CN1105111C - 吡嗪化合物 - Google Patents
吡嗪化合物 Download PDFInfo
- Publication number
- CN1105111C CN1105111C CN98804593A CN98804593A CN1105111C CN 1105111 C CN1105111 C CN 1105111C CN 98804593 A CN98804593 A CN 98804593A CN 98804593 A CN98804593 A CN 98804593A CN 1105111 C CN1105111 C CN 1105111C
- Authority
- CN
- China
- Prior art keywords
- compound
- formula
- pyrazine
- preparation
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/20—Nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/24—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D241/26—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with nitrogen atoms directly attached to ring carbon atoms
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Addiction (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Detergent Compositions (AREA)
- Nitrogen- Or Sulfur-Containing Heterocyclic Ring Compounds With Rings Of Six Or More Members (AREA)
Abstract
式(I)化合物中,R1选自一个或多个卤原子取代的苯基、萘基和一个或多个卤原子取代的萘基,R2选自-NH2和-NHC(=O)Ra;R3选自-NRbRc、-NHC(=O)Ra和氢原子,R4选自氢原子、-C1-4烷基、一个或多个卤原子取代的-C1-4烷基、-CN、-CH2OH、-CH2ORd和-CH2S(O)xRd;其中Ra代表C1-4烷基或C3-7环烷基,此外Rb和Rc可能相同或不同,它们选自氢原子和C1-4烷基,或与它们所结合的氮原子形成一个含氮的六元杂环,此杂环可进一步含有一个或多个C1-4烷基取代基;Rd选自C1-4烷基或有一个或多个卤原子取代的C1-4烷基;X为0、1或2的整数;及其药学上可接受的衍生物;条件是:当R2是-NH2、R3和R4均为氢原子时R1不能代表结构式(a)。
Description
本发明涉及一类用于治疗中枢神经系统(CNS)疾病和紊乱的吡嗪类化合物,以及这些化合物的药学上可接受的衍生物、含有这些化合物的药用组合物,涉及这些化合物在治疗此类紊乱方面的应用、以及这些化合物的制备方法。
先有技术已报道了众多的苯基吡嗪类衍生物。例如,synthesis(1987),(10),914~915,报道了苯基吡嗪类衍生物,包括,尤其是3-(4-氯苯基)-吡嗪胺。但先有技术文献中没有关于药学应用的报道。
本发明涉及一系列作为钠通道阻断剂的吡嗪衍生物。这些化合物具很好的抗惊厥作用,因而适用于治疗诸如癫痫的中枢神经系统疾病。
为此,本发明提供式(I)所示的化合物及其药学上可接受的衍生物 其中:R1选自一个或多个卤原子取代的苯基、萘基和一个或多个卤原子取代的萘基;R2选自-NH2和-NHC(=O)Ra;R3选自-NRbRc、-NHC(=O)Ra和氢;R4选自氢原子、-C1-4烷基(优选甲基)、一个或多个卤原子取代的-C1-4烷基(优选甲基)、-CN、-CH2OH、-CH2ORd和-CH2S(O)xRd。其中:Ra代表C1-4烷基或C3-7环烷基,及Rb和Rc可以相同或不同,它们选自氢和与C1-4烷基,或与它们所连结的氮原子一起形成含氮的六元杂环,此杂环可进一步由一个或多个C1-4烷基取代;Rd选自C1-4烷基或由一个或多个卤原子取代的C1-4烷基;X为0、1或2的整数;条件是当R2是-NH2且R3和R4均为氢时,R1不能代表:
其中:R1选自一个或多个卤原子取代的苯基、萘基和一个或多个卤原子取代的萘基;R2选自-NH2和-NHC(=O)Ra;R3选自-NRbRc、-NHC(=O)Ra和氢;R4选自氢原子、-C1-4烷基(优选甲基)、一个或多个卤原子取代的-C1-4烷基(优选甲基)、-CN、-CH2OH、-CH2ORd和-CH2S(O)xRd。其中:Ra代表C1-4烷基或C3-7环烷基,及Rb和Rc可以相同或不同,它们选自氢和与C1-4烷基,或与它们所连结的氮原子一起形成含氮的六元杂环,此杂环可进一步由一个或多个C1-4烷基取代;Rd选自C1-4烷基或由一个或多个卤原子取代的C1-4烷基;X为0、1或2的整数;条件是当R2是-NH2且R3和R4均为氢时,R1不能代表:
药学上可接受的衍生物指式(I)化合物的任何药学上可接受的盐或溶剂化物,或其它任何通过服药者服用而能直接或间接地提供式(I)化合物或其活性代谢物或残基(例如药物前体)的化合物。下文所述的式(I)化合物包括式(I)化合物以及它们的药学上可接受的衍生物。
适当的药物前体在本领域内已所知甚详,包括N-酰基衍生物(比如连于式(I)化合物的任何一个氮原子上),例如简单的酰基衍生物(如乙酰酚、丙酰基等)或基团如R-O-CH2-N或R-O-C(O)-N。
这里采用的术语卤原子包括氟、氯、溴、碘。
这里采用的术语C1-4烷基包括含1到4个碳原子的直链和支链烷基,尤其包括甲基和异丙基。
这里采用的术语C3-7环烷基包括含3到7个碳原子的环烷基,尤其包括环丙基。
这里采用的术语杂环包括至少含有一个氮杂原子的6元杂环,优选含2个氮杂原子。尤其适合的杂环是哌嗪基。
R1适当地选自未取代的萘基和由1个或多个卤原子取代的苯基。R1尤指被一个以上卤原子取代的苯基,如二-或三-卤代苯基。优选R1中的卤取代基为氯代。R1适合选自2,3,5-三氯苯基、2,3-二氯苯基基、2,5-二氯苯基基、1-萘基和2-萘基。R1尤指2,3,5-三氯苯基。
R2适合选自-NH2、异丙基碳酰氨基和环丙碳酰氨基。R2优选-NH2。
R3适当地选自氢、-NH2、二甲基氨基、4-甲基-1-哌嗪基、乙酰氨基、异丙碳酰氨基、环丙碳酰氨基。R3优选-NH2。
R4适当地选自氢原子、-CN、-CH2OH或甲基。R4优选-CH2OH或更优选氢。
更优选R2和R3同为-NH2。
一类优选的式(I)化合物包括那些R1,R2和R3如上定义,而R4选自氢、-C1-4烷基(优选甲基)和由一或多个卤原子取代的-C1-4烷基(优选甲基)的化合物。
优选的式(I)化合物为其中R1为2,3,5-三氯苯基;R2为-NH2;R3是-NH2;且R4为氢的化合物。
根据本发明实施方案的特例,提供式(1a)化合物以及它们的药学上可接受的衍生物 其中Hal代表选自氟、氯、溴和碘的卤原子;n为2或3;R2选自-NH2或-NHC(=O)Ra;R3选自-NRbRc、-NHC(=O)Ra和氢;R4选自氢、-C1-4烷基(优选甲基)、1个或多个卤原子取代的-C1-4烷基(优选甲基)、-CN、-CH2OH、CH2ORd和-CH2S(O)xRd;其中Ra代表C1-4烷基或C3-7环烷基,及Rb和Rc可以相同或不同,选自氢和C1-4烷基,或和它们所连接的氮原子形成一个含氮的六元杂环,其杂环可进一步由一个或多个C1-4烷基取代;Rd选自C1-4烷基或1个或多个卤原子取代的C1-4烷基。X为0、1或2的整数。
其中Hal代表选自氟、氯、溴和碘的卤原子;n为2或3;R2选自-NH2或-NHC(=O)Ra;R3选自-NRbRc、-NHC(=O)Ra和氢;R4选自氢、-C1-4烷基(优选甲基)、1个或多个卤原子取代的-C1-4烷基(优选甲基)、-CN、-CH2OH、CH2ORd和-CH2S(O)xRd;其中Ra代表C1-4烷基或C3-7环烷基,及Rb和Rc可以相同或不同,选自氢和C1-4烷基,或和它们所连接的氮原子形成一个含氮的六元杂环,其杂环可进一步由一个或多个C1-4烷基取代;Rd选自C1-4烷基或1个或多个卤原子取代的C1-4烷基。X为0、1或2的整数。
