WO2012018126A1 - 相容化樹脂の製造法、熱硬化性樹脂組成物、プリプレグ及び積層板 - Google Patents
相容化樹脂の製造法、熱硬化性樹脂組成物、プリプレグ及び積層板 Download PDFInfo
- Publication number
- WO2012018126A1 WO2012018126A1 PCT/JP2011/067985 JP2011067985W WO2012018126A1 WO 2012018126 A1 WO2012018126 A1 WO 2012018126A1 JP 2011067985 W JP2011067985 W JP 2011067985W WO 2012018126 A1 WO2012018126 A1 WO 2012018126A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin
- mass
- thermosetting resin
- reaction
- parts
- Prior art date
Links
- 229920005989 resin Polymers 0.000 title claims abstract description 220
- 239000011347 resin Substances 0.000 title claims abstract description 220
- 229920001187 thermosetting polymer Polymers 0.000 title claims abstract description 106
- 239000011342 resin composition Substances 0.000 title claims abstract description 48
- 238000000034 method Methods 0.000 title claims abstract description 13
- 230000008569 process Effects 0.000 title abstract description 4
- 238000006243 chemical reaction Methods 0.000 claims abstract description 156
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 claims abstract description 45
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims abstract description 41
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 35
- 239000005350 fused silica glass Substances 0.000 claims abstract description 30
- -1 cyanate compound Chemical class 0.000 claims abstract description 25
- XLJMAIOERFSOGZ-UHFFFAOYSA-M cyanate group Chemical group [O-]C#N XLJMAIOERFSOGZ-UHFFFAOYSA-M 0.000 claims description 61
- 150000001875 compounds Chemical class 0.000 claims description 55
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 54
- 238000004519 manufacturing process Methods 0.000 claims description 48
- 239000000463 material Substances 0.000 claims description 23
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical group C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 claims description 17
- 239000002904 solvent Substances 0.000 claims description 12
- ICGQLNMKJVHCIR-UHFFFAOYSA-N 1,3,2-dioxazetidin-4-one Chemical group O=C1ONO1 ICGQLNMKJVHCIR-UHFFFAOYSA-N 0.000 claims description 11
- AUHZEENZYGFFBQ-UHFFFAOYSA-N mesitylene Substances CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 claims description 8
- 125000001827 mesitylenyl group Chemical group [H]C1=C(C(*)=C(C([H])=C1C([H])([H])[H])C([H])([H])[H])C([H])([H])[H] 0.000 claims description 8
- 239000003960 organic solvent Substances 0.000 claims description 8
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims description 7
- 239000008096 xylene Substances 0.000 claims description 7
- 125000002947 alkylene group Chemical group 0.000 claims description 6
- 125000004432 carbon atom Chemical group C* 0.000 claims description 6
- 125000003700 epoxy group Chemical group 0.000 claims description 5
- 125000002524 organometallic group Chemical group 0.000 claims description 5
- 125000005529 alkyleneoxy group Chemical group 0.000 claims description 3
- 125000000732 arylene group Chemical group 0.000 claims description 3
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 abstract description 57
- 239000003822 epoxy resin Substances 0.000 abstract description 29
- 229920000647 polyepoxide Polymers 0.000 abstract description 29
- 239000011889 copper foil Substances 0.000 abstract description 28
- 239000000203 mixture Substances 0.000 abstract description 11
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 54
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 30
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 28
- 229910052802 copper Inorganic materials 0.000 description 28
- 239000010949 copper Substances 0.000 description 28
- 239000000047 product Substances 0.000 description 28
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 27
- 238000005259 measurement Methods 0.000 description 26
- 230000008034 disappearance Effects 0.000 description 25
- 230000000052 comparative effect Effects 0.000 description 20
- 239000007787 solid Substances 0.000 description 20
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 18
- 238000002156 mixing Methods 0.000 description 18
- 238000011156 evaluation Methods 0.000 description 17
- WSFQLUVWDKCYSW-UHFFFAOYSA-M sodium;2-hydroxy-3-morpholin-4-ylpropane-1-sulfonate Chemical compound [Na+].[O-]S(=O)(=O)CC(O)CN1CCOCC1 WSFQLUVWDKCYSW-UHFFFAOYSA-M 0.000 description 17
- 239000004793 Polystyrene Substances 0.000 description 16
- 229920002223 polystyrene Polymers 0.000 description 16
- 239000000758 substrate Substances 0.000 description 16
- 229910052500 inorganic mineral Inorganic materials 0.000 description 15
- 239000011707 mineral Substances 0.000 description 15
- 238000010992 reflux Methods 0.000 description 15
- 238000003756 stirring Methods 0.000 description 15
- 239000003480 eluent Substances 0.000 description 14
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 14
- 238000010438 heat treatment Methods 0.000 description 13
- 239000004305 biphenyl Substances 0.000 description 10
- 235000010290 biphenyl Nutrition 0.000 description 10
- 229910000679 solder Inorganic materials 0.000 description 10
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 9
- 239000004593 Epoxy Substances 0.000 description 9
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 9
- 239000011256 inorganic filler Substances 0.000 description 9
- 229910003475 inorganic filler Inorganic materials 0.000 description 9
- 239000012046 mixed solvent Substances 0.000 description 9
- 239000002994 raw material Substances 0.000 description 9
- 229930185605 Bisphenol Natural products 0.000 description 8
- 229910052751 metal Inorganic materials 0.000 description 8
- 239000002184 metal Substances 0.000 description 8
- 238000007747 plating Methods 0.000 description 8
- 238000001816 cooling Methods 0.000 description 7
- 239000012796 inorganic flame retardant Substances 0.000 description 7
- 230000014759 maintenance of location Effects 0.000 description 7
- 239000002966 varnish Substances 0.000 description 7
- 239000011248 coating agent Substances 0.000 description 6
- 238000000576 coating method Methods 0.000 description 6
- 238000010828 elution Methods 0.000 description 6
- 238000005530 etching Methods 0.000 description 6
- 239000011521 glass Substances 0.000 description 6
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 6
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 5
- 239000003063 flame retardant Substances 0.000 description 5
- 239000011810 insulating material Substances 0.000 description 5
- KBJFYLLAMSZSOG-UHFFFAOYSA-N n-(3-trimethoxysilylpropyl)aniline Chemical compound CO[Si](OC)(OC)CCCNC1=CC=CC=C1 KBJFYLLAMSZSOG-UHFFFAOYSA-N 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 238000005979 thermal decomposition reaction Methods 0.000 description 5
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 4
- 239000005062 Polybutadiene Substances 0.000 description 4
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 4
- 238000005553 drilling Methods 0.000 description 4
- 229920003986 novolac Polymers 0.000 description 4
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 4
- 229920002857 polybutadiene Polymers 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 238000007363 ring formation reaction Methods 0.000 description 4
- 150000003839 salts Chemical class 0.000 description 4
- 230000007704 transition Effects 0.000 description 4
- RNFJDJUURJAICM-UHFFFAOYSA-N 2,2,4,4,6,6-hexaphenoxy-1,3,5-triaza-2$l^{5},4$l^{5},6$l^{5}-triphosphacyclohexa-1,3,5-triene Chemical compound N=1P(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP=1(OC=1C=CC=CC=1)OC1=CC=CC=C1 RNFJDJUURJAICM-UHFFFAOYSA-N 0.000 description 3
- 229910002706 AlOOH Inorganic materials 0.000 description 3
- 0 CN(*1=CC1)N Chemical compound CN(*1=CC1)N 0.000 description 3
- 229910001593 boehmite Inorganic materials 0.000 description 3
- 229930003836 cresol Natural products 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- FAHBNUUHRFUEAI-UHFFFAOYSA-M hydroxidooxidoaluminium Chemical compound O[Al]=O FAHBNUUHRFUEAI-UHFFFAOYSA-M 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 239000011229 interlayer Substances 0.000 description 3
- 238000010030 laminating Methods 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 238000000465 moulding Methods 0.000 description 3
- 230000035515 penetration Effects 0.000 description 3
- 150000002989 phenols Chemical class 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- XAEWLETZEZXLHR-UHFFFAOYSA-N zinc;dioxido(dioxo)molybdenum Chemical compound [Zn+2].[O-][Mo]([O-])(=O)=O XAEWLETZEZXLHR-UHFFFAOYSA-N 0.000 description 3
- ARXJGSRGQADJSQ-UHFFFAOYSA-N 1-methoxypropan-2-ol Chemical compound COCC(C)O ARXJGSRGQADJSQ-UHFFFAOYSA-N 0.000 description 2
- KJCVRFUGPWSIIH-UHFFFAOYSA-N 1-naphthol Chemical compound C1=CC=C2C(O)=CC=CC2=C1 KJCVRFUGPWSIIH-UHFFFAOYSA-N 0.000 description 2
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 2
- QTWJRLJHJPIABL-UHFFFAOYSA-N 2-methylphenol;3-methylphenol;4-methylphenol Chemical compound CC1=CC=C(O)C=C1.CC1=CC=CC(O)=C1.CC1=CC=CC=C1O QTWJRLJHJPIABL-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 2
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- 239000006087 Silane Coupling Agent Substances 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- ADCOVFLJGNWWNZ-UHFFFAOYSA-N antimony trioxide Chemical compound O=[Sb]O[Sb]=O ADCOVFLJGNWWNZ-UHFFFAOYSA-N 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 239000003849 aromatic solvent Substances 0.000 description 2
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 2
- 239000007809 chemical reaction catalyst Substances 0.000 description 2
- 229910017052 cobalt Inorganic materials 0.000 description 2
- 239000010941 cobalt Substances 0.000 description 2
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 2
- 238000013329 compounding Methods 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- 239000000806 elastomer Substances 0.000 description 2
- 230000007613 environmental effect Effects 0.000 description 2
- 230000003628 erosive effect Effects 0.000 description 2
- 239000004744 fabric Substances 0.000 description 2
- 239000000835 fiber Substances 0.000 description 2
- 239000011888 foil Substances 0.000 description 2
- 239000003365 glass fiber Substances 0.000 description 2
- 230000009477 glass transition Effects 0.000 description 2
- 239000003999 initiator Substances 0.000 description 2
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 2
- 239000000347 magnesium hydroxide Substances 0.000 description 2
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 2
- GEMHFKXPOCTAIP-UHFFFAOYSA-N n,n-dimethyl-n'-phenylcarbamimidoyl chloride Chemical compound CN(C)C(Cl)=NC1=CC=CC=C1 GEMHFKXPOCTAIP-UHFFFAOYSA-N 0.000 description 2
- 239000012766 organic filler Substances 0.000 description 2
- 229920000573 polyethylene Polymers 0.000 description 2
- 229920001721 polyimide Polymers 0.000 description 2
- 229920001955 polyphenylene ether Polymers 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 2
- 239000004810 polytetrafluoroethylene Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 238000003825 pressing Methods 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 238000004381 surface treatment Methods 0.000 description 2
- 229920005992 thermoplastic resin Polymers 0.000 description 2
- HCNHNBLSNVSJTJ-UHFFFAOYSA-N 1,1-Bis(4-hydroxyphenyl)ethane Chemical compound C=1C=C(O)C=CC=1C(C)C1=CC=C(O)C=C1 HCNHNBLSNVSJTJ-UHFFFAOYSA-N 0.000 description 1
- ZVNPWFOVUDMGRP-UHFFFAOYSA-N 4-methylaminophenol sulfate Chemical compound OS(O)(=O)=O.CNC1=CC=C(O)C=C1.CNC1=CC=C(O)C=C1 ZVNPWFOVUDMGRP-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Nc1ccccc1 Chemical compound Nc1ccccc1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 239000004642 Polyimide Substances 0.000 description 1
- 208000034189 Sclerosis Diseases 0.000 description 1
- YSMRWXYRXBRSND-UHFFFAOYSA-N TOTP Chemical compound CC1=CC=CC=C1OP(=O)(OC=1C(=CC=CC=1)C)OC1=CC=CC=C1C YSMRWXYRXBRSND-UHFFFAOYSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- NOHQTLHHNIKWBA-UHFFFAOYSA-N [SiH4].NC(=O)N Chemical compound [SiH4].NC(=O)N NOHQTLHHNIKWBA-UHFFFAOYSA-N 0.000 description 1
- 239000006096 absorbing agent Substances 0.000 description 1
- 229920000122 acrylonitrile butadiene styrene Polymers 0.000 description 1
- 150000008360 acrylonitriles Chemical class 0.000 description 1
- 238000007259 addition reaction Methods 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000002723 alicyclic group Chemical group 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 239000002635 aromatic organic solvent Substances 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 239000012965 benzophenone Substances 0.000 description 1
- 150000008366 benzophenones Chemical class 0.000 description 1
- 150000001565 benzotriazoles Chemical class 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910001902 chlorine oxide Inorganic materials 0.000 description 1
- 239000004020 conductor Substances 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- GYZLOYUZLJXAJU-UHFFFAOYSA-N diglycidyl ether Chemical class C1OC1COCC1CO1 GYZLOYUZLJXAJU-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000006081 fluorescent whitening agent Substances 0.000 description 1
- 238000001879 gelation Methods 0.000 description 1
- 229910001679 gibbsite Inorganic materials 0.000 description 1
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012784 inorganic fiber Substances 0.000 description 1
- 239000012774 insulation material Substances 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910000000 metal hydroxide Inorganic materials 0.000 description 1
- 150000004692 metal hydroxides Chemical class 0.000 description 1
- 239000010445 mica Substances 0.000 description 1
- 229910052618 mica group Inorganic materials 0.000 description 1
- VLAPMBHFAWRUQP-UHFFFAOYSA-L molybdic acid Chemical compound O[Mo](O)(=O)=O VLAPMBHFAWRUQP-UHFFFAOYSA-L 0.000 description 1
- 150000002790 naphthalenes Chemical class 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 239000004745 nonwoven fabric Substances 0.000 description 1
- AFEQENGXSMURHA-UHFFFAOYSA-N oxiran-2-ylmethanamine Chemical class NCC1CO1 AFEQENGXSMURHA-UHFFFAOYSA-N 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- 229920006287 phenoxy resin Polymers 0.000 description 1
- 239000013034 phenoxy resin Substances 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 229920006122 polyamide resin Polymers 0.000 description 1
- 229920005668 polycarbonate resin Polymers 0.000 description 1
- 239000004431 polycarbonate resin Substances 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920001225 polyester resin Polymers 0.000 description 1
- 239000004645 polyester resin Substances 0.000 description 1
- 239000009719 polyimide resin Substances 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 229920002050 silicone resin Polymers 0.000 description 1
- PJANXHGTPQOBST-UHFFFAOYSA-N stilbene Chemical class C=1C=CC=CC=1C=CC1=CC=CC=C1 PJANXHGTPQOBST-UHFFFAOYSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- BFKJFAAPBSQJPD-UHFFFAOYSA-N tetrafluoroethene Chemical group FC(F)=C(F)F BFKJFAAPBSQJPD-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000002341 toxic gas Substances 0.000 description 1
- 238000005829 trimerization reaction Methods 0.000 description 1
- BPSIOYPQMFLKFR-UHFFFAOYSA-N trimethoxy-[3-(oxiran-2-ylmethoxy)propyl]silane Chemical compound CO[Si](OC)(OC)CCCOCC1CO1 BPSIOYPQMFLKFR-UHFFFAOYSA-N 0.000 description 1
- XZZNDPSIHUTMOC-UHFFFAOYSA-N triphenyl phosphate Chemical compound C=1C=CC=CC=1OP(OC=1C=CC=CC=1)(=O)OC1=CC=CC=C1 XZZNDPSIHUTMOC-UHFFFAOYSA-N 0.000 description 1
- 239000006097 ultraviolet radiation absorber Substances 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- 239000002759 woven fabric Substances 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L83/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon only; Compositions of derivatives of such polymers
- C08L83/04—Polysiloxanes
- C08L83/06—Polysiloxanes containing silicon bound to oxygen-containing groups
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B15/00—Layered products comprising a layer of metal
- B32B15/14—Layered products comprising a layer of metal next to a fibrous or filamentary layer
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B15/00—Layered products comprising a layer of metal
- B32B15/20—Layered products comprising a layer of metal comprising aluminium or copper
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/02—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by structural features of a fibrous or filamentary layer
- B32B5/022—Non-woven fabric
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/02—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by structural features of a fibrous or filamentary layer
- B32B5/024—Woven fabric
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/02—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by structural features of a fibrous or filamentary layer
- B32B5/08—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by structural features of a fibrous or filamentary layer the fibres or filaments of a layer being of different substances, e.g. conjugate fibres, mixture of different fibres
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B7/00—Layered products characterised by the relation between layers; Layered products characterised by the relative orientation of features between layers, or by the relative values of a measurable parameter between layers, i.e. products comprising layers having different physical, chemical or physicochemical properties; Layered products characterised by the interconnection of layers
- B32B7/04—Interconnection of layers
- B32B7/06—Interconnection of layers permitting easy separation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/0622—Polycondensates containing six-membered rings, not condensed with other rings, with nitrogen atoms as the only ring hetero atoms
- C08G73/0638—Polycondensates containing six-membered rings, not condensed with other rings, with nitrogen atoms as the only ring hetero atoms with at least three nitrogen atoms in the ring
- C08G73/0644—Poly(1,3,5)triazines
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/0622—Polycondensates containing six-membered rings, not condensed with other rings, with nitrogen atoms as the only ring hetero atoms
- C08G73/0638—Polycondensates containing six-membered rings, not condensed with other rings, with nitrogen atoms as the only ring hetero atoms with at least three nitrogen atoms in the ring
- C08G73/065—Preparatory processes
- C08G73/0655—Preparatory processes from polycyanurates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/42—Block-or graft-polymers containing polysiloxane sequences
- C08G77/452—Block-or graft-polymers containing polysiloxane sequences containing nitrogen-containing sequences
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/241—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres
- C08J5/244—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres using glass fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/249—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs characterised by the additives used in the prepolymer mixture
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K9/00—Use of pretreated ingredients
- C08K9/04—Ingredients treated with organic substances
- C08K9/06—Ingredients treated with organic substances with silicon-containing compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L79/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen or carbon only, not provided for in groups C08L61/00 - C08L77/00
- C08L79/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K1/00—Printed circuits
- H05K1/02—Details
- H05K1/03—Use of materials for the substrate
- H05K1/0313—Organic insulating material
- H05K1/032—Organic insulating material consisting of one material
- H05K1/0326—Organic insulating material consisting of one material containing O
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2260/00—Layered product comprising an impregnated, embedded, or bonded layer wherein the layer comprises an impregnation, embedding, or binder material
- B32B2260/02—Composition of the impregnated, bonded or embedded layer
- B32B2260/021—Fibrous or filamentary layer
- B32B2260/023—Two or more layers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2260/00—Layered product comprising an impregnated, embedded, or bonded layer wherein the layer comprises an impregnation, embedding, or binder material
- B32B2260/04—Impregnation, embedding, or binder material
- B32B2260/046—Synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2262/00—Composition or structural features of fibres which form a fibrous or filamentary layer or are present as additives
- B32B2262/02—Synthetic macromolecular fibres
- B32B2262/0276—Polyester fibres
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2262/00—Composition or structural features of fibres which form a fibrous or filamentary layer or are present as additives
- B32B2262/10—Inorganic fibres
- B32B2262/101—Glass fibres
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/30—Properties of the layers or laminate having particular thermal properties
- B32B2307/306—Resistant to heat
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/30—Properties of the layers or laminate having particular thermal properties
- B32B2307/306—Resistant to heat
- B32B2307/3065—Flame resistant or retardant, fire resistant or retardant
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/50—Properties of the layers or laminate having particular mechanical properties
- B32B2307/51—Elastic
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/714—Inert, i.e. inert to chemical degradation, corrosion
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/732—Dimensional properties
- B32B2307/734—Dimensional stability
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/748—Releasability
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2457/00—Electrical equipment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/14—Polysiloxanes containing silicon bound to oxygen-containing groups
- C08G77/16—Polysiloxanes containing silicon bound to oxygen-containing groups to hydroxyl groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2383/00—Characterised by the use of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon with or without sulfur, nitrogen, oxygen, or carbon only; Derivatives of such polymers
- C08J2383/10—Block- or graft-copolymers containing polysiloxane sequences
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K1/00—Printed circuits
- H05K1/02—Details
- H05K1/03—Use of materials for the substrate
- H05K1/0313—Organic insulating material
- H05K1/0353—Organic insulating material consisting of two or more materials, e.g. two or more polymers, polymer + filler, + reinforcement
- H05K1/0366—Organic insulating material consisting of two or more materials, e.g. two or more polymers, polymer + filler, + reinforcement reinforced, e.g. by fibres, fabrics
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K1/00—Printed circuits
- H05K1/02—Details
- H05K1/03—Use of materials for the substrate
- H05K1/0313—Organic insulating material
- H05K1/0353—Organic insulating material consisting of two or more materials, e.g. two or more polymers, polymer + filler, + reinforcement
- H05K1/0373—Organic insulating material consisting of two or more materials, e.g. two or more polymers, polymer + filler, + reinforcement containing additives, e.g. fillers
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K2201/00—Indexing scheme relating to printed circuits covered by H05K1/00
- H05K2201/02—Fillers; Particles; Fibers; Reinforcement materials
- H05K2201/0203—Fillers and particles
- H05K2201/0206—Materials
- H05K2201/0209—Inorganic, non-metallic particles
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K2201/00—Indexing scheme relating to printed circuits covered by H05K1/00
- H05K2201/02—Fillers; Particles; Fibers; Reinforcement materials
- H05K2201/0275—Fibers and reinforcement materials
- H05K2201/029—Woven fibrous reinforcement or textile
Definitions
- the present invention is compatible to obtain a thermosetting resin composition excellent in low thermal expansion, copper foil adhesion, heat resistance, flame resistance, heat resistance with copper (T-300), dielectric properties, and drilling workability.
