WO2015111187A1 - 電気デバイス - Google Patents
電気デバイス Download PDFInfo
- Publication number
- WO2015111187A1 WO2015111187A1 PCT/JP2014/051526 JP2014051526W WO2015111187A1 WO 2015111187 A1 WO2015111187 A1 WO 2015111187A1 JP 2014051526 W JP2014051526 W JP 2014051526W WO 2015111187 A1 WO2015111187 A1 WO 2015111187A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- active material
- electrode active
- negative electrode
- positive electrode
- material layer
- Prior art date
Links
Images


Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/058—Construction or manufacture
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/131—Electrodes based on mixed oxides or hydroxides, or on mixtures of oxides or hydroxides, e.g. LiCoOx
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/133—Electrodes based on carbonaceous material, e.g. graphite-intercalation compounds or CFx
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/134—Electrodes based on metals, Si or alloys
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/362—Composites
- H01M4/364—Composites as mixtures
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/38—Selection of substances as active materials, active masses, active liquids of elements or alloys
- H01M4/386—Silicon or alloys based on silicon
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/483—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides for non-aqueous cells
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/50—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese
- H01M4/505—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese of mixed oxides or hydroxides containing manganese for inserting or intercalating light metals, e.g. LiMn2O4 or LiMn2OxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/52—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron
- H01M4/525—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron of mixed oxides or hydroxides containing iron, cobalt or nickel for inserting or intercalating light metals, e.g. LiNiO2, LiCoO2 or LiCoOxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/583—Carbonaceous material, e.g. graphite-intercalation compounds or CFx
- H01M4/587—Carbonaceous material, e.g. graphite-intercalation compounds or CFx for inserting or intercalating light metals
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/42—Methods or arrangements for servicing or maintenance of secondary cells or secondary half-cells
- H01M2010/4292—Aspects relating to capacity ratio of electrodes/electrolyte or anode/cathode
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P70/00—Climate change mitigation technologies in the production process for final industrial or consumer products
- Y02P70/50—Manufacturing or production processes characterised by the final manufactured product
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02T—CLIMATE CHANGE MITIGATION TECHNOLOGIES RELATED TO TRANSPORTATION
- Y02T10/00—Road transport of goods or passengers
- Y02T10/60—Other road transportation technologies with climate change mitigation effect
- Y02T10/70—Energy storage systems for electromobility, e.g. batteries
Definitions
- the present invention relates to an electrical device.
- the electric device according to the present invention is used, for example, as a secondary battery, a capacitor or the like as a driving power source or auxiliary power source for motors of vehicles such as electric vehicles, fuel cell vehicles, and hybrid electric vehicles.
- Motor drive secondary batteries are required to have extremely high output characteristics and high energy compared to consumer lithium ion secondary batteries used in mobile phones and notebook computers. Therefore, lithium ion secondary batteries having the highest theoretical energy among all the batteries are attracting attention, and are currently being developed rapidly.
- a lithium ion secondary battery includes a positive electrode in which a positive electrode active material or the like is applied to both surfaces of a positive electrode current collector using a binder, and a negative electrode in which a negative electrode active material or the like is applied to both surfaces of a negative electrode current collector using a binder.
- a positive electrode in which a positive electrode active material or the like is applied to both surfaces of a positive electrode current collector using a binder
- a negative electrode in which a negative electrode active material or the like is applied to both surfaces of a negative electrode current collector using a binder.
- it has the structure connected through an electrolyte layer and accommodated in a battery case.
- a battery using a silicon-containing negative electrode active material such as a SiO x (0 ⁇ x ⁇ 2) material that forms a compound with Li is improved in energy density as compared with a conventional carbon / graphite negative electrode material. It is expected as a negative electrode material for vehicle use.
- a silicon oxide having a chemical composition represented by SiO x when viewed microscopically, Si (single crystal nanoparticles) and amorphous SiO 2 exist in phase separation.
- Silicon oxide has a tetrahedral structure as a unit structure, and silicon oxides (intermediate oxides) other than SiO 2 correspond to the number of oxygen at the apex of the tetrahedron, 1, 2 and 3, respectively. Although they can be expressed as 2 O, SiO and Si 2 O 3 , these intermediate oxides are thermodynamically unstable and are extremely difficult to exist as single crystals. Therefore, SiO x is composed of an amorphous structure in which unit structures are irregularly arranged, and this amorphous structure is an amorphous structure in which a plurality of amorphous compounds are formed without forming an interface. The structure is mainly composed of a homogeneous amorphous structure portion. Therefore, SiO x has a structure in which Si nanoparticles are dispersed in amorphous SiO 2 .
- Li y SiO x such as Li 4 SiO 4 , Li 2 SiO 3 , Li 2 Si 2 O 5 , Li 2 Si 3 O 8 , Li 6 Si 4 O 11, etc. (0 ⁇ y, 0 ⁇ x ⁇ 2)
- Li y SiO x has extremely low electron conductivity, and furthermore, since SiO 2 does not have electron conductivity, the resistance of the negative electrode increases. There is. As a result, it is extremely difficult to desorb and insert lithium ions into the negative electrode active material.
- a lithium ion secondary battery using a material that is alloyed with Li for the negative electrode has a large expansion and contraction in the negative electrode during charge and discharge.
- the volume expansion when lithium ions are occluded is about 1.2 times in graphite materials, whereas in Si materials, when Si and Li are alloyed, the amorphous state transitions to the crystalline state, resulting in a large volume change. (Approximately 4 times), there was a problem of reducing the cycle life of the electrode.
- the Si negative electrode active material the battery capacity and the cycle durability are in a trade-off relationship, and there is a problem that it is difficult to improve the high cycle durability while exhibiting a high capacity.
- Patent Document 1 a negative electrode for a lithium ion secondary battery containing SiO x and a graphite material has been proposed (see, for example, Patent Document 1).
- paragraph “0018” describes that, by minimizing the content of SiO x , good cycle life is exhibited in addition to high capacity.
- the lithium ion secondary battery using a negative electrode containing SiO x and a carbon material described in Patent Document 1 is said to be able to exhibit good cycle characteristics.
- An object of the present invention is to provide means capable of further improving cycle durability in an electric device such as a lithium ion secondary battery having a negative electrode containing a silicon-containing negative electrode active material.
- the present inventors have conducted intensive research to solve the above problems. As a result, it has been found that the above problem can be solved by taking good care of the ratio of the area of the positive electrode active material layer and the area of the negative electrode active material layer constituting the single battery layer of the electric device to a value within a predetermined range.
- the present invention has been completed.
- a positive electrode in which a positive electrode active material layer containing a positive electrode active material is formed on the surface of a positive electrode current collector, and a negative electrode active material layer containing a silicon-containing negative electrode active material on the surface of the negative electrode current collector are formed.
- the present invention relates to an electric device having a power generation element including a single battery layer including a negative electrode and a separator.
- the area of the negative electrode active material layer is set to A [m 2 ] and the area of the positive electrode active material layer is set to C [m 2 ] in at least one of the single cell layers constituting the power generation element. 2 ] is characterized in that the expression (1): 0.91 ⁇ C / A ⁇ 1 is satisfied.
- FIG. 1 is a schematic cross-sectional view showing the basic configuration of a non-aqueous electrolyte lithium ion secondary battery that is not a flat type (stacked type) bipolar type, which is an embodiment of the electrical device according to the present invention. It is a perspective view showing the appearance of a flat lithium ion secondary battery which is a typical embodiment of an electric device according to the present invention.
- a positive electrode in which a positive electrode active material layer containing a positive electrode active material is formed on the surface of a positive electrode current collector, and a negative electrode active material containing a silicon-containing negative electrode active material on the surface of the negative electrode current collector
- a lithium ion secondary battery will be described as an example of an electric device.
- the lithium ion secondary battery using the electric device according to the present invention the voltage of the cell (single cell layer) is large, and high energy density and high output density can be achieved. Therefore, the lithium ion secondary battery of the present embodiment is excellent as a vehicle driving power source or an auxiliary power source. As a result, it can be suitably used as a lithium ion secondary battery for a vehicle driving power source or the like. In addition to this, the present invention can be sufficiently applied to lithium ion secondary batteries for portable devices such as mobile phones.
- the lithium ion secondary battery When the lithium ion secondary battery is distinguished by its form / structure, it can be applied to any conventionally known form / structure such as a stacked (flat) battery or a wound (cylindrical) battery. Is. By adopting a stacked (flat) battery structure, long-term reliability can be secured by a sealing technique such as simple thermocompression bonding, which is advantageous in terms of cost and workability.
- a solution electrolyte type battery using a solution electrolyte such as a nonaqueous electrolyte solution for the electrolyte layer, a polymer battery using a polymer electrolyte for the electrolyte layer, etc. It can be applied to any conventionally known electrolyte layer type.
- the polymer battery is further divided into a gel electrolyte type battery using a polymer gel electrolyte (also simply referred to as gel electrolyte) and a solid polymer (all solid) type battery using a polymer solid electrolyte (also simply referred to as polymer electrolyte). It is done.
- FIG. 1 schematically shows the overall structure of a flat (stacked) lithium ion secondary battery (hereinafter also simply referred to as “stacked battery”), which is a typical embodiment of the electrical device of the present invention.
- stacked battery a flat (stacked) lithium ion secondary battery
- the stacked battery 10 of the present embodiment has a structure in which a substantially rectangular power generation element 21 in which a charge / discharge reaction actually proceeds is sealed inside a laminate sheet 29 that is an exterior body.
- the positive electrode in which the positive electrode active material layer 13 is disposed on both surfaces of the positive electrode current collector 11, the electrolyte layer 17, and the negative electrode active material layer 15 is disposed on both surfaces of the negative electrode current collector 12. It has a configuration in which a negative electrode is laminated. Specifically, the negative electrode, the electrolyte layer, and the positive electrode are laminated in this order so that one positive electrode active material layer 13 and the negative electrode active material layer 15 adjacent thereto face each other with the electrolyte layer 17 therebetween. .
- the adjacent positive electrode, electrolyte layer, and negative electrode constitute one unit cell layer 19. Therefore, it can be said that the stacked battery 10 shown in FIG. 1 has a configuration in which a plurality of single battery layers 19 are stacked and electrically connected in parallel.
- the positive electrode current collector 13 on the outermost layer located on both outermost layers of the power generating element 21 is provided with the positive electrode active material layer 13 only on one side, but the active material layer may be provided on both sides. . That is, instead of using a current collector dedicated to the outermost layer provided with an active material layer only on one side, a current collector having an active material layer on both sides may be used as it is as an outermost current collector.
- the outermost negative electrode current collector is positioned on both outermost layers of the power generation element 21, and one side of the outermost negative electrode current collector or A negative electrode active material layer may be disposed on both sides.
- the positive electrode current collector 11 and the negative electrode current collector 12 are attached to a positive electrode current collector plate 25 and a negative electrode current collector plate 27 that are electrically connected to the respective electrodes (positive electrode and negative electrode), and are sandwiched between end portions of the laminate sheet 29. Thus, it has a structure led out of the laminate sheet 29.
- the positive electrode current collector plate 25 and the negative electrode current collector plate 27 are ultrasonically welded to the positive electrode current collector 11 and the negative electrode current collector 12 of each electrode via a positive electrode lead and a negative electrode lead (not shown), respectively, as necessary. Or resistance welding or the like.
- the lithium ion secondary battery according to this embodiment is characterized by the configuration of the positive electrode and the negative electrode.
- main components of the battery including the positive electrode and the negative electrode will be described.
- the active material layers (13, 15) contain an active material, and further contain other additives as necessary.
- the positive electrode active material layer 13 includes a positive electrode active material.
- a positive electrode active material There is no restriction
- the positive electrode active material preferably includes a positive electrode active material made of a solid solution material (also referred to as “solid solution positive electrode active material” in the present specification) as a positive electrode active material capable of achieving a high electric capacity and energy density.
- Li ⁇ Co-based composite oxide such as LiCoO 2
- Li ⁇ Ni-based composite oxide such as LiNiO 2
- Li ⁇ Mn-based composite oxide such as spinel LiMn 2 O 4
- LiFeO 2 Li / Fe-based composite oxides such as can be used.
- transition metal and lithium phosphate compounds and sulfate compounds such as LiFePO 4
- transition metal oxides and sulfides such as V 2 O 5 , MnO 2 , TiS 2 , MoS 2 , and MoO 3 , PbO 2 , AgO, NiOOH or the like can be used.
- the solid solution positive electrode active material which is a preferable positive electrode active material is demonstrated in detail.
- Solid solution positive electrode active material has a composition represented by the following formula (3) as a basic structure.
- the solid solution positive electrode active material has the composition represented by the formula (3) as a basic structure
- the active material itself having the composition represented by the formula (3) is used as the solid solution positive electrode active material.
- the latter form for example, the following three forms (A) to (C) are exemplified.
- (A) 1 selected from the group consisting of Al, Zr, Ti, Nb, B, S, Sn, W, Mo and V on the particle surface of the solid solution positive electrode active material having the composition represented by formula (3)
- the form in which the element M exists is not particularly limited, and in addition to the form of the oxide, a form of a compound with Li can be assumed, but the form of the oxide is preferable.
- the average particle diameter of the material (such as oxide) containing the element M is preferably 5 to 50 nm.
- the oxide is scattered on the particle surface of the solid solution positive electrode active material.
- the average particle diameter of the oxides scattered in this manner is preferably 5 to 50 nm, but may be aggregated on the particle surface of the solid solution positive electrode active material to form secondary particles.
- the average particle diameter of such secondary particles is preferably 0.1 ⁇ m (100 nm) to 1 ⁇ m (1000 nm).
- the oxide itself containing the element M to be doped or a sol of the oxide is defined as the active material.
- a method may be used in which the mixture is mixed at a temperature of about 100 to 150 ° C. for about 5 to 20 hours and further processed at a temperature of about 200 to 300 ° C. for about 3 to 10 hours.
- a coating layer made of a metal oxide or composite oxide selected from the group consisting of Al, Zr and Ti is formed on the particle surface of the solid solution positive electrode active material having the composition represented by formula (3).
- the content of the oxide or composite oxide in the solid solution positive electrode active material after coating is 0.1 to 3.0% by weight in terms of oxide
- the specific configuration of the metal oxide present on the particle surface of the solid solution positive electrode active material is not particularly limited, and any of the theoretically possible oxides or composite oxides containing the metal elements described above is used. Can be.
- Al 2 O 3 , ZrO 2 or TiO 2 is used.
- a (composite) oxide containing one or more elements selected from the group consisting of Nb, Sn, W, Mo, and V may be further included in the coating layer.
- the solid solution positive electrode active material after substitution is 20-23 °, 35-40 ° (101), 42-45 ° (104) and 64-65 (108) in X-ray diffraction (XRD) measurement.
- XRD X-ray diffraction
- ) / 65-66 (110) preferably has a diffraction peak indicating a rock salt type layered structure. At this time, in order to surely obtain the effect of improving the cycle characteristics, those having substantially no peak attributed to other than the diffraction peak of the rock salt type layered structure are preferable. More preferably, one having three diffraction peaks at 35-40 ° (101) and one diffraction peak at 42-45 ° (104) is suitable.
- the X-ray diffraction measurement shall employ the measurement method described in the examples described later.
- the notation of 64-65 (108) / 65-66 (110) has two peaks close to 64-65 and 65-66.
- one peak is broadly separated without being clearly separated. It is meant to include.
- the solid solution positive electrode active material after substitution in the form (C) has a plurality of specific diffraction peaks in the X-ray diffraction (XRD) measurement.
- the solid solution positive electrode active material having the above composition formula is a solid solution system of Li 2 MnO 3 and LiMnO 2.
- the diffraction peak at 20-23 ° is characteristic of Li 2 MnO 3 .
- the diffraction peaks of 36.5-37.5 ° (101), 44-45 ° (104) and 64-65 (108) / 65-66 (110) are usually in the rock salt type layered structure of LiMnO 2. It is characteristic.
- the solid solution positive electrode active material of the present embodiment does not include those having a peak other than a diffraction peak showing a rock salt type layered structure, for example, other peaks derived from impurities or the like, in these angular ranges.