应该明确的是上述式(Ia)定义的R2,R3和R4基本上与前文所述的式(I)中对应的基团相同。
结构式(1a)中尤其优选R2和R3均代表-NH2。R4代表-CN、甲基或更适合为-CH2OH、或最适合为氢。
在结构式(1a)中Hal适合为氯。N适合为3,适当的三-取代化合物为下式(1b)化合物以及它们的药学上可接受的衍生物其中R2,R3和R4基本上与结构式(1a)所定义的相同。
本发明中优选的化合物是:2,6-二氨基-3-(2,3-二氯苯基基)吡嗪;2,6-二氨基-3-(2,5-二氯苯基基)吡嗪;2,6-二氨基-3-(1-萘基)吡嗪;2,6-二氨基-3-(2-萘基)吡嗪;2-氨基-6-(4-甲基-1-哌嗪基)-3-(2,3,5-三氯苯基)吡嗪;2-氨基-6-二甲基氨基-3-(2,3-二氯苯基基)吡嗪;2-氨基-6-二甲基氨基-3-(1-萘基)吡嗪;2,6-二环丙基碳酰氨基-3-(2,3,5-三氯苯基)吡嗪;2-氨基-6-环丙基碳酰氨基-3-(2,3,5-三氯苯基)吡嗪;2,6-二异丙基碳酰氨基-3-(2,3,5-三氯苯基)吡嗪;2-氨基-6-异丙基碳酰氨基-3-(2,3,5-三氯苯基)吡嗪;2-异丙基碳酰氨基-6-氨基-3-(2,3,5-三氯苯基)吡嗪;2-环丙基碳酰氨基-6-氨基-3-(2,3,5-三氯苯基)吡嗪;2-氨基-6-乙酰基氨基-3-(2,3,5-三氯苯基)吡嗪;2-氨基-6-乙酰基氨基-3-(2,5-二氯苯基基)吡嗪;2-氨基-6-乙酰基氨基-3-(2-萘)吡嗪;5-甲基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪;5-氰基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪;及它们的药学上可接受的衍生物。
更优选的化合物是5-羟甲基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪及其药学上可接受的衍生物。
根据本发明的特别优选的化合物是2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪及其药学上可接受的衍生物。
应该明确,本发明涵盖含上述特定和优选基团的所有组合。
式(I)化合物特别用作抗癫痫剂。它们因此而用于治疗癫痫。它们可用于改善患者,特别是人类癫痫病患者的病情。它们可被用来减轻患者的癫痫症状。“癫痫”包括下述发作形式:单纯的局部发作,复合的局部发作,继发的全身发作,包括癫痫小发作、肌阵挛性发作、阵挛性发作、强直性发作、强直阵挛性发作和驰缓性发作的全身发作。
此外,式(I)化合物在治疗双相性精神障碍或称躁狂抑郁症方面也有作用。可治疗I型或II型双相性精神障碍。因此这些化合物可用于改善双相性精神障碍患者的病情。它们可用于减缓双相性精神障碍患者的症状。式(I)化合物也可用于治疗单相精神抑郁症。
式(I)化合物是有用的镇痛剂。因而它们可用于治疗或预防疼痛。它们可用于改善患者,尤其是人类患者的疼痛状况。它们可用于减轻患者疼痛。因此,式(I)化合物可作为预防性止疼剂用于治疗急性疼痛诸如肌肉骨骼疼痛、手术后疼痛和外科疼痛,慢性疼痛诸如慢性炎症性疼痛(例如:类风湿性关节炎和骨关节炎)、神经性疼痛(例如疱疹后神经痛、三叉神经痛和交感神经持续性疼痛)以及与癌症和肌纤维疼痛相关的疼痛。式(I)化合物还可用于治疗或预防与偏头痛相关的疼痛。
式(I)化合物还可用于治疗肠机能紊乱,包括非溃疡性消化不良、非心源性胸痛、特别是肠过敏性综合征。肠过敏性综合征是一种胃肠机能紊乱,其特征是腹部疼痛和没有任何器质性病变症状的肠习惯改变。式(I)化合物因此可用于减轻因过敏性肠综合征引起的疼痛。过敏性肠综合征患者的疼痛症状可能因此而改善。
式(I)化合物也可用于治疗神经变性性疾病如:阿尔海默氏病,ALS,运动神经元疾病和帕金森氏病。式(I)化合物还可用于神经保护和治疗由休克、心脏停搏、肺分流术、脑外伤、脊髓损伤等引发的神经变性性疾病。
式(I)化合物尚可用于预防或减缓对成瘾性物质的依赖性,或预防、减缓或逆转对成瘾性物质的耐受性。成瘾性物质的实例包括阿片类(如吗啡)、中枢神经系统抑制剂(如乙醇)、神经兴奋剂(如可卡因)和尼古丁。
因此本发明进一步提供式(I)化合物以用于制备具有治疗上述疾病的用途的药物。本发明还包括治疗患有或易患上述病症的患者的方法,该方法包括给患者服用有效量的式(I)化合物。这里采用的术语“治疗”包括治疗已确诊的病症,也包括预防这些病症。
本发明化合物尤其适用于治疗癫痫和双相性精神障碍,特别是癫痫。
给患者,尤其是人类患者,服用式(I)化合物或其盐类的准确剂量应由助理医生决定。而且,决定给药剂量的因素包括病人的年龄、性别、需治疗的具体病症及其严重程度、给药途径等。
按游离碱计算,式(I)化合物和其盐的给药剂量应在0.1-10mg/kg体重/天,优选在0.5-5mg/kg体重/天范围内。按游离碱计算,成人的剂量范围一般为5-1000mg/天,比如5-200mg/天,优选10-50mg/天。
尽管式(I)化合物或其药学上可接受的衍生物可以原药给药,但最好使用药物制剂。本发明的制剂由式(I)化合物或其药学上可接受的衍生物与一种或多种可接受的载体或稀释剂以及任选其它治疗成份组成。载体必须是‘可接受的’,意为可与制剂中的其它成份共存并且对服用者无毒。
制剂包括适用于口服、胃肠道外给药(包括皮下给药如注射或包埋片剂给药、皮内给药、鞘内给药、肌内给药如包埋及静注给药)、直肠给药和局部给药(包括皮肤、颊和舌下给药)的剂型,尽管最适给药途径由例如服药者的病症和病情等因素决定。制剂可以简便地为单位剂量的形式及采用已知药学技术中的任何一种方法制备。所有方法均包括这样的步骤:使式(I)化合物或其药学上可接受的酸或盐(“活性成份”)与载体(由一个或多个辅助成分组成)相结合。一般来说,制剂的制备应使活性成份与液态载体或精细固体载体或两者均匀而细密地结合,然后根据需要将产物制成所希望的制剂形式。
本发明的口服制剂可以为分散的单位,诸如含预定量活性成份的胶囊剂、扁囊剂或片剂(例如特制的适于儿童服药的咀嚼片剂);也可以是粉剂或颗粒剂;可以是水或非水的溶液或悬浊液;还可以是水包油或油包水的乳剂。活性成份也可以丸剂、糖剂或糊剂形式存在。
片剂可选择一种或多种辅料采用压制或模制方法制成。压制的片剂可将诸如粉状或粒状的能流动的活性成分任选与粘合剂、润滑剂、惰性稀释剂、表面活性剂或分散剂混合后,采用适当的机器压制成片。模制片剂可以适当的机器把惰性液体稀释剂稀释的粉状化合物铸模制成。这些片剂可任选包衣或划痕,并可制成能从中缓慢或控制性地释放活性成份的制剂。
适于胃肠道外给药的制剂包括:水和非水的灭菌注射液,其中可含有抗氧化剂、缓冲剂、抑菌剂和使制剂与用药者的血液等渗的溶液;水或非水的无菌悬液,其中可含有悬浮剂或增稠剂。这些制剂可以单位剂量或多剂量的形式如封装于安瓿瓶或小玻瓶等容器中,也可在冷冻干燥(冻干)的条件下贮存,仅需在临用前加入无菌液体,例如无菌注射用水。临时的注射溶液和悬液可以用前述种类的无菌粉剂、粒剂和片剂制备。
用于直肠给药的制剂可为栓剂,其常用载体有可可脂,硬脂或者聚乙二醇等。
口腔局部给药(如颊部或舌下给药)的制剂包括将活性成份包含于诸如蔗糖和阿拉伯胶或西黄蓍胶的调味基质的锭剂,和将活性成份包含于诸如白明胶和甘油或蔗糖和阿拉伯胶的基质的软锭剂。
本发明化合物还可制成包埋制剂,这些长效制剂的给药可采用植入法(如皮下或肌内)或肌内注射。因此,举例来说,本发明化合物可与适当的聚合物或疏水物(如制成可接受的油性乳剂)或离子交换树脂,或作为难溶的衍生物(如难溶的盐)配制成制剂。
除上述特别提到的组分外,制剂还可含有其它与所用剂型有关的本领域常用的物质,例如,适于口服给药的制剂可含调味剂。
优选的单位剂量制剂是那些含有上文所述的有效日剂量或适当分剂量活性成分的制剂。以游离碱计算,常用量可为5mg-1000mg,更常用的量是从5mg-200mg(如5、25和100mg),最常用的是从10mg-50mg。
当式(I)化合物与其它治疗药物联合给药时,此类化合物可以任一种方便的途径与其它药物按顺序给药或同时给药。
式(I)化合物(当然也包括结构式(1a)和(1b)所示化合物)和其药学上可接受的盐及其溶剂化物可采用本领域已知的类似实例的制备方法制备。尤其是,式(I)化合物可以下文概略叙述方法制备,这些方法形成本发明的另一个方面。若无特别说明,在下述制备方法中的R1,R2,R3和R4与上述结构式(I)所定义的相同。