- the present invention relates to a resin production method, a thermosetting resin composition, a prepreg, and a laminate.
- thermosetting resin composition has a cross-linked structure and exhibits high heat resistance and dimensional stability, and thus is widely used in the field of electronic equipment and the like.
- it is used as a printed wiring board on which wirings and circuit patterns are printed, a prepreg constituting a copper-clad laminate in which printed wiring boards are multilayered, and an interlayer insulating material.
- the copper foil peel strength is desirably 1.0 kN / m or more, and more desirably 1.2 kN / m or more.
- heat resistance with copper (T- 300) is preferably free from blistering for 30 minutes or more.
- the base material tends to be thinner, and it is necessary that the base material bend less during heat treatment. In order to reduce warpage, it is effective that the surface direction of the substrate has a low thermal expansion, and the linear expansion coefficient is preferably 7 ppm / ° C. or less, more preferably 5 ppm / ° C. or less.
- the base material is in a direction that requires more reliability, and the inner wall roughness of the drill hole during drilling is required to be small.
- the evaluation of the inner wall roughness of the drill hole is evaluated by the penetration property of the plated copper, and the maximum plating penetration length is desirably 20 ⁇ m or less, and more desirably 15 ⁇ m or less.
- the demand for high-speed response continues to increase, and it is desirable that the relative dielectric constant of the base material is 4.7 or less and the dielectric loss tangent is 0.010 or less.
- thermosetting resin composition used as the insulating resin is required to have a high level of properties because of the demand for higher density and high reliability of the laminated board material, and the resin composition satisfying all these characteristics is required. Development is underway. Moreover, due to recent environmental problems, mounting of electronic parts using lead-free solder and flame resistance using halogen-free are required, and therefore higher heat resistance and flame resistance than conventional ones are required. Furthermore, in order to improve the safety of the product and the working environment, there is a demand for a thermosetting resin composition that is composed only of low-toxic components and does not generate toxic gases.
- Cyanate compound which is a thermosetting resin
- Patent Documents 1, 2, and 3 disclose resin compositions that exhibit low thermal expansibility composed of a cyanate compound and an inorganic filler. However, since these exhibit low thermal expansibility, the amount of inorganic filler used is large, and when used as a copper-clad laminate or an interlayer insulating material, drill workability and formability are insufficient.
- Patent Documents 4 and 5 disclose thermosetting resins containing a cyanate resin and an aralkyl-modified epoxy resin as essential components in order to develop low thermal expansibility.
- the cyanate resin which is an essential component, is a resin with poor toughness and curing reactivity, improvement in curing reactivity and toughness is still insufficient, and when these are used as copper-clad laminates and interlayer insulation materials However, heat resistance, reliability, workability, etc. are insufficient.
- the object of the present invention is to solve the above-mentioned problems when using a cyanate compound that is a thermosetting resin, low thermal expansion, copper foil adhesion, heat resistance, flame resistance, and heat resistance with copper.
- a thermosetting resin composition excellent in all of dielectric properties and drill workability and a prepreg, a laminate and a wiring board using the same.
- the present invention is a compatibilizing resin (A1) obtained by reacting a cyanate compound and a siloxane resin having a hydroxyl group at the terminal, or further an epoxy resin at a specific reaction rate.
- thermosetting resin (A2) and an excellent thermosetting resin composition having the above-mentioned characteristics by using a resin composition containing fused silica (B) surface-treated with a trimethoxysilane compound Has been obtained, and the present invention has been completed.
- the present invention has been completed based on such findings. That is, the present invention provides the following method for producing a compatibilizing resin, a thermosetting resin composition, a prepreg, a laminate and a wiring board.
- a process for producing a compatibilizing resin having an imino carbonate structure and a triazine structure is produced by the following general formula (I) (a), a compound (b) having at least two cyanate groups in one molecule, and a compound having at least two epoxy groups in one molecule
- R 1 is each independently an alkylene group or alkyleneoxy group having 1 to 5 carbon atoms
- Ar 1 is each independently a single bond, an arylene group or an alkylene group having 1 to 5 carbon atoms; It is an integer of 100.
- thermosetting resin comprising a compatibilizing resin (A1) produced by the method 1 and a fused silica (B) surface-treated with a trimethoxysilane compound represented by the following formula (II): Resin composition.
- thermosetting resin composition containing a thermosetting resin (A2) and fused silica (B) surface-treated with a trimethoxysilane compound represented by the formula (II),
- the thermosetting resin (A2) is A product obtained by reacting a siloxane resin (a) having a hydroxyl group at the terminal represented by the general formula (I) with a compound (b) having at least two cyanate groups in one molecule in an organic solvent.
- thermosetting resin composition wherein the reaction rate of the compound (b) is 40 to 70 mol%. 4).
- a prepreg that has been B-staged after impregnating or coating the thermosetting resin composition of 2 or 3 above into a substrate. 5).
- a prepreg obtained by impregnating or coating a base material with the thermosetting resin composition of the present invention, and a laminate produced by laminating the prepreg, have low thermal expansion, copper foil adhesion, heat resistance. , Flame retardant, heat resistance with copper (T-300), dielectric properties, drilling workability, no environmental problems, and excellent product safety It is.
- the method for producing a compatibilizing resin (A1) according to the present invention includes a siloxane resin (a) having a hydroxyl group at the terminal represented by the following general formula (I), a compound having at least two cyanate groups in one molecule.
- component (B) and a compound (c) having at least two epoxy groups in one molecule (a) 10 to 50 parts by mass of component (a) to (c) per 100 parts by mass of the total amount of components (b) 40) to 80 parts by mass of component (c) and 10 to 50 parts by mass of component (c), in the presence of the organometallic salt (d), in an solvent selected from toluene, xylene and mesitylene at 80 to 120 ° C.
- a triazine ring formation reaction is performed, and the reaction rate of the component (b) is 30 to 70 mol%.
- R 1 is each independently an alkylene group or alkyleneoxy group having 1 to 5 carbon atoms
- Ar 1 is each independently a single bond, an arylene group or an alkylene group having 1 to 5 carbon atoms; It is an integer of 100.
- the iminocarbonation reaction is a reaction in which an iminocarbonate bond (—O— (C ⁇ NH) —O—) is generated by the addition reaction of a hydroxyl group and a cyanate group, and the triazine cyclization reaction is performed using a cyanate group.
- a three-dimensional network structure is formed by a reaction in which the cyanate group is trimerized to form a triazine ring.
- the compound (c) having at least two epoxy groups in one molecule is converted into a three-dimensional network structure.
- a compatibilized resin in which the components (a), (b) and (c) are uniformly dispersed is produced.
- the siloxane resin of component (a) used for the production of the compatibilizing resin (A1) of the present invention is not particularly limited as long as it is a siloxane resin containing a hydroxyl group having a structure represented by the above general formula (I).
- trade names X-22-1821 hydroxyl value: 35 KOHmg / g
- trade names X-22-1822 hydroxyl value: 20 KOHmg / g
- Shin-Etsu Chemical Co., Ltd. which are phenolic hydroxyl groups at both ends
- Toray -Trade name BY16-752A hydroxyl value: 30 KOHmg / g manufactured by Dow Corning Co., Ltd.
- trade name X-22-160AS (hydroxyl value: manufactured by Shin-Etsu Chemical Co., Ltd.) where both ends are alcoholic hydroxyl groups.
- 112 KOH mg / g) trade name KF-6001 (hydroxyl value: 62 KOH mg / g), trade name KF-6002 (hydroxyl value: 35 KOH mg / g), trade name KF-6003 (hydroxyl value: 20 KOH mg / g), trade name X -22-4015 (hydroxyl value: 27 KOH mg / g).
- Examples of the compound having at least two cyanate groups in one molecule of the component (b) used in the production of the compatibilizing resin (A1) include novolak type cyanate resin, bisphenol A type cyanate resin, and bisphenol E type cyanate. Resins, bisphenol F-type cyanate resins, tetramethylbisphenol F-type cyanate resins and the like can be mentioned, and one kind or two or more kinds can be mixed and used. Among these, bisphenol A type cyanate resins and novolak type cyanate resins represented by the following general formula (III) are particularly preferred from the viewpoints of dielectric properties, heat resistance, flame retardancy, low thermal expansibility, and low cost.
- N is 0 or an integer of 1 or more.
- n is the average number of repeats of the novolak cyanate resin, and is not particularly limited, but an average value of 0.1 to 30 is preferable. If n is 0.1 or more, crystallization does not become difficult to handle, and if it is 30 or less, the cured product does not become brittle.
- Examples of the compound having at least two epoxy groups in one molecule of the component (c) used for the production of the compatibilizing resin (A1) include bisphenol A, bisphenol F, biphenyl, novolac, Examples thereof include glycidyl ethers such as functional phenols, naphthalenes, alicyclics, and alcohols, glycidylamines, glycidyl esters, and the like, which can be used alone or in combination.
- naphthalene type epoxy resin naphthol aralkyl type epoxy resin, dihydroxynaphthalene aralkyl type epoxy resin, naphthol aralkyl cresol and naphthol aralkyl / cresol are used in terms of high rigidity, dielectric properties, heat resistance, flame resistance, moisture resistance and low thermal expansion.
- Naphthalene ring-containing epoxy resins such as polymerization type epoxy resins, biphenyl type epoxy resins, biphenyl group-containing epoxy resins such as biphenyl aralkyl type epoxy resins are preferred, and naphthol aralkyl type epoxy resins from the viewpoint of solubility in aromatic organic solvents, A naphthol aralkyl / cresol copolymer type epoxy resin and a biphenyl type epoxy resin are more preferable, and a biphenyl type epoxy resin represented by the following formula (IV) is particularly preferable because it is inexpensive and has a small epoxy equivalent and may contain a small amount.
- formula (IV) is particularly preferable because it is inexpensive and has a small epoxy equivalent and may contain a small amount.
- the raw material composition is 10 to 50 parts by mass of component (a) and 40 to 80 parts by mass of component (b) per 100 parts by mass of the total amount of components (a) to (c).
- the component (c) is reacted at 80 to 120 ° C. in a solvent selected from toluene, xylene and mesitylene in the presence of the organometallic salt (d) as 10 to 50 parts by mass, and the reaction rate of the component (b) is 30. ⁇ 70 mol%.
- the organometallic salt of component (d) serves as a reaction catalyst, and examples thereof include zinc naphthenate, cobalt naphthenate, tin octylate, and cobalt octylate.
- An amine-based or imidazole-based nitrogen atom-containing reaction catalyst is not preferable because a cured resin obtained is brittle and heat resistance and adhesiveness are lowered.
- the components (a) to (c) are uniformly dissolved in advance in a solvent selected from toluene, xylene and mesitylene, and the reaction temperature is 80 to 120 ° C.
- An iminocarbonation reaction and a triazine cyclization reaction are performed, and a pre-reaction is performed so that the reaction rate of the compound (b) having a cyanate group is 30 to 70 mol%.
- using an aromatic solvent selected from toluene, xylene and mesitylene in the reaction solvent A small amount of other solvent may be used if necessary, but the desired reaction does not proceed with the other solvent, and heat resistance and the like are lowered.
- benzene is highly toxic, and an aromatic solvent having a molecular weight larger than that of mesitylene is not preferable because it tends to be a residual solvent during prepreg production coating.
- the raw material composition in the production of the compatibilizing resin (A1) is (a) component 10 to 50 parts by mass, (b) component 40 to 80 parts by mass per 100 parts by mass of the total amount of components (a) to (c) c) 10 to 50 parts by mass of component, (a) 10 to 30 parts by mass, (b) 50 to 70 parts by mass, and (c) 10 to 40 parts by mass of component are preferable.
- component (a) is less than 10 parts by mass, the low thermal expansion in the surface direction of the obtained base material may be reduced, and when the component (a) exceeds 50 parts by mass, Chemical resistance may decrease.
- the compatibility of the obtained resin may be reduced, and when the component (b) exceeds 80 parts by mass, the low thermal expansion in the surface direction of the obtained substrate is obtained. May decrease.
- the component (c) is less than 10 parts by mass, the moisture and heat resistance may be reduced, and when the component (c) is more than 50 parts by mass, the copper foil adhesion and dielectric properties may be reduced.
- the amount of the organic metal salt (d) used is preferably 0.0001 to 0.004 parts by mass with respect to 100 parts by mass of the total amount of the components (a) to (c).
- a desired reaction rate can be achieved without requiring a long time for the reaction.
- reaction rate is not too fast and end point management becomes difficult.
