WO2011007900A1 - 生体組織から単離できる多能性幹細胞 - Google Patents
生体組織から単離できる多能性幹細胞 Download PDFInfo
- Publication number
- WO2011007900A1 WO2011007900A1 PCT/JP2010/062480 JP2010062480W WO2011007900A1 WO 2011007900 A1 WO2011007900 A1 WO 2011007900A1 JP 2010062480 W JP2010062480 W JP 2010062480W WO 2011007900 A1 WO2011007900 A1 WO 2011007900A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- cell
- negative
- muse
- pluripotent stem
- Prior art date
Links
Images









































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0607—Non-embryonic pluripotent stem cells, e.g. MASC
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0603—Embryonic cells ; Embryoid bodies
- C12N5/0605—Cells from extra-embryonic tissues, e.g. placenta, amnion, yolk sac, Wharton's jelly
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0696—Artificially induced pluripotent stem cells, e.g. iPS
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/28—Bone marrow; Haematopoietic stem cells; Mesenchymal stem cells of any origin, e.g. adipose-derived stem cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/48—Reproductive organs
- A61K35/50—Placenta; Placental stem cells; Amniotic fluid; Amnion; Amniotic stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
- C12N15/1138—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing against receptors or cell surface proteins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/13—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells
- C12N2506/1346—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells
- C12N2506/1353—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells from bone marrow mesenchymal stem cells (BM-MSC)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2509/00—Methods for the dissociation of cells, e.g. specific use of enzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0662—Stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0662—Stem cells
- C12N5/0663—Bone marrow mesenchymal stem cells (BM-MSC)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0662—Stem cells
- C12N5/0667—Adipose-derived stem cells [ADSC]; Adipose stromal stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0662—Stem cells
- C12N5/0668—Mesenchymal stem cells from other natural sources
Definitions
- the present invention relates to pluripotent stem cells derived from living tissue.
- ICM inner cell mass
- pluripotent stem cells such as planarians and newts are present in adult mammals.
- MSC bone marrow mesenchymal cell fraction
- the bone marrow mesenchymal cell fraction is a group of cells containing various cells, and its differentiation ability is diverse, but the body is not clear, and in order to differentiate into specific cells, stimulation with specific compounds and genes It was necessary to introduce a differentiation induction system.
- iPS cells induced pluripotent stem cells
- Patent Document 1 Patent Document 2, Non-Patent Document 3, etc.
- an induction operation using a specific substance is required to introduce a specific gene or a specific compound into a somatic cell into a dermal fibroblast fraction (dermal fibroblast) that is a mesenchymal cell.
- Dermal fibroblast dermal fibroblast fraction
- An object of the present invention is to provide a method for directly obtaining pluripotent stem cells from a living tissue and to provide pluripotent stem cells obtained by the method.
- MSC bone marrow mesenchymal cell fraction
- the present inventors have found that characteristic cell clusters are formed at a very low frequency from untreated human MSC cells. It was. The appearance of the initial cell mass was very similar to ES cells. However, unlike ES cells, the cells stopped growing indefinitely when they reached a certain size for a certain period of time, and then the cells stopped growing and became a heterogeneous population containing various cells such as hair and pigment cells.
- the inventor gives stress stimulation by various methods when culturing mesenchymal cells or mesodermal cells such as bone marrow mesenchymal cell fraction and skin fibroblast fraction (for example, serum-free Culture, Hank's Balanced Salt Solution (HBSS), hypoxic culture, intermittent short-time trypsin culture for a total of 3 hours, long-time trypsin culture for 8 hours or 16 hours, etc.), collecting living cells Then, suspension culture (referred to as MC culture) was performed in a methylcellulose (MC) -containing medium. As a result, formation of embryoid body-like cell clusters having various sizes up to a maximum diameter of 150 ⁇ m was observed.
- mesenchymal cells or mesodermal cells such as bone marrow mesenchymal cell fraction and skin fibroblast fraction
- HBSS Hank's Balanced Salt Solution
- hypoxic culture intermittent short-time trypsin culture for a total of 3 hours, long-time trypsin culture for 8 hours or 16 hours,
- the present inventors investigated the characteristics of cells in the obtained embryoid body-like cell cluster, and found that the cells had characteristics of pluripotent stem cells. Furthermore, the present inventors have found that the cells in the obtained embryoid body-like cell cluster have characteristics that the pluripotent stem cells previously reported do not have, and further, the cells in the obtained cell cluster The present inventors have investigated the expression protein of, and found that the expression pattern is different from that of pluripotent stem cells such as ES cells and iPS cells which have been reported conventionally.
- the present inventors have further found that SSEA-3 is expressed as a surface antigen of the pluripotent stem cells, and that the pluripotent stem cells can be isolated from living tissues using the expression of SSEA-3 as an index. I found.
- the present inventors have described that the pluripotent stem cell is a novel pluripotent stem cell, which is different from the conventionally reported pluripotent stem cells such as ES cells and iPS cells, without directing manipulation from a living tissue.
- the present inventors have found that it is an obtained pluripotent stem cell and have completed the present invention.
- the present inventors have termed pluripotent stem cells Muse cells (Mu ltilineage-differentiating S tress E nduring cells). That is, the present invention is as follows.
- [1] SSEA-3-positive pluripotent stem cells that can be isolated from living tissue.
- the pluripotent stem cells can be isolated from a culture of a living tissue such as cultured fibroblasts or bone marrow stem cells, or can be isolated as a single cell.
- pluripotent stem cells [5] CD1 negative, CD117 negative, CD146 negative, CD271 negative, NG2 negative, vWF negative, Sox10 negative, Snai1 negative, Slug negative, Tyrp1 negative and Dct negative [1] or [2] pluripotent stem cells.
- the pluripotent stem cell according to any one of [1] to [6] which has the ability to differentiate into three germ layers.
- the pluripotent stem cell of the present invention has the ability to differentiate into three germ layers by in vitro adhesion culture, and can differentiate into skin, liver, nerve, muscle, bone, fat, etc. by induction culture in vitro .
- the pluripotent stem cell according to any one of [1] to [7], which does not show neoplastic growth.
- the pluripotent stem cell of the present invention has the property that it grows in suspension culture at a growth rate of about 1.3 days but stops growing in about 10 days, and further, when transplanted to the testis, it does not become cancerous for at least half a year. Has properties.
- the pluripotent stem cell of the present invention can be proliferated by repeating the operations of suspension culture and adhesion culture. Moreover, the pluripotent stem cell of this invention carries out asymmetric division like other somatic stem cells.
- the pluripotent stem cell of any one of [1] to [10] which has a high phagocytic ability.
- olfactory receptor family 8, subfamily G, member 2 (OR8G2); olfactory receptor, family 7, subfamily G, member 3 (OR7G3); olfactory receptor, family 4, subfamily D, member 5 (OR4D5); olfactory receptor, family 5, subfamily AP, member 2 (OR5AP2); olfactory receptor, family 10, subfamily H, member 4 (OR10H4); olfactory receptor, family 10, subfamily T, member 2 (OR10T2); olfactory receptor, family 2, subfamily M, member 2 (OR2M2); olfactory receptor, family 2, subfamily T, member 5 (OR2T5); olfactory receptor, family 7, subfamily D, member 4 (OR7D4); olfactory receptor, family 1, subfamily L, member 3 (OR1L3); olfactory receptor, family 8, subfamily G, member 2 (OR8G2); olfactory receptor, family 7, subfamily G, member 3 (OR7G3); o
- the pluripotent stem cell according to any one of [1] to [12], wherein at least one of the following five chemokine receptors is positive: chemokine (C-C motif) receptor 5 (CCR5); chemokine (C-X-C motif) receptor 4 (CXCR4); chemokine (C-C motif) receptor 1 (CCR1); Duffy blood group, chemokine receptor (DARC); and chemokine (C-C-C motif) receptor 7 (CXCR7).
- the pluripotent stem cell according to any one of [1] to [13], which is derived from a mesodermal or mesenchymal tissue.
- [17] A method for isolating a pluripotent stem cell or a pluripotent cell fraction, comprising exposing living tissue-derived cells to cell stress and recovering surviving cells.
- Cell stress is protease treatment, culture under hypoxic conditions, culture under low phosphate conditions, culture under serum starvation, culture under sugar starvation, culture under radiation exposure, heat shock
- the pluripotency of [17] selected from culture under exposure to, culturing in the presence of harmful substances, culturing in the presence of active oxygen, culturing under mechanical stimulation and culturing under pressure treatment
- a pluripotent stem cell that is a derivative or derived cell of the pluripotent stem cell of any one of [1] to [14].
- Examples of the derived cells or induced cells include cells induced by gene introduction or compound addition.
- the iPS cell derived from the stem cell of this invention is mentioned.
- a differentiated cell which is a derivative cell or induced cell of the pluripotent stem cell of any one of [1] to [14].
- a pharmaceutical composition comprising the pluripotent stem cell of any one of [1] to [14] and [20].
- a pharmaceutical composition comprising the differentiated cell of [21].
- This specification includes the contents described in the specification and / or drawings of US Provisional Application 61 / 213,788 and US Provisional Application 61 / 290,159, which are the basis of priority of the present application.
- FIG. 1-1 is a figure which shows the relationship between the mesenchymal cell fraction, Muse cells, and embryoid body-like cell clusters derived from Muse cells.
- SSEA-3-positive cells are directly separated and suspended culture without applying stress for a long time, thereby obtaining an analid body-like cell mass derived from Muse cells.
- FIG. 1-2 is a diagram showing a method of growing Muse cells in large quantities.
- FIG. 2 is a graph showing factors having a high expression ratio in the embryoid body-like cell cluster derived from Muse cells / the untreated cell fraction.
- FIG. 3 is a diagram showing factors having a high expression ratio in embryoid body-like cell clusters derived from Muse cells / human ES cells.
- FIG. 4 is a diagram showing a protocol for MACS sorting.
- FIG. 5 shows trypan blue after 16 hours of long-term trypsin treatment of human fibroblast (H-fibroblast) fraction (FIG. 5a) and after vortexing for 3 minutes at 1800-2200 rpm / min (FIG. 5b). It is a photograph which shows the removal of dead cells by a staining image.
- FIG. 6 is a photograph of various cells.
- FIG. 6a shows one cell (bar is 10 ⁇ m) in the Muse cell-rich fraction
- FIG. 6b shows an embryoid somatic cell mass derived from human ES cells (bar is 25 ⁇ m)
- FIG. 6c shows an embryo derived from a Muse cell having a diameter of about 25 ⁇ m.
- FIG. 6d is an alkaline phosphatase-stained image of the human ES cell-derived cell mass on the 4th day (bar is 25 ⁇ m), and Figs. 6e to 6g are Muse cell-derived embryoid body-like cell masses. It is a photograph which shows the immuno-staining image of Oct3 / 4 (e), Sox2 (f), and PAR4 (g).
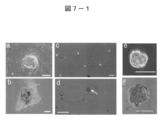
- FIG. 7-1 is a photograph showing characteristics of cell clusters derived from the H-fibroblast fraction and the human MSC (H-MSC) fraction.
- FIGS. 7-1a and b show cell clusters (bars are 100 ⁇ m) spontaneously generated in normal adhesion culture of untreated human MSC fractions.
- FIG. 7-1c and d show the state of MC culture after long-time trypsin treatment of the H-fibroblast-1 fraction on day 0 (c) and day 7 (d) (bar is 100 ⁇ m), FIG. The 1d arrow indicates a Muse cell-derived embryoid body-like cell mass.
- FIGS. 7-1e and f show Muse cell-derived embryoid body-like cell clusters formed from the H-fibroblast-1 fraction after 7 days of MC culture (bars are 50 ⁇ m).
- FIG. 7-2 is a photograph showing characteristics of cell clusters derived from the H-fibroblast fraction and the human MSC (H-MSC) fraction.
- FIG. 7-2g-l shows a Muse cell-derived embryoid body-like cell cluster (FIGS.
- FIG. 7-2g, i and k formed from the H-fibralst fraction and a Muse cell-derived embryoid body-like body formed from the H-MSC fraction.
- Nanog Fig. 7-2g and j
- Oct3 / 4 Fig. 7-2h
- SSEA-3 Fig. 7-2i
- PAR4 Fig. 7-2k
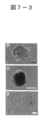
- FIG. 7-3 is a photograph showing the characteristics of the cell mass derived from the H-fibroblast fraction and the human MSC (H-MSC) fraction.
- FIG. 7-3m-o shows human ES cells (Fig. 7-3m), Muse cell-derived embryoid body-like cell mass derived from H-fibroblast fraction (Fig. 7-3n) and untreated H-fibroblast-1 fraction (Fig. 7-3 shows the result of alkaline phosphatase staining in o) (bar is 50 ⁇ m).
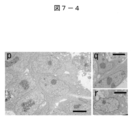
- FIG. 7-4 is an electron micrograph showing the characteristics of cell clusters derived from the H-fibroblast fraction and the human MSC (H-MSC) fraction.
- FIG. 7-4p-r shows human ES cell embryoid bodies (FIG. 7-4p, MC culture day 3), Muse cell-derived embryoid body-like cell mass derived from H-fibroblast-1 fraction (FIGS.
- FIG. 8-1 is a diagram showing the clonality and self-renewal of Muse cell-derived embryoid body-like cell clusters (M-cluster), and is an overview of experiments conducted to determine the clonality and self-renewal of Muse cells.
- FIG. 8-2 is a diagram showing the proliferation rate of Muse cells in floating cells.
- FIG. 8-3 is a diagram showing a normal karyotype of cells (clonally expanded cells) grown from a single Muse cell-derived embryoid body-like cell cluster (derived from H-fibroblast-1, first generation (cycle)). .
- FIG. 8-1 is a diagram showing the proliferation rate of Muse cells in floating cells.
- FIG. 8-3 is a diagram showing a normal karyotype of cells (clonally expanded cells) grown from a single Muse cell-derived embryoid body-like cell cluster (derived from H-fibroblast-1, first generation (cycle)). .
- FIG. 9-1 is a diagram showing differentiation of Muse cell-derived embryoid body-like cell clusters.
- a-c is an immunostaining showing the localization of ⁇ -smooth muscle actin and neurofilament (FIGS. 9-1a and b) and ⁇ -fetoprotein (FIG. 9-1c) of a cluster of differentiated cells derived from the H-fibroblas-1 fraction Image is the bar ( Figure 9-1a is 500 ⁇ m, Figures 9-1b and c are 50 ⁇ m).
- the arrowhead in FIG. 9-1a shows the adhered Muse cell-derived embryoid body-like cell mass.
- FIG. 9-2 shows cells in which spontaneous differentiation was induced by culturing the first and third generation Muse cell-derived embryoid body-like cell clusters (Cluster) derived from the untreated cell fraction and the H-fibroblast fraction on gelatin.
- Cluster Muse cell-derived embryoid body-like cell clusters
- ⁇ -FP ⁇ -fetoprotein
- GATA6, MAP-2 and Nkx2.5 expression are shown.
- human fetal liver was used for ⁇ -FP
- whole human embryos were used for GATA6, MAP-2 and Nkx2.5.
- Fig. 9-3e-l shows the testis of an immunodeficient mouse administered with the rich Muse cell fraction.
- Fig. 9-3e-l shows the testis of an immunodeficient mouse administered with the rich Muse cell fraction.
- FIG. 9-3e shows intact testis as a control, mouse ES cells (mES cells) (8 weeks), MEF (feeder cells) (8 weeks), Muse cell-derived embryoid body-like cell mass (M-cluster). ) (6 months) and Muse cell fraction (Muse) (6 months).
- Fig. 9-3f-i shows testicular tissue neurofilament M (Fig. 9-3f, stained green in the photograph), ⁇ -fetoprotein (Fig. 9) to which the Muse cell fraction or Muse cell-derived embryoid body-like cell mass was administered.
- -3 g stained in green in the photograph
- immunostained images of smooth muscle actin FIG. 9-3h, stained in red in the figure
- FIG. 9-3i show double-stained images of human mitochondria (stained in green) and smooth muscle actin (stained in red) (bar is 20 ⁇ m).
- Fig. 9-3j-l shows a histological image of the testis to which the Muse cell fraction was administered (Fig. 9j and k).
- the tube-like structure observed in FIG. 9k is stained with an antibody against human mitochondria (the bars are 500 ⁇ m in FIG. 9-3j and 50 ⁇ m in FIG. 9-3k-1).
- FIG. 10a is a diagram showing the results of quantitative PCR of factors involved in pluripotency and undifferentiated cell states of H-fibroblast (Fibro-1, Fibro-2) and H-MSC (MSC-1, MSC-2). (Part 2).
- the column pattern in the figure shows the gene expression level compared to the untreated cell fraction of the rich Muse cell fraction and the Muse cell-derived embryoid body-like cell mass (day 7).
- White indicates the ratio of the rich Muse cell group or Muse cell-derived embryoid body-like cell mass / untreated cell fraction greater than 1/3 and less than 3
- gray indicates the rich Muse cell fraction or Muse cell-derived embryoid-like
- the ratio of the somatic cell mass / untreated cell fraction is greater than 3
- the hatched line indicates that the ratio of the rich Muse cell fraction or Muse cell-derived embryoid somatic cell mass / untreated cell fraction is 1/3 Indicates a small one.
- FIG. 10b shows the telomerase activity of H-MSC-derived untreated cell fraction (Naive), rich Muse cell fraction (Muse), and Muse cell-derived embryoid body-like cell mass (M-cluster) (day 7).
- FIG. A heat inactivated sample (Heat) was used as a negative control.
- FIG. 11 is a diagram showing the results of DNA microarray analysis of an untreated cell fraction derived from the H-fibroblast fraction and the H-MSC fraction, a rich Muse cell fraction, and a Muse cell-derived embryoid body-like cell mass.
- FIG. 12 is a photograph showing an embryoid body-like cell mass formed by MC culture of Muse cells directly collected as SSEA-3 / CD105 double positive cells from the mononuclear cell component of human bone marrow.
- FIG. 12a shows a Muse cell-derived embryoid body-like cell mass formed by MC culture of a mononuclear cell fraction isolated from human bone marrow and treated with trypsin for 8 hours for a long time (8 hr-hBM-MC, 7 days). Shown (bar is 100 ⁇ m).
- FIG. 12 b shows an alkaline phosphatase-stained image of the Muse cell-derived embryoid body-like cell mass formed by 8 hr-hBM-MC (7 days) (bar is 50 ⁇ m).
- FIG. 12a shows a Muse cell-derived embryoid body-like cell mass formed by MC culture of Muse cells directly collected as SSEA-3 / CD105 double positive cells from the mononuclear cell component of human bone marrow
- FIG. 13 shows untreated H-MSC-1 fraction (naive 1), untreated H-MSC-2 fraction (naive 2) (both negative controls), and human bone marrow-derived mononuclear treated with trypsin for 8 hours.
- Muse cell-derived embryoid body-like cell mass formed from cell fraction (8hr-hBM) or human bone marrow-derived mononuclear cell fraction (naive hBM) without trypsin treatment is cultured on gelatin to induce spontaneous differentiation.
- It is a figure which shows the result of RT-PCR analysis of (alpha) -fetoprotein ((alpha) -FP), GATA6, MAP-2, and Nkx2.5 in the induced cell group.
- FIG. 14 is a diagram showing the results of FACS analysis of the H-fibroblast fraction (untreated cells) and the H-MSC fraction (untreated cells).
- FIG. 15-1 shows an SSEA-3 positive cell (15-1a left) in the untreated cell fraction and a single Muse cell-derived embryoid body-like cell cluster derived from SSEA-3 positive cells collected by FACS sorting. It is a photograph which shows the dyeing
- FIG. 15-2 is a photograph showing a stained image showing the localization of Numblike (green), which is a factor involved in asymmetric division during cell division of Muse cells (H-fibroblast). The bar in the figure is 10 ⁇ m.
- Fig. 15-3 is an electron micrograph of SSEA-3 negative cells (Fig. 15-3c) and SSEA-3 positive cells (Fig. 15-3d) derived from H-fibroblast. The bar in the figure is 5 ⁇ m.
- Fig. 15-4 shows the results of Oct3 / 4 (green) (Fig. 15-4e), Sox2 (green) (Fig. 15-4f) and SSEA-3 (red) (Fig. 15-4g) of H-fibroblast-derived Muse cells. It is a photograph which shows a dyeing
- FIG. 16-1 is a photograph showing differentiation of a GFP-labeled SSEA-3-positive Muse cell fraction in a hyperimmune-deficient mouse (Nog mouse) damaged tissue.
- FIG. 16-1 N and O are GFP positive cells in the compression-injured spinal cord (after 4 weeks), and express neurofilament (red) and human Golgi complex (white). O is an enlarged image of a portion surrounded by a square N.
- FIG. 16-1P is a GFP positive labeled cell of damaged liver (after 4 weeks), and expresses human albumin (red) and human Golgi complex (white).
- FIG. 16-2 is a photograph showing the expression of human albumin in the liver transplanted with SSEA-3-positive Muse cells examined by RT-PCR.
- FIG. 16-3 is a photograph showing differentiation of a GFP-labeled SSEA-3-positive Muse cell fraction in a hyperimmune-deficient mouse (Nog mouse) damaged tissue, and a muscle expressing human dystrophin (red) (after 3 weeks) ) Is a photograph showing GFP positive cells.
- FIG. 17-1 is a photograph showing differentiation of cells grown from a Muse cell-derived embryoid body-like cell cluster formed from a single Muse cell.
- FIGS. 17A to 17D show the results of nerve induction, A shows the formed sphere, and, as the immunostaining data of shperle, A shows nestin, B shows Musashi, and C shows NuroD expression.
- E is a MAP-2 positive cell obtained by further differentiating these spheres into neural cells.
- FIGS. 17-1F to G show the results of bone cell induction and show the expression of osteocalcin (F) and ALP (G).
- FIGS. 17-1 H and I show the results of adipocyte induction, H shows cells containing oil droplets, and I shows the results of oil red staining.
- FIG. 17-1J shows the results of liver cell induction and shows ⁇ -fetoprotein positive cells.
- FIG. 17-2 is a photograph showing the expression of human albumin and human ⁇ -fetoprotein in hepatocyte-derived cells examined by RT-PCR.
- FIG. 18-1 is a photograph showing expression of Sox10, Snail1, Slug, Tyrp1 and Dct in SSEA-3-positive Muse cells examined by RT-PCR.
- FIG. 18-2 is a diagram showing expression of NG2, CD34, vWF, CD117, CD146 and CD271 analyzed by FACS.
- pericyte marker NG2 pericyte marker NG2
- endothelial progenitor cell markers CD34 and vWF were negative, and SSEA-3 positive cells were also negative.
- CD117, a melanoblast marker, CD146, a pericyte marker (CD146), and NCSC marker, CD271 were found to be slightly positive in untreated human skin fibroblasts (0.2%, respectively). 0.2% and 0.8%), which are SSEA-3 negative cells and are not considered Muse cells.
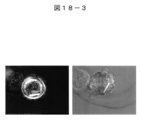
- FIG. 18-3 is a diagram showing that Muse cells phagocytosed ferrite.
- FIG. 19 is a photograph showing the formation of iPS cells prepared from Muse cells
- FIG. 19a shows the morphology of human iPS cells derived from skin fibroblast (NHDF) -derived Muse cells
- FIGS. Shows the expression of pluripotent cell markers (b is Nonog, c is Oct3 / 4, d is Sox2, e is SSEA-3, and f is Tra-1-60).
- FIG. 20 is a photograph showing the results of immunohistochemistry of Nonog (E), Oct3 / 4 (F), Sox2 (G) and Tra-1-81 (H).
- FIG. 19a shows the morphology of human iPS cells derived from skin fibroblast (NHDF) -derived Muse cells
- FIGS. Shows the expression of pluripotent cell markers (b is Nonog, c is Oct3 / 4, d is Sox2, e is SSEA-3, and f is Tra-1-60).
- FIG. 20 is a photograph showing the results of immunohisto
- FIG. 21 shows pluripotency examined by RT-PCR of colonies (( ⁇ )-1, ( ⁇ )-2) grown from Muse-derived iPS cells (Mi-1, Mi-2) and SSEA-3 negative cells. It is a photograph which shows expression of a marker.
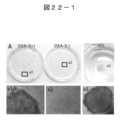
- Fig. 22-1 shows Tra- of colonies formed from SSEA-3 positive and negative cells 30 days after Oct3 / 4, Sox2, Klf4, c-Myse introduced with retrovirus and then cultured on feeder cells MEF. It is a photograph which shows the result of 1-81 immuno-staining. Human ES cells are used as a control. Colonies (a1) and human ES cells (a2) from SSEA-3 positive cells are Tra-1-81 positive, but all colonies from SSEA-3 negative cells are negative.
- FIG. 222-2 shows SSEA-3 positive and negative cell pluripotency markers (endogenous Oct3 / 4 (endo Oct), endogenous Sox2) at the stage of 30-day culture with MEF as in 22-1 It is a photograph which shows expression of Nanog, endogenous Klf4 (endo Klf4), Rex1, and UTF1). In the SSEA-3 negative cell group, signals of Sox2 and Nanog are not seen.
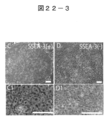
- FIG. 22-3 shows colonies of iPS cells derived from Muse cells (Muse cell-derived iPS cells) (FIGS. 22-3C and C1) and colonies grown from SSEA-3 negative cells (FIGS. 22-3D and D1). It is a photograph.
- FIG. 22-3C and C1 colonies of iPS cells derived from Muse cells (Muse cell-derived iPS cells)
- FIG. 22-3D and D1 It is a photograph.
- FIG. 23-1 is a photograph showing the in vitro differentiation of iPS cells derived from skin fibroblast (NHDF) -derived Muse cells.
- Fig. 23-1i shows the expression of ⁇ -fetoprotein (green) as an endoderm marker and smooth muscle actin (red, blue as DNA) as a mesoderm marker, and
- Fig. 23-1j shows a nerve as an ectoderm marker. The expression of the filament (green) is shown.
- FIG. 23-2 is a diagram showing the results of RT-PCR analysis of in vitro differentiation of iPS cells derived from Muse cells, and shows the expression of three germ layer markers.
- FIG. 23-3 is a photograph showing the tissue structure of teratoma formed from iPS cells derived from skin fibroblast (NHDF) -derived Muse cells, and various iPS cells were obtained by staining with HE (Hematoxylin and eosin). It shows that it has differentiated into an organization.
- Fig. 23-3m is cartilage
- Fig. 23-3n is muscle
- Fig. 23-3o is neuroepithelium
- Fig. 23-3p is pigmented epithelium
- Fig. 23-3q is A columnar epithelium is shown.
- FIG. 24 is a diagram showing the results of Bisulfite (bisulfite) sequence of Nanog gene and Oct3 / 4 gene of SSEA-3 negative cell fraction, Muse cell-derived embryoid body-like cell cluster, and Muse-derived iPS cell.
- the numerical value in each column indicates the position of CpG relative to the downstream of the transcription start site (TSS).
- White circles indicate unmethylated cytosine, and black circles indicate methylated cytosine.
- FIG. 25 is a diagram showing the results of quantitative PCR of factors related to the cell cycle in untreated fibroblasts (Naive), Muse cell-derived embryoid body-like cell clusters (Cluster), and iPS cells.
- the white column indicates that the ratio of the Muse fraction to the untreated cells or the Muse cell-derived embryoid body-like cell mass is less than 2 and greater than 1/2.
- a filled column indicates that the ratio is greater than 2, and a shaded column indicates that the ratio is less than 1/2.
- “*” indicates that the amount of the expressed gene is larger in the Muse cell-derived embryoid body-like cell mass than iPS, and “**” indicates the amount of the expressed gene. The amount is larger in iPS than the Muse cell-derived embryoid body-like cell mass.
- FIG. 26 is a diagram showing the results of quantitative PCR of factors related to pluripotent and undifferentiated cell states in untreated fibroblasts (Naive), Muse cell-derived embryoid body-like cell clusters (Cluster), and iPS cells. It is. The meaning of each column is the same as in FIG.
- FIG. 27 is a diagram summarizing the papers on the induction efficiency of iPS cell lines prepared in human and mouse models.
- FIG. 27 shows a combination of transcription factors that induce nuclear reprogramming.
- the present invention relates to a pluripotent stem cell or pluripotent stem cell fraction obtainable directly from living body tissue, a method for isolating the pluripotent stem cell or the pluripotent stem cell fraction, and It is a pluripotent stem cell or pluripotent stem cell fraction derived from a biological tissue obtained by the method.
- the pluripotent stem cells of the present invention are referred to as Muse cells (multilineage differentiating stress extending cells).
- the term “cell fraction” refers to a cell group containing at least a certain amount of cells to be isolated.
- the pluripotent stem cell fraction includes a group of cells containing 1% or more, 10% or more, 30% or more, 50% or more, 70% or more, 90% or more, or 95% or more of pluripotent stem cells.
- a cell cluster obtained by culturing pluripotent stem cells and a cell group enriched with pluripotent stem cells are included.
- the cell fraction may be referred to as a substantially uniform cell fraction.
- a living body refers to a living body of a mammal, and refers to an animal body that has developed to some extent.
- the living body does not include embryos whose developmental stage is earlier than the fertilized egg or blastocyst stage, but includes embryos in the developmental stage after the blastocyst stage including the fetus and blastocyst.
- Mammals include, but are not limited to, primates such as humans and monkeys, rodents such as mice, rats, rabbits, guinea pigs, cats, dogs, sheep, pigs, cows, horses, donkeys, goats, ferrets, etc. .
- the pluripotent stem cells of the present invention are clearly distinguished from embryonic stem cells (ES cells) and embryonic germ stem cells (EG cells) in that they are derived from living tissue.
- Mesodermal tissue refers to tissue of mesoderm origin that appears in the early development of animals, and includes muscular tissue, connective tissue, circulatory tissue, excretory tissue, reproductive tissue, and the like.
- the pluripotent stem cell of the present invention can be obtained from skin tissues such as bone marrow fluid and dermal connective tissue.
- Mesenchymal tissue refers to tissues such as bone, cartilage, fat, blood, bone marrow, skeletal muscle, dermis, ligament, tendon, and heart.
- the pluripotent stem cell of the present invention can be obtained from bone marrow or skin. It can also be obtained from the umbilical cord.
- That cells can be obtained directly from tissue means that they can be isolated from tissue and obtained without artificial induction procedures such as compound treatment such as introduction of foreign genes or proteins, or administration of compounds.
- the foreign gene is not limited, but refers to a gene that can initialize the nucleus of a somatic cell, for example, an Oct family gene such as an Oct3 / 4 gene, a Klf family gene such as a Klf gene, a c-Myc gene, etc.
- Examples include Sox family genes such as Myc family genes and Sox2 genes.
- foreign proteins include proteins and cytokines encoded by these genes.
- examples of the compound include a low molecular weight compound that induces expression of a gene that can reprogram the somatic cell nucleus, DMSO, a compound that functions as a reducing agent, a DNA methylating agent, and the like.
- the pluripotent stem cells of the present invention are clearly distinguished from iPS (Induced Primitive Stem Cell) cells and ES cells in that they can be obtained directly from living bodies or tissues.
- iPS Induced Primitive Stem Cell
- ES cells in that they can be obtained directly from living bodies or tissues.
- the pluripotent cell of the present invention may be obtained without requiring reprogramming or induction of dedifferentiation.
- the pluripotent stem cells of the present invention are considered to be present in living mesodermal or mesenchymal tissues and the like. In the present invention, cells or cell fractions existing in these tissues are simply isolated. Release.
- the pluripotent stem cell of the present invention is present, for example, in the bone marrow, and may be supplied from the bone marrow to each tissue of the living body through blood or the like. For this reason, it can be isolated from bone marrow, living body tissues such as skin, and blood.
- a pluripotent stem cell means a cell having pluripotency, and has the following characteristics. (1) It expresses pluripotent markers such as Nanog, Oct3 / 4, SSEA-3, PAR-4, and Sox2.
- the pluripotent stem cells of the present invention are clearly distinguished from adult stem cells and tissue stem cells in that they have pluripotency.
- the pluripotent stem cells of the present invention are clearly distinguished from cell fractions such as bone marrow mesenchymal cells in that they are isolated as single cells or multiple cells having pluripotency. . Furthermore, the pluripotent stem cell of this invention has the following characteristics.
- the growth rate is relatively slow and the division cycle is 1 day or longer, for example, 1.2 to 1.5 days. However, it does not show infinite proliferation as shown by ES cells and iPS cells.
- Ii shows differentiation into endoderm, mesoderm and ectoderm when transplanted into immunodeficient mice. In ES cells and iPS cells, teratoma does not become cancerous for more than half a year compared to canceration in a short period of time.
- Low telomerase activity means, for example, telomerase activity comparable to that of human fibroblasts, or telomerase activity of 1/5 or less, preferably 1/10 or less that of Hela cells. That means.
- Viii Regarding the methylation status, the methylation levels of the Nanog and Oct3 / 4 promoter regions are low for iPS cells derived from Muse cells.
- Ix High phagocytic ability.
- X Does not show neoplastic growth. Here, not exhibiting neoplastic growth means that when suspension culture is performed, the growth stops when the cell mass (cluster) of a certain size is reached, and does not grow infinitely.
- the cell of the present invention is, for example, the following pluripotent stem cell.
- B A pluripotent stem cell having the characteristic (1) above, wherein the mesodermal or mesenchymal tissue of a living body is selected from the group consisting of bone marrow, skin, blood, umbilical cord, fat and the like.
- D The pluripotent stem cell according to (A) or (B), which does not become cancerous for at least half a year when transplanted to the testis.
- E The pluripotent stem cell according to (A) or (B), which does not exhibit infinite proliferation like ES cells and iPS cells.
- F A pluripotent stem cell derived from a mesodermal or mesenchymal tissue in a living body, which survives when cells such as a mesodermal or mesenchymal tissue in a living body are treated with a protease. Pluripotent stem cells that are resistant.
- the pluripotent stem cell of the present invention can be isolated by applying cell stress to cells such as mesodermal or mesenchymal tissue in a living body and collecting surviving cells.
- cell stress refers to external stress, including protease treatment, culture under low oxygen conditions, culture under low phosphate conditions, culture under serum starvation conditions, culture under sugar starvation conditions, under radiation exposure Exposure to stress by culturing in the presence of heat shock, culturing in the presence of toxic substances, culturing in the presence of harmful substances, culturing in the presence of active oxygen, culturing under mechanical stimulation, culturing under pressure treatment, etc. That means.
- protease treatment that is, culture in the presence of protease is preferable.