- a structure other than the rock salt type layered structure is included in the positive electrode active material. If the structure other than the rock salt type layered structure is not included, the effect of improving the cycle characteristics can be surely obtained.
- the average particle diameter of the positive electrode active material contained in the positive electrode active material layer 13 is not particularly limited, but is preferably 1 to 30 ⁇ m and more preferably 5 to 20 ⁇ m from the viewpoint of increasing the output.
- the “particle diameter” refers to the outline of the active material particles (observation surface) observed using an observation means such as a scanning electron microscope (SEM) or a transmission electron microscope (TEM). It means the maximum distance among any two points.
- the value of “average particle diameter” is the value of particles observed in several to several tens of fields using observation means such as a scanning electron microscope (SEM) or a transmission electron microscope (TEM). The value calculated as the average value of the particle diameter shall be adopted.
- the particle diameters and average particle diameters of other components can be defined in the same manner.
- the positive electrode active material layer preferably contains a solid solution positive electrode active material, and preferably contains a positive electrode active material (solid solution positive electrode active material) represented by the following formula (2).
- e represents the weight% of each component in the positive electrode active material layer, and 80 ⁇ e ⁇ 98.
- the content of the solid solution positive electrode active material in the positive electrode active material layer is essential to be 80 to 98% by weight, preferably 84 to 98% by weight. It is.
- the positive electrode active material layer preferably contains a binder and a conductive aid in addition to the solid solution positive electrode active material described above. Further, if necessary, it further contains other additives such as an electrolyte (polymer matrix, ion-conductive polymer, electrolyte solution, etc.) and a lithium salt for increasing the ion conductivity.
- a binder and a conductive aid in addition to the solid solution positive electrode active material described above. Further, if necessary, it further contains other additives such as an electrolyte (polymer matrix, ion-conductive polymer, electrolyte solution, etc.) and a lithium salt for increasing the ion conductivity.
- Binder Although it does not specifically limit as a binder used for a positive electrode active material layer, for example, the following materials are mentioned. Polyethylene, polypropylene, polyethylene terephthalate (PET), polyether nitrile, polyacrylonitrile, polyimide, polyamide, cellulose, carboxymethyl cellulose (CMC) and its salts, ethylene-vinyl acetate copolymer, polyvinyl chloride, styrene-butadiene rubber (SBR) ), Isoprene rubber, butadiene rubber, ethylene / propylene rubber, ethylene / propylene / diene copolymer, styrene / butadiene / styrene block copolymer and hydrogenated product thereof, styrene / isoprene / styrene block copolymer and hydrogenated product thereof.
- Thermoplastic polymers such as products, polyvinylidene fluoride (P
- the binder content in the positive electrode active material layer is preferably 1 to 10% by weight, more preferably 1 to 8% by weight.
- the conductive assistant refers to an additive that is blended in order to improve the conductivity of the positive electrode active material layer or the negative electrode active material layer.
- Examples of the conductive assistant include carbon black such as ketjen black and acetylene black.
- the content of the conductive auxiliary in the positive electrode active material layer is preferably 1 to 10% by weight, more preferably 1 to 8% by weight.
- electrolyte salt examples include Li (C 2 F 5 SO 2 ) 2 N, LiPF 6 , LiBF 4 , LiClO 4 , LiAsF 6 , LiCF 3 SO 3 and the like.
- Examples of the ion conductive polymer include polyethylene oxide (PEO) and polypropylene oxide (PPO) polymers.
- the positive electrode (positive electrode active material layer) can be applied by any one of a kneading method, a sputtering method, a vapor deposition method, a CVD method, a PVD method, an ion plating method, and a thermal spraying method in addition to a method of applying (coating) a normal slurry. Can be formed.
- the negative electrode active material layer 15 includes a silicon-containing negative electrode active material.
- a silicon-containing negative electrode active material There is no restriction
- two or more negative electrode active materials may be used in combination.
- the silicon-containing negative electrode active material preferably contains, as a negative electrode active material capable of achieving a high electric capacity and energy density, SiO x or a Si-containing alloy (also collectively referred to as “Si material”). More preferably, the material includes a carbon material.
- Si material that is a preferable silicon-containing negative electrode active material and the carbon material that is preferably used in combination will be described in more detail.
- the Si material means SiO x (x represents the number of oxygen satisfying the valence of Si) and Si-containing alloy which are a mixture of amorphous SiO 2 particles and Si particles. Only 1 type of these may be used as Si material, and 2 or more types may be used together. Hereinafter, these Si materials will be described in detail.
- SiO x is a mixture of amorphous SiO 2 particles and Si particles, and x represents the number of oxygen satisfying the valence of Si. There is no restriction
- the SiO x may be an electrically conductive SiO x particles the surface of the SiO x particulate is coated with a conductive material by mechanical surface fusion treatment.
- Si in the SiO x particles can easily desorb and insert lithium ions, and the reaction in the active material can proceed more smoothly.
- the content of the conductive substance in the conductive SiO x particles is preferably 1 to 30% by weight, and more preferably 2 to 20% by weight.
- the average particle diameter of the SiO x is not particularly limited as long as it is approximately the same as the average particle diameter of the negative electrode active material contained in the existing negative electrode active material layer 15. From the viewpoint of higher output, it is preferably in the range of 1 to 20 ⁇ m. However, it is not limited at all to the above range, and it goes without saying that it may be outside the above range as long as the effects of the present embodiment can be effectively expressed.
- the shape of SiO x is not particularly limited, and may be spherical, elliptical, cylindrical, polygonal, flaky, indefinite, or the like.
- SiO x in accordance with the manufacturing method according to this embodiment of SiO x is not particularly limited, it can be produced by utilizing the production of conventionally known various. That is, since there is almost no difference in the amorphous state / characteristics depending on the manufacturing method, various manufacturing methods can be applied.
- Si powder and SiO 2 powder are blended at a predetermined ratio as raw materials, and mixed, granulated and dried mixed granulated raw materials are heated in an inert gas atmosphere (830 ° C. or higher) or heated in vacuum (1 , 100 ° C. or higher and 1,600 ° C. or lower) to generate (sublimate) SiO.
- Gaseous SiO generated by sublimation is vapor-deposited on the deposition substrate (substrate temperature is 450 ° C. or more and 800 ° C. or less) to deposit SiO precipitates.
- the SiO x powder is obtained by removing the SiO deposit from the deposition substrate and pulverizing it using a ball mill or the like.
- X value can be determined by X-ray fluorescence analysis. For example, it can be obtained by using a fundamental parameter method in fluorescent X-ray analysis using O-K ⁇ rays.
- RIX3000 manufactured by Rigaku Corporation
- conditions for the fluorescent X-ray analysis for example, rhodium (Rh) may be used as a target, the tube voltage may be 50 kV, and the tube current may be 50 mA. Since the x value obtained here is calculated from the intensity of the O-K ⁇ ray detected in the measurement region on the substrate, it becomes an average value in the measurement region.
- Si-containing alloy is not particularly limited as long as it is an alloy with another metal containing Si, and conventionally known knowledge can be appropriately referred to.
- Si-containing alloy Si x Ti y Ge z A a , Si x Ti y Zn z A a , Si x Ti y Sn z A a , Si x Sn y Al z A a , and Si x Sn y V z A a , Si x Sn y C z A a , Si x Zn y V z A a , Si x Zn y Sn z A a , Si x Zn y Al z A a , Si x Zn y C zA a, Si x Al y C z a a and Si x Al y Nb z a a ( wherein, a is unavoidable impurities.
- the carbon material that can be used in the present invention is not particularly limited, but graphite (graphite), which is a highly crystalline carbon such as natural graphite or artificial graphite; low crystalline carbon such as soft carbon or hard carbon; ketjen black, acetylene Carbon black such as black, channel black, lamp black, oil furnace black, and thermal black; and carbon materials such as fullerene, carbon nanotube, carbon nanofiber, carbon nanohorn, and carbon fibril. Of these, graphite is preferably used.
- the use of the Si material or the carbon material as the negative electrode active material and further the combined use of these materials, while exhibiting higher cycle durability and high initial capacity and well-balanced characteristics. Can be shown.
- SiO x may not be uniformly arranged in the negative electrode active material layer.
- the potential and capacity that each SiO x develops are different.
- SiO x of the negative electrode active material layer includes a SiO x that react excessively lithium ion, SiO x is produced that does not react with lithium ions. That is, non-uniformity of the reaction between SiO x and lithium ions in the negative electrode active material layer occurs.
- SiO x that reacts with lithium ions excessively acts among the alloys described above, and the decomposition of the electrolytic solution due to a significant reaction with the electrolytic solution or the destruction of the structure of SiO x due to excessive expansion may occur.
- the cycle characteristics can be deteriorated as a negative electrode for an electric device.
- the SiO x when the SiO x is mixed with a carbon material, the above problem can be solved. More specifically, by mixing SiO x with a carbon material, it may be possible to uniformly dispose SiO x in the negative electrode active material layer. As a result, it is considered that any SiO x in the negative electrode active material layer exhibits the same reactivity and can prevent deterioration of cycle characteristics.
- the initial capacity can be reduced by reducing the content of SiO x in the negative electrode active material layer.
- the carbon material itself has reactivity with lithium ions, the degree of decrease in the initial capacity is relatively small. That is, the negative electrode active material according to the present embodiment has a large effect of improving the cycle characteristics as compared with the effect of reducing the initial capacity.
- the carbon material is unlikely to undergo a volume change when reacting with lithium ions as compared with SiO x . Therefore, even when the volume change of SiO x is large, when the negative electrode active material is taken as a whole, the influence of the volume change of the negative electrode active material associated with the lithium reaction can be made relatively minor. Such an effect can also be understood from the results of Examples in which the cycle characteristics increase as the carbon material content rate increases (the SiO x content rate decreases).
- the amount of electricity consumed (Wh) can be improved by containing a carbon material. More specifically, the carbon material has a relatively low potential compared with SiO x . As a result, the relatively high potential of SiO x can be reduced. Then, since the electric potential of the whole negative electrode falls, power consumption (Wh) can be improved. Such an action is particularly advantageous when used in, for example, a vehicle application among electric devices.
- the shape of the carbon material is not particularly limited, and may be spherical, elliptical, cylindrical, polygonal, scaly, indefinite, or the like.
- the average particle diameter of the carbon material is not particularly limited, but is preferably 5 to 25 ⁇ m, and more preferably 5 to 10 ⁇ m.
- the average particle diameter of the carbon material may be the same as or different from the average particle diameter of SiO x , but is preferably different. .
- the average particle diameter of the SiO x is more preferably smaller than the average particle diameter of the carbon material.
- the ratio of the average particle diameter of the carbon material to the average particle diameter of SiO x is preferably 1/250 to less than 1, More preferably, it is 100 to 1/4.
- negative electrode active materials other than the two types of negative electrode active materials (Si material and carbon material) described above may be used in combination.
- examples of the negative electrode active material that can be used in combination include lithium-transition metal composite oxides (for example, Li 4 Ti 5 O 12 ), metal materials, lithium alloy negative electrode materials, and the like. Of course, other negative electrode active materials may be used.
- the negative electrode active material layer preferably contains both a Si material and a carbon material, and preferably contains a negative electrode active material represented by the following formula (4).
- the Si material is selected from the group consisting of SiO x (x represents the number of oxygen satisfying the valence of Si), which is a mixture of amorphous SiO 2 particles and Si particles, and a Si-containing alloy.
- ⁇ and ⁇ represent the weight percentage of each component in the negative electrode active material layer, and 80 ⁇ ⁇ + ⁇ ⁇ 98, 3 ⁇ ⁇ ⁇ 40, and 40 ⁇ ⁇ ⁇ 95.
- the content of the Si material as the negative electrode active material in the negative electrode active material layer is 3 to 40% by weight.
- the content of the carbon material negative electrode active material is 40 to 95% by weight. Furthermore, the total content thereof is 80 to 98% by weight.
- the mixing ratio of the Si material and the carbon material of the negative electrode active material is not particularly limited as long as the above-described content specification is satisfied, and can be appropriately selected according to a desired application.
- the content of the Si material in the negative electrode active material is preferably 3 to 40% by weight.
- the content ratio of the Si material in the negative electrode active material is more preferably 4 to 30% by weight.
- the Si material content in the negative electrode active material is more preferably 5 to 20% by weight.
- the content of the Si material is 3% by weight or more because a high initial capacity can be obtained.
- the content of the Si material is 40% by weight or less because high cycle characteristics can be obtained.
- the negative electrode active material layer preferably contains a binder and a conductive additive in addition to the negative electrode active material described above. Further, if necessary, it further contains other additives such as an electrolyte (polymer matrix, ion conductive polymer, electrolytic solution, etc.) and a lithium salt for increasing the ion conductivity.
- an electrolyte polymer matrix, ion conductive polymer, electrolytic solution, etc.
- a lithium salt for increasing the ion conductivity.
- each active material layer (active material layer on one side of the current collector) is not particularly limited, and conventionally known knowledge about the battery can be appropriately referred to.
- the thickness of each active material layer is usually about 1 to 500 ⁇ m, preferably 2 to 100 ⁇ m, taking into consideration the intended use of the battery (emphasis on output, energy, etc.) and ion conductivity.
- the current collectors (11, 12) are made of a conductive material.
- the size of the current collector is determined according to the intended use of the battery. For example, if it is used for a large battery that requires a high energy density, a current collector having a large area is used.
- the thickness of the current collector is usually about 1 to 100 ⁇ m.
- the shape of the current collector is not particularly limited.
- a mesh shape (such as an expanded grid) can be used.
- the negative electrode active material is formed directly on the negative electrode current collector 12 by sputtering or the like, it is preferable to use a current collector foil.
- a metal or a resin in which a conductive filler is added to a conductive polymer material or a non-conductive polymer material can be employed.
- examples of the metal include aluminum, nickel, iron, stainless steel, titanium, and copper.
- a clad material of nickel and aluminum, a clad material of copper and aluminum, or a plating material of a combination of these metals can be preferably used.
- covered on the metal surface may be sufficient.
- aluminum, stainless steel, copper, and nickel are preferable from the viewpoints of electronic conductivity, battery operating potential, and adhesion of the negative electrode active material by sputtering to the current collector.
- examples of the conductive polymer material include polyaniline, polypyrrole, polythiophene, polyacetylene, polyparaphenylene, polyphenylene vinylene, polyacrylonitrile, and polyoxadiazole. Since such a conductive polymer material has sufficient conductivity without adding a conductive filler, it is advantageous in terms of facilitating the manufacturing process or reducing the weight of the current collector.
- Non-conductive polymer materials include, for example, polyethylene (PE; high density polyethylene (HDPE), low density polyethylene (LDPE), etc.), polypropylene (PP), polyethylene terephthalate (PET), polyether nitrile (PEN), polyimide (PI), polyamideimide (PAI), polyamide (PA), polytetrafluoroethylene (PTFE), styrene-butadiene rubber (SBR), polyacrylonitrile (PAN), polymethyl acrylate (PMA), polymethyl methacrylate (PMMA) , Polyvinyl chloride (PVC), polyvinylidene fluoride (PVdF), or polystyrene (PS).
- PE polyethylene
- HDPE high density polyethylene
- LDPE low density polyethylene
- PP polypropylene
- PET polyethylene terephthalate
- PEN polyether nitrile
- PI polyimide
- PAI polyamideimide
- PA polyamide
- PTFE polytetraflu
- a conductive filler may be added to the conductive polymer material or the non-conductive polymer material as necessary.
- a conductive filler is inevitably necessary to impart conductivity to the resin.
- the conductive filler can be used without particular limitation as long as it has a conductivity.
- metals, conductive carbon, etc. are mentioned as a material excellent in electroconductivity, electric potential resistance, or lithium ion barrier
- the metal is not particularly limited, but at least one metal selected from the group consisting of Ni, Ti, Al, Cu, Pt, Fe, Cr, Sn, Zn, In, Sb, and K, or these metals It is preferable to contain an alloy or metal oxide containing.