- the reaction rate of the compound (b) having a cyanate group is determined by comparing the peak area of the compound having the cyanate group (b) at the start of the reaction with the peak area after reaction for a predetermined time by GPC measurement. It is calculated from the disappearance rate.
- the reaction temperature of this pre-reaction is 80 ⁇ 120 ° C., preferably 100 ⁇ 110 ° C.. If the reaction temperature is less than 80 ° C., the production time (reaction time) may be too long, and if it exceeds 120 ° C., a side reaction of the epoxy resin occurs, which may cause gelation.
- the reaction rate of the pre-reaction is such that the reaction rate (disappearance rate) of the compound (b) having a cyanate group is 30 to 70 mol%, preferably 40 to 68 mol%. If the reaction rate is less than 30 mol%, the resulting resin is not compatibilized, the resin separates and becomes cloudy, and the B-stage coated fabric is not manufactured.
- thermosetting resin composition When the reaction rate exceeds 70 mol%, the resulting thermosetting resin is insolubilized in the solvent, and the A-stage varnish (thermosetting resin composition) cannot be produced, or the gel time of the prepreg becomes too short. Formability may be reduced during pressing.
- thermosetting resin composition (I) of the present invention comprises a compatibilized resin (A1) produced by the above method and a fused silica (Silica treated with a trimethoxysilane compound represented by the following formula (II) ( B) is contained.
- the surface-treated fused silica of component (B) is obtained by surface-treating the fused silica using the trimethoxysilane compound represented by the above formula (II).
- component for example, after adding fused silica to a ketone organic solvent such as methyl ethyl ketone, methyl isobutyl ketone, cyclohexanone, or alcohol organic solvent such as ethylene glycol monomethyl ether, propylene glycol monomethyl ether, and the like,
- the trimethoxysilane compound represented by the formula (II) is added and subjected to surface treatment (wet treatment) at 60 to 120 ° C. with stirring for about 0.5 to 5 hours.
- the fused silica is also commercially available from Admatechs and others, for example, trade names SC-2050KNK and SC-2050HNK manufactured by Admatechs.
- the amount of the component (B) fused silica used is preferably 10 to 300 parts by weight, more preferably 100 to 250 parts by weight, based on 100 parts by weight of the compatibilizing resin (A1) in terms of solid content.
- the amount is preferably 150 to 250 parts by mass.
- thermosetting resin composition (II) of the present invention comprises a thermosetting resin (A2) and a thermosetting resin containing fused silica (B) surface-treated with a trimethoxysilane compound represented by the formula (II).
- a resin composition comprising:
- the thermosetting resin (A2) is A product obtained by reacting a siloxane resin (a) having a hydroxyl group at the terminal represented by the general formula (I) with a compound (b) having at least two cyanate groups in one molecule in an organic solvent.
- thermosetting resin composition having a reaction rate of the compound (b) of 40 to 70 mol%.
- the siloxane resin (a) having a hydroxyl group at the terminal used for the thermosetting resin (A2), the compound (b) having at least two cyanate groups in one molecule, and the organic solvent are the compatibilizing resins.
- the thing similar to what is used for manufacture of (A1) is used.
- the thing similar to what is used for the thermosetting resin composition (I) is used for the fused silica (B) used for the thermosetting resin composition (II).
- the blending amount of the siloxane resin (a) and the compound (b) used for the thermosetting resin (A2) is preferably as follows. That is, the blending amount of the siloxane resin (a) is in the range of 10 to 70 parts by mass with respect to 100 parts by mass of the total of the siloxane resin (a) and the compound (b). The compounding amount of the compound (b) is in the range of 30 to 90 parts by mass. When the amount of the siloxane resin (a) is in the range of 10 to 70 parts by mass, a sufficiently low thermal expansion property in the surface direction of the substrate to which the thermosetting resin (A) is applied can be obtained.
- the amount of the siloxane resin (a) is in the range of 10 to 70 parts by mass, sufficient heat resistance and chemical resistance can be obtained.
- the compounding amount of the compound (b) is in the range of 30 to 90 parts by mass, sufficient low thermal expansion in the surface direction of the substrate obtained using the thermosetting resin (A) can be obtained. Moreover, sufficient heat resistance is obtained.
- thermosetting resin (A2) is prepared by mixing the siloxane resin (a) and the compound (b) in an organic solvent so that the reaction rate (disappearance rate) of the compound (b) is 40 to 70 mol%. It is preferable to pre-react in advance. Examples of the pre-reaction include the above-mentioned imino carbonate reaction and triazine cyclization reaction. When the reaction rate of the compound (b) is 40 to 70 mol%, the compatibility between the thermosetting resin (A2) obtained by reacting the siloxane resin (a) and the compound (b) with a general-purpose organic solvent is sufficient. Since sufficient sclerosis
- thermosetting resin (A) is hard to crystallize, it is easy to make a varnish, and a moldability is good. Further, the occurrence of tack can be reduced in a state where glass fiber or the like is impregnated with a solution of the thermosetting resin (A) and is semi-cured (so-called B stage state). In addition, the reaction rate of a compound (b) is calculated
- the peak of the cyanate resin that appears in the vicinity of a predetermined holding time between a solution before the reaction in which the siloxane resin (a) and the compound (b) are mixed and a solution after the reaction of the solution. Compare areas. Disappearance rate of the peak area of the solution after the reaction to the peak area of the reaction before the solution is equivalent to the reaction rate.
- the blending amount of the fused silica (B) in the thermosetting resin composition (II) is preferably 10 to 300 parts by mass with respect to 100 parts by mass of the thermosetting resin (A) in terms of solid content. More preferably, the content is set to ⁇ 250 parts by mass, and particularly preferably 150 to 250 parts by mass. If the amount is 10 to 300 parts by mass, sufficient rigidity of the substrate, heat and heat resistance, flame resistance, resistance to erosion by the plating solution, and the like can be obtained.
- thermosetting resin composition (I) and the thermosetting resin composition (II) (hereinafter collectively referred to as the thermosetting resin composition of the present invention)
- inorganic fillers other than the component (B) ( C) may be used.
- the inorganic filler (C) include crushed silica, mica, talc, short glass fiber or fine powder, hollow glass, calcium carbonate, quartz powder, and metal hydrate.
- metal hydrates such as aluminum hydroxide and magnesium hydroxide are preferable from the viewpoint of low thermal expansion, high elasticity, heat resistance, and flame retardancy.
- metal hydrates having a thermal decomposition temperature of 300 ° C.
- boehmite type aluminum hydroxide such as boehmite type aluminum hydroxide (AlOOH), or gibbsite type aluminum hydroxide, because both high heat resistance and flame retardancy are compatible. More preferred is a compound in which the thermal decomposition temperature of (Al (OH) 3) is adjusted to 300 ° C. or higher by heat treatment, magnesium hydroxide, etc., particularly inexpensive, a particularly high thermal decomposition temperature of 350 ° C. or higher, and a high resistance to resistance. Boehmite type aluminum hydroxide (AlOOH) having chemical properties is particularly preferable.
- the amount of the inorganic filler (C) used in the thermosetting resin composition (I) is preferably 0 to 200 parts by mass with respect to 100 parts by mass of the compatibilizing resin (A1) in terms of solid content. more preferably to to 150 parts by weight, particularly preferably 50 to 150 parts by weight. If it is 10 parts by mass or more, the flame retardancy will not be insufficient, and if it is 200 parts by mass or less, chemical resistance such as plating solution resistance and moldability will not be reduced.
- the amount of the inorganic filler (C) used in the thermosetting resin composition (II) is preferably 10 to 200 parts by mass based on 100 parts by mass of the thermosetting resin (A2) in terms of solid content. more preferably 10 to 150 parts by mass, particularly preferably 50 to 150 parts by mass. If it is in the range of 10 to 200 parts by mass, sufficient flame retardancy, resistance to erosion by the plating solution, and formability can be obtained.
- thermosetting resin composition of the present invention it is desirable to use a curing accelerator for improving heat resistance, flame retardancy, copper foil adhesion, etc.
- examples of the curing accelerator include zinc naphthenate, Examples include organic metal salts such as cobalt naphthenate, tin octylate, and cobalt octylate, imidazoles and derivatives thereof, tertiary amines, and quaternary ammonium salts.
- an inorganic flame retardant aid (D) other than the components (B) and (C) can be arbitrarily used.
- halogen-containing flame retardants containing bromine and chlorine and metal hydroxides having a thermal decomposition temperature of less than 300 ° C. are not suitable for the purpose of the present invention.
- inorganic flame retardant aids (D) include triphenyl phosphate, tricresyl phosphate, trisdichloropropyl phosphate, phosphoric ester compounds, phosphazenes, phosphorous flame retardants such as red phosphorus, antimony trioxide, molybdic acid
- examples include inorganic flame retardant aids such as zinc.
- an inorganic flame retardant aid in which zinc molybdate is supported on an inorganic filler such as talc is a particularly preferred inorganic flame retardant aid because it significantly improves not only the flame retardancy but also the drill workability.
- the amount of zinc molybdate used is preferably 5 to 20 parts by mass with respect to 100 parts by mass of the compatibilizing resin (A1) or thermosetting resin (A2). By setting it to 5 parts by mass or more, flame retardancy and drilling workability are improved, and by setting it to 20 parts by mass or less, the gel time of the varnish becomes too short and the moldability is improved when a laminate is formed by pressing. There is no decline.
- thermosetting resin composition of the present invention can optionally contain a known thermoplastic resin, elastomer, flame retardant, and organic filler.
- thermoplastic resin include polytetrafluoroethylene, polyethylene, polypropylene, polystyrene, polyphenylene ether resin, phenoxy resin, polycarbonate resin, polyester resin, polyamide resin, polyimide resin, xylene resin, petroleum resin, and silicone resin.
- elastomer include polybutadiene, ABS resin, epoxy-modified polybutadiene, maleic anhydride-modified polybutadiene, phenol-modified polybutadiene, and carboxy-modified acrylonitrile.
- flame retardant include the inorganic filler (C).
- organic fillers include organic powders such as silicone powder, polytetrafluoroethylene, polyethylene, polypropylene, polystyrene, and polyphenylene ether.
- an ultraviolet absorber for the thermosetting resin composition of the present invention, an ultraviolet absorber, an antioxidant, a photopolymerization initiator, a fluorescent whitening agent, an adhesion improver, and the like can be arbitrarily added.
- UV absorbers such as benzotriazoles, antioxidants such as hindered phenols and styrenated phenols, photopolymerization initiators such as benzophenones, benzyl ketals, and thioxanthones, and fluorescence such as stilbene derivatives.
- Examples include brighteners, urea compounds such as urea silane, and adhesion improvers such as silane coupling agents.
- the prepreg of the present invention is obtained by impregnating or coating the above-described thermosetting resin composition of the present invention in a base material and then forming a B-stage.
- the prepreg of the present invention will be described in detail. That is, the prepreg of the present invention can be produced by impregnating or coating the base material with the thermosetting resin composition of the present invention and semi-curing (B-stage) by heating or the like. .
- the base material used for the prepreg known materials used for various types of laminated sheets for electrical insulating materials can be used.
- the material include inorganic fibers such as E glass, D glass, S glass, and Q glass, organic fibers such as polyimide, polyester, and tetrafluoroethylene, and mixtures thereof.
- These base materials have, for example, shapes such as woven fabric, non-woven fabric, robink, chopped strand mat, and surfacing mat, but the material and shape are selected depending on the intended use and performance of the molded product, and if necessary, A single material or two or more materials and shapes can be combined.
- the thickness of the substrate is not particularly limited.
- a substrate having a thickness of about 0.03 to 0.5 mm can be used, and the substrate is surface-treated with a silane coupling agent or the like or mechanically subjected to fiber opening treatment. Is suitable from the viewpoints of heat resistance, moisture resistance and processability.
- the prepreg of the present invention is usually used after impregnating or coating the base material so that the amount of the thermosetting resin composition attached to the base material is 20 to 90% by mass as the resin content of the prepreg after drying. It can be obtained by heating and drying at a temperature of 100 to 200 ° C. for 1 to 30 minutes and semi-curing (B stage).
- the laminate of the present invention is formed using the prepreg of the present invention, and can be formed by laminate molding using the prepreg described above. That is, the laminate of the present invention can be produced by laminating the above-described prepreg, for example, by stacking 1 to 20 sheets, and laminating a metal foil such as copper and aluminum on one or both sides thereof.
- the metal foil is not particularly limited as long as it is used for electrical insulating material applications.
- As the molding conditions for example, a method of a laminated plate for an electrical insulating material and a multilayer plate can be applied. Molding can be performed in a range of ⁇ 100 kg / cm 2 (0.2 ⁇ 10 MPa) and heating time of 0.1 ⁇ 5 hours. Further, the prepreg of the present invention and the inner layer wiring board can be combined and laminated to produce a multilayer board.
- the present invention will be described in more detail with reference to the following examples, but these examples do not limit the present invention in any way.
- the copper clad laminated board obtained in the following examples and comparative examples was measured by the following method and evaluated.
- Copper foil adhesion (copper foil peel strength) A 1 cm wide copper foil was formed by immersing the copper clad laminate in a copper etching solution to produce an evaluation substrate, and the adhesion (peel strength) of the copper foil was measured using a tensile tester.
- Tg Glass transition temperature
- solder heat resistance A 5 cm square evaluation board from which the copper foil has been removed by immersing a copper clad laminate in a copper etching solution is prepared and 121 ° C. using a pressure cooker test apparatus manufactured by Hirayama Seisakusho. After performing the pressure-cooker treatment for up to 4 hours under the condition of 2 atm, the evaluation substrate was immersed in a solder bath at a temperature of 288 ° C. for 20 seconds, and then the solder heat resistance was evaluated by observing the appearance.
- a test piece cut out to a length of 127 mm and a width of 12.7 mm was prepared from an evaluation substrate obtained by removing a copper foil by immersing a copper clad laminate in a copper etching solution, and a UL94 test method ( Evaluation was made according to V method.
- Dielectric properties (dielectric constant and dielectric loss tangent)
- the obtained copper-clad laminate was immersed in a copper etching solution to prepare an evaluation substrate from which the copper foil was removed, and a relative dielectric constant measuring apparatus (product name: HP4291B) manufactured by Hewlett-Packard Company was used at a frequency of 1 GHz. The relative dielectric constant and dielectric loss tangent of were measured.
- Production Example 1 Production of compatibilized resin (A1-1) A bisphenol A type cyanate resin (manufactured by Lonza Japan Co., Ltd.) was placed in a 3 liter reaction vessel equipped with a thermometer, a stirrer, and a reflux condenser and capable of heating and cooling.
- a bisphenol A type cyanate resin manufactured by Lonza Japan Co., Ltd.
- Trade name Primaset BADCy 600.0 g, siloxane resin represented by the following formula (V) (manufactured by Shin-Etsu Chemical Co., Ltd .; trade name X-22-1821, hydroxyl equivalent: 1,600): 200.0 g, biphenyl type epoxy Resin (manufactured by Japan Epoxy Resin; trade name YX-4000, epoxy equivalent: 186): 200.0 g and toluene: 1000.0 g were charged. Next, the temperature was raised to 120 ° C.
- V siloxane resin represented by the following formula (V) (manufactured by Shin-Etsu Chemical Co., Ltd .; trade name X-22-1821, hydroxyl equivalent: 1,600): 200.0 g, biphenyl type epoxy Resin (manufactured by Japan Epoxy Resin; trade name YX-4000, epoxy equivalent: 186): 200.0 g and toluene: 1000.0 g were charged. Next, the temperature was raised to 120
- the peak area of the bisphenol A type cyanate resin which is a synthetic raw material and the elution time appears around 12.4 minutes Compared with the peak area of the bisphenol A type cyanate resin at the time, the disappearance rate of the peak area [reaction rate of the component (b)] was 68%.
- the peak of the thermosetting resin product appearing around about 10.9 minutes and around 8.0 to 10.0 was confirmed.
- the reaction solution taken out in a small amount was dropped into a mixed solvent of methanol and benzene (mixing weight ratio 1: 1) and reprecipitated to take out the purified solid, and FT-IR measurement was performed. imino carbonates due to group 1700 cm -1 vicinity of the peak, also around 1560 cm -1 due to the triazine ring, and 1380cm strong peak to check the vicinity of -1, compatibilizing resin (A1-1) is produced I confirmed.
- Production Example 2 Production of compatibilizing resin (A1-2) A novolak-type cyanate resin (manufactured by Lonza Japan Co., Ltd.) was placed in a reaction vessel having a volume of 3 liters that can be heated and cooled with a thermometer, a stirrer, and a reflux condenser.
- Trade name Primaset PT-15 mass average molecular weight 500 to 1,000
- 800.0 g and a siloxane resin represented by the following formula (VI) (manufactured by Shin-Etsu Chemical Co., Ltd .; trade name KF-6003, hydroxyl group equivalent: 2800): 100.0 g, naphthol aralkyl-cresol copolymer epoxy resin (manufactured by Nippon Kayaku Co., Ltd .; trade name NC-7000L, epoxy equivalent; 230): 100.0 g and toluene: 1000.0 g were added. Next, the temperature was raised to 120 ° C.
- the peak area of the novolac-type cyanate resin which is a synthetic raw material with an elution time of about 12.1 minutes.
- the disappearance rate of the peak area [reaction rate of the component (b)] was 43%.
- the peak of the thermosetting resin product appearing around about 10.9 minutes and around 8.0 to 10.0 was confirmed.
- the reaction solution taken out in a small amount was dropped into a mixed solvent of methanol and benzene (mixing weight ratio 1: 1) and reprecipitated to take out the purified solid, and FT-IR measurement was performed. imino carbonates due to group 1700 cm -1 vicinity of the peak, also around 1560 cm -1 due to the triazine ring, and 1380cm strong peak to check the vicinity of -1, compatibilizing resin (A1-2) is produced I confirmed.
- VII siloxane resin represented by the following formula (VII) (manufactured by Shin-Etsu Chemical Co., Ltd .; product name X-22-160AS, hydroxyl group equivalent: 500) ): 100.0 g, biphenyl aralkyl type epoxy resin (manufactured by Nippon Kayaku Co., Ltd
- the disappearance rate of the peak area [reaction rate of the component (b)] was 43%.
- the peak of the thermosetting resin product appearing around about 10.9 minutes and around 8.0 to 10.0 was confirmed.
- the reaction solution taken out in a small amount was dropped into a mixed solvent of methanol and benzene (mixing mass ratio 1: 1) and reprecipitated to take out the purified solid, and FT-IR measurement was performed. imino carbonates due to group 1700 cm -1 vicinity of the peak, also around 1560 cm -1 due to the triazine ring, and 1380cm strong peak to check the vicinity of -1, compatibilizing resin (A1-3) is produced I confirmed.
- Production Example 4 Production of compatibilizing resin (A1-4) A bisphenol A type cyanate resin (manufactured by Lonza Japan Co., Ltd.) was placed in a 3 liter reaction vessel equipped with a thermometer, a stirrer, and a reflux condenser.
- Trade name Primaset BADCy 400.0 g and siloxane resin represented by the above formula (V) (manufactured by Shin-Etsu Chemical Co., Ltd .; trade name X-22-1821, hydroxyl equivalent: 1,600): 500.0 g, naphthalene Type epoxy resin (Dainippon Ink Chemical Co., Ltd .; trade name Epicron HP-4032, epoxy equivalent: 150): 100.0 g and toluene: 1000.0 g were charged. Next, the temperature was raised to 120 ° C.
- the peak area of the bisphenol A type cyanate resin which is a synthetic raw material and the elution time appears around 12.4 minutes Compared with the peak area of the bisphenol A type cyanate resin at the time, the disappearance rate of the peak area [reaction rate of the component (b)] was 55%.
- the peak of the thermosetting resin product appearing around about 10.9 minutes and around 8.0 to 10.0 was confirmed.
- the reaction solution taken out in a small amount was dropped into a mixed solvent of methanol and benzene (mixing weight ratio 1: 1) and reprecipitated to take out the purified solid, and FT-IR measurement was performed. imino carbonates due to group 1700 cm -1 vicinity of the peak, also around 1560 cm -1 due to the triazine ring, and 1380cm strong peak to check the vicinity of -1, compatibilizing resin (A1-4) is produced I confirmed.
- Production Example 5 Production of fused silica (B-1) surface-treated with a trimethoxysilane compound
- a fused silica Admatechs Co., Ltd .; trade name SO-25R
- 700.0 g and propylene glycol monomethyl ether 1000.0 g were mixed and stirred with N-phenyl-3-aminopropyltrimethoxysilane (Shin-Etsu Chemical Co., Ltd .; product) Name KBM-573): 7.0 g was added.
- the temperature was raised to 80 ° C., reacted at 80 ° C.
- Comparative production example 1 Production of (resin (A1-5): (b) component reaction rate 18%) In a reaction vessel with a volume of 3 liters capable of heating and cooling with a thermometer, a stirrer and a reflux condenser, Bisphenol A type cyanate resin (Lonza Japan Co., Ltd .; trade name Primaset BADCy): 600.0 g and siloxane resin represented by the above formula (V) (Shin-Etsu Chemical Co., Ltd .; trade name X-22-1821, hydroxyl equivalent: 1 , 600): 200.0 g and biphenyl type epoxy resin (manufactured by Japan Epoxy Resin; trade name YX-4000, epoxy equivalent; 186): 200.0 g and toluene: 1000.0 g.
- Bisphenol A type cyanate resin Lionza Japan Co., Ltd .; trade name Primaset BADCy
- siloxane resin represented by the above formula (V) Shin-Etsu Chemical Co.
- the peak area of the bisphenol A type cyanate resin which is a synthetic raw material and the elution time appears around 12.4 minutes Compared with the peak area of the bisphenol A type cyanate resin at the time, the disappearance rate of the peak area [reaction rate of the component (b)] was 18%. In addition, a precipitate was formed in the solution by crystallization the next day.
- Comparative production example 2 Production of (resin (A1-6): reaction rate of component (b) 76%) In a reaction vessel having a volume of 3 liters capable of being heated and cooled with a thermometer, a stirrer and a reflux condenser, Bisphenol A type cyanate resin (Lonza Japan Co., Ltd .; trade name Primaset BADCy): 600.0 g, siloxane resin represented by the above formula (V) (Shin-Etsu Chemical Co., Ltd .; trade name X-22-1821, hydroxyl group equivalent; 1, 600): 200.0 g, biphenyl type epoxy resin (manufactured by Japan Epoxy Resin; trade name YX-4000, epoxy equivalent; 186): 200.0 g and toluene: 1000.0 g were charged.
- Bisphenol A type cyanate resin Lionza Japan Co., Ltd .; trade name Primaset BADCy
- V siloxane resin represented by the above formula (V) (Shin-E
- the peak area of the bisphenol A type cyanate resin which is a synthetic raw material and the elution time appears around 12.4 minutes Compared with the peak area of the bisphenol A type cyanate resin at the time, the disappearance rate of the peak area [reaction rate of the component (b)] was 76%.
- Comparative production example 3 Production of (resin (A1-7): (b) component reaction rate 53%, (c) no component) Heating and cooling volume 2 with thermometer, stirrer and reflux condenser 2 In a reaction vessel of 1 liter, bisphenol A type cyanate resin (Lonza Japan Co., Ltd .; trade name Primaset BADCy): 600.0 g and a siloxane resin represented by the above formula (V) (Shin-Etsu Chemical Co., Ltd .; trade name X-22- 1821, hydroxyl equivalent: 1,600): 200.0 g and toluene: 800.0 g. Next, the temperature was raised to 120 ° C.
- bisphenol A type cyanate resin Lionza Japan Co., Ltd .; trade name Primaset BADCy
- V siloxane resin represented by the above formula (V) (Shin-Etsu Chemical Co., Ltd .; trade name X-22- 1821, hydroxyl equivalent: 1,600): 200.0 g
- the peak area of the bisphenol A type cyanate resin which is a synthetic raw material and the elution time appears around 12.4 minutes Compared with the peak area of the bisphenol A type cyanate resin at the time, the disappearance rate of the peak area [reaction rate of the component (b)] was 53%.
- thermosetting resin (A2-1) In a reaction vessel having a thermometer, a stirrer, and a reflux condenser with a capacity of 3 liters that can be heated and cooled, the above formula (V) ) (Made by Shin-Etsu Chemical Co., Ltd .; trade name X-22-1821, hydroxyl equivalent: 1,600 g / eq.): 500 g and toluene: 1000 g, and bisphenol A type cyanate resin (b) as a compound (b) Made by Lonza Japan Co., Ltd .; trade name Arocy B-10): 500, heated while stirring, and after reaching 120 ° C., 0.01 g of 8% by mass mineral spirit solution of zinc naphthenate was added, The mixture was refluxed at about 115 to 125 ° C. for 4 hours and then cooled to room temperature (25 ° C.) to obtain a thermosetting resin (A2-1) solution.
- V thermosetting resin
- the reaction rate of the compound (b) in the thermosetting resin (A-1) was 65 mol%.
- the peak of the thermosetting resin product appearing around about 10.9 minutes and around 8.0 to 10.0 was confirmed.
- the reaction solution taken out in a small amount was dropped into a mixed solvent of methanol and benzene (mixing mass ratio 1: 1) and reprecipitated to take out the purified solid, and FT-IR measurement was performed.
- imino carbonate group peak near 1700 cm -1 due to, also, around 1560 cm -1 due to the triazine ring, and 1380cm can strong peaks confirmed the vicinity of -1, a thermosetting resin (A2-1) is produced I confirmed.
- thermosetting resin product appearing around about 10.9 minutes and around 8.0 to 10.0 was confirmed. Further, the reaction solution taken out in a small amount was dropped into a mixed solvent of methanol and benzene (mixing mass ratio 1: 1) and reprecipitated to take out the purified solid, and FT-IR measurement was performed. imino carbonate group peak near 1700 cm -1 due to, also, around 1560 cm -1 due to the triazine ring, and 1380cm can strong peaks confirmed the vicinity of -1, a thermosetting resin (A2-2) is produced I confirmed.
- thermosetting resin (A2-3) A novolak-type cyanate resin (compound (b)) was added to a reaction vessel having a volume of 3 liters that can be heated and cooled with a thermometer, a stirrer, and a reflux condenser. Lonza Japan Co., Ltd.
- thermosetting resin product appearing around about 10.9 minutes and around 8.0 to 10.0 was confirmed. Further, the reaction solution taken out in a small amount was dropped into a mixed solvent of methanol and benzene (mixing mass ratio 1: 1) and reprecipitated to take out the purified solid, and FT-IR measurement was performed. imino carbonate group peak near 1700 cm -1 due to, also, around 1560 cm -1 due to the triazine ring, and 1380cm can strong peaks confirmed the vicinity of -1, a thermosetting resin (A2-3) is produced I confirmed.
- thermosetting resin (A2-4) Bisphenol A type cyanate resin (Lonza Japan Co., Ltd.) was placed in a 3 liter reaction vessel equipped with a thermometer, a stirrer and a reflux condenser and capable of heating and cooling.
- thermosetting resin (A2-4) The solution of the thermosetting resin (A2-4) is the precipitate by crystallization occurred.
- GPC measurement polystyrene conversion, eluent: tetrahydrofuran
- thermosetting resin A 2-5) A bisphenol A type cyanate resin (Lonza Japan Co., Ltd.) was placed in a 3 liter reaction vessel equipped with a thermometer, a stirrer and a reflux condenser.
- the solution of the thermosetting resin (A2-5) has turbidity caused by insoluble components.
- a solution before the reaction in which the siloxane resin (a) and the compound (b) were blended and a solution after the reaction were taken out little by little, and GPC measurement (polystyrene conversion, eluent: tetrahydrofuran) was performed on each.
- thermosetting resin A bisphenol A type cyanate resin (Lonza Japan Co., Ltd.) was placed in a 3 liter reaction vessel equipped with a thermometer, a stirrer, and a reflux condenser and capable of heating and cooling.
- the reaction rate of the compound (b) in the thermosetting resin (A2-6) was 65 mol%.
- the peak of the thermosetting resin product appearing around about 10.9 minutes and around 8.0 to 10.0 was confirmed.
- the reaction solution taken out in a small amount was dropped into a mixed solvent of methanol and benzene (mixing mass ratio 1: 1) and reprecipitated to take out the purified solid, and FT-IR measurement was performed. imino carbonates due to group 1700 cm -1 vicinity of the peak, also confirmed around 1560 cm -1 due to the triazine ring, and a strong peak at around 1380 cm -1.
- thermosetting resin (A2-7) A bisphenol A type cyanate resin (Lonza Japan Co., Ltd.) was placed in a 3 liter reaction vessel equipped with a thermometer, a stirrer, and a reflux condenser and capable of heating and cooling.
- reaction solution taken out in a small amount was dropped into a mixed solvent of methanol and benzene (mixing mass ratio 1: 1) and reprecipitated to take out the purified solid, and FT-IR measurement was performed. around 1560 cm -1 due to the triazine ring, and confirmed the strong peak around 1380 cm -1.
- Fused silica (B-2): Fused silica surface-treated with 1.0% by mass of N-phenyl-3-aminopropyltrimethoxysilane based on fused silica (manufactured by Admatech; trade name SC-2050KNK) , Diluent solvent; methyl isobutyl ketone)
- Fused silica (B-3): Fused silica surface-treated with 1.0% by mass of N-phenyl-3-aminopropyltrimethoxysilane based on fused silica (manufactured by Admatech; trade name SC-2050HNK, diluent solvent; Cyclohexanone)
- Fused silica (B-4): fused silica (Admatechs Co., Ltd.; trade name SO-25R) Fused silica (B-5): 1.0% by mass of fused silica represented by the following formula (VIII) and surface-treated with ⁇ -glycidoxyprop
- Inorganic filler AlOOH: Boehmite type aluminum hydroxide (manufactured by Kawai Lime Co., Ltd .; trade name BMT-3L, thermal decomposition temperature: 400 ° C.)
- Inorganic flame retardant assistant KG-1100: Inorganic flame retardant assistant with zinc molybdate supported on talc (manufactured by Sherwin Williams; trade name Chemguard 1100) Curing accelerator: 8% by mass mineral spirit solution of zinc naphthenate Epoxy resin (YX-4000) used in Comparative Example 3: Biphenyl type epoxy resin (manufactured by Japan Epoxy Resin; trade name YX-4000, epoxy equivalent; 186)
- Comparative Examples 1-3 In Table 2 could not be evaluated performance of the laminate for the following reasons. Comparative Example 1: A thermosetting resin precipitated and a varnish could not be produced. Comparative Example 2: The moldability was poor and a laminate could not be produced. Comparative Example 3: The resin separated and a prepreg and a laminate could not be produced. In Table 4, in Comparative Examples 6 to 7, the thermosetting resin composition could not be evaluated because the thermosetting resin (A) was precipitated and the varnish could not be produced.
- Examples 1 to 6 of the present invention have copper foil peel strength, crow transition temperature (Tg), solder heat resistance, low thermal expansion, flame resistance, and heat resistance with copper (T- 300), which is excellent in all of low dielectric properties, low dielectric loss tangent properties, and drill workability, and satisfies the reference values described in the background art.
- Comparative Examples 1 to 5 are copper foil peel strength, crow transition temperature (Tg), solder heat resistance, low thermal expansion, flame resistance, and heat resistance with copper (T-300). ), None of the low dielectric properties, low dielectric loss tangent properties, and drillability are all inferior.
- Examples 7 to 10 of the present invention have copper foil peel strength, crow transition temperature (Tg), solder heat resistance, low thermal expansion, flame resistance, copper It can be seen that it has excellent heat resistance (T-300), low dielectric properties, low dielectric loss tangent, and drillability, and satisfies the above-mentioned standard value.
- Tg copper foil peel strength
- Tg crow transition temperature
- T-300 heat resistance with copper
- T-300 low dielectric properties, low dielectric loss tangent
- All drill workability is below the standard value.
- thermosetting resin composition of the present invention is not only excellent in solder heat resistance and flame retardancy, but also has the copper foil adhesion (copper foil peel strength) described in the background art, with copper. All the properties of heat resistance (T-300), drill workability, relative permittivity, and dielectric loss tangent have reached the level required for recent high density and high reliability. Therefore, by using the thermosetting resin composition of the present invention, high density and high reliability of the wiring board required today are achieved, and the thermosetting resin composition of the present invention is manufactured for electronic devices and the like. Can be widely used.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Textile Engineering (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Inorganic Chemistry (AREA)
- Reinforced Plastic Materials (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
例えば、銅張積層板では、プリント配線及び回路パターンが微細化された場合であっても、プリント配線及び回路パターンを構成する導体層が剥がれないようなピール強度が求められる。
また、近年の環境問題から、鉛フリーはんだによる電子部品の搭載やハロゲンフリーによる難燃化が要求され、そのため従来のものよりも高い耐熱性及び難燃性が必要とされる。さらに、製品の安全性や作業環境の向上化のため、毒性の低い成分のみで構成され、毒性ガス等が発生しない熱硬化性樹脂組成物が望まれている。
このため、シアネート化合物と無機充填剤からなる低熱膨張性を発現させる樹脂組成物が特許文献1、2および3に開示されている。しかし、これらは低熱膨張性を発現させるため無機充填剤の配合使用量が多く、銅張積層板や層間絶縁材料として使用した場合にドリル加工性や成形性が不足する。
即ち本発明は、以下の相容化樹脂の製造方法、熱硬化性樹脂組成物、プリプレグ、積層板及び配線板を提供するものである。
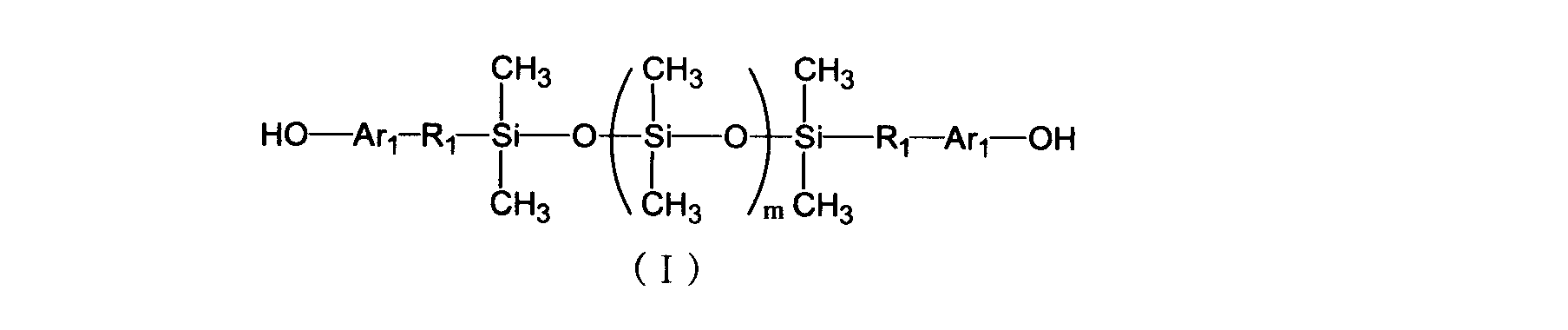
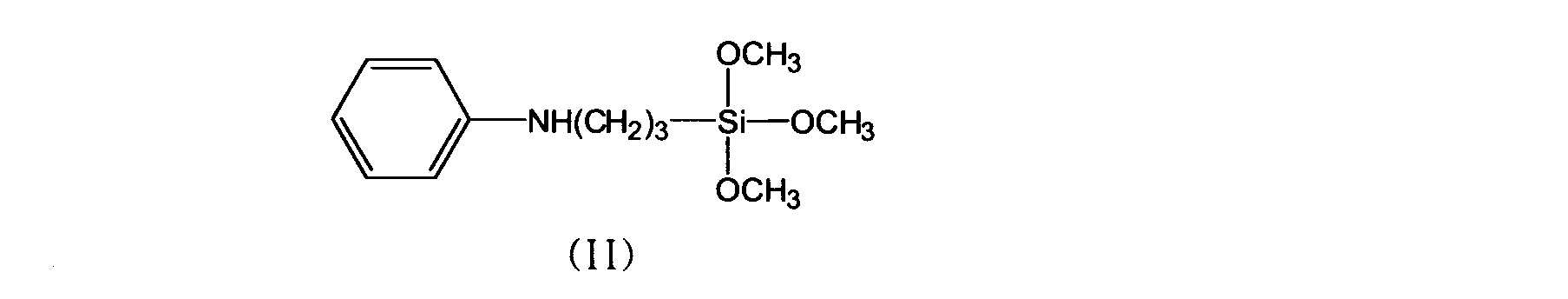
前記熱硬化性樹脂(A2)は、
一般式(I)で示される末端に水酸基を有するシロキサン樹脂(a)と、1分子中に少なくとも2個以上のシアネート基を有する化合物(b)とが有機溶媒中で反応して得られたものであり、
該シロキサン樹脂(a)と該化合物(b)との総和100質量部に対し、該シロキサン樹脂(a)10~70質量部及び該化合物(b)30~90質量部が含まれており、
該化合物(b)の反応率が40~70モル%である
ことを特徴とする熱硬化性樹脂組成物。
4.上記2又は3の熱硬化性樹脂組成物を基材中に含侵又は塗工した後、Bステージ化したプリプレグ。
5.上記4のプリプレグを用いて形成された積層板。
先ず、本発明の相容化樹脂(A1)の製造方法は、下記一般式(I)で示される末端に水酸基を有するシロキサン樹脂(a)、1分子中に少なくとも2個のシアネート基を有する化合物(b)及び1分子中に少なくとも2個のエポキシ基を有する化合物(c)を、(a)~(c)成分の合計量100質量部当たり、(a)成分10~50質量部、(b)成分40~80質量部、(c)成分10~50質量部として、有機金属塩(d)の存在下、トルエン、キシレン及びメシチレンから選ばれる溶媒中で80~120℃でイミノカーボネート化反応及びトリアジン環形成反応をさせ、(b)成分の反応率が30~70モル%であることを特徴とする方法である。
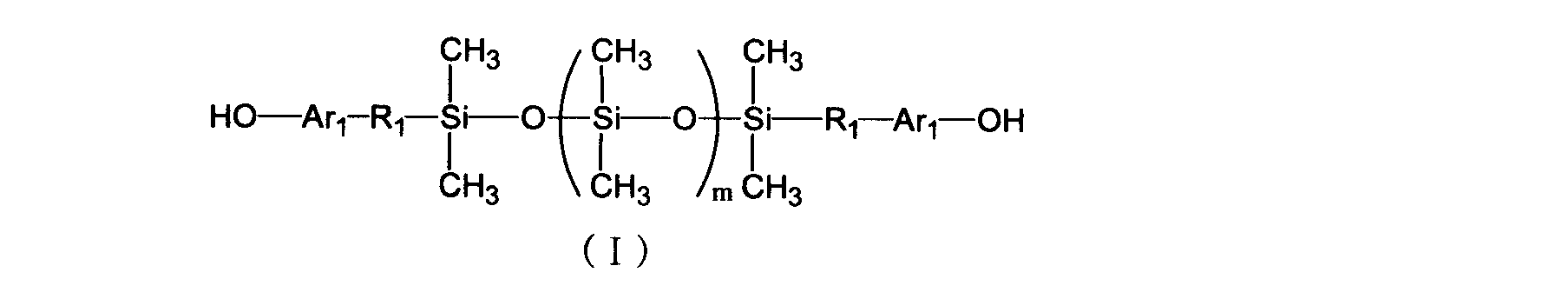

これらの中で、高剛性、誘電特性、耐熱性、難燃性、耐湿性及び低熱膨張性の点からナフタレン型エポキシ樹脂、ナフトールアラルキル型エポキシ樹脂、ジヒドロキシナフタレンアラルキル型エポキシ樹脂、ナフトールアラルキル・クレゾール共重合型エポキシ樹脂等のナフタレン環含有エポキシ樹脂、ビフェニル型エポキシ樹脂、ビフェニルアラルキル型エポキシ樹脂等のビフェニル基含有エポキシ樹脂が好ましく、芳香族系有機溶剤への溶解性の点からナフトールアラルキル型エポキシ樹脂、ナフトールアラルキル・クレゾール共重合型エポキシ樹脂、ビフェニル型エポキシ樹脂がより好ましく、安価であることやエポキシ当量が小さく少量の配合でよいことから、下記式(IV)に示すビフェニル型エポキシ樹脂が特に好ましい。
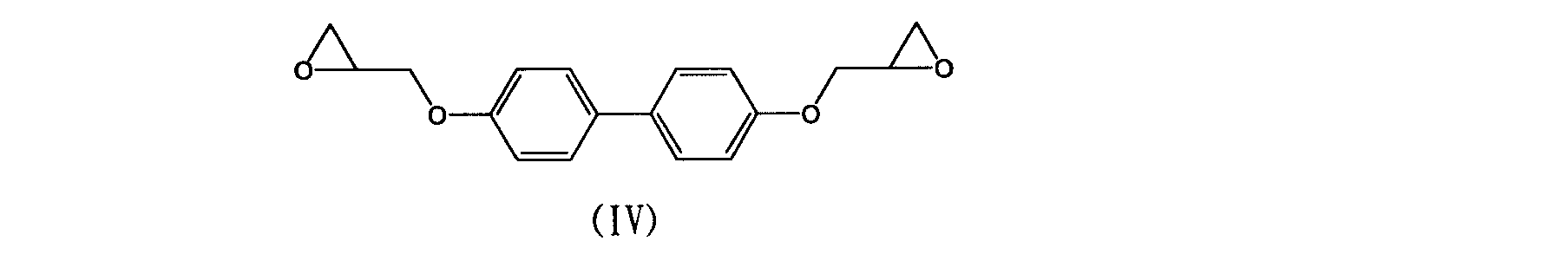
(d)成分の有機金属塩は反応触媒となるものであり、例えば、ナフテン酸亜鉛、ナフテン酸コバルト、オクチル酸錫、オクチル酸コバルト等が挙げられる。アミン系やイミダゾール系の窒素原子含有反応触媒は得られる樹脂の硬化物が脆くなり、耐熱性や接着性が低下するので好ましくない。
ここで、反応溶媒にはトルエン、キシレン及びメシチレンから選ばれる芳香族系溶媒を用いる。必要により少量の他の溶剤を用いてもよいが、他の溶剤では所望の反応が進行せず、耐熱性等が低下する。また、ベンゼンは毒性が強く、メシチレンよりも分子量の大きい芳香族系溶媒はプリプレグの製造塗工時に残溶剤となりやすいので好ましくない。
プレ反応の反応率は、シアネート基を有する化合物(b)の反応率(消失率)を30~70モル%となるようし、好ましくは40~68モル%となるようする。反応率が30モル%未満であると、得られる樹脂が相容化されておらず、樹脂が分離して白濁し、Bステージの塗工布が製造でない。また、反応率が70モル%を超えると、得られる熱硬化性樹脂が溶剤に不溶化し、Aステージのワニス(熱硬化性樹脂組成物)が製造できなくなったり、プリプレグのゲルタイムが短くなり過ぎ、プレスの際に成形性が低下する場合がある。

前記熱硬化性樹脂(A2)は、
一般式(I)で示される末端に水酸基を有するシロキサン樹脂(a)と、1分子中に少なくとも2個以上のシアネート基を有する化合物(b)とが有機溶媒中で反応して得られたものであり、
該シロキサン樹脂(a)と該化合物(b)との総和100質量部に対し、該シロキサン樹脂(a)10~70質量部及び該化合物(b)30~90質量部が含まれており、
該化合物(b)の反応率が40~70モル%である
ことを特徴とする熱硬化性樹脂組成物である。
また、熱硬化性樹脂組成物(II)に使用される溶融シリカ(B)は、熱硬化性樹脂組成物(I)に使用されるものと同様のものが用いられる。
即ち、シロキサン樹脂(a)と化合物(b)との総和100質量部に対して、シロキサン樹脂(a)の配合量を10~70質量部の範囲とする。また、化合物(b)の配合量を30~90質量部の範囲とする。
シロキサン樹脂(a)の配合量が10~70質量部の範囲であれば、熱硬化性樹脂(A)が塗布される基材の面方向の十分な低熱膨張性が得られる。また、シロキサン樹脂(a)の配合量が10~70質量部の範囲であれば、十分な耐熱性及び耐薬品性を得ることができる。また、化合物(b)の配合量が30~90質量部の範囲であれば、熱硬化性樹脂(A)を用いて得られる基材の面方向の十分な低熱膨張性が得られる。また、十分な耐熱性が得られる。
化合物(b)の反応率が40~70モル%であると、シロキサン樹脂(a)と化合物(b)が反応して得られる熱硬化性樹脂(A2)と汎用の有機溶媒との相溶性がよく、十分な硬化性が得られるため、得られる熱硬化性樹脂(A2)の耐熱性を高めることができる。また、上記範囲であると、銅箔接着性がよい。また、上記範囲であれば、熱硬化性樹脂(A)が結晶化しにくく、ワニスが作りやすく、成形性がよい。また、熱硬化性樹脂(A)の溶液を、ガラス繊維等に含浸させて半硬化した状態(いわゆるBステージ状態)においてタックの発生を低減できる。
なお、化合物(b)の反応率は、GPC測定の測定結果から求められる。具体的に、シロキサン樹脂(a)と化合物(b)とが配合された反応前の溶液と、この溶液を反応させた後の溶液とで、所定の保持時間付近に出現するシアネート樹脂のピークの面積を比較する。反応前の溶液のピーク面積に対する反応後の溶液のピーク面積の消失率が反応率に相当する。
熱硬化性樹脂組成物(II)における無機充填剤(C)の使用量は、固形分換算で、熱硬化性樹脂(A2)100質量部に対し、10~200質量部とすることが好ましく、10~150質量部とすることがより好ましく、50~150質量部とすることが特に好ましい。10~200質量部の範囲であれば、十分な難燃性、めっき溶液による浸食に対する耐性、成形性が得られる。
無機難燃助剤(D)の例としては、トリフェニルホスフェート、トリクレジルホスフェート、トリスジクロロプロピルホスフェート、リン酸エステル系化合物、ホスファゼン、赤リン等のリン系難燃剤、三酸化アンチモン、モリブデン酸亜鉛等の無機難燃助剤等が挙げられる。特に、モリブデン酸亜鉛をタルク等の無機充填剤に担持した無機難燃助剤は、難燃性のみならずドリル加工性をも著しく向上化させるので、特に好ましい無機難燃助剤である。モリブデン酸亜鉛の使用量は相容化樹脂(A1)又は熱硬化性樹脂(A2)100質量部に対し、5~20質量部とすることが好ましい。5質量部以上とすることにより、難燃性やドリル加工性が向上し、また20質量部以下とすることにより、ワニスのゲルタイムが短くなり過ぎてプレスにより積層板を成形する際に成形性が低下することがない。
熱可塑性樹脂の例としては、ポリテトラフルオロエチレン、ポリエチレン、ポリプロピレン、ポリスチレン、ポリフェニレンエーテル樹脂、フェノキシ樹脂、ポリカーボネート樹脂、ポリエステル樹脂、ポリアミド樹脂、ポリイミド樹脂、キシレン樹脂、石油樹脂及びシリコーン樹脂等が挙げられる。
エラストマーの例としては、ポリブタジエン、ABS樹脂、エポキシ変性ポリブタジエン、無水マレイン酸変性ポリブタジエン、フェノール変性ポリブタジエン及びカルボキシ変性アクリロニトリル等が挙げられる。
難燃剤の例としては、前記の無機充填剤(C)が挙げられる。
有機充填剤の例としては、シリコーンパウダー、ポリテトラフルオロエチレン、ポリエチレン、ポリプロピレン、ポリスチレン、並びにポリフェニレンエーテル等の有機物粉末等が挙げられる。
即ち、本発明のプリプレグは、本発明の熱硬化性樹脂組成物を、基材に含浸又は塗工し、加熱等により半硬化(Bステージ化)して本発明のプリプレグを製造することができる。
本発明のプリプレグは、該基材に対する熱硬化性樹脂組成物の付着量が、乾燥後のプリプレグの樹脂含有率で20~90質量%となるように基材に含浸又は塗工した後、通常、100~200℃の温度で1~30分加熱乾燥し、半硬化(Bステージ化)させて得ることができる。
即ち、本発明の積層板は前述のプリプレグを、例えば1~20枚重ね、その片面又は両面に銅及びアルミニウム等の金属箔を配置した構成で積層成形することにより製造することができる。金属箔は、電気絶縁材料用途で用いるものであれば特に制限されない。
また、成形条件は、例えば、電気絶縁材料用積層板及び多層板の手法が適用でき、例えば多段プレス、多段真空プレス、連続成形、オートクレーブ成形機等を使用し、温度100~250℃、圧力2~100kg/cm2(0.2~10MPa)、加熱時間0.1~5時間の範囲で成形することができる。また、本発明のプリプレグと内層用配線板とを組合せ、積層成形して、多層板を製造することもできる。
なお、以下の実施例および比較例において得られた銅張積層板を以下の方法により測定し、評価を行った。
銅張積層板を銅エッチング液に浸漬することにより1cm幅の銅箔を形成して評価基板を作製し、引張り試験機を用いて銅箔の接着性(ピール強度)を測定した。
銅張積層板を銅エッチング液に浸漬することにより銅箔を取り除いた5mm角の評価基板を作製し、TMA試験装置(デュポン社製、TMA2940)を用い、評価基板の面方向の熱膨張特性を観察することにより評価した。
銅張積層板を銅エッチング液に浸漬することにより銅箔を取り除いた5cm角の評価基板を作製し、平山製作所(株)製プレッシャー・クッカー試験装置を用いて、121℃、2atmの条件で4時間までプレッシャー・クッカー処理を行った後、温度288℃のはんだ浴に、評価基板を20秒間浸漬した後、外観を観察することによりはんだ耐熱性を評価した。
銅張積層板を銅エッチング液に浸漬することにより銅箔を取り除いた5mm角の評価基板を作製し、TMA試験装置(デュポン社製、TMA2940)を用い、評価基板の面方向の30℃~100℃の線熱膨張率を測定した。
銅張積層板を銅エッチング液に浸漬することにより銅箔を取り除いた評価基板から、長さ127mm、幅12.7mmに切り出した試験片を作製し、UL94の試験法(V法)に準じて評価した。
銅張積層板から5mm角の評価基板を作製し、TMA試験装置(デュポン社製、TMA2940)を用い、300℃で評価基板の膨れが発生するまでの時間を測定することにより評価した。
得られた銅張積層板を銅エッチング液に浸漬することにより銅箔を取り除いた評価基板を作製し、Hewllet・Packerd社製比誘電率測定装置(製品名:HP4291B)を用いて、周波数1GHzでの比誘電率及び誘電正接を測定した。
ドリルに径0.105mm(ユニオンツールMV J676)を用い、回転数:160,000rpm、送り速度:0.8m/分、重ね枚数:1枚でドリル加工を行い、6000ヒットさせて評価基板を作製し、ドリル穴の内壁粗さを評価した。内壁粗さの評価は、無電解銅めっきを行い(めっき厚:15μm)、穴壁へのめっき染み込み長さの最大値を測定することにより評価した。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、ビスフェノールA型シアネート樹脂(ロンザジャパン社製;商品名Primaset BADCy):600.0g、下記の式(V)に示すシロキサン樹脂(信越化学社製;商品名X-22-1821、水酸基当量;1,600):200.0g、ビフェニル型エポキシ樹脂(ジャパンエポキシレジン社製;商品名YX-4000、エポキシ当量;186):200.0g及びトルエン:1000.0gを投入した。次いで、攪拌しながら120℃に昇温し、樹脂固形分が溶解し均一な溶液になっていることを確認した後、ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.01g添加し、約110℃で4時間反応を行った。その後、室温に冷却し相容化樹脂(A1-1)の溶液を得た。
この反応溶液を少量取り出し、GPC測定(ポリスチレン換算、溶離液:テトラヒドロフラン)を行ったところ、溶出時間が約12.4分付近に出現する合成原料のビスフェノールA型シアネート樹脂のピーク面積が、反応開始時のビスフェノールA型シアネート樹脂のピーク面積と比較し、ピーク面積の消失率〔(b)成分の反応率〕が68%であった。また、約10.9分付近、及び8.0~10.0付近に出現する熱硬化性樹脂の生成物のピークが確認された。さらに、少量取り出した反応溶液を、メタノールとベンゼンの混合溶媒(混合重量比1:1)に滴下して再沈殿させることにより、精製された固形分を取り出し、FT-IR測定を行ったところ、イミノカーボネート基に起因する1700cm-1付近のピーク、また、トリアジン環に起因する1560cm-1付近、及び1380cm-1付近の強いピークが確認でき、相容化樹脂(A1-1)が製造されていることを確認した。

温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、ノボラック型シアネート樹脂(ロンザジャパン社製;商品名Primaset PT-15,質量平均分子量500~1,000):800.0gと、下記の式(VI)に示すシロキサン樹脂(信越化学社製;商品名KF-6003、水酸基当量;2800):100.0g、ナフトールアラルキル・クレゾール共重合型エポキシ樹脂(日本化薬社製;商品名NC-7000L、エポキシ当量;230):100.0g及びトルエン:1000.0gを投入した。次いで、攪拌しながら120℃に昇温し、樹脂固形分が溶解し均一な溶液になっていることを確認した後、ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.01g添加し、約110℃で4時間反応を行った。その後、室温に冷却し相容化樹脂(A-2)の溶液を得た。
この反応溶液を少量取り出し、GPC測定(ポリスチレン換算、溶離液:テトラヒドロフラン)を行ったところ、溶出時間が約12.1分付近に出現する合成原料のノボラック型シアネート樹脂のピーク面積が、反応開始時のノボラック型シアネート樹脂のピーク面積と比較し、ピーク面積の消失率〔(b)成分の反応率〕が43%であった。また、約10.9分付近、及び8.0~10.0付近に出現する熱硬化性樹脂の生成物のピークが確認された。さらに、少量取り出した反応溶液を、メタノールとベンゼンの混合溶媒(混合重量比1:1)に滴下して再沈殿させることにより、精製された固形分を取り出し、FT-IR測定を行ったところ、イミノカーボネート基に起因する1700cm-1付近のピーク、また、トリアジン環に起因する1560cm-1付近、及び1380cm-1付近の強いピークが確認でき、相容化樹脂(A1-2)が製造されていることを確認した。

温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、ジシクロペンタジエン型シアネート樹脂(ロンザジャパン社製;商品名Primaset DT-4000,質量平均分子量500~1,000):400.0g、下記式(VII)に示すシロキサン樹脂(信越化学社製;商品名X-22-160AS、水酸基当量;500):100.0g、ビフェニルアラルキル型エポキシ樹脂(日本化薬社製;商品名NC-3000H、エポキシ当量;280):500.0g及びシチレン:1000.0gを投入した。
次いで、攪拌しながら120℃に昇温し、樹脂固形分が溶解し均一な溶液になっていることを確認した後、ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.30g添加し、約110℃で4時間反応を行った。その後、室温に冷却し、相容化樹脂(A1-3)の溶液を得た。
この反応溶液を少量取り出し、GPC測定(ポリスチレン換算、溶離液:テトラヒドロフラン)を行ったところ、溶出時間が約12.0分付近に出現する合成原料のノボラック型シアネート樹脂のピーク面積が、反応開始時のノボラック型シアネート樹脂のピーク面積と比較し、ピーク面積の消失率〔(b)成分の反応率〕が43%であった。また、約10.9分付近、及び8.0~10.0付近に出現する熱硬化性樹脂の生成物のピークが確認された。さらに、少量取り出した反応溶液を、メタノールとベンゼンの混合溶媒(混合質量比1:1)に滴下して再沈殿させることにより、精製された固形分を取り出し、FT-IR測定を行ったところ、イミノカーボネート基に起因する1700cm-1付近のピーク、また、トリアジン環に起因する1560cm-1付近、及び1380cm-1付近の強いピークが確認でき、相容化樹脂(A1-3)が製造されていることを確認した。
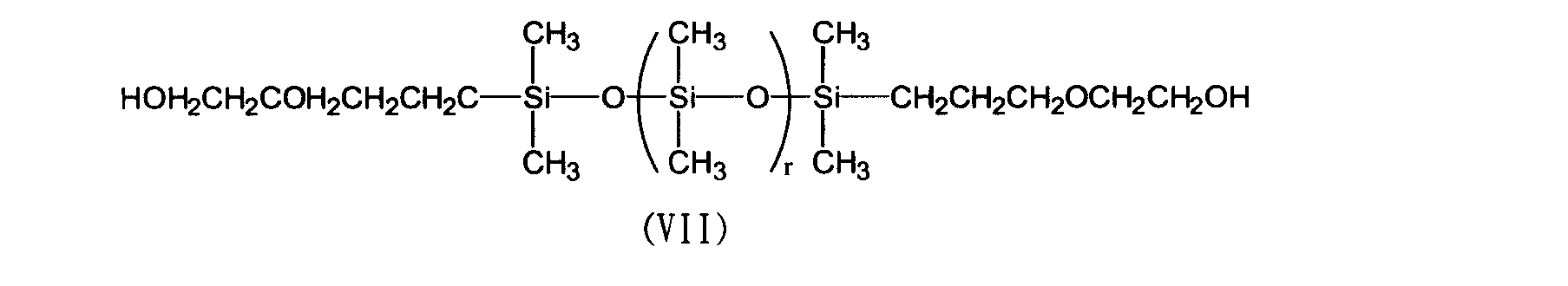
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、ビスフェノールA型シアネート樹脂(ロンザジャパン社製;商品名Primaset BADCy):400.0gと、前記の式(V)に示すシロキサン樹脂(信越化学社製;商品名X-22-1821、水酸基当量;1,600):500.0gと、ナフタレン型エポキシ樹脂(大日本インキ化学社製;商品名エピクロンHP-4032、エポキシ当量;150):100.0gと、トルエン:1000.0gを投入した。次いで、攪拌しながら120℃に昇温し、樹脂固形分が溶解し均一な溶液になっていることを確認した後、ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.01g添加し、約110℃で4時間反応を行った。その後、室温に冷却し相容化樹脂(A1-4)の溶液を得た。
この反応溶液を少量取り出し、GPC測定(ポリスチレン換算、溶離液:テトラヒドロフラン)を行ったところ、溶出時間が約12.4分付近に出現する合成原料のビスフェノールA型シアネート樹脂のピーク面積が、反応開始時のビスフェノールA型シアネート樹脂のピーク面積と比較し、ピーク面積の消失率〔(b)成分の反応率〕が55%であった。また、約10.9分付近、及び8.0~10.0付近に出現する熱硬化性樹脂の生成物のピークが確認された。さらに、少量取り出した反応溶液を、メタノールとベンゼンの混合溶媒(混合重量比1:1)に滴下して再沈殿させることにより、精製された固形分を取り出し、FT-IR測定を行ったところ、イミノカーボネート基に起因する1700cm-1付近のピーク、また、トリアジン環に起因する1560cm-1付近、及び1380cm-1付近の強いピークが確認でき、相容化樹脂(A1-4)が製造されていることを確認した。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、溶融シリカ(アドマテックス社製;商品名SO-25R):700.0gと、プロピレングリコールモノメチルエーテル:1000.0gを配合し、攪拌しながらN-フェニル-3-アミノプロピルトリメトキシシラン(信越化学社製;商品名KBM-573):7.0gを添加した。次いで80℃に昇温し、80℃で1時間反応を行い溶融シリカの表面処理(湿式処理)を行った後、室温に冷却し、N-フェニル-3-アミノプロピルトリメトキシシランにより表面処理(湿式処理)された溶融シリカ(B-1)の溶液を得た。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、ビスフェノールA型シアネート樹脂(ロンザジャパン社製;商品名Primaset BADCy):600.0gと、前記の式(V)に示すシロキサン樹脂(信越化学社製;商品名X-22-1821、水酸基当量;1,600):200.0gと、ビフェニル型エポキシ樹脂(ジャパンエポキシレジン社製;商品名YX-4000、エポキシ当量;186):200.0gと、トルエン:1000.0gを投入した。次いで、攪拌しながら120℃に昇温し、樹脂固形分が溶解し均一な溶液になっていることを確認した後、ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.01g添加し、約110℃で1時間反応を行った。その後、室温に冷却し、樹脂(A1-5)の溶液を得た。
この反応溶液を少量取り出し、GPC測定(ポリスチレン換算、溶離液:テトラヒドロフラン)を行ったところ、溶出時間が約12.4分付近に出現する合成原料のビスフェノールA型シアネート樹脂のピーク面積が、反応開始時のビスフェノールA型シアネート樹脂のピーク面積と比較し、ピーク面積の消失率〔(b)成分の反応率〕が18%であった。また、この溶液は翌日結晶化により沈殿物が生じた。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、ビスフェノールA型シアネート樹脂(ロンザジャパン社製;商品名Primaset BADCy):600.0g、前記の式(V)に示すシロキサン樹脂(信越化学社製;商品名X-22-1821、水酸基当量;1,600):200.0g、ビフェニル型エポキシ樹脂(ジャパンエポキシレジン社製;商品名YX-4000、エポキシ当量;186):200.0g及びトルエン:1000.0gを投入した。次いで、攪拌しながら120℃に昇温し、樹脂固形分が溶解し均一な溶液になっていることを確認した後、ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.01g添加し、約120℃で6時間反応を行った。その後、室温に冷却し、樹脂(A1-6)の溶液を得た。
この反応溶液を少量取り出し、GPC測定(ポリスチレン換算、溶離液:テトラヒドロフラン)を行ったところ、溶出時間が約12.4分付近に出現する合成原料のビスフェノールA型シアネート樹脂のピーク面積が、反応開始時のビスフェノールA型シアネート樹脂のピーク面積と比較し、ピーク面積の消失率〔(b)成分の反応率〕が76%であった。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積2リットルの反応容器に、ビスフェノールA型シアネート樹脂(ロンザジャパン社製;商品名Primaset BADCy):600.0gと、前記の式(V)に示すシロキサン樹脂(信越化学社製;商品名X-22-1821、水酸基当量;1,600):200.0gと、トルエン:800.0gを投入した。次いで、攪拌しながら120℃に昇温し、樹脂固形分が溶解し均一な溶液になっていることを確認した後、ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.01g添加し、約110℃で4時間反応を行った。その後、室温に冷却し、樹脂(A1-7)の溶液を得た。
この反応溶液を少量取り出し、GPC測定(ポリスチレン換算、溶離液:テトラヒドロフラン)を行ったところ、溶出時間が約12.4分付近に出現する合成原料のビスフェノールA型シアネート樹脂のピーク面積が、反応開始時のビスフェノールA型シアネート樹脂のピーク面積と比較し、ピーク面積の消失率〔(b)成分の反応率〕が53%であった。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、シロキサン樹脂(a)として前記式(V)に示すシロキサン樹脂(信越化学工業株式会社製;商品名X-22-1821、水酸基当量;1,600g/eq.):500gとトルエン:1000gと、化合物(b)としてビスフェノールA型シアネート樹脂(ロンザジャパン株式会社製;商品名Arocy B-10):500とを配合し、攪拌しながら昇温し、120℃に到達後、ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.01g添加し、約115~125℃で4時間還流反応を行った後、室温(25℃)に冷却し、熱硬化性樹脂(A2-1)の溶液を得た。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、化合物(b)としてノボラック型シアネート樹脂(ロンザジャパン株式会社製;商品名プリマセットPT-30,重量平均分子量500~1,000):800gと、シロキサン樹脂(a)として前記式(VI)に示すシロキサン樹脂(信越化学工業株式会社製;商品名KF-6003、水酸基当量;2800g/eq.):200gとトルエン:1000gを配合し、攪拌しながら昇温し、120℃に到達後ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.01g添加し、約115~125℃で4時間還流反応を行った後、室温(25℃)に冷却し、熱硬化性樹脂(A2-2)の溶液を得た。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、化合物(b)としてノボラック型シアネート樹脂(ロンザジャパン株式会社製;商品名プリマセットPT-60,重量平均分子量2,000~3,000):300gと、シロキサン樹脂(a)として前記式(VII)に示すシロキサン樹脂(信越化学工業株式会社製;商品名X-22-160AS、水酸基当量;500g/eq.):700gとメシチレン:1000gを配合し、攪拌しながら昇温し、120℃に到達後ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.30g添加し、約115~125℃で4時間還流反応を行った後、室温(25℃)に冷却し、熱硬化性樹脂(A2-3)の溶液を得た。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、ビスフェノールA型シアネート樹脂(ロンザジャパン株式会社製;商品名Arocy B-10):500gと、前記式(V)に示すシロキサン樹脂(信越化学工業株式会社製;商品名X-22-1821、水酸基当量;1,600g/eq.):500gとトルエン:1000gを配合し、攪拌しながら昇温し、120℃に到達後ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.01g添加し、約100℃で2時間還流反応を行った後、室温(25℃)に冷却し、熱硬化性樹脂(A2-4)の溶液を得た。
シロキサン樹脂(a)と化合物(b)とが配合された反応前の溶液と、反応後の溶液とを少量ずつ取り出し、それぞれについてGPC測定(ポリスチレン換算、溶離液:テトラヒドロフラン)を行った。反応前の溶液と反応後の溶液とで、保持時間が約12.4分付近に出現するビスフェノールA型シアネート樹脂のピークの面積を比較し、反応前の溶液のピーク面積に対する反応後の溶液のピーク面積の消失率を算出した。その結果、反応後の溶液におけるピーク面積の消失率が35%であった。よって、熱硬化性樹脂(A2-4)における化合物(b)の反応率は、35モル%であった。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、ビスフェノールA型シアネート樹脂(ロンザジャパン株式会社製;商品名Arocy B-10):500gと、前記式(V)に示すシロキサン樹脂(信越化学工業株式会社製;商品名X-22-1821、水酸基当量;1,600g/eq.):500gとトルエン:1000gを配合し、攪拌しながら昇温し、120℃に到達後ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.05g添加し、約120℃で8時間還流反応を行った後、室温(25℃)に冷却し、熱硬化性樹脂(A2-5)の溶液を得た。
シロキサン樹脂(a)と化合物(b)とが配合された反応前の溶液と、反応後の溶液とを少量ずつ取り出し、それぞれについてGPC測定(ポリスチレン換算、溶離液:テトラヒドロフラン)を行った。反応前の溶液と反応後の溶液とで、保持時間が約12.4分付近に出現するビスフェノールA型シアネート樹脂のピークの面積を比較し、反応前の溶液のピーク面積に対する反応後の溶液のピーク面積の消失率を算出した。その結果、反応後の溶液におけるピーク面積の消失率が79%であった。よって、熱硬化性樹脂(A2-5)における化合物(b)の反応率は、79mol%であった。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、ビスフェノールA型シアネート樹脂(ロンザジャパン株式会社製;商品名Arocy B-10):200gと、前記式(V)に示すシロキサン樹脂(信越化学工業株式会社製;商品名X-22-1821、水酸基当量;1,600g/eq.):800gとトルエン:1000gを配合し、攪拌しながら昇温し、120℃に到達後ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.05g添加し、約120℃で8時間還流反応を行った後、室温(25℃)に冷却し、熱硬化性樹脂(A2-6)の溶液を得た。
また、約10.9分付近、及び8.0~10.0付近に出現する熱硬化性樹脂の生成物のピークが確認された。さらに、少量取り出した反応溶液を、メタノールとベンゼンの混合溶媒(混合質量比1:1)に滴下して再沈殿させることにより、精製された固形分を取り出し、FT-IR測定を行ったところ、イミノカーボネート基に起因する1700cm-1付近のピーク、また、トリアジン環に起因する1560cm-1付近、及び1380cm-1付近の強いピークを確認した。
温度計、攪拌装置、還流冷却管の付いた加熱及び冷却可能な容積3リットルの反応容器に、ビスフェノールA型シアネート樹脂(ロンザジャパン株式会社製;商品名Arocy B-10):1000gと、トルエン:1000gを配合し、攪拌しながら昇温し、120℃に到達後ナフテン酸亜鉛の8質量%ミネラルスピリット溶液を0.01g添加し、約120℃で4時間還流反応を行った後、室温(25℃)に冷却し、熱硬化性樹脂(A2-7)の溶液を得た。
さらに、少量取り出した反応溶液を、メタノールとベンゼンの混合溶媒(混合質量比1:1)に滴下して再沈殿させることにより、精製された固形分を取り出し、FT-IR測定を行ったところ、トリアジン環に起因する1560cm-1付近、及び1380cm-1付近の強いピークを確認した。
(A)成分として、製造例1~4により得られた相容化樹脂、比較製造例1~3で得られた樹脂、又は製造例6~8及び比較製造例4~7で得られた熱硬化性樹脂、製造例5又は商業的に入手した(B)成分、また必要により(C)成分、(D)成分、及び硬化促進剤に、希釈溶剤としてメチルエチルケトンを使用して、第1表及び第2表に示した配合割合(質量部)で混合して樹脂分60質量%の均一なワニスを得た。
次に、得られたワニスを厚さ0.2mmのSガラスクロスに含浸塗工し、160℃で10分加熱乾燥して樹脂含有量55質量%のプリプレグを得た。
このプリプレグを4枚重ね、18μmの電解銅箔を上下に配置し、圧力25kg/cm2(2.45MPa)、温度185℃で90分間プレスを行って、銅張積層板を得た。
このようにして得られた銅張積層板を用いて、銅箔接着性(銅箔ピール強度)、ガラス転移温度、はんだ耐熱性、線膨張係数、難燃性、銅付き耐熱性(T-300)、比誘電率(1GHz)、誘電正接(1GHz)及びドリル加工性について前記の方法で測定・評価した。評価結果を第1表~第4表に示す。



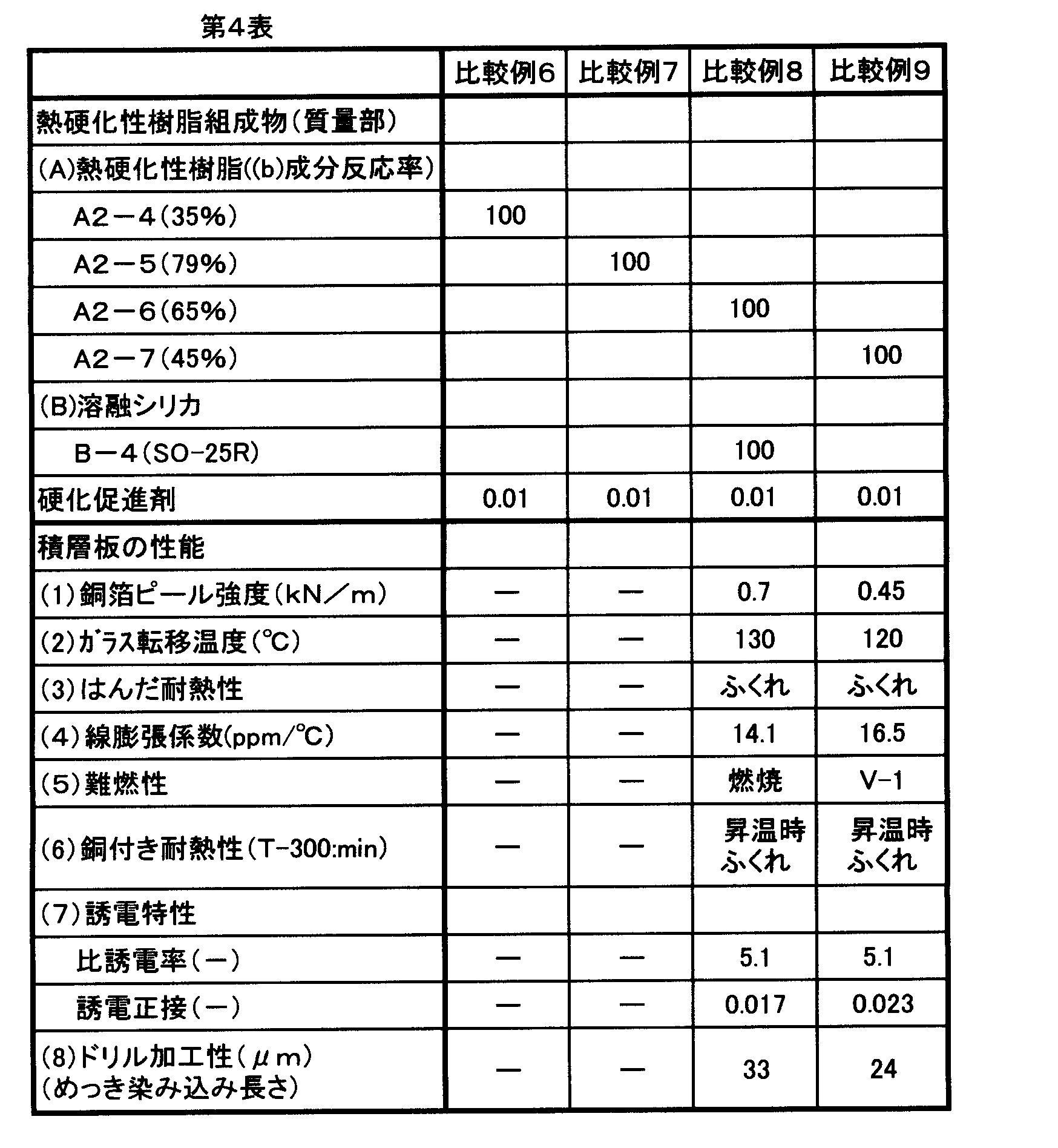
溶融シリカ(B-2):溶融シリカに対し1.0質量%のN-フェニル-3-アミノプロピルトリメトキシシランにより表面処理された溶融シリカ(アドマテック社製;商品名SC-2050KNK,希釈溶剤;メチルイソブチルケトン)
溶融シリカ(B-3):溶融シリカに対し1.0質量%のN-フェニル-3-アミノプロピルトリメトキシシランにより表面処理された溶融シリカ(アドマテック社製;商品名SC-2050HNK,希釈溶剤;シクロヘキサノン)
溶融シリカ(B-4):溶融シリカ(アドマテック社製;商品名SO-25R)
溶融シリカ(B-5):溶融シリカに対し1.0質量%の下記式(VIII)に示しγ-グリシドキシプロピルトリメトキシシランにより表面処理された溶融シリカ(アドマテック社製;商品名SC1030-MJA、希釈溶剤;メチルエチルケトン)
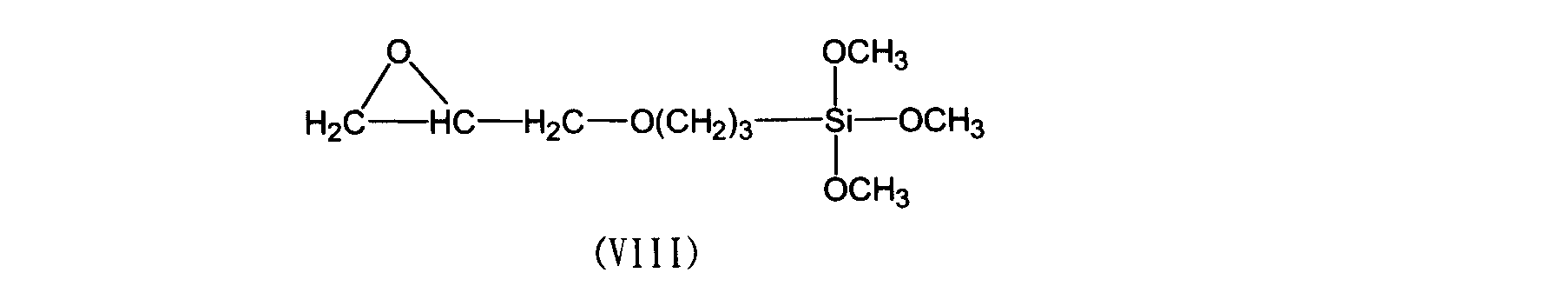
(D)無機難燃助剤(KG-1100):モリブデン酸亜鉛をタルクに担持した無機難燃助剤(シャーウィン・ウィリアムス社製;商品名 ケムガード1100)
硬化促進剤:ナフテン酸亜鉛の8質量%ミネラルスピリット溶液
比較例3で用いたエポキシ樹脂(YX-4000):ビフェニル型エポキシ樹脂(ジャパンエポキシレジン社製;商品名YX-4000、エポキシ当量;186)
比較例1:熱硬化性樹脂が析出しワニスを製造できなかった。
比較例2:成形性が不良であり積層板を作製できなかった。
比較例3:樹脂が分離し、プリプレグ及び積層板を作製できなかった。
また、第4表において比較例6~7では、熱硬化性樹脂組成物は熱硬化性樹脂(A)が析出し、ワニスを製造することができなかったため、評価できなかった。
一方、第2表から明らかなように、比較例1~5は、銅箔ピール強度、カラス転移温度(Tg)、はんだ耐熱性、低熱膨張性、難燃性、銅付き耐熱性(T-300)、低誘電特性、低誘電正接性、ドリル加工性の全てを満たすものは無く、いずれかの特性に劣っている。
従って、本発明の熱硬化性樹脂組成物を使用することにより、今日要求される配線板の高密度化や高信頼性が達成され、本発明の熱硬化性樹脂組成物を電子機器などの製造に広く用いることができる。
Claims (5)
- 下記一般式(I)で示される末端に水酸基を有するシロキサン樹脂(a)、1分子中に少なくとも2個のシアネート基を有する化合物(b)及び1分子中に少なくとも2個のエポキシ基を有する化合物(c)を、(a)~(c)成分の合計量100質量部当たり、(a)成分10~50質量部、(b)成分40~80質量部、(c)成分10~50質量部として、有機金属塩(d)の存在下、トルエン、キシレン及びメシチレンから選ばれる溶媒中で80℃~120℃で反応させ、(b)成分の反応率が30~70モル%であることを特徴とするイミノカーボネート構造及びトリアジン構造を有する相容化樹脂の製造方法。

- 請求項1に記載の方法により製造された相容化樹脂(A1)及び、下記式(II)で示されるトリメトキシシラン化合物により表面処理された溶融シリカ(B)を含有することを特徴とする熱硬化性樹脂組成物。

- 熱硬化性樹脂(A2)及び、式(II)で示されるトリメトキシシラン化合物により表面処理された溶融シリカ(B)を含有する熱硬化性樹脂組成物であって、
前記熱硬化性樹脂(A2)は、
一般式(I)で示される末端に水酸基を有するシロキサン樹脂(a)と、1分子中に少なくとも2個以上のシアネート基を有する化合物(b)とが有機溶媒中で反応して得られたものであり、
該シロキサン樹脂(a)と該化合物(b)との総和100質量部に対し、該シロキサン樹脂(a)10~70質量部及び該化合物(b)30~90質量部が含まれており、
該化合物(b)の反応率が40~70モル%である
ことを特徴とする熱硬化性樹脂組成物。 - 請求項2又は請求項3に記載の熱硬化性樹脂組成物を基材中に含侵又は塗工した後、Bステージ化したプリプレグ。
- 請求項4記載のプリプレグを用いて形成された積層板。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201180038869.XA CN103189418B (zh) | 2010-08-06 | 2011-08-05 | 相容性树脂的制造方法、热固化性树脂组合物、预浸料及层叠板 |
| KR1020137002782A KR20130095730A (ko) | 2010-08-06 | 2011-08-05 | 상용화 수지의 제조 방법, 열경화성 수지 조성물, 프리프레그 및 적층판 |
| US13/814,350 US20130172459A1 (en) | 2010-08-06 | 2011-08-05 | Process for producing compatibilized resin, thermosetting resin composition, prepreg, and laminate |
| EP11814751.1A EP2602277B1 (en) | 2010-08-06 | 2011-08-05 | Process for producing compatibilized resin, thermosetting resin composition, prepreg, and laminate |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-177380 | 2010-08-06 | ||
| JP2010177380 | 2010-08-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012018126A1 true WO2012018126A1 (ja) | 2012-02-09 |
Family
ID=45559621
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/067985 WO2012018126A1 (ja) | 2010-08-06 | 2011-08-05 | 相容化樹脂の製造法、熱硬化性樹脂組成物、プリプレグ及び積層板 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20130172459A1 (ja) |
| EP (1) | EP2602277B1 (ja) |
| JP (1) | JP5857514B2 (ja) |
| KR (1) | KR20130095730A (ja) |
| CN (1) | CN103189418B (ja) |
| TW (1) | TWI512008B (ja) |
| WO (1) | WO2012018126A1 (ja) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013189579A (ja) * | 2012-03-14 | 2013-09-26 | Hitachi Chemical Co Ltd | 熱硬化性樹脂組成物、およびこれを用いたプリプレグ、積層板 |
| JP2013189581A (ja) * | 2012-03-14 | 2013-09-26 | Hitachi Chemical Co Ltd | 相溶化樹脂、およびそれを用いたプリプレグ、積層板 |
| JP2013189578A (ja) * | 2012-03-14 | 2013-09-26 | Hitachi Chemical Co Ltd | 相容化樹脂、及びこれを用いた熱硬化性樹脂組成物、プリプレグ、積層板 |
| JP2013189577A (ja) * | 2012-03-14 | 2013-09-26 | Hitachi Chemical Co Ltd | 熱硬化性樹脂組成物、プリプレグ及び積層板 |
| JP2013189580A (ja) * | 2012-03-14 | 2013-09-26 | Hitachi Chemical Co Ltd | 樹脂組成物、これを用いたプリプレグ及び積層板 |
| JP2013216882A (ja) * | 2012-03-14 | 2013-10-24 | Hitachi Chemical Co Ltd | 熱硬化性樹脂組成物、プリプレグ及び積層板 |
| JP2013216880A (ja) * | 2012-03-14 | 2013-10-24 | Hitachi Chemical Co Ltd | 相容化樹脂、熱硬化性樹脂組成物、プリプレグ及び積層板 |
| JP2013216881A (ja) * | 2012-03-14 | 2013-10-24 | Hitachi Chemical Co Ltd | 相容化樹脂の製造法、熱硬化性樹脂組成物、プリプレグ及び積層板 |
| JP2014084441A (ja) * | 2012-10-26 | 2014-05-12 | Sumitomo Bakelite Co Ltd | 樹脂基板、金属張積層板、プリント配線基板、および半導体装置 |
| JP2014122339A (ja) * | 2012-11-26 | 2014-07-03 | Hitachi Chemical Co Ltd | 熱硬化性樹脂組成物、プリプレグ、積層板、プリント配線板、及び実装基板、並びに熱硬化性樹脂組成物の製造方法 |
| JP2015505063A (ja) * | 2012-02-20 | 2015-02-16 | エルジー・ケム・リミテッド | 光硬化性および熱硬化性を有する樹脂組成物と、ドライフィルムソルダレジスト |
| JP2015052081A (ja) * | 2013-09-09 | 2015-03-19 | 日立化成株式会社 | 相容化樹脂の製造方法、熱硬化性樹脂組成物、プリプレグ、積層板、及びプリント配線板 |
| JP2015063585A (ja) * | 2013-09-24 | 2015-04-09 | 日立化成株式会社 | 熱硬化性樹脂組成物並びにそれを用いたプリプレグ、積層板、プリント配線板、及び実装基板 |
| WO2015155982A1 (ja) * | 2014-04-08 | 2015-10-15 | パナソニックIpマネジメント株式会社 | プリント配線板用樹脂組成物、プリプレグ、金属張積層板、プリント配線板 |
| JP2015207753A (ja) * | 2014-04-08 | 2015-11-19 | パナソニックIpマネジメント株式会社 | プリント配線板用樹脂組成物、プリプレグ、金属張積層板、プリント配線板 |
| JP2016147986A (ja) * | 2015-02-13 | 2016-08-18 | パナソニックIpマネジメント株式会社 | プリント配線板用樹脂組成物、プリプレグ、金属張積層板およびプリント配線板 |
| US9775239B2 (en) | 2014-04-08 | 2017-09-26 | Panasonic Intellectual Property Management Co., Ltd. | Resin composition for printed wiring board, prepreg, metal-clad laminate, and printed wiring board |
| JP2019194345A (ja) * | 2019-07-11 | 2019-11-07 | パナソニックIpマネジメント株式会社 | プリント配線板用樹脂組成物、プリプレグ、金属張積層板およびプリント配線板 |
| JPWO2019176074A1 (ja) * | 2018-03-15 | 2021-04-08 | 昭和電工マテリアルズ株式会社 | エポキシ樹脂、エポキシ樹脂組成物、樹脂シート、bステージシート、cステージシート、硬化物、樹脂付金属箔、金属基板、及びパワー半導体装置 |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5909916B2 (ja) * | 2011-08-05 | 2016-04-27 | 日立化成株式会社 | 樹脂の製造法、熱硬化性樹脂組成物、プリプレグ及び積層板 |
| JP6074943B2 (ja) * | 2012-08-08 | 2017-02-08 | 日立化成株式会社 | 熱硬化性樹脂組成物、及びこれを用いたプリプレグ、積層板 |
| JP2014111719A (ja) | 2012-11-12 | 2014-06-19 | Panasonic Corp | 積層板、金属張積層板、プリント配線板、多層プリント配線板 |
| US9332637B2 (en) * | 2013-11-04 | 2016-05-03 | Novoset Llc | Ultra low loss dielectric thermosetting resin compositions and high performance laminates manufactured therefrom |
| JP6458978B2 (ja) * | 2014-04-17 | 2019-01-30 | 日立化成株式会社 | 積層板 |
| KR101792755B1 (ko) | 2014-10-28 | 2017-11-01 | 주식회사 엘지화학 | 광경화성 및 열경화성을 갖는 수지 조성물 및 드라이 필름 솔더 레지스트 |
| JP2016199644A (ja) * | 2015-04-08 | 2016-12-01 | 日立化成株式会社 | 変性シアネートエステルワニスの製造方法、それを用いたプリプレグ、積層板及び配線板 |
| JP6723788B2 (ja) * | 2016-03-31 | 2020-07-15 | 太陽インキ製造株式会社 | 硬化性樹脂組成物、ドライフィルム、硬化物およびプリント配線板 |
| KR102489450B1 (ko) * | 2017-03-30 | 2023-01-16 | 쇼와덴코머티리얼즈가부시끼가이샤 | 프리프레그 및 그의 제조 방법, 적층판, 프린트 배선판 그리고 반도체 패키지 |
| JP6896591B2 (ja) * | 2017-11-14 | 2021-06-30 | Eneos株式会社 | プリプレグ、繊維強化複合材料及び成形体 |
| JP7285343B2 (ja) * | 2019-09-04 | 2023-06-01 | サムスン エスディアイ カンパニー,リミテッド | 硬化型樹脂組成物、それから形成された硬化膜、および前記硬化膜を有する電子装置 |
| EP4067077A4 (en) * | 2019-11-29 | 2024-01-03 | Nippon Soda Co., Ltd. | FINALLY MODIFIED POLYBUTADIENE, RESIN COMPOSITION FOR METAL BACKED LAMINATES, PREPREG AND METAL BACKED LAMINATES |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10287809A (ja) * | 1997-04-14 | 1998-10-27 | Mitsubishi Gas Chem Co Inc | シアン酸エステル液状樹脂組成物及び半導体封止装置 |
| JP2000034345A (ja) * | 1998-05-13 | 2000-02-02 | Sumitomo Chem Co Ltd | 多官能シアン酸エステル樹脂組成物および硬化物 |
| JP2001339130A (ja) * | 2000-03-21 | 2001-12-07 | Hitachi Chem Co Ltd | 誘電特性に優れる樹脂組成物並びにこれを用いて作製されるワニス、ワニスの製造方法、プリプレグ及び金属張積層板 |
| JP2002285015A (ja) | 2001-03-26 | 2002-10-03 | Sumitomo Bakelite Co Ltd | 樹脂組成物、プリプレグ及びそれを用いた銅張積層板 |
| JP2002309085A (ja) | 2001-04-13 | 2002-10-23 | Sumitomo Bakelite Co Ltd | 耐熱性樹脂組成物、これを用いたプリプレグ及び積層板 |
| JP2002348469A (ja) | 2001-05-25 | 2002-12-04 | Sumitomo Bakelite Co Ltd | 耐熱性樹脂組成物、これを用いたプリプレグ及び積層板 |
| JP2003073543A (ja) | 2001-09-03 | 2003-03-12 | Sumitomo Bakelite Co Ltd | 樹脂組成物、プリプレグ及びそれを用いたプリント配線板 |
| JP2003268136A (ja) | 2002-03-20 | 2003-09-25 | Sumitomo Bakelite Co Ltd | プリプレグおよび積層板 |
| JP2009035728A (ja) * | 2007-07-12 | 2009-02-19 | Mitsubishi Gas Chem Co Inc | プリプレグ及び積層板 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS61228015A (ja) * | 1985-04-01 | 1986-10-11 | Yokohama Rubber Co Ltd:The | エポキシ樹脂組成物 |
| US4847154A (en) * | 1987-05-29 | 1989-07-11 | Basf Corporation | Thermosetting resin systems containing secondary amine-terminated siloxane modifiers |
| US4797454A (en) * | 1987-09-24 | 1989-01-10 | Basf Corporation | Curable resin systems containing cyanate ester functional oxazolinylpolysiloxanes |