- the protease is not limited, and serine proteases such as trypsin and chymotrypsin, aspartic proteases such as pepsin, cysteine proteases such as papain and chymopapain, metalloproteases such as thermolysin, glutamate protease, N-terminal threonine protease, and the like can be used.
- the concentration at which protease is added to the culture is not limited, and it may be used at a concentration generally used when peeling adherent cells cultured in a petri dish or the like.
- the pluripotent stem cell of the present invention can be said to be a stem cell resistant to the above external stress, for example, a cell resistant to trypsin.
- Biological mesodermal or mesenchymal tissues are not limited, and include bone marrow mononuclear cells, fibroblast fractions such as skin cells, dental pulp tissues, eyeball tissues, hair root tissues, and the like.
- As the cells cultured cells or cells collected from tissues can be used.
- bone marrow cells and skin cells are desirable, and examples thereof include a human bone marrow mesenchymal cell (MSC) fraction and a human skin fibroblast fraction.
- the bone marrow mesenchymal cell fraction can be obtained by culturing a bone marrow puncture solution for 2 to 3 weeks. Most of the cells of the above-mentioned various stressed tissues are killed, and the pluripotent stem cells of the present invention are contained in the surviving cells.
- the pluripotent stem cell or pluripotent cell fraction of the present invention can be further isolated from the cells thus obtained using the following surface marker as an index.
- the pluripotent stem cell or pluripotent cell fraction of the present invention can be obtained. It can be isolated. Since cells of the injured tissue are exposed to stress, in the present invention, injured mesodermal tissue or mesenchymal tissue or the like may be cultured in the living organism. It is said that cell stress is applied to systemic tissue cells. As an example, a method for treating these cells with trypsin will be described.
- the trypsin concentration at this time is not limited.
- the trypsin concentration may be used within the concentration range used when peeling the adherent culture adhered to the culture vessel, and is 0.1 to 1%, preferably 0. 1 to 0.5% is exemplified.
- cells derived from mesodermal or mesenchymal tissue containing 100 to 500,000 cells can be exposed to external stress by incubation in 5 ml of a trypsin solution having the above concentration.
- the trypsin treatment time is about 5 to 24 hours, preferably about 5 to 20 hours.
- trypsin treatment for 8 hours or more, for example, treatment for 8 hours or 16 hours is referred to as long-time trypsin treatment.
- the pluripotent stem cells (Muse cells) of the present invention are contained in a concentrated state in the cell population that survived this external stress. This cell population is referred to as the Muse enriched population.
- the abundance of Muse cells in the rich Muse cell fraction varies depending on the stress treatment method.
- the medium and culture conditions used for culturing cells derived from mesodermal or mesenchymal tissues of a living body may be those employed for normal animal cell culture. Further, a known stem cell culture medium may be used.
- the present invention also includes pluripotent stem cells that are derived or derived cells of pluripotent stem cells that can be obtained directly from the mesodermal or mesenchymal tissues of the living body of the present invention.
- Derived cells or induced cells refer to cells or cell groups obtained by culturing the pluripotent stem cells, or cells obtained by performing artificial induction operations such as introduction of foreign genes into the pluripotent stem cells, and progeny Includes cells.
- the iPS cells reported at the time of the present invention are said to be cells induced by pluripotent stem cells as a result of reprogramming by introducing a foreign gene into a differentiated cell of biological tissue such as skin fibroblasts.
- a differentiated cell of biological tissue such as skin fibroblasts.
- Embryoid body-like (EB body) -like cell clusters can be obtained by suspension culture of the pluripotent stem cells of the present invention, and the present invention provides the embryoid body-like cell clusters and embryoid body-like bodies.
- the cells contained in the cell mass are also included.
- the embryoid body is formed as a cell mass by suspension culture of the pluripotent stem cells of the present invention.
- an embryoid body obtained by culturing the pluripotent stem cell of the present invention may be referred to as a Muse cell-derived embryoid body-like cell mass (referred to as an M cluster).
- M cluster Muse cell-derived embryoid body-like cell mass
- As a suspension culture method for forming embryoid body-like cell clusters culture using a medium containing a water-soluble polymer such as methylcellulose (Nakahata, T. et al., Blood 60, 352-361 (1982)). And hanging drop culture (Keller, J. Physiol. (Lond) 168: 131-139, 1998).
- the present invention also includes an embryoid body-like cell cluster obtained by self-renewal from the embryoid body-like cell cluster, cells contained in the embryoid body-like cell cluster, and pluripotent stem cells.
- self-renewal refers to culturing cells contained in an embryoid body-like cell cluster to form an embryoid body-like cell cluster again. The self-renewal may be repeated one to several times.
- the present invention also includes cells and tissues differentiated from any of the embryoid body-like cell clusters and cells contained in the embryoid body-like cell cluster. FIG.
- 1-1 shows the relationship between the mesenchymal cells (human fibroblasts, human bone marrow mesenchymal cells, fresh bone marrow fluid) fraction, Muse cells, and Muse cell-derived embryoid body-like cell masses.
- stress stimulation such as long term trypsin treatment (LTT) is applied to a mesenchymal cell-like cell mass for a long time, a cell fraction containing a large number of Muse cells is obtained (Muse cell-rich fraction).
- LTT long term trypsin treatment
- an embryoid body-like cell cluster derived from Muse cells can be obtained by directly separating SSEA-3 positive cells and subjecting them to suspension culture without applying long-term stress. Muse cells start to proliferate when transferred to adherent cultures even if the growth is once stopped in suspension culture. It is possible to proliferate Muse cells in large quantities by repeating the separation using suspension culture-adhesion culture-SSEA-3 expression as an index (FIG. 1-2).
- the pluripotent stem cell or pluripotent cell fraction of the present invention can also be isolated directly from living tissue without exposure to cell stress.
- the pluripotent stem cell or pluripotent stem cell fraction of the present invention is isolated by the following method without introducing a foreign gene from a mesodermal or mesenchymal tissue or the like of a living body, without an induction operation. be able to.
- the biological tissue is not limited, and examples thereof include mesodermal or mesenchymal tissue such as bone marrow, skin, and umbilical cord.
- the mononuclear cell fraction of bone marrow can be used. Isolation can be performed using cell surface markers that are highly expressed on the surface of Muse cells. For example, the expression of SSEA-3 can be used as an indicator.
- the pluripotent stem cell of the present invention is sometimes referred to as an SSEA-3-positive Muse cell.
- Muse cells express CD105, which is a pluripotent stem cell marker, are SSEA-3 positive, and are CD105 positive. Therefore, the expression of both SSEA-3 and CD105 can be isolated using as an index.
- the pluripotent stem cell of the present invention can be isolated as a single cell, and the isolated single cell can be grown by culture.
- this invention shall also include the pluripotent stem cell which can be isolated from the biological tissue of mammals other than a human by the marker equivalent to SSEA-3.
- Muse cells are negative for NG2, CD34, vWF (von Willebrand factor), c-kit (CD117), CD146, CD271 (NGFR). Furthermore, Sox10, Snai1, Slug, Tyrp1, and Dct are negative. Whether surface antigens such as NG2, CD34, vWF, CD117, CD146, and CD271 are negative or weakly expressed is an antibody against these antigens. Cells are labeled with an antibody labeled with a chromogenic enzyme, a fluorescent compound, or the like. Whether it is stained or not can be determined by microscopic observation or the like.
- cells can be immunostained using these antibodies to determine the presence or absence of surface antigens, or can be determined using magnetic beads to which the antibodies are bound. It can also be determined whether there is a surface antigen using a FACS or flow cytometer.
- a FACS or flow cytometer for example, FACSAria (manufactured by Becton Dickinson), FACS vantage (manufactured by Becton Dickinson), FACS Calibur (manufactured by Becton Dickinson) and the like can be used.
- expression of transcription factors such as Sox10, Snai1, Slug, Tyrp1, and Dct can be examined by a technique such as RT-PCR.
- these surface antigens are negative, when analyzed using FACS as described above, it means that they are not sorted as positive cells, or expression is not observed when expression is examined by RT-PCR, Even if it is expressed to such an extent that it cannot be detected by these techniques, it is considered negative in the present invention.
- measurement is performed simultaneously with cells such as hematopoietic stem cells that are known to be positive for the above-mentioned marker, and compared to these positive cells, it is hardly detected or may be negative when the expression level is significantly low. .
- the cells of the present invention can be isolated based on the antigenic properties of these cell surfaces.
- Muse cells can be isolated using SSEA-3 positivity as an indicator, and further can be isolated using CD105 expression as an indicator, but also NG2, CD34, vWF (von Willebrand factor) ), C-kit (CD117), CD146, CD271 (NGFR), Sox10, Snai1, Slug, Tyrp1 and Dct, at least one marker, for example, 2, 3, Non-expression of 4, 5, 6, 7, 8, 9, 10, or 11 markers can be isolated using as an indicator.
- CD117 and CD146 non-expression can be isolated as an index
- CD117, CD146, NG2, CD34, vWF and CD271 non-expression can be isolated as an index.
- the non-expression of the marker can be isolated using as an indicator.
- one or a plurality of pluripotent stem cells of the present invention can be directly isolated from a mesodermal or mesenchymal tissue or the like of a living body without culturing or the like. Is possible.
- the pluripotent stem cells of the present invention can be identified and isolated by visual observation of the cell morphology using a microscope or the like. After applying cell stress to mesodermal tissue or mesenchymal tissue in a living body, it may be isolated from a surviving cell group using a surface marker.
- the pluripotent stem cell or pluripotent cell fraction of the present invention is also characterized by high expression of other specific factors.
- Muse cells which are the pluripotent stem cells of the present invention, are obtained from the untreated bone marrow mesenchymal cell fraction or dermal fibroblast fraction, and further, the Muse cell-derived embryoid body (EB) is obtained by culturing the Muse cell. A cell-like mass is obtained.
- the factors expressed in Muse cells untreated cells, Muse-derived embryoid body-like cell clusters, and human ES cells, the factors highly expressed in Muse cells can be found.
- factors include gene transcripts, proteins, lipids, and sugars.
- FIG. 2 shows factors having a high expression ratio in Muse cell-derived embryoid body-like cell clusters / untreated cells. In particular, the ratio of the following 18 factors is high.
- FIG. 3 shows factors having a high expression ratio in Muse cell-derived embryoid body-like cell clusters / human ES cells. In particular, the ratio of the following 20 factors is high.
- A matrix metallopeptidase 1 (interstitial collagenase)
- B epiregulin
- C chitinase 3-like 1 (cartile glycoprotein-39)
- D Transscribed locus
- E chitinase 3-like 1 (cartile glycoprotein-39)
- F serlycin
- G MRNA full length insert cDNA clone EUROIMAGE 1913076 (H) Ras and Rab interactor 2
- I lumican
- J CLCA family member 2
- Chloride channel regulator K
- interleukin 8 L
- M dermatopontin
- N EGF, latrophilin and seven transmembrane domain containing 1
- O Insulin-like growth factor binding protein 1
- P solid carrier family 16, member 4 (monocarboxic acid transporter 5)
- Q serlycin
- R gremlin 2
- cysteine knot superfamily homolog
- Xenopus laevis insulin-like growth factor binding protein
- the pluripotent stem cell or pluripotent stem cell fraction of the present invention expresses factors of the odorant receptor (olfactory receptor) group and the chemokine receptor group other than the pluripotency marker. That is, it is characterized by being positive for a specific odorant receptor or chemokine receptor.
- Examples of the odorant receptor expressed in the pluripotent stem cell or pluripotent stem cell fraction of the present invention include the following 22 receptors. olfactory receptor, family 8, subfamily G, member 2 (OR8G2); olfactory receptor, family 7, subfamily G, member 3 (OR7G3); olfactory receptor, family 4, subfamily D, member 5 (OR4D5); olfactory receptor, family 5, subfamily AP, member 2 (OR5AP2); olfactory receptor, family 10, subfamily H, member 4 (OR10H4); olfactory receptor, family 10, subfamily T, member 2 (OR10T2); olfactory receptor, family 2, subfamily M, member 2 (OR2M2); olfactory receptor, family 2, subfamily T, member 5 (OR2T5); olfactory receptor, family 7, subfamily D, member 4 (OR7D4); olfactory receptor, family 1, subfamily L, member 3 (OR1L3); olfactory receptor, family
- the pluripotent stem cell or pluripotent stem cell fraction of the present invention expresses at least one of the olfactory receptors, or expresses at least one of the chemokine receptors.
- the pluripotent stem cell of the present invention migrates to the damaged tissue, engrafts, and differentiates in situ by the action of the migratory factor that binds to these odorant receptors and chemokine receptors.
- the migratory factor that binds to these odorant receptors and chemokine receptors.
- the specific migratory factor and the odorant receptor expressed on the cell surface migrate to and engraft each tissue, the liver (endoderm), Differentiated into skin (ectodermal), spinal cord (ectodermal), muscle (mesoderm) cells, and tissue can be regenerated.
- Rex1, Sox2, KLF-4, c-Myc, DPPA2, ERAS, GRB7, SPAG9, TDGF1, etc. are up-regulated.
- DAZL, DDX4, DPPA4, Stella, Hoxb1, PRDM1, SPRY2 and the like are up-regulated in the mass of Muse cells.
- expression of CD34 and CD117, which are hematopoietic stem cell markers, is not observed or is very low.
- the present invention includes not only Muse cells but also cell populations enriched with Muse cells, cell populations with expanded Muse cells, and cell populations with differentiated Muse cells, and further include Muse cells and cells derived from Muse cells. Includes research kits, cell chips, and therapeutic devices.
- the pluripotent stem cell of the present invention has pluripotency, and can differentiate into any tissue.
- the pluripotent stem cell or pluripotent cell fraction can be used for regenerative medicine and the like. For example, it can be used for regeneration of various tissues and various organs. Specific examples include skin, cerebral spinal cord, liver, and muscle.
- the pluripotent stem cell By administering the pluripotent stem cell or pluripotent stem cell fraction of the present invention directly or nearby to a damaged or damaged tissue, organ, etc., the pluripotent stem cell enters the tissue, organ, It can differentiate into cells peculiar to the tissue and contribute to regeneration and reconstruction of the tissue and organs. Further, systemic administration may be performed by intravenous administration or the like. In this case, for example, the pluripotent stem cell is directed to a damaged tissue or organ by homing or the like, reaches / invades, and then differentiates into a cell of the tissue or organ to regenerate the tissue or organ. , Can contribute to reconstruction.
- Administration can be performed, for example, by parenteral or oral administration such as subcutaneous injection, intravenous injection, intramuscular injection, intraperitoneal injection, or intrauterine injection into an embryo. Moreover, local administration or systemic administration may be sufficient. Local administration can be performed using a catheter, for example. The dose can be appropriately determined depending on the type and size of the organ or tissue to be regenerated.
- the organ to be regenerated is not limited, but bone marrow, spinal cord, blood, spleen, liver, lung, intestinal tract, eye, brain, immune system, circulatory system, bone, connective tissue, muscle, heart, blood vessel, pancreas, central nervous system , Peripheral nervous system, kidney, bladder, skin, epithelial appendages, breast-mammary gland, adipose tissue, and mucous membranes including mouth, esophagus, vagina, anus and the like.
- Diseases to be treated include cancer, cardiovascular disease, metabolic disease, liver disease, diabetes, hepatitis, hemophilia, blood system disease, degenerative or traumatic neurological diseases such as spinal cord injury, autoimmune disease, genetic Examples include defects, connective tissue diseases, anemia, infections, transplant rejection, ischemia, inflammation, and skin and muscle damage.
- the cells may be administered with a pharmaceutically acceptable substrate.
- the substrate is made of, for example, collagen, a highly biocompatible substance, or a biodegradable substance, and may be in the form of particles, plates, cylinders, containers, and the like. What is necessary is just to couple
- the pluripotent stem cell of the present invention may be induced to differentiate in vitro, a tissue may be constructed using the differentiated cell, and the differentiated cell or the tissue may be transplanted. Since the pluripotent stem cell of the present invention does not become a tumor, even if the transplanted differentiated cell or the tissue contains the pluripotent stem cell of the present invention undifferentiated, the possibility of canceration is low and safe. is there.
- mesoderm tissue or mesenchymal tissue is collected from a patient who is going to receive regenerative medicine, and the present invention is obtained from the tissue.
- the pluripotent stem cell or pluripotent stem cell fraction of the present invention can be used for the treatment of diseases caused by tissue degeneration or dysfunction.
- the pluripotent stem cell or pluripotent stem cell fraction of the present invention may be concentrated ex vivo, proliferated, or differentiated and returned to the body. What is necessary is just to differentiate into a cell and to transplant this cell to the tissue which is going to be treated. In situ cell therapy can also be performed by cell transplantation.
- target cells include liver cells, nervous cells such as nerve cells and glial cells, muscle cells such as skin cells and skeletal muscle cells, and the pluripotent stem cells of the present invention are differentiated into these cells.
- Parkinson's disease, cerebral infarction, spinal cord injury, muscle degenerative disease and the like can be treated.
- the pluripotent stem cell of the present invention does not become a tumor, even if it is used for such treatment, the possibility of canceration is low and it is safe.
- blood and blood components can be formed ex vivo and in vitro by differentiating the pluripotent stem cells of the present invention to form blood and blood components.
- red blood cells red blood cells, white blood cells, and platelets Etc.
- the blood and blood components formed in this way can be used for autologous blood transfusion or transfusion.
- the pluripotent stem cell or pluripotent stem cell fraction of the present invention when used for treatment, it may be differentiated ex vivo, in vivo, or in vitro.
- the pluripotent stem cells of the present invention include, for example, osteoblasts, chondrocytes, adipocytes, fibroblasts, bone marrow stroma, skeletal muscle, smooth muscle, myocardium, eyes, endothelium, epithelium, liver, pancreas, hematopoiesis, glia Differentiate into neurons, oligodendrocytes, etc. Differentiation of the pluripotent stem cell of the present invention can be achieved by culturing in the presence of a differentiation factor.
- Differentiation factors include basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), dimethyl sulfoxide (DMSO) and isoproterenol; or fibroblast growth factor 4 (FGF4), hepatocyte growth factor (HGF) and the like.
- the present invention also includes cells differentiated from the pluripotent stem cells of the present invention.
- a gene encoding a proteinaceous anticancer substance or physiologically active substance may be introduced.
- the pluripotent stem cell of this invention also has the delivery function of a therapeutic agent.
- An example of such a substance is an anti-angiogenic drug.
- the present invention relates to a cell transplantation treatment material comprising a Muse cell, an embryoid body-like cell mass made of Muse cells, and a cell or tissue / organ obtained by differentiation from the Muse cell or the embryoid body-like cell mass Alternatively, it includes a composition for cell transplantation treatment, or a material for regenerative medicine or a composition for regenerative medicine.
- the composition is pharmaceutically acceptable in addition to Muse cells, embryoid body-like cell masses made from Muse cells, or cells or tissues / organs obtained by differentiation from Muse cells or the embryoid body-like cell masses. Including buffer solution and diluent.
- cells can be collected from a patient, Muse cells can be isolated, and used for various diagnoses using the Muse cells.
- a patient gene from Muse cells it is possible to collect a patient gene from Muse cells, obtain genetic information, and perform an accurate diagnosis reflecting the information. For example, by differentiating Muse cells derived from a subject's cells, cells of each tissue / organ having the same genetic background as the subject can be obtained. And diagnosis of side effects, etc., it is possible to make an appropriate diagnosis according to the characteristics of each subject.
- Muse cells, embryoid body-like cell masses made from Muse cells, and cells or tissues / organs obtained by differentiation from Muse cells or the embryoid body-like cell masses can be used as diagnostic materials,
- the present invention diagnoses a disease or the like of a subject using a tissue or organ having the same genetic background as the subject obtained by isolating a Muse cell from the subject and differentiating the Muse cell or the Muse cell.
- somatic cells can be obtained in large quantities by differentiating Muse cells, basic research such as elucidation of disease mechanisms, therapeutic drug development, screening of drug effects and toxicity, drug evaluation, etc. can be performed. .
- Muse cells, embryoid body-like cell masses made from Muse cells, and cells or tissues / organs obtained by differentiation from Muse cells or the embryoid body-like cell masses are used as materials for drug evaluation and drug screening.
- the present invention includes a method for screening and evaluating drugs by differentiating and proliferating Muse cells, obtaining somatic cells, administering a candidate drug to the somatic cells, and examining the response of the somatic cells.
- a Muse cell bank in which various (for example, various HLA types) Muse cells are made into a library, a system capable of providing cells in the above Muse cell utilization scene as needed can be realized.
- the present invention includes a method for producing a library of Muse cells having different gene characteristics, that is, a Muse cell bank, by isolating and collecting Muse cells having various gene characteristics.
- a Muse cell bank a method for producing a library of Muse cells having different gene characteristics.
- Muse cells embryoid body-like cell masses made from Muse cells, and cells or tissues / organs obtained by differentiation from Muse cells or the embryoid body-like cell masses are obtained in libraries or banks. Can also be built.
- an embryoid body-like cell mass made from these Muse cells, and cells or tissues / organs obtained by differentiating from the Muse cells and the embryoid body-like cell mass, libraries and banks are also obtained. It is called a cell library or cell bank.
- the present invention includes the cell library or cell bank thus prepared.
- the cell library or cell bank is composed of, for example, a container such as a plurality of tubes in which cells having different genetic characteristics are stored, and the cells may be frozen. For example, when it becomes necessary to transplant or regenerate a tissue or organ in a subject, a cell suitable for genetic background etc. is selected from the cell library or cell bank, and the cell Can be used for transplantation and regenerative treatment.
- the present invention provides administration of a therapeutically effective amount of the pluripotent stem cells of the present invention, the cell fraction, derivative cells derived from the cells, or induced cells to a patient in need of treatment for the treatment of a disease.
- a therapeutic method comprising:
- the effective amount can be specified by, for example, the number of cells to be administered, and can be appropriately determined depending on the type and severity of the disease.
- the pluripotent stem cells of the present invention do not form teratomas (teratomas), and thus teratomas are not formed in patients.
- Muse cells can be a source of iPS cells (induced pluripotent stem cells). The production efficiency of iPS cells using Muse cells as a source is much higher (at least 25 times or more) than when other cells (for example, skin fibroblasts not fractionated using SSEA-3 expression) are used as a source. high.
- An iPS cell can be produced by introducing a specific gene into a Muse cell or changing a cytoplasm by introducing a specific compound.
- the change in cytoplasm includes reprogramming and canceration, and any currently known method or any method established in the future can be used.
- a gene can be introduced into Muse cells as described in Japanese Patent No. 4182742, or iPS cells can be established from Muse cells as described in FIG.
- iPS cells can be established by introducing a chemical substance, a foreign gene or a foreign protein.
- Establishment of iPS cells from Muse cells can be performed, for example, by the method described in Examples described later.
- the iPS cell obtained from the Muse cell may be referred to as a Muse-derived iPS cell (Muse-iPSC), and the present invention also includes the Muse-derived iPS cell.
- Muse-derived iPS cells can be referred to as Muse cell-derived proliferative pluripotent stem cells. The present invention will be specifically described by the following examples, but the present invention is not limited to these examples.
- H-fibroblast-1 normal human fibroblasts (NHDF), obtained from Lonza)
- H-fibroblast-2 adult human skin fibroblasts (HDFA), (Obtained from Science Cell).
- H-MSC-1, H-MSC-2, H-MSC-3 and H-MSC-4 were obtained from Lonza and ALLCELLS.
- H-MSC-1, H-MSC-2, H-MSC-3 and H-MSC-4 were obtained from Lonza and ALLCELLS.
- the cells were treated with ⁇ -MEM (alpha-minimum essential medium) containing 10% FBS and 0.1 mg / ml kanamycin at 37 ° C, 5% CO2. 2 Cultured under conditions. The first culture of the obtained one was the first generation, and when the cells became 95% confluent, the cell culture medium: medium ratio was passaged at 1: 2, and the passage was used for passages 4-10. .
- ⁇ -MEM alpha-minimum essential medium
- ES cells As human ES cells (hESC), Kyoto hESC-1 (KhES-1) obtained from Kyoto University was used. Mouse ES cells (TT2 cells) and human ES cells (KhES-1) were maintained in the presence of mouse-derived feeder cells established from day 12.5 embryos of C57BL / 6 mice. The experiment was conducted by the following method. 1. Stress stimulation of mesenchymal cells As stress stimulation, the following culture conditions were employed for culture under low nutrient conditions, culture at low serum concentration, culture at low oxygen concentration, repeated trypsin treatment, and long-time trypsin treatment.
- MC culture In this example, cells were cultured in suspension in a methylcellulose-containing medium. Culture in a methylcellulose-containing medium is called MC culture. For MC culture, see Nakahata, T .; et al. , Blood 60, 352-361 (1982). In order to prevent the cells from attaching to the bottom of the container, the culture dish was coated with Poly-HEMA (Poly (2-hydroxyethyl methacrylate). That is, 600 mg of poly-HEMA (SIGMA) was added to 40 ml of 95% ethanol at 37 ° C.
- Poly-HEMA Poly (2-hydroxyethyl methacrylate
- the Muse cell-derived embryoid body-like cell mass because the cell mass is a cell mass derived from the pluripotent stem cell Muse cell of the present invention
- Cloning of the cell mass was performed on the 7th day. 0.01M PBS was added to the medium, centrifuged at 2000 rpm for 20 minutes, and the supernatant was discarded. This treatment was repeated three times to wash the cells. The collected cell pellet was suspended in 10 ⁇ l of 0.01M-containing trypan blue solution, placed on a slide glass, and photographed using a phase contrast microscope. Only trypan blue negative cell masses having a diameter greater than 25 ⁇ m and similar in appearance to human ES cells were counted as Muse cell-derived embryoid body-like cell masses.
- the formation rate of Muse cell-derived embryoid body-like cell clusters was calculated by the number of cell clusters / total number of living cells (all trypan blue negative cells). When counting cells, the cell mass was counted as one cell regardless of its size. This is because it is difficult to accurately measure the number of cells contained in the Muse cell-derived embryoid body-like cell cluster. In human ES cells, the cells were carefully isolated so as not to contain feeder cells, and MC culture was performed by the method described above. A phase contrast microscope observation image was obtained on the third day of the culture. 3.
- Single cell suspension culture A 96-well dish was coated with poly-HEMA by the above method, single cells were seeded in each well by limiting dilution using ⁇ -MEM containing 10% FBS, and the actual state in the well was examined using a phase contrast microscope. The number of cells was counted, and wells containing no cells or multiple wells were excluded from the measurement. On day 10 of culture, embryoid body (EB) (Muse cell-derived embryoid body-like cell mass) formation was counted. Three experiments were performed on each cell line, and at least 250 wells were observed in one experiment. 4).
- EB embryoid body
- Muse cell-derived embryoid body-like cell clusters derived from the H-fibroblast fraction and the H-MSC fraction were collected, washed several times with physiological saline, and stained with Leukocyte Alkaline Phosphatase kit (Sigma). 5.
- Leukocyte Alkaline Phosphatase kit (Sigma). 5.
- embryoid body-like cell clusters derived from H-fibroblast fraction and H-MSC fraction were collected with a glass micropipette and placed on a gelatin-coated culture dish or cover glass. When transferred and further cultured for 7 days, the cells spread from the cell mass.
- karyotype Muse cell-derived embryoid body-like cell mass derived from H-fibroblast fraction and H-MSC fraction single cells are taken from the Muse cell-derived embryoid body-like cell mass to form a Muse cell-derived embryoid body-like cell mass again.
- the karyotype of the expanded cells from the cells that were repeated 1 to 3 times) was determined by quinacrine-Hoechst staining. 8).
- Cell injection into immunodeficient mouse testis The untreated cell fraction and the rich Muse cell fraction and the Muse cell-derived embryoid body-like cell mass derived from the H-fibroblast and H-MSC fractions were used.
- Muse cell-derived embryoid body-like cell mass 50 Muse cell-derived embryoid body-like cell masses are taken using a 3D graphic analysis technique of a laser confocal microscope, and Muse cell-derived embryoid body-like cell masses are obtained.
- the average volume of each constituent cell was measured by measuring the total volume of the mass and dividing by the number of nuclei. As a result of the measurement, 1.5 ⁇ 10 5 The number of cells was collected according to this calculation and injected into NOG mouse testis. Mice were subjected to experiments 6 months after injection.
- Muse cell-derived embryoid body-like cell mass In order to measure the doubling time of the cell group for the Muse cell-derived embryoid body-like cell mass derived from the H-fibroblast fraction and the H-MSC fraction, 96 times each of the Muse cell-derived embryoid body-like cell mass was 96. The plate was transferred to a well plate, treated with trypsin for 15 minutes, and then pipetted using a glass micropipette. The number of cells was counted, and at least 20 to 30 Muse cell-derived embryoid body-like cell clusters were analyzed every predetermined time.
- RNA from untreated cell fraction, H-fibroblast-1, H-fibroblast-2, H-MSC-1 and H-MSC-2 rich Muse cell fraction and Muse cell derived embryoid body-like cell mass was collected by Rneasy Mini Kit (Qiagen GmbH) and cDNA was 2 Synthesized using First Strand Kit (SA Biosciences). Primers were custom-made from SA Biosciences, and quantitative PCR was performed using a 7300 real time PCR system (Applied Biosystems).
- telomere activity was detected using TRAPEZE XL telomerase detection kit (Millipore) and Ex Taq polymerase (Takara Bio).
- the fluorescence intensity was measured using a microplate reader (TECAN). 16.
- Bisulfite (bisulfite) sequence 1 ⁇ g of genomic DNA derived from the untreated cell fraction, H-fibroblast fraction and H-MSC fraction-rich Muse cell fraction and Muse cell-derived embryoid body-like cell mass were obtained using the CpGenome DNA modification kit (chemicon). Processed. DNA was purified with QIAquick column (Qiagen). Promoter regions of human Oct3 / 4 and Nanog genes were amplified by PCR, PCR products were subcloned into pCR2.1-TOPO, and each sample was sequenced up to 10 clones using M13 universal primers, and the promoter region methylated. The state of conversion was examined.
- Muse cell-derived embryoid body-like cell mass formation from human bone marrow aspirate Mononuclear cell fractions were collected from human bone marrow aspiration solution (obtained from ALLCELLS) from 3 healthy individuals using Lymphoprep Tube (Axis-Shield PoC AS) and subjected to trypsin treatment for 8 hours. MC culture was performed. In addition, it was directly brought into the MC culture without being treated with trypsin. The cell number was measured on the 7th day. 18.
- the collected samples were treated with trypsin for a long period of 8 hours, and then the formation of Muse cell-derived embryoid body-like cell mass was measured. 19.
- Immunohistochemistry Mouse testes were fixed with 4% paraformaldehyde in 0.02M PBS. Sections were prepared as frozen sections with a thickness of 10 ⁇ m. The sample was washed with 0.02 M PBS, blocked with a buffer containing 20% BlockAce (Snow Brand), and then incubated with a primary antibody for immunohistochemical analysis.
- the primary antibodies used were anti-smooth muscle actin antibody (1: 200, Lab Vision), anti-MAP-2 antibody (1: 200, Biogenesis) and anti- ⁇ -fetoprotein antibody (1:10, DAKO). there were.
- anti-rabbit IgG antibody conjugated with Alexa488 or Alexa568 and anti-mouse IgG antibody conjugated with Alexa568 were used and reacted in the presence of DAPI.
- the sample was observed using a Nikon confocal microscope system C1si (Nikon). 20.
- Flow cytometry and cell sorting Cells are phycoerythrin-labeled anti-CD11c antibody, anti-CD29 antibody, anti-CD34 antibody, anti-CD44 antibody, anti-CD45 antibody, anti-CD49f antibody, anti-CD54 antibody, anti-CD71 antibody, anti-CD90 antibody, anti-CD105 antibody, anti-CD166 antibody, anti-CD271 Incubated with antibody or anti-vWF antibody (Beckton Dickinson) or anti-SSEA-3 antibody (Millipore). When anti-SSEA-3 antibody was used, the cells were further reacted with FITC-conjugated anti-rat IgM antibody.
- FACS antibody dilution 0.02M PBS without calcium and magnesium supplemented with 2 mM EDTA and 0.5% bovine serum albumin was used as the FACS antibody dilution.
- Data analysis was performed using CellQuest software by FACSCalibur (Becton Dickinson) or DIA software by FACSAria. Cells were incubated with anti-SSEA-3 antibody in FACS antibody diluent and sorted by FACSAria (Becton Dickinson) at low flow rate and 4 way purity sorting mode to sort the cells. 21.
- Statistical analysis Data are expressed as mean ⁇ SEM. Data were compared using ANOVA by pairwise comparison by Bonferroni method. result A.
- Table 1 shows an example of stress stimulation results of the H-fibroblast fraction and the H-MSC fraction. After stress stimulation and vortex treatment, the number of viable cells was counted by trypan blue staining, and the survival rate was calculated. Viable cells were collected and subjected to MC culture for 7 days. In the case of the condition (2), a large number of dead cells existed and the viable cell collection efficiency was low, so that the Muse cell-derived embryoid body-like cell mass could not be measured well. In Table 1, “ND (not determined)” is shown. Among the six conditions, 16 hours trypsin treatment of the H-fibroblast fraction and 8 hours trypsin treatment of the H-MSC fraction were the most efficient stimuli.
- LTT long-time trypsinization
- B. Criteria for Muse cell-derived embryoid body-like cell mass In this example, criteria for determining Muse cell-derived embryoid body-like cell clusters were provided.
- the average diameter of single cells in the rich Muse cell fraction derived from the H-fibroblast fraction and the H-MSC fraction was 10-13 ⁇ m (FIG. 6a). When these cells were cultured in MC, the cells started dividing.
- each cell When dividing, the size of each cell was reduced, and a multicellular mass composed of 8 to 10 ⁇ m cells was gradually formed (FIGS. 7e and 7f). The size and appearance of each cell was similar to MC cultured human ES cells (FIGS. 6b and 6c). On the seventh day, most multicellular masses were larger than 25 ⁇ m and were 100-150 ⁇ m in diameter. The cell mass had an appearance similar to ES cells. By using a filter with a diameter of 25 ⁇ m, a mass larger than 25 ⁇ m was recovered (FIG. 6 b).
- Muse cell-derived embryoid body-like cell masses are pluripotent markers Nanog, Oct3 / 4, Sox2, PAR4 and SSEA-3 were found to be positive, and alkaline phosphatase staining was also positive (FIGS. 6e-g).
- markers may or may not be detected, and localization of pluripotent markers may not show a pattern typical of these markers. Furthermore, the appearance of the cells may rather resemble the rich Muse cell fraction.
- MC culture suspension culture in a methylcellulose (MC) -containing medium, and further MC culture at a density of 8000 cells / mL for 7 days.