根据通用的制备方法(A),式(I)化合物可采用适当的反应条件由式(II)的化合物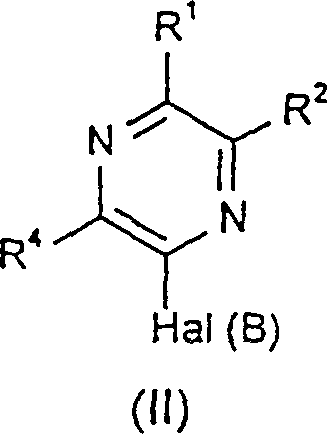 制备。其中Hal(B)代表卤原子,优选氯。例如,通过在适当的溶剂(比如乙醇溶液)中的胺的反应可以使Hal(B)转换成-NRbRc。
制备。其中Hal(B)代表卤原子,优选氯。例如,通过在适当的溶剂(比如乙醇溶液)中的胺的反应可以使Hal(B)转换成-NRbRc。
结构式(II)所示化合物可以通过结构式(III)所示的化合物 与式(IV)R1B(OH)2的化合物的适当反应制备。结构式(IV)R1B(OH)2的实例包括2,3,5-三氯苯硼酸,2,3-二氯苯基硼酸,2,5-二氯苯基硼酸,1-萘硼酸和2-萘苯酸。应该说,上述式(III)中的Hal(A)的反应活性比Hal(B)强,Hal(A)适合选自溴和碘,而Hal(B)适合选择氯。式(IV)化合物可从商业途径购得,或适合由商业购得的苯的同系物诸如1-溴代-2,3-二氯苯基或2-溴代-4,6-二氯苯基胺制备,制备方法在下文实施例中有更详细叙述。
与式(IV)R1B(OH)2的化合物的适当反应制备。结构式(IV)R1B(OH)2的实例包括2,3,5-三氯苯硼酸,2,3-二氯苯基硼酸,2,5-二氯苯基硼酸,1-萘硼酸和2-萘苯酸。应该说,上述式(III)中的Hal(A)的反应活性比Hal(B)强,Hal(A)适合选自溴和碘,而Hal(B)适合选择氯。式(IV)化合物可从商业途径购得,或适合由商业购得的苯的同系物诸如1-溴代-2,3-二氯苯基或2-溴代-4,6-二氯苯基胺制备,制备方法在下文实施例中有更详细叙述。
结构式(III)所示化合物可以通过适当的反应由式(V)的化合物 制备,例如与卤化剂(如N-溴代丁二酰亚胺)于室温搅拌数小时进行反应。
制备,例如与卤化剂(如N-溴代丁二酰亚胺)于室温搅拌数小时进行反应。
结构式(V)所示化合物可以通过结构式(VI)所示的二-卤代化合物与R2H反应而制备,其中Hal(B)和Hal(C)可以是相同或不同的卤代基团。优选Hal(B)和Hal(C)均为氯。式(VI)化合物可从商业途径获得。
根据通用的方法(B),式(I)化合物可以通过式(VII)化合物,与上述式(IV)化合物反应而制备其中Hal代表卤原子,优选氯和碘。
当R3和R4均为氢时,结构式(VII)所示化合物可从商业途径获得。此外,结构式(VII)所示化合物可以由式(VIII)化合物制备。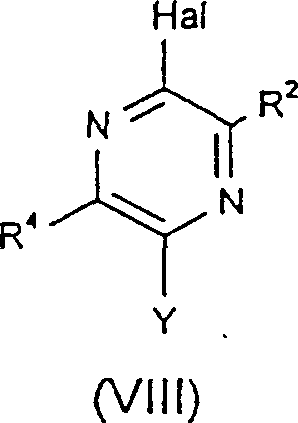 其中的Y是一种易转化成R3的基团。例如,当Y代表NH2时,它可在乙酰化试剂(如醋酸酐)存在时,通过回流转化为-NHC(=O)CH2。
其中的Y是一种易转化成R3的基团。例如,当Y代表NH2时,它可在乙酰化试剂(如醋酸酐)存在时,通过回流转化为-NHC(=O)CH2。
结构式(VIII)所示化合物可通过式(IX)化合物 与R2H在适当反应条件下制备。例如结构式(IX)所示化合物与氨气在高压釜中反应数小时。结构式(IX)所示化合物可由式(X)化合物
与R2H在适当反应条件下制备。例如结构式(IX)所示化合物与氨气在高压釜中反应数小时。结构式(IX)所示化合物可由式(X)化合物 制备。后者则可由前述的结构式(VI)所示的化合物制备,结构式(VI)所示的化合物可由商业途径获得。
制备。后者则可由前述的结构式(VI)所示的化合物制备,结构式(VI)所示的化合物可由商业途径获得。
按照另一种方法(C),R2代表NH2的式(I)化合物可按常规步骤通过结晶和氧化结构式(XI)所示的化合物 或其盐类制备,例如使结构式(XI)所示化合物的盐类中和,比如在适当的溶剂如醇溶液(如甲醇溶液)中与氢氧化锂中和,在自然氧化的条件下生成结构式(I)所示的化合物。
或其盐类制备,例如使结构式(XI)所示化合物的盐类中和,比如在适当的溶剂如醇溶液(如甲醇溶液)中与氢氧化锂中和,在自然氧化的条件下生成结构式(I)所示的化合物。
结构式(XI)所示的化合物可由结构式(XII)R1C(O)H表示的化合物在氰化物(如氰化钠)存在的情况下,与结构式(XIII)所示的化合物 或其盐类反应来制备。R1为三-卤代苯基如2,3,5-三氯苯甲醛时,结构式(XII)所示的化合物即为已知化合物,并可按WO95/07877中所述的方法制备。R1为其它基团时化合物为已知的,或可按制备已知化合物的已知方法制备。
或其盐类反应来制备。R1为三-卤代苯基如2,3,5-三氯苯甲醛时,结构式(XII)所示的化合物即为已知化合物,并可按WO95/07877中所述的方法制备。R1为其它基团时化合物为已知的,或可按制备已知化合物的已知方法制备。
结构式(XIII)所示的化合物,例如氨基乙脒,可按已知步骤(例如Chem.Berichte,
89,1185(1956)中叙述的方法)制备。
此外,按照方法D,式(I)化合物可通过适当的反应转化成相应的式(I)化合物。例如,其中R3代表NHC(=O)Ra的结构式(I)所示的化合物通过水解反应(如与盐酸水溶液反应)转化成R3代表-NH2的化合物。另外,其中R4代表氢的式(I)化合物可先与卤化剂(如N-溴代丁二酰亚胺)进行卤代反应,然后和适当的含氰离子的化合物(如氰化钠和氰化铜(I)混合物)反应而转变为R4代表-CN的化合物。进一步地,其中R4代表CN的式(I)化合物可由其甲酰化衍生物转化成R4代表-CH2OH的化合物,其甲酰化衍生物可由含-CN化合物与氢化二异丁基铝的甲苯溶液反应,而后再进行水解来制备。例如采用硼氢化钠的乙醇溶液可使此甲酰化衍生物再还原成-CH2OH化合物。R4代表-CH2OH的化合物可通过烷化反应转化为R4代表-CH2ORd的化合物。而且,R4代表-CN的式(I)化合物还可通过其甲酰基衍生物而转变为R4代表甲基的化合物,即以上述方法制备甲酰基衍生物后,与对-甲苯磺酰肼反应转化为甲苯磺酰腙,后者再与儿茶酚硼烷的氯仿/四氢呋喃溶液反应。
上述几种通用方法在逐步地合成所需化合物的任何步骤中,对于引入所希望的基团都是非常有用的,此外,应该知道,在多步骤的合成过程中,这几类通用方法可以不同方式联合使用。当然,应该选择多步骤的合成过程中各步反应的顺序,以使所采用的反应条件不会影响所需终产物的分子中的基团。
提供下列实施例以说明本发明,但不应把它们当成是一种限制。实施例1: 2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪1.
2,3,5-三氯溴苯
将亚硝酸钠(3.88g,0.056mol)分批加入在10℃以下搅拌的浓硫酸(28.16ml)中。加入2-溴-4,6-二氯苯基胺(12g,0.05mol,Lancaster)的冰醋酸(126ml)溶液,使温度保持在10℃以下。此混合物在10℃以下搅拌1小时,然后在室温下缓慢地加入到不停搅拌的氯化亚铜(10.11g,0.10mol)的浓盐酸(101.05ml)溶液中。接着将此混合物在室温下搅拌17小时。过滤产物,水(3×50ml)洗,再溶于氯仿(150ml)中,以无水硫酸镁干燥,过滤,真空蒸发滤液得到所需产物。产量为10g(77%),M.P.55~57℃。2.
2,3,5-三氯苯硼酸
将2,3,5-三氯溴苯(8.60g,0.033mol)的无水乙醚(33ml)溶液和溴乙烷(4.73ml,7.31g,0.067mol)在室温下逐滴加入含镁粉(2.80g,0.12mol)的无水乙醚(21.50ml)悬液中。将此混合物回流0.5小时并冷却至室温。混合物在低于-60℃及氮气中逐滴加入到三甲基硼酸酯(5.16ml,5.16g,0.05mol)的无水乙醚(8.60ml)溶液中。将其温热至室温过夜,然后冰浴中冷却并以2M盐酸(10ml)处理。分离醚层,用水(2×20ml)洗,以无水硫酸镁干燥,过滤,真空蒸发滤液。残留物在40-60℃石油醚中研磨,过滤后真空蒸发。产量4.57g(61%),M.P.257-260℃。路线A3.
2-氯-6-氨基-吡嗪
20个大气压和150℃下,将2,6-二氯吡嗪(100g,0.67mol,Lancaster)的0.880氨水(50ml)悬液在玻璃管高压釜中搅拌16小时。混合物冷却后过滤,充分水(200ml)洗后干燥。产物在氯仿中重结晶。产量41.98g(48%),M.P.150-152℃。4.