| JP3855074B2 (ja) * | 1995-03-14 | 2006-12-06 | ナガセケムテックス株式会社 | オルガノポリシロキサン誘導体 |
| TW541323B (en) * | 1998-05-13 | 2003-07-11 | Sumitomo Chemical Co | Cyanate ester composition and cured product thereof |
| CN100506917C (zh) * | 2000-03-21 | 2009-07-01 | 日立化成工业株式会社 | 介电特性优良的树脂组合物和该树脂组合物的制备方法及用其生产的树脂清漆和树脂清漆的制备方法及用它们生产的层压材料和金属贴合的层压板 |
| JP5114816B2 (ja) * | 2001-04-12 | 2013-01-09 | 日立化成工業株式会社 | シロキサン変性シアネート樹脂組成物、ならびにそれを用いる接着フィルム、樹脂付き金属箔および多層プリント配線板 |
| JP2006131743A (ja) * | 2004-11-05 | 2006-05-25 | Hitachi Chem Co Ltd | 熱硬化性樹脂組成物及びそれを用いたプリプレグ、金属張積層板、プリント配線板 |
| JP4888147B2 (ja) * | 2007-02-13 | 2012-02-29 | 住友ベークライト株式会社 | 樹脂組成物、フィルム付きまたは金属箔付き絶縁樹脂シート、多層プリント配線板、多層プリント配線板の製造方法および半導体装置 |
| JP2009007531A (ja) * | 2007-06-29 | 2009-01-15 | Kaneka Corp | 樹脂/フィラー複合材料とそれを用いたプリント配線板 |
| EP2192167A4 (en) * | 2007-09-19 | 2013-07-03 | Toray Industries | ADHESIVE COMPOSITION FOR ELECTRONIC COMPONENTS AND ADHESIVE SHEET FOR ELECTRONIC COMPONENTS USING THE SAME |
-
2011
- 2011-08-05 JP JP2011172362A patent/JP5857514B2/ja not_active Expired - Fee Related
- 2011-08-05 KR KR1020137002782A patent/KR20130095730A/ko not_active Application Discontinuation
- 2011-08-05 WO PCT/JP2011/067985 patent/WO2012018126A1/ja active Application Filing
- 2011-08-05 CN CN201180038869.XA patent/CN103189418B/zh not_active Expired - Fee Related
- 2011-08-05 EP EP11814751.1A patent/EP2602277B1/en not_active Not-in-force
- 2011-08-05 US US13/814,350 patent/US20130172459A1/en not_active Abandoned
- 2011-08-05 TW TW100128117A patent/TWI512008B/zh not_active IP Right Cessation
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10287809A (ja) * | 1997-04-14 | 1998-10-27 | Mitsubishi Gas Chem Co Inc | シアン酸エステル液状樹脂組成物及び半導体封止装置 |
| JP2000034345A (ja) * | 1998-05-13 | 2000-02-02 | Sumitomo Chem Co Ltd | 多官能シアン酸エステル樹脂組成物および硬化物 |
| JP2001339130A (ja) * | 2000-03-21 | 2001-12-07 | Hitachi Chem Co Ltd | 誘電特性に優れる樹脂組成物並びにこれを用いて作製されるワニス、ワニスの製造方法、プリプレグ及び金属張積層板 |
| JP2002285015A (ja) | 2001-03-26 | 2002-10-03 | Sumitomo Bakelite Co Ltd | 樹脂組成物、プリプレグ及びそれを用いた銅張積層板 |
| JP2002309085A (ja) | 2001-04-13 | 2002-10-23 | Sumitomo Bakelite Co Ltd | 耐熱性樹脂組成物、これを用いたプリプレグ及び積層板 |
| JP2002348469A (ja) | 2001-05-25 | 2002-12-04 | Sumitomo Bakelite Co Ltd | 耐熱性樹脂組成物、これを用いたプリプレグ及び積層板 |
| JP2003073543A (ja) | 2001-09-03 | 2003-03-12 | Sumitomo Bakelite Co Ltd | 樹脂組成物、プリプレグ及びそれを用いたプリント配線板 |
| JP2003268136A (ja) | 2002-03-20 | 2003-09-25 | Sumitomo Bakelite Co Ltd | プリプレグおよび積層板 |
| JP2009035728A (ja) * | 2007-07-12 | 2009-02-19 | Mitsubishi Gas Chem Co Inc | プリプレグ及び積層板 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2602277A4 |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015505063A (ja) * | 2012-02-20 | 2015-02-16 | エルジー・ケム・リミテッド | 光硬化性および熱硬化性を有する樹脂組成物と、ドライフィルムソルダレジスト |
| US9389504B2 (en) | 2012-02-20 | 2016-07-12 | Lg Chem, Ltd. | Photo-curable and thermo-curable resin composition, and dry film solder resist |
| US9134609B2 (en) | 2012-02-20 | 2015-09-15 | Lg Chem, Ltd. | Photo-curable and thermo-curable resin compostion, and dry film solder resist |
| JP2013216882A (ja) * | 2012-03-14 | 2013-10-24 | Hitachi Chemical Co Ltd | 熱硬化性樹脂組成物、プリプレグ及び積層板 |
| JP2013189578A (ja) * | 2012-03-14 | 2013-09-26 | Hitachi Chemical Co Ltd | 相容化樹脂、及びこれを用いた熱硬化性樹脂組成物、プリプレグ、積層板 |
| JP2013189579A (ja) * | 2012-03-14 | 2013-09-26 | Hitachi Chemical Co Ltd | 熱硬化性樹脂組成物、およびこれを用いたプリプレグ、積層板 |
| JP2013216880A (ja) * | 2012-03-14 | 2013-10-24 | Hitachi Chemical Co Ltd | 相容化樹脂、熱硬化性樹脂組成物、プリプレグ及び積層板 |
| JP2013216881A (ja) * | 2012-03-14 | 2013-10-24 | Hitachi Chemical Co Ltd | 相容化樹脂の製造法、熱硬化性樹脂組成物、プリプレグ及び積層板 |
| JP2013189580A (ja) * | 2012-03-14 | 2013-09-26 | Hitachi Chemical Co Ltd | 樹脂組成物、これを用いたプリプレグ及び積層板 |
| JP2013189581A (ja) * | 2012-03-14 | 2013-09-26 | Hitachi Chemical Co Ltd | 相溶化樹脂、およびそれを用いたプリプレグ、積層板 |
| JP2013189577A (ja) * | 2012-03-14 | 2013-09-26 | Hitachi Chemical Co Ltd | 熱硬化性樹脂組成物、プリプレグ及び積層板 |
| JP2014084441A (ja) * | 2012-10-26 | 2014-05-12 | Sumitomo Bakelite Co Ltd | 樹脂基板、金属張積層板、プリント配線基板、および半導体装置 |
| JP2014122339A (ja) * | 2012-11-26 | 2014-07-03 | Hitachi Chemical Co Ltd | 熱硬化性樹脂組成物、プリプレグ、積層板、プリント配線板、及び実装基板、並びに熱硬化性樹脂組成物の製造方法 |
| JP2015052081A (ja) * | 2013-09-09 | 2015-03-19 | 日立化成株式会社 | 相容化樹脂の製造方法、熱硬化性樹脂組成物、プリプレグ、積層板、及びプリント配線板 |
| JP2015063585A (ja) * | 2013-09-24 | 2015-04-09 | 日立化成株式会社 | 熱硬化性樹脂組成物並びにそれを用いたプリプレグ、積層板、プリント配線板、及び実装基板 |
| WO2015155982A1 (ja) * | 2014-04-08 | 2015-10-15 | パナソニックIpマネジメント株式会社 | プリント配線板用樹脂組成物、プリプレグ、金属張積層板、プリント配線板 |
| JP2015207753A (ja) * | 2014-04-08 | 2015-11-19 | パナソニックIpマネジメント株式会社 | プリント配線板用樹脂組成物、プリプレグ、金属張積層板、プリント配線板 |
| US9775239B2 (en) | 2014-04-08 | 2017-09-26 | Panasonic Intellectual Property Management Co., Ltd. | Resin composition for printed wiring board, prepreg, metal-clad laminate, and printed wiring board |
| JP2016147986A (ja) * | 2015-02-13 | 2016-08-18 | パナソニックIpマネジメント株式会社 | プリント配線板用樹脂組成物、プリプレグ、金属張積層板およびプリント配線板 |
| JPWO2019176074A1 (ja) * | 2018-03-15 | 2021-04-08 | 昭和電工マテリアルズ株式会社 | エポキシ樹脂、エポキシ樹脂組成物、樹脂シート、bステージシート、cステージシート、硬化物、樹脂付金属箔、金属基板、及びパワー半導体装置 |
| JP7115538B2 (ja) | 2018-03-15 | 2022-08-09 | 昭和電工マテリアルズ株式会社 | エポキシ樹脂、エポキシ樹脂組成物、樹脂シート、bステージシート、cステージシート、硬化物、樹脂付金属箔、金属基板、及びパワー半導体装置 |
| US11795293B2 (en) | 2018-03-15 | 2023-10-24 | Resonac Corporation | Epoxy resin, epoxy resin composition, resin sheet, B-stage sheet, C-stage sheet, cured product, metal foil with resin, metal substrate, and power semiconductor device |
| JP2019194345A (ja) * | 2019-07-11 | 2019-11-07 | パナソニックIpマネジメント株式会社 | プリント配線板用樹脂組成物、プリプレグ、金属張積層板およびプリント配線板 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20130172459A1 (en) | 2013-07-04 |
| KR20130095730A (ko) | 2013-08-28 |
| JP5857514B2 (ja) | 2016-02-10 |
| EP2602277B1 (en) | 2014-11-05 |
| EP2602277A4 (en) | 2013-07-17 |
| JP2012052110A (ja) | 2012-03-15 |
| EP2602277A1 (en) | 2013-06-12 |
| TW201224001A (en) | 2012-06-16 |
| CN103189418A (zh) | 2013-07-03 |
| TWI512008B (zh) | 2015-12-11 |
| CN103189418B (zh) | 2015-08-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2012018126A1 (ja) | 相容化樹脂の製造法、熱硬化性樹脂組成物、プリプレグ及び積層板 | |
| JP2013216884A (ja) | 熱硬化性樹脂組成物、プリプレグ及び積層板 | |
| JP2010106150A (ja) | 熱硬化性樹脂組成物、及びこれを用いたプリプレグ,積層板及びプリント配線板 | |
| JP2014122339A (ja) | 熱硬化性樹脂組成物、プリプレグ、積層板、プリント配線板、及び実装基板、並びに熱硬化性樹脂組成物の製造方法 | |
| JP6028349B2 (ja) | 熱硬化性樹脂組成物、プリプレグ及び積層板 | |
| JP6163804B2 (ja) | 相容化樹脂の製造法、熱硬化性樹脂組成物、プリプレグ及び積層板 | |
| JP5736944B2 (ja) | 熱硬化性樹脂組成物、プリプレグ及び積層板 | |
| JP6106931B2 (ja) | 相容化樹脂、及びこれを用いた熱硬化性樹脂組成物、プリプレグ、積層板 | |
| JP5909916B2 (ja) | 樹脂の製造法、熱硬化性樹脂組成物、プリプレグ及び積層板 | |
| JP6221203B2 (ja) | 樹脂組成物、これを用いたプリプレグ及び積層板 | |
| JP2013108067A (ja) | 相溶化樹脂の製造方法、相溶化樹脂、熱硬化性樹脂組成物、プリプレグ及び積層板 | |
| JP6353633B2 (ja) | 相容化樹脂、熱硬化性樹脂組成物、プリプレグ及び積層板 | |
| JP6194603B2 (ja) | 熱硬化性樹脂組成物、プリプレグ及び積層板 | |
| JP6408752B2 (ja) | 相溶化樹脂、およびそれを用いたプリプレグ、積層板 | |
| JP6183081B2 (ja) | 相容化樹脂の製造方法、熱硬化性樹脂組成物、プリプレグ、積層板、及びプリント配線板 | |
| JP2015063608A (ja) | 熱硬化性樹脂組成物、これを用いたプリプレグ及びそれを用いた積層板 | |
| JP2013189579A (ja) | 熱硬化性樹脂組成物、およびこれを用いたプリプレグ、積層板 | |
| JP2010106149A (ja) | 熱硬化性樹脂組成物、及びこれを用いたプリプレグ,積層板 | |
| JP6425873B2 (ja) | 熱硬化性樹脂組成物、プリプレグ及び積層板 | |
| JP6217280B2 (ja) | 熱硬化性樹脂組成物並びにそれを用いたプリプレグ、積層板、プリント配線板、及び実装基板 | |
| JP2015063613A (ja) | プリプレグ及び、それを用いた積層板、プリント配線板 | |
| JP2015063609A (ja) | 熱硬化性樹脂組成物、これを用いたプリプレグ及び積層板 | |
| JP2015063614A (ja) | 相容化樹脂を用いたプリプレグ及びこれを用いた積層板、配線板 | |
| JP2015063605A (ja) | 有機繊維基材を用いたプリプレグ、及びそれを用いた積層板 | |
| JP2015063606A (ja) | 有機繊維基材を用いたプリプレグ、及びそれを用いた積層板 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11814751 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20137002782 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011814751 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13814350 Country of ref document: US |