- FIGS. 7-1e and f show the state of the cell mass (Muse cell-derived embryoid body-like cell mass) formed from the H-fibroblast-1 fraction.
- Fig. 7-1e shows the state of MC culture on the 7th day
- Fig. 7-1f shows the state of single cell suspension culture on the 10th day.
- Cell clumps were fractionated by size using a filter, and immunocytochemical analysis was performed.
- Nanog, Oct3 / 4, SSEA-3, PAR-4 and Sox2 pluripotent stem cell markers are positive (FIG. 7-2g-l), and further positive by alkaline phosphatase staining Cells (Figure 7-3mo) were detected.
- Figure 7-3mo alkaline phosphatase staining Cells
- Muse cells (2nd cycle) were generated again, and Muse cell-derived embryoid body-like cell clusters were formed at a rate of about 10% (FIG. 8-1).
- trypsin-suspension culture-adhesion culture was repeated 5 times, the same characteristics and Muse cell-derived embryoid body-like cell mass formation rate were shown in each generation.
- the pluripotency marker and alkaline phosphatase were also positive in the 5-cycle Muse cell-derived embryoid body-like cell cluster. In order to confirm that these phenomena were not caused by abnormal cells with mutations, karyotype tests were performed.
- Muse cell-derived embryoid body-like cell clusters had a normal karyotype, and no chromosomal abnormality was observed (FIG. 8-3). This indicates a phenomenon caused by normal cells.
- the above results show that Muse cells have the ability to self-renew (self-renewal) and clonal expansion. Muse cells grow through a series of cycles of Muse cells-Muse cell-derived embryoid body-like cell mass-clonal expansion. Therefore, it is expected that a large amount of Muse cells can be obtained from the mesenchymal cell population. D.
- Muse cell-derived embryoid body-like cell mass was transferred to a gelatin-coated dish and differentiated.
- alpha smooth muscle actin (mesoderm marker), desmin (mesoderm marker), neurofilament-M (ectoderm marker), alpha fetoprotein (endoderm marker) and cytokeratin 7 (endoderm marker) were detected.
- a gelatin-coated dish On day 7, alpha smooth muscle actin (mesoderm marker), desmin (mesoderm marker), neurofilament-M (ectoderm marker), alpha fetoprotein (endoderm marker) and cytokeratin 7 (endoderm marker) were detected. (FIGS. 9-1a-c).
- Muse cell-derived embryoid body-like cell clusters (first to third cycles) cultured for 1 to 3 cycles by RT-PCR are ⁇ -fetoprotein and GATA6 (endoderm marker), microtubule-associated protein 2: MAP-2 ( It was confirmed that Nkx2.5 (mesoderm marker) and Nkx2.5 (mesoderm marker) were expressed, but no differentiation was observed in untreated H-fibroblast or MSC group even when cultured on gelatin-coated dishes (FIG. 9-2). Furthermore, the Muse cell fraction, Muse cell-derived embryoid body-like cell mass, and ES cells were injected into the testis of immunodeficient mice, and it was confirmed whether or not teratomas (teratomas) were formed (FIGS. 9-3e).
- the rich Muse cell fraction contains only 9-10% Muse cells as described above, but Rex1 (ZFP42), Sox2, KLF-4, c-Myc, DPPA2 (developmental pluripotency associated 2), ERAS , GRB7 (Growth factor receptor-bound protein 7), SPAG9 (Sperm associated antigen), TDGF1 (teratocarcinoma-derived growth factor 1) was moderately increased or untreated.
- DAZL azospermia-like
- DDX4 VASA
- DPPA4 evelopmental pluripotency associated 4
- Stella Hoxb1, PRDM1 and SPRY2 (sprouty homolog2)
- Fig. 10a The total gene expression of the untreated cell fraction, the rich Muse cell fraction, and the Muse cell-derived embryoid body-like cell mass derived from the H-fibroblast fraction and the H-MSC fraction is expressed as a human peripheral blood mononuclear cell fraction.
- the expressed odorant receptor and chemokine receptor were picked up by DNA microarray.
- H. Presence of Muse cells in living body The examination shown above was performed using stable cultured cells. When cells are removed from an adult and cultured, they may have different characteristics from those in the living body, and therefore it is undeniable that Muse cells and Muse cell-derived embryoid body-like cell clusters are artifact products. Therefore, an attempt was made to obtain a Muse cell-derived embryoid body-like cell mass directly from a human living body, ie, human bone marrow cells, without culturing.
- Mononuclear cell fraction is isolated from human bone marrow aspiration solution and directly subjected to MC culture (naive hBM-MC) or subjected to MC culture after 8 hours of trypsin treatment (8 hr-hBM-MC, trypsin)
- the survival rate of the mononuclear cell fraction after treatment was about 3.5%).
- the formation of Muse cell-derived embryoid body-like cell clusters was observed with an efficiency of about 0.004% in the untreated naive hBM-MC, and about 0.3%, or about 75 times, with 8hr-hBM-MC. It was efficient (Figure 12a).
- Muse cell-derived embryoid body-like cell clusters were positive for alkaline phosphatase staining (FIG. 12b).
- RT-PCR of cells cloned from untreated naive hBM-MC and 8 hr-hBM-MC-derived single Muse cell-derived embryoid body-like cell mass, ⁇ -fetoprotein, GATA6, MAP-2 and Nkx2.5 was observed (FIG. 13).
- Muse cells belong to the CD105-positive mesenchymal cell fraction in many types of bone marrow cells.
- the formation efficiency of untreated hBM-MC in the mononuclear cell fraction directly isolated from human bone marrow aspirate was as low as 0.004%. Since the composition of the cell population may change in cells in culture, cells that are stably cultured are Muse cell-derived embryoid body-like cell clusters different from untreated mononuclear cells isolated from bone marrow It is thought that it shows a tendency to form.
- human bone marrow aspiration solution was then cultured, primary MSCs were collected, and MC culture was performed.
- Muse cell-derived embryoid body-like cell mass formation As a result, about 0.2% of Muse cell-derived embryoid body-like cell mass formation was observed. When these primary MSCs were further subcultured two or five times, the efficiency of Muse cell-derived embryoid body-like cell mass formation increased by about 0.5% to 1.0% with respect to the untreated cell fraction, respectively. . About 1.2% of the untreated H-fibroblast fraction and the human MSC fraction form Muse cell-derived embryoid body-like cell masses. These results show that stress-resistant cells such as Muse cells remain in an in vitro culture environment such as subculture, and thus stable subculture cell fractions were isolated directly from bone marrow aspirates. This suggests that the Muse cell-derived embryoid body-like cell mass formation efficiency was higher than that of the fraction.
- Bone marrow contains a variety of mononuclear cells including MSCs, hematopoietic cells, and endothelial cells.
- MSCs mononuclear cells
- hematopoietic cells hematopoietic cells
- endothelial cells hematopoietic cells
- CD34 both hematopoietic stem cell markers
- CD105 meenchymal cell markers
- Muse cell-derived embryoid body-like cell clusters were hardly detected in the CD34-positive and CD117-positive fractions, but in the CD34-negative, CD117-negative and CD105-positive fractions, the CD34-negative, CD117-negative and CD105-negative fractions were 50. Double cluster formation was observed. This result suggests that Muse cells are mainly present in the CD105 positive mesenchymal cell fraction.
- I. MACS sorting The yield of each fraction from the bone marrow mononuclear cell fraction was 1.8% for fraction 1 (CD105 positive fraction), 8.5% for fraction 2 (CD34 positive / CD117 positive fraction), and fraction 3 (CD34 negative The CD117 negative / CD105 negative fraction) was 89.7%.
- fractions 1, 2 and 3 were 0.5%, 0% and 0.01%, respectively. Therefore, the cell mass formation rate of fraction 1 was about 50 times that of fraction 3.
- FACS sorting was performed using SSEA-3 as a marker.
- SSEA-3 positive cells and SSEA-3 negative cells were separated by FACS sorting and subjected to single cell suspension culture. About 50 to 60% of SSEA-3 positive cells formed Muse cell-derived embryoid body-like cell clusters. On the other hand, the SSEA-3 negative cells did not form Muse cell-derived embryoid body-like cell clusters.
- SSEA-3 is one of pluripotency markers
- the rate of formation of Muse cell-derived embryoid body-like cell mass in the untreated H-fibroblast and H-MSC fractions (about 1.2%)
- the formation rate (9-10%) in the Muse-rich cell fraction is the positive rate of each SSEA-3 (approximately 0.7-0.9% in the untreated cell fraction, 7 in the Muse-rich cell fraction). ⁇ 8.3%).
- a positive rate of SSEA-3 may indicate a certain state of Muse cells. According to immunohistochemistry, the number of SSEA-3 positive cells in untreated H-fibroblast and H-MSC fractions was less than 1%.
- SSEA-3 positive cells were sorted from the H-fibroblast fraction and the Huse MSC fraction-derived Muse cell fraction, and single suspension culture was performed. As a result, 50-60% of SSEA-3 positive cells formed Muse cell-derived embryoid body-like cell clusters. This is about 6 to 7 times the Muse cell-derived embryoid body-like cell mass formation of the rich Muse cell fraction and about 60 times the Muse cell-derived embryoid body-like cell mass formation of the untreated cell fraction. On the other hand, formation of Muse cell-derived embryoid body-like cell mass was not observed from the cell fraction negative for SSEA-3.
- the SSEA-3 positive rate of cells (3,000 to 5,000 cells) obtained by clonal expansion from a single cell mass derived from the fraction of SSEA-3 positive cells obtained by FACS sorting was about 45%.
- FIG. 15-1a This suggests that asymmetric division is involved in the formation process of the Muse cell-derived embryoid body-like cell mass, and the same is true for the clonal expansion of a single Muse cell-derived embryoid body-like cell mass. ing.
- Numblike which is known to be involved in asymmetric division, exists only in one cell during the process of dividing into two cells (FIG. 15-2b).
- Muse cells from human adult bone marrow aspirates as double positive cells of SSEA-3 and CD105.
- Double positive cells were present in bone marrow-derived mononuclear cells in an amount of 0.025% to 0.05%, and were directly subjected to single cell suspension culture without prolonged trypsin treatment. Seven days later, 11.4 ⁇ 1.2% of cells (corresponding to 0.003 to 0.005% of mononuclear cells) formed Muse cell-derived embryoid body-like cell mass and were positive for alkaline phosphatase. It was. Subsequently, a single Muse cell-derived embryoid body-like cell mass was grown to 3,000 cells by adhesion culture and subjected to single suspension culture.
- Muse cells expressed pluripotency markers were positive for alkaline phosphatase staining, and formed Muse cell-derived embryoid body-like cell masses that could differentiate into ectoderm, mesoderm and endoderm cells. Moreover, it did not have the characteristic regarding tumorigenic growth in the measurement of the growth rate, and did not have telomerase activity. This indicates that Muse cells have multiple security systems to prevent explosive growth. The non-tumorigenic properties of Muse cells were also observed in experiments where Muse cells were injected into mouse testis. This characteristic is convenient for maintaining the balance of biological functions. Without this characteristic, the living body is destroyed by abnormal growth and dysplasia, leading to tumor formation and teratoma formation.
- Muse cells The pluripotency of Muse cells did not become apparent in the adhesion culture system, but was observed in suspension culture. Usually, Muse cells are in an inactive dormant state, and when a living body falls into a critical state or suffers a serious injury, or when an overstressed state such as starvation or ischemia is maintained, a certain signal is generated. It is thought to be transmitted and activated. Thereafter, Muse cells contribute to tissue regeneration and contribute to cell-cell interaction and organization.
- the SSEA-3 positive cell fraction obtained by FACS has the characteristics of pluripotent stem cells, that is, Muse cells
- the isolated SSEA-3 positive cells were examined using the isolated SSEA-3-positive cells to examine the in vitro differentiation ability and the in vivo differentiation ability. Cell establishment was performed. 1. Study GFP differentiation ability in in vivo by implantation into damaged tissue (green fluorescent protein) - release the SSEA-3 positive Muse cells labeled with lentivirus single, spinal cord (compression injury), liver (CCl 4 intraperitoneal injection , Fulminant hepatitis model) or immunodeficient mice (NOG mice) injured in the gastrocnemius muscle (cardiotoxin injection).
- Muse cells derived from human skin cells are labeled with GFP (green fluorescent protein) -lentivirus (Hayase M et al., J Cereb Blood Flow Metab. 29 (8): 1409-20, 2009), Muse cell-derived embryo-like It was confirmed by GFP that the somatic cell mass was derived from labeled cells.
- Spinal cord injury was performed on NOG mice at level Th9 (Faroque M et al., Acta Neuropathol., 100; 13-22, 2000), and cardiotoxin was administered to the gastrocnemius of NOG mice to induce muscle degeneration and carbon tetrachloride. Was intraperitoneally administered to NOG mice to induce liver degeneration.
- mice After 2 days for muscle and liver and 7 days for spinal cord, 1 ⁇ 10 5 Muse cells were transplanted intravenously (using 6 mice each). As a control, mice injected intravenously with a GFP-labeled MEC population were used. Three or four weeks after transplantation, mice were fixed with paraformaldehyde and subjected to immunohistochemistry and confocal laser microscopy. Four weeks after spinal cord injury, GFP and human Golgi complex positive cells formed neurofilaments. (FIGS. 16-1 N and O). Further, after 4 weeks, GFP and human Golgi complex positive cells expressed human albumin in the regenerating liver (FIG. 16-1P).
- ⁇ 10 5 / ml For nerve induction, cells were cultured for 7 days in a NEUROBASAI medium (Gibco) containing B-27 supplement at a density of 1 ⁇ 10 5 / ml using a dish coated with poly-HEMA, and sphere (spherical shape) Cell mass) was formed.
- spheres were transferred onto glass coated with poly-L-lysine and cultured for 10 days in 2% FBS containing 25 ng / ml FGF and 25 ng / ml BDNF.
- the cells were cultured for 14 days at a density of 4.2 ⁇ 10 3 cells / cm 2 using a human mesenchymal stem cell functional identification kit (R & D Systems).
- adipocyte induction cells were cultured at a density of 2.1 ⁇ 10 4 cells / cm 2 for 14 days using an adipocyte induction medium from Human Mechanical Stem Cell Functional Identification Kit (R & D Systems).
- adipocyte induction medium from Human Mechanical Stem Cell Functional Identification Kit (R & D Systems).
- liver cell induction cells were DMEM (10 ⁇ ITS (GIBOCO) containing 10% FBS, 10 nM dexamethasone and 100 ng / ml HGF) at a density of 2.0 ⁇ 10 4 cells / cm 2 , 50 ng / ml FGF4.
- DMEM 10 ⁇ ITS (GIBOCO) containing 10% FBS, 10 nM dexamethasone and 100 ng / ml HGF
- DMEM 10 ⁇ ITS (GIBOCO) containing 10% FBS, 10 nM dexamethasone and 100 ng / ml HGF
- FGF4 ng / ml FGF
- Skin cells digested by filtration were collected, centrifuged at 1500 rpm for 20 minutes, washed with ⁇ -MEM, and incubated with 0.25% trypsin-HBSS for 5 minutes. The cells were further incubated with Chitose magazine SSEA-3 in FACS buffer and SSEA-3 positive cells were sorted using FACS. It was possible to recover 1.3 ⁇ 0.3 ⁇ 10 4 single cells from about 7 cm 2 of skin tissue. SSEA-3 positive cells were 1.7 ⁇ 0.2% of the recovered single cells. 21.0 ⁇ 1.7% of SSEA-3 positive cells formed Muse cell-derived embryoid body-like cell clusters in 7 days in single cell suspension culture by limiting dilution.
- the Muse cell-derived embryoid body-like cell mass is ALP positive, and the cells grown from a single Muse cell-derived embryoid body-like cell mass using a gelatin-coated dish were confirmed by RT-PCR. MAP-2, Brachyury, Nkx2.5, GATA6 and ⁇ -fetoprotein were expressed. This result indicates that cells having the same properties as Muse cells are present in adult human skin as well as the above-described adult human bone marrow fluid.
- SKP skin-derived progenitor cells
- NCSC neural crest stem cells
- melanoblast MB
- PC perivascular cells
- EP endothelial progenitor cells
- ADSC adipose-derived stem cells
- Snai1 Slug (SKP marker), Sox10 (NCSC marker), CD271 (NCSC marker) in Muse cells
- Tyrp1 MB marker
- Dct MB marker
- CD117 c-Kit
- CD146 PC and ADSC marker
- NG2 PC marker
- EP and ADSC marker von Willebrand factor expression
- None of these markers was observed in SSEA-3 positive cells by FACS analysis or RT-PCR (FIGS. 18-1 and 18-2). This result indicates that Muse cells are different from stem cells and progenitor cells known to exist in human adult skin.
- Muse-derived iPS cells (Muse-iPSCs) iPS cells are prepared by introducing Oct3 / 4 gene, Sox2 gene, Klf4 gene, c-Myc gene, Nanog gene, Lin28 gene and the like. Muse cells have characteristics similar to iPS cells in that they express pluripotency markers and can differentiate into ectoderm, endoderm and mesoderm cells. Therefore, it was examined whether Muse cells could be a good material for iPS cells. The method was as follows.
- the medium was replaced with fresh medium. After 3 days, the supernatant was collected, passed through a 0.45 ⁇ m filter, and 4 ⁇ g / ml polybrene was added.
- the virus solution was infected with NHDF (skin fibroblasts) spread at a density of 1 ⁇ 10 5 cells in a 60 mm dish. After 24 hours, the medium was replaced with fresh medium without virus. Cells detached by trypsin 4 days after virus infection were spread on MEF (feeder cells) at a density of 3 ⁇ 10 4 cells. The next day, the medium was replaced with Primate ES medium supplemented with 4 ng / ml bFGF.
- teratoma 60 mm dish iPS cells were treated with a Rock inhibitor, collected by Accutase (registered trademark), collected in a tube, centrifuged, and suspended in PBS. This was injected into the testis of NOG mouse (registered trademark) (Central Laboratory for Experimental Animals). After 12 weeks, it was fixed with 4% paraformaldehyde. The paraffin sections were stained with HE (Hematoxylin & Eosin). The following results were obtained. SSHA-3 positive cells and negative cells derived from the H-fibroblast fraction were added to Takahashi et al.
- RT-PCR and Q-PCR Factor was up-regulated or newly expressed (FIGS. 19 and 21). Further, Nanog, Oct3 / 4, Sox2 and TRA-1-81 were expressed in the obtained Muse cell-derived iPS cells (FIG. 19). RT-PCR revealed that Nanog, Oct3 / 4 and Sox2 were expressed in Muse cell-derived iPS cells but not in SSEA-3 negative cell-derived colonies (FIG. 21).
- the introduction efficiency of Oct3 / 4 gene, Sox2 gene, Klf4 gene and c-Myc gene was almost the same in the SSEA-3 positive cell fraction and the SSEA-3 negative cell fraction.
- the introduced cells were cultured at a density of 1 ⁇ 10 5 per dish on mouse feeder cells. Although colonies were formed in both groups, the number of colonies formed from the SSEA-3 negative cell fraction was about one-seventh of the SSEA-3 positive cell fraction. Furthermore, unlike the SSEA-3 positive cell fraction, in the SSEA-3 negative cell fraction, the TRA-1-81 positive colony, which is a human pluripotent stem cell marker, is also observed after 30 days of culture just before collecting the colonies. There was not (FIG. 22-1).
