ES2873875T3 - Quinazolinonas como inhibidores de fosfatidilinositol 3-quinasa delta humano - Google Patents
Quinazolinonas como inhibidores de fosfatidilinositol 3-quinasa delta humano Download PDFInfo
- Publication number
- ES2873875T3 ES2873875T3 ES16185213T ES16185213T ES2873875T3 ES 2873875 T3 ES2873875 T3 ES 2873875T3 ES 16185213 T ES16185213 T ES 16185213T ES 16185213 T ES16185213 T ES 16185213T ES 2873875 T3 ES2873875 T3 ES 2873875T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- methyl
- phenyl
- quinazolin
- pi3k5
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 *C1=NC2=N**C2=C(*C(*c2c(*34)c(*)c(*)c(*)c2*)C3=NC2=CC=*=CC=C2C4=O)*1 Chemical compound *C1=NC2=N**C2=C(*C(*c2c(*34)c(*)c(*)c(*)c2*)C3=NC2=CC=*=CC=C2C4=O)*1 0.000 description 12
- JSMRMEYFZHIPJV-UHFFFAOYSA-N C1C2CCC1C2 Chemical compound C1C2CCC1C2 JSMRMEYFZHIPJV-UHFFFAOYSA-N 0.000 description 1
- HRORNJNAUXHWIP-UHFFFAOYSA-N CC(C(N1c(c(Cl)ccc2)c2Cl)=Nc(cccc2C)c2C1=O)Nc1ncnc2c1nc[nH]2 Chemical compound CC(C(N1c(c(Cl)ccc2)c2Cl)=Nc(cccc2C)c2C1=O)Nc1ncnc2c1nc[nH]2 HRORNJNAUXHWIP-UHFFFAOYSA-N 0.000 description 1
- YAFJINVASNOMDF-UHFFFAOYSA-N CC(C(N1c2cccc(N3CCOCC3)c2)=Nc(cccc2C)c2C1=O)Nc1ncnc2c1nc[nH]2 Chemical compound CC(C(N1c2cccc(N3CCOCC3)c2)=Nc(cccc2C)c2C1=O)Nc1ncnc2c1nc[nH]2 YAFJINVASNOMDF-UHFFFAOYSA-N 0.000 description 1
- OHTRQLWMEVACLS-UHFFFAOYSA-N CC(CI)C(N1C(C=C(C2)F)=CC2F)=Nc2cccc(O)c2C1=O Chemical compound CC(CI)C(N1C(C=C(C2)F)=CC2F)=Nc2cccc(O)c2C1=O OHTRQLWMEVACLS-UHFFFAOYSA-N 0.000 description 1
- DGAHCFAEMMPTKZ-UHFFFAOYSA-N CC1S(C)C1CC[O](CN1c2c(C)cnc(C3C(C4)C4C3)c2O[CH]1)=C Chemical compound CC1S(C)C1CC[O](CN1c2c(C)cnc(C3C(C4)C4C3)c2O[CH]1)=C DGAHCFAEMMPTKZ-UHFFFAOYSA-N 0.000 description 1
- RFKCHADEWBVTLO-UHFFFAOYSA-N CCC(C(N1c2ccccc2)=NC2=CCCC(C)=C2C1=O)NC1(C)N=CN=C2NC=CC12 Chemical compound CCC(C(N1c2ccccc2)=NC2=CCCC(C)=C2C1=O)NC1(C)N=CN=C2NC=CC12 RFKCHADEWBVTLO-UHFFFAOYSA-N 0.000 description 1
- VWSHORDBCNRQOE-UHFFFAOYSA-N CCC1=NCC1 Chemical compound CCC1=NCC1 VWSHORDBCNRQOE-UHFFFAOYSA-N 0.000 description 1
- ZSVBDDOMSONPSF-UHFFFAOYSA-N CCNc1c(C(Nc2cc(F)cc(F)c2)=O)c(N)ccc1 Chemical compound CCNc1c(C(Nc2cc(F)cc(F)c2)=O)c(N)ccc1 ZSVBDDOMSONPSF-UHFFFAOYSA-N 0.000 description 1
- GTVPFTDPYCAGLU-UHFFFAOYSA-N [O-][N+](C1=CCSC(F)=C1C(Nc1ccccc1)=O)=O Chemical compound [O-][N+](C1=CCSC(F)=C1C(Nc1ccccc1)=O)=O GTVPFTDPYCAGLU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/16—Central respiratory analeptics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/14—Decongestants or antiallergics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
- A61P39/06—Free radical scavengers or antioxidants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/28—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/88—Oxygen atoms
- C07D239/91—Oxygen atoms with aryl or aralkyl radicals attached in position 2 or 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/16—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Diabetes (AREA)
- Physical Education & Sports Medicine (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Rheumatology (AREA)
- Endocrinology (AREA)
- Ophthalmology & Optometry (AREA)
- Biomedical Technology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Dermatology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Urology & Nephrology (AREA)
- Communicable Diseases (AREA)
- Pain & Pain Management (AREA)
- Epidemiology (AREA)
- Gastroenterology & Hepatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Emergency Medicine (AREA)
- Biochemistry (AREA)
- Otolaryngology (AREA)
- Vascular Medicine (AREA)
- Obesity (AREA)
Abstract
Una composición farmacéutica que comprende el compuesto **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo.
Description
DESCRIPCIÓN
Quinazolinonas como inhibidores de fosfatidilinositol 3-quinasa delta humano
CAMPO DE LA INVENCIÓN
[0001] La presente invención se refiere en general a las enzimas 3-quinasa fosfatidilinositol (PI3K), y más particularmente a los inhibidores selectivos de la actividad de PI3K y métodos de uso de tales inhibidores.
ANTECEDENTES DE LA INVENCIÓN
[0002] La señalización celular a través de fosfoinositidos 3’-fosforilados se ha implicado en una variedad de procesos celulares, por ejemplo, la transformación maligna, señalización del factor de crecimiento, la inflamación y la inmunidad (véase Rameh et al., J. Biol. Chem., 274: 8347-8350 (1999) para una revisión). La enzima responsable de generar estos productos de señalización fosforilados es la fosfatidilinositol 3-quinasa (PI 3-quinasa; PI3K). PI3K se identificó originalmente como una actividad asociada con oncoproteínas virales y tirosina quinasas receptoras del factor de crecimiento que fosforila el fosfatidilinositol (PI) y sus derivados fosforilados en el 3'-hidroxilo del anillo de inositol (Panayotou et al., Trends Cell Biol 2: 358- 60 (1992)).
[0003] Los niveles de fosfatidilinositol-3,4,5-trifosfato (PIP3), el producto principal de la activación de la PI 3-quinasa, aumentan con el tratamiento de las células con una variedad de agonistas. Por lo tanto, se cree que la activación de la PI 3-quinasa participa en una variedad de respuestas celulares que incluyen el crecimiento celular, la diferenciación y la apoptosis (Parker et al., Curr. Biol., 5: 577-99 (1995); Yao et al., Science, 267: 2003-05 (1995)). Aunque los objetivos posteriores de los lípidos fosforilados generados tras la activación de la PI 3-quinasa no se han caracterizado bien, la evidencia emergente sugiere que las proteínas que contienen el dominio de homología de pleckstrina y el dominio de dedo FYVE se activan cuando se unen a varios lípidos de fosfatidilinositol (Sternmark et al., J Cell. Sci., 112: 4175-83 (1999); Lemmon y col., Trends Cell Biol., 7: 237-42 (1997)). In vitro, algunas isoformas de la proteína quinasa C (PKC) son activadas directamente por PIP3, y se ha demostrado que la proteína quinasa relacionada con PKC, PKB, es activada por PI 3-quinasa (Burgering et al., Nature, 376: 599-602 (1995)).
[0004] Actualmente, la familia de la enzima PI 3-quinasa se divide en tres clases basándose en sus especificidades de sustrato. Los PI3K de clase I pueden fosforilar fosfatidilinositol (PI), fosfatidilinositol-4-fosfato y fosfatidilinositol-4,5-bifosfato (PIP2) para producir fosfatidilinositol-3-fosfato (PIP), fosfatidilinositol-3-fosfato (PIP), fosfatidilinositol-3,4-bifatolfosfato, 4,5-trifosfato, respectivamente. Las PI3K de clase II fosforilan PI y fosfatidilinositol-4-fosfato, mientras que las PI3K de clase III solo pueden fosforilar PI.
[0005] La purificación inicial y la clonación molecular de PI 3-quinasa reveló que era un heterodímero que consiste en subunidades p85 y p110 (Otsu et al, Cell, 65: 91-104 (1991); Hiles et al, Cell, 70: 419 - 29 (1992)). Desde entonces, se han identificado cuatro PI3K de Clase I distintas, denominadas PI3K a, p, 5 y y, cada una de las cuales consta de una subunidad catalítica distinta de 110 kDa y una subunidad reguladora. Más específicamente, tres de las subunidades catalíticas, es decir, p110a, p110p y p110y, interactúan cada una con la misma subunidad reguladora, es decir, p85, mientras que p110y interactúa con una subunidad reguladora p101 distinta. Como se describe a continuación, los patrones de expresión de cada una de estas PI3K en células y tejidos humanos también son distintos. Aunque se ha acumulado una gran cantidad de información sobre las funciones celulares de las PI 3-quinasas en general, las funciones que desempeñan las isoformas individuales se desconocen en gran medida.
[0006] Se ha descrito la clonación de p110a bovino. Esta proteína se identificó como relacionada con la proteína Saccharomyces cerevisiae: Vps34p, una proteína involucrada en el procesamiento de proteínas vacuolares. También se demostró que el producto p110a recombinante se asociaba con p85a, para producir una actividad PI3K en células COS-1 transfectadas. Véase Hiles y col., Cell, 70, 419 - 29 (1992).
[0007] La clonación de una segunda isoforma humana p110, designado p110p, se describe en Hu et al., Mol. Cell. Biol., 13: 7677 - 88 (1993). Se dice que esta isoforma se asocia con p85 en las células y se expresa de manera ubicua, ya que se ha encontrado ARNm de p110p en numerosos tejidos humanos y de ratón, así como en células endoteliales de la vena umbilical humana, células T leucémicas humanas Jurkat, 293 células renales embrionarias humanas, fibroblastos 3T3 de ratón, células HeLa y células de carcinoma de vejiga de rata NBT2. Una expresión tan amplia sugiere que la isoforma p110B es muy importante en las vías de señalización.
[0008] La identificación de la isoforma p1105 de PI 3-quinasa se describe en Chantry et al., J. Biol. Chem., 272: 19236 -41 (1997). Se observó que la isoforma p1105 humana se expresa de una manera restringida al tejido. Se expresa en niveles elevados en linfocitos y tejidos linfoides, lo que sugiere que la proteína podría desempeñar un papel en la señalización mediada por PI 3-quinasa en el sistema inmunológico. Los detalles concernientes a la isoforma p1105 también se pueden encontrar en las Patentes de Estados Unidos N° 5,858,753; 5,822,910; y 5,985,589.
Véase también, Vanhaesebroeck et al., Proc. Natl. Acad. Sci. EE. UU., 94: 4330-5 (1997), y Publicación Internacional N° WO 97/46688.
[0009] En cada una de los subtipos PI3Ka, p, y 5, la subunidad p85 actúa para localizar PI 3-quinasa a la membrana plasmática mediante la interacción de su dominio SH2 con residuos de tirosina fosforilados (presente en un contexto de secuencia apropiado) en proteínas diana (Rameh et al., Cell, 83: 821-30 (1995)). Se han identificado dos isoformas de p85, p85a, que se expresa de forma ubicua, y p85p, que se encuentra principalmente en el cerebro y los tejidos linfoides (Volinia et al., Oncogene, 7: 789-93 (1992)). La asociación de la subunidad p85 a las subunidades catalíticas a, p o 5 de PI 3-quinasa p110 parece ser necesaria para la actividad catalítica y la estabilidad de estas enzimas. Además, la unión de las proteínas Ras también regula al alza la actividad de PI 3-quinasa.
[0010] La clonación de p110y reveló aún más complejidad dentro de la familia de PI3K de enzimas (Stoyanov et al, Science,. 269: 690-93 (1995)). La isoforma p110y está estrechamente relacionada con p110a y p110p (45-48% de identidad en el dominio catalítico), pero, como se ha señalado, no utiliza p85 como subunidad de direccionamiento. En cambio, p110y contiene un dominio adicional denominado "dominio de homología de pleckstrina" cerca de su extremo amino. Este dominio permite la interacción de p110y con las subunidades pY de las proteínas G heterotriméricas y esta interacción parece regular su actividad.
[0011] La subunidad reguladora p101 para PI3Kgamma se clonó originalmente en los cerdos, y el ortólogo humano identificado posteriormente (Krugmann et al, J. Biol Chem., 274: 17152-8 (1999)). La interacción entre la región N-terminal de p101 con la región N-terminal de p110y parece ser crítica para la activación de PI3Ky a través de GpY mencionado anteriormente.
[0012] Un polipéptido constitutivamente activo PI3K se describe en la Publicación Internacional N° WO 96/25488. Esta publicación describe la preparación de una proteína de fusión quimérica en donde un fragmento de 102 residuos de p85 conocido como la región interSH2 (iSH2) se fusiona a través de una región enlazadora al extremo N-terminal de p110 murino. El dominio iSH2 de p85 aparentemente es capaz de activar la actividad de PI3K de una manera comparable a la de p85 intacta (Klippel y col., Mol. Cell. Biol., 14: 2675-85 (1994)).
[0013] Por lo tanto, PI 3-quinasas se pueden definir por su identidad de aminoácidos o por su actividad. Los miembros adicionales de esta familia de genes en crecimiento incluyen lípidos y quinasas de proteína más distantes, como Vps34 TOR1, TOR2 de Saccharomyces cerevisiae (y sus homólogos de mamíferos como FRAP y mTOR), el producto del gen de ataxia telangiectasia (ATR) y la subunidad catalítica de quinasa de proteína dependiente de ADN (ADN-PK). Véase, en general, Hunter, Cell, 83: 1-4 (1995).
[0014] La PI 3-quinasa también parece estar implicada en varios aspectos de la activación de leucocitos. Se ha demostrado que una actividad de PI 3-quinasa asociada a p85 se asocia físicamente con el dominio citoplásmico de CD28, que es una molécula coestimuladora importante para la activación de células T en respuesta al antígeno (Pages et al., Nature, 369: 327-29 (1994); Rudd, Immunity, 4: 527-34 (1996)). La activación de las células T a través de CD28 reduce el umbral de activación por el antígeno y aumenta la magnitud y duración de la respuesta proliferativa. Estos efectos están relacionados con aumentos en la transcripción de varios genes, incluida la interleucina-2 (IL2), un importante factor de crecimiento de células T (Fraser et al., Science, 251: 313-16 (1991)). La mutación de CD28 de tal manera que ya no puede interactuar con la PI 3-quinasa conduce a una falla para iniciar la producción de IL2, lo que sugiere un papel crítico para la PI 3-quinasa en la activación de las células T.
[0015] Los inhibidores específicos contra miembros individuales de una familia de enzimas proporcionan herramientas valiosas para descifrar las funciones de cada enzima. Dos compuestos, LY294002 y wortmanina, se han utilizado ampliamente como inhibidores de la PI 3-quinasa. Sin embargo, estos compuestos son inhibidores inespecíficos de PI3K, ya que no distinguen entre los cuatro miembros de las PI 3-quinasas de Clase I. Por ejemplo, los valores CI50 de wortmanina contra cada uno de las diversas PI 3-quinasas de Clase I están en el intervalo de 1-10 nM. Del mismo modo, los valores CI50 para LY294002 contra cada una de estas PI 3-quinasas es de aproximadamente 1 pm (Fruman et al, Ann Rev. Biochem., 67: 481-507 (1998)). Por tanto, la utilidad de estos compuestos en el estudio de las funciones de las PI 3-quinasas de Clase I individuales es limitada.

[0016] Sobre la base de los estudios que utilizan la wortmanina, existe evidencia de que la función PI 3-quinasa también es necesaria para algunos aspectos de la señalización de leucocitos a través de receptores acoplados a proteína G (Thelen et al., Proc. Natl. Acad. Sci. EE.UU., 91: 4960 - 64 (1994)). Además, se ha demostrado que la wortmanina y el LY294002 bloquean la migración de neutrófilos y la liberación de superóxido. Sin embargo, debido a que estos compuestos no distinguen entre las diversas isoformas de PI3K, no está claro qué isoforma o isoformas de PI3K en particular están implicadas en estos fenómenos.
[0017] En vista de las consideraciones anteriores, es claro que el conocimiento existente es deficiente con respecto a la estructura y características funcionales de las enzimas PI 3-quinasa, incluyendo la localización subcelular, estados de activación, las afinidades de sustrato, y similares. Además, las funciones que realizan estas enzimas tanto en tejidos normales como enfermos quedan por dilucidar. En particular, la función de PI3K5 en leucocitos no se ha caracterizado previamente y el conocimiento sobre su función en fisiología humana sigue siendo limitado. La coexpresión en estos tejidos de otras isoformas de PI3K ha confundido hasta ahora los esfuerzos para segregar las actividades de cada enzima. Además, la separación de las actividades de las diversas isoenzimas PI3K puede no ser posible sin la identificación de inhibidores que demuestren características de inhibición selectiva. De hecho, los solicitantes actualmente no son conscientes de que se hayan demostrado tales inhibidores selectivos, o mejor, específicos, de las isoenzimas PI3K.
[0018] Por lo tanto, existe una necesidad en la técnica para la caracterización adicional estructural del polipéptido PI3K5. También existe la necesidad de una caracterización funcional de PI3K5. Además, la comprensión de PI3K5 requiere una mayor elaboración de las interacciones estructurales de p1105, tanto con su subunidad reguladora como con otras proteínas en la célula. También persiste la necesidad de inhibidores selectivos o específicos de las isoenzimas PI3K, de modo que las funciones de cada isoenzima se puedan caracterizar mejor. En particular, los inhibidores selectivos o específicos de PI3K5 son deseables para explorar el papel de esta isoenzima y para el desarrollo de productos farmacéuticos para modular la actividad de la isozima.
RESUMEN DE LA INVENCIÓN
[0019] Un aspecto de la presente invención es proporcionar compuestos capaces de inhibir la actividad biológica de PI3K5 humana. Otro aspecto de la presente invención es proporcionar compuestos que inhiban PI3K5 selectivamente en comparación con las otras isoformas de PI3K. Otro aspecto más de la divulgación es proporcionar un método para modular selectivamente la actividad de PI3K5 humana, y de ese modo promover el tratamiento médico de enfermedades mediadas por la disfunción de PI3K5. Otro aspecto más de la divulgación es proporcionar un método para caracterizar la función de la PI3K5 humana.
[0020] Otro aspecto de la presente descripción es proporcionar un método de interrumpir la función de leucocitos que comprende leucocitos en contacto con un compuesto que inhibe selectivamente la actividad fosfatidilinositol 3-quinasa delta (PI3K5) en los leucocitos. Los leucocitos pueden comprender células seleccionadas del grupo que consiste en neutrófilos, linfocitos B, linfocitos T y basófilos.
[0021] Por ejemplo, en casos en los que los leucocitos comprenden neutrófilos, el método comprende la interrupción de al menos una función de los neutrófilos seleccionados del grupo que consiste en la liberación de superóxido estimulada, exocitosis estimulada y migración quimiotáctica. Preferiblemente, el método no altera sustancialmente la fagocitosis bacteriana o la muerte bacteriana por los neutrófilos. En los casos en los que los leucocitos comprenden linfocitos B, el método comprende interrumpir la proliferación de los linfocitos B o la producción de anticuerpos por parte de los linfocitos B. En los casos en los que los leucocitos comprenden linfocitos T, el método comprende interrumpir la proliferación de los
linfocitos T. En los casos en los que los leucocitos comprenden basófilos, el método comprende interrumpir la liberación de histamina por parte de los basófilos.
[0022] Se prefiere que el inhibidor PI3K5 sea selectivo. Se prefiere que el inhibidor de PI3K5 sea al menos aproximadamente 100 veces selectivo para la inhibición de p1105 en relación con p110a, al menos aproximadamente 40 veces selectivo en relación con p110p, y al menos aproximadamente 10 veces selectivo en relación con p120Y en un ensayo bioquímico.
[0023] La presente invención proporciona (i) una composición farmacéutica como se define en la reivindicación anexa 1 o 2, comprendiendo dicha composición farmacéutica un compuesto como se define en la reivindicación anexa 1; (ii) dicho compuesto o composición farmacéutica para uso terapéutico, como se define en la reivindicación 3 adjunta; y (iii) dicho compuesto para su uso en los métodos o usos específicos definidos en cualquiera de las reivindicaciones adjuntas 4 a 15. Cualquier tema mencionado a continuación en el presente documento y que no esté dentro del alcance de las reivindicaciones adjuntas no forma parte de la presente invención.
[0024] También se describen compuestos que son capaces de inhibir actividad de PI3K5 y tienen una fórmula estructural (I):

en donde
X e Y, independientemente, son N o CRc;
Z es N-R7 u O;
R1 son los mismos y son hidrógeno, halo, o C1-3 alquilo;
R2 y R3, independientemente, son hidrógeno, halo, o C1-3 alquilo;
R4 es hidrógeno, halo, ORa, Cn , C2-6 alquinilo, C(=O)Ra, C(=O)NRaRb, C3-6 heterocicloalquilo, C1-3 alquilenoC3-6 heterocicloalquilo, OC1-3 alquilenoORa, OC1-3 alquilenoNRaRb, OC1-3 alquilenoC3-6 cicloalquilo, OC3-6 heterocicloalquilo, OC1-3 alquilenoCECH, o OC1-3 alquilenoC(=O)NRaRb;
R5 es C1-3 alquilo, CH2CF3 , fenilo, CH2CECH, C1-3 alquilenoORe, C1-4 alquilenoNRaRb, o C1-4 alquilenoNHC(=O) ORa,
R6 es hidrógeno, halo, o NRaRb;
R7 es hidrógeno o R5 y R7 se toman junto con los átomos a los que están unidos para formar un anillo saturado de cinco o seis miembros;
R8 es C1-3 alquilo, halo, CF3 , o CH2C3-6 heterocicloalquilo;
n es 0, 1 o 2;
Ra es hidrógeno, C1-4 alquilo, o CH2C6H5 ;
Rb es hidrógeno o C1-3 alquilo; y
Rc es hidrógeno, C1-3 alquilo, o halo,
en donde cuando los grupos R1 son diferentes de hidrógeno, R2 y R4 son los mismos;
o una sal farmacéuticamente aceptable, profármaco o solvato (p. ej., hidrato) de la misma.
[0025] También se describen compuestos de fórmula estructural (II) y capaz de inhibir actividad de PI3K5:

en donde X, Y, Z, R1 a través de R8, Ra, Rb, Rc y n son como se definen anteriormente,
o una sal farmacéuticamente aceptable, profármaco o solvato (p. ej., hidrato) de la misma.
[0026] Otro aspecto de la presente descripción es proporcionar un método de tratamiento de una condición médica mediada por los neutrófilos, que comprende administrar a un mamífero en necesidad de la misma una cantidad terapéuticamente eficaz de un compuesto de fórmulas estructurales (I) o (II). Las afecciones médicas ejemplares que pueden tratarse de acuerdo con el método incluyen aquellas afecciones caracterizadas por una función de neutrófilos indeseable seleccionada del grupo que consiste en liberación estimulada de superóxido, exocitosis estimulada y migración quimiotáctica. Preferiblemente, de acuerdo con el método, la actividad fagocítica o la destrucción bacteriana por neutrófilos está sustancialmente desinhibida.
[0027] Todavía otro aspecto de la presente descripción es proporcionar un método de alterar una función de los osteoclastos que comprenden osteoclastos en contacto con un compuesto de fórmulas estructurales (I) o (II).
[0028] Otro aspecto de la presente descripción es proporcionar un método de mejorar un trastorno de resorción ósea en un mamífero en necesidad del mismo que comprende administrar al mamífero una cantidad terapéuticamente eficaz de un compuesto de fórmulas estructurales (I) o (II). Un trastorno de resorción ósea preferido susceptible de tratamiento según el método es la osteoporosis.
[0029] Todavía otro aspecto de la presente descripción es proporcionar un método para inhibir el crecimiento o proliferación de células cancerosas de origen hematopoyético comprende poner en contacto las células cancerosas con un compuesto de fórmulas estructurales (I) o (II). El método puede ser ventajoso para inhibir el crecimiento o la proliferación de cánceres seleccionados del grupo que consiste en linfomas, mielomas múltiples y leucemias.
[0030] Otro aspecto de la presente descripción es proporcionar un método para inhibir la actividad quinasa de un polipéptido PI3K5 comprende poner en contacto el polipéptido PI3K5 con un compuesto de fórmulas estructurales (I) o (II).
[0031] Todavía otro aspecto de la presente descripción es proporcionar un método de interrumpir la función de leucocitos que comprende leucocitos en contacto con un compuesto de fórmulas estructurales (I) o (II).
[0032] En el presente documento se describen compuestos de fórmulas estructurales (I) o (II) que inhiben la actividad de PI3K5 en ensayos bioquímicos y basados en células, y exhiben un beneficio terapéutico en el tratamiento de afecciones médicas en las que la actividad de PI3K5 es excesiva o indeseable.
[0033] Otro aspecto de la presente descripción es proporcionar composiciones farmacéuticas que comprenden uno o más compuestos de fórmulas estructurales (I) o (II), y el uso de las composiciones en un tratamiento terapéutico, en donde la inhibición del polipéptido PI3K5, in vivo o ex vivo, proporciona un beneficio terapéutico o es de interés para la investigación o el diagnóstico.
[0034] Otro aspecto de la presente descripción es proporcionar un artículo de fabricación para uso farmacéutico humano que comprende:
(a) una composición farmacéutica que comprende un compuesto de fórmulas estructurales (I) o (II); y (b) un recipiente, que opcionalmente comprende además un prospecto, siempre que la composición sea útil en el tratamiento de una enfermedad o trastorno mediado por la actividad de PI3K5.
[0035] Otro aspecto de la presente descripción es proporcionar:
(a) composición farmacéutica que comprende un compuesto de fórmulas estructurales (I) o (II); y (b) un recipiente, que opcionalmente comprende además un prospecto, siempre que la composición sea útil en el tratamiento de un trastorno de resorción ósea o un cáncer de origen hematopoyético.
[0036] Estos y otros aspectos y ventajas de la presente invención resultarán evidentes de la siguiente descripción detallada de las formas de realización preferidas, que se proporcionan para mejorar la comprensión de la invención sin limitar el alcance de la invención.
DESCRIPCIÓN DETALLADA DE LAS REALIZACIONES PREFERIDAS
[0037] La presente divulgación proporciona compuestos que inhiben selectivamente la actividad de PI3K5. La presente divulgación proporciona además métodos para usar los compuestos para inhibir la actividad de PI3K5, incluidos métodos para modular selectivamente la actividad de la isoenzima PI3K5 en células, especialmente leucocitos, osteoclastos y células cancerosas. Los métodos incluyen aplicaciones in vitro, in vivo y ex vivo.
[0038] De particular beneficio son métodos de uso de los compuestos de la invención para modular selectivamente actividad de PI3K5 en entornos clínicos para mejorar enfermedades o trastornos mediados por actividad de PI3K5. Por tanto, el tratamiento de enfermedades o trastornos caracterizados por una actividad de PI3K5 excesiva o inapropiada puede tratarse mediante la administración de moduladores selectivos de PI3K5.
[0039] Por otra parte, la descripción proporciona composiciones farmacéuticas que comprenden un inhibidor PI3K5 selectiva de fórmulas estructurales (I) o (II). También se proporcionan artículos de fabricación que comprenden un compuesto inhibidor selectivo de PI3K5 (o una composición farmacéutica que comprende el compuesto) e instrucciones para usar el compuesto. Otros métodos de la divulgación incluyen permitir la caracterización adicional del papel fisiológico de la isoenzima.
[0040] Los métodos descritos en el presente documento se benefician del uso de compuestos que inhiben selectivamente, y preferiblemente específicamente inhiben la actividad de PI3K5 en las células, incluyendo las células in vitro, in vivo, o ex vivo. Las células tratadas mediante métodos descritos en este documento incluyen aquellas que expresan PI3K5 endógena, donde endógeno indica que las células expresan PI3K5 sin introducción recombinante en las células de uno o más polinucleótidos que codifican un polipéptido PI3K5 o un fragmento biológicamente activo del mismo. Los métodos descritos en el presente documento también abarcan el uso de células que expresan PI3K5 exógena, en donde uno o más polinucleótidos que codifican PI3K5 o un fragmento biológicamente activo del mismo se han introducido en la célula usando procedimientos recombinantes.
[0041] Las células pueden ser in vivo, es decir, en un sujeto vivo, por ejemplo, un mamífero, incluyendo seres humanos, en donde un inhibidor PI3K5 se puede utilizar terapéuticamente para inhibir actividad de PI3K5 en el sujeto. Alternativamente, las células pueden aislarse como células discretas o en un tejido, para métodos ex vivo o in vitro. Los métodos in vitro abarcados por la invención pueden comprender la etapa de poner en contacto una enzima PI3K5 o un fragmento biológicamente activo de la misma con un compuesto inhibidor de la divulgación. La enzima PI3K5 puede incluir una enzima purificada y aislada, en donde la enzima se aísla de una fuente natural (p. ej., células o tejidos que normalmente expresan un polipéptido PI3K5 sin modificación mediante tecnología recombinante) o se aísla de células modificadas mediante técnicas recombinantes para expresar enzima exógena.
[0042] Los compuestos de la divulgación inhiben potentemente p1105. La potencia se expresa típicamente como la concentración de un compuesto necesaria para lograr un resultado determinado. Cuanto mayor sea la potencia, menos compuesto se requiere para realizar su función prevista. La potencia in vitro típicamente se expresa en términos de valores CI50 medidos utilizando un ensayo de dosis-respuesta. Los valores CI50 pueden medirse poniendo en contacto un sistema de ensayo sensible con un compuesto de interés en un intervalo de concentraciones, incluyendo concentraciones a las que no se observa un efecto o un efecto mínimo, a través de las concentraciones más altas en las que se observa un efecto parcial, a concentraciones de saturación en donde se observa el efecto máximo. Teóricamente, tales ensayos del efecto dosis-respuesta de los compuestos inhibidores pueden describirse como una curva sigmoidea que expresa un grado de inhibición en función de la concentración cuando se representa en una escala logarítmica. La curva también pasa teóricamente por un punto en donde la concentración es suficiente para reducir la actividad de la enzima p1105 a un nivel que es 50% del de la diferencia entre la actividad enzimática mínima y máxima observada en el ensayo. Esta concentración se define como la concentración inhibidora al 50% de inhibición o valor CI50.
[0043] Los valores CI50 se pueden determinar usando ya sea técnicas de ensayo bioquímico convencional (acelular) o técnicas de ensayo basadas en células bien conocidas por personas de experiencia ordinaria en la técnica. En los ejemplos siguientes se proporciona un ejemplo de dicho ensayo. Preferiblemente, valores CI50 se obtienen realizando el ensayo pertinente al menos dos veces, con el valor CI50 expresado como el promedio (media aritmética, o "media") de los valores individuales obtenidos. Más preferiblemente, el ensayo se repite de 3 a 10 (o más) veces, con el valor CI50 expresado como la media de los valores obtenidos. Aún más preferiblemente, el ensayo se realiza un número de veces suficiente para generar un valor CI50 medio estadísticamente fiable, utilizando métodos estadísticos conocidos por personas de experiencia ordinaria en la técnica.
[0044] Los compuestos de las fórmulas (I) y (II) presentan inesperadamente bajos valores CI50 relativos a PI3K5, correspondientes a potencia in vitro inesperadamente alta. En diversas realizaciones, los compuestos de fórmulas (I) y (II), cuando se ensayan como se describe en el Ejemplo 14 a continuación, exhiben valores CI50 PI3K5 de menos de aproximadamente 250 nM, menos de aproximadamente 200 nM, menos de aproximadamente 150 nM, menos de aproximadamente 125 nM, menos de aproximadamente 100 nM, menos de aproximadamente 75 nM, menos de aproximadamente 50 nM, menos de aproximadamente 25 nM, menos de aproximadamente 10 nM, y en otros menos de aproximadamente 5 nM. En otras realizaciones, los compuestos de la divulgación exhiben valores CI50 de aproximadamente 0,1 nM a aproximadamente 5 nM.
[0045] Los compuestos de fórmulas (I) y (II) son inhibidores de PI3K5 selectivos. El término "inhibidor selectivo de PI3K5”, como se usa en este documento, se refiere a un compuesto que inhibe la isoenzima PI3K5 más eficazmente que otras isoenzimas de la familia PI3K. Se entiende que un compuesto "inhibidor selectivo de PI3K5 " es más selectivo para PI3K5 que los compuestos denominados convencional y genéricamente inhibidores de PI3K, por ejemplo, wortmanina o LY294002. Al mismo tiempo, la wortmanina y el LY294002 se consideran "inhibidores de PI3K no selectivos". Además, los compuestos de la presente divulgación regulan de forma selectiva y negativa la expresión o actividad de PI3K5 y poseen propiedades farmacológicas aceptables para su uso en los métodos terapéuticos de la invención.
[0046] Por consiguiente, un inhibidor selectivo alternativamente puede ser entendido para referirse a al menos un compuesto que exhibe una concentración inhibidora del 50% (CI50) con respecto a PI3K5 que es al menos aproximadamente 50 veces, al menos aproximadamente 100 veces, al menos aproximadamente 150 veces, al menos aproximadamente 200 veces, al menos aproximadamente 250 veces, al menos aproximadamente 300 veces, al menos aproximadamente 350 veces, al menos aproximadamente 400 veces, al menos aproximadamente 500 veces, al menos aproximadamente 600 veces, al menos aproximadamente 700 veces, al menos aproximadamente 800 veces, al menos aproximadamente 900 veces, o al menos aproximadamente 1000 veces menor que el valor CI50 para PI3Ka. En realizaciones alternativas, el término inhibidor selectivo puede ser entendido para referirse a al menos un compuesto que exhibe una CI50 con respecto a PI3K5 que es al menos aproximadamente 5 veces, al menos aproximadamente 10 veces, al menos aproximadamente 20 veces, al menos aproximadamente 30 veces, al menos aproximadamente 40 veces, al menos aproximadamente 50 veces, al menos aproximadamente 60 veces, al menos aproximadamente 70 veces, al menos aproximadamente 80 veces, al menos aproximadamente 90 veces, o al menos aproximadamente 100 veces menor que el CI50 para PI3Ky. En realizaciones adicionales, el término inhibidor selectivo puede ser entendido para referirse a al menos un compuesto que exhibe una CI50 con respecto a PI3K5 que es al menos aproximadamente 5 veces, al menos aproximadamente 10 veces, al menos aproximadamente 20 veces, al menos aproximadamente 30 veces, al menos aproximadamente 40 veces, al menos aproximadamente 50 veces, al menos aproximadamente 75 veces, al menos aproximadamente 100 veces, al menos aproximadamente 125 veces, al menos aproximadamente 150 veces, al menos aproximadamente 175 veces, al menos aproximadamente 200 veces, al menos aproximadamente 250 veces, al menos aproximadamente 300 veces, o al menos aproximadamente 350 veces menor que el CI50 para PI3Kp. Los inhibidores selectivos se administran típicamente en una cantidad tal que inhiben selectivamente PI3K5, como se describió anteriormente.
[0047] Los compuestos más preferidos de la presente descripción, por lo tanto, tener un bajo valor CI50 vs. PI3K5 (es decir, el compuesto es un inhibidor potente), y son selectivos con respecto a la inhibición de PI3K5 con respecto a al menos una de PI3Ka, PI3Kp, y PI3Ky.
[0048] ”in vivo” significa dentro de un sujeto vivo, como dentro de un animal o humano. En este contexto, los agentes pueden usarse terapéuticamente in vivo para retardar o eliminar la proliferación de células que se replican de forma aberrante. Los agentes también pueden usarse in vivo como profilácticos para prevenir la proliferación celular aberrante o la manifestación de síntomas asociados con la misma.
[0049] ”Ex vivo”significa fuera de un sujeto vivo. Los ejemplos de poblaciones de células ex vivo incluyen cultivos celulares y muestras biológicas tales como muestras de fluidos o tejidos de seres humanos o animales. Estas muestras se pueden obtener mediante métodos bien conocidos en la técnica. Las muestras de fluido biológico ejemplares incluyen sangre, fluido cerebroespinal, orina, saliva. Las muestras de tejido ejemplares incluyen tumores y biopsias. En este contexto, los presentes compuestos pueden tener numerosas aplicaciones, tanto terapéuticas como experimentales.
[0050] El término "contenedor" significa cualquier receptáculo y cierre del mismo adecuado para el almacenamiento, envío, distribución, y/o manejo de un producto farmacéutico.
[0051] El término "prospecto", la información que acompaña al producto que proporciona una descripción de cómo administrar el producto, junto con los datos de seguridad y eficacia requeridos para que el médico, farmacéutico o paciente adopte una decisión informada en relación con el uso del producto. En el caso de un producto farmacéutico aprobado para su uso en seres humanos o animales, la autoridad de aprobación puede especificar el contenido del prospecto, y dicho prospecto puede denominarse informalmente la "etiqueta" del producto.
[0052] En una realización, la presente descripción proporciona un método para inhibir la función leucocitaria. Más particularmente, la presente divulgación proporciona métodos para inhibir o suprimir funciones de neutrófilos y linfocitos
T y B. Con respecto a los neutrófilos, se ha descubierto inesperadamente que la inhibición de la actividad de PI3K5 inhibe las funciones de los neutrófilos. Por ejemplo, se ha observado que los compuestos de la presente divulgación provocan la inhibición de las funciones clásicas de los neutrófilos, tales como la producción de superóxido estimulada, la exocitosis estimulada y la migración quimiotáctica. Sin embargo, se ha observado además que un método de la presente divulgación permite la supresión de ciertas funciones de los neutrófilos, sin afectar sustancialmente a otras funciones de estas células. Por ejemplo, se ha observado que los compuestos inhibidores selectivos de PI3K5 de la presente invención no inhiben sustancialmente la fagocitosis de bacterias por neutrófilos.
[0053] Así, la presente divulgación incluye procedimientos de inhibición de funciones de los neutrófilos, sin inhibir sustancialmente la fagocitosis de las bacterias. Las funciones de los neutrófilos adecuadas para la inhibición según el presente método incluyen cualquier función mediada por la actividad o expresión de PI3K5. Tales funciones incluyen, sin limitación, liberación estimulada de superóxido, exocitosis o desgranulación estimuladas, migración quimiotáctica, adhesión al endotelio vascular (p. ej., inmovilización/balanceo de neutrófilos, activación de la actividad de neutrófilos y/o fijación de neutrófilos al endotelio), diapédesis transmural, o emigración a través del endotelio a tejidos periféricos. En general, estas funciones pueden denominarse colectivamente "funciones inflamatorias", ya que normalmente están relacionadas con la respuesta de los neutrófilos a la inflamación. Las funciones inflamatorias de los neutrófilos se pueden distinguir de las funciones de destrucción bacteriana exhibidas por estas células, por ejemplo, fagocitosis y destrucción de bacterias. Por consiguiente, la presente divulgación incluye además métodos para tratar estados patológicos en los que una o más de las funciones inflamatorias de los neutrófilos son anormales o indeseables.
[0054] Los compuestos de la presente divulgación pueden usarse para inhibir una respuesta inmune endógena estimulada por al menos un factor endógeno sin inhibir sustancialmente una respuesta inmune exógena estimulada por al menos un factor exógeno tal como se describe en el documento US 2005/0043239 A1.
[0055] Los compuestos de la presente divulgación también se pueden usar para inhibir una respuesta inmune endógena estimulada por al menos un factor endógeno sin inhibir sustancialmente la capacidad de respuesta inmune, como se describe en el documento US 2005/0043239 A1. En consecuencia, los compuestos de la divulgación permiten ventajosamente el tratamiento de afecciones asociadas con una respuesta inmune endógena indeseable estimulada por al menos un factor endógeno sin comprometer la capacidad para combatir infecciones.
[0056] Los compuestos de la presente divulgación también se pueden usar para inhibir la acumulación de leucocitos como se describe en los US 2005/0054,614 A1. Los compuestos también pueden usarse para la inmovilización de inhibición de leucocitos a las células endoteliales y para inhibir la transmigración de leucocitos en el tejido inflamado, como se describe en el documento US 2005/0043239 A1. Por consiguiente, los compuestos de la divulgación permiten ventajosamente el tratamiento de individuos que tienen una afección inflamatoria en donde se encuentra que los leucocitos se acumulan en el sitio de la lesión o tejido inflamado.
[0057] Además, se ha establecido que PI3K5 desempeña un papel en la proliferación estimulada de los linfocitos, incluyendo las células B y las células T. Además, PI3K5 parece desempeñar un papel en la secreción estimulada de anticuerpos por las células B. Se han empleado compuestos inhibidores selectivos de PI3K5 de la presente divulgación para establecer que estos fenómenos pueden ser anulados por inhibición de PI3K5. Por tanto, la presente divulgación incluye métodos de uso de compuestos de fórmulas estructurales (I) o (II) para inhibir la proliferación de linfocitos o para inhibir la producción de anticuerpos por linfocitos B. Otros métodos habilitados por la presente divulgación incluyen métodos para tratar estados de enfermedad en los que una o más de estas funciones de los linfocitos son anormales o indeseables.
[0058] Los métodos de la presente divulgación se pueden practicar utilizando compuestos de fórmulas estructurales (I) o (II). Los métodos se pueden practicar usando una mezcla racémica de los compuestos o un enantiómero específico. En realizaciones preferidas, el enantiómero S de los compuestos se utilizan en los presentes métodos.
[0059] Los métodos se pueden practicar utilizando, por ejemplo, los siguientes compuestos:
Los compuestos ejemplares incluyen 5-metil-3-fenil-2-[1-(9H-purin-6-ilamino)propil]-3H-quinazolin-4-ona; 2-[1-(2-fluoro-9H-purin-6-ilamino)-etil]-5-metil-3-fenil-3H-quinazolin-4-ona; 3-(2,6-difluoro-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-(2,6-difluoro-fenil)-5-metil-3H-quinazolin-4-ona; 3-(2,6-difluoro-fenil)-2-[1-(2-fluoro-9H-purin-6-ilamino)-etil]-5-metil-3H-quinazolin-4-ona; 3-(2,6-difluoro-fenil)-5-metil-2-[1-(7H-pirrolo[2.3-d]pirimidin-4-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-propil]-5-metil-3-fenil-3H-quinazolin-4-ona; 5-metil-3-fenil-2-[1-(7H-pirrolo[2.3-d]pirimidin-4-ilamino)-propil]-3H-quinazolin-4-ona; 2-[1-(2-fluoro-9hpurin-6-ilamino)-propil]-5-metil-3-fenil-3H-quinazolin-4-ona; 5-metil-3-fenil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-5-metil-3-fenil-3H-quinazolin-4-ona; 2-[2-benciloxi-1-(9H-purin-6-ilamino)-etil]-5-metil-3-fenil-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-2-benciloxi-etil]-5-metil-3-fenil-3H-quinazolin-4-ona; 2-[2-benciloxi-1-(7H-pirrolo[2.3-d]pirimidin-4-ilamino)-etil]-5-metil-3-fenil-3H-quinazolin-4-ona; 2-[2-benciloxi-1-(2-fluoro-9H-purin-6-ilamino)-etil]-5-metil-3-fenil-3H-quinazolin-4-ona; 3-(4-fluoro-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-(4-fluoro-fenil)-5-metil-3H-quinazolin-4-ona; 3-(4-fluorofenil)-2-[1-(2-fluoro-9H-purin-6-ilamino)-etil]-5-metil-3H-quinazolin-4-ona; 3-(4-fluoro-fenil)-5-metil-2-[1-(7H-pirrolo[2.3-d]pirimidin-4-ilamino)-etil]-3H-quinazolin-4-ona; 5-metil-3-fenil-2-[1-(7H-pirrolo[2.3-d]pirimidin-4-ilamino)-etil]-3H-quinazolin-4-ona; 3-(3-fluoro-fenil)-5-metil-2-[1 -(9Hpurin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1 -(2-amino-9H-purin-6ilamino)-etil]-3-(3-fluoro-fenil)-5-metil-3H-quinazolin-4-ona; 3-(3-fluorofenil)-5-metil-2-[1-(7H-pirrolo[2.3-d]pirimidin-4-ilamino)-etil]-3H-quinazolin-4-ona; 5-metil-3-fenil-2-[1-(9H-purin-6-il)-pirrolidin-2-il]-3H-quinazolin-4-ona; 2-[2-hidroxi-1-(9H-purin-6-ilamino)-etil]-5-metil-3-fenil-3H-quinazolin-4-ona; 5-metil-3-fenil-2-[fenil-(9H-purin-6-ilamino)-metil]-3hquinazolin-4-ona; 2-[(2-amino-9H-purin-6-ilamino)-fenil-metil]-5-metil-3-fenil-3H-quinazolin-4-ona; 2-[(2 -fluoro-9H-purin-6-ilamino)-fenil-metil]-5-metil-3-fenil-3H-quinazolin-4-ona; 5-metil-3-fenil-2-[fenil-(7Hpirrolo[2.3-d]pirimidin-4-ilamino)-metil]-3H-quinazolin-4-ona; 5-fluoro-3-fenil-2-[1 -(9H-purin-6-ilamino)-etil]-3h-quinazolin-4-ona; 2-[1 -(2-amino-9H-purin-6-ilamino)-etil]-5-fluoro-3-fenil-3H-quinazolin-4-ona; 2-[1-(2-amino-9Hpurin-6-ilamino)-etil]-5-cloro-3-fenil-3H-quinazolin-4-ona; éster bencílico del ácido (5-(5-metil-4-oxo-3-fenil-3,4-dihidro-quinazolin-2-il)-5-(9H-purin-6-ilamino)-pentil]-carbámico; éster bencílico del ácido [5-(2-amino-9H-purin-6-ilamino)-5-(5-metil-4-oxo-3-fenil-3,4-dihidro-quinazolin-2-il)-pentil]-carbámico; éster bencílico del ácido [4-(5-metil-4-oxo-3-fenil-3,4-dihidro-quinazolin-2-il)-4-(9H-purin-6-ilamino)-butil]-carbámico; éster bencílico de ácido [4-(2-amino-9H-purin-6-ilamino)-4-(5-metil-4-oxo-3-fenil-3,4-dihidro-quinazolin-2-il)-butil]-carbámico; 3-fenil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[5-amino-1-(9H-purin-6-ilamino)-pentil]-5-metil-3-fenil-3H-quinazolin-4-ona); 2-[5-amino-1-(2-amino-9H-purin-6-ilamino)-pentil]-5-metil-3-fenil-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-(2,6-dimetil-fenil)-5-metil-3H-quinazolin-4-ona; 3-(2,6-dimetil-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 5-morfolin-4-ilmetil-3-fenil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1 -(2-amino-9H-purin-6-ilamino)-etil]-5-morfolin-4ilmetil-3-fenil-3H-quinazolin-4-ona; 2-[4-amino-1 -(2-amino-9H-purin-6-ilamino)-butil]-5-metil-3-fenil-3H-quinazolin-4-ona; 6-fluoro-3-fenil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1 -(2-amino-9H-purin-6-ilamino)-etil]-6-fluoro-3-fenil-3H-quinazolin-4-ona; 2-[2-terc-butoxi-1 -(9H-purin-6-ilamino)-etil]-5-metil-3-fenil-3H-quinazolin-4-ona; 3-(3-metil-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-(3-metil-fenil)-5-metil-3H-quinazolin-4-ona; 3-(3-cloro-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-(3-clorofenil)-5-metil-3H-quinazolin-4-ona; 2-[1 -(2-amino-9H-purin-6-ilamino)-2-hidroxi-etil]-5-metil-3-fenil-3H-quinazolin-4-ona; 2-[1 -(2-amino-9H-purin-6-ilamino)-etil]-3-(3-fluoro-fenil)-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-(2,6-difluorofenil)-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-propil]-5-fluoro-3-fenil-3H-quinazolin-4-ona; 5-cloro-3-(3-fluoro-fenil)-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona;
2-[1-(2-amino-9H-purin-6-ilamino)-etil]-5-cloro-3-(3-fluorofenil)-3H-quinazolin-4-ona; 3-fenil-2-[1-(9H-purin-6-ilamino)-etil]-5- trifluorometil-3H-quinazolin-4-ona; 3-(2,6-difluorofenil)-5-metil-2-[1-(9H-purin-6-ilamino)-propil]-3H-quinazolin-4-ona; 3-(2,6-difluoro-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-propil]-3-(2,6-difluorophenil)-5-metil-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-(2,6-difluoro-fenil)-5-metil-3H quinazolin-4-ona; 3-(3,5-dicloro-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 3-(2,6-dicloro-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-(2,6-dicloro-fenil)-5-metil-3H-quinazolin-4-ona; 5-cloro-3-fenil-2-[1 -(9H-purin-6-ilamino)-propil]-3H-quinazolin-4-ona; 2-[1 -(2-amino-9H-purin-6- ilamino)-propil]-5-cloro-3-fenil-3H-quinazolin-4-ona; 5-metil-3-fenil-2-[1-(9H-purin-6-ilamino)-butil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-butil]-5-metil-3-fenil-3h-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-(3,5-diclorofenil)-5-metil-3H-quinazolin-4-ona; 5-metil-3-(3-morfolin-4-ilmetil-fenil)-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1 -(2-amino-9H-purin-6-ilamino)-etil]-5-metil-3-(3-morfolin-4-ilmetil-fenil)-3H-quinazolin-4-ona; 2-[1 -(5-bromo-7H-pirrolo[2.3-d]pirimidin-4-ilamino)-etil]-5-metil-3-fenil-3H-quinazolin-4-ona; 5-metil-2-[1-(5-metil-7H-pirrolo[2.3-d]pirimidin-4-ilamino)-etil]-3-fenil-3H-quinazolin-4-ona; 2-[1-(5-fluoro-7H-pirrolo[2.3-d]pirimidin-4-ilamino)-etil]-5-metil-3-fenil-3H-quinazolin-4-ona; 2-[2-hidroxi-1-(9H-purin-6-ilamino)-etil]-3-fenil-3H-quinazolin-4-ona; 3-(3,5-difluoro-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-propil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-propil]-3-(3,5-difluoro-fenil)-5-metil-3H-quinazolin-4-ona; 3-(3,5-difluoro-fenil)-2-[1 -(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1 -(5-bromo-7H-pirrolo[2.3-d]pirimidin-4-ilamino)-etil]-3-(3-fluoro-fenil)-5-metil-3H-quinazolin-4-ona; 3-(3-fluoro-fenil)-5-metil-2-[1-(5-metil-7H-pirrolo[2.3-dlpirimidin-4-ilamino)-etil]-3h-quinazolin-4-ona; 3-fenil-2-[1-(9H-purin-6-ilamino)-propil]-3H-quinazolin-4-ona; 2-[1 -(2-amino-9H-purin-6-ilamino)-etil]-3-(3,5-difluoro-fenil)-3H-quinazolin-4-ona; 2-[1 -(2-amino-9H-purin-6-ilamino)-propil]-3-fenil-3H-quinazolin-4-ona; 6,7-difluoro-3-fenil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 6-fluoro-3-(3-fluoro-fenil)-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[4-dietilamino-1-(9H-purin-6-ilamino)-butil]-5-metil-3-fenil-3H-quinazolin-4-ona; 5-fluoro-3-fenil-2-[1 -(9H-purin-6-ilamino)-propil]-3H-quinazolin-4-ona; 3-fenil-2-[1 -(9Hpurin-6-ilamino)-etil]-3H-quinazolin-4-ona; 6-fluoro-3-fenil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 3-(3,5-difiuorofenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 5-fluoro-2-[1-(2-fluoro-9H-purin-6-ilamino)-etil]-3-fenil-3H-quinazolin-4-ona; 3-(3-fluoro-fenil)-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 5-cloro-3-(3,5-difluoro-fenil)-2-[1-(9H-purin-6-ilamino)-propil]-3H-quinazolin-4-ona; 3-(2,6-difluoro-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 3-(2,6-difluoro-fenil)-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 5-metil-3-fenil-2-[3,3,3-trifluoro-1-(9H-purin-6-ilamino)-propil]-3H-quinazolin-4-ona; 3-(3-hidroxi-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 3-(3-metoxi-fenil)-5-metil-2-{1-[9H-purin-6-ilamino]-etil}-3H-quinazolin-4-ona; 3-[3-(2-dimetilamino-etoxi)-fenil]-5-metil-2-{1-[9H-purin-6-ilamino]-etil}-3H-quinazolin-4-ona; 3-(3-ciclopropilmetoxi-fenil)-5-metil-2-{1-[9H-purin-6-ilamino]-etil}-3h-quinazolin-4-ona; 5-metil-3-(3-prop-2-iniloxi-fenil)-2-{1-[9H-purin-6-ilamino]-etil}-3H-quinazolin-4-ona; 2-(1 -[2-amino-9H-purin-6-ilamino]etil}-3-(3-hidroxifenil)-5-metil-3H-quinazolin-4-ona; 2-{1-[2-amino-9H-purin-6-ilamino]etil}-3-(3-metoxifenil)-5-metil-3H-quinazolin-4-ona; 2-{1-[2-amino-9H-purin-6-ilamino]etil}-3-(3-ciclopropilmetoxi-fenil)-5-metil-3H-quinazolin-4-ona; 2-{1-[2-amino-9H-purin-6-ilamino]etil}-5-metil-3-(3-prop-2-iniloxi-fenil)-3H-quinazolin-4-ona; 3-(3-etinilfenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 3-(5-metil-4-oxo-2-[1-(9H-purin-6-ilamino)-etil]-4H-quinazolin-3-il}-benzonitrilo; 3-{5-metil-4-oxo-2-{1-[9Hpurin-6-ilamino)-etil]-4H-quinazolin-3-il}-benzamida; 3-(3-acetilfenil)-5-metil-2-{1-[9H-purin-6-ilamino]-etil}-3H-quinazolin-4-ona; 2-(3-(5-metil-4-oxo-2-{1-[9H-purin-6-ilamino]-etil}-4H-quinazolin-3-il-fenoxiacetamida; 5-metil-2-{1-[9H-purin-6-ilamino]-etil)-3-[3-(tetrahidropuran-4-iloxi)-fenil]-3H-quinazolin-4-ona; 3-[3-(2-metoxietoxi)-fenil]-5-metil-2-[1 -(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 6-fluoro-2-[1 -(9H-purin-6-ilamino)etil]-3-[3-(tetrahidro-piran-4-iloxi)-fenil]-3H-quinazolin-4-ona; 3-[3-(3-dimetilamino-propoxi)-fenil]-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-(3-etinil-fenil)-5-metil-3H-quinazolin-4ona; 3-(2-[1-(2-amino-9H-purin-6-ilamino)-etil]-5-metil-4-oxo-4H-quinazolin-3-il}-benzonitrilo; 3-(2-[1-(2-amino-9H-purin-6-ilamino)-etil]-5-metil-4-oxo-4H-quinazolin-3-il}-benzamida; 3-{2-[1-(2-amino-9H-purin-6-ilamino)-etil]-5-metil-4-oxo-4H-quinazolin-3-il}-benzamida; 5-metil-3-(3-morfolin-4-il-fenil)-2-[1 -(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1 -(2-amino-9H-purin-6-ilamino)-etil]-5-metil-3-(3-morfolin-4-il-fenil)-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-[3-(2-metoxi-etoxi)-fenil]-5-metil-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-3-(3-(2-dimetilaminoetoxi)-fenil]-5-metil-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-but-3-inil]-5-metil-3-fenil-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-but-3-inil)-5-metil-3-fenil-3H-quinazolin-4-ona; 5-cloro-3-(3,5-difluoro-fenil)-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-propil]-5-cloro-3-(3,5-difluoro-fenil)-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-etil]-5-cloro-3-(3,5-difluoro-fenil)-3h-quinazolin-4-ona; 3-(3,5-difluorofenil)-6-fluoro-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona; 5-cloro-3-(2,6-difluorofenil)-2-[1-(9H-purin-6-ilamino)-propil)-3H-quinazolin-4-ona; 2-[1-(2-amino-9H-purin-6-ilamino)-propil]-5-cloro-3-(2,6-difluoro-fenil)-3H-quinazolin-4-ona; 5-metil-3-fenil-2-[1-(9H-purin-6-iloxi)-etil)-3H-quinazolin-4-ona.
[0060] Los métodos descritos en el presente documento se pueden practicar utilizando compuestos que exhiben actividad de PI3K5 inhibidora. En particular, los métodos descritos en este documento se pueden practicar usando compuestos que tienen la fórmula estructural general (I):