2-氯-3-溴-6氨基吡嗪和2-氨基-3-溴-6-氯吡嗪
在-5至0℃下搅拌2-氯-6-氨基吡嗪(20g,0.15mol)的氯仿(1940ml)溶液。分批加入N-溴丁二酰亚胺(27.58g,0.15mol)上述溶液中,并使温度保持在-5至0℃间。将混合物温热至室温并搅拌3.50小时。然后依次用饱和的碳酸氢钠(1×300ml)和水(1×500ml)洗涤该混合物,无水硫酸镁干燥,过滤,真空蒸发滤液。残留物以氯仿为洗脱剂,用“快速闪脱层析法”纯化。2-氯-3-溴-6-氨基吡嗪的产量为13.89g(43%),M.P.146-147℃,2-氨基-3-溴-6-氯吡嗪的产量为4.90g(15%),M.P.124-125℃。5.
2-氨基-6-氯-3-(2,3,5-三氯苯基)吡嗪
将2,3,5-三氯苯硼酸(1.62g,7.18×10-3mol)的纯乙醇(2.05ml)溶液缓慢加入2-氨基-3-溴-6-氯吡嗪(1g,5.1×10-3mol)和四-(三苯膦)钯(O)(0.334g,2.89×10-4mol)的苯(10.20ml)/2M碳酸钠水溶液(5.50ml)中。将混合物回流17小时。冷却后的反应混合物经真空蒸发并用氯仿(50ml)抽提。氯仿层水(2×30ml)洗后,用无水硫酸镁干燥,过滤并将滤液真空蒸发。残留物在40-60℃的石油醚中研磨,过滤后真空干燥。产量0.205g(14%),M.P.211-214℃。6.
2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪
将2-氨基-6-氯-3-(2,3,5-三氯苯基)吡嗪(0.3g,9.71×10-4mol)的纯乙醚(4ml)悬液和0.880氨水(8.24ml)在180℃的高压釜中搅拌44小时。混合物冷却后真空蒸发,残留物以氯仿(3×30ml)抽提。合并氯仿抽提物,以无水硫酸镁干燥,过滤,真空蒸发滤液。此残留物用‘快速闪脱层析法’纯化,以氯仿至98∶2的氯仿∶甲醇为洗脱剂。产物在40-60℃的石油醚中研磨,过滤后真空蒸发。产量0.155g(56%),M.P.178-180℃。路线B7.
2,6-二氨基-3-溴吡嗪
将2-氯-3-溴-6-氨基吡嗪(15g,0.072mol)的无水乙醇(150ml)悬液和0.880氨水(375ml)在20个大气压和160℃的高压釜中搅拌并加热16个小时。混合物冷却后真空蒸发并用热甲醇(3×100ml)抽提。合并甲醇抽提液后真空蒸发。残留物溶于热氯仿,以无水硫酸镁干燥,过滤并将滤液真空蒸发。残留物在40-60℃石油醚中研磨,过滤后真空蒸发。产量5.51g(40%),M.P.176-178℃。8.
2-氨基-3-溴-6-乙酰胺基吡嗪
在氮气中,将2,6-二氨基-3-溴吡嗪(10.50g,0.056mol)的无水1,1-二甲氧基乙烷(168ml)溶液和乙酸酐(7.91ml,8.56g,0.084mol)的混合物回流2.50小时。冷却混合物后真空蒸发。残留物在乙醚中研磨、过滤后真空蒸发。产量10.31g(80%),M.P.218-221℃。9.
2-氨基-6-乙酰胺基-3-(2,3,5-三氯苯基)吡嗪
在室温下,将2-氨基-3-溴-6-乙酰胺基吡嗪(7.00g,0.03mol)的苯(60.90ml)溶液与四-(三苯膦)钯(O)的混合物在氮气中搅拌10分钟。依次加入2M碳酸钠溶液(30.24ml)和2,3,5-三氯苯硼酸溶液(6.83g,0.03mol)的无水乙醇(7.07ml)溶液后,使混合物中在氮气中回流17小时。然后再加入等量的2,3,5-三氯苯硼酸的无水乙醇溶液,使混合物再回流7.50小时。最后,再加入等量的2,3,5-三氯苯硼酸的无水乙醇溶液至混合物中并继续回流17小时。混合物冷却后真空蒸发。残留物溶于氯仿(150ml)后,依次用饱和的碳酸氢钠(1×100ml)和水(1×100ml)洗涤,用无水硫酸镁干燥,过滤,滤液真空蒸发。残留物以‘快速闪脱层析法’纯化,以氯仿至98∶2的氯仿∶甲醇作为洗脱剂。产量3.02g(30%),M.P.200-203℃。10.
2.6-二氨基-3-(2,3,5-三氯苯)吡嗪
将2-氨基-6-乙酰胺基-3-(2,3,5-二氯苯基基)吡嗪(2.97g,8.96×10-3mol)的12M盐酸(1.31ml)和水(4.04ml)的悬液回流1.75小时。混合物冷却后以0.880氨水(5ml)碱化并用氯仿(3×50ml)抽提。合并氯仿抽提液,用无水硫酸镁干燥,过滤,滤液于80℃下真空蒸发。产量2.29g(88%),M.P.178-180℃。11.
2-{[氰基-(2,3,5-三氯苯基)-甲基]-氨基}-乙脒氢溴酸盐
在室温下,将氨基乙脒二氢溴酸盐(162.1g,0.774mol)分批加入2,3,5-三氯苯甲醛(200.0g,0.851mol)的甲醇(2.43升)溶液中。在每次加入后,向生成的混合物中,一次性加入氰化钾(50.4g,0.774mol)。将此悬液在25℃下搅拌4小时后,再加热至50℃。在50℃下搅拌混合物24小时。然后真空除去甲醇,所得固体与水(1.5升)和乙酸乙酯(2.5升)混合后过滤收集。于50℃过夜真空干燥该固体后得到所需产物。产量96.31g(33.4%)。1H nmr(d-6 DMSO)δ/ppm 8.72(3H,br,NH);7.99(1H,d,J 2.3 Hz,ArH);7.79(1H,d,J 2.3 Hz,ArH);5.39(1H,d,J10.6Hz,ArCH(CH)NH);4.35(1H,m,ArCH(CN)NH);3.56(2H,d,J 6.4Hz,ArCH(CN)NHCH2C(=NH)NH2)。12.
2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪
在室温下,将2-{[氰基-(2,3,5-三氯苯基)-甲基]-氨基}乙脒氢溴酸盐(95.36g,0.256mol)分批加入氢氧化锂一水合物(16.11g,0.384mol)的甲醇(1.9升)溶液中。所得溶液在室温下搅拌3个小时后真空蒸发。所得固体于水(1.15升)中制成淤浆并过滤收集。50℃真空干燥后,所得粗产物在甲苯中经重结晶纯化得到所希望的产物。产量69.51g(93.8%),M.P.178-180℃。实施例2: 2,6-二氨基-3-(2,3-二氯苯基基)吡嗪1.
2,3-二氯苯基硼酸
在维持低于-65℃的温度下,将1-溴-2,3-二氯苯基(20g,0.088mol,Aldrich)的无水四氢呋喃(44.24ml)溶液逐滴加入正丁基锂(1.6M的己烷溶液,66.36ml,0.11mol)的无水四氢呋喃(16ml)溶液中。所得淡黄色悬液于-78℃下搅拌75分钟。在维持低于-55℃下逐滴加入三甲基硼酸酯(13.28ml,12.16g,0.12mol),所得淡黄色液体温热至室温下过夜。以水(30ml)分解过量的正丁基锂,然后将反应混合物真空蒸发。所得残留物悬溶于水中并以2M盐酸(10ml)酸化。过滤不溶固体,用水充分洗涤后干燥。将此固体悬溶于60-80℃石油醚中,在室温下搅拌10分钟,过滤后真空干燥。产量8.42g(50%),M.P.235-238℃。2.
2-氨基-6-氯-3-(2,3-二氯苯基)吡嗪
该化合物可采用类似实施例1中路线A的方法由2-氨基-3-溴-6-氯吡嗪制得。产量0.343g(26%),M.P.179-181℃。2.
2,6-二氨基-3-(2,3-二氯苯基)吡嗪
此化合物可采用与实施例1中路线A类似的方法,由2-氨基-6-氯-3-(2,3-二氯苯基)吡嗪与0.880氨水反应制得。产量0.195g,M.P.169-170℃。实施例3: 2,6,-二氨基-3-(2,5-二氯苯基基)吡嗪1.
2,5,-二氯苯基硼酸
该化合物由2,5,-二氯溴苯(Aldrich)制备,制备方法类似实施例2。产量2.19g(55%),熔点278-280℃。2.
2-氨基-6-乙酰胺基-3-(2,5-二氯苯基基)吡嗪
该化合物由2-氨基-3-溴-6-乙酰氨基吡嗪制备,制备方法类似实施例1中的路线B。产量0.45g,熔点152-154℃。3.
2,6-二氨基-3-(2,5-二氯苯基基)吡嗪
该化合物可由2-氨基-6-乙酰胺基-3-(2,5-二氯苯基基)吡嗪制备,制备方法类似实施例1中的路线B。产量0.123g,熔点159-160℃。实施例4: 2,6,-二氨基-(1-萘)吡嗪1.
2-氨基-6-氯-3-(1-萘)吡嗪
该化合物由2-氨基-3-溴-6-氯吡嗪和1-萘硼酸(Lancaster)制备,制备方法类似实施例1中的路线A。产量0.709g(58%),熔点138-139℃。2.
2,6-二氨基-(1-萘)吡嗪
该化合物由2-氨基-6-氯-3-(1-萘)吡嗪与0.880氨反应制备,制备方法类似实施例1中的路线A。产量0.167g(50%),熔点180-183℃。
实施例5:2,6,-二氨基-3-(2-萘)吡嗪1.
2-萘硼酸
该化合物由2-溴萘(Aldrich)制备,制备方法类似实施例2。产量1.71g(40%),熔点280-282℃。2.
2-氨基-6-乙酰胺基-3-(2-萘)吡嗪
该化合物由2-氨基-3-溴-6-乙酰胺基吡嗪制备,制备方法类似实施例1中的路线B。产量0.46g,熔点235-237℃。3.
2,6-二氨基-3-(2-萘)吡嗪
该化合物由2-氨基-6-乙酰胺基-3-(2-萘)吡嗪制备,制备方法类似实施例1中的路线B。产量0.121g,熔点168-170℃。实施例6: 2-氨基-6-(4-甲基-1-哌嗪)-3-(2,3,5,-三氯苯基)吡嗪
将实施例1路线A中的2-氨基-6-氯-3-(2,3,5-三氯苯基)吡嗪(0.215g,7.0×10-3mol)与1-甲基哌嗪(5ml,Aldrich)于140℃加热1小时。混合物真空蒸发,残留物以“快速闪脱层析法”纯化,洗脱液为0-4%的乙醇:二氯甲烷。产物以微量二氯甲烷溶解,缓慢加入己烷(10ml)得到黄色针晶。产量0.247g(95%),熔点185℃。实施例7: 2-氨基-6-二甲基氨基-3-(2,3-二氯苯基基)吡嗪
该化合物以2-氨基-6-氯-3-(2,3-二氯甲基)吡嗪(实施例2)和二甲基胺(Aldrich)制备,方法与实施例6类似。熔点147-148℃。实施例8: 2-氨基-6-二甲基氨基-3-(1-萘)吡嗪
该化合物以2-氨基-6-氯-3-(1-萘)吡嗪(实施例4)制备,制备方法类似于实施例6。熔点131℃。实施例9: 2,6-二环丙基碳酰氨基-3-(2,3,5-三氯苯基)吡嗪和2-氨基-6-环丙基碳 酰氨基-3-(2,3,5-三氯苯基)吡嗪
将无水吡啶(0.137ml),4-二甲氨基吡啶(0.114g)和环丙基碳酰氯(0.33g,0.286ml,3.14×10-3mol,Aldrich)加入2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪(实施例1)的无水呋喃(12ml)溶液中,所得反应混合物于90℃回流2.50小时。所得悬液冷却至室温,真空蒸发。残留物以乙酸乙酯(3×20ml)萃取,水(2×20ml)洗,无水硫酸镁干燥,过滤,真空蒸发滤液。残留物以“快速闪脱层析法”纯化,以己烷∶乙醚(15∶25至5∶35)洗脱。2,6-二
环丙基碳酰氨基-3-(2,3,5-三氯苯基)吡嗪的产量为0.340g(51%)。熔点212-214℃。2-氨基-6-
环丙基碳酰氨基-3-(2,3,5-三氯苯基)吡嗪的产量为0.174g(31%),熔点240-244℃。实施例10: 2,6-二异丙基碳酰氨基-3-(2,3,5-三氯苯基)吡嗪和2-氨基-6-异丙基碳 酰氨基-3-(2,3,5-三氯苯基)吡嗪
按照与实施例9类似的制备方法。从2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪(实施例1)与异丁酰氯(Aldrich)反应制备这些化合物。2,6-二异丙基碳酰氨基-3-(2,3,5-三氯苯基)吡嗪的产量为0.313g(46%),熔点227-229℃。2-氨基6-异丙基碳酰氨基-3-(2,3,5-三氯苯基)吡嗪的产量为0.166g(29%),熔点230-232℃。实施例11: 2-异丙基碳酰氨基-6-氨基-3-(2,3,5-三氯苯基)吡嗪 1. 2-异丙基碳酰氨基-6-乙酰胺-3-(2,3,5-三氯苯基)吡嗪
该化合物的制备方法与实施例9类似,以2-二氨基-6-乙酰胺基-3-(2,3,5-三氯苯基)吡嗪(实施例1)与1当量的异丁酰氯(Aldrich)反应制备。产量为0.120g,熔点230-232℃。2.
2-异丙基碳酰氨基-6-氨基-3-(2,3,5-三氯苯基)吡嗪
将氯化锡(0.182g,0.0096mol)加入2-异丙基碳酰氨基-6-乙酰胺基-3-(2,3,5-三氯苯基)吡嗪(0.13g,3.24×10-4mol)的无水乙醇(6.50ml)悬液中,生成的混合物在50-60℃下搅拌1小时20分钟,冷却反应混合物,真空蒸发。残留物以乙酸乙脂(3×20ml)提取,水(2×20ml)洗,无水硫酸镁干燥,过滤,滤液真空蒸发。残留物以“快速闪脱层析法”纯化,以氯仿∶甲醇(99∶1)洗脱。产量0.046g(40%)。熔点105-108℃。实施例12: 2-环丙基碳酰氨基-6氨基-3-(2,3,5-三氯苯基)吡嗪1.
2-环丙基碳酰氨基-6-乙酰胺基-3-(2,3,5-三氯苯基)吡嗪
以与实施例9类似方法由2-氨基-6-乙酰胺基-3-(2,3,5-三氯苯基)吡嗪(实施例1)与环丙碳酰氯(Aldrich)反应制备该化合物,。产量0.387g(61%)。N.m.r.(D6DMSO)δ:0.53(2H,m),0.7(2H,m),1.75(1H,m),2.18(3H,s),7.43(1H,d),7.87(1H,d),9.22(1H,s),10.36(1H,b),10.83(1H,b)。2.
环丙基碳酰氨基-6氨基-3-(2,3,5-三氯苯基)吡嗪
以与实施例11类似方法由2-环丙基碳酰氨基-6-乙酰胺基-3-(2,3,5-三氯苯基)吡嗪制备该化合物。。产量0.120g(35%)。熔点188-190℃。实施例13: 2-氨基-3-(2,3,5-三氯苯基)吡嗪
将2,3,5-三氯苯硼酸(1.54g,6.82mmol)的无水乙醇溶液(1.5ml)缓慢加入2-氨基-3-氯吡嗪(0.589g,4.54mmol)和四-(三苯基膦)钯(0)(0.299g,0.259mmol)的苯(10.5ml)/2M碳酸钠水溶液(4.54ml)的混合液中。使混合物回流17小时。反应混合物冷却后在水和乙酸乙酯(50ml)间分配。水(2×30ml)洗有机层,干燥(硫酸镁),蒸发。残留物以“快速闪脱层析法”纯化,以氯仿至0.5%甲醇/氯仿液洗脱。然后使产物在40-60汽油中结晶。产量为0.15g(12%)。熔点142-143℃。实施例14: 2-氨基-6-乙酰胺基-3-(2,3,5-三氯苯基)吡嗪
制备方法同前文所述(见实施例1.9)。实施例15: 2-氨基-6-乙酰胺基-3-(2,5-二氯苯基基)吡嗪
制备方法同前文所述(见实施例3.2)。实施例16: 2-氨基-6-乙酰胺基-3-(2-萘)吡嗪
制备方法同前文所述(见实施例5.2)。实施例17: 5-氰基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪1.
5-溴-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪
用20分钟的时间,将N-溴化琥珀酰亚胺(0.194g,1.09×10-3mol)加入低于15℃的2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪(0.3g,1.04×10-3mol)的二甲亚砜(10ml)和水(0.25ml)的混合液中。反应混合物在低于15℃的温度下搅拌1小时,倒入冰水(150ml)后以乙酸乙酯(2×75ml)抽提。提取物以2M碳酸钠(50ml)和水(100ml)洗涤,无水硫酸镁干燥,过滤,滤液真空蒸发。残留物以“快速闪脱层析法”纯化,以5-13%乙酸乙酯的环己烷溶液洗脱。产量0.183g(48%)。熔点222-224℃。2.
5-氰基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪
将97%氰化钠(0.064g,1.306×10-3mol)和90%氰化铜(I)(0.135g,1.306×10-3mol)的无水二甲亚砜(5ml)混合溶液搅拌加热至130℃。向所得清澈溶液中少量分批加入5-溴-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪(0.35g,0.95×10-3mol),溶液在140-150℃下保温16小时。反应混合物冷却后真空蒸发。残留物以乙酸乙脂(100ml)提取,水(100ml)和盐水(100ml)洗涤,无水硫酸镁干燥,过滤,真空蒸发滤液。残留物以“快速闪脱层析法”纯化,以5-17%乙酸乙酯的环己烷溶液洗脱。产量0.152g(51%)。熔点277-279℃。元素分析,计算值C11H6N5Cl30.02C6H12:C,42.23;H,1.99;N,22.15。实验值:C,42.36;H,1.78;N,21.79。实施例18: 5-羟甲基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪1.
5-甲酰基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪
将氢化二异丁基铝(1.5M的甲苯溶液)(2.12ml,3.18×10-3mol)在0℃氮气中逐滴加入5-氰基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪(0.5g,1.59×10-3mol)的无水甲苯溶液(70ml)中,0℃下搅拌1小时进行反应。然后再加入1当量的氢化二异丁基铝,混合物在0℃下再搅拌1小时。0℃氮气中小心加入甲醇(1ml)消除过量的氢化物,反应物升至室温。加入乙酸乙酯(100ml)后,以5%柠檬酸溶液(2×100ml)洗涤。分离有机层,盐水(100ml)洗涤,无水硫酸镁干燥,过滤,滤液真空蒸发得到所需产物。产量0.340g(67%)。核磁共振谱(N.m.r)(D6DMSO)δ:7.53(1H,d),7.91(1H,d),6.90-7.80(4H,b),9.49(1H,s)2.
5-羟甲基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪
室温下向搅拌的5-甲酰基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪(0.198g,6.24×10-4mol)的乙醇溶液(100ml)中加入硼氢化钠(0.035g,9.35×10-4mol)。室温下氮气中搅拌该反应物1小时,加入水(1ml),溶液进行真空蒸发。残留物溶于乙酸乙酯(200ml),盐水洗涤,硫酸镁干燥,过滤后真空蒸发。将该产物以11-40%乙酸乙酯的环己烷溶液为洗脱液进行“快速闪脱层析”纯化。产量0.104g(52%)。熔点185-186℃。核磁共振谱(N.m.r)(D6DMSO)δ:4.32(2H,d),4.98(1H,t),5.56(2H,s),5.85(2H,s),7.30(1 H,d),7.76(1H,d)。实施例19: 5-甲基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪1.
5-甲酰基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪,对甲苯磺酰腙
将5-甲酰基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪(0.330g,1.04×10-3mol)加入对-甲苯磺酰肼(0.30g,1.61×10-3mol)的甲醇(50ml)溶液中。溶液在氮气中回流4小时后,冷却至室温,真空蒸发溶剂。残留物以0-30%乙酸乙酯的环己烷溶液为洗脱液进行“快速闪脱层析”纯化。产量0.270g(53%)。质谱(Mass):(电子喷雾)487(MH+)保留时间 3.33分钟Micromass Platform Series 25min Grad.(2mmABZ)仪器:Red 流速:0.8ml/min洗脱液:A-0.1%V/V甲酸+10mmol乙酸铵
B-95%MeCN+0.05%V/V甲酸色谱柱:5cm×2.1mm ID ABZ+PLUS进样量:5μl 温度:室温(RT)时间: A% B%0.00 100 03.50 0.0 1005.00 0.0 1005.50 100 02.
5-甲基-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪
将邻苯二酚硼烷(1.0M的四氢呋喃溶液)(1.09ml,1.09×10-3mol)于0℃氮气中逐滴加入5-对甲苯磺酰腙-2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪(0.265g,5.46×10-4mol)的无水氯仿(15ml)和四氢呋喃溶液(20ml)溶液中。0℃搅拌1小时进行反应,淬灭后加入三水乙酸钠(g,5.46×10-4mol)。使混合物升至室温,搅拌1小时,真空蒸发溶剂。残留物溶于乙酸乙酯(100ml),用5%碳酸钠溶液洗涤,再以水洗涤,硫酸镁干燥,过滤后真空蒸发滤液。残留物以10-25%乙酸乙酯的环己烷溶液为洗脱液进行“快速闪脱层析”纯化。产量0.017g(10%)。核磁共振谱(N.m.r)(CDCl3)δ:2.35(3H,s),4.10(2H,b),4.45(2H,b),7.35(1H,d),7.52(1H,d)。质谱(Mass):(电子喷雾)305(MH+)保留时间 2.89分钟(条件同实施例19.1)药学实施例 无菌制剂 实施例A
mg/ml
本发明化合物 0.1mg
氯化钠USP 9.0mg
注射用水USP适量至 1ml
使各组分溶于一份注射用水中,并使该溶液的终体积含本发明化合物0.1mg/ml。采用所述化合物的盐时,化合物的用量增加至可产生0.1mg/ml的游离碱。溶液应分装以便注射,例如灌装并密封于安瓿瓶、小玻瓶或注射器中。可以是无菌灌装和/或灌装后灭菌,例如,121℃高压灭菌。
此外,可以类似方法制备含不同浓度的化合物的其它无菌制剂。实施例B
mg/ml
本发明化合物 0.5mg
甘露醇 50.0mg
注射用水适量至 1.0ml
使各组分溶于一份注射用水中,使至所需终体积并混匀。制剂通过无菌滤膜过滤灭菌并装入小玻瓶。冷冻干燥并封瓶。使用前再以适当的溶剂重新配制。口服制剂
以常用方法制备片剂,例如直接压片或湿粒制片。片剂可以适当的制膜物质,例如Opadry,采用标准技术进行包膜。亦可选择糖包衣。实施例C直接压制片剂
mg/片
本发明化合
物 5.0mg
硬脂酸镁 4.0mg
微晶纤维素(Avicel pH 102)适量至 400.0mg
本发明化合物过30目筛后,与微晶纤维素及硬脂酸镁混合。以带有直径为11.0mm冲头的适当的压片机将混合物压制成片,使每片中含本发明化合物5mg。亦可以类似方法制备其它规格的片剂,例如含本发明化合物25或100mg/片。实施例D 湿粒片剂
mg/片
本发明化合物 5.0mg
预胶凝淀粉 28.0mg
羟乙酸淀粉钠 16.0mg
硬脂酸镁 4.0mg
乳糖适量 至 400.0mg
将本发明化合物与乳糖、预胶凝淀粉及羟乙酸淀粉钠干燥混合后以适量的纯水制粒。成粒干燥后混以硬脂酸镁。采用带有直径为11.0mm冲头的适当的压片机将干粒压制成片,以使每片含本发明化合物5mg。
可制备其它规格的片剂,例如含本发明化合物25或100mg/片。实施例E硬明胶胶囊剂
mg/粒胶囊
本发明中化合物 5.0mg
微晶纤维素(Avicel PH 102)适量至 700.0mg
将本发明化合物过30目筛后与微晶纤维素混合均匀。将混合物装入0EL硬明胶胶囊壳中,使胶囊含本发明化合物5.0mg/粒。以类似方法制备其它规格的胶囊剂,例如含本发明化合物25或100mg/粒。实施例F软明胶胶囊剂
mg/粒
本发明化合物 10.0mg
聚乙二醇 90.0mg
丙二醇适量 至 200.0mg
将聚乙二醇和丙二醇混合,必要时加热。搅拌均匀。加入本发明化合物并混合均匀。装入适当的软胶囊使每粒胶囊含制剂200mg,含本发明化合物10.0mg/粒。
以类似方法制备其它规格的软胶囊剂,例如含本发明化合物5或25mg/粒。实施例G糖浆剂
本发明化合物 5.0mg
山梨醇溶液 1500.0mg
甘油 1000.0mg
苯甲酸钠 5.0mg
香料 12.5mg
纯水适量 至 5.0ml
将苯甲酸钠溶入1份纯水并加入山梨醇溶液。加入本发明化合物、香料和甘油并混合均匀。混合物以纯水调至需要的体积。其它制剂实施例H栓剂
mg/粒栓制
本发明化合物 10.0mg
硬脂Witepsol W32, 适量 至 2000.0mg
约36℃的温度下将Witepsol W32熔化。取一部分熔化的WitepsolW32,加入本发明的化合物并混合之。合并剩余熔化的Witepsol W32并混合均匀。向栓模中注入2000mg制剂物,使栓剂中含本发明化合物10.0mg/粒。实施例I透皮剂
本发明化合物 5.0mg
液态聚硅氧烷 90.9mg
胶体二氧化硅 5.0mg
将液态聚硅氧烷与活性物混合并加入胶体二氧化硅。而后将该物质分入随后热封的聚合膜,聚合膜由下列物质组成:聚合脂构成的释放衬垫、由聚硅氧烷或丙烯酸聚合物组成的皮肤胶粘剂、聚合烯烃(例如聚乙烯或聚乙烯乙酸酯)或聚氨基甲酸酯组成的控释膜、及多层聚合脂构成的不透水的反向(backing)膜。生物学资料
本发明化合物已表明具有抗癫痫活性,例如在超限度的电休克模型可以抑制后肢伸展。雄性Han Wistar大鼠(150-200mg)在实验前2小时腹腔注射0.25%的实验化合物的甲基纤维素悬浮液。在共济失调出现前做肉眼观察。使用心房电极,给予200mA,200毫秒的电流刺激,观察是否出现后肢伸展。
在上述实验中,本发明化合物的ED50的范围为1至20mg/kg。
在上述活体实验中,未发现由本发明的化合物给药引起的毒性作用。
Claims (13)
1.式(I)化合物及其药学上可接受的盐或溶剂化物 其中R1选自一个或多个卤原子取代的苯基、萘基和一个或多个卤原子取代的萘基;R2选自-NH2和-NHC(=O)Ra;R3选自-NRbRc、-NHC(=O)Ra和氢;R4选自氢、-C1-4烷基、一个或多个卤原子取代的-C1-4烷基、-CN、-CH2OH、-CH2ORd和-CH2S(O)xRd,其中Ra表示C1-4烷基或C3-7环烷基,此外Rb和Rc,可以相同或不同,并选自氢原子与C1-4烷基;或者与它们所连接的氮原子一起形成六元含氮杂环,该杂环可进一步被一个或多个C1-4烷基取代;Rd选自C1-4烷基或由一个或多个卤原子取代的C1-4烷基;X是0、1或2的整数;条件是:当R2为-NH2而且R3和R4均为氢原子时,R1不代表:
其中R1选自一个或多个卤原子取代的苯基、萘基和一个或多个卤原子取代的萘基;R2选自-NH2和-NHC(=O)Ra;R3选自-NRbRc、-NHC(=O)Ra和氢;R4选自氢、-C1-4烷基、一个或多个卤原子取代的-C1-4烷基、-CN、-CH2OH、-CH2ORd和-CH2S(O)xRd,其中Ra表示C1-4烷基或C3-7环烷基,此外Rb和Rc,可以相同或不同,并选自氢原子与C1-4烷基;或者与它们所连接的氮原子一起形成六元含氮杂环,该杂环可进一步被一个或多个C1-4烷基取代;Rd选自C1-4烷基或由一个或多个卤原子取代的C1-4烷基;X是0、1或2的整数;条件是:当R2为-NH2而且R3和R4均为氢原子时,R1不代表: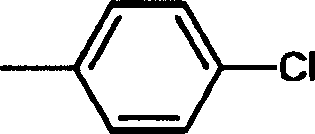
2.权利要求1的化合物,其中R1为由一个或多个卤原子取代的苯基。
3.权利要求2的化合物,其中R1为2,3,5-三氯苯基。
4.权利要求1至3中任一项的化合物,其中R2和R3是-NH2。
5.权利要求1至4中任一项的化合物,其中R4为氢或-CH2OH。
6.权利要求5的化合物,其中R4为氢。
7.化合物,它是2,6-二氨基-3-(2,3,5-三氯苯基)吡嗪及其药学上可接受的盐或溶剂化物。
8.一种药物组合物,含有权利要求1至7中任一项的化合物以及药学上可接受的载体。
9.权利要求1至7中任一项的化合物在神经保护或用于治疗下列疾病的药物制备中的用途:癫痫、双相性精神障碍或躁狂性抑郁、疼痛、功能性肠道紊乱、神经源性疾病、神经退化,预防或减少对成瘾性药物的依赖,预防或减少或逆转对成瘾性药物的耐受。
10.权利要求1中定义的式(I)化合物的制备方法,该方法包括:
(A)在溶剂如乙醇中,使式(II)化合物与适当的胺反应 其中Hal(B)表示卤原子。
其中Hal(B)表示卤原子。
11.权利要求1中定义的式(I)化合物的制备方法,该方法包括:
(B)使式(VII)化合物 其中Hal表示卤原子,与式(IV)R1B(OH)2表示的化合物反应。
其中Hal表示卤原子,与式(IV)R1B(OH)2表示的化合物反应。
12.权利要求1中定义的式(I)化合物的制备方法,该方法包括:;
(C)在适当的溶剂如醇中,在自然氧化反应发生的条件下,通过用如氢氧化锂中和,使式(XI)化合物 或其盐经环化和氧化。
或其盐经环化和氧化。
13.制备式(I)化合物的方法,该方法包括:
(i)通过水解使其中R3代表NHC(=O)Ra的式(I)化合物转化为R3代表NH2的式(I)化合物;或
(ii)通过卤化、接着与适当来源的氰离子反应使其中R4代表氢的式(I)化合物转化为R4代表-CN的式(I)化合物;或
(iii)通过在甲苯中与氢化二异丁基铝反应、接着水解、然后还原,使其中R4代表-CN的式(I)化合物转化为R4代表-CH2OH的式(I)化合物;或
(iv)通过烷基化,使其中R4代表-CH2OH的式(I)化合物转化为R4代表-CH2ORd的式(I)化合物;或
(v)通过在甲苯中与氢化二异丁基铝反应、接着水解、然后与对-甲苯磺酰肼反应、随后在氯仿/四氢呋喃中儿茶酚硼烷反应,使其中R4代表-CN的式(I)化合物转化为R4代表甲基的式(I)化合物。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB9704275.8A GB9704275D0 (en) | 1997-03-01 | 1997-03-01 | Pharmacologically active compound |
| GBGB9708183.0A GB9708183D0 (en) | 1997-04-23 | 1997-04-23 | Pharmacologically active compound |
| GB9708183.0 | 1997-04-23 | ||
| GB9704275.8 | 1997-04-23 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1253551A CN1253551A (zh) | 2000-05-17 |
| CN1105111C true CN1105111C (zh) | 2003-04-09 |
Family
ID=26311097
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN98804593A Expired - Fee Related CN1105111C (zh) | 1997-03-01 | 1998-02-26 | 吡嗪化合物 |
Country Status (46)
Families Citing this family (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6420354B1 (en) | 1998-06-08 | 2002-07-16 | Advanced Medicine, Inc. | Sodium channel drugs and uses |
| GB9818881D0 (en) | 1998-08-28 | 1998-10-21 | Glaxo Group Ltd | Compounds |
| GB9907965D0 (en) * | 1999-04-09 | 1999-06-02 | Glaxo Group Ltd | Medical use |
| US6479498B1 (en) | 1999-06-04 | 2002-11-12 | Theravance, Inc. | Sodium channel drugs and uses |
| EP1500653A1 (en) * | 2000-02-16 | 2005-01-26 | Neurogen Corporation | Substituted arylpyrazines |
| EE200200453A (et) | 2000-02-16 | 2003-12-15 | Neurogen Corporation | Asendatud arüülpürasiinid |
| GB0010971D0 (en) * | 2000-05-05 | 2000-06-28 | Glaxo Group Ltd | Process |
| GB0016040D0 (en) * | 2000-06-29 | 2000-08-23 | Glaxo Group Ltd | Novel process for preparing crystalline particles |
| SE0102438D0 (sv) * | 2001-07-05 | 2001-07-05 | Astrazeneca Ab | New compounds |
| SE0102439D0 (sv) * | 2001-07-05 | 2001-07-05 | Astrazeneca Ab | New compounds |
| US6992087B2 (en) | 2001-11-21 | 2006-01-31 | Pfizer Inc | Substituted aryl 1,4-pyrazine derivatives |
| BR0214309A (pt) * | 2001-11-21 | 2004-10-13 | Upjohn Co | Derivados aril-1,4-pirazina substituìdos |
| DE60219687T2 (de) * | 2001-12-27 | 2007-12-27 | Ortho-Mcneil Pharmaceutical, Inc. | Aroylpyrrolheteroeryl und methanole zur behandlung von störungen des zentralnervensystems |
| JP2005533014A (ja) * | 2002-04-26 | 2005-11-04 | ファルマシア・アンド・アップジョン・カンパニー・エルエルシー | 置換ピラジン誘導体 |
| JP2006509729A (ja) | 2002-08-20 | 2006-03-23 | ニューロジェン・コーポレーション | Crf受容体モジュレータとしての5−置換−2−アリールピラジン類 |
| SE0203754D0 (sv) * | 2002-12-17 | 2002-12-17 | Astrazeneca Ab | New compounds |
| SE0203753D0 (sv) * | 2002-12-17 | 2002-12-17 | Astrazeneca Ab | New compounds |
| WO2004066987A2 (en) * | 2003-01-30 | 2004-08-12 | Dynogen Pharmaceuticals, Inc. | Use of sodium channel modulators for treating gastrointestinal tract disorders |
| TW200510373A (en) * | 2003-07-14 | 2005-03-16 | Neurogen Corp | Substituted quinolin-4-ylamine analogues |
| CN101163478B (zh) | 2005-01-25 | 2013-11-27 | 幸讬制药公司 | 用于炎症及免疫相关用途之化合物 |
| EP1853230A2 (en) * | 2005-02-15 | 2007-11-14 | Jazz Pharmaceuticals Inc. | Dosage form and method for sustained release of a substituted pyrazine compound |
| NL2000284C2 (nl) * | 2005-11-04 | 2007-09-28 | Pfizer Ltd | Pyrazine-derivaten. |
| CA2633329A1 (en) * | 2006-01-23 | 2007-07-26 | Pfizer Limited | Pyridine derivatives as sodium channel modulators |
| WO2008135830A1 (en) * | 2007-05-03 | 2008-11-13 | Pfizer Limited | N- [6-amino-s- (phenyl) pyrazin-2-yl] -isoxazole-4-carboxamide derivatives and related compounds as nav1.8 channel modulators for the treatment of pain |
| GB0800741D0 (en) | 2008-01-16 | 2008-02-20 | Univ Greenwich | Cyclic triazo and diazo sodium channel blockers |
| GB201111712D0 (en) | 2011-07-08 | 2011-08-24 | Gosforth Ct Holdings Pty Ltd | Pharmaceutical compositions |
| FR3001151B1 (fr) | 2013-01-21 | 2016-04-08 | Pf Medicament | Association d'un bloqueur de courant sodique lent et d'un inhibiteur du courant if sinusal et les compositions pharmarceutiques la contenant |
| JO3517B1 (ar) * | 2014-01-17 | 2020-07-05 | Novartis Ag | ان-ازاسبيرو الكان حلقي كبديل مركبات اريل-ان مغايرة وتركيبات لتثبيط نشاط shp2 |
| RU2744988C2 (ru) | 2016-06-14 | 2021-03-17 | Новартис Аг | Соединения и композиции для подавления активности shp2 |
| US11504347B1 (en) | 2016-07-22 | 2022-11-22 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11602512B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11986451B1 (en) | 2016-07-22 | 2024-05-21 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| UY37341A (es) | 2016-07-22 | 2017-11-30 | Flamel Ireland Ltd | Formulaciones de gamma-hidroxibutirato de liberación modificada con farmacocinética mejorada |
| US11602513B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| CN108101857B (zh) * | 2016-11-24 | 2021-09-03 | 韶远科技(上海)有限公司 | 制取2-氨基-3-溴-6-氯吡嗪的可放大工艺 |
| CN107011273A (zh) * | 2017-05-04 | 2017-08-04 | 无锡捷化医药科技有限公司 | 一种合成6‑碘‑3‑(2,3‑二氯苯基)吡嗪‑2‑胺的方法 |
| CA3127871A1 (en) | 2019-03-01 | 2020-09-10 | Flamel Ireland Limited | Gamma-hydroxybutyrate compositions having improved pharmacokinetics in the fed state |
| US11779557B1 (en) | 2022-02-07 | 2023-10-10 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11583510B1 (en) | 2022-02-07 | 2023-02-21 | Flamel Ireland Limited | Methods of administering gamma hydroxybutyrate formulations after a high-fat meal |
| CN115611700B (zh) * | 2022-10-11 | 2024-06-14 | 辽宁东大光明化工科技有限责任公司 | 一种1-溴-2,5-二氯-3-氟苯的制备方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4402958A (en) * | 1981-11-19 | 1983-09-06 | American Cyanamid Company | Novel (substituted phenyl)-1,2,4-triazolo (4,3-A)pyrazines and novel 2-hydrazino-(substituted phenyl)pyrazine intermediates |
| JPH08238778A (ja) * | 1995-03-07 | 1996-09-17 | Brother Ind Ltd | インク噴射装置の電極形成方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0288431B1 (de) | 1987-04-07 | 1992-08-19 | Ciba-Geigy Ag | 3H-1,2,3-Triazolo[4,5-d]pyrimidine |
-
1998
- 1998-02-23 MA MA24973A patent/MA26473A1/fr unknown
- 1998-02-24 PE PE1998000134A patent/PE58299A1/es not_active Application Discontinuation
- 1998-02-26 PT PT98913592T patent/PT966448E/pt unknown
- 1998-02-26 CA CA002282585A patent/CA2282585C/en not_active Expired - Fee Related
- 1998-02-26 SK SK1173-99A patent/SK117399A3/sk unknown
- 1998-02-26 IL IL13129398A patent/IL131293A/xx not_active IP Right Cessation
- 1998-02-26 HU HU0001802A patent/HU225852B1/hu not_active IP Right Cessation
- 1998-02-26 WO PCT/EP1998/001077 patent/WO1998038174A1/en active IP Right Grant
- 1998-02-26 CZ CZ19993111A patent/CZ295618B6/cs not_active IP Right Cessation
- 1998-02-26 ES ES98913592T patent/ES2205469T3/es not_active Expired - Lifetime
- 1998-02-26 DK DK98913592T patent/DK0966448T3/da active
- 1998-02-26 ID IDW990951A patent/ID22850A/id unknown
- 1998-02-26 YU YU40799A patent/YU40799A/sh unknown
- 1998-02-26 EE EEP199900376A patent/EE9900376A/xx unknown
- 1998-02-26 JO JO19982035A patent/JO2035B1/en active
- 1998-02-26 AU AU68237/98A patent/AU732915B2/en not_active Ceased
- 1998-02-26 JP JP53731098A patent/JP3369189B2/ja not_active Expired - Fee Related
- 1998-02-26 NZ NZ337121A patent/NZ337121A/xx not_active IP Right Cessation
- 1998-02-26 SI SI9830578T patent/SI0966448T1/xx unknown
- 1998-02-26 MY MYPI98000852A patent/MY118612A/en unknown
- 1998-02-26 EP EP98913592A patent/EP0966448B1/en not_active Expired - Lifetime
- 1998-02-26 EA EA199900687A patent/EA002102B1/ru not_active IP Right Cessation
- 1998-02-26 GT GT199800046A patent/GT199800046A/es unknown
- 1998-02-26 AT AT98913592T patent/ATE251143T1/de active
- 1998-02-26 CN CN98804593A patent/CN1105111C/zh not_active Expired - Fee Related
- 1998-02-26 US US09/380,062 patent/US6255307B1/en not_active Expired - Fee Related
- 1998-02-26 TR TR1999/02082T patent/TR199902082T2/xx unknown
- 1998-02-26 GE GEAP19984976A patent/GEP20012555B/en unknown
- 1998-02-26 DE DE69818643T patent/DE69818643T2/de not_active Expired - Lifetime
- 1998-02-26 PL PL335441A patent/PL192864B1/pl unknown
- 1998-02-26 AP AP9901632A patent/AP9901632A0/xx unknown
- 1998-02-26 BR BRPI9807814-3A patent/BR9807814B1/pt not_active IP Right Cessation
- 1998-02-26 CO CO98010500A patent/CO4950513A1/es unknown
- 1998-02-26 KR KR1019997007937A patent/KR100544263B1/ko not_active IP Right Cessation
- 1998-02-27 HR HR9708183.0A patent/HRP980107A2/hr not_active Application Discontinuation
- 1998-02-27 PA PA19988448201A patent/PA8448201A1/es unknown
- 1998-02-27 AR ARP980100906A patent/AR011174A1/es active IP Right Grant
- 1998-02-27 SV SV1998000029A patent/SV1998000029A/es not_active Application Discontinuation
- 1998-02-27 HN HN1998000036A patent/HN1998000036A/es unknown
- 1998-02-27 UY UY24911A patent/UY24911A1/es unknown
- 1998-03-03 TW TW087103079A patent/TW513416B/zh not_active IP Right Cessation
-
1999
- 1999-02-11 HN HN1999000019A patent/HN1999000019A/es unknown
- 1999-08-24 IS IS5163A patent/IS5163A/is unknown
- 1999-08-27 OA OA9900196A patent/OA11151A/en unknown
- 1999-08-31 NO NO19994213A patent/NO313383B1/no not_active IP Right Cessation
- 1999-09-09 BG BG103723A patent/BG103723A/bg unknown
-
2000
- 2000-04-10 HK HK00102173A patent/HK1023116A1/xx not_active IP Right Cessation
-
2001
- 2001-05-16 US US09/855,703 patent/US6599905B2/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4402958A (en) * | 1981-11-19 | 1983-09-06 | American Cyanamid Company | Novel (substituted phenyl)-1,2,4-triazolo (4,3-A)pyrazines and novel 2-hydrazino-(substituted phenyl)pyrazine intermediates |
| JPH08238778A (ja) * | 1995-03-07 | 1996-09-17 | Brother Ind Ltd | インク噴射装置の電極形成方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1105111C (zh) | 吡嗪化合物 | |
| CN1102596C (zh) | 5H-噻唑并[3,2-a]嘧啶衍生物 | |
| CN85105301A (zh) | (S)-α-乙基-2-氧代-1-吡咯烷乙酰胺的制备方法 | |
| JP2003512454A (ja) | 悪性腫瘍治療薬 | |
| CN1195741C (zh) | 嘧啶酮化合物,包含该化合物的药物组合物以及制备该化合物的方法 | |
| CN1324349A (zh) | 吡嗪类化合物 | |
| CN87104099A (zh) | 抗变态反应和消炎药剂 | |
| CN1812968A (zh) | 4-[6-乙酰基-3-[3-(4-乙酰基-3-羟基-2-丙基苯硫基)丙基]-2-丙基苯氧基]丁酸的多晶型物a | |
| AU753874B2 (en) | Triazine compounds for treatment of CNS disorders | |
| HU199440B (en) | Process for producing morpholine derivatives and pharmaceutical compositions comprising the compounds | |
| CN1378528A (zh) | 羰基氨基衍生物对抗cns障碍的应用 | |
| CN1042935C (zh) | 2-氰基-3-羟基丙烯酰胺类化合物及其制法、用途和药物组合物 | |
| CN1060838A (zh) | 一种新的5h-苯并二氮杂衍生物,含有它的药物组合物,以及制备它的方法 | |
| CN1040798A (zh) | 苯并二氮杂草化合物及其制备方法和在药物上的应用 | |
| CN1014992B (zh) | 喹唑啉二酮和吡啶并嘧啶二酮的制备方法 | |
| CN1052305A (zh) | 新吡啶衍生物、含它们的药物组合物及其制备方法 | |
| CN86108627A (zh) | 亚磺酰和磺酰取代的3-苯并吖庚因 | |
| CN1064962C (zh) | 芳烷基-噻二嗪酮类化合物 | |
| CN1047076A (zh) | 苯乙醇胺衍生物的制备 | |
| CN1030229A (zh) | 丁烯酸酰胺类及它们的盐类和包含它们的医药组成物及其制备方法 | |
| CN1104637A (zh) | 2-氨基吡嗪-5-甲酰胺衍生物,其制备方法及其药用 | |
| CN1025997C (zh) | 异噁唑衍生物的制备方法 | |
| CN1049504A (zh) | 新型丙烯酸盐、其制备方法、含有它们的药用组合物及其在医学中的用途 | |
| CN87104206A (zh) | 安息香酸衍生物及其制造方法 | |
| CN1096028A (zh) | 新的环己基磺酰基丙烯酸和其衍生物及其制备方法与用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: GR Ref document number: 1033956 Country of ref document: HK |
|
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20030409 Termination date: 20130226 |