- RT-PCR endogenous Sox2 and Nanog were expressed only in the SSEA-3 positive cell-derived fraction, but not in the SSEA-3 negative cell fraction (FIG. 22-2). Furthermore, SSEA-3 positive and negative cell fractions were subcultured to establish an iPS cell line. After 3 passages, RT-PCR was performed on each colony (FIGS. 22-3C and C1) showing human ES cell-like morphology (flat colonies) to express endogenous oct3 / 4, Sox2 and Nanog. Colonies were counted as iPS colonies. As a result, iPS cells were formed only from SSEA-3 positive cell-derived colonies, and the efficiency was 0.03%. It was not formed from colonies derived from SSEA-3 negative cells (FIGS.
- Muse cells established from Muse cells differentiated into ectoderm, endoderm and mesoderm cells, and formed teratomas in mouse testis (FIGS. 23-1 to 23-3).
- the proliferation activity of Muse cells is not so high in terms of proliferation rate and telomerase activity. This corresponds to the fact that Muse cells differentiate into tridermal cells in mouse testis, but do not form teratomas. If Muse cells are maintained in human adult tissues such as skin and bone marrow, their proliferation should be tightly regulated, otherwise Muse cells form tumors in all parts of the body It is reasonable to consider what will happen.
- Muse cells show the characteristics of pluripotent cells from the beginning in terms of pluripotency marker expression and differentiation ability, so Muse cells become iPS cells alone, proliferating activity is increased, and teratomas are formed in mouse testis It is predicted that it will become.
- the induction mechanism of iPS has not been elucidated, but there is a possibility of acquiring neoplastic growth in Muse cells in mesenchymal cells.
- IPS cells could be established from the untreated human skin fibroblast fraction with an efficiency of about 0.001%. Takahashi et al. , Cell 131, 861 (2007). Therefore, iPS cell production efficiency from SSEA-3 positive cells was 30 times higher than that of untreated fibroblasts. This suggests that Muse cells mainly contribute to the formation of iPS cells. Embryoid bodies obtained from Muse-derived iPS cells by immunohistochemistry and RT-PCR are in vitro ectoderm (neurofilament and MAP-2), mesoderm (SMA, Brachyury and Nkx2.5), and endoderm. It was found to differentiate into cells ( ⁇ -fetoprotein and GATA-6).
- Muse-derived iPS cells were administered to immunodeficient mouse testis, formation of teratomas was observed.
- Muse cell-derived embryoid body-like cell mass was injected into the testis, teratoma formation was not observed even after 6 months, and the size was not so large compared to the control injected with MEF.
- human mitochondrial and cells positive for SMA, ⁇ -fetoprotein and neurofilament were identified. This result shows that unlike Muse-derived iPS cells, the original Muse cells do not form teratomas but can differentiate into ectoderm, mesoderm and endoderm in immunodeficient mice.
- Quantitative PCR was performed on Muse cell-derived embryoid body-like cell clusters and Muse-derived iPS cells. The results are shown in FIGS. The expression patterns of genes related to cell cycle regulation differed greatly. Most of the genes associated with cell cycle progression were down-regulated in Muse cell-derived embryoid body-like cell clusters, but up-regulated in Muse-derived iPS cells. On the other hand, in Muse cell-derived embryoid body-like cell clusters and Muse-derived iPS cells, the expression of genes involved in pluripotency and undifferentiated state showed the same tendency.
- pluripotent stem cells can be obtained from living tissue without using germ cells or early embryos and without undergoing artificial induction operations such as introduction of foreign genes or introduction of specific compounds. . Since no artificial manipulation such as introduction of a foreign gene is required, the pluripotent stem cell of the present invention can be efficiently produced, and can be used safely even when used for treatment. Furthermore, the pluripotent stem cells of the present invention can be used for regenerative medicine, treatment of dysfunctional tissues, and the like, and further can be used for studies of cell differentiation and tissue regeneration. All publications, patents and patent applications cited herein are incorporated herein by reference in their entirety.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Developmental Biology & Embryology (AREA)
- Cell Biology (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Transplantation (AREA)
- Immunology (AREA)
- Reproductive Health (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Physical Education & Sports Medicine (AREA)
- Neurology (AREA)
- Gynecology & Obstetrics (AREA)
- Dermatology (AREA)
- Diabetes (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Cardiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Heart & Thoracic Surgery (AREA)
Abstract
Description
近年、組織再生に貢献し得る成人幹細胞又は組織幹細胞が注目されている。しかし、哺乳類の成体に、プラナリアやイモリのように多能性(pluripotent)幹細胞が存在するか否かは不明であった。
成体から得られる分化能を有する細胞として、例えば骨、軟骨、脂肪細胞、神経細胞、骨格筋等への分化能を有する骨髄間葉系細胞画分(MSC:Bone marrow stromal cell)が報告されている(非特許文献1及び2を参照)。しかしながら、骨髄間葉系細胞画分は様々な細胞を含む細胞群であり、その分化能は多様でありながら本体がはっきりせず、また特定の細胞に分化させるために特定の化合物による刺激や遺伝子導入等が必要であり、分化誘導システムを構築する必要があった。
さらに、成体由来の多能性幹細胞としてiPS細胞(induced pluripotent stem cell)(特許文献1、特許文献2、非特許文献3等を参照)が報告されていた。しかしながら、iPS細胞の樹立には、間葉系細胞である皮膚線維芽細胞画分(dermal fibroblast)に特定の遺伝子や特定の化合物を体細胞に導入するという特定の物質を用いた誘導操作が必要であった。
本発明者らは、骨髄間葉系細胞画分(MSC:Bone marrow stromal cell)についての研究過程において、無処理のヒトMSC細胞から極めて低い頻度で特徴的な細胞塊が形成されることを見出した。初期の細胞塊の外見はES細胞に酷似していた。しかしながら、ES細胞とは異なり無限増殖をせずに、ある一定の期間である大きさに到達すると増殖が停止し、さらに毛、色素細胞などの種々の細胞を含む不均一な集団となった。また、この細胞塊について免疫細胞化学(immunocytochemistry)を行ったところ、外胚葉、中胚葉及び内胚葉マーカーにそれぞれ陽性の細胞が細胞塊内に混在して検出された。本発明者らは、この結果より、無処理の通常に維持・培養されている(naive)ヒトMSC細胞画分中に多能性細胞に相当する細胞が存在する可能性を考え、さらに鋭意検討を行った。
生体がストレスに曝されたり、傷害を受けると休眠状態の組織幹細胞が活性化され、組織再生に寄与することが知られている。本発明者は、骨髄間葉系細胞画分や皮膚線維芽細胞画分等の間葉系細胞又は中胚葉系細胞を培養している際に種々の方法でストレス刺激を与え(例えば、無血清培養、Hank’s Balanced Salt Solution(HBSS)による培養、低酸素培養、トータル3時間の間欠的短時間トリプシン培養、8時間若しくは16時間の長時間のトリプシン培養等)、生存している細胞を集め、メチルセルロース(MC)含有培地中で浮遊培養(MC培養という)を行った。その結果、最大直径150μmまでの種々の大きさの胚様体様(embryoid body−like)細胞塊の形成が認められた。特に長時間のトリプシン処理を行ったヒト皮膚線維芽細胞画分及びヒトMSC画分において、最も多くの胚様体様細胞塊の形成率が認められた。
本発明者らは、得られた胚様体様細胞塊中の細胞の特性を調べ、該細胞が多能性幹細胞の特性を有していることを見出した。さらに、本発明者らは、得られた胚様体様細胞塊中の細胞が従来報告されていた多能性幹細胞が有しない特性を有することを見出し、さらに、得られた細胞塊中の細胞の発現タンパク質を調べ、従来報告されていたES細胞、iPS細胞などの多能性幹細胞とは異なる発現パターンを示すことを見出した。
本発明者らは、さらに上記多能性幹細胞の表面抗原としてSSEA−3が発現していることを見出し、SSEA−3の発現を指標に上記多能性幹細胞を生体組織からも単離し得ることを見出した。
本発明者らは、上記多能性幹細胞は、新規な多能性幹細胞であり、従来報告されていたES細胞やiPS細胞などの多能性幹細胞と異なり、生体組織から誘導操作なしに、直接得られる多能性幹細胞であることを見出し、本発明を完成させるに至った。本発明者らは、該多能性幹細胞をMuse細胞(Multilineage−differentiating Stress Enduring cells)と名付けた。
すなわち、本発明は以下のとおりである。
[1] 生体組織から単離できるSSEA−3陽性の多能性幹細胞。
該多能性幹細胞は、培養線維芽細胞や骨髄幹細胞等の生体組織の培養物からも単離することができ、また単一細胞として単離することができる。
[2] CD105陽性の[1]の多能性幹細胞。
[3] CD117(c−Kit)陰性及びCD146陰性の[1]又は[2]の多能性幹細胞。
[4] CD117陰性、CD146陰性、NG2陰性、CD34陰性、vWF陰性及びCD271陰性の[1]又は[2]の多能性幹細胞。
[5] CD34陰性、CD117陰性、CD146陰性、CD271陰性、NG2陰性、vWF陰性、Sox10陰性、Snai1陰性、Slug陰性、Tyrp1陰性及びDct陰性の[1]又は[2]の多能性幹細胞。
[6] テロメラーゼ活性が低いか又は無い、[1]~[5]のいずれかの多能性幹細胞。
[7] 三胚葉に分化する能力を持つ、[1]~[6]のいずれかの多能性幹細胞。
本発明の多能性幹細胞は、in vitroの接着培養で三胚葉に分化する能力を有し、in vitroで誘導培養することにより、皮膚、肝、神経、筋、骨、脂肪等に分化し得る。また、in vivoで精巣に移植した場合にも三胚葉に分化する能力を有する。さらに、静注により生体に移植することで損傷を受けた臓器(皮膚、脊髄、肝、筋肉等)に生着し分化する能力を有する。
[8] 腫瘍性増殖を示さない、[1]~[7]のいずれかの多能性幹細胞。
本発明の多能性幹細胞は、浮遊培養で増殖速度約1.3日で増殖するが10日間程度で増殖が止まるという性質を有し、さらに精巣に移植した場合少なくとも半年間は癌化しないという性質を有する。
[9] セルフリニューアル能を持つ、[1]~[8]のいずれかの多能性幹細胞。
本発明の多能性幹細胞は、浮遊培養と接着培養の操作を繰り返すことにより増殖させることができる。また、本発明の多能性幹細胞は他の体性幹細胞と同じく、非対称分裂をする。
[10] ストレス耐性である、[1]~[9]のいずれかの多能性幹細胞。
[11] 貪食能が高い、[1]~[10]のいずれかの多能性幹細胞。
[12] 以下に示す22個のオドラント受容体の少なくとも一つが陽性の[1]~[11]のいずれかの多能性幹細胞:
olfactory receptor,family 8,subfamily G,member 2(OR8G2);
olfactory receptor,family 7,subfamily G,member 3(OR7G3);
olfactory receptor,family 4,subfamily D,member 5(OR4D5);
olfactory receptor,family 5,subfamily AP,member 2(OR5AP2);
olfactory receptor,family 10,subfamily H,member 4(OR10H4);
olfactory receptor,family 10,subfamily T,member 2(OR10T2);
olfactory receptor,family 2,subfamily M,member 2(OR2M2);
olfactory receptor,family 2,subfamily T,member 5(OR2T5);
olfactory receptor,family 7,subfamily D,member 4(OR7D4);
olfactory receptor,family 1,subfamily L,member 3(OR1L3);
olfactory receptor,family 4,subfamily N,member 4(OR4N4);
olfactory receptor,family 2,subfamily A,member 7(OR2A7);
guanine nucleotide binding protein(G protein),alpha activating activity polypeptide,olfactory type(GNAL);
olfactory receptor,family 6,subfamily A,member 2(OR6A2);
olfactory receptor,family 2,subfamily B,member 6(OR2B6);
olfactory receptor,family 2,subfamily C,member 1(OR2C1);
olfactory receptor,family 52,subfamily A,member 1(OR52A1);
olfactory receptor,family 10,subfamily H,member 3(OR10H3);
olfactory receptor,family 10,subfamily H,member 2(OR10H2);
olfactory receptor,family 51,subfamily E,member 2(OR51E2);
olfactory receptor,family 5,subfamily P,member 2(OR5P2);及び
olfactory receptor,family 10,subfamily P,member 1(OR10P1)。
[13] 以下に示す5個のケモカイン受容体の少なくとも一つが陽性の[1]~[12]のいずれかの多能性幹細胞:
chemokine(C−C motif)receptor 5(CCR5);
chemokine(C−X−C motif)receptor 4(CXCR4);
chemokine(C−C motif)receptor 1(CCR1);
Duffy blood group,chemokine receptor(DARC);及び
chemokine(C−X−C motif)receptor 7(CXCR7)。
[14] 中胚葉系組織または間葉系組織由来である、[1]~[13]のいずれかの多能性幹細胞。
[15] [1]~[14]のいずれかの多能性幹細胞を含む細胞塊または細胞画分。
[16] 生体組織から、以下の(i)~(vi)の特性の少なくとも1つの特性を指標に多能性幹細胞又は多能性細胞画分を単離する方法:
(i) SSEA−3陽性;
(ii) CD105陽性;
(iii) CD117陰性及びCD146陰性;
(iv) CD117陰性、CD146陰性、NG2陰性、CD34陰性、vWF陰性及びCD271陰性;
(v) CD34陰性、CD117陰性、CD146陰性、CD271陰性、NG2陰性、vWF陰性、Sox10陰性、Snai1陰性、Slug陰性、Tyrp1陰性及びDct陰性;並びに
(vi) テロメラーゼ活性が低いか又は無い。
[17] 生体組織由来細胞を細胞ストレスに暴露し生き残った細胞を回収することを含む多能性幹細胞又は多能性細胞画分を単離する方法。
[18] 細胞ストレスが、プロテアーゼ処理、低酸素条件下での培養、低リン酸条件下での培養、血清飢餓状態での培養、糖飢餓状態での培養、放射線曝露下での培養、熱ショックへの曝露下での培養、有害物質存在下での培養、活性酸素存在下での培養、機械的刺激下での培養及び圧力処理下での培養から選択される、[17]の多能性幹細胞又は多能性細胞画分を単離する方法。
[19] 細胞ストレスが、トリプシン処理である、[18]の多能性幹細胞又は多能性細胞画分を単離する方法。
[20] [1]~[14]のいずれかの多能性幹細胞の派生細胞又は誘導細胞である多能性幹細胞。
該派生細胞又は誘導細胞として、例えば、遺伝子の導入や、化合物を添加により誘導した細胞が挙げられる。また、本発明の幹細胞由来のiPS細胞が挙げられる。
[21] [1]~[14]のいずれかの多能性幹細胞の派生細胞又は誘導細胞である分化した細胞。
[22] [1]~[14]及び[20]のいずれかの多能性幹細胞を含む医薬組成物。
[23] [21]の分化した細胞を含む医薬組成物。
本明細書は本願の優先権の基礎である米国仮出願61/213,788号及び米国仮出願61/290,159号の明細書および/または図面に記載される内容を包含する。
図1−2は、Muse細胞を大量に増殖させる方法を示す図である。
図2は、Muse細胞由来の胚様体様細胞塊/無処理細胞画分での発現量の比が高い因子を示す図である。
図3は、Muse細胞由来の胚様体様細胞塊/ヒトES細胞での発現量の比が高い因子を示す図である。
図4は、MACSソーティングのプロトコールを示す図である。
図5は、ヒト線維芽細胞(H−fibroblast)画分の16時間の長時間トリプシン処理後(図5a)及び1800~2200rpm/分で3分間ボルテックス処理を行った後(図5b)のトリパンブルー染色像にて死細胞の除去を示す写真である。
図6は、各種細胞の写真である。図6aは富Muse細胞画分の中の1細胞(バーは10μm)、図6bはヒトES細胞由来の胚様体細胞塊(バーは25μm)、図6cは直径約25μmのMuse細胞由来の胚様体様細胞塊(バーは25μm)、図6dはヒトES細胞由来細胞塊4日目のアルカリフォスファターゼ染色像(バーは25μm)、図6e~gはMuse細胞由来の胚様体様細胞塊のOct3/4(e)、Sox2(f)及びPAR4(g)の免疫染色像を示す写真である。
図7−1は、H−fibroblast画分及びヒトMSC(H−MSC)画分由来の細胞塊の特徴を示す写真である。図7−1a及びbは無処理ヒトMSC画分の通常の接着培養において自発的に生じた細胞塊(バーは100μm)を示す。図7−1c及びdは0日目(c)及び7日目(d)のH−fibroblast−1画分の長時間トリプシン処理後のMC培養の状態を示し(バーは100μm)、図7−1dの矢印はMuse細胞由来胚様体様細胞塊を示す。図7−1e及びfはMC培養7日後のH−fibroblast−1画分から形成されたMuse細胞由来胚様体様細胞塊を示す(バーは50μm)。
図7−2は、H−fibroblast画分及びヒトMSC(H−MSC)画分由来の細胞塊の特徴を示す写真である。図7−2g−lはH−fibrobalst画分から形成されたMuse細胞由来胚様体様細胞塊(図7−2g、i及びk)及びH−MSC画分から形成されたMuse細胞由来胚様体様細胞塊(図7−2h、j及びl)のNanog(図7−2g及びj)、Oct3/4(図7−2h)、SSEA−3(図7−2i)、PAR4(図7−2k)及びSox2(図7−2l)の局在を示す免疫染色の結果を示す(バーは50μm)。
図7−3は、H−fibroblast画分及びヒトMSC(H−MSC)画分由来の細胞塊の特徴を示す写真である。図7−3m−oはヒトES細胞(図7−3m)、H−fibroblast画分由来のMuse細胞由来胚様体様細胞塊(図7−3n)及び無処理H−fibroblast−1画分(図7−3o)のアルカリフォスファターゼ染色の結果を示す(バーは50μm)。
図7−4は、H−fibroblast画分及びヒトMSC(H−MSC)画分由来の細胞塊の特徴を示す電子顕微鏡写真である。図7−4p−rはヒトES細胞胚様体(図7−4p、MC培養3日目)、H−fibroblast−1画分由来のMuse細胞由来胚様体様細胞塊(図7−4q及びr、MC培養5日目)の電子顕微鏡像を示す(バーは5μm)。
図8−1は、Muse細胞由来胚様体様細胞塊(M−cluster)のクローナリティー及びセルフリニューアルを示す図であり、Muse細胞のクローナリティー及びセルフリニューアルを決定するために行った実験の概要を示す図である。
図8−2は、Muse細胞の浮遊細胞における増殖速度を示す図である。
図8−3は単一のMuse細胞由来胚様体様細胞塊(H−fibroblast−1由来、第1世代(サイクル))から増殖した細胞(clonally expanded cells)の正常核型を示す図である。
図9−1は、Muse細胞由来胚様体様細胞塊の分化を示す図である。a−cはH−fiboroblas−1画分由来の分化細胞の塊のα平滑筋アクチン及び神経フィラメント(図9−1a及びb)並びにα−フェトプロテイン(図9−1c)の局在を示す免疫染色像である(バーは図9−1aが500μm、図9−1b及びcが50μm)。図9−1aの矢頭は接着したMuse細胞由来胚様体様細胞塊を示す。
図9−2は無処理細胞画分、H−fibroblast画分由来の第1及び第3世代のMuse細胞由来胚様体様細胞塊(Cluster)をゲラチン上で培養し自発的分化を誘導した細胞群におけるα−フェトプロテイン(α−FP)、GATA6、MAP−2及びNkx2.5発現のRT−PCR分析の結果を示す。陽性コントロールとしてはα−FPに対してヒト胎児肝臓を、GATA6、MAP−2及びNkx2.5に対してはヒト胚全体を用いた。
図9−3e−lは富Muse細胞画分を投与した免疫不全マウスの精巣を示す。図9−3eはコントロール(intact)となる無傷の精巣並びにマウスES細胞(mES cells)(8週)、MEF(フィーダー細胞)(8週)、Muse細胞由来胚様体様細胞塊(M−cluster)(6ヶ月)及び富Muse細胞画分(Muse)(6ヶ月)を投与した精巣を示す。図9−3f−iは富Muse細胞画分又はMuse細胞由来胚様体様細胞塊を投与した精巣組織の神経フィラメントM(図9−3f、写真では緑に染色)、α−フェトプロテイン(図9−3g、写真では緑に染色)及び平滑筋アクチン(図9−3h、図では赤に染色)の免疫染色像を示す(バーは50μm)。図9−3iの3枚のパネルはヒトミトコンドリア(緑に染色)及び平滑筋アクチン(赤に染色)の二重染色像を示す(バーは20μm)。図9−3j−lは富Muse細胞画分を投与した精巣の組織像を示す(図9j及びk)。図9kに認められるチューブ様の構造はヒトミトコンドリアに対する抗体で染色されている(バーは図9−3jが500μm、図9−3k−lが50μm)。
図10aは、H−fibroblast(Fibro−1,Fibro−2)及びH−MSC(MSC−1,MSC−2)のpluripotency及び未分化細胞状態に関与する因子の定量PCRの結果を示す図である(その2)。図中のカラムの模様は富Muse細胞画分及びMuse細胞由来胚様体様細胞塊(7日目)の無処理細胞画分に比較した遺伝子発現レベルを示す。白は富Muse細胞群又はMuse細胞由来胚様体様細胞塊/無処理細胞画分の比が1/3より大きく3より小さいものを示し、グレーは富Muse細胞画分又はMuse細胞由来胚様体様細胞塊/無処理細胞画分の比が3より大きいものを示し、斜線は富Muse細胞画分又はMuse細胞由来胚様体様細胞塊/無処理細胞画分の比が1/3より小さいものを示す。
図10bは、H−MSC由来の無処理細胞画分(Naive)、富Muse細胞画分(Muse)及びMuse細胞由来胚様体様細胞塊(M−cluster)(7日目)のテロメラーゼ活性を示す図である。熱不活性化サンプル(Heat)を陰性コントロールとして用いた。
図11は、H−fibroblast画分及びH−MSC画分由来の無処理細胞画分、富Muse細胞画分及びMuse細胞由来胚様体様細胞塊のDNAマイクロアレイ分析の結果を示す図である。
図12は、ヒト骨髄の単核球成分からSSEA−3/CD105二重陽性細胞として直接採取したMuse細胞をMC培養し、形成された胚様体様細胞塊を示す写真である。図12aはヒト骨髄から単離し8時間の長時間トリプシン処理を行った単核細胞画分をMC培養し(8hr−hBM−MC、7日)形成されたMuse細胞由来胚様体様細胞塊を示す(バーは100μm)。図12bは8hr−hBM−MC(7日)により形成されたMuse細胞由来胚様体様細胞塊のアルカリフォスファターゼ染色像を示す(バーは50μm)。
図13は、無処理H−MSC−1画分(naive1)、無処理H−MSC−2画分(naive2)(いずれもネガティブコントロール)、並びに8時間のトリプシン処理を行ったヒト骨髄由来単核細胞画分(8hr−hBM)あるいはトリプシン処理を行わないヒト骨髄由来単核細胞画分(naive hBM)から形成されたMuse細胞由来胚様体様細胞塊をゲラチンの上で培養し自発的分化を誘導した細胞群におけるα−フェトプロテイン(α−FP)、GATA6、MAP−2及びNkx2.5のRT−PCR分析の結果を示す図である。
図14は、H−fibroblast画分(無処理細胞)及びH−MSC画分(無処理細胞)のFACS分析の結果を示す図である。
図15−1は、無処理細胞画分中のSSEA−3陽性細胞(15−1a左)、及びFACSソーティングにより採取したSSEA−3陽性細胞由来の単一Muse細胞由来胚様体様細胞塊からクローン増殖したSSEA−3陽性細胞(15−1a右)の染色像を示す写真である。図中のバーは100μmである。
図15−2は、Muse細胞(H−fibroblast)の細胞分裂中の非対称分裂に関わる因子であるNumblike(緑色)の局在を示す染色像を示す写真である。図中のバーは10μmである。
図15−3は、H−fibroblast由来のSSEA−3陰性細胞(図15−3c)及びSSEA−3陽性細胞(図15−3d)の電子顕微鏡写真である。図中のバーは5μmである。
図15−4は、H−fibroblast由来Muse細胞のOct3/4(緑色)(図15−4e)、Sox2(緑色)(図15−4f)及びSSEA−3(赤色)(図15−4g)の染色像を示す写真である。
図16−1は、超免疫不全マウス(Nogマウス)損傷組織におけるGFP標識SSEA−3陽性Muse細胞画分の分化を示す写真である。図16−1N及びOは圧迫損傷脊髄(4週間後)におけるGFP陽性細胞であり、ニューロフィラメント(赤色)及びヒトゴルジ複合体(白色)を発現している。OはNの四角で囲んだ部分の拡大像である。図16−1Pは損傷肝臓(4週間後)のGFP陽性標識細胞であり、ヒトアルブミン(赤色)及びヒトゴルジ複合体(白色)を発現している。
図16−2は、RT−PCRで調べた、SSEA−3陽性Muse細胞を移植した肝臓におけるヒトアルブミンの発現を示す写真である。
図16−3は、超免疫不全マウス(Nogマウス)損傷組織におけるGFP標識SSEA−3陽性Muse細胞画分の分化を示す写真であり、ヒトジストロフィン(赤色)を発現している筋肉(3週間後)のGFP陽性細胞を示す写真である。
図17−1は、単一Muse細胞から形成されたMuse細胞由来胚様体様細胞塊から増殖させた細胞の分化を示す写真である。図17−1A~Dは、神経誘導の結果を示し、Aは形成されたsphereを示し、さらにshpereの免疫染色データとしてAはネスチン、BはMusashi、CはNuroDの発現を示す。Eはこれらのsphereをさらに神経系細胞への分化をさせたものであり、MAP−2陽性細胞を示す。図17−1F~Gは、骨細胞誘導の結果を示し、オステオカルシン(F)及びALP(G)の発現を示す。図17−1H及びIは、脂肪細胞誘導の結果を示し、Hは油滴を含む細胞を示し、Iはオイルレッド染色の結果を示す。図17−1Jは、肝臓細胞誘導の結果を示し、α−フェトプロテイン陽性細胞を示す。
図17−2は、RT−PCRで調べた、肝細胞誘導した細胞におけるヒトアルブミン及びヒトα−フェトプロテインの発現を示す写真である。
図18−1は、RT−PCRで調べた、SSEA−3陽性Muse細胞におけるSox10、Snail1、Slug、Tyrp1及びDctの発現を示す写真である。
図18−2は、FACSで分析したNG2、CD34、vWF、CD117、CD146及びCD271の発現を示す図である。無処理ヒト皮膚線維芽細胞において、周皮細胞(pericyte)マーカーであるNG2、内皮前駆細胞マーカーであるCD34及びvWFは陰性であり、SSEA−3陽性細胞でも陰性であった。メラノブラストマーカーであるCD117、周皮細胞(pericyte)マーカーであるCD146、NCSCマーカーであるCD271は、無処理ヒト皮膚線維芽細胞では、わずかに陽性細胞が認められたが(それぞれ0.2%、0.2%及び0.8%)、それらはSSEA−3陰性細胞であったためMuse細胞ではないと考えられる。
図18−3は、Muse細胞がフェライトを貪食したことを示す図である。
図19は、Muse細胞から作製したiPS細胞の形成を示す写真であり、図19aは、皮膚線維芽細胞(NHDF)由来Muse細胞から誘導されたヒトiPS細胞の形態を示し、図19b~fは、多能性細胞マーカー(bがNonog、cがOct3/4、dがSox2、eがSSEA−3、fがTra−1−60)の発現を示す。
図20は、Nonog(E)、Oct3/4(F)、Sox2(G)及びTra−1−81(H)の免疫組織化学の結果を示す写真である。
図21は、Muse由来iPS細胞(Mi−1,Mi−2)及びSSEA−3陰性細胞から増殖したコロニー((−)−1,(−)−2)のRT−PCRにより調べた多能性マーカーの発現を示す写真である。
図22−1は、Oct3/4,Sox2,Klf4,c−Myseをレトロウイルスで導入し、その後フィーダー細胞MEF上で培養した30日後のSSEA−3陽性及び陰性細胞から形成されたコロニーのTra−1−81免疫染色の結果を示す写真である。ヒトES細胞をコントロールとして用いている。SSEA−3陽性細胞からのコロニー(a1)及びヒトES細胞(a2)はTra−1−81陽性であるが、SSEA−3陰性細胞からのコロニーはすべて陰性である。
図22−2は、22−1と同じくMEFで30日培養した段階におけるSSEA−3陽性及び陰性細胞の多能性マーカー(内因性Oct3/4(endo Oct)、内因性Sox2(endo Sox2)、Nanog、内因性Klf4(endo Klf4)、Rex1及びUTF1)の発現を示す写真である。SSEA−3陰性細胞群ではSox2,Nanogのシグナルが見られない。
図22−3は、Muse細胞から誘導したiPS細胞(Muse細胞由来iPS細胞)(図22−3C及びC1)及びSSEA−3陰性細胞から増殖したコロニー(図22−3D及びD1)のコロニーを示す写真である。
図23−1は、皮膚線維芽細胞(NHDF)由来Muse細胞から誘導されたiPS細胞のin vitroでの分化の様子を示す写真である。図23−1iは、内胚葉マーカーであるα−フェトプロテイン(緑色)及び中胚葉マーカーである平滑筋アクチン(赤色、青色はDNA)の発現を示し、図23−1jは、外胚葉マーカーである神経フィラメント(緑色)の発現を示す。
図23−2は、Muse細胞から誘導されたiPS細胞のin vitroでの分化のRT−PCR分析結果を示す図であり、3胚葉マーカーの発現を示す。
図23−3は、皮膚線維芽細胞(NHDF)由来Muse細胞から誘導されたiPS細胞から形成されたテラトーマの組織構造を示す写真であり、HE(Hematoxylin及びeosin)染色により、iPS細胞が種々の組織に分化していることを示している。図23−3mは、軟骨(cartilage)、図23−3nは筋肉(Muscle)、図23−3oは神経上皮(neural epithelium)、図23−3pは色素上皮(pigmented epithelium)、図23−3qは円柱上皮(columnar epithelium)を示す。
図24は、SSEA−3陰性細胞画分、Muse細胞由来胚様体様細胞塊及びMuse由来iPS細胞のNanog遺伝子及びOct3/4遺伝子のBisulfite(亜硫酸水素塩)シーケンスの結果を示す図である。それぞれのカラムの数値は、転写開始部位(TSS)の下流に対すCpGの位置を示す。白丸はメチル化されていないシトシンを、黒丸は、メチル化されたシトシンを示す。
図25は、無処理線維芽細胞(Naive)、Muse細胞由来胚様体様細胞塊(Cluster)及びiPS細胞中の細胞周期に関連した因子の定量PCRの結果を示す図である。「/Naive」で示されるカラム中、白カラムは、無処理細胞に対するMuse画分又はMuse細胞由来胚様体様細胞塊との比が2未満で1/2より大きいことを示す。また、塗りつぶされたカラムは比が2より大きいことを示し、斜線が引かれたカラムは、比が1/2より小さいことを示す。「/iPS」で示されるカラム中、「*」は、発現された遺伝子の量がiPSよりMuse細胞由来胚様体様細胞塊で大きいことを示し、「**」は、発現された遺伝子の量がMuse細胞由来胚様体様細胞塊よりiPSで大きいことを示す。
図26は、無処理線維芽細胞(Naive)、Muse細胞由来胚様体様細胞塊(Cluster)及びiPS細胞中の多能性及び未分化細胞状態に関連した因子の定量PCRの結果を示す図である。各カラムの意味は図25と同じである。
図27は、ヒト及びマウスモデルにおいて作製されたiPS細胞株の誘導効率に関する論文報告をまとめた図である。図27は核のリプログラミングを誘導する転写因子の組合せを示している。
本発明は、生体の生体組織から直接得ることができる多能性(pluripotent)幹細胞又は多能性幹細胞画分及び該多能性幹細胞又は該多能性幹細胞画分を単離する方法、並びに該方法により得られた生体組織由来の多能性幹細胞又は多能性幹細胞画分である。本発明の多能性幹細胞をMuse細胞(multilineage differentiating stress enduring cells)という。
本発明において、細胞画分というときは、単離したい細胞を少なくとも一定量含む細胞群のことをいう。例えば、多能性幹細胞画分とは、多能性幹細胞を1%以上、10%以上、30%以上、50%以上、70%以上、90%以上、又は95%以上含む細胞群が挙げられ、多能性幹細胞の培養によって得られる細胞塊や多能性幹細胞を濃縮した細胞群を含む。また、前記細胞画分を実質的に均一な細胞画分ということもある。
生体は、哺乳動物の生体をいい、ある程度発生が進んだ動物体をいう。本発明において、生体には、受精卵や胞胚期より発生段階が前の胚は含まれないが、胎児や胞胚を含む胞胚期以降の発生段階の胚は含まれる。哺乳動物は限定されないが、例えばヒト、サル等の霊長類、マウス、ラット、ウサギ、モルモット等のげっ歯類、ネコ、イヌ、ヒツジ、ブタ、ウシ、ウマ、ロバ、ヤギ、フェレット等が含まれる。本発明の多能性幹細胞は、生体の組織由来である点で、胚性幹細胞(ES細胞)や胚性生殖幹細胞(EG細胞)と明確に区別される。
中胚葉系組織とは、動物の初期発生途上で現れる中胚葉起源の組織をいい、筋肉系組織、結合組織、循環系組織、排泄系組織、生殖系組織等が含まれる。例えば、本発明の多能性幹細胞は、骨髄液や真皮結合組織等の皮膚組織から得ることができる。
間葉系組織とは、骨、軟骨、脂肪、血液、骨髄、骨格筋、真皮、靭帯、腱、心臓、などの組織をいう。例えば、本発明の多能性幹細胞は、骨髄や皮膚から得ることができる。また、臍帯から得ることもできる。
細胞が組織から直接得ることができるとは、組織から単離することができ、外来遺伝子や外来タンパク質の導入又は化合物の投与などの化合物処理等の人為的な誘導操作を経ずに得られることを意味する。ここで、外来遺伝子は、限定されないが、例えば体細胞の核を初期化し得る遺伝子をいい、例えば、Oct3/4遺伝子等のOctファミリー遺伝子、Klf遺伝子等のKlfファミリー遺伝子、c−Myc遺伝子等のMycファミリー遺伝子、Sox2遺伝子等のSoxファミリー遺伝子が挙げられる。また、外来タンパク質としてはこれらの遺伝子がコードするタンパク質やサイトカインが挙げられる。さらに、化合物としては、例えば、上記の体細胞の核を初期化し得る遺伝子の発現を誘導する低分子化合物やDMSO、還元剤として機能する化合物,DNAメチル化剤等が挙げられる。本発明の多能性幹細胞は、生体あるいは組織から直接得ることができるという点で、iPS(induced pluripotent stem cell)細胞及びES細胞とは明確に区別される。なお、本発明においては、細胞の培養、細胞の表面マーカーを指標に細胞又は細胞画分を単離すること、細胞を細胞ストレスに曝露すること、及び細胞に物理的衝撃を与えることは、人為的な誘導操作には含まれない。また、本発明の多能性細胞は、リプログラミング又は脱分化の誘導を必要とせずに得られることを特徴としてもよい。
本発明の多能性幹細胞は、生体の中胚葉系組織又は間葉系組織等に存在していると考えられ、本発明においては、これらの組織に存在している細胞又は細胞画分を単離する。本発明の多能性幹細胞は、例えば、骨髄に存在しており、骨髄から血液等を介して生体の各組織に供給される可能性がある。このため骨髄や、皮膚等の生体の各組織、さらには血液から単離することが可能である。
多能性幹細胞とは、pluripotencyを有している細胞をいい、以下の特性を有する。
(1) Nanog、Oct3/4、SSEA−3、PAR−4及びSox2等の多能性マーカー(Pluripotent marker)を発現する。
(2) 1細胞から増殖し、自己のクローンを作り続けるクローナリティー(Clonality)を有する。
(3) 自己複製(セルフリニューアル)能を有する。
(4) 3胚葉系(内胚葉系、中胚葉系及び外胚葉系)へin vitro及びin vivoで分化し得る。
(5) マウスの精巣や皮下に移植した場合、3胚葉系への分化を呈する。
(6) アルカリフォスファターゼ染色で陽性となる。
本発明の多能性幹細胞は、pluripotencyを有している点で、成人幹細胞、組織幹細胞とは明確に区別される。また、本発明の多能性幹細胞は、pluripotencyを有している単一の又は複数の細胞として単離されている点で、骨髄間葉系細胞等の細胞画分とは明確に区別される。
さらに、本発明の多能性幹細胞は、以下の特性を有する。
(i) 増殖速度が比較的緩やかで、分裂周期が1日以上、例えば1.2~1.5日である。ただし、ES細胞やiPS細胞が示すような無限増殖は示さない。
(ii) 免疫不全マウスに移植した場合に内胚葉系、中胚葉系及び外胚葉系への分化を示す。ES細胞やiPS細胞ではテラトーマが短期間で癌化するのに比べ、半年以上癌化しないことを特徴とする。
(iii) 浮遊培養により胚様体様細胞塊を形成する。
(iv) 浮遊培養にて胚様体様細胞塊を形成し、10日程度で増殖が停止する。その後、接着培養に移動させることにより再増殖する。
(v) 増殖の際に非対称分裂を伴う。
(vi) 核型は正常である。