en donde
X e Y, independientemente, son N o CRc;
Z es N-R7 u O;
R1 son los mismos y son hidrógeno, halo, o C1-3 alquilo;
R2 y R3, independientemente, son hidrógeno, halo, o C1-3 alquilo;
R4 es hidrógeno, halo, ORa, Cn , C2-6 alquinilo, C(=O)Ra, C(=O)NRaRb, C3-6 heterocicloalquilo, C1-3 alquilenoC3-6 heterocicloalquilo, OC1-3 alquilenoORa, OC1-3 alquilenoNRaRb, OC1-3 alquilenoC3-6 cicloalquilo, OC3-6 heterocicloalquilo, OC1-3 alquilenoCECH, o OC1-3 alquilenoC(=O)NRaRb;
R5 es alquilo C1-3, CH2CF3 , fenilo, CH2CECH, C1-3 alquilenoORe, C1-4 alquilenoNRaRb, o C1-4 alquilenoNHC(=O)ORa,
R6 es hidrógeno, halo, o NRaRb;
R7 es hidrógeno o R5 y R7 se toman junto con los átomos a los que están unidos para formar un anillo saturado de cinco o seis miembros;
R8 es C1-3 alquilo, halo, CF3 , o CH2C3-6 heterocicloalquilo;
n es 0, 1 o 2;
Ra es hidrógeno, C1-4 alquilo, o CH2C6H5 ;
Rb es hidrógeno o C1-3 alquilo; y
Rc es hidrógeno, C1-3 alquilo, o halo,
en donde cuando los grupos R1 son diferentes de hidrógeno, R2 y R4 son los mismos;
o una sal farmacéuticamente aceptable, profármaco o solvato (p. ej., hidrato) de la misma.
[0061] Los compuestos de la presente descripción son inhibidores selectivos de actividad de PI3K5. Los compuestos exhiben inhibición de PI3K5 en ensayos bioquímicos y alteran selectivamente la función de las células que expresan P13K5 en ensayos basados en células. Como se describe en el presente documento, los presentes compuestos han demostrado una capacidad para inhibir ciertas funciones en neutrófilos y otros leucocitos, así como funciones de los osteoclastos.
[0062] En general, los compuestos descritos en este documento tienen las fórmulas estructurales generales (I) o (II), o una sal farmacéuticamente aceptable de los mismos, o profármaco, o solvato de la misma:

en donde X, Y, Z, R1 a través de R8, Ra, Rb, Rc y n son como se definen anteriormente.
[0063] En diversas realizaciones que presentan una mayor potencia con respecto a otros compuestos de acuerdo con la divulgación, R8 es C1-3 alquilo, F, Cl, o CF3. Como alternativa, en tales realizaciones, n es 0 (de manera que no hay sustituyente R8).
[0064] En otras formas de realización que exhiben tal aumento de la potencia, X e Y son, independientemente, N o CH. En una realización adicional que exhibe una potencia aumentada, X es N e Y es CH. Alternativamente, X e Y también pueden ser CH. En realizaciones adicionales que exhiben una potencia incrementada, R6 es hidrógeno, halo o NH2.
[0065] Inesperadamente, la potencia contra PI3K5 se conserva cuando R1 es la misma. En las fórmulas estructurales (I) y (II), R2 y R4 pueden diferir siempre que R1 sea H. Cuando R1 es H, se permite inesperadamente la rotación libre alrededor del enlace que conecta el sustituyente del anillo fenilo con el anillo de quinazolina, y ventajosamente, los compuestos no exhiben atropisomería (es decir, se evita la formación de múltiples diastereómeros). Alternativamente, R2 y R4 pueden ser iguales de manera que los compuestos, ventajosamente, no exhiban atropisomería.
[0066] Tal como se utiliza aquí, el término "alquilo" se define como grupos hidrocarbonados de cadena lineal y ramificada que contiene el número indicado de átomos de carbono, por ejemplo, metilo, etilo, y de cadena lineal y grupos propilo y butilo ramificados.
[0067] Los términos "C1-3 alquileno'' y "C1-4 alquileno'' se definen como grupos hidrocarburo que contienen el número indicado de átomos de carbono y un hidrógeno menos que el grupo alquilo correspondiente.
[0068] El término "C2-6 alquinilo" se define como un grupo hidrocarburo que contiene el número indicado de átomos de carbono y un triple enlace carbono-carbono.
[0069] El término "C3-6 cicloalquilo" se define como un grupo hidrocarburo cíclico que contiene el número indicado de átomo de carbonos.
[0070] El término "C2-6 heterocicloalquilo" se define de manera similar como cicloalquilo excepto el anillo contiene uno o
dos heteroátomos seleccionados del grupo que consiste en O, NRa, y S.
[0071] El término "halo" se define como fluoro, bromo, cloro y yodo.
[0072] En realizaciones preferidas, Z es N-R7, y el sistema de anillo bicíclico que contiene X e Y es

[0073] En otras realizaciones preferidas, R1 es hidrógeno, fluoro, cloro, metilo, o

R2 es hidrógeno, metilo, cloro o flúor; R3 es hidrógeno o fluoro; R6 es NH2 , hidrógeno o fluoro; R7 es hidrógeno o R5 y R7 se toman juntos para formar

R8 es metilo, trifluorometilo, cloro o flúor; R4 es hidrógeno, flúor, cloro, OH, OCH3 , OCH2C=CH, O(CH2)2N(CH3)2 , C(=O)CH3, C=CH, CN, C(=O)NH2 , OCH2C(=O) NH2 , O(CH2)2OCH3, O(CH2)2N (CH3)2,

o
/ \
N O\_y
y R5 es metilo, etilo, propilo, fenilo, CH2OH, CH2OCH2C6H5 , CH2CF3 , CH2OC (CH3)3, CH2CECH, (CH2)3N ^ 5)2 , (CH2)3NH2, (CH2)4NH2, (CH2)3NHC (=O)OCH2C6H5, O(CH2)4NHC (=O)OCH2C6H5; Rc es hidrógeno, metilo, fluoro, o bromo; y n es 0 o 1.
[0074] En general se acepta que los sistemas biológicos pueden exhibir respuestas que son muy sensibles a la absoluta naturaleza estereoquímica de los compuestos para los que están expuestos. Véase, EJ Ariens, Medicinal Research Reviews, 6: 451-66 (1986); EJ Ariens, Medicinal Research Reviews, 7: 367-87 (1987); KW Fowler, Handbook of Stereoisomers: Therapeutic Drugs, CRC Press, editado por Donald P. Smith, págs. 35 - 63 (1989); y SC Stinson, Chemical and Engineering News, 75: 38-70 (1997).
[0075] Por lo tanto, los compuestos de la presente descripción incluyen todos los posibles estereoisómeros e isómeros geométricos de los compuestos de fórmula estructural (I), e incluyen no sólo los compuestos racémicos, sino también los isómeros ópticamente activos. En realizaciones preferidas, un compuesto de la presente divulgación es el enantiómero S de un compuesto (I), como se representa en la fórmula estructural (II).
[0076] Cuando un compuesto de fórmula estructural (I) se desea como un enantiómero individual, se puede obtener ya sea por resolución del producto final o por síntesis estereoespecífica a partir de material de partida ya sea isoméricamente puro o el uso de un reactivo auxiliar quiral. Por ejemplo, véase Z. Ma et al., Tetrahedron: Asymmetry, 8 (6), páginas 883 88 (1997). La resolución del producto final, un intermedio o un material de partida se puede lograr mediante cualquier método adecuado conocido en la técnica. Estereoisómeros específicos, en particular, enantiómeros S de los compuestos de la divulgación, exhiben una excelente capacidad para inhibir la actividad quinasa de PI3K5.
[0077] El término "profármaco" tal como se utiliza aquí, se refiere a compuestos que se transforman rápidamente in vivo a un compuesto que tiene las fórmulas estructurales (I) o (II), por ejemplo, por hidrólisis. El diseño de profármacos se discute generalmente en Hardma et al. (Eds.), The Pharmacological Basis of Therapeutics de Goodman y Gilman, 9a ed., Págs. 11 - 6 (1996). Se proporciona una discusión detallada de los profármacos en Higuchi et al., Prodrugs as Novel Delivery Systems, vol. 14, ASCD Symposium Series, y en Roche (ed.), "Bioreversible Carriers in Drug Design," American Pharmaceutical Association and Pergamon Press (1987).
[0078] En resumen, la administración de un fármaco es seguido por eliminación del cuerpo o alguna biotransformación mediante el cual la actividad biológica del fármaco se reduce o elimina. Alternativamente, un proceso de biotransformación puede conducir a un subproducto metabólico, que en sí mismo es más activo o igualmente activo en comparación con el fármaco administrado inicialmente. Una mayor comprensión de estos procesos de biotransformación permite el diseño de los denominados "profármacos" que, tras una biotransformación, se vuelven más activos fisiológicamente en su estado alterado. Los profármacos, por lo tanto, abarcan compuestos farmacológicamente inactivos que se convierten en metabolitos biológicamente activos.
[0079] Para ilustrar, los profármacos se pueden convertir en una forma farmacológicamente activa a través de hidrólisis de, por ejemplo, un éster o amida de ligamiento, la introducción o la exposición de un grupo funcional en el producto resultante de este modo. Se pueden diseñar profármacos para que reaccionen con un compuesto endógeno para formar un conjugado soluble en agua que mejore aún más las propiedades farmacológicas del compuesto, por ejemplo, una vida media circulatoria aumentada. Alternativamente, los profármacos pueden diseñarse para sufrir una modificación covalente en un grupo funcional con, por ejemplo, ácido glucurónico, sulfato, glutatión, aminoácidos o acetato. El conjugado resultante puede inactivarse y excretarse en la orina, o volverse más potente que el compuesto original. Los conjugados de alto peso molecular también pueden excretarse en la bilis, someterse a escisión enzimática y liberarse de nuevo a la circulación, aumentando así eficazmente la vida media biológica del compuesto administrado originalmente.
Métodos para identificar reguladores negativos de actividad de PI3K5
[0080] Las proteínas PI3K5, así como fragmentos de los mismos que poseen la actividad biológica, se pueden utilizar para el cribado de compuestos inhibidores putativos en cualquiera de una variedad de técnicas de cribado de fármacos. Un inhibidor de PI3K5 es un compuesto que disminuye o anula la capacidad de PI3K5 para realizar cualquiera de sus funciones biológicas. Un ejemplo de tales compuestos es un agente que disminuye la capacidad de un polipéptido PI3K5 para fosforilar el fosfatidilinositol o dirigirse a estructuras apropiadas dentro de una célula. La selectividad de un compuesto que regula negativamente la actividad de PI3K5 puede evaluarse comparando su actividad sobre PI3K5 con su actividad sobre otras proteínas. Los inhibidores selectivos incluyen, por ejemplo, anticuerpos y otras proteínas o péptidos que se unen específicamente a un polipéptido PI3K5, oligonucleótidos que se unen específicamente a polipéptidos PI3K5 y otros compuestos no peptídicos (p. ej., moléculas orgánicas sintéticas o aisladas) que interactúan específicamente con polipéptido PI3K5s. Los inhibidores también incluyen compuestos como los descritos anteriormente, pero que interactúan con una pareja de unión específica de los polipéptidos PI3K5.
[0081] Las dianas actualmente preferidas para el desarrollo de inhibidores selectivos de PI3K5 incluyen, por ejemplo:
(1) regiones citoplasmáticas de polipéptidos de PI3K5 que contactan con otras proteínas y/o localizan PI3K5 dentro de una célula;
(2) regiones de polipéptidos de PI3K5 que se unen a compañeros de unión específicos;
(3) regiones de los polipéptidos PI3K5 que se unen al sustrato;
(4) sitios reguladores alostéricos de los polipéptidos PI3K5 que pueden o no pueden interactuar directamente con el sitio activo en la señal reguladora;
(5) regiones de los polipéptidos PI3K5 que median la multimerización.
Por ejemplo, una diana para el desarrollo de moduladores es la interacción reguladora identificada de p85 con p1105, que puede estar implicada en la activación y/o localización subcelular del resto p1105. Otros moduladores selectivos más incluyen aquellos que reconocen secuencias de nucleótidos reguladoras específicas o que codifican PI3K5. Los moduladores de la actividad de PI3K5 pueden ser terapéuticamente útiles en el tratamiento de una amplia gama de enfermedades y afecciones fisiológicas en las que está implicada la actividad aberrante de PI3K5.
[0082] Por consiguiente, en el presente documento se describen métodos para caracterizar la potencia de un compuesto de prueba como inhibidor del polipéptido PI3K5, comprendiendo dicho método las etapas de (a) medir la actividad de un polipéptido PI3K5 en presencia de un compuesto de prueba; (b) comparar la actividad del polipéptido PI3K5 en presencia del compuesto de prueba con la actividad del polipéptido PI3K5 en presencia de una cantidad equivalente de un compuesto de referencia (p. ej., un compuesto que tiene una potencia conocida contra PI3K5), donde una menor actividad del polipéptido PI3K5 en presencia del compuesto de prueba que en presencia de la referencia indica que el compuesto de prueba es un inhibidor más potente que el compuesto de referencia, y una mayor actividad del polipéptido PI3K5 en la presencia del compuesto de prueba que en presencia de la referencia indica que el compuesto de prueba es un inhibidor menos potente que el compuesto de referencia.
[0083] Por otra parte, en el presente documento se divulgan métodos para caracterizar la potencia de un compuesto de ensayo como un inhibidor de polipéptido PI3K5, que comprende las etapas de (a) determinar una cantidad de un compuesto de referencia (p. ej., un inhibidor PI3K5 de compuesto de la presente divulgación) que inhibe una actividad de un polipéptido PI3K5 mediante un porcentaje de inhibición de referencia, definiendo así una cantidad inhibidora de referencia para el compuesto de referencia; (b) determinar una cantidad de un compuesto de prueba que inhibe una actividad de un polipéptido PI3K5 mediante un porcentaje de inhibición de referencia, definiendo de ese modo una cantidad inhibidora de referencia para el compuesto de prueba; (c) comparar la cantidad inhibidora de referencia para el compuesto de prueba con la cantidad inhibidora de referencia para el compuesto de referencia, en donde una cantidad inhibitoria de referencia más baja para el compuesto de prueba que para el compuesto de referencia indica que el compuesto de prueba es un inhibidor más potente que el compuesto de referencia, y una cantidad inhibidora de referencia más alta para el compuesto de prueba que para el compuesto de referencia indica que el compuesto de prueba es un inhibidor menos potente que el compuesto de referencia. En un aspecto, el método usa una cantidad inhibidora de referencia que es la cantidad del compuesto que inhibe la actividad del polipéptido PI3K5 en un 50%, 60%, 70% u 80%. En otro aspecto, el método emplea una cantidad inhibidora de referencia que es la cantidad del compuesto que inhibe la actividad del polipéptido PI3K5 en un 90%, 95% o 99%. Estos métodos comprenden determinar la cantidad inhibidora de referencia de los compuestos en un ensayo bioquímico in vitro, en un ensayo basado en células in vitro o en un ensayo in vivo.
[0084] Por otra parte, en el presente documento se divulgan métodos de identificación de un inhibidor de actividad de PI3K5, que comprende las etapas de (i) medir la actividad de un polipéptido PI3K5 en la presencia y ausencia de un compuesto de ensayo, e (ii) identificar como un inhibidor un compuesto de prueba que disminuye la actividad de PI3K5 y que compite con un compuesto de la divulgación por unirse a PI3K5. Además, se describen métodos para identificar compuestos que inhiben la actividad de PI3K5, que comprenden las etapas de (i) poner en contacto un polipéptido de PI3K5 con un compuesto de la presente divulgación en presencia y ausencia de un compuesto de prueba, e (ii) identificar un compuesto de prueba como inhibidor de la actividad de PI3K5 en donde el compuesto compite con un compuesto de la divulgación por unirse a PI3K5. Por lo tanto, la divulgación proporciona un método para seleccionar inhibidores candidatos de la actividad de PI3K5 y/o confirmar el modo de acción de dichos inhibidores candidatos. Dichos métodos se pueden emplear contra otras isoformas de PI3K en paralelo para establecer la actividad comparativa del compuesto de prueba a través de las isoformas y/o en relación con un compuesto de la divulgación.
[0085] En estos métodos, el polipéptido PI3K5 puede ser un fragmento de p1105 que exhibe actividad de la quinasa, es decir, un fragmento que comprende el sitio catalítico de p1105. Alternativamente, el polipéptido PI3K5 puede ser un fragmento del dominio de unión a p1105 de p85 y proporciona un método para identificar moduladores alostéricos de PI3K5. Los métodos se pueden emplear en células que expresan células que expresan PI3K5 o sus subunidades, ya sea de forma endógena o exógena. Por consiguiente, el polipéptido empleado en tales métodos puede estar libre en solución, fijado a un soporte sólido, modificado para mostrarse en una superficie celular o localizado intracelularmente. Entonces se puede medir la modulación de la actividad o la formación de complejos de unión entre el polipéptido PI3K5 y el agente que se está probando.
[0086] Los polipéptidos PI3K humanos son susceptibles de ensayos de cribado de alto rendimiento bioquímicos o basados en células (HTS) de acuerdo con métodos conocidos y practicados en la técnica, incluyendo sistemas de ensayo de melanóforos para investigar las interacciones de ligando-receptor, sistemas de ensayo basados en levadura, y sistemas de expresión de células de mamífero. Para una revisión, véase Jayawickreme et al., Curr Opin Biotechnol, 8: 629-34 (1997). Los ensayos de HTS automatizados y miniaturizados también se comprenden como se describe, por ejemplo, en Houston et al., Curr Opin Biotechnol, 8: 734-40 (1997).
[0087] Dichos ensayos de HTS se utilizan para cribar bibliotecas de compuestos para identificar compuestos particulares que exhiben una propiedad deseada. Puede usarse cualquier biblioteca de compuestos, incluidas bibliotecas químicas, bibliotecas de productos naturales y bibliotecas combinatorias que comprenden oligopéptidos, oligonucleótidos u otros compuestos orgánicos aleatorios o diseñados. Las bibliotecas químicas pueden contener compuestos conocidos, análogos estructurales patentados de compuestos conocidos o compuestos que se identifican a partir de la selección de productos naturales.
[0088] Bibliotecas de productos naturales son colecciones de materiales aislados de fuentes naturales, típicamente, microorganismos, animales, plantas, u organismos marinos. Los productos naturales se aíslan de sus fuentes por
fermentación de microorganismos seguida del aislamiento y extracción de los caldos de fermentación o por extracción directa de los propios microorganismos o tejidos (plantas o animales). Las bibliotecas de productos naturales incluyen policétidos, péptidos no ribosómicos y variantes (incluidas variantes de origen no natural) de los mismos. Para una revisión, véase Cane et al., Science, 2 8 2 : 63-68 (1998).
[0089] Las bibliotecas combinatorias se componen de un gran número de compuestos relacionados, tales como péptidos, oligonucleótidos u otros compuestos orgánicos como una mezcla. Dichos compuestos son relativamente sencillos de diseñar y preparar mediante protocolos de síntesis automatizados tradicionales, PCR, clonación o métodos sintéticos patentados. Son de particular interés las bibliotecas combinatorias de péptidos y oligonucleótidos.
[0090] Todavía otras bibliotecas de interés incluyen péptidos, proteínas, peptidomimético, colección sintética multiparalela, recombinación, y bibliotecas de polipéptidos. Para una revisión de la química combinatoria y las bibliotecas creadas de esta manera, véase Myers, Curr Opin Biotechnol, 8 : 701 -07 (1997).
Usos terapéuticos de los inhibidores de actividad de PI3K5
[0091] La descripción proporciona un método para inhibir selectivamente o específicamente actividad de PI3K5 terapéutica o profilácticamente usando compuestos de la invención. El método comprende administrar un inhibidor selectivo o específico de la actividad de PI3K5 a un individuo que lo necesite en una cantidad suficiente para inhibir la actividad de PI3K5. El método puede emplearse para tratar seres humanos o animales que padecen o están sujetos a una afección cuyos síntomas o patología están mediados por la expresión o actividad de PI3K5.
[0092] "Tratar", como se usa aquí se refiere a la prevención de que un trastorno se produzca en un animal que puede estar predispuesto a la enfermedad, pero que aún no se ha diagnosticado que la tiene; inhibir el trastorno, es decir, detener su desarrollo; aliviar el trastorno, es decir, provocar su regresión; o mejorar el trastorno, es decir, reducir la gravedad de los síntomas asociados con el trastorno. "Trastorno" pretende abarcar trastornos médicos, enfermedades, afecciones, síndromes y similares, sin limitación.
[0093] Los métodos de la descripción abarcan diversos modos de tratamiento de un sujeto animal, preferiblemente un mamífero, más preferiblemente un primate, y aún más preferiblemente un ser humano. Entre los animales mamíferos que pueden tratarse se encuentran, por ejemplo, seres humanos; animales de compañía (mascotas), incluidos perros y gatos; animales de granja, incluidos bovinos, caballos, ovejas, cerdos y cabras; animales de laboratorio, incluidos ratas, ratones, conejos, cobayas y primates no humanos; y especímenes de zoológicos. Los animales no mamíferos incluyen, por ejemplo, aves, peces, reptiles y anfibios.
[0094] Un método de la presente descripción puede ser empleado para tratar los sujetos que padecen, terapéutica o profilácticamente, o estén sujetos a un trastorno inflamatorio. Un aspecto de la presente divulgación se deriva de la participación de PI3K5 en la mediación de aspectos del proceso inflamatorio. Sin pretender ceñirse a ninguna teoría, se teoriza que, debido a que la inflamación involucra procesos típicamente mediados por la activación de leucocitos (p. ej., neutrófilos o linfocitos) y transmigración quimiotáctica, y debido a que PI3K5 puede mediar tales fenómenos, se pueden usar antagonistas de PI3K5 para suprimir las lesiones asociadas con la inflamación.
[0095] "Trastorno inflamatorio" como se usa en el presente documento puede referirse a cualquier enfermedad, trastorno, o síndrome en donde una respuesta inflamatoria excesiva o no regulada conduce a los síntomas excesivos inflamatorios, daño de tejido anfitrión, o pérdida de tejido función. "Trastorno inflamatorio" también se refiere a un estado patológico mediado por la afluencia de leucocitos y/o quimiotaxis de neutrófilos.
[0096] "Inflamación", como se usa aquí, se refiere a una respuesta localizada protectora provocada por lesión o destrucción de tejidos, que sirve para destruir, diluir o separar (secuestrar) tanto el agente perjudicial como el tejido lesionado. La inflamación se asocia con un influjo de leucocitos y/o quimiotaxis de neutrófilos. La inflamación puede resultar de la infección con organismos patógenos y virus, y de medios no infecciosos como traumatismo o reperfusión después de un infarto de miocardio o accidente cerebrovascular, respuesta inmune a antígenos extraños y respuestas autoinmunes. Por consiguiente, los trastornos inflamatorios susceptibles de la invención abarcan trastornos asociados con reacciones del sistema de defensa específico así como con reacciones del sistema de defensa inespecífico.
[0097] Como se usa en este documento, el término "sistema de defensa específico" se refiere al componente del sistema inmune que reacciona a la presencia de antígenos específicos. Los ejemplos de inflamación resultante de una respuesta del sistema de defensa específico incluyen la respuesta clásica a antígenos extraños, enfermedades autoinmunes y respuesta de hipersensibilidad de tipo retardada mediada por células T. Las enfermedades inflamatorias crónicas, el rechazo de tejidos y órganos sólidos trasplantados, por ejemplo, trasplantes de riñón y médula ósea, y enfermedad de injerto contra huésped (GVHD), son ejemplos adicionales de reacciones inflamatorias del sistema de defensa específico.
[0098] El término "sistema de defensa inespecífico", como se usa en este documento, se refiere a trastornos inflamatorios mediados por leucocitos que son incapaces de memoria inmunológica (p. ej., granulocitos y macrófagos). Los ejemplos de inflamación que resultan, al menos en parte, de una reacción del sistema de defensa inespecífico incluyen inflamación asociada con afecciones tales como síndrome de dificultad respiratoria (ARDS) del adulto (agudo) o síndromes de lesiones
de múltiples órganos; lesión por reperfusión; glomerulonefritis aguda; artritis reactiva; dermatosis con componentes inflamatorios agudos; meningitis purulenta aguda u otros trastornos inflamatorios del sistema nervioso central tales como apoplejía; lesión térmica; enfermedad inflamatoria del intestino; síndromes asociados a transfusiones de granulocitos; y toxicidad inducida por citocinas.
[0099] "Enfermedad autoinmunitaria" como se usa en el presente documento se refiere a cualquier grupo de trastornos en los que la lesión del tejido está asociada con respuestas humorales o mediadas por células a los propios constituyentes del cuerpo. "Enfermedad alérgica", como se usa en este documento, se refiere a cualquier síntoma, daño tisular o pérdida de la función tisular resultante de la alergia. "Enfermedad artrítica", como se usa en este documento, se refiere a cualquier enfermedad que se caracteriza por lesiones inflamatorias de las articulaciones atribuibles a una variedad de etiologías. "Dermatitis", como se usa en este documento, se refiere a cualquiera de una gran familia de enfermedades de la piel que se caracterizan por una inflamación de la piel atribuible a una variedad de etiologías. "Rechazo al trasplante" como se usa en el presente documento se refiere a cualquier reacción inmunológica dirigida contra tejido injertado, tal como órganos o células (p. ej., médula ósea), caracterizada por una pérdida de función de los tejidos injertados y circundantes, dolor, hinchazón, leucocitosis y trombocitopenia.
[0100] Los métodos terapéuticos de la presente descripción incluyen métodos para el tratamiento de trastornos asociados con la activación de células inflamatorias. "Activación de células inflamatorias" se refiere a la inducción por un estímulo (que incluye, entre otros, citocinas, antígenos o autoanticuerpos) de una respuesta celular proliferativa, la producción de mediadores solubles (que incluyen, entre otros, citocinas, radicales de oxígeno, enzimas, prostanoides o aminas vasoactivas), o la expresión en la superficie celular de un número nuevo o aumentado de mediadores (incluidos, entre otros, antígenos de histocompatibilidad principales o moléculas de adhesión celular) en células inflamatorias (incluidos, entre otros, monocitos, macrófagos, linfocitos T, linfocitos B, granulocitos (es decir, leucocitos polimorfonucleares como neutrófilos, basófilos y eosinófilos), mastocitos, células dendríticas, células de Langerhans y células endoteliales). Los expertos en la técnica apreciarán que la activación de uno o una combinación de estos fenotipos en estas células puede contribuir al inicio, perpetuación o exacerbación de un trastorno inflamatorio.
[0101] Se ha encontrado que los compuestos de la presente descripción inhiben la liberación de superóxido por los neutrófilos. Los neutrófilos liberan superóxido en respuesta a una variedad de estímulos, incluidas las señales de infección, como mecanismo de muerte celular. Por ejemplo, se sabe que la liberación de superóxido es inducida por el factor de necrosis tumoral alfa (TNFa), que es liberado por macrófagos, mastocitos y linfocitos al entrar en contacto con componentes de la pared celular bacteriana como el lipopolisacárido (LPS). El TNFa es un activador extraordinariamente potente y promiscuo de los procesos inflamatorios, que participa en la activación de neutrófilos y varios otros tipos de células, la inducción de la adhesión de leucocitos/células endoteliales, la pirexia, la producción mejorada de MHC de clase I y la estimulación de la angiogénesis. Alternativamente, la liberación de superóxido puede ser estimulada por formil-Met-Leu-Phe (fMLP) u otros péptidos bloqueados en el extremo N-terminal por metionina formilada. Estos péptidos normalmente no se encuentran en eucariotas, pero son fundamentalmente característicos de las bacterias y señalan la presencia de bacterias al sistema inmunológico. Los leucocitos que expresan el receptor fMLP, por ejemplo, neutrófilos y macrófagos, son estimulados para migrar gradientes ascendentes de estos péptidos (es decir, quimiotaxis) hacia loci de infección. Como se demuestra en el presente documento, los compuestos de la presente divulgación inhiben la liberación de superóxido estimulada por los neutrófilos en respuesta a TNFa o fMLP. También se ha demostrado que otras funciones de los neutrófilos, incluida la exocitosis estimulada y la migración quimiotáctica dirigida, son inhibidas por los inhibidores de PI3K5 de la divulgación. Por consiguiente, se puede esperar que los compuestos de la presente divulgación sean útiles en el tratamiento de trastornos, tales como trastornos inflamatorios, que están mediados por cualquiera o todas estas funciones de los neutrófilos.
[0102] La presente invención permite procedimientos de tratamiento de enfermedades tales como enfermedades artríticas, tales como artritis reumatoide, artritis monoarticular, osteoartritis, artritis gotosa, espondilitis; enfermedad de Behcet; sepsis, choque séptico, choque endotóxico, sepsis por gramnegativa, sepsis grampositiva y síndrome de choque tóxico; síndrome de lesión de múltiples órganos secundario a septicemia, traumatismo o hemorragia; trastornos oftálmicos, tales como conjuntivitis alérgica, conjuntivitis primaveral, uveítis y oftalmopatía asociada a tiroides; granuloma eosinofílico; trastornos pulmonares o respiratorios, como asma, bronquitis crónica, rinitis alérgica, SDRA, enfermedad pulmonar inflamatoria crónica (p. ej., enfermedad pulmonar obstructiva crónica), silicosis, sarcoidosis pulmonar, pleuresía, alveolitis, vasculitis, enfisema, neumonía, bronquiectasia y toxicidad de oxígeno pulmonar; lesión por reperfusión del miocardio, el cerebro o las extremidades; fibrosis, como fibrosis quística; formación de queloides o formación de tejido cicatricial; aterosclerosis; enfermedades autoinmunes, tales como lupus eritematoso sistémico (LES), tiroiditis autoinmune, esclerosis múltiple, algunas formas de diabetes y síndrome de Reynaud; trastornos de rechazo de trasplantes tales como GVHD y rechazo de aloinjertos; glomerulonefritis crónica; enfermedades inflamatorias del intestino, tales como enfermedad inflamatoria crónica del intestino (CIBD), enfermedad de Crohn, colitis ulcerosa y enterocolitis necrotizante; dermatosis inflamatorias, tales como dermatitis de contacto, dermatitis atópica, psoriasis o urticaria; fiebre y mialgias por infección; trastornos inflamatorios del sistema nervioso central o periférico, tales como meningitis, encefalitis y lesión cerebral o de la médula espinal debida a un traumatismo menor; Síndrome de Sjogren; enfermedades que implican diapédesis de leucocitos; hepatitis alcohólica; neumonía bacteriana; enfermedades mediadas por complejos antígenoanticuerpo; choque hipovolémico; Diabetes mellitus tipo I; hipersensibilidad aguda y retardada; estados de enfermedad debidos a discrasia leucocitaria y metástasis; lesión térmica; síndromes asociados a transfusiones de granulocitos; y toxicidad inducida por citocinas.
[0103] El método puede tener utilidad en el tratamiento de sujetos que padecen o están sujetos a lesión por reperfusión, es decir, lesión resultante de situaciones en las que un tejido u órgano experimenta un período de isquemia seguida por reperfusión. El término "isquemia" se refiere a la anemia tisular localizada debido a la obstrucción del flujo de entrada de sangre arterial. La isquemia transitoria seguida de reperfusión resulta característicamente en la activación y transmigración de neutrófilos a través del endotelio de los vasos sanguíneos en el área afectada. La acumulación de neutrófilos activados a su vez da como resultado la generación de metabolitos reactivos del oxígeno, que dañan componentes del tejido u órgano afectado. Este fenómeno de "lesión por reperfusión" se asocia comúnmente con afecciones tales como accidente cerebrovascular vascular (que incluye isquemia global y focal), choque hemorrágico, isquemia o infarto de miocardio, trasplante de órganos y vasoespasmo cerebral. A modo de ilustración, la lesión por reperfusión se produce al finalizar los procedimientos de derivación cardíaca o durante un paro cardíaco cuando el corazón, una vez que se le impide recibir sangre, comienza a reperfundir. Se espera que la inhibición de la actividad de PI3K5 dé como resultado cantidades reducidas de lesión por reperfusión en tales situaciones.
[0104] Con respecto al sistema nervioso, la isquemia global se produce cuando el flujo sanguíneo a todo el cerebro cesa durante un periodo. La isquemia global puede resultar de un paro cardíaco. La isquemia focal se produce cuando una parte del cerebro se ve privada de su suministro de sangre normal. La isquemia focal puede resultar de la oclusión tromboembolítica de un vaso cerebral, traumatismo craneoencefálico, edema o tumor cerebral. Incluso si es transitoria, tanto la isquemia global como la focal pueden causar daño neuronal generalizado. Aunque el daño al tejido nervioso ocurre durante horas o incluso días después del inicio de la isquemia, se puede desarrollar algo de daño permanente en el tejido nervioso en los minutos iniciales posteriores al cese del flujo sanguíneo al cerebro.
[0105] La isquemia también puede ocurrir en el corazón en el infarto de miocardio y otros trastornos cardiovasculares en los que las arterias coronarias se han obstruido como resultado de aterosclerosis, trombo o espasmo. Por consiguiente, se cree que la invención es útil para tratar el daño del tejido cardíaco, particularmente el daño resultante de la isquemia cardíaca o causado por la lesión por reperfusión en mamíferos.
[0106] En otro aspecto, inhibidores de PI3K5 selectivos de la presente descripción se pueden emplear en métodos de tratamiento de enfermedades de huesos, especialmente enfermedades en las que la función de los osteoclastos es anormal o indeseable. Como se muestra a continuación, los compuestos de la presente divulgación inhiben la función de los osteoclastos in vitro. Por consiguiente, el uso de tales compuestos y otros inhibidores selectivos de PI3K5 puede ser valioso en el tratamiento de la osteoporosis, la enfermedad de Paget y los trastornos relacionados con la resorción ósea.
[0107] En un aspecto adicional, la presente descripción incluye métodos de uso de compuestos inhibidores PI3K5 para inhibir el crecimiento o proliferación de células cancerosas de origen hematopoyético, preferiblemente células de cáncer de origen linfoide, y más preferiblemente células de cáncer relacionadas con o derivadas de linfocitos B o progenitores de linfocitos B. Los cánceres susceptibles de tratamiento usando el método de la invención incluyen, sin limitación, linfomas, por ejemplo, neoplasias malignas de tejidos linfoides y reticuloendoteliales, tales como linfoma de Burkitt, linfoma de Hodgkin, linfomas no Hodgkin, linfomas linfocíticos y similares; mielomas múltiples; leucemias, tales como leucemias linfocíticas, leucemias mieloides (mielógenas) crónicas y similares. En una realización preferida, los presentes compuestos inhibidores de PI3K5 pueden usarse para inhibir o controlar el crecimiento o la proliferación de células de leucemia mieloide (mielógena) crónica. Otras células cancerosas, de origen hematopoyético o no, que expresan p1105 también pueden tratarse mediante la administración de un inhibidor de PI3K5 de la presente invención (C. Sawyer et al., Cancer Research, 63 (7), 1667-75 (2003)).
[0108] En otro aspecto, la descripción incluye un método para suprimir una función de los basófilos y/o mastocitos, permitiendo con ello el tratamiento de enfermedades o trastornos caracterizados por actividad de basófilos y/o mastocitos excesivos o indeseables. De acuerdo con el método, se puede usar un presente compuesto para inhibir selectivamente la expresión o actividad de PI3K5 en los basófilos y/o mastocitos. Preferiblemente, el método emplea un inhibidor de PI3K5 en una cantidad suficiente para inhibir la liberación de histamina estimulada por los basófilos y/o mastocitos. Por consiguiente, el uso de inhibidores selectivos de PI3K5 presentes puede ser valioso en el tratamiento de enfermedades caracterizadas por liberación de histamina, es decir, trastornos alérgicos, incluidos trastornos tales como enfermedad pulmonar obstructiva crónica (EPOC), asma, ARDS, enfisema y trastornos relacionados.
Composiciones farmacéuticas de inhibidores de actividad de PI3K5
[0109] Un compuesto de la presente descripción se puede administrar como el producto químico puro, pero es típico, y preferible, para administrar el compuesto en la forma de una composición o formulación farmacéutica. Por consiguiente, la presente invención proporciona composiciones farmacéuticas que comprenden un modulador presente de la actividad de PI3K5 y un portador, adyuvante o vehículo farmacéutico biocompatible. La composición puede incluir la modulación de la actividad de PI3K5 como único agente activo o en combinación con otros agentes, tales como oligo o polinucleótidos, oligo o polipéptidos, fármacos u hormonas mezcladas con un excipiente u otros vehículos farmacéuticamente aceptables. Los vehículos y otros ingredientes pueden considerarse farmacéuticamente aceptables en la medida en que sean compatibles con otros ingredientes de la formulación y no sean perjudiciales para el receptor de los mismos.
[0110] Las técnicas para la formulación y administración de composiciones farmacéuticas se pueden encontrar en
Remington Pharmaceutical Sciences, 18a Ed., Mack Publishing Co, Easton, PA, 1990. Las composiciones farmacéuticas de la presente invención se pueden fabricar usando cualquier método convencional, por ejemplo, procesos de mezcla, disolución, granulación, grageado, levigación, emulsión, encapsulación, atrapamiento, hilado en fusión, secado por aspersión o liofilización. Un experto en la técnica puede determinar una formulación farmacéutica óptima dependiendo de la vía de administración y la dosis deseada. Tales formulaciones pueden influir en el estado físico, la estabilidad, la velocidad de liberación in vivo y la velocidad de eliminación in vivo del agente administrado. Dependiendo de la afección que se esté tratando, estas composiciones farmacéuticas se pueden formular y administrar sistémica o localmente.
[0111] Las composiciones farmacéuticas se formulan para contener vehículos farmacéuticamente aceptables adecuados, y opcionalmente pueden comprender excipientes y auxiliares que facilitan el procesamiento de los compuestos activos en preparaciones que pueden usarse farmacéuticamente. El modo de administración generalmente determina la naturaleza del portador. Por ejemplo, las formulaciones para administración parenteral pueden comprender soluciones acuosas de los compuestos activos en forma soluble en agua. Los vehículos adecuados para la administración parenteral pueden seleccionarse entre solución salina, solución salina tamponada, dextrosa, agua y otras soluciones fisiológicamente compatibles. Los vehículos preferidos para la administración parenteral son tampones fisiológicamente compatibles tales como solución de Hanks, solución de Ringer o solución salina tamponada fisiológicamente. Para la administración tisular o celular, se utilizan en la formulación penetrantes apropiados para la barrera particular a permear. Dichos penetrantes son generalmente conocidos en la técnica. Para preparaciones que comprenden proteínas, la formulación puede incluir materiales estabilizantes, tales como polioles (p. ej., sacarosa) y/o tensioactivos (p. ej., tensioactivos no iónicos) y similares.
[0112] Alternativamente, las formulaciones para uso parenteral pueden comprender dispersiones o suspensiones de los compuestos activos preparados como suspensiones de inyección aceitosas apropiadas. Los disolventes o vehículos lipófilos adecuados incluyen aceites grasos, como aceite de sésamo, y ésteres de ácidos grasos sintéticos, como oleato de etilo o triglicéridos, o liposomas. Las suspensiones acuosas para inyección pueden contener sustancias que aumentan la viscosidad de la suspensión, tales como carboximetilcelulosa sódica, sorbitol, dextrano y mezclas de los mismos. Opcionalmente, la suspensión también puede contener estabilizadores o agentes adecuados que aumentan la solubilidad de los compuestos para permitir la preparación de soluciones altamente concentradas. Los polímeros acuosos que proporcionan solubilización sensible al pH y/o liberación sostenida del agente activo también pueden usarse como recubrimientos o estructuras de matriz, por ejemplo, polímeros metacrílicos, tales como la serie EUDRAGIT® disponible de Rohm America Inc. (Piscataway, NJ). También se pueden usar emulsiones, por ejemplo, dispersiones de aceite en agua y agua en aceite, opcionalmente estabilizadas por un agente emulsionante o dispersante (materiales tensioactivos; tensioactivos). Las suspensiones pueden contener agentes de suspensión tales como alcoholes isoestearílicos etoxilados, ésteres de polioxietilenosorbitol y sorbitán, celulosa microcristalina, metahidróxido de aluminio, bentonita, agar-agar, goma de tragacanto y mezclas de los mismos.
[0113] Los liposomas que contienen el agente activo también se pueden emplear para la administración parenteral. Los liposomas generalmente se derivan de fosfolípidos u otras sustancias lipídicas. Las composiciones en forma de liposomas también pueden contener otros ingredientes, tales como estabilizantes, conservantes, excipientes y similares. Los lípidos preferidos incluyen fosfolípidos y fosfatidil colinas (lecitinas), tanto naturales como sintéticos. Se conocen en la técnica métodos para formar liposomas. Véase, por ejemplo, Prescott (Ed.), Methods in Cell Biology, vol. XIV, pág. 33, Academic Press, Nueva York (1976).
[0114] Las composiciones farmacéuticas que comprenden el agente en dosificaciones adecuadas para la administración oral se pueden formular usando vehículos farmacéuticamente aceptables bien conocidos en la técnica. Las preparaciones formuladas para la administración oral pueden estar en forma de comprimidos, píldoras, cápsulas, sellos, grageas, pastillas, líquidos, geles, jarabes, pastas, elixires, suspensiones o polvos. Para ilustrar, pueden obtenerse preparaciones farmacéuticas para uso oral combinando los compuestos activos con un excipiente sólido, triturando opcionalmente la mezcla resultante y procesando la mezcla de gránulos, después de añadir auxiliares adecuados si se desea, para obtener comprimidos o núcleos de grageas. Las formulaciones orales pueden emplear vehículos líquidos de tipo similar a los descritos para uso parenteral, por ejemplo, soluciones acuosas tamponadas, suspensiones y similares.
[0115] Las formulaciones orales preferidas incluyen tabletas, grageas, y cápsulas de gelatina. Estas preparaciones pueden contener uno o más excipientes, que incluyen, sin limitación:
a) diluyentes, tales como azúcares, que incluyen lactosa, dextrosa, sacarosa, manitol o sorbitol;
b) aglutinantes, tales como silicato de magnesio y aluminio, almidón de maíz, trigo, arroz, patata, etc.;
c) materiales de celulosa, tales como metilcelulosa, hidroxipropilmetilcelulosa y carboximetilcelulosa de sodio, polivinilpirrolidona, gomas, tales como goma arábiga y goma tragacanto, y proteínas, tales como gelatina y colágeno;
d) agentes disgregantes o solubilizantes tales como polivinilpirrolidona reticulada, almidones, agar, ácido algínico o una sal del mismo, tal como alginato de sodio, o composiciones efervescentes;
e) lubricantes, tales como sílice, talco, ácido esteárico o su sal de magnesio o calcio y polietilenglicol;
f) aromatizantes y edulcorantes;
g) colorantes o pigmentos, por ejemplo, para identificar el producto o para caracterizar la cantidad (dosis) de compuesto activo; y
h) otros ingredientes, tales como conservantes, estabilizadores, agentes de hinchamiento, agentes emulsionantes, promotores de solución, sales para regular la presión osmótica y tampones.
[0116] Las cápsulas de gelatina incluyen cápsulas de ajuste a presión hechas de gelatina, así como cápsulas blandas selladas hechas de gelatina y un recubrimiento tal como glicerol o sorbitol. Las cápsulas de ajuste rápido pueden contener el ingrediente activo mezclado con rellenos, aglutinantes, lubricantes y/o estabilizadores, etc. en las cápsulas blandas, los compuestos activos se pueden disolver o suspender en fluidos adecuados, como aceites grasos, parafina líquida o polietilenglicol líquido con o sin estabilizadores.
[0117] Los núcleos de grageas pueden proporcionarse con recubrimientos adecuados tales como soluciones concentradas de azúcar, que también pueden contener goma árabe, talco, polivinilpirrolidona, gel carbopol, polietilenglicol, y/o dióxido de titanio, soluciones de laca y disolventes orgánicos adecuados o mezclas de disolvente.
[0118] La composición farmacéutica se puede proporcionar como una sal del agente activo. Las sales son más solubles en disolventes acuosos o protónicos que las correspondientes formas de ácido o base libres. Las sales farmacéuticamente aceptables son bien conocidas en la técnica. Los compuestos que contienen restos ácidos pueden formar sales farmacéuticamente aceptables con cationes adecuados. Los cationes farmacéuticamente aceptables adecuados incluyen, por ejemplo, cationes de metales alcalinos (p. ej., sodio o potasio) y alcalinotérreos (p. ej., calcio o magnesio).
[0119] Los compuestos de fórmula estructural (I) y (II) que contienen restos básicos pueden formar sales farmacéuticamente aceptables de adición de ácido con ácidos adecuados. Por ejemplo, Berge et al., J. Pharm. Sci., 66: 1 (1977), describen sales farmacéuticamente aceptables en detalle. Las sales se pueden preparar in situ durante el aislamiento final y la purificación de los compuestos de la divulgación o por separado haciendo reaccionar una función de base libre con un ácido adecuado.
[0120] Las sales farmacéuticamente aceptables de compuestos de la descripción generalmente se prefieren en los métodos descritos en este documento. Como se usa en el presente documento, el término "sales farmacéuticamente aceptables" se refiere a sales o formas bipolares de los compuestos de fórmulas estructurales (I) o (II). Cationes farmacéuticamente aceptables adecuados incluyen cationes de metal alcalino (p. ej., sodio o potasio) y metal alcalinotérreo (p. ej., calcio o magnesio). Además, las sales farmacéuticamente aceptables de compuestos de fórmulas estructurales (I) o (II) que contienen un centro básico son sales de adición de ácido formadas con ácidos farmacéuticamente aceptables. Los ejemplos de ácidos que pueden emplearse para formar sales farmacéuticamente aceptables incluyen ácidos inorgánicos tales como clorhídrico, bromhídrico, sulfúrico y fosfórico, y ácidos orgánicos tales como oxálico, maleico, succínico, malónico y cítrico. Los ejemplos no limitantes de sales de compuestos de la divulgación incluyen, pero no se limitan a hidrocloruro, hidrobromuro, hidroyoduro, sulfato, bisulfato, 2-hidroxietansulfonato, fosfato, hidrogenofosfato, acetato, adipato, alginato, aspartato, benzoato, butirato, canforato, canforsulfonato, citrato, digluconato, glicerolfosfato, hemisulfato, heptanoato, hexanoato, formiato, succinato, malonato, fumarato, maleato, metanosulfonato, mesitilenosulfonato, naftilenosulfonato, nicotinato, 2-naftalenosulfonato, oxalato, pamoato, 3-pectinato, picpropronato sales de pivalato, propionato, tricloroacetato, trifluoroacetato, glutamato, bicarbonato, paratoluensulfonato, undecanoato, lactato, citrato, tartrato, gluconato, bencenosulfonato y p-toluenosulfonato. Además, los grupos amino disponibles presentes en los compuestos de la divulgación se pueden cuaternizar con cloruros, bromuros y yoduros de metilo, etilo, propilo y butilo; sulfatos de dimetilo, dietilo, dibutilo y diamilo; cloruros, bromuros y yoduros de decilo, laurilo, miristilo y esterilo; y bromuros de bencilo y fenetilo.
[0121] En vista de lo anterior, cualquier referencia a los compuestos de la presente descripción que aparece en este documento pretende incluir sales farmacéuticamente aceptables, solvatos, y derivados cuaternarios de los mismos.
[0122] Las composiciones que comprenden un compuesto de la divulgación formulado en un vehículo farmacéuticamente aceptable pueden prepararse, colocarse en un recipiente apropiado y etiquetarse para el tratamiento de una afección indicada. Por consiguiente, también se contempla un artículo de fabricación, tal como un recipiente que comprende una forma de dosificación de un compuesto de la divulgación y una etiqueta que contiene instrucciones para el uso del compuesto. También se contemplan kits. Por ejemplo, un kit puede comprender una forma de dosificación de una composición farmacéutica y un prospecto que contiene instrucciones para el uso de la composición en el tratamiento de una afección médica. En cualquier caso, las condiciones indicadas en la etiqueta pueden incluir el tratamiento de trastornos inflamatorios, cáncer y similares.
Métodos de administración de inhibidores de actividad de PI3K5
[0123] Las composiciones farmacéuticas que comprenden un inhibidor de actividad de PI3K5 se pueden administrar al sujeto por cualquier método convencional, incluyendo técnicas parenteral y enteral. Las modalidades de administración parenteral incluyen aquellas en las que la composición se administra por una vía distinta a la del tracto gastrointestinal, por ejemplo, inyecciones intravenosas, intraarteriales, intraperitoneales, intramedulares, intramusculares, intraarticulares, intratecales e intraventriculares. Las modalidades de administración enteral incluyen, por ejemplo, administración oral, bucal, sublingual y rectal. Las modalidades de administración transepitelial incluyen, por ejemplo, administración transmucosa y administración transdérmica. La administración transmucosa incluye, por ejemplo, la administración enteral así como la administración nasal, por inhalación y pulmonar profunda; administración vaginal; y administración bucal y
sublingual. La administración transdérmica incluye modalidades transdérmicas o transcutáneas pasivas o activas, que incluyen, por ejemplo, parches y dispositivos de iontoforesis, así como la aplicación tópica de pastas, pomadas o ungüentos. La administración parenteral también se puede lograr usando una técnica de alta presión, por ejemplo, POWDERJECT®.
[0124] Las técnicas quirúrgicas incluyen la implantación de composiciones de depósito, bombas osmóticas, y similares. Una vía de administración preferida para el tratamiento de la inflamación puede ser la administración local o tópica para trastornos localizados tales como artritis, o administración sistémica para trastornos distribuidos, por ejemplo, administración intravenosa para lesión por reperfusión o para afecciones sistémicas tales como septicemia. Para otras enfermedades, incluidas las que afectan al tracto respiratorio, por ejemplo, enfermedad pulmonar obstructiva crónica, asma y enfisema, la administración puede realizarse mediante inhalación o administración pulmonar profunda de pulverizadores, aerosoles, polvos y similares.
[0125] Para el tratamiento de enfermedades neoplásicas, especialmente leucemias y otros cánceres distribuidos, la administración parenteral se prefiere típicamente. Serían deseables formulaciones de los compuestos para optimizarlos para la biodistribución después de la administración parenteral. Los compuestos inhibidores de PI3K5 se pueden administrar antes, durante o después de la administración de quimioterapia, radioterapia y/o cirugía.
[0126] Por otra parte, el índice terapéutico de lis compuestos inhibidores PI3K5 se puede mejorar mediante la modificación o derivatización de los compuestos para la administración dirigida a células cancerosas que expresan un marcador que identifica las células como tales. Por ejemplo, los compuestos se pueden unir a un anticuerpo que reconoce un marcador que es selectivo o específico para las células cancerosas, de modo que los compuestos se acercan a las células para ejercer sus efectos localmente, como se describió anteriormente (ver por ejemplo, Pietersz y col., Immunol. Rev., 129: 57 (1992); Trail y col., Science, 261: 212 (1993); y Rowlinson-Busza y col., Curr. Opin. Oncol., 4: 1142 (1992)). La administración dirigida al tumor de estos compuestos mejora el beneficio terapéutico, entre otras cosas, minimizando las posibles toxicidades inespecíficas que pueden resultar del tratamiento con radiación o quimioterapia. En otro aspecto, los compuestos inhibidores de PI3K5 y los radioisótopos o agentes quimioterapéuticos se pueden conjugar con el mismo anticuerpo antitumoral.
[0127] Para el tratamiento de trastornos de resorción ósea o trastornos mediada por los osteoclastos, los inhibidores PI3K5 se pueden administrar por cualquier procedimiento adecuado. Puede ser deseable la administración focal, por ejemplo mediante inyección intraarticular. En algunos casos, puede ser deseable acoplar los compuestos a un resto que pueda dirigir los compuestos al hueso. Por ejemplo, un inhibidor PI3K5 se puede acoplar a compuestos con alta afinidad por la hidroxiapatita, que es un componente principal del hueso. Esto se puede lograr, por ejemplo, adaptando un método de acoplamiento de tetraciclina desarrollado para la entrega dirigida de estrógeno al hueso (Orme et al., Bioorg. Med. Chem. Lett., 4 (11): 1375-80 (1994)).
[0128] Para ser terapéuticamente eficaces en la modulación de dianas del sistema nervioso central, los agentes usados deben penetrar fácilmente la barrera hematoencefálica cuando se administran periféricamente. Sin embargo, los compuestos que no pueden atravesar la barrera hematoencefálica pueden administrarse eficazmente por vía intravenosa.
[0129] Como se señaló anteriormente, las características del propio agente y la formulación del agente pueden influir en la física del estado, estabilidad, velocidad de liberación in vivo, y la tasa de aclaramiento in vivo del agente administrado. Dicha información farmacocinética y farmacodinámica se puede recopilar mediante estudios preclínicos in vitro e in vivo, que luego se confirman en humanos durante el transcurso de los ensayos clínicos. Por tanto, para cualquier compuesto utilizado en el método descrito en este documento, una dosis terapéuticamente eficaz puede estimarse inicialmente a partir de ensayos bioquímicos y/o basados en células. Luego, la dosis se puede formular en modelos animales para lograr un rango de concentración circulante deseable que modula la expresión o actividad de PI3K5. A medida que se realicen estudios en humanos, surgirá más información sobre los niveles de dosis apropiados y la duración del tratamiento para diversas enfermedades y afecciones.
[0130] La toxicidad y eficacia terapéutica de dichos compuestos puede determinarse mediante procedimientos farmacéuticos estándar en cultivos celulares o animales experimentales, por ejemplo, para determinar el DL50 (la dosis letal para el 50% de la población) y la DE50 (la dosis terapéuticamente eficaz en el 50% de la población). La relación de dosis entre los efectos tóxicos y terapéuticos es el "índice terapéutico", que normalmente se expresa como la relación DL50/DE50. Se prefieren los compuestos que exhiben índices terapéuticos grandes, es decir, la dosis tóxica es sustancialmente más alta que la dosis efectiva. Los datos obtenidos de tales ensayos de cultivo celular y estudios adicionales en animales pueden usarse para formular un rango de dosis para uso humano. La dosificación de tales compuestos se sitúa preferiblemente dentro de una gama de concentraciones circulantes que incluye la DE50 con poca o ninguna toxicidad.
[0131] De acuerdo con la presente invención, cualquier régimen de administración eficaz de regulación de la sincronización y la secuencia de las dosis pueden ser utilizados. Los compuestos y composiciones farmacéuticas adecuadas para su uso en la presente invención incluyen aquellos en los que el ingrediente activo se administra en una cantidad eficaz para lograr su propósito pretendido. Más específicamente, una "cantidad terapéuticamente eficaz" significa una cantidad suficiente para modular la expresión o actividad de PI3K5 y, de ese modo, tratar a un individuo que padece
una indicación, o para aliviar los síntomas existentes de la indicación. La determinación de una cantidad terapéuticamente eficaz está dentro de la capacidad de los expertos en la técnica, especialmente a la luz de la descripción detallada proporcionada en este documento.
[0132] Los niveles de dosificación ejemplares para un sujeto humano son del orden de aproximadamente 0,001 miligramos de agente activo por kilogramo de peso corporal (mg/kg) a aproximadamente 1000 mg/kg. Normalmente, las unidades de dosificación del agente activo comprenden de aproximadamente 0,01 mg a aproximadamente 1000 mg, preferiblemente de aproximadamente 0,1 mg a aproximadamente 100 mg, dependiendo de la indicación, vía de administración y gravedad de la afección, por ejemplo. Dependiendo de la vía de administración, se puede calcular una dosis adecuada según el peso corporal, la superficie corporal o el tamaño de los órganos. El régimen de dosificación final lo determina el médico tratante en vista de la buena práctica médica, considerando varios factores que modifican la acción de los medicamentos, por ejemplo, la actividad específica del compuesto, la identidad y gravedad del estado de la enfermedad, la capacidad de respuesta del paciente, la edad, la condición, el peso corporal, el sexo y la dieta del paciente, y la gravedad de cualquier infección. Los factores adicionales que pueden tenerse en cuenta incluyen el tiempo y la frecuencia de administración, las combinaciones de fármacos, las sensibilidades a las reacciones y la tolerancia/respuesta a la terapia. El médico experto realiza rutinariamente un refinamiento adicional de la dosis apropiada para el tratamiento que involucra cualquiera de las formulaciones mencionadas en este documento sin experimentación indebida, especialmente a la luz de la información de dosis y los ensayos descritos, así como los datos farmacocinéticos observados en ensayos clínicos en humanos. Se pueden determinar las dosis apropiadas mediante el uso de ensayos establecidos para determinar la concentración del agente en un fluido corporal u otra muestra junto con los datos de respuesta a la dosis.
[0133] La frecuencia de dosificación dependerá de los parámetros farmacocinéticos del agente y la vía de administración. La dosis y la administración se ajustan para proporcionar niveles suficientes del resto activo o para mantener el efecto deseado. Por consiguiente, las composiciones farmacéuticas se pueden administrar en una sola dosis, múltiples dosis discretas, infusión continua, depósitos de liberación sostenida o combinaciones de los mismos, según se requiera para mantener el nivel mínimo deseado del agente. Las composiciones farmacéuticas de acción corta (es decir, semivida corta) se pueden administrar una vez al día o más de una vez al día (p. ej., dos, tres o cuatro veces al día). Las composiciones farmacéuticas de acción prolongada se pueden administrar cada 3 a 4 días, cada semana o una vez cada dos semanas. Las bombas, como las subcutáneas, intraperitoneales o subdurales, se pueden utilizar para la infusión continua.
[0134] Los siguientes ejemplos se proporcionan para ayudar en la comprensión de la invención, y presuponen una comprensión de los métodos convencionales bien conocidos por aquellas personas que tienen experiencia ordinaria en la técnica a la que los ejemplos se refieren, por ejemplo, la construcción de vectores y plásmidos, la inserción de genes que codifican polipéptidos en dichos vectores y plásmidos, o la introducción de vectores y plásmidos en células huésped. Dichos métodos se describen en detalle en numerosas publicaciones que incluyen, por ejemplo, Sambrook et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press (1989), Ausubel et al. (Eds.), Current Protocols in Molecular Biology, John Wiley & Sons, Inc. (1994); y Ausubel et al. (Eds.), Short Protocols in Molecular Biology, 4a ed., John Wiley & Sons, Inc. (1999). Los materiales y condiciones particulares descritos a continuación están destinados a ejemplificar aspectos particulares de la invención y no deben interpretarse como limitantes del alcance razonable de la misma.
EJEMPLO 1
Preparación y purificación de PI3Ka, p, y 5 recombinante
[0135] Complejos heterodiméricos PI3K recombinantes que consisten en una subunidad catalítica p110 y una subunidad reguladora p85 se sobreexpresan usando el sistema de expresión de baculovirus BAC-TO-BAC® HT (GIBCO/BRL) y luego se purifican para su uso en ensayos bioquímicos. Las cuatro PI 3-quinasas de Clase I se clonaron en vectores de baculovirus de la siguiente manera:
p1105: Una versión etiquetada con FLAG® de p1105 humano (SEQ ID NOS: 1 y 2) (véase Chantry et al., J. Biol. Chem., 272: 19236-41 (1997)) se subclonó usando técnicas estándar de ADN recombinante en el sitio BamH1-Xba1 del vector de expresión de células de insecto pFastbac HTb (Life Technologies, Gaithersburg, MD), de manera que el clon estaba en el marco con su etiqueta del vector. El sistema FLAG® se describe en las Patentes de Estados Unidos N° 4,703,004; 4,782,137; 4,851,341; y 5,011,912, y los reactivos están disponibles en Eastman Kodak Co.
p110a: Similarmente al método utilizado para p1105, descrito anteriormente, una versión etiquetada con FLAG® de p110a (ver Volinia et al., Genomics, 24 (3): 427-77 (1994)) se subclonó en sitios BamH1-HindIII de pFastbac HTb (Life Technologies) de manera que el clon estaba en marco con la etiqueta His del vector.
p110p: Se amplificó un clon p110p (véase Hu et al., Mol. Cell. Biol., 13: 7677-88 (1993)) a partir de la biblioteca de cADN de bazo humano MARATHON® Ready (Clontech, Palo Alto CA) según el protocolo del fabricante que utiliza los siguientes cebadores:
Cebador 5’
[0136] 5 ' -
GATCGAATTCGGCGCCACCATGGACTACAAGGACGACGATGACAAGTGCTTCAGTTT
CATAATGCCTCC-3’ (SEQ ID NO:3)
Cebador 3'
[0137] 5'-GATCGCGGCCGCTTAAGATCTGTAGTCTTTCCGAACTGTGTG-3' (SEQ ID NO: 4)
[0138] El cebador 5' se construyó para contener una etiqueta FLAG® en marco con la secuencia p110p. Después de la amplificación, la secuencia FLAG®-p110p se subclonó usando técnicas recombinantes estándar en los sitios £coR1 -Not1 de pFastbac HTa (Life Technologies), de manera que el clon estaba en el marco con la etiqueta His del vector.
[0139] p110y: El p110y cADN (ver Stoyanov et al, Science, 269: 690-93 (1995)) se amplificó a partir de una biblioteca de ADNc humano Marathon Ready bazo (Clontech) de acuerdo con el protocolo del fabricante usando los siguientes cebadores:
Cebador 5’
[0140] 5'-AGAATGCGGCCGCATGGAGCTGGAGAACTATAAACAGCCC-3' (SEQ ID NO: 5)
Cebador 3’
[0141] 5'-CGCGGATCCTTAGGCTGAATGTTTCTCTCCTTGTTTG-3' (SEQAG ID NO: 6) se adjuntó posteriormente al extremo 5' de la secuencia p110y y se clonó en los sitios fíamH1-Spe1 de pFastbac HTb (Life Technologies) usando técnicas estándar de ADN recombinante, con la secuencia FLAG®-110y en marco con la etiqueta His del vector. p85a: Se subclonó un fragmento BamH1-EcoR1 de ADNc de p85 etiquetado con FLAG® (véase Skolnik et al., Cell, 65: 83-89 (1991)) en los sitios fíamH1-£coR1 del vector pFastbac dual (Life Tecnologías).
[0142] Los baculovirus recombinantes que contienen los clones anteriores se generaron utilizando recomendada por el fabricante de protocolo (Life Technologies). Los baculovirus que expresan la subunidad catalítica p110a, p110p o p1105 etiquetados con His y la subunidad p85 se coinfectaron en células de insecto Sf21. Para enriquecer el complejo enzimático heterodimérico, se infectó una cantidad en exceso de baculovirus que expresa la subunidad p85, y la subunidad catalítica p110 marcada con His complejada con p85 se purificó en una columna de afinidad de níquel. Dado que p11 üy no se asocia con p85, las células Sf21 se infectaron con baculovirus recombinantes que expresan p110y marcado con His solamente. En un enfoque alternativo, p101 se puede clonar en baculovirus, para permitir la coexpresión con su compañero de unión preferido p110y.
[0143] Se recogieron y se homogeneizaron en un tampón hipotónico las células Sf21 infectadas 72 horas después (3 litros (20 mM HEPES-KOH, pH 7,8, KCl 5 mM, cóctel completo de inhibidor de proteasa (Roche Biochemicals, Indianapolis, IN), utilizando un homogeneizador Dounce. Los homogeneizados se centrifugaron a 1.000 x g durante 15 min. Los sobrenadantes se centrifugaron adicionalmente a 10.000 x g durante 20 min, seguido de ultracentrifugación a 100.000 x g durante 60 min. La fracción soluble se cargó inmediatamente en 10 ml de columna de afinidad de níque1HITRAP® (Pharmacia, Piscataway, NJ) equilibrada con 50 ml de tampón A (HEPES-KOH 50 mM, pH 7,8, NaCl 0,5 M, imidazol 10 mM). La columna se lavó extensamente con tampón A y se eluyó con un gradiente lineal de imidazol 10-500 mM. La subunidad p85 libre se eliminó de la columna durante la etapa de lavado y solo el complejo enzimático heterodimérico se eluyó en imidazol 250 mM. Se analizaron alícuotas de fracciones de níquel mediante electroforesis en gel de poliacrilamida-SDS al 10% (SDS- PAGE), teñido con SYPRO® Red (Molecular Probes, Inc., Eugene, OR) y cuantificado con STORM® PhosphoImager (Molecular Dynamics, Sunnyvale, CA). Las fracciones activas se combinaron y se cargaron directamente en una columna de heparina Hi-trap de 5 ml preequilibrada con Tampón B que contenía HEPES-KOH 50 mM, pH 7,5, NaCl 50 mM, ditiotreitol (DTT) 2 mM. La columna se lavó con 50 ml de Tampón B y se eluyó con un gradiente lineal de NaCl 0,05-2 M. Un solo pico que contenía el complejo enzimático PI3K eluyó en NaCl 0,8 M. El análisis en gel de SDS-poliacrilamida mostró que las fracciones de enzima PI3K purificadas contenían un complejo estequiométrico 1:1 de subunidades p110 y p85. El perfil de proteínas del complejo enzimático durante la cromatografía con heparina correspondió al de la actividad de la quinasa lipídica. Las fracciones activas se combinaron y congelaron bajo nitrógeno líquido.
EJEMPLOS 2-6
[0144] Debido a que PI3K5 se expresa a niveles significativos en los leucocitos, es importante para estudiar los efectos del inhibidor PI3K5 selectivo en funciones de los leucocitos. Por consiguiente, se examinaron los efectos de la inhibición de PI3K5 en varios tipos de leucocitos. Se examinaron los neutrófilos para determinar los efectos que podrían provocar la inhibición selectiva de PI3K5 (Ejemplo 2, a continuación). Sorprendentemente, se encontró que la inhibición selectiva
de la actividad de PI3K5 parece estar significativamente asociada con la inhibición de algunas pero no todas las funciones características de los neutrófilos activados. Además, también se ensayaron los efectos de la inhibición de PI3K5 sobre la función de las células B y T (Ejemplos 3-4, más adelante). Además, como PI3K5 también se expresa en osteoclastos, se estudió el efecto de la inhibición de PI3K5 sobre la función de estas células especializadas (Ejemplo 5, a continuación).
EJEMPLO 2
Caracterización del papel de PI3K5 en la función de neutrófilos
[0145] Los efectos de un inhibidor PI3K5 de la divulgación en funciones de los neutrófilos, tales como la generación de superóxido, exocitosis de elastasa, la quimiotaxis, y la destrucción bacteriana puede ser probada.
A. Preparación de los neutrófilos de sangre humana
[0146] Alícuotas (8 ml) de sangre heparinizada de voluntarios sanos se colocan en capas en cojines de 3 ml de 7,3% Ficoll® (Sigma, St. Louis, MO) y 15,4% Hypaque® (Sigma) y se centrifugó a 900 rpm durante 30 min a temperatura ambiente en una centrífuga de mesa (Beckman). La banda rica en neutrófilos justo encima del cojín FICOLL®-HYPAQUE® se recoge y se lava con la solución salina equilibrada de Hanks (HBSS) que contiene gelatina al 0,1%. Los eritrocitos residuales se eliminan mediante lisis hipotónica con NaCl al 0,2%. La preparación de neutrófilos se lava dos veces con HBSS que contiene gelatina al 0,1% y se usa inmediatamente.
B. Medición de la producción de superóxido a partir de neutrófilos
[0147] La generación de superóxido es una de las características distintivas de la activación de neutrófilos. Diversos activadores potencian la generación de superóxido por los neutrófilos. Se mide el efecto de un inhibidor de PI3K5 presente sobre la generación de superóxido por tres agonistas diferentes: TNF1a, IgG y fMLP, cada uno de los cuales representa clases separadas de activador. El superóxido generado por los neutrófilos se mide controlando un cambio en la absorbancia tras la reducción del citocromo C mediante la modificación del método descrito por Green et al., (Págs. 14,5,1 14,5,11 en Supp. 12, Curr. Protocols Immunol. (Eds., Colligan et al.) (1994)), como sigue. Los pocillos individuales de una placa de 96 pocillos se recubren durante la noche a 4°C con 50 pl de solución 2 mg/ml de fibrinógeno humano o IgG. Los pocillos se lavaron con PBS y se añadieron los siguientes reactivos a cada pocillo: 50 pl de HBSS o superóxido dismutasa (1 mg/ml), 50 pl de HBSS o TNF1a (50 ng/ml), 50 pl citocromo C (2,7 mg/mL), y 100 pl de suspensión de neutrófilos humanos purificada (2 x 106 células/mL). La placa se centrifuga durante 2 min a 200 rpm y se monitoriza la absorbancia a 550 nm durante 2 h. Para medir las cantidades relativas de superóxido generadas, los valores obtenidos de los pocillos que contienen superóxido dismutasa se restan de todos y se normalizan a los valores obtenidos de los pocillos sin ningún inhibidor.
[0148] Los compuestos de la presente divulgación inhiben la generación de superóxido inducida por TNF por los neutrófilos de una manera dependiente de la concentración. Además, los compuestos de la presente divulgación no inhibieron significativamente la generación de superóxido inducida por IgG.
[0149] El efecto de los compuestos de la presente divulgación en la generación de superóxido inducida por otro potente inductor, el péptido bacteriano formilado-Met-Leu-Phe (fMLP), también pueden ser estudiados. Al igual que la generación de superóxido inducida por TNF, la generación de superóxido inducida por fMLP también son compuestos inhibidos de la presente divulgación. Estos resultados muestran que los compuestos inhibidores de PI3K5 de la presente divulgación pueden prevenir la inducción específica de estímulos de la generación de superóxido por neutrófilos, lo que indica que PI3K5 está involucrado en este proceso.
C. Medición de la exocitosis de elastasa a partir de neutrófilos
[0150] Además de la generación de superóxido, neutrófilos activados también responden por la liberación de varias proteasas que son responsables de la destrucción de los tejidos y el cartílago durante la inflamación. Como indicación de la liberación de proteasa, se mide el efecto del presente compuesto sobre la exocitosis de elastasa. La exocitosis de elastasa se cuantifica mediante la modificación del procedimiento descrito por Ossanna et al. (J. Clin. Invest., 77: 1939 51 (1986)), como sigue. Los neutrófilos humanos purificados (0,2 x 106) (tratados con DMSO o una dilución en serie de un compuesto presente en DMSO) se estimulan con fMLP en PBS que contiene 0,01 mg/ml de citocalasina B, azida de sodio 1,0 pm (NaN3), 5 pg/mL de L-metionina y 1 pm de fMLP durante 90 min a 37°C en una placa de 96 pocillos. Al final del período de incubación, la placa se centrifuga durante 5 min a 1000 rpm y se transfieren 90 ml del sobrenadante a 10 ml de una solución 10 mM de un péptido de sustrato de elastasa, MeO-suc-Ala-AlaPro-Val-pNA, en donde MeO-suc = metoxi-succinilo; pNA = p-nitroanilida (Calbiochem, San Diego, CA). La absorbancia a 410 nm se controla durante 2 horas en un lector de placas de 96 pocillos. Para medir las cantidades relativas de elastasa excitadas, todos los valores de absorbancia se normalizan a los valores sin ningún inhibidor. Los compuestos inhibidores de PI3K5 de la presente divulgación inhiben significativamente la exocitosis de elastasa inducida por fMLP, y lo hacen de una manera dependiente de la dosis.
D. Medición de la migración de neutrófilos humanos inducida por fMLP
[0151] Los neutrófilos tienen la capacidad intrínseca de migrar a través de los tejidos, y son uno de los primeros tipos de células para llegar a los sitios de inflamación o lesión tisular. Se mide el efecto de los presentes compuestos sobre la migración de neutrófilos hacia un gradiente de concentración de fMLP. El día antes de que se realicen los ensayos de migración, las placas de 6 pocillos se recubren con proteína de fusión ICAM-1/Fc recombinante (Van der Vieren et al., Immunity, 3: 683-90 (1995)) (25 pg/ml en tampón de bicarbonato, pH 9,3) y se dejó durante la noche a 4°C. Después del lavado, se agrega una solución de agarosa al 1%, en RPMI-1640 con albúmina de suero bovino (BSA) al 0,5%, a los pocillos con o sin un inhibidor, y las placas se colocan en un refrigerador antes de perforar agujeros en la agarosa gelificada para crear placas (1 agujero central rodeado de 6 periféricos por pocillo).
[0152] Los neutrófilos humanos se obtienen como se describe anteriormente, y se resuspendieron en medio RPMI suplementado con 0,5% de BSA a 5 x 106 células/ml. Después de combinar volúmenes iguales de suspensión de neutrófilos y medio (ya sea con DMSO o una dilución en serie del compuesto de prueba en DMSO), los neutrófilos se colocan en alícuotas en los orificios periféricos, mientras que el orificio central recibió fMLP (5 pm). Las placas se incubaron a 37°C en presencia de 5% de CO2 durante 4 h, seguido de la terminación de la migración mediante la adición de solución de glutaraldehído al 1 % en D-PBS. Después de eliminar la capa de agarosa, los pocillos se lavan con agua destilada y se secan.
[0153] El análisis de la migración de neutrófilos se realiza en una estación de trabajo de video con microscopio invertido Nikon DIAPHOT® (objetivo 1x) usando el programa NIH 1,61. Utilizando los programas Microsoft Excel y Table Curve 4 (SSPS Inc., Chicago IL), se obtiene un índice de migración para cada una de las condiciones estudiadas. El índice de migración se define como el área bajo una curva que representa el número de neutrófilos migrados frente a la distancia neta de migración por célula.
[0154] Los compuestos inhibidores de PI3K5 de la presente divulgación tienen un efecto sobre la migración de neutrófilos, inhibiendo esta actividad de una manera dependiente de la dosis.
E. Medición de la capacidad bactericida de los neutrófilos
[0155] Dado que los compuestos inhibidores PI3K5 de la presente descripción afectan ciertas funciones de los neutrófilos, si los compuestos afectan la destrucción bacteriana mediada por neutrófilos es de interés. El efecto de los compuestos sobre la muerte de Staphylococcus aureus mediada por neutrófilos se estudia de acuerdo con el método descrito por Clark y Nauseef (págs. 7,23,4-7,23,6 en Vol. 2, Sup. 6, Curr. Protocols Immunol. (Eds., Colligan). et al.) (1994)). Se mezclan neutrófilos humanos purificados (5 x 106 células/ml) (tratados con DMSO o con una dilución en serie del presente compuesto en DMSO) con suero autólogo. Las células de S. aureus cultivadas durante la noche se lavan, se resuspenden en HBSS y se añaden a los trófilos neuopsonizados en suero en una proporción de 10:1. Se permite que los neutrófilos internalicen las bacterias mediante fagocitosis mediante incubación a 37°C durante 20 min. Las bacterias no internalizadas se destruyen con 10 unidades/ml de lisostafina a 37°C durante 5 min y la mezcla total se rota a 37°C. Las muestras se extraen en varios momentos durante hasta 90 min y los neutrófilos se lisan mediante dilución en agua. Bacterias viables se cuentan en placas diluciones apropiadas en la placa de tripticasa-soja-agar y contando el S. aureus después del crecimiento durante la noche.
[0156] La muerte mediada por neutrófilos de S. aureus es similar en muestras tratadas con DMSO (control) y con un compuesto presente. Por lo tanto, un inhibidor PI3K5 no afecta significativamente a la capacidad de los neutrófilos para matar S. aureus, lo que sugiere que PI3K5 no está involucrado en esta vía de función de los neutrófilos.
EJEMPLO 3
Caracterización del papel de PI3K5 en función de los linfocitos B
[0157] Los efectos de un inhibidor de la PI3-quinasa en las funciones de células B, incluyendo los índices clásicos, tales como la producción de anticuerpos y la proliferación inducida por estímulos específicos también se estudian.
A. Preparación y estimulación de células B de sangre periférica humana
[0158] La sangre heparinizada (200 ml) de voluntarios sanos se mezcla con un volumen igual de D-PBS, en capas sobre 10 x 10 ml de Ficoll-PAQUE® (Pharmacia), y centrifugado a 1600 rpm durante 30 min a temperatura ambiente. Se recogen células mononucleares de sangre periférica (PBMC) de la interfaz FICOLL®/suero, se superponen en 10 ml de suero fetal bovino (FBS) y se centrifugan a 800 rpm durante 10 min para eliminar las plaquetas. Después del lavado, las células se incuban con DYNAL® Antibody Mix (kit de células B) (Dynal Corp., Lake Success, NY) durante 20 min a 4-8°C. Después de la eliminación del anticuerpo no unido, los PBL se mezclan con perlas magnéticas recubiertas de IgG anti-ratón (Dynal) durante 20 min a 4-8°C con agitación suave seguida de la eliminación de las células no B marcadas en el separador de perlas magnéticas. Este procedimiento se repite una vez más. Las células B se resuspenden en RPMI-1640 con FBS al 10% y se mantienen en hielo hasta su uso posterior.
B. Medición de la producción de anticuerpos por las células B humanas
[0159] Para estudiar la producción de anticuerpos, las células B se dividieron en alícuotas en 50-75 x 103 células/pocillo en placa de 96 pocillos con o sin inhibidor, a la que IL-2 (100 U/ml) y PANSORBIN® (Calbiochem) se añadieron células de Staphylococcus aureus (1:90.000). Parte del medio se retira después de 24-36 horas y se añade medio fresco (con o sin inhibidor) e IL-2. Los cultivos se incuban a 37°C, en presencia de una incubadora de CO2 durante 7 días adicionales. Se extraen muestras de cada condición (por triplicado) y se analizan en busca de IgG e IgM, según se mide mediante ELISA. Brevemente, las placas IMMULON® 4 de 96 pocillos se recubren (50 pL/pocillo) con 150 ng/ml de IgG antihumana de burro (H L) (Jackson ImmunoResearch, West Grove PA) o 2 pg/ml de IgG antihumana de burro IgM (H L) (Jackson ImmunoResearch) en tampón de bicarbonato y se dejó durante la noche a 4°C. Después de lavar tres veces con solución salina tamponada con fosfato que contiene 0,1% TWEEN®-80 (PBST) (350 pL/pocillo), y bloquear con suero de cabra al 3% en PBST (100 pL/pocillo) durante 1 hora a temperatura ambiente, se añaden muestras (100 pl/pocillo) de medio gastado de células B diluido en PBST. Para las placas de IgG, el rango de dilución es de 1:500 a 1:10000 y para IgM de 1:50 a 1:1000. Después de 1 hora, las placas se exponen a IgG antihumana conjugada con biotina (100 ng/ml) o IgM antihumana (200 ng/ml) (Jackson ImmunoResearch) durante 30 minutos, seguido de estreptavidina-HRP (1:20000) durante 30 minutos, y finalmente, a una solución de TMB (1:100) con H2O2 (1:10000) durante 5 min, con 3 x PBST lavados entre pasos. El desarrollo del color se detiene con una solución de H2SO4 y las placas se leen en un lector de placas ELISA.
[0160] Los compuestos de la presente divulgación inhibieron la producción de anticuerpos.
C. Medición de la proliferación de células B en respuesta a la estimulación de superficie de la célula IgM
[0161] En el experimento anterior, las células B son estimuladas usando PANSORBIN®. También se midió el efecto de los compuestos de la presente divulgación sobre la respuesta de proliferación de células B cuando se estimulan a través de su IgM de superficie celular usando anticuerpo anti-IgM. Los esplenocitos murinos (Balb/c) se sembraron en pocillos de microtitulación de 96 placas a 2 x 105 células por pocillo en 10% FBS/RPMI. Se añaden a las células diluciones apropiadas de inhibidor de prueba en medio completo y las placas se incuban durante 30-60 minutos antes de la adición del estímulo. Después de la preincubación con el inhibidor de la prueba, se añade a los pocillos una preparación F(ab’ )2 de anticuerpo de cabra específico para la cadena p de IgM de ratón a una concentración final de 25 pg/ml. Las placas se incuban a 37°C durante 3 días y se añade 1 pCi de [3H]-timidina a cada pocillo durante las últimas cuatro horas de cultivo. Las placas se recolectan en filtros de fibra, se lavan y se determina la incorporación de radiomarcaje usando un contador beta (Matrix 96, Packard Instrument Co., Downers Grove, IL) y se expresa como recuentos por minuto (CPM).
[0162] Los compuestos de la presente divulgación inhiben la proliferación de células B estimulada por anti-IgM de una manera dependiente de la dosis. Debido a que los compuestos de la presente divulgación inhiben la proliferación de células B, se prevé que estos compuestos y otros inhibidores de PI3K5 podrían usarse para suprimir la proliferación indeseable de células B en entornos clínicos. Por ejemplo, en la neoplasia maligna de células B, las células B de diversas etapas de diferenciación muestran una proliferación no regulada. Basándose en los resultados mostrados anteriormente, se puede inferir que los inhibidores selectivos de PI3K5 podrían usarse para controlar, limitar o inhibir el crecimiento de tales células.
EJEMPLO 4
Caracterización del papel de PI3K5 en función de los linfocitos T
[0163] Se mide la proliferación de células T en respuesta a la coestimulación de células CD3+CD28. Las células T se purifican a partir de sangre humana sana mediante selección negativa utilizando perlas magnéticas recubiertas de anticuerpo de acuerdo con el protocolo del fabricante (Dynal) y se resuspenden en RPMI. Las células se tratan con DMSO o una dilución en serie de un compuesto presente en DMSO y se sembraron a 1 x 105 células/pocillo en una placa de 96 pocillos previamente recubierta con anticuerpo de cabra anti-ratón IgG. A continuación, se añaden anticuerpos monoclonales de ratón anti-CD3 y anti-CD28 a cada pocillo a 0,2 ng/ml y 0,2 pg/ml, respectivamente. La placa se incuba a 37°C durante 24 horas y se añade [3H]-timidina (1 pCi/pocillo). Después de otra incubación de 18 horas, las células se recogen con un recolector de células automático, se lavan y se cuantifica la radiactividad incorporada.
[0164] Aunque los presente compuestos de inhibidor PI3K5 inhibieron la proliferación de T células inducida por anti-CD3 y por anti-CD28, un efecto no es tan fuerte como un efecto sobre las células B o en algunas de las funciones de los neutrófilos. Por consiguiente, los presentes compuestos no son tóxicos para las células en general.
EJEMPLO 5
Caracterización del papel de PI3K5 en la función de osteoclastos
[0165] Para analizar el efecto de los presentes compuestos inhibidores PI3K5 sobre los osteoclastos, las células de médula ósea de ratón se aislan y se diferencian para los osteoclastos por tratamiento de las células con factor-1 estimulante de colonias de macrófagos (mCSF-1) y ligando de osteoprotegerina (OPGL) en medio que contiene suero (aMEM con FBS inactivado por calor al 10%; Sigma) durante 3 días. El día cuatro, cuando se han desarrollado los osteoclastos, se retira
el medio y se recolectan las células. Los osteoclastos se siembran en rodajas de dentina en 105 células/pocillo en medio de crecimiento, es decir, aMEM que contiene 1% de suero y 2% de BSA con 55 pg/ml OPGL y 10 ng/ml M-CSF-1. Después de 3 horas, el medio se cambia a suero al 1% y BSA al 1%, con o sin osteopontina (25 pg/ml) y los inhibidores de PI3K (100 nM). El medio se cambia cada 24 horas con osteopontina fresca y los inhibidores. a las 72 h, se retira el medio y las superficies de dentina se lavan con agua para eliminar los restos celulares y se tiñen con hematoxilina ácida. Se lava el exceso de tinción y se cuantifican las profundidades de las fosas mediante microscopía confocal.
[0166] Los presentes inhibidores de la PI3-quinasa tuvieron un efecto inhibidor sobre la función de los osteoclastos. Tanto los inhibidores inespecíficos LY2940C2 como la wortmanina inhibieron la actividad de los osteoclastos. Sin embargo, los presentes compuestos inhibidores de PI3K5 tuvieron un efecto mayor y, en algunos casos, inhibieron casi por completo la actividad de los osteoclastos.
EJEMPLO 6
Caracterización del papel de PI3K5 en la función de los basófilos
[0167] La evaluación del efecto de un compuesto de la divulgación en la función de los basófilos se prueba utilizando un ensayo convencional de liberación de histamina, generalmente de acuerdo con el método descrito en Miura et al., J Immunol., 162: 4198 - 206 (1999). Brevemente, se preincuban basófilos enriquecidos con compuestos de prueba a varias concentraciones de 0,1 nM a 1000 nM durante 10 min a 37°C. A continuación, se añade IgE policlonal antihumana de cabra (0,1 pg/ml) o fMLP y se deja incubar durante 30 min más. La histamina liberada en el sobrenadante se mide usando una técnica fluorométrica automatizada.
[0168] Se observó una disminución dependiente de la dosis en la liberación de histamina para los presentes compuestos cuando los basófilos se estimulan con anti-IgE. Esta supresión de la liberación de histamina fue esencialmente del 100% a 1.000 nM. El presente compuesto no provocó ningún efecto cuando los basófilos se estimulan con fMLP. A modo de comparación, el inhibidor no selectivo de PI3K LY294002 se prueba a 0,1 nM y 10.000 nM, mostrando una inhibición cercana al 100% de la liberación de histamina a la concentración más alta.
[0169] Esto indica que los presentes inhibidores de actividad de PI3K5 se pueden usar para la liberación de supresión de histamina, que es uno de los mediadores de la alergia. Dado que la actividad de varias PI 3-quinasas es necesaria para el tráfico, secreción y exocitosis de proteínas en muchos tipos de células, lo anterior sugiere que la liberación de histamina por otras células, como los mastocitos, también puede ser interrumpida por inhibidores de PI 3-quinasa delta selectivos.
EJEMPLOS DE SÍNTESIS QUÍMICA
[0170] Los ejemplos específicos no limitantes se proporcionan a continuación. Se entiende en la técnica que se pueden emplear grupos protectores cuando sea necesario de acuerdo con los principios generales de la química sintética. Estos grupos protectores se eliminan en las etapas finales de la síntesis en condiciones básicas, ácidas o hidrogenolíticas fácilmente evidentes para los expertos en la técnica. Empleando la manipulación y protección apropiadas de cualesquiera funcionalidades químicas, la síntesis de compuestos de fórmulas estructurales (I) o (II) no especificadas en el presente documento puede lograrse mediante métodos análogos a los esquemas y procedimientos sintéticos demostrados que se exponen a continuación.
[0171] A menos que se indique lo contrario, todos los materiales de partida se obtuvieron de proveedores comerciales y se utilizaron sin más purificación. Todas las reacciones y fracciones de cromatografía se analizaron mediante cromatografía en capa fina (TLC) en placas de gel de sílice de 250 mm, visualizadas con luz ultravioleta (UV) o tinción con yodo (I2). Los productos e intermedios se purificaron típicamente mediante cromatografía flash o cromatografía líquida de alta resolución de fase inversa.
[0172] Las siguientes abreviaturas se usan en los ejemplos sintéticos: aq (acuoso), h (horas), min (minuto), saturado (saturado), eq (equivalentes), THF (tetrahidrofurano), RT (temperatura ambiente), Et3N (trietilamina), Zn (zinc en polvo metálico), n-BuOH (alcohol n-butílico), n-BuLi (n-butilo litio), t-BuOH (alcohol butílico terciario), NaCl (cloruro sódico), MgSO4 (sulfato de magnesio), BOC (C(=O) OtBu), CDCh (cloroformo deuterado), MtBe (metil terc-butilo éter), H2O (agua), CHCl3 (cloroformo), HCl (ácido clorhídrico)), MeOH (metanol), NaOH (hidróxido de sodio), NaOMe (metóxido de sodio), TFA (ácido trifluoroacético), K2CO3 (carbonato de potasio), SOCl2(cloruro de tionilo), CH2Cl2(cloruro de metileno), EtOAC (acetato de etilo), DMF (dimetilformamida), EtOH (etanol), DMSO (dimetilsulfóxido), NaHCO3 (bicarbonato de sodio), TLC (cromatografía en capa fina), HPLC (cromatografía líquida de alta resolución), espectrometría de masas por ionización por electrospray (ESI-MS) o MS (ES), HOBT (hidroxibenzotriazol), EDC
(etildietilaminopropilcarbodiimida), DIEA (diisopropiletilamina), HOAc (ácido acético), ACCUFLUOR® NFSi (N-fluorobis(fenilsulfonil)amina) y otras abreviaturas estándar similares aquí se utilizan.
PREPARACIÓN DE COMPUESTOS INTERMEDIOS
4-cloro-5-fluoro-7H-p¡rrolof2.3-dlp¡rim¡d¡na (1)
[0173] Una solución de 4-cloro-5-bromo-7H-pirrolo[2.3-d]pirimidina (800 mg, 3,45 mmol) en THF (50 ml) a -78°C se trató con n-BuLi (1,6 M en hexano, 2,2 eq, 7,6 mmol, 4,7 ml) gota a gota y se agitó durante 30 minutos a la misma temperatura. La mezcla se trató con una solución de ACCu Fl UOR® NFSi (2,0 eq, 7 mmol, 2,2 g) en THF (10 ml). La mezcla de reacción se dejó calentar a temperatura ambiente, se agitó durante 10 horas y luego se concentró hasta sequedad. El residuo se disolvió en EtOAc (100 ml), se lavó con agua (3 x 15 ml) y salmuera (15 ml), se secó con Na2SO4, y se purificó por HPLC de fase inversa (columna C18 Luna 10 x 250 mm, 4,7 ml/min, acetonitrilo al 10-90% en agua durante 20 min) para proporcionar el compuesto intermedio 1. ESI-MS m/z = 172,1 (MH+).
[0174] La reacción descrita anteriormente y el compuesto intermedio 1 se muestran abajo.