- it includes at least one selected from the group consisting of acetylene black, vulcan, black pearl, carbon nanofiber, ketjen black, carbon nanotube, carbon nanohorn, carbon nanoballoon, and fullerene.
- the amount of the conductive filler added is not particularly limited as long as it is an amount capable of imparting sufficient conductivity to the current collector, and is generally about 5 to 35% by weight.
- the area of the negative electrode active material layer is A [m 2 ] and the area of the positive electrode active material layer is C [m 2]. ]
- the formula (1) 0.91 ⁇ C / A ⁇ 1 is satisfied, and preferably 0.95 ⁇ C / A ⁇ 0.99.
- the size of the negative electrode active material layer is improved in order to improve the device performance by suppressing defects such as generation of lithium dendrite in the negative electrode and facing deviation between the positive and negative electrodes. Is designed to be one size larger. According to the study by the present inventors, according to the design conventionally performed in this manner, the size of the negative electrode active material layer is relatively large compared to the positive electrode active material layer, so that the silicon-containing negative electrode active material is used. It was found that the cycle durability was reduced when This is because the irreversible capacity of the silicon-containing negative electrode active material is large.
- Such irreversible means that Li is consumed by the irreversible capacity in the positive electrode non-facing region that should not originally contribute to the charge / discharge capacity. For this reason, it has been found that even if a silicon-containing negative electrode active material having a potentially large electric capacity is used, the battery capacity that can be finally taken out decreases.
- the single battery layer that satisfies the above-described formula (1) that defines the C / A ratio is composed of a combination having a relatively large irreversible capacity for both positive and negative electrodes. It has also been found. That is, the irreversible capacity in the negative electrode active material is overwhelmingly larger than that of the positive electrode, but the negative electrode has a large irreversible capacity (for example, a solid solution positive electrode active material).
- the irreversible capacity per unit area of the active material layer and I a, the irreversible capacity per unit area of the positive electrode active material layer is taken as I C, defined as when "a I C / I a ⁇ 0.40" can do.
- the C / A ratio described above preferably satisfies 0.91 ⁇ C / A ⁇ 0.99, and more preferably satisfies 0.95 ⁇ C / A ⁇ 0.99. .
- the cycle durability can be further improved by setting the upper limit value of C / A to a smaller value.
- the separator has a function of holding an electrolytic solution (liquid electrolyte) to ensure lithium ion conductivity between the positive electrode and the negative electrode, and a function as a partition between the positive electrode and the negative electrode.
- separator examples include a separator made of a porous sheet made of a polymer or fiber that absorbs and holds the electrolyte and a nonwoven fabric separator.
- a microporous (microporous film) can be used as the separator of the porous sheet made of polymer or fiber.
- the porous sheet made of the polymer or fiber include polyolefins such as polyethylene (PE) and polypropylene (PP); a laminate in which a plurality of these are laminated (for example, three layers of PP / PE / PP) And a microporous (microporous membrane) separator made of a hydrocarbon resin such as polyimide, aramid, polyvinylidene fluoride-hexafluoropropylene (PVdF-HFP), glass fiber, and the like.
- PE polyethylene
- PP polypropylene
- a microporous (microporous membrane) separator made of a hydrocarbon resin such as polyimide, aramid, polyvinylidene fluoride-hexafluoropropylene (PVdF-HFP), glass fiber, and the like.
- the thickness of the microporous (microporous membrane) separator cannot be uniquely defined because it varies depending on the intended use. For example, in applications such as secondary batteries for driving motors such as electric vehicles (EV), hybrid electric vehicles (HEV), and fuel cell vehicles (FCV), it is 4 to 60 ⁇ m in a single layer or multiple layers. Is desirable.
- the fine pore diameter of the microporous (microporous membrane) separator is desirably 1 ⁇ m or less (usually a pore diameter of about several tens of nm).
- nonwoven fabric separator cotton, rayon, acetate, nylon, polyester; polyolefins such as PP and PE; conventionally known ones such as polyimide and aramid are used alone or in combination.
- the bulk density of the nonwoven fabric is not particularly limited as long as sufficient battery characteristics can be obtained by the impregnated polymer gel electrolyte.
- the thickness of the nonwoven fabric separator may be the same as that of the electrolyte layer, and is preferably 5 to 200 ⁇ m, particularly preferably 10 to 100 ⁇ m.
- the separator includes an electrolytic solution (liquid electrolyte).
- the liquid electrolyte has a function as a lithium ion carrier and has a form in which a lithium salt is dissolved in an organic solvent.
- organic solvent include carbonates such as ethylene carbonate (EC), propylene carbonate (PC), dimethyl carbonate (DMC), diethyl carbonate (DEC), and ethyl methyl carbonate.
- the lithium salt Li (CF 3 SO 2) 2 N, Li (C 2 F 5 SO 2) 2 N, LiPF 6, LiBF 4, LiClO 4, LiAsF 6, LiTaF such 6, LiCF 3 SO 3
- a compound that can be added to the active material layer of the electrode can be similarly employed.
- the electrolytic solution (liquid electrolyte) preferably contains an additive.
- the electrolytic solution preferably contains 1,5,2,4-dioxadithian-2,2,4,4-tetraoxide (DDTO) and lithium difluorophosphate (LiPO 2 F 2 ).
- the concentration of the DDTO and LiPO 2 F 2 in the electrolytic solution is not particularly limited.
- the concentration of DDTO in the electrolytic solution is preferably 0.5 to 2.5% by weight, more preferably 1.0 to 2.0% by weight.
- the concentration of LiPO 2 F 2 in the electrolytic solution is preferably 1.8 to 3.0% by weight, more preferably 1.8 to 2.5% by weight.
- the electrolytic solution may further contain additives other than the components described above.
- additives include, for example, vinylene carbonate, methyl vinylene carbonate, dimethyl vinylene carbonate, phenyl vinylene carbonate, diphenyl vinylene carbonate, ethyl vinylene carbonate, diethyl vinylene carbonate, vinyl ethylene carbonate, 1,2-divinyl ethylene.
- vinylene carbonate, methyl vinylene carbonate, and vinyl ethylene carbonate are preferable, and vinylene carbonate and vinyl ethylene carbonate are more preferable.
- vinylene carbonate and vinyl ethylene carbonate are more preferable.
- these additives only 1 type may be used independently and 2 or more types may be used together.
- the separator is preferably a separator in which a heat-resistant insulating layer is laminated on a porous substrate (a separator with a heat-resistant insulating layer).
- the heat resistant insulating layer is a ceramic layer containing inorganic particles and a binder.
- a highly heat-resistant separator having a melting point or a heat softening point of 150 ° C. or higher, preferably 200 ° C. or higher is used.
- the separator is less likely to curl in the battery manufacturing process due to the effect of suppressing thermal shrinkage and high mechanical strength.
- the inorganic particles in the heat resistant insulating layer contribute to the mechanical strength and heat shrinkage suppressing effect of the heat resistant insulating layer.
- the material used as the inorganic particles is not particularly limited. Examples thereof include silicon, aluminum, zirconium, titanium oxides (SiO 2 , Al 2 O 3 , ZrO 2 , TiO 2 ), hydroxides and nitrides, and composites thereof. These inorganic particles may be derived from mineral resources such as boehmite, zeolite, apatite, kaolin, mullite, spinel, olivine and mica, or may be artificially produced. Moreover, only 1 type may be used individually for these inorganic particles, and 2 or more types may be used together. Of these, silica (SiO 2 ) or alumina (Al 2 O 3 ) is preferably used, and alumina (Al 2 O 3 ) is more preferably used from the viewpoint of cost.
- the basis weight of the heat-resistant particles is not particularly limited, but is preferably 5 to 15 g / m 2 . If it is this range, sufficient ion conductivity will be acquired and it is preferable at the point which maintains heat resistant strength.
- the binder in the heat-resistant insulating layer has a role of adhering the inorganic particles and the inorganic particles to the resin porous substrate layer.
- the heat resistant insulating layer is stably formed, and peeling between the porous substrate layer and the heat resistant insulating layer is prevented.
- the binder used for the heat-resistant insulating layer is not particularly limited.
- a compound such as butadiene rubber, polyvinylidene fluoride (PVDF), polytetrafluoroethylene (PTFE), polyvinyl fluoride (PVF), or methyl acrylate can be used as the binder.
- PVDF polyvinylidene fluoride
- PTFE polytetrafluoroethylene
- PVF polyvinyl fluoride
- methyl acrylate methyl acrylate
- PVDF polyvinylidene fluoride
- these compounds only 1 type may be used independently and 2 or more types may be used together.
- the binder content in the heat-resistant insulating layer is preferably 2 to 20% by weight with respect to 100% by weight of the heat-resistant insulating layer.
- the binder content is 2% by weight or more, the peel strength between the heat-resistant insulating layer and the porous substrate layer can be increased, and the vibration resistance of the separator can be improved.
- the binder content is 20% by weight or less, the gap between the inorganic particles is appropriately maintained, so that sufficient lithium ion conductivity can be ensured.
- the thermal contraction rate of the separator with a heat-resistant insulating layer is preferably 10% or less for both MD and TD after holding for 1 hour at 150 ° C. and 2 gf / cm 2 .
- a current collector plate (tab) electrically connected to a current collector is taken out of a laminate film as an exterior material for the purpose of taking out current outside the battery.
- the material constituting the current collector plate is not particularly limited, and a known highly conductive material conventionally used as a current collector plate for a lithium ion secondary battery can be used.
- a constituent material of the current collector plate for example, metal materials such as aluminum, copper, titanium, nickel, stainless steel (SUS), and alloys thereof are preferable. From the viewpoint of light weight, corrosion resistance, and high conductivity, aluminum and copper are more preferable, and aluminum is particularly preferable. Note that the same material may be used for the positive electrode current collector plate (positive electrode tab) and the negative electrode current collector plate (negative electrode tab), or different materials may be used.
- the tabs 58 and 59 shown in FIG. 2 are not particularly limited.
- the positive electrode tab 58 and the negative electrode tab 59 may be drawn out from the same side, or the positive electrode tab 58 and the negative electrode tab 59 may be divided into a plurality of parts and taken out from each side, as shown in FIG. It is not limited to.
- a terminal may be formed using a cylindrical can (metal can).
- the seal portion is a member unique to the serially stacked battery and has a function of preventing leakage of the electrolyte layer. In addition to this, it is possible to prevent current collectors adjacent in the battery from coming into contact with each other and a short circuit due to a slight unevenness at the end of the laminated electrode.
- the constituent material of the seal part is not particularly limited, but polyolefin resin such as polyethylene and polypropylene, epoxy resin, rubber, polyimide and the like can be used. Among these, it is preferable to use a polyolefin resin from the viewpoints of corrosion resistance, chemical resistance, film-forming property, economy, and the like.
- ⁇ Positive terminal lead and negative terminal lead> As a material for the negative electrode and the positive electrode terminal lead, a lead used in a known laminated secondary battery can be used.
- the parts removed from the battery exterior material should be heat-insulating so that they do not affect products (for example, automobile parts, especially electronic devices) by touching peripheral devices or wiring and causing leakage. It is preferable to coat with a heat shrinkable tube or the like.
- Laminate film A conventionally known metal can case can be used as the exterior material.
- the power generation element 17 may be packed using a laminate film 22 as shown in FIG.
- the laminate film can be configured as a three-layer structure in which, for example, polypropylene, aluminum, and nylon are laminated in this order.
- the manufacturing method in particular of a lithium ion secondary battery is not restrict
- a lithium ion secondary battery is not limited to this.
- the electrode (positive electrode and negative electrode) is prepared, for example, by preparing an active material slurry (positive electrode active material slurry or negative electrode active material slurry) and applying the active material slurry onto a current collector. It can be made by drying, then pressing.
- the active material slurry includes the above-described active material (positive electrode active material or negative electrode active material), a binder, a conductive additive, and a solvent.
- the solvent is not particularly limited, and N-methyl-2-pyrrolidone (NMP), dimethylformamide, dimethylacetamide, methylformamide, cyclohexane, hexane, water and the like can be used.
- NMP N-methyl-2-pyrrolidone
- the method for applying the active material slurry to the current collector is not particularly limited, and examples thereof include a screen printing method, a spray coating method, an electrostatic spray coating method, an ink jet method, and a doctor blade method.
- the method for drying the coating film formed on the surface of the current collector is not particularly limited as long as at least a part of the solvent in the coating film is removed.
- An example of the drying method is heating. Drying conditions (drying time, drying temperature, etc.) can be appropriately set according to the volatilization rate of the solvent contained in the applied active material slurry, the coating amount of the active material slurry, and the like. A part of the solvent may remain. The remaining solvent can be removed by a press process described later.
- the pressing means is not particularly limited, and for example, a calendar roll, a flat plate press, or the like can be used.
- the single cell layer can be produced by laminating the electrodes (positive electrode and negative electrode) produced in (1) via an electrolyte layer.
- the power generation element can be produced by laminating the single cell layers in consideration of the output and capacity of the single cell layer, the output and capacity required for the battery, and the like.
- the structure of the battery various shapes such as a square, a paper, a laminated, a cylindrical, and a coin can be adopted.
- the current collector and insulating plate of the component parts are not particularly limited, and may be selected according to the above shape.
- a stacked battery is preferable.
- a lead is joined to the current collector of the power generation element obtained above, and the positive electrode lead or the negative electrode lead is joined to the positive electrode tab or the negative electrode tab.
- a power generation element is placed in a laminate sheet so that the positive electrode tab and the negative electrode tab are exposed to the outside of the battery, and an electrolytic solution is injected with a liquid injector and then sealed in a vacuum to produce a stacked battery. sell.
- the initial charge treatment, gas removal treatment and activation treatment are further performed under the following conditions.
- it is done (see Example 1).
- the three sides of the laminate sheet (exterior material) are completely sealed in a rectangular shape by thermocompression when sealing in the production of the laminated battery of (4) so that the gas removal treatment can be performed. Stop (main sealing), and the remaining one side is temporarily sealed by thermocompression bonding.
- the remaining one side may be freely opened and closed by, for example, clip fastening, but from the viewpoint of mass production (production efficiency), it is preferable to temporarily seal the side by thermocompression bonding.
- thermocompression it is only necessary to adjust the temperature and pressure for pressure bonding.
- it can be opened by lightly applying force, and after degassing, it may be sealed again by thermocompression, or finally completely sealed by thermocompression ( Main sealing).
- the battery aging treatment is preferably performed as follows. At 25 ° C., a constant current charging method is used for 0.05 C and charging for 4 hours (SOC about 20%), and the state is maintained for about 1 day. Next, after charging to 4.45 V at a 0.1 C rate at 25 ° C., the charging is stopped, and the state (SOC about 70%) is maintained for about 1 day, and then discharged to 2.0 V at 0.1 C. After being left in that state for 1 hour, it is discharged to 2.0 V at 0.05C.
- thermocompression bonding Next, the following process is performed as the first (first) gas removal process. First, one side temporarily sealed by thermocompression bonding is opened, gas is removed at 10 ⁇ 3 hPa for 5 minutes, and then thermocompression bonding is performed again to perform temporary sealing. Further, pressurization with a roller (surface pressure 0.5 ⁇ 0.1 MPa) is performed, and the electrode and the separator are sufficiently adhered.
- activation process when using a solid solution positive electrode active material, as an activation treatment method, for example, the following electrochemical pretreatment method is performed.
- the constant current charging method is used as the activation processing method, and the electrochemical pretreatment method when the voltage is set as the termination condition is described as an example, but the charging method is a constant current constant voltage charging method. You may use. Further, as the termination condition, a charge amount or time may be used in addition to the voltage.
- thermocompression bonding Next, the following process is performed as the first (first) gas removal process. First, one side temporarily sealed by thermocompression bonding is opened, gas is removed at 10 ⁇ 3 hPa for 5 minutes, and then thermocompression bonding is performed again to perform main sealing. Further, pressurization with a roller (surface pressure 0.5 ⁇ 0.1 MPa) is performed, and the electrode and the separator are sufficiently adhered.