(vii) テロメラーゼ活性が無いか又は低い。ここで、テロメラーゼ活性が無いか又は低いとは、例えばTRAPEZE XL telomerase detection kit(Millipore社)を用いてテロメラーゼ活性を検出した場合に検出できないか又は低いことをいう。テロメラーゼ活性が低いとは、例えば、ヒト線維芽細胞と同程度のテロメラーゼ活性を有しているか、あるいはHela細胞に比べて1/5以下、好ましくは1/10以下のテロメラーゼ活性を有していることをいう。
(viii) メチル化の状態については、Muse細胞から誘導したiPS細胞に関してはNanogおよびOct3/4のプロモータ領域のメチル化レベルが低い。
(ix) 貪食能が高い。
(x) 腫瘍性増殖を示さない。ここで、腫瘍性増殖を示さないとは、浮遊培養を行った場合、一定の大きさの細胞塊(クラスター)に達すると増殖が止まり、無限増殖しないことをいう。また免疫不全マウスの精巣に移植しても奇形腫を形成しないことである。なお、上記(i)~(iv)等も腫瘍性増殖を示さないことに関連する。
すなわち、本発明の細胞は、例えば以下の多能性幹細胞である。
(A) 生体の中胚葉系組織又は間葉系組織等から得られる細胞であって、当該細胞内に化学物質、外来遺伝子又は外来タンパク質を導入することなく直接得ることができる多能性幹細胞。
(B) 生体の中胚葉系組織又は間葉系組織等が、骨髄、皮膚、血液、臍帯、脂肪などからなる群から選択される上記(1)の特性を有する多能性幹細胞。
(C) リプログラミングまたは脱分化を誘導することなく得ることができる、上記(A)又は(B)の多能性幹細胞。
(D) 精巣へ移植した場合に、少なくとも半年間は癌化しない、上記(A)又は(B)の多能性幹細胞。
(E) ES細胞、iPS細胞のように無限増殖を示さない、上記(A)又は(B)の多能性幹細胞。
(F) 生体の中胚葉系組織又は間葉系組織等由来の多能性幹細胞であって、生体の中胚葉系組織又は間葉系組織等の細胞をプロテアーゼで処理したときに生き残る、プロテアーゼに耐性である多能性幹細胞。
さらに、本発明の多能性幹細胞は、生体の中胚葉系組織又は間葉系組織等の細胞に細胞ストレスをかけ、生き残った細胞を回収することにより単離することができる。ここで、細胞ストレスとは外的ストレスをいい、プロテアーゼ処理、低酸素条件下での培養、低リン酸条件下での培養、血清飢餓状態での培養、糖飢餓状態での培養、放射線曝露下での培養、熱ショックへの曝露下での培養、有害物質存在下での培養、活性酸素存在下での培養、機械的刺激下での培養、圧力処理下での培養等によりストレスに曝露することをいう。この中でもプロテアーゼ処理、すなわちプロテアーゼ存在下での培養が好ましい。プロテアーゼは限定されず、トリプシン、キモトリプシン等のセリンプロテアーゼ、ペプシン等のアスパラギン酸プロテアーゼ、パパイン、キモパパイン等のシステインプロテアーゼ、サーモリシン等の金属プロテアーゼ、グルタミン酸プロテアーゼ、N−末端スレオニンプロテアーゼなどを用いることができる。プロテアーゼを培養に添加する際の添加濃度は限定されず、一般的にシャーレ等で培養した付着細胞を剥がすときに用いる濃度で用いればよい。本発明の多能性幹細胞は、上記外的ストレスに耐性を有する幹細胞、例えば、トリプシンに耐性を有する細胞ということができる。
生体の中胚葉系組織又は間葉系組織等は限定されず、骨髄単核細胞、皮膚細胞等の線維芽細胞画分、歯髄組織、眼球組織、毛根組織等が含まれる。細胞としては、培養細胞も組織から採取した細胞も用いることもできる。この中でも、骨髄細胞、皮膚細胞が望ましく、例えば、ヒト骨髄間葉系細胞(MSC)画分又はヒト皮膚線維芽細胞画分が挙げられる。骨髄間葉系細胞画分は、骨髄穿刺液を2~3週間培養することにより得ることができる。
上記の各種のストレスを受けた組織の細胞の大部分は死滅し、生き残った細胞中に本発明の多能性幹細胞が含まれる。細胞にストレスをかけたのち、死細胞を除去する必要があるが、プロテアーゼを用いた場合は、これらの死細胞はプロテアーゼの作用により分解される。
また、細胞にストレスをかけた後に、細胞に物理的衝撃を与え壊れ易くなった細胞を除去してもよい。物理的衝撃は、例えば激しいピペッティング、激しい攪拌、ボルテックス等により与えることができる。
細胞に細胞ストレスをかけ、必要に応じて物理的衝撃を与えた後に、細胞群を遠心分離にかけ、生き残った細胞をペレットとして得て回収することにより、本発明の多能性幹細胞を単離することができる。また、このようにして得られた細胞からさらに、下記の表面マーカーを指標に本発明の多能性幹細胞又は多能性細胞画分を単離することもできる。
また、外傷や火傷等のストレスを受けた生体の中胚葉系組織又は間葉系組織等を培養し、遊走した細胞を回収しても本発明の多能性幹細胞又は多能性細胞画分を単離することができる。傷害を受けた組織の細胞はストレスに曝露されるので、本発明においては、傷害を受けた生体の中胚葉系組織又は間葉系組織等を培養することも生体の中胚葉系組織又は間葉系組織細胞等に細胞ストレスをかけるという。
一例として、これらの細胞をトリプシン処理する方法について説明する。このときのトリプシン濃度は、限定されないが、例えば接着細胞の通常の培養において、培養容器に接着した接着培養を剥がすときに用いられる濃度範囲で用いればよく、0.1~1%、好ましくは0.1~0.5%が例示される。例えば、10~50万個の細胞を含む生体の中胚葉系組織又は間葉系組織等由来の細胞を上記濃度のトリプシン溶液5ml中でインキュベーションすることにより外的ストレスに曝すことができる。トリプシン処理時間は、5~24時間、好ましくは5~20時間程度である。本発明においては、8時間以上のトリプシン処理、例えば8時間又は16時間の処理を長時間トリプシン処理という。
トリプシン処理後、上記のように、ピペッティング、攪拌、ボルテックス等により物理的衝撃を与えることが望ましい。それは死んだ細胞あるいは死にかけている細胞を除去するためである。
トリプシン処理後の浮遊培養の際には細胞同士の凝集を防ぐために、例えば、メチルセルロースゲル等のゲル中でインキュベーションするのが望ましい。また、細胞の培養容器への付着を防ぎ浮遊状態を維持するために、容器をPoly(2−hydroxyethyl methacrylate)等でコートしておくことが望ましい。
外的ストレスに曝した細胞を遠心分離により集め培養を行うと細胞塊(細胞クラスター)を形成する。この細胞塊の大きさは直径25μmから150μm程度である。本発明の多能性幹細胞(Muse細胞)は、この外的ストレスに曝して生き残った細胞集団中に濃縮した状態で含まれる。この細胞集団を富Muse細胞画分(Muse enriched population)と呼ぶ。富Muse細胞画分中のMuse細胞の存在割合は、ストレス処理の方法により異なる。
このように本発明の多能性幹細胞又は多能性幹細胞画分がストレスをかけた後も生存することは、本発明の多能性幹細胞又は多能性幹細胞画分がストレス耐性であることを示している。
生体の中胚葉系組織又は間葉系組織等由来の細胞の培養に用いる培地、培養条件は通常の動物細胞の培養で用いる培地、培養条件を採用すればよい。また、公知の幹細胞培養用培地を用いてもよい。培地には、適宜ウシ胎児血清等の血清やペニシリン、ストレプトマイシン等の抗生物質及び種々の生理活性物質を添加してもよい。
さらに、本発明は、本発明の生体の中胚葉系組織又は間葉系組織等から直接得ることができる多能性幹細胞の派生細胞又は誘導細胞である多能性幹細胞も含む。派生細胞又は誘導細胞とは前記多能性幹細胞を培養して得られる細胞又は細胞群、あるいは前記多能性幹細胞に外来遺伝子の導入等の人為的な誘導操作を行い得られる細胞をいい、子孫細胞も含む。なお、本発明時点において報告されているiPS細胞は、皮膚線維芽細胞などの生体組織の分化した細胞に外来遺伝子導入等することによりリプログラミングした結果、多能性幹細胞に誘導された細胞といわれており、本発明の組織から直接得ることができ、すでに多能性幹細胞としての性質を有する細胞に外来遺伝子導入等の人為的な誘導操作を行い得られた細胞は、iPS細胞と区別される。
本発明の多能性幹細胞を浮遊培養することにより、胚様体様(Embryoid body(EB body)−like)細胞塊が得られるが、本発明はこの胚様体様細胞塊及び胚様体様細胞塊に含まれる細胞も包含する。胚様体は、本発明の多能性幹細胞を浮遊培養することにより、細胞塊として形成される。この際、本発明においては、本発明の多能性幹細胞を培養することにより得られる胚様体をMuse細胞由来胚様体様細胞塊と呼ぶことがある(Mクラスター(M−cluster)と呼ぶこともある)。胚様体様細胞塊を形成するための浮遊培養の方法として、メチルセルロース等の水溶性ポリマーを含有した培地を用いた培養(Nakahata,T.et al.,Blood 60,352−361(1982))やハンギングドロップ培養(Keller,J.Physiol.(Lond)168:131−139,1998)等が挙げられる。本発明は前記胚様体様細胞塊からセルフリニューアルして得られる胚様体様細胞塊及び胚様体様細胞塊に含まれる細胞及び多能性幹細胞も包含する。ここで、セルフリニューアルとは、胚様体様細胞塊に含まれる細胞を培養し、再度胚様体様細胞塊を形成させることをいう。セルフリニューアルは1~複数回のサイクルを繰り返せばよい。また、本発明は前記いずれかの胚様体様細胞塊及び胚様体様細胞塊に含まれる細胞から分化した細胞及び組織も包含する。
図1−1に間葉系細胞(ヒト線維芽細胞、ヒト骨髄間葉系細胞、新鮮骨髄液)画分とMuse細胞及びMuse細胞由来の胚様体様細胞塊の関連を示す。間葉系細胞様細胞塊に長時間トリプシン処理(Long term trypsin incubation;LTT)等のストレス刺激を与えると、Muse細胞が濃縮されMuse細胞を多く含む細胞画分が得られ(富Muse細胞画分と呼ぶ)、該細胞画分中のMuse細胞を浮遊培養すると胚様体様細胞塊(Muse細胞由来胚様体様細胞塊)が得られる。胚様体様細胞塊をゼラチンでコートした培養皿で培養すると3胚葉の細胞に分化する。また、図1−1に示すように、SSEA−3陽性細胞を直接分離し、長時間のストレスをかけることなく浮遊培養することによりMuse細胞由来の胚様体様細胞塊を得ることができる。
Muse細胞は、浮遊培養で一旦増殖が停止しても、接着培養に移すことにより増殖を開始する。浮遊培養−接着培養−SSEA−3発現を指標にした分離を繰り返すことにより、Muse細胞を大量に増殖させることが可能である(図1−2)。
さらに、本発明の多能性幹細胞又は多能性細胞画分は、生体組織から細胞ストレスに暴露せずに直接単離することもできる。すなわち、本発明の多能性幹細胞又は多能性幹細胞画分は、生体の中胚葉系組織又は間葉系組織等から外来遺伝子を導入することなく、誘導操作なしに以下の方法で単離することができる。
生体組織は、限定されないが、生体の中胚葉系組織又は間葉系組織、例えば、骨髄、皮膚、臍帯等が挙げられる。骨髄を用いる場合、骨髄の単核球分画を用いることができる。単離は、Muse細胞の表面に多く発現している細胞表面マーカーを利用して行うことができ、例えばSSEA−3の発現を指標に単離することができる。本発明の多能性幹細胞をSSEA−3陽性Muse細胞ということもある。さらに、Muse細胞は多能性幹細胞マーカーであるCD105を発現しており、SSEA−3陽性であり、CD105陽性である。従って、SSEA−3及びCD105の両方の発現を指標に単離することができる。これらの細胞表面マーカーを利用することにより、本発明の多能性幹細胞を単一細胞として単離でき、単離した単一細胞を、培養により増殖させることができる。なお、本発明は、ヒト以外の哺乳動物の生体組織からSSEA−3に相当するマーカーによって単離できる多能性幹細胞をも含むものとする。
一方、Muse細胞は、NG2、CD34、vWF(フォンビルブランド因子)、c−kit(CD117)、CD146、CD271(NGFR)が陰性である。さらに、Sox10、Snai1、Slug、Tyrp1、Dctが陰性である。
NG2、CD34、vWF、CD117、CD146、CD271などの表面抗原が陰性かどうか、発現が弱いかどうかはこれらの抗原に対する抗体であって、発色酵素、蛍光化合物等で標識した抗体を用いて細胞が染色されたか否かを顕微鏡観察等により決定することができる。例えば、これらの抗体を用いて細胞を免疫染色して、表面抗原の有無を決定することができ、また該抗体を結合させた磁性ビーズを用いても決定することができる。また、FACS又はフローサイトメーターを用いても表面抗原があるかどうか決定することができる。フローサイトメーターとしては例えばFACSAria(ベクトン・ディッキンソン社製)、FACS vantage(ベクトン・ディッキンソン社製)、FACS Calibur(ベクトン・ディッキンソン社製)等を用いることができる。
また、Sox10、Snai1、Slug、Tyrp1、Dctなどの転写因子に関してはRT−PCR等の手法により発現を調べることもできる。
これらの表面抗原が陰性とは、上記のようにFACSを用いて分析した場合に、陽性細胞としてソーティングされないこと、あるいはRT−PCRにより発現を調べた場合に、発現が認められないことをいい、これらの手法により検出できない程度発現していたとしても、本発明においては陰性とする。また、上記マーカーが陽性であることが公知の造血幹細胞等の細胞と同時に測定を行い、これらの陽性細胞と比較して、ほとんど検出されないか、あるいは有意に発現量が低い場合に陰性としてもよい。
本発明の細胞は、これらの細胞表面の抗原特性に基づいて単離することができる。
上記のように、Muse細胞は、SSEA−3陽性を指標に単離することができ、さらにCD105の発現を指標に単離することができるが、さらに、NG2、CD34、vWF(フォンビルブランド因子)、c−kit(CD117)、CD146、CD271(NGFR)、Sox10、Snai1、Slug、Tyrp1及びDctからなる群から選択される11個のマーカーのうち少なくとも1個、例えば、2個、3個、4個、5個、6個、7個、8個、9個、10個又は11個のマーカーの非発現を指標に単離することができる。例えば、CD117及びCD146の非発現を指標に単離することができ、さらに、CD117、CD146、NG2、CD34、vWF及びCD271の非発現を指標に単離することができ、さらに、上記の11個のマーカーの非発現を指標に単離することができる。
表面マーカーを用いて単離する場合、生体の中胚葉系組織又は間葉系組織等から1個又は複数個の本発明の多能性幹細胞を、培養等を経ることなく直接単離することが可能である。また、本発明の多能性幹細胞を、細胞形態を顕微鏡等を使って目視することにより同定して単離することが可能である。
生体の中胚葉系組織又は間葉系組織等に細胞ストレスをかけた後に、生存した細胞群から表面マーカーを用いて単離してもよい。
また、上記のマーカーに加えて、本発明の多能性幹細胞又は多能性細胞画分は、他の特定の因子の高発現によっても特徴付けられる。
無処理の骨髄間葉系細胞画分又は皮膚線維芽細胞画分から本発明の多能性幹細胞であるMuse細胞が得られ、さらにMuse細胞を培養することによりMuse細胞由来の胚様体(EB)様細胞塊が得られる。Muse細胞、無処理細胞、Muse由来胚様体様細胞塊及びヒトES細胞において発現している因子を比較検討することにより、Muse細胞で高発現している因子がわかる。ここで、因子とは遺伝子転写産物、タンパク質、脂質、糖を含む。
図2に、Muse細胞由来胚様体様細胞塊/無処理細胞での発現量の比が高い因子を示す。特に以下の18個の因子の比が高い。
(i)SSEA−3
(ii)v−fos FBJ murine osteosarcoma viral oncogene homolog
(iii)solute carrier family 16,member 6(monocarboxylic acid transporter 7)
(iv)tyrosinase−related protein 1
(v)Calcium channel,voltage−dependent,P/Q type,alpha 1A subunit
(vi)chromosome 16 open reading frame 81
(vii)chitinase 3−like 1(cartilage glycoprotein−39)
(viii)protease,serine,35
(ix)kynureninase(L−kynurenine hydrolase)
(x)solute carrier family 16,member 6(monocarboxylic acid transporter 7)
(xi)apolipoprotein E
(xii)synaptotagmin−like 5
(xiii)chitinase 3−like 1(cartilage glycoprotein−39)
(xiv)ATP−binding cassette,sub−family A(ABC1),member 13
(xv)angiopoietin−like 4
(xvi)prostaglandin−endoperoxide synthase 2(prostaglandin G/H synthase and cyclooxygenase)
(xvii)stanniocalcin 1
(xviii)coiled−coil domain containing 102B
本発明の多能性幹細胞又は多能性幹細胞画分は、上記因子の少なくとも2つ、3つ、4つ、5つ、6つ、7つ、8つ、9つ、10、11、12、13、14、15、16、17又は18が高発現していることを特徴として、少なくとも2つの因子が高発現していることを指標に単離することができる。
図3に、Muse細胞由来胚様体様細胞塊/ヒトES細胞での発現量の比が高い因子を示す。特に以下の20個の因子の比が高い。
(a)matrix metallopeptidase 1(interstitial collagenase)
(b)epiregulin
(c)chitinase 3−like 1(cartilage glycoprotein−39)
(d)Transcribed locus
(e)chitinase 3−like 1(cartilage glycoprotein−39)
(f)serglycin
(g)MRNA full length insert cDNA clone EUROIMAGE 1913076
(h)Ras and Rab interactor 2
(i)lumican
(j)CLCA family member 2,chloride channel regulator
(k)interleukin 8
(l)Similar to LOC166075
(m)dermatopontin
(n)EGF,latrophilin and seven transmembrane domain containing 1
(o)insulin−like growth factor binding protein 1
(p)solute carrier family 16,member 4(monocarboxylic acid transporter 5)
(q)serglycin
(r)gremlin 2,cysteine knot superfamily,homolog(Xenopus laevis)
(s)insulin−like growth factor binding protein 5
(t)sulfide quinone reductase−like(yeast)
本発明の多能性幹細胞又は多能性幹細胞画分は、上記因子の少なくとも2つ、3つ、4つ、5つ、6つ、7つ、8つ、9つ、10、11、12、13、14、15、16、17、18、19又は20が高発現していることを特徴として、少なくとも2つの因子が高発現していることを指標に単離することができる。
さらに、本発明の多能性幹細胞又は多能性幹細胞画分は、上記(i)~(xviii)の因子の少なくとも2つと上記(a)~(t)の因子の少なくとも2つが同時に高発現していてもよく、これらの遺伝子が高発現していることを指標に単離することができる。
さらに、本発明の多能性幹細胞又は多能性幹細胞画分は多能性マーカー以外のオドラント(odorant)受容体(オルファクトリーレセプター;olfactory receptor)群及びケモカイン(chemokine)受容体群の因子を発現していること、すなわち特定のオドラント受容体やケモカイン受容体陽性であることを特徴とする。
本発明の多能性幹細胞又は多能性幹細胞画分で発現しているオドラント受容体として例えば、以下の22個の受容体が挙げられる。
olfactory receptor,family 8,subfamily G,member 2(OR8G2);
olfactory receptor,family 7,subfamily G,member 3(OR7G3);
olfactory receptor,family 4,subfamily D,member 5(OR4D5);
olfactory receptor,family 5,subfamily AP,member 2(OR5AP2);
olfactory receptor,family 10,subfamily H,member 4(OR10H4);
olfactory receptor,family 10,subfamily T,member 2(OR10T2);
olfactory receptor,family 2,subfamily M,member 2(OR2M2);
olfactory receptor,family 2,subfamily T,member 5(OR2T5);
olfactory receptor,family 7,subfamily D,member 4(OR7D4);
olfactory receptor,family 1,subfamily L,member 3(OR1L3);
olfactory receptor,family 4,subfamily N,member 4(OR4N4);
olfactory receptor,family 2,subfamily A,member 7(OR2A7);
guanine nucleotide binding protein(G protein),alpha activating activity polypeptide,olfactory type(GNAL);
olfactory receptor,family 6,subfamily A,member 2(OR6A2);
olfactory receptor,family 2,subfamily B,member 6(OR2B6);
olfactory receptor,family 2,subfamily C,member 1(OR2C1);
olfactory receptor,family 52,subfamily A,member 1(OR52A1);
olfactory receptor,family 10,subfamily H,member 3(OR10H3);
olfactory receptor,family 10,subfamily H,member 2(OR10H2);
olfactory receptor,family 51,subfamily E,member 2(OR51E2);
olfactory receptor,family 5,subfamily P,member 2(OR5P2);及び
olfactory receptor,family 10,subfamily P,member 1(OR10P1)
本発明の多能性幹細胞又は多能性幹細胞画分で発現しているケモカイン受容体としては以下の5個の受容体が挙げられる。
chemokine(C−C motif)receptor 5(CCR5);
chemokine(C−X−C motif)receptor 4(CXCR4);
chemokine(C−C motif)receptor 1(CCR1);
Duffy blood group,chemokine receptor(DARC);及び
chemokine(C−X−C motif)receptor 7(CXCR7)
本発明の多能性幹細胞又は多能性幹細胞画分は、上記の嗅覚受容体の少なくとも1個を発現しており、あるいは、上記のケモカイン受容体の少なくとも1個を発現している。
これらのオドラント受容体やケモカイン受容体と受容体に結合する遊走因子の作用で本発明の多能性幹細胞は、損傷組織へ遊走し、生着し、その場で分化する。例えば、肝臓、皮膚、脊髄、筋肉が損傷した場合、特定の遊走因子と細胞表面に発現しているオドラント受容体の働きで、それぞれの組織に遊走し、生着し、肝臓(内胚葉)、皮膚(外胚葉)、脊髄(外胚葉)、筋肉(中胚葉)細胞に分化し、組織を再生することができる。
さらに、本発明の多能性幹細胞であるMuse細胞が豊富に含まれる富Muse細胞画分において、Rex1、Sox2、KLF−4、c−Myc、DPPA2、ERAS、GRB7、SPAG9、TDGF1等がアップレギュレートされており、Muse細胞の細胞塊において、DAZL、DDX4、DPPA4、Stella、Hoxb1、PRDM1、SPRY2等がアップレギュレートされている。
また、本発明の多能性幹細胞又は多能性幹細胞画分においては、造血幹細胞マーカーであるCD34及びCD117の発現は認めらないかもしくは発現が極めて低い。
本発明は、Muse細胞のみならず、Muse細胞を濃縮した細胞集団、Muse細胞を増殖させた細胞集団、Muse細胞を分化させた細胞集団を含み、さらに、Muse細胞やMuse細胞由来の細胞を含む研究用キット、細胞チップ、治療用デバイスも含む。
本発明の多能性幹細胞は、pluripotencyを有しており、あらゆる組織へと分化し得る。該多能性幹細胞又は多能性細胞画分は、再生医療等に用いることができる。例えば、各種組織、各種器官等の再生に用いることができる。具体的には皮膚、脳脊髄、肝臓、筋肉等が挙げられる。本発明の多能性幹細胞又は多能性幹細胞画分を損傷あるいは障害を受けた組織、器官等に直接あるいは近傍に投与することにより、該多能性幹細胞はその組織、器官内に侵入し、その組織特有の細胞に分化し、組織、器官の再生、再建に貢献し得る。また、静脈投与等により全身投与してもよい。この場合、該多能性幹細胞は、例えば、損傷を受けた組織や器官をホーミング等により指向し、そこに到達・侵入した上で、その組織や器官の細胞に分化し、組織、器官の再生、再建に貢献し得る。
投与は、例えば皮下注、静注、筋注、腹腔内注等の非経口投与や経口投与、あるいは胚への子宮内注射等により行うことができる。また、局所投与でも全身投与でもよい。局所投与は例えばカテーテルを利用して行うことができる。投与量は、再生しようとする器官、組織の種類や、サイズにより適宜決定することができる。
再生しようとする器官は限定されず、骨髄、脊髄、血液、脾臓、肝臓、肺、腸管、眼、脳、免疫系、循環系、骨、結合組織、筋、心臓、血管、膵臓、中枢神経系、末梢神経系、腎臓、膀胱、皮膚、上皮付属器、乳房−乳腺、脂肪組織、および口、食道、膣、肛門を含む粘膜等を含む。また、治療対象となる疾患として、癌、心血管疾患、代謝疾患、肝疾患、糖尿病、肝炎、血友病、血液系疾患、脊髄損傷等の変性または外傷性神経疾患、自己免疫疾患、遺伝的欠陥、結合組織疾患、貧血、感染症、移植拒絶、虚血、炎症、皮膚や筋肉の損傷等が挙げられる。
細胞は医薬として許容される基材と共に投与してもよい。該基材は例えばコラーゲン等でできた生体親和性が高い物質や、生分解性の物質できており、粒子状、板状、筒状、容器状等の形状とすればよく、細胞を該基材に結合させあるいは該基材中に収容して投与すればよい。
また、本発明の多能性幹細胞をin vitroで分化誘導し、さらに分化した細胞を用いて組織を構築させ、該分化した細胞又は該組織を移植してもよい。本発明の多能性幹細胞は、腫瘍化しないので、移植した前記分化した細胞又は該組織に本発明の多能性幹細胞が未分化のまま含まれていても癌化の可能性が低く安全である。これらの再生医療において、移植した細胞又は組織のレシピエントによる拒絶を避けるためには、再生医療を受けようとする患者から中胚葉系組織又は間葉系組織等を採取し、該組織から本発明の多能性幹細胞又は多能性細胞画分を単離し、利用することが望ましい。さらに、本発明の多能性幹細胞又は多能性幹細胞画分を組織の変性や機能不全を原因とする疾患の治療に用いることができる。この場合、例えば、本発明の多能性幹細胞又は多能性幹細胞画分をex vivoで濃縮し、増殖させ、あるいは分化させて体内に戻せばよく、例えば、多能性幹細胞を特定の組織の細胞に分化させ、該細胞を治療しようとする組織に移植すればよい。また、細胞の移植により、in situ細胞治療を行うこともできる。この場合、対象細胞の例として、肝臓細胞、神経細胞やグリア細胞などの神経系細胞、皮膚細胞、骨格筋細胞などの筋肉細胞が挙げられ、本発明の多能性幹細胞をこれらの細胞に分化させ、移植し、in situで治療を行うことができる。該治療により、例えば、パーキンソン病、脳梗塞、脊髄損傷、筋変性疾患などを治療することができる。本発明の多能性幹細胞は、腫瘍化しないので、このような治療に用いても癌化の可能性が低く安全である。
また、本発明の多能性幹細胞を、分化させて血液や血液成分を形成させることにより、血液や血液成分をex vivo、in vitroで形成させることができる、血液成分として、赤血球、白血球、血小板等が挙げられる。このようにして形成させた血液や血液成分を、自家輸血や他家輸血に用いることができる。
上記のように、本発明の多能性幹細胞又は多能性幹細胞画分を治療に用いる場合、ex vivo、in vivo、in vitroのいずれで分化させてもよい。本発明の多能性幹細胞は、例えば、骨芽細胞、軟骨細胞、脂肪細胞、線維芽細胞、骨髄間質、骨格筋、平滑筋、心筋、眼、内皮、上皮、肝、膵、造血、グリア、神経細胞、稀突起膠細胞等に分化する。本発明の多能性幹細胞の分化は、分化因子の存在下で、培養することにより達成することができる。分化因子としては、塩基性繊維芽細胞成長因子(bFGF)、血管内皮成長因子(VEGF)、ジメチルスルホキシド(DMSO)およびイソプロテレノール;あるいは繊維芽細胞成長因子4(FGF4)、肝細胞成長因子(HGF)等が挙げられる。本発明は、本発明の多能性幹細胞から分化した細胞も包含する。
本発明の多能性幹細胞を治療に用いる場合、タンパク質性の抗癌物質や生理活性物質等をコードする遺伝子を導入してもよい。これにより、本発明の多能性幹細胞は、治療薬のデリバリー機能も有することになる。このような物質として例えば、抗血管新生薬が挙げられる。
本発明は、Muse細胞、Muse細胞からできた胚様体様細胞塊、及びMuse細胞や前記胚様体様細胞塊から分化させて得られた細胞若しくは組織・器官を含む、細胞移植治療用材料若しくは細胞移植治療用組成物、又は再生医療用材料若しくは再生医療用組成物を包含する。該組成物はMuse細胞、Muse細胞からできた胚様体様細胞塊、又はMuse細胞や前記胚様体様細胞塊から分化させて得られた細胞若しくは組織・器官に加えて、医薬的に許容される緩衝液や希釈液等を含む。
さらに、患者から細胞を採取し、Muse細胞を単離し、該Muse細胞を用いて種々の診断に用いることができる。例えば、Muse細胞から患者の遺伝子を採取し、遺伝子情報を得て、該情報を反映させた正確な診断が可能になる。例えば、被験体の細胞由来のMuse細胞を分化させることで、被験者と同じ遺伝子背景などの性質を持った各組織・器官の細胞を得ることができるため、疾病の診断や病態解明、薬剤の効果や副作用の診断、などに関し、各々の被験者の性質に合わせ適切な診断を行うことができる。すなわち、Muse細胞、Muse細胞からできた胚様体様細胞塊、及びMuse細胞や前記胚様体様細胞塊から分化させて得られた細胞若しくは組織・器官は診断用材料として用いることができ、例えば、本発明は、被験体からMuse細胞を単離し、該Muse細胞又はMuse細胞から分化させて得られた、被験体と同じ遺伝子背景を有する組織や器官を用いて被験体の疾病等を診断する方法を包含する。
また、Muse細胞を分化させることで体細胞を大量に得ることができるため、疾病のメカニズム解明等の基礎的研究、治療薬開発、薬剤の効果や毒性に関するスクリーニング、薬剤評価などを行うことができる。すなわち、Muse細胞、Muse細胞からできた胚様体様細胞塊、及びMuse細胞や前記胚様体様細胞塊から分化させて得られた細胞若しくは組織・器官を薬剤評価や薬剤スクリーニングの材料として用いることができる。例えば、本発明はMuse細胞を分化・増殖させ、体細胞を得て、該体細胞に候補薬剤を投与し、体細胞の応答を調べることにより、薬剤のスクリーニングや薬剤評価を行う方法を包含する。
また、種々の(例えば様々なHLA型の)Muse細胞をライブラリー化したMuse細胞バンクを構築することで、上記のMuse細胞利用場面における細胞を必要に応じて提供できる体制を実現でき、例えば、上記に挙げた目的の他、緊急に要する細胞移植治療のための拒絶反応の無い(少ない)細胞提供、などを行うことができる。すなわち、本発明は種々の遺伝子特性を有するMuse細胞を単離し、集めることにより、異なる遺伝子特性を有するMuse細胞のライブラリー、すなわちMuse細胞バンクを作製する方法を包含する。また、Muse細胞だけでなく、Muse細胞からできた胚様体様細胞塊、及びMuse細胞や前記胚様体様細胞塊から分化させて得られた細胞若しくは組織・器官を得てライブラリーやバンクを構築することもできる。本発明においては、これらのMuse細胞からできた胚様体様細胞塊、及びMuse細胞や前記胚様体様細胞塊から分化させて得られた細胞若しくは組織・器官を得てライブラリーやバンクも細胞ライブラリー又は細胞バンクと称する。本発明は、このようにして作製した細胞ライブラリー又は細胞バンクを包含する。該細胞ラブラリー又は細胞バンクは例えば、異なる遺伝的特性を有する細胞等が収納された複数のチューブ等の容器からなり、該細胞は凍結されていてもよい。例えば、被験体において、組織や器官を移植し、あるいは再生する必要が生じた場合に、上記細胞ライブラリー又は細胞バンクから、前記被験体に遺伝的背景等に関して適合した細胞を選択し、該細胞を用いて移植や再生治療を行うことができる。
本発明は、疾患の治療のために、本発明の多能性幹細胞や該細胞画分や該細胞由来の派生細胞や誘導細胞の治療上有効な量を治療を必要としている患者に投与することを含む治療方法を包含する。ここで、有効な量とは、例えば、投与する細胞数で特定することができ、疾患の種類や重篤度により適宜決定することができる。上記治療法においては、本発明の多能性幹細胞は、テラトーマ(奇形腫)を形成しないため、患者にテラトーマが形成されない。また、自己細胞由来のMuse細胞を投与する場合、患者を放射線照射や化学療法等の処置により骨髄機能を欠損させる必要はないが、自己細胞ではないMuse細胞を用いる場合は、上記処置を行えばよい。
さらに、Muse細胞は、iPS細胞(induced pluripotent stem cell)のソースとなり得る。Muse細胞をソースとしたiPS細胞の作製効率は他の細胞(例えば、SSEA−3発現を指標に分画していない皮膚線維芽細胞)をソースとした場合に比べはるかに(少なくとも25倍以上)高い。
Muse細胞に特定の遺伝子を導入し、あるいは特定の化合物を導入すること等により細胞形質を変化させることによりiPS細胞を作製することができる。細胞形質の変化は、リプログラミングや癌化を含み、現在知られている方法、あるいは将来的に確立されるあらゆる方法を用いることができる。
例えば、特許4182742号の記載に従って遺伝子をMuse細胞に導入し、あるいは図27の記載に従って、Muse細胞からiPS細胞を確立することができる。また、図27に記載の方法以外に、化学物質、外来遺伝子又は外来タンパク質を導入して、iPS細胞を樹立することが可能であるといえる。Muse細胞からのiPS細胞の確立は、例えば後述の実施例に記載の方法で行うことができる。
このようにしてMuse細胞から得られたiPS細胞をMuse由来iPS細胞(Muse−iPSC)と呼ぶことがあり、本発明は該Muse由来iPS細胞をも包含する。Muse由来iPS細胞は、Muse細胞由来の増殖性を有する多能性幹細胞ということができる。
本発明を以下の実施例によって具体的に説明するが、本発明はこれらの実施例によって限定されるものではない。
材料及び方法
本実施例において、以下の細胞を用いた。
間葉系細胞として、2株のヒト皮膚線維芽細胞画分及び4株のヒトMSC(bone marrow stromal cell)画分を用いた。ヒト線維芽細胞画分は、(1)H−fibroblast−1(正常ヒト線維芽細胞(NHDF)、Lonza社より入手)及び(2)H−fibroblast−2(成人ヒト皮膚線維芽(HDFA)、ScienCell社より入手)を用いた。ヒトMSC画分としては、H−MSC−1、H−MSC−2、H−MSC−3及びH−MSC−4をLonza社及びALLCELLS社より入手して用いた。ヒトMSC画分については、Pittenger,M.F.et al.Science 284,143−147(1999);Dezawa,M.et al.J Clin Invest 113,1701−1710(2004);Dezawa,M.et al.Science 309,314−317(2005)に詳細に記載されている。
細胞は10%FBS及び0.1mg/mlカナマイシン含有α−MEM(alpha−minimum essential medium)を用いて、37℃、5%CO2条件下で培養した。入手したものの最初の培養を第1代とし、その後細胞が95%コンフルエントになったところで、細胞培養液:培地の比を1:2として継代し、4~10代継代したものを用いた。
ヒトES細胞(hESC)は京都大学より入手したkyoto hESC−1(KhES−1)を用いた。
マウスES細胞(TT2細胞)及びヒトES細胞(KhES−1)はC57BL/6マウスの12.5日胚より確立したマウス由来フィーダー細胞存在下で維持した。
以下の方法で実験を行った。
1.間葉系細胞のストレス刺激
ストレス刺激として低栄養条件での培養、低血清濃度での培養、低酸素濃度での培養、繰返しトリプシン処理、長時間トリプシン処理を行うため以下の培養条件を採用した。
(1)無血清培地(STEMPRO MSC SFM(Invitrogen社)を用いた2日間の培養(無血清)
(2)Hanks’ Balanced Salt Solution(HBSS)バッファー(Invitrogen社)を用いた2日間の培養(HBSS)
(3)10%FBS含有α−MEMを用いての低酸素濃度(1%)での2日間の培養(10%FBS+LowO2)
(4)トリプシン中(0.25%トリプシン−HBSS)での1時間インキュベーション3回の処理(トータル3時間のトリプシン処理)(Try 3×1hr)
(5)トリプシン中での8時間インキュベーション(LTT 8hr)
(6)トリプシン中での16時間インキュベーション(LTT 16hr)
陰性対照として、ヒト末梢血単核細胞画分を用いた。
上記(4)、(5)及び(6)の条件の場合、100,000個~500,000個の細胞を5mlトリプシン溶液中に浮遊させて培養を行った。上記の(1)~(3)のストレス刺激の場合、刺激後、細胞を5分間のトリプシン処理により集め、(4)~(6)のストレス刺激の場合、細胞を直接チューブに集めた。
ストレス刺激後に生じる大量の死細胞はボルテックス処理することにより破壊した。すなわち、最大500,000個の細胞を含む5mlの培地を15mlのファルコンチューブに移し、ボルテックスミキサー(IKA Works社)を用いて1800~2200rpm/minで3分間ボルテックス処理を行った。その後、2000rpmで15分間遠心分離を行い、上清を除去した。生細胞の回収率は約70~80%であった。
2.MC培養
本実施例においては、細胞をメチルセルロース含有培地中で浮遊培養した。メチルセルロース含有培地中での培養をMC培養と呼ぶ。MC培養については、Nakahata,T.et al.,Blood 60,352−361(1982)に記載されている。
細胞が容器底に付着するのを防ぐために、培養ディッシュをPoly−HEMA(Poly(2−hydroxyethyl methacrylate)でコートした。すなわち、600mgのpoly−HEMA(SIGMA社)を95%エタノール40mlに37℃で攪拌して溶解し、ディッシュに添加し(96ウェル培養ディッシュの場合40μl/ウェル、12ウェルディッシュの場合200μl/ウェル)、一晩乾燥させた。
MC(MethoCult H4100)(StemCell technologies社より入手)を20%FBS含有α−MEMに最終濃度2%で懸濁した。ゲル状のMC培地中の細胞濃度は細胞同士の凝集を抑える十分な細胞間距離を確保できる8000細胞/mlとした。細胞とMC培地を穏やかなピペッティングにより十分混合し、poly−HEMAコートした培養ディッシュに移した。乾燥を防ぐために、10%FBS含有α−MEMを3日毎に最初のMC培養の容積の1/10量、ゆっくりと添加した。
細胞塊(細胞塊は、本発明の多能性幹細胞Muse細胞由来の細胞塊であるためMuse細胞由来胚様体様細胞塊と呼ぶ)のクローニングは7日目に行った。0.01MのPBSを培地に添加し、2000rpmで20分間遠心し、上清を捨てた。この処理を3回繰返し、細胞を洗浄した。回収した細胞ペレットを10μlの0.01M含有トリパンブルー溶液に懸濁し、スライドガラスに載せ位相差顕微鏡を用いて写真を撮った。直径25μmより大きい、ヒトES細胞と外観が似ているトリパンブルー陰性細胞塊のみをMuse細胞由来胚様体様細胞塊として数えた。Muse細胞由来胚様体様細胞塊の形成率は細胞塊の数/すべての生細胞数(すべてのトリパンブルー陰性細胞)により算出した。細胞計数の際、細胞塊はその大きさにかかわらず1細胞として数えた。これは、Muse細胞由来胚様体様細胞塊中に含まれる細胞の数の正確な計測は困難だからである。
ヒトES細胞においては、細胞を注意深くフィーダー細胞が含まれないように単離し、上記の方法でMC培養を行った。培養3日目に位相差顕微鏡観察像を得た。
3.単一細胞浮遊培養
96ウェルディッシュを上記の方法でpoly−HEMAでコートし、10%FBS含有α−MEMを用いて限界希釈法にて、単一細胞をそれぞれのウェルに播き、位相差顕微鏡にてウェル中の実際の細胞数を計数し、細胞が入っていないウェル、あるいは複数入っているウェルは計測から除外した。培養10日目に胚様体(EB)(Muse細胞由来胚様体様細胞塊)形成を計数した。それぞれの細胞株に対して3回の実験を行い、一回の実験では最低250ウェル以上の観察を行った。
4.アルカリフォスファターゼ染色
H−fibroblast画分及びH−MSC画分由来のMuse細胞由来胚様体様細胞塊を集め生理食塩水で数回洗浄し、Leukocyte Alkaline Phosphatase kit(sigma社)を用いて染色した。
5.Muse細胞由来胚様体様細胞塊のin vitroでの分化
MC培養又は単一細胞浮遊培養の7~10日後、H−fibroblast画分及びH−MSC画分由来の胚様体様細胞塊をガラスマイクロピペットにより採取し、ゼラチンコート培養ディッシュ又はカバーガラス上に移しさらに7日間培養すると細胞塊から細胞が広がる。かかる細胞における分化の有無を免疫組織化学分析及びRT−PCR分析に供した。
6.免疫組織化学
細胞を0.01M PBS中4%パラホルムアルデヒドで固定し、H−fibroblast画分及びH−MSC画分由来の富Muse細胞画分又はMuse細胞由来胚様体様細胞塊を遠心分離により集め、OCTコンパウンド中に埋め込み8μmの凍結切片を作製した。細胞塊は、ゼラチンコートスライドガラス上で固定し、免疫組織化学分析に供した。
1次抗体として、Nanog(1:500,Chemicon社)、Oct3/4(1:800,大阪大学 Dr.H.Hamadaより入手)、Sox2(1:1000,Abcam社)、PAR4(1:100,Santa Cruz社)、SSEA−3(1:20,DSHB社)、平滑筋アクチン(1:100,Lab Vision社)、neurofilamentM(1:200,Chemicon社)、α−フェトプロテイン(1:100,DAKO社)、マウスNumblike(1:500,カリフォルニア大学サンフランシスコ校のDr.Yuh−Nung Janより入手)及びtype1コラーゲン(1:20,Southern Biotech社)に対する抗体を用いた。2次抗体として、Alexa488又は568とコンジュゲートした抗ウサギIgG、抗マウスIgG又は抗マウスIgM抗体(Molecular Probes社)を用いて、免疫組織化学分析を行った。
7.核型の決定
H−fibroblast画分及びH−MSC画分由来のMuse細胞由来胚様体様細胞塊(Muse細胞由来胚様体様細胞塊から単一細胞を取り再度Muse細胞由来胚様体様細胞塊を形成させることを1~3回繰り返したもの)からの増殖細胞(clonally expanded cells)の核型をquinacrine−Hoechst染色により決定した。
8.免疫不全マウス精巣への細胞の注入
無処理細胞画分、並びにH−fibroblast画分及びH−MSC画分由来の富Muse細胞画分及びMuse細胞由来胚様体様細胞塊を用いた。富Muse細胞画分の場合、長時間トリプシン処理後血清を添加し、0.01M PBSで3回洗浄した。Muse細胞由来胚様体様細胞塊もMC培養から採取後に同PBSで3回洗浄した。1×105個の細胞をPBSに浮遊し、NOGマウス(登録商標)(NOD/Shi−scid,IL−2RγKO Jic、8週齢、財団法人 実験動物中央研究所より入手)の精巣にガラスマイクロチューブを用いて注入した。Muse細胞由来胚様体様細胞塊を用いた場合、レーザ共焦点顕微鏡の3Dグラフィック分析手法を用いて、50個のMuse細胞由来胚様体様細胞塊を取り、Muse細胞由来胚様体様細胞塊のトータル体積を測定し核の数で除することにより、それぞれの構成細胞の平均体積を測定した。測定の結果、1.5×105細胞/μlであることが算定され、この算定にしたがって上記数の細胞を集め、NOGマウス精巣に注入した。マウスは注入6ヵ月後に実験に供した。
対照試験として、1×106のマウスES細胞(陽性対照)及びマイトマイシンC処理MEF(マウス胚性フィーダー細胞)(陰性対照)をSCIDマウス精巣に注入し、注入後8週後に実験に供した。
9.光学顕微鏡により細胞の高解像度分析
H−fibroblast画分及びH−MSC画分由来の富Muse細胞画分及びMuse細胞由来胚様体様細胞塊について、安定な高解像度光学顕微鏡を用いてヒトMSC、線維芽細胞及び神経細胞などの細胞タイプの観察を行った。
10.電子顕微鏡による極薄切片観察
Muse細胞由来胚様体様細胞塊、H−fibroblast画分及びH−MSC画分由来のSSEA−3陽性細胞及びSSEA−3陰性細胞、並びにヒトES細胞の細胞塊を遠心分離により集め、100mMリン酸バッファー(pH7.2)中2.5%グルタルアルデヒドで30分間固定し、1%寒天中に包埋し、1mm3に切り、PBSで洗浄後、100mMリン酸バッファー(pH7.2)中2%OsO4で4℃で10分間染色した。サンプルを蒸留水で洗浄後、5滴の2%酢酸ウラニルで4℃で20分間染色した。蒸留水で洗浄後、染色サンプルを50%、70%又は90%エタノールを用いて4℃で10分間脱水し、次いで100%エタノールを3回交換することにより完全に脱水した。得られたサンプルを酸化プロピレンで5分間置換し、酸化プロピレン中50%エポキシレジン中に60分間包埋した。これを純粋なエポキシレジンに包埋し、60℃オーバーナイトで硬化させた。超薄切片は70~80nmの厚さで作製し、CCDカメラ付きの100kV電子顕微鏡で観察した。
11.Muse細胞由来胚様体様細胞塊の増殖速度
H−fibroblast画分及びH−MSC画分由来のMuse細胞由来胚様体様細胞塊について細胞群の倍化時間を測定するために、それぞれのMuse細胞由来胚様体様細胞塊をひとつずつ96ウェルプレートに移し、15分間のトリプシン処理を行った後にガラスマイクロピペットを用いてピペッティングを行った。細胞の数を計測し、所定の時間後ごとに少なくとも20~30個のMuse細胞由来胚様体様細胞塊について分析した。
12.RT−PCR
無処理細胞画分(24ウェルスケール中の約10,000細胞)及びH−fibroblast画分及びH−MSC画分(24ウェルスケール中の約10,000細胞)由来のin vitroで単一Muse細胞由来胚様体様細胞塊から分化した細胞(1~3サイクル)を用いた。トータルRNAを、NucleoSpin RNA XS(Macherey−Nagel社)を用いて抽出、精製し、第1鎖cDNAをSuperScript VILO cDNA Synthesis Kit(Invitrogen)を用いて調製した。PCR反応は適切なプライマーを設計し、Ex Taq DNA polymerase(タカラバイオ社)を用いて行った。用いたプライマーは以下のとおりであった。
陽性対照としては、α−フェトプロテインプライマーについてはヒト胎児肝臓(Clonetech社)を用い、それ以外はヒト完全胚(Clonetech社)を用いた。
13.定量的PCR(Q−PCR)
無処理細胞画分、H−fibroblast−1、H−fibroblast−2、H−MSC−1及びH−MSC−2由来の富Muse細胞画分及びMuse細胞由来胚様体様細胞塊からのトータルRNAをRneasy Mini Kit(Qiagen GmbH社)により集め、cDNAをRT2 First Strand Kit(SA Biosciences社)を用いて合成した。プライマーはSA Biosciences社より特注し、7300real time PCRシステム(Applied Biosystems社)を用いて定量的PCRを行った。得られたデータはΔΔCT方法(Livak KJ et al.,Methods 25:402−408,2001)により解析した。
14.DNAマイクロアレイ分析
無処理細胞画分、H−fibroblast−1、H−fibroblast−2、H−MSC−1及びH−MSC−2由来の富Muse細胞画分及びMuse細胞由来胚様体様細胞塊、並びに4名の健常者由来ヒト末梢血単核細胞画分混合物を用いた。totalRNAをRneasy Mini Kit(Qiagen GmbH社)により集め、DNAマイクロアレイにより分析した(タカラバイオ社)。アレイシグナルは、Affimetrix Expression Console V1.1ソフトウェア)により処理しノーマライズした。Pathway Studio 6.0(Ariadne genomics社)を用いて発現変動が認められた遺伝子を遺伝子オントロジーの機能的カテゴリーに割り当てた。階層的クラスタリングは、MeV4による群平均クラスタリングの手法を用いて、遺伝子の発現変動に基づいてユークリッド距離を用いて行った(Saeed AI et al.,Biotechniques 34(2):374−378,2003)。
15.テロメラーゼ活性の検出
H−fibroblast画分及びH−MSC画分由来の富Muse細胞画分、Muse細胞由来胚様体様細胞塊及びHela細胞を用いた。テロメラーゼ活性は、TRAPEZE XL telomerase detection kit(Millipore社)とEx Taqポリメラーゼ(タカラバイオ社)を用いて検出した。蛍光強度はマイクロプレートリーダー(TECAN社)を用いて測定した。
16.Bisulfite(亜硫酸水素塩)シーケンス
無処理細胞画分、H−fibroblast画分及びH−MSC画分由来の富Muse細胞画分及びMuse細胞由来胚様体様細胞塊由来の1μgのゲノムDNAをCpGenome DNA modification kit(chemicon社)で処理した。DNAをQIAquick column(Qiagen社)で精製した。ヒトOct3/4及びNanog遺伝子のプロモータ領域をPCRにより増幅し、PCR産物をpCR2.1−TOPO中にサブクローニングしそれぞれのサンプルについて10クローンまでをM13ユニバーサルプライマーを用いて配列決定し、プロモータ領域のメチル化の状態を調べた。PCR増幅には、Shimazaki T et al.,EMBO J,12:4489−4498,1993に記載のプライマーを用いた。
17.ヒト骨髄穿刺液からのMuse細胞由来胚様体様細胞塊形成
3人の健常人からのヒト骨髄穿刺液(ALLCELLS社より入手)から単核細胞画分をLymphoprep Tube(Axis−Shield PoC AS社)を用いて集め、8時間の長時間トリプシン処理を行った後にMC培養を行った。またトリプシン処理をせずに直接そのままでMC培養にも持って行った。7日目に細胞数を測定した。
18.MACSソーティング
3人の健常人からのヒト骨髄穿刺液(ALLCELLS社より入手)由来の単核細胞画分を抗CD105抗体とマイクロビーズのコンジュゲートと反応させ、MSカラム(Miltenyi Biotech社)を用いてソーティングした。CD105陽性細胞をフラクション1(間葉系細胞群)として集め、さらにCD105陰性細胞を抗CD34抗体及び抗CD117抗体の混合物とマイクロビーズのコンジュゲートとインキュベートし、再度ソーティングし、CD34陽性・CD117陽性細胞(フラクション2;造血幹細胞群にあたる)及びCD105陰性・CD34陰性・CD117陰性細胞(フラクション3)を得た(図4)。集めたサンプルを8時間の長時間トリプシン処理を行った後にMuse細胞由来胚様体様細胞塊の形成を測定した。
19.免疫組織化学
マウス精巣を0.02MPBS中4%パラホルムアルデヒドを用いて固定した。切片は凍結切片として10μmの厚さで作製した。サンプルを0.02MPBSで洗浄し、20%BlockAce(雪印社)含有バッファーを用いてブロッキングした後に免疫組織化学分析用の1次抗体とインキュベートした。用いた1次抗体は、抗平滑筋アクチン抗体(1:200、Lab Vision社)、抗MAP−2抗体(1:200、Biogenesis社)及び抗α−フェトプロテイン抗体(1:10、DAKO社)であった。
2次抗体としては、Alexa488又はAlexa568を結合した抗ウサギIgG抗体、Alexa568を結合した抗マウスIgG抗体を用い、DAPI存在下で反応させた。サンプルはニコン共焦点顕微鏡システムC1si(ニコン)を用いて観察した。
20.フローサイトメトリー及び細胞ソーティング
細胞をフィコエリトリン標識抗CD11c抗体、抗CD29抗体、抗CD34抗体、抗CD44抗体、抗CD45抗体、抗CD49f抗体、抗CD54抗体、抗CD71抗体、抗CD90抗体、抗CD105抗体、抗CD166抗体、抗CD271抗体又は抗vWF抗体(Beckton Dickinson社)又は抗SSEA−3抗体(Millipore社)とインキュベートした。抗SSEA−3抗体を用いる場合、細胞をさらにFITC結合抗ラットIgM抗体と反応させた。2mM EDTA及び0.5%ウシ血清アルブミンを添加したカルシウム及びマグネシウムを含まない0.02M PBSをFACS抗体希釈液として用いた。FACSCalibur(Becton Dickinson)によりCellQuestソフトウェア又はFACSAriaによりDIAソフトウェアを用いてデータ解析を行った。細胞をFACS抗体希釈液中で抗SSEA−3抗体とインキュベートしFACSAria(Becton Dickinson)により低流速及び4way purityソーティングモードでソーティングし、細胞のソーティングを行った。
21.統計解析
データは、平均±SEMで表す。データは、Bonferroni法による一対比較によりANOVAを用いて比較した。
結果
A.H−fibroblast画分及びH−MSC画分のストレス刺激
H−fibroblast画分とH−MSC画分のストレス刺激の結果の一例を表1に示す。
ストレス刺激とボルテックス処理後、トリパンブルー染色により生細胞数を計測し、生存率を計算した。生細胞を回収し、7日間のMC培養に供した。条件(2)の場合、大量の死細胞が存在し、生細胞回収効率が低かったため、Muse細胞由来胚様体様細胞塊はうまく計測できなかった。表1には「ND(not determined)」で示した。
6つの条件中で、H−fibroblast画分の16時間トリプシン処理及びH−MSC画分の8時間のトリプシン処理が最も効率的な刺激であった。2株のH−fibroblast画分及び4株のH−MSC画分を用いて繰返しこの実験を行ったところ、同様の傾向が認められた。陰性対照(ヒト末梢血単核細胞画分)においては、Muse細胞由来胚様体様細胞塊は認められなかった。代表的な観察値を表1に示す。


Muse細胞由来胚様体様細胞塊形成効率から判断すると、16時間(H−fibroblast画分)及び8時間(H−MSC画分)のトリプシン処理が6つの条件の中で最も効率的であると考えられた。16時間又は8時間トリプシン処理、1800~2000rpm/minでの3分間のボルテックス及び2000rpm15分間の遠心分離の一連の操作をMuse細胞の濃縮のための長時間トリプシン処理(LTT)と名付けた。ボルテックス処理後の細胞の回収率は約70~80%であった(図5)。
B.Muse細胞由来胚様体様細胞塊の判定基準
本実施例において、Muse細胞由来胚様体様細胞塊の判定基準を設けた。H−fibroblast画分及びH−MSC画分由来の富Muse細胞画分中の単一細胞の平均直径は10~13μmであった(図6a)。これらの細胞をMC培養すると、細胞は分裂を開始した。分裂するとそれぞれの細胞のサイズは小さくなり、8~10μmの細胞から構成される多細胞塊が徐々に形成された(図7e及び7f)。それぞれの細胞のサイズと外観は、MC培養したヒトES細胞に類似していた(図6b及び6c)。7日目に、ほとんどの多細胞塊は25μmより大きくなり、直径100~150μmになった。該細胞塊はES細胞と類似した外観を有していた。Φ25μmのフィルターを用いることにより、25μmより大きい塊を回収した(図6b)。H−fibroblast画分及びH−MSC画分の100個のMuse細胞由来胚様体様細胞塊までの免疫細胞化学によりほとんどのMuse細胞由来胚様体様細胞塊は多能性マーカーNanog、Oct3/4、Sox2、PAR4及びSSEA−3が陽性であることがわかり、アルカリフォスファターゼ染色でも陽性であった(図6e−g)。25μmより小さい細胞塊の場合、マーカーは検出される場合とされない場合があり、また多能性マーカーの局在はこれらのマーカーに典型的なパターンを示さない場合もあった。さらに、細胞の外観はむしろ富Muse細胞画分に類似している場合があった。
上記結果より、直径25μを超える多細胞塊を、Muse細胞由来胚様体様細胞塊とした。
C.ヒト間葉系細胞画分から形成される細胞塊の分析
生体がストレスに曝されたり、傷害を受けると休眠状態の組織幹細胞が活性化されることが知られている。本実施例において、H−MSC画分及びH−fibroblast画分に種々の方法でストレスをかけ(無血清処理、Hank’s Balanced Salt Solution(HBSS)による処理、低酸素処理、トータル3時間、8時間若しくは16時間のトリプシン処理等)、生存した細胞を集め、その後メチルセルロース(MC)含有培地中で浮遊培養(MC培養という)を行い、さらに8000細胞/mLの密度で7日間MC培養を行った(図7−1d)。その結果、最大で約直径150μmまでの種々の大きさの細胞塊の生成が認められた(図7−1e及びf)。図7−1cはH−fibroblast−1画分のMC培養の0日目の状態を、図7−1dは7日目の状態を示す。16時間のトリプシン処理を行ったH−fibroblast画分及び8時間のトリプシン処理を行ったH−MSC画分において、最も多くの細胞塊の形成が認められた。図7−1e及びfにH−fibroblast−1画分から形成された細胞塊(Muse細胞由来胚様体様細胞塊)の状態を示す。図7−1eはMC培養7日目の状態であり、図7−1fは単一細胞の浮遊培養10日目の状態である。細胞塊をサイズごとにフィルターを用いて分画し、免疫細胞化学分析を行った。直径25μmを超える細胞塊中に、Nanog、Oct3/4、SSEA−3、PAR−4及びSox2の多能性幹細胞マーカーが陽性であり(図7−2g−l)、さらにアルカリフォスファターゼ染色で陽性の細胞(図7−3m−o)が検出された。これらの細胞について電子顕微鏡を用いて観察したところ、H−fibroblast画分及びH−MSC画分から形成した細胞塊において、ES細胞と同様の核/細胞質比、細胞内器官の減少、核中の1つ若しくは2つの巨大な核小体の存在という特徴が認められた(図7−4p−r)。
生体のH−MSC画分及びH−fibroblast画分から、多能性マーカー陽性及びアルカリフォスファターゼ染色陽性を示す細胞塊を浮遊培養で形成し得る細胞を見出した。本発明者らは、これらの細胞をMuse細胞(multilineage differentiating stress enduring cells)と名付けた。16時間のトリプシン処理を行ったH−fibroblast画分及び8時間のトリプシン処理を行ったH−MSC画分から形成した細胞集団を「富Muse細胞画分(Muse−enriched population)」と呼び、該集団から得た単一細胞を浮遊細胞したところ、富Muse細胞画分の9~10%において、細胞塊の形成(Muse細胞由来細胞塊:Muse細胞由来胚様体様細胞塊)が認められた。このことは、富Muse細胞画分には約9~10%のMuse細胞が含まれることを示している。
単離したMuse細胞の増殖について調べたところ、培養開始後1~2日目で細胞分裂が観察され、細胞は10日目頃までは約1.3日の分裂周期で増殖した(図8−2)。しかし、11~12日目には増殖速度は低下し、14日目に直径150μmの大きさの細胞塊が形成された時点で増殖が停止した。Muse細胞由来胚様体様細胞塊を5分のトリプシン処理でばらばらにし単一細胞とし、単一浮遊培養を行ったところ、細胞は生存したが、増殖速度は分裂周期が5~7日と非常に低いままであり、細胞によっては細胞増殖の停止も観察された(図8−1の(1))。このことは、これらの細胞塊の増殖が制限され、一旦速度が低下すると、浮遊培養において、増殖速度は再上昇しにくいことを示す。しかしながら、単一のMuse細胞由来胚様体様細胞塊を接着培養に移すと、増殖を再開した。5~7日後、比較的小規模の増殖細胞群(3000~5000細胞)を5分のトリプシン処理でばらばらにし、MC培養すると、約40%の効率で細胞塊の形成が認められた(図8−1の(2))。細胞群のスケールをさらに上げて約5~10×104に達したところで、トリプシンで長時間処理すると、再びMuse細胞(2nd cycle)を生成し、約10%の率でMuse細胞由来胚様体様細胞塊を形成した(図8−1)。トリプシンでの長時間処理−浮遊培養−接着培養を5回繰り返したところ、それぞれの世代において、同様の特性及びMuse細胞由来胚様体様細胞塊形成率を示した。5cycle目のMuse細胞由来胚様体様細胞塊においても、多能性マーカー及びアルカリフォスファターゼは陽性であった。
これらの現象が変異などを起こした異常な細胞によるものでないことを確認するために核型検査を行った。Muse細胞由来胚様体様細胞塊由来の細胞(clonally expanded cells)においては多くは正常な核型を有し、染色体の異常は認められなかった(図8−3)。このことにより正常な細胞による現象であることを示す。
上記の結果は、Muse細胞が自己複製(セルフリニューアル)能を有し、クローン増殖することを示している。Muse細胞は、Muse細胞−Muse細胞由来胚様体様細胞塊−クローン増殖の一連のサイクルにより増殖する。従って、間葉系細胞集団から大量のMuse細胞を得ることが可能であると予測される。
D.Muse細胞由来胚様体様細胞塊の3胚葉細胞への分化
分化能を確かめるため、単一のMuse細胞由来胚様体様細胞塊をゼラチンでコートしたディッシュに移し分化させた。7日目にα平滑筋アクチン(中胚葉マーカー)、デスミン(中胚葉マーカー)、神経フィラメント−M(外胚葉マーカー)、αフェトプロテイン(内胚葉マーカー)及びサイトケラチン7(内胚葉マーカー)が検出された(図9−1a−c)。RT−PCRにより、1から3サイクルの培養を行ったMuse細胞由来胚様体様細胞塊(第1から3cycle)はαフェトプロテイン及びGATA6(内胚葉マーカー)、微小管結合タンパク質2:MAP−2(中胚葉マーカー)及びNkx2.5(中胚葉マーカー)を発現していることが確認されたが、無処理のH−fibroblast又はMSC群はゼラチンコートしたディッシュ上での培養でも分化が認められなかった(図9−2)。
さらに、富Muse細胞画分、Muse細胞由来胚様体様細胞塊及びES細胞を免疫不全マウスの精巣に注入し、テラトーマ(奇形腫)を形成するか否かを確かめた(図9−3e)。組織学検査の結果、ES細胞ではすべてのマウスにおいて8週以内にテラトーマの形成が認められた。しかしながら、富Muse細胞画分では13頭中10頭で、Muse細胞由来胚様体様細胞塊では11頭中10頭で、図9−3eに示すように、移植されたヒト細胞の残存と、種々の細胞種への分化が認められた。テラトーマは少なくとも6ヶ月まで富Muse細胞画分及びMuse細胞由来胚様体様細胞塊の移植群において形成が全く認められなかった。移植したヒト細胞は抗ヒトミトコンドリア抗体によって標識されるが、それらの細胞は同時に外胚葉マーカー(神経フィラメント)、内胚葉マーカー(α−フェトプロテイン)及び中胚葉マーカー(平滑筋アクチン)を発現することが確認された(図9−3f−i)。
これらのデータは、H−fibroblast画分、H−MSC画分由来Muse細胞及びMuse細胞由来胚様体様細胞塊はin vitroでもin vivoでも3胚様に分化し得ることを示している。
E.定量的PCR
多能性及び分化状態に関連したマーカーの発現を図10に示す。Nanogの発現は、富Muse細胞画分及び細胞クラスターにおいては、無処理細胞に比べそれほど高くなかった。多能性幹細胞の幾つかにおいては、Nanogはそれほど発現しない(Chou YF et al.,Cell 135,449−461(2008);Bui HT et al.,Development.135(23):3935−3945(2008))。Nanogと同様に、Q−PCRでのOct−4はマウスES細胞に比較するとリプログラミング体細胞において低い(Bui HT et al.,Development.135(23):3935−3945(2008))。従って、Nanog及び他の多能性(pluripotency)マーカーの発現量はpluripotencyにとってそれほど重要ではない。
F.富Muse細胞画分及びMuse細胞由来胚様体様細胞塊の遺伝子発現
定量PCRにより多能性及び非分化状態に関連するいくつかのマーカーが富Muse細胞画分及びMuse細胞由来胚様体様細胞塊でアップレギュレートされていることが示された。富Muse細胞画分は上記のように9~10%のMuse細胞を含んでいるに過ぎないが、Rex1(ZFP42)、Sox2、KLF−4、c−Myc、DPPA2(developmental pluripotency associated 2)、ERAS、GRB7(Growth factor receptor−bound protein 7)、SPAG9(Sperm associated antigen)、TDGF1(teratocarucinoma−derived growth factor1が、無処理細胞画分に比べて高度に又は適度にアップレギュレートされていた。Muse細胞由来胚様体様細胞塊においては、DAZL(azoospermia−like)、DDX4(VASA)、DPPA4(developmental pluripotency associated 4)、Stella、Hoxb1、PRDM1及びSPRY2(sprouty homolog2)が無処理細胞に比べてアップレギュレートされていた(図10a)。
H−fibroblast画分及びH−MSC画分由来の、無処理細胞画分、富Muse細胞画分及びMuse細胞由来胚様体様細胞塊の全体的な遺伝子発現を、ヒト末梢血単核細胞画分をコントロールとして比較したところ、無処理細胞画分、富Muse細胞画分及びMuse細胞由来胚様体様細胞塊の間で幾つかの遺伝子において発現パターンの変化が認められた(図10a)。
富Muse細胞画分及びMuse細胞由来胚様体様細胞塊におけるテロメラーゼ活性は低かった。このことは、テロメラーゼ活性がMuse細胞の増殖活性にそれほど関与していないことを示している(図10b)。
G.DNAマイクロアレイによる全体的遺伝子発現
108プローブのピアソン相関分析を、ヒト末梢血単核細胞(陰性対照)、無処理細胞画分、H−fibroblast画分及びH−MSC画分由来の富Muse細胞画分及びMuse細胞由来胚様体様細胞塊について行った(図11)。
また、DNAマイクロアレイにより、発現しているオドラント受容体及びケモカイン受容体をピックアップした。
H.生体におけるMuse細胞の存在
上記に示した検討は、安定な培養細胞を用いて行った。細胞を成体から取り出して培養した場合、生体内とは異なる特性を持ち得るので、Muse細胞やMuse細胞由来胚様体様細胞塊がアーティファクトな産物である可能性は否定できない。そこで、ヒト生体すなわちヒト骨髄細胞から培養を経ないで直接Muse細胞由来胚様体様細胞塊を得ることを試みた。ヒト骨髄穿刺液から単核細胞画分を単離し、直接MC培養に供するか(naive hBM−MC)、あるいは8時間のトリプシン処理を行った後にMC培養に供した(8hr−hBM−MC、トリプシン処理後の単核細胞画分の生存率は約3.5%であった)。7日後、Muse細胞由来胚様体様細胞塊の形成が無処理のnaive hBM−MCでは約0.004%の効率で認められ、8hr−hBM−MCでは約0.3%すなわち約75倍の効率であった(図12a)。これらのMuse細胞由来胚様体様細胞塊はアルカリフォスファターゼ染色陽性であった(図12b)。無処理のnaive hBM−MC及び8hr−hBM−MC由来の単一Muse細胞由来胚様体様細胞塊からクローン増殖した細胞のRT−PCRにより、α−フェトプロテイン、GATA6、MAP−2及びNkx2.5の発現が認められた(図13)。これらの結果は、ヒト骨髄中にin vivoにおいてもMuse細胞が存在し、8時間のトリプシン処理で富化しMuse細胞由来胚様体様細胞塊を形成させることができることを示している。さらに、骨髄の多くのタイプの細胞中で、Muse細胞がCD105陽性間葉系細胞画分に属していることが確認された。
上記のように、ヒト骨髄穿刺液から直接単離した単核細胞画分中の無処理hBM−MCの形成効率は0.004%と著しく低かった。培養中の細胞においては、細胞集団の構成が変化し得るため、安定的な培養を行っている細胞は、骨髄から単離した無処理単核球とは異なるMuse細胞由来胚様体様細胞塊形成傾向を示すと考えられる。これを確認するため、次いで、ヒト骨髄穿刺液を培養し、初代MSCを回収しMC培養を行ったところ、約0.2%のMuse細胞由来胚様体様細胞塊形成が認められた。これらの初代MSCをさらに2回又は5回継代培養したところ、Muse細胞由来胚様体様細胞塊形成効率は無処理細胞画分に対して約0.5%~1.0%それぞれ上昇した。無処理のH−fibroblast画分及びヒトMSC画分の約1.2%がMuse細胞由来胚様体様細胞塊を形成する。これらの結果は、Muse細胞のようなストレス耐性を有する細胞は継代培養等のin vitro培養環境で残り、このため安定な継代培養細胞画分が骨髄穿刺液から直接単離した単核細胞画分よりもより高いMuse細胞由来胚様体様細胞塊形成効率を示したことを示唆する。
骨髄はMSC、造血系細胞、内皮細胞を含む多種の単核細胞を含む。どの画分がMuse細胞を含むかを調べるために、ヒト骨髄穿刺液から単離した単核細胞画分を、CD34、CD117(いずれも造血幹細胞マーカー)及びCD105(間葉系細胞のマーカー)に対する抗体を用いて直接MACSソーティングに供し、各々の画分を8時間トリプシン処理し、7日間MC培養を行った。CD34陽性、CD117陽性画分にはMuse細胞由来胚様体様細胞塊はほとんど検出されなかったが、CD34陰性、CD117陰性、CD105陽性画分にはCD34陰性、CD117陰性、CD105陰性画分の50倍のクラスターの形成が認められた。この結果は、Muse細胞は主にCD105陽性間葉系細胞画分中に存在することを示唆している。
I.MACSソーティング
骨髄単核細胞画分からのそれぞれのフラクションの収率は、フラクション1(CD105陽性フラクション)で1.8%、フラクション2(CD34陽性・CD117陽性フラクション)で8.5%、フラクション3(CD34陰性・CD117陰性・CD105陰性フラクション)で89.7%であった。フラクション1、2及び3の細胞塊形成率は、それぞれ0.5%、0%及び0.01%であった。従って、フラクション1の細胞塊形成率はフラクション3の約50倍であった。
J.FACSソーティング
一例として、SSEA−3をマーカーとしてFACSソーティングを行った。
H−fibroblast及びHMSCの両方について、SSEA−3陽性細胞及びSSEA−3陰性細胞をFACSソーティングにより分離し、単一細胞懸濁培養に供した。SSEA−3陽性細胞の約50~60%がMuse細胞由来胚様体様細胞塊を形成した。一方、SSEA−3陰性細胞ではMuse細胞由来胚様体様細胞塊を形成しなかった。
K.Muse細胞の特徴
FACS分析により、無処理H−fibroblast画分及びH−MSC画分は間葉系細胞に発現するCD44、CD49f、CD54、CD90、CD105陽性の画分を含むことがわかった。しかし、CD11c、CD34、CD45、CD71、CD166、CD271及びフォンビルブランド(vWF)因子は陰性であった。富Muse細胞画分においては、CD44及びCD54が陰性となったSSEA−3陽性画分は約0.7~1.9%であった(図14)。
SSEA−3は多能性のマーカーの1つであるので、無処理のH−fibroblast画分及びH−MSC画分中のMuse細胞由来胚様体様細胞塊形成率(約1.2%)及び富Muse細胞画分での形成率(9~10%)は、それぞれのSSEA−3の陽性率(無処理細胞画分は約0.7~0.9%、富Muse細胞画分では7~8.3%)に似ていた。SSEA−3の陽性率は、Muse細胞のある状態を示している可能性がある。免疫組織化学によると、無処理のH−fibroblast画分及びH−MSC画分中のSSEA−3陽性細胞の数は1%未満であった。そこで、H−fibroblast画分及びH−MSC画分由来の富Muse細胞画分からSSEA−3陽性細胞をソーティングし、単一浮遊培養を行った。その結果、SSEA−3陽性細胞の50~60%がMuse細胞由来胚様体様細胞塊を形成した。これは富Muse細胞画分のMuse細胞由来胚様体様細胞塊形成の約6~7倍、無処理細胞画分のMuse細胞由来胚様体様細胞塊形成の約60倍である。一方SSEA−3陰性の細胞画分からはMuse細胞由来胚様体様細胞塊の形成は認められなかった。FACSによるソーティングにより得られたSSEA−3陽性細胞画分由来の単一細胞塊からクローン増殖により得られた細胞(3,000~5,000細胞)のSSEA−3陽性率は約45%であった(図15−1a)。このことは、Muse細胞由来胚様体様細胞塊の形成過程において、非対称性分裂が関与しており、単一Muse細胞由来胚様体様細胞塊のクローン増殖においても同様であることを示唆している。実際、非対称性分裂に関与していることが知られているNumblikeは、2つの細胞への分裂の過程で1つの細胞中にのみ存在する(図15−2b)。これらの結果は、非対称性細胞分裂がMuse細胞の増殖に関与していることを示唆する。
電子顕微鏡観察により、H−fibroblast画分及び長時間トリプシン処理後にソーティングしたH−MSC画分由来SSEA−3陰性細胞において核の変形と細胞質中の小胞が観察され、細胞が障害を受けていることが示された。しかし、SSEA−3陰性細胞と陽性細胞の間で明らかな形態的な差は認められなかった(図15−3c及びd)。
SSEA−3陽性細胞の重要性は、移植実験においても示された。SSEA−3陰性細胞画分を移植した場合、SSEA−3陽性細胞画分を移植した場合に比べて、組織マーカーの発現はほとんどの細胞で認められなかった。
富Muse細胞画分において、ほとんどのSSEA−3陽性細胞はOct3/4及びSox2を共発現しており、細胞質中に検出された(図15−4e及びg)。しかし、核中で発現している細胞は極めて少なかった(図15−4f)。この結果は、SSEA−3がMuse細胞の良いマーカーとなり得ることを示している。一方、Muse細胞由来胚様体様細胞塊の細胞においては、Oct3/4及びSox2は核中に主に局在していた(図7−2h及びl)。この2つのマーカーの細胞内局在の差異は細胞状態の差異を反映している可能性がある。
Muse細胞が長時間のトリプシン処理により人工的に誘導された可能性も否定できない。多くのMuse細胞が骨髄中のCD105陽性細胞画分に存在する。さらに、SSEA−3陽性細胞は、Muse細胞の特性を示す。そこで、ヒト成体骨髄穿刺液からSSEA−3及びCD105の二重陽性(double positive)細胞として、Muse細胞を直接得ることを試みた。二重陽性細胞は、骨髄由来単核球中に0.025%~0.05%存在し、それを長時間のトリプシン処理なしに、直接単一細胞浮遊培養に供した。7日後、11.4±1.2%の細胞(単核球の0.003~0.005%に相当する)が、Muse細胞由来胚様体様細胞塊を形成し、アルカリフォスファターゼ陽性であった。次いで、単一Muse細胞由来胚様体様細胞塊を接着培養により3,000細胞まで増殖させ、単一浮遊培養に供した。これらの細胞中、33.5±3.1%の細胞が第2世代のMuse細胞由来胚様体様細胞塊を形成した。また、単一Muse細胞由来胚様体様細胞塊からゼラチンでコートした培養ディッシュで増殖させた細胞についてRT−PCRを行ったところ、α−フェトプロテイン、GATA6、MAP−2及びNkx2.5の発現が認められた。これは、ヒト成体骨髄中にMuse細胞の特性を有する細胞が存在することを示唆する。
上記のように、ストレス刺激により非幹細胞を除去することにより幹細胞を富化することができた。Muse細胞を長時間トリプシン処理とその後のSSEA−3陽性細胞のソーティングにより効率的に集めることができた。Muse細胞は、多能性マーカーを発現しアルカリフォスファターゼ染色陽性であり、外胚葉、中胚葉及び内胚葉系細胞に分化し得るMuse細胞由来胚様体様細胞塊を形成した。また、増殖速度の測定において腫瘍形成性増殖に関する特徴を有しておらず、テロメラーゼ活性も有していなかった。このことは、Muse細胞は爆発的増殖を防ぐための多重のセキュリティーシステムを有していることを示している。Muse細胞の非腫瘍形成性特性はMuse細胞をマウス精巣に注入する実験においても認められた。この特性は生体機能のバランスを維持する上で都合がよく、この特性がなければ、異常増殖や異形成により生体は破壊され、腫瘍形成やテラトーマ形成をもたらす。
Muse細胞の多能性は接着培養系では顕在化せず、浮遊培養で観察された。
通常、Muse細胞は不活性休眠状態にあり生体が、危機的状態に陥ったり、あるいは重篤な傷害を被り、又は飢餓若しくは虚血状態等のストレス過多状態が維持されるとある種のシグナルが伝達されて活性化すると考えられる。その後、Muse細胞は組織再生に貢献し、細胞間相互作用や組織化に貢献する。
実施例1の検討において、FACSにより得たSSEA−3陽性細胞画分が多能性幹細胞の特性を有していること、すなわちMuse細胞であることが判明したが(上記J、K等)、さらに、単離したSSEA−3陽性細胞を用いて、in vitroでの分化能及びin vivoでの分化能を検討し、さらにMuse由来iPS細胞の確立を行った。
1.損傷組織への移植によるin vivoでの分化能の検討
GFP(緑色蛍光タンパク質)−レンチウイルスで標識したSSEA−3陽性Muse細胞を単離し、脊髄(圧迫損傷)、肝臓(CCl4を腹腔内注射、劇症肝炎モデル)又は腓腹筋(cardiotoxin注射)に損傷を与えた免疫不全マウス(NOGマウス)に静脈注射により移植した。ヒト皮膚細胞由来のMuse細胞をGFP(緑色蛍光タンパク質)−レンチウイルスで標識し(Hayase M et al.,J Cereb Blood Flow Metab.29(8):1409−20,2009)、Muse細胞由来胚様体様細胞塊が標識細胞由来であることをGFPにより確認した。脊髄損傷はNOGマウスに対してレベルTh9で行い(Farooque M et al.,Acta Neuropathol.,100;13−22,2000)、NOGマウスの腓腹筋にcardiotoxinを投与し筋肉変性を誘発し、四塩化炭素をNOGマウスに腹腔内投与し、肝臓変性を誘発した。筋肉及び肝臓については2日後、脊髄については7日後に1×105のMuse細胞を静脈注射により移植した(各6頭のマウスを使用)。コントロールとして、GFP標識MEC集団を静脈注射したマウスを用いた。移植の3又は4週間後に、マウスをパラホルムアルデヒドで固定し、免疫組織化学及び共焦点レーザ顕微鏡観察に供した。
脊髄損傷の4週間後、GFP及びヒトゴルジ複合体陽性細胞がニューロフィラメントを形成していた。(図16−1N及びO)。また、4週間後、再生している肝臓において、GFP及びヒトゴルジ複合体陽性細胞がヒトアルブミンを発現していた(図16−1P)。RT−PCRの結果によりMuse細胞を移植したNOGマウス肝臓においてヒトアルブミンの形成が示された(図16−2)。GFP陽性細胞を再生している筋肉に注射した場合、3週間でヒトジストロフィンを発現した(図16−3)。一方、SSEA−3陰性ヒト皮膚線維芽細胞画分を移植した場合、細胞のインテグレートは顕著に少なく、組織マーカーが陽性の細胞はほとんど認められなかった。これらの結果は、Muse細胞が損傷組織に統合され、それぞれ、in vivoで外胚葉、中胚葉及び内胚葉に分化し得ることを示す。
2.単一Muse細胞由来のMuse細胞由来胚様体様細胞塊からの増殖細胞の分化
Muse細胞の分化調節に誘導システムが有効かどうかを検討した。単一SSEA−3陽性Muse細胞由来のMuse細胞由来胚様体様細胞塊を接着培養に供し、増殖させた。単一Muse細胞由来の増殖細胞を回収し、4つの群に分け、それぞれの群に対して、神経、骨細胞、脂肪細胞及び幹細胞への分化誘導を行った(n=5)。
神経誘導のためには、細胞をpoly−HEMAでコートしたディッシュを用いて1×105/mlの密度でB−27supplementを含むNEUROBASAI培地(Gibco社)中で7日間培養し、sphere(球状の細胞塊)を形成させた。分化させるためにsphereをポリ−L−リシンでコートしたガラス上に移し、25ng/ml FGF及び25ng/ml BDNFを含む2%FBS中で10日間培養した。
骨細胞誘導のためには、細胞を4.2×103細胞/cm2の密度でHuman Mesenchymal Stem Cell Functional Identification Kit(R&D Systems)の骨細胞誘導培地を用いて14日間培養した。
脂肪細胞誘導のためには、細胞を2.1×104細胞/cm2の密度でHuman Mesenchymal Stem Cell Functional Identification Kit(R&D Systems)の脂肪細胞誘導培地を用いて14日間培養した。
肝臓細胞誘導のためには、細胞を2.0×104細胞/cm2の密度でDMEM(10%FBS、10nMデキサメタゾン及び100ng/ml HGFを含む10×ITS(GIBOCO社)、50ng/ml FGF4を含む)を用いてコラーゲンコートしたディッシュ上で14日間培養した。
神経誘導によりsphereが形成され、sphereは神経幹細胞マーカーであるネスチン、Musashi及びNeuroD陽性であることが確認された(図17−1A~D)。これらのsphereを分化培地中で培養するとMAP−2又はGFAP陽性細胞に分化した(図17−1E、MAP−2、GFAPいずれも89±5.7%の陽性率)。細胞を骨細胞誘導したとき、オステオカルシン陽性細胞(97±3.5%)及びアルカリフォスファターゼが認められた(図17−1F及びG)。脂肪細胞分化については、オイルレッドで染色される脂肪滴を有する細胞が形成された(90±4.9%)(図17−1H~I)。肝細胞誘導にヒトαフェトプロテイン陽性細胞(図17−1J、87±7.6%)並びにヒトアルブミンとヒトαフェトプロテイン陽性細胞(図17−2)の形成がRT−PCRにより認められた。これらの結果は、Muse細胞の3胚葉への分化が、非常に高い効率で誘導により調節できることを示している。
3.ヒト成人皮膚からのSSEA−3陽性細胞の採取
培養細胞を培養したり、Muse細胞由来胚様体様細胞塊を形成させることなく、ヒト成人皮膚から直接Muse細胞を単離することを試みた。
ヒト健常人(n=3)からの皮膚(BIOPREDIC Internationalより入手)の上皮及び脂肪組織を除き真皮を得、真皮を10% FBSを含むα−MEM中コラゲナーゼ/ジスパーゼと共に37℃で36時間インキュベートした。濾過により消化された皮膚細胞を回収し、1500rpmで20分間遠心分離し、α−MEMで洗浄し、0.25%トリプシン−HBSSで5分間インキュベートした。細胞をさらにFACSバッファーで千兆誌、SSEA−3とインキュベートし、FACSを用いてSSEA−3陽性細胞をソーティングした。約7cm2の皮膚組織から1.3±0.3×104の単一細胞を回収することができた。SSEA−3陽性細胞は、回収した単一細胞の1.7±0.2%であった。
SSEA−3陽性細胞の21.0±1.7%が、限界希釈による単一細胞浮遊培養で7日でMuse細胞由来胚様体様細胞塊を形成した。Muse細胞由来胚様体様細胞塊はALP陽性であり、単一のMuse細胞由来胚様体様細胞塊からゼラチンでコートしたディッシュを用いて増殖させた細胞について、RT−PCRで確認したところ、MAP−2、Brachyury、Nkx2.5、GATA6及びα−フェトプロテインを発現していた。この結果は、Muse細胞と同じ性質を有する細胞が上述の成人ヒト骨髄液と同様に、成人ヒト皮膚にも存在することを示している。
ヒト成人皮膚は、SKP(皮膚由来前駆細胞)、NCSC(神経堤幹細胞)、メラノブラスト(MB)、血管周囲細胞(PC)、内皮前駆細胞(EP)、脂肪由来幹細胞(ADSC)等の種々のタイプの幹細胞や前駆細胞を含んでいる。Muse細胞がこれらの幹細胞等と同じである可能性を排除するために、Muse細胞におけるSnai1(SKPのマーカー)、Slug(SKPのマーカー)、Sox10(NCSCのマーカー)、CD271(NCSCのマーカー)、Tyrp1(MBのマーカー)、Dct(MBのマーカー)、CD117(c−Kit)(MBのマーカー)、CD146(PC及びADSCのマーカー)、NG2(PCのマーカー)、CD34(EP及びADSCのマーカー)及びフォンビルブランド因子(EPのマーカー)の発現を分析した。これらのマーカーのいずれもSSEA−3陽性細胞において、FACS分析又はRT−PCRで認められなかった(図18−1及び図18−2)。この結果は、Muse細胞はヒト成人皮膚に存在することが公知である幹細胞や前駆細胞と異なる細胞であることを示している。
さらに、フェライト粒子を用いてMuse細胞の貪食能を調べたところ、Muse細胞はフェライト粒子を容易に取り込み、高い貪食能を有していた(図18−3)。
4.Muse由来iPS細胞(Muse−iPSC)の確立
iPS細胞は、Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子、c−Myc遺伝子、Nanog遺伝子、Lin28遺伝子等を導入し作製されている。Muse細胞は、多能性(pluripotency)マーカーを発現し、外胚葉、内胚葉及び中胚葉系細胞に分化し得るという点で、iPS細胞と類似した特性を有している。そこで、Muse細胞がiPS細胞のよい材料となり得るかを検討した。
方法は以下のとおりであった。
H−fibroblast画分由来のSSEA−3陽性細胞及び陰性細胞に、Takahashi et al.,Cell,131,861−872(2007)の記載に従って、レトロウイルスベクターを用いて、Nanog、Oct3/4、KLF4及びc−Mycの4つの因子を導入し、培養した。以下に、方法の詳細を示す。
プラスミドの確立
pMXsレトロウイルスベクター(Cell Biolabs)にヒトOct3/4、Sox2、Klf4、c−Mycのopen reading frameを組み込んだ。
レトロウイルスの感染及びiPS細胞の確立
PLAT−A細胞を100mm dishに5×106細胞の密度でまき、一晩培養した。次の日に、Fugene HDを用いてトランスフェクションをを行った。トランスフェクションから24時間後、新しい培地に交換した。3日後に上清を回収し、0.45μmのフィルターに通し、4μg/mlのポリブレンを加えた。60mm dishに1×105細胞の密度でまいたNHDF(皮膚線維芽細胞)にウイルス溶液を感染させた。24時間後、ウイルスの入っていない新しい培地に交換した。ウイルス感染から4日後にトリプシンにより剥がした細胞を、MEF(フィーダー細胞)の上に3×104細胞の密度でまいた。翌日、培地を4ng/ml bFGFを加えたPrimate ES mediumに交換した。その後2日に1回培地交換を行い、30日後、コロニーをピックアップし、24ウェルプレートにまいた。
PCR分析
RNeasy mini kit(QIAGEN)によりRNAを精製した。500ngのRNAをSuperScriptIIを用いて逆転写した。内因性のOct、Sox2、Klf4、Myc、NanogのプライマーとPCRの条件等はakahashi et al.,Cell,131,861−872(2007)に記載の通りで行った。
in vitroにおけるiPS細胞の分化
コラゲナーゼによりiPS細胞を採取した。細胞の塊をPoly−HEMAでコーティングしたdishに、20% Knockout serum replacement(Invitrogen)、2mM L−Glutamine、1×10−4M nonessential amino acid、1×10−4M 2−mercaptoethanol(Nacalai)、0.5% Penicillin/Streptomycinを含むDMEM/F12培地にて培養した。培地は2日に一回交換した。7日後、EBをゼラチンコートしたdishにまき、同じ培地で一週間培養した。
テラトーマの形成
60mm dishのiPS細胞をRock inhibitorにより処理し、Accutase(登録商標)により採取しチューブに集めて遠心後、PBSに浮遊させた。これをNOG mouse(登録商標)(財団法人 実験動物中央研究所)の精巣に注射した。12週後に4%パラホルムアルデヒドにより固定した。パラフィン切片はHE(Hematoxylin & Eosin)染色を行った。
以下の結果が得られた。
H−fibroblast画分由来のSSEA−3陽性細胞及び陰性細胞に、Takahashi et al.,Cell,131,861−872(2007)の記載に従って、レトロウイルスベクターを用いて、Nanog、Oct3/4、KLF4及びc−Mycの4つの因子を導入し、5日後にMEF上に再度まきなおし、培養した。colony pickupを行う直前、すなわちMEF上での培養30日目においては、SSEA−3陰性細胞中で形成されたコロニーは非ES細胞様コロニーであり、ES細胞のマーカーであるTra−1−80陽性のコロニーはまったく見られなかった。一方、多くのSSEA−3陽性細胞においては陰性の細胞群の約7倍ほどの数のコロニーを形成し、それらはTra−1−80は陽性であった。注目すべきは、SSEA−3陰性細胞(コロニーやコロニーを形成していない細胞すべてを回収)では、RT−PCRで測定したところ、MEF上30日目のcolony pick up直前でもNonog、Sox2などの多能性に密接に関連した重要な遺伝子は陰性であった。一方、SSEA−3陽性細胞において、内因性のOct3/4、KLF4及びRex1がアップレギュレーションされており、さらにNanog、Sox2が発現していた。予測されていたとおり、SSEA−3陽性細胞をcolony pick upし、新たなMEF(フィーダー細胞)上に移した場合、無処理H−fibroblast画分細胞を用いた場合よりも30倍ほど高い効率でiPSを作製することができた。これらのiPS細胞において、免疫細胞化学、RT−PCR及びQ−PCRにより、Tra−1−60、Tra−a−80、Rex1、UTF−1、テロメラーゼ逆転写酵素(TERT)及びヒトES細胞において発現している因子がアップレギュレーションしており、あるいは新たに発現していた(図19、21)。また、得られたMuse細胞由来iPS細胞において、Nanog、Oct3/4、Sox2及びTRA−1−81が発現していた(図19)。RT−PCRにより、Muse細胞由来iPS細胞ではNanog、Oct3/4及びSox2が発現しているが、SSEA−3陰性細胞由来コロニーでは発現していないことがわかった(図21)。
SSEA−3陽性細胞とSSEA−3陰性細胞画分とで、Oct3/4遺伝子、Sox2遺伝子、Klf4遺伝子およびc−Myc遺伝子の導入効率はほとんど同じであった。導入した細胞をマウスフィーダー細胞上でディッシュ当たり1×105の密度で培養した。両方の各群でコロニーを形成したが、SSEA−3陰性細胞画分から形成されたコロニー数は、SSEA−3陽性細胞画分の7分の1程度であった。さらに、SSEA−3陽性細胞画分と異なり、SSEA−3陰性細胞画分では、ヒト多能性幹細胞マーカーであるTRA−1−81陽性コロニーは、コロニーを採取する直前の培養30日後でも認められなかった(図22−1)。RT−PCRにより、内因性Sox2及びNanogはSSEA−3陽性細胞由来画分のみで発現しており、SSEA−3陰性細胞画分では発現していなかった(図22−2)。
さらに、SSEA−3陽性及び陰性細胞画分をそれぞれ継代し、iPS細胞株を確立した。3継代後、ヒトES細胞様の形態(平らなコロニー)を示したそれぞれのコロニー(図22−3C及びC1)についてRT−PCRを行い、内因性oct3/4、Sox2及びNanogを発現しているコロニーをiPSコロニーとしてカウントした。その結果、iPS細胞は、SSEA−3陽性細胞由来コロニーのみから形成され、効率は0.03%であった。SSEA−3陰性細胞由来コロニーからは形成されなかった(図22−3D及びD1)。
さらに、Muse細胞から確立したiPS細胞は、外胚葉、内胚葉及び中胚葉系細胞に分化し、マウス精巣にてテラトーマを形成した(図23−1~23−3)。
Muse細胞の増殖活性は、増殖速度及びテロメラーゼ活性の観点からそれほど高くない。このことは、Muse細胞は、マウス精巣中で3胚葉性の細胞に分化するが、テラトーマは形成しないことと対応している。もし、Muse細胞が皮膚や骨髄等のヒト成人組織において維持されている場合、その増殖は厳密に調節されているはずであり、そうでなければ、Muse細胞は生体のすべての部分で腫瘍を形成してしまうであろうことを考えても妥当である。さらに、ある条件下で培養した杯盤葉上層(epiblast)幹細胞がマウス精巣中でテラトーマを形成しないことが示されていることを考慮すると(Chou et al.,Cell,135,449−461(2008))、多能性細胞が常にテラトーマを形成するとは言えない。Muse細胞は、最初から多能性マーカーの発現や分化能等において多能性細胞の特性を示すので、Muse細胞は単独でiPS細胞になり、増殖活性が上昇し、マウス精巣中でテラトーマを形成するようになることが予測される。iPSの誘導メカニズムは解明されていないが、間葉系細胞中のMuse細胞における腫瘍性増殖性の獲得の可能性がある。
無処理ヒト皮膚線維芽細胞画分から約0.001%の効率でiPS細胞が確立できたが、これはK.Takahashi et al.,Cell 131,861(2007)の報告と一致する。従って、SSEA−3陽性細胞からのiPS細胞作製効率は、無処理線維芽細胞に対して30倍高かった。このことは、iPS細胞の形成に主にMuse細胞が寄与していることを示唆している。
免疫組織化学及びRT−PCRにより、Muse由来iPS細胞から得た胚様体は、in vitroで外胚葉(ニューロフィラメント及びMAP−2)、中胚葉(SMA、Brachyury及びNkx2.5)、及び内胚葉細胞(α−フェトプロテイン及びGATA−6)に分化することがわかった。さらに、免疫不全マウス精巣にMuse由来iPS細胞を投与したところ、テラトーマの形成が観察された。一方、Muse細胞由来胚様体様細胞塊を精巣に注射した場合、6ヶ月後においてもテラトーマの形成は認められず、MEFを注射したコントロールと比べて大きさもそれほど大きくならなかった。しかし、ヒトミトコンドリア並びにSMA、α−フェトプロテイン及びニューロフィラメントが陽性の細胞が同定された。この結果は、Muse由来iPS細胞と異なり、元々のMuse細胞はテラトーマを形成しないが、免疫不全マウス中で外胚葉、中胚葉及び内胚葉に分化し得ることを示す。
Muse細胞由来胚様体様細胞塊とMuse由来iPS細胞について定量PCR(Q−PCR)を行った。結果を図25及び26に示す。細胞周期調節に関連した遺伝子の発現パターンは、大きく異なっていた。細胞周期進行に関連した遺伝子のほとんどは、Muse細胞由来胚様体様細胞塊ではダウンレギュレートされていたが、Muse由来iPS細胞ではアップレギュレートされていた。一方、Muse細胞由来胚様体様細胞塊とMuse由来iPS細胞において、多能性及び未分化状態に関与した遺伝子の発現は同様の傾向を示した。ただし、Muse細胞由来胚様体様細胞塊におけるNanog、Oct3/4及びSox2の発現レベルはMuse由来iPS細胞よりはるかに低かった。Nanog遺伝子及びOct3/4遺伝子のプロモータ領域のシトシン・グアニンジヌクレオチド(CpG)はMuse由来iPS細胞でMuse細胞由来胚様体様細胞塊に比べてメチル化の程度は小さかった。なお、Nanog遺伝子のプロモータ領域のCpGのメチル化はSSEA−3陰性無処理細胞画分に比べて、Muse細胞由来胚様体様細胞塊で低かった(図24)。この結果は、Muse細胞とMuse由来iPS細胞の間の多能性マーカーの発現レベルの差を説明する。
本明細書で引用した全ての刊行物、特許および特許出願をそのまま参考として本明細書にとり入れるものとする。
Claims (23)
- 生体組織から単離できるSSEA−3陽性の多能性幹細胞。
- CD105陽性の請求項1記載の多能性幹細胞。
- CD117陰性及びCD146陰性の請求項1又は2に記載の多能性幹細胞。
- CD117陰性、CD146陰性、NG2陰性、CD34陰性、vWF陰性及びCD271陰性の請求項1又は2に記載の多能性幹細胞。
- CD34陰性、CD117陰性、CD146陰性、CD271陰性、NG2陰性、vWF陰性、Sox10陰性、Snai1陰性、Slug陰性、Tyrp1陰性及びDct陰性の請求項1又は2に記載の多能性幹細胞。
- テロメラーゼ活性が低いか又は無い、請求項1~5のいずれか1項に記載の多能性幹細胞。
- 三胚葉に分化する能力を持つ、請求項1~6のいずれか1項に記載の多能性幹細胞。
- 腫瘍性増殖を示さない、請求項1~7のいずれか1項に記載の多能性幹細胞。
- セルフリニューアル能を持つ、請求項1~8のいずれか1項に記載の多能性幹細胞。
- ストレス耐性である、請求項1~9のいずれか1項に記載の多能性幹細胞。
- 貪食能が高い、請求項1~10のいずれか1項に記載の多能性幹細胞。
- 以下に示す22個のオドラント受容体の少なくとも一つが陽性の請求項1~11のいずれか1項に記載の多能性幹細胞:
olfactory receptor,family 8,subfamily G,member 2(OR8G2);
olfactory receptor,family 7,subfamily G,member 3(OR7G3);
olfactory receptor,family 4,subfamily D,member 5(OR4D5);
olfactory receptor,family 5,subfamily AP,member 2(OR5AP2);
olfactory receptor,family 10,subfamily H,member 4(OR10H4);
olfactory receptor,family 10,subfamily T,member 2(OR10T2);
olfactory receptor,family 2,subfamily M,member 2(OR2M2);
olfactory receptor,family 2,subfamily T,member 5(OR2T5);
olfactory receptor,family 7,subfamily D,member 4(OR7D4);
olfactory receptor,family 1,subfamily L,member 3(OR1L3);
olfactory receptor,family 4,subfamily N,member 4(OR4N4);
olfactory receptor,family 2,subfamily A,member 7(OR2A7);
guanine nucleotide binding protein(G protein),alpha activating activity polypeptide,olfactory type(GNAL);
olfactory receptor,family 6,subfamily A,member 2(OR6A2);
olfactory receptor,family 2,subfamily B,member 6(OR2B6);
olfactory receptor,family 2,subfamily C,member 1(OR2C1);
olfactory receptor,family 52,subfamily A,member 1(OR52A1);
olfactory receptor,family 10,subfamily H,member 3(OR10H3);
olfactory receptor,family 10,subfamily H,member 2(OR10H2);
olfactory receptor,family 51,subfamily E,member 2(OR51E2);
olfactory receptor,family 5,subfamily P,member 2(OR5P2);及び
olfactory receptor,family 10,subfamily P,member 1(OR10P1)。 - 以下に示す5個のケモカイン受容体の少なくとも一つが陽性の請求項1~12のいずれか1項に記載の多能性幹細胞:
chemokine(C−C motif)receptor 5(CCR5);
chemokine(C−X−C motif)receptor 4(CXCR4);
chemokine(C−C motif)receptor 1(CCR1);
Duffy blood group,chemokine receptor(DARC);及び
chemokine(C−X−C motif)receptor 7(CXCR7)。 - 中胚葉系組織または間葉系組織由来である、請求項1~13のいずれか1項に記載の多能性幹細胞。
- 請求項1~14のいずれか1項に記載の多能性幹細胞を含む細胞塊または細胞画分。
- 生体組織から、以下の(i)~(vi)の特性の少なくとも1つの特性を指標に多能性幹細胞又は多能性細胞画分を単離する方法:
(i) SSEA−3陽性;
(ii) CD105陽性;
(iii) CD117陰性及びCD146陰性;
(iv) CD117陰性、CD146陰性、NG2陰性、CD34陰性、vWF陰性及びCD271陰性;
(v) CD34陰性、CD117陰性、CD146陰性、CD271陰性、NG2陰性、vWF陰性、Sox10陰性、Snai1陰性、Slug陰性、Tyrp1陰性及びDct陰性;並びに
(vi) テロメラーゼ活性が低いか又は無い。 - 生体組織由来細胞を細胞ストレスに暴露し生き残った細胞を回収することを含む多能性幹細胞又は多能性細胞画分を単離する方法。
- 細胞ストレスが、プロテアーゼ処理、低酸素条件下での培養、低リン酸条件下での培養、血清飢餓状態での培養、糖飢餓状態での培養、放射線曝露下での培養、熱ショックへの曝露下での培養、有害物質存在下での培養、活性酸素存在下での培養、機械的刺激下での培養及び圧力処理下での培養から選択される、請求項17に記載の多能性幹細胞又は多能性細胞画分を単離する方法。
- 細胞ストレスが、トリプシン処理である、請求項18に記載の多能性幹細胞又は多能性細胞画分を単離する方法。
- 請求項1~14のいずれか1項に記載の多能性幹細胞の派生細胞又は誘導細胞である多能性幹細胞。
- 請求項1~14のいずれか1項に記載の多能性幹細胞の派生細胞又は誘導細胞である分化した細胞。
- 請求項1~14及び20のいずれか1項に記載の多能性幹細胞を含む医薬組成物。
- 請求項21に記載の分化した細胞を含む医薬組成物。
Priority Applications (16)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020157013171A KR101651795B1 (ko) | 2009-07-15 | 2010-07-15 | 생체조직으로부터 단리 할 수 있는 다능성 줄기세포 |
| EP20160074.9A EP3680323A1 (en) | 2009-07-15 | 2010-07-15 | Pluripotent stem cell that can be isolated from body tissue |
| JP2011522886A JP5185443B2 (ja) | 2009-07-15 | 2010-07-15 | 生体組織から分離できる多能性幹細胞画分 |
| PL17163323T PL3214170T3 (pl) | 2009-07-15 | 2010-07-15 | Pluripotencjalna komórka macierzysta, którą można wyizolować z tkanki ciała |
| ES10799956T ES2647360T5 (es) | 2009-07-15 | 2010-07-15 | Célula madre pluripotente que puede aislarse de tejido corporal |
| CN201080041908.7A CN102858951B (zh) | 2009-07-15 | 2010-07-15 | 可从机体组织分离的多能干细胞 |
| SG2012003836A SG177705A1 (en) | 2009-07-15 | 2010-07-15 | Pluripotent stem cell that can be isolated from body tissue |
| AU2010271722A AU2010271722B8 (en) | 2009-07-15 | 2010-07-15 | Pluripotent stem cell that can be isolated from body tissue |
| KR1020177020765A KR101945343B1 (ko) | 2009-07-15 | 2010-07-15 | 생체조직으로부터 분리 할 수 있는 만능성 줄기세포 |
| EP10799956.7A EP2455452B2 (en) | 2009-07-15 | 2010-07-15 | Pluripotent stem cell that can be isolated from body tissue |
| NO10799956A NO2455452T3 (ja) | 2009-07-15 | 2010-07-15 | |
| DK10799956.7T DK2455452T4 (da) | 2009-07-15 | 2010-07-15 | Pluripotent stamcelle, der kan isoleres fra kropsvæv |
| PL10799956T PL2455452T5 (pl) | 2009-07-15 | 2010-07-15 | Pluripotencjalna komórka macierzysta, którą można wyizolować z tkanki ciała |
| EP17163323.3A EP3214170B1 (en) | 2009-07-15 | 2010-07-15 | Pluripotent stem cell that can be isolated from body tissue |
| CA2768238A CA2768238C (en) | 2009-07-15 | 2010-07-15 | Pluripotent stem cell that can be isolated from body tissue |
| KR1020167008209A KR101764080B1 (ko) | 2009-07-15 | 2010-07-15 | 생체조직으로부터 분리 할 수 있는 만능성 줄기세포 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US21378809P | 2009-07-15 | 2009-07-15 | |
| US61/213,788 | 2009-07-15 | ||
| US29015909P | 2009-12-24 | 2009-12-24 | |
| US61/290,159 | 2009-12-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011007900A1 true WO2011007900A1 (ja) | 2011-01-20 |
Family
ID=43449509
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/062480 WO2011007900A1 (ja) | 2009-07-15 | 2010-07-15 | 生体組織から単離できる多能性幹細胞 |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US9550975B2 (ja) |
| EP (3) | EP2455452B2 (ja) |
| JP (7) | JP5185443B2 (ja) |
| KR (4) | KR20120069663A (ja) |
| CN (2) | CN107012117B (ja) |
| AU (1) | AU2010271722B8 (ja) |
| CA (1) | CA2768238C (ja) |
| DK (2) | DK3214170T3 (ja) |
| ES (2) | ES2647360T5 (ja) |
| HU (2) | HUE052559T2 (ja) |
| NO (1) | NO2455452T3 (ja) |
| PL (2) | PL3214170T3 (ja) |
| PT (2) | PT3214170T (ja) |
| SG (1) | SG177705A1 (ja) |
| WO (1) | WO2011007900A1 (ja) |
Cited By (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012133942A1 (ja) * | 2011-03-30 | 2012-10-04 | 株式会社Clio | 生体の臍帯又は脂肪組織から単離できる多能性幹細胞 |
| WO2012133948A1 (ja) * | 2011-03-30 | 2012-10-04 | 株式会社Clio | 生体組織から単離できるssea-3陽性の多能性幹細胞を含む他家移植用細胞治療用組成物 |
| WO2013015457A1 (en) | 2011-07-27 | 2013-01-31 | Kyoto University | Novel markers for dopaminergic neuron progenitor cells |
| WO2013058403A1 (ja) | 2011-10-21 | 2013-04-25 | 国立大学法人京都大学 | 層流による多能性維持単一分散細胞培養法 |
| WO2014027474A1 (ja) * | 2012-08-17 | 2014-02-20 | 株式会社Clio | 心筋梗塞の修復再生を誘導する多能性幹細胞 |
| WO2014123242A1 (ja) | 2013-02-08 | 2014-08-14 | 国立大学法人京都大学 | 巨核球及び血小板の製造方法 |
| JP2014519825A (ja) * | 2011-06-06 | 2014-08-21 | レゲネシス ベーフェーベーアー | 中空繊維バイオリアクター中での幹細胞の増幅 |
| WO2014133090A1 (ja) * | 2013-02-28 | 2014-09-04 | 国立大学法人浜松医科大学 | 脳腫瘍治療のための多能性幹細胞 |
| WO2014133170A1 (ja) | 2013-03-01 | 2014-09-04 | 株式会社Clio | 多能性幹細胞を損傷部位に誘導する遊走因子を含む医薬組成物 |
| WO2014148646A1 (ja) | 2013-03-21 | 2014-09-25 | 国立大学法人京都大学 | 神経分化誘導用の多能性幹細胞 |
| WO2014157257A1 (ja) | 2013-03-25 | 2014-10-02 | 公益財団法人先端医療振興財団 | 細胞の選別方法 |
| WO2014168264A1 (ja) | 2013-04-12 | 2014-10-16 | 国立大学法人京都大学 | 肺胞上皮前駆細胞の誘導方法 |
| JP2014528710A (ja) * | 2011-08-24 | 2014-10-30 | 株式会社アモーレパシフィックAmorepacific Corporation | ヒトの皮膚真皮由来成体幹細胞 |
| WO2014185358A1 (ja) | 2013-05-14 | 2014-11-20 | 国立大学法人京都大学 | 効率的な心筋細胞の誘導方法 |
| WO2015020113A1 (ja) | 2013-08-07 | 2015-02-12 | 国立大学法人京都大学 | 膵ホルモン産生細胞の製造法 |
| WO2015030149A1 (ja) * | 2013-08-29 | 2015-03-05 | 国立大学法人鳥取大学 | 細胞のアンチエイジングに関連する生体分子群 |
| WO2015034012A1 (ja) | 2013-09-05 | 2015-03-12 | 国立大学法人京都大学 | 新規ドーパミン産生神経前駆細胞の誘導方法 |
| WO2015033558A1 (ja) * | 2013-09-04 | 2015-03-12 | 株式会社大塚製薬工場 | 多能性幹細胞の調製方法 |
| WO2015064754A1 (ja) | 2013-11-01 | 2015-05-07 | 国立大学法人京都大学 | 新規軟骨細胞誘導方法 |
| JP2015516812A (ja) * | 2012-04-24 | 2015-06-18 | ザ ブリガム アンド ウィメンズ ホスピタル インコーポレイテッドThe Brigham and Women’s Hospital, Inc. | 多能性細胞のデノボ生成 |
| JP2015159895A (ja) * | 2014-02-26 | 2015-09-07 | 株式会社Clio | 脳梗塞治療のための多能性幹細胞 |
| WO2016035419A1 (ja) * | 2014-09-05 | 2016-03-10 | 国立大学法人 東京大学 | 糖尿病性皮膚潰瘍治療のための多能性幹細胞 |
| JP2017052731A (ja) * | 2015-09-11 | 2017-03-16 | 二村 芳弘 | 皮膚幹細胞の活性化作用を呈するフラボノイド誘導体 |
| JP2017518772A (ja) * | 2014-06-13 | 2017-07-13 | ビービーエイチシー・カンパニー・リミテッドBbhc Co., Ltd. | 脂肪由来間葉系幹細胞から誘導多能性幹細胞を製造する方法およびその方法により製造された誘導多能性幹細胞 |
| JP2017151060A (ja) * | 2016-02-26 | 2017-08-31 | 国立大学法人岐阜大学 | 急性心筋梗塞の予後マーカー及びその利用 |
| WO2017183736A1 (ja) | 2016-04-22 | 2017-10-26 | 国立大学法人京都大学 | ドーパミン産生神経前駆細胞の製造方法 |
| WO2017199976A1 (ja) * | 2016-05-16 | 2017-11-23 | 国立大学法人名古屋大学 | 多能性幹細胞による周産期脳障害の改善及び治療 |
| WO2018021515A1 (ja) | 2016-07-29 | 2018-02-01 | 国立大学法人東北大学 | 血管障害の予防又は治療剤 |
| WO2018025975A1 (ja) * | 2016-08-03 | 2018-02-08 | 株式会社生命科学インスティテュート | インビトロで多能性幹細胞を分化誘導する方法 |
| US20180161378A1 (en) * | 2015-05-29 | 2018-06-14 | Nippon Zoki Pharmaceutical Co., Ltd. | Agent for promoting migration of pluripotent stem cells |
| JP2018111722A (ja) * | 2018-04-09 | 2018-07-19 | 株式会社生命科学インスティテュート | 脳梗塞治療のための多能性幹細胞 |
| WO2018235834A1 (ja) | 2017-06-19 | 2018-12-27 | 国立大学法人北海道大学 | 表皮水疱症の治療剤 |
| WO2018235878A1 (ja) | 2017-06-20 | 2018-12-27 | 国立大学法人名古屋大学 | 多能性幹細胞による胎児発育不全に伴う脳障害の改善及び治療 |
| KR20190039102A (ko) | 2016-08-03 | 2019-04-10 | 고쿠리츠 다이가쿠 호우징 나고야 다이가쿠 | 다능성 간세포에 따른 만성폐질환의 개선 및 치료 |
| KR20190039700A (ko) | 2016-08-03 | 2019-04-15 | 가부시키가이샤 세이메이카가쿠 인스티튜트 | 다능성 간세포에 의한 허혈재관류 폐장애의 경감 및 치료 |
| WO2019078262A1 (ja) | 2017-10-17 | 2019-04-25 | 国立大学法人広島大学 | 骨軟骨修復を誘導する多能性幹細胞 |
| WO2019092939A1 (ja) * | 2017-11-10 | 2019-05-16 | 株式会社リジェネシスサイエンス | 培養細胞の製造方法,脊髄損傷疾患の治療剤の製造方法 |
| US10293003B2 (en) | 2014-02-26 | 2019-05-21 | Clio, Inc. | Multilineage-differentiating stress enduring (MUSE) cells for treatment of chronic kidney disease |
| US10472610B2 (en) | 2014-05-21 | 2019-11-12 | Kyoto University | Method for generating pancreatic bud cells and therapeutic agent for pancreatic disease containing pancreatic bud cells |
| WO2019216380A1 (ja) | 2018-05-09 | 2019-11-14 | 株式会社生命科学インスティテュート | 脊髄損傷の治療剤 |
| CN110494559A (zh) * | 2017-03-28 | 2019-11-22 | 味之素株式会社 | 不分化地维持用培养基添加剂 |
| WO2020017575A1 (ja) | 2018-07-19 | 2020-01-23 | 国立大学法人京都大学 | 多能性幹細胞由来の板状軟骨およびその製造方法 |
| JPWO2020111249A1 (ja) * | 2018-11-30 | 2020-06-04 | ||
| WO2020130147A1 (ja) | 2018-12-21 | 2020-06-25 | 国立大学法人京都大学 | ルブリシン局在軟骨様組織、その製造方法及びそれを含む関節軟骨損傷治療用組成物 |
| AU2017207154B2 (en) * | 2016-01-15 | 2020-09-17 | Mari Dezawa | Mobilization of pluripotent stem cells for ischemic cerebral infarction |
| WO2021029346A1 (ja) | 2019-08-09 | 2021-02-18 | 国立大学法人東北大学 | 脳血管性認知症の治療または予防剤 |
| WO2021045190A1 (ja) | 2019-09-05 | 2021-03-11 | 国立大学法人東北大学 | 心筋炎の治療剤 |
| WO2021066076A1 (ja) | 2019-10-01 | 2021-04-08 | 国立大学法人京都大学 | 尿管芽先端部細胞の単離方法 |
| WO2021085639A1 (ja) | 2019-10-31 | 2021-05-06 | 株式会社生命科学インスティテュート | 多能性幹細胞による間質性膀胱炎の治療 |
| WO2021117886A1 (ja) | 2019-12-12 | 2021-06-17 | 国立大学法人千葉大学 | 巨核球および血小板を含む凍結乾燥製剤 |
| WO2021187601A1 (ja) | 2020-03-19 | 2021-09-23 | 国立大学法人京都大学 | 心筋細胞の精製方法 |
| WO2021187602A1 (ja) | 2020-03-19 | 2021-09-23 | 国立大学法人京都大学 | 心筋細胞の精製方法 |
| WO2021201286A1 (ja) | 2020-04-02 | 2021-10-07 | 国立大学法人東北大学 | ハイポテンシャル多能性幹細胞 |
| WO2022014681A1 (ja) | 2020-07-15 | 2022-01-20 | 国立大学法人 岡山大学 | 運動ニューロン疾患(mnd)の治療に有効な多能性幹細胞 |
| WO2022014604A1 (ja) | 2020-07-13 | 2022-01-20 | 国立大学法人京都大学 | 骨格筋前駆細胞及びその精製方法、筋原性疾患を治療するための組成物、並びに骨格筋前駆細胞を含む細胞群の製造方法 |
| WO2023153464A1 (ja) | 2022-02-09 | 2023-08-17 | 住友ファーマ株式会社 | 多能性幹細胞から中脳底板領域の神経系細胞への分化における、培養液中の細胞の分化能を判定する方法 |
Families Citing this family (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9550975B2 (en) * | 2009-07-15 | 2017-01-24 | Mari Dezawa | SSEA-3 pluripotent stem cell isolated from body tissue |
| US9399758B2 (en) | 2009-07-15 | 2016-07-26 | Mari Dezawa | SSEA3(+) pluripotent stem cell that can be isolated from body tissue |
| EP2467469A4 (en) | 2009-08-21 | 2013-07-24 | Univ Leland Stanford Junior | INCREASED EFFICIENCY OF INDUCED GENERATION OF PLURIPOTENTAL STEM CELLS FROM HUMAN BODIES |
| US8883210B1 (en) | 2010-05-14 | 2014-11-11 | Musculoskeletal Transplant Foundation | Tissue-derived tissuegenic implants, and methods of fabricating and using same |
| US10130736B1 (en) | 2010-05-14 | 2018-11-20 | Musculoskeletal Transplant Foundation | Tissue-derived tissuegenic implants, and methods of fabricating and using same |
| US9352003B1 (en) | 2010-05-14 | 2016-05-31 | Musculoskeletal Transplant Foundation | Tissue-derived tissuegenic implants, and methods of fabricating and using same |
| US20150056170A1 (en) * | 2012-03-06 | 2015-02-26 | Sct&B Inc. | Placental stem cells, methods for isolating same and use thereof |
| CA2880200A1 (en) * | 2012-07-27 | 2014-01-30 | Bioquark, Inc. | Extracts isolated from electroporated ambhibian oocytes and use thereof in treating diseases and disorders |
| WO2014100806A1 (en) * | 2012-12-21 | 2014-06-26 | Rutgers, The State University Of New Jersey | Muse cells isolation and expansion |
| US9709554B2 (en) | 2013-01-31 | 2017-07-18 | Rutgers, The State University Of New Jersey | In vitro model of macrosteatotic (fatty) liver |
| EP2970892A1 (en) * | 2013-03-15 | 2016-01-20 | The Jackson Laboratory | Isolation of non-embryonic stem cells and uses thereof |
| EP2999780B1 (en) | 2013-05-22 | 2019-10-09 | The Regents of the University of California | Pluripotent human adipose adult stem cells: isolation, characterization and clinical implications |
| MX2016001247A (es) | 2013-07-30 | 2016-08-17 | Musculoskeletal Transplant Foundation | Matrices derivadas de tejido suave acelular y metodos para preparar las mismas. |
| WO2015081094A1 (en) * | 2013-11-27 | 2015-06-04 | University Of Louisville Research Foundation, Inc. | Cardiac progenitor cells and methods of use therefor |
| RU2711942C2 (ru) | 2014-03-19 | 2020-01-23 | Виселл Терапеутикс, Инк. | Способы, относящиеся к плюрипотентным клеткам |
| JP6391364B2 (ja) * | 2014-08-25 | 2018-09-19 | 国立大学法人 鹿児島大学 | 認知機能障害改善用細胞製剤 |
| KR101700598B1 (ko) * | 2014-11-11 | 2017-01-31 | 한국원자력의학원 | Cd105를 포함하는 줄기세포능을 가진 섬유아세포 분리용 바이오마커 조성물 및 이를 이용한 줄기세포능을 가진 섬유아세포 분리 방법 |
| EP3297694A1 (en) | 2015-05-21 | 2018-03-28 | Musculoskeletal Transplant Foundation | Modified demineralized cortical bone fibers |
| US10912864B2 (en) | 2015-07-24 | 2021-02-09 | Musculoskeletal Transplant Foundation | Acellular soft tissue-derived matrices and methods for preparing same |
| US11052175B2 (en) | 2015-08-19 | 2021-07-06 | Musculoskeletal Transplant Foundation | Cartilage-derived implants and methods of making and using same |
| EP4324915A3 (en) * | 2016-03-17 | 2024-06-19 | Synova Life Sciences, Inc. | Separation, dissociation and/or disaggregation of cells using shockwaves or mechanical impacts |
| SG11201811806TA (en) | 2016-07-01 | 2019-02-27 | Univ Tohoku | Prophylactic or therapeutic agent for organ fibrosis |
| WO2018143378A1 (ja) | 2017-02-03 | 2018-08-09 | 国立大学法人九州大学 | 歯髄組織由来細胞から歯髄幹細胞を調製する方法 |
| WO2019107917A1 (ko) | 2017-11-28 | 2019-06-06 | 주식회사 셀투인 | 실시간 글루타치온 측정을 통한 치료용 세포의 품질 측정 방법 |
| JP7072777B2 (ja) * | 2018-07-03 | 2022-05-23 | 株式会社生命科学インスティテュート | 慢性腎障害治療のための多能性幹細胞 |
| EP3882340A4 (en) * | 2018-11-14 | 2022-08-17 | Kataoka Corporation | METHOD FOR PRODUCING INSULIN-PRODUCING CELLS |
| JP2021073305A (ja) * | 2021-02-09 | 2021-05-13 | 株式会社生命科学インスティテュート | 慢性腎障害治療のための多能性幹細胞 |
| TWI832070B (zh) * | 2021-07-15 | 2024-02-11 | 國璽幹細胞應用技術股份有限公司 | 幹細胞製劑用於治療關節炎的用途及用於製備幹細胞製劑的方法 |
| CN117187174B (zh) * | 2023-11-08 | 2024-02-06 | 广州正源生物技术有限公司 | 一种Muse细胞培养基以及一种脂肪Muse细胞的提取方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4182742B2 (ja) | 2002-12-12 | 2008-11-19 | 日産自動車株式会社 | 自動車の前部車体構造 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL147990A0 (en) | 1999-08-05 | 2002-09-12 | Mcl Llc | Multipotent adult stem cells and methods for isolation |
| US7575921B2 (en) | 1999-12-30 | 2009-08-18 | Vbi Technologies, L.L.C. | Spore-like cells and uses thereof |
| US20020151050A1 (en) | 2000-10-30 | 2002-10-17 | Vacanti Charles A. | Isolation of spore-like cells from tissues exposed to extreme conditions |
| US20050042595A1 (en) | 2003-08-14 | 2005-02-24 | Martin Haas | Banking of multipotent amniotic fetal stem cells |
| EP1660644A1 (en) * | 2003-08-29 | 2006-05-31 | Regents Of The University Of Minnesota | Kidney derived stem cells and methods for their isolation, differentiation and use |
| US20060216821A1 (en) | 2004-02-26 | 2006-09-28 | Reliance Life Sciences Pvt. Ltd. | Pluripotent embryonic-like stem cells derived from corneal limbus, methods of isolation and uses thereof |
| JP2009517001A (ja) * | 2005-05-17 | 2009-04-30 | リライアンス ライフ サイエンシーズ プライベイト リミテッド | 哺乳動物細胞を使用するヒト胚性幹細胞株の構築 |
| US9388382B2 (en) | 2005-10-05 | 2016-07-12 | The Board Of Trustees Of The University Of Illinois | Isolation of CD14 negative, CD45 positive and CD117 positive embryonic-like stem cells free of monocytes from human umbilical cord blood mononuclear cells |
| WO2007047581A2 (en) | 2005-10-17 | 2007-04-26 | Academia Sinica | Pulmonary stem cells, related methods and kits |
| EP3418297B1 (en) | 2005-12-13 | 2023-04-05 | Kyoto University | Nuclear reprogramming factor |
| US20090155225A1 (en) | 2006-11-02 | 2009-06-18 | Mariusz Ratajczak | Uses and isolation of very small of embryonic-like (vsel) stem cells |
| WO2008080200A1 (en) | 2006-12-29 | 2008-07-10 | Irina Kerkis | Process for obtaining stem cells |
| JP2008307007A (ja) | 2007-06-15 | 2008-12-25 | Bayer Schering Pharma Ag | 出生後のヒト組織由来未分化幹細胞から誘導したヒト多能性幹細胞 |
| US20090172832A1 (en) * | 2007-12-28 | 2009-07-02 | Timothy Caspar | Expression of rubisco enzyme from a non-rubisco locus |
| ITRM20080342A1 (it) * | 2008-06-26 | 2009-12-27 | Univ Degli Studi Udine | Cellule di polpa dentale midollo-simili, metodi per isolamento ed uso. |
| US9550975B2 (en) * | 2009-07-15 | 2017-01-24 | Mari Dezawa | SSEA-3 pluripotent stem cell isolated from body tissue |
-
2010
- 2010-07-14 US US12/836,264 patent/US9550975B2/en active Active
- 2010-07-15 EP EP10799956.7A patent/EP2455452B2/en active Active
- 2010-07-15 SG SG2012003836A patent/SG177705A1/en unknown
- 2010-07-15 PT PT171633233T patent/PT3214170T/pt unknown
- 2010-07-15 CN CN201611060144.1A patent/CN107012117B/zh active Active
- 2010-07-15 CN CN201080041908.7A patent/CN102858951B/zh active Active
- 2010-07-15 PL PL17163323T patent/PL3214170T3/pl unknown
- 2010-07-15 DK DK17163323.3T patent/DK3214170T3/da active
- 2010-07-15 EP EP20160074.9A patent/EP3680323A1/en active Pending
- 2010-07-15 KR KR1020127003879A patent/KR20120069663A/ko active Search and Examination
- 2010-07-15 NO NO10799956A patent/NO2455452T3/no unknown
- 2010-07-15 KR KR1020157013171A patent/KR101651795B1/ko active IP Right Grant
- 2010-07-15 CA CA2768238A patent/CA2768238C/en active Active
- 2010-07-15 EP EP17163323.3A patent/EP3214170B1/en active Active
- 2010-07-15 PT PT107999567T patent/PT2455452T/pt unknown
- 2010-07-15 AU AU2010271722A patent/AU2010271722B8/en active Active
- 2010-07-15 HU HUE17163323A patent/HUE052559T2/hu unknown
- 2010-07-15 KR KR1020167008209A patent/KR101764080B1/ko active IP Right Grant
- 2010-07-15 HU HUE10799956A patent/HUE037448T2/hu unknown
- 2010-07-15 WO PCT/JP2010/062480 patent/WO2011007900A1/ja active Application Filing
- 2010-07-15 ES ES10799956T patent/ES2647360T5/es active Active
- 2010-07-15 PL PL10799956T patent/PL2455452T5/pl unknown
- 2010-07-15 KR KR1020177020765A patent/KR101945343B1/ko active IP Right Grant
- 2010-07-15 ES ES17163323T patent/ES2831030T3/es active Active
- 2010-07-15 JP JP2011522886A patent/JP5185443B2/ja active Active
- 2010-07-15 DK DK10799956.7T patent/DK2455452T4/da active
-
2012
- 2012-05-01 JP JP2012104994A patent/JP5852503B2/ja active Active
-
2015
- 2015-12-04 JP JP2015237186A patent/JP6092357B2/ja active Active
-
2017
- 2017-02-08 JP JP2017021036A patent/JP6416298B2/ja active Active
-
2018
- 2018-10-03 JP JP2018188234A patent/JP6641441B2/ja active Active
-
2019
- 2019-12-27 JP JP2019239248A patent/JP6937821B2/ja active Active
-
2021
- 2021-08-31 JP JP2021140656A patent/JP7134317B2/ja active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4182742B2 (ja) | 2002-12-12 | 2008-11-19 | 日産自動車株式会社 | 自動車の前部車体構造 |
Non-Patent Citations (27)
| Title |
|---|
| BUI HT ET AL., DEVELOPMENT, vol. 135, no. 23, 2008, pages 3935 - 3945 |
| BUI HT ET AL., DEVELOPMENT., vol. 135, no. 23, 2008, pages 3935 - 3945 |
| CHOU ET AL., CELL, vol. 135, 2008, pages 449 - 461 |
| CHOU YF ET AL., CELL, vol. 135, 2008, pages 449 - 461 |
| DEZAWA, M. ET AL., J CLIN INVEST, vol. 113, 2004, pages 1701 - 1710 |
| DEZAWA, M. ET AL., SCIENCE, vol. 309, 2005, pages 314 - 317 |
| FAROOQUE M ET AL., ACTA NEUROPATHOL., vol. 100, 2000, pages 13 - 22 |
| GANG, E J ET AL.: "SSEA-4 identifies mesenchymal stem cells from bone marrow.", BLOOD., vol. 109, no. 4, 15 February 2007 (2007-02-15), pages 1743 - 1751, XP008152783 * |
| HAYASE M ET AL., J CEREB BLOOD FLOW METAB., vol. 29, no. 8, 2009, pages 1409 - 20 |
| HUANG, Y C ET AL.: "Isolation of mesenchymal stem cells from human placental decidua basalis and resistance to hypoxia and serum deprivation.", STEM CELL REV., vol. 5, no. 3, September 2009 (2009-09-01), pages 247 - 255, XP002635213 * |
| KELLER, J. PHYSIOL. (LOND, vol. 168, 1998, pages 131 - 139 |
| KURODA, Y ET AL.: "Unique multipotent cells in adult human mesenchymal cell populations.", PROC NATL ACAD SCI U S A., vol. 107, no. 19, 11 May 2010 (2010-05-11), pages 8639 - 8643, XP008152782 * |
| LIVAK KJ ET AL., METHODS, vol. 25, 2001, pages 402 - 408 |
| M. DEZAWA ET AL., SCIENCE, vol. 309, 2005, pages 314 - 317 |
| M. DEZAWA ET AL., THE JOURNAL OF CLINICAL INVESTIGATION, vol. 113, no. 12, 2004, pages 1701 - 1710 |
| MAUNEY, J R ET AL.: "Matrix-mediated retention of in vitro osteogenic differentiation potential and in vivo bone-forming capacity by human adult bone marrow-derived mesenchymal stem cells during ex vivo expansion.", J BIOMED MATER RES A., vol. 79, no. 3, 1 December 2006 (2006-12-01), pages 464 - 475, XP008152786 * |
| MORISCOT, C ET AL.: "Human bone marrow mesenchymal stem cells can express insulin and key transcription factors of the endocrine pancreas developmental pathway upon genetic and/or microenvironmental manipulation in vitro.", STEM CELLS., vol. 23, no. 4, April 2005 (2005-04-01), pages 594 - 603, XP008152792 * |
| NAKAHATA, T. ET AL., BLOOD, vol. 60, 1982, pages 352 - 361 |
| OKITA K. ET AL., SCIENCE, vol. 322, no. 5903, 7 November 2008 (2008-11-07), pages 949 - 953 |
| PITTENGER, M. F. ET AL., SCIENCE, vol. 284, 1999, pages 143 - 147 |
| SAEED AI ET AL., BIOTECHNIQUES, vol. 34, no. 2, 2003, pages 374 - 378 |
| SART, S ET AL.: "Ear mesenchymal stem cells: an efficient adult multipotent cell population fit for rapid and scalable expansion.", J BIOTECHNOL., vol. 139, no. 4, 23 February 2009 (2009-02-23), pages 291 - 299, XP025987454 * |
| See also references of EP2455452A1 |
| SHIMAZAKI T ET AL., EMBO J, vol. 12, 1993, pages 4489 - 4498 |
| TAKAHASHI ET AL., CELL, vol. 131, 2007 |
| TAKAHASHI ET AL., CELL, vol. 131, 2007, pages 861 - 872 |
| YOON, Y S ET AL.: "Clonally expanded novel multipotent stem cells from human bone marrow regenerate myocardium after myocardial infarction.", J CLIN INVEST., vol. 115, no. 2, February 2005 (2005-02-01), pages 326 - 338, XP002458476 * |
Cited By (112)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012133948A1 (ja) * | 2011-03-30 | 2012-10-04 | 株式会社Clio | 生体組織から単離できるssea-3陽性の多能性幹細胞を含む他家移植用細胞治療用組成物 |
| CN103442724A (zh) * | 2011-03-30 | 2013-12-11 | 株式会社克里奥 | 含有可从机体组织中分离的ssea-3阳性的多能干细胞的同种异体移植用细胞治疗用组合物 |
| WO2012133942A1 (ja) * | 2011-03-30 | 2012-10-04 | 株式会社Clio | 生体の臍帯又は脂肪組織から単離できる多能性幹細胞 |
| JP2017121257A (ja) * | 2011-06-06 | 2017-07-13 | レゲネシス ベーフェーベーアー | 中空繊維バイオリアクター中での幹細胞の増幅 |
| JP2014519825A (ja) * | 2011-06-06 | 2014-08-21 | レゲネシス ベーフェーベーアー | 中空繊維バイオリアクター中での幹細胞の増幅 |
| WO2013015457A1 (en) | 2011-07-27 | 2013-01-31 | Kyoto University | Novel markers for dopaminergic neuron progenitor cells |
| JP2014528710A (ja) * | 2011-08-24 | 2014-10-30 | 株式会社アモーレパシフィックAmorepacific Corporation | ヒトの皮膚真皮由来成体幹細胞 |
| US10865386B2 (en) | 2011-08-24 | 2020-12-15 | Amorepacific Corporation | Adult stem cells derived from human skin dermis |
| WO2013058403A1 (ja) | 2011-10-21 | 2013-04-25 | 国立大学法人京都大学 | 層流による多能性維持単一分散細胞培養法 |
| US11963977B2 (en) | 2012-04-24 | 2024-04-23 | Vcell Therapeutics Inc. | Generating pluripotent cells de novo |
| JP2015516812A (ja) * | 2012-04-24 | 2015-06-18 | ザ ブリガム アンド ウィメンズ ホスピタル インコーポレイテッドThe Brigham and Women’s Hospital, Inc. | 多能性細胞のデノボ生成 |
| US10376544B2 (en) | 2012-08-17 | 2019-08-13 | Clio, Inc. | Pluripotent stem cell that induces repair and regeneration after myocardial infarction |
| JPWO2014027684A1 (ja) * | 2012-08-17 | 2016-07-28 | 株式会社Clio | 心筋梗塞の修復再生を誘導する多能性幹細胞 |
| WO2014027474A1 (ja) * | 2012-08-17 | 2014-02-20 | 株式会社Clio | 心筋梗塞の修復再生を誘導する多能性幹細胞 |
| US9844570B2 (en) | 2012-08-17 | 2017-12-19 | Clio, Inc. | Pluripotent stem cell that induces repair and regeneration after myocardial infarction |
| WO2014027684A1 (ja) | 2012-08-17 | 2014-02-20 | 株式会社Clio | 心筋梗塞の修復再生を誘導する多能性幹細胞 |
| KR101730052B1 (ko) * | 2012-08-17 | 2017-04-25 | 가부시키가이샤 클리오 | 심근경색의 수복 재생을 유도하는 다능성 간세포 |
| US10639335B2 (en) | 2012-08-17 | 2020-05-05 | Clio, Inc. | Pluripotent stem cell that induces repair and regeneration after myocardial infarction |
| AU2013303492B2 (en) * | 2012-08-17 | 2017-02-16 | Clio, Inc. | Pluripotent stem cell that induces repair and regeneration after myocardial infarction |
| EP3659612A1 (en) | 2012-08-17 | 2020-06-03 | Clio, Inc. | Pluripotent stem cell that induces repair and regeneration after myocardial infarction technical field |
| WO2014123242A1 (ja) | 2013-02-08 | 2014-08-14 | 国立大学法人京都大学 | 巨核球及び血小板の製造方法 |
| WO2014133090A1 (ja) * | 2013-02-28 | 2014-09-04 | 国立大学法人浜松医科大学 | 脳腫瘍治療のための多能性幹細胞 |
| JPWO2014133090A1 (ja) * | 2013-02-28 | 2017-02-02 | 国立大学法人浜松医科大学 | 脳腫瘍治療のための多能性幹細胞 |
| US10034889B2 (en) | 2013-03-01 | 2018-07-31 | Clio, Inc. | Pharmaceutical composition including migratory factor for guiding pluripotent stem cells to damage |
| JP2018111734A (ja) * | 2013-03-01 | 2018-07-19 | 株式会社生命科学インスティテュート | 多能性幹細胞を損傷部位に誘導する遊走因子を含む医薬組成物 |
| WO2014133170A1 (ja) | 2013-03-01 | 2014-09-04 | 株式会社Clio | 多能性幹細胞を損傷部位に誘導する遊走因子を含む医薬組成物 |
| US9446033B2 (en) | 2013-03-01 | 2016-09-20 | Clio, Inc. | Pharmaceutical composition including migratory factor for guiding pluripotent stem cells to injury |
| JPWO2014133170A1 (ja) * | 2013-03-01 | 2017-02-09 | 株式会社Clio | 多能性幹細胞を損傷部位に誘導する遊走因子を含む医薬組成物 |
| US10369162B2 (en) | 2013-03-01 | 2019-08-06 | Clio, Inc. | Pharmaceutical composition including migratory factor for guiding pluripotent stem cells to damage |
| WO2014148646A1 (ja) | 2013-03-21 | 2014-09-25 | 国立大学法人京都大学 | 神経分化誘導用の多能性幹細胞 |
| WO2014157257A1 (ja) | 2013-03-25 | 2014-10-02 | 公益財団法人先端医療振興財団 | 細胞の選別方法 |
| WO2014168264A1 (ja) | 2013-04-12 | 2014-10-16 | 国立大学法人京都大学 | 肺胞上皮前駆細胞の誘導方法 |
| WO2014185358A1 (ja) | 2013-05-14 | 2014-11-20 | 国立大学法人京都大学 | 効率的な心筋細胞の誘導方法 |
| WO2015020113A1 (ja) | 2013-08-07 | 2015-02-12 | 国立大学法人京都大学 | 膵ホルモン産生細胞の製造法 |
| US9796962B2 (en) | 2013-08-07 | 2017-10-24 | Kyoto University | Method for generating pancreatic hormone-producing cells |
| US10265347B2 (en) | 2013-08-29 | 2019-04-23 | Norimasa Miura | Biomolecular group related to cell anti-aging |
| JPWO2015030149A1 (ja) * | 2013-08-29 | 2017-03-02 | 国立大学法人鳥取大学 | 細胞のアンチエイジングに関連する生体分子群 |
| WO2015030149A1 (ja) * | 2013-08-29 | 2015-03-05 | 国立大学法人鳥取大学 | 細胞のアンチエイジングに関連する生体分子群 |
| JPWO2015033558A1 (ja) * | 2013-09-04 | 2017-03-02 | 株式会社大塚製薬工場 | 多能性幹細胞の調製方法 |
| US9765296B2 (en) | 2013-09-04 | 2017-09-19 | Otsuka Pharmaceutical Factory, Inc. | Method for preparing pluripotent stem cells |
| US10370639B2 (en) | 2013-09-04 | 2019-08-06 | Otsuka Pharmaceutical Factory, Inc. | Method for preparing pluripotent stem cells |
| US11155782B2 (en) | 2013-09-04 | 2021-10-26 | Otsuka Pharmaceutical Factory, Inc. | Method for preparing pluripotent stem cells |
| WO2015033558A1 (ja) * | 2013-09-04 | 2015-03-12 | 株式会社大塚製薬工場 | 多能性幹細胞の調製方法 |
| JP2018023401A (ja) * | 2013-09-04 | 2018-02-15 | 株式会社大塚製薬工場 | 多能性幹細胞の調製方法 |
| WO2015034012A1 (ja) | 2013-09-05 | 2015-03-12 | 国立大学法人京都大学 | 新規ドーパミン産生神経前駆細胞の誘導方法 |
| WO2015064754A1 (ja) | 2013-11-01 | 2015-05-07 | 国立大学法人京都大学 | 新規軟骨細胞誘導方法 |
| US10293003B2 (en) | 2014-02-26 | 2019-05-21 | Clio, Inc. | Multilineage-differentiating stress enduring (MUSE) cells for treatment of chronic kidney disease |
| JP2015159895A (ja) * | 2014-02-26 | 2015-09-07 | 株式会社Clio | 脳梗塞治療のための多能性幹細胞 |
| US10993966B2 (en) | 2014-02-26 | 2021-05-04 | Life Science Institute, Inc. | Pluripotent stem cell for treatment of cerebral infarction |
| US10472610B2 (en) | 2014-05-21 | 2019-11-12 | Kyoto University | Method for generating pancreatic bud cells and therapeutic agent for pancreatic disease containing pancreatic bud cells |
| JP2017518772A (ja) * | 2014-06-13 | 2017-07-13 | ビービーエイチシー・カンパニー・リミテッドBbhc Co., Ltd. | 脂肪由来間葉系幹細胞から誘導多能性幹細胞を製造する方法およびその方法により製造された誘導多能性幹細胞 |
| AU2015310286B2 (en) * | 2014-09-05 | 2018-11-08 | The University Of Tokyo | Pluripotent stem cell for treating diabetic skin ulcer |
| US11000552B2 (en) | 2014-09-05 | 2021-05-11 | The University Of Tokyo | Pluripotent stem cell for treating diabetic skin ulcer |
| KR20170055477A (ko) | 2014-09-05 | 2017-05-19 | 고쿠리츠다이가쿠호우진 도쿄다이가쿠 | 당뇨병성 피부 궤양 치료를 위한 다능성 줄기 세포 |
| WO2016035419A1 (ja) * | 2014-09-05 | 2016-03-10 | 国立大学法人 東京大学 | 糖尿病性皮膚潰瘍治療のための多能性幹細胞 |
| JP2016056109A (ja) * | 2014-09-05 | 2016-04-21 | 国立大学法人 東京大学 | 糖尿病性皮膚潰瘍治療のための多能性幹細胞 |
| US20180161378A1 (en) * | 2015-05-29 | 2018-06-14 | Nippon Zoki Pharmaceutical Co., Ltd. | Agent for promoting migration of pluripotent stem cells |
| JP2017052731A (ja) * | 2015-09-11 | 2017-03-16 | 二村 芳弘 | 皮膚幹細胞の活性化作用を呈するフラボノイド誘導体 |
| AU2017207154B2 (en) * | 2016-01-15 | 2020-09-17 | Mari Dezawa | Mobilization of pluripotent stem cells for ischemic cerebral infarction |
| JP2017151060A (ja) * | 2016-02-26 | 2017-08-31 | 国立大学法人岐阜大学 | 急性心筋梗塞の予後マーカー及びその利用 |
| WO2017183736A1 (ja) | 2016-04-22 | 2017-10-26 | 国立大学法人京都大学 | ドーパミン産生神経前駆細胞の製造方法 |
| KR20200127039A (ko) | 2016-05-16 | 2020-11-09 | 고쿠리츠 다이가쿠 호우징 도우카이 고쿠리츠 다이가쿠 기코우 | 다능성 간세포에 의한 주산기 뇌장애의 개선 및 치료 |
| WO2017199976A1 (ja) * | 2016-05-16 | 2017-11-23 | 国立大学法人名古屋大学 | 多能性幹細胞による周産期脳障害の改善及び治療 |
| AU2017267809B2 (en) * | 2016-05-16 | 2020-01-30 | Life Science Institute, Inc. | Amelioration and treatment of perinatal brain damage with pluripotent stem cells |
| WO2018021515A1 (ja) | 2016-07-29 | 2018-02-01 | 国立大学法人東北大学 | 血管障害の予防又は治療剤 |
| US11419899B2 (en) * | 2016-07-29 | 2022-08-23 | Tohoku University | Method of treating aortic aneurysm using muse cells |
| KR20190039700A (ko) | 2016-08-03 | 2019-04-15 | 가부시키가이샤 세이메이카가쿠 인스티튜트 | 다능성 간세포에 의한 허혈재관류 폐장애의 경감 및 치료 |
| JP7148402B2 (ja) | 2016-08-03 | 2022-10-05 | 株式会社生命科学インスティテュート | インビトロで多能性幹細胞を分化誘導する方法 |
| US12083149B2 (en) * | 2016-08-03 | 2024-09-10 | Tohoku University | Amelioration and treatment of chronic lung disease using pluripotent stem cells |
| KR20190039102A (ko) | 2016-08-03 | 2019-04-10 | 고쿠리츠 다이가쿠 호우징 나고야 다이가쿠 | 다능성 간세포에 따른 만성폐질환의 개선 및 치료 |
| KR20190034552A (ko) | 2016-08-03 | 2019-04-02 | 가부시키가이샤 세이메이카가쿠 인스티튜트 | 인비트로로 다능성 간세포를 분화유도하는 방법 |
| US11920180B2 (en) | 2016-08-03 | 2024-03-05 | Tohoku University | Method for inducing differentiation of pluripotent stem cells in vitro |
| US20190240262A1 (en) * | 2016-08-03 | 2019-08-08 | National University Corporation Nagoya University | Amelioration and treatment of chronic lung disease using pluripotent stem cells |
| JPWO2018025975A1 (ja) * | 2016-08-03 | 2019-06-06 | 株式会社生命科学インスティテュート | インビトロで多能性幹細胞を分化誘導する方法 |
| WO2018025975A1 (ja) * | 2016-08-03 | 2018-02-08 | 株式会社生命科学インスティテュート | インビトロで多能性幹細胞を分化誘導する方法 |
| CN110494559B (zh) * | 2017-03-28 | 2023-10-31 | 味之素株式会社 | 不分化地维持用培养基添加剂 |
| CN110494559A (zh) * | 2017-03-28 | 2019-11-22 | 味之素株式会社 | 不分化地维持用培养基添加剂 |
| JPWO2018235834A1 (ja) * | 2017-06-19 | 2020-04-16 | 国立大学法人北海道大学 | 表皮水疱症の治療剤 |
| WO2018235834A1 (ja) | 2017-06-19 | 2018-12-27 | 国立大学法人北海道大学 | 表皮水疱症の治療剤 |
| KR20200016871A (ko) | 2017-06-20 | 2020-02-17 | 고쿠리츠 다이가쿠 호우징 나고야 다이가쿠 | 다능성 간세포에 의한 태아발육부전에 따르는 뇌장애의 개선 및 치료 |
| WO2018235878A1 (ja) | 2017-06-20 | 2018-12-27 | 国立大学法人名古屋大学 | 多能性幹細胞による胎児発育不全に伴う脳障害の改善及び治療 |
| KR20200070247A (ko) | 2017-10-17 | 2020-06-17 | 고쿠리츠다이가쿠호진 히로시마다이가쿠 | 골연골 수복을 유도하는 다능성 간세포 |
| JPWO2019078262A1 (ja) * | 2017-10-17 | 2020-11-05 | 国立大学法人広島大学 | 骨軟骨修復を誘導する多能性幹細胞 |
| WO2019078262A1 (ja) | 2017-10-17 | 2019-04-25 | 国立大学法人広島大学 | 骨軟骨修復を誘導する多能性幹細胞 |
| JP7489016B2 (ja) | 2017-10-17 | 2024-05-23 | 国立大学法人東北大学 | 骨軟骨修復を誘導する多能性幹細胞 |
| CN111566204A (zh) * | 2017-11-10 | 2020-08-21 | 再生医科学股份有限公司 | 培养细胞的制造方法、脊髓损伤疾病的治疗剂的制造方法 |
| JPWO2019092939A1 (ja) * | 2017-11-10 | 2021-01-21 | 株式会社リジェネシスサイエンス | 培養細胞の製造方法,脊髄損傷疾患の治療剤の製造方法 |
| WO2019092939A1 (ja) * | 2017-11-10 | 2019-05-16 | 株式会社リジェネシスサイエンス | 培養細胞の製造方法,脊髄損傷疾患の治療剤の製造方法 |
| CN111566204B (zh) * | 2017-11-10 | 2024-03-26 | 再生医科学股份有限公司 | 培养细胞的制造方法、脊髓损伤疾病的治疗剂的制造方法 |
| JP7207742B2 (ja) | 2017-11-10 | 2023-01-18 | 株式会社リジェネシスサイエンス | 培養細胞の製造方法,脊髄損傷疾患の治療剤の製造方法 |
| JP2018111722A (ja) * | 2018-04-09 | 2018-07-19 | 株式会社生命科学インスティテュート | 脳梗塞治療のための多能性幹細胞 |
| JPWO2019216380A1 (ja) * | 2018-05-09 | 2021-05-13 | 株式会社生命科学インスティテュート | 脊髄損傷の治療剤 |
| WO2019216380A1 (ja) | 2018-05-09 | 2019-11-14 | 株式会社生命科学インスティテュート | 脊髄損傷の治療剤 |
| WO2020017575A1 (ja) | 2018-07-19 | 2020-01-23 | 国立大学法人京都大学 | 多能性幹細胞由来の板状軟骨およびその製造方法 |
| JP7473207B2 (ja) | 2018-11-30 | 2024-04-23 | 京都府公立大学法人 | 末梢血流障害の治療剤 |
| KR20210099014A (ko) | 2018-11-30 | 2021-08-11 | 교토후고리츠다이가쿠호진 | 말초 혈류 장애의 치료제 |
| EP3888751A4 (en) * | 2018-11-30 | 2022-10-05 | Kyoto Prefectural Public University Corporation | THERAPEUTIC AGENT FOR PERIPHERAL BLOOD FLOW DISORDER |
| WO2020111249A1 (ja) | 2018-11-30 | 2020-06-04 | 京都府公立大学法人 | 末梢血流障害の治療剤 |
| CN113195054A (zh) * | 2018-11-30 | 2021-07-30 | 京都府公立大学法人 | 外周血流紊乱的治疗剂 |
| JPWO2020111249A1 (ja) * | 2018-11-30 | 2020-06-04 | ||
| WO2020130147A1 (ja) | 2018-12-21 | 2020-06-25 | 国立大学法人京都大学 | ルブリシン局在軟骨様組織、その製造方法及びそれを含む関節軟骨損傷治療用組成物 |
| WO2021029346A1 (ja) | 2019-08-09 | 2021-02-18 | 国立大学法人東北大学 | 脳血管性認知症の治療または予防剤 |
| WO2021045190A1 (ja) | 2019-09-05 | 2021-03-11 | 国立大学法人東北大学 | 心筋炎の治療剤 |
| WO2021066076A1 (ja) | 2019-10-01 | 2021-04-08 | 国立大学法人京都大学 | 尿管芽先端部細胞の単離方法 |
| WO2021085639A1 (ja) | 2019-10-31 | 2021-05-06 | 株式会社生命科学インスティテュート | 多能性幹細胞による間質性膀胱炎の治療 |
| WO2021117886A1 (ja) | 2019-12-12 | 2021-06-17 | 国立大学法人千葉大学 | 巨核球および血小板を含む凍結乾燥製剤 |
| WO2021187602A1 (ja) | 2020-03-19 | 2021-09-23 | 国立大学法人京都大学 | 心筋細胞の精製方法 |
| WO2021187601A1 (ja) | 2020-03-19 | 2021-09-23 | 国立大学法人京都大学 | 心筋細胞の精製方法 |
| WO2021201286A1 (ja) | 2020-04-02 | 2021-10-07 | 国立大学法人東北大学 | ハイポテンシャル多能性幹細胞 |
| WO2022014604A1 (ja) | 2020-07-13 | 2022-01-20 | 国立大学法人京都大学 | 骨格筋前駆細胞及びその精製方法、筋原性疾患を治療するための組成物、並びに骨格筋前駆細胞を含む細胞群の製造方法 |
| WO2022014681A1 (ja) | 2020-07-15 | 2022-01-20 | 国立大学法人 岡山大学 | 運動ニューロン疾患(mnd)の治療に有効な多能性幹細胞 |
| WO2023153464A1 (ja) | 2022-02-09 | 2023-08-17 | 住友ファーマ株式会社 | 多能性幹細胞から中脳底板領域の神経系細胞への分化における、培養液中の細胞の分化能を判定する方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7134317B2 (ja) | 生体組織から単離できる多能性幹細胞 | |
| US11261426B2 (en) | Pluripotent stem cell that can be isolated from body tissue | |
| WO2012133948A1 (ja) | 生体組織から単離できるssea-3陽性の多能性幹細胞を含む他家移植用細胞治療用組成物 | |
| WO2012133942A1 (ja) | 生体の臍帯又は脂肪組織から単離できる多能性幹細胞 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080041908.7 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10799956 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2011522886 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2768238 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010271722 Country of ref document: AU |
|
| REEP | Request for entry into the european phase |
Ref document number: 2010799956 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010799956 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20127003879 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1395/CHENP/2012 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2010271722 Country of ref document: AU Date of ref document: 20100715 Kind code of ref document: A |