2-am¡no-6-morfol¡n-4-ilmet¡l-N-fen¡l-benzam¡da (2)
[0175] El compuesto intermedio 2 se preparó de acuerdo con los procedimientos expuestos en los pasos A-F a continuación.
Éster metílico del ác¡do 6-n¡tro-benzo¡co (3)
[0176] Paso A: Una solución de ácido 6-nitro-benzoico en benceno se trató con cloruro de tionilo (2,5 eq) y se agitó a reflujo durante 8 h. Después de la evaporación, el residuo se disolvió en cloroformo y luego se trató con metanol. Después de agitar a reflujo durante 3 h, la mezcla se evaporó para proporcionar el compuesto 3.
Éster metílico del ác¡do 2-bromomet¡l-6-n¡tro-benzo¡co (4)
[0177] Paso B: Una mezcla del compuesto 3 (2 g), N-bromosuccinimida (1,93 g) y peróxido de benzoílo (0,124 g) en tetracloruro de carbono (30 ml) se agitaron a reflujo durante la noche. Después de enfriar, los sólidos se separaron por filtración y el filtrado se concentró para producir el compuesto 4 como un aceite amarillo crudo.
Éster metílico del ácido 2-morfol¡n-4-¡lmetil-6-n¡tro-benzo¡co (5)
[0178] Paso C: Una mezcla del compuesto 4 (3,5 g, material bruto), morfolina (0,99 g), carbonato de potasio (2,89 g), y yoduro de potasio (1,66 g) en DMF (22 ml) se agitó a temperatura ambiente durante la noche. La mezcla de reacción se vertió en agua (30 ml) y se extrajo con acetato de etilo (3 x 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (30 ml), se secaron sobre sulfato de sodio y se concentraron hasta dar un residuo. El material bruto se purificó mediante cromatografía flash (gel de sílice, acetato de etilo al 30-50%/hexanos) para producir el compuesto 5 en forma de un sólido amarillo.
Ácido 2-morfol¡n-4-¡lmetil-6-n¡tro-benzo¡co (6)
[0179] Paso D: Una solución de compuesto 5 (0,85 g) y NaOH (0,304 g) en una mezcla de agua (9 ml), metanol (5 ml) y THF (45 ml) se agitó a reflujo durante 24 h y se concentró hasta sequedad. El residuo se disolvió en agua y la solución se enfrió a 0°C y se ajustó a pH 7 con hidrogenosulfato de potasio al 10%. La mezcla se concentró a sequedad, el residuo se trató con metanol y se filtró para eliminar los sólidos. El filtrado se concentró para producir el compuesto 6 como un sólido amarillo.
2-morfol¡n-4-¡lmet¡l-6-n¡tro-N-fenil-benzam¡da (7)
[0180] Paso E: Una solución de compuesto 6 (0,62 g, 2,3 mmol) en THF (50 ml) se trató con cloruro de tionilo (0,94 ml), se agitó a temperatura ambiente durante 4 h y se evaporó hasta sequedad. El residuo se disolvió en THF (50 ml) y se trató con anilina (0,58 ml, 6,4 mmol) y diisopropiletilamina (1,12 ml, 6,4 mmol). La mezcla de reacción se agitó durante 4 h, se concentró, se disolvió en diclorometano (50 ml), se lavó con bicarbonato de sodio acuoso saturado (25 ml) y salmuera (25 ml), se secó con sulfato de sodio y se concentró. A continuación, se purificó el residuo bruto del compuesto 7 mediante cromatografía flash (metanol al 5% en diclorometano).
2-am¡no-6-morfol¡n-4-¡lmetil-N-fen¡l-benzam¡da (2)
[0181] Paso F: Una solución de compuesto 7 (0,3 g) y 10% Pd/C (30 mg) en metanol (35 ml) se agitó bajo hidrógeno (1 atm) durante 1,5 h. La mezcla se filtró a través de un lecho de CELITE® y el filtrado se concentró para proporcionar un producto sólido amarillo, compuesto intermedio 2. ESI-MS m/z = 312 (MH+).
[0182] Los pasos A-F y los compuestos 2-7 se muestran en el siguiente esquema de reacción.

3- morfol¡n-4-¡lmetil-fen¡lam¡na (8)
[0183] El compuesto intermedio 8 (que se muestra a continuación) se preparó de acuerdo con los procedimientos establecidos en los pasos A y B.
4- (3-n¡trobenc¡l)morfol¡na (9)
[0184] Paso A: Se cargó un matraz de fondo redondo de una boca de 250 ml equipado con un agitador magnético y un condensador de reflujo con 1-clorometil-3-nitrobenceno (10,0 g, 58,3 mmol), morfolina (15,0 g, 175 mmol) y tolueno (75 ml) y la solución se calentó a reflujo durante 2,5 h. La mezcla de reacción se dejó enfriar a temperatura ambiente y luego se lavó con hidróxido de sodio acuoso 1N (2 x 50 ml). La capa acuosa se extrajo con acetato de etilo (3 x 50 ml) y los extractos combinados se secaron sobre sulfato de sodio y se concentraron a presión reducida para producir el compuesto 9 como un sólido blanquecino. 1H RMN (CDCh) 5 (ppm) 8,22 (s, 1H), 8,12 (d, 1H, J = 8,2 Hz), 7,68 (d, 1H, J = 7,6 Hz), 7,49 (t, 1H, J = 7,9 Hz), 3,73 (m, 4H), 3:59 (s, 2H), 2,46 (m, 4H).
3-morfol¡n-4-¡lmet¡l-fen¡lam¡na (8)
[0185] Paso B: A 250 ml, de tres bocas, de fondo redondo equipado con un agitador y condensador de reflujo mecánico se cargó con el compuesto 9 (12,7 g, 57,4 mmol), polvo de hierro (40,0 g, 71,7 mmol), ácido clorhídrico 2N (20 ml) y etanol (75 ml). La suspensión resultante se calentó a reflujo durante 2 h. Después de este tiempo, la mezcla se dejó enfriar y se filtró a través de un lecho de CELITE® 521. El filtrado se concentró a presión reducida, el residuo se diluyó con agua (100 mL) y se basificó con carbonato de potasio sólido a pH 10. La mezcla se extrajo con acetato de etilo (3 x 75 ml) y los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro y se concentraron a presión reducida para producir el compuesto 8 como un aceite viscoso oscuro. 1H RMN (CDCh) 5 (ppm) 7,09 (t, 1H, J = 8,1 Hz), 6,70 (m, 2H), 6,58 (m, 1H), 3,70 (m, 4H), 3,40 (bs, 2H), 2,44 (m, 4H). El compuesto intermedio 8 se muestra a continuación.

6-bromo-9-(2-tr¡met¡l-etox¡met¡l)-9H-pur¡na (10)
[0186] A 500 ml, de una boca, matraz de fondo redondo equipado con un agitador magnético, se cargó con 6-bromopurina (10,4 g, 52,0 mmol), carbonato de potasio (21,5 g, 156 mmol), tamices moleculares 4Á (22,6 g), dimetilformamida (200 ml) y cloruro de 2-(trimetilsilil)etoximetilo (13,2 g, 78,0 mmol). La mezcla de reacción se agitó durante 18 h a temperatura ambiente, luego se filtró con la ayuda de CELITE®521. La concentración del filtrado a alto vacío seguido de cromatografía en columna dio un rendimiento del 57% del compuesto intermedio 10 en forma de un sólido blanquecino. pf 49-52°C; 1H RMN (DMSO-afe) 5 (ppm) 8,95 (s, 1H), 8,86 (s, 1H), 3,69 (t, 2H, J = 8,0 Hz), 0,93 (t, 2H, J = 8,2 Hz), 0,08 (s, 9H); m/z = 330 (M+H). El compuesto intermedio 10 se muestra a continuación.

2-amino-6-bromo-9-(2-trimetilsililetoxi)metilpurina (11)
[0187] El compuesto intermedio 11 se preparó usando el método descrito anteriormente con respecto a intermedio compuesto 10, pero 2-amino-6-bromopurina se utilizó en lugar de 6-bromopurina. El compuesto intermedio 11 se muestra a continuación.

2-di-ferc-butiloxicarbonilamino-6-bromo-9-(2-trimetilsililetoxi)metil-9H-purina (12)
[0188] Un matraz de fondo redondo de 250 ml, de una boca, equipado con un agitador magnético se purgó con nitrógeno y cargado con compuesto intermedio 11 (11,7 g, 34,0 mmol), dicarbonato de di-ferc-butilo (22,2 g, 102 mmol), DMAP (581 mg, 4,76 mmol) y tetrahidrofurano anhidro (150 ml). La mezcla de reacción se agitó durante 18 h a temperatura ambiente y luego se evaporó hasta sequedad. La cromatografía en columna del residuo resultante proporcionó el compuesto intermedio 12 como un sólido blanco. p.f. 9395°C; 1H RMN (DMSO-afe) 5 (ppm) 8,99 (s, 1H), 5,71 (s, 2H), 3,66 (t, 2h , J = 8,0 Hz), 1,47 (s, 18H), 0,92 (t, 2H, J = 8,2 Hz); m/z = 546 (M+H). El compuesto intermedio 12 se muestra a continuación.

6-cloro-9-(2-tr¡met¡ls¡lanil-etox¡met¡l)-9H-pur¡na (13)
[0189] El compuesto intermedio 13 se preparó usando el método descrito anteriormente con respecto al compuesto intermedio 10, pero se usó 6-cloropurina en lugar de 6-bromopurina. El compuesto intermedio 13 se muestra a continuación.

PROCEDIMIENTOS GENERALES
[0190] Los compuestos de la presente divulgación se pueden preparar por los métodos siguientes. Se preparan compuestos adicionales usando uno de los siguientes métodos y seleccionando materiales de partida y reactivos apropiados. Los procedimientos generales y los procedimientos específicos para sintetizar los presentes compuestos también se establecen en la Patente de Estados Unidos N° 6,518,277.
EJEMPLO 8
PREPARACIÓN DE COMPUESTOS
[0191] Los compuestos de acuerdo con la fórmula general I (se muestra más arriba) se han preparado de acuerdo con los ejemplos de los procedimientos sintéticos descritos a continuación.
5-met¡l-3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)prop¡l]-3H-qu¡nazol¡n-4-ona (14)
[0192] El compuesto (14) se preparó de acuerdo con el procedimientos establecidos en los pasos A-D a continuación.
2-am¡no-6-met¡l-N-fen¡l-benzam¡da (15)
[0193] Paso A: cloruro de tionilo (14,5 ml, 198 mmol) se añadió a una solución de ácido 2-amino-6-metilbenzoico (10,0 g, 66,1 mmol) en benceno (250 mL). La suspensión resultante se calentó a reflujo y se agitó durante la noche. Después de enfriar, la reacción se concentró al vacío y el residuo resultante se disolvió en cloroformo (300 ml). Se añadió anilina (15 ml, 165 mmol) y la mezcla se calentó a reflujo. Después de tres horas, se dejó enfriar la reacción y se filtró la suspensión resultante. El filtrado se sometió a cromatografía flash y el producto bruto se recristalizó en isopropanol para proporcionar el compuesto 15 en forma de un sólido cristalino amarillo. MS (EN): m/z 227 (M+H), 134. 1H Rm N (300 MHz, DMSO-d6) 5 10,26 (s, 1H), 7,75 (d, 2H, J = 7,9 Hz), 7,32 (t, 2H, J = 7,8 Hz), 7,07 (t, 1H, J = 7,3 Hz), 7,00 (t, 1H, J = 7,8 Hz), 6,58 (d, 1H, J = 8,1 Hz), 6,46 (d, 1H, J = 7,4 Hz), 4,98 (br. s, 2H), 2,21 (s, 3H). La reacción descrita anteriormente y el compuesto 15 se muestran a continuación.

Éster bencílico de ác¡do [1-(5-met¡l-4-oxo-3-fen¡l-3.4-d¡h¡droqu¡nazol¡n-2-¡l)prop¡n-carbám¡co (16)
[0194] Paso B: El compuesto 15 (1,20 g, 5,30 mmol) se combinó con ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo (2,13 g, 6,36 mmol), dimetilaminopiridina (915 mg, 7,49 mmol) y diisopropiletilamina (1,10 ml, 6,36 mmol) en tolueno (15 ml). La mezcla se calentó a 110°C y se agitó a esa temperatura durante 22 horas. Después de enfriar, la reacción se purificó mediante cromatografía flash para proporcionar la quinazolinona (16) como un sólido amarillo pálido. MS (EN): m/z 428 (M+H). 1H RMN (300 MHz, DMSO-d6) 5 10,26 (s, 1H), 7,75 (d, 2H, J = 7,9 Hz), 7,32 (t, 2H, J = 7,8 Hz), 7,07 (t, 1 H, J = 7,3 Hz), 7,00 (t, 1H, J = 7,8 Hz), 6,58 (d, 1H, J = 8,1 Hz), 6,46 (d, 1H, J = 7,4 Hz), 4,98 (br. s, 2H), 2,21 (s, 3H). La reacción descrita anteriormente y el compuesto 16 se muestran a continuación.

2-(1-am¡no-prop¡l)-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (17)
[0195] Paso C: Se añadió una cantidad catalítica de 10% de Pd/C a una solución de compuesto 16 (691 mg, 1,62 mmol) en etanol (8 ml). La mezcla resultante se colocó bajo una atmósfera de hidrógeno (presión de balón) y se dejó agitar a temperatura ambiente durante la noche. La reacción se filtró y el filtrado se concentró al vacío. El residuo bruto resultante se purificó mediante cromatografía flash para producir la amina libre, compuesto 17, como un aceite amarillo pálido. (ES): m/z 294 (M+H), 237. 1H RMN (300 MHz, DMSO-d6) 5 7,68 (t, 1H, J = 7,7 Hz), 7,49 - 7,63 (m, 3H), 7,43 - 7,49 (m, 1H), 7,36 - 7,43 (m, 1H), 7,19 - 7,34 (m, 2H), 4,03 - 4,19 (m, 1H), 2,72 (s, 3H), 2,04 (br. s, 2H), 1,63 -1,79 (m, 1H), 1,29 - 1,44 (m, 1H), 0,68 (t, 3H, J = 7,3 Hz). La reacción descrita anteriormente y el compuesto 17 se muestran a continuación.

[0196] Paso C (procedimiento alternativo): Ácido trifluoroacético se añadió a una solución del compuesto de amina
protegida con Boc 16 en diclorometano. La solución resultante se dejó agitar a temperatura ambiente hasta que LCMS o TLC indicaron el consumo completo del material de partida. La reacción se concentró a vacío, y el residuo se purificó por cromatografía flash para proporcionar la amina libre 17.
5-metil-fen¡l-2-3 [1-(9H-pur¡n-6-¡lam¡no)prop¡l]-3H-qu¡nazol¡n-4-ona (14)
[0197] Paso D: El compuesto 17 (100 mg, 0,341 mmol) se combinó con purina 6-bromo (75 mg, 0,375 mmol) y diisopropiletilamina (65uL, 0,375 mmol) en n-butanol (1,0 ml). La reacción se selló, se calentó a 120°C y se agitó durante 18 horas. Se dejó enfriar la solución y luego se concentró al vacío. El residuo resultante se purificó mediante HPLC preparativa para proporcionar el compuesto 14 como un sólido de oliva. (ES): m/z 412 (M+H), 206. 1H RMN (300 MHz, DMSO-d6) 58,96 - 9,06 (br m, 1H.), 8,53 (br s, 1H.), 8,51 (s, 1H), 7,68 (t, 1 H, J = 7,8 Hz), 7,45 - 7,62 (m, 6H), 7,31 (d, 1 H, J = 7,3 Hz), 4,76 - 4,86 (m, 1H), 2,73 (s, 3H), 1,98 - 2,11 (m, 1H), 1,76 - 1,94 (m, 1H), 0,79 (t, 3H, J = 7,2 Hz). La reacción descrita anteriormente y el compuesto 14 se muestran a continuación.

2-[1-(2-fluoro-9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (18)
[0198] El compuesto 18 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero se usó 2-fluoro-6-cloropurina en lugar de 6-bromopurina en el paso D. ESI-MS m/z 416,1 (Mh ). El compuesto 18 se muestra a continuación.

3-(2.6-d¡fluoro-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (19)
[0199] Compuesto 19 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero se usó 2,6-difluorolanilina en lugar de anilina en el paso A, se utilizó el ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1 -ilo sustituido por éster de 2,5-dioxo-pirrolidin-1 -ilo del ácido 2-benciloxicarbonilaminobutírico en el paso B, y se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C. ESI-MS m/z 434,1 (MH+). El compuesto 19 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-(2.6-d¡fluorofen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (20)
[0200] En el compuesto 20 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero 2,6-difluoroanilina fue sustituida por anilina en el paso A, ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-amino-6-bromopurina por 6-bromopurina en Paso D. ESI-MS m/z 449,5 (MH+). El compuesto 20 se muestra a continuación.

3-(2.6-d¡fluoro-fen¡l)-2-[1-(2-fluoro-9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-3H-qu¡nazol¡n-4-ona (21)
[0201] El compuesto 21 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero la anilina se sustituyó por 2,6-difluoroanilina en el paso A, ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por el éster de 2,5-dioxo-pirrolidin-1-ilo del ácido 2-benciloxicarbonilaminobutírico en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó la 2-fluoro-6-cloropurina por 6-bromopurina en el paso D. 1H RMN (MeOH-d4): 5 8,22-8,17 (m, 1H); 7,76-7,62 (m, 2H); 7,47-7,45 (m, 1H); 7,38 7,33 (m, 1,2 H); 7,30-7,17 (m, 1H); 6,88-6,75 (m, 1H); 5,30-5,27 (m, 0,5 H); 5,09-5,07 (m, 0,5 H); 2,778 (s, 3H); 1,62-1,50 (m, 3H). El compuesto 21 se muestra a continuación.

3-(2.6-d¡fluoro-fen¡l)-5-met¡l-2-[1-(7 H -p¡rrolo[2.3-d]p¡r¡m¡d¡n-4-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (22)
[0202] El compuesto 22 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero 2,6-difluoroanilina fue sustituida por anilina en el paso A, ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y 4-cloro-7H-pirrolo[2.3-d]pirimidina se sustituyó por 6-bromopurina en el paso D. ESI-MS m/z 433 (MH+). El compuesto 22 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)prop¡l]-5-met¡l-3-fenil-3H-qu¡nazol¡n-4-ona (23)
[0203] El compuesto 23 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero 2-amino-6-bromopurina se sustituyó por 6-bromopurina en el paso D. MS (ES): m/z 427 (M+H), 214. El compuesto 23 es mostrado a continuación.

5-metil-3-fenil-2-[1-(7H-pirrolo[2.3-d]pirimid¡n-4-ilamino)propin-3H-quinazolin-4-ona (24)
[0204] El Compuesto 24 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero se sustituyó 7-deaza-6-cloropurina por 6-bromopurina en el paso D. MS (ES): m/z 411 (M+H), 206. El compuesto 24 se muestra a continuación.

2-[1-(2-fluoro-9H-pur¡n-6-¡lam¡no)-prop¡n-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (25)
[0205] El compuesto 25 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero se sustituyó 2-fluoro-6-cloropurina por 6-bromopurina en el paso D. MS (ES): m/z 430 (M+H), 446. El compuesto 25 se muestra a continuación.

5-met¡l-3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-quinazol¡n-4-ona (26)
[0206] El compuesto 26 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1 -ilo fue sustituido por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxopirrolidin-1-ilo en el paso B, y se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C. MS (ES): m/z 398 (M+H). El compuesto 26 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (27)
[0207] Se preparó el compuesto 27 usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1-ilo fue sustituido por éster 2,5-dioxopirrolidin-1-ilo ácido 2-benciloxicarbonilaminobutírico en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS m/z 413,1 (MH+). El compuesto 27 se muestra a continuación.

2-[2-benc¡lox¡-1-(9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-3-fen¡l-3H-quinazol¡n-4-ona (28)
[0208] El compuesto 28 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 3-benciloxi-2-terc-butoxicarbonilaminopropiónico éster 2,5-dioxo-pirrolidin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se utilizó en el paso C. MS (ES): m/z 504 (M+H), 396, 261. El compuesto 28 se muestra a continuación.

2-[1-(2-amino-9H-purin-6-ilamino)-2-bencilox¡-etin-5-metil-3-fenil-3H-quinazolin-4-ona (29)
[0209] El compuesto 29 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 3-benciloxi-2-terc-butoxicarbonilaminopropiónico éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y 2-amino-6-bromopurina se sustituyó por 6-bromopurina en el paso D. MS (ES): m/z 519 (M+H), 411,261. El compuesto 29 se muestra a continuación.

2-[2-benc¡lox¡-1-(7H-p¡rrolo[2.3-d]p¡r¡m¡d¡n-4-¡lam¡no)-et¡l]-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (30)
[0210] El compuesto 30 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 3-benciloxi-2-terc-butoxicarbonilaminopropiónico éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 4-cloro-7H-pirrolo[2.3-d]pirimidina por 6-bromopurina en el paso D. MS (EN): m/z 503 (M+H), 395. El compuesto 30 se muestra a continuación.

2-[(2-fluoro-9H-1-2-benciloxi-purin-6-ilamino)-etin-5-metil-3-fenil-3H-quinazolin-4-ona (31)
[0211] El compuesto 31 se preparó utilizando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 3-benciloxi-2-terc-butoxicarbonilaminopropiónico éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y 2-fluoro-6-cloropurina se sustituyó por 6-bromopurina en el paso D. MS (ES): m/z 522 (M+H), 414, 261. El compuesto 31 se muestra a continuación.

3-(4-fluoro-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (32)
[0212] El compuesto 32 fue preparado usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero, con los siguientes cambios: 4-fluoroanilina se sustituyó por anilina en el paso A, ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1 -ilo fue sustituido por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y se usó el procedimiento alternativo (desprotección de TFA) en el paso C. ESI-MS m/z 416,1 (MH+). El compuesto 32 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-(4-fluorofen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (33)
[0213] El compuesto 33 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero se sustituyó 4-fluoroanilina por anilina en el paso A, 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS m/z 431,1 (MH+). El compuesto 33 se muestra a continuación.

3-(4-fluoro-fen¡l)-2-[1-(2-fluoro-9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-3H-qu¡nazol¡n-4-ona (34)
[0214] El compuesto 34 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero, en el paso A, se sustituyó 4-fluoroanilina por anilina, se sustituyó el ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1 -ilo por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-fluoro-6-cloropurina por 6-bromopurina en el paso D. ESI-MS m/z 434,1 (MH+). El compuesto 34 se muestra a continuación.

3-(4-fluoro-fen¡l)-5-met¡l-2-[1-(7H-p¡rrolo[2.3-d]p¡r¡m¡d¡n-4-¡lam¡no)-et¡n-3H-qu¡nazol¡n-4-ona (35)
[0215] El compuesto 35 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero en el paso A se sustituyó 4-fluoroanilina por anilina, ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y 4-cloro-7H-pirrolo[2.3-d]pirimidina sustituido por 6-bromopurina en el paso D. ESI-MS m/z 415,1 (MH+). El compuesto 35 se muestra a continuación.

5-met¡l-3-fen¡l-2-[1-(7H-p¡rrolo[2.3-d]p¡r¡m¡d¡n-4-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (36)
[0216] El compuesto 36 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y 4-cloro-7H-pirrolo[2.3-d]pirimidina se sustituyó por 6-bromopurina en el paso D.

3-(3-fluoro-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (37)
[0217] El compuesto 37 fue preparado usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero se sustituyó la anilina por 3-fluoroanilina en el paso A, el ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se utilizó en el paso C. 1H RMN (dmso-d6): 8,30-8,28 (m, 2h ); 7,70 7,49 (m, 3H); 7,44-7,30 (m, 4h ); 4,91-4,88 (m, 1H); 2,72-2,68 (m, 3H); 1,50-1,48 (m, 3H). El compuesto 37 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡n-3-(3-fluorofen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (38)
[0218] El compuesto 38 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero se sustituyó 3-fluoroanilina por anilina en el paso A, el ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se usó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. 1H RMN (DMSO-d6): 8,16-8,12 (m, 1H); 7,74-7,69 (m, 1H); 7,62-7,53 (m, 2H); 7,46-7,12 (m, 6H); 4,96 (bs, 1H); 2,74-2,67 (m, 3H); 1,47-1,45 (m, 3H). El compuesto 38 se muestra a continuación.

3-(3-fluoro-fen¡l)-5-met¡l-2-[1-(7H-p¡rrolo[2.3-d]p¡r¡m¡d¡n-4-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (39)
[0219] El compuesto 39 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero en el paso A se sustituyó 3-fluoroanilina por anilina, ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y 4-cloro-7H-pirrolo[2.3-d]pirimidina se sustituyó por 6-bromopurina en el paso D. ESI-MS m/z 415,1 (MH+). El compuesto 39 se muestra a continuación.

5-met¡l-3-fen¡l-2-[1-(9H-pur¡n-6-¡l)-p¡rrol¡d¡n-2-¡n-3H-qu¡nazol¡n-4-ona (40)
[0220] El compuesto 40 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero ácido pirrolidin-1,2-dicarboxí’lico éster 1-ferc-butílico éster 2-(2,5-dioxo-pirrolidin-1 -ilo) sustituido por ácido 2-benciloxicarbonilaminobutírico, éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y se usó el procedimiento alternativo (desprotección de TFA) en el paso C. MS (ES): m/z 424 (M+H), 212. El compuesto 40 se muestra a continuación.

2-[2-h¡drox¡-1-(9H-pur¡n-6-¡lam¡no)-et¡n-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (41)
[0221] Una suspensión del compuesto 28 (60 mg, 0,097 mmol), Pd(OH)2 (cat.), y Na2CO3 acuosa (0,5 ml) en etanol (2,5 ml) se hidrogenó a 45psi durante 14 días. La mezcla se filtró para eliminar los disolventes y el filtrado resultante se purificó por HPLC para proporcionar el producto 41 como un sólido blanco. MS (EN): m/z 414 (M+H), 396, 261. El compuesto 41 se muestra a continuación.

5-met¡l-3-fen¡l-2-[fen¡l-(9H-pur¡n-6-¡lam¡no)-met¡l]-3H-qu¡nazol¡n-4-ona (42)
[0222] El compuesto 42 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido ferc-butoxicarbonilamino-fenil-acético éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, y se usó el procedimiento alternativo (desprotección de TFA) en el paso C. MS (ES): m/z 460 (M+H), 325. El compuesto 42 se muestra a continuación.

2-[(2-am¡no-9H-pur¡n-6-¡lam¡no)-phonyl-met¡n-5-met¡l-3-fen¡l-3H-quinazol¡n-4-ona (43)
[0223] El compuesto 43 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido ferc-butoxicarbonilamino-fenil-acético éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. MS (ES): m/z 475 (M+H). El compuesto 43 se muestra a continuación.

2-[(2-fluoro-9H-pur¡n-6-¡lam¡no)-fen¡l-met¡n-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (44)
[0224] El compuesto 44 se preparó utilizando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero se sustituyó el ácido ferc-butoxicarbonilamino-fenil-acético éster 2,5-dioxo-pirrolidin-1-ilo por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-fluoro-6-cloropurina por 6-bromopurina en el paso D. MS (ES): m/z 478 (M+H), 325. El compuesto 44 se muestra a continuación.

5-met¡l-3-fen¡l-2-[fen¡l-(7H-p¡rrolo[2.3-d]p¡r¡m¡d¡n-4-¡lam¡no)-met¡l]-3H-qu¡nazol¡n-4-ona (45)
[0225] El compuesto 45 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido ferc-butoxicarbonilamino-fenil-acético éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidina-1-ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y 4-cloro-7H-pirrolo[2.3-d]pirimidina se sustituyó por 6-bromopurina en el paso D. MS (ES): m/z 459 (M+H), 230. El compuesto 45 se muestra a continuación.

5-fluoro-3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡n-3H-qu¡nazol¡n-4-ona (46)
[0226] El compuesto 46 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-amino-6-fluoro-benzoico se sustituyó por ácido 2-amino-6-metil-benzoico en el paso A, ácido 2-fercbutoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C. ESI-MS m/z 402,3 (MH+). El compuesto 46 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡n-5-fluoro-3-fen¡l-3H-qu¡nazol¡n-4-ona (47)
[0227] Se preparó el compuesto 47 usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-amino-6-fluoro-benzoico se sustituyó por ácido 2-amino-6-metil-benzoico en el paso A, ácido 2-fercbutoxicarbonilaminopropiónico éster 2,5-dioxopirrolidina-1-ilo sustituido por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo se utilizó en el paso B, el procedimiento alternativo (TFA desprotección) en el paso C, y 2-amino-6-bromopurina se sustituyó por 6-bromopurina en el paso D. ESI-MS m/z 417,2 (MH+). El compuesto 47 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l1-5-cloro-3-fen¡l-3H-qu¡nazol¡n-4-ona (48)
[0228] El compuesto 48 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-amino-6-clorobenzoico se sustituyó por ácido 2-amino-6-metilbenzoico en el paso A, ácido 2-tercbutoxicarbonilaminopropiónico 2,5-dioxopirrolidin-1-ilo. El ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó la 2-amino-6-bromopurina por 6-bromopurina en el paso D. MS (EN): m/z 433 (M+H), 177. El compuesto 48 se muestra a continuación.

Éster bencílico de ác¡do [5-(5-met¡l-4-oxo-3-fen¡l-3.4-d¡h¡dro-qu¡nazol¡n-2-¡l)-5-(9H-pur¡n-6-¡lam¡no)-pent¡l1-carbám¡co (49)
[0229] El compuesto 49 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 6-benciloxicarbonilamino-2-terc-butoxicarbonilamino-hexanoico éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se usó en el paso C. ESI-MS m/z 589 (MH+). El compuesto 49 se muestra a continuación.

Éster bencílico del ác¡do [5-(2-am¡no-9H-pur¡n-6-¡lam¡no)-5-(5-met¡l-4-oxo-3-fen¡l-3.4-d¡h¡dro-qu¡nazol¡n-2-¡l)-pent¡l1-carbám¡co (50)
[0230] El compuesto 50 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero ácido 6-benciloxicarbonilamino-2-terc-butoxicarbonilamino-hexanoico 2,5-dioxo-pirrolidin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo
(desprotección de TFA) en el paso C, y se sustituyó la 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS

Éster bencílico de ácido [4-(5-met¡l-4-oxo-3-fen¡l-3.4-d¡h¡dro-qu¡nazol¡n-2-¡l)-4-(9H-pur¡n-6-¡lam¡no)but¡l1-carbámico (51)
[0231] El compuesto 51 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 6-benciloxicarbonilamino-2-terc-butoxicarbonilamino-pentanoico éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se usó en el paso C. ESI-MS m/z 575 (MH+). El compuesto 51 se muestra a continuación.

Éster bencílico de ácido f4-(2-am¡no-9H-pur¡n-6-Mam¡no)-4-(5-met¡l-4-oxo-3-fenM-3.4-d¡h¡dro-qu¡nazol¡n-2-¡l)-but¡l1-carbamjco (52)
[0232] El compuesto 52 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero éster de ácido 6-benciloxicarbonilamino-2-terc-butoxicarbonilamino-pentanoico 2,5-dioxo-pirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS m/z 590 (MH+). El compuesto 52 se muestra a continuación.

3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l1-3H-qu¡nazol¡n-4-ona (53)
[0233] El compuesto 53 se preparó usando el procedimiento general descrito anteriormente con respecto a compuesto 14, pero el ácido 2-amino-6-metilbenzoico se sustituyó por ácido 2-amino-6-metilbenzoico en el paso A, el ácido 2-tercbutoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se utilizó en el paso C. MS
(ES): m/z 384,1 (M+H). El compuesto 53 se muestra a continuación.

2-[5-am¡no-1-(9H-pur¡n-6-¡lam¡no)-pent¡l]-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona) (54)
[0234] Una mezcla del compuesto 49 (28,5 mg) y paladio sobre carbono (10 mg, Pd al 10%) en etanol (2 ml) se agitaron con hidrógeno (40 psi) durante 48 h. La mezcla se filtró a través de CELITE® y el filtrado se concentró para producir el producto 54. ESI-MS m/z 455 (MH+). El compuesto 54 se muestra a continuación.

2-[5-am¡no-1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-pent¡n-5-met¡l-3-fen¡l-3H-qu¡nazolin-4-ona (55)
[0235] El compuesto 55 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 54, pero el compuesto 50 se sustituyó por el compuesto 49. ESI-MS m/z 470 (MH+). El compuesto 55 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-(2.6-D¡met¡lfen¡l)-5-met¡l-3H-¡nazol¡n-4-ona (56)
[0236] El compuesto 56 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero 2,6-dimetilanilina sustituido por anilina en el paso A, ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-amino-6-bromopurina por 6-bromopurina en Paso D. ESI-MS m/z 441 (MH+). El compuesto 56 se muestra a continuación.

3-(2.6-d¡met¡l-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (57)
[0237] El compuesto 57 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero 2,6-dimetilanilina se sustituyó por anilina en el paso A, ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por Ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se usó en el paso C. ESI-MS m/z 426 (MH+). El compuesto 57 se muestra a continuación.

5-morfol¡n-4-¡lmet¡l-3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (58)
[0238] El compuesto 58 se preparó usando pasos B, C y D del procedimiento general descrito anteriormente con respecto al compuesto 14, pero el compuesto intermedio 2 se sustituyó por el producto del paso A en el paso B (el paso A no fue necesario), ácido 2-terc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, y se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C. ESI- MS m/z 483 (MH+). El compuesto 58 se muestra a continuación.

2-[1-(2-amino-9H-purin-6-ilamino)-etin-5-morfolin-4ilmetil-3-fenil-3H-quinazolin-4-ona (59)
[0239] El c o m p u e s to 59 se p re p a ró u sa n d o el p ro c e d im ie n to g e n e ra l d e sc rito a n te rio rm e n te con re sp e c to al c o m p u e s to 58, pe ro se su s titu yó 2 -a m in o -6 -b ro m o p u rin a p o r 6 -b ro m o p u rin a en el pa so D. E S I-M S m/z 498 (MH+). El c o m p u e s to 59 se m u e s tra a c o n tin u a c ió n .

2-[4-am¡no-1-(2-am¡no-9H-pur¡n-6-¡lam¡no)but¡l]-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (60)
[0240] El compuesto 60 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 54, pero el compuesto 52 se sustituyó por el compuesto 49. ESI-MS m/z 456 (MH+). El compuesto 60 se muestra a continuación.

6-fluoro-3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡n-3H-qu¡nazol¡n-4-ona (61)
[0241] El compuesto 61 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-amino-5-fluorobenzoico se sustituyó por ácido 2-amino-5-metilbenzoico en el paso A, se sustituyó el ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1 -ilo por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso C, y el procedimiento alternativo (desprotección de TFA) se usó en el paso C. ESI-MS m/z 402 (MH+). El compuesto 61 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-6-fluoro-3-fen¡l-3H-qu¡nazol¡n-4-ona (62)
[0242] El c o m p u e s to 62 se p re p a ró u sa n d o el p ro c e d im ie n to g e n e ra l d e sc rito a n te rio rm e n te con re sp e c to al c o m p u e s to 61, pe ro se su s titu yó 2 -a m in o -6 -b ro m o p u rin a p o r 6 -b ro m o p u rin a en el pa so D. E S I-M S m/z 417 (MH+). El c o m p u e s to 62 se m u e s tra a c o n tin u a c ió n .

2-[2-terc-butox¡-1-(9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (63)
[0243] El compuesto 63 fue preparado usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-benciloxicarbonilamino-3-ferc-butoxipropiónico éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B. m S (ES): m/z 470 (M+H), 396, 261. El compuesto 63 se muestra a continuación.

3-(3-met¡l-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (64)
[0244] El compuesto 64 fue preparado usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero m-toluidina se sustituyó por anilina en el paso A, el ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, y el suplente procedimiento (TFA desprotección) se utilizó en el paso C. 1H RMN (DMsO-d6): 8,41-8,36 (m, 2H); 7,71-7,66 (m, 1H); 7,53-7,51 (m, 5H); 4,97 (m, 1H); 2,72 (s, 3H); 2,39 (s, 1,5 H); 2,09 (s, 1,5 H); 1,50-1,48 (m, 3H). El compuesto 64 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡n-3-(3-met¡lfen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (65)
[0245] El compuesto 65 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero en el paso A se sustituyó m-toluidina por anilina, ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. 1H RMN (DMSO): 8,94-8,92 (m, 1H); 8,18 (s, 1H); 7,75-7,68 (m, 1H); 7,58-7,51 (m, 1H); 5,07 4,96 (m, 1H); 2,79-2,73 (m, 3H); 2,40 (s, 1,5H); 1,91 (s, 1,5H); 1,48-1,43 (m, 3H). El compuesto 65 se muestra a continuación.

3-(3-cloro-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (66)
[0246] El compuesto 66 fue preparado usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero se sustituyó la anilina por 3-cloroanilina en el paso A, el ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se utilizó en el paso C. 1H RMN (dmso-d6): 8,40-8,31 (m, 2H); 7,73 7,66 (m, 1H); 7,55-7,32 (m, 6H); 5,04-4,86 (m, 1H); 2,72 (s, 3H); 1,52-1,50 (m, 3H). El compuesto 66 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-(3-clorofen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (67)
[0247] El compuesto 67 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 66, pero se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS m/z 447,2 (MH+). El compuesto 67 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-2-h¡drox¡-et¡n-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (68)
[0248] El compuesto 68 se preparó utilizando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-benciloxicarbonilamino-3-ferc-butoxipropiónico éster 2,5-dioxo-pirrolidin-1 -ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, y 2-amino-6-bromopurina se sustituyó por 6-bromopurina en el paso D. El obtuvo 2-[1-(2-amino-9H-purin-6-ilamino)-2-ferc-butoxi-etil]-5-metil-3-fenil-3H-quinazolin-4-ona a continuación, se disolvió en ácido trifluoroacético y se dejó en agitación a temperatura ambiente durante 5 horas. La purificación por LC proporcionó el producto 68 como un sólido blanco. MS (EN): m/z 429 (M+H), 215, 206, 151. El compuesto 68 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-(3-fluorofen¡l)-3H-quinazol¡n-4-ona (69)
[0249] El compuesto 69 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero la anilina se sustituyó por 3-fluoroanilina en el paso A, el ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS m/z 417,1 (MH+). El compuesto 69 se muestra a continuación.

2-[1-(2-amino-9H-purin-6-ilamino)-etin-3-(2.6-difluorofenil)-3H-quinazolin-4-ona (70)
[0250] El compuesto 70 fue preparado usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero la anilina se sustituyó por 2,6-difluoroanilina en el paso A, el ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS m/z 435,1 (MH+). El compuesto 70 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)prop¡l]-5-fluoro-3-fenil-3H-qu¡nazol¡n-4-ona (71)
[0251] Se preparó el compuesto 71 usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-amino-5-fluorobenzoico se sustituyó por el ácido 2-amino-5-metilbenzoico en el paso A, y la 6-bromopurina se sustituyó por 2-amino-6-bromopurina en Paso D. ESI-MS m/z 431 (MH+). El compuesto 71 se muestra a continuación.

5-cloro-3-(3-fluoro-fen¡l)-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (72)
[0252] El compuesto 72 fue preparado utilizando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-amino-6-clorobenzoico se sustituyó por ácido 2-amino-6-metilbenzoico y la anilina se sustituyó por 3-fluoroanilina en el paso A, ácido 2-terc-butoxicarbonilamino-propiónico éster 2,5-dioxopirro1 idin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, y se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C. ESI-MS m/z 436,1 (MH+). El compuesto 72 se muestra a continuación.

2- [1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-5-cloro-3-(3-fluoro-fen¡l)-3H-qu¡nazol¡n-4-ona (73)
[0253] El compuesto 73 se preparó utilizando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-amino-6-clorobenzoico se sustituyó por el ácido 2-amino-6-metilbenzoico y la anilina se sustituyó por 3- fluoroanilina en el paso A, ácido 2-terc-butoxicarbonilamino-propiónico éster 2,5-diaxopirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y 2-amino-6-bromopurina se sustituyó por 6-bromopurina en el paso D. ESI-MS m/z 451,1 (MH+). El compuesto 73 se muestra a continuación.

3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡n-5-tr¡fluoromet¡l-3H-qu¡nazol¡n-4-ona (74)
[0254] El compuesto 74 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero 2-amino-W-fenil-6-trifluorometil-benzamida se utilizó en lugar del compuesto 15 en el paso A, ácido 2-tercbutoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1-ilo se se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y se utilizó el procedimiento alternativo (desprotección de TFA). MS (EN): m/z 452 (M+H). El compuesto 74 se muestra a continuación.

[0255] Se preparó 2-amino-W-fenil-6-trifluorometil-benzamida mediante la adición de anilina (1,0 eq.) a una suspensión de trihidrato de ácido 2-amino-6-(trifluorometil)-benzoico (1,0 g, 4,0 mmol, 1,3 eq.) y poliestireno-carbodiimida (3,6 g, 1,1 1,7 eq.) en THF (40 ml). La reacción se agitó a temperatura ambiente durante 18 horas y luego se filtró. El filtrado se concentró al vacío y se purificó mediante cromatografía flash para proporcionar 2-amino-W-fenil-6-trifluorometil-benzamida como un sólido amarillo.
3-(2.6-d¡fluoro-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)prop¡l]-3H-qu¡nazol¡n-4-ona (75)
[0256] El compuesto 75 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero la anilina se sustituyó por 2,6-difluoroanina en el paso A. ESI-MS m/z 448 (MH+). El compuesto 75 se muestra a continuación.

3-(2.6-d¡fluoro-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (76)
[0257] El compuesto 76 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero 2,6-difluoroanilina sustituido por anilina en el paso A, ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C. ESI-MS m/z 434,1 (MH+). El compuesto 76 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-prop¡n-3-(2.6-d¡fluoro-fen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (77)
[0258] El c o m p u e s to 77 se p re p a ró u sa n d o el p ro c e d im ie n to g e n e ra l d e sc rito a n te rio rm e n te con re sp e c to al c o m p u e s to 75, pe ro se su s titu yó 2 -a m in o -6 -b ro m o p u rin a p o r 6 -b ro m o p u rin a en el p a so D. E S I-M S m/z 463 (MH+). El c o m p u e s to 77 se m u e s tra a co n tin u a c ió n .

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-(2.6-d¡fluorofen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (78)
[0259] El compuesto 78 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 76, pero se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS m/z 449 (MH+). El compuesto 78 se muestra a continuación.

3-(3.5-dicloro-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etin-3H-quinazolin-4-ona (79)
[0260] El compuesto 79 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero 3,5-dicloroanilina se sustituyó por anilina en el paso 1, y se utilizó el ácido 2-ferc-butoxicarbcnilaminopropiónico éster 2,5-dioxopirrolidin-1 -ilo sustituido por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C. ESI-MS m/z 467 (MH+). El compuesto 79 se muestra a continuación.

3-(2.6-d¡cloro-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡n-3H-qu¡nazol¡n-4-ona (80)
[0261] El compuesto 80 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero 2,6-dicloroanilina se sustituyó por anilina en el paso A, ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1-il sustituido por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se utilizó en el paso C. ESI-MS m/z 467 (MH+). El compuesto 80 se muestra a continuación.
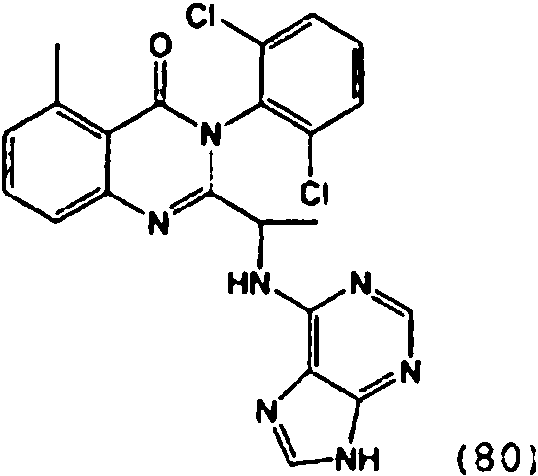
2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-(2.6-d¡clorofen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (81)
[0262] El compuesto 81 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 80, pero se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS m/z 482 (MH+). El compuesto 81 se muestra a continuación.

5-cloro-3-fenil-2-[1-(9H-purin-6-ilam¡no)propin-3H-quinazolin-4-ona (82)
[0263] El compuesto 82 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-amino-6-cloro-benzoico se sustituyó por ácido 2-amino-6-metil-benzoico en el paso A, ácido 2-tercbutoxicarbonilamino propiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C. ESI-MS m/z 432 (MH+). El compuesto 82 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)prop¡l]-5-cloro-3-fen¡l-3H-qu¡nazol¡n-4-ona (83)
[0264] El c o m p u e s to 83 se p re p a ró u sa n d o el p ro c e d im ie n to g e n e ra l d e sc rito a n te rio rm e n te con re sp e c to al c o m p u e s to 14, pe ro se su s titu yó 2 -a m in o -6 -b ro m o p u rin a p o r 6 -b ro m o p u rin a en el pa so D. E S I-M S m/z 447 (MH+). El c o m p u e s to 83 se m u e s tra a c o n tin u a c ió n .

5-met¡l-3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)but¡l]-3H-qu¡nazol¡n-4-ona (84)
[0265] El compuesto 84 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-benciloxicarbonilaminopentanoico éster 2,5-dioxo-pirrolidin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B. MS (ES): m/z 426 (M+H), 213. El compuesto 84 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)but¡l]-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (85)
[0266] El compuesto 85 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-benciloxicarbonilaminopentanoico éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en paso B, y 2-amino-6-bromopurina se sustituyó por 6-bromopurina en el paso D. MS (ES): m/z 441 (M+H), 221. El compuesto 85 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-(3.5-d¡clorofen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (86)
[0267] El c o m p u e s to 86 se p re p a ró u sa n d o el p ro c e d im ie n to g e n e ra l d e sc rito a n te rio rm e n te con re sp e c to al c o m p u e s to 14, pe ro se su s titu yó 2 -a m in o -6 -b ro m o p u rin a p o r 6 -b ro m o p u rin a en el pa so D. E S I-M S m/z 482 (MH+). El c o m p u e s to 86 se m u e s tra a c o n tin u a c ió n .

5-met¡l-3-(3-morfol¡n-4-¡lmet¡l-fen¡l)-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (87)
[0268] El compuesto 87 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el compuesto intermedio 8 se sustituyó por anilina en el paso A, el ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se utilizó en el paso C. ESI-MS m/z 497 (MH+). El compuesto 87 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-3-(3-morfol¡n-4-¡lmet¡l-fen¡l)-3H-qu¡nazol¡n-4-ona (88)
[0269] El compuesto 88 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 87, pero se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS m/z 498 (MH+). El compuesto 88 se muestra a continuación.

2-[1-(5-bromo-7H-p¡rrolo[2.3-d]p¡r¡m¡d¡n-4-¡lam¡no)-et¡n-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (89)
[0270] El compuesto 89 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero ácido 2-ferc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1-ilo sustituido por ácido 2-benciloxicarbonilaminobutírico 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y 4-cloro-5-bromo-7H-pirrolo[2.3-d]pirimidina (preparado como en J Med. Chem. 1988, 31, 2086 2092) se sustituyó por 6-bromopurina en el paso D. ESI-MS m/z 411,1 (MH+). El compuesto 89 se muestra a continuación.

5-metil-2-[1-(5-metil-7n-pirrolo[2.3-d]pirim idin-4-ilamino)-etil]-3-fenil-3n-quinazolin-4-ona (90)
[0271] El compuesto 90 se preparó utilizando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-terc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por 2,5-dioxo del ácido 2-benciloxicarbonilaminobutírico-pirrolidin-1-ilo éster en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y 4-cloro-5-metil-7H-pirrolo[2.3-d]pirimidina (preparado como en J. Med. Chem. 1990, 33, 1984 1992) se sustituyó por 6-bromopurina en el paso D. ESI-Ms m/z 411,1 (MH+). El compuesto 90 se muestra a continuación.

2-[1-(5-fluoro-7H-p¡rrolo[2.3-d]p¡r¡m¡d¡n-4-¡lam¡no)-et¡n-5-met¡l-3-fen¡l-3H-quinazol¡n-4-ona (91)
[0272] El compuesto 91 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-terc-butoxicarbonilamino-propiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C, y el compuesto intermedio 1 se sustituyó por 6-bromopurina. ESI-MS m/z 415,1 (MH+). El compuesto 91 se muestra a continuación.

2-[2-hidroxi-1-(9H-purin-6-ilamino)etil]-3-fenil-3H-quinazolin-4-ona (92)
[0273] El compuesto 92 se preparó mediante la adición de ácido trifluoroacético a una solución de 2-[2-terc-butoxi-1-(9H-purin-6-ilamino)etil]-3-fenil-3H-quinazolin-4-ona en diclorometano. La reacción se agitó a temperatura ambiente durante 18 horas y luego se concentró al vacío. La purificación por LC proporcionó el producto como un sólido blanco. MS (EN): m/z 400 (M+H), 382, 200, 136. El compuesto 92 se muestra a continuación.

[0274] 2-[2-ferc-butoxi-1-(9H-purin-6-ilamino)etil]-3-fenil-3H-quinazolin-4-ona se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-amino-6-clorobenzoico se sustituyó por ácido 2-amino-6-metilbenzoico en el paso A, ácido 2-benciloxicarbonilamino-3-ferc-butoxi-propiónico éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y las condiciones utilizadas en el paso C eliminaron tanto el grupo protector bencilo como el sustituyente cloro del anillo A.
3-(3.5-d¡fluoro-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)prop¡l]-3H-quinazol¡n-4-ona (93)
[0275] El compuesto 93 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero se sustituyó 3,5-difluoroanilina por anilina en el paso A. ESI-MS m/z 448 (MH+). El compuesto 93 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)prop¡n-3-(3.5-d¡fluoro-fen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (94)
[0276] El compuesto 94 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 90, pero 2-amino-6-bromopurina se sustituyó por 6-bromopurina en el paso D. ESI-MS m/z 463 (MH+). El compuesto 94 se muestra a continuación.

3-(3.5-difluoro-fenil)-2-[1-(9ti-purin-6-¡lamino)-et¡n-3H-quinazolin-4-ona (95)
[0277] El compuesto 95 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido antranílico se sustituyó por ácido 2-amino-6-metilbenzoico y 3,5-difluoroanilina por anilina en el paso A, ácido 2-tercbutoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, y se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C. ESI-MS m/z 420 (MH+). El compuesto 95 se muestra a continuación.

2-[1-(5-bromo-7H-p¡rrolo[2.3-d]p¡r¡m¡d¡n-4-¡lam¡no)-et¡l]-3-(3-fluoro-fen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (96)
[0278] El compuesto 96 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero en el paso A se sustituyó 3-fluoroanilina por anilina, ácido 2-terc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidina-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y se utilizó el procedimiento alternativo (desprotección de TFA) en el paso C. ESI-MS m/z 494 (MH+). El compuesto 96 se muestra a continuación.

3-(3-fluoro-fen¡l)-5-met¡l-2-[1-(5-met¡l-7H-p¡rrolo[2.3-d]p¡r¡m¡d¡n-4-¡lam¡no)-et¡n-3H-qu¡nazol¡n-4-ona (97)
[0279] El compuesto 97 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero en el paso A se sustituyó 3-fluoroanilina por anilina, ácido 2-terc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se utilizó en el paso C. ESI-MS m/z 429 (MH+). El compuesto 97 se muestra a continuación.

3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)prop¡l]-3H-qu¡nazol¡n-4-ona (98)
[0280] El c o m p u e s to 98 se p re p a ró u sa n d o el p ro c e d im ie n to g e n e ra l d e s c rito a n te rio rm e n te con re sp e c to a c o m p u e s to 14, p e ro el á c ido 2 -a m in o -6 -m e til-b e n z o ic o se su s titu yó p o r á c ido 2 -a m in o -6 -m e til-b e n z o ic o en el pa so A. M S (E S ): m /z 398 (M H ), 199. El c o m p u e s to 98 se m u e s tra a co n tin u a c ió n .

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-(3.5-d¡fluorofen¡l)-3H-qu¡nazolin-4-ona (99)
[0281] El compuesto 99 fue preparado usando el procedimiento general descrito anteriormente con respecto al compuesto 95, pero se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS m/z 435 (MH+). El compuesto 96 se muestra a continuación.

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)prop¡l]-3-fen¡l-3H-qu¡nazolin-4-ona (100)
[0282] El compuesto 100 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido antranílico se sustituyó por ácido 2-amino-6-metil-benzoico en el paso A, y 2-amino-6-bromopurina se sustituyó por 6-bromopurina en el paso D. MS (ES): m/z 413 (M+H), 207. El compuesto 100 se muestra a continuación.

6.7-difluoro-3-fenil-2-[1-(9H-purin-6-ilamino)-etin-3H-quinazolin-4-ona (101)
[0283] El compuesto 101 se preparó utilizando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-amino-4,5-difluoro-benzoico se sustituyó por ácido 2-amino-6-metil-benzoico en el paso A, ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1 -ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1 -ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se utilizó en el paso C. ESI-MS m/z 420 (MH+). El compuesto 101 se muestra a continuación.

6-fluoro-3-(3-fluoro-fen¡l)-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazolin-4-ona (102)
[0284] El compuesto 102 era preparado utilizando el procedimiento general descrito anteriormente con respecto al compuesto 14, pero el ácido 2-amino-5-fluoro-benzoico se sustituyó por ácido 2-amino-6-metil-benzoico en el paso A, ácido 2-ferc-butoxicarbonilaminopropiónico éster 2,5-dioxopirrolidin-1-ilo se sustituyó por el ácido 2-benciloxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo en el paso B, y el procedimiento alternativo (desprotección de TFA) se utilizó en el paso C. ESI-MS m/z 420 (MH+). El compuesto 102 se muestra a continuación.

2-[4-dietilamino-1-(9H-purin-6-ilamino)but¡n-5-metil-3-fenil-3H-quinazolin-4-ona (103)
[0285] El compuesto 103 fue preparado siguiendo los pasos A-D a continuación.
Éster terc-butílico del ácido [4-am¡no-1-(5-met¡l-4-oxo-3-fen¡l-3.4-d¡h¡dro-qu¡nazol¡n-2-¡l)-but¡l]carbámico (104)
[0286] Paso A: Se purgó con nitrógeno un matraz de fondo redondo de una boca, de 10 ml, equipado con un agitador magnético y se cargó con paladio al 10% sobre carbono (30 mg, 50% húmedo). Una solución de éster terc-butílico del ácido [4-benciloxicarbonilamino-1-(5-metil-4-oxo-3-fenil-3,4-dihidro-quinazolin-2-il)-butil]-carbámico (producto del paso B del procedimiento para el compuesto 51) (100 mg, 0,18 mmol) en etanol (2 ml) y luego se añadieron secuencialmente polimetilhidrosiloxano (130 mg). La mezcla de reacción se agitó a 50°C durante 3 h y luego se enfrió a temperatura ambiente. La mezcla se filtró a través de un lecho corto de CELITE® y el filtrado se evaporó a sequedad. La posterior purificación del producto bruto resultante mediante cromatografía en columna proporcionó un rendimiento del 62% del compuesto 101 en forma de un sólido blanco. 1H RMN (CD 3 OD) 57,55-7,69 (m, 5H), 7,33-7,49 (m, 2H), 7,30 (d, 1H, J = 7,3 Hz), 4,29 (m, 1H) 2,77 (s, 3H), 2,38-2,47 (m, 2H), 1,72-1,88 (m, 1H), 1,57-1,79 (m, 1H), 1,13-1,55 (m, 4H), 1,40 (s, 9H). La reacción descrita anteriormente y el compuesto 104 se muestran a continuación.

Éster terc-butílico del ácido [4-d¡et¡lam¡no-1-(5-met¡l-4-oxo-3-fen¡l-3.4-d¡h¡dro-qu¡nazol¡n-2-¡l)-butil]carbám¡co (105)
[0287] Paso B: Se purgó con nitrógeno un matraz de fondo redondo de una boca de 10 ml, equipado con un agitador magnético, y luego se cargó con el compuesto 104 (100 mg, 0,24 mmol) y 1,2-dicloroetano (2 ml). Posteriormente se añadieron acetaldehído (42 mg, 0,85 mmol) y triacetoxiborohidruro de sodio (400 mg, 1,90 mmol). La mezcla de reacción se agitó durante 18 h a temperatura ambiente, se evaporó hasta sequedad y el residuo resultante se disolvió en metanol
(20 ml). Esta solución metanólica se trató con paladio sobre carbón al 10% (5 mg, 50% húmedo), se agitó durante 30 min, se evaporó a sequedad y se repartió entre carbonato de potasio acuoso al 10% (20 ml) y cloruro de metileno (20 ml). La capa orgánica se separó y se secó sobre sulfato de sodio. La filtración de la capa orgánica seguida de concentración dio el compuesto 105 como un aceite amarillo, que se usó sin purificación adicional. 1H RMN (CDCh) 5 7,51-7,63 (m, 5H), 7,41 (d, 1H, J = 7,4 Hz), 7,29 (d, 1H, J = 7,15 Hz), 7,22 (d, 1H, J = 7,1 Hz), 6,10 (d, 1 H, J = 8,8 Hz), 4,38-4,52 (m, 1H), 2,81 (s, 3H), 2,32-2,50 (m, 4H), 2,08-2,27 (m, 2H), 1,47 -1,73 (m, 4H), 1,42 (s, 9H, 1,25-1,38 (m, 1H), 0,96 (t, 6H, J = 7,2 Hz); ESI-MS m /z= 479 (MH+). La reacción descrito anteriormente y compuesto 105 se muestran a continuación.

2-(1-am¡no-4-d¡et¡lam¡no-but¡l)-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (106)
[0288] El procedimiento de desprotección alternativo descrito anterior con respecto a la preparación del compuesto 14 (y específicamente la preparación del compuesto 17) se usó para desproteger el compuesto 105. A continuación se muestran la reacción y el compuesto 106.

2-[4-D¡et¡lam¡no-1-(9H-pur¡n-6-¡lam¡no)but¡l]-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (103)
[0289] El compuesto 103 se preparó entonces siguiendo el procedimiento general anteriormente proporcionado en el paso D del procedimiento para el compuesto 14, pero como amina libre se utilizó el compuesto 106. ESI-MS m/z 497 (MH+). la reacción y el compuesto 103 se muestran a continuación.

EJEMPLO 9
PREPARACIÓN DE COMPUESTOS
[0290] Los compuestos de acuerdo con la fórmula general I (imagen superior), incluyendo compuestos quirales que tienen una fórmula general II (mostrada anteriormente), se han preparado de acuerdo con los pasos A-E del esquema sintético que se muestra a continuación.

(S)-5-fluoro-3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-prop¡l]-3H-qu¡nazol¡n-4-ona (107)
[0291] La síntesis de un compuesto de acuerdo con con la fórmula I se ejemplifica primero usando los pasos A-E a continuación, que proporcionan un procedimiento sintético para el compuesto 107, cuya estructura se muestra a continuación.

2-fluoro-6-nitro-N-fen¡l-benzam¡da (108)
[0292] Paso A: Una solución de ácido 2-fluoro-6-nitrobenzoico (100 g, 0,54 mol) y dimetilformamida (5 ml) en diclorometano (600 ml) se trató gota a gota con cloruro de oxalilo (2 M en diclorometano, 410 ml, 0,8 mol, 1,5 eq) durante 30 min. Después de agitar 2 h a temperatura ambiente, la reacción se concentró hasta un jarabe de naranja con algunos sólidos presentes. El jarabe se disolvió en dioxano seco (80 ml) y se añadió lentamente a una suspensión de anilina (49 ml, 0,54 mol, 1 eq) y bicarbonato de sodio (90 g, 1,08 mol, 2 eq) en una mezcla de dioxano (250 ml) y agua (250 mL) a 6°C. La temperatura alcanzó los 27°C al final de la adición. Después de 30 min, la mezcla de reacción se trató con agua
(1,2 L). El precipitado se recogió mediante filtración al vacío, se lavó con agua (300 ml), se secó al aire en el embudo y se secó al vacío a 50°C durante 24 h para dar un producto sólido blanquecino (139 g, 99%). 1H RMN (300 MHz, DMSO-d6) 5 10,82 (s, 1H), 8,12 (d, J = 7,7 Hz, 1H), 7,91-7,77 (m, 2H), 7,64 (d, J = 7,7 Hz, 2H), 7,38 (t, J = 7,9 Hz, 2H), 7,15 (t, J = 7,4 Hz, 1H), ESI-MS m/z 261 (MH+). La reacción descrita anteriormente y el compuesto 108 se muestran a continuación.
1 cloruro de oxalilo

a
dioxano/HzO
5-10 degC inicio
10 min
Éster terc-butílico del ácido (S)-[1-(2-fluoro-6-n¡tro-benzo¡l)-fen¡l-am¡nocarbon¡l]prop¡l-carbám¡co (109)
[0293] Paso B: Una suspensión del compuesto 108 (0,5 mol) y se agitó dimetilformamida (5 ml) en cloruro de tionilo (256 ml, 2,5 mol, 5 eq) a 85°C durante 5 horas. La mezcla de reacción se concentró al vacío hasta un jarabe marrón. El jarabe se disolvió en diclorometano (200 ml) y se añadió lentamente a una solución de ácido N-BOC-L-2-aminobutírico (112 g, 0,55 mol, 1,1 eq) y trietilamina (77 ml, 0,55 mol, 1,1 eq) en diclorometano (600 mL) a 10°C. Después de agitar a temperatura ambiente durante 3 h, las sales se eliminaron por filtración y la solución se lavó con 100 ml de agua, bicarbonato de sodio saturado, agua, ácido cítrico al 5% y cloruro de sodio saturado. La fase orgánica se secó con sulfato de magnesio y se concentró hasta un jarabe rojo. El jarabe se disolvió en diclorometano (450 ml) y se purificó mediante cromatografía flash en un tapón de gel de sílice (15 x 22 cm, 4 l de sílice seca) eluido con hexanos/acetato de etilo (10%, 8 L; 15%, 8 L; 20%, 8 L; 25%, 4 L) para producir el compuesto 109 como un sólido blanquecino (147 g, 66%). 1H RMN (300 MHz, DMSO-d6) 58,13 (d, J = 8,0 Hz, 1H), 7,84 (t, J = 8,6 Hz, 1H), 7,78-7,67 (m, 1H), 7,65-7,49 (m, 3H), 7,40-7,28 (m, 2H), 7,19 (d, J = 7,5 Hz, 1H), 4.05 (br. s, 1H), 1,75-1,30 (m, 2H), 1,34 (s, 9H), 0,93 (br. s, 3H). ESI-MS m/z 446,3 (MH+). La reacción descrita anteriormente y el compuesto 109 se muestran a continuación.

Éster terc-butíl¡co del ác¡do (S)-[1-(5-fluoro-4-oxo-3-fen¡l-3.4-d¡h¡dro-qu¡nazol¡n-2-¡l)prop¡l]carbám¡co (110)
[0294] Paso C: Se trató una solución del compuesto 109 (125 mmol, 1 eq) en ácido acético (500 mL) con polvo de zinc (48,4g, 740 mmol, 6 eq) agregado en 3 porciones, y la mezcla de reacción se dejó enfriar hasta por debajo de 35°C entre adiciones. Después de agitar durante 2 h a temperatura ambiente, los sólidos se filtraron mediante filtración al vacío y se lavaron con ácido acético (50 ml). El filtrado se concentró al vacío, se disolvió en EtOAc (400 ml), se lavó con agua (300 ml) y la capa de agua se extrajo con EtOAc (300 ml). Las capas orgánicas combinadas se lavaron con agua (200 ml), bicarbonato de sodio saturado (2 x 200 ml), NaCl saturado (100 ml), se secaron con MgSO4 y se concentraron hasta un jarabe. El jarabe se disolvió en tolueno (200 ml) y se purificó mediante cromatografía flash en un tapón de gel de sílice (13 x 15 cm, 2 L de sílice seca) eluido con hexanos/acetato de etilo (10%, 4 L; 15%, 4 L; 17,5%, 8 L; 25%, 4 L) para producir el compuesto 110 como un sólido espumoso blanquecino (33,6 g, 69%). 1H Rm N (300 MHz, DMSO-d6) 5 7,83 (td, J = 8,2, 5,7 Hz, 1H), 7,64-7,48 (m, 5H), 7,39 (ancho d, J = 7,6 Hz, 1H), 7,30 (dd, J = 8,3 Hz, 1H), 7,23 (d, J = 7,6 Hz, 1H), 4,02-3,90 (m, 1H), 1,76-1,66 (m, 1H), 1,62-1,46 (m, 1H), 1,33 (s, 9H), 0,63 (t, J = 7,3 Hz, 3H). ESI-MS m/z 398,3 (MH+). La reacción descrita anteriormente y el compuesto 110 se muestran a continuación.

(S)-2-(1-am¡no-prop¡l)-5-fluoro-3-fen¡l-3H-qu¡nazol¡n-4-ona (111)
[0295] Paso D: Una solución de compuesto 110 (85 mmol) en diclorometano (60 ml) se trató con ácido trifluoroacético (60 ml). La mezcla de reacción se agitó durante 1 h, se concentró al vacío y se repartió entre diclorometano (150 ml) y K2CO3
al 10% (cantidad suficiente para mantener el pH superior a 10). La capa acuosa se extrajo con más diclorometano (100 ml) y las capas orgánicas combinadas se lavaron con agua (50 ml) y salmuera (50 ml). Después de secar con Mg SO4 , la solución se concentró para proporcionar el compuesto 111 como un sólido blanquecino (22 g, 88%). 1H RMN (300 MHz, CDCh) 57,73-7,65 (m, 1H), 7,62-7,49 (m, 4H), 7,32-7,22 (m, 2H), 7,13 - 7,06 (m, 1H), 3,42 (dd, J = 7,5, 5,2 Hz, 1H), 1,87 1,70 (m, 1H), 1,58-1,43 (m, 1H), 0,80 (t, J = 7,4 Hz, 3H). eSi-m S m/z 298,2 (m H+). La reacción descrita anteriormente y el compuesto 111 se muestran a continuación.

iS)-5-fluoro-3-feml-2-ri-(9H-purm-6-ilammo)propin-3H-quinazolin-4-ona (107)
[0296] Paso E: Una suspensión de compuesto 111 (65,6 mmol, 1 eq), 6-bromopurina (14,6 g, 73,4 mmol, 1,1 eq) y DIEA (24,3 mL, 140 mmol, 2 eq) en terc-butanol (40 mL) se agitó durante 24 h a 80°C. La mezcla de reacción se concentró al vacío y se trató con agua para producir un producto bruto sólido que se recogió mediante filtración al vacío, se lavó con agua y se secó al aire. La mitad del producto bruto sólido obtenido se disolvió en MeOH (600 ml), se concentró sobre gel de sílice (300 ml seco) y se purificó mediante cromatografía flash (7,5 x 36 cm, eluida con 10 L de MeOH al 4%/CH2Cl2) para producir un producto sólido. Después, el producto sólido se disolvió en EtOH (250 ml) y se concentró al vacío para dar el compuesto 107 en forma de un sólido de color amarillo claro (7,2 g, 50%). 1H RMN (300 MHz, 80°C, DMSO-d6) 5 12,66 (br. s, 1H), 8,11 (s, 1H), 8,02 (br. s, 1H), 7,81 -7,73 (m, 1H), 7,60- 7,42 (m, 6H), 7,25-7,15 (m, 2H), 4,97 (br. s, 1H), 2,02-1,73 (m, 2H), 0,79 (t, J = 7,3 Hz, 3H). ESI-Ms m/z 416,2 (MH+). Análisis elemental de C, H, N (C22H18N7OF-EtOH-0,4H2O). Pureza quiral 99,8: 0,2 (S:R) usando HPLC quiral (columna Chiralpak ODH de 4,6 x 250 mm, 20°C, 85:15 hexanos: EtOH, 1 ml/min, muestra cargada a una concentración de 1 mg/ml en EtOH). La reacción descrita anteriormente y el compuesto 107 se muestran a continuación.

(S)-3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡n-3H-quinazol¡n-4-ona (112)
[0297] El compuesto 112 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 107, pero el ácido 2-nitrobenzoico se sustituyó por ácido 2-fluoro-6-nitrobenzoico en el paso A, y N-BOC-L-alanina se sustituyó por ácido N-BOC-L-2-aminobutírico en Paso B. ESI-MS m/z 384,3 (MH+). Pureza quiral 99,5:0,5 (S:R). El compuesto 112 se muestra a continuación.

(S)-6-fluoro-3-fenll-2-[1-(9H-purln-6-llamlno)-etl^-3H-qulnazolln-4-ona (113)
[0298] El compuesto 113 se preparó usando el método procedimiento general descrito anteriormente con respecto al compuesto 107, pero el ácido 2-nitro-5-fluorobenzoico se sustituyó por ácido 2-fluoro-6-nitrobenzoico en el paso A, y N-BOC-L-alanina se sustituyó por ácido N-BOC-L-2-aminobutírico en el paso B. ESI-MS m/z 402,3 (MH+). Pureza quiral 99,9:0,1 (S: R). El compuesto 113 se muestra a continuación.

(S)-3-(3.5-d¡fluoro-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (114)
[0299] El compuesto 114 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 107, pero el ácido 2-nitro-5-metilbenzoico se sustituyó por ácido 2-fluoro-6-nitrobenzoico y la 3,5-difluoroanilina se sustituyó por anilina en el paso A y N-BOC-L-alanina se sustituyó por ácido N-BOC-L-2-aminobutírico en el paso B. ESI-MS m/z 434,3 (MH+). El compuesto 114 se muestra a continuación.

(S)-5-fluoro-2-[1-(2-fluoro-9H-pur¡n-6-¡lam¡no)-et¡l]-3-fen¡l-3H-qu¡nazol¡n-4-ona (115)
[0300] El compuesto 115 fue preparado usando el procedimiento general descrito anteriormente con respecto al compuesto 107, pero el ácido 2-nitro-6-fluorobenzoico en lugar del ácido 2-fluoro 6-nitrobenzoico en el paso A, N-BOC-L-alanina era substituido por ácido n-BOC-L-2-aminobutírico en el paso B, y 6-cloro-2-fluoropurina se sustituyó por 6-bromopurina en el paso E. ESI-MS m/z 420,3 (MH+). Pureza quiral 100:0 (S:R). El compuesto 115 se muestra a continuación.

(S)-3-(3-fluoro-fen¡l)-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡n-3H-qu¡nazol¡n-4-ona (116)
[0301] El compuesto 116 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 107, pero el ácido 2-nitrobenzoico se sustituyó por el ácido 2-fluoro-6-nitrobenzoico y la 3-fluoroanilina se sustituyó por anilina en el paso A, y la N-BOC-L-alanina se sustituyó por ácido N-BOC-L-2-aminobutírico en el paso B. ESI-MS m/z 402,3 (MH+). Pureza quiral 96: 4 (S:R). El compuesto 116 se muestra a continuación.

(S)-5-cloro-3-(3.5-d¡fluoro-fen¡l)-2-[1-(9H-pur¡n-6-¡lam¡no)-prop¡n-3H-qu¡nazol¡n-4-ona (117)
[0302] El compuesto 117 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 107, pero el ácido 2-nitro-5-clorobenzoico se sustituyó por ácido 2-fluoro-6-nitrobenzoico, y la 3,5-difluoroanilina se sustituyó por anilina en el paso A. ESI-MS m/z 468,3 (MH+). Pureza quiral 100:0 (S:R). El compuesto 117 se muestra a
continuación.

(S)-3-(2.6-d¡fluoro-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (118)
[0303] El compuesto 118 se preparó usando un paso B alternativo en relación con la preparación del compuesto 107, pero el procedimiento general descrito anteriormente para el compuesto 107 se siguió generalmente para cada uno de los pasos A, C y D.
N-(2.6-d¡fluoro-fen¡l)-2-met¡l-6-n¡tro-benzam¡da (118a)
[0304] Paso A: 118 se preparó el compuesto siguiendo el procedimiento de preparación descrito anteriormente con respecto a compuesto 107, pero el ácido 2-nitro-5-metilbenzoico fue sustituido por la anilina por ácido 2-fluoro-6-nitrobenzoico y 2,6-difluoroanilina.
Éster terc-butíl¡co del ác¡do L-í2-[(2.6-d¡fluoro-fen¡l)-(2-met¡l-6-n¡tro-benzo¡l)-am¡no]-1-met¡l-2-oxo-et¡ll-carbám¡co (118b)
[0305] Paso B: El compuesto 118b se preparó por tratamiento de una solución de compuesto 118a (13,8 g, 47 mmol) en THF (200ml gota a gota) con una solución de hexametildisilazida de potasio (KHMDS) (0,5 M en tolueno, 95 ml, 47 mmol, 1 eq) a 0°C y agitando la mezcla de reacción durante 30 min a la misma temperatura. Después, la mezcla de reacción se trató con éster 2,5-dioxo-pirrolidin-1 -ilo del ácido L-2-terc-butoxicarbonilamino-propiónico (13,5 g, 47 mmol, 1 eq) y se agitó a la misma temperatura durante 30 min más. La mezcla de reacción se inactivó con agua (50 ml) y se concentró al vacío. El residuo se disolvió en acetato de etilo (300 ml) y se lavó con 100 ml de cada uno de los siguientes: (1) agua, (2) bicarbonato de sodio saturado, (3) agua, (4) ácido cítrico al 5%, (5) agua y (6) salmuera. La capa orgánica se secó con MgSÜ4 y se concentró hasta un jarabe. El material bruto se disolvió en diclorometano (75 ml) y se purificó mediante cromatografía flash en gel de sílice (7,5 x 40 cm), se eluyó con EtOAc al 20% en hexanos (10 l del 20%, luego 6 L del 33%).
[0306] Los pasos C y D se realizaron como se describe en relación con la preparación del compuesto 107. ESI-MS m/z 434,3 (MH+). Pureza quiral 100:0 (S:R)’. El compuesto 118 se muestra a continuación.

(S)-3-(2.6-d¡fluoro-fen¡l)-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡n-3H-qu¡nazol¡n-4-ona (119)
[0307] El compuesto 119 fue preparado usando el procedimiento general descrito anteriormente con respecto al compuesto 118, pero el ácido 2-nitrobenzoico se sustituyó por ácido 2-nitro-5-metilbenzoico. ESI-MS m/z 420,3 (MH+). Pureza quiral 100:0 (S:R). El compuesto 119 se muestra a continuación.

5-Met¡l-3-fen¡l-2-[3,3,3-tr¡fluoro-1-(9H-pur¡n-6-¡lam¡no)prop¡l]-3H-qu¡nazol¡n-4-ona (120)
[0308] El compuesto 120 se preparó usando el procedimiento general descrito anteriormente con respecto al compuesto 107, pero el ácido 2-metil-6-nitro-benzoico se sustituyó por ácido 2-fluoro-6-nitro-benzoico en el paso A y ácido 2-tercbutoxicarbonilamino-4,4,4-trifluoro-butírico se sustituyó por ácido 2-terc-butoxicarbonilamino-butírico en el paso B. ESI-MS m/z 466 (MH+). El compuesto 120 se muestra a continuación.

EJEMPLO 10
PREPARACIÓN DE COMPUESTOS
[0309] Los compuestos que tienen la fórmula general I (se muestra más arriba) han sido preparados de acuerdo con los pasos A-D del esquema sintético titulado "Procedimiento C" mostrado a continuación. Un esquema sintético alternativo titulado "Esquema D" ilustra rutas sintéticas adicionales para compuestos que tienen la fórmula I.
Procedimiento C:
[0310]


[0311] La síntesis de compuestos de acuerdo con el Procedimiento C y el Esquema D se ejemplifica en primer lugar mediante el procedimiento sintético para 3-(3-hidroxi-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona, también denominado compuesto 121, cuya estructura se muestra a continuación.

3-(3-Hidroxi-fenil)-5-metil-2-[1 -(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona (121)
[0312] El compuesto 121 era preparado siguiendo los pasos A-D a continuación, y usando el compuesto 122 (a continuación) en el paso A.
3-(terc-butilo dim etilsililoxi)fenilam ina (122)
[0313] Se utilizó un matraz de fondo redondo de tres bocas de 250 ml equipado con un agitador magnético se purgó con nitrógeno y se cargó con cloruro de terc-butildimetilsililo (12,5 g, 82,8 mmol), imidazol (7,64 g, 112 mmol) y DMF anhidra (60 ml). Se añadió 3-aminofenol (10,0 g, 91,7 mmol) a la solución resultante. Después de agitar durante 12 h a temperatura ambiente, la mezcla de reacción se vertió en agua (300 ml). La suspensión resultante se extrajo con hexanos (3 x 300 ml)
y los extractos se combinaron, se secaron sobre sulfato de sodio y se filtraron. La concentración del filtrado seguida de cromatografía en columna dio el compuesto 122 en forma de un aceite de color amarillo claro. 1H RMN (DMSO-afe) 5 (ppm), 6,84 (t, 1H, J = 7,9 Hz), 6,16 (d, 1H, J = 6,7 Hz), 6,10 (m, 1H), 5,97 (d, 1H, J = 9,1 Hz), 4,98 (s, 2H), 0,95 (s, 9H), 0,16 (s, 6H); m/z = 224 (M+H).
2-am¡no-6-met¡l-N-[(3-terc-but¡l-d¡met¡l-s¡lanox¡)-fen¡l]-benzam¡da (123)
[0314] Paso A: Un frasco de fondo redondo 5-L, de tres bocas, equipado con un burbujeador de gas, un agitador mecánico y un condensador de reflujo se cargó con ácido 6-amino-2-metilbenzoico (25 g, 16,8 mmol), tolueno (300 ml) y cloruro de tionilo (50 ml). La mezcla de reacción se calentó a reflujo durante 1 h hasta que cesó el desprendimiento de gas. Después, la mezcla resultante se enfrió y se concentró a presión reducida a 50°C. Se añadieron THF anhidro (400 ml) y DIEA (90 ml) al residuo resultante, seguido del compuesto 122 (37 g, 1,0 eq). La mezcla de reacción resultante se agitó a temperatura ambiente durante 2 horas, luego se inactivó mediante la adición de carbonato de potasio acuoso al 20% (250 ml). La capa orgánica se separó y se concentró hasta sequedad a presión reducida. La trituración del residuo con MfBE (70 ml), la filtración y el secado proporcionaron el compuesto 123. La preparación del compuesto 123 se muestra generalmente arriba como el paso A del Procedimiento C.
Éster terc-butíl¡co de ác¡do 1-í[3-(3-h¡drox¡-fen¡l)-5-met¡l-4-oxo-3.4-d¡h¡dro-qu¡nazol¡n-2-¡l]-et¡ll-carbám¡co (124)
[0315] Paso B: Un matraz de fondo redondo de 100 ml, de tres bocas, equipado con un agitador magnético y un condensador de reflujo se purgó con nitrógeno y se cargó con el compuesto 123 (10 g, 28 mmol), éster de N-hidroxisuccinimida de N-ferc-butiloxicarbonilalanina (9,60 g, 1,0 eq), DMAP (1,90 g), DIEA (6 ml), tamices moleculares de 4Á (1,20 g) y tolueno anhidro (100 ml). La mezcla resultante se calentó en un baño de aceite a 80°C durante 24 h. Se añadió 1-hidroxibenzotriazol (3,78 g) a la reacción y se continuó calentando durante 48 h más. Después de enfriar, se añadieron tolueno (10 ml) y CELITE® (0,70 g) a la mezcla de reacción caliente, seguido de filtración y evaporación del filtrado para dar un residuo marrón. El residuo se disolvió en CH2Cl2 (15 ml) y se trató con una solución de fluoruro de tetrabutilamonio (5,12 g) en MeOH (15 ml). Después de agitar durante 1 h a temperatura ambiente, la mezcla de reacción se evaporó hasta sequedad a presión reducida y el residuo se purificó por cromatografía en columna para dar un rendimiento del 81% del compuesto 124 como un sólido blanquecino. La preparación del compuesto 124 generalmente se muestra arriba como el paso B del Procedimiento C.
2- (1-am¡no-et¡l)-3-(3-h¡drox¡-fen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (125)
[0316] Paso C1: Un matraz de fondo redondo de 100 ml, de tres bocas, equipado con un agitador magnético, se cargó con el compuesto 124 (4,50 g, 11,18 mmol) en CH2Cl2 (15 ml) y TFA (15 ml). Después de agitar durante 1 hora a temperatura ambiente, la mezcla se concentró a presión reducida para producir el compuesto 125, que se usó de la misma manera en el siguiente paso.
3- (3-h¡drox¡-fen¡l)-5-met¡l-2-í1-[9-(2-tr¡met¡ls¡lan¡l-etox¡met¡l)-9H-pur¡n-6-¡lam¡no]-et¡ll-3H-qu¡nazol¡n-4-ona (126)
[0317] Paso C2: Un matraz de fondo redondo, de 50 ml, de una boca, purgado con nitrógeno, equipado con un agitador y el reflujo magnético condensador, se cargó con el compuesto 125 (2,4 g, 8,16 mmol), compuesto intermedio 10 (2,70 g, 1,0 eq), n-butanol (20 ml) y DIEA (4,2 ml). La mezcla se calentó a 100°C durante 4 h y luego se enfrió a temperatura ambiente. La concentración de la mezcla de reacción a alto vacío seguida de cromatografía en columna dio el compuesto como un sólido blanco. La preparación del compuesto 126 generalmente se muestra arriba como el paso C del Procedimiento C.
3-(3-h¡drox¡-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡n-3H-qu¡nazol¡n-4-ona (121)
[0318] Paso D: Un matraz de fondo redondo de 100-ml, de una boca, equipado con un agitador magnético se cargó con el compuesto 126 (294 mg, 0,5 mmol), MeOH (15 ml) y se calentó el ácido clorhídrico 4 N (15 ml) y la mezcla de reacción se calentó durante 5 h a 40°C. La evaporación del metanol a presión reducida seguida de basificación a pH 10 con carbonato de potasio acuoso al 10% dio un precipitado blanco. El precipitado se filtró, se lavó con agua y se secó al vacío durante la noche a temperatura ambiente para producir el compuesto 121 en forma de un sólido blanco. m/z = 414 (M+H). La preparación del compuesto 121 generalmente se muestra arriba como el paso D del Procedimiento C.
3-(3-metox¡-fen¡l)-5-met¡l-2-f1-[9-(2-tr¡met¡ls¡lan¡l-etox¡met¡l)-9H-pur¡n-6-¡lam¡no]-et¡l1-3H-qu¡nazol¡n-4-ona (127)
[0319] Un matraz de fondo redondo de 100-ml, de una boca, equipado con un agitador magnético se cargó con el compuesto 126 (370 mg, 0,68 mmol), carbonato de potasio (235 mg, 1,70 mmol) y DMF (4 ml). La mezcla resultante se agitó durante 5 min y luego se añadió yoduro de metilo (435 mg, 3,10 mmol). Después de agitar durante 1 h a temperatura ambiente, la mezcla de reacción se evaporó a sequedad a presión reducida. El residuo se purificó mediante cromatografía en columna para dar un rendimiento del 82% del compuesto 127 en forma de un aceite de color amarillo claro. 1H RMN (CD3OD) 5 (ppm) 8,20 (m, 2H), 6,99-7,66 (m, 7H), 5,58 (s, 2H), 5,15 (bs, 1H), 3,62 (dt, 2H, J = 1,8, 8,0 Hz), 3,32 (s, 3H), 2,78 (s, 3H), 1,55 (m, 3H), 0,88 (m, 2H), -0,07 (s 9 H); m/z = 558 (M+H).
3-(3-metox¡-fen¡l)-5-met¡l-2-f1-f9H-pur¡n-6-¡lam¡no1-et¡l1-3H-quinazol¡n-4-ona (128)
[0320] El compuesto 127 se hizo reaccionar de acuerdo con el procedimiento descrito anteriormente para el compuesto 121 (paso D) para proporcionar el compuesto 128. m/z = 428 (M+H). La estructura del compuesto 128 se muestra a continuación.

3-(3-metoxi-fenil)-5-metil-2-[1 -(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona (128)
3-[3-(2-d¡met¡lam¡no-etox¡)-fen¡l1-5-met¡l-2-f1-[9H-pur¡n-6-¡lam¡no1-et¡l1-3H-qu¡nazol¡n-4-ona (129)
[0321] El compuesto 126 (300 mg, 0,54 mmol) con sal de hidrocloruro de 2-cloro etil dimetilamina, a 90°C durante 17 h, utilizando el procedimiento descrito anteriormente para el compuesto 127. El compuesto resultante se trató luego con 4N HCl, en MeOH, utilizando el procedimiento descrito para compuesto 121 (paso D). Se obtuvo el compuesto 129. m/z = 485 (M+H). La estructura del compuesto 129 se muestra a continuación.

3-[3-(2-dimetilamino-etoxi)-fenil]-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona (129)
3-(3-c¡cloprop¡lmetox¡-fen¡l)-5-met¡l-2-f1-[9H-pur¡n-6-¡lam¡no)-et¡l1-3H-qu¡nazol¡n-4-ona (130)
[0322] Compuesto 126 (300 mgs, 0,54 mmol) con bromometil ciclopropano usando el procedimiento descrito para el compuesto 127, a temperatura ambiente durante 24 horas. Este intermedio se trató con 4N HCl de acuerdo con el procedimiento descrito para el compuesto 121 (Paso D). m/z = 468 (M+H). La estructura del compuesto 130 se muestra a continuación.

3-(3-ciclopropilmetoxi-fenil)-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona (130)
5-met¡l-3-(-2-¡n¡lox¡-fen¡l 3-prop) 2-f1-[9H-pur¡n-6-¡lam¡no1-et¡H-3H-qu¡nazol¡n-4-ona (131)
[0323] El compuesto 126 (300 mg, 0,54 mmol) se trató con bromuro de propargilo a temperatura ambiente durante 24 horas usando el procedimiento descrito anteriormente para el compuesto 127. Este intermedio se trató luego con 4N HCl de acuerdo con el procedimiento descrito para el compuesto 121 (Paso D). m/z = 467 (M+H). La estructura del compuesto 131 se muestra a continuación.

5-metil-3-(3-prop-2-iniloxi-fenil)-2-[1 -(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona (131)
2-f1-f2-am¡no-9H-pur¡n-6-¡lam¡no1et¡l1-3-(3-h¡drox¡fen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (132)
[0324] El compuesto 132 se preparó de acuerdo con los procedimientos establecidos en los pasos A y B a continuación.
2-f1-12-D¡terc-but¡lox¡carbon¡lam¡no-9-(2-tr¡met¡ls¡l¡letox¡met¡l)-9H-pur¡n-6-¡lam¡no1et¡l1-3-(3-h¡drox¡fen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (133)
[0325] Paso A: Un matraz de fondo redondo de 50 ml, de una boca, purgado con nitrógeno, equipado con un agitador y el reflujo magnético condensador, se cargó con el compuesto 125 (3,02 g, 10,2 mmol), compuesto intermedio 12 (5,56 g, 1,0 eq), n-butanol (20 ml) y DIPEA (6,0 ml). La mezcla se calentó a 100°C durante 1 hora y luego se enfrió a temperatura ambiente. La concentración de la mezcla de reacción a alto vacío seguida de cromatografía en columna dio el compuesto 133 como un sólido blanco.
2-f1-[2-am¡no-9H-pur¡n-6-¡lam¡no1et¡l1-3-(3-h¡drox¡fen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (132)
[0326] Paso B: El compuesto 133 se disolvió en MeOH (3 ml), se trató con 4N HCl (3 ml) y se calentó a 40°C durante 6 h. Después, la mezcla de reacción se concentró hasta aproximadamente la mitad del volumen y se repartió entre agua (5 ml) y acetato de etilo (10 ml). La capa acuosa se separó, se basificó con carbonato de potasio a pH 10 y se filtró. Después de lavar la torta del filtro con agua (5 ml) y secar al vacío, se obtuvo el compuesto 132 en forma de un sólido blanco. m/z = 429 (M+H). La estructura del compuesto 132 se muestra a continuación.

2-[1-(2-Amino-9H-purin-6-ilamino)-etil]-3-(3-hidroxi-fenil)-5-metil-3H-quinazolin-4-ona (132)
2-f1-[2-am¡no-9H-pur¡n-6-¡lam¡no1et¡l1-3-(3-metox¡fen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (134)
[0327] El compuesto 133 (300 mgs, 0,39 mmol) se trató con yoduro de metilo usando el procedimiento descrito anteriormente para el compuesto 127. A continuación, este intermedio se trató con 4N HCl en metanol de acuerdo con el procedimiento descrito anteriormente para el compuesto 132 (Paso B). m/z = 443 (M+H). La estructura del compuesto 134 se muestra a continuación.

2-[1 -(2-Amino-9H-purin-6-ilamino)-etil]-3-(3-metoxi-fenil)-5-metil-3H-quinazolin-4-ona (134)
2-f1-f2-am¡no-9H-pur¡n-6-¡lam¡no1et¡l1-3-(3-c¡cloprop¡lmetox¡fen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (135)
[0328] El compuesto 133 (231 mg, 0,30 mmol) con bromuro de ciclopropil metilo usando el procedimiento descrito anteriormente para el compuesto 127. El intermedio generado se trató luego con 4N HCl en MeOH de acuerdo con el procedimiento descrito anteriormente para el compuesto 132 (Paso B). m/z = 483 (M+H). La estructura del compuesto 135 se muestra a continuación.

2-[1-(2-Amino-9H-purin-6-ilamino)-etil]-3-(3-ciclopropilmetoxi-fenil)-5-metil-3H-quinazolin-4-ona (135)
2-f1-[2-am¡no-9H-pur¡n-6-¡lam¡no1et¡l1-5-met¡l-3-(3-prop-2-¡n¡lox¡-fen¡l)-3H-qu¡nazol¡n-4-ona (136)
[0329] El compuesto 133 (231 mg, 0,30 mmol) se trató con bromuro de propargilo usando el procedimiento descrito anteriormente para el compuesto 127. El intermedio generado se trató luego con 4N HCl en MeOH de acuerdo con el procedimiento descrito anteriormente para el compuesto 132 (Paso B). m/z = 467 (M+H). La estructura del compuesto 136 se muestra a continuación.

2- [1-(2-Amino-9H-purin-6-ilamino)-etil]-5-metil-3-(3-prop-2-iniloxi-fenil)-3H-quinazolin-4-ona (136)
3- (3-et¡n¡l-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l1-3H-qu¡nazol¡n-4-ona (137)
[0330] El compuesto 137 se preparó de acuerdo con los procedimientos establecidos en los pasos A-C a continuación.
Ác¡do tr¡fluorom etanosulfón¡co 3-(5-met¡l-4-oxo-2-í1-[9-(2-tr¡met¡ls¡lan¡l-etox¡met¡l)-9H-pur¡n-6-¡lam¡no]-et¡lMH-qu¡nazol¡n-3-¡l)-éster feníl¡co (138)
[0331] Paso A: Un matraz de fondo redondo de 50 ml, de tres bocas, equipado con un agitador magnético se purgó con nitrógeno y se cargó con el compuesto 126 (500 mg, 0,92 mmol), trietilamina (218 mg, 2,16 mmol), cloruro de metileno anhidro (10 ml) y N-feniltrifluorometanosulfonimida (496 mg, 1,39 mmol). Después de agitar durante 2 h a temperatura ambiente, la mezcla de reacción se repartió entre cloruro de metileno (50 ml) y carbonato de potasio acuoso al 10% (50 ml). La fase orgánica se separó, se secó sobre sulfato de sodio y se filtró. La concentración del filtrado seguida de cromatografía en columna dio un rendimiento del 77% del compuesto 138 en forma de un sólido blanquecino. 1H RMN (DMSO-dfe) 5 (ppm) 8,32 (bs, 1H), 7,48-8,18 (m, 7H), 7,30 (d, 1H, J = 8,0 Hz), 5,51 (s, 2H), 4,75-4,85 (m, 1H), 3,55 (t, 2H, J = 8,0 Hz), 2,72 (s, 3H), 1,46 (d, 3H, J = 6,6 Hz), 0,83 (dt, 2H, J = 1,6, 8,1 Hz), -0,09 (s, 9H).
5-Met¡l-2-f1-[9-(2-tr¡met¡ls¡l¡letox¡met¡l)-9H-pur¡n-6-¡lam¡nolet¡l}-3-(3-tr¡met¡ls¡l¡let¡n¡lfen¡l)-3H-qu¡nazol¡n-4-ona (139)
[0332] Paso B: Un vial de reacción de 5 ml equipado con un agitador magnético se purgó con nitrógeno y se cargó con el compuesto 138 (220 mg, 0,33 mmol), diclorobis(trifenilfosfina)paladio (II) (27,1 mg, 0,039 mmol) y DMF anhidra. (1 mL). Se añadieron trietilamina (146 mg, 1,44 mmol) y (trimetilsilil)acetileno (102 mg, 1,04 mmol) y la mezcla de reacción se agitó durante 10 horas a 90°C y 8 horas más a 100°C. La evaporación de la mezcla de reacción a sequedad seguida de purificación por cromatografía en columna proporcionó un rendimiento del 63% del compuesto 139 en forma de un sólido blanquecino. 1H RMN (CDaOA) 5 (ppm) 7,43-8,33 (m, 8H), 7,30 (d, 1H, J = 6,6 Hz), 5,64 (s, 2H), 5,14 (bs, 1H), 3,68 (t,
2H, J = 8,0 Hz), 2,81 (s, 3H), 1,59-1,64 (m, 3H), 0,94 (m, 2H), 0,32 (s, 9H), -0,09 (s, 9H).
3-(3-et¡n¡l-fen¡l)-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (137)
[0333] Paso C: El compuesto 139 (113 mg, 0,18 mmol) se trató con 4N HCl en MeOH de acuerdo con el procedimiento descrito para el compuesto 121 (Paso D). Esto proporcionó el compuesto 137. m/z = 422 (M+H). La estructura del compuesto 137 se muestra a continuación.

3-(3-Etinil-fenil)-5-metil-2-[1 -(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona (137)
3-{5-met¡l-4-oxo-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-4H-qu¡nazol¡n-3-¡l}-benzon¡tr¡lo (140)
[0334] El compuesto 140 se preparó de acuerdo con los procedimientos establecidos en los pasos A y B a continuación.
3-(5-met¡l-4-oxo-2-í1-[9-(2-tr¡met¡ls¡l¡letox¡met¡l)-9H-pur¡n-6-¡lam¡no]et¡l}-4H-qu¡nazol¡n-3-¡l)benzon¡tr¡lo (141) [0335] Paso A: Se cargó un vial de reacción de 5 ml equipado con un agitador magnético con compuesto 138 (200 mg, 0,300 mmol), tetraquis(trifenilfosfina)paladio (34,0 mg, 0,029 mmol), cianuro de zinc (70 mg, 0,60 mmol) y DMF anhidra (1 ml). El vial se purgó con nitrógeno, se calentó a 120°C durante 3,5 h, luego se enfrió a temperatura ambiente y se vertió en una solución acuosa saturada de bicarbonato de sodio (25 ml). La suspensión resultante se extrajo con cloruro de metileno (3 x 20 ml) y los extractos orgánicos se combinaron, se secaron sobre sulfato de sodio y se filtraron. La concentración del filtrado seguida de purificación por cromatografía en columna dio un rendimiento del 66% del compuesto 141 en forma de un sólido blanco. 1H RMN (CDCla) 5 (ppm) 8,30 (d, 1H, J = 6,9 Hz), 7,58-8,02 (m, 7H), 7,30 (d, 1H, J = 7,0 Hz), 5,60 (s, 2H), 5,07 (bs, 1H), 3,65 (m, 2H), 2,84 (s, 3H), 1,58 (d, 3H, J = 6,7 Hz), 0,96 (m, 2H), 0,02 (s, 9H). 3-í5-met¡l-4-oxo-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-4H-qu¡nazol¡n-3-¡l}-benzon¡tr¡lo (140)
[0336] Paso B: El compuesto 141 se trató con 4N HCl en MeOH durante 1 hora usando el procedimiento descrito para el compuesto 121 (Paso B) para proporcionar el compuesto 140. m/z = 423 (M+H). La estructura del compuesto 140 se muestra a continuación.

3-{5-metil-4-oxo-2-[1 -(9H-purin-6-ilamino)-etil]-4H-quinazolin-3-il}-benzonitrilo (140)
3-í5-Met¡l-4-oxo-2-í1-[9H-pur¡n-6-¡lam¡no)-et¡l]-4H-qu¡nazol¡n-3-¡l}-benzam¡da (142)
[0337] El compuesto 142 se preparó de acuerdo con los procedimientos establecidos en pasos A y B a continuación. 3-(5-met¡l-4-oxo-2-f1-[9-(2-tr¡met¡ls¡l¡letox¡met¡l)-9H-pur¡n 6-¡lam¡no]et¡l}-4H-qu¡nazol¡n-3-¡l)benzam¡da (143) [0338] Paso A: Un matraz de fondo redondo de tres bocas de 100 ml equipado con un agitador magnético y un condensador de reflujo se purgó con nitrógeno y se cargó con el compuesto 141 (219 mg, 0,40 mmol) y cloruro de metileno anhidro (15 ml). Se añadió W,W-dietilhidroxilamina (146 mg, 1,64 mmol) a la solución resultante y la mezcla de reacción se calentó durante 16 h a 50°C, se enfrió a temperatura ambiente y luego se evaporó hasta sequedad a presión reducida. El residuo resultante se purificó mediante cromatografía en columna para dar un rendimiento del 97% del compuesto 143 en forma de un sólido blanco. pf 195-197°C; 1H RMN (CDCl3) 5 (ppm) 8,34 (d, 1H, J = 10,9 Hz), 7,59-8,06 (m, 7H), 7,28
(m, 1H), 6,95 (bs, 1H), 6,00 (bs, 2H), 5,60 (s, 2H), 5,28 (bs, 1H), 3,62 (t, 2H, J = 8,4 Hz), 2,85 (s, 3H), 1,53 (dd, 3H, J = 6,7, 10,8 Hz), 0,96 (t, 2H, J = 8,3 Hz), 0,01 (s, 9H); m/z = 571 (M+H).
3-í5-met¡l-4-oxo-2-í1-[9H-pur¡n-6-¡lam¡no)-et¡l]-4H-qu¡nazol¡n-3-¡l}-benzam¡da (142)
[0339] Paso B: Compuesto 143 se trató con 4N HCl en MeOH durante 1,5 horas usando el procedimiento descrito para el compuesto 121 (Paso D) para proporcionar el compuesto 142. m/z = 441 (M+H). La estructura del compuesto 142 se muestra a continuación.

3-{5-metil-4-oxo-2-[1-(9H-purin-6-ilamino)-etil]-4H-quinazolin-3-il}-benzamida (142)
3-(3-acet¡l-fen¡l)-5-met¡l-2-f1-[9H-pur¡n-6-¡lam¡no]-et¡l1-3H-qu¡nazol¡n-4-ona (144)
[0340] El compuesto 139 se trató con 4N HCl en MeOH a 70°C durante 16 horas de acuerdo con el procedimiento descrito para el compuesto 121 (paso D). Esta reacción proporcionó el compuesto 144, cuya estructura se muestra a continuación. m/z = 440 (M+H)

3-(3-acetil-fenil)-5-metil-2-[1 -(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona (144)
2-(3-(5-met¡l-4-oxo-2-í1-[9H-2ur¡n-6-¡lam¡no]-et¡lMH-qu¡nazol¡n-3-¡l-fenox¡acetam¡da (145)
[0341] El compuesto 126 (300 mg, 0,54 mmol) se trató con 2 bromo acetamida usando el procedimiento descrito anteriormente para el compuesto 127. La reacción se llevó a reflujo durante 24 h en CH3CN. Este intermedio se trató con 4N HCl en MeOH durante 1 hora, siguiendo el procedimiento descrito para el compuesto 121 (paso D), para proporcionar el compuesto 145. m/z = 471 (M+H). A continuación se muestra la estructura del compuesto 145.

2-(3-{5-Metil-4-oxo-2-[1-(9H-purin-6-ilamino)-etil]-4H-quinazolin-3-il}-fenoxi)acetamida (145)
S-met¡l-2-f1-[9H-pur¡n-6-¡lam¡no]-et¡l1-3-[3-(tetrah¡dropuran-4-¡lox¡)-fen¡n-3H-qu¡nazol¡n-4-ona (146)
[0342] Un matraz de fondo redondo, tres bocas y 25 ml equipado con un agitador magnético se purgó con nitrógeno y se cargó con el compuesto 126 (270 mg, 0,5 mmol), tetrahidro piran-4-ol (60 uL), trifenilfosfina (560 mg), THF (5 ml) y
azodicarboxilato de dietilo (340 ul). Después de agitar durante 16 h a temperatura ambiente, la mezcla de reacción se evaporó hasta sequedad y el residuo se disolvió en metanol (3 ml), se trató con ácido clorhídrico 4N (3 ml) y se calentó a 40°C durante 6 h. Después, la mezcla de reacción se concentró hasta aproximadamente la mitad del volumen y se repartió entre agua (5 ml) y acetato de etilo (10 ml). La capa acuosa se separó, se basificó con carbonato de potasio a pH 10 y se filtró. Después de lavar la torta del filtro con agua (5 ml) y secar al vacío, se obtuvo el compuesto 146. m/z = 498 (M+H). La estructura del compuesto 146 se muestra a continuación.

5-Metil-2-[1-(9H-purin-6-ilamino)-etil]-3-[3-(tetrahidro-piran-4-iloxi)-fenil]-3H-quinazolin-4-ona (146)
3-[3-(2-metox¡-etox¡)-fen¡l]-5-met¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (147)
[0343] El compuesto 126 (300 mg, 0,54 mmol) se trató con éster etílico de ácido 4-sulfónico de tolueno 2-metoxi a 50°C durante 42 h utilizando el procedimiento descrito anteriormente para el compuesto 127. El intermedio generado después se trató con 4N HCl en MeOH, usando el procedimiento descrito para el compuesto 121 (paso D). m/z = 487 (M+H):

3-[3-(2-metoxi-etoxi)-fenil]-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona (147)
6-fluoro-2-[1-(9H-pur¡n-6-¡lam¡no)et¡l]-3-[3-(tetrah¡dro-p¡ran-4-¡lox¡)-fen¡l]-3H-qu¡nazol¡n-4-ona (148)
[0344] El compuesto 148 se preparó de acuerdo con los procedimientos expuestos en los pasos A y B siguientes. 6-fluoro-3-(3-h¡drox¡-fen¡l)-2-f1-[9-(2-tr¡met¡ls¡lan¡l-etox¡met¡l)-9H-pur¡n-6¡lam¡no-et¡l1-3H-qu¡nazol¡n-4-ona (149) [0345] Paso A: El compuesto 149 se obtuvo a partir del ácido 6-amino 3-fluorobenzoico usando los procedimientos descritos anteriormente para los compuestos 123, 124, 125 y 126.
6-fluoro-2-[1-(9H-pur¡n-6-¡lam¡no)et¡n-3-[3-(tetrah¡dro-p¡ran-4-¡lox¡)-fen¡n-3H-qu¡nazol¡n-4-ona (148)
[0346] Paso B: El compuesto 148 se obtuvo a partir del compuesto 149 usando el procedimiento descrito para el compuesto 146, m/z = 502 (M+H). La estructura del compuesto 148 se muestra a continuación.

6-Fluoro-2-[1-(9H-purin-6-ilamino)-etil]-3-[3-(tetrahidro-piran-4-iloxi)-fenil]-3H-quinazolin-4-ona (148)
3-f3-(3-d¡met¡lam¡no-propox¡)-fen¡n -5-rnet¡l-2-f1-(9H-pur¡n-6-¡lam¡no)-et¡n -3H-qu¡nazol¡n-4-ona (150)
[0347] El compuesto 150 se obtuvo siguiendo el procedimiento general descrito para el compuesto 146, pero se usó 3-dimetilamino-1 -propanol en lugar de tetrahidropiran-4-ol. m/z = 499 (M+H). La estructura del compuesto 150 se muestra a continuación.

3-[3-(3-dimetilamino-propoxi)-fenil]-5-metil-2-[1-(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona (150)
2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡n-3-(3-et¡l-fen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (151)
[0348] El compuesto 151 se preparó de acuerdo con los procedimientos establecidos en los pasos A y B siguientes. Ác¡do tr¡fluorom etanosulfón¡co 3-í2-[1-(2-d¡terc-but¡lox¡carbon¡lam¡no-9-(2-tr¡met¡ls¡l¡letox¡met¡l)-pur¡n-6¡lam¡no)-et¡l]-5-met¡l-4-oxo-4H-qu¡nazol¡n-3-¡l}-éster feníl¡co (152)
[0349] Paso A: El compuesto 152 se obtuvo a partir del compuesto 133, que se hizo reaccionar de acuerdo con el procedimiento descrito para el compuesto 138.
2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡n-3-(3-et¡n¡l-fen¡l)-5-met¡l-3H-qu¡nazol¡n-4-ona (151)
[0350] Paso B: El compuesto 151 se obtuvo a partir del compuesto 152, que se hizo reaccionar de acuerdo con los procedimientos para los compuestos 139 y 137 (Paso C). m/z = 437 (M+H). La estructura del compuesto 151 se muestra a continuación.

2- [1 -(2-Amino-9H-purin-6-ilamino)-etil]-3-(3-etinil-fenil)-5-metil-3H-quinazolin-4-ona (151)
3- f2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-4-oxo-4H-qu¡nazol¡n-3-¡ll-benzon¡tr¡lo (153)
[0351] El compuesto 153 fue obtenido a partir del compuesto 152, que se hizo reaccionar de acuerdo con el procedimiento para los compuestos 141 y 140 (Paso D) descrito anteriormente. m/z = 438 (M+H). La estructura del compuesto 153 se muestra a continuación.

3-{2-[1-(2-Amino-9H-purin-6-ilamino)-etil]-5-metil-4-oxo-4H-quinazolin-3-il}-benzonitrilo (153)
3-í2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-4-oxo-4H-qu¡nazol¡n-3-¡ll-benzam¡da (154)
[0352] El compuesto 154 se obtuvo mediante la primera reacción del compuesto 152 de acuerdo con el procedimiento para el compuesto 141. Este producto de reacción se hizo reaccionar luego adicionalmente de acuerdo con el procedimiento para los compuestos 143 y 142 (Paso B). m/z = 456 (M+H). La estructura del compuesto 154 se muestra a continuación.

3-{2-[1 -(2-Amino-9H-purin-6-ilamino)-etil]-5-metil-4-oxo-4H-quinazolin-3-il}-benzamida (154)
3-í2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-4-oxo-4H-qu¡nazol¡n-3-¡ll-benzam¡da (155)
[0353] El compuesto 155 fue obtenido haciendo reaccionar primero el compuesto 151 de acuerdo con el procedimiento descrito para el compuesto 139. Este producto de reacción se trató de acuerdo con el procedimiento descrito para el compuesto 144. m/z = 455 (M+H). La estructura del compuesto 155 se muestra a continuación.

3-(3-acetil-fenil)-2-[1 -(2-amino-9H-purin-6-ilamino)-etil]-5-metil-3H-quinazolin4-ona (155)
5-met¡l-3-(3-morfol¡n-4-¡l-fen¡l)-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (156)
[0354] Se preparó el compuesto 156 de acuerdo con los procedimientos establecidos en los pasos A y B a continuación.
5-met¡l-3-(3-morfol¡no-4-¡l-fen¡l)-2-f1-[9-(2-tr¡met¡ls¡lan¡letox¡met¡l)-9H-pur¡n-6-¡lam¡no]-et¡l1-3h-qu¡nazol¡n-4-ona (157)
[0355] Paso A: Un vial de reacción de 3-mL se cargó con el compuesto 138 (96,1 mg, 0,142 mmol), paladio (II) acetato de metilo (3,20 mg, 0,014 mmol), carbonato de cesio (84,2 mg, 0,258 mmol) y (+)-BINAP (es decir, 2,2'-bis(difenilfosfino)
1,1 ’-binaftil) (13,8 mg, 0,022 mmol). A continuación, el vial se lavó abundantemente con nitrógeno durante 10 min. Tolueno (0,3 ml) y morfolina (18 gL) entonces se añadieron, y la solución se calentó a 100°C durante 6 h. Posteriormente, la solución se diluyó con diclorometano (5 ml), se filtró y el filtrado se concentró a presión reducida. La HPLC preparativa del residuo proporcionó el compuesto 157 en forma de un aceite amarillo. 1H RMN (CH3OD) 5 (ppm) 8,24-8,30 (m, 2H), 7,48 7,72 (m, 3H), 7,33 (m, 2H), 6,94-7,13 (m, 3H), 5,56 y 5,64 (dos s, relación de rotámero de CH21:10), 5,15-5,30 (m, 1H), 3,90 (m, 2H), 3,76 (m, 2H), 3,67 (m, 2H), 3,28 (m, 1H), 2,97-3,15 (m, 3H), 2,84 (s, 3H), 1,62 (m, 3H), 0,96 (m, 2H), -0,03 (s, 9H).
5-met¡l-3-(3-morfol¡n-4-¡l-fen¡l)-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (156)
[0356] Paso B: El compuesto 156 se preparó haciendo reaccionar el compuesto 157 de acuerdo con el procedimiento para la preparación del compuesto 137 (paso final C). m/z = 483 (M+H). La estructura del compuesto 156 se muestra a continuación.

5-metil-3-(3-morfolin-4-il-fenil)-2-[1 -(9H-purin-6-ilamino)-etil]-3H-quinazolin-4-ona (156)
2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-3-(3-morfol¡n-4-¡l-fen¡l)-3H-qu¡nazol¡n-4-ona (158)
[0357] El compuesto 158 se preparó de acuerdo con el procedimiento descrito para el compuesto 129, pero se usó el compuesto 133 en lugar del compuesto 126. m/z = 498 (M+H). La estructura del compuesto 158 se muestra a continuación.

2-[1-(2-Am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-5-met¡l-3-(3-morfol¡n-4-¡l-fen¡l)-3H-qu¡nazol¡n-4-ona (158)
2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-[3-(2-metox¡-etox¡)-fen¡l]-5-met¡l-3H-qu¡nazol¡n-4-ona (159)
[0358] El compuesto 159 se preparó haciendo reaccionar el compuesto 133 de acuerdo con el procedimiento para la preparación del compuesto 147. m/z = 487 (M+H). La estructura del compuesto 159 se muestra a continuación.

2-[1-(2-Am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-[3-(2-metox¡-etox¡)-fen¡l]-5-met¡l-3H-qu¡nazol¡n-4-ona (159)
2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-[3-(2-d¡met¡lam¡no-etox¡)-fen¡l]-5-met¡l-3H-qu¡nazol¡n-4-ona (160)
[0359] El compuesto 160 se preparó haciendo reaccionar el compuesto 133 de acuerdo con el procedimiento para la preparación del compuesto 146. m/z = 500 (M+H). La estructura del compuesto 160 se muestra a continuación.

2-[1-(2-Am¡no-9H-pur¡n-6-¡lam¡no)-et¡l]-3-[3-(2-d¡met¡lam¡no-etox¡)-fen¡l]-5-met¡l-3H-qu¡nazol¡n-4-ona (160) EJEMPLO 11
PREPARACIÓN DE COMPUESTOS
[0360] Los compuestos que tienen la fórmula general I (mostrada más arriba) han sido preparados de acuerdo con los pasos A-D del esquema sintético titulado "Procedimiento E" se muestra a continuación. El procedimiento E proporciona un método alternativo adicional para preparar compuestos con una variedad de cadenas laterales unidas al enlazador entre los anillos de quinazolinona y purina de los compuestos de la invención. Aunque se ejemplifica un grupo funcional propargilo, el método ilustrado en el Procedimiento E es aplicable a muchos grupos funcionales conocidos.
Procedimiento E
[0361]

2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-but-3-¡n¡l]-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (161)
[0362] El compuesto 161 se preparó de acuerdo con los procedimientos expuestos en los pasos A-D a continuación.
2-[(benzh¡dr¡l¡deno-am¡no)-met¡l]-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (162)
[0363] Paso A: Un matraz de fondo redondo de 100 ml, de una boca, equipado con un agitador magnético y un condensador de reflujo se cargó con 2-aminometil-5-metil-3-fenil-3H-quinazolin-4-ona (2,91 g, 11,0 mmol), benzhidrilideneamina (2,39 g, 13,2 mmol) y 1,2 -dicloroetano (15 ml). La mezcla de reacción se calentó a reflujo durante 4 h en atmósfera de nitrógeno y luego se enfrió a temperatura ambiente. La concentración a presión reducida seguida de purificación por cromatografía en columna proporcionó el compuesto 162 como un sólido naranja. pf 49°C (desc.); 1H RMN (DMSO-afe) 5 (ppm) 7,22-7,78 (m, 17H), 6,74 (m, 1H), 4,17 (s, 2H), 2,74 (s, 3H); m/z = 430 (M+H). La reacción descrita anteriormente y el compuesto 162 se muestran a continuación.

2-[1-(Benzh¡dr¡l¡den-am¡no)-but-3-¡n¡n-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (163)
[0364] Paso B: Un matraz de fondo redondo de 25 ml, de una boca, equipado con un agitador magnético se purgó con nitrógeno y se cargó con el compuesto 162 (500 mg, 1,20 mmol) y THF anhidro (4 ml). Se añadió en una porción una solución 1 M de ferc-butóxido de potasio en THF (1,40 ml, 1,40 mmol). Después de agitar durante 20 min a temperatura ambiente, se añadió una solución al 80% de bromuro de propargilo en tolueno (210 ml, 1,89 mmol) y la reacción se agitó durante 15 min más a temperatura ambiente. Luego se añadió bicarbonato de sodio acuoso saturado (5 ml), las capas se separaron y la capa acuosa se extrajo con acetato de etilo (2 x 10 ml). Los extractos orgánicos combinados se secaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. La purificación del residuo por cromatografía en columna proporcionó el producto como un sólido amarillo. 1H RMN (CD3OD) 5 7,72 (m, 2H), 7,24-7,67 (m, 3H), 6,77 (m, 3H), 4,61 (t, 1 H, J = 7,1 Hz), 3,01-3,11 (m, 1H), 2,76 (s, 3H), 2,57 (m, 1H), 2,23 (t, 1 H, J = 2,5 Hz). La reacción descrita anteriormente y el compuesto 163 se muestran a continuación.

2-(1-am¡no-but-3-¡n¡l)-5-met¡l-3-fen¡l-3H-qu¡nazol¡n-4-ona (164)
[0365] Paso C: Un matraz de fondo redondo de una boca y 50 ml equipado con un agitador magnético se cargó con el compuesto 163 (193 mg, 0,41 mmol) y éter dietílico (5 ml). Se añadió una solución 2N de ácido clorhídrico (5 ml) en una porción. Después de agitar durante 1,5 h a temperatura ambiente, se añadió cloruro de sodio (750 mg, 12,8 mmol) a la mezcla de reacción y se continuó agitando durante 10 min más. El precipitado resultante se filtró, se lavó secuencialmente con éter dietílico (0,5 ml), ácido clorhídrico 2N (1 ml) y MfBE (2 X1 ml). El secado al vacío a 45°C durante 2 h proporcionó el producto como un sólido rosa. 1H RMN (DMSO-ds) 58,82 (s, 3H), 7,79 (t, 1 H, J = 7,7 Hz), 7,41-7,66 (m, 6H), 7,42 (d, 1 H, J = 7,3 Hz), 3,91 9 s, 1H), 3,11 (m, 1H), 2,87 (m, 1H), 2,75 (s, 3H), 2,54-2,67 (m, 1H). La reacción descrita anteriormente y el compuesto 164 se muestran a continuación.

5-met¡l-3-fen¡l-2-[1-(9H-pur¡n-6-¡lam¡no)-but-3-¡n¡l]-3H-quinazol¡n-4-ona (161)
[0366] Paso D: El compuesto 161 se preparó haciendo reaccionar el compuesto 164 preparado siguiendo el procedimiento para la preparación del compuesto 14 (Paso D) usando tres equivalentes de diisopropiletilamina en lugar de uno. ESI-MS m/z = 422 (MH+). La reacción descrita anteriormente y el compuesto 161 se muestran a continuación.

2-[1-(2-amino-9H-purin-6-ilamino)-but-3-in¡n-5-metil-3-fenil-3H-quinazolin-4-ona (165)
[0367] El compuesto 165 se preparó siguiendo el procedimiento general descrito anteriormente para la preparación del compuesto 161, pero se sustituyó 2-amino-6-bromopurina por 6-bromopurina en el paso D. ESI-MS m/z = 437 (MH+). Ejemplo 12
Preparación de compuestos
[0368] Los compuestos que tienen la fórmula general I (mostrada más arriba) han sido preparados de acuerdo con los pasos A-E del esquema sintético titulado "Procedimiento K" mostrado a continuación. El procedimiento K proporciona un método alternativo adicional para preparar tales compuestos mediante un intermedio de oxazina (paso C).
Procedimiento K
[0369]


5-cloro-3-(3.5-d¡fluoro-fen¡l)-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l1-3H-qu¡nazol¡n-4-ona (166)
[0370] El compuesto 166 se preparó de acuerdo con los procedimientos expuestos en los pasos A-E a continuación. 2-am¡no-6-cloro-N-(3.5-d¡fluoro-fen¡l)-benzam¡da (167)
[0371] Paso A: Compuesto 167 se preparó siguiendo el procedimiento siguiendo el procedimiento para la preparación de compuesto 15 (Paso A), pero el ácido 2-amino-6-clorobenzoico se sustituyó por ácido 2-amino-6-metilbenzoico y 3,5-difluoroanilina se sustituyó por anilina. La reacción descrita anteriormente y el compuesto 167 se muestran a continuación.

Éster terc-butílico del ácido f1-f3-cloro-2-(3.5-d¡fluoro-fen¡l-carbamo¡l)-fen¡l-carbamo¡l1-et¡l}carbám¡co (168) [0372] Paso B: Un matraz de fondo redondo redondo de 100 ml, de una boca, se cargó equipado con un agitador
magnético y un condensador de reflujo con el compuesto 167 (10,6 mmol), éster de n-terc-butiloxicarbonil-D, L-alanina N-hidroxisuccinimida (3,64 g, 12,7 mmol), 4-N,N-dimetilaminopiridina (710 mg, 5,82 mmol) y tamices moleculares de 4Á (3,00 g). El matraz se purgó con nitrógeno y se añadieron tolueno anhidro (15 ml) y N,N-diisopropiletilamina (1,64 g, 2,22 mmol). La mezcla de reacción se calentó a 90°C durante 7 h y la suspensión resultante se filtró en caliente. La concentración del filtrado a presión reducida proporcionó un sólido marrón claro, que se purificó mediante cromatografía en columna (gel de sílice, EtOAc/hexanos). Esto proporcionó un rendimiento del 86% del compuesto 168 en forma de un sólido blanco. pf 194-196°C (desc.); 1H RMN (DMSO-ds) 5 (ppm) 10,96 (s, 1H), 9,46 (s, 1H), 7,84 (d, 1H, J = 7,8 Hz), 7,51 -7,36 (m, 4H), 7,19 (d, 1H, J = 6,8 Hz), 6,99 (t, 1H, J = 9,3 Hz), 4,10 (t, 1H, J = 7,0 Hz), 1,31 (s, 9H), 1,16 (d, 3H, J = 6,9 Hz); m/z = 454 (M+H). La reacción descrita anteriormente y el compuesto 168 se muestran a continuación.

Éster terc-butílico del ácido f1-[5-cloro-4-(3.5-d¡fluoro-fen¡l¡m¡no)-4H-benzo[d1[1.31oxaz¡n-2-¡l1-et¡l)-carbám¡co (169)
[0373] Paso C: un matraz de fondo redondo de 100 ml, de tres bocas, equipado con un agitador magnético y termómetro, se purgó con nitrógeno y se cargó con el compuesto 168 (3,30 mmol), cloruro de metileno anhidro (25 ml), N,N-diisopropiletilamina (4,60 g, 35,7 mmol) y trifenilfosfina (3,98 g, 15,2 mmol). Después, la mezcla de reacción se enfrió a 0-5°C en un baño de hielo/agua. A continuación, se añadió yodo (3,61 g, 14,2 mmol) en porciones a la mezcla de reacción durante 1 h. Una vez que se completó la adición, se retiró el baño de enfriamiento y la mezcla se agitó a temperatura ambiente durante 30 min más. Después, la reacción se inactivó con carbonato de potasio acuoso al 10% (25 ml), la capa orgánica se separó, se secó sobre sulfato de sodio, se filtró y se concentró a presión reducida. La cromatografía en columna del sólido resultante (gel de sílice, EtOAc/hexanos) dio un rendimiento del 52% del compuesto 169 en forma de un sólido blanco. pf 117-118°C; 1H RMN (DMSO-ds) 5 (ppm) 7,72-7,60 (m, 2H), 7,42 (dd, 1H, J = 7,6 Hz, 1,3 Hz), 7,31 (d, 1H, J = 7,0 Hz), 6,95 (t, 1 H, J = 9,3 Hz), 6,82 (m, 2H), 4,27 (m, 1H), 1,33 (s, 9H), 1,28 (d, 3H, J = 7,2 Hz); m/z = 436 (M+H). La reacción descrita anteriormente y el compuesto 169 se muestran a continuación.

2-(1-am¡no-et¡l)-5-cloro-3-(3.5-d¡fluoro-fen¡l)-3H-qu¡nazol¡n-4-ona (170)
[0374] Paso D: Se agitó una solución del compuesto 169 (1,68 mmol) en piperidina (2 ml) durante 3 h a temperatura ambiente. La evaporación de la mezcla de reacción a sequedad a alto vacío dio una espuma amarilla. Esta espuma se disolvió en una solución 4 M de cloruro de hidrógeno en 1,4-dioxano (4 ml) y se agitó durante 17 h a temperatura ambiente. Después, la mezcla de reacción se concentró a sequedad, se basificó con carbonato de potasio acuoso al 10% (40 ml) y se extrajo con MTBE (3 x 20 ml). La combinación de los extractos orgánicos, el secado sobre sulfato de sodio y la concentración a sequedad proporcionó un residuo sólido. Este residuo se disolvió en d-cloroformo (5 ml) y se calentó durante 15 h a 50°C. Después de enfriar a temperatura ambiente, la mezcla de reacción se lavó con agua (3 x 10 ml), se secó sobre sulfato de sodio y se evaporó hasta sequedad, proporcionando un rendimiento del 98% del compuesto 170 en forma de un sólido blanco. pf 200-202°C; 1H RMN (CDCh) 5 (ppm) 7,65 (m, 2H), 7,49 (m, 1H), 7,01 (t, 1 H, J = 6,6 Hz), 6,89 (m, 2H), 3,68 (m, 1H), 1,33 (d, 3H, J = 6,6 Hz), 1,25 (s, 2 H); m/z = 336 (M+H). La reacción descrita anteriormente y el compuesto 170 se muestran a continuación.

5-cloro-3-(3.5-d¡fluoro-fen¡l)-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡l]-3H-qu¡nazol¡n-4-ona (166)
[0375] Paso E: El compuesto 166 se preparó haciendo reaccionar el compuesto 170 de acuerdo con el procedimiento para la preparación del compuesto 107 (procedimiento final). ESI-MS m/z 454,3 (MH+). La reacción descrita anteriormente y el compuesto 166 se muestran a continuación.

2-[1-(2-amino-9H-purin-6-ilamino)-propin-5-cloro-3-(3.5-difluoro-fenil)-3H-quinazolin-4-ona (171)
[0376] El compuesto 171 se preparó siguiendo el procedimiento general para el compuesto 161 (pasos A-E), pero el éster 2,5-dioxo-pirrolidin-1 -ilo del ácido 2-terc-butoxicarbonilamino-butírico se sustituyó por N-terc-butiloxicarbonil-D,L-alanina éster de N-hidroxisuccinimida en el paso B, y 2-amino-6-bromopurina se sustituyó por 6-bromopurina en el paso E. ESI-MS m/z 454,3 (MH+). La estructura del compuesto 171 se muestra a continuación.

(1 71 )
2-[1-(2-am¡no-9H-pur¡n-6-¡lam¡no)-et¡n-5-cloro-3-(3.5-d¡fluoro-fen¡l)-3H-qu¡nazol¡n-4-ona (172)
[0377] El compuesto 172 se preparó siguiendo el procedimiento general para el compuesto 161 (pasos A-E), pero 2-amino-6-bromopurina fue sustituido por 6-bromopurina en el paso E. La estructura del compuesto 172 se muestra a continuación.

3-(3.5-d¡fluoro-fen¡l)-6-fluoro-2-[1-(9H-pur¡n-6-¡lam¡no)-et¡n-3H-qu¡nazol¡n-4-ona (173)
[0378] El compuesto 173 fue preparado siguiendo el procedimiento general para el compuesto 161 (pasos A-E), pero el ácido 2-amino-5-fluorobenzoico se sustituyó por ácido 2-amino-6-clorobenzoico en el paso A. ESI-MS m/z 438,2 (MH+).
La estructura del compuesto 173 se muestra a continuación.

5-cloro-3-(2.6-d¡fluoro-fen¡l)-2-[1-(9H-pur¡n-6-¡lam¡no)-prop¡l]-3H-qu¡nazol¡n-4-ona (174)
[0379] El compuesto 174 fue preparado siguiendo el procedimiento general para el compuesto 161 (pasos A-E), pero 2,6-difluoroanilina sustituido por anilina en el paso A, y ácido 2-terc-butoxicarbonilamino-butírico éster 2,5-dioxo-pirrolidin-1-ilo sustituido para éster de N-terc-butiloxicarbonil-D,L-alanina N-hidroxisuccinimida en el paso B. ESI-MS m/z 468,2 (MH+). La estructura del compuesto 174 se muestra a continuación.

2-[1-(2-amino-9H-purin-6-ilamino)-propin-5-cloro-3-(2.6-difluoro-fenil)-3H-quinazolin-4-ona (175)
[0380] El compuesto 175 se preparó siguiendo el procedimiento general para el compuesto 161 (pasos A-E), pero en el paso A se sustituyó la anilina por 2,6-difluoroanilina, ácido 2-terc-butoxicarbonilaminobutírico éster 2,5-dioxo-pirrolidin-1-ilo se sustituyó por N-terc-butiloxicarbonil-D,L-alanina éster de N-hidroxisuccinimida de en el paso B, y 2-amino-6-bromopurina se sustituyó por 6-bromopurina en el paso E. ESI-MS m/z 483,2 (MH+). La estructura del compuesto 175 se muestra a continuación.

EJEMPLO 13
PREPARACIÓN DE COMPUESTOS
[0381] Los compuestos que tienen la fórmula general I (mostrada más arriba) han sido preparados de acuerdo con los pasos A-G del esquema sintético titulado "Procedimiento L" mostrado a continuación.

5-met¡l-3-fen¡l-2-[1-(9H-pur¡n-6-¡lox¡)-et¡l]-3H-qu¡nazol¡n-4-ona (176)
[0382] El compuesto 176 se preparó de acuerdo con los procedimientos establecidos en pasos A-G a continuación.
Ác¡do acético 1-(3-met¡l-2-fen¡l-carbamo¡l-fen¡l-carbamo¡l)-éster etílico (177)
[0383] Paso A: Cloruro de (S)-2-acetoxipropionilo (5,469 g, 36,32 mmol) se añadió a una solución del compuesto 15 (6,788 g, 30 mmol) en diclorometano (150 ml). Inmediatamente se formó un precipitado. La reacción se agitó durante 25 h y el precipitado se filtró. El filtrado se lavó con una solución saturada de bicarbonato de sodio (3 x 50 ml) y se secó (MgSO4). La filtración y concentración del filtrado dio un sólido marrón (7,6 g). La purificación por cromatografía flash (EtOAc: hexanos 1:2 ->EtOAc->EtOAc: MeOH 10:1) seguida de recristalización de las fracciones impuras en EtOAc:hexanos dio el compuesto 177 como un sólido. ESI-MS m/z = 341 (MH+). La reacción descrita anteriormente y el compuesto 177 se muestran a continuación.

Ác¡do acético 1-(5-met¡l-4-fen¡l¡m¡no-4H-benzo[d][1.3]oxaz¡n-2-¡l)-éster etílico (178)
[0384] Paso B: El compuesto 177 (0,34 g, 1,0 mmol) se disolvió en diclorometano (25 ml). Se añadió trifenilfosfina (1,311 g, 5 mmol) a la solución, seguido de yodo (1,269 g, 5 mmol) y DIEA (1,9 ml, 11 mmol). La reacción se tapó y se agitó durante 4 días. La reacción se detuvo mediante la adición de una solución acuosa saturada de bicarbonato de sodio (25 ml). La capa orgánica se separó, se secó (MgSO4), se filtró y el filtrado se concentró a presión reducida para dar una goma de color marrón oscuro (2,886 g). La purificación por cromatografía flash (CH2Cl2) dio el compuesto de imino-1,3 -oxazina 178 en forma de un aceite amarillo. ESI-MS m/z = 323 (MH+). La reacción descrita anteriormente y el compuesto 178 se muestran a continuación.

Ácido acético 1-met¡l-2-(3-met¡l-2-fen¡l-carbamo¡l-fen¡l¡m¡no)-2-p¡per¡d¡n-1-¡l-éster etílico (179)
[0385] Paso C: La piperidina (1 ml) se añadió al compuesto 178 (0,161 g, 0,5 mmol) y la mezcla de reacción se agitó durante 19,5 h. Después, la mezcla de reacción se concentró a presión reducida para dar una goma amarilla. La trituración con EtOAc:hexanos 1:4 dio una pequeña cantidad del compuesto 179 (0,041 g). La cromatografía flash (EtOAc:hexanos 1:4) del filtrado dio solo una mezcla parcialmente separable de los productos esperados, el compuesto acetoxiquinazolinona 179 y la hidroxiquinazolinona (masa total 0,122 g). ESI-MS m/z = 408 (MH+). La reacción descrita anteriormente y el compuesto 179 se muestran a continuación.

Ácido acético 1-(5-met¡l-4-oxo-3-fen¡l-3.4-d¡h¡dro-qu¡nazolin-2-¡l)éster etílico (180)
[0386] Paso D: El compuesto 179 (0,037 g, 0,09 mmol) se disolvió en acetonitrilo (10 ml) y la mezcla de reacción se calentó a reflujo durante 3 h. El disolvente se eliminó a presión reducida y el residuo se disolvió en una mezcla de acetato de etilo (10 ml) y 1M HCl (5 ml). Después de separar la capa acuosa, la capa orgánica se lavó con 1M HCl adicional (2 x 5 ml), solución saturada de bicarbonato de sodio (3 x 5 ml), agua (2 x 5 ml) y salmuera saturada (5 ml). La solución se secó (MgSO4), se filtró, y el filtrado se concentró a presión reducida para dar el compuesto 180. ESI-MS m/z = 323 (MH+). El producto se había racemizado casi por completo en este punto (la pureza quiral era 46:54 S:R). La reacción descrita anteriormente y el compuesto 180 se muestran a continuación.

2-(1-hidroxi-etil)-5-metil-3-fenil-3H-quinazolin-4-ona (181)
[0387] Paso E: El compuesto 180 (0,011 g, 0,034 mmol) se disolvió en metanol (2 mL) y se añadió carbonato de potasio (0,012 g, 0,085 mmol). La mezcla de reacción se agitó durante 20 min y luego se añadió agua (20 ml). La mezcla se extrajo con acetato de etilo (3 x 10 ml) y los extractos orgánicos combinados se lavaron con salmuera saturada (10 ml). La solución orgánica se secó (MgSO4), se filtró y se concentró a presión reducida para dar el compuesto 181. ESI-MS m/z

5-met¡l-3-fen¡l-2-í1-[9-(2-tr¡met¡ls¡lan¡l-etox¡met¡l)-9H-pur¡n-6-¡lox¡]-et¡l}-3H-qu¡nazol¡n-4-ona (182)
[0388] Paso F: Se trató una solución del compuesto 181 (0,069 g, 0,25 mmol) en THF (5 ml) con hidruro de sodio (0,007 g, 0,27 mmol) y se agitó durante 10 min. Se añadió a la mezcla de reacción una solución del compuesto intermedio 13 (0,077 g, 0,27 mmol) en THF (1 ml). El matraz que originalmente contenía el compuesto intermedio 13 se lavó con THF adicional (1 ml) y también se añadieron los lavados a la mezcla de reacción. Se dejó que prosiguiera la reacción y se añadió más hidruro de sodio (0,005 g, 0,21 mmol) a las 21,5 h y 23 h. La reacción se detuvo después de un total de 24 h mediante la adición de una solución saturada de cloruro de amonio (5 ml). La mezcla se extrajo con diclorometano (3 x 5 ml) y los extractos orgánicos combinados se secaron sobre MgSO4. Después de la filtración, el filtrado se concentró a presión reducida y el residuo se purificó mediante cromatografía flash (EtOAc:hexanos 1:1 - EtOAc:hexanos 3:2) para dar el compuesto 182. ESI-MS m/z = 529 (MH+). La reacción descrita anteriormente y el compuesto 182 se muestran a continuación.

5-Met¡l-3-fen¡l-2-[1-(9H-pur¡n-6-¡lox¡)-et¡l]-3H-qu¡nazol¡n-4-ona (176)
[0389] Se disolvió el compuesto 182 (0,053 g, 0,1 mmol) en una mezcla de metanol (2 ml) y 4M HCl (2 ml). La mezcla se agitó y se calentó a 40°C durante 3 h. La mezcla de reacción se retiró de la fuente de calor y se dejó enfriar. Después, la mezcla de reacción se filtró a través de un lecho corto de papel de filtro GFA (fibra de vidrio) y el filtrado se concentró para eliminar únicamente el metanol. El residuo se ajustó a pH 10 mediante la adición de una solución de carbonato de potasio al 10%. El sólido resultante se recogió por filtración y se purificó mediante RP-HPLC (columna C18 Luna, 10x250 mm, 4,7 ml/min, CH3CN al 10-90% en agua en 18 min, con todos los disolventes que contenían ácido fórmico al 0,05%) para dar compuesto 176 como un sólido blanco esponjoso después de la liofilización. 1H RMN (400 MHz, d6-DMSO) 5 13,40, br s, 1H; 8,40, s, 1 H; 8,35, s, 1 H; 7,66, t, J = 7,8 Hz, 1 H; 7,48 - 7,58, m, 4H; 7,31-7,36, m, 2H; 7,20 - 7,23, m, 1H; 5,65, q, J = 6,6 Hz, 1H; 2,73, s, 3 H; 1,65, d, J = 6,6 Hz, 3H. eS i-MS m/z = 399 (MH+). La reacción descrita anteriormente y el compuesto 176 se muestran a continuación.

EJEMPLO 14
Ensayos bioquím icos de potencia y selectividad PI3K
Ensayo bioquím ico utilizando 20 pM ATP
[0390] Usando el método descrito en el Ejemplo 2, los compuestos de la divulgación se ensayaron para la actividad inhibidora y potencia contra PI3K5, y para la selectividad para PI3K5 versus otras isoenzimas PI3K de clase I. En la Tabla 1, valores CI50 (gm) se dan para PI3K5 ("Delta"), y se pueden calcular para las otras isoformas utilizando las proporciones de valores CI50 discutidas a continuación. Para ilustrar la selectividad de los compuestos, las proporciones de los valores CI50 de los compuestos para PI3Ka, PI3Kp, y PI3Ky relativos a PI3K5 se dan, respectivamente, como "Relación de Alfa/Delta", "Relación de Beta/Delta,” y “Relación Gamma/Delta”.
[0391] Los ensayos de selectividad iniciales se realizaron de forma idéntica al protocolo del ensayo de selectividad en el Ejemplo 2, excepto usando 100 gl Ecoscint para la detección del radiomarcador. Los ensayos de selectividad subsiguientes se realizaron de manera similar utilizando las mismas reservas de sustrato 3X, excepto que contenían 0,05 mCi/ml de y[32P]ATP y PIP2 3 mM. Los ensayos de selectividad posteriores también utilizaron las mismas reservas de enzimas 3X, excepto que ahora contenían 3 nM de cualquier isoforma de PI3K dada.
[0392] Para todos los ensayos de selectividad, los compuestos de ensayo se pesaron y se disolvieron en reservas de 10 50 mM en 100% DMSO (en función de sus respectivas solubilidades) y se almacenaron a -20°C. Los compuestos se descongelaron (a temperatura ambiente o 37°C), se diluyeron a 300 gm en agua a partir de la cual se realizó una serie de diluciones de 3 veces en agua. A partir de estas diluciones, se añadieron 20 gl a los pocillos de ensayo junto con los blancos de agua utilizados para el control de enzima (positivo) y el control sin enzima (de fondo). El resto del ensayo se realizó esencialmente de acuerdo con el protocolo de ensayo de selectividad en el Ejemplo 2.
Claims (15)
1. Una composición farmacéutica que comprende el compuesto

o una sal farmacéuticamente aceptable del mismo.
2. Una composición farmacéutica según la reivindicación 1, que además comprende un portador, adyuvante o vehículo farmacéutico biocompatible.
3. El compuesto o una sal farmacéuticamente aceptable del mismo,

o una composición farmacéutica según la reivindicación 1 o 2, para uso terapéutico.
4. El compuesto

o una sal farmacéuticamente aceptable del mismo:
(A) para su uso en un método de alteración de la función leucocitaria, que comprende poner en contacto leucocitos con el compuesto en una cantidad suficiente para inhibir la actividad 3-quinasa delta de fosfatidilinositol en dichos leucocitos; o
(B) para su uso en un método de alteración de la función de los leucocitos que comprende poner en contacto los
leucocitos con una cantidad eficaz del compuesto; o
(C) para su uso en un método para interrumpir una función de los osteoclastos que comprende poner en contacto los osteoclastos con el compuesto en una cantidad suficiente para inhibir PI3K5 en los osteoclastos; o (D) para su uso en un método para mejorar un trastorno de resorción ósea en un mamífero que comprende administrar al mamífero una cantidad terapéuticamente eficaz del compuesto; o
(E) para su uso en un método para inhibir el crecimiento o la proliferación de células cancerosas de origen hematopoyético que comprende poner en contacto las células cancerosas con una cantidad eficaz del compuesto; o
(F) para su uso en el tratamiento de un cáncer de origen hematopoyético; o
(G) para su uso en la inhibición de PI3K5; o
(H) para su uso en el tratamiento de artritis reumatoide, artritis monoarticular, osteoartritis, artritis gotosa o espondilitis.
5. Un compuesto para su uso de acuerdo con la reivindicación 4 para su uso en un método de alteración de la función leucocitaria que comprende poner en contacto los leucocitos con una cantidad eficaz del compuesto, en donde los leucocitos comprenden células seleccionadas del grupo que consiste en neutrófilos, linfocitos B, linfocitos T, y basófilos.
6. Un compuesto para su uso según la reivindicación 5, en donde los leucocitos comprenden neutrófilos y en donde se interrumpe una o más funciones de neutrófilos seleccionados del grupo que consiste en liberación estimulada de superóxido, exocitosis estimulada y migración quimiotáctica.
7. Un compuesto para su uso según la reivindicación 5, en donde
(A) los leucocitos comprenden linfocitos B y en donde se interrumpe la proliferación de linfocitos B, la producción de anticuerpos por los linfocitos B o ambos; o
(B) los leucocitos comprenden linfocitos T y en donde se interrumpe la proliferación de linfocitos T ; o
(C) los leucocitos comprenden basófilos y en donde se interrumpe la liberación de histamina por los basófilos.
8. Un compuesto para su uso según la reivindicación 4 para su uso en el tratamiento de un cáncer de origen hematopoyético, en donde el cáncer de origen hematopoyético es linfoma, mieloma múltiple o leucemia.
9. Un compuesto para su uso de acuerdo con la reivindicación 4 para su uso en el tratamiento de un cáncer de origen hematopoyético, en donde el cáncer se selecciona del grupo que consiste en linfoma de Burkitt, linfoma de Hodgkin, linfomas no Hodgkin, linfomas linfocíticos, leucemia linfocítica y leucemia mieloide crónica.
10. Un compuesto para su uso de acuerdo con la reivindicación 4 para su uso en un método de tratamiento de trastornos inflamatorios o lesiones asociadas con la inflamación.
11. Un compuesto para su uso de acuerdo con la reivindicación 4 para su uso en un método de tratamiento de una enfermedad autoinmune.
12. Un compuesto para su uso de acuerdo con la reivindicación 4 para su uso en un método de tratamiento de lesión por reperfusión.
13. Un compuesto para su uso de acuerdo con la reivindicación 4 para su uso en un método de tratamiento de la osteoporosis.
14. Un compuesto para su uso de acuerdo con la reivindicación 4 para su uso en un método de tratamiento de la enfermedad de Paget.
15. Un compuesto para su uso de acuerdo con la reivindicación 4 para su uso en un método de tratamiento de trastornos alérgicos.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US57078404P | 2004-05-13 | 2004-05-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2873875T3 true ES2873875T3 (es) | 2021-11-04 |
Family
ID=34969603
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES05752122.1T Active ES2607804T3 (es) | 2004-05-13 | 2005-05-12 | Quinazolinones como inhibidores de la fosfatidilinositol humana 3-quinasa delta |
| ES16185213T Active ES2873875T3 (es) | 2004-05-13 | 2005-05-12 | Quinazolinonas como inhibidores de fosfatidilinositol 3-quinasa delta humano |
| ES13150110.8T Active ES2605792T3 (es) | 2004-05-13 | 2005-05-12 | Quinazolinona usada como inhibidor de la fosfatidilinositol 3-quinasa delta humana |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES05752122.1T Active ES2607804T3 (es) | 2004-05-13 | 2005-05-12 | Quinazolinones como inhibidores de la fosfatidilinositol humana 3-quinasa delta |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13150110.8T Active ES2605792T3 (es) | 2004-05-13 | 2005-05-12 | Quinazolinona usada como inhibidor de la fosfatidilinositol 3-quinasa delta humana |
Country Status (22)
| Country | Link |
|---|---|
| US (14) | USRE44638E1 (es) |
| EP (4) | EP3153514B1 (es) |
| JP (1) | JP2007537291A (es) |
| CN (2) | CN102229609A (es) |
| AU (2) | AU2005245875C1 (es) |
| BE (1) | BE2017C002I2 (es) |
| CA (1) | CA2566609C (es) |
| CY (3) | CY2016046I1 (es) |
| DK (2) | DK2612862T3 (es) |
| ES (3) | ES2607804T3 (es) |
| HR (2) | HRP20161657T1 (es) |
| HU (3) | HUE030950T2 (es) |
| IL (4) | IL179176A (es) |
| LT (3) | LT1761540T (es) |
| LU (1) | LUC00005I2 (es) |
| ME (2) | ME02688B (es) |
| NL (1) | NL300867I2 (es) |
| PL (3) | PL2612862T3 (es) |
| PT (3) | PT3153514T (es) |
| RS (2) | RS55546B1 (es) |
| SI (2) | SI3153514T1 (es) |
| WO (2) | WO2005113554A2 (es) |
Families Citing this family (385)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6667300B2 (en) * | 2000-04-25 | 2003-12-23 | Icos Corporation | Inhibitors of human phosphatidylinositol 3-kinase delta |
| WO2005016348A1 (en) * | 2003-08-14 | 2005-02-24 | Icos Corporation | Method of inhibiting immune responses stimulated by an endogenous factor |
| HUE030950T2 (en) | 2004-05-13 | 2017-06-28 | Icos Corp | Quinazolinones as 3-kinase delta inhibitors of human phosphatidylinositol |
| CA2566436C (en) * | 2004-05-13 | 2011-05-10 | Vanderbilt University | Phosphoinositide 3-kinase delta selective inhibitors for inhibiting angiogenesis |
| US9512125B2 (en) | 2004-11-19 | 2016-12-06 | The Regents Of The University Of California | Substituted pyrazolo[3.4-D] pyrimidines as anti-inflammatory agents |
| WO2006089106A2 (en) * | 2005-02-17 | 2006-08-24 | Icos Corporation | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| GB2431156A (en) * | 2005-10-11 | 2007-04-18 | Piramed Ltd | 1-cyclyl-3-substituted- -benzenes and -azines as inhibitors of phosphatidylinositol 3-kinase |
| DK2004654T3 (da) * | 2006-04-04 | 2013-07-22 | Univ California | Pyrazolopyrimidin derivater til anvendelse som kinase antagonister |
| KR20090087027A (ko) | 2006-11-13 | 2009-08-14 | 일라이 릴리 앤드 캄파니 | 염증 질환 및 암의 치료를 위한 티에노피리미디논 |
| EP1953163A1 (en) * | 2007-02-01 | 2008-08-06 | Boehringer Ingelheim Pharma GmbH & Co. KG | Pteridinone derivatives as PI3-kinases inhibitors |
| WO2009002553A1 (en) * | 2007-06-25 | 2008-12-31 | Prolexys Pharmaceuticals, Inc. | Methods of treating multiple myeloma and resistant cancers |
| FR2918668B1 (fr) * | 2007-07-09 | 2009-08-21 | Arkema France | Procede ameliore de preparation d'alcoxyamines issues de nitroxydes beta-phosphores. |
| WO2009046448A1 (en) * | 2007-10-04 | 2009-04-09 | Intellikine, Inc. | Chemical entities and therapeutic uses thereof |
| GEP20125635B (en) * | 2007-11-13 | 2012-09-10 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| AU2014203687B2 (en) * | 2008-01-04 | 2016-04-14 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US8703777B2 (en) * | 2008-01-04 | 2014-04-22 | Intellikine Llc | Certain chemical entities, compositions and methods |
| RU2537549C2 (ru) * | 2008-01-04 | 2015-01-10 | Интелликайн ЭлЭлСи | Некоторые химические структуры, композиции и способы |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| JP5547099B2 (ja) * | 2008-03-14 | 2014-07-09 | インテリカイン, エルエルシー | キナーゼ阻害剤および使用方法 |
| WO2009114874A2 (en) | 2008-03-14 | 2009-09-17 | Intellikine, Inc. | Benzothiazole kinase inhibitors and methods of use |
| EP2444403A1 (en) * | 2008-04-18 | 2012-04-25 | Shionogi Co., Ltd. | Heterocyclic compound having inhibitory activity on PI3K |
| WO2009132202A2 (en) | 2008-04-24 | 2009-10-29 | Incyte Corporation | Macrocyclic compounds and their use as kinase inhibitors |
| US20110224223A1 (en) | 2008-07-08 | 2011-09-15 | The Regents Of The University Of California, A California Corporation | MTOR Modulators and Uses Thereof |
| AU2009268611B2 (en) | 2008-07-08 | 2015-04-09 | Intellikine, Llc | Kinase inhibitors and methods of use |
| EA201100255A1 (ru) * | 2008-07-29 | 2011-08-30 | Бёрингер Ингельхайм Интернациональ Гмбх | 5-алкинилпиримидины |
| JP5731978B2 (ja) | 2008-09-26 | 2015-06-10 | インテリカイン, エルエルシー | 複素環キナーゼ阻害剤 |
| ES2570429T3 (es) | 2008-10-16 | 2016-05-18 | Univ California | Inhibidores de heteroaril quinasa de anillo condensado |
| US8476282B2 (en) * | 2008-11-03 | 2013-07-02 | Intellikine Llc | Benzoxazole kinase inhibitors and methods of use |
| CA3092449A1 (en) * | 2008-11-13 | 2010-05-20 | Gilead Calistoga Llc | Therapies for hematologic malignancies |
| US9492449B2 (en) | 2008-11-13 | 2016-11-15 | Gilead Calistoga Llc | Therapies for hematologic malignancies |
| WO2010092962A1 (ja) | 2009-02-12 | 2010-08-19 | アステラス製薬株式会社 | へテロ環誘導体 |
| EP2411391A1 (en) | 2009-03-24 | 2012-02-01 | Gilead Calistoga LLC | Atropisomers of2-purinyl-3-tolyl-quinazolinone derivatives and methods of use |
| ES2548253T3 (es) | 2009-04-20 | 2015-10-15 | Gilead Calistoga Llc | Métodos para el tratamiento de tumores sólidos |
| JP5789252B2 (ja) | 2009-05-07 | 2015-10-07 | インテリカイン, エルエルシー | 複素環式化合物およびその使用 |
| SI2448938T1 (sl) | 2009-06-29 | 2014-08-29 | Incyte Corporation Experimental Station | Pirimidinoni kot zaviralci pi3k |
| MX2012000817A (es) * | 2009-07-21 | 2012-05-08 | Gilead Calistoga Llc | Tratamiento para desordenes del higado con inhibidores pi3k. |
| SG178454A1 (en) | 2009-08-17 | 2012-03-29 | Intellikine Inc | Heterocyclic compounds and uses thereof |
| NZ598808A (en) * | 2009-09-09 | 2014-07-25 | Celgene Avilomics Res Inc | Pi3 kinase inhibitors and uses thereof |
| US8980899B2 (en) | 2009-10-16 | 2015-03-17 | The Regents Of The University Of California | Methods of inhibiting Ire1 |
| GB0918249D0 (en) | 2009-10-19 | 2009-12-02 | Respivert Ltd | Compounds |
| WO2011075643A1 (en) | 2009-12-18 | 2011-06-23 | Incyte Corporation | Substituted heteroaryl fused derivatives as pi3k inhibitors |
| BR112012019635A2 (pt) | 2010-02-22 | 2016-05-03 | Hoffmann La Roche | compostos inibidores de pirido[3,2-d] pirimidina pi3k delta e métodos de uso |
| US9193721B2 (en) | 2010-04-14 | 2015-11-24 | Incyte Holdings Corporation | Fused derivatives as PI3Kδ inhibitors |
| AU2011255218B2 (en) | 2010-05-21 | 2015-03-12 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US9062055B2 (en) | 2010-06-21 | 2015-06-23 | Incyte Corporation | Fused pyrrole derivatives as PI3K inhibitors |
| JP5555378B2 (ja) | 2010-07-14 | 2014-07-23 | エフ.ホフマン−ラ ロシュ アーゲー | Pi3kp110デルタに選択的なプリン化合物とその使用の方法 |
| ES2570177T3 (es) | 2010-08-10 | 2016-05-17 | Astellas Pharma Inc | Compuesto heterocíclico |
| UY33337A (es) | 2010-10-18 | 2011-10-31 | Respivert Ltd | DERIVADOS SUSTITUIDOS DE 1H-PIRAZOL[ 3,4-d]PIRIMIDINA COMO INHIBIDORES DE LAS FOSFOINOSITIDA 3-QUINASAS |
| EP2637669A4 (en) | 2010-11-10 | 2014-04-02 | Infinity Pharmaceuticals Inc | Heterocyclic compounds and their use |
| ES2764848T3 (es) | 2010-12-20 | 2020-06-04 | Incyte Holdings Corp | N-(1-(fenilo sustituido)etilo)-9H-purina-6-aminas como inhibidores de PI3K |
| JP2014501790A (ja) | 2011-01-10 | 2014-01-23 | インフィニティー ファーマシューティカルズ, インコーポレイテッド | イソキノリノンの調製方法及びイソキノリノンの固体形態 |
| US9295673B2 (en) | 2011-02-23 | 2016-03-29 | Intellikine Llc | Combination of mTOR inhibitors and P13-kinase inhibitors, and uses thereof |
| US20140213630A1 (en) | 2011-03-08 | 2014-07-31 | Thomas Diacovo | Methods and pharmaceutical compositions for treating lymphoid malignancy |
| US9108984B2 (en) | 2011-03-14 | 2015-08-18 | Incyte Corporation | Substituted diamino-pyrimidine and diamino-pyridine derivatives as PI3K inhibitors |
| WO2012135009A1 (en) | 2011-03-25 | 2012-10-04 | Incyte Corporation | Pyrimidine-4,6-diamine derivatives as pi3k inhibitors |
| CN102719517B (zh) * | 2011-03-29 | 2015-08-19 | 中国科学院上海药物研究所 | 一种检测化合物对人Ⅰ型PI3Ks抑制活性的方法 |
| EA024842B9 (ru) * | 2011-05-04 | 2017-08-31 | Ризен Фармасьютикалз Са | Соединения в качестве модуляторов протеинкиназы pi3k |
| CN102838600A (zh) * | 2011-06-24 | 2012-12-26 | 山东亨利医药科技有限责任公司 | 苯基喹唑啉类PI3Kδ抑制剂 |
| CN102838601A (zh) * | 2011-06-24 | 2012-12-26 | 山东亨利医药科技有限责任公司 | 选择性磷酰肌醇3-激酶δ抑制剂 |
| EP2734520B1 (en) | 2011-07-19 | 2016-09-14 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| AR088218A1 (es) | 2011-07-19 | 2014-05-21 | Infinity Pharmaceuticals Inc | Compuestos heterociclicos utiles como inhibidores de pi3k |
| RU2014111823A (ru) | 2011-08-29 | 2015-10-10 | Инфинити Фармасьютикалз, Инк. | Гетероциклические соединения и их применения |
| JP6342805B2 (ja) | 2011-09-02 | 2018-06-13 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 置換ピラゾロ[3,4−d]ピリミジンおよびその用途 |
| HUE030869T2 (en) | 2011-09-02 | 2017-06-28 | Incyte Holdings Corp | Heterocyclic amines as inhibitors of PI3K |
| US9630979B2 (en) | 2011-09-29 | 2017-04-25 | Infinity Pharmaceuticals, Inc. | Inhibitors of monoacylglycerol lipase and methods of their use |
| EP2578582A1 (en) | 2011-10-03 | 2013-04-10 | Respivert Limited | 1-Pyrazolyl-3-(4-((2-anilinopyrimidin-4-yl)oxy)napththalen-1-yl)ureas as p38 MAP kinase inhibitors |
| WO2013050757A1 (en) | 2011-10-03 | 2013-04-11 | Respivert Limited | 1-pyrazolyl-3- (4- ( (2 -anilinopyrimidin- 4 - yl) oxy) napththalen- 1 - yl) ureas as p38 map kinase inhibitors |
| WO2013082540A1 (en) * | 2011-12-02 | 2013-06-06 | Gilead Calistoga Llc | Compositions and methods of treating a proliferative disease with a quinazolinone derivative |
| EP2790705B1 (en) | 2011-12-15 | 2017-12-06 | Novartis AG | Use of inhibitors of the activity or function of pi3k |
| AU2015252058A1 (en) * | 2012-03-05 | 2015-11-19 | Gilead Calistoga Llc | Polymorphic forms of (S)-2-(1-(9H-purin-6-ylamino)propyl)-5-fluoro-3-phenylquinazolin-4(3H)-one |
| PE20141792A1 (es) * | 2012-03-05 | 2014-12-07 | Gilead Calistoga Llc | Formas polimorficas de (s)-2-(1-(9h-purin-6-ilamino)propil)-5-fluor-3-fenilquinazolin-4(3h)-ona |
| MX355299B (es) | 2012-03-13 | 2018-04-11 | Respivert Ltd | Inhibidores de cinasa de fosfoinositida 3 cristalina. |
| AR090548A1 (es) | 2012-04-02 | 2014-11-19 | Incyte Corp | Azaheterociclobencilaminas biciclicas como inhibidores de pi3k |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| WO2014015523A1 (en) * | 2012-07-27 | 2014-01-30 | Hutchison Medipharma Limited | Novel heteroaryl and heterocycle compounds, compositions and methods |
| SG11201501173SA (en) * | 2012-08-08 | 2015-05-28 | Kbp Biosciences Co Ltd | PI3Kδ INHIBITOR |
| EP2884980A1 (en) * | 2012-08-14 | 2015-06-24 | Gilead Calistoga LLC | Combination therapies for treating cancer |
| RU2015115631A (ru) | 2012-09-26 | 2016-11-20 | Дзе Риджентс Оф Дзе Юниверсити Оф Калифорния | Модулирование ire1 |
| SG11201502032VA (en) * | 2012-10-16 | 2015-05-28 | Almirall Sa | Pyrrolotriazinone derivatives as pi3k inhibitors |
| DK2914296T4 (da) | 2012-11-01 | 2022-01-03 | Infinity Pharmaceuticals Inc | Behandling af cancere under anvendelse af PI3-kinase-isoform-modulatorer |
| EA035391B1 (ru) * | 2012-11-08 | 2020-06-05 | Ризен Фармасьютикалз Са | Фармацевтические композиции, содержащие ингибитор pde4 и ингибитор pi3-дельта или двойной ингибитор pi3-дельта-гамма киназы |
| MX2015006192A (es) | 2012-11-16 | 2015-08-10 | Merck Sharp & Dohme | Inhibidores de purina de fosfatidilinositol 3-quinasa delta humana. |
| JP6207100B2 (ja) | 2012-12-21 | 2017-10-04 | ギリアード カリストガ エルエルシー | イソキノリノンまたはキナゾリノンホスファチジルイノシトール3−キナーゼ阻害剤 |
| CA2895782C (en) | 2012-12-21 | 2017-08-22 | Gilead Calistoga Llc | Substituted pyrimidine aminoalkyl-quinazolones as phosphatidylinositol 3-kinase inhibitors |
| JP6229896B2 (ja) * | 2013-01-21 | 2017-11-15 | 国立大学法人大阪大学 | フェノキシアルキルアミン化合物 |
| WO2014128612A1 (en) * | 2013-02-20 | 2014-08-28 | Novartis Ag | Quinazolin-4-one derivatives |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| NZ629037A (en) | 2013-03-15 | 2017-04-28 | Infinity Pharmaceuticals Inc | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| TW201522341A (zh) * | 2013-03-15 | 2015-06-16 | Respivert Ltd | 化合物 |
| AR095353A1 (es) | 2013-03-15 | 2015-10-07 | Respivert Ltd | Compuesto |
| CA2907726A1 (en) | 2013-03-22 | 2014-09-25 | Millennium Pharmaceuticals, Inc. | Combination of catalytic mtorc1/2 inhibitors and selective inhibitors of aurora a kinase |
| AU2014273946B2 (en) | 2013-05-30 | 2020-03-12 | Infinity Pharmaceuticals, Inc. | Treatment of cancers using PI3 kinase isoform modulators |
| JP6030783B2 (ja) | 2013-06-14 | 2016-11-24 | ギリアード サイエンシーズ, インコーポレイテッド | ホスファチジルイノシトール3−キナーゼ阻害剤 |
| UY35675A (es) | 2013-07-24 | 2015-02-27 | Novartis Ag | Derivados sustituidos de quinazolin-4-ona |
| WO2015014315A1 (zh) * | 2013-08-01 | 2015-02-05 | 杭州普晒医药科技有限公司 | 一种抑制剂的晶型及其制备方法和用途 |
| CN104418858B (zh) | 2013-08-30 | 2018-12-11 | 浙江医药股份有限公司新昌制药厂 | 2,6-二含氮取代的嘌呤衍生物及其制备方法和其药物组合物与应用 |
| KR20160060100A (ko) | 2013-09-22 | 2016-05-27 | 칼리토르 사이언시즈, 엘엘씨 | 치환된 아미노피리미딘 화합물 및 이용 방법 |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| PL3052485T3 (pl) | 2013-10-04 | 2022-02-28 | Infinity Pharmaceuticals, Inc. | Związki heterocykliczne i ich zastosowania |
| ES2818933T3 (es) | 2013-10-10 | 2021-04-14 | Acetylon Pharmaceuticals Inc | Inhibidores de la HDAC en combinación con los inhibidores de la pi3k, para el tratamiento del linfoma no Hodgkin |
| WO2015061204A1 (en) * | 2013-10-21 | 2015-04-30 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| IN2013MU03641A (es) | 2013-11-20 | 2015-07-31 | Cadila Healthcare Ltd | |
| AU2014354769A1 (en) | 2013-11-26 | 2016-05-26 | Gilead Sciences, Inc. | Therapies for treating myeloproliferative disorders |
| WO2015083008A1 (en) | 2013-12-05 | 2015-06-11 | Acerta Pharma B.V. | Therapeutic combination of a pi3k inhibitor and a btk inhibitor |
| CA2934531C (en) | 2013-12-20 | 2020-02-25 | Gilead Calistoga Llc | Process methods for phosphatidylinositol 3-kinase inhibitors |
| EP3083623A1 (en) | 2013-12-20 | 2016-10-26 | Gilead Calistoga LLC | Polymorphic forms of a hydrochloride salt of (s) -2-(9h-purin-6-ylamino) propyl) -5-fluoro-3-phenylquinazolin-4 (3h) -one |
| WO2015109009A1 (en) * | 2014-01-15 | 2015-07-23 | The Trustees Of Columbia University In The City Of New York | Carbonyl erastin analogs and their use |
| US20160331754A1 (en) | 2014-01-20 | 2016-11-17 | Gilead Sciences, Inc. | Therapies for treating cancers |
| JOP20200094A1 (ar) | 2014-01-24 | 2017-06-16 | Dana Farber Cancer Inst Inc | جزيئات جسم مضاد لـ pd-1 واستخداماتها |
| CN104817559B (zh) * | 2014-01-30 | 2021-05-25 | 苏州泽璟生物制药股份有限公司 | 氘代喹唑啉酮化合物以及包含该化合物的药物组合物 |
| JOP20200096A1 (ar) | 2014-01-31 | 2017-06-16 | Children’S Medical Center Corp | جزيئات جسم مضاد لـ tim-3 واستخداماتها |
| WO2015131080A1 (en) | 2014-02-28 | 2015-09-03 | Nimbus Lakshmi, Inc. | Tyk2 inhibitors and uses thereof |
| CU24481B1 (es) | 2014-03-14 | 2020-03-04 | Immutep Sas | Moléculas de anticuerpo que se unen a lag-3 |
| SG11201607705XA (en) | 2014-03-19 | 2016-10-28 | Infinity Pharmaceuticals Inc | Heterocyclic compounds for use in the treatment of pi3k-gamma mediated disorders |
| US20150320755A1 (en) | 2014-04-16 | 2015-11-12 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US20150320754A1 (en) | 2014-04-16 | 2015-11-12 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| WO2015168079A1 (en) | 2014-04-29 | 2015-11-05 | Infinity Pharmaceuticals, Inc. | Pyrimidine or pyridine derivatives useful as pi3k inhibitors |
| WO2015179772A1 (en) | 2014-05-23 | 2015-11-26 | Concert Pharmaceuticals, Inc. | Deuterated phenylquinazolinone and phenylisoquinolinone compounds |
| TW201613916A (en) | 2014-06-03 | 2016-04-16 | Gilead Sciences Inc | TANK-binding kinase inhibitor compounds |
| WO2015191677A1 (en) | 2014-06-11 | 2015-12-17 | Incyte Corporation | Bicyclic heteroarylaminoalkyl phenyl derivatives as pi3k inhibitors |
| TWI729644B (zh) | 2014-06-12 | 2021-06-01 | 美商西爾拉癌症醫學公司 | N-(氰基甲基)-4-(2-(4-𠰌啉基苯基胺基)嘧啶-4-基)苯甲醯胺 |
| NZ726052A (en) * | 2014-06-13 | 2018-04-27 | Gilead Sciences Inc | Phosphatidylinositol 3-kinase inhibitors |
| CN106459005A (zh) * | 2014-06-13 | 2017-02-22 | 吉利德科学公司 | 磷脂酰肌醇3‑激酶抑制剂 |
| ES2763104T3 (es) | 2014-06-13 | 2020-05-27 | Gilead Sciences Inc | Derivados de quinazolinona como inhibidores de fosfatidilinositol 3-quinasa |
| AU2015274635B2 (en) | 2014-06-13 | 2018-04-19 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| MA40240B1 (fr) | 2014-06-19 | 2019-03-29 | Ariad Pharma Inc | Composés hétéroaryle d'inhibition de la kinase |
| SG11201610770PA (en) | 2014-07-04 | 2017-01-27 | Lupin Ltd | Quinolizinone derivatives as pi3k inhibitors |
| CN104130261B (zh) * | 2014-08-04 | 2016-03-02 | 山东康美乐医药科技有限公司 | 艾德利布的合成方法 |
| TW201618775A (zh) | 2014-08-11 | 2016-06-01 | 艾森塔製藥公司 | Btk抑制劑、pi3k抑制劑、jak-2抑制劑、pd-1抑制劑及/或pd-l1抑制劑之治療組合物 |
| WO2016024232A1 (en) | 2014-08-11 | 2016-02-18 | Acerta Pharma B.V. | Therapeutic combinations of a btk inhibitor, a pi3k inhibitor, a jak-2 inhibitor and/or a cdk 4/6 inhibitor |
| HRP20211813T1 (hr) | 2014-08-11 | 2022-03-04 | Acerta Pharma B.V. | Terapeutske kombinacije inhibitora btk i inhibitora bcl-2 |
| CN117164657A (zh) | 2014-08-12 | 2023-12-05 | 莫纳什大学 | 定向淋巴的前药 |
| CN105330699B (zh) * | 2014-08-13 | 2018-12-04 | 山东汇睿迪生物技术有限公司 | 一种含磷吡啶并[2,3-d]嘧啶-7-酮类化合物或其药学上可接受的盐、药物组合物及其应用 |
| CN104262344B (zh) * | 2014-08-22 | 2015-11-04 | 苏州明锐医药科技有限公司 | 艾德拉尼的制备方法 |
| JP6681905B2 (ja) | 2014-09-13 | 2020-04-15 | ノバルティス アーゲー | Alk阻害剤の併用療法 |
| CN105503877A (zh) | 2014-09-24 | 2016-04-20 | 和记黄埔医药(上海)有限公司 | 咪唑并哒嗪类化合物及其用途 |
| CN106715419A (zh) | 2014-09-26 | 2017-05-24 | 吉利德科学公司 | 用作tank‑结合激酶抑制剂化合物的氨基三嗪衍生物 |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| AU2015327868A1 (en) | 2014-10-03 | 2017-04-20 | Novartis Ag | Combination therapies |
| MA41044A (fr) | 2014-10-08 | 2017-08-15 | Novartis Ag | Compositions et procédés d'utilisation pour une réponse immunitaire accrue et traitement contre le cancer |
| CR20170143A (es) | 2014-10-14 | 2017-06-19 | Dana Farber Cancer Inst Inc | Moléculas de anticuerpo que se unen a pd-l1 y usos de las mismas |
| ES2749679T3 (es) | 2014-10-22 | 2020-03-23 | Bristol Myers Squibb Co | Compuestos de pirrolotriazina amina sustituidos como inhibidores de PI3k |
| EP3209664B1 (en) | 2014-10-22 | 2020-06-03 | Bristol-Myers Squibb Company | Bicyclic heteroaryl amine compounds as pi3k inhibitors |
| ES2879809T3 (es) | 2014-11-01 | 2021-11-23 | Shanghai Fochon Pharmaceutical Co Ltd | Ciertos inhibidores de proteínas quinasas |
| EP3031808B1 (en) | 2014-12-09 | 2018-06-27 | ratiopharm GmbH | Salt of idelalisib |
| EP3048104A1 (en) | 2015-01-20 | 2016-07-27 | Sandoz AG | Amorphous and crystalline forms of idelalisib and process for forming the same |
| WO2016097314A1 (en) * | 2014-12-19 | 2016-06-23 | Sandoz Ag | Amorphous and crystalline forms of idelalisib and process for forming the same |
| WO2016100882A1 (en) | 2014-12-19 | 2016-06-23 | Novartis Ag | Combination therapies |
| WO2016108206A2 (en) * | 2014-12-31 | 2016-07-07 | Dr. Reddy’S Laboratories Limited | Processes for preparation of idelalisib and intermediates thereof |
| KR101710461B1 (ko) * | 2015-01-16 | 2017-02-27 | 순천향대학교 산학협력단 | 키랄 2-플루오로-4-나이트로 부탄산 유도체의 제조방법 |
| US9637488B2 (en) | 2015-01-29 | 2017-05-02 | Fuqiang Ruan | Heterocyclic compounds as inhibitors of class I PI3KS |
| TW201639573A (zh) | 2015-02-03 | 2016-11-16 | 吉李德科學股份有限公司 | 有關治療癌症之合併治療 |
| TWI788655B (zh) | 2015-02-27 | 2023-01-01 | 美商林伯士拉克許米公司 | 酪胺酸蛋白質激酶2(tyk2)抑制劑及其用途 |
| TWI748941B (zh) | 2015-02-27 | 2021-12-11 | 美商英塞特公司 | Pi3k抑制劑之鹽及製備方法 |
| CA2978188C (en) | 2015-03-04 | 2020-05-12 | Gilead Sciences, Inc. | Toll-like receptor modulating 4,6-diamino-pyrido[3,2-d]pyrimidine compounds |
| US20180064714A1 (en) | 2015-03-13 | 2018-03-08 | Mylan Laboratories Ltd | Process for the Preparation of Amorphous Idelalisib and its Premix |
| EP3277667A1 (en) * | 2015-03-31 | 2018-02-07 | Synthon B.V. | Improved process for preparing idelalisib |
| WO2016157136A1 (en) | 2015-04-02 | 2016-10-06 | Mylan Laboratories Ltd | Crystalline forms of idelalisib |
| CN106146502B (zh) * | 2015-04-09 | 2019-01-04 | 上海医药工业研究院 | 艾代拉里斯的合成方法及制备中间体 |
| CN108033961A (zh) | 2015-04-15 | 2018-05-15 | 上海方楠生物科技有限公司 | 一种艾德力布的无定型物及其制备方法 |
| CN106146352A (zh) * | 2015-04-16 | 2016-11-23 | 上海医药工业研究院 | Idelalisib中间体及其制备方法 |
| CN106146411A (zh) * | 2015-04-16 | 2016-11-23 | 上海医药工业研究院 | (s)-2-(1-氨基-丙基)-5-氟-3-苯基-3h-喹唑啉-4-酮的制备方法 |
| CN106146503A (zh) * | 2015-04-16 | 2016-11-23 | 上海医药工业研究院 | 一种Idelalisib的制备方法 |
| GB201506786D0 (en) * | 2015-04-21 | 2015-06-03 | Ucb Biopharma Sprl | Therapeutic use |
| WO2016183060A1 (en) | 2015-05-11 | 2016-11-17 | Incyte Corporation | Process for the synthesis of a phosphoinositide 3-kinase inhibitor |
| WO2016183063A1 (en) | 2015-05-11 | 2016-11-17 | Incyte Corporation | Crystalline forms of a pi3k inhibitor |
| CZ2015347A3 (cs) | 2015-05-22 | 2016-11-30 | Zentiva, K.S. | Pevné formy 5-fluor-3-fenyl-2-[(1S)-1-(9H-purin-6-ylamino)propyl]chinazolin-4-onu a jejich příprava |
| CN106279171A (zh) * | 2015-06-09 | 2017-01-04 | 南京安源生物医药科技有限公司 | 一种Idelalisib的制备方法 |
| WO2016209961A1 (en) | 2015-06-23 | 2016-12-29 | Gilead Sciences, Inc. | Combination therapies for treating b-cell malignancies |
| WO2017004134A1 (en) | 2015-06-29 | 2017-01-05 | Nimbus Iris, Inc. | Irak inhibitors and uses thereof |
| CN112062778B (zh) | 2015-07-02 | 2024-04-19 | 豪夫迈·罗氏有限公司 | 苯并氧氮杂䓬噁唑烷酮化合物及其使用方法 |
| CN111848643A (zh) | 2015-07-02 | 2020-10-30 | 豪夫迈·罗氏有限公司 | 苯并氧氮杂*噁唑烷酮化合物及其使用方法 |
| US20180207273A1 (en) | 2015-07-29 | 2018-07-26 | Novartis Ag | Combination therapies comprising antibody molecules to tim-3 |
| PT3317301T (pt) | 2015-07-29 | 2021-07-09 | Novartis Ag | Terapias de associação compreendendo moléculas de anticorpo contra lag-3 |
| WO2017019896A1 (en) | 2015-07-29 | 2017-02-02 | Novartis Ag | Combination therapies comprising antibody molecules to pd-1 |
| WO2017032679A1 (en) | 2015-08-21 | 2017-03-02 | Morphosys Ag | Combinations and uses thereof |
| CZ2015575A3 (cs) * | 2015-08-26 | 2017-03-08 | Zentiva, K.S. | Soli 5-fluoro-3-fenyl-2-[(1S)-1-(9H-purin-6-ylamino)propyl]quinazolin-4-onu a jejich příprava |
| JP6802263B2 (ja) | 2015-09-02 | 2020-12-16 | ニンバス ラクシュミ, インコーポレイテッド | Tyk2阻害剤およびその使用 |
| WO2017041139A1 (en) | 2015-09-08 | 2017-03-16 | Monash University | Lymph directing prodrugs |
| WO2017044720A1 (en) | 2015-09-11 | 2017-03-16 | Navitor Pharmaceuticals, Inc. | Rapamycin analogs and uses thereof |
| US10160761B2 (en) | 2015-09-14 | 2018-12-25 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| WO2017059224A2 (en) | 2015-10-01 | 2017-04-06 | Gilead Sciences, Inc. | Combination of a btk inhibitor and a checkpoint inhibitor for treating cancers |
| CA3234750A1 (en) | 2015-10-23 | 2017-04-27 | Navitor Pharmaceuticals, Inc. | Modulators of sestrin-gator2 interaction and uses thereof |
| SG11201803520PA (en) | 2015-11-03 | 2018-05-30 | Janssen Biotech Inc | Antibodies specifically binding pd-1 and their uses |
| WO2017087235A1 (en) | 2015-11-20 | 2017-05-26 | Senhwa Biosciences, Inc. | Combination therapy of tetracyclic quinolone analogs for treating cancer |
| HU231016B1 (hu) * | 2015-11-30 | 2019-11-28 | Egis Gyógyszergyár Zrt. | Idelalisib új polimorf és szolvát formája |
| US11370792B2 (en) | 2015-12-14 | 2022-06-28 | Raze Therapeutics, Inc. | Caffeine inhibitors of MTHFD2 and uses thereof |
| AU2016369537B2 (en) | 2015-12-17 | 2024-03-14 | Novartis Ag | Antibody molecules to PD-1 and uses thereof |
| CA3006772A1 (en) | 2015-12-17 | 2017-06-22 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| WO2017130221A1 (en) * | 2016-01-29 | 2017-08-03 | Sun Pharmaceutical Industries Limited | Improved process for the preparation of idelalisib |
| WO2017134607A1 (en) | 2016-02-03 | 2017-08-10 | Lupin Limited | A process for the preparation of phosphatidylinositol 3-kinase inhibitor |
| CN107033145B (zh) * | 2016-02-04 | 2019-11-22 | 浙江大学 | 苯并噻嗪和苯并噻二嗪类化合物及制备和应用 |
| EP3423057A1 (en) | 2016-03-04 | 2019-01-09 | Gilead Sciences, Inc. | Compositions and combinations of autotaxin inhibitors |
| ES2965264T3 (es) | 2016-03-09 | 2024-04-11 | Raze Therapeutics Inc | Inhibidores de la 3-fosfoglicerato deshidrogenasa y sus usos |
| EP4234552A3 (en) | 2016-03-09 | 2023-10-18 | Raze Therapeutics, Inc. | 3-phosphoglycerate dehydrogenase inhibitors and uses thereof |
| WO2017161116A1 (en) | 2016-03-17 | 2017-09-21 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as pi3k kinase inhibitors |
| WO2017177230A1 (en) | 2016-04-08 | 2017-10-12 | X4 Pharmaceuticals, Inc. | Methods for treating cancer |
| WO2017177179A1 (en) | 2016-04-08 | 2017-10-12 | Gilead Sciences, Inc. | Compositions and methods for treating cancer, inflammatory diseases and autoimmune diseases |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| CN106074430A (zh) * | 2016-06-13 | 2016-11-09 | 佛山市腾瑞医药科技有限公司 | 一种艾代拉利司泡腾片及其制备方法 |
| PH12018502623A1 (en) | 2016-06-13 | 2019-10-07 | I Mab | Anti-pd-l1 antibodies and uses thereof |
| WO2017223229A1 (en) | 2016-06-21 | 2017-12-28 | X4 Pharmaceuticals, Inc. | Cxcr4 inhibitors and uses thereof |
| ES2870920T3 (es) | 2016-06-21 | 2021-10-28 | X4 Pharmaceuticals Inc | Inhibidores de CXCR4 y usos de los mismos |
| JP6994767B2 (ja) | 2016-06-21 | 2022-01-14 | エックス4 ファーマシューティカルズ, インコーポレイテッド | Cxcr4阻害剤およびその使用 |
| WO2017221272A1 (en) * | 2016-06-23 | 2017-12-28 | Sun Pharmaceutical Industries Limited | Process for the preparation of idelalisib |
| SG10201912456RA (en) | 2016-06-24 | 2020-02-27 | Infinity Pharmaceuticals Inc | Combination therapies |
| KR101932146B1 (ko) * | 2016-07-14 | 2018-12-24 | 주식회사 바이오웨이 | Pi3k를 억제하는 신규한 퀴나졸리논 유도체 및 이를 포함하는 약학적 조성물 |
| EP3272348A1 (en) | 2016-07-21 | 2018-01-24 | LEK Pharmaceuticals d.d. | Pharmaceutical composition comprising idelalisib |
| WO2018026835A1 (en) | 2016-08-04 | 2018-02-08 | Gilead Sciences, Inc. | Cobicistat for use in cancer treatments |
| US10640499B2 (en) | 2016-09-02 | 2020-05-05 | Gilead Sciences, Inc. | Toll like receptor modulator compounds |
| WO2018045144A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | Toll like receptor modulator compounds |
| TW201813963A (zh) | 2016-09-23 | 2018-04-16 | 美商基利科學股份有限公司 | 磷脂醯肌醇3-激酶抑制劑 |
| TW201815787A (zh) | 2016-09-23 | 2018-05-01 | 美商基利科學股份有限公司 | 磷脂醯肌醇3-激酶抑制劑 |
| TW201825465A (zh) | 2016-09-23 | 2018-07-16 | 美商基利科學股份有限公司 | 磷脂醯肌醇3-激酶抑制劑 |
| CN109952303B (zh) | 2016-10-14 | 2022-10-21 | 林伯士拉克许米公司 | Tyk2抑制剂及其用途 |
| CN106632337B (zh) * | 2016-10-18 | 2018-11-20 | 湖北生物医药产业技术研究院有限公司 | 艾代拉里斯的晶型、药物组合物、制备方法和用途 |
| WO2018075937A1 (en) | 2016-10-21 | 2018-04-26 | Nimbus Lakshmi, Inc. | Tyk2 inhibitors and uses thereof |
| US20180133212A1 (en) | 2016-11-03 | 2018-05-17 | Gilead Sciences, Inc. | Combination of a bcl-2 inhibitor and a bromodomain inhibitor for treating cancer |
| EP3538091A4 (en) | 2016-11-08 | 2020-06-10 | Navitor Pharmaceuticals, Inc. | PHENYL AMINO PIPERIDINE MTORC INHIBITORS AND USES THEREOF |
| US20180141939A1 (en) | 2016-11-22 | 2018-05-24 | Gilead Sciences, Inc. | Solid forms of a bet inhibitor |
| AU2017368050A1 (en) | 2016-11-29 | 2019-06-20 | Puretech Lyt, Inc. | Exosomes for delivery of therapeutic agents |
| US11091451B2 (en) | 2016-12-05 | 2021-08-17 | Raze Therapeutics, Inc. | SHMT inhibitors and uses thereof |
| JP2020502238A (ja) | 2016-12-23 | 2020-01-23 | バイスクルアールディー・リミテッド | 新規連結構造を有するペプチド誘導体 |
| EP3565638B8 (en) | 2017-01-06 | 2024-04-10 | BicycleRD Limited | Bicycle conjugate for treating cancer |
| PT3383916T (pt) | 2017-01-24 | 2022-03-30 | I Mab Biopharma Us Ltd | Anticorpos anti-cd73 e seus usos |
| AR110998A1 (es) | 2017-02-24 | 2019-05-22 | Gilead Sciences Inc | Inhibidores de la tirosina cinasa de bruton |
| JP2020508326A (ja) | 2017-02-24 | 2020-03-19 | ギリアド サイエンシズ, インコーポレイテッド | ブルトン型チロシンキナーゼの阻害剤 |
| TWI783978B (zh) | 2017-03-08 | 2022-11-21 | 美商林伯士拉克許米公司 | Tyk2抑制劑、其用途及生產方法 |
| EP3375784A1 (en) | 2017-03-14 | 2018-09-19 | Artax Biopharma Inc. | Aza-dihydro-acridone derivatives |
| EP3375778A1 (en) | 2017-03-14 | 2018-09-19 | Artax Biopharma Inc. | Aryl-piperidine derivatives |
| US11339144B2 (en) | 2017-04-10 | 2022-05-24 | Navitor Pharmaceuticals, Inc. | Heteroaryl Rheb inhibitors and uses thereof |
| JOP20180040A1 (ar) | 2017-04-20 | 2019-01-30 | Gilead Sciences Inc | مثبطات pd-1/pd-l1 |
| WO2018198131A1 (en) | 2017-04-24 | 2018-11-01 | Natco Pharma Limited | Process for the preparation of amorphous idelalisib |
| CN116370448A (zh) | 2017-04-26 | 2023-07-04 | 纳维托制药有限公司 | Sestrin-gator2相互作用的调节剂及其用途 |
| US10857196B2 (en) | 2017-04-27 | 2020-12-08 | Bicycletx Limited | Bicyclic peptide ligands and uses thereof |
| WO2018237173A1 (en) | 2017-06-22 | 2018-12-27 | Novartis Ag | ANTIBODY MOLECULES DIRECTED AGAINST CD73 AND CORRESPONDING USES |
| SG11201912473PA (en) | 2017-06-22 | 2020-01-30 | Novartis Ag | Antibody molecules to cd73 and uses thereof |
| CN109111447A (zh) | 2017-06-23 | 2019-01-01 | 中国科学院上海药物研究所 | 7-位取代吡咯并三嗪类化合物或其药学上可用的盐,及其制备方法和用途 |
| JP7301757B2 (ja) | 2017-06-26 | 2023-07-03 | バイスクルアールディー・リミテッド | 検出可能部分を持つ二環式ペプチドリガンドおよびその使用 |
| PT3658557T (pt) | 2017-07-28 | 2024-09-11 | Nimbus Lakshmi Inc | Inibidores de tyk2 e usos dos mesmos |
| JP2020529418A (ja) * | 2017-07-31 | 2020-10-08 | ザ トラスティーズ オブ コロンビア ユニバーシティ イン ザ シティ オブ ニューヨークThe Trustees Of Columbia University In The City Of New York | T細胞急性リンパ芽球性白血病を治療するための化合物、組成及び方法 |
| JP2020529427A (ja) | 2017-08-04 | 2020-10-08 | バイスクルテクス・リミテッド | Cd137に対して特異的な二環式ペプチドリガンド |
| US20200283482A1 (en) | 2017-08-14 | 2020-09-10 | Bicyclerd Limited | Bicyclic peptide ligand prr-a conjugates and uses thereof |
| EP3668887A1 (en) | 2017-08-14 | 2020-06-24 | Bicyclerd Limited | Bicyclic peptide ligand sting conjugates and uses thereof |
| US11883497B2 (en) | 2017-08-29 | 2024-01-30 | Puretech Lyt, Inc. | Lymphatic system-directing lipid prodrugs |
| AU2018324037A1 (en) | 2017-08-29 | 2020-04-16 | Monash University | Lymphatic system-directing lipid prodrugs |
| AU2018329840A1 (en) | 2017-09-07 | 2020-03-19 | Augusta University Research Institute, Inc. | Specific Akt3 activator and uses thereof |
| WO2019060693A1 (en) | 2017-09-22 | 2019-03-28 | Kymera Therapeutics, Inc. | CRBN LIGANDS AND USES THEREOF |
| JP7366031B2 (ja) | 2017-09-22 | 2023-10-20 | カイメラ セラピューティクス, インコーポレイテッド | タンパク質分解剤およびそれらの使用 |
| WO2019092253A1 (en) * | 2017-11-10 | 2019-05-16 | Synthon B.V. | Process for preparing idelalisib |
| US11304954B2 (en) | 2017-12-19 | 2022-04-19 | Puretech Lyt, Inc. | Lipid prodrugs of mycophenolic acid and uses thereof |
| EP3727362A4 (en) | 2017-12-19 | 2021-10-06 | PureTech LYT, Inc. | MYCOPHENOLIC ACID LIPID MEDICINAL PRODUCTS AND THEIR USES |
| GB201721265D0 (en) | 2017-12-19 | 2018-01-31 | Bicyclerd Ltd | Bicyclic peptide ligands specific for EphA2 |
| US11608345B1 (en) | 2017-12-19 | 2023-03-21 | Puretech Lyt, Inc. | Lipid prodrugs of rapamycin and its analogs and uses thereof |
| TWI825046B (zh) | 2017-12-19 | 2023-12-11 | 英商拜西可泰克斯有限公司 | Epha2特用之雙環胜肽配位基 |
| AU2018392213B2 (en) | 2017-12-20 | 2021-03-04 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3' cyclic dinucleotides with phosphonate bond activating the STING adaptor protein |
| CN111511754B (zh) | 2017-12-20 | 2023-09-12 | 捷克共和国有机化学与生物化学研究所 | 活化sting转接蛋白的具有膦酸酯键的2’3’环状二核苷酸 |
| IL315310A (en) | 2017-12-26 | 2024-10-01 | Kymera Therapeutics Inc | IRAK joints and used in them |
| WO2019136373A1 (en) * | 2018-01-05 | 2019-07-11 | The Regents Of The University Of Colorado, A Body Corporate | P110-delta inhibitors treat and prevent autoimmunity while sparing the ability to mount an immune response to exogenous immunogens |
| US11512080B2 (en) | 2018-01-12 | 2022-11-29 | Kymera Therapeutics, Inc. | CRBN ligands and uses thereof |
| WO2019140380A1 (en) | 2018-01-12 | 2019-07-18 | Kymera Therapeutics, Inc. | Protein degraders and uses thereof |
| US10751339B2 (en) | 2018-01-20 | 2020-08-25 | Sunshine Lake Pharma Co., Ltd. | Substituted aminopyrimidine compounds and methods of use |
| CN111918651B (zh) | 2018-01-29 | 2024-01-30 | 默克专利股份有限公司 | Gcn2抑制剂及其用途 |
| AU2019212969A1 (en) | 2018-01-29 | 2020-08-13 | Merck Patent Gmbh | GCN2 inhibitors and uses thereof |
| CN108409674A (zh) * | 2018-02-09 | 2018-08-17 | 南京法恩化学有限公司 | 一种艾代拉利司中间体的制备方法 |
| AU2019222644B2 (en) | 2018-02-13 | 2021-04-01 | Gilead Sciences, Inc. | PD-1/PD-L1 inhibitors |
| WO2019168688A2 (en) | 2018-02-15 | 2019-09-06 | Senhwa Biosciences, Inc. | Quinolone analogs and their salts, compositions, and method for their use |
| AU2019229258B2 (en) | 2018-02-27 | 2023-09-14 | Artax Biopharma Inc. | Chromene derivatives as inhibitors of TCR-Nck interaction |
| CN108409740B (zh) * | 2018-03-14 | 2020-05-08 | 盐城师范学院 | 一种艾代拉里斯制备方法 |
| WO2019178596A1 (en) | 2018-03-16 | 2019-09-19 | Johnson Matthey Public Limited Company | Pyridine or n,n-dimethyl acetamide solvated solid state forms of solvated idelalisib, their use and preparation |
| TW202005654A (zh) | 2018-04-06 | 2020-02-01 | 捷克科學院有機化學與生物化學研究所 | 2,2,─環二核苷酸 |
| TWI833744B (zh) | 2018-04-06 | 2024-03-01 | 捷克科學院有機化學與生物化學研究所 | 3'3'-環二核苷酸 |
| TWI818007B (zh) | 2018-04-06 | 2023-10-11 | 捷克科學院有機化學與生物化學研究所 | 2'3'-環二核苷酸 |
| US10442799B1 (en) | 2018-04-07 | 2019-10-15 | Fuqiang Ruan | Heterocyclic compounds and uses thereof |
| CN112041311B (zh) | 2018-04-19 | 2023-10-03 | 吉利德科学公司 | Pd-1/pd-l1抑制剂 |
| CN112236428A (zh) | 2018-04-24 | 2021-01-15 | 默克专利股份有限公司 | 抗增殖化合物及其用途 |
| CN112218865B (zh) | 2018-04-24 | 2024-03-12 | 沃泰克斯药物股份有限公司 | 喋啶酮化合物及其用途 |
| WO2019211799A1 (en) | 2018-05-03 | 2019-11-07 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide |
| JP2021523151A (ja) | 2018-05-11 | 2021-09-02 | ホスホレックス、インコーポレイテッド | 負の表面電荷を有するマイクロ粒子及びナノ粒子 |
| PL3793565T3 (pl) | 2018-05-14 | 2022-05-02 | Gilead Sciences, Inc. | Inhibitory MCL-1 |
| JP7361727B2 (ja) | 2018-05-24 | 2023-10-16 | ヤンセン バイオテツク,インコーポレーテツド | Psma結合剤及びその使用 |
| AR126019A1 (es) | 2018-05-30 | 2023-09-06 | Novartis Ag | Anticuerpos frente a entpd2, terapias de combinación y métodos de uso de los anticuerpos y las terapias de combinación |
| US20210214459A1 (en) | 2018-05-31 | 2021-07-15 | Novartis Ag | Antibody molecules to cd73 and uses thereof |
| IL279329B1 (en) | 2018-06-15 | 2024-09-01 | Navitor Pharm Inc | Rapamycin analogs and their uses |
| GB201810316D0 (en) | 2018-06-22 | 2018-08-08 | Bicyclerd Ltd | Peptide ligands for binding to EphA2 |
| US11180531B2 (en) | 2018-06-22 | 2021-11-23 | Bicycletx Limited | Bicyclic peptide ligands specific for Nectin-4 |
| EP3817748A4 (en) | 2018-07-06 | 2022-08-24 | Kymera Therapeutics, Inc. | TRICYCLIC CRBN LIGANDS AND USES THEREOF |
| EP4234030A3 (en) | 2018-07-13 | 2023-10-18 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| US10548889B1 (en) | 2018-08-31 | 2020-02-04 | X4 Pharmaceuticals, Inc. | Compositions of CXCR4 inhibitors and methods of preparation and use |
| EP3846793B1 (en) | 2018-09-07 | 2024-01-24 | PIC Therapeutics, Inc. | Eif4e inhibitors and uses thereof |
| EP3860717A1 (en) | 2018-10-03 | 2021-08-11 | Gilead Sciences, Inc. | Imidozopyrimidine derivatives |
| EP3866789A4 (en) | 2018-10-15 | 2022-07-06 | Nimbus Lakshmi, Inc. | TYK2 INHIBITORS AND USES THEREOF |
| CN112955459A (zh) | 2018-10-23 | 2021-06-11 | 拜斯科技术开发有限公司 | 双环肽配体和其用途 |
| US11345654B2 (en) | 2018-10-24 | 2022-05-31 | Navitor Pharmaceuticals, Inc. | Polymorphic compounds and uses thereof |
| WO2020086556A1 (en) | 2018-10-24 | 2020-04-30 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| EP3873903B1 (en) | 2018-10-31 | 2024-01-24 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| US11071730B2 (en) | 2018-10-31 | 2021-07-27 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| MX2021006154A (es) | 2018-11-30 | 2021-08-24 | Kymera Therapeutics Inc | Degradadores de cinasas asociadas al receptor de interleucina 1 (irak) y usos de los mismos. |
| US11053241B2 (en) | 2018-11-30 | 2021-07-06 | Nimbus Lakshmi, Inc. | TYK2 inhibitors and uses thereof |
| EP3670659A1 (en) | 2018-12-20 | 2020-06-24 | Abivax | Biomarkers, and uses in treatment of viral infections, inflammations, or cancer |
| CN109593066B (zh) * | 2018-12-21 | 2020-06-19 | 上海交通大学 | 一种用于治疗肠道细菌感染的叶酸拮抗剂及其制备与应用 |
| JP2022518505A (ja) | 2019-01-23 | 2022-03-15 | ニンバス ラクシュミ, インコーポレイテッド | Tyk2阻害剤およびその使用 |
| WO2020165600A1 (en) | 2019-02-14 | 2020-08-20 | Bicycletx Limited | Bicyclic peptide ligand sting conjugates and uses thereof |
| KR20210137518A (ko) | 2019-03-07 | 2021-11-17 | 인스티튜트 오브 오가닉 케미스트리 앤드 바이오케미스트리 에이에스 씨알 브이.브이.아이. | 3'3'-사이클릭 다이뉴클레오티드 및 이의 프로드럭 |
| US11766447B2 (en) | 2019-03-07 | 2023-09-26 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| CN113543851A (zh) | 2019-03-07 | 2021-10-22 | 捷克共和国有机化学与生物化学研究所 | 2’3’-环二核苷酸及其前药 |
| AU2020253990A1 (en) | 2019-04-02 | 2021-10-28 | Bicycletx Limited | Bicycle toxin conjugates and uses thereof |
| US11485750B1 (en) | 2019-04-05 | 2022-11-01 | Kymera Therapeutics, Inc. | STAT degraders and uses thereof |
| TW202210480A (zh) | 2019-04-17 | 2022-03-16 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| TW202212339A (zh) | 2019-04-17 | 2022-04-01 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| JP2022529985A (ja) | 2019-04-19 | 2022-06-27 | ヤンセン バイオテツク,インコーポレーテツド | 抗psma/cd3抗体で前立腺癌を治療する方法 |
| WO2020237025A1 (en) | 2019-05-23 | 2020-11-26 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| JP2022534425A (ja) | 2019-05-31 | 2022-07-29 | イケナ オンコロジー, インコーポレイテッド | Tead阻害剤およびその使用 |
| WO2020255038A1 (en) | 2019-06-18 | 2020-12-24 | Janssen Sciences Ireland Unlimited Company | Combination of hepatitis b virus (hbv) vaccines and pyridopyrimidine derivatives |
| CA3142513A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| KR20220047277A (ko) | 2019-07-16 | 2022-04-15 | 길리애드 사이언시즈, 인코포레이티드 | Hiv 백신, 및 이의 제조 및 사용 방법 |
| TW202110485A (zh) | 2019-07-30 | 2021-03-16 | 英商拜西可泰克斯有限公司 | 異質雙環肽複合物 |
| AU2020345962A1 (en) | 2019-09-11 | 2022-03-31 | Vincere Biosciences, Inc. | USP30 inhibitors and uses thereof |
| BR112022004451A2 (pt) | 2019-09-13 | 2022-06-21 | Nimbus Saturn Inc | Antagonistas de hpk1 e usos dos mesmos |
| EP4031578A1 (en) | 2019-09-18 | 2022-07-27 | Novartis AG | Entpd2 antibodies, combination therapies, and methods of using the antibodies and combination therapies |
| MX2022003658A (es) | 2019-09-30 | 2022-04-25 | Gilead Sciences Inc | Vacunas contra el virus de la hepatitis b (vhb) y metodos para tratar el vhb. |
| EP4045083B1 (en) | 2019-10-18 | 2024-01-10 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| CN114599392A (zh) | 2019-10-31 | 2022-06-07 | 四十七公司 | 基于抗cd47和抗cd20的血癌治疗 |
| WO2021087432A1 (en) | 2019-11-01 | 2021-05-06 | Navitor Pharmaceuticals, Inc. | Methods of treatment using an mtorc1 modulator |
| TWI778443B (zh) | 2019-11-12 | 2022-09-21 | 美商基利科學股份有限公司 | Mcl1抑制劑 |
| US11819476B2 (en) | 2019-12-05 | 2023-11-21 | Janssen Pharmaceutica Nv | Rapamycin analogs and uses thereof |
| US11591332B2 (en) | 2019-12-17 | 2023-02-28 | Kymera Therapeutics, Inc. | IRAK degraders and uses thereof |
| KR20220145325A (ko) | 2019-12-17 | 2022-10-28 | 카이메라 쎄라퓨틱스 인코포레이티드 | Irak 분해제 및 이의 용도 |
| CN115297931A (zh) | 2019-12-23 | 2022-11-04 | 凯麦拉医疗公司 | Smarca降解剂和其用途 |
| CN117736207A (zh) | 2019-12-24 | 2024-03-22 | 卡尔那生物科学株式会社 | 二酰基甘油激酶调节化合物 |
| KR20220149534A (ko) | 2020-02-05 | 2022-11-08 | 퓨어테크 엘와이티, 아이엔씨. | 신경스테로이드의 지질 전구약물 |
| CN117964757A (zh) | 2020-02-14 | 2024-05-03 | 吉利德科学公司 | 与ccr8结合的抗体和融合蛋白及其用途 |
| WO2021178488A1 (en) | 2020-03-03 | 2021-09-10 | PIC Therapeutics, Inc. | Eif4e inhibitors and uses thereof |
| JP2023518423A (ja) | 2020-03-19 | 2023-05-01 | カイメラ セラピューティクス, インコーポレイテッド | Mdm2分解剤およびそれらの使用 |
| CN118373824A (zh) * | 2020-04-09 | 2024-07-23 | 成都赜灵生物医药科技有限公司 | 取代喹唑啉-4-酮类化合物及其制备方法和用途 |
| US12110294B2 (en) | 2020-05-01 | 2024-10-08 | Gilead Sciences, Inc. | CD73 compounds |
| WO2021233227A1 (en) * | 2020-05-16 | 2021-11-25 | Fochon Pharmaceuticals, Ltd. | Compounds as protein kinase inhibitors |
| TW202210483A (zh) | 2020-06-03 | 2022-03-16 | 美商凱麥拉醫療公司 | Irak降解劑之結晶型 |
| WO2022038158A1 (en) | 2020-08-17 | 2022-02-24 | Bicycletx Limited | Bicycle conjugates specific for nectin-4 and uses thereof |
| CN111840297B (zh) * | 2020-08-24 | 2023-06-16 | 天津济坤医药科技有限公司 | 艾代拉里斯在制备用于治疗肝纤维化疾病的药物中的应用 |
| CA3200814A1 (en) | 2020-12-02 | 2022-06-09 | Alfredo C. Castro | Tead inhibitors and uses thereof |
| WO2022120353A1 (en) | 2020-12-02 | 2022-06-09 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| MX2023008909A (es) | 2021-01-28 | 2023-10-23 | Janssen Biotech Inc | Proteínas de unión a psma y usos de estas. |
| US20230091528A1 (en) | 2021-02-02 | 2023-03-23 | Liminal Biosciences Limited | Gpr84 antagonists and uses thereof |
| EP4288427A1 (en) | 2021-02-02 | 2023-12-13 | Liminal Biosciences Limited | Gpr84 antagonists and uses thereof |
| CN116867494A (zh) | 2021-02-15 | 2023-10-10 | 凯麦拉医疗公司 | Irak4降解剂和其用途 |
| WO2022187856A1 (en) | 2021-03-05 | 2022-09-09 | Nimbus Saturn, Inc. | Hpk1 antagonists and uses thereof |
| JP2024513011A (ja) | 2021-03-29 | 2024-03-21 | ニンバス サターン, インコーポレイテッド | Hpk1アンタゴニスト及びその使用 |
| TW202302145A (zh) | 2021-04-14 | 2023-01-16 | 美商基利科學股份有限公司 | CD47/SIRPα結合及NEDD8活化酶E1調節次單元之共抑制以用於治療癌症 |
| AU2022258968A1 (en) | 2021-04-16 | 2023-10-19 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| TW202309030A (zh) | 2021-05-07 | 2023-03-01 | 美商凱麥拉醫療公司 | Cdk2降解劑及其用途 |
| TW202313094A (zh) | 2021-05-18 | 2023-04-01 | 美商基利科學股份有限公司 | 使用FLT3L—Fc融合蛋白之方法 |
| CN117396478A (zh) | 2021-06-23 | 2024-01-12 | 吉利德科学公司 | 二酰基甘油激酶调节化合物 |
| EP4359411A1 (en) | 2021-06-23 | 2024-05-01 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| EP4359415A1 (en) | 2021-06-23 | 2024-05-01 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| US11932634B2 (en) | 2021-06-23 | 2024-03-19 | Gilead Sciences, Inc. | Diacylglycerol kinase modulating compounds |
| JP2024532276A (ja) | 2021-08-25 | 2024-09-05 | ピク セラピューティクス, インコーポレイテッド | eIF4E阻害剤及びその使用 |
| TW202315621A (zh) | 2021-08-25 | 2023-04-16 | 美商皮克醫療公司 | Eif4e抑制劑及其用途 |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| MX2024005066A (es) | 2021-10-29 | 2024-05-24 | Gilead Sciences Inc | Compuestos de cd73. |
| US20230220106A1 (en) | 2021-12-08 | 2023-07-13 | Dragonfly Therapeutics, Inc. | Antibodies targeting 5t4 and uses thereof |
| WO2023107956A1 (en) | 2021-12-08 | 2023-06-15 | Dragonfly Therapeutics, Inc. | Proteins binding nkg2d, cd16 and 5t4 |
| WO2023114984A1 (en) | 2021-12-17 | 2023-06-22 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| TW202340168A (zh) | 2022-01-28 | 2023-10-16 | 美商基利科學股份有限公司 | Parp7抑制劑 |
| AU2023214044A1 (en) | 2022-01-31 | 2024-08-08 | Kymera Therapeutics, Inc. | Irak degraders and uses thereof |
| WO2023173057A1 (en) | 2022-03-10 | 2023-09-14 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| WO2023173053A1 (en) | 2022-03-10 | 2023-09-14 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| IL315083A (en) | 2022-03-17 | 2024-10-01 | Gilead Sciences Inc | The IKAROS family of zinc fingers degrades and uses them |
| US20230355796A1 (en) | 2022-03-24 | 2023-11-09 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| TW202345901A (zh) | 2022-04-05 | 2023-12-01 | 美商基利科學股份有限公司 | 用於治療結腸直腸癌之組合療法 |
| AU2023256670A1 (en) | 2022-04-21 | 2024-10-17 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2023211889A1 (en) | 2022-04-25 | 2023-11-02 | Ikena Oncology, Inc. | Polymorphic compounds and uses thereof |
| CN117088867A (zh) * | 2022-05-19 | 2023-11-21 | 杭州百诚医药科技股份有限公司 | 可吸入型芳环并噻嗪及类似物、含其的药物组合物及其在抗炎、抗肿瘤中的应用 |
| WO2023230205A1 (en) | 2022-05-25 | 2023-11-30 | Ikena Oncology, Inc. | Mek inhibitors and uses thereof |
| US20240116928A1 (en) | 2022-07-01 | 2024-04-11 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2024015741A1 (en) | 2022-07-12 | 2024-01-18 | Gilead Sciences, Inc. | Hiv immunogenic polypeptides and vaccines and uses thereof |
| WO2024028365A1 (en) | 2022-08-02 | 2024-02-08 | Liminal Biosciences Limited | Substituted pyridone gpr84 antagonists and uses thereof |
| TW202415650A (zh) | 2022-08-02 | 2024-04-16 | 英商利米那生物科技有限公司 | 芳基-三唑基及相關gpr84拮抗劑及其用途 |
| TW202416950A (zh) | 2022-08-02 | 2024-05-01 | 英商利米那生物科技有限公司 | 雜芳基甲醯胺及相關gpr84拮抗劑及其用途 |
| US20240091351A1 (en) | 2022-09-21 | 2024-03-21 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPa DISRUPTION ANTICANCER COMBINATION THERAPY |
| US20240208961A1 (en) | 2022-11-22 | 2024-06-27 | PIC Therapeutics, Inc. | Eif4e inhibitors and uses thereof |
| US20240254118A1 (en) | 2022-12-22 | 2024-08-01 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
Family Cites Families (117)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US35862A (en) * | 1862-07-08 | Improved portable evaporator for saccharine jutces | ||
| DE1249281B (es) | 1963-05-18 | |||
| US3691016A (en) | 1970-04-17 | 1972-09-12 | Monsanto Co | Process for the preparation of insoluble enzymes |
| DE2027645A1 (de) | 1970-06-05 | 1971-12-09 | Byk Gulden Lomberg Chemische Fa bnk GmbH, 7750 Konstanz | Piperazinylalkyl chinazolon (4) den vate, Verfahren zu deren Herstellung und sie enthaltende Arzneimittel |
| NL7204972A (es) | 1971-04-21 | 1972-10-24 | ||
| US3897432A (en) | 1971-04-21 | 1975-07-29 | Merck & Co Inc | Substituted benzimidazole derivatives |
| CA1023287A (en) | 1972-12-08 | 1977-12-27 | Boehringer Mannheim G.M.B.H. | Process for the preparation of carrier-bound proteins |
| US4179337A (en) | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| JPS52120848A (en) | 1976-04-03 | 1977-10-11 | Nippon Telegr & Teleph Corp <Ntt> | Welding method for optical fiber |
| US4195128A (en) | 1976-05-03 | 1980-03-25 | Bayer Aktiengesellschaft | Polymeric carrier bound ligands |
| DE2644265C2 (de) | 1976-09-30 | 1983-02-10 | Bayer Ag, 5090 Leverkusen | Chinazoline |
| US4330440A (en) | 1977-02-08 | 1982-05-18 | Development Finance Corporation Of New Zealand | Activated matrix and method of activation |
| CA1093991A (en) | 1977-02-17 | 1981-01-20 | Hideo Hirohara | Enzyme immobilization with pullulan gel |
| US4183931A (en) | 1977-09-08 | 1980-01-15 | Research Corporation | 2-Ketoalkyl-4(3H)-quinazolinones |
| US4229537A (en) | 1978-02-09 | 1980-10-21 | New York University | Preparation of trichloro-s-triazine activated supports for coupling ligands |
| DE2812635A1 (de) | 1978-03-22 | 1979-09-27 | Bayer Ag | Heterocyclische verbindungen |
| JPS6124020Y2 (es) | 1979-01-19 | 1986-07-18 | ||
| JPS55118918U (es) | 1979-01-19 | 1980-08-22 | ||
| JPS55118918A (en) | 1979-03-06 | 1980-09-12 | Mitsubishi Electric Corp | Production of quinazolone ring-containing epoxy resin |
| JPS55118917A (en) | 1979-03-06 | 1980-09-12 | Mitsubishi Electric Corp | Production of quinazolone ring-containing epoxy resin |
| US4289872A (en) | 1979-04-06 | 1981-09-15 | Allied Corporation | Macromolecular highly branched homogeneous compound based on lysine units |
| JPS6017375B2 (ja) | 1979-06-20 | 1985-05-02 | 三菱電機株式会社 | ポリアミド樹脂の製造方法 |
| JPS6023084B2 (ja) | 1979-07-11 | 1985-06-05 | 味の素株式会社 | 代用血液 |
| US4640835A (en) | 1981-10-30 | 1987-02-03 | Nippon Chemiphar Company, Ltd. | Plasminogen activator derivatives |
| US4496689A (en) | 1983-12-27 | 1985-01-29 | Miles Laboratories, Inc. | Covalently attached complex of alpha-1-proteinase inhibitor with a water soluble polymer |
| US4703004A (en) | 1984-01-24 | 1987-10-27 | Immunex Corporation | Synthesis of protein with an identification peptide |
| US4782137A (en) | 1984-01-24 | 1988-11-01 | Immunex Corporation | Synthesis of protein with an identification peptide, and hybrid polypeptide incorporating same |
| EP0206448B1 (en) | 1985-06-19 | 1990-11-14 | Ajinomoto Co., Inc. | Hemoglobin combined with a poly(alkylene oxide) |
| US4791192A (en) | 1986-06-26 | 1988-12-13 | Takeda Chemical Industries, Ltd. | Chemically modified protein with polyethyleneglycol |
| JPS6310777A (ja) | 1986-07-01 | 1988-01-18 | Hokuriku Seiyaku Co Ltd | ピペラジンアセトアミド誘導体 |
| AU610083B2 (en) | 1986-08-18 | 1991-05-16 | Clinical Technologies Associates, Inc. | Delivery systems for pharmacological agents |
| USRE35862E (en) | 1986-08-18 | 1998-07-28 | Emisphere Technologies, Inc. | Delivery systems for pharmacological agents encapsulated with proteinoids |
| US6696250B1 (en) | 1986-12-03 | 2004-02-24 | Competitive Technologies, Inc. | RNA ribozyme polymerases, dephosphorylases, restriction endoribonucleases and methods |
| US5011912A (en) | 1986-12-19 | 1991-04-30 | Immunex Corporation | Hybridoma and monoclonal antibody for use in an immunoaffinity purification system |
| US4851341A (en) | 1986-12-19 | 1989-07-25 | Immunex Corporation | Immunoaffinity purification system |
| US5229490A (en) | 1987-05-06 | 1993-07-20 | The Rockefeller University | Multiple antigen peptide system |
| US5225347A (en) | 1989-09-25 | 1993-07-06 | Innovir Laboratories, Inc. | Therapeutic ribozyme compositions and expression vectors |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| FR2675803B1 (fr) | 1991-04-25 | 1996-09-06 | Genset Sa | Oligonucleotides fermes, antisens et sens et leurs applications. |
| DE69209183T2 (de) | 1991-06-18 | 1996-08-08 | American Home Prod | Verwendung von Rapamycin zur Behandlung von T-Zellen Lymphom/Leukämie bei Erwachsenen |
| ATE156158T1 (de) | 1992-04-14 | 1997-08-15 | Cornell Res Foundation Inc | Makromoleküle auf basis von dendritischen polymeren und verfahren zur herstellung |
| US5658780A (en) | 1992-12-07 | 1997-08-19 | Ribozyme Pharmaceuticals, Inc. | Rel a targeted ribozymes |
| GB9301000D0 (en) | 1993-01-20 | 1993-03-10 | Glaxo Group Ltd | Chemical compounds |
| US5378725A (en) | 1993-07-19 | 1995-01-03 | The Arizona Board Of Regents | Inhibition of phosphatidylinositol 3-kinase with wortmannin and analogs thereof |
| GB9404485D0 (en) | 1994-03-09 | 1994-04-20 | Cancer Res Campaign Tech | Benzamide analogues |
| FI951367A (fi) | 1994-03-28 | 1995-09-29 | Japan Energy Corp | Puriinijohdannaiset ja tulehdustautien tukahduttajat (suppressantit) |
| US5480906A (en) | 1994-07-01 | 1996-01-02 | Eli Lilly And Company | Stereochemical Wortmannin derivatives |
| EP0771204A4 (en) | 1994-08-12 | 1999-10-20 | Pro Neuron Inc | METHODS OF TREATING SEPSIA OR INFLAMMATORY DISEASES WITH OXYPURINE-BASED NUCLEOSIDES |
| US5561138A (en) | 1994-12-13 | 1996-10-01 | American Home Products Corporation | Method of treating anemia |
| US6043062A (en) | 1995-02-17 | 2000-03-28 | The Regents Of The University Of California | Constitutively active phosphatidylinositol 3-kinase and uses thereof |
| US6096871A (en) | 1995-04-14 | 2000-08-01 | Genentech, Inc. | Polypeptides altered to contain an epitope from the Fc region of an IgG molecule for increased half-life |
| US5948664A (en) | 1996-02-29 | 1999-09-07 | The Regents Of The University Of California | PI 3-kinase polypeptides |
| EP0904107B1 (en) | 1996-03-18 | 2004-10-20 | Board Of Regents, The University Of Texas System | Immunoglobin-like domains with increased half lives |
| KR100375155B1 (ko) | 1996-05-15 | 2003-08-19 | 화이자 인코포레이티드 | 신규한2,3-이치환된-4(3에이치)-퀴나졸리논 |
| GB9611460D0 (en) | 1996-06-01 | 1996-08-07 | Ludwig Inst Cancer Res | Novel lipid kinase |
| WO1997041097A2 (en) | 1996-12-31 | 1997-11-06 | Dr. Reddy's Research Foundation | Novel heterocyclic compounds process for their preparation and pharmaceutical compositions containing them and their use in the treatment of diabetes and related diseases |
| US5858753A (en) | 1996-11-25 | 1999-01-12 | Icos Corporation | Lipid kinase |
| TW528755B (en) | 1996-12-24 | 2003-04-21 | Glaxo Group Ltd | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| GB9702701D0 (en) | 1997-02-01 | 1997-04-02 | Univ Newcastle Ventures Ltd | Quinazolinone compounds |
| KR100358636B1 (ko) | 1997-02-28 | 2002-10-31 | 화이자 프로덕츠 인코포레이티드 | 3-아릴-4(3에이치)-퀴나졸리논의 회전장애이성질체 및 에이엠피에이-수용체 길항물질로서 그의 용도 |
| ATE303997T1 (de) | 1997-06-09 | 2005-09-15 | Pfizer Prod Inc | Chinazolin-4-on ampa antagonisten |
| US6627755B1 (en) | 1997-06-09 | 2003-09-30 | Pfizer Inc | Quinazolin-4-one AMPA antagonists |
| AU8280798A (en) | 1997-07-03 | 1999-01-25 | Thomas Jefferson University | An improved method for design and selection of efficacious antisense oligonucleotides |
| IL125950A0 (en) | 1997-09-05 | 1999-04-11 | Pfizer Prod Inc | Methods of administering ampa receptor antagonists to treat dyskinesias associated with dopamine agonist therapy |
| US5822910A (en) | 1997-10-02 | 1998-10-20 | Shewmake; I. W. | Fishing line tensioning device |
| AU2108299A (en) | 1998-01-08 | 1999-07-26 | University Of Virginia Patent Foundation | A2A adenosine receptor agonists |
| AU1120599A (en) | 1998-04-23 | 1999-03-08 | Dr. Reddy's Research Foundation | New heterocyclic compounds and their use in medicine, process for their reparation and pharmaceutical compositions containing them |
| US6048970A (en) | 1998-05-22 | 2000-04-11 | Incyte Pharmaceuticals, Inc. | Prostate growth-associated membrane proteins |
| US6100090A (en) | 1999-06-25 | 2000-08-08 | Isis Pharmaceuticals Inc. | Antisense inhibition of PI3K p85 expression |
| AUPQ136299A0 (en) | 1999-07-02 | 1999-07-22 | Microsystem Controls Pty Ltd | Coin validation |
| US6046049A (en) | 1999-07-19 | 2000-04-04 | Isis Pharmaceuticals Inc. | Antisense modulation of PI3 kinase p110 delta expression |
| EP1686120A3 (en) * | 1999-10-27 | 2007-05-30 | Cytokinetics, Inc. | Methods and compositions utilizing quinazolinones |
| DK1257537T3 (da) | 2000-01-24 | 2007-10-01 | Astrazeneca Ab | Terapeutiske morpholino-substituerede forbindelser |
| WO2001057034A1 (en) | 2000-02-07 | 2001-08-09 | Bristol-Myers Squibb Co. | 3-aminopyrazole inhibitors of cyclin dependent kinases |
| US6667300B2 (en) | 2000-04-25 | 2003-12-23 | Icos Corporation | Inhibitors of human phosphatidylinositol 3-kinase delta |
| SI2223922T1 (sl) | 2000-04-25 | 2016-04-29 | Icos Corporation | Inhibitorji humane fosfatidil-inositol 3-kinazne delta izoforme |
| US6518227B2 (en) * | 2001-02-13 | 2003-02-11 | Robert Woosley | Solvent composition for denture adhesive |
| US7026330B2 (en) | 2002-05-30 | 2006-04-11 | The Children's Hospital Of Philadelphia | Methods for treatment of acute lymphocytic leukemia |
| US20040092561A1 (en) | 2002-11-07 | 2004-05-13 | Thomas Ruckle | Azolidinone-vinyl fused -benzene derivatives |
| BR0312752A (pt) | 2002-07-10 | 2005-04-26 | Applied Research Systems | Derivados de benzeno fundido de azolidinona-vinil |
| GB0217777D0 (en) | 2002-07-31 | 2002-09-11 | Novartis Ag | Organic compounds |
| US20040023390A1 (en) | 2002-08-05 | 2004-02-05 | Davidson Beverly L. | SiRNA-mediated gene silencing with viral vectors |
| CA2400254A1 (en) | 2002-09-19 | 2004-03-19 | University Health Network | Compositions and methods for treating heart disease |
| SI1549652T1 (sl) | 2002-09-30 | 2009-04-30 | Bayer Healthcare Ag | Kondenzirani azolpirimidinski derivati |
| BR0316386A (pt) | 2002-12-06 | 2005-09-27 | Warner Lambert Co | Benzoxazin-3-onas e seus derivados como inibidores de p13k |
| EP1581529A1 (en) | 2002-12-20 | 2005-10-05 | Warner-Lambert Company | Benzoxazines and derivatives thereof as inhibitors of pi3ks |
| NZ542475A (en) | 2003-04-03 | 2009-04-30 | Semafore Pharmaceuticals Inc | PI-3 kinase inhibitor prodrugs |
| MXPA05012953A (es) | 2003-06-05 | 2006-02-13 | Warner Lambert Co | Benzo[b]tiofenos 3-arilsulfanil y 3-heteroarilsulfanil sustituidos como agentes terapeuticos. |
| DE602004007268D1 (de) | 2003-06-05 | 2007-08-09 | Warner Lambert Co | CYCLOALKYLSULFANYL-SUBSTITUIERTE BENZOiBöTHIOPHENE ALS THERAPEUTISCHE MITTEL |
| EP1644364A1 (en) | 2003-06-05 | 2006-04-12 | Warner-Lambert Company LLC | Tetrazol benzofurancarboxamides with pi3k activity as therapeutic agents |
| BRPI0410913A (pt) | 2003-06-05 | 2006-06-27 | Warner Lambert Co | benzotiofenos substituìdos com cicloalquil e heterocicloalquil como agentes terapêuticos |
| US20040259926A1 (en) | 2003-06-05 | 2004-12-23 | Bruendl Michelle M. | 3-Aryloxy and 3-heteroaryloxy substituted benzo[b]thiophenes as therapeutic agents |
| WO2004108708A1 (en) | 2003-06-05 | 2004-12-16 | Warner-Lambert Company Llc | 3-substituted indoles and derivatives thereof as therapeutic agents |
| US20050054614A1 (en) | 2003-08-14 | 2005-03-10 | Diacovo Thomas G. | Methods of inhibiting leukocyte accumulation |
| WO2005016348A1 (en) * | 2003-08-14 | 2005-02-24 | Icos Corporation | Method of inhibiting immune responses stimulated by an endogenous factor |
| WO2005067901A2 (en) * | 2004-01-08 | 2005-07-28 | Michigan State University | Methods for treating and preventing hypertension and hypertension-related disorders |
| GB0400452D0 (en) * | 2004-01-09 | 2004-02-11 | Norton Healthcare Ltd | A pharmaceutical composition |
| CA2566436C (en) | 2004-05-13 | 2011-05-10 | Vanderbilt University | Phosphoinositide 3-kinase delta selective inhibitors for inhibiting angiogenesis |
| HUE030950T2 (en) | 2004-05-13 | 2017-06-28 | Icos Corp | Quinazolinones as 3-kinase delta inhibitors of human phosphatidylinositol |
| CA2567883A1 (en) | 2004-05-25 | 2005-12-15 | Icos Corporation | Methods for treating and/or preventing aberrant proliferation of hematopoietic cells |
| CN101123968A (zh) | 2004-06-04 | 2008-02-13 | 艾科斯有限公司 | 肥大细胞病的治疗方法 |
| MX2007002819A (es) | 2004-09-09 | 2007-08-14 | Natco Pharma Ltd | Nuevos derivados de fenilaminopirimidina como inhibidores de bcl-abl cinasa. |
| WO2006089106A2 (en) | 2005-02-17 | 2006-08-24 | Icos Corporation | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| US20090131512A1 (en) | 2007-10-31 | 2009-05-21 | Dynavax Technologies Corp. | Inhibition of type I in IFN production |
| CA3092449A1 (en) | 2008-11-13 | 2010-05-20 | Gilead Calistoga Llc | Therapies for hematologic malignancies |
| WO2010065923A2 (en) | 2008-12-04 | 2010-06-10 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Phosphatidylinositol-3-kinase p110 delta-targeted drugs in the treatment of cns disorders |
| JP5719779B2 (ja) | 2008-12-24 | 2015-05-20 | プラナ バイオテクノロジー リミティッド | キナゾリノン化合物 |
| EP2411391A1 (en) | 2009-03-24 | 2012-02-01 | Gilead Calistoga LLC | Atropisomers of2-purinyl-3-tolyl-quinazolinone derivatives and methods of use |
| ES2548253T3 (es) | 2009-04-20 | 2015-10-15 | Gilead Calistoga Llc | Métodos para el tratamiento de tumores sólidos |
| JP2013504604A (ja) | 2009-09-18 | 2013-02-07 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | ウイルスポリメラーゼ阻害剤としてのキナゾリノン誘導体 |
| AU2011255218B2 (en) | 2010-05-21 | 2015-03-12 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| PE20141792A1 (es) | 2012-03-05 | 2014-12-07 | Gilead Calistoga Llc | Formas polimorficas de (s)-2-(1-(9h-purin-6-ilamino)propil)-5-fluor-3-fenilquinazolin-4(3h)-ona |
| SG11201501173SA (en) | 2012-08-08 | 2015-05-28 | Kbp Biosciences Co Ltd | PI3Kδ INHIBITOR |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| EP3083623A1 (en) | 2013-12-20 | 2016-10-26 | Gilead Calistoga LLC | Polymorphic forms of a hydrochloride salt of (s) -2-(9h-purin-6-ylamino) propyl) -5-fluoro-3-phenylquinazolin-4 (3h) -one |
| CA2934531C (en) | 2013-12-20 | 2020-02-25 | Gilead Calistoga Llc | Process methods for phosphatidylinositol 3-kinase inhibitors |
| CN106459005A (zh) | 2014-06-13 | 2017-02-22 | 吉利德科学公司 | 磷脂酰肌醇3‑激酶抑制剂 |
-
2005
- 2005-05-12 HU HUE13150110A patent/HUE030950T2/en unknown
- 2005-05-12 PL PL13150110T patent/PL2612862T3/pl unknown
- 2005-05-12 ES ES05752122.1T patent/ES2607804T3/es active Active
- 2005-05-12 PL PL05752122T patent/PL1761540T3/pl unknown
- 2005-05-12 LT LTEP05752122.1T patent/LT1761540T/lt unknown
- 2005-05-12 WO PCT/US2005/016661 patent/WO2005113554A2/en active Application Filing
- 2005-05-12 ES ES16185213T patent/ES2873875T3/es active Active
- 2005-05-12 SI SI200532295T patent/SI3153514T1/sl unknown
- 2005-05-12 CN CN2011101010187A patent/CN102229609A/zh active Pending
- 2005-05-12 EP EP16185213.2A patent/EP3153514B1/en active Active
- 2005-05-12 RS RS20161005A patent/RS55546B1/sr unknown
- 2005-05-12 HU HUE05752122A patent/HUE030839T2/en unknown
- 2005-05-12 ES ES13150110.8T patent/ES2605792T3/es active Active
- 2005-05-12 DK DK13150110.8T patent/DK2612862T3/en active
- 2005-05-12 PT PT161852132T patent/PT3153514T/pt unknown
- 2005-05-12 SI SI200532115A patent/SI2612862T1/sl unknown
- 2005-05-12 PL PL16185213T patent/PL3153514T3/pl unknown
- 2005-05-12 CA CA2566609A patent/CA2566609C/en active Active
- 2005-05-12 DK DK05752122.1T patent/DK1761540T3/en active
- 2005-05-12 EP EP13150110.8A patent/EP2612862B1/en active Active
- 2005-05-12 US US13/730,256 patent/USRE44638E1/en active Active
- 2005-05-12 EP EP05752122.1A patent/EP1761540B1/en active Active
- 2005-05-12 JP JP2007513402A patent/JP2007537291A/ja active Pending
- 2005-05-12 CN CN2005800234499A patent/CN101031569B/zh active Active
- 2005-05-12 AU AU2005245875A patent/AU2005245875C1/en active Active
- 2005-05-12 US US11/596,092 patent/US7932260B2/en not_active Ceased
- 2005-05-12 ME MEP-2016-297A patent/ME02688B/me unknown
- 2005-05-12 EP EP21169213.2A patent/EP3943494A1/en not_active Withdrawn
- 2005-05-12 ME MEP-2016-284A patent/ME02695B/me unknown
- 2005-05-12 LT LTEP13150110.8T patent/LT2612862T/lt unknown
- 2005-05-12 RS RS20161180A patent/RS55551B1/sr unknown
- 2005-05-12 WO PCT/US2005/016778 patent/WO2005113556A1/en active Application Filing
- 2005-05-12 PT PT131501108T patent/PT2612862T/pt unknown
- 2005-05-12 PT PT57521221T patent/PT1761540T/pt unknown
-
2006
- 2006-11-09 IL IL179176A patent/IL179176A/en active IP Right Grant
-
2009
- 2009-10-23 AU AU2009230739A patent/AU2009230739A1/en not_active Abandoned
-
2010
- 2010-03-25 US US12/732,128 patent/US8207153B2/en active Active
- 2010-03-25 US US12/732,124 patent/US20100256167A1/en not_active Abandoned
-
2011
- 2011-06-17 US US13/163,597 patent/US20120015964A1/en not_active Abandoned
-
2012
- 2012-12-27 US US13/728,807 patent/US8586597B2/en active Active
- 2012-12-28 US US13/730,276 patent/USRE44599E1/en active Active
-
2013
- 2013-02-12 US US13/765,610 patent/US8779131B2/en active Active
- 2013-10-08 US US14/049,163 patent/US8980901B2/en active Active
- 2013-10-08 US US14/049,154 patent/US8993583B2/en active Active
-
2014
- 2014-05-21 US US14/284,331 patent/US9149477B2/en active Active
-
2015
- 2015-08-13 US US14/826,096 patent/US10336756B2/en active Active
- 2015-09-20 IL IL24173215A patent/IL241732B/en active IP Right Grant
-
2016
- 2016-11-24 CY CY2016046C patent/CY2016046I1/el unknown
- 2016-12-06 HR HRP20161657TT patent/HRP20161657T1/hr unknown
- 2016-12-14 CY CY20161101299T patent/CY1118493T1/el unknown
- 2016-12-20 HR HRP20161751TT patent/HRP20161751T1/hr unknown
- 2016-12-28 CY CY20161101359T patent/CY1118516T1/el unknown
-
2017
- 2017-02-16 BE BE2017C002C patent/BE2017C002I2/fr unknown
- 2017-02-17 LU LU00005C patent/LUC00005I2/en unknown
- 2017-02-22 HU HUS1700006C patent/HUS1700006I1/hu unknown
- 2017-02-23 LT LTPA2017004C patent/LTC1761540I2/lt unknown
- 2017-03-24 NL NL300867C patent/NL300867I2/nl unknown
-
2019
- 2019-05-21 US US16/418,558 patent/US10906907B2/en active Active
- 2019-10-15 IL IL269976A patent/IL269976B/en active IP Right Grant
-
2020
- 2020-11-24 US US17/103,631 patent/US20210332047A1/en not_active Abandoned
-
2021
- 2021-03-31 IL IL281945A patent/IL281945A/en unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2873875T3 (es) | Quinazolinonas como inhibidores de fosfatidilinositol 3-quinasa delta humano | |
| ES2788383T3 (es) | Inhibidores de delta fosfatidilo-inositol 3-quinasa humana | |
| ES2358785T3 (es) | Inhibidores de fosfatidilinositol 3-quinasa delta. |