ligada ao átomo de nitrogênio do anel pirrolidina de fórmula I através do grupo arila. Em algumas modalidades, a Cn-m heterociclo- alquilarila fundida tem de 6 a 16, 6 a 15, 6 a 14, 6 a 13, 6 a 12, ou 6 a 10 átomos de carbono.
Como usado aqui, o aparecimento do termo "bicíclico" antes do nome de uma porção indica que a porção tem dois anéis fundidos.
Como usado aqui, o aparecimento do termo "monociclico" antes do nome de uma porção indica que a porção tem um único anel.
A menos que de outro modo indicado aqui, o ponto de ligação de um substituinte é geralmente na última porção do nome (por exemplo, arilal- quila é ligada através da porção alquileno do grupo).
Como usado aqui, em que Ar é indicado como "um anel tiazol", "um anel piridina", "um anel piridimina", etc., o anel pode ser ligado em qual-quer posição do anel, contanto que a valência do átomo no ponto de ligação não seja excedida. Ao contrário, em algumas modalidades, o ponto exato de ligação é claramente indicado no nome (por exemplo, "tiazol-2-ila, "piridin-2- ila, "piridin-3-ila", "piridin-4-ila", "piridimin-2-ila" e "pirimidin-4-ila"). Por exem-plo, o ponto de ligação para "tiazol-2-ila" é a posição 2 do anel.
Como usado aqui, a frase "farmaceuticamente aceitável" é em-pregado aqui para se referir àqueles compostos, materiais, composições, e/ou formas de dosagem que são, dentro do escopo de diagnóstico médico seguro, adequados para uso em contato com os tecidos de seres humanos e animais sem toxicidade excessiva, irritação, resposta alérgica, ou outro problema ou complicação, comensurável com uma relação risco/benefício razoável.
Como usado aqui, as expressões, "temperatura ambiente” e "temperatura local," como usado aqui, são entendidas na técnica, e se referem geralmente a uma temperatura, por exemplo, uma temperatura de reação, que é de cerca da temperatura do local em que a reação é realizada, por exemplo, uma temperatura de cerca de 20 °C a cerca de 30 °C.
Como usado aqui, o termo "contatar" refere-se à junção de por-ções indicadas em um sistema in vitro ou um sistema in vivo. Por exemplo, "contatar" uma JAK com um composto da invenção inclui a administração de um composto da presente invenção a um indivíduo ou paciente, tal como um humano, tendo uma JAK, bem como, por exemplo, introdução de um composto da invenção em uma amostra contendo uma preparação celular ou purificada contendo a JAK.
Como usado aqui, os termos "indivíduo" ou "paciente," usados intercambiavelmente, refere-se a qualquer animal, incluindo mamíferos, preferivelmente camundongos, ratos, outros roedores, coelhos, cachorros, gatos, suínos, gado vacum, ovelhas, cavalos, ou primatas, e mais preferivelmente humanos.
Como usado aqui, a frase "quantidade terapeuticamente eficaz" refere-se à quantidade de composto ativo ou agente farmacêutico que produz a resposta biológica ou médica que está sendo procurada em um tecido, sistema, animal, indivíduo ou humano por um pesquisador, veterinário, médico ou outro clínico.
Como usado aqui, o termo "tratrando" ou "tratamento" refere-se a um ou mais de (1) prevenção da doença; por exemplo, prevenção da doença, condição ou distúrbio em um indivíduo que pode ser predisposto à doença, condição ou distúrbio, porém ainda não experimenta ou exibe a patologia ou sintomatologia da doença; (2) inibição da doença; por exemplo, inibição de uma doença disease, condição ou distúrbio em um indivíduo que está experimentando ou exibindo a patologia ou sintomatologia da doença, condição ou distúrbio; e (3) melhora da doença; por exemplo, melhora de uma doença, condição ou distúrbio em um indivíduo que está experimentando ou exibindo a patologia ou sintomatologia da doença, condição ou distúrbio (isto é, reversão da patologia e/ou sintomatologia) tal como diminuição da gravidade da doença.
A presente invenção fornece, entre outras coisas, um composto de fórmula I:

ou um sal farmaceuticamente aceitável ou N-óxido do mesmo; em que: X é ciano ou halogênio; YéCH ou N; Z é hidrogênio, C1-4 alquila, C1-4 alquila fluorada, ou flúor; Ar é C6-14 arila, C1-14 heteroarila, C7-14 cicloalquilarila fundida, C6-14 heterocicloalquilarila fundida, C2-14 cicloalquil-heterocicloarila fundida, ou C2-14 heterocicloalquil-heteroarila fundida, cada das quais é opcionalmente substituída por 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, C2-6 alquinila, C3-14 cicloalquila, C3-14 cicloalquil-C1-4-alquila, C2-14 heterocicloalquila, C2-14 heterocicloalquil-C1-4-alquila, C6-14 arila, C6-14 aril-C1-4-alquila, C1-13 heteroarila, C1-13 heteroaril-C1-4-alquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, 15 -S(=O)NReRf, -C(=O)Rb, -C(=O)ORb, -C(=O)NReRf, -OC(=O)Rb, -OC(=O)NReRf, -NReRf, -NRcC(=O)Rd, -NRcC(=O)ORd, -NRcC(=O)NRd, -NRcS(=O)2Rd, e -NRbS(=O)2NReRf; em que as referidas C1-6 alquila, C1- 6 haloalquila, C2-6 alquenila, e C2-6 alquinila são cada qual opcionalmente ! substituídas por 1, 2, 3, ou 4 grupos R1a independentemente selecionados; I e em que as referidas C3-14 cicloalquila, C3-14 cicloalquil-C1-4-alquila, C2- i 14 heterocicloalquila, C2-14 heterocicloalquil-C1-4-alquila, C6-14 arila, C6- | 14 aril-C1-4-alquila, C1-13 heteroarila, e C1-13 heteroaril-C1-4-alquila são | cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos R2a indepen- I dentemente selecionados; | cada R1a é independentemente selecionado de halogênio, cia- ! no, nitro, hidroxila, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1-4 alquilsul- finila, C1-4 alquilsulfonila, amino, C1-4 alquilamino, did-4-alquilamino, C1-4 alquilcarbonila, carbóxi, C1-4 alcoxicarbonila, C1-4-alquilcarbonilamino, did- 4-alquilcarbonilamino, C1-4-alcoxicarbonilamino, C1-4-alcoxicarbonil-(C1-4 alquil)amino, carbamila, C1-4 alquilcarbamila, e did-4-alquilcarbamila; cada R2a é independentemente selecionado de halogênio, ciano, nitro, hidroxila, C1-4 alquila, C1-4 haloalquila, C2-4 alquenila, C2-4 alqui- nila, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1-4 alquilsulfinila, C1-4 alquilsulfonila, amino, C1-4 alquilamino, dicl-4-alquilamino, C1-4 alquilcar- bonila, carbóxi, C1-4 alcoxicarbonila, C1-4-alquilcarboπilamino, did-4-alquil- carbonilamino, C1-4-alcoxicarbonilamino, C1-4-alcoxicarbonil-(C1-4 al- quiljamino, carbamila, C1-4 alquilcarbamila, e did-4-alquílcarbamila; cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, C2-6 alquinila, C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-7 heterocicloalquila, C2-7 heterocicloalquil-C1-4-alquila, fenila, fenil-C 1 -4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1 -4-alquila; em que as referidas C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, e C2-6 alquinila são cada qual opcionalmente substituídas por 1,2, 3, ou 4 grupos Rx independentemente selecionados; e em que as referidas C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-7 heterocicloalquila, C2-7 heterocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos Ry independentemente selecionados; ou qualquer Rc e Rd, juntamente com a porção a qual eles são ligados, podem formar um anel heterocicloalquila de 3, 4, 5, 6, ou 7 membros, em que o referido anel heterocicloalquila é opcionalmente substituído com 1, 2, 3, ou 4 grupos independentemente selecionados de hidroxila, C1-4 alquila, C1-4 haloalquila, C1-4 alcóxi, C1-4 haloalcóxi, amino, C1-4 alquilamino, e did-4-alquilamino; ou qualquer Re e Rf, juntamente com o átomo de nitrogênio ao qual eles são ligados, podem formar um anel heterocicloalquila ou anel heteroarila de 3, 4, 5, 6, ou 7 membros, em que o referido anel heterocicloalquila ou heteroarila é opcionalmente substituído com 1, 2, 3, ou 4 grupos independentemente selecionados de hidroxila, C1-4 alquila, C1-4 haloalquila, C1-4 alcóxi, C1-4 haloalcóxi, amino, C1-4 alquilamino, e did-4-alquilamino; cada Rx é independentemente selecionado de hidroxila, C1-4 alcóxi, C1-4 haloalcóxi, amino, C1-4 alquilamino, e did-4-alquilamino; e cada Ry é independentemente selecionado de hidroxila, halogen, ciano, nitro, C1^4 alquila, C1-4 haloalquila, C1-4 alcóxi, C1-4 haloalcóxi, amino, C1-4 alquilamino, e did-4-alquilamino.
Entende-se que a valência de cada átomo nas porções opcionalmente substituídas não é excedida.
Em algumas modalidades, Yé N. Em algumas modalidades, Y é CH.
Em algumas modalidades, X é ciano, flúor, ou cloro. Em algumas modalidades, X é ciano ou flúor. Em algumas modalidades, X é ciano. Em algumas modalidades, X é cloro ou flúor. Em algumas modalidades, X é flúor.
Em algumas modalidades, Z é hidrogênio ou flúor. Em algumas modalidades, Z é hidrogênio. Em algumas modalidades, Z é flúor.
Em algumas modalidades, Ar é selecionado de fenila, C1-6 heteroarila monocíclica, C1-9 heteroarila bicíclica, C7-14 cicloalquilarila fundida bicíclica, C6-14 heterocicloalquilarila fundida bicíclica, C2-14 cicloalquil- heterocicloarila fundida bicíclica, e C2-14 heterocicloalquil-heteroarila fundida bicíclica; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados. Em algumas modalidades, Ar é selecionado de fenila, C1-6 heteroarila monocíclica, C1-9 heteroarila bicíclica, e C2-14 heterocicloalquil-heteroarila fundida bicíclica; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados. Em algumas modalidades, Ar é fenila, que é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados. Em algumas modalidades, Ar é C1-6 heteroarila monocíclica, que é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados. Em algumas modalidades, Ar é C1-9 heteroarila bicíclica, que é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados. Em algumas modalidades, Ar é C2-14 heterocicloalquil-heteroarila fundida bicícfila, que é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados. Em algumas modalidades, Ar é C2-14 cicloalquil-heterocicloarila fundida bicíclica, que é opcíonalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados. Em algumas modalidades, Ar é sele- cionado de fenila, um anel tiazol, um anel piridina, um anel piridimina, um anel pirazina, um anel benzo[d]oxazol, um anel oxazolo[4,5-c]piridina, um anel oxazolo[5,4-b]piridina, um anel oxazolo[5,4-d]pirimidina, um anel 7H- pirrolo[2,3-d]pirimidina, um anel 2,3-di-hidrotieno[2,3-b]piridina, um anel S- oxo-2,3-di-hidrotieno[2,3-b]piridina, um anel S,S-dioxo-2,3-di-hidrotieno[2,3- b]piridina, um anel quinazolina, um anel quinolina, e um anel quinoxalina; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados. Em algumas modalidades, Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2- 10 ila, pirimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b] piridin-2-ila, oxazolo[5,4-d]pirimidin-2-ila, 7H-pirrolo[2,3- d]pirimidin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S-oxo-2,3-di-hidrotie- no[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, quinazo- lin-2-ila, quinolin-2-ila, e quinoxalin-2-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados. Em algumas modalidades, Ar é selecionado de fenila, um anel tiazol, um anel piridina, um anel piridimina, um anel pirazina, um anel benzo[d]oxa- zol, um anel oxazolo[4,5-c]piridina, um anel oxazolo[5,4-b]piridina, um anel oxazolo[5,4-d]pirimidina, um anel 7H-pirrolo[2,3-d]pirimidina, um anel 2,3-di- hidrotieno[2,3-b]piridina, um anel S-oxo-2,3-di-hidrotieno[2,3-b]piridina, um anel S,S-dioxo-2,3-di-hidrotieno [2,3-b]piridina, um anel quinazolina, um anel quinolina, um anel pirrolo[2,3-b]piridina, um anel oxazolo[4,5-b]piridina, um anel 3-oxo-3,4-di-hidropirazina, e um anel quinoxalina; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemen- te selecionados. Em algumas modalidades, Ar é selecionado de fenila, tia- zol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]pirídin-2-ila, oxazolo[5,4- b]piridin-2-ila, oxazolo[5,4-d]pirimidin-2-ila, 7H-pirrolo[2,3-d]pirimidin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S-oxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, quinazolin-2-ila, quinolin-2-ila, pirrolo[2,3-b]piridin-6-ila, oxazolo[4,5-b]piridin-2-ila, 3-oxo-3,4-di-hidropirazin- 2-ila, e quinoxalin-2-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados. Em algumas modalidades, Ar é selecionado de fenila, um anel tiazol, um anel piridina, um anel piridimina, um anel pirazina, um anel benzo[d]oxazol, um anel oxazo- lo[4,5-c]piridina, um anel oxazolo[5,4-b]piridina, um anel oxazolo[5,4-d]piri- midina, um anel 7H-pirrolo[2,3-d]pirimidina, um anel 2,3-dÍ-hidrotieno[2,3- bjpiridina, um anel S-oxo-2,3-di-hidrotÍeno[2,3-b] piridina, um anel S,S-dioxo- 2,3-di-hidrotieno[2,3-b]piridina, um anel quinazolina, um anel quinolina, um anel pirrolo[2,3-b]piridina, um anel oxazolo[4,5-b]piridina, um anel 3-oxo-3,4- di-hidropirazina, um anel quinoxalina, um anel oxazolo[5,4-d]pirimidina, um anel tieno[3,2-b]piridina, um anel tieno[2,3-c]piridina, um anel tiofeno, um anel tiazolo[5,4-d]pirimidina, um anel tieno[2,3-b]piridina, um anel 2,3-di-hi- drofuro[2,3-b]piridina, um anel 6,7-di-hidro-5H-ciclopenta[b]piridina, um anel furo[3,2-c]piridina, um anel 2,3-di-hidrotÍeno[3,2-c]piridina, um anel S-oxo- 2,3-di-hidrotieno[3,2-c]piridina, um anel S,S-dioxo-2,3-dÍ-hidrotieno[3,2-c] piridina, um anel tieno[3,2-c]piridina, e um anel 1 H-pirrolo[3,2-c]piridina; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados. Em algumas modalidades, Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pi- rimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirimidin-2-ila, 7H-pirrolo[2,3-d] piri- midin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S-oxo-2,3-di-hidrotieno[2,3-b] pirÍdin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, quinazolin-2-ila, quinolin-2-ila, pirrolo[2,3-b]piridin-6-ila, oxazolo[4,5-b]piridin-2-ila, 3-oxo-3,4- di-hidropirazin-2-ila, quinoxalin-2-ila, tiazol-4-ila, tiazol-5-ila, pirimidin-5-ila, oxazolo[5,4-d]pirimidin-2-ila, tieno[3,2-b]piridin-5-ila, tieno[2,3-c]piridin-5-ila, tiofen-2-ila, tiofen-3-ila, tiazolo[5,4-d]pirimidin-5-ila, tieno[2,3-b]piridin-6-ila, 2,3-di-hidrofuro[2,3-b]piridin-6-ila, 6,7-di-hidro-5H-ciclopenta[b]piridin-2-ila, furo[3,2-c] piridin-6-ila, 2,3-di-hidrotieno[3,2-c]piridin-6-ila, S-oxo-2,3-di-hi- drotieno[3,2-c] piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[3,2-c]piridin-6-ila, tie- no[3,2-c]piridin-6-ila, e 1H-pirrolo[3,2-c]piridin-6-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados.
Em algumas modalidades, cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, C2-6 alquinila, C3-7 cicloalquila, C3-7 cÍcloalquil-C1-4-alquila, C2-6 heterocicloalquila, C2-6 heterocicloalquil-C1-4-alquila, fenila, fenil-C1-4-al- quila, C1-6 heteroarila, C1-6 heteroaril-C1-4-alquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)NReRf, -NReRf, -C(=O)Rb, -C(=O)ORb, -C(=O)NReRf, -NRcC(=O)Rd, e -NRcC(=O)ORd; em que a C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, e C2-6 alquinila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos R1a independentemente selecionados; em que a C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-6 heterocicloalquila, C2-6 heterocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-6 heteroarila, e C1-6 heteroaril-C1-4-alquila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos R2a independentemente selecionados. Em algumas modalidades, cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)NReRf, -NReRf, -C(=O)Rb, -C(=O)ORb, -C(=O)NReRf, e -NRcC (=O)Rd. Em algumas modalidades, cada R1 é independentemente selecionado de C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, e -NReRf. Em algumas modalidades, cada R1 é independentemente selecio- nado de flúor, bromo, cloro, ciano, hidroxila, metila, trifluorometila, metóxi, isopropilamino, dimetilamino, metiltio, metilsulfinila, e metilsulfonila.
Em algumas modalidades, cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, C1-6 haloalquila, C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-7 heterocicloalquila, C2-7 hete- rocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila. Em algumas modalidades, cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, C1-6 haloalquila, C3-7 cicloalquila, C2-7 heterocicloalquila, fenila, e C1-7 heteroarila. Em algumas modalidades, cada Ra, Rb, Rc, Rd, Re, e Rf é independente- mente selecionado de horas, C1-6 alquila, e C1-6 haloalquila. Em algumas modalidades, cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de H e C1-6 alquila. Em algumas modalidades, cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de H e metila. Em algumas modalidades: cada R1a é independentemente selecionado de flúor, cloro, bromo, ciano, nitro, hidroxila, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1- 4 alquilsulfinila, C1-4 alquilsulfonila, amino, C1-4 alquilamino, e did-4- alquilamino; e cada R2a é independentemente selecionado de flúor, cloro, bromo, ciano, nitro, hidroxila, C1-4 alquila, C1-4 haloalquila, C2-4 alquinila, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1-4 alquilsulfinila, C1-4 alquilsulfonila, amino, C1-4 alquilamino, e did-4-alquilamino.
Em algumas modalidades: X é ciano ou flúor Y é CH ou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, C1-6 heteroarila monociclica, C1-9 heteroarila bicíclíca, C7-14 cicloalquilarila fundida bicíclica, C6-14 heteroci- cloalquilarila fundida bicíclica, C2-14 cicloalquil-heterocicloarila fundida bicíclica, e C2-14 heterocicloalquil-heteroarila fundida bicíclica; cada das quais é opcionalmente substituída com 1,2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, C2-6 alquinila, C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-6 heterocicloalquila, C2-6 hete- rocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-6 heteroarila, C1-6 he- teroaril-C1 -4-alquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)NReRf, -NReRf, -C(=O)Rb, -C(=O)ORb, -C(=O)NReRf, -NRcC(=O)Rd, e -NRcC(=O)ORd; em que a C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, e C2-6 alquinila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 independentemente selecionados R1a groups; em que a C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-6 heterocicloalquila, C2-6 heterocicloalquil- C1-4-alquila, fenila, fenil-C1-4-alquila, C1-6 heteroarila, e C1-6 heteroaril-C1- 4-alquila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos R2a independentemente selecionados; cada R1a é independentemente selecionado de flúor, cloro, bromo, ciano, nitro, hidroxila, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1- 4 alquilsulfinila, C1-4 alquilsulfonila, amino, C1-4 alquilamino, e did-4-al- quilamino; cada R2a é independentemente selecionado de flúor, cloro, bromo, ciano, nitro, hidroxila, C1-4 alquila, C1-4 haloalquila, C2-4 alquinila, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1-4 alquilsulfinila, C1-4 alquilsulfonila, amino, C1-4 alquilamino, e did-4-alquilamino; cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, C2-6 alquinila, C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-7 heterocicloalquila, C2-7 heterocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila; em que a C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, e C2-6 alquinila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos Rx independentemente selecionados; e em que a C3-7 cicloalquila, C3-7 cicloalquil-C1 -4-alquila, C2-7 heterocicloalquila, C2-7 hete- rocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos Ry independentemente selecionados; cada Rx é independentemente selecionado de hidroxila, C1-4 alcóxi, amino, C1-4 alquilamino, e did-4-alquilamino; e cada Ry é independentemente selecionado de hidroxila, halogen, ciano, nitro, C1-4 alquila, C1-4 haloalquila, C1-4 alcóxi, C1-4 haloalcóxi, amino, C1-4 alquilamino, e dic'M-alquilamino.
Em algumas modalidades: X é ciano ou flúor; YéCH ou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, C1-6 heteroarila monocíclica, C1-9 heteroarila bicíclica, C7-14 cicloalquilarila fundida bicíclica, C6-14 heteroci- cloalquilarila fundida bicíclica, C2-14 cicloalquil-heterocicloarila fundida bicí- clica, C2-14 cicloalquil-heterocicloarila fundida bicíclica, e C2-14 heterociclo- alquil-heteroarila fundida bicíclica; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, C2-6 alquinila, C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-6 heterocicloalquila, C2-6 hete- rocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-6 heteroarila, C1-6 heteroaril-C1-4-alquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)NReRf, -NReRf, -C(=O)Rb, -C(=O)ORb, -C(=O)NReRf, -NRcC(=O)Rd, e -NRcC(=O)ORd; em que as referidas C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, e C2-6 alquinila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 independentemente selecionados R1a groups; em que as referidas C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-6 heterocicloalquila, C2-6 heterocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-6 heteroarila, e C1-6 hete roa ri I-C1-4-alquila são cada qual opcíonalmente substituídas por 1, 2, 3, ou 4 grupos R2a independentemente selecionados; cada R1a é independentemente selecionado de flúor, cloro, bromo, ciano, nitro, hidroxila, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1- 4 alquilsulfinila, C1-4 alquilsulfonila, amino, C1-4 alquilamino, e did-4-al- quilamino; cada R2a é independentemente selecionado de flúor, cloro, bromo, ciano, nitro, hidroxila, C1-4 alquila, C1-4 haloalquila, C2-4 alquinila, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1-4 alquilsulfinila, C1-4 alquilsulfonila, amino, C1-4 alquilamino, e did-4-alquilamino; cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, C2-6 alquinila, C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-7 heterocicloalquila, C2-7 heterocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila; em que as referidas C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, e C2-6 alquinila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos Rx independentemente selecionados; e em que as referidas C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-7 heteroci- cloalquila, C2-7 heterocicloalquil-C1-4-alquila, fenila, fenÍI-C1-4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila sâo cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos Ry independentemente selecionados; cada Rx é independentemente selecionado de hidroxila, C1-4 alcóxi, amino, C1-4 alquilamino, e did-4-alquilamino; e cada Ry é independentemente selecionado de hidroxila, halogen, ciano, nitro, C1^4 alquila, C1-4 haloalquila, C1-4 alcóxi, C1-4 haloalcóxi, amino, C1-4 alquilamino, e did-4-alquilamino.
Em algumas modalidades: X é ciano ou flúor; Yé CH ou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, C1-6 heteroarila monocíclica, C1-9 heteroarila bicíclica, e C2-14 heterocicloalquil-heteroarila fundida bicíclica; cada das quais é opcionalmente substituída com 1,2,3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, C2-6 alquinila, C3-7 cicloalquila, C3-7 cícloalquií-C 1 -4-alquila, C2-6 heterocicloalquila, C2-6 hete- rocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-6 heteroarila, C1-6 he- teroaril-C1-4-alquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)NReRf, -NReRf, -C(=O)Rb, -C(=O)ORb, -C(=O)NReRf, -NRcC(=O)Rd, e -NRcC(=O)ORd; em que a C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, e C2-6 alquinila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 independentemente selecionados R1a groups; em que a C3-7 cicloalquila, C3-7 cicloalquil-C1 -4-alquila, C2-6 heterocicloalquila, C2-6 heterocicloalquil- C1-4-alquila, fenila, fenil-C1-4-alquila, C1-6 heteroarila, e C1-6 heteroaril-C1- 4-alquila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos R2a independentemente selecionados; cada R1a é independentemente selecionado de flúor, cloro, bromo, ciano, nitro, hidroxila, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1- 4 alquilsulfinila, C1-4 alquilsulfonila, amino, C1-4 alquilamino, e did-4-al- quilamino; cada R2a é independentemente selecionado de flúor, cloro, bromo, ciano, nitro, hidroxila, C1-4 alquila, C1-4 haloalquila, C2-4 alquinila, C1-4 alcóxi, C1-4 haloalcóxi, C1-4 alquiltio, C1-4 alquilsulfinila, C1-4 alquil- sulfonila, amino, C1-4 alquilamino, e did-4-alquilamino; cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, C2-6 alquinila, C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-7 heterocicloalquila, C2-7 heterocicloalquil-C1 -4-alquila, fenila, fenil-C1 -4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila; em que a C1-6 alquila, C1-6 haloalquila, C2-6 alquenila, e C2-6 alquinila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos Rx independentemente selecionados; e em que a C3-7 cicloalquila, C3-7 cicloalquil-C1-4-alquila, C2-7 heterocicloalquila, C2-7 hete- rocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila são cada qual opcionalmente substituídas por 1, 2, 3, ou 4 grupos Ry independentemente selecionados; cada Rx é independentemente selecionado de hidroxila, C1-4 alcóxi, amino, C1-4 alquilamino, e did-4-alquilamino; e cada Ry é independentemente selecionado de hidroxila, halogen, ciano, nitro, C1-4 alquila, C1-4 haloalquila, C1-4 alcóxi, C1-4 haloalcóxi, amino, C1-4 alquilamino, e did-4-alquilamino.
Em algumas modalidades: X é ciano ou flúor; YéCH ou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, C1-6 heteroarila monocíclica, C1-9 heteroarila bicíclica, C2-14 cicloalquil-heterocicloarila fundida bicíclica, e C2- 14 heterocicloalquil-heteroarila fundida bicíclica; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)NReRf, -NReRf, -C(=O)Rb, -C(=O)ORb, -C(=O)NReRf, e -NRcC(=O)Rd; cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, C1-6 haloalquila, C3-7 cicloalquila, C3-7 cicloal- 5 quil-C1 -4-alquila, C2-7 heterocicloalquila, C2-7 heterocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila.
Em algumas modalidades: X é ciano ou flúor; YéCH ou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, C1-6 heteroarila monocíclica, C1-9 heteroarila bicíclica, e C2-14 heterocicloalquil-heteroarila fundida bicíclica; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)NReRf, -NReRf, -C(=O)Rb, -C(=O)ORb, -C(=O)NReRf, e -NRcC(=O)Rd; cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente seleciona- do de horas, C1-6 alquila, C1-6 haloalquila, C3-7 cicloalquila, C3-7 cicloal- quil-C1 -4-alquila, C2-7 heterocicloalquila, C2-7 heterocicloalquil-C1-4-alquila, fenila, fenil-C1-4-alquila, C1-7 heteroarila, e C1-7 heteroaril-C1-4-alquila.
Em algumas modalidades: X é ciano ou flúor; YéCHouN; Z é hidrogênio ou flúor; Ar é selecionado de fenila, um anel tiazol, um anel piridina, um anel piridimina, um anel pirazina, um anel benzo[d]oxazol, um anel oxazo- lo[4,5-c]piridina, um anel oxazolo[5,4-b]piridina, um anel oxazolo[5,4-d]pirimi- 30 dina, um anel 7H-pirrolo[2,3-d]pirimidina, um anel 2,3-di-hidrotieno[2,3-b]piri- dina, um anel S-oxo-2,3-di-hidrotieno[2,3-b]piridina, um anel S,S-dioxo-2,3- di-hidrotieno[2,3-b]piridina, um anel quinazolina, um anel quinolina, e um a- nel quinoxalina; cada das quais é opcionafmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)NReRf, -NReRf, -C(=O)Rb, -C(=O)ORb, -C(=O)NReRf, e -NRcC(=O)Rd; e cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, C1-6 haloalquila, C3-7 cicloalquila, C2-7 heterocicloalquila, fenila, e C1-7 heteroarila.
Em algumas modalidades: X é ciano ou flúor; YéCHou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirimidin- 2-ila, 7H-pirrolo[2,3-d]pirimidin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S- oxo-2,3-di-hidrotieno[2,3-b]píridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piri- din-6-ila, quinazolin-2-ila, quinolin-2-ila, e quinoxalin-2-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, e -NReRf; e cada Ra, Rb, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, e C1-6 haloalquila.
Em algumas modalidades: X é ciano ou flúor; YéCH ou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, pirídin-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-Íla, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridrn-2-ila, oxazolo[5,4-d]pirimidin- 2-ila, 7H-pirrolo[2,3-d]pirimidin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S- oxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piri- din-6-ila, quinazolin-2-ila, quinolin-2-ila, e quinoxalin-2-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=0)2Rb, e -NReRf; e cada Ra, Rb, Re, e Rf é independentemente selecionado de H e C1-6 alquila.
Em algumas modalidades: X é ciano ou flúor; YéCHou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirimidin- 2-ila, 7H-pirrolo[2,3-d]pirimidin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S- oxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piri- diπ-6-ila, quinazolin-2-ila, quiπolin-2-ila, e quinoxalin-2-ila, cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=0)2Rb, e -NReRf; e cada Ra, Rb, Re, e Rf é independentemente selecionado de H e metila.
Em algumas modalidades: X é ciano ou flúor; YéCHou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirimidin- 2-ila, 7H-pirrolo[2,3-d]pirimidin-2-ila, 2,3-di-hidrotieno[2,3-b]piπdin-6-ila, S- oxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piri- din-6-ila, quinazolin-2-ila, quinolin-2-ila, e quinoxalin-2-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; e cada R1 é independentemente selecionado de flúor, bromo, cloro, ciano, hidroxila, metila, trifluorometila, metóxi, isopropilamino, dimetilami- no, metiltio, metilsulfinila, e metilsulfonila.
Em algumas modalidades: X é ciano ou flúor; YéCH ou N; Z é hidrogénio ou flúor; Ar é selecionado de fenila, um anel tiazol, um anel piridina, um anel piridimina, um anel pirazina, um anel benzo[d]oxazol, um anel oxazo- lo[4,5-c]piridina, um anel oxazolo[5,4-b]piridina, um anel oxazolo[5,4-d]pirimi- dina, um anel 7H-pirrolo[2,3-d]pirimidina, um anel 2,3-di-hidrotieno[2,3-b]pi- ridina, um anel S-oxo-2,3-di-hidrotieno[2,3-b]piridina, um anel S,S-dioxo-2,3- di-hidrotieno[2,3-b]piridina, um anel quinazolina, um anel quinolina, um anel pirrolo[2,3-b]piridina, um anel oxazolo[4,5-b]piridina, um anel 3-oxo-3,4-di-hi- dropirazina, e um anel quinoxalina; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=0)2Rb, -S(=O)NReRf, -NReRf, -C(=O)Rb, -C(=O)ORb, -C(=O)NReRf, e -NRcC(=O)Rd; e cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, C1-6 haloalquila, C3-7 cicloalquila, C2-7 heterocicloalquila, fenila, e C1-7 heteroarila.
Em algumas modalidades: X é ciano ou flúor; Yé CH ou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]pindín-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirimidin- 2-ila, 7H-pirrolo[2,3-d]pirimidin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S- oxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piri- din-6-ila, quinazolin-2-ila, quinolin-2-ila, pirrolo[2,3-b]piridin-6-ila, oxazolo[4,5- b]piridin-2-ila, 3-oxo-3,4-di-hidropirazin-2-ila, e quinoxalin-2-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, e -NReRf; e cada Ra, Rb, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, e C1-6 haloalquila.
Em algumas modalidades: X é ciano ou flúor; YéCHou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirimidin- 2-ila, 7H-prrrolo[2,3-d]pirimídin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S- oxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piri- din-6-ila, quinazolin-2-ila, pirrolo[2,3-b]piridin-6-ila, oxazolo[4,5-b]piridin-2-ila, 3-oxo-3,4-di-hidropirazin-2-ila, quinolin-2-ila, e quinoxalin-2-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=0)2Rb, e -NReRf; e cada Ra, Rb, Re, e Rf é independentemente selecionado de H e C1-6 alquila.
Em algumas modalidades: X é ciano ou flúor; YéCH ou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pirimídin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-íla, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirímidin- 2-ila, 7H-pirrolo[2,3-d]pirimidin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S- oxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piri- din-6-ila, quinazolin-2-ila, quinolin-2-ila, pirrolo[2,3-b]piridin-6-ila, oxazolo[4,5- b]piridin-2-ila, 3-oxo-3,4-di-hidropirazin-2-ila, e quinoxalin-2-ila; cada das quais é opcionalmente substituída com 1,2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, e -NReRf; e cada Ra, Rb, Re, e Rf é independentemente selecionado de H e metila.
Em algumas modalidades: X é ciano ou flúor; YéCHou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirimidin- 2-ila, 7H-pirrolo[2,3-d]pirimidin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S- oxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piri- din-6-ila, quinazolin-2-ila, quiπolin-2-ila, pirrolo[2,3-b]piridin-6-Íla, oxazolo[4,5- b]piridin-2-ila, 3-oxo-3,4-di-hidropirazin-2-ila, e quinoxalin-2-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de flúor, bromo, cloro, ciano, hidroxila, metila, trifluorometila, metóxi, isopropilamino, dimetilami- no, metiltio, metilsulfinila, e metilsulfonila.
Em algumas modalidades: X é ciano ou flúor; YéCHou N; Zé hidrogênio ou flúor; Ar é selecionado de fenila, um anel tiazol, um anel piridina, um anel piridimina, um anel pirazina, um anel benzo[d]oxazol, um anel oxazo- lo[4,5-c]piridina, um anel oxazolo[5,4-b]piridina, um anel oxazolo[5,4-d] piri- midina, um anel 7H-pirrolo[2,3-d]pirimidina, um anel 2,3-di-hidrotieno[2,3-b] 10 piridina, um anel S-oxo-2,3-di-hidrotieno[2,3-b]piridina, um anel S,S-dioxo- 2,3-di-hidrotieno[2,3-b]piridina, um anel quinazolina, um anel quinolina, um anel pirrolo[2,3-b]piridina, um anel oxazolo[4,5-b]piridina, um anel 3-oxo-3,4- di-hidropirazina, um anel quinoxalina, um anel oxazolo[5,4-d]pirimidina, um anel tieno[3,2-b]piridina, um anel tieno[2,3-c]piridina, um anel tiofeno, um 15 anel tiazolo[5,4-d]pirimidina, um anel tieno[2,3-b]piridina, um anel 2,3-di-hi- drofuro[2,3-b]piridina, um anel 6,7-di-hidro-5H-ciclopenta[b]piridina, um anel furo[3,2-c]piridina, um anel 2,3-di-hidrotieno[3,2-c]piridina, um anel S-oxo- 2,3-di-hidrotieno[3,2-c]piridina, um anel S,S-dioxo-2,3-di-hidrotieno[3,2-c]piri- dina, um anel tieno[3,2-c]piridina, e um anel 1 H-pirrolo[3,2-c]piridina; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)NReRf, -NReRf, -C(-O)Rb, -C(=O)ORb, -C(=O)NReRf, e 25 -NRcC(=O)Rd; e cada Ra, Rb, Rc, Rd, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, C1-6 haloalquila, C3-7 cicloalquila, C2-7 heterocicloalquila, fenila, e C1-7 heteroarila. Em algumas modalidades: X é ciano ou flúor; YéCH ou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridÍn-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirimídin- 2-ila, 7H-pirrolo[2,3-d]pirimidin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S- oxo-2,3-di-hÍdrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotíeno[2,3-b]piri- din-6-ila, quinazolin-2-ila, pirrolo[2,3-b]piridin-6-ila, oxazolo[4,5-b]piridin-2-ila, 3-oxo-3,4-di-hidropirazin-2-Íla, quinolin-2-ila, quinoxalin-2-ila, tiazol-4-ila, tia- zol-5-ila, pirimidin-5-ila, oxazolo[5,4-d]pirimidin-2-ila, tieno[3,2-b]piridin-5-ila, tieno[2,3-c]piridin-5-ila, tiofen-2-ila, tiofen-3-ila, tiazolo[5,4-d]pirimidin-5-ila, tieno[2,3-b] piridin-6-ila, 2,3-di-hidrofuro[2,3-b]piridin-6-ila, 6,7-di-hidro-5H- ciclopentafb] piridÍn-2-ila, furo[3,2-c]piridin-6-ila, 2,3-di-hidrotieno[3,2-c]piri- din-6-ila, S-oxo-2,3-di-hidrotieno[3,2-c]piridin-6-ila, S,S-díoxo-2,3-di-hidro- tieno[3,2-c]piridin-6-ila, tieno[3,2-c]piridin-6-ila, e 1 H-pirrolo[3,2-c]piridin-6-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, ciano, nitro, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, e -NReRf; e cada Ra, Rb, Re, e Rf é independentemente selecionado de horas, C1-6 alquila, e C1-6 haloalquila. Em algumas modalidades: X é ciano ou flúor; YéCHou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-íla, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirimidin- 2-ila, 7H-pirrolo[2,3-d]pirimÍdin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S- oxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piri- din-6-ila, quinazolin-2-ila, pirrolo[2,3-b]piridin-6-ila, oxazolo[4,5-b]piridin-2-ila, 3-oxo-3,4-di-hidropirazin-2-Íla, quinolin-2-ila, quinoxalin-2-ila, tiazol-4-ila, tia- zol-5-ila, pirimidin-5-ila, oxazolo[5,4-d]pirimidin-2-ila, tieno[3,2-b]piridin-5-ila, tieno[2,3-c]piridin-5-ila, tiofen-2-ila, tiofen-3-ila, tiazolo[5,4-d]pirimidín-5-ila, tieno[2,3-b] piridin-6-ila, 2,3-di-hidrofuro[2,3-b]piridin-6-ila, 6,7-di-hidro-5H- ciclopenta[b] piridin-2-ila, furo[3,2-c]piridin-6-ila, 2,3-di-hidrotieno[3,2-c]piri- din-6-ila, S-oxo-2,3-di-hidrotieno[3,2-c]piridin-6-ila, S,S-dioxo-2,3-di-hidrotie- no[3,2-c]piridin-6-ila, tieno[3,2-c]piridin-6-ila, e 1 H-pirrolo[3,2-c]piridin-6-ila; cada das quais é opcionalmente substituída com 1,2,3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, e -NReRf; e cada Ra, Rb, Re, e Rf é independentemente selecionado de H e C1-6 alquila.
Em algumas modalidades: X é ciano ou flúor; YéCHou N; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirimidin- 2-ila, 7H-pirrolo[2,3-d]pirimidin-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S- oxo-2,3-di-hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3-di-hidrotieno[2,3-b]piri- din-6-ila, quinazolin-2-ila, pirrolo[2,3-b]piridin-6-ila, oxazolo[4,5-b]piridin-2-ila, 3-oxo-3,4-di-hidropirazin-2-ila, quinolin-2-ila, quinoxalin-2-ila, tiazol-4-ila, tia- zol-5-ila, pirimidin-5-ila, oxazolo[5,4-d]pirimidin-2-ila, tieno[3,2-b]piridin-5-ila, tieno[2,3-c]piridin-5-ila, tiofen-2-ila, tiofen-3-ila, tiazolo[5,4-d]pirimidin-5-ila, tieno[2,3-b] piridin-6-ila, 2,3-di-hidrofuro[2,3-b]piridin-6-ila, 6,7-di-hidro-5H- ciclopentafb] piridin-2-ila, furo[3,2-c]piridin-6-ila, 2,3-di-hidrotieno[3,2-c]pi- ridin-6-ila, S-oxo-2,3-di-hidrotieno[3,2-c]piridin-6-ila, S,S-dioxo-2,3-di-hidrotie- no[3,2-c]piridin-6-ila, tieno[3,2-c]piridin-6-ila, e 1 H-pirrolo[3,2-c]piridin-6-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de halogênio, C1-6 alquila, C1-6 haloalquila, -ORa, -SRa, -S(=O)Rb, -S(=0)2Rb, e -NReRf; e cada Ra, Rb, Re, e Rf é independentemente selecionado de H e metila.
Em algumas modalidades: X é ciano ou flúor; YéCHouN; Z é hidrogênio ou flúor; Ar é selecionado de fenila, tiazol-2-ila, piridin-2-ila, piridin-3-ila, piridin-4-ila, piridimin-2-ila, pirimidin-4-ila, pirazin-2-ila, benzo[d]oxazol-2-ila, oxazolo[4,5-c]piridin-2-ila, oxazolo[5,4-b]piridin-2-ila, oxazolo[5,4-d]pirimidin- 2-ila, 7H-pirrolo[2,3-d]pirimidín-2-ila, 2,3-di-hidrotieno[2,3-b]piridin-6-ila, S- oxo-2,3-di“hidrotieno[2,3-b]piridin-6-ila, S,S-dioxo-2,3~di-hidrotieno[2,3-b]piri- din-6-ila, quinazolin-2-ila, quinolin-2-ila, pirrolo[2,3-b]piridin-6-ila, oxazolo[4,5- b]piridin-2-ila, 3-oxo-3,4-di-hidropirazin-2-ila, quinoxalin-2-ila, tiazol-4-ila, tia- zol-5-ila, pirimidin-5-ila, oxazolo[5,4-d]pirimidin-2-ila, tieno[3,2-b]piridin-5-ila, tieno[2,3-c]piridin-5-ila, tiofen-2-ila, tiofen-3-ila, tiazolo[5,4-d]pirimidin-5-ila, tieno[2,3-b] piridin-6-ila, 2,3-di-hidrofuro[2,3-b]piridin-6-ila, 6,7-di-hidro-5H-ci- clopenta[b] piridin-2-ila, furo[3,2-c]piridin-6-ila, 2,3-di-hidrotieno[3,2-c]piridin- 6-ila, S-oxo-2,3-di-hidrotieno[3,2-c]piridin-6-ila, S,S-dioxo-2,3-di-hidrotie- no[3,2-c]piridin-6-ila, tieno[3,2-c]piridin-6-ila, e 1 H-pirrolo[3,2-c]piridin-6-ila; cada das quais é opcionalmente substituída com 1, 2, 3, 4, 5, ou 6 grupos R1 independentemente selecionados; cada R1 é independentemente selecionado de flúor, bromo, cloro, ciano, hidroxila, metila, trifluorometila, metóxi, isopropilamino, dimetilami- no, metiltio, metilsulfinila, e metilsulfonila.
Em algumas modalidades, o composto é um composto tendo a fórmula la:
ou um sal farmaceuticamente aceitável ou N-óxido do mesmo.
Em algumas modalidades, o composto é um composto tendo a fórmula lb:
ou um sal farmaceuticamente aceitável ou N-óxido do mesmo.
Em algumas modalidades, o composto é um composto tendo a fórmula lc:
ou um sal farmaceuticamente aceitável ou N-óxido do mesmo.
Em algumas modalidades, o composto é um composto tendo a fórmula Id:
ou um sal farmaceuticamente aceitável ou N-óxido do mesmo.
Em algumas modalidades, o composto é um composto tendo a fórmula le:
ou um sal farmaceuticamente aceitável ou N-óxido do mesmo.
Em algumas modalidades, o composto é um composto tendo a fórmula If:
ou um sal farmaceuticamente aceitável ou N-óxido do mesmo.
Em algumas modalidades, o composto é um composto de fórmula Ha, llb, llc, lld, He, llf, Hg, llh, Hi, llj, Hm, Hn, ou Ho:
ou um sal farmaceuticamente aceitável ou N-óxido do mesmo, em que cada n é um número inteiro selecionado de 0 a 5; e m é um número inteiro selecionado de 0 a 2; contanto que a valência de cada átomo nas porções opcionalmente substituídas não seja excedida. Cada uma das fórmulas precedentes pode representar uma modalidade individual. Em algumas das modalidades precedentes, Z é hidrogênio. Em algumas das modalidades precedentes, Z é flúor. Em algumas das modalidades precedentes, X é ciano. Em algumas das modalidades precedentes, X é flúor. Em algumas das modalidades precedentes, Z é hidrogênio e X é ciano. Em algumas das modalidades precedentes, Z é hidrogênio e X é flúor. Em algumas das modalidades precedentes, Z é flúor e X é ciano.
Em algumas modalidades, o composto é um composto de fórmula llp, llq, ou llr:
ou um sal farmaceuticamente aceitável ou N-óxido do mesmo, em que cada n é um número inteiro selecionado de 0 a 5; contanto que a valência de cada átomo nas porções opcionalmente substituídas não seja excedida. Cada uma das fórmulas precedentes pode representar uma modalidade individual. Em algumas das modalidades precedentes, Z é hidrogênio. Em algumas das modalidades precedentes, Z é flúor. Em algumas das modalidades precedentes, X é ciano. Em algumas das modalidades precedentes, X é flúor. Em algumas das modalidades precedentes, Z é hidrogênio e X é ciano. Em algumas das modalidades precedentes, Z é hidrogênio e X é flúor. Em algu- mas das modalidades precedentes, Z é flúor e X é ciano.
Em algumas modalidades, o composto é um composto de fórmula llla ou lllb:
ou um sal farmaceuticamente aceitável ou N-óxido do mesmo, em que cada n é um número inteiro selecionado de 0 a 5; e m é um número inteiro sele-cionado de 0 a 2; contanto que a valência de cada átomo nas porções op-cionalmente substituídas não seja excedida. Cada uma das fórmulas prece-dentes pode representar uma modalidade individual. Em algumas das moda-lidades precedentes, m é 1 ou 2. Em algumas das modalidades precedentes, Z é hidrogênio. Em algumas das modalidades precedentes, Z é flúor. Em algumas das modalidades precedentes, X é ciano. Em algumas das modali-dades precedentes, X é flúor. Em algumas das modalidades precedentes, Z é hidrogênio e X é ciano. Em algumas das modalidades precedentes, Z é hidrogênio e X é flúor. Em algumas das modalidades precedentes, Z é flúor e X é ciano.
Em algumas modalidades, Ar é opcionalmente substituído com 1, 2, 3, ou 4 grupos R1 independentemente selecionados. Em algumas mo-dalidades, Ar é opcionalmente substituído com 1, 2, ou 3 grupos R1 inde-pendentemente selecionados. Em algumas modalidades, Ar é opcionalmente substituído com 1 ou 2 grupos R1 independentemente selecionados.
Em algumas modalidades, o composto é selecionado de: 3-[1-(6-cloropirazin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pi- rimidin-4-il)-1 H-pirazol-1-il]propanonitrila; 3-[1 -(6-cloropiridin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pi- rimidin-4-il)-1H-pirazol-1-il]propanonitrila; 3-[1-(2-cloropirimidin-4-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pi- nmidin-4-il)-1 H-pirazol-1 -iljpropanon itrila; J 3-[1-(4-cloropirimidin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d] p i rim id i n-4-i I)-1 H-pirazol-1 -iljpropanonitrila; 3-[1-(4-bromo-1,3-tiazol-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- djpirimid in-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-{1-[4-(dimetilamino)pirrmidin-2-il]pirrolidin-3-il}-3-[4-(7H- i pirrolo[2 , 3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 3-{1-[4-(isopropilamino)pirimidin-2-il]pirrolidin-3-il}-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila; 3-[1 -(1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-[1 -(5-cloro-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 3-(1-[1,3]oxazolo[4,5-c]piridin-2-ilpirrolidin-3-il)-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-(1-[1,3]oxazolo[4,5-b]pirid in-2-ilpirrolidin-3-il)-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 3-(1-[1,3]oxazolo[5,4-b]piridin-2-ilpirrolidin-3-il)-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 3-[1-(6-metil[1,3]oxazolo[5,4-b]piridin-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 3-[1-(6-fluoro[1>3]oxazolo[5,4-b]piridin-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirim id in-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]-3-[1 -(7H- pirrolo[2,3-d]pirimidin-2-il)pirrolidin-3-il]propanonitrila; 3-[1-(7-metil-7H-pirrolo[2,3-d]pirimidin-2-il)pirrolidin-3-ilJ-3-[4-(7H- pirrolo[2,3-dJpirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 3-(1-[1,3]oxazolo[5,4-d]pirimidin-2-ilpirrolidin-3-il)-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 3-[ 1 -(5-fluoro-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-[1-(4-fluoro-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-dJpirimid in-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-[1-(7-fluoro-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]p i rim id i n-4-i I)-1 H-pirazol-1 -i I] propa n on itrila; 3-[1-(5,7-difluoro-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirim id in-4-iI)-1 H-pirazol-1 -il]propanonitrila; 3-{1 -[2-(metiltio)pirimidin-4-il]pirrolidin-3-il}-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]p ropanon itrila; 3-{1-[2-(metilsulfinil)pirimidin-4-il]pirrolidin-3-il}-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il]propanonitrila; 3-{1-[2-(metilsulfonil)pirimidin-4-il]pirrolidin-3-il}-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila; 3-{1-[6-(metilsulfonil)piridin-2-il]pirrolidin-3-il}-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1H-pirazol-1-il]propanon itrila; 3-{1 -[2-(metilsulfonil)piridin-4-il]pirrolidin-3-il}-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-[1-(1-óxido-2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidín-3-il]-3-[4- (7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il]propanonitrila; 3-[1-(2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila; 3-[1 -(1,1-dióxido-2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidin-3-il]- 3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila; 3-(3-fluoro-1-[1,3]oxazolo[5,4-b]piridin-2-ilpirrolidin-3-il)-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila; 3-(1 -[1,3]oxazolo[5,4-b]piridin-2-ilpirrolidin-3-il)-3-[3-(7H- p irrolo[2,3-d]p i ri m id i n-4-i I)-1 H-pirrol-1 -i l]p ropa n on itri la; 3-[1-(1,1 -dióxido-2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidin-3-il]- 3-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 -il]propanonitrila; 3-[1-(1-óxido-2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[3- (7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1-il]propanonitrila; 3-(1 -(6-cloro-4-metil-3-oxo-3,4-di-hidropirazin-2-il)pirrolidin-3-il]-3- [4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila; 3-[1-(4-metil-3-oxo-3,4-di-hidropirazin-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -ii]propanonitrila; 3-cloro-2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H- pirazol-1-il]etil}pirrolidin-1-il)isonicotinonitrila; 2-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirim id in-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1 -il)piridina-3,4-dicarbonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- ilJetil}pirrolidin-1-il)-6-(metiltio)benzonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pírrolidin-1 -il)-6-(metilsulfonil)benzonitrila; 3-[1-(8-cloroquinolin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirimid in-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-[1-(3-hidroxiquinoxalin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila; 3-[1 -(8-cloroquinazolin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila; 3-[1-(6-cloro-1-oxidopiridin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d] pi ri m id i n-4-i I)-1 H-pirazol-1 -iljpropanonitrila; 3-[1-(8-fluoroquinazolin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d] p iri m id i n-4-i I)-1 H-pirazol-1 -iljpropanonitrila; 3-[1-(5-bromo-1,3-tiazol-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 2-cloro-6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1 -il]etil}pirrolidin-1 -i I )benzon itri la; 3-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-dJpirimidm-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)ftalonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1 -il)-4-(trifluorometil)nicotinonitrila; 3-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1 -il)pirazina-2-carbonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirim id in-4-il)-1 H-pirazol-1- i IJeti l}p í rrol id i n-1 -il)benzonitrila; 2-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolídin-1 -il)-6-metilbenzonitrila; 2-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1 -il)-6-fluorobenzonitrila; 2-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1 -il)-6-metoxibenzonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)-6-(trifluorometil)benzonitrila; 2-bromo-6-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirim idin-4-il)-1 H- pirazol-1 -il]etil}pirrolidin-1 -il)benzonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)-3-fluorobenzonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolid in-1 -il)isoftalonitrila; 6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolid in-1 -il)-2,3-difluorobenzonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin^-il)-1 H-pirazol-1- il]etil}pirrolidin-1 -il)-3,5,6-trifluorobenzonitrila; 2-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1-il)nicotinonitrila; 3-cloro-5-(3-{2-ciano-1-[4-(7H-pirrolo[2>3-d]pirimidin-4-il)-1H- pirazol-1 -il]etil}pirrolidiπ-1 -il)isonicotinonitrila; 3-(3-{2-ciano-1 -[4-(7 H-p irro lo[2,3-d]pi ri m id i n-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1-il)-2,5,6-trifluoroisonicotinonitrila; 3-{1 -[3-fluoro-4-(trif luorometil)pirid in-2-il]pirrolid in-3-il}- 3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il>-1 H-pirazol-1-il]-3-[1 - (3,5,6-trifluoropiridin-2-il)pirrolidin-3-il]propanonitrila; 3-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolid in-1 -il)piridina-2-carbon itrila; 2-cloro-6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)- 1 H-pirazol-1-il]etil}pirrolidin-1-il)nicotinonitrila; 2-(3-{2-fluoro-1-[4-(7H-pirrolo[2,3-d]pirimidin- 4-il)-1 H-pirazol-1- il]etil}pirrolidiπ-1-il)[1,3]oxazolo[5, 4-b]piridina; 2-(3-(1 -(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-2- fluoroetil)pirrolidin-1-il)oxazolo[5,4-b]piridina; e 3-[1-(1H-pirrolo[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila, ou um sal farmaceuticamente aceitável ou N-óxido do mesmo. Em algumas modalidades, o composto é selecionado de: 5-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pÍrimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1 -il)tieno[2,3-c]piridina-4-carbonitrila; 5-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - 10 il]etil}pirrolidin-1 -il)tieno[3,2-b]piridina-6-carbonitrila; 2-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1-il)-4-hidroxitiofeno-3-carbonitrila; 4-bromo-2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1 -il]etil}pirrolidin-1 -il)tiofeno-3-carbonitrila; 4-cloro-2-(3-{2-ciano-1 -[4-(7 H-pi rro lo[2,3-d]p i ri m id i n-4-i I)-1H- pirazol-1-il]etil}pirrolidin-1-il)tiofeno-3-carbonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidrn-1-il)tiofeno-3,4-dicarbonitrila; 2-(3-{(1 R)-2-fluoro-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H- pirazol-1 -il]etil}pirrolidin-1-il)tiofeno-3,4-dicarbonitrila; 2-(3-{2-ciano-1 -[3-(7 H-p i rro lo[2,3-d] p i rim id i n-4-i I)-1 H-pirrol-1 - il]etil}pirrolidin-1 -il)tiofeno-3,4-dicarbonitrila; 4-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1 -il)-1,3-tiazol-5-carbonitrila; 5-(3-{2-fluoro-1 -[3-(7H-pirrolo[2,3-d]pirim id in-4-il)-1 H-pirrol-1 - il]etil}pirrolidin-1 -il)-1,3-tiazol-4-carbonitrila; 4-(3-(1 -(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-2- I cianoetil)pirrolidin-1 -il)pirimidina-5-carbonitrila; 4-(1-{2-fluoro-1-[1-(5-fluoro-2,3-di-hidrotieno[2,3-b]piridin-6- il)pirrolidin-3-il]etil}-1 H-pirazol-4-il)-7H-pirrolo[2,3-d]pirimidina; 4-(1-í2-fluoro-1-[1-(5-fluoro-1,1-dióxido-2,3-di-hidrotieno[2,3- b]piridin-6-il)pirrolidin-3-il]etil}-1 H-pirazol-4-il)-7H-pirrolo[2,3-d]pÍnmidina; 3-[1 -(5-fluoro-2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidin-3-il]-3- [4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanon itrila; 3-[1-(6-bromo-3-fluoropiridin-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il]propanonitrila; 3-[1-(5,6-difluoropiridin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- djpirimidi n-4-il)-1 H-pirazol-1 -il]propanonitrila; 3-{1-[6-cloro-3-fluoro-5-(hidroximetil)piridin-2-il]pirrolidin-3-il}-3-[4- (7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pi rimidin-4-il)-1 H-pirazol-1 -il)-3-(1 -(5- amino-6-cloro-3-fluoropiridin-2-il)pirrolidin-3-il)propanonitrila; N-(2-(3-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-2- cianoetil)pirrolidin-1-il)-6-cloro-5-fluoropiridin-3-il)formamida; 3-{1-[6-(etilsulfonil)-3-fluoropiridin-2-il]pirrolidin-3-il}-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila; 3-[1-(6-cloro-3-fluoropiridin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d ]p i rim id i n-4-i I)-1 H-pirazol-1 -il]propanonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2>3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1 -il)-5-fluoro-4-(metoximetil)nicotinonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1-il)-4-(metoximetil)πicotinon itrila; 4-(3-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-2- cianoetil)pirrolidin-1-il)-6-metoxipirimidina-5-carboπitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-3-(1 -(6- (etilsulfonil)-3-fluoropiridin-2-il)pirrolidin-3-il)propanonitrila; 2-(3-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-2- ciaπoetil)pirrolidin-1-il)-4-metilnicotinon itrila; 3-{1-[3,5-difluoro-4-(metoximetil)piridin-2-il]pirrolidin-3-il}-3-[4- (7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila; 3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il]-3-[1- [1,3]tiazolo[5,4-d]pirimidin-5-ilpirrolidin-3-il]propanonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)-4-(difluorometil)nicotinon itrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-rl)-1 H-pirazol-1 -il)-3-(1-(5-fluoro- 2-metoxipirimidin-4-il)pirrolidin-3-il)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-3-(1-(3- amino-6-cloropiridin-2-il)pirrolidin-3-il)propanonitrila; 4-(3-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-2- I cianoetil)pirrolidin-1 -il)piridazina-3-carbonitπla; 6-(3-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-2- cianoetil)pirrolidin-1-il)-5-fluoronicotinonitrila; 2-(3-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-2- cianoetil)pirrolidin-1-il)-5-fluoronicotinonitrila; 2(3(1 (4(7 H-p irro!o[2,3-d]p i rim id in-4-il)-1 H-pirazol-1 -il)-2- ciaπoetil)pirrolidin-1 -il)-5-metilnicotinonitrila; 4-(3-(1 -(4-(7H-pirrolo[2t3-d]pirimidin-4-il)-1 H-pirazol-1-il)-2- cianoetil)pirrolidin-1-il)-6-(difluorometil)pirimidina-5-carbonitrila; 2-(3-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-2- cianoetil)pirrolidin-1 -il)-6-(difluorometil)benzonitrila; 2-(3-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-2- cia n oetil) p i rrol id i n-1 -il)-6-(metoximetil)benzonitrila; 4-(3-(1-(3-(7H-pirrolo[2,3-d]piπmidin-4-ii)-1 H-pirrol-1-il)-2- cianoetil)pirrolid in-1 -il)piridazina-3-carbonitrila; 2-(3-( 1 -(3-(7 H-pi rrolo[2,3-d]p i rim id iπ-4-il)-1 H-pirrol-1 -i l)-2- cianoetil)pirrolidin-1-il)nicotinonitrila; 3-(3-(1-(3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1-il)-2- cianoetil)pirrolidin-1-il)pirazina-2-carbonitrila; 4-(3-(1 -(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-2- fluoroetil)pirrolidin-1-il)piridazina-3-carbonitrila, 3-(3-{2-fluoro-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1 -il)piridina-2-carbonitrila; 2-(3-{2-fluoro-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)nicotinonitrila; 4-(1-{1-[1-(1,1-dióxido-2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidin- 3-il]-2-fluoroetil}-1 H-pirazol-4-il)-7H-pirrolo[2,3-d]pirimidina; 2-(3-( 1 -(4-(7H-pirrolo[2,3-d]pirimid in-4-il)-1 H-pirazol-1 -il)-2- fluoroetil)pirrolidin-1-il)piridina-3,4-dicarbonitrila; 3-(3-{2-fluoro-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)ftalonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1-il)-4-iodonicotinonitrila; 2-cloro-4-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1 -il]etil}pirrolidin-1 -i I) n icot i non rtrrla; 4-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1- il]etil}pirrolidin-1 -il)piridina-2,3-dicarbonitrila; 3-[1-(2,6-dicloropiridin-3-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 5-(3-{2-fluoro-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljetiljpirrolid in-1 -il)-1,3-tiazol-4-carbonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1-il)-4-(metiltio)nicotinonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1- il]etil}pirrolidin-1-il)-4-(metilsulfonil)nicotinonitrila; 3-{1-[3,5-difluoro-6-(metiltio)piridin-2-il]pirrolidin-3-il}-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 3-{1-[3,5-difluoro-6-(metilsulfonil)piridin-2-il]pirrolidin-3-il}-3-[4- (7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 3-(1-{3,5-difluoro-6-[(2J2,2-trifluoroetil)-sulfonil]piridin-2- il}pirrolidin-3-il)-3-[4-(7H-pirrolo[2,3-d]pirimidin^l-il)-1H-pirazol-1- iljpropanonitrila; 4-[1-(1-{1-[3,5-difluoro-6-(metilsulfonil)piridin-2-il]pirrolidin-3-il}-2- fluoroetil)-1 H-pirazol-4-il]-7H-pirrolo-[2,3-d]pirimidina; 3-{1-[3-fluoro-6-(metilsulfonil)piridin-2-il]pirrolidin-3-il}-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-{1-[2,5-difluoro-6-(metilsulfonil)piridin-3-il]pirrolidin-3-il}-3-[4- (7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila 2-(3-{2-ciano-1-[4-(7H-pirrolo[2l3-d]pirimidin-4-il)-1 H-pirazol-1- i l]etil}p i rrol id in-1 -i l)-4-( 1 -fluoroeti I) n icoti non itrila; 3-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1 -il)-6-(difluorometil)pirazina-2-carbonitrila; 3-(3-i2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil]pirrolidin-1-il)-6-(2,2-difluoroetil)pirazina-2-carbonitrila; 3-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)-6-(hidroximetil)pirazina-2-carbonitrila; 3-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1- il]etil}pirrolidin-1 -il)-6-(metoximetil)pirazina-2-carbonitrila; 6-bromo-3-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirim id in-4-il)-1 H- pirazol-1-il]etil}pirrolidiπ-1-il)pirazina-2-carbonitrila; 3-(3-{2-ciano-1 -[4-(7 H-p i rrolo[2,3-d] p iri m id i n-4-i I)-1 H-pirazol-1 - il]etil]pirrolidin-1 -il)-6-etinilpirazina-2-carbonitrila; 3-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil]pirrolidin-1 -il)-6-etilpirazina-2-carbonitrila; 3-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1- il]etil}pirrolidin-1 -il)-6-metilpirazina-2-carbonitrila; 3-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)-6-metilpirazina-2-carbonitrila; 3-fluoro-5-(3-{2-fluoro-1 -[4-(7 H-p i rrolo[2,3-d ] pi ri m id i n-4-il)-1H- pirazol-1 -il]etil}pirrolidin-1 -il)piridina-2-carbonitrila; 3-{1-[2-(etilsulfonil)piridin-4-il]pirrolidin-3-il}-3-[3-(7H-pirrolo[2,3- d] p i rim id i n-4-i I)-1 H-pirrol-1 -il] p ropanon itri la; 5-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 - il]etil}pirrolidin-1 -il)-1,3-tiazol-4-carbonitrila; 3-[1-(2-mercaptopirimidin-4-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila; N-[4-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)pirimidin-2-il]-N,N-dimetilsulfonamida; 4-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1 -il)-N-metilpiridina-2-carboxamida; 4-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)-N,N-dimetilpiridina-2-carboxamida; 4-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1 -il)-N-fenilpiridina-2-carboxamida; 3-[1-(2,3-di-hidrofuro[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]piπmid in-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-[4-(7H-pirrolo[2,3-d]pirimid in-4-il)-1 H-pirazol-1 -il]-3-(1 -tieno[2,3- b]piridin-6-ilpirrolidin-3-il)propanonitrila; 3-[1-(7,7-difluoro-6,7-di-hidro-5H-ciclopenta[b]piridin-2- il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - 10 iljpropanonitrila; 3-[1-(7-fluoro-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[3-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 -iljpropanonitrila; 3-[1-(7-bromo-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila; 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1 -il)-1,3-benzoxazol-7-carbonitrila; 3-[1-(7-hidróxi-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 3-[1 -(7-metóxi-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)-3-(1-(7- etoxibenzo[d]oxazol-2-il)pirrolidin-3-il)propanonitrila; 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il)-3-(1 -(7- (difluorometóxi)benzo[d]oxazol-2-il)pirrolidin-3-il)propanonitrila; 3-[1-(4-hidróxi-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirim id in-4-il)-1 H-pirazol-1 -iljpropanonitrila; 3-{1-[7-(hidroximetil)-1,3-benzoxazol-2-il]pirrolidin-3-il}-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila; 6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il jetiljpirrolid in-1 -il)furo[3,2-c]piridina-7-carbonitrila; 6-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 - il]etil}pirrolidin-1 -il)furo[3,2-c]piridina-7-carbonitrila; 1,1-dióxido de 6-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidiπ-4-il)- 1 H-pirrol-1 -i IJeti l}p i rrolid in-1 -il)-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila; 1,1-dióxido de 6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)- 1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila; 6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1- il]etil}pirrolidin-1 -il)-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila; 6-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 - il]etil}pirrolidin-1-il)-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila; 6-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirim id in-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1 -il)tieno[3,2-c]piridiπa-7-carbonitrila; 6-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1- il]etii}pirrolidin-1-il)tieno[3,2-c]piridina-7-carbonitrila; 6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1 -il)-1 H-pirrolo[3,2-c]piridina-7-carbonitrila; e 1,1-óxido de 6-((3S)-3-{2-fluoro-1-[4-(7H-pirrolo[2,3-d]pirimidin-4- il)-1 H-pirazol-1-il]etil}pirrolidin-1 -il)-2,3-di-hidrotieno[3,2-c]piridina-7- carbonitrila, ou um sal farmaceuticamente aceitável ou N-óxido do mesmo.
Em algumas modalidades, o composto é 6-(3-(1-(4-(7H-pir- rolo[2,3-d]pirimídin-4-il)-1 H-pirazol-1-il)-2-cianoetil)pirrolidin-1-il)-2-cloro-5- fluoronicotinonitrila, ou um sal farmaceuticamente aceitável ou N-óxido do mesmo.
Em algumas modalidades, o composto é o sal de trifluoroacetato ou fosfato.
A presente invenção inclui sais farmaceuticamente aceitável e N-óxidos dos compostos descritos aqui. Como usado aqui, "sais farmaceuticamente aceitáveis" refere-se aos derivados dos compostos descritos em que o composto origem é modificado por conversão de uma porção de ácido ou base existente em sua forma de sal. Exemplos de sais farmaceuticamen- te aceitáveis incluem, porém não estão limitados a sais de ácido minerais ou orgânicos de resíduos básicos tais como aminas; sais de álcali ou orgânicos de resíduos acídicos tal como ácidos carboxílicos; e similares. Os sais far- maceuticamente aceitáveis da presente invenção incluem os sais não tóxicos convencionais do composto origem formado, por exemplo, de ácidos não tóxicos inorgânicos ou orgânicos. Os sais farmaceuticamente aceitáveis da presente invenção podem ser sintetizados a partir do composto origem que contém uma porção básica ou acídica por métodos químicos convencionais. Geralmente, tais sais podem ser preparados reagindo as formas de base ou de ácido livre destes compostos com uma quantidade estequiomé- trica da base ou ácido apropriado em água ou em solvente orgânico, ou na mistura dos dois; geralmente, meios não aquosos como éter, acetato de eti- la, etanol, isopropanol, ou acetonitrila (ACN) são preferidos. As listas de sais adequados são encontradas em Remington's Pharmaceutical Sciences, 17a edição, Mack Publishing Company, Easton, Pa., 1985, página 1418 e Journal of Pharmaceutical Science, 66, 2 (1977), cada dos quais é incorporado aqui por referência em sua totalidade.
Os compostos descritos aqui podem ser assimétricos (por e- xemplo, tendo um ou mais estereocentros). Todos os estereoisômeros, tais como enantiômeros e diastereômeros, são pretendidos a menos que de outro modo indicado. Compostos descritos aqui que contêm átomos de carbono assimetricamente substituídos podem ser isolados em formas oticamente ativas ou racêmicas. Métodos sobre como preparar formas oticamente ativas de materiais de partida oticamente ativos são conhecidos na técnica, tais como por resolução de misturas racêmicas ou por síntese estereosseletiva. A maioria dos isómeros geométricos de olefinas, ligações duplas de C=N, e similares pode também estar presente nos compostos descritos aqui, e todos os tais isómeros estáveis são contemplados na presente invenção. Isô- meros geométricos cis e trans dos compostos descritos aqui podem ser isolados como a mistura de isómeros ou formas isoméricas separadas. Onde um composto capaz de estereoisomerismo ou isomerismo geométrico é designado em sua estrutura ou nome sem referência às configurações R/S ou cis/trans específicas, entende-se que todos os tais isómeros são contemplados.
A resolução de misturas racêmicas de compostos pode ser reali- zada por qualquer um dos numerosos métodos conhecidos na técnica. Um método exemplo inclui a recristalização fracionai usando um ácido de resolução quiral que é um ácido de formação de sal, oticamente ativo. Agentes de resolução adequados para métodos de recristlização fracionai são, por 5 exemplo, ácidos oticamente ativos, tais como as formas D e L de ácido tartá- rico, ácido diacetiltartárico, ácido dibenzoiltartárico, ácido mandélico, ácido málico, ácido láctico ou os vários ácidos canforsulfônicos oticamente ativos tal como ácido β-canforsulfonico. Outros agentes de resolução adequados para métodos de cristalização fracionai incluem formas estereoisomerica- 10 mente puras a-metilbenzilamina (por exemplo, formas S e R, ou formas dias- tereomericamente puras), 2-fenilglicinol, norefedrina, efedrina, N-metilefe- drina, ciclo-hexiletilamina, 1,2-diaminociclo-hexano, e similares.
A resolução de misturas racêmicas pode também ser realizada por eluição em uma coluna empacotada com um agente de resolução otica- 15 mente ativo (por exemplo, dinitrobenzoilfenilglicina). Composição de solvente de eluição adequada pode ser determinada por alguém versado na técnica.
Compostos descritos aqui também incluem formas tautoméricas.
Formas tautoméricas resultam da troca de uma ligação simples com uma ligação dupla adjacente juntamente com a migração concomitante de um 20 próton. Formas tautoméricas incluem tautômeros prototrópicos que são estates de protonação isomérica tendo a mesma fórmula empírica e carga total. Exemplo tautômeros prototrópicos incluem cetona - pares de enol, amida - pares de ácido imimico, lactam - pares de lactim, amida - pares de ácido imídico, enamina - pares de imina, e formas anulares onde um próton pode 25 ocupar duas ou mais aposições de um sistema heterocíclico, por exemplo,1H- e 3H-imidazol, 1H-, 2H- e 4H- 1,2,4-triazol, 1H- e 2H- isoindol, e 1H- e 2H-pirazol. Formas tautoméricas podem estar em equilíbrio ou estericamen- te fechadas em uma forma por substituição apropriada.
Compostos descritos aqui podem também incluir todos os isóto- 30 pos de seus átomos constituintes. Os isótopos incluem aqueles átomos tendo o mesmo número atômico, porém diferem os números de massa. Por e- xemplo, isótopos de hidrogênio incluem trício e deutério.
O termo, "composto" como usado aqui significa incluir todos os estereoisômeros, tautômeros, e isótopos das estruturas descritas ou nomes químicos fornecidos, a menos que de outro modo indicado.
Todos os compostos, e sais farmaceuticamente aceitáves dos 5 mesmos, podem ser encontrados juntamente com outras substâncias tais como água e solvents (por exemplo, hidratos e solvatos) ou podem ser encontrados.
Em algumas modalidades, os compostos da invenção, e sais dos mesmos, são substancialmente isolados. Por "substancialmente isolado" 10 entende-se que o composto é pelo menos parcialmente ou substancialmente separado do ambiente em que ele foi formado ou detectado. Separação parcial pode incluir, por exemplo, uma composição enriquecida no composto da invenção. A separação substancial pode incluir composições contendo pelo menos cerca de 50%, pelo menos cerca de 60%, pelo menos cerca de 70%, 15 pelo menos cerca de 80%, pelo menos cerca de 90%, pelo menos cerca de 95%, pelo menos cerca de 97%, ou pelo menos cerca de 99% em peso do composto da invenção, ou sal, ou N-óxido do mesmo.
Métodos
Compostos da invenção são inibidores de JAK, e a maioria dos 20 compostos da invenção refere-se aos inibidores seletivos de JAK1. Um inibidor seletivo de JAK1 é um composto que inibe a atividade de JAK1 preferivelmente mais do que outras Janus quinases. Por exemplo, os compostos da invenção preferivelmente inibem JAK1 além de uma ou mais de JAK2, JAK3, e TYK2. Em algumas modalidades, os compostos inibem JAK1 prefe- 25 rivelmente mais do que JAK2 (por exemplo, têm uma relação de IC50 de JAK1/JAK2 >1).
JAK1 desempenha um papel central em diversas séries de reação de sinalização de citocina e fator de crescimento que, quando desregu- ladas, podem resultar em ou contribuir para os estados de doença. Por e- 30 xemplo, os níveis de IL-6 são elevados em artrite reumatoide, uma doença na qual foi sugerido ter efeitos prejudiciais (Fonesca, J.E. e outro, Autoimmunity Reviews, 8:538-42, 2009). Por que sinais de IL-6, pelo menos em parte, por meio de JAK1, antagonizam IL-6 direta ou indiretamente por meio de inibição de JAK1, espera-se fornecer benefício clínico (Guschin, D., N., e outro, Embo J 14:1421, 1995; Smolen, J. S., e outro, Lancet 371:987, 2008). Entretanto, em alguns cânceres JAK1 é mutada resultando em sobrevivência e crescimento de célula de tumor indesejável constitutiva (mullighan CG, Proc Natl Acad Sei U S A.106:9414-8, 2009; Flex E., e outro, J Exp Med. 205:751-8, 2008). Em outras doenças autoimunes e cânceres, níveis sistêmicos elevados citocinas inflamatórias que ativam JAK1 podem também contribuir para a doença e/ou sintomas associados. Portanto, pacientes com tais doenças podem se beneficiar da inibição de JAK1. Inibidores seletivos de JAK1 podem ser eficazes ao mesmo tempo que evitando efeitos desnecessários e potencialmente indesejáveis de inibição de outras JAK quinases.
Inibidores seletivos de JAK1, com relação às JAK quinases, podem ter múltiplas vantagens terapêuticas mais do que de inibidores menos 15 seletivos. Com respeito à seletividade contra JAK2, diversas citocinas importantes e sinal de fatores de crescimento por meio de JAK2 incluindo, por e- xemplo, eritropoietina (Epo) e trombopoietina (Tpo) (Parganas E, e outro, Cell. 93:385-95, 1998). Epo é um fator de crescimento essencial para produção de células sanguíneas vermelhas; portanto, uma escassez de sinaliza- ção dependente de Epo pode resultar em números reduzidos de células sanguíneas vermelhas e anemia (Kaushansky K, NEJM 354:2034-45, 2006). Tpo, outro exemplo de um fator crescimento dependente de JAK2, desempenha um papel central no controle da proliferação e maturação de megaca- riócitos - as células das quais as plaquetas são produzidas (Kaushansky K, NEJM 354:2034-45, 2006). Como tal, sinalização de Tpo reduzida diminuiria os números de megacariócitos (megacariocitopenia) e reduziria as contagens de plaquetas circulantes (trombocitopenia). Isto pode resultar em hemorragia indesejável e/ou incontrolável. Inibição reduzida de outras JAKs, tais como JAK3 e Tyk2, pode também ser desejável quando humanos sem versão funcional destas quinases mostraram sofrer de numerosas enfermidades tal como síndome de hiperimunoglobina E ou imunodeficiência combinada severa (minegishi, Y, e outro, Immunity 25:745-55, 2006; Macchi P, e outro, Nature. 377:65-8, 1995). Portanto, um inibidor de JAK1 com afinidade reduzida quanto às outras JAKs teria vantagens significantes mais do que um inibidor menos seletivo com respeito aos efeitos colaterais reduzidos envolvendo a supressão imune, anemia e trombocitopenia.
Outro aspecto da presente invenção pertence aos métodos de tratamento de uma doença associada com JAK ou distúrbio em um indivíduo (por exemplo, paciente) administrando-se ao indivíduo em necessidade de tal tratamento a quantidade terapeuticamente eficaz ou dose of um composto, salt do mesmo, ou N-óxido do mesmo, da presente invenção ou uma composição farmacêutica de qualquer um dos anteriormente mencionados. Uma doença associada com JAK pode incluir qualquer doença, distúrbio ou condição que está direta ou indiretamente ligado à expressão ou atividade da JAK, incluindo superexpressão e/ou níveis de atividade anormais. Uma doença associada com JAK pode também incluir qualquer doença, distúrbio ou condição que pode ser prevenido, melhorado, ou curado por modulação da atividade de JAK. Em algumas modalidades, a doença associada com JAK é uma doença associada com JAK1.
Exemplos de doenças associadas com JAK incluem doenças envolvendo o sistema imune incluindo, por exemplo, rejeição ao transplante de órgão (por exemplo, rejeição ao aloenxerto e doença do hospedeiro versus enxerto).
Outros exemplos de doenças associadas com JAK incluem doenças autoimunes tais como esclerose múltipla, artrite reumatoide, artrite juvenil, artrite psoriática, diabetes tipo I, lúpus, psoríase, doença inflamatória do intestino, colite ulcerativa, doença de Crohn, miastenia grave, nefropatias de ímunoglobulina, distúrbios de tiroide autoimunes, doença pulmonar obstrutiva crônica (COPD), e similares. Em algumas modalidades, a doença au- toimune é distúrbio de pele bolhoso autoimune tal como penfigoide vulgar (PV) ou penfigoide bolhoso (BP).
Outros exemplos de doenças associadas com JAK incluem condições alérgicas tal como asma, alergias alimentares, dermatite eczematoso, dermatite de contato, dermatite atópica e rinite. Outros exemplos de doenças associadas com JAK incluem doenças virais tais como Vírus Epstein Barr (EBV), Hepatite B, Hepatite C, HIV, HTLV 1, vírus Varicella-Zoster (VZV) e Papilomavírus humana (HPV).
Outros exemplos de doença associada com JAK incluem doen- 5 ças associadas com regeneração de cartilagem, por exemplo, artrite gotosa, artrite séptica ou infecciosa, artrite reativa, distrofia simpática reflexa, algo- distrofia, síndrome de Tietze, atropatia costal, osteoartrite deformante endêmica, doença de Mseleni, doença de Handigodu, degeneração resultante de fibromialgia, lúpus eritematoso sistêmico, escleroderma, ou espondilite anci- 10 losante.
Outros exemplos de doença associada com JAK incluem malformações de cartilagem congênitas, incluindo condrólise hereditária, con- drodisplasias, e pseudocondrodisplasias (por exemplo, microtia, enotia, e condrodisplasia metafisária).
Outros exemplos de doenças ou condições associadas com JAK incluem distúrbios de pele tais como psoriase (por exemplo, psoríase vulgar), dermatite atópica, brotoeja da pele, irritação da pele, senbilídade da pele (por exemplo, dermatite de contato ou dermatite de contato alérgica). Por exemplo, certas substâncias incluindo alguns produtos farmacêuticos quando topicamente aplicados podem causar senbilidade da pele. Em algumas modalidades, a coadministração ou administração sequencial de pelo menos um inibidor de JAK da invenção juntamente com o agente causando sensibilização indesejada pode ser útil no tratamento de tal sensibilização indesejada ou dermatite. Em algumas modalidades, o distúrbio de pele é tratado por administração tópica de pelo menos um inibidor de JAK da invenção.
Em outras modalidades, a doença associada com JAK é câncer incluindo aqueles caracterizados por tumores sólidos (por exemplo, câncer de próstata, câncer renal, câncer hepático, câncer pancreático, câncer gás- 30 trico, câncer de mama, câncer de pulmão, cânceres da cabeça e pescoço, câncer de tireoide, glioblastoma, sarcoma de Kaposi, doença de Castleman, leiomiossarcoma uterino, melanoma etc.), cânceres hematológicos (por e- xemplo, linfoma, leucemia tal como leucemia linfoblástica aguda, leucemia mielogenosa aguda (AML) ou mieloma múltiplo), e câncer de pele tai como linfoma de célula T cutâneo (CTCL) e linfoma de célula B cutâneo. Exemplos de CTCLs incluem sindrome de Sézary e micose fungoides.
Em algumas modalidades, os inibidores de JAK descritos aqui,bem como outros inibidores de JAK, tais como aqueles reportados em U.S. Ser. n° 11/637.545, podem ser usados para tratar cânceres associados com inflamação. Em algumas modalidades, o câncer está associado com doença inflamatória do intestino. Em algumas modalidades, a doença inflamatória do 10 intestino é colite ulcerativa. Em algumas modalidades, a doença inflamatória do intestino é doença de Crohn. Em algumas modalidades, o câncer associado com inflamação é câncer associado com colite. Em algumas modalidades, o câncer associado com inflamação é câncer de cólon ou câncer color- retal. Em algumas modalidades, o câncer é câncer gástrico, tumor carcinoide 15 gastrointestinal, tumor estromal gastrointestinal (GIST), adenocarcinoma, câncer do intestino delgado, ou câncer retal.
Doenças associadas com JAK podem também incluir aquelas caracterizadas por exepressão de uma JAK2 mutante tal como aquelas tendo pelo menos uma mutação no domínio de pseudo-quinase (por exemplo, 20 JAK2V617F).
Doenças associadas com JAK podem também incluir distúrbios mieloproliferativos (MPDs) tais como policitemia vera (PV), trombocitemia essencial (ET), mielofibrose com metaplasia mieloide (MMM), mielofibrose primária (PMF), leucemia crônica mielogenosa (CML), leucemia mielomono- 25 cítica crônica (CMML), síndrome hipereosinofílica (HES), doença de mastóci- to sistêmica (SMCD), e similares. Em algumas modalidades, a distúrbio mie- loproliferativo é mielofibrose primária (PMF) ou pós-policitemia vera/trombo- citemia essencial mielofibrose (Pós-PV/ET MF).
A presente invenção também fornece métodos de tratamento de 30 psoríase ou outros distúrbios de pele por administração de uma formulação tópica contendo um composto da invenção.
A presente invenção também fornece um método de tratamento de efeitos colaterais dermatológicos de outros produtos farmacêuticos por administração do composto da invenção. Por exemplo, numerosos agentes farmacêuticos resultam em reações alérgicas indesejadas que podem se manifestar como brotoeja acneiforme ou dermatite relacionada. Exemplos de agentes farmacêuticos que têm tais efeitos colaterais indesejados incluem fármacos anticâncer tais como gefitinibe, cetuximabe, erlotinibe, e similares. Os compostos da invenção podem ser administrados sistemicamente ou topicamente (por exemplo, localizados nas vizinhaças da dermatite) em combinação com (por exemplo, simultaneamente ou sequencialmente) o agente farmacêutico tendo o efeito colateral dermatológico indesejado. Em algumas modalidades, o composto da invenção pode ser administrado topicamente juntamente com um ou mais outros produtos farmacêuticos, onde os outros produtos farmacêuticos quando topicamente aplicados na ausência de um composto da invenção causam dermatite de contato, sensibilização de contato alérgico, ou similar distúrbio de pele. Consequentemente, composições da invenção incluem formulações tópicas contendo o composto da invenção e um outro agente farmacêutico que podem causar dermatite, distúrbio de peles, ou efeitos colaterais relacionados.
Outras doenças associadas com JAK incluem inflamação e doenças inflamatórias. Exemplo doenças inflamatórias incluem doenças inflamatórias do olho (por exemplo, irite, uveíte, esclerite, conjuntivite, ou doença relacionada), doenças inflamatórias do trato respiratório (por exemplo, o trato respiratório superior incluindo o nariz e sinos tais como rinite ou sinusite ou o trato respiratório inferior incluindo bronquite, doença pulmonar obstrutiva crônica, e similares), miopatia inflamatória tal como miocardite, e outras doenças inflamatórias.
Os inibidores de JAK descritos aqui podem também ser usados para tratar lesões de reperfusão de isquemia ou uma doença ou condição relacionada a um evento isquêmico inflamatório tal como acidente vascular cerebral ou parada cardíaca. Os inibidores de JAK descritos aqui podem também ser usados para tratar estado de doença impulSiOnado por endoxi- na (por exemplo, compliações após cirurgia de desvio ou estados de endo- toxina crônicos contribuindo para insuficiência cardíaca crônica). Os inibidores de JAK descritos aqui podem também ser usados para tratar anorexia, caquexia, ou fatiga tal como aquela resultante de ou associada com câncer. Os inibidores de JAK descritos aqui podem também ser usados para tratar restenose, esclerodermite, ou fibrose. Os inibidores de JAK descritos aqui podem também ser usados para tratar condições associadas com hipoxia ou astrogliose tal como, por exemplo, retinopatia diabética, câncer, ou neurode- generação. Veja, por exemplo, Dudley, A.C. e outro, Biochem. J. 2005, 390(Pt 2):427-36 e Sriram, K. e outro, J. Biol. Chem. 2004, 279(19): 19936- 47. Epub 2 de Março de 2004. Os inibidores de JAK descritos aqui podem ser usados para tratar doença de Alzheimer.
Os inibidores de JAK descritos aqui podem também ser usados para tratar outras doenças inflamatórias tais como síndrome de resposta inflamatória sistêmica (SIRS) e choque séptico.
Os inibidores de JAK descritos aqui podem também ser usados para tratar gota e tamanho de próstata aumentado devido à, por exemplo, hipertrofia prostática benigna ou hioerplasia prostática benigna.
Em algumas modalidades, inibidores de JAK descrito aqui podem também ser usados para tratar um distúrbio de olho seco. Como usado aqui, "distúrbio de olho seco" destina-se a abranger os estados de doença sumariados em um relato oficial do Olho seco Workshop (DEWS), que define olho seco como "uma doença multifatorial das lágrimas e superfície ocular que resulta em sintomas de desconforto, distúrbio visual, e instabilidade de película de lágrima com dano potencial à superfície ocular. Ele é acompanhado por osmolaridade aumentada da película de lágrima da superfície ocular." Lemp, "The Definition and Classification of Dry Eye Disease: Report of the Definition and Classification Subcommittee of the International Olho seco Workshop", The Ocular Superficie, 5(2), 75-92 Abril de 2007. Em algumas modalidades, o distúrbio de olho seco é selecionado de olho seco deficiente de lágrima aquosa (ADDE) ou distúrbio do olho seco evaporative, ou combinações apropriadas dos mesmos.
Em um outro aspecto, a presente invenção fornece um método de tratamento de conjuntivite, uveíte (incluindo uveíte crônica), coriodite, retinite, ciclite, esclerite, episclerite, ou irite; tratamento de inflamação ou dor relacionada ao transplante de córnea, LASIK (ceratomileuse in situ assistida por laser), ceratoctomia fotorefrativa, ou LASEK (ceratomileuse sub-epitelial 5 assistida por laser); inibição de perda de acuidade visual relacionada com o transplante de córnea, LASIK, ceratectomia fotorefrativo, ou LASEK; ou ini-bição de rejeição de transplante em um paciente em necessidade dos mesmos, que compreende administrar ao paciente uma quantidade terapeutica- mente eficaz do composto da invenção, ou um sal farmaceuticamente acei- 10 tável do mesmo.
Adicionalmente, o composto da invenção, bem como outros inibidores de JAK tal como aqueles reportados em U.S. Ser. nO 11/637.545, podem ser usados para tratar disfunção respiratória ou insuficiência associada com infecção virai, tal como influenza e SARS.
Em algumas modalidades, a presente invenção fornece um composto de fórmula I, sal farmaceuticamente aceitável do mesmo, ou N- óxido do mesmo, como descrito em qualquer uma das modalidades aqui, para uso em um método de tratamento de qualqur uma das doenças ou distúrbios descritos aqui. Em algumas modalidades, a presente invenção forne- 20 ce o uso de um composto de fórmula I como descrito em qualquer uma das modalidades aqui, para a preparação de um medicamento para uso em um método de tratamento de qalquer uma das doenças ou distúrbios descritos aqui.
Em algumas modalidades, a presente invenção fornece um com- 25 posto de fórmula I como descrito aqui, ou um sal farmaceuticamente aceitável ou N-óxido do mesmo, para uso em um método para modulação de uma JAK1. Em algumas modalidades, a presente invenção também fornece uso de um composto de fórmula I como descrito aqui, ou um sal farmaceuticamente aceitável ou N-óxido do mesmo, para a preparação de um medica- 30 mento para uso em um método para modulação de uma JAK1.
Terapias de Combinação
Um ou mais agentes farmacêuticos adicionais tais como, por exemplo, quimioterapêuticos, agentes anti-inflamatórios, esteroides, imunos- supressores, bem como inibidores de Bcr-abl, Flt-3, RAF e FAK quinase tais como, por exemplo, aqueles descritos no WO 2006/056399, ou outros agentes podem ser usados em combinação com os compostos descritos aqui para tratamento de doenças, distúrbios ou condições associados com JAK,. Um ou mais agentes farmacêuticos adicionais podem ser administrados a um paciente simultaneamente ou sequencialmente.
Exemplos de quimioterapêuticos incluem inibidores de proteos- soma (por exemplo, bortezomibe), talidomida, revlimida, e agentes de danificação de DNA tais como melfalano, doxorubicina, ciclofosfamída, vincristina, etoposídeo, carmustina, e similares.
Esteroides exemplos incluem coriticosteroides tais como dexa- metasona ou prednisona.
Inibidores de Bcr-abl do exemplo incluem os compostos, e sais farmaceuticamente aceitáveis dos mesmos, dos gêneros e espécies descritos na Patente dos Estados Unidos n0 5,521,184, WO 04/005281, e U.S. Ser. no 60/578.491.
Inibidores de Flt-3 adequados do exemplo incluem compostos, e seus sais farmaceuticamente aceitáveis, como descrito no WO 03/037347, WO 03/099771, e WO 04/046120.
Inibidores de RAF adequados do exemplo incluem compostos, e seus sais farmaceuticamente aceitáveis, como descrito no WO 00/09495 e WO 05/028444.
Inibidores de FAK adequados do exemplo incluem compostos, e seus sais farmaceuticamente aceitáveis, como descrito nos WO 04/080980, WO 04/056786, WO 03/024967, WO 01/064655, WO 00/053595, e WO 01/014402.
Em algumas modalidades, um ou mais dos compostos da invenção podem ser usados em combinação com um ou mais outros inibidores de quinase incluindo imatinibe, particularmente para o tratamento de pacientes resistantes á imatinibe ou outros inibidores de quinase.
Em algumas modalidades, um ou mais inibidores de JAK da in- venção podem ser usados em combinação com um quimioterapèutico no tratamento de câncer, tal como mieloma múltiplo, e pode melhorar a respos-ta de tratamento quando comparado á resposta ao agente quimioterapèutico sozinho, sem exacerbação de seus efeitos tóxicos. Exemplos de agentes farmacêuticos adicionais usados no tratamento de mieloma múltiplo, por e- xemplo, podem incluir, sem limitação, melfalanp, melfalano mais prednisona [MP], doxorubicina, dexametasona, e Velcade (bortezomibe). Outros agentes adicionais usados no tratamento de mieloma múltiplo incluem inibidores de Bcr-abl, Flt-3, RAF e FAK quinase. Efeitos aditivos ou sinérgicos são resul- tados desejados de combinação de um inibidor de JAK da presente invenção com um agente adicional. Além disso, resistência de células de mieloma múltiplo aos agentes tal como dexametasona pode ser reversível no tratamento com um inibidor de JAK da presente invenção. Os agentes podem ser combinados com os presentes compostos em uma forma de dosagem sim- pies ou contínua, ou os agentes podem ser administrados simultaneamente ou sequencialmente como formas de dosagem separadas.
Em algumas modalidades, um corticosteroide tal como dexametasona é administrado com um paciente em combinação com pelo menos um inibidor de JAK onde a dexametasona é administrada intermitentemente quando em oposição a continuamente.
Em algumas outras modalidades, combinações de um ou mais inibidores de JAK da invenção com outros agentes terapêuticos podem ser administradas com um paciente antes de, durante, e/ou após o transplante de medula óssea ou transplante de célula tronco.
Em algumas modalidades, o agente terapêutico adicional é fluo-cinolona acetonida (Retisert®), ou rimexolona (AL-2178, Vexol, Alcon).
Em algumas modalidades, o agente terapêutico adicional é ci- closporina (Restasis®).
Em algumas modalidades, o agente terapêutico adicional é um corticosteroide. Em algumas modalidades, a corticosteroide é triamcinolona, dexametasona, fluocinolona, cortisona, prednisolona, ou flumetolona.
Em algumas modalidades, o agente terapêutico adicional é sele- cionado de Dehidrex™ (Holies Labs), Civamida (Opko), hialuronato de sódio (Vismed, Laπtibio/TRB Chemedia), ciclosporina (ST-603, Sirion Therapeutics), ARG101(T) (testosterone, Argentis), AGR1012(P) (Argentis), ecabete sódico (Senju-lsta), gefarnato (Santen), ácido 15-(s)-hidroxieicosatetraenoico (15(S)-HETE), cevilemina, doxiciclina (ALTY-0501, Alacrity), minociclina, i- Destrin™ (NP50301, Nascent Pharmaceuticals), ciclosporina A (Nova22007, Novagali), oxitetraciclina (Duramycin, MOLI1901, Lantibio), CF101 (2S,3S,4R,5R)-3,4-diidróxi-5-[6-[(3-iodofenil)metilamino]purin-9-ÍI]-N-metil- oxolano-2-carbamila, Can-Fite Biopharma), voclosporina (LX212 ou LX214, Lux Biosciences), ARG 103 (Agentis), RX-10045 (análogo de resolvina sintético, Resolvyx), DYN15 (Dyanmis Therapeutics), rivoglitazona (DE011, Daiichi Sanko), TB4 (RegeneRx), OPH-01 (Ophtalmis Monaco), PCS101 (Peri- cor Science), REV1-31 (Evolutec), Lacritina (Senju), rebamipida (Otsuka- Novartis), OT-551 (Othera), PAI-2 (University of Pennsilvania and Temple University), pilocarpina, tacrolimo, pimecrolimo (AMS981, Novartis), etabona- to de loteprednol, rituximabe, tetrassódio de diquafosol (INS365, Inspire), KLS-0611 (Kissei Pharmaceuticals), desidroepiandrosterona, anacinra, efali- zumabe, sódio de micofenolato, etanorcepte (Embrel®), hidroxicloroquina, NGX267 (TorreyPines Therapeutics), ou talidomida.
Em algumas modalidades, o agente terapêutico adicional é um agente antiangiogênico, agonista colinérgico, modulador de receptor TRP-1, um bloqueador de canal de cálcio, um secretagogo de mucina, estimulante de MUC1, um inibidor de calcineurina, um corticosteroide, um agonista de receptor de P2Y2, um agonista de receptor muscarínico, outro inibidor de JAK, inibidor de Bcr-abl quinase, inibidor de Fít-3 quinase, inibidor de RAF quinase, e inibidor de FAK quinase tal como, por exemplo, aqueles descritos no WO 2006/056399. Em algumas modalidades, o agente terapêutico adicional é um derivado de tetraciclina (por exemplo, minociclina ou doxiclina).
Em algumas modalidades, o agente(s) terapêutico adicional são colírios demulcentes (também conhecidos como "lágrimas artificiais"), que incluem, porém não estão limitados às composições contendo polivinilálcool, hidroxipropil metilcelulose, glicerina, polietileno glicol (por exemplo, PEG400), ou carboximetil celulose. Lágrimas artificiais podem auxiliar no tratamento de olho seco por compensação por umidade e capacidade de lubrificação da película de lágrima reduzidas. Em algumas modalidades, o agente terapêutico adicional é um fármaco mucolítico, tal como N-acetil-cis- 5 teína, que pode interagir com as mucoproteínas e, portanto, para diminuir a viscosidade da película de lágrima.
Em algumas modalidades, o agente terapêutico adicional inclui um antibiótico, antiviral, antifúngico, anestésico, agentes anti-inflamatórios incluindo ati-inflamatórios esteroidais e não estereoidais, e agentes antialér- 10 gicos. Exemplos de medicamentos adequados incluem aminoglcosídeos tais como amicacina, gentamícina, tobramicina, estreptomicina, netilmicina, e ca- namicina; fluorquinolonas tais como ciprofloxacina, norfloxacina, ofloxacina, trovafloxacina, lomefloxacina, levofloxacina, e enoxacina; naftiridina; sulfo- namidas; polimixina; cloranfenicol; neomicina; paramomicina; colistimetato; bacitracina; vancomicina; tetraciclinas; rifampina e seus derivados ("rifampi- nas"); cicloserina; beta-lactans; cefalosporinas; anfotericinas; fluconazol; flu- citosina; natamicina; miconazol; cetoconazol; corticosteroides; diclofenac; flurbiprofeno; cetorolac; suprofeno; cromolina; lodoxamida; levocabastina; nafazolina; antazolina; feniramina; ou azalida antibiótico.
Formulações Farmacêuticas e Formas de Dosagem
Quando empregados como produtos farmacêuticos, os compostos da invenção podem ser administrados na forma de composições farmacêuticas. Estas composições podem ser preparadas de uma maneira bem conhecida na técnica farmacêutica, e podem ser administradas por uma va- 25 riedade de rotinas, dependendo de se tratamento local ou sistêmico é desejado e da área a ser tratado. A administração pode ser tópica (incluindo transdérmica, epidérmica, oftálmica e para membranas mucosas incluindo liberação retal, vaginal e intranasal), pulmonar (por exemplo, por inalação ou insulflação de pós ou aerossóis, incluindo por nebulizador; intratraqueal ou 30 intranasal), oral ou parenteral. Administração parenteral inclui administração intravenosa, intraarterial, subcutânea, intraperitoneal intramuscular ou injeção ou infusão; ou intracranial, por exemplo, intratecal ou intraventricular. A administração parenteral pode ser na forma de uma única dose de bolo, ou pode ser, ou pode ser, por exemplo, por uma bomba de perfusão contínua. Composições farmacêuticas e formulações para administração tópica podem incluir emplastros transdérmicos, unguentos, loções, cremes, géis, gotas, 5 supositórios, sprays, líquidos e pós. Veículos farmacêuticos convencionais, bases aquosas, em pó ou oleosas, espessantes e similares podem ser necessários ou desejáveis. Luvas, condons revestidos e similares podem também ser úteis. Em algumas modalidades, a composição é para liberação oral. Em outras modalidades, a composição é para aplicação tópica.
Esta invenção também inclui composições farmacêuticas que contêm, como o ingrediente ativo, um ou mais dos compostos da invenção acima em combinação com um ou mais veículos farmaceuticamente aceitáveis (excipientes). Na fabricação das composições da invenção, o ingrediente ativo é tipicamente misturado com um excipiente, diluído por um excipien- te ou incluso em tal veículo na forma de, por exemplo, uma cápsula, sachê, papel, ou outro recipiente. Quando o excipiente serve como um diluente, ele pode ser um material sólido, semissólido, ou líquido, que age como um veículo, portador, ou meio para o ingrediente ativo. Desse modo, as composições podem ser na forma de comprimidos, pílulas, pós, lozangos, sachês, 20 selos, elixires, suspensões, emulsões, soluções, xaropes, aerossóis (como um sólido ou em um meio líquido), unguentos contendo, por exemplo, até 10% em peso do composto ativo, cápsulas de gelatina macias e duras, supositórios, soluções injetáveis estéreis, e pós empacotados estéreis.
Na preparação da formulação, o composto ativo pode ser moído para fornecer o tamanho de partícula apropriado antes de combinar com outros ingredientes. Se o composto ativo é substancialmente insolúvel, ele pode ser moído para um tamanho de partícula de menos ddo que 200 malhas. Se o composto ativo é substancialmente solúvel em água, o tamanho de partícula pode ser ajustado moendo-se para fornecer uma distribuição substan- cialmente uniforme na formulação, por exemplo, cerca de 40 malhas.
Os compostos da invenção podem ser moídos usando procedimentos de moagem conhecidos tal como moagem úmida para obter um ta- manho de partícula apropriada para a formação de comprimido e para outros tipos de formulação. Preparações finomente divididas (nanoparticulados) dos compostos da invenção podem ser peraparadas por processos conhecidos na técnica, por exemplo, veja o Pedido de Patente Internacional n° WO 2002/000196.
Alguns exemplos de excipientes adequados incluem lactose, dextrose, sacarose, sorbitol, manitol, amidos, goma acácia, fosfato de cálcio, alginatos, tragacanto, gelatina, silicato de cálcio, celulose microcristalina, polivinilpirrolidona, celulose, água, xarope, e metil celulose. As formulações podem adicionalmente incluir: agentes de lubrificação tais como talco, estea- rato de magnésio, e óleo mineral; agentes umectantes; agentes de emulsifi- cação e suspensão; agentes conservantes tal como metil- e propilhidróxi- benzoatos; agentes adoçantes; e agentes aromatizantes. As composições da invenção podem ser formuladas a fim de fornecer liberação rápida, prolongada ou retardada do ingrediente ativo após a administração ao paciente empregando-se os procedimentos conhecidos na técnica.
As composições podem ser formuladas em uma forma de dosagem unitária, cada dosagem contendo de cerca de 5 a cerca de 1000 mg (1 g), mais gralmente cerca de 100 a cerca de 500 mg, do ingrediente ativo. O termo "formas de dosagem unitária" refere-se às unidades fisicamente discretas adequadas como dosagens unitárias para pacientes humanos e outros animais, cada unidade contendo uma quantidade predeterminada de material ativo calculado para produzir o efeito terapêutico desejado, em associação com um excipiente farmacêutico adequado.
O composto ativo pode ser eficaz sobre uma ampla faixa de dosagem e é geralmente administrado em uma quantidade farmaceuticamente eficaz. Será entendido, entretanto, que a quantidade do composto realmente administrada será usualmente determinada por um médico, de acordo com as circunstâncias relevantes, incluindo a condição a ser tratado, a rotina escolhida de administração, o composto real administrado, a idade, peso, e a resposta do paciente individual, a gravidade dos sintomas de paciente, e similares.
Para a preparação de composições sólidas tal como comprimidos, o ingrediente ativo principal é misturado com um excipiente farmacêutico para formar uma composição de pré-formulação sólida contendo uma mistura homogênea de um composto da presente invenção. Quando se referindo a estas composições de pré-formulação como homogêneas, o ingrediente ativo é tipicamente disperso uniformemente por toda a composição de modo que a composição possa ser facilmente subdividida em formas de dosagem unitárias igualmente eficazes tais como comprimidos, pílulas e cápsulas. Esta pré-formulação sólida é em seguida subdivida em formas de dosagem unitárias do tipo descrito acima contendo de, por exemplo, cerca de 0,1 a cerca de 1000 mg do ingrediente ativo da presente invenção.
Os comprimidos ou as pílulas da presente invenção podem ser revestidos ou de outro modo compostos para fornecer uma forma de dosagem fornecendo a vantagem de ação prolongada. Por exemplo, o comprimido ou pílula pode compreender uma dosagem interna e um componente de dosagem externa, o último sendo na forma de um envelope sobre o formador. Os dois componentes podem ser separados por uma camada entérica que serve para resistir a desintegração no estômago e permitir o componente interno de passer intacto no duodenum ou ser retardado na liberação. Uma variedade de materiais pode ser usada para tais revestimento ou camadas enteréricas, tais materiais incluindo diversos ácidos poliméricos e misturas de ácidos poliméricos com tais materiais como verniz, cetil álcool, e acetato de celulose.
As formas líquidas em que os compostos e composições da presente invenção podem ser incorporados para administração oralmente ou por injeção incluem soluções aquosas, xaropes adequadamente aromatizados, suspensões aquosas ou oleosas, e emulsões aromatizadas com óleos comestíveis tais como óleo de semente de algodão, óleo de gergelim, óleo de coco, ou óleo de amendoim, bem como elixires e veículos farmacêuticos.
Composições para inalação ou insuflação incluem soluções e suspensões em solventes farmaceuticamente aceitáveis, aquosos ou orgânicos, ou misturas dos mesmos, e pós. As composições líquidas ou sólidas podem conter excipientes farmaceuticamente aceitáveis adequados como descrito supra. Em algumas modalidades, as composições são administradas pela rotina respiratória oral e nasal para efeito local ou sistêmico. Composições podem ser nebulizadas por uso de gases inertes. Soluções nebuli- zadas podem ser respiradas diretamente do dispositivo de nebulização ou o dipositivo de nebulização pode ser ligado a uma de tenda de máscaras de face, ou máquina de respiração de pressão positiva intermitente. Composições de solução, suspensão, ou pó podem ser administradas oralmente ou nasalmente de dispositivos que liberam a formulação de uma maneira apropriada.
A quantidade de composto ou composição administrada com um paciente variará dependendo do que está sendo administrado, o propósito da administração, tal como profilaxia ou terapia, do estado do paciente, da maneira de administração, e similares. Em aplicações terapêuticas, composições podem ser administradas com um paciente já sofrendo de uma doença em uma quantidade suficiente para curar ou pelo menos parcialmente interromper os sintomas da doença e suas complicações. Doses eficazes dependerão da condição de doença sendo tratado bem como pelo diagnóstico do médico atendente dependendo de fatores tais como a gravidade da doença, da idade, peso, e condição geral do paciente, e similares.
As composições administradas com um paciente podem ser na forma de composições farmacêuticcomo descrito acima. Estas composições podem ser esterilizadas por técnicas de esterilização convencional, ou podem ser filtradas estéreis. Soluções aquosas podem ser empacotadas para usar no estado em que se encontram, ou liofilizadas, a preparação liofilízada sendo combinada com um veículo aquoso estéril antes da administração. O pH das preparações do composto tipicamente sera entre 3 e 11, mais Preferivelmente de 5 a 9 e mais preferivelmente de 7 a 8. Será entendido que o uso de certos dos excipientes, veículos, ou estabilizantes precedentes resultará na formação de sais farmacêuticos.
A dosagem terapêutica dos compostos descritos aqui pode variar de acordo com, por exemplo, o uso particular para qual o tratamento é feito, a maneira e administração do composto, a saúde e condição do paciente, e o diagnóstico do médico receitista. A proporção ou concentração de um composto da invenção em uma composição farmacêutica pode variar dependendo de diversos fatores incluindo dosagem, características químicas (por exemplo, hidrofobicidade), e da rotina de administração. Por exemplo, os compostos da invenção podem ser fornecidos em uma solução de tapão fisiológica aquosa contendo cerca de 0,1 a cerca de 10% de peso/volume do composto para administração parenteral. Algumas faixas de dose típicas são de cerca de 1 g/kg a cerca de 1 g/kg de peso corporal por dia. Em algumas modalidades, a faixa de dose é de cerca de 0,01 mg/kg a cerca de 100 mg/kg de peso corporal por dia. A dosagem provavelmente depende de tais variáveis como o tipo e extensão de progressão da doença ou distúrbio, o estado de saúde total do paciente particular, a eficácia biológica relativa do composto selecionado, formulação do excipiente, e sua rotina de administração. Doses eficazes podem ser extrapoladas das curvas de resposta de dose derivadas de sistemas de teste de modelo in vitro ou animal.
As composições da invenção podem também incluir um ou mais agentes farmacêuticos adicionais tal como um composto anti-inflamatório, esteroide, quimioterapêutico, ou imunossupressor, exemplos de que são listados aqui acima.
Em algumas modalidades, o composto, ou sal farmaceuticamente aceitável ou N-óxido do mesmo, é administrado como uma composição oftálmica. Consequentemente, em algumas modalidades, os métodos compreendem a administração do composto, ou sal farmaceuticamente aceitável ou N-óxido do mesmo, e um veículo oftalmicamente aceitável. Em algumas modalidades, a composição oftálmica é uma composição líquida, composição semissólida, inserção, película, microparticulas ou nanopartículas.
Em algumas modalidades, a composição oftálmica é uma composição liquida. Em algumas modalidades, a composição oftálmica é a composição semissólida. Em algumas modalidades, a composição oftálmica é uma composição tópica. As composições tópicas incluem, porém não estão limitadas às composições líquidas e semissólidas. Em algumas modalida- des, a composição oftálmica é uma composição tópica. Em algumas modalidades, a composição tópica compreende solução aquosa, uma suspensão aquosa, um unguento ou um gel. Em algumas modalidades, a composição oftálmica é topicamente aplicada na frente do olho, sob a pálpebra superior, 5 na pálpebra inferior e no fundo do saco. Em algumas modalidades, a composição oftálmica é esterilizada. A esterilização pode ser realizada por técnicas conhecidas como filtragem por esterilização da solução ou por aquecimento da solução na ampola pronta para uso. As composições oftálmicas da invenção podem também conter excipientes farmacêuticos adequados para a preparação de formulações oftálmicas. Exemplos de tais excipientes são agentes conservantes, agentes de tamponomento, agentes de quelação, agentes antioxidantes e sais para a regulação da pressão osmótica.
Como usado aqui, o termo "veículo oftalmicamente aceitável" refere-se a qualquer material que pode conter e liberar o composto, ou sal far- 15 maceuticamente aceitável ou N-óxido do mesmo, e que é compatível com o olho. Em algumas modalidades, o veículo oftalmicamente aceitável é água ou um solução ou suspensão aquosa, porém também inclui óleos tais como aqueles usados para fazer unguentos e matrizes polímeras tais como usados em inserções oculares. Em algumas modalidades, a composição pode 20 ser uma suspensão aquosa que compreende o composto, ou sal farmaceuticamente aceitável ou N-óxido do mesmo. Composições oftálmicas líquidas, incluindo tanto unguentos quanto suspensões, podem ter uma viscosidade que é adaptada para a rotina selecionada de administração. Em algumas modalidades, a composição oftálmica tem uma viscosidade na faixa de cerca de 1000 a cerca de 30.000 centipoise.
Em algumas modalidades, as composições oftálmicas podem também compreender um ou mais de tensoativo, adjuvantes, tampões, antioxidantes, tonicidade, ajustadores, conservantes (por exemplo, EDTA, BAK (cloreto de benzalcônio), cloreto de sódio, perborato de sódio, poliquatério- 30 1), espessantes ou modificadores de viscosidade (por exemplo, carboximetil celulose, hidroximetil celulose, polivinil álcool, polietileno glicol, glicol 400, propileno glicol hidroximetil celulose, hidroxpropil-guar, ácido hialurônico, e hidroxipropil celulose) e similares. Aditivos na formulação podem incluir, porém não estão limitados a cloreto de sódio, bicarbonato de sódio, ácido sór- bico, metil parabeno, propil parabeno, clorexidina, óleo de rícino, e perborato de sódio.
Composições oftálmicas aquosas (soluções ou suspensões) geralmente não contêm constituintes fisiologicamente ou oftalmicamente prejudiciais. Em algumas modalidades, água purificada ou deSiOnizada é usada na composição. O pH pode ser adjustado adicionando-se quaisquer ácidos de ajuste de pH, bases ou tampões fisiologicamente e oftalmicamente acei- 10 táveis dentro da faixa de cerca de 5,0 a 8,5. Exemplos oftalmicamente aceitáveis de ácidos incluem acéticos, bóricos, cítricos, lácticos, fosfóricos, hi- droclóricos, e similares, e exemplos de bases incluem hidróxido de sódio, fosfato de sódio, borato de sódio, citrato de sódio, acetato de sódio, lactato de sódio, trometamina, trisidroximetilamino-metano, e similares. Sais e tam- 15 pões incluem citrato/dextrose, bicarbonato de sódio, cloreto de amónio e misturas dos ácidos e bases anteriormente mencionados.
Em algumas modalidades, os métodos envolvem a formação ou fornecimento de um depósito do agente terapêutico em contato com a superficie externa do olho. Um depósito refere-se a uma fonte de agente terapêu- 20 tico que não é rapidamente removido por lágrimas ou outros mecanismos de purificação do olho. Isto permite concentrações elevadas sustentadas, continuadas de agente terapêutico estarem no presente no fluído na superfície externa do olho por uma única aplicação. Sem querer ser ligado à qualquer teoria, acredita-se que a absorção e penetração podem ser dependentes 25 tanto da concentração de fármaco dissolvido quanto a duração de contato do tecido externo com o fármaco contendo fluido. Quando o fármaco é removido por purificação do fluido ocular e/ou absorção no tecido de olho, mais fármaco é fornecido, por exemplo, dissolvido, fluido ocular reaproviSiOnado do depósito. Consequentemente, o uso de um depósito pode mais facilmen- 30 te facilitar a carga do tecido ocular para agentes terapêuticos mais insolú-veis. Em algumas modalidades, o depósito pode permanecer durante até oito horas ou mais. Em algumas modalidades, as formas de depósito oftál- mico incluem, porém não estão limitados às suspensões poliméricas, unguentos, e inserções sólidas.
Em algumas modalidades, a composição oftálmica é um unguento ou gel. Em alguma modalidade, a composição oftálmica é um veículo de liberação com base em óleo. Em algumas modalidades, a composição compreende uma de petróleo ou lanolina a qual é adicionada o ingrediente ativo, geralmente como 0,1 a 2%, e excipientes. Bases comuns podem incluir, porém não estão limitados a óleo mineral, petróleo e combinações dos mesmos. Em algumas modalidades, o unguento é aplicado como uma fita na pálpebra inferior.
Em alguma modalidade, a composição oftálmica é uma inserção oftálmica. Em algumas modalidades, a inserção oftálmica é biologicamente inerte, macia, bioerosível, viscoelática, estável à esterilização após exposição aos agentes terapêuticos, resistantes às infecções a partir de bactérias aerotransportadas, bioerosíveis, biocompatíveis, e/ou viscoelásticos. Em algumas modalidades, a inserção compreende uma matriz oftalmicamente aceitável, por exemplo, uma matriz polímera. A matriz é tipicamente um polímero e o agente terapêutico é geralmente disperso nele ou ligado à matriz polímera. Em algumas modalidades, o agente terapêutico pode ser lenta-mente liberado da matriz através da dissolução ou hidrólise da ligação cova- lente. Em algumas modalidades, o polímero é bioerosível (solúvel) e a taxa de dissolução do mesmo pode controlar a taxa de liberação do agente terapêutico disperso nele. Em outra forma, a matriz polímera é uma biodegradável que se decompõe, tal como por hidrólise, para desse modo liberar o a- gente terapêutico ligado a ela ou disperso nela. Em outras modalidades, a matriz e agente terapêutico pode ser circundada com um revestimento poli- mérico adicional para também controlar a liberação. Em algumas modalidades, a inserção compreende um polímero biodegradável tal como policapro- lactona (PCL), um copolímero de etileno/vinila (EVA), cianoacrilato de polial- quila, poliuretano, um náilon, ou poli (dl-lactide-co-glicolídeo) (PLGA), ou um copolímero de qualquer um destes. Em algumas modalidades, o agente terapêutico é disperso no material matriz ou disperso no meio da composição monômera usada para fazer o material matriz antes da polimerização. Em algumas modalidades, a quantidade de agente terapêutico é de cerca de 0,1 a cerca de 50%, ou de cerca de 2 a cerca de 20%. Em outras modalidades, a matriz polímera biodegradável ou bioerosivel é usada de modo que a inserção gasta não tenha que ser removida. Quando o polímero biodegradável ou bioerosivel é degradado ou dissolvido, o agente terapêutico é liberado.
Em outras modalidades, a inserção oftálmica compreende um polímero, incluindo, porém não está limitada àquelas descritas em Wagh, e outro, "Polimers usado in ocular dosage form and drug delivery systems", Asian J. Pharm., páginas 12 a 17 (Jan. 2008), que é incorporado aqui por referência em sua totalidade. Em algumas modalidades, a inserção compreende um polímero selecionado de polivinilpirrolidona (PVP), um polímero ou copolímero de acrilato ou metacrilato (por exemplo, família de Eudragit® de polímeros de Rohm ou Degussa), hidroximetil celulose, ácido poliacrílico, dendrímeros de poli(amidoamina), polifsiloxano de dimetila), óxido de polieti- leno, poli(lactídeo-co-glicolídeo), poli(2-hidroxietilmetacrilato), poli(vinil álcool), ou poli(fumarato de propileno). Em algumas modalidades, a inserção compreende Gelfoam® R. Em algumas modalidades, a inserção é um ácido poliacrílico de conjugado de 450 kDa-cisteína.
Em algumas modalidades, a composição oftálmica é uma película oftálmica. Polímeros adequados para tais películas incluem, porém não estão limitados àqueles descritod em Wagh, e outro, "Polimers usado in ocular dosage form and drug delivery systems", Asian J. Pharm., páginas 12 a 17 (Janeiro 2008), Em algumas modalidades, a película é uma lente de contato macia, tal como aquelas feitas de copolímeros N,N-dietilacrilamida e ácido metacrilico reticulado com etilenoglicol dimetacrilato.
Em algumas modalidades, a composição oftálmica compreende microesferas ou nanopartículas. Em alguma modalidade, as microesferas compreendem gelatina. Em algumas modalidades, as microesferas são injetadas no segmento posterior do olho, no espaço coroidal, na esclera, intravi- treamente ou sub-retinalmente. Em algumas modalidades, as microesferas ou nanopartículas compreende um polímero incluindo, porém não limitado àqueles descritos em Wagh, e outro, "Polimers usado in ocular dosage form and drug delivery systems", Asian J. Pharm., páginas 12 a 17 (Janeiro de 2008), que é incorporado aqui por referência em sua totalidade. Em algumas modalidades, o polímero é quitosana, um ácido policarboxílico tal como áci- 5 do poliacrílico, partículas de albumina, ésteres de ácido hialurônico, ácido poliitacôníco, poli(butil)cianoacrilato, policaprolactona, poli(isobutil) caprolac- tona, poli(ácido láctico-ácido co-glicólico), ou poli(ácido láctico). Em algumas modalidades, as microesferas ou nanopartículas compreendem partículas de lipídeo sólidas.
Em algumas modalidades, a composição oftálmica compreende uma resina de permuta de íon. Em algumas modalidades, a resina de permuta de íon é um zeólito inorgânico ou resina orgânica sintética. Em algumas modalidades, a resina de permuta de íon inclui, porém não está limitada àquelas descritas em Wagh, e outro, "Polimers usado in ocular dosage form and drug delivery systems", Asian J. Pharm., páginas 12 a 17 (Janeiro de 2008), que é incorporado aqui por referência em sua totalidade. Em algumas modalidades, a resina de permuta de íon é um ácido poliacrílico parcialmente neutralizado.
Em algumas modalidades, a composição oftálmica é uma sus- 20 pensão polimérica aquosa. Em algumas modalidades, o agente terapêutico ou um agente de suspensão polimérico é suspenso em um meio aquoso. Em algumas modalidades, as suspensões poliméricas aquosas podem ser formuladas de modo que eles retenham a mesma ou substancialmente a mesma viscosidade no olho que elas tinham antes da administração ao olho. Em 25 algumas modalidades, elas podem ser formuladas de modo que exista gelifi- cação no contato com fluido de lágrima.
A invenção também fornece uma formulação farmacêutica para aplicação de pele tópica, que compreende uma quantidade terapeuticamente eficaz de um composto da invenção, ou um sal farmaceuticamente aceitável 30 do mesmo.
Em algumas modalidades, uma formulação farmacêutica compreende: uma emulsão óleo-eM-água; e uma quantidade terapeuticamente eficaz de um composto da invenção, ou um sal farmaceuticamente aceitável do mesmo.
Em algumas modalidades, a emulsão compreende água, um 5 componente de óleo, e um compoente emulsificante.
Como usado aqui, o termo "componente emulsificante" refere- se, em um aspecto, a uma substância, ou misturas de substâncias que mantêm um elemento ou partícula em suspensão dentro de um meio fluido. Em algumas modalidades, o componente emulsificante permite uma fase de ó- 10 leo formar uma emulsão quando combinado com água. Em algumas modalidades, o componente emulsificante refere-se a um ou mais tensoativo não iônicos.
Em algumas modalidades, o componente de óleo está presente em uma quantidade de cerca de 10% a cerca de 40% em peso da formula- 15 çâo.
Em algumas modalidades, o componente de óleo compreende uma ou mais substâncias independentemente selecionadas de petróleos, álcoois graxos, óleos minerais, triglicerídeos, e óleos de silicone.
Em algumas modalidades, o componente de óleo compreende 20 uma ou mais substâncias independentemente selecionadas de petróleo branco, cetil álcool, estearil álcool, óleo mineral leve, triglicerídeos de cadeia média, e dimeticona.
Em algumas modalidades, o componente de óleo compreende um componente de agente oclusivo.
Em algumas modalidades, o componente de agente oclusivo está presente em uma quantidade de cerca de 2% a cerca de 15% em peso da formulação.
Como usado aqui, o termo "componente de agente oclusivo" refere-se a um agente hidrofóbico ou misturas de agentes hidrofóbicos que 30 formam uma película oclusiva na pele que reduz perda de água transepi- dérmica (TEWL) prevenindo a evoparação de água do estrato córneo.
Em algumas modalidades, o componente de agente oclusivo compreende uma ou mais substâncias selecionadas de ácidos graxos (por exemplo, ácido de lanolina), álcoois graxos (por exemplo, lanolin álcool), ó- leos de hidrocarboneto & ceras (por exemplo, petróleo), álcoois poliídricos (por exemplo, propileno glicol), silicones (por exemplo, dimeticona), esteróis (por exemplo, colesterol). Gordura vegetal ou animal (por exemplo, manteiga de cacau), cera de abelha (por exemplo, cera de carnaúba), e éster de cera (por exemplo, ceras de abelha).
Em algumas modalidades, o componente de agente oclusivo compreende uma ou mais substâncias selecionadas de ácido de lanolina álcoois graxos, lanolin álcool, petróleo, propileno glicol, dimeticona, colesterol, manteiga de cacau, cera de carnaúba, e ceras de abelha.
Em algumas modalidades, o componente de agente oclusivo compreende petróleo.
Em algumas modalidades, o componente de agente oclusivo compreende petróleo branco.
Em algumas modalidades, o componente de óleo compreende um componente de agente de endurecimento.
Em algumas modalidades, o componente de agente de endurecimento está presente em uma quantidade de cerca de 2% a cerca de 8% 20 em peso da formulação.
Como usado aqui, o termo "componente de agente de endurecimento" refere-se a uma substância ou mistura de substâncias que aumenta a viscosidade e/ou consistência da formulação ou melhora a reologia da formulação.
Em algumas modalidades, o componente de agente de endurecimento compreende uma ou mais substâncias independentemente selecio-nadas de álcoois graxos.
Em algumas modalidades, o componente de agente de endurecimento compreende uma ou mais substâncias independentemente selecio- nadas de C12-20 álcoois graxos.
Em algumas modalidades, o componente de agente de endurecimento compreende uma ou mais substâncias independentemente selecio- nadas de C16-18 álcoois graxos.
Em algumas modalidades, o componente de agente de endurecimento compreende uma ou mais substâncias índependentemente selecionadas de cetil álcool e estearil álcool.
Em algumas modalidades, o componente de óleo compreende um componente emoliente.
Em algumas modalidades, o componente emoliente está presente em uma quantidade de cerca de 5% a cerca de 15% em peso da formulação.
Como usado aqui, o termo "componente emoliente" refere-se a um agente que amacia ou suaviza a pele ou suaviza uma superfície interna irritada.
Em algumas modalidades, o componente emoliente compreende uma ou mais substâncias independentemente selecionadas de óleos mine- 15 rais e triglicerídeos.
Em algumas modalidades, o componente emoliente compreende uma ou mais substâncias independentemente selecionadas de óleo mineral leve e triglicerídeos de cadeia média.
Em algumas modalidades, o componente emoliente compreende uma ou mais substâncias independentemente selecionadas de óleo mineral leve, triglicerídeos de cadeia média, e dimeticona.
Em algumas modalidades, a água está presente em uma quantidade de cerca de 35% a cerca de 65% em peso da formulação.
Em algumas modalidades, o componente emulsificante está pre- sente em uma quantidade de cerca de 1 % a cerca de 9% em peso da formulação. |
Em algumas modalidades, o componente emulsificante compreende uma ou mais substâncias independentemente selecionadas de ésteres graxos de glicerila e ésteres graxos de sorbitano.
Em algumas modalidades, o componente emulsificante compreende uma ou mais substâncias independentemente selecionadas de estea- rato de glicerila, e polissorbato 20.
Em algumas modalidades, a formulação farmacêutica também compreende um componente de agente de estabilização.
Em algumas modalidades, o componente de agente de estabilização está presente em uma quantidade de cerca de 0,05% a cerca de 5% em peso da formulação.
Como usado aqui, o termo "componente de agente de estabilização" refere-se a uma substância ou mistura de substâncias que melhora da estabilidade da formulação farmacêutica e/ou a compatibilidade dos componentes na formulação. Em algumas modalidades, o componente de agente de estabilização impede a aglomeração da emulsão e estabiliza as gotícu- las na emulsão óleo-eM-água.
Em algumas modalidades, o componente de agente de estabilização compreende uma ou mais substâncias independentemente selecionadas de polissacarídeos.
Em algumas modalidades, o componente de agente de estabilização compreende goma xantana.
Em algumas modalidades, a formulação farmacêutica também compreende um componente de solvente.
Em algumas modalidades, o componente de solvente está presente em uma quantidade de cerca de 10% a cerca de 35% em peso da formulação.
Como usado aqui, o termo "compoente de solvente" é uma substância líquida ou mistura de substâncias líquidas capazes de dissolver um composto da invenção ou outras substâncias na formulação. Em algumas modalidades, o componente de solvente é uma substância liquida ou mistura de substâncias liquidas em que um composto da invenção, ou um sal farmaceuticamente aceitável do mesmo, tem solubilidade razoável.
Em algumas modalidades, o componente de solvente compreende uma ou mais substâncias independentemente selecionadas de alquileno glicóis e polialquileno glicóis.
Em algumas modalidades, o componente de solvente compreende uma ou mais substâncias independentemente selecionadas de propile- no glicol e polietileno glicol.
Em algumas modalidades, o composto da invenção está presente em uma quantidade de cerca de 0,5% a cerca de 2,0% em peso da formulação em uma em uma base de base livre.
Em algumas modalidades, o composto da invenção está presente em uma quantidade de cerca de 0,5% em peso da formulação em uma base de base livre.
Em algumas modalidades, o composto da invenção está presente em uma quantidade de cerca de 1% em peso da formulação em uma base de base livre.
Em algumas modalidades, o composto da invenção está presente em uma quantidade de cerca de 1,5% em peso da formulação em uma base de base livre.
Em algumas modalidades, o composto da invenção está presente em uma quantidade selecionada de cerca de 0,5, 0,6, 0,7, 0,8, 09, 1,0, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, e 2,0% em peso da formulação em uma base de base livre.
Em algumas modalidades, a formulação farmacêutica compreende: água; um componente de óleo; um compoente emulsificante; um componente de solvente; um componente de agente de estabilização; e de cerca de 0,5% a cerca de 2,0% de um composto da invenção, ou um sal farmaceuticamente aceitável do mesmo, por peso da formulação em uma base de base livre.
Em algumas modalidades, a formulação farmacêutica compreende: de cerca de 35% a cerca de 65% de água por peso da formulação; de cerca de 10% a cerca de 40% de um componente de óleo por peso da formulação; de cerca de 1% a cerca de 9% de um compoente emulsificante por peso da formulação; de cerca de 10% a cerca de 35% de um componente de solven- te por peso da formulação; de cerca de 0,05% a cerca de 5% de um componente de agente de estabilização por peso da formulação; e de cerca de 0,5% a cerca de 2,0% de um composto da invenção, ou um sal farmaceuticamente aceitável do mesmo, por peso da formulação em uma base de base livre.
Em algumas modalidades: o componente de óleo compreende uma ou mais substâncias independentemente selecionadas de petróleos, álcoois graxos, óleos minerais, triglicerídeos, e dimeticonas; o componente emulsificante compreende uma ou mais substâncias independentemente selecionadas de ésteres graxos de glicerila e ésteres graxos de sorbitano; o componente de solvente compreende uma ou mais substâncias independentemente selecionadas de alquileno glicóis e polialquileno gli- cóis; e o componente de agente de estabilização compreende uma ou mais substâncias independentemente selecionadas de polissacarídeos.
Em algumas modalidades: o componente de óleo compreende uma ou mais substâncias independentemente selecionadas de petróleo branco, cetil álcool, estearil álcool, óleo mineral leve, triglicerídeos de cadeia média, e dimeticona; o componente emulsificante compreende uma ou mais substâncias independentemente selecionadas de estearato de glicerila e polissorba- to 20; o componente de solvente compreende uma ou mais substâncias independentemente selecionadas de propileno glicol e polietileno glicol; e o componente de agente de estabilização compreende goma xantana.
Em algumas modalidades, a formulação farmacêutica compre- ende: de cerca de 35% a cerca de 65% de água por peso da formulação; de cerca de 2% a cerca de 15% de um componente agente o- clusivopor peso da formulação; de cerca de 2% a cerca de 8% de um componente de agente de endurecimento por peso da formulação; de cerca de 5% a cerca de 15% de um componente emoliente por peso da formulação; de cerca de 1% a cerca de 9% de um compoente emulsificante por peso da formulação; de cerca de 0,05% a cerca de 5% de um componente de agente de estabilização por peso da formulação; de cerca de 10% a cerca de 35% de um componente de solvente por peso da formulação; e de cerca de 0,5% a cerca de 2,0% de um composto da invenção, ou um sal farmaceuticamente aceitável do mesmo, por peso da formulação em uma base de base livre.
Em algumas modalidades: o componente de agente oclusivo compreende a petróleo; o componente de agente de endurecimento compreende uma ou mais substâncias independentemente selecionadas de um ou mais álcoois graxos; o componente emoliente compreende uma ou mais substâncias independentemente selecionadas de óleos minerais e triglicerideos; o componente emulsificante compreende uma ou mais substâncias independentemente selecionadas de ésteres graxos de glicerila e ésteres graxos de sorbitano; o componente de agente de estabilização compreende uma ou mais substâncias independentemente selecionadas de polissacarídeos; e o componente de solvente compreende uma ou mais substâncias independentemente selecionadas de alquileno glicóis e polialquileno gli- cóis.
Em algumas modalidades: o componente de agente oclusivo compreende petróleo branco; o componente de agente de endurecimento compreende uma ou mais substâncias independentemente selecionadas de cetil álcool e estearil álcool; o componente emoliente compreende uma ou mais substâncias independentemente selecionadas de óleo mineral leve, triglicerídeos de cadeia média, e dimeticona; o componente emulsificante compreende uma ou mais substân- cias independentemente selecionadas de estearato de glicerila e polissorba- to 20; o componente de agente de estabilização compreende goma xantana; e o componente de solvente compreende uma ou mais substân- cias independentemente selecionadas de propileno glicol e polietileno glicol.
Em algumas modalidades, a formulação farmacêutica também compreende um componente conservante antimicrobiano.
Em algumas modalidades, o componente conservante antimicrobiano está presente em uma quantidade de cerca de 0,05% a cerca de 20 3% em peso da formulação.
Como usado aqui, a frase "componente conservante antimicrobiano" é uma substância ou misturas de substâncias que inibem o desenvolvimento microbiano na formulação.
Em algumas modalidades, o componente conservante antimicro- biano compreende uma ou mais substâncias independentemente selecionadas de alquil parabenos e fenoxietanol.
Em algumas modalidades, o componente conservante antimicrobiano compreende uma ou mais substâncias independentemente selecionadas de metil parabeno, propil parabeno, e fenoxietanol.
Em algumas modalidades, a formulação farmacêutica também compreende um componente de agente de quelação.
Como usado aqui, a frase "componente de agente de quelação" refere-se a um composto ou misturas de compostos que têm a capacidade de ligar-se fortemente com íons de metal.
Em algumas modalidades, o componente de agente de quelação compreende dissódio de edetato.
Como usado aqui, "% em peso da formulação" significa que o percentual de concentração do componente da formulação é com base em peso/peso. Por exemplo, 1 % de peso/peso de componente A = [(massa de componente A) / (massa total da formulação)] x 100.
Como usado aqui, "% em peso da formulação em uma base de 10 base livre" de um composto da invenção, ou um sal farmaceuticamente aceitável do mesmo" significa que a % de peso/peso é calculada com base no peso da base livre do composto da invenção na formulação total.
Em algumas modalidades, os componentes estão presentes e- xatamente nas faixas específicas (por exemplo, o termo "cerca de" não está 15 presente). Em algumas modalidades, "cerca de" significa mais ou menos 10% do valor.
Como será apreciado, alguns componentes das formulações farmacêuticas descrito aqui podem possuir múltiplas funções. Por exemplo, uma determinada substância pode agir tanto como um componente emulsifi- 20 cante quanto um agente de estabilização. Em alguns tais casos, a função de um determinado componente pode ser considerada singular, mesmo se suas propriedades puderem permitir funcionalidade múltipla. Em algumas modalidades, cada componente da formulação compreende uma diferente substância ou mistura de substâncias.
Como usado aqui, o termo "componente" pode significar uma substância ou uma mistura de substâncias.
Como usado aqui, o termo "ácido graxo" refere-se a um ácido alifático que é saturado ou insaturado. Em algumas modalidades, o ácido graxo é uma mistura de ácidos graxos diferentes. Em algumas modalidades, 30 o ácido graxo tem entre cerca de oito a cerca de trinta carbonos em média.
Em algumas modalidades, o ácido graxo tem cerca de 12 a 20, 14 a 20, ou 16 a 18 carbonos em média. Ácidos graxos incluem, porém não estão limita- dos a ácido de cetila, ácido esteárico, ácido laúrico, ácido mirístico, ácido erúcico, ácido palmítico, ácido palmitoléico, ácido cáprico, ácido caprílico, ácido oléico, ácido linoléico, ácido linolênico, ácido hidroxisteárico, ácido 12- hidroxisteárico, ácido cetosteárico, ácido isosteárico, ácido sesquioléico, áci- 5 do sesqui-9-octadecanoico, ácido sesquiisooctadecanoico, ácido beênico, ácido isobeênico, e ácido araquidônico, ou misturas dos mesmos.
Como usado aqui, o termo "álcool graxo" refere-se a um álcool alifático que é saturado ou insaturado. Em algumas modalidades, o álcool graxo é uma mistura de diferentes álcoois graxos. Em algumas modalidades, 10 o álcool graxo tem entre cerca de 12 a cerca de 20, cerca de 14 a cerca de 20, ou cerca de 16 a cerca de 18 carbonos em média. Álcoois graxos adequados incluem, porém não estão limitados a estearil álcool, lauril álcool, palmitil álcool, cetil álcool, capril álcool, caprilil álcool, oleil álcool, linolenil álcool, álcool araquidônico, beenil álcool, isobeenil álcool, selaquil álcool, qui- 15 mil álcool, e linoleil álcool, ou misturas dos mesmos.
Como usado aqui, o termo "polialquileno glicol", empregado sozinho ou em combinação com outros termos, refere-se a um polímero contendo unidades monômeras de oxialquileno, ou copolímero de diferentes unidades monômeras de oxialquileno, em que o grupo alquileno tem de 2 a 20 6, 2 a 4, ou 2 a 3 átomos de carbono. Como usado aqui, o termo "oxialquileno", empregado sozinho ou em combinação com outros termos, refere-se a um grupo de fórmula -O-alquileno-. Em algumas modalidades, o polialquileno glicol é polietileno glicol.
Como usado aqui, o termo, "éster graxo de sorbitano" inclui pro- 25 dutos derivados de unidades de sorbitano ou sorbitol e ácidos graxos e, opcionalmente, poli(etileno glicol), incluindo ésteres de sorbitano e ésteres de sorbitano polietoxilados. Em algumas modalidades, o éster graxo de sorbitano é um éster de sorbitano polietoxilado.
Como usado aqui, o termo "éster de sorbitano" refere-se a um 30 composto, ou mistura de compostos, derivado da esterificação de sorbitol e pelo menos um ácido graxo. Ácidos graxos úteis para derivação dos ésteres de sorbitano incluem, porém não estão limitados àqueles descritos aqui. És- teres de sorbitano adequados incluem, porém não estão limitados às séries de Span™ (disponíveis de Uniqema), que incluem Span 20 (monolaurato de sorbitano), 40 (monopalmitato de sorbitano), 60 (monostearato de sorbitano), 65 (tristearato de sorbitano), 80 (monooleato de sorbitano), e 85 (trioleato de sorbitano). Outros ésteres de sorbitano adequados incluem aqueles listados em R. C. Rowe e P. J. Shesky, Handbook of pharmaceutical excipients, (2006), 5a edição, que é incorporado aqui por referência em sua totalidade.
Como usado aqui, o termo "éster de sorbitano polietoxilado" refere-se a um composto, ou mistura do mesmo, derivado da etoxilação de um éster de sorbitano. A porção polioxetileno do composto pode ser entre a porção éster graxo e sorbitano. Como usado aqui, o termo "éster de sorbitano" refere-se a um composto, ou mistura de compostos, derivado da esterifica- ção de sorbitol e pelo menos one ácido graxo. Ácidos graxos úteis para derivação dos ésteres de sorbitano polietoxilados esters incluem, porém não estão limitados àqueles descritos aqui. Em algumas modalidades, a porção polioxietileno do composto ou mistura tem cerca de 2 a cerca de 200 unidades de oxietileno. Em algumas modalidades, a porção polioxietileno do composto ou mistura tem cerca de 2 a cerca de 100 unidades de oxietileno. Em algumas modalidades, a porção polioxietileno do composto ou mistura tem cerca de 4 a cerca de 80 unidades de oxietileno. Em algumas modalidades, a porção polioxietileno do composto ou mistura tem cerca de 4 a cerca de 40 unidades de oxietileno. Em algumas modalidades, a porção polioxietileno do composto ou mistura tem cerca de 4 a cerca de 20 unidades de oxietileno. Ésteres de sorbitano polietoxilados adequados incluem, porém não estão limitados às séries de Tween™ (disponíveis de Uniqema), que incluem Tween 20 (POE(20) monolaurato de sorbitano), 21 (POE(4) monolaurato de sorbitano), 40 (POE(20) monopalmitato de sorbitano), 60 (POE(20) monoestea- rato de sorbitano), 60K (POE(20) monoestearato de sorbitano), 61 (POE(4) monoestearato de sorbitano), 65 (POE(20) tristearato de sorbitano), 80 (PO- E(20) monooleato de sorbitano), 80K (POE(20) monooleato de sorbitano), 81 (POE(5) monooleato de sorbitano), e 85 (POE(20) trioleato de sorbitano). Como usado aqui, a abreviação "POE" refere-se ao polioxietileno. O número seguindo a abreviação de POE refere-se ao número de unidades de repetição de oxietileno no composto. Outros ésteres de sorbitano polietoxilados incluem os ésteres de ácido graxo de polioxietileno sorbitano listados em R. C. Rowe e P. J. Shesky, Handbook of pharmaceutical excipients, (2006), 5a edição, que é incorporado aqui por referência em sua totalidade. Em algumas modalidades, o éster de sorbitano polietoxilado é a polissorbato. Em algumas modalidades, o éster de sorbitano polietoxilado é polissorbato 20.
Como usado aqui, o termo "ésteres graxos de glicerila" refere-se a mono-, di- ou triglicerídeos de ácidos graxos. Os ésteres graxos de glicerila podem ser opcionalmente substituídos com grupos ácidos sulfônicos, ou sais farmaceuticamente aceitáveis dos mesmos. Ácidos graxos adequados para derivação de glicerídeos de ácidos graxos incluem, porém não estão limitados àqueles descritos aqui. Em algumas modalidades, o éster graxo de glicerila é um mono-glicerídeo de um ácido graxo tendo 12 a 18 átomos de carbono. Em algumas modalidades, o éster graxo de glicerila é estearato de glicerila.
Como usado aqui, o termo "triglicerídeos" refere-se a um triglice- rídeo de um ácido graxo. Em algumas modalidades, o triglicerídeo é triglicerídeos de cadeia média.
Como usado aqui, o termo "alquileno glicol" refere-se a um grupo de fórmula -O-alquileno-, em que o grupo alquileno tem 2 a 6, 2 a 4, ou 2 a 3 átomos de carbono. Em algumas modalidades, o alquileno glicol é propi- leno glicol (1,2-propanodiol).
Como usado aqui, o termo "polietileno glicol" refere-se a um polímero contendo unidades monômeras de etileno glicol de fórmula -O-CH2- CH2-. Polietileno glicóis adequados podem ter um grupo hidroxila livre em cada extremidade da molécula polímera, ou podem ter um ou mais grupos hidroxila eterificados com uma alquila inferior, por exemplo, um grupo metila. São também adequados derivados de polietileno glicóis tendo grupos carbó- xi esterificáveis. Polietileno glicóis úteis na presente invenção podem ser polímeros de qualquer comprimento de cadeia ou peso molecular, e podem incluir ramificação. Em algumas modalidades, o peso molecular médio do polietileno glicol é de cerca de 200 a cerca de 9000. Em algumas modalidades, o peso molecular médio do polietileno glicol é de cerca de 200 a cerca de 5000. Em algumas modalidades, o peso molecular médio do polietileno glicol é de cerca de 200 a cerca de 900. Em algumas modalidades, o peso molecular médio do polietileno glicol é cerca de 400. Polietileno glicóis incluem, porém não estão limitados a polietileno glicol-200, polietileno glicol-300, polietileno glicol-400, polietileno glicol-600, e polietileno glicol-900. O número seguindo o nome refere-se ao peso molecular médio do polímero.
As formulações de creme oleo-eM-água podem ser sintetizadas de acordo com o uso de um misturador aéreo com lâminas de mistura de alto e baixo cisalhamento. Por exemplo, em algumas modalidades, a formulação pode ser sintetizada pelo seguinte procedimento. 1. Uma fase conservante antimicrobiana pode ser preparada misturando pelo menos uma porção do componente conservante antimicrobiano com uma porção de um componente de solvente. 2. A seguir, uma fase de agente de estabilização é preparada misturando um componente de agente de estabilização com uma porção do componente de solvente. 3. Uma fase de óleo é em seguida preparada misturando um componente emoliente, um compoente emulsificante, um componente oclusivo, e um componente de agente de endurecimento. A fase de óleo é aquecida para 70 a 80°C para fundir e formar uma mistura uniforme. 4. Uma fase aquosa é em seguida preparada misturando água purificada, o restante do componente de solvente, e um componente de a- gente de quelação. A fase é aquecida para 70 a 80°C. 5. A fase aquosa de etapa 4, fase conservante antimicrobiana de etapa 1, e o composto da invenção, ou um sal farmaceuticamente aceitável do mesmo, são combinados para formar uma mistura. 6. A fase de agente de estabilização de etapa 2 foi em seguida adicionada à mistura de etapa 5. 7. A fase de óleo de etapa 3 é em seguida combinada sob mistura de cisalhamento elevado com a mistura da etapa 6 para formar uma e mulsão. 8. Finalmente, o componente conservante antimicrobiano pode ser em seguida adicionado á emulsão de etapa 7. A mistura é continuada, e em seguida o produto é resfriado sob misturade de baixo cisalhamento.
Síntese
Os compostos da invenção, incluindo sais e N-óxidos dos mesmos, podem ser preparados usando técnicas de síntese orgânica e podem ser sintetizadas de acordo com qualquer uma das numerosas possíveis rotinas sintéticas.
As reações para a preparação de compostos da invenção podem ser realizadas em solventes adequados que podem ser facilmente selecionados por alguém versado na técnica de síntese orgânica. Solventes a- dequados podem ser substancialmente não reativos com os materiais de partida (reagentes), os intermediários, ou produtos nas temperaturas em que as reações são realizadas, por exemplo, temperaturas que podem variar da temperatura de congelamento de solvente à temperatura de ebulição de solvente. Uma determinada reação pode ser realizada em um solvente ou uma mistura de mais do que um solvente. Dependendo da etapa de reação particular, solventes adequados para uma etapa de reação particular podem ser selecionados pelo técnico versado.
A preparação de compostos da invenção pode envolver a proteção e desproteção de vários grupos químicos. A necessidade de proteção e desproteção, e da seleção de grupos de proteção apropriados, podem ser facilmente determinadas por alguém versado na técnica. A química de grupos de proteção pode ser encontrada, por exemplo, em Wuts e Greene, Protective Groups in Organic Synthesis, 4a edição, John Wiley & Sons: New Jersey,(2007), que é incorporado aqui por referência em sua totalidade.
As reações podem ser monitoradas de acordo com qualquer método conhecido na técnica. Por exemplo, a formação de produto pode ser monitorada por métodos espectroscópicos, tais como espectroscopia de ressonância magnética nuclear (por exemplo, 1H ou 13C), espectroscopia de infravermelho, espectrofotometria (por exemplo, UV visível), espectrometria de massa, ou por métodos cromatográficos tais como cromatografia líquida de desempenho elevado (HPLC) ou cromatografia de camada fina (TLC).
Compostos de fórmula I, em que X é ciano, podem ser feitos por métodos análogos àqueles descritos no Esquema I. Consequentemente, 5 uma pirazol-4-il-pirrolo[2,3-d]pirimidina protegida ou pirrol-3-il-pirrolo[2,3- d]pirimidina de fórmula (a) é reagida com um alcano protegido de fórmula (b) em uma dição de Michael na presença de um agente de acoplamento. Os grupos de proteção, P1 e R, podem ser qualquer grupo de proteção apropri-ado, incluindo, porém não limitado aos grupos de proteção para aminas deli- neadas em Wuts e Greene, Protective Groups in Organic Synthesis, 4a edição, John Wiley & Sons: New Jersey, páginas 696 a 887 (e em particular, páginas 872 a 887) (2007), que é incorporado aqui por referência em sua totalidade. Em algumas modalidades, P1 é 2-(trimetilsilil)etoximetila (SEM). Em algumas modalidades, o grupo de proteção de R é um que pode ser se- letivamente removido na presença do grupo de proteção de P1. Em algumas modalidades, o grupo de proteção de R é t-butoxicarbonila (BOC) ou benziloxicarbonila (Cbz). O agente de acoplamento pode ser qualquer agente de acoplamento apropriado útil para adição de Michael, incluindo, porém não limitado a um haleto de tetraalquilamônio, hidróxido de tetraalquilamônio, 20 guanidina, amidina, hidróxido, alcóxido, silicato, fosfato de metal de álcali, óxido, amina terciária, carbonato de metal de álcali, bicarbonato de metal de álcali, fosfato de hidrogênio de metal de álcali, fosfina, ou sal de metal de álcali de um ácido carboxílico. Em algumas modalidades, o agente de acoplamento é tetrametil guanidina, 1,8-diazabiciclo[5,4.0]undec-7-eno, 1,5-dia- zabiciclo(4,3.0)non-5-eno, 1,4-diazabicíclo(2,2.2)octano, hidróxido de amónio de terc-butila, hidróxido de sódio, hidróxido de potássio, metóxido de sódio, etóxido de sódio, fosfato de tripotássio, silicato de sódio, óxido de cálcio, trie- tilamina, carbonato de sódio, carbonato de potássio, bicarbonato de sódio, bicarbonato de potássio, fosfato de hidrogênio de potássio, trifenil fosfina, trietil fosfina, acetato de potássio, ou acrilato de potássio. Em algumas modalidades, o agente de acoplamento é 1,8-diazabiciclo[5,4,0]undec-7-eno (DBU). A adição Michael dos compostos de fórmulas (a) e (b) pode ser con- duzida em um solvente apropriado (por exemplo, acetonitrila).Esquema I

O produto de adição Michael (c) pode em seguida ser desprote- gido para remover o grupo de proteção de R para formar a base pirrolidina (d). Por exemplo, quando R é BOC, o grupo de proteção pode ser removido através de tratamento com HCI em dioxano, ao mesmo tempo que quando R é Cbz, o grupo de proteção pode ser removido sob condições de hidrogena- ção (por exemplo, gás de hidrogênio na presença de 10% de paládio sobre carbono). A base pirrolidina (d) pode em seguida ser reagida com uma porção aromática de fórmula Ar-X1 para formar a aril-pirrolidina de fórmula (e). Grupos de saída apropriados para X1 incluem, porém não estão limitados a cloro, bromo, flúor, -OSO2CF3, e tio (SH). A reação pode ser realizada na presença de uma base (tal como uma amina terciária, por exemplo, di- isopropiletilamina) em um solvente tal como N-metilpirrolidona (NMP), dioxano, ou etanol (EtOH) em uma temperatura elevada (por exemplo, 60 a 135°C). O composto de fórmula (e) pode em seguida ser desprotegido para fornecer o composto de fórmula I.
Compostos de fórmula (a) podem ser formados por métodos a- nálogos àqueles descritos no esquema II. Consequentemente, acoplamento Suzuki de uma 4-cloro-pirrolo[2,3-d]pirimidina protegida de fórmula (f) (em que R" é hidrogênio, alquila ou dois R" ligaM-se juntamente com os átomos de oxigénio e boro para formar um anel heterocicloalquila opcionalmente substituída tal como um anel pinacol) com um ácido borônico de pirrol-3-ila ou pirazol-4-ila protegido ou não protegido de fórmula (g) (por exemplo, em que R' é H ou um grupo de proteção) na presença de um catalisador de paládio (por exemplo, tetracis(trifenilfosfína)paládio(0) ou tetracis(tri(o-tolilfosfi- na))paládio(0)) e uma base (por exemplo, carbonato de potássio) fornece o material de partida desejado de fórmula (a) (veja, por exemplo, Exemplo 65 de US 20070135461, que é incorporado aqui por referência em sua totalidade). O éster ou ácido borônico de pirrol-3-ila ou pirazol-4-ila pode ser protegido com qualquer grupo de proteção apropriado. Similarmente, o grupo de proteção P1 pode ser qualquer grupo de proteção apropriado (por exemplo, dietoximetila (DEM) ou 2-(trimetilsilil)etoximetila (SEM)).Esquema II

O alqueno de fórmula (b) pode ser formado por reação do aldeído de pirrolidina de fórmula (h) com um reagente Horner-Wadsworth- Emmons como mostrado no esquema III. O grupo de proteção de R pode ser qualquer um dos grupos de proteção sumariados acima (por exemplo, BOC ou Cbz). Esquema III
Os compostos de Ar-X1 podem ser formados por métodos conhecidos na técnica. Compostos em que Ar é um grupo heterocicloalquil(He- tero)arila fundido com uma porção tioéter de S-oxo ou S,S-dioxo (v) ou (vi) pode ser formado por métodos análogos àqueles mostrados no esquema IV. Consequentemente, uma porção dicloro(hetero)arilaldeído apropriada (por exemplo, um composto de fórmula (i)) é reagido com um reagente Wittig para fornecer uma porção β-metoxialquenila (por exemplo, um composto de fórmula (ii)), que pode em seguida ser hidrolisada e reduzida para fornecer uma porção β-hidroxietano (por exemplo, um composto de fórmula (iii)). O 5 grupo hidroxila na porção β-hidroxietano pode em seguida ser convertido em um grupo tio (por exemplo, um composto de fórmula (iv) e ciclizado para formar um tioéter. O tioéter pode em seguida ser oxidado usando um agente oxidante apropriado (por exemplo, ácido M-cloroperbenzoico, mCPBA) para fornecer o S-oxotioéter (por exemplo, um composto de fórmula (v)) e o S,S- dioxotioéter (por exemplo, um composto de fórmula (vi)). Esquema IV

Certos compostos de fórmula Ar-X1 em que Ar é um compost de heteroarila ou ariloxazol pode ser formado por métodos análogos àqueles mostrados no esquema V. Consequentemente uma porção 1-nitro-2-hidroxi- larila ou heteroarila de fórmula (i) é reduzida para fornecer uma porção 1- amino-2-hidroxilarila ou heteroarila de fórmula (ii), que pode ser ciclizada para fornecer o composto de benzo[d]oxazol-2(3H)-tiona de fórmula (iii). Certos compostos de fórmula (iii) podem em seguidos ser reagidos para fornecer o composto de oxazol fundido por 2-cloro de fórmula (iv).Esquema V
Em alguns casos, o composto de fórmula (iii) pode ser reagido diretamente com o composto de fórmula (d) de esquema I por métodos análogos àqueles descritos no esquema VI. Consequentemente, o composto de fórmula (d) é reagido com o composto de fórmula (iii) na presença de uma base tai como uma amina terciária (por exemplo, di-isopropiletilamina) e um solvente (por exemplo, dioxano) em uma temperatura elevada (por exemplo, 70°C), seguido por tratamento com nitrato de prata na presença de hidróxido de amónio e etanol e desproteção para formar o composto desejado. Esquema VI
Altemativamente, compostos de fórmula Ar-X1 em que Ar é um composto de heteroarila ou aril-oxazol fundido pode ser formado por métodos análogos àqueles mostrados no esquema VII. Consequentemente, o 10 composto de fórmula (i) é reagido com clorotionocarbonato de fenila e piridi- na, seguido por reação com a estrutura de núcleo de pirrolidina de fórmula (ii) para fornecer o composto de fórmula (iii). O composto de fórmula (iii) pode em seguida ser ciclizado e desprotegido para fornecer o composto desejado de fórmula (iv). Esquema VII
Composto de fórmula I, em que X é flúor, pode ser feito pelos métodos mostrados no esquema VIII. Consequentemente, uma 3-carboxi- pirrolidina protegida (R é um grupo de proteção) de fórmula 1 é reduzida para fornecer um derivado de metilol de fórmula 2. O derivado de metilol pode em seguida ser oxidado por meio de uma procedimento de oxidação Swern para fornecer o aldeido de fórmula 3. O aldeído pode em seguida ser reagido com sulfona de flurometilfenila na presença de di-isopropilamida de lítio (LDA) para fornecer o composto de fórmula 4. O composto de fórmula 4 pode em seguida ser reagido para remover o grupo -SO2Ph para fornecer o composto de hidroxila de fórmula 5. O grupo hidroxila do composto de fórmula 5 pode ser convertido em um grupo mesilato (composto de fórmula 6), que pode em seguida ser reagida com o composto de pirrolo[2,3-d]pirimidina para fornecer o composto de fórmula 7. O composto de fórmula 7 pode em seguida ser desprotegido para fornecer a pirrolidina base de fórmula 8. O composto de fórmula 8 pode em seguida ser substituída pelo composto de fórmula (d) no esquema I, o composto de fórmula (d) no esquema VI, ou o composto de fórmula (ii) no esquema VII para fornecer o composto desejado de fórmula I. Alternativamente, o composto de hidroxila de fórmula 5 de Esquema VIII pode ser formado pelo processo mostrado no esquema IX. Esquema VIII


Outras rotinas nos compostos de fórmula I sao mencionadas nos Exemplos. Os exemplos destinaM-se a descrever a invenção em maiores detalhes. Os exemplos são oferecidos para propósitos ilustrativos, e não se destinam a limitar a invenção de modo algum. Aqueles versados na técnica facilmente reconhecerá uma variedade de parâmetros não críticos que podem ser mudados ou modificados para produzir essencialmente os mesmos resultados.
EXEMPLOS
As seguintes abreviações são usadas por todo o texto: NMP (N- metilpirrolidona), EtOH (etanol), HOAc (ácido acético), TFA (ácido trifluoroa- cético), DCM (cloreto de metileno ou diclorometano), MeOH (metanol), EDA (etilenodiamina), DIPEA (N,N-di-isopropiletilamina), LDA (di-isopropilamida de litio), MsCI (cloreto de mesila), DMF (N.N-dimetilformamida), HATU (he- xafluorofosfato de 3-óxido de 1-[Bis(dimetilamino)metileno]-1H-1,2,3-triazo- lo[4,5-b]piridínio), THF (tetraidrofurano), LCMS (cromatografia liquida -es- pectrometria de massa), MS (espectrometria de massa), HPLC (cromatografia de líquida de desempenho elevado), LC (cromatografia líquida), TMS (trimetilsilil), MeCN ou ACN (acetonitrila), IPA (isopropanol), EtOAc (acetato de etila), DMSO (dimetilsulfóxido), tBu (terc-butila), SEM (2-(trimetilsiíil)eto- ximetila), h (hora ou horas), e minuto (minuto ou minutos). Exemplo 1. 3-í1-(6-cloropirazin-2-il)pirrolidin-3-ill-3-[4-(7H-pirrolo[2,3-d1pirimi- din-4-il)-1 H-pirazol-1-illpropanomtrila (racemato de um único diastereõmero)

Etapa 1. 3-{2-cíano-1-[4-(7-{[2-(tnmetilsilil)etôxi]metil}-7H-pirrolo[2,3-d]pirími- din-4-il) ~ 1 H-pirazol-1 -HJetilJpirrolidina-1 -carboxilato de benzi la 3-[2-cianovinil]pirrolidina-1-carboxilato de benzila (4,3 g, 0,017 mol, mistura de isómeros E e Z preparada como descrito no WO 2007/070514 Ex.742) foi dissolvido em acetonitrila (270 mL). 1,8-diaza- biciclo[5,4,0]undec-7-eno (5,02 mL, 0,0336 mol) foi adicionado, seguido por 4-(1H-pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidina (5,6 g, 0,017 mol, preparado como descrito no WO 2007/070514, Ex. 65). A mistura foi agitada em temperatura ambiente durante a noite. O solvente foi removido por evaporação giratória, e o resíduo foi redissolvido em acetato de etila. A solução foi lavada sucessivamente com HCI a 1N, água, bicarbonato de sódio saturado, e salmoura, secada sobre sulfato de sódio e concentrada em vácuo. O produto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 0 a 100% de acetato de etila em he- xanos para fornecer diastereômero 1 (primeiro a eluir) (3,5 g, 36%) e diaste- 5 reômero 2 (segundo a eluir) (2,5 g, 25%). LCMS (M+H)+: 572,2. Etapa 2. 3-pirrolidin-3-il-3-[4-(7-{[2-(trímetilsilil)etóxi]metilj-7H-pirrolo[2,3-d] pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila 3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d] pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidina-1-carboxilato de benzila (3,5 g, 6,1 10 mmol) (diastereômero 1 de exemplo 1, etapa 1) foi dissolvido em 100 ml_ de metanol, e uma quantidade catalítica de 10% de Pd-C foi adicionada. A mis-tura foi agitada sob 3,51 kg/cm2 de hidrogênio durante 24 horas. A mistura foi em seguida filtrada e o solvente removido em vácuo. O produto foi usado sem outra purificação. LCMS (M+H)+: 438,2. Etapa 3. 3-[1-(6-cloropirazin-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi] metil}- 7H-pirrolo[2,3-d]pirimidin-4-il) -1 H-pirazol-1 -iljpropanonitrila
A mistura de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il]propanonitrila (150 mg, 0,27 mmol) e 2,6-dicloropirazina (49,0 mg, 0,329 mmol) em etanol (1,2 mL), e 20 N,N-di-isopropiletilamina (96 μL, 0,55 mmol) foi aquecida em um frasco selado em um banho de óleo mantido a 85°C durante uma hora. Cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 0 a 100% de acetato de etila em hexanos forneceu o produto (49 mg, 27%). LCMS (M+H)+: 550,0. Etapa 4. 3-[1-(6-cloropirazin-2-il)pirrolidin-3-ilj-3-[4-(7H-pirrolo[2,3-djpirimidin- 4-il)-1 H-pirazol-1-iljpropanonitrila 3-[1-(6-cloropirazin-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi] metil}-7H-pirrolo[2,3-d]pirimidin-4-ii)-1 H-pirazol-1-il]propanonitrila (15 mg, 0,027 mmol) foi dissolvida em DCM (1 mL), e 0,2 mL de TFAfoi adicionada.
A mistura foi agitada durante 2 horas, em seguida concentrada. O resíduo foi dissolvido em MeOH (1 mL), e 0,2 mL de EDA foram adicionados. Purificação por HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto (7 mg, 61%). 1H RMN (400 MHz, CD3OD): δ 8,66 (s, 2H), 8,41 (s, 1H), 7,77 (s, 1H), 7,67 (s, 1H), 7,50 (d, 1H), 6,93 (d, 1H), 4,85-4,76 (m, 1H), 3,86 (dd, 1H), 3,61-3,54 (m, 1H), 3,43-3,16 (m, 4H), 3,11-2,99 (m, 1H), 1,89-1,81 (m, 2H); LCMS (M+H)+: 420,0. Exemplo 2. 3-f1-(6-cloropiridin-2-il)pirrolidin-3-ill-3-[4-(7H-pirrolo[2,3-d1 pirimi- din-4-il)-1 H-pirazol-1-illpropanonitrila (dois diferentes enantiômeros isolados)
Etapa 1. 3-[ 1-(6-cloropindin-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(tnmetilsilil)etóxi] metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il]propanonitríla
A mistura de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (150 mg, 0,27 mmol, de exemplo 1, etapa 2) e 2,6-dicloropiridina (48,7 mg, 0,329 mmol) em NMP (1,6 ml_) e N,N-di-isopropiletilamina (96 microL, 0,55 mmol) foi aquecida para 135°C durante 20 minutos no micro-ondas. Purificação por cromato- grafia de coluna rápida em sílica-gel, eluindo com um gradiente de 0 a 80% de acetato de etila em hexanos, forneceu o produto título (28 mg, 18%). 1H RMN (400 MHz, CDCI3): δ 8,85 (s, 1H), 8,36 (s, 1H), 8,36 (s, 1H), 7,41 (d, 1H), 7,37 (dd, 1H), 6,79 (d, 1H), 6,57 (d, 1H), 6,22 (d, 1H), 5,68 (s, 2H), 4,45 (dt, 1H), 3,91 (dd, 1H), 3,57-3,46 (m, 3H), 3,39-3,29 (m, 2H), 3,24 (dd, 1H), 3,13-3,01 (m, 1H), 3,01 (dd, 1H), 1,98-1,88 (m, 1H), 1,82-1,69 (m, 1H), 0,95- 0,88 (m, 2H), -0,06 (s, 9H); LCMS (M+H)+: 549,1.
Este produto racêmico foi separado em seus enantiômeros por HPLC quiral (Chiral Technologies Chiralcel OJ-H, 5 μ, 30 x 250 mm, 45% de EtOH/Hexanos, 20 mL/minuto) para fornecer enantiômero 1 (primeiro a eluir, tempo de retenção de 40,7 minutos) e enantiômero 2 (segundo a eluir, tempo de retenção de 51,6 minutos), que foram desprotegidos separadamente na Etapa 2. Etapa 2a. 3-[1-(6-cloropirídin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d] pirimi- din-4-il)-1 H-pirazol-1-iljpropanonitrila (enantiômero 1) 3-[1-(6-cloropiridin-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi] metil}-7H-pirrolo[2,3-d]pirimídrn-4-il)-1 H-pirazol-1-iljpropanonitrila (enantiôme-ro 1 de etapa 1) foi agitado em uma solução contendo 1:1 de TFA/DCM (2 mL) durante 2 horas, e em seguida concentrada. O resíduo foi dissolvido em 1 mL de MeOH, e 0,2 mL de EDA foi adicionado. Purificação por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto. 1H RMN (400 MHz, CD- CI3): δ 9,44 (br s, 1H), 8,84 (s, 1H), 8,37 (s, 2H), 7,39 (dd, 1H), 7,38 (dd, 1H), 6,79 (dd, 1H), 6,58 (d, 1H), 6,22 (d, 1H), 4,46 (dt, 1H), 3,92 (dd, 1H), 3,55- 3,48 (m, 1H), 3,39-3,31 (m, 2H), 3,25 (dd, 1H), 3,13-3,02 (m, 1H), 3,02 (dd, 1H), 2,00-1,88 (m, 1H), 1,84-1,71 (m, 1H); LCMS (M+H)+: 419,1. Etapa 2b. 3-[1-(6-cloropindin-2-il)pirrolidin-3-ilJ-3-[4-(7H-pirrolo[2,3-d] pirimi- din-4-il)-1 H-pirazol-1 -iljpropanonitrila (enantiômero 2)
Realizada como no etapa 2a, usando enantiômero 2 de etapa 1: 1H RMN (400 MHz, CDCI3): δ 9,59 (brs, 1H), 8,84 (s, 1H), 8,37 (s, 2H), 7,40 (dd, 1H), 7,38 (dd, 1H), 6,79 (dd, 1H), 6,58 (d, 1H), 6,22 (d, 1H), 4,46 (dt, 1H), 3,92 (dd, 1H), 3,55-3,48 (m, 1H), 3,39-3,31 (m, 2H), 3,25 (dd, 1H), 3,14- 3,02 (m, 1H), 3,02 (dd, 1H), 1,99-1,90 (m, 1H), 1,83-1,72 (m, 1H); LCMS (M+H)+: 419,1. Exemplo 3. 3-[1-(2-cloropinmidin-4-il)pirrolidin-3-il1-3-(4-(7H-pirrolo[2,3-dl pi- rimidin-4-il)-1H-pirazol-1-il]propanonitrila (dois diferentes enantiômeros isola-dos)
Etapa 1. 3-[1 -(2-cloropirimidin-4-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi] metil}-7H-pirrolo[2,3-dJpirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila e 3-( 1 -(4- cloropirimidin-2-il)pirrolidin-3-il)-3-(4-(7-((2-(trimetilsilil)etóxi)metil)-7H-pirrolo [2,3-d]pinmidin-4-il)-1 H-pirazol-1-il)propanonitnla
A mistura de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxÍ]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila (181 mg, 0,331 mmol, preparada no exemplo 1, etapa 2) e 2,4-dicloropirimidina (59 mg, 0,40 mmol) em 1,4-díoxano (1,1 mL) e N,N-di-isopropiletilamina (115 microL) foi aquecida para 70°C durante 110 minutos. A mistura foi concentrada, em seguida purificada por cromatografia de coluna rápida em silica-gel, eluindo com 0 a 5% de metanol em DCM para fornecer dois produtos regioisoméri- cos. A reação foi repetida na mesma escala, substituindo etanol (1,1 mL) por 1,4-dioxano e foi aquecida para 80°C durante 1 hora. Os produtos deste ciclo foram submetidos à cromatografia similarmente e combinados com os produtos do ciclo anterior. 3-[1 -(2-cloropirimidin-4-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil) etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila: 1H RMN (500 MHz, d6-dMSO, 90“C): δ 8,81 (s, 1H), 8,76 (s, 1H), 8,40 (s, 1H), 8,02 (d, 1H), 7,71 (d, 1H), 7,84 (d, 1H), 6,45 (d, 1H), 5,65 (s, 2H), 4,85 (dt, 1H), 3,80 (br s, 1H), 3,61-3,57 (m, 2H), 3,53 (br s, 1H), 3,42-3,25 (m, 4H), 3,03-2,90 (m, 1H), 1,83-1,69 (m, 2H), 0,89-0,84 (m, 2H), -0,07 (s, 9H); LCMS (M+H)+: 550,1.
Separação de 3-[1-(2-cloropirimidin-4-il)pirrolidin-3-il]-3-[4-(7-{[2- (trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila por HPLC quiral (Chiral Technologies Chiralpak IA, 5 μ, 20 x 250 mm, 40% de EtOH/Hexanos, 10 mUminuto) forneceu enantiômero 1 (primeiro a eluir, tempo de retenção de 26,5 minutos), 42 mg, 9%; e enanti-ômero 2 (segundo a eluir, tempo de retenção de 32,7 minutos), 37 mg, 8%.
Separação do isômero [3-(1-(4-cloropirimidin-2-il)pirrolidin-3-il)-3- (4-(7-((2-(trimetilsilil)etóxi)metil)-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila] por HPLC quiral (Chiral Technologies Chiralcel OJ-H, 5 μ, 30 x 250 mm, 45% de EtOH/Hexanos, 22 mL/minuto) forneceu enantiômero 1 (primeiro a eluir, tempo de retenção de 32,6 minutos), 12 mg, 2,6%; e enan-tiômero 2 (segundo a eluir, tempo de retenção de 39,6 minutos), 12 mg, 2,6%. Etapa 2a. 3-[1-(2-cloropirímidin-4-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-dJpirimi- din-4-il)-1 H-pirazol-1-iljpropanonitrila (enantiômero 1) 3-[1 -(2-cloropirimidin-4-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etó- xi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (42 mg, 0,076 mmol, Exemplo 3, etapa 1, enantiômero 1) foi dissolvido em 3 mL de 20% de TFA/DCM. A mistura foi agitada durante 2 horas, em seguida con-centrada. O resíduo foi dissolvido em MeOH (2 mL), e 0,3 mL de EDA foram adicionados. Seguinte à conclusão da reação de desproteção, HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usada para purificar o produto (18 mg, 56%). 1H RMN (400 MHz, d6-dMSO): δ 12,13 (br s, 1H), 8,87 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 8,08 (d, 0,5 H), 8,02 (d. 0,5H), 7,61 (d, 1H), 6,99 (d, 1H), 6,51-6,45 (m, 1H), 4,86-4,78 (m, 1H), 3,90-2,81 (m, 7H), 1,79-1,58 (m, 2H); LCMS (M+H)+: 420,0. Etapa 2b. 3-[1-(2-cloropirimidin-4-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-dJpirimi- din-4-il)-1 H-pirazol-1-iljpropanonitrila (enantiômero 2)
Enantiômero 2 de 3-[1-(2-cloropirimidin-4-il)pirrolidin-3-il]-3-[4-(7- {[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirÍmidin-4-il)-1 H-pirazol-1 - iljpropanonitrila de exemplo 3, etapa 1 (37 mg) foi desprotegido e purificado como descrito na etapa 2a (17 mg, 60%). 1H RMN (400 MHz, d6-dMSO): δ 12,13 (br s, 1H), 8,87 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 8,08 (d, 0,5 H), 8,02 (d, 0,5H), 7,61 (dd, 1H), 6,99 (d, 1H), 6,48 (dd, 1H), 4,87-4,78 (m, 1H), 3,88- 2,78 (m, 7H), 1,79-1,56 (m, 2H); LCMS (M+H)+: 420,0. Exemplo 4a. 3-[1-(4-cloropirimidín-2-il)pirrolidin-3-il1-3-[4-(7H-pirrolo[2,3-d]pi- rimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (um enantiômero isolado)

3-[1-(4-cloropirimidin-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(tnmetilsilil)etó- xi]metil}-7H-pirro1o[2,3-d]pirimidin"4-il)-1 H-pirazol-1-iljpropanonitrila (12 mg, 0,022 mmol, enantiômero 1 de exemplo 3, etapa 1) foi dissolvido em 20% de TFA/DCM (2 mL). A mistura foi agitada durante 2 horas, em seguida concentrada. O resíduo foi dissolvido em MeOH (2 mL), e 0,3 mL de EDA foram adicionados. Seguindo completa reação, HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi u- sada para purificar o produto (6 mg, 65%). 1H RMN (400 MHz, CDCI3): δ 10,03 (br s, 1H), 8,85 (s, 1H), 8,39 (s, 1H), 8,38 (s, 1H), 8,18 (d, 1H), 7,41 (dd, 1H), 6,79 (dd, 1H), 6,56 (d, 1H), 4,47 (dt, 1H), 4,00 (dd, 1H), 3,81-3,73 (m, 1H), 3,52-3,44 (m, 1H), 3,38 (dd, 1H), 3,26 (dd, 1H), 3,10 (dq, 1H), 3,00 (dd, 1H), 1,97-1,89 (m, 1H), 1,82-1,70 (m, 1H); LCMS (M+H)+: 420,0.
Enantiômero 2 de 3-[1-(4-cloropirimidin-2-il)pirrolidin-3-il]-3-[4-(7- {[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila de exemplo 3, etapa 1 (12 mg) foi desprotegido e purificado in the same manner como descrito por exemplo 4a (8 mg, 87%). 1H RMN (400 MHz, CDCI3): δ 10,24 (brs, 1H), 8,86 (s, 1H), 8,39 (s, 1H), 8,38 (s, 1H), 8,18 (d, 1H), 7,45-7,41 (m, 1H), 6,82-6,78 (m, 1H), 6,56 (d, 1H), 4,47 (dt, 1H), 4,00 (dd, 1H), 3,82-3,73 (m, 1H), 3,54-3,44 (m, 1H), 3,42-3,34 (m, 1H), 3,26 (dd, 1H), 3,16-3,05 (m, 1H), 3,04-2,96 (m, 1H), 1,98-1,88 (m, 1H), 1,82-1,70 (m, 1H); LCMS (M+H)+: 420,0. Exemplo 5. Trifluoroacetato de 3-[1-(4-bromo-1,3-tiazol-2-il)pirrolidin-3-ilJ-3- f4-(7H-pirrolo[2,3-dlpirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila
Etapa 1. 3-[1-(4-bromo-1,3-tiazol-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(tnmetilsilil)etó- xi]me til}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila
A mistura de 3-prrrolrdin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il]propanonitrila (151 mg, 0,345 mmol, de exemplo 1, etapa 2) e 2,4-dibromo-1,3-tiazol (126 mg, 0,518 mmol) em NMP (0,50 mL), e N,N-di-isopropiletilamina (0,12 mL) foi aquecida para 135°C no micro-oπdas durante 50 minutos. O produto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 0 a 60% de acetato de etila em hexanos para fornecer o produto como um sólido branco (66 mg, 32%). 1H RMN (400 MHz, CDCI3): δ 8,85 (s, 1H), 8,36 (s, 1H), 8,35 (s, 1H), 7,42 (d, 1H), 6,79 (d, 1H), 6,38 (s, 1H), 5,68 (s, 2H), 4,47 (dt, 1H), 3,89 (dd, 1H), 3,57-3,52 (m, 2H), 3,51-3,33 (m, 1H), 3,22 (dd, 1H), 3,20-3,10 (m, 1H), 2,96 (dd, 1H), 2,04-1,77 (m, 2H), 0,95-0,90 (m, 2H), -0,06 (s, 9H); LCMS (M+H)+: 599,0, 601,0. Etapa 2. Trifluoroacetato de 3-[1-(4-bromo-1;3-tiazol-2-il)pirrolidin-3-il]-3-[4- (7H-pirrolo[2,3-d]pirimÍdin-4-il)-1 H-pira zol-1 -il]propanonitrila 3-[1 -(4-bromo-1,3-tiazol-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsi- lil)etóxÍ]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila (10,0 mg, 0,0167 mmol) foi dissolvido em 20% de TFA/DCM e agitada durante 2 horas. Os solventes foram evaporados, O resíduo foi re-dissolvido em 1 mL de MeOH, e 0,2 mL de EDA foram adicionados. Após a reação ser determinada completa, HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,1% de TFA) foi usada para purificar o produto, que foi obtido como o sal de trifluoroacetato (6 mg). 1H RMN (400 MHz, d6-dMSO): δ 12,63 (br s, 1H), 9,00 (br s, 1H), 8,84 (br s, 1H), 8,55 (br s, 1H), 7,91 (br s, 1H), 7,79 (br s, 1H), 7,15 (br s, 1H), 4,96^,79 (m, 1H), 4,01-3,94 (m, 1H), 3,70-3,64 (m, 1H), 3,54-3,19 (m, 4H), 3,09-2,90 (m, 1H), 1,82-1,61 (m, 2H); LCMS (M+H)+: 469,0, 470,9. Exemplo 6. 3-(1-[4-(dimetilamino)pirimidin-2-il1pirrolidin-3-il'|-3-í4-(7H-pirro-lo[2.3-d]pirimidÍn-4-il)-1H-pirazol-1-illpropanonitrila

3-[1-(4-cloropirimidin-2-i|)pirro|idin-3-i|]-3-[4-(71{2-(trimeti|si|i|)etó-xi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazok1-il]propanonitrila (11 mg,0,020 mmol, racèmico, de exemplo 3, etapa 1) foi misturado com 2,0 M de dimetilamina em THF (0,10 mL). A mistura foi aquecida para 70°C durante 1,5 hora e em seguida concentrada. O produto bruto foi desprotegido por agitação em uma solução de 1:1 de TFNDCM durante 1,5 hora. Após a remoção do solvente em vácuo, o reslduo foi dissolvido em metanol, e 0,2 mL de EDA foi adicionado. O produto foi purificado por meio de HPLC/MS prepa- rativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15°/, de NH4OH). 1H RMN (400 MHz, CDCl3): çj 9,72 (br s, IH), 8,85 (s, IH), 8,38 (s, IH), 8,37(S, 1H),7,91 (d, 1H),7,39(dd, 1H),6,80(dd, IH), 5.82(d,1H), 4,44 (dt, IH), 3,96 (dd, IH), 3,74 (ddd, IH), 3,45 (dddd, IH), 3,39 (dd, IH), 3,25 (dd, IH), 3,05 (s, 6H), 2,98 (dd, IH), 1,91-1,82 (m, IH), 1,74-1,63 (m, IH); LCMS (M+H)+: 429,1. Exemplo 7. 3-{1-[4-(isopropi|amino)pirimidin-2-i|]pirro|idin-3-il¥3-[4-(7H-pirro-1o[2,3-d]pirimidin-4-il)-1 H-pirazo|-1-i|Ípropanonitrila

3-[1 -(4-cloropirimidin-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etó- xi]metil}-7H-pirrolo[2,3-d]pirimidin-4-i I)-1 H-pirazol-1 -il]propanon itrila (7 mg, 0,01 mmol; racêmico, de exemplo 3, etapa 1) foi misturado com 2-propa- namina (0,011 mL, 0,127 mmol) em 0,1 mL THF. A mistura foi aquecida para 70°C durante quantro dias. Seguinte à remoção do solvente, o produto foi desprotegido por agitação em uma solução de 1:1 de TFA/DCM durante 1,5 hora, seguido por evaporação de solvente, e agitação subsequente com 0,2 mL de EDA em metanol. HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usada para purificar 4 o produto (1 mg, 18%). LCMS (M+H)+: 443,2. Exemplo 9. 3-[1-(1,3-benzoxazol-2-il)pirrolidin-3-ill-3-[4-(7H-pirrolo[2,3-d]piri- midin-4-il)-1 H-pirazol-1-illpropanonitrila (dois diferentes enantiômeros isola-dos)

Etapa 1. Separação de enantiômeros de diastereômero 1 de 3-{2-ciano-1-[4- 15 (7-{[2-(trimetilsÍIÍI)etóxi]metil}-7H-pirrolo[2,3-d]pinmidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidina-1-carboxilato de benzila 3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pi- rimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidina-1-carboxilato de metila (4,2 g, 7,3 mmols, de exemplo 1, etapa 1, diastereômero 1) foi separado em seus enan- tiômeros por meio de HPLC quiral (Chiral Technologies Chiralcel OJ-H, 5 μ, 30 x 250 mm, 45% de EtOH/Hexanos, 20 mUminuto) para fornecer 1,6 g de enantiômero 1 (primeiro a eluir, tempo de retenção de 46,1 minutos) e 1,6 g de enantiômero 2 (segundo a eluir, tempo de retenção de 57,5 minutos). Cada qual foi desprotegido separadamente de acordo com o seguinte procedi- mento na etapa 2. Etapa 2a. 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pÍrimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (enantiômero 1) 3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidina-1-carboxilato de benzila (1,6 g, 2,8 mmols, enantiômero 1, de etapa 1) foi dissolvido em metanol (60 mL), e uma quantidade catalítica de 10% de Pd-C foi adicionada. A mistura foi agi-tada sob hidrogênio (3,51 kg/cm2) durante 3,5 horas. A mistura foi filtrada e o solvente removido por evaporação giratória para fornecer o produto (1g, 80%). LCMS (M+H)+: 438,2. Etapa 2b. 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxiJmetil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (enantiômero 2)
Desproteção de enantiômero 2 (de etapa 1) de 3-{2-ciano-1-[4- (7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidina-1 -carboxilato de benzila (1,6 g, 2,8 mmols) foi realizada como descrito na etapa 2a, exceto que a hidrogenação prosseguiu durante 4 horas. LCMS (M+H)+: 438,1. Etapa 3a. 3-[1-(1,3-benzoxazol-2-il)pirrolidin-3-ilJ-3-[4-(7H-pirrolo[2,3-dJpirimi- din-4-il)-1 H-pirazol-1-iljpropanonitrila (enantiômero 1) 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (10,0 mg, 0,0228 mmol, enanti-ômero 1, de etapa 2a) e 2-clorobenzoxazol (4,2 mg, 0,027 mmol) foram dis-solvidos em 1,4-dioxano (0,20 mL), e N,N-di-isopropiletilamina (8,0 microL, 0,046 mmol) foi adicionada. A mistura foi aquecida para 70°C durante 1,5 hora. O solvente foi removido em vácuo e o resíduo foi sequencialmente agi-tado com 50% de TFA/DCM durante 1,5 hora, concentrado, e agitado com 0,3 mL de EDA em metanol durante 30 minutos. Purificação por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto. 1H RMN (400 MHz, d6- dMSO): Õ 12,13 (br s, 1H), 8,89 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 7,61 (d, 1H), 7,39 (d, 1H), 7,26 (d, 1H), 7,13 (t, 1H), 7,82-6,95 (m, 2H), 4,86 (dt, 1H), 3,87 (dd, 1H), 3,68-3,60 (m, 1H), 3,53-3,29 (m, 4H), 3,03-2,91 (m, 1H), 1,78- 1,64 (m, 2H); LCMS (M+H)+: 425,1. Etapa 3b. 3-[1-(1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-dJpirimÍ- din-4-il)-1 H-pirazol-1-iljpropanonitrila (enantiômero 2) 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila (10,0 mg, 0,0228 mmol; enantiômero 2, de etapa 2b) e 2-clorobenzoxazol (4,2 mg, 0,027 mmol) foram dissolvidos em 1,4-dioxano (0,20 mL) e N,N-di-isopropiletilamina (8,0 microL, 0,046 mmol) foi adicionada. A mistura foi aquecida para 70°C durante 1,5 hora. O solvente foi removido em vácuo e o resíduo foi sequencialmente agitado com 50% de TFA/DCM durante 1,5 hora, concentrado, e agitado com 0,3 mL de EDA em metanol durante 30 minutos. Purificação por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto. 1H RMN (400 MHz, d6- dMSO): δ 12,13 (br s, 1H), 8,89 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 7,61 (d, 1H), 7,39 (d, 1H), 7,26 (dd, 1H), 7,13 (dt, 1H), 7,81-6,96 (m, 2H), 4,86 (dt, 1H), 3,87 (dd, 1H), 3,68-3,61 (m, 1H), 3,53-3,28 (m, 4H), 3,03-2,91 (m, 1H), 1,79-1,64 (m, 2H); LCMS (M+H)+: 425,0. Exemplo 10. 3-[1-(5-cloro-1,3-benzoxazol-2-il)pirrolidin-3-il1-3-[4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)

A uma solução de 5-cloro-1,3-benzoxazol-2-tiol (0,50 g, 2,7 mmols, Aldrich) em tolueno (10 mL) foi adicionado cloreto de tionila (0,59 mL, 8,1 mmols) seguido por uma gota de DMF. A reação foi aquecida até o refluxo durante 30 minutos e o solvente removido em vácuo. Uma porção deste produto bruto (17 mg) e 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila (20,0 mg, 0,0457 mmol; enantiômero 2 de exemplo 9, etapa 2b) foram dissolvidos em 1,4-dioxano (0,40 mL) e N,N-di-isopropiletilamina (16 microL, 0,091 mmol) foi adicionada. A mistura foi aquecida para 70°C durante 1,5 hora. O solvente foi removido em vácuo e o resíduo foi sequencialmente agitado com 50% de TFA/DCM durante 1,5 hora, concentrado, em seguida agitado com 0,3 mL de EDA em metanol durante 30 minutos. Purificação por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto. 1H RMN (400 MHz, d6-dMSO): δ 12,13 (brs, 1H), 8,88 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 7,60 (d, 1H), 7,41 (d, 1H), 7,31 (d, 1H), 7,82-6,97 (m, 2H), 4,86 (dt, 1H), 3,86 (dd, 1H), 3,68-3,59 (m, 1H), 3,54-3,28 (m, 4H), 3,03-2,90 (m, 1H), 1,77-1,67 (m, 2H); LCMS (M+H)+: 459,0, 461,0. Exemplo 11, 3-(1 -[1,31oxazolo[4,5-c]piridin-2-ilpirrolidin-3-il)-3-[4-(7H-pirro- lof2,3-dlpirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)

Uma solução de 3-aminopiridin-4-ol (0,250 g, 2,27 mmol, Bosche Scientific) e ditiocarbonato de O-etila de potássio (0,400 g, 2,50 mmols) em etanol (1 mL) foi aquecida ao refluxo. Quando a reação foi determinada completa, ela foi resfriada para temperatura ambiente e dividida entre HCI a 1N e acetato de etila. A camada orgânica foi lavada com água, secada sobre sulfato de sódio, decantada e concentrada. Este produto bruto foi dissolvido em tolueno (6 mL) e cloreto de tionila (0,365 mL, 5,01 mmols) seguido por DMF (3 microL) foi adicionado. A mistura foi aquecida até o refluxo durante 1 hora, resfriada e o solvente removido em vácuo. Uma porção deste produto bruto (14 mg) foi dissolvido em 1,4-dioxano (0,40 mL), junto com 3-pirrolidin- 3-il-3-[4-(7-{[2-(trimetílsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pira- zol-1-iljpropanonitrila (20,0 mg, 0,0457 mmol, enantiômero 2 de exemplo 9, etapa 2b), e N,N-di-isopropiletilamina (16 microL, 0,091 mmol) foi adicionada. A mistura foi aquecida para 70°C durante 1,5 hora. O solvente foi remo-vido em vácuo e o resíduo foi sequencialmente agitado com 50% de TFA/DCM durante 1,5 hora, concentrada, e agitada com 0,3 mL de EDA em metanol durante 30 minutos. Purificação por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto. 1H RMN (400 MHz, d6-dMSO): δ 8,89 (s, 1H), 8,68 (s, 1H). 8,44 (s, 1H), 8,11 (dd, 1H), 7,72 (dd, 1H), 7,60 (d, 1H), 6,99 (d, 1H), 6,96 (dd, 1H), 4,88 (dt, 1H), 3,90 (dd, 1H), 3,71-3,63 (m, 1H), 3,58-3,30 (m, 4H), 3,04-2,93 (m, 1H), 1,80-1,66 (m, 2H); LCMS (M+H)+: 426,1. Exemplo 12. 3-(1-M,3loxazolo[4,5-b]piridin-2-ilpirrolidin-3-il)-3-[4-(7H-pirro- lor2.3-d1pirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)

Uma solução de 2-aminopiridin-3-ol (0,250 g, 2,27 mmols, Aldrich) e ditiocarbonato de O-etila de potássio (0,400 g, 2,50 mmols) em etanol (1 mL) foi aquecida para 120°C no micro-ondas durante 10 minutos. A mistura reacional foi dividida entre HCI a 1N e acetato de etila. A camada orgânica foi lavada com água, secada sobre sulfato de sódio, decantada e concentrada. Este produto bruto foi dissolvido em tolueno (6 mL) e cloreto de tionila (0,365 mL, 5,01 mmols) seguido por DMF (3 microL) foi adicionado. A mistura foi aquecida até o refluxo durante 1 hora, resfriada e o solvente removido em vácuo. Uma porção deste produto bruto (14 mg) e 3-pirrolidin-3-il-3-[4-(7- {[2-(trimetilsilil)etóxí]metil}-7H-pÍrrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]pro- panonitrila (20,0 mg, 0,0457 mmol; enantiômero 2, de exemplo 9, etapa 2b) foram dissolvidos em 1,4-dioxano (0,40 mL), e N,N-di-isopropiletilamina (16 microL, 0,091 mmol) foi adicionada. A mistura foi aquecida para 70°C durante 1,5 hora. O solvente foi removido em vácuo e o resíduo foi sequencialmente agitado com 50% de TFA/DCM durante 1,5 hora, concentrado, e agitado com 0,3 mL de EDA em metanol durante 30 minutos. Purificação por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto. 1H RMN (400 MHz, d6-dMSO): δ 12,08 (br s, 1H), 8,89 (s, 1H), 8,68 (s, 1H), 8,44 (s, 1H), 8,11 (dd, 1H), 7,72 (dd, 1H), 7,60 (d, 1H), 6,99 (d, 1H), 6,96 (dd, 1H), 4,88 (dt, 1H), 3,90 (dd, 1H), 3,72-3,63 (m, 1H), 3,58-3,31 (m, 4H), 3,04-2,92 (m, 1H), 1,80-1,66 (m, 2H); LCMS (M+H)+: 426,1. Exemplo 13a. 3-(1-[1,31oxazoloF5,4-blpirídin-2-ilpirrolidin-3-il)-3-[4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)

A uma solução de 3-aminopiridin-2-ol (0,500 g, 4,54 mmols, 3B Scientific) em THF (4 mL) foi adicionado tiocarbonildiimidazol (1,21 g, 6,81 mmols). Após a reação ser determinada completa, o THF foi removido em vácuo. O produto foi dividido entre acetato de etila e HCI a 1N suficiente para ajustar o pH para 4 a 5. A porção aquosa foi extraída mais duas vezes com acetato de etila. Os extratos combinados foram lavados com salmoura, secados sobre sulfato de sódio, decantados e concentrados. Uma suspensão de oxazolo[5,4-b]piridina-2(1H)-tiona (0,65 g, 4,3 mmols) preparada nesta maneira em tolueno (13 mL) foi tratado com cloreto de tionila (0,94 mL, 12,9 mmols) e uma gota de DMF A reação foi aquecida até o refluxo durante 1 hora, e o solvente foi em seguida removido por evaporação giratória. Este produto bruto (0,018 g) e 3-pirrolidin-3-ÍI-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il]propanonÍtrila (0,020 g, 0,046 mmol, enantiômero 2 de exemplo 9, etapa 2b) em 1,4-dioxano (0,2 mL) contendo N,N-di-isopropiletilamina (32 microL, 0,183 mmol) foram aquecidos para 70°C durante 1,5 hora. No resfriamento, o produto foi purificado aplican- do-se a um tampão de sílica, primeiro eluindo com acetato de etila, em seguida com metanol. O eluente de metanol foi concentrado para fornecer cerca de 30 mg de material bruto tão desejado quanto o componente principal. O produto foi desprotegido agitando-se sequencialmente com 20% de TFA em DCM durante 2 horas, evaporação de solvente, em seguida agitação com EDA em metanol. Purificação por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto (5 mg, 25%). 1H RMN (400 MHz, d6-dMSO): δ 11,97 (br s, 1H), 8,82 (s, 1H), 8,61 (s, 1H), 8,37 (s, 1H), 7,79 (dd, 1H), 7,56-7,52 (m, 2H), 7,12 (dd, 1H), 6,92 (d, 1H), 4,80 (dt, 1H), 3,82 (dd, 1H), 3,63-3,54 (m, 1H), 3,50-3,24 (m, 4H), 2,97-2,85 (m, 1H), 1,73-1,61 (m, 2H); LCMS (M+H)+: 426,1. Exemplo 13b. 3-(1-[1,3loxazolof5,4-b]piridin-2-ilpirrolidin-3-il)-3-f4-(7H- pirrolo[2,3-dlpirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)

A uma solução de 3-aminopiridin-2-ol (0,500 g, 4,54 mmols, 3B Scientific) em THF (4 mL) foi adicionado tiocarbonildiimidazol (1,21 g, 6,81 mmol). Após a reação ser determinada completa, o THF foi removido em vácuo. O produto foi dividido entre acetato de etila e HCI a 1N suficiente para ajustar o pH para 4 a 5. A porção aquosa foi extraída mais duas vezes com acetato de etila. Os extratos combinados foram lavados com salmoura, secados sobre sulfato de sódio, decantados e concentrados. Uma suspensão de oxazolo[5,4-b]piridina-2(1H)-tiona (0,65 g, 4,3 mmols) preparada nesta maneira em tolueno (13 mL) foi tratado com cloreto de tionila (0,94 mL, 12,9 mmols) e uma gota de DMF. A reação foi aquecida até o refluxo durante 1 hora, e o solvente foi em seguida removido por evaporação giratória. Este produto bruto (0,018 g) e 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (0,020 g, 0,046 mmol, enantiômero 1 de exemplo 9, etapa 2a) em 1,4-dioxano (0,2 mL) contendo N,N-di-isopropÍletilamina (32 microL, 0,183 mmol) foram aquecidos para 70°C durante 1,5 hora e em seguida solvente evaporado. Desprotegido por agitação com 20% de TFA/DCM durante 2 horas, seguido por evaporação e agitação com EDA (0,3 mL) em metanol durante 1 hora. Purificado por HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) para fornecer o produto (5 mg, 25%). 1H RMN (400 MHz, d6-dMSO): δ 12,10 (br s, 1H), 8,89 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 7,85 (dd, 1H), 7,62-7,59 (m, 2H), 7,19 (dd, 1H), 6,99 (dd, 1H), 4,87 (dt, 1H), 3,89 (dd, 1H), 3,69-3,61 (m, 1H), 3,57-3,30 (m, 4H), 3,03-2,91 (m, 1H), 1,78-1,68 (m, 2H); LCMS (M+H)+: 426,1. Exemplo 13c. 3-(141,31oxazoloi5,4-blpiridin-2-ilpirrolidin-3-il)-3-[4-(7H-pirro- lof2,3-dlpirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)

Etapa 1. 3]oxazolo[5,4-b]pindin-2-ilpirrolidin-3-il)-3-[4-(7-{[2- (trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila Oxazolo[5,4-b]piridina-2(1H)-tiona (1,17 g, 7,68 mmols, preparada no exemplo 33, etapa 4) e 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (2,80 g, 6,40 mmols de exemplo 15, etapa 3) em 1,4-dioxano (30 mL) foram aquecidos para 70°C durante 2 horas. O solvente foi removido em vácuo. O produto bruto foi reconstituído em etanol (40 mL) e tratado com nitrato de prata (3 g, 15 mmols) e hidróxido de amónio aquoso (6 mL) em porções durante o curso de 20 horas. Na reação foi adicionada água, NaOH a 1N e salmoura. O material insolúvel foi removido por filtragem. As camadas do filtrado foram separadas. A porção aquosa foi extraída com três porções de acetato de etila. Os extratos foram secados sobre sulfato de sódio, decantados e concentrados. O produto bruto foi purificado por cromatografia de coluna rápida em sílica- gel, eluindo com 10% de MeOH/DCM para fornecer o produto como uma espuma não totalmente branca (2,84 g, 80%). 1H RMN (300 MHz, CDCI3): δ 8,83 (s, 1H), 8,37 (s, 1H), 8,36 (s, 1H), 7,92 (dd, 1H), 7,57 (dd, 1H), 7,40 (d, 1H), 7,13 (dd, 1H), 6,78 (d, 1H), 5,67 (s, 2H), 4,52 (dt, 1H), 4,05 (dd, 1H), 3,82 (ddd, 1H), 3,67-3,44 (m, 4H), 3,25 (dd, 1H), 3,24-3,09 (m, 1H), 2,98 (dd, 1H), 2,06-1,74 (m, 2H), 0,97-0,88 (m, 2H), -0,06 (s, 9H); LCMS (M+H)+: 556,1. Etapa 2. 3-(1-[1,3]oxazolo[5,4-b]piridin-2-ilpirrolidin-3-il)-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila 3-(1-[1,3]oxazolo[5,4-b]piridin-2-ilpirrolidin-3-il)-3-[4-(7-{[2-(trime- tilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (5,35 g, 9,63 mmols, preparado pelo método de etapa 1) foi agitado em uma mistura de 2:1 de DCM e TFA (60 mL) durante 6 horas. Os solventes foram removidos por evaporação giratória. O resíduo bruto foi dissolvido em metanol (50 mL) contendo EDA (5,15 mL, 77,8 mmol) e foi agitado durante a noite. Após remoção de solvente, o produto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 0 a 15% de Me- OH/DCM (3,59 g, 88%). 1H RMN (300 MHz, CDCI3): δ 8,72 (s, 1H), 8,40 (s, 1H), 8,34 (s, 1H), 7,89 (dd, 1H), 7,54 (dd, 1H), 7,36 (d, 1H), 7,12 (dd, 1H), 6,75 (d, 1H), 4,56 (dt, 1H), 4,01 (dd, 1H), 3,80 (ddd, 1H), 3,60 (ddd, 1H), 3,48 (dd, 1H), 3,26 (dd, 1H), 3,21-3,06 (m, 1H), 3,02 (dd, 1H), 2,03-1,76 (m, 2H); LCMS (M+H)+: 426,1. Exemplo 14. 3-[1-(6-metil[1,3]oxazolo[5,4-blpiridin-2-il)pirrolidin-3-il1-3-[4-(7H- pirrolo[2,3-dlpirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)

Etapa 1. 6-metil[1,3]oxazolo[5,4-b]piridina-2(1 H)-tiona 3-amino-5-metilpiridin-2-ol (0,21 g, 1,7 mmol) foi dissolvido em THF (5 mL), e 1,T-tiocarbonildiimidazol (0,48 g, 2,7 mmols) foi adicionado. A mistura foi agitada em temperatura ambiente durante 20 minutos. A reação foi diluída com água, tratada com HCI a 1N para ajustar o pH para a faixa de 4 a 5. O produto foi em seguida extraído com acetato de etila, os extratos foram lavados com salmoura, e secados sobre sulfato de sódio, filtrados e concentrados para fornecer o produto, usado sem outra purificação na etapa seguinte. LCMS (M+H)+: m/z = 167,0. Etapa 2. 2-cloro-6-metil[1,3]oxazolo[5,4-b]pindina
A uma solução de 6-metil[1,3]oxazolo[5,4-b]piridina-2(1H)-tiona (0,23 g, 1,4 mmol) em tolueno (6,0 mL) foi adicionado cloreto de tionila (0,36 mL, 5,0 mmols), seguido por uma gota catalítica de DMF. A mistura foi em seguida aquecida até o refluxo durante 1 hora. A mistura reacional foi resfri-ada e o solvente removido em vácuo. LCMS (M+H)+: 168,9, 170,9. Etapa 3. 3-[1-(6-metil[1,3]oxazolo[5,4-b]piridin-2-il)pirrolidin-3-il]-3-[4-(7H-pir- rolo[2,3-d]pinmidin-4-il)-1 H-pirazol-1 -iljpropanonitrila 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (20,0 mg, 0,0457 mmol, enanti- õmero 2, de exemplo 9, etapa 2b) e 2-cloro-6-metil[1,3]oxazolo[5,4-b]pÍridina (15,4 mg, 0,0914 mmol) foram dissolvidos em 1,4-dioxano (0,40 mL), e N,N- di-isopropiletilamina (16 microL, 0,091 mmol) foi adicionada. A mistura foi aquecida para 70°C durante 1,5 hora. A mistura foi concentrada, em seguida sequencialmente agitada com 50% de TFA/DCM durante 1,5 hora, concen-trada, e agitada com 0,3 mL de EDA em metanol durante 30 minutos. O pro-duto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente deACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6-dMSO): δ 12,11 (br s, 1H), 8,88 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 7,66 (dd, 1H), 7,60 (d, 1H), 7,42 (dd, 1H), 6,98 (d, 1H), 4,86 (dt, 1H), 3,87 (dd, 1H), 3,67-3,60 (m, 1H), 3,55-3,30 (m, 4H), 3,02-2,90 (m, 1H), 2,30 (s, 3H), 1,77-1,67 (m, 2H); LCMS (M+H)+: 440,1. Exemplo 15. 3-[1-(6-fluoro[1,31oxazolo[5,4-b1piridin-2-il)pirrolidin-3-il1-3-[4- (7H-pirroloí2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (um enantiômero isolado)

Etapa 1. 3-[2-cianovinil]pirrolidina-1-carboxilato de terc-butila
A uma solução de 0,10 M de terc-butóxido de potássio em THF (190 mL, 0,19 mol) a 0°C foi adicionada uma solução de cianometilfosfonato de dietila (30,0 mL, 0,185 mol) em THF (400 mL) gota a gota. O banho foi removido e a reação foi aquecida para temperatura ambiente durante o curso de aproximadamente duas horas. A mistura foi re-resfriada para 0°C e 5 uma solução de 3-formilpirrolidina-1-carboxilato de terc-butila (35,00 g, 0,1757 mol, Adesis) em THF (300 mL) foi adicionada gota a gota. O banho foi removido e a reação foi deixada aquecer para temperatura ambiente e agitar durante 16 horas. A mistura foi em seguida diluída com acetato de etila e água, a solução aquosa foi extraída com mais duas porções de acetato de 10 etila, os extratos combinados foram lavados com salmoura, secados sobre sulfato de sódio, filtrados e concentrados. O produto foi usado sem outra pu-rificação na etapa seguinte (39 g, 100%). 1H RMN (300 MHz, CDCI3): δ 6,65 (dd, 1H, trans), 6,38 (t, 1H, cis), 5,41 (dd, 1H, trans), 5,37 (dd, 1H, cis), 4,30- 2,68 (m, 10H), 2,21-2,00 (m, 2H), 1,84-1,65 (m, 2H), 1,45 (s, 9H), 1,45 (s, 15 9H). Etapa 2. 3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimi- din-4-il)-1 H-pirazol-1 -il]etil}pirrolidina-1-carboxilato de terc-butila
A uma solução de 3-[2-cianovinil]pirrolidina-1-carboxilato de terc- butila (39 g, 180 mmols) e 4-(1 H-pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H- 20 pirrolo[2,3-d]pirimidina (55 g, 180 mmols, preparado como descrito no WO 2007/070514, Ex. 65) em acetonitrila (500 mL) foi adicionado 1,8-diazabi- ciclo[5,4,0]undec-7-eno (26 mL) e a reação foi agitada durante três dias. A maioria do solvente foi removida por evaporação giratória antes da divisão da mistura reacional entre solução de bicarbonato de sódio saturada e ace- tato de etila. O produto foi extraído com mais duas porções de acetato de etila. Os extratos combinados foram secados sobre sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida (em 2,5 Kg de sílica- gel) usando 7,5% de isopropanol/25% de acetato de etila/67,5% de hexanos como eluente forneceu 27,73 g de diastereômero 1 puro (primeiro a eluir).
Recoluna de frações misturadas, eluindo com um gradiente de 5% de iso- propanol/5% de acetato de etila/90% de hexanos a 10% de isopropanol/50% de acetato de etila/40% de hexanos forneceu 9,84 g de produto adicional (diastereômero 1). Os enantiômeros foram separados por HPLC quiral (Chiral Technologies Chiralcel OD-H, 5 μ, 30x 250 mm, 20% de EtOH/Hexanos, 22 mL/minuto). Enantiômero desejado 2 (segundo a eluir, tempo de retenção de 22,9 minutos) foi coletado (17,3 g, 18%). 1H RMN (300 MHz, CDCI3): δ 5 8,84 (s, 1H), 8,34 (s, 1H), 8,33 (s, 1H), 7,40 (d, 1H), 6,78 (d, 1H), 5,67 (s, 2H), 4,37 (dt, 1H), 3,76-2,80 (m, 9H), 1,85-1,52 (m, 2H), 1,45 (s, 9H), 0,95- 0,87 (m, 2H), -0,07 (s, 9H); LCMS (M+H)+: 538,1. Etapa 3. 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila
A uma solução de 3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7 H-pi rrolo[2,3-d]piri m id in-4-i I)-1 H-pirazol-1 -i IJeti l]p irrol id in a-1 -carboxilato de terc-butila (8,9 g, 16 mmols) (de etapa 2, diastereômero 1, enantiômero 2) em 1,4-dioxano (200 mL) foi adicionado 4 M de cloreto de hidrogênio em 1,4- dioxano (32 mL, 130 mmols) e a reação foi agitada durante 16 horas. O sol- vente foi removido em vácuo. O resíduo foi dividido entre 500 mL de NaOH a 1N e acetato de etila. As camadas foram separadas, e a camada aquosa foi extraída com acetato de etila quatro vezes. Os extratos combinados foram lavados com salmoura, secados sobre sulfato de sódio, decantados e con-centrados para fornecer o produto como um sólido amarelo (7,12 g, 98%). 1H RMN (300 MHz, CDCI3): δ 8,84 (s, 1H), 8,34 (s, 1H), 8,33 (s, 1H), 7,39 (d, 1H), 6,79 (d, 1H), 5,67 (s, 2H), 4,38 (dt, 1H), 3,57-3,49 (m, 2H), 3,26 (dd, 1H), 3,13 (dd, 1H), 2,98-2,77 (m, 4H), 2,73 (dd, 1H), 1,83-1,70 (m, 1H), 1,55- 1,38 (m, 1H), 0,95-0,87 (m, 2H), -0,07 (s, 9H); LCMS (M+H)+: 438,1. Etapa 4. 3-amÍno-5-fíuoropiridin-2-ol
A mistura de 5-fluoro-3-nitropiridin-2-ol (73 mg, 0,46 mmol; preparado de acordo com o procedimento reportado WO 2006/114706) e ferro (130 mg, 2,3 mmols), em etanol (0,1 mL), ácido acético (0,76 mL), água (0,38 mL) e HCI concentrado (1 gota) foi aquecida para 100°C durante 20 minutos. Após resfriamento para a temperatura ambiente, a solução foi dilui- da com água (10 mL), filtrada, e o filtrado foi concentrado. O resíduo foi em seguida tratado com solução de bicarbonato de sódio saturada para próximo de pH 7, e o produto foi extraído com 6 x 40 mL de 20% de isopropa- nol/DCM. Os extratos foram secados sobre sulfato de sódio, filtrados e con-centrados e o produto foi usado sem outra purificação na etapa seguinte. LCMS (M+H)+: 129,0. Etapa 5. 3-[1-(6-fluoro[1,3]oxazolo[5,4-b]piridÍn-2-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila 3-amino-5-fluoropiridin-2-ol (24 mg, 0,19 mmol) foi dissolvido em THF (0,66 mL), e dicloreto carbonotioico (21 microL, 0,28 mmol) foi adicionado gota a gota. A mistura foi agitada em temperatura ambiente durante duas horas, em seguida o solvente foi removido em vácuo para fornecer um óleo marrom escuro. 1,4-dioxano (0,50 mL) e N,N-dr-isopropiletilamina (98 microL, 0,56 mmol) foram adicionados, seguido por 3-pirrolidin-3-il-3-[4-(7- {[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpro- panonitrila (41 mg, 0,094 mmol, enantiômero simples, de etapa 3). A mistura foi agitada a 60°C durante 16 horas para produzir o piridil oxazol desejado bem como tioureia. O solvente foi removido em vácuo e a mistura foi despro-tegida por agitação sequencialmente em uma solução de 1:1 de TFA/DCM durante 1,5 hora, evaporação de solvente, e agitação em uma solução de 0,4 mL de EDA em 1,5 mL de metanol durante 30 minutos. O produto foi pu-rificado por HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6-dMSO): δ 12,11 (br s, 1H), 8,88 (s, 1H), 8,67 (s, 1H), 8,43 (s, 1H), 7,81 (dd, 1H), 7,61- 7,57 (m, 2H), 6,98 (d, 1H), 4,87 (dt, 1H), 3,88 (dd, 1H), 3,67-3,29 (m, 5H), 3,03-2,91 (m, 1H), 1,81-1,69 (m, 2H); LCMS (M+H)+: 444,0. Exemplo 16. 3-(4-(7H-pirrolo[2,3-dlpirimidin-4-il)-1 H-pirazol-1-il]-3-[1-(7H-pir- roloí2.3-dlpirimidin-2-il)pirrolidin-3-il1propanonitrila (um enantiômero isolado)

3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (21 mg, 0,048 mmol de exemplo 15, etapa 3) e 2-cloro-7H-pirrolo[2,3-d]pirimidina (16 mg, 0,058 mmol, prepa-rado como reportado em Bioorganic and Medicinal Chemistry Letters, 16(22), 5778-5783; 2006) foram dissolvidos em NMP (0,1 mL), e N,N-di-isopropile- tilamina (41 microL, 0,23 mmol) foi adicionada. A mistura foi aquecida para135°C durante 40 minutos no micro-ondas. A mistura foi concentrada, tratada com 1:1 de TFA/DCM durante 2 horas, concentrada novamente, e agitada em uma solução de metanol (1 mL) contendo 0,2 mL de EDA durante 30 minutos. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6-dMSO): Õ 12,12 (br s, 1H), 11,32 (s, 1H), 8,88 (s, 1H), 8,69 (s, 1H), 8,57 (s, 1H), 8,43 (s, 1H), 7,61 (d, 1H), 7,84 (dd, 1H), 7,80 (d, 1H), 6,30 (d, 1H), 4,84 (dt, 1H), 3,86 (dd, 1H), 3,68-3,60 (m, 1H), 3,46- 3,22 (m, 4H), 2,97-2,84 (m, 1H), 1,77-1,58 (m, 2H); LCMS (M+H)+: 425,1. Exemplo 17. 3-[1-(7-metil-7H-pirrolo[2,3-dlpirimidÍn-2-il)pirrolidin-3-il]-3-[4- (7H-pirrolo[2,3-d]pirimidin^-il)-1 H-pirazol-1 -illpropanonitrila (um enantiômero isolado)

Etapa 1. 2-cloro-7-metil-7H-pirrolo[2,3-d]pirimidina
A uma solução de 2-cloro-7H-pirrolo[2,3-d]pirimidina (27 mg, 20 0,16 mmol, preparado como reportado em Bioorganic and Medicinal Chemis try Letters, 16(22), 5778-5783 (2006)) em DMF (0,15 mL) foi adicionado car-bonato de potássio (67 mg, 0,48 mmol), seguido por iodeto de metila (10 microL, 0,16 mmol). A mistura foi agitada em um frasco selado em temperatura ambiente durante 3 horas. A reação foi diluída com DCM e acetonitrila, filtra- 25 da e concentrada. O produto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com 0 a 50% de acetato de etila em hexanos para fornecer o produto como um sólido branco (13 mg, 47%). LCMS (M+H)+: 167,9, 169,9. Etapa 2. 3-[1-(7-metil-7H-pirrolo[2,3-d]pinmidin-2-il)pirrolidin-3-il]-3-[4-(7H-pir- rolo[2,3-d]pÍrimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (26 mg, 0,060 mmol; de exem- pio 15, etapa 3) e 2-cloro-7-metil-7H-pirrolo[2,3-d]pirimidina (12,0 mg, 0,0716 mmol) foram dissolvidos em NMP (0,050 mL) e N,N-di-isopropiletilamina (42 microL, 0,24 mmol) foi adicionada. A mistura foi aquecida para 135°C durante 60 minutos no micro-ondas. Após remoção de solvente, o resíduo foi agitado sequencialmente em 1:1 de TFA/DCM durante 1,5 hora, concentrado, em seguida em uma solução em 0,1 mL de metanol contendo 0,2 mL de EDA durante 30 minutos. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6-dMSO): δ 12,13 (br s, 1H), 8,89 (s, 1H), 8,69 (s, 1H), 8,55 (s, 1H), 8,43 (s, 1H), 7,61 (d, 1H), 7,88 (d, 1H), 7,80 (d, 1H), 6,33 (d, 1H), 4,84 (dt, 1H), 3,91 (dd, 1H), 3,67 (dd, 1H), 3,62 (s, 3H), 3,46-3,27 (m, 4H), 2,96-2,84 (m, 1H), 1,77-1,59 (m, 2H); LCMS (M+H)+:439,1. Exemplo 18. 3-(1-[1,31oxazolo[5,4-d1pirimidin-2-ilpirrolidin-3-il)-3-|4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il1propanonitrila (um enantiômero isolado)

Etapa 1. 5-aminopirimidin-4-ol
A uma mistura desgaseificada de 5-amino-6-cloropirimidin-4-ol (227 mg, 1,56 mmol, Matrix) e trietilamina (0,19 mL, 7,80 mmols) em etanol (15,0 mL) foram adicionados 10% de paládio sobre carbono (51 mg) e a mis- 25 tura foi agitada sob 3,51 kg/cm2 hidrogênio durante duas horas. A mistura foi filtrada e concentrada para fornecer o produto. LCMS (M+H)+: 112,1. Etapa 2. 3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxiJmetil}-7H-pirrolo[2,3-d]pirimi- din-4-il) -1 H-pirazol-1 -HJetil}-N-(4-hidroxipirimidin-5-il) pirrolidin a-1 -carbotioami- da 5-aminopirimidin-4-ol (52,4 mg, 0,203 mmol) foi dissolvido em piridina (0,55 mL) e clorotionocarbonato de fenila (33 microL, 0,24 mmol) foi adicionado. A mistura foi agitada em temperatura ambiente durante uma ho- ra. A reação foi diluída com DCM e lavada com água e salmoura, a fase orgânica foi secada sobre sulfato de sódio e concentrada. O resíduo foi dissolvido em clorofórmio (1,7 mL), e trietilamina (141 microL, 0,11 mmol) e 3-pir- rolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)- 1 H-pirazol-1-iljpropanonitrila (75 mg, 0,17 mmol; de exemplo 15, etapa 3) foram adicionados. A mistura foi agitada durante 30 minutos a 70°C. Os solventes foram removidos em vácuo e o produto foi purificado por HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) (15 mg, 15%). LCMS (M+H)+: 591,1. ! Etapa 3. 3-(1-[1,3]oxazolo[5,4-dJpirimidin-2-ilpirrolidin-3-il)-3-[4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila
Uma solução de 3-{2-ciano-1 -[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}-N-(4-hidroxipirimidin-5-il)pirroli- dina-1-carbotioamida (15 mg, 0,025 mmol) em etanol (0,50 mL), foi tratada com nitrato de prata (17,2 mg, 0,10 mmol) e solução de hidróxido de amónio (24 microL). A mistura foi em seguida aquecida para 60°C durante uma hora.
Seguindo completa reação, a mistura foi diluída com acetonitrila, filtrada e j concentrada. O resíduo foi agitado sequencialmente com 1:1 de TFA/DCM durante 1,5 hora, concentrado, em seguida com 0,1 mL de MeOH contendo । 0,2 mL de EDA durante 30 minutos. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). LCMS (M+H)+: 427,0. Exemplo 19. 3-ri-(5-fluoro-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H-pirro- lo[2,3-dlpirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)

2-amino-4-fluorofenol (24 mg, 0,19 mmol, Matrix) foi dissolvido em THF (0,67 mL), e dicloreto carbonotioico (21 microL, 0,28 mmol) foi adicionada. A mistura foi agitada em temperatura ambiente durante duas horas, 5 em seguida concentrada para fornecer um óleo marrom escuro. O resíduo foi redissolvido em 1,4-dioxano (0,50 mL) e N,N-di-isopropiletilamina (98 microL, 0,56 mmol) foi adicionado, seguido por 3-pirrolidin-3-il-3-[4-(7-{[2-(tri- metilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (40,1 mg, 0,0937 mmol, de exemplo 15, etapa 3). A mistura foi agitada a 70°C durante 1,5 hora, em seguida a 80°C durante 2,5 horas. A mistura foi concentrada. O produto bruto foi desprotegido por agitação sequencialmente em uma mistura de 1:1 de TFA/DCM durante 1,5 hora, em seguida concentrada e agitada com 0,3 mL de EDA em 1,5 mL de metanol durante 30 minutos. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6-dMSO): δ 12,12 (br s, 1H), 8,88 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 7,61 (d, 1H), 7,38 (dd, 1H), 7,89 (dd, 1H), 6,99 (d, 1H), 6,78 (ddd, 1H), 4,86 (dt, 1H), 3,86 (dd, 1H), 3,67-3,59 (m, 1H), 3,53-3,28 (m, 4H), 3,02-2,90 (m, 1H), 1,78-1,66 (m, 2H); LCMS (M+H)+: 443,0. Exemplo 20. 3-[1-(4-fluoro-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)

Etapa 1. 2-amino-3-fíuorofenol Cloreto estanhoso, di-hidrato (0,724 g, 3,18 mmols) foram adicionados a uma solução de 3-fluoro-2-nitrofenol (0,100 g, 0,636 mmol, Syn- Quest) em THF (5,0 mL), e água (5,0 mL) e a mistura foi aquecida para 80°C durante 40 minutos. Sob resfriamento para temperatura ambiente, a reação 5 foi diluída com acetato de etila e solução de bicarbonato de sódio saturada.
A mistura foi em seguida filtrada para remover o material insolúvel e as camadas foram separadas. A camada aquosa foi extraída com acetato de etila três vezes. Os extratos foram lavados com salmoura, secados sobre sulfato de sódio, decantados e concentrados para fornecer o produto que foi usado 10 sem outra purificação (65 mg, 80%). LCMS (M+H)+: 128,0. Etapa 2. 3-[1-(4-fluoro-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila
Uma solução de 2-amino-3-fluorofenol (24 mg, 0,19 mmol) em THF (0,67 mL) foi tratada com dicloreto carbonotioico (21 microL, 0,28 15 mmol). A mistura foi agitada em temperatura ambiente durante duas horas e o solvente foi removido em vácuo. O resíduo foi dissolvido em 1,4-dioxano (0,50 mL) e N,N-di-isopropiletilamina (98 microL, 0,56 mmol) foi adicionada, seguido por 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila (40,1 mg, 0,0937 mmol; de e- 20 xemplo 15, etapa 3). A mistura foi agitada a 60°C durante a noite e em seguida concentrada. O produto bruto foi agitado sequencialmente em uma solução de 1:1 de TFA/DCM durante 1,5 hora, concentrado, e em uma solução de 0,3 mL de EDA em 1,5 mL de metanol durante 30 minutos. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um 25 gradiente de ACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6- dMSO): δ 12,13 (br s, 1H), 8,89 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 7,61 (d, 1H), 7,28 (dd, 1H), 7,86-6,95 (m, 3H), 4,86 (dt, 1H), 3,88 (dd, 1H), 3,69-3,61 (m, 1H), 3,56-3,31 (m, 4H), 3,03-2,91 (m, 1H), 1,79-1,66 (m, 2H); LCMS (M+H)+: 443,1. Exemplo 21. 3-[1-(7-fluoro-1,3-benzoxazol-2-il)pirrolidín-3-il1-3-[4-(7H-pirro- lof2,3-d]pirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)
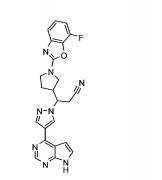
Etapa 1. 2-amino-6-fluorofenol
A 2-fluoro-6-nitrofenol (SynQuest) (2,4 g, 15 mmols) em THF (70 mL), e água (70 mL) foi adicionado dicloreto de estanho (14,6 g, 76,4 mmols). A mistura foi em seguida aquecida a 80°C durante 2 horas. O THF foi removido em vácuo. Uma solução de NaHCO3 saturada foi adicionada, seguido por EtOAc. O material insolúvel foi removido por filtragem. As ca-madas do filtrado foram separadas e a porção aquosa foi extraída com ace- 10 tato de etila trés vezes. Os extratos combinados foram lavados com salmoura, secados sobre sulfato de sódio, filtrados através de um tampão de sílica- gel e concentrados. O produto foi purificado por cromatografia de sílica-gel, eluindo com 0-60% de acetato de etila em hexanos (230 mg, 12%). 1H RMN (300 MHz, CD3OD): δ 6,56 (ddd, 1H), 6,51 (ddd, 1H), 6,41 (ddd, 15 1H); 19F RMN (300 MHz, CD3OD): δ -141,27 (dd, 1F); LCMS (M+H)+: 128,1. Etapa 2. 7-fluorobenzo[dJoxazol-2(3H)-tiona
A uma solução de 2-amino-6-fluorofenol (8,2 g, 64 mmols) em THF (100 mL) a 0°C foi adicionado dicloreto carbonotioico (6,15 mL, 80,6 mmols) gota a gota. A reação foi aquecida para temperatura ambiente e agitada durante 16 horas. O THF foi evaporado em vácuo e o resíduo foi dividido entre água e acetato de etila. O extrato foi lavado com salmoura, secado sobre sulfato de sódio, decantado e concentrado. O produto foi usado sem outra purificação. 1H RMN (500 MHz, d6-dMSO): δ 11,4 (brs, 1H), 7,28 (ddd, 1H), 7,17 (ddd, 1H), 7,86 (dd, 1H); 13C RMN (500 MHz, d6-dMSO): δ 180,12 (s), 144,43 (d), 134,88 (d), 131,47 (d), 126,15 (d), 110,80 (d), 106,85 (d); LCMS (M+H)+: 169,9. Etapa 3. 3-[ 1 -(7-fíuoro-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(tnmetilsi- lil)etóxi]metil}-7H-pirrolo[2!3-d]pinmidin-4-i!)-1 H-pirazol-1 –iljpropanonitrila

3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (0,1 g, 2,3 mmols, preparada no 15 exemplo 15, etapa 3) foi adicionada a uma mistura de 7-fluorobenzo[d]oxa- zol-2(3H)-tiona (0,773 g, 4,57 mmols) e N,N-di-isopropiletilamina (1,59 mL, 9,14 mmols) em 1,4-dioxano (12 mL). A mistura foi aquecida para 60°C durante 16 horas, em seguida para 80°C durante 1,5 hora. O dioxano foi removido em vácuo e substituído com etanol (12 mL). Nitrato de prata (0,776 g, 4,57 mmol) e solução de hidróxido de amónio (29% de água, 1,35 mL) foram adicionados, e a mistura foi agitada durante 16 horas. A reação foi diluída com água e acetato de etila e filtrada para remover o material insolúvel. As camadas foram separadas e a orgânica foi lavada com água e salmoura, secada sobre sulfato de sódio, filtrada e concentrada. O produto foi purifica- do por cromatografia de coluna rápida, eluindo primeiro com 0 a 100% de acetato de etila/hexanos seguido por um gradiente de 0 a 5% de metanol em acetato de etila (0,1 g, 76%). 1H RMN (300 MHz, CDCI3): δ 8,85 (s, 1H), 8,37 (s, 2H), 7,41 (d, 1H), 7,16-7,85 (m, 2H), 6,84-6,76 (m, 2H), 5,68 (s, 2H), 4,51 (dt, 1H), 1,404,01 (m, 1H), 3,87-3,78 (m, 1H), 3,68-3,45 (m, 4H), 3,32-3,10 (m, 2H), 2,99 (dd, 1H), 2,07-1,80 (m, 2H), 0,97-0,88 (m, 2H), -0,06 (s, 9H); LCMS (M+H)+: 573,1. Etapa 4. 3-[1-(7-fluoro-1,3-benzoxazol-2~il)pirroiidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)- 1 H-pirazol-1 -iljpropanonitrila 3-[1-(7-fluoro-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(trime- tilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (0,1 g, 1,7 mmol) foi dissolvida em DCM (33 mL) e TFA (8,3 mL) foi adicionado. A mistura foi agitada em temperatura ambiente durante 5 horas e os solventes foram removidos em vácuo. O resíduo foi dissolvido em metanol (33 mL), e EDA (2,2 mL, 0,033 mol) foi adicionado. Após agitação durante 1 hora, a mistura foi concentrada em vácuo. O produto foi purificado por cromatografia de coluna rápida eluindo com 0 a 5% de metanol em acetato de etila (500 mg, 65%). 1H RMN (400 MHz, d6-dMSO): δ 12,13 (br s, 1H), 8,89 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 7,60 (d, 1H), 7,16-7,89 (m, 2H), 6,99 (d, 1H), 6,91 (ddd, 1H), 4,86 (dt, 1H), 3,89 (dd, 1H), 3,70-3,62 (m, 1H), 3,57-3,30 (m, 4H), 3,03-2,91 (m, 1H), 1,79-1,66 (m, 2H); LCMS (M+H)+: 443,1. Onde desejado, o produto final pode ser purificado também por HPLC/MS (C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH), congelado e liofilizado para fornecer o composto origem. Além disso, o composto pode ser convertido no sal de ácido fosfórico pelo seguinte procedimento: a base livre foi dissolvida em refluxo 3:1 de MeOH:CH2CI2 em uma concentração de aproximadamente 27 mg/mL. Um equivalente de H3PO4 dissolvido em uma pequena quantidade de IPA foi adicionado. O aquecimento foi descontinuado e a mistura resfriada para temperatura ambiente, em seguida o volume de solvente foi reduzido por evaporação giratória até a mistura tornar-se turva. A mistura foi em seguida agitada em temperatura ambiente durante 3 dias. O sólido foi isolado por filtragem e em seguida secado sob vácuo a 50 a 60°C durante a noite. Exemplo 22. 3-[1-(5,7-difluoro-1,3-benzoxazol-2-il)pÍrrolidin-3-ill-3-[4-(7H- pirrolo[2,3-dlpirimidin-4-il)-1H-pirazol-1-illpropanonitrila (um enantiômero isolado)

Preparado de acordo com o método de exemplo 20, etapa 2, partindo com 2-amino-4,6-difluorofenol (Apollo Scientific), com a exceção de que a substituição foi realizada a 60°C durante a noite, em seguida a 80°C durante 3 horas: 1H RMN (400 MHz, d6-dMSO): δ 8,88 (s, 1H), 8,67 (s, 1H), 8,43 (s, 1H), 7,60 (d, 1H), 7,80 (dd, 1H), 6,98 (d, 1H), 6,93 (dt, 1H), 4,86 (dt, 1H), 3,88 (dd, 1H), 3,68-3,60 (m, 1H), 3,58-3,30 (m, 4H), 3,03-2,90 (m, 1H), 1,80-1,67 (m, 2H); LCMS (M+H)+: 461,0. Exemplo 23. 3-41-f2-(metiltio)pirimidin-4-il]pirrolidin-3-il}-3-f4-(7H-pirrolo[2,3- dlpirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)
Uma solução de 3-pirrolidin-3-ÍI-3-[4-(7-{[2-(trimetilsilil)etóxi] me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (51 mg, 0,12 mmol; de exemplo 15, etapa 3) e 4-cloro-2-(metiltio)pÍrimidina (22,5 mg, 0,140 mmol, Aldrich) em 1,4-dioxano (0,20 mL), e contendo N,N-di-isopro- piletilamina (40 microL, 0,233 mmol) foi aquecida para 70°C durante uma hora. A mistura foi concentrada e desprotegida por agitação sequencialmente em 1:1 de TFA/DCM durante 1,5 hora, concentrada, em seguida com 0,3 mL de EDA em 1,5 mL de metanol durante 30 minutos. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6-dMSO): δ 12,12 (brs, 1H), 8,87 (s, 1H), 8,69 (s, 1H), 8,43 (s, 1H), 8,00 (brs, 1H), 7,61 (d, 1H), 6,99 (d, 1H), 6,19 (br s, 1H), 4,81 (dt, 1H), 3,94-2,32 (10H), 1,74- 1,57 (m, 2H); LCMS (M+H)+: 432,0. Exemplo 24. 3-{1-[2-(metilsulfinil)pirimidin-4-il]pirrolídin-3-il}-3-[4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-ilipropanonitrila (um enantiômero isolado)

3-{1 -[2-(metiltio)pirimidin-4-il]pirrolidin-3-il}-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (6,1 mg, 0,014 mmol, de exemplo 23) foi dissolvida em DCM (0,1 mL) e resfriada para -10°C. Ácido M- cloroperbenzoico (3,2 mg, 0,014 mmol) em DCM foi adicionado gota a gota. A mistura foi lentamente aquecida para temperatura ambiente durante 3 horas. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6-dMSO): δ 12,13 (br s, 1H), 8,87 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H). 8,30 (d, 0,5H), 8,24 (dd, 0,5H), 7,61 (d, 1H), 6,98 (d, 1H), 6,54 (br t, 1H), 4,88-4,80 (m, 1H), 3,96-2,82 (m, 7H), 2,80 (s, 1,5H), 2,73 (d, 1,5H), 1,81-1,58 (m, 2H); LCMS (M+H)+: 448,0. Exemplo 25. 3-{ 1 -[2-(metilsulfonil)pirimidin-4-inpirrolidin-3-il}-3-[4-(7H-pírro- lo[2,3-dlpirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (um enantiômero isolado)
A uma solução de ácido M-cloroperbenzoico (8,2 mg, 0,036 mmol) em DCM (1,2 mL) a -5°C foi adicionada 3-{1 -[2-(metiltio)pirimidin-4- il]pirrolidÍn-3-il}-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanoni- trila (7,5 mg, 0,017 mmol, de exemplo 23) em DCM (0,6 mL), gota a gota. A mistura foi deixada aquecer lentamente para 0°C, em seguida o banho foi removido e a mistura foi agitada em temperatura ambiente durante 50 minutos. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6-dMSO): δ 12,14 (br s, 1H), 8,88 (d, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 8,34 (d, 0,5H), 8,28 (d, 0,5H), 7,63-7,59 (m, 1H), 6,99 (dd, 1H), 6,69 (dd, 1H), 4,89-4,80 (m, 1H), 3,96-3,30 (6H), 3,32 (s, 1,5H), 3,25 (s, 1,5H), 3,02-2,84 (m, 1H), 1,82-1,59 (m, 2H); LCMS (M+H)+: 464,0. Exemplo 26. 34146-(metilsulfonil)piridin-2-illpirrolidin-3-il}-3-[4-(7H-pirro- lo(213-d1pirimidin-4-il)-1 H-pirazol-1-illpropanomtrila (um enantiômero isolado)

Etapa 1. 2-cloro-6-(metilsulfonil)pindina
Ácido M-cloroperbenzoico (326 mg, 1,46 mmol) em DCM (35 mL) foi resfriada para -5°C. 2-cloro-6-(metiltio)piridina (101 mg, 0,633 mmol, preparado de acordo com o método reportado em J. Org. Chem., 67(1), 234237; 2002) em DCM (5,0 mL) foi adicionado gota a gota. A mistura foi deixa-da aquecer para 0°C lentamente, em seguida o banho foi removido e a mis- tura alcançou a temperatura ambiente e foi agitada durante mais 2 horas. A solução de reação foi em seguida lavada com NaHCO3 saturada, salmoura, secada sobre sulfato de sódio, filtrada e concentrada para fornecer o produto (120 mg, 99%). 1H RMN (400 MHz, CDCI3): δ 8,03 (dd, 1H), 7,94 (t, 1H), 5 7,59 (dd, 1H), 3,26 (s, 3H); LCMS (M+H)+: 191,9, 194,0. Etapa 2. 3-{ 1 -[6-(metilsulfonil)piridin-2-il]pirrolidin-3-il}-3-[4-(7H-pirrolo[2,3- d]pirímidin-4-il)-1 H-pirazol-1-iljpropanonitrila
Uma solução de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila (21 mg, 0,048 10 mmol; de exemplo 15, etapa 3) e 2-cloro-6-(metilsulfonil)piridina (10 mg, 0,053 mmol) em etanol (0,050 mL) e N,N-di-isopropiletilamina (17 microL, 0,096 mmol) foi aquecida em um frasco selado por meio de um banho de óleo mantido a 120°C durante 4 horas. A mistura foi resfriada e concentrada.
O produto bruto foi desprotegido por agitação sequencialmente em 1:1 de 15 TFA/DCM durante 1,5 hora, em seguida concentrado e agitado com 0,2 mL de EDA em 1,5 mL de metanol durante 30 minutos. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6-dMSO): δ 12,10 (br s, 1H), 8,88 (s, 1H), 8,69 (s, 1H), 8,44 (s, 1H), 7,76 (dd, 1H), 7,61 (d, 1H), 7,13 (d, 1H), 6,99 (d, 1H), 6,74 (d, 1H), 4,84 (dt, 1H), 3,83-3,71 (m, 1H), 3,60-3,48 (m, 1H), 3,42 (dd, 1H), 3,38-3,24 (m, 3H), 3,21 (s, 3H), 3,002,87 (m, 1H), 1,75-1,61 (m, 2H); LCMS (M+H)+: 463,0. Exemplo 27. 3-(1-[2-(metilsulfonil)pirÍdin-4-il1pÍrrolidin-3-il}-3-í4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1H-pirazol-1-iilpropanonitrila (um enantiômero isolado)
Preparado de acordo com o método de exemplo 26, usando 4- cloro-2-(metiltio)piridina (preparado de acordo com o procedimento reportado em Tetrahedron, 62(26), 6166-6171, 2006) como material de partida na etapa 1 e a modificação à etapa 2 cuja reação de substituição foi realizada a 120°C durante 1 hora. 1H RMN (400 MHz, d6-dMSO): δ 12,11 (br s, 1H), 8,88 (s, 1H), 8,69 (s, 1H), 8,44 (s, 1H), 8,24 (d, 1H), 7,61 (d, 1H), 7,84 (brs, 5 1H), 6,98 (d, 1H), 6,70-6,65 (m, 1H), 4,87-4,77 (m, 1H), 3,70-3,61 (m, 1H), 3,50-3,22 (m, 5H), 3,19 (s, 3H), 3,06-2,89 (m, 1H), 1,77-1,64 (m, 2H); LCMS (M+H)+: 463,1. Exemplo 28. 3-[1-(1-óxido-2,3-di-hidrotienoí2,3-blpiridin-6-il)pirrolidin-3-il1-3- [4-(7H-pirrolo[2,3-dlpirimidin-4-il)-1 H-pirazol-1 -illpropanonitrila (um enantiõ- 10 mero isolado)
Etapa 1. 2,6-dicloro-3-[2-metoxivinil]piridina
A uma solução de cloreto de (metoximetil)(trifenil)fosfônio (9,97 g, 29,1 mmols) em THF (80 mL) a 0°C sob uma atmosfera de nitrogênio foi 15 adicionado 0,1 M de terc-butóxido de potássio em THF (29,1 mL, 29,1 mmols). Após agitação durante 30 minutos, uma solução de 2,6-dicloroni- cotinaldeído (3,01 g, 17,1 mmol, Aldrich) em THF (22 mL) foi adicionada gota a gota. A solução resultante foi agitada a 0°C durante 20 minutos, em seguida para temperatura ambiente durante 1 hora.
A reação foi extinguida pela adição de água e o produto foi extraído com três porções de acetato de etila. Os extratos orgânicos combinados foram lavados com salmoura, secados sobre Na2SO4, filtrados e concentrados. O produto bruto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 0 a 10% de acetato de etila em he- xanos para fornecer o produto como uma mistura de isômeros de olefina (3,1 g, 80%). 1H RMN, uma mistura de 1:1 de isômeros de olefina (300 MHz, CDCI3): δ 8,34 (d, 1H), 7,60 (d, 1H), 7,19 (d, 1H), 7,17 (d, 1H), 7,84 (d, 1H), 6,36 (d, 1H), 5.94 (d, 1H), 5,53 (d, 1H), 3,83 (s, 3H), 3,75 (s, 3H); LCMS (M+H)+: 204,0. Etapa 2. 2-(2,6-dicloropiridin-3-il)etanol 2,6-dicloro-3-[2-metoxivinil]piridina (3,1 g, 14 mmols) foi dissolvido em THF (41 mL), e cloreto de hidrogênio a 4,0 M em água (12 mL) foi adicionado. A mistura foi aquecida até o refluxo durante 3 horas. A reação foi resfriada para temperatura ambiente e os solventes foram removidos em vácuo. O resíduo foi dissolvido em acetato de etila, lavado com NaHCO3 saturado, salmoura, secado sobre sulfato de sódio e concentrado para fornecer um óleo amarelo claro. O produto bruto foi dissolvido em metanol (51 mL) e foi resfriado para 0°C. Boroidreto de sódio (0,517 g, 13,7 mmols) foi adicionado e a reação agitada durante 30 minutos nesta temperatura. A mistura foi extinguida pela adição de cloreto de amónio saturado, o metanol foi removido por evaporação giratória, em seguida solução aquosa restante foi extraída com acetato de etila três vezes. Os extratos combinados foram secados sobre sulfato de sódio e concentrados. O produto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 0 a 50% de acetato de etila em hexanos para fornecer a incolor oil (1,47 g, 56%). 1H RMN (300 MHz, CDCI3): δ 7,62 (d, 1H), 7,23 (d, 1H), 3,92 (q, 2H), 2,97 (t, 2H), 1,55 (t, 1H); LCMS (M+H)+: 192,0. Etapa 3. 2-(2,6-dicloropindin-3-ÍI)etanotiol
A uma solução de 2-(2,6-dicloropiridin-3-il)etanol (0,500 g, 2,60 mmol) e trifenilfosfina (0,12 g, 3,90 mmols) em THF (20 mL) a 0°C foi adicionada azodicarboxilato de dietila (615 microL, 3,90 mmols). Após 10 minutos, ácido tioacético (279 microL, 3,90 mmols) foi adicionado. A mistura foi agitada durante duas horas em temperatura ambiente. A reação foi diluída com hexanos e o precipitado branco foi filtrado. O filtrado foi concentrado e o tio- acetato bruto resultante foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 0 a 10% de acetato de etila em hexanos. 1H RMN (300 MHz, CDCI3): δ 7,96 (d, 1H), 7,24 (d, 1H), 3,13 (dd, 2H), 2,98 (dd, 2H), 2,34 (s, 3H); LCMS (M+H)+: 250,0.
O tioacetato foi agitado durante a noite em uma solução de cio- reto de acetila (4 eq.) em metanol (20 mL). O solvente foi removido em vácuo para fornecer o produto como um óleo viscoso (320 mg, 59%). 1H RMN (300 MHz, CDCI3): δ 7,57 (d, 1H), 7,24 (d, 1H), 3,01 (t, 2H), 2,82 (q, 2H), 1,39 (t, 1H); LCMS (M+H)+: 208,0. Etapa 4. 1-óxido de 6-cloro-2,3-di-hidrotieno[2,3-b]piridina e 1,1-dióxido de 6- cloro-2,3-dÍ-hidrotieno[2,3-b]pirídina 2-(2,6-dicloropiridin-3-il)etanotiol (0,25 g, 0,85 mmol) foi dissolvido em DMF (8,7 mL). A solução foi desgaseificada passando um sistema de nitrogênio através da solução durante 15 minutos. A solução foi em seguida resfriada para 0°C e hidreto de sódio (60% de óleo mineral, 68 mg, 1,7 mmol) foi adicionada e a reação foi agitada nesta temperatura durante 1,5 hora. A reação foi extinguida pela adição de água (80 mL) e o produto foi extraído com acetato de etila (100 mL). A camada orgânica foi lavada com água (3x), salmoura (1x), secada sobre sulfato de sódio e concentrada. O produto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 0 a 30% de acetato de etila em hexanos para fornecer o produto como um óleo amarelo claro (150 mg, 92%). LCMS (M+H)+: 172,0.
Ácido M-cloroperbenzoico (110 mg, 0,49 mmol) em DCM (5,0 mL) foi adicionado a uma solução de 6-cloro-2,3-di-hidrotieno[2,3-b]piridina (71 mg, 0,38 mmol) em DCM (21 mL) a 0°C. A solução clara foi deixada alcançar a temperatura ambiente e agitar durante uma hora. A mistura foi extinguida com solução de Na2S2O3 saturada seguido por solução de NaH- CO3 saturada. A camada orgânica foi lavada com água, salmoura, secada sobre sulfato de sódio e concentrada. O produto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 0 a 100% de acetato de etila em hexanos para fornecer sulfona (17 mg, 22% de rendimento), seguido por uma mudança no eluente para 5% de metanol em acetato de etila para fornecer sulfóxido (29 mg, 41% de rendimento). 1-óxido de 6-cloro-2,3-di-hidrotieno[2,3-b]piridina: 1H RMN (300 MHz, CDCI3): δ 7,80 (d, 1H), 7,45 (d, 1H), 3,91-3,76 (m, 1H), 3,46-3,28 (m, 3H); LCMS (M+H)+: 188,1. 1,1-dióxido de 6-cloro-2,3-di-hidrotieno[2,3-b]piridina: 1H RMN (400 MHz, CDCI3): δ 7,75 (d, 1H), 7,53 (d, 1H), 3,56 (t, 2H), 3,36 (t, 2H); LCMS (M+H)+: 204,0. Etapa 5. 3-[ 1-(1 -óxido-2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4- (7H-pirrolo[2,3-d]pirimidin-4-il)- 1 H-pirazol-1 -iljpropanonitrila 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirÍmidin-4-il)-1 H-pirazol-1-iljpropanonitrila (21 mg, 0,048 mmol; de exemplo 15, etapa 3) e 1-óxido de 6-cloro-2,3-di-hidrotieno[2,3-b]piridina (10,0 mg, 0,0533 mmol) foram dissolvidos em etanol (0,050 mL) e N,N-di-isopropiletila- mina (17 microL, 0,1 mmol). A mistura foi aquecida em um frasco selado por meio de um banho de óleo mantido a 120°C durante 5,5 horas. A mistura foi concentrada. O resíduo foi dissolvido em uma mistura de 1:1 de TFA/DCM, agitado durante 1,5 hora, em seguida concentrado novamente. O resíduo foi dissolvido em 1,5 mL de metanol contendo 0,2 mL de EDA e foi agitado durante 30 minutos. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6-dMSO): δ 12,04 (br s, 1H), 8,81 (s, 1H), 8,62 (s, 1H), 8,36 (s, 1H), 7,63 (dd, 1H), 7,53 (d, 1H), 6,92 (d, 1H), 6,56 (dd, 1H), 4,81-4,76 (m, 1H), 3,75-3,67 (m, 1H), 3,49-2,78 (10H), 1,72-1,55 (m, 2H); LCMS (M+H)+: 459,1. Exemplo 29. 3-[1-(2,3-di-hidrotieno[2,3-b1piridin-6-il)pirrolidin-3-il1-3-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il1propanonitrila (um enantiômero iso-

A uma solução de 3-[1-(1-óxido-2,3-di-hidrotieno[2,3-b]piridin-6- il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4- il)-1 H-pirazol-1-iljpropanonitrila (36 mg, 0,061 mmol, preparada no exemplo 28) em isopropil álcool (0,1 mL) foi adicionado índio (21 mg, 0,18 mmol) e cloreto de 2,2-dimetilpropanoíla (45 microL, 0,37 mmol). A mistura foi agitada em temperatura ambiente durante 3 horas. A reação foi diluída com solução de Na2CO3 saturada, extraída três vezes com DCM e os extratos combinados foram concentrados até a secura. O resíduo foi dissolvido em uma mistura de 1:1 de TFA/DCM e foi agitado durante 1,5 hora, e em seguida os solventes foram removidos em vácuo. O resíduo foi agitado em uma mistura de 1,5 mL de metanol e 0,2 mL de EDA durante 30 minutos. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). 1H RMN (400 MHz, d6- dMSO): δ 12,11 (br s, 1H), 8,87 (s, 1H), 8,69 (s, 1H), 8,42 (s, 1H), 7,61 (d, 1H), 7,29 (d, 1H), 6,99 (d, 1H), 6,04 (d, 1H), 4,79 (dt, 1H), 3,67 (dd, 1H), 3,44-3,25 (m, 5H), 3,23-3,14 (m, 2H), 3,13-3,06 (dd, 2H), 2,92-2,80 (m, 1H), 1,75-1,56 (m, 2H); LCMS (M+H)+: 443,2. Exemplo 30. 3-[1-(1 ,1-dióxído-2,3-dí-hidrotieno[2.3-b]piridin-6-il’)pirrolidin-3-il1- 3-[4-(7H-pirroloí2,3-d1pirimidin-4-il)-1 H-pirazol-1 -illpropanonitrila (um enantiômero isolado)

Preparado no exemplo 28, etapa 5 usando 3-pirroíidin-3-il-3-[4- (7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin^-il)-1 H-pirazol-1 - iljpropanonitrila (21 mg, 0,048 mmol; de exemplo 15, etapa 3) e 1,1-dióxido de 6-cloro-2,3-di-hidrotieno[2,3-b]piridina (10,7 mg, 0,0528 mmol, de exemplo 28, etapa 4) em etanol (0,050 mL) e N,N-di-isopropiletilamina (17 microL, 0,1 mmol) com a exceção de que o tempo de reação de substituição foi de 2,5 horas. 1H RMN (400 MHz, CDCI3): δ 8,88 (s, 1H), 8,69 (s, 1H), 8,43 (s, 1H), 7,68 (d, 1H), 7,61 (d, 1H), 6,99 (d, 1H), 6,74 (d, 1H), 4,84 (dt, 1H), 3,78 (dd, 1H), 3,58-3,22 (m, 7H), 3,11 (dd, 2H), 2,98-2,86 (m, 1H), 1,78-1,61 (m, 2H); LCMS (M+H)+: 475,0. Exemplo 31. 3-(3-fluoro-1-[1,31oxazolo[5,4-b]piridin-2-ilpirrolidin-3-il)-3-f4- (7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -illpropanonitrila (quatro estereoi- sómeros isolados)
Etapa 1. 3-[2-cianovini!]-3-fluoropirrolidina-1-carboxilato de terc-butila
A uma solução de 0,10 M de terc-butóxido de potássio em THF (6,4 mL, 6,4 mmols) a 0°C foi adicionada uma solução de cianometilfosfona- to de dietila (0,11 mL, 6,27 mmols) em THF (10 mL) gota a gota. O banho foi removido e a reação foi aquecida para temperatura ambiente e agitada durante 30 minutos. A mistura foi re-resfriada para 0°C e uma solução de 3- fluoro-3-formilpirrolidina-1-carboxilato de terc-butila (1,29 g, 5,94 mmols, preparada como descrito em US2007/0037853) em THF (10 mL) foi adicionada gota a gota. A reação foi agitada durante a noite com aquecimento para temperatura ambiente. A mistura reacional foi diluída com acetato de etila e água, a solução aquosa foi extraída com acetato de etila três vezes e os extratos combinados foram lavados com salmoura, secados sobre sulfato de sódio, filtrados e concentrados. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 60% de acetato de etila em hexanos forneceu cis- e trans- olefinas, combinadas e usadas na etapa subsequente (950 mg, 66%). 1H RMN trans- olefina (300 MHz, CDCI3): δ 6,66 (dd, 1H), 5,78 (d, 1H), 3,81-3,30 (m, 4H), 2,27-1,94 (m, 2H), 1,43 (s, 9H). 19F RMN trans- olefina (300 MHz, CDCI3): δ -158,3 (m, 1F). 1H RMN cis- olefina (300 MHz, CDCI3): δ 6,45 (ddd, 1H), 5,56 (dd, 1H), 3,92-3,34 (m, 4H), 2,44-2,02 (m, 2H), 1,44 (s, 9H). 19F RMN cis- olefina (300 MHz, CDCI3): δ -151,9 (m, 1F). Etapa 2. 3-{2-ciano- 1-[4-(7-{[2-(tnmetilsilil)etóxiJmetil}-7H-pirrolo[2,3-d]pirimi- din-4-il) -1 H-pirazol-1 -HJetil}-3-fluoropirrolidina-1 -carbox Ha to de terc-butila 3-[2-cianovinil]-3-fluoropÍrrolidina-1 -carboxilato de terc-butila (0,95 g, 4,0 mmols) (como uma mistura de isómeros de olefina) e 4-(1H- pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidina (1,2 g, 4,0 mmols, preparada como descrito no WO 2007/070514, Ex. 65, ou US2007/135461) em acetonitrila (10 mL) foram tratados com 1,8-diazabici- clo[5,4,0]undec-7-eno (0,59 mL, 4,0 mmols) e agitados em temperatura ambiente durante 30 minutos. O solvente foi removido em vácuo e o resíduo foi purificado por cromatografia de coluna rápida em silica-gel, eluindo com um gradiente de 5% de IPA/5% de acetato de etila/90% de hexanos a 10% de IPA/50% de acetato de etila/40% de hexanos. Diastereômero 1 (primeiro a eluir) (0,92 g, 42%), e diastereômero 2 (segundo a eluir) (0,91 g, 41%). 1H RMN diastereômero 1 (400 MHz, CDCI3): δ 8,86 (s, 1H), 8,39 (s, 1H), 8,34 (s, 1H), 7,42 (d, 1H), 6,81-6,78 (m, 1H), 5,68 (s, 2H), 4,93-4,81 (m, 1H), 3,84- 3,34 (m, 7H), 3,08 (dt, 1H), 2,37-2,13 (m, 1H), 1,93 (dt, 1H), 1,45 (s, 9H), 0,95-0,89 (m, 2H), -0,06 (s, 9H) 19F RMN diastereômero 1 (400 MHz, CD- CI3): -158,8 (m, 1F); LCMS (M+H)+: 556,2. 1H RMN diastereômero 2 (400 MHz, CDCI3): δ 8,86 (s, 0,5H), 8,85 (s, 0,5H), 8,39 (s, 0,5H), 8,37 (s, 0,5H), 8,35 (s, 0,5H), 8,30 (s, 0,5H), 7,44-7,40 (m, 1H), 6,81-6,77 (m, 1H), 5,68 (s, 2H), 4,87 (ddd, 1H), 3,83-3,38 (m, 6H), 3,34 (dd, 1H), 3,17 (dd, 1H), 2,34- 2,18 (m, 1H), 2,07-1,79 (m, 1H), 1,42 (s, 9H), 0,96-0,88 (m, 2H), -0,06 (s, 9H); 19F RMN diastereômero 2 (400 MHz, CDCI3): δ -157,6 (m, 1F); LCMS (M+H)+: 556,2. Etapa 3a. 3-(3-fluoropirrolidin-3-il)-3-[4-(7-{[2-(tnmetilsiiil)etóxi]metil}-7H-pirro- lo[2,3-d]pirimidin-4-i!)-1 H-pirazoi-1-iljpropanonitrila
A uma solução de 3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}-3-fluoropirrolidina-1-carbo- xilato de terc-butila (0,200 g, 0,360 mmol) (diastereômero 1 de etapa 2) em 1,4-dioxano (10 mL) foi adicionado cloreto de hidrogênio a 4,0 M em 1,4- dioxano (0,70 mL, 2,8 mmols) e agitado em temperatura ambiente até a reação ser concluída. O solvente foi removido em vácuo. O resíduo foi dividido entre NaOH a 1N e acetato de etila, as camadas foram separadas e a porção aquosa foi extraída com mais três porções de acetato de etila. Os extratos combinados foram lavados com salmoura, secados sobre sulfato de só- dio, decantados e concentrados. O produto foi usado sem outra purificação. LCMS (M+H)+: 456,0. Etapa 3b. 3-(3-fluoropirrolidin-3-il)-3-[4-(7-{[2-(trimetilsilil)etóxiJmetilJ-7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila O procedimento descrito para a etapa 3a foi seguido, usando 3- {2-ciano-1 -[4-(7-{[2-(tri met i Isil i l)etóxi] metil}-7 H-p irro lo[2,3-d]pi ri m id i n-4-i I)-1H- pirazol-1 -il]etil}-3-fluoropirrolidina-1 -carboxilato de terc-butila (0,480 g, 0,864 mmol) (diastereômero 2 de etapa 2). LCMS (M+H)+: 456,0. Etapa 4a. 3-(3-fluoro-1 -[1,3Joxazolo[5,4-b]piridin-2-ilpirrolidin-3-il)-3-[4-(7H- pirrolo[2,3-dJpirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila
Uma solução de oxazolo[5,4-b]piridina-2(1 H)-tiona (0,042 g, 0,27 mmol, de exemplo 33, etapa 4) e 3-(3-fluoropirrolidin-3-il)-3-[4-(7-{[2-(trime- tilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (0,100 g, 0,219 mmol, de etapa 3a) em 1,4-dioxano (2 mL) contendo N,N-di- isopropiletilamina (153 microL, 0,878 mmol) foi aquecida para 70°C durante 2 horas. A mistura reacional resfriada foi concentrada em vácuo. O resíduo foi reconstituído em etanol (2 mL) e a suspensão resultante foi tratada com nitrato de prata (0,149 g, 0,878 mmol) e hidróxido de amónio aquoso (0,5 j mL) e agitada em temperatura ambiente durante a noite. Água e NaOH a 1N foram adicionados na reação que foi em seguida agitada durante 15 minutos e subsequentemente filtrada. O filtrado foi extraído com acetato de etila três vezes e os extratos combinados foram lavados com salmoura, secados sobre sulfato de sódio, filtrados e concentrados. O produto foi desprotegido por agitação com 25% de TFA/DCM durante 3 horas, seguido por evaporação dos solventes e agitação do resíduo com excesso de EDA em metanol. Quando a etapa de desproteção foi concluída, o produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) para fornecer o racemato purificado (20 mg, 20%), uma porção da qual foi separada em seus enantiômeros por HPLC quiral (Coluna Phenomenex Lux-celulose-1, 5 μ, 20 x 250 mm, 70% de EtOH/ Hexanos, 8 mL/minuto) para fornecer enantiômero 1 (primeiro a eluir, tempo de retenção de 26,9 minutos) e enantiômero 2 (segundo a eluir, tempo de retenção de 31,7 minutos). 1H RMN (300 MHz, d6-dMSO): δ 12,11 (br s, 1H), 8,90 (s, 1H), 8,70 (s, 1H), 8,46 (s, 1H), 7,90 (dd, 1H), 7,66 (dd, 1H), 7,63 (d, 1H), 7,22 (dd, 1H), 7,82 (d, 1H), 5,43 (ddd, 1H), 1,44-3,55 (m, 5H), 3,50 (dd, 1H), 2,50-2,25 (m, 1H), 1,88-1,73 (m, 1H); 19F RMN (300 MHz, d6-dMSO): δ -159,6; LCMS (M+H)+: 444,0. Etapa 4b. 3-(3-fíuoro-1-[1,3Joxazolo[5,4-bJpiridin-2-ilpirrolidin-3-il)-3-[4-(7H- pirrolo[2,3-d]pirímidin-4-il)-1 H-pirazol-1 -iljpropanonitrila
O procedimento descrito para a etapa 4a foi seguido, usando o produto de etapa 3b. O produto foi submetido à HPLC quiral para separar os enantiômeros (Coluna Phenomenex Lux-celulose-1, 5μ, 20 x 250 mm, 60% de EtOH/Hexanos, 10 mL/minuto) para fornecer enantiômero 1 (primeiro a eluir, tempo de retenção de 18,0 minutos) e enantiômero 2 (segundo a eluir, tempo de retenção de 25,7 minutos). 1H RMN (300 MHz, d6-dMSO): δ 12,12 (brs, 1H), 8,92 (s, 1H), 8,72 (s, 1H), 8,49 (s, 1H), 7,87 (dd, 1H), 7,63 (d, 1H), 7,61 (dd, 1H), 7,19 (dd, 1H), 7,83 (d, 1H), 5,44 (ddd, 1H), 1,42-3,30 (m, 6H), 2,54-2,26 (m, 2H); 19F RMN (300 MHz, d6-dMSO): δ -160,2; LCMS (M+H)+: 444,0. Exemplo 32. 3-(1-[1,31oxazolo[5,4-blpíridin-2-ilpirrolidin-3-il)-3-[3-(7H-pirro- lo[2.3-dlpirimidin-4-il)-1H-pirrol-1-inpropanonitrila (dois enantiômeros e um
Etapa 1. 3-(2-ciano-1 ~{3-[7-(dietoximetil) - 7H-pirrolo[2,3-dJpirimidin-4-ilJ-1H- pirrol-1-il}etil)pirrolidina-1 -carboxilato de terc-butila
Uma solução de 3-[2-cianovinil]pirrolidina-1 -carboxilato de terc- butila (0,480 g, 2,16 mmols, preparada no exemplo 15, etapa 1) e 7-(dieto- ximetil)-4-(1 H-pirrol-3-il)-7H-pirrolo[2,3-d]pirimidina (0,72 g, 2,2 mmols, preparada como no WO 2007/070514 Ex. 500) em acetonitrila (5 mL) foi tratada com 1,8-diazabiciclo[5,4,0]undec-7-eno (0,323 mL, 2,16 mmols) e agitada durante 5 dias. O solvente foi removido por evaporação giratória e o produto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 50 a 90% de acetato de etila em hexanos para fornecer di- astereômeros separados. Diastereômero 1 (primeiro a eluir): 279 mg, 25%.
Diastereômero 2 (segundo a eluir): 352 mg, 32%. 1H RMN (300 MHz, CDCI3) diastereômero 1: Õ 8,78 (s, 1H), 7,68 (t, 1H), 7,53 (d, 1H), 6,98 (dd, 1H), 6,91 (t, 1H), 6,83 (d, 1H), 6,77 (s, 1H), 1,42-4,03 (m, 1H), 3,80-3,63 (m, 3H), 3,60-3,38 (m, 3H), 3,34-3,20 (m, 1H), 3,17-3,05 (m, 1H), 2,89 (d, 2H), 2,91-2,78 (m, 1H), 1,89-1,76 (m, 1H), 1,68- 10 1,52 (m, 1H), 1,47 (s,9H), 1,23 (t,6H); LCMS (M+H)+: 509,1. 1H RMN (300 MHz, CDCI3) diastereômero 2: Õ 8,78 (s, 1H), 7,65 (br s, 1H), 7,53 (br d, 1H), 7,80-6,79 (m, 3H), 6,77 (s, 1H), 1,47-4,05 (m, 1H), 3,79-3,28 (m, 7H), 3,09-2,80 (m, 4H), 2,27-2,13 (m, 1H), 1,79-1,60 (m, 1H), 1,44-1,34 (m, 9H), 1,23 (t, 6H); LCMS (M+H)+: 509,1. Etapa 2a. 3-(1-[1,3Joxazolo[5,4-b]pindin-2-ilpirrolidin-3-il)-3-[3-(7H-pirrolo[2,3- d]pirimidin-4-il)- 1 H-pirrol-1 -iljpropanonitrila
A uma solução de 3-(2-ciano-1-{3-[7-(dietoximetil)-7H-pirrolo[2,3- d]pirimidin-4-il]-1 H-pirrol-1-il}etil)pirrolidina-1 -carboxilato de terc-butila (0,060 g, 0,12 mmol, diastereômero 1 de etapa 1) em 1,4-dioxano (2 mL) foi adicio- 20 nado cloreto de hidrogênio a 4,00 M em 1,4-dioxano (0,24 mL, 0,94 mmol). A reação foi agitada durante 16 horas. O solvente foi removido por evaporação giratória e o produto globalmente desprotegido, 3-pirrolidin-3-il-3-[3-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 -iljpropanonitrila, foi usado sem outra purificação. LCMS (M+H)+: 307,1.
A uma solução de 3-aminopiridÍn-2-ol (2,00 g, 18,2 mmols, 3B Scientific) em THF (40 mL) foi adicionada tiofosgênio (1,52 mL, 20 mmols). Água foi adicionada e o pH ajustado para 4 a 5. O produto foi extraído com acetato de etila, secado sobre sulfato de sódio e concentrado. O sólido material foi triturado com éter durante a noite, e o produto foi filtrado e secado 30 por ar. (6,25 g, 41,1 mmols) de oxazolo[5,4-b]piridina-2(1H)-tiona preparada nesta maneira foi misturado com tolueno (100 mL) e foi tratado com cloreto de tionila (9,0 mL, 120 mmols) e algumas gotas de DMF. A reação foi aque- cida até o refluxo durante 1 hora. Sob resfriamento para temperatura ambiente e repouso durante a noite, um precipitado formou-se que foi isolado por filtragem e secado por ar. Este produto bruto (23 mg) e 3-pirrolidin-3-il-3-[3- (7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1-íl]propanonitrila (0,036 g) em 1,4- dioxano (0,6 mL) contendo N,N-di-isopropiletÍlamina (82 microL, 0,47 mmol) foram aquecidos para 70°C durante 2 horas. O solvente foi removido em vácuo. O resíduo foi reconstituído em etanol (0,8 mL) e a suspensão resultante foi tratada com nitrato de prata (0,040 g, 0,23 mmol) e solução de hidróxido de amónio (36 microL). Após agitação durante 16 horas, a reação foi preparada por divisão entre água e acetato de etila. As camadas foram separadas e a porção aquosa foi extraída com acetato de etila três vezes. Os extratos combinados foram secados sobre sulfato de sódio, filtrados e concentrados. O produto foi purificado por meio de HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH). Uma porção deste produto foi purificada por HPLC quiral (Chiral Technologies Chiral- pak IA, 5p, 20 x 250 mm, eluida com 45% de EtOH/Hexanos) para fornecer enantiômero 1 (primeiro a eluir, tempo de retenção de 47,8 minutos) e enantiômero 2 (segundo a eluir, tempo de retenção de 53,4 minutos). 1H RMN (300 MHz, d6-dMSO) enantiômero 1: δ 11,97 (br s, 1H), 8,61 (s, 1H), 8,01 (dd, 1H), 7,87 (dd, 1H), 7,62 (dd, 1H), 7,51 (d, 1H), 7,20 (dd, 1H), 7,16 (dd, 1H), 6,96-6,90 (m, 2H), 4,57 (dt, 1H), 3,85 (dd, 1H), 3,713,60 (m, 1H), 3,53-3,23 (m, 4H), 2,99-2,83 (m, 1H), 1,76-1,56 (m, 2H); LCMS (M+H)+: 425,1. 1H RMN (300 MHz, d6-dMSO) enantiômero 2: δ 11,97 (br s, 1H), 8,61 (s, 1H), 8,01 (dd, 1H), 7,87 (dd, 1H), 7,62 (dd, 1H), 7,51 (d, 1H), 7,20 (dd, 1H), 7,16 (dd, 1H), 6,97-6,93 (m, 2H), 4,59 (dt, 1H), 3,86 (dd,1H), 3,733,62 (m, 1H), 3,56-3,24 (m, 4H), 3,01-2,85 (m, 1H), 1,75-1,61 (m, 2H); LCMS (M+H)+: 425,1. Etapa 2b. 3-(1-[ 1,3Joxazolo[5,4-b]pindin-2-ilpirrolidin-3-il)-3-[3-(7H-pirrolo[2,3- d]pirimidin-4-il) -1 H-pirrol-1 -iljpropanonitrila
O procedimento para a etapa 2a foi seguido, usando diastereô- mero 2 de etapa 1 como material de partida para fornecer o produto como o racemato. 1H RMN (400 MHz, d6-dMSO): δ 11,97 (br s, 1H), 8,61 (s, 1H), 8,00 (dd, 1H), 7,81 (dd, 1H), 7,53 (dd, 1H), 7,50 (dd, 1H), 7,17-7,13 (m, 2H), 6,96 (dd, 1H), 6,93 (dd, 1H), 4,60 (dt, 1H), 3,80-3,73 (m, 1H), 3,65-3,56 (m, 1H), 3,47 (dd, 1H), 3,39-3,23 (m, 3H), 3,05-2,94 (m, 1H), 2,31-2,20 (m, 1H), 1,98-1,88 (m, 1H); LCMS (M+H)+: 425,0. Exemplo 33. Fosfato de 3-(1T1,3]oxazolof5.4-b1piridin-2-ilpirrolidin-3-il)-3-[3- (7H-pirrolo[2,3-d1pirimidin-4-il)-1 H-pirrol-1 -illpropanonitrila (um enantiômero convertido na forma de sal)
Etapa 1. 4-(1 H-pirrol-3-il)-7-{[2-(trimetilsilil)etóxijmetil}- 7H-pirrolo[2,3-d]piri- midina
A mistura de 4-cloro-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidina (12,9 g, 45,4 mmols) (preparada como no WO 2007/070514, Exemplo 65) e ácido [1-(tri-isopropilsilil)-1H-pirrol-3-il]borônico (Frontier Scientific) (10,4 g, 38,9 mmols) e carbonato de sódio (4,36 g, 41,2 mmols) em 1,2-dimetoxietano (100 mL) e água (35 mL) foi desgaseificada purgando com um sistema de nitrogênio durante 20 minutos. Tetracis(trifenilfosfina)palá- dio(0) (2,25 g, 1,94 mmol) foi em seguida adicionado e a reação foi aquecida até o refluxo durante 9 horas. Quando a reação de acoplamento prosseguiu, o grupo de proteção de TIPS foi também lentamente removido. No momento em que a reação foi interrompida, os materiais de partida foram quase consumidos, entretanto a desproteção de TIPS não foi concluída. O solvente foi removido por evaporação giratória e o produto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 10 a 50% de acetato de etila em hexanos. Material protegido por TIPS foi também coleta- do (3,8 g, 21%), na adição à 4-(1 H-pirrol-3-il)-7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidina desejada (7 g, 57%). 1H RMN (300 MHz, CDCI3): δ 8,93 (br s, 1H), 8,84 (s, 1H), 7,73-7,69 (m, 1H), 7,33 (d, 1H), 7,84-7,80 (m, 1H), 6,94 (dd, 1H), 6,85 (d, 1H), 5,66 (s, 2H), 3,55 (m, 2H), 0,92 (m, 2H), - 0,06 (s, 9H). LCMS (M+H)+: 315,2. Etapa 2. 3-{2-ciano-1-[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pÍrrolo[2,3-d]pirimi- din-4-il)-1 H-pirrol-1-il]etil}pirrolidina-1-carboxilato de terc-butila
A uma mistura de 4-(1 H-pirrol-3-il)-7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidina (7,69 g, 24,4 mmols) e 3-[2-cianovinil]pirrolidina-1 - carboxilato de terc-butila como uma mistura de isômeros de olefina (5,70 g, 25,7 mmols, preparada no exemplo 15, etapa 1) em acetonitrila (70 mL) foi adicionado 1,8-diazabiciclo[5,4,0]undec-7-eno (3,66 mL, 24,4 mmols) e a reação foi agitada durante a noite. O volume foi em seguida reduzido à metade em vácuo e a reação foi aquecida para 60°C durante 4,5 horas. O solvente foi removido por evaporação giratória. O produto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente inicialmente de 0 a 65% de B em A, (A: 5% de isopropanol/5% de acetato de etila/90% de hexanos, B: 10% de isopropanol/50% de acetato de etíla/40% de hexanos), até o segundo diastereômero começar a eluir, em seguida rapidamente aumentou para 90% de B em A. O primeiro diastereômero a eluir (diastereômero 1) foi coletado (4,45 g, 34%). 1H RMN (400 MHz, CDCI3) diastereômero 1: δ 8,82 (s, 1H), 7,70-7,68 (m, 1H), 7,35 (d, 1H), 7,80 (dd, 1H), 6,92 (t, 1H), 6,84 (d, 1H), 5,66 (s, 2H), 1,43-4,04 (m, 1H), 3,80-3,65 (m, 1H), 3,57-3,38 (m, 3H), 3,33-3,20 (m, 1H), 3,16-3,05 (m, 1H), 2,93-2,80 (m, 3H), 1,89-1,78 (m, 1H), 1,63-1,52 (m, 1H), 1,47 (s, 9H), 0,92 (m, 2H), -0,06 (s, 9H); LCMS (M+H)+: 537,3. 1H RMN (500 MHz, d6-dMSO, 90’C) diastereômero 2: δ 8,69 (s, 1H), 7,92 (t, 1H), 7,62 (d, 1H), 7,89 (t, 1H), 6,99 (d, 1H), 6,94 (dd, 1H), 5,63 (s, 2H), 4,47 (dt, 1H), 3,58 (m, 2H), 3,43 (ddd, 1H), 3,30 (dd, 1H), 3,28-3,22 (m, 1H), 3,19 (dd, 1H), 3,11 (dd, 1H), 2,94 (dd, 1H), 2,87- 2,77 (m, 1H), 2,13-2,04 (m, 1H), 1,73 (dq, 1H), 1,34 (s, 9H), 0,86 (m, 2H), - 0,07 (s, 9H); LCMS (M+H)+: 537,3. Etapa 3. 3-pirrolidin-3-il-3-[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pi- rimidin-4-il)-1 H-pirrol-1-iljpropanonitrila
A uma solução de 3-{2-ciano-1-[3-(7-{[2-(trimetilsÍlil)etóxi]metil}- 7H-pirrolo[2,3-d]pirim idin-4-il)-1 H-pirrol-1 -il]etil}pirrolidina-1 -carboxilato de terc-butila (4,45 g, 8,29 mmols) (diastereômero 1 de etapa 2) em 1,4-dioxano (50 mL) foi adicionado cloreto de hidrogénio a 4 M em 1,4-dioxano (31 mL). A reação foi agitada durante 16 horas. O produto foi filtrado e enxaguado com uma pequena quantidade de dioxano. O sólido úmido foi dissolvido e dividido entre NaOH a 1N e acetato de etila. A porção aquosa foi extraída mais duas vezes com acetato de etila. Os extratos combinados foram lavados com salmoura, secados sobre sulfato de sódio, decantados e concentrados (3,6 g, 99%). 1H RMN (300 MHz, CDCI3): Ô 8,81 (s, 1H), 7,69 (dd, 1H), 7,34 (d, 1H), 6,98 (dd, 1H), 6,92 (dd, 1H), 6,85 (d, 1H), 5,66 (s, 2H), 1,45-4,03 (m, 1H), 3,53 (m, 2H), 3,28 (dd, 1H). 2,95 (t, 2H), 2,86 (d, 2H), 2,82-2,61 (m, 2H), 1,89-1,73 (m, 1H), 1,50-1,35 (m, 1H), 0,91 (m, 2H), -0,06 (s, 9H); (M+H)+: 437,3. Etapa 4. oxazolo[5,4-b]piridina-2(1H)-tiona
Preparação similar àquela referenciada em J. Org. Chem. 1995, 60(17), 5721-5725. A uma mistura de 3-aminopiridin-2-ol (3B Scientific Corporation) (10,12 g, 91,9 mmol) em THF (200 mL) a 0’C em um banho de gelo foi adicionado dicloreto carbonotioico (7,71 mL, 101 mmol) gota a gota. A reação foi agitada em temperatura ambiente durante 3 horas. Água foi adicionada e o pH foi ajustado para a faixa de 4 a 5. O produto foi obtido por extração com acetato de etila. Os extratos foram secados sobre sulfato de sódio, filtrados e concentrados. O produto foi usado sem outra purificação. 1H RMN (300 MHz, d6-dMSO): δ 14,08 (br s, 1H), 8,14 (d, 1H), 7,67 (d, 1H), 7,35 (dd, 1H); (M+H)+: 152,9. Etapa 5. 3-(1-[1,3]oxazolo[5,4-b]piridin-2-ilpirrolidin-3-il)-3-[3-(7-{[2-(trimetil- silil)etóxÍ]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 -iljpropanonitrila
A mistura de 3-pirrolidin-3-il-3-[3-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1-iljpropanonitrila (1,2 g, 2,7 mmols, de etapa 3) e oxazolo[5,4-b]piridina-2(1H)-tiona (0,50 g, 3,3 mmols) em 1,4- dioxano (20 mL) e N,N-di-isopropiletilamina (0,96 mL, 5,5 mmols) foi aqueci- da para 70°C durante 2 horas. O solvente foi evaporado e substituído com etanol (20 mL) e nitrato de prata (1,4 g, 8,2 mmols) e solução de hidróxido de amónio (3,8 mL) foram adicionados. A reação foi agitada durante 16 horas. Água, NaOH a 1N e acetato de etila foram adicionados na reação e os sólidos foram filtrados, enxaguados com acetato de etila, e as camadas do filtrado foram separadas. A camada aquosa foi extraída com mais três porções de acetato de etila. Os extratos combinados foram lavados com salmoura, secados sobre sulfato de sódio, decantados e concentrados. O produto foi purificado por cromatografia de coluna rápida em sílica-gel, eluindo com um gradiente de 0 a 10% de acetato de etila em hexanos (930 mg, 61%). 1H RMN (300 MHz, CDCI3): δ 8,83 (s, 1H), 7,96 (dd, 1H), 7,73 (t, 1H), 7,60 (dd, 1H), 7,36 (d, 1H), 7,15 (dd, 1H), 7,83 (dd, 1H), 6,96 (t, 1H), 6,85 (d, 1H), 5,67 (s, 2H), 4,27-1,47 (m, 1H), 4,09 (dd, 1H), 3,91-3,81 (m, 1H), 3,70- 3,60 (m, 1H), 3,55 (m, 2H), 3,52-3,45 (m, 1H), 3,17-3,04 (m, 1H), 2,98 (d, 2H), 2,12-2,00 (m, 1H), 1,91-1,74 (m, 1H), 0,92 (m, 2H), -0,06 (s, 9H); LCMS (M+H)+: 555.2. Etapa 6. Fosfato de 3-(1-[1,3Joxazolo[5,4-b]piridin-2-ilpirrolidin-3-ÍI)-3-[3-(7H- pirrolo[2,3-d]pirimidÍn-4-il)-1 H-pirrol-1-iljpropanonitrila
Uma solução de 3-(1 -[1,3]oxazolo[5,4-b]piridin-2-ilpirrolidin-3-il)- 3-[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1- iljpropanonitrila racêmica (0,93 g, 1,7 mmol, produzida na etapa 5) em DCM (30 mL) e TFA (10 mL) foi agitada durante 2,5 horas. Os solventes foram e- vaporados e o resíduo foi agitado com solução de hidróxido de amónio em metanol durante 16 horas. O solvente foi novamente evaporado. O resíduo foi dividido entre água e 10% de IPA em CHCI3. As camadas foram separadas e a porção aquosa foi extraída mais duas vezes com 10% de IPA em CHCI3. Os extratos foram secados sobre sulfato de sódio, decantados e concentrados. Os enantiômeros foram separados por HPLC quiral (Pheno- menex Lux-celulose-1, 5 μ, 20 x 250 mm, 80% de Etanol/hexanos, 10 mL/mi- nuto), para fornecer enantiômero 1 (primeiro a eluir, tempo de retenção de 19,3 minutos), e enantiômero 2 (segundo a eluir, tempo de retenção de 21,4 minutos). Enantiômero 1 foi obtido por remoção de solvente em vácuo, em uma quantidade de 314 mg. Este produto foi dissolvido em isopropanol quente e um equivalente de ácido fosfórico foi adicionado. Houve formação imediata de um precipitado e a mistura foi deixada resfriar lentamente para temperatura ambiente, com agitação. O produto foi isolado por filtragem e foi secado por ar, em seguida secado também a 60°C sob vácuo (289 mg, 33%). 1H RMN (300 MHz, d6-dMSO): δ 11,97 (s, 1H), 8,61 (s, 1H), 8,01 (t, 1H), 7,87 (dd, 1H), 7,62 (dd, 1H), 7,52 (dd, 1H), 7,20 (dd, 1H), 7,16 (t, 1H), 6,97-6,92 (m, 2H), 4,59 (dt, 1H), 3,87 (dd, 1H), 3,72-3,62 (m, 1H), 3,56-3,40 (m, 3H), 3,30 (dd, 1H), 3,00-2,85 (m, 1H), 1,75-1,63 (m, 2H); LCMS (M+H)+: 425,1. Exemplo 34. 3-[1-(1,1-dióxido-2,3-di-hidrotienoí2l3-blpiridin-6-il)pirrolidin-3-ill- 3-[3-(7H-pirroloí2.3-dlpirimidin-4-il)-1 H-pirrol-1 -il]propanonitrila (dois enanti-

3-pirrolidin-3-il-3-[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirrol-1-iljpropanonitrila (80 mg, 0,18 mmol, de exemplo 33, etapa 3) e 1,1-dióxido de 6-cloro-2,3-di-hidrotieno[2,3-b]pirídina (37 mg, 0,18 mmol, preparado como descrito no exemplo 28, etapa 4) foram dissolvidos em etanol (190 microL) e N,N-dÍ-isopropiletilamina (64 microL, 0,37 mmol). Em um frasco selado, a mistura foi aquecida em um banho de óleo mantido a 120°C durante 3 horas. A mistura foi em seguida concentrada em vácuo. O produto foi purificado por cromatografia de coluna rápida, eluindo com um gradiente de 0 a 5% de metanol em acetato de etila para fornecer o produto racêmico. Este material foi separado por HPLC quiral (Chiral Technologies Chiralpak AD-H, 5μ, 20 x 250 mm, eluindo com 80% de EtOH/He- xanos, 8 mUminuto) para fornecer enantiômero 1 (primeiro a eluir, tempo de retenção de 35,0 minutos) e enantiômero 2 (segundo a eluir, tempo de re- tenção de 55,6 minutos).
Após a remoção de solvente em vácuo, cada enantiômero foi desprotegido separadamente por agitação sequencialmente em uma mistura de 1:1 de TFA/DCM durante 1 hora, remoção de solvente, em seguida agitação em metanol (1,5 mL) contendo EDA (0,2 mL) durante 30 minutos. H- PLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usada para purificar os produtos. Enantiômero 1: (6 mg, 6%). 1H RMN (400 MHz, d6-dMSO): δ 11,96 (br s, 1H), 8,61 (s, 1H), 8,01 (t, 1H), 7,69 (d, 1H), 7,51 (d, 1H), 7,16 (dd, 1H), 6,96-6,93 (m, 2H), 6,75 (d, 1H), 4,56 (dt, 1H), 3,76 (dd, 1H), 3,57- 3,22 (m, 7H), 3,15-3,08 (m, 2H), 2,94-2,81 (m, 1H), 1,71-1,63 (m, 2H); LCMS (M+H)+: 471,4. Enantiômero 2: (4 mg, 4%). 1H RMN (400 MHz, d6-dMSO): δ 11,96 (br s, 1H), 8,61 (s, 1H), 8,01 (t, 1H), 7,69 (d, 1H), 7,51 (d, 1H), 7,16 (dd, 1H), 6,96-6,93 (m, 2H), 6,75 (d, 1H), 4,56 (dt, 1H), 3,76 (dd, 1H), 3,57- 3,22 (m, 7H), 3,15-3,08 (m, 2H), 2,94-2,81 (m, 1H), 1,71-1,63 (m, 2H); LCMS (M+H)+: 471,4. Exemplo 35. 3-í1-(1-óxido-2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidin-3-ill-3- [3-(7H-pirrolo[2,3-d1pirímidin-4-il)-1 H-pirrol-1-illpropanonitríla

A mistura de 3-pirrolidin-3-il-3-[3-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1-iljpropanonitrila (26 mg, 0,053 mmol, de exemplo 33, etapa 3) e 1-óxido de 6-cloro-2,3-di-hidrotieno[2,3- b]piridina (10,0 mg, 0,0533 mmol, preparada como descrito no exemplo 28, etapa 4) em etanol (56 microL) e N,N-dÍ-isopropiletilamina (19 microL) foi aquecida para 120°C durante 1,5 hora no micro-ondas. A mistura foi concentrada em vácuo. A mistura crua foi dissolvida em 1:1 de TFA/DCM, agitada durante 1,5 hora, e em seguida concentrada novamente. O residuo foi em seguida dissolvido em metanol (1,5 mL) e EDA (0,2 mL) foi adicionado. Após agitação durante 30 minutos, HPLC/MS preparativa (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usada para purificar o produto (3 mg, 12%). 1H RMN (400 MHz, d6-dMSO): δ 11,96 (br s, 1H), 8,61 (s, 1H), 8,02-8,00 (m, 1H), 7,71 (d, 1H), 7,51 (d, 1H), 7,16 (t, 1H), 6,96-6,92 (m, 2H), 6,64 (d, 1H), 4,56 (tt, 1H), 3,75 (dd, 1H), 3,56-2,98 (m, 9H), 2,94-2,82 (m, 1H), 1,73-1,61 (m, 2H); LCMS (M+H)+: 458,1. Exemplo 36. Trifluoroacetato de ±3-ri-(6-cloro-4-metil-3-oxo-3,4-di-hidropi- razin-2-il)pirrolidin-3-in-3-[4-(7H-pirrolo[2,3-d1pirimidin-4-il)-1 H-pirazol-1-illpro- panonitrila

Etapa 1. 3,5-dicloro-1-metilpirazin-2(1H)-ona
Cloridrato de (metilamino)acetonitrila (2,55 g, 23,9 mmols) foi dissolvido em clorofórmio em um frasco de fundo redondo de 1 gargalo (38,93 mL, 486,5 mmols) e cloreto de oxalila (6,07 mL, 71,8 mmols) foi adicionado. A reação foi aquecida até o refluxo durante a noite e foi transferida em um frasco diferente, e o solvente removido por evaporação giratória. A reação foi submetida à cromatografia em sílica-gel usando 1:1 de EtO- Ac/hexanos para fornecer o produto. Espectro de massa: [M+1]: 179. 1H RMN(CDCI3): 7,25 (s, 1H), 3,60 (s, 3H). Etapa 2. ±3-[1-(6-cloro-4-metil-3-oxo-3,4-di-hidropirazin-2-il)pirrolidin-3-il]-3- [4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (50,00 mg, 0,1142 mmol, preparada no exemplo 15, etapas 1 a 3, omitindo a separação quiral realizada na etapa 2) foi misturada com 3,5-dicloro-1-metilpirazin-2(1H)-ona (32,00 mg, 0,1788 mmol) e foi dissolvida em 1,4-dioxano (0,5 mL). A reação foi aquecida a 100°C durante 2 horas, em cujo tempo a análise de LCMS mostrou principalmente o produto. Os resíduos foram submetidos à cromatografia em sílica-gel usando EtOAc e 5% de MeOH/EtOAc para fornecer o produto. Espectro de massa: [M+1 ]: 580. Etapa 3. Trifluoroacetato de ±3-[1-(6-cloro-4-metil-3-oxo-3,4-di-hidropirazin-2- il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-dJpirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila
O produto de etapa 2 foi dissolvido em DCM (0,50 mL) em um frasco de fundo redondo de 1 gargalo, e TFA (0,30 mL, 3,9 mmol) foi adicionado. A reação foi agitada a 25°C durante 1 hora, e o solvente removido por evaporação giratória. O resíduo foi dissolvido em metanol (2,00 mL) e amónia a 16 M em água (0,30 mL, 4,9 mmols) foi adicionada. A mistura foi agitada durante 1 hora, e o solvente removido por evaporação giratória. O produto foi isolado por HPLC-MS preparativa usando um instrumento Waters Frac- tion-Linx e uma coluna 19 mm x 100 mm Sunfire C18; eluindo com um gradiente de ACN/H2O (0,1% de TFA), 30 mL/minuto; Espectro de massa: [M+1]: 450. 1H RMN(CD3OD): δ 8,95 (s, 1H), 8,87 (s, 1H), 8,54 (s, 1H), 7,83 (d, 1H), 7,25 (d, 1H), 6,77 (s, 1H), 4,83 (m, 1H), 3,60-4,2 (m, 4H), 3,35 (s, 3H), 3,40 (m, 1H), 3,20 (m, 1H), 2,94 (m, 1H), 1,76 (m, 2H). Exemplo 37. Trifluoroacetato de ±3-[1-(4-metil-3-oxo-3,4-di-hidropirazin-2- il)pirrolidin-3-ill-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-illpropanoni- trila

Etapa 1. ± 3-[1-(4-metil-3-oxo-3,4-di-hidropirazin-2-il)pirrolidÍn-3-il]-3-[4-(7-{[2- (trimetilsilil)etóxijmetil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila 3-[1-(6-cloro-4-metil-3-oxo-3,4-di-hidropirazin-2-il)pirrolidin-3-il]-3- [4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila (de exemplo 36, 27 mg, 0,046 mmol) foi dissolvida em iso- propil álcool (0,10 mL) e metanol (0,10 mL) com bicarbonato de sódio (12 mg, 0,14 mmol) em um frasco de fundo redondo de 1 gargalo. 10% de paládio sobre carbono (10:90, paládio negro defumo, 15,0 mg, 0,0141 mmol) foram adicionados. A reação foi agitada sob uma atmosfera de hidrogênio durante a noite em cujo tempo a análise de LCMS mostrou uma mistura de 1:1 de material de partida e produto. Na reação foram adicionados mais 10% de paládio sobre carbono (10:90, paládio:negro de fumo, 15,0 mg, 0,0141 mmol) e foi agitada sob uma atmosfera de hidrogênio durante 48 horas, em cujo tempo a análise de LCMS não mostrou nenhum material de partida presente. A reação foi filtrada, e o solvente removido por evaporação giratória. O resíduo foi usado na reação seguinte sem purificação. Espectro de massa: [M+1]: 546. Etapa 2. Trifluoroacetato de + 3-[1-(4-metil-3-oxo-3,4-di-hidropirazin-2-il)pir- rolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila
O produto de etapa 1 foi dissolvido em DCM (0,50 mL) em um frasco de fundo redondo de 1 gargalo, e TFA (0,30 mL, 3,9 mmols) foi adicionado. A reação foi agitada a 25°C durante 2 horas, e o solvente removido por evaporação giratória. O resíduo foi dissolvido em metanol (2,00 mL) e amónia a 16 M em água (0,30 mL, 4,9 mmols) foi adicionada e foi agitada durante 2 horas. O solvente foi em seguida removido por evaporação giratória. O produto foi isolado por HPLC-MS preparativa usando um instrumento Waters Fraction-Linx e uma coluna 19 mm x 100 mm Sunfire C18; eluindo com um gradiente de ACN/H2O (0,1% de TFA), 30 mL/minuto; detector fixado a m/z 415; para fornecer o produto como o sal de TFA . Espectro de massa: [M+1]: 416; 1H RMN(CD3OD): δ 8,94 (s, 1H), 8,86 (s, 1H), 8,54 (s, 1H), 7,82 (d, 1H), 7,23 (d, 1H), 6,88 (d, 1H), 6,68 (d, 1H), 4,90 (m, 1H), 3,70^1,5 (m, 4H), 3,43 (s, 3H), 3,40 (m, 1H), 3,20 (m, 1H), 3,10 (m, 1H), 1,88 (m, 2H). Exemplo 38. Trifluoroacetato de ±3-cloro-2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3- dlpirimidin-4-il)-1H-pirazol-1-inetil}pirrolidin-1-il)isonicotinonitrila

Etapa 1. 2,3-dicloroisonicotinaldeído N,N-di-isopropilamina (1,14 mL, 8,11 mmols) foi dissolvida em THF em um frasco de fundo redondo de 1 gargalo (15,00 mL, 184,9 mmols). A solução foi em seguida resfriada a -78°C e n-butil-litio a 1,60 M em hexano (4,64 mL, 7,43 mmols) foi adicionado. A reação foi deixada aquecer a 0°C e foi re-resfriada para -78°C. Na reação foi adicionada 2,3-dicloropiridina (0,10 g, 6,76 mmols) em THF (5,00 mL, 61,6 mmol). A reação foi em seguida agitada a -78°C durante 2 horas e DMF (0,146 mL, 13,51 mmols) foi adicionado e foi agitado a -78°C durante 30 minutos e foi deixado aquecer para 0°C. A análise de TLC (20% de EtOAc/hexanos) não mostrou nenhum material de partida presente. Análise de LCMS mostrou pico de M+1+CH3OH. A análise de RMN mostrou um próton de aldeído, embora a maior parte do produto tenha se solidificado e a RMN é mais indicativa da composição do material não cristalizado. A reação foi submetida à cromatografia em sílica-gel usando 25% de EtOAc/hexanos para fornecer o produto. 1H RMN(CDCI3): Õ 10,49 (s, 1H), 8,49 (d, 1H), 7,67 (d, 1H). Etapa 2. 2,3-dicloroisonicotinaldeído oxima 2,3-dicloroisonicotinaldeído (0,50 g, 2,8 mmol) foi dissolvido em metanol (10,0 mL, 247 mmols) com bicarbonato de potássio (0,35 g, 3,5 mmols) em um frasco de fundo redondo de 1 gargalo, e cloridrato de hidroxi- lamina (0,22 g, 3,2 mmols) foi adicionado, A reação foi agitada a 25°C duran- te 2 dias. Análise de LCMS não mostrou nenhum material de partida e os dois isômeros de oxima principais. A mistura reacional foi dividida entre EtO- Ac e água e extrato de EtOAc foi lavado com salmoura, secado (mgSO4), e removido em vácuo. O produto foi usado na reação seguinte sem outrapurifi- cação. Espectro de massa: [M+1]: 191; Etapa 3. 2,3-dicloroisonicotinonitrila 2,3-dicloroisonicotinaldeido oxima(0,617 g, 0,00323 mol) foi dissolvido em piridina (6,0 mL, 0,074 mol) em um frasco de fundo redondo de 1 gargalo. Cloreto de metanossulfonila (0,1 mL, 0,013 mol) foi adicionado gota a gota, e a reação foi aquecida a 60°C durante 2 horas. Após resfriamento, a reação foi extraída com acetato de etila e os extratos orgânicos foram lavados com água, HCI a 0,5 N, solução de NaCI saturada, secados (mgSO4) e removidos em vácuo. O resíduo foi submetido à cromatografia em silica-gel usando 20% de EtOAc/hexanos para fornecer o produto. 1H RMN(CDCI3): δ 8,48 (d, 1H), 7,53 (d, 1H). Etapa 4. ±3-cloro-2-(3-{2-ciano-1 -[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol- 1-il]etil}pirrolidin-1 -il) isonicotinonitrila 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxÍ]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (100,00 mg, 0,22851 mmol, preparada no exemplo 15, etapas 1 a 3, omitindo a separação quiral realizada na etapa 2) foi misturada com 2,3-dicloroisonicotinonitrila (57,99 mg, 0,3352 mmol) e em seguida foi dissolvida em NMP (0,60 mL, 6,2 mmols). A reação foi aquecida a 130°C durante 2 horas, em cujo tempo a análise de LCMS mostrou principalmente o produto. Os resíduos foram submetidos à cromatografia em sílica-gel usando EtOAc e 5% de MeOH/EtOAc para fornecer o produto. Espectro de massa: [M+1]: 574. Etapa 5. Trifluoroacetato de ±3-cloro-2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidÍn-4-H)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)isonicotinonitrila 3-cloro-2-(3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etíl}pirrolidin-1 -iljisonicotínonitríla (25,00 g, 43,54 mmols) foi dissolvida em DCM (0,50 mL, 7,8 mmols) em um frasco de fundo redondo de 1 gargalo, e TFA (0,30 mL, 3,9 mmols) foi adi cionada. A reação foi agitada a 25°C durante 1 hora, e o solvente foi removido por evaporação giratória. O resíduo foi dissolvido em metanol (2,00 mL, 49,4 mmols) e amónia a 16 M em água (0,30 mL, 4,9 mmols) foi adicionada. A mistura foi agitada durante 1 hora, e o solvente foi removido por evaporação giratória. O produto foi isolado por HPLC-MS preparativa usando um instrumento Waters Fraction-Linx e uma coluna 19 mm x 100 mm Sunfire C18; eluindo com um gradiente de ACN/H2O (0,1% de TFA), 30 mL/minuto; detector fixado a m/z 443 para fornecer o produto como o sal de TFA . Es-pectro de massa: [M+1]: 444; 1H RMN(CD3OD): δ 8,96 (s, 1H), 8,87 (s, 1H), 8,54 (s, 1H), 8,16 (d, 1H), 7,84 (d, 1H), 7,25 (d, 1H), 6,96 (d, 1H), 4,83 (m, 1H), 3,70-4,00 (m, 4H), 3,40 (m, 1H), 3,20 (m, 1H), 3,00 (m, 1H), 1,80 (m, 2H). Exemplo 39. Trifluoroacetato de í2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-dlpiri- midin-4-il)-1 H-pirazol-1-il1etil}pirrolidin-1-il)piridina-3,4-dicarbonitrila

Etapa 1. ±2-(3-{2-ciano~ 1 -[4-(7-{[2-(trimetilsilil) etóxijmetil}-7H-pirrolo[2,3- d]pirímidin-4-il) -1 H-pirazol-1 -il]etil}pirrolidin-1 -il)pindina-3,4-dicarbonitrila 3-cloro-2-(3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- p irrolo[2,3-d]p i rim id in-4-il)-1 H-pirazol-1 - i l]etil}p i rrol id i n-1 -il) ison icoti n on itri la (65,0 mg, 0,113 mmol, de exemplo 38, etapa 4) foi dissolvido em NMP (0,8 mL, 8 mmols) em um frasco de fundo redondo de 1 gargalo, e Cianeto de zinco (39,9 mg, 0,340 mmol) e zinco (22,2 mg, 0,340 mmol) foram adicionados. A reação foi desgaseificada e bis(tri-t-butilfosfina)paládio (28,9 mg, 0,0566 mmol) foi adicionado e a reação foi aquecida a 130°C durante 100 minutos em cujo tempo a análise de LCMS mostrou que ela foi principalmen- te uma mistura de 1:1 de produto e material de partida desclorado. A reação foi filtrada e o produto foi isolado por HPLC-MS preparativa usando um instrumento Waters Fraction-Linx e uma coluna 19 mm x 100 mm Sunfire C18; eluindo com um gradiente de ACN/H2O (0,1% de TFA), 30 mL/minuto; detector fixado a m/z 564; Espectro de massa: [M+1] m/z: 565. Etapa 2. Trífluoroacetato de ±2-(3-{2-cÍano-1-[4-(7H-pirrolo[2,3-dJpirímidin-4- il)-1 H-pirazol-1-il]etil}pirrolidin-1 -il)piridina-3,4-dicarbonitrila
O produto de etapa 1 foi dissolvido em DCM (0,50 mL, 7,8 mmols) em um frasco de fundo redondo de 1 gargalo, e TFA (0,30 mL, 3,9 mmols) foi adicionado. A reação foi agitada a 25°C durante 2 horas, e o solvente foi removido por evaporação giratória. O resíduo foi dissolvido em metanol (2,00 mL, 49,4 mmols) e amónia a 16 M em água (0,30 mL, 4,9 mmols) foi adicionada. A mistura foi em seguida agitada durante 2 horas, e o solvente foi removido por evaporação giratória. O resíduo foi purificado por prep. O produto foi isolado por HPLC-MS preparativa usando um instrumento Waters Fraction-Linx e uma coluna 19 mm x 100 mm Sunfire C18; eluindo com um gradiente de ACN/H2O (0,1% de TFA), 30 mL/minuto; detector fixado a m/z 434; para fornecer o produto como o sal de TFA. Espectro de massa: [M+1]: 435; 1H RMN(CD3OD): δ 9,00 (s, 1H), 8,90 (s, 1H), 8,56 (s, 1H), 8,44 (d, 1H), 7,90 (d, 1H), 7,29 (d, 1H), 6,99 (d, 1H), 4,91 (m, 1H), 1,41 (m, 1H), 3,9 (m, 1H), 3,75 (m, 2H), 3,40 (m, 1H), 3,30 (m, 1H), 3,05 (m, 1H), 1,89 (m, 2H). Exemplo 40. ± 2-(3-{2-ciano-1-í4-(7H-pirrolo[2,3-d]pirímidin-4-ií)-1H-pirazol-1- illetíl}pirroiidin-1-il)-6-(metiltio)benzonitrila

Etapa 1. 2-f!uoro-6-(metiltio)benzonitnla
A uma solução a 0°C de 2,6-difluorobenzonitrila (0,20 g, 1,4 mmol) em DMF (2 mL, 20 mmols) foi adicionado mercaptideo de metila de sódio (0,13 g, 1,7 mmol). A reação foi deixada aquecer para temperatura ambiente e foi agitada durante a noite. Ela foi em seguida filtrada e purificada por LCMS (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH a 5 mL/minuto) para fornecer 41 mg de sólido amarelo claro (17% de rendimento). 1H RMN (400 MHz, CDCI3): δ 7,55 (1H, m); 7,87 (1H, m); 6,96 (1H, t); 2,59 (3H, s). LCMS (M+1): 168. Etapa 2. 2-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - H]etil}pÍrrolidin-1-il)-6-(metiltio)benzonitríla
Uma solução de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetiisilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (0,075 g, 0,17 mmol, preparada no exemplo 15, etapas 1 a 3, omitindo a separação quiral realizada na etapa 2), 2-fluoro-6-(metiltÍo)benzonitrila (0,040 g, 0,24 mmol) e N,N-di-isopropiletilamina (60 microL, 0,3 mmol) em fluoreto/trifluoroborano de 1-butil-3-metil-1H-imidazol-3-io (1:1) (0,15 g, 0,66 mmol) foi aquecida a 120°C durante 3,2 horas, em seguida a 150°C durante 2 horas. Ela foi purificada por LCMS (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH a 5 mUminuto) para fornecer 30 mg 2-(3-{2-ciano-1- [4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1-il)-6-(metiltio)benzonitrila (A, 30% de rendimento). LCMS (M+1): 585. mg A foram agitados em 1mL de DCE e 1 mL de TFA durante 1 hora. Ele foi concentrado e o resíduo foi agitado em 50 uL de EDA e 1 mL de MeOH durante 1 hora. A reação foi purificada por LCMS (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH a 5 mL/minuto) forneceu 3,5 mg de sólido branco. 1H RMN (400 MHz, DMSO): δ 12,1 (1H, br); 8,85 (1H, s); 8,65 (1H, s); 8,0 (1H, s); 7,6 (1H, m); 7,18 (1H, m); 6,98 (1H, m); 6,6 (2H, m); 4,81 (1H, m); 3,68 (1H, m); 3,6-3,2 (5H, m); 2,9 (1H, m); 2,5 (3H, s); 1,62 (2H, br). LCMS (M+1): 455. Exemplo 41. ± 2-(3-{2-ciano-1-[4-(7H-pirroloí2,3-d]pirímidín-4-il)-1 H-pirazol-1-illetil}pirrolidin-1-il)-6-(metilsulfonil)benzonitrila

Uma solução de 15 mg A (de exemplo 40, etapa 2, 26 mmol) e ácido M-cloroperbenzoico (0,018 g, 0,080 mmol) em DCM (1 mL, 20 mmols) foi agitada durante 1,5 hora. MeOH e DMF foram adicionados. A mistura foi purificada por LCMS para fornecer 5 mg de sólido branco. O composto foi em seguida desprotegido para remover o grupo SEM. Ele foi agitado em 1 mL de DCE e 1 mL de TFA durante 1 hora. Ele foi concentrado e o resíduo foi agitado em 50 uL de EDA e 1 mL de MeOH durante 1 hora. A reação foi purificada por LCMS (coluna C18 eluindo com um gradiente ACN/H2O contendo 0,15% de NH4OH at 5 mL/minuto) e forneceu 1,4 mg de sólido branco. 1H RMN (400 MHz, DMSO): δ 12,13 (1H, s); 8,88 (1H, s); 8,68 (1H, s); 8,41 (1H, s); 7,61(1H, m); 7,6 (1H, m); 7,35 (1H, m); 7,19 (1H, m); 6,98 (1H, m); 4,82 (1H, m); 3,67 (1H, m); 3,6- 3,2 (5H, m); 2,92 (1H, m); 2,5 (3H, s); 1,63 (2H, m). LCMS (M+1): 487. Exemplo 42. ± 3-ri-(8-cloroquinolin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- dlpirimidin-4-il)-1H-pirazol-1-inpropanonitríla

Uma solução de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7 H-pirrolo[2,3-d]pirim id in-4-il)-1 H-pirazol-1 -iljpropanonitrila (0,020 g, 0,000046 mol, preparada no exemplo 15, etapas 1 a 3, omitindo a separação quiral realizada na etapa 2) e 2,8-dicloroquinolina (0,020 g, 0,00010 mol) em etanol (0,020 mL, 0,00034 mol) e N,N-di-isopropiletilamina (20,0 microL, 0,000115 mol) foi aquecida a 120°C durante 1,3 horas. O bruto foi purificado por LCMS (coluna C18 eluindo com um gradiente ACN/H2O contendo 0,15% de NH4OH at 5 mL/minuto) para fornecer 16 mg. LCMS (M+1): 599.
O composto foi em seguida desprotegido para remover o grupo SEM. Ele foi agitado em 1 mL de DCE e 1 mL de TFA durante 1 hora. Ele foi concentrado e o resíduo foi agitado em 50 μL EDA e 1 mL de MeOH durante 1 hora. A reação foi purificada por LCMS (coluna C18 eluindo com um gradiente ACN/H2O contendo 0,15% de NH4OH at 5 mL/minuto) para fornecer 9,7 mg (45% de rendimento). 1H RMN (400 MHz, DMSO): δ 12,1 (1H, br); 8,9 (1H, s); 8,68 (1H, s); 8,42 (1H, s); 8,15 (1H, d); 7,65 (2H, m); 7,6 (1H, m); 7,15 (1H, t); 7,8 (1H, m); 6,95 (1H, d); 4,85 (1H, m); 3,85 (1H, br); 3,75 (1H, br); 3,45 (2H, m); 3,40-3,26 (2H, m); 2,98 (1H, m); 1,7 (2H, br). LCMS (M+1): 469.
Os exemplos 43 a 46 na tabela abaixo foram preparados pelo método geral de exemplo 42, com a exceção de que para o exemplo 45, o material de partida usado foi um enantiômero simples de 3-pirrolidin-3-il-3-[4- (7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]propanonitrila, preparado pelo método de exemplo 15, etapas 1 a 3. Adicionalmente, para os exemplos 43 e 44, o produto final foi purificado por LCMS (coluna C18 eluindo com um gradiente de ACN/H2O contendo 0,1% de TFA) para fornecer o sal de trifluoroacetato do composto origem.
Exemplo 47. Trifluoroacetato de (3S)-3-((3S)-1-(5-bromo-1,3-tiazol-2-il)pir-rolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin~4-il)-1 H-pirazol-1-illpropanonitrila

(3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (175 mg, 0,400 mmol; de exemplo 15, etapa 3) (base livre) e 2,5-dibromo-1,3-tiazol (290 mg, 1,2 mmol) foram misturado em isopropil álcool (2,2 mL, 29 mmol). 4-metilmor- folina (130 microL, 1,2 mmol) foi em seguida adicionada. A mistura foi aquecida a 80°C. Após 16 horas, LCMS mostrou a reação, M+H 599/601. O in- termediário foi isolado por HPLC-MS preparativa usando um instrumento Waters Fraction-Linx e uma coluna 30 mm x 100 mm Sunfire C18; 37% de CH3CN-H2O (0,1% de TFA), 0,5 minuto, gradiente de 6 minutos para 54%; 60 mL/minuto; tempo de retenção de 5,6 minutos. O composto foi em seguida secado por congelamento para fornecer 101 mg. O composto foi em seguida desprotegido usando TFA seguido por NH4OH. A desproteção geral-mente envolveu os seguintes: O composto foi dissolvido em DCM (1 mL) a 21 °C, e TFA (1 mL) foi adicionado. A mistura reacional foi em seguida agitada durante 1 hora. A solução foi concentrada para remover o TFA. O resíduo foi dissolvido em acetonitrila ou metanol (1 mL), e hidróxido de amónio a 15,0 M em água (0,25 mL) adicionado. A solução foi agitada a 21 °C durante 2 a 18 horas. Após LCMS mostrou que a desproteção foi concluída, a solução foi concentrada por evaporação giratória. O produto foi isolado por H- PLCMS preparativa usando um instrumento Waters Fraction-Linx e uma coluna de 19 mm x 100 mm Sunfire C18; 30 mL/minuto; 12% de CH3CN-H2O (0,1% de TFA), 0,5 minuto, gradiente a 30% at 6min; detector fixado a m/z 471; tempo de retenção, 5,5 minutos (3 runs). O composto foi em seguida secado por congelamento para fornecer o sal de TFA (47 mg). 1H RMN (400 MHz, DMSO-d6): Õ 12,8 (s, 1H); 9,03 (s, 1H); 8,87 (s, 1H); 8,56 (s, 1H); 7,84 (s, 1H); 7,18 (s, 1H); 7,17 (s, 1H); 4,88 (m, 1H); 3,64 (m, 1H); 3,22-3,42 (m, 5H); 2,96 (m, 1H); 1,71 (m, 2H); LCMS (M+H)+: 469. Exemplo 48. Trifluoroacetato de 2-cloro-6-((3S)-3-{(1S)-2-ciano-1-r4-(7H- pirrolo[2,3-d1pirimidin-4-il)-1 H-pirazol-1-il1etil}pirrolidin-1-il)benzonitrila

Uma solução de (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trime- tilsilil)etóxi]metÍI}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (92 mg, 0,21 mmol; de exemplo 15, etapa 3), NMP (1,5 mL, 16 mmols), 4- metilmorfolina (69 microL, 0,63 mmol) e 2-cloro-6-fluorobenzonitrila (65 mg, 0,42 mmol) foi aquecida a 90°C durante 50 minutos em um reator de microondas. LCMS mostrou cerca de 80% de reação concluída, e mostrou o esperado, [M+H] 573. O intermediário foi isolado por HPLCMS preparativa usando um instrumento Waters Fraction-Linx e uma coluna de 30 mm x 100 mm Sunfire C18; 49% de CH3CN-H2O (0,1% de TFA), 0,5 minuto, para 67% a 6 minutos; 60 mL/minuto; tempo de retenção de 5,3 minutos. O solvente foi em seguida removido por evaporação giratória. O composto foi em seguida desprotegido usando TFA seguido por NH4OH (veja o procedimento geral para o exemplo 47). O produto foi isolado por LCMS preparativa usando um ins-trumento Waters Fraction-Linx e uma coluna Sunfire C18 de 19 mm x 100 mm; 30mL/minuto; 29% de CH3CN-H2O (0,1% de TFA), 0,5 minuto, gradiente a 47% a 6 minutos; tempo de retenção de 5,1 minutos. O composto foi secado por congelamento para fornecer o sal de TFA, 24 mg,. 1H RMN (400 MHz, DMSO-d6): δ 12,8 (s, 1H); 8,99 (s, 1H); 8,82 (s, 1H); 8,53 (s, 1H); 7,77 (s, 1H); 7,37 (t, 1H); 7,12 (s, 1H); 6,86 (d, 1H); 6,74 (d, 1H); 4,87 (m, 1H); 3,21-3,74 (m, 6H); 2,91 (m, 1H); 1,66 (m, 2H); LCMS (M+H)+: 443. Exemplo 49. Trifluoroacetato de 3-((3S)-3-f(1S)-2-ciano-1-[4-(7H-pirrolof2.3- d]pirimidin-4-il)-1H-pirazol-1-illetil}pirrolidin-1-il)ftalonitrila

2-bromo-6-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)benzonitrila (20,0 mg, 0,0324 mmol, preparada de uma maneira similar àquela descrita no exemplo 48) foi agitada em NMP (0,75 mL, 7,8 mmols). Cianeto de zinco (57,8 mg, 0,486 mmol) foi em seguida adicionado. Tetracis(trifenilfosfi- na)paládio(O) (37,4 mg, 0,0324 mmol) foi em seguida adicionado, e a solução foi estimulada com nitrogênio (sub-superficie). O frasconete foi em seguida selado e aquecido a 150°C durante 20 minutos em um reator de micro-ondas. A reação foi preparada com NaHCO3 a 5% e EtOAc e filtrada. O in- termediário foi isolado por LCMS preparativa usando um instrumento Waters Fraction-Linx e uma coluna Sunfire C18 de 30 mm x 100 mm; 46% de CH3CN-H2O (0,1% de TFA), 0,5 minuto, para 64% em 6 minutos; 60 mL/minuto; detector fixado a m/z 564; tempo de retenção de 5,3 minutos. O solvente foi em seguida removido por evaporação giratória. O composto foi em seguida desprotegido usando TFA seguido por NH4OH (veja o procedimento geral para o exemplo 47). O produto foi isolado por LCMS preparativa usando um instrumento Waters Fraction-Linx e uma coluna Sunfire C18de 19 mm x 100 mm; 24% de CH3CN-H2O (0,1% de TFA), 0,5 minuto, para 42% em 6 minutos; 30 mL/minuto; tempo de retenção de 5,5 minutos, secado por congelamento para fornecer um sal de TFA de sólido branco. LCMS (M+H)+: 431,4. 1H RMN (300 MHz, DMSO-d6): δ 12,4 (s, 1H); 8,93 (s, 1H); 8,75 (s, 1H); 8,48 (s, 1H); 7,69 (s, 1H); 7,55 (dd, 1H); 7,25 (d, 1H); 7,10 (d, 1H); 7,85 (s, 1H); 4,86 (m, 1H); 3,2-3,8 (m, 6H); 2,93 (m, 1H); 1,69 (m, 2H). Exemplo 50. Trifluoroacetato de 2-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- dlpirímidin-4-il)-1 H-pirazol-1-il1etil}pírrolidin-1-il)-4-(trifluorometil)nicotinonitrila

6-cloro-2-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-4- (trifluorometil)nicotinonitrila (7,8 mg, 0,011 mmol, preparada de uma maneira similar aquela descrita no exemplo 51) foi dissolvida em metanol (0,1 mL, 25 mmol), e 6 mg de Pd/C a 10% adicionados. Agitada a 21 C. Um balão contendo hidrogênio foi ligado ao frasco durante 0,5 horas. LCMS mostrou o [M+H] 608 desejado, subproduto de amina, [M+H] 612, outros subprodutos de super-redução, e um traço de material de partida restante. O produto foi isolado por HPLC-MS preparativa usando um instrumento Waters Fraction- Linx e uma coluna Sunfire C18 de 19 mm x 100 mm; 51% de CH3CN-H2O (0,1% de TFA), para 69% em 6 minutos; 30 mL/minuto; detector fixado a m/z 608; tempo de retenção, 5,0 minutos. O composto foi em seguida desprotegido usando TFA seguido por NH4OH (veja o procedimento geral para o e- xemplo 47). O produto foi isolado por HPLC-MS preparativa usando um instrumento Waters Fraction-Linx e uma coluna Sunfire C18 de 19 mm x 100 mm; 30% de CH3CN-H2O (0,1% de TFA), para 48% em 6 minutos; 30 mL/minuto; tempo de retenção, 1,4 minutos. LCMS (M+H)+: 478. Exemplo 51. Trifluoroacetato de 3-((3S)-3-f(1S)-2-ciano-1-f4-(7H-pirrolof2.3- d]pirimidin-4-il)-1 H-pirazol-1-il]etil]pirrolidin-1-il)pirazina-2"Carbonitrila

(3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila (38 mg, 0,087 mmol; de exemplo 15, etapa 3) foi dissolvido em NMP (0,7 mL, 7 mmols) e N,N-di- isopropiletilamina (3,0E1 microL, 0,17 mmol). 3-cloropirazina-2-carbonitrila (18 mg, 0,13 mmol) foi adicionada. A solução foi agitada a 80°C durante 20 minutos (ou 21 °C durante 16 horas). LCMS mostrou zzz fairly conversão completa no o intermediário esperado, e mostrou [M+Hj: 541. O intermediário foi isolado por HPLC-MS preparativa usando um instrumento Waters Fraction-Linx e uma coluna 30 mm x 100 mm Sunfire C18; 44% de CH3CN- H2O (0,1% de TFA), 0,5 minuto; gradiente de 6 minutos para 62 %; 60 mL/minuto; tempo de retenção, 4,9 minutos. O solvente foi em seguida removido por evaporação giratória. O composto foi em seguida desprotegido usando TFA seguido por NH4OH (veja o procedimento geral para o exemplo 47). O produto foi isolado por LCMS preparativa usando um instrumento Waters Fraction-Linx e uma coluna Sunfire C18 de 19 mm x 100 mm; 30ml_/minuto; 20% de CH3CN-H2O (0,1% de TFA), 0,5 minuto, gradiente a 38% em minutos. Secado por congelamento para fornecer um sal de TFA sólido amarelo. 1H RMN (400 MHz, DMSO-d6): δ 12,7 (s, 1H); 9,01 (s, 1H); 8,83 (s, 1H); 8,54 (s, 1H); 8,36 (d, 1H); 7,95 (d, 1H); 7,79 (s, 1H); 7,14 (s, 1H); 4,89 (m, 1H); 3,92 (m, 1H); 3,80 (m, 1H); 3,61 (m, 2H); 3,40 (m, 1H); 3,29 (m, 1H); 2,91 (m, 1H); 1,71 (m, 2H); LCMS (M+H)+: 411.
Os exemplos na tabela abaixo foram feitos por procedimentos análogos àqueles para produção d e exemplos 47-51.
Exemplo 70. Sal de ácido fosfórico de 2-((3S)-342-fluoro-1-[4-(7H-pirrolo[2l3- dlpirimidin- 4-il)-1 H-pirazol-1-iHetil)pirrolidin-1-il)[1,31oxazolo[5, 4-blpíridina
Etapa 1. (3S)-3-(hidroximetil)pirrolidina-1-carboxilato de terc-butila
Uma solução de ácido (3S)-1-(terc-butoxicarboníl)pirrolidina-3- carboxilico (CheM-Impex; 2,5 g, 12 mmols) em THF (54 mL) foi resfriada para 0°C e BH3 a 0,1 M em THF (14 mL, 14 mmols) foi lentamente adicionado. A reação foi deixada aquecer para temperatura ambiente e foi agitada durante 4 horas, em cujo tempo a análise de LCMS mostrou redução completa em álcool. A solução foi resfriada para 0°C e extinguida por adição cuidadosa de HCI a 1N. EtOAc foi adicionada e as fases foram separadas, a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com NaHCO3 saturado, em seguida NaCl saturado, secada sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto como um óleo incolor, 2,15 g. 1H RMN (300 MHz, CDCI3): δ 3,7-3,2 (m, 5H), 3,1 (m, 1H), 2,4 (m, 1H), 1,95 (m, 1H), 1,7 (s, 2H), 1,45 (s, 9H). MS (El): 146,0 (M-tBu + 2H), 128,1 (M-OtBu). Etapa 2: (3S)-3-formilpirrolidina-1-carboxilato de terc-butila
Cloreto de oxalila (1,4 mL, 16 mmols) foi dissolvida em DCM (28 mL) e esta solução foi resfriada para -78°C, em seguida DMSO (1,8 mL, 26 mmols) foi adicionada. A esta foi em seguida adicionada uma solução de (3S)-3-(hidroximetil)pirrolidina-1-carboxilato de terc-butila (2,15 g, 10,7 mmols) em DCM (28 mL), seguindo em 20 minutos por adição de 4- metilmorfolina (5,9 mL, 53 mmols). A reação foi mantida a -78°C durante 20 minutos, em seguida aquecida para 0°C durante 1 hora, em cujo tempo a análise de TLC mostrou oxidação completa em aldeído. A reação foi extinguida por adição de água e CHCI3, as fases foram separadas e a fase aquosa foi extraída com CHCI3 adicional. A fase orgânica combinada foi lavada com água, em seguida HCI a 1N, em seguida NaHCO3 saturado, NaCl saturado, secada sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto que foi usado sem outra purificação, 2,1 g. 1H RMN (300 MHz, CD- CI3): δ 9,68 (s, 1H), 3,68 (m, 1H), 3,50 (m, 1H), 3,37 (m, 2H), 3,0 (m, 1H), 2,13 (m, 2H), 1,42 (s, 9H). MS (El): 141,4 (M-tBu + 2H), 126,0 (M-OtBu). Etapa 3: (3S)-3-[2-fíuoro-1 -hidróxi-2-(fenilsulfonil)etil]pirrolidina- 1-carboxilato de terc-butila N,N-di-isopropilamina (1,62 mL, 11,59 mmols) foi adicionada a THF (9,89 mL) e esta solução foi resfriada para -78°C, em seguida n-butil- lítio a 1,60 M em hexano (6,59 mL, 10,54 mmols) foi adicionado. A reação foi mantida a -78°C durante 5 minutos, em seguida aquecida para 0°C durante 15 minutos, em seguida resfriada novamente para -78°C. A esta foi adicionada uma solução de fiuormetil fenil sulfona (2,02 g, 11,59 mols) em THF (14 mL), a reação foi mantida durante 20 minutos, em seguida uma solução de (3S)-3-formilpirrolidina-1-carboxilato de terc-butila (2,1 g, 10,0 mmols) em THF (14 mL) foi adicionada. A reação foi mantida a -78°C durante 1,5 hora, em cujo tempo a análise de LCMS indicou a conclusão da reação. A reação foi extinguida a -78°C por adição de solução de NH4CI saturada e extraída em EtOAc. As fases foram separadas e a fase orgânica foi lavada com água, em seguida NaCl saturado, e secada sobre MgSO4. O solvente foi removido em vácuo e o resíduo foi purificado (120 g de cartucho de SiO2 empacotado, 85 mL/minuto, gradiente de 0 a 65% de EtOAc/hexanos durante 25 minutos) para obter o composto desejado, 2,96 g. 1H RMN (300 MHz, CDCI3): δ 7,91 (m, 2H), 7,75 (m, 1H), 7,66 (m, 2H), 5,06 (m, 0.5H), 4,91 (m, 0,5H), 4,31 (m, 1H), 3,51 (m, 2H), 3,24 (m, 2H), 2,99 (m, 1H), 2,54 (m, 1H), 1,92 (m, 2H), 1,42 (s, 9H). MS (El): 318,1 (M-tBu + 2H), 300,0 (M-OtBu), 271,4 (M-BOC + H). Etapa 4: (3S)-3-(2-fluoro-1-hidroxietil)pirrolidina-1-carboxilato de terc-butila (3S)-3-[2-fluoro-1 -hidróxi-2-(fenilsulfonil)etil]pirrolidina-1 - carboxilato de terc-butila (2,96 g, 7,93 mmols) foi dissolvido em CH3OH (290 mL) e Na2HPO4 (6,8 g, 48 mmols) foi adicionado, a reação foi resfriada para -5°C e almágama de mercúrio-sódio (10% Na) (11 g, 48 mmols) foi adicionado. A reação foi mantida a -5°C durante 1 a 3 horas, em cujo tempo a análise de LCMS indicou a conclusão da reação. Agitação foi descontinuada e os sólidos foram deixados assentar-se. A fase metanólica de sobrenadante foi decantada e o residuo sólido foi enxaguado com metanol, a fase de metanol combinada foi mantida a 0°C. O pH foi ajustado para neutro por adição cuidadosa de HCI a 1N, em seguida a solução foi reduzida em vácuo, manten- 5 do a temperatura abaixo de 15°C. O residuo foi dividido entre NaCl saturado e EtOAc, as fases foram separadas e a fase aquosa foi extraída novamente com EtOAc. A fase de EtOAc combinada foi secada sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto, 1,8 g, que continuou sem outra purificação. MS (El): 178,0 (M-tBu + 2H), 160,0 (M-OtBu). Etapa 5: (3S)-3-{2-fluoro-1-[(metilsulfonil)óxi]etil}pirrolidina-1-carboxilato de terc-butila
Uma solução de (3S)-3-(2-fluoro-1 -hidroxietil)pirrolidina-1 - carboxilato de terc-butila (1,80 g, 7,72 mmols) em DCM (35 mL) foi resfriada para 0°C e cloreto de metanossulfonila (657 microL, 8,49 mmols) foi adicio- 15 nado seguido por Et3N (2,15 mL, 15,4 mmols), a reação foi mantida a 0°C durante 1 hora em cujo tempo a análise de LCMS indicou a conclusão da reação. A mistura reacional foi dividida entre água e CHCI3, as fases foram separadas e a fase aquosa foi extraída com CHCI3 adicional. A fase orgânica combinada foi lavada com HCI a 1N, NaHCO3 saturado, água, em segui- 20 da NaCl saturado, secados sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto, que foi purificado (120 g de cartucho de SiO2 pré- empacotado, 85 mL/minuto, hexanos for 2 minutos, em seguida gradiente de 0 a 70% de EtOAc/hexanos durante 12 minutos, mantida a 70% de EtO- Ac/hexanos durante 10 minutos, produto foi visualizado em placa de tic com 25 manchamento por KMnO4) para fornecer o composto desejado, 1,6 g. 1H RMN (300 MHz, CDCI3): Õ 4,9-4,4 (m, 3H), 3,50 (m, 2H), 3,22 (m, 2H), 3,09 (s, 3H), 2,50 (m, 1H), 2,2-1,7 (m, 2H), 1,43 (s, 9H). 19F RMN (300 MHz, CDCI3): δ -226,39 (td, J = 49,5, 17,4 Hz), -227,34 (td, J = 47,3, 19,0 Hz), - 227,59 (td, J = 47,3, 19,5 Hz). MS (El): 256,0 (M-tBu + 2H), 238,0 (M-OtBu). Etapa 6: (3S)-3-{2-fluoro-1-[4-(7-{[2-(tnmetilsihl)etoxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pírrolidina-1 -carboxilato de terc-butila
À 4-(1H-pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidina (1,71 g, 5,41 mmols, preparada como descrito no WO i 2007/070514) foi adicionado hidreto de sódio (60% de óleo mineral, 60%, 247 mg, 6,17 mmols) seguido por DMF (1,4 mL). Após evolução de gás cessar, uma solução de (3S)-3-{2-fluoro-1-[(metilsulfonil)óxi]etil}pirrolidina-1- carboxilato de terc-butila (1,6 g, 5,1 mmols) em DMF (17,1 mL) foi adicionada e a reação foi aquecida para 60°C durante 16 a 36 horas. A reação foi resfriada para temperatura ambiente e dividida entre água e EtOAc, as fases ,foram separadas e a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida NaCI saturado, secada sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto. A mistura foi separada (120 g de cartucho de SiO2 pré-empacotado, 85 mL/minuto, solvente A = 92/5/3 hexanos/EtOAc/IPA, solvente B = 49/45/6 hexanos/EtOAc/IPA. Eluída com gradiente de 0 a 40% de B durante 20 minutos, e mantida a 40% de B durante 10 minutos. Frações misturadas foram recombinadas e purificadas pela mesma maneira para fornecer o isómero (primeira eluição) desejado, 0,8g. 1H RMN (300 MHz, CDCI3): δ 8,91 (s, 1H), 8,374 (s, 1H), 8,367 (s, 1H), 7,46 (d, 1H, J = 3,68 Hz), 6,86 (d, 1H, J = ( 3,78 Hz), 5,74 (s, 2H), 5,0-4,6 (m, 2H), 4,4 (m, 1H), 1,4 (m, 1H), 3,76 (m, । 1H), 3,60 (m, 2H), 3,54 (m, 1H), 3,25 (m, 2H), 2,92 (m, 1H), 1,9-1,6 (m, 1H), 1,52 (s, 9H), 0,97 (m, 2H). 19F RMN (300 MHz, CDCI3): δ -225,12 (td, J = 49,5, 19,0 Hz), -225,52 (td, J = 49,5, 19,8 Hz). MS (El): 531,2 (M+H). Etapa 7: 4-( 1 -{2-fluoro-1-[(3S)-pirrolidin-3-il]etil}-1 H-pirazol-4-il)-7-{[2- (trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pinmidina (3S)-3-{2-fluoro-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d Jpiri m idin-4-il)-1 H-pirazol-1 -il]etil}pirrolid ina-1 -carboxilato de terc-butila (800,0 mg, 1,507 mmol) foi dissolvido em THF (12 mL), em seguida HCI a 12,0 M (1,40 mL, 16,7 mmols) foi adicionado. A reação foi deixada agitar em temperatura ambiente durante 16 horas, em cujo tempo a análise de LCMS indicou desproteção de BOC completa. A reação foi neutralizada por adição de NaHCO3 saturado, seguido por NaHCO3 sólido até o pH tornar-se básico. A solução de reação foi extraída com EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada | foi lavada com água, em seguida NaCl saturado, secada sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto. MS (El): 431,2 (M+H). Etapa 8: 2-((3S)-3-{2-fluoro-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidÍn-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)[1,3]oxazolo[5,4-b]pirídina [1,3]oxazolo[5,4-b]piridina-2(1H)-tiona (233 mg, 1,53 mmol, preparada no exemplo 33, etapa 4) e 4-(1-{2-fluoro-1-[(3S)-pirrolidin-3-il]etil}-1H- pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidina (550 mg, 1,3 mmol) foram misturadas em 1,4-dioxano (6,0 mL) e a reação foi aquecida para 70°C durante 2 horas em cujo ponto a análise de LCMS indicou a conclusão da reação para o intermediário hidroxipiridinil tioureia. A reação foi resfriada para temperatura ambiente e foi concentrada em vácuo, em seguida EtOH (7,4 mL) foi adicionada e a mistura foi bem agitada até a tioureia ser livremente suspensa no solvente. Esta mistura foi em seguida tratada com AgNO3 (434 mg, 2,55 mmols) e NH4OH (790 microL, 11,5 mmols), e a reação foi agitada em temperatura ambiente durante 16 horas, em cujo ponto a análise de LCMS indicou conversão completa no composto desejado. A reação foi filtrada através de um tampão de celite um funil de vidro sinteriza- do e enxaguada com dioxano (20 mL) e EtOAc (20 mL). O filtrado foi reduzido em vácuo e o resíduo foi dividido entre água e EtOAc, as fases foram separadas e a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida NaCl saturado, secada sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto que foi purificado (40 g de cartucho de SÍO2 pré-empacotado, 40 mL/minuto, DCM durante 3 minutos, em seguida eluição isocrática com 5% de MeOH/DCM durante 15 minutos) para recuperar o produto, 592 mg. MS (El): 549,2 (M+H). Etapa 9: trifluoroacetato de 2-((3S)-3-{2-fluoro-1-[4-(7H-pirrolo[2,3-d]piri- midin- 4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)[ 1,3]oxazolo[5, 4-b]pÍridina
A 2-((3S)-3-{2-fluoro-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)[1,3]oxazolo[5,4- b]piridina (592 mg, 0,17 mmol) foi adicionado DCM (5,0 mL) e TFA (5,0 mL). A reação foi agitada em temperatura ambiente durante 1 hora, em seguida os solventes foram removidos em vácuo e metanol (5,0 mL) e NH4OH (5,0 mL) foram adicionados. Após 30 minutos a análise de LCMS indicou remoção completa de grupo SEM. Os solventes foram removidos em vácuo e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters FractionLynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 μM, 30x100 mm, fase móvel A: água (0,1% de TFA), B: acetonitrila (0,1% de TFA), gradiente 17 a 37% de B durante 5 minutos, taxa de fluxo de 60 mL/minuto), o produto foi isolado como um sal de trís-TFA, 455 mg. 1H RMN (300 MHz, CD3OD): δ 8,97 (s, 1H), 8,90 (s, 1H), 8,55 (s, 1H), 7,88 (d, 1H, J = 3,88 Hz), 7,86 (dd, 1H, J = 5,40, 1,45 Hz), 7,58 (dd, 1H, J = 7,64, 1,38 Hz), 7,31 (d, 1H, J = 3,58 Hz), 7,20 (dd, 1H, J = 7,83, 4,90 Hz), 5,09 (m, 1H), 5,0-4,8 (m, 2H), 4,02 (m, 1H), 3,76 (m, 1H), 3,61 (m, 2H), 3,15 (m, 1H), 1,96 (m, 2H). MS (El): 419,1 (M+H). Etapa 10: fosfato de 2-((3S)-3-{2-fíuoro-1-[4-(7H-pÍrrolo[2,3-d]pirimidin-4-il)- 1 H-pirazol-1 -il]etil}pirrolidin-1 -il)[ 1,3]oxazolo[5,4-b]piridina
Tris-trifluoroacetato de 2-((3S)-3-{2-fluoro-1-[4-(7H-pirrolo[2,3- d]pÍrimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)[1,3]oxazolo[5, 4-b]piridina (455 mg, 0,60 mmol) foi dividido entre NaHCO3 saturado e 3:1 CHCI3/IPA, as fases foram separadas e a fase aquosa foi extraída com solvente adicional. A fase orgânica combinada foi lavada com NaCl saturado, secada sobre MgSO4 e reduzida em vácuo para fornecer a base livre. A este material foram adicionados IPA (10 mL) e EtOH (3 mL) e a reação foi aquecida para 90°C sob um condensador de refluxo até a dissolução completa ocorrer. Uma solução de H3PO4 (61 mg, 0,63 mmol) em um IPA de 200 microL foi em seguida adicionada, e a reação foi removida do banho de óleo e deixada repousar e resfriar. No resfriamento um sólido foi precipitado, e este foi coletado por filtragem. Os sólidos foram secados em vácuo para fornecer o produto como o sal de fosfato, 204 mg. 1H RMN (300 MHz, DMSO-d6): δ 12,1 (bs, 1H), 8,80 (s, 1H), 8,65 (s, 1H), 8,38 (s, 1H), 7,84 (dd, 1H, J = 5,22, 1,32 Hz), 7,58 (m, 2H), 7,17 (dd, 1H, J = 7,63, 5,15 Hz), 6,96 (m, 1H), 5,00 (m, 1H), 4,81 (m, 2H), 3,88 (m, 1H), 3,65 (m, 1H), 3,47 (m, 2H), 2,95 (m, 1H), 1,72 (m, 2H). 19F RMN (300 MHz, DMSO-d6): δ -223,176 (td, J = 46,1, 15.9 Hz). MS (El): 419,1 (M+H). Esquema 1. O ácido carboxilico 1 fol reduzido em álcool 2 por ação de borano, que foi subsequentemente oxidado por meio de uma oxidação Swern ao aldeído 3 correspondente. O ânion de fluormetil fenil sulfona 5 foi adicionado a 3 para fornecer o intermediário 4, que foi desulfonilado sob condições redutivas usando amálgama de sódio para fornecer o fluorálcool 5. Este composto foi em seguida convertido em mesilato 6, que foi adicionado ao ânion do núcleo de pirazol, os diastereômeros resultantes foram separados por cromatografia de sílica-gel para fornecer o único isômero desejado do derivado de pirrolidina de N-BOC 7. Este foi completamente desprotegido sob condições acídicas para fornecer o intermediário avançado 8, que foi submetido à reação com o oxazolopiridina-2-tiol indicado em um procedimento de duas etapas, seguido por um procedimento de desproteção de duas etapas para fornecer o produto final 9 em boa produção e excesso di- astereomérico elevado.

Exemplo 71. Sal de ácido fosfórico 2-((3R)-3-{2-fluoro-1-j4-(7H-pirrolo[2,3- dlpirimidin- 4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)[1,31oxazolo[5, 4-blpiridina
Este exemplo pode ser preparado seguindo as etapas 1 a 10 para o exemplo 70, substituindo ácido (3R)-1-(terc-butoxicarbonil)pirrolidina- 3-carboxílico para o isómero (S) na etapa 1. Todas as seguintes etapas são realizadas analogamente. 1H RMN (300 MHz, DMSO-d6): δ 12,1 (bs, 1H), 8,80 (s, 1H), 8,65 (s, 1H), 8,38 (s, 1H), 7,84 (dd, 1H, J = 5.22, 1,32 Hz), 7,58 (m, 2H), 7,17 (dd, 1H, J = 7,63, 5,15 Hz), 6,96 (m, 1H), 5,00 (m, 1H), 4,81 (m, 2H), 3,88 (m, 1H), 3,65 (m, 1H), 3,47 (m, 2H), 2,95 (m, 1H), 1,72 (m, 2H). 19F RMN (300 MHz, DMSO-d6): δ -223,176 (td, J = 46,1, 15.9 Hz). MS (El). 419,1 (M+H). Exemplo 72. Trifluoroacetato de 2-(3-(1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1-il)-2-fluoroetil)pirrolidin-1-il)oxazoloí5,4-blpiridina
O produto racêmico acima pode ser preparado seguindo as etapas 1 a 9 para o exemplo 70, utilizando ácido l-(terc-butoxicarbonil) pirrolidi- na-3-carboxilico racêmico na etapa 1.
Alternativamente, a seguinte sequência pode ser usada para acessar o produto racêmico. Etapa 1: 3-{[metóxi(metil)amino]carbonil}pirrolidina-1-carboxilato de terc-butila Ao ácido 1-(terc-butoxicarbonil)pirrolidina-3-carboxílico (0,1 g, 4,6 mmols) foi adicionado DMF (26 mL) seguido por hexafluorofosfato de N,N,N',N'-tetrametil-O-(7-azabenzotriazol-1-il)urônio (2,6 g, 7,8 mmols) e N,N-di-isopropiletilamina (3,9 mL, 22 mmols). HCI de N,O-dimetilhidroxila- mina (910 mg, 9,3 mmols) foi em seguida adicionado e a reação foi agitada em temperatura ambiente durante 16 horas em cujo ponto LCMS e tlc indr- cram a conversão completa na amida Weinreb. A mistura reacional foi dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida NaCl saturado, secada sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto, que foi em seguida purificado por cromatografia de coluna (40 g de cartucho de SÍO2 pré-empacotado, 40 mL/minuto, gradiente de 0 a 90% de EtOAc/hexanos durante 20 minutos) para fornecer o composto desejado, 0,15 g. 1H RMN (300 MHz, CDCI3): δ 3,71 (s, 3H), 3,7-3,3 (m, 5H), 3,20 (s, 3H), 2,1 (m, 2H), 1,45 (s, 9H). MS (El): 203,1 (M-tBu + 2H), 185,1 (M-OtBu). Etapa 2: 3-acetilpirrolidina-1-carboxilato de terc-butila
Uma solução de 3-{[metóxi(metil)amino]carbonil}pirrolidina-1- carboxilato de terc-butila (0,10 g, 3,87 mmols) em THF (11 mL) foi resfriada para -78°C, em seguida CH3MgBr a 3,0 M em éter (3,87 mL, 11,6 mmols) foi adicionado. A reação foi mantida a -78°C durante 1,5 hora, em seguida a- quecida para 0°C durante 1 hora, em cujo ponto análise de cromatografia de camada fina indicou conversão completa em cetona. A reação foi extinguida com NH4CI saturado e extraída com EtOAc. As fases foram separadas e a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com NaCl saturada, secada sobre MgSO4, e reduzida em vácuo para fornecer o produto bruto, que foi usado diretamente na reação seguinte. 1H RMN (300 MHz, CDCI3):δ 3,6-3,4 (m, 3H), 3,33 (s, 1H), 3,12 (m, 1H), 2,20 (s, 3H), 2,06 (m, 2H), 1,45 (s, 9H). MS (El): 158,1 (M-tBu + 2H), 140,1 (M- OtBu). Etapa 3: 3-{1-[(trimetilsilil)óxi]vinÍI}pÍrrolidina-1 -carboxilato de terc-butila N,N-di-isopropilamina (631 microL, 4,50 mmol) foi adicionada a THF (4,2 mL) e esta solução foi resfriada para -78°C, em seguida n-butil-lítio a 1,60 M em hexano (2,81 mL, 4,50 mmols) foi adicionado. A reação foi mantida a -78°C durante 5 minutos, em seguida aquecida para 0°C durante 15 minutos, em seguida resfriada novamente para -78°C. A esta foi adicionada uma solução de 3-acetilpirrolidina-1-carboxilato de terc-butila (800 mg, 3,75 mmols) em THF (10 mL). Este foi mantido a -40°C durante 20 minutos, em seguida TMSCI (714 microL, 5,63 mmols) foi adicionado. A reação foi aque- cida de -40°C para -10°C durante 1,5 hora, em seguida a reação foi resfriada novamente para -40°C, extinguida por adição de NaHCO3 saturado e extraída em EtOAc. As fases foram separadas e a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com água seguido por solução de HCI a 0,1 N até o pH de lavagem acídica ser acidica. A fase orgânica foi em seguida lavada duas vezes com água, em seguida NaCI saturado, secada sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto, 0,11 g. 1H RMN (300 MHz, CDCI3):δ 3,95 (s, 1H), 3,91 (s, 1H), 3,40 (m, 2H), 3,20 (m, 2H), 2,72 (m, 1H), 1,90 (m, 2H), 1,30 (s, 9H), 0,05 (m, 9H). MS (El): 230,1 (M-tBu + 2H). Etapa 4: 3-(fluoroacetil)pirrolidina-1-carboxilato de terc-butila
A uma solução de 3-{1 -[(trimetilsilil)óxi]vinil}pirrolidina-1- carboxilato de terc-butila (847 mg, 2,97 mmols) em CH3CN (26 mL) foi adicionado SelectFluor® (1,38 g, 3,88 mmols). A mistura foi agitada em temperatura ambiente durante 1,5 hora, em cujo ponto a análise de LCMS indicou a conversão em fluormetil cetona. A mistura reacional foi dividida entre NaHCO3 saturado e EtOAc, as fases foram separadas e a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida NaCI saturado, secada sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto. O produto foi purificado (40g de cartucho de SiO2 pré-empacotado, 40 mL/minuto, gradiente de 0 a 80% de EtO- Ac/hexanos durante 20 minutos) para recuperar a fluormetil cetona, 351 mg. 1H RMN (300 MHz, CDCI3): δ 4,89 (d, 2H, J = 47,4 Hz), 3,35-3,15 (m, 5H), 2,10 (m, 2H), 1,45 (s, 9H). MS (El): 176,0 (M-tBu + 2H), 158,1 (M-OtBu). Etapa 5: 3-(2-fíuoro-1-hidroxietil)pirrolidina-1-carboxilato de terc-butila
Uma solução de 3-(fluoroacetil)pirrolidina-1-carboxilato de terc-butila (245 mg, 0,16 mmol) em CH3OH (0,61 mL) foi resfriada para 0°C, e NaBH4 (28 mg, 0,74 mmol) foi adicionada. A reação foi agitada a 0°C duran- te 30 minutos, em seguida uma amostra foi retirada e extinguida em H- CI/EtOAc a 0,1 N e a análise subsequente de TLC indicou redução completa de cetona. A reação foi extinguida por adição de 0,HCI a 1 N e EtOAc, as fases foram separadas e a fase aquosa foi extraída com EtOAc adicional. A 5 fase orgânica combinada foi lavada com NaHCO3 saturado, em seguida NaCl saturado, secada sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto, 240 mg, que continuou sem outra purificação. MS (El): 178,0 (M-tBu + 2H), 160,0 (M-OtBu).
A síntese é concluída pelas seguintes etapas restantes mostra- 10 das pelo exemplo 70. Esquema 2. O ácido carboxílico 1 foi convertido na amida Wein- reb 2, que em seguida foi tratada com metil Grignard para fornecer uma boa produção de metil cetona 3. A cetona foi convertida no silil enol éter 4, que em seguida reagiu com Selectfluor® para fornecer fluormetil cetona 5. Redu- 15 ção de 5 com boroidreto de sódio forneceu o fluorálcool 6, que foi subsequentemente absorvido para formar os compostos de fluormetil pirrolidina racêmicos por procedimentos ilustrados no esquema 1.
Exemplo 73. Trifluoroacetato de 3-(1-(1H-pirrolo[2,3-blpiridin-6-il)pirrolidin-3-20 il)-3-(4-(7H-pirrolo[2,3-d1pirimidin-4-il)-1 H-pirazol-1-il)propanomtrila

Etapa 1: 3-(1-(1 H-pirrolo[2,3-b]piridin-6-il)pirrolidin-3-il)-3-(4-(7-( (2-(trime- tilsilil)etóxi)metil)-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila Hemí-hidrato de 1H-pirrolo[2,3-b]piridina-7-óxido (31 mg, 0,22 mmol, Sigma-aldrich) foi suspenso em CH3CN (0,4 mL) e sulfato de dimetila (25 microL, 0,27 mmol) foi adicionado. A reação foi aquecida para 55°C em cujo ponto a solução tornou-se homogênea. A reação foi mantida a 55°C durante 16 horas. À solução foi em seguida adicionada 3-pirrolidin-3-il-3-[4-(7- {[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (98 mg, 0,22 mmol, preparada no exemplo 15, etapas 1 a 3, omitindo a separação quiral realizada na etapa 2) e 2,2,6,6-tetrametilpiperidina (75 microL, 0,45 mmol). A reação foi em seguida aquecida para 50°C durante 4 horas, em cujo ponto a análise de LCMS indicou a conclusão da reação. A reação foi resfriada para temperatura ambiente e dividida entre água e E- tOAc, as fases foram separadas e a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida NaCI saturado, secada sobre MgSO4 e reduzida em vácuo para fornecer o produto bruto. Este foi purificado (4 g de cartucho de SiO2 pré-empacotado, 20 mL/minuto, gradiente de 0 a 100% de EtOAc/hexanos durante 16 minutos) para recuperar o produto, 31 mg. MS (El): 554,2 (M+H). Etapa 2: trifluoroacetato de 3-[1-(1H-pirrolo[2,3-b]piridin-6-il)pirrolidin-3-il]-3- [4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila
À 3-1 -(1 H-pirrolo[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4-(7-{[2-(trime- tilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (31 mg, 0,056 mmol) foram adicionados DCM (500 microL) e TFA (500 microL). A reação foi agitada em temperatura ambiente durante 1 hora, em se- guida os solventes foram removidos e metanol (500 microL) e NH4OH (500 microL) foram adicionados. Após 30 minutos, análise de LCMS indicou remoção completa de grupo SEM. Os solventes foram removidos em vácuo e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters FractionLynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 microM, 19 x 100 mm, fase móvel A: água (0,1% de TFA), B: acetonitrila (0,1% de TFA), taxa de fluxo de 30 mL/minuto, para fornecer o produto como um sal de TFA. 1H RMN (300 MHz, DMSO-d6): õ 12,61 (bs, 1H), 11,10 (bs, 1H), 8,97 (s, 1H), 8,78 (s, 1H), 8,50 (s, 1H), 7,79 (d, 1H, J = 8,2 Hz), 7,73 (m, 1H), 7,10 (m, 1H), 6,97 (m, 1H), 6,31 (d, 1H, J = 8,8 Hz), 6,24 (m, 1H), 4,84 (m, 1H), 3,72 (m, 1H), 3,48 (m, 1H), 3,4-3,2 (m, 4H), 2,90 (m, 1H), 1,67 (m, 2H). MS (El): 424,0 (M+H). Esquema 3. Pirrolopiridina 1 foi tratada com sulfato de dimetila 15 para formar o intermediário pirrolopiridina de 7-metóxi 2, que não foi isolado, porém diretamente reagido com o núcleo de pirrolidina 3 para fornecer o intermediário avançado 4. O grupo de proteção SEM foi removido em um procedimento de duas etapas para fornecer o alvo desejado 5.
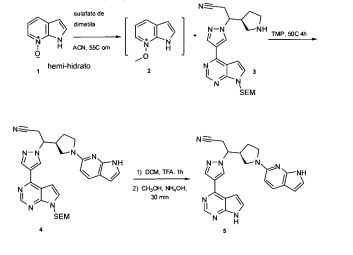
Exemplo 74. 5-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3-dlpirimidin-4-il)-1H-pirazol-1-illetil}pirrolidin-1-il)tienoí2,3-clpiridina-4-carbonitrila
Etapa 1. 5-clorotieno[2:3-c]piridina-4-carbonitnla
A uma mistura de 3-tienilacetonitrila (1,16 g, 9,42 mmols), cloreto de fosforila (10,1 g, 65,9 mmols) foi adicionado DMF (2,2 mL, 28 mmol). A mistura resultante foi aquecida a 100°C durante 2,5 horas. Cloridrato de hi- droxilamina (1,28 g, 18,4 mmols) foi adicionado em porções e agitado durante mais 20 minutos. A solução de reação foi resfriada para temperatura ambiente e o precipitado resultante foi filtrado e lavado com acetona para fornecer o composto desejado como um sólido branco (26% de rendimento). LCMS (M+H)+: 194,9. Etapa 2. 5-((3S)-3-{(1S)-2-cÍano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1 -il]etil}pirrolidin-1 -il) tieno[2,3-c]piridina-4-carbonitrila
Uma mistura de (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetil- silil)etóxi]metil}-7H-pirrolo[2,3-d]pinmidin-4-il)-1 H-pirazol-1-iljpropanonitrila (de exemplo 15, etapa 3; 18 mg, 0,041 mmol), 5-clorotieno[2,3-c]piridina-4- carbonitrila (18 mg, 0,095 mmol) e DIPEA (20 pL, 0,1 mmol) em etanol (0,09 mL) foi aquecida até o refluxo durante 2,5 horas. Ela foi purificada por LCMS (coluna Sunfire C18 de 19*100 mm), eluindo com um gradiente de acetoni- trila/água contendo 0,1% de TFA, em taxa de fluxo de 30 mL/minuto) para fornecer 7 mg de sólido amarelo claro (30% de rendimento). LCMS (M+1): 596,0.
O sólido amarelo claro acima (7 mg) foi agitado em 0,5 mL de DCM e 0,5 mL de TFA durante 1 hora. A mistura foi concentrada e o resíduo foi agitado em 50 μL EDA e 1 mL de MeOH durante 1 hora. A solução de reação foi purificada por LCMS (coluna C18 19*100 mm) eluindo com um gra- diente ACN/H2O contendo 0,15% de NH4OH a 30 mL/minuto) para fornecer o composto desejado como um sólido branco (1,6 mg, 8% de rendimento). LCMS (M+H)+: 466,1. Exemplo 75. 5-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirroloi2,3-dlpirimidin-4-il)-1H- pirazol-1-illetil}pirrolidin-1-il)tienof3,2-blpiridina-6-carbonitrila
Etapa 1. 3-acetiltiofeno oxima
A uma solução de 1-(3-tienil)etanona (1,49 g, 11,8 mmols) em etanol (43 mL) e água (13 mL), cloridrato de /V-hidroxiamina (1,96 g, 28,2 mmols) e acetato de sódio (2,32 g, 28,3 mmols) foram adicionados sequencialmente. A solução resultante foi refluxada durante 1 hora, em seguida 100 mL de água fria foram adicionados e precipitado resultante foi coletado para produzir o composto desejado como pó branco (816 mg, 49%). LCMS (M+H)+: 142,0. Etapa 2. 5-clorotieno[3,2-b]pindina-6-carbonitn!a
A uma solução de oxima de 3-acetiltiofeno (0,816 g, 5,78 mmols) em éter (20 mL) foi adicionado cloreto de fosforila (5,2 mL, 56 mmols) gota a gota a 10°C durante 20 minutos. A mistura resultante foi agitada a 10°C durante 2 horas. DMF (1,1 mL, 14,5 mmols) foi em seguida adicionado gota a gota e a mistura foi aquecida para ebulição do éter. O aquecimento foi continuado até todo o éter ter sido removido e temperatura da mistura reacional alcançar 110°C. A mistura reacional foi aquecida a 110°C durante 1 hora. Cloridrato de hidroxilamina (0,800 g, 11,5 mmols) foi adicionado em porções durante 15 minutos e agitado durante mais 20 minutos. A solução de reação foi resfriada para temperatura ambiente e a mistura resultante foi vertida em uma mistura de 40 g de gelo e 60 g de água com agitação. O precipitado amarelo que se formou foi coletado por filtragem e secado para fornecer o composto desejado (449 mg, 40%). LCMS (M+H)+: 195,0. Etapa 3. 5-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H’ pirazol-1-il]etil}pirrolidin-1-il)tieno[3,2--b]piridina-6-carbonitnla
Uma mistura de (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsi- Iil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (18 mg, 0,041 mmol; de exemplo 15, etapa 3), 5-clorotieno[3,2-b]piridina-6- carbonitrila (18 mg, 0,095 mmol) e DIPEA (20 μL, 0,1 mmol) em etanol (0,1 mL) foi aquecida até o refluxo durante 2,5 horas. Ela foi purificada por LCMS (coluna Sunfire C18 de 19x100 mm), eluindo com um gradiente de acetoni- trila/água contendo 0,1% de TFA, em taxa de fluxo de 30 mL/minuto) para fornecer 7 mg de amarelo sólido (30% de rendimento). LCMS (M+1): 596,2.
O sólido amarelo (7 mg) foi agitado em 1 mL de DCM e 1 mL de TFA durante 1 hora. Ele foi concentrado e o resíduo foi agitado em 50 μL de EDA e 1 mL de MeOH durante 1 hora. A solução de reação foi purificada por LCMS (coluna C18 de 19x100 mm) eluindo com um gradiente ACN/H2O contendo 0,15% de NH4OH at 30 mL/minuto) para fornecer o composto desejado como um sólido branco (2,0 mg, 10% de rendimento). LCMS (M+H)+: 466,0. Exemplo 76. 2-((3S)-3-{(1S)-2-ciano-144-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H- pirazol-1-illetil}pirrolidin-1-il)-4-hidroxitiofeno-3-carbonitrila
Etapa 1. 2,4-dibromo-3~metiltiofeno
Uma suspensão de zinco (1,94 g, 29,7 mmols) em ácido acético (5,20 mL, 91,4 mmols), água (14,0 mL) e THF (2,0 mL) foi conduzida a um refluxo suave, e em seguida o aquecimento foi removido. 2,3,5-tribromo-4- metiltiofeno (de TCI; 10,0 g, 29,9 mmols) em THF (0,1 mL) foi em seguida adicionado gota a gota em uma taxa que a mistura reacional manteve-se refluxante. Após a adição ser concluída, a mistura foi refluxada durante a noite, e em seguida resfriada para temperatura ambiente e extraída com a- cetato de etila. Um extrato orgânico foi lavado com água, NaHCO3 saturado, e salmoura, secado sobre Na2SO4 e concentrado. Destilação do produto bruto sob vácuo elevado forneceu o composto desejado como liquido incolor (4 g, 52%). 1H RMN (300 MHz, CDCI3): δ 7,22 (s, 1H), 2,20 (s, 3H). Etapa 2. 2,4-dibromo-3-(bromometil)tiofeno
Uma suspensão de 2,4-dibromo-3-metiltiofeno (1,89 g, 7,38 mmols) e A/-bromossuccinimida (1,42 g, 7,98 mmols) e 2,2'-azobis(isobuti- ronitrila) (de Aldrich 10,3 mg, 0,0626 mmol) em tetracloreto de carbono (15,4 mL) foi aquecida para 80°C durante 2 horas. A suspensão de reação foi resfriada para temperatura ambiente. O precipitado foi filtrado e lavado com pequena quantidade de DCM. O filtrado foi concentrado para fornecer a solid. O bruto material foi usado na etapa seguinte sem purificação. Etapa 3. Acetato de (2,4-dÍbromo-3-tienii)metila
A uma solução de 2,4-dibromo-3-(bromometil)tiofeno (2,47 g, 7,38 mmols) em DMF (20 mL) foi adicionado acetato de sódio (3,02 g, 36,9 mmol). A mistura foi aquecida a 110°C durante 3 horas. A solução de reação foi resfriada para temperatura ambiente e diluída com acetato de etila e á- gua. A camada aquosa foi extraída com acetato de etila uma vez. Os extratos orgânicos combinados foram lavados com água, salmoura e secados sobre Na2SO4, filtrada e concentrada. O resíduo foi purificado por coluna de sílica-gel (0% a 10% de acetato de etila/hexanos) para fornecer o composto desejado como óleo claro (1,89 g, 81%). 1H RMN (300 MHz, CD3OD): δ 7,57 (s, 1H), 5,06 (s, 2H), 2,05 (s, 3H); LCMS (M+Na)+: 336,7. Etapa 4. (2l4-dÍbromo-3-tienil)metanol
A uma solução de acetato de (2,4-dibromo-3-tienil)metila (1,89 g, 6,02 mmols) em acetonitrila (10 mL) e água (10 mL) foi adicionado NaOH a 50% em água (50:50, água:hidróxido de sódio, 0,651 mL, 18,0 mmols). A solução resultante foi agitada em temperatura ambiente durante a noite. A solução de reação foi diluída com H2SO4 a 2 N e acetato de etila. A camada aquosa foi extraída com acetato de etila uma vez. Os extratos orgânicos combinados foram lavados com salmoura, secados sobre Na2SO4, filtrados e concentrados. O resíduo foi purificado em uma coluna de sílica-gel (0% a 15% de acetato de etila/hexanos) para fornecer o composto desejado como um sólido branco (1,46 g, 89%). LCMS (M+H-H2O)+: 254,9. Etapa 5. 2,4-dibromotiofeno-3-carbaldeído
A uma solução de (2,4-dibromo-3-tienil)metanol (1,46 g, 5,37 mmols) em DCM (50 mL) foi adicionado períodinano de Dess-Martin (2,5 g, 5,9 mmols). A solução de reação foi agitada em temperatura ambiente durante 2 horas. A solução de reação foi diluída com éter e NaHCO3 saturado. Após agitação durante 1 hora, a mistura reacional foi filtrada através de uma almofada de celite. A camada aquosa foi extraída com acetato de etila uma vez. Os extratos orgânicos combinados foram lavados com salmoura, secados sobre Na2SO4, filtrados e concentrados em vácuo para fornecer o composto desejado (1,45 g, 100%). O produto bruto foi usado na etapa seguinte sem outra purificação Etapa 6. Oxima de 2,4-dibromotiofeno-3-carbaldeído
A uma solução de 2,4-dibromotiofeno-3-carbaldeído (1,45 g, 5,37 mmols) em etanol (19,5 mL) e água (5,9 mL) foram adicionados clori- drato de /V-hidroxiamina (0,410 g, 5,91 mmols) e acetato de sódio (0,617 g, 7,52 mmols) sequencialmente. A solução resultante foi refluxada durante 1 hora, o solvente orgânico foi removido em vácuo e a solução foi diluída com água. O precipitado resultante foi coletado e secado sob vácuo para fornecer o composto desejado como um sólido branco (1,38 g, 90%). LCMS (M+H)+: 285,8. Etapa 7. 2,4-dibromotiofeno-3-carbonitnla
A uma solução de oxima de 2,4-dibromotiofeno-3-carbaldeido (1,37 g, 4,81 mmols) em piridina (15 mL) foi adicionado cloreto de metanos- sulfonila (1,5 mL, 19 mmols). Ela foi aquecida a 60°C durante 2 horas. A solução de reação foi diluída com acetato de etila e solução de CuSO4 saturada. A camada orgânica foi lavada com CuSO4 duas vezes, seguido por solução de HCI a 1 N e salmoura, secada sobre Na2SO4, filtrada e concentrada. O resíduo foi purificado por coluna de sílica-gel (0 a 10% de acetato de etila/hexanos) para fornecer o composto desejado como um sólido branco (1,18 g, 92%). 1H RMN (300 MHz, CD3OD): δ 7,73 (s, 1H). Etapa 8. 4-bromo-2-((3S)-3-{(1 S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)~ 1 H-pirazol-1 -il]etil}pirrolidin-1 -il)tiofeno-3- carbonitrila
Uma mistura de (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsi- lil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (122 mg, 0,279 mmol; de exemplo 15, etapa 3), 2,4-dibromotiofeno-3- carbonitrila (83,0 mg, 0,311 mmol) e DIPEA (53,4 μl_, 0,307 mmol) em tetra- fluoroborato de 1-butil-3-metil-1H-imidazol-3-io (315 mg) foi aquecida a 120°C durante 2 horas. A solução de reação foi resfriada para temperatura ambiente e diluída com acetato de etila e água. A camada aquosa foi extraída com acetato de etila. Os extratos foram lavados com salmoura, secados sobre Na2SO4, filtrados e concentrados. O resíduo foi purificado com coluna de sílica-gel para fornecer o composto desejado como um sólido amarelo (88 mg, 50%). LCMS (M+H)+: 623,0, 625,1. Etapa 9. 2-((3S)-3-{( 1 S)-2-ciano-1 -[4-(7H-pirrolo[2:3-d]pirimidin-4-il)-1 H-pira-zol-1 -il]etil}pirrolidin-1 -il)-4-hidroxitiofeno-3-carbonitn'la
A uma solução de 4-bromo-2-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2- (trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirro- lidin-1 -il)tiofeno-3-carbonitrila (16,1 mg, 0,0258 mmol) em DCM (0,50 mL) foi adicionado TFA (0,50 mL). A solução de reação foi agitada em temperatura ambiente durante 2 horas e o solvente foi removido em vácuo. O resíduo foi dissolvido em metanol (1 mL) e tratado com (100 μL, 1,50 mmol). A solução de reação foi agitada durante 1 hora e diluída com metanol de EDA e purificada por LCMS (coluna C18 19*100 mm) eluindo com um gradiente ACN/H2O contendo 0,15% de NH4OH a 30 mL/min para fornecer o composto desejado como um sólido branco (5,7 mg, 51%). LCMS (M+H)+: 431,1. Exemplo 77. 4-bromo-2-((3S)-3-[(1S)-2-ciano-1-í4-(7H-pirrolo[2,3-dlpirimidin- 4-il)-1 H-pirazol-1-il]etíl}pirrolidin-1-il)tiofeno-3-carbonitrila
A uma suspensão agitada de 4-bromo-2-((3S)-3-{(1S)-2-ciano-1- [4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimÍdin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1-il)tÍofeno-3-carbonitrila (de exemplo 76, etapa 8; 24,2 mg, 0,039 mmol) em acetonitrila (0,2 mL) para temperatura ambiente foi adicionado eterato de trifluoreto de boro (12,3 μL, 0,097 mmol). A solução marrom clara resultante foi agitada durante 2 horas. A solução de reação foi diluída com metanol (0,5 mL) e tratada com EDA (50 μL, 0,75 mmol). A solução de reação foi agitada durante 1 hora e diluída com metanol e purificada com LCMS preparativa (coluna C18 19*100 mm eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH em 30 mL/minuto) para fornecer o composto desejado (5,3 mg, 27%). 1H RMN (300 MHz, DMSO-d6): δ 12,1 (s, 1H); 8,86 (s, 1H); 8,67 (s, 1H); 8,42 (s, 1H); 7,59 (d, 1H); 6,97 (d, 1H); 6,75 (s, 1H); 4,84 (m, 1H); 3,72-3,20 (m, 6H); 2,96 (m, 1H); 1,74 (m, 2H); LCMS (M+H)+: 493,0, 495,0. Exemplo 78. 4-cloro-2-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2.3-dlpirimidin- 4-ÍD-1 H-pirazol-1-il]etil}pirrolidin-1-il)tiofeno-3-carbonitrila

Etapa 1. 4-cloro-2-((3S)-3-{( 1 S)-2-ciano-1 -[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-dJpirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il) tiofeno-3-car- bonitrila
A uma solução de 4-bromo-2-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2- (trimetilsilil)etóxi]metrl}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirro- lidin-1-il)tiofeno-3-carbonitrila (de exemplo 76, etapa 8; 21 mg, 0,034 mmol) em piridina (100 μL) foi adicionado monocloreto cuproso (16,7 mg, 0,168 mmol). A mistura resultante foi aquecida a 120°C durante a noite. A solução de reação foi diluída com metanol e purificada com LCMS preparativa (coluna Sunfire C18 de 19x100 mm eluindo com um gradiente ACN/H2O contendo 0,1% de TFA em 30 mL/minuto) para fornecer o composto desejado (3,2 mg, 16%). LCMS (M+H)+: 579,2. Etapa 2. 4-cloro-2-((3S)-3-{( 1 S)-2-ciano-1-[4-(7H-pirrolo[2,3-d]pinmidin-4-il)- 1 H-pirazol-1 -il]etil}pirrolidin-1 -il) tiofeno-3-carbonitrila
Este composto foi preparado de acordo com o procedimento de exemplo 77, usando 4-cloro-2-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsi- lil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1- il)tiofeno-3-carbonitrila como o material de partida. LCMS (M+H)+: 449,1. Exemplo 79. 2-((3S)-3-í( 1 S)-2-ciano-1 -r4-(7H-pirroloí2,3-dlpirimidin-4-il)-1 H- pirazol-1-il1etil}pírrolidin-1-il)tiofeno-3,4-dicarbonitrila
Etapa 1. 2-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -H)tiofeno-3,4- dicarbonitrila
A uma solução de 4-bromo-2-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2- (trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirímidin-4-il)-1 H-pirazol-1-iljetiljpirro- lidin-1 -il)tiofeno-3-carbonitrila (de exemplo 76, etapa 8; 30,0 mg, 0,0481 mmol) em NMP (0,4 mL) foi adicionado cianeto de zinco (28,2 mg, 0,240 mmol). Tetracis(trifenilfosfina)paládío(0) (13,9 mg, 0,012 mmol) e a solução foi estimulada com nitrogênio. A solução foi aquecida a 150°C durante 15 minutos em um reator de micro-ondas. A solução de reação foi diluída com metanol e purificada por LCMS preparativa (coluna Sunfire C18 de 19x100 mm eluindo com um gradiente ACN/H2O contendo 0,1% de TFA em 30 mL/minuto) para fornecer o composto desejado (8,3 mg, 30%). LCMS (M+H)+: 570,2. Etapa 2. 2-((3S)-3-{(1 S)-2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-ÍI)-1 H- pirazol-1 -il]etil}pirrolidin-1 -il) tiofeno-3,4-dicarb onitrila
Este composto foi preparado de acordo com o procedimento de exemplo 77, usando 2-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)tiofeno-3,4- dicarbonitrila como o material de partida. 1H RMN (300 MHz, DMSO-d6): δ 12,06 (s, 1H); 8,81 (s, 1H), 8,62 (s, 1H); 8,36 (s, 1H); 7,59 (s, 1H); 7,54 (d, 1H); 6,92 (d, 1H); 4,80 (m, 1H); 3,69-3,16 (m, 6H); 2,96 (m, 1H); 1,72 (m, 2H); LCMS (M+H)+: 440,1. Exemplo 80. 2-((3S)-3-{2-fluoro-1-r4-(7H-pirrolo[2,3-dlpirimidin-4-il)-1 H-pira- zol-1-il]etil}pirrolidin-1-il)tiofeno-3,4-dicarbonitrila
Etapa 1. 4-bromo-2-((3S)-3-{2-fluoro-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il) tiofeno-3- carbonitrila
Este composto foi preparado de acordo com o procedimento de exemplo 76, etapa 8, usando 4-(1 -{2-fluoro-1 -[(3S)-pirrolidin-3-il]etil}-1 H- pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidina (de e- xemplo 70, etapa 7) e 2,4-dibromotiofeno-3-carbonitrila como o material de partida. LCMS (M+H)+: 616,2, 618,2. Etapa 2. 2-((3S)-3-{2-fluoro-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirímidin-4-il) -1 H-pirazol-1 -il]e til}piirolidin-1 -il) tiofeno-3,4-dicarbonitrila
Este composto foi preparado de acordo com o procedimento de exemplo 79, etapa 1, usando 4-bromo-2-((3S)-3-{2-fluoro-1-[4-(7-{[2-(trime- tilsilil)etóxi]metÍI}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1- il)tiofeno-3-carbonitrila como o material de partida. LCMS (M+H)+: 563,2. Etapa 3. 2-((3S)-3-{2-fíuoro-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)tiofeno-3,4-dicarbonitrila
Este composto foi preparado de acordo com o procedimento de exemplo 77, usando 2-((3S)-3-(2-fluoro-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1 -il)tiofeno-3,4- dicarbonitrila como o material de partida. 1H RMN (300 MHz, DMSO-d6): δ 8,73 (s, 1H); 8,61 (s, 1H); 8,33 (s, 1H); 7,61 (s, 1H); 7,53 (d, 1H); 6,91 (d, 1H); 4,97-4,69 (m, 3H); 3,73 (m, 1H); 3,55 (m, 1H), 3,45 (m, 2H), 2,94 (m, 1H); 1,72 (m, 2H) LCMS (M+H)+: 433,1. Exemplo 81. 2-(3-f2-ciano-1-[3-(7H-pirrolo[2,3-d1pinmidin-4-il)-1 H-pirrol-1- il]etil}pirrolidin-1-il)tiofeno-3,4-dicarbonitrila (dois enantiômeros isolados)
Etapa 1. 4-bromo-2-(3-{2-ciano-1-[3-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirrol-1-il]etil}pirrolidin-1 -il)tiofeno-3-carbonitrila
Este composto foi preparado de acordo com o procedimento de exemplo 76, etapa 8, usando 3-pirrolidin-3-il-3-[3-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1-il]propanonitrila (de exemplo 33, etapa 3) e 2,4-dibromotiofeno-3-carbonÍtrila como os materiais de partida. LCMS calculada para C28H33BrN7OSSi(M+H)+: m/z = 622,1, 624,1. Etapa 2. 2-(3-{2-ciano-1 -[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]piri- midin-4-il) -1 H-pirrol-1 -il]etil}pirrolidin-1 -il) tiofeno-3,4-dicarbonitrila
Este composto foi preparado de acordo com o procedimento de exemplo 79, etapa 1, usando 4-bromo-2-(3-{2-ciano-1-[3-(7-{[2-(trimetil- s i I i l)etóxi] meti l}-7H-pi rrolo[2,3-d]p i ri m id i n-4-il)-1 H-pirrol-1 -il]etil}pirrolidin-1 - il)tiofeno-3-carbonitrila como o material de partida. Este material foi separado por HPLC quiral (Chiral Technologies Chiralpak AD-H, 5μ, 20 x 250 mm, eluindo com 80% de EtOH/Hexanos, 8 mL/minuto) para fornecer enantiômero 1 (primeiro a eluir) e enantiômero 2 (segundo a eluir). LCMS (M+H)+: 569,2. Etapa 3. 2-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 - il]etil}pirrolidin-1-il)tiofeno-3,4-dicarbonitrila (dois enantiômeros isolados) Cada enantiômero da última etapa foi desprotegido separadamente por agitação sequencialmente em uma mistura de 1:1 de TFA/DCM durante 1 hora, remoção de solvente, em seguida agitação em metanol (1,5 mL) contendo EDA (0,2 mL) durante 30 minutos. HPLC/MS preparativa (coluna C18 (19x100 mm) eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usada para purificar os produtos.
Enantiômero 1 LCMS (M+H)+: 439,0; Enantiômero 2 LCMS (M+H)+: 439,1. Exemplo 82. 4-((3S)-3-{(1 S)-2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H- pirazol-l-illetillpirrolidin-l-iD-l^-tiazol-δ-carbonitrila
Etapa 1. 2,4-dicloro-1,3-tiazol-5-carbaldeído
A uma suspensão de 2,4-tiazolidinadiona (10,0 g, 85,4 mmols) em cloreto de fosforila (48,0 mL, 515 mmols) a 0°C foi adicionado DMF (7,3 mL, 94 mmols) gota a gota. A mistura reacional foi deixada aquecer para temperatura ambiente e agitada durante 1 hora. A mistura foi em seguida aquecida a 85°C durante 1 hora antes da agitação a 115°C durante 3,5 horas. Após resfriamento para temperatura ambiente, a mistura foi cuidadosa- mente vertida em gelo com agitação lenta. A camada aquosa foi extraída com DCM três vezes. Os extratos orgânicos combinados foram lavados com NaHCO3 saturado, água, secados sobre MgSO4, filtrados, e concentrados. O resíduo foi purificado por coluna de sílica-gel (0% a 20% de acetato de 5 etila/hexanos) para fornecer o composto desejado como sólido esbranquiçado (8,1 g, 52%). 1H RMN (300 MHz, DMSO-d6): δ 9,92 (s, 1H); LCMS (M+H- CO)+: 153,9 Etapa 2. 2,4-dicloro-5-(1,3-dioxolan-2-il)-1,3-tiazol
A uma mistura de 2,4-dicloro-1,3-tiazol-5-carbaldeído (4,0 g, 22 10 mmols) e 1,2-etanodiol (3,6 mL, 64 mmols) em tolueno anidroso (50 mL) foi adicionado monoidrato de ácido p-toluenossulfônico (0,31 g, 1,6 mmol). O frasco foi ajustado com uma armadilha Dean-Stark e a mistura aquecida até o refluxo durante 3,5 horas. Após resfriamento para temperatura ambiente, a reação foi extinguida com solução de Na2CO3 a 10%. A camada aquosa foi 15 extraída com acetato de etila três vezes. Os extratos combinados foram lavados com salmoura, secados sobre MgSO4, filtrados, e concentrados. O resíduo foi purificado por coluna de sílica-gel (0% a 30% de acetato de etila/hexanos) para fornecer o composto desejado como óleo amarelo (1,47 g, 84%). 1H RMN (300 MHz, CDCI3): δ 6,03 (s, 1H); 1,40 (m, 4H); LCMS (M+H)+: 225,9. Etapa 3. 4-cloro-5-(1,3-dioxolan-2-il)-1,3-tiazol
A uma solução de 2,4-dicloro-5-(1,3-dioxolan-2-il)-1,3-tiazol (0,1 g, 4,4 mmols) em THF(20 mL) a -78°C foi adicionado n-butil-lítio a 2,5 M em hexano (2,28 mL, 5,69 mmols) gota a gota. A solução escura resultante foi agitada a -78°C durante 75 minutos. A reação foi extinguida com água e em seguida vertida em salmoura. A camada aquosa foi extraída com acetato de etila três vezes. Os extratos combinados foram secados sobre MgSO4, filtrados, e concentrados. O resíduo foi purificado por coluna de sílica-gel (0% a 30% de acetato de etila/hexano) para fornecer o composto desejado como um óleo amarelo (770 mg, 91%). 1H RMN (300 MHz, CDCI3): δ 8,74 (s, 1 H), 6,18 (s, 1H); 1,40 (m, 4H); LCMS (M+H)+: 191,9. Etapa 4. 4-cloro-1,3-tiazol-5-carbaldeído
A uma solução de 4-cloro-5-(1,3-dioxolan-2-il)-1,3-tiazol (0,75 g, 3,9 mmols) em THF (10 mL) foi adicionada solução de HCI a 5,0 M em água (2 mL, 10 mmols). A mistura resultante foi agitada em temperatura ambiente durante 2 horas. A solução de reação foi vertida em salmoura e extraída com acetato de etila três vezes. Os extratos combinados foram lavados com bicarbonato de sódio saturado, salmoura, secados sobre MgSO4, filtrados, e concentrados para fornecer o composto desejado como um sólido esbranquiçado (0,53 g, 92%). 1H RMN (300 MHz, CDCI3): δ 10,16 (s, 1 H), 9,03 (s, 1H). Etapa 5. 4-cloro-1,3-tiazol-5-carbaldeído oxima
A uma solução agitada de bicarbonato de sódio (0,17 g, 2,0 mmols) em água (6,4 mL) foi adicionado cloridrato de hidroxilamina (0,14 g, 2,0 mmols) em porções. A uma mistura foi adicionada uma solução de 4- cloro-1,3-tiazol-5-carbaldeido (0,30 g, 2,0 mmols) em etanol (2,0 mL). A mistura foi agitada durante 1 hora em temperatura ambiente. A solução de reação foi diluída com água. O precipitado resultante foi coletado e secado sob vácuo para fornecer o composto desejado como um sólido branco (0,25 g, 76%). 1H RMN (300 MHz, CD3OD): δ 9,02 (s, 1 H), 7,83 (s, 1H); LCMS (M+H)+: 162,9. Etapa 6. 4-cloro-1,3-tiazol-5-carbonitrila
A mistura de oxima de 4-cloro-1,3-tiazol-5-carbaldeído (0,24 g, 1,5 mmol) e anidrido acético (1,2 mL, 13 mmol) foi aquecida a 140°C durante 3 horas. A mistura foi concentrada em vácuo para fornecer um sólido marrom (65 mg, 30%). O bruto material foi usado na etapa seguinte sem outra purificação. Etapa 7. 4-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il) -1 H-pirazol-1 - il]etil}pirrolidin-1-il)-1,3-tiazol-5-carbonitrila (enantiômero simples)
Este composto foi preparado de acordo com o procedimento de exemplo 74, etapa 2, usando (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetil- silil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (de exemplo 15, etapa 3) e 4-cloro-1,3-tiazol-5-carbonitrila como os materiais de partida. LCMS (M+H)+: 416,1. Exemplo 83. 5-(3-{2-fluoro-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1-illetil}pirrolídin-1-il)-1,3-tiazol-4-carbonitrila (enantiômero simples)
Etapa 1. 3-[(E)-2-fluoro-2-(fenilsulfonil)vinil]pirrolidina-1-carboxÍlato de terc- butila
A uma mistura de 3-[2-fluoro-1-hidróxi-2-(fenilsulfonil)etil] pirroli- dina-1-carboxilato de terc-butila (sintetizado de acordo com o procedimento de exemplo 70 etapa 3, usando ferc-butil-3-formilpirrolidina-1-carboxilato como o material de partida, 0,53 g, 1,4 mmol) e trietilamina (0,80 mL, 5,7 10 mmols) em DCM (7,8 mL) a 0°C foi adicionado cloreto de metanossulfonila (132 pL, 1,70 mmol). A mistura foi agitada a 0°C durante 1 hora em seguida aquecida para temperatura ambiente. Após 4 horas, outra porção de trietilamina (2,0 eq) foi adicionada e agitada durante a noite. A solução de reação foi diluída com salmoura e a camada aquosa foi extraída com DCM três ve- 15 zes. Os extratos combinados foram secados sobre MgSO4, filtrados, e concentrados. O resíduo foi purificado por coluna de sílica-gel (0% a 40% de acetato de etila/hexanos) para fornecer o composto desejado (350 mg, 69%). LCMS (M+Na)+: 378,1. Etapa 2 3-{2-fluoro-2-(fenilsulfonil)-1-[3-(7-{[2-(tnmetilsilil)etóxi]metil}-7H- 20 pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1-il]etil}pirrolidina-1-carboxilato de terc-butila
A uma mistura de 4-(1 H-pirrol-3-il)-7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidina (de exemplo 33, etapa 1; 0,33 g, 0,1 mmol) e 3- [(E)-2-fluoro-2-(fenilsulfonil)vinil]pirrolidina-1 -carboxilato de terc-butila (0,35 25 g, 0,98 mmol) em acetonitrila (6,0 mL) foi adicionado 1,8-diazabici- clo[5,4,0]undec-7-eno (180 pL, 1,2 mmol) e a solução de reação foi agitada a 65°C durante 36 horas. O solvente foi removido em vácuo e o resíduo foi purificado por coluna de sílica-gel (0% a 50% de acetato de etila/hexanos) para fornecer o composto desejado como 1: 1 de dois diastereômeros (134 mg, 20%). LCMS (M+H)+: 670,3. Etapa 3. 3-{2-fluoro-1-[3-(7-{[2-(trimetilsilil)etóxi]metÍI}-7H-pirrolo[2,3-d]piri- midin-4-il)-1 H-pirrol-1 -il]etil}pirrolidina-1 -carboxilato de terc-butila
Este composto foi preparado de acordo com o procedimento de exemplo 70, etapa 4, usando 3-{2-fluoro-2-(fenilsulfonil)-1 -[3-(7-{[2-(trimetil- silil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1-il]etil}pirrolidina-1- carboxilato de terc-butila como o material de partida. Dois diastereômeros foram isolados usando coluna de sílica-gel eluindo com 5 a 60% de acetato de etila/hexanos. LCMS (M+H)+: 530,1. Etapa 4. 4-[1-(2-fluoro-1-pirrolidin-3-iletil)-1 H-pirrol-3-il]-7-{[2-(trimetilsilil)etó- xi]metil}- 7H-pirrolo[2,3-d]pirimidina
A uma solução de 3-{2-fluoro-1-[3-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-rl)-1 H-pirrol-1 -il]etil}pirrolidina-1 -carboxilato de terc-butila (104 mg, 0,196 mmol;) em DCM (0,5 mL) foi adicionado cloreto de hidrogênio a 4,0 M em 1,4-dioxano(0,5 mL, 2,0 mmols). A solução de reação foi agitada em temperatura ambiente durante 90 minutos. O solvente foi removido em vácuo e o resíduo foi dissolvido em acetato de etila. A camada orgânica foi lavada com solução de NaOH a 0,1 N. O aquoso foi extraído com acetato de etila. Os extratos combinados foram secados sobre MgSO4, filtrados, e concentrados para fornecer o composto desejado como uma goma pegajosa marrom (86 mg, 100%). LCMS (M+H)+: 430,1. Etapa 5. 5-(3-{2-fluoro-1-[3-(7-{[2-(trimetilsilil)etóxi]metÍI}-7H-pirrolo[2,3-d]pi- rimidin-4-il) -1 H-pirrol-1 -il]etil}pirrolidin-1 -il) -1,3-tiazol-4-carbonitrila
A uma mistura de 4-[1 -(2-fluoro-1 -pirrolidin-3-iletil)-1 H-pirrol-3-il]- 7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidina (diastereômero 2 de etapa 3) (54 mg, 0,12 mmol) e 5-bromo-1,3-tiazol-4-carbonitrila (29,5 mg, 0,156 mmol) foi adicionado tetrafluoroborato de 1-butil-3-metil-1H-imidazol-3- io (0,2 mL) e DIPEA (32,4 μL, 0,186 mmol). A mistura resultante foi agitada a 120°C durante 3 horas em seguida resfriada para temperatura ambiente. A solução de reação foi diluída com acetato de etila e lavada com água. A ca- mada orgânica foi secada sobre MgSO4, filtrada, e concentrada. O resíduo foi diluído com metanol e purificado por LCMS preparativa (coluna C18 (19*100 mm) eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) para fornecer o composto desejado (20 mg, 28%). Os enantiômeros foram separados por HPLC quiral (Chiral Technologies Chiralcel OD-H, 5μ, 20 x 250 mm, 20% de EtOH/Hexanos, 12 mL/minuto). Enantiômero desejado 1 (primeiro a eluir) foi coletado (12,7 mg, 18%). LCMS (M+H)+: 538,2. Another enantiômero 2 (segundo a eluir) foi coletado (6,2 mg, 9%). LCMS (M+H)+: 538,2 Etapa 6. 5-(3-{2-fluoro-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirrol-1 -iljetil}- pirrolidin-1-il)-1,3-tiazol-4-carbonitrila (enantiômero simples)
O enantiômero desejado 1 (de etapa 5) foi tratado com 1:1 de TFA/DCM durante 1 hora, concentrado novamente, e agitado em uma solução de metanol (1 mL) contendo 0,2 mL de EDA durante 30 minutos. O produto foi purificado por meio de LCMS preparativa (coluna C18 (19*100 mm) eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) para fornecer o composto desejado. LCMS (M+H)+: 408,1. Exemplo 84. Trifluoroacetato de 4-((S)-3-((S)-1-(4-(7H-pirrolof2,3-d]pirimidin- 4-íl)-1H-pirazol-1-il)-2-cianoetil)pirrolídin-1-il)pirimidina-5-carbonítrila

Etapa 1: (3S)-3-[(3S)-1 -(5-iodopirimidin-4-il)pirrolidin-3-il]-3-[4-(7-{[2-(trime- tilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (60,0 mg, 0,1371 mmol; de exemplo 15, etapa 3) foi misturada com 4-cloro-5-iodopirimidina (WO 2008/079965; 48,35 mg, 0,2011 mmol) e DIPEA (36,0 μL, 0,2067 mmol) e foi dissolvida em NMP (0,40 mL). A reação foi aquecida a 130°C durante 2 horas, em cujo tempo a análise de LCMS mostrou principalmente o produto. O resíduo foi purificado em LC preparativa para fornecer o produto. Este foi dividido entre EtOAc e NaHCO3 saturado e o extrato de EtOAc foi lavado com salmoura, secado (mgSO4), e removido em vácuo e foi transportado na reação seguinte. MS (El): 642 (M+H) Etapa 2: 4-((S)-3-((S)-2-ciano-1 -(4-(7-((2-(tnmetilsihl)etóxi)metil)- 7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)etil)pirrolÍdin-1-il)pirimidina-5-carbonitrila
Em um frasco de fundo redondo de 1 gargalo 3-[1-(5-iodopiri- midin-4-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (32,0 mg, 0,0499 mmol) foi dissolvida em DMF (0,3 mL) e cianeto de zinco (17,6 mg, 0,150 mmol) foi adicionado. A reação foi desgaseificada, tetracis(trifenilfosfina)paládio(0) (11,5 mg, 0,00998 mmol) foi adicionado e aquecido a 100°C durante 4 horas, em cujo tempo a análise de LCMS mostrou que existia principalmente o produto. A reação foi filtrada e o produto foi purificado por LC preparativa. MS (El): 541 (M+H) Etapa 3: trifluoroacetato de 4-((S)-3-((S)-1-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)- 1 H-pirazol-1 -il)-2-cianoetÍI)pirrolidin-1-il)pirimidina-5-carbonitrila
O produto de etapa 2 foi desprotegido (CH2CI2/TFA; Me- OH/NH4OH) como no exemplo 1, e o produto foi purificado por LC (método de ACN/água/TFA como no exemplo 5). MS (El): 411 (M+H). Exemplo 85. bis(trifluoroacetato) de 4-(1-{2-fluoro-1-[(3S)-1-(5-fluoro-2,3-di- hidrotieno[2,3-blpiridin-6-il)pirrolidin-3-il]etil}-1H-pirazol-4-il)-7H-pirrolo[2.3-djpirimidina
Etapa 1:3-[(E)-2-etoxivinil]-2,5,6-trifíuoropiridina
Em um frasco de fundo redondo de 1 gargalo 3-cloro-2,5,6- trifluoropiridina (de Lancaster Synthesis Inc.; 0,1 g, 5,97 mmols) foi dissolvida em tolueno (6,7 mL) com (2-etoxietenil)tri-n-butiltina (de Synthonix Corpo- ration; 2,01 g, 5,57 mmols) e tetracis(trifenilfosfina) paládio(O) (348,0 mg, 0,3012 mmol) e foi desgaseificada. A reação foi aquecida até o refluxo durante 4 horas após cujo tempo a análise de TLC mostrou que a maioria do material de partida foi consumida. A mistura reacional foi submetida à cromatografia usando 3% de EtOAc/hexanos para fornecer 3-[(E)-2-etoxivinil]- 2,5,6-trifluoropiridina contaminada com algum cloreto de butiltina. 1H RMN (400 MHz, CDCI3): δ 8,41 (m, 1H), 6,40 (dd, 1H0, 5,35 (dd, 1H), 1,40 (q, 2H), 1,30 (t, 3H). Etapa 2: 1-etóxi-2-(2,5,6-trifluoropiridin-3-il)etanol A 3-[(E)-2-etoxivinil]-2,5,6-trifluoropiridina de etapa 1 em THF (26,6 mL) e HCI a 5,0 M em água (17 mL, 83 mmols) foi adicionada e agitada a 25°C durante 20 horas, em cujo tempo a análise de TLC de uma preparação de amostra (EtOAc/NaHCO3) mostrou a ausência de material de partida. A reação foi neutralizada com NaHCO3 e foi dividida entre éter e água e o extrato de éter foi lavado com salmoura, secado (mgSO4), e removido em vácuo. A análise de RMN não mostrou nenhum pico de aldeído e foi consistente com o 1-etóxi-2-(2,5,6-trifluoropiridin-3-il)etanol de hemiacetal de etila. O produto foi purificado por cromatografia em sílica-gel usando 30% de éter/hexanos para fornecer o produto (0,75 g, 61% para as duas etapas). Análise de HPLC mostrou um pico. 1H RMN (400 MHz, CDCI3): δ 7,72 (m, 1H), 5,42 (m, 1H), 5,02 (m, 1H), 4,38 (m, 1H), 3,70-3,85 (m, 2H), 2,90 (m, 2H), 1,35 (m, 2H), 1,22 (m, 3H). Etapa 3: 2-(2,5,6-trifíuoropindin-3-il)etanol
Em um tubo selado de 10 mL 1-etóxi-2-(2,5,6-trifluoropiridin-3- il)etanol (250,0 mg, 1,130 mmol) foi dissolvido em THF (10,0 mL) e HCI a 0,1 M em água (5,0 mL, 5,0 mmols) foi adicionada a reação foi aquecida a 75°C durante 90 minutos e foi neutralizada com NaHCO3 e foi extraída com éter. A mistura reacional foi evaporada até a secura e a análise de RMN do bruto indicou que foi o 2-(2,5,6-trifluoropiridin-3-il)etano-1,1 -diol de hidrato de aldeído.
Em um tubo selado de 10 mL o bruto 2-(2,5,6-trifluoropiridin-3- il)etano-1,1 -diol (138,0 mg, 0,7146 mmol) foi dissolvido em isopropil álcool (6,0 mL) e tetra id robo rato de sódio (16,22 mg, 0,4287 mmol) foi adicionado. A reação foi agitada a 0°C durante 2 horas e foi extinguida com NH4CI e foi extraída com éter. O produto foi purificado por cromatografia de silica-gel para fornecer 2-(2,5,6-trifluoropiridin-3-il)etanol (80 mg). 1H RMN (400 MHz, CD3OD): δ7,90 (m, 1H), 3,75 (m, 2H), 2,80 (m, 2H). Etapa 4: etanotioato de S-[2-(2,5,6-tπfluoropiπdin-3-il)etila]
Em um frasco de fundo redondo de 1 gargalo 2-(2,5,6-trifluoro- piridin-3-il)etanol (0,520 g, 2,94 mmols) foi dissolvido em THF (13,0 mL) com trifenilfosfina (0,770 g, 2,94 mmols). A solução foi resfriada a 0°C, azodicar- boxilato de di-isopropila (0,578 mL, 2,94 mmols) foi adicionado, e 10 minutos depois, ácido tioacético (0,210 mL, 2,94 mmols) foi adicionado. A mistura foi agitada a 0°C durante 60 minutos. TLC, LC e LCMS mostraram -50% de conversão no produto. A reação foi extinguida com NaHCO3 saturado e foi dividida entre EtOAc e água e extrato de EtOAc foi lavado com salmoura, secado (mgSO4), e removido em vácuo. A reação foi submetida à cromatografia em sílica-gel usando 2% de EtOAc/hexanos para fornecer o produto contaminada com pequena quantidade de impurezas (0,30g). 1H RMN (400 MHz, CDCI3): δ 7,60 (m, 1H), 3,10 (m, 2H), 2,85 (m, 2H), 2,30 (s, 3H). Etapa 5: 5,6-difíuoro-2,3-di-hÍdrotieno[2,3-b]piridina
Em um frasco de fundo redondo de 1 gargalo S-[2-(2,5,6-tri- fluoropiridin-3-il)etil] etanotioato (190,0 mg, 0,80773 mmol) foi dissolvido em THF (30,0 mL) e água (30,0 mL) e foi desgaseificado. Na reação foi adicionado hidróxido de sódio a 0,1 M em água (7,8 mL) e foi agitada a 25°C durante 1 hora em cujo tempo a análise de LCMS mostrou principalmente 2- (2,5,6-trifluoropiridin-3-il)etanotiol. A reação foi agitada durante 2 dias em cujo tempo a análise de LCMS mostrou dissulfeto, e algum produto. A mistura reacional foi dividida entre éter e água e extrato de éter foi lavado com salmoura, secado (mgSO4), e removido em vácuo. Em seguida ele foi submetido à cromatografia em sílica-gel usando 3% de EtOAc/hexanos para fornecer o produto (13 mg). 1H RMN (400 MHz, CDCI3): δ7,29 (m, 1H), 3,50 (m, 2H), 3,25 (m, 2H). Etapa 6: 4-(1-{2-fíuoro-1-[(3S)-1-(5-fiuoro-2,3-di-hidrotieno[2,3-b]piridin-6- il)pin-olidin-3-il]etil}-1H-pirazol-4-il)-7-{[2-(trímetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidina 4-(1-{2-Fluoro-1 -[(3S)-pirrolidin-3-il]etil}-1 H-pirazol-4-il)-7-{[2- (trimetílsilil)etóxi]metil}-7H-pirrolo{2J3-d]pirimidina (de exemplo 70, etapa 7; 31,58 mg, 0,073332 mmol) foi misturada com 5,6-difluoro-2,3-di- hidrotieno[2,3-b]piridina (12,7 mg, 0,0733 mmol) e DIPEA (21,88 μL, 0,1256 mmol) e foi dissolvida em NMP (0,24 mL). A reação foi aquecida a 130°C durante 5 h em cujo tempo a análise de LCMS mostrou algum produto presente. O produto foi purificado por LC (método de ACN/TFA/água como no exemplo 5) para fornecer the purificado material. MS (El): 584 (M+H). Etapa 7: bis(trifluoroacetato) de 4-(1-{2-fluoro-1-[(3S)-1-(5-fluoro-2.3-di-hidro- tieno[2,3-b]pindin-6-il)pirrolidin-3-il]etil}-1H-pirazol-4-il)-7H-pirrolo[2,3-d]pirimi- dina 4-(1-{2-Fluoro-1-[(3S)-1-(5-fluoro-2,3-di-hidrotieno[2,3-b]piridin-6- il)pirrolidin-3-il]etil}-1H-pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirro- lo[2,3-d]pirimidina foi desprotegida (TFA/CH2CI2; MeOH/NH4OH) como no exemplo 1, e o composto desprotegido foi purificado em LC preparativa (método ACN/TFA como no exemplo 5) para fornecer o produto. MS (El): 454 (M+H). 1H RMN(CD3OD): δ 8,92 (m, 1H), 8,86 (m, 1H), 8,55 (m, 1H), 7,85 (m, 1H), 7,30 (m, 1H), 7,10 (m, 1H), 5,05 (m, 1H), 4,80 (m, 2H), 3,85 (m, 1H), 3,62 (m, 1H), 3,50 (m, 2H), 3,35 (m, 2H), 3,10 (m, 2H), 2,95 (m, 1H), 1,80 (m, 2H). Exemplo 86. tetracis(trifluoroacetato) de 4-(1-f2-fluoro-1-f(3S)-1-(5-fluoro-1,1- dióxido-2,3-di-hidrotieno[2,3-blpiridin-6-il)pirrolidin-3-il]etil}-1H-pÍrazol-4-il)- 7H-pirrolo[2,3-d1pirimidina

Em um frasco de fundo redondo de 1 gargalo bis(trifluoroace- tato) de 4-(1-{2-fluoro-1-[(3S)-1-(5-fluoro-2,3-di-hidrotieno[2,3-b]piridin-6- il)pirrolidin-3-il]etil}-1 H-pirazol-4-il)-7H-pirrolo[2,3-d]pirimidina (de exemplo 85; 3,0 mg, 0,0044 mmol) foi dissolvida em metanol (0,1 mL) e água (0,30 mL) foi adicionada. Na reação foi adicionado Oxone® (5,4 mg, 0,0088 mmol) e foi agitado a 25°C durante a noite em cujo tempo a análise de LCMS mostrou principalmente sulfona e sulfona super-oxidada. A reação foi filtrada e o produto foi purificado por LC preparativa (método ACN/TFA como no exemplo 5) para fornecer tetracis(trifluoroacetato) de 4-(1-{2-fluoro-1-[(3S)-1-(5- fluoro-1,1 -d ióxid o-2,3-di-h id rotieno[2,3-b]piridin-6-il)pirrolidin-3-il]etil}-1H- pirazol-4-il)-7H-pirrolo[2,3-d]pirimidina. MS(EI) 486 (M+1). 1H RMN(DMSO- d6): δ12,2 (brs, 1H), δ 8,82 (s, 1H), 8,70 (s, 1H), 8,40 (s, 1H), 7,60 (m, 2H), 7,80 (m, 1H), 5,00 (m, 1H), 4,90 (m, 2H), 4,80 (m, 2H), 3,90 (m, 1H), 3,70 (m, 1H), 3,50 (m, 2H), 3,10 (m, 2H), 2,9 (m, 1H), 1,65 (m, 2H). Exemplo 87, bis(trifluoroacetato) de (3S)-3-r(3S)-1-(5-fluoro-2,3-di-hidrotie- no[2,3-b1piridin-6-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-ilM H-pirazol-1 –illpropanonitrila

Etapa 1: metanossulfonato de but-3-in-1-ila
Em um frasco de fundo redondo de 1 gargalo 3-butin-1-ol (0,50 mL, 6,6 mmols) foi dissolvido em DCM (7,8 mL) e DIPEA (1,6 mL, 9,2 mmols) foi adicionado e foi resfriado a 0°C. Na reação foi adicionado cloreto de metanossulfonila (0,61 mL, 7,9 mmols) e foi agitada a 0°C durante 1 hora em cujo tempo a análise de TLC mostrou ausência de material de partida. A mistura reacional foi dividida entre EtOAc e água e extrato de EtOAc foi lavado com água, HCI a 1 N, NaHCO3, salmoura, secado (mgSO4), e removido em vácuo. O metanossulfonato de but-3-in-1 -ila resultante foi usado na reação seguinte sem purificação. 1H RMN (300 MHz, CDCI3): δ 4,52 (m, 2H), 3,10 (s, 3H), 2,65 (m, 2H), 2,05 (m, 1H). Etapa 2: etanotioato de S-but-3-in-1-ila
Em um frasco de fundo redondo de 1 gargalo carbonato de césio (0,57 g, 1,8 mmol) foi dissolvido em metanol (5,0 mL, 123 mmols) e ácido tioacético (0,241 mL, 3,37 mmols) foi adicionada. A reação foi agitada durante 30 minutos, e but-3-in-1 -il metanosulfonate (0,50 g, 3,4 mmols) em meta- nol (4,0 mL) foi adicionada e foi agitada a 25°C durante a noite em cujo tempo a análise de TLC mostrou material de partida e produto. A reação foi evaporada até a secura e DMF (5,0 mL) foi adicionada e foi agitada a 25°C durante a noite em cujo tempo a análise de TLC não mostrou nenhum material de partida. A mistura reacional foi dividida entre EtOAc e água e extrato de EtOAc foi lavado com água, salmoura, secado (mgSO4), e removido em vácuo. Usado na reação seguinte sem purificação. 1H RMN (300 MHz, CDCI3): δ 3,01 (m, 2H), 2,45 (s, 3H), 2,35 (m, 2H), 2,02 (m, 1H). Etapa 2a: 2-cloro-5-fluoro-4-[(4-metoxibenzil)óxi]pirimidina
Em um frasco de fundo redondo de 1 gargalo 2,4-dicloro-5- fluoropirimidina (de Frontier Scientific, Inc.; 0,80 g, 4,8 mmols) foi misturada com hidreto de sódio (60% de óleo mineral, 0,23 g, 5,7 mmols) e foi secada. A reação foi resfriada a 0°C e THF (9,0 mL) foi adicionado seguido por 4- metoxibenzenometanol (0,60 mL, 4,8 mmols). A reação foi agitada a 25°C durante a noite. Em seguida ela foi dividida entre EtOAc e água e extrato de EtOAc foi lavado com salmoura, secado (mgSO4), e removido em vácuo. O resíduo foi submetido à cromatografia em sílica-gel usando 5% de EtOAc/hexanos para fornecer o produto (1,2 g). 1H RMN (400 MHz, CDCI3): Õ 8,20 (s, 1H), 7,42 (d, 2H), 6,91 (d, 2H), 5,42 (s, 2H), 3,80 (s, 3H) Etapa 3: 2-(but-3-in-1-iltio)-5-fíuoropirimidin-4-ol
Em um frasco de fundo redondo de 1 gargalo etanotioato de S-but-3-in-1 -ila (0,69 g, 5,4 mmols) foi dissolvido em DMF (4,0 mL) com 2- cloro-5-fluoro-4-[(4-metoxibenzil)óxi]pirimidina (1,4 g, 5,4 mmols) e hidróxido de litio (0,259 g, 10,8 mmols) e água (0,5 mL) foi adicionada e agitada a 60°C durante a noite em cujo tempo análise de HPLC e análise de LCMS mostraram o produto desbenzilado e o material de partida. A reação foi continuada durante 24 horas sem muita mudança. Em seguida ela foi dividida entre EtOAc e água e extrato de EtOAc foi lavado com salmoura, secado (mgSO4), e removido em vácuo. Um extrato orgânico não conteria qualquer produto. A camada de água foi evaporada até a secura e foi lavada com metanol e foi filtrada. A lavagem de metanol foi evaporada e foi submetida à cromatografia usando 1:1 de EtOAc/hexanos e EtOAc como eluente para 5 fornecer o produto 2-(but-3-in-1 -iltÍo)-5-fluoropirimidín-4-ol (0,2 g). 1H RMN (300 MHz, DMSO D6): δ 7,90 (m, 1H), 3,30 (m, 2H), 2,60 (m, 2H), 2,35 (m, 1H). Etapa 4: 5-fluoro-2,3-di-hidrotieno[2,3-b]pirídin-6-ol
Em um tubo selado de 10 mL 2-(but-3-in-1-iltio)-5-fluoropirimidin- 4-ol (185 mg, 0,931 mmol) foi dissolvido em NMP (0,1 mL) e foi aquecido a 200°C durante 3 horas, em cujo tempo a análise de LCMS mostrou principalmente o produto. A mistura reacional foi purificada por LC preparativa para fornecer 5-fluoro-2,3-di-hidrotieno[2,3-b]piridin-6-ol (81 mg). LCMS: 172(M+1). 1H RMN (300 MHz, DMSO D6): δ 7,36 (d, 1H), 3,55 (m, 2H), 3,16 (m, 2H). Etapa 5: 5-fluoro-2,3-di-hidrotieno[2,3-b]píndin-6-il trifluorometanosulfonate 5-fluoro-2,3-di-hidrotieno[2,3-b]piridin-6-ol (40,0 mg, 0,234 mmol) foi dissolvido em DCM (1,72 mL) e trietilamina (48,85 pL, 0,3505 mmol) foi adicionada, a solução foi resfriada para 0°C e N-fenilbis(trifluorometano- sulfonimida) (0,1043 g, 0,2921 mmol) foi adicionada. A reação foi agitada a 25°C durante 48 horas, em cujo tempo a análise de LCMS mostrou ausência de material de partida. A reação foi submetida à cromatografia em sílica-gel usando 20% de EtOAc/hexanos para fornecer o produto trifluorometanosul- fonato de 5-fluoro-2,3-di-hidrotieno[2,3-b]piridin-6-ila contaminada com uma pequena quantidade de reagente. 1H RMN (300 MHz, CDCI3): <57,55 (m, 1H), 3,50 (m, 2H), 3,30 (m, 2H). Etapa 6: (3S)-3-[(3S)~ 1-(5-fluoro-2,3-di-hidrotieno[2,3~b]pindin-6-il)pirrolidin- 3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metii}-7H-pirrolo[2,3-d]pÍrímidin-4-il)-1H- pirazol-1-iljpropanonitrila
Em um tubo selado de 10 mL (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7- {[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin~4-il)-1 H-pirazol-1 - iljpropanonitrila (0,08246 g, 0,1884 mmol; de exemplo 15, etapa 3) foi mistu- rada com 5-fluoro-2,3-di-hidrotieno[2,3-b]piridin-6-il trifluorometanosulfonate (0,067 g, 0,22 mmol) em NMP (0,24 mL) com DIPEA (21,88 μL, 0,1256 mmol) e foi aquecida a 130°C durante 2 horas, em cujo tempo a análise de LCMS mostrou principalmente o produto. O produto foi purificado por LC (método de ACN/TFA/água como no exemplo 5) para fornecer (3S)-3-[(3S)- 1-(5-fluoro-2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4-(7-{[2- (trimetilsílil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-ÍI)-1 H-pirazol-1 - iljpropanonitrila. MS(EI):591(M+1) Etapa 7: bis(trifluoroacetato) de (3S)-3-[(3S)-1-(5-fluoro-2,3-di-hidrotieno[2,3- 10 b]pihdin-6-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila (3S)-3-[(3S)-1-(5-fluoro-2,3-di-hidrotieno[2,3-b]piridin-6- il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4- il)-1 H-pirazol-1-iljpropanonitrila foi desprotegida (TFA/CH2CI2; Me- OH/NH4OH) como no exemplo 1, e o composto desprotegido foi purificado em LC preparativa (método de ACN/TFA/água como no exemplo 5) para fornecer o produto bís(trifluoroacetato) de (3S)-3-[(3S)-1-(5-fluoro-2,3-di- hidrotieno[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin^4-il)- 1 H-pirazol-1-iljpropanonitrila. Espectro de massa (El):461 (M+1). 1HRMN(400 MHz CD3OD): δ 9,00 (s, 1H), 8,90 (m, 1H), 8,55 (m, 1H), 7,85 (d, 1H), 7,26 (d, 1H), 7,88 (d, 1H), 4,85 (m, 1H), 3,85 (m, 1H), 3,40-3,60 (m, 2H), 3,20-3,40 (m, 4H), 3,10 (m, 2H), 2,95 (m, 1H), 1,80 (m, 2H). Exemplo 88. bis(trifluoroacetato) de (3S)-3-[(3S)-1-(6-bromo-3-fluoropiridin-2- il)pirrolidin-3-in-3-[4-(7H-pirrolo[2,3-dlpirimidin-4-il)-1H-pirazol-1-il]propano- 25 nítríla e Exemplo 89. Trifluoroacetato de (3S)-3-[(3S)-1-(5,6-difluoropiridin-2-il)pirro- lidin-3-il1-3-[4-(7H-pirrolo[2,3-d1pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila

Etapa 1:2:3-difluoro-6-hidrazinopiridina
Em um frasco de fundo redondo de 1 gargalo, 2,3,6-trifluoro- piridina (de Alfa Aesar; 0,40 mL, 4,5 mmols) foi dissolvida em THF (5,0 mL) e hidrato de hidrazina (0,44 mL, 9,012 mmols) foi adicionado e foi agitado a 25°C durante a noite e foi aquecido até o refluxo durante 2 horas. A mistura reacional foi evaporada até a secura para fornecer 2,3-difluoro-6-hidrazino- piridina que foi usada na reação seguinte sem purificação. Etapa 2: 6-bromo-2,3-difluoropiridina
Em um frasco de fundo redondo de 1 gargalo 2,3-difluoro-6- hidrazinopiridina (0,65 g, 4,5 mmols) foi suspensa em clorofórmio (5,0 mL) e bromina (0,46 mL, 9,0 mmols) foi adicionada gota a gota. A reação foi aquecida até o refluxo durante 3 horas com uma armadilha ácida e foi extinguida com NaHSO3 e neutralizada com NaHCO3. Em seguida ela foi dividida entre éter e água e o extrato de éter foi lavado com salmoura, secado (mgSO4), e removido em vácuo. A análise de RMN da mistura crua indicou que ela con- sistinha em uma mistura de 2:1 de 6-bromo-2,3-difluoropiridina e 2,3- dibromo-5,6-difluoropiridina. A mistura reacional foi submetida à cromatografia em sílica-gel usando 5% de éter/hexanos para fornecer o produto. 1H RMN (300 MHz, CDCI3): δ 7,55 (m, 1H), 6,90 (m, 1H). Etapa 3: trifluoroacetato de (3S)-3-[(3S)-1-(6-bromo-3-fluoropiridÍn-2-il)pir- rolidin-3-ilJ-3-[4-(7H-pirrolo[2,3-dJpirimidin-4-il) -1 H-pirazol-1 -iljpropanonitrila e Trifluoroacetato de (3S)-3-[(3S)-1-(5,6-difluoropiridin-2-il)pirrolidin-3-il]-3-[4- (7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsílil)etóxiJmetil}-7H- pÍrrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (100,0 mg, 0,22851 mmol; de exemplo 15, etapa 3) foi misturada com 6-bromo-2,3-difluoropi- ridina (53,2 mg, 0,27421 mmol) e DIPEA (50,0 μl_, 0,2870 mmol) e foi dissolvida em NMP (0,62 mL). A reação foi aquecida a 130°C durante 2 horas, em cujo tempo a análise de LCMS mostrou principalmente os dois produtos (3S)-3-[(3S)-1-(6-bromo-3-fluoropiridin-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(trime- tilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila.
A mistura reacional foi purificada por LC preparativa (método de ACN/TFA/água como no exemplo 5) para fornecer os dois compostos: (3S)- 3-[(3S)-1-(6-bromo-3-fluoropiridin-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsi- lil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila e (3S)-3-[(3S)-1-(5,6-difluoropiridin-2-il)pirrolidÍn-3-il]-3-[4-(7-{[2-(trimetilsilil)etó-. xi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila nue fo ram desprotegidosdesprotegido (TFA/CH2CI2; MeOH/NH4OH) como no e-xemplo 1, e o composto desprotegidos foram purificado em LC preparativa (método ACN/TFA como no exemplo 5) para fornecer (3S)-3-[(3S)-1 -(5,6 d ifluoropirid in-2-il)pirrolid in-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimid in-4-il)-1 H-pira-' zol-1 -iljpropanonitrila como o sal de TFA {m/z: 421 (M+1). 1H RMN(400 MHz CD3OD): δ 8,95 (s, 1H), 8,85 (s, 1H), 8,55 (s, 1H), 7,80 (d, 1H), 7,35 (m, 1H), 7,20 (d, 1H), 6,04 (m, 1H), 4,85 (m, 1H), 3,93 (m, 1H), 3,70 (m, 1H), 3,51 (m, ■ 2H), 3,40 (m, 1H), 2,95 (m, 1H), 1,80 (m, 2H)} e (3S)-3-[(3S)-1-(6-bromo-3- fluoropiridin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol- 1-iljpropanonitrila como o sal de bisTFA {m/z: 482, 484 (M+1). 1H RMN(400 MHz CD3OD): δ 9,00 (s, 1H), 8,90 (s, 1H), 8,55 (s, 1H), 7,85 (d, 1H), 7,35 (m, 1H), 7,25 (d, 1H), 6,39 (m, 1H), 4,85 (m, 1H), 3,80 (m, 1H), 3,20-3,50 (m, 5H), 3,04 (m, 1H), 1,85 (m, 2H)}. Exemplo 90. Trifluoroacetato de (3S)-3-{(3S)-1-[6-cloro-3-fluoro-5-(hidroxime- til)piridin-2-ilJpirrolidin-3-il)-3-r4-(7H-pirrolo[2.3-dlpirimidin-4-il)-1 H-pirazol-1- iljpropanonitrila

Etapa 1: (2,6-dicloro-5-fluoropiridin-3-il)metanol
Uma solução de ácido 2,6-dicloro-5-fluoronicotínico (de Aldrich; 0,50 g, 2,4 mmols) em THF (10,0 mL) foi resfriada para 0°C e borano a 0,1 M em THF (2,8 mL) foi adicionado lentamente, a reação foi deixada aquecer a 25°C e foi agitada durante a noite. Análise de LCMS da mistura reacional mostrou material de partida presente e borano a 0,1 M em THF (1,50 mL) foi adicionado e foi agitado a 25°C durante a noite em cujo tempo a análise de LCMS mostrou principalmente o produto. A reação foi extinguida com água e HCI a 1 N e foi dividido entre EtOAc e água e extrato de EtOAc foi lavado com salmoura, secado (mgSO4), e removido em vácuo para fornecer (2,6- dicloro-5-fluoropiridin-3-il)metanol. Usado na reação seguinte sem purificação. m/z 197 (M+1) Etapa 2: (3S)-3-{(3S)-1-[6-cloro-3-fluoro-5-(hidroximetil)piridin-2-il]pirrolidin-3- il}-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirímidin-4-il)-1 H-pirazol- 1 -iljpropanonitrila (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (52,0 mg, 0,1188 mmol; de exemplo 15, etapa 3) foi misturada com (2,6-dicloro-5-fluoropiridín- 3-il)metanol (62,0 mg, 0,316 mmol) e DIPEA (25,0 pL, 0,1435 mmol) e foi dissolvida em NMP (0,31 mL). A reação foi aquecida a 130°C durante 2 horas, em cujo tempo a análise de LCMS mostrou algum produto. Este foi purificado por LC (método de ACN/TFA/água como no exemplo 5) give o produto (3S)-3-{(3S)-1-[6-cloro-3-fluoro-5-(hidroximetil)piridin-2-il]pirrolidin-3-il}-3- [4-(7-{[2-(trimetÍlsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- iljpropanonitrila. Etapa 3. trifluoroacetato de (3S)-3-{(3S)-1-[6-cloro-3-fluoro-5-(hidroxime- til)pindin-2-il]pirrolidin-3-il}-3-[4-(7H-pirrolo[2,3-d]pinmidin-4-il)-1 H-pirazol-1- iljpropanonitrila (3S)-3-{(3S)-1-[6-cloro-3-fluoro-5-(hidroximetil)piridin-2-il]pirroli- d in-3-il}-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimid in-4-il)-1H- pirazol-1 -iljpropanonitrila foi desprotegida (TFA/CH2CI2; MeOH/NH4OH) como no exemplo 1, e o composto desprotegido foi purificado em LC prepa-rativa (método ACN/TFA como no exemplo 5) para fornecer trifluoroacetato de (3S)-3-{(3S)-1-[6-cloro-3-fluoro-5-(hidroximetil)piridin-2-il]pirrolidin-3-il}-3- [4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila. MS(EI): 466(M+1). 1H RMN(300 MHz, CD3OD): δ 8,95 (s, 1H), 8,85 (s, 1H), 8,55 (s, 1H), 7,80 (d, 1H), 7,40 (m, 1H), 7,20 (d, 1H), 4,85 (m, 1H), 4,46 (s, 2H), 2,90- 4,00 (m, 7H), 1,80 (m, 2H) Exemplo 91. bis(trifluoroacetato) de (S)-3-(4-(7H-pirrolo[2,3-d1pirimidin-4-il)-1H-pirazol-1-il)-3-((S)-1-(5-amino-6-cloro-3-fluoropiridin-2-il)pirrolidin-3-5 il)propanonitrila

Etapa 1: trifluoroacetato de ácido 2-cloro-6-((3S)-3-{(1 S)-2-ciano-1-[4-(7-{[2- (trimetilsililjetóxijmetil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - HJetilJpirrolidin-1 -il)-5-fluoronicotínico (3S)-3-[(3S)-pirrolidin-3-ilJ-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (250,0 mg, 0,57128 mmol; de exemplo 15, etapa 3) foi misturada com ácido 2,6-dicloro-5- fluoronicotínico (167,95 mg, 0,79979 mmol) e DIPEA (125,0 μL, 0,7176 mmol) e foi dissolvida em NMP (1,5 mL). A reação foi aquecida a 130°C du- rante 3 horas, em cujo tempo a análise de LCMS mostrou principalmente o produto em uma mistura regiométrica de *“5:1. Purificada por LC preparativa como no exemplo 5 para fornecer o produto trifluoroacetato de ácido 2-cloro- 6-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- djpirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-5-fluoronicotínico (248 mg). Etapa 2: (3S)-3-[(3S)-1 -(5-amino-6-cloro-3-fluoropiridin-2-il)pirrolidin-3-ilj-3- [4-(7-{[2-(trimetilsilil)etóxijmetil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1- iljpropanonitrila
Em um frasco de fundo redondo de 1 gargalo, ácido 2-cloro-6- ((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metÍI}-7H-pirrolo[2,3-d]piri- midin-4-il)-1 H-pirazol-1-iljetil}pirrolidin-1-il)-5-fluoronicotínico (50,0 mg, 0,08181 mmol) foi dissolvido em THF (0,1 mL) e trietilamina (30,0 μL, 0,2152 mmol) foi adicionado, seguido por azida difenilfosfônica (19,39 μL, 0,090 10 mmol). A reação foi agitada a 25°C durante 3 horas, em cujo tempo a análise de LCMS mostrou principalmente o intermediário de isocianato: (3S)-3-[(3S)- 1-(6-cloro-3-fluoro-5-isocyanatopiridin-2-il)pirrolidin-3-íl]-3-[4-(7-{[2-(trimetil- silil)etóxiJmetil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il]propanonitrila.
Na mistura reacional foi adicionada água (150,0 μL, 8,3263 mmols) e foi a- 15 quecida até o refluxo durante 2 horas, em cujo tempo a análise de LCMS mostrou principalmente amina (3S)-3-[(3S)-1-(5-amino-6-cloro-3-fluoropiridin- 2-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin- 4-il)-1 H-pirazol-1-iljpropanonitrila. O produto foi purificado por LC preparativa (método de ACN/TFA/água como no exemplo 5) e foi levado para uma etapa 20 de desproteção. MS(EI): 582 (M+1) Etapa 3. bis(trifluoroacetato) de 3-[(3S)-1-(5-amino-6-cloro-3-fluoropiridin-2- il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila. (3S)-3-[(3S)-1-(5-amino-6-cloro-3-fluoropiridin-2-il)pirrolidin-3-il]- 25 3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila foi desprotegida (TFA/CH2CI2; MeOH/NH4OH) como no e- xemplo 1, e o composto desprotegido foi purificado em LC preparativa (método ACN/TFA como no exemplo 5) para fornecer o produto bis(trif]uoroa- cetato) de 3-[(3S)-1 -(5-am i no-6-cloro-3-fl u o rop i rid i n-2-i t) pi rrol id i n-3-i l]-3-[4- 30 (7H-prrrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila MS(EI): 452 (M+1). 1H RMN(300 MHz CD3OD): δ 9,00 (s, 1H), 8,90 (s, 1H), 8,55 (s, 1H), 7,85 (m, 1H), 7,30 (m, 1H), 7,80 (m, 1H), 4,85 (m, 1H), 3,90 (m, 1H), 3,20- 3,50 (m, 5H), 3,00 (m, 1H), 1,85 (m, 2H).
O bis(trifluoroacetato) de (3S)-3-[(3S)-1-(3-amino-6-cloro-5- fluoropirídín-2-il)pírrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol- 1-iljpropanonitrila de amina isomérica foi também isolado MS(EI): 452 (M+1). Exemplo 92. Trifluoroacetato de N-(2-((S)-3-((S)-1-(4-(7H-pirrolo[2,3-dlpiri- midin-4-il)-1 H-pirazol-1 -il)-2-cianoetil)pirrolidin-1 -il)-6-cloro-5-fluoropiridin-3- iDformamida
Em um frasco de fundo redondo de 1 gargalo, ácido 2-cloro-6- ((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]piri- midin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)-5-fluoronicotínico (de exemplo 91, etapa 1; 200,0 mg, 0,32726 mmol) foi dissolvido em THF (4,5 mL) e trietila- mina (120,0 μL, 0,8610 mmol) foi adicionado, seguido por azida difenilfosfô- nica (77,58 μL, 0,36 mmol). A reação foi agitada a 25°C durante 3 horas, em cujo tempo a análise de LCMS mostrou principalmente o intermediário de isocianato.
A reação foi hidrogenada sob uma atmosfera de hidrogênio (1 atm) durante 30 minutos, em cujo tempo a análise de LCMS mostrou princi-palmente formamida e algum subproduto desclorado. Este foi purificado por LC (método de ACN/TFA/água como no exemplo 5) e desprotegido como no exemplo 1, e purificado por LC preparativa (método de ACN/TFA/água como no exemplo 5) para fornecer ambos os regiômeros de amina.
Trifluoroacetato de N-[2-cloro-6-((3S)-3-{2-ciano-1-[4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1 -il)-5-fluoropiridin-3-il]for- mamida MS(EI): 481 (M+1), 1H RMN(300 MHz ,CD3OD): δ 8,95 (s, 1H), 8,85 (s, 1H), 8,55 (s, 1H), 7,80 (d, 1H), 7,58 (d, 1H), 7,20 (d, 1H), 4,85 (m, 1H), 4,46 (s, 2H), 2,90-4,00 (m, 7H), 1,80 (m, 2H); e trifluoroacetato de N-[6- cloro-2-((3S)-3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1 -il)-5-fluoropiridin-3-il]formamida MS(EI): 481 (M+1) Exemplo 93. Trifluoroacetato de (3S)-3-{(3S)-1-[6-(etilsulfonil)-3-fluoropiridin-2-il1pirrolidin-3-il}-3-r4-(7H-pirrolo|2,3-d1pirimidin-4-il)-1 H-pirazol-1 – illpropanonitrila
Etapa 1: 6-(etilsulfonil)-2,3-difluoropiridina
Em um frasconete, 2,3,6-trifluoropiridina (0,1 mL, 1,13 mmol) foi dissolvida em THF (2,0 mL) e hidreto de sódio (60% de óleo mineral, 0,050 g, 1,2 mmol) foi adicionado e foi resfriado a 0°C. Etanotiol (0,077 g, 1,2 mmol) foi adicionado e foi agitado a 25°C durante 16 horas e evaporado até a secura para fornecer 6-(etiltio)-2,3-difluoropiridina.
Este foi dissolvido em metanol (10,0 mL) e água (5,0 mL) e Oxo-ne® (1,38 g, 2,25 mmols) foi adicionado e foi agitado a 25°C durante 16 horas. Em seguida a mistura reacional foi dividida entre EtOAc e água e extrato de EtOAc foi lavado com salmoura, secado (mgSO4), e removido em vácuo. Análise de LCMS mostrou principalmente produto MS(EI): 207 (M+1). Etapa 2: trifluoroacetato de (3S)-3-{(3S)-1-[6-(etilsulfonil)-3-fluoropindin-2~ il]pirrolidin-3-il}-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropa-nonitrila (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (50,0 mg, 0,1142 mmol; de exemplo 15, etapa 3) foi misturada com 6-(etilsulfonÍI)-2,3-difluoro- pindina (33,143 mg, 0,15996 mmol) e DIPEA (25,0 μL, 0,1435 mmol) e foi dissolvida em NMP (0,3 mL). A reação foi aquecida a 130°C durante 2 horas, em cujo tempo a análise de LCMS mostrou principalmente produto. Este foi purificado por LC preparativa como no exemplo 5 para fornecer (3S)-3-{(3S)- 1-[6-(etilsulfonil)-3-fluoropiridin-2-il]pirrolidin-3-il}-3-[4-(7-{[2-(trimetilsilil)etó- xi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila. O grupo SEM foi clivado como no exemplo 1 e purificado por LC preparativa (método de ACN/TFA/água como no exemplo 5) para fornecer trifluoroacetato de (3S)-3-{(3S)-1 -[6-(etilsulfonil)-3-fluoropiridin-2-il]pirrolid in-3-il}-3-[4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila contaminado com ~10% do regiômero. MS(EI): 495 (M+1), 1H RMN(300 MHz, CD3OD): Õ 8,95 (s, 1H), 8,85 (s, 1H), 8,55 (s, 1H), 7,82 (d, 1H), 7,58 (m, 1H), 7,20 (d, 1H), 6,75 (dd, 1H), 4,85 (m, 1H), 4,46 (s, 2H), 2,90-4,00 (m, 9H), 1,90 (m, 2H), 1,30 (t, 3H). Exemplo 94. Trifluoroacetato de (3S)-3-[(3S)-1-(6-cloro-3-fluoropiridin-2- il)pirrolidin-3-ill-3-[4-(7H-pirrolof2,3-dlpirimidin-4-il)-1H-pirazol-1- illpropanonitrila

Em um frasco de fundo redondo de 1 gargalo bistrifluoroacetato de (3S)-3-[(3S)-1-(5-amino-6-cloro-3-fluoropiridin-2-il)pirrolidin-3-il]-3-[4-(7- {[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- iljpropanonitrila (Exemplo 91; 60,02 mg, 0,08621 mmol) foi dissolvido em THF (2,0 mL) e nitrito de terc-butila (15,0 pL, 0,1135 mmol) foi adicionado. A reação foi aquecida até o refluxo durante 3 horas, em cujo tempo a análise de LCMS mostrou (3S)-3-[(3S)-1-(6-cloro-3-fluoropÍridÍn-2-il)pirrolidin-3-il]-3- [4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila produto e nenhum material de partida. O produto foi purificado por LC preparativa (método de ACN/TFA/água como no exemplo 5) e foi desprotegido como no exemplo 1, e purificado como no exemplo 5 para for-necer trifluoroacetato de (3S)-3-[(3S)-1-(6-cloro-3-fluoropiridin-2-il)pirrolidin- 3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila. MS(EI): 437 (M+1). Exemplo 95. bis(trifluoroacetato) de 2-((3S)-3-f(1S)-2-ciano-1-[4-(7H-pir- rolo[2,3-djpirimidin-4-il)-1H-pirazol-1-illetil}pirrolidin-1-il)-5-fluoro-4- (metoximetil)nicotinonitrila

Etapa 1: 2,3-dibromo-5-fluoro-4-(metoximetil)piridina
Em um frasco de fundo redondo de 1 gargalo, N,N-dÍ-isopro- pilamina (0,09898 mL, 0,7062 mmol) foi dissolvida em THF (2,14 mL) e res-friada a -78°C. Na reação foi adicionado n-butil lítio a 1,6 M em hexano (0,3825 mL, 0,6120 mmol) e foi agitada a -78°C durante 30 minutos e uma solução de 2,3-dibromo-5-fluoropiridina (de Matrix Scientific; 120,0 mg, 0,4708 mmol) em THF (2,0 mL) foi adicionada e foi agitada a -78°C durante 2 horas e bromometil metil éter (0,079 mL, 0,96 mmol) foi adicionada e foi agitada a -78°C durante 1 hora. A reação foi extinguida com NH4CI saturado e dividida entre EtOAc e água e extrato de EtOAc foi lavado com salmoura, secado (mgSO4), e removido em vácuo. A análise de RMN mostrou principalmente o produto 2,3-dibromo-5-fluoro-4-(metoximetil)piridina. Usado na reação seguinte sem purificação. 1H RMN (400 MHz, CDCI3): δ 8,24 (s, 1H), 4,65 (d, 2H), 3,02 (s, 3H). Etapa 2: (3S)-3-{(3S)-1 -[3-bromo-5-fluoro-4-(metoximetil)piridin-2-!!]pirrolidin- 3-il}-3-[4-(7-{[2-(trimetilsilil)etóxiJmetil}-7H-pirrolo[2,3-dJpirimidin-4-ÍI)-1H- pirazol-1-iljpropanonitrila (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (80,0 mg, 0,1828 mmol; de exemplo 15, etapa 3) foi misturada com 2,3-dibromo-5-fluoro-4- (metoximetil)piridina (100,0 mg, 0,3345 mmol) e DIPEA (60,0 μL, 0,3445 mmol) e foi dissolvida em NMP (0,40 mL). A reação foi aquecida a 130°C durante 3 horas. O resíduo foi purificado por LC preparativa (método de ACN/TFA/água como no exemplo 5) para fornecer o produto (3S)-3-{(3S)-1 - [3-bromo-5-fluoro-4-(metoximetil)piridin-2-il]pirrolidin-3-il}-3-[4-(7-{[2-(trime- tilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila.
Este foi evaporado e foi dividido entre EtOAc e NaHCO3 saturado e extrato de EtOAc foi lavado com salmoura, secado (mgSO4), e removido em vácuo (33 mg). LCMS(EI): 656 (M+1). Etapa 3: 2-((3S)-3-{( 1 S)-2-ciano-1 -[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirro- lo[2,3-d]pinmidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-5-fíuoro-4-(metoxime- til)nicotinonitríla
Em um frasco de fundo redondo de 1 gargalo, (3S)-3-{(3S)-1-[3- bromo-5-fluoro-4-(rnetoximetil)piridin-2-il]pirrolidin-3-il}-3-[4-(7-{[2-(trimetil- silil)etóxi]-metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (33,0 mg, 0,0503 mmol) foi dissolvida em NMP (0,4 mL) e cianeto de zinco (17,7 mg, 0,151 mmol) e pó de zinco (9,87 mg, 0,151 mmol) foram adicionados. A reação foi desgaseificada e bis(tri-t-butilfosfina)paládio (12,9 mg, 0,0252 mmol) foi adicionado, desgaseificado e aquecido a 130°C durante 100 minutos, em cujo tempo a análise de LCMS mostrou que ela consistia principalmente no produto. A reação foi filtrada e o produto foi purificado por LC preparativa (método de ACN/TFA/água como no exemplo 5) para fornecer 2-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)-5-fluoro-4-(metoximetil)ni- cotinonitrila. MS(EI): 602 (M+1). Etapa 3: bis(trifíuoroacetato) de 2-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-5-fluoro-4-(metoximetil)ni- cotinonitrila 2-((3S)-3-{(1S)-2-Ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-5-fluoro-4-(meto- ximetil)nicotinonitrila foi desprotegida como no exemplo 1. O produto despro-tegido foi purificado por LC (método de ACN/TFA/água como no exemplo 5) para fornecer o produto título. MS(EI): 472 (M+1). 1H RMN(400 MHz CD3OD): δ 8,95 (s, 1H), 8,86 (s, 1H), 8,55 (s, 1H), 8,22 (d, 1H), 7,83 (d, 1H), 7,23 (d, 1H), 4,85 (m, 1H), 4,52 (s, 2H), 4,00 (m, 1H), 3,70-3,80 (m, 3H), 3,40 (s, 3H), 3,00-3,40 (m, 5H) Exemplo 96. tris(trifluoroacetato) de 2-((3S)-34(1S)-2-ciano-1-[4-(7H-pirro- lo[2,3-d1pirimidin-4-il)-1 H-pirazol-1-il1etil}pirrolidin-1-il)-4-(metoximetil)nicotinonitrila

Etapa 1: 2,3-dibromo-4-(metoximetil)piridina
Em um frasco de fundo redondo de 1 gargalo, N,N-di-isopro- pilamina (3,550 mL, 25,33 mmols) foi dissolvida em THF (60,0 mL) e foi res-friada a -78°C e n-butil litio a 1,6 M em hexano (14,51 mL, 23,22 mmols) foi adicionado e foi agitado durante 30 minutos. Na reação foi adicionada 2,3- dibromopiridina (5,0 g, 21,1 mmols) em THF (33 mL) e foi agitada a -78°C durante 1 hora e bromometil metil éter (1,895 mL, 23,22 mmols) foi adicionado e foi agitado durante 30 minutos a -78°C. A análise de LCMS mostrou uma mistura de -3:1 de 2,3-dibromo-4-(metoximetil)piridina e 2,4-dibromo-3- (metoximetil)piridina. A reação foi extinguida com NH4CI saturado e foi divi-dida entre EtOAc e água e extrato de EtOAc foi lavado com salmoura, secado (mgSO4), e removido em vácuo. O resíduo foi submetido á cromatografia em sílica-gel usando 5% de EtOAc/hexanos para fornecer o produto 2,3- dibromo-4-(metoximetil)piridina. MS: 282 (M+1). 1H RMN (400 MHz, CDCI3): δ 8,36 (d, 1H), 7,80 (d, 1H), 4,48 (s, 2H), 3,50 (s, 3H). Etapa 2: (3S)-3-{(3S)-1-[3-bromo-4-(metoximetil)piridin-2-il]pirrolidin-3-il}-3- [4-(7-{[2-(trímetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (600,0 mg, 1,371 mmol; de exemplo 15, etapa 3) foi misturada com 2,3-dibromo-4-(metoxi- metil)piridina (610,0 mg, 2,17 mmols) e DIPEA (235 μL, 1,35 mmol) e foi dis-solvida em NMP (1,6 mL). A reação foi aquecida a 140°C durante 3 horas, em cujo tempo a análise de LCMS mostrou principalmente o produto. O resíduo foi purificado por cromatografia para fornecer o produto (3S)-3-{(3S)-1- [3-bromo-4-(metoximetil)pÍridin-2-il]pirrolidin-3-il}-3-[4-(7-{[2-(trimetilsílil)etó- xi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila. (391 mg). MS(EI): 637, 639 (M+1). Etapa 3: 2-((3S)-3-{(1 S)-2-ciano-1-[4-(7-{[2-(tnmetilsihl)etóxi]metil}-7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-4-(metoximetil)nicoti- nonitrila
Em um frasco de fundo redondo de 1 gargalo, (3S)-3-{(3S)-1-[3- bromo-4-(metoximetil)-píridin-2-il]pirrolidin-3-il}-3-[4-(7-{[2-(trimetilsilil)etó- xi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (390,0 mg, 0,6116 mmol) foi dissolvida em NMP (4,0 mL) e cianeto de zinco (215 mg, 1,83 mmol) e pó de zinco (120 mg, 1,83 mmol) foram adicionados. A reação foi desgaseificada e bis(tri-t-butilfosfina)paládio (50,0 mg, 0,09784 mmol) foi adicionado e foi aquecido a 130°C durante 100 minutos, em cujo tempo a análise de LCMS mostrou que foi principalmente material de partida e algum produto em uma relação de ~3:1. Na reação foi adicionado bis(tri-t- butilfosfina)paládio (80,0 mg, 0,156 mmol) e foi aquecido a 130°C durante 100 minutos, em cujo tempo a análise de LCMS mostrou principalmente produto. A mistura foi filtrada e foi purificada por cromatografia para fornecer o produto (190 mg). MS(EI): 584 (M+1) Etapa 4: tris(trifíuoroacetato) de 2-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-4-(metoximetil)nicotinonitrila
Em um frasco de fundo redondo de 1 gargalo 2-((3S)-3-{(1S)-2- ciano-1 -[4-(7-{[2-(trimetÍlsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirÍmidin-4-il)-1 H- pirazol-1 -il]etil}pirrolidin-1-il)-4-(metoximetil)nicotinonitrila (0,380 g, 0,651 mmol) foi dissolvida em DCM (2,0 mL) e TFA (0,1 mL, 13,0 mmols) foi adi-cionado. A reação foi agitada a 25°C durante 2 horas, em cujo tempo a análise de LCMS mostrou uma -3:1 de produto e material de partida. Em seguida uma quantidade adicional de TFA (0,1 mL, 13,0 mmols) foi adicionada e foi agitada durante 1 hora em cujo tempo a análise de LCMS mostrou apenas o produto. A mistura reacional foi evaporada até a secura e foi dissolvida em metanol (4,0 mL) e amónia a 16 M em água (0,1 mL, 16,4 mmols) foi a- dicionada e foi agitada a 25°C durante 1 hora em cujo tempo a análise de LCMS mostrou principalmente o produto. A mistura reacional foi evaporada e foi purificada por LCMS preparativa como no exemplo 5 para fornecer o pro-duto. MS(EI): 454 (M+1). 1H RMN(400 MHz CD3OD): δ 9,00 (s, 1H), 8,90 (s, 1H), 8,57 (s, 1H), 8,22 (d, 1H), 7,85 (d, 1H), 7,25 (d, 1H), 6,80 (d, 1H), 4,90 (m, 1H), 4,50 (s, 2H), 4,02 (m, 1H), 3,82 (m, 1H), 3,75 (m, 2H), 3,42 (s, 3H), 5 3,40 (m, 1H), 3,22 (m, 1H), 3,03 (m,1 H), 1,86 (m, 2H).
Os exemplos na tabela abaixo foram feitos por procedimentos análogos àqueles para a produção de exemplos 84 a 96.
Exemplo 102. bis (trifluoroacetato) de (3S)-3-í4-(7H-pirrolo[2,3-d]pírimidin-4-il)-1 H-pirazol-1 -il]-3-[(3S)-1-[1,3]tiazolo[5,4-d]pirimidin-5-ilpirrolidin-3-illpropanonitrila
Etapa 1. 5-amino-2-cloropirimidina-4-tiol
Sulfeto de hidrogênio de sódio (0,1 g, 18 mmols) foi adicionado a uma solução de 2,4-dicloropirimidin-5-amina (1 g, 6 mmols), em etanol (40 mL), sob N2 agitada a 60°C durante 2 horas. LCMS mostrou a reação quase completa, e mostrou o produto esperado (M+H: 162), e também mostrou algum dissulfeto (M+H: 321). A mistura reacional foi evaporada, adicionada água (25 mL) seguido por ácido acético (5 mL, 90 mmols) para ajustar o pH para 3. A mistura foi agitada durante 2 dias, filtrada, enxaguada com água, secada por ar, em seguida secada sob vácuo elevado. O produto isolado (0,6g, 60% de rendimento) provavelmente contém algum enxofre. LCMS calculada para C4H5CIN3S(M+H)+: m/z = 161,989. Etapa 2. 5-cloro[1,3]tiazolo[5,4-d]pirímidina 5-amino-2-cloropirimidina-4-tiol (0,3 g, 2 mmols) foi agitado em ortoformiato de etila (3 mL, 20 mmols) durante 2 horas a 21 °C. LCMS mostrou-se quase completa (M+H 172 muito fraco). A mistura reacional foi evaporada até a secura. O resíduo foi extraído com ACN e filtrado para remover enxofre, etc. O produto foi isolado por HPLC preparativa usando um instru-mento Waters Fraction-Lynx e uma coluna Xbridge C18 de 30 mm x 10 mm; 25% de CH3OH-H2O (0,1% de TFA), 0,6 minutos; gradiente de 6 minutos para 45%; 60 mL/minuto; detector fixado a 220 nm; tempo de retenção de 3,7 minutos. As frações coletadas foram evaporadas até a secura para fornecer um sólido amarelo em 5% de rendimento. HPLC mostrou produto UVmax 220 nm. LCMS calculada para C5H3CIN3S(M+H)+: m/z = 171,974. Etapa 3. bis (trifluoroacetato) de (3S)-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1-il]-3-[(3S)-1-[1,3]tiazolo[5,4-d]pirimidin-5rilpirrolidin-3-il]propanoni- trila (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirÍmidin-4-il)-1 H-pirazol-1-iljpropanonitrila (38 mg, 0,087 mmol; de exemplo 15, etapa 3), foi dissolvida em NMP (0,41 mL) e 4-metilmorfolÍna (24 μL, 0,22 mmol). 5-cloro[1,3]tiazolo[5,4-d]pirimidina (15 mg, 0,087 mmol) foi adicionada, agitada a 120°C em um reator de micro-ondas durante 10 minutos. LCMS mostrou a reação quase completa para o intermediário espe-rado (M+H, 573). O produto foi isolado por HPLC/MS preparativa usando um instrumento Waters Fraction-Lynx e uma coluna Sunfire C18 de 30 mm x 100 mm; 30% ACN-H2O (0,1% de TFA), 2,0 minutos; gradiente de 10 minutos a 60%; 60mL/minuto; tempo de retenção de 10,9 minutos. O produto fractions foram secado por congelamento para fornecer 20 mg (sal de TFA).
Desproteção: O resíduo acima foi dissolvido em CH2CI2 (0,4mL) a 21 °C, e TFA (0,34 mL, 4,4 mmols) foi adicionado e agitado durante 1,2 horas. A solução foi concentrada para remover o TFA. O resíduo foi dissolvido em acetonitrila (0,8 mL) e hidróxido de amõnio a 15,0 M em água (0,20 mL, 2,9 mmols) foi adicionado. A solução foi agitada a 21 °C durante 3 horas. LCMS mostrou a reação a ser concluída. A mistura reacional foi concentrada. O produto foi isolado por HPLC/MS preparativa usando um instrumento Waters Fraction-Lynx e uma coluna Sunfire C18 de 19 mm x 100 mm; 9% de ACN-H2O (0,1% de TFA), 2,5 minutos; gradiente de 10 minutos a 35%; 30 mL/minuto; tempo de retenção de 11,8 minutos. As frações coletadas foram secado por congelamento para fornecer sólido branco (11 mg; sal de bis- TFA presumido). HPLC mostrou UVmax a 228, 268, 288, e 330nm. 1H RMN (300 MHz, DMSO-d6): δ 12,5 (s, 1H); 8,98 (s, 1H); 8,95 (s, 2H), 8,77 (s, 1H); 8,48 (s, 1H); 7,72 (s, 1H); 7,88 (s, 1H); 4,84 (m, 1H); 3,86 (m, 1H); 3,61 (m, 1H); 3,35 (m, 4H); 2,88 (m, 1H); 1,64 (m, 2H); LCMS calculada para C21H19N10S(M+H)+: m/z = 443,151; encontrado 443. Exemplo 103. bis (trifluoroacetato) de 2-((3S)-3-{(1S)-2-ciano-1-(4-(7H-pir- rolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-ilietil)pirrolidin-1-il)-4-(difluorometii)nico-

Etapa 1. 2,3-dicloro-4-(difluorometil)piridina 2,3-dicloroisonicotinaldeído (146 mg, 0,830 mmol), foi agitado em 2-metóxi-N-(2-metoxietil)-N-(trifluoro-À(4)-sulfanil)etanamina (Aldrich; 0,30 mL, 1,6 mmol) a 21 °C. Etanol (10 μL, 0,2 mmol) foi adicionado para fornecer catalisador de HF. Após 1,5 hora, LCMS mostrou a conversão completa no produto (não ionizou). A reação foi extinguida vertendo em solução de NaHCO3 a 5% seguido por extração com EtOAc. A camada de EtOAc foi agitada com ácido cítrico a 5% para remover bis(metoxietil)amina. Os extratos orgânicos foram evaporados até a secura para fornecer 130 mg de óleo, que lentamente cristalizado. O produto foi limpo o suficiente para usar sem purificação. HPLC mostrou UVmax 216 & 280 nm. A FMR mostrou um du- pleto para o CHF2 a -118,9 ppm. 1H RMN (300 MHz, CDCI3): δ 8,46 (d, J = 4,9 Hz, 1H); 7,53 (d, J = 4,9 Hz, 1H); 6,90 (t, J = 53,8 Hz, 1H); LCMS calculada para C6H4CI2F2N(M+H)+: m/z = 197,969. Etapa 2. (3S)-3-{(3S)- 1-[3-cloro-4-(difíuorometil)piridin-2-il]pirrolidin-3-il}-3-[4- (7-{[2-(trimetilsilil) etóxijmetil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila
Em um frasconete foi adicionada (3S)-3-[(3S)-pirrolidin-3-il]-3-[4- (7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidm-4-il)-1 H-pirazol-1- iljpropanonitrila (68 mg, 0,16 mmol; de exemplo 15, etapa 3), NMP (0,75 mL); 4-metilmorfolina (34 μL, 0,31 mmol), e 2,3-dicloro-4-(difluorometil)pi- ridina (46 mg, 0,23 mmol). Agitada a 150°C durante 15 minutos em um reator de micro-ondas. LCMS & HPLC mostrou 80% de reação, com cerca de 60% de conversão no produto (M+H 599). O produto foi isolado por HPLC preparativa usando um instrumento Waters Fraction-Lynx e uma coluna X- bridge C18 de 30 mm x 10 mm; 67% de CH3OH-H2O (0,1% de TFA), 0,5 minuto; em seguida gradiente de 5 minutos para 85%; 60mL/minuto; detector fixado a 254 nm; tempo de retenção de 5,6 minutos. O eluído coletado foi evaporado até a secura para fornecer 40 mg (36% de rendimento; provavelmente sal de TFA). HPLC forneceu UVmax 208, 226, 260, & 314 nm. LCMS calculada para C28H34CIF2N8OSi(M+H)+: m/z = 599,228. Etapa 3. bis (trifluoroacetato) de 2-((3S)-3-{(1 S^-ciano-l-ft-ÍVH-pirrolo^S- dJpirimidin^-ity-l H-pirazol-1 -il]etil}pirrolidin-1 -il)-4-(difluorometil)nicotÍnonitrila (3S)-3-{(3S)-1-[3-cloro-4-(difluorometil)piridin-2-il]pirrolidin-3-il}-3- [4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pÍrimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila (34 mg, 0,057 mmol; 40mg de sal de TFA), foi agitada em NMP (0,1 mL). Cianeto de zinco (21 mg, 0,18 mmol) e tetracis(trifenilfos- fina)paládio(O) (14 mg, 0,012 mmol) foram adicionados e a solução foi esti-mulada com nitrogênio (sub-superfície). O frasconete foi selado. A solução foi aquecida a 180°C durante 15 minutos em um reator de micro-ondas. LCMS mostrou cerca de 50% de reação para fornecer M+H 590. A mistura reacional foi diluída com MeOH e filtrada. O produto foi isolado por LCMS preparativa usando um instrumento Waters Fraction-Lynx e uma coluna Sun- fire C18 de 30 mm x 100 mm; 40% de ACN-H2O (0,1% de TFA), 2,0 minutos; gradiente de 10 minutos a 65%; 60 mL/minuto; detector fixado a m/z 590 & 599; tempo de retenção, 10,5 & 11,8 minutos. As frações coletadas foram evaporadas até a secura.
Desproteção: O acima foi dissolvido em DCM (0,35 mL) e TFA (0,35 mL, 4,5 mmols) e foi agitado durante 1,1 horas. A solução foi concen-trada para remover TFA. Ao resíduo foram adicionados acetonitrila (0,8 mL) e hidróxido de amónio a 15,0 M em água (0,21 mL, 3,2 mmols). A reação foi agitada a 20°C durante 2 horas. LCMS mostrou a reação a ser concluída. A mistura reacional foi concentrada. O produto foi isolado por HPLC/MS prepa-rativa usando um instrumento Waters Fraction-Lynx e uma coluna Sunfire C18 de 30 mm x 100 mm; 18% de ACN-H2O (0,1% de TFA), 2,5 minutos; gradiente de 10 minutos a 44%; 60 mL/minuto; detector fixado a m/z 460; tempo de retenção de 11,8 minutos. O produto fractions foram coletado e secado por congelamento para fornecer 6 mg de sólido branco. HPLC: UVmax 220, 266, 292, e 330nm. O FMR mostrou o produto a ser o sal de di- TFA, e mostrou dois dupletos para o CHF2 (a -116,8 ppm), de dois rotôme- ros. 1H RMN (400 MHz, DMSO-d6): δ 12,5 (s, 1H); 8,98 (s, 1H); 8,81 (s, 1H); 8,52 (s, 1H); 8,46 (d, J = 5,0 Hz, 1H); 7,75 (s, 1H); 7,13 (t, J = 53,8 Hz, 1H); 7,11 (s, 1H); 6,93 (d, J = 5,0 Hz, 1H); 4,90 (m, 1H); 3,95 (m, 1H); 3,81 (m, 1H); 3,66 (m, 2H); 3,35 (m, 2H); 2,90 (m, 1H); 1,72 (m, 2H); LCMS calculada para C23H20F2N9(M+H)+: m/z = 460,181; encontrado 460.
Exemplos 104 a 116.
Os exemplos na tabela abaixo foram feitos por procedimentos análog os àqueles para a produção dos exemplos 47 a 50.
Exemplo 117. Trifluoroacetato de 3-((3S)-3-{2-fluoro-1-[4-(7H-pirrolof2,3- dlpirimidin-4-il)-1H-pirazol-1-il]etil}pirrolidin-1-il)piridina-2-carbonitrila
Uma solução de 4-(1-{2-fluoro-1-[(3S)-pirrolidin-3-il]etil}-1 H-pira- zol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidina (de exemplo 70, etapa 7; 37 mg, 0,087 mmol), e DIPEA (30,0 μL, 0,17 mmol), em NMP (0,7 mL) com 3-fluoropiridina-2-carbonitrila (de Alfa Aesar; 16 mg, 0,13 mmol), foi aquecida para 130°C durante 2 horas. LCMS mostrou a conversão no intermediário esperado. A mistura reacional foi dividida entre água e EtOAc, a fase aquosa foi extraída mais 2x com EtOAc. As fases orgânicas combinadas foram lavadas com salmoura, secadas sobre MgSO4, filtradas e concentradas em vácuo. O resíduo bruto foi dissolvido em 5 mL de Me- OH/ACN com uma pequena quantidade de água e foram purificados por LCMS preparativa como no exemplo 5 em pH 2 para recuperar o produto. O produto purificado foi concentrado em vácuo e levado para desproteção.
Ao resíduo foram adicionados DCM (0,5 mL) e TFA (0,5 mL), a reação foi agitada em temperatura ambiente durante 1 hora, evaporada até a secura, em seguida adicionados metanol (0,5 mL) e hidróxido de amónio (0,5 mL), após 45 minutos LCMS mostrou desproteção completa. Os solventes foram removidos e o resíduo foi dissolvido em MeOH/ACN/água e purificado por LCMS preparativa como no exemplo 5 no pH 2, os tubos de produto foram combinados e liofilizados até a secura para fornecer o produto como um sal de TFA. 1H RMN (300 MHz, DMSO-d6): δ 12,8 (s, 1H); 8,9 (s, 1H); 8,8 (s, 1H); 8,5 (s, 1H); 7,9 (m, 1H); 7,8 (s, 1H); 7,4 (m, 1H); 7,25 (m, 1H); 7,2 (s, 1H); 4,8 (m, 3H); 3,7 (m, 1H); 3,5 (m, 3H); 2,9 (m, 1H); 1,7 (m, 2H). LCMS calculada para C21H20FN8(M+H)+: m/z = 403,179, observado 403,2. Exemplo 118. Trifluoroacetato de 2-((3S)-3-(2-fluoro-1-[4-(7H-pirrolo[2,3- d]pirimidín-4-il)-1 H-pirazol-1-illetil}pirrolidin-1-il)nicotinonitrila
Uma solução foi preparada dissolvendo 4-(1-{2-fluoro-1-[(3S)- pirrolidin-3-il]etil}-1H-pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidina (de exemplo 70, etapa 7; 37 mg, 0,087 mmol) e DIPEA (30 μL, 0,17 mmol) em NMP (0,7 mL) com 2-fluoronicotinonitrila (de Alfa Aesar; 16 mg, 0,13 mmol) foi aquecida para 130°C durante 2 horas. A mistura reacio- nal foi dividida entre água e EtOAc, a fase aquosa foi extraída mais 2x com EtOAc. A fase orgânica combinada foi lavada com salmoura, secada sobre MgSO4, filtrada e concentrada em vácuo e foi purificada por LCMS preparativa como no exemplo 5 em pH 2 para recuperar o produto. Este foi concentrado em vácuo e o grupo SEM foi removido como no exemplo 1. O solvente foi evaporado e o resíduo foi dissolvido em MeOH/ACN/água e purificado por LCMS preparativa como no exemplo 5 em pH 2, os tubos de produto foram combinados e liofilizados até a secura para fornecer o produto como um sal de TFA. 1H RMN (300 MHz, DMSO-d6): δ 12,8 (s, 1H); 8,95 (s, 1H); 8,85 (s, 1H); 8,5 (s, 1H); 8,3 (m, 1H); 7,9 (m, 1H); 7,8 (s, 1H); 7,2 (s, 1H); 6,7 (m, 1H); 4,9 (m, 3H); 3,9 (m, 1H); 3,8 (m, 1H); 3,6 (m, 2H); 2,9 (m, 1H); 1,7 (m, 2H). LCMS calculada para C21H20FN8(M+H)+: m/z = 403,179, observado 403,1. Exemplo 119. Trifluoroacetato de 4-(1-{1-[(3S)-1-(1,1-dióxido-2,3-di- hidrotieno[2,3-blpiridín-6-il)pirrolidin-3-il]-2-fluoroetil}-1H-pirazol-4-i1)-7H-

Uma solução foi preparada dissolvendo 4-(1-{2-fluoro-1-[(3S)- pirrolidin-3-il]etil}-1 H-pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidina (de exemplo 70, etapa 7; 25 mg, 0,058 mmol) e DIPEA (2,0E1 μL, 0,12 mmol) em NMP (0,2 mL). A esta solução foi adicionado 1,1 -dióxido de 6-cloro-2,3-di-hidrotieno[2,3-b]piridina (de exemplo 28, etapa 4; 18 mg, 0,087 mmol) e a reação foi aquecida para 100°C durante 2 horas, em cujo tempo a análise de LCMS mostrou a conversão no composto desejado. A reação foi resfriada para temperatura ambiente e dividida entre água e EtO- Ac. As fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com salmoura, secada sobre Mg- SO4, filtrada e concentrada em vácuo para fornecer o produto bruto, que foi em seguida dissolvido em ACN/MeOH e purificado por LCMS preparativa, em pH 2 método MeOH/água como no exemplo 5, para recuperar o produto.
Os tubos de produto foram evaporados até a secura, e o resíduo foi tratado com DCM (0,4 mL) e TFA (0,4 mL) durante 30 minutos. Os solventes foram removidos e hidróxido de amónio (0,4 mL) e metanol (0,4 mL) foram adicio-nados e a reação foi agitada em temperatura ambiente durante 45 minutos. Os solventes foram evaporados até a secura, o resíduo foi dissolvido em MeOH/ACN/água e purificado por LCMS preparativa como no exemplo 5 em pH 2. Os tubos de produto foram combinados e liofilizados até a secura para fornecer o produto como um sal de TFA. 1H RMN (300 MHz, DMSO-d6): δ 12,5 (s, 1H); 8,95 (s, 1H); 8,75 (s, 1H); 8,4 (s, 1H); 7,5 (s, 1H); 7,4 (d, 1H); 7,1 (s, 1H); 6,7 (d, 1H); 4,95 (m, 1H); 4,8 (m, 2H); 3,75 (m, 1H); 3,5 (m, 1H); 3,45 (t, 2H); 3,25 (m, 2H); 3,05 (t, 2H); 2,85 (m, 1H); 1,65 (m, 2H). LCMS calculada para C22H23FN7O2S(M+H)+: m/z = 468,162, observado 468,15. Exemplo 120. trifluoroacetato de 2-((3S)-3-(1-(4-(7H-pirrolo[2,3-dlpirimidin-4- il)-1 H-pirazol-1-il)-2-fluoroetil)pirrolidin-1-il)pindina-3,4-dicarbonitrila

Etapa 1. 3-cloro-2-((3S)-3-{2-fluoro-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)isonicotinonitnla
Uma solução foi preparada dissolvendo 4-(1-{2-fluoro-1-[(3S)- pirrolidin-3-il]etil}-1 H-pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidina (de exemplo 70, etapa 7; 60 mg, 0,1 mmol) e DIPEA (48 μL, 0,28 mmol) em NMP (0,5 mL). A esta solução foi adicionada 2,3-dicloroiso- 5 nicotinonitrila (36 mg, 0,21 mmol) e a reação foi aquecida para 130°C durante 1,5 hora. LCMS mostrou a conversão completa no composto desejado, (m/z = 567/569). A reação foi resfriada para temperatura ambiente e dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com salmoura, 10 secada sobre MgSO4, filtrada e concentrada em vácuo para fornecer o produto bruto. LCMS calculada para C27H33CIFN8OSÍ (M+H)+: m/z = 567,222, observado 567,15. Etapa 2. Trifluoroacetato de 2-(3-{2-fluoro-1-[4-(7H-pirrolo[2,3-d]pirimidin-4- il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)piridina-3,4-dicarbonitrila
Em um frasco de fundo redondo de 1 gargalo, 3-cloro-2-(3-{2- fluoro-1 -[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H- pirazol-1 -il]etil}pirrolidin-1 -il)isonicotinonitrila (35,7 mg, 0,0629 mmol) foi dissolvida em NMP (0,4 mL) e cianeto de zinco (22,2 mg, 0,189 mmol) e zinco (12,3 mg, 0,189 mmol) foram adicionados. A reação foi desgaseificada com 20 vácuo/N2 e bis(tri-t-butilfosfina)paládio (16,1 mg, 0,0315 mmol) foi adicionado. A reação foi desgaseificada novamente e foi em seguida aquecida para 130 °C durante 3 horas. A reação foi resfriada para temperatura ambiente e dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com 25 água, em seguida salmoura, secada sobre MgSO4, filtrada e concentrada em vácuo para fornecer o produto bruto, que foi dissolvido em MeOH/ACN e purificado por LCMS preparativa como no exemplo 5. O eluído foi concentrado em vácuo e tratado com DCM (0,5 mL) e TFA (0,2 mL), a reação foi agitada em temperatura ambiente durante 1 hora, evaporada até a secura, e em seguida adicionados metanol (0,5 mL) e hidróxido de amónio (0,5 mL) e agitada durante 1 hora. Os solventes foram evaporados e o resíduo foi dis-solvido em MeOH/ACN/água e purificado por LCMS preparativa em pH 2 como no exemplo 5. Os tubos de produto foram liofilizados até a secura para fornecer o produto como sal de TFA. LCMS calculada para C22H18FN9(M+H)+: m/z = 428,175, observado 428,10. Exemplo 121. Trifluoroacetato de 3~((3S)-3-{2-fluoro-1-[4-(7H-pirrolo[2,3- dlpirimidin-4-il)-1 H-pirazol-1-inetil}pirrolidin-1-il)ftalonitrila

Etapa 1. 2-((3S)-3-{2-fIuoro-1 -[4-(7-{[2-(tnmetiisilil)etóxi]metil}- 7H-pirrolo[2,3- d]pirímidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -H)-6-iodobenzonÍtríla
Uma solução foi preparada dissolvendo 4-(1-{2-fluoro-1-[(3S)- pirrolidin-3-il]etil}-1 H-pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- djpirimidina (de exemplo 70, etapa 7; 45 mg, 0,10 mmol) e DIPEA (36 μL, 0,21 mmol) em NMP (0,4 mL). A esta solução foi adicionada 2-fluoro-6- iodobenzonitrila (39 mg, 0,16 mmol) e a solução foi aquecida para 100°C durante 2 horas. A reação foi resfriada para temperatura ambiente, dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida salmoura, secada sobre MgSO4, filtrada e concentrada em vácuo para fornecer o produto bruto, que foi purificado por cromatografia de sílica- gel eluindo com hexanos --> 5% de MeOH/CH2CI2. LCMS calculada para C28H34FIN7OSÍ (M+H)+: m/z = 658,68, observado 658,15. Etapa 2. 3-((3S)-3-{2-fiuoro-1-[4-(7-{[2-(tnmetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)ftalonitrila
A uma solução de 2-((3S)-3-{2-fluoro-1 -[4-(7-{[2-(trimetil- silil)etóxi]metil}-7H-pirrolo[2,3-d]pÍrimidin-4-il)-1 H-pirazol-1-il]etíl}pirrolídin-1 - il)-6-iodobenzonitrila (22 mg, 0,033 mmol) em NMP (0,764 mL) foi adicionado cianeto de zinco (58,1 mg, 0,495 mmol). A mistura foi desgaseificada com dois ciclos de vácuo/N2, em seguida tetracis(trifenilfosfina) paládio(O) (38,1 mg, 0,0330 mmol) foi adicionado, a reação foi novamente desgaseificada com dois ciclos de vácuo/N2. A reação foi aquecida para 130°C durante 2 horas, em seguida LCMS mostrou a formação de produto desejado. A reação foi resfriada para temperatura ambiente e filtrada para remover os sólidos, em seguida foi dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida salmoura, secada sobre MgSO4, filtrada e concentrada em vácuo para fornecer o produto bruto. O produto bruto foi purificado por LCMS preparativa como no exemplo 5, os tubos de produto foram evaporados até a secura para fornecer 3-((3S)-3-{2-fluoro-1 -[4-(7-{[2- (trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1-il)ftalonitrila. LCMS calculada para C29H34FN8OSÍ (M+H)+: m/z = 557,261, observado 557,25. Etapa 3. Trifluoroacetato de 3-((3S)-3-{2-fíuoro-1-[4-(7H-pirrolo[2,3-d]pirimi- din-4-H)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)ftalonitrila
Ao resíduo de etapa 3 foram adicionados DCM (0,5 mL) e TFA (0,5 mL), a reação foi agitada em temperatura ambiente durante 1 hora, e- vaporada até a secura, em seguida adicionados metanol (0,5 mL) e hidróxido de amónio (0,5 mL). Após 30 minutos, LCMS mostrou a desproteção completa. Os solventes foram removidos e o resíduo foi dissolvido em Me- OH/ACN/água e purificado por LCMS preparativa em pH 2 como no exemplo 5. Os tubos de produto foram combinados e liofilizados até a secura para fornecer o produto 3-((3S)-3-{2-fluoro-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H- pirazol-1 -il]etil}pirrolidin-1-ii)ftalonitrila como um sal de TFA. LCMS calculada para C23H20FN8 (M+H)+: m/z = 427,179, observado 427,85. Exemplo 122. Trifluoroacetato de 2-((3S)-3-((1S)-2-ciano-1-[4-(7H-pirrolo[2,3- dlpirimidin-4-il)-1 H-pirazol-1-illetil}pirrolidin-1-il)-4-iodonicotinonitrila

Etapa 1. 2-cloro-4-iodonicotinonitrila Ao 2-cloro-4-iodonicotinaldeido (0,1 g, 3,7 mmols) dissolvido em THF (11 mL) foi adicionado hidróxido de amónio (11 mL, 280 mmols) seguido por iodo (1040 mg, 1,41 mmol), a reação foi mantida em temperatura ambiente 3,5 horas, a cor visivilmente clareia quando a reação progride até o término quando ela está quase incolor. LCMS indica que a reação deve ser concluída. A reação foi extinguida por adição de NaHSO3 saturado, extraída em EtOAc. A fase orgânica foi lavada com salmoura, secada sobre MgSO4, filtrada e concentrada em vácuo para fornecer o produto bruto, 926 mg. Dissolvido em CHCI3/MeOH e aplicado na coluna de sílica-gel de 120 g, as frações do produto foram concentradas em vácuo para fornecer 728 mg do produto. O material purificado foi levado diretamente para a etapa seguinte. Etapa 2. 2-((3S)-3-{(1S)-2-c/ano-1-[4-(7-{[2-(tnmetilsilil)etóxi]metil}--7H- pirrolo[2,3-d]pÍrimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-4- iodonicotinonitrila e 2-doro-4-((3S)-3-{( 1 S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H- pirrolo[2,3-d]pirimidin-4-ÍI)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)nicotinonitrila
A uma solução de 2-cloro-4-iodonicotinonitrila (50,8 mg, 0,192 mmol) em NMP (0,112 mL) foi adicionada (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7- {[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila (56,0 mg, 0,128 mmol; de exemplo 15, etapa 3) seguido por DIPEA (31,9 μL, 0,183 mmol), a reação foi tampada e aquecida para 100°C em um bloco de aquecimento durante 3 horas. LCMS mostrou a formação do produto resultante do deslocamento de iodo (maior) com o produto menor do deslocamento de cloro. A reação foi resfriada para temperatura ambiente e diluída com ACN/MeOH e os produtos foram separados por LCMS preparativa como no exemplo 5 para recuperar os dois produtos 2-cloro-4-((3S)-3- {(1 S)-2-ciano-1 -[4-(7-{[2-(trirnetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4- il)-1H-pirazol-1-il]etil}pirrolidin-1-il)nicotinonitrila (LCMS calculada para C28H33IN9OSÍ (M+H)+: m/z = 666,162, observado 666,20) e 2-((3S)-3- {(1 S)-2-ciano-1 -[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4- il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-4-iodonicotinonitrila (LCMS calculada para C28H33CIN9OSÍ (M+H)+: m/z = 574,227, observado 574,20). Etapa 3. Trifluoroacetato de 2-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]p/rimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)-4-iodonicotinonitrila 2-((3S)-3-{(1S)-2-Ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-4- iodonicotinonitrila foi dissolvida em DCM (400 μL) e TFA (400 μL). Após 1 hora, LCMS mostra a reação completa, solvente evaporado, e metanol (800 μL) e hidróxido de amónio adicionados (400 μL), LCMS mostra desproteção completa. Evaporado o solvente e apreendeu o resíduo em MeOH/ACN, pu-rificado por LCMS preparativa como no exemplo 5 (ACN/água, em pH 2) para fornecer o produto como sal de TFA. 1H RMN (300 MHz, DMSO-d6): δ 12,6 (s, 1H); 9,0 (s, 1H); 8,8 (s, 1H); 8,5 (s, 1H); 7,9 (d, 1H); 7,75 (s, 1H); 7,25 (d, 1H); 7,1 (s, 1H); 4,9 (m, 1H); 3,9 (m, 1H); 3,7 (m, 1H); 3,6 (m, 2H); 3,3 (m, 2H); 2,9 (m, 1H); 1,7 (m, 2H); LCMS calculada para C22H19IN9 (M+H)+: m/z = 536,081, observado 535,85. Exemplo 123. Trifluoroacetato de 2-cloro-4-((3S)-3-((1S)-2-ciano-1-[4-(7H- pirroloí2,3-dlpirimidin-4-il)-1 H-pirazol-1 -il]etíl}pirrolidin-1-il)nicotinonitrila

2-cloro-4-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1 -il)nícotinonitrila (de exemplo 122, etapa 2) foi desprotegida e purificada como no exemplo 5 122, etapa 3, para fornecer o produto como sal de TFA. 1H RMN (300 MHz, DMSO-d6): δ 12,5 (s, 1H); 8,95 (s, 1H); 8,8 (s, 1H); 8,5 (s, 1H); 8,0 (d, 1H); 7,75 (s, 1H); 7,1 (s, 1H); 6,65 (d, 1H); 4,9 (m, 1H); 3,8 (m, 1H); 3,7 (m, 1H); 3,6 (m, 2H); 3,3 (m, 2H); 2,9 (m, 1H); 1,7 (m, 1H); 1,6 (m, 1H); LCMS calculada para C22H19CIN9 (M+H)+: m/z = 441,45, observado 443,90. Exemplo 124. trifluoroacetato de 4-((3S)-3-{(1S)-2-ciano-1-(4-(7H-pirrolor2,3- d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)piridina-2,3-dicarbonitrila
A uma solução de 2-cloro-4-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2- (trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)nicotinonitrila (Exemplo 123; 32 mg, 0,056 mmol) em NMP (0,10 mL) foi adicionado cianeto de zinco (26,2 mg, 0,223 mmol). A mistura foi desgaseificada com dois ciclos de vácuo/N2, em seguida tetracis(tri- fenilfosfina)paládio(O) (51,5 mg, 0,0446 mmol) foi adicionado, a reação foi novamente desgaseificada com dois ciclos de vácuo/N2. A reação foi aque-cida para 120°C durante 16 horas. LCMS mostrou a conversão quaso com-pleta no produto. Rxn resfriado para temperatura ambiente e dividido entre água e EtOAc, as fases foram separadas e a fase aquosa foi extraída com EtOAc. A fase orgânica combinada foi lavada com água, em seguida sal-moura, secada sobre MgSO4, filtrada e concentrada em vácuo para fornecer o produto bruto. O produto bruto foi purificado por LCMS preparativa como no exemplo 5. Os tubos de produto foram evaporados até a secura. O produto foi dissolvido em DCM (800 μL) e TFA (800 pL); após evaporados até a secura e adicionado metanol (800 pL) e hidróxido de amónio (800 pL). Após 45 minutos, LCMS mostrou a desproteção completa. Os solventes foram evaporados e o produto purificado por LCMS preparativa em pH 2 como no exemplo 5. Os tubos de produto foram combinados e liofilizados até a secura para fornecer (4-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)- 1 H-pirazol-1 -il]etil}pirrolidin-1-il)pindina-2,3-dicarbonitrila como um sal de TFA. 1H RMN (300 MHz, DMSO-d6): δ 12,5 (s, 1H); 8,9 (s, 1H); 8,8 (s, 1H); 8,5 (s, 1H); 8,3 (d, 1H); 7,75 (s, 1H); 7,1 (s, 1H); 6,9 (d, 1H); 4,85 (m, 1H); 3,8 (m, 1H); 3,7 (m, 1H); 3,6 (m, 2H); 3,3 (m, 2H); 2,95 (m, 1H); 1,75 (m, 1H); 1,6 (m, 1H); LCMS calculada para C23H19N10 (M+H)+: m/z = 435,179, observado 434,90. Exemplo 125. Trifluoroacetato de (3S)-3-f(3S)-1-(2,6-dicloropirtdin-3-il)pirroli-din-3-ill-3-|4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-illpropanonitrila

Etapa 1. (3S)-3-[(3S)-1-(2,6-dicloropiridin-3-il)pirrolidin-3-il]-3-[4-(7-{[2-(trime- tilsililjetóxijmetil}- 7H-pirrolo[2,3-dJpirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila
A uma solução de (3S)~3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trime- tilsilil)etóxiJmetil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (18,4 mg, 0,0422 mmol; de exemplo 15, etapa 3) e DIPEA (10,1 μl_, 0,0633 mmol) em NMP (0,1 mL) foi adicionada 2,6-dicloro-3-fluoropiridina (1 equiva-lente) e a reação foi aquecida para 100°C durante 24 horas. A reação foi diluída com MeOH/ACN/água e purificada por LCMS preparativa como no exemplo 5 para separar os dois produtos (3S)-3-[(3S)-1-(6-cloro-5-fluoro- piridin-2-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila e (3S)-3-[(3S)-1-(2,6-dicloro- piridin-3-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila. As frações foram misturadas e evaporadas até a secura para fornecer o produo de dicloro. LCMS calculada para C27H33CI2N8OSÍ (M+H)+: m/z = 583,192, observado 583,05. Etapa 2. trifluoroacetato de (3S)-3-[(3S)-1-(2,6-dicloropiridin-3-il)pirrolidin-3- ilJ-3-[4-(7H-pirrolo[2,3-dJpirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila
À (3S)-3-[(3S)-1-(2,6-dicloropiridín-3-il)pirrolidin-3-il]-3-[4-(7-{[2- (trimetilsilil)etóxiJmetil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila foram adicionados DCM (300 pL) e TFA (200 μL), a reação foi agitada em temperatura ambiente durante 1 hora, evaporada até a secura, em seguida adicionados metanol (300 pL) e hidróxido de amónio (300 pL). Após 45 minutos, LCMS mostrou a desproteção completa. Os solventes foram evaporados e o resíduo foi dissolvido em MeOH/ACN/água e purifica- do por LCMS preparativa em pH 2 como no exemplo 5. Os tubos de produto foram combinados e liofilizados até a secura para fornecer o produto como um sal de TFA. LCMS calculada para C22H19CIN9 (M+H)+: m/z = 453,111, observado 453,00. Exemplo 126. Trifluoroacetato de 5-((3S)-3-{2-fluoro-1-[4-(7H-pirroloí2,3- dJpirimidin-4-il)-1 H-pirazol-1-il1etil)pirrolidin-1-il)-1,3-tiazol-4-carbonitrila
Etapa 1. 5-bromo-1:3-tiazol-4-carbonitπla
A uma mistura de 5-bromo-1,3-tiazol-4-carboxamida (preparada de acordo com o procedimento reportado em W02008/057336 de ácido 5- bromo-1,3-tiazol-4-carboxílico obtido de SynChem; 714 mg, 3,45 mmols)) e trietilamina (7,21 mL, 51,7 mmols) em DCM (10 mL) foi adicionado tricloroa- nidrido acético (6,30 mL, 34,5 mmols) gota a gota a 0°C. A mistura foi agitada a 0°C durante 1 hora. A reação foi extinguida com solução de NaHCO3 aquosa saturada, extraída com DCM, secada sobre MgSO4, filtrada e concentrada em vácuo. O produto bruto foi dissolvido em CHCI3 e aplicado em coluna de sílica-gel de 120 g, eluída para recuperar 593 mg de produto. 1H RMN (300 MHz, CDCI3): δ 8,82 (s, 1H). Etapa 2. 5-((3S)-3-{2-fluoro-1-[4-(7-{[2-(trímetilsilil)etóxÍ]metil}-7H-pirrolo[2,3- d]pirímÍdin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-1,3-tiazol-4-carbonitrila
A mistura de 4-(1-{2-fluoro-1 -[(3S)-pirrolidin-3-il]etil}-1 H-pirazol-4- ÍI)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidina (de exemplo 70, etapa 7; 84,8 mg, 0,000197 mol), 5-bromo-1,3-tiazol-4-carbonitrila (66 mg, 0,00035 mol) e DIPEA (62 pL, 0,00035 mol) em tetrafluoroborato de 1-butil- 3-metil-1H-imidazol-3-io (350 μL, 0,0019 mol) foi aquecida a 120 °C durante 3 horas. Resfriada para temperatura ambiente, dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi extraída com EtOAc. A fase orgânica combinada foi lavada com água, em seguida salmoura, secada sobre MgSO4, filtrada e concentrada em vácuo para fornecer o produto bruto. Este foi dissolvido em CHCI3/hexanos e aplicado em coluna de sílica-gel de 4 g; recuperados 29,5 mg de 5-((3S)-3-{2-fluoro-1-[4-(7-{[2-(trimetil- silil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 - il)-1,3-tiazol-4-carbonitrila. LCMS calculada para C25H32FN8OSSÍ (M+H)+: m/z = 539,217, observado 539,05. Etapa 3. trifluoroacetato de 5-((3S)-3-{2-fluoro-1-[4-(7H-pirrolo[2,3-d]pirimi- din-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-1,3-tiazol-4-carbonitrila
À 5-((3S)-3-{2-fluoro-1-[4-(7-{[2-(trimetilsilil)etóxi]metíl}-7H-pÍrro- lo[2,3-d]piπm id rn-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-1,3-tiazol-4-carbonitrila submetida à cromatografia foram adicionados DCM (0,50 mL) e TFA (0,50 mL), a reação foi agitada em temperatura ambiente durante 2 horas, evaporada até a secura; em seguida adicionados metanol (1 mL) e hidróxido de amónio (1 mL). Deixada agitar durante a noite. Os solventes foram evapora-dos e o resíduo foi dissolvido em MeOH/ACN/água e purificado por LCMS preparativa em pH 2 como no exemplo 129 (meOH/water/TFA). Os tubos de produto foram combinados e liofilizados até a secura para fornecer 5-((3S)-3- {2-fluoro-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)- 1,3-tiazoM-carbonitrila como o sal de bis-TFA (19F RMN). 1H RMN (300 MHz, DMSO-d6): Õ 12,7 (s, 1H); 8,95 (s, 1H); 8,85 (s, 1H); 8,55 (s, 1H); 8,15 (s, 1H); 7,8 (s, 1H); 7,2 (s, 1H); 4,9 (m, 3H); 3,75 (m, 1H); 3,6 (m, 1H); 3,5 (m, 2H); 3,0 (m, 1H); 1,75 (m, 2H); LCMS calculada para C19H18FN8S (M+H)+: m/z = 409,136, observado 409,00. Exemplo 127. Trifluoroacetato de 2-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- dlpirimídin-4-il)-1 H-pirazol-1-il1etil}pirrolidin-1-ilM-(metiltio)nicotinonitrila

Etapa 1. 2-cloro-4-(metiltio)nicotinonitrila 2-cloro-4-iodonicotinonitrila (de exemplo 122, etapa 1; 209,5 mg, 0,7922 mmol) foi dissolvido em 1,4-dioxano (1,85 mL) e mercaptideo de metila de sódio (60,1 mg, 0,871 mmol) foi adicionado. A reação foi agitada em temperatura ambiente. Após 40 horas a mistura reacional foi dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi extraída com EtOAc. A fase orgânica combinada foi lavada com água, em seguida sal-moura, secada sobre MgSO4, filtrada e concentrada em vácuo para fornecer 180 mg do produto bruto. Dissolvido em CHCI3/hexanos e aplicado em coluna de sílica-gel de 40 g; produto recuperado: 52 mg. LCMS calculada para C7H6CIN2S (M+H)+: m/z = 184,994, observado 184,90. Etapa 2. trifluoroacetato de 2-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-4-(metiltio)nicotinonitrila A uma solução de (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsílil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (62 mg, 0,14 mmol; de exemplo 15, etapa 3) em NMP (0,23 mL) foi adicionada 2-cloro-4- (metiltio)nicotinonitrila (52 mg, 0,28 mmol) seguido por DIPEA (35,1 μL, 0,202 mmol), a reação foi tampada e aquecida para 130°C em um banho de óleo durante 3 horas. LCMS mostrou a reação quase completa para o produto desejado. Isolado por LCMS preparativa método acteona/água/em pH 2 (instrumento Waters Fraction-Lynx, coluna C18 de 20 x 100 mm, aceto- na/água (0,1% de TFA), 30 mL/minuto). Os tubos de produto foram evaporados para próximo da secura. Adicionado NaHCO3 em seguida extraído com EtOAc. A fase orgânica foi lavada com água, em seguida salmoura, secada sobre MgSO4, filtrada e concentrada em vácuo para fornecer a 2-((3S)-3- {(1 S)-2-ciano-1 -[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pinmidin-4- il)-1H-pirazol-1-íl]etil}pirrolidin-1-il)-4-(metiltio)nicotinonitrila.
Ao resíduo foi adicionado DCM (400 pL) e TFA (400 pL), a reação foi agitada em temperatura ambiente durante 1 hora, evaporada até a secura, em seguida adicionados metanol (600 pL) e hidróxido de amónio (600 pL), deixada agitar durante a noite. LCMS mostrou desproteção completa. Os solventes foram evaporados e o residuo foi dissolvido em Me- OH/ACN/água e purificado por LCMS preparativa como no exemplo 129 em pH 2, os tubos de produto foram combinados e liofilizados até a secura para fornecer o produto 2-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4- il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)-4-(metiltio)nicotinonitrila como um sal de TFA. LCMS calculada para C23H22N9S (M+H)+: m/z = 456,172, observado 456,00. Exemplo 128. 2-((3S)-3-f(1S)-2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-4-(metilsulfonil)nicotinonitrila
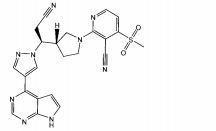
2-((3S)-3-{(1 S)-2-Ciano-1 -[4(7 H-p irrolo[2,3-d]p i rim id i n-4-i I)-1H- pirazol-1 -il]etil}pirrolidin-1 -il)-4-(metiltio)nicotinonitrila (de exemplo 127; 30,0 mg, 0,0658 mmol) foi dissolvida em metanol (0,2 mL) e água (0,2 mL). Oxone® (80,1 mg, 0,132 mmol) foi adicionado e a solução foi agitada em temperatura ambiente durante 4 horas. LCMS mostrou a oxidação quase completa em sulfona (algum sulfóxido restante), e nenhuma evidência de superoxida- çâo. A reação foi concentrada em vácuo e resíduo foi dissolvido em DM- SO/MeOH; insolúveis foram removidos por filtragem, em seguida o filtrado foi purificado por LCMS preparativa como no exemplo 5, ACN/água em pH 2 para recuperar a 2-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)- 1 H-pirazol-1-il]etil}pirrolidin-1-il)-4-(metilsulfonil)nicotinonitrila. 1H RMN (500 MHz, DMSO-d6): δ 12,5 (s, 1H); 8,95 (s, 1H); 8,8 (s, 1H); 8,55 (d, 1H); 8,5 (s, 1H); 7,7 (s, 1H); 7,2 (d, 1H); 7,1 (s, 1H); 4,9 (m, 1H); 4,0 (m, 1H); 3,8 (m, 1H); 3,7 (m, 2H); 3,4 (m, 1H); 3,35 (s, 3H); 3,3 (m, 1H); 2,9 (m, 1H); 1,7 (m, 2H); LCMS calculada para C23H22N9O2S (M+H)+: m/z = 488,162, observado 487,90. Exemplo 129. Trifluoroacetato de (3S)-3-f(3S)-143,5-difluoro-6- (metiltio)piπdin-2-iljpirrolidin-3-il}-3-[4-(7H-pirrolo[2,3-dlpirimidin-4-il)-1H-pirazol-1 –iljpropanonitrila

Etapa 1. 2,3,5-trifluoro-6-(metiltio)pindina 2,3,5,6-tetrafluoropiridina (310 μL, 3,0 mmols) foi dissolvida em THF (2,0 mL) e resfriada para 0°C. A esta solução foi gradualmente adicionado mercaptídeo de metila de sódio (227,8 mg, 3,25 mmols) em MeOH (1 mL). A reação foi mantida a 0°C durante 70 minutos em cujo tempo, a análise de HPLC indicou a conclusão da reação. A mistura reacional foi dividida entre água e Et20, as fases foram separadas e a fase aquosa foi lavada com Et20 adicional. A fase orgânica combinada foi lavada com água seguido por salmoura, em seguida secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto, 424 mg. Este material foi usado diretamente na etapa seguinte . 1H RMN (300 MHz, CDCI3): δ 7,29 (m, 1H), 2,53 (s, 3H). Etapa 2. (3S)-3-{(3S)-1-[3,5-difluoro-6-(metiltio)piridin-2-il]pirrolidin-3-il}-3-[4- (7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidín-4-il)-1 H-pirazol-1 - iljpropanonitrila
A uma solução de (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(tnme- tilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (86 mg, 0,20 mmol; de exemplo 15, etapa 3) em NMP (0,32 mL) foi adicionada 2,3,5-trifluoro-6-(metiltio)piridina (70,1 mg, 0,391 mmol) seguido por DIPEA (48,8 μL, 0,280 mmol), a reação foi tampada e aquecida para 100°C em um banho de óleo durante 3,5 horas, em cujo tempo a análise de LCMS indicou a conclusão da reação. A reação foi resfriada para temperatura ambiente e diluída com metanol e acetonitrila (solvente total adicionado: 2 mL) e purificada por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 μm, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), o produto foi isolado como um sal de TFA, 43 mg. LCMS calculada para C28H35F2N8OSSÍ (M+H)+: m/z = 597,239, observado 597,30. Etapa 3. trifluoroacetato de (3S)-3-{(3S)-1-[3,5-difluoro-6-(metiltio)piridin-2- il]pirrolidin-3-il}-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropano-nitrila À (3S)-3-{(3S)-1 -[3,5-dífluoro-6-(metiltio)pindín-2-il]pirrolidin-3-il}- 3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]propanonitrila (43 mg, 0,072 mmol) foram adicionados DCM (0,5 mL) e TFA (0,5 mL). A reação foi agitada em temperatura ambiente durante 1 hora, em seguida os solventes foram removidos em vácuo e metanol (0,5 mL) e NH4OH (0,5 mL) foram adicionados. Após 30 minutos, análise de LCMS indicou remoção completa de grupo SEM. Os solventes foram removidos e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 μ, 30 x100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), o produto foi isolado como um sal de TFA, 18,9 mg. 1H RMN (300 MHz, DMSO-d6): δ 12,58 (bs, 1H), 8,92 (s, 1H), 8,75 (s, 1H), 8,47 (s, 1H), 7,71 (s, 1H), 7,58 (m, 1H), 7,88 (m, 1H), 4,77 (m, 1H), 3,75 (m, 1H), 3,50 (m, 1H), 3,40 (m, 3H), 3,31 (m, 2H), 2,82 (m, 1H), 2,42 (s, 3H), 1,57 (m, 2H); LCMS calculada para C22H21F2N8S (M+H)+: m/z = 467,158, observado 466,95. Exemplo 130. Trifluoroacetato de (3S)-3-{(3S)-1-[3,5-difluoro-6-(metilsul- fonil)piridin-2-il]pirrolidin-3-il}-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il]propanonitrila

(3S)-3-{(3S)-1-[3,5-difluoro-6-(metiltio)piridin-2-il]pirrolidin-3-il}-3- [4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (de exemplo 129; 45 mg, 0,096 mmol) foi dissolvida em água (0,3 mL) e Oxone® (119 mg, 0,193 mmol) foi adicionado, a solução foi agitada em temperatura ambiente durante 3,5 horas, em cujo tempo a análise de LCMS indicou a presença de sulfóxido e sulfona superoxidada em N-óxido. A mistura reacional foi dividida entre água e 3:1 de CHCI3/IPA. A fase orgânica foi concentrada em vácuo e o resíduo foi dissolvido em MeOH e DMSO (~4 mL total). A mistura foi filtrada para remover sólidos insolúveis e o filtrado foi purificado por HPLC preparativa de fase reversa para recuperar o produto. O produto foi dissolvido em EtOH (3,0 mL) e Pd/C a 10% (15 mg) foi adicionado e a reação foi hidrogenada em um agitador Parr a 1,4 kg/cm2 de H2 durante 1 hora em cujo tempo a análise de LCMS indicou redução de N-óxido. A reação foi filtrada e concentrada em vácuo para fornecer o produto bruto, que foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 p, 30 x100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), para recuperar a sulfona produto como um sal de TFA, 2,0 mg. LCMS calculada para C22H21F2N8O2S (M+H)+: m/z = 499,147, observado 499,20.
Os exemplos na seguinte tabela foram feitos por procedimentos análogos àqueles usados para preparar o exemplo 130.
Exemplo 134. Trifluoroacetato de (3S)-3-{(3S)-1-[2,5-difluoro-6-(metilsulfo- nil)piridin-3-il]pirrolidin-3-il}-344-(7H-pirrolo|2,3-d]pirimidin-4-il)-1 H-pirazol-1- iljpropanonitrila

Etapa 1. 2,3:5-trifluoro-6-(metilsulfonil)piridina 2,3,5-trifluoro-6-(metiltio)piridina (74 mg, 0,42 mmol) foi dissolvida em DCM (5 mL) e ácido m--cloroperbenzoico (208 mg, 0,904 mmol) foi adicionado. A solução foi agitada em temperatura ambiente durante 4 horas, em cujo tempo a análise de HPLC indicou a conclusão da reação. A mistura reacional foi dividida entre água e Et20, as fases foram separadas e a fase aquosa foi lavada com Et20 adicional. A fase orgânica combinada foi lavada com NaHSO3 saturado, NaHCO3 saturado, água, em seguida salmoura, e foi secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto como um sólido branco, 80 mg. O produto foi usado na etapa seguinte sem outra purificação. Etapa 2. (S)-3-((S)-1-(2,5-difluoro-6-(metilsulfonil)piridin-3-il)pirrolidin-3-il)-3- (4-(7-((2-(trimetilsilil)etóxi)metil)-7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1- iljpropanonitrila
A uma solução de (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trime- tilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (26 mg, 0,059 mmol; de exemplo 15, etapa 3) em NMP (0,096 mL) foi adi-cionada 2,3,5-trifluoro-6-(metilsulfonil)piridina (25,1 mg, 0,119 mmol) seguido por DIPEA (14,8 μL, 0,0851 mmol). A reação foi tampada e aquecida para 100°C em um banho de óleo durante 1,5 hora, em cujo tempo a análise de LCMS indicou a conclusão da reação. A reação foi resfriada para temperatura ambiente e diluída com metanol e acetonitrila (solvente total adicionado: 2 mL) e purificada por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), para fornecer o produto. LCMS calculada para C28H35F2N8O3SSÍ (M+H)+: m/z = 629,229, observado 629,00. Etapa 3. Trifluoroacetato de (3S)-3-{(3S)-1-[2,5-difluoro-6-(metilsulfonil)piri- din-3-il]pirrolidin-3-il}-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila
À (S)-3-((S)-1-(2,5-difluoro-6-(metilsulfonil)piridin-3-il)pirrolidin-3- il)-3-(4-(7-((2-(trimetilsilil)etóxi)metil)-7H-pirrolo[2,3-d]pirimidin-4-il)-1H- Y <■ pirazol-1-iljpropanonitrila foram adicionados DCM (0,5 mL) eTFA (0,5 mL). A reação foi agitada em temperatura ambiente durante 1 hora, em seguida os solventes foram removidos em vácuo e metanol (0,5 mL) e NH4OH (0,5 mL) foram adicionados. Após 30 minutos, análise de LCMS indicou remoção completa de grupo SEM. Os solventes foram removidos e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Waters Sun-Fire C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), o produto foi isolado como urn sal de TFA, 14 mg. 1H RMN (300 MHz, DMSO- d6): δ 12,60 (bs, 1H), 8,98 (s, 1H), 8,80 (s, 1H), 8,52 (s, 1H), 7,74 (s, 1H), 7,11 (s, 1H), 7,85 (m, 1H), 4,82 (m, 1H), 3,75 (m, 1H), 3,56 (m, 1H), 3,20 (m, 4H), 3,19 (s, 3H), 2,90 (m, 1H), 1,65 (m, 2H); LCMS calculada para C22H21F2N8O2S (M+H)+: m/z = 499,148, observado 498,95. Exemplo 136. Trifluoroacetato de 2-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- dlpirimidiπ-4-il)-1H-pirazol-1-il]etil}pirrolidin-1-il)-4-(1-fluoroetil)nicotinonitrila

Etapa 1. 2-cloro-4-(1-etoxivinil)nicotinonitrila
Uma solução de 2-cloro-4-iodonicotinonitrila (de exemplo 122, etapa 1; 240,0 mg, 0,9075 mmol) e tributil(1-etoxivinil)estanho (398,58 μL, 1,1798 mmol) em tolueno (3,2 mL) foi desgaseificada, e tetracis(trifenilfos- fina)paládio(O) (104,9 mg, 0,09075 mmol) foi adicionado. A reação foi desgaseificada novamente e aquecida para 100°C durante 16 horas, A mistura reacional foi dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com salmoura, secada sobre MgSO4 e filtrada e concentrada em vácuo para fornecer o produto bruto, 195,6 mg. O produto bruto foi submetido à cromatografia em coluna de 40 g, recuperados 143 mg de produto. 1H RMN (300 MHz CDCI3): δ 8,51 (d, 1H), 7,44 (d, 1H), 4,85 (d, 1H), 4,60 (d, 1H), 3,97 (q, 2H), 1,42 (t, 3H). Etapa 2. 4-acetíl-2-cloronicotinonitríla 2-cloro-4-(1-etoxivinil)nicotinonitrila (143 mg, 0,685 mmol) foi dissolvida em THF (9,1 mL) e cloreto de hidrogênio a 3,0 M em água (5,7 mL, 17 mmols) foi adicionado, a reação foi agitada a 25 °C durante 20 horas, em cujo tempo a análise de LCMS mostrou o progresso da hidrólise, porém nâo conclusão. A mistura reacional foi dividida entre água e EtOAc, e a fase aquosa foi extraída com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida salmoura, secada sobre MgSO4 e filtrada e concentrada em vácuo para fornecer o produto bruto. Este foi em seguida 5 dissolvido em CHCI3/hexanos e aplicado em coluna ISCO de 12 g e submetido à cromatografia para fornecer o produto bruto, 95,6 mg. 1H RMN (300 MHz CDCI3): δ 8,76 (d, 1H), 2,71 (s, 3H). Etapa 3. 4-acetil-2-((3S)-3-{( 1 S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1 -il)nicotinonitríla
A uma solução de 4-acetil-2-cloronicotinonitrila (95 mg, 0,53 mmol) em NMP (0,85 mL) foi adicionada (3S)-3-[(3S)-pirrolidin-3-il]-3-[4-(7- {[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila (230 mg, 0,53 mmol; de exemplo 15, etapa 3) seguido por DIPEA (131 μL, 0,754 mmol), a reação foi tampada e aquecida para 100°C em um banho de óleo durante 2 horas. A reação foi resfriada para temperatura ambiente e dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida salmoura, secada sobre MgSO4 e filtrada e concentrada em vácuo para fornecer o produto bruto, 320 mg. O produto bruto foi dissolvido em CHCI3/hexanos e aplicado em coluna ISCO de 40 g, e foi submetido à cromatografia para fornecer o produto (98,5 mg), 4-acetil- 2-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)nicotinonitrila. MS(EI): 582 (M+1). Etapa 4. 2-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirro- lo[2,3-d]pirimidin-4-il)~ 1 H-pirazol- 1-il]etil}pirrolidin-1-il)-4-( 1 -fluoroetil)nicotino- nitrila
À 4-acetil-2-((3S)-3-{(1 S)-2-ciano-1 -[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)nicotinonitri- la (50,0 mg, 0,0859 mmol) em metanol (0,5 mL) resfriada para 0°C foi adicionado tetraidroborato de sódio (6,50 mg, 0,172 mmol), e foi agitada durante 10 minutos. A reação foi extinguida com HCI a 1N para o pH acídico (lotes de desprendimento de gás), em seguida neutralizada com NaHCO3 sólido para o pH 8, extraída 2x com EtOAc. A fase de EtOAc foi lavada com salmoura, secada sobre MgSO4 e filtrada. A fase de EtOAc foi evaporada até a secura para fornecer álcool bruto MS(EI): 584 (M+1). A uma mistura de 25 ((3S)-3-{(1 S)-2-ciano-1 -[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pi-rimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-4-(1-hidroxietil)nicotinonitrila em DCM (0,89 mL) foi adicionada 2-metóxi-N-(2-metoxietil)-N-(trifluoro-À(4)- sulfanil)etanamina (47,5 μL, 0,258 mmol) seguido por uma gota de etanol (9,4 μL, 0,16 mmol). A reação foi agitada em temperatura ambiente durante 10 5,5 horas e foi dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida salmoura, secada sobre MgSO4 e filtrada e concentrada em vácuo para fornecer o produto bruto, 41,3 mg. O produto bruto foi dissolvido em MeOH/ACN e purificada por LCMS preparativa como 15 no exemplo 129, para fornecer 26,6 mg de fluoreto purificado. Etapa 5. trifluoroacetato de 2-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-4-( 1 -fíuoroetil)nicotinonitrila
À 2-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolídin-1 -il)-4-(1- fluoroetil)nicotinonitrila purificada foram adicionados DCM (0,1 mL) e TFA (0,1 mL, 0,02 mol), após 1 hora o solvente foi removido e adicionados metanol (0,1 mL) e hidróxido de amónio (0,1, 0,03 mol), e foi agitada durante 20 minutos, e o resíduo após remoção do solvente foi dissolvida em ACN/MeOH e purificada por LMCS preparativa para recuperar o produto como um sal de TFA. MS(EI): 456 M+1). 1H RMN (300 MHz DMSO-d6): δ 12,5 (brs, 1H), 9,02 (s, 1H), 8,83 (s, 1H), 8,55 (s, 1H), 8,35 (dd, 1H), 7,82 (m, 1H), 7,18 (m, 1H), 6,80 (dd, 1H), 5.88 (m, 1H), 4,80 (m, 1H), 3,2,00^,00 (m, 6H), 2,90 (m, 1H), 1,70 (m, 2H), 1,60 (m, 3H). Exemplo 138. trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-ir]etil}pirrolidin-1-il)-6-(difluorornetil)pirazina-2-carbonitrila

Etapa 1. 3-amino-6-bromopirazina-2-carbonitrila
Uma mistura de NaCN (140 mg, 2,85 mmols) e cianeto de cobre (I) (255 mg, 2,85 mmols) em DMF anidroso (13 mL) foi agitada a 120°C du- 5 rante 20 minutos sob uma atmosfera de N2. À solução clara resultante foi adicionada gota a gota uma solução de 3,5-dibromopirazin-2-amina (de Aldrich; 800 mg, 3,16 mmols) em DMF (4,8 mL) e agitação foi continuada a 120°C. A reação foi mantida a 120°C durante 40 h em cujo tempo a análise de LCMS indicou conversão completa. A reação foi resfriada para tempera- * 10 tura ambiente e dividida entre água e EtOAc, as fases foram separadas e a ,♦ fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com água seguido por salmoura, em seguida secada sobre MgSO4 e • concentrada em vácuo para fornecer o produto bruto, 597,1 mg, que foi usado sem qualquer outra purificação. Etapa 2. 6-bromo-3-cloropirazina-2-carbonitríla
A uma solução de 3-amino-6-bromopirazina-2-carbonitrila (587 mg, 2,95 mmols) em acetonitrila (29,4 mL) foi adicionada cloreto de cobre (I) (470 mg, 3,5 mmols). A reação foi aquecida para 60°C durante 10 minutos, em seguida nitrito de t-butila (510 μL, 4,3 mmols) foi adicionado gota a gota.
A reação foi mantida a 60°C durante 16 horas em cujo ponto LCMS indicou a conclusão da reação. A reação foi resfriada para temperatura ambiente e dividida entre HCI a 1N e EtOAc e as fases foram separadas. A fase orgânica foi lavada 2x com água seguido por salmoura, em seguida secado sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto que se crista- 25 lizou sob repouso. O produto foi purificado (120 g de cartucho de SÍO2 pré- empacotado, 85 mL/minuto, gradiente de 0 a 20% de EtOAc/hexanos duran- te 12 minutos) para recuperar o composto desejado, 442 mg. 1H RMN (300 MHz, CDCI3): õ 8,68 (s, 1H). Etapa 3. 3-cloro-6-[(E)-2-etoxivinil]pírazina-2-carbonitríla
Uma solução de 6-bromo-3-cloropirazina-2-carbonitrila (220 mg, 5 0,11 mmol) e (2-etoxietenil)tri-n-butiltina (434 pL, 1,31 mmol) em tolueno (1,8 mL) foi desgaseificada com N2 e tetracis(trifenilfosfina)paládio(0) (67,8 mg, 0,0587 mmol) foi adicionado. A reação foi desgaseificada novamente e a- quecida para 100°C durante 16 horas em cujo tempo LCMS indicou conversão completa no produto desejado. A reação foi resfriada para temperatura 10 ambiente e dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com água seguido por salmoura, em seguida secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto. Este foi purificado (40 g de cartucho de SiO2 pré-empacotado, 40 mL/minuto, gradiente de 0 a 15 50% de EtOAc/hexanos durante 20 minutos) para recuperar o produto, 134 mg. 1H RMN (300 MHz, CDCI3): δ 9,19 (s, 1H), 6,69 (d, 1H), 5,52 (d, 1H), 1,44 (m, 2H), 1,41 (t, 3H); LCMS calculada para C9H9CIN3O(M+H)+: m/z = 210,043, observado 209,9. Etapa 4. 3-cloro-6-formilpirazina-2-carbonitrila
À 3-cloro-6-[(E)-2-etoxivinil]pirazina-2-carbonitrila (70,5 mg, 0,303 mmol) foi adicionado 1,4-dioxano (8,8 mL), água (2,2 mL), e periodato de sódio (190 mg, 0,91 mmol), seguido por uma solução a 4% de tetraóxido de ósmio em água (66,7 pL, 0,0105 mmol). A reação foi agitada em temperatura ambiente durante 16 horas, em cujo tempo a análise de TLC indicou a 25 conclusão da reação. A mistura reacional foi dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida salmoura, secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto. Este foi purificado (12 g de cartucho de SiO2 pré-empacotado, 30 mL/mi- 30 nuto, gradiente de 0 a 50% de EtOAc/hexanos durante 16 minutos) para recuperar o produto como a crystalline solid, 43,2 mg. 1H RMN (300 MHz, CDCI3). δ 10,12 (s, 1H), 9,11 (s, 1H). Etapa 5. 3-cloro-6-(difluorometil)pirazina-2-carbonitríla À 3-cloro-6-formilpirazina-2-carbonitrila (30,1 mg, 0,185 mmol) em DCM (1,9 mL) foi adicionada 2-metóxi-N-(2-metoxietil)-N-(trifluoro-À(4)- sulfanil)etanamina (100 μL, 0,55 mmol) seguida por uma gota de etanol. A 5 reação foi mantida em temperatura ambiente durante 16 horas, em cujo tempo a análise de TLC (3:1 hexanos:EtOAc) indicou a conclusão da reação. A mistura reacional foi dividida entre água e CHCI3. As fases foram separadas e a fase aquosa foi lavada com CHCI3 adicional. A fase orgânica combinada foi lavada com salmoura, secada sobre MgSO4 e concentrada em vá- 10 cuo para fornecer o produto, 43 mg. O produto foi usado sem outra purificação. 1H RMN (400 MHz, CDCI3): δ 8,92 (s, 1H), 6,73 (t, 1H). Etapa 6. 3-((3S)-3-{(1 S)-2-ciano-1 -[4-(7-{[2-(trímetilsilil)etóxi]metil}-7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)-6-(difluorometil) pirazi- na-2-carbonitríla
A uma solução de 3-cloro-6-(difluorometil)pirazina-2-carbonitrila (30,0 mg, 0,158 mmol) em NMP (257 μL) foi adicionada (3S)-3-[(3S)- pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2>3-d]pirimidin-4- il)-1 H-pirazol-1-iljpropanonitrila (69 mg, 0,16 mmol; de exemplo 15, etapa 3) seguido por DIPEA (39,5 μL, 0,227 mmol). A reação foi selada e aquecida 20 para 100°C durante 1 hora em cujo tempo a análise de LCMS indicou a conclusão da reação. A reação foi resfriada para temperatura ambiente e dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com água, em seguida salmoura, secada sobre MgSO4 e concentrada em vácuo para for- 25 necer o produto bruto. Este foi purificado (4 g de cartucho de SiO2 pré- empacotado, 20 mL/minuto, gradiente de 10 a 90% de EtOAc/hexanos durante 16 minutos) para recuperar o produto, 49 mg. LCMS calculada para C28H33F2N10OSÍ (M+H)+: m/z = 591,257, observado 591,05. Etapa 7. Trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3-d]pi- 30 rimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-6-(difluorometil)pirazina-2-car-bonitrila
À 3-((3S)-3-{( 1 S)-2-ciano-1 -[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-ÍI)-6-(difluorometil) pirazina-2-carbonitrila (49 mg, 0,083 mmol) foram adicionados DCM (0,5 mL) e TFA (0,5 mL). A reação foi agitada em temperatura ambiente durante 1 hora, em seguida os solventes foram removidos em vácuo e metanol (0,5 mL) e NH4OH (0,5 mL) foram adicionados. Após 30 minutos análise de LCMS indicou a remoção completa de grupo SEM. Os solventes foram removidos e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), o produto foi isolado como um sal de TFA, 34 mg. 1H RMN (400 MHz, CD3OD). δ 9,00 (s, 1H), 8,89 (s, 1H), 8,57 (s, 1H), 8,51 (s, 1H), 7,86 (d, 1H), 7,28 (d, 1H), 6,63 (t, 1H), 4,90 (m, 2H), 1,45 (m, 1H), 3,97 (m, 1H), 3,75 (m, 2H), 3,22 (m, 1H), 3,12 (m, 1H), 3,09 (m, 1H), 1,91 (m, 2H). LCMS calculada para C22H19F2N10 (M+H)+: m/z = 461,18, observado 460,90. Exemplo 139. Trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin- 4-íl)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-6-(2, 2-difluoroetil)pirazina-2-

Etapa 1. 3-cloro-6-(2-oxoetil)pirazina-2-carbonitríla 3-cloro-6-[(E)-2-etoxivinil]pirazina-2-carbonitrila (de exemplo 138, etapa 3; 395 mg, 1,88 mmol) foi dissolvida em THF (25 mL) e HCI a 3,0 M (16 mL, 47 mmols) foi adicionado. A reação foi aquecida para 60°C durante 25 3,5 horas em cujo tempo LCMS indicou a conclusão da reação. A mistura reacional foi dividida entre NaHCO3 aquoso saturado e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com água seguido por salmoura, em seguida secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bru- 5 to, 417 mg. Este material foi usado diretamente na reação seguinte no estado em que foi encontrado para decompor-se sob repouso. 1H RMN (300 MHz, CDCI3): δ 9,91 (s, 1H), 8,53 (s, 1H), 4,08 (s, 2H). Etapa 2. 3-cloro-6-(2,2-difluoroetil)pirazina-2-carbonitrila 3-cloro-6-(2-oxoetil)pirazina-2-carbonitrila recentemente prepa- 10 rada (90,0 mg, 0,496 mmol; foi dissolvida em DCM (5,1 mL) e 2-metóxi-N-(2- metoxietil)-N-(trifluoro-À(4)-sulfanil)etanamina (274 pL, 1,49 mmol) foi adicionada seguido por uma gota de etanol. A reação foi agitada em temperatura ambiente durante 16 horas em cujo tempo TLC e análise de LCMS indicaram a conclusão da reação. A mistura reacional foi dividida entre água e 15 CHCI3. As fases foram separadas e a fase aquosa foi lavada com CHCI3 adicional. A fase orgânica combinada foi lavada com salmoura, secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto, 109 mg. Este material foi usado diretamente na etapa seguinte. 1H RMN (300 MHz, CDCI3): δ 8,49 (s, 1H), 6,12 (tt, 1H), 3,38 (m, 2H). Etapa 3. Trifluoroacetato de 3-((3S)-3-{(1S)~2-ciano-1-[4-(7-{[2-(trimetilsilil) etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1-il]etil}pirrolidin-1-il)-6- (2,2-difluoroetil) pirazina-2-carbonitríla
A uma solução de 3-cloro-6-(2,2-difluoroetil)pirazina-2-carbo- nitrila (100 mg, 0,49 mmol) em NMP (400 μL) foi adicionada (3S)-3-[(3S)- 25 pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4- il)-1 H-pirazol-1-iljpropanonitrila (110 mg, 0,24 mmol; de exemplo 15, etapa 3) seguido por DIPEA (61,3 pL, 0,352 mmol). A reação foi tampada e aquecida para 100°C em um banho de óleo durante 1,5 hora em cujo tempo a análise de LCMS indicou a conclusão da reação. A reação foi resfriada para tempe- 30 ratura ambiente e diluída com metanol e acetonitrila (solvente total adicionada: 2 mL) e purificada por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Wa- ters SunFire C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/mi-nuto), o produto foi isolado como um sal de TFA, 19 mg. LCMS calculada para C29H35F2N10OSÍ (M+H)+: m/z = 605.273, observado 605,00. Exemplo 140. trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- dlpirimidin-4-il)-1H-pirazol-1-il]etil}pirrolidin-1-il)-6-(hidroximetil)pirazina-2-carbonitrila

Etapa 1. cloro-6-(hidroximetil)pirazina-2-carbonitrila
À 3-cloro-6-formilpirazina-2-carbonitrila (de exemplo 138, etapa 4; 20,0 mg, 0,12 mmol) foi adicionado éter (0,79 mL). A mistura foi resfriada para -78°C e borano a 0,1 M em THF (140 μL, 0,14 mmol) foi adicionado. A reação foi mantida a -78°C durante 1,5 hora, e foi em seguida extinguida por adição de HCI a 0,1N a -78°C. A solução foi deixada aquecer e EtOAc foi adicionado. As fases foram separadas e a fase aquosa foi neutralizada com NaHCO3 e lavada com EtOAc adicional. A fase orgânica combinada foi lavada com água seguido por salmoura, em seguida secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto, 20 mg. Este material foi levado diretamente para a próxima reação. 1H RMN (300 MHz, CD3OD): δ 8,78 (s, 1H), 4,77 (s, 2H). Etapa 2. 3-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirro- lo[2,3-d]pirimidin-4-il)-1H- pirazol-1-il]etil}pirrolidin-1-il)-6-(hidroximetil) pirazi- na-2-carbonitrila
A uma solução de 3-cloro-6-(hidroximetil)pirazina-2-carbonitrila (20,0 mg, 0,106 mmol) em NMP (172 μL) foi adicionada (3S)-3-[(3S)- pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4- il)-1 H-pirazol-1-iljpropanonitrila (46 mg, 0,11 mmol; de exemplo 15, etapa 3) seguido por DIPEA (26,5 μL, 0,152 mmol), a reação foi tampada e aquecida para 100°C em um banho de óleo durante 1 hora em cujo tempo a análise de LCMS indicou a conclusão da reação. A solução de reação crua foi diluída com MeOH/ACN (solvente total adicionado: 2 mL) e purificada por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto). O produto purificado foi liofilizado até a secura para recuperar o produto como um sal de TFA, 34 mg. LCMS calculada para C28H32N10O2SÍ (M+H)+; m/z = 571,271, observado 570,10. Etapa 3. trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-ii)-6-(hidroximetil)pirazina-2- carbonitríla
À 3-((3S)-3-{( 1 S)-2-ciano-1 -[4-(7-{[2-(trimetiIsilíl) etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1H- pirazol-1 -il]etil}pirrolidin-1-il)-6-(2,2-difluoroetil) pirazina-2-carbonitrila (12 mg, 0,02 mmol) foram adicionados DCM (0,5 mL) e TFA (0,5 mL). A reação foi agitada em temperatura ambiente durante 1 hora, em seguida os solventes foram removidos em vácuo e metanol (0,5 mL) e NH4OH (0,5 mL) foram adicionados. Após 30 minutos análise de LCMS indicou remoção completa de grupo SEM. Os solventes foram removidos e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), o produto foi isolado como um sal de TFA, 4,9 mg. LCMS calculada para C22H21N10O (M+H)+; m/z = 441,190, observado 441,00. Exemplo 141. Trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirroloF2,3- dJpirimidin-4-il)-1 H-pirazol-1-ilJetil}pirrolidin-1-il)-6-(metoximetil)pirazina-2- carbonitrila

3-((3S)-3-{(1S)-2-Ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pi rrolo[2,3-d]p i rim id i n-4-i I)-1 H-pirazol-1 -i l]eti l}p i rrol id in-1 -í l)-6- (hidroximetil)pirazina-2-carbonitrila (Exemplo 140; 24,3 mg, 0,0426 mmol) foi combinada com óxido de prata (II) (52,7 mg, 0,426 mmol) e iodometano (53,0 μL, 0,852 mmol) em THF (0,2 mL). A reação foi aquecida em vaso selado durante 3,5 horas em cujo tempo a análise de LCMS indicou a conclusão da reação. A reação foi resfriada para temperatura ambiente e filtrada através de um filtro de Teflon de 0,45. O filtro foi enxaguado com metanol e o filtrado orgânico resultante foi concentrado em vácuo para fornecer o produto bruto. Ao produto bruto foram adicionados DCM (0,5 mL) e TFA (0,5 mL). A reação foi agitada em temperatura ambiente durante 1 hora, em seguida os solventes foram removidos em vácuo e metanol (0,5 mL) e NH4OH (0,5 mL) foram adicionados. Após 30 minutos, análise de LCMS indicou remoção completa de grupo SEM. Os solventes foram removidos e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), o produto foi isolado como um sal de TFA, 5,5 mg. LCMS calculada para C23H23N10O (M+H)+: m/z = 455,206, observado 455,0. Exemplo 142. trifluoroacetato de 6-bromo-3-((3S)-3-{(1S)-2-ciano-1-[4-(7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)pirazina-2-carbo- nitrile

Etapa 1. 6-bromo-3-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trímetilsilil) etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol- 1-il]etil}pirrolidin-1-il)pirazina-2- carbonitrila
A uma solução de 6-bromo-3-cloropirazina-2-carbonitrila (Exemplo 138 Etapa 2; 29 mg, 0,13 mmol) em NMP (216 μL) foi adicionada (3S)-3- [(3S)-pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pírimi- din-4-il)-1 H-pirazol-1 -iljpropanonitrila (58 mg, 0,13 mmol; de exemplo 15, etapa 3) seguido por DIPEA (33,1 μL, 0,190 mmol). A reação foi tampada e aquecida para 100°C em um banho de óleo durante 1 hora em cujo tempo a análise de LCMS indicou a conclusão da reação. A reação foi resfriada para temperatura ambiente e foi dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com água, seguido por salmoura, em seguida secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto. O produto foi purificado (4 g de cartucho de SiO2 pré-empacotado, 20 mL/minuto, gradiente de 0 a 75% de EtOAc/hexanos durante 18 minutos) para recuperar o composto desejado, 51 mg. LCMS calculada para C27H32BrN10OSi (M+H)+: m/z = 619,17, observado 618,90. Etapa 2. trifluoroacetato de 6-bromo-3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pir- rolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)pirazina-2-carbonitrila
À 6-bromo-3-((3S)-3-{(1 S)-2-ciano-1 -[4-(7-{[2-(trimetilsilil) etó- xi]metil}-7H-pirrolo[2,3-d]p irim id i n-4-i I)-1 H-pirazol-1 -i l]etil}pirrol id i n-1 -il)pira- zina-2-carbonitrila (51 mg, 0,08 mmol) foram adicionados DCM (0,5 mL) e TFA (0,5 mL). A reação foi agitada em temperatura ambiente durante 1 hora, em seguida os solventes foram removidos em vácuo e metanol (0,5 mL) e NH4OH (0,5 mL) foram adicionados. Após 30 minutos análise de LCMS indi-cou remoção completa de grupo SEM. Os solventes foram removidos e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (co-luna Waters SunFíre C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), o produto foi isolado como um sal de TFA, 34 mg. LCMS cal-culada para C21H18BrN10(M+H)+: m/z = 489,090, observado 489,10. Exemplo 143. Trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimídin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-6-etinilpirazina-2-carbonitrila

Etapa 1. 3-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirro- lo[2f3-d]pirímidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-6-[(trimetilsilil)etinil]pi- razina-2-carbonitrila 6-bromo-3-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etó- xi]metil}-7H-pirrolo[2,3-d]pirimid in-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 - il)pirazina-2-carbonitrila (Exemplo 142; 40,0 mg, 0,0646 mmol) foi dissolvida em DMF (0,323 mL), e Cul (0,969 mg, 0,00509 mmol) foram adicionados. A mistura reacional foi desgaseificada com três ciclos de vácuo/purga de N2, em seguida Et3N (13,34 μL, 0,09568 mmol) e trimetilsililacetileno (18,2 μL, 0,129 mmol) foram adicionado, seguido por cloreto de bis(trifenilfosfi- na)paládio(ll) (1,93 mg, 0,00276 mmol). A reação foi mantida em temperatura ambiente durante 1,5 hora em cujo tempo a análise de LCMS indicou a conclusão da reação. A reação foi tratado com éter e uma solução de 9:1 de NH4CI/NH4OH aquoso saturado e as fases foram separadas. A fase aquosa foi extraída com éter adicional, a solução de éter combinada foi lavada com H2O seguido por salmoura, em seguida secada sobre MgSO4 e concentrada em vácuo fornece o produto bruto. O produto foi purificado (4 g de cartucho de SÍO2 pré-empacotado, 20 mL/minuto, gradiente de 0 a 75% de EtOAc/hexanos durante 18 minutos) para recuperar o composto desejado, 41 mg. LCMS calculada para C32H41N10OSi2(M+H)+: m/z = 637,300, observado 637,10. Etapa 2. 3-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol- 1-il]etil}pirrolidin-1-il)-6-etinilpirazina-2- carbonitrila 3-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pir- rolo[2,3-d]pi rim id i n-4-i I)-1 H-pirazol-1 -i Ijeti l}p i rrolid in-1 -i l)-6-[(trimeti Isili l)eti- nil]pirazina-2-carbonitrila (41 mg, 0,064 mmol) foi dissolvida em THF (50,1 pL), esta solução foi resfriada para 0°C, e água (3,4 pL) foi adicionado, se-guido por adição gota a gota de TBAF a 0,1 M em THF (75 pL, 0,075 mmol). A reação foi deixada aquecer de 0°C a 15°C durante 1 hora em cujo tempo a análise de LCMS indicou a conclusão da reação. A mistura reacional foi divi-dida entre água e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com 3x água, seguido por salmoura, em seguida secados sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto, 44 mg. LCMS calculada para C29H33N10OSi(M+H)+: m/z = 565,261, observado 565,00. Etapa 3. Trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)~ 1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-6-etinilpirazina-2-carbonitrila
À 3-((3S)-3-{(1 S)-2-ciano-1 -[4-(7-{[2-(trimetilsi I il)etóxi]metil}-7 H- pirrolo[2,3-d] p irim id i n-4-i I)-1 H-pirazol-1 -il]etil}pirrolid in-1 -ÍI)-6-etin i Ipi razina-2- carbonitrila (10,8 mg, 0,0191 mmol) foram adicionados DCM (0,5 mL) e TFA (0,5 mL), a reação foi agitada em temperatura ambiente durante 3 horas. A reação foi evaporada até a secura, em seguida metanol (0,5 mL) e NH4OH (0,5 mL) foram adicionados. A reação foi agitada 30 minutos, em cujo tempo LCMS indicou a desproteção completa. Os solventes foram removidos e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (co-luna Waters SunFire C18, tamanho de partícula de 5 p, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), o produto foi isolado como um sal de TFA, 4,8 mg. 1H RMN (300 MHz, DMSO-d6): δ 12,40 (bs, 1H), 8,90 (s, 1H), 8,73 (s, 1H), 8,43 (m, 2H), 7,70 (s, 1H), 7,83 (s, 1H), 4,82 (m, 1H), 3,89 (m, 1H), 3,78 (m, 1H), 3,60 (m, 2H), 3,29 (m, 3H), 2,85 (m, 1H), 1,68 (m, 2H); LCMS calculada para C23H19N10(M+H)+: m/z = 435,179, observado 434,95. Exemplo 144. Trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolídin-1-il)-6-etilpirazina-2-carbonitrila

3-((3S)-3-{(1S)-2-Ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-6-etinilpirazina-2- carbonitrila (Exemplo 143; 33 mg, 0,058 mmol) foi dissolvida em EtOH (2,3 mL) e Pd/C a 10% (8,7 mg, 0,0082 mmol) foi adicionada. A mistura foi hidro- genada em 2,03 kg/cm2 em um agitador Parr durante 3 horas, em cujo tempo a análise de LCMS indicou a redução completa. A reação foi filtrada através de um filtro de Teflon de 0,45 pm e o filtro foi enxaguado com MeOH. O filtrado foi concentrado em vácuo para fornecer o produto. Ao resíduo foram adicionados DCM (0,5 mL) e TFA (0,5 mL). A reação foi agitada em temperatura ambiente durante 1 hora, em seguida os solventes foram removidos em vácuo e metanol (0,5 mL) e NH4OH (0,5 mL) foram adicionados. Após 30 minutos análise de LCMS indicou remoção completa de grupo SEM. Os solventes foram removidos e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), o produto foi isolado como um sal de TFA, 9,8 mg. 1H RMN (300 MHz, DMSO-d6): δ 12,62 (bs, 1H), 8,99 (s, 1H), 8,82 (s, 1H), 8,53 (s, 1H), 8,29 (s, 1H), 7,76 (s, 1H), 7,12 (s, 1H), 4,90 (m, 1H), 3,90 (m, 1H), 3,74 (m, 1H), 3,59 (m, 2H), 3,36 (m, 3H), 2,90 (m, 1H), 2,61 (m, 2H), 1,70 (m, 2H), 1,11 (m, 3H); LCMS calculada para C23H23N10(M+H)+: m/z = 439,211, observado 438,95. Exemplo 145. Trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iπetil}pirrolidin-1-il)-6-metilpirazina-2-carbonitrila

Etapa 1. 3-((3S)-3~{(1S)-2-ciano-1-[4-(7-{[2-(tπmetilsilil) etóxi]metil}-7H-pir- rolo[2,3-d]pirimidin-4-il)-TH- pirazol-1 -il]etil}pirrolidin-1 -il)-6-metilpirazina- 2- carbonitrila 6-bromo-3-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)pirazina-2- carbonitrila (Exemplo 142; 55.8 mg, 0,09 mmol) e tetracis(trifenilfosfina)pa- ládio(O) (1,46 mg, 0,00360 mmol) foram dissolvidos em THF (0,28 mL) e tri- metilaluminio a 2,0 M em tolueno (90,0 μL, 0,180 mmol) foi adicionada gota a gota. A reação foi aquecida para 70°C durante 3,5 horas em cujo tempo a análise de LCMS indicou a conclusão da reação. A reação foi resfriada para temperatura ambiente e foi diluída com tolueno (0,28 mL) e MeOH (71 μL) foi adicionado gota a gota até a reatividade ceder. A reação foi em seguida reaquecida para 70°C durante 10 minutos, em seguida 2 mL de NH4CI satu-rado aquoso foram adicionados, o aquecimento continuou a 70°C durante 10 minutos. A reação foi resfriada para temperatura ambiente e dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com água, seguido por salmoura, em seguida secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto. O produto foi purificado (4 g de cartucho de SiO2 pré-empacotado, 20 mL/minuto, gradiente de 0 a 90% de EtOAc/hexanos durante 20 minutos) para recuperar o composto desejado, 44 mg. LCMS calculada para C28H35N10OSÍ (M+H)+: m/z = 555.276, observado 555,05. Etapa 2. Trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)-6-metilpirazina-2-carbonitrila
À 3-((3S)-3-{(1S)-2-ciano-1-[4-(7-[[2-(trimetilsilil) etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H- pirazol-1 -il]etil}pirrolidin-1-il)-6-metilpirazina- 2- carbonitrila (44 mg, 0,08 mmol) foram adicionados DCM (0,5 mL) e TFA (0,5 mL). A reação foi agitada em temperatura ambiente durante 1 hora, em se-guida os solventes foram removidos em vácuo e metanol (0,5 mL) e NH4OH (0,5 mL) foram adicionados. Após 30 minutos análise de LCMS indicou re-moção completa de grupo SEM. Os solventes foram removidos e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Wa-ters SunFire C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/mi-nuto), o produto foi isolado como um sal de TFA, 25,8 mg. LCMS calculada para C22H21N10(M+H)+: m/z = 425,195, observado 425,20. Exemplo 146. Trifluoroacetato de 3-((3S)-3-{(1S)-2-ciano-1-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-il]etil)pirrolidin-1-il)-6-metilpirazina-2-carbonitrila

Etapa 1. 3-cloro-6-(1-etoxivinil)pirazina-2-carbonitrila
Uma solução de 6-bromo-3-cloropirazina-2-carbonitrila (Exemplo 138 Etapa 2; 39,4 mg, 0,18 mmol) e tributil(1-etoxivinil)estanho (79,2 μL, 0,23 mmol) em tolueno (0,64 mL) foi desgaseificada com três ciclos de vá- cuo/N2, e tetracis(trifenilfosfma)paládio(0) (20,84 mg, 0,01804 mmol) foi adicionado. A reação foi desgaseificada novamente e aquecida para 100°C durante 3 horas, em cujo tempo a análise de LCMS indicou a conclusão da re- ação. A reação foi resfriada para temperatura ambiente e foi dividida entre água e EtOAc, as fases foram separadas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica combinada foi lavada com salmoura, secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto, 119,4 mg. O produto foi purificado (4 g de cartucho de SiO2 pré- empacotado, 20 mL/minuto, gradiente de 0 a 20% de EtOAc/hexanos durante 10 minutos) para recuperar o composto desejado, 19 mg. 1H RMN (300 MHz, CDCI3): δ 8,87 (s, 1H), 5,54 (d, 1H), 4,59 (d, 1H), 4,01 (m, 2H), 1,48 (t, 3H); LCMS calculada para C9H9CIN3O(M+H)+: m/z = 209,85; encontrado 209,85. Etapa 2. 6-acetil-3-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)pirazina-2- carbonitrila
A uma solução de 3-cloro-6-(1-etoxivinil)pirazina-2-carbonitrila (9,0 mg, 0,043 mmol) em NMP (0,0806 mL) foi adicionada (3S)-3-[(3S)- pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4- il)-1 H-pirazol-1-iljpropanonitrila (22 mg, 0,050 mmol; de exemplo 15, etapa 3) seguido por DIPEA (12,4 μL, 0,0710 mmol). A reação foi tampada e aquecida para 100°C em um banho de óleo durante 2 horas, em cujo tempo a análise de LCMS indicou a conclusão da reação. A reação foi resfriada para temperatura ambiente e dividida entre água e EtOAc, as fases foram sepa-radas e a fase aquosa foi lavada com EtOAc adicional. A fase orgânica com-binada foi lavada com água seguido por salmoura, em seguida secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto, 32,8 mg. Este foi purificado (4 g de cartucho de SiO2 pré-empacotado, 20 mL/minuto, gradiente de 0 a 90% de EtOAc/hexanos durante 14 minutos) para recuperar o composto desejado, 22 mg. LCMS calculada para C29H35N10O2Si(M+H)+: m/z = 583,271, observado 583,05. Etapa 3. Trifluoroacetato de 3-((3S)-3-{(1 S)-2-ciano-1 -[4-(7H-pirrolo[2,3-d]pi- rimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-6-( 1 -hidroxietil)pirazina-2-carbo- nitrila
Uma solução de 6-acetil-3-((3S)-3-{(1S)-2-ciano-1-[4-(7-{[2-(tri- metilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pir- rolidin-1-il)pirazína-2-carbonitrila (22 mg, 0,038 mmol) em MeOH (219,6 μL,) foi resfriada para 0°C e tetra id roborato de sódio (2,86 mg, 0,0755 mmol) foi adicionada. A reação foi mantida a 0°C durante 10 minutos, em cujo tempo a análise de TLC indicou redução completa de cetona. A reação foi extinguida com adição gota a gota de HCI a 1N para fornecer pH ~3, em seguida a reação foi neutralizada por adição gradual de NaHCO3 sólido para pH 8. A reação foi extraída 2x com EtOAc e a solução orgânica resultante foi lavada com salmoura, secada sobre MgSO4 concentrada em vácuo para fornecer o produto bruto. LCMS calculada para C29H37N10O2SÍ (M+H)+: m/z = 585,287, observado 585,05. A esta produto foram adicionados DCM (0,5 mL) e TFA (0,5 mL). A reação foi agitada em temperatura ambiente durante 1 hora, em seguida os solventes foram removidos em vácuo e metanol (0,5 mL) e NH4OH (0,5 mL) foram adicionados. Após 30 minutos análise de LCMS indicou remoção completa de grupo SEM. Os solventes foram removidos e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fracionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B: metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), o produto foi isolado como um sal de TFA, 5.2 mg. 1H RMN (400 MHz, CDCI3): δ RMN (300 MHz, CD3OD): δ 8,98 (s, 1H), 8,89 (s, 1H), 8,57 (s, 1H), 8,42 (s, 1H), 7,85 (d, 1H), 7,26 (d, 1H), 4,77 (m, 2H), 4,08 (m, 1H), 3,90 (m, 1H), 3,71 (m, 2H), 3,40 (m, 1H), 3,22 (m, 2H), 3,08 (m, 1H), 1,90 (m, 2H), 1,42 (m, 3H); LCMS calculada para C23H23N10O(M+H)+: m/z = 455.206, observado 454,95. Exemplo 147. Trifluoroacetato de 3-fluoro-5-((3S)-3-{2-fluoro-1-[4-(7H-pir- rolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljetillpirrolidin-l -iDpiridina- 2-carbonitrila

A uma solução de 4-(1 -{2-fluoro-1-[(3S)-pirrolidin-3-íl]etil}-1 H- pirazol-4-il)-7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidina (de exemplo 70, etapa 7; 37 mg, 0,087 mmol) e DIPEA (3,0E1 μL, 0,17 mmol) em NMP (0,7 mL) foi adicionada 3-cloropirazina-2-carbonitrila e a reação foi aquecida para 130°C durante 2 horas, em cujo tempo a análise de LCMS indicou a conclusão da reação. A mistura reacional foi dividida entre água e EtOAc, a fase aquosa foi extraída mais 2x com EtOAc. A fase orgânica combinada foi lavada com salmoura, secada sobre MgSO4 e concentrada em vácuo para fornecer o produto bruto. LCMS calculada para C26H33FN9OSÍ (M+H)+: m/z = 534,256, observado 531,45. O produto bruto foi tratado com DCM (0,5 mL) e TFA (0,5 mL). A reação foi agitada em temperatura ambiente durante 1 hora, em seguida os solventes foram removidos em vácuo e metanol (0,5 mL) e NH4OH (0,5 mL) foram adicionados. Após 30 minutos, análise de LCMS indicou remoção completa de grupo SEM. Os solventes foram removidos e o material residual foi purificado por LCMS preparativa de fase reversa em um sistema Waters Fraction-Lynx usando fra- cionação direcionada à massa (coluna Waters SunFire C18, tamanho de partícula de 5 μ, 30 x 100 mm, fase móvel A: água (0,1% de TFA), B; metanol (0,1% de TFA), taxa de fluxo de 60 mL/minuto), o produto foi isolado como um sal de TFA, 13,4 mg. LCMS calculada para C21H20FN8(M+H)+: m/z = 403,179, observado 401,40. Exemplo 148. 3-{1-[2-(etilsulfonil)piridin-4-il]pirrolidin-3-íl}-3-[3-(7H-pirrolo[2,3- d]pírimidin-4-il)-1 H-pirrol-1-il]propanonitrila (racemato)

Etapa 1. 4-cloro-2-(etilsulfonil)piridina
A uma solução de 2,4-dicloropiridina (0,20 mL, 1,8 mmol) e eta- notiol (0,14 mL, 1,8 mmol) em 1,4-dioxano (1 mL), foi adicionado hidreto de sódio (60% de óleo mineral, 0,074 g, 1,8 mmol), in one portion. A mistura foi agitada em temperatura ambiente durante 3 dias. Água foi adicionada na reação e o produto foi extraído com dietil éter. Os extratos foram secados sobre sulfato de sódio, decantados e o solvente removido por evaporação giratória. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 10% de acetato de etila em hexanos, foi usado para purificação parcial de produto. O produto foi dissolvido em DCM (10 mL) e tratado com ácido M- cloroperbenzoico (0,28 g, 1,1 mmol) e agitada durante 2 horas. A mistura foi lavada com solução de NaHCO3e a camada de DCM foi secada sobre sulfato de sódio, decantada e concentrada. Purificação por cromatografia de coluna rápida, eluindo com um gradiente de 0 a 50% de acetato de etila em hexanos forneceu o produto (48 mg, 12%). 1H RMN (400 MHz, CDCI3): δ 8,65 (d, 1H), 8,11 (d, 1H), 7,56 (dd, 1H), 3,44 (q, 2H), 1,32 (t, 3H); LCMS (M+H)+: 206,0. Etapa 2. 3-{1 -[2-(etilsulfonil)pindin-4-il]pirrolidin-3-il}-3-[3-(7H-pirrolo[2,3- d]pirímidin-4-il)- 1 H-pirrol-1 -iljpropanonitrila
Uma solução de 4-cloro-2-(etilsulfonil)piridina (15 mg, 0,073 mmol) e 3-pirrolidin-3-il-3-[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pi- rimidin-4-il)-1 H-pirrol-1 -iljpropanonitrila (32 mg, 0,073 mmol, de exemplo 33, etapa 3) em NMP (0,20 mL) e 4-metilmorfolina (16 μL) foi aquecida para 120°C no micro-ondas durante 15 minutos. A mistura reacional foi dividida entre água e acetato de etila. As camadas foram separadas e a camada a-quosa foi extraída um total de três vezes com acetato de etila. Os extratos foram secados sobre sulfato de sódio, decantados e concentrados. O produto bruto foi desprotegido por agitação em 50% de TFA/DCM durante 1 hora, evaporado e em seguida agitado com excesso de EDA em metanol. O produto foi purificado por HPLC-MS preparativa, eluindo com um gradiente de ACN e H2O contendo 0,15% de NH4OH, congelado e liofilizado para fornecer o produto como a base livre (11 mg, 31%). 1H RMN (400 MHz, DMSO- d6): δ 11,96 (br s, 1H), 8,61 (s, 1H), 8,25 (d, 1H), 8,01 (t, 1H), 7,52 (d, 1H), 7,16 (t, 1H), 7,86 (br s, 1H), 6,95 (dd, 1H), 6,94 (d, 1H), 6,68 (dd, 1H), 4,54 (td, 1H), 3,63 (dd, 1H), 3,52-3,22 (m, 7H), 2,97-2,85 (m, 1H), 1,76-1,59 (m, 2H), 1,10 (t, 3H); LCMS (M+H)+: 476,1. Exemplo 149. 5-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d)pirimidin-4-il)-1H-pirrol-1- il]etil}pirrolidin-1-il)-1,3-tiazol-4-carbonitrila (tanto enantiômeros racêmicos quanto simples isolados)

Etapa 1. 5-bromo-1,3-tiazol-4-carbonitrila
A uma mistura de 5-bromo-1,3-tiazol-4-carboxamida (preparado de acordo com o procedimento reportado em W02008/057336 de ácido 5- bromo-1,3-tiazol-4-carboxílico obtido de SynChem; 2,75 g, 13,3 mmols) e trietilamina (9,26 mL, 66,4 mmols) em DCM (50 mL) foi adicionado anidrido tricloroacético (7,28 mL, 39,8 mmols) gota a gota a 0°C. A mistura foi agitada a 0°C durante 1 hora. A reação foi extinguida pela adição de solução de NaHCO3 aquosa saturada, extraída com DCM, secada sobre MgSO4, concentrada e purificado em sílica-gel (eluindo com um gradiente de 0 a 20% de EtOAc/Hexanos) para fornecer o produto (2,19 g, 87%). LCMS (M+H)+: 190,9/188,9. Etapa 2. 5-(3-{2-ciano-1-[3-(7-{[2-(tnmetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirrol-1 -il]etil}pirrolidin-1-il)-1,3-tiazol-4-carbonitrila 5-bromo-1,3-tiazol-4-carbonitrila (24 mg, 0,13 mmol) e 3- pirrolidin-3-il-3-[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)- 1 H-pirrol-1 -iljpropanonitrila (51 mg, 0,12 mmol, de exemplo 33, etapa 3) foram misturados com tetrafluoroborato de 1-butil-3-metil-1H-imidazol-3-io (0,15 g, 0,66 mmol), e 4-metilmorfolina (14 μL, 0,13 mmol) foi adicionado. A mistura foi aquecida a 120°C durante 2 horas, em seguida resfriada para temperatura ambiente, dividida entre EtOAc e salmoura, e a camada de salmoura foi extraída com EtOAc três vezes. Os extratos orgânicos combinados foram secados sobre sulfato de sódio e concentrada. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 100% de acetato de etila em hexanos forneceu o produto (27 mg, 42%). 1H RMN (300 MHz, CDCI3): δ . 8,82 (s, 1H), 7,91 (s, 1H), 7,72 (s, 1H), 7,36 (d, 1H), 7,84-6,99 (m, 1H), 6,94 (t, 1H), 6,84 (d, 1H), 5,66 (s, 2H), 4,28-1,46 (m, 1H), 3,96 (dd, 1H), 3,68-3,41 (m, 5H), 3,20-3,02 (m, 1H), 2,97 (m, 2H), 2,09-1,73 (m, 2H), 0,99-0,85 (m, 2H), -0,06 (s, 9H); LCMS (M+H)+: 545,2.
Uma porção deste material foi desprotegida para fornecer o ra-cemato no seguinte procedimento, etapa 3.
Uma porção (20 mg) deste produto protegido por SEM foi sepa-rada em seus enantiômeros por cromatografia quiral (Chiral Technologies ChiralCel OD-H: 30 x 250 mm, 5 pm, 45% de EtOH/55% de hexanos a 15 mL/minuto; enantiômero 1: tempo de retenção de 41,8 minutos; enantiômero 2: tempo de retenção de 47,4 minutos). Cada enantiômero foi evaporado e desprotegido separadamente de acordo com os seguintes procedimentos na etapa 4 e etapa 5. Etapa 3. 5-(3-{2-ciano-1 -[3-(7H-pirrolo[2,3-d]pirimidin-4-íl)-1 H-pirrol-1 - il]etil}pirrolidin-1 -il)-1,3-tiazol-4-carbonitríla (racêmica) 5-(3-{2-ciano-1-[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirrol-1-il]etil}pirrolidin-1-il)-1,3-tiazol-4-carbonitrila racê- mica (7,8 mg, 0,013 mmol) foi dissolvida em uma mistura de 1:1 de DCM/TFA, agitada durante 1 hora, e concentrada. O resíduo foi dissolvido em MeOH (1 mL), e 0,2 mL de EDA foram adicionados e a reação foi agitada durante 15 minutos. HPLC-MS preparativa, eluindo com um gradiente de ACN e H2O contendo 0,15% de NH4OH, seguido por liofilização forneceu o produto como uma base livre (3,5 mg, 66%). 1H RMN (400 MHz, DMSO-d6): δ 11,97 (brs, 1H), 8,61 (s, 1H), 8,20 (s, 1H), 7,99 (t, 1H), 7,52 (d, 1H), 7,14 (t, 1H), 6,96-6,92 (m, 2H), 4,58 (td, 1H), 3,68 (dd, 1H), 3,66-3,59 (m, 1H), 3,54-3,37 (m, 3H), 3,22 (dd, 1H), 3,02-2,91 (m, 1H), 1,80-1,63, (m, 2H); LCMS (M+H)+: m/z = 415,0. Etapa 4. 5-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1-il]etil}- pirrolidin-1-il)-1,3-tiazol-4-carbonitrila (enantiômero simples 1) 5-(3-{2-ciano-1-[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d] pi rim id i n-4-il)-1 H-pirrol-1 -il]etil}pirrolidin-1 -il)-1,3-tiazol-4-carbonitrila (6,0 mg, 0,011 mmol; Pico 1 de etapa 2) foi dissolvida em uma mistura de 1:1 de DCM/TFA, a mistura foi agitada durante 1 hora, em seguida concentrada. O resíduo foi redissolvido em 1 mL de MeOH, e 0,2 mL de EDA foram adicio-nados. HPLC-MS preparativa, eluindo com um gradiente de ACN e H2O contendo 0,15% de NH4OH, seguido por liofilização forneceu o produto como uma base livre (2,5 mg, 54%). 1H RMN (400 MHz, DMSO-d6): õ 11,97 (br s, 1H), 8,61 (s, 1H), 8,19 (s, 1H), 7,99 (t, 1H), 7,52 (dd, 1H), 7,14 (dd, 1H), 6,96-6,93 (m, 2H), 4,58 (td, 1H), 3,68 (dd, 1H), 3,66-3,59 (m, 1H), 3,543,36 (m, 3H), 3,22 (dd, 1H), 3,03-2,90 (m, 1H), 1,80-1,64 (m, 2H); LCMS (M+H)+: 415,0. Etapa 5. 5-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-íl)-1 H-pirrol-1- il]etil}pirrolidin-1-il)-1,3-tiazol-4-carbonitrila (enantiômero simples 2) 5-(3-{2-Ciano-1-[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimídin-4-il)-1 H-pirrol-1-il]etil}pirrolidin-1 -il)-1,3-tiazol-4-carbonitrila (6,0 mg, 0,011 mmol; Pico 2 de etapa 2) foi dissolvida em uma mistura de 1:1 de DCM/TFA, a mistura foi agitada durante 1 hora, em seguida concentrada. O resíduo foi redissolvido em 1 mL de MeOH, 0,2 mL de EDA foram adicionados. HPLC-MS preparativa, eluindo com um gradiente de ACN e H2O con- tendo 0,15% de NH4OH, seguido por liofilização forneceu o produto como a base livre (2,5 mg, 54%). 1H RMN (400 MHz, DMSO-d6): δ 11,97 (br s, 1H), 8,61 (s, 1H), 8,19 (s, 1H), 7,99 (t, 1H), 7,52 (dd, 1H), 7,14 (t, 1H), 6,96-6,93 (m, 2H), 4,58 (td, 1H), 3,68 (dd, 1H), 3,66-3,60 (m, 1H), 3,54-3,38 (m, 3H), 3,22 (dd, 1H), 3,02-2,90 (m, 1H), 1,81-1,62 (m, 2H); LCMS (M+H)+: 415,0. Exemplo 150. 3-[ 1 -(2-mercaptopirimidin-4-il)pirrolidin-3-il]-3-[4-(7H-pirro-lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]propanonitrila (enantiômero simples)

Etapa 1. 3-[1-(2-mercaptopirimidin-4-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsi- lil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (150 mg, 0,34 mmol; de exemplo 15, etapa 3) e 2,4-dicloropirimidina (61 mg, 0,41 mmol) foram dissolvidas em 1,4-dioxano (0,30 mL) e DIPEA (119 μL, 0,686 mmol) foi adicionado. A solução foi aquecida para 100°C durante 30 minutos. A mistura foi concentrada, etanol (0,1 mL) foi adicionado, seguido por di-hidrato de sulfeto de hidrogênio de sódio (82 mg, 0,9 mmol). A suspensão foi em seguida agitada em temperatura ambiente durante 24 horas. Di-hidrato sulfeto de hidrogênio de sódio adicional (31 mg, 0,34 mmol) foi adicionado e agitado em temperatura ambiente durante mais 3 dias. A mistura foi diluída com acetonitrila e filtrada. HPLC-MS preparativa, eluindo com um gradiente de ACN e H2O contendo 0,15% de NH4OH, seguido por liofilização forneceu o produto como a base livre , 75 mg. LCMS (M+H)+: 548,1. Etapa 2. 3-[1-(2-mercaptopirimidin-4-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila 3-[1-(2-mercaptopirimidin-4-il)pirrolidin-3-il]-3-[4-(7-{[2- (trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila (19 mg, 0,024 mmol) foi dissolvida em uma mistura de 1:1 de TFA/DCM, agitada durante 1 hora em temperatura ambiente, em seguida concentrada. O resíduo foi dissolvido em 1 mL de metanol, e 0,2 mL de EDA foram adicionados e a reação agitada durante 30 minutos. HPLC-MS preparativa, eluindo com um gradiente de ACN e H2O contendo 0,15% de 5 NH4OH, seguido por liofilização forneceu o produto como uma base livre. 1H RMN (400 MHz, DMSO-d6): δ 8,87 (s, 1H), 8,69 (s, 1H), 8,43 (s, 1H), 7,61 (d, 1H), 7,50 (br d, 1H), 6,99 (d, 1H), 6,01 (br d, 1H), 4,82 (br m, 1H), 4,00- 2,73 (m, 7H), 1,79-1,47 (m, 2H); LCMS (M+H)+: 418,0. Exemplo 151. N-[4-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pira- 10 zol-1-illetil}pirrolidin-1-il)pirimidin-2-il]-N,N-dimetilsulfonamida (enantiômero simples)

Etapa 1: 3-[ 1 -(2-aminopirimidin-4-il)pirrolidin-3-il]-3-[4-(7-{[2-(trimetilsilil)etó- xi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila
Uma solução de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrila (0,100 g, 0,228 mmol, de exemplo 15, etapa 3) e 4-cloropirimidin-2-amina (0,031 g, 0,24 mmol, SynChem) em etanol (0,1 mL) e DIPEA (0,1 mL, 0,6 mmol) foi aquecida para 120°C durante 1,5 hora. A mistura reacional foi dividida entre água e acetato de etila, e a fase aquosa foi extraída três vezes. Os extratos foram secados sobre sulfato de sódio, decantados e concentrados. O produto foi usado sem outra purificação na etapa seguinte (120 mg, 99%). LCMS (M+H)+: 531,2. Etapa 2. N-[4-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - 2 5 il]etil}pirrolidin-1 -il)pirimidin-2-il]-N, N-dimetilsulfonamida 3-[1-(2-aminopirimidín^4-il)pirrolidin-3-il]-3-[4-(7-{[2- (trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- iljpropanonitrila (20 mg, 0,04 mmol) foi dissolvida em DCM (0,20 mL), e DIPEA (13 μL, 0,075 mmol) foi adicionado seguido por cloreto de dimetilsulfa- moíla (4,0 μL, 0,038 mmol). A reação foi agitada durante 16 horas. Cloreto de dimetilsulfamoíla adicional (2,0 μL, 0,019 mmol) foi adicionado, agitado durante algumas horas, em seguida concentrada. O resíduo foi agitado com 50% de TFA/DCM durante 1 hora, concentrado, em seguida re-dissolvido em metanol e tratado com excesso de EDA. HPLC-MS preparativa, eluindo com um gradiente de ACN e H2O contendo 0,15% de NH4OH, seguido por liofili-zação forneceu o produto como a base livre . 1H RMN (500 MHz, CD3OD) (rotamers): δ 8,82 (s, 1H), 8,77 (s, 1H), 8,49-8,45 (m, 1H), 7,68 (d, 1H), 7,67 e 7,54 (each as d, together = 1H), 7,10 (d, 1H), 6,21 e 6,03 (cada qual como d, juntamente = 1H), 4,86 (td, 1H), 4,04-2,95 (m, 7H), 2,81-2,77 (br singletos, juntamente 6H), 2,00-1,77 (m, 2H); LCMS (M+H)+: 508,1. Exemplo 152, 4-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-dJpirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)-N-metilpiridina-2-carboxamida (enantiômero simples)

Etapa 1. Ácido 4-(3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirímidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)piridina-2-carboxílico 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (250 mg, 0,57 mmol; de exemplo 15, etapa 3) e ácido 4-cloropiridina-2-carboxílico (135 mg, 0,857 mmol) foram combinados em etanol (1,2 mL) e DIPEA (0,20 mL, 1,14 mmol) e a- quecidos para 120°C em um frasco selado durante 2 horas, com ácido 4- cloropiridina-2-carboxílico adicional (0,135 g, 0,857 mmol) adicionado após 1 hora para conduzir a reação à conclusão. HPLC-MS preparativa (eluindo com um gradiente de MeOH/H2O contendo 0,15% de NH4OH) foi usada para purificar o produto. As frações eluídas foram evaporadas para fornecer o produto como o sal de carboxilado de amónio (150 mg, 47%). LCMS (M+H)+: 559,2, Etapa 2. 4-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)-N-metilpiridina-2-carboxamida
Ácido 4-(3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)piridina-2-carboxílico (20 mg, 0,036 mmol) foi dissolvido em DCM (1 mL). DIPEA (20 μL, 0,1 mmol) seguido por hexafluorofosfato de N,N,N',N'-tetrametil-O-(7-azaben- zotriazol-1-il)urônio (0,020 g, 0,054 mmol) foram adicionados, e agitados durante 1 hora. Este foi seguido por adição de metilamina a 2 M em THF (36 μL, 0,072 mmol). A reação foi continuada durante 16 horas. O solvente foi removido em vácuo. O produto bruto foi desprotegido por agitação em uma mistura de 2:1 DCM/TFA for 3 horas, evaporação, em seguida agitação na mistura de metanol (1,4 mL) e EDA (0,1 mL). HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usada para purificar o produto. 1H RMN (300 MHz, DMSO-d6): δ 12,10 (br s, 1H), 8,88 (s, 1H), 8,69 (s, 1H), 8,66-8,59 (m, 1H), 8,44 (s, 1H), 8,14 (d, 1H), 7,61 (d, 1H), 7,17-7,14 (m,1H), 6,99 (d, 1H), 6,58 (dd, 1H), 4,88-4,76 (m, 1H), 3,683,57 (m, 1H), 3,48-3,19 (m, 5H), 3,01-2,87 (m, 1H), 2,79 (d, 3H), 1,75-1,63 (m, 2H); LCMS (M+H)+: 442,0. Exemplo 153. 4-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-dlpirimidin~4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1-il)-N,N-dimetilpiridina-2-carboxamida (enantiômero simples)

Preparada no exemplo 152, etapa 2, substituindo dimetilamina a 2 M em THF (36 pL, 0,072 mmol) no lugar de metilamina. 1H RMN (300 MHz, DMSO-d6): δ 12,05 (br s, 1H), 8,80 (s, 1H), 8,62 (s, 1H), 8,36 (s, 1H), 8,10-7,98 (m, 1H), 7,54 (d, 1H), 6,92 (d, 1H), 6,54-6,46 (m, 1H), 6,42 (dd, 1H), 4,79-4,69 (m, 1H), 3,61-2,78 (m, 7H), 2,89 (s, 3H), 2,82 (s, 3H), 1,671,56 (m, 2H); LCMS (M+H)+: 456,0. Exemplo 154. 4-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljetiljpirrolidin-1 -il)-N-fenilpiridina-2-carboxamida (enantiômero simples)
Preparada no exemplo 152, etapa 2, substituindo anilina (6,5 μL, 0,072 mmol) no lugar de metilamina. 1H RMN (300 MHz, CD3OD): õ 8,68 (s, 1H), 8,66 (s, 1H), 8,42 (s, 1H), 8,19 (d, 1H), 7,80-7,72 (m, 2H), 7,50 (d, 1H), 7,42-7,30 (m, 3H), 7,18-7,10 (m, 1H), 6,94 (d, 1H), 6,62 (dd, 1H), 4,89^,76 (m, 1H), 3,73 (dd, 1H), 3,55-3,17 (m, 5H), 3,16-3,01 (m, 1H), 1,92-1,80 (m, 2H); LCMS (M+H)+: 501,4. Exemplo 155. 3-[1-(2l3-di-hidrofuro[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4-(7H- pirrolo[2,3-dlpirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (enantiômero simples)

Etapa 1. 3-[1-(2,3-di-hidrofuro[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4-(7-{[2- (trimetilsilil)etó)d]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila 3-pirrolidin-3-il-3-[4-(7-{[2-(tnmetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (86,6 mg, 0,198 mmol, de e-xemplo 15, etapa 3) foi misturada com NMP (0,20 mL, 2,1 mmols), DIPEA (53,0 μL, 0,305 mmol), e trifluorometanossulfonato de 2,3-di-hidrofuro[2,3- b]piridin-6-ila (preparado como descrito em Org. Lett. 2006, 8(17), 37773779; 50,0 mg, 0,152 mmol) foi adicionado. A mistura foi aquecida no microondas a 130°C durante um total de 1 hora. A mistura reacional foi diluída com EtOAc, lavada com água e salmoura, secada sobre sulfato de sódio e concentrada. O produto foi purificado por cromatografia de coluna rápida, eluindo com um gradiente de 0 a 100% de acetato de etila em hexanos para fornecer o produto purificado (16 mg, 17%). 1H RMN (300 MHz, CDCI3): δ . 8,85 (s, 1H), 8,36 (s, 2H), 7,40 (d, 1H), 7,28 (d, 1H), 6,80 (d, 1H), 5.81 (d, 1H), 5,68 (s, 2H), 4,57 (t, 2H), 4,43 (td, 1H), 3,88 (dd, 1H), 3,55 (dd, 2H), 3,55-3,38 (m, 1H), 3,38-3,26 (m, 2H), 3,22 (dd, 1H), 3,12 (t, 2H), 3,09-2,99 (m, 1H), 2,97 (dd, 1H), 1,98-1,85 (m, 1H), 1,82-1,64 (m, 1H), 0,92 (dd, 2H), - 0,06 (s, 9H); LCMS (M+H)+: 557,2. Etapa 2. 3-[ 1 -(2,3-di-hidrofuro[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila 3-[1 -(2,3-di-hidrofuro[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4-(7-{[2- (trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila (8 mg, 0,01 mmol) foi dissolvida em uma mistura de 1:1 de DCM:TFA, agitada em temperatura ambiente durante 1 hora, em seguida concentrada. O resíduo foi dissolvido em 1,5 mL de MeOH, e 0,2 mL de EDA foram adicionados. A mistura foi agitada durante 30 minutos em temperatura ambiente, em seguida HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usada para purificar o produto (3 mg, 49%). 1H RMN (400 MHz, DMSO-d6): δ 12,08 (br s, 1H), 8,86 (s, 1H), 8,68 (s, 1H), 8,42 (s, 1H), 7,60 (d, 1H), 7,35 (d, 1H), 6,98 (d, 1H), 5.85 (d, 1H), 4,79 (td, 1H), 4,46 (t, 2H), 3,64 (dd, 1H), 3,43-3,12 (m, 5H), 3,04 (t, 2H), 2,93-2,81 (m, 1H), 1,74-1,56 (m, 2H); LCMS (M+H)+: 427,2. Exemplo 156. 3-[4-(7H-pirrolo[2,3-d1pirimidin~4-il)-1 H-pirazol-1-il]-3-(1- tieno[2,3-b]piridin-6-ilpirrolidin-3-il)propanonitrila (enantiômero simples)

Etapa 1. 3-[1-(1-óxido-2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4-(7- {[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxijmetil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (0,140 g, 0,320 mmol; de exemplo 15, etapa 3) e 6-cloro-2,3-di-hidrotieno[2,3-b]piridina-1-óxido (50,0 mg, 0,266 mmol, de exemplo 28, etapa 4) foram dissolvidos em etanol (0,20 mL) e DIPEA (83,6 μL, 0,480 mmol) foi adicionado. A mistura foi aquecida no micro-ondas at 125°C durante 100 minutos. LCMS mostrou mais do que 60% de conversão no composto desejado. A mistura foi concentrada e purificada por cromatografia de coluna rápida (eluindo com um gradiente primeiro de 0 a 100% de acetato de etila em hexanos, seguido por 5% de MeOH em acetato de etila) para fornecer o produto como como um sólido amarelo. (46 mg, 29%). LCMS (M+H)+: 589,2. Etapa 2. 3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]-3-( 1 -tieno[2,3- b]piridin-6-ilpirrolidin-3-il)propanonitrila 3-[1-(1 -óxido-2,3-di-hidrotieno[2,3-b]piridin-6-il)pirrolidin-3-il]-3-[4- (7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimídin-4-il)-1 H-pirazol-1 - iljpropanonitrila (15,0 mg, 0,0255 mmol) foi aquecida para 140°C em anidrido acético (0,50 mL, 5,3 mmols) durante 3 horas. A mistura foi concentrada, o resíduo foi dissolvido em uma mistura de 1:1 de TFA/DCM, agitado durante 1 hora em temperatura ambiente, e foi concentrado novamente. O resíduo foi em seguida agitado com 0,2 mL de EDA em 0,1 mL de MeOH. HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usada para purificar o produto (3 mg, 26%). 1H RMN (300 MHz, CDCI3): δ 9,16 (br s, 1H), 8,84 (s, 1H), 8,38 (s, 2H), 7,81 (d, 1H), 7,37 (dd, 1H), 7,86 (s, 1H), 6,79 (dd, 1H), 6,44 (d, 1H), 4,49 (td, 1H), 4,01 (dd, 1H), 3,66-3,54 (m, 1H), 3,51-3,38 (m, 2H), 3,27 (dd, 1H), 3,18-3,03 (m, 1H), 3,04 (dd, 1H), 2,05-1,91 (m, 1H), 1,88-1,72 (m, 1H); LCMS (M+H)+: 441,1. Exemplo 157. bis(trifluoroacetato) de 3-[1-(7,7-difluoro-6,7-di-hidro-5H-ciclo- penta[b]piridin-2-il)pirrolidin-3-il]-3-f4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pira-zol-1-illpropanonitrila (enantiômero simples)

Etapa 1. 2-cloro-5,6-di-hidro-7H-ciclopenta[b]piridin-7-ona
Periodinano de Dess-Martin (0,550 g, 1,30 mmol) foi adicionado a uma solução de 2-cloro-6,7-di-hidro-5H-ciclopenta[b]piridin-7-ol (preparado como descrito no W02006/103511; 0,200 g, 1,18 mmol) em DCM (5 mL). A solução foi agitada durante 2 horas, em seguida foi lavada com NaOH a 1N, secada sobre sulfato de sódio, decantada e concentrada. O produto foi usado sem outra purificação (180 mg, 91%). 1H RMN (400 MHz, em CDCI3 e CD3OD): δ 7,64 (d, 1H), 7,39 (d, 1H), 3,06-2,96 (m, 2H), 2,74-2,58 (m, 2H); LCMS (M+H)+: 168,1. Etapa 2. 2-cloro-7,7-difluoro-6,7-di-hidro-5H-ciclopenta[b]piridina 2-metóxi-N-(2-metoxietil)-N-(trifluoro-À(4)-sulfanil)etanamina (0,3 mL, 1,5 mmol) foi adicionada a uma solução de 2-cloro-5,6-di-hidro-7H- ciclopenta[b]piridin-7-ona (0,071 g, 0,42 mmol) em DCM (0,8 mL) e etanol (4 μL) e a reação foi agitada durante 4 dias. Acetato de etila e água foram adi-cionados, as camadas separadas, e a fase aquosa foi extraída com mais duas porções de acetato de etila. Os extratos combinados foram secados sobre sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida (eluindo com um gradiente de 10 a 50% de acetato de etila em hexanos) forneceu o produto como um sólido cristalino incolor (26 mg, 32%). 1H RMN (300 MHz, CDCI3): δ 7,64 (d, 1H), 7,39 (d, 1H), 3,05-2,96 (m, 2H), 2,74-2,58 (m, 2H); 19F RMN (300 MHz, CDCI3): δ -92,88 (t); LCMS (M+H)+: 190,1/192,0. Etapa 3. Bis(trifluoroacetato) de 3-[1-(7,7-difluoro-6,7-di-hidro-5H-ciclopen- ta[b]piridin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila
Uma solução de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (0,050 g, 0,11 mmol, de exemplo 15, etapa 3) e 2-cloro-7,7-difluoro-6,7-di-hidro-5H-ciclo- penta[b]piridina (0,026 g, 0,14 mmol) em tetrafluoroborato de 1 -butil-3-metil- 1 H-imidazol-3-io (0,3 mL) contendo 4-metilmorfolina (0,038 mL, 0,34 mmol) foi aquecida para 120°C durante um total de 15 horas. A mistura reacional foi dividida entre água e acetato de etila, e a fase aquosa foi extraída em um total de 3 vezes. Os extratos foram secados sobre sulfato de sódio e concentrados. O resíduo foi agitado com 1:1 de TFA:DCM durante 2 horas e em seguida concentrada. A mistura foi reconstituída em metanol e excesso de of EDA foi adicionada. Após agitação durante 16 horas, o sólido presente na mistura reacional foi filtrado e o filtrado foi purificado por HPLC-MS preparativa (eluindo com um gradiente de ACN e H2O contendo 0,1% de TFA) para fornecer o produto como o sal de trifluoroacetato de (2 mg, 2%). 1H RMN (400 MHz, DMSO-d6): δ 12,69 (br s, 1H), 9,03 (s, 1H), 8,86 (s, 1H), 8,56 (s, 1H), 7,81 (br s, 1H), 7,56 (d, 1H), 7,17 (br s, 1H), 6,60 (d, 1H), 4,88 (td, 1H), 3,77 (dd, 1H), 3,54-3,23 (m, 6H), 2,91 (dd, 1H), 2,85-2,77 (m, 2H), 2,59-2,49 (m, 1H), 1,78-1,60 (m, 2H); LCMS (M+H)+: 461,2. Exemplo 158. 3-[1-(7-fluoro-1,3-benzoxazol-2-il)pirrolidin-3-ilj-3-[3-(7H-pirro- lo[2,3-d]pirimidin-4-íl)-1 H-pirrol-1 -iljpropanonitrila (enantiômeros simples iso-lados)

Uma mistura de 7-fluorobenzo[d]oxazol-2(3H)-tiona (0,076 g, 0,45 mmol, preparada no exemplo 21), 3-pirrolidin-3-il-3-[3-(7-{[2-(trime- tilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirímidin-4-íl)-1 H-pirrol-1-iljpropanonitrila (0,150 g, 0,240 mmol, de exemplo 33, etapa 3) e DIPEA (250 μL, 1,4 mmol) em 1,4-dioxano (1 mL) foi aquecida para 80°C durante 3 horas. O dioxano foi removido em vácuo e foi substituído com etanol (1 mL). Nitrato de prata (0,0817 g, 0,481 mmol) e solução de hidróxido de amónio (0,2 mL) foram adicionados. Após agitação durante a noite, a mistura foi filtrada e enxaguada com metanol. Após evaporação, o resíduo foi purificado por cromatografia de coluna rápida, eluindo com 0 a 10% de MeOH/DCM. Após evaporação das as frações contendo o produto, o resíduo foi dissolvido em acetato de etila e foi lavado com NaOH a 1N, secado sobre sulfato de sódio, e evaporado novamente. O produto foi desprotegido por agitação em 25% de TFA/DCM durante 3 horas, em seguida evaporação e agitação com excesso de EDA em metanol durante a noite. HPLC-MS preparativa (eluindo com um gradiente de MeOH e H2O contendo NH4OH) foi usada para fornecer o produto como a base livre . 1H RMN (300 MHz, CDCI3): δ 9.37 (br s, 1H), 8,81 (s, 1H), 7,75 (t, 1H), 7,34 (d, 1H), 7,18-7,87 (m, 2H), 7,83 (dd, 1H), 6,97 (t, 1H), 6,84 (d, 1H), 6,82 (dd, 1H), 4,22 (td, 1H), 4,09 (dd, 1H), 3,85 (ddd, 1H), 3,65 (ddd, 1H), 3,49 (dd, 1H), 3,16-3,04 (m, 1H), 2,98 (app d, 2H), 2,13-1,99 (m, 1H), 1,91-1,74 (m, 1H); LCMS (M+H)+: 442,2. HPLC quiral (coluna Phe- nomenex Lux Celulose-1, 21,2 x 250 mm, 5pm, eluindo com 45% de E- tOH/55% de hexanos em uma taxa de 20 mL/minuto) foi usado para separar a mistura racêmica nos enantiômeros simples (enantiômero 1 tempo de re-tenção: 17,8 minutos; enantiômero 2 tempo de retenção: 20,1 minutos). Na remoção de solvente, os produtos de enantiômero simples foram separada-mente reconstituídos em ACN/H2O e liofilizado. Pico 1 (primeiro a eluir): 1H RMN (500 MHz, DMSO-d6, 90°C): δ 11,71 (br s, 1H), 8,61 (s, 1H), 7,94 (t, 1H), 7,45 (dd, 1H), 7,17-7,89 (m, 3H), 6,94 (dd, 1H), 6,91-6,86 (m, 2H), 4,59 (td, 1H), 3,91 (dd, 1H), 3,69 (ddd, 1H), 3,57-3,49 (m, 2H), 3,40 (dd, 1H), 3,26 (dd, 1H), 3,03-2,94 (m, 1H), 1,84-1,69 (m, 2H); LCMS (M+H)+: 442,2. Pico 2 (segundo a eluir): 1H RMN (400 MHz, DMSO-d6): δ 11,96 (br s, 1H), 8,61 (s, 1H), 8,01 (t, 1H), 7,51 (dd, 1H), 7,18-7,10 (m, 3H), 6,96-6,89 (m, 3H), 4,58 (td, 1H), 3,86 (dd, 1H), 3,71-3,63 (m, 1H), 3,54-3,10 (m, 4H), 2,99-2,87 (m, 1H), 1,73-1,63 (m, 2H); LCMS (M+H)+: 442,2. Exemplo 159. Trifluoracetato de 3-[1-(7-bromo-1,3-benzoxazol-2-il)pirrolidin- 3-il1-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -illpropanonitrila (enanti-ômero simples)

Etapa 1. 2-amino-6-bromofenol 2-bromo-6-nitrofenol (Aldrich, 0,25 g, 1,1 mmol) foi dissolvido em THF (6,4 mL), água (6,4 mL) e di-hidrato de cloreto estanhoso (1,3 g, 5,7 mmols) foram adicionados. A mistura foi aquecida para 80°C durante 1 hora. Sob resfriamento para temperatura ambiente, bicarbonato de sódio saturado foi adicionado, seguido por acetato de etila. O material insolúvel foi filtrado.
As camadas foram separadas e a fase aquosa foi extraída com mais duas porções de acetato de etila. Os extratos orgânicos combinados foram lavados com salmoura, secados sobre sulfato de sódio e concentrados para fornecer o composto desejado como um sólido cristalino esbranquiçado, usado sem outra purificação (200 mg, 93%). LCMS (M+H)+: 188,0/190,0. Etapa 2. 7-bromobenzo[d]oxazol-2(3H)-tiona
Dicloreto carbonotioico (0,122 mL, 1,60 mmol) foi adicionado gota a gota a uma solução de 2-amino-6-bromofenol (0,20 g, 1,1 mmol) em THF (2,8 mL) a 0°C. A mistura foi deixada aquecer para temperatura ambiente e agitar durante 2 horas. O solvente foi removido em vácuo, e o sólido bruto foi usado na etapa seguinte sem outra purificação. LCMS (M+H)+: m/z = 229,9/231,9. Etapa 3. 3-[ 1 -(7-bromo-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7-{[2- (trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - iljpropanonitrila
Uma mistura de 7-bromobenzo[d]oxazol-2(3H)-tiona (0,105 g, 0,457 mmol), DIPEA (0,159 mL, 0,914 mmol), e 3-pirrolidin-3-il-3-[4-(7-{[2- (trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- iljpropanonitrila (0,10 g, 0,23 mmol; de exemplo 15, etapa 3) em 1,4-dioxano (0,20 mL) foi agitada a 80°C durante 3 horas. Uma mistura foi em seguida concentrada. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 100% de acetato de etila em hexanos forneceu o produto como como um sólido amarelo (47 mg, 32%). 1H RMN (300 MHz, CDCI3): δ 8,86 (s, 1H), 8,46 (s, 1H), 8,36 (s, 1H), 7,43 (d, 1H), 7,26 (dd, 1H), 7,14 (dd, 1H), 7,84 (t, 1H), 6,81 (d, 1H), 5,68 (s, 2H), 4,55 (td, 1H), 4,05 (dd, 1H), 3,88-3,78 (m, 1H), 3,69-3,45 (m, 4H), 3,27 (dd, 1H), 3,25-3,09 (m, 1H), 3,00 (dd, 1H), 2,07- 1,77 (m, 2H), 0,92 (dd, 2H), -0,06 (s, 9H); LCMS (M+H)+: 633,1/635,1. Etapa 4. Trifluoracetato de 3-[1-(7-bromo-1,3-benzoxazol-2-il)pirrolidin-3-il]-3- [4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila
Uma solução de 3-[1-(7-bromo-1,3-benzoxazol-2-il)pirrolidin-3-il]- 3-[4-(7-{[2-(trímetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- iljpropanonitrila (10 mg, 0,016 mmol) em 1:1 de TFA/DCM foi agitada durante 1 hora em temperatura ambiente, concentrada, em seguida agitada em 1 mL de MeOH, contendo 0,2 mL de EDA até a desproteção ser concluída. HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,1 % de TFA), seguido por liofilização forneceu o produto como o sal de trifluo- roacetato de (5.8 mg, 59%). 1H RMN (300 MHz, DMSO-d6): δ 12,45 (br s, 1H), 8,98 (s, 1H), 8,78 (s, 1H), 8,51 (s, 1H), 7,77-7,68 (m, 1H), 7,24 (dd, 1H), 7,17 (dd, 1H), 7,13-7,84 (m, 2H), 4,90 (td, 1H), 3,89 (dd, 1H), 3,70-2,86 (m, 6H), 1,81-1,63 (m, 2H); LCMS (M+H)+: m/z = 503,0/505,1. Exemplo 160. 2-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin^4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)-1,3-beπzoxazol-7-carbonitrila (enantiômero simples)
Uma mistura de 3-[1-(7-bromo-1,3-benzoxazol-2-il)pirrolidin-3-il]- 3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- iljpropanonitrila (22 mg, 0,035 mmol, de exemplo 159, etapa 3), Cianeto de zinco (8,2 mg, 0,069 mmol), e tetracis(trifenilfosfina)palàdio(0) (8,0 mg, 0,0069 mmol) em DMF (0,3 mL) foi aquecida no micro-ondas para 120°C durante 60 minutos. Uma porção adicionai de tetracis(trifenilfosfina)pa- ládio(O) (24 mg, 0,020 mmol) foi adicionada e foi aquecida para 120°C em um banho de óleo durante 2 horas. A mistura foi em seguida diluída com EtOAc, lavada com água, salmoura, secada sobre sulfato de sódio e concentrada. O produto bruto foi agitado com 1:1 de TFA/DCM durante 1 hora, concentrado, em seguida agitado em 1 mL de MeOH contendo 0,2 mL de EDA durante 15 minutos. HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15 % de NH4OH) forneceu o produto como a base livre (9 mg, 57%). 1H RMN (300 MHz, DMSO-d6): δ 8,89 (s, 1H), 8,67 (s, 1H), 8,43 (s, 1H), 7,60 (d, 1H), 7,57 (dd, 1H), 7,41 (dd, 1H), 7,28 (t, 1H), 6,98 (d, 1H), 4,87 (td, 1H), 3,91 (dd, 1H), 3,73-3,62 (m, 1H), 3,61-3,26 (m, 4H), 3,06-2,89 (m, 1H), 1,82-1,66 (m, 2H); LCMS (M+H)+: 450,1. Exemplo 161. 3-[1-(7-hidróxi-1,3-benzoxazol-2-il)pirrolidin-3-il1-3-|4-(7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -illpropanonitrila (enantiômero simples)

Etapa 1. 7-hidroxibenzo[d]oxazol-2(3H)-tiona
Uma mistura de 3-aminobenzeno-1,2-diol (preparado como descrito no W02007/071434; 0,5 g, 4 mmol) e O-etil ditiocarbonato de potássio (0,80 g, 5,0 mmols) em etanol (5.2 mL) foi aquecida até o refluxo durante 1,5 hora, em seguida para temperatura ambiente durante 3 dias. HCI diluído foi adicionado na reação, e o produto foi extraído com acetato de etila. Os extratos combinados foram secados sobre sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 100% de acetato de etila, foi usada para purificar o produto (80 mg, 12%). 1H RMN (400 MHz, CD3OD): δ 7,84 (t, 1H), 6,69 (dd, 1H), 6,64 (dd, 1H); LCMS (M+H)+: 167,9. Etapa 2. 3-[ 1-(7-hidróxi-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirímidin-4-il)-1 H-pirazol-1 -iljpropanonitrila 7-Hidroxibenzo[d]oxazol-2(3H)-tiona (0,080 g, 0,48 mmol) e 3- pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)- 1 H-pirazol-1-iljpropanonitrila (0,17 g, 0,38 mmol, de exemplo 15, etapa 3) em 1,4-dioxano (1 mL, 10 mmol) foram aquecidos para 80°C durante diversas horas até os materiais de partida serem consumidos. O solvente foi removido em vácuo e substituído com etanol (1 mL). Nitrato de prata (0,065 g, 0,38 mmol) e solução de hidróxido de amónio (0,12 mL) foram adicionados e a reação foi continuada durante 16 horas. A mistura reacional foi dividida entre água e acetato de etila, as camadas separadas e a fase aquosa extraída em um total de 3 vezes com acetato de etila. Os extratos foram filtrados para remover o resíduo insolúvel, secados sobre sulfato de sódio, decantados e concentrados. HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15 % de NH4OH) foi usada para purificar o produto bruto antes da etapa de desproteção. A etapa de desproteção foi realizada com 1:1 de TFA:DCM durante 2 horas, seguido por evaporação, em seguida agitação com excesso de EDA em metanol durante 1 hora. HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15 % de NH4OH) forneceu o produto como a base livre (12 mg, 7%). 1H RMN (400 MHz, DM- SO-d6): õ 8,89 (s, 1H), 8,69 (s, 1H), 8,43 (s, 1H), 7,61 (d, 1H), 6,99 (d, 1H), 6,90 (t, 1H), 6,71 (dd, 1H), 6,50 (dd, 1H), 4,86 (td, 1H), 3,85 (dd, 1H), 3,67- 3,60 (m, 1H), 3,51-3,19 (m, 4H), 3,02-2,91 (m, 1H), 1,79-1,63 (m, 2H); LCMS (M+H)+: 441,0. Exemplo 162. 3-[1-(7-metóxi-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H-pirro- lo[2,3-dlpirimidin-4-il)-1H-pirazol-1-illpropanonitrila (enantiômero simples)

Etapa 1. 7-metóxi-1,3-benzoxazol-2(3H)-tiona
Uma mistura de 2-amino-6-metoxifenol (preparado como descrito no eP333176; 1,2 g, 8,6 mmol) e ditiocarbonato de O-etila de potássio (1,7 g, 11 mmols) em etanol (11 mL) foi aquecida até o refluxo durante 3 horas, em seguida resfriada para temperatura ambiente, seguido por resfriamento em um banho de gelo. HCI diluído foi adicionado à reação, o precipitado branco foi isolado por filtragem e lavada com água. O sólido pegajoso resultante foi azeotropeado com benzeno (700 mg, 45%). 1H RMN (300 MHz, DMSO-d6): δ 13,86 (brs, 1H), 7,22 (t, 1H), 6,93 (dd, 1H), 6,82 (dd, 1H), 3,93 (s, 3H); LCMS (M+H)+: 182,0. Etapa 2. 3-[ 1 -(7-metóxi-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirímidin-4-il)-1 H-pirazol-1 -iljpropanonitrila
Uma mistura de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (0,050 g, 0,11 mmol; de exemplo 15, etapa 3) e 4-metóxi-1,3-benzoxazol-2(3H)-tiona (0,031 g, 0,17 mmol) em 1,4-dioxano (0,6 mL, 8 mmol) foi aquecida para 80°C durante 3 horas. O solvente foi removido em vácuo e substituído com etanol (0,6 mL). Nitrato de prata (0,019 g, 0,11 mmol) e solução de hidróxido de amónio (0,036 mL) foram adicionados e a reação foi agitada durante 4 horas. A mistura foi filtrada através de uma seringa de filtro PTFE, enxaguada com metanol. O metanol foi evaporado em vácuo. O resíduo foi dividido entre acetato de etila e água, as camadas separadas e a fase aquosa foi extraída mais duas vezes. Os extratos combinados foram secados sobre sulfato de sódio, decantados e concentrados. O produto foi purificado por HPLC-MS preparativa, eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH. Este produto foi desprotegido por agitação com 1:1 de TFA/DCM durante 1 hora, seguido por remoção de solventes, em seguida agitação com excesso de EDA em metanol até a desproteção ser concluída. Purificação por meio de HPLC-MS preparativa, eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH, forneceu o produto como uma base livre (10 mg, 19%). 1H RMN (300 MHz, DMSO-d6): δ 8,89 (s, 1H), 8,68 (s, 1H), 8,43 (s, 1H), 7,60 (d, 1H), 7,86 (t, 1H), 6,99 (d, 1H), 6,88 (dd, 1H), 6,69 (dd, 1H), 4,85 (td, 1H), 3,89 (s, 3H), 3,86 (dd, 1H), 3,67-3,59 (m, 1H), 3,52- 3,29 (m, 4H), 3,02-2,90 (m, 1H), 1,79-1,63 (m, 2H); LCMS (M+H)+: 455,1. Exemplo 163. 3-(4-(7H-pirrolo[2,3-d]pirimidín-4-il)-1 H-pirazol-1-il)-3-(1-(7- etoxibenzo[d]oxazol-2-il)pirrolidin-3-il)propanonítrila (enantiômero simples)

Etapa 1. 2-etóxi-6-nitrofenol
Ácido nítrico (3,89 mL, 60 mmols) foi adicionado gota a gota a 2- etóxi-fenol, (Aldrich, 5,00 mL, 39,4 mmols) em água (20 mL) e dietil éter (49 mL). A mistura resultante tornou-se quente ao ponto do refluxo do éter, e em seguida foi deixada resfriar para temperatura ambiente e agitar durante 3 horas. A mistura reacional foi vertida em água e foi extraída três vezes com dietil éter. Os extratos foram secados sobre sulfato de sódio, decantados e concentrados. O resíduo foi dissolvido em um pequeno volume de DCM e 5 hexanos, e o material que não foi dissolvido foi excluído ao carregar a coluna de sílica-gel para cromatografia rápida. O produto foi eluído com um gradiente de 20-50% clorofórmio em hexanos, para fornecer o produto como um sólido laranja (1,36 g, 19%). 1H RMN (300 MHz, CDCI3): δ 10,73 (br s, 1H), 7,68 (dd, 1H), 7,12 (dd, 1H), 6,88 (dd, 1H), 1,44 (q, 2H), 1,50 (t, 3H); LCMS 10 (M+H)+: 183,9. Etapa 2. 2-amino-6-etoxifenol
A uma suspensão de 2-etóxi-6-nitrofenol (1,36 g, 7,42 mmols) em água (30 mL) e metanol (30 mL) foi adicionado ditionita de sódio (~85%, 9,58 g, 46,8 mmols). A reação foi aquecida para 60°C durante 30 minutos, 15 até ela tornar-se incolor. Sob resfriamento para temperatura ambiente, salmoura foi adicionada, e o produto foi extraído com acetato de etila. Os extratos foram secados sobre sulfato de sódio, decantados e concentrados (0,11 g, 89%). 1H RMN (400 MHz, CD3OD): δ 6,58 (t, 1H), 6,40 (dd, 1H), 6,38 (dd, 1H), 4,04 (q, 2H), 1,39 (t, 3H); LCMS (M+H)+: 154,1. Etapa 3. 7-etóxi-1,3-benzoxazol-2(3H)-tiona
Preparado de 2-amino-6-etoxifenol (0,11 g, 6,59 mmols) pelo método de exemplo 162, etapa 1 (1 g, 77%). 1H RMN (400 MHz, DMSO-d6): δ 13,86 (br s, 1H), 7,21 (t, 1H), 6,93 (dd, 1H), 6,81 (dd, 1H), 4,21 (q, 2H), 1,38 (t, 3H); LCMS (M+H)+: 196,1. Etapa 4. 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-3-(1-(7-etoxi- benzo[d]oxazol-2-il)pirrolidin-3-il)propanonitrila
À 3-pirrolidín-3-il-3-[4-(7-{[2-(trimetílsilil)etóxi]metil}-7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (0,070 g, 0,16 mmol, de exemplo 15, etapa 3) em 1,4-dioxano (1 mL) foi adicionada 7-etóxi-1,3- 30 benzoxazol-2(3H)-tiona e a solução foi aquecida para 80°C durante 3,5 horas. O solvente foi removido em vácuo e substituído com etanol (1 mL). Nitrato de prata (0,014 g, 0,080 mmol) e solução de hidróxido de amónio (50 μL foram adicionados e a reação agitada durante 16 horas. Mais nitrato de prata (0,019 g, 0,11 mmol) e solução de hidróxido de amónio (50 μl_) foram adicionados e a reação foi continuada durante mais 7 horas. NaOH a 1N foi adicionado na reação, seguido por acetato de etila. A mistura bifásica foi filtrada e as camadas separadas. A fase aquosa foi extraída com mais duas porções de acetato de etila. Os extratos combinados foram secados sobre sulfato de sódio, filtrados através de uma almofada curta de sílica, em seguida concentrada. O produto foi desprotegido por agitação com 1:1 de TFA/DCM durante 1 hora, seguido por evaporados e agitação com EDA (0,1 mL) em uma pequena quantidade de MeOH. Purificação por meio de HPLC- MS preparativa, eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH, forneceu o produto como a base livre . 1H RMN (300 MHz, CD3OD): δ 8,68 (s, 1H), 8,64 (d, 1H), 8,41 (s, 1H), 7,48 (dd, 1H), 7,83 (dt, 1H), 6,93 (dd, 1H), 6,83 (d, 1H), 6,64 (d, 1H), 4,91-4,79 (m, 1H), 1,48 (q, 2H), 3,96 (dd, 1H), 3,74-3,64 (m, 1H), 3,60-3,03 (m, 5H), 1,97-1,84 (m, 2H), 1,41 (t, 3H); LCMS (M+H)+: 469,2. Exemplo 164. 3-(4-(7H-pirrolo(2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-3-(1-(7-(di- fluorometóxi)benzofd]oxazol-2-il)pirrolidin-3-il)propanonitrila (enantiômero simples)

Etapa 1. 2-(difluorometóxi)-6-nitrofenol
A uma solução de 2-(difluorometóxi)fenol (preparado como des-crito na Patente dos Estados Unidos 4.512.984; 0,90 g, 5,6 mmols) em ácido acético (1 mL) a 0°C foi adicionado gota a gota ácido nítrico branco (65%, 0,43 mL, 6,7 mmols). A mistura reacional foi em seguida vertida em água e extraída com dietil éter três vezes. Os extratos combinados foram lavados com salmoura, secados sobre sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida, eluindo com um gradiente de 20 a 50% de CHCI3 em hexanos forneceu o produto como um xarope amarelo (250 mg, 22%). 1H RMN (300 MHz, CDCI3): δ 10,77 (s, 1H), 8,03 (dd, 1H), 7,56-7,51 (m, 1H), 6,99 (t, 1H), 6,67 (t, 1H). Etapa 2. 2-amino-6-(difluorometóxi)fenol
Preparado de 2-(difluorometóxi)-6-nitrofenol (0,25 g, 1,2 mmol) pelo método descrito por exemplo 163, etapa 2 (170 mg, 79%). 1H RMN (400 MHz, CD3OD): δ 6,67 (t, 1H), 6,63-6,60 (m, 2H), 6,50-6,47 (m, 1H); 19F RMN (400 MHz, CD3OD): δ -83,03 (d); LCMS (M+H)+: 176,1. Etapa 3. 7-(difluorometóxi)-1,3-benzoxazol-2(3H)-tíona
Preparado de 2-amino-6-(difluorometóxi)fenol (0,17 g, 0,97 mmol) pelo método descrito por exemplo 162, etapa 1 (120 mg, 57%). 1H RMN (400 MHz, DMSO-d6): δ 11,44 (br s, 1H), 7,38 (t, 1H), 7,32 (t, 1H), 7,16-7,11 (m, 2H); 19F RMN (400 MHz, DMSO-d6): δ -82,65 (d); LCMS (M+H)+: 218,0. Etapa 4. 3-(4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il)-3-( 1-(7-(difluoro- metóxi)benzo[d]oxazol-2-il)pirrolidin-3-il)propanonitrila
Preparado de 7-(difluorometóxi)-1,3-benzoxazol-2(3H)-tiona (de etapa 3) pelo método de exemplo 163, etapa 4. 1H RMN (300 MHz, DMSO- d6): δ 8,88 (s, 1H), 8,69 (s, 1H), 8,43 (s, 1H), 7,59 (d, 1H), 7,30 (t, 1H), 7,157,11 (m, 2H) 6,99 (d, 1H), 6,85 (t, 1H), 4,86 (td, 1H), 3,89 (dd, 1H), 3,71-3,60 (m, 1H), 3,57-3,26 (m, 4H), 3,04-2,91 (m, 1H), 1,80-1,66 (m, 2H); LCMS (M+H)+: 491,2. Exemplo 165. 3-[1-(4-hidróxi-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-|4-(7H-pir- rolo[2,3-dlpirimidin-4-il)-1 H-pirazol-1-il]propanonitrila (enantiômero simples)
Etapa 1. 2-aminobenzeno-1,3-diol
Tribrometo de boro a 0,1 M em DCM (12 mL, 12 mmols) foi adi-cionado lentamente gota a gota a uma solução de 2,6-dimetoxianiline (Alfa Aesar, 0,5 g, 3 mmols) em DCM (5 mL) a -45°C sob nitrogênio. A mistura foi agitada, com aquecimento para temperatura ambiente, durante 3 dias. A 5 mistura foi resfriada em um banho de gelo e água foi adicionada gota a gota.
Solução de bicarbonato de sódio saturada foi adicionada para ajustar o pH para 5 a 6 e a fase aquosa foi extraída com DCM. A camada aquosa, que continha o produto, foi evaporada para fornecer uma mistura sólida. O sólido foi suspenso em etanol e os sólidos foram filtrados. A solução de etanol foi 10 usada na etapa seguinte. 1H RMN (400 MHz, CD3OD): δ 6,68 (t, 1H), 6,37 (d, 2H). Etapa 2. 4-hidroxibenzo[d]oxazol-2(3H)-tiona
Uma solução de 2-aminobenzeno-1,3-diol (0,37 g, 3,0 mmols) e O-etil ditiocarbonato de potássio (0,59 g, 3,7 mmols) em etanol (3,8 mL) foi 15 aquecida até o refluxo durante 3 horas, em seguida resfriada para temperatura ambiente. HCI diluído foi adicionado na reação, e o produto foi extraído com acetato de etila. Os extratos combinados foram secados sobre sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida (eluindo com um gradiente de 0 a 100% de acetato de etila-hexanos) forneceu o 20 produto (300 mg, 60%). 1H RMN (400 MHz, CD3OD): δ 7,84 (t, 1H), 6,83 (dd, 1H), 6,70 (dd, 1H), 4,92 (brs, 2H); LCMS (M+H)+: 168,0. Etapa 3. 3-[ 1-(4-hidróxi-1,3-benzoxazol-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila
Uma mistura de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]me- 25 til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (0,080 g, 0,18 mmol, de exemplo 15, etapa 3) e 4-hidroxibenzo[d]oxazol-2(3H)-tiona (0,046 g, 0,27 mmol) em 1,4-dioxano (1 mL, 10 mmols) e DIPEA (excesso) foi a- quecida para 100°C durante a 3 horas. O solvente foi removido em vácuo e foi substituído com etanol (1 mL). Nitrato de prata (0,031 g, 0,18 mmol) e 30 hidróxido de amónio (0,057 mL, 1,5 mmol) foram adicionados e a reação foi agitada durante 16 horas. NaOH a 1N foi adicionado na reação, e a mistura foi filtrada através de uma seringa de filtro PVDF (Whatman), enxaguada com metanol. O solvente foi evaporado. O resíduo foi dividido entre acetato de etila e água, a camadas separadas, e a fase aquosa extraída em um total de três vezes. Os extratos combinados foram secados sobre sulfato de sódio, decantados e concentrados. O produto foi purificado por meio de HPLC- MS preparativa, eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH e o eluente foi evaporado. O produto foi agitado com 1:1 de TFA/DCM durante 1 hora, evaporado, em seguida agitado com excesso de EDA em metanol até a desproteção ser concluída. O produto foi isolado por meio de HPLC-MS preparativa, eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH, para fornecer o produto como a base livre (5 mg, 6%). 1H RMN (400 MHz, DMSO-d6): δ 8,89 (s, 1H), 8,69 (s, 1H), 8,44 (s, 1H), 7,61 (d, 1H), 6,99 (d, 1H), 6,86 (dd, 1H), 6,82-6,77 (m, 1H), 6,58 (dd, 1H), 4,86 (td, 1H), 3,85 (dd, 1H), 3,67-3,60 (m, 1H), 3,50-3,25 (m, 4H), 3,022,90 (m, 1H), 1,79-1,62 (m, 2H); LCMS (M+H)+: m/z = 441,1. Exemplo 166. 3-{1-[7-(hidroximetil)-1,3-benzoxazol-2-il]pirrolidin-3-il}-3-[4- (7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-illpropanonitrila (enantiômero simples)

Etapa 1. 2-tioxo-2,3-di-hidro-1,3-benzoxazol-7-carboxilato de metila
Preparado de 3-amino-2-hidroxibenzoato de metila (Apollo, 0,1 g, 6,0 mmols) de acordo com o método de exemplo 162, etapa 1 (830 mg, 66%). 1H RMN (400 MHz, DMSO-d6): δ 7,73 (dd, 1H), 7,49 (dd, 1H), 7,40 (t, 1H), 3,92 (s, 3H); LCMS (M+H)+: 210,0. Etapa 2. 7-(hidroximetil)-1,3-benzoxazol-2(3H)-tiona
Hidreto de di-isobutilalumínio a 0,1 M em hexano (1,4 mL, 1,4 mmol) foi adicionado a uma solução de 2-tioxo-2,3-di-hidro-1,3-benzoxazol- 7-carboxilato de metila (0,430 g, 2,06 mmols) em THF (8 mL) a 0°C. Após 2 horas, uma outra porção de hidreto de di-isobutilalumínio a 0,1 M em hexano (1,4 mL, 1,4 mmol) foi adicionada, e a reação foi deixada alcançar a tempe-ratura ambiente. Uma solução saturada de sal de Rochelle e acetato de etila foram adicionados e agitados até as camadas separareM-se. A fase aquosa foi extraída uma outra vez com acetato de etila, e os extratos orgânicos combinados foram secados sobre sulfato de sódio, decantados e concentra-dos (320 mg, 86%). LCMS (M+H)+: 182,0. Etapa 3. 3-{1-[7-(hidroximetil)-1,3-benzoxazol-2-il]pirrolidin-3-il}-3-[4-(7H-pir- rolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila
Uma mistura de 7-(hidroximetil)-1,3-benzoxazol-2(3H)-tiona (0,32 g, 1,8 mmol) e 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H- pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-iljpropanonitrila (0,64 g, 1,5 mmol, de exemplo 15, etapa 3) em 1,4-dioxano (4 mL) foi aquecida para 80°C durante 24 horas. O produto protegido por SEM desejado foi isolado por HPLC-MS preparativa (gradiente de ACN/H2O contendo 0,1% de TFA). O eluente contendo produto foi desejado basificado usando NaOH a 1N e o produto foi extraído com acetato de etila. O produto foi desprotegido por agitação em 1.1 de TFA/DCM durante 1 hora, evaporação de solventes, e agitação com EDA (0,1 mL) em MeOH até a desproteção ser concluída. HPLC-MS preparativa (gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usado para fornecer o produto purificado como a base livre (10 mg, 2%). 1H RMN (300 MHz, CD3OD): δ 8,68 (s, 1H), 8,64 (s, 1H), 8,42 (s, 1H), 7,49 (d, 1H), 7,19- 7,88 (m, 2H), 7,86-7,81 (m, 1H), 6,93 (d, 1H), 4,93-4,79 (m, 1H), 4,77 (s, 2H), 3,97 (dd, 1H), 3,77-3,67 (m, 1H), 3,62-3,49 (m, 2H), 3,40 (dd, 1H), 3,22 (dd, 1H), 3,15-3,04 (m, 1H), 1,96-1,84 (m, 2H); LCMS (M+H)+: 455,2. Exemplo 169. 6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-djpirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)furo[3,2-c]piridina-7-carbonitrila (enantiômero simples)

Etapa 1. 5-íodo-4-metóxi-2-oxo-1,2-di-hidropiridina-3-carbonitrila N-iodossuccinimida (22 g, 0,10 mol) foi adicionada a uma solu-ção de 4-metóxi-2-oxo-1,2-di-hidropiridina-3-carbonitrila (10,0 g, 0,0666 mol, Ryan Scientific) em 1,2-dicloroetano (200 mL). A mistura foi aquecida até o refluxo durante a noite. Para completar a reação, N-iodosuccinimida adicio-nal (11,2 g, 0,0500 mol) foi adicionada e o refluxo continuada durante 5 ho-ras. O solvente foi removido em vácuo. O resíduo foi triturado com metanol para fornecer o produto como um pó branco (16,4 g, 89%). 1H RMN (400 MHz, DMSO-d6): δ 12,29 (br s, 1H), 8,01 (s, 1H), 4,28 (s, 3H); LCMS (M+H)+: 277,0. Etapa 2. 4-hidróxi-5-iodo-2-oxo-1,2-di-hidropiridina-3-carbonitríla lodotrimetilsilano (2,1 mL, 14 mmols) foi adicionado a uma solu-ção de 5-iodo-4-metóxi-2-oxo-1,2-di-hidropiridina-3-carbonitrila (2,0 g, 7,2 mmols) em acetonitrila (80 mL). A mistura foi agitada em temperatura ambi-ente durante 1,5 hora. O solvente foi removido em vácuo. O produto foi tritu-rado com DCM durante a noite, em seguida filtrado e lavado com éter para fornecer produto (1,69 g, 89%). 1H RMN (400 MHz, DMSO-d6): δ 11,56 (br s, 1H), 7,81 (s, 1H); LCMS (M+H)+: 262,9. Etapa 3. 4-hidróxi-2-oxo-5-[(trimetilsilil)etinil]-1,2-di-hidropiridina-3-carbonitrila
Uma solução de 4-hidróxi-5-iodo-2-oxo-1,2-di-hidropiridina-3- carbonitrila (1,18 g, 4,50 mmols) em acetonitrila (15 mL), foi desgaseificada. Trietilamina (0,942 mL, 6,76 mmols) foi adicionada, seguido por (trimetilsi- lil)acetileno (0,955 mL, 6,76 mmolss), cloreto de bis(trifenilfosfina)paládio(ll) (0,190 g, 0,271 mmol) e iodo de cobre (I) (69 mg, 0,36 mmol). A mistura foi desgaseificada novamente, em seguida foi agitada em temperatura ambiente durante 1 hora. A mistura foi absorvida em sílica-gel. Cromatografia de colu-na rápida, eluindo com 0 a 15% de metanol em DCM forneceu o produto como o sal de trietilamina (890 mg). 1H RMN (400 MHz, DMSO-d6): δ 10,01 (d, 1H), 9,04 (br s, 1H), 2,95 (dd, 6H), 0,13 (t, 9H), 0,14 (s, 9H); LCMS (M+H)+: 233,1. Etapa 4. 6-oxo-5,6-di-hidrofuro[3,2-c]piridina-7-carbonitríla
Ácido metanossulfônico (0,64 mL, 9,9 mmols) foi adicionado a uma solução de 4-hidróxi-2-oxo-5-[(trimetilsilil)etinil]-1,2-di-hidropiridina-3- carbonitrila*TEA (1,32 g, de etapa 3) em THF (25 mL). A reação foi agitada em temperatura ambiente durante 16 horas, em seguida foi aquecida para 40°C durante 8 horas seguido por 35°C durante 16 horas. O solvente foi removido em vácuo. A purificação por cromatografia de coluna rápida, eluindo com um gradiente de 0 a 10% de metanol em DCM forneceu o produto desejado (150 mg, 23%). 1H RMN (400 MHz, DMSO-d6): δ 12,77 (br s, 1H), 8,29 (s, 1H), 7,89 (d, 1H), 6,90 (d, 1H); LCMS (M+H)+: 161,1. Etapa 5. Trifluorometanossulfonato de 7-cianofuro[3,2-c]piridin-6-ila 6-oxo-5,6-di-hÍdrofuro[3,2-c]piridina-7-carbonitrila (10,0 mg, 0,062 mmol) e N-Fenilbis(trifluorometanossulfonimida) (27,9 mg, 0,078 mmol) foram dissolvidos em acetonitrila (0,35 mL), e trietilamina (17 pL, 0,12 mmol) foi adicionada. A mistura foi aquecida para 50°C durante 40 minutos. O solvente foi removido em vácuo, e o produto foi usado diretamente na etapa seguinte. LCMS (M+H)+: 293,0. Etapa 6. 6-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirímidin-4-il)-1 H-pirazol-1 - il]etil}pirrolídin-1-il)furo[3,2-c]pirídina-7-carbonitríla 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (40,0 mg, 0,092 mmol, de e-xemplo 15, etapa 3), foi dissolvida em NMP (0,20 mL) e 4-metilmorfolina (14 pL, 0,12 mmol) e trifluorometanossulfonato de 7-cianofuro[3,2-c]piridin-6-ila bruto (18 mg, 0,062 mmol, formado na etapa 5) foi adicionado. A mistura reacional foi aquecida para 90°C durante 1 hora. O solvente foi removido em vácuo. O resíduo foi apreendido em água e acetato de etila. As camadas foram separadas e a fase aquosa foi extraída com acetato de etila três vezes. Os extratos orgânicos combinados foram secados sobre sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 100% de acetato de etila em hexanos forneceu o produto desejado. O produto foi tratado com 1:1 de DCM:TFA durante 1,5 hora, em seguida concentrado. O resíduo foi dissolvido em 1,5 mL de Me- OH, e 0,2 mL de EDA foram adicionados para completar a etapa de desproteção. O produto foi purificado por meio de HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) (5,9 mg, 21%). 1H RMN (400 MHz, DMSO-d6): δ 12,13 (br s, 1H), 8,88 (s, 1H), 8,68 (s, 1H), 8,65 (s, 1H), 8,43 (s, 1H), 7,91 (d, 1H), 7,60 (d, 1H), 6,99 (d, 1H), 6,98 (d, 1H), 4,87 (td, 1H), 3,98 (dd, 1H), 3,86-3,79 (m, 1H), 3,72-3,58 (m, 2H), 3,42 (dd, 1H), 3,28 (dd, 1H), 2,97-2,84 (m, 1H), 1,79-1,67 (m, 2H); LCMS (M+H)+: 450,2. Exemplo 170. 6-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1- il]etil}pirrolidin-1-il)furo[3,2-c]piridina-7-carbonitrila (racemato)

A uma solução de 3-(2-ciano-1-{3-[7-(dietoximetil)-7H-pirrolo[2,3- d]pirimidin-4-il]-1 H-pirrol-1-il}etil)pirrolidina-1-carboxilato de terc-butila (0,62 g, 0,98 mmol, diastereômero racêmico 1 de exemplo 32, etapa 1) em 1,4- dioxano (20 mL) foi adicionado HCI a 4 M em 1,4-dioxano (6,9 mL, 28 mmols) e a reação foi agitada durante a noite. O solvente foi removido em vácuo. Purificação por meio de HPLC-MS preparativa forneceu o produto como um sólido amarelo claro (0,17 g, 56%). LCMS (M+H)+: 307,1. Uma porção deste produto (28 mg, 0,092 mmol), foi dissolvida em NMP (0,20 mL) e 4-metilmorfolina (14 μL, 0,12 mmol). Trifluorometanossulfonato de 7- cianofuro[3,2-c]piridin-6-ila bruto (18 mg, 0,062 mmol, de exemplo 169, etapa 5), foi adicionado. A reação foi aquecida para 60°C durante 1 hora. Purifica-ção por meio de HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto como uma base livre (5,5 mg, 20%). 1H RMN (400 MHz, DMSO-d6): δ 11,96 (br s, 1H), 8,67 (s, 1H), 8,61 (s, 1H), 8,00 (t, 1H), 7,93 (d, 1H), 7,51 (d, 1H), 7,15 (dd, 1H), 7,81 (d, 1H), 6,96-6,93 (m, 2H), 4,59 (td, 1H), 3,96 (dd, 1H), 3,89-3,81 (m, 1H), 3,74-3,65 (m, 1H), 3,56 (dd, 1H), 3,47 (dd, 1H), 3,23 (dd, 1H), 2,93-2,81 (m, 1H), 1,79-1,60 (m, 2H); LCMS (M+H)+: 449,2. Exemplo 171. 1,1-dióxido de 6-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidin-4- il)-1 H-pirrol-1-il]etil}pirrolidin-1-il)-2,3-di-hidrotienof3,2-c]piridina-7-carbonitrila

Etapa 1:2-(benzilóxi)-5-iodo-4-metoxinicotinonitrila
Uma mistura de 5-iodo-4-metóxi-2-oxo-1,2-di-hidropiridina-3- carbonitrila (3,7 g, 13 mmols, de exemplo 169, etapa 1), óxido de prata (I) (3,4 g, 15 mmols) e cloreto de benzila (2,00 mL, 17,4 mmols) em tolueno (74 mL) foi aquecida para 107°C durante 4,5 horas. Cloreto de benzila adicional (1,54 mL, 13,4 mmols) foi adicionado e a reação foi aquecida para 120°C durante 16 horas. A mistura foi resfriada para temperatura ambiente, e filtrada. O solvente foi removido do filtrado em vácuo, e o produto foi triturado com dietil éter, azeotropado com tolueno, em seguida secado sob vácuo elevado para fornecer o produto como um sólido branco (4,30 g, 87%). 1H RMN (300 MHz, CDCI3): δ 8,43 (s, 1H), 7,49-7,43 (m, 2H), 7,42-7,30 (m, 3H), 5,47 (s, 2H), 4,35 (s, 3H); LCMS (M+H)+: 367,8. Etapa 2: 2-(benzilóxi)-5-(2-hidroxietil)-4-metoxinicotinonitrila n-butil-litio a 2,5 M em hexano (4,85 mL, 12,1 mmols) foi adicio-nado gota a gota a uma solução de 2-(benzilóxi)-5-iodo-4-metoxinico- tinonitrila (3,7 g, 10 mmols) em THF (150 mL) a -78°C. A reação foi mantida a -78°C durante 1,5 hora, em cujo tempo 2,2-dióxido de 1,3,2-dioxatiolano (1,25 g, 10,1 mmols, Aldrich) em THF (5,0 mL) foi introduzido gota a gota. A mistura foi deixada aquecer para temperatura ambiente e agitar durante 16 horas. HCI concentrado foi adicionado (1,85 mL) e a mistura foi agitada durante 30 minutos. Solução de NaHCO3 saturada foi adicionada para ajustar o pH para 7. Alguma água foi adicionada e o produto foi extraído com três porções de EtOAc. Os extratos foram combinados, secados sobre sulfato de sódio e concentrados. Cromatografia de coluna rápida, eluindo com um gra-diente de 0 a 70% de acetato de etila em hexanos forneceu o produto como um sólido branco (1,62 g, 56%). 1H RMN (300 MHz, CDCI3): δ 8,00 (s, 1H), 7,51-7,45 (m, 2H), 7,41-7,30 (m, 3H), 5,47 (s, 2H), 4,33 (s, 3H), 3,77 (dd, 2H), 2,77 (t, 2H); LCMS (M+H)+: 285,1. Etapa 3. 4-metilbenzenossulfonato de 2-[6-(benzilóxi)-5-ciano-4-meto- xipiridin-3-il]etila
A uma solução de 2-(benzilóxi)-5-(2-hidroxietil)-4-metoxinico- tinonitrila (1,21 g, 4,26 mmols) em DCM (50 mL) foi adicionada trietilamina (0,652 mL, 4,68 mmols) seguido por cloreto de p-toluenossulfonila (0,811 g, 4,26 mmols) e 4-dimetilaminopiridina (52 mg, 0,426 mmol). A reação foi agitada durante 8 horas. Para conduzir a reação à conclusão, cloreto de p- toluenossulfonila adicional (0,243 g, 1,28 mmol) foi adicionado e a reação continuada durante 16 horas. O volume de solvente foi reduzido em vácuo e o produto foi purificado por cromatografia de coluna rápida, eluindo com um gradiente de 0 a 50% de acetato de etila em hexanos para fornecer o produto como um sólido branco (1,36 g, 73%). 1H RMN (300 MHz, CDCI3): δ 7,86 (s, 1H), 7,56 (d, 2H), 7,56-7,48 (m, 2H), 7,44-7,30 (m, 3H), 7,14 (d, 2H), 5,47 (s, 2H), 1,48 (s, 3H), 1,45 (t, 2H), 2,79 (t, 2H), 2,42 (s, 3H); LCMS (M+H)+: 438,9. Etapa 4. Etanotioato de S-{2-[6-(benzilóxi)-5-cíano-4-metoxipiridin-3-il]etila} Uma solução de 4-metilbenzenossulfonato de 2-[6-(benzilóxi)-5- ciano-4-metoxipiridin-3-il]etila (1,36 g, 3,10 mmols) em acetonitrila (30 mL) e DMF (30 mL) foi tratada com tioacetato de potássio (0,50 g, 4,4 mmols) e agitada durante 16 horas. Água foi adicionada e o produto foi extraído com três porções de acetato de etila. Os extratos combinados foram secados sobre sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 50% de acetato de etila em hexanos, foi usada para purificar o produto (698 mg, 66%). 1H RMN (300 MHz, CDCI3): δ 7,93 (s, 1H), 7,51-7,27 (m, 5H), 5,46 (s, 2H), 4,36 (s, 3H), 3,03 (t, 2H), 2,75 (t, 2H), 2,30 (s, 3H); LCMS (M+H)+: 343,1. Etapa 5. 6-(benzilóxi)-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila
A uma solução de S-{2-[6-(benzilóxi)-5-ciano-4-metoxipiridin-3- iljetil} etanotioato (0,698 g, 2,04 mmols) em metanol (90 mL) foi adicionada 5 solução de hidróxido de amónio (30 mL, 400 mmols, e a reação foi agitada durante 8 horas. O solvente foi removido em vácuo para fornecer um sólido branco, Produção teórica assumida e usado sem outra purificação. 1H RMN (400 MHz, CD3OD): δ 7,98 (s, 1H), 7,48-7,25 (m, 5H), 5,43 (s, 2H), 3,56 (t, 2H), 3,36-3,27 (m, 2H); LCMS (M+H)+: 268,9. Etapa 6. 6-hidróxi-2,3-di-hidrotieno[3,2-c]pindina-7-carbonitrila
Uma solução de cloreto de acetila (0,43 mL, 6,1 mmol) em me-tanol (90 mL) foi agitada durante 1,5 hora, em seguida esta solução foi adicionada a 6-(benzilóxi)-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila (0,547 g, 2,04 mmols). A reação foi agitada durante 3 dias, e o solvente foi removido 15 em vácuo. Produção teórica foi assumida e o produto foi usado sem outra purificação. 1H RMN (400 MHz, CD3OD): δ 7,37 (s, 1H), 7,19 (s, 1H), 3,49 (t, 2H), 3,18 (t, 2H); LCMS (M+H)+: 179,1. Etapa 7. 6-cloro-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitríla 6-hidróxi-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila (70 mg, 20 0,39 mmol) foi aquecida em cloreto de fosforila (2 mL, 20 mmol) para 110°C durante 1 hora. O excesso de reagente foi removido em vácuo. O resíduo foi dissolvido em DCM e lavado com NaOH a 0,1 N, A fase aquosa foi extraída com acetato de etila três vezes e estes extratos foram combinados com a camada de DCM. Os orgânicos combinados foram secados sobre sulfato de 25 sódio, decantados e concentrados para fornecer o produto como um sólido cristalino bege (34 mg, 44%). 1H RMN (300 MHz, CDCI3): δ 8,14 (s, 1H), 3,59 (dd, 2H), 3,41 (dd, 2H); LCMS (M+H)+: 197,8/199,0. Etapa 8. 1,1-dióxido de 6-cloro-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila
A uma solução de 6-cloro-2,3-di-hidrotieno[3,2-c]piridina-7- 30 carbonitrila (34 mg, 0,17 mmol) em DCM (2 mL) a 0°C foi adicionado ácido M-cloroperbenzoico (88 mg, 0,38 mmol). A reação foi agitada com aquecimento para temperatura ambiente durante a noite. A reação foi diluída com NaOH a 0,2 N e acetato de etila. NaCl sólida foi adicionada para auxiliar na separação de camada. A fase aquosa foi extraída com acetato de etila três vezes e os extratos foram secados sobre sulfato de sódio, decantados e concentrados. O produto foi usado sem outra purificação na etapa 9. 1H RMN (300 MHz, CD3OD): δ 8,78 (s, 1H), 3,71 (dd, 2H), 3,47 (dd, 2H); LCMS (M+H)+: 228,9/230,8. Etapa 9. 1,1-dióxido de 6-(3-{2-ciano-1 -[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H- pirrol- 1-il]etil}pirrolidin- 1-il)-2,3-di-hidrotieno[3,2-c]pirídina- 7-carbonitríla (ra- cêmico) 3-pirrolidin-3-il-3-[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirrol-1 -iljpropanonitrila (66 mg, 0,11 mmol, de exemplo 33, etapa 3) e 1,1-dióxido de 6-cloro-2,3-di-hidrotieno[3,2-c]piridina-7-carbo- nitrila (de etapa 8) foram dissolvidos em DMF (0,5 mL). 4-metilmorfolina (0,037 mL, 0,34 mmol) foi adicionada e a reação foi aquecida para 80°C durante 1 hora. Sob resfriamento para temperatura ambiente, a mistura reacional foi dividida entre água e acetato de etila. A fase aquosa foi extraída com acetato de etila três vezes. Os extratos combinados foram lavados com salmoura, secados sobre sulfato de sódio, decantados e concentrados. O produto bruto foi agitado com 1:1 de TFA/DCM durante 1 hora, evaporado, em seguida agitado com 0,4 mL de EDA em metanol (4 mL) até a desproteção ser concluída. Purificação por meio de HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto como a base livre (15 mg, 28%). 1H RMN (300 MHz, DMSO-d6): δ 11,96 (br s, 1H), 8,61 (s, 1H), 8,57 (s, 1H), 7,99 (br t, 1H), 7,51 (d, 1H), 7,15 (t, 1H), 6,97-6,91 (m, 2H), 4,59 (td, 1H), 3,95 (dd, 1H), 3,90-3,79 (m, 1H), 3,77-3,14 (m, 8H), 2,93-2,78 (m, 1H), 1,80-1,57 (m, 2H); LCMS (M+H)+: 499,2. Exemplo 172. 1,1-dióxido de 6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirímídin-4- íl)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-2,3-di-hidrotieno[3,2-cjpiridina-7- carbonitrila (enantiômero simples)

Ácido m-cloroperbenzoico (4,74 mg, 0,0212 mmol) em DCM (0,14 mL) foi adicionado a uma solução de 6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3- d]p irim id in-4-il)-1 H-pirazol-1 -i l]etil}pi rrol id in-1 -i l)-2,3-di-h id rotieno[3,2- c]piridina-7-carbonitrila (3,3 mg, 0,0070 mmol, de exemplo 173) em DCM (0,60 mL) a 0°C. A reação foi agitada com aquecimento para temperatura ambiente durante 1,5 hora, em seguida o solvente foi removido em vácuo. Purificação por meio de HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto como a base livre (800 μg, 22%). 1H RMN (500 MHz, DMSO-d6): δ 12,09 (br s, 1H), 8,85 (s, 1H), 8,68 (s, 1H), 8,54 (s, 1H), 8,41 (s, 1H), 7,59 (d, 1H), 6,96 (d, 1H), 4,87 (td, 1H), 3,98 (dd, 1H), 3,85-3,79 (m, 1H), 3,72-3,63 (m, 4H), 3,41 (dd, 1H), 3,33-3,23 (m, 1H), 3,23-3,13 (m, 2H), 2,95-2,86 (m, 1H), 1,80-1,67 (m, 2H); LCMS (M+H)+: 500,0. Exemplo 173. 6-(3-{2-ciano-1-|4-(7H-pirrolo[2,3-d1pirimidin-4-il)-1H-pirazol-1- il]etil}pirrolidin-1-il)-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrÍla (enantiômero simples)

Etapa 1. 2,4-dicloro-5-iodonicotinamida
Ao ácido 2,4-dicloro-5-iodonicotínico (preparado como descrito no european Journal of Organic Chemistry, (7), 1371-1376; 2001; 2,95 g, 7,33 mmol) em benzeno (20 mL) foi adicionado cloreto de oxalila (1,24 mL, 14,6 mmols), seguido por uma quantidade catalítica de DMF (10 μL). A mistura foi agitada em temperatura ambiente durante 2 horas. O solvente foi removido em vácuo. O resíduo foi dissolvido em THF (34 mL) e gás de amónia foi borbolhado através de uma mistura durante 5 minutos. A suspensão foi agitada, bem selada, durante mais 20 minutos. O solvente foi em seguida removido em vácuo. O sólido foi dissolvido em DCM (200 mL) e água (75 mL). As camadas foram separadas e a fase aquosa foi extraída com outra porção de DCM. Os extratos combinados foram secados sobre sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 100% de acetato de etila em hexanos foi usada para purificar o produto (1,64 g, 70%). 1H RMN (300 MHz, CDCI3 e CD3OD): δ 8,67 (s, 1H); LCMS (M+H)+: 316,9/318,9. Etapa 2. 2,4-dicloro-5-iodonicotinonitrila
A uma mistura de 2,4-dícloro-5-iodonicotinamida (2,43 g, 7,67 mmols) e DCM (122 mL) a 0°C, foi adicionada trietilamina (10,7 mL, 76,7 mmols), seguido por tricloroanidrido acético (14,0 mL, 76,7 mmols). Seguinte à adição, a solução foi agitada a 0°C durante 20 minutos. A mistura foi extin-guida pela adição de água nesta temperatura, e foi agitada durante 30 minu-tos antes de ser diluída com acetato de etila. A mistura bifásica foi separada. A camada orgânica foi lavada sucessivamente com NaHCO3 saturado, água, e salmoura, em seguida secada sobre sulfato de sódio, decantada e concentrada. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 15% de acetato de etila em hexanos forneceu o produto como um sólido amarelo (1,94 g, 84%). 1H RMN (300 MHz, CDCI3): δ 8,88 (s, 1H). Etapa 3. 2,4-dicloro-5-[(Z)-2-etoxivinil]nicotinonitrila
Uma mistura de 2,4-dicloro-5-iodonicotinonitrila (1,94 g, 6,49 mmols) e (2-etoxietenil)tn-n-butiltina (2,58 g, 7,14 mmols) em tolueno (16 mL) foi desgaseificada. Tetracís(trifenilfosfina)paládio(0) (750 mg, 0,649 mmol) foi adicionado, e a reação foi aquecida para 110°C durante 5 horas. O solvente foi removido em vácuo. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 20% de acetato de etila em hexanos foi usado para purificar o produto produto (590 mg, 37%). 1H RMN (300 MHz, CDCI3): δ . 9,26 (s, 1H), 6,55 (d, 1H), 5,47 (d, 1H), 4,09 (q, 2H), 1,37 (t, 3H). Etapa 4. 2,4-dicloro-5-(2-oxoetil)nicotinonitrila
Uma solução de 2,4-dicloro-5-[(Z)-2-etoxivinil]nicotinonitrila (0,670 g, 2,76 mmols) em THF (10,0 mL) e 4,0 M de HCI em água (2,75 mL, 5 10,1 mmols) foi aquecida até o refluxo durante 1,5 hora. A reação foi resfria da para temperatura ambiente e vertida em solução de bicarbonato de sódio saturada e o produto foi extraído com DCM. Os extratos orgânicos foram lavados com salmoura, secados sobre sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida, eluindo com um gradiente de 50- 10 100% de acetato de etila em hexanos forneceu o produto como an oil (500 mg, 84%). 1H RMN (300 MHz, CDCI3): δ 9,83 (br t, 1H), 8,39 (s, 1H), 4,00 (s, 2H). Etapa 5. 2,4-dicloro-5-(2-hidroxietil)nicotinonitrila
Hidreto de di-isobutilalumínio a 0,1 M em DCM (2,4 mL, 2,4 15 mmols) foi adicionado em porções durante o curso de 30 minutos a uma solução de 2,4-dicloro-5-(2-oxoetil)nicotinonitrila (500 mg, 2,4 mmols) em DCM (30 mL) a -78°C. Quando a reação foi considerada concluída por TLC e LCMS, ela foi extinguida a -78°C pela adição de água, em seguida foi deixada aquecer para temperatura ambiente. Uma solução saturada de sal de 20 Rochelle foi adicionada e a mistura foi agitada até as camadas separarem- se, O produto foi extraído com DCM três vezes. Os extratos foram secados sobre sulfato de sódio, decantados e concentrados. Cromatografia de coluna rápida, eluindo com um gradiente de 50 a 100% de acetato de etila em hexanos, foi usada para purificar o produto (120 mg, 23%). 1H RMN (300 MHz, 25 CDCI3): δ. 8,49 (s, 1H), 3,93 (dd, 2H), 3,04 (t, 2H); LCMS (M+H)+: 216,9/218,9. Etapa 6. 6-cloro-2,3-di-hidrotieno[3,2-c]piridina- 7-carbonitríla 2,4-dicloro-5-(2-hidroxietil)nicotinonitrila (0,060 g, 0,28 mmol) e trifenilfosfina (0,109 g, 0,415 mmol) foram dissolvidas em THF (2,12 mL). A 30 solução foi resfriada a 0°C, e azodicarboxilato de dietila (65,3 pL, 0,415 mmol) foi adicionado. Após agitação durante 10 minutos, ácido tioacético (29,6 μL, 0,415 mmol) foi adicionado. A mistura reacional foi agitada durante 2 horas a 0°C, em seguida durante 2 horas em temperatura ambiente. Cro-matografia de coluna rápida, eluindo com um gradiente de 0 a 10% de acetato de etila em hexanos foi used, para fornecer o produto como um óleo. Uma solução deste produto em metanol (1,5 mL) foi tratada com cloreto de acetila 5 (59 μL, 0,829 mmol), agitada em temperatura ambiente durante 7 horas, em seguida mantida no congelador durante 3 dias. O solvente foi removido em vácuo. O resíduo foi em seguida agitado durante 10 minutos em metanol (6,0 mL) e solução de hidróxido de amónio (0,50 mL, 3,7 mmol). O solvente foi removido em vácuo e o resíduo foi dividido entre água e acetato de etila.
A camada aquosa foi extraída com mais três porções de acetato de etila. Os extratos foram secados sobre sulfato de sódio e concentrada para fornecer o produto como um sólido branco, usado diretamente na etapa 7 (11 mg, 10%). LCMS (M+H)+: 196,9/199,0. Etapa 7. 6-(3-{2-ciano- 1-[4-(7-{[2-(trimetilsilil)etóxi]metil}- 7H-pirrolo[2,3-d]pi- 15 rimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)-2,3-di-hidrotieno[3,2-c]piridina-7- carbonitrila
Uma mistura de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (24 mg, 0,056 mmol, de exemplo 15, etapa 3) e 6-cloro-2,3-di-hidrotieno[3,2-c]piridina-7- 20 carbonitrila (11 mg, 0,028 mmol) em NMP (100 μL) e DIPEA (9,7 μL, 0,056 mmol) foi aquecida no micro-ondas a 135°C durante 15 minutos. 3-pirrolidin- 3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pira- zol-1-iljpropanonitrila adicional (18 mg, 0,042 mmol) foi adicionada e a reação submetida a micro-ondas na mesma temperatura durante mais 10 minu- 25 tos. A mistura reacional foi em seguida diluída com água, extraída com acetato de etila quatro vezes, os extratos secados sobre sulfato de sódio e concentrada. Cromatografia de coluna rápida, primeiro eluindo com um gradiente de 0 a 100% de acetato de etila em hexanos, em seguida 0-5% de metanol em acetato de etila, foi usada para purificar o produto (10 mg, 60%). 1H 30 RMN (300 MHz, CDCI3): δ 8,85 (s, 1H), 8,36 (s, 1H), 8,356 (s, 1H), 7,89 (s, 1H), 7,41 (d, 1H), 6,80 (d, 1H), 5,68 (s, 2H), 4,45 (td, 1H), 4,02 (dd, 1H), 3,92-3,82 (m, 1H), 3,81-3,68 (m, 1H), 3,65-1,63 (m, 12H), 0,92 (dd, 2H), - 0,06 (s, 9H); LCMS (M+H)+: 598,2. Etapa 8. 6-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1-il)-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila 6-(3-{2-Ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- 5 d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1-il)-2,3-di-hidrotieno[3,2- c]piridina-7-carbonitrila (10,0 mg, 0,0167 mmol) foi dissolvida em DCM (1,5 mL) e TFA (0,8 mL) foi adicionada. A mistura foi agitada em temperatura ambiente durante 1 hora, em seguida os solventes foram removidos em vá-cuo. O resíduo foi dissolvido em metanol (1 mL) e EDA (0,2 mL) foi adicio- nado e agitado durante 30 minutos. Purificação por meio de HPLC-MS pre-parativa (eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) forneceu o produto como uma base livre (5,4 mg, 69%). 1H RMN (300 MHz, CDCI3): δ 9,42 (br s, 1H), 8,85 (s, 1H), 8,38 (s, 1H), 8,37 (s, 1H), 7,90 (s, 1H), 7,39 (d, 1H), 6,80 (d, 1H), 4,46 (td, 1H), 4,02 (dd, 1H), 3,93-3,84 15 (m, 1H), 3,81-3,70 (m, 1H), 3,61 (dd, 1H), 3,50-3,41 (m, 2H), 3,31-3,19 (m, 3H), 3,14-2,98 (m, 1H), 2,99 (dd, 1H), 1,98-1,85 (m, 1H), 1,82-1,64 (m, 1H); LCMS (M+H)+: 468,0. Exemplo 174, 6-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirrol-1- inetil}pirrolidin-1-il)-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila (enantiôme- 20 ros simples isolados)

Etapa 1. trifluorometanossulfonato de 7-ciano-2,3-di-hidrotieno[3,2-c]piridin- 6-ila
Uma solução de 6-hidróxi-2,3-di-hidrotieno[3,2-c]piridina-7-car- bonitrila (24 mg, 0,13 mmol, de exemplo 171, etapa 6) e N-fenilbis(trifluoro- metanossulfonimida) (60 mg, 0,168 mmol) em acetonitrila (3 mL) e trietilamina (0,038 mL, 0,27 mmol) foi aquecida para 50°C durante 3 horas, em seguida deixada agitar em temperatura ambiente durante a noite. O solvente 5 foi removido em vácuo e o produto foi usado sem outra purificação na etapa de substituição. LCMS (M+H)+: 311,0. Etapa 2. 6-(3-{2-ciano- 1-[3-(7H-pirrolo[2,3-d]pirímidin-4-il)-1 H-pirrol-1 - il]etil}pirrolidin-1-il)-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitríla (enantiômeros simples isolados) 3-pirrolidin-3-il-3-[3-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirrol-1 -iljpropanonitrila (59 mg, 0,14 mmol, de exemplo 33, etapa 3) foi adicionada a uma solução de trifluorometanossulfonato de 7- ciano-2,3-di-hidrotieno[3,2-c]piridin-6-ila (40 mg, 0,13 mmol) em 4-metilmor- folina (45 μL, 0,41 mmol) e DMF (2 mL). A solução foi aquecida para 60°C 15 durante 45 minutos. HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usado para pré-purificar o aduzido protegido por SEM. O eluente foi removido em vácuo. O grupo de proteção SEM foi removido por agitação em 25% de TFA em DCM, seguido por evaporação e agitação com excesso de EDA em metanol. O produto despro- 20 tegido foi purificado por meio de HPLC-MS preparativa (eluindo com um gra diente de ACN/H2O contendo 0,15% de NH4OH). HPLC quiral foi usado para separar o produto racêmico nos enantiômeros simples (Phenomenex Lux Celulose-1 21,2 x 250 mm, 5 μm, eluindo com 30% de EtOH/70% de hexanos at 16 mL/minuto). Pico 1, (primeiro a eluir, tempo de retenção de 17,6 minutos), e Pico 2 (segundo a eluir, tempo de retenção de 37,1 minutos) foram separadamente evaporados. Pico 1: (2,1 mg, 3%), Pico 2: (2,3 mg, 3%). Pico 1: 1H RMN (500 MHz, CD3OD): δ 8,59 (s, 1H), 7,88 (br m, 1H), 7,86 (t, 1H), 7,44 (d, 1H), 7,11 (t, 1H), 7,81 (dd, 1H), 6,94 (d, 1H), 4,50 (td, 1H), 3,99 (dd, 1H), 3,81 (ddd, 1H), 3,69 (m, 1H), 3,60 (dd, 1H), 3,49-3,44 (m, 2H), 3,28-3,23 (m, 3H), 3,11 (dd, 1H), 2,97-2,87 (m, 1H), 1,87 (pd, 1H), 1,76 (dq, 1H); LCMS (M+H)+: 467,1. Pico 2: 1H RMN (500 MHz, CD3OD): δ 8,58 (s, 1H), 7,88 (br m, 1H), 7,85 (t, 1H), 7,42 (d, 1H), 7,10 (dd, 1H), 7,81 (dd, 1H), 6,93 (d, 1H), 4,49 (td, 1H), 3,99 (dd, 1H), 3,80 (ddd, 1H), 3,69 (m, 1H), 3,59 (dd, 1H), 3,49-3,43 (m, 2H), 3,29-3,21 (m, 3H), 3,10 (dd, 1H), 2,97-2,88 (m, 1H), 1,87 (pd, 1H), 1,76 (dq, 1H); LCMS (M+H)+: 467,1. Exemplo 175. 6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1- inetil}pirrolidin-1-il)tieno[3,2-c]piridina-7-carbonitrila (enantiômero simples)

Etapa 1. 6-(3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]piri- midin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-2,3-di-hidrotieno[3,2-c]piridina-7- carbonitrila
Uma mistura de 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (56 mg, 0,13 mmol, de exemplo 15, etapa 3), trifluorometanosulfonato de 7-ciano-2,3-di- hidrotieno[3,2-c]piridin-6-ila (40 mg, 0,13 mmol, de exemplo 174, etapa 1) e 4-metilmorfolina (42 μL, 0,39 mmol) em DMF (2 mL) foi aquecida para 60°C durante 3 horas. HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usada para fornecer produto puri-ficado (33 mg, 43%). LCMS (M+H)+: 598,2. Etapa 2. 6-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 - il]etil}pirrolidin-1 -il) tieno[3,2-c]piridina- 7-carbonitrila
Ácido M-cloroperbenzoico (0,017 g, 0,074 mmol) foi adicionado a uma solução de 6-(3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirro- lo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]etil}pirrolidin-1-il)-2,3-di-hidrotieno[3,2- c]piridina-7-carbonitrila (33 mg, 0,055 mmol) em DCM (2 mL) a 0°C. A reação foi agitada nesta temperatura durante 2 horas. A reação foi diluída com DCM e lavada com NaOH a 0,1 N. A camada orgânica foi secada sobre sulfato de sódio, decantada e concentrada. O produto bruto foi dissolvido em anidrido acético (0,5 mL, 5 mmol) e em seguida aquecido para 140°C durante 24 horas, para 150°C durante 2 horas, em seguida para 160°C durante 2 horas, em seguida no micro-ondas para 200°C durante 70 minutos. O sol-vente foi em seguida removido em vácuo. A mistura reacional bruto foi divi-dida entre NaOH a 0,1 Ne acetato de etila. A porção aquosa foi extraída com mais duas porções de acetato de etila. Os extratos combinados foram secados sobre sulfato de sódio, decantados e concentrados. A desproteção do grupo SEM foi realizada por agitação com 1:1 de TFA em DCM seguido por evaporação e agitação com excesso de EDA em metanol. HPLC-MS preparativa (gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usada para fornecer produto purificado (6 mg, 23%). 1H RMN (300 MHz, DMSO- d6): δ 12,03 (br s, 1H), 8,81 (s, 1H), 8,79 (s, 1H), 8,61 (s, 1H), 8,37 (s, 1H), 7,53 (d, 1H), 7,46 (d, 1H), 7,37 (d, 1H), 6,92 (d, 1H), 4,86-4,76 (m, 1H), 3,94 (dd, 1H), 3,85-3,72 (m, 1H), 3,69-3,52 (m, 2H), 3,42-3,16 (m, 2H), 2,93-2,79 (m, 1H), 1,74-1,60 (m, 2H); LCMS (M+H)+: 466,2. Exemplo 176. 6-(3-{2-ciano-1-[3-(7H-pirrolo[2,3-d]pirimidin-4-íl)-1 H-pirrol-1- il]etil}pirrolidin-1-il)tieno[3,2-c]piridina-7-carbonitrila (racêmica)

Etapa 1. 6-clorotieno[3,2-c]piridina-7-carbonitrila
A uma solução de 6-cloro-2,3-di-hidrotieno[3,2-c]piridina-7-car- bonitrila (72 mg, 0,37 mmol, preparada no exemplo 171, etapa 7) em DCM (10 mL) a 0°C foi adicionado ácido M-cloroperbenzoico (0,11 g, 0,49 mmol) e a reação foi agitada durante 2 horas. A reação foi também diluída com DCM, e lavada com 0,1 N NaOH. A fase aquosa foi novamente extraída com três porções de acetato de etila e estes foram combinados com a solução de DCM. Os extratos combinados foram secados sobre sulfato de sódio, decan- tados e concentrados. O produto bruto foi dissolvido em anidrido acético (3 mL) e aquecido para 140°C durante 16 horas. A mistura foi concentrada e o resíduo foi dissolvida em acetona (2,0 mL), e carbonato de sódio a 0,1 M em água (2,0 mL) foi adicionado. A mistura foi aquecida para 40°C durante 3,5 5 horas. A acetona foi removida em vácuo, e o produto foi extraído da fase aquosa com 3 porções de DCM. Os extratos foram secados sobre sulfato de sódio e concentrados. Cromatografia de coluna rápida, eluindo com 0 a 30% de acetato de etila em hexanos, forneceu o produto como um sólido branco que foi usado na etapa seguinte sem outra purificação. 1H RMN (300 MHz, 10 CDCI3): δ 9,03 (s, 1H), 7,68 (d, 1H), 7,53 (d, 1H); LCMS (M+H)+: 195,0. Etapa 2. 6-(3-{2-ciano-1 -[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 - il]etil}pirrolidin-1 -il) tieno[3,2-c]piridina- 7-carbonitrila
Uma mistura de 3-pirrolidin-3-il-3-[3-(7-{[2-(trimetilsilil)etóxi]me- til}-7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 -iljpropanonitrila (26 mg, 0,059 15 mmol; de exemplo 33, etapa 3), 6-clorotieno[3,2-c]piridina-7-carbonitrila (23 mg, 0,059 mmol) e 4-metilmorfolina (0,019 mL, 0,18 mmol) em DMF (0,3 mL) foi aquecida para 80°C durante 2 horas. Sob resfriamento para temperatura ambiente, a mistura reacional foi dividida entre água e acetato de etila. A camada aquosa foi extraída com mais duas porções de acetato de etila. Os 20 extratos orgânicos combinados foram lavados com salmoura, secados sobre sulfato de sódio, decantados e concentrados. O produto foi agitado com 1:1 de TFA/DCM durante 1 hora, evaporado, em seguida agitado com EDA (0,2 mL) em metanol (1,5 mL). Quando desproteção foi concluída, o produto foi purificado por HPLC-MS preparativa (gradiente de ACN/H2O contendo 25 0,15% de NH4OH) (11 mg, 40%). 1H RMN (400 MHz, DMSO-d6): δ 11,96 (br s, 1H), 8,88 (s, 1H), 8,61 (s, 1H), 8,01 (t, 1H), 7,54 (d, 1H), 7,51 (dd, 1H), 7,46 (d, 1H), 7,16 (t, 1H), 6,97-6,93 (m, 2H), 4,60 (td, 1H), 3,99 (dd, 1H), 3,91-3,83 (m, 1H), 3,77-3,68 (m, 1H), 3,60 (dd, 1H), 3,48 (dd, 1H), 3,25 (dd, 1H), 3,95-2,82 (m, 1H), 1,80-1,60 (m, 2H); LCMS (M+H)+: 464,9. Exemplo 177. 6-(3-{2-ciano-1-[4-(7H-pirrolo[2,3-dJpirimidin-4-il)-1 H-pirazol-1- il]etil}pirrolidin-1-il)-1 H-pirrolo[3,2-clpiridina-7-carbonitrila (enantiômero sim-ples)

Etapa 1. 5-(2-aminoetil)-2-(benzilóxi)-4-metoxinicotinonitríla
Azida de sódio (330 mg, 5,1 mmol) foi adicionada a uma solução de 4-metilbenzenossulfonato de 2-[6-(benzilóxi)-5-ciano-4-metoxipiridin-3- il]etila (1,5 g, 3,4 mmol, preparada no exemplo 171, etapa 3) em DMF (15 mL). A mistura foi aquecida para 60°C durante um total de 85 minutos. Sob resfriamento para temperatura ambiente, a mistura reacional foi dividida entre EtOAc e água. A camada orgânica foi lavada com água duas vezes, NaHCO3 saturado duas vezes, água novamente uma vez, salmoura, secada sobre sulfato de sódio e concentrada para fornecer um óleo amarelo claro. O óleo foi dissolvido em uma mistura de THF (27 mL) e água (3,0 mL), e trife- nilfosfina (0,99 g, 3,8 mmol) foi adicionada. A reação foi agitada durante 16 horas, e o solvente foi em seguida removido em vácuo. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 10% de metanol em DCM contendo 1% trietilamina, forneceu o produto como um óleo amarelo claro (780 mg, 80%). 1H RMN (300 MHz, CDCI3): δ 7,96 (s, 1H), 7,50-7,45 (m, 2H), 7,41-7,27 (m, 3H), 5,46 (s, 2H), 4,32 (s, 3H), 2,86 (t, 2H), 2,63 (t, 2H); LCMS (M+H)+: 284,0. Etapa 2. 6-(benzilóxi)-2,3-di-hidro-1 H-pirrolo[3,2-c]piridina-7-carbonithla
Uma solução de 5-(2-aminoetil)-2-(benzilóxi)-4-metoxinicotinoni- trila (0,78 g, 2,8 mmols) em metanol (80 mL) foi tratada com solução de hi-dróxido de amónio (40 mL, 600 mmol) e foi agitada em temperatura ambiente durante 6 dias. A remoção do solvente em vácuo forneceu o produto como um sólido branco (700 mg, 100%). LCMS (M+H)+: 252,1. Etapa 3. 6-hidróxi-2,3-di-hidro-1 H-pirrolo[3,2-c]pindina-7-carbonitrila
Uma solução de cloreto de acetila (0,50 mL, 7,8 mmols) em me-tanol (100 mL) foi preparada e foi agitada durante 3 horas. A solução foi mis- turada com 6-(benzilóxi)-2,3-di-hidro-1 H-pirrolo[3,2-c]piridina-7-carbonitrila (0,59 g, 2,3 mmols), e foi agitada em temperatura ambiente durante 3 dias. O solvente foi removido em vácuo para fornecer o produto como um pó branco, produção teórica assumida. LCMS (M+H)+: 162,1. Etapa 4. 6-cloro-2,3-di-hidro-1H-pirrolo[3,2-c]piridina-7-carbonitrila
Uma solução de 6-hidróxi-2,3-di-hidro-1 H-pirrolo[3,2-c]piridina-7- carbonitrila (0,38 g, 2,4 mmols) em cloreto de fosforila (12 mL, 130 mmols) foi aquecida para 110°C durante 2 horas. A mistura foi resfriada para tempe-ratura ambiente e vertida em gelo esmagado. NaOH sólido foi adicionado lentamente para a solução resfriada para obter um pH entre 6 e 7. A solução foi extraída com DCM três vezes. Os extratos combinados foram secados sobre sulfato de sódio, decantados e concentrados. O produto bruto foi absorvido em sílica-gel. Cromatografia de coluna rápida, eluindo com um gradiente de 0 a 10% de MeOH em DCM, forneceu o produto como um sólido amarelo claro (230 mg, 49%). 1H RMN (300 MHz, DMSO-d6): δ 8,31 (br s, 1H), 7,76 (s, 1H), 3,73 (t, 2H), 3,02 (dt, 2H); (M+H)+: 180,0/182,1. Etapa 5. 6-(3-{2-ciano-1-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3-d]piri- midin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-2,3-di-hidro-1 H-pirrolo[3,2- c]piridina-7-carbonitrila 3-pirrolidin-3-il-3-[4-(7-{[2-(trimetilsilil)etóxi]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -iljpropanonitrila (0,14 g, 0,32 mmol, de exemplo 15, etapa 3), 6-cloro-2,3-di-hidro-1H-pirrolo[3,2-c]piridina-7-carbonitrila (0,045 g, 0,25 mmol) e 4-metilmorfolina (0,083 mL, 0,75 mmol) foram combinados em NMP (0,10 mL) e a reação foi aquecida a 90°C durante 15 horas.
Sob resfriamento para temperatura ambiente, a mistura reacional foi dividida entre água e acetato de etila. A fase aquosa foi extraída com três porções adicionais de acetato de etila. Os extratos orgânicos combinados foram la-vados com salmoura, secados sobre sulfato de sódio, decantados e concen-trados. Cromatografia de coluna rápida, eluindo com uma mistura de gradi-ente de hexanos:EtOAc:MeOH (100:0:0) a (0:98:2), forneceu o composto desejado (20 mg, 14%). LCMS (M+H)+: 581,1. Etapa 6. 6-(3-{2-ciano-1 -[4-(7H-pirrolo[2,3-d]pirímidin-4-il)-1 H-pirazol-1- il]etil}piirolidin-1 -il) -1 H-pirrolo[3,2-c]piridina- 7-carbonitrila 6-(3-{2-Ciano-1-[4-(7-{[2-(trimetilsilil)etóxí]metil}-7H-pirrolo[2,3- d]pirimidin-4-il)-1 H-pirazol-1 -il]etil}pirrolidin-1 -il)-2,3-di-hidro-1 H-pirrolo[3,2- c]piridina-7-carbonitrila (12 mg, 0,021 mmol) foi tratada com óxido de man ganês (IV) (12 mg, 0,14 mmol) em THF (0,37 mL). A mistura foi agitada em temperatura ambiente durante 1 hora, e em seguida foi aquecida a 68°C du-rante 16 horas. Óxido de manganês (IV) adicional (18 mg, 0,21 mmol) foi adicionado e o aquecimento continuou nesta temperatura durante 24 horas. No resfriamento, a reação foi filtrada, enxaguada com metanol. O filtrado foi concentrado, e o resíduo foi agitado com 1:1 de TFA/DCM durante 1 hora. Os solventes foram novamente removidos em vácuo, e o resíduo foi agitado em MeOH (1 mL) contendo EDA (0,2 mL). HPLC-MS preparativa (eluindo com um gradiente de ACN/H2O contendo 0,15% de NH4OH) foi usada para fornecer o produto como a base livre (2,2 mg, 24%). 1H RMN (400 MHz, CD3OD): δ 8,68 (d, 1H), 8,65 (s, 1H), 8,45 (s, 1H), 8,42 (s, 1H), 7,49 (d, 1H), 7,88 (d, 1H), 6,93 (d, 1H), 6,48 (d, 1H), 4,87-4,79 (m, 1H), 4,05 (dd, 1H), 3,87-3,69 (m, 3H), 3,39 (dd, 1H), 3,18 (dd, 1H), 3,08-2,96 (m, 1H), 1,89-1,82 (m, 2H); LCMS (M+H)+: 449,1. Exemplo 178, 1,1-dióxído de 6-((3S)-3-{2-fluoro-1-[4-(7H-pirrolo[2,3-d]piri- midin-4-il)-1 H-pirazol-1-il1etil}pirrolidin-1-il)-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila (enantiômero simples)

Uma solução de 1,1-dióxido de 6-cloro-2,3-di-hidrotieno[3,2-c]piridina-7-carbonitrila (de exemplo 171, etapa 8; 0,015 g, 0,065 mmol) e 4- (1 -{2-f I uoro-1 -[(3S)-pirrol idin-3-il]eti I}-1 H-pirazol-4-il)-7-{[2-(trimetilsi I i l)etó- xi]metil}-7H-pirrolo[2,3-d]pirimidina (de exemplo 70, etapa 7; 0,020 g, 0,046 mmol) em DMF (1 mL) contendo 4-metilmorfolina (15 μL, 0,14 mmol) foi aquecida para 80°C durante 2 horas. A mistura reacional crua foi dividida en- 5 tre acetato de etila e água. A camada aquosa foi extraída com três porções de acetato de etila. Os extratos combinados foram secados sobre sulfato de sódio, decantados e concentrados. O produto foi desprotegido por agitação em uma solução de TFA e DCM (1:1) durante uma hora, seguido por evaporação e agitação com excesso de etilenodiamina em metanol durante 20 mi- 10 nutos. HPLC-MS preparativa, eluindo com um gradiente de ACN e H2O contendo 0,15% de NH4OH, seguido por liofilização forneceu o produto como a base livre. 1H RMN (300 MHz, CDCI3): δ 10,01 (br s, 1H), 8,85 (s, 1H), 8,38 (s, 1H), 8,36 (s, 1H), 8,34 (s, 1H), 7,42 (d, 1H), 6,81 (d, 1H), 5,04-4,69 (m, 2H), 4,57-4,40 (m, 1H), 1,45 (dd, 1H), 4,04-3,94 (m, 1H), 3,86-3,65 (m, 2H), 15 3,56 (t, 2H), 3,24 (t, 2H), 3,16-2,98 (m, 1H), 2,03-1,72 (m, 2H); LCMS (M+H)+: 493,2.
Exemplo A: Ensaio de JAK Quinase In vitro
Os compostos aqui foram testados quanto à atividade inibidora de JAK alvo de acordo com o seguinte ensaio in vitro descritos em Park e 20 outro, Analytical Biochemistry 1999, 269, 94-104. Os domínios catalíticos de JAK1 humana (a.a. 837-1142), Jak2 (a.a. 828-1132) e Jak3 (a.a. 781-1124) com um rótulo His de terminal N foram expressos usando baculovírus em células de insetos e purificados. A atividade catalítica de JAK1, JAK2 ou JAK3 foi ensaiada avaliando-se a fosforilação de um peptídeo biotinilado. O 25 peptídeo fosforilado foi detectado por Fluorescência Resolvida com o Tempo Homogênea (HTRF). IC50s de compostos foram avaliados para cada quinase nas reações de 40 microL que contêm a enzima, ATP e peptídeo a 500 nM em tampão de Tris a 50 mM (pH 7,8) com NaCl a 100 mM, DTT a 5 mM, e 0,1 mg/mL (0,01%) de BSA. A concentração de ATP nas reações foi de 90 30 μM quanto à Jak1, 30 μM quanto à Jak2 e 3 μM quanto à Jak3 quanto às condições de Km. Para as avaliações de IC50 a 1 mM, a concentração de ATP nas reações fói de 1 mM. As reações foram realizadas em temperatura ambiente durante 1 hora e em seguida interrompidas com 20 μL de EDTA a 45 mM, SA-aPC a 300 nM, Eu-py20 a 6 nM em tampão de ensaio (Perkin Elmer, Boston, MA). Ligação ao anticorpo rotulado por Európio ocorreu durante 40 minutos e o sinal HTRF foi medido em um leitor de placa Fusion (Perkin Elmer, Boston, MA).
Os compostos aqui foram testados quanto à atividade inibidora de JAK1 e JAK2 alvos de acordo com o ensaio de exemplo A (experimentos realizados em Km ou 1 mM como indicado). Os dados são mostrados nas tabelas A a E abaixo. O símbolo "+" indica um IC50 < 50 nM; O símbolo "++" indica um IC50 > 50 e < 100 nM; e o símbolo "+++" indica um IC50 >100 e < 500 nM.Tabela A
Linhagens celulares de câncer dependem das citocinas e portan-to transdução de sinal de JAK/STAT, para crescimento, podem ser semeadas em 6000 células por cavidade (formato de placa de 96 cavidades) em RPMI 1640, 10% de FBS, e 1 nG/mL de citocina apropriada. Os compostos podem ser adicionados às células em DMSO/meio (concentração final de 0,2% de DMSO) e incubados durante 72 horas a 37°C, 5 % de CO2. O efeito do composto sobre a viabilidade celular é avaliado usando o CelITiter-Glo Luminescent Cell Viability Assay (Promega) seguido por quantificação Top-Count (Perkin Elmer, Boston, MA). Efeitos fora de alvo potenciais de com-postos são medidos em paralelo usando uma linhagem celular controlada por não JAK com a mesma leitura de ensaio. Todos os experimentos são tipicamente realizados em duplicata.
As linhagens celulares acima podem também ser usadas para examinar os efeitos de compostos sobre fosforilação de JAK quinases ou substratos ajusantes potenciais tais como proteínas STAT, Akt, Shp2, ou Erk. Estes experimentos podem ser realizados seguindo um jejum de citocina durante a noite, seguido por uma breve pré-incubação com composto (2 horas ou menos) e estimulação de citocina de aproximadamente 1 hora ou menos. As proteínas são em seguida extraídas de células e analisadas por técnicas familiares àquelas ensinadas na técnica incluindo Western blotting ou ELISAs usando anticorpos que podem diferenciar entre proteína fosforila- da e total. Estes experimentos podem utilizar células normais ou de câncer para investigar a atividade de compostos na biologia de sobrevivência de 5 célula de tumor ou nos mediadores de doença inflamatória. Por exemplo, com respeito aos últimos, citocinas tal como IL-6, IL-12, IL-23, ou IFN podem ser usadas para estimular ativação de JAK resultando em fosforilação de proteína(s) STAT e potencialmente em perfis transcricionais (avaliados por tecnologia de disposição ou qPCR) ou produção e/ou secreção de proteínas, 10 tal como IL-17. A habilidade de compostos para inibir estes efeitos mediados por citocina pode ser medida usando técnicas comuns àquelas ensinadas na técnica.
Os compostos aqui podem também ser testados em modelos celulares designados para avaliar sua potência e atividade contra JAKs mu- 15 tantes, por exemplo, a mutação JAK2V617F encontrada em distúrbios proli- ferativos de mieloide. Estes experimentos frequentemente utilizam células dependentes de citocina de linhagem hematológica (por exemplo, BaF/3) em que as JAK quinases selvagens ou mutantes são ectopicamente expressas (James, C., e outro, Nature 434:1144-1148; Staerk, J., e outro, JBC 20 280:41893-41899). As finalidades incluem os efeitos de compostos sobre a sobrevivência celular, proliferação, e proteínas JAK, STAT, Akt, ou Erk fosfo- riladas.
Certos compostos aqui foram ou podem ser avaliados por sua inibição de atividade de proliferação de célula T. Tal como o ensaio pode ser 25 considerado um segundo ensaio de proliferação controlado por citocina (isto é JAK) e também um ensaio simplístico de inibição ou supressão imune de ativação imune. O seguinte é um breve esboço de como tais experimentos podem ser realizados. Células mononucleares de sangue periférico (PBMCs) são preparadas de amostras de sangue todo humano usando método de 30 separação Ficoll Hipaque e células T (fração 2000) podem ser obtidas de PBMCs por elutriação. Células T humanas recentemente isoladas podem ser mantidas em meio de cultura (RPMI 1640 suplementado com 10 % de soro bovino fetal, 100 U/mL de penicilina, 100 μg/mL de estreptomicina) a uma densidade de 2 x 106 células/mL a 37°C durante até 2 dias. Para análise de proliferação de célula estimulada por IL-2, células T são primeiro tratadas com fitoemaglutinína (PHA) a uma concentração final de 10 μg/mL durante 72 h. Após lavar uma vez com PBS, 6000 células/cavidade são semeadas em placas de 96 cavidades e tratadas com compostos em diferentes concentrações no meio de cultura na presença de 100 U/mL de IL-2 humana (ProSpec-Tany TechnoGene; Rehovot, Israel). As placas são incubadas a 37°C durante 72 h e o índice de proliferação é avaliado usando reagentes CelITiter-Glo Luminescent seguindo o protocolo sugerido pela fábrica (Pro- mega; Madison, Wl).
Exemplo C: Eficácia Antitumor In vivo
Os compostos aqui podem ser avaliados em modelos de xeno- enxerto de tumor humano em camundongos imuno-comprometidos. Por e-xemplo, uma variante tumorigênica da linhagem celular de plasmacitoma INA-6 pode ser usada para inocular camundongos SCID subcutaneamente (Burger, R., e outro, Hematol J. 2:42-53, 2001). Animais transportando tumor podem em seguida ser randomizados em grupos de tratamento com fármaco ou veículo e diferentes doses de compostos podem ser administradas por qualquer número das rotinas usuais incluindo oral, i.p., ou infusão contínua usando bombas implantáveis. O crescimento do tumor é seguido em tempo prolongado usando calipers. Além disso, amostras de tumor podem ser co-lhidas em qualquer tempo após o início do tratamento para análises como descrito acima (Exemplo B) para avaliar efeitos compostos na atividade de JAK e trilhas de sinalização ajusantes. Além disso, a seletividade do(s) com-posto^) pode ser avaliada usando modelos de tumor de xenoenxerto que são controlados por outras quinases conhecidas (por exemplo, Bcr-abl) tal como o modelo de tumor K562.
Exemplo D: Teste de Resposta de Hipersensibilidade Retardada de Contato de Pele de Murino
Os compostos aqui podem também ser testados para sua eficácia (de inibir alvos de JAK) no modelo de teste de hipersensibilidade retar- dada de murina controlado por célula T. A resposta de hipersensibilidade de tipo retardada de contato de pele de murina (DTH) é considerada ser um modelo válido de dermatite de contato clínica, e outros distúrbios imunes mediados por linfócito T da pele, tal como psoriase (Immunol Today. 1998 Jan;19(1):37-44), DTH de murina compartilha características múltiplas com psoríase, incluindo o infiltrado imune, o aumento acompanhante em citocinas inflamatórias, e hiperproliferação de ceratinócito. Além disso, muitas classes de agentes que são eficazes no tratamento de psoríase na clínica são também inibidores eficazes da resposta de DTH em camundongos (Agents Actions. 1993 Jan;38(1-2):116-21).
Nos dias 0 e 1, camundongos Balb/c são sensibilizados com uma aplicação tópica, em seu abdomem barbeado com o antígeno 2,4,dinitro-fluorobenzeno (DNFB). No dia 5, as orelhas são medidas quanto a espessura usando um micrometro de engenheiro. Esta medição é registrada e usada como uma referência. Ambas as orelhas dos animais são em seguida desafiadas por uma aplicação tópica de DNFB em um total de 20 μL (10 μL na aurícula interna e 10 pL na aurícula externa) em uma concentração de 0,2 %. Vinte e quatro a setenta e duas horas após o desafio, as orelhas são medidas novamente. O tratamento com os compostos teste foi dado em todas as fases de sensibilização e desafio (dia -1 a dia 7) ou antes de e em toda a fase de desafio (frequentemente a tarde do dia 4 ao dia 7). Tratamento dos compostos teste (em diferente concentração) foi administrado sistemicamente ou topicamente (aplicação tópica do tratamento às orelhas). Eficácia dos compostos teste são indicadas por uma redução na inchação da orelha comparando à situação sem tratamento. Compostos causando a redução de 20% ou mais foram considerados eficazes. Em alguns experimentos, os camundongos são desafiados porém não sensibilizados (controle negativo).
O efeito inibitório (ativação de inibição das trilhas de JAK-STAT) dos compostos teste pode ser confirmado por análise imunoistoquímica. A ativação da(s) trilha(s) de JAK-STAT resulta na formação e translocação de fatores de transcrição funcionais. Além disso, o influxo de células imunes e a proliferação aumentada de ceratinócitos devem também fornecer mudanças de perfil de expressão única na orelha que podem ser investigadas e quanti-ficadas. Seções de orelhas fixadas por formalina e embutidas por parafina (colhidas após a fase de desafio no modelo DTH) são submetidas à análise imunoistoquímica usando um anticorpo que especificamente interage com STAT3 fosforilado (clone 58E12, Cell Signaling Technologies). As orelhas de camundongos são tratadas com compostos teste, veículo, ou dexametasona (um tratamento clinicamente eficaz para psoríase), ou sem qualquer trata-mento, no modelo DTH para comparações. Os compostos teste e a dexame-tasona podem produzir mudanças transcricionais similares tanto qualitativa-mente quanto quantitativamente, e tanto os compostos teste quanto a dexa-metasona podem reduzir o número de células de infiltração. Administração tanto sistemicamente quanto tópica dos compostos teste pode produzir efeitos inibitórios, isto é, redução no número de células de infiltração e inibição das mudanças transcricionais.
Exemplo E: Atividade anti-inflamatória In vivo
Os compostos aqui podem ser avaliados em modelos de roedores ou não roedores designados para replicar uma resposta de inflamação simples ou complexa. Por exemplo, os modelos de roedores de artrite podem ser usados para avaliar o potencial terapêutico de compostos dosados preventivamente ou terapeuticamente. Estes modelos incluem porém não estão limitados à artrite induzida por colágeno de rato ou camundongo, artrite induzida por adjuvante de rato, e artrite induzida por anticorpo de colágeno. Doenças autoimunes incluindo, porém não limitadas à esclerose múltipla, diabetes melito tipo I, uveoretinite, tireôidite, miastenia grave, nefropatias de imunoglobulina, miocardite, sensibilidade das vias aéreas (asma), lúpus, ou colite podem também ser usadas para avaliar o potencial terapêutico dos compostos aqui. Estes modelos são bem estabilizados na comunidade de pesquisa e são familiares àqueles ensinados na técnica (Current Protocols in Immunology, Vol 3., Coligan, J.E. et al, Wiley Press.; Methods in Molecular Biology. Vol. 225, Inflamation Protocols., Winyard, P.G. e Willoughby, D.A., Humana Press, 2003.).
Exemplo F: Modelos de Animal para o Tratamento de Olho Seco, Uveíte e Conjuntivite
Os agentes podem ser avaliados em um ou mais modelos pré- clínicos de olho seco conhecidos por aqueles versados na técnica incluindo, porém não limitados ao modelo de glândula lacrimal de concanavalina A (ConA) de coelho, o modelo de camundongo de escopolamina (subcutâneo ou transdérmico), o modelo de glândula lacrimal de camundongo de botulina, ou quaisquer dos diversos modelos autoimunes de roedores espontâneos que resultam em disfunção da glândula ocular (por exemplo, NOD-SCID, MRL/lpr, ou NZB/NZW) (Barabino e outro, Experimental Eye Research 2004, 79, 613-621 e Schrader e outro, Developmental Opthalmology, Karger 2008, 41, 298-312, cada dos quais é incorporado aqui por referência em sua totali-dade). As finalidades nestes modelos podem incluir histopatologia das glân-dulas oculares e olho (córnea, etc.) e possivelmente o teste Schirmer clássico ou versões modificadas do mesmo (Barabino e outro) que medem a produção de lágrima. A atividade pode ser avaliada dosando por meio de rotinas múltiplas de administração (por exemplo, sistêmica ou tópica) que podem começar antes de ou após doença mensurável existir.
Os agentes podem ser avaliados em um ou mais modelos pré- clínicos de uveíte conhecidos por aqueles versados na técnica. Estes inclu-em, porém não estão limitados a modelos de uveíte autoimune experimental (EAU) e uveíte induzida por endotoxina (EIU). Experimentos de EAU podem ser realizados no coelho, rato, ou camundongo e podem envolver imunização passiva ou ativa. Por exemplo, quaisquer de diversos antígenos retinais podem ser usados para sensibilizar animais para um imunógeno relevante após o qual os animais podem ser desafiados ocularmente com o mesmo antígeno. O modelo EIU é mais agudo e envolve administração local ou sis-têmica de lipopolisacarídeo em doses sub-letais. As finalidades para ambos os modelos EIU e EAU podem incluir exame fundoscópico, histopatologia entre outros. Estes modelos são revistos por Smith e outro, (Immunology e Cell Biology 1998, 76, 497-512, que é incorporado aqui por referência em sua totalidade). A atividade é avaliada dosando por meio de rotinas múltiplas de administração (por exemplo, sistêmica ou tópica) que podem começar antes de ou após doença mensurável existir. Alguns modelos listados acima podem também desenvolver esclerite/episclerite, choriodite, ciclite, ou irite e são portanto úteis em investigar a atividade potencial de compostos para o 5 tratamento terapêutico destas doenças.
Os agentes podem também ser avaliados em um ou mais mode-los pré-clínicos de conjuntivite conhecidos por aqueles ensinados na técnica. Estes incluem, porém não estão limitados a modelos de roedores utilizando cobaia, rato, ou camundongo. Os modelos de cobaia incluem aqueles utili- 10 zando imunização ativa ou passiva e/ou protocolos de desafio imunes com antígenos tal como ovalbumina ou ambrósia americana (revisto em Grone- berg, D.A., e outro, Allergy 2003, 58, 1101-1113, que é incorporado aqui por referência em sua totalidade). Modelos de rato e camundongo são similares de forma geral àqueles na cobaia (também revistos por Groneberg). A ativi- 15 dade pode ser avaliada dosando por meio de rotinas múltiplas de administração (por exemplo, sistêmica ou tópica) que podem começar antes de ou após doença mensurável existir. As finalidades para tais estudos podem incluir, por exemplo, análise histológica, imunológica, bioquímica, ou molecular de tecidos oculares tal como a conjuntiva.
Diversas modalidades da invenção foram descritas. Apesar disso, será entendido que várias modificações podem ser feitas sem divergir do espí-rito e escopo da invenção. Cada das referências mencionadas aqui, incluindo patente, pedido de patente, e literatura de não patente, é incorporada aqui por referência em sua totalidade.