- the performance and durability of the obtained battery can be improved by performing the initial charging process, the gas removal process, and the activation process described above.
- the assembled battery is configured by connecting a plurality of batteries. Specifically, at least two or more are used, and are configured by serialization, parallelization, or both. Capacitance and voltage can be freely adjusted by paralleling in series.
- a small assembled battery that can be attached and detached by connecting a plurality of batteries in series or in parallel. Then, a plurality of small assembled batteries that can be attached and detached are connected in series or in parallel to provide a large capacity and large capacity suitable for vehicle drive power supplies and auxiliary power supplies that require high volume energy density and high volume output density.
- An assembled battery having an output can also be formed. How many batteries are connected to make an assembled battery, and how many small assembled batteries are stacked to make a large-capacity assembled battery depends on the battery capacity of the mounted vehicle (electric vehicle) It may be determined according to the output.
- the electric device of the present invention including the lithium ion secondary battery according to the present embodiment maintains a discharge capacity even when used for a long time, and has good cycle characteristics. Furthermore, the volume energy density is high. Vehicle applications such as electric vehicles, hybrid electric vehicles, fuel cell vehicles, and hybrid fuel cell vehicles require higher capacity, larger size, and longer life than electric and portable electronic devices. . Therefore, the lithium ion secondary battery (electric device) can be suitably used as a vehicle power source, for example, as a vehicle driving power source or an auxiliary power source.
- a battery or an assembled battery formed by combining a plurality of these batteries can be mounted on the vehicle.
- a plug-in hybrid electric vehicle having a long EV mileage or an electric vehicle having a long charge mileage can be formed by mounting such a battery.
- a car a hybrid car, a fuel cell car, an electric car (four-wheeled vehicles (passenger cars, trucks, buses, commercial vehicles, light cars, etc.) This is because it can be used for motorcycles (including motorcycles) and tricycles) to provide a long-life and highly reliable automobile.
- the application is not limited to automobiles.
- it can be applied to various power sources for moving vehicles such as other vehicles, for example, trains, and power sources for mounting such as uninterruptible power supplies. It is also possible to use as.
- the dried powder was pulverized in a mortar and then calcined at 500 ° C. for 5 hours.
- Lithium hydroxide monohydrate (molecular weight 41.96 g / mol) 10.67 g was mixed with the calcined powder and pulverized and mixed for 30 minutes.
- This powder was calcined at 500 ° C. for 2 hours and then calcined at 900 ° C. for 12 hours to obtain a solid solution positive electrode active material C1.
- composition of the solid solution positive electrode active material C1 thus obtained was as follows.
- composition C1 Li 1.5 [Ni 0.45 Mn 0.85 [Li] 0.20 ] O 3
- composition of slurry for positive electrode had the following composition.
- Positive electrode active material solid solution positive electrode active material C1 obtained above 9.4 parts by weight
- Conductive aid flake graphite 0.15 parts by weight
- Acetylene black 0.15 parts by weight
- Binder polyvinylidene fluoride (PVDF) 0.3 Part by weight
- Solvent 8.2 parts by weight of N-methyl-2-pyrrolidone (NMP).
- a positive electrode slurry having the above composition was prepared as follows. First, 4.0 parts by weight of a solvent (NMP) is added to 2.0 parts by weight of a 20% binder solution obtained by dissolving a binder in a solvent (NMP) into a 50 ml disposable cup, and a stirring defoaming machine (spinning revolving mixer: Awatori) A binder diluted solution was prepared by stirring for 1 minute with Rentaro AR-100).
- NMP solvent
- NMP spinning revolving mixer
- the positive electrode slurry was applied to one side of an aluminum current collector with a thickness of 20 ⁇ m using an automatic coating apparatus (Doctor blade manufactured by Tester Sangyo: PI-1210 automatic coating apparatus). Subsequently, the current collector coated with the positive electrode slurry was dried on a hot plate (100 ° C. to 110 ° C., drying time 30 minutes), and the amount of NMP remaining in the positive electrode active material layer was 0.02 wt%.
- a sheet-like positive electrode was formed as follows.
- the sheet-like positive electrode was compression-molded by applying a roller press and cut to produce a positive electrode having a density of 2.65 g / cm 3 .
- composition of slurry for negative electrode had the following composition.
- a negative electrode slurry having the above composition was prepared as follows. First, 5 parts by weight of a solvent (NMP) was added to 1.75 parts by weight of a 20% binder solution obtained by dissolving a binder in a solvent (NMP), and the mixture was stirred for 1 minute with a stirring deaerator to prepare a binder diluted solution. To this binder diluted solution, 0.2 parts by weight of conductive auxiliary agent, 9.45 parts by weight of negative electrode active material powder, and 3.6 parts by weight of solvent (NMP) are added, and the mixture is stirred for 3 minutes with a stirring defoaming machine. A slurry (solid content concentration 50 wt%) was obtained.
- NMP solvent
- the negative electrode slurry was applied to one side of a 10 ⁇ m thick electrolytic copper current collector using an automatic coating apparatus. Subsequently, the current collector coated with the negative electrode slurry was dried on a hot plate (100 ° C. to 110 ° C., drying time 30 minutes), and the amount of NMP remaining in the negative electrode active material layer was 0.02 wt% or less. A sheet-like negative electrode was formed.
- the obtained sheet-like negative electrode was compression-molded by a roller press and cut to prepare a negative electrode having a weight of about 8.54 mg / cm 2 and a density of 1.45 g / cm 3 of the negative electrode active material layer on one side. When the surface of this negative electrode was observed, no cracks were observed.
- the positive electrode C1 obtained above was cut out so as to have an active material layer area of 2.5 cm in length and 2.0 cm in width, and the two current collectors faced each other, so that the uncoated surface (aluminum current collector)
- the current collector portion was spot welded together with the surface not coated with the foil slurry.
- an aluminum positive electrode tab positive electrode current collector plate
- the negative electrode A1 obtained above was cut out so as to have an active material layer area of 2.7 cm in length and 2.2 cm in width, and then a negative electrode tab of electrolytic copper was further welded to the current collector portion to form a negative electrode A11.
- the negative electrode A11 has a structure in which a negative electrode active material layer is formed on one surface of a current collector.
- a porous polypropylene separator (S) (length 3.0 cm ⁇ width 2.5 cm, thickness 25 ⁇ m, porosity 55%) is sandwiched between the negative electrode A11 to which these tabs are welded and the positive electrode C11.
- a laminated power generation element was produced.
- the structure of the stacked type power generation element is the structure of negative electrode (single side) / separator / positive electrode (both sides) / separator / negative electrode (single side), that is, A11- (S) -C11- (S) -A11. The configuration.
- both sides of the power generation element were sandwiched with an aluminum laminate film exterior material (length 3.5 cm ⁇ width 3.5 cm), and the above power generation element was accommodated by thermocompression sealing at three sides.
- the electrolyte After injecting 0.8 cm 3 of the electrolyte into this power generation element (in the case of the above five-layer configuration, the two-cell configuration is used and the amount of liquid injection per cell is 0.4 cm 3 ), the remaining one side is temporarily bonded by thermocompression bonding. Sealed to produce a laminate type battery. In order to sufficiently infiltrate the electrolyte into the electrode pores, the electrolyte was held at 25 ° C. for 24 hours while being pressurized at a surface pressure of 0.5 Mpa.
- LiPF 6 electrolyte
- EC ethylene carbonate
- DEC diethyl carbonate
- fluoro lithium phosphate those lithium difluorophosphate the (LiPO 2 F 2) was dissolved at a concentration of 1.8 wt% was used as the electrolyte.
- Comparative Example 2 A battery was prepared in the same manner as in Comparative Example 1 except that the active material layer area of the negative electrode A1 was 2.65 cm long and 2.2 cm wide.
- Example 1 A battery was fabricated in the same manner as Comparative Example 1 described above except that the active material layer area of the negative electrode A1 was 2.6 cm long and 2.1 cm wide.
- Example 2 A battery was fabricated in the same manner as Comparative Example 1 described above except that the active material layer area of the negative electrode A1 was 2.55 cm long and 2.05 cm wide.
- Example 3 A battery was fabricated in the same manner as Comparative Example 1 described above except that the area of the active material layer of the negative electrode A1 was 2.525 cm long and 2.02 cm wide.
- Comparative Example 3 A battery was fabricated in the same manner as in Comparative Example 1 except that the active material layer area of the negative electrode A1 was 2.5 cm long and 2.0 cm wide.
- Comparative Example 4 A battery was fabricated in the same manner as Comparative Example 1 described above, except that Si 42 Ti 7 Sn 51 , which is a Si-containing alloy, was used instead of SiO x as the Si material used for the preparation of the negative electrode slurry.
- the negative electrode created in this comparative example be negative electrode A2.
- the Si-containing alloy was produced by a mechanical alloy method. Specifically, using a planetary ball mill device P-6 manufactured by Fricht, Germany, zirconia pulverized balls and alloy raw material powders were charged into a zirconia pulverized pot and alloyed at 600 rpm for 48 hours.
- Si-containing alloy prepared in the above Si 42 Ti 7 Sn 51
- other alloys that may be used in the present invention Si x Ti y Ge z A a, Si x Ti y Zn z A a, and Si of x Ti y Sn z a, Si 42 Ti 7 Sn 51 except one
- Si x Ti y Ge z A a, Si x Ti y Zn z A a, and Si of x Ti y Sn z a, Si 42 Ti 7 Sn 51 except one also, since those having the same characteristics as Si 42 Ti 7 Sn 51, experiments with Si 42 Ti 7 Sn 51 Results that are identical or similar to the examples are obtained.
- Example 4 A battery was fabricated in the same manner as Comparative Example 4 described above except that the area of the active material layer of the negative electrode A2 was 2.55 cm long and 2.05 cm wide.
- Comparative Example 5 A battery was obtained in the same manner as in Comparative Example 4 except that Si 34 Sn 21 C 45 was used instead of Si 42 Ti 7 Sn 51 as the Si material (Si-containing alloy) used for the preparation of the slurry for the negative electrode. Was made. Here, let the negative electrode created in this comparative example be negative electrode A3.
- Si 34 Sn 21 C 45 a Si-containing alloy prepared above, other alloys that may be used in the present invention (Si x Sn y Al z A a, Si x Sn y V z A a, and Si of x Sn y C z a, Si 34 Sn 21 C 45 other things) also, since those having the same characteristics as Si 34 Sn 21 C 45, experiments with Si 34 Sn 21 C 45 Results that are identical or similar to the examples are obtained.
- Example 5 A battery was fabricated in the same manner as Comparative Example 5 described above except that the area of the active material layer of the negative electrode A3 was 2.55 cm long and 2.05 cm wide.
- Comparative Example 6 A battery was fabricated in the same manner as Comparative Example 1 described above except that LiCoO 2 was used in place of the solid solution positive electrode active material C1 as the positive electrode active material used for the preparation of the positive electrode slurry.
- the positive electrode prepared in this comparative example is defined as a positive electrode C2.
- Example 6 A battery was fabricated in the same manner as in Comparative Example 6 described above except that the area of the active material layer of the negative electrode A1 was 2.6 cm long and 2.1 cm wide.
- Example 7 A battery was fabricated in the same manner as Comparative Example 6 described above except that the area of the active material layer of the negative electrode A1 was 2.55 cm long and 2.05 cm wide.
- Example 8 A battery was fabricated in the same manner as in Comparative Example 6 described above except that the area of the active material layer of the negative electrode A1 was 2.525 cm long and 2.02 cm wide.
- Comparative Example 7 A battery was fabricated in the same manner as Comparative Example 6 described above except that the area of the active material layer of the negative electrode A1 was 2.5 cm long and 2.0 cm wide.
- the power generation element of each battery obtained above was set on an evaluation cell mounting jig, and a positive electrode lead and a negative electrode lead were attached to each tab end of the power generation element, and a test was performed.
- the battery aging treatment was performed as follows. At 25 ° C., 0.05 C charging for 4 hours (SOC about 20%) was performed by a constant current charging method, and the state was maintained for about 1 day. Subsequently, after charging to 4.45V at a 0.1C rate at 25 ° C., the charging was stopped, and the state (SOC about 70%) was maintained for about 1 day, and then discharged to 2.0V at 0.1C. After being left in that state for 1 hour, it was discharged to 2.0 V at 0.05C.
- thermocompression bonding One side temporarily sealed by thermocompression bonding was opened, gas was removed at 10 ⁇ 3 hPa for 5 minutes, and then thermocompression bonding was performed again to perform temporary sealing. Furthermore, pressure (surface pressure 0.5 ⁇ 0.1 MPa) was shaped with a roller, and the electrode and the separator were sufficiently adhered.
- thermocompression bonding One side temporarily sealed by thermocompression bonding was opened, gas was removed at 10 ⁇ 3 hPa for 5 minutes, and then thermocompression bonding was performed again to perform main sealing. Furthermore, pressure (surface pressure 0.5 ⁇ 0.1 MPa) was shaped with a roller, and the electrode and the separator were sufficiently adhered.
- Capacity maintenance rate (%) 100th cycle discharge capacity / first cycle discharge capacity ⁇ 100 [Evaluation of battery characteristics 2]
- the batteries of Comparative Examples 6 to 7 and Examples 6 to 8 using the positive electrode C2 containing LiCoO 2 as the positive electrode active material were subjected to initial charge treatment and activation treatment under the following conditions, Battery capacity confirmation and cycle durability evaluation were performed.
- thermocompression bonding One side temporarily sealed by thermocompression bonding was opened, gas was removed at 10 ⁇ 3 hPa for 5 minutes, and then thermocompression bonding was performed again to perform temporary sealing. Furthermore, pressure (surface pressure 0.5 ⁇ 0.1 MPa) was shaped with a roller, and the electrode and the separator were sufficiently adhered.
- thermocompression bonding One side temporarily sealed by thermocompression bonding was opened, gas was removed at 10 ⁇ 3 hPa for 5 minutes, and then thermocompression bonding was performed again to perform main sealing. Furthermore, pressure (surface pressure 0.5 ⁇ 0.1 MPa) was shaped with a roller, and the electrode and the separator were sufficiently adhered.
- Capacity retention rate (%) 100th cycle discharge capacity / 1st cycle discharge capacity ⁇ 100
- the lithium ion secondary batteries of Examples 1 to 9 which are electrical devices according to the present invention, have excellent cycle durability (100th cycle) as compared with Comparative Examples 1 to 6. Capacity retention rate).
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Inorganic Chemistry (AREA)
- Manufacturing & Machinery (AREA)
- Composite Materials (AREA)
- Battery Electrode And Active Subsutance (AREA)
- Secondary Cells (AREA)
Abstract
Description

図1は、本発明の電気デバイスの代表的な一実施形態である、扁平型(積層型)のリチウムイオン二次電池(以下、単に「積層型電池」ともいう)の全体構造を模式的に表した断面概略図である。
活物質層(13、15)は活物質を含み、必要に応じてその他の添加剤をさらに含む。
正極活物質層13は、正極活物質を含む。正極活物質の種類について特に制限はなく、従来公知の知見が適宜参照されうる。また、場合によっては、二種以上の正極活物質を併用してもよい。正極活物質としては、高い電気容量およびエネルギー密度を達成可能な正極活物質として、固溶体材料からなる正極活物質(本明細書中、「固溶体正極活物質」とも称する)を含むことが好ましい。また、その他の正極活物質として、LiCoO2などのLi・Co系複合酸化物、LiNiO2などのLi・Ni系複合酸化物、スピネルLiMn2O4などのLi・Mn系複合酸化物、LiFeO2などのLi・Fe系複合酸化物などを用いることができる。この他、LiFePO4などの遷移金属とリチウムのリン酸化合物や硫酸化合物、V2O5、MnO2、TiS2、MoS2、MoO3などの遷移金属酸化物や硫化物、PbO2、AgO、NiOOHなどを用いることができる。以下では、好ましい正極活物質である固溶体正極活物質について、より詳細に説明する。
固溶体正極活物質は、下記式(3)で表される組成を基本構造として有する。
形態(B)において、固溶体正極活物質の粒子表面に存在する金属酸化物の具体的な構成は特に制限されず、上述した金属元素を含む理論上可能な酸化物または複合酸化物のいずれも用いられうる。好ましくは、Al2O3、ZrO2またはTiO2が用いられる。なお、Nb、Sn、W、MoおよびVからなる群から選択される1種または2種以上のような他の元素を含む(複合)酸化物が被覆層にさらに含まれていてもよい。
正極活物質層に用いられるバインダとしては、特に限定されないが、例えば、以下の材料が挙げられる。ポリエチレン、ポリプロピレン、ポリエチレンテレフタレート(PET)、ポリエーテルニトリル、ポリアクリロニトリル、ポリイミド、ポリアミド、セルロース、カルボキシメチルセルロース(CMC)およびその塩、エチレン-酢酸ビニル共重合体、ポリ塩化ビニル、スチレン・ブタジエンゴム(SBR)、イソプレンゴム、ブタジエンゴム、エチレン・プロピレンゴム、エチレン・プロピレン・ジエン共重合体、スチレン・ブタジエン・スチレンブロック共重合体およびその水素添加物、スチレン・イソプレン・スチレンブロック共重合体およびその水素添加物などの熱可塑性高分子、ポリフッ化ビニリデン(PVdF)、ポリテトラフルオロエチレン(PTFE)、テトラフルオロエチレン・ヘキサフルオロプロピレン共重合体(FEP)、テトラフルオロエチレン・パーフルオロアルキルビニルエーテル共重合体(PFA)、エチレン・テトラフルオロエチレン共重合体(ETFE)、ポリクロロトリフルオロエチレン(PCTFE)、エチレン・クロロトリフルオロエチレン共重合体(ECTFE)、ポリフッ化ビニル(PVF)等のフッ素樹脂、ビニリデンフルオライド-ヘキサフルオロプロピレン系フッ素ゴム(VDF-HFP系フッ素ゴム)、ビニリデンフルオライド-ヘキサフルオロプロピレン-テトラフルオロエチレン系フッ素ゴム(VDF-HFP-TFE系フッ素ゴム)、ビニリデンフルオライド-ペンタフルオロプロピレン系フッ素ゴム(VDF-PFP系フッ素ゴム)、ビニリデンフルオライド-ペンタフルオロプロピレン-テトラフルオロエチレン系フッ素ゴム(VDF-PFP-TFE系フッ素ゴム)、ビニリデンフルオライド-パーフルオロメチルビニルエーテル-テトラフルオロエチレン系フッ素ゴム(VDF-PFMVE-TFE系フッ素ゴム)、ビニリデンフルオライド-クロロトリフルオロエチレン系フッ素ゴム(VDF-CTFE系フッ素ゴム)等のビニリデンフルオライド系フッ素ゴム、エポキシ樹脂等が挙げられる。これらのバインダは、単独で用いてもよいし、2種以上を併用してもよい。
導電助剤とは、正極活物質層または負極活物質層の導電性を向上させるために配合される添加物をいう。導電助剤としては、ケッチェンブラック、アセチレンブラック等のカーボンブラックが挙げられる。活物質層が導電助剤を含むと、活物質層の内部における電子ネットワークが効果的に形成され、電池の出力特性の向上に寄与しうる。
電解質塩(リチウム塩)としては、Li(C2F5SO2)2N、LiPF6、LiBF4、LiClO4、LiAsF6、LiCF3SO3等が挙げられる。
負極活物質層15は、ケイ素含有負極活物質を含む。ケイ素含有負極活物質の種類について特に制限はなく、従来公知の知見が適宜参照されうる。また、場合によっては、二種以上の負極活物質を併用してもよい。ケイ素含有負極活物質としては、高い電気容量およびエネルギー密度を達成可能な負極活物質として、SiOxまたはSi含有合金(これらをまとめて「Si材料」とも称する)を必須に含むことが好ましく、Si材料を炭素材料とともに含むことがより好ましい。以下では、好ましいケイ素含有負極活物質であるSi材料と、好ましく併用される炭素材料について、より詳細に説明する。
SiOxは、アモルファスSiO2粒子とSi粒子との混合体であり、xはSiの原子価を満足する酸素数を表す。xの具体的な値について特に制限はなく、適宜設定されうる。
本形態に係るSiOxの製造方法としては、特に制限されるものではなく、従来公知の各種の製造を利用して製造することができる。すなわち、作製方法によるアモルファス状態・特性の違いはほとんどないため、ありとあらゆる作製方法が適用できる。
Si含有合金は、Siを含有する他の金属との合金であれば特に制限されず、従来公知の知見が適宜参照されうる。ここでは、Si含有合金の好ましい実施形態として、SixTiyGezAa、SixTiyZnzAa、SixTiySnzAa、SixSnyAlzAa、SixSnyVzAa、SixSnyCzAa、SixZnyVzAa、SixZnySnzAa、SixZnyAlzAa、SixZnyCzAa、SixAlyCzAaおよびSixAlyNbzAa(式中、Aは、不可避不純物である。さらに、x、y、z、およびaは、重量%の値を表し、0<x<100、0<y<100、0<z<100、および0≦a<0.5であり、x+y+z+a=100である)が挙げられる。これらのSi含有合金を負極活物質として用いることで、所定の第1添加元素および所定の第2添加元素を適切に選択することによって、Li合金化の際に、アモルファス-結晶の相転移を抑制してサイクル寿命を向上させることができる。また、これによって、従来の負極活物質、例えば炭素系負極活物質よりも高容量のものとなる。
本発明に用いられうる炭素材料は、特に制限されないが、天然黒鉛、人造黒鉛等の高結晶性カーボンである黒鉛(グラファイト);ソフトカーボン、ハードカーボン等の低結晶性カーボン;ケッチェンブラック、アセチレンブラック、チャンネルブラック、ランプブラック、オイルファーネスブラック、サーマルブラック等のカーボンブラック;フラーレン、カーボンナノチューブ、カーボンナノファイバー、カーボンナノホーン、カーボンフィブリル等の炭素材料が挙げられる。これらのうち、黒鉛を用いることが好ましい。
集電体(11、12)は導電性材料から構成される。集電体の大きさは、電池の使用用途に応じて決定される。例えば、高エネルギー密度が要求される大型の電池に用いられるのであれば、面積の大きな集電体が用いられる。
本実施形態に係る電気デバイスでは、上述した発電要素を構成する単電池層の少なくとも1つにおいて、負極活物質層の面積をA[m2]とし、正極活物質層の面積をC[m2]としたときに、式(1):0.91≦C/A<1を満足することが必須であり、好ましくは0.95≦C/A≦0.99である。
本実施形態において、セパレータは、電解液(液体電解質)を保持して正極と負極との間のリチウムイオン伝導性を確保する機能、および正極と負極との間の隔壁としての機能を有する。
リチウムイオン二次電池においては、電池外部に電流を取り出す目的で、集電体に電気的に接続された集電板(タブ)が外装材であるラミネートフィルムの外部に取り出されている。
シール部は、直列積層型電池に特有の部材であり、電解質層の漏れを防止する機能を有する。このほかにも、電池内で隣り合う集電体同士が接触したり、積層電極の端部の僅かな不ぞろいなどによる短絡が起こったりするのを防止することもできる。
負極および正極端子リードの材料は、公知の積層型二次電池で用いられるリードを用いることができる。なお、電池外装材から取り出された部分は、周辺機器や配線などに接触して漏電したりして製品(例えば、自動車部品、特に電子機器等)に影響を与えないように、耐熱絶縁性の熱収縮チューブなどにより被覆するのが好ましい。
外装材としては、従来公知の金属缶ケースを用いることができる。そのほか、図1に示すようなラミネートフィルム22を外装材として用いて、発電要素17をパックしてもよい。ラミネートフィルムは、例えば、ポリプロピレン、アルミニウム、ナイロンがこの順に積層されてなる3層構造として構成されうる。このようなラミネートフィルムを用いることにより、外装材の開封、容量回復材の添加、外装材の再封止を容易に行うことができる。
リチウムイオン二次電池の製造方法は特に制限されず、公知の方法により製造されうる。具体的には、(1)電極の作製、(2)単電池層の作製、(3)発電要素の作製、および(4)積層型電池の製造を含む。以下、リチウムイオン二次電池の製造方法について一例を挙げて説明するが、これに限定されるものではない。
電極(正極または負極)は、例えば、活物質スラリー(正極活物質スラリーまたは負極活物質スラリー)を調製し、当該活物質スラリーを集電体上に塗布、乾燥し、次いでプレスすることにより作製されうる。前記活物質スラリーは、上述した活物質(正極活物質または負極活物質)、バインダ、導電助剤および溶媒を含む。
単電池層は、(1)で作製した電極(正極および負極)を、電解質層を介して積層させることにより作製されうる。
発電要素は、単電池層の出力および容量、電池として必要とする出力および容量等を適宜考慮し、前記単電池層を積層して作製されうる。
電池の構成としては、角形、ペーパー型、積層型、円筒型、コイン型等、種々の形状を採用することができる。また構成部品の集電体や絶縁板等は特に限定されるものではなく、上記の形状に応じて選定すればよい。しかし、本実施形態では積層型電池が好ましい。積層型電池は、上記で得られた発電要素の集電体にリードを接合し、これらの正極リードまたは負極リードを、正極タブまたは負極タブに接合する。そして、正極タブおよび負極タブが電池外部に露出するように、発電要素をラミネートシート中に入れ、注液機により電解液を注液してから真空に封止することにより積層型電池が製造されうる。
さらに、本実施形態では、上記により得られた積層型電池の性能および耐久性を高める観点から、さらに、以下の条件で初充電処理、ガス除去処理および活性化処理を行うことが好ましい(実施例1参照)。この場合には、ガス除去処理ができるように、上記(4)の積層型電池の製造において、封止する際に、矩形形状にラミネートシート(外装材)の3辺を熱圧着により完全に封止(本封止)し、残る1辺は、熱圧着で仮封止しておく。残る1辺は、例えば、クリップ留め等により開閉自在にしてもよいが、量産化(生産効率)の観点からは、熱圧着で仮封止するのがよい。この場合には、圧着する温度、圧力を調整するだけでよいためである。熱圧着で仮封止した場合には、軽く力を加えることで開封でき、ガス抜き後、再度、熱圧着で仮封止してもよいし、最後的には熱圧着で完全に封止(本封止)すればよい。
例えば、固溶体正極活物質を用いる場合、電池のエージング処理は、以下のように実施することが好ましい。25℃にて、定電流充電法で0.05C、4時間の充電(SOC約20%)を行い、その状態で約1日間保持する。次いで、25℃にて0.1Cレートで4.45Vまで充電した後、充電を止め、その状態(SOC約70%)で約1日間保持したのち、0.1Cで2.0Vまで放電する。その状態のまま1時間放置したのち、0.05Cにて、2.0Vまで放電する。
次に、最初(1回目)のガス除去処理として、以下の処理を行う。まず、熱圧着で仮封止した1辺を開封し、10±3hPaで5分間ガス除去を行った後、再度、熱圧着を行って仮封止を行う。さらに、ローラーで加圧(面圧0.5±0.1MPa)整形し電極とセパレータとを十分に密着させる。
次に、固溶体正極活物質を用いる場合には、活性化処理法として、例えば以下の電気化学前処理法を行う。
次に、最初(1回目)のガス除去処理として、以下の処理を行う。まず、熱圧着で仮封止した一辺を開封し、10±3hPaで5分間ガス除去を行った後、再度、熱圧着を行って本封止を行う。さらに、ローラーで加圧(面圧0.5±0.1MPa)整形し電極とセパレータとを十分に密着させる。
組電池は、電池を複数個接続して構成した物である。詳しくは少なくとも2つ以上用いて、直列化あるいは並列化あるいはその両方で構成されるものである。直列、並列化することで容量および電圧を自由に調節することが可能になる。
本実施形態に係るリチウムイオン二次電池をはじめとした本発明の電気デバイスは、長期使用しても放電容量が維持され、サイクル特性が良好である。さらに、体積エネルギー密度が高い。電気自動車やハイブリッド電気自動車や燃料電池車やハイブリッド燃料電池自動車などの車両用途においては、電気・携帯電子機器用途と比較して、高容量、大型化が求められるとともに、長寿命化が必要となる。したがって、上記リチウムイオン二次電池(電気デバイス)は、車両用の電源として、例えば、車両駆動用電源や補助電源に好適に利用することができる。
(固溶体正極活物質C1の調製)
1.硫酸マンガン・1水和物(分子量223.06g/mol)28.61g、
硫酸ニッケル・6水和物(分子量262.85g/mol)17.74g、
を純水200gに加え、攪拌溶解し、混合溶液を調製した。
固溶体正極活物質C1の組成を式(3)に当てはめると、a+b+c+d=1.5、d=0.20、a+b+c=1.3、z;原子価を満足する酸素数となり、式(3)の要件を満足する。
(正極用スラリーの組成)
正極用スラリーは下記組成とした。
導電助剤: 燐片状黒鉛 0.15重量部
アセチレンブラック 0.15重量部
バインダ: ポリフッ化ビニリデン(PVDF) 0.3重量部
溶媒: N-メチル-2-ピロリドン(NMP) 8.2重量部。
上記組成の正極用スラリーを次のように調製した。まず、50mlのディスポカップに、溶媒(NMP)にバインダを溶解した20%バインダ溶液2.0重量部に溶媒(NMP)4.0重量部を加え、攪拌脱泡機(自転公転ミキサー:あわとり錬太郎AR-100)で1分間攪拌してバインダ希釈溶液を作製した。次に、このバインダ希釈液に、導電助剤0.4重量部と固溶体正極活物質C1 9.2重量部、および溶媒(NMP)2.6重量部を加え、攪拌脱泡機で3分間攪拌して正極用スラリー(固形分濃度55重量%)とした。
20μm厚のアルミニウム集電体の片面に、上記正極用スラリーを自動塗工装置(テスター産業製ドクターブレード:PI-1210自動塗工装置)により塗布した。続いて、この正極用スラリーを塗布した集電体について、ホットプレートにて乾燥(100℃~110℃、乾燥時間30分)を行い、正極活物質層に残留するNMP量を0.02重量%以下として、シート状正極を形成した。
上記シート状正極を、ローラープレスをかけて圧縮成形し、切断して、密度2.65g/cm3の正極を作製した。
次に、上記手順で作製した正極を用い真空乾燥炉にて乾燥処理を行った。乾燥炉内部に正極C1を設置した後、室温(25℃)にて減圧(100mmHg(1.33×104Pa))し乾燥炉内の空気を除去した。続いて、窒素ガスを流通(100cm3/分)しながら、10℃/分で120℃まで昇温し、120℃で再度減圧して炉内の窒素を排気したまま12時間保持した後、室温まで降温した。こうして正極表面の水分を除去した正極C1を得た。
(負極用スラリーの組成)
負極用スラリーは下記組成とした。
炭素材料(日立化成製、黒鉛) 8.45重量部
導電助剤: SuperP 0.20重量部
バインダ: ポリフッ化ビニリデン(PVDF) 0.35重量部
溶媒: N-メチル-2-ピロリドン(NMP) 10.0重量部。
上記組成の負極用スラリーを次のように調製した。まず、溶媒(NMP)にバインダを溶解した20%バインダ溶液1.75重量部に溶媒(NMP)5重量部を加えて、攪拌脱泡機1分間攪拌してバインダ希釈溶液を作製した。このバインダ希釈液に、導電助剤0.2重量部、負極活物質粉末9.45重量部、および溶媒(NMP)3.6重量部を加え、攪拌脱泡機で3分間攪拌して負極用スラリー(固形分濃度50重量%)とした。
10μm厚の電解銅集電体の片面に、上記負極用スラリーを自動塗工装置により塗布した。続いて、この負極スラリーを塗布した集電体について、ホットプレートにて乾燥(100℃~110℃、乾燥時間30分)を行い、負極活物質層に残留するNMP量を0.02重量%以下として、シート状負極を形成した。
得られたシート状負極を、ローラープレスをかけて圧縮成形し、切断して、片面の負極活物質層の重量約8.54mg/cm2、密度1.45g/cm3の負極を作製した。この負極の表面を観察したところ、クラックの発生は見られなかった。
次に、上記手順で作製した負極を用い真空乾燥炉にて乾燥処理を行った。乾燥炉内部に負極を設置した後、室温(25℃)にて減圧(100mmHg(1.33×104Pa))し乾燥炉内の空気を除去した。続いて、窒素ガスを流通(100cm3/分)しながら、10℃/分で135℃まで昇温し、135℃で再度減圧して炉内の窒素を排気したまま12時間保持した後、室温まで降温した。こうして負極表面の水分を除去して、負極A1を得た。
上記で得られた正極C1を、活物質層面積;縦2.5cm×横2.0cmになるように切り出し、これら2枚を集電体同士が向き合うようにして、未塗工面(アルミニウム集電箔のスラリーを塗工していない面)を合わせて集電体部分をスポット溶接した。これにより、外周部をスポット溶接により一体化された2枚重ねの集電箔の両面に正極活物質層を有する正極を形成した。その後、さらに集電体部分にアルミニウムの正極タブ(正極集電板)を溶接して正極C11を形成した。すなわち、正極C11は、集電箔の両面に正極活物質層が形成された構成である。
負極A1の活物質層面積;縦2.65cm×横2.2cmとしたこと以外は、上述した比較例1と同様にして、電池を作成した。
負極A1の活物質層面積;縦2.6cm×横2.1cmとしたこと以外は、上述した比較例1と同様にして、電池を作成した。
負極A1の活物質層面積;縦2.55cm×横2.05cmとしたこと以外は、上述した比較例1と同様にして、電池を作成した。
負極A1の活物質層面積;縦2.525cm×横2.02cmとしたこと以外は、上述した比較例1と同様にして、電池を作成した。
負極A1の活物質層面積;縦2.5cm×横2.0cmとしたこと以外は、上述した比較例1と同様にして、電池を作成した。
負極用スラリーの調製に用いたSi材料として、SiOxに代えてSi含有合金であるSi42Ti7Sn51を用いたこと以外は、上述した比較例1と同様にして、電池を作製した。ここで、本比較例において作成された負極を負極A2とする。なお、上記Si含有合金は、メカニカルアロイ法により製造した。具体的には、ドイツ フリッチュ社製遊星ボールミル装置P-6を用いて、ジルコニア製粉砕ポットにジルコニア製粉砕ボールおよび合金の各原料粉末を投入し、600rpmで48時間かけて合金化させた。
負極A2の活物質層面積;縦2.55cm×横2.05cmとしたこと以外は、上述した比較例4と同様にして、電池を作成した。
負極用スラリーの調製に用いたSi材料(Si含有合金)として、Si42Ti7Sn51に代えてSi34Sn21C45を用いたこと以外は、上述した比較例4と同様にして、電池を作製した。ここで、本比較例において作成された負極を負極A3とする。
負極A3の活物質層面積;縦2.55cm×横2.05cmとしたこと以外は、上述した比較例5と同様にして、電池を作成した。
正極用スラリーの調製に用いた正極活物質として、固溶体正極活物質C1に代えてLiCoO2を用いたこと以外は、上述した比較例1と同様にして、電池を作製した。ここで、本比較例において作成された正極を正極C2とする。
負極A1の活物質層面積;縦2.6cm×横2.1cmとしたこと以外は、上述した比較例6と同様にして、電池を作成した。
負極A1の活物質層面積;縦2.55cm×横2.05cmとしたこと以外は、上述した比較例6と同様にして、電池を作成した。
負極A1の活物質層面積;縦2.525cm×横2.02cmとしたこと以外は、上述した比較例6と同様にして、電池を作成した。
負極A1の活物質層面積;縦2.5cm×横2.0cmとしたこと以外は、上述した比較例6と同様にして、電池を作成した。
上記で作製したラミネート型電池について、性能を評価した。この際、固溶体正極活物質を含む正極C1を用いた比較例1~5および実施例1~5の電池に対しては、以下の条件で、初充電処理および活性化処理を行ったのち、電池容量確認およびサイクル耐久性評価を行った。
電池のエージング処理は、以下のように実施した。25℃にて、定電流充電法で0.05C、4時間の充電(SOC約20%)を行い、その状態で約1日間保持した。次いで、25℃にて0.1Cレートで4.45Vまで充電した後、充電を止め、その状態(SOC約70%)で約1日間保持したのち、0.1Cで2.0Vまで放電した。その状態のまま1時間放置したのち、0.05Cにて、2.0Vまで放電した。
熱圧着で仮封止した一辺を開封し、10±3hPaで5分間ガス除去を行った後、再度、熱圧着を行い仮封止を行った。さらに、ローラーで加圧(面圧0.5±0.1MPa)整形し電極とセパレータとを十分に密着させた。
25℃にて、定電流充電法で0.1Cで電圧が4.45Vとなるまで充電した後、その状態で1日間放置したのち、0.1Cで2.0Vまで放電したのち、1時間放置して、0.05Cで2.0Vまで放電するサイクルを1回行った。同様に、25℃にて、定電流充電法で0.1Cで4.55Vとなるまで充電した後、その状態で1日間放置したのち、0.1Cで2.0Vまで放電したのち、1時間放置して、0.05Cで2.0Vまで放電するサイクルを1回行った。同様に、0.1Cで4.65Vとなるまで充電した後、その状態で1日間放置したのち、0.1Cで2.0Vまで放電したのち、1時間放置して、0.05Cで2.0Vまで放電するサイクルを1回行った。更に、25℃にて、定電流充電法で、0.1Cで4.75Vとなるまで充電した後した後、その状態で1日間放置したのち、0.1Cで2.0Vまで放電したのち、1時間放置して、0.05Cで2.0Vまで放電するサイクルを1回行った。
熱圧着で仮封止した一辺を開封し、10±3hPaで5分間ガス除去を行った後、再度、熱圧着を行い本封止を行った。さらに、ローラーで加圧(面圧0.5±0.1MPa)整形し電極とセパレータとを十分に密着させた。
各電池について充放電試験を行い、電池容量について調査した。すなわち、30℃の雰囲気下、定電流定電圧充電方式にて、電流密度0.1C相当、上限電圧4.45Vとして充電し、1分間休止させた後、定電流放電方式にて電流密度0.1C相当にて2Vまで放電した。この充放電サイクルを3回繰り返し、3サイクル目の放電容量を電池容量と規定した。結果を下記の表1に示す。なお、表1に示す「電池容量」の値は、比較例1~3および実施例1~3については比較例3の電池容量を100%としたときの相対値であり、比較例4および実施例4については実施例4の電池容量を100%としたときの相対値であり、比較例5および実施例5については実施例5の電池容量を100%としたときの相対値である。
各電池について充放電サイクル試験を行い、放電容量保持率について調査した。すなわち、30℃の雰囲気下、定電流定電圧充電方式にて、電流密度1C相当、上限電圧4.45Vとして充電し、1分間休止させた後、定電流放電方式にて電流密度1C相当にて2Vまで放電した。この充放電サイクルを100回繰り返した。そして、1サイクル目の放電容量に対する100サイクル目の放電容量の割合を「容量維持率(%)」として評価した。結果を下記の表1に示す。
[電池特性の評価2]
一方、正極活物質としてLiCoO2を含む正極C2を用いた比較例6~7および実施例6~8の電池に対しては、以下の条件で、初充電処理および活性化処理を行ったのち、電池容量確認およびサイクル耐久性評価を行った。
電池のエージング処理は、以下のように実施した。25℃にて、定電流充電法で0.05C、4時間の充電(SOC約20%)を行い、その状態で約1日間保持した。次いで、25℃にて0.1Cレートで4.2Vまで充電した後、充電を止め、その状態(SOC約70%)で約1日間保持したのち、0.1Cで2.5Vまで放電した。その状態のまま1時間放置したのち、0.05Cにて、2.5Vまで放電した。
熱圧着で仮封止した一辺を開封し、10±3hPaで5分間ガス除去を行った後、再度、熱圧着を行い仮封止を行った。さらに、ローラーで加圧(面圧0.5±0.1MPa)整形し電極とセパレータとを十分に密着させた。
25℃にて、定電流充電法で0.1Cで電圧が4.2Vとなるまで充電した後、その状態で1日間放置したのち、0.1Cで2.5Vまで放電したのち、1時間放置して、0.05Cで2.5Vまで放電するサイクルを1回行った。
熱圧着で仮封止した一辺を開封し、10±3hPaで5分間ガス除去を行った後、再度、熱圧着を行い本封止を行った。さらに、ローラーで加圧(面圧0.5±0.1MPa)整形し電極とセパレータとを十分に密着させた。
各電池について充放電試験を行い、電池容量について調査した。すなわち、30℃の雰囲気下、定電流定電圧充電方式にて、電流密度0.1C相当、上限電圧4.2Vとして充電し、1分間休止させた後、定電流放電方式にて電流密度0.1C相当にて2.5Vまで放電した。この充放電サイクルを3回繰り返し、3サイクル目の放電容量を電池容量と規定した。結果を下記の表1に示す。なお、表1に示す「電池容量」の値は、比較例6~7および実施例6~8については比較例7の電池容量を100%としたときの相対値である。
各電池について充放電サイクル試験を行い、放電容量保持率について調査した。すなわち、30℃の雰囲気下、定電流定電圧充電方式にて、電流密度1C相当、上限電圧4.2Vとして充電し、1分間休止させた後、定電流放電方式にて電流密度1C相当にて2.5Vまで放電した。この充放電サイクルを100回繰り返した。そして、1サイクル目の放電容量に対する100サイクル目の放電容量の割合を「容量維持率(%)」として評価した。結果を下記の表1に示す。
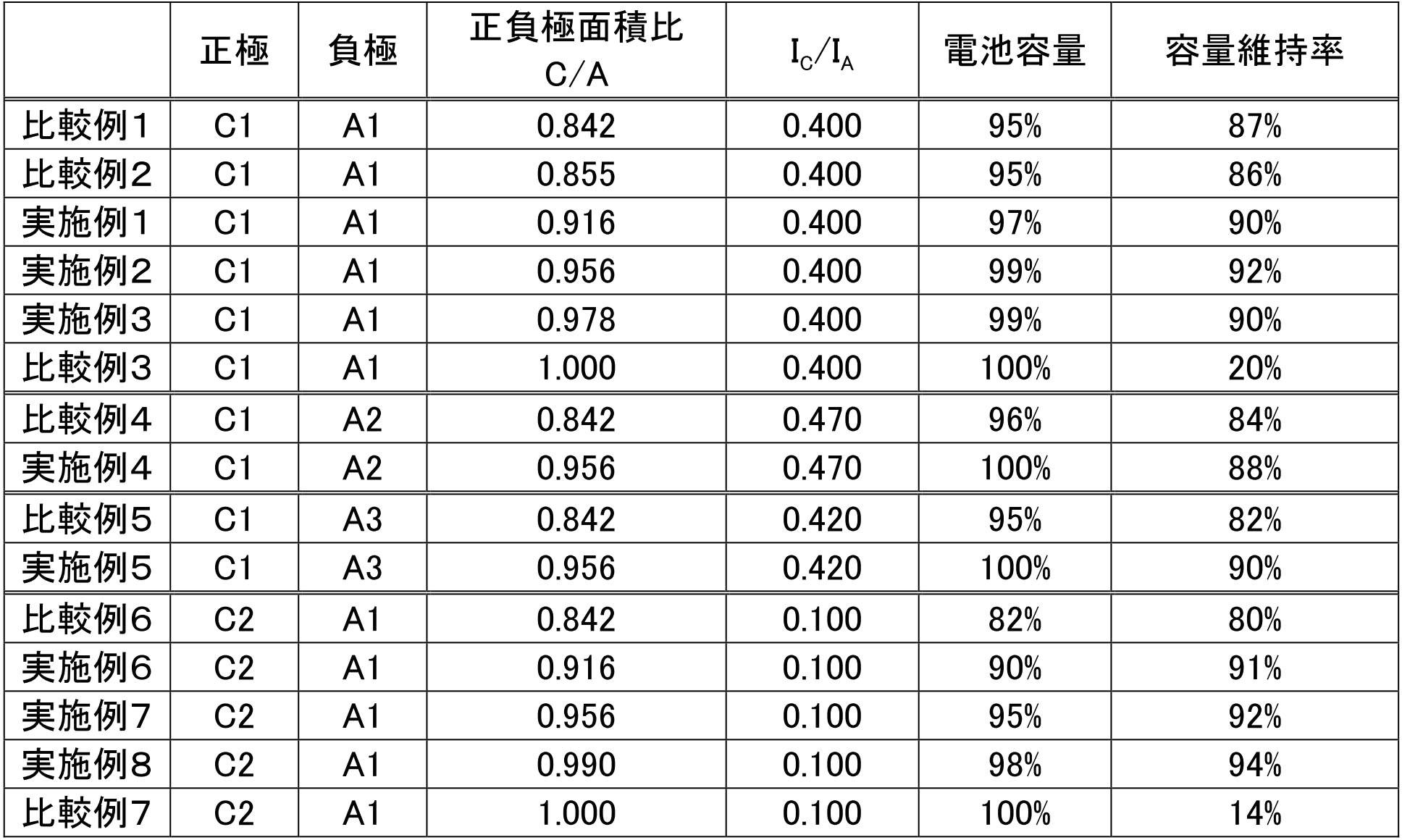
11 負極集電体、
12 正極集電体、
13 負極活物質層、
15 正極活物質層、
17 セパレータ、
19 単電池層、
21、57 発電要素、
25 負極集電板、
27 正極集電板、
29、52 電池外装材、
58 正極タブ、
59 負極タブ。
Claims (6)
- 正極集電体の表面に正極活物質を含む正極活物質層が形成されてなる正極と、
負極集電体の表面にケイ素含有負極活物質を含む負極活物質層が形成されてなる負極と、
セパレータと、
を含む単電池層を含む発電要素を有する電気デバイスであって、
前記発電要素を構成する前記単電池層の少なくとも1つにおいて、
前記負極活物質層の面積をA[m2]とし、前記正極活物質層の面積をC[m2]としたときに、式(1):0.91≦C/A<1を満足する、電気デバイス。 - 前記式(1)を満足する前記単電池層において、
前記負極活物質層の単位面積当たりの不可逆容量をIAとし、前記正極活物質層の単位面積当たりの不可逆容量をICとしたときに、IC/IA≧0.40であり、かつ、0.91≦C/A<0.99である、請求項1に記載の電気デバイス。 - 前記式(1)を満足する前記単電池層において、
前記正極活物質層が、下記式(2):

で表される正極活物質を含有し、この際、前記固溶体正極活物質は、下記式(3):

で表される組成を基本構造として有する、請求項1または2に記載の電気デバイス。 - 前記式(1)を満足する前記単電池層において、
前記負極活物質層が、下記式(4):

で表される負極活物質を含有する、請求項1~3のいずれか1項に記載の電気デバイス。 - 前記Si含有合金が、SixTiyGezAa、SixTiyZnzAa、SixTiySnzAa、SixSnyAlzAa、SixSnyVzAa、SixSnyCzAa、SixZnyVzAa、SixZnySnzAa、SixZnyAlzAa、SixZnyCzAa、SixAlyCzAaおよびSixAlyNbzAa(式中、Aは、不可避不純物である。さらに、x、y、z、およびaは、重量%の値を表し、0<x<100、0<y<100、0<z<100、および0≦a<0.5であり、x+y+z+a=100である)からなる群から選択される1種または2種以上である、請求項4に記載の電気デバイス。
- リチウムイオン二次電池である、請求項1~5のいずれか1項に記載の電気デバイス。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020167019128A KR20160100348A (ko) | 2014-01-24 | 2014-01-24 | 전기 디바이스 |
| JP2015558664A JP6187602B2 (ja) | 2014-01-24 | 2014-01-24 | 電気デバイス |
| US15/113,250 US10476101B2 (en) | 2014-01-24 | 2014-01-24 | Electrical device |
| EP14879986.9A EP3098892B1 (en) | 2014-01-24 | 2014-01-24 | Electrical device |
| CN201480073896.4A CN105934847B (zh) | 2014-01-24 | 2014-01-24 | 电器件 |
| PCT/JP2014/051526 WO2015111187A1 (ja) | 2014-01-24 | 2014-01-24 | 電気デバイス |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2014/051526 WO2015111187A1 (ja) | 2014-01-24 | 2014-01-24 | 電気デバイス |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015111187A1 true WO2015111187A1 (ja) | 2015-07-30 |
Family
ID=53681016
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/051526 WO2015111187A1 (ja) | 2014-01-24 | 2014-01-24 | 電気デバイス |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10476101B2 (ja) |
| EP (1) | EP3098892B1 (ja) |
| JP (1) | JP6187602B2 (ja) |
| KR (1) | KR20160100348A (ja) |
| CN (1) | CN105934847B (ja) |
| WO (1) | WO2015111187A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017041407A (ja) * | 2015-08-21 | 2017-02-23 | 日産自動車株式会社 | リチウムイオン二次電池 |
| WO2018003843A1 (ja) * | 2016-06-30 | 2018-01-04 | 三洋電機株式会社 | 二次電池及びその製造方法 |
| JP2018055901A (ja) * | 2016-09-28 | 2018-04-05 | 日産自動車株式会社 | 非水電解質二次電池の製造方法 |
| CN109075377A (zh) * | 2016-04-28 | 2018-12-21 | 日产自动车株式会社 | 非水电解质二次电池 |
| EP3451430A4 (en) * | 2016-04-28 | 2019-04-03 | Nissan Motor Co., Ltd. | NONAQUEOUS ELECTROLYTE SECONDARY BATTERY |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20160102026A (ko) | 2014-01-24 | 2016-08-26 | 닛산 지도우샤 가부시키가이샤 | 전기 디바이스 |
| JP6187602B2 (ja) | 2014-01-24 | 2017-08-30 | 日産自動車株式会社 | 電気デバイス |
| US20170110725A1 (en) * | 2014-04-03 | 2017-04-20 | Sony Corporation | Secondary battery, battery pack, electronic device, electrically driven vehicle, storage device, and power system |
| JP7169763B2 (ja) * | 2018-04-09 | 2022-11-11 | 日産自動車株式会社 | 非水電解質二次電池 |
| DE102018003328B4 (de) * | 2018-04-24 | 2020-12-17 | Envites Energy Gesellschaft für Umwelttechnik und Energiesysteme mbH | Verfahren zur Herstellung einer Batteriezelle, vorzugsweise einer Li-lonen-Bi-Stapelzelle mit Feststoff |
| JP6846490B1 (ja) * | 2019-09-25 | 2021-03-24 | 積水化学工業株式会社 | 蓄電素子及び蓄電素子の製造方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005011699A (ja) * | 2003-06-19 | 2005-01-13 | Canon Inc | リチウム二次電池 |
| JP2008004535A (ja) * | 2006-05-23 | 2008-01-10 | Sony Corp | 負極および電池 |
| JP2009517850A (ja) | 2005-12-01 | 2009-04-30 | スリーエム イノベイティブ プロパティズ カンパニー | ケイ素含有量が高いアモルファス合金に基づく電極組成物 |
| JP2009099328A (ja) * | 2007-10-15 | 2009-05-07 | Sony Corp | 負極および電池 |
| JP2009158415A (ja) * | 2007-12-27 | 2009-07-16 | Mitsui Mining & Smelting Co Ltd | 非水電解液二次電池用正極活物質及びそれを有する非水電解液二次電池 |
| JP2011060471A (ja) * | 2009-09-08 | 2011-03-24 | Nec Energy Devices Ltd | 非水系電解質二次電池 |
| JP2012142154A (ja) * | 2010-12-28 | 2012-07-26 | Sony Corp | リチウムイオン二次電池、電動工具、電動車両および電力貯蔵システム |
| WO2013094465A1 (ja) * | 2011-12-19 | 2013-06-27 | 日立マクセル株式会社 | リチウム二次電池 |
Family Cites Families (130)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4560524A (en) | 1983-04-15 | 1985-12-24 | Smuckler Jack H | Method of manufacturing a positive temperature coefficient resistive heating element |
| JPS62134243A (ja) | 1985-12-06 | 1987-06-17 | Mitsui Toatsu Chem Inc | アイソタクテイクポリスチレン成形体 |
| JPH08250117A (ja) | 1995-03-09 | 1996-09-27 | Shin Kobe Electric Mach Co Ltd | リチウム二次電池負極用炭素材料及びその製造方法 |
| JP4029235B2 (ja) | 1998-10-02 | 2008-01-09 | 大阪瓦斯株式会社 | リチウム二次電池用負極 |
| JP2000299108A (ja) | 1999-04-14 | 2000-10-24 | Sony Corp | 非水電解質電池 |
| CA2305837C (en) | 1999-04-14 | 2011-05-31 | Sony Corporation | Material for negative electrode and nonaqueous-electrolyte battery incorporating the same |
| US6685804B1 (en) | 1999-10-22 | 2004-02-03 | Sanyo Electric Co., Ltd. | Method for fabricating electrode for rechargeable lithium battery |
| JP2002083594A (ja) | 1999-10-22 | 2002-03-22 | Sanyo Electric Co Ltd | リチウム電池用電極並びにこれを用いたリチウム電池及びリチウム二次電池 |
| WO2001031723A1 (fr) | 1999-10-22 | 2001-05-03 | Sanyo Electric Co., Ltd. | Electrode pour accumulateur au lithium et accumulateur au lithium |
| WO2001029918A1 (fr) | 1999-10-22 | 2001-04-26 | Sanyo Electric Co., Ltd | Electrode pour accumulateur au lithium et accumulateur au lithium |
| JP3733071B2 (ja) | 1999-10-22 | 2006-01-11 | 三洋電機株式会社 | リチウム電池用電極及びリチウム二次電池 |
| CN1260841C (zh) | 1999-10-22 | 2006-06-21 | 三洋电机株式会社 | 锂电池和可再充电锂电池中用的电极 |
| JP2001196052A (ja) | 2000-01-12 | 2001-07-19 | Sony Corp | 負極及び非水電解質電池 |
| US6699336B2 (en) | 2000-01-13 | 2004-03-02 | 3M Innovative Properties Company | Amorphous electrode compositions |
| US6680143B2 (en) | 2000-06-22 | 2004-01-20 | The University Of Chicago | Lithium metal oxide electrodes for lithium cells and batteries |
| JP3848065B2 (ja) | 2000-08-08 | 2006-11-22 | キヤノン株式会社 | 画像形成装置 |
| CN1278438C (zh) | 2000-09-25 | 2006-10-04 | 三星Sdi株式会社 | 用于可充电锂电池的正电极活性材料及其制备方法 |
| EP1313158A3 (en) | 2001-11-20 | 2004-09-08 | Canon Kabushiki Kaisha | Electrode material for rechargeable lithium battery, electrode comprising said electrode material, rechargeable lithium battery having said electrode , and process for the production thereof |
| JP4225727B2 (ja) | 2001-12-28 | 2009-02-18 | 三洋電機株式会社 | リチウム二次電池用負極及びリチウム二次電池 |
| JP3744462B2 (ja) | 2002-05-08 | 2006-02-08 | ソニー株式会社 | 非水電解質電池 |
| EP1536499B1 (en) | 2002-06-26 | 2012-02-29 | Sanyo Electric Co., Ltd. | Negative electrode for lithium secondary cell and lithium secondary cell |
| JP2004119199A (ja) * | 2002-09-26 | 2004-04-15 | Japan Storage Battery Co Ltd | 非水電解質二次電池 |
| WO2004049473A2 (en) | 2002-11-26 | 2004-06-10 | Showa Denko K.K. | Electrode material comprising silicon and/or tin particles and production method and use thereof |
| JP4385589B2 (ja) | 2002-11-26 | 2009-12-16 | 昭和電工株式会社 | 負極材料及びそれを用いた二次電池 |
| JP3750117B2 (ja) | 2002-11-29 | 2006-03-01 | 三井金属鉱業株式会社 | 非水電解液二次電池用負極及びその製造方法並びに非水電解液二次電池 |
| AU2003302519A1 (en) | 2002-11-29 | 2004-06-23 | Mitsui Mining And Smelting Co., Ltd. | Negative electrode for non-aqueous electrolyte secondary cell and method for manufacture thereof, and non-aqueous electrolyte secondary cell |
| CN100365849C (zh) | 2002-11-29 | 2008-01-30 | 三井金属矿业株式会社 | 非水电解液二次电池用负极及其制造方法以及非水电解液二次电池 |
| JP3643108B2 (ja) | 2003-07-23 | 2005-04-27 | 三井金属鉱業株式会社 | 非水電解液二次電池用負極及び非水電解液二次電池 |
| JP4046601B2 (ja) | 2002-12-03 | 2008-02-13 | 大阪瓦斯株式会社 | リチウム二次電池用負極材及びそれを用いたリチウム二次電池 |
| US20040137327A1 (en) * | 2003-01-13 | 2004-07-15 | Gross Karl J. | Synthesis of carbon/silicon composites |
| JP2004296412A (ja) | 2003-02-07 | 2004-10-21 | Mitsui Mining & Smelting Co Ltd | 非水電解液二次電池用負極活物質の製造方法 |
| JP4366222B2 (ja) | 2003-03-26 | 2009-11-18 | キヤノン株式会社 | リチウム二次電池用の電極材料、該電極材料を有する電極構造体、該電極構造体を有する二次電池 |
| US7378041B2 (en) | 2003-03-26 | 2008-05-27 | Canon Kabushiki Kaisha | Electrode material for lithium secondary battery, electrode structure comprising the electrode material and secondary battery comprising the electrode structure |
| JP4464173B2 (ja) | 2003-03-26 | 2010-05-19 | キヤノン株式会社 | リチウム二次電池用の電極材料、該電極材料を有する電極構造体、及び該電極構造体を有する二次電池 |
| WO2004086539A1 (en) | 2003-03-26 | 2004-10-07 | Canon Kabushiki Kaisha | Electrode material for lithium secondary battery and electrode structure having the electrode material |
| JP4366101B2 (ja) | 2003-03-31 | 2009-11-18 | キヤノン株式会社 | リチウム二次電池 |
| WO2004095612A1 (ja) | 2003-04-23 | 2004-11-04 | Mitsui Mining & Smelting Co., Ltd. | 非水電解液二次電池用負極及びその製造方法並びに非水電解液二次電池 |
| JP2005011650A (ja) | 2003-06-18 | 2005-01-13 | Sony Corp | 負極材料およびそれを用いた電池 |
| JP4029291B2 (ja) | 2003-09-02 | 2008-01-09 | 福田金属箔粉工業株式会社 | リチウム二次電池用負極材料及びその製造方法 |
| US7479351B2 (en) | 2003-10-09 | 2009-01-20 | Samsung Sdi Co., Ltd. | Electrode material for a lithium secondary battery, lithium secondary battery, and preparation method for the electrode material for a lithium secondary battery |
| JP3746501B2 (ja) | 2003-10-09 | 2006-02-15 | 三星エスディアイ株式会社 | リチウム二次電池用電極材料及びリチウム二次電池及びリチウム二次電池用電極材料の製造方法 |
| JP4625672B2 (ja) | 2003-10-30 | 2011-02-02 | 株式会社東芝 | 非水電解質二次電池 |
| CN100413122C (zh) | 2004-11-03 | 2008-08-20 | 深圳市比克电池有限公司 | 含锰的多元金属氧化物、锂离子二次电池的正极材料及其制备方法 |
| US7635540B2 (en) | 2004-11-15 | 2009-12-22 | Panasonic Corporation | Negative electrode for non-aqueous electrolyte secondary battery and non-aqueous electrolyte secondary battery comprising the same |
| CN101103475B (zh) | 2005-01-14 | 2011-08-10 | 松下电器产业株式会社 | 用于锂离子二次电池的负极、其制备方法、锂离子二次电池及其制备方法 |
| JP2006216277A (ja) | 2005-02-01 | 2006-08-17 | Canon Inc | リチウム二次電池用電極材料の製造方法、電極構造体および二次電池 |
| JP4794893B2 (ja) * | 2005-04-12 | 2011-10-19 | パナソニック株式会社 | 非水電解液二次電池 |
| JP5076288B2 (ja) | 2005-07-14 | 2012-11-21 | 日本電気株式会社 | 二次電池用負極およびそれを用いた二次電池 |
| JP2007026926A (ja) | 2005-07-19 | 2007-02-01 | Nec Corp | 二次電池用負極およびこれを用いた二次電池 |
| WO2007015508A1 (ja) | 2005-08-02 | 2007-02-08 | Showa Denko K.K. | リチウム二次電池負極用合金 |
| JPWO2007015508A1 (ja) | 2005-08-02 | 2009-02-19 | 昭和電工株式会社 | リチウム二次電池負極用合金 |
| JP2007149604A (ja) | 2005-11-30 | 2007-06-14 | Sanyo Electric Co Ltd | リチウム二次電池用負極及びリチウム二次電池 |
| US7906238B2 (en) | 2005-12-23 | 2011-03-15 | 3M Innovative Properties Company | Silicon-containing alloys useful as electrodes for lithium-ion batteries |
| JP2007305424A (ja) | 2006-05-11 | 2007-11-22 | Sony Corp | 負極活物質およびそれを用いた電池 |
| WO2007136046A1 (ja) | 2006-05-23 | 2007-11-29 | Sony Corporation | 負極およびその製造方法、ならびに電池およびその製造方法 |
| US8080335B2 (en) | 2006-06-09 | 2011-12-20 | Canon Kabushiki Kaisha | Powder material, electrode structure using the powder material, and energy storage device having the electrode structure |
| JP2008016446A (ja) | 2006-06-09 | 2008-01-24 | Canon Inc | 粉末材料、粉末材料を用いた電極構造体及び該電極構造体を有する蓄電デバイス、並びに粉末材料の製造方法 |
| KR101375455B1 (ko) | 2006-08-29 | 2014-03-26 | 강원대학교산학협력단 | 이차 전지용 전극 활물질 |
| WO2008038798A1 (fr) * | 2006-09-29 | 2008-04-03 | Mitsui Mining & Smelting Co., Ltd. | Batterie secondaire à électrolyte non aqueux |
| US10665892B2 (en) | 2007-01-10 | 2020-05-26 | Eocell Limited | Lithium batteries with nano-composite positive electrode material |
| KR20090109570A (ko) | 2007-02-06 | 2009-10-20 | 쓰리엠 이노베이티브 프로퍼티즈 컴파니 | 신규한 결합제를 포함하는 전극과, 그의 제조 방법 및 사용 방법 |
| US7875388B2 (en) | 2007-02-06 | 2011-01-25 | 3M Innovative Properties Company | Electrodes including polyacrylate binders and methods of making and using the same |
| US9070935B2 (en) | 2007-06-06 | 2015-06-30 | Asahi Kasei E-Materials Corporation | Multilayer porous film |
| JP5343342B2 (ja) | 2007-06-26 | 2013-11-13 | 大同特殊鋼株式会社 | リチウム二次電池用負極活物質およびリチウム二次電池 |
| KR101406013B1 (ko) | 2008-03-17 | 2014-06-11 | 신에쓰 가가꾸 고교 가부시끼가이샤 | 비수 전해질 2차 전지용 부극재 및 그것의 제조 방법, 및 비수 전해질 2차 전지용 부극 및 비수 전해질 2차 전지 |
| JP2009224239A (ja) | 2008-03-18 | 2009-10-01 | Nissan Motor Co Ltd | 電池用電極 |
| JP5046302B2 (ja) * | 2008-03-28 | 2012-10-10 | 日立マクセルエナジー株式会社 | 非水二次電池 |
| JP5357565B2 (ja) | 2008-05-27 | 2013-12-04 | 株式会社神戸製鋼所 | リチウムイオン二次電池用負極材、および、その製造方法、ならびに、リチウムイオン二次電池 |
| US9012073B2 (en) | 2008-11-11 | 2015-04-21 | Envia Systems, Inc. | Composite compositions, negative electrodes with composite compositions and corresponding batteries |
| JP2010205609A (ja) | 2009-03-04 | 2010-09-16 | Nissan Motor Co Ltd | 電極およびこれを用いた電池 |
| JPWO2010110205A1 (ja) | 2009-03-24 | 2012-09-27 | 古河電気工業株式会社 | リチウムイオン二次電池、該電池用電極、該電池電極用電解銅箔 |
| EP2239803A1 (fr) | 2009-04-10 | 2010-10-13 | Saft Groupe Sa | Composition de matiere active pour electrode negative d'accumulateur lithium-ion. |
| US20100288077A1 (en) | 2009-05-14 | 2010-11-18 | 3M Innovative Properties Company | Method of making an alloy |
| WO2010150513A1 (ja) | 2009-06-23 | 2010-12-29 | キヤノン株式会社 | 電極構造体及び蓄電デバイス |
| WO2011010421A1 (ja) | 2009-07-21 | 2011-01-27 | パナソニック株式会社 | 角形の非水電解質二次電池及びその製造方法 |
| JP2011048969A (ja) | 2009-08-26 | 2011-03-10 | Toyobo Co Ltd | リチウムイオン二次電池用負極及びこれを用いた二次電池 |
| WO2011031546A2 (en) | 2009-08-27 | 2011-03-17 | Envia Systems, Inc. | Layer-layer lithium rich complex metal oxides with high specific capacity and excellent cycling |
| JP5673545B2 (ja) | 2009-09-25 | 2015-02-18 | 日本ゼオン株式会社 | リチウムイオン二次電池負極及びリチウムイオン二次電池 |
| MX2012002805A (es) | 2009-11-27 | 2012-04-02 | Nissan Motor | Material activo del electrodo negativo, de aleacion de si, para dispositivos electricos. |
| KR101399685B1 (ko) | 2009-11-27 | 2014-05-27 | 닛산 지도우샤 가부시키가이샤 | 전기 디바이스용 Si 합금 부극 활물질 |
| US9876221B2 (en) | 2010-05-14 | 2018-01-23 | Samsung Sdi Co., Ltd. | Negative active material for rechargeable lithium battery and rechargeable lithium battery including same |
| CN102576901A (zh) * | 2010-05-18 | 2012-07-11 | 松下电器产业株式会社 | 锂二次电池 |
| JP5128695B2 (ja) | 2010-06-28 | 2013-01-23 | 古河電気工業株式会社 | 電解銅箔、リチウムイオン二次電池用電解銅箔、該電解銅箔を用いたリチウムイオン二次電池用電極、該電極を使用したリチウムイオン二次電池 |
| CA2800929C (en) | 2010-06-29 | 2014-09-16 | Umicore | Submicron sized silicon powder with low oxygen content |
| WO2012000854A1 (en) | 2010-06-29 | 2012-01-05 | Umicore | Negative electrode material for lithium-ion batteries |
| JP5472041B2 (ja) | 2010-10-28 | 2014-04-16 | 三菱化学株式会社 | 非水系電解液およびそれを用いた非水系電解液二次電池 |
| JP5850611B2 (ja) | 2010-11-17 | 2016-02-03 | 三井金属鉱業株式会社 | リチウムイオン二次電池負極集電体用の銅箔、リチウムイオン二次電池負極材及びリチウムイオン二次電池負極集電体選定方法。 |
| JP5652161B2 (ja) | 2010-11-26 | 2015-01-14 | 日産自動車株式会社 | 電気デバイス用Si合金負極活物質 |
| JP5276158B2 (ja) | 2010-12-27 | 2013-08-28 | 古河電気工業株式会社 | リチウムイオン二次電池、該電池用負極電極、該電池負極集電体用電解銅箔 |
| KR20120090594A (ko) | 2011-02-08 | 2012-08-17 | 삼성전자주식회사 | 고분자 전극의 제조방법 및 고분자 전극을 채용한 고분자 구동기 |
| JP5614729B2 (ja) | 2011-03-03 | 2014-10-29 | 日産自動車株式会社 | リチウムイオン二次電池用正極活物質、リチウムイオン二次電池用正極及びリチウムイオン二次電池 |
| JP5768968B2 (ja) | 2011-03-08 | 2015-08-26 | 日産自動車株式会社 | リチウムイオン二次電池用負極活物質 |
| JP5741908B2 (ja) | 2011-03-09 | 2015-07-01 | 日産自動車株式会社 | リチウムイオン二次電池用正極活物質 |
| WO2012132153A1 (ja) * | 2011-03-28 | 2012-10-04 | 日本電気株式会社 | 二次電池 |
| JP2012238581A (ja) | 2011-04-28 | 2012-12-06 | Nichia Chem Ind Ltd | 非水電解液二次電池用正極活物質 |
| JP5751449B2 (ja) | 2011-05-25 | 2015-07-22 | 日産自動車株式会社 | リチウムイオン二次電池用負極活物質 |
| JP5751448B2 (ja) | 2011-05-25 | 2015-07-22 | 日産自動車株式会社 | リチウムイオン二次電池用負極活物質 |
| JP5776931B2 (ja) | 2011-05-25 | 2015-09-09 | 日産自動車株式会社 | リチウムイオン二次電池用負極活物質 |
| JP5776888B2 (ja) * | 2011-05-25 | 2015-09-09 | 日産自動車株式会社 | 電気デバイス用負極活物質 |
| JP5970978B2 (ja) * | 2011-07-04 | 2016-08-17 | 日産自動車株式会社 | 電気デバイス用正極活物質、電気デバイス用正極及び電気デバイス |
| US9905838B2 (en) | 2011-08-30 | 2018-02-27 | Gs Yuasa International Ltd. | Electrode and method of manufacturing the same |
| KR20130037091A (ko) | 2011-10-05 | 2013-04-15 | 삼성에스디아이 주식회사 | 음극 활물질 및 이를 채용한 리튬 전지 |
| CN103843177A (zh) | 2011-10-10 | 2014-06-04 | 3M创新有限公司 | 用于锂离子电化学电池的非晶态合金负极组合物 |
| JP5945903B2 (ja) | 2011-12-16 | 2016-07-05 | 日産自動車株式会社 | 電気デバイス用負極活物質 |
| JP5903556B2 (ja) | 2011-12-22 | 2016-04-13 | パナソニックIpマネジメント株式会社 | 非水電解液二次電池 |
| JP5904364B2 (ja) | 2011-12-27 | 2016-04-13 | 日産自動車株式会社 | 電気デバイス用負極活物質 |
| JP5904363B2 (ja) | 2011-12-27 | 2016-04-13 | 日産自動車株式会社 | 電気デバイス用負極活物質 |
| KR101649808B1 (ko) | 2012-02-01 | 2016-08-19 | 닛산 지도우샤 가부시키가이샤 | 고용체 리튬 함유 전이 금속 산화물, 비수전해질 이차 전지용 정극 및 비수전해질 이차 전지 |
| US20130202967A1 (en) | 2012-02-07 | 2013-08-08 | Jae-Hyuk Kim | Negative active material for rechargeable lithium battery and rechargeable lithium battery including same |
| JP5830419B2 (ja) * | 2012-03-21 | 2015-12-09 | 古河電気工業株式会社 | 多孔質シリコン粒子及び多孔質シリコン複合体粒子並びにこれらの製造方法 |
| JP6268729B2 (ja) | 2012-03-23 | 2018-01-31 | 三菱ケミカル株式会社 | 非水系二次電池用負極材、非水系二次電池用負極及び非水系二次電池 |
| CN104205434B (zh) | 2012-03-26 | 2017-01-18 | 索尼公司 | 正极活性材料、正极、二次电池、电池组、电动车辆、电力存储系统、电动工具、以及电子设备 |
| JP5999968B2 (ja) | 2012-05-02 | 2016-09-28 | セイコーインスツル株式会社 | 扁平形一次電池、扁平形一次電池用負極合剤及びその製造方法 |
| WO2014080893A1 (ja) | 2012-11-22 | 2014-05-30 | 日産自動車株式会社 | 電気デバイス用負極、及びこれを用いた電気デバイス |
| US20150295228A1 (en) | 2012-11-22 | 2015-10-15 | Nissan Motor Co., Ltd. | Negative electrode for electric device and electric device using the same |
| KR101709027B1 (ko) | 2012-11-22 | 2017-02-21 | 닛산 지도우샤 가부시키가이샤 | 전기 디바이스용 부극, 및 이것을 사용한 전기 디바이스 |
| JP6052299B2 (ja) | 2012-11-22 | 2016-12-27 | 日産自動車株式会社 | 電気デバイス用負極、及びこれを用いた電気デバイス |
| JP6040994B2 (ja) | 2012-11-22 | 2016-12-07 | 日産自動車株式会社 | リチウムイオン二次電池用負極、及びこれを用いたリチウムイオン二次電池 |
| JP6040997B2 (ja) | 2012-11-22 | 2016-12-07 | 日産自動車株式会社 | リチウムイオン二次電池用負極、及びこれを用いたリチウムイオン二次電池 |
| JP6040996B2 (ja) | 2012-11-22 | 2016-12-07 | 日産自動車株式会社 | リチウムイオン二次電池用負極、及びこれを用いたリチウムイオン二次電池 |
| CN104813516B (zh) | 2012-11-22 | 2017-12-01 | 日产自动车株式会社 | 电气设备用负极、及使用其的电气设备 |
| WO2014080895A1 (ja) | 2012-11-22 | 2014-05-30 | 日産自動車株式会社 | 電気デバイス用負極、及びこれを用いた電気デバイス |
| WO2014080891A1 (ja) | 2012-11-22 | 2014-05-30 | 日産自動車株式会社 | 電気デバイス用負極、及びこれを用いた電気デバイス |
| US20150303466A1 (en) | 2012-11-22 | 2015-10-22 | Nissan Motor Co., Ltd. | Negative electrode for electric device and electric device using the same |
| EP2924774B1 (en) | 2012-11-22 | 2018-04-25 | Nissan Motor Co., Ltd | Negative electrode for electric device and electric device using the same |
| KR101891014B1 (ko) | 2014-01-24 | 2018-08-22 | 닛산 지도우샤 가부시키가이샤 | 전기 디바이스 |
| JP6187602B2 (ja) | 2014-01-24 | 2017-08-30 | 日産自動車株式会社 | 電気デバイス |
| KR20160102026A (ko) * | 2014-01-24 | 2016-08-26 | 닛산 지도우샤 가부시키가이샤 | 전기 디바이스 |
| US9954252B2 (en) | 2014-01-24 | 2018-04-24 | Nissan Motor Co., Ltd. | Electrical device |
| US9680150B2 (en) | 2014-01-24 | 2017-06-13 | Nissan Motor Co., Ltd. | Electrical device |
-
2014
- 2014-01-24 JP JP2015558664A patent/JP6187602B2/ja not_active Expired - Fee Related
- 2014-01-24 WO PCT/JP2014/051526 patent/WO2015111187A1/ja active Application Filing
- 2014-01-24 KR KR1020167019128A patent/KR20160100348A/ko active IP Right Grant
- 2014-01-24 US US15/113,250 patent/US10476101B2/en not_active Expired - Fee Related
- 2014-01-24 EP EP14879986.9A patent/EP3098892B1/en not_active Not-in-force
- 2014-01-24 CN CN201480073896.4A patent/CN105934847B/zh not_active Expired - Fee Related
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005011699A (ja) * | 2003-06-19 | 2005-01-13 | Canon Inc | リチウム二次電池 |
| JP2009517850A (ja) | 2005-12-01 | 2009-04-30 | スリーエム イノベイティブ プロパティズ カンパニー | ケイ素含有量が高いアモルファス合金に基づく電極組成物 |
| JP2008004535A (ja) * | 2006-05-23 | 2008-01-10 | Sony Corp | 負極および電池 |
| JP2009099328A (ja) * | 2007-10-15 | 2009-05-07 | Sony Corp | 負極および電池 |
| JP2009158415A (ja) * | 2007-12-27 | 2009-07-16 | Mitsui Mining & Smelting Co Ltd | 非水電解液二次電池用正極活物質及びそれを有する非水電解液二次電池 |
| JP2011060471A (ja) * | 2009-09-08 | 2011-03-24 | Nec Energy Devices Ltd | 非水系電解質二次電池 |
| JP2012142154A (ja) * | 2010-12-28 | 2012-07-26 | Sony Corp | リチウムイオン二次電池、電動工具、電動車両および電力貯蔵システム |
| WO2013094465A1 (ja) * | 2011-12-19 | 2013-06-27 | 日立マクセル株式会社 | リチウム二次電池 |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017041407A (ja) * | 2015-08-21 | 2017-02-23 | 日産自動車株式会社 | リチウムイオン二次電池 |
| CN109075377A (zh) * | 2016-04-28 | 2018-12-21 | 日产自动车株式会社 | 非水电解质二次电池 |
| EP3451430A4 (en) * | 2016-04-28 | 2019-04-03 | Nissan Motor Co., Ltd. | NONAQUEOUS ELECTROLYTE SECONDARY BATTERY |
| EP3451431A4 (en) * | 2016-04-28 | 2019-04-03 | Nissan Motor Co., Ltd. | NONAQUEOUS ELECTROLYTE SECONDARY BATTERY |
| US20190157664A1 (en) * | 2016-04-28 | 2019-05-23 | Nissan Motor Co., Ltd. | Non-Aqueous Electrolyte Secondary Battery |
| US10374225B2 (en) | 2016-04-28 | 2019-08-06 | Nissan Motor Co., Ltd. | Non-aqueous electrolyte secondary battery |
| CN109075377B (zh) * | 2016-04-28 | 2020-08-25 | 远景Aesc日本有限公司 | 非水电解质二次电池 |
| WO2018003843A1 (ja) * | 2016-06-30 | 2018-01-04 | 三洋電機株式会社 | 二次電池及びその製造方法 |
| JPWO2018003843A1 (ja) * | 2016-06-30 | 2019-04-18 | 三洋電機株式会社 | 二次電池及びその製造方法 |
| US10916760B2 (en) | 2016-06-30 | 2021-02-09 | Sanyo Electric Co., Ltd. | Secondary battery and method of manufacturing same |
| JP7088008B2 (ja) | 2016-06-30 | 2022-06-21 | 三洋電機株式会社 | 二次電池及びその製造方法 |
| JP2018055901A (ja) * | 2016-09-28 | 2018-04-05 | 日産自動車株式会社 | 非水電解質二次電池の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3098892A1 (en) | 2016-11-30 |
| JP6187602B2 (ja) | 2017-08-30 |
| EP3098892A4 (en) | 2016-11-30 |
| US10476101B2 (en) | 2019-11-12 |
| JPWO2015111187A1 (ja) | 2017-03-23 |
| CN105934847A (zh) | 2016-09-07 |
| EP3098892B1 (en) | 2018-11-14 |
| CN105934847B (zh) | 2019-11-05 |
| KR20160100348A (ko) | 2016-08-23 |
| US20170012316A1 (en) | 2017-01-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6252602B2 (ja) | 電気デバイス | |
| JP6187602B2 (ja) | 電気デバイス | |
| JP6202106B2 (ja) | 電気デバイス | |
| WO2016098212A1 (ja) | 電気デバイス | |
| JP6252600B2 (ja) | 電気デバイス | |
| JP6202107B2 (ja) | 電気デバイス | |
| JP6252604B2 (ja) | 電気デバイス | |
| JP6737091B2 (ja) | 非水電解質二次電池用負極及びこれを用いた非水電解質二次電池 | |
| JP6380553B2 (ja) | 電気デバイス | |
| JP6380554B2 (ja) | 電気デバイス | |
| JP6252603B2 (ja) | 電気デバイス | |
| WO2015111195A1 (ja) | 電気デバイス用負極およびこれを用いた電気デバイス | |
| JP6252601B2 (ja) | 電気デバイス | |
| JP2018078052A (ja) | 非水電解質二次電池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14879986 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20167019128 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15113250 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2015558664 Country of ref document: JP Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014879986 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014879986 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |