JP4492826B2 - アデノウイルスベクターの産生および精製のための改良された方法 - Google Patents
アデノウイルスベクターの産生および精製のための改良された方法 Download PDFInfo
- Publication number
- JP4492826B2 JP4492826B2 JP52396598A JP52396598A JP4492826B2 JP 4492826 B2 JP4492826 B2 JP 4492826B2 JP 52396598 A JP52396598 A JP 52396598A JP 52396598 A JP52396598 A JP 52396598A JP 4492826 B2 JP4492826 B2 JP 4492826B2
- Authority
- JP
- Japan
- Prior art keywords
- cells
- cell
- virus
- adenovirus
- medium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images










































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/66—Microorganisms or materials therefrom
- A61K35/76—Viruses; Subviral particles; Bacteriophages
- A61K35/761—Adenovirus
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4746—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used p53
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10041—Use of virus, viral particle or viral elements as a vector
- C12N2710/10045—Special targeting system for viral vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10051—Methods of production or purification of viral material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10322—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10332—Use of virus as therapeutic agent, other than vaccine, e.g. as cytolytic agent
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10341—Use of virus, viral particle or viral elements as a vector
- C12N2710/10343—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10351—Methods of production or purification of viral material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2810/00—Vectors comprising a targeting moiety
- C12N2810/50—Vectors comprising as targeting moiety peptide derived from defined protein
- C12N2810/60—Vectors comprising as targeting moiety peptide derived from defined protein from viruses
- C12N2810/6009—Vectors comprising as targeting moiety peptide derived from defined protein from viruses dsDNA viruses
- C12N2810/6018—Adenoviridae
Description
本出願は、1997年11月20に提出された、米国特許分割出願第60/031,329号の一部継続出願である。上記に参照される開示の全体の内容は放棄せずに、本明細書中で参考として特に援用される。
1.発明の分野
本発明は、一般的に、細胞培養およびウイルス産生の分野に関する。より詳細には、哺乳動物細胞の培養のための改良された方法、これらの細胞のアデノウイルスでの感染、およびそれらからの感染性アデノウイルス粒子の産生に関与する。
2.関連技術の説明
アデノウイルスベクター(これは、治療用タンパク質を発現する)は、現在、種々のガン徴候(肺および頭部および頚部ガン)の処置のための診療において評価される。診療試行発展のための、診療用グレードのアデノウイルスベクターについての需要は、劇的に増加している。300人の患者の臨床試行のために計画された一年間の需要は、約6×1014PFUに達し得た。
伝統的には、アデノウイルスは、市販の組織培養フラスコまたは「セルファクトリー(cellfactory)」において産生される。細胞に感染したウイルスは採集され、そして凍結融解されて、粗細胞溶解物の形態の細胞からウイルスを放出する。次いで、産生した粗細胞溶解物(CCL)は、二重CsCl勾配超遠心分離によって精製される。代表的に報告される100シングルトレイセルファクトリーから得られるウイルスは、約6×1012FPUである。明らかに、この伝統的なプロセスを用いて、必要な量のウイルスを産生することが実行できないようになる。新規な大スケール化可能(scaleable)ならびに確認可能(validatable)産生および精製プロセスが、増加する需要に見合うように開発されなければならない。
CsCl勾配超遠心分離の精製処理能力は限られているので、遺伝子治療適用のためのアデノウイルスベクターについての需要に応じ得ない。それゆえ、大規模アデノウイルスベクター産生を達成するために、CsCl勾配超遠心分離以外の精製方法が開発されなければならない。ウイルスのクロマトグラフィー精製の報告は、組換えタンパク質の精製のためのクロマトグラフィーの幅広い適用にもかかわらず、非常に限られている。サイズ排除、イオン交換、およびアフィニティークロマトグラフィーは、レトロウイルス、ダニ媒介性脳炎ウイルス、および植物ウイルスの精製について、変動する程度の成功とともに評価されている(Crooksら、1990;Aboudら、1982;McGrathら、1978、SmithおよびLee,1978;O’NeilおよびBalkovic,1993)。より少ない調査も、アデノウイルスのクロマトグラフィー精製においてなされている。この調査活性の不足は、アデノウイルスのための有効な(スケールアップ可能でないにもかかわらず)CsCl勾配超遠心分離精製法の存在に部分的に起因し得る。
近年、Huygheら(1996)は、金属キレートアフィニティクロマトグラフィーと組み合わせてイオン交換クロマトグラフィーを用いる、アデノウイルスベクター精製を報告した。CsCl勾配超遠心分離と同様のウイルス純度が報告された。不運なことに、23%のウイルスのみが、二重カラム精製プロセスの後に回収された。この低いウイルス回収に寄与するプロセス因子は、著者らによって利用される、細胞からウイルスを放出するために、細胞を溶解する凍結/融解工程、および2つのカラム精製手順である。
明らかに、このような手順のための増加する需要に応じる、高収量の産物を回収するアデノウイルスベクター産生の有効な、そしてスケールアップ可能方法についての需要が存在する。
発明の要旨
本発明は、アデノウイルスの産生および精製のための新規なプロセスを記載する。この新規な産生プロセスは、スケーラビリティおよび有効性能(validatability)のみでなく、CsCl勾配超遠心分離を用いて達成されるものに匹敵するウイルス純度を提供する。
本発明は、アデノウイルスを産生するための方法を提供し、これは、低灌流速度にて培地中で宿主細胞を増殖させる工程、宿主細胞をアデノウイルスで感染させる工程、宿主細胞を採集および溶解して、粗細胞溶解物を産生する工程、粗細胞溶解物を濃縮する工程、粗細胞溶解物の緩衝液を交換する工程、および粗細胞溶解物中に含有する核酸の濃度を低減する工程を含む。
特定の実施態様において、この方法はさらに、クロマトグラフィーを用いて、溶解物からアデノウイルス粒子を単離する工程を含む。特定の実施態様において、単離工程は、本質的には、シングルクロマトグラフィー工程からなる。他の実施態様において、クロマトグラフィー工程は、イオン交換クロマトグラフィーである。特定の好ましい実施態様において、イオン交換クロマトグラフィーは、約7.0〜約10.0の範囲のpHで行われる。より好ましい実施態様において、イオン交換クロマトグラフィーは、陰イオン交換クロマトグラフィーである。特定の実施態様において、陰イオン交換クロマトグラフィーは、DEAE、TMAE、QAE、またはPEIを利用する。他の好ましい実施態様において、陰イオン交換クロマトグラフィーは、Toyoperal Super Q 650M、MonoQ、Source Q、またはFrantogel TMAEを利用する。
本発明の特定の実施態様において、培地中のグルコース濃度は、約0.7〜約1.7g/Lに維持される。特定の他の実施態様において、交換緩衝液は、ダイアフィルトレーション工程に関与する。
本発明の好ましい実施態様において、アデノウイルスは、外因性遺伝子構築物をコードするアデノウイルスベクターを含む。特定のこのような実施態様において、遺伝子構築物は、プロモーターに作動可能に連結される。特定の実施態様において、プロモーターは、SV40 IE、RSV LTR、βアクチンもしくはCMV IE、アデノウイルス主要後期、ポリオーマF9-1、またはチロシナーゼである。本発明の特定の実施態様において、アデノウイルスは、複製コンピテントなアデノウイルスである。他の実施態様において、アデノウイルスは、少なくともE1領域の一部を欠く。特定の局面において、アデノウイルスは、少なくともE1Aおよび/またはE1B領域の一部を欠く。他の実施態様において、宿主細胞は、複製を完了し得る。特定の好ましい実施態様において、宿主細胞は、293細胞である。
本発明の好ましい実施態様において、外因性遺伝子構築物が、治療用遺伝子をコードすることが意図される。例えば、治療用遺伝子は、アンチセンスras、アンチセンスmyc、アンチセンスraf、アンチセンスerb、アンチセンスsrc、アンチセンスfms、アンチセンスjun、アンチセンスtrk、アンチセンスret、アンチセンスgsp、アンチセンスhst、アンチセンスbcl、アンチセンスabl、Rb、CFTR、p16、p21、p27、p57、p73、C-CAM、APC、CTS-1、zac1、scFV ras、DCC、NF-1、NF-2、WT-1、MEN-I、MEN-II、BRCA1、VHL、MMAC1、FCC、MCC、BRCA2、IL-1、IL-2、IL-3、IL-4、IL-5、IL-6、IL-7、IL-8、IL-8、IL-9、IL-10、IL-11、IL-12、GM-CSF G-CSF、チミジンキナーゼ、またはp53をコードし得る。
本発明の特定の局面において、低張液、高張液、凍結溶解、超音波処理、衝撃噴出(impinging jet)、微小流動化(microfluidization)、または界面活性剤を用いて、エキソサイチュで(ex situ)細胞は採集されそして溶解され得る。他の局面において、低張液、高張液、または界面活性剤を用いて、インサイチュで細胞は採集されそして溶解される。本明細書中で使用されるように、用語「インサイチュ」は、CellCubeTMのような組織培養装置内に位置する細胞をいい、そして「エキソサイチュ」は、組織培養装置から取り出された細胞をいう。
特定の実施態様において、細胞は、界面活性剤を用いて溶解されそして採集される。好ましい実施態様において、界面活性剤は、Thesit▲R▼、NP-40▲R▼、Tween-20▲R▼、Brij-58▲R▼、Triton▲R▼X-100、またはオクチルグルコシドであり得る。本発明の他の局面において、溶解は、感染細胞の自己溶解を通じて達成される。本発明の特定の他の局面において、細胞溶解物は、Benzinase▲R▼、またはPulmozyme▲R▼で処理される。
特定の実施態様において、方法はさらに、膜濾過を使用する濃縮工程を含む。特定の実施態様において、濾過は、接線方向流動(tangential flow)濾過である。好ましい実施態様において、濾過は、100〜300K NMWC、再生セルロース、またはポリエーテルスルホン膜を利用し得る。
本発明はまた、培地中で低灌流速度にて宿主細胞を増殖させる工程、宿主細胞をアデノウイルスで感染させる工程、宿主細胞を採取および溶解して、粗細胞溶解物を産生する工程、および粗細胞溶解物に夾雑する核酸の濃度を低減する工程を含むプロセスに従って産生される、アデノウイルスを提供する。
本発明の他の局面は、アデノウイルスの精製のための方法を提供し、これは、宿主細胞を増殖させる工程、宿主細胞をアデノウイルスで感染させる工程、細胞を界面活性剤に接触させることによって、宿主細胞を採取および溶解して、粗細胞溶解物を産生する工程、粗細胞溶解物を濃縮する工程、粗細胞溶解物の緩衝液を交換する工程、および粗細胞溶解物に夾雑する核酸の濃度を低減する工程を含む。
特定の実施態様において、界面活性剤は、Thesit▲R▼、NP-40▲R▼、Tween-20▲R▼、Brij-58▲R▼、TritonX-100▲R▼、またはオクチルグルコシドであり得る。より好ましい実施態様において、界面活性剤は、溶解溶液中に、約1%(w/v)の濃度で存在する。
本発明の他の局面において、宿主細胞を増殖させる工程、宿主細胞をアデノウイルスで感染させる工程、細胞を界面活性剤に接触させることによって、宿主細胞を採取および溶解して、粗細胞溶解物を産生する工程、粗細胞溶解物を濃縮する工程、粗細胞溶解物の緩衝液を交換する工程、および粗細胞溶解物に夾雑する核酸の濃度を低減する工程を含むプロセスに従って産生される、アデノウイルスが提供される。
なお別の実施態様において、本発明は、アデノウイルスを精製するための方法を提供し、これは血清非含有培地において宿主細胞を増殖させる工程;宿主細胞をアデノウイルスで感染させる工程;宿主細胞を採取および溶解して、粗細胞溶解物を産生する工程;粗細胞溶解物を濃縮する工程;粗細胞溶解物の緩衝液を交換する工程;および粗細胞溶解物に夾雑する核酸の濃度を低減する工程を含む。好ましい実施態様において、細胞は、細胞懸濁物または足場依存性培養として独立して増殖され得る。
好ましい実施態様において、宿主細胞は、血清非含有培地における増殖のために順応される。より好ましい実施態様において、血清非含有培地における増殖のための改作は、増殖培地のウシ胎児血清含有量における一連の減少を含む。より好ましくは、血清非含有培地は、0.03%v/v未満のウシ胎児血清含有量を含む。
他の実施態様において、この方法はさらに、アデノウイルスを溶解物からクロマトグラフィーによって単離する工程を含む。好ましい実施態様において、単離は、本質的に一回のクロマトグラフィー工程からなる。より好ましくは、クロマトグラフィー工程は、イオン交換クロマトグラフィーである。
宿主細胞を血清非含有培地において増殖させる工程;宿主細胞をアデノウイルスで感染させる工程;宿主細胞を採取および溶解して、粗細胞溶解物を産生する工程;粗細胞溶解物を濃縮する工程;粗細胞溶解物の緩衝液を交換する工程;および粗細胞溶解物に夾雑する核酸の濃度を低減する工程を含むプロセスに従って産生されるアデノウイルスもまた、本発明によって意図される。
本発明はさらに、血清非含有培地における増殖に順応した293宿主細胞を提供する。特定の局面において、血清非含有培地における増殖のための改作は、増殖培地のウシ血清含有量における一連の減少を含む。特定の好ましい実施態様において、細胞は、懸濁培養における増殖に順応される。特定の実施態様において、本発明の細胞は、IT293SF細胞と称される。これらの細胞は、特許手順の目的のための微生物の寄託の国際認識に基づくブタペスト条約の要求に合わせるために、アメリカン組織培養コレクション(ATCC)に寄託された。細胞は、Dr.Shuyuan Zhangによって、Introgen Therapeutics,Inc.(Houston,Tx.)の利益に基づいて、1997年11月17日に寄託された。IT293SF細胞株は、293細胞株の本明細書中に記載の血清非含有懸濁物への順応に由来する。細胞は、100mg/Lヘパリンおよび0.1%pluonic F-68を補充したIS 293血清非含有培地(Irvine Scientific.Santa Ana,Ca.)において培養され得、そしてヒトアデノウイルス感染に許容的である。
本発明の他の目的、性質、および利点は、以下の詳細な説明から明らかになる。しかし、詳細な説明および特定の実施例は、本発明の好ましい実施態様を示すが、例証のためにのみ与えられる。なぜなら、本発明の精神および範囲内の種々の変更および改変は、この詳細な説明から、当業者に明らかになるからである。
図面の詳細な説明
以下の図面は、本明細書の一部を形成し、そして本発明の特定の局面のさらなる実証が含まれる。本発明は、1つ以上のこれらの図面を、本明細書中に示される特定の実施態様の詳細な説明と見あわせて参照することによって、より理解され得る。
図1Aおよび図1B。「高」(図1A)および「低」(図1B)として特徴づけられる培地灌流速度を用いる産生実行からのウイルス溶液のHPLCプロフィール。
図2。CellCubeTMからの粗細胞溶解物(CCL)のHPLCプロフィール(実線A260;破線A280)。
図3A、図3B、図3C、図3D、および図3E。異なる界面活性剤を用いたCellCubeTMからの溶解溶液のHPLCプロフィール。図3A Thesit▲R▼。図3B Triton▲R▼X-100。図3C NP-40▲R▼。図3D Brij-58▲R▼。図3E Tween▲R▼20。界面活性剤濃度:1%(w/v)。溶解温度:室温。(実線A260;破線A280)。
図4Aおよび図4B。Benzonase処理の前(図4A)および後(図4B)のウイルス溶液のHPLCプロフィール。(実線A260;破線A280)。
図5。1M NaClの存在下のBenzonase処理後のウイルス溶液のHPLCプロフィール。(実線A260;破線A280)。
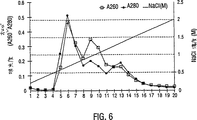
図6。20mM Tris+1mM MgCl2+0.2M NaCl(pH=7.5)の緩衝液A条件下のAdCMVp53ウイルスの精製。
図7。20mM Tris+1mM MgCl2+0.2M NaCl(pH=9.0)の緩衝液A条件下のAdCMVp53ウイルスの精製。
図8A、図8B、および図8C。精製の間に得られた画分のHPLC分析。図8A 画分3。図8B 画分4。図8C 画分8。(実線A260;破線A280)。
図9。20mM Tris+1mM MgCl2+0.3M NaCl(pH=9)の緩衝液A条件下のAdCMVp53ウイルスの精製。
図10A、図10B、図10C、図10D、および図10E。粗対精製の間に得られた画分およびCsCl勾配精製したウイルスのHPLC分析。図10A 粗ウイルス溶液。図10B フロースルー。図10C ピーク数1。図10D ピーク数2。図10E CsCl精製したウイルス。(実線A260;破線A280)。
図11。5cm idカラムからのHPLC精製プロフィール。
図12。SDS-PAGEで検出された主要なアデノウイルス構造タンパク質。
図13。ウエスタンブロットアッセイの検出されたレベルとしての、精製ウイルスにおけるBSA濃度。
図14。CellCubeTMから作製された粗細胞溶解物材料についてのクロマトグラム。
図15。Toyopearl Super Q樹脂を用いる本発明の方法を用いて精製した処理ウイルス溶液の溶出プロフィール。
図16Aおよび図16B。精製プロトコルからのウイルス画分のHPLC分析。図16A 第1精製工程からのウイルス画分のHPLCプロフィール。図16B 第2精製からのウイルス画分のHPLCプロフィール。(実線A260;破線A280)。
図17。低培地灌流速度下での1%Twee▲R▼採集ウイルス溶液の精製。
図18。低培地灌流速度下で産生したウイルス画分のHPLC分析。
図19A、図19B、および図19C。カラム精製したウイルスの分析。図19A SDS-PAGE分析。図19B BSAについてのウエスタンブロット。図19C 夾雑する核酸濃度を決定するための核酸スロットブロット。
図20A、図20B、図20C、図20D、図20E、および図20F。Toyopearl Super Q 350M樹脂の行為能力研究。図20A 1:1のロード比からのフロースルー。図20 B1:1のロード比からの精製ウイルス。図20C 2:1のロード比からのフロースルー。図20D 2:1のロード比からの精製ウイルス。図20E 3:1のロード比からのフロースルー。図20F 3:1のロード比からの精製ウイルス。(実線A260;破線A280)。
図21。等密度CsCl超遠心分離カラム精製したウイルス。
図22。カラム精製したウイルスAに存在するインタクトなウイルスのHPLCプロフール。インタクトなウイルスB。欠損ウイルス。(実線A260;破線A280)。
図23。AdCMVp53についての産生および精製フローチャート。
例示的な実施態様の説明
アデノウイルスは、真核生物遺伝子発現およびワクチン開発に首尾良く使用され得ることが示されている。近年、動物研究は、組換えアデノウイルスが、遺伝子治療に使用され得ることを実証している。組換えアデノウイルスの異なる組織への投与における首尾良い研究は、治療におけるアデノウイルスベクターの有効性を提供している。この成功は、このようなベクターのヒト治療試行における使用を導いている。現在、種々の治療に使用されるアデノウイルスベクターの産生のための増加した需要が存在する。現在入手可能な技術は、このような需要に見合うには不十分である。本発明は、このような治療に使用するための大量のアデノウイルスの産生のための方法を提供する。
本発明は、複製欠損組換えアデノウイルスの産生および精製のために開発されているプロセスに関する。産生プロセスは、細胞増殖およびウイルス産生のためのCellcibeTMバイオリアクターの使用に基づいている。細胞増殖および培養のウイルス産生段階に使用される所定の灌流速度は、ウイルスの下流精製における有意な効果を有する。より詳細には、低培地灌流速度は、ウイルス産生を改良する。さらに、ウイルス産生段階の最後にCellcibeTM中の細胞を溶解するのに使用される、緩衝化界面活性剤からなる溶解溶液もまた、プロセスを改良する。これらの2つの利点とともに、回収した粗ウイルス溶液は、粗ウイルス溶液中に夾雑する核酸を減少するための濃縮/ダイアフィルトレーションおよびヌクレアーゼ処理の後、シングルイオン交換クロマトグラフィー実行を用いて精製され得る。カラム精製したウイルスは、二重CsCl勾配精製ウイルスのものと相対的に等しい純度を有する。ウイルス産物の全プロセス回収率は、70%±10%である。これは、Huygheら(1996)によって報告された結果を越える、有意な改良である。CsCl勾配超遠心分離と比較して、カラム精製は、より一貫した、スケール化可能、有効可能、より早い、そしてあまり高価でないという利点を有する。この新規なプロセスは、遺伝子治療のためのアデノウイルスベクターの製造のための技術における有意な改良を示す。
それゆえ、本発明は、アデノウイルスベクターを産生および精製する目的のための大規模培養系および精製におけるこれらの改良の利点を得るために設計される。このような系についての種々の成分、およびそれとともにアデノウイルスを産生する方法は、以下に詳細に示される。
1.宿主細胞
A)細胞
好ましい実施態様において、アデノウイルスベクターの作製および増殖は、唯一のヘルパー細胞株(293と称される)に依存する。この細胞は、ヒト胎児性腎臓細胞から、アデノウイルス血清型5(Ad5)DNAフラグメントによって形質転換されており、そしてE1タンパク質を構成的に発現する(Grahamら、1977)。E3領域は、Adゲノムから不必要であるので(JonesおよびShenk,1978)、最新のAdベクター(293細胞の補助を伴う)は、E1、E3、またはその両方の領域において外来DNAを保有する(GrahamおよびPrevec,1991;Bettら、1994)。
本発明の最初の局面は、アデノウイルスゲノムの部分を発現する組換え細胞株である。これらの細胞株は、アデノウイルス組換えベクターおよび特定のアデノウイルスにおいて欠損を有する(すなわち、これらのウイルスおよびベクターの増殖に「許容的」である)ヘルパーウイルスの複製を支持し得る。組換え細胞はまた、複製非コンピテントなアデノウイルスベクターにおいて欠損を完了しそして複製を支持する能力のために、ヘルパー細胞ともいわれる。アデノウイルスヘルパー細胞のための原型は、293細胞株であり、これは、アデノウイルスE1領域を含む。293細胞は、トランスで複製に必要なE1活性エレメントを提供することにより、E1機能を欠くアデノウイルスベクターの複製を指示する。
本発明のヘルパー細胞は、哺乳動物細胞に由来し、そして好ましくは、ヒト胎児性腎臓細胞のような霊長類細胞に由来する。種々の霊長類細胞が好ましく、そしてヒトまたはヒト胎児性腎臓細胞が最も好ましいが、ウイルスの複製を支持し得る任意の型の細胞が、本発明の実施において容認される。他の細胞型としては、Vero細胞、CHO細胞、または細胞がアデノウイルス許容性である細胞である限りは細胞培養技術が確立されている任意の真核生物細胞が挙げられ得るがこれらに限定されない。用語「アデノウイルス許容性」は、アデノウイルス(単数または複数)ベクターが、細胞環境内で全体の細胞内ウイルス生存周期を完了し得ることを意味する。
ヘルパー細胞は、存在する細胞株(例えば、293細胞株)に由来し得るか、または新規に開発され得る。このようなヘルパー細胞は、トランスでアデノウイルスゲノムにおける欠損に相補するのに必要な、または別の欠損アデノウイルスベクターの複製を指示するアデノウイルス遺伝子(例えば、E1、E2、E4、E5、および後期機能)を発現する。アデノウイルスゲノムの特定の部分(E1領域)は、相補する細胞株を作製するのに既に使用されている。取り込まれているかまたはエピソームのいずれであっても、ウイルス複製起点を欠くアデノウイルスゲノムの部分は、細胞株が導入される場合、細胞が野生型アデノウイルスで重感染された場合でさえ、複製されない。さらに、主要後期ユニットの転写は、ウイルスDNA複製の後であるので、アデノウイルスの後期機能は、細胞株から有意に発現され得ない。従って、E2領域(これは、後期機能(L1-5)と重複する)は、ヘルパーウイルスによって提供されるが、細胞株によっては提供されない。代表的には、本発明の細胞株は、E1および/またはE4を発現する。
本明細書中で使用されるように、用語「組換え」細胞は、遺伝子(例えば、アデノウイルスゲノムまたは別の細胞からの遺伝子)が導入されている細胞をいうことが意図される。それゆえ、組換え細胞は、組換え的に導入された遺伝子を含まない天然に存在する細胞から区別可能である。従って、組換え細胞は、「人の手」を介して導入された遺伝子(単数または複数)を有する細胞である。
複製は、未感染細胞、または1つ以上のヘルパーウイルスで感染した細胞の層を、ウイルス粒子と接触させ、続いて細胞をインキュベーションすることによって決定される。ウイルスプラーク、または細胞層における無細胞領域の形成は、特定のウイルス産物の発現によって引き起こされる細胞溶解の結果である。細胞溶解は、ウイルス複製の指標である。
複製可能なウイルスとともに使用される得るか、または複製欠損ウイルスとともに使用するために相補する宿主細胞に変換され得る、他の有用な哺乳動物細胞株の例は、VeroおよびHeLa細胞、ならびにチャイニーズハムスター卵巣の細胞株、W138、BHK、COS-7、HepG2、3T3、RIN、およびMDCK細胞である。
B)選択培地における増殖
特定の実施態様において、所望でない細胞の増殖を防止する選択系を使用することが有用であり得る。これは、細胞株を選択可能なマーカーで持続的に形質転換することによって、または選択可能なマーカーをコードするウイルスベクターで細胞株を形質導入または感染することによって達成され得る。いずれの場合でも、適切な薬物または選択的化合物で形質転換/形質導入した細胞の培養は、細胞集団において、マーカーを保有するこれらの細胞の増強を生じる。
マーカーの例としては、それぞれ、tk-、hgprt-、またはaprt-細胞における、HSVチミジンキナーゼ、ヒポキサンチン-グアニンホスホリボシルトランスフェラーゼ、およびアデニンホスホリボシルトランスフェラーゼ遺伝子が挙げられるが、これらに限定されない。また、抗代謝産物耐性は、dhfr(メトトレキサートへの耐性を付与する);gpt(ミコフェノール酸への耐性を付与する);neo(アミノグリコシドG418への耐性を付与する);およびhygro(ハイグロマイシンへの耐性を付与する)についての選択の基準として使用され得る。
C)血清ウィーニングの増殖
足場依存性細胞の無血清懸濁培養への血清ウィーニング順応は、組換えタンパク質(Berg,1993)およびウイルスワクチン(Perrin,1995)の産生のために使用されている。293A細胞の無血清懸濁培養への順応は、近年まであまり報告されていなかった。Gilbertは、アデノウイルスおよび組換えタンパク質産生のための、293A細胞の無血清懸濁培養への順応を報告した(Gilbert,1996)。同様の順応方法は、アデノウイルス産生のためのA549細胞の無血清懸濁培養への順応のために使用されていた(Morrisら、1996)。細胞特異的ウイルスは、順応した懸濁物細胞を生じるが、親付着細胞において達成されたものより約5〜10倍低い。
同様の血清ウィーニング手順を用いて、本発明者らは293A細胞を無血清懸濁培養に首尾良く順応させている(293SF細胞)。この手順において、293細胞を、Tフラスコにおいて連続希釈したFBS濃度によって、市販の293培地に順応させた。簡単には、培地中の最初の血清濃度は、T-75フラスコ中約10%FBS DMEM培地であり、そして細胞を、続いて培地中のFBS濃度を連続的に低くすることによって、Tフラスコ中の無血清IS 293培地に順応させた。T-75フラスコにおける6継代の後、FBS%は、約0.019%および293細胞であることが推定された。細胞は、撹拌フラスコに移される前に、Tフラスコにおいてさらに2回継代培養された。本明細書中以下に記載される結果は、細胞が、無血清培地(IS293培地、Irvine Scientific,Santa Ana,CA)において満足に増殖することを示す。細胞の平均倍加時間は、18〜24時間であり、培地の変更なしで、4〜10×106細胞/mlのオーダーの定常細胞濃度に達した。
D)細胞の懸濁培養への順応
2つの方法論が、293細胞を懸濁培養に順応させるのに使用されてきている。Grahamは、ヌードマウスにおける3連続継代によって、293A細胞を懸濁培養(293N3S細胞)に順応させた(Graham,1987)。懸濁物293N3S細胞は、E1-アデノウイルスベクターを支持し得ることが見いだされた。しかし、Garnierら(1994)は、293N35細胞は、懸濁物における比較的長い開始誘導期、低い増殖速度、および強力に群がる傾向を有することを観察した。
使用されている第2の方法は。293A細胞の懸濁物増殖への逐次的順応である(Cold Spring HarborLaboratories,、293S細胞)。Garnierら(1994)は、アデノウイルスベクターからの組換えタンパク質の産生のための293S細胞の使用を報告した。著者らは、293S細胞が、無カルシウム培地においてほとんど塊にならず、そしてウイルス感染の時点での新鮮な培地への交換が、タンパク質産生を有意に増加し得ることを見いだした。グルコースが、培地の交換なしでは、培養における制限因子であることが見いだされた。
本発明において、無血清状態における増殖に順応した293細胞は、懸濁培養に順応した。細胞は、懸濁培養のために、約1.18E+5vc/mL〜約5.22E+5vc/mLの最初の細胞密度で、無血清の250mL撹拌懸濁培養(100mL作動容量)に移された。培地は、細胞の凝集を防ぐために、ヘパリンを補充され得る。この細胞培養系は、細胞密度の何らかの増加を可能にする一方、細胞生存度が維持される。一旦、これらの細胞が培養において増殖されると、これらの細胞は、撹拌フラスコにおいて、さらに約7継代、継代培養される。細胞の倍加時間は、倍加時間が約1.3日である首尾良い継代の終点(すなわち付着細胞培養において10%FBS培地の細胞の1.2日と匹敵する)まで、段階的に減少される。ヘパリンを補充した無血清IS 293培地において、ほとんど全ての細胞は、懸濁培養において細胞の凝集を形成しない個別の細胞として存在した。
2.細胞培養系
感染性ウイルスベクターを産生する能力は、製薬産業、特に遺伝子治療の文脈において、ますます重要である。ここ十年にわたって、バイオテクノロジーにおける進歩は、治療法、ワクチン、およびタンパク質産生機として潜在的に使用されている多数の重要なウイルスベクターの産生をもたらした。哺乳動物培養におけるウイルスベクターの使用は、ジスルフィド依存性折り畳みおよびグリコシル化のような複合タンパク質構造を転写後プロセスするそれらの能力において、細菌または他の下等生命体(lifeform)宿主において産生されるタンパク質を越える利点を有する。
ウイルスベクターの産生のための細胞培養の開発は、哺乳動物細胞培養において非常に有効なベクター系の設計および構築のための技術の分子生物学における開発、一連の有用な選択マーカー、遺伝子増幅体系、および誘導したベクターからの最終的に生物学的に活性な分子を確保することに関与する生化学および細胞機構のより総合的な理解によって、非常に助けられている。
頻繁に、製造のスケールアップの下流(この場合、細胞溶解以降)側に影響する因子は、宿主としての細胞株を発現系について選択する前に考慮されなかった。また、長時間非常に高密度の培養物を持続し得るバイオリアクター系の開発は、低コストの増加した産生についての増加する需要に応えていない。
本発明は、近年入手可能なバイオリアクター技術の利点を利用する。バイオリアクターにおいて増殖する本発明の細胞は、本発明のアデノウイルスベクターによって感染され得る完全に生物学的に活性な細胞の大規模産生を可能にする。低灌流速度で系を操作し、そして感染する粒子の精製に異なる体系を適用することによって、本発明は、大量の高度に精製した産物を産生するのに容易にスケール化可能である精製ストラテジーを提供する。
バイオリアクターは、懸濁および足場依存性細胞培養の両方からの生物学的産物の産生のために、広範に使用されている。アデノウイルスベクター産生のために最も広範に使用される手順は、足場依存性ヒト胎児性腎臓細胞(293細胞)である。アデノウイルスベクター産生のために開発されるバイオリアクターは、高いプロデューサー細胞密度および高いウイルス収率を達成するために、高い容量特異的培養表面積の特徴を有さなければならない。滅菌したタンクバイオリアクターにおけるマイクロキャリア細胞培養は、非常に高い容量特異的培養表面積を提供し、そしてウイルスワクチンの産生のために使用されている(Griffiths,1986)。さらに、滅菌したタンクバイオリアクターは、スケール化可能であることが工業的に証明されている。また、Corning-Costarによって製造されるマルチプレートCellcubeTM細胞培養系は、非常に高い容量特異的培養表面積を提供する。細胞は、コンパクトキューブの形状でともに密封封入された培養プレートの両側で増殖する。滅菌したバイオリアクターとは異なり、CellcubeTM培養ユニットは、使い捨てである。これは、使い捨て系に関連して資源の消費費用、品質制御費用、および品質保証費用の削減のために、診療用産物の初期段階の産生で非常に所望される。異なる系によって提供される利点の考慮において、滅菌したタンクバイオリアクターおよびCellcubeTM系の両方は、アデノウイルスの産生について評価された。
A)足場依存性培養物対非足場依存性培養物。
動物およびヒト細胞は、2つの態様においてインビトロで増殖され得る:非足場依存性細胞は、培養物の空間中に懸濁物中で自由に増殖する;または足場依存性細胞は、それらの増殖のために、固体基質への接着を必要とする(すなわち、単層型の細胞増殖)。
続いて確立された細胞株からの非足場依存性すなわち懸濁培養は、細胞および細胞生成物の大規模な産生の最も広範に使用される手段である。微生物(細菌および酵母)発酵技術に基づく大規模懸濁培養は、哺乳動物細胞産物の製造のための明らかな利点を有する。プロセスは、比較的簡単に操作され、そしてスケールアップに向いている。均一な条件は、温度、溶存酸素、およびpHの正確なモニタリングおよび制御を可能にし、そして培養の代表的なサンプルが得られ得ることを保証にするリアクターにおいて提供され得る。
しかし、懸濁培養した細胞は、生物学的製剤の産生に、いつも使用され得るとは限らない。懸濁培養物はさらに、腫瘍原生能力を有すると考えられ、従って、産生のための基質としてのそれらの使用は、ヒトおよび獣医学適用において得られる産物の使用において制限を加える(Petricciani,1985;Larsson,1987)。足場依存性培養に反対の懸濁培養において増殖したウイルスは、ウイルスマーカーにおける迅速な変化を時々引き起こし得、減少した免疫原性を導く(Bahnemann,1980)。最終的に、時々、組換え細胞株でさえ、懸濁における同じ細胞株と比較して、足場依存性培養で増殖させた場合、相当高い量の産物を分泌し得る(NilssonおよびMosbach,1987)。これらの理由のために、異なる型の足場依存性細胞は、異なる生物学的製剤産物の産生において、広範に使用される。
B)懸濁のためのリアクターおよびプロセス。
滅菌したタンクにおける哺乳動物培養の大規模懸濁培養が試みられた。バイオリアクターのための計測および制御は、発酵の設計に沿って、関連した微生物適用から順応させた。しかし、より遅く増殖する哺乳動物培養において、夾雑制御の増大する需要を認識したので、改良された無菌設計は、迅速に実行され、これらのリアクターの信頼性を改良させた。計測および制御は、他の発酵槽において見いだされるのと基本的に同様であり、そして撹拌速度、温度、溶存酸素、およびpH制御を含む。濁度(存在粒子の関数)、電気容量(存在生存細胞の関数)、グルコース/乳酸、炭酸/重炭酸、および二酸化炭素のオンラインおよびオフライン測定のためのより進歩したプローブおよび自動分析器が入手可能である。懸濁培養において得られ得る最大細胞密度は、比較的低く約2〜4×106細胞/mlの培地(これは、1mlあたり1mg未満の乾燥細胞重量である)であり、微生物発酵において達成され数値未満である。
2つの懸濁培養リアクター設計は、操作のそれらの単純さおよび堅牢さに起因して、産業においてより広範に使用される(撹拌リアクターおよびエアリフトリアクター)。撹拌リアクター設計は、インターフェロンの産生のために、8000リットル容量において首尾よく使用されている(Phillipsら、1985;Mizrahi,1983)。細胞は、1:1〜3:1の高さ-直径比のステンレス鋼タンクにおいて増殖される。培養物は、通常は、葉片ディスクまたは船舶プロペラパターンに基づいて、1つ以上の撹拌装置で混合される。葉より小さい剪断焦点を提供する撹拌系が記載されている。撹拌は、磁性的に結合したドライブによって、直接的または間接的のいずれかで駆動される。間接的な駆動は、スターラーバー(stirrer shaft)におけるシールを介する微生物夾雑のリスクを低減する。
エアリフトリアクター(微生物発酵について最初に記載され、そして後に哺乳動物培養に順応される)は、培養を混合および酸素添加の両方をする気体流に関連する。気体流は、リアクターのけ上げ区画に入り、そして循環を駆動する。気体は、培養表面で開放し、これは、リアクターの降水管区画において下向きに移動する気体泡を離れて濃密(denser)を生じる。この設計の主な利点は、単純性、および機械的混合の必要性の欠失である。代表的には、高さ-直径比は、10:1である。エアリフトリアクターは、比較的容易にスケールアップし、気体の良好な手段移入を有し、そして比較的低い剪断焦点を作製する。
最も大規模な懸濁培養は、バッチまたはフェッドバッチプロセスとして操作される。なぜなら、それらは、操作およびスケールアップするのに最も向いているからである。しかし、ケモスタットまたは灌流方式に基づく連続プロセスが入手可能である。
バッチプロセスは、代表的な増殖プロフィールが見られる閉鎖系である。誘導期は、指数期、定常期、および減衰期に続く。このような系において、環境は、連続的に変化する。なぜなら、栄養素が枯渇し、そして代謝産物が蓄積するからである。これは、細胞増殖および産生性に影響する要因の分析をなし、それゆえプロセス(複合タスク)の至適化をなす。バッチプロセスの生産性は、増殖周期を延長するために、鍵栄養素の供給を制御することによって増加され得る。このようなフェッドバッチプロセスは、依然として閉鎖系である。なぜなら、細胞、産物、および消耗産物が、除去されないからである。
依然として閉鎖系において、培養物を通じる新鮮な培地の灌流は、細胞を種々のデバイス(例えば、微細メッシュ回転フィルター、中空繊維または平厚板膜フィルター、沈澱管)で維持することによって達成され得る。回転フィルター培養は、約5×107細胞/mlの細胞密度を産生する。真の開放系および最も単純な灌流プロセスは、培地のインフローならびに細胞および産物のアウトフローが存在するケモスタットである。培養培地は、細胞の最大特異的増殖速度より低い値で培養の希釈濃度を維持する(リアクターからの細胞集団の洗い出しを防ぐため)、予め決定されそして一定の速度で、リアクターに供給される。細胞および細胞産物および副産物を含む培養液は、同じ速度で除去される。
C)非灌流付着系。
伝統的には、足場依存性細胞培養は、小さなガラスまたはプラスチック容器の底部で増殖される。研究室規模に適切な、古典的で伝統的な技術によって提供される制限された表面-容量比は、大規模な細胞および細胞産物の産生の問題を生じる。小さな培養容器における細胞増殖のための大きなアクセス可能な表面を提供する系を提供するために、多数の技術が報告されている:ローラーボトル系、堆積プレート増殖器(stack plate propagator)、らせんフィルムボトル、中空繊維系、圧縮化ベッド(packed bed)、プレート交換系(plate exchanger system)、および膜チューブリール(membrane tubing reel)。これらの系は、これらの性質に非相同であり、そして時々複数のプロセスに基づくので、以下の短所を提供する−スケールアップのための限定された能力、細胞サンプルを得ることにおける困難、鍵プロセスパラメーターを測定および制御するための限定された能力、および培養を通して相同な環境条件を維持することにおける困難。
これらの欠点にもかかわらず、大規模足場依存性細胞産生に一般的に使用されたプロセスは、ローラーボトルである。大きな異なる形状のTフラスコより小さいので、系の単純性は、非常に信頼でき、従って魅力的になる。1日あたり何千ものローラーボトルを操作し得る、完全な自動化ロボットが入手可能であり、従って、夾雑の危険および別に要求された強烈なヒト操作と関連した矛盾を排除する。頻繁な培地変更で、ローラーボトル培養は、0.5×106細胞/cm2に近い細胞密度を達成し得る(約109細胞/ボトルまたは細胞培地のほとんどは107細胞/mlに対応する)。
D)マイクロキャリアでの培養
伝統的な足場依存性培養プロセスの短所を克服するための努力の結果、van Wezel(1967)は、マイクロキャリア培養系の概念を開発した。この系において、細胞は、増殖培地に懸濁された小さな固体粒子の表面で、遅い撹拌によって増殖される。細胞はマイクロキャリアに付着し、そしてマイクロキャリアの表面でコンフルエンシーまで段階的に増殖する。実際、この大規模培養系は、単層および懸濁培養の両方がともにもたらされている、単一ディスクプロセスからユニットプロセスまでの、付着依存性培養をアップグレードする。従って、増殖する細胞に必要な表面と、相同な懸濁培養の利点との組み合わせは、産生を増加する。
ほとんどの他の足場依存性大規模培養法に対するマイクロキャリア培養の利点は、何倍にもなる。第1に、マイクロキャリア培養は、高い表面-容量比(キャリア濃度を変化させることによって変動する)を提供し、これは、高い細胞密度収率、および高度に濃縮した細胞産物を得るための能力を導く。細胞収率は、培養が灌流リアクター態様において増殖される場合、1〜2×107細胞/mlまでである。第2に、細胞は、多くの小さな低産生性容器(すなわち、フラスコまたは皿)を用いるかわりに、1ユニットのプロセス容器において増殖され得る。これは、さらに良好な栄養素利用、および培養培地の相当な節約を生じる。さらに、単一リアクターにおける増殖は、設備空間の必要性、および1細胞あたりに必要な操作工程の数における減少を導き、従って、労働コストおよび夾雑の危険性を低減する。第3に、よく混合されそして相同なマイクロキャリア懸濁培養は、環境条件(例えば、pH、pO2、および培地成分の濃度)をモニターおよび制御することを可能にし、従ってより再現性の細胞増殖および産物回収性を導く。第4に、顕微鏡観察、化学試験、または列挙のための代表的なサンプルを得ることが可能である。第5に、マイクロキャリアは迅速に懸濁物に沈澱するので、フェッドバッチプロセスの使用または細胞の採集が、比較的容易になされ得る。第6に、マイクロキャリアにおける足場依存性培養増殖の態様は、他の細胞操作(例えば、タンパク質分解性酵素の使用を伴わない細胞移入、細胞の同時培養、動物への移植、およびデカンター、カラム、流動床、またはマイクロキャリア維持のための中空繊維を使用する培養の灌流)のためにこの系を使用するのを可能にする。第7に、マイクロキャリア培養は、懸濁物における微生物および動物細胞の培養のために使用された従来の装置を用いて、比較的容易にスケールアップされる。
E)哺乳動物細胞のマイクロカプセル化
哺乳動物細胞を培養するのに特に有用であることが示されている1つの方法は、マイクロカプセル化である。哺乳動物細胞は、半透性ヒドロゲル膜内に保持される。多孔性膜は、栄養素、気体、および代謝産物の、カプセルの周りのバルクの培地との交換を許容する細胞を囲んで形成される。穏和で、迅速な、そして非毒性のいくつかの方法が開発されており、そしてここで、得られる膜は、培養の期間を通じて増殖する細胞塊を維持するのに、十分多孔性かつ強力である。これらの方法は全て、カルシウム含有溶液との液滴接触によってゲル化された可溶性アルギン酸に基づいている。Lim(1982,米国特許第4,352,883号、本明細書中に参考として援用される)は、小さな開口部を介して影響される、約1%アルギン酸ナトリウム溶液に濃縮される細胞を記載し、これは液滴を形成し、そして約1%塩化カルシウム溶液に逃げ出す。次いで、液滴は、アルギン酸の表面にイオン結合するポリアミノ酸の層に注入される。最後に、アルギン酸は、キレート試薬中で液滴を処理して、カルシウムイオンを除去することによって、再液化される。他の方法は、アルギン酸溶液に滴下されて、従って中空アルギン酸球を作製するカルシウム溶液中の細胞を使用する。同様のアプローチは、アルギン酸に滴下され、また中空球を作製するキトサン溶液中の細胞に関する。
マイクロカプセル化細胞は、撹拌タンクリアクターにおいて、容易に増殖され、そして、150〜1500μmの直径の範囲のビーズサイズを有して、微細メッシュスクリーンを用いて灌流リアクターに容易に保持される。カプセル容量の全培地容量に対する比は、1:2〜1:10の密度で維持され得る。108までのカプセル内細胞密度で、培養において有効な細胞密度は、1〜5×107である。
マイクロカプセル化の他のプロセスを越える利点としては、散布および撹拌から生じる剪断応力の有害な効果からの保護、灌流系を用いる目的のためにビーズを容易に保持する能力(スケールアップは比較的簡単である)、および移植のためにそのビーズを使用する能力が挙げられる。
本発明は、天然において足場依存性である細胞を含む。293細胞は、例えば、足場依存性であり、そして懸濁において増殖する場合、細胞は、互いに接着し、そして凝集塊において増殖し、最終的には各凝集塊の内部コア中の細胞を窒息させる。なぜなら、それらは、培養条件によって維持できないコア細胞を残すサイズに達するからである。それゆえ、足場依存性細胞の大規模培養の有効な手段は、これらの細胞を、大量のアデノウイルスを作製するのに有効に使用するために必要とされる。
F)灌流付着系
灌流付着系は、本発明の好ましい形態である。灌流は、(生理学的栄養溶液の)細胞の集団を介してまたは越えて、一定速度で連続する流れを意味する。細胞を回収された培地で洗い流す連続フロー培養(例えば、ケモスタット)とは反対の、培養単位内の細胞の保持をいう。灌流の考え方は、今世紀初め以来公知であり、そして拡張された顕微鏡観察に実行可能な組織の小片を保存するために適用されている。この技術は、細胞が、血液、リンパ、または他の体液で連続的に供給される、インビボでの細胞環境を模倣することを開始した。灌流なしでは、培養における細胞は、流加(fed)および飢餓の交互段階を通過し、従って、それらの増殖および代謝能力の完全な発現を制限する。
灌流培養の現在の使用は、高密度(すなわち、0.1〜5×108細胞/ml)で細胞を増殖する投与に対して応答する。2〜4×106細胞/mlを越えて密度を増加させるために、培地は、栄養欠乏を補い、そして毒性産物を除去するために、絶え間なく新鮮な培地で置換されなければならない。灌流は、培養環境(pH、pO2、栄養レベルなど)。のよりよい制御を可能にし、そして細胞付着のために、培養内の表面積の利用を有意に増加させる手段である。
不織布のベッドマトリックスを用いる灌流充填ベッドリアクターの開発は、108細胞/mlの総容積を越える密度で灌流培養を維持するための手段を提供している(CelliGenTM、New Brunswick Scietific,Edison,NJ;Wangら、1992;Wangら、1993;Wangら、1994)。簡潔に記載すると、このリアクターは、足場および非足場依存性細胞の両方の培養のために改良されたリアクターを含む。リアクターは、内部循環を提供するための手段で充填されたベッドとして設計される。好ましくは、繊維マトリックスキャリアが、リアクター容器内のバスケットに配置される。バスケットの頂部および底部は孔を有し、培地がバスケットを介して流れるのを可能にする。特別に設計された羽根(impeller)は、栄養分の単一供給および消耗物の除去を確実にするための繊維マトリックスによって占有される空間を介する培地の再循環を提供する。これは同時に、全細胞塊のごく少量が培地に懸濁されることを確実にする。バスケットおよび再循環の組合せはまた、繊維マトリックスを介して酸化された培地の泡なしの流れを提供する。繊維マトリックスは、10μm〜100μmの「孔」直径を有する不織布であり、これは、個々の細胞の容量の1〜20倍に対応する孔容量を有する高い内部容量を提供する。
他の培養系と比較して、このアプローチは、いくつかの有意な利点を提供する。繊維マトリックスキャリアを用いて、細胞は、撹拌および起泡からの機械的ストレスに対して保護される。バスケットを介する自由な培地の流れは、至適に調節されたレベルの酸素、pH、および栄養素を細胞に提供する。産物は、培養から連続的に除去され、そして採集した産物は細胞から離れ、そして続く精製工程を容易にする低タンパク質培地において産生され得る。また、このリアクター系の唯一の設計は、リアクターをスケールアップするのにより簡単な方法を提供する。現在、30リットルまでのサイズが利用可能である。100リットルから300リットルのバージョンは、開発中であり、そして理論的な計算は、1000リットルまでのリアクターを支持する。この技術は、WO 94/17178(1994年8月4日、Freedmanら)に詳細に説明され、これは、本明細書中でその全体を参考として援用される。
CellcubeTM(Corning-Costar)モジュールは、基質に接着した細胞の固定化および増殖のための、大きなスチレン(styrenic)表面積を提供する。それは、隣接するプレート間の薄くシールされた薄層の流れ空間を作製するためにつながれた一連の平行培養プレートを有する、完全にカプセル化された滅菌使い捨てデバイスである。
CellcubeTMモジュールは、互いに対角線上の反対にあり、そして培地の流れを調節するのを助ける注入口および放出口を有する。最初の数日の増殖の間、培養は一般的に、最初の播種の後に系内に含まれている培地によって満たされる。最初の播種と培地灌流の開始との間の時間は、播種接種物における細胞密度および細胞増殖速度に依存する。循環する培地における栄養素濃度の測定は、培養状態の良い指標である。手順が確立された場合、栄養素組成を、種々の異なる灌流速度でモニターして、最も経済的かつ生産性の高い操作パラメーターを決定することが必要であり得る。
この系内の細胞は、伝統的な培養系におけるものより高い溶液密度(細胞/ml)に達する。多くの代表的に使用される基本培地は、1〜2×106細胞/ml/日を支持するように設計される。代表的なCellcubeTM(85,000cm2表面で実行)は、モジュール内に約6Lの培地を含む。細胞密度はしばしば、培養容器において107細胞/mLを越える。コンフルエンスでは、培地の2〜4リアクター容量が、1日あたりに必要である。
培養の産生相のタイミングおよびパラメーターは、特定の細胞株の型および使用に依存する。多くの培養物は、産生相では、培養の増殖相で必要とされる培地とは異なる培地を必要とする。1つの相から他の相への移行は、伝統的培養においては複数の洗浄工程を必要とするようである。しかし、CellcubeTM系は、灌流系を用いる。このような系の利点の1つは、種々の操作相の間で穏やかな移行を提供する能力である。灌流系は、増殖培地中の血清成分を除去しようとする伝統的洗浄工程の必要性がない。
本発明の例示的な実施態様では、CellCubeTM系は、AdCMVp53でトランスフェクトした細胞を増殖するために用いられる。293細胞を、製造者の推奨に従ってCellcubeTMに接種した。接種細胞密度は、1〜1.5×104/cm2の範囲であった。細胞を、7日間37℃にて、pH=7.20、DO=60%の空気飽和の培養条件下で増殖させた。培地灌流速度を、CellcubeTM中のグルコース濃度によって調節した。ウイルス感染の1日前に、灌流のための培地を、10%FBSを含む緩衝液から、2%FBSを含む緩衝液へと交換した。8日目に、細胞を、5の感染多重度(MOI)でウイルスに感染させた。培地灌流を、感染直後に1時間の間停止し、次いで残りの期間はウイルス産生相を再開した。培養物を、感染後45〜48時間で回収した。もちろん、これらの培養条件は例示的であり、そして特定の細胞株の栄養要求性および増殖要件によって変更され得る。このような変更は、過度の実験を伴わずに実施され得、そして充分に当業者の技術範囲内である。
G)無血清懸濁培養
特定の実施態様において、遺伝子治療のためのアデノウイルスベクターは、上記のように、293細胞(293A細胞)の足場依存性培養物から産生される。アデノウイルスベクター産生のスケールアップは、293A細胞の足場依存性によって束縛される。スケールアップを容易にし、そしてアデノウイルスベクターについての将来の需用を満たすために、かなりの努力が、スケールアップしやすい別の産生プロセスの開発のために費やされている。方法は、293A細胞をマイクロキャリア培養において増殖させ、そして293Aプロデューサー細胞を懸濁培養に順応させることを含む。マイクロキャリア培養技術は、上記に記載されている。この技術は、プロデューサー細胞が、機械的撹拌によって培養培地に懸濁されるマイクロキャリアの表面に接着することに頼る。細胞接着の必要要件は、マイクロキャリア培養のスケール変更能力(scaleability)に対するいくらかの制限を示し得る。
本願までに、遺伝子治療用のアデノウイルスベクター産生のための293懸濁細胞の使用の報告はされていない。さらに、報告された懸濁293細胞は、至適な細胞増殖およびウイルス産生のために、培養培地において5〜10%のFBSの存在を必要とする。歴史的に、細胞培養培地におけるウシ供給源タンパク質の存在は、特に近年は、いくつかの国におけるウシ海綿状脳障害(BSE)の大発生のために、規制の要件である。厳しくかつ複雑な下流の精製プロセスが、夾雑タンパク質および任意の外来性ウイルスを最終産物から除去するために、開発されなければならない。無血清293懸濁培養の開発は、遺伝子治療用のアデノウイルスベクターの産生のための、主要なプロセスの改良であると判断される。
スピナーフラスコおよび3L撹拌タンクバイオリアクターにおけるウイルス産生の結果は、293SF細胞の細胞特異的ウイルス産生性が、約2.5×104vp/細胞であったことを示し、これは、293A細胞からの産生性の約60〜90%である。しかし、より高い定常細胞濃度のために、293SF培養物からの容積測定のウイルス産生性は、293A細胞培養物のウイルス産生性と、本質的に等価であった。本発明者らはまた、ウイルス産生が、ウイルス感染時に新鮮培地の交換を行うことによって有意に増加したことを観察した。本発明者らは、培地における制限要因の評価を始める。
これらの知見は、アデノウイルスベクターを産生するための、スケール変更可能な、効率的な、かつ簡単に確認可能なプロセスを可能にする。この順応方法は、293A細胞のみに限定されず、そして他のアデノウイルスベクタープロデューサー細胞に適用される場合に、等しく有用である。
3.細胞採集および溶解の方法
アデノウイルス感染は、感染した細胞の溶解を生じる。アデノウイルス感染の溶解特徴は、ウイルス産生の2つの異なる態様を許容する。一方は、細胞溶解の前に、感染した細胞を採集する。他方の態様は、産生されたウイルスによる完全な細胞溶解の後に、ウイルス上清を採集する。後者の態様については、完全な細胞溶解を達成するためにより長いインキュベーション時間が必要である。ウイルス感染後のこの長期のインキュベーション時間は、複製可能アデノウイルス(RCA)の生成の可能性の増加について、特に、最新の第一世代アデノウイルスベクター(E1欠失ベクター)についての深刻な懸念を生じる。それゆえ、感染細胞を細胞溶解前に採集することが、選り抜きの産生態様として選択された。表1は、細胞採集後に細胞を溶解するために使用されている最も一般的な方法を列挙する。

細胞は、膜によって結合される。細胞の成分を放出するために、細胞をこじ開けることが必要である。本発明に従ってこれを達成し得る最も有利な方法は、界面活性剤の使用により膜を可溶化することである。界面活性剤は、脂肪族または芳香族の性質の無極性末端と、荷電していても荷電していなくてもよい極性末端とを有する、両親媒性分子である。界面活性剤は、脂質より親水性であり、従って、脂質より高い水溶性を有する。界面活性剤は、水性媒体への水不溶性化合物の分散を可能にし、そして天然の形態のタンパク質を単離および精製するのに使用される。
界面活性剤は、変性性または非変性性であり得る。前者は、ドデシル硫酸ナトリウムのような陰イオン性またはエチルトリメチルアンモニウムブロミドのような陽イオン性であり得る。これらの界面活性剤は全体的に膜を破壊し、そしてタンパク質-タンパク質相互作用を壊すことによってタンパク質を変性させる。非変性性界面活性剤は、Triton▲R▼X-100のような非陰イオン性界面活性剤、コール酸塩のような胆汁酸塩、およびCHAPSのような両イオン性界面活性剤に分類され得る。両性イオン性物(zwitterionics)は、同じ分子中に陽イオン基および陰イオン基の両方を含み、正電荷は、同じまたは隣接する分子の負電荷によって中和される。
SDSのような変性剤は、モノマーとしてタンパク質に結合し、そしてその反応は、飽和するまで平衡に働く。従って、モノマーの遊離濃度は、必要な界面活性剤濃度を決定する。SDS結合は、協同性(すなわち、SDSの1つの分子の結合が、そのタンパク質への別の分子結合の可能性を増加する)であり、そしてタンパク質を、その長さがそれらの分子量に比例する桿状に変更させる。
Triton▲R▼X-100のような非変性性剤は、天然のコンホメーションに結合せず、これらは協同性結合機構も有さない。これらの界面活性剤は、水可溶性タンパク質に貫入しない堅固でかさ高い無極性部分を有する。これらは、タンパク質の疎水性部分に結合する。Triton▲R▼X-100および他のポリオキシエチレン非陰イオン性界面活性剤は、タンパク質-タンパク質相互作用を壊すのには効果がなく、そしてタンパク質の人為的な凝集を引き起こし得る。しかし、これらの界面活性剤は、タンパク質-脂質相互作用を破壊するが、ずっと穏和であり、そしてタンパク質の天然の形態および機能的能力を維持し得る。
界面活性剤除去は、多数の方法において試みられ得る。透析は、モノマーとして存在する界面活性剤では良好に作用する。透析は、容易に凝集して、ミセルを形成する界面活性剤では、いくらか効果がない。なぜなら、それらのミセルは、大きすぎて、透析を通過しないからである。イオン交換クロマトグラフィーは、この問題を回避するのに利用され得る。破壊されたタンパク質の溶液は、イオン交換クロマトグラフィーカラムにアプライされ、次いでカラムは、界面活性剤を含まない緩衝液で洗浄される。界面活性剤は、緩衝液と界面活性剤溶液との平衡化の結果として除去される。あるいは、タンパク質溶液は、密度勾配を介して通過し得る。タンパク質が勾配を介して沈降するにつれ、界面活性剤は、化学ポテンシャルに起因して離れる。
しばしば、単一の界面活性剤は、細胞中に見いだされるタンパク質の環境の可溶化および分析に十分には融通が利かない。タンパク質は、ある界面活性剤において可溶化され得、次いで、タンパク質分析のために、別の適切な界面活性剤に入れられる。最初の工程で形成したタンパク質界面活性剤ミセルは、純粋な界面活性剤ミセルから分離するべきである。これらが、分析のために過剰の界面活性剤に添加される場合、このタンパク質は、両方の界面活性剤とのミセルにおいて見いだされる。界面活性剤-タンパク質ミセルの分離は、イオン交換もしくはゲル濾過クロマトグラフィー、透析、または浮遊密度型分離を用いて達成され得る。
Triton▲R▼X-100界面活性剤:このファミリーの界面活性剤(Triton▲R▼X-100、X114X、およびNP-40)は、同様の基本的特徴を有するが、それらの特異的疎水性-親水性性質において異なる。これらの異種起源の界面活性剤の全ては、芳香環に付着した分岐した8炭素鎖を有する。分子のこの部分は、界面活性剤の疎水性性質の大部分に寄与する。Triton▲R▼X-100界面活性剤は、非変性条件下で、膜タンパク質を可溶化するのに使用される。タンパク質を可溶化するための界面活性剤の選択は、可溶化されるタンパク質の疎水性性質に依存する。疎水性タンパク質は、それらを効果的に可溶化するために疎水性界面活性剤を必要とする。
Triton▲R▼X-100およびNP-40は、構造および疎水性において非常に類似しており、そして細胞溶解、脱脂タンパク質解離、ならびに膜タンパク質および脂質可溶化を含むほとんどの適用において交換可能である。一般的に、2mgの界面活性剤が、1mgの膜タンパク質または10mgの界面活性剤/1mgの脂質膜を可溶化するのに使用される。Triton▲R▼X-114は、親水性タンパク質から疎水性タンパク質を分離するために有用である。
Brij▲R▼界面活性剤:これらは、疎水性鎖に付着した種々の長さのポリオキシエチレン鎖を有する点で、Triton▲R▼X界面活性剤と、構造において類似する。しかし、Triton▲R▼X界面活性剤とは異なり、Brij▲R▼界面活性剤は、芳香環を有さず、そしてその炭素鎖の長さが変化し得る。Brij▲R▼界面活性剤は、透析を用いて溶液から除去するのは困難であるが、界面活性剤除去ゲルを用いて除去され得る。Brij▲R▼58は、その疎水性/親水性特徴の点で、Triton▲R▼X-100と最も類似する。Brij▲R▼35は、HPLC適用において通常使用される界面活性剤である。
透析可能な非イオン性界面活性剤:η-オクチル-β-D-グルコシド(オクチルグルコピラノシド)およびη-オクチル-β-D-チオグルコシド(オクチルチオグルコピラノシド、OTG)は、非変性性陰イオン界面活性剤であり、これらは溶液から容易に透析される。これらの界面活性剤は、膜タンパク質を可溶化するために有用であり、そして280nmでの低いUV吸収を有する。オクチルグルコシドは、23〜25mMの高いCMCを有し、そして膜タンパク質を可溶化するのに、1.1〜1.2%の濃度で使用されている。
オクチルチオグルコシドは、オクチルグルコシドの代替物を提供するために最初に合成された。オクチルグルコシドは、製造するには高価であり、そして生物学的系においていくつかの固有の問題が存在する。なぜなら、β-グルコシダーゼによって加水分解され得るからである。
Tween▲R▼界面活性剤:Tween▲R▼界面活性剤は、非変性性の非イオン性界面活性剤である。それらは、脂肪酸のポリオキシエチレンソルビタンエステルである。Tween▲R▼20およびTween▲R▼80界面活性剤は、生物学的適用においてブロッキング剤として使用され、そして通常、タンパク質溶液に添加されて、プラスチックまたはニトロセルロースのような疎水性材料への非特異的結合を防ぐ。それらは、ELISAおよびブロッティング適用におけるブッロキング剤として使用されている。一般的には、これらの界面活性剤は、0.01〜1.0%の濃度で使用されて、疎水性材料への非特異的結合を防ぐ。
Tween▲R▼20および他の非イオン性界面活性剤は、いくつかのタンパク質をニトロセルロースの表面から除去することが示されている。Tween▲R▼80は、マルチウェルプラスチック組織培養プレートへのタンパク質の非特異的結合を示す膜タンパク質を可溶化するため、そして血清タンパク質およびビオチン化プロテインAによる、ELISAにおけるポリスチレンプレートへの非特異的結合を減少させるために使用されている。
これらの界面活性剤の間の差異は、脂肪酸鎖の長さである。Tween▲R▼80は、C18鎖を有するオレイン酸から誘導されるが、一方、Tween▲R▼20は、C12鎖を有するラウリン酸から誘導される。より長い脂肪酸鎖によって、Tween▲R▼20界面活性剤よりも親水性の低いTween▲R▼80界面活性剤が作成される。両方の界面活性剤が、水に非常に可溶性である。
Tween▲R▼界面活性剤は、透析による溶液からの除去が困難であるが、Tween▲R▼20は界面活性剤除去ゲルによって除去され得る。これらの界面活性剤中に見出されるポオリオキシエチレン鎖によって、これらの界面活性剤が酸化(過酸化物の形成)に供され、そしてTriton▲R▼XおよびBrij▲R▼シリーズの界面活性剤の場合もこれにあてはまる。
双イオン性界面活性剤:双イオン性界面活性剤であるCHAPSは、コール酸のスルホベタイン誘導体である。この双イオン性界面活性剤は、タンパク質活性が重要である場合に、膜タンパク質の可溶化に有用である。この界面活性剤は、広範なpHの範囲(pH2〜12)にわたって有用であり、そして高CMC(8〜10mM)による透析によって溶液から容易に除去される。この界面活性剤は、280nmで低い吸光度を有し、この波長でのタンパク質のモニターリングが必要な場合にこれを有用にする。CHAPSは、BCAタンパク質アッセイに適合性であり、そして界面活性剤除去ゲルによって溶液から除去され得る。タンパク質は、CHAPSの存在下でヨウ素化され得る。
CHAPSは、内在性の膜タンパク質およびレセプターを可溶化するために良好に使用されており、そしてタンパク質の機能的能力を維持する。チトクロムP-450がTriton▲R▼X-100またはコール酸ナトリウムのいずれかで可溶化される場合、凝集が形成される。
B)非界面活性剤法
種々の非界面活性剤法(好ましくはないが)が、本発明の他の有利な局面と組み合わせて使用され得る:
凍結−融解:これは、穏やかなおよび効果的な様式で細胞を溶解させるために広範に使用されている技術である。一般には、細胞は、例えばドライアイス/エタノール浴中で完全に凍結するまで迅速に凍結され、次いで完全に融解するまで37℃浴中に移される。このサイクルが、複数回、細胞の完全な溶解が達成されるまで繰り返される。
超音波処理:高周波の超音波振動が細胞の破壊に有用であることが見出されている。超音波で細胞を崩壊させるこの方法は完全には理解されていないが、懸濁物が超音波振動に供された場合に一時的に高い圧力が生じることが知られている。この技術の主要な欠点は、相当量の熱が発生することである。熱の影響を最小にするために、特別に設計されたガラス容器が、細胞懸濁物を入れるために使用される。このような設計によって、懸濁物を超音波処理チューブからフラスコが氷中におかれていて冷却されている容器の外側へ循環させることが可能である。
高圧放出:これは、微生物細胞を破壊するために頻繁に使用される方法である。フレンチプレッシャー細胞(French pressure cell)は、細胞を破壊させて開くために10.4×107Pa(16,000p.s.i)の圧力を使用する。これらの装置は、針状のバルブによって外側に開いているステンレス鋼チャンバーからなる。細胞懸濁物を、閉じられた部位に針状バルブを備えたチャンバー中に置く。チャンバーを反転させた後、バルブを開き、そしてピストンを押してチャンバー内に空気を送り込む。閉じられた部位中にバルブを備えたチャンバーを、その元の部位に戻し、固体の基板上に置き、そして必要な圧力を、水圧によってピストンにかけた。圧力が到達すると、針状のバルブがほんのわずかに開いて、わずかに圧力を放出し、そして細胞を膨らませてそれらを破裂させる。バルブを開いたままにし、一方、圧力はそのままにし、その結果、回収され得る破裂した細胞の破片が生じる。
固体剪断法:研磨剤を用いる機械的剪断を、Mickleシューカー中で行い得る。これは、500nmの直径のガラスビーズの存在下で激しく懸濁物を振動(300〜3000回/分)させる。この方法は、細胞小器官に損傷を生じ得る。さらに制御された方法は、ピストンがほとんどの細胞を、細胞の剥離または深い凍結ペーストを伴って圧力チャンバー中の0.25mmの直径のスロットを通って押し出す、Hughes pressを使用することである。5.5×107Pa(8000p.s.i)までの圧力が細菌調製物を溶解させるために使用され得る。
液体剪断法:これらの方法は、高速の往復または回転羽のホモジナイザーを使用する破砕機を使用する。これは、プランジャー、ボール、および微量流動化装置の上方/下方の動き、または小さい直径のチューブの高速での通過または2つの液体の流れの高速での衝突を使用する衝突ジェットを使用する。破砕機の羽は、効率的な混合が可能にするために種々の角度の傾斜がつけられている。ホモジナイザーは、通常は、局部的な加熱を最小にするために数秒の短い高速での破裂で通常作動させられる。これらの技術は、一般には、微生物細胞には適切ではないが、非常に穏やかな液体剪断が、通常は、動物細胞を破壊するために適切である。
低張/高張法:細胞は、はるかに低い(低張)または高い(高張)の溶質濃度を有する溶液に曝される。溶質濃度の差異は、浸透圧勾配を生成する。低張環境では細胞中に向かって水流が生じ、細胞の膨張および破裂を生じる。高張環境では細胞の外側への水流によって、細胞を収縮およびそれに続いて破裂が生じる。
4.濃縮および濾過の方法
本発明の1つの局面は、細胞溶解物からのアデノウイルスの粗精製の方法を使用する。これらの方法は、明澄化、濃縮、および透析を含む。この精製プロセスの最初の工程は、細胞の溶解物から大きな粒子物質(特に、細胞成分)を除去するための細胞溶解物の明澄化である。溶解物の明澄化は、深部(depth)フィルターを使用して、または接線流量濾過(tangential flow filtration)によって達成され得る。本発明の好ましい実施態様において、細胞溶解物は、比較的非吸着性の物質(例えば、ポリエステル樹脂、砂、珪藻土、コロイド、ゲルなど)のパッケージされたカラムからなる深部フィルターを通過させられる。接線流量濾過(TFF)において、溶解物の液体は、膜表面を通過して流れ、これによって、バルクの溶液への膜表面からの溶質の逆方向の拡散を促進する。膜は、一般的には、種々の型のフィルター装置(オープンチャンネルプレートおよびフレーム、中空ファイバー、および細管)中に配置される。
細胞溶解物の明澄化および予備濾過後、得られるウイルス上清を最初に濃縮し、次いで、緩衝液をダイアフィルトレーションによって交換する。ウイルス上清を100〜300Kの名目上の分子量カットオフの限外濾過膜を通した接線流量濾過によって、濃縮する。限外濾過は、分子の大きさ、形状、および/または電荷で種類を分離する半透膜を使用する、圧改変対流プロセスである。これによって、溶質分子の大きさから独立して、種々の大きさの溶質から溶媒を分離する。限外濾過は穏やかであり、効率的であり、そして同時に溶液を濃縮しそして脱塩するために使用され得る。限外濾過膜は一般に、以下の2つの異なる層を有する:薄い(0.1〜1.5μm)、10〜400オングストロームの孔直径を有する高密度の外板、および限外濾過膜の浸透側に大きく開いている次第に大きくなる隙間の開放構造。したがって、外板の孔を通過し得る任意の種が、膜を自由に通過し得る。溶質の最大の保持のために、保持される種よりも十分に低い名目上の分子量カットオフを有する膜が選択される。巨大分子の濃縮において、膜は、所望の生物学的種の含量を富化し、そして保持された物質の明澄化された濾過物を提供する。ミクロ溶質は、溶媒での対流によって除去される。保持される溶質の濃度を上昇させる場合、限外濾過速度は低下する。
ダイアフィルトレーション、または限外濾過膜を使用する緩衝液交換は、塩、糖の除去および交換、種に結合されていない非水溶性の溶媒の分離、低分子量の物質の除去、またはイオン環境およびpH環境の迅速な交換のための理想的な方法である。ミクロ溶質は、限外濾過速度と同じ速度での限外濾過される溶液への溶媒の添加によって最も効率よく除去される。これによって、一定の容量で溶液からミクロ種を洗浄し、保持された種を精製する。本発明は、Benzonase▲R▼処理の前のウイルス上清の緩衝液の交換のために、ダイアフィルトレーション工程を利用する。
5.ウイルス感染
本発明は、1つの例において、治療的に有意なベクターを作成するために細胞のアデノウイルス感染を使用する。代表的には、ウイルスは、ウイルスの取り込みが可能である生理学的条件下で、適切な宿主細胞に単純に曝される。アデノウイルスが具体的に説明されるが、本方法は、以下に考察される他のウイルスベクタートともに有利に使用され得る。
A)アデノウイルス
アデノウイルスは、その中程度の大きさのDNAゲノム、操作の容易さ、高力価、広範な標的細胞の範囲、および高い感染性によって、遺伝子移入ベクターとしての使用に特に適切である。およそ36kBのウイルスゲノムが、100〜200塩基対(bp)の逆末端反復(ITR)によって結合され、これには、ウイルスDNAの複製およびパッケージングに必要なシス作用エレメントが含まれる。種々の転写ユニットを含むゲノムの初期(E)および後期(L)領域が、ウイルスDNAの複製の開始によって分けられる。
E1領域(E1AおよびE1B)は、ウイルスゲノムおよび数個の細胞性遺伝子の転写の調節を担うタンパク質をコードする。E2領域(E2AおよびE2B)の発現は、ウイルスDNAの複製のためのタンパク質の合成を生じる。これらのタンパク質は、DNAの複製、後期遺伝子の発現、および宿主細胞のシャットオフに関与する(Renan、1990)。後期遺伝子(L1、L2、L3、L4、およびL5)の産物(ウイルスキャプシドタンパク質の大部分を含む)は、主要後期プロモーター(MLP)によって誘導される単一の一次転写産物の有意なプロセシング後にのみ発現される。MLP(16.8マップユニットに位置する)は、感染の後期の間に特に効果的である。そしてこのプロモータから誘導される全てのmRNAが、それらを翻訳のために好ましいmRNAにする5’三部分リーダー(TL)配列を有する。
アデノウイルスを遺伝子治療について最適化するために、DNAの大きなセグメントが含まれ得るように保有する能力を最大にすることが必要である。特定のアデノウイルス産物に関連する毒性および免疫学的反応を減少させることもまた非常に所望される。アデノウイルスゲノムの大部分の除去、ヘルパーウイルスおよび/またはヘルパー細胞によるトランスでの遺伝子産物の除去を提供することによって、ベクター中への異種DNAの大きな部分の挿入を可能にする。このストラテジーはまた、アデノウイルス遺伝子産物の毒性および免疫原性の低下を生じる。
DNAの大きな置換は、ウイルスDNAの複製に必要とされるシスエレメントが線状のウイルスゲノムのいずれかの末端の逆末端反復(ITR)(100〜200bp)中にすべて局在するので、可能である。ITRを含有するプラスミドは、非欠損アデノウイルス(Hayら、1984)の存在下で複製され得る。したがって、アデノウイルスベクター中へのこれらのエレメントの包含は、複製を可能にするはずである。
さらに、ウイルスのキャプシド化のためのパッケージングシグナルは、ウイルスゲノムの左側末端での194〜385bp(0.5〜1.1マップユニット)の間に局在する(Hearingら、1987)。このシグナルは、バクテリオファージλDNA中のタンパク質認識部位を模倣している。ここで、特異的配列は左側の末端に隣接しているが、凝集性の末端配列の外側が、頭部構造へのDNAの挿入のために必要であるタンパク質への結合を媒介する。AdのE1置換ベクターは、ウイルスゲノム左側末端の450bp(0〜1.25マップユニット)のフラグメントが293細胞においてパッケージングを指向し得ることを実証した(Levreroら、1991)。
以前に、アデノウイルスゲノムの特定の領域が哺乳動物細胞のゲノム中に取り込まれ得ること、およびそれによってコードされる遺伝子が発現されることが示されている。これらの細胞株は、細胞株によってコードされるアデノウイルス機能を欠損している、アデノウイルスベクターの複製を支持し得る。複製欠損アデノウイルスベクターの「ヘルピング」ベクター(例えば、野生型ウイルスまたは条件的欠損変体)の相補体の報告もまた存在する。
複製欠損アデノウイルスベクターは、ヘルパーウイルスによってトランスで相補され得る。この観察のみでは、複製欠損ベクターの単離は可能ではないが、しかし、ヘルパーウイルスの存在が複製機能を提供するために必要とされるので、任意の調製物を混入する。したがって、複製欠損ベクターの複製および/またはパッケージングに対して特異性を付与するさらなるエレメントが、必要とされた。本発明において提供されるようなそのエレメントは、アデノウイルスのパッケージング機能に由来する。
アデノウイルスのパッケージングシグナルが従来のアデノウイルスマップの左側末端に存在することが示されている(Tibbetts、1977)。後の研究において、ゲノムのE1A(194〜358bp)領域に欠失を有する変異体が、初期(E1A)機能を意図された細胞株中でもなおわずかに増殖することが示された(HearingおよびShenk、1983)。補償(compensating)アデノウイルスDNA(0〜353bp)を変異体の右末端に再結合させた場合、ウイルスは正常にパッケージングされた。さらなる変異分析によって、Ad5ゲノムの左側末端の短い、反復する、部位依存性エレメントを同定した。反復の1つのコピーは、ゲノムのいずれかの末端に存在する場合に効率的なパッケージングに十分であるが、Ad5 DNA分子の内部に移動する場合はそうではないことが見出された(Hearingら、1987)。
パッケージングシグナルの変異したバージョンを使用することによって、種々の効率でパッケージングされるヘルパーウイルスを作成することが可能である。代表的には、変異体は点変異体または欠失変異体である。低効率のパッケージングを有するヘルパーウイルスをヘルパー細胞中で増殖させる場合、ウイルスは、野生型ウイルスと比較して低下した割合ではあるがパッケージされ、それによってヘルパーの増殖を可能にする。しかし、これらのヘルパーウイルスが野生型のパッケージングシグナルを含むウイルストともに細胞中で増殖される場合、野生型パッケージングシグナルは変異したバージョンにわたって優先的に認識される。パッケージング因子の限定量を仮定すると、野生型シグナルを含むウイルスは、ヘルパーと比較した場合に選択的にパッケージングされる。選択が極めて十分である場合、均一に近づけたストックが達成されるはずである。
B)レトロウイルス
治療的に有意なベクターの生成のための細胞のアデノウイルス感染が本発明の好ましい実施態様であるが、本発明がこのようなベクターを生成する目的のために細胞のレトロウイルス感染を使用し得ることが意図される。レトロウイルスは、逆転写のプロセスによって感染させられた細胞中でそれらのRNAを二本鎖のDNAに転換する能力によって特徴付けられる一本鎖RNAウイルスの群である(Coffin、1990)。次いで、得られるDNAが、プロウイルスとして細胞の染色体に安定に組み込まれ、そしてウイルスタンパク質の合成を導く。組み込みによって、レシピエント細胞およびその子孫におけるウイルス遺伝子の配列の保持を生じる。レトロウイルスゲノムは、3つの遺伝子(gag、pol、およびenv−それぞれ、キャプシドタンパク質、ポリメラーゼ酵素、およびエンベロープ成分をコードする)を含む。gag遺伝子の上流に見出される、Yと呼ばれる配列は、ビリオンへのゲノムのパッケージングのためのシグナルとして機能する。2つの長末端反復(LTR)配列が、ウイルスゲノムの5’および3’末端に存在する。これらは、強力なプロモーターおよびエンハンサー配列を含み、そして宿主細胞ゲノムへの組み込みにもまた必要とされる(Coffin、1990)。
レトロウイルスベクターを構築するために、プロモーターをコードする核酸は、複製欠損ウイルスを産生するために特定のウイルス配列の代わりにウイルスゲノム中に挿入される。ビリオンを産生するために、gag、pol、およびenv遺伝子を含むが、LTRおよびY成分を含まないパッケージング細胞株が、構築される(Mannら、1983)。レトロウイルスLTRおよびY配列とともにヒトcDNAを含む組換えプラスミドがこの細胞株に導入される(例えば、リン酸カルシウム沈殿によって)場合、Y配列は、ウイルス粒子中にパッケージングされる組換えプラスミドのRNAの転写を可能にし、次いで、これは、培養培地に分泌される(NicolasおよびRubenstein、1988;Temin、1986;Mannら、1983)。次いで、組換えレトロウイルスを含有する培地が回収され、必要に応じて濃縮され、そして遺伝子移入に使用される。レトロウイルスベクターは、広範な種々の細胞型を感染させ得る。しかし、組み込みおよび安定な発現は、宿主細胞の分裂を必要とする(Paskindら、1975)。
レトロウイルスベクターの特異的標的を可能にするために設計された新規のアプローチが、最近、ウイルスエンベロープへのガラクトース残基の化学的付加によるレトロウイルスの化学的改変に基づいて開発された。この改変は、アシアロ糖タンパク質レセプターを介する肝細胞のような細胞の特異的感染を可能にし得、これが所望されるはずである。
組換えレトロウイルスを標的化する異なるアプローチが設計され、ここで、レトロウイルスエンベロープタンパク質および特異的細胞レセプターに対するビオチン化抗体は使用される。この抗体は、ストレプトアビジンを使用することによって、ビオチン成分を介して結合された(Rouxら、1989)。主要組織適合性複合体クラスIおよびクラスII抗原に対する抗体を使用して、その表面抗原を有する種々のヒト細胞の感染が、インビトロで同種指向性ウイルスを用いて実証された(Rouxら、1989)。
C)他のウイルスベクター
他のウイルスベクターは、本発明の発現構築物として使用され得る。ウイルス(例えば、ワクシニアウイルス(Ridgeway、1988;BaichwalおよびSugden、1986;Couparら、1988)、アデノ隋半ウイルス(AAV)(Ridgeway、1988;BaichwalおよびSugden、1986;HermonatおよびMuzycska、1984)、およびヘルペスウイルス)由来のベクターが使用され得る。これらのウイルスは、種々の哺乳動物細胞への遺伝子の移入における使用のためのいくつかの特徴を付与する。
6.ウイルスベクターの操作
特定の実施態様において、本発明はさらに、ウイルスベクターの操作に関する。このような方法は、例えば、目的の遺伝子をコードする異種DNAを含むベクター構築物の使用、およびその発現の手段、適切なヘルパー細胞中でのベクターの複製、そこから産生されるウイルス粒子の得る工程、および細胞を組換えウイルス粒子で感染させる工程を包含する。遺伝子は、タンパク質を単純にコードし得、ここで、大量のタンパク質が所望される(すなわち、大規模なインビトロでの産生方法が所望される)。あるいは、遺伝子は、例えば、ガン細胞を処置するため、ウイルス感染に抵抗するための免疫調節遺伝子を発現するため、または遺伝的欠損の結果として遺伝子の機能を置換するための、治療用遺伝子であり得る。遺伝子治療ベクターとは対照的に、遺伝子は異種DNAであり、これは、ベクターの骨格を提供するウイルスゲノム以外の供給源から誘導されるDNAを含むことを意味する。最終的に、ウイルスは、生ウイルスワクチンとして作用し得、そしてそれらが対抗する抗体の産生のための目的の抗原を発現する。遺伝子は、例えば、細菌、ウイルス、酵母、寄生虫、植物、またはさらに動物のような、原核生物または真核生物供給源に由来し得る。異種DNAはまた、1つ以上の供給源(すなわち、複合遺伝子構築物または融合タンパク質)由来であり得る。異種DNAはまた、1つの供給源に由来し得る調節配列および種々の供給源由来の遺伝子を含み得る。
A)治療用遺伝子
現在、p53が、腫瘍サプレッサー遺伝子として認識されている(Montenarh、1992)。高レベルの変異体p53が、化学的発ガン、紫外線照射、およびいくつかのウイルス(SV40を含む)によって形質転換した多くの細胞中に見出されている。p53遺伝子は、広範な種々のヒトの腫瘍において変異の不活化の頻繁な標的であり、そして一般的なヒトのガンにおいて最も頻繁に変異する遺伝子であることがすでに報告されている(Mercer、1992)。これは、ヒトのNSCLCの50%以上において変異しており(Hollesteinら、1991)、そして他の腫瘍の広範な範囲において変異している。
p53遺伝子は、ラージT抗原およびE1Bのような宿主タンパク質と複合体を形成し得る393-アミノ酸のリンタンパク質をコードする。このタンパク質は、正常な組織および細胞中で、形質転換された細胞または腫瘍組織と比較して一般に少ない濃度であるが、見出されている。興味深いことに、野生型p53は、細胞の増殖および分裂を制御するために重要であるようである。野生型のp53の過剰発現は、いくつかの場合、ヒト腫瘍細胞株において坑増殖性であることが示されている。したがって、p53は、細胞増殖のネガティブレギュレーターとして作用し得(Weinberg、1991)、そして制御されていない細胞の増殖を直接抑制し得るか、またはこの増殖を抑制する遺伝子を直接もしくは間接的に活性化する。したがって、野生型p53の欠損または不活化は、形質転換に寄与し得る。しかし、いくつかの研究は、変異体p53の存在が遺伝子の形質転換能力の完全な発現に必要であり得ることを示す。
野生型のp53は、多くの細胞の型において重要な増殖のレギュレーターとして認識されている。ミスセンス変異がp53遺伝子について一般的であり、そして少なくとも30個の異なるコドン中に生じることが公知であり、しばしば、ホモ接合性を損なうことなく細胞の表現系のシフトを生じる優勢の対立遺伝子を作成する。さらに、これらの優勢のネガティブな対立遺伝子の多くが、生物中で寛容であるようであり、そして生殖系を通じて継代される。種々の変異対立遺伝子は、最小の機能不全から強力に浸透する優勢のネガティブな対立遺伝子までの範囲にあるようである(Weinberg,1991)。
Caseyおよび共同研究者らは、2つのヒト乳ガン細胞株への野生型のp53をコードするDNAのトランスフェクションが,このような細胞中での増殖抑制制御を回復することを報告した(Caseyら、1991)。同様の効果がまた野生型のトランスフェクションについて示されているが、ヒト肺ガン細胞株への変異p53のトランスフェクションはそうではない(Takahasiら、1992)。p53は、変異遺伝子にわたって明らかに優性であり、そして変異体遺伝子を有する細胞にトランスフェクトされた場合に増殖に対して選択される。トランスフェクトされたp53の正常な発現は、内因性の野生型p53を有する正常な細胞に対して有害ではない。したがって、このような構築物は、正常な細胞によって、有害な影響を伴わずに取り込まれ得る。したがって、野生型のp53発現構築物でのp53関連ガンの処置によって悪性細胞の数またはそれらの増殖速度を減少させることが提案されている。さらに、最近の研究では、いくつかのp53野生型腫瘍がまた、外因性のp53の発現の効果に敏感であることを示唆する。
真核生物の細胞周期の主要な転移は、サイクリン依存性キナーゼ、すなわち、CDKによって誘発される。1つのCDKである、サイクリン依存性キナーゼ4(CDK4)は、G1期を介する進行を調節する。この酵素の活性は、G1後期でRbをリン酸化することであり得る。CDK4の活性は、サブユニット、D型サイクリンを活性化させること、および阻害サブユニット(例えば、p16INK4、これは、CDK4に特異的に結合しそして阻害するタンパク質として、生物化学的に特徴付けられている)によって制御され、したがって、Rbのリン酸化を調節し得る(Serranoら、1993;Serranoら、1995)。p16INK4タンパク質がCDK4インヒビターであるので(Serrano、1993)、この遺伝子の欠損は、CDK4の活性を増大し得、それによってRbタンパク質の過リン酸化を生じる。p16はまた、CDK6の機能を調節することが知られている。
p16INK4はまた、p16B、p21WAF1、CIP1、SDI1、およびp27KIP1を含むCDK阻害タンパク質の新たに記載されたクラスに属する。p16INK4遺伝子は、多くの腫瘍型において頻繁に欠失される染色体領域である、9p21にマップされる。ホモ接合性の欠損およびp16INK4遺伝子の変異体は、ヒトの腫瘍細胞株中に頻繁に存在する。この証拠は、p16INK4遺伝子が腫瘍サプレッサー遺伝子であることを示唆する。しかし、p16INK4遺伝子の変化の頻度が、培養された細胞株よりも最初の培養されていない細胞株においてはるかに低いという観察によって、この解釈が試みられている(Caldasら、1994;Chengら、1994;Hussussianら、1994;Kambら、1994a;Kambら、1994b、Moriら、1994;Okamotoら、1994;Noboriら、1995;Orlowら、1994;Arapら、1995)。プラスミド発現ベクターでのトランスフェクションによる野生型p16INK4の回復は、いくつかのヒトガン細胞株によるコロニー形成を減少させた(Okamoto、1994;Arap、1995)。
C-CAMは、実質的に全ての上皮細胞で発現される(OdinおよびObrink、1987)。105kDの見かけの分子量を有するC-CAMは、ラットの肝細胞の原形質膜から、細胞の凝集を中和する特異的抗体とのその反応によって最初に単離された(Obrink、1991)。最近の研究は、構造的にはC-CAMは、免疫グロブリン(Ig)スーパーファミリーに属し、そしてその配列は、ガン胎児抗原(CEA)に高く相同であることを示す(LinおよびGuidotti、1989)。バキュロウイルス発現系を使用して(Cheungら(1993a;1993b、および1993c))、C-CAMの第1Igドメインが細胞の接着活性に重要であることが示された。
細胞接着分子またはCAMは、器官の発達および細胞の分化を調節する複雑な分子の相互作用のネットワークに関連することが知られている(Edelman、1985)。最近のデータは、CAMの異常な発現が、いくつかの新生物の腫瘍形成に関与し得ることを示す;例えば、上皮細胞で優先的に発現されるEカドヘリンの発現の減少は、新生物のいくつかの種類の発達に関連する(EdelmanおよびCrossin、1991;Frixenら、1991;Bussemakersら、1992;Matsuraら、1992;Umbasら、1992)。また、GiancottiおよびRuoslahti(1990)は、遺伝子導入によるα5β1インテグリンの発現の増大が、インビボでチャイニーズハムスター卵巣細胞の腫瘍形成を減少し得ることを示した。C-CAMは現在、インビトロおよびインビボで腫瘍の増殖を抑制することが示されている。
本発明に従って使用され得る他の腫瘍サプレッサーとして、RB、APC、DCC、NF-1、NF-2、WT-1、MEN-I、MEN-II、zac1、p73、BRCA1、VHL、FCC、MMAC1、MCC、p16、p21、p57、C-CAM、p27、およびBRCA2が含まれる。アポトーシスのインデューサー(例えば、Bax、Bak、Bcl-Xs、Bik、Bid、Harakiri、Ad E1B、BadおよびICE-CED3プロテアーゼ)も同様に、本発明にしたがって使用されることが見出され得る。
種々の酵素遺伝子が、本発明の目的である。このような酵素として、シトシンデアミナーゼ、ヒポキサンチン-グアニンホスホリボシルトランスフェラーゼ、ガラクトース-1-リン酸ウリジルトランスフェラーゼ、フェニルアラニンヒドロキシラーゼ、グルコセレブロシダーゼ、スフィンゴミエリナーゼ、α-L-イズロニダーゼ、グルコース-6-リン酸デヒドロゲナーゼ、HSVチミジンキナーゼおよびヒトチミジンキナーゼが挙げられる。
ホルモンは、本明細書中で記載されるベクター中で使用され得る遺伝子の別の群である。以下が含まれる:成長ホルモン、プロラクチン、胎盤性ラクトゲン、黄体形成ホルモン、卵胞刺激ホルモン、絨毛性性腺刺激ホルモン、甲状腺刺激ホルモン、レプチン(leptin)、副腎皮質刺激ホルモン(adenocorticotropin、ACTH)、アンギオテンシンIおよびII、β-エンドルフィン、β-メラニン細胞刺激ホルモン(β-MSH)、コレシストキニン、エンドセリンI、ガラニン、胃抑制ペプチド(GIP)、グルカゴン、インシュリン、リポトロフィン、ニューロフィシン、ソマトスタチン、カルシトニン、カルシトニン遺伝子関連ペプチド(CGRP)、β-カルシトニン遺伝子関連ペプチド、高カルシウム血症の悪性因子(1-40)、副甲状腺ホルモン関連タンパク質(107-139)(PTH-rP)、副甲状腺ホルモン関連タンパク質(107-111)(PTH-rP)、グルカゴン様ペプチド(GLP-1)、パンクレアチン(pancreastatin)、膵臓ペプチド、ペプチドYY、PHM、セクレチン、血管作用性腸ペプチド(VIP)、オキシトシン、バソプレシン(AVP)、バソトシン、エンケファリンアミド、メトロフィンアミド、αメラニン細胞刺激ホルモン(α-MSH)、心房性ナトリウム利尿因子(5-28)(ANF)、アミリン、アミロイドP成分(SAP-1)、副腎皮質刺激ホルモン放出ホルモン(CRH)、成長ホルモン放出因子(GHRH)、黄体形成ホルモン放出ホルモン(LHRH)、神経ペプチドY、サブスタンスK(ニューロキニンA)、サブスタンスPおよび甲状腺刺激ホルモン放出ホルモン(TRH)。
本発明のベクターへの挿入が意図される他の遺伝子のクラスとして、インターロイキンおよびサイトカインが挙げられる。インターロイキン1(IL-1)、IL-2、IL-3、IL-4、IL-5、IL-6、IL-7、IL-8、IL-9、IL-10、IL-11、IL-12、GM-CSF、およびG-CSF。
本発明のウイルスベクターが有用である疾患の例として、以下が挙げられるが、これらに限定されない:アデノシンデアミナーゼ欠損症、血友病Bにおけるヒト血液凝固第IX因子欠損症、および嚢胞性繊維症膜貫通レセプター遺伝子の置換に関与する嚢胞性繊維症。本発明で具体化されるベクターはまた、リウマチ様関節炎または再狭窄のような過増殖性障害の処置に、血管形成インヒビターまたは細胞周期インヒビターをコードする遺伝子の移入によって、使用され得る。HSV-TK遺伝子のようなプロドラッグ活性化因子の導入もまた、ガンを含む過増殖性障害の処置で使用され得る。
B)アンチセンス構築物
ras、myc、neu、raf、erb、src、fms、jun、trk、ret、gsp、hst、bcl、およびablのような癌遺伝子もまた、適切な標的である。しかし、治療の利点について、これらの癌遺伝子は、アンチセンス核酸を発現し、その結果、癌遺伝子の発現を阻害する。用語「アンチセンス核酸」は、癌遺伝子をコードするDNAおよびRNAの塩基配列に相補的なオリゴヌクレオチドを意味することが意図される。アンチセンスオリゴヌクレオチドは、標的細胞に導入された場合、それらの標的核酸に特異的に結合し、そして転写、RNAのプロセシング、輸送、および/または翻訳を妨害する。オリゴヌクレオチドでの二本鎖(ds)DNAの標的化は、三重ヘリックスの形成を導き;RNAの標的化は二本鎖ヘリックスの形成を導く。
アンチセンス構築物は、プロモーターおよび他の制御領域、エキソン、イントロン、またはさらに遺伝子のエキソン−イントロンの境界に結合するように設計され得る。アンチセンスRNA構築物、またはこのようなアンチセンスRNAをコードするDNAは、インビトロまたはインビボのいずれにおいても、宿主(例えば、ヒト被験体を含む宿主動物)細胞内で、遺伝子の転写もしくは翻訳、または両方を阻害するために使用され得る。「相補的なヌクレオチド」を含む核酸配列は、標準Watson-Crick相補規則に従って塩基対を形成し得るヌクレオチドである。すなわち、より大きなプリンがより小さいピリミジンと塩基対合し、DNAの場合には、シトシンと対合したグアニン(G:C)およびチミンと対合したアデニン(A:T)、またはRNAの場合には、ウラシルと対合したアデニン(A:U)の組合せのみを形成する。
本明細書中で使用される場合、用語「相補的」または「アンチセンス配列」は、それらの全長にわたって実質的に相補的であり、そして非常にわずかな塩基ミスマッチを有する核酸配列を意味する。例えば、15塩基長の核酸配列は、それらが、わずかに1つまたは2つのみのミスマッチを有する、13または14位での相補的核酸を有する場合、相補的と呼ばれ得る。本来は、「完全に相補的」である核酸配列は、その全長にわたって完全に相補的であり、そして塩基ミスマッチを有さない核酸配列である。
遺伝子配列の全てまたは一部がアンチセンス構築物の状況において使用され得るが、統計学的に、17塩基長の任意の配列がヒトゲノムにおいて一度だけ生じる。したがって、独特の標的配列を特定するに十分である。より短いオリゴマーは作製が容易であり、そしてインビボでの接触可能性が増大するが、多数の他の因子がハイブリダイゼーションの特異性の決定に関与する。その相補的標的に対するオリゴヌクレオチドの結合親和性および配列特異性の両方が、長さの増大にしたがって増大する。8、9、10、11、12、13、14、15、16、17、18、19、20、またはそれ以上の塩基対が使用されることが意図される。所定のアンチセンス核酸が、対応する宿主遺伝子の標的化に有効であるかどうかは、内因性の遺伝子の機能が影響を受けるかどうか、または相補的な配列を有する関連する遺伝子の発現が影響を受けるかどうかを決定するために、インビトロで構築物を単に試験することによって容易に決定され得る。
特定の実施態様において、他のエレメント(例えば、C-5プロピンピリミジンを含むエレメント)を含むアンチセンス構築物を使用することを所望し得る。ウリジンおよびシチジンのC-5プロピンのアナログを含むオリゴヌクレオチドが、高い親和性でRNAに結合し、そして遺伝子発現の強力なアンチセンスインヒビターであることが示されている(Wagnerら、1993)。
標的化アンチセンス送達の代替えとして、標的化リボザイムが使用され得る。用語「リボザイム」は、癌遺伝子DNAおよびRNA中の特定の塩基配列を標的化し、そして切断し得るRNAに基づく酵素をいう。リボザイムは、リボザイム配列を取り込むRNAオリゴヌクレオチドの形態で、細胞に対して直接標的化され得るか、または所望のリボザイムRNAをコードする発現構築物として細胞中に導入され得るかのいずれかである。リボザイムは、アンチセンス核酸について記載されているものとほとんど同じ方法で使用され、そして適用され得る。
C)ワクチンのための抗原
他の治療用遺伝子は、ウイルス抗原、細菌抗原、真菌抗原、または寄生虫抗原のような抗原をコードする遺伝子を含み得る。ウイルスは以下を含む:ピコルナウイルス、コロナウイルス、トガウイルス、フラビウイルス(flavirviru)、ラブドウイルス、パラミクソウイルス、オルトミクソウイルス、ブンヤウイルス、アレンウイルス(arenvirus)、レオウイルス、レトロウイルス、パポバウイルス、パルボウイルス、ヘルペスウイルス、ポックスウイルス、ヘパドナウイルス、およびスポンジ型ウイルス。好ましいウイルス標的として、インフルエンザ、単純ヘルペスウイルス1および2、麻疹、小痘ポリオ、またはHIVが挙げられる。病原体として、トリパノソーマ、サナダムシ、回虫、ぜん虫が挙げられる。胎児抗原または前立腺特異的抗原のような腫瘍マーカーもまた、この様式で標的化され得る。好ましい例として、HIVエンベロープタンパク質およびB型肝炎表面抗原が挙げられる。ワクチンのための本発明のベクターの投与は、ベクターに結合する抗原が、導入遺伝子の長期間の発現を可能にするために十分に非免疫原性であることを必要とする。このために、強力な免疫応答が所望される。好ましくは、個々のワクチン接種は、たまに(例えば、1年ごとまたは2年ごとのように)必要とされるのみであり、そして感染性の薬剤に対する長期間の免疫原性の保護が提供される。
D)制御領域
ウイルスベクターが治療用遺伝子をコードする転写の発現に影響を及ぼすために、治療用遺伝子をコードするポリヌクレオチドは、プロモーターおよびポリアデニル化シグナルの転写制御下にある。「プロモーター」は、宿主細胞の合成機構によって認識されるDNA配列、または遺伝子の特異的な転写を開始するために必要とされる導入された合成機構をいう。ポリアデニル化シグナルは、宿主細胞の合成機構によって認識されるDNA配列、または翻訳のための細胞質への細胞核の転写後の適切なプロセシングおよび輸送のために、mRNA転写物の末端への一連のヌクレオチドの付加を導入することに必要とされる、導入された合成機構をいう。句「転写制御下」は、プロモーターがRNAポリメラーゼの開始およびポリヌクレオチドの発現を制御するためにポリヌクレオチドに関して適切な位置に存在することを意味する。
用語プロモーターは、本明細書中で、RNAポリメラーゼIIの開始部位の周辺に集まる転写制御モジュールの群をいうために使用される。プロモーターが組織化される方法についてのほとんどの考察は、いくつかのウイルスプロモーター(HSVチミジンキナーゼ(tk)およびSV40初期転写ユニットのプロモーターを含む)の分析に由来する。さらに最近の研究によって議論されるこれらの研究は、各プロモーターが約7〜20bpのDNAからなり、そして転写活性化因子または抑制タンパク質の1つ以上の認識部位を含む、不連続の機能的モジュールからなる。
各プロモーターの少なくとも1つのモジュールは、RNA合成の開始部位を位置づけるために機能する。最も公知のこの例はTATAボックスであるが、例えば哺乳動物末端デオキシヌクレオチジルトランスフェラーゼ遺伝子のプロモーター、およびSV40後期遺伝子のプロモーター、それ自体が開始の配置を固定することを補助する開始部位に含まれる別々のエレメントのようないくつかのプロモーターは、TATAボックスを欠いている。
さらなるプロモーターエレメントは、転写開始の頻度を調節する。多数のプロモーターが最近、開始部位の下流の機能的エレメントを含むことも示されているが、代表的には、これらは、開始部位の領域30〜110bp上流に存在する。プロモーターエレメント間の間隔はしばしば可撓性であり、その結果、プロモーター機能は、エレメントが逆転または互いに相対的に移動している場合に、保存される。tkプロモーターにおいて、プロモーターエレメント間の間隔は、活性が減少し始める前は、50bp離れるまで増大し得る。プロモーターに依存して、個々のエレメントは、共作動性または独立のいずれかで機能し、転写を活性化し得ると思われる。
治療用遺伝子の発現を制御するために使用される特定のプロモーターは、それが標的化された細胞中でポリヌクレオチドを発現し得る限りは、重要でないと考えられている。したがって、ヒトの細胞が標的化される場合、ヒト細胞中で発現され得るプロモーターに近接してまたはその制御下に、ポリヌクレオチドコード領域を位置づけることが好ましい。一般的に言えば、このようなプロモーターは、ヒトまたはウイルスプロモーターのいずれかを含み得る。プロモーターのリストを表2に提供する。
周知の特性を有するプロモーターを使用することによって、トランスフェクション後のポリヌクレオチドの発現のレベルおよびパターンを最適化し得る。例えば、特異的細胞中で活性である(例えば、チロシナーゼ(黒色腫)、α-フェトプロテイン、およびアルブミン(肝腫瘍)、CC10(肺腫瘍)、および前立腺特異的抗原(前立腺腫瘍)プロモーターの選択により、治療用遺伝子の組織特異的発現が可能である。
エンハンサーは、DNAの同じ分子上の異なる位置に配置されたプロモーターによる転写を増大する遺伝子エレメントとして、最初に検出された。広い範囲にわたって作用するこの能力は、原核生物の転写の調節の伝統的な研究においてほとんど前例がなかった。続く研究は、エンハンサー活性を有するDNAの領域が、ほとんどプロモーターのように組織化されることを示した。すなわち、これらは、多くの個々のエレメント(そのそれぞれが1つ以上の転写タンパク質に結合する)からなる。
エンハンサーとプロモーターとの間の塩基の距離は、作動可能である。全体としてのエンハンサー領域は、一定の距離で転写を刺激し得なければならない;これは、プロモーター領域またはその相補エレメントにはあてはまる必要はない。一方、プロモーターは、RNA合成の開始を導く1つ以上のエレメントを特定の部位および特定の方向で有さなければならないにもかかわらず、エンハンサーはこれらの特異性を欠いている。プロモーターおよびエンハンサーはしばしば、重複し、そして連続的であり、しばしば、非常に類似した調節機構を有するようである。
さらに、任意のプロモーター/エンハンサーの組み合わせ(真核生物のプロモーターデータベース(EPDB)として)もまた、特定の構築物の発現を駆動するために使用され得る。T3、T7、またはSP6細胞質発現系の使用は、別の可能性のある実施態様である。真核生物細胞は、適切なバクテリオファージポリメラーゼが、送達複合体の一部として、またはさらなる遺伝子発現ベクターとして提供される場合、特定のバクテリオファージプロモーターからの細胞質の転写を支持し得る。
cDNAインサートが用いられる場合、代表的には、遺伝子転写物の適切なポリアデニル化をもたらすためにポリアデニル化シグナルを含むことが所望される。ポリアデニル化シグナルの性質は、本発明の好結果の実施に重要ではないと考えられ、そしてこのような任意の配列が用いられ得る。SV40、ウシ成長ホルモン、および単純ヘルペスウイルスチミジンキナーゼ遺伝子からのようなポリアデニル化シグナルは、多くの標的細胞において十分に機能することが見いだされている。
7.遺伝子導入の方法
本発明のヘルパー細胞株を作製するために、およびその使用のための組換えアデノウイルスベクターを作製するために、種々の遺伝的(すなわち、DNA)構築物は細胞に送達されなければならない。これを達成するための1つの方法は、例えば、本発明のアデノウイルスベクターでの形質転換によって、感染性ウイルス粒子を使用するウイルス形質導入による。あるいは、レトロウイルスまたはウシパピローマウイルスが用いられ得、その両方とも、目的の1つまたは複数の遺伝子での宿主細胞の永久的形質転換を可能にする。他の状況では、導入されるべき核酸は感染性ではなく、すなわち、感染性ウイルス粒子に含まれない。この遺伝物質は、導入のための非ウイルス方法に頼らなければならない。
培養した哺乳動物細胞への発現構築物の導入のためのいくつかの非ウイルス方法はまた、本発明によって意図される。これらには、リン酸カルシウム沈降法(GrahamおよびVan Der Eb,1973;ChenおよびOkayama,1987;Rippeら,1990)、DEAE-デキストラン(Gopal,1985)、エレクトロポレーション(Tur-Kaspaら,1986;Potterら,1984)、直接マイクロインジェクション(HarlandおよびWeintraub,1985)、DNA積載リポソーム(NicolauおよびSene,1982;Fraleyら,1979)、細胞超音波処理(Fechheimerら,1987)、高速マイクロプロジェクタイルを使用する遺伝子ボンバードメント(Yangら,1990)、およびレセプター媒介されるトランスフェクション(WuおよびWu,1987;WuおよびWu,1988)が挙げられる。
一旦構築物が細胞に送達されると、治療遺伝子をコードする核酸は、異なる位置に配置されそして発現され得る。ある実施態様では、治療遺伝子をコードする核酸は、細胞のゲノムに安定に組み込まれ得る。この組込みは、相同組換え(遺伝子置換)によって同種の位置および配向であり得るか、またはランダムな非特異的位置(遺伝子増強(augmentation))に組み込まれ得る。なおさらなる実施態様では、核酸は、DNAの別々のエピソームセグメントとして細胞に安定に維持され得る。このような核酸セグメントまたは「エピソーム」は、宿主細胞周期と独立したまたは同調して維持および複製を可能にするために十分な配列をコードする。発現構築物を細胞に送達する方法および細胞内で核酸が維持される場所は、用いられる発現構築物のタイプに依存する。
本発明の1つの実施態様では、発現構築物は、単に、裸の組換えDNAまたはプラスミドからなり得る。構築物の導入は、細胞膜を物理的または化学的に透過性にする上記の方法のいずれかによって行われ得る。これは、インビトロでの導入に特に適用可能であるが、同様にインビボでの使用にも適用され得る。Dubenskyら(1984)は、成体および新生仔マウスの肝臓および脾臓に、CaPO4沈殿物の形態でポリオーマウイルスDNAを首尾良く注射した。これは、活性なウイルス複製および急性感染を示す。BenvenistyおよびNeshif(1986)はまた、CaPO4沈殿したプラスミドの直接的腹腔内注射が、トランスフェクトした遺伝子の発現を生じることを示した。CAMをコードするDNAがまた、インビボで同様の様式で導入されそしてCAMを発現し得ることが想像される。
裸のDNA発現構築物を細胞に導入するための本発明の別の実施態様は、パーティクルボンバードメントを含み得る。この方法は、細胞膜を貫通させそして細胞を殺さずに細胞に侵入するのを可能にする高速に、DNAコートしたマクロプロジェクタイルを加速する能力に依存する(Kleinら,1987)。小粒子を加速するためのいくつかのデバイスが開発されている。このようなデバイスの1つは、電流を生じるために高圧放電に頼り、これは次に動力を提供する(Yangら,1990)。使用したマイクロプロジェクタイルは、タングステンまたは金のビーズのような生物学的に不活性な物質からなった。
本発明のさらなる実施態様では、発現構築物は、リポソームに封入され得る。リポソームは、リン脂質二重層膜および内部水性媒体によって特徴づけられる小胞構造である。多重ラメラリポソームは、水性媒体によって分離された複数の脂質層を有する。これらは、リン脂質が過剰の水性溶液に懸濁される場合に、自然に形成される。脂質成分は、閉鎖構造の形成前に自己再配置を受け、そして脂質二重層間に水および溶解した溶質を封入する(GhoshおよびBachhawat,1991)。
インビトロでの外来DNAのリポソーム媒介された核酸送達および発現は、非常に成功している。β-ラクタマーゼ遺伝子を使用して、Wongら(1980)は、培養したニワトリ胚、HeLa、および肝ガン腫細胞における、外来DNAのリポソーム媒介性送達および発現の可能性を証明した。Nicolauら(1987)は、静脈内注射後、ラットで好結果なリポソーム媒介された遺伝子導入を成し遂げた。「リポフェクション」技術を含む種々の市販のアプローチも含まれる。
本発明のある実施態様では、リポソームは、血球凝集性ウイルス(HVJ)と複合体化され得る。これは、細胞膜との融合を容易にし、そしてリポソームカプセル化したDNAの細胞侵入を促進することが示されている(Kanedaら,1989)。他の実施態様では、リポソームは、核非ヒストン染色体タンパク質(HMG-1)とともに複合体化されるかまたは用いられ得る(Katoら,1991)。なおさらなる実施態様では、リポソームは、HVJおよびHMG-1の両方とともに複合体化されるかまたは用いられ得る。このような発現構築物が、インビトロおよびインビボでの核酸の導入および発現に好結果に用いられているので、これらは本発明に適用可能である。
治療遺伝子をコードする核酸を細胞に送達するために用いられ得る他の発現構築物は、レセプター媒介される送達ビヒクルである。これらは、ほとんどすべての真核生物細胞においてレセプター媒介されるエンドサイトーシスによる巨大分子の選択的取り込みを利用する。種々のレセプターの細胞タイプ特異的分布のため、送達は非常に特異的であり得る(WuおよびWu,1993)。
レセプター媒介される遺伝子標的化ビヒクルは、一般的に、2つの成分からなる:細胞レセプター特異的リガンドおよびDNA結合剤。いくつかのリガンドは、レセプター媒介される遺伝子導入に使用されている。最も詳細に特徴づけられたリガンドは、アシアロオロソムコイド(asialoorosomusoid)(ASOR)(WuおよびWu,1987)およびトランスフェリン(transferring)(Wagnerら,1990)である。最近、ASORと同じレセプターを認識する合成ネオ糖タンパク質(neoglycoprotein)は、遺伝子送達ビヒクル(Ferkolら,1993;Peralesら,1994)として使用されており、そして上皮増殖因子(EGF)はまた扁平上皮ガン細胞に遺伝子を送達するために使用されている(Myers,EPO 0273085)。
他の実施態様では、送達ビヒクルは、リガンドおよびリポソームを含み得る。例えば、Nicolauら(1987)は、ラクトシル-セラミド、ガラクトース末端アシアロガングリオシド(asialganglioside)を用い、リポソームに組み込まみ、そして肝細胞によるインスリン遺伝子の取り込みの上昇を観察した。したがって、治療遺伝子をコードする核酸がまた、リポソームとともにまたはなしで任意の数のレセプター-リガンド系によって、前立腺、上皮、または腫瘍細胞のような細胞タイプに特異的に送達され得ることが可能である。例えば、ヒト前立腺特異的抗原(Wattら,1986)は、前立腺組織における核酸の媒介された送達についてレセプターとして使用され得る。
8.核酸夾雑物を除去すること
本発明は、夾雑する核酸を除去するためにヌクレアーゼを用いる。代表的ヌクレアーゼには、Benzonase▲R▼、Pulmozyme▲R▼;あるいは当該技術分野内で普通に使用される任意の他のDNaseまたはRNaseが挙げられる。
Benzonase▲R▼のような酵素は、核酸を分解し、そしてタンパク質分解活性を有さない。Benzonase▲R▼が核酸を迅速に加水分解する能力は、細胞溶解物粘度を減少させるためにこの酵素を理想的にする。核酸がウイルスのような細胞由来粒子に接着し得ることは周知である。接着は、凝塊形成により分離を妨害し得るか、粒子のサイズを変化し得るか、または粒子の電荷を変化させ得、任意の産物が所定の精製スキームで回収される場合にはほとんど生じない。Benzonase▲R▼は、精製中に核酸負荷を減少させるために十分適切であり、そのため妨害を排除しそして収量を改良する。
すべてのエンドヌクレアーゼについてのように、Benzonase▲R▼は、特定のヌクレオチド間の内部ホスホジエステル結合を加水分解する。完全な切断の際、溶液中に存在するすべての遊離核酸は、2〜4塩基長のオリゴヌクレオチドまで減少する。
9.精製技術
本発明は、本発明のアデノウイルスベクターを精製するために多くの異なる精製法を用いる。このような技術としては、沈降およびクロマトグラフィーに基づく技術が挙げられ、そして本明細書中以下でより詳細に説明する。
A)密度勾配遠心分離
密度勾配遠心分離の2つの方法(速度帯技術(rate zonal technique)および等密度(等しい密度)技術)があり、そして両方とも、粒子の混合物のすべての成分の定量的分離が必要とされる場合に使用され得る。これらは、浮遊密度の決定のためにおよび沈降係数の評価のためにも使用される。
速度帯技術による粒子分離は、サイズまたは沈降速度の差に基づく。この技術は、予め形成された液体密度勾配の上部に試料溶液を注意深く積層する工程を包含し、この最高密度は、分離されるべき最も密度の高い粒子の密度を越える。次いで、試料を、所望の程度の分離がもたらされるまで、すなわち、粒子が、勾配を通って動いて粒子の相対速度に従って間隔をあけられた分離した帯またはバンドを形成するために十分な時間、遠心分離する。この技術は時間依存的であるので、遠心分離は、分離された帯のいずれかがチューブの底部でペレットになる前に終了されなければならない。この方法は、酵素、ホルモン、RNA-DNAハイブリッド、リボソームサブユニット、非細胞性(subcellular)オルガネラの分離のために、ポリソームの試料のサイズ分布の分析のために、およびリポタンパク質分画のために使用されている。
試料は、分離されるべき粒子密度の全範囲にわたる連続的密度勾配の上部に積層される。それゆえ、勾配の最大密度は、最も密な粒子の密度をいつも超えなければならない。遠心分離中、粒子の沈降は、粒子の浮遊密度と勾配の密度とが等しくなるまで生じる(すなわち、ここでは等式2.12においてpp=pm)。この時点で、粒子はそれ自体の密度よりも大きな密度を有する物質のクッションに浮かんでいるので、遠心分離をどんなに長く続けたとしても、さらなる沈降は生じない。
速度帯技術とは対照的に、等密度遠心分離は、平衡化方法であり、粒子は、バンドを形成して、それ自体の特徴的浮遊密度でそれぞれ帯を形成する。おそらく、粒子の混合物中のすべての成分が必要とされるとは限らない場合、混合物の望ましくない成分は遠心チューブの底に沈降するが、目的の粒子がそれぞれの等密度位置に沈降する勾配範囲が選択され得る。このような技術は、速度帯および等密度アプローチの両方の組み合わせを包含する。
等密度遠心分離は、粒子の浮遊密度にのみ依存し、そしてその形状またはサイズには依存せず、そして時間とは関係ない。したがって、非常に類似の密度(例えば、スクロース溶液においてp=1.3g cm-3)を有する可溶性タンパク質は、通常はこの方法によって分離され得ないが、非細胞性オルガネラ(例えば、スクロース溶液において、ゴルジ装置、p=1.11g cm-3、ミトコンドリア、p=1.19g cm-3、およびペルオキシソーム、p=1.23g cm-3)は、効果的に分離され得る。
予め形成された勾配上で分離されるべき粒子混合物を積層することの代わりとして、試料は、最初に、均一密度の溶液を得るために勾配培地と混合され、遠心分離中に沈降平衡化によって、勾配が「自己形成する」。この方法(平衡化等密度方法という)では、一般的に、重金属(例えば、セシウムまたはルビジウム)の塩、スクロース、コロイド状シリカ、またはMetrizamideが使用される。
試料(例えば、DNA)は、例えば、塩化セシウムの濃縮された溶液と均質に混合される。濃縮された塩化セシウム溶液の遠心分離は、CsCl分子の沈降を生じて、濃度勾配、従って密度勾配を形成する。チューブ全体を通して最初は均一に分布した試料分子(DNA)は、ここで、溶液密度がそれ自体の浮遊密度に等しい領域、すなわち、その等密度位置に達するまで上昇するかまたは沈降するかのいずれかであり、この位置で試料分子はバンド形成して帯になる。この技術はしばしば、平衡を確立するために非常に長い遠心分離時間(例えば、36〜48時間)が必要とされるという不利益がある。しかし、これは、粒子の浮遊密度、二本鎖DNAの塩基組成を決定するために、および環状形態のDNAから線状形態のDNAを分離するために、分析遠心分離で普通に使用される。
分離の多くは、生合成の間に重い同位元素(例えば、15N)の組込みによりDNAの異なる形態間の密度の差を増加させること(Esherichia coliにおけるDNA複製のメカニズムを解明するためにLeselsonおよびStahlによって使用された技術)によって、あるいは重金属イオンまたはエチジウムブロミドのような染料の結合によって改良され得る。等密度勾配はまた、ウイルスを分離および精製するため、およびヒト血漿リポタンパク質を分析するために使用されている。
B)クロマトグラフィー
本発明のある実施態様では、精製したアデノウイルスを産生することが所望される。精製技術は、当該技術分野で周知である。これらの技術は、混合物の他の成分からアデノウイルス粒子を分離するための細胞環境の分画化を包含する傾向がある。他の成分からアデノウイルス粒子が分離されると、アデノウイルスは、完全な精製を達成するために、クロマトグラフィーおよび電気泳動技術を使用して精製され得る。本発明の純粋なアデノウイルス粒子の調製に特に適切な分析方法は、イオン交換クロマトグラフィー、サイズ排除クロマトグラフィー;ポリアクリルアミドゲル電気泳動である。本発明に関連して用いられるべき特に効率的な精製方法は、HPLCである。
本発明のある局面は、アデノウイルス粒子の精製に関し、そして特定の実施態様では、その実質的な精製に関する。本明細書で使用される用語「精製された」は、他の成分から単離可能な組成物をいうことが意図され、ここで、アデノウイルス粒子は、天然に得られ得る形態に対して任意の程度まで精製される。したがって、精製されたアデノウイルス粒子はまた、その粒子が天然に存在し得た環境から免れたアデノウイルス成分をいう。
一般的に、「精製された」とは、種々の他の成分を除去するための分画化を受けているアデノウイルス粒子をいい、そしてこの組成物は、その発現された生物学的活性を実質的に保持する。用語「実質的に精製された」が使用される場合、この名称は、粒子、タンパク質、またはペプチドが、組成物中の約50%以上の構成成分を構成するような、組成物の主要成分を形成する組成物をいう。
タンパク質またはペプチドの精製の程度を定量するための種々の方法は、本明細書の開示を考慮すれば当業者に公知である。これらには、例えば、活性画分の比活性を決定すること、またはSDS/PAGE分析によって画分内のポリペプチドの量を評価することが包含される。画分の純度を評価するために好ましい方法は、画分の比活性を算出すること、これを最初の抽出物の比活性と比較すること、および純度の程度を算出することであり、ここでは「-倍精製数」によって評価される。活性の量を表すために使用される実際の単位は、もちろん、精製に続いて選択される特定のアッセイ技術、ならびに、発現されたタンパク質またはペプチドが検出可能な活性を示すかどうかに依存する。
アデノウイルスが、常に最も精製された状態で提供されるという一般的な要件は存在しない。実際、実質的にあまり精製されていない産物が、ある実施態様で有用性を有することが予想される。部分精製は、少数の精製工程を組み合わせて使用することによって、または同じ一般的精製スキームの種々の形態を利用することによって、達成され得る。例えば、HPLC装置を利用して行われる陽イオン交換カラムクロマトグラフィーが、一般的に、低圧クロマトグラフィーシステムを利用する同じ技法よりも大きな倍率の精製を生じることが理解される。より低い程度の相対精製を示す方法は、タンパク質産物の総回収に、または発現されたタンパク質の活性を維持することに利点を有し得る。
もちろん、当業者に公知のクロマトグラフィー技法および他の精製技法もまた、本発明のアデノウイルスベクターによって発現されるタンパク質を精製するために用いられ得ることが理解される。イオン交換クロマトグラフィーおよび高速液体クロマトグラフィーは、アデノウイルス粒子の精製に用いられる代表的精製技法であり、そして本明細書で以下にさらに詳細に記載される。
イオン交換クロマトグラフィー。イオン交換クロマトグラフィーの基本原理は、交換体に対する物質の親和性が、材料の電気的特性および溶媒中の他の荷電物質の相対親和性の両方に依存する。したがって、結合した材料は、pHを変化させること、そのため材料の電荷を変化させることによって、あるいは競合する材料(その塩は1例にすぎない)を添加することによって、溶出され得る。異なる物質は異なる電気的特性を有するので、遊離についての条件は、各結合した分子種で変化宇する。一般的に、良好な分離を得るために、選択方法は、連続的イオン強度グラジエント溶出または段階的溶出のいずれかである。(イオン強度を同時に増加させることなくpHグラジエントを設定することは困難であるので、pHのみのグラジエントはしばしば使用されない。)陰イオン交換体については、pHおよびイオン強度が徐々に増加するか、またはイオン強度のみが増加するかのいずれかである。陽イオン交換体については、pHおよびイオン強度の両方とも増加する。溶出手順の実際の選択は、通常、試行錯誤、および安定性の考慮の結果である。例えば、不安定な材料については、かなり一定のpHを維持することが最良である。
イオン交換体は、イオンが静電的に結合する、化学的に結合した荷電基を有する固体であり;水性溶液中のイオンに対してこれらのイオンを交換し得る。イオン交換体は、電荷に従って分子を分離するためにカラムクロマトグラフィーで使用され得;実際には、分子の他の特徴は、通常、クロマトグラフの挙動が、分子の電荷密度、電荷分布、およびサイズに感受性であるために重要である。
イオン交換クロマトグラフィーの原理は、荷電分子が可逆的にイオン交換体に吸着し、その結果、分子がイオン環境を変化させることによって結合または溶出され得ることである。イオン交換体における分離は、通常、2つの段階で達成される:第1は、分離されるべき物質が、安定で強固な結合を与える条件を使用して、交換体に結合される;次いで、カラムが、異なるpH、イオン強度、または組成の緩衝液で溶出され、そして緩衝液の成分が、結合部位に対して、結合した材料と競合する。
イオン交換体は、通常、共有結合した荷電基を含む三次元ネットワークまたはマトリクスである。基は、負に荷電している場合、陽イオンを交換しそして陽イオン交換体である。陽イオン交換体で使用される代表的な基は、スルホン基SO3 -である。H+がこの基に結合されるならば、交換体は、酸形態であるといい;例えば、1つのNa+に対してH+を、または1つのCa2+に対して2つのH+を交換し得る。スルホン酸基は、強酸性陽イオン交換体と呼ばれる。他の通常使用される基は、フェノール性ヒドロキシルおよびカルボキシルであり、両方とも弱酸性陽イオン交換体である。荷電基が正、例えば、4級アミノ基であるならば、これは強塩基性陰イオン交換体である。最も通常の弱塩基性陰イオン交換体は、芳香族または脂肪族アミノ基である。
マトリクスは、種々の材料から作製され得る。普通に使用される材料は、デキストラン、セルロース、アガロース、ならびにスチレンとビニルベンゼンとのコポリマーであり、ここでジビニルベンゼンはポリスチレン鎖を架橋し、しかも荷電基を含む。表4は、多くのイオン交換体の組成を示す。
イオン交換体の総容量が、乾燥重量のミリグラム当たりの交換可能な基を取り込む能力の尺度となる。この数は、製造者によって提供され、そして容量を超過してしまうと、イオンが結合せずにカラムを通過するので、重要である。
マトリクスの多孔性は、荷電基がマトリクスの内側および外側の両方にあるので、そしてマトリクスがまた分子ふるいとして作用するので、重要な特徴である。大きな分子は、孔を通過することが不可能であり得;そのため、容量は分子の大きさの増加とともに減少する。ポリスチレンベースの樹脂の多孔性は、ジビニルベンゼンによる架橋の量によって決定される(多孔性は、ジビニルベンゼンの量の増加とともに減少する)。DowexおよびAGシリーズでは、ジビニルベンゼンの割合は、Xの後の数字によって示され、したがって、Dowex 50-X8は、8%ジビニルベンゼンである。
イオン交換体は、メッシュサイズと呼ばれる、種々の粒子サイズにはいる。より細かいメッシュは、増加した表面対容量比(ration)、したがって交換体の所定の容量について生じる交換についての増加した容量および減少した時間を意味する。一方、細かいメッシュは、拡散的な広がりを増加し得るゆっくりとした流速を意味する。非常に細かい(約10μm直径の)粒子および適切な流れを維持するための高圧の使用は、高速または高圧液体クロマトグラフィー、すなわち単にHPLCと呼ばれる。
このような異なる特性−電荷、容量、多孔性、メッシュ−を有する交換体のこのようなコレクションは、特定の分離を達成するために適切な交換体の選択を困難にする。カラム材料のタイプならびに結合および溶出のための条件をどのように決定するかを、以下の実施例に記載する。
技法としてイオン交換クロマトグラフィーを用いる場合、行われるべき多くの選択がある。行われるべき第1の選択は、交換体が陰イオン性または陽イオン性のどちらであるべきかである。カラムに結合されるべき材料が単一電荷(すなわち、正または負のいずれか)を有するならば、選択は明らかである。しかし、多くの物質(例えば、タンパク質、ウイルス)は、負電荷および正電荷の両方を有し、そして正味の電荷はpHに依存する。このような場合、主要な因子は、種々のpH値での物質の安定性である。ほとんどのタンパク質は、正または負のいずれかに荷電する安定性のpH範囲(すなわち、この範囲でタンパク質は変性しない)を有する。したがって、タンパク質が等電点より上のpH値で安定であるならば、陰イオン交換体が使用されるべきであり;等電点以下の値で安定ならば、陽イオン交換体が必要とされる。
強イオン交換体と弱イオン交換体との間の選択はまた、電荷および安定性に対するpHの効果に基づく。例えば、イオン化のために非常に低いまたは高いpHを必要とする弱くイオン化された物質がクロマトグラフされるならば、全pH範囲にわたり機能するので、強イオン交換体が要求される。しかし、物質が不安定であるならば、強交換体はしばしば分子が変性するように分子をゆがめ得るので、弱イオン交換体が好ましい。物質が安定であるpHは、もちろん、特定の弱交換体が荷電されるpHの狭い範囲にマッチさせなければならない。弱く荷電したイオンは通常結合しないので、弱イオン交換体はまた、小さな電荷を有する分子からの高い電荷を有する分子の分離に関して優れている。弱交換体はまた、電荷の差が非常に小さいならば、物質のより大きな分解能を示す。巨大分子が非常に強い電荷を有するならば、強交換体から溶出することが不可能であり得、そして弱交換体がまた好ましくあり得る。一般に、弱交換体は、強交換体よりも有用である。
SephadexおよびBio-gel交換体は、低イオン強度で不安定である巨大分子について特に利点を提供する。これらの材料における架橋は、たとえマトリクスが非常に極性であっても、マトリクスの不溶性を維持するので、イオン化可能な基の密度は、セルロースイオン交換体で可能であるよりも数倍大きくさせ得る。増加した電荷密度は、増加した親和性を意味し、その結果、より高いイオン強度で吸着が行われ得る。一方、これらの交換体は、分子ふるい特性をいくつか保持し、そのため、ときどき、分子量の差は、電荷の差によって引き起こされる分配を無効にする。分子ふるい効果はまた、分離を増強し得る。
小分子は、利用可能容量が大きいので、小さい孔サイズ(高い程度の架橋)のマトリクスで最良に分離されるが、巨大分子は大きな孔サイズを必要とする。しかし、Sephadexタイプを除いて、ほとんどのイオン交換体は、多孔性を分子量とマッチさせる機会がない。
セルロースイオン交換体は、タンパク質およびポリヌクレオチドのような大きな分子を精製するために最良であることが証明されている。これは、マトリクスが繊維状であり、したがって、すべての官能基は表面上にあり、そして最大の分子でさえも利用可能であるからである。しかし、多くの場合には、DEAE-SephacelおよびDEAE-Biogel Pのようなビーズ化形態は、分離におけるより良好な流速および分子ふるい効果の補助があるため、より有用である。
メッシュサイズを選択することは常に難しい。小さいメッシュサイズは、分解能を改良するが、流速を減少させ、これはゾーンの広がりを増加させ、そして分解能を減少させる。したがって、適切なメッシュサイズは、通常、経験的に決定される。
緩衝液自体はイオンからなるので、これらはまた交換し得、そしてpH平衡が影響され得る。これらの問題を避けるために、陰イオン交換体には陽イオン緩衝液を使用し、そして陽イオン交換体には陰イオン緩衝液を使用せよという、緩衝液の規則が採用される。イオン強度は結合における因子であるので、そのイオン強度が高すぎる必要がないように、高い緩衝能を有する緩衝液を選択すべきである。さらに、最良の解析について、一般的に、カラムに試料を適用するために使用されるイオン条件(いわゆる開始条件)は、カラムを溶出するために使用される条件に近くあるべきである。
高速液体クロマトグラフィー(HPLC)は、ピークの並外れた分解能を有する非常に迅速な分離によって特徴づけられる。これは、非常に細かい粒子および適切な流速を維持するための高圧の使用によって達成される。分離は、数分またはせいぜい1時間で達成され得る。さらに、粒子が非常に小さくそして密にパックされ、ボイドボリュームがベッド容量の非常に小さな画分であるので、非常に小さな容量の試料のみが必要とされる。また、バンドが狭く試料はほとんど希釈されないので、試料の濃度は非常に大きい必要はない。
10.薬学的組成物および処方物。
上記の方法によって精製されると、本発明のウイルス粒子は投与され、インビトロ、エクスビボ、またはインビボが意図される。したがって、目的の適用に適切な薬学的組成物として複合体を調製することが所望される。一般に、これは、発熱物質、ならびにヒトまたは動物に有害であり得る任意の他の不純物を、本質的に含まない薬学的組成物を調製することを伴う。一般に、複合体を安定にしそして標的細胞による複合体の取り込みを可能にするために適切な塩および緩衝液を用いることも所望される。
本発明の水性組成物は、薬学的に受容可能なキャリアまたは水性媒体に溶解または分散された、有効量の発現構築物および核酸を含む。このような組成物はまた、接種物ともいわれ得る。句「薬学的または薬理学的に受容可能」とは、動物または適切にはヒトに投与される場合、有害な、アレルギー性の、または他の望ましくない反応を生じない分子実体および組成物をいう。本明細書で使用される場合、「薬学的に受容可能なキャリア」は、任意のおよびすべての溶媒、分散媒体、コーティング剤、抗菌および抗真菌剤、等張および吸収遅延剤などを含む。薬学的に活性な物質のためのこのような媒体および薬剤の使用は、当該分野で周知である。任意の従来の媒体または薬剤が活性成分と適合不可能である限りを除いて、治療組成物におけるその使用が意図される。追加の活性成分もまた、組成物に組み込まれ得る。
遊離塩基または薬理学的に受容可能な塩としての活性化合物の溶液は、ヒドロキシプロピルセルロースのような界面活性剤と適切に混合された水において調製され得る。分散物はまた、グリセロール、液体ポリエチレングリコール、およびその混合物中で、および油中で調製され得る。貯蔵および使用の通常の条件下で、これらの調製物は、微生物の増殖を防止するための保存剤を含む。
本発明のウイルス粒子は、ヒトへの投与を含む治療レジメにおける使用のための古典的な薬学的調製物を含み得る。本発明による治療組成物の投与は、標的組織がその経路によって利用可能である限り、任意の普通の経路による。これには、経口、鼻内、頬内、直腸内、膣内、または局所を含む。あるいは、投与は、患部に直接(orthotopic)、皮内、皮下、筋肉内、腹腔内、または静脈内注射による。このような組成物は、正常には、生理学的に受容可能なキャリア、緩衝液、または他の賦形剤を含む薬学的に受容可能な組成物として投与される。腫瘍に対する適用については、腫瘍内直接注入、切除された腫瘍床への注入、局所的(すなわち、リンパ)または全身的投与が企図される。疾患部位、例えば、腫瘍または腫瘍部位へカテーテルによって数時間から数日にわたって持続灌流を行うことも所望され得る。
本発明の治療組成物は、液体溶液または懸濁液のいずれかとしての注射可能組成物の形態で有利に投与される。液体における注射前の溶解または懸濁に適切な固体形態もまた調製され得る。これらの調製物はまた乳化され得る。このような目的のための代表的な組成物は、薬学的に受容可能なキャリアを含む。例えば、組成物は、リン酸緩衝化生理食塩水の1ミリリットル当たり約100mgのヒト血清アルブミンを含み得る。他の薬学的に受容可能なキャリアは、水性溶液を含み、塩、保存剤、緩衝剤などを含む非毒性賦形剤が使用され得る。非水性溶媒の例は、プロピレングリコール、ポリエチレングリコール、植物油、およびエチルオレエートのような注射可能な有機エステルである。水性キャリアには、水、アルコール性/水性溶液、生理食塩溶液、塩化ナトリウムのような非経口ベヒクル、リンゲルデキストロースなどが挙げられる。静脈内ベヒクルには、液体および栄養補充物が挙げられる。保存剤には、抗菌剤、抗酸化剤、キレート剤、および不活性ガスが挙げられる。薬学的組成物の種々の成分のpHおよび正確な濃度は、周知のパラメータによって調節される。
経口投与に適切な追加の処方物。経口処方物には、例えば、薬学的グレードのマンニトール、ラクトース、スターチ、ステアリン酸マグネシウム、サッカリンナトリウム、セルロース、炭酸マグネシウムなどのような代表的賦形剤が挙げられる。組成物は、溶液、懸濁液、錠剤、丸剤、カプセル剤、持続放出処方物、または粉剤の形態をとる。経路が局所である場合、形態は、クリーム、軟膏、膏薬、またはスプレーであり得る。
有効量の治療剤は、意図された目的に基づいて決定され、例えば、(i)腫瘍細胞増殖の阻害、(ii)腫瘍細胞の排除または殺傷、(iii)ワクチン接種、または(iv)治療遺伝子の長期発現のための遺伝子導入である。用語「単位用量」とは、被験体における使用のために適切な物理学的に離散的単位をいい、各単位は、投与に関連して、すなわち、適切な経路および処置レジメで、上記の所望の応答を生じるために算出された所定量の治療組成物を含む。処置の数および単位用量の両方に従って投与される量は、処置される被験体、被験体の状態、および所望される結果に依存する。複数の遺伝子治療レジメが、特にアデノウイルスについて期待される。
本発明のある実施態様では、腫瘍サプレッサー遺伝子をコードするアデノウイルスベクターは、ガン患者を処置するために使用される。ガンの遺伝子治療に使用されるアデノウイルスベクターの代表的量は、103〜1015PFU/用量(103、104、105、106、107、108、109、1010、1011、1012、1013、1014、1015)であり、ここで用量は、固体腫瘍内の異なる部位で数回の注射に分けられ得る。処置レジメはまた、3〜10週間にわたる、数サイクルの遺伝子移入ベクターの投与を含み得る。数カ月から数年のより長期間のベクターの投与は、継続的な治療利益に必要であり得る。
本発明の別の実施態様では、治療遺伝子をコードするアデノウイルスベクターは、ヒトまたは他の哺乳動物にワクチン接種するために使用され得る。代表的には、この場合のワクチン接種において、所望の効果を生じるために効果的なウイルスの量は、トランスジーンの長期発現が達成されそして強い宿主免疫応答が発生するように、ヒトまたは哺乳動物に投与される。一連の注射、例えば、最初の注射、および続く2回の追加免疫注射は、長期の免疫応答を誘導するために十分である。代表的用量は、所望の結果に依存して106〜1015PFU/注射である。低用量の抗原は、一般に、強い細胞媒介応答を誘導するが、高用量の抗原は、一般に、抗体媒介免疫応答を誘導する。治療組成物の正確な量はまた、主治医の判断に依存し、そして各個体に固有である。
11.実施例
以下の実施例は、本発明の好ましい実施態様を証明するために含まれる。以下の実施例に開示される技法が、本発明の実施において十分に機能するために発明者によって発見された技法を示し、そのため実施について好ましい態様を構成すると見なされ得ることが、当業者に理解されるべきである。しかし、当業者は、本明細書の開示に照らせば、開示される特定の実施態様において多くの変更が行われ得、そして本発明の精神および範囲から逸脱することなく同様または類似の結果をなお獲得し得ることを理解すべきである。
実施例1
材料および方法
A)細胞
Master Cell Bankからの293細胞(ヒト上皮胚性腎細胞)を研究に使用した。
B)培地
ダルベッコ改変イーグル培地(DMEM、4.5g/Lグルコース)+10%ウシ胎児血清(FBS)を、細胞増殖相に使用した。ウイルス産生相については、DMEM中のFBS濃度を2%まで低下させた。
C)ウイルス
AdCMVp53は、サイトメガロウイルス(CMV)即時プロモーターの制御下でヒト野生型p53タンパク質を発現する、遺伝子操作した複製不可能なヒト5型アデノウイルスである。
D)Celligenバイオリアクター
5Lの総容量(3.5Lの作業用量)を有するCelligenバイオリアクター(New Brunswick Scientific,Co.Inc.)を使用して、マイクロキャリア培養を用いてウイルス上清を産生した。13g/Lのガラスコーティングされたマイクロキャリア(SoloHill)を、バイオリアクターで細胞を培養するために使用した。
E)Celligenバイオリアクターにおけるウイルス上清の産生
マスターセルバンク(MCB)からの293細胞を融解し、そしてCellfactories(Nunc)に拡げた。一般に、細胞を、約85〜90%のコンフルエンスに分割した。細胞を、1×105細胞/mlの接種濃度でバイオリアクターに接種した。細胞を、中程度の撹拌によってマイクロキャリアに付着させた。30rpmの速度での連続撹拌を、細胞接種後6〜8時間で開始した。細胞を、pH=7.20、溶存酸素(DO)=60%の空気飽和、温度=37℃に設定したプロセスパラメータで7日間培養した。8日目に、細胞を、MOI=5でAdCMVp53に感染させた。ウイルス感染の50時間後、撹拌速度を、30rpmから150rpmに増加して、細胞溶解および上清へのウイルスの放出を容易にした。ウイルス上清を、感染後74時間で採取した。次いで、ウイルス上清を、さらに濃縮/ダイアフィルトレーションのために濾過した。
F)CellcubeTMバイオリアクターシステム
CellcubeTMバイオリアクターシステム(Corning-Costar)もまた、AdCMVp53ウイルスの産生のために使用した。これは、使い捨ての細胞培養モジュール、酸素発生装置、培地再循環ポンプ、および潅流用の培地ポンプから構成される。使用される細胞培養モジュールは、21,550cm2(1マー)の培養表面積を有する。
G)CellcubeTMにおけるウイルスの産生
マスターセルバンク(MCB)からの293細胞を融解し、そしてCellfactories(Nunc)に拡げた。一般に、細胞を、約85〜90%のコンフルエンスで分割した。細胞を、製造者の推奨に従ってCellcubeTMに接種した。接種細胞密度は、1〜1.5×104/cm2の範囲であった。細胞を、pH=7.20、DO=60%空気飽和の培養条件下で37℃にて7日間増殖させた。培地灌流速度を、CellcubeTM中のグルコース濃度に従って調節した。ウイルス感染の1日前に、灌流用の培地を、DMEM+10%FBSからDMEM+2%FBSに変更した。8日目に、細胞を、5の感染多重度(MOI)でAdCMVp53ウイルスに感染させた。培地灌流を、感染直後1時間で停止し、次いでウイルス産生相の残りの期間に再開した。培養物を感染後45〜48時間に採取した。
H)溶解溶液
20mM Tris+0.25M NaCl+1mM MgCl2、pH=7.50緩衝液中1%(v/v)の濃度でTween-20(Fisher Chemicals)を使用して、CellcubeTMにおいてウイルス産生相の最後に細胞を溶解した。
I)清澄化および濾過
Celligenバイオリアクターからのウイルス上清およびCellcubeTMからのウイルス溶液を、最初に、深層(depth)フィルター(Preflow,GelmanSciences)を用いて清澄化し、次いで、0.8/0.22μmフィルター(SuporCap 100,GelmanSciences)に通して濾過した。
J)濃縮/ダイアフィルトレーション
接流濾過(tangential flow filtration)(TFF)を使用して、Celligenバイオリアクターからのウイルス上清およびCellcubeTMからのウイルス溶液を濃縮および緩衝液交換した。300K名目分子量カットオフ(NMWC)のPellicon IIミニカセット(Millipore)を、濃縮およびダイアフィルトレーションに使用した。ウイルス溶液を最初に10倍濃縮した。これに続いて、一定容量ダイアフィルトレーション法を用いて、20mM Tris+1.0M NaCl+1mM MgCl2(pH=9.00)緩衝液に対して4サンプル容量の緩衝液交換を行った。
同様の濃縮/ダイアフィルトレーションを、カラム精製したウイルスについて行った。100K NMWCのPellicon IIミニカセットを、300K NMWCカセットの代わりに使用した。ダイアフィルトレーションを、20mM Tris+0.25M NaCl+1mM MgCl2(pH=9.00)緩衝液またはダルベッコリン酸緩衝化生理食塩水(DPBS)に対して行った。
K)Benzonase処理
濃縮/ダイアフィルトレーションされたウイルス溶液を、ウイルス溶液中の夾雑する核酸の濃度を低減するために、室温で一晩、100u/ml濃度のBenzonaseTM(American International Chemicals)を用いて処理した。
L)CsCl勾配超遠心分離
粗ウイルス溶液を、Beckman超遠心分離機(XL-90)においてSW40ローターを用いる2重CsCl勾配超遠心分離を用いて精製した。最初に、7mlの粗ウイルス溶液を、それぞれ、1.25g/mlおよび1.40g/ml CsCl溶液の2.5ml等容量で作られる段階CsCl勾配の上部に重層した。CsCl勾配を、室温で1時間、35,000rpmで遠心分離した。勾配界面のウイルスバンドを回収した。次いで、回収されたウイルスを、等密度(isopicnic)CsCl勾配に通してさらに精製した。これは、ウイルス溶液を少なくとも1.5倍容量の1.33g/ml CsCl溶液と混合することによりなされた。CsCl溶液を、室温で少なくとも18時間35,000rpmで遠心分離した。低部のバンドをインタクトなウイルスとして回収した。直ぐに、CsClを取り除くために、ウイルスを20mM Tris+1mM MgCl2(pH7.50)緩衝液に対して透析した。透析されたウイルスを、将来の使用のために-70℃で貯蔵した。
M)イオン交換クロマトグラフィー(IEC)精製
Benzonase処理されたウイルス溶液を、IECを用いて精製した。強陰性樹脂Toyopearl SuperQ 650M(Tosohaas)を、精製のために使用した。XK16カラム(Pharmacia)を備えたFPLCシステム(Pharmacia)を、最初の方法開発のために使用した。さらなるスケールアップ研究を、XK50カラム(Pharmacia)を備えたBioPilotシステム(Pharmacia)を用いて行った。簡単には、樹脂をカラムにパックし、そして1N NaOHで清浄し、次いで緩衝液Bを充填し、これに続いて緩衝液Aで調節した。緩衝液AおよびBは、それぞれ、20mM Tris+0.25M NaCl+1mM MgCl2(pH9.00)および20mM Tris+2M NaCl+1mM MgCl2(pH9.00)からなる。ウイルス溶液サンプルを調節されたカラムにロードし、続いてUV吸収がベースラインに達するまで緩衝液Aでカラムを洗浄した。精製されたウイルスを、10カラム容量の直線NaCl勾配を用いることによりカラムから溶出させた。
N)HPLC分析
HPLC分析手順を、ウイルス生成および精製の効率を評価するために開発した。トリス(ヒドロキシメチル)アミノメタン(tris)を、FisherBiotech(カタログ番号BP154-1;Fair Lawn,New Jersey,U.S.A.)から得;塩化ナトリウム(NaCl)を、Sigma(カタログ番号S-7653,St.Louis,MO,U.S.A)から得た。両方を、さらなる精製なしで直接使用した。HPLC分析を、TosoHaasからの陰イオン交換カラム(7.5cm×7.5mm ID、10μm粒子サイズ、カタログ番号18257)を備えたGold Workstation Software(126バイナリーポンプおよび168ダイオードアレイ検出器)を用いる、BeckmanからのAnalytical Gradient Systemにおいて行った。1-ml Resource Q(Pharmacia)陰イオン交換カラムを使用して、それらのHEPES緩衝系を用いて、Huygheらにより開発された方法を評価した。この方法は、Bioreactorシステムのみについて試みた。
本発明のHPLCシステムにおいて使用された緩衝液は、緩衝液A:10mM tris緩衝液(pH9.0)、緩衝液B:緩衝液A中の1.5M NaCl(pH9.0)であった。緩衝液を、Corning(カタログ番号25970-33)による0.22μmの瓶上部(bottle top)フィルターに通して濾過した。全てのサンプルを、注入前にGelman Sciences(カタログ番号4187)からの0.8/0.22μm Acrodisc PFに通して濾過した。
サンプルを、60〜100μl容量でHPLCカラムに注入する。注入後、カラム(TosoHaas)を、0.75ml/分の流速で3分間、20%Bで洗浄する。次いで、勾配を開始し、ここでBを6分間にわたって20%から50%に増大させる。次いで、勾配を、3分間にわたって50%から100%Bに変え、続いて6分間100%Bにする。次いで、塩濃度を、4分間にわたって再び20%に段階的にもとに戻し、そしてさらに6分間20%Bで維持する。Adp53の保持時間は、
実施例2
ウイルス生成および精製に対するCellcubeTMにおける培地灌流速度の効果
灌流細胞培養システム(例えば、CellcubeTM)について、培地灌流速度は、産物の収率および品質に重要な役割を果たす。2つの異なる培地灌流ストラテジーを調べた。一方のストラテジーは、CellcubeTMにおいてグルコース濃度を
培養パラメーター(例えば、pH、DO)における有意な変化は、2つの異なる灌流速度間で観察されなかった。ほとんど等量の粗ウイルス(精製前)は、表5に示されるように、1% Tween-20溶解溶液を用いて採集した後に生成された。しかし、劇的な差異が、高および低培地灌流速度の生成作業からのウイルス溶液のHPLCプロフィールにおいて見られた。
実施例3
細胞採集および溶解の方法
以前の経験に基づいて、本発明者らは、最初に凍結解凍法を評価した。細胞を、感染の45〜48時間後にCellcubeTMから採集した。最初に、CellcubeTMを培養系から単離し、そして消費された培地を排水した。次いで、50mM EDTA溶液をCubeにポンプでくみ上げて、培養表面から細胞を取り外した。このように得られた細胞懸濁物を、10分間、1,500rpm(Beckman GS-6KR)で遠心分離した。得られた細胞ペレットを、ダルベッコリン酸緩衝化生理食塩水(DPBS)に再懸濁した。細胞懸濁物を、37℃水浴とドライアイスエタノール浴との間の5サイクルの凍結/解凍に供して、細胞からウイルスを放出した。このように作製された粗細胞溶解物(CCL)を、HPLCにおいて分析した。
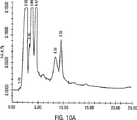
図2は、HPLCプロフィールを示す。ウイルスピークは、9.32分の保持時間で観察されなかった。代わりに、9.11分および9.78分の保持時間の2つのピークを生じた。このプロフィールは、ウイルスと同様の溶出時間を有する他方の夾雑物がCCLに存在し、そしてウイルス精製を妨げることを示唆する。結果として、CCLをFPLCを用いたIECにより精製した場合、非常に低い精製効率が観察された。
低い精製効率に加えて、表6に示されるように、EDTA溶液への細胞採集工程の間の有意な産物損失があった。約20%の産物は、処分したEDTA溶液中で失われた。さらに、約24%の粗ウイルス産物は、消費された培地(これもまた捨てられた)に存在した。従って、56%の粗ウイルス産物しかCCLになかった。さらに、凍結解凍は、変化の大きいプロセスであり、測定の可能が非常に制限される。産物の損失がより少ない、より効率的な細胞溶解プロセスは、開発される必要があった。
細胞溶解を、室温または氷上のいずれかで30分間行った。澄んだ溶解溶液を、沈殿物および細胞破片を除去するための遠心分離後に得た。溶解溶液をBenzonaseで処理し、次いでHPLCにより分析した。図3は、異なる界面活性剤に由来する溶解溶液のHPLCプロフィールを示す。ThesitおよびNP-40は、Triton X-100と同様に振る舞った。1% Tween-20から生成された溶解溶液は、最も良いウイルス分離を生じ、最も少ないウイルス分離はBrij-58で観察された。より効率的な細胞溶解は、1%(w/v)の界面活性剤濃度で見出された。溶解温度は、調べられた界面活性剤濃度下でウイルス分離に有意に寄与しなかった。プロセス平易の目的のために、室温での溶解が推奨される。溶解溶液は、20mM Tris+0.25M NaCl+1mM MgCl2中の1% Tween-20からなり、細胞溶解のためにpH=7.50を使用し、そしてウイルス採集をCelltubeTMにおいて行った。
実施例4
ウイルス回収に対する濃縮/ダイアフィルトレーションの効果
溶解工程からのウイルス溶液を清澄化し、そして濃縮/ダイアフィルトレーション前に濾過した。異なるNMWC(100K、300K、500K、および1000Kを含む)のTFF膜を、効率的な濃縮/ダイアフィルトレーションについて評価した。濾過物に対して最小のウイルス損失を伴う最も高い培地流動(medium flux)を、300K NMWC膜を用いて得た。より大きなNMWC膜はより高い培地流動を提供するが、濾過物に対してより大きなウイルス損失をもたらし、一方より小さなNMWC膜は不十分な培地流動を達成した。ウイルス溶液を最初に10倍濃縮し、続いて、一定容量法を用いて、20mM Tris+0.25M NaCl+1mM MgCl2(pH9.00)緩衝液に対する4サンプル容量のダイアフィルトレーションを行った。濃縮/ダイアフィルトレーションプロセス間、膜を横切る圧力の低下を
Benzonase処理に対する塩添加の効果
濃縮/ダイアフィルトレーション後のウイルス溶液を、ウイルス溶液中の夾雑核酸の濃度を低減するためにBenzonase(ヌクレアーゼ)で処理した。異なる作用濃度のBenzonase(50、100、200、300単位/mlを含んだ)を、核酸濃度の低減について評価した。プロセス平易の目的のために、処理を室温で一晩行った。ヒトゲノムDNAプローブにハイブリダイズ可能な夾雑核酸の有意な低減は、Benzonase処理後に見られた。
表9は、Benzonase処理前後の核酸濃度の低減を示す。ウイルス溶液を、Benzonase処理前後にHPLCにおいて分析した。図4Aおよび図4Bに示されるように、夾雑核酸ピークの劇的な低減は、Benzonase処理後に観察された。これは、核酸ハイブリダイゼーションアッセイの結果と一致する。有効性のために、100u/mlのBenzonase濃度を、粗ウイルス溶液処理に使用した。
Benzonase処理間の可能な核酸ウイルス相互作用を最小にするために、異なる濃度のNaClを、Benzonase処理前にウイルス溶液に添加した。HPLCプロフィールにおける劇的な変化は、ウイルス溶液中1M NaClの存在下でのBenzonase処理後に起こらなかった。図5は、1M NaClの存在下でのBenzonase処理後のウイルス溶液のHPLCプロフィールを示す。図4Bに示されるものとは異なり、9.35分の保持時間でのウイルスピークは、Benzonase処理後になお存在する。この結果は、1M NaClの存在が、Benzonase処理間に核酸とウイルスとの相互作用を妨げ、そして夾雑核酸からのウイルスのさらなる精製を容易にすることを示す。
実施例6
イオン交換クロマトグラフィー精製
生理的なpH条件でのアデノウイルス表面上の負電荷の存在は、アデノウイルス精製のための陰イオン交換体の評価を促進した。強い陰イオン交換体Toyopearl Super Q 650Mを、精製方法の開発のために使用した。ウイルス精製に対するローディング緩衝液(緩衝液A)におけるNaCl濃度およびpHの効果を、FPLCシステムを用いて評価した。
A)方法開発
イオン交換クロマトグラフィーについて、緩衝液pHは最も重要なパラメーターの1つであり、そして精製効率に対して劇的な影響を有し得る。ウイルス生成を通じて使用される培地のpHおよび伝導性に関して、本発明者らは、緩衝液Aとして20mM Tris+1mM MgCl2+0.2M NaCl(pH7.50)を処方した。5cmの長さを有するToyopearl Super Q 650MでパックされたXK16カラムを緩衝液Aで調節した。
CelligenバイオリアクターからのBenzonase処理濃縮/ダイアフィルトレーションウイルス上清の5mlサンプルを、カラムにロードした。カラムを洗浄した後、溶出を、mM Tris+1mM MgCl2+2M NaCl(pH7.50)に達するように、10カラム容量以上の緩衝液B処方物の直線勾配で行った。
図6は溶出プロフィールを示す。3つのピークが、それらの間での満足な分離なく溶出間で観察された。293細胞馴化培地(ウイルスなし)を用いて行われたコントロール研究は、最初の2つのピークがウイルス関連であることを示した。分離効率をさらに改善するために、緩衝液pHの効果を評価した。緩衝液pHを9.00まで増大させたが、一方他の条件を一定に保った。図7に示されるように、7.50の緩衝液pHのものと比較して、非常に改善した分離が観察された。画分#3、#4、および#8をHPLCにおいて分析した。
図8に示されるように、ウイルスの大部分は画分#4において見出され、ウイルスは画分#3および#8において検出されなかった。画分#8は、主に夾雑核酸からなると見出された。しかし、精製はまだ最適ではなかった。画分#3と#4との間に重複があり、夾雑物は画分#4においてなお検出された。
図7におけるクロマトグラムに基づいて、ウイルス精製のさらなる改善は、緩衝液A中の塩濃度を増大させることにより達成され得ることが推論された。結果として、ウイルスピークの前である画分#3に存在する夾雑物を、フロースルー(flow through)画分に移し得る。緩衝液A中のNaCl濃度を0.3Mに増大させたが、一方他の条件を一定に保った。図9は、緩衝液A中の0.3M NaCl条件下での溶出プロフィールを示す。
精製効率における劇的な改善が達成された。予想されたように、図7において観察された夾雑物ピークを、増大した塩条件下で排除した。粗ウイルス上清、フロースルー、ピーク#1、およびピーク#2からのサンプルをHPLCにおいて分析し、そして結果を図10に示す。ウイルスは、フロースルー画分において検出されなかった。粗材料中に存在する夾雑物の大部分が、フロースルー中に見出された。ピーク#1のHPLC分析は、ウイルスピークと定義された単一ウェルを示した。このHPLCプロフィールは、2重CsCl勾配精製ウイルスから得られるものと等しい。CsCl勾配精製ウイルスにおいて3.14分および3.61分の保持時間で観察されたピークは、グリセロール関連ピークである。精製ウイルスは、1.27±0.03のA260/A280比を有する。これは、2重CsCl勾配精製ウイルスならびにHuygheら(1996)により報告された結果の値と同様である。ピーク#2は、主に夾雑核酸からなる。精製結果に基づいて、本発明者らは、バイオリアクターからのアデノウイルス上清のIEC精製のために以下の方法を提案した。
緩衝液A:20mM Tris+1mM MgCl2+0.3M NaCl、pH=9.00
緩衝液B:20mM Tris+1mM MgCl2+2M NaCl、pH=9.00
溶出:10カラム容量直線勾配
B)方法スケールアップ
方法開発の後、精製を、同じ精製方法を用いてXK16カラム(1.6cm I.D.)からXK50カラム(5cm I.D.、10倍スケールアップ)にスケールアップした。同様の溶出プロフィールを、図11に示されるようにXK50カラムにおいて達成した。ウイルス画分をHPLCにおいて分析し、これはXK16カラムから得られたものと等しいウイルス純度を示した。
スケールアップ研究の間に、緩衝液Aの塩濃度を定量するために、伝導率を用いることが、より好都合であり確実であることが認められた。緩衝液Aの最適伝導率は、ほぼ室温(21℃)で、25±2mS/cmの範囲である。二重のCsCl精製ウイルスとともに精製工程で産生されるサンプルを、SDS-PAGEで分析した。
図12に示されているように、主要アデノウイルス構造タンパク質の全てを、SDS-PAGEで検出した。IEC精製ウイルスは、二重のCsCl精製ウイルスと同じ染色を示した。ウシ血清アルブミン(BSA)濃度の有意な減少が、精製中に達成された。精製ウイルス中のBSA濃度は、図13に示されているように、ウエスタンブロットアッセイの検出レベルを下まわった。
精製工程中のウイルス溶液中の夾雑核酸濃度の減少は、核酸スロットブロットを用いて測定された。32P標識ヒトゲノムDNAを、(293細胞はヒト胚腎細胞であるので)ハイブリダイゼーションプローブとして使用した。表10は、精製プロセスの異なる段階での核酸濃度を示す。最終精製ウイルス溶液の核酸濃度は、60pg/mlに減少され、最初のウイルス上清に比較して約3.6 x 106倍の減少であった。ウイルス力価および総粒子の比に対して感染性を、精製ウイルスに対して測定し、表9において、二重のCsCl精製からの結果と比較した。両ウイルス回収率および粒子/PFU比は、2つの精製方法間で非常に類似する。カラム精製ウイルス溶液の力価は、濃縮工程を実施することによってさらに増大され得る。
他の精製方法
強陰イオン交換クロマトグラフィーに加えて、クロマトグラフ法の他の態様(例えば、サイズ排除クロマトグラフィー、疎水相互作用クロマトグラフィー、陽交換クロマトグラフィー、または金属イオンアフィニティークロマトグラフィー)もまた、AdCMVp53ウイルス精製について評価した。Toyopearl Super Qに比較して、精製のこれら全態様が、産物の低回収率をともなって、より非効率的な精製を示した。従って、Toyopearl Super Q樹脂が、AdCMVp53の精製に対して推薦される。しかし、他の四級アンモニウム化学に基づく強陰イオン交換体は、いくらかのプロセスの改変によりAdCMVp53精製に適するようである。
実施例8
CellcubeTMから産生された粗AdCMVp53ウイルスの精製
2つの異なる産生方法を、AdCMVp53ウイルスの産生のために開発した。一方の方法は、撹拌されたタンクバイオリアクター中のマイクロタイター培養物に基づいた。他方は、CellcubeTMバイオリアクターに基づいた。上記のように、精製方法は、撹拌タンクバイオリアクターにより生産された粗ウイルス上清を使用して、開発した。バイオリアクターおよびCellcubeTMの両方において、ウイルス生産のために、同じ培地、細胞、およびウイルスを使用したが、細胞が接着する培養物表面が異なることが認識された。
バイオリアクターでは、細胞を、マイクロキャリアに被覆されたガラス上で増殖し、一方、CellcubeTM細胞を、独自に処理したポリスチレン培養表面上で増殖した。CellcubeTMでは、定常的な培地潅流を使用したが、一方、バイオリアクターでは、培地潅流を使用しなかった。CellcubeTMでは、粗ウイルス産物をウイルス感染細胞形態で回収し、これはバイオリアクターから回収されたウイルス上清とは異なる。
回収されたウイルス感染細胞を5サイクルの凍結-融解した後に産生された粗ウイルス細胞溶解物(CCL)を、上記の方法を使用したIECによって精製した。バイオリアクターからのウイルス上清とは異なり、CellcubeTMから産生されたCCL物質に対して満足される精製は達成されなかった。図14は、そのクロマトグラムである。結果は、凍結-融解回収された細胞による、CellcubeTMから産生された粗ウイルス溶液は、IEC方法では容易に精製されないことを示唆する。
他の精製方法(疎水相互作用および金属キレートクロマトグラフィーを含む)を、CCLでのウイルス精製について試験した。不運にも、どちらの方法によっても精製の改善は認められなかった。CCLでのウイルス精製の困難さおよび生産プロセスにおける凍結-融解工程に関連する不利益を考慮して、発明者は他の細胞溶解方法を探求することにした。
A)溶解緩衝液中の粗ウイルス溶液の精製
実施例1および3に記載されているように、異なる界面活性剤溶解方法をスクリーニングするために、HPLC分析を使用した。HPLCの結果に基づき、20mM Tris+0.25M NaCl+1mM MgCl2、pH=7.50緩衝液中の1% Tween-20を、溶解緩衝液として使用した。ウイルス産生期の最終段階で、感染細胞の回収にかわって、使用済み培地を取り出した後に、溶解緩衝液をCellcubeTMへポンプで注入した。細胞を溶解し、30分間インキュベートして溶解緩衝液へウイルスを放出した。
浄化および濾過後に、ウイルス溶液を濃縮/透析濾過し、Benzonase処理によって夾雑アミノ酸濃度を減少した。処理されたウイルス溶液をToyopearl SuperQ樹脂を使用して、上記改良された方法によって精製した。バイオリアクターからのウイルス上清を用いて得られたのと同様に、溶出中に満足な分離が達成された。図15は、溶出プロフィールを示す。しかし、ウイルス画分がHPLCで分析されたとき、ウイルスピークに加えて、別のピークが検出された。結果は図16Aに示されている。
ウイルスをさらに精製するために、回収ウイルス画分を、同じ方法を用いて再度精製した。図16Bに示されているように、ウイルス画分の純度は、第二精製後にかなり改善された。金属キレートクロマトグラフィーもまた、第二精製の候補として評価した。ウイルス純度において第二IECで認められるのと同様の改良が達成された。しかし、その簡便さの理由から、IECが第二精製に対して選択される方法として好ましい。
実施例2で上述されているように、細胞増殖およびウイルス産生期に用いられた培地灌流速度は、Tween-20粗ウイルス回収のHPLC分離プロフィールにかなりの影響を有する。高速培地灌流下で生産される粗ウイルス溶液に対しては、必要なウイルス純度を達成するために2つのイオン交換カラムが必要である。
低速培地灌流下で生産されるウイルス溶液に対してHPLCで観察される、非常に改良された分離に基づいて、1つのイオン交換カラムによる精製が、必要なウイルス純度を達成し得るようである。図17は、低速培地灌流下で生産された粗ウイルス溶液を用いた、溶出プロフィールを示す。鋭いウイルスピークが溶出の間に達成された。ウイルス画分のHPLC分析は、1回のイオン交換クロマトグラフィー工程後のCsCl濃度勾配精製ウイルスの純度と同程度のウイルス純度を示す。図18は、HPLC分析結果を示す。
精製されたウイルスを、SDS-PAGE、BSAに対するウエスタンブロット、および核酸スロットブロットによって更に分析し、夾雑核酸濃度を測定した。分析結果は、それぞれに図19A、図19B、および図19Cに示されている。これらの全分析は、二重CsCl濃度勾配精製ウイルスに比較して、同程度の純度を有することを示す。表11は、カラム精製前および後のウイルス力価および回収率を示す。比較目的のために、二重CsCl濃度勾配精製によって達成された典型的なウイルス回収率もまた含まれている。両方法によって、同じウイルス回収率が達成された。
Toyopearl Super Q樹脂の動力学的許容量を、低速培地灌流下で生産された、Tween-20回収ウイルス溶液の精製について評価した。樹脂100mlをXK50カラムに詰めた。個別の粗ウイルス溶液量を、本明細書に記載されている方法を用い、カラムに通して精製した。
ウイルス突破(breakthrogh)および精製効率を、HPLCで分析した。図20は、HPLC分析結果を示す。2:1の試料/カラム容積比を超えるカラム負荷係数で、ウイルス画分の純度が減少された。夾雑物がウイルスとともに溶出した。3:1を超える負荷係数で、ウイルスのフロースルーへの突破が観察された。従って、樹脂の作用負荷許容量は、1:1の試料/カラム容積比の範囲であると提案される。
B)精製後の濃縮/透析濾過
カラム精製後の濃縮/透析濾過工程は、ウイルス力価を増加するのみでなく、必要に応じて、ウイルス産物に対して特定される緩衝系を交換するように作用する。300K NMWC TFF膜を、濃縮工程で使用した。精製されたウイルス中にタンパク質分解性および核酸夾雑物が存在しないので、膜を通しての顕著な圧力降下をともなわずに、非常に高い緩衝液流動が達成された。
ほぼ100%のウイルス回収が、20mM Tris+1mM MgCl2+0.15M NaCl,pH=7.50に緩衝液を変化させるてことによって、この工程の間に達成された。精製ウイルスもまた、濃縮/透析濾過工程間に、DPBSへ首尾よく緩衝液交換された。濃縮係数は、最終産物において所望されるウイルス力価、およびカラムから溶出されたウイルス溶液の力価によって決定され得る。この可撓性(flexibility)は、最終精製ウイルス産物一貫性を維持する助けとなる。
C)IEC精製AdCMVp53中の欠陥アデノウイルスの評価
プロデューサー細胞における100%未満のアデノウイルスパッケージング効率に起因して、概して、いくらかの欠陥アデノウイルスが粗ウイルス溶液中に存在する。欠陥ウイルスは、ウイルスキャプシド内にパッケージされたDNAを有さず、従って、密度差に基づいてCsCl濃度勾配超遠心分離で、完全ウイルスから分離され得る。イオン交換クロマトグラフィーに基づいて、完全ウイルスから欠陥ウイルスを分離することは困難であるようであり、両ウイルスは類似の表面化学特性を有すると思われる。過剰量の欠陥ウイルスの存在は、精製産物の性質に影響する。
存在する欠陥ウイルス粒子のパーセントを評価するために、精製および濃縮されたウイルスを、等圧(isopicnic)CsCl超遠心分離に供した。図21に示されているように、完全ウイルスバンドの頂上のかすかなバンドが、遠心分離後に認められた。両バンドを回収し、20mM Tris+1mM MgCl2、pH=7.50緩衝液で透析し、CsClを除去した。透析されたウイルスをHPLCで分析し、結果を図22に示す。両ウイルスは、同様の保持時間を示す。しかし、欠陥ウイルスは、完全ウイルスよりも小さなA260/A280比を有する。これは、欠陥ウイルスにおいてウイルスDNAがより少ないことを示している。
3.02分から3.48分の保持時間に認められるピークは、-70℃で凍結される前にウイルスに添加される(10% v/v)グリセロールによって生じる。欠陥ウイルスパーセントは、全ウイルスの1%未満であった。欠陥ウイルスのこの低いパーセントは、精製ウイルス産物中の感染ウイルス(PFU)に対する全粒子比に影響しないようである。両ウイルスを、SDS-PAGEで分析した(図19Aに示されている)。完全ウイルスに比較して、欠陥ウイルスは24および48.5KDのバンドの、DNA結合コアタンパク質を欠如する。この結果は、欠陥ウイルスでのDNA非存在に一致する。
D)AdCMVp53ウイルスの生産および精製のプロセス概説
上記プロセスの改良結果に基づいて、発明者は、図23に示されているような、AdCMVp53についての生産および精製フローチャートを提供する。段階的および累積ウイルス回収が含まれ、対応ウイルス収量は1mer CellcubeTMに基づく。最終ウイルス回収率は、約70±10%である。これは、p53タンパク質コーディングアデノウイルスの精製のために、DEAEイオン交換および金属キレートクロマトグラフィー精製手順を用いた、Huygheら(1996)によって報告されたウイルス回収率より約3倍高い。最終精製ウイルス産物の約3×1012PFUが、1mer CellcubeTMから産生された。これは、精製のための二重CsCl濃度勾配超遠心分離を用いる現在の生産方法に比較して、同様の最終産物収量を示す。
E)規模拡大
良好な拡大規模研究は、4mer CellcubeTMシステムによって実施され、16mer CellcubeTMシステムでのウイルス生産でウイルス産生を評価することが、最近進行中である。産生された粗ウイルス溶液を、濾過し、濃縮して、より大きなPelliconカセットを用いて透析濾過する。ウイルスの性質および回収量を決定する。Benzonase処理後に、粗ウイルス溶液を、20cmおよび30cm BioProcessカラムを用いて、4merおよび16merのそれぞれについて精製する。
実施例9
無血清懸濁培地での改良されたAd-p53生産
293細胞の適応
293細胞を、Tフラスコ中でFBS濃度を順次下げることにより、市販のIS293無血清培地(Irvine Scientific; Santa Ana,CA)に適応させた。PDWB1バイアル中の凍結細胞を解凍し、Tフラスコ中の10% FBS DMEM培地に入れ、その細胞を、培地中のFBS濃度を順次下げることにより、Tフラスコ中の無血清IS 293培地に適応させた。T-75フラスコ中での6継代後に、FBS%を約0.019%と評価した。撹拌フラスコに移す前に、その細胞をTフラスコ中でさらに2回継代培養した。
Tフラスコ中の無血清適応293細胞を懸濁培地に適応させた
Tフラスコ中の上記無血清適応細胞を、懸濁培養のために血清非含有の250mL撹拌懸濁培地(100mL 操作容積)移した。開始細胞密度は1.18E+5vc/mLであった。細胞培養中に、生存率が減少し、細胞の大きな凝塊が認められた。Tフラスコ中でさらに2継代した後に、懸濁培地への適応を再度試みた。二回目の試みで、培地に100mg/L濃度のヘパリンを補充し、細胞の凝集を防ぎ、開始細胞密度を5.22E+5vc/mLに増加した。細胞培養中に、細胞密度のなんらかの増加があり、細胞生存率が維持された。それ以降、その細胞を、撹拌フラスコ中でさらに7継代培養し、その継代中に細胞の倍化時間はめざましく減少され、7継代の最終には(付着細胞培養での10% FBS培地での1.2日細胞に匹敵する)約1.3日であった。ヘパリンを補充された無血清IS 293培地では、ほとんど全ての細胞が、懸濁培養で細胞凝塊を形成せずに、個別細胞として存在した(表12)。
無血清懸濁培養でのAd5-CMVp53ベクターの産生をテストするために、無血清懸濁培養に適応された上記細胞を、250mL撹拌フラスコ中で、0.1% Pluronic F-68およびヘパリン(100mg/L)を補充した100mL無血清IS293培地にて増殖させた。3日目に、細胞が1.36E+06生存細胞/mLに達したときに、5MOIに感染していた。その上清を、感染後毎日、HPLCウイルス粒子/mLについて分析した。3日目の試料以外は、ウイルスは検出されなかった。3日目には、ウイルスは2.2E+09vps/mLであった。6日目のpfu/mLは、2.6+/-0.6E+07pfu/mLであった。細胞あたりのpfu産生は、血清補充培地での付着培養の約46倍下回る、19であると評価した。コントロールとして、感染なしに細胞増殖を確認した。
上記のように、細胞がAd-p53ベクターを産生したことが実証された後に、その細胞を、撹拌フラスコ中で、0.1% F-68および100mg/Lヘパリンを含有する無血清IS293培地で増殖し、1.0E+07生存細胞/mL/バイアルを有する、無血清懸濁適応細胞バンクを作製した。細胞を回収するために、細胞が中期対数増殖期(mid-log phase growth)にあり、生存率が90%を超えるときに、その細胞を遠心分離し、無血清のIS293補充培地に再懸濁させ、再度遠心分離沈降させてその細胞を洗浄した。次に、0.1% F-68、100mg/mLヘパリン、10% DMSO、および0.1%メチルセルロースを含有する冷却IS293である、凍結保存培地に再度懸濁させた(1E+07生存細胞/mLをもたらす)。細胞懸濁物を滅菌凍結保存バイアルに移し、封印して-70℃で一晩、凍結容器中で凍結した。バイアルを液体窒素貯蔵装置へ移した。マイコプラズマテストは、陰性であった。
凍結細胞を再生するために、1バイアルを、T-150中で、0.1% F-68および100mg/Lヘパリンを含有する50mLの無血清IS293培地へ解凍した。次に、培養物を、250mL撹拌フラスコ中で3回継代培養した。もう一方の研究で、1バイアルを、250mL撹拌フラスコ中で、100mLの無血清補充IS293培地へ解凍した。それ以降、これらを、無血清撹拌フラスコ中で2回継代培養した。この両方の研究では、細胞は非常に良好に増殖した。
撹拌フラスコ中の無血清懸濁培養での培地交換およびウイルス生産
以前の撹拌フラスコでの懸濁培養による無血清ウイルス生産では、細胞あたりのウイルス産生は非常に少なすぎて、無血清懸濁生産に実用的でない。これは、栄養の枯渇および/または阻害副産物の産生に起因すると考えられた。消費された培地を新鮮な無血清補充IS293培地と交換するために、細胞を3日目に遠心沈降させ、F-68およびヘパリン(100mg/L)を補充した、新鮮な無血清IS293培地に再懸濁させ、得られた細胞密度は1.20E+06vc/mLであり、細胞を、5 MOIでAd5-CMVp53ベクターに感染させた。細胞外HPLC vps/mLは、3日目で7.7E+09vps/mLであり、4日目で1.18E+10vps/mLであり、5日目で1.2E+10vps/mLであり、6日目で1.3E+10vps/mLであり、6日目のpfu/mLは、2.75+/-0.86E+08tvps/mLであった。pfuに対するHPLCウイルス粒子の比は、約47であった。また、細胞を遠心沈降させ、CellCubeの回収で使用した同じタイプの界面活性溶解緩衝液を用いて溶解した。細胞HPLC vps/mLは、2日目で1.6E+10vps/mLであり、3日目で6.8E+09vps/mLであり、4日目で2.2E+09vps/mLであり、5日目で2.24E+09vps/mLであり、6日目で2.24E+09vps/mLであった。
消費された培地の、新鮮な血清比含有補充IS293培地との交換は、Ad-p53ベクターの産生を有意に増加した。培地交換は、細胞外HPLCウイルス粒子の産生を、3日目の以前のレベルより3.6倍高くに増加し、細胞外pfu力価の産生を6日目の以前のレベルより10倍高く増加した。Ad-p53ベクターの細胞あたりの産生は、約1.33E+04 HPLC vpsと概算した。
細胞内HPLCウイルス粒子は、感染後2日目にピークに達し、次に粒子数は減少した。かわりに、細胞外ウイルス粒子は、次第に、回収6日目まで増加した。Ad-p53ベクターのほとんど全てが、感染後2日間に産生され、細胞内局在化され、次にウイルスが細胞外へ放出された。ウイルスのほぼ半数が、感染後2日目および3日目の間に細胞の外側で上清に放出され、放出速度は、時間経過にともなって減少された。
Af-p53ベクターで感染された全細胞が、感染後6日の終わりにその生存性を失い、一方、感染していない細胞は97%生存率を有した。感染が存在するときの、培地交換を伴わないおよび伴う消費培地のpHは、それぞれに6.04および5.97であっが、一方、感染が存在しないものは、7.00であった(表12)。
培地交換およびガスオーバーレイをともなう撹拌バイオリアクターでのウイルス生産および細胞培養
Ad-p53ベクターの生産を増加するために、より制御された環境を提供するために、5L CelliGenバイオリアクターを使用した。5L CelliGenバイオリアクターでは、pHおよび溶存酸素ならびに温度を制御した。酸素および二酸化炭素ガスを、酸素供給およびpH調整のために、それぞれソレノイドバルブに接続した。低剪断環境をつくっている間、より良好に混合するために、marine-blade羽根を装備した。バイオリアクター内の加圧状態を維持するために、操作中の時間の間ずっと空気を供給した。
バイオリアクターに接種するために、細胞1バイアルを、250mL撹拌フラスコ中で100mL無血清培地へ解凍し、その細胞を250または500mL撹拌フラスコ中で拡大させた。500mLフラスコ中で増殖された、800mL細胞接種物を、10Lカルボイ中で2700mL新鮮培地と混合し、ガス圧によってCelliGenバイオリアクターへ移した。CelliGenバイオリアクターの開始操作容積は、約3.5L培養であった。marine-blade羽根の回転速度を80rpmに設定し、温度37℃、開始pH7.1、感染後pH7.0、そして操作中全時間にわたりDO 40%であった。
開始細胞密度は、4.3E+5vc/mL(97%生存率)、細胞密度が2.7E+6vc/mL(93%生存率)に達した4日後に、細胞を遠心沈降させ、そして細胞を新鮮培地に再懸濁させて、CelliGenバイオリアクターへ移した。培地交換後の細胞密度は2.1E+6vc/mLであり、その細胞を10 MOIで感染させた。それ以降、DOが40%未満に減少した。DOを40%を超えるように維持するために、約500mLの培養物をCelliGenバイオリアクターから回収し、細胞培養に要求された酸素を減少させ、上部marine-bladeを気相および液相の間の界面近くに設置して、表面回復を増大することによって酸素移動を改善した。それ以降、DOは、操作終了まで40%を超えて維持された。
pH制御のために、CO2ガスを使用して細胞培養物を酸性化し、1N NaHCO3溶液を用いて細胞培養物をアルカリにした。pH制御は、最初は7.10に設定した。細胞培養の開始pHは、約pH7.41であった。細胞培養pHがpH7.1周辺に安定化されるまで、1N NaHCO3溶液を約280mL消費した。細胞培養物のウイルス感染後、pH制御をpH7.0に下げ、CO2ガス供給ラインを閉じてNaHCO3溶液の消費を減少した。pH調整のための過剰NaHCO3溶液の消費は、望ましくない細胞培養容積を増加する。それ以降、1N NaHCO3溶液70mLを消費して、操作中のほとんどの時間、pHを7.0および7.1間であった。温度は、35℃および37℃間に制御した。
感染後、細胞生存率は、感染後6日目の回収まで一定して減少した。回収日には、生存細胞はなかった。CelliGenバイオリアクターのウイルス生産容積は、撹拌フラスコでの1.3E+10vps/mLに比較して、5.1E+10 HPLC vps/mLであった。CelliGenバイオリアクターでの制御環境は、Ad-p53ベクターの産生を、培地交換を伴う撹拌フラスコに比較して4倍増加した。これは、感染時の細胞密度の1.2E+6から2.1E+6vc/mLへの増加、および細胞あたりのウイルス産生の1.3E+4から2.5E+4vps/mLへの増加の両方に原因する。2.5E+4vps/mLは、血清補充付着細胞培養での3.5E+4vps/細胞に匹敵する。
撹拌および散布バイオリアクターでのウイルス生産および細胞培養
最初の研究で、細胞を、ウイルス生産のために、撹拌バイオリアクター中で首尾よく増殖し、酸素およびCO2を、バイオリアクターの上部にガスオーバーレイによって供給した。しかし、この方法は、ガス輸送が非効率であるという理由によって、細胞培養系の規模拡大を制限する。従って、第二研究では、散布バイオリアクターにおける、無血清懸濁培養の規模拡大の可能性をテストし、細胞増殖およびAd-p53産生を調べるために、純粋酸素およびCO2ガスを、F-68(0.1%)およびヘパリン(100mg/L)を補充した無血清IS293培地に吹き込んで、供給した。
純粋酸素を液体培地に吹き込んで、溶存酸素を細胞に供給し、純粋酸素の供給を、溶存酸素を40%を超えて維持するようにソレノイドバルブによって制御した。細胞に対する損傷を最少にする効率的な酸素供給のために、公称孔サイズ約0.22マイクロメーターのステンレス鋼焼結散気装置を、純粋酸素送達に使用した。CO2ガスもまた、純粋酸素と同じ散気装置およびチューブから吹き込むことにより液体培地に供給し、pHをpH7.0付近に維持した。pH制御のために、Na2CO3溶液(106g/L)もまた、バイオリアクターにつないだ。空気をバイオリアクターの上部に供給して、バイオリアクター内部を陽圧に維持した。その他のバイオリアクターの配置は、最初の研究と同じであった。
接種細胞を、凍結バイアルから発育させた。凍結細胞1バイアル(1.0E+7vc)を、T-150フラスコ中で50mL培地に解凍し、500mL撹拌フラスコ中で200mL培地に3回継代培養した。2つの500mL撹拌フラスコで増殖された接種細胞400mLを、10Lカルボイ中で、F-68およびヘパリンを含有したIS293培地と混合して、3.5L細胞懸濁物を作製し、5L CelliGEnバイオリアクターへ移した。
バイオリアクターでの開始細胞密度は、3.0E+4vc/mLであった。開始細胞密度は、第一の研究より低い。第一研究では、500mL撹拌フラスコの4つを、接種物として使用した。より低い開始細胞密度でさえ、細胞は、散布環境で7日目に1.8E+6vc/mLに増殖され、生存率は98%であった。7日間の増殖中に、グルコース濃度は、5.4g/Lから3.0g/Lに減少し、乳酸は、0.3g/Lから1.8g/Lに増加した。
7日目に、細胞密度は1.8E+6vc/mLに達し、バイオリアクター中の細胞を遠心沈降させ、10Lカルボイ中で、F-68およびヘパリン含有する、3.5Lの新鮮な無血清IS293培地に再懸濁させた。293細胞を1.25E+11 pfu Ad-p53で感染させ、CelliGenバイオリアクターへ移した。バイオリアクターでは、細胞生存率は100%であったが、細胞密度は7.2E+5vc/mLのみであった。培地交換操作中に細胞損失があった。培地中のウイルス力価は、回収の2日目に2.5E+10 HPLC vps/mLであり、3日目に2.0E+10であり、4日目に2.8E+10であり、5日目に3.5E+10であり、6日目に3.9E+10 HPLC vps/mLであった。ガスオーバーレイによる第一CelliGenバイオリアクター研究は、5.1E+10 HPLC vps/mLを生産した。第二研究操作でのより低いウイルス濃度は、感染時のより低い細胞密度に原因するようである。第二操作での7.2E+5vc/mLに比較して、第一操作で2.1E+6vc/mLを使用した。事実、第二散布CelliGenバイオリアクターでの細胞あたりAd-p53生産は、これまで達成された最も高い細胞あたりの産生である5.4E+4vps/細胞と概算される。散布をともなわない、第一の無血清CelliGenバイオリアクター、および血清補充T-フラスコでの細胞あたりの生産は、それぞれに、2.5E+4vps/細胞および3.5E+4vps/細胞であった。
ウイルス感染後に、細胞生存率は、回収の6日目に100%から13%に減少した。感染後のこの6日間に、グルコース濃度は、5.0g/Lから2.1g/Lに減少し、乳酸は、0.3g/Lから2.9g/Lに増加した。操作の全期間中、約20mLのNa2CO3(106g/L)溶液を消費した。
実験結果は、散布および振盪バイオリアクターでAd-p53の生産が、技術的および経済的に実行可能であることを示す。散布および撹拌バイオリアクターの拡大および多量規模単位の操作は、十分に確立される。
***
本明細書に開示および請求されている全ての組成物および/または方法は、本開示を考慮して過度の実験を行うことなく調製および実施され得る。一方、本発明の組成物および方法は、好ましい実施態様によって記載されてきたが、変更が、組成物および/または方法、および、本明細書に記載されている方法の工程または一連の工程に、本発明の概念、精神、および範囲を逸脱することなく、適用され得ることは、当行者に明白である。より詳細には、化学的および生理学的に関連するある種の因子が、同一または同類の結果が達成される限り、本明細書に記載されている因子と置換され得ることは、明白である。当業者に明白であるこのような同類の置換および改変は、添付の請求の範囲によって定義されるように、本発明の精神、範囲、および概念内であると考えられる。
参考文献
以下の参考文献は、本明細書に記載されている文献に補充して、例示的な手順または他の詳細を提供する程度に、詳細に本明細書に参考として援用されている。
Claims (22)
- 以下の工程を包含するアデノウイルス組成物を作製する方法であって:
a)グルコースを含む培地中で宿主細胞を増殖させる工程;
b)該細胞を該培地中で灌流する工程;
c)該宿主細胞をアデノウイルスに感染させる工程;
d)該アデノウイルス組成物を含む溶解物を作製するために、該宿主細胞を採取し、そして界面活性剤を用いて溶解させる工程;及び
e)前記細胞溶解液からアデノウイルスを精製する工程
を包含する、方法。 - 界面活性剤が非変性性界面活性剤である、請求項1の方法。
- 前記細胞が、約0.7〜1.7g/Lのグルコース濃度を提供する速度で灌流される、請求項1に記載の方法。
- 前記アデノウイルスが、外因の遺伝子構築物を含むアデノウイルスベクターを含む、請求項1に記載の方法。
- 前記遺伝子構築物が、プロモーターに作動可能に連結されている、請求項4に記載の方法。
- 前記アデノウイルスが、複製不能アデノウイルスである、請求項1に記載の方法。
- 前記アデノウイルスが、E1-領域の少なくとも一部分を欠失している、請求項6に記載の方法。
- 前記アデノウイルスが、E1Aおよび/またはE1B領域の少なくとも一部分を欠失している、請求項7に記載の方法。
- 前記宿主細胞が複製を補完し得る、請求項1に記載の方法。
- 前記宿主細胞が293細胞である、請求項1に記載の方法。
- 前記外因の遺伝子構築物が治療遺伝子をコードする、請求項4の記載の方法。
- 前記界面活性剤が、Thesit(登録商標)、NP-40(登録商標)、Tween-20(登録商標)、Brij-58(登録商標)、Triton X(登録商標)-100、またはオクチルグルコシドである、請求項1に記載の方法。
- 前記精製工程が、ヌクレアーゼを用いる細胞溶解物の処理を含む、請求項1に記載の方法。
- 前記ヌクレアーゼが、Benzonase(登録商標)、またはPulmozyme(登録商標)である、請求項13に記載の方法。
- 前記精製工程に、1以上のクロマトグラフィー工程を用いるクロマトグラフィーを含む、請求項1に記載の方法。
- 前記クロマトグラフィーが、本質的に1回のクロマトグラフィー工程を含む、請求項15に記載の方法。
- 前記クロマトグラフィー工程が、イオン交換クロマトグラフィーを含む、請求項16に記載の方法。
- 前記イオン交換クロマトグラフィーが、陰イオン交換クロマトグラフィーである、請求項17に記載の方法。
- 膜濾過を用いる濃縮工程をさらに包含する、請求項1に記載の方法。
- 前記濾過が、接線流動(tangential flow)濾過である、請求項19の記載の方法。
- 前記溶解物を濃縮する工程、前記溶解物の緩衝液を交換する工程、および前記溶解物の夾雑する核酸の濃度を減少させる工程をさらに包含する、請求項1に記載の方法。
- 前記培地が無血清培地である、請求項1に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US3132996P | 1996-11-20 | 1996-11-20 | |
| US60/031,329 | 1996-11-20 | ||
| PCT/US1997/021504 WO1998022588A2 (en) | 1996-11-20 | 1997-11-20 | An improved method for the production and purification of adenoviral vectors |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2001504701A JP2001504701A (ja) | 2001-04-10 |
| JP2001504701A5 JP2001504701A5 (ja) | 2005-09-08 |
| JP4492826B2 true JP4492826B2 (ja) | 2010-06-30 |
Family
ID=21858843
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP52396598A Expired - Lifetime JP4492826B2 (ja) | 1996-11-20 | 1997-11-20 | アデノウイルスベクターの産生および精製のための改良された方法 |
Country Status (15)
| Country | Link |
|---|---|
| US (7) | US6194191B1 (ja) |
| EP (3) | EP0968284B1 (ja) |
| JP (1) | JP4492826B2 (ja) |
| KR (1) | KR100503701B1 (ja) |
| CN (1) | CN1244215B (ja) |
| AT (2) | ATE550429T1 (ja) |
| AU (1) | AU732703B2 (ja) |
| BR (1) | BR9713368A (ja) |
| CA (1) | CA2272820C (ja) |
| DE (1) | DE69737107T2 (ja) |
| ES (2) | ES2278399T3 (ja) |
| HK (1) | HK1107116A1 (ja) |
| NO (1) | NO992389L (ja) |
| NZ (1) | NZ335947A (ja) |
| WO (1) | WO1998022588A2 (ja) |
Families Citing this family (254)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2625279A1 (en) * | 1995-08-30 | 1997-03-06 | Genzyme Corporation | Chromatographic purification of adenovirus and aav |
| US20020088014A1 (en) * | 1996-05-31 | 2002-07-04 | Xiangming Fang | Minimal adenovirus mediated recombinant vaccine |
| US6485958B2 (en) | 1996-07-01 | 2002-11-26 | Gencell Sa | Method for producing recombinant adenovirus |
| EP0968284B1 (en) | 1996-11-20 | 2006-12-13 | Introgen Therapeutics, Inc. | An improved method for the production and purification of adenoviral vectors |
| US7732129B1 (en) * | 1998-12-01 | 2010-06-08 | Crucell Holland B.V. | Method for the production and purification of adenoviral vectors |
| US6544769B1 (en) | 1996-12-13 | 2003-04-08 | Schering Corporation | Compostions comprising viruses and methods for concentrating virus preparations |
| US6261823B1 (en) | 1996-12-13 | 2001-07-17 | Schering Corporation | Methods for purifying viruses |
| US6995006B2 (en) * | 1997-09-05 | 2006-02-07 | Targeted Genetics Corporation | Methods for generating high titer helper-free preparations of released recombinant AAV vectors |
| CA2307016A1 (en) * | 1997-10-24 | 1999-05-06 | Life Technologies, Inc. | Recombinational cloning using nucleic acids having recombination sites |
| US7705602B2 (en) * | 1997-11-03 | 2010-04-27 | Midtronics, Inc. | Automotive vehicle electrical system diagnostic device |
| US20040204381A1 (en) * | 1997-11-12 | 2004-10-14 | Moskal Joseph R | Detection and treatment of glyco-enzyme-related disease |
| US6899870B1 (en) * | 1998-03-11 | 2005-05-31 | Board Of Regents, The University Of Texas System | Induction of apoptic or cytotoxic gene expression by adenoviral mediated gene codelivery |
| MXPA00010338A (es) * | 1998-04-22 | 2005-06-17 | Genvec Inc | Purificacion eficiente de adenovirus. |
| US6416992B1 (en) * | 1998-10-13 | 2002-07-09 | Avigen, Inc. | Compositions and methods for producing recombinant adeno-associated virus |
| US7691370B2 (en) * | 1998-10-15 | 2010-04-06 | Canji, Inc. | Selectivity replicating viral vector |
| ATE421337T1 (de) * | 1998-11-16 | 2009-02-15 | Introgen Therapeutics Inc | Adenovirus-formulierungen zur gentherapie |
| US6689600B1 (en) * | 1998-11-16 | 2004-02-10 | Introgen Therapeutics, Inc. | Formulation of adenovirus for gene therapy |
| US7148035B1 (en) * | 1998-11-18 | 2006-12-12 | Oxford Biomedica (Uk) Limited | Polypeptide |
| US6210922B1 (en) * | 1998-11-30 | 2001-04-03 | National Research Council Of Canada | Serum free production of recombinant proteins and adenoviral vectors |
| AU779267B2 (en) * | 1998-12-31 | 2005-01-13 | Centelion S.A.S. | Method for separating viral particles |
| FR2788064B1 (fr) * | 1998-12-31 | 2003-01-31 | Aventis Pharma Sa | Methode de separation de particules virales |
| EP1155120B1 (fr) | 1999-02-22 | 2006-07-05 | Transgene S.A. | Procede d'obtention d'une preparation virale purifiee |
| US6795585B1 (en) | 1999-07-16 | 2004-09-21 | Eastman Kodak Company | Representing digital images in a plurality of image processing states |
| US20010043916A1 (en) * | 1999-12-29 | 2001-11-22 | Mcneilly David S. | Method using filtration aids for the separation of virus vectors from nucleic acids and other cellular contaminants |
| US6168941B1 (en) * | 2000-04-07 | 2001-01-02 | Genvec, Inc. | Method of producing adenoviral vector stocks |
| JP2004509065A (ja) * | 2000-04-28 | 2004-03-25 | ゼンジェン インコーポレイテッド | α−MSHおよびその誘導体を用いる遺伝子治療システムおよび方法 |
| US6783939B2 (en) * | 2000-07-07 | 2004-08-31 | Alphavax, Inc. | Alphavirus vectors and virosomes with modified HIV genes for use in vaccines |
| TWI289158B (en) * | 2000-08-10 | 2007-11-01 | Oncolytics Biotech Inc | Method of producing infectious reovirus |
| US7198924B2 (en) | 2000-12-11 | 2007-04-03 | Invitrogen Corporation | Methods and compositions for synthesis of nucleic acid molecules using multiple recognition sites |
| US20020064860A1 (en) * | 2000-11-29 | 2002-05-30 | Schering Corporation | Method for purifying adenoviruses |
| EP2253701B1 (en) | 2001-03-16 | 2014-08-13 | Oncolytics Biotech Inc. | Method of extracting virus from cell culture |
| US20040146590A1 (en) * | 2001-03-22 | 2004-07-29 | Iadarola Michael J | Molecular neurochirurgie for pain control administering locally capsaicin or resinferatoxin |
| JP4499990B2 (ja) * | 2001-03-27 | 2010-07-14 | バーテックス ファーマシューティカルズ インコーポレイテッド | Hcv感染に有用な組成物および方法 |
| JP2004538005A (ja) | 2001-08-08 | 2004-12-24 | ザ・トラステイーズ・オブ・ザ・ユニバーシテイ・オブ・ペンシルベニア | シアル酸に結合するタンパク質を有するウイルスベクターの精製法 |
| AU2002332736A1 (en) * | 2001-08-30 | 2003-03-18 | The Government Of The United States Of America, As Represented By The Secretary Of The Department Of | New adenovirus type 7 vectors |
| DE60233061D1 (de) * | 2001-09-06 | 2009-09-03 | Alphavax Inc | Alphavirus replikon-vektorsysteme |
| AU2002348151A1 (en) * | 2001-11-05 | 2003-05-19 | Genvec, Inc. | Viral vector production methods and compositions |
| NZ533586A (en) * | 2001-12-20 | 2005-02-25 | Bavarian Nordic As | Method for the recovery and purification of poxviruses from infected cells using high pressure homogenisation |
| KR100451308B1 (ko) * | 2001-12-28 | 2004-10-06 | 선바이오(주) | 단백질 의약품 정제 방법에서의 바이러스 제거 방법 |
| US20040063188A1 (en) * | 2002-02-14 | 2004-04-01 | Novavax, Inc. | Kit for treating gastrointestinal tract |
| CA2476447A1 (en) * | 2002-02-20 | 2003-08-28 | Merck & Co., Inc. | Method of determining adenovirus particle concentration |
| US20030175688A1 (en) * | 2002-03-15 | 2003-09-18 | Rukmini Pennathur-Das | Method for the purification and production of oncolytic adenoviruses |
| US20030186832A1 (en) * | 2002-03-15 | 2003-10-02 | Unilever Home & Personal Care Usa, Division Of Conopco, Inc. | Isotropic liquid detergents with improved anti-redeposition |
| US20030180936A1 (en) * | 2002-03-15 | 2003-09-25 | Memarzadeh Bahram Eric | Method for the purification, production and formulation of oncolytic adenoviruses |
| EP1492890A4 (en) * | 2002-03-29 | 2006-10-18 | Merck & Co Inc | METHODS OF LARGE SCALE PRODUCTION OF ADENOVIRUS AND ADENOVIRUS SEMEN COLLECTIONS |
| ES2323454T5 (es) | 2002-04-30 | 2012-11-14 | Oncolytics Biotech Inc. | Procedimiento mejorado de purificación de virus |
| AU2003229060A1 (en) * | 2002-05-14 | 2003-12-02 | Merck And Co., Inc. | Methods of adenovirus purification |
| US20030224354A1 (en) * | 2002-05-30 | 2003-12-04 | Introgen Therapeutics Inc. | Quantifying viral particles with intrinsic fluorescence |
| EP1371723A1 (en) * | 2002-06-12 | 2003-12-17 | Procorde GmbH | Process for preparing an adenovirus-containing preparation |
| WO2004009768A2 (en) * | 2002-07-18 | 2004-01-29 | Invitrogen Corporation | Viral vectors containing recombination sites |
| US20040106184A1 (en) * | 2002-08-28 | 2004-06-03 | Introgen Therapeutics Inc. | Chromatographic methods for adenovirus purification |
| US7208493B2 (en) * | 2002-10-03 | 2007-04-24 | Targegen, Inc. | Vasculostatic agents and methods of use thereof |
| US20050282814A1 (en) * | 2002-10-03 | 2005-12-22 | Targegen, Inc. | Vasculostatic agents and methods of use thereof |
| CA2504129A1 (en) * | 2002-11-01 | 2004-05-21 | Promega Corporation | Cell lysis compositions, methods of use, apparatus, and kit |
| NZ540657A (en) * | 2002-12-13 | 2006-09-29 | Alphavax Inc | High-yielding, GMP-compatible, commercially feasible process for producing highly purified alphavirus replicon particle (ARP) preparations for use in human and veterinary medicine |
| ES2618309T3 (es) * | 2002-12-13 | 2017-06-21 | Alphavax, Inc. | Partículas de replicón de alfavirus multi-antigénico y métodos |
| US20040225077A1 (en) | 2002-12-30 | 2004-11-11 | Angiotech International Ag | Drug delivery from rapid gelling polymer composition |
| CA2802143C (en) | 2003-01-14 | 2018-06-19 | Dana-Farber Cancer Institute | Sparc encoding polynucleotide as a cancer therapy sensitizer |
| KR101454842B1 (ko) * | 2003-03-20 | 2014-11-04 | 알파벡스, 인크. | 개선된 알파바이러스 레플리콘 및 헬퍼 구축물 |
| KR20060015568A (ko) * | 2003-05-01 | 2006-02-17 | 디에스엠 아이피 어셋츠 비.브이. | 현탁된 동물 세포의 관류 배양에 의한 생물학적 물질의제조방법 |
| US20040229335A1 (en) * | 2003-05-15 | 2004-11-18 | Introgen Therapeutics, Inc. | Methods and compositions for the production of adenoviral vectors |
| CA2529053A1 (en) * | 2003-06-18 | 2004-12-29 | Onyx Pharmaceuticals, Inc. | Method for purifying virus |
| AU2004257214B2 (en) * | 2003-07-11 | 2010-04-22 | Alphavax, Inc. | Alphavirus-based cytomegalovirus vaccines |
| ES2359473T3 (es) | 2003-07-21 | 2011-05-23 | Transgene S.A. | Citoquinas multifuncionales. |
| ATE469984T1 (de) * | 2003-12-01 | 2010-06-15 | Life Technologies Corp | Rekombinationsstellen enthaltende nukleinsäuremoleküle und verfahren zur verwendung davon |
| CA2551026A1 (en) * | 2003-12-23 | 2005-07-14 | Schering Corporation | Methods for producing a549 cell lines stable in serum-free medium suspension culture |
| DK1780269T3 (da) * | 2004-02-23 | 2009-10-12 | Crucell Holland Bv | Virusrensningsmetoder |
| CN100333797C (zh) * | 2005-01-26 | 2007-08-29 | 彭朝晖 | 重组腺病毒p53制品在肿瘤治疗中的新用途 |
| CA2567574C (en) * | 2004-04-08 | 2013-01-08 | Targegen, Inc. | Benzotriazine inhibitors of kinases |
| US20050266550A1 (en) * | 2004-05-18 | 2005-12-01 | Alphavax, Inc. | TC-83-derived alphavirus vectors, particles and methods |
| BRPI0511764B8 (pt) * | 2004-06-01 | 2021-05-25 | Avigen Inc | método de prevenção de agregação de vírions de vírus adeno-associado recombinante (raav) em uma preparação purificada de virions raav |
| US20050287616A1 (en) * | 2004-06-29 | 2005-12-29 | Gross Adam F | Method of assaying denaturation of proteins |
| NZ553492A (en) | 2004-08-25 | 2010-11-26 | Targegen Inc | Triazole derivatives and methods of use |
| DE202004014949U1 (de) * | 2004-09-25 | 2004-11-18 | Techno-Grafica Gmbh | Vorrichtung zum Wärmebehandeln einer Beschichtung von Flachoffset-Druckplatten |
| DE102004049290A1 (de) * | 2004-10-09 | 2006-04-20 | Bayer Healthcare Ag | Verfahren zur Herstellung von Virusmaterial |
| US7901921B2 (en) * | 2004-10-22 | 2011-03-08 | Oncolytics Biotech Inc. | Viral purification methods |
| WO2006052302A2 (en) | 2004-11-03 | 2006-05-18 | Introgen Therapeutics Inc. | Method of producing and purifying adenoviral vectors |
| CN100497640C (zh) * | 2004-11-26 | 2009-06-10 | 中国人民解放军军事医学科学院生物工程研究所 | 一种高效生产重组腺病毒载体的方法 |
| US20060166364A1 (en) * | 2004-12-22 | 2006-07-27 | Introgen, Inc. | Use of flexible bag containers for viral production |
| ATE547511T1 (de) | 2004-12-23 | 2012-03-15 | Medimmune Llc | Nichttumorbildende mdck-zellinie zur vermehrung von viren |
| US20060205080A1 (en) * | 2005-03-01 | 2006-09-14 | David Frey | Formulations for therapeutic viruses having enhanced storage stability |
| SG10201912554TA (en) | 2005-03-23 | 2020-02-27 | Genmab As | Antibodies against cd38 for treatment of multiple myeloma |
| DE602006003420D1 (de) | 2005-04-11 | 2008-12-11 | Crucell Holland Bv | Virusreinigung mit ultrafiltration |
| WO2006121803A1 (en) * | 2005-05-05 | 2006-11-16 | Sensient Flavors Inc. | Production of beta-glucans and mannans |
| EP1893216A4 (en) * | 2005-06-08 | 2012-08-08 | Targegen Inc | METHOD AND COMPOSITIONS FOR THE TREATMENT OF EYE DISEASES |
| US20090216452A1 (en) * | 2005-07-05 | 2009-08-27 | Develop Tech Resources | Energy recovery within a fluid distribution network using geographic information |
| PT1951684T (pt) * | 2005-11-01 | 2016-10-13 | Targegen Inc | Inibidores de cinases de tipo biaril-meta-pirimidina |
| US8133900B2 (en) * | 2005-11-01 | 2012-03-13 | Targegen, Inc. | Use of bi-aryl meta-pyrimidine inhibitors of kinases |
| US8604042B2 (en) * | 2005-11-01 | 2013-12-10 | Targegen, Inc. | Bi-aryl meta-pyrimidine inhibitors of kinases |
| AU2007227218A1 (en) * | 2006-03-22 | 2007-09-27 | Viral Logic Systems Technology Corp. | Methods for identifying polypeptide targets and uses thereof for treating immunological diseases |
| US7691858B2 (en) * | 2006-04-25 | 2010-04-06 | Targegen, Inc. | Kinase inhibitors and methods of use thereof |
| EP2615167A1 (en) | 2006-09-15 | 2013-07-17 | MedImmune, LLC | Method for eliminating DNA contaminants from viral preparations |
| EP2390340A3 (en) | 2007-01-30 | 2012-02-22 | Transgene SA | vector encoding Papillomavirus E1 and E2 polypeptides with reduced percentage of identity |
| US20080299624A1 (en) * | 2007-06-01 | 2008-12-04 | Edward Heslop | Continuous fermentation apparatus and method |
| WO2008156829A2 (en) * | 2007-06-21 | 2008-12-24 | Alphavax, Inc. | Promoterless cassettes for expression of alphavirus structural proteins |
| RU2484132C2 (ru) * | 2007-10-08 | 2013-06-10 | Интрексон Корпорейшн | Модифицированные дендритные клетки и их применение в лечении злокачественных опухолей |
| EP2612868B1 (en) | 2007-11-01 | 2018-08-15 | Astellas Pharma Inc. | Immunosuppressive polypeptides and nucleic acids |
| US20090253184A1 (en) * | 2008-01-23 | 2009-10-08 | Introgen Therapeutics, Inc. | Compositions and methods related to an adenoviral trans-complementing cell line |
| EP2806037B1 (en) * | 2008-05-13 | 2016-09-21 | Gen-Probe Incorporated | Inactivatable target capture oligomers for use in the selective hybridization and capture of target nucleic acid sequences |
| PL2307551T3 (pl) * | 2008-06-18 | 2017-07-31 | Oxford Biomedica (Uk) Limited | Oczyszczanie wektorów retrowirusowych |
| RU2015103990A (ru) | 2008-09-24 | 2015-10-27 | Медиммун, Ллк | Способы культивирования клеток, размножения и очистки вирусов |
| CN102215865B (zh) * | 2008-09-24 | 2013-10-30 | 米迪缪尼有限公司 | 病毒纯化方法 |
| KR101504392B1 (ko) | 2008-11-03 | 2015-03-19 | 크루셀 홀란드 비.브이. | 아데노바이러스 벡터의 제조방법 |
| CA2742295A1 (en) * | 2008-11-03 | 2010-06-03 | Ligocyte Pharmaceuticals, Inc. | Improved methods for isolating enveloped virus-based vlps free of infectious agents |
| CN102449269A (zh) * | 2009-05-26 | 2012-05-09 | 利维坦能源空气动力学有限公司 | 水电式管内涡轮机的用途 |
| CN103398936A (zh) * | 2009-07-07 | 2013-11-20 | 索尼公司 | 微流体装置 |
| KR20120052352A (ko) | 2009-08-07 | 2012-05-23 | 트랜스진 에스.에이. | Hbv 감염을 치료하는 조성물 |
| CN102791852B (zh) | 2009-10-15 | 2014-05-07 | 克鲁塞尔荷兰公司 | 纯化腺病毒颗粒的方法 |
| EP2488635B1 (en) | 2009-10-15 | 2013-11-20 | Crucell Holland B.V. | Process for adenovirus purification from high cell density cultures |
| CA2786374C (en) | 2010-01-05 | 2019-01-22 | Vascular Biogenics Ltd. | Methods for use of a specific anti-angiogenic adenoviral agent |
| CA2786377C (en) | 2010-01-05 | 2018-02-27 | Vascular Biogenics Ltd. | Compositions and methods for treating malignant gliomas employing viral vectors encoding a fas-chimera |
| EP2523681A1 (en) * | 2010-01-12 | 2012-11-21 | Vascular Biogenics Ltd. | Methods of producing adenovirus vectors and viral preparations generated thereby |
| AU2011214262B2 (en) | 2010-02-15 | 2015-05-21 | Crucell Holland B.V. | Method for the production of Ad26 adenoviral vectors |
| CN102985536B (zh) * | 2010-04-14 | 2017-12-05 | Emd密理博公司 | 生产高效价、高纯度的病毒母液的方法及使用其的方法 |
| CN103052399B (zh) | 2010-08-12 | 2016-06-29 | 依生生物制药控股有限公司 | 用于减少病毒组合物中dna杂质的方法 |
| WO2012038367A1 (en) | 2010-09-20 | 2012-03-29 | Crucell Holland B.V. | Therapeutic vaccination against active tuberculosis |
| BR112013004582A2 (pt) | 2010-09-27 | 2016-09-06 | Crucell Holland Bv | método para induzir uma resposta imune em um sujeito contra um antígeno de um parasita que causa a malária |
| US8892184B2 (en) | 2010-10-18 | 2014-11-18 | Siemens Medical Solutions Usa, Inc. | Systems and methods for reducing interference in a dual modality imaging system |
| AU2010363329A1 (en) | 2010-11-07 | 2013-05-09 | Targegen, Inc. | Compositions and methods for treating myelofibrosis |
| EP2461162A1 (en) | 2010-12-03 | 2012-06-06 | Texcell, . | Method for determining the titre of viruses by using infectious standards |
| EP3202897B1 (en) | 2010-12-09 | 2020-04-15 | Institut Pasteur | Diagnostic use of a fusion polypeptide comprising a viral protein and a mgmt enzyme |
| TWI623618B (zh) | 2011-07-12 | 2018-05-11 | 傳斯堅公司 | Hbv聚合酶突變體 |
| WO2013045658A1 (en) | 2011-09-29 | 2013-04-04 | Transgene Sa | Immunotherapy composition and regimen for treating hepatitis c virus infection |
| TW201321016A (zh) | 2011-09-29 | 2013-06-01 | Transgene Sa | 免疫療法組成物及用於治療c型肝炎病毒感染之療程(二) |
| US20130122038A1 (en) | 2011-11-14 | 2013-05-16 | The United States Of America As Represented By The Secretary Of The Department | Heterologous prime-boost immunization using measles virus-based vaccines |
| US8932607B2 (en) | 2012-03-12 | 2015-01-13 | Crucell Holland B.V. | Batches of recombinant adenovirus with altered terminal ends |
| DK2825640T3 (en) | 2012-03-12 | 2016-08-01 | Crucell Holland Bv | BATCHES OF RECOMBINANT ADENOVIRUS WITH CHANGED TERMINAL END |
| US9125870B2 (en) | 2012-03-22 | 2015-09-08 | Crucell Holland B.V. | Vaccine against RSV |
| AP2014007993A0 (en) | 2012-03-22 | 2014-10-31 | Crucell Holland Bv | Vaccine against RSV |
| US9567634B2 (en) * | 2012-07-26 | 2017-02-14 | Vectalys | Method for detecting or measuring the impact of a viral vector composition on eukaryotic cells and biomarkers used thereof |
| EP2912069B1 (en) | 2012-10-23 | 2019-07-31 | Emory University | Gm-csf and il-4 conjugates, compositions, and methods related thereto |
| CN105188745B (zh) | 2013-04-25 | 2019-10-18 | 扬森疫苗与预防公司 | 稳定化的可溶性融合前rsv f多肽 |
| EP2992553A4 (en) | 2013-05-03 | 2017-03-08 | Honeywell International Inc. | Lead frame construct for lead-free solder connections |
| EP3010931B1 (en) | 2013-06-17 | 2018-06-13 | Janssen Vaccines & Prevention B.V. | Stabilized soluble pre-fusion rsv f polypeptides |
| US9956166B2 (en) | 2013-09-18 | 2018-05-01 | Sorrento Therapeutics, Inc. | Methods for administration and methods for treating cardiovascular diseases with resiniferatoxin |
| JP6466437B2 (ja) * | 2013-10-30 | 2019-02-06 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツングMerck Patent Gesellschaft mit beschraenkter Haftung | 微生物を複雑な試料から単離する方法 |
| EP3107683B1 (en) | 2014-02-20 | 2021-12-08 | Honeywell International Inc. | Method of forming a solder wire |
| WO2015148704A1 (en) | 2014-03-25 | 2015-10-01 | Terumo Bct, Inc. | Passive replacement of media |
| EA201691696A1 (ru) | 2014-04-25 | 2017-03-31 | Шир Хьюман Дженетик Терапис, Инк. | Способы очистки матричной рнк |
| WO2015191508A1 (en) | 2014-06-09 | 2015-12-17 | Voyager Therapeutics, Inc. | Chimeric capsids |
| CN105316296A (zh) * | 2014-06-13 | 2016-02-10 | 亚宝药业太原制药有限公司 | 一种纯化腺病毒颗粒的方法 |
| MA40595A (fr) | 2014-09-09 | 2021-05-26 | Unum Therapeutics | Récepteurs chimériques et utilisations de ceux-ci en thérapie immunitaire |
| GB201417042D0 (en) | 2014-09-29 | 2014-11-12 | Fkd Therapies Oy | Method |
| MX2017005834A (es) | 2014-11-05 | 2017-11-17 | Voyager Therapeutics Inc | Polinucleotidos aad para el tratamiento de la enfermedad de parkinson. |
| AU2015346162B2 (en) | 2014-11-14 | 2022-02-10 | Voyager Therapeutics, Inc. | Compositions and methods of treating amyotrophic lateral sclerosis (ALS) |
| CN112375760A (zh) | 2014-11-14 | 2021-02-19 | 沃雅戈治疗公司 | 调节性多核苷酸 |
| WO2016094783A1 (en) | 2014-12-12 | 2016-06-16 | Voyager Therapeutics, Inc. | Compositions and methods for the production of scaav |
| CN107428799A (zh) * | 2015-01-13 | 2017-12-01 | 阿尔法韦士曼公司 | 纯化腺相关病毒(AAV)及/或重组腺相关病毒(rAAV)之方法及其梯度和流通式缓冲液 |
| EP3054007A1 (en) | 2015-02-09 | 2016-08-10 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | Recombinant adeno-associated virus particle purification comprising an immuno-affinity purification step |
| WO2016131945A1 (en) | 2015-02-20 | 2016-08-25 | Transgene Sa | Combination product with autophagy modulator |
| AU2016249798B2 (en) | 2015-04-14 | 2022-05-26 | Janssen Vaccines And Prevention B.V. | Recombinant adenovirus expressing two transgenes with a bidirectional promoter |
| WO2017004592A1 (en) | 2015-07-02 | 2017-01-05 | Terumo Bct, Inc. | Cell growth with mechanical stimuli |
| US10457708B2 (en) | 2015-07-07 | 2019-10-29 | Janssen Vaccines & Prevention B.V. | Stabilized soluble pre-fusion RSV F polypeptides |
| PL3319633T3 (pl) | 2015-07-07 | 2021-04-19 | Janssen Vaccines & Prevention B.V. | Szczepionka przeciwko rsv |
| US9663766B2 (en) | 2015-07-24 | 2017-05-30 | Bio-Rad Laboratories, Inc. | Methods for purifying adenovirus vectors |
| CA3001859A1 (en) | 2015-10-16 | 2017-04-20 | The Trustees Of Columbia University In The City Of New York | Compositions and methods for inhibition of lineage specific antigens |
| US20180230489A1 (en) | 2015-10-28 | 2018-08-16 | Voyager Therapeutics, Inc. | Regulatable expression using adeno-associated virus (aav) |
| AU2017248021B2 (en) | 2016-04-05 | 2021-08-12 | Janssen Vaccines & Prevention B.V. | Stabilized soluble pre-fusion RSV F proteins |
| WO2017174564A1 (en) | 2016-04-05 | 2017-10-12 | Janssen Vaccines & Prevention B.V. | Vaccine against rsv |
| CN116144500A (zh) * | 2016-04-14 | 2023-05-23 | 崔泽尔有限公司 | 具有恒流泵/管道系统的固定床生物反应器 |
| CN109071594A (zh) * | 2016-04-14 | 2018-12-21 | 崔泽尔有限公司 | 具有恒流泵/管道系统的固定床生物反应器 |
| WO2017189959A1 (en) | 2016-04-29 | 2017-11-02 | Voyager Therapeutics, Inc. | Compositions for the treatment of disease |
| EP3448987A4 (en) | 2016-04-29 | 2020-05-27 | Voyager Therapeutics, Inc. | COMPOSITIONS FOR THE TREATMENT OF DISEASES |
| CA3023022A1 (en) | 2016-05-04 | 2017-11-09 | Transgene Sa | Combination therapy with cpg tlr9 ligand |
| SG11201809699XA (en) | 2016-05-18 | 2018-12-28 | Voyager Therapeutics Inc | Modulatory polynucleotides |
| RU2764587C2 (ru) | 2016-05-18 | 2022-01-18 | Вояджер Терапьютикс, Инк. | Способы и композиции для лечения хореи гентингтона |
| WO2017205667A1 (en) | 2016-05-25 | 2017-11-30 | Terumo Bct, Inc. | Cell expansion |
| JP2019523644A (ja) | 2016-05-30 | 2019-08-29 | ヤンセン ファッシンズ アンド プリベンション ベーフェーJanssen Vaccines & Prevention B.V. | 安定化された融合前rsv fタンパク質 |
| US11685883B2 (en) | 2016-06-07 | 2023-06-27 | Terumo Bct, Inc. | Methods and systems for coating a cell growth surface |
| US11104874B2 (en) | 2016-06-07 | 2021-08-31 | Terumo Bct, Inc. | Coating a bioreactor |
| EP3472327B1 (en) | 2016-06-20 | 2020-08-19 | Janssen Vaccines & Prevention B.V. | Potent and balanced bidirectional promoter |
| JP7229151B2 (ja) | 2016-07-14 | 2023-02-27 | ヤンセン ファッシンズ アンド プリベンション ベーフェー | Hpvワクチン |
| US11298041B2 (en) | 2016-08-30 | 2022-04-12 | The Regents Of The University Of California | Methods for biomedical targeting and delivery and devices and systems for practicing the same |
| US20190328869A1 (en) | 2016-10-10 | 2019-10-31 | Transgene Sa | Immunotherapeutic product and mdsc modulator combination therapy |
| US20180119141A1 (en) | 2016-10-28 | 2018-05-03 | Massachusetts Institute Of Technology | Crispr/cas global regulator screening platform |
| US10294452B2 (en) | 2016-11-22 | 2019-05-21 | Dao-Yao He | Lysis, extraction and purification of adeno-associated virus and adenovirus from host cells |
| CN108085301B (zh) * | 2016-11-22 | 2021-10-26 | 贺道耀 | 从宿主细胞提取和纯化腺相关病毒和腺病毒的方法及其组分和试剂盒 |
| US20210405031A1 (en) * | 2016-12-08 | 2021-12-30 | Temple University - Of The Commonwealth System Of Higher Education | Small animal models for in vivo testing of polyomavirus therapeutics |
| WO2018148246A1 (en) | 2017-02-07 | 2018-08-16 | Massachusetts Institute Of Technology | Methods and compositions for rna-guided genetic circuits |
| US11629332B2 (en) | 2017-03-31 | 2023-04-18 | Terumo Bct, Inc. | Cell expansion |
| SG11201909868YA (en) | 2017-05-05 | 2019-11-28 | Voyager Therapeutics Inc | Compositions and methods of treating huntington's disease |
| JP2020518258A (ja) | 2017-05-05 | 2020-06-25 | ボイジャー セラピューティクス インコーポレイテッドVoyager Therapeutics,Inc. | 筋萎縮性側索硬化症(als)治療組成物および方法 |
| CA3061278A1 (en) | 2017-05-17 | 2018-11-22 | Janssen Vaccines & Prevention B.V. | Methods and compositions for inducing protective immunity against rsv infection |
| JOP20190269A1 (ar) | 2017-06-15 | 2019-11-20 | Voyager Therapeutics Inc | بولي نوكليوتيدات aadc لعلاج مرض باركنسون |
| CN111132626B (zh) | 2017-07-17 | 2024-01-30 | 沃雅戈治疗公司 | 轨迹阵列引导系统 |
| KR20200044793A (ko) | 2017-08-03 | 2020-04-29 | 보이저 테라퓨틱스, 인크. | Aav의 전달을 위한 조성물 및 방법 |
| CN107384873A (zh) * | 2017-09-05 | 2017-11-24 | 成都汇宇生物技术有限公司 | 重组腺病毒的纯化方法 |
| WO2019053109A1 (en) | 2017-09-15 | 2019-03-21 | Janssen Vaccines & Prevention B.V. | METHOD FOR SAFE INDUCTION OF IMMUNITY AGAINST RSV |
| JP2021502060A (ja) | 2017-10-16 | 2021-01-28 | ボイジャー セラピューティクス インコーポレイテッドVoyager Therapeutics,Inc. | 筋萎縮性側索硬化症(als)の治療 |
| WO2019079242A1 (en) | 2017-10-16 | 2019-04-25 | Voyager Therapeutics, Inc. | TREATMENT OF AMYOTROPHIC LATERAL SCLEROSIS (ALS) |
| US11339377B2 (en) * | 2017-11-08 | 2022-05-24 | Bluesky Immunotherapies Gmbh | SO3 chromatography for use in a method for virus purification |
| EP3762500A1 (en) | 2018-03-06 | 2021-01-13 | Voyager Therapeutics, Inc. | Insect cell manufactured partial self-complementary aav genomes |
| AU2019257755A1 (en) | 2018-04-27 | 2020-11-12 | Voyager Therapeutics, Inc. | Methods for measuring the potency of AADC viral vectors |
| SG11202011296VA (en) | 2018-05-15 | 2020-12-30 | Voyager Therapeutics Inc | Compositions and methods for the treatment of parkinson's disease |
| US20210230632A1 (en) | 2018-05-15 | 2021-07-29 | Voyager Therapeutics, Inc. | Compositions and methods for delivery of aav |
| US20210214749A1 (en) | 2018-05-16 | 2021-07-15 | Voyager Therapeutics, Inc. | Directed evolution |
| WO2019222441A1 (en) | 2018-05-16 | 2019-11-21 | Voyager Therapeutics, Inc. | Aav serotypes for brain specific payload delivery |
| CN112567035A (zh) | 2018-07-02 | 2021-03-26 | 沃雅戈治疗公司 | 肌萎缩侧索硬化症及脊髓相关病症的治疗 |
| US20210355454A1 (en) | 2018-07-24 | 2021-11-18 | Voyager Therapeutics, Inc. | Systems and methods for producing gene therapy formulations |
| US11389485B2 (en) | 2018-08-28 | 2022-07-19 | Vor Biopharma Inc. | Genetically engineered hematopoietic stem cells and uses thereof |
| CN113166208A (zh) | 2018-10-02 | 2021-07-23 | 沃雅戈治疗公司 | 重新定向aav衣壳的嗜性 |
| US20210348242A1 (en) | 2018-10-04 | 2021-11-11 | Voyager Therapeutics, Inc. | Methods for measuring the titer and potency of viral vector particles |
| US20210371470A1 (en) | 2018-10-12 | 2021-12-02 | Voyager Therapeutics, Inc. | Compositions and methods for delivery of aav |
| EP3876953A4 (en) * | 2018-12-11 | 2022-10-19 | ERBI Biosystems, Inc. | METHODS OF MAKING CELL PRODUCTS USING LOW VOLUME PERFUSION METHODS |
| AU2020208467A1 (en) | 2019-01-18 | 2021-08-05 | Voyager Therapeutics, Inc. | Methods and systems for producing AAV particles |
| EP3927833A4 (en) | 2019-02-21 | 2022-11-30 | Unleash Immuno Oncolytics, Inc. | ONCOLYTIC ADENOVIRAL VECTOR AND METHODS OF USE |
| CN110093455B (zh) * | 2019-04-27 | 2020-03-17 | 中国医学科学院病原生物学研究所 | 一种呼吸道病毒的检测方法 |
| TW202106879A (zh) | 2019-04-29 | 2021-02-16 | 美商航海家醫療公司 | 於生物反應器中生產經桿狀病毒感染之昆蟲細胞(biic)之系統及方法 |
| WO2020227515A1 (en) | 2019-05-07 | 2020-11-12 | Voyager Therapeutics, Inc. | Compositions and methods for the vectored augmentation of protein destruction, expression and/or regulation |
| CN112175996A (zh) * | 2019-07-05 | 2021-01-05 | 杭州荣泽生物科技有限公司 | 一种基因治疗载体的制备方法和用途 |
| EP4010465A1 (en) | 2019-08-09 | 2022-06-15 | Voyager Therapeutics, Inc. | Cell culture medium for use in producing gene therapy products in bioreactors |
| US20220364114A1 (en) | 2019-08-26 | 2022-11-17 | Voyager Therapeutics, Inc. | Controlled expression of viral proteins |
| WO2021046155A1 (en) | 2019-09-03 | 2021-03-11 | Voyager Therapeutics, Inc. | Vectorized editing of nucleic acids to correct overt mutations |
| WO2021097366A1 (en) * | 2019-11-15 | 2021-05-20 | City Of Hope | System, device and method for production of bioproduct including high density cell respirator for intensified production of adeno-associated viruses |
| CN110894494B (zh) * | 2019-11-22 | 2022-09-27 | 广西梧州制药(集团)股份有限公司 | 一种大规模高密度悬浮培养293细胞高产腺病毒的方法 |
| US20230131352A1 (en) | 2020-04-01 | 2023-04-27 | Voyager Therapeutics, Inc. | Redirection of tropism of aav capsids |
| BR112022020753A2 (pt) | 2020-04-15 | 2022-12-20 | Voyager Therapeutics Inc | Compostos de ligação a tau |
| GB202005732D0 (en) | 2020-04-20 | 2020-06-03 | Synpromics Ltd | Regulatory nucleic acid sequences |
| WO2021230987A1 (en) | 2020-05-13 | 2021-11-18 | Voyager Therapeutics, Inc. | Redirection of tropism of aav capsids |
| EP4192514A1 (en) | 2020-08-06 | 2023-06-14 | Voyager Therapeutics, Inc. | Cell culture medium for use in producing gene therapy products in bioreactors |
| GB202013940D0 (en) | 2020-09-04 | 2020-10-21 | Synpromics Ltd | Regulatory nucleic acid sequences |
| CN112226418B (zh) * | 2020-09-25 | 2022-07-08 | 阜外华中心血管病医院 | 重组腺相关病毒纯化方法 |
| US20230374542A1 (en) | 2020-10-07 | 2023-11-23 | Asklepios Biopharmaceutical, Inc. | Therapeutic adeno-associated virus delivery of fukutin related protein (fkrp) for treating dystroglycanopathy. disorders including limb girdle 21 (lgmd21) |
| CN112293394B (zh) * | 2020-10-30 | 2022-11-25 | 海南德安欣有害生物防治科技有限公司 | 一种捕鼠装置及捕鼠方法 |
| AU2021372262A1 (en) | 2020-11-02 | 2023-06-01 | Biomarin Pharmaceutical Inc. | Process for enriching adeno-associated virus |
| GB202019454D0 (en) * | 2020-12-10 | 2021-01-27 | Chancellor Masters And Scholars Of The Univ Of Oxford | Method for purifying virus |
| WO2022187473A2 (en) | 2021-03-03 | 2022-09-09 | Voyager Therapeutics, Inc. | Controlled expression of viral proteins |
| US20240141378A1 (en) | 2021-03-03 | 2024-05-02 | Voyager Therapeutics, Inc. | Controlled expression of viral proteins |
| WO2023034997A1 (en) | 2021-09-03 | 2023-03-09 | Biomarin Pharmaceutical Inc. | Aav capsid compositions and methods for delivery |
| WO2023034980A1 (en) | 2021-09-03 | 2023-03-09 | Bomarin Pharmaceutical Inc. | Aav capsid compositions and methods for delivery |
| WO2023034989A1 (en) | 2021-09-03 | 2023-03-09 | Biomarin Pharmaceutical Inc. | Aav capsid compositions and methods for delivery |
| WO2023034996A1 (en) | 2021-09-03 | 2023-03-09 | Biomarin Pharmaceutical Inc. | Aav capsid compositions and methods for delivery |
| WO2023034994A1 (en) | 2021-09-03 | 2023-03-09 | Biomarin Pharmaceutical Inc. | Aav capsid compositions and methods for delivery |
| WO2023034990A1 (en) | 2021-09-03 | 2023-03-09 | Biomarin Pharmaceutical Inc. | Aav capsid compositions and methods for delivery |
| WO2023044483A2 (en) | 2021-09-20 | 2023-03-23 | Voyager Therapeutics, Inc. | Compositions and methods for the treatment of her2 positive cancer |
| AU2022349103A1 (en) | 2021-09-27 | 2024-03-28 | Sotio Biotech Inc. | Chimeric receptor polypeptides in combination with trans metabolism molecules that re-direct glucose metabolites out of the glycolysis pathway and therapeutic uses thereof |
| TW202334436A (zh) | 2021-11-02 | 2023-09-01 | 美商航海家醫療公司 | Aav衣殼變異體及其用途 |
| CA3237201A1 (en) | 2021-11-16 | 2023-05-25 | Sotio Biotech Inc. | Treatment of myxoid/round cell liposarcoma patients |
| WO2023091948A1 (en) | 2021-11-17 | 2023-05-25 | Voyager Therapeutics, Inc. | Aav capsid variants and uses thereof |
| WO2023111580A1 (en) | 2021-12-16 | 2023-06-22 | University Of Dundee | Targeted degradation of alpha-synuclein |
| WO2023147374A2 (en) | 2022-01-25 | 2023-08-03 | Voyager Therapeutics, Inc. | Baculovirus expression system |
| TW202346599A (zh) | 2022-02-08 | 2023-12-01 | 美商航海家醫療公司 | Aav衣殼變異體及其用途 |
| GB202201713D0 (en) | 2022-02-10 | 2022-03-30 | Univ Dundee | An affinity directed phosphatase system for targeted protein dephosphorylation |
| CN115354033A (zh) * | 2022-03-25 | 2022-11-18 | 上海碧博生物医药科技有限公司 | 一种细胞裂解液、细胞裂解方法及该方法在收获aav的应用 |
| WO2023235791A1 (en) | 2022-06-02 | 2023-12-07 | Voyager Therapeutics, Inc. | Aav capsid variants and uses thereof |
| WO2024006741A1 (en) | 2022-06-28 | 2024-01-04 | Voyager Therapeutics, Inc. | Aav capsid variants and uses thereof |
| WO2024011112A1 (en) | 2022-07-06 | 2024-01-11 | Voyager Therapeutics, Inc. | Aav capsid variants and uses thereof |
| WO2024040207A1 (en) | 2022-08-19 | 2024-02-22 | Sotio Biotech Inc. | Genetically engineered natural killer (nk) cells with chimeric receptor polypeptides in combination with trans metabolism molecules and therapeutic uses thereof |
| WO2024040208A1 (en) | 2022-08-19 | 2024-02-22 | Sotio Biotech Inc. | Genetically engineered immune cells with chimeric receptor polypeptides in combination with multiple trans metabolism molecules and therapeutic uses thereof |
| WO2024054983A1 (en) | 2022-09-08 | 2024-03-14 | Voyager Therapeutics, Inc. | Controlled expression of viral proteins |
| WO2024059739A1 (en) | 2022-09-15 | 2024-03-21 | Voyager Therapeutics, Inc. | Tau binding compounds |
| WO2024064856A1 (en) | 2022-09-22 | 2024-03-28 | Biomarin Pharmaceutical Inc. | Treatment of cardiomyopathy with aav gene therapy vectors |
| WO2024064863A2 (en) | 2022-09-22 | 2024-03-28 | Biomarin Pharmaceutical Inc. | Treatment of arrhythmogenic cardiomyopathy with aav gene therapy vectors |
| CN117551624A (zh) * | 2024-01-11 | 2024-02-13 | 深圳源兴基因技术有限公司 | 一种基于无血清悬浮扩增的高效回收腺病毒的方法 |
Family Cites Families (85)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US789244A (en) * | 1902-10-01 | 1905-05-09 | Hans Thormeyer | Steam-turbine. |
| US824544A (en) * | 1905-12-04 | 1906-06-26 | George Hossege | Pipe-hanger. |
| US4222743A (en) * | 1978-07-20 | 1980-09-16 | Wang Wei Kung | Method and apparatus for detecting biological particles by fluorescent stain |
| US4352883A (en) | 1979-03-28 | 1982-10-05 | Damon Corporation | Encapsulation of biological material |
| US4539020A (en) * | 1983-07-10 | 1985-09-03 | Kawasaki Steel Corporation | Methods for obtaining high-purity carbon monoxide |
| JPS6147187A (ja) | 1984-08-10 | 1986-03-07 | Chemo Sero Therapeut Res Inst | 狂犬病ウイルスの精製方法 |
| US4797368A (en) * | 1985-03-15 | 1989-01-10 | The United States Of America As Represented By The Department Of Health And Human Services | Adeno-associated virus as eukaryotic expression vector |
| US5173418A (en) * | 1985-05-10 | 1992-12-22 | Benzon Pharma, A/S | Production in Escherichia coli of extracellular Serratia spp. hydrolases |
| US5139941A (en) * | 1985-10-31 | 1992-08-18 | University Of Florida Research Foundation, Inc. | AAV transduction vectors |
| US5106841A (en) * | 1986-05-13 | 1992-04-21 | Chai-Tech Corporation | Antiviral compositions and method for their use |
| EP0273085A1 (en) | 1986-12-29 | 1988-07-06 | IntraCel Corporation | A method for internalizing nucleic acids into eukaryotic cells |
| US5591624A (en) * | 1988-03-21 | 1997-01-07 | Chiron Viagene, Inc. | Retroviral packaging cell lines |
| JPH0761955B2 (ja) | 1988-04-28 | 1995-07-05 | 国立予防衛生研究所長 | 凍結乾燥a型肝炎ワクチン |
| US5219752A (en) * | 1988-05-25 | 1993-06-15 | Teijin, Limited | Process for continuously culturing adherent animal cells |
| KR910700593A (ko) | 1989-01-16 | 1991-03-15 | 종케 보이젠, 로베르트 아인젤 | 양립성 주파수 분할 다중 텔레비젼 시스템 |
| US5221958A (en) * | 1989-05-01 | 1993-06-22 | Wolfram Bohnenkamp | Reflection fluorometer |
| DE69023488T2 (de) | 1989-12-12 | 1996-05-09 | Lidak Pharmaceuticals | BESTIMMUNG FREIER FETTSäUREN. |
| IT1238231B (it) | 1989-12-18 | 1993-07-12 | Consiglio Nazionale Ricerche | Impiego di immunomodulanti come agenti sinergici di chemioterapici nella terapia dei tumori |
| US5670488A (en) * | 1992-12-03 | 1997-09-23 | Genzyme Corporation | Adenovirus vector for gene therapy |
| NO312681B1 (no) | 1990-08-24 | 2002-06-17 | Univ California | Fremgangsmåte for fremstilling av en farmasöytisk blanding med suppresiv virkning/aktivitet |
| US6410010B1 (en) * | 1992-10-13 | 2002-06-25 | Board Of Regents, The University Of Texas System | Recombinant P53 adenovirus compositions |
| IT1248075B (it) | 1991-06-18 | 1995-01-05 | Sclavo Spa | Processo per la purificazione del virus dell'epatite a (hav), virus cosi` purificato e composizioni vaccinali che lo contengono. |
| EP0552108B1 (en) * | 1992-01-17 | 1999-11-10 | Lakowicz, Joseph R. | Energy transfer phase-modulation fluoro-immunoassay |
| US5323008A (en) * | 1992-03-23 | 1994-06-21 | Diatron Corporation | Fluorometer detection system |
| ES2188593T3 (es) | 1992-03-24 | 2003-07-01 | United Cancer Res Inst | Vacuna que contiene un virus vivo. |
| US5428011A (en) | 1992-06-16 | 1995-06-27 | Procyon Biopharma, Inc. | Pharmaceutical preparations for inhibiting tumours associated with prostate adenocarcinoma |
| US5733720A (en) * | 1992-06-18 | 1998-03-31 | Washington University | Genetically engineered cell lines for detecting infectious herpesvirus and methods therefor |
| AU4774093A (en) * | 1992-07-17 | 1994-02-14 | Aprogenex, Inc. | Free radical scavengers useful for reducing autofluorescence in fixed cells |
| AU676204B2 (en) | 1992-09-18 | 1997-03-06 | Canji, Inc. | Gene therapy by retroviral vector with tumor suppressive gene |
| US5733721A (en) * | 1992-11-20 | 1998-03-31 | The Board Of Regents Of The University Of Oklahoma | Cell analysis method using quantitative fluorescence image analysis |
| EP0682697A4 (en) | 1993-01-29 | 1997-07-30 | New Brunswick Scientific Co | METHOD AND APPARATUS FOR CULTURING ANCHOR AND SUSPENSION CELLS. |
| US5604096A (en) * | 1993-03-04 | 1997-02-18 | Schaeffer; Warren I. | Fluorometric quantitation of mycoplasmas |
| US5994314A (en) | 1993-04-07 | 1999-11-30 | Inhale Therapeutic Systems, Inc. | Compositions and methods for nucleic acid delivery to the lung |
| CA2139623A1 (en) | 1993-05-06 | 1994-11-24 | Baxter Diagnostics Inc. | Human papillomavirus detection assay |
| US5421339A (en) * | 1993-05-12 | 1995-06-06 | Board Of Regents, The University Of Texas System | Diagnosis of dysplasia using laser induced fluoroescence |
| US6686200B1 (en) * | 1993-08-31 | 2004-02-03 | Uab Research Foundation | Methods and compositions for the large scale production of recombinant adeno-associated virus |
| JPH09504429A (ja) | 1993-10-12 | 1997-05-06 | チロン ビアジーン,インコーポレイティド | 組換えウイルスの保存方法 |
| TW442569B (en) * | 1993-10-25 | 2001-06-23 | Canji Inc | Recombinant adenoviral vector |
| JPH09508017A (ja) * | 1994-01-12 | 1997-08-19 | ジェネティック セラピー,インコーポレイテッド | レトロウイルスベクターの精製 |
| HRP950097A2 (en) | 1994-03-08 | 1997-06-30 | Merck & Co Inc | Hepatitis a virus culture process |
| US5498537A (en) * | 1994-03-09 | 1996-03-12 | Cellco, Inc. | Serum-free production of packaged viral vector |
| ATE189697T1 (de) * | 1994-03-22 | 2000-02-15 | Immune Response Corp Inc | Die hocheffiziente herstellung und isolierung von viruspartikeln |
| US5521083A (en) * | 1994-05-13 | 1996-05-28 | The Research Foundation Of State University Of New York Et Al. | Large granular lymphocyte leukemia associated virus |
| AU3551195A (en) * | 1994-09-23 | 1996-04-09 | Somatix Therapy Corporation | Chimeric adenovirus for gene delivery |
| US5552309A (en) * | 1994-09-30 | 1996-09-03 | Indiana University Foundation | Use of polyols for improving the introduction of genetic material into cells |
| US6348343B2 (en) * | 1995-02-24 | 2002-02-19 | Genentech, Inc. | Human DNase I variants |
| US5837520A (en) * | 1995-03-07 | 1998-11-17 | Canji, Inc. | Method of purification of viral vectors |
| US5707618A (en) * | 1995-03-24 | 1998-01-13 | Genzyme Corporation | Adenovirus vectors for gene therapy |
| US5744304A (en) * | 1995-05-30 | 1998-04-28 | Board Of Regents, The University Of Texas System | Inflammation-induced expression of a recombinant gene |
| US5772888A (en) * | 1995-07-27 | 1998-06-30 | Massachusetts Institute Of Technology | Separation and/or concentration of an analyte from a mixture using a two-phase aqueous micellar system |
| AU709584B2 (en) * | 1995-08-01 | 1999-09-02 | Pasteur Merieux Serums Et Vaccins | Industrial production process for a vaccine against Japanese encephalitis, and vaccine obtained |
| CA2625279A1 (en) | 1995-08-30 | 1997-03-06 | Genzyme Corporation | Chromatographic purification of adenovirus and aav |
| KR19990071795A (ko) * | 1995-11-30 | 1999-09-27 | 파라비 레이 | 암 진단 및 치료를 위한 방법과 조성물 |
| US5910434A (en) * | 1995-12-15 | 1999-06-08 | Systemix, Inc. | Method for obtaining retroviral packaging cell lines producing high transducing efficiency retroviral supernatant |
| US5789244A (en) * | 1996-01-08 | 1998-08-04 | Canji, Inc. | Compositions and methods for the treatment of cancer using recombinant viral vector delivery systems |
| AR006240A1 (es) * | 1996-03-14 | 1999-08-11 | Fuller H B Licensing Financ | Adhesivo de fusion caliente que comprende interpolimeros, articulo no tejido que lo comprende, procedimiento de polimerizacion para preparlo y caja, envase, bandeja y libro unidos con dicho adhesivo |
| JPH09262099A (ja) * | 1996-03-28 | 1997-10-07 | Aisin Seiki Co Ltd | フルオレセイン誘導体リン酸エステルによる核酸等の検出方法 |
| US6485958B2 (en) * | 1996-07-01 | 2002-11-26 | Gencell Sa | Method for producing recombinant adenovirus |
| EP0968284B1 (en) * | 1996-11-20 | 2006-12-13 | Introgen Therapeutics, Inc. | An improved method for the production and purification of adenoviral vectors |
| US7732129B1 (en) | 1998-12-01 | 2010-06-08 | Crucell Holland B.V. | Method for the production and purification of adenoviral vectors |
| AU5370598A (en) | 1996-12-13 | 1998-07-03 | Schering Corporation | Methods for purifying viruses |
| US6261823B1 (en) * | 1996-12-13 | 2001-07-17 | Schering Corporation | Methods for purifying viruses |
| US6168944B1 (en) | 1997-01-31 | 2001-01-02 | Schering Corporation | Methods for cultivating cells and propagating viruses |
| IL131447A0 (en) | 1997-02-18 | 2001-01-28 | Canji Inc | Combined tumor supressor gene therapy and chemotherapy in the treatment of neoplasms |
| US6210683B1 (en) | 1997-09-05 | 2001-04-03 | Merck & Co., Inc. | Stabilizers containing recombinant human serum albumin for live virus vaccines |
| US6130313A (en) | 1997-10-02 | 2000-10-10 | Clontech Laboratories, Inc. | Rapidly degrading GFP-fusion proteins |
| ID28298A (id) | 1998-02-17 | 2001-05-10 | Schering Corp | Komposisi yang mengandung virus dan metode untuk memekatkan preparat-preparat virus |
| US5922576A (en) | 1998-02-27 | 1999-07-13 | The John Hopkins University | Simplified system for generating recombinant adenoviruses |
| JP2002505866A (ja) | 1998-03-10 | 2002-02-26 | ラージ・スケール・プローティオーミックス・コーポレイション | 微生物の検出および特性付与 |
| US6265151B1 (en) * | 1998-03-27 | 2001-07-24 | Seroptix, Inc. | Apparatus and method for infectious disease detection |
| MXPA00010338A (es) | 1998-04-22 | 2005-06-17 | Genvec Inc | Purificacion eficiente de adenovirus. |
| US6171790B1 (en) * | 1998-05-01 | 2001-01-09 | Incyte Pharmaceuticals, Inc. | Human protease associated proteins |
| AU4188599A (en) | 1998-05-15 | 1999-12-06 | Geron Corporation | Method and apparatus for detecting hazardous agents |
| DE69917779T2 (de) * | 1998-06-30 | 2005-06-16 | Lamina, Inc. | Zytologische und histologische fixier-zusammensetzung und verfahren zur verwendung |
| JP2002522040A (ja) | 1998-08-08 | 2002-07-23 | インペリアル キャンサー リサーチ テクノロジー リミテッド | 改変されたグリーン蛍光タンパク質 |
| US5965358A (en) | 1998-08-26 | 1999-10-12 | Genvec, Inc. | Method for assessing the relative purity of viral gene transfer vector stocks |
| US6689600B1 (en) * | 1998-11-16 | 2004-02-10 | Introgen Therapeutics, Inc. | Formulation of adenovirus for gene therapy |
| US6225289B1 (en) | 1998-12-10 | 2001-05-01 | Genvec, Inc. | Methods and compositions for preserving adenoviral vectors |
| AU779267B2 (en) | 1998-12-31 | 2005-01-13 | Centelion S.A.S. | Method for separating viral particles |
| US6316185B1 (en) * | 1999-09-29 | 2001-11-13 | Mountain View Pharmaceuticals, Inc. | Quantitation of viruses by light scattering |
| JP2002054561A (ja) * | 2000-08-08 | 2002-02-20 | Toyota Industries Corp | 容量可変型圧縮機の制御弁及び容量可変型圧縮機 |
| US6447995B1 (en) * | 2000-10-04 | 2002-09-10 | Genvec, Inc. | Utilizing intrinsic fluorescence to detect adenovirus |
| DE10050114A1 (de) * | 2000-10-09 | 2002-04-25 | Wolf Gmbh Richard | Verwendung einer Gelmasse |
| US20040106184A1 (en) * | 2002-08-28 | 2004-06-03 | Introgen Therapeutics Inc. | Chromatographic methods for adenovirus purification |
| US20040229335A1 (en) * | 2003-05-15 | 2004-11-18 | Introgen Therapeutics, Inc. | Methods and compositions for the production of adenoviral vectors |
-
1997
- 1997-11-20 EP EP97950677A patent/EP0968284B1/en not_active Expired - Lifetime
- 1997-11-20 ES ES97950677T patent/ES2278399T3/es not_active Expired - Lifetime
- 1997-11-20 EP EP06025694A patent/EP1760151B1/en not_active Expired - Lifetime
- 1997-11-20 CA CA2272820A patent/CA2272820C/en not_active Expired - Lifetime
- 1997-11-20 DE DE69737107T patent/DE69737107T2/de not_active Expired - Lifetime
- 1997-11-20 AU AU53617/98A patent/AU732703B2/en not_active Expired
- 1997-11-20 AT AT06025694T patent/ATE550429T1/de active
- 1997-11-20 EP EP06012869A patent/EP1707631A3/en not_active Withdrawn
- 1997-11-20 KR KR10-1999-7004446A patent/KR100503701B1/ko not_active IP Right Cessation
- 1997-11-20 CN CN971812543A patent/CN1244215B/zh not_active Expired - Lifetime
- 1997-11-20 JP JP52396598A patent/JP4492826B2/ja not_active Expired - Lifetime
- 1997-11-20 NZ NZ335947A patent/NZ335947A/xx not_active IP Right Cessation
- 1997-11-20 BR BR9713368-0A patent/BR9713368A/pt not_active Application Discontinuation
- 1997-11-20 US US08/975,519 patent/US6194191B1/en not_active Expired - Lifetime
- 1997-11-20 WO PCT/US1997/021504 patent/WO1998022588A2/en active IP Right Grant
- 1997-11-20 ES ES06025694T patent/ES2383640T3/es not_active Expired - Lifetime
- 1997-11-20 AT AT97950677T patent/ATE348155T1/de active
-
1999
- 1999-05-19 NO NO992389A patent/NO992389L/no not_active Application Discontinuation
-
2000
- 2000-04-24 US US09/556,570 patent/US6726907B1/en not_active Expired - Lifetime
-
2001
- 2001-12-27 US US10/033,491 patent/US20030008375A1/en not_active Abandoned
- 2001-12-27 US US10/033,571 patent/US7510875B2/en not_active Expired - Fee Related
-
2004
- 2004-07-30 US US10/909,066 patent/US7445930B2/en not_active Expired - Fee Related
-
2006
- 2006-08-23 US US11/508,780 patent/US20070155008A1/en not_active Abandoned
-
2007
- 2007-07-19 HK HK07107808.0A patent/HK1107116A1/xx not_active IP Right Cessation
-
2009
- 2009-10-02 US US12/572,943 patent/US20100055763A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4492826B2 (ja) | アデノウイルスベクターの産生および精製のための改良された方法 | |
| WO1998022588A9 (en) | An improved method for the production and purification of adenoviral vectors | |
| US9428768B2 (en) | Method for the production and purification of adenoviral vectors | |
| US7125706B2 (en) | Method for the production and purification of adenoviral vectors | |
| US20060166364A1 (en) | Use of flexible bag containers for viral production | |
| KR20060028388A (ko) | 아데노바이러스 벡터의 생산을 위한 방법과 조성물 | |
| US20040106184A1 (en) | Chromatographic methods for adenovirus purification | |
| WO1999027123A2 (en) | Modified sv40 viral vectors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20041122 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20041122 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20041122 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20041122 |
|
| A072 | Dismissal of procedure [no reply to invitation to correct request for examination] |
Free format text: JAPANESE INTERMEDIATE CODE: A072 Effective date: 20050809 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20050930 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20071023 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080121 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080303 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080220 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20080227 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080331 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080319 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080512 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080423 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20090825 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20091225 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100222 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20100311 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100324 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100331 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130416 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130416 Year of fee payment: 3 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130416 Year of fee payment: 3 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130416 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140416 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |