ES2933694T3 - Procesos para la preparación de un inhibidor de PI3K - Google Patents
Procesos para la preparación de un inhibidor de PI3K Download PDFInfo
- Publication number
- ES2933694T3 ES2933694T3 ES20201979T ES20201979T ES2933694T3 ES 2933694 T3 ES2933694 T3 ES 2933694T3 ES 20201979 T ES20201979 T ES 20201979T ES 20201979 T ES20201979 T ES 20201979T ES 2933694 T3 ES2933694 T3 ES 2933694T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- reaction
- carried out
- salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims abstract description 138
- 230000008569 process Effects 0.000 title claims abstract description 99
- 238000002360 preparation method Methods 0.000 title description 12
- 239000012828 PI3K inhibitor Substances 0.000 title description 6
- 229940043441 phosphoinositide 3-kinase inhibitor Drugs 0.000 title description 6
- 150000003839 salts Chemical group 0.000 claims abstract description 120
- 150000001875 compounds Chemical class 0.000 claims description 315
- 238000006243 chemical reaction Methods 0.000 claims description 102
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 65
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 45
- 239000002904 solvent Substances 0.000 claims description 38
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 35
- -1 C1-6 alkylsulfonyl halide Chemical class 0.000 claims description 33
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 29
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 claims description 22
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical group CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 claims description 18
- 150000003512 tertiary amines Chemical class 0.000 claims description 18
- BOSVWXDDFBSSIZ-UHFFFAOYSA-N 2-(1-ethoxyethylidene)propanedinitrile Chemical compound CCOC(C)=C(C#N)C#N BOSVWXDDFBSSIZ-UHFFFAOYSA-N 0.000 claims description 17
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 15
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 claims description 15
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-diisopropylethylamine Substances CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 14
- 239000003054 catalyst Substances 0.000 claims description 14
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 13
- XPOLVIIHTDKJRY-UHFFFAOYSA-N acetic acid;methanimidamide Chemical compound NC=N.CC(O)=O XPOLVIIHTDKJRY-UHFFFAOYSA-N 0.000 claims description 13
- LYXHWHHENVLYCN-QMDOQEJBSA-N (1z,5z)-cycloocta-1,5-diene;rhodium;tetrafluoroborate Chemical group [Rh].F[B-](F)(F)F.C\1C\C=C/CC\C=C/1.C\1C\C=C/CC\C=C/1 LYXHWHHENVLYCN-QMDOQEJBSA-N 0.000 claims description 6
- 238000005984 hydrogenation reaction Methods 0.000 claims description 6
- 239000007800 oxidant agent Substances 0.000 claims description 6
- LLJITAAISCMRAR-UHFFFAOYSA-N bis[4-(trifluoromethyl)phenyl]phosphane Chemical compound C1=CC(C(F)(F)F)=CC=C1PC1=CC=C(C(F)(F)F)C=C1 LLJITAAISCMRAR-UHFFFAOYSA-N 0.000 claims description 5
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 claims description 4
- 230000003647 oxidation Effects 0.000 claims description 4
- 238000007254 oxidation reaction Methods 0.000 claims description 4
- 239000012359 Methanesulfonyl chloride Substances 0.000 claims description 3
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 claims description 2
- 238000006646 Dess-Martin oxidation reaction Methods 0.000 claims description 2
- QARBMVPHQWIHKH-KHWXYDKHSA-N methanesulfonyl chloride Chemical group C[35S](Cl)(=O)=O QARBMVPHQWIHKH-KHWXYDKHSA-N 0.000 claims description 2
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 claims description 2
- 230000001590 oxidative effect Effects 0.000 claims description 2
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 2
- 238000002955 isolation Methods 0.000 claims 1
- 239000003112 inhibitor Substances 0.000 abstract description 22
- ZQPDJCIXJHUERQ-QWRGUYRKSA-N (4r)-4-[3-[(1s)-1-(4-amino-3-methylpyrazolo[3,4-d]pyrimidin-1-yl)ethyl]-5-chloro-2-ethoxy-6-fluorophenyl]pyrrolidin-2-one Chemical compound CCOC1=C([C@H](C)N2C3=NC=NC(N)=C3C(C)=N2)C=C(Cl)C(F)=C1[C@@H]1CNC(=O)C1 ZQPDJCIXJHUERQ-QWRGUYRKSA-N 0.000 abstract description 9
- 239000000543 intermediate Substances 0.000 abstract description 6
- 150000003906 phosphoinositides Chemical class 0.000 abstract description 4
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 94
- 239000000203 mixture Substances 0.000 description 87
- 239000011541 reaction mixture Substances 0.000 description 82
- 239000000243 solution Substances 0.000 description 79
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 73
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical class Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 65
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 60
- 108091007960 PI3Ks Proteins 0.000 description 53
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 52
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 52
- 201000010099 disease Diseases 0.000 description 48
- 239000007787 solid Substances 0.000 description 46
- 239000000047 product Substances 0.000 description 44
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical group [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 42
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 36
- 235000019439 ethyl acetate Nutrition 0.000 description 36
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 36
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 35
- 238000004128 high performance liquid chromatography Methods 0.000 description 34
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 32
- 210000003719 b-lymphocyte Anatomy 0.000 description 30
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 29
- 239000010410 layer Substances 0.000 description 28
- 239000012044 organic layer Substances 0.000 description 27
- 239000007858 starting material Substances 0.000 description 27
- 108091000080 Phosphotransferase Proteins 0.000 description 24
- 102000020233 phosphotransferase Human genes 0.000 description 24
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 23
- 210000004027 cell Anatomy 0.000 description 23
- 238000001914 filtration Methods 0.000 description 23
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 23
- 238000005160 1H NMR spectroscopy Methods 0.000 description 21
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 21
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 20
- 238000011282 treatment Methods 0.000 description 19
- 230000000694 effects Effects 0.000 description 18
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 18
- 238000000634 powder X-ray diffraction Methods 0.000 description 17
- 239000002002 slurry Substances 0.000 description 17
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 16
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 14
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 14
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 14
- 230000002829 reductive effect Effects 0.000 description 14
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 13
- 239000004480 active ingredient Substances 0.000 description 13
- 239000012043 crude product Substances 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 12
- 238000003556 assay Methods 0.000 description 12
- 208000035475 disorder Diseases 0.000 description 12
- 238000000746 purification Methods 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 206010028980 Neoplasm Diseases 0.000 description 11
- 208000031981 Thrombocytopenic Idiopathic Purpura Diseases 0.000 description 11
- 239000002585 base Substances 0.000 description 11
- 239000003795 chemical substances by application Substances 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- 239000003921 oil Substances 0.000 description 11
- 235000019198 oils Nutrition 0.000 description 11
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 10
- 206010021245 Idiopathic thrombocytopenic purpura Diseases 0.000 description 10
- 201000003710 autoimmune thrombocytopenic purpura Diseases 0.000 description 10
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 10
- 239000000546 pharmaceutical excipient Substances 0.000 description 10
- 229960004641 rituximab Drugs 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 9
- 206010061218 Inflammation Diseases 0.000 description 9
- 229940116839 Janus kinase 1 inhibitor Drugs 0.000 description 9
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 9
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 9
- 230000004913 activation Effects 0.000 description 9
- 238000004458 analytical method Methods 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 9
- 239000012267 brine Substances 0.000 description 9
- 239000002552 dosage form Substances 0.000 description 9
- 230000004054 inflammatory process Effects 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- 239000008194 pharmaceutical composition Substances 0.000 description 9
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 9
- 239000000843 powder Substances 0.000 description 9
- 239000000741 silica gel Substances 0.000 description 9
- 229910002027 silica gel Inorganic materials 0.000 description 9
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 9
- 239000006188 syrup Substances 0.000 description 9
- 235000020357 syrup Nutrition 0.000 description 9
- 229940121730 Janus kinase 2 inhibitor Drugs 0.000 description 8
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 8
- 241000699670 Mus sp. Species 0.000 description 8
- 206010047115 Vasculitis Diseases 0.000 description 8
- 206010012818 diffuse large B-cell lymphoma Diseases 0.000 description 8
- 239000006260 foam Substances 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical group [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 229910052938 sodium sulfate Inorganic materials 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- 238000002411 thermogravimetry Methods 0.000 description 8
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 8
- 208000003950 B-cell lymphoma Diseases 0.000 description 7
- 208000035186 Hemolytic Autoimmune Anemia Diseases 0.000 description 7
- 208000031671 Large B-Cell Diffuse Lymphoma Diseases 0.000 description 7
- 102000001708 Protein Isoforms Human genes 0.000 description 7
- 108010029485 Protein Isoforms Proteins 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 238000000113 differential scanning calorimetry Methods 0.000 description 7
- 238000009472 formulation Methods 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 6
- 239000007832 Na2SO4 Substances 0.000 description 6
- 108091008611 Protein Kinase B Proteins 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- ZSZXYWFCIKKZBT-ZVDPZPSOSA-N [(2r)-3-[[(2s,3s,5r,6s)-2,6-dihydroxy-3,4,5-triphosphonooxycyclohexyl]oxy-hydroxyphosphoryl]oxy-2-hexadecanoyloxypropyl] hexadecanoate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCCCCCCCCCC)COP(O)(=O)OC1[C@H](O)[C@H](OP(O)(O)=O)C(OP(O)(O)=O)[C@H](OP(O)(O)=O)[C@H]1O ZSZXYWFCIKKZBT-ZVDPZPSOSA-N 0.000 description 6
- 229960000583 acetic acid Drugs 0.000 description 6
- 201000000448 autoimmune hemolytic anemia Diseases 0.000 description 6
- 239000011324 bead Substances 0.000 description 6
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical group NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 6
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 6
- 201000011510 cancer Diseases 0.000 description 6
- 239000013078 crystal Substances 0.000 description 6
- 150000002576 ketones Chemical class 0.000 description 6
- LCEDQNDDFOCWGG-UHFFFAOYSA-N morpholine-4-carbaldehyde Chemical compound O=CN1CCOCC1 LCEDQNDDFOCWGG-UHFFFAOYSA-N 0.000 description 6
- 230000035772 mutation Effects 0.000 description 6
- 239000008177 pharmaceutical agent Substances 0.000 description 6
- 238000000926 separation method Methods 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- FMCAFXHLMUOIGG-JTJHWIPRSA-N (2s)-2-[[(2r)-2-[[(2s)-2-[[(2r)-2-formamido-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,5-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoic acid Chemical compound O=CN[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(=O)N[C@@H](CCSC)C(O)=O)CC1=CC(C)=C(O)C=C1C FMCAFXHLMUOIGG-JTJHWIPRSA-N 0.000 description 5
- UMCMPZBLKLEWAF-BCTGSCMUSA-N 3-[(3-cholamidopropyl)dimethylammonio]propane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 UMCMPZBLKLEWAF-BCTGSCMUSA-N 0.000 description 5
- RBDZXYMSVYEWSL-QWRGUYRKSA-N 5-amino-1-[(1S)-1-[5-chloro-2-ethoxy-4-fluoro-3-[(3R)-5-oxopyrrolidin-3-yl]phenyl]ethyl]-3-methylpyrazole-4-carbonitrile Chemical compound NC1=C(C(=NN1[C@@H](C)C1=C(C(=C(C(=C1)Cl)F)[C@@H]1CNC(C1)=O)OCC)C)C#N RBDZXYMSVYEWSL-QWRGUYRKSA-N 0.000 description 5
- 208000023275 Autoimmune disease Diseases 0.000 description 5
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 5
- 206010025323 Lymphomas Diseases 0.000 description 5
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 5
- 206010035226 Plasma cell myeloma Diseases 0.000 description 5
- 229940124639 Selective inhibitor Drugs 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 239000002246 antineoplastic agent Substances 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 239000012455 biphasic mixture Substances 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 238000004113 cell culture Methods 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 238000004296 chiral HPLC Methods 0.000 description 5
- 229940127089 cytotoxic agent Drugs 0.000 description 5
- 229960003957 dexamethasone Drugs 0.000 description 5
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 5
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- 229940043355 kinase inhibitor Drugs 0.000 description 5
- 150000002632 lipids Chemical class 0.000 description 5
- 229960001924 melphalan Drugs 0.000 description 5
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 5
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 5
- 238000005457 optimization Methods 0.000 description 5
- 230000007170 pathology Effects 0.000 description 5
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 230000004044 response Effects 0.000 description 5
- 206010039073 rheumatoid arthritis Diseases 0.000 description 5
- 230000004083 survival effect Effects 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 238000004809 thin layer chromatography Methods 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- WVSLNFBNTNTPJI-UHFFFAOYSA-N 2-(5-chloro-2-ethoxy-4-fluorophenyl)-2-methyl-1,3-dioxolane Chemical compound ClC=1C(=CC(=C(C=1)C1(OCCO1)C)OCC)F WVSLNFBNTNTPJI-UHFFFAOYSA-N 0.000 description 4
- LMPKUDJPQHRXIR-UHFFFAOYSA-N 5-chloro-2-ethoxy-6-fluoro-3-(2-methyl-1,3-dioxolan-2-yl)benzaldehyde Chemical compound ClC=1C(=C(C=O)C(=C(C=1)C1(OCCO1)C)OCC)F LMPKUDJPQHRXIR-UHFFFAOYSA-N 0.000 description 4
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 4
- 201000001320 Atherosclerosis Diseases 0.000 description 4
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 4
- 208000017604 Hodgkin disease Diseases 0.000 description 4
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 4
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 4
- 208000005777 Lupus Nephritis Diseases 0.000 description 4
- 239000007993 MOPS buffer Substances 0.000 description 4
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 4
- 208000034578 Multiple myelomas Diseases 0.000 description 4
- 201000004681 Psoriasis Diseases 0.000 description 4
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 description 4
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 4
- 206010069351 acute lung injury Diseases 0.000 description 4
- 206010003246 arthritis Diseases 0.000 description 4
- 208000006673 asthma Diseases 0.000 description 4
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 4
- 208000029742 colonic neoplasm Diseases 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 229960004679 doxorubicin Drugs 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 238000003818 flash chromatography Methods 0.000 description 4
- 230000014509 gene expression Effects 0.000 description 4
- 239000001963 growth medium Substances 0.000 description 4
- 239000005457 ice water Substances 0.000 description 4
- 230000001771 impaired effect Effects 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 4
- 235000019341 magnesium sulphate Nutrition 0.000 description 4
- 238000002844 melting Methods 0.000 description 4
- 230000008018 melting Effects 0.000 description 4
- OWIUPIRUAQMTTK-UHFFFAOYSA-M n-aminocarbamate Chemical compound NNC([O-])=O OWIUPIRUAQMTTK-UHFFFAOYSA-M 0.000 description 4
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 4
- 239000002245 particle Substances 0.000 description 4
- 239000006187 pill Substances 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 229910000160 potassium phosphate Inorganic materials 0.000 description 4
- 235000011009 potassium phosphates Nutrition 0.000 description 4
- 238000010791 quenching Methods 0.000 description 4
- 230000000171 quenching effect Effects 0.000 description 4
- 238000002821 scintillation proximity assay Methods 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 239000003643 water by type Substances 0.000 description 4
- RVMXTRGDMDINDQ-QMMMGPOBSA-N (4R)-4-(3-acetyl-5-chloro-2-ethoxy-6-fluorophenyl)pyrrolidin-2-one Chemical compound C(C)(=O)C=1C(=C(C(=C(C=1)Cl)F)[C@H]1CC(NC1)=O)OCC RVMXTRGDMDINDQ-QMMMGPOBSA-N 0.000 description 3
- PBKONEOXTCPAFI-UHFFFAOYSA-N 1,2,4-trichlorobenzene Chemical compound ClC1=CC=C(Cl)C(Cl)=C1 PBKONEOXTCPAFI-UHFFFAOYSA-N 0.000 description 3
- RFFLAFLAYFXFSW-UHFFFAOYSA-N 1,2-dichlorobenzene Chemical compound ClC1=CC=CC=C1Cl RFFLAFLAYFXFSW-UHFFFAOYSA-N 0.000 description 3
- VOQMFPPAYMCUTJ-UHFFFAOYSA-N 1-(5-chloro-4-fluoro-2-hydroxyphenyl)ethanone Chemical compound CC(=O)C1=CC(Cl)=C(F)C=C1O VOQMFPPAYMCUTJ-UHFFFAOYSA-N 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 3
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 208000020084 Bone disease Diseases 0.000 description 3
- 208000003174 Brain Neoplasms Diseases 0.000 description 3
- 206010006187 Breast cancer Diseases 0.000 description 3
- 208000026310 Breast neoplasm Diseases 0.000 description 3
- 208000011691 Burkitt lymphomas Diseases 0.000 description 3
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- 108090000695 Cytokines Proteins 0.000 description 3
- 206010012689 Diabetic retinopathy Diseases 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 206010014759 Endometrial neoplasm Diseases 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 206010018364 Glomerulonephritis Diseases 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 3
- 206010033645 Pancreatitis Diseases 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 3
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 3
- 102000001253 Protein Kinase Human genes 0.000 description 3
- 102100033810 RAC-alpha serine/threonine-protein kinase Human genes 0.000 description 3
- 239000007868 Raney catalyst Substances 0.000 description 3
- 229910000564 Raney nickel Inorganic materials 0.000 description 3
- 206010038389 Renal cancer Diseases 0.000 description 3
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 3
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 208000011341 adult acute respiratory distress syndrome Diseases 0.000 description 3
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 3
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 3
- 150000008041 alkali metal carbonates Chemical class 0.000 description 3
- 208000026935 allergic disease Diseases 0.000 description 3
- 150000001413 amino acids Chemical group 0.000 description 3
- 230000033115 angiogenesis Effects 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 229910000085 borane Inorganic materials 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 3
- 239000006227 byproduct Substances 0.000 description 3
- 238000001516 cell proliferation assay Methods 0.000 description 3
- 230000005754 cellular signaling Effects 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 210000001072 colon Anatomy 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- BEPAFCGSDWSTEL-UHFFFAOYSA-N dimethyl malonate Chemical compound COC(=O)CC(=O)OC BEPAFCGSDWSTEL-UHFFFAOYSA-N 0.000 description 3
- 229910001873 dinitrogen Inorganic materials 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 210000001102 germinal center b cell Anatomy 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 201000005787 hematologic cancer Diseases 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- 239000011630 iodine Substances 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 201000010982 kidney cancer Diseases 0.000 description 3
- 208000017169 kidney disease Diseases 0.000 description 3
- 206010025135 lupus erythematosus Diseases 0.000 description 3
- 230000036210 malignancy Effects 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 238000010172 mouse model Methods 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 229960002340 pentostatin Drugs 0.000 description 3
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 3
- 235000011181 potassium carbonates Nutrition 0.000 description 3
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 3
- 229960004618 prednisone Drugs 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 108060006633 protein kinase Proteins 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 208000037803 restenosis Diseases 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- 238000007789 sealing Methods 0.000 description 3
- 239000012265 solid product Substances 0.000 description 3
- 150000003431 steroids Chemical class 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 239000012049 topical pharmaceutical composition Substances 0.000 description 3
- KWGRBVOPPLSCSI-WPRPVWTQSA-N (-)-ephedrine Chemical compound CN[C@@H](C)[C@H](O)C1=CC=CC=C1 KWGRBVOPPLSCSI-WPRPVWTQSA-N 0.000 description 2
- RCQWEKFYYAZQPG-SFYZADRCSA-N (4R)-4-[5-chloro-2-ethoxy-6-fluoro-3-[(1R)-1-hydroxyethyl]phenyl]pyrrolidin-2-one Chemical compound ClC=1C(=C(C(=C(C=1)[C@@H](C)O)OCC)[C@H]1CC(NC1)=O)F RCQWEKFYYAZQPG-SFYZADRCSA-N 0.000 description 2
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 2
- UBOXGVDOUJQMTN-UHFFFAOYSA-N 1,1,2-trichloroethane Chemical compound ClCC(Cl)Cl UBOXGVDOUJQMTN-UHFFFAOYSA-N 0.000 description 2
- HKWJHKSHEWVOSS-MRQSADPDSA-N 1,2-dihexadecanoyl-sn-glycero-3-phospho-(1D-myo-inositol-4,5-bisphosphate) Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCCCCCCCCCC)COP(O)(=O)O[C@@H]1[C@H](O)[C@H](O)[C@@H](OP(O)(O)=O)[C@H](OP(O)(O)=O)[C@H]1O HKWJHKSHEWVOSS-MRQSADPDSA-N 0.000 description 2
- IVSZLXZYQVIEFR-UHFFFAOYSA-N 1,3-Dimethylbenzene Natural products CC1=CC=CC(C)=C1 IVSZLXZYQVIEFR-UHFFFAOYSA-N 0.000 description 2
- CTBNZCXPULIMIZ-UHFFFAOYSA-N 1-(5-chloro-2-ethoxy-4-fluoro-3-iodophenyl)ethanone Chemical compound CCOC1=C(I)C(F)=C(Cl)C=C1C(C)=O CTBNZCXPULIMIZ-UHFFFAOYSA-N 0.000 description 2
- QTKBGYUBVPKNNY-UHFFFAOYSA-N 1-(5-chloro-2-ethoxy-4-fluorophenyl)ethanone Chemical compound ClC=1C(=CC(=C(C=1)C(C)=O)OCC)F QTKBGYUBVPKNNY-UHFFFAOYSA-N 0.000 description 2
- HRJLBPBABHQLKQ-UHFFFAOYSA-N 1-(5-chloro-4-fluoro-2-hydroxy-3-iodophenyl)ethanone Chemical compound CC(=O)C1=CC(Cl)=C(F)C(I)=C1O HRJLBPBABHQLKQ-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- VQSJAWPFQCXIOB-VODLGYORSA-N 2,3-dihydroxypropyl [(1r,2r,3s,4r,5r,6s)-2,3,6-trihydroxy-4,5-diphosphonooxycyclohexyl] hydrogen phosphate Chemical compound OCC(O)COP(O)(=O)O[C@@H]1[C@H](O)[C@H](O)[C@@H](OP(O)(O)=O)[C@H](OP(O)(O)=O)[C@H]1O VQSJAWPFQCXIOB-VODLGYORSA-N 0.000 description 2
- NNZBNWVYPGIQAP-UHFFFAOYSA-N 2-(5-chloro-2-ethoxy-4-fluoro-3-iodophenyl)-2-methyl-1,3-dioxolane Chemical compound CCOC1=C(I)C(F)=C(Cl)C=C1C1(C)OCCO1 NNZBNWVYPGIQAP-UHFFFAOYSA-N 0.000 description 2
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 2
- DVLFYONBTKHTER-UHFFFAOYSA-N 3-(N-morpholino)propanesulfonic acid Chemical compound OS(=O)(=O)CCCN1CCOCC1 DVLFYONBTKHTER-UHFFFAOYSA-N 0.000 description 2
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 2
- XPYQFIISZQCINN-QVXDJYSKSA-N 4-amino-1-[(2r,3e,4s,5r)-3-(fluoromethylidene)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one;hydrate Chemical compound O.O=C1N=C(N)C=CN1[C@H]1C(=C/F)/[C@H](O)[C@@H](CO)O1 XPYQFIISZQCINN-QVXDJYSKSA-N 0.000 description 2
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 2
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 2
- 206010005003 Bladder cancer Diseases 0.000 description 2
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- GATVIKZLVQHOMN-UHFFFAOYSA-N Chlorodibromomethane Chemical compound ClC(Br)Br GATVIKZLVQHOMN-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- 206010014733 Endometrial cancer Diseases 0.000 description 2
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 2
- 102100037813 Focal adhesion kinase 1 Human genes 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 2
- 206010018372 Glomerulonephritis membranous Diseases 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 2
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- 238000004566 IR spectroscopy Methods 0.000 description 2
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 2
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 2
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 2
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 2
- ATHHXGZTWNVVOU-UHFFFAOYSA-N N-methylformamide Chemical compound CNC=O ATHHXGZTWNVVOU-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- 108700020796 Oncogene Proteins 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 description 2
- 102000038030 PI3Ks Human genes 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- URLKBWYHVLBVBO-UHFFFAOYSA-N Para-Xylene Chemical group CC1=CC=C(C)C=C1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 description 2
- 241000721454 Pemphigus Species 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 208000007048 Polymyalgia Rheumatica Diseases 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- 206010060862 Prostate cancer Diseases 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- 208000021386 Sjogren Syndrome Diseases 0.000 description 2
- 208000000453 Skin Neoplasms Diseases 0.000 description 2
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 description 2
- 208000004732 Systemic Vasculitis Diseases 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- 206010043540 Thromboangiitis obliterans Diseases 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 2
- 208000002495 Uterine Neoplasms Diseases 0.000 description 2
- 108010046516 Wheat Germ Agglutinins Proteins 0.000 description 2
- XMYKNCNAZKMVQN-NYYWCZLTSA-N [(e)-(3-aminopyridin-2-yl)methylideneamino]thiourea Chemical compound NC(=S)N\N=C\C1=NC=CC=C1N XMYKNCNAZKMVQN-NYYWCZLTSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 230000007815 allergy Effects 0.000 description 2
- 229960000473 altretamine Drugs 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 2
- 230000001028 anti-proliverative effect Effects 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 2
- 229960001467 bortezomib Drugs 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- JPOXNPPZZKNXOV-UHFFFAOYSA-N bromochloromethane Chemical compound ClCBr JPOXNPPZZKNXOV-UHFFFAOYSA-N 0.000 description 2
- DIKBFYAXUHHXCS-UHFFFAOYSA-N bromoform Chemical compound BrC(Br)Br DIKBFYAXUHHXCS-UHFFFAOYSA-N 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 229960005243 carmustine Drugs 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 239000003246 corticosteroid Substances 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 201000003278 cryoglobulinemia Diseases 0.000 description 2
- DMEGYFMYUHOHGS-UHFFFAOYSA-N cycloheptane Chemical compound C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 230000001086 cytosolic effect Effects 0.000 description 2
- 238000005831 deiodination reaction Methods 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 239000002702 enteric coating Substances 0.000 description 2
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 2
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 2
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 2
- 229960005420 etoposide Drugs 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 2
- 201000003444 follicular lymphoma Diseases 0.000 description 2
- 238000010575 fractional recrystallization Methods 0.000 description 2
- 229960002258 fulvestrant Drugs 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 206010017758 gastric cancer Diseases 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- ZQBFAOFFOQMSGJ-UHFFFAOYSA-N hexafluorobenzene Chemical compound FC1=C(F)C(F)=C(F)C(F)=C1F ZQBFAOFFOQMSGJ-UHFFFAOYSA-N 0.000 description 2
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229960003445 idelalisib Drugs 0.000 description 2
- YKLIKGKUANLGSB-HNNXBMFYSA-N idelalisib Chemical compound C1([C@@H](NC=2[C]3N=CN=C3N=CN=2)CC)=NC2=CC=CC(F)=C2C(=O)N1C1=CC=CC=C1 YKLIKGKUANLGSB-HNNXBMFYSA-N 0.000 description 2
- 229960001101 ifosfamide Drugs 0.000 description 2
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 229960002411 imatinib Drugs 0.000 description 2
- 229960003444 immunosuppressant agent Drugs 0.000 description 2
- 239000003018 immunosuppressive agent Substances 0.000 description 2
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical compound C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 2
- 208000027866 inflammatory disease Diseases 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 239000013067 intermediate product Substances 0.000 description 2
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 238000005567 liquid scintillation counting Methods 0.000 description 2
- DBTNVRCCIDISMV-UHFFFAOYSA-L lithium;magnesium;propane;dichloride Chemical compound [Li+].[Mg+2].[Cl-].[Cl-].C[CH-]C DBTNVRCCIDISMV-UHFFFAOYSA-L 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- 201000007919 lymphoplasmacytic lymphoma Diseases 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- 201000007924 marginal zone B-cell lymphoma Diseases 0.000 description 2
- 208000021937 marginal zone lymphoma Diseases 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 201000008350 membranous glomerulonephritis Diseases 0.000 description 2
- 231100000855 membranous nephropathy Toxicity 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 238000003801 milling Methods 0.000 description 2
- 201000006417 multiple sclerosis Diseases 0.000 description 2
- 206010028417 myasthenia gravis Diseases 0.000 description 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 2
- KPSSIOMAKSHJJG-UHFFFAOYSA-N neopentyl alcohol Chemical compound CC(C)(C)CO KPSSIOMAKSHJJG-UHFFFAOYSA-N 0.000 description 2
- BKIMMITUMNQMOS-UHFFFAOYSA-N nonane Chemical compound CCCCCCCCC BKIMMITUMNQMOS-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N o-dimethylbenzene Natural products CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- 201000008482 osteoarthritis Diseases 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- AQIXEPGDORPWBJ-UHFFFAOYSA-N pentan-3-ol Chemical compound CCC(O)CC AQIXEPGDORPWBJ-UHFFFAOYSA-N 0.000 description 2
- 229940124531 pharmaceutical excipient Drugs 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 229920002223 polystyrene Polymers 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 238000003345 scintillation counting Methods 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- 201000000849 skin cancer Diseases 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000008247 solid mixture Substances 0.000 description 2
- 201000011549 stomach cancer Diseases 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 2
- DKACXUFSLUYRFU-UHFFFAOYSA-N tert-butyl n-aminocarbamate Chemical compound CC(C)(C)OC(=O)NN DKACXUFSLUYRFU-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 238000001757 thermogravimetry curve Methods 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 229960000575 trastuzumab Drugs 0.000 description 2
- 229960005526 triapine Drugs 0.000 description 2
- 201000005112 urinary bladder cancer Diseases 0.000 description 2
- 206010046766 uterine cancer Diseases 0.000 description 2
- 229940099039 velcade Drugs 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- FMCGSUUBYTWNDP-ONGXEEELSA-N (1R,2S)-2-(dimethylamino)-1-phenyl-1-propanol Chemical compound CN(C)[C@@H](C)[C@H](O)C1=CC=CC=C1 FMCGSUUBYTWNDP-ONGXEEELSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- DNISEZBAYYIQFB-PHDIDXHHSA-N (2r,3r)-2,3-diacetyloxybutanedioic acid Chemical compound CC(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(C)=O DNISEZBAYYIQFB-PHDIDXHHSA-N 0.000 description 1
- VMKAFJQFKBASMU-KRWDZBQOSA-N (3as)-1-methyl-3,3-diphenyl-3a,4,5,6-tetrahydropyrrolo[1,2-c][1,3,2]oxazaborole Chemical compound C([C@H]12)CCN1B(C)OC2(C=1C=CC=CC=1)C1=CC=CC=C1 VMKAFJQFKBASMU-KRWDZBQOSA-N 0.000 description 1
- UHPOZFYNJIMRCT-FBMWCMRBSA-N (3r)-3-[1-(6-chloropyridin-2-yl)pyrrolidin-3-yl]-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical group ClC1=CC=CC(N2CC(CC2)[C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)=N1 UHPOZFYNJIMRCT-FBMWCMRBSA-N 0.000 description 1
- UHPOZFYNJIMRCT-JRZJBTRGSA-N (3s)-3-[1-(6-chloropyridin-2-yl)pyrrolidin-3-yl]-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile Chemical compound ClC1=CC=CC(N2CC(CC2)[C@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)=N1 UHPOZFYNJIMRCT-JRZJBTRGSA-N 0.000 description 1
- RCQWEKFYYAZQPG-MQWKRIRWSA-N (4R)-4-[5-chloro-2-ethoxy-6-fluoro-3-(1-hydroxyethyl)phenyl]pyrrolidin-2-one Chemical compound ClC=1C(=C(C(=C(C=1)C(C)O)OCC)[C@H]1CC(NC1)=O)F RCQWEKFYYAZQPG-MQWKRIRWSA-N 0.000 description 1
- ZQPDJCIXJHUERQ-MNOVXSKESA-N (4r)-4-[3-[(1r)-1-(4-amino-3-methylpyrazolo[3,4-d]pyrimidin-1-yl)ethyl]-5-chloro-2-ethoxy-6-fluorophenyl]pyrrolidin-2-one Chemical compound CCOC1=C([C@@H](C)N2C3=NC=NC(N)=C3C(C)=N2)C=C(Cl)C(F)=C1[C@@H]1CNC(=O)C1 ZQPDJCIXJHUERQ-MNOVXSKESA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- AVQQQNCBBIEMEU-UHFFFAOYSA-N 1,1,3,3-tetramethylurea Chemical compound CN(C)C(=O)N(C)C AVQQQNCBBIEMEU-UHFFFAOYSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- LZDKZFUFMNSQCJ-UHFFFAOYSA-N 1,2-diethoxyethane Chemical compound CCOCCOCC LZDKZFUFMNSQCJ-UHFFFAOYSA-N 0.000 description 1
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 1
- VDFVNEFVBPFDSB-UHFFFAOYSA-N 1,3-dioxane Chemical compound C1COCOC1 VDFVNEFVBPFDSB-UHFFFAOYSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical compound CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- RRQYJINTUHWNHW-UHFFFAOYSA-N 1-ethoxy-2-(2-ethoxyethoxy)ethane Chemical compound CCOCCOCCOCC RRQYJINTUHWNHW-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- RQEUFEKYXDPUSK-UHFFFAOYSA-N 1-phenylethylamine Chemical compound CC(N)C1=CC=CC=C1 RQEUFEKYXDPUSK-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- GCKMFJBGXUYNAG-UHFFFAOYSA-N 17alpha-methyltestosterone Natural products C1CC2=CC(=O)CCC2(C)C2C1C1CCC(C)(O)C1(C)CC2 GCKMFJBGXUYNAG-UHFFFAOYSA-N 0.000 description 1
- 238000004293 19F NMR spectroscopy Methods 0.000 description 1
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 1
- SBASXUCJHJRPEV-UHFFFAOYSA-N 2-(2-methoxyethoxy)ethanol Chemical compound COCCOCCO SBASXUCJHJRPEV-UHFFFAOYSA-N 0.000 description 1
- UZYQSNQJLWTICD-UHFFFAOYSA-N 2-(n-benzoylanilino)-2,2-dinitroacetic acid Chemical compound C=1C=CC=CC=1N(C(C(=O)O)([N+]([O-])=O)[N+]([O-])=O)C(=O)C1=CC=CC=C1 UZYQSNQJLWTICD-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- IUVCFHHAEHNCFT-INIZCTEOSA-N 2-[(1s)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one Chemical compound C1=C(F)C(OC(C)C)=CC=C1C(C1=C(N)N=CN=C11)=NN1[C@@H](C)C1=C(C=2C=C(F)C=CC=2)C(=O)C2=CC(F)=CC=C2O1 IUVCFHHAEHNCFT-INIZCTEOSA-N 0.000 description 1
- KTBSXLIQKWEBRB-UHFFFAOYSA-N 2-[1-[1-[3-fluoro-2-(trifluoromethyl)pyridine-4-carbonyl]piperidin-4-yl]-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]azetidin-3-yl]acetonitrile Chemical group C1=CN=C(C(F)(F)F)C(F)=C1C(=O)N1CCC(N2CC(CC#N)(C2)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CC1 KTBSXLIQKWEBRB-UHFFFAOYSA-N 0.000 description 1
- RVJLMNKUMXOVED-SNAWJCMRSA-N 2-[5-chloro-2-ethoxy-4-fluoro-3-[(E)-2-nitroethenyl]phenyl]-2-methyl-1,3-dioxolane Chemical compound ClC=1C(=C(C(=C(C=1)C1(OCCO1)C)OCC)\C=C\[N+](=O)[O-])F RVJLMNKUMXOVED-SNAWJCMRSA-N 0.000 description 1
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 1
- IJXJGQCXFSSHNL-UHFFFAOYSA-N 2-amino-2-phenylethanol Chemical compound OCC(N)C1=CC=CC=C1 IJXJGQCXFSSHNL-UHFFFAOYSA-N 0.000 description 1
- KMGUEILFFWDGFV-UHFFFAOYSA-N 2-benzoyl-2-benzoyloxy-3-hydroxybutanedioic acid Chemical compound C=1C=CC=CC=1C(=O)C(C(C(O)=O)O)(C(O)=O)OC(=O)C1=CC=CC=C1 KMGUEILFFWDGFV-UHFFFAOYSA-N 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- 229940093475 2-ethoxyethanol Drugs 0.000 description 1
- GGDYAKVUZMZKRV-UHFFFAOYSA-N 2-fluoroethanol Chemical compound OCCF GGDYAKVUZMZKRV-UHFFFAOYSA-N 0.000 description 1
- AUVALWUPUHHNQV-UHFFFAOYSA-N 2-hydroxy-3-propylbenzoic acid Chemical class CCCC1=CC=CC(C(O)=O)=C1O AUVALWUPUHHNQV-UHFFFAOYSA-N 0.000 description 1
- MSXVEPNJUHWQHW-UHFFFAOYSA-N 2-methylbutan-2-ol Chemical compound CCC(C)(C)O MSXVEPNJUHWQHW-UHFFFAOYSA-N 0.000 description 1
- KIPMDPDAFINLIV-UHFFFAOYSA-N 2-nitroethanol Chemical compound OCC[N+]([O-])=O KIPMDPDAFINLIV-UHFFFAOYSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- UHEIPWYAZVECGS-ZDUSSCGKSA-N 4-[3-(cyanomethyl)-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]azetidin-1-yl]-2,5-difluoro-n-[(2s)-1,1,1-trifluoropropan-2-yl]benzamide Chemical group C1=C(F)C(C(=O)N[C@@H](C)C(F)(F)F)=CC(F)=C1N1CC(CC#N)(N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)C1 UHEIPWYAZVECGS-ZDUSSCGKSA-N 0.000 description 1
- ZQPDJCIXJHUERQ-UHFFFAOYSA-N 4-[3-[1-(4-amino-3-methylpyrazolo[3,4-d]pyrimidin-1-yl)ethyl]-5-chloro-2-ethoxy-6-fluorophenyl]pyrrolidin-2-one Chemical compound CCOC1=C(C(C)N2C3=NC=NC(N)=C3C(C)=N2)C=C(Cl)C(F)=C1C1CNC(=O)C1 ZQPDJCIXJHUERQ-UHFFFAOYSA-N 0.000 description 1
- ZHSKUOZOLHMKEA-UHFFFAOYSA-N 4-[5-[bis(2-chloroethyl)amino]-1-methylbenzimidazol-2-yl]butanoic acid;hydron;chloride Chemical compound Cl.ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 ZHSKUOZOLHMKEA-UHFFFAOYSA-N 0.000 description 1
- XLHYAEBESNFTCA-UHFFFAOYSA-N 4-chloro-3-fluorophenol Chemical compound OC1=CC=C(Cl)C(F)=C1 XLHYAEBESNFTCA-UHFFFAOYSA-N 0.000 description 1
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 208000011776 Aggressive B-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 208000032671 Allergic granulomatous angiitis Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- ITPDYQOUSLNIHG-UHFFFAOYSA-N Amiodarone hydrochloride Chemical compound [Cl-].CCCCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(I)=C(OCC[NH+](CC)CC)C(I)=C1 ITPDYQOUSLNIHG-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 206010002412 Angiocentric lymphomas Diseases 0.000 description 1
- 102100034613 Annexin A2 Human genes 0.000 description 1
- 108090000668 Annexin A2 Proteins 0.000 description 1
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 1
- GOLCXWYRSKYTSP-UHFFFAOYSA-N Arsenious Acid Chemical compound O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 108091008875 B cell receptors Proteins 0.000 description 1
- 208000037914 B-cell disorder Diseases 0.000 description 1
- 230000003844 B-cell-activation Effects 0.000 description 1
- OLCWFLWEHWLBTO-HSZRJFAPSA-N BMS-214662 Chemical compound C=1C=CSC=1S(=O)(=O)N([C@@H](C1)CC=2C=CC=CC=2)CC2=CC(C#N)=CC=C2N1CC1=CN=CN1 OLCWFLWEHWLBTO-HSZRJFAPSA-N 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 208000033386 Buerger disease Diseases 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- CEIFLXZQZIUBTK-JOCHJYFZSA-N CN(C)Cc1cc(F)cc(OC2CCN(C[C@@H](CC#N)n3cc(cn3)-c3ncnc4[nH]ccc34)CC2)c1 Chemical compound CN(C)Cc1cc(F)cc(OC2CCN(C[C@@H](CC#N)n3cc(cn3)-c3ncnc4[nH]ccc34)CC2)c1 CEIFLXZQZIUBTK-JOCHJYFZSA-N 0.000 description 1
- CEIFLXZQZIUBTK-QFIPXVFZSA-N CN(C)Cc1cc(F)cc(OC2CCN(C[C@H](CC#N)n3cc(cn3)-c3ncnc4[nH]ccc34)CC2)c1 Chemical compound CN(C)Cc1cc(F)cc(OC2CCN(C[C@H](CC#N)n3cc(cn3)-c3ncnc4[nH]ccc34)CC2)c1 CEIFLXZQZIUBTK-QFIPXVFZSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 208000006344 Churg-Strauss Syndrome Diseases 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- 108091007958 Class I PI3Ks Proteins 0.000 description 1
- 208000010007 Cogan syndrome Diseases 0.000 description 1
- 208000019707 Cryoglobulinemic vasculitis Diseases 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 239000012623 DNA damaging agent Substances 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 208000018428 Eosinophilic granulomatosis with polyangiitis Diseases 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- BFPYWIDHMRZLRN-SLHNCBLASA-N Ethinyl estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 BFPYWIDHMRZLRN-SLHNCBLASA-N 0.000 description 1
- PLUBXMRUUVWRLT-UHFFFAOYSA-N Ethyl methanesulfonate Chemical compound CCOS(C)(=O)=O PLUBXMRUUVWRLT-UHFFFAOYSA-N 0.000 description 1
- 229940124783 FAK inhibitor Drugs 0.000 description 1
- VQDFOOLMNPPTBD-LJQANCHMSA-N Fc1cc(ccc1C(=O)N1CCN(C[C@@H](CC#N)n2cc(cn2)-c2ncnc3[nH]ccc23)CC1)C#N Chemical compound Fc1cc(ccc1C(=O)N1CCN(C[C@@H](CC#N)n2cc(cn2)-c2ncnc3[nH]ccc23)CC1)C#N VQDFOOLMNPPTBD-LJQANCHMSA-N 0.000 description 1
- PHXVZSVYYRTVHM-HXUWFJFHSA-N Fc1cc(ccc1C(=O)N1CCN(C[C@@H](CC#N)n2ccc(c2)-c2ncnc3[nH]ccc23)CC1)C#N Chemical compound Fc1cc(ccc1C(=O)N1CCN(C[C@@H](CC#N)n2ccc(c2)-c2ncnc3[nH]ccc23)CC1)C#N PHXVZSVYYRTVHM-HXUWFJFHSA-N 0.000 description 1
- VQDFOOLMNPPTBD-IBGZPJMESA-N Fc1cc(ccc1C(=O)N1CCN(C[C@H](CC#N)n2cc(cn2)-c2ncnc3[nH]ccc23)CC1)C#N Chemical compound Fc1cc(ccc1C(=O)N1CCN(C[C@H](CC#N)n2cc(cn2)-c2ncnc3[nH]ccc23)CC1)C#N VQDFOOLMNPPTBD-IBGZPJMESA-N 0.000 description 1
- PHXVZSVYYRTVHM-FQEVSTJZSA-N Fc1cc(ccc1C(=O)N1CCN(C[C@H](CC#N)n2ccc(c2)-c2ncnc3[nH]ccc23)CC1)C#N Chemical compound Fc1cc(ccc1C(=O)N1CCN(C[C@H](CC#N)n2ccc(c2)-c2ncnc3[nH]ccc23)CC1)C#N PHXVZSVYYRTVHM-FQEVSTJZSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 208000007465 Giant cell arteritis Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 1
- 108010069236 Goserelin Proteins 0.000 description 1
- 206010072579 Granulomatosis with polyangiitis Diseases 0.000 description 1
- DOJXGHGHTWFZHK-UHFFFAOYSA-N Hexachloroacetone Chemical compound ClC(Cl)(Cl)C(=O)C(Cl)(Cl)Cl DOJXGHGHTWFZHK-UHFFFAOYSA-N 0.000 description 1
- 101000577121 Homo sapiens Monocarboxylate transporter 3 Proteins 0.000 description 1
- 101000844245 Homo sapiens Non-receptor tyrosine-protein kinase TYK2 Proteins 0.000 description 1
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 description 1
- 101000779418 Homo sapiens RAC-alpha serine/threonine-protein kinase Proteins 0.000 description 1
- 101000798015 Homo sapiens RAC-beta serine/threonine-protein kinase Proteins 0.000 description 1
- 101000798007 Homo sapiens RAC-gamma serine/threonine-protein kinase Proteins 0.000 description 1
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 101000595531 Homo sapiens Serine/threonine-protein kinase pim-1 Proteins 0.000 description 1
- 101000934996 Homo sapiens Tyrosine-protein kinase JAK3 Proteins 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 208000031814 IgA Vasculitis Diseases 0.000 description 1
- 208000028622 Immune thrombocytopenia Diseases 0.000 description 1
- 208000011821 Indolent B-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 108010024121 Janus Kinases Proteins 0.000 description 1
- 102000015617 Janus Kinases Human genes 0.000 description 1
- 208000011200 Kawasaki disease Diseases 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 208000034624 Leukocytoclastic Cutaneous Vasculitis Diseases 0.000 description 1
- 208000032514 Leukocytoclastic vasculitis Diseases 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- HLFSDGLLUJUHTE-SNVBAGLBSA-N Levamisole Chemical compound C1([C@H]2CN3CCSC3=N2)=CC=CC=C1 HLFSDGLLUJUHTE-SNVBAGLBSA-N 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- GCKMFJBGXUYNAG-HLXURNFRSA-N Methyltestosterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)CC2 GCKMFJBGXUYNAG-HLXURNFRSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 102100025275 Monocarboxylate transporter 3 Human genes 0.000 description 1
- HTGGWBJDPLPEPS-IBYPIGCZSA-N N#CC[C@@H](C1CCN(C1)c1nc2cccnc2o1)n1cc(cn1)-c1ncnc2[nH]ccc12 Chemical compound N#CC[C@@H](C1CCN(C1)c1nc2cccnc2o1)n1cc(cn1)-c1ncnc2[nH]ccc12 HTGGWBJDPLPEPS-IBYPIGCZSA-N 0.000 description 1
- HTGGWBJDPLPEPS-XPKAQORNSA-N N#CC[C@H](C1CCN(C1)c1nc2cccnc2o1)n1cc(cn1)-c1ncnc2[nH]ccc12 Chemical compound N#CC[C@H](C1CCN(C1)c1nc2cccnc2o1)n1cc(cn1)-c1ncnc2[nH]ccc12 HTGGWBJDPLPEPS-XPKAQORNSA-N 0.000 description 1
- QJMCKEPOKRERLN-UHFFFAOYSA-N N-3,4-tridhydroxybenzamide Chemical compound ONC(=O)C1=CC=C(O)C(O)=C1 QJMCKEPOKRERLN-UHFFFAOYSA-N 0.000 description 1
- FMCGSUUBYTWNDP-UHFFFAOYSA-N N-Methylephedrine Natural products CN(C)C(C)C(O)C1=CC=CC=C1 FMCGSUUBYTWNDP-UHFFFAOYSA-N 0.000 description 1
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 1
- OHLUUHNLEMFGTQ-UHFFFAOYSA-N N-methylacetamide Chemical compound CNC(C)=O OHLUUHNLEMFGTQ-UHFFFAOYSA-N 0.000 description 1
- 102100032028 Non-receptor tyrosine-protein kinase TYK2 Human genes 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 239000002033 PVDF binder Substances 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 208000007452 Plasmacytoma Diseases 0.000 description 1
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 206010065857 Primary Effusion Lymphoma Diseases 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 208000033766 Prolymphocytic Leukemia Diseases 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 229940079156 Proteasome inhibitor Drugs 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 102100032315 RAC-beta serine/threonine-protein kinase Human genes 0.000 description 1
- 102100032314 RAC-gamma serine/threonine-protein kinase Human genes 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- 208000002200 Respiratory Hypersensitivity Diseases 0.000 description 1
- 241000219061 Rheum Species 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 102100036077 Serine/threonine-protein kinase pim-1 Human genes 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 208000004346 Smoldering Multiple Myeloma Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- 208000001106 Takayasu Arteritis Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- 101150115851 Tff2 gene Proteins 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 1
- MSLJYSGFUMYUDX-UHFFFAOYSA-N Trimidox Chemical compound ON=C(N)C1=CC(O)=C(O)C(O)=C1 MSLJYSGFUMYUDX-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 102100025387 Tyrosine-protein kinase JAK3 Human genes 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- HNSXCKBVOZPUIE-UHFFFAOYSA-N [Mg][I]I Chemical group [Mg][I]I HNSXCKBVOZPUIE-UHFFFAOYSA-N 0.000 description 1
- 238000002679 ablation Methods 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 229940081735 acetylcellulose Drugs 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 150000001278 adipic acid derivatives Chemical class 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 208000037884 allergic airway inflammation Diseases 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000005875 antibody response Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000006286 aqueous extract Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 238000003149 assay kit Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 201000004988 autoimmune vasculitis Diseases 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 229940120638 avastin Drugs 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 125000004567 azetidin-3-yl group Chemical group N1CC(C1)* 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 239000003637 basic solution Substances 0.000 description 1
- 229960002707 bendamustine Drugs 0.000 description 1
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000002798 bone marrow cell Anatomy 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- FMWLUWPQPKEARP-UHFFFAOYSA-N bromodichloromethane Chemical compound ClC(Cl)Br FMWLUWPQPKEARP-UHFFFAOYSA-N 0.000 description 1
- 229950005228 bromoform Drugs 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000000378 calcium silicate Substances 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- 229960003340 calcium silicate Drugs 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- 229940112129 campath Drugs 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical class C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- RBHJBMIOOPYDBQ-UHFFFAOYSA-N carbon dioxide;propan-2-one Chemical compound O=C=O.CC(C)=O RBHJBMIOOPYDBQ-UHFFFAOYSA-N 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 230000011712 cell development Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 238000012054 celltiter-glo Methods 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 208000010353 central nervous system vasculitis Diseases 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- BFPSDSIWYFKGBC-UHFFFAOYSA-N chlorotrianisene Chemical compound C1=CC(OC)=CC=C1C(Cl)=C(C=1C=CC(OC)=CC=1)C1=CC=C(OC)C=C1 BFPSDSIWYFKGBC-UHFFFAOYSA-N 0.000 description 1
- 229960002559 chlorotrianisene Drugs 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 229960000928 clofarabine Drugs 0.000 description 1
- WDDPHFBMKLOVOX-AYQXTPAHSA-N clofarabine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1F WDDPHFBMKLOVOX-AYQXTPAHSA-N 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- SSJXIUAHEKJCMH-UHFFFAOYSA-N cyclohexane-1,2-diamine Chemical compound NC1CCCCC1N SSJXIUAHEKJCMH-UHFFFAOYSA-N 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- KWGRBVOPPLSCSI-UHFFFAOYSA-N d-ephedrine Natural products CNC(C)C(O)C1=CC=CC=C1 KWGRBVOPPLSCSI-UHFFFAOYSA-N 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 229960002448 dasatinib Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 238000000432 density-gradient centrifugation Methods 0.000 description 1
- CFCUWKMKBJTWLW-UHFFFAOYSA-N deoliosyl-3C-alpha-L-digitoxosyl-MTM Natural products CC=1C(O)=C2C(O)=C3C(=O)C(OC4OC(C)C(O)C(OC5OC(C)C(O)C(OC6OC(C)C(O)C(C)(O)C6)C5)C4)C(C(OC)C(=O)C(O)C(C)O)CC3=CC2=CC=1OC(OC(C)C1O)CC1OC1CC(O)C(O)C(C)O1 CFCUWKMKBJTWLW-UHFFFAOYSA-N 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- FJBFPHVGVWTDIP-UHFFFAOYSA-N dibromomethane Chemical compound BrCBr FJBFPHVGVWTDIP-UHFFFAOYSA-N 0.000 description 1
- 229940019778 diethylene glycol diethyl ether Drugs 0.000 description 1
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 1
- 229940075557 diethylene glycol monoethyl ether Drugs 0.000 description 1
- GLUUGHFHXGJENI-UHFFFAOYSA-N diethylenediamine Natural products C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 1
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 1
- 229960000452 diethylstilbestrol Drugs 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- NKDDWNXOKDWJAK-UHFFFAOYSA-N dimethoxymethane Chemical compound COCOC NKDDWNXOKDWJAK-UHFFFAOYSA-N 0.000 description 1
- KFAYDGPGOZCGAF-JTQLQIEISA-N dimethyl 2-[(1R)-1-(3-acetyl-5-chloro-2-ethoxy-6-fluorophenyl)-2-nitroethyl]propanedioate Chemical compound C(C)(=O)C=1C(=C(C(=C(C=1)Cl)F)[C@H](C[N+](=O)[O-])C(C(=O)OC)C(=O)OC)OCC KFAYDGPGOZCGAF-JTQLQIEISA-N 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- DLNKOYKMWOXYQA-UHFFFAOYSA-N dl-pseudophenylpropanolamine Natural products CC(N)C(O)C1=CC=CC=C1 DLNKOYKMWOXYQA-UHFFFAOYSA-N 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- NOTIQUSPUUHHEH-UXOVVSIBSA-N dromostanolone propionate Chemical compound C([C@@H]1CC2)C(=O)[C@H](C)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](OC(=O)CC)[C@@]2(C)CC1 NOTIQUSPUUHHEH-UXOVVSIBSA-N 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 229950004683 drostanolone propionate Drugs 0.000 description 1
- 239000013583 drug formulation Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 239000008157 edible vegetable oil Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 231100001129 embryonic lethality Toxicity 0.000 description 1
- 238000004945 emulsification Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 230000002357 endometrial effect Effects 0.000 description 1
- RDYMFSUJUZBWLH-UHFFFAOYSA-N endosulfan Chemical compound C12COS(=O)OCC2C2(Cl)C(Cl)=C(Cl)C1(Cl)C2(Cl)Cl RDYMFSUJUZBWLH-UHFFFAOYSA-N 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 239000012055 enteric layer Substances 0.000 description 1
- 238000007824 enzymatic assay Methods 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- 229960002179 ephedrine Drugs 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 1
- 229940082789 erbitux Drugs 0.000 description 1
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- WRWJHZAQLXGJQX-UHFFFAOYSA-N ethyl acetate;formic acid Chemical compound OC=O.CCOC(C)=O WRWJHZAQLXGJQX-UHFFFAOYSA-N 0.000 description 1
- 125000000219 ethylidene group Chemical group [H]C(=[*])C([H])([H])[H] 0.000 description 1
- 230000005713 exacerbation Effects 0.000 description 1
- 229960000255 exemestane Drugs 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 201000006569 extramedullary plasmacytoma Diseases 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000020375 flavoured syrup Nutrition 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- 229960005304 fludarabine phosphate Drugs 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- YLRFCQOZQXIBAB-RBZZARIASA-N fluoxymesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)C[C@@H]2O YLRFCQOZQXIBAB-RBZZARIASA-N 0.000 description 1
- 229960001751 fluoxymesterone Drugs 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- 230000003325 follicular Effects 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 229940014259 gelatin Drugs 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 210000001280 germinal center Anatomy 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 229960002913 goserelin Drugs 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000003862 health status Effects 0.000 description 1
- 230000002489 hematologic effect Effects 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 238000006698 hydrazinolysis reaction Methods 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical group I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 239000012051 hydrophobic carrier Substances 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 230000004047 hyperresponsiveness Effects 0.000 description 1
- 201000006362 hypersensitivity vasculitis Diseases 0.000 description 1
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 208000037880 idiopathic vasculitis Diseases 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical group O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 208000026876 intravascular large B-cell lymphoma Diseases 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 229940084651 iressa Drugs 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 1
- ULYZAYCEDJDHCC-UHFFFAOYSA-N isopropyl chloride Chemical compound CC(C)Cl ULYZAYCEDJDHCC-UHFFFAOYSA-N 0.000 description 1
- 210000001503 joint Anatomy 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 229960001614 levamisole Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000011344 liquid material Substances 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- DHMTURDWPRKSOA-RUZDIDTESA-N lonafarnib Chemical compound C1CN(C(=O)N)CCC1CC(=O)N1CCC([C@@H]2C3=C(Br)C=C(Cl)C=C3CCC3=CC(Br)=CN=C32)CC1 DHMTURDWPRKSOA-RUZDIDTESA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 208000003747 lymphoid leukemia Diseases 0.000 description 1
- 210000003563 lymphoid tissue Anatomy 0.000 description 1
- 208000006116 lymphomatoid granulomatosis Diseases 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 229940124302 mTOR inhibitor Drugs 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229960002985 medroxyprogesterone acetate Drugs 0.000 description 1
- PSGAAPLEWMOORI-PEINSRQWSA-N medroxyprogesterone acetate Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 PSGAAPLEWMOORI-PEINSRQWSA-N 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- 229960004296 megestrol acetate Drugs 0.000 description 1
- 102000006240 membrane receptors Human genes 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 238000003328 mesylation reaction Methods 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- 229960001566 methyltestosterone Drugs 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 206010063344 microscopic polyangiitis Diseases 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 201000005328 monoclonal gammopathy of uncertain significance Diseases 0.000 description 1
- PYLWMHQQBFSUBP-UHFFFAOYSA-N monofluorobenzene Chemical compound FC1=CC=CC=C1 PYLWMHQQBFSUBP-UHFFFAOYSA-N 0.000 description 1
- 229940126619 mouse monoclonal antibody Drugs 0.000 description 1
- 208000001725 mucocutaneous lymph node syndrome Diseases 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 206010028537 myelofibrosis Diseases 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- MBHINSULENHCMF-UHFFFAOYSA-N n,n-dimethylpropanamide Chemical compound CCC(=O)N(C)C MBHINSULENHCMF-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- AGVKXDPPPSLISR-UHFFFAOYSA-N n-ethylcyclohexanamine Chemical compound CCNC1CCCCC1 AGVKXDPPPSLISR-UHFFFAOYSA-N 0.000 description 1
- 229940073569 n-methylephedrine Drugs 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 229960002450 ofatumumab Drugs 0.000 description 1
- 231100000590 oncogenic Toxicity 0.000 description 1
- 230000002246 oncogenic effect Effects 0.000 description 1
- 230000006548 oncogenic transformation Effects 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- PUPAWTXNPAJCHR-UHFFFAOYSA-N oxazaborole Chemical compound O1C=CB=N1 PUPAWTXNPAJCHR-UHFFFAOYSA-N 0.000 description 1
- 238000010979 pH adjustment Methods 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- JYVLIDXNZAXMDK-UHFFFAOYSA-N pentan-2-ol Chemical compound CCCC(C)O JYVLIDXNZAXMDK-UHFFFAOYSA-N 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- DLNKOYKMWOXYQA-APPZFPTMSA-N phenylpropanolamine Chemical compound C[C@@H](N)[C@H](O)C1=CC=CC=C1 DLNKOYKMWOXYQA-APPZFPTMSA-N 0.000 description 1
- 229960000395 phenylpropanolamine Drugs 0.000 description 1
- 238000003566 phosphorylation assay Methods 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 1
- 229960000952 pipobroman Drugs 0.000 description 1
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 208000031223 plasma cell leukemia Diseases 0.000 description 1
- 210000003720 plasmablast Anatomy 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 238000005498 polishing Methods 0.000 description 1
- 201000006292 polyarteritis nodosa Diseases 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 238000011176 pooling Methods 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 description 1
- 239000003207 proteasome inhibitor Substances 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 239000003586 protic polar solvent Substances 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 229960003876 ranibizumab Drugs 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 229940120975 revlimid Drugs 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 238000003118 sandwich ELISA Methods 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 150000003333 secondary alcohols Chemical class 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 239000012056 semi-solid material Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 201000009295 smoldering myeloma Diseases 0.000 description 1
- 208000010721 smoldering plasma cell myeloma Diseases 0.000 description 1
- QJQRNDGUWQVAEV-AAFSJPGBSA-M sodium bisulfite adduct Chemical compound [Na+].[O-]S(=O)(=O)C([C@H]1N(C(C2=C3)=O)C=C(C1)/C=C/C(=O)N(C)C)NC2=CC1=C3OCO1 QJQRNDGUWQVAEV-AAFSJPGBSA-M 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 206010062113 splenic marginal zone lymphoma Diseases 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000012058 sterile packaged powder Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 230000004960 subcellular localization Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229940120982 tarceva Drugs 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- 206010043207 temporal arteritis Diseases 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- YQPJFHSRVUXUAS-MOSLJXCSSA-N tert-butyl N-[(E)-1-[5-chloro-2-ethoxy-4-fluoro-3-[(3R)-5-oxopyrrolidin-3-yl]phenyl]ethylideneamino]carbamate Chemical compound ClC=1C(=C(C(=C(C=1)\C(\C)=N\NC(=O)OC(C)(C)C)OCC)[C@@H]1CNC(C1)=O)F YQPJFHSRVUXUAS-MOSLJXCSSA-N 0.000 description 1
- ZLCLJRWHJYMKFE-QWRGUYRKSA-N tert-butyl N-[[(1S)-1-[5-chloro-2-ethoxy-4-fluoro-3-[(3R)-5-oxopyrrolidin-3-yl]phenyl]ethyl]amino]carbamate Chemical compound ClC=1C(=C(C(=C(C=1)[C@H](C)NNC(=O)OC(C)(C)C)OCC)[C@@H]1CNC(C1)=O)F ZLCLJRWHJYMKFE-QWRGUYRKSA-N 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- OULAJFUGPPVRBK-UHFFFAOYSA-N tetratriacontan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCO OULAJFUGPPVRBK-UHFFFAOYSA-N 0.000 description 1
- 229950006410 tezacitabine Drugs 0.000 description 1
- 229960003433 thalidomide Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 230000002992 thymic effect Effects 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 1
- 229960005026 toremifene Drugs 0.000 description 1
- 229960005267 tositumomab Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 229940066958 treanda Drugs 0.000 description 1
- 229950001353 tretamine Drugs 0.000 description 1
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- 229960005294 triamcinolone Drugs 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- YFNKIDBQEZZDLK-UHFFFAOYSA-N triglyme Chemical compound COCCOCCOCCOC YFNKIDBQEZZDLK-UHFFFAOYSA-N 0.000 description 1
- 229940086984 trisenox Drugs 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 238000009827 uniform distribution Methods 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 229960001055 uracil mustard Drugs 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 238000001238 wet grinding Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000003871 white petrolatum Substances 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/14—Vasoprotectives; Antihaemorrhoidals; Drugs for varicose therapy; Capillary stabilisers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/18—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D207/22—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/24—Oxygen or sulfur atoms
- C07D207/26—2-Pyrrolidones
- C07D207/263—2-Pyrrolidones with only hydrogen atoms or radicals containing only hydrogen and carbon atoms directly attached to other ring carbon atoms
- C07D207/267—2-Pyrrolidones with only hydrogen atoms or radicals containing only hydrogen and carbon atoms directly attached to other ring carbon atoms with only hydrogen atoms or radicals containing only hydrogen and carbon atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Pulmonology (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Diabetes (AREA)
- Urology & Nephrology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pain & Pain Management (AREA)
- Oncology (AREA)
- Epidemiology (AREA)
- Neurology (AREA)
- Transplantation (AREA)
- Dermatology (AREA)
- Vascular Medicine (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Obesity (AREA)
- Ophthalmology & Optometry (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyrrole Compounds (AREA)
Abstract
La presente solicitud proporciona procesos para preparar (R)-4-(3-((S)-1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil) -5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona, que es útil como inhibidor de la fosfoinositida 3-quinasa-delta (PI3Kδ), así como una forma de sal e intermedios relacionados con la misma. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Procesos para la preparación de un inhibidor de PI3K
Esta solicitud reivindica el beneficio de la U.S. N.° de serie 62/121,697, presentada el 27 de febrero de 2015.
CAMPO TÉCNICO
La presente solicitud proporciona un proceso para preparar (R)-4-(3-((S)-1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona, que es útil como inhibidor de la fosfoinosítido 3-quinasa-delta (PI3K5), así como una forma de sal y productos intermedios relacionados con la misma.
ANTECEDENTES
Las fosfoinosítido 3-quinasas (PI3K) pertenecen a una familia grande de quinasas señalizadoras de lípidos que fosforilan fosfoinosítidos en la posición D3 del anillo de inositol (Cantley, Science, 2002, 296(5573): 1655-7). Las PI3K se dividen en tres clases (clase I, II y III) de acuerdo con su estructura, regulación y especificidad de sustrato. Las PI3K de clase I, que incluyen PI3Ka, PI3Kp, PI3Ky y PI3K5, son una familia de quinasas de lípidos y proteínas de especificidad dual que catalizan la fosforilación de fosfatidilinosito-4,5-bisfosfato (PIP2) dando lugar a fosfatidilinosito-3,4,5-trisfosfato (PIP3). El PIP3 funciona como un segundo mensajero que controla una serie de procesos celulares, incluyendo el crecimiento, la supervivencia, la adhesión y la migración. Las cuatro isoformas de PI3K de clase I existen como heterodímeros compuestos por una subunidad catalítica (p110) y una subunidad reguladora estrechamente asociada que controla su expresión, activación y localización subcelular. PI3Ka, PI3Kp y PI3K5 se asocian con una subunidad reguladora conocida como p85 y son activados por factores de crecimiento y citoquinas a través de un mecanismo dependiente de tirosinas quinasas (Jiménez, et al., J Biol Chem., 2002, 277(44): 41556-62) mientras que PI3Ky se asocia con dos subunidades reguladoras (p101 y p84) y su activación es impulsada por la activación de receptores acoplados a proteína G (Brock, et al., J Cell Biol., 2003, 160(1):89-99). PI3Ka y PI3Kp se expresan ubicuamente. Por el contrario, PI3Ky y PI3K5 se expresan predominantemente en leucocitos (Vanhaesebroeck, et al., Trends Biochem Sci., 2005, 30(4):194-204).
La distribución tisular diferencial de las isoformas de PI3K influye en sus distintas funciones biológicas. La ablación genética de PI3Ka o PI3Kp da como resultado la letalidad embrionaria, lo que indica que PI3Ka y PI3Kp tienen funciones esenciales y no redundantes, por lo menos durante el desarrollo (Vanhaesebroeck, et al., 2005). Por el contrario, los ratones que carecen de PI3Ky y PI3K5 son viables, fértiles y tienen una duración de vida normal, aunque muestran un sistema inmunológico alterado. La deficiencia de PI3Ky lleva a un reclutamiento deteriorado de macrófagos y neutrófilos a los sitios de inflamación así como a la activación de células T deteriorada (Sasaki, et al., Science, 2000, 287(5455):1040-6). Los ratones mutantes en PI3K5 tienen defectos específicos en la señalización de las células B que llevan a un desarrollo deteriorado de las células B y respuestas de anticuerpos reducidas después de la estimulación del antígeno (Clayton, et al., J Exp Med. 2002, 196(6):753-63; Jou, et al., Mol Cell Biol. 2002, 22(24):8580-91; Okkenhaug et al., Science, 2002, 297(5583):1031-4).
Los fenotipos de los ratones mutantes en PI3Ky y PI3K5 sugieren que estas enzimas pueden desempeñar una función en la inflamación y otras enfermedades de base inmunitaria y esto se confirma en modelos preclínicos. Los ratones mutantes en PI3Ky están ampliamente protegidos de la enfermedad en modelos de ratón de artritis reumatoide (AR) y asma (Camps, et al., Nat Med. 2005, 11(9):936-43; Thomas, et al., Eur. J Immunol. 2005, 35(4):1283-91). Además, se demostró que el tratamiento de ratones de tipo salvaje con un inhibidor selectivo de PI3Ky reduce la glomerulonefritis y prolonga la supervivencia en el modelo MRL-lpr de nefritis lúpica sistémica (SEL) y suprime la inflamación y el daño articular en modelos de AR (Barber, et al., Nat Med.2005, 11 (9):933-5; Camps, et al., 2005). De manera similar, se ha demostrado que tanto los ratones mutantes en PI3K5 como los ratones de tipo salvaje tratados con un inhibidor selectivo de PI3Kó han atenuado la inflamación alérgica de las vías respiratorias y la hipersensibilidad en un modelo de asma de ratón (Ali, et al., Nature. 2004, 431 (7011):1007-11; Lee, et al., FASeB J. 2006, 20(3):455-65) y han atenuado la enfermedad en un modelo de AR (Randis, et al., Eur. J. Immunol., 2008, 38(5) :1215-24).
Se ha demostrado que la proliferación de células B desempeña un papel importante en el desarrollo de enfermedades autoinmunes inflamatorias (Puri, Frontiers in Immunology (2012), 3(256), 1-16; Walsh, Kidney International (2007) 72, 676-682). Por ejemplo, las células B respaldan la autorreactividad de las células T, un componente importante de las enfermedades autoinmunes inflamatorias. Una vez activadas y maduradas, las células B pueden transitar hacia los sitios de inflamación y reclutan células inflamatorias o se diferencian en plasmablastos. Por tanto, la actividad de las células B puede verse afectada dirigiéndose a citoquinas estimuladoras de células B, receptores de superficie de células B, o mediante el agotamiento de las células B. Se ha demostrado que el rituximab, un anticuerpo monoclonal quimérico de ratón/humano de IgG1 k dirigido contra el receptor CD20 de superficie de células B, agota las células B CD20+. Se ha demostrado que el uso de rituximab tiene eficacia en el
tratamiento de púrpura trombocitopénica idiopática, anemia hemolítica autoinmune o vasculitis. Por ejemplo, el tratamiento con rituximab dio como resultado la remisión de la enfermedad en pacientes que padecían vasculitis sistémica asociada a anticuerpos del citoplasma antineutrófilos (ANCA) (AASV) con agotamiento demostrado de células B periféricas (Walsh, 2007; Lovric, Nephrol Dial Transplant (2009) 24:179-185). De manera similar, se ha informado de una respuesta completa de un tercio a dos tercios de los pacientes que tenían vasculitis por crioglobulinemia mixta después del tratamiento con rituximab, incluyendo los pacientes que presentaban una forma grave de vasculitis que era resistente o intolerante a otros tratamientos (Cacoub, Ann Rheum Dis 2008;67: 283-287). De manera similar, se ha demostrado que el rituximab tiene eficacia en el tratamiento de pacientes con púrpura trombocitopénica idiopática (o púrpura trombocitopénica inmune) (Garvey, British Journal of Hematology, (2008) 141, 149-169; Godeau, Blood (2008), 112(4), 999-1004; Medeo, European Journal of Hematology, (2008) 81, 165-169) y anemia hemolítica autoinmune (Garvey, British Journal of Hematology, (2008) 141, 149-169).
La señalización de PI3K5 se ha relacionado con la supervivencia, migración y activación de células B (Puri, Frontiers in Immunology, 2012, 3(256), 1-16, en las páginas 1-5; y Clayton, J Exp Med, 2002, 196(6):753-63). Por ejemplo, se requiere PI3K5 para la activación de células B dependiente de antígeno impulsada por el receptor de células B. Al bloquear la adhesión, supervivencia, activación y proliferación de las células B, la inhibición de PI3K5 puede deteriorar la capacidad de las células B para activar las células T, previniendo su activación y reduciendo la secreción de autoanticuerpos y citoquinas proinflamatorias. Por tanto, por su capacidad para inhibir la activación de las células B, se esperaría que los inhibidores de PI3K5 trataran enfermedades mediadas por células B que se podrían tratar mediante métodos similares, como el agotamiento de las células B por rituximab. En efecto, Se ha demostrado que los inhibidores de PI3K5 son modelos de ratón útiles de varias enfermedades autoinmunes que también pueden tratarse con rituximab, como la artritis (Puri (2012)). Además, las células B de tipo innato, que están vinculadas a la autoinmunidad son sensibles a la actividad de PI3K5, ya que las células MZ y B-1 están casi ausentes en ratones que carecen del gen p1105 (Puri (2012). Los inhibidores de PI3K5 pueden reducir el tránsito y la activación de células MZ y B-1, que están implicadas en enfermedades autoinmunes.
Además de su función potencial en las enfermedades inflamatorias, las cuatro isoformas de PI3K de clase I pueden desempeñar un papel en el cáncer. El gen que codifica p110a está mutado con frecuencia en cánceres comunes, incluyendo los de mama, próstata, colon y endometrio (Samuels, et al., Science, 2004, 304(5670):554; Samuels, et al., Curr Opin Oncol. 2006, 18(1):77-82). El ochenta por ciento de estas mutaciones están representadas por una de las tres sustituciones de aminoácidos en los dominios helicoidales o de quinasas de la enzima y llevan a una regulación por incremento significativa de la actividad de quinasas que da como resultado una transformación oncogénica en cultivos celulares y en modelos animales (Kang, et al., Proc Natl Acad Sci USA. 2005, 102(3):802-7; Bader et al., Proc Natl Acad Sci USA. 2006, 103(5):1475-9). No se han identificado tales mutaciones en las otras isoformas de PI3K, aunque hay evidencias de que pueden contribuir al desarrollo y progresión de enfermedades malignas. Se observa una sobreexpresión consistente de PI3K5 en la leucemia mieloblástica aguda (Sujobert, et al., Blood, 2005, 106(3):1063-6) y los inhibidores de PI3K5 pueden evitar el crecimiento de células leucémicas (Billottet et al., Oncogene. 2006, 25(50):6648-59). Se observa una expresión elevada de PI3Ky en la leucemia mieloide crónica (Hickey et al., J Biol. Chem. 2006, 281(5):2441-50). También se han observado alteraciones en la expresión de PI3Kp, PI3Ky y PI3K5 en cánceres de cerebro, colon y vejiga (Benistant, et al., Oncogene, 2000, 19(44):5083-90; Mizoguchi et al., Brain Pathol. 2004, 14(4):372-7; Knobbe et al., Neuropathol Appl Neurobiol. 2005, 31(5):486-90). Además, se ha demostrado que todas estas isoformas son oncogénicas en cultivos celulares (Kang, et al., 2006).
Por estas razones, hay una necesidad de desarrollar nuevos inhibidores de PI3K que puedan usarse en trastornos inflamatorios, enfermedades autoinmunes y cáncer. Esta invención está dirigida a esta y otras necesidades.
SUMARIO
La presente solicitud proporciona procesos para preparar un compuesto de Fórmula I:

o una sal farmacéuticamente aceptable del mismo, que es útil como un inhibidor de PI3K5.
En particular, la presente solicitud proporciona un proceso para preparar un compuesto de Fórmula I, que comprende hacer reaccionar un compuesto de Fórmula XVI:

con acetato de formamidina para formar dicho compuesto de Fórmula I:

La presente solicitud proporciona además un compuesto de Fórmula XIV:

o una sal farmacéuticamente aceptable del mismo.
La presente solicitud también proporciona un compuesto de Fórmula XV:

farmacéuticamente aceptable del mismo.
La presente solicitud proporciona adicionalmente un compuesto de Fórmula XVI:

farmacéuticamente aceptable del mismo.
La presente solicitud también proporciona un compuesto de Fórmula XX:

farmacéuticamente aceptable del mismo.
La presente solicitud también proporciona un compuesto de Fórmula XXI:

o una sal farmacéuticamente aceptable del mismo.
DESCRIPCIÓN DE LOS DIBUJOS FIG. 1 muestra un termograma DSC representativo de la sal del Ejemplo 3.
FIG. 2 muestra datos de TGA representativos de la sal del Ejemplo 3.
FIG. 3 muestra un patrón de XRPD representativo de la sal del Ejemplo 3.
DESCRIPCIÓN DETALLADA
Compuestos y sales
La presente solicitud proporciona procesos para preparar un compuesto de Fórmula I:

o una sal farmacéuticamente aceptable del mismo, que es útil como inhibidor de PI3K5, donde Et es etilo.
El proceso de la presente invención puede ser útil en la preparación de una sal del compuesto de Fórmula I. Por consiguiente, en algunos realizaciones, el proceso de la invención puede usarse para la preparación de sal de ácido clorhídrico de 4-(3-(1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona. En algunas realizaciones, el proceso de la invención puede usarse para la preparación de sal de ácido clorhídrico de (R)-4-(3-((S)-1-(4-amino-3-metil-1H-pirazolo[3,4-d ]pirimidin-1 -il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona. La sal puede ser una relación estequiométrica 1:1 de (R)-4-(3-((S)-1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1 -il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona a ácido clorhídrico.
Diferentes formas de la misma sustancia tienen diferentes propiedades intensivas relacionadas, por ejemplo, con la higroscopicidad, solubilidad, estabilidad y similares. Las formas con altos puntos de fusión a menudo tienen una buena estabilidad termodinámica, lo que es ventajoso para prolongar la vida útil de las formulaciones de fármacos que contienen la forma sólida. Las formas con puntos de fusión más bajos a menudo son menos estables termodinámicamente, pero son ventajosas ya que tienen una mayor solubilidad en agua, lo que se traduce en una mayor biodisponibilidad del fármaco. Las formas que son débilmente higroscópicas son deseables por su estabilidad al calor y la humedad y son resistentes a la degradación durante un almacenamiento prolongado.
La sal de ácido clorhídrico del compuesto de Fórmula I pude ser cristalina. Como se usa en la presente, "cristalino" o "forma cristalina" se refiere a una determinada configuración de entramado de una sustancia cristalina. Las diferentes formas cristalinas de la misma sustancia suelen tener diferentes entramados cristalinos (por ejemplo, celdas unitarias) que se atribuyen a diferentes propiedades físicas que son características de cada una de las formas cristalinas. En algunos casos, diferentes configuraciones de entramado tienen diferente contenido de agua o solvente.
Las diferentes formas de sal pueden identificarse mediante métodos de caracterización de estado sólido, como por difracción de rayos X en polvo (XRPD). Otros métodos de caracterización, como calorimetría diferencial de barrido (DSC), análisis termogravimétrico (TGA), sorción dinámica de vapor (DVS) y similares, ayudan adicionalmente a identificar la forma, así como a determinar la estabilidad y el contenido de agua/solvente.
Un patrón XRPD de reflexiones (picos) se considera generalmente una huella de una forma cristalina particular. Es bien sabido que las intensidades relativas de los picos de XRPD pueden variar ampliamente dependiendo, entre otras cosas, de la técnica de preparación de la muestra, la distribución del tamaño del cristal, los
varios filtros usados, el procedimiento de montaje de la muestra, y el instrumento particular empleado. En algunos casos, pueden observarse nuevos picos o pueden desaparecer picos existentes, dependiendo del tipo de instrumento o la configuración. Como se usa en la presente, el término "pico" se refiere a una reflexión que tiene una altura/intensidad relativa de por lo menos aproximadamente el 4% de la altura/intensidad del pico máxima. Además, la variación del instrumento y otros factores pueden afectar los valores 2-theta. Por tanto, las asignaciones de picos, como las que se informan en la presente, pueden variar en más o menos aproximadamente 0,2° (2-theta), y el término "sustancialmente" y "aproximadamente" como se usan en el contexto de XRPD en la presente se entiende que abarca las variaciones mencionadas anteriormente.
En algunas realizaciones, la sal de ácido clorhídrico del compuesto de Fórmula I tiene por lo menos un pico de XRPD, en términos de 2-theta, seleccionado de aproximadamente 11,3°, aproximadamente 16,4°, aproximadamente 21,0°, aproximadamente 23,0°, aproximadamente 28,1°, aproximadamente 31,2° y aproximadamente 32,8°. En algunas realizaciones, la sal de ácido clorhídrico del compuesto de Fórmula I tiene por lo menos dos picos de XRPD, en términos de 2-theta, seleccionados de aproximadamente 11,3°, aproximadamente 16,4°, aproximadamente 21,0°, aproximadamente 23,0°, aproximadamente 28,1°, aproximadamente 31,2° y aproximadamente 32,8°. En algunas realizaciones, la sal de ácido clorhídrico del compuesto de Fórmula I tiene por lo menos tres picos de XRPD, en términos de 2-theta, seleccionados de aproximadamente 11,3°, aproximadamente 16,4°, aproximadamente 21,0°, aproximadamente 23,0°, aproximadamente 28,1°, aproximadamente 31,2° y aproximadamente 32,8°. En algunas realizaciones, la sal de ácido clorhídrico del compuesto de Fórmula I tiene por lo menos cuatro picos de XRPD, en términos de 2-theta, seleccionados de aproximadamente 11,3°, aproximadamente 16,4°, aproximadamente 21,0°, aproximadamente 23,0°, aproximadamente 28,1°, aproximadamente 31,2° y aproximadamente 32,8°. En algunas realizaciones, la sal de ácido clorhídrico del compuesto de Fórmula I tiene por lo menos cinco picos de XRPD, en términos de 2-theta, seleccionados de aproximadamente 11,3°, aproximadamente 16,4°, aproximadamente 21,0°, aproximadamente 23,0°, aproximadamente 28,1°, aproximadamente 31,2° y aproximadamente 32,8°. En algunas realizaciones, la sal de ácido clorhídrico del compuesto de Fórmula I tiene un perfil de XRPD sustancialmente como se muestra en la Figura 3.
De la misma manera, las lecturas de temperatura en relación con DSC, TGA u otros experimentos térmicos pueden variar aproximadamente en ± 3° C dependiendo del instrumento, las configuraciones particulares, preparación de la muestra, etc. Por consiguiente, una forma cristalina informada en la presente tiene un termograma de DSC "sustancialmente" como se muestra en cualquiera de las figuras o se entiende que el término "aproximadamente" acomoda tal variación. En algunas realizaciones, la sal de ácido clorhídrico del compuesto de Fórmula I tiene un termograma de DSC que tiene un pico endotérmico a aproximadamente 207° C. En algunas realizaciones, la sal de ácido clorhídrico del compuesto de Fórmula I tiene un termograma de DSC sustancialmente como se muestra en la Figura 1. En algunas realizaciones, la sal de ácido clorhídrico del compuesto de Fórmula I tiene un termograma de TGA sustancialmente como se muestra en la Figura 2.
En algunas realizaciones, las sales y compuestos descritos en la presente (por ejemplo, el compuesto de Fórmula I o la sal de ácido clorhídrico del compuesto de Fórmula I) están sustancialmente aislados. Por "sustancialmente aislado" se entiende que la sal o el compuesto está por lo menos parcial o sustancialmente separado del entorno en el que se formó o detectó. La separación parcial puede incluir, por ejemplo, una composición enriquecida en las sales descritas en la presente. La separación sustancial puede incluir composiciones que contienen por lo menos aproximadamente un 50%, por lo menos aproximadamente un 60%, por lo menos aproximadamente un 70%, por lo menos aproximadamente un 80%, por lo menos aproximadamente un 90%, por lo menos aproximadamente un 95%, por lo menos aproximadamente un 97%, o por lo menos aproximadamente un 99% en peso de las sales descritas en la presente, o sal de las mismas. Los métodos para aislar compuestos y sus sales son rutinarios en la técnica.
Productos intermedios
La presente solicitud proporciona además productos intermedios que son útiles en la preparación del compuesto de Fórmula I.
Por consiguiente, en algunas realizaciones, la presente solicitud proporciona un compuesto de Fórmula XIV:

farmacéuticamente aceptable del mismo.
La presente solicitud proporciona además un compuesto de Fórmula XV:

farmacéuticamente aceptable del mismo.
La presente solicitud proporciona además un compuesto de Fórmula XVI:

farmacéuticamente aceptable del mismo.
La presente solicitud proporciona además un compuesto de fórmula XIX:

farmacéuticamente aceptable del mismo.
La presente solicitud proporciona además un compuesto de Fórmula XX:

o una sal farmacéuticamente aceptable del mismo.
La presente solicitud proporciona además un compuesto de Fórmula XXI:

o una sal farmacéuticamente aceptable del mismo.
Procesos
La presente solicitud proporciona además un proceso para preparar una sal de Fórmula I:

En algunas realizaciones, el proceso comprende hacer reaccionar un compuesto de Fórmula I:

con ácido clorhídrico para formar dicha sal.
En algunas realizaciones, dicho ácido clorhídrico es ácido clorhídrico acuoso 1 M.
En algunas realizaciones, se usan de aproximadamente 3,3 a aproximadamente 3,7 equivalentes de ácido clorhídrico en base a 1 equivalente del compuesto de fórmula I.
En algunas realizaciones, dicha reacción se realiza a una temperatura de aproximadamente 45° C a aproximadamente 55° C.
En algunas realizaciones, el proceso comprende:
añadir ácido clorhídrico al compuesto de Fórmula I a temperatura ambiente para formar una lechada; calentar dicha lechada a una temperatura de aproximadamente 45° C a aproximadamente 55° C para formar una solución; y
enfriar la solución a una temperatura de aproximadamente 0° C a aproximadamente 5° C para cristalizar dicha sal.
En algunas realizaciones, el proceso comprende hacer reaccionar un compuesto de Fórmula XVI:

con acetato de formamidina para formar un compuesto de Fórmula I:

En algunas realizaciones, dicha reacción del compuesto de Fórmula XVI con acetato de formamidina se realiza en un componente solvente que comprende 1,2-etanodiol.
En algunas realizaciones, dicha reacción del compuesto de Fórmula XVI con acetato de formamidina se realiza a una temperatura de aproximadamente 100°C a aproximadamente 105°C.
En algunas realizaciones, se usan de aproximadamente 8 a aproximadamente 10 equivalentes de acetato de formamidina basado en 1 equivalente del compuesto de Fórmula XVI.
En algunas realizaciones, el proceso comprende además preparar el compuesto de Fórmula XVI mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula XV:

con (1-etoxietilideno)malononitrilo en presencia de una amina terciaria.
En algunas realizaciones, dicha amina terciaria es N-metilpirrolidinona.
En algunas realizaciones, dicha reacción del compuesto de Fórmula XV con (1-etoxietiliden)malononitrilo se realiza aproximadamente a temperatura ambiente.
En algunas realizaciones, el proceso comprende además preparar el compuesto de Fórmula XV mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula XlV-a:

con hidrazina en presencia de una amina terciaria, en donde P1 es alquilsulfonilo C1-6.
En algunas realizaciones, dicha amina terciaria es N-metilpirrolidinona.
En algunas realizaciones, dicha reacción del compuesto de Fórmula XlV-a con hidrazina se realiza a una temperatura de aproximadamente 35° C a aproximadamente 60° C.
En algunas realizaciones, dicha reacción del compuesto de Fórmula XlV-a con hidrazina se realiza en un componente solvente que comprende diclorometano.
En algunas realizaciones, P1 es un grupo metanosulfonilo.
En algunas realizaciones, el proceso comprende además preparar el compuesto de Fórmula XIV mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula XIII:

con haluro de alquilsulfonilo Ci-6 en presencia de una amina terciaria.
En algunas realizaciones, dicho haluro de alquilsulfonilo Ci-6 es cloruro de metanosulfonilo.
En algunas realizaciones, dicha amina terciaria es N,N-diisopropiletilamina.
En algunas realizaciones, se usa de aproximadamente 1,1 a aproximadamente 1,5 equivalentes de haluro de alquilsulfonilo en base a 1 equivalente del compuesto de Fórmula XIII.
En algunas realizaciones, dicha reacción de dicho compuesto de Fórmula XIII con haluro de alquilsulfonilo C1-6 se realiza a una temperatura de aproximadamente -10° C a aproximadamente 5° C.
En algunas realizaciones, dicha reacción de dicho compuesto de fórmula XIII con haluro de alquilsulfonilo C1-6 se realiza en un componente solvente que comprende diclorometano.
En algunas realizaciones, los pasos de: (i) hacer reaccionar dicho compuesto de Fórmula XIII con haluro de alquilsulfonilo C1-6; (ii) hacer reaccionar dicho compuesto de Fórmula XlV-a con hidrazina en presencia de una amina terciaria para formar un compuesto de Fórmula XV; y (iii) hacer reaccionar dicho compuesto de Fórmula XV con acetato de formamidina para formar un compuesto de Fórmula XVI se lleva a cabo en el mismo recipiente sin aislamiento del compuesto de Fórmula XlV-a o el compuesto de Fórmula XV.
En algunas realizaciones, el proceso comprende además preparar el compuesto de Fórmula XVI mediante un proceso que comprende hacer reaccionar una sal de Fórmula XV-a:

con (1-etoxietiliden)malononitrilo en presencia de una amina terciaria, en donde TsOH es ácido p-toluenosulfónico.
En algunas realizaciones, dicha amina terciaria es N,N-diisopropiletilamina.
En algunas realizaciones, dicha reacción de una sal de Fórmula XV-a con (1-etoxietiliden)malononitrilo se realiza aproximadamente a temperatura ambiente.
En algunas realizaciones, se usan de aproximadamente 1,3 a aproximadamente 1,6 equivalentes de (1-etoxietiliden)malononitrilo en base a 1 equivalente de la sal de Fórmula XV-a.
En algunas realizaciones, dicha reacción de la sal de Fórmula XV-a con (1-etoxietiliden)malononitrilo se realiza en un componente solvente que comprende etanol.
En algunas realizaciones, el proceso comprende además preparar la sal de Fórmula XV-a mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula XXI:

con ácido p-toluenosulfónico, en donde Boc es terc-butoxicarbonilo.
En algunas realizaciones, dicho ácido p-toluenosulfónico es ácido p-toluenosulfónico monohidrato.
En algunas realizaciones, se usan de aproximadamente 1,3 a aproximadamente 1,6 equivalentes de ácido p-toluenosulfónico en base a 1 equivalente del compuesto de Fórmula XXI.
En algunas realizaciones, dicha reacción de dicho compuesto de fórmula XXI con ácido p-toluenosulfónico se realiza a una temperatura de aproximadamente 45°C a aproximadamente 65°C.
En algunas realizaciones, la reacción de dicho compuesto de Fórmula XXI con ácido p-toluenosulfónico se realiza en un componente solvente que comprende etanol.
En algunas realizaciones, los pasos de: (i) hacer reaccionar dicho compuesto de Fórmula XXI con ácido ptoluenosulfónico para formar una sal de Fórmula XV-a; y (ii) hacer reaccionar dicha sal de Fórmula XV-a con (1-etoxietiliden)malononitrilo se realizan en el mismo recipiente sin aislamiento de la sal de Fórmula XV-a.
En algunas realizaciones, el proceso comprende además preparar el compuesto de Fórmula XXI mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula XX:

con hidrógeno gaseoso en presencia de uno o más catalizadores de hidrogenación seleccionados independientemente, en donde el Boc es ferc-butoxicarbonilo.
En algunas realizaciones, dicha reacción del compuesto de fórmula XX con gas de hidrógeno se realiza en
presencia de dos catalizadores de hidrogenación seleccionados independientemente.
En algunas realizaciones, un catalizador de hidrogenación es bis(1,5-ciclooctadieno)rodio(I)tetrafluoroborato y el otro es (R)-(-)-1 -{(S)-2-[bis(4-trifluorometilfenil)fosfina]ferrocenil}etil-di-t-butilfosfina.
En algunas realizaciones, se usan de aproximadamente 13,5 a aproximadamente 14,5 equivalentes de compuesto de Fórmula XX en base a 1 equivalente de bis(1,5-ciclooctadieno)rodio(I)tetrafluoroborato.
En algunas realizaciones, se usan de aproximadamente 12 a aproximadamente 13 equivalentes del compuesto de Fórmula XX en base a 1 equivalente de (R)-(-)-1-{(S)-2-[bis(4-trifluorometilfenil)fosfina]ferrocenil}etildi-t-butilfosfina.
En algunas realizaciones, dicha reacción del compuesto de fórmula XX con gas de hidrógeno se realiza aproximadamente a temperatura ambiente.
En algunas realizaciones, dicha reacción del compuesto de fórmula XX con gas de hidrógeno se realiza en un componente solvente que comprende metanol.
En algunas realizaciones, el proceso comprende además preparar el compuesto de Fórmula XX mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula XIX:

con carbazato de f-butilo.
En algunas realizaciones, dicha reacción del compuesto de Fórmula XIX con carbazato de t-butilo se realiza a una temperatura de aproximadamente 60° C a aproximadamente 70° C.
En algunas realizaciones, dicha reacción del compuesto de Fórmula XIX con carbazato de t-butilo se realiza en un componente solvente que comprende metanol.
En algunas realizaciones, el proceso comprende además preparar el compuesto de Fórmula XIX mediante un proceso que comprende oxidar un compuesto de Fórmula XIII-a:

en presencia de un agente oxidante.
En algunas realizaciones, dicho agente oxidante es peryodinano de Dess-Martin.
En algunas realizaciones, se usan de aproximadamente 1,2 a aproximadamente 1,7 equivalentes de dicho agente oxidante en base a 1 equivalente del compuesto de Fórmula XIII-a.
En algunas realizaciones, dicha oxidación del compuesto de Fórmula XIII-a se realiza aproximadamente a
temperatura ambiente.
En algunas realizaciones, dicha oxidación del compuesto de Fórmula XlII-a se realiza en un componente solvente que comprende diclorometano.
En algunas realizaciones, dicho compuesto de Fórmula XIII se prepara mediante un proceso que comprende calentar un compuesto de Fórmula XII:

en presencia de un componente solvente.
En algunas realizaciones, dicho calentamiento se realiza a una temperatura de aproximadamente 95° C a aproximadamente 105° C.
En algunas realizaciones, dicho componente solvente comprende 1,4-dioxano.
En algunas realizaciones, dicho componente solvente comprende tolueno.
En algunas realizaciones, dicho componente solvente comprende 1,4-dioxano y tolueno.
En algunas realizaciones, dicho compuesto de Fórmula XII se prepara mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula XI:

en presencia de una base fuerte.
En algunas realizaciones, dicha base fuerte es hidróxido de sodio.
En algunas realizaciones, dicha base fuerte es hidróxido de sodio acuoso 1 M.
En algunas realizaciones, dicha reacción se realiza a aproximadamente la temperatura ambiente.
En algunas realizaciones, dicho compuesto de Fórmula XI se prepara mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula X :

con gas hidrógeno en presencia de níquel Raney.
En algunas realizaciones, dicha reacción se realiza a una temperatura de aproximadamente 50° C a aproximadamente 70° C.
En algunas realizaciones, dicho compuesto de Fórmula X se prepara mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula IX:

en presencia de eterato de trifluoruro de boro, catalizador de (3aS)-1-metil-3,3-difeniltetrahidro-3H-pirrolo[1,2-c][1,3,2]oxazaborol ((S)-MeCBS), y un complejo de borano.
En algunas realizaciones, se usan de aproximadamente 0,03 a aproximadamente 0,07 equivalentes de eterato de trifluoruro de boro en base a 1 equivalente del compuesto de Fórmula IX.
En algunas realizaciones, se usan de aproximadamente 0,05 a aproximadamente 0,15 equivalentes de catalizador (3aS) -1-metil- 3,3-difeniltetrahidro-3H-pirrolo[1,2-c][1,3,2]oxazaborol ((S)-MeCBS) en base a 1 equivalente del compuesto de Fórmula IX.
En algunas realizaciones, dicho complejo de borano es un complejo de borano-THF 1,0 M en
THF.
En algunas realizaciones, se usan de aproximadamente 0,9 a aproximadamente 1,1 equivalentes de complejo de borano en base a 1 equivalente del compuesto de Fórmula IX.
En algunas realizaciones, dicha reacción se realiza aproximadamente a temperatura ambiente.
En algunas realizaciones, dicho compuesto de Fórmula IX se prepara mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula VIII:

con yodo en presencia de un componente solvente.
En algunas realizaciones, dicho componente solvente comprende acetona.
En algunas realizaciones, se usan de aproximadamente 0,75 a aproximadamente 1,25 equivalentes de yodo en base a 1 equivalente del compuesto de Fórmula VIII.
En algunas realizaciones, dicho compuesto de Fórmula VIII se prepara mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula VII:

con malonato de dimetilo en presencia de un catalizador.
En algunas realizaciones, dicho catalizador es (1S,2S)-N,N'-dibencilciclohexano-1,2-diamina-dibromoníquel (catalizador de Evans).
En algunas realizaciones, se usan de aproximadamente 1,1 a aproximadamente 1,3 equivalentes de malonato de dimetilo en base a 1 equivalente del compuesto de Fórmula VII.
En algunas realizaciones, se usan de aproximadamente 0,02 a aproximadamente 0,03 equivalentes de catalizador de metal de transición en base a 1 equivalente del compuesto de Fórmula VII.
En algunas realizaciones, dicho compuesto de Fórmula VII se prepara mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula VI:

con nitrometano en presencia de un ácido orgánico para formar una primera mezcla.
En algunas realizaciones, dicho ácido orgánico es ácido acético glacial.
En algunas realizaciones, se usan de aproximadamente 9,5 a aproximadamente 10,5 equivalentes de nitrometano en base a 1 equivalente del compuesto de Fórmula VI.
En algunas realizaciones, dicha reacción comprende además añadir una base de amina a dicha primera mezcla para formar una segunda mezcla.
En algunas realizaciones, dicha base de amina es bencilamina.
En algunas realizaciones, se usan de aproximadamente 0,2 a aproximadamente 0,3 equivalentes de base amina en base a 1 equivalente del compuesto de Fórmula VI.
En algunas realizaciones, dicha segunda mezcla se calienta de aproximadamente 55° C a aproximadamente 65°C.
En algunas realizaciones, dicho compuesto de Fórmula VI se prepara mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula V :

con un complejo de haluro de (alquilo C1-4)magnesio para formar una primera mezcla.
En algunas realizaciones, dicho complejo de haluro de (alquilo C1-4)magnesio es un complejo de cloruro de litio cloruro de isopropilmagnesio 1,3 M.
En algunas realizaciones, se usan de aproximadamente 1,1 a aproximadamente 1,3 equivalentes de dicho complejo de haluro de (alquilo C1-4)magnesio en base a 1 equivalente del compuesto de Fórmula V.
En algunas realizaciones, dicha reacción comprende además añadir N-formilmorfolina a dicha primera mezcla para formar una segunda mezcla.
En algunas realizaciones, se usan de aproximadamente 1,8 a aproximadamente 2,2 equivalentes de N-formilmorfolina en base a 1 equivalente del compuesto de Fórmula V.
En algunas realizaciones, dicha reacción se realiza a una temperatura de aproximadamente -5°C a aproximadamente 10°C.
En algunas realizaciones, dicho compuesto de Fórmula V se prepara de acuerdo con los procedimientos descritos en la Publicación de Estados Unidos N° 2013-0059835A1.
En algunas realizaciones, dicho compuesto de Fórmula VI se prepara mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula V-a:

con N,N-dimetilformamida en presencia de diisopropilamida de litio.
En algunas realizaciones, dicha diisopropilamida de litio se prepara haciendo reaccionar N,N-diisopropilamina en presencia de n-butillitio.
En algunas realizaciones, dicha diisopropilamida de litio se prepara a una temperatura de aproximadamente -75 °C a aproximadamente 5° C.
En algunas realizaciones:
(ii) dicho compuesto de fórmula V-a se hace reaccionar con diisopropilamida de litio para formar una primera mezcla; y
(ii) se añade N,N-dimetilformamida a dicha primera mezcla para formar una segunda mezcla.
En algunas realizaciones, se usan de aproximadamente 1,2 a aproximadamente 1,3 equivalentes de base amina en base a 1 equivalente del compuesto de fórmula V-a.
En algunas realizaciones, se usan de aproximadamente 1,4 a aproximadamente 1,6 equivalentes de N,N-dimetilformamida en base a 1 equivalente del compuesto de fórmula V-a.
En algunas realizaciones, dicho compuesto de Fórmula V-a se prepara mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula IV-a:

con 1,2-etanodiol en presencia de ácido p-toluenosulfónico.
En algunas realizaciones, dicho ácido p-toluenosulfónico es monohidrato de ácido p-toluenosulfónico. En algunas realizaciones, se usan de aproximadamente 2,2 a aproximadamente 2,7 equivalentes de 1,2-etanodiol en base a 1 equivalente del compuesto de Fórmula IV-a.
En algunas realizaciones, se usan de aproximadamente 0,05 a aproximadamente 0,1 equivalentes de ácido p-toluenosulfónico en base a 1 equivalente del compuesto de Fórmula IV-a.
En algunas realizaciones, dicha reacción se realiza aproximadamente a reflujo.
En algunas realizaciones, dicho compuesto de Fórmula IV-a se prepara mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula II:

con CH2CH2-X1 en presencia de una base de carbonato de metal alcalino, en donde:
X1 es haluro.
En algunas realizaciones, X1 es yoduro.
En algunas realizaciones, dicha base de carbonato de metal alcalino es carbonato de potasio.
En algunas realizaciones, se usan de aproximadamente 1,1 a aproximadamente 1,3 equivalentes de CH2CH2-X1 en base a 1 equivalente del compuesto de Fórmula II.
En algunas realizaciones, se usan de aproximadamente 1,8 a aproximadamente 2,2 equivalentes de base de carbonato de metal alcalino en base a 1 equivalente del compuesto de Fórmula II.
En algunas realizaciones, dicha reacción se realiza a de aproximadamente 55° C a aproximadamente 65° C.
En algunas realizaciones, dicho compuesto de Fórmula II se prepara de acuerdo con los procedimientos descritos en la Publicación de Estados Unidos N° 2013-0059835A1.
Se aprecia que ciertas características de la invención, que, para mayor claridad, se describen en el contexto de realizaciones separadas, también pueden proporcionarse en combinación en una única realización (aunque se pretende que las realizaciones se combinen como si estuvieran escritas en forma dependiente múltiple). A la inversa, varias características de la invención que, por brevedad, se describen en el contexto de una única realización, también pueden proporcionarse por separado o en cualquier subcombinación adecuada.
Las sales y compuestos descritos en la presente pueden ser asimétricos (por ejemplo, tener uno o más estereocentros). Si no se indica estereoquímica, entonces se pretenden todos los estereoisómeros, como enantiómeros y diastereómeros, a menos que se indique lo contrario por la estructura o el nombre. Las sales y compuestos de la presente solicitud que contienen átomos de carbono sustituidos asimétricamente pueden aislarse en formas ópticamente activas o racémicas. En la técnica se conocen métodos sobre cómo preparar formas ópticamente activas a partir de materiales de partida ópticamente inactivos, como por resolución de mezclas
racémicas o por síntesis estereoselectiva. Muchos isómeros geométricos de olefinas, enlaces dobles C=N y similares también pueden estar presentes en las sales y compuestos descritos en la presente, y todos estos isómeros estables se contemplan en la presente solicitud. Se describen los isómeros geométricos cis y trans de las sales y compuestos de la presente solicitud y pueden aislarse como una mezcla de isómeros o como formas isoméricas separadas.
La resolución de mezclas racémicas de sales y compuestos puede llevarse a cabo mediante cualquiera de los numerosos métodos conocidos en la técnica. Un método de ejemplo incluye la recristalización fraccionada usando un ácido de resolución quiral que es un ácido orgánico formador de sales, ópticamente activo. Los agentes de resolución adecuados para métodos de recristalización fraccionada son, por ejemplo, ácidos ópticamente activos, como las formas D y L del ácido tartárico, ácido diacetiltartárico, ácido dibenzoiltartárico, ácido mandélico, ácido málico, ácido láctico o los varios ácidos canforsulfónicos ópticamente activos como ácido p-canforsulfónico. Otros agentes de resolución adecuados para los métodos de cristalización fraccionada incluyen formas estereoisoméricamente puras de a-metilbencilamina (por ejemplo, formas S y R, o formas diastereoisoméricas puras), 2-fenilglicinol, norefedrina, efedrina, N-metilefedrina, ciclohexiletilamina, 1,2-diaminociclohexano, y similares.
La resolución de mezclas racémicas también puede llevarse a cabo mediante elución en una columna empaquetada con un agente de resolución ópticamente activo (por ejemplo, dinitrobenzoilfenilglicina). Un experto en la técnica puede determinar la composición de solvente de elución adecuada.
Las sales de la invención y los compuestos también pueden incluir todos los isótopos de átomos que se producen en los productos intermedios o sales o compuestos finales. Los isótopos incluyen aquellos átomos que tienen el mismo número atómico pero diferentes números de masa. Por ejemplo, los isótopos de hidrógeno incluyen tritio y deuterio.
En algunas realizaciones, los compuestos o sales pueden encontrarse junto con otras sustancias como agua y solventes (por ejemplo, hidratos y solvatos) o pueden aislarse.
En algunas realizaciones, los compuestos descritos en la presente, o sales de los mismos (por ejemplo, la sal de ácido clorhídrico del compuesto de Fórmula I), están sustancialmente aislados. Por "sustancialmente aislado" se entiende que el compuesto está por lo menos parcial o sustancialmente separado del entorno en el que se formó o detectó. La separación parcial puede incluir, por ejemplo, una composición enriquecida en los compuestos de la invención. La separación sustancial puede incluir composiciones que contienen por lo menos aproximadamente el 50%, por lo menos aproximadamente el 60%, por lo menos aproximadamente el 70%, por lo menos aproximadamente el 80%, por lo menos aproximadamente el 90%, por lo menos aproximadamente el 95%, por lo menos aproximadamente el 97%, o por lo menos aproximadamente el 99% en peso de los compuestos, o sal de los mismos. Los métodos para aislar compuestos y sus sales son rutinarios en la técnica.
La frase "farmacéuticamente aceptable" se emplea en la presente para referirse a aquellos compuestos, materiales, composiciones y/o formas de dosificación que, dentro del alcance del buen juicio médico, son adecuados para su uso en contacto con los tejidos de seres humanos y animales sin toxicidad, irritación, respuesta alérgica u otro problema o complicación, excesivas acorde con una relación beneficio/riesgo razonable.
Como se apreciará, los compuestos proporcionados en la presente, incluyendo las sales del mismo, pueden prepararse usando técnicas de síntesis orgánica conocidas y pueden sintetizarse de acuerdo con cualquiera de las numerosas rutas sintéticas posibles. Los procesos descritos en la presente pueden monitorizarse de acuerdo con cualquier método adecuado conocido en la técnica. Por ejemplo, la formación del producto puede monitorizarse por medios espectroscópicos, como espectroscopía de resonancia magnética nuclear (por ejemplo, 1H o 13C), espectroscopía de infrarrojos, o espectrofotometría (por ejemplo, visible a UV); o por cromatografía como cromatografía líquida de alto rendimiento (HPLC) o cromatografía en capa fina (TLC) u otras técnicas relacionadas.
Como se usa en la presente, el término "reaccionar" se usa como se conoce en la técnica y generalmente se refiere a la unión de reactivos químicos de tal manera que permita su interacción a nivel molecular para lograr una transformación química o física. En algunas realizaciones, la reacción implica dos reactivos, en donde se usan uno o más equivalentes del segundo reactivo con respecto al primer reactivo. Los pasos de reacción de los procesos descritos en la presente pueden realizarse durante un tiempo y en condiciones adecuadas para preparar el producto identificado.
Las reacciones de los procesos descritos en la presente pueden llevarse a cabo en solventes adecuados que pueden ser seleccionados fácilmente por un experto en la técnica de síntesis orgánica. Los solventes adecuados pueden ser sustancialmente no reactivos con los materiales de partida (reactivos), los productos intermedios, o los productos a las temperaturas a las que se llevan a cabo las reacciones, por ejemplo, temperaturas que pueden variar de la temperatura de congelación del solvente a la temperatura de ebullición del solvente. Una reacción dada puede llevarse a cabo en un solvente o una mezcla de más de un solvente. Dependiendo del paso de reacción particular, pueden seleccionarse solventes adecuados para un paso de reacción particular.
Los solventes adecuados pueden incluir solventes halogenados como tetracloruro de carbono, bromodiclorometano, dibromoclorometano, bromoformo, cloroformo, bromoclorometano, dibromometano, cloruro de butilo, diclorometano, tetracloroetileno, tricloroetileno, 1,1,1-tricloroetano, 1,1,2-tricloroetano, 1,1- dicloroetano, 2-cloropropano, 1,2-dicloroetano, 1,2-dibromoetano, hexafluorobenceno, 1,2,4-triclorobenceno, 1,2-diclorobenceno, clorobenceno, fluorobenceno, mezclas de los mismos y similares.
Los solventes de éter adecuados incluyen: dimetoximetano, tetrahidrofurano, 1,3-dioxano, 1,4-dioxano, furano, éter dietílico, éter dimetílico de etilenglicol, éter dietílico de etilenglicol, éter dimetílico de dietilenglicol, éter dietílico de dietilenglicol, éter dimetílico de trietilenglicol, anisol, t-butil metil éter, mezclas de los mismos y similares.
Los solventes próticos adecuados pueden incluir, a modo de ejemplo y sin limitación, agua, metanol, etanol, 2-nitroetanol, 2-fluoroetanol, 2,2,2-trifluoroetanol, etilenglicol, 1-propanol, 2-propanol, 2-metoxietanol, 1-butanol, 2-butanol, alcohol i-butílico, alcohol t-butílico, 2-etoxietanol, dietilenglicol, 1-, 2- o 3- pentanol, alcohol neo-pentílico, alcohol t-pentílico, éter monometílico de dietilenglicol, éter monoetílico de dietilenglicol, ciclohexanol, alcohol bencílico, fenol o glicerol.
Los solventes apróticos adecuados pueden incluir, a modo de ejemplo y sin limitación, tetrahidrofurano (THF), N,N-dimetilformamida (DMF), N,N-dimetilacetamida (DMA), 1,3-dimetil-3,4,5,6 -tetrahidro-2(1H)-pirimidinona (DMPU), 1,3-dimetil-2-imidazolidinona (DMI), N metilpirrolidinona (NMP), formamida, N-metilacetamida, N-metilformamida, acetonitrilo, dimetilsulfóxido, propionitrilo, formiato de etilo, acetato de metilo, hexacloroacetona, acetona, etil metil cetona, acetato de etilo, sulfolano, N,N-dimetilpropionamida, tetrametilurea, nitrometano, nitrobenceno o hexametilfosforamida.
Los solventes de hidrocarburos adecuados incluyen benceno, ciclohexano, pentano, hexano, tolueno, cicloheptano, metilciclohexano, heptano, etilbenceno, m-, o- o p-xileno, octano, indano, nonano o naftaleno.
Las reacciones de los procesos descritos en la presente pueden llevarse a cabo a temperaturas apropiadas que pueden ser determinadas fácilmente por el experto en la técnica. Las temperaturas de reacción dependerán, por ejemplo, de los puntos de fusión y ebullición de los reactivos y el solvente, si los hay; la termodinámica de la reacción (por ejemplo, puede ser necesario llevar a cabo reacciones exotérmicas vigorosas a temperaturas reducidas); y la cinética de la reacción (por ejemplo, una barrera de alta energía de activación puede necesitar temperaturas elevadas).
Las expresiones "temperatura ambiente" o "rt" como se usan en la presente, se entienden en la técnica y se refieren generalmente a una temperatura, por ejemplo, una temperatura de reacción, que es aproximadamente la temperatura de la habitación en la que se lleva a cabo la reacción, por ejemplo, a una temperatura de aproximadamente 20° C a aproximadamente 30° C.
Las reacciones de los procesos descritos en la presente pueden realizarse al aire o bajo atmósfera inerte. Típicamente, las reacciones que contienen reactivos o productos que son sustancialmente reactivos con el aire pueden llevarse a cabo usando técnicas sintéticas sensibles al aire que son bien conocidas por los expertos en la técnica.
Métodos de uso
Las sales y los compuestos proporcionados por los procesos de la invención pueden modular la actividad de una o más de varias quinasas que incluyen, por ejemplo, fosfoinosítido 3-quinasas (PI3K). Se entiende que el término "modular" se refiere a la capacidad de aumentar o disminuir la actividad de uno o más miembros de la familia PI3K. Por consiguiente, las sales de la invención pueden usarse en métodos para modular una PI3K poniendo en contacto la PI3K con una cualquiera o más de las sales, compuestos o composiciones descritas en la presente. En algunas realizaciones, las sales de la invención y los compuestos de la presente solicitud pueden actuar como inhibidores de una o más PI3K. En realizaciones adicionales, las sales de la invención pueden usarse para modular la actividad de una PI3K en un individuo que necesite modular el receptor mediante la administración de una cantidad moduladora de un compuesto, o una sal farmacéuticamente aceptable del mismo. En algunas realizaciones, modular es inhibir.
Dado que el crecimiento y la supervivencia de las células cancerosas se ven afectados por múltiples vías de señalización, los compuestos proporcionados por los procesos de la presente solicitud son útiles para tratar estados patológicos caracterizados por mutantes de quinasas resistentes a fármacos. Además, pueden usarse en combinación diferentes inhibidores de quinasas, que muestran diferentes preferencias en las quinasas cuyas actividades modulan. Este enfoque podría resultar muy eficaz en el tratamiento de estados patológicos al dirigirse a múltiples vías de señalización, reducir la probabilidad de que surja resistencia a los fármacos en una célula, y reducir la toxicidad de los tratamientos para la enfermedad.
Las quinasas a las que las presentes sales y compuestos se unen y/o modulan (por ejemplo, inhiben) incluyen cualquier miembro de la familia PI3K. En algunas realizaciones, la PI3K es PI3Ka, PI3Kp, PI3K5 o PI3Ky. En algunas realizaciones, la PI3K es PI3K5 o PI3Ky. En algunas realizaciones, la PI3K es PI3K5. En algunas realizaciones, la PI3K es PI3Ky. En algunas realizaciones, la PI3K incluye una mutación. Una mutación puede ser el reemplazo de un aminoácido por otro, o la deleción de uno o más aminoácidos. En tales realizaciones, la mutación puede estar presente en el dominio quinasa de la PI3K.
En algunas realizaciones, puede usarse usa más de una sal o compuesto proporcionado por los procesos de la invención para inhibir la actividad de una quinasa (por ejemplo, PI3K5 o PI3Ky).
En algunas realizaciones, puede usarse más de una sal o compuesto proporcionado por los procesos de la invención para inhibir más de una quinasa, como por lo menos dos quinasas (por ejemplo, PI3K5 y PI3Ky).
En algunas realizaciones, puede usarse una o más de las sales o compuestos proporcionados por los procesos de la invención en combinación con otro inhibidor de quinasas para inhibir la actividad de una quinasa (por ejemplo, PI3K5 o PI3Ky).
En algunas realizaciones, puede usarse una o más de las sales o compuestos proporcionados por los procesos de la invención en combinación con otro inhibidor de quinasas para inhibir las actividades de más de una quinasa (por ejemplo, PI3K5 o PI3Ky), como por lo menos dos quinasas.
Las sales y los compuestos proporcionados por los procesos de la invención pueden ser selectivos. Por "selectivo" se entiende que el compuesto se une o inhibe una quinasa con mayor afinidad o potencia, respectivamente, en comparación con por lo menos otra quinasa. En algunas realizaciones, las sales y los compuestos de la invención son inhibidores selectivos de PI3K5 o PI3Ky sobre PI3Ka y/o PI3Kp. En algunas realizaciones, las sales y los compuestos proporcionados por los procesos de la invención son inhibidores selectivos de PI3K5 (por ejemplo, sobre PI3Ka, PI3Kp y PI3Ky). En algunas realizaciones, las sales y los compuestos proporcionados por los procesos de la invención son inhibidores selectivos de PI3K5 (por ejemplo, sobre PI3Ka, PI3Kp y PI3Ky). En algunas realizaciones, la selectividad puede ser por lo menos aproximadamente 2 veces, 5 veces, 10 veces, por lo menos aproximadamente 20 veces, por lo menos aproximadamente 50 veces, por lo menos aproximadamente 100 veces, por lo menos aproximadamente 200 veces, por lo menos aproximadamente 500 veces o por lo menos aproximadamente 1000 veces. La selectividad puede medirse mediante métodos rutinarios en la técnica. En algunas realizaciones, la selectividad puede probarse a la concentración de Km ATP de cada enzima. En algunas realizaciones, la selectividad de las sales de la invención y los compuestos puede determinarse mediante ensayos celulares asociados con una actividad quinasa de PI3K particular.
En otro aspecto los compuestos proporcionados por los procesos de la presente solicitud pueden ser útiles en métodos para tratar una enfermedad o trastorno asociado a quinasas (como PI3K) en un individuo (por ejemplo, paciente) administrando al individuo con necesidad de dicho tratamiento una cantidad o dosis terapéuticamente eficaz de una o más sales o compuestos de la presente solicitud o una composición farmacéutica de los mismos. Una enfermedad asociada a PI3K puede incluir cualquier enfermedad, trastorno o afección que esté directa o indirectamente relacionada con la expresión o actividad de PI3K, incluyendo la sobreexpresión y/o niveles de actividad anormales. En algunas realizaciones, la enfermedad puede estar vinculada a Akt (proteína quinasa B), objetivo mamífero de rapamicina (mTOR) o quinasa 1 dependiente de fosfoinosítido (PDK1). En algunas realizaciones, la enfermedad relacionada con mTOR puede ser inflamación, aterosclerosis, psoriasis, reestenosis, hipertrofia prostática benigna, trastornos óseos, pancreatitis, angiogénesis, retinopatía diabética, aterosclerosis, artritis, trastornos inmunológicos, enfermedad renal o cáncer. Una enfermedad asociada a PI3K también puede incluir cualquier enfermedad, trastorno o afección que se pueda prevenir, mejorar o curar modulando la actividad de PI3K. En algunas realizaciones, la enfermedad se caracteriza por la actividad anormal de PI3K. En algunas realizaciones, la enfermedad se caracteriza por PI3K mutante. En tales realizaciones, la mutación puede estar presente en el dominio quinasa de PI3K.
Ejemplos de enfermedades asociadas a PI3K incluyen enfermedades de base inmunitaria que involucran al sistema incluyendo, por ejemplo, artritis reumatoide, alergia, asma, glomerulonefritis, lupus o inflamación relacionada con cualquiera de los anteriores.
Ejemplos adicionales de enfermedades asociadas a PI3K incluyen cánceres como cáncer de mama, próstata, colon, endometrio, cerebro, vejiga, piel, útero, ovario, pulmón, páncreas, riñón, gástrico o hematológico.
Ejemplos adicionales de enfermedades asociadas a PI3K incluyen enfermedades pulmonares como lesión pulmonar aguda (ALI) y síndrome de dificultad respiratoria del adulto (SDRA).
Ejemplos adicionales de enfermedades asociadas a PI3K incluyen osteoartritis, reestenosis, aterosclerosis, trastornos óseos, artritis, retinopatía diabética, psoriasis, hipertrofia prostática benigna, inflamación, angiogénesis, pancreatitis, enfermedad renal, enfermedad inflamatoria intestinal, miastenia gravis, esclerosis múltiple o síndrome
de Sjogren, y similares.
Ejemplos adicionales de enfermedades asociadas a PI3K incluyen púrpura trombocitopénica idiopática (PTI), anemia hemolítica autoinmune (AIHA), vasculitis, lupus eritematoso sistémico, nefritis lúpica, pénfigo, nefropatía membranosa, leucemia linfocítica crónica (CLL), linfoma no de Hodgkin (LNH), linfoma de células pilosas, linfoma de células del manto, linfoma de Burkitt, linfoma linfocítico pequeño, linfoma folicular, linfoma linfoplasmocítico, linfoma de zona marginal extraganglionar, linfoma de células B grandes difuso de tipo células B activadas (ABC) o linfoma de células B grandes difuso del centro germinal (GCB).
En algunas realizaciones, la enfermedad se selecciona de púrpura trombocitopénica idiopática (ITP), anemia hemolítica autoinmune, vasculitis, lupus eritematoso sistémico, nefritis lúpica, pénfigo, anemia hemolítica autoinmune (AIHA), nefropatía membranosa, leucemia linfocítica crónica (CLL), linfoma no de Hodgkin (LNH), leucemia de células pilosas, linfoma de células del manto, linfoma de Burkitt, linfoma linfocítico pequeño, linfoma folicular, linfoma linfoplasmocítico, linfoma de zona marginal extraganglionar, linfoma de Hodgkin, macroglobulinemia de Waldenstrom, leucemia prolinfocítica, leucemia linfoblástica aguda, mielofibrosis, linfoma de tejido linfático asociado a mucosas (MALT), linfoma de células B, linfoma mediastínico (tímico) de células B grandes, granulomatosis linfomatoide, linfoma de zona marginal esplénica, linfoma de derrame primario, linfoma de células B grandes intravascular, leucemia de células plasmáticas, plasmocitoma extramedular, mieloma latente (también conocido como mieloma asintomático), gammapatía monoclonal de significado indeterminado (MGUS) y linfoma de células B.
Los compuestos preparados de acuerdo con los procesos de la invención, y sus sales pueden ser útiles en un método para tratar púrpura trombocitopénica idiopática (ITP) seleccionada de ITP recidivante e ITP refractaria.
Los compuestos preparados de acuerdo con los procesos de la invención, y sus sales pueden ser útiles en un método para tratar la vasculitis seleccionada de enfermedad de Behget, síndrome de Cogan, arteritis de células gigantes, polimialgia reumática (PMR), arteritis de Takayasu, enfermedad de Buerger (tromboangitis obliterante), vasculitis del sistema nervioso central, enfermedad de Kawasaki, poliarteritis nodosa, síndrome de Churg-Strauss, vasculitis mixta por crioglobulinemia (esencial o inducida por el virus de la hepatitis C (VHC)), púrpura de Henoch-Schonlein (HSP), vasculitis por hipersensibilidad, poliangeítis microscópica, granulomatosis de Wegener y vasculitis sistémica asociada a anticuerpos del citoplasma anti-neutrófilos (ANCA) (AASV).
Los compuestos preparados de acuerdo con los procesos de la invención, y sus sales pueden ser útiles en un método para tratar el linfoma no de Hodgkin (NHL) seleccionado de NHL recidivante, NHL refractario y NHL folicular recurrente.
Los compuestos preparados de acuerdo con los procesos de la invención, y sus sales pueden ser útiles en un método para tratar el linfoma de células B, en donde dicho linfoma de células B es linfoma difuso de células B grandes (dLbCL).
Los compuestos preparados de acuerdo con los procesos de la invención, y sus sales pueden ser útiles en un método para tratar el linfoma de células B, en donde dicho linfoma de células B es linfoma de células B grandes difuso del tipo activado por células B (ABC) o linfoma difuso de células B grandes de células B de centro germinal (GCB).
En algunas realizaciones, dicha enfermedad es osteoartritis, reestenosis, aterosclerosis, trastornos óseos, artritis, retinopatía diabética, psoriasis, hipertrofia prostática benigna, inflamación, angiogénesis, pancreatitis, enfermedad renal, enfermedad inflamatoria intestinal, miastenia gravis, esclerosis múltiple o síndrome de Sjogren.
En algunas realizaciones, dicha enfermedad es artritis reumatoide, alergia, asma, glomerulonefritis, lupus o inflamación relacionada con cualquiera de las mencionadas anteriormente.
En algunas realizaciones, dicho lupus es lupus eritematoso sistémico o nefritis lúpica.
En algunas realizaciones, dicha enfermedad es cáncer de mama, cáncer de próstata, cáncer de colon, cáncer de endometrio, cáncer de cerebro, cáncer de vejiga, cáncer de piel, cáncer de útero, cáncer de ovario, cáncer de pulmón, cáncer de páncreas, cáncer renal, cáncer gástrico, o un cáncer hematológico.
En algunas realizaciones, dicho cáncer hematológico es leucemia mieloblástica aguda o leucemia mieloide crónica.
En algunas realizaciones, dicho cáncer hematológico son enfermedades malignas linfoides de origen de células B que incluyen linfoma no de Hodgkin (NHL) de células B indolente/agresivo y linfoma de Hodgkin (HL).
En algunas realizaciones, dicha enfermedad es lesión pulmonar aguda (ALI) o síndrome de dificultad
respiratoria del adulto (ARDS).
Como se usa en la presente, el término "poner en contacto" se refiere a reunir las fracciones indicadas en un sistema in vitro o en un sistema in vivo. Por ejemplo, "poner en contacto" una PI3K con un compuesto de la invención incluye la administración de un compuesto de la presente solicitud a un individuo o paciente, como un humano, que tiene una PI3K, así como, por ejemplo, introducir un compuesto de la invención en una muestra que contiene una preparación celular o purificada que contiene la PI3K.
Como se usa en la presente, el término "individuo" o "paciente", usado indistintamente, se refiere a cualquier animal, incluyendo mamíferos, preferiblemente ratones, ratas, otros roedores, conejos, perros, gatos, cerdos, vacas, ovejas, caballos o primates, y lo más preferible humanos.
Como se usa en la presente, la frase "cantidad terapéuticamente eficaz" se refiere a la cantidad de compuesto activo o agente farmacéutico que provoca la respuesta biológica o medicinal que se busca en un tejido, sistema, animal, individuo o humano por un investigador, veterinario, doctor médico u otro practicante clínico. En algunas realizaciones, la dosificación del compuesto, o una sal farmacéuticamente aceptable del mismo, administrada a un paciente o individuo es de aproximadamente 1 mg a aproximadamente 2 g, de aproximadamente 1 mg a aproximadamente 1000 mg, de aproximadamente 1 mg a aproximadamente 500 mg, de aproximadamente 1 mg a aproximadamente 100 mg, de aproximadamente 1 mg a 50 mg, o de aproximadamente 50 mg a aproximadamente 500 mg.
Como se usa en la presente, el término "tratar" o "tratamiento" se refiere a uno o más de (1) prevenir la enfermedad; por ejemplo, prevenir una enfermedad, afección o trastorno en un individuo que puede estar predispuesto a la enfermedad, afección o trastorno pero que todavía no experimenta ni muestra la patología o sintomatología de la enfermedad; (2) inhibir la enfermedad; por ejemplo, inhibir una enfermedad, afección o trastorno en un individuo que está experimentando o mostrando la patología o sintomatología de la enfermedad, afección o trastorno (es decir, detener el desarrollo adicional de la patología y/o sintomatología); y (3) mejorar la enfermedad; por ejemplo, mejorar una enfermedad, condición o trastorno en un individuo que está experimentando o mostrando la patología o sintomatología de la enfermedad, afección o trastorno (es decir, revertir la patología y/o sintomatología), como disminuir la gravedad de la enfermedad.
Terapias de combinación
Uno o más agentes farmacéuticos adicionales como, por ejemplo, agentes quimioterapéuticos, agentes antiinflamatorios, esteroides, inmunosupresores, así como inhibidores de quinasas Bcr-Abl, Flt-3, EGFR, HER2, JAK (por ejemplo, JAK1 o JAK2), c-MET, VEGFR, PDGFR, cKit, IGF-1R, RAF, FAK, Akt mTOR, PIM y AKT (por ejemplo, AKT1, AKT2 o AKT3) como, por ejemplo, los descritos en la WO 2006/056399, u otros agentes como anticuerpos terapéuticos que pueden usarse en combinación con las sales o compuestos proporcionados por los procesos de la presente solicitud para el tratamiento de enfermedades, trastornos o afecciones asociadas a PI3K. El uno o más agentes farmacéuticos adicionales pueden administrarse a un paciente simultánea o secuencialmente.
En algunas realizaciones, el agente farmacéutico adicional es un inhibidor de JAK1 y/o JAK2. Los compuestos proporcionados por los procesos de la presente solicitud, y sus sales, pueden ser útiles en un método para tratar una enfermedad descrita en la presente (por ejemplo, una enfermedad maligna de células B, como linfoma difuso de células B) en un paciente que comprende administrar al paciente el compuesto descrito en la presente o una sal farmacéuticamente aceptable del mismo, y un inhibidor de JAK1 y/o JAK2. Las enfermedades malignas de células B pueden incluir las descritas en la presente y en la U.S. N° de serie 61/976.815, presentada el 8 de abril de 2014.
En algunas realizaciones, el inhibidor de JAK1 y/o JAK2 es un compuesto de la Tabla 1, o una sal farmacéuticamente aceptable del mismo. Los compuestos de la Tabla 1 son inhibidores selectivos de JAK1 (selectivos sobre JAK2, JAK3 y TYK2). Las IC50 obtenidas por el método del Ensayo A a ATP 1 mM se muestran en la Tabla 1.
Tabla 1

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

En algunas realizaciones, el inhibidor de JAK1 y/o JAK2 es {1-{1-[3-fluoro-2-(trifluorometil)isonicotinoil]piperidin-4-il}-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]azetidin-3-il}acetonitrilo, o una sal farmacéuticamente aceptable del mismo.
En algunas realizaciones, el inhibidor de JAK1 y/o JAK2 es sal del ácido adípico de {1-{1-[3-fluoro-2-(trifluorometil)isonicotinoil]piperidin-4-il}-3-[4-(7H-pirrolo[2, 3-d]pirimidin-4-il)-1 H-pirazol-1 -il]azetidin-3-il}acetonitrilo.
En algunas realizaciones, el inhibidor de JAK1 y/o JAK2 es 4-{3-(cianometil)-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]azetidin-1 -il}-2,5-difluoro-N-[(1 S)-2,2,2-trifluoro-1 -metiletil]benzamida, o una sal farmacéuticamente aceptable de la misma.
En algunas realizaciones, el inhibidor de JAK1 y/o JAK2 se selecciona de (R)-3-[1-(6-cloropiridin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1H-pirazol-1-il]propanonitrilo, (R)-3-(1-[1,3]oxazolo[5,4-b]piridin-2ilpirrolidin-3-il)-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrilo, (R)-4-[(4-{3-ciano-2-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propil}piperazin-1 -il)carbonil]-3-fluorobenzonitrilo, (R)-4-[(4-{3-ciano-2-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 -il]propil}piperazin-1-il)carbonil]-3-fluorobenzonitrilo, o (R)-4-(4-{3-[(dimetilamino)metil]-5-fluorofenoxi}piperidin-1-il)-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 il]butanonitrilo, (S)-3-[1 -(6-cloropiridin-2-il)pirrolidin-3-il]-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propanonitrilo, (S)-3-(1 -[1,3]oxazolo[5,4-b]piridin-2-ilpirrolidin-3-il)-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 il]propanonitrilo, (S)-4-[(4-{3-ciano-2-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1 -il]propil}piperazin-1 -il)carbonil]-3-fluorobenzonitrilo, (S)-4-[(4-{3-ciano-2-[3-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirrol-1 -il]propil}piperazin-1 -il)carbonil]-3-fluorobenzonitrilo, (S)-4-(4-{3-[(dimetilamino)metil]-5-fluorofenoxi}piperidin-1-il)-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)-1 H-pirazol-1-il]butanonitrilo; y sales farmacéuticamente aceptables de cualquiera de los mencionados anteriormente.
En algunas realizaciones, el inhibidor de JAK1 y/o JAK2 se selecciona de los compuestos de la publicación de Patente de Estados Unidos N° 2010/0298334, presentada el 21 de mayo de 2010, publicación de Patente de Estados Unidos N° 2011/0059951, presentada el 31 de agosto de 2010, publicación de Patente de Estados Unidos N° 2011/0224190, presentada el 9 de marzo de 2011, publicación de Patente de Estados Unidos N° 2012/0149681, presentada el 18 de noviembre de 2011, publicación de Patente de Estados Unidos N° 2012/0149682, presentada el 18 de noviembre de 2011, publicación de patente de EE.UU. 2013/0018034, presentada el 19 de junio de 2012, publicación de Patente de Estados Unidos N° 2013/0045963, presentada el 17 de agosto de 2012, y la publicación de Patente de Estados Unidos N° 2014/0005166, presentada el 17 de mayo de 2013.
Los anticuerpos ejemplares para su uso en terapia de combinación incluyen, entre otros, trastuzumab (por ejemplo, anti-HER2), ranibizumab (por ejemplo, anti-VEGF-A), bevacizumab (Avastin™, por ejemplo, anti-VEGF), panitumumab (por ejemplo, anti-EGFR), cetuximab (por ejemplo, anti-EGFR), rituximab (Rituxan™, anti-CD20) y anticuerpos dirigidos a c-MET.
Pueden usarse uno o más de los siguientes agentes en combinación con las sales o compuestos proporcionados por los procesos de la presente solicitud y se presentan como una lista no limitativa: un agente citostático, cisplatino, doxorrubicina, taxotere, taxol, etopósido, irinotecán, camptostar, topotecán, paclitaxel, docetaxel, epotilonas, tamoxifeno, 5-fluorouracilo, metoxtrexato, temozolomida, ciclofosfamida, SCH 66336, R115777, L778, 123, BMS 214662, Iressa, Tarceva, anticuerpos contra EGFR, Gleevec™, intrón, ara-C, adriamicina, Citoxano, gemcitabina, uracilo mostaza, clormetina, ifosfamida, melfalan, clorambucilo, pipobroman, trietilenmelamina, trietilentiofosforamina, busulfano, carmustina, lomustina, estreptozocina, dacarbazina, floxuridina, citarabina, 6-mercaptopurina, 6-tioguanina, fosfato de fludarabina, oxaliplatino, leucovirina, ELOXATIN™, pentostatina, vinblastina, vincristina, vindesina, bleomicina, dactinomicina, daunorrubicina, doxorrubicina, epirrubicina, idarubicina, mitramicina, desoxicoformicina, mitomicina-C, L-asparaginasa, teniposide 17.alfa.-Etinilestradiol, dietilestilbestrol, testosterona, prednisona, fluoximesterona, propionato de dromostanolona, testolactona, megestrolacetato, metilprednisolona, metiltestosterona, prednisolona, triamcinolona, clorotrianiseno, hidroxiprogesterona, aminoglutetimida, estramustina, acetato de medroxiprogesterona, leuprolida, flutamida, toremifeno, goserelina, cisplatino, carboplatino, hidroxiurea, amsacrina, procarbazina, mitotano, mitoxantrona, levamisol, navelbeno, anastrazol, letrazol, capecitabina, reloxafina, droloxafina, hexametilmelamina, avastina, herceptina, Bexxar, Velcade, Zevalin, Trisenox, Xeloda, vinorelbina, Porfimer, Erbitux, Liposomal, Thiotepa, altretamina, Melfalan, Trastuzumab, lerozol, Fulvestrant, exemestano, Fulvestrant, ifosfomida, Rituximab, C225, Campath, clofarabina, cladribina, afidicolon, rituxan, sunitinib, dasatinib, tezacitabina, Sml1, fludarabina, pentostatina, triapina, didox, trimidox, amidox, 3-AP, MDL-101,731, bendamustina (Treanda), ofatumumab y GS-1101 (también conocido como CAL-101).
Los agentes quimioterapéuticos de ejemplo incluyen inhibidores de proteosomas (por ejemplo, bortezomib), talidomida, revlimid y agentes que dañan el ADN como melfalán, doxorrubicina, ciclofosfamida, vincristina, etopósido, carmustina y similares.
Los esteroides de ejemplo incluyen coriticosteroides como dexametasona o prednisona.
Los inhibidores de Bcr-Abl de ejemplo incluyen los compuestos, y sales farmacéuticamente aceptables de los mismos, de los géneros y especies divulgados en la Patente de Estados Unidos N° 5.521.184, WO 04/005281 y N° de serie de Estados Unidos 60/578.491.
Los inhibidores de Flt-3 de ejemplo adecuados incluyen compuestos y sus sales farmacéuticamente aceptables, como se divulga en la WO 03/037347, WO 03/099771 y WO 04/046120.
Los inhibidores de RAF de ejemplo adecuados incluyen compuestos, y sus sales farmacéuticamente aceptables, como se divulga en la WO 00/09495 y WO 05/028444.
Los de inhibidores de FAK adecuados de ejemplo incluyen compuestos, y sus sales farmacéuticamente aceptables, como se divulga en la WO 04/080980, WO 04/056786, WO 03/024967, WO 01/064655, WO 00/053595 y WO 01/014402.
Los de inhibidores de mTOR adecuados de ejemplo incluyen compuestos, y sus sales farmacéuticamente aceptables, como se divulga en la WO 2011/025889.
En algunas realizaciones, las sales y compuestos proporcionados por los procesos de la invención pueden usarse en combinación con uno o más de otros inhibidores de quinasas, incluyendo imatinib, particularmente para tratar pacientes resistentes a imatinib u otros inhibidores de quinasas.
En algunas realizaciones, las sales y compuestos proporcionados por los procesos de la invención pueden usarse en combinación con un agente quimioterapéutico en el tratamiento del cáncer, como el mieloma múltiple, y pueden mejorar la respuesta al tratamiento en comparación con la respuesta al agente quimioterapéutico solo, sin exacerbación de sus efectos tóxicos. Ejemplos de agentes farmacéuticos adicionales usados en el tratamiento del mieloma múltiple, por ejemplo, pueden incluir, sin limitación, melfalán, melfalán más prednisona [MP], doxorrubicina, dexametasona y Velcade (bortezomib). Otros agentes adicionales usados en el tratamiento del mieloma múltiple incluyen inhibidores de quinasas Bcr-Abl, Flt-3, RAF y FAK. Los efectos aditivos o sinérgicos son resultados deseables de la combinación de un inhibidor de PI3K de la presente solicitud con un agente adicional. Además, la resistencia de las células de mieloma múltiple a agentes como dexametasona puede ser reversible tras el tratamiento con el inhibidor de PI3K de la presente solicitud. Los agentes pueden combinarse con el presente compuesto en una forma de dosificación única o continua, o los agentes pueden administrarse simultánea o secuencialmente como formas de dosificación separadas.
Puede administrase un corticosteroide como dexametasona a un paciente en combinación con las sales y compuestos proporcionados por los procesos de la invención donde la dexametasona se administra intermitentemente en lugar de continuamente.
Pueden administrarse combinaciones de las sales y compuestos proporcionados por los procesos de la invención con otros agentes terapéuticos a un paciente antes, durante y/o después de un trasplante de médula ósea o de células madre.
Formulaciones farmacéuticas y formas de dosificación
Cuando se emplean como productos farmacéuticos, los compuestos y sales proporcionados por los procesos de la invención pueden administrarse en forma de composiciones farmacéuticas. Estas composiciones pueden prepararse de una manera bien conocida en la técnica farmacéutica y pueden administrarse mediante una variedad de vías, dependiendo de si se desea un tratamiento local o sistémico y del área a tratar. La administración puede ser tópica (incluyendo transdérmica, epidérmica, oftálmica y a membranas mucosas incluyendo administración intranasal, vaginal y rectal), pulmonar (por ejemplo, por inhalación o insuflación de polvos o aerosoles, incluyendo por nebulizador; intratraqueal o intranasal), oral o parenteral. La administración parenteral incluye administración intravenosa, intraarterial, subcutánea, intraperitoneal, intramuscular o inyección o infusión; o intracraneal, por ejemplo, intratecal o intraventricular. La administración parenteral puede ser en forma de una sola dosis en bolo o puede ser, por ejemplo, mediante una bomba de perfusión continua. Las composiciones farmacéuticas y las formulaciones para administración tópica pueden incluir parches transdérmicos, pomadas, lociones, cremas, geles, gotas, supositorios, espráis, líquidos y polvos. Pueden ser necesarios o deseables portadores farmacéuticos convencionales, bases acuosas, en polvo o aceitosas, espesantes y similares.
Las composiciones farmacéuticas pueden contener, como ingrediente activo, el compuesto o la sal proporcionados por los procesos de la invención (por ejemplo, la sal de ácido clorhídrico del compuesto de Fórmula I), en combinación con uno o más portadores (excipientes) farmacéuticamente aceptables. En algunas realizaciones, la composición es adecuada para administración tópica. Al elaborar las composiciones el ingrediente activo se mezcla típicamente con un excipiente, se diluye por un excipiente o se encierra dentro de dicho portador en forma de, por ejemplo, una cápsula, bolsita, papel u otro recipiente. Cuando el excipiente sirve como diluyente, puede ser un material sólido, semisólido o líquido, que actúa como vehículo, portador o medio para el ingrediente activo. Por tanto, las composiciones pueden estar en forma de comprimidos, píldoras, polvos, pastillas, bolsitas, obleas, elixires, suspensiones, emulsiones, soluciones, jarabes, aerosoles (como un sólido o en un medio líquido), pomadas que contienen, por ejemplo, hasta un 10% en peso del compuesto activo, cápsulas de gelatina duras y blandas, supositorios, soluciones inyectables estériles, y polvos envasados estériles.
Al preparar una formulación, el compuesto activo o la sal puede molerse para proporcionar el tamaño de partícula apropiado antes de combinarlo con los otros ingredientes. Si el compuesto activo o la sal es
sustancialmente insoluble, puede molerse hasta un tamaño de partícula de menos de 200 mesh. Si el compuesto activo o la sal es sustancialmente soluble en agua, el tamaño de partícula puede ajustarse moliendo para proporcionar una distribución sustancialmente uniforme en la formulación, por ejemplo, aproximadamente 40 mesh.
Los compuestos y las sales proporcionados por los procesos de la invención pueden molerse usando procedimientos de molienda conocidos como molienda en húmedo para obtener un tamaño de partícula apropiado para la formación de comprimidos y para otros tipos de formulaciones. Las preparaciones finamente divididas (nanoparticuladas) de las sales de la invención pueden prepararse mediante procesos conocidos en la técnica, por ejemplo, ver la Solicitud Internacional N° WO 2002/000196.
Algunos ejemplos de excipientes adecuados incluyen lactosa, dextrosa, sacarosa, sorbitol, manitol, almidones, goma arábiga, fosfato de calcio, alginatos, tragacanto, gelatina, silicato de calcio, celulosa microcristalina, polivinilpirrolidona, celulosa, agua, jarabe y metilcelulosa. Las formulaciones pueden incluir adicionalmente: agentes lubricantes como talco, estearato de magnesio y aceite mineral; agentes humectantes; agentes emulsionantes y suspensores; agentes conservantes como metil- y propilhidroxi-benzoatos; agentes edulcorantes; y agentes aromatizantes. Las composiciones pueden formularse para proporcionar una liberación rápida, sostenida o retardada del ingrediente activo después de la administración al paciente empleando procedimientos conocidos en la técnica.
Las composiciones pueden formularse en una forma de dosificación unitaria, cada dosificación conteniendo de aproximadamente 5 a aproximadamente 1000 mg (1 g), más habitualmente de aproximadamente 100 a aproximadamente 500 mg, del ingrediente activo. El término "formas de dosificación unitaria" se refiere a unidades físicamente discretas adecuadas como dosis unitarias para sujetos humanos y otros mamíferos, cada unidad conteniendo una cantidad predeterminada de material activo calculada para producir el efecto terapéutico deseado, en asociación con un excipiente farmacéutico adecuado.
Las composiciones pueden contener de aproximadamente 5 a aproximadamente 50 mg del ingrediente activo. Un experto en la técnica apreciará que esto incluye composiciones que contienen de aproximadamente 5 a aproximadamente 10, de aproximadamente 10 a aproximadamente 15, de aproximadamente 15 a aproximadamente 20, de aproximadamente 20 a aproximadamente 25, de aproximadamente 25 a aproximadamente 30, de aproximadamente 30 a aproximadamente 35, de aproximadamente 35 a aproximadamente 40, de aproximadamente 40 a aproximadamente 45, o de aproximadamente 45 a aproximadamente 50 mg del ingrediente activo.
Las composiciones pueden contener aproximadamente 2,5, aproximadamente 5, aproximadamente 7,5, aproximadamente 10, aproximadamente 12,5, aproximadamente 15, aproximadamente 17,5, aproximadamente 20, aproximadamente 22,5 o aproximadamente 25 mg del ingrediente activo. En algunas realizaciones, las composiciones de la invención contienen aproximadamente 5 mg del ingrediente activo. En algunas realizaciones, las composiciones de la invención contienen aproximadamente 10 mg del ingrediente activo.
Pueden usarse dosificaciones similares de los compuestos y sales descritos en la presente en los métodos y usos de los compuestos y sales proporcionados por los procesos de la invención.
El compuesto activo o la sal puede ser eficaz en un amplio intervalo de dosificaciones y generalmente se administra en una cantidad farmacéuticamente eficaz. Se entenderá, sin embargo, que la cantidad de compuesto o sal realmente administrada será habitualmente determinada por un médico, de acuerdo con las circunstancias relevantes, incluyendo la afección a tratar, la vía elegida de administración, el compuesto o la sal real administrada., la edad, el peso y la respuesta del paciente individual, la gravedad de los síntomas del paciente y similares.
Para preparar composiciones sólidas como comprimidos, el ingrediente activo principal se mezcla con un excipiente farmacéutico para formar una composición de preformulación sólida que contiene una mezcla homogénea de un compuesto o sal de la presente solicitud. Cuando se hace referencia a estas composiciones de preformulación como homogéneas, el ingrediente activo se dispersa típicamente uniformemente por toda la composición de tal manera que la composición puede subdividirse fácilmente en formas de dosificación unitaria igualmente eficaces tales como comprimidos, píldoras y cápsulas. Esta preformulación sólida se subdivide luego en formas de dosificación unitaria del tipo descrito anteriormente que contienen, por ejemplo, de aproximadamente 0,1 a aproximadamente 1000 mg del ingrediente activo de la presente solicitud.
Los comprimidos o píldoras pueden recubrirse o componerse de otro modo para proporcionar una forma de dosificación que ofrezca la ventaja de una acción prolongada. Por ejemplo, el comprimido o píldora puede comprender un componente de dosificación interior y un componente de dosificación exterior, este último estando en forma de una envoltura sobre el primero. Los dos componentes pueden estar separados por una capa entérica que sirve para resistir la disgregación en el estómago y permitir que el componente interno pase intacto al duodeno o se retrase en su liberación. Pueden usarse una variedad de materiales para tales capas o recubrimientos entéricos, tales materiales incluyendo una serie de ácidos poliméricos y mezclas de ácidos poliméricos con materiales como goma laca, alcohol cetílico y acetato de celulosa.
Las formas líquidas en las que pueden incorporarse los compuestos, las sales, y las composiciones proporcionadas por los procesos de la presente solicitud para la administración oral o por inyección incluyen soluciones acuosas, jarabes adecuadamente aromatizados, suspensiones acuosas u oleosas y emulsiones aromatizadas con aceites comestibles como aceite de semilla de algodón, aceite de sésamo, aceite de coco o aceite de cacahuete, así como elixires y vehículos farmacéuticos similares.
Las composiciones para inhalación o insuflación incluyen soluciones y suspensiones en solventes acuosos u orgánicos farmacéuticamente aceptables, o mezclas de los mismos, y polvos. Las composiciones líquidas o sólidas pueden contener excipientes farmacéuticamente aceptables adecuados como se ha descrito anteriormente. Las composiciones pueden administrarse por vía respiratoria oral o nasal para un efecto local o sistémico. Las composiciones pueden nebulizarse mediante el uso de gases inertes. Las soluciones nebulizadas pueden respirarse directamente desde el dispositivo nebulizador o el dispositivo nebulizador puede conectarse a una máscara facial, mascarilla, o respirador de presión positiva intermitente. Las composiciones en solución, suspensión o polvo pueden administrarse por vía oral o nasal desde dispositivos que administran la formulación de una manera apropiada.
Las formulaciones tópicas pueden contener uno o más portadores convencionales. Las pomadas pueden contener agua y uno o más portadores hidrófobos seleccionados de, por ejemplo, parafina líquida, polioxietilen alquil éter, propilenglicol, vaselina blanca y similares. Las composiciones portadoras de cremas pueden basarse en agua en combinación con glicerol y uno o más de otros componentes, por ejemplo, monoestearato de glicerina, monoestearato de PEG-glicerina y alcohol cetilestearílico. Los geles pueden formularse usando alcohol isopropílico y agua, adecuadamente en combinación con otros componentes como, por ejemplo, glicerol, hidroxietilcelulosa y similares. Las formulaciones tópicas contienen por lo menos aproximadamente 0,1, por lo menos aproximadamente 0,25, por lo menos aproximadamente 0,5, por lo menos aproximadamente 1, por lo menos aproximadamente 2, o por lo menos aproximadamente un 5% en peso del compuesto o sal de la invención. Las formulaciones tópicas pueden envasarse adecuadamente en tubos de, por ejemplo, 100 g que se asocian opcionalmente con instrucciones para el tratamiento de la indicación seleccionada, por ejemplo, psoriasis u otra afección de la piel.
La cantidad de compuesto, sal o composición administrada a un paciente variará dependiendo de lo que se esté administrando, el propósito de la administración, como profilaxis o terapia, el estado del paciente, la manera de administración y similares. En aplicaciones terapéuticas, las composiciones pueden administrarse a un paciente que ya padece una enfermedad en una cantidad suficiente para curar o por lo menos detener parcialmente los síntomas de la enfermedad y sus complicaciones. Las dosis efectivas dependerán del estado de enfermedad que se esté tratando así como del juicio del médico que lo atiende, dependiendo de factores como la gravedad de la enfermedad, la edad, el peso y el estado general del paciente, y similares.
Las composiciones administradas a un paciente pueden estar en forma de las composiciones farmacéuticas descritas anteriormente. Estas composiciones pueden esterilizarse mediante técnicas de esterilización convencionales o pueden esterilizarse por filtración. Las soluciones acuosas pueden envasarse para su uso tal cual, 0 liofilizarse, la preparación liofilizada combinándose con un portador acuoso estéril antes de la administración. El pH de las preparaciones de compuestos estará típicamente entre 3 y 11, más preferiblemente entre 5 y 9 y lo más preferiblemente entre 7 y 8. Se entenderá que el uso de algunos de los excipientes, portadores o estabilizantes anteriores dará como resultado la formación de sales farmacéuticas.
La dosificación terapéutica de un compuesto o sal proporcionados por los procesos de la presente solicitud puede variar de acuerdo con, por ejemplo, el uso particular para el que se realiza el tratamiento, la manera de administración del compuesto o sal, la salud y el estado del paciente, y el juicio del médico que prescribe. La proporción o concentración de un compuesto o sal proporcionado por los procesos de la invención en una composición farmacéutica puede variar dependiendo de una serie de factores que incluyen dosificación, características químicas (por ejemplo, hidrofobicidad) y la vía de administración. Por ejemplo, los compuestos y sales proporcionados por los procesos de la invención se pueden proporcionar en una solución de tampón fisiológica acuosa que contiene de aproximadamente un 0,1 a aproximadamente un 10% p/v del compuesto para administración parenteral. Algunos intervalos de dosis típicos son de aproximadamente 1 pg/kg a aproximadamente 1 g/kg de peso corporal por día. En algunas realizaciones, el intervalo de dosis es de aproximadamente 0,01 mg/kg a aproximadamente 100 mg/kg de peso corporal por día. Es probable que la dosificación dependa de variables como el tipo y grado de progresión de la enfermedad o trastorno, el estado de salud general del paciente en particular, la eficacia biológica relativa del compuesto seleccionado, la formulación del excipiente y su vía de administración. Las dosis eficaces pueden extrapolarse a partir de curvas de respuesta a la dosis derivadas de sistemas de ensayo de modelos animales o in vitro.
Las composiciones pueden incluir además uno o más agentes farmacéuticos adicionales como agentes quimioterapéuticos, esteroides, compuestos antiinflamatorios o inmunosupresores, ejemplos de los cuales se proporcionan en la presente.
Kits
Los kits farmacéuticos útiles, por ejemplo, en el tratamiento o prevención de enfermedades o trastornos asociados a PI3K, como el cáncer, pueden incluir uno o más recipientes que contienen una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de un compuesto. Tales kits pueden incluir además, si se desea, uno o más de varios componentes de kits farmacéuticos convencionales como, por ejemplo, recipientes con uno o más portadores farmacéuticamente aceptables, recipientes adicionales, etc., como será fácilmente evidente para los expertos en la técnica. También pueden incluirse en el kit instrucciones, ya sea en forma de prospectos o etiquetas, que indican las cantidades de los componentes que se van a administrar, las pautas de administración y/o las pautas para mezclar los componentes.
La invención se describirá con mayor detalle a modo de ejemplos específicos. Los siguientes ejemplos se ofrecen con propósitos ilustrativos y no se pretende que limiten la invención de ninguna manera. Los expertos en la técnica reconocerán fácilmente una variedad de parámetros no críticos que pueden cambiarse o modificarse para producir esencialmente los mismos resultados. Se ha descubierto que la sal de ácido clorhídrico del compuesto de Fórmula I y el compuesto de Fórmula I son inhibidores de PI3K de acuerdo con por lo menos un ensayo descrito en la presente.
EJEMPLOS
La invención se describirá con mayor detalle mediante ejemplos específicos. Los siguientes ejemplos se ofrecen con propósitos ilustrativos y no se pretende que limiten la invención de ninguna manera. Los compuestos de ejemplo, o sales de los mismos, que contienen uno o más centros quirales se obtuvieron en forma de racemato o como mezclas isoméricas, a menos que se especifique lo contrario.
Métodos generales
Las purificaciones LC-MS preparativas de algunos de los compuestos preparados se realizaron en sistemas de fraccionamiento dirigido de masa de Waters. La configuración básica del equipo, los protocolos y el software de control para el funcionamiento de estos sistemas se han descrito en detalle en la literatura. Ver, por ejemplo, "Two-Pump At Column Dilution Configuration for Preparative LC-MS", K. Blom, J. Combi. Chem., 4, 295 (2002); "Optimizing Preparative LC-MS Configurations and Methods for Parallel Synthesis Purification'', K. Blom, R. Sparks, J. Doughty, G. Everlof, T. Haque, A. Combs, J. Combi. Chem., 5, 670 (2003); y "Preparative LC-MS Purification: Improved Compound Specific Method Optimization", K. Blom, B. Glass, R. Sparks, A. Combs, J. Combi. Chem., 6, 874-883 (2004). Los compuestos separados se sometieron típicamente a espectrometría de masas de cromatografía líquida analítica (LCMS) para análisis de pureza en las siguientes condiciones: Instrumento, serie Agilent 1100, LC/MSD, columna: Waters Sunfire™ C185 pm, 2,1 x 50 mm, tampones: fase móvil A: TFA al 0,025% en agua y fase móvil B: acetonitrilo; gradiente del 2% al 80% de B en 3 minutos con caudal de 2,0 ml/minuto.
Algunos de los compuestos preparados también se separaron a una escala preparativa mediante cromatografía líquida de alto rendimiento de fase inversa (RP-HPLC) con detector de MS o cromatografía ultrarrápida (gel de sílice) como se indica en los Ejemplos. Las condiciones típicas de la columna de cromatografía líquida de alto rendimiento de fase inversa preparativa (RP-HPLC) son las siguientes:
pH = 2 purificaciones: columna Waters Sunfire™ C185 pm, 19 x 100 mm, eluyendo con fase móvil A: TFA (ácido trifluoroacético)al 0,1% en agua y fase móvil B: acetonitrilo; el caudal fue de 30 ml/minuto, el gradiente de separación se optimizó para cada compuesto usando el protocolo de optimización del método específico del compuesto como se describe en la literatura [ver " Preparative LCMS Purification: Improved Compound Specific Method Optimization", K. Blom, B. Glass, R. Sparks, A. Combs, J. Comb. Chem., 6, 874-883 (2004)]. Típicamente, el caudal usado con la columna de 30 x 100 mm fue de 60 ml/minuto.
pH = 10 purificaciones: columna Waters XBridge C185 pm, 19 x 100 mm, eluyendo con fase móvil A: 0,15% de NH4OH en agua y fase móvil B: acetonitrilo; el caudal fue de 30 ml/minuto, el gradiente de separación se optimizó para cada compuesto usando el protocolo de optimización del método específico del compuesto como se describe en la literatura [Ver "Preparative LCMS Purification: Improved Compound Specific Method Optimization", K. Blom, B. Glass, R. Sparks, A. Combs, J. Comb. Chem., 6, 874-883 (2004)]. Típicamente, el caudal usado con una columna de 30 x 100 mm fue de 60 ml/minuto.
Algunos de los compuestos preparados también se analizaron mediante calorimetría diferencial de barrido (DSC). Las condiciones típicas del instrumento DSC son las siguientes:
Calorimetría diferencial de barrido de TA instrument, modelo Q200 con automuestreador: 30-350° C a 10° C/min; Plato y tapa para muestras de aluminio T-cero; flujo de gas nitrógeno a 50 ml/min.
Instrumento 822 de calorimetría diferencial de barrido (DSC) de Mettler Toledo: 40-340° C a una velocidad de calentamiento de 10° C/min.
Algunos de los compuestos preparados también se analizaron mediante análisis termogravimétrico (TGA). Las condiciones típicas del instrumento de TGA son las siguientes:
Analizador termogravimétrico de TA Instrument, modelo Pyris: rampa de temperatura de 25° C a 300° C a 10° C/min; flujo de gas de purga de nitrógeno a 60 ml/min; Portamuestras de crisol de cerámica TGA.
TA Instrument Q500: rampa de temperatura de 20° C a 300° C a 10° C/min.
Algunos de los compuestos preparados también se analizaron mediante difracción de rayos X de polvo (XRPD). Las condiciones típicas del instrumento de XRPD son las siguientes:
Instrumento difractómetro de rayos X de polvo Bruker D2 PHASER: Longitud de onda de radiación de rayos X: 1,05406 A CuKAI; potencia de rayos X: 30 KV, 10 mA; polvo de muestra: disperso en un portamuestras de fondo cero; condiciones generales de medición: ángulo de inicio - 5 grados, ángulo de parada - 60 grados, muestreo -0,015 grados, velocidad de escaneo - 2 grados/min.
Difractómetro de polvo Rigaku Miniflex: Cu a 1.054056 Á con filtro Kp; condiciones generales de medición: ángulo de inicio - 3 grados, ángulo de parada - 45 grados, muestreo - 0,02 grados, velocidad de escaneo - 2 grados/min.
Ejemplo 1. Síntesis de (R)-4-(3-cloro-6-etox¡-2-fluoro-5-((R)-1-h¡drox¡et¡l)fen¡l)pirrol¡d¡n-2-ona

Paso 1. 1-(5-Cloro-4-fluoro-2-hidroxifenil)etanona (ii)

Se cargaron 4-cloro-3-fluorofenol (i, 166 g, 1,11 mol) y cloruro de acetilo (107 ml, 1,50 mol) en un matraz de 5 l a temperatura ambiente. La mezcla de la reacción se agitó y se convirtió en una solución transparente mientras se registraba que la temperatura del lote disminuía a 6° C. Después, la mezcla de la reacción se calentó a 60° C durante 2 h. La mezcla de la reacción se cargó con nitrobenceno (187,5 ml, 1,82 mol) y posteriormente se enfrió a temperatura ambiente. Luego, se añadió tricloruro de aluminio (160 g, 1,2 mmol) a la mezcla en tres porciones (50 g, 50 g y 60 g a intervalos de 5 minutos). La temperatura del lote aumentó a 78° C una vez completada la adición. Luego, la mezcla de la reacción se calentó a 100-120°C durante 3 h, momento en el que el análisis de HPLC mostró que la reacción se había completado. Luego, la mezcla de la reacción se enfrió a 0° C y se cargó con hexanos (0,45 l), acetato de etilo (0,55 l), y luego se cargó lentamente con ácido clorhídrico acuoso 1,0 N (1,0 l) a temperatura ambiente. La adición de ácido clorhídrico acuoso fue exotérmica y la temperatura del lote aumentó de 26° C a 60° C. La mezcla resultante se agitó a temperatura ambiente durante 20 min. Las capas se separaron y la capa orgánica se lavó secuencialmente con ácido clorhídrico acuoso 1,0 N (2 x 600 ml) y agua (400 ml). Luego, la capa orgánica se extrajo con solución de hidróxido de sodio acuosa 1,0 N (2 x 1,4 l). Las soluciones básicas combinadas se
acidificaron a pH 2 mediante la adición de ácido clorhídrico acuoso 12 N hasta que no se separó más precipitado. El sólido resultante se recogió por filtración, se lavó con agua y se secó en el embudo de filtración bajo succión para dar el compuesto ii como un sólido amarillo (187,4 g, 89,5%). 1H NMR (400 MHz, CDCta) 512.44 (d, J=1.4 Hz, 1H), 7.78 (d, J=8.1 Hz, 1H), 6.77 (d, J=10.2 Hz, 1H), 2.61 (s, 3H).
Paso 2. 1-(5-cloro-4-fluoro-2-hidroxi-3-yodofenil)etanona (iii)

Se disolvió 1-(5-cloro-4-fluoro-2-hidroxifenil)etanona (ii, 100,0 g, 530,3 mmol) en ácido acético (302 ml) y se añadió N-yodosuccinimida (179,2 g, 796,5 mmol) a la solución. La mezcla de la reacción se agitó entre aproximadamente 61° C y aproximadamente 71° C durante 2 h, momento en el que el análisis HPLC indicó que la reacción se había completado. Luego, la mezcla de la reacción se enfrió a temperatura ambiente, se añadió agua (613 ml) y la suspensión resultante se agitó a temperatura ambiente durante 30 min. El producto se recogió por filtración y se lavó con agua para producir sólidos marrones. El producto húmedo se disolvió en ácido acético (400 ml) a 60°C. Se añadió agua (800 ml) (durante 15 min) a la solución para precipitar el producto puro. El producto se recogió por filtración y se lavó con agua (100 ml). El producto se secó en el embudo de filtración bajo succión durante 18 h para dar el compuesto iii en forma de un sólido marrón (164,8 g, rendimiento del 95,0%). 1H NMR (300 MHz, DMSO-ds) 513.34 (s, 1H), 8.26 (d, J=8.4 Hz, 1H), 2.68 (s, 3H).
Paso 3. 1-(5-cloro-2-etoxi-4-fluoro-3-yodofenil)etanona (iv)

En un matraz de fondo redondo de tres bocas de 5 l equipado con un condensador y un termómetro, se disolvió 1-(5-cloro-4-fluoro-2-hidroxi-3-yodofenil)etanona (iii, 280 g, 840 mmol) en N,N-dimetilformamida (600 ml). Durante la disolución, la temperatura interna descendió de 19,3° C a 17,0° C. Se añadió yodoetano (81,2 ml, 1020 mmol) a la mezcla resultante. Luego, se añadió carbonato de potasio (234 g, 1690 mmol) durante 2 min a la mezcla de la reacción y no se observó ningún cambio en la temperatura del lote. La mezcla de la reacción se calentó a 60° C durante 3 h, momento en el que el análisis HPLC indicó que la reacción se había completado. La mezcla de la reacción se dejó enfriar a temperatura ambiente y el producto se recogió por filtración. Los sólidos se disolvieron en una mezcla de DCM (1,0 l), hexano (500 ml) y agua (2,1 l). El sistema bifásico se agitó a 20° C durante 20 min. Las capas se separaron y la capa acuosa se extrajo con DCM (1,0 l). La capa orgánica combinada se lavó con agua (2 x 250 ml) y salmuera (60 ml). La fase orgánica se separó, se secó sobre Na2SO4 anhidro, se filtró y se concentró al vacío hasta la sequedad para dar el compuesto iv como un sólido amarillo (292 g, rendimiento del 94%). 1H NMR (400 MHz, DMSO-ds) 57.69 (d, J=8.4 Hz, 1H), 3.95 (q, J=7.0 Hz, 2H), 2.62 (s, 3H), 1.49 (t, J=7.0 Hz, 3H). LCMS para C10H10ClFIO2 (M+H)+: m/z=342.9.
Paso 4.2-(5-cloro-2-etoxi-4-fluor-3-yodofenil)-2-metil-1,3-dioxolano (v)

Se trató una solución de 1-(5-cloro-2-etoxi-4-fluoro-3-yodofenil)etanona (iv, 250,0 g, 693,4 mmol) y 1,2-etanodiol (58,0 ml, 1040 mmol) en tolueno (1.5 l) con monohidrato de ácido p-toluenosulfónico (10,6 g, 55,5 mmol). El matraz de reacción se equipó con una trampa Dean-Stark y la mezcla se calentó a reflujo durante 7 h. El análisis LCMS indicó que la mezcla de la reacción contenía un 8,3% de material de partida y un 91,7% de producto. La mezcla de la reacción se enfrió a 106° C y se introdujo una cantidad adicional de 1,2-etanodiol (11,6 ml, 208 mmol)
mediante una jeringuilla. Luego, la mezcla de la reacción se calentó a reflujo durante 8 h adicionales. El análisis LCMS indicó que la mezcla de la reacción contenía un 3,6% de material de partida y un 96,4% de producto. La mezcla de la reacción se enfrió a 106°C y se introdujo 1,2-etanodiol adicional (7,73 ml, 139 mmol) mediante una jeringuilla. La mezcla de la reacción se calentó a reflujo durante 15,5 h adicionales. El análisis LCMS indicó que la mezcla de la reacción contenía un 2,2% de material de partida y 97,8% de producto.
Luego, la mezcla de la reacción se enfrió a 0° C y se añadieron agua (200 ml) y NaHCÜ3 acuoso saturado (300 ml) para ajustar la mezcla a un pH de 9. Se añadió DCM (200 ml) y el lote se agitó durante 10 min. Las capas se separaron y la capa acuosa se extrajo con tolueno (300 ml). La capa orgánica combinada se lavó secuencialmente con una mezcla de agua (200 ml) y NaHCÜ3 acuoso saturado (200 ml), agua (300 ml), salmuera (300 ml), se secó sobre Na2SÜ4 anhidro, se filtró y se concentró al vacío hasta la sequedad para proporcionar el compuesto bruto como un sólido marrón claro (268 g, rendimiento del 100%). 1H NMR (400 m Hz, c Dc I3) 57.59 (d, J=8.6 Hz, 1H), 4.26-3.96 (m, 4H), 3.92-3.72 (m, 2H), 1.74 (s, 3H), 1.50 (t, J=7.0 Hz, 3H). LCMS para C12H14CIFIO3 (M+H)+: m/z=387.0.
Paso 5. 3-cloro-6-etoxi-2-fluoro-5-(2-metil-1,3-dioxolan-2-il)benzaldehído (vi)

A una solución agitada de 2-(5-cloro-2-etoxi-4-fluoro-3-yodofenil)-2-metil-1,3-dioxolano (v, 135,0 g, 349,2 mmol) (86,8% de pureza por HPLC con 5,5% de la cetona) en tetrahidrofurano anhidro (300 ml) a aproximadamente de 0° C a aproximadamente 3° C se le añadió lentamente complejo de cloruro de litio cloruro de isopropilmagnesio 1,3 M en THF (322,3 ml, 419,0 mmol) durante 1 h. La mezcla de la reacción se agitó a de aproximadamente 0° C a aproximadamente 5° C durante 30 min, momento en el cual el análisis LCMS indicó que la reacción de intercambio de yodo-magnesio se había completado. Luego, se añadió a la mezcla de la reacción N-formilmorfolina (71,1 ml, 700 mmol) durante 1 h a de aproximadamente 0° C a aproximadamente 8° C. La mezcla de la reacción se agitó a de aproximadamente 0° C a aproximadamente 8° C durante 1 hora adicional, momento en el que los análisis LCMS y HPLC mostraron que el material de partida se había consumido y se observó una cantidad significativa de subproducto de la desyodación 2-(5-cloro-2-etoxi-4-fluorofenil)-2-metil-1,3-dioxolano. La reacción se inactivó con una solución acuosa de ácido cítrico (120,8 g, 628,6 mmol) en agua (1,20 l) a 0° C. Luego, la mezcla de la reacción inactivada se extrajo con EtOAc (2 x 600 ml). Las fases se separaron fácilmente. La capa orgánica combinada se lavó secuencialmente con agua (300 ml) y salmuera (500 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida sobre gel de sílice con EtOAc/hexano al 0-10% para dar el producto bruto compuesto vi como un sólido amarillo pálido, que era una mezcla del producto deseado, 3-cloro-6-etoxi-2-fluoro-5-(2-metil-1,3-dioxolan-2-il)benzaldehído (vi, 80 g, 80%) que contenía un 36% en moles del subproducto de desyodación, 2-(5-cloro-2-etoxi-4-fluorofenil)-2-metil-1,3-dioxolano como se indica por análisis de NMR. El producto bruto compuesto vi se purificó adicionalmente mediante la formación del correspondiente aducto de bisulfito de sodio.
Se disolvió bisulfito de sodio (36,91 g, 354,7 mmol) en agua (74,3 ml, 4121 mmol). A una solución agitada de 3-cloro-6-etoxi-2-fluoro-5-(2-metil-1,3-dioxolan-2-il)benzaldehído bruto (vi, 80,00 g, 177,3 mmol) en acetato de etilo (256,0 ml), la solución de bisulfito de sodio recién preparada se añadió en una porción. La solución se agitó durante aproximadamente 10 min y se observaron precipitados. Luego, la lechada se agitó durante 1 h adicional. El aducto de aldehído-bisulfito se recogió por filtración, se lavó con EtOAc y se secó al vacío y en atmósfera de nitrógeno durante 20 h para dar un sólido blanco (58,2 g, rendimiento del 83,6%). Al aducto de aldehído-bisulfito (58,2 g, 148 mmol) mezclado en hidróxido de sodio acuoso 1,0 M (296 ml, 296 mmol) se le añadió metil t-butil éter (600 ml) (MTBE). La mezcla de la reacción se agitó a temperatura ambiente durante 6 min para dar una mezcla bifásica transparente y se continuó agitando durante 5 min adicionales. Se recogió la fase orgánica y se extrajo la fase acuosa con MTBE (2 x 300 ml). Las capas orgánicas combinadas se lavaron con salmuera (300 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron al vacío para dar el compuesto vi puro como un sólido cristalino blanco (31,4 g, rendimiento del 73,4%). 1H NMR (400 MHz, CDCI3) 510.27 (s, 1H), 7.78 (d, J=8.5 Hz, 1H), 4.10-3.96 (m, 4H), 3.87-3.76 (m, 2H), 1.72 (s, 3H), 1.44 (t, J=7.0 Hz, 3H). LCMS para C13H15CIFO4 (M+H)+: m/z=289.0.
Paso 6. (E)-2-(5-cloro-2-etoxi-4-fluoro-3-(2-nitrovinil)fenil)-2-metil-1,3-dioxolano (vii)

En un matraz de fondo redondo de 4 bocas de 5 l equipado con agitador superior, tabiques, termopar, entrada de nitrógeno y condensador se cargó 3-cloro-6-etoxi-2-fluoro-5-(2-metil-1,3-dioxolan-2-il)benzaldehído (vi, 566,2 g, 1961 mmol), nitrometano (1060 ml, 19600 mmol) y ácido acético glacial (1120 ml). Luego, la mezcla de la reacción se cargó con bencilamina (53,6 ml, 490 mmol) y la mezcla resultante se calentó a 60° C y la reacción se monitorizó mediante LCMS durante 5,5 h. Se realizó un análisis de referencia inicial en t = 0. La reacción se comprobó después de 2 h y 5 h. Después de 2 h, había aproximadamente un 20% del material de partida aldehido sin reaccionar. Después de 5 h, el perfil de reacción fue el siguiente: material de partida compuesto vi (<2%), producto intermedio imina (<4%), producto compuesto vii (>93%) y aducto de Michael de bencilamina (no detectado). En el tiempo de reacción de 5,5 h, la reacción se consideró completa. La mezcla de la reacción se enfrió a temperatura ambiente y se diluyó con acetato de etilo (3,0 l). La mezcla se dividió por la mitad para su procesamiento debido al gran volumen involucrado.
Cada mitad se trató de acuerdo con el siguiente procedimiento: La mezcla de la reacción se lavó primero con NaCl 1,5 M en agua (2 x 1500 ml; después de cada lavado, la producción de volumen acuoso aumentó en comparación con la entrada, lo que sugiere que se estaban eliminando el ácido acético y/o el nitrometano.). Luego, la mezcla se enfrió a aproximadamente 15°C y se lavó con una solución acuosa de NaOH 4 M (4 x 300 ml) hasta que el extracto acuoso alcanzó un pH de 8-9. Durante los lavados iniciales, la capa acuosa permaneció ácida, pero a medida que la acuosa se volvió ligeramente básica durante los lavados posteriores, la mezcla se calentó y el extracto se volvió oscuro. Las capas se separaron. La capa orgánica después del ajuste de pH se lavó luego con cloruro de sodio 1,5 M en agua (1000 ml) y agua (500 ml). Se observaron emulsión y partición lenta durante estos lavados finales. La fase orgánica se secó sobre MgSO4 anhidro, se filtró y se concentró a presión reducida. El jarabe color ámbar resultante se colocó a vacío alto durante la noche. El jarabe solidificó y se obtuvieron 740 g de producto bruto.
Se añadió heptano (1,3 l) al producto bruto, formando una lechada, que se calentó en un baño de agua a 60° C hasta que se disolvieron todos los sólidos. La solución resultante se filtró para pulir en un matraz de fondo redondo de 4 bocas de 3 l limpio equipado con un agitador superior y entrada de nitrógeno. El filtro se enjuagó con heptano (40 ml). La solución filtrada se enfrió a temperatura ambiente y se agitó durante 5 h. Se observaron precipitados sólidos y la suspensión se enfrió a 0° C en un baño de hielo durante 1 h. El producto se recogió por filtración y la torta húmeda resultante se lavó con 500 ml de heptano enfriado con hielo. El producto se secó parcialmente sobre el filtro bajo succión al vacío y se secó adicionalmente bajo vacío alto durante la noche. El filtrado y la solución de lavado se concentraron a presión reducida y el residuo resultante se mantuvo para purificación en columna.
Los sólidos de la cristalización de heptano se disolvieron en un pequeño volumen de DCM y EtOAc/hexano al 20% y se cargaron en una columna que contenía aproximadamente 1 kg de gel de sílice. La columna se eluyó con EtOAc/hexano al 20%. Las fracciones deseadas se combinaron y concentraron a presión reducida para dar un sólido amarillo. El sólido se secó a vacío alto durante la noche para dar 497 g del producto compuesto vii como un sólido cristalino amarillo pálido.
El filtrado concentrado y el lavado de la cristalización de heptano se cargaron en la misma columna que antes usando EtOAc/hexano al 20%. La columna se eluyó usando el mismo sistema solvente que antes para eliminar las impurezas de la referencia y el ácido acético residual. Las fracciones deseadas se combinaron y concentraron a presión reducida para dar un aceite color ámbar rojizo. El aceite se colocó a vacío alto y se obtuvieron aproximadamente 220 g de producto bruto. Este producto bruto se disolvió en heptano (500 ml) y se sembró con una pequeña cantidad del producto sólido de la primera cosecha. La lechada se agitó a temperatura ambiente durante 16 h y luego se enfrió en un baño de hielo durante 3 h. El segundo producto de cosecha se recogió mediante filtración. El producto se secó a vacío alto y se obtuvieron 110 g de producto en forma de un sólido amarillo. La cantidad total de producto compuesto vii obtenido fue de 607 g (rendimiento del 93,3%). 1H NMR (400 MHz, DMSO-ds) 57.94 (s, 2H), 7.68 (d, J=8.9 Hz, 1H), 4.07-3.95 (m, 4H), 3.82-3.73 (m, 2H), 1.65 (s, 3H), 1.39 (t, J=7.0 Hz, 3H). LCMS para CuH16ClFNO5 (M+H)+: m/z=332.0.
Paso 7. (R)-dimetil-2-(1-(3-cloro-6-etoxi-2-fluoro-5-(2-metil-1,3-dioxolan-2-il)fenil)-2-nitroetil)malonato (viii)

En un matraz de fondo redondo de 2 l con barra de agitación magnética y entrada de nitrógeno que contenía (E)-2-(5-cloro-2-etoxi-4-fluoro-3-(2-nitrovinil)fenil)-2-metil-1,3-dioxolano (vii, 352,8 g, 1064 mmol) se añadió tetrahidrofurano anhidro (1,06 l) y malonato de dimetilo (146 ml, 1280 mmol). Se cargó (1S,2S)-NN’-dibencilciclohexano-1,2-diamina-dibromoníquel (Catalizador de Evans, 21,4 g, 26,6 mmol) a la mezcla de la reacción. La mezcla de la reacción era de color marrón y se observó una solución homogénea. La mezcla de la reacción se agitó a temperatura ambiente durante 18,5 h. La reacción se analizó después de 18,5 h mediante HPLC. El material de partida sin reaccionar, compuesto vii, estaba presente al 2%. La mezcla de la reacción se concentró a presión reducida para eliminar el THF y el residuo resultante se purificó mediante cromatografía ultrarrápida (DCM/hexanos usados para la carga, se usaron 1422 g de gel de sílice para esta columna y la columna se eluyó con EtOAc/hexano del 10% al 20%; las fracciones de la columna se monitorizaron mediante TLC usando EtOAc/hexano al 30% como eluyente y se visualizaron mediante UV). Las fracciones deseadas se combinaron y concentraron a presión reducida. El residuo se secó a vacío alto. Con el tiempo, el jarabe se solidificó a sólidos amarillo claro (503,3 g) y se tomó una muestra para análisis por HPLC quiral. La pureza quiral era del 95,7% del enantiómero (R) deseado y del 4,3%) del enantiómero (S) no deseado. Se añadió etanol (1,0 l) a los sólidos y la mezcla se calentó en un baño de agua a 60° C hasta que se disolvieron los sólidos. La solución se filtró para pulir en un fondo redondo de 4 bocas de 3 l limpio a través de papel de filtro Whatman N° 1. La solución filtrada se enfrió a temperatura ambiente mientras se agitaba. Se observó cristalización después de 30 min de agitación y la lechada se agitó a temperatura ambiente durante 16 h. La lechada se enfrió en un baño de hielo durante 1 h. El producto se recogió por filtración y la torta de filtración resultante se lavó con etanol helado (500 ml) y se secó parcialmente sobre el filtro. Los sólidos se secaron a vacío alto para dar el producto viii deseado (377,5 g) como sólidos cristalinos blancos. Se determinó mediante HPLC quiral que la pureza quiral era del 100% del enantiómero (R) deseado y del 0% del enantiómero (S) no deseado.
El filtrado y el lavado se combinaron y se concentraron a presión reducida hasta un aceite (118,9 g). El aceite se disolvió en etanol (475,0 ml) (4 ml/g) y se sembró con los cristales de la primera cosecha. La lechada resultante se agitó durante 16 h a temperatura ambiente y luego se enfrió en un baño de hielo durante 5,5 h. El segundo producto de cosecha se aisló por filtración y se secó parcialmente sobre el filtro. Se secó a vacío alto para dar el segundo producto de cosecha (31,0 g). Se determinó por HPLC quiral que la pureza quiral era del 98,3% del enantiómero (R) deseado y del 1,7% del enantiómero (S) no deseado. El rendimiento combinado de la primera y segunda cosecha de producto fue de 408,5 g con un rendimiento del 82,8%. 1H NMR (500 MHz, DMSO-d6) 57.51 (d, J=9.0 Hz, 1H), 5.20 4.81 (m, 2H), 4.62 (m, 1H), 4.14-4.03 (m, 2H), 4.03-3.97 (m, 2H), 3.95-3.88 (m, 1H), 3.84-3.72 (m, 2H), 3.70 (s, 3H), 3.38 (s, 3H), 1.61 (s, 3H), 1.39 (t, J=6.9 Hz, 3H). LCMS para C1gH24ClFOg (M+H)+: m/z=463.9.
Paso 8.2-(1-(3-acetil-5-cloro-2-etoxi-6-fluorofenil)-2-nitroetiljmalonato de (R)-dimetilo (ix)

A una solución agitada de (R)-dimetil-2-(1-(3-cloro-6-etoxi-2-fluoro-5-(2-metil-1,3-dioxolan-2-il)fenil)-2-nitroetil)malonato (viii, 244,0 g, 526,0 mmol) en acetona (1,2 l) en un matraz de fondo redondo de 5 l de tres bocas, equipado con un agitador mecánico, se añadió yodo (13,4 g, 52,6 mmol) a temperatura ambiente. La solución marrón resultante se calentó a 50° C en un baño de agua durante 30 min, momento en el que el análisis LCMS mostró que la reacción se había completado. La mezcla de la reacción se enfrió a temperatura ambiente y luego se inactivó con una solución de tiosulfato de sodio (17,0 g, 108 mmol) en agua (160 ml) para dar una solución transparente amarillo pálido. En este momento, se cargó una cantidad adicional de agua (1,2 l) a la solución inactivada y la lechada blanca resultante se agitó a temperatura ambiente durante 1 h. Luego, el sólido se recogió mediante filtración y se volvió a disolver en acetona (1,4 l) a 40° C. La solución se enfrió a temperatura ambiente y
luego se añadió agua adicional (1,4 l). La lechada blanca resultante se agitó a temperatura ambiente durante 1 h. El sólido se recogió por filtración y se lavó con agua (3 x 100 ml). El producto sólido se secó en un embudo de filtración bajo succión con un flujo de nitrógeno durante 46 h para dar el compuesto ix como un sólido blanco (212 g, rendimiento del 96%). 1H NMR (300 MHz, CDCla) 57.54 (d, J=8.6 Hz, 1H), 5.09-4.67 (m, 3H), 4.10-3.83 (m, 3H), 3.90 (s, 3H), 3.57 (s, 3H), 2.57 (s, 3H), 1.46 (t, J=7.0 Hz, 3H). LCMS para C17H20CFNO8 (M+H)+: m/z=420.1.
Paso 9.2-((R)-1-(3-cloro-6-etoxi-2-fluoro-5-((R)-l-hidroxietil)fenil)-2-nitroetiljmalonato de dimetilo (x)

A una solución agitada de (3a,S)-1-metil-3,3-difeniltetrahidro-3H-pirrolo[1,2-c][1,3,2]oxazaborol, ((S)-MeCBS, 16,39 g, 59,12 mmol, 0,1 eq.) En THF anhidro (100 ml) en un matraz de fondo redondo de 5 l a temperatura ambiente, se le añadió una solución de complejo de borano-THF 1,0 M en THF (591 ml, 591 mmol, 1 eq.) seguido de eterato de trifluoruro de boro (3,75 ml, 29,6 mmol, 0,05 eq). La solución resultante se agitó a temperatura ambiente durante 30 min. Luego se añadió gota a gota una solución de [(1R)-1-(3-acetil-5-cloro-2-etoxi-6-fluorofenil)-2-nitroetil]malonato de dimetilo (ix, 253,0 g, 591,2 mmol) en THF anhidro (1,7 l) mediante un embudo de adición durante 60 min. El matraz que contenía la cetona ix se enjuagó con THF anhidro (135 ml) y la solución se añadió gota a gota mediante el embudo de adición a la mezcla de la reacción. La solución resultante se agitó a temperatura ambiente durante 10 min adicionales, momento en el que el análisis LCMS mostró la conversión completa de la cetona en alcohol. La reacción se inactivó mediante la adición gota a gota de metanol (71,8 ml, 1770 mmol) a 0°C. La mezcla de la reacción inactivada se agitó a temperatura ambiente durante 15 min antes de concentrarse al vacío para dar el producto bruto. Los productos brutos de este lote y un lote similar (usando 200 g de material de partida) se combinaron y se purificaron por cromatografía en columna de gel de sílice usando del 0 al 5% de MeOH/DCM como eluyente para dar el compuesto x como un sólido blanco (437 g, rendimiento del 97,9%). 1H NMR (300 MHz, CDCla) 57.50 (d, J=8.8 Hz, 1H), 5.13 (q, J=6.3 Hz, 1H), 5.01-4.65 (m, 3H), 4.14-3.89 (m, 3H), 3.79 (s, 3H), 3.57 (s, 3H), 1.57-1.42 (m, 6H). LCMS para C17H21ClFNNaOe (M+Na)+: m/z=444.0.
Paso 10.4-(3-cloro-6-etoxi-2-fluoro-5-((R)-1-hidroxietil)fenil)-2-oxopirrolidin-3-carboxilato de (4R)-metilo (xi)

Un matraz de fondo redondo Morton de 3 bocas que contenía ((1R)-1-{3-cloro-6-etoxi-2-fluoro-5-[(1R)-1-hidroxietil]fenil}-2-nitroetil)malonato de dimetilo (x, 100,0 g, 237,1 mmol) en tetrahidrofurano (800,0 ml) y níquel Raney (120 g después de eliminar el agua con una pipeta) se equipó con un condensador, un agitador mecánico (varilla de agitación de vidrio y rodamiento de teflón) y dos globos de gas de hidrógeno (después de la purga al vacío). El matraz se colocó en un baño de aceite a 65° C. El lote se agitó vigorosamente durante 16 horas y los globos se retiraron periódicamente y se volvieron a llenar con hidrógeno. Se tomó una muestra y se analizó por HPLC. El producto, compuesto xi, estaba presente al 83%. Había 7,8% de amina no ciclada y 5,5% de subproducto de hidroxilamina en la mezcla de la reacción. El catalizador se eliminó por filtración (debe tenerse cuidado de no permitir que el níquel Raney se seque para exponerlo al aire). El filtrado se evaporó hasta la sequedad para dar 91 g de producto bruto como una espuma blanca. El producto bruto (91 g, pureza del área del 82,6%) se combinó con otro lote similar de producto bruto (93 g, pureza del 72,8%) para la purificación. El producto bruto combinado (184 g) se purificó mediante cromatografía en columna sobre gel de sílice (EtOAc/hexano como eluyente) para dar el compuesto xi (101,1 g, pureza por HPLC del 93%, rendimiento bruto del 59,3%). El material bruto se usó para el siguiente paso sin purificación adicional. LCMS para C16H20CFNO5 (M+H)+: m/z=360.0.
Paso 11. Ácido (4R)-4-(3-cloro-6-etoxi-2-fluoro-5-((R)-1-hidroxietil)fenil)-2-oxopirrolidin-3-carboxílico (xii)

En un matraz de fondo redondo de 4 bocas y 5 l equipado con un agitador superior y una entrada de nitrógeno se cargó una solución de 4-(3-cloro-6-etoxi-2-fluoro-5-((R)-1-hidroxietil)fenil)-2-oxopirrolidin-3-carboxilato de (R)-metilo (xi, 268 g, 581 mmol) en tetrahidrofurano (2150 ml, 26500 mmol). A la solución se le cargó hidróxido de sodio 1,0 M en agua (1420 ml, 1420 mmol). La solución turbia resultante se volvió transparente en 1 min. La mezcla de la reacción se agitó durante la noche a temperatura ambiente. La reacción se analizó por LCMS después de 15 horas y pareció completarse ya que no se observó el material de partida. La mezcla de la reacción se enfrió en un baño de hielo a una temperatura interna de 9,52C y la mezcla se acidificó a pH 1-2 mediante la adición de ácido clorhídrico acuoso 6,0 M (237,0 ml, 1422 mmol) mediante un embudo de goteo durante 30 min. La mezcla de la reacción se dividió por la mitad y cada mitad se extrajo con acetato de etilo (2 x 1 l). Las capas acuosas combinadas se extrajeron adicionalmente con acetato de etilo (500 ml). Las dos capas orgánicas se lavaron con salmuera (20% p/p de NaCl/agua, 2 x 1000 ml), se secaron sobre MgSÜ4 anhidro, se filtraron y se concentraron para dar el ácido intermedio xii bruto como una espuma amarillenta que se usó directamente en la siguiente reacción. 1H NMR (400 MHz, DMSO-ds) 512.68 (br s, 1H), 8.26 (s, 1H), 7.52 (d, J=8.0 Hz, 1H), 5.27 (br s, 1H), 4.90 (q, J=6.3 Hz, 1H), 4.28 (q, J=8.8 Hz, 1H), 3.92-3.81 (m, 1H), 3.76-3.65 (m, 1H), 3.57 (t, J=9.6 Hz, 1H), 3.46 (d, J=9.4 Hz, 1H), 3.23 (q, J=9.5 Hz, 1H), 1.33 (t, J=6.9 Hz, 3H), 1.28 (d, J=6.4 Hz, 3H). LCMS para C15H17ClFNNaO5 (M+Na)+: m/z=368.0.
Paso 12. (R)-4-(3-cloro-6-etoxi-2-fluoro-5-((R)-1-hidroxietil)fenil)pirrolidin-2-ona (xiii)

El compuesto xii bruto se disolvió en 1,4-dioxano (976 ml) y tolueno (976 ml) y la solución amarilla resultante se calentó a 100° C. El color de la solución se volvió marrón a medida que avanzaba la reacción. Las muestras se extrajeron en puntos temporales de 1 h, 2 h y 2,5 h. A las 2,5 h, el análisis por HPLC mostró que el ácido, compuesto 12, estaba al 0,38% y el producto deseado, compuesto xiii, al 78,8%. La mezcla de la reacción se enfrió a temperatura ambiente y se filtró por pulido en un matraz limpio de fondo redondo de 3 l. Luego, la solución se concentró a presión reducida y el residuo resultante se colocó a vacío alto para dar una espuma marrón (254 g).
Se cargó acetonitrilo (350 ml) en el jarabe marrón y se calentó en un baño de agua a 65° C hasta que se observó la disolución. La solución se enfrió a temperatura ambiente y se agitó durante 16 h. Los sólidos se separaron de la solución. La lechada resultante se enfrió en un baño de hielo durante 1 h. El producto se recogió por filtración y la torta del filtro se enjuagó con 400 ml de acetonitrilo enfriado con hielo. Los sólidos parecían ser higroscópicos. El sólido se disolvió en DCM (2,0 l) y se concentró hasta un jarabe que se colocó a vacío alto para producir el compuesto xiii como una espuma blanca (106,4 g).
El filtrado se concentró hasta un jarabe oscuro (aproximadamente 120 g) que se purificó mediante cromatografía ultrarrápida (4 columnas de gel de sílice de 330 g, cargadas con DCM, eluidas con del 50% al 100% de EtOAc/hexano, y monitorizadas por TLC usando 100% de EtOAc como eluyente). Las fracciones de la cromatografía se concentraron a presión reducida y se colocaron a vacío alto para producir una espuma de color marrón claro (54,4 g). La espuma se cargó con MTBE (400 ml) y MeOH (10 ml) y se calentó en un baño de agua a 56° C durante 15 minutos y quedaron algunos sólidos. La lechada se enfrió a temperatura ambiente con agitación. La lechada se filtró para eliminar las sustancias insolubles. El filtrado se concentró a un jarabe y se colocó a vacío alto para producir una espuma. La espuma se cargó con acetonitrilo (72 ml, 1,5 ml/g) y se calentó en un baño de agua a 60°C hasta que la solución se volvió homogénea. La solución se enfrió a temperatura ambiente con agitación y los sólidos precipitaron de la solución y se volvieron muy espesos. Se cargó acetonitrilo adicional (24 ml) para ajustar la dilución a 2 ml/g. La lechada se agitó a temperatura ambiente durante 16 h y luego se enfrió en un baño de
hielo durante 1 hora. El producto se recogió por filtración y se enjuagó con acetonitrilo. Se obtuvo el compuesto xiii (25 g) como segunda cosecha. Se obtuvieron un total de 131,4 g del compuesto xiii con un rendimiento del 74,9% a partir del compuesto xi.1H NMR (400 MHz, DMSO-d6) 57.83 (s, 1H), 7.47 (d, J=8.7 Hz, 1H), 5.24 (d, J=4.5 Hz, 1H), 4.96-4.85 (m, 1H), 4.08-3.92 (m, 1H), 3.80 (qt, J=6.9, 3.5 Hz, 2H), 3.61-3.51 (m, 1H), 3.25 (t, J=9.1 Hz, 1H), 2.61 -2.50 (m, 1H), 2.35-2.26 (m, 1H), 1.33 (t, J=7.0 Hz, 3H), 1.27 (d, J=6.4 Hz, 3H). LCMS para C14H18CFNO3 (M+H)+: m/z=302.0.
Ejemplo 2. Síntesis alternativa de (R)-4-(3-cloro-6-etoxi-2-fluoro-5-((R)-1-hidroxietil)fenil)pirrolidin-2-ona

Paso 1. 1-(5-cloro-2-etoxi-4-fluorofenil)etanona (iv-a)

Se mezclaron 1-(5-cloro-4-fluoro-2-hidroxifenil)etanona (Compuesto ii del ejemplo 1, paso 1, 1350 g, 7160 mmol), N,N-dimetilformamida (3,32 l), yodoetano (1340 g, 8590 mmol)) y carbonato de potasio (1980 g, 14300 mmol) y se agitaron a temperatura ambiente durante 45 min. La temperatura del lote subió a 55° C desde 22° C. La mezcla de la reacción se calentó a 60° C durante 1 h (la temperatura del lote alcanzó 67° C en 30 min y luego bajó a 60° C). El análisis HPLC indicó que se consumió todo el material de partida. Se añadió agua (10 l) en una porción (la agitación cesará si se añade agua en porciones). La lechada resultante se agitó a temperatura ambiente durante 30 min. El producto se recogió por filtración y se lavó con agua (3 l). El producto se secó sobre el filtro al vacío durante 5 días para dar el compuesto iv-a como un sólido de color tostado (1418 g). 1H NMR (400 MHz, DMSO-d6) 57.69 (d, J=8.9 Hz, 1H), 7.30 (d, J=11.6 Hz, 1H), 4.15 (q, J=7.0 Hz, 2H), 2.51 (s, 3H), 1.37 (t, J=7.0 Hz, 3H).
Paso 2.2-(5-cloro-2-etoxi-4-fluorofenil)-2-metil-1,3-dioxolano (va)

Se disolvió una solución de 1-(5-cloro-2-etoxi-4-fluorofenil)etanona (iv-a, 1481,0 g, 6836,3 mmol) en tolueno (6 l). Se añadieron a la solución 1,2-etanodiol (953 ml, 17100 mmol) y monohidrato de ácido p-toluenosulfónico (104 g, 547 mmol). La mezcla de la reacción se calentó a reflujo a 104-110° C con el uso de una trampa Dean-Stark para eliminar el agua durante 17,4 h. El análisis de HPLC indicó que el 37% del material de partida permaneció sin reaccionar. Se eliminaron aproximadamente 600 ml de destilado y la mezcla de la reacción se calentó a reflujo
durante 5 h adicionales (22 h en total). El análisis por HPLC indicó que no hubo reacción adicional.
Se especuló que el K2CO3 residual en el material de partida compuesto iv-a puede haber detenido la reacción. Por tanto, la mezcla de la reacción se enfrió a temperatura ambiente y se lavó con ácido clorhídrico acuoso 1 N (3 x 6,66 l). Después del lavado con ácido acuoso, la capa orgánica se transfirió de nuevo al recipiente de reacción. Se añadieron 1,2-etanodiol (381 ml, 6840 mmol) y monohidrato de ácido p-toluenosulfónico (104 g, 547 mmol) y la mezcla de la reacción se calentó a reflujo durante 16 h. El análisis por HPLC indicó que aproximadamente el 20% del material de partida permaneció sin reaccionar. Se eliminaron aproximadamente 100 ml de destilado. Se añadió 1,2-etanodiol (380 ml, 6800 mmol) y se calentó a reflujo durante 6 h (22 h en total). La HPLC indicó que el 7% del material de partida permaneció sin reaccionar. Se eliminaron aproximadamente 125 ml de destilado. La mezcla de la reacción se calentó a reflujo durante 6 h (total 28 h). La HPLC indicó que el 5,4% del material de partida permaneció sin reaccionar. Se eliminaron aproximadamente 125 ml de destilado. La mezcla de la reacción se calentó a reflujo durante 7 h adicionales. El análisis de HPLC indicó que el 3,5% del material de partida permaneció sin reaccionar. Se eliminaron aproximadamente 80 ml de destilado. La reacción se consideró completa en este punto.
La mezcla de la reacción se lavó con una solución acuosa de hidróxido de sodio 1 N (2 x 5,5 l). El primer lavado básico se extrajo con tolueno (2,1 l). La solución de tolueno combinada se lavó con agua (7 l) y se concentró para dar 2153 g de aceite oscuro. El análisis por HPLC indicó una pureza del producto al 93,8% con un 1,90% de material de partida y un 0,79% de producto de desyodo. El análisis por 1HMR indicó que permanecieron en el producto aproximadamente 0,5 equivalentes de tolueno (aproximadamente 256 g). El rendimiento corregido de compuesto v-a fue del 88,0%. 1H NMR (300 MHz, CDCla) 57.51 (d, J=8.8 Hz, 1H), 6.70 (d, J=11.0 Hz, 1H), 4.17 3.92 (m, 4H), 3.91 -3.80 (m, 2H), 1.75 (s, 3H), 1.46 (t, J=7.0 Hz, 3H).
Paso 3. 3-doro-6-etoxi-2-fluoro-5-(2-metil-1,3-dioxolan-2-il)benzaldehído (vi)

Se cargó N,N-diisopropilamina (87,8 ml, 626 mmol) y tetrahidrofurano anhidro (1090 ml, 13500 mmol) en un matraz de fondo redondo de 4 bocas de 3 l secado al horno equipado con un agitador superior, un embudo de adición de 500 ml, entrada de nitrógeno, tabiques y termopar. Esta solución se enfrió a -72° C y se cargó con 2,5 M de n-butillitio en hexanos (261 ml, 652 mmol). La solución de n-butillitio se añadió durante 18 min. La temperatura interna máxima durante la adición fue de -65° C. El baño de hielo seco-acetona se reemplazó por un baño de agua con hielo y la mezcla de la reacción se calentó a aproximadamente de -5° C a aproximadamente 0° C y se mantuvo durante 15 min. Luego, la mezcla de la reacción se enfrió a -74,5°C.
A un matraz de fondo redondo de 1 l separado que contenía 2-(5-cloro-2-etoxi-4-fluorofenil)-2-metil-1,3-dioxolano (v-a, 136,1 g, 522,1 mmol) se añadió tetrahidrofurano anhidro (456 ml) para disolver los sólidos. La solución resultante se enfrió en un baño de hielo a aproximadamente 0° C. La solución que contenía el compuesto v a se transfirió a la solución de LDA durante 40 minutos mediante una cánula mientras se mantenía la temperatura de reacción entre -70° C y -72,5° C. La mezcla de la reacción se volvió una lechada amarilla y se agitó durante 37 min a -74° C. Se cargó N,N-dimetilformamida (60,6 ml, 783 mmol) en una porción mediante una jeringuilla y esta adición produjo una exotermia de -74,5° C a -66,5° C. La reacción se monitorizó por HPLC a los 90 minutos después de la adición. El material de partida estaba presente al 2,9%. Se retiró el baño frío y se calentó la mezcla de la reacción a temperatura ambiente. Se tomaron muestras de la mezcla de la reacción y se analizaron después de 3 horas y el material de partida sin reaccionar estaba presente al 1,5%. La reacción se consideró completa y se inactivó mediante la adición de la solución de reacción a agua con hielo (1,4 l) y se diluyó con acetato de etilo (1,5 l). La capa acuosa se extrajo con acetato de etilo (1,5 l) y las capas orgánicas se combinaron y se lavaron con salmuera (20% p/p de NaCl acuoso, 2 x 600 ml) y se secaron sobre MgSO4 anhidro. El MgSO4 se eliminó por filtración y el filtrado se concentró a un aceite con algunos sólidos presentes. Este residuo se disolvió en cloruro de metileno y se cargó en una almohadilla de gel de sílice (586 g). La almohadilla de sílice se eluyó con EtOAc/DCM al 2% (monitorizado por TLC usando DCM al 100% como eluyente). Las fracciones deseadas se recogieron y concentraron a presión reducida para dar un aceite de color ámbar claro. El aceite se colocó a vacío alto para dar el compuesto vi como un sólido amarillo (146,5 g, rendimiento del 95,1%). 1H NMR (400 MHz, CDCh) 510.27 (s, 1H), 7.78 (d, J=8.5 Hz, 1H), 4.10-3.96 (m, 4H), 3.87-3.76 (m, 2H), 1.72 (s, 3H), 1.44 (t, J=7.0 Hz, 3H). LCMS para C13H15CFO4 (M+H)+: m/z=289.1.
Pasos 4-10. (R)-4-(3-cloro-6-etoxi-2-fluoro-5-((R)-1-hidroxietil)fenil)pirrolidin-2-ona (xiii)
El compuesto del título se preparó usando procedimientos análogos a los descritos en el Ejemplo 1, Pasos 6-12.
Ejemplo 3. Clorhidrato de (R)-4-(3-((S)-1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona

Paso 1. (R)-1-(5-doro-2-etoxi-4-fluoro-3-((R)-5-oxopirrolidin-3-il)fenil)etilmetanosulfonato (xiv)

Se disolvió (R)-4-(3-cloro-6-etoxi-2-fluoro-5-((R)-1-hidroxietil)fenil)pirrolidin-2-ona (xiii, 172,0 g, 570,0 mmol) (consistía en 147 g al 99,83%: 0,09% de pureza quiral, 99,33% de pureza química; y 25 g, 87,46%: 12,54% de pureza quiral, 86,74% de pureza química) en cloruro de metileno (860 ml). Se añadió N,N-diisopropiletilamina (149 ml, 855 mmol) a la solución a una temperatura de aproximadamente -7° C a aproximadamente 2°C. Se añadió gota a gota cloruro de metanosulfonilo (57,4 ml, 741 mmol) a la mezcla de la reacción durante 25 min. La suspensión se volvió una solución transparente. En el punto temporal de reacción de 30 min, la HPLC indicó que la reacción se había completado. Esta mezcla de la reacción que contiene el compuesto xiv se usó directamente en la siguiente reacción.
Paso 2. (R)-4-(S-cloro-6-etoxi-2-fluoro-5-((S)-1-hidraziniletil)fenil)pirrolidin-2-ona (xv)

A 0° C, se añadió hidrazina (178,9 ml, 5,7 mol) en una porción seguida de N-metilpirrolidinona (860 ml) a la mezcla de la reacción que contenía el compuesto xiv del Paso 1. La mezcla de la reacción se volvió turbia y se formaron algunos precipitados. La mezcla se calentó a 40-57° C bajo nitrógeno durante 90 min. La HPLC indicó que
se había consumido todo el mesilato.
La mezcla de la reacción se enfrió a temperatura ambiente y se añadió una solución saturada de bicarbonato de sodio (28,3 g) en agua (300 ml). La mezcla se agitó durante 20 min, momento en el que se añadió diclorometano (300 ml). La capa orgánica se separó y se agitó con una solución de bicarbonato de sodio (14,2 g) en agua (150 ml). La capa acuosa se extrajo con diclorometano (200 ml x 2). Las capas orgánicas combinadas se lavaron con salmuera (80 ml), se secaron sobre Na2SO4 anhidro (311 g), se concentraron y se destilaron azeotrópicamente con tolueno (250 ml) para dar una solución de N-metilpirrolidinona incolora que contenía el compuesto xv que se usó directamente en la siguiente reacción. Se purificó una muestra para análisis de NMR. 1H NMR (400 MHz, DMSO-d6), 57.88 (s, 1H), 7.66 (d, J=8.5 Hz, 1H), 4.42 (q, J=6.7 Hz, 1H), 4.06-3.88 (m, 2H), 3.79 3.66 (m, 1H), 3.65-3.51 (m, 1H), 3.24 (t, J=8.8 Hz, 1H), 2.60-2.46 (m, 1H), 2.36-2.25 (m, 1H), 1.37 (t, J=6.9 Hz, 3H), 1.26 (d, J=6.8 Hz, 3H). LCMS para CuH19ClFN3O2(M+H)+: m/z=316.1.
Paso 3. 5-amino-1-((S)-1-(5-cloro-2-etoxi-4-fluoro-3-((R)-5-oxopirrolidin-3-il)fenil)etil)-3-metil-1H-pirazol-4-carbonitrílo (xvi)

Con agitación, se añadió (1-etoxietiliden)malononitrilo (101 g, 741 mmol) a la solución en N-metilpirrolidinona del compuesto xv del Paso 2, en porciones y la mezcla se agitó a temperatura ambiente bajo nitrógeno. Después de 15 min, el análisis por HPLC indicó un 11% de material de partida de hidrazina, compuesto xv, con respecto al producto compuesto xvi. Se añadió N,N-diisopropiletilamina (15 ml, 86 mmol) y la mezcla de la reacción se agitó a temperatura ambiente durante 15 h. El análisis por HPLC indicó que quedaba un 5,6% de material de partida. Se añadió N,N-diisopropiletilamina (5 ml, 30 mmol) y la mezcla de la reacción se agitó a temperatura ambiente durante 5 h. La HPLC indicó que quedaba un 5,6% de material de partida. La mezcla de la reacción se agitó durante 2,5 días y se combinó con dos lotes similares y se trataron juntos.
Se combinaron las mezclas de reacción de tres lotes del compuesto xvi. Se añadió una solución acuosa de hidróxido de sodio 0,5 M (3,8 l) a 10-20° C y se agitó durante 5 min. La HPLC indicó que se había consumido todo el material de partida (1-etoxietiliden)malononitrilo. Se añadió acetato de etilo (4,0 l) y la mezcla se agitó durante 15 min. Las capas se separaron. La capa orgánica se lavó con hidróxido de sodio 0,5 M en agua (2,38 l). Las capas se separaron. La capa acuosa combinada se extrajo con acetato de etilo (2 x 2 l). Las capas orgánicas combinadas se lavaron con ácido clorhídrico acuoso 1,0 M (3,56 l) y el pH de la capa acuosa resultante fue de 2-3. La capa orgánica se lavó con salmuera (5 l), se secó sobre Na2SO4 anhidro, se concentró y se secó a vacío alto durante 40 h para dar el compuesto xvi en forma de un sólido espumoso de color marrón claro (702,7 g). 1H NMR (500 MHz, DMSO-d6) 5 7.78 (s, 1H), 7.44 (d, J=8.4 Hz, 1H), 6.53 (s, 2H), 5.64 (q, J=6.7 Hz, 1H), 3.96 (m, 1H), 3.74 (m, 1H), 3.34 (m, 1H), 3.58 (m, 2H), 2.59-2.50 (m, 1H), 2.29 (m, 1H), 2.04 (s, 3H), 1.57 (d, J=6.8 Hz, 3H), 1.37 (t, J=6.9 Hz, 3H). LCMS para C1gH22ClFN5O2(M+H)+: m/z=406.1.
Se calculó que el rendimiento total del compuesto xvi en tres pasos (mesilación, hidrazinolisis y formación de pirazol) era del 72,8% a partir de la entrada total del compuesto xiii. Se determinó por HPLC que la pureza era aproximadamente del 80%. El análisis por HPLC indicó que existía algo de producto en la capa acuosa básica que se extrajo posteriormente con EtOAc (2 l), se lavó con ácido clorhídrico acuoso 1,0 M y salmuera, se secó con sulfato de sodio anhidro, se concentró y se secó en una bomba de vacío alto durante 40 h para proporcionar el compuesto xvi en forma de un aceite marrón (134 g, 13,9%).
Paso 4. (R)-4-(3-((S)-1-(4-amino-3-metil-1H-yrazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona (xvii)

Se añadió 5-amino-1 -((S)-1 -(5-cloro-2-etoxi-4-fluoro-3-((R)-5-oxopirrolidin-3-il)fenil)etil)-3-metil-1 H-pirazol-4-carbonitrilo (xvi, 702,7 g, 1731 mmol) a un recipiente de reacción con acetato de formamidina (1802 g, 17,31 mol) y 1,2-etanodiol (3,51 l). La mezcla de la reacción se calentó a 102-103° C con agitación durante 18 h. La mezcla de la reacción se enfrió a temperatura ambiente y se añadieron acetato de etilo (7 l) y agua (6 l) y la mezcla bifásica se agitó durante 15 min. La capa orgánica se separó y la capa acuosa se diluyó con agua adicional (4,5 l) y acetato de etilo (3 l) y se agitó durante 10 min. Se separó la capa orgánica. La capa acuosa se extrajo adicionalmente con acetato de etilo (2 l). Las capas orgánicas se combinaron y se agitaron con agua (4,5 l). La capa acuosa se separó y la capa orgánica se filtró a través de una almohadilla de celite (aproximadamente 1 kg). La capa orgánica se extrajo con ácido clorhídrico acuoso 1,0 M (7 l) agitando la mezcla durante 10 min. Se separó la capa acuosa. La capa orgánica marrón claro se agitó con ácido clorhídrico acuoso 1,0 M adicional (3 l) durante 10 min. Se separó la capa acuosa. Las capas ácidas acuosas se combinaron y se lavaron con tolueno (500 ml). La solución ácida acuosa se enfrió con un baño de agua con hielo y se añadió cloruro de metileno (4 l). A 5-15° C, se añadió lentamente una solución de hidróxido de sodio (530 g) en agua (530 ml) (solución de NaOH al 50%) hasta un pH de solución de 11 12. Se observaron precipitados sólidos. Se añadieron más cloruro de metileno (3,5 l) y metanol (300 ml) y la mezcla se agitó durante 10-15 min. El producto sólido se recogió por filtración y se secó en el filtro bajo succión durante 16 h para dar el compuesto xvii (289,7 g) como un sólido marrón. 1H NMR (400 MHz, DMSO-d6) 58.11 (s, 1H), 7.82 (s, 1H), 7.52 (d, J=8.5 Hz, 1H), 7.30 (br s, 2H), 6.23 (q, J=7.0 Hz, 1H), 3.97 (p, J=9.2 Hz, 1H), 3.90-3.73 (m, 2H), 3.57 (t, J=9.9 Hz, 1H), 3.25 (dd, J=9.2, 8.7 Hz, 1H), 2.48 (s, 3H), 2.60-2.50 (m, 1H), 2.36-2.20 (m, 1H), 1.69 (d, J=7.1 Hz, 3H), 1.39 (t, J=6.9 Hz, 3H). LCMS para C2üH23ClFN6O2 (M+H)+: m/z=433.3.
El filtrado se transfirió a un embudo de separación y se separó la capa orgánica. La capa acuosa se agitó con cloruro de metileno (5 l) y metanol (200 ml). La capa orgánica combinada se secó sobre sulfato de sodio anhidro, se concentró, se secó en una bomba de vacío alto durante 16 h para dar una cantidad adicional de 259,3 g como un sólido marrón. El rendimiento total de xvii fue de 548,3 g con un rendimiento del 73,2%.
Paso 5. Sal de clorhidrato de (R)-4-(3-((S)-1-(4-amino-3-metil-1H-piyrazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona (xviii)

Se añadió una solución de ácido clorhídrico acuosa 1,0 M (HCl, 5,0 l, 5,0 mol) a (R)-4-(3-((S-1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona (xvii, 609,8 g, 1,409 mol) a temperatura ambiente. La lechada espesa resultante se calentó luego a 50° C para proporcionar una solución transparente. Se añadieron 1,82 l adicionales de solución de ácido clorhídrico acuosa 1,0 M (HCl, 1,82 l, 1,82 mol;
6,82 l totales, 6,82 mol, 4,84 equiv.) a la solución transparente a 50° C y luego la solución se filtró a través de un filtro de pulido aproximadamente a 50° C. La mezcla de la reacción filtrada con pulido enfrió gradualmente a temperatura ambiente durante 2 h antes de que se enfriase adicionalmente a 0-5°C. La mezcla de la reacción se agitó a 0-5° C durante por lo menos 20 min para iniciar la precipitación. Los sólidos resultantes se recogieron por filtración, se enjuagaron con una porción de solución madre fría, seguido de ácido clorhídrico acuoso 1,0 M (HCl, 200 ml), y se secaron en el embudo de filtración a temperatura ambiente bajo succión a peso constante (en aproximadamente 39 h) para producir la sal de ácido clorhídrico del compuesto de Fórmula I: Clorhidrato de (R)-4-(3-((S)-1-(4-amino-3-metil-1 H-pirazolo[3,4-i]pirimidin-1 -il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona (xviii, 348,7 g, 661,2 g teóricos, 52,7%) como un polvo cristalino blanco. 1H NMR (400 MHz, DMSü-ds) 59.39 (br s, 1H), 9.05 (br s, 1H), 8.50 (s, 1H), 7.84 (s, 1H), 7.59 (d, J=8.4 Hz, 1H), 6.28 (q, J=6.9 Hz, 1H), 3.95 (m, 1H), 3.79 (m, 2H), 3.55 (m, 1H), 3.22 (m, 1H), 2.59 (s, 3H), 2.55 (ddd, J=16.8, 10.3, 2.3 Hz, 1H), 2.28 (ddd, J=16.8, 8.6, 1.5 Hz, 1H), 1.73 (d, J=7.0 Hz, 3H), 1.38 (t, J=6.9 Hz, 3H) ppm. 13C NMR (100 MHz, DMSO-ds) 5175.3, 156.4 (Jcf=249.8 Hz), 153.8 (Jcf=7.0 Hz), 152.4, 150.8, 147.3, 144.3, 131.4 (Jcf=3.5 Hz), 127.3, 126.4 (Jcf=12.6 Hz), 116.1 (Jcf=18.4 Hz), 98.0, 72.1, 49.1, 46.6, 36.0, 29.4, 21.0, 15.4, 14.6 ppm. 19F NMR (376 MHz, DMSO-ds) 5 -113.6 (d, Jfh=7.7 Hz) ppm. C20H23Cl2FNsO2(MW 469.34); LCMS (EI) m/e 433.2 (M+H; masa exacta de xvii: 432,15). Contenido de agua por KF: 3,63% en peso; contenido de cloruro (CI-) por titulación: 7,56% en peso (7,56% teórico).
El intervalo de fusión/descomposición de la forma cristalina de la sal de clorhidrato de (R)-4-(3-((S)-1-(4-amino-3-metil-1 H-pirazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona se determinó mediante DSC, desde una temperatura inicial de 30° C hasta una temperatura final de 350° C usando una tasa de calentamiento de 10° C/min. El termograma de DSC reveló un evento endotérmico con un inicio a 194,37° C y el pico a 206,55° C, como se muestra en la FIG 1.
El termograma de TGA mostró una pérdida de peso total del 4,3% hasta 210° C. Por encima de 210° C, la sal comienza a descomponerse, como se muestra en la FIG. 2.
En la FIG. 3 se muestra un patrón representativo de Difracción de Rayos X de Polvo (XRPD) y la Tabla 2 muestra los picos e intensidades correspondientes.
Tabla 2.

continuación

Ejemplo 4. Síntesis alternativa de clorhidrato de (R)-4-(3-((S)-1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil)--cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona

Paso 1. (R)-4-(3-acetil-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona (xix)

Se disolvió (4R)-4-[3-cloro-6-etoxi-2-fluoro-5-(l-hidroxietil)fenil]pirrolidin-2-ona (como una mezcla de dos diastereómeros con configuración R en la pirrolidinona y configuraciones R o S en el alcohol secundario) (xiii, 16,7 g, 55,3 mmol) en diclorometano (167 ml). La solución se enfrió en un baño de agua con hielo y se añadió peryodinano de Dess-Martin (35,2 g, 83,0 mmol) en pequeñas porciones. La mezcla de la reacción se agitó a temperatura ambiente durante 2 h, momento en el que el análisis por HPLC mostró que la reacción se había completado. Se añadió una solución de sulfito de sodio (28 g, 220 mmol) en agua (70 ml) a la mezcla de la reacción y la mezcla se agitó durante 20 min. Se añadió una solución de hidróxido de sodio 1,0 M a la mezcla y se agitó durante 10 min. Las capas se dejaron sedimentar y la capa orgánica se separó y se lavó secuencialmente con una solución acuosa de hidróxido de sodio 1 M (66 ml) y agua (60 ml). La capa orgánica se secó sobre sulfato de sodio anhidro. El agente de secado se eliminó por filtración y el filtrado se concentró para dar (R)-4-[3-acetil-5-cloro-2-etoxi-6-fluorofenil]pirrolidin-2-ona como un aceite que se usó en el siguiente reacción sin purificación adicional.
Paso 2.2-(1-(5-cloro-2-etoxi-4-fluoro-3-(5-oxopirrolidin-3-il)fenil)etiliden)hidrazinacarboxilato de (R,E)-terc-butilo (xx)

Se disolvió (R)-4-[3-acetil-5-cloro-2-etoxi-6-fluorofenil]pirrolidin-2-ona bruta (compuesto xix del paso 1) en metanol (60 ml) y se añadió carbazato de t-butilo (8,04 g, 60,8 mmol) a la solución. La mezcla de la reacción se agitó a 65° C durante 3,5 días, momento en el que el análisis por HPLC mostró que la reacción se había completado. La mezcla se concentró a presión reducida y el residuo se purificó por cromatografía en gel de sílice eluyendo con una mezcla del 0-5% de metanol en acetato de etilo para dar 2-(1-(5-cloro-2-etoxi-4-fluoro-3-(5-oxopirrolidin-3-il)fenil)etiliden)hidrazinacarboxilato de (R,E)-terc-butilo (xx, 19,5 g, 85%). 1H NMR (500 MHz, DMSo-da) 59.83 (s, 1H), 7.78 (s, 1H), 7.36 (d, J=8.6 Hz, 1H), 4.07 (p, J=9.1 Hz, 1H), 3.84-3.69 (m, 2H), 3.59 (t, J=9.5 Hz, 1H), 3.28 (t, J=9.5 Hz, 1H), 2.54 (m, 1H), 2.33 (m, 1H), 2.14 (s, 3H), 1.46 (s, 9H), 1.25 (t, J=7.0 Hz, 3H). LCMS para C19H25ClFN3NaÜ4 (M+Na)+: m/z=436.1.
Paso 3.2-((S)-1-(5-cloro-2-etoxi-4-fluoro-3-((R)-5-oxopirrolidin-3-il)fenil)etil)hidrazinacarboxilato de terc-butilo (xxi)
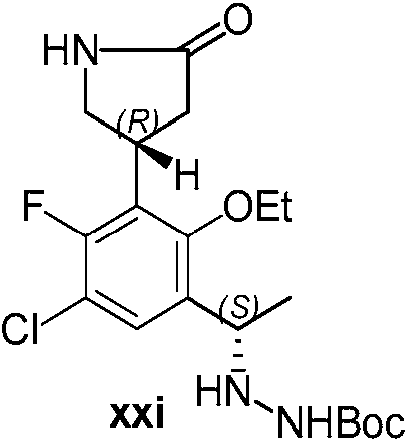
Se disolvió 2-(1-(5-cloro-2-etoxi-4-fluoro-3-(5-oxopirrolidin-3-il)fenil)etiliden)hidrazinacarboxilato de (R,E)-terc-butilo (xx, 0,5 g, 1,2 mmol) en metanol (25 ml) y la solución se burbujeó con gas nitrógeno durante 5 min. Se añadieron Bis(1,5-ciclooctadieno)rodio(I)tetrafluoroborato (35 mg, 0,086 mmol) y (R)-(-)-1-{(S)-2-[bis(4-trifluorometilfenil)fosfina]ferrocenil}etilo-di-t-butilfosfina (64 mg, 0,094 mmol) a la solución y la mezcla de la reacción resultante se burbujeó con gas nitrógeno durante 30 min. Luego, la mezcla de la reacción se agitó bajo una presión de gas hidrógeno (56 psi) durante 2,5 días. La mezcla de la reacción se concentró a presión reducida y el residuo resultante se purificó mediante cromatografía en columna de gel de sílice eluyendo con una mezcla de metanol (0-10%) en acetato de etilo. Las fracciones deseadas se concentraron para dar 2-((S)-1-(5-cloro-2-etoxi-4-fluoro-3-((R)-5-oxopirrolidin-3-il)fenil)etil)hidrazinacarboxilato de terc-butilo (xxi, 428 mg, 85% de rendimiento). 1H NMR (500 MHz, DMSO-da) 58.18 (s, 1H), 7.78 (s, 1H), 7.53 (d, J=8.2 Hz, 1H), 4.73 (s, 1H), 4.41 (br s, 1H), 3.98 (m, 1H), 3.75 (m, 2H), 3.61 (m, 1H), 3.26 (m, 1H), 2.53 (m, 1H), 2.29 (dd, J=17.6, 8.6 Hz, 1H), 1.32 (s, 12H), 1.10 (d, J=6.5 Hz, 1H). LCMS para C1gH27ClFN3NaO4 (M+Na)+: m/z=437.9. El análisis de HPLC quiral indicó que el producto contenía el diastereómero deseado -2-((S)-1-(5-cloro-2-etoxi-4-fluoro-3-((R)-5-oxopirrolidin-3-il)fenil)etil)hidracina carboxilato de terc-butil (xxi) al 85,6% y el diastereómero no deseado -2-((R)-1-(5-cloro-2-etoxi-4-fluoro-3-((R))-5-oxopirrolidin-3-il)fenil)etil)hidrazina carboxilato de terc-butilo al 14,3%.
Paso 4. 5-amino-1-((S)-1-(5-cloro-2-etoxi-4-fluoro-3-((R)-5-oxopirrolidin-3-il)fenil)etil)-3-metil-1H-pirazol-4-carbonitrilo (xvi)

Se añadieron 2-((S)-1 -(5-cloro-2-etoxi-4-fluoro-3-((R)-5-oxopirrolidin-3-il)fenil)etil)hidrazinacarboxilato de terc-butilo (xxi, 130 mg, 0,31 mmol) y monohidrato de ácido p-toluensulfónico (86 mg, 0,45 mmol) a etanol (3 ml) y la mezcla de la reacción se calentó a 50° C durante 20 h. El análisis de HPLC mostró que había aproximadamente un 88% de material de partida sin reaccionar. Se cargó una cantidad adicional de ácido p-toluenosulfónico (86 mg, 0,45 mmol) y la mezcla de la reacción se calentó a 60° C durante 24 h. El análisis por HPLC mostró una desprotección completa de Boc. Esta mezcla de la reacción se añadió con (1-etoxietiliden)malononitrilo (61 mg, 0,45 mmol) y N,N-diisopropiletilamina (260 pl, 1,5 mmol). La mezcla de la reacción se agitó a temperatura ambiente durante 2 h. La HPLC mostró la finalización de la formación del anillo de pirazol. Se añadió una solución acuosa de hidróxido de sodio 1,0 M a la mezcla de la reacción y se agitó durante 20 min. Se añadió acetato de etilo (20 ml) a la mezcla y se agitó. Se dejó sedimentar la mezcla bifásica. Se recogió la capa de acetato de etilo y se extrajo la capa acuosa con acetato de etilo (10 ml). La solución de acetato de etilo combinada se añadió con ácido clorhídrico acuoso 1 M (5 ml) y se agitó durante 15 min. Se dejó sedimentar la mezcla bifásica y se recogió la capa orgánica y se secó sobre sulfato de sodio anhidro. El sulfato de sodio se eliminó por filtración y el filtrado se concentró para dar 5-amino-1-((S)-1 -(5-cloro-2-etoxi-4-fluoro-3-((R)-5-oxopirrolidin-3-il)fenil)etil)-3-metil-1 H-pirazol-4-carbonitrilo (xvi, 126 mg, rendimiento cuantitativo de producto bruto) y se usó en el siguiente paso sin purificación adicional.
Paso 5. (R)-4-(3-((S)-1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona (xvii)

Se añadió 5-amino-1 -((S)-1 -(5-cloro-2-etoxi-4-fluoro-3-((R)-5-oxopirrolidin-3-il)fenil)etil)-3-metil-1 H-pirazol-4-carbonitrilo (xvi, 126 mg, 0,31 mmol) con acetato de formamidina (323 mg, 3,1 mmol) y 1,2-etanodiol (2 ml). La mezcla de la reacción se calentó a 104-105° C con agitación. Luego de 18 h, el análisis por HPLC mostró que quedaba aproximadamente un 44% del compuesto xvi de material de partida. La mezcla de la reacción se calentó a 115° C durante 24 h. El análisis de HPLC mostró que la reacción se había completado. La mezcla de la reacción se enfrió a temperatura ambiente y se añadieron acetato de etilo (10 ml) y agua (5 ml). Se agitó la mezcla bifásica. Se permitió que las capas se separaran. Se recogió la capa orgánica y se extrajo la capa acuosa con acetato de etilo (5 ml). La solución de acetato de etilo combinada se lavó con agua (5 ml), se secó sobre sulfato de sodio anhidro. El sulfato de sodio se eliminó por filtración y el filtrado se concentró hasta dar un residuo. El residuo se purificó mediante cromatografía en gel de sílice. La columna se eluyó con una mezcla de metanol (0-5%) en cloruro de
metileno. Las fracciones deseadas se combinaron y evaporaron para dar (R)-4-(3-((S)-1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona (xvii, 94 mg, rendimiento del 69,9%). 1H NMR (400 MHz, DMSO-d6) 58.11 (s, 1H), 7.82 (s, 1H), 7.52 (d, J=8.5 Hz, 1H), 7.30 (br s, 2H), 6.23 (q, J=7.0 Hz, 1H), 3.97 (p, J=9.2 Hz, 1H), 3.90-3.73 (m, 2H), 3.57 (t, J=9.9 Hz, 1H), 3.25 (dd, J=9.2, 8.7 Hz, 1H), 2.48 (s, 3H), 2.60 2.50 (m, 1H), 2.36-2.20 (m, 1H), 1.69 (d, J=7.1 Hz, 3H), 1.39 (t, J=6.9 Hz, 3H). LCMS para C20H23CFN6O2 (M+H)+: m/z=433.3.
El análisis de HPLC quiral del producto indicó que contenía el diastereoisómero deseado, (R)-4-(3-((S)-1 -(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona (xvii), al 87% y el diastereoisómero no deseado (R)-4-(3-((R)-1 -(4-amino-3-metil-1 H-pirazolo[3,4-d]pirimidin-1 -il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona al 13%.
Paso 6. Clorhidrato de (R)-4-(3-((S)-1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona
El producto del título se preparó de acuerdo con el procedimiento descrito en el Ejemplo 3, Paso 5. La sal de clorhidrato resultante coincide bien con el material elaborado a partir del proceso sintético descrito en el Ejemplo 3, en todos los aspectos comparables, incluyendo las características de pureza química, pureza quiral y estado sólido.
Ejemplo A1: ensayo enzimático de PI3K
El kit de ensayo luminiscente de PI3-Quinasa que incluye sustrato de lípido quinasa, D-mio-fosfatidilinositol 4,5-bisfosfato (PtdIns (4,5)P2)D (+)-sn-1,2-di-O-octanoilglicerilo, proteína detectora biotinilada I(1,3,4,5)P4, PI(3,4,5)P3 ligada a 3-O-fosfo (PIP2), se adquirío de Echelon Biosciences (Salt Lake City, UT). El kit de detección AlphaScreen™ GST, que incluye perlas donantes y aceptoras, se adquirió de PerkinElmer Life Sciences (Waltham, MA). PI3K5 (p1105/p85a) se adquirió de Millipore (Bedford, Ma ). ATP, MgCh, DTT, EDTA, HEPES y CHAPS se adquirieron de Sigma-Aldrich (St. Louis, MO).
Ensayo AlphaScreen™ para PI3K5
La reacción de la quinasa se realiza en una placa REMP de 384 pocillos de Thermo Fisher Scientific en un volumen final de 40 pl. Los inhibidores se diluyen primero en serie en DMSO y se añaden a los pocillos de la placa antes de la adición de otros componentes de reacción. La concentración final de DMSO en el ensayo es del 2%. Los ensayos de PI3K se llevan a cabo a temperatura ambiente en HEPES 50 mM, pH 7,4, MgCl25 mM, NaCl 50 mM, DTT 5 mM y CHAPS 0,04%. Las reacciones se inician mediante la adición de ATP, la mezcla de la reacción final consistía de 20 pM de PIP2, 20 pM de ATP, PI3K51,2 nM y se incuban durante 20 minutos. Luego, se transfieren 10 pl de la mezcla de la reacción a 5 pI de I(1,3,4,5)P4 biotinilada 50 nM en tampón de inactivación: HEPES 50 mM pH 7,4, NaCl 150 mM, EDTA 10 mM, DTT 5 mM, Tween-20 al 0,1%, seguido de la adición de 10 pl de perlas donantes y aceptoras AlphaScreen™ suspendidas en tampón de inactivación contiene proteína detectora PI(3,4,5)P325 nM. La concentración final de perlas tanto donantes como aceptoras es de 20 mg/ml. Después del sellado de la placa, la placa se incubó en un lugar oscuro a temperatura ambiente durante 2 horas. La actividad del producto se determina en un lector de microplacas Fusion-alpha (Perkin-Elmer). La determinación de IC50 se realiza ajustando la curva del porcentaje de actividad de control frente al logaritmo de la concentración de inhibidor usando el software GraphPad Prism 3.0.
Ejemplo A2: Ensayo enzimático de PI3K
Materiales: sustrato de lípido quinasa, fosfoinositol-4,5-bifosfato (PIP2), se adquirieron de Echelon Biosciences (Salt Lake City, UT). Las isoformas a, p, 5 y y de PI3K se adquirieron de Millipore (Bedford, MA). ATP, MgCl2, DTT, EDTA, MOPS y CHAPS se adquirieron de Sigma-Aldrich (St. Louis, MO).
Las reacciones de quinasa se llevan a cabo en una placa de 96 pocillos de fondo transparente de Thermo Fisher Scientific en un volumen final de 24 pl. Los inhibidores se diluyen primero en serie en DMSO y se añaden a los pocillos de la placa antes de la adición de otros componentes de la reacción. La concentración final de DMSO en el ensayo es del 0,5%. Los ensayos de PI3K se llevan a cabo a temperatura ambiente en MOPS 20 mM, pH 6,7, MgCl210 mM, DTT 5 mM y CHAPS al 0,03%. La mezcla de la reacción se prepara conteniendo PIP250 pM, quinasa y concentraciones variables de inhibidores. Las reacciones se inician mediante la adición de ATP que contiene 2,2 pCi [y-33P]ATP hasta una concentración final de 1000 pM. La concentración final de las isoformas a, p, 5 y y de PI3K en el ensayo fueron 1,3, 9,4, 2,9 y 10,8 nM, respectivamente. Las reacciones se incuban durante 180 minutos y se terminan mediante la adición de 100 pl de fosfato de potasio 1 M pH 8,0, tampón de inactivación de EDTA 30 mM. A continuación, se transfiere una alícuota de 100 pl de la solución de reacción a una placa de filtro de PVDF de 0,45 pm Millipore MultiScreen IP de 96 pocillos (la placa de filtro se humedece previamente con 200 pl de etanol al 100%, agua destilada y fosfato de potasio 1 M, pH 8,0, respectivamente). La placa de filtro se aspira en un colector Millipore al vacío y se lava con tampón de lavado 18 x 200 pl que contiene fosfato de potasio 1 M, pH 8,0 y ATP 1 mM.
Después de secar por aspiración y transferencia, la placa se seca al aire en una incubadora a 37° C durante la noche. Luego se acopla un adaptador Packard TopCount (Millipore) a la placa seguido de la adición de 120 pl de cóctel de centelleo Microscint 20 (Perkin Elmer) en cada pocillo. Después del sellado de la placa, se determina la radiactividad del producto mediante recuento de centelleo en Topcount (Perkin-Elmer). La determinación de IC50 se realiza ajustando la curva del porcentaje de actividad de control frente al logaritmo de la concentración de inhibidor usando el software GraphPad Prism 3.0.
La sal de ácido clorhídrico de (R)-4-(3-((S)-1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona se probó en el ensayo del Ejemplo A2 y se determinó que era un inhibidor selectivo de PI3K5.
La sal de ácido clorhídrico de (R)-4-(3-((S)-1-(4-amino-3-metil-1H-pirazolo[3,4-d]pirimidin-1-il)etil)-5-cloro-2-etoxi-6-fluorofenil)pirrolidin-2-ona se probó en el ensayo del Ejemplo A2 y se determinó que era un inhibidor selectivo >100 veces mayor para PI3K5 sobre cada uno de PI3Ka, PI3Kp y PI3Ky.
Ejemplo A3: Ensayo de proximidad de centelleo de PI3K5
Materiales
Se adquirió [y-33P]ATP (10 mCi/ml) de Perkin-Elmer (Waltham, MA). El sustrato de lípido quinasa, D-miofosfatidilinositol 4,5-bisfosfato (PtdIns(4,5)P2)D (+)-sn-1,2-di-O-octanoilglicerilo, 3-O-fosfo enlazado (PIP2), CAS 204858-53-7, se adquirió de Echelon Biosciences (Salt Lake City, UT). PI3K5 (p1105/p85a) se adquirió de Millipore (Bedford, mA). Se adquirieron ATP, MgCl2, DTT, EDTA, MOPS y CHAPS de Sigma-Aldrich (St. Louis, MO). Las perlas de centelleo YSi SPA de aglutinina de germen de trigo (WGA) se adquirieron de GE healthcare life sciences (Piscataway, NJ).
La reacción de la quinasa se llevó a cabo en una placa blanca de matriz de 384 pocillos de poliestireno de Thermo Fisher Scientific en un volumen final de 25 pl. Los inhibidores se diluyeron primero en serie en DMSO y se añadieron a los pocillos de la placa antes de la adición de otros componentes de reacción. La concentración final de DMSO en el ensayo fue del 0,5%. Los ensayos de PI3K se llevaron a cabo a temperatura ambiente en MOPS 20 mM, pH 6,7, MgCl210 mM, DTT 5 mM y ChApS al 0,03%. Las reacciones se iniciaron mediante la adición de ATP, la mezcla de la reacción final consistió en PIP220 pM, ATP 20 pM, 0,2 pCi [y-33P]ATP, PI3K54 nM. Las reacciones se incubaron durante 210 min y se terminaron mediante la adición de 40 pl de perlas de SPA suspendidas en tampón de inactivación: fosfato de potasio 150 mM pH 8,0, glicerol al 20%. eDt A 25 mM, ATP 400 pM. La concentración final de perlas de SPA fue de 1,0 mg/ml. Después del sellado de la placa, las placas se agitaron durante la noche a temperatura ambiente y se centrifugaron a 1800 rpm durante 10 minutos, la radiactividad del producto se determinó mediante recuento de centelleo en Topcount (Perkin-Elmer). La determinación de IC50 se realizó ajustando la curva del porcentaje de actividad de control frente al logaritmo de la concentración de inhibidor usando el software GraphPad Prism 3.0. Se descubrió que el compuesto de Fórmula I tenía una IC50 de <10 nM en el ensayo del Ejemplo A3.
Ejemplo B1: ensayo de proliferación de células B
Para adquirir células B, se aíslan PBMC humanas de la sangre periférica de donantes normales libres de fármaco mediante centrifugación en gradiente de densidad estándar en Ficoll-Hypague (GE Healthcare, Piscataway, NJ) y se incubaron con microperlas anti-CD 19 (Miltenyi Biotech, Auburn, CA). Luego, las células B se purifican mediante inmunoclasificación positiva usando un autoMacs (Miltenyi Biotech) de acuerdo con las instrucciones del fabricante.
Las células B purificadas (2x10Vpocillo/200 pl) se cultivan en placas de unión ultrabaja de 96 pocillos (Corning, Corning, NY) en RPMI1640, FBS al 10% e IgM anti-humana F(ab')2 de cabra (10 pg/ml) (Invitrogen, Carlsbad, CA) en presencia de diferentes cantidades de compuestos de prueba durante tres días. Luego se añade [3H]-timidina (1 pCi/pocillo) (PerkinElmer, Boston, MA) en PBS a los cultivos de células B durante 12 horas adicionales antes de que la radiactividad incorporada se separe mediante filtración con agua a través de filtros GF/B (Packard Bioscience, Meriden, CT) y se mida mediante recuento de centelleo líquido con un TopCount (Packard Bioscience).
Ejemplo B2: ensayo de proliferación celular de Pfeiffer
La línea celular Pfeiffer (linfoma de células B grandes difuso) se adquirió de ATCC (Manassas, VA) y se mantuvo en el medio de cultivo recomendado (RPMI y FBS al 10%). Para medir la actividad antiproliferación de los compuestos, las células Pfeiffer se colocaron en placas con el medio de cultivo (2x103 células/pocillo/por 200 pl) en placas de unión ultrabaja de 96 pocillos (Corning, Corning, NY), en el presencia o ausencia de un intervalo de concentración de compuestos de prueba. Después de 3-4 días, se añade [3H]-timidina (1 pCi/pocillo) (PerkinElmer, Boston, MA) en PBS al cultivo celular durante 12 horas adicionales antes de que la radiactividad incorporada se
separe por filtración con agua a través de filtros GF/B (Packard Bioscience, Meridenj, CT) y se mida mediante recuento de centelleo líquido con un TopCount (Packard Bioscience).
Ejemplo B3: ensayo de proliferación celular SUDHL-6
La línea celular SUDHL-6 (linfoma de células B grandes difuso) se adquirió de ATCC (Manassas, VA) y mantuvo en el medio de cultivo recomendado (RPMI y FBS al 10%). Para medir la actividad antiproliferación de los compuestos mediante cuantificación de ATP, las células SUDHL-6 se colocaron en placas con el medio de cultivo (5000 células/pocillo/por 200 gl) en una placa de cultivo de tejido de poliestireno negra transparente de 96 pocillos (Greiner-bio-one a través de VWR, NJ) en presencia o ausencia de un intervalo de concentración de compuestos de prueba. Después de 3 días, se añadió agente de cultivo celular Cell Titer-GLO Luminescent (Promega, Madison, WI) a cada pocillo durante 10 minutos a temperatura ambiente para estabilizar la señal luminiscente. Esto determina el número de células viables en cultivo en base a la cuantificación del ATP presente, que señala la presencia de células metabólicamente activas. La luminiscencia se midió con TopCount 384 (Packard Bioscience de Perkin Elmer, Boston, MA).
Ejemplo C: ensayo de fosforilación de Akt
Las células Ramos (linfocitos B de linfoma de Burkitt) se obtuvieron de ATCC (Manassas, VA) y se mantuvieron en RPMI1640 y FBS al 10%. Las células (3x107 células/tubo/3 ml en RPMI) se incuban con diferentes cantidades de los compuestos de prueba durante 2 horas a 37° C y luego se estimularon con IgM anti-humana F(ab')2 de cabra (5 g/ml) (Invitrogen) durante 17 minutos en un baño de agua a 37° C. Las células estimuladas se centrifugan a 4° C con centrifugación y los extractos de células completas se preparan usando tampón de lisis 300 gl (Cell Signaling Technology, Danvers, MA). Los lisados resultantes se sonican y se recogen los sobrenadantes. El nivel de fosforilación de Akt en los sobrenadantes se analiza usando kits de ELISA en sándwich PathScan phospho-Aktl (Ser473) (Cell Signaling Technology) de acuerdo con las instrucciones del fabricante.
Claims (16)
1. Un proceso, que comprende hacer reaccionar un compuesto de Fórmula XVI:

con acetato de formamidina para formar un compuesto de Fórmula I:

opcionalmente, en donde:
(a) dicha reacción del compuesto de Fórmula XVI con acetato de formamidina se lleva a cabo en un componente solvente que comprende 1,2-etanodiol; y/o
(b) dicha reacción del compuesto de Fórmula XVI con acetato de formamidina se realiza a una temperatura de aproximadamente 100° C a aproximadamente 105° C; y/o
(c) se usan de aproximadamente 8 a aproximadamente 10 equivalentes de acetato de formamidina en base a 1 equivalente del compuesto de Fórmula XVI.
2. El proceso de la reivindicación 1, que comprende además preparar el compuesto de Fórmula XVI mediante un proceso que comprende hacer reaccionar un compuesto de Ffórmula XV:

con (1 -etoxietiliden)malononitrilo en presencia de una amina terciaria; opcionalmente, en donde:
(a) dicha amina terciaria es N-metilpirrolidinona; y/o
(b) dicha reacción del compuesto de Fórmula XV con (l-etoxietiliden)malononitrilo se realiza a aproximadamente temperatura ambiente.
3. El proceso de la reivindicación 2, que comprende además preparar el compuesto de Fórmula XV mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula XlV-a:

con hidrazina en presencia de una amina terciaria, en donde P1 es alquilsulfonilo Ci-6; opcionalmente, en donde:
(a) dicha amina terciaria es N-metilpirrolidinona; y/o
(b) dicha reacción del compuesto de Fórmula XlV-a con hidrazina se realiza a una temperatura de aproximadamente 35° C a aproximadamente 60° C; y/o
(c) dicha reacción del compuesto de Fórmula XlV-a con hidrazina se lleva a cabo en un componente solvente que comprende diclorometano; y/o
(d) P1 es un grupo metanosulfonilo.
4. El proceso de la reivindicación 3, que comprende además preparar el compuesto de Fórmula XlV-a mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula Xlll:

con haluro de alquilsulfonilo C1-6 en presencia de una amina terciaria; opcionalmente, en donde:
(a) dicho haluro de alquilsulfonilo C1-6 es cloruro de metanosulfonilo; y/o
(b) dicha amina terciaria es NN-diisopropiletilamina; y/o
(c) se usan de aproximadamente 1,1 a aproximadamente 1,5 equivalentes de haluro de alquilsulfonilo en base a 1 equivalente del compuesto de Fórmula XIII.
5. El proceso de la reivindicación 4, en donde dicha reacción de dicho compuesto de Fórmula XIII con haluro de alquilsulfonilo C1-6 se realiza:
(a) a una temperatura de aproximadamente -10° C a aproximadamente 5° C; y/o
(b) en un componente solvente que comprende diclorometano.
6. El proceso de una cualquiera de las reivindicaciones 1-5, en donde los pasos de: (i) hacer reaccionar dicho compuesto de Fórmula XIII con haluro de alquilsulfonilo C1-6; (ii) hacer reaccionar dicho compuesto de Fórmula XIV-a con hidrazina en presencia de una amina terciaria para formar un compuesto de Fórmula XV; y (iii) hacer reaccionar dicho compuesto de Fórmula XV con acetato de formamidina para formar un compuesto de Fórmula XVI se realizan en el mismo recipiente sin aislar el compuesto de Fórmula XIV-a o el compuesto de Fórmula XV.
7. El proceso de la reivindicación 1, que comprende además preparar el compuesto de Fórmula XVI mediante un proceso que comprende hacer reaccionar una sal de Fórmula XV-a:

con (1-etoxietiliden)malononitrilo en presencia de una amina terciaria, en donde TsOH es ácido p-toluenosulfónico; opcionalmente, en donde:
(a) dicha amina terciaria es /V,N-diisopropiletilamina; y/o
(b) dicha reacción de una sal de Fórmula XV-a con (1-etoxietiliden)malononitrilo se realiza a aproximadamente temperatura ambiente; y/o
(c) se usan de aproximadamente a 1,3 a aproximadamente 1,6 equivalentes de (l-etoxietiliden)malononitrilo basado en 1 equivalente de la sal de Fórmula XV-a; y/o
(d) dicha reacción de la sal de Fórmula XV-a con (1-etoxietiliden)malononitrilo se lleva a cabo en un componente solvente que comprende etanol.
8. El proceso de la reivindicación 7, que comprende además preparar la sal de Fórmula XV-a mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula XXI:

con ácido p-toluenosulfónico, en donde Boc es terc-butoxicarbonilo; opcionalmente, en donde:
(a) dicho ácido p-toluenosulfónico es monohidrato de ácido p-toluenosulfónico; y/o
(b) se usan de aproximadamente 1,3 a aproximadamente 1,6 equivalentes de ácido p-toluenosulfónico en base a 1 equivalente del compuesto de Fórmula XXI; y/o
(c) dicha reacción de dicho compuesto de Fórmula XXI con ácido p-toluenosulfónico se realiza a una temperatura de aproximadamente 45° C a aproximadamente 65° C; y/o
(d) dicha reacción de dicho compuesto de Fórmula XXI con ácido p-toluenosulfónico se lleva a cabo en un componente solvente que comprende etanol.
9. El proceso de la reivindicación 8, en el que los pasos de: (i) hacer reaccionar dicho compuesto de Fórmula XXI con ácido p-toluenosulfónico para formar una sal de Fórmula XV-a; y (ii) hacer reaccionar dicha sal de Fórmula XV-a con (1-etoxietiliden)malononitrilo se lleva a cabo en el mismo recipiente sin aislamiento de la sal de Fórmula XV-a.
10. El proceso de cualquiera de las reivindicaciones 8-9, que comprende además preparar el compuesto de Fórmula XXI mediante un proceso que comprende hacer reaccionar un compuesto de fórmula XX:

con hidrógeno gaseoso en presencia de uno o más catalizadores de hidrogenación seleccionados independientemente, en dondee Boc es f-butoxicarbonilo.
11. El proceso de la reivindicación 10, en donde dicha reacción del compuesto de Fórmula XX con gas hidrógeno se realiza en presencia de dos catalizadores de hidrogenación seleccionados independientemente; opcionalmente en donde un catalizador de hidrogenación es tetrafluoroborato de bis(1,5-ciclooctadieno)rodio(I) y el otro es (R)-(-)-1-{(S)-2-[bis(4-trifluorometilfenil)fosfina]ferrocenilo}etil-di-f-butilfosfina.
12. El proceso de la reivindicación 11 en donde:
(a) se usan de aproximadamente 13,5 a aproximadamente 14,5 equivalentes del compuesto de Fórmula XX en base a 1 equivalente de tetrafluoroborato de bis(1,5-ciclooctadieno)rodio(I); y/o
(b) se usan de aproximadamente 12 a aproximadamente 13 equivalentes del compuesto de Fórmula XX en base a 1 equivalente de (R )-(-)-1-{(S)-2-[bis(4-trifluorometilfenil)fosfina]ferrocenil}etil-di-f-butilfosfina.
13. El proceso de cualquiera de las reivindicaciones 10-12, en donde dicha reacción del compuesto de Fórmula XX con gas de hidrógeno realiza aproximadamente a temperatura ambiente; y/o dicha reacción del compuesto de Fórmula XX con gas de hidrógeno se lleva a cabo en un componente solvente que comprende metanol.
14. El proceso de cualquiera de las reivindicaciones 10-13, que comprende además preparar el compuesto de Fórmula XX mediante un proceso que comprende hacer reaccionar un compuesto de Fórmula XIX:

con carbazato de f-butilo; opcionalmente, en donde:
(a) dicha reacción del compuesto de Fórmula XIX con carbazato de f-butilo se realiza a una temperatura de aproximadamente 60° C a aproximadamente 70° C; y/o
(b) dicha reacción del compuesto de Fórmula XIX con carbazato de f-butilo se lleva a cabo en un componente solvente que comprende metanol.
15. El proceso de la reivindicación 14, que comprende además preparar el compuesto de Fórmula XIX mediante un proceso que comprende oxidar un compuesto de Fórmula XIII-a:

en presencia de un agente oxidante; opcionalmente, en donde:
(a) dicho agente oxidante es peryodinano de Dess-Martin; y/o
(b) se usan de aproximadamente 1,2 a aproximadamente 1,7 equivalentes de dicho agente oxidante en base a 1 equivalente del compuesto de Fórmula Xlll-a; y/o
(c) dicha oxidación del compuesto de Fórmula Xlll-a se realiza a aproximadamente temperatura ambiente; y/o (d) dicha oxidación del compuesto de Fórmula Xlll-a se lleva a cabo en un componente disolvente que comprende diclorometano.
16. Un compuesto seleccionado de:
(a) Fórmula XlV:

o una sal farmacéuticamente aceptable del mismo; o
(b) Fórmula XV:

o una sal farmacéuticamente aceptable del mismo; o
(c) Fórmula XVl:

o una sal farmacéuticamente aceptable del mismo; o
(d) Fórmula XX:

o una sal farmacéuticamente aceptable del mismo; o
(e) Fórmula XXI:

o una sal farmacéuticamente aceptable del mismo.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562121697P | 2015-02-27 | 2015-02-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2933694T3 true ES2933694T3 (es) | 2023-02-13 |
Family
ID=55854770
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES20201979T Active ES2933694T3 (es) | 2015-02-27 | 2016-02-26 | Procesos para la preparación de un inhibidor de PI3K |
| ES16718921T Active ES2843522T3 (es) | 2015-02-27 | 2016-02-26 | Sales del inhibidor de PI3K y procesos para su preparación |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16718921T Active ES2843522T3 (es) | 2015-02-27 | 2016-02-26 | Sales del inhibidor de PI3K y procesos para su preparación |
Country Status (36)
| Country | Link |
|---|---|
| US (3) | US10336759B2 (es) |
| EP (3) | EP3831833B1 (es) |
| JP (2) | JP6816005B2 (es) |
| KR (2) | KR20240132104A (es) |
| CN (4) | CN117800973A (es) |
| AR (1) | AR103804A1 (es) |
| AU (3) | AU2016222556B2 (es) |
| BR (1) | BR122021004509B1 (es) |
| CA (1) | CA2977659A1 (es) |
| CL (2) | CL2017002179A1 (es) |
| CO (1) | CO2017008924A2 (es) |
| CR (2) | CR20170389A (es) |
| CY (1) | CY1123729T1 (es) |
| DK (2) | DK3262046T3 (es) |
| EA (1) | EA201791923A1 (es) |
| EC (2) | ECSP17064131A (es) |
| ES (2) | ES2933694T3 (es) |
| HK (1) | HK1248229A1 (es) |
| HR (2) | HRP20221444T1 (es) |
| HU (2) | HUE053025T2 (es) |
| IL (1) | IL254093B (es) |
| LT (2) | LT3262046T (es) |
| MA (2) | MA41607B1 (es) |
| MD (2) | MD3831833T2 (es) |
| MX (2) | MX2017010918A (es) |
| MY (1) | MY187502A (es) |
| PE (2) | PE20180129A1 (es) |
| PH (2) | PH12017501538A1 (es) |
| PL (2) | PL3262046T3 (es) |
| PT (2) | PT3262046T (es) |
| RS (2) | RS61233B1 (es) |
| SG (3) | SG10201907576SA (es) |
| SI (2) | SI3831833T1 (es) |
| TW (2) | TWI764392B (es) |
| UA (1) | UA122332C2 (es) |
| WO (1) | WO2016138363A1 (es) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BRPI1015135B1 (pt) | 2009-06-29 | 2021-08-03 | Incyte Holdings Corporation | Pirimidinonas inibidoras de pi3k, composição compreendendo tais compostos, bem como usos dos mesmos |
| WO2012087881A1 (en) | 2010-12-20 | 2012-06-28 | Incyte Corporation | N-(1-(substituted-phenyl)ethyl)-9h-purin-6-amines as pi3k inhibitors |
| ES2722524T3 (es) | 2011-09-02 | 2019-08-13 | Incyte Holdings Corp | Heterociclaminas como inhibidores de pi3k |
| AR090548A1 (es) | 2012-04-02 | 2014-11-19 | Incyte Corp | Azaheterociclobencilaminas biciclicas como inhibidores de pi3k |
| TWI657090B (zh) * | 2013-03-01 | 2019-04-21 | 英塞特控股公司 | 吡唑并嘧啶衍生物治療PI3Kδ 相關病症之用途 |
| NZ763326A (en) | 2014-04-08 | 2023-04-28 | Incyte Holdings Corp | Treatment of b-cell malignancies by a combination jak and pi3k inhibitor |
| WO2015191677A1 (en) | 2014-06-11 | 2015-12-17 | Incyte Corporation | Bicyclic heteroarylaminoalkyl phenyl derivatives as pi3k inhibitors |
| MD3831833T2 (ro) | 2015-02-27 | 2023-04-30 | Incyte Holdings Corp | Procedee pentru prepararea unui inhibitor PI3K |
| US9732097B2 (en) | 2015-05-11 | 2017-08-15 | Incyte Corporation | Process for the synthesis of a phosphoinositide 3-kinase inhibitor |
| US9988401B2 (en) | 2015-05-11 | 2018-06-05 | Incyte Corporation | Crystalline forms of a PI3K inhibitor |
| JP6860267B2 (ja) | 2015-12-11 | 2021-04-14 | シチュアン ケルン−バイオテック バイオファーマシューティカル カンパニー リミテッド | アゼチジン誘導体、その調製方法、及びその使用 |
| LT3762368T (lt) | 2018-03-08 | 2022-06-10 | Incyte Corporation | Aminopirazindiolio junginiai, kaip pi3k-γ inhibitoriai |
| JP7455763B2 (ja) * | 2018-06-01 | 2024-03-26 | インサイト・コーポレイション | Pi3k関連障害の処置のための投薬レジメン |
| WO2022115120A1 (en) | 2020-11-30 | 2022-06-02 | Incyte Corporation | Combination therapy with an anti-cd19 antibody and parsaclisib |
| US20220241411A1 (en) | 2020-11-30 | 2022-08-04 | Incyte Corporation | Combination therapy with an anti-cd19 antibody and parsaclisib |
| TW202329976A (zh) | 2021-12-16 | 2023-08-01 | 美商英塞特公司 | P13K—δ抑制劑之局部調配物 |
| CN114409654A (zh) * | 2021-12-30 | 2022-04-29 | 安徽普利药业有限公司 | 一种btk抑制剂的中间体合成方法 |
Family Cites Families (291)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3169967A (en) | 1957-11-14 | 1965-02-16 | Ciba Geigy Corp | Methyl o-lower alkanoyl-reserpates |
| US3037980A (en) | 1955-08-18 | 1962-06-05 | Burroughs Wellcome Co | Pyrrolopyrimidine vasodilators and method of making them |
| US3506643A (en) | 1966-12-09 | 1970-04-14 | Max Thiel | N**6-aralkyl-adenosine derivatives |
| DE2139107A1 (de) | 1971-08-04 | 1973-02-15 | Merck Patent Gmbh | Heterocyclisch substituierte adenosinverbindungen |
| US3962443A (en) | 1972-08-14 | 1976-06-08 | Dainippon Pharmaceutical Co., Ltd. | Antibacterial pharmaceutical compositions and processes for preparation thereof |
| DE2248232A1 (de) | 1972-10-02 | 1974-04-11 | Basf Ag | 4-thiopyrimido eckige klammer auf 4,5-d eckige klammer zu pyrimidine |
| AR204003A1 (es) | 1973-04-03 | 1975-11-12 | Dainippon Pharmaceutical Co | Procedimiento para preparar compuestos derivados del acido 2-(1-piperazinil)-5-oxopirido-(2,3-d)pirimidino-6-carboxilico y sus sales farmaceuticamente aceptables |
| US3862189A (en) | 1973-08-14 | 1975-01-21 | Warner Lambert Co | Aralkyl-substituted purines and pyrimidines as antianginal bronchodilator agents |
| US3936454A (en) | 1973-08-14 | 1976-02-03 | Warner-Lambert Company | 5-Amino-4-chloro-6-(substituted amino)-pyrimidines |
| DK3375A (es) | 1974-01-25 | 1975-09-15 | Ciba Geigy Ag | |
| JPS587626B2 (ja) | 1974-02-13 | 1983-02-10 | 大日本製薬株式会社 | ナフチリジン オヨビ キノリンユウドウタイノセイホウ |
| JPS5359663A (en) | 1976-11-09 | 1978-05-29 | Sumitomo Chem Co Ltd | 2-halogeno methyl indole derivatives and process for praparation of the same |
| JPS52106897A (en) | 1977-01-10 | 1977-09-07 | Dainippon Pharmaceut Co Ltd | Synthesis of piperazine derivatives |
| JPS5392767A (en) | 1977-01-27 | 1978-08-15 | Sumitomo Chem Co Ltd | Preparation of 2-phthalimidomethylindole derivatives |
| JPS5625234A (en) | 1979-08-02 | 1981-03-11 | Hitachi Denshi Ltd | Dropout display system |
| JPS56123981A (en) | 1981-02-23 | 1981-09-29 | Dainippon Pharmaceut Co Ltd | Preparation of 1,4-disubstituted piperazine |
| JPS5883698A (ja) | 1981-11-13 | 1983-05-19 | Takeda Chem Ind Ltd | キノン化合物およびその製造法 |
| JPS58162949A (ja) | 1982-03-20 | 1983-09-27 | Konishiroku Photo Ind Co Ltd | ハロゲン化銀カラ−写真感光材料 |
| JPS6067709U (ja) | 1983-10-14 | 1985-05-14 | 三洋電機株式会社 | ヘア−ドライヤ− |
| JPS60140373U (ja) | 1984-02-28 | 1985-09-17 | 東洋ハ−ネス株式会社 | ワイヤハ−ネスのア−ス構造 |
| JPS6190153A (ja) | 1984-10-09 | 1986-05-08 | Fuji Photo Film Co Ltd | ハロゲン化銀写真感光材料の処理方法 |
| JPS6263591U (es) | 1985-06-29 | 1987-04-20 | ||
| JPS62103640A (ja) | 1985-07-18 | 1987-05-14 | Fuji Photo Film Co Ltd | ハロゲン化銀カラ−写真感光材料 |
| JPS635783Y2 (es) | 1985-10-17 | 1988-02-17 | ||
| JPH07119970B2 (ja) | 1986-04-18 | 1995-12-20 | 富士写真フイルム株式会社 | 画像形成方法 |
| JPS6310746A (ja) | 1986-07-01 | 1988-01-18 | Tanabe Seiyaku Co Ltd | ナフタレン誘導体 |
| CA1324609C (en) | 1986-07-30 | 1993-11-23 | Eastman Kodak Company | Photographic element and process |
| JPS6427257U (es) | 1987-08-11 | 1989-02-16 | ||
| US4861701A (en) | 1987-10-05 | 1989-08-29 | Eastman Kodak Company | Photographic element and process comprising a compound which comprises two timing groups in sequence |
| AT388372B (de) | 1987-10-08 | 1989-06-12 | Tanabe Seiyaku Co | Neue naphthalinderivate und sie enthaltende pharmazeutika |
| JPH01250316A (ja) | 1987-12-28 | 1989-10-05 | Tanabe Seiyaku Co Ltd | 抗脂血剤 |
| EP0364598A4 (en) | 1988-03-02 | 1992-01-15 | Yoshitomi Pharmaceutical Industries, Ltd. | 3,4-dihydrothieno 2,3-d¨pyrimidine compounds and pharmaceutical application thereof |
| US5208250A (en) | 1988-05-25 | 1993-05-04 | Warner-Lambert Company | Known and selected novel arylmethylenyl derivatives of thiazolidinones, imidazolidinones and oxazolidinones useful as antiallergy agents and anti-inflammatory agents |
| JPH054831Y2 (es) | 1988-11-22 | 1993-02-08 | ||
| CA2053863C (en) | 1990-04-25 | 1996-10-29 | Keizo Tanikawa | Pyridazinone derivatives |
| SU1712359A1 (ru) | 1990-05-07 | 1992-02-15 | Уфимский Нефтяной Институт | Гидрохлорид 8 @ -гидроксихинолинового эфира 8-гидроксихинолин-7-карбоновой кислоты, в качестве бактерицида дл подавлени сульфатвосстанавливающих бактерий и культур РSеUDомоNаS и АRтнRовастеR |
| EP0464612B1 (en) | 1990-06-28 | 1998-05-13 | Fuji Photo Film Co., Ltd. | Silver halide photographic materials |
| EP0481614A1 (en) | 1990-10-01 | 1992-04-22 | Merck & Co. Inc. | Substituted pyridopyrimidinones and related heterocycles as angiotensin II antagonists |
| JPH04190232A (ja) | 1990-11-26 | 1992-07-08 | Fuji Photo Film Co Ltd | ハロゲン化銀カラー写真感光材料 |
| US5480883A (en) | 1991-05-10 | 1996-01-02 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Bis mono- and bicyclic aryl and heteroaryl compounds which inhibit EGF and/or PDGF receptor tyrosine kinase |
| IL101860A0 (en) | 1991-05-31 | 1992-12-30 | Ici Plc | Heterocyclic derivatives |
| JP3108483B2 (ja) | 1991-09-30 | 2000-11-13 | 日清製粉株式会社 | インドール誘導体およびこれを有効成分とする抗潰瘍薬 |
| HUT64064A (en) | 1992-02-13 | 1993-11-29 | Chinoin Gyogyszer Es Vegyeszet | Process for producing puyrido/1,2-a/pyrimidine derivatives and pharmaceutical compositions comprising same as active ingredient |
| US5521184A (en) | 1992-04-03 | 1996-05-28 | Ciba-Geigy Corporation | Pyrimidine derivatives and processes for the preparation thereof |
| AU3933493A (en) | 1992-04-24 | 1993-11-29 | E.I. Du Pont De Nemours And Company | Arthropodicidal and fungicidal aminopyrimidines |
| TW229140B (es) | 1992-06-05 | 1994-09-01 | Shell Internat Res Schappej B V | |
| FR2714907B1 (fr) | 1994-01-07 | 1996-03-29 | Union Pharma Scient Appl | Nouveaux dérivés de l'Adénosine, leurs procédés de préparation, compositions pharmaceutiques les contenant. |
| US6342501B1 (en) | 1994-02-25 | 2002-01-29 | The Regents Of The University Of Michigan | Pyrrolo[2,3-d] pyrimidines as antiviral agents |
| JPH0987282A (ja) | 1995-09-21 | 1997-03-31 | Kyowa Hakko Kogyo Co Ltd | チアゾール誘導体 |
| JPH09176162A (ja) | 1995-12-22 | 1997-07-08 | Toubishi Yakuhin Kogyo Kk | チアゾリジンジオン誘導体及びその製造法並びにそれを含む医薬組成物 |
| JPH09176116A (ja) | 1995-12-27 | 1997-07-08 | Toray Ind Inc | 複素環誘導体およびその医薬用途 |
| JPH1025294A (ja) | 1996-03-26 | 1998-01-27 | Akira Matsuda | 縮合ヘテロ環誘導体、その製造法及びそれを含有する悪性腫瘍治療剤 |
| JP4373497B2 (ja) | 1996-06-19 | 2009-11-25 | ローン−プーラン・ロレ・リミテツド | 置換されたアザビシクロ化合物、ならびにtnfおよびサイクリックampホスホジエステラーゼ産生の阻害剤としてのそれらの使用 |
| CA2230808C (en) | 1996-07-03 | 2006-08-15 | Japan Energy Corporation | A novel purine derivative |
| US5866702A (en) | 1996-08-02 | 1999-02-02 | Cv Therapeutics, Incorporation | Purine inhibitors of cyclin dependent kinase 2 |
| US6630496B1 (en) | 1996-08-26 | 2003-10-07 | Genetics Institute Llc | Inhibitors of phospholipase enzymes |
| JPH10231297A (ja) | 1997-02-20 | 1998-09-02 | Japan Energy Corp | 新規なアデニン−1−n−オキシド誘導体およびその医薬用途 |
| KR100540046B1 (ko) | 1997-11-12 | 2006-01-10 | 미쓰비시 가가꾸 가부시키가이샤 | 퓨린유도체 및 이를 유효성분으로 함유하는 의약 |
| TW572758B (en) | 1997-12-22 | 2004-01-21 | Sumitomo Pharma | Type 2 helper T cell-selective immune response inhibitors comprising purine derivatives |
| EE200000486A (et) | 1998-02-25 | 2002-02-15 | Genetics Institute, Inc. | Fosfolipaasensüümide inhibiitorid, neid sisaldav farmatseutiline kompositsioon ning meetod põletikulise reaktsiooni raviks imetajal |
| EP1062216A1 (en) | 1998-02-25 | 2000-12-27 | Genetics Institute, Inc. | Inhibitors of phospholipase a2 |
| US6828344B1 (en) | 1998-02-25 | 2004-12-07 | Genetics Institute, Llc | Inhibitors of phospholipase enzymes |
| US6479487B1 (en) | 1998-02-26 | 2002-11-12 | Aventis Pharmaceuticals Inc. | 6, 9-disubstituted 2-[trans-(4-aminocyclohexyl)amino] purines |
| EP1109785B1 (de) | 1998-05-04 | 2003-01-02 | Zentaris AG | Indolderivate und deren verwendung zur behandlung von malignen und anderen, auf pathologischen zellproliferationen beruhenden erkrankungen |
| US6673797B1 (en) | 1998-05-26 | 2004-01-06 | Chugai Seiyaku Kabushiki Kaisha | Heterocyclic indole derivatives and mono- or diazaindole derivatives |
| JP3997651B2 (ja) | 1998-06-24 | 2007-10-24 | コニカミノルタホールディングス株式会社 | 新規色素及び画像記録材料及び感熱転写材料及びインクジェット記録液 |
| AU5620299A (en) | 1998-08-11 | 2000-03-06 | Novartis Ag | Isoquinoline derivatives with angiogenesis inhibiting activity |
| EP1109805B1 (en) | 1998-08-25 | 2003-12-17 | The Uab Research Foundation | Inhibitors of bacterial nad synthetase |
| US6133031A (en) | 1999-08-19 | 2000-10-17 | Isis Pharmaceuticals Inc. | Antisense inhibition of focal adhesion kinase expression |
| MXPA01007851A (es) | 1999-02-01 | 2003-06-04 | Cv Therapeutics Inc | Inhibidores purina de cinasa 2 e 1kb-alfa dependientes del ciclin. |
| GB9905075D0 (en) | 1999-03-06 | 1999-04-28 | Zeneca Ltd | Chemical compounds |
| JP2000281654A (ja) | 1999-03-26 | 2000-10-10 | Tanabe Seiyaku Co Ltd | イソキノリン誘導体 |
| DE19932571A1 (de) | 1999-07-13 | 2001-01-18 | Clariant Gmbh | Verfahren zur Herstellung von Biarylen unter Palladophosphacyclobutan-Katalyse |
| JP2001151771A (ja) | 1999-09-10 | 2001-06-05 | Kyowa Hakko Kogyo Co Ltd | 含窒素芳香族複素環誘導体 |
| GB0004890D0 (en) | 2000-03-01 | 2000-04-19 | Astrazeneca Uk Ltd | Chemical compounds |
| US6436965B1 (en) | 2000-03-02 | 2002-08-20 | Merck Frosst Canada & Co. | PDE IV inhibiting amides, compositions and methods of treatment |
| EP1138328A1 (en) | 2000-03-29 | 2001-10-04 | Eli Lilly And Company Limited | Naphthalene derivatives as CNS drugs |
| US6667300B2 (en) | 2000-04-25 | 2003-12-23 | Icos Corporation | Inhibitors of human phosphatidylinositol 3-kinase delta |
| MXPA03000051A (es) | 2000-06-28 | 2003-08-19 | Smithkline Beecham Plc | Procedimiento de molido en humedo. |
| CN1331340A (zh) | 2000-06-30 | 2002-01-16 | 上海博德基因开发有限公司 | 一种新的多肽——人拓扑异构酶12.1和编码这种多肽的多核苷酸 |
| ATE322485T1 (de) | 2000-07-05 | 2006-04-15 | Astellas Pharma Inc | Propan-1,3-dion-derivate |
| FR2814073B1 (fr) | 2000-09-21 | 2005-06-24 | Yang Ji Chemical Company Ltd | Composition pharmaceutique antifongique et/ou antiparasitaire et nouveaux derives de l'indole a titre de principes actifs d'une telle composition |
| DOP2002000334A (es) | 2001-02-14 | 2002-08-30 | Warner Lambert Co | Pirimidinas biciclicas como inhibidores de metaloproteinasas de matriz |
| SE0100568D0 (sv) | 2001-02-20 | 2001-02-20 | Astrazeneca Ab | Compounds |
| ATE411292T1 (de) | 2001-03-01 | 2008-10-15 | Shionogi & Co | Stickstoffhaltige heteroarylverbindungen mit hiv- integrase inhibierender wirkung |
| UA76977C2 (en) | 2001-03-02 | 2006-10-16 | Icos Corp | Aryl- and heteroaryl substituted chk1 inhibitors and their use as radiosensitizers and chemosensitizers |
| US20050107400A1 (en) | 2001-03-30 | 2005-05-19 | Boyd Leslie F. | Use of pyrazolopyridines as therapeutic compounds |
| US7034030B2 (en) | 2001-03-30 | 2006-04-25 | Smithkline Beecham Corporation | Pyralopyridines, process for their preparation and use as therapeutic compounds |
| ES2242028T3 (es) | 2001-04-27 | 2005-11-01 | Smithkline Beecham Corporation | Derivados de pirazolo(1,5-a)piridina. |
| CZ294535B6 (cs) | 2001-08-02 | 2005-01-12 | Ústav Experimentální Botaniky Avčr | Heterocyklické sloučeniny na bázi N6-substituovaného adeninu, způsoby jejich přípravy, jejich použití pro přípravu léčiv, kosmetických přípravků a růstových regulátorů, farmaceutické přípravky, kosmetické přípravky a růstové regulátory tyto sloučeniny obsahující |
| GB0121033D0 (en) | 2001-08-30 | 2001-10-24 | Novartis Ag | Organic compounds |
| US8124625B2 (en) | 2001-09-14 | 2012-02-28 | Shionogi & Co., Ltd. | Method of enhancing the expression of apolipoprotein AI using olefin derivatives |
| PL369530A1 (en) | 2001-09-19 | 2005-05-02 | Aventis Pharma S.A. | Chemical compounds |
| JP2005504807A (ja) | 2001-09-26 | 2005-02-17 | バイエル・フアーマシユーチカルズ・コーポレーシヨン | 抗糖尿病薬としての1,8−ナフチリジン誘導体 |
| DE60228103D1 (de) | 2001-10-02 | 2008-09-18 | Smithkline Beecham Corp | Chemische verbindungen |
| CN1582150B (zh) | 2001-10-30 | 2011-09-07 | 诺瓦提斯公司 | 作为flt3受体酪氨酸激酶活性抑制剂的星形孢菌素衍生物 |
| US7262164B2 (en) | 2001-11-09 | 2007-08-28 | Enzon, Inc. | Polymeric thiol-linked prodrugs employing benzyl elimination systems |
| EP1314733A1 (en) | 2001-11-22 | 2003-05-28 | Aventis Pharma Deutschland GmbH | Indole-2-carboxamides as factor Xa inhibitors |
| ES2291543T3 (es) | 2001-12-06 | 2008-03-01 | MERCK & CO., INC. | Inhibicion de kinesina mitotica. |
| EP1551812B1 (en) | 2001-12-06 | 2009-03-04 | Merck & Co., Inc. | Mitotic kinesin inhibitors |
| TW200301135A (en) | 2001-12-27 | 2003-07-01 | Otsuka Maryland Res Inst Inc | Pharmaceutical compositions comprising a multifunctional phosphodiesterase inhibitor and an adenosine uptake inhibitor |
| EP1484320A1 (en) | 2002-02-13 | 2004-12-08 | Takeda Chemical Industries, Ltd. | Jnk inhibitor |
| WO2003074497A1 (en) | 2002-03-01 | 2003-09-12 | Pintex Pharmaceutical, Inc. | Pin1-modulating compounds and methods of use thereof |
| WO2003084959A1 (en) | 2002-04-03 | 2003-10-16 | Bristol-Myers Squibb Company | Thiopene-based tricyclic compounds and pharmaceutical compositions comprising same |
| US20040063658A1 (en) | 2002-05-06 | 2004-04-01 | Roberts Christopher Don | Nucleoside derivatives for treating hepatitis C virus infection |
| AR037647A1 (es) | 2002-05-29 | 2004-12-01 | Novartis Ag | Derivados de diarilurea utiles para el tratamiento de enfermedades dependientes de la cinasa de proteina |
| GB0215676D0 (en) | 2002-07-05 | 2002-08-14 | Novartis Ag | Organic compounds |
| WO2004024693A1 (ja) | 2002-08-13 | 2004-03-25 | Shionogi & Co., Ltd. | Hivインテグラーゼ阻害活性を有するヘテロ環化合物 |
| JP4190232B2 (ja) | 2002-08-26 | 2008-12-03 | 富士通株式会社 | 機械研磨を行う方法 |
| KR101111085B1 (ko) | 2002-09-27 | 2012-04-12 | 다이닛본 스미토모 세이야꾸 가부시끼가이샤 | 신규 아데닌 화합물 및 그 용도 |
| CL2003002353A1 (es) | 2002-11-15 | 2005-02-04 | Vertex Pharma | Compuestos derivados de diaminotriazoles, inhibidores d ela proteina quinasa; composicion farmaceutica; procedimiento de preparacion; y su uso del compuesto en el tratamiento de enfermedades de desordenes alergicos, proliferacion, autoinmunes, condic |
| ES2412273T3 (es) | 2002-11-21 | 2013-07-10 | Novartis Ag | Inhibidores de 2-morfolín-4-pirimidinas como inhibidores de fosfotidilinositol (PI) 3-quinasa y su uso en el tratamiento del cáncer. |
| WO2004048326A1 (ja) | 2002-11-25 | 2004-06-10 | Mochida Pharmaceutical Co., Ltd. | 4−ヒドロキシピペリジン誘導体を有効成分とする呼吸器疾患治療剤 |
| UA80767C2 (en) | 2002-12-20 | 2007-10-25 | Pfizer Prod Inc | Pyrimidine derivatives for the treatment of abnormal cell growth |
| JP4500689B2 (ja) | 2002-12-26 | 2010-07-14 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 選択的エストロゲン受容体モジュレーター |
| CZ294538B6 (cs) | 2002-12-30 | 2005-01-12 | Ústav Experimentální Botaniky Akademie Vědčeské Re | Substituční deriváty N6-benzyladenosinu, způsob jejich přípravy, jejich použití pro přípravu léčiv, kosmetických přípravků a růstových regulátorů, farmaceutické přípravky, kosmetické přípravky a růstové regulátory tyto sloučeniny obsahující |
| RU2233842C1 (ru) | 2003-01-13 | 2004-08-10 | Петров Владимир Иванович | Производные пурина, обладающие противовирусной активностью |
| AR043692A1 (es) | 2003-02-06 | 2005-08-10 | Novartis Ag | 2-cianopirrolopirimidinas y sus usos farmaceuticos |
| GB0304640D0 (en) | 2003-02-28 | 2003-04-02 | Novartis Ag | Organic compounds |
| GB0305929D0 (en) | 2003-03-14 | 2003-04-23 | Novartis Ag | Organic compounds |
| SE0300908D0 (sv) | 2003-03-31 | 2003-03-31 | Astrazeneca Ab | Azaindole derivatives, preparations thereof, uses thereof and compositions containing them |
| US7129264B2 (en) | 2003-04-16 | 2006-10-31 | Bristol-Myers Squibb Company | Biarylmethyl indolines and indoles as antithromboembolic agents |
| JP2007501272A (ja) | 2003-05-05 | 2007-01-25 | ニューロジェン・コーポレーション | 置換イミダゾロピラジンおよびトリアゾロピラジン誘導体類:gabaaレセプタリガンド類 |
| EP1636225B1 (en) | 2003-06-20 | 2010-02-24 | Novartis Vaccines and Diagnostics, Inc. | Pyridino 1,2-a pyrimidin-4-one compounds as anticancer agents |
| WO2005000309A2 (en) | 2003-06-27 | 2005-01-06 | Ionix Pharmaceuticals Limited | Chemical compounds |
| JP4570015B2 (ja) | 2003-07-14 | 2010-10-27 | クミアイ化学工業株式会社 | 2−イソオキサゾリン誘導体及びそれを有効成分として含有する除草剤 |
| JP2007502776A (ja) | 2003-08-15 | 2007-02-15 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Rtk阻害剤としての6−置換アニリノプリン類 |
| US7148228B2 (en) | 2003-09-18 | 2006-12-12 | Conforma Therapeutics Corporation | Pyrazolopyrimidines and related analogs as HSP90-inhibitors |
| CA2539853C (en) | 2003-09-23 | 2011-04-19 | Merck & Co., Inc. | Isoquinolinone potassium channel inhibitors |
| PE20050952A1 (es) | 2003-09-24 | 2005-12-19 | Novartis Ag | Derivados de isoquinolina como inhibidores de b-raf |
| CA2553724A1 (en) | 2004-02-03 | 2005-08-18 | Abbott Laboratories | Aminobenzoxazoles as therapeutic agents |
| US20070191395A1 (en) | 2004-02-16 | 2007-08-16 | Katsuhiro Kawakami | Heterocyclic compounds having antifungal activity |
| CN1560035A (zh) | 2004-03-12 | 2005-01-05 | 沈阳药科大学 | 5-羟基吲哚-3-羧酸脂类衍生物 |
| WO2005091857A2 (en) | 2004-03-12 | 2005-10-06 | Bayer Pharmaceuticals Corporation | 1,6-naphthyridine and 1,8-naphthyridine derivatives and their use to treat diabetes and related disorders |
| JPWO2005092892A1 (ja) | 2004-03-26 | 2008-02-14 | 大日本住友製薬株式会社 | 8−オキソアデニン化合物 |
| US7217702B2 (en) | 2004-04-02 | 2007-05-15 | Adenosine Therapeutics, Llc | Selective antagonists of A2A adenosine receptors |
| LT2612862T (lt) | 2004-05-13 | 2017-01-25 | Icos Corporation | Chinazolinonai kaip žmogaus fosfatidilinozitol-3-kinazės delta inhibitoriai |
| EP1753428A4 (en) | 2004-05-14 | 2010-09-15 | Abbott Lab | INHIBITORS OF KINASES AS THERAPEUTIC AGENTS |
| KR20070044458A (ko) | 2004-07-22 | 2007-04-27 | 아스트라제네카 아베 | 암의 예방 및 치료에 유용한 융합 피리미돈 |
| CA2575188A1 (en) | 2004-08-18 | 2006-02-23 | Astrazeneca Ab | Enantiomers of selected fused pyrimidones and uses in the treatment and preventi on of cancer |
| DE102004044221A1 (de) | 2004-09-14 | 2006-03-16 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Neue 3-Methyl-7-butinyl-xanthine, deren Herstellung und deren Verwendung als Arzneimittel |
| GB0420719D0 (en) | 2004-09-17 | 2004-10-20 | Addex Pharmaceuticals Sa | Novel allosteric modulators |
| AU2005301133A1 (en) | 2004-10-19 | 2006-05-11 | Novartis Vaccines And Diagnostics Inc. | Indole and benzimidazole derivatives |
| US20080004269A1 (en) | 2004-11-04 | 2008-01-03 | Yuelian Xu | Pyrazolylmethy Heteroaryl Derivatives |
| WO2006068760A2 (en) | 2004-11-19 | 2006-06-29 | The Regents Of The University Of California | Anti-inflammatory pyrazolopyrimidines |
| WO2006056399A2 (en) | 2004-11-24 | 2006-06-01 | Novartis Ag | Combinations of jak inhibitors and at least one of bcr-abl, flt-3, fak or raf kinase inhibitors |
| CA2598409A1 (en) | 2005-02-17 | 2006-08-24 | Icos Corporation | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| US7799795B2 (en) | 2005-06-27 | 2010-09-21 | Amgen Inc. | Aryl nitrile compounds and compositions and their uses in treating inflammatory and related disorders |
| FR2889192A1 (fr) | 2005-07-27 | 2007-02-02 | Cytomics Systems Sa | Composes antifongiques, compositions contenant ces composes et leurs utilisations |
| CA2619881A1 (en) | 2005-08-16 | 2007-02-22 | Genzyme Corporation | Chemokine receptor binding compounds |
| US7642270B2 (en) | 2005-09-14 | 2010-01-05 | Janssen Pharmaceutica N.V. | 5-oxo-5,8-dihydro-pyrido-pyrimidine as inhibitors of c-fms kinase |
| EP1939202A4 (en) | 2005-09-22 | 2010-06-16 | Dainippon Sumitomo Pharma Co | NEW COMPOUND OF ADENINE |
| GB0520657D0 (en) | 2005-10-11 | 2005-11-16 | Ludwig Inst Cancer Res | Pharmaceutical compounds |
| MY167260A (en) | 2005-11-01 | 2018-08-14 | Targegen Inc | Bi-aryl meta-pyrimidine inhibitors of kinases |
| EP1783114A1 (en) | 2005-11-03 | 2007-05-09 | Novartis AG | N-(hetero)aryl indole derivatives as pesticides |
| ATE486875T1 (de) | 2005-11-10 | 2010-11-15 | Chemocentryx Inc | Substituierte chinolone und verwendungsverfahren |
| ES2970354T3 (es) | 2005-12-13 | 2024-05-28 | Incyte Holdings Corp | Derivados de pirrolo[2,3-d]pirimidina como inhibidores de Janus quinasas |
| US7989461B2 (en) | 2005-12-23 | 2011-08-02 | Amgen Inc. | Substituted quinazolinamine compounds for the treatment of cancer |
| US20100227880A1 (en) | 2006-01-25 | 2010-09-09 | Kristjan Gudmundsson | Chemical compounds |
| PE20071025A1 (es) | 2006-01-31 | 2007-10-17 | Mitsubishi Tanabe Pharma Corp | Compuesto amina trisustituido |
| PE20070978A1 (es) | 2006-02-14 | 2007-11-15 | Novartis Ag | COMPUESTOS HETEROCICLICOS COMO INHIBIDORES DE FOSFATIDILINOSITOL 3-QUINASAS (PI3Ks) |
| WO2007102392A1 (ja) | 2006-03-03 | 2007-09-13 | Shionogi & Co., Ltd. | Mmp-13選択的阻害剤 |
| CN101460161A (zh) | 2006-03-29 | 2009-06-17 | 弗尔德里克斯制药股份有限公司 | α-突触核蛋白毒性的抑制 |
| PT2004654E (pt) | 2006-04-04 | 2013-08-27 | Univ California | Derivados de pirazolopirimidina para utilização como antagonistas da quinase |
| US8933130B2 (en) | 2006-06-23 | 2015-01-13 | Radius Health, Inc. | Treatment of vasomotor symptoms with selective estrogen receptor modulators |
| US20080004253A1 (en) | 2006-06-30 | 2008-01-03 | Bryan James Branstetter | Thiazolopyrimidine modulators of TRPV1 |
| US20080009508A1 (en) | 2006-07-10 | 2008-01-10 | Lucie Szucova | 6,9-Disubstituted Purine Derivatives And Their Use For Treating Skin |
| EP2046788A1 (en) | 2006-07-12 | 2009-04-15 | Syngeta Participations AG | Triazolopyridine derivatives as herbicides |
| ES2657635T3 (es) | 2006-08-08 | 2018-03-06 | Chugai Seiyaku Kabushiki Kaisha | Derivado de pirimidina como inhibidor de PI3K y uso del mismo |
| AU2007291190A1 (en) | 2006-08-30 | 2008-03-06 | Cellzome Limited | Triazole derivatives as kinase inhibitors |
| WO2008032033A1 (en) | 2006-09-14 | 2008-03-20 | Astrazeneca Ab | 4-benzimidazolyl-2-morpholino-6-piperazinylpyrimidine derivatives as pi3k and mtor inhibitors for the treatment of proliferative disorders |
| EP1972631A1 (en) | 2007-03-23 | 2008-09-24 | Novartis AG | Imidazopyridazines as PI3K lipid kinase inhibitors |
| WO2008055013A2 (en) | 2006-10-31 | 2008-05-08 | Janssen Pharmaceutica N.V. | 5-oxo-5,8 - dihydro - pyrido - pyrimidines as inhibitors of c-fms kinase |
| CA2669399A1 (en) | 2006-11-13 | 2008-05-29 | Eli Lilly & Co. | Thienopyrimidinones for treatment of inflammatory disorders and cancers |
| HUE026659T2 (en) | 2006-11-22 | 2016-07-28 | Incyte Holdings Corp | Imidazotriazines and imidazopyrimidines as kinase inhibitors |
| WO2008082490A2 (en) | 2006-12-20 | 2008-07-10 | Schering Corporation | Novel jnk inhibitors |
| MX2009006921A (es) | 2006-12-29 | 2009-07-06 | Hoffmann La Roche | Derivados azaespiro. |
| JP2010518026A (ja) | 2007-02-05 | 2010-05-27 | ゼノン・ファーマシューティカルズ・インコーポレイテッド | ナトリウムチャネルが介在する疾患または状態の治療に有用なピリドピリミジノン化合物 |
| CN101687789A (zh) | 2007-02-12 | 2010-03-31 | 因特蒙公司 | C型肝炎病毒复制的新颖抑制剂 |
| TW200902018A (en) | 2007-03-20 | 2009-01-16 | Dainippon Sumitomo Pharma Co | Novel adenine compound |
| US20080233127A1 (en) | 2007-03-21 | 2008-09-25 | Wyeth | Imidazolopyrimidine analogs and their use as pi3 kinase and mtor inhibitors |
| ES2563458T3 (es) | 2007-03-23 | 2016-03-15 | Amgen Inc. | Compuestos heterocíclicos y sus usos |
| WO2008118454A2 (en) | 2007-03-23 | 2008-10-02 | Amgen Inc. | Derivatives of quinoline or benzopyrazine and their uses for the treatment of (inter alia) inflammatory diseases, autoimmune diseases or various kinds of cancer |
| US8039505B2 (en) | 2007-04-11 | 2011-10-18 | University Of Utah Research Foundation | Compounds for modulating T-cells |
| US8633186B2 (en) | 2007-06-08 | 2014-01-21 | Senomyx Inc. | Modulation of chemosensory receptors and ligands associated therewith |
| US9603848B2 (en) | 2007-06-08 | 2017-03-28 | Senomyx, Inc. | Modulation of chemosensory receptors and ligands associated therewith |
| AU2008266856A1 (en) | 2007-06-18 | 2008-12-24 | University Of Louisville Research Foundation, Inc. | Family of PFKFB3 inhibitors with anti-neoplastic activities |
| BRPI0813952A2 (pt) | 2007-06-29 | 2017-05-09 | Gilead Sciences Inc | derivados de purina e seu emprego como moduladores e receptor 7 semelhante ao dobre de sino |
| EP2190466A4 (en) | 2007-08-10 | 2011-12-21 | Burnham Inst Medical Research | Tissue-specific alkaline phosphatase (TNAP) activators and their use |
| TW200916447A (en) | 2007-08-29 | 2009-04-16 | Methylgene Inc | Sirtuin inhibitors |
| JP2009076865A (ja) | 2007-08-29 | 2009-04-09 | Fujifilm Corp | 有機電界発光素子 |
| WO2009034386A1 (en) | 2007-09-13 | 2009-03-19 | Astrazeneca Ab | Derivatives of adenine and 8-aza-adenine and uses thereof-796 |
| JP2009080233A (ja) | 2007-09-26 | 2009-04-16 | Kyocera Mita Corp | 電子写真感光体 |
| CZ300774B6 (cs) | 2007-10-05 | 2009-08-05 | Univerzita Palackého | Substituované 6-(alkylbenzylamino)purinové deriváty pro použití jako antagonisté cytokininových receptoru a prípravky obsahující tyto slouceniny |
| JP2011503103A (ja) | 2007-11-07 | 2011-01-27 | フォールドアールエックス ファーマシューティカルズ インコーポレーティッド | タンパク質輸送の調節方法 |
| WO2009063235A1 (en) | 2007-11-13 | 2009-05-22 | Astrazeneca Ab | Derivatives of 1,9-dihydro-6h-purin-6-one and uses thereof-018 |
| JP2009120686A (ja) | 2007-11-14 | 2009-06-04 | Toyo Ink Mfg Co Ltd | 光重合開始剤、重合性組成物、および重合物の製造方法。 |
| AU2008343813B2 (en) | 2007-12-19 | 2012-04-12 | Amgen Inc. | Inhibitors of PI3 kinase |
| US8399483B2 (en) | 2007-12-21 | 2013-03-19 | Ucb Pharma S.A. | Quinoxaline and quinoline derivatives as kinase inhibitors |
| JP2011507900A (ja) | 2007-12-21 | 2011-03-10 | ワイス・エルエルシー | イミダゾ[1,2−a]ピリジン化合物 |
| US8642660B2 (en) | 2007-12-21 | 2014-02-04 | The University Of Rochester | Method for altering the lifespan of eukaryotic organisms |
| US8410098B2 (en) | 2007-12-28 | 2013-04-02 | Topharman Shanghai Co., Ltd. | N-{1-[3-(2-ethoxy-5-(4-ethylpiperazinyl)sulfonylphenyl)-4,5-dihydro-5-OXO-1,2,4-triazin-6-yl]ethyl}butyramide, the preparation method and use thereof |
| US7960397B2 (en) | 2007-12-28 | 2011-06-14 | Institute Of Experimental Botany, Academy Of Sciences Of The Czech Republic | 6,9-disubstituted purine derivatives and their use as cosmetics and cosmetic compositions |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| AP2010005347A0 (en) | 2008-01-11 | 2010-08-31 | Natco Pharma Ltd | Novel pyrazolo [3,4-D] pyrimidine derivatives as anti-cancer agents |
| US9089572B2 (en) | 2008-01-17 | 2015-07-28 | California Institute Of Technology | Inhibitors of p97 |
| CN101981037B (zh) | 2008-01-30 | 2013-09-04 | 吉宁特有限公司 | 吡唑并嘧啶pi3k抑制剂化合物及使用方法 |
| EP2277881A4 (en) | 2008-04-18 | 2011-09-07 | Shionogi & Co | HETEROCYCLIC COMPOUND HAVING INHIBITORY ACTIVITY ON P13K |
| US8119647B2 (en) | 2008-04-23 | 2012-02-21 | Glenmark Pharmaceuticals S.A. | Fused pyrimidineone compounds as TRPV3 modulators |
| WO2009140215A2 (en) | 2008-05-11 | 2009-11-19 | Geraghty, Erin | Method for treating drug-resistant bacterial and other infections with clioquinol, phanquinone, and related compounds |
| WO2009151972A1 (en) | 2008-05-28 | 2009-12-17 | , The United States Of America, As Represented By The Secretary Of The Army, On Behalf Of U.S. Army Medical Research And Materiel Command | Small molecule inhibitors of botulinum neurotoxins |
| CA2727174A1 (en) | 2008-06-20 | 2010-01-21 | Jiangao Song | Aryl gpr119 agonists and uses thereof |
| US8026271B2 (en) | 2008-07-11 | 2011-09-27 | National Health Research Institutes | Formulations of indol-3-yl-2-oxoacetamide compounds |
| WO2010018458A2 (en) | 2008-08-12 | 2010-02-18 | Crystalgenomics, Inc. | Phenol derivatives and methods of use thereof |
| CA2738429C (en) | 2008-09-26 | 2016-10-25 | Intellikine, Inc. | Heterocyclic kinase inhibitors |
| JP5794919B2 (ja) | 2008-11-13 | 2015-10-14 | ギリアード カリストガ エルエルシー | 血液学的な悪性疾患のための療法 |
| US20110245209A1 (en) | 2008-12-16 | 2011-10-06 | Schering Corporation | Pyridopyrimidine derivatives and methods of use thereof |
| BR122013027950A2 (pt) | 2008-12-24 | 2019-12-10 | BIAL PORTELA & Cª S A | compostos farmacêuticos |
| US8563579B2 (en) | 2009-01-15 | 2013-10-22 | Anvyl Llc | α-7 nicotinic acetylcholine receptor allosteric modulators, their derivatives and uses thereof |
| EP2396315B1 (en) | 2009-02-13 | 2016-08-31 | UCB Biopharma SPRL | Quinoline derivatives as pi3k kinase inhibitors |
| WO2010114900A1 (en) | 2009-03-31 | 2010-10-07 | Arqule, Inc. | Substituted indolo-piperidine compounds |
| WO2010118367A2 (en) | 2009-04-10 | 2010-10-14 | Progenics Pharmaceuticals, Inc. | Antiviral pyrimidines |
| US8546409B2 (en) | 2009-04-20 | 2013-10-01 | Gilead Calistoga Llc | Methods of treatment for solid tumors |
| WO2010127208A1 (en) | 2009-04-30 | 2010-11-04 | Forest Laboratories Holdings Limited | Inhibitors of acetyl-coa carboxylase |
| EP2427195B1 (en) | 2009-05-07 | 2019-05-01 | Intellikine, LLC | Heterocyclic compounds and uses thereof |
| KR101771401B1 (ko) | 2009-05-22 | 2017-08-25 | 인사이트 홀딩스 코포레이션 | 야누스 키나제 억제제로서 피라졸4일피롤로[2,3d]피리미딘 및 피롤3일피롤로[2,3d]피리미딘의 N(헤테로)아릴피롤리딘 유도체 |
| WO2010151735A2 (en) | 2009-06-25 | 2010-12-29 | Amgen Inc. | Heterocyclic compounds and their uses |
| AU2010265974B2 (en) | 2009-06-25 | 2014-09-11 | Amgen Inc. | Polycyclic derivatives of pyridine and their use in the treatment of (inter alia) rheumatoid arthritis and similar diseases |
| CN102481300B (zh) | 2009-06-29 | 2015-04-15 | 安吉奥斯医药品有限公司 | 治疗性化合物和组合物 |
| BRPI1015135B1 (pt) | 2009-06-29 | 2021-08-03 | Incyte Holdings Corporation | Pirimidinonas inibidoras de pi3k, composição compreendendo tais compostos, bem como usos dos mesmos |
| AR077252A1 (es) | 2009-06-29 | 2011-08-10 | Xenon Pharmaceuticals Inc | Enantiomeros de compuestos de espirooxindol y sus usos como agentes terapeuticos |
| FR2947269B1 (fr) | 2009-06-29 | 2013-01-18 | Sanofi Aventis | Nouveaux composes anticancereux |
| JP2013500257A (ja) | 2009-07-21 | 2013-01-07 | ギリアード カリストガ エルエルシー | Pi3kインヒビターでの肝障害の処置 |
| SI2470546T1 (sl) | 2009-08-28 | 2013-12-31 | Takeda Pharmaceutical Company Limited | Spojine heksahidrooksazinopteridina za uporabo kot MTOR inhibitorji |
| US9249145B2 (en) | 2009-09-01 | 2016-02-02 | Incyte Holdings Corporation | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| CN102666545B (zh) | 2009-10-20 | 2016-04-06 | 塞尔卓姆有限公司 | 作为jak抑制剂的杂环吡唑并嘧啶类似物 |
| MX2012004706A (es) | 2009-10-22 | 2012-06-08 | Gilead Sciences Inc | Derivados de purina o deazapurina utiles para el tratamiento de (inter alia) infecciones virales. |
| KR101821768B1 (ko) | 2009-11-05 | 2018-01-24 | 리젠 파마슈티컬스 소시에떼 아노님 | 신규한 벤조피란 키나제 조절제 |
| DK2499139T3 (da) | 2009-11-10 | 2014-01-27 | Pfizer | N1-pyrazolospiroketon-acetyl-coa-carboxylasehæmmere |
| WO2011058113A1 (en) | 2009-11-12 | 2011-05-19 | Ucb Pharma S.A. | Fused bicyclic pyridine and pyrazine derivatives as kinase inhibitors |
| WO2011058111A1 (en) | 2009-11-12 | 2011-05-19 | Ucb Pharma S.A. | Aminopurine derivatives as kinase inhibitors |
| JP2013513551A (ja) | 2009-12-10 | 2013-04-22 | 中国医学科学院葯物研究所 | N6−置換アデノシン誘導体とn6−置換アデニン誘導体の鎮静、催眠,抗うつ,抗痙攣,抗てんかん,抗パーキンソン病と認知証予防・治療の用途 |
| AR079529A1 (es) | 2009-12-18 | 2012-02-01 | Incyte Corp | Derivados arilo y heteroarilo sustituidos y fundidos como inhibidores de la pi3k |
| JP2013514989A (ja) | 2009-12-18 | 2013-05-02 | アムジエン・インコーポレーテツド | 複素環式化合物およびその使用法 |
| WO2011075643A1 (en) | 2009-12-18 | 2011-06-23 | Incyte Corporation | Substituted heteroaryl fused derivatives as pi3k inhibitors |
| JP2011136925A (ja) | 2009-12-28 | 2011-07-14 | Dainippon Sumitomo Pharma Co Ltd | 含窒素二環性化合物 |
| KR101814835B1 (ko) | 2010-01-07 | 2018-01-04 | 다우 아그로사이언시즈 엘엘씨 | 티아졸로[5,4-d]피리미딘 및 그의 농약으로서의 용도 |
| UY33199A (es) | 2010-01-26 | 2011-08-31 | Boehringer Ingelheim Int | 5-alquinil-pirimidinas. |
| DK2545045T3 (en) | 2010-03-10 | 2016-01-25 | Incyte Holdings Corp | PIPERIDINE-4-YL-azetidine derivatives AS JAK1 INHIBITORS |
| KR20130028081A (ko) | 2010-03-22 | 2013-03-18 | 그렌마크 파머수티칼스 에스. 아. | 피리미딘온 유도체를 포함하는 약제학적 조성물 |
| UY33304A (es) | 2010-04-02 | 2011-10-31 | Amgen Inc | Compuestos heterocíclicos y sus usos |
| EP2558463A1 (en) | 2010-04-14 | 2013-02-20 | Incyte Corporation | Fused derivatives as i3 inhibitors |
| AU2011255218B2 (en) | 2010-05-21 | 2015-03-12 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| JP2013528228A (ja) | 2010-06-11 | 2013-07-08 | ギリアード カリストガ エルエルシー | キナゾリノン化合物による選択した患者における血液系障害を処置する方法 |
| US9062055B2 (en) | 2010-06-21 | 2015-06-23 | Incyte Corporation | Fused pyrrole derivatives as PI3K inhibitors |
| US8759371B2 (en) | 2010-07-01 | 2014-06-24 | Amgen Inc. | Heterocyclic compounds and their uses |
| CA2803009A1 (en) | 2010-07-01 | 2012-01-05 | Amgen Inc. | Heterocyclic compounds and their use as inhibitors of pi3k activity |
| EP2588469A1 (en) | 2010-07-02 | 2013-05-08 | Amgen Inc. | Heterocyclic compounds and their use as inhibitors of pi3k activity |
| WO2012040634A1 (en) | 2010-09-24 | 2012-03-29 | Gilead Calistoga Llc | Atropisomers of pi3k-inhibiting compounds |
| EP2635565A1 (en) | 2010-11-04 | 2013-09-11 | Amgen Inc. | 5 -cyano-4, 6 -diaminopyrimidine or 6 -aminopurine derivatives as pi3k- delta inhibitors |
| EP2637669A4 (en) | 2010-11-10 | 2014-04-02 | Infinity Pharmaceuticals Inc | Heterocyclic compounds and their use |
| CA2816144A1 (en) | 2010-11-17 | 2012-05-24 | Amgen Inc. | Quinoline derivatives as pik3 inhibitors |
| US9034884B2 (en) | 2010-11-19 | 2015-05-19 | Incyte Corporation | Heterocyclic-substituted pyrrolopyridines and pyrrolopyrimidines as JAK inhibitors |
| US8933085B2 (en) | 2010-11-19 | 2015-01-13 | Incyte Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| EP2651404B1 (en) | 2010-12-14 | 2015-10-14 | Electrophoretics Limited | Casein kinase 1delta (ck1delta) inhibitors |
| WO2012087881A1 (en) | 2010-12-20 | 2012-06-28 | Incyte Corporation | N-(1-(substituted-phenyl)ethyl)-9h-purin-6-amines as pi3k inhibitors |
| EP2655342A1 (en) | 2010-12-23 | 2013-10-30 | Amgen Inc. | Heterocyclic compounds and their uses |
| CA2824197C (en) | 2011-01-10 | 2020-02-25 | Michael Martin | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| CA2825599C (en) | 2011-02-01 | 2021-07-13 | The Board Of Trustees Of The University Of Illinois | 4-methyl-n-hydroxybenzamide compounds as histone deacetylase (hdac) inhibitors |
| US9108984B2 (en) | 2011-03-14 | 2015-08-18 | Incyte Corporation | Substituted diamino-pyrimidine and diamino-pyridine derivatives as PI3K inhibitors |
| US9126948B2 (en) | 2011-03-25 | 2015-09-08 | Incyte Holdings Corporation | Pyrimidine-4,6-diamine derivatives as PI3K inhibitors |
| MX344479B (es) | 2011-06-20 | 2016-12-16 | Incyte Holdings Corp | Derivados de azetidinil fenil, piridil o pirazinil carboxamida como inhibidores de cinasa janus (jak). |
| AU2012284088B2 (en) | 2011-07-19 | 2015-10-08 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| TW201313721A (zh) | 2011-08-18 | 2013-04-01 | Incyte Corp | 作為jak抑制劑之環己基氮雜環丁烷衍生物 |
| ES2722524T3 (es) | 2011-09-02 | 2019-08-13 | Incyte Holdings Corp | Heterociclaminas como inhibidores de pi3k |
| CN104024257A (zh) | 2011-10-04 | 2014-09-03 | 吉利德卡利斯托加有限责任公司 | Pi3k的新的喹喔啉抑制剂 |
| AR090548A1 (es) | 2012-04-02 | 2014-11-19 | Incyte Corp | Azaheterociclobencilaminas biciclicas como inhibidores de pi3k |
| AR091079A1 (es) | 2012-05-18 | 2014-12-30 | Incyte Corp | Derivados de pirrolopirimidina y pirrolopiridina sustituida con piperidinilciclobutilo como inhibidores de jak |
| US9181271B2 (en) | 2012-11-01 | 2015-11-10 | Incyte Holdings Corporation | Tricyclic fused thiophene derivatives as JAK inhibitors |
| TWI657090B (zh) * | 2013-03-01 | 2019-04-21 | 英塞特控股公司 | 吡唑并嘧啶衍生物治療PI3Kδ 相關病症之用途 |
| EA201591685A1 (ru) | 2013-03-15 | 2016-01-29 | Янссен Фармацевтика Нв | Способы и промежуточные соединения для получения лекарственного препарата |
| PE20160126A1 (es) | 2013-05-17 | 2016-02-24 | Incyte Corp | Derivados del bipirazol como inhibidores jak |
| NZ763326A (en) | 2014-04-08 | 2023-04-28 | Incyte Holdings Corp | Treatment of b-cell malignancies by a combination jak and pi3k inhibitor |
| AU2015266191A1 (en) | 2014-05-27 | 2016-09-15 | Almirall, S.A. | Medical use |
| WO2015191677A1 (en) | 2014-06-11 | 2015-12-17 | Incyte Corporation | Bicyclic heteroarylaminoalkyl phenyl derivatives as pi3k inhibitors |
| TWI698436B (zh) | 2014-12-30 | 2020-07-11 | 美商佛瑪治療公司 | 作為泛素特異性蛋白酶7抑制劑之吡咯并及吡唑并嘧啶 |
| MD3831833T2 (ro) | 2015-02-27 | 2023-04-30 | Incyte Holdings Corp | Procedee pentru prepararea unui inhibitor PI3K |
| US9732097B2 (en) | 2015-05-11 | 2017-08-15 | Incyte Corporation | Process for the synthesis of a phosphoinositide 3-kinase inhibitor |
| US20160362424A1 (en) | 2015-05-11 | 2016-12-15 | Incyte Corporation | Salts of (s)-7-(1-(9h-purin-6-ylamino)ethyl)-6-(3-fluorophenyl)-3-methyl-5h-thiazolo[3,2-a]pyrimidin-5-one |
| US9988401B2 (en) | 2015-05-11 | 2018-06-05 | Incyte Corporation | Crystalline forms of a PI3K inhibitor |
| JP7455763B2 (ja) | 2018-06-01 | 2024-03-26 | インサイト・コーポレイション | Pi3k関連障害の処置のための投薬レジメン |
-
2016
- 2016-02-26 MD MDE20220030T patent/MD3831833T2/ro unknown
- 2016-02-26 SI SI201631644T patent/SI3831833T1/sl unknown
- 2016-02-26 MA MA41607A patent/MA41607B1/fr unknown
- 2016-02-26 SG SG10201907576SA patent/SG10201907576SA/en unknown
- 2016-02-26 DK DK16718921.6T patent/DK3262046T3/da active
- 2016-02-26 US US15/054,474 patent/US10336759B2/en active Active
- 2016-02-26 EP EP20201979.0A patent/EP3831833B1/en active Active
- 2016-02-26 HU HUE16718921A patent/HUE053025T2/hu unknown
- 2016-02-26 EP EP22202130.5A patent/EP4183789A1/en active Pending
- 2016-02-26 HU HUE20201979A patent/HUE060953T2/hu unknown
- 2016-02-26 CN CN202311539089.4A patent/CN117800973A/zh active Pending
- 2016-02-26 JP JP2017544953A patent/JP6816005B2/ja active Active
- 2016-02-26 KR KR1020247027829A patent/KR20240132104A/ko active Search and Examination
- 2016-02-26 RS RS20201563A patent/RS61233B1/sr unknown
- 2016-02-26 AU AU2016222556A patent/AU2016222556B2/en active Active
- 2016-02-26 MA MA55193A patent/MA55193B1/fr unknown
- 2016-02-26 MD MDE20180031T patent/MD3262046T2/ro unknown
- 2016-02-26 TW TW109141640A patent/TWI764392B/zh active
- 2016-02-26 CR CR20170389A patent/CR20170389A/es unknown
- 2016-02-26 CN CN202311505572.0A patent/CN117736209A/zh active Pending
- 2016-02-26 ES ES20201979T patent/ES2933694T3/es active Active
- 2016-02-26 PL PL16718921T patent/PL3262046T3/pl unknown
- 2016-02-26 SG SG10201912302VA patent/SG10201912302VA/en unknown
- 2016-02-26 CR CR20210055A patent/CR20210055A/es unknown
- 2016-02-26 KR KR1020177027583A patent/KR102698327B1/ko active IP Right Grant
- 2016-02-26 WO PCT/US2016/019741 patent/WO2016138363A1/en active Application Filing
- 2016-02-26 CN CN202311519531.7A patent/CN117777139A/zh active Pending
- 2016-02-26 SG SG11201706917WA patent/SG11201706917WA/en unknown
- 2016-02-26 PT PT167189216T patent/PT3262046T/pt unknown
- 2016-02-26 DK DK20201979.0T patent/DK3831833T3/da active
- 2016-02-26 ES ES16718921T patent/ES2843522T3/es active Active
- 2016-02-26 UA UAA201709412A patent/UA122332C2/uk unknown
- 2016-02-26 LT LTEP16718921.6T patent/LT3262046T/lt unknown
- 2016-02-26 TW TW105106000A patent/TWI748941B/zh active
- 2016-02-26 CN CN201680011760.XA patent/CN107580597A/zh not_active Withdrawn
- 2016-02-26 MX MX2017010918A patent/MX2017010918A/es active IP Right Grant
- 2016-02-26 PE PE2017001461A patent/PE20180129A1/es unknown
- 2016-02-26 LT LTEP20201979.0T patent/LT3831833T/lt unknown
- 2016-02-26 EP EP16718921.6A patent/EP3262046B1/en active Active
- 2016-02-26 CA CA2977659A patent/CA2977659A1/en active Pending
- 2016-02-26 PT PT202019790T patent/PT3831833T/pt unknown
- 2016-02-26 PL PL20201979.0T patent/PL3831833T3/pl unknown
- 2016-02-26 PE PE2022000834A patent/PE20230156A1/es unknown
- 2016-02-26 AR ARP160100515A patent/AR103804A1/es unknown
- 2016-02-26 MY MYPI2017001264A patent/MY187502A/en unknown
- 2016-02-26 HR HRP20221444TT patent/HRP20221444T1/hr unknown
- 2016-02-26 BR BR122021004509-7A patent/BR122021004509B1/pt active IP Right Grant
- 2016-02-26 EA EA201791923A patent/EA201791923A1/ru unknown
- 2016-02-26 RS RS20221159A patent/RS63963B1/sr unknown
- 2016-02-26 SI SI201630994T patent/SI3262046T1/sl unknown
-
2017
- 2017-08-22 IL IL254093A patent/IL254093B/en unknown
- 2017-08-24 MX MX2019012927A patent/MX2019012927A/es unknown
- 2017-08-25 PH PH12017501538A patent/PH12017501538A1/en unknown
- 2017-08-25 CL CL2017002179A patent/CL2017002179A1/es unknown
- 2017-08-30 CO CONC2017/0008924A patent/CO2017008924A2/es unknown
- 2017-09-26 EC ECIEPI201764131A patent/ECSP17064131A/es unknown
-
2018
- 2018-06-14 HK HK18107731.9A patent/HK1248229A1/zh unknown
-
2019
- 2019-06-21 US US16/448,815 patent/US11084822B2/en active Active
- 2019-11-07 CL CL2019003188A patent/CL2019003188A1/es unknown
-
2020
- 2020-05-07 PH PH12020550575A patent/PH12020550575A1/en unknown
- 2020-08-11 AU AU2020217339A patent/AU2020217339B2/en active Active
- 2020-09-09 JP JP2020151255A patent/JP7080941B2/ja active Active
-
2021
- 2021-01-08 HR HRP20210036TT patent/HRP20210036T1/hr unknown
- 2021-01-22 CY CY20211100051T patent/CY1123729T1/el unknown
- 2021-03-02 EC ECSENADI202114448A patent/ECSP21014448A/es unknown
- 2021-07-01 US US17/365,888 patent/US12024522B2/en active Active
- 2021-12-10 AU AU2021282550A patent/AU2021282550A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2933694T3 (es) | Procesos para la preparación de un inhibidor de PI3K | |
| US11999751B2 (en) | Bicyclic heteroarylaminoalkyl phenyl derivatives as PI3K inhibitors | |
| ES2873001T3 (es) | Heterociclaminas como inhibidores de PI3K | |
| US9732097B2 (en) | Process for the synthesis of a phosphoinositide 3-kinase inhibitor | |
| WO2012135009A1 (en) | Pyrimidine-4,6-diamine derivatives as pi3k inhibitors | |
| BR112017018312B1 (pt) | Sais de inibidor de pi3k, composições que as compreende, seu uso, processos para seu preparo e método de inibição de uma atividade de uma quinase pi3k |