WO2016204302A1 - ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 - Google Patents
ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 Download PDFInfo
- Publication number
- WO2016204302A1 WO2016204302A1 PCT/JP2016/068311 JP2016068311W WO2016204302A1 WO 2016204302 A1 WO2016204302 A1 WO 2016204302A1 JP 2016068311 W JP2016068311 W JP 2016068311W WO 2016204302 A1 WO2016204302 A1 WO 2016204302A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- water
- weight
- gel
- salt
- particles
- Prior art date
Links
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 title claims abstract description 352
- 239000002250 absorbent Substances 0.000 title claims abstract description 272
- 230000002745 absorbent Effects 0.000 title claims abstract description 158
- 150000003839 salts Chemical class 0.000 title claims description 128
- 238000004519 manufacturing process Methods 0.000 title claims description 50
- 229920002845 Poly(methacrylic acid) Polymers 0.000 title claims description 27
- 239000002245 particle Substances 0.000 claims abstract description 326
- 229920006037 cross link polymer Polymers 0.000 claims abstract description 234
- 238000006116 polymerization reaction Methods 0.000 claims abstract description 160
- 238000010521 absorption reaction Methods 0.000 claims abstract description 113
- 239000007788 liquid Substances 0.000 claims abstract description 109
- 238000004132 cross linking Methods 0.000 claims abstract description 73
- 238000001035 drying Methods 0.000 claims abstract description 73
- 230000035699 permeability Effects 0.000 claims abstract description 33
- 239000011347 resin Substances 0.000 claims description 316
- 229920005989 resin Polymers 0.000 claims description 316
- 239000000499 gel Substances 0.000 claims description 280
- 239000000017 hydrogel Substances 0.000 claims description 254
- 239000006096 absorbing agent Substances 0.000 claims description 237
- 239000000843 powder Substances 0.000 claims description 158
- 239000000178 monomer Substances 0.000 claims description 141
- -1 polysiloxane Polymers 0.000 claims description 133
- 239000007864 aqueous solution Substances 0.000 claims description 119
- 238000010298 pulverizing process Methods 0.000 claims description 117
- 150000002430 hydrocarbons Chemical class 0.000 claims description 109
- 238000000227 grinding Methods 0.000 claims description 108
- 239000003795 chemical substances by application Substances 0.000 claims description 85
- 239000000126 substance Substances 0.000 claims description 75
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 claims description 67
- 150000001875 compounds Chemical class 0.000 claims description 60
- 239000007863 gel particle Substances 0.000 claims description 48
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 44
- 239000007787 solid Substances 0.000 claims description 44
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 40
- 125000002947 alkylene group Chemical group 0.000 claims description 39
- 150000005846 sugar alcohols Polymers 0.000 claims description 39
- 229920005862 polyol Polymers 0.000 claims description 35
- 150000003077 polyols Chemical class 0.000 claims description 35
- 239000002994 raw material Substances 0.000 claims description 27
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 22
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 21
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 claims description 21
- 238000006386 neutralization reaction Methods 0.000 claims description 21
- 229920002125 Sokalan® Polymers 0.000 claims description 19
- 239000004584 polyacrylic acid Substances 0.000 claims description 19
- 239000010419 fine particle Substances 0.000 claims description 17
- 229920001296 polysiloxane Polymers 0.000 claims description 16
- 125000002091 cationic group Chemical group 0.000 claims description 15
- 235000014113 dietary fatty acids Nutrition 0.000 claims description 14
- 239000000194 fatty acid Substances 0.000 claims description 14
- 229930195729 fatty acid Natural products 0.000 claims description 14
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 claims description 14
- 239000000463 material Substances 0.000 claims description 14
- 239000004215 Carbon black (E152) Substances 0.000 claims description 13
- 150000003863 ammonium salts Chemical class 0.000 claims description 13
- 229930195733 hydrocarbon Natural products 0.000 claims description 13
- 125000000129 anionic group Chemical group 0.000 claims description 12
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 claims description 12
- 125000001424 substituent group Chemical group 0.000 claims description 12
- 150000003973 alkyl amines Chemical class 0.000 claims description 11
- 235000011187 glycerol Nutrition 0.000 claims description 11
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Natural products C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 claims description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 9
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 claims description 8
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical compound OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 claims description 7
- 230000000379 polymerizing effect Effects 0.000 claims description 7
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 claims description 6
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 claims description 6
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 claims description 5
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 claims description 5
- 239000000600 sorbitol Substances 0.000 claims description 5
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 claims description 3
- 150000003141 primary amines Chemical class 0.000 claims description 3
- 150000001720 carbohydrates Chemical class 0.000 claims description 2
- 230000002829 reductive effect Effects 0.000 abstract description 43
- 238000000034 method Methods 0.000 description 220
- 230000008569 process Effects 0.000 description 147
- 239000000047 product Substances 0.000 description 98
- 238000007792 addition Methods 0.000 description 94
- 229920001223 polyethylene glycol Polymers 0.000 description 74
- 239000002202 Polyethylene glycol Substances 0.000 description 70
- 239000003431 cross linking reagent Substances 0.000 description 70
- 238000010438 heat treatment Methods 0.000 description 60
- 239000000243 solution Substances 0.000 description 52
- 230000000704 physical effect Effects 0.000 description 44
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 39
- 229920000642 polymer Polymers 0.000 description 38
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 36
- 239000004480 active ingredient Substances 0.000 description 34
- 238000002360 preparation method Methods 0.000 description 33
- 238000011282 treatment Methods 0.000 description 32
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 31
- 235000019441 ethanol Nutrition 0.000 description 27
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 24
- 230000000052 comparative effect Effects 0.000 description 24
- 125000004386 diacrylate group Chemical group 0.000 description 24
- 230000000694 effects Effects 0.000 description 24
- 235000013372 meat Nutrition 0.000 description 24
- 238000003756 stirring Methods 0.000 description 23
- 150000001768 cations Chemical class 0.000 description 22
- 238000006243 chemical reaction Methods 0.000 description 21
- 230000007423 decrease Effects 0.000 description 21
- 229910052751 metal Inorganic materials 0.000 description 21
- 239000002184 metal Substances 0.000 description 21
- 125000003277 amino group Chemical group 0.000 description 20
- 125000004432 carbon atom Chemical group C* 0.000 description 20
- 125000000524 functional group Chemical group 0.000 description 20
- 238000002156 mixing Methods 0.000 description 20
- 239000000654 additive Substances 0.000 description 19
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 18
- 238000009826 distribution Methods 0.000 description 17
- 238000005259 measurement Methods 0.000 description 17
- 239000003505 polymerization initiator Substances 0.000 description 17
- 239000007789 gas Substances 0.000 description 16
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 16
- 125000005037 alkyl phenyl group Chemical group 0.000 description 15
- 125000004122 cyclic group Chemical group 0.000 description 15
- 239000011361 granulated particle Substances 0.000 description 15
- 229930195734 saturated hydrocarbon Natural products 0.000 description 15
- 239000011780 sodium chloride Substances 0.000 description 15
- 125000006177 alkyl benzyl group Chemical group 0.000 description 14
- 239000011521 glass Substances 0.000 description 14
- 125000001624 naphthyl group Chemical group 0.000 description 14
- 125000005575 polycyclic aromatic hydrocarbon group Chemical group 0.000 description 14
- 238000010528 free radical solution polymerization reaction Methods 0.000 description 13
- 239000011164 primary particle Substances 0.000 description 13
- 229910052799 carbon Inorganic materials 0.000 description 12
- 229940048053 acrylate Drugs 0.000 description 11
- 238000011276 addition treatment Methods 0.000 description 11
- 125000003118 aryl group Chemical group 0.000 description 11
- 229920006317 cationic polymer Polymers 0.000 description 11
- 239000003960 organic solvent Substances 0.000 description 11
- 239000002504 physiological saline solution Substances 0.000 description 11
- 239000004094 surface-active agent Substances 0.000 description 11
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 10
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 10
- 230000000996 additive effect Effects 0.000 description 10
- 230000008859 change Effects 0.000 description 10
- 239000003245 coal Substances 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 9
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 9
- 239000002585 base Substances 0.000 description 9
- 238000005520 cutting process Methods 0.000 description 9
- 230000007246 mechanism Effects 0.000 description 9
- DVEKCXOJTLDBFE-UHFFFAOYSA-N n-dodecyl-n,n-dimethylglycinate Chemical compound CCCCCCCCCCCC[N+](C)(C)CC([O-])=O DVEKCXOJTLDBFE-UHFFFAOYSA-N 0.000 description 9
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 8
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 description 8
- 239000006185 dispersion Substances 0.000 description 8
- 238000005516 engineering process Methods 0.000 description 8
- 230000002209 hydrophobic effect Effects 0.000 description 8
- 239000000523 sample Substances 0.000 description 8
- 210000002700 urine Anatomy 0.000 description 8
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- 238000004364 calculation method Methods 0.000 description 7
- 238000011156 evaluation Methods 0.000 description 7
- 230000006872 improvement Effects 0.000 description 7
- 239000011148 porous material Substances 0.000 description 7
- 238000012545 processing Methods 0.000 description 7
- 230000002441 reversible effect Effects 0.000 description 7
- 230000008961 swelling Effects 0.000 description 7
- 238000012546 transfer Methods 0.000 description 7
- UWFRVQVNYNPBEF-UHFFFAOYSA-N 1-(2,4-dimethylphenyl)propan-1-one Chemical compound CCC(=O)C1=CC=C(C)C=C1C UWFRVQVNYNPBEF-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 238000007906 compression Methods 0.000 description 6
- 230000006835 compression Effects 0.000 description 6
- 230000006866 deterioration Effects 0.000 description 6
- 239000000835 fiber Substances 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 229960003330 pentetic acid Drugs 0.000 description 6
- 229920001983 poloxamer Polymers 0.000 description 6
- 238000001179 sorption measurement Methods 0.000 description 6
- 239000007921 spray Substances 0.000 description 6
- LCPVQAHEFVXVKT-UHFFFAOYSA-N 2-(2,4-difluorophenoxy)pyridin-3-amine Chemical compound NC1=CC=CN=C1OC1=CC=C(F)C=C1F LCPVQAHEFVXVKT-UHFFFAOYSA-N 0.000 description 5
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical group NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 5
- 239000004593 Epoxy Chemical class 0.000 description 5
- 206010040880 Skin irritation Diseases 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 229910052783 alkali metal Inorganic materials 0.000 description 5
- 229910052782 aluminium Inorganic materials 0.000 description 5
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 5
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 5
- 239000002738 chelating agent Substances 0.000 description 5
- 238000009833 condensation Methods 0.000 description 5
- 230000005494 condensation Effects 0.000 description 5
- 239000008367 deionised water Substances 0.000 description 5
- 229910021641 deionized water Inorganic materials 0.000 description 5
- 238000001125 extrusion Methods 0.000 description 5
- 238000005469 granulation Methods 0.000 description 5
- 230000003179 granulation Effects 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 229910052739 hydrogen Inorganic materials 0.000 description 5
- 238000000691 measurement method Methods 0.000 description 5
- 150000007524 organic acids Chemical class 0.000 description 5
- 229920001451 polypropylene glycol Polymers 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 230000036556 skin irritation Effects 0.000 description 5
- 231100000475 skin irritation Toxicity 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- CHQMHPLRPQMAMX-UHFFFAOYSA-L sodium persulfate Substances [Na+].[Na+].[O-]S(=O)(=O)OOS([O-])(=O)=O CHQMHPLRPQMAMX-UHFFFAOYSA-L 0.000 description 5
- 239000003643 water by type Substances 0.000 description 5
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical class O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 4
- AOBIOSPNXBMOAT-UHFFFAOYSA-N 2-[2-(oxiran-2-ylmethoxy)ethoxymethyl]oxirane Chemical compound C1OC1COCCOCC1CO1 AOBIOSPNXBMOAT-UHFFFAOYSA-N 0.000 description 4
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- 101100439211 Caenorhabditis elegans cex-2 gene Proteins 0.000 description 4
- 241000047703 Nonion Species 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 150000001767 cationic compounds Chemical class 0.000 description 4
- 230000018044 dehydration Effects 0.000 description 4
- 238000006297 dehydration reaction Methods 0.000 description 4
- 229910001873 dinitrogen Inorganic materials 0.000 description 4
- DDXLVDQZPFLQMZ-UHFFFAOYSA-M dodecyl(trimethyl)azanium;chloride Chemical compound [Cl-].CCCCCCCCCCCC[N+](C)(C)C DDXLVDQZPFLQMZ-UHFFFAOYSA-M 0.000 description 4
- 239000005297 pyrex Substances 0.000 description 4
- 238000010008 shearing Methods 0.000 description 4
- 125000006850 spacer group Chemical group 0.000 description 4
- 238000010557 suspension polymerization reaction Methods 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 238000005979 thermal decomposition reaction Methods 0.000 description 4
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 description 3
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 3
- PQUXFUBNSYCQAL-UHFFFAOYSA-N 1-(2,3-difluorophenyl)ethanone Chemical compound CC(=O)C1=CC=CC(F)=C1F PQUXFUBNSYCQAL-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 239000002211 L-ascorbic acid Substances 0.000 description 3
- 235000000069 L-ascorbic acid Nutrition 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- VBIIFPGSPJYLRR-UHFFFAOYSA-M Stearyltrimethylammonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCC[N+](C)(C)C VBIIFPGSPJYLRR-UHFFFAOYSA-M 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- DIZPMCHEQGEION-UHFFFAOYSA-H aluminium sulfate (anhydrous) Chemical compound [Al+3].[Al+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O DIZPMCHEQGEION-UHFFFAOYSA-H 0.000 description 3
- 150000001450 anions Chemical class 0.000 description 3
- 229960005070 ascorbic acid Drugs 0.000 description 3
- 229920001400 block copolymer Polymers 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000002781 deodorant agent Substances 0.000 description 3
- GDVKFRBCXAPAQJ-UHFFFAOYSA-A dialuminum;hexamagnesium;carbonate;hexadecahydroxide Chemical compound [OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Al+3].[Al+3].[O-]C([O-])=O GDVKFRBCXAPAQJ-UHFFFAOYSA-A 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 238000005187 foaming Methods 0.000 description 3
- 150000002431 hydrogen Chemical class 0.000 description 3
- 229910001701 hydrotalcite Inorganic materials 0.000 description 3
- 229960001545 hydrotalcite Drugs 0.000 description 3
- 230000005499 meniscus Effects 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 239000004745 nonwoven fabric Substances 0.000 description 3
- 150000002918 oxazolines Chemical class 0.000 description 3
- 150000002921 oxetanes Chemical class 0.000 description 3
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 3
- JRKICGRDRMAZLK-UHFFFAOYSA-L persulfate group Chemical group S(=O)(=O)([O-])OOS(=O)(=O)[O-] JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 3
- 229920000166 polytrimethylene carbonate Polymers 0.000 description 3
- 230000003405 preventing effect Effects 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- XWIHRGFIPXWGEF-UHFFFAOYSA-N propafenone hydrochloride Chemical compound Cl.CCCNCC(O)COC1=CC=CC=C1C(=O)CCC1=CC=CC=C1 XWIHRGFIPXWGEF-UHFFFAOYSA-N 0.000 description 3
- 125000001453 quaternary ammonium group Chemical group 0.000 description 3
- 230000009257 reactivity Effects 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000007873 sieving Methods 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 229940047670 sodium acrylate Drugs 0.000 description 3
- 159000000000 sodium salts Chemical class 0.000 description 3
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 3
- 238000004381 surface treatment Methods 0.000 description 3
- SOBHUZYZLFQYFK-UHFFFAOYSA-K trisodium;hydroxy-[[phosphonatomethyl(phosphonomethyl)amino]methyl]phosphinate Chemical compound [Na+].[Na+].[Na+].OP(O)(=O)CN(CP(O)([O-])=O)CP([O-])([O-])=O SOBHUZYZLFQYFK-UHFFFAOYSA-K 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 239000004925 Acrylic resin Substances 0.000 description 2
- 229920000178 Acrylic resin Polymers 0.000 description 2
- 101100439208 Caenorhabditis elegans cex-1 gene Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- 241000579895 Chlorostilbon Species 0.000 description 2
- 229920000742 Cotton Polymers 0.000 description 2
- RUPBZQFQVRMKDG-UHFFFAOYSA-M Didecyldimethylammonium chloride Chemical compound [Cl-].CCCCCCCCCC[N+](C)(C)CCCCCCCCCC RUPBZQFQVRMKDG-UHFFFAOYSA-M 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 2
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 2
- 239000005977 Ethylene Substances 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 229920002873 Polyethylenimine Polymers 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 2
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- XYUQYRXTKKPYEF-UHFFFAOYSA-L [K+].[Zr+4].[O-]C([O-])=O Chemical compound [K+].[Zr+4].[O-]C([O-])=O XYUQYRXTKKPYEF-UHFFFAOYSA-L 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 125000005211 alkyl trimethyl ammonium group Chemical group 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 235000015278 beef Nutrition 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 150000007942 carboxylates Chemical class 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- WOWHHFRSBJGXCM-UHFFFAOYSA-M cetyltrimethylammonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCC[N+](C)(C)C WOWHHFRSBJGXCM-UHFFFAOYSA-M 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 239000004927 clay Substances 0.000 description 2
- 239000011362 coarse particle Substances 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000004040 coloring Methods 0.000 description 2
- 238000010924 continuous production Methods 0.000 description 2
- 150000001923 cyclic compounds Chemical group 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- GPLRAVKSCUXZTP-UHFFFAOYSA-N diglycerol Chemical compound OCC(O)COCC(O)CO GPLRAVKSCUXZTP-UHFFFAOYSA-N 0.000 description 2
- SYELZBGXAIXKHU-UHFFFAOYSA-N dodecyldimethylamine N-oxide Chemical compound CCCCCCCCCCCC[N+](C)(C)[O-] SYELZBGXAIXKHU-UHFFFAOYSA-N 0.000 description 2
- 239000000428 dust Substances 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 229910052876 emerald Inorganic materials 0.000 description 2
- 239000010976 emerald Substances 0.000 description 2
- JBKVHLHDHHXQEQ-UHFFFAOYSA-N epsilon-caprolactam Chemical compound O=C1CCCCCN1 JBKVHLHDHHXQEQ-UHFFFAOYSA-N 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 230000003631 expected effect Effects 0.000 description 2
- 239000003337 fertilizer Substances 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 239000004088 foaming agent Substances 0.000 description 2
- 239000003205 fragrance Substances 0.000 description 2
- 229910021485 fumed silica Inorganic materials 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 238000001879 gelation Methods 0.000 description 2
- 239000003349 gelling agent Substances 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 238000011221 initial treatment Methods 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 238000004898 kneading Methods 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 235000010746 mayonnaise Nutrition 0.000 description 2
- 239000008268 mayonnaise Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 230000003472 neutralizing effect Effects 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 239000003973 paint Substances 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 239000000049 pigment Substances 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229920001515 polyalkylene glycol Polymers 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 2
- 150000003242 quaternary ammonium salts Chemical group 0.000 description 2
- 229920005604 random copolymer Polymers 0.000 description 2
- 238000007717 redox polymerization reaction Methods 0.000 description 2
- 238000010334 sieve classification Methods 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 239000010703 silicon Substances 0.000 description 2
- 235000012239 silicon dioxide Nutrition 0.000 description 2
- 238000005507 spraying Methods 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000003760 tallow Substances 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- 239000010936 titanium Substances 0.000 description 2
- 229910052719 titanium Inorganic materials 0.000 description 2
- VXYADVIJALMOEQ-UHFFFAOYSA-K tris(lactato)aluminium Chemical compound CC(O)C(=O)O[Al](OC(=O)C(C)O)OC(=O)C(C)O VXYADVIJALMOEQ-UHFFFAOYSA-K 0.000 description 2
- 229920003176 water-insoluble polymer Polymers 0.000 description 2
- OERNJTNJEZOPIA-UHFFFAOYSA-N zirconium nitrate Chemical compound [Zr+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O OERNJTNJEZOPIA-UHFFFAOYSA-N 0.000 description 2
- WYTZZXDRDKSJID-UHFFFAOYSA-N (3-aminopropyl)triethoxysilane Chemical compound CCO[Si](OCC)(OCC)CCCN WYTZZXDRDKSJID-UHFFFAOYSA-N 0.000 description 1
- BJYGGFGTOTUNJA-UHFFFAOYSA-N (3-butyloxetan-3-yl)methanol Chemical compound CCCCC1(CO)COC1 BJYGGFGTOTUNJA-UHFFFAOYSA-N 0.000 description 1
- UNMJLQGKEDTEKJ-UHFFFAOYSA-N (3-ethyloxetan-3-yl)methanol Chemical compound CCC1(CO)COC1 UNMJLQGKEDTEKJ-UHFFFAOYSA-N 0.000 description 1
- NLQMSBJFLQPLIJ-UHFFFAOYSA-N (3-methyloxetan-3-yl)methanol Chemical compound OCC1(C)COC1 NLQMSBJFLQPLIJ-UHFFFAOYSA-N 0.000 description 1
- FFJCNSLCJOQHKM-CLFAGFIQSA-N (z)-1-[(z)-octadec-9-enoxy]octadec-9-ene Chemical compound CCCCCCCC\C=C/CCCCCCCCOCCCCCCCC\C=C/CCCCCCCC FFJCNSLCJOQHKM-CLFAGFIQSA-N 0.000 description 1
- ORTVZLZNOYNASJ-UPHRSURJSA-N (z)-but-2-ene-1,4-diol Chemical compound OC\C=C/CO ORTVZLZNOYNASJ-UPHRSURJSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- ZZXUZKXVROWEIF-UHFFFAOYSA-N 1,2-butylene carbonate Chemical compound CCC1COC(=O)O1 ZZXUZKXVROWEIF-UHFFFAOYSA-N 0.000 description 1
- KOMNUTZXSVSERR-UHFFFAOYSA-N 1,3,5-tris(prop-2-enyl)-1,3,5-triazinane-2,4,6-trione Chemical compound C=CCN1C(=O)N(CC=C)C(=O)N(CC=C)C1=O KOMNUTZXSVSERR-UHFFFAOYSA-N 0.000 description 1
- KATAXDCYPGGJNJ-UHFFFAOYSA-N 1,3-bis(oxiran-2-ylmethoxy)propan-2-ol Chemical compound C1OC1COCC(O)COCC1CO1 KATAXDCYPGGJNJ-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- VEGNIXCUDMQGFZ-UHFFFAOYSA-N 1-[3-[3-[2,3-bis(oxiran-2-ylmethoxy)propoxy]-2-hydroxypropoxy]-2-(oxiran-2-ylmethoxy)propoxy]-3-(oxiran-2-ylmethoxy)propan-2-ol Chemical compound C1OC1COCC(OCC1OC1)COCC(O)COCC(OCC1OC1)COCC(O)COCC1CO1 VEGNIXCUDMQGFZ-UHFFFAOYSA-N 0.000 description 1
- HBXWUCXDUUJDRB-UHFFFAOYSA-N 1-octadecoxyoctadecane Chemical compound CCCCCCCCCCCCCCCCCCOCCCCCCCCCCCCCCCCCC HBXWUCXDUUJDRB-UHFFFAOYSA-N 0.000 description 1
- CBQFBEBEBCHTBK-UHFFFAOYSA-N 1-phenylprop-2-ene-1-sulfonic acid Chemical compound OS(=O)(=O)C(C=C)C1=CC=CC=C1 CBQFBEBEBCHTBK-UHFFFAOYSA-N 0.000 description 1
- RAXXELZNTBOGNW-UHFFFAOYSA-N 1H-imidazole Chemical group C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 1
- JCTXKRPTIMZBJT-UHFFFAOYSA-N 2,2,4-trimethylpentane-1,3-diol Chemical compound CC(C)C(O)C(C)(C)CO JCTXKRPTIMZBJT-UHFFFAOYSA-N 0.000 description 1
- IVIDDMGBRCPGLJ-UHFFFAOYSA-N 2,3-bis(oxiran-2-ylmethoxy)propan-1-ol Chemical compound C1OC1COC(CO)COCC1CO1 IVIDDMGBRCPGLJ-UHFFFAOYSA-N 0.000 description 1
- BJELTSYBAHKXRW-UHFFFAOYSA-N 2,4,6-triallyloxy-1,3,5-triazine Chemical compound C=CCOC1=NC(OCC=C)=NC(OCC=C)=N1 BJELTSYBAHKXRW-UHFFFAOYSA-N 0.000 description 1
- NQIGSEBFOJIXSE-UHFFFAOYSA-N 2-(3-ethyloxetan-3-yl)ethanol Chemical compound OCCC1(CC)COC1 NQIGSEBFOJIXSE-UHFFFAOYSA-N 0.000 description 1
- BDKLKNJTMLIAFE-UHFFFAOYSA-N 2-(3-fluorophenyl)-1,3-oxazole-4-carbaldehyde Chemical compound FC1=CC=CC(C=2OC=C(C=O)N=2)=C1 BDKLKNJTMLIAFE-UHFFFAOYSA-N 0.000 description 1
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 1
- NFMOAAFJCIYUQR-UHFFFAOYSA-N 2-(3-methyloxetan-3-yl)ethanol Chemical compound OCCC1(C)COC1 NFMOAAFJCIYUQR-UHFFFAOYSA-N 0.000 description 1
- HSDVRWZKEDRBAG-UHFFFAOYSA-N 2-[1-(oxiran-2-ylmethoxy)hexoxymethyl]oxirane Chemical compound C1OC1COC(CCCCC)OCC1CO1 HSDVRWZKEDRBAG-UHFFFAOYSA-N 0.000 description 1
- HDPLHDGYGLENEI-UHFFFAOYSA-N 2-[1-(oxiran-2-ylmethoxy)propan-2-yloxymethyl]oxirane Chemical compound C1OC1COC(C)COCC1CO1 HDPLHDGYGLENEI-UHFFFAOYSA-N 0.000 description 1
- KFNAHVKJFHDCSK-UHFFFAOYSA-N 2-[2-(4,5-dihydro-1,3-oxazol-2-yl)ethyl]-4,5-dihydro-1,3-oxazole Chemical compound N=1CCOC=1CCC1=NCCO1 KFNAHVKJFHDCSK-UHFFFAOYSA-N 0.000 description 1
- SEFYJVFBMNOLBK-UHFFFAOYSA-N 2-[2-[2-(oxiran-2-ylmethoxy)ethoxy]ethoxymethyl]oxirane Chemical compound C1OC1COCCOCCOCC1CO1 SEFYJVFBMNOLBK-UHFFFAOYSA-N 0.000 description 1
- VSRMIIBCXRHPCC-UHFFFAOYSA-N 2-[2-[2-[2-[2-(oxiran-2-ylmethoxy)ethoxy]ethoxy]ethoxy]ethoxymethyl]oxirane Chemical compound C1OC1COCCOCCOCCOCCOCC1CO1 VSRMIIBCXRHPCC-UHFFFAOYSA-N 0.000 description 1
- WFUGQJXVXHBTEM-UHFFFAOYSA-N 2-hydroperoxy-2-(2-hydroperoxybutan-2-ylperoxy)butane Chemical compound CCC(C)(OO)OOC(C)(CC)OO WFUGQJXVXHBTEM-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- AIFLGMNWQFPTAJ-UHFFFAOYSA-J 2-hydroxypropanoate;titanium(4+) Chemical compound [Ti+4].CC(O)C([O-])=O.CC(O)C([O-])=O.CC(O)C([O-])=O.CC(O)C([O-])=O AIFLGMNWQFPTAJ-UHFFFAOYSA-J 0.000 description 1
- QENRKQYUEGJNNZ-UHFFFAOYSA-N 2-methyl-1-(prop-2-enoylamino)propane-1-sulfonic acid Chemical compound CC(C)C(S(O)(=O)=O)NC(=O)C=C QENRKQYUEGJNNZ-UHFFFAOYSA-N 0.000 description 1
- NWIIFBPIDORBCY-UHFFFAOYSA-N 2-methylprop-2-enoic acid;propane-1,2,3-triol;prop-2-enoic acid Chemical compound OC(=O)C=C.CC(=C)C(O)=O.OCC(O)CO NWIIFBPIDORBCY-UHFFFAOYSA-N 0.000 description 1
- AGBXYHCHUYARJY-UHFFFAOYSA-N 2-phenylethenesulfonic acid Chemical compound OS(=O)(=O)C=CC1=CC=CC=C1 AGBXYHCHUYARJY-UHFFFAOYSA-N 0.000 description 1
- QGBLCIBATKETJC-UHFFFAOYSA-N 3,7-dioxido-2,4,6,8,9-pentaoxa-1,3,5,7-tetraborabicyclo[3.3.1]nonane;manganese(2+) Chemical compound [Mn+2].O1B([O-])OB2OB([O-])OB1O2 QGBLCIBATKETJC-UHFFFAOYSA-N 0.000 description 1
- GOXNUYXRIQJIEF-UHFFFAOYSA-N 3-(2-hydroxyethyl)-1,3-oxazolidin-2-one Chemical compound OCCN1CCOC1=O GOXNUYXRIQJIEF-UHFFFAOYSA-N 0.000 description 1
- UKLWXKWTXHHMFK-UHFFFAOYSA-N 3-(chloromethyl)-3-ethyloxetane Chemical compound CCC1(CCl)COC1 UKLWXKWTXHHMFK-UHFFFAOYSA-N 0.000 description 1
- MCZYEFODKAZWIH-UHFFFAOYSA-N 3-(chloromethyl)-3-methyloxetane Chemical compound ClCC1(C)COC1 MCZYEFODKAZWIH-UHFFFAOYSA-N 0.000 description 1
- WOAMRAPSJUZQJV-UHFFFAOYSA-N 3-oxopent-4-ene-2-sulfonic acid Chemical compound OS(=O)(=O)C(C)C(=O)C=C WOAMRAPSJUZQJV-UHFFFAOYSA-N 0.000 description 1
- PUEFXLJYTSRTGI-UHFFFAOYSA-N 4,4-dimethyl-1,3-dioxolan-2-one Chemical compound CC1(C)COC(=O)O1 PUEFXLJYTSRTGI-UHFFFAOYSA-N 0.000 description 1
- LWLOKSXSAUHTJO-UHFFFAOYSA-N 4,5-dimethyl-1,3-dioxolan-2-one Chemical compound CC1OC(=O)OC1C LWLOKSXSAUHTJO-UHFFFAOYSA-N 0.000 description 1
- UHIIHYFGCONAHB-UHFFFAOYSA-N 4,6-dimethyl-1,3-dioxan-2-one Chemical compound CC1CC(C)OC(=O)O1 UHIIHYFGCONAHB-UHFFFAOYSA-N 0.000 description 1
- JFMGYULNQJPJCY-UHFFFAOYSA-N 4-(hydroxymethyl)-1,3-dioxolan-2-one Chemical compound OCC1COC(=O)O1 JFMGYULNQJPJCY-UHFFFAOYSA-N 0.000 description 1
- OVDQEUFSGODEBT-UHFFFAOYSA-N 4-methyl-1,3-dioxan-2-one Chemical compound CC1CCOC(=O)O1 OVDQEUFSGODEBT-UHFFFAOYSA-N 0.000 description 1
- AEYSASDBPHWTGR-UHFFFAOYSA-N 4-oxohex-5-ene-3-sulfonic acid Chemical compound CCC(S(O)(=O)=O)C(=O)C=C AEYSASDBPHWTGR-UHFFFAOYSA-N 0.000 description 1
- ZMGMDXCADSRNCX-UHFFFAOYSA-N 5,6-dihydroxy-1,3-diazepan-2-one Chemical class OC1CNC(=O)NCC1O ZMGMDXCADSRNCX-UHFFFAOYSA-N 0.000 description 1
- DUFCMRCMPHIFTR-UHFFFAOYSA-N 5-(dimethylsulfamoyl)-2-methylfuran-3-carboxylic acid Chemical compound CN(C)S(=O)(=O)C1=CC(C(O)=O)=C(C)O1 DUFCMRCMPHIFTR-UHFFFAOYSA-N 0.000 description 1
- WIGIPJGWVLNDAF-UHFFFAOYSA-N 8-methyl-1-(8-methylnonoxy)nonane Chemical compound CC(C)CCCCCCCOCCCCCCCC(C)C WIGIPJGWVLNDAF-UHFFFAOYSA-N 0.000 description 1
- 229910002012 Aerosil® Inorganic materials 0.000 description 1
- 229910002016 Aerosil® 200 Inorganic materials 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- SGHZXLIDFTYFHQ-UHFFFAOYSA-L Brilliant Blue Chemical compound [Na+].[Na+].C=1C=C(C(=C2C=CC(C=C2)=[N+](CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=2C(=CC=CC=2)S([O-])(=O)=O)C=CC=1N(CC)CC1=CC=CC(S([O-])(=O)=O)=C1 SGHZXLIDFTYFHQ-UHFFFAOYSA-L 0.000 description 1
- WRAGBEWQGHCDDU-UHFFFAOYSA-M C([O-])([O-])=O.[NH4+].[Zr+] Chemical compound C([O-])([O-])=O.[NH4+].[Zr+] WRAGBEWQGHCDDU-UHFFFAOYSA-M 0.000 description 1
- 229920003043 Cellulose fiber Polymers 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- 235000013162 Cocos nucifera Nutrition 0.000 description 1
- 244000060011 Cocos nucifera Species 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000982822 Ficus obtusifolia Species 0.000 description 1
- 108700042658 GAP-43 Proteins 0.000 description 1
- 108010090155 GLM-R cytokine receptor Proteins 0.000 description 1
- CTKINSOISVBQLD-UHFFFAOYSA-N Glycidol Chemical compound OCC1CO1 CTKINSOISVBQLD-UHFFFAOYSA-N 0.000 description 1
- 101000618467 Hypocrea jecorina (strain ATCC 56765 / BCRC 32924 / NRRL 11460 / Rut C-30) Endo-1,4-beta-xylanase 2 Proteins 0.000 description 1
- 206010021639 Incontinence Diseases 0.000 description 1
- 102100021594 Interleukin-31 receptor subunit alpha Human genes 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- ALQSHHUCVQOPAS-UHFFFAOYSA-N Pentane-1,5-diol Chemical compound OCCCCCO ALQSHHUCVQOPAS-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 229920002025 Pluronic® F 88 Polymers 0.000 description 1
- 229920002057 Pluronic® P 103 Polymers 0.000 description 1
- 229920002070 Pluronic® P 84 Polymers 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 229920001131 Pulp (paper) Polymers 0.000 description 1
- 229920000297 Rayon Polymers 0.000 description 1
- 239000008156 Ringer's lactate solution Substances 0.000 description 1
- 239000006087 Silane Coupling Agent Substances 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 239000004147 Sorbitan trioleate Substances 0.000 description 1
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical compound ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 1
- 229920002978 Vinylon Polymers 0.000 description 1
- 229910021536 Zeolite Inorganic materials 0.000 description 1
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 1
- LXEKPEMOWBOYRF-UHFFFAOYSA-N [2-[(1-azaniumyl-1-imino-2-methylpropan-2-yl)diazenyl]-2-methylpropanimidoyl]azanium;dichloride Chemical compound Cl.Cl.NC(=N)C(C)(C)N=NC(C)(C)C(N)=N LXEKPEMOWBOYRF-UHFFFAOYSA-N 0.000 description 1
- JSIVLBGUDVQDHZ-UHFFFAOYSA-J [Cl-].[Cl-].[Cl-].O[Zr+3] Chemical compound [Cl-].[Cl-].[Cl-].O[Zr+3] JSIVLBGUDVQDHZ-UHFFFAOYSA-J 0.000 description 1
- 239000011358 absorbing material Substances 0.000 description 1
- 150000008062 acetophenones Chemical class 0.000 description 1
- HDYRYUINDGQKMC-UHFFFAOYSA-M acetyloxyaluminum;dihydrate Chemical compound O.O.CC(=O)O[Al] HDYRYUINDGQKMC-UHFFFAOYSA-M 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000002390 adhesive tape Substances 0.000 description 1
- 239000002156 adsorbate Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- ILRRQNADMUWWFW-UHFFFAOYSA-K aluminium phosphate Chemical compound O1[Al]2OP1(=O)O2 ILRRQNADMUWWFW-UHFFFAOYSA-K 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 229940009827 aluminum acetate Drugs 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- KRMDCWKBEZIMAB-UHFFFAOYSA-N amitriptyline Chemical compound C1CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 KRMDCWKBEZIMAB-UHFFFAOYSA-N 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- ROOXNKNUYICQNP-UHFFFAOYSA-N ammonium peroxydisulfate Substances [NH4+].[NH4+].[O-]S(=O)(=O)OOS([O-])(=O)=O ROOXNKNUYICQNP-UHFFFAOYSA-N 0.000 description 1
- VAZSKTXWXKYQJF-UHFFFAOYSA-N ammonium persulfate Chemical compound [NH4+].[NH4+].[O-]S(=O)OOS([O-])=O VAZSKTXWXKYQJF-UHFFFAOYSA-N 0.000 description 1
- 229910001870 ammonium persulfate Inorganic materials 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 description 1
- 239000007869 azo polymerization initiator Substances 0.000 description 1
- WAKZZMMCDILMEF-UHFFFAOYSA-H barium(2+);diphosphate Chemical compound [Ba+2].[Ba+2].[Ba+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O WAKZZMMCDILMEF-UHFFFAOYSA-H 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- YSJGOMATDFSEED-UHFFFAOYSA-M behentrimonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCCCCCC[N+](C)(C)C YSJGOMATDFSEED-UHFFFAOYSA-M 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- ISAOCJYIOMOJEB-UHFFFAOYSA-N benzoin Chemical class C=1C=CC=CC=1C(O)C(=O)C1=CC=CC=C1 ISAOCJYIOMOJEB-UHFFFAOYSA-N 0.000 description 1
- 150000008366 benzophenones Chemical class 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- OCBHHZMJRVXXQK-UHFFFAOYSA-M benzyl-dimethyl-tetradecylazanium;chloride Chemical compound [Cl-].CCCCCCCCCCCCCC[N+](C)(C)CC1=CC=CC=C1 OCBHHZMJRVXXQK-UHFFFAOYSA-M 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- LBSPZZSGTIBOFG-UHFFFAOYSA-N bis[2-(4,5-dihydro-1h-imidazol-2-yl)propan-2-yl]diazene;dihydrochloride Chemical compound Cl.Cl.N=1CCNC=1C(C)(C)N=NC(C)(C)C1=NCCN1 LBSPZZSGTIBOFG-UHFFFAOYSA-N 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 238000012662 bulk polymerization Methods 0.000 description 1
- CDQSJQSWAWPGKG-UHFFFAOYSA-N butane-1,1-diol Chemical compound CCCC(O)O CDQSJQSWAWPGKG-UHFFFAOYSA-N 0.000 description 1
- MKJXYGKVIBWPFZ-UHFFFAOYSA-L calcium lactate Chemical compound [Ca+2].CC(O)C([O-])=O.CC(O)C([O-])=O MKJXYGKVIBWPFZ-UHFFFAOYSA-L 0.000 description 1
- 239000001527 calcium lactate Substances 0.000 description 1
- 235000011086 calcium lactate Nutrition 0.000 description 1
- 229960002401 calcium lactate Drugs 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- RLGQACBPNDBWTB-UHFFFAOYSA-N cetyltrimethylammonium ion Chemical compound CCCCCCCCCCCCCCCC[N+](C)(C)C RLGQACBPNDBWTB-UHFFFAOYSA-N 0.000 description 1
- 239000012986 chain transfer agent Substances 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- MRUAUOIMASANKQ-UHFFFAOYSA-N cocamidopropyl betaine Chemical compound CCCCCCCCCCCC(=O)NCCC[N+](C)(C)CC([O-])=O MRUAUOIMASANKQ-UHFFFAOYSA-N 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000007334 copolymerization reaction Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 238000013480 data collection Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- LSXWFXONGKSEMY-UHFFFAOYSA-N di-tert-butyl peroxide Chemical compound CC(C)(C)OOC(C)(C)C LSXWFXONGKSEMY-UHFFFAOYSA-N 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 229960004670 didecyldimethylammonium chloride Drugs 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 229940105990 diglycerin Drugs 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- UAKOZKUVZRMOFN-JDVCJPALSA-M dimethyl-bis[(z)-octadec-9-enyl]azanium;chloride Chemical compound [Cl-].CCCCCCCC\C=C/CCCCCCCC[N+](C)(C)CCCCCCCC\C=C/CCCCCCCC UAKOZKUVZRMOFN-JDVCJPALSA-M 0.000 description 1
- REZZEXDLIUJMMS-UHFFFAOYSA-M dimethyldioctadecylammonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCC[N+](C)(C)CCCCCCCCCCCCCCCCCC REZZEXDLIUJMMS-UHFFFAOYSA-M 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 239000004664 distearyldimethylammonium chloride (DHTDMAC) Substances 0.000 description 1
- 238000007580 dry-mixing Methods 0.000 description 1
- 238000010894 electron beam technology Methods 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- GKIPXFAANLTWBM-UHFFFAOYSA-N epibromohydrin Chemical compound BrCC1CO1 GKIPXFAANLTWBM-UHFFFAOYSA-N 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000012632 extractable Substances 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000013373 food additive Nutrition 0.000 description 1
- 239000002778 food additive Substances 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 125000003827 glycol group Chemical group 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- ACCCMOQWYVYDOT-UHFFFAOYSA-N hexane-1,1-diol Chemical compound CCCCCC(O)O ACCCMOQWYVYDOT-UHFFFAOYSA-N 0.000 description 1
- XXMIOPMDWAUFGU-UHFFFAOYSA-N hexane-1,6-diol Chemical compound OCCCCCCO XXMIOPMDWAUFGU-UHFFFAOYSA-N 0.000 description 1
- 238000007602 hot air drying Methods 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- YAMHXTCMCPHKLN-UHFFFAOYSA-N imidazolidin-2-one Chemical compound O=C1NCCN1 YAMHXTCMCPHKLN-UHFFFAOYSA-N 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 238000007603 infrared drying Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- DDSZSJDMRGXEKQ-UHFFFAOYSA-N iron(3+);borate Chemical compound [Fe+3].[O-]B([O-])[O-] DDSZSJDMRGXEKQ-UHFFFAOYSA-N 0.000 description 1
- 229940035429 isobutyl alcohol Drugs 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 229910052743 krypton Inorganic materials 0.000 description 1
- DNNSSWSSYDEUBZ-UHFFFAOYSA-N krypton atom Chemical compound [Kr] DNNSSWSSYDEUBZ-UHFFFAOYSA-N 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 150000004668 long chain fatty acids Chemical class 0.000 description 1
- 230000001050 lubricating effect Effects 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 150000002736 metal compounds Chemical class 0.000 description 1
- 229910001463 metal phosphate Inorganic materials 0.000 description 1
- 125000005641 methacryl group Chemical group 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000003595 mist Substances 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- UPHWVVKYDQHTCF-UHFFFAOYSA-N octadecylazanium;acetate Chemical compound CC(O)=O.CCCCCCCCCCCCCCCCCCN UPHWVVKYDQHTCF-UHFFFAOYSA-N 0.000 description 1
- OEIJHBUUFURJLI-UHFFFAOYSA-N octane-1,8-diol Chemical compound OCCCCCCCCO OEIJHBUUFURJLI-UHFFFAOYSA-N 0.000 description 1
- 238000013021 overheating Methods 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical class [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- UWJJYHHHVWZFEP-UHFFFAOYSA-N pentane-1,1-diol Chemical compound CCCCC(O)O UWJJYHHHVWZFEP-UHFFFAOYSA-N 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- ACVYVLVWPXVTIT-UHFFFAOYSA-N phosphinic acid Chemical compound O[PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-N 0.000 description 1
- PWGIEBRSWMQVCO-UHFFFAOYSA-N phosphono prop-2-enoate Chemical compound OP(O)(=O)OC(=O)C=C PWGIEBRSWMQVCO-UHFFFAOYSA-N 0.000 description 1
- 238000000053 physical method Methods 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 238000005498 polishing Methods 0.000 description 1
- 229920001993 poloxamer 188 Polymers 0.000 description 1
- 229920000083 poly(allylamine) Polymers 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920000223 polyglycerol Polymers 0.000 description 1
- 229920000151 polyglycol Polymers 0.000 description 1
- 239000010695 polyglycol Substances 0.000 description 1
- 239000002952 polymeric resin Substances 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920000259 polyoxyethylene lauryl ether Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 239000000249 polyoxyethylene sorbitan monopalmitate Substances 0.000 description 1
- 235000010483 polyoxyethylene sorbitan monopalmitate Nutrition 0.000 description 1
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 description 1
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 description 1
- 239000001816 polyoxyethylene sorbitan tristearate Substances 0.000 description 1
- 235000010988 polyoxyethylene sorbitan tristearate Nutrition 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- USHAGKDGDHPEEY-UHFFFAOYSA-L potassium persulfate Chemical compound [K+].[K+].[O-]S(=O)(=O)OOS([O-])(=O)=O USHAGKDGDHPEEY-UHFFFAOYSA-L 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000012673 precipitation polymerization Methods 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- ULWHHBHJGPPBCO-UHFFFAOYSA-N propane-1,1-diol Chemical compound CCC(O)O ULWHHBHJGPPBCO-UHFFFAOYSA-N 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 239000007870 radical polymerization initiator Substances 0.000 description 1
- 239000002964 rayon Substances 0.000 description 1
- 238000007670 refining Methods 0.000 description 1
- 239000011342 resin composition Substances 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000001542 size-exclusion chromatography Methods 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229940087562 sodium acetate trihydrate Drugs 0.000 description 1
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical compound [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 235000019337 sorbitan trioleate Nutrition 0.000 description 1
- 229960000391 sorbitan trioleate Drugs 0.000 description 1
- 238000004544 sputter deposition Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 239000002344 surface layer Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000010558 suspension polymerization method Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 239000012085 test solution Substances 0.000 description 1
- VDWRUZRMNKZIAJ-UHFFFAOYSA-N tetradecylazanium;acetate Chemical compound CC(O)=O.CCCCCCCCCCCCCCN VDWRUZRMNKZIAJ-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- KRDJTDULHZPJPB-UHFFFAOYSA-N titanium(4+);tetraborate Chemical compound [Ti+4].[Ti+4].[Ti+4].[O-]B([O-])[O-].[O-]B([O-])[O-].[O-]B([O-])[O-].[O-]B([O-])[O-] KRDJTDULHZPJPB-UHFFFAOYSA-N 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- VPYJNCGUESNPMV-UHFFFAOYSA-N triallylamine Chemical compound C=CCN(CC=C)CC=C VPYJNCGUESNPMV-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- VLCLHFYFMCKBRP-UHFFFAOYSA-N tricalcium;diborate Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]B([O-])[O-].[O-]B([O-])[O-] VLCLHFYFMCKBRP-UHFFFAOYSA-N 0.000 description 1
- ZIBGPFATKBEMQZ-UHFFFAOYSA-N triethylene glycol Chemical compound OCCOCCOCCO ZIBGPFATKBEMQZ-UHFFFAOYSA-N 0.000 description 1
- NFMWFGXCDDYTEG-UHFFFAOYSA-N trimagnesium;diborate Chemical compound [Mg+2].[Mg+2].[Mg+2].[O-]B([O-])[O-].[O-]B([O-])[O-] NFMWFGXCDDYTEG-UHFFFAOYSA-N 0.000 description 1
- BPSIOYPQMFLKFR-UHFFFAOYSA-N trimethoxy-[3-(oxiran-2-ylmethoxy)propyl]silane Chemical compound CO[Si](OC)(OC)CCCOCC1CO1 BPSIOYPQMFLKFR-UHFFFAOYSA-N 0.000 description 1
- XHGIFBQQEGRTPB-UHFFFAOYSA-N tris(prop-2-enyl) phosphate Chemical compound C=CCOP(=O)(OCC=C)OCC=C XHGIFBQQEGRTPB-UHFFFAOYSA-N 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 238000009834 vaporization Methods 0.000 description 1
- 230000008016 vaporization Effects 0.000 description 1
- NLVXSWCKKBEXTG-UHFFFAOYSA-N vinylsulfonic acid Chemical compound OS(=O)(=O)C=C NLVXSWCKKBEXTG-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 238000011041 water permeability test Methods 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
- ZXAUZSQITFJWPS-UHFFFAOYSA-J zirconium(4+);disulfate Chemical compound [Zr+4].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O ZXAUZSQITFJWPS-UHFFFAOYSA-J 0.000 description 1
Images







Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/04—Acids; Metal salts or ammonium salts thereof
- C08F220/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/22—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons containing macromolecular materials
- A61L15/24—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/42—Use of materials characterised by their function or physical properties
- A61L15/60—Liquid-swellable gel-forming materials, e.g. super-absorbents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F20/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride, ester, amide, imide or nitrile thereof
- C08F20/02—Monocarboxylic acids having less than ten carbon atoms, Derivatives thereof
- C08F20/10—Esters
- C08F20/12—Esters of monohydric alcohols or phenols
- C08F20/16—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms
- C08F20/18—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms with acrylic or methacrylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F6/00—Post-polymerisation treatments
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/02—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques
- C08J3/03—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques in aqueous media
- C08J3/075—Macromolecular gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
- C08J3/245—Differential crosslinking of one polymer with one crosslinking type, e.g. surface crosslinking
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/01—Use of inorganic substances as compounding ingredients characterized by their specific function
- C08K3/013—Fillers, pigments or reinforcing additives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/06—Ethers; Acetals; Ketals; Ortho-esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/15—Heterocyclic compounds having oxygen in the ring
- C08K5/151—Heterocyclic compounds having oxygen in the ring having one oxygen atom in the ring
- C08K5/1535—Five-membered rings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/17—Amines; Quaternary ammonium compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/17—Amines; Quaternary ammonium compounds
- C08K5/175—Amines; Quaternary ammonium compounds containing COOH-groups; Esters or salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/17—Amines; Quaternary ammonium compounds
- C08K5/19—Quaternary ammonium compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/32—Compounds containing nitrogen bound to oxygen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/36—Sulfur-, selenium-, or tellurium-containing compounds
- C08K5/41—Compounds containing sulfur bound to oxygen
- C08K5/42—Sulfonic acids; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2300/00—Characterised by the use of unspecified polymers
- C08J2300/14—Water soluble or water swellable polymers, e.g. aqueous gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/02—Homopolymers or copolymers of acids; Metal or ammonium salts thereof
Definitions
- the present invention relates to a particulate water-absorbing agent mainly composed of poly (meth) acrylic acid (salt) -based water-absorbing resin particles. More specifically, the present invention relates to a particulate water-absorbing agent that can improve the performance of absorbent articles such as paper diapers.
- a water-absorbing resin (SAP / Super Absorb Polymer) is a water-swelling, water-insoluble polymer gelling agent, and exhibits excellent characteristics of absorbing body fluids. For this reason, water-absorbing agents mainly composed of water-absorbing resins are widely used for absorbent articles such as disposable diapers and sanitary napkins, as well as agricultural and horticultural water retention agents and industrial water-stopping materials. Many monomers and hydrophilic polymers have been proposed as raw materials for the water-absorbing resin constituting such a water-absorbing agent. From the viewpoint of price and performance, (meth) acrylic acid and / or its salts are mainly used. The poly (meth) acrylic acid (salt) water-absorbing resin used as a component is most frequently used industrially.
- Non-Patent Document 1 Such a water-absorbent resin is produced through a polymerization process, a drying process, if necessary, a undried product removal process, a pulverization process, a classification process, a surface crosslinking process, and the like.
- a paper diaper which is the main use of a water-absorbing resin (water-absorbing agent) as an example, improvements in urine leakage and skin irritation are required.
- a method for evaluating these a method of measuring a return amount from a paper diaper under pressure and a method of measuring a liquid absorption time of a paper diaper under pressure are proposed. It is estimated that urine leakage and skin irritation occur when urine becomes difficult to be taken into a paper diaper when the weight of the wearer of the paper diaper is applied, or when absorption by the water-absorbing agent is slow even when taken. And improving the water absorption rate of the water absorbent while improving the absorbability of the water absorbent under pressure reduces the return amount of the diaper and the liquid absorption time, which can lead to urine leakage and skin irritation. It is thought to lead to reduction.
- the water-absorbing agent is used in a mixed form with pulp.
- absorbents with a so-called low water-absorbing agent concentration (the water-absorbing resin concentration is less than 50% by weight), in which the weight of the pulp is greater than the weight of the water-absorbing agent, are preferred.
- Absorbers having a so-called high water-absorbing agent concentration (with a water-absorbing resin concentration of 50% by weight or more) greater than the weight are preferably used, and absorbers using only the water-absorbing resin without using pulp are also preferably used.
- Patent Document 1 a technology that uses a water-absorbing agent having high total water absorption capacity (PAI) under four different pressures (PAI) in a paper diaper (Patent Document 1), in the SAP layer under pressure, only in the vertical direction Technology that improves the diffusibility of the liquid in the horizontal direction (Patent Documents 2 and 3), technology that improves the water absorption capacity under pressure when the amount of SAP per unit area is large (Patent Documents 4 and 5), Techniques (Patent Documents 6 and 7) and the like for improving the water absorption magnification under pressure measured with a difference in height between the glass filter in contact with the liquid surface on the liquid supply side have been proposed.
- PAI total water absorption capacity
- Patent Document 7 introduces a new parameter, GCA (Gel Capillary Absorption, Gel Capillary Absorption), which evaluates the ability to suck liquid in a short time, and suggests that the higher the value, the lower the return amount. .
- GCA Gel Capillary Absorption, Gel Capillary Absorption
- Patent Documents 8 to 10 a technology for pulverizing the polymer gel with specific energy to form gel particles
- meet chopper a technology for pulverizing the polymer gel with a specific shape
- Patent Documents 8 to 14 a technique related to a gel pulverizer and pulverization conditions
- Patent Documents 15 to 12 a technique of adding additives such as polyethylene glycol and hydrophobic substances during polymerization or pulverization of a polymer gel
- the pulverization conditions are insufficient to improve GCA, and the surface tension is greatly reduced.
- Techniques for producing granulated particles by the reverse phase suspension polymerization method have also been proposed, but there are problems with complicated processes, problems due to remaining organic solvent, and return due to a decrease in surface tension. The effect of improving the performance of disposable diapers, such as increasing the amount, was insufficient.
- the present invention aims to solve at least one of the following problems.
- An object of the present invention is to provide a poly (meth) acrylic acid (salt) -based particulate water-absorbing agent having a reduced return amount and an excellent liquid uptake rate.
- a sanitary material for example, a paper diaper
- a reduced return amount at a high absorbent concentration and has an excellent liquid uptake rate is to provide.
- Patent Document 7 focusing on GCA (Gel Capillary Absorption) is still insufficient in liquid permeability.
- FGBP Free Gel Bed Permeability
- the present invention includes (i) a step of preparing a (meth) acrylic acid (salt) monomer aqueous solution, and (ii) a step of polymerizing the (meth) acrylic acid (salt) monomer aqueous solution.
- (Iv) a step of drying the hydrogel particles to obtain a dried product
- a step of obtaining a water absorbent resin powder by pulverizing and / or classifying the dried product and
- a step of obtaining the water absorbent resin powder is a step of obtaining the water absorbent resin powder.
- the weight average particle diameter of the water-containing gel particles when converted into a dry product is adjusted to be 50 to 650 ⁇ m, and the surface tension of the poly (meth) acrylic acid (salt) -based particulate water-absorbing agent is 60 mN / m or more, under no pressure
- a poly (meth) acrylic acid (salt) -based particulate water-absorbing material composed mainly of poly (meth) acrylic acid (salt) -based water-absorbing resin particles, adjusted so that the yield (CRC) is 28 g / g or more.
- a method for producing the agent is provided.
- a polyacrylic acid (salt) -based particulate water-absorbing agent comprising polyacrylic acid (salt) -based water-absorbing resin particles as a main component, wherein the following (1) to (5) :( 1) Absorption capacity under no pressure (CRC) is 28 g / g or more, (2) GCA is 28.0 g / g or more, (3) The relationship between FGBP and GCA is GCA of 28.0 g / g or more, 36.0 g FGBP ⁇ ⁇ 10 ⁇ 10 ⁇ 9 ⁇ GCA + 380 ⁇ 10 ⁇ 9 cm 2 is satisfied in a range less than / g, and FGBP ⁇ 30 ⁇ 10 ⁇ 9 cm 2 is satisfied when GCA is 36.0 g / g or more.
- a polyacrylic acid (salt) -based particulate water-absorbing agent satisfying a weight-average particle size (D50) of 300 to 500 ⁇ m and (5) a surface tension of 60
- the present invention has at least one of the following effects.
- a poly (meth) acrylic acid (salt) -based particulate water-absorbing agent having a reduced return amount and an excellent liquid uptake rate can be provided.
- a sanitary material for example, a paper diaper
- a reduced return amount at a high absorbent concentration and has an excellent liquid uptake rate.
- FIG. 1 is a schematic view of an apparatus used for the GCA measurement of the present invention.
- FIG. 2 is an SEM photograph (magnification 30 times) of the water-absorbent resin powder of Example 9.
- FIG. 3 is an SEM photograph (magnification 130 times) of the water-absorbent resin powder of Example 9.
- 4 is an SEM photograph (magnification 30 times) of the water-absorbent resin powder of Comparative Example 1.
- FIG. FIG. 5 is an SEM photograph (magnification 130 times) of the water-absorbent resin powder of Comparative Example 5.
- FIG. 6 is a correlation diagram showing the relationship between Examples and Comparative Examples.
- FIG. 7 is a schematic view of an apparatus used for the absorber evaluation of the present invention.
- the particulate water-absorbing agent and the method for producing the same according to the present invention will be described in detail.
- the scope of the present invention is not limited to these descriptions, and the scope of the present invention is not impaired except for the following examples. And can be changed as appropriate.
- the present invention is not limited to the following embodiments, and various modifications are possible within the scope of the claims, and technical means disclosed in different embodiments are appropriately combined. Embodiments obtained in this manner are also included in the technical scope of the present invention.
- the “particulate water-absorbing agent” is an aqueous liquid containing water-absorbent resin particles as a main component (preferably 60% by weight or more, more preferably 80% by weight or more, most preferably 90% by weight or more).
- optional ingredients include water, inorganic fine particles, moisture absorption blocking inhibitor, cationic polymer compound, water-soluble polyvalent metal cation-containing compound, surfactant, dust generation inhibitor, anti-coloring agent Urine resistance improver, deodorant, fragrance, antibacterial agent, foaming agent, pigment, dye, fertilizer, oxidizing agent, reducing agent, etc., 0 to 10 wt%, preferably 0.1 wt% to 1 wt. % May be contained.
- the “particulate water absorbing agent” may be simply referred to as “water absorbing agent”.
- Water Absorbent Resin The water-absorbing resin in the present invention means a water-swellable water-insoluble polymer gelling agent. “Water swellability” means that the CRC (water absorption capacity under no pressure) specified by ERT441.2-02 is 5 g / g or more, and “water-insoluble” means ERT470.2- Ext (soluble matter) specified by 02 is 0 to 50% by weight.
- the total amount (100% by weight) of the water-absorbent resin is not limited to the polymer, and may contain additives and the like within the range of maintaining the above performance, and the water-absorbent resin composition containing a small amount of additives is also included.
- it is generically called a water-absorbing resin.
- the shape of the water-absorbent resin is preferably powder, and particularly preferably a powder-form water-absorbent resin having a particle size described later.
- the “water absorbent resin” may be referred to as “water absorbent resin powder” or “water absorbent resin particles”.
- the “poly (meth) acrylic acid (salt) -based water-absorbing resin” in the present invention optionally includes a graft component, and (meth) acrylic acid and / or a salt thereof (hereinafter referred to as (meth) acrylic acid) as a repeating unit. (Referred to as “salt”).
- a polymer containing 50 to 100 mol% of (meth) acrylic acid (salt) among total monomers (excluding the crosslinking agent) used in the polymerization preferably 70 to 100 mol%, More preferably, it refers to a water-absorbing resin containing 90 to 100 mol%, particularly preferably substantially 100 mol%.
- poly (meth) acrylate type (neutralization type) polymers are also collectively referred to as poly (meth) acrylic acid (salt) water-absorbing resins.
- EDANA European Disposables and Nonwovens Associations
- ERT is an abbreviation for a method for measuring water-absorbent resin of the European standard (almost world standard) (EDANA Recommended Test Methods).
- the physical properties of the water-absorbent resin are measured according to the original ERT (revised in 2002 / known literature).
- CRC Centrifugation Retention Capacity (centrifugal retention capacity), and means the water absorption capacity of the water absorbent resin under no pressure (sometimes referred to as “water absorption capacity”). Specifically, 0.2 g of the water-absorbing resin was put in a non-woven bag, and then immersed in a large excess of 0.9 wt% sodium chloride aqueous solution for 30 minutes to freely swell, and then centrifuged (250 G ) Is the water absorption rate (unit: g / g) after draining.
- AAP is an abbreviation for Absorption against Pressure, which means the water absorption capacity of a water absorbent resin under pressure. Specifically, after swelling 0.9 g of water-absorbing resin with a large excess of 0.9 wt% sodium chloride aqueous solution for 1 hour under a load of 2.06 kPa (21 g / cm 2 , 0.3 psi) Of water absorption (unit: g / g). In addition, ERT 442.2-02 describes “Absorption Under Pressure”, which has substantially the same content.
- PSD is an abbreviation for Particle Size Distribution, and means a particle size distribution of a water-absorbent resin measured by sieving classification.
- the weight average particle diameter (D50) and the logarithmic standard deviation ( ⁇ ) of the particle size distribution are described in US Pat. No. 7,638,570, “(3) Mass-Average Particle Diameter (D50) and Logical Standard Deviation ( ⁇ ) of”. It measures by the method similar to "Particle Diameter Distribution.”
- Extractables which means the water-soluble component (water-soluble component amount) of the water-absorbent resin. Specifically, it refers to the amount of dissolved polymer (unit:% by weight) after adding 1.0 g of water-absorbing resin to 200 ml of 0.9 wt% aqueous sodium chloride solution and stirring at 500 rpm for 16 hours. The amount of dissolved polymer is measured using pH titration.
- X to Y indicating a range means “X or more and Y or less”.
- t (ton) as a unit of weight means “Metricton”, and “ppm” means “weight ppm” unless otherwise noted.
- weight and “mass”, “wt%” and “mass%”, “part by weight” and “part by mass” are treated as synonyms.
- ⁇ acid (salt) means “ ⁇ acid and / or salt thereof”
- (meth) acryl means “acryl and / or methacryl”.
- it is measured at room temperature (20 to 25 ° C.) and relative humidity 40 to 50% RH.
- the manufacturing method of the particulate water-absorbing agent according to the present invention includes (i) a step of preparing a (meth) acrylic acid (salt) -based monomer aqueous solution, (Ii) a step of polymerizing the aqueous solution of the (meth) acrylic acid (salt) monomer, and (iii) a step of gel-pulverizing the hydrogel crosslinked polymer during or after polymerization to obtain hydrogel particles; (Iv) drying the hydrogel particles to obtain a dried product, (v) obtaining a water absorbent resin powder by pulverizing and / or classifying the dried product, and (vi) the water absorbent resin powder.
- the hydrated gel-like crosslinked polymer obtained in the polymerization step is pulverized to a specific weight average particle size (the weight average particle size of the hydrated gel particles when converted to a dry product is 50 to 650 ⁇ m) and dried. Then, by adding a liquid permeation improver during or after surface cross-linking, a water absorbing agent excellent in GCA (Gel Capillary Absorption) and FGBP (Free Gel Bed Permeability) can be obtained. Can solve the problem.
- GCA Gel Capillary Absorption
- FGBP Free Gel Bed Permeability
- a step of preparing a (meth) acrylic acid (salt) monomer aqueous solution and (ii) polymerizing the (meth) acrylic acid (salt) monomer aqueous solution.
- a step of gel pulverizing the hydrogel crosslinked polymer during or after polymerization to obtain hydrogel particles and (iv) a step of drying the hydrogel particles to obtain a dried product, v) pulverizing and / or classifying the dried product to obtain a water-absorbent resin powder, (vi) surface-crosslinking the water-absorbent resin powder to obtain water-absorbent resin particles, and (vii) the water-absorbent resin powder.
- the hydrogel crosslinked polymer and / or the hydrogel particles are Adding an adhesion control agent to control the adhesion
- the poly (meth) acrylic acid (salt) -based particles are adjusted so that the solid content of the water-containing gel particles is 10 to 80% by weight and the weight-average particle diameter of the water-containing gel particles is 100 to 900 ⁇ m.
- CRC absorption capacity without pressure
- the hydrogel crosslinked polymer obtained in the polymerization step is pulverized to a specific weight average particle size (the weight average particle size of the hydrogel particles is 100 to 900 ⁇ m) and dried, and then during surface cross-linking or the surface
- a liquid permeation improver after cross-linking a water absorbing agent excellent in GCA (Gel Capillary Absorption) and FGBP (Free Gel Bed Permeability) can be obtained, and the intended problem of the present invention can be solved.
- the weight average particle diameter of the hydrogel particles when converted into a dry product is 50 to 650 ⁇ m” and “the weight average particle diameter of the hydrogel particles is 100 to 900 ⁇ m”. At least one of these may be simply referred to as “the particle size after gel pulverization is significantly small”.
- steps (i) to (vii) are preferably performed “sequentially” in this order, but each step may be performed before or after the previous step.
- the time between the above steps is appropriately determined including transportation time and storage time, and the time is preferably 0 second or more and 2 hours or less, more preferably 1 second or more and 1 hour or less.
- the manufacturing method of the particulate water-absorbing agent according to the present invention will be described mainly in the order of time.
- the manufacturing method only has to include the essential steps described above, and deviates from the gist of each manufacturing method. Other steps may be included as long as they are not.
- (2-1) Step of preparing (meth) acrylic acid (salt) monomer aqueous solution is an aqueous solution of a monomer mainly composed of (meth) acrylic acid (salt), and if necessary, a crosslinking agent, This refers to a mixture of components that constitute a water-absorbing resin, such as graft components and trace components (chelating agents, surfactants, dispersants, etc.), and is used for polymerization by adding a polymerization initiator as it is. Say things.
- the (meth) acrylic acid (salt) may be unneutralized or salt type (completely neutralized type or partially neutralized type), and the monomer aqueous solution may exceed the saturation concentration, Even a supersaturated aqueous solution or aqueous slurry (aqueous dispersion) of (meth) acrylic acid (salt) is treated as the (meth) acrylic acid (salt) monomer aqueous solution of the present invention.
- water is preferable as the solvent for dissolving the monomer, and the (meth) acrylic acid (salt) monomer is treated as an aqueous solution.
- the “aqueous solution” is not limited to the case where 100% by weight of the solvent is water, and when the total amount of the solvent is 100% by weight of the water-soluble organic solvent (for example, alcohol) in addition to water. May be used in an amount of 0 to 30% by weight, preferably 0 to 5% by weight. In the present invention, these are treated as an aqueous solution.
- the “(meth) acrylic acid (salt) -based monomer aqueous solution under preparation” described later refers to all monomer aqueous solutions containing the above (meth) acrylic acid (salt) as the main component. This refers to an aqueous solution of (meth) acrylic acid (salt) before the components are mixed. Specifically, (meth) acrylic acid aqueous solution, fully neutralized or partially neutralized (meth) acrylic acid (salt) aqueous solution Applicable.
- the final (meth) Acrylic acid (salt) monomer aqueous solution By further neutralizing the (meth) acrylic acid (salt) monomer aqueous solution being prepared, mixing water as a solvent, or mixing the above trace components, the final (meth) Acrylic acid (salt) monomer aqueous solution.
- the state before the polymerization is started before being charged into the polymerization apparatus or after being charged into the polymerization apparatus, It is referred to as “(meth) acrylic acid (salt) monomer aqueous solution after preparation”.
- (2-1-1) Monomer A monomer mainly composed of (meth) acrylic acid (salt) is used in the water absorbent resin of the present invention.
- the main component means that (meth) acrylic acid (salt) is usually at least 50 mol%, preferably at least 70 mol%, more preferably at least 80 mol%, based on the entire monomer (excluding the internal crosslinking agent), More preferably, it refers to a state of 90 mol% or more, particularly preferably 95 mol% or more (the upper limit is 100 mol%).
- poly (meth) acrylic acid (salt) is not limited to non-neutralized (neutralization rate 0 mol%), but includes a concept of partial neutralization or complete neutralization (neutralization rate 100 mol%). It is.
- (meth) acrylic acid (salt) as the main component of the monomer, it may also contain a monomer that becomes a water-absorbing resin by polymerization, such as (anhydrous) maleic acid, itaconic acid, Cinnamic acid, vinylsulfonic acid, allyltoluenesulfonic acid, vinyltoluenesulfonic acid, styrenesulfonic acid, 2- (meth) acrylamido-2-methylpropanesulfonic acid, 2- (meth) acryloylethanesulfonic acid, 2- (meta ) Anionic unsaturated monomers (salts) such as acryloylpropane sulfonic acid and 2-hydroxyethyl (meth) acryloyl phosphate; mercapto group-containing unsaturated monomers; phenolic hydroxyl group-containing unsaturated monomers; ) Amido groups such as acrylamide, N-ethyl (meth)
- the neutralization rate of the (meth) acrylic acid (salt) monomer or the water-containing gel-like crosslinked polymer after polymerization is not particularly limited, but the physical properties of the resulting water-absorbent resin and the reaction of the surface crosslinking agent From the viewpoint of properties, it is preferably 40 to 90 mol%, more preferably 50 to 85 mol%, still more preferably 65 to 80 mol%. Therefore, according to a preferred embodiment of the present invention, the neutralization rate of the acrylic acid (salt) monomer aqueous solution in step (i) is 40 to 90 mol%.
- the neutralization rate When the neutralization rate is low, the water absorption rate tends to decrease (for example, the water absorption time by the Vortex method increases). Conversely, when the neutralization rate is high, poly (meth) acrylic acid (salt) water absorption Because the reactivity between the resin and the surface cross-linking agent (especially the dehydration reactive surface cross-linking agent described later) decreases, productivity tends to decrease, and the water absorption capacity under pressure (for example, AAP) tends to decrease. A neutralization rate within the above range is preferred.
- the neutralization may be performed on the hydrated gel after polymerization (hydrated gel-like crosslinked polymer) in addition to the monomer and / or the monomer aqueous solution before polymerization. Also good. In addition, when performing several times, it is preferable to adjust to the range of the said neutralization rate in consideration of the addition amount of all the basic compounds.
- the (meth) acrylic acid (salt) monomer or the hydrogel crosslinked polymer is Part or all may be in a salt form, preferably one or more of monovalent salts such as sodium salt, lithium salt, potassium salt, ammonium salt, amines, etc., more preferably one or more alkali metal salts, more preferably sodium Salts and / or potassium salts are preferable, and sodium salts are particularly preferable from the viewpoints of cost and physical properties.
- monovalent salts such as sodium salt, lithium salt, potassium salt, ammonium salt, amines, etc.
- alkali metal salts more preferably sodium Salts and / or potassium salts are preferable
- sodium salts are particularly preferable from the viewpoints of cost and physical properties.
- an internal cross-linking agent is used as necessary in the above polymerization.
- the internal cross-linking agent known ones can be used. For example, N, N′-methylenebis (meth) acrylamide, (poly) ethylene glycol di (meth) acrylate, (poly) propylene glycol di (meth) acrylate, trimethyl Roll propane tri (meth) acrylate, glycerin tri (meth) acrylate, glycerin acrylate methacrylate, ethylene oxide modified trimethylolpropane tri (meth) acrylate, pentaerythritol hexa (meth) acrylate, triallyl cyanurate, triallyl isocyanurate, tri Allyl phosphate, triallylamine, poly (meth) allyloxyalkane, (poly) ethylene glycol diglycidyl ether, glycerol diglycidyl ether,
- the amount of the internal crosslinking agent used can be appropriately determined depending on the desired properties of the water-absorbing resin, but is appropriately adjusted so that the CRC of the water-absorbing agent of the present invention is 28 g / g or more.
- 0.001 to 1 mol% is preferable, 0.005 to 0.5 mol% is more preferable, and 0.005 to 0.5 mol% is preferable with respect to 100 mol% of the (meth) acrylic acid (salt) monomer. It is more preferably from 01 to 0.3 mol%, particularly preferably from 0.01 to 0.1 mol%.
- the amount used is less than 0.001 mol%, the water-absorbing resin obtained has a high soluble content, and it becomes impossible to secure a sufficient amount of water absorption under pressure, and the values of GCA and FGBP also deteriorate. It is not preferable.
- the amount used exceeds 1 mol%, the crosslinking density of the resulting water-absorbent resin becomes too high, the water absorption amount cannot be sufficiently secured, and as a result, the GCA becomes low, which is not preferable.
- the internal cross-linking agent is added to the reaction system before or during polymerization of the monomer, after polymerization, or after neutralization. You can do it.
- the internal cross-linking agent may be added all at once or dividedly to the reaction system.
- adhesion control agent In order to highly solve the problems of the present invention, an adhesion control agent (also referred to as a fusion control agent) described in detail in (2-3-2) is added, and (meth) acrylic acid is added. It may be added during or after the preparation step of the (salt) monomer aqueous solution. Specifically, it is added before step (iii) to step (iii).
- step (ii)) Examples of the polymerization method for obtaining the water-absorbent resin particles (water-containing gel-like crosslinked polymer) of the present invention include spray polymerization, droplet polymerization, bulk polymerization, precipitation polymerization, aqueous solution polymerization, and reverse phase suspension polymerization. However, in order to solve the problems of the present invention, aqueous solution polymerization or reverse phase suspension polymerization using a monomer as an aqueous solution is preferable.
- the aqueous solution polymerization is a method of polymerizing an aqueous monomer solution without using a dispersion solvent.
- the reverse phase suspension polymerization is a method in which an aqueous monomer solution is suspended in a hydrophobic organic solvent for polymerization.
- a hydrophobic organic solvent for polymerization For example, U.S. Pat. Nos. 4,093,764, 4,367,323, 4,446,261, Nos. 4,683,274 and 5,244,735.
- Monomers, polymerization initiators and the like disclosed in these patent documents can also be applied to the present invention.
- the concentration of the aqueous monomer solution during the polymerization is not particularly limited, but is preferably 20% by weight to saturated concentration, more preferably 25 to 80% by weight, and further preferably 30 to 70% by weight. A concentration of 20% by weight or more is preferable because high productivity can be achieved.
- the polymerization in the monomer slurry shows a decrease in physical properties, and therefore, it is preferable to carry out the polymerization at a saturated concentration or less (see Japanese Patent Laid-Open No. 1-318021). Publication).
- the polymerization step in the present invention can be carried out at normal pressure, reduced pressure, or increased pressure, but is preferably carried out at normal pressure (or in the vicinity thereof, usually ⁇ 10 mmHg).
- the temperature at the start of the polymerization is preferably 15 to 130 ° C., more preferably 20 to 120 ° C., although it depends on the type of polymerization initiator used.
- the polymerization initiator used in the present invention is appropriately determined depending on the polymerization form and is not particularly limited, and examples thereof include a photodegradable polymerization initiator, a thermal decomposition polymerization initiator, and a redox polymerization initiator. The polymerization of the present invention is initiated by these polymerization initiators.
- photodegradable polymerization initiator examples include benzoin derivatives, benzyl derivatives, acetophenone derivatives, benzophenone derivatives, and azo compounds.
- thermal decomposition polymerization initiator examples include persulfates such as sodium persulfate, potassium persulfate, and ammonium persulfate; peroxides such as hydrogen peroxide, t-butyl peroxide, and methyl ethyl ketone peroxide; 2 Azo compounds such as 2,2′-azobis (2-amidinopropane) dihydrochloride and 2,2′-azobis [2- (2-imidazolin-2-yl) propane] dihydrochloride.
- persulfates such as sodium persulfate, potassium persulfate, and ammonium persulfate
- peroxides such as hydrogen peroxide, t-butyl peroxide, and methyl ethyl ketone peroxide
- 2 Azo compounds such as 2,2′-azobis (2-amidinopropane) dihydrochloride and 2,2′-azobis [2- (2-imidazolin-2-yl) propane]
- examples of the redox polymerization initiator include a system in which a reducing compound such as L-ascorbic acid or sodium bisulfite is used in combination with the persulfate or peroxide. It is also a preferred embodiment to use the photodegradable polymerization initiator and the thermal decomposition polymerization initiator in combination.
- a reducing compound such as L-ascorbic acid or sodium bisulfite
- the photodegradable polymerization initiator and the thermal decomposition polymerization initiator in combination.
- an azo polymerization initiator that generates N 2 by thermal decomposition may be used to promote foaming.
- active energy rays such as ultraviolet rays, electron beams, and ⁇ rays may be used alone or in combination with the above polymerization initiator.
- the amount of the polymerization initiator used is preferably 0.0001 to 1 mol%, more preferably 0.0005 to 0.5 mol%, based on 100 mol% of the monomer. If the amount used is 1 mol% or less, the color tone deterioration of the water-absorbing resin is preferably suppressed, and if it is 0.5 mol% or less, the color tone deterioration is further suppressed, which is more preferable. Moreover, if this usage-amount is 0.0001 mol% or more, it is preferable since the increase in a residual monomer is suppressed, and if it is 0.0005 mol% or more, it is more preferable because the increase in a residual monomer is suppressed more.
- adhesion control agent In order to solve the problems of the present invention to a high degree, an adhesion control agent described in detail in (2-3-2) is added.
- the adhesion control agent may be added before, during or after the polymerization step. Specifically, it is added before step (iii) to step (iii).
- aqueous solution polymerization is performed from the viewpoint of physical properties of the water absorbent resin (for example, water absorption speed and liquid permeability) and ease of polymerization control. Is adopted. Of these, kneader polymerization or belt polymerization is preferable, and continuous aqueous solution polymerization is more preferably employed.
- aqueous solution polymerization examples include continuous belt polymerization (disclosed in US Pat. Nos. 4,893,999 and 6,241,928, US Patent Application Publication No. 2005/215734, etc.), continuous kneader polymerization, batch kneader polymerization (US Pat. No. 6,987,151). , And the like are disclosed in No. 6710141).
- the method described in JP-A No. 2002-212204 can shorten the polymerization step by setting the polymerization initiation temperature to a high temperature of 50 ° C. or higher.
- the use of the latent heat of vaporization of water makes it possible to suppress the maximum temperature, which is particularly preferable because a water-absorbing resin having a high molecular weight of the main chain and a narrow molecular weight distribution can be obtained.
- the monomer concentration in the (meth) acrylic acid (salt) monomer aqueous solution is preferably 10 to 80% by weight, more preferably 20 to 60% by weight, and still more preferably. 30 to 50% by weight.
- the Example of this application was 38 weight% and 39 weight%.
- the load on the machine in the next gel pulverization step is not excessively high, and the weight average particle diameter of the particulate hydrous gel can be efficiently controlled within the desired range.
- the maximum temperature during polymerization specifically 70 ° C. to 130 ° C., more preferably 80 ° C. to 120 ° C. 85 ° C. to 110 ° C. is more preferable.
- a chain transfer agent such as hypophosphorous acid (salt), for example, a chelating agent such as trisodium diethylenetriaminepentaacetic acid, pentasodium diethylenetriaminepentaacetic acid, or the like is added to the reaction system before or during the polymerization. May be added in an amount of preferably 0 to 3% by weight, more preferably 0.001 to 1% by weight based on 100% by weight of the monomer.
- step (iii)) a water-containing gel-like crosslinked polymer (hereinafter sometimes referred to as “water-containing gel”) obtained through the polymerization step and the like is pulverized to form a particulate water-containing gel (hereinafter “water-containing gel particles”). Or “particulate hydrous gel”). GCA and vortex are improved by refining the particulate hydrous gel to a specific particle size range by gel pulverization of the hydrated gel, particularly gel pulverization by kneading.
- the solid content of the hydrogel crosslinked polymer is preferably 10 to 80% by weight from the viewpoint of ease of gel grinding and drying of the particulate hydrogel after gel grinding. It is more preferably 20 to 70% by weight, further preferably 20 to 60% by weight, still more preferably 30 to 50% by weight, and particularly preferably 35 to 48% by weight.
- the solid content is lower than 10% by weight, the moisture content of the pulverized particulate hydrogel increases, and there is a concern that the cost increases due to the increase in energy required for drying, and the production efficiency decreases due to the longer drying time. Is done.
- the solid content exceeds 80% by weight, the gel becomes hard, the load on the pulverizer increases, and the pulverizer may be broken.
- the method for setting the solid content of the hydrogel crosslinked polymer to 10 to 80% by weight is not particularly limited. For example, it may be performed by limiting the total monomer amount during polymerization.
- the “weight average particle diameter of the particulate hydrogel (hydrated gel particles)” after the gel pulverization step is 100 ⁇ m to 900 ⁇ m, preferably 120 to 870 ⁇ m, more preferably 130 to 860 ⁇ m, still more preferably 140 to 850 ⁇ m, It may be 150 to 800 ⁇ m, 160 to 700 ⁇ m, 170 to 600 ⁇ m, or 100 to 350 ⁇ m.
- the “weight average particle diameter of the water-containing gel particles when converted into a dry product” is 50 to 650 ⁇ m, preferably 80 to 640 ⁇ m, more preferably 100 to 630 ⁇ m, and more preferably 110 to 600 ⁇ m. Even more preferably, 120 to 500 ⁇ m is particularly preferable, and 130 to 460 ⁇ m is particularly preferable, and 140 to 400 ⁇ m may be used.
- the weight average particle diameter (RB) of the water-containing gel particles when converted as a dried product is “weight average particle diameter (RA) of the water-containing gel particles” ⁇ “(water-containing The solid content of gel particles) 1/3 ".
- the hydrogel particles change in volume in a similar shape depending on the amount of water absorption (water absorption magnification), and the particle diameter also changes in a similar shape as the volume changes.
- water-containing gel particles and the dried body thereof will be described as spheres.
- the volume V of the sphere can be represented by ( ⁇ ⁇ R 3 ) / 6 where R is the diameter of the sphere.
- the volume VA can be expressed as ⁇ ⁇ (RA) 3 ) / 6.
- the volume VA and VB of the hydrogel particles are proportional to the reciprocal of the solid content.
- a particulate hydrogel of 100 ⁇ m to 900 ⁇ m becomes 74 ⁇ m to 663 ⁇ m in terms of a dry product.
- the weight average particle diameter of the particulate hydrogel (hydrated gel particles) is controlled in the above range, and any means for achieving it is not limited.
- examples thereof include a gel crusher equipped with a plurality of rotary stirring blades such as a double-arm kneader, a single-screw extruder, a twin-screw extruder, and a meat chopper.
- a screw type extruder (for example, a meat chopper) having a perforated plate at the tip is preferable, and examples thereof include a screw type extruder disclosed in Japanese Patent Application Laid-Open No. 2000-063527.
- the gel crusher used in this step is more preferably a screw-type extruder, more preferably a screw-type extruder (for example, a meat chopper) in which a porous plate is installed at one end of a casing
- a screw-type extruder for example, a meat chopper
- a porous plate is installed at one end of a casing
- screw type extruders disclosed in Japanese Patent Application Laid-Open No. 2000-63527, International Publication No. 2015/030129 and International Publication No. 2015/030130.
- the screw-type extruder used in this step is composed of, for example, a casing, a base, a screw, a supply port, a hopper, an extrusion port, a perforated plate, a rotary blade, a ring, a reverse prevention member, a motor, a streak, and the like.
- the casing has a cylindrical shape, and a screw is disposed therein.
- One end of the casing is provided with an extrusion port for extruding the water-containing gel-like crosslinked polymer and pulverizing the gel, and a perforated plate is installed in front of it, and the other end is a motor for rotating the screw.
- a drive system etc. are arranged.
- a stand is provided below the casing, whereby the screw type extruder can be stably installed.
- a supply port for supplying the hydrogel crosslinked polymer above the casing there is a supply port for supplying the hydrogel crosslinked polymer above the casing, and a hopper is provided to facilitate the supply of the hydrogel crosslinked polymer.
- the shape and size of the casing need only have a cylindrical inner surface corresponding to the shape of the screw, and are not particularly limited.
- the rotation speed of a screw changes with shapes of a screw extruder, it is not specifically limited, As mentioned later, it is preferable that the rotation speed of a screw can be changed.
- a reverse prevention member, a streaky protrusion disposed on the screw, and the like can be provided in the vicinity of the extrusion port.
- the reversion preventing member is not particularly limited as long as it is a structure that can prevent the water-containing gel-like cross-linked polymer from reversing in the vicinity of the extrusion port, and is a spiral or concentric belt-shaped protrusion installed on the inner wall of the casing, Alternatively, a streak-like, granular, spherical or angular projection placed in parallel with the screw can be used.
- a streak-like, granular, spherical or angular projection placed in parallel with the screw can be used.
- the particle size after gel pulverization is significantly small.
- the method to do so when using a screw type extruder in which a perforated plate is installed at one end of the casing, according to the gel grinding treatment amount of the hydrogel crosslinked polymer, The device size needs to be changed. In that case, adjust by appropriately adjusting the thickness of the perforated plate, the hole diameter of the perforated plate, the aperture ratio of the perforated plate, the rotational speed of the screw shaft, the charging speed of the hydrogel crosslinked polymer, etc. can do.
- the perforated plate provided at the outlet of the casing of the gel crusher its thickness, pore diameter, and open area ratio are appropriately selected depending on the processing amount per unit time of the gel crusher and the properties of the hydrogel crosslinked polymer.
- the thickness of the porous plate is preferably in the range of 3.5 mm to 100 mm, and more preferably in the range of 6 mm to 80 mm.
- the diameter is preferably 30 to 1500 mm, and more preferably 40 to 1000 mm.
- the hole diameter of the perforated plate is preferably in the range of 1.0 mm to 50 mm, more preferably 2.0 mm to 30 mm.
- the aperture ratio of the perforated plate is preferably 10% to 80%, preferably 20% to 60%, and more preferably 25% to 55%.
- the number of holes is preferably 10 to 2000, more preferably 20 to 1000.
- the simple average value of the pore diameters of the perforated plates is used as the pore size of the perforated plate in the gel crusher.
- the shape of the hole is preferably circular, but when the shape is other than a circle, for example, a quadrangle, an ellipse, a slit, etc., the hole area is converted to a circle and the hole diameter (mm) To do.
- the outer diameter of the screw shaft is preferably 10 to 2000 mm, more preferably 20 to 1000 mm.
- the charging rate of the hydrogel crosslinked polymer is preferably 0.10 to 550 kg / min, and more preferably 0.12 to 500 kg / min.
- the hydrogel crosslinked polymer is in the form of particles having a desired particle diameter. It may not be possible to grind the hydrogel.
- the thickness of the porous plate is more than 100 mm, the pore diameter is less than 1.0 mm, and the open area ratio is less than 10%, excessive shearing / compression is applied to the hydrogel crosslinked polymer. It may give power and cause deterioration of physical properties.
- the charging rate of the hydrated gel-like crosslinked polymer is 0.10 kg / min or less, excessive shearing / compressing force may be applied to the hydrated gel-like crosslinked polymer, leading to a decrease in physical properties. On the contrary, if it exceeds 550 kg, the hydrogel crosslinked polymer may not be pulverized into a particulate hydrogel having a desired particle size.
- the particle size after gel pulverization is significantly small.
- the method of doing so it may be performed by setting the gel grinding energy to an appropriate value.
- the gel grinding energy (GGE (1)) is preferably 10 to 500 J / g, more preferably 15 to 400 J / g, still more preferably 20 to 300 J / g, and still more preferably 45 to 250 J. / G, particularly preferably 25 to 200 J / g.
- the hydrogel crosslinked polymer may not be pulverized into a particulate hydrogel having a desired particle size.
- the gel grinding energy (GGE (1)) exceeds 500 J / g, the load on the grinding apparatus increases, and there is a risk of breaking during continuous operation. A compressive force may be applied, resulting in a decrease in physical properties such as an increase in the amount of water-soluble components and a decrease in CRC and AAP.
- the gel grinding energy (GGE (2)) is preferably 5 to 300 J / g, more preferably 6 to 280 J / g, 8 to 260 J / g, 9 to 250 J / g, 10 to 240 J / g.
- the hydrogel crosslinked polymer may not be pulverized into a particulate hydrogel having a desired particle size.
- the gel grinding energy (GGE (2)) exceeds 300 J / g, excessive shear / compression force is applied to the hydrogel crosslinked polymer, resulting in an increase in the amount of water-soluble component generated or CRC. In some cases, physical properties such as AAP decrease.
- GGE calculation method "Gel grinding energy” (GGE (1), GGE (2))
- the “gel grinding energy” in the present invention refers to mechanical energy per unit weight (unit weight of the water-containing gel-like crosslinked polymer) required by the gel grinding device when gelling the water-containing gel-like crosslinked polymer. It does not include the energy to heat and cool the jacket and the energy of water and steam to be charged.
- “Gel grinding energy” is abbreviated as “GGE (1)” from “Gel Grinding Energy” in English. GGE is calculated by the following formula (1) when the gel crusher is driven by three-phase AC power.
- the above “power factor” and “motor efficiency” are values unique to the apparatus that vary depending on the operating conditions of the gel crushing apparatus, and take values from 0 to 1. These values can be obtained by inquiries to the device manufacturer.
- GGE When the gel crusher is driven by single-phase AC power, GGE can be calculated by changing “ ⁇ 3” in the above equation to “1”.
- the unit of voltage is [V]
- the unit of current is [A]
- the unit of weight of the hydrogel crosslinked polymer is [g / s].
- the mechanical energy applied to the hydrogel crosslinked polymer is important, it is preferable to calculate the gel grinding energy by subtracting the current value when the gel grinding device is idling.
- the gel grinding energy in this case is calculated by the following formula (2).
- GGE (1) in order to distinguish from said GGE (1), it describes with GGE (2).
- the hydrogel crosslinked polymer is gel-pulverized to obtain hydrogel particles, the hydrogel particles are dried to obtain a dried product, and then the dried product is “pulverized”.
- the gel is formed by performing multiple pulverization of the “hydrated gel-like crosslinked polymer” that is difficult to pulverize or by setting the gel pulverization energy to a predetermined value or more. It was thought that there was no need to make the particle size after grinding significantly smaller.
- the present invention is such that the particle size after gel pulverization is significantly small, and by adopting a unique configuration that uses an adhesion control agent, the return amount is reduced and excellent liquid uptake is achieved.
- a poly (meth) acrylic acid (salt) -based particulate water-absorbing agent having a speed and a method for producing the same can be provided.
- the hydrogel crosslinked polymer during or after polymerization preferably the hydrogel crosslinked polymer after polymerization is preferably tens of centimeters in size. Can be cut or crushed.
- the gel cutting or gel crushing is a primary treatment (for example, a primary treatment of 1000 cm 3 or less to 1000 cm 2 or less on a flat surface) to a size that can be continuously charged into a grinder. Is differentiated in that it is finely divided (particularly, finely divided to a weight average particle diameter of 50 to 650 microns converted to a dry product).
- the gel grinding device makes it easy to fill the gel grinding device with the hydrogel crosslinked polymer, and the gel grinding process can be carried out more smoothly.
- a method capable of cutting or crushing so as not to knead the hydrogel crosslinked polymer is preferable, and examples thereof include cutting with a guillotine cutter or crushing.
- the size and shape of the hydrogel crosslinked polymer obtained by the above cutting or crushing are not particularly limited as long as the gel pulverizing apparatus can be filled, and the shape is preferably a block shape.
- the gel pulverization is performed during and / or after polymerization, and is preferably performed on the hydrogel crosslinked polymer after polymerization.
- kneader polymerization is mentioned as superposition
- Preferred examples of the polymerization for gel pulverization after polymerization include belt polymerization and standing aqueous solution polymerization in a tank (aqueous solution polymerization without substantial stirring), but are not particularly limited to these polymerizations.
- the polymerization rate of the hydrogel crosslinked polymer to be crushed is preferably 90 mol% or more, more preferably 93 mol% or more, still more preferably 95 mol% or more, and particularly preferably 97 mol% or more.
- the upper limit is preferably 99.5 mol%. If the polymerization rate of the hydrogel crosslinked polymer to be crushed by gel is 90 mol% or more, the residual monomer contained in the water absorbent resin can be reduced, which is preferable. In the case of gel pulverization during the polymerization, the polymerization may be continued as described below until the polymerization rate is shown.
- the polymerization rate is also referred to as a conversion rate, and is a value calculated from the polymer amount of the hydrogel crosslinked polymer and the unreacted monomer amount.
- the polymerization rate of the hydrogel crosslinked polymer to be gel crushed is preferably within the above range, but in the case of gel pulverization during polymerization, such as kneader polymerization, the monomer aqueous solution is “sufficiently gelled”. It is set as a gel grinding
- the monomer aqueous solution changes to a hydrogel crosslinked polymer as the polymerization time elapses. That is, the stirring region of the monomer aqueous solution at the start of polymerization, the stirring region of the low-polymerized hydrogel crosslinked polymer having a constant viscosity during the polymerization, and a part of the hydrogel crosslinked polymer as the polymerization proceeds
- the gel pulverization start region and the gel pulverization region in the latter half of the polymerization or the final stage are continuously performed.
- the above “sufficient gelation” means a state in which the hydrogel crosslinked polymer can be subdivided by applying a shearing force after the polymerization temperature reaches the maximum (polymerization peak temperature).
- the time when the polymerization rate of the monomer in the aqueous monomer solution is preferably 90 mol% or more, more preferably 93 mol% or more, still more preferably 95 mol% or more, particularly preferably 97 mol% or more It means a state in which a shearing force can be applied to subdivide the hydrogel crosslinked polymer.
- the water-containing gel-like crosslinked polymer having a monomer polymerization rate within the above range is gel-pulverized.
- the polymerization rate of the monomer is preferably 90 mol% or more, more Preferably, it is judged to be “sufficiently gelled” when it is 93 mol% or more, more preferably 95 mol% or more, particularly preferably 97 mol% or more.
- the screw shaft rotation speed of the screw extruder depends on the inner diameter of the casing, the outer diameter of the screw shaft, etc.
- the shaft rotational speed is preferably 80 rpm to 500 rpm, more preferably 90 rpm to 400 rpm, and still more preferably 100 rpm to 300 rpm.
- the inner diameter of the casing is preferably about 15 to 2500 mm, more preferably about 25 to 1500 mm.
- the shaft rotational speed is less than 80 rpm, it is difficult to obtain the shear / compression force necessary for gel crushing.
- the shaft rotational speed exceeds 500 rpm, the shear / compressive force applied to the hydrogel crosslinked polymer is excessive. As a result, the physical properties may be deteriorated, the load applied to the gel crushing device is increased, and the device may be damaged.
- the outer peripheral speed of the rotary blade at this time is preferably 0.5 m / s to 10 m / s, more preferably 0.5 m / s to 8 m / s.
- the temperature of the gel crusher in the present invention is preferably heated or kept at 30 ° C. to 120 ° C., more preferably 40 ° C. to 100 ° C. in order to prevent adhesion of the hydrogel crosslinked polymer.
- the temperature of the gel crusher in the present invention is also preferable to set the temperature of the gel crusher in the present invention within the temperature range of the gel temperature described below.
- the number of gel pulverization treatments is not particularly limited.
- a method of processing a plurality of times with a single gel pulverizer may be used, or a plurality of gel pulverizers may be installed in series and continuously processed.
- the weight average particle diameter of the particulate hydrous gel can be brought into a desired range by gel pulverization under relatively mild conditions, and it is difficult to cause deterioration in the physical properties of the water absorbent resin.
- the load on the gel crusher is small and there is no risk of damage to the device.
- the gel pulverizer for multiple treatments need not be the same type of machine, and different types of machines may be combined, or the setting conditions and operating conditions may be changed even for the same type of machine.
- the gel crushing step is performed using a plurality of gel crushers. According to this embodiment, it has the technical effect of reducing the load applied to one pulverizer.
- the preferred number is 2 to 5 times, more preferably 2 to 4 times, and even more preferably 2 to 3 times.
- the gel pulverization performed twice means that the hydrogel particles discharged from the outlet of the gel pulverizer are again introduced into the gel pulverizer and the gel pulverization is performed. The same applies to the third and subsequent times.
- gel pulverization may be performed once.
- the cost of introducing and maintaining a gel pulverizer can be suppressed, and the space for manufacturing equipment can also be suppressed.
- the energy load can be reduced, which is preferable.
- the gel temperature that is, the temperature of the hydrogel crosslinked polymer before gel pulverization is preferably 40 to 120 ° C., more preferably 50 to 120 ° C., and still more preferably 52 to 110 ° C., from the viewpoints of particle size control and physical properties. It is more preferably 48 to 80 ° C., particularly preferably 56 to 70 ° C. It may be 65 ° C. to 110 ° C.
- the numerical value of the gel temperature may be applied to the temperature of the gel crusher.
- the adhesive property is relatively high due to the characteristics of the hydrogel crosslinked polymer, and it becomes difficult to control the particle shape and particle size distribution during gel pulverization.
- the gel temperature is higher than 120 ° C., the evaporation of water from the gel becomes remarkable and the solid content of the hydrogel changes, so that the pulverization becomes difficult, and the control of the particle diameter and the particle shape of the particulate hydrogel is possible. It becomes difficult.
- Such a gel temperature can be appropriately controlled by the polymerization temperature, the post-polymerization heating, the heat retaining or the cooling.
- the CRC of the hydrogel crosslinked polymer before gel pulverization and the particulate hydrogel after gel pulverization (hydrogel particles) are preferably either one, more preferably both values are 25 to 50 g / g, The amount is preferably 26 to 45 g / g, more preferably 27 to 40 g / g.
- the gel CRC is within the above range, it is preferable because the particle shape and particle size distribution during gel pulverization can be easily controlled.
- Such gel CRC can be appropriately controlled by the amount of crosslinking agent added during polymerization, the polymerization concentration, and the like.
- the water absorbing resin which has high CRC is preferable, when the said gel CRC is high exceeding the said range, it may become difficult to control particle shape and a particle size distribution.
- the CRC change in the gel pulverization step for pulverizing the hydrogel crosslinked polymer to obtain hydrogel particles (before gel pulverization) The value obtained by subtracting the CRC of the hydrogel particles after gel grinding from the CRC of the hydrogel crosslinked polymer, the unit is g / g) is preferably ⁇ 10 to +10, more preferably ⁇ 8 to +8. More preferably, it is ⁇ 6 to +6, particularly preferably ⁇ 5 to +5, and most preferably ⁇ 4 to +4.
- the gel CRC is a hydrated gel-like crosslinked polymer, which is cut and finely divided using scissors, a cutter, etc. so that the hydrated gel-like crosslinked polymer before gel grinding is 1 to 3 mm on a side.
- the CRC of the particulate hydrogel (hydrogel particles) after gel pulverization can be obtained by the measurement method described in the following [Examples] without the need to cut and refine.
- the resin solid content of the hydrogel crosslinked polymer before pulverization of the gel is preferably 10 to 80% by weight, more preferably 20 to 60% by weight, and further preferably 30 to 55% by weight from the viewpoint of physical properties. More preferably, it is 33 to 50% by weight, and particularly preferably 36 to 46% by weight.
- the resin solid content of the particulate hydrogel (hydrogel particles) after gel pulverization is 10 to 80% by weight, preferably 20 to 60% by weight, more preferably 30 to 55% from the viewpoint of physical properties. % By weight, more preferably 33 to 50% by weight, and still more preferably 36 to 46% by weight.
- the resin solid content of the particulate hydrogel (hydrogel particle) after gel pulverization is in the above range, preferably the water-containing gel-like crosslinked polymer resin solid content before gel pulverization is in the above range, thereby causing damage due to drying ( Less increase in water-soluble content).
- the resin solid content of the hydrous gel particles after gel pulverization can be appropriately controlled by adding water before gel pulverization or during gel pulverization, or by evaporating water by heating during gel pulverization, if necessary. it can.
- the change in solid content in the gel pulverization process (gel) by pulverizing the hydrogel crosslinked polymer to obtain hydrogel particles is preferably ⁇ 10 to +10, more preferably ⁇ 8 to +8. More preferably, it is ⁇ 6 to +6, particularly preferably ⁇ 5 to +5, and most preferably ⁇ 4 to +4.
- the minus sign here means that the solid content is low (the water content is high), and the + sign means that the solid content is high (the water content is low).
- the change in solid content is greater than -10, the moisture content of the hydrogel particles will increase, increasing the load on the drying process, making it difficult to dry sufficiently and requiring more heat energy. Decreases.
- the change in solid content is larger than +10, the gel is damaged in the gel crushing process, the eluted components are increased, and the water absorbing agents GCA and FGBP may be lowered.
- gel pulverization may be performed by adding water to the hydrogel crosslinked polymer.
- water includes at least one of solid, liquid, and gas.
- the addition of water there is no limitation on the addition method and the addition timing, and it is sufficient that water is supplied into the apparatus while the hydrogel crosslinked polymer is retained in the gel crushing apparatus. Moreover, you may throw into the gel grinding
- the water is not limited to “water alone”, and other additives (for example, surfactant, neutralizing base, etc.) and solvents other than water may be added.
- the water content is preferably 90 to 100% by weight, more preferably 99 to 100% by weight, and still more preferably substantially 100% by weight.
- the water can be used in at least one of solid, liquid, and gas, but liquid and / or gas are preferable from the viewpoint of handling.
- the amount of water supplied is preferably 0 to 4 parts by weight, more preferably 0 to 2 parts by weight with respect to 100 parts by weight of the hydrogel crosslinked polymer. When the supply amount of water exceeds 4 parts by weight, there is a risk that problems such as generation of undried material during drying occur.
- the temperature of the water at the time of supply is preferably 10 to 100 ° C., more preferably 40 to 100 ° C. Liquid water is appropriately added by spraying, mist, showering, droplets, straight pipes or the like.
- the temperature of the water at the time of supply is preferably 100 to 220 ° C., more preferably 100 to 160 ° C., and further preferably 100 to 130 ° C.
- the preparation method is not particularly limited. For example, a method using water vapor generated by heating of a boiler, a gaseous state generated from a water surface by vibrating water with ultrasonic waves. The method of using the water of this is mentioned.
- steam having a pressure higher than atmospheric pressure is preferable, and steam generated in a boiler is more preferable.
- aqueous solution polymerization rather than reverse phase suspension polymerization that does not require gel grinding, and particularly during polymerization (for example, kneader polymerization) or after polymerization (for example, It is preferable to employ aqueous solution polymerization in which gel pulverization is employed for belt polymerization and further, if necessary, kneader polymerization.
- the gel contains an adhesion control agent during gel pulverization.
- the adhesion control agent may be added before the gel grinding is completely finished.
- adhesion is performed in at least one of the above-mentioned step (i) (meth) acrylic acid (salt) monomer aqueous solution adjustment step, step (ii) polymerization step, and step (iii) gel grinding step.
- a control agent is added, and a step of adding between step (i) and step (ii) or between step (ii) and step (iii) may be provided.
- Examples of the process between the process (i) and the process (ii) include a process of storing and transporting the prepared (meth) acrylic acid (salt) monomer aqueous solution, and the process (ii) and the process ( Examples of the step during iii) include a aging step of the hydrogel polymer.
- the desired effect of the present invention can be achieved by including an adhesion control agent in and / or on the surface of the hydrogel particles before the steps (iii) to (iii). .
- the step (iii) is a step of gel-pulverizing the hydrogel crosslinked polymer during or after polymerization to obtain hydrogel particles.
- the “hydrogel-crosslinked polymer” before gel pulverization or The addition of an adhesion control agent to “the hydrated gel-like crosslinked polymer cut or crushed” is also included in the concept of “adding an adhesion control agent in the step (iii)”. To do.
- the adhesion control agent may be liquid or solid, but may be added as it is, or may be added in a solution state or a suspended state.
- the radical polymerizable adhesion control agent having an unsaturated bond may be consumed by the reaction during the polymerization in the step (i) or the step (ii) and may not remain during the gel pulverization. ) May not be sufficiently consumed and may remain in the final product and cause coloring, and the adhesion control agent is preferably non-radically polymerizable.
- the solvent when added in the solution state and the dispersion medium when added in the suspended state are not particularly limited, but water and alcohol are preferable, and water is particularly preferable.
- the concentration of the adhesion control agent is preferably 0.1 to 99% by weight, more preferably 0.1 to 75% by weight, and even more preferably 0.1 to 50% by weight.
- the temperature at the time of addition in the solution is from the melting point to the boiling point and further from 0 to 100 ° C. and from 20 to 50 ° C. It may be heated if necessary to improve the solubility.
- the addition amount of the adhesion control agent is not particularly limited, and may be determined in consideration of the type of the adhesion control agent to be added.
- the addition amount of the adhesion control agent is preferably 0.01 to 5% by weight, preferably 0.02 to 3% by weight based on the raw material monomer of the hydrogel crosslinked polymer, although it depends on the kind of the adhesion control agent to be added. More preferred is 0.03 to 2% by weight.
- this raw material monomer is not a residual raw material monomer, but the raw material monomer used when it adjusts by process (i) Indicates the amount.
- the addition amount of the adhesion control agent is 0.01 to 5% by weight based on the raw material monomer of the hydrogel crosslinked polymer. Below these lower limits, the adhesion control effect is difficult to be confirmed, and when these upper limits are exceeded, the improvement in the adhesion prevention effect is not commensurate with the amount added, which is uneconomical.
- the amount of the adhesion control agent may be within the range of the raw material monomer of the hydrogel crosslinked polymer. 0.01 to 5% by weight, preferably 0.02 to 3% by weight, more preferably 0.03 to 2% by weight, still more preferably 0.1 to 1.8% by weight, ⁇ 1.5% by weight is particularly preferred.
- the adhesion control effect is difficult to be confirmed. If the upper limit is exceeded, the improvement in the adhesion preventing effect is not commensurate with the amount added, which is uneconomical.
- (b-2) an alkylene oxide adduct of a higher alcohol
- (b-3) an alkylene oxide adduct of a polyhydric alcohol fatty acid ester
- (c) a side chain and / or a terminal polyether-modified polysiloxane
- the ammonium salt may be within the above range with respect to the hydrogel crosslinked polymer, but is preferably 0.01 to 5% by weight, more preferably 0.01 to 2% by weight, and It is more preferably from 01 to 1% by weight, particularly preferably from 0.01 to 0.5% by weight.
- the adhesion control effect is difficult to be confirmed. If the upper limit is exceeded, the improvement in the adhesion preventing effect is not commensurate with the amount added, which is uneconomical.
- the raw material monomers are acrylic acid and sodium acrylate.
- the adhesion control agent referred to in the present invention means that the gel water-containing gel after gel pulverization or drying after the gel pulverization is present on the surface of the water-containing gel-like crosslinked polymer and / or the water-containing gel particles.
- the water-absorbent resin powder after gel pulverization has the same particle size (water-absorbent resin powder that passes through a sieve opening of 500 ⁇ m and does not pass through a sieve opening of 425 ⁇ m) as compared to the case of no addition.
- the BET specific surface area is increased, or the vortex is shortened by 3 seconds or more, more preferably 5 seconds or more, and even more preferably 7 seconds or more, compared to the case where vortex is not added. . In this embodiment, comparison is made with “same particle size”.
- the weight average particle size of the hydrogel particles after gel pulverization is smaller than the weight average particle size of the water-absorbing agent.
- the water-absorbing agent to be obtained has a larger shape formed by the primary particles. That is, each of the primary particles comes into gentle contact (the contact area is relatively small). Thereby, the surface area can be increased and desired physical properties can be obtained.
- the value obtained by dividing the weight average particle diameter of the water-absorbing agent by the weight average particle diameter converted to the dried product of the hydrous gel particles after the gel pulverization is preferably 0.40 to 10.0, more preferably 0.00. It is 45 to 5.0, more preferably 0.50 to 4.0.
- the water-containing gel particles obtained by adding the adhesion control agent are controlled for adhesion between the water-containing gel particles, thereby improving the slip. For this reason, it becomes easy to pour the hydrogel particle which passed through the gel grinding
- it is more preferable to use an adhesion control agent because it becomes easier to pour the gel, thereby reducing the thickness unevenness in the dryer, reducing the drying unevenness at the time of drying, and stabilizing the physical properties of the obtained dried product. .
- the strength of the gel dried product obtained through the drying step (iv) is too high, a heavy load is applied to the pulverizer in the pulverizing and classification step (step v) in the pulverizing step of the dried product using a pulverizer.
- Equipment life may be shortened. For this reason, it is better that the strength of the dried product is low.
- the dried product of the hydrogel particles produced by adding the adhesion control agent can also control the adhesion between the water absorbent resin powders, the strength of the dried product of the hydrogel particles can be reduced. From this point of view, it is preferable to use an adhesion control agent.
- the present adhesion control agent is an additive that suppresses excessive adhesion of particles (also referred to as primary particles) constituting the granulated particles, in other words, appropriate adhesion of primary particles occurs and granulation occurs. Is done.
- an adhesion control agent may exist not only near the surface but also inside.
- the distribution of the adhesion control agent may be examined by analyzing the cut surface of the water-absorbing agent particles, or the water-absorbing agent particles by polishing or sputtering. And the change in the content of the adhesion control agent component may be examined.
- the number of particles to be analyzed at that time is preferably evaluated by taking an average of 10 or more of the particles in the vicinity of the weight-average particle diameter.
- the inside said here refers to a part having a depth of 50 ⁇ m or more from the surface of the water-absorbing agent particles.
- the adhesion control agent is one or more compounds selected from a nonionic substance, an amphoteric substance, an anionic substance and a cationic substance
- the nonionic substance is ( a) polyols, (b) modified hydroxyl group of polyols, (c) side chain and / or terminal polyether-modified polysiloxane, (d) alkylene oxide adduct of higher aliphatic amine
- the active substance is (e) an alkylaminobetaine or (f) an alkylamine oxide
- the anionic substance is (g) a sulfate ester salt of a higher alcohol alkylene oxide adduct or (h) an alkyl diphenyl ether disulfonate.
- the cationic substance is (i) an ammonium salt.
- adhesion control agent As a nonionic substance, (A) polyols, (B) a modified hydroxyl group of polyols, (C) side chain and / or terminal polyether-modified polysiloxane, (D) an alkylene oxide adduct of a higher aliphatic amine, As an amphoteric substance, (E) alkylaminobetaine, (F) Alkylamine oxide As an anionic substance, (G) a sulfate ester salt of a higher alcohol alkylene oxide adduct, (H) Alkyl diphenyl ether disulfonate As a cationic substance, (I) An ammonium salt is mentioned.
- polyols having a plurality of hydroxy groups examples include (a-1) non-polymeric polyols and (a-2) polymer polyols.
- Non-polymeric polyol Specifically, as non-polymeric polyols having a plurality of hydroxy groups, ethylene glycol, diethylene glycol, triethylene glycol, glycerin, diglycerin, propanediol, butanediol, pentanediol, hexanediol, octanediol and the like, And triol and tetraol.
- examples of the polymer polyol having a plurality of hydroxy groups include polyethylene glycol, polypropylene glycol, and polyalkylene glycol such as block copolymer or random copolymer of polyethylene glycol and polypropylene glycol.
- the carbon number of the alkylene unit as the repeating unit is preferably C1 to C6, more preferably C2 to C4, and particularly preferably C2 to C3.
- the number of carbon atoms may be expressed by describing a number after C. For example, if the number of carbon atoms is 1, C1 may be described, and if the number of carbon atoms is 10, it may be described as C10).
- polyalkylene glycols such as block copolymers or random copolymers of polyethylene glycol and polypropylene glycol can be easily obtained from the market.
- the following products are preferably exemplified.
- the (a) polyol is (poly) alkylene glycol.
- the adhesion between the crushed hydrogel particles is controlled.
- (B) Modified product of hydroxy group of polyols It is preferable that one or more hydroxy groups are ester-modified and / or ether-modified with a modified hydroxyl group of polyols.
- the ether and / or ester modification is preferably a hydrocarbon group, and the hydrocarbon group is preferably C1 to C30, more preferably C2 to C28, still more preferably C3 to C26, particularly preferably C4 to C24, and C6 to C22. Is most preferred. When the carbon number exceeds C30, the hydrophobicity becomes strong and the surface tension may be lowered, which is not preferable.
- the hydrocarbon group is not limited to a straight chain, but may be a branched or cyclic saturated hydrocarbon group and / or an aromatic hydrocarbon group such as an unsaturated hydrocarbon group, a phenyl group or an alkylphenyl group.
- the hydrocarbon group may have a reactive functional group such as a hydroxy group, an amino group, or a glycidyl group.
- a compound having two or more unsaturated hydrocarbon bonds at the structural end of the compound is not included.
- a di (meth) acrylate compound at both ends of polyethylene glycol is not included.
- the radical-polymerizable adhesion control agent having two or more unsaturated bonds at the structure terminal of the compound is consumed by the reaction during the polymerization in the step (i) or the step (ii) and does not remain during the gel grinding.
- the addition in step (iii) may not be sufficiently consumed and may remain in the final product and cause coloring.
- adhesion control agent having a substituent such as a hydroxyl group, amino group, or glycidyl group having reactivity with a carboxyl group, which is a functional group of the water-absorbent resin
- the adhesion control agent is present on the gel particle surface. While reducing excessive adhesion of the gel during pulverization, cross-linking between the gel particles occurs due to the reaction of the functional group during drying, and the collapse of the granulated particles during swelling can be further suppressed.
- modified hydroxyl group of polyols examples include (b-1) glycidyl-modified polyols, (b-2) alkylene oxide adducts of higher alcohols, and (b-3) alkylene oxide adducts of polyhydric alcohol fatty acid esters. It is done.
- the hydroxy-modified product of the polyol (b) is (b-1) a glycidyl-modified polyol, (b-2) an alkylene oxide adduct of a higher alcohol, or (b -3) an alkylene oxide adduct of a polyhydric alcohol fatty acid ester, wherein (b-1) is one in which at least one terminal of the (poly) alkylene glycol is modified with a glycidyl group, (b-2) Is one in which one end of (poly) alkylene glycol is modified with a substituent having a C1-C30 hydrocarbon, and (b-3) is an alkylene oxide in at least one of the hydroxy groups of the polyhydric alcohol.
- water-soluble (poly) alkylene glycol diglycidyl ether such as ethylene glycol diglycidyl ether, diethylene glycol diglycidyl ether, polyethylene glycol diglycidyl ether, propylene glycol diglycidyl ether, polypropylene glycol diglycidyl ether, hexanediol diglycidyl ether
- water-soluble polyglycidyl ethers of polyols such as ether, glycerol polyglycidyl ether, trimethylolpropane polyglycidyl ether, pentaerythritol polyglycidyl ether, diglycerol polyglycidyl ether, polyglycerol polyglycidyl ether, sorbitol polyglycidyl ether and the like.
- the adhesion control agent which is a compound in which two or more of the hydroxy groups of (poly) alkylene glycol are modified with glycidyl groups, is added in the step (i) or step (ii) and consumed by the reaction during polymerization. There is a possibility that it may decrease or not remain during pulverization, and it is preferably added after step (ii).
- the addition amount of the glycidyl-modified polyol may be in the range of the addition amount of the above adhesion control agent, but is 0.01 to 5% by weight based on the raw material monomer of the hydrogel crosslinked polymer. Is preferable, 0.02 to 3% by weight is more preferable, and 0.03 to 2% by weight is further preferable.
- Glycidyl-modified polyols can be easily obtained from the market.
- the following products are preferably exemplified.
- the alkylene oxide adduct of a higher alcohol is one in which one end of (poly) alkylene glycol is modified with a substituent having a C1-C30 hydrocarbon, and the general formula is shown in “Chemical Formula 1”.
- the hydrocarbon group having C1-C30 is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or an unsaturated hydrocarbon group, an aromatic carbon group such as an alkylphenyl group or an alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as a hydrogen group or a naphthyl group. Furthermore, the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C2 to C28, further preferably C3 to C26, particularly preferably C4 to C24, and most preferably C6 to C22. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- AO is a repeating unit that can also be represented by C n H 2n O (n is a natural number), and the carbon number is preferably C1 to C6, more preferably C1 to C3, still more preferably C2 to C3, and C2 or ethylene. It is particularly preferred that CH 2 CH 2 O, which is a structure derived from oxide addition or ethylene glycol condensation, is a repeating unit.
- the repeating unit may be a polymer having the same carbon number, or may be a block polymer or a random polymer having different units.
- A is the number of repeating units of AO, preferably 1 to 1000, more preferably 2 to 500, and even more preferably 2 to 300. If the number of repeating units exceeds 1000, the viscosity increases and the addition becomes non-uniform.
- the HLB of the alkylene oxide adduct of higher alcohol is preferably 10 to 20, more preferably 12 to 20 and even more preferably 14 to 20 by the Griffin method. If it is below the above range, the hydrophobicity becomes strong, so that the GCA is lowered, the absorption rate is slow, or the surface tension is significantly lowered, which is not preferable. In the above HLB calculation method, 20 is the upper limit value.
- the addition amount of the alkylene oxide adduct of the higher alcohol may be in the range of the addition amount of the above adhesion control agent, but is 0.01% relative to the raw material monomer of the hydrogel crosslinked polymer. Is preferably 5 to 5% by weight, more preferably 0.01 to 2% by weight, and still more preferably 0.01 to 0.5% by weight.
- the alkylene oxide adduct of higher alcohol can be easily obtained from the market.
- the following products are preferably exemplified.
- polyhydric alcohol fatty acid ester alkylene oxide adduct The ethylene oxide adduct of polyhydric alcohol fatty acid ester is modified with a substituent having at least one polyhydric alcohol modified with (poly) alkylene glycol and at least one having a C1-C30 hydrocarbon via an ester bond It is what has been.
- the polyhydric alcohol include glycerin, pentaerythritol, sorbitol, sorbitan, and saccharides.
- Preferred examples include alkylene oxide adducts of glycerin fatty acid monoesters and alkylene oxide adducts of sorbitan fatty acid monoesters.
- the general formula of alkylene oxide adducts of glycerin fatty acid monoesters is shown in “Chemical Formula 2”. Since the alkylene oxide adduct of sorbitan fatty acid monoester contains structural isomers, it is divided into general formulas “Chemical Formula 3” and “Chemical Formula 4”.
- the hydrocarbon group having C1-C30 is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or an unsaturated hydrocarbon group, an aromatic carbon group such as an alkylphenyl group or an alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as a hydrogen group or a naphthyl group. Furthermore, the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C2 to C28, further preferably C3 to C26, particularly preferably C4 to C24, and most preferably C6 to C22. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- a 1 O and A 2 O are repeating units that can also be represented by C n H 2n O (n is a natural number), and the carbon number is preferably C1 to C6, more preferably C1 to C3, and C2 to C3 is Further preferred is CH 2 CH 2 O, which is a structure derived from C2, that is, ethylene oxide addition or ethylene glycol condensation.
- the repeating unit may be a polymer having the same carbon number, or may be a block polymer or a random polymer having different units.
- a 1 O and A 2 O may be different or the same.
- a and b are the number of repeating units of A 1 O and A 2 O, and the sum of a + b is preferably from 1 to 1000, more preferably from 2 to 500, and even more preferably from 2 to 300. a and b may be different or the same. If the sum of a + b exceeds 1000, the viscosity increases and the addition becomes non-uniform, which is not preferable.
- the hydrocarbon group having C1-C30 is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or an unsaturated hydrocarbon group, an aromatic carbon group such as an alkylphenyl group or an alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as a hydrogen group or a naphthyl group. Furthermore, the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C2 to C28, further preferably C3 to C26, particularly preferably C4 to C24, and most preferably C6 to C22. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- a 1 O, A 2 O, and A 3 O are repeating units that can also be represented by C n H 2n O (n is a natural number), and the carbon number is preferably C1 to C6, more preferably C1 to C3, more preferably C2 ⁇ C3, and particularly preferably C2 clogging ethylene oxide adduct or a structure derived from the condensation of ethylene glycol CH 2 CH 2 O.
- the repeating unit may be a polymer having the same carbon number, or may be a block polymer or a random polymer having different units.
- a 1 O, A 2 O, and A 3 O may be different or the same.
- A, b and c are the number of repeating units of the above repeating unit, and the sum of a + b + c is preferably from 1 to 1000, more preferably from 2 to 500, and even more preferably from 2 to 300. a and b may be different or the same. When the sum of a + b + c exceeds 1000, the viscosity increases and the addition becomes non-uniform, which is not preferable.
- A, b and c are average repeating units of polyethylene glycol, and the total amount of a + b + c is preferably 1 to 300, more preferably 2 to 200, and even more preferably 2 to 100. a, b and c may be different or the same.
- the HLB of the alkylene oxide adduct of polyhydric alcohol fatty acid ester is preferably 10 to 20, more preferably 12 to 20, and still more preferably 14 to 20 by the Griffin method. If it is below the above range, the hydrophobicity becomes strong, so that the GCA is lowered, the absorption rate is slow, or the surface tension is significantly lowered, which is not preferable. In the above HLB calculation method, 20 is the upper limit value.
- the addition amount of the alkylene oxide adduct of the polyhydric alcohol fatty acid ester may be within the range of the addition amount of the above adhesion control agent, but with respect to the raw material monomer of the hydrogel crosslinked polymer, 0.01 to 5% by weight is preferred, 0.01 to 2% by weight is more preferred, and 0.01 to 0.5% by weight is even more preferred.
- the alkylene oxide adduct of a polyhydric alcohol fatty acid ester can be easily obtained from the market.
- the following products are preferably exemplified.
- the HLB of the polyether-modified polysiloxane is preferably 10 to 20, more preferably 12 to 20, more preferably 14 to 20 by the Griffin method. If it is below the above range, the hydrophobicity becomes strong, so that the GCA is lowered, the absorption rate is slow, or the surface tension is significantly lowered, which is not preferable. In the above HLB calculation method, 20 is the upper limit value.
- the addition amount of the side chain and / or terminal polyether-modified polysiloxane may be within the range of the addition amount of the above-mentioned adhesion control agent, but is 0 with respect to the raw material monomer of the hydrogel crosslinked polymer. 0.01 to 5% by weight is preferred, 0.01 to 2% by weight is more preferred, and 0.01 to 0.5% by weight is even more preferred.
- the polyether-modified siloxane can be easily obtained from the market.
- the following products are preferably exemplified.
- the alkylene oxide adduct of (d) a higher aliphatic amine is obtained by adding an alkylene oxide to two hydrogens of a primary amine having a C1-C30 hydrocarbon. . With this configuration, the adhesion between the crushed water-containing gels can be controlled.
- An alkylene oxide adduct of a higher aliphatic amine is a compound in which two hydrogens of a primary amine having a C1-C30 hydrocarbon group are modified with (poly) alkylene glycol. Show.
- the hydrocarbon group having C1-C30 is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or an unsaturated hydrocarbon group, an aromatic carbon group such as an alkylphenyl group or an alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as a hydrogen group or a naphthyl group. Furthermore, the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C2 to C28, further preferably C3 to C26, particularly preferably C4 to C24, and most preferably C6 to C22. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- a 1 O and A 2 O are repeating units that can also be represented by C n H 2n O (n is a natural number), and the carbon number is preferably C1 to C6, more preferably C1 to C3, and C2 to C3 is Further preferred is CH 2 CH 2 O, which is a structure derived from C2, that is, ethylene oxide addition or ethylene glycol condensation.
- the alkylene unit may be a polymer having the same carbon number, or may be a block polymer or a random polymer having different units.
- a 1 O and A 2 O may be different or the same.
- a and b are repeating units of an alkylene glycol unit, and the sum of a + b is preferably from 1 to 1000, more preferably from 2 to 500, and even more preferably from 2 to 300. a and b may be different or the same. If the sum of a + b exceeds 1000, the viscosity increases and the addition becomes non-uniform, which is not preferable.
- the HLB of the alkylene oxide adduct of a higher aliphatic amine is preferably 10 to 20, more preferably 12 to 20 and even more preferably 14 to 20 by the Griffin method. If it is below the above range, the hydrophobicity becomes strong, so that the GCA is lowered, the absorption rate is slow, or the surface tension is significantly lowered, which is not preferable. In the above HLB calculation method, 20 is the upper limit value.
- the addition amount of the alkylene oxide adduct of the higher aliphatic amine may be within the range of the addition amount of the above adhesion control agent, but is 0.01% with respect to the raw material monomer of the hydrogel crosslinked polymer. Is preferably 5 to 5% by weight, more preferably 0.01 to 2% by weight, and still more preferably 0.01 to 0.5% by weight.
- Alkylene oxide adducts of higher aliphatic amines can be easily obtained from the market.
- the following products are preferably exemplified.
- the hydrocarbon group having 1 to 30 carbon atoms is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or unsaturated hydrocarbon group, an aromatic group such as an alkylphenyl group or an alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as a hydrocarbon group or a naphthyl group.
- the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C2 to C28, further preferably C3 to C26, particularly preferably C4 to C24, and most preferably C6 to C22. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- R 2 and R 3 are hydrogen, or the hydrocarbon group having 1 to 30 carbon atoms is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or unsaturated hydrocarbon group, alkylphenyl group or alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as an aromatic hydrocarbon group such as naphthyl group. Furthermore, the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C1 to C25, and still more preferably C1 to C20. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- R 1 , R 2 and R 3 may be different or the same.
- the structure of X is not particularly limited except that it contains C1 or more carbon atoms.
- anion portion (Z) examples include carboxylate, sulfonate, and phosphate.
- the hydrocarbon group having 1 to 30 carbon atoms is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or unsaturated hydrocarbon group, an aromatic group such as an alkylphenyl group or an alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as a hydrocarbon group or a naphthyl group.
- the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C2 to C28, further preferably C3 to C26, particularly preferably C4 to C24, and most preferably C6 to C22. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- R 2 is hydrogen, or the hydrocarbon group having 1 to 30 carbon atoms is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or unsaturated hydrocarbon group, an aromatic group such as an alkylphenyl group or an alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as an aromatic hydrocarbon group or a naphthyl group.
- the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C1 to C25, and still more preferably C1 to C20. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- R 1 and R 2 may be different or the same.
- the structure of X is not particularly limited except that it contains C1 or more carbon atoms.
- anion portion (Z) examples include carboxylate, sulfonate, and phosphate.
- Alkylamine oxide has a cationic group and an anionic group at adjacent positions in the same molecule, the cationic group is a secondary to quaternary ammonium, At least one of them is modified with a substituent having a C1-C30 hydrocarbon group, and the general formula is shown in “Chemical Formula 8”.
- the hydrocarbon group having 1 to 30 carbon atoms is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or unsaturated hydrocarbon group, an aromatic group such as an alkylphenyl group or an alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as a hydrocarbon group or a naphthyl group.
- the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C2 to C28, further preferably C3 to C26, particularly preferably C4 to C24, and most preferably C6 to C22. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- R 2 and R 3 are hydrogen, or the hydrocarbon group having 1 to 30 carbon atoms is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or unsaturated hydrocarbon group, alkylphenyl group or alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as an aromatic hydrocarbon group such as naphthyl group. Furthermore, the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C1 to C25, and still more preferably C1 to C20. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- R 1 , R 2 and R 3 may be different or the same.
- the addition amount of (e) alkylaminobetaine and (f) alkylamine oxide may be in the range of the addition amount of the above-mentioned adhesion control agent. It is preferably from 01 to 5% by weight, more preferably from 0.01 to 2% by weight, still more preferably from 0.01 to 0.5% by weight.
- Alkylaminobetaine and (f) Alkylamine oxide can be easily obtained from the market.
- the following products are preferably exemplified.
- Amphitol 20BS Amphitol 24B (desalted product of 20BS), Amphitol 86B, Amphitol 20N, Amphitol 20YB, Amphitol 20AB, Amphitol 55AB, Amphitol 20HD Made by Daiichi Kogyo Seiyaku Co., Ltd .: Amogen S-H, Amogen K, Amogen LB-C, Amogen CB-H, Amogen HB-C, Amogen AOL Made by Adeka Corporation: Adeka Anhout PB-30L, Adeka Anhout AB-35L NOF Corporation: Nissan Anon BF, Nissan Anon BL, Nissan Anon BL-SF, Nissan Anon BDF-R, Nissan Anon BDF-SF, Nissan Anon BDC-SF, Nissan Anon BDL-SF, Nissan Anon GLM-R, Unisafe A-LM, Unisafe A-SM, Unisafe A-LE Made in Japan emulsifier: Texnol R2.
- (G) Sulfuric acid ester salt of higher alcohol ethylene oxide adduct) According to a preferred embodiment of the present invention, (g) the sulfate ester salt of a higher alcohol alkylene adduct is modified with a substituent having one end of (poly) alkylene glycol having a C1-C30 hydrocarbon, and The other end is a sulfate ester salt. With this configuration, the adhesion between the crushed water-containing gels can be controlled.
- the sulfate ester salt of a higher alcohol ethylene oxide adduct is one in which (poly) alkylene glycol is modified with a substituent having a C1-C30 hydrocarbon and the other is a sulfate ester salt. 9 ”.
- the hydrocarbon group having C1-C30 is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or an unsaturated hydrocarbon group, an aromatic carbon group such as an alkylphenyl group or an alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as a hydrogen group or a naphthyl group. Furthermore, the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C2 to C28, further preferably C3 to C26, particularly preferably C4 to C24, and most preferably C6 to C22. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- AO is a repeating unit that can also be represented by C n H 2n O (n is a natural number), and the carbon number is preferably C1 to C6, more preferably C1 to C3, still more preferably C2 to C3, and C2 or ethylene. Particularly preferred is CH 2 CH 2 O, which is a structure derived from oxide addition or condensation of ethylene glycol.
- the repeating unit may be a polymer having the same carbon number, or may be a block polymer or a random polymer having different units.
- A is the number of repeating units of AO, preferably 1 to 1000, more preferably 2 to 500, and even more preferably 2 to 300. If the number of repeating units exceeds 1000, the viscosity increases and the addition becomes non-uniform.
- M includes alkali metals (Li, Na, K) and ammonium ions.
- the addition amount of the sulfate ester salt of the higher alcohol alkylene oxide adduct may be within the range of the addition amount of the above-mentioned adhesion control agent. It is preferably from 01 to 5% by weight, more preferably from 0.01 to 2% by weight, still more preferably from 0.01 to 0.5% by weight.
- the sulfate ester salt of the higher alcohol alkylene oxide adduct can be easily obtained from the market.
- the following products are preferably exemplified.
- the hydrocarbon group having C1-C30 is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or an unsaturated hydrocarbon group, an aromatic carbon group such as an alkylphenyl group or an alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as a hydrogen group or a naphthyl group. Furthermore, the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C2 to C28, further preferably C3 to C26, particularly preferably C4 to C24, and most preferably C6 to C22. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- M includes alkali metals (Li, Na, K) and ammonium ions.
- the addition amount of the alkyl diphenyl ether disulfonate may be within the range of the addition amount of the above adhesion control agent, but is 0.01 to 5% by weight based on the raw material monomer of the hydrogel crosslinked polymer. It is preferably 0.01 to 2% by weight, more preferably 0.01 to 0.5% by weight.
- Alkyl diphenyl ether disulfonate can be easily obtained from the market.
- the following products are preferably exemplified.
- the ammonium salt is one in which at least one hydrogen of the ammonium salt is modified with a substituent having a C1-C30 hydrocarbon, and the general formula is shown in “Chemical Formula 11”.
- the hydrocarbon group having 1 to 30 carbon atoms is not limited to a straight chain, but is a branched or cyclic saturated hydrocarbon group and / or unsaturated hydrocarbon group, an aromatic group such as an alkylphenyl group or an alkylbenzyl group. It may be a polycyclic aromatic hydrocarbon such as a hydrocarbon group or a naphthyl group.
- the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C2 to C28, further preferably C3 to C26, particularly preferably C4 to C24, and most preferably C6 to C22. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- R 2 , R 3 and R 4 are hydrogen or the above-mentioned C1-C30 hydrocarbon group is not limited to a straight chain, but is branched or cyclic saturated hydrocarbon group and / or unsaturated hydrocarbon group, alkylphenyl group, It may be an aromatic hydrocarbon group such as an alkylbenzyl group or a polycyclic aromatic hydrocarbon such as a naphthyl group. Furthermore, the said hydrocarbon group may have reactive functional groups, such as a hydroxyl group, an amino group, and a glycidyl group, and may have an ether bond, an ester bond, a urethane bond, and an amide bond.
- the hydrocarbon group preferably has C1 to C30, more preferably C1 to C25, and still more preferably C1 to C20. If the hydrocarbon group exceeds C30, the hydrophobicity becomes too strong and the surface tension of the water-absorbing agent is significantly reduced, which is not preferable.
- R 1 , R 2 , R 3 , and R 4 may be different or the same.
- N is a counter anion of an ammonium cation, and examples thereof include halogen ions, carboxylate ions, sulfonate ions, hydroxy ions, BF 4 ⁇ , PF 6 ⁇ , ClO 4 ⁇ , AsF 6 ⁇ , and SbF 6 ⁇ .
- the addition amount of the ammonium salt may be in the range of the addition amount of the above-mentioned adhesion control agent, but is preferably 0.01 to 5% by weight with respect to the raw material monomer of the hydrogel crosslinked polymer, 0.01 to 2% by weight is more preferable, and 0.01 to 0.5% by weight is more preferable.
- ammonium salt can be easily obtained from the market, and for example, the following products are preferably exemplified.
- hydrophilic unit of the adhesion control agent interacts with the inside or / and the surface of the particulate absorbent that is hydrophilic, and is difficult to elute from the absorbent. For this reason, it is thought that the fall of the surface tension of an absorber is suppressed and the return amount of the liquid from a water absorption body accompanying a surface tension fall can be suppressed.
- the hydrophobic unit of the adhesion control agent collects on the surface layer of the hydrogel particles after gel pulverization (portion close to the air layer), thereby suppressing the adhesion between the crushed gels It is considered that the gel particles can be prevented from re-adhering to each other. On the other hand, it is considered that the hydrophilic unit acts as an anchor for staying on the surface of the gel particle by the interaction with the gel.
- the adhesion control agent used in the present application is preferably polyethylene glycol having only a hydrophilic unit without having a hydrophobic unit.
- polyethylene glycol consisting only of hydrophilic units interacts with the inside or / and the surface of the hydrophilic particulate absorbent and is particularly difficult to elute from the absorbent.
- the optimum range of the addition amount is wide and insensitive to the fluctuation of the addition amount because the operation control range at the time of manufacture can be widened.
- the carboxyl groups that are functional groups of the water-absorbent resin and the hydroxyl groups at both ends of the polyethylene glycol react to form primary particles constituting the granulated particles.
- This is preferable because it is crosslinked and has an effect of suppressing the collapse of the granulated particles when they absorb water and swell.
- the main effect of using the adhesion control agent is the adhesion control between the primary particles of the hydrogel particles. Also.
- the incidental effects include an improvement in the fluidity of the hydrogel and a reduction in the strength of the dried product of the hydrogel particles.
- an adhesion control agent because the load of the gel crusher can be reduced due to its lubricating effect, the productivity is further increased, and the deterioration of the gel during gel crushing is suppressed.
- the HLB of the nonionic substance is preferably 10 to 20, more preferably 12 to 20, and still more preferably 14 to 20 according to the Griffin method. If it is below the above range, the hydrophobicity becomes strong, so that the GCA is lowered, the absorption rate is slow, or the surface tension is significantly lowered, which is not preferable. In the above HLB calculation method, 20 is the upper limit value.
- the molecular weight (weight average molecular weight) of the adhesion control agent is not particularly limited, but in order to avoid adverse effects such as the effect being exerted with a smaller addition amount or the absorption capacity being lowered, the molecular weight (hereinafter referred to as adhesion control).
- the weight average molecular weight) is preferably in the range of 100 to 1,000,000, more preferably 150 to 500,000, further preferably 200 to 500,000, 300 to 300,000, and more preferably 500 to More preferably, it is 200,000, particularly preferably 1,000 to 50,000 or less, and most preferably 30,000 or less.
- polyethylene glycol is used as an adhesion control agent, the desired effect of the present invention can be efficiently achieved by having a weight average molecular weight of 500 or more.
- the weight average molecular weight of the nonionic substance or the sulfate ester salt of the (g) higher alcohol ethylene oxide adduct is independently 200 to 200,000. According to this embodiment, adhesion between the crushed water-containing gels can be controlled. In this embodiment, it is particularly preferably 50,000 or less, and most preferably 30,000 or less.
- step (iv) This step is a step of obtaining a dry polymer (dried product) by drying the hydrogel (hydrated gel particles) obtained through the polymerization step and the like.
- the said polymerization process is aqueous solution polymerization
- pulverization fine-graining
- the dried polymer (aggregate) (dried product) obtained in the drying step may be supplied to the pulverization step as it is.
- the drying method in the present invention is not particularly limited, and various methods can be employed. Specific examples include heat drying, hot air drying, vacuum drying, infrared drying, microwave drying, azeotropic dehydration drying with a hydrophobic organic solvent, and high humidity drying using high-temperature steam. Or 2 types can also be used together.
- the finely pulverized particulate hydrogel adheres to each other, and granulated particles are formed.
- granulation refers to forming particles larger than the original particles (primary particles) by adhering the particles by physical and chemical techniques, and adhering the primary particles to each other. Means that it is loose or point contact.
- the granulated product or granulated particle of the present invention is a particle to which primary particles are attached in a state that can be clearly distinguished, as shown in the drawings of the examples.
- the degree of adhesion is controlled by the presence or absence and type of the adhesion control agent, and GCA, FGBP, vortex, AAP, and the like can be controlled.
- the drying temperature in the present invention is preferably 100 to 300 ° C, more preferably 150 to 250 ° C. Further, the drying time depends on the surface area and water content of the water-containing gel particles, the type of dryer, etc., and for example, 1 minute to 5 hours is preferable, and 5 minutes to 1 hour is more preferable.
- the solid content of the water-absorbent resin (dried product) obtained from loss on drying (1 g of powder or particles is dried at 180 ° C. for 3 hours) is preferably 80% by weight or more, more preferably 85 to 99% by weight, 98% by weight is more preferable, and 92 to 97% by weight is particularly preferable.
- This step is a step of pulverizing and / or classifying the dried polymer (dried product) obtained in the drying step to obtain a water absorbent resin powder having a specific particle size.
- the (2-3) gel pulverization step is different in that the object to be pulverized has undergone a drying step.
- This step is performed before and / or after the (2-6) surface crosslinking step, preferably (2-6) before the surface crosslinking step, and (2-6) before and after the surface crosslinking step. It may be performed at least twice.
- Examples of the equipment (pulverizer) used in the pulverization process of the present invention include a high-speed rotary pulverizer such as a roll mill, a hammer mill, a screw mill, and a pin mill, a vibration mill, a knuckle type pulverizer, and a cylindrical mixer. , Used as needed.
- the weight average particle diameter (D50) of the water-absorbent resin powder before surface crosslinking is 300 to 500 ⁇ m from the viewpoints of handleability (especially handleability under moisture absorption), GCA, FGBP, water absorption rate, water absorption magnification under pressure, and the like. Preferably, it is 310 to 480 ⁇ m, more preferably 320 to 450 ⁇ m.
- the content of fine particles having a particle diameter of less than 150 ⁇ m as defined by the standard sieve classification is preferably as small as possible, preferably 0 to 5% by weight, more preferably 0 to 3% by weight, and more preferably 0 to 2% with respect to the entire water-absorbent resin powder. More preferred is weight percent.
- 0 to 5% by weight is preferable with respect to the entire water-absorbent resin powder, more preferably 0 to 3% by weight.
- 0 to 1% by weight is more preferable.
- the proportion of particles having a particle diameter of 150 ⁇ m or more and less than 850 ⁇ m is preferably 90% by weight or more, and 95% by weight or more with respect to the entire water-absorbent resin powder in terms of GCA, FGBP, water absorption speed, water absorption capacity under pressure, and the like. Is more preferably 98% by weight or more, and particularly preferably 99% by weight or more (the upper limit is 100% by weight).
- the logarithmic standard deviation ( ⁇ ) of the particle size distribution is preferably 0.20 to 0.50, more preferably 0.25 to 0.45, and still more preferably 0.30 to 0.40.
- this range is finer in the gel pulverization stage, and the dried water-absorbent resin particles are highly granulated.
- the surface area can be based on the BET specific surface area described later in the examples.
- the BET specific surface area of the water-absorbent resin powder of the same particle size is preferably 29 m 2 / kg or more, more preferably 30 m 2 / kg or more Preferably, 31 m 2 / kg or more is more preferable.
- the water absorbent resin powder having a particle size fraction of 300 ⁇ m or more and less than 425 ⁇ m was represented as a water absorbent resin powder (425/300).
- the particle size can be controlled at the time of polymerization, gel pulverization, pulverization after drying, or classification, and particularly preferably at the time of pulverization and / or classification after drying.
- the particle size is measured using a JIS standard sieve according to the method defined in International Publication No. 2004/69915 pamphlet and EDANA-ERT420.2-02.
- the above particle size can be applied to the water-absorbent resin particles after surface crosslinking and the particulate water-absorbing agent as the final product.
- the fine particles generated by controlling the particle size may be discarded or, as is conventionally known, a method for recovering into an aqueous monomer solution before polymerization (International Publication No. 92 / No. 001008, No. 92/020723) and a method of recovering to a hydrous gel during polymerization (International Publication Nos. 2007/074167, 2009/109563, 2009/153196, 2010/006937) ).
- the shape of the water-absorbent resin powder of the present invention is not particularly limited, such as a spherical shape, a fiber shape, a rod shape, a substantially spherical shape, a flat shape, an indefinite shape, a granulated particle shape, and a particle having a porous structure.
- the water-absorbing agent of the present invention preferably has CRC ⁇ 28 g / g
- the CRC of the water-absorbent resin powder before surface cross-linking is also preferably 28 g / g or more, more preferably 30 g / g or more.
- the amount of the crosslinking agent at the time of the polymerization may be appropriately adjusted.
- the CRC of the water-absorbent resin powder before surface crosslinking is suitably adjusted in the range of preferably 30 to 60 g / g, more preferably 32 to 55 g / g and 33 to 50 g / g, such as the amount of crosslinking agent, polymerization temperature and drying temperature.
- high temperature polymerization and drying tend to improve the CRC, and an increase in the amount of the crosslinking agent results in a decrease in the CRC.
- the CRC of the inventive water-absorbing agent can be controlled.
- step (vi) This step further includes a surface cross-linking agent addition step shown in the following (2-6-1) and a heat treatment step shown in (2-6-2).
- the water-absorbing resin powder containing the surface cross-linking agent used in the surface cross-linking step is prepared by mixing the water-absorbing resin powder and the surface cross-linking agent. It is a process to do.
- surface cross-linking is performed by adding an organic surface cross-linking agent described later, polymerizing a monomer (polymerizable surface cross-linking agent) on the surface of the water-absorbent resin powder, or a radical polymerization initiator such as persulfate (in a broad sense). It is carried out by addition of a surface cross-linking agent), heating, ultraviolet irradiation, or the like. In the present invention, it is preferable to add an organic surface cross-linking agent to the water absorbent resin powder obtained above.
- Organic surface cross-linking agent As the organic surface cross-linking agent that can be used in the present invention, from the viewpoint of physical properties of the water-absorbing resin particles obtained, carboxyl groups that are functional groups of poly (meth) acrylic acid (salt) -based water-absorbing resin particles, and dehydration esterification An organic compound having a reactive group such as a hydroxy group and / or an amino group that undergoes a reaction or dehydration amidation reaction is preferable.
- the organic compound is not limited to an alcohol compound or an amine compound having a hydroxy group or an amino group directly, and even if it is a cyclic compound such as an alkylene carbonate compound or an oxazolidinone compound, a reactive group that generates a hydroxy group or an amino group and A compound having a reactive group that directly reacts with the carboxyl group is also included.
- organic surface crosslinking agent examples include polyhydric alcohol compounds, epoxy compounds, polyvalent amine compounds or condensates thereof with haloepoxy compounds, oxazoline compounds, (mono, di, or poly) oxazolidinone compounds, oxetane compounds, alkylene carbonate compounds, and the like.
- a polyhydric alcohol compound, an alkylene carbonate compound, and an oxazolidinone compound are more preferable.
- organic surface crosslinking agent examples include (di, tri, tetra, poly) ethylene glycol, (di, poly) propylene glycol, 1,3-propanediol, 2,2,4-trimethyl-1,3-pentanediol.
- Poly glycerin, 2-butene-1,4-diol, 1,4-butanediol, 1,3-butanediol, 1,5-pentanediol, 1,6-hexanediol, trimethylolpropane, di- or tri Polyalcohol compounds (polyhydric alcohol) such as ethanolamine, pentaerythritol, sorbitol; Epoxy compounds such as (poly) ethylene glycol diglycidyl ether, (di, poly) glycerol polyglycidyl ether, glycidol; Oxazoline compounds such as 2-oxazolidone, N-hydroxyethyl-2-oxazolidone, 1,2-ethylenebisoxazoline; 1,3-dioxolan-2-one (ie ethylene carbonate), 4-methyl-1,3-dioxolan-2-one, 4,5-dimethyl-1,3-dioxolan-2-one,
- the polyhydric alcohol is preferably a polyhydric alcohol having 2 to 8 carbon atoms, more preferably a polyhydric alcohol having 3 to 6 carbon atoms, and further preferably a polyhydric alcohol having 3 or 4 carbon atoms.
- diols are preferred, and examples include ethylene glycol, propylene glycol, 1,3-propanediol, and 1,4-butanediol, and a polyvalent selected from propylene glycol, 1,3-propanediol, and 1,4-butanediol. Alcohol is preferred.
- epoxy compound a polyglycidyl compound is preferable, ethylene glycol diglycidyl ether is preferably used, As the oxazoline compound, 2-oxazolidinone is preferably used, As the alkylene carbonate compound, 1,3-dioxolan-2-one (that is, ethylene carbonate) is preferably used.
- a combination of two or more compounds selected from polyhydric alcohol compounds, epoxy compounds, oxazoline compounds, and alkylene carbonate compounds is preferable, a combination of a polyhydric alcohol and the organic surface crosslinking agent other than the polyhydric alcohol is preferable, a combination of a polyhydric alcohol and an epoxy compound or an alkylene carbonate compound is more preferable, and a polyhydric alcohol and an alkylene carbonate. More preferred are combinations with compounds.
- the ratio (weight ratio) is 1 except for the polyhydric alcohol: polyhydric alcohol. : 100 to 100: 1 is preferable, 1:50 to 50: 1 is more preferable, and 1:30 to 30: 1 is more preferable.
- the total amount of the organic surface cross-linking agent added is preferably 0.001 to 15 parts by weight, and 0.01 to 5 parts by weight with respect to 100 parts by weight of the water absorbent resin powder before addition. More preferably it is.
- the total amount of the polyhydric alcohol compound is preferably 0.001 to 10 parts by weight, and more preferably 0.01 to 5 parts by weight with respect to 100 parts by weight of the water absorbent resin powder before addition.
- the total amount of compounds other than polyhydric alcohol is preferably 0.001 to 10 parts by weight, and more preferably 0.01 to 5 parts by weight with respect to 100 parts by weight of the water-absorbent resin powder.
- the organic surface cross-linking agent is preferably added as an aqueous solution.
- the amount of water used in the aqueous solution is preferably 0.5 to 20 parts by weight, more preferably 0.5 to 10 parts by weight, based on 100 parts by weight of the water absorbent resin powder before the addition treatment. Note that the amount of water includes crystallization water, hydration water, and the like of the surface cross-linking agent.
- a hydrophilic organic solvent may be added to the organic surface crosslinking agent aqueous solution.
- the amount of the hydrophilic organic solvent is 0% by weight with respect to 100 parts by weight of the water absorbent resin powder before the addition treatment. More than 10 parts by weight and preferably less than 5 parts by weight, more preferably more than 0 parts by weight and 5 parts by weight.
- the hydrophilic organic solvent include primary alcohols having 1 to 4 carbon atoms, further 2 to 3 carbon atoms, and other lower ketones having 4 or less carbon atoms such as acetone. Volatile alcohols having a temperature of less than 100 ° C., more preferably less than 100 ° C. are more preferable because they do not leave a residue because they volatilize during the surface crosslinking treatment.
- lower alcohols such as methyl alcohol, ethyl alcohol, n-propyl alcohol, isopropyl alcohol, n-butyl alcohol, isobutyl alcohol and t-butyl alcohol; ketones such as acetone; dioxane, tetrahydrofuran, methoxy (poly ) Ethers such as ethylene glycol; Amides such as ⁇ -caprolactam and N, N-dimethylformamide; Sulphoxides such as dimethyl sulfoxide; Polyhydric alcohols such as polyoxypropylene and oxyethylene-oxypropylene block copolymers Is mentioned.
- the water-insoluble fine particles and the surfactant are added to 100 parts by weight of the water-absorbent resin powder before the addition treatment within a range not impeding the effects of the present invention.
- more than 0 parts by weight and 10 parts by weight or less preferably more than 0 parts by weight and 5 parts by weight or less, more preferably more than 0 parts by weight and 1 part by weight or less can coexist.
- the surfactant used is disclosed in US Pat. No. 7,473,739.
- the concentration of the surface cross-linking agent in the surface cross-linking agent aqueous solution is appropriately determined. From the viewpoint of physical properties, 1-80 wt%, further 5-60 wt%, 10-40 wt%, 15-30 wt% aqueous solution and Is done. The remainder contains the hydrophilic organic solvent and other components.
- the temperature of the organic surface cross-linking agent aqueous solution is appropriately determined from the solubility of the organic surface cross-linking agent used, the viscosity of the aqueous solution, etc., but is preferably ⁇ 10 to 100 ° C., more preferably 5 to 70 ° C., and more preferably 10 to 65. ° C is more preferable, and a range of 25 to 50 ° C is particularly preferable.
- the cyclic compound may hydrolyze (for example, decomposition from ethylene carbonate to ethylene glycol, decomposition from oxazolidinone to ethanolamine), water or hydrophilic organics before mixing or reacting with the water absorbent resin powder. This is not preferable because the solvent may be volatilized and the mixing property may decrease.
- the surface cross-linking agent solution may solidify or the surface cross-linking agent may precipitate, which is not preferable.
- the surface crosslinking agent solution contains an acid or a base in addition to the organic surface crosslinking agent, the hydrophilic organic solvent, the surfactant and the water-insoluble fine particles in order to promote the reaction and uniform mixing of the surface crosslinking agent. You may go out.
- an organic acid or a salt thereof, an inorganic acid or a salt thereof, or an inorganic base is used, and is 0 to 10 parts by weight, more preferably 0 with respect to 100 parts by weight of the water absorbent resin powder before the addition treatment. 0.001 to 5 parts by weight, and more preferably 0.01 to 3 parts by weight.
- the organic acid is a water-soluble organic acid having 1 to 6 carbon atoms, more preferably 2 to 4 carbon atoms, a water-soluble saturated organic acid, particularly a saturated organic acid containing a hydroxy group.
- non-crosslinkable water-soluble inorganic bases preferably alkali metal salts, ammonium salts, alkali metal hydroxides, and ammonia or hydroxides thereof
- non-reducing alkali metal salt pH buffering agents Preferably bicarbonate, dihydrogen phosphate, hydrogen phosphate, etc.
- the organic surface cross-linking agent is added to the water absorbent resin powder.
- the method of the addition treatment is not particularly limited, for example, a method in which the water-absorbing resin powder is immersed in a hydrophilic organic solvent and the added cross-linking agent is adsorbed; The method of dropping and mixing can be exemplified, and the latter is preferable from the viewpoint of uniformly adding a predetermined amount. Furthermore, in order to add uniformly, it is preferable to perform the addition treatment while stirring the water-absorbent and water-absorbent resin powder, and it is preferable to spray the organic surface crosslinking agent solution.
- two or more kinds of the additive crosslinking agents having different compositions may be added simultaneously using, for example, different spray nozzles, but a single composition is preferable from the viewpoint of uniformity. Moreover, if it is a single composition, you may use several spray nozzles in consideration of the magnitude
- Examples of the apparatus used for the addition treatment include, for example, a cylindrical mixer, a double wall conical mixer, a V-shaped mixer, a ribbon mixer, and a screw-type mixer. Suitable are a machine, a fluidized-type furnace, a rotary disk mixer, an airflow-type mixer, a double-arm kneader, an internal mixer, a pulverizing kneader, a rotary mixer, a screw-type extruder, a turbuler, a pro-share mixer, etc. . Furthermore, in large-scale production such as commercial production, an apparatus capable of continuous mixing is preferable. Moreover, the same apparatus may be used for each addition process, and a different apparatus may be used.
- the water-absorbent resin powder used in this step is preferably heated and kept warm, and the temperature is preferably in the range of 10 to 100 ° C., more preferably 15 to 80 ° C., and further preferably 20 to 70 ° C. It is.
- this temperature is 10 ° C. or higher, precipitation of the surface cross-linking agent and moisture absorption of the water-absorbent resin powder are suppressed, and the surface treatment is preferably performed sufficiently and uniformly. Moreover, if this temperature is 100 degrees C or less, since evaporation of the water from surface crosslinking agent aqueous solution is suppressed and fear of precipitation of a surface crosslinking agent etc. is reduced, it is preferable.
- Heat treatment step In this step, heat treatment is performed to cross-link the surface of the water-absorbent resin powder or in the vicinity of the surface in order to improve the water absorption capacity under pressure and the GCA of the water-absorbent resin particles. It is a process. However, since excessive surface cross-linking treatment may reduce the CRC too much, it is preferable to have a step of performing the surface cross-linking treatment until the CRC is 28 g / g or more.
- the preferable degree of surface cross-linking can be confirmed by the CRC decrease before and after surface cross-linking, and the CRC decrease due to surface cross-linking is 0.5 g / g or more, more preferably 1 to 20 g / g, 2 to 15 g / g.
- the amount of the surface cross-linking agent and the reaction temperature time may be appropriately selected so as to reduce the temperature.
- the heat treatment step may be performed simultaneously with the surface cross-linking agent addition step, or may be performed after the surface cross-linking agent addition step, but is preferably performed after the surface cross-linking agent addition step. In addition, this step may be performed once, or may be performed a plurality of times under the same conditions or different conditions.
- the dried water-absorbent resin particles obtained up to the above step (2-5) have a granulated shape, but the primary particles constituting the granulated particles are physically attached before surface crosslinking. In this state, the granulated shape collapses and becomes disintegrated at the time of swelling, and the water absorption performance and liquid passing performance may be lowered.
- Heating device As the heating device used in the present invention, a continuous or batch type (batch type) heating device provided with a gas discharge mechanism and / or a gas supply mechanism for setting a predetermined atmosphere in a known dryer or heating furnace, A continuous heating device is preferable.
- a conduction heat transfer type, a radiation heat transfer type, a hot air heat transfer type, and a dielectric heating type are suitable. More preferred is a conductive heat transfer and / or hot air heat transfer type heating method, and still more preferred is a conductive heat transfer type method.
- control temperature of the heating device is not limited as long as the water-absorbent resin powder can be heated to a temperature described later, and is not necessarily constant from the beginning to the end of the step.
- the temperature is preferably 50 ° C to 300 ° C.
- the physical properties of the resulting water-absorbent resin particles and water-absorbing agent when importance is attached to damage resistance, it is preferably 250 ° C. or lower, more preferably 70 ° C. to 230 ° C., and still more preferably 90 ° C. to 220 ° C.
- it is more preferably 120 ° C. to 280 ° C., further preferably 150 ° C. to 250 ° C., and particularly preferably 170 ° C. to 230 ° C.
- the heating time is preferably 1 minute to 180 minutes, more preferably 5 minutes to 120 minutes, further preferably 10 minutes to 120 minutes, and further preferably 15 to 60 minutes.
- the heat treatment time is shorter than 1 minute, the surface cross-linking treatment becomes insufficient and the absorption capacity under pressure (AAP) decreases.
- the heat treatment time is long, the color is increased, or the absorption capacity without pressure (CRC) is excessively decreased.
- an apparatus equipped with a mechanism for continuously stirring and / or flowing the object to be heated in order to increase the heating efficiency and perform uniform heat treatment is preferable.
- a stirring and / or fluidizing method a grooved stirring method, a screw type, a rotary type, a disk type, a kneading type, a fluidized tank type, etc. are preferable, such as a stirring method using a stirring blade (paddle) or a rotary retort furnace.
- a stirring method by movement of the heat transfer surface itself is more preferable.
- the agitation and / or flow mechanism is intended to perform a uniform heat treatment, and therefore is not used when the amount of treatment is small, for example, when the thickness of an object to be dried is less than 1 cm. It doesn't matter.
- the discharge mechanism When the gas is discharged from the outlet of the heat treatment product as well as a simple exhaust port, the discharge mechanism also corresponds to the discharge mechanism. Further, it is preferable to adjust the amount of gas discharged and the pressure using a blower or the like. Further, the number of exhaust locations is not limited to one, and a plurality of exhaust locations can be provided in consideration of the size of the heating device and the adjustment state of the dew point and temperature.
- the heating apparatus includes a gas supply mechanism, and the dew point and temperature of the atmosphere of the heating unit can be controlled by adjusting the mechanism, for example, the supply amount.
- the gas pressure in the heating part is slightly reduced from normal pressure.
- the differential pressure with respect to atmospheric pressure is preferably 0 kPa to ⁇ 10 kPa, more preferably 0 kPa to ⁇ 5 kPa, and further preferably 0 kPa to ⁇ 2 kPa.
- a batch processing system or a continuous processing system heating apparatus having the above-described mechanism can be used.
- the addition process when the additive addition process is performed before or after the heat treatment, or both, the addition process may be performed using the same apparatus as the addition process or using a different apparatus.
- a continuous production apparatus it may be preferable in terms of production efficiency to use the same apparatus for the addition treatment before heating and the heat treatment, and use a separate apparatus for the addition treatment after heating.
- the water-absorbent resin particles taken out from the heating device as necessary are preferably less than 100 ° C., more preferably 0 ° C. to 95 ° C., 40 for the purpose of suppressing excessive crosslinking reaction and improving the handleability in the subsequent process. It may be cooled to 0 ° C to 90 ° C.
- step (vii) Addition of a fluid flow improver This step is a step of adding a liquid permeation improver to improve FGBP, and is preferably performed during or after the surface cross-linking step.
- the liquid permeation improver in the present invention refers to an additive selected from an insoluble fine particle compound and a polyvalent cationic compound, or an additive that improves FGBP compared to a case where the liquid permeation improver is not used.
- the water-insoluble fine particle compound and the cationic compound act as a steric spacer or an electrostatic spacer on the surface of the water-absorbent resin particles, and “improve liquid passage (for example, unused).
- it is an agent exhibiting an improvement of 10 ⁇ 10 ⁇ 9 cm 2 or more, preferably 30 ⁇ 10 ⁇ 9 cm 2 or more, and more preferably 50 ⁇ 10 ⁇ 9 cm 2 or more.
- the liquid permeation improver that is essentially added in the production method according to the present invention may be selected from water-insoluble inorganic fine particles and polyvalent cationic compounds (cationic polymer compounds or water-soluble polyvalent metal cation-containing compounds). preferable.
- the liquid permeability improving agent is water-insoluble inorganic fine particles. Such an embodiment has a technical effect of improving liquid permeability.
- the “water-soluble” compound refers to a compound that dissolves 1 g or more, more preferably 5 g or more, with respect to 100 g of water at 25 ° C.
- the “water-insoluble” compound refers to 100 g of water at 25 ° C. Refers to a compound that dissolves less than 1 g, further less than 0.5 g, and less than 0.1 g.
- the organic surface cross-linking agent is covalently cross-linked with the functional group of the water-absorbent resin powder, whereas the polyvalent cationic compound (cationic polymer) preferably used as a liquid permeation improver in the present invention.
- a compound or a water-soluble polyvalent metal cation-containing compound) is crosslinked with water-absorbent resin powder or water-absorbent resin particles by ionic crosslinking, or acts as a three-dimensional spacer or an electrostatic spacer to improve liquid permeability. Guessed.
- inorganic fine particles examples include silicon dioxide, titanium dioxide, aluminum oxide, magnesium oxide, zinc oxide, talc, metal phosphate (eg, calcium phosphate, barium phosphate, aluminum phosphate), metal borate (eg, titanium borate, boron Water-insoluble particulate inorganic powders such as aluminum oxide, iron borate, magnesium borate, manganese borate, and calcium borate), silicic acid or its salts, clay, diatomaceous earth, zeolite, bentonite, kaolin, hydrotalcite, activated clay, etc.
- Organic fine powder powder such as body, calcium lactate, aluminum lactate, metal soap (polyvalent metal salt of long chain fatty acid).
- the inorganic fine particles preferably have a volume average particle diameter of 10 ⁇ m or less, more preferably 1 ⁇ m or less.
- the inorganic fine particles may be mixed with the water-absorbent resin powder or the water-absorbent resin particles in the form of powder, or may be mixed with the water-absorbent resin powder or the water-absorbent resin particles with an aqueous dispersion (slurry, for example, colloidal silica). They may be mixed, or dispersed in a surface cross-linking agent or an aqueous solution thereof and mixed with water-absorbent resin particles.
- aqueous dispersion slurry, for example, colloidal silica
- cationic polymer compound Although the cationic polymer compound is not particularly limited, the cationic polymer compounds described in US Pat. Nos. 5,382,610, 7098284, WO2009 / 110645, WO2009 / 041731, and WO2009 / 041727 can be preferably used.
- the cationic polymer compound in the present invention is preferably a polyethylenimine, polyvinylamine, polyallylamine, or a dimethylamine / ammonia / epichlorohydrin condensate among the above-mentioned documents.
- the molecular weight of the cationic polymer compound is preferably a weight average molecular weight of 1,000 to 5,000,000, more preferably 2,000 to 1,000,000, and even more preferably 10,000 to 500,000.
- the cationic polymer compound is preferably water-soluble from the viewpoint of easy mixing.
- water-soluble means that 1 g or more dissolves in 100 g of water at 25 ° C.
- the cationic polymer compound may be mixed directly with the water-absorbent resin particles, may be mixed with a solution, particularly an aqueous solution, or may be mixed after being dissolved in a surface cross-linking agent or an aqueous solution thereof.
- the water-soluble polyvalent metal cation-containing compound refers to a compound containing a metal cation that is divalent or higher, preferably trivalent or higher.
- Examples of the trivalent or higher metal cation include aluminum, zirconium and titanium, and among these, aluminum is preferable.
- polyvalent metal cation-containing compound examples include inorganic surface cross-linking agents such as aluminum sulfate, aluminum chloride, chlorinated zirconium oxide, ammonium zirconium carbonate, zirconium carbonate potassium, zirconium carbonate potassium, zirconium sulfate, zirconium acetate, and zirconium nitrate.
- inorganic salts of valent metals examples include inorganic salts of valent metals, polyvalent metal compounds such as organic salts of polyvalent metals such as aluminum acetate, aluminum lactate, hydroxy zirconium chloride, titanium triethanolamate, and titanium lactate.
- a compound containing aluminum as a polyvalent metal cation is preferable.
- water-absorbent resin particles may be mixed directly with the water-absorbent resin particles as a powder, or may be a solution or a dispersion, may be mixed in an aqueous solution, or may be mixed in a surface cross-linking agent or an aqueous solution thereof.
- the water-soluble polyvalent metal cation-containing compound may be added a plurality of times.
- the addition ratio (first time / second time) is specified in the range of 1/99 to 99/1, preferably 10/90 to 90/10. Is done. Exceeding the above range is not preferable because it is very close to the same situation as the one-time addition and the effect of the plurality of additions becomes poor.
- non-metallic ionic crosslinking agents such as cationic polymer compounds may exhibit adhesiveness during the above-mentioned mixing, it is preferable to add them after the final heat treatment.
- water or an aqueous solution of a crosslinking agent is preferable, and if necessary, a hydrophilic organic solvent (alcohol or polyglycol) or a surfactant is used in combination with water. Dispersibility, solubility, and mixing properties may be improved.
- the amount of water to be used is appropriately determined depending on the type of additive and the addition method. For example, 0 part by weight (dry mixing) to 50 parts by weight, and further 0.1 -10 parts by weight, 0.5 parts by weight to 5 parts by weight.
- liquid permeation improver other than the above, water-soluble polysiloxanes described in International Publication No. 2009/093708, primary to tertiary amine compounds described in International Publication No. 2008/108343, etc. are preferably used.
- the amount of the liquid permeation improver is preferably 0.001 to 5 parts by weight, and preferably 0.002 to 2 parts by weight with respect to 100 parts by weight of the water-absorbing resin particles to be added. More preferred is 0.005 to 1 part by weight.
- a water-soluble polyvalent metal cation-containing compound it is a value converted to the amount of polyvalent metal cation (for example, in the case of aluminum sulfate, the value is defined by the amount of Al 3+ ).
- the addition time is after pulverization and is appropriately added before surface crosslinking, during surface crosslinking, or after surface crosslinking.
- Addition process of other additives This process is a process of adding other additives to impart various functions to the water absorbent resin powder or surface-crosslinked water absorbent resin particles. Or it consists of a plurality of processes.
- the additives include deodorants, fragrances, antibacterial agents, foaming agents, chelating agents such as trisodium diethylenetriaminepentaacetic acid and pentasodium diethylenetriaminepentaacetic acid, surfactants, anti-coloring agents, pigments, dyes, fertilizers and oxidizing agents. It is an agent that can impart or enhance the function, such as a reducing agent.
- the additive may be added in a solution or by dry blending.
- This step may be performed between any of the steps (i) to (vii), or may be performed simultaneously with any of the steps (i) to (vii). Preferably, it is carried out during the step (vi) or after the step (vi).
- the use ratio of these additives is less than 10% by weight, preferably less than 5% by weight, more preferably less than 1% by weight of the water-absorbent resin powder or the surface-crosslinked water-absorbent resin particles.
- These additives may be added simultaneously with the surface cross-linking step or may be added separately.
- the polyacrylic acid (salt) water-absorbing agent obtained is adjusted so that the surface tension is 60 mN / m or more and the absorption capacity without pressure is 28 g / g or more.
- the crosslinking density is controlled by the drying temperature and time of the hydrogel and the strength of surface crosslinking of the water-absorbent resin powder.
- the surface tension is preferably controlled by the type of additive and the amount of additive added.
- the present invention is excellent in GCA (Gel Capillary Absorption) and FGBP (Free Gel Bed Permeability) by adjusting the absorption capacity (CRC) and surface tension of the water-absorbing agent to a specific range. It is possible to solve the desired problem efficiently.
- GCA Gel Capillary Absorption
- FGBP Free Gel Bed Permeability
- D50
- the water absorbing agent of the present invention is not limited to the production method of the present invention as long as the above (1) to (5) are satisfied.
- the water absorption capacity without load (CRC) of the particulate water-absorbing agent of the present invention is 28 g / g or more, more preferably 29 g / g or more, more preferably 30 g by appropriately producing internal crosslinking or surface crosslinking by the above production method.
- / G or more particularly preferably 31 g / g or more, and most preferably 32 g / g or more.
- CRC can be controlled by adjusting the type and amount of the crosslinking agent during polymerization or surface crosslinking within the ranges described in (2-1) to (2-6) above.
- the value of GCA of the particulate water-absorbing agent of the present invention is calculated by the method described in Examples described later, and the higher the value, the better the performance. 28.0 g / g or more, 29.0 g / g g, 30.0 g / g or higher is preferable, and 31.0 g / g or higher is more preferable, 31.5 g / g or higher is more preferable, 33 / g or higher is further preferable, and 34 g / g or higher is the most. preferable.
- the upper limit of GCA is preferably as high as possible, but usually about 50.0 g / g is preferable from the balance with other physical properties.
- the GCA is 31.0 g / g or more. According to this embodiment, the return amount of the liquid can be reduced, and the technical effect of improving the liquid uptake speed is obtained.
- the GCA is within the above range, and it is preferable that the water absorption capacity under pressure is high and the water absorption speed is high (the water absorption time by the Vortex method is short).
- FGBP Water-absorbing agent (3)
- FGBP FGBP is a solution in which a saline solution is injected from the top of a gel layer in a state where a load of 0.3 psi is applied to a water absorbent layer freely swollen in a cell having a mesh structure on the bottom surface. This is a method for evaluating the transmission ability. The higher the value of FGBP, the lower the liquid uptake speed and return amount at the high absorbent concentration absorber.
- FGBP ⁇ ⁇ 10 ⁇ 10 ⁇ 9 ⁇ GCA + 380 ⁇ 10 ⁇ 9 cm 2 (Formula 1) is satisfied when GCA is in the range of 28.0 g / g or more and less than 35.0 g / g.
- GCA ⁇ 35.0 g / g it is preferable to satisfy FGBP ⁇ 30 ⁇ 10 ⁇ 9 cm 2 , more preferably FGBP ⁇ 50 ⁇ 10 ⁇ 9 cm 2 , and FGBP ⁇ 75 ⁇ 10 ⁇ 9. More preferably, cm 2 is satisfied.
- FGBP it is more preferable to satisfy FGBP ⁇ 100 ⁇ 10 ⁇ 9 cm 2 , more preferable to satisfy FGBP ⁇ 120 ⁇ 10 ⁇ 9 cm 2 , and FGBP ⁇ 140 ⁇ 10 ⁇ 9 cm 2 . More preferably, FGBP ⁇ 160 ⁇ 10 ⁇ 9 cm 2 is more preferable, FGBP ⁇ 200 ⁇ 10 ⁇ 9 cm 2 is particularly preferable, and FGBP ⁇ 300 ⁇ 10 ⁇ 9 cm 2 is satisfied. Is most preferred.
- the upper limit of FGBP is preferably higher, but is usually preferably about 500 ⁇ 10 ⁇ 9 cm 2 in view of balance with other physical properties.
- the FGBP is within the above range.
- the water absorption capacity under pressure is high and the water absorption speed is high (the water absorption time by the Vortex method is short).
- the weight average particle diameter (D50) of the particulate water absorbing agent of the present invention is preferably 300 to 500 ⁇ m, more preferably 310 to 480 ⁇ m, and even more preferably 320 ⁇ m to 450 ⁇ m. preferable.
- the polyacrylic acid (salt) -based particulate water-absorbing agent has a weight average particle diameter (D50) of 300 to 500 ⁇ m. According to this embodiment, it is possible to improve GCA and FGBP, and to increase the water absorption capacity under pressure.
- the content of fine particles having a particle diameter of less than 150 ⁇ m in the particulate water-absorbing agent is preferably 0 to 5% by weight, more preferably 0 to 3% by weight in 100% by weight. More preferably, it is 2% by weight.
- the coarse particles having a particle size of 850 ⁇ m or more in the particulate water-absorbing agent are preferably 0% by weight to 5% by weight, more preferably 0% by weight to 3% by weight, and still more preferably 0% by weight to 1% by weight.
- the proportion of particles having a particle diameter of 150 ⁇ m or more and less than 850 ⁇ m in the particulate water-absorbing agent is preferably 90% by weight or more, more preferably 95% by weight or more, still more preferably 98% by weight or more, and 99% by weight or more. Is particularly preferable (the upper limit is 100% by weight).
- the logarithmic standard deviation ( ⁇ ) of the particle size distribution of the particulate water-absorbing agent is preferably 0.20 to 0.50, more preferably 0.25 to 0.45, and still more preferably 0.30 to 0.40. .
- the surface tension (specified by the measurement method in the examples) of the particulate water absorbing agent of the present invention is 60 mN / m or more, more preferably 61 mN / m or more. More preferably, it may be 62 mN / m or more, 63 mN / m or more, or 64 mN / m or more. As an upper limit, 75 mN / m is usually sufficient.
- water absorption capacity under pressure of the particulate water-absorbing agent of the present invention is defined as the water absorption capacity with respect to a 0.90% by weight sodium chloride aqueous solution under a pressure of 2.06 kPa, as shown in Examples described later, preferably 24 g. / G or more, more preferably 25 g / g or more, still more preferably 26 g / g or more, particularly preferably 27 g / g or more, and most preferably 28 g / g or more.
- the balance with other physical properties is usually preferably about 40 g / g.
- the performance of the disposable diaper can be further improved.
- the water absorption time (Vortex method) of the particulate water-absorbing agent of the present invention is preferably 40 seconds or less, more preferably 35 seconds or less, more preferably 30 seconds or less, more preferably 28 seconds or less, more preferably 26 seconds or less, 24 It is more preferably 2 seconds or less, particularly preferably 22 seconds or less, and most preferably 19 seconds or less.
- the performance of the disposable diaper can be further improved.
- water content of the particulate water-absorbing agent of the present invention (specified by loss on drying at 180 ° C. for 3 hours) is not particularly limited as long as the above physical properties are satisfied. % To 20%, further 1% to 15%, especially 2% to 10%. When the water content is high, it becomes difficult to satisfy the physical properties. When the water content is low, the water absorption rate is lowered and the wear resistance of the particles tends to be inferior.
- the nonionic substance is contained in one or more compounds selected from nonionic substances, zwitterionic substances, anionic substances and cationic substances in the interior and / or on the surface.
- the substance is (a) a polyol, (b) a modified product of a hydroxyl group of the polyol, (c) a side chain and / or a terminal polyether-modified polysiloxane, and (d) an alkylene oxide adduct of a higher aliphatic amine.
- the zwitterionic substance is (e) an alkylaminobetaine or (f) an alkylamine oxide, and the anionic substance is (g) a sulfate ester salt of a higher alcohol alkylene oxide adduct or (h) an alkyl diphenyl ether disulfone. It is an acid salt, and the cationic substance is (i) an ammonium salt.
- the polyacrylic acid (salt) -based particulate water-absorbing agent further contains a liquid permeability improving agent.
- a liquid permeability improving agent Such an embodiment has a technical effect of improving liquid permeability.
- the use of the particulate water-absorbing agent of the present invention is not particularly limited, it is preferably used for an absorbent used in a paper diaper or a sanitary napkin.
- the absorbent body in the present invention is an absorbent material formed mainly of the particulate water-absorbing agent of the present invention and hydrophilic fibers.
- the particulate water-absorbing agent and hydrophilic fibers are used.
- the content (core concentration) of the particulate water-absorbing agent with respect to the total weight is preferably 20% to 100% by weight, more preferably 30% to 95% by weight, particularly preferably 50% to 90% by weight.
- an absorber with less water absorbing agent content exhibits an effect of reducing the return amount, but does not use high concentration or pulp.
- the absorber has a problem that the expected effect cannot always be confirmed in terms of the uptake speed and the return amount of the liquid.
- the present invention by making GCA and FGBP highly compatible with each other, it is possible to improve the liquid uptake speed and reduce the liquid return amount even in an absorbent body that does not use pulp at a high concentration.
- the absorber of the present invention when the absorber of the present invention is thin, it is preferable that the absorber has a thin thickness of 1 mm to 5 mm.
- a thin absorbent article By using such a thin absorbent body, a thin absorbent article can be obtained.
- it is set as an absorbent article provided with the above-mentioned thin absorber of the present invention, a top sheet having liquid permeability, and a back sheet having liquid impermeability.
- the method for producing a thin absorbent article according to the present invention includes, for example, an absorbent body (absorbing core) prepared by blending or sandwiching a fiber base material and a particulate water absorbing agent, and a base material such as a surface sheet having liquid permeability. And absorbent material, especially disposable diapers and sanitary items by sandwiching the absorbent body with a base material such as a back sheet having liquid impermeability and, if necessary, equipped with an elastic member, diffusion layer, adhesive tape, etc. Use a napkin.
- Such an absorbent article is compression molded to a density of 0.06 g / cc to 0.50 g / cc and a basis weight of 0.01 g / cm 2 to 0.20 g / cm 2 .
- the fiber base used include hydrophilic fibers such as pulverized wood pulp, cotton linters and cross-linked cellulose fibers, rayon, cotton, wool, acetate, and vinylon. Preferably, they are airlaid.
- the particulate water-absorbing agent of the present invention exhibits excellent absorption characteristics. Accordingly, the absorbent article of the present invention specifically includes adult diapers that have been growing rapidly in recent years, sanitary materials such as diapers for children, sanitary napkins, so-called incontinence pads, and the like. Since the particulate water-absorbing agent of the present invention present in the absorbent article reduces the amount of leakage and reduces skin irritation, the burden on the wearer and the caregiver can be greatly reduced.
- the sanitary material contains the polyacrylic acid (salt) -based particulate water absorbing agent.
- FGBP Free Gel Bed Permeability
- Patent Documents 1 to 6 and Prior Patent Document 7 suggest the water-absorbing agent of the present invention that is compatible with both GCA and FGB. If not.
- gel pulverization after polymerization or during polymerization in the production process of the water-absorbent resin has been proposed in many ways such as in Patent Documents 10 to 21 described above.
- the particle size after gel pulverization is used after using an adhesion control agent. Does not disclose the use of a liquid permeability improver.
- the present invention provides a novel production method in which the particle size after gel pulverization is significantly small and an adhesion control agent and a liquid permeability improver are used. It is as described in the said specification and the following Example that this novel said manufacturing method provides the said novel water absorbing agent of this invention.
- each step in each example is carried out at a substantially normal pressure ( ⁇ 5% of atmospheric pressure, more preferably within 1%). In the same step, pressure by intentional pressurization or reduced pressure is used. It was carried out without any changes.
- Gel CRC was operated in the same manner as above except that 0.6 g of the hydrogel crosslinked polymer or hydrogel particles were used as a sample and the free swelling time was 24 hours. Furthermore, separately, the resin solid content of the water-containing gel-like crosslinked polymer or the water-containing gel particles is measured, and the weight of the water-absorbing resin in the 0.6 g of the water-containing gel-like crosslinked polymer or the water-containing gel particles is determined. ) To calculate the gel CRC. In addition, it measured 5 times per sample and employ
- msi weight (g) of the hydrogel crosslinked polymer or hydrogel particles before measurement
- mb Weight (g) of Blank (nonwoven fabric only) after free swelling and draining
- mwi Total weight (g) of the hydrogel crosslinked polymer and nonwoven fabric after free swelling and draining
- Wn water-containing gel-like crosslinked polymer or solid content of water-containing gel particles (% by weight) It is.
- AAP Absorption capacity under pressure
- ERT442.2-02 The water absorption capacity under load (AAP) of the particulate water-absorbing agent according to the present invention was measured according to ERT442.2-02. That is, 0.900 g (weight W3 (g)) of the particulate water-absorbing agent was put into a measuring device, and the weight (W4 (g)) of the measuring device set was measured. Next, a 0.90 wt% sodium chloride aqueous solution adjusted to 23 ⁇ 2 ° C. was absorbed under a load of 2.06 kPa (0.3 psi, 21 g / cm 2 ).
- the end point is the time from when the water-absorbing agent absorbs physiological saline until the test solution covers the stirrer chip in accordance with the criteria described in JISK 7224-1996 “Explanation of water absorption rate test method for highly water-absorbent resin”. Was measured as the water absorption time (seconds).
- a JIS standard sieve having a mesh opening of 850 ⁇ m, 600 ⁇ m, 500 ⁇ m, 425 ⁇ m, 300 ⁇ m, 150 ⁇ m, 45 ⁇ m (The IIDA TESTING SIEVE: inner diameter 80 mm; JIS Z8801-1 (2000)) or a sieve corresponding to a JIS standard sieve is used.
- 10.00 g of the sample was classified.
- the weight of each sieve was measured, and the weight percentage (% by weight) having a particle diameter of less than 150 ⁇ m was calculated.
- the “percentage by weight with a particle diameter of less than 150 ⁇ m” is the weight ratio (%) of the particles passing through the JIS standard sieve having an opening of 150 ⁇ m to the entire sample.
- a weight average particle diameter (D50) means the particle diameter corresponding to 50 weight% of the whole particulate water absorbing agent (sample).
- the logarithmic standard deviation ( ⁇ ) of the particle size distribution is expressed by the following (Equation 4), and the smaller the value of ⁇ , the narrower the particle size distribution.
- the amount of the supernatant necessary for measurement does not remain after the particulate water-absorbing agent has settled because the water-absorbing rate by the particulate water-absorbing agent is high or the absorption capacity is high, 0.90% by weight sodium chloride
- the amount of 50 ml of the aqueous solution was appropriately adjusted within the minimum range necessary for the measurement.
- a plate method using a platinum plate is adopted, and the plate is sufficiently washed with deionized water before each measurement, and heated and washed with a gas burner.
- the glass filter 2 used in this measurement method is a 500 ml glass filter as defined in ISO 4793 (1980), has a pore diameter of P40 (16 to 40 ⁇ m) and a thickness of 7 mm. For example, it is made of Schott Duran glass. Grade 3 of the filter. Also, a 30 cm radius filter at 20 ° C. must have a water flow capacity of 50 ml / min with a pressure difference of 50 mbar.
- the silicon tube 3 is connected to the lower part of the filter 1 with the glass filter, and further connected to the lower part of the tank 6 equipped with the glass tube 5 and the stopcock 4.
- the upper surface of the glass filter is fixed at a position 10 cm higher than the meniscus below the glass tube in the tank.
- Fill the system with 0.90 wt% aqueous sodium chloride solution.
- a high wet strength cellulose tissue 8 cut into an 8 cm square is fixed to the bottom of a plastic support cylinder 7 having an inner diameter of 60 mm with a metal ring.
- the tissue has a basis weight max of 24.6 g / m 2 , wet tensile strength Min 0.32 N / cm (CD direction), 0.8 N / cm (MD direction) (the flow direction when paper is made on a paper machine is MD direction, The vertical direction is the CD direction) and is available, for example, from Fripa, Germany. Under conditions of room temperature (20 ° C.
- the FGBP of the present invention does not select a water-absorbing agent in the range of 300 ⁇ m to 600 ⁇ m, and measures with a particle size as it is with a water-absorbing agent.
- the data collection time is from 1 second for at least 20 seconds to 5 seconds for 180 seconds Except for the above, the gel bed water permeability test under the “free swelling” condition described in International Publication WO 2004/096304 is performed.
- Emar aqueous solution sodium chloride aqueous solution
- a stirrer chip having a length of 50 mm ⁇ diameter 7 mm was stirred at 300 rpm for 16 hours (cylindrical polypropylene having a height of 21 cm and a diameter of 8 cm) Made using about 1.14L container).
- the water pouring operation was repeated four times so that the water pouring range (50 cm 2 ) spread over the entire sieve, and the hydrogel crosslinked polymer (hydrogel particles) Was classified.
- the hydrated gel-like crosslinked polymer (hydrated gel particles) on the classified first stage sieve was drained for about 2 minutes and weighed.
- the second and subsequent sieves were classified in the same manner, and the hydrogel crosslinked polymers (hydrogel particles) remaining on each sieve after draining were weighed.
- X Weight% (%) of hydrogel cross-linked polymer (hydrogel particles) remaining on each sieve after classification and draining
- w Weight (g) of each of the hydrogel crosslinked polymers (hydrogel particles) remaining on each sieve after classification and draining
- W Total weight (g) of the hydrogel crosslinked polymer (hydrogel particles) remaining on each sieve after classification and draining
- R sieve opening when converted to a hydrogel crosslinked polymer having a solid content of ⁇ wt% (mm)
- r Opening (mm) of a sieve in which a hydrogel crosslinked polymer (hydrogel particles) swollen in a 20% by weight sodium chloride aqueous solution is classified.
- Measurement conditions Apparatus: Waters Alliance, Waters Alliance (2695) Analysis software: Waters, Empor professional + GPC option Column used: Tosoh, TSK guard column SWXL + TSKgel G4000SWXL + G3000SWXL + G2000SWXL Detector: differential refractometer (RI) detector (Waters 2414, manufactured by Waters) Eluent: 115.6 g of sodium acetate trihydrate dissolved in a mixed solvent of 10,999 g of water and 6,001 g of acetonitrile, and further adjusted to pH 6.0 with acetic acid.
- RI differential refractometer
- Standard substance for preparing a calibration curve polyethylene glycol [peak Top molecular weight (Mp) 300000, 200000, 107000, 50000, 27700, 11840, 6450, 1470, 472]
- Calibration curve Created by cubic equation based on Mp value and elution time of polyethylene glycol Flow rate: 1.0 mL / min Column temperature: 40 ° C Measurement time: 45 minutes
- Sample solution injection amount 100 ⁇ L (eluent preparation solution with sample concentration of 0.5 wt%) (L)
- BET specific surface area of the water absorbent resin powder of the present invention was measured based on the following method.
- a high-accuracy gas / vapor adsorption amount measuring device (manufactured by Nippon Bell Co., Ltd., BELSORP-max) is used.
- a pretreatment device for adsorption measurement (manufactured by Nippon Bell Co., Ltd., BELSORP-vac II) was used.
- the Pyrex (registered trademark) glass rod attached to the Pyrex (registered trademark) test tube attached to the BET specific surface area measuring device was put in, and degassed under reduced pressure using a pretreatment device for adsorption measurement. After reaching the predetermined pressure of the apparatus, the water-absorbent resin powder is visually observed in the Pyrex (registered trademark) test tube using the attached BELSORP-max sampling funnel (Pyrex (registered trademark)). About 80%. At this time, the weight of the water absorbent resin powder was recorded. Thereafter, the test tube was deaerated under reduced pressure using a pretreatment apparatus for adsorption measurement.
- Example 1 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- a reaction vessel consisting of a thermometer, a nitrogen gas inlet tube and a lid provided with an exhaust hole and a vat with a bottom of 300 mm ⁇ 220 mm and a depth of 60 mm, 170 g of acrylic acid, 1800 g of a 37 wt% aqueous sodium acrylate solution, polyethylene glycol diacrylate (weight) An average molecular weight 523) 0.99 g, polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) 6.688 g (0.8% by weight with respect to the monomer component) and 216 g of deionized water were fed and mixed at 20 ° C. Was immersed in a water bath to a height of 10 mm from the bottom.
- Nitrogen gas was introduced into this aqueous solution and deaerated for 20 minutes. After confirming that this solution had reached 20 ° C., 6.61 g of a 20 wt% aqueous sodium persulfate solution and 6.33 g of a 0.1 wt% L-ascorbic acid aqueous solution were added and mixed under stirring in a nitrogen stream atmosphere. The monomer concentration was 38% by weight. Polymerization started after 1 minute, and the reaction system temperature at that time was 20 ° C. After the polymerization was started, the polymerization system was not stirred, and the reaction vessel was subsequently immersed in a 20 ° C. water bath for cooling. After 17 minutes, the polymerization system showed a maximum temperature of 89 ° C. Thereafter, the temperature of the water bath was set to 70 ° C., and the polymerization reaction was carried out for 20 minutes to obtain a hydrogel crosslinked polymer (GK1).
- GK1 hydrogel crosslinked polymer
- hydrogel crosslinked polymer (GK1) was cut into blocks, put into Unipack (manufactured by Production Nippon Co., Ltd.), left in a thermostat for 1 hour, and kept constant at 60 ° C.
- Meat chopper manufactured by Remacom Co., Ltd., model: HL-
- a hydrogel crosslinked polymer (GK1) kept at 60 ° C. was heated to 60 ° C. using a sheet heater with a 3.5 mm die diameter plate installed G22SN) was passed twice to obtain hydrogel particles (also referred to as “hydrous gel-like crosslinked polymer pulverized product”) (GKF1).
- the screw centrifuge speed of the meat chopper was 210 rpm, and the hydrogel crosslinked polymer (GK1) was supplied at 360 g / min, and the obtained gel pulverized product was similarly supplied at 360 g / min and passed twice.
- the physical properties of the hydrogel crosslinked polymer pulverized product (GKF1) are shown in the following table.
- the following hydrogel crosslinked polymer pulverized product (GKF) is also shown in the following table.
- the obtained hydrated gel-like crosslinked polymer pulverized product (GKF1) was dried with a hot air dryer at 160 ° C. for 45 minutes to obtain a dried product, and then pulverized with a roll mill (manufactured by Inoguchi Giken Co., Ltd.), with an opening of 850 ⁇ m. After sieving with a sieve having 600 ⁇ m, 500 ⁇ m, 300 ⁇ m, and 150 ⁇ m, 3% by weight of particles that pass through 850 ⁇ m and not pass through 600 ⁇ m, and 10% by weight of particles that pass through 600 ⁇ m and not pass through 500 ⁇ m pass through to 300 ⁇ m.
- the water-absorbent resin powder (B1) is prepared by blending the particles that do not pass through 54% by weight, particles that pass through 300 ⁇ m and do not pass through 150 ⁇ m into 31% by weight, and particles that pass through 150 ⁇ m and do not pass through 45 ⁇ m into 2% by weight. Obtained. The preparation was carried out in the same manner as in Example 15.
- the weight average particle diameter D50, logarithmic standard deviation and CRC of the water absorbent resin powder (B1) are shown in the following table.
- the following water absorbent resin powder (B) is also shown in the following table.
- Example 2 Except that the amount of polyethylene glycol used was changed to 3.344 g (0.4% by weight based on the monomer component), the same operation as in Example 1 was performed, and the water absorbent resin powder (B2) was surface-treated. Water-absorbing resin particles (S2) and a particulate water-absorbing agent (EX-2) were obtained.
- Example 3 Except that the amount of polyethylene glycol used was changed to 10.03 g (1.2% by weight with respect to the monomer component), the same operation as in Example 1 was performed, and the water absorbent resin powder (B3) was surface-treated. Water-absorbing resin particles (S3) and a particulate water-absorbing agent (EX-3) were obtained.
- Example 4 Except that the weight average molecular weight of the polyethylene glycol used was changed to 400, the same operation as in Example 1 was performed to obtain the water absorbent resin powder (B4), the surface treated water absorbent resin particles (S4), and the particulate water absorbent. (EX-4) was obtained.
- Example 5 Except that the weight average molecular weight of polyethylene glycol used was changed to 20000, the same operation as in Example 1 was performed to obtain a water absorbent resin powder (B5), surface treated water absorbent resin particles (S5), and a particulate water absorbent. (EX-5) was obtained.
- Example 6 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- a reaction vessel consisting of a thermometer, a lid equipped with a nitrogen gas inlet tube and an exhaust hole, and a vat with a bottom of 300 mm ⁇ 220 mm and a depth of 60 mm
- 170 g of acrylic acid, 1800 g of a 37 wt% sodium acrylate aqueous solution, polyethylene glycol diacrylate (average 5.9 g of molecular weight 523) and 216 g of deionized water were supplied and mixed, and immersed in a 20 ° C. water bath to a height of 10 mm from the bottom.
- Nitrogen gas was introduced into this aqueous solution and deaerated for 20 minutes. After confirming that the solution reached 20 ° C., 6.61 g of a 20 wt% aqueous sodium persulfate solution and 6.33 g of a 0.1 wt% L-ascorbic acid aqueous solution were added and mixed under stirring in a nitrogen stream atmosphere. The monomer concentration was 38% by weight. Polymerization started after 1 minute, and the reaction system temperature at that time was 20 ° C. After the polymerization was started, the polymerization system was not stirred, and the reaction vessel was subsequently immersed in a 20 ° C. water bath for cooling. After 17 minutes, the polymerization system showed a maximum temperature of 89 ° C. Thereafter, the temperature of the water bath was set to 70 ° C., and the polymerization reaction was carried out for 20 minutes to obtain a hydrogel crosslinked polymer (GK6). The obtained hydrogel crosslinked polymer (GK6) was cut into blocks.
- the obtained water-containing gel-like crosslinked polymer (GK6) was cut into blocks, put into Unipack (manufactured by Nippon Nihon Co., Ltd.), and allowed to stand at 60 ° C. for 1 hour in a thermostat. 66.9 g of a methanol solution containing 10% by weight of polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) in the hydrogel crosslinked polymer (GK6) kept constant at 60 ° C.
- Meat Chopper (Remacom Co., Ltd., Model: HL-G22SN), sprinkled uniformly over the surface of 2200 g of a crosslinked polymer (GK6), placed on a 3.5 mm die caliber plate, and heated to 60 ° C. using a sheet heater The hydrated gel-like cross-linked polymer pulverized product (GKF6) was obtained by passing twice through.
- GK6 crosslinked polymer
- the screw centrifuge speed of the meat chopper was 210 rpm, and the hydrogel crosslinked polymer (GK6) was supplied at 360 g / min, and the obtained gel pulverized product was similarly supplied at 360 g / min and passed twice.
- the obtained water-containing gel-like crosslinked polymer pulverized product (GKF6) was dried with a hot air dryer at 160 ° C. for 45 minutes to obtain a dried product, and then pulverized with a roll mill (manufactured by Inoguchi Giken Co., Ltd.), with an opening of 850 ⁇ m. , 600 ⁇ m, 500 ⁇ m, 300 ⁇ m, and 150 ⁇ m, and then sieving to prepare a water absorbent resin powder (B6).
- the performance of the water absorbent resin powder (B6) obtained is shown in Table 1.
- Example 7 To 100 parts by weight of the surface-crosslinked water-absorbent resin particles (S1) in Example 1, 1 part by weight of a 1% by weight DTPA aqueous solution was added with stirring, mixed for 1 minute, and then 27.5% by weight. A solution comprising 1.17 parts by weight of an aqueous aluminum sulfate solution (8% by weight in terms of aluminum oxide), 0.196 parts by weight of a 60% by weight aqueous sodium lactate solution and 0.029 parts by weight of propylene glycol was added and mixed for 1 minute, followed by hot air After leaving it in the dryer for 30 minutes, it was passed through a wire mesh having an opening of 850 ⁇ m to obtain a particulate water-absorbing agent (EX-7). The performance of the obtained particulate water-absorbing agent (EX-7) is shown in the following table.
- Example 8 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step) Except having changed the quantity of polyethyleneglycol diacrylate into 1.73g, operation similar to Example 1 was performed and the hydrogel crosslinked polymer (GK8) was obtained.
- the obtained water-containing gel-like crosslinked polymer (GK8) was cut into blocks, put into Unipack (manufactured by Production Japan Co., Ltd.), allowed to stand in a thermostat for 1 hour, and kept constant at 60 ° C.
- the hydrogel crosslinked polymer (GK8) kept at a constant temperature of 60 ° C. was supplied to a screw extruder heated to 60 ° C. using a sheet heater, and gel pulverized.
- the screw extruder is provided with a perforated plate having a diameter of 100 mm, a hole diameter of 3.2 mm, a hole number of 316, an opening ratio of 32.3%, and a thickness of 10 mm.
- a meat chopper having an inner diameter of 88 mm was used.
- the hydrogel crosslinked polymer (GK8) was supplied at 1680 g / min at a screw shaft rotation speed of the meat chopper of 126 rpm, and passed once to obtain a hydrogel crosslinked polymer pulverized product (GKF8).
- the gel grinding energy GGE (1) at this time was 174.8 J / g, and GGE (2) was 40.4 J / g.
- the water-absorbent resin powder (B8) was obtained in the same manner as in Example 1 except that the obtained hydrogel crosslinked polymer (GKF8) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- the obtained water-absorbing resin powder (B8) was treated in the same manner as in Example 1 to obtain surface-crosslinked water-absorbing resin particles (S8) and particulate water-absorbing agent (EX-8).
- the performance of the obtained particulate water-absorbing agent (EX-8) is shown in the following table.
- Example 9 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step) A hydrogel crosslinked polymer (GK9) was obtained in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol was not used.
- the obtained hydrogel crosslinked polymer (GK9) was supplied at 1680 g / min, and at the same time, 6.688 g of polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) (0.8 weight relative to the monomer concentration). %) was supplied as a 20 wt% aqueous solution at 20.5 g / min, and the same operation as in Example 8 was performed to obtain a hydrated gel-like crosslinked polymer pulverized product (GKF9).
- the gel grinding energy GGE (1) at this time was 175.7 J / g, and GGE (2) was 41.3 J / g.
- the water-absorbent resin powder (B9) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF9) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- the obtained water-absorbing resin powder (B9) was treated in the same manner as in Example 1 to obtain surface-crosslinked water-absorbing resin particles (S9) and particulate water-absorbing agent (EX-9).
- the performance of the obtained particulate water-absorbing agent (EX-9) is shown in the following table.
- Example 9 the adhesion control agent is not added at the time of polymerization, but the adhesion control agent is added while performing the meat chopper (at the time of gel grinding).
- Example 10 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step) Except having changed the quantity of polyethyleneglycol diacrylate into 1.73g, operation similar to Example 1 was performed and the hydrogel crosslinked polymer (GK10) was obtained.
- the gel grinding energy GGE (1) at this time was 174.8 J / g, and GGE (2) was 40.4 J / g.
- the water-absorbent resin powder (B10) was obtained in the same manner as in Example 1 except that the obtained hydrogel crosslinked polymer (GKF10) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 11 has a relatively long heat treatment time in the surface cross-linking step. This has the effect of improving FGBP.
- Example 11 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step) 380 g of acrylic acid, 158 g of 48% by weight sodium hydroxide aqueous solution, 1.53 g of polyethylene glycol diacrylate (weight average molecular weight 523), 3.74 g of polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) 0.8 wt%) and a monomer solution consisting of 23.4 g of 0.1 wt% diethylenetriaminepentaacetic acid trisodium aqueous solution.
- the gel grinding energy GGE (1) at this time was 69.4 J / g, and GGE (2) was 24.0 J / g.
- the water-absorbent resin powder (B11) was obtained in the same manner as in Example 1 except that the obtained hydrogel crosslinked polymer (GKF11) was dried with a hot air dryer at 190 ° C. for 30 minutes. .
- Example 12 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the amount of polyethylene glycol diacrylate was changed to 1.39 g, and instead of polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.), Amphitol 20BS (Kao Corporation, active ingredient 30% by weight) 1.17 g
- a hydrogel crosslinked polymer (GK12) was obtained in the same manner as in Example 11 except that 0.075% by weight of the active ingredient was used relative to the monomer component.
- the screw extruder has a perforated plate with a diameter of 100 mm, a hole diameter of 6.4 mm, a number of holes of 83, an aperture ratio of 34.0%, a thickness of 10 mm, an outer diameter of the screw shaft of 86 mm, and an inner diameter of the casing of 88 mm.
- I used a meat chopper.
- the water-containing gel-like crosslinked polymer (GK12) was supplied to the meat chopper at a screw shaft rotation speed of 130 rpm and 4640 g / min.
- a pulverized product (GKF12) was obtained.
- the gel grinding energy GGE (1) at this time was 57.5 J / g, and GGE (2) was 13.3 J / g.
- the water-absorbent resin powder (B12) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF12) was dried with a hot air dryer at 190 ° C. for 30 minutes. .
- the surface-treated water-absorbing resin particles (S12) were subjected to the same operation as in Example 1 to obtain a particulate water-absorbing agent (EX-12).
- the performance of the obtained particulate water-absorbing agent (EX-12) is shown in the following table.
- Example 12 the particle diameter of the hydrogel crosslinked polymer pulverized product (GKF12) is increased by lowering GGE.
- Example 13 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step) 1.17 g of amphital 20BS (manufactured by Kao Corporation, 30% by weight of active ingredient) instead of polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) (0.075% by weight of active ingredient relative to monomer component) Except that was used, the same operation as in Example 11 was performed to obtain a hydrogel crosslinked polymer (GK13).
- amphital 20BS manufactured by Kao Corporation, 30% by weight of active ingredient
- polyethylene glycol weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.
- the gel grinding energy GGE (1) at this time was 65.5 J / g, and GGE (2) was 24.4 J / g.
- the water-absorbent resin powder (B13) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF13) was dried with a hot air dryer at 190 ° C. for 30 minutes. .
- Example 14 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the amount of polyethylene glycol diacrylate was changed to 1.73 g, and instead of polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.), Amphitol 20BS (manufactured by Kao Corporation, active ingredient 30% by weight) 2.090 g
- a hydrogel crosslinked polymer (GK14) was obtained in the same manner as in Example 1 except that (0.075% by weight as an active ingredient with respect to the monomer component) was used.
- the gel grinding energy GGE (1) at this time was 182.2 J / g, and GGE (2) was 58.3 J / g.
- Example 15 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the amount of polyethylene glycol diacrylate was changed to 1.73 g, and instead of polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.), Amphitol 20BS (manufactured by Kao Corporation, active ingredient 30% by weight) 2.090 g
- a hydrogel crosslinked polymer (GK15) was obtained in the same manner as in Example 1 except that (0.075% by weight as an active ingredient with respect to the monomer component) was used.
- the water-absorbent resin powder (B15) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF15) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- the surface-treated water-absorbing resin particles (S15) were subjected to the same operation as in Example 1 to obtain a particulate water-absorbing agent (EX-15).
- the performance of the obtained particulate water-absorbing agent (EX-15) is shown in the following table.
- Example 16 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used.
- a cross-linked polymer (GK16) was obtained.
- the obtained hydrogel crosslinked polymer (GK16) was cut into blocks, put into Unipack (manufactured by Production Japan Co., Ltd.), allowed to stand in a thermostat for 1 hour, and kept constant at 60 ° C. 62.7 g of methanol solution containing 1% by weight of Amphital 20BS (manufactured by Kao Corporation, 30% by weight of active ingredient) uniformly on the surface of the hydrogel crosslinked polymer (GK16) kept at 60 ° C.
- step (i) Meat chopper (manufactured by Iizuka Kogyo Co., Ltd.), sprinkled with 0.075% by weight of active ingredient with respect to the amount of raw material monomer component, installed with a 4.7 mm die caliber plate and heated to 60 ° C. using a sheet heater By passing twice through a model (ROYAL, type: VR-400K), a hydrated gel-like crosslinked polymer pulverized product (GKF16) was obtained.
- ROYAL type: VR-400K
- the screw centrifuge speed of the meat chopper was set to 170 rpm, and the hydrogel crosslinked polymer (GK17) was supplied at 150 g / min.
- the obtained gel pulverized product was similarly supplied at 150 g / min and passed twice.
- the water-absorbent resin powder (B16) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF16) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 17 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used.
- a cross-linked polymer (GK17) was obtained.
- the water-absorbent resin powder (B17) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF17) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 18 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used.
- a cross-linked polymer (GK18) was obtained.
- the water-absorbent resin powder (B18) was obtained in the same manner as in Example 1 except that the obtained hydrogel crosslinked polymer (GKF18) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 19 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used.
- a cross-linked polymer (GK19) was obtained.
- the water-absorbent resin powder (B19) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF19) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 20 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used.
- a cross-linked polymer (GK20) was obtained.
- the water-absorbent resin powder (B20) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF20) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- the surface-treated water-absorbing resin particles (S20) were subjected to the same operation as in Example 1 to obtain a particulate water-absorbing agent (EX-20).
- the performance of the obtained particulate water-absorbing agent (EX-20) is shown in the following table.
- Example 21 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used.
- a cross-linked polymer (GK21) was obtained.
- the water-absorbent resin powder (B21) was obtained in the same manner as in Example 1 except that the obtained hydrogel crosslinked polymer (GKF21) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 22 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used.
- a cross-linked polymer (GK22) was obtained.
- the water-absorbent resin powder (B22) was obtained in the same manner as in Example 1 except that the obtained hydrogel crosslinked polymer (GKF22) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 23 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used.
- a cross-linked polymer (GK23) was obtained.
- the water-absorbent resin powder (B23) was obtained in the same manner as in Example 1 except that the obtained hydrogel crosslinked polymer (GKF23) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 24 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used.
- a cross-linked polymer (GK24) was obtained.
- the water-absorbent resin powder (B24) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF24) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 25 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used.
- a cross-linked polymer (GK25) was obtained.
- Example 26 (Gel polymerization process) The hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used. A cross-linked polymer (GK26) was obtained.
- the water-absorbent resin powder (B26) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF26) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 27 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the hydrogel was carried out in the same manner as in Example 1 except that the amount of polyethylene glycol diacrylate was changed to 1.73 g and polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.) was not used.
- a cross-linked polymer (GK27) was obtained.
- the water-absorbent resin powder (B27) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF27) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 28 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step) Except changing the quantity of polyethyleneglycol diacrylate to 1.73g, operation similar to Example 1 was performed and the water-containing gel-like crosslinked polymer (GK28) was obtained.
- the screw extruder has a perforated plate with a diameter of 100 mm, a hole diameter of 8.0 mm, a number of holes of 54, an aperture ratio of 34.5%, and a thickness of 10 mm, an outer diameter of the screw shaft of 86 mm, and an inner diameter of the casing of 88 mm.
- the same operation as in Example 8 was performed except that the meat chopper was used, and a hydrogel-like crosslinked polymer pulverized product (GKF28) was obtained.
- the gel grinding energy GGE (1) at this time was 108.0 J / g, and GGE (2) was 14.2 J / g.
- the water-absorbent resin powder (B28) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF28) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 29 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the amount of polyethylene glycol diacrylate was changed to 1.73 g, and instead of polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.), Amphitol 20BS (manufactured by Kao Corporation, active ingredient 30% by weight) 2.090 g
- a hydrogel crosslinked polymer (GK29) was obtained in the same manner as in Example 1 except that (0.075% by weight as an active ingredient with respect to the monomer component) was used.
- the gel grinding energy GGE (1) at this time was 182.2 J / g, and GGE (2) was 58.3 J / g.
- the water-absorbent resin powder (B29) was obtained in the same manner as in Example 1 except that the obtained hydrogel crosslinked polymer (GKF29) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- Example 30 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step)
- the amount of polyethylene glycol diacrylate was changed to 1.73 g, and instead of polyethylene glycol (weight average molecular weight 2000, manufactured by Wako Pure Chemical Industries, Ltd.), Amphitol 20BS (manufactured by Kao Corporation, active ingredient 30% by weight) 2.090 g
- a hydrogel crosslinked polymer (GK30) was obtained in the same manner as in Example 1 except that (0.075% by weight as an active ingredient with respect to the monomer component) was used.
- Example 8 (Gel grinding process) Except that the screw shaft rotation speed was 225 rpm, the same operation as in Example 8 was performed to obtain a hydrogel-like crosslinked polymer pulverized product (GKF30).
- the gel grinding energy GGE (1) at this time was 182.2 J / g, and GGE (2) was 58.3 J / g.
- the water-absorbent resin powder (B30) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF30) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- the surface-treated water-absorbing resin particles (S30) were subjected to the same operation as in Example 1 to obtain a particulate water-absorbing agent (EX-30).
- the performance of the obtained particulate water-absorbing agent (EX-30) is shown in the following table.
- Example 31 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step) The same operation as in Example 11 was performed to obtain a hydrogel crosslinked polymer (GK31).
- the screw extruder has a perforated plate with a diameter of 100 mm, a hole diameter of 6.4 mm, a number of holes of 83, an aperture ratio of 34.0%, a thickness of 10 mm, an outer diameter of the screw shaft of 86 mm, and an inner diameter of the casing of 88 mm.
- the same procedure as in Example 8 was carried out except that the meat chopper was used and the screw shaft rotation speed was 130 rpm and the hydrogel crosslinked polymer (GK32) was supplied to the meat chopper at 4640 g / min. A polymer pulverized product (GKF31) was obtained.
- the gel grinding energy GGE (1) at this time was 56.4 J / g, and GGE (2) was 13.1 J / g.
- the water-absorbent resin powder (B31) was obtained in the same manner as in Example 1 except that the obtained hydrated gel-like crosslinked polymer pulverized product (GKF31) was dried with a hot air dryer at 190 ° C. for 30 minutes. .
- Example 32 ((Meth) acrylic acid (salt) monomer aqueous solution preparation, polymerization step) The same operation as in Example 13 was performed to obtain a hydrogel crosslinked polymer (GK32).
- the screw extruder has a perforated plate with a diameter of 100 mm, a hole diameter of 6.4 mm, a number of holes of 83, an opening ratio of 34.0%, and a thickness of 10 mm, an outer diameter of the screw shaft of 86 mm, and a casing inner diameter of
- a meat chopper of 88 mm the same procedure as in Example 8 was performed, except that the screw shaft rotation speed was 130 rpm and the hydrogel crosslinked polymer (GK33) was supplied to the meat chopper at 4640 g / min. A crushed cross-linked polymer (GKF32) was obtained.
- the gel grinding energy GGE (1) at this time was 56.7 J / g, and GGE (2) was 12.6 J / g.
- Example 1 In Example 1, the same treatment as in Example 1 was performed except that the die diameter of the meat chopper used in the gel pulverization process was changed from 3.5 mm to 9.0 mm, and the hydrous gel particles (CGKF-1), water absorption Resin powder (CB-1), surface-treated water-absorbing resin particles (CS1), and particulate water-absorbing agent (CEX-1) were obtained.
- CKF-1 hydrous gel particles
- CB-1 water absorption Resin powder
- CS1 surface-treated water-absorbing resin particles
- CEX-1 particulate water-absorbing agent
- Example 2 S1 obtained in Example 1 was used as a comparative water-absorbing agent (CEX-2).
- CEX-2 The performance of the obtained particulate water-absorbing agent (CEX-2) is shown in the following table.
- Example 3 In the same manner as in Example 1 of application number PCT / JP2015 / 56110, a water absorbent resin powder (CB-3) having a granulated shape and a water absorbent for comparison (CEX-3) were obtained. The performance of the obtained water-absorbing resin powder (CB-3) and particulate water-absorbing agent (CEX-3) is shown in the following table.
- Example 4 In the same manner as in Example 2 of application number PCT / JP2015 / 56110, a water absorbent resin powder (CB-4) having a granulated shape and a water absorbent for comparison (CEX-4) were obtained. The performance of the obtained water absorbent resin powder (CB-4) and particulate water absorbing agent (CEX-4) is shown in the following table.
- Hydrogel particles (CGKF-5), water-absorbing resin powder (CB-5), and a particulate water-absorbing agent for comparison (CEX-5) were prepared by the method described in Example 6 of WO 2011/126079 pamphlet.
- the performances of the obtained hydrogel particles (CGKF-5), water-absorbing resin powder (CB-5), water-absorbing resin powder (CEX-5), and particulate water-absorbing agent (CEX-6) are shown in the following table.
- the water-absorbent resin powder (CB8) was obtained in the same manner as in Example 1 except that the obtained hydrogel crosslinked polymer (GKF8) was dried with a hot air dryer at 185 ° C. for 30 minutes. .
- FIG. 2 shows an SEM photograph of the water-absorbent resin powder (particle size cut 500/425) of Example 9, and the measurement conditions are 30 times magnification and applied voltage: 1.3 kV.
- FIG. 3 shows an SEM photograph of the water-absorbent resin powder (particle size cut 500/425) of Example 9, and the measurement conditions are a magnification of 130 times and an applied voltage of 1.3 kV.
- FIG. 4 shows an SEM photograph of the water-absorbent resin powder (particle size cut 500/425) of Comparative Example 1, and the measurement conditions are 30 times magnification and applied voltage: 1.3 kV.
- FIG. 5 shows an SEM photograph of the water-absorbent resin powder (particle size cut 500/425) of Comparative Example 6, and the measurement conditions are a magnification of 130 times and an applied voltage of 1.3 kV.
- the absorber to be measured was prepared by the following method. That is, first, water-absorbing paper 12 of 80 mm ⁇ 80 mm and thickness of about 0.1 mm is laid in an acrylic resin container 11 (inner dimensions 80 mm ⁇ 80 mm, height 4 cm). .4 g uniformly dispersed, 80 mm ⁇ 80 mm, a water absorbent paper 14 having a thickness of about 0.1 mm is laid thereon, and a surface sheet 15 having a liquid permeability (80 mm ⁇ 80 mm, 80 mm ⁇ 80 mm, A model diaper absorber 18 was prepared by placing a thickness of about 0.1 mm. (Water absorbent concentration including water absorbent paper: about 82%).
- a liquid charging device 16 weight 80 g, load to the absorber 1.25 g / cm 2 (0.1 kPa), and liquid is applied to the central portion so that the load is uniformly applied on the absorber 18 of the model diaper.
- a cylinder having a diameter of 30 mm and a height of 120 mm that can be charged was placed.
- 48 g of physiological saline (0.90% sodium chloride aqueous solution) at 37 ° C. was quickly poured into the cylinder. The time from the start of pouring physiological saline to the time when the physiological saline was completely absorbed by the absorbent was measured and taken as the first physiological saline uptake rate (seconds).
- the liquid charging device 16 and the weight 17 placed on the absorber 18 are removed, and a paper towel (manufacturer: Wang Napier Co., Ltd., kitchen towel, cut into 80 mm ⁇ 80 mm and stacked 30 sheets) is absorbed. It was placed on the body 18 and a load of 30 g / cm 2 (3.0 kPa) was placed thereon and left for 1 minute. By measuring the change in weight of the paper towel, the amount of liquid absorbed by the paper towel was determined, and this was defined as the return amount (g).
- the water absorbing body using the particulate water absorbing agent (EX-1) in which GCA and FGBP are highly compatible exhibits excellent uptake speed at the first time and particularly at the second time, and the return amount is also reduced. It becomes possible to do.
- Comparative Example 2 in the water absorbent using the particulate water-absorbing agent (CEX-2) that is high in GCA but low in FGBP, the amount of return can be reduced. In particular, the second capturing speed becomes slow, and the performance of the return amount and the capturing speed cannot be compatible.
- Comparative Example 6 in the water absorbent using the particulate water-absorbing agent (CEX-6) having a low GCA and a high FGBP outside the scope of the invention, the first and particularly the second time show an excellent uptake rate. However, the return amount greatly increases, and the performance of the return amount and the capture speed cannot be compatible.
- Comparative Example 8 in the water absorbent using the particulate water-absorbing agent (CEX-8) that is high in FGBP but low in CRC and GCA and low in surface tension, the first and particularly 2 The second time shows an excellent take-in speed, but the return amount increases greatly, and the performance of the return amount and the take-in speed cannot be compatible.
- CEX-8 particulate water-absorbing agent
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Polymers & Plastics (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Materials Engineering (AREA)
- General Health & Medical Sciences (AREA)
- Dispersion Chemistry (AREA)
- Hematology (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Absorbent Articles And Supports Therefor (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
(iv)前記含水ゲル粒子を乾燥し、乾燥物を得る工程と、(v)前記乾燥物を粉砕及び/又は分級して吸水性樹脂粉末を得る工程と、(vi)前記吸水性樹脂粉末を表面架橋し、吸水性樹脂粒子を得る工程と、(vii)前記吸水性樹脂粉末または前記吸水性樹脂粒子に通液性向上剤を添加する工程と、を含み、工程(iii)ないし工程(iii)以前に、前記含水ゲル状架橋重合体及び/又は前記含水ゲル粒子が接着することを制御する接着制御剤を添加することを有し、前記含水ゲル粒子の固形分が10~80重量%となり、かつ、乾燥物として換算した際の前記含水ゲル粒子の重量平均粒子径が50~650μmとなるように調整し、ポリ(メタ)アクリル酸(塩)系粒子状吸水剤の表面張力が60mN/m以上となり、無加圧下吸収倍率(CRC)が28g/g以上となるように調整する、ポリ(メタ)アクリル酸(塩)系吸水性樹脂粒子を主成分とする、ポリ(メタ)アクリル酸(塩)系粒子状吸水剤の製造方法が提供される。
(1-1)「粒子状吸水剤」
本発明において、「粒子状吸水剤」とは、吸水性樹脂粒子を主成分(好ましくは全体の60重量%以上、さらに好ましくは80重量%以上、最も好ましくは90重量%以上)とする水性液のゲル化剤であり、その他に任意の成分として水、無機微粒子、吸湿ブロッキング抑制剤、カチオン性高分子化合物、水溶性多価金属カチオン含有化合物、界面活性剤、発塵抑制剤、着色防止剤、耐尿性向上剤、消臭剤、香料、抗菌剤、発泡剤、顔料、染料、肥料、酸化剤、還元剤等を、それぞれ0~10重量%、好ましくは0.1重量%~1重量%含有してもよい。なお、本明細書において、「粒子状吸水剤」を単に「吸水剤」と称する場合もある。
本発明における吸水性樹脂とは、水膨潤性水不溶性の高分子ゲル化剤を意味する。なお、「水膨潤性」とは、ERT441.2-02で規定するCRC(無加圧下吸水倍率)が5g/g以上であることをいい、また、「水不溶性」とは、ERT470.2-02で規定するExt(可溶分)が0~50重量%であることをいう。
本発明における「ポリ(メタ)アクリル酸(塩)系吸水性樹脂」とは、任意にグラフト成分を含み、繰り返し単位として、(メタ)アクリル酸及び/又はその塩(以下、(メタ)アクリル酸(塩)と称する)を主成分とする重合体を意味する。
「EDANA」は、欧州不織布工業会(European Disposables and Nonwovens Associations)の略称であり、「ERT」は、欧州標準(ほぼ世界標準)の吸水性樹脂の測定法(EDANA Recommended Test Methods)の略称である。本発明では、特に断りのない限り、ERT原本(2002年改定/公知文献)に準拠して、吸水性樹脂の物性を測定する。
「CRC」は、Centrifuge Retention Capacity(遠心分離保持容量)の略称であり、吸水性樹脂の無加圧下吸水倍率(「吸水倍率」と称する場合もある)を意味する。具体的には、吸水性樹脂0.2gを不織布製の袋に入れた後、大過剰の0.9重量%塩化ナトリウム水溶液中に30分間浸漬して自由膨潤させ、その後、遠心分離機(250G)で水切りした後の吸水倍率(単位;g/g)のことをいう。
「AAP」は、Absorption Against Pressureの略称であり、吸水性樹脂の加圧下吸水倍率を意味する。具体的には、吸水性樹脂0.9gを大過剰の0.9重量%塩化ナトリウム水溶液に対して、1時間、2.06kPa(21g/cm2、0.3psi)荷重下で膨潤させた後の吸水倍率(単位;g/g)のことをいう。また、ERT442.2-02には、Absorption Under Pressureと表記されているが、実質的に同一内容である。
「PSD」は、Particle Size Distributionの略称であり、篩分級により測定される、吸水性樹脂の粒度分布を意味する。なお、重量平均粒子径(D50)及び粒度分布の対数標準偏差(σζ)は、米国特許第7638570号に記載された「(3)Mass-Average Particle Diameter (D50) and Logarithmic Standard Deviation (σζ) of Particle Diameter Distribution」と同様の方法で測定する。
「Ext」は、Extractablesの略称であり、吸水性樹脂の水可溶分(水可溶成分量)を意味する。具体的には、吸水性樹脂1.0gを0.9重量%塩化ナトリウム水溶液200mlに添加し、500rpmで16時間攪拌した後の溶解ポリマー量(単位;重量%)のことをいう。溶解ポリマー量の測定は、pH滴定を用いて行う。
特許文献7で初めて着目され記載の新規パラメーター(ゲル毛管吸収力)であり、ガラスフィルターの上面とマリオット管の下部のメニスカスとの間に高さ10cmの差をつけた状態で0.05psiの荷重下、10分間の液吸収能力を評価するものである。
吸水剤0.9gをそのままセル中で生理食塩水に対して自由膨潤させた後に荷重下で、ゲル層の通液性を測定するものである。特許文献WO2004/096304など「GBP」として知られている従来技術のGBPが全粒子中の特定粒度(300μm~600μmのみを篩い分けしてする)の測定を行うことに対して、FGBPは篩分けを行わずにあり姿のままの粒度(即ち全吸収剤粒子)での測定であり、吸水剤本来の通液性が評価できる。
本明細書において、範囲を示す「X~Y」は、「X以上Y以下」であることを意味する。又、重量の単位である「t(トン)」は、「Metricton(メトリックトン)」であることを意味し、さらに、特に注釈のない限り、「ppm」は「重量ppm」を意味する。又、「重量」と「質量」、「重量%」と「質量%」、「重量部」と「質量部」は同義語として扱う。さらに、「~酸(塩)」は「~酸及び/又はその塩」を意味し、「(メタ)アクリル」は「アクリル及び/又はメタクリル」を意味する。又、物性等の測定に関しては、特に断りのない限り、室温(20~25℃)、相対湿度40~50%RHで測定する。
上述したように、本発明に係る粒子状吸水剤の製造方法は、(i)(メタ)アクリル酸(塩)系単量体水溶液を調製する工程と、(ii)前記(メタ)アクリル酸(塩)系単量体水溶液を重合する工程と、(iii)重合中又は重合後の含水ゲル状架橋重合体をゲル粉砕し、含水ゲル粒子を得る工程と、(iv)前記含水ゲル粒子を乾燥し、乾燥物を得る工程と、(v)前記乾燥物を粉砕及び/又は分級して吸水性樹脂粉末を得る工程と、(vi)前記吸水性樹脂粉末を表面架橋し、吸水性樹脂粒子を得る工程と、(vii)前記吸水性樹脂粉末または前記吸水性樹脂粒子に通液性向上剤を添加する工程と、を含み、工程(iii)ないし工程(iii)以前に、前記含水ゲル状架橋重合体及び/又は前記含水ゲル粒子が接着することを制御する接着制御剤を添加することを有し、前記含水ゲル粒子の固形分が10~80重量%となり、かつ、乾燥物として換算した際の前記含水ゲル粒子の重量平均粒子径が50~650μmとなるように調整し、ポリ(メタ)アクリル酸(塩)系粒子状吸水剤の表面張力が60mN/m以上となり、無加圧下吸収倍率(CRC)が28g/g以上となるように調整する、ポリ(メタ)アクリル酸(塩)系吸水性樹脂粒子を主成分とする、ポリ(メタ)アクリル酸(塩)系粒子状吸水剤の製造方法である。
本明細書において、「(メタ)アクリル酸(塩)系単量体水溶液」とは、(メタ)アクリル酸(塩)を主成分とする単量体の水溶液であって、必要により架橋剤、グラフト成分や微量成分(キレート剤、界面活性剤、分散剤等)等の吸水性樹脂を構成する成分が調合されたものを指し、そのままの状態で重合開始剤を添加して重合に供されるものをいう。
本発明の吸水性樹脂には、(メタ)アクリル酸(塩)を主成分とする単量体が使用される。主成分とは、単量体(内部架橋剤を除く)全体に対して、(メタ)アクリル酸(塩)が通常50モル%以上、好ましくは70モル%以上、より好ましくは80モル%以上、さらに好ましくは90モル%以上、特に好ましくは95モル%以上(上限は100モル%)含まれる状態を指す。
本発明では、上記重合に際して、必要に応じて内部架橋剤が用いられる。該内部架橋剤としては、公知のものが使用でき、例えば、N,N’-メチレンビス(メタ)アクリルアミド、(ポリ)エチレングリコールジ(メタ)アクリレート、(ポリ)プロピレングリコールジ(メタ)アクリレート、トリメチルロールプロパントリ(メタ)アクリレート、グリセリントリ(メタ)アクリレート、グリセリンアクリレートメタクリレート、エチレンオキサイド変性トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリトールヘキサ(メタ)アクリレート、トリアリルシアヌレート、トリアリルイソシアヌレート、トリアリルホスフェート、トリアリルアミン、ポリ(メタ)アリロキシアルカン、(ポリ)エチレングリコールジグリシジルエーテル、グリセロールジグリシジルエーテル、エチレングリコール、ポリエチレングリコール、プロピレングリコール、グリセリン、1,4-ブタンジオール、ペンタエリスリトール、エチレンジアミン、エチレンカーボネート、プロピレンカーボネート、ポリエチレンイミン、グリシジル(メタ)アクリレート等を挙げることができる。これらの中から、反応性を考慮して、1種又は2種以上を使用することができ、中でも2個以上の重合性不飽和基を有する化合物を使用することが好ましい。
本発明の課題を高度に解決するためには、(2-3-2)で詳述する接着制御剤(融着制御剤と呼称することもできる)を添加し、それは、(メタ)アクリル酸(塩)系単量体水溶液の調製工程中又は後に添加してもよく、具体的には、具体的には、工程(iii)ないし工程(iii)以前に添加する。
本発明の吸水性樹脂粒子(含水ゲル状架橋重合体)を得るための重合方法としては、噴霧重合、液滴重合、バルク重合、沈殿重合、水溶液重合又は逆相懸濁重合等を挙げることができるが、本発明の課題解決には、単量体を水溶液とする水溶液重合や逆相懸濁重合が好ましい。
本発明において使用される重合開始剤は、重合形態によって適宜決定され、特に限定されないが、例えば、光分解型重合開始剤、熱分解型重合開始剤、レドックス系重合開始剤等が挙げられる。これらの重合開始剤によって、本発明の重合が開始される。
本発明の課題を高度に解決するためには、(2-3-2)で詳述する接着制御剤を添加する。接着制御剤は、重合工程の前、中、後のいずれかで添加してもよく、具体的には、工程(iii)ないし工程(iii)以前に添加する。
本発明において、(メタ)アクリル酸(塩)系単量体水溶液の重合方法として、吸水性樹脂の物性(例えば、吸水速度や通液性)や重合制御の容易性等の観点から、水溶液重合が採用される。中でもニーダー重合又はベルト重合が好ましく、連続水溶液重合がより好ましく採用される。
本工程は、上記重合工程等を経て得られる、含水ゲル状架橋重合体(以下、「含水ゲル」と称する場合がある)を粉砕することによって、粒子状の含水ゲル(以下、「含水ゲル粒子」または「粒子状含水ゲル」と称する場合がある)を得る工程である。上記含水ゲルのゲル粉砕、特に混練によるゲル粉砕によって粒子状含水ゲルを特定の粒子径範囲まで細粒化されることで、GCA、vortexが向上する。
本発明においては、粒子状含水ゲル(含水ゲル粒子)の重量平均粒子径が上記範囲に制御されることが重要であり、それを達成する手段は問わないが、例えば、バッチ式又は連続式の双腕型ニーダー等、複数の回転撹拌翼を備えたゲル粉砕機、1軸押出機、2軸押出機、ミートチョッパー等が挙げられる。中でも、先端に多孔板を有するスクリュー型押出機(例えば、ミートチョッパー)が好ましく、例えば、特開2000-063527号公報に開示されたスクリュー型押出機が挙げられる。
「ゲル粉砕エネルギー」(GGE(1),GGE(2))
本発明における「ゲル粉砕エネルギー」とは、含水ゲル状架橋重合体をゲル粉砕する際、ゲル粉砕装置が必要とする単位重量(含水ゲル状架橋重合体の単位重量)あたりの機械的エネルギーをいい、ジャケットを加熱冷却するエネルギーや投入する水・スチームのエネルギーは含まれない。尚、「ゲル粉砕エネルギー」は、英語表記の「Gel Grinding Energy」から「GGE(1)」と略称する。GGEは、ゲル粉砕装置が三相交流電力で駆動する場合、以下の式(1)によって算出される。


また、上記重合工程がベルト重合の場合、ゲル粉砕を行う前に、重合中又は重合後の含水ゲル状架橋重合体、好ましくは重合後の含水ゲル状架橋重合体を数10cm程度の大きさに切断又は粗砕することができる。ここで、ゲル切断又はゲル粗砕とは、粉砕機への連続投入できる大きさへの一次処理(例えば1000cm3以下ないし平面で1000cm2以下の一次処理)のことであり、いっぽう、ゲル粉砕とは、細粒化(特に乾燥物に換算した50~650ミクロンの重量平均粒子径等への細粒化)である点において区別される。
本発明において上記ゲル粉砕は、重合中及び/又は重合後に行われ、好ましくは重合後の含水ゲル状架橋重合体に対してに行われる。なお、重合中でのゲル粉砕を行う重合としてニーダー重合が挙げられるが、さらに重合後にゲル粉砕を行ってもよい。また重合後にゲル粉砕を行う重合としてベルト重合やタンク中での静置水溶液重合(実質無攪拌での水溶液重合)が好ましく挙げられるが、特にこれら重合に限定されない。
本発明のゲル粉砕工程で使用されるゲル粉砕装置が、スクリュー押出機(例えば、ミートチョッパー)である場合、そのスクリュー押出機のスクリュー軸回転数は、そのケーシングの内径、スクリュー軸外径等によって適宜調整すればよいが、軸回転数は、好ましくは80rpm~500rpm、より好ましくは90rpm~400rpm、さらに好ましくは100rpm~300rpmである。
本発明においては、ゲル粉砕後の粒径が有意に小さいものとなるのであれば、ゲル粉砕の処理回数は特に限定されないが、本発明の一形態によれば、複数回である。
ゲル温度、即ち、ゲル粉砕前の含水ゲル状架橋重合体の温度は、粒度制御や物性の観点から、好ましくは40~120℃、より好ましくは50~120℃、さらに好ましくは52~110℃、よりさらに好ましくは48~80℃であり、特に好ましくは56~70℃である。なお、65℃~110℃であってもよい。
ゲル粉砕前の含水ゲル状架橋重合体、及び、ゲル粉砕後の粒子状含水ゲル(含水ゲル粒子)のCRCは、好ましくは何れか一方、より好ましくは両方の値が25~50g/g、より好ましくは26~45g/g、さらに好ましくは27~40g/gである。上記ゲルCRCが上記範囲内である場合、ゲル粉砕時の粒子形状や粒度分布の制御が容易になるため好ましい。かようなゲルCRCは、重合時の架橋剤の添加量、その他重合濃度等で適宜制御することができる。なお、高CRCを有する吸水性樹脂が好ましいことは周知の事実であるが、上記ゲルCRCが上記範囲を超えて高い場合、粒子形状や粒度分布の制御がしにくくなる場合がある。
本発明において、ゲル粉砕前の含水ゲル状架橋重合体の樹脂固形分は、物性の観点から、好ましくは10~80重量%、より好ましくは20~60重量%、さらに好ましくは30~55重量%であり、よりさらに好ましくは33~50重量%であり、特に好ましくは36~46重量%である。
本発明のゲル粉砕工程においては、含水ゲル状架橋重合体に水を添加してゲル粉砕することもできる。なお、本発明において、「水」には、固体、液体、及び気体の少なくとも1つの形態を含むものとする。
本発明の課題をより高度に解決するためには、ゲル粉砕時にゲルが接着制御剤を含んでいる。換言すれば、ゲル粉砕が完全に終わりきる前に、接着制御剤を添加すればよい。そのためには、上記工程(i)の(メタ)アクリル酸(塩)系単量体水溶液の調整工程、工程(ii)の重合工程、工程(iii)のゲル粉砕工程の少なくとも一つの工程で接着制御剤が加えられ、また工程(i)と工程(ii)の間、或いは、工程(ii)と工程(iii)の間に添加する工程を設けてもよい。工程(i)と工程(ii)の間の工程としては、例えば、調製した(メタ)アクリル酸(塩)系単量体水溶液の貯蔵や輸送の工程が挙げられ、工程(ii)と工程(iii)の間の工程としては、例えば、含水ゲル状重合体の熟成工程が挙げられる。このように、工程(iii)ないし工程(iii)以前に、含水ゲル粒子の内部及び/又は表面に接着制御剤が含まれるようにすることによって、本発明の所期の効果を奏することができる。
接着制御剤の添加量は、特に制限されず、添加する接着制御剤の種類を考慮して決定すればよい。
非イオン性物質として、
(a)ポリオール類、
(b)ポリオール類のヒドロキシ基の変性物、
(c)側鎖及び/又は末端ポリエーテル変性ポリシロキサン、
(d)高級脂肪族アミンのアルキレンオキサイド付加物、
両イオン性物質として、
(e)アルキルアミノベタイン、
(f)アルキルアミンオキサイド
アニオン性物質として、
(g)高級アルコールアルキレンオキサイド付加物の硫酸エステル塩、
(h)アルキルジフェニルエーテルジスルホン酸塩
カチオン性物質として、
(i)アンモニウム塩
が挙げられる。
複数のヒドロキシ基を有するポリオール類として、(a-1)非高分子ポリオール類、(a-2)高分子ポリオール類が挙げられる。
具体的には、複数のヒドロキシ基を有する非高分子ポリオール類として、エチレングリコール、ジエチレングリコール、トリエチレングリコール、グリセリン、ジグリセリン、プロパンジオール、ブタンジオール、ペンタンジオール、ヘキサンジオール、オクタンジオール等のジ、トリ、テトラオールが挙げられる。
具体的には、複数のヒドロキシ基を有する高分子ポリオール類として、ポリエチレングリコールやポリプロピレングリコール、及びポリエチレングリコールとポリプロピレングリコールのブロック共重合体またはランダム共重合体等のポリアルキレングリコールを挙げることが出来る。ここで繰り返し単位のアルキレンユニットの炭素数はC1~C6が好ましく、C2~C4がより好ましく、C2~C3が特に好ましい。(本明細書において、炭素数の事をCのあとに数字を記載して表すことがある。例えば炭素数1であればC1、炭素数10であればC10と記載することがある)。
・プルロニックシリーズ
プルロニックL-34、プルロニックL-44、プルロニックL-64、プルロニックP-84、プルロニックP-85、プルロニックP-103、プルロニックF-68、プルロニックF-88、プルロニックF-108、プルロニック17R-3、プルロニック17R-4、プルロニックTR-704、プルロニックTR-913R
日油株式会社製
プロノン#104、プロノン#204、プロノン#208、ユニルーブ70DP-600B、ユニルーブ70DP-950B
第一工業製薬株式会社製
エパン450、エパン485、エパン680、エパン740、エパン750、エパン785、エパンU-103、エパンU-105、エパンU-108。
ポリオール類のヒドロキシ基の変性物とは、1つ以上のヒドロキシ基がエステル変性及び/又はエーテル変性されていることが好ましい。エ-テル及び/又はエステルの変性は炭化水素基が好ましく、炭化水素基はC1~C30が好ましく、C2~C28がより好ましく、C3~C26がさらに好ましく、C4~C24が特に好ましく、C6~C22が最も好ましい。炭素数がC30を超えると疎水性が強くなり、表面張力が低下する恐れがあり好ましくない。
グリシジル変性ポリオール類は、(ポリ)アルキレングリコールの末端の少なくとも1つがグリシジル基で修飾されているものである。具体的には、エチレングリコールジグリシジルエーテル、ジエチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテルなどの水溶性(ポリ)アルキレングリコールジグリシジルエーテル、プロピレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、ヘキサンジオールジグリシジルエーテル、グリセロールポリグリシジルエーテル、トリメチロールプロパンポリグリシジルエーテル、ペンタエリスリトールポリグリシジルエーテル、ジグリセロールポリグリシジルエーテル、ポリグリセロールポリグリシジルエーテル、ソルビトールポリグリシジルエーテル等のポリオールの水溶性ポリグリシジルエーテル類が挙げられる。
デナコールEX-145、デナコールEX-171、デナコールEX-211、デナコールEX-212、デナコールEX-252、デナコールEX-810、デナコールEX-811、デナコールEX-850、デナコールEX-851、デナコールEX-821、デナコールEX-830、デナコールEX-832、デナコールEX-841、デナコールEX-861、デナコールEX-911、デナコールEX-941、デナコールEX-920、デナコールEX-931、デナコールEX-313、デナコールEX-314、デナコールEX-321、デナコールEX-411、デナコールEX-421、デナコールEX-512、デナコールEX-521、デナコールEX-612、デナコールEX-614、デナコールEX-614B。
高級アルコールのアルキレンオキサイド付加物は、(ポリ)アルキレングリコールの片末端がC1~C30の炭化水素を有する置換基で修飾されているものであり、一般式を「化1」に示す。
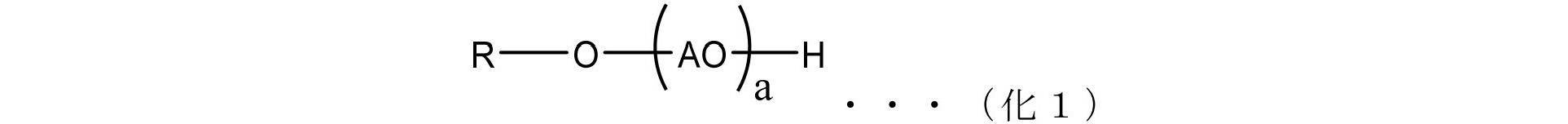
・ポリオキシエチレンラウリルエーテル
エマルゲン106(HLB=10.5)、エマルゲン108(HLB=12.1)、エマルゲン109P(HLB=13.6)、エマルゲン120(HLB=15.3)、エマルゲン123P(HLB=16.9)、エマルゲン130K(HLB=18.1)、エマルゲン147(HLB=16.3)、エマルゲン150(HLB=18.4)
・ポリオキシエチレンセチルエーテル
エマルゲン210P(HLB=10.7)、エマルゲン220(HLB=14.2)
・ポリオキシエチレンステアリルエーテル
エマルゲン320P(HLB=13.9)、エマルゲン350(HLB=17.8)
・ポリオキシエチレンオレイルエーテル
エマルゲン408(HLB=10.0)、エマルゲン409PV(HLB=12.0)、エマルゲン420(HLB=13.6)、エマルゲン430(HLB=16.2)
・ポリオキシエチレンミリスチルエーテル
エマルゲン4085(HLB=18.9)
・ポリオキシエチレンオクチルドデシルエーテル
エマルゲン2020G-HA(HLB=13.0)、エマルゲン2025G(HLB=15.7)
日油株式会社製
・ポリオキシエチレンイソデシルエーテル
ノニオンID-203(HLB=12.5)、ノニオンID-209(HLB=14.3)
・ポリオキシエチレン-2-エチルヘキシルエーテル
ノニオンEH-204(HLB=11.5)、ノニオンEH-208(HLB=14.6)
日本乳化剤株式会社性
・ポリオキシエチレンノニルフェニルエーテル
ニューコール560(HLB=10.9)、ニューコール564(HLB=12.3)、ニューコール565(HLB=13.3)、ニューコール566(HLB=14.1)、ニューコール568(HLB=15.2)、ニューコール504(HLB=16.0)、ニューコール506(HLB=17.2)、ニューコール509(HLB=18.0)、ニューコール516(HLB=18.8)。
多価アルコール脂肪酸エステルのエチレンオキサイド付加物は多価アルコールの少なくとも1つが(ポリ)アルキレングリコールで修飾されており、かつ少なくとも1つがエステル結合を介してC1~C30の炭化水素を有する置換基で修飾されているものである。多価アルコールとしては、グリセリン、ペンタエリスリトール、ソルビトール、ソルビタンおよび糖類などが挙げられる。

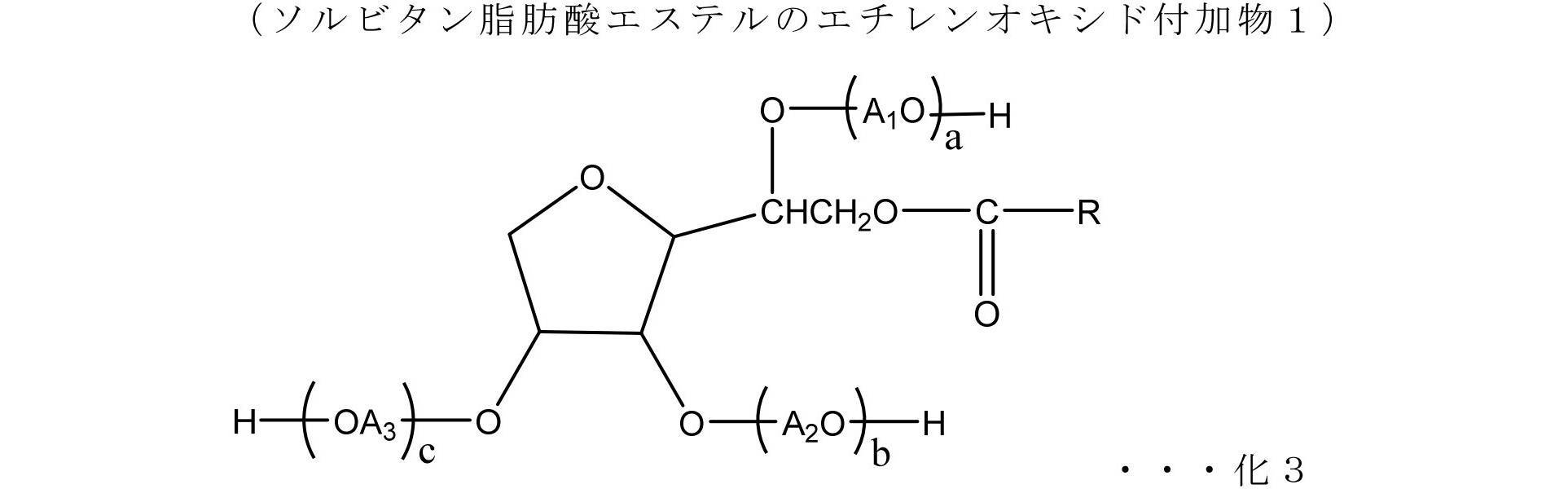

・ポリオキシエチレンソルビタンモノラウレート
レオドールTW-L120(HLB=16.7)、レオドールTW-L106(HLB=13.3)、レオドールスーパーTW-L120
・ポリオキシエチレンソルビタンモノパルミテート
レオドールTW-P120(HLB=15.6)
・ポリオキシエチレンソルビタンモノステアレート
レオドールTW-S120V(HLB=14.9)
・ポリオキシエチレンソルビタントリステアレート
レオドールTW-S320V(HLB=10.5)
・ポリオキシエチレンソルビタンモノオレート
レオドールTW-O120V(HLB=15.0)、レオドールTW-O106V(HLB=10.0)
・ポリオキシエチレンソルビタントリオレート
レオドールTW-O320V(HLB=11.0)
日油株式会社製
・ポリオキシエチレンヤシ脂肪酸グリセリル
ユニグリMK-207(HLB=13.0)、ユニグリMK-230(HLB=17.4)
((c)側鎖及び/又は末端ポリエーテル変性ポリシロキサン)
ポリシロキサンのポリエーテル変性部位は特に限定はしないが、ポリシロキサンの側鎖、ポリシロキサンの両末端、ポリシロキサンの片末端、ポリシロキサンの側鎖と両末端との両方のいずれでもよい。ポリエーテル変性基としては、ポリオキシエチレン基、ポリオキシプロピレン基、ポリオキシエチレン基およびポリオキシプロピレン基の両方を有するものが含まれる。
KF-351A(HLB=12)、KF-353(HLB=10)、KF-354L(HLB=16)、KF-355A(HLB=12)、KF-615A(HLB=10)、KF-640(HLB=14)、KF-642(HLB=12)、KF-643(HLB=14)、KF-6011(HLB=12)
東レ・ダウコーニング株式会社製
FZ-77(HLB=11)、L-7604(HLB=11)
((d)高級脂肪族アミンのアルキレンオキサイド付加物)
本発明の好ましい実施形態によれば、前記(d)高級脂肪族アミンのアルキレンオキサイド付加物が、C1~C30の炭化水素を有する1級アミンの2つの水素にアルキレンオキサイドが付加されたものである。かかる構成によって、粉砕された含水ゲル同士の接着を制御できる。

・ポリオキシエチレンラウリルアミン
ナイミーンL-207(HLB=12.5)、
・ポリオキシエチレンアルキル(ヤシ)アミン
ナイミーンF-215(HLB=15.4)
・ポリオキシエチレンステアリルアミン
ナイミーンS-210(HLB=12.5)、ナイミーンS-215(HLB=14.5)、ナイミーンS-220(HLB=15.4)
・ポリオキシエチレン牛脂アルキルアミン
ナイミーンT2-210(HLB=12.5)、ナイミーンT2-230(HLB=16.7)
・ポリオキシエチレンアルキルプロピレンジアミン
ナイミーンDT-208(HLB=10.7)
花王株式会社製
アミート105A(HLB=10.8)、アミート320(HLB=15.4)
((e)アルキルアミノベタイン)
アルキルアミノベタインは同一分子内の隣接しない位置に、カチオン性基とアニオン性基を有しており、カチオン性基が2~4級アンモニウムであり、2~4級アンモニウムの少なくとも1つがC1~C30の炭化水素基を有する置換基で修飾されているものであり、一般式を「化6」に示す。


アルキルアミンオキシドは同一分子内の隣接する位置に、カチオン性基とアニオン性基を有しており、カチオン性基が2~4級アンモニウムであり、2~4級アンモニウムの少なくとも1つがC1~C30の炭化水素基を有する置換基で修飾されているものであり、一般式を「化8」に示す。

アンヒトール20BS、アンヒトール24B(20BSの脱塩品)、アンヒトール86B、アンヒトール20N、アンヒトール20YB、アンヒトール20AB、アンヒトール55AB、アンヒトール20HD
第一工業製薬株式会社製:
アモーゲンS-H、アモーゲンK、アモーゲンLB-C、アモーゲンCB-H、アモーゲンHB-C、アモーゲンAOL
株式会社アデカ製:
アデカアンホートPB-30L、アデカアンホートAB-35L
日油株式会社製:
ニッサンアノンBF、ニッサンアノンBL、ニッサンアノンBL-SF、ニッサンアノンBDF-R、ニッサンアノンBDF-SF、ニッサンアノンBDC-SF、ニッサンアノンBDL-SF、ニッサンアノンGLM-R、ユニセーフA-LM、ユニセーフA-SM、ユニセーフA-LE
日本乳化剤製:
テクスノールR2。
本発明の好ましい実施形態によれば、前記(g)高級アルコールアルキレン付加物の硫酸エステル塩が、(ポリ)アルキレングリコールの1つの末端がC1~C30の炭化水素を有する置換基で修飾され、かつ、もう1つの末端が硫酸エステル塩である。かかる構成によって、粉砕された含水ゲル同士の接着を制御できる。
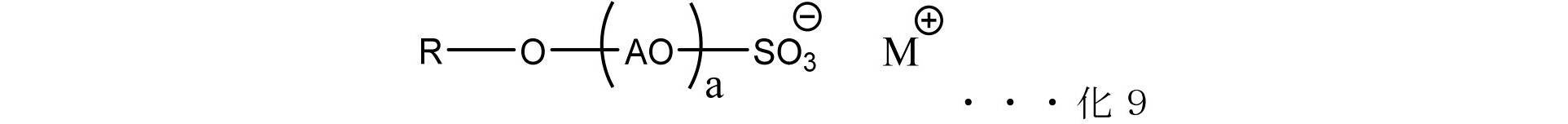
・ポリオキシエチレンラウリルエーテル硫酸ナトリウム
エマール20C、エマールE-27C、エマール270J、エマール20CM
日本乳化剤株式会社製
・ポリオキシエチレンアルキルエーテル硫酸エステル塩
ニューコール1020-SN、ニューコール2308-SF、ニューコール2320-SN、ニューコール2360-SN、ニューコール1305-SN、ニューコール1330-SF、ニューコール1703-SFD、ニューコール1525-SFC
日油株式会社製
・ポリオキシエチレンアルキルエーテル硫酸エステルナトリウム
パーソフトEP、ニッサントラックスK-40、ニッサントラックスK-300、パーソフトEF、パーソフトEDO、パーソフトEL、パーソフトEK
((h)アルキルジフェニルエーテルジスルホン酸塩)
アルキルジフェニルエーテルジスルホン酸塩の一般式を「化10」に示す。

・アルキルジフェニルエーテルジスルホン酸ナトリウム
ペレックスSS-L、ペレックスSS-H
竹本油脂株式会社製
・アルキルジフェニルエーテルジスルホン酸ナトリウム
パイオニンA-43-D、タケサーフA-43-NQ
((i)アンモニウム塩)
アンモニウム塩は、アンモニウム塩の少なくとも1つの水素がC1~C30の炭化水素を有する置換基で修飾されているものであり、一般式を「化11」に示す。
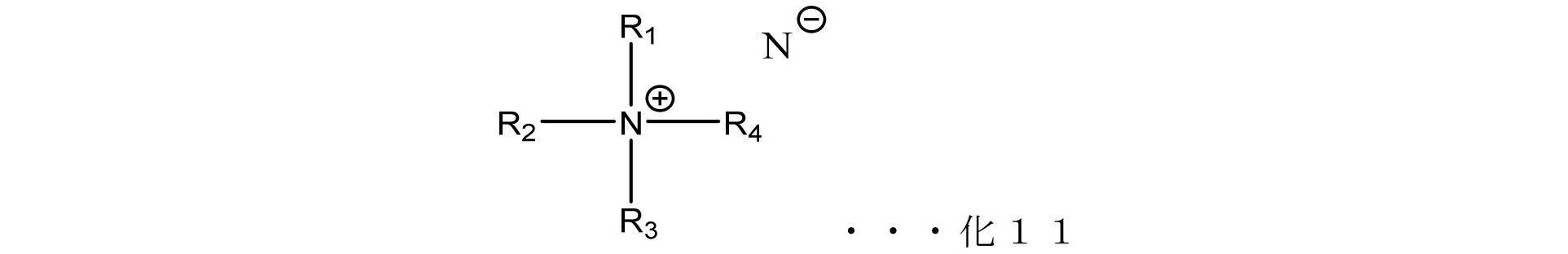
・ココナットアミンアセテート
アセタミン24
・ステアリルアミンアセテート
アセタミン86
・ラウリルトリメチルアンモニウムクロライド
コータミン24P
・ステアリルトリメチルアンモニウムクロライド
コータミン86W
・セチルトリメチルアンモニウムクロライド
コータミン60W
・ジステアリルジメチルアンモニウムクロライド
コータミンD86P
・アルキルベンジルジメチルアンモニウムクロライド
サニゾールC、サニゾールB-50
日油株式会社製
・テトラデシルアミンアセテート
ニッサンカチオンMA
・ドデシルトリメチルアンモニウムクロライド
ニッサンカチオンBB
・ヤシアルキルトリメチルアンモニウムクロライド
ニッサンカチオンFB
・ヘキサデシルトリメチルアンモニウムクロライド
ニッサンカチオンPB-300
・牛脂アルキルトリメチルアンモニウムクロライド
ニッサンカチオンABT2-500
・オクタデシルトリメチルアンモニウムクロライド
ニッサンカチオンAB、ニッサンカチオンAB-600
・ベヘニルトリメチルアンモニウムクロライド
ニッサンカチオンVB-Mフレーク、ニッサンカチオンVB-F
・ジデシルジメチルアンモニウムクロライド
ニッサンカチオン2-DB-500E
・ジオレイルジメチルアンモニウムクロライド
ニッサンカチオン2-OLR
・ヤシアルキルジメチルベンジルアンモニウムクロライド
ニッサンカチオンF2-50R
・テトラデシルジメチルベンジルアンモニウムクロライド
ニッサンカチオンM2-100R
本願で用いる接着制御剤は、親水性ユニット(4級アンモニウム塩等のカチオン性基、スルホン酸塩、アミン、ポリエチレングリコール鎖)と疎水性ユニット(炭化水素基)を同一化合物内に有することが好ましい。親水性ユニットとしては、特に4級アンモニウム塩、ポリエチレングリコール鎖が好ましい。
本工程は、上記重合工程等を経て得られる含水ゲル(含水ゲル粒子)を乾燥して乾燥重合体(乾燥物)を得る工程である。なお、上記重合工程が水溶液重合である場合、含水ゲルの乾燥前に、ゲル粉砕(細粒化)が行われる。また、乾燥工程で得られる乾燥重合体(凝集物)(乾燥物)はそのまま粉砕工程に供給されてもよい。
本工程は、上記乾燥工程で得られた乾燥重合体(乾燥物)を粉砕及び/又は分級して、好ましくは特定粒度の吸水性樹脂粉末を得る工程である。なお、上記(2-3)ゲル粉砕工程とは、粉砕対象物が乾燥工程を経ている点で異なる。
表面架橋前の吸水性樹脂粉末の重量平均粒子径(D50)は、取り扱い性(特に吸湿下での取り扱い性)、GCA、FGBP、吸水速度、加圧下吸水倍率等の観点から、300~500μmが好ましく、310~480μmがより好ましく、320~450μmがさらに好ましい。
本発明の吸水剤は、CRC≧28g/gであることが好ましいため、表面架橋前の吸水性樹脂粉末のCRCも28g/g以上、さらには30g/g以上であることが好ましく、その範囲で上記重合時の架橋剤量などが適宜調整されるとよい。表面架橋前の吸水性樹脂粉末のCRCは好ましくは30~60g/g、さらには32~55g/g、33~50g/gの範囲に架橋剤量や重合温度や乾燥温度など適宜調整される。
本工程は、さらに細かくは、以下の(2-6-1)に示す表面架橋剤添加工程と、(2-6-2)に示す加熱処理工程とからなる。
本工程は、上記吸水性樹脂粉末と、表面架橋剤とを混合することで、表面架橋工程に供する表面架橋剤を含有する吸水性樹脂粉末を調製する工程である。
本発明で使用できる有機表面架橋剤としては、得られる吸水性樹脂粒子の物性の観点から、ポリ(メタ)アクリル酸(塩)系吸水性樹脂粒子の官能基であるカルボキシル基と、脱水エステル化反応、あるいは脱水アミド化反応する、ヒドロキシ基及び/又はアミノ基等の反応性基を有する有機化合物が好ましい。
(ポリ)エチレングリコールジグリシジルエーテル、(ジ、ポリ)グリセロールポリグリシジルエーテル、グリシドール等のエポキシ化合物;
2-オキサゾリドン、N-ヒドロキシエチル-2-オキサゾリドン、1,2-エチレンビスオキサゾリン等のオキサゾリン化合物;
1,3-ジオキソラン-2-オン(つまり、エチレンカーボネート)、4-メチル-1,3-ジオキソラン-2-オン、4,5-ジメチル-1,3-ジオキソラン-2-オン、4,4-ジメチル-1,3-ジオキソラン-2-オン、4-エチル-1,3-ジオキソラン-2-オン、4-ヒドロキシメチル-1,3-ジオキソラン-2-オン、1,3-ジオキサン-2-オン、4-メチル-1,3-ジオキサン-2-オン、4,6-ジメチル-1,3-ジオキサン-2-オン、1,3-ジオキソパン-2-オン等のアルキレンカーボネート化合物;
エピクロロヒドリン、エピブロムヒドリン、α-メチルエピクロロヒドリン等のハロエポキシ化合物、及び、その多価アミン付加物(例えばハーキュレス製カイメン;登録商標);
γ-グリシドキシプロピルトリメトキシシラン、γーアミノプロピルトリエトキシシラン等のシランカップリング剤;
3-メチル-3-オキセタンメタノール、3-エチル-3-オキセタンメタノール、3-ブチル3-オキセタンメタノール、3-メチル-3-オキセタンエタノール、3-エチル-3-オキセタンエタノール、3-ブチル3-オキセタンエタノール、3-クロロメチル-3-メチルオキセタン、3-クロロメチル-3-エチルオキセタン、多価オキセタン化合物などのオキセタン化合物、2-イミダゾリジノン等の環状尿素化合物等が挙げられる。
オキサゾリン化合物としては2-オキサゾリジノンが好適に使用され、
アルキレンカーボネート化合物としては1,3-ジオキソラン-2-オン(つまり、エチレンカーボネート)が好適に使用される。
前記有機表面架橋剤の添加量は、その総量が、添加前の前記吸水性樹脂粉末100重量部に対して、0.001~15重量部であることが好ましく、0.01~5重量部であることがさらに好ましい。
添加前の前記吸水性樹脂粉末100重量部に対して、多価アルコール化合物の総量が0.001~10重量部であることが好ましく、0.01~5重量部であることがさらに好ましい。
前記表面架橋剤溶液は、表面架橋剤の反応や均一な混合を促進するため、前記有機表面架橋剤、前記親水性有機溶媒、前記界面活性剤及び前記水不溶性微粒子以外に、酸又は塩基を含んでいてもよい。
添加処理により、前記有機表面架橋剤は吸水性樹脂粉末に添加される。該添加処理の方法は特に限定されず、例えば、吸水性樹脂粉末を親水性有機溶剤に浸漬し、添加架橋剤を吸着させる方法、吸水性樹脂粉末に直接、添加し、架橋剤溶液を噴霧又は滴下して混合する方法等が例示でき、所定量を均一に添加する観点から、後者が好ましい。さらに、均一に添加するために、吸吸水性樹脂粉末を攪拌しながら添加処理を行うのが好ましく、さらに前記有機表面架橋剤溶液を噴霧するのが好ましい。
本工程は、吸水性樹脂粒子の加圧下吸水倍率やGCAを向上させるために、吸水性樹脂粉末の表面又は表面近傍を架橋処理するために加熱処理を行う工程である。ただし、過度の表面架橋処理はCRCを低下させすぎることがあることから、CRCが28g/g以上まで表面架橋処理する工程を有することが好ましい。
本発明で用いられる加熱装置としては、公知の乾燥機又は加熱炉に所定の雰囲気とするための気体排出機構及び/又は気体供給機構を具備せしめた連続式又は回分式(バッチ式)加熱装置、好ましくは連続式加熱装置が好適である。該加熱装置の加熱方式としては、伝導伝熱型、輻射伝熱型、熱風伝熱型、誘電加熱型が好適である。より好ましくは、伝導伝熱及び/又は熱風伝熱型の加熱方式であり、さらに好ましくは伝導伝熱型の方式である。
本工程は、FGBPを向上させるため通液向上剤を添加する工程であり、好ましくは上記表面架橋工程中又は後に行われる。
本発明における通液向上剤は、不溶性微粒子化合物及び多価カチオン性化合物から選択される添加剤、又は、通液向上剤を未使用の場合に比べてFGBPを向上させる添加剤をさす。
上記無機微粒子としては、二酸化ケイ素、二酸化チタン、酸化アルミニウム、酸化マグネシウム、酸化亜鉛、タルク、金属リン酸塩(例えばリン酸カルシウム、リン酸バリウム、リン酸アルミニウム)、金属硼酸塩(例えばホウ酸チタン、ホウ酸アルミニウム、ホウ酸鉄、ホウ酸マグネシウム、ホウ酸マンガン、及びホウ酸カルシウム)、珪酸又はその塩、粘土、珪藻土、ゼオライト、ベントナイト、カオリン、ハイドロタルサイト、活性白土等の水不溶性微粒子状無機粉体、乳酸カルシウム、乳酸アルミニウム、金属石鹸(長鎖脂肪酸の多価金属塩)等の有機微紛末が挙げられる。上記無機微粒子は、体積平均粒子径は10μm以下が好ましく、1μm以下がより好ましい。
カチオン性高分子化合物は、特に限定されないが、米国特許5382610号、同7098284号、WO2009/110645号、WO2009/041731号、WO2009/041727号に記載のカチオン性高分子化合物が好適に使用できる。
上記水溶性多価金属カチオン含有化合物は、2価以上、好ましく3価以上の、金属カチオンを含有する化合物を指す。該3価以上の金属カチオンとしては、アルミニウム、ジルコニウム、チタニウムが例示され、これらの中でもアルミニウムが好ましい。
本工程は吸水性樹脂粉末または表面架橋された吸水性樹脂粒子に種々の機能を付与するために、その他の添加剤を添加する工程であり、一つ又は複数の工程から構成される。上記添加剤としては、消臭剤、香料、抗菌剤、発泡剤、ジエチレントリアミン5酢酸3ナトリウム、ジエチレントリアミン五酢酸五ナトリウムなどのキレート剤、界面活性剤、着色防止剤、顔料、染料、肥料、酸化剤、還元剤等、機能を付与あるいは高めることが出来る剤である。また上記添加剤の添加においては、溶液で添加しても、ドライブレンドによる添加でもよい。
本発明では、ポリアクリル酸(塩)系吸水性樹脂粒子を主成分とする粒子状吸水剤であって、以下の(1)~(5)を満たす粒子状吸水剤も提供される。
本発明の粒子状吸水剤の無加圧下吸水倍率(CRC)は、上記製造方法で内部架橋又は表面架橋を適宜製造することで28g/g以上、より好ましくは29g/g以上、さらに好ましくは30g/g以上、特に好ましくは31g/g以上、最も好ましくは32g/g以上に制御される。
GCAとは、ガラスフィルターの上面とマリオット管の下部のメニスカスとの間に高さ10cmの差をつけた状態で0.05psiの荷重下、10分間の液吸収能力を評価するものである。GCAは10分間という短時間での吸収性能を評価しており、従来公知の加圧下吸水倍率(AAP)や、米国特許第7108916号に記載のFHAの場合、1時間での飽和状態での吸収性能を評価していることから、本発明に係るGCAとは思想を異にする評価方法である。粒子状吸水剤のGCAの値が高いほど、紙おむつにおいてパルプからの尿の吸い取り能力に優れ、戻り量を低減でき、肌かぶれや尿漏れを抑制できるようになる。
FGBPとは、底面がメッシュ構造となっているセル中で自由膨潤させた吸水剤層に0.3psiの荷重をかけた状態でゲル層上部から生理食塩水を注水し、ゲル層の生理食塩水透過能力を評価する方法である。FGBPの値が高いほど高吸収剤濃度吸収体での液の取り込み速度や戻り量が低減できる。
本発明の粒子状吸水剤の重量平均粒子径(D50)は、300~500μmが好ましく、310~480μmがさらに好ましく、320μm~450μmがよりさらに好ましい。
本発明の粒子状吸水剤の表面張力(実施例の測定法で規定)は、60mN/m以上であり、より好ましくは61mN/m以上、さらに好ましくは62mN/m以上、63mN/m以上であってもよく、64mN/m以上であってもよい。上限としては通常75mN/mで十分である。
本発明の粒子状吸水剤の加圧下吸水倍率は、後述の実施例で示すように、2.06kPaの圧力下における0.90重量%塩化ナトリウム水溶液に対する吸水倍率として規定されるが、好ましくは24g/g以上、より好ましくは25g/g以上、さらに好ましくは26g/g以上、特に好ましくは27g/g以上、最も好ましくは28g/g以上に制御される。
本発明の粒子状吸水剤の吸水時間(Vortex法)は40秒以下が好ましく、35秒以下がより好ましく、30秒以下がより好ましく、28秒以下がより好ましく、26秒以下がより好ましく、24秒以下がさらに好ましく、22秒以下が特に好ましく、19秒以下が最も好ましい。
本発明の粒子状吸水剤の含水率(180℃×3時間の乾燥減量で規定)は上記物性を満たす限り特に問わないが、0.1%~20%、さらには1%~15%、特に2%~10%に調整される。含水率が高いと物性を満たすのが困難になり、低いと吸水速度の低下や粒子の耐摩耗性が劣る傾向にある。
本発明の粒子状吸水剤の用途は特定に限定されないが、好ましくは、紙おむつや生理用ナプキンに使用される吸収体に好適に使用される。
以上、多くのパラメーター制御の吸水性樹脂があり、特許文献1~6などの従来技術に対して本発明者らは新規パラメーターであるGCA(Gel Capillary Absorption。ゲル毛管吸収)に着目した特許文献7を出願したが、いまだ不十分な点が見出された。そこで、上記課題を解決するために、本発明者らは鋭意検討した結果、新規パラメーターであるGCA(Gel Capillary Absorption ゲル毛管吸収)に着目した特許文献7では通液性がいまだ不十分であり、本発明者らは、GCAに加えて、通液性の新たな指標してFGBP(Free Gel Bed Permeability)が高いことで、上記課題を解決できること見出し、上記新規な吸水剤及びその製造方法を提供する。
無加圧下吸水倍率(CRC)は、ERT441.2-02に準じて測定した。即ち、試料0.200g(重量W0(g))を秤量し、不織布製の袋(60×85mm)に均一に入れヒートシールした後、23±2℃に調温した0.90重量%塩化ナトリウム水溶液500mL中に浸漬した。30分経過後、袋を引上げ、遠心分離機(株式会社コクサン社製遠心機:形式H-122)を用いて、250G、3分間の条件で水切りを行った。その後、袋の重量(W1(g))を測定した。同様の操作を、試料を入れずに行い、そのときの袋の重量(W2(g))を測定した。得られたW0(g)、W1(g)、W2(g)から下記(式1)にしたがって、無加圧下吸水倍率(CRC)を算出した。
msi:測定前の含水ゲル状架橋重合体、または含水ゲル粒子の重量(g)
mb :自由膨潤して水切り後のBlank(不織布のみ)の重量(g)
mwi:自由膨潤して水切り後の含水ゲル状架橋重合体および不織布の合計重量(g)
Wn :含水ゲル状架橋重合体、または含水ゲル粒子の固形分(重量%)
である。
本発明に係る粒子状吸水剤の加圧下吸水倍率(AAP)は、ERT442.2-02に準じて測定した。即ち、粒子状吸水剤0.900g(重量W3(g))を測定装置に投入し、測定装置一式の重量(W4(g))を測定した。次に、23±2℃に調温した0.90重量%塩化ナトリウム水溶液を2.06kPa(0.3psi,21g/cm2)の荷重下で吸収させた。1時間経過後、測定装置一式の重量(W5(g))を測定し、得られたW3(g)、W4(g)、W5(g)から下記(式3)にしたがって、加圧下吸水倍率(AAP)を算出した。
予め調製された0.90重量%塩化ナトリウム水溶液の1000重量部に食品添加物である食用青色1号0.02重量部を添加し、液温30℃に調整した。青色に着色した0.90重量%塩化ナトリウム水溶液50mlを100mlビーカーに計り取り、長さ40mmで太さ8mmの円筒型攪拌子で、600rpmで攪拌する中に、粒子状吸水剤2.00gを投入し、吸水時間(秒)を測定した。終点は、JISK 7224-1996年度「高吸水性樹脂の吸水速度試験方法解説」に記載されている基準に準じ、吸水剤が生理食塩水を吸液して試験液がスターラーチップを覆うまでの時間を吸水時間(秒)として測定した。
本発明に係る粒子状吸水剤(吸水性樹脂粉末)の粒度(PSD)及び粒度分布の対数標準偏差(σζ)は、米国特許出願公開第2006/204755号に開示された測定方法に準じて測定した。
(e)表面張力
十分に洗浄された100mlのビーカーに20℃に調整された0.90重量%塩化ナトリウム水溶液50mlを入れ、まず、0.90重量%塩化ナトリウム水溶液の表面張力を表面張力計(KRUSS社製のK11自動表面張力計)を用いて測定する。この測定において表面張力の値が71mN/m~75mN/mの範囲でなくてはならない。次に、20℃に調整した表面張力測定後の0.90重量%塩化ナトリウム水溶液50mlを含んだビーカーに、十分に洗浄された長さ25mmの円筒型攪拌子及び粒子状吸水剤0.500gを投入し、350rpmの条件で3分間攪拌する。3分後、攪拌を止め、2分間静置させて含水した粒子状吸水剤が沈降した後に、上澄み液の表面張力を再度同様の操作を行い測定した。ただし、粒子状吸水剤による吸水速度が速いため、もしくは吸収倍率が高いために粒子状吸水剤が沈降した後に、測定に必要な量の上澄み液が残らない場合は、0.90重量%塩化ナトリウム水溶液50mlの量を測定に必要な最低限の範囲で適宜調整して測定した。なお、本発明では白金プレートを用いるプレート法を採用し、プレートは各測定前に十分脱イオン水にて洗浄し、かつガスバーナーで加熱洗浄して使用する。
底面の直径が約5cmのアルミカップ(重量W8(g))に、約1gの吸水性樹脂(吸水剤)(粒子状含水ゲル)を量り取り(重量W9(g))、180℃の無風乾燥機中において3時間静置し、乾燥させた。乾燥後のアルミカップと吸水性樹脂(吸水剤)との合計重量(W10(g))を測定し、下記(式5)より固形分を求めた。また、含水率は、下記(式6)より求められる。
図1を参照してGCAを測定する装置及び方法を記載する。この測定法で使用されるガラスフィルター2はISO4793(1980)で規定される通りの500mlガラス濾過器であり、孔径がP40(16~40μm)、厚さ7mmであり、例えばSchott社のDuranガラス製濾過器のグレード3である。また20℃で30cm半径のフィルターが50mbarの圧力差にて50ml/minの水流能力を持たなければならない。このガラスフィルター付きの濾過器1の下部にシリコン製チューブ3をつなぎ、さらにガラス管5及びストップコック4を完備しているタンク6の下部につなぐ。このとき、ガラスフィルターの上面を、タンク内のガラス管の下部のメニスカスより10cm高い位置で固定する。系に0.90重量%塩化ナトリウム水溶液を満たす。内径60mmのプラスチックの支持円筒7の底に、8cmの正方形に切断された高湿潤強度セルロースティッシュ8を金属リングにより固定する。該ティッシュは坪量max24.6g/m2、湿潤引っ張り強度Min0.32N/cm(CD方向)、0.8N/cm(MD方向)(抄紙機で抄かれる際の流れ方向をMD方向、これに垂直な方向をCD方向)であり、例えばドイツのフリパ社(Fripa)から入手可能である。室温(20℃~25℃)、湿度50RH%の条件下で、該ティッシュ上に粒子状吸水剤100.2g(重量W11(g)を均一に散布し、その上に、吸水剤に対して0.39kPa(0.05psi)の荷重を均一に加えることができるよう調整された、外径が60mmよりわずかに小さく支持円筒との隙間が生じず、かつ上下の動きが妨げられないピストン9を載置し、この測定装置一式の重量(W12[g])を測定した。上記測定装置一式をガラスフィルター上に載せ、マリオット管付き流体貯槽のバルブを開けて、10分間吸収させる。その後測定装置一式を持ち上げ、その重量(W13(g))を測定した。W11、W12、W13から下記(式7)に従って、GCA(g/g)を算出した。
本発明のFGBPは、300μm~600μmの範囲の吸水剤を選別せず、吸水剤ありのままの粒子径で測定し、データ採取の時間を少なくとも20秒間にわたって1秒ごとから、180秒間にわたって5秒ごととした以外は国際公開WO2004/096304に記載の「自由膨潤」条件下でのゲルベッド透水性試験に準拠して行われる。
温度20℃~25℃の含水ゲル状架橋重合体(含水ゲル粒子)(固形分α重量%)20gを、0.08重量%エマール20C(界面活性剤、花王株式会社製)を含む20重量%塩化ナトリウム水溶液(以下、「エマール水溶液」と称する)1000g中に添加して分散液とし、長さ50mm×直径7mmのスターラーチップを300rpmで16時間攪拌した(高さ21cm、直径8cmの円柱のポリプロピレン製 約1.14L容器を使用)。
X;分級、水切り後に各篩上に残留した含水ゲル状架橋重合体(含水ゲル粒子)の重量%(%)
w;分級、水切り後に各篩上に残留した含水ゲル状架橋重合体(含水ゲル粒子)のそれぞれの重量(g)
W;分級、水切り後に各篩上に残留した含水ゲル状架橋重合体(含水ゲル粒子)の総重量(g)
R(α);固形分α重量%の含水ゲル状架橋重合体に換算したときの篩の目開き(mm)
r;20重量%塩化ナトリウム水溶液中で膨潤した含水ゲル状架橋重合体(含水ゲル粒子)が分級された篩の目開き(mm)である。
GelD50:含水ゲル粒子の重量平均粒子径(μm)
GS:含水ゲル粒子の固形分(重量%)
SolidD50:含水ゲル粒子の乾燥物に換算した重量平均粒子径(μm)
とすると次式で定義される。
(式)SolidD50=GelD50×(GS/100)1/3
(k)重量平均分子量
ポリエチレングリコール換算によるサイズ排除クロマトグラフィ(GPC)によって以下の測定条件で測定した。
装置:Waters社製、Waters Alliance(2695)
解析ソフト:Waters社製、Empowerプロフェッショナル+GPCオプション
使用カラム:東ソー社製、TSK guard column SWXL+TSKgel G4000SWXL+G3000SWXL+G2000SWXL
検出器:示差屈折率計(RI)検出器(Waters社製、Waters 2414)
溶離液:水10,999g、アセトニトリル6,001gの混合溶媒に酢酸ナトリウム三水和物115.6gを溶解し、さらに酢酸でpH6.0に調整した溶液
較正曲線作成用標準物質:ポリエチレングリコール[ピークトップ分子量(Mp)300000、200000、107000、50000、27700、11840、6450、1470、472]
較正曲線:上記ポリエチレングリコールのMp値と溶出時間とを基礎にして3次式で作成
流量:1.0mL/min
カラム温度:40℃
測定時間:45分
試料液注入量:100μL(試料濃度0.5wt%の溶離液調製溶液)
(l)BET比表面積
本発明の吸水性樹脂粉末のBET比表面積は、以下の手法に基づいて測定した。該BET比表面積の測定には、高精度ガス/蒸気吸着量測定装置(日本ベル株式会社製、BELSORP-max)を、また、前処理には吸着測定用前処理装置(日本ベル株式会社製、BELSORP-vacII)を用いた。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
温度計、窒素ガス導入管および排気孔を備えた蓋と底面300mm×220mm、深さ60mmのバットからなる反応容器に、アクリル酸170g、37重量%アクリル酸ナトリウム水溶液1800g、ポリエチレングリコールジアクリレート(重量平均分子量523)0.99g、ポリエチレングリコール(重量平均分子量2000、和光純薬株式会社製)6.688g(モノマー成分に対して0.8重量%)および脱イオン水216gを供給混合し、20℃の水浴に底から10mmの高さまで浸した。
得られた含水ゲル状架橋重合体(GK1)をブロックに切り分け、ユニパック(株式会社生産日本社製)に入れ、恒温器内に1時間静置し60℃に恒温させた。60℃に恒温させた含水ゲル状架橋重合体(GK1)を、3.5mmのダイス口径プレートを設置し、シートヒーターを用いて60℃に暖めたミートチョッパー(レマコム株式会社製、モデル:HL-G22SN)に2回通すことにより、含水ゲル粒子(「含水ゲル状架橋重合体粉砕物」とも称する)(GKF1)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF1)を、160℃、45分間熱風乾燥機で乾燥し、乾燥物を得、その後ロールミル(有限会社井ノ口技研社製)で粉砕し、目開き850μm、600μm、500μm、300μm、150μmを有する篩で篩い分けた後、850μmを通過し600μmを通過しない粒子が3重量%、600μmを通過し500μmを通過しない粒子が10重量%、500μmを通過し300μmを通過しない粒子が54重量%、300μmを通過し150μmを通過しない粒子が31重量%、150μmを通過し45μmを通過しない粒子が2重量%に調合することにより、吸水性樹脂粉末(B1)を得た。なお、当該調合は、実施例15以外同様に行った。
吸水性樹脂粉末(B1)100重量部に、エチレングリコールジグリシジルエーテル0.025重量部と、エチレンカーボネート0.3重量部と、プロピレングリコール0.5重量部と、脱イオン水2.0重量部とからなる表面架橋剤溶液を噴霧することによって混合した。上記の混合物を200℃で35分間加熱処理することにより、表面架橋された吸水性樹脂粒子(S1)を得た。
表面架橋された吸水性樹脂粒子(S1)100重量部に対して、キレート剤として、1重量%のDTPA(ジエチレントリアミン五酢酸五ナトリウム)水溶液1重量部を攪拌しながら添加し、1分間混合した。キレート剤を含有させることで、耐尿性が向上する。
使用したポリエチレングリコールの量を3.344g(モノマー成分に対して0.4重量%)に変更した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B2)、表面処理された吸水性樹脂粒子(S2)、粒子状吸水剤(EX-2)を得た。
使用したポリエチレングリコールの量を10.03g(モノマー成分に対して1.2重量%)に変更した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B3)、表面処理された吸水性樹脂粒子(S3)、粒子状吸水剤(EX-3)を得た。
使用したポリエチレングリコールの重量平均分子量を400に変更した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B4)、表面処理された吸水性樹脂粒子(S4)、粒子状吸水剤(EX-4)を得た。
使用したポリエチレングリコールの重量平均分子量を20000に変更した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B5)、表面処理された吸水性樹脂粒子(S5)、粒子状吸水剤(EX-5)を得た。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
温度計、窒素ガス導入管及び排気孔を備えた蓋と底面300mm×220mm、深さ60mmのバットからなる反応容器に、アクリル酸170g、37重量%アクリル酸ナトリウム水溶液1800g、ポリエチレングリコールジアクリレート(平均分子量523)0.99g、及び脱イオン水216gを供給混合し、20℃の水浴に底から10mmの高さまで浸した。この水溶液に窒素ガスを導入し20分間脱気した。この溶液が20℃になったのを確認後、窒素気流雰囲気下20重量%過硫酸ナトリウム水溶液6.61g、及び0.1重量%L-アスコルビン酸水溶液6.33gを添加し、攪拌混合した。単量体濃度は38重量%であった。1分後に重合が開始し、その時の反応系温度は20℃であった。重合開始後、重合系は攪拌せず、引き続き20℃の水浴に反応容器を浸して冷却を行った。17分後に重合系は最高到達温度の89℃を示した。この後、水浴の温度を70℃にし、20分間重合反応を行い、含水ゲル状架橋重合体(GK6)を得た。得られた含水ゲル状架橋重合体(GK6)をブロック状に切り分けた。
得られた含水ゲル状架橋重合体粉砕物(GKF6)を、160℃、45分間熱風乾燥機で乾燥し、乾燥物を得、その後ロールミル(有限会社井ノ口技研社製)で粉砕し、目開き850μm、600μm、500μm、300μm、150μmを有する篩で篩い分けた後調合することにより、吸水性樹脂粉末(B6)を得た。得られた吸水性樹脂粉末(B6)の性能を表1に示した。
吸水性樹脂粉末(B6)100重量部に、エチレングリコールジグリシジルエーテル0.025重量部と、エチレンカーボネート0.3重量部と、プロピレングリコール0.5重量部と、脱イオン水2.0重量部とからなる表面架橋剤溶液を混合した。上記の混合物を200℃で35分間加熱処理することにより、表面架橋された吸水性樹脂粒子(S6)を得た。
表面架橋された吸水性樹脂粒子(S6)100重量部に対して、1重量%のDTPA水溶液1重量部を攪拌しながら添加し、1分間混合した。次いで60℃の熱風乾燥機中に30分間放置してから、目開き850μmの金網を通過させ、ハイドロタルサイト(DHT-6、協和化学工業株式会社)0.6重量部を混合した。混合は表面架橋された吸水性樹脂粒子(S6)30gを容量225mlのマヨネーズ瓶にハイドロタルサイトと共に入れ、ペイントシェーカーを用いて3分間振とうし、粒子状吸水剤(EX-6)を得た。得られた粒子状吸水剤(EX-6)の性能を下記表に示した。
実施例1における表面架橋された吸水性樹脂粒子(S1)100重量部に対して、1重量%のDTPA水溶液1重量部を攪拌しながら添加して1分間混合後、さらに、27.5重量%硫酸アルミニウム水溶液(酸化アルミニウム換算で8重量%)1.17重量部、60重量%乳酸ナトリウム水溶液0.196重量部及びプロピレングリコール0.029重量部からなる溶液を添加して1分間混合後、熱風乾燥機中に30分間放置してから、目開き850μmの金網を通過させ、粒子状吸水剤(EX-7)を得た。得られた粒子状吸水剤(EX-7)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更した以外は実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK8)を得た。
得られた含水ゲル状架橋重合体(GK8)をブロック状に切り分け、ユニパック(株式会社生産日本社製)に入れ、恒温器内に1時間静置し60℃に恒温させた。60℃に恒温させた含水ゲル状架橋重合体(GK8)を、シートヒーターを用いて60℃に暖めたスクリュー押出機に供給しゲル粉砕した。該スクリュー押出機としては、先端部に直径100mm、孔径3.2mm、孔数316個、開口率32.3%、厚さ10mmの多孔板が供えられた、スクリュー軸の外径が86mm、ケーシング内径が88mmであるミートチョッパーを使用した。
得られた含水ゲル状架橋重合体粉砕物(GKF8)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B8)を得た。
得られた吸水性樹脂粉末(B8)を実施例1と同様の操作を行い、表面架橋された吸水性樹脂粒子(S8)、粒子状吸水剤(EX-8)を得た。得られた粒子状吸水剤(EX-8)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコールを使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK9)を得た。
得られた含水ゲル状架橋重合体(GK9)を1680g/minで供給すると同時に、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)6.688g(モノマー濃度に対して0.8重量%)を20重量%水溶液として20.5g/minで供給した以外は実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF9)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF9)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B9)を得た。
得られた吸水性樹脂粉末(B9)を実施例1と同様の操作を行い、表面架橋された吸水性樹脂粒子(S9)、粒子状吸水剤(EX-9)を得た。得られた粒子状吸水剤(EX-9)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更した以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK10)を得た。
得られた含水ゲル状架橋重合体(GK10)を、実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF11)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF10)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B10)を得た。
吸水性樹脂粉末(B10)の加熱処理時間を45分とすること以外は、実施例1と同様の操作を行い表面処理された吸水性樹脂粒子(S10)を得た。
表面処理された吸水性樹脂粒子(S10)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-10)を得た。得られた粒子状吸水剤(EX-10)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
アクリル酸380g、48重量%水酸化ナトリウム水溶液158g、ポリエチレングリコールジアクリレート(重量平均分子量523)1.53g、ポリエチレングリコール(重量平均分子量2000、和光純薬株式会社製)3.74g(モノマー成分に対して0.8重量%)、0.1重量%ジエチレントリアミン5酢酸3ナトリウム水溶液23.4gからなる単量体溶液を作成する。次に、45℃に調温した該単量体溶液を攪拌しながら、48重量%水酸化ナトリウム水溶液162gを加える。この時の中和熱によって80℃まで上昇する。さらに、4重量%過硫酸ナトリウム水溶液18.58gを加えたのち、雰囲気温度を60℃の中で、あらかじめ50℃に底面を加温しておいたテフロン(登録商標)シート(四方を1.5cmの高さを有する堰で囲われた30cm×30cmの反応容器)上に流し込み重合を行った。過硫酸ナトリウム水溶液を加えてから1分後に、重合系は最高到達温度が105℃を示した。さらに4分経過後に得られた重合物を取り出し、含水ゲル状架橋重合体(GK11)を得た。
スクリュー軸回転数を130rpm、含水ゲル状架橋重合体(GK11)を4640g/minでミートチョッパーに供給した以外は、実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF11)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF11)を、190℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B11)を得た。
吸水性樹脂粉末(B11)の加熱温度処理時間を25分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S11)を得た。
表面処理された吸水性樹脂粒子(S11)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-11)を得た。得られた粒子状吸水剤(EX-11)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.39gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)の代わりにアンヒトール20BS(花王株式会社、有効成分30重量%)1.17g(モノマー成分に対して有効成分で0.075重量%)を使用した以外は、実施例11と同様の操作を行い、含水ゲル状架橋重合体(GK12)を得た。
スクリュー押出機として、先端部に直径100mm、孔径6.4mm、孔数83個、開口率34.0%、厚さ10mmの多孔板を有し、スクリュー軸の外径が86mm、ケーシング内径が88mmであるミートチョッパーを使用した。ミートチョッパーを用い、含水ゲル状架橋重合体(GK12)を、スクリュー軸回転数130rpm、4640g/minでミートチョッパーに供給した以外は、実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF12)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF12)を、190℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B12)を得た。
吸水性樹脂粉末(B12)の加熱温度処理時間を30分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S12)を得た。
表面処理された吸水性樹脂粒子(S12)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-12)を得た。得られた粒子状吸水剤(EX-12)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)の代わりにアンヒトール20BS(花王株式会社製、有効成分30重量%)1.17g(モノマー成分に対し有効成分で0.075重量%)を使用した以外は、実施例11と同様の操作を行い、含水ゲル状架橋重合体(GK13)を得た。
スクリュー軸回転数を130rpmとし、含水ゲル状架橋重合体(GK13)を4640g/minでミートチョッパーに供給した以外は、実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF13)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF13)を、190℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B13)を得た。
吸水性樹脂粉末(B13)の加熱温度処理時間を25分とした以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S13)を得た。
表面処理された吸水性樹脂粒子(S13)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-13)を得た。得られた粒子状吸水剤(EX-13)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)の代わりにアンヒトール20BS(花王株式会社製、有効成分30重量%)2.090g(モノマー成分に対して有効成分で0.075重量%)を使用した以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK14)を得た。
スクリュー軸回転数を225rpmとした以外は、実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF14)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF14)を、185℃、30分間熱風乾燥機で乾燥し、850μmを通過し600μmを通過しない粒子が12重量%、600μmを通過し500μmを通過しない粒子が25重量%、500μmを通過し300μmを通過しない粒子が41重量%、300μmを通過し150μmを通過しない粒子が21重量%、150μmを通過し45μmを通過しない粒子が1重量%に調合すること以外は実施例1と同様の操作を行い、吸水性樹脂粉末(B14)を得た。
吸水性樹脂粉末(B14)の加熱温度処理時間を25分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S14)を得た。
表面処理された吸水性樹脂粒子(S14)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-14)を得た。得られた粒子状吸水剤(EX-14)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)の代わりにアンヒトール20BS(花王株式会社製、有効成分30重量%)2.090g(モノマー成分に対して有効成分で0.075重量%)を使用した以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK15)を得た。
スクリュー軸回転数を225rpmとした以外は、実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF15)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF15)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B15)を得た。
吸水性樹脂粉末(B15)の加熱温度処理時間を25分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S15)を得た。
表面処理された吸水性樹脂粒子(S15)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-15)を得た。得られた粒子状吸水剤(EX-15)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK16)を得た。
得られた含水ゲル状架橋重合体(GK16)をブロックに切り分け、ユニパック(株式会社生産日本社製)に入れ、恒温器内に1時間静置し60℃に恒温させた。60℃に恒温させた含水ゲル状架橋重合体(GK16)の表面に均一に1重量%のアンヒトール20BS(花王株式会社製、有効成分30重量%)を含むメタノール溶液62.7g(工程(i)の原料モノマー成分量に対して有効成分で0.075重量%)を振りかけ、4.7mmのダイス口径プレートを設置し、シートヒーターを用いて60℃に暖めたミートチョッパー(飯塚工業株式会社製、モデル:ROYAL、タイプ:VR-400K)に2回通すことにより、含水ゲル状架橋重合体粉砕物(GKF16)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF16)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B16)を得た。
吸水性樹脂粉末(B16)の加熱温度処理時間を30分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S16)を得た。
表面処理された吸水性樹脂粒子(S16)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-16)を得た。得られた粒子状吸水剤(EX-16)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK17)を得た。
1重量%のアンヒトール20HD(花王株式会社製、有効成分30重量%)を含むメタノール溶液62.7g(工程(i)の原料モノマー成分量に対して有効成分で0.075重量%)を、得られた含水ゲル状架橋重合体(GK17)を切り分けたブロックの表面に均一に振りかけた以外は、実施例16と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF17)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF17)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B17)を得た。
吸水性樹脂粉末(B17)の加熱温度処理時間を30分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S17)を得た。
表面処理された吸水性樹脂粒子(S17)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-17)を得た。得られた粒子状吸水剤(EX-17)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK18)を得た。
1重量%のアンヒトール20N(花王株式会社製、有効成分30重量%)を含むメタノール溶液62.7g(工程(i)の原料モノマー成分量に対して有効成分で0.075重量%)を、得られた含水ゲル状架橋重合体(GK18)を切り分けたブロックの表面に均一に振りかけた以外は、実施例17と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF18)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF18)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B18)を得た。
吸水性樹脂粉末(B18)の加熱温度処理時間を30分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S18)を得た。
表面処理された吸水性樹脂粒子(S18)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-18)を得た。得られた粒子状吸水剤(EX-18)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK19)を得た。
0.75重量%のアミート105A(花王株式会社製、有効成分100重量%)を含むメタノール溶液55.7g(工程(i)の原料モノマー成分量に対して有効成分で0.050重量%)を、得られた含水ゲル状架橋重合体(GK19)を切り分けたブロックの表面に均一に振りかけた以外は、実施例16と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF19)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF19)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B19)を得た。
吸水性樹脂粉末(B19)の加熱温度処理時間を30分とした以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S19)を得た。
表面処理された吸水性樹脂粒子(S19)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-19)を得た。得られた粒子状吸水剤(EX-19)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK20)を得た。
1重量%のエマール20C(花王株式会社製、有効成分25重量%)を含むイソプロピルアルコール溶液62.7g(工程(i)の原料モノマー成分量に対して有効成分で0.075重量%)を、得られた含水ゲル状架橋重合体(GK20)を切り分けたブロックの表面に均一に振りかけた以外は、実施例16と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF20)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF20)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B20)を得た。
吸水性樹脂粉末(B20)の加熱温度処理時間を30分とした以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S20)を得た。
表面処理された吸水性樹脂粒子(S20)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-20)を得た。得られた粒子状吸水剤(EX-20)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK21)を得た。
1重量%のアセタミン24(花王株式会社製、有効成分98重量%)を含むメタノール溶液62.7g(工程(i)の原料モノマー成分量に対して有効成分で0.075重量%)を、得られた含水ゲル状架橋重合体(GK21)を切り分けたブロックの表面に均一に振りかけた以外は、実施例16と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF21)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF21)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B21)を得た。
吸水性樹脂粉末(B21)の加熱温度処理時間を20分とした以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S21)を得た。
表面処理された吸水性樹脂粒子(S21)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-21)を得た。得られた粒子状吸水剤(EX-21)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK22)を得た。
1重量%のアデカプルロニックL-44(株式会社ADEKA製、有効成分100重量%)を含むメタノール溶液62.7g(工程(i)の原料モノマー成分量に対して有効成分で0.075重量%)を、得られた含水ゲル状架橋重合体(GK22)を切り分けたブロックの表面に均一に振りかけた以外は、実施例16と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF22)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF22)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B22)を得た。
吸水性樹脂粉末(B22)の加熱温度処理時間を30分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S22)を得た。
表面処理された吸水性樹脂粒子(S22)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-22)を得た。得られた粒子状吸水剤(EX-22)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK23)を得た。
1重量%のエマルゲン430(花王株式会社製、有効成分100重量%)を含むメタノール溶液62.7g(工程(i)の原料モノマー成分量に対して有効成分で0.075重量%)を、得られた含水ゲル状架橋重合体(GK23)を切り分けたブロックの表面に均一に振りかけた以外は、実施例16と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF23)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF23)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B23)を得た。
吸水性樹脂粉末(B23)の加熱温度処理時間を30分とした以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S23)を得た。
表面処理された吸水性樹脂粒子(S23)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-23)を得た。得られた粒子状吸水剤(EX-23)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK24)を得た。
1重量%のペレックスSS-L(花王株式会社製、有効成分50重量%)を含むメタノール溶液62.7g(工程(i)の原料モノマー成分量に対して有効成分で0.075重量%)を、得られた含水ゲル状架橋重合体(GK24)を切り分けたブロックの表面に均一に振りかけた以外は、実施例16と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF24)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF24)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B24)を得た。
吸水性樹脂粉末(B24)の加熱温度処理時間を30分とした以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S24)を得た。
表面処理された吸水性樹脂粒子(S24)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-24)を得た。得られた粒子状吸水剤(EX-24)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK25)を得た。
10重量%のデナコールEX-861(ナガセケムテックス株式会社製、有効成分100重量%)を含むメタノール溶液66.9g(工程(i)の原料モノマー成分量に対して有効成分で0.8重量%)を、得られた含水ゲル状架橋重合体(GK25)を切り分けたブロックの表面に均一に振りかけた以外は、実施例16と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF25)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF25)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B25)を得た。
吸水性樹脂粉末(B25)の加熱温度処理時間を20分とした以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S25)を得た。
表面処理された吸水性樹脂粒子(S25)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-25)を得た。得られた粒子状吸水剤(EX-25)の性能を下記表に示した。
(ゲル重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK26)を得た。
1重量%のレオドールTW-S120V(花王株式会社製、有効成分100重量%、カタログHLB値14.9)を含むメタノール溶液62.7g(工程(i)の原料モノマー成分量に対して有効成分で0.050重量%)を、得られた含水ゲル状架橋重合体(GK26)を切り分けたブロックの表面に均一に振りかけた以外は、実施例16と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF26)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF26)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B26)を得た。
吸水性樹脂粉末(B26)の加熱温度処理時間を20分とした以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S26)を得た。
表面処理された吸水性樹脂粒子(S26)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-26)を得た。得られた粒子状吸水剤(EX-26)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)を使用しなかった以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK27)を得た。
1重量%のKF-354L(信越シリコーン株式会社製、有効成分100重量%、カタログHLB値16)を含むメタノール溶液62.7g(工程(i)の原料モノマー成分量に対して有効成分で0.050重量%)を、得られた含水ゲル状架橋重合体(GK27)を切り分けたブロックの表面に均一に振りかけた以外は、実施例16と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF27)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF27)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B27)を得た。
吸水性樹脂粉末(B27)の加熱温度処理時間を20分とした以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S27)を得た。
表面処理された吸水性樹脂粒子(S27)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-27)を得た。得られた粒子状吸水剤(EX-27)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更する以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK28)を得た。
スクリュー押出機として、先端部に直径100mm、孔径8.0mm、孔数54個、開口率34.5%、厚さ10mmの多孔板を有し、スクリュー軸の外径が86mm、ケーシング内径が88mmであるミートチョッパーを使用した以外は、実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF28)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF28)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B28)を得た。
得られた吸水性樹脂粉末(B28)を、実施例1と同様の操作を行い、表面架橋された吸水性樹脂粒子(S28)、粒子状吸水剤(EX-28)を得た。得られた粒子状吸水剤(EX-28)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)の代わりにアンヒトール20BS(花王株式会社製、有効成分30重量%)2.090g(モノマー成分に対して有効成分で0.075重量%)を使用した以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK29)を得た。
スクリュー軸回転数を225rpmとした以外は、実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF29)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF29)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B29)を得た。
吸水性樹脂粉末(B29)の加熱温度処理時間を40分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S29)を得た。
表面処理された吸水性樹脂粒子(S29)を実施例1と同様の操作を行い、粒子状吸水剤(EX-29)を得た。得られた粒子状吸水剤(EX-29)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールジアクリレートの量を1.73gに変更し、ポリエチレングリコール(重量平均分子量2000、和光純薬工業株式会社製)の代わりにアンヒトール20BS(花王株式会社製、有効成分30重量%)2.090g(モノマー成分に対して有効成分で0.075重量%)を使用した以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(GK30)を得た。
スクリュー軸回転数を225rpmとした以外は、実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF30)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF30)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B30)を得た。
吸水性樹脂粉末(B30)の加熱温度処理時間を20分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S30)を得た。
表面処理された吸水性樹脂粒子(S30)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-30)を得た。得られた粒子状吸水剤(EX-30)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
実施例11と同様の操作を行い、含水ゲル状架橋重合体(GK31)を得た。
スクリュー押出機として、先端部に直径100mm、孔径6.4mm、孔数83個、開口率34.0%、厚さ10mmの多孔板を有し、スクリュー軸の外径が86mm、ケーシング内径が88mmであるミートチョッパーを用い、スクリュー軸回転数を130rpm、含水ゲル状架橋重合体(GK32)を4640g/minでミートチョッパーに供給した以外は、実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF31)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF31)を、190℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(B31)を得た。
吸水性樹脂粉末(B31)の加熱温度処理時間を20分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S31)を得た。
表面処理された吸水性樹脂粒子(S31)を実施例1と同様の操作を行い、粒子状吸水剤(EX-31)を得た。得られた粒子状吸水剤(EX-31)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
実施例13と同様の操作を行い、含水ゲル状架橋重合体(GK32)を得た。
スクリュー押出機としては、先端部に直径100mm、孔径6.4mm、孔数83個、開口率34.0%、厚さ10mmの多孔板を有し、スクリュー軸の外径が86mm、ケーシング内径が88mmであるミートチョッパーを用い、スクリュー軸回転数を130rpm、含水ゲル状架橋重合体(GK33)を4640g/minでミートチョッパーに供給した以外は、実施例8と同様の操作を行い、含水ゲル状架橋重合体粉砕物(GKF32)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF32)を、190℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末((B32)を得た。
吸水性樹脂粉末(B32)の加熱温度処理時間を20分としたこと以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(S32)を得た。
表面処理された吸水性樹脂粒子(S32)を、実施例1と同様の操作を行い、粒子状吸水剤(EX-32)を得た。得られた粒子状吸水剤(EX-32)の性能を下記表に示した。
実施例1において、ゲル粉砕工程で使用したミートチョッパーのダイス口径を3.5mmから9.0mmに変更した以外は、実施例1と同様の処理を行い、含水ゲル粒子(CGKF-1)、吸水性樹脂粉末(CB-1)、表面処理された吸水性樹脂粒子(CS1)、粒子状吸水剤(CEX-1)を得た。
実施例1で得られたS1を比較用吸水剤(CEX-2)とした。得られた粒子状吸水剤(CEX-2)の性能を下記表に示した。
出願番号PCT/JP2015/56110の実施例1と同様に行い、造粒形状を有する吸水性樹脂粉末(CB-3)、比較用吸水剤(CEX-3)を得た。得られた吸水性樹脂粉末(CB-3)、粒子状吸水剤(CEX-3)の性能を下記表に示した。
出願番号PCT/JP2015/56110の実施例2と同様に行い、造粒形状を有する吸水性樹脂粉末(CB-4)、比較用吸水剤(CEX-4)を得た。得られた吸水性樹脂粉末(CB-4)、粒子状吸水剤(CEX-4)の性能を下記表に示した。
国際公開第2011/126079号パンフレット実施例6に記載の方法により含水ゲル粒子(CGKF-5)、吸水性樹脂粉末(CB-5)、比較用粒子状吸水剤(CEX-5)を作成した。得られた含水ゲル粒子(CGKF-5)、吸水性樹脂粉末(CB-5)、吸水性樹脂粉末(CEX-5)、粒子状吸水剤(CEX-6)の性能を下記表に示した。
国際公開第2015/030130の実施例13と同様に行い、含水ゲル粒子(CGKF-6)、吸水性樹脂粉末(CB-6)、比較用吸水剤(CEX-6)を得た。得られた含水ゲル粒子(CGKF-6)、吸水性樹脂粉末(CB-6)、粒子状吸水剤(CEX-6)の性能を下記表に示した。
国際公開第2008/096713の実施例6と同様に行い、含水ゲル粒子(CGKF-6)、比較用吸水剤(CEX-7)を得た。得られた含水ゲル粒子(CGKF-6)、粒子状吸水剤(CEX-7)の性能を下記表に示した。
((メタ)アクリル酸(塩)系単量体水溶液の調製、重合工程)
ポリエチレングリコールの代わりに、ジ(2-エチルヘキシル)スルホコハク酸ナトリウムを0.418g(モノマー成分に対して0.05重量%)に変更した以外は、実施例1と同様の操作を行い、含水ゲル状架橋重合体(CGK8)を得た。
実施例1と同様の操作を行い、含水ゲル状架橋重合体粉砕物(CGKF8)を得た。
得られた含水ゲル状架橋重合体粉砕物(GKF8)を、185℃、30分間熱風乾燥機で乾燥した以外は、実施例1と同様の操作を行い、吸水性樹脂粉末(CB8)を得た。
吸水性樹脂粉末(CB8)の加熱温度処理時間を60分とした以外は、実施例1と同様の操作を行い、表面処理された吸水性樹脂粒子(CS8)を得た。
表面処理された吸水性樹脂粒子(CS8)を、実施例1と同様の操作を行い、粒子状吸水剤(CEX-8)を得た。得られた粒子状吸水剤(CEX-8)の性能を下記表に示した。

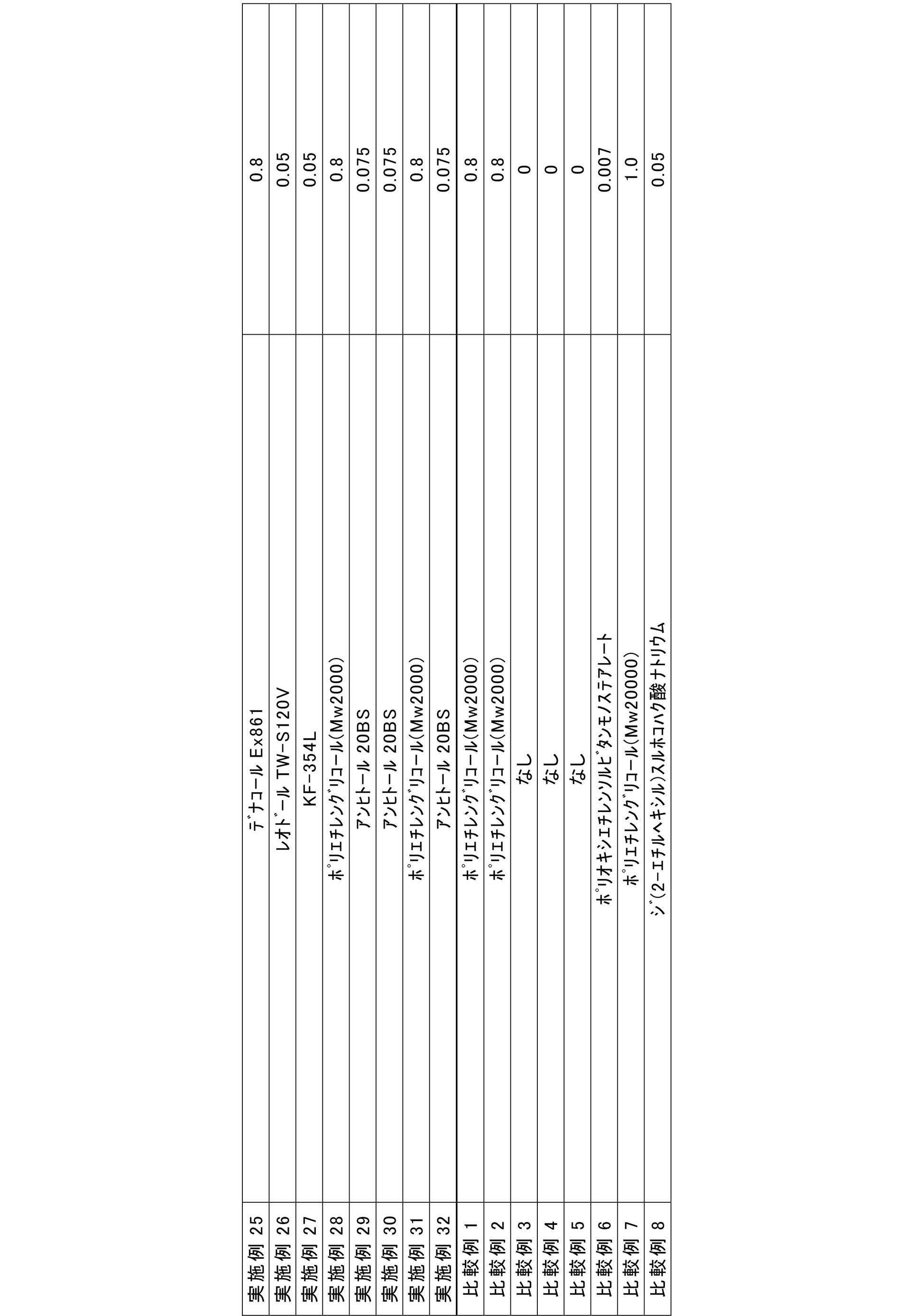
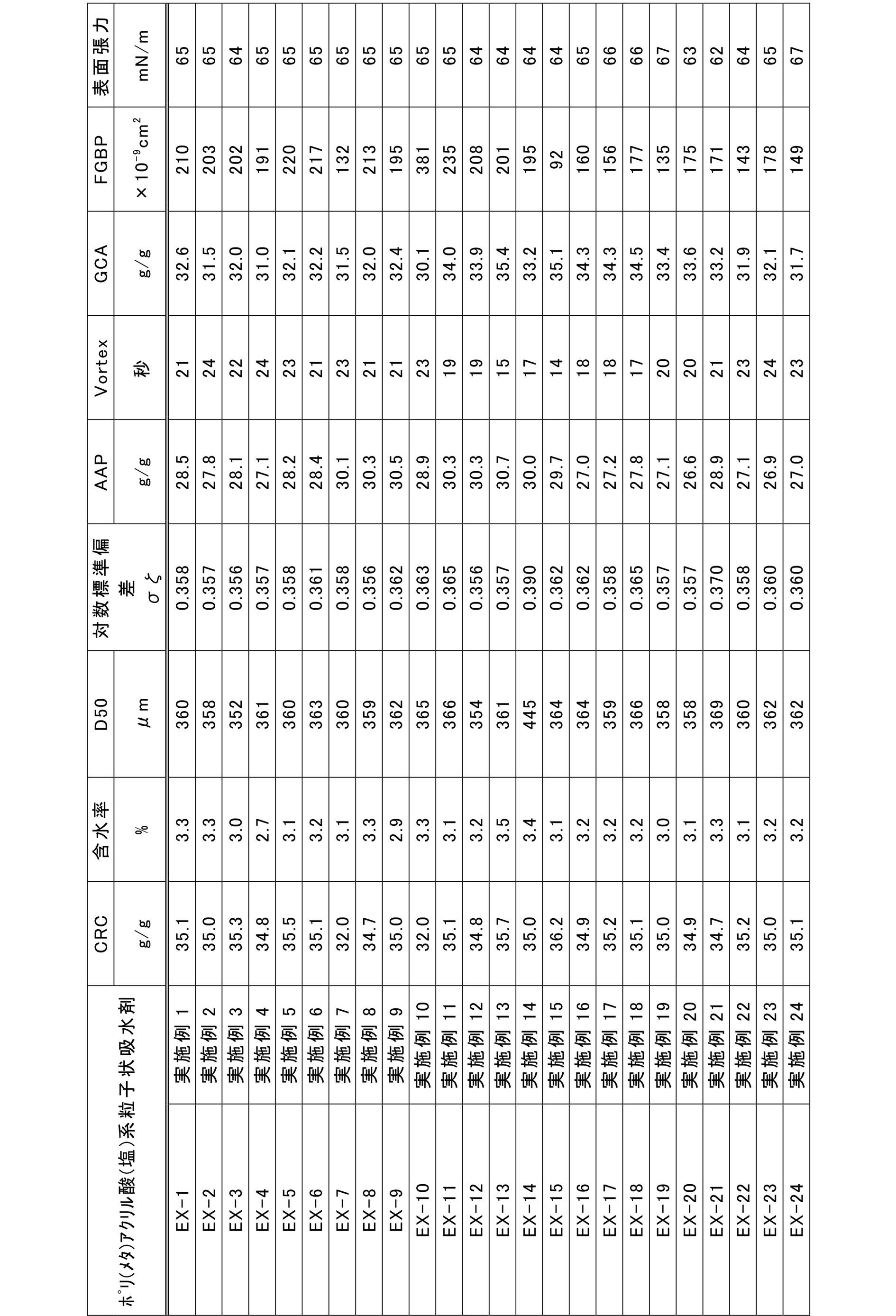




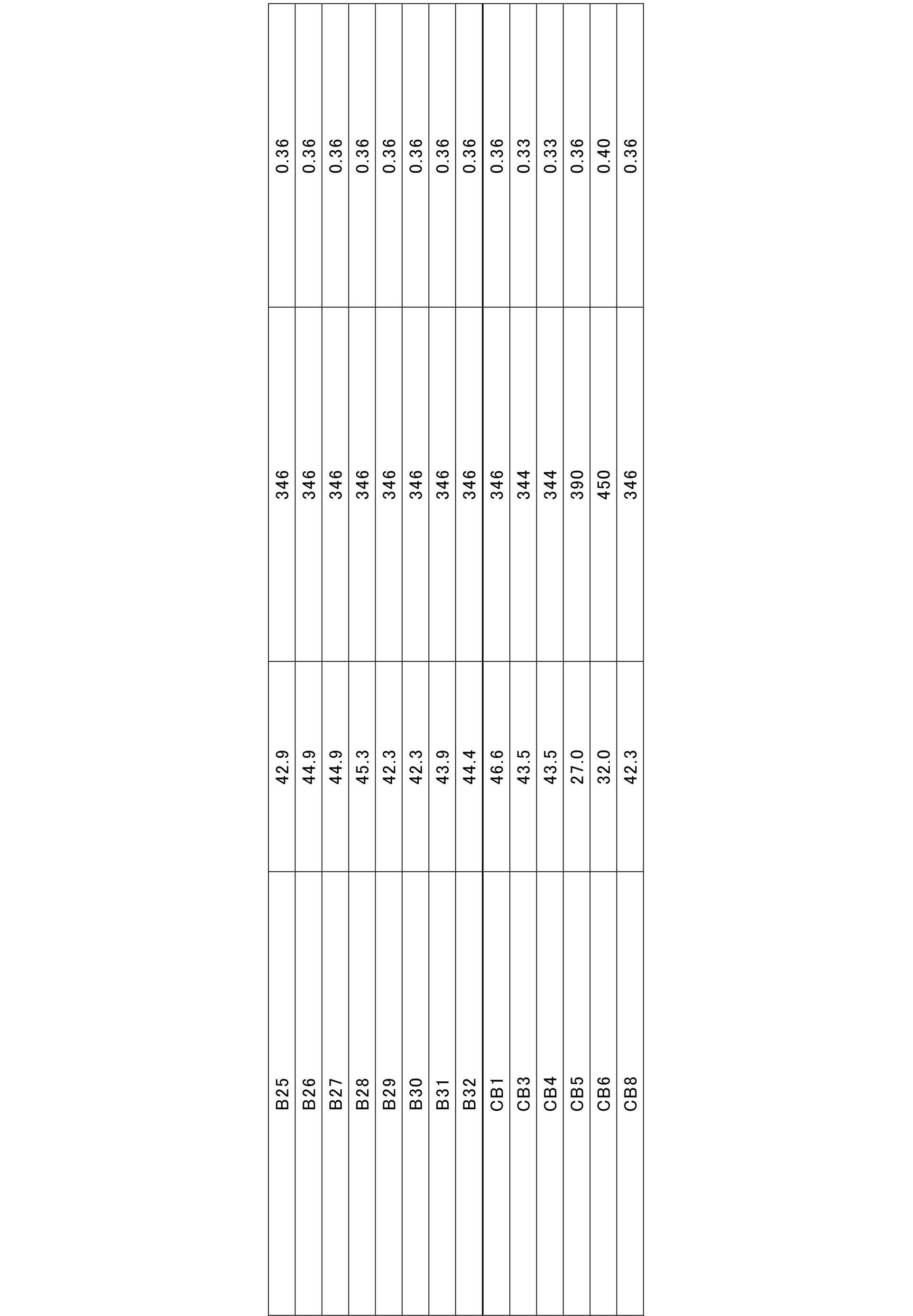
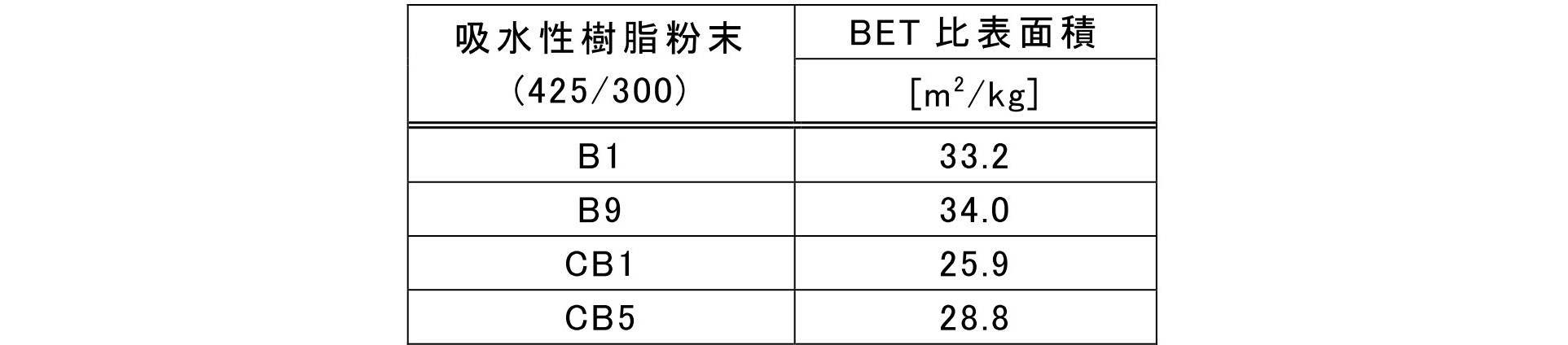
図2は、実施例9の吸水性樹脂粉末(粒度カット500/425)のSEM写真を示すものであり、測定条件は、倍率30倍、印加電圧:1.3kVである。
(吸収体の生理食塩水取り込み速度(コア・アクイジション)・戻り量の評価方法)
測定すべき吸収体は、下記の方法により作製した。即ち、先ず、アクリル樹脂製容器11(内寸80mm×80mm、高さ4cm)の中へ、80mm×80mm、厚さ約0.1mmの吸水紙12を敷いて、次に、吸水剤13を2.4g均一に散布し、この上に80mm×80mm、厚さ約0.1mmの吸水紙14を敷き、さらにこの上に市販のオムツより採取した液透過性を有する表面シート15(80mm×80mm、厚さ約0.1mm)を載せてモデルオムツの吸収体18を作成した。(吸水紙を含めた吸水剤濃度:約82%)。
実施例1、比較例2、比較例6、比較例8で得られた粒子状吸水剤(EX-1)、(CEX-2)、(CEX-6)、(CEX-8)それぞれについて、生理食塩水の吸収体への取り込み速度(コア・アクイジション)、吸収体からの戻り量の評価の測定を行った。これら測定あるいは評価の方法は、上記の吸収体性能評価に従って測定した。結果を下記表に示す。
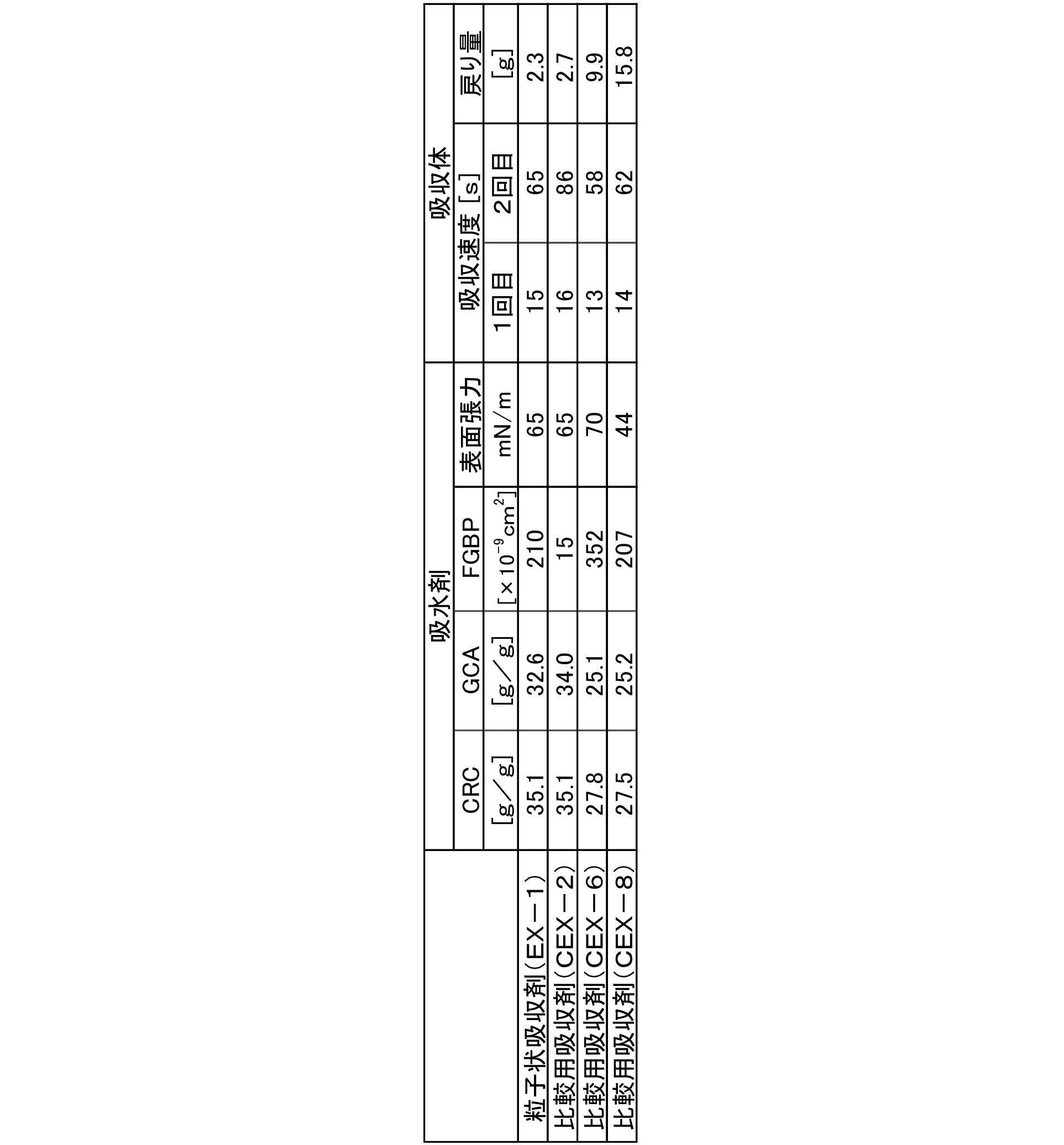
2…ガラスフィルター、
3…シリコン製チューブ、
4…ストップコック、
5…ガラス管、
6…タンク、
7…支持円筒、
8…高湿潤強度セルロースティッシュ、
9…ピストン、
10…金属リング、
11…アクリル樹脂製容器、
12…吸水紙、
13…吸水剤、
14…吸水紙、
15…表面シート、
16…液投入装置、
17…錘、
18…吸収体。
Claims (18)
- (i)(メタ)アクリル酸(塩)系単量体水溶液を調製する工程と、
(ii)前記(メタ)アクリル酸(塩)系単量体水溶液を重合する工程と、
(iii)重合中又は重合後の含水ゲル状架橋重合体をゲル粉砕し、含水ゲル粒子を得る工程と、
(iv)前記含水ゲル粒子を乾燥し、乾燥物を得る工程と、
(v)前記乾燥物を粉砕及び/又は分級して吸水性樹脂粉末を得る工程と、
(vi)前記吸水性樹脂粉末を表面架橋し、吸水性樹脂粒子を得る工程と、
(vii)前記吸水性樹脂粉末または前記吸水性樹脂粒子に通液性向上剤を添加する工程と、を含み、
工程(iii)ないし工程(iii)以前に、前記含水ゲル状架橋重合体及び/又は前記含水ゲル粒子が接着することを制御する接着制御剤を添加することを有し、
前記含水ゲル粒子の固形分が10~80重量%となり、かつ、乾燥物として換算した際の前記含水ゲル粒子の重量平均粒子径が50~650μmとなるように調整し、
ポリ(メタ)アクリル酸(塩)系粒子状吸水剤の表面張力が60mN/m以上となり、無加圧下吸収倍率(CRC)が28g/g以上となるように調整する、ポリ(メタ)アクリル酸(塩)系吸水性樹脂粒子を主成分とする、ポリ(メタ)アクリル酸(塩)系粒子状吸水剤の製造方法。 - 前記接着制御剤が、非イオン性物質、両イオン性物質、アニオン性物質およびカチオン性物質から選ばれる1種以上の化合物であり、
前記非イオン性物質が、(a)ポリオール類、(b)ポリオール類のヒドロキシ基の変性物、(c)側鎖及び/又は末端ポリエーテル変性ポリシロキサン、(d)高級脂肪族アミンのアルキレンオキサイド付加物であり、
前記両イオン性物質が、(e)アルキルアミノベタインまたは(f)アルキルアミンオキサイドであり、
前記アニオン性物質が、(g)高級アルコールアルキレンオキサイド付加物の硫酸エステル塩または(h)アルキルジフェニルエーテルジスルホン酸塩であり、
前記カチオン性物質が、(i)アンモニウム塩である、
請求項1に記載の製造方法。 - 前記接着制御剤の添加量が、前記含水ゲル状架橋重合体の原料モノマーに対して0.01~5重量%である、請求項1または2に記載の製造方法。
- 前記非イオン性物質または前記(g)高級アルコールエチレンオキサイド付加物の硫酸エステル塩の重量平均分子量が、それぞれ独立して、200~200,000である、請求項2または3のいずれか1項に記載の製造方法。
- 前記(a)ポリオール類が、(ポリ)アルキレングリコールである、請求項2~4のいずれか1項に記載の製造方法。
- 前記(b)ポリオール類のヒドロキシ基の変性物が、(b-1)グリシジル変性ポリオール類、(b-2)高級アルコールのアルキレンオキサイド付加物、または(b-3)多価アルコール脂肪酸エステルのアルキレンオキサイド付加物であり、
(b-1)が、(ポリ)アルキレングリコールの末端の少なくとも一つがグリシジル基で修飾されているものであり、
(b-2)が、(ポリ)アルキレングリコールの片末端がC1~C30の炭化水素を有する置換基で修飾されたものであり、
(b-3)が、多価アルコールのヒドロキシ基の少なくとも1つにアルキレンオキサイドが付加され、かつ、多価アルコールのヒドロキシ基の少なくとも1つにエステル結合を介してC1~C30の炭化水素を有する置換基で修飾されたものであり、該多価アルコールが、グリセリン、ペンタエリスリトール、ソルビトール、ソルビタンまたは糖類である、
請求項2~5のいずれか1項に記載の製造方法。 - 前記(d)高級脂肪族アミンのアルキレンオキサイド付加物が、C1~C30の炭化水素を有する1級アミンの2つの水素にアルキレンオキサイドが付加されたものである、請求項2~6のいずれか1項に記載の製造方法。
- 前記(g)高級アルコールアルキレン付加物の硫酸エステル塩が、(ポリ)アルキレングリコールの1つの末端がC1~C30の炭化水素を有する置換基で修飾され、かつ、もう1つの末端が硫酸エステル塩である、請求項2~7のいずれか1項に記載の製造方法。
- 工程(i)における、前記アクリル酸(塩)系単量体水溶液の中和率が、40~90モル%である、請求項1~8のいずれか1項に記載の製造方法。
- 前記ゲル粉砕の工程を、複数のゲル粉砕機を用いて行う、請求項1~9のいずれか1項に記載の製造方法。
- 前記ポリアクリル酸(塩)系粒子状吸水剤の重量平均粒子径(D50)が、300~500μmである、請求項1~10のいずれか1項に記載の製造方法。
- 前記通液性向上剤が、水不溶性無機微粒子である、請求項1~11のいずれか1項に記載の製造方法。
- 前記乾燥物として換算した際の前記含水ゲル粒子の重量平均粒子径が、130~460μmである、請求項1~12のいずれか1項に記載の製造方法。
- ポリアクリル酸(塩)系吸水性樹脂粒子を主成分とする、ポリアクリル酸(塩)系粒子状吸水剤であって、以下(1)~(5):
(1)無加圧下吸収倍率(CRC)が28g/g以上、
(2)GCAが28.0g/g以上、
(3)FGBPとGCAの関係が、
GCAが28.0g/g以上、35.0g/g未満の範囲においては
FGBP≧-10×10-9×GCA+380×10-9cm2を満たし、
GCAが35.0g/g以上においては、FGBP≧30×10-9cm2を満たし、
(4)粒子状吸水剤の重量平均粒子径(D50)が300~500μm、
(5)表面張力が60mN/m以上、
を満たす、ポリアクリル酸(塩)系粒子状吸水剤。 - GCAが31.0g/g以上である、請求項14に記載のポリアクリル酸(塩)系粒子状吸水剤。
- 内部及び/又は表面に、非イオン性物質、両イオン性物質、アニオン性物質およびカチオン性物質から選ばれる1種以上の化合物に含まれ、
前記非イオン性物質が、(a)ポリオール類、(b)ポリオール類のヒドロキシ基の変性物、(c)側鎖及び/又は末端ポリエーテル変性ポリシロキサン、(d)高級脂肪族アミンのアルキレンオキサイド付加物であり、
前記両イオン性物質が、(e)アルキルアミノベタインまたは(f)アルキルアミンオキサイドであり、
前記アニオン性物質が、(g)高級アルコールアルキレンオキサイド付加物の硫酸エステル塩または(h)アルキルジフェニルエーテルジスルホン酸塩であり、
前記カチオン性物質が、(i)アンモニウム塩である、
請求項14または15に記載のポリアクリル酸(塩)系粒子状吸水剤。 - 通液性向上剤をさらに含む、請求項14~16のいずれか1項に記載のポリアクリル酸(塩)系粒子状吸水剤。
- 請求項14~17のいずれか1項に記載のポリアクリル酸(塩)系粒子状吸水剤を含む、衛生材料。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202410048363.6A CN118005960A (zh) | 2015-06-19 | 2016-06-20 | 聚(甲基)丙烯酸(盐)系颗粒状吸水剂及制造方法 |
| EP16811776.0A EP3312218A4 (en) | 2015-06-19 | 2016-06-20 | Poly (meth) acrylic acid (salt) granular water absorbent and method for producing same |
| CN201680036057.4A CN107709415A (zh) | 2015-06-19 | 2016-06-20 | 聚(甲基)丙烯酸(盐)系颗粒状吸水剂及制造方法 |
| JP2017524903A JP6460495B2 (ja) | 2015-06-19 | 2016-06-20 | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 |
| CN202410048849.XA CN118005961A (zh) | 2015-06-19 | 2016-06-20 | 聚(甲基)丙烯酸(盐)系颗粒状吸水剂及制造方法 |
| KR1020177036018A KR102717404B1 (ko) | 2015-06-19 | 2016-06-20 | 폴리(메트)아크릴산(염)계 입자상 흡수제 및 제조 방법 |
| US15/737,884 US11535689B2 (en) | 2015-06-19 | 2016-06-20 | Poly (meth) acrylic acid (salt)-based particulate water-absorbing agent and production method therefor |
| US17/988,581 US11958921B2 (en) | 2015-06-19 | 2022-11-16 | Poly (meth) acrylic acid (salt)-based particulate water-absorbing agent and production method therefor |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-123529 | 2015-06-19 | ||
| JP2015123529 | 2015-06-19 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/737,884 A-371-Of-International US11535689B2 (en) | 2015-06-19 | 2016-06-20 | Poly (meth) acrylic acid (salt)-based particulate water-absorbing agent and production method therefor |
| US17/988,581 Division US11958921B2 (en) | 2015-06-19 | 2022-11-16 | Poly (meth) acrylic acid (salt)-based particulate water-absorbing agent and production method therefor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016204302A1 true WO2016204302A1 (ja) | 2016-12-22 |
Family
ID=57545990
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/068311 WO2016204302A1 (ja) | 2015-06-19 | 2016-06-20 | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US11535689B2 (ja) |
| EP (1) | EP3312218A4 (ja) |
| JP (1) | JP6460495B2 (ja) |
| CN (3) | CN107709415A (ja) |
| WO (1) | WO2016204302A1 (ja) |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017170501A1 (ja) | 2016-03-28 | 2017-10-05 | 株式会社日本触媒 | 吸水剤およびその製造方法、並びに吸水剤を用いた吸収性物品 |
| WO2018092864A1 (ja) | 2016-11-16 | 2018-05-24 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法及びその製造装置 |
| WO2018147317A1 (ja) * | 2017-02-10 | 2018-08-16 | Sdpグローバル株式会社 | 吸水性樹脂粒子並びにこれを用いた吸収体及び吸収性物品 |
| WO2018155591A1 (ja) | 2017-02-22 | 2018-08-30 | 株式会社日本触媒 | 吸水性シート、長尺状吸水性シートおよび吸収性物品 |
| JP2018203997A (ja) * | 2017-05-30 | 2018-12-27 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| WO2019064634A1 (ja) * | 2017-09-28 | 2019-04-04 | 第一工業製薬株式会社 | 水系樹脂組成物 |
| WO2019111812A1 (ja) * | 2017-12-08 | 2019-06-13 | Sdpグローバル株式会社 | 吸収性樹脂粒子、吸収体及び吸収性物品、並びに吸収性樹脂粒子の製造方法 |
| JP2019141754A (ja) * | 2018-02-16 | 2019-08-29 | Sdpグローバル株式会社 | 吸水性樹脂粒子、これを用いた吸収体及び吸収性物品、並びに吸水性樹脂粒子の製造方法 |
| WO2019198821A1 (ja) | 2018-04-13 | 2019-10-17 | 株式会社日本触媒 | 吸水性シート、吸水性シートの製造方法および吸収性物品 |
| WO2020032281A1 (ja) * | 2018-08-09 | 2020-02-13 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸水性物品 |
| WO2020032283A1 (ja) * | 2018-08-09 | 2020-02-13 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸水性物品 |
| WO2020032282A1 (ja) * | 2018-08-09 | 2020-02-13 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸水性物品 |
| WO2020115988A1 (ja) * | 2018-12-04 | 2020-06-11 | Sdpグローバル株式会社 | 吸水性樹脂粒子およびその製造方法 |
| WO2020189539A1 (ja) * | 2019-03-15 | 2020-09-24 | 株式会社日本触媒 | 粒子状吸水剤の製造方法 |
| WO2021049466A1 (ja) * | 2019-09-09 | 2021-03-18 | 住友精化株式会社 | 架橋重合体粒子の製造方法、吸水性樹脂粒子の製造方法、及び、荷重下吸水量の向上方法 |
| JPWO2019188648A1 (ja) * | 2018-03-26 | 2021-03-18 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| US20210147637A1 (en) * | 2018-05-16 | 2021-05-20 | Nippon Shokubai Co., Ltd. | Method for producing water-absorbent resin |
| WO2021201177A1 (ja) * | 2020-03-31 | 2021-10-07 | 株式会社日本触媒 | 粒子状吸水剤 |
| JP2021181507A (ja) * | 2018-11-21 | 2021-11-25 | 三洋化成工業株式会社 | 脱水処理が容易な吸水性樹脂粒子及びその製造方法 |
| JPWO2020144948A1 (ja) * | 2019-01-11 | 2021-11-25 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| JP2021534314A (ja) * | 2019-05-07 | 2021-12-09 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法および高吸水性樹脂 |
| WO2022014550A1 (ja) | 2020-07-13 | 2022-01-20 | 株式会社日本触媒 | 吸水剤組成物およびその製造方法 |
| WO2022025003A1 (ja) * | 2020-07-28 | 2022-02-03 | 住友精化株式会社 | 吸水性樹脂粒子及び吸水性樹脂粒子を製造する方法 |
| WO2022065365A1 (ja) | 2020-09-25 | 2022-03-31 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法 |
| JPWO2022075392A1 (ja) * | 2020-10-08 | 2022-04-14 | ||
| WO2022080367A1 (ja) | 2020-10-13 | 2022-04-21 | 株式会社日本触媒 | 予測方法、予測装置 |
| JP2022533035A (ja) * | 2019-12-20 | 2022-07-21 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| JP2022533561A (ja) * | 2019-12-20 | 2022-07-25 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| WO2022181771A1 (ja) | 2021-02-26 | 2022-09-01 | 株式会社日本触媒 | 粒子状吸水剤、該吸水剤を含む吸収体及び該吸収体を用いた吸収性物品 |
| WO2022196763A1 (ja) | 2021-03-18 | 2022-09-22 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2022197991A1 (en) | 2021-03-18 | 2022-09-22 | The Procter & Gamble Company | Method for producing absorbent articles comprising water-absorbing resin |
| US11559784B2 (en) | 2018-12-11 | 2023-01-24 | Lg Chem, Ltd. | Superabsorbent polymer and preparation method thereof |
| JP7339253B2 (ja) | 2018-07-19 | 2023-09-05 | Sdpグローバル株式会社 | 吸水性樹脂粒子、これを含む吸収体及び吸収性物品 |
| WO2023190494A1 (ja) | 2022-03-30 | 2023-10-05 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法 |
| JP7493862B2 (ja) | 2021-06-18 | 2024-06-03 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| US12024577B2 (en) | 2020-01-20 | 2024-07-02 | Lg Chem, Ltd. | Preparation method of super absorbent polymer |
| WO2024204126A1 (ja) * | 2023-03-27 | 2024-10-03 | 株式会社日本触媒 | 粒子状吸水剤、当該粒子状吸水剤を含む吸収体および当該吸収体を含む衛生製品 |
| JP7570758B2 (ja) | 2020-12-07 | 2024-10-22 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017221911A1 (ja) * | 2016-06-20 | 2017-12-28 | 株式会社日本触媒 | 吸水剤の製造方法 |
| KR102086052B1 (ko) | 2016-12-27 | 2020-03-06 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| KR102086058B1 (ko) | 2017-01-24 | 2020-03-06 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR102417078B1 (ko) * | 2017-09-05 | 2022-07-04 | 주식회사 엘지화학 | 고흡수성 수지 |
| KR102568226B1 (ko) | 2017-12-11 | 2023-08-18 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| KR102418591B1 (ko) | 2018-11-13 | 2022-07-07 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| WO2020122426A1 (ko) | 2018-12-11 | 2020-06-18 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| JP6681492B1 (ja) * | 2018-12-12 | 2020-04-15 | 住友精化株式会社 | 吸水性樹脂粒子 |
| KR102556373B1 (ko) * | 2018-12-13 | 2023-07-17 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR102452567B1 (ko) | 2019-01-07 | 2022-10-06 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| JP7181948B2 (ja) * | 2019-01-11 | 2022-12-01 | 株式会社日本触媒 | 吸水剤、及び吸水剤の製造方法 |
| KR102457690B1 (ko) * | 2019-01-17 | 2022-10-21 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| CN109772254A (zh) * | 2019-03-06 | 2019-05-21 | 广东产品质量监督检验研究院(国家质量技术监督局广州电气安全检验所、广东省试验认证研究院、华安实验室) | 一种新型化工生产设备 |
| US20220192899A1 (en) * | 2019-05-07 | 2022-06-23 | Kimberly-Clark Worldwide, Inc. | Absorbent article |
| CN113214501A (zh) * | 2020-11-30 | 2021-08-06 | 长春理工大学 | 一种环境监测防潮组合结构及其制备工艺 |
| CN116368174A (zh) * | 2021-06-18 | 2023-06-30 | 株式会社Lg化学 | 超吸收性聚合物的制备方法 |
| CN115386186A (zh) * | 2022-04-21 | 2022-11-25 | 宜兴丹森科技有限公司 | 一种聚丙烯酸系高吸水性树脂及其制备方法 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS55133413A (en) * | 1979-04-06 | 1980-10-17 | Nippon Shokubai Kagaku Kogyo Co Ltd | Preparation of crosslinked alkali metal acrylate polymer |
| JPH10119042A (ja) * | 1996-10-24 | 1998-05-12 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造方法 |
| US20060204755A1 (en) | 2003-02-10 | 2006-09-14 | Kazushi Torii | Walter-absorbing agent |
| JP2010088993A (ja) | 2008-10-07 | 2010-04-22 | Akushii:Kk | エアフィルタ |
| WO2011078298A1 (ja) | 2009-12-24 | 2011-06-30 | 株式会社日本触媒 | ポリアクリル酸系吸水性樹脂粉末及びその製造方法 |
| WO2011126079A1 (ja) * | 2010-04-07 | 2011-10-13 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法及びポリアクリル酸(塩)系吸水性樹脂粉末 |
| US20140193641A1 (en) | 2011-06-29 | 2014-07-10 | Nippon Shokubai Co., Ltd. | Polyacrylic Acid (Salt)-Based Water Absorbent Resin Powder and Method for Producing the Same |
| JP2014198853A (ja) * | 2007-03-23 | 2014-10-23 | エボニック コーポレイションEvonik Corporation | 高透過率高吸収性ポリマー組成物 |
| US20150225514A1 (en) | 2012-09-11 | 2015-08-13 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water absorbing agent, and water absorbing agent |
| US20150273433A1 (en) | 2012-10-03 | 2015-10-01 | Nippon Shokubai Co., Ltd. | Water absorbing agent and method for producing the same |
Family Cites Families (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4286082A (en) | 1979-04-06 | 1981-08-25 | Nippon Shokubai Kagaku Kogyo & Co., Ltd. | Absorbent resin composition and process for producing same |
| JP2938920B2 (ja) | 1990-01-31 | 1999-08-25 | 住友精化株式会社 | 吸水性樹脂の製造方法 |
| TW241279B (ja) | 1991-02-01 | 1995-02-21 | Catalyst co ltd | |
| JP3175791B2 (ja) | 1992-06-10 | 2001-06-11 | 株式会社日本触媒 | 吸水剤の製造方法 |
| CA2114815C (en) | 1993-02-24 | 2005-06-14 | Mark Kevin Melius | Absorbent composite |
| DE69524928T2 (de) | 1994-06-13 | 2002-08-29 | Nippon Shokubai Co. Ltd., Osaka | Wasser-absorbent, verfahren zu dessen herstellung und den enthaltender absorbent-gegenstand |
| TW522024B (en) | 1995-09-01 | 2003-03-01 | Nippon Catalytic Chem Ind | Absorbing agent composite, absorbent material, and absorbent product containing absorbent material |
| DE19646484C2 (de) | 1995-11-21 | 2000-10-19 | Stockhausen Chem Fab Gmbh | Flüssigkeitsabsorbierende Polymere, Verfahren zu deren Herstellung und deren Verwendung |
| CN100345891C (zh) | 1996-10-24 | 2007-10-31 | 株式会社日本触媒 | 吸水性树脂的制造方法 |
| JP3415036B2 (ja) | 1998-08-12 | 2003-06-09 | 株式会社日本触媒 | 含水ゲル状架橋重合体の細粒化方法 |
| US6297335B1 (en) | 1999-02-05 | 2001-10-02 | Basf Aktiengesellschaft | Crosslinked, hydrophilic, highly swellable hydrogels, production thereof and use thereof |
| JP4341143B2 (ja) | 2000-06-01 | 2009-10-07 | 東亞合成株式会社 | 親水性重合体塊または粒子の製造方法 |
| US7169843B2 (en) | 2003-04-25 | 2007-01-30 | Stockhausen, Inc. | Superabsorbent polymer with high permeability |
| EP2221068A2 (en) | 2003-06-30 | 2010-08-25 | The Procter & Gamble Company | Absorbent structures comprising coated super-absorbent polymer articles |
| AU2005210411B2 (en) | 2004-02-05 | 2008-01-31 | Nippon Shokubai Co., Ltd. | Particulate water absorbing agent and method for production thereof, and water absorbing article |
| EP2116571B1 (en) | 2007-02-05 | 2019-05-01 | Nippon Shokubai Co., Ltd. | Granular water absorber and method of producing the same |
| US9517289B2 (en) | 2007-12-12 | 2016-12-13 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and method for producing the same |
| WO2010073658A1 (ja) | 2008-12-26 | 2010-07-01 | サンダイヤポリマー株式会社 | 吸収性樹脂粒子、この製造方法、これを含む吸収体及び吸収性物品 |
| JP5600670B2 (ja) | 2009-02-17 | 2014-10-01 | 株式会社日本触媒 | ポリアクリル酸系吸水性樹脂粉末およびその製造方法 |
| CN102548654A (zh) | 2009-09-29 | 2012-07-04 | 株式会社日本触媒 | 颗粒状吸水剂及其制造方法 |
| CN105363421A (zh) | 2009-09-30 | 2016-03-02 | 株式会社日本触媒 | 颗粒状吸水剂及其制造方法 |
| JP5487864B2 (ja) | 2009-09-30 | 2014-05-14 | 富士通株式会社 | データ収集装置、データ収集方法およびデータ収集プログラム |
| US8510631B2 (en) | 2009-11-24 | 2013-08-13 | Mediatek Inc. | Multi-channel memory apparatus and method thereof |
| EP2589613B1 (en) | 2010-06-30 | 2015-05-13 | Nippon Shokubai Co., Ltd. | Polyacrylic acid-based water-absorbing resin and process for producing same |
| US20130130017A1 (en) | 2010-08-19 | 2013-05-23 | Sumitomo Seika Chemicals Co., Ltd. | Water-absorbing resin |
| CN104936989B (zh) | 2013-01-29 | 2019-04-16 | 巴斯夫欧洲公司 | 制备具有高自由溶胀率、高离心保留容量和高溶胀凝胶床渗透性的吸水性聚合物颗粒的方法 |
| CN109608661B (zh) | 2013-08-28 | 2021-09-10 | 株式会社日本触媒 | 凝胶粉碎装置、及聚丙烯酸(盐)系吸水性树脂粉末的制造方法、以及吸水性树脂粉末 |
| WO2015030129A1 (ja) | 2013-08-28 | 2015-03-05 | 株式会社日本触媒 | ゲル粉砕装置、及びポリアクリル酸(塩)系吸水性樹脂粉末の製造方法、並びに吸水性樹脂粉末 |
| KR20160127742A (ko) | 2014-02-28 | 2016-11-04 | 가부시키가이샤 닛폰 쇼쿠바이 | 폴리(메트)아크릴산(염)계 입자상 흡수제 및 제조 방법 |
| JP6441374B2 (ja) | 2014-09-29 | 2018-12-19 | 株式会社日本触媒 | 吸水性樹脂粉末及び吸水性樹脂粉末の弾性率の測定方法 |
-
2016
- 2016-06-20 WO PCT/JP2016/068311 patent/WO2016204302A1/ja active Application Filing
- 2016-06-20 CN CN201680036057.4A patent/CN107709415A/zh active Pending
- 2016-06-20 EP EP16811776.0A patent/EP3312218A4/en active Pending
- 2016-06-20 JP JP2017524903A patent/JP6460495B2/ja active Active
- 2016-06-20 CN CN202410048363.6A patent/CN118005960A/zh active Pending
- 2016-06-20 US US15/737,884 patent/US11535689B2/en active Active
- 2016-06-20 CN CN202410048849.XA patent/CN118005961A/zh active Pending
-
2022
- 2022-11-16 US US17/988,581 patent/US11958921B2/en active Active
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS55133413A (en) * | 1979-04-06 | 1980-10-17 | Nippon Shokubai Kagaku Kogyo Co Ltd | Preparation of crosslinked alkali metal acrylate polymer |
| JPH10119042A (ja) * | 1996-10-24 | 1998-05-12 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造方法 |
| US20060204755A1 (en) | 2003-02-10 | 2006-09-14 | Kazushi Torii | Walter-absorbing agent |
| JP2014198853A (ja) * | 2007-03-23 | 2014-10-23 | エボニック コーポレイションEvonik Corporation | 高透過率高吸収性ポリマー組成物 |
| JP2010088993A (ja) | 2008-10-07 | 2010-04-22 | Akushii:Kk | エアフィルタ |
| US20120258851A1 (en) | 2009-12-24 | 2012-10-11 | Nippon Shokubai Co., Ltd | Water-absorbable polyacrylic acid resin powder, and process for production thereof |
| JP2014098172A (ja) * | 2009-12-24 | 2014-05-29 | Nippon Shokubai Co Ltd | ポリアクリル酸系吸水性樹脂粉末及びその製造方法 |
| WO2011078298A1 (ja) | 2009-12-24 | 2011-06-30 | 株式会社日本触媒 | ポリアクリル酸系吸水性樹脂粉末及びその製造方法 |
| WO2011126079A1 (ja) * | 2010-04-07 | 2011-10-13 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法及びポリアクリル酸(塩)系吸水性樹脂粉末 |
| US20130026412A1 (en) | 2010-04-07 | 2013-01-31 | Nippon Shokubai Co. Ltd | Method for producing water absorbent polyacrylic acid (salt) resin powder, and water absorbent polyacrylic acid (salt) resin powder |
| US20140193641A1 (en) | 2011-06-29 | 2014-07-10 | Nippon Shokubai Co., Ltd. | Polyacrylic Acid (Salt)-Based Water Absorbent Resin Powder and Method for Producing the Same |
| US20150225514A1 (en) | 2012-09-11 | 2015-08-13 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water absorbing agent, and water absorbing agent |
| US20150273433A1 (en) | 2012-10-03 | 2015-10-01 | Nippon Shokubai Co., Ltd. | Water absorbing agent and method for producing the same |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3312218A4 |
Cited By (79)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017170501A1 (ja) | 2016-03-28 | 2017-10-05 | 株式会社日本触媒 | 吸水剤およびその製造方法、並びに吸水剤を用いた吸収性物品 |
| US11602577B2 (en) | 2016-03-28 | 2023-03-14 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and method for producing same, and absorbent article produced using water-absorbing agent |
| WO2018092864A1 (ja) | 2016-11-16 | 2018-05-24 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法及びその製造装置 |
| WO2018092863A1 (ja) | 2016-11-16 | 2018-05-24 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法、並びに粒子状含水ゲルの乾燥装置及び乾燥方法 |
| US11465126B2 (en) | 2016-11-16 | 2022-10-11 | Nippon Shokubai Co., Ltd. | Method for producing water-absorbent resin powder and production apparatus therefor |
| US11766659B2 (en) | 2016-11-16 | 2023-09-26 | Nippon Shokubai Co., Ltd. | Method for producing water-absorbent resin powder, and drying device and drying method for particulate hydrous gel |
| JPWO2018147317A1 (ja) * | 2017-02-10 | 2019-12-26 | Sdpグローバル株式会社 | 吸水性樹脂粒子並びにこれを用いた吸収体及び吸収性物品 |
| WO2018147317A1 (ja) * | 2017-02-10 | 2018-08-16 | Sdpグローバル株式会社 | 吸水性樹脂粒子並びにこれを用いた吸収体及び吸収性物品 |
| WO2018155591A1 (ja) | 2017-02-22 | 2018-08-30 | 株式会社日本触媒 | 吸水性シート、長尺状吸水性シートおよび吸収性物品 |
| JP2018203997A (ja) * | 2017-05-30 | 2018-12-27 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| JP7239276B2 (ja) | 2017-05-30 | 2023-03-14 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| WO2019064634A1 (ja) * | 2017-09-28 | 2019-04-04 | 第一工業製薬株式会社 | 水系樹脂組成物 |
| CN111051437A (zh) * | 2017-09-28 | 2020-04-21 | 第一工业制药株式会社 | 水系树脂组合物 |
| JP2019059884A (ja) * | 2017-09-28 | 2019-04-18 | 第一工業製薬株式会社 | 水系樹脂組成物 |
| CN111433258A (zh) * | 2017-12-08 | 2020-07-17 | 三大雅株式会社 | 吸收性树脂颗粒、吸收体和吸收性物品、以及吸收性树脂颗粒的制造方法 |
| WO2019111812A1 (ja) * | 2017-12-08 | 2019-06-13 | Sdpグローバル株式会社 | 吸収性樹脂粒子、吸収体及び吸収性物品、並びに吸収性樹脂粒子の製造方法 |
| JPWO2019111812A1 (ja) * | 2017-12-08 | 2020-12-24 | Sdpグローバル株式会社 | 吸収性樹脂粒子、吸収体及び吸収性物品、並びに吸収性樹脂粒子の製造方法 |
| JP7194695B2 (ja) | 2017-12-08 | 2022-12-22 | Sdpグローバル株式会社 | 吸収性樹脂粒子、吸収体及び吸収性物品、並びに吸収性樹脂粒子の製造方法 |
| CN111433258B (zh) * | 2017-12-08 | 2023-06-20 | 三大雅株式会社 | 吸收性树脂颗粒、吸收体和吸收性物品、以及吸收性树脂颗粒的制造方法 |
| JP2019141754A (ja) * | 2018-02-16 | 2019-08-29 | Sdpグローバル株式会社 | 吸水性樹脂粒子、これを用いた吸収体及び吸収性物品、並びに吸水性樹脂粒子の製造方法 |
| JP7108422B2 (ja) | 2018-02-16 | 2022-07-28 | Sdpグローバル株式会社 | 吸水性樹脂粒子、これを用いた吸収体及び吸収性物品、並びに吸水性樹脂粒子の製造方法 |
| JP7291686B2 (ja) | 2018-03-26 | 2023-06-15 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| JPWO2019188648A1 (ja) * | 2018-03-26 | 2021-03-18 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| WO2019198821A1 (ja) | 2018-04-13 | 2019-10-17 | 株式会社日本触媒 | 吸水性シート、吸水性シートの製造方法および吸収性物品 |
| US11970585B2 (en) * | 2018-05-16 | 2024-04-30 | Nippon Shokubai Co., Ltd. | Method for producing water-absorbent resin |
| US20210147637A1 (en) * | 2018-05-16 | 2021-05-20 | Nippon Shokubai Co., Ltd. | Method for producing water-absorbent resin |
| JP7339253B2 (ja) | 2018-07-19 | 2023-09-05 | Sdpグローバル株式会社 | 吸水性樹脂粒子、これを含む吸収体及び吸収性物品 |
| JPWO2020032283A1 (ja) * | 2018-08-09 | 2021-08-10 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸収性物品 |
| WO2020032283A1 (ja) * | 2018-08-09 | 2020-02-13 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸水性物品 |
| JPWO2020032282A1 (ja) * | 2018-08-09 | 2021-08-12 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸収性物品 |
| JPWO2020032280A1 (ja) * | 2018-08-09 | 2021-08-10 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸収性物品 |
| JPWO2020032281A1 (ja) * | 2018-08-09 | 2021-08-10 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸収性物品 |
| WO2020032284A1 (ja) * | 2018-08-09 | 2020-02-13 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸水性物品 |
| WO2020032281A1 (ja) * | 2018-08-09 | 2020-02-13 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸水性物品 |
| JP7244523B2 (ja) | 2018-08-09 | 2023-03-22 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸収性物品 |
| WO2020032282A1 (ja) * | 2018-08-09 | 2020-02-13 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸水性物品 |
| JP7174759B2 (ja) | 2018-08-09 | 2022-11-17 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸収性物品 |
| WO2020032280A1 (ja) | 2018-08-09 | 2020-02-13 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸水性物品 |
| JP7085627B2 (ja) | 2018-08-09 | 2022-06-16 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸収性物品 |
| JP7174760B2 (ja) | 2018-08-09 | 2022-11-17 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸収性物品 |
| JPWO2020032284A1 (ja) * | 2018-08-09 | 2021-08-10 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸収性物品 |
| JP7174761B2 (ja) | 2018-08-09 | 2022-11-17 | 株式会社日本触媒 | 吸水性シートおよびそれを含む吸収性物品 |
| JP7200323B2 (ja) | 2018-11-21 | 2023-01-06 | 三洋化成工業株式会社 | 衛生用品の処理方法 |
| JP7234101B2 (ja) | 2018-11-21 | 2023-03-07 | 三洋化成工業株式会社 | 脱水処理が容易な吸水性樹脂粒子及びその製造方法 |
| JP2022009235A (ja) * | 2018-11-21 | 2022-01-14 | 三洋化成工業株式会社 | 衛生用品の処理方法 |
| JP2021181507A (ja) * | 2018-11-21 | 2021-11-25 | 三洋化成工業株式会社 | 脱水処理が容易な吸水性樹脂粒子及びその製造方法 |
| WO2020115988A1 (ja) * | 2018-12-04 | 2020-06-11 | Sdpグローバル株式会社 | 吸水性樹脂粒子およびその製造方法 |
| JPWO2020115988A1 (ja) * | 2018-12-04 | 2021-10-28 | Sdpグローバル株式会社 | 吸水性樹脂粒子およびその製造方法 |
| JP7453918B2 (ja) | 2018-12-04 | 2024-03-21 | Sdpグローバル株式会社 | 吸水性樹脂粒子およびその製造方法 |
| US11559784B2 (en) | 2018-12-11 | 2023-01-24 | Lg Chem, Ltd. | Superabsorbent polymer and preparation method thereof |
| JPWO2020144948A1 (ja) * | 2019-01-11 | 2021-11-25 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| JP7165753B2 (ja) | 2019-01-11 | 2022-11-04 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| WO2020189539A1 (ja) * | 2019-03-15 | 2020-09-24 | 株式会社日本触媒 | 粒子状吸水剤の製造方法 |
| US12083497B2 (en) | 2019-05-07 | 2024-09-10 | Lg Chem, Ltd. | Preparation method of super absorbent polymer and super absorbent polymer prepared therefrom |
| JP2021534314A (ja) * | 2019-05-07 | 2021-12-09 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法および高吸水性樹脂 |
| WO2021049466A1 (ja) * | 2019-09-09 | 2021-03-18 | 住友精化株式会社 | 架橋重合体粒子の製造方法、吸水性樹脂粒子の製造方法、及び、荷重下吸水量の向上方法 |
| JP7448553B2 (ja) | 2019-09-09 | 2024-03-12 | 住友精化株式会社 | 架橋重合体粒子の製造方法、吸水性樹脂粒子の製造方法、及び、荷重下吸水量の向上方法 |
| JP2022533035A (ja) * | 2019-12-20 | 2022-07-21 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| JP7321627B2 (ja) | 2019-12-20 | 2023-08-07 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| JP7321628B2 (ja) | 2019-12-20 | 2023-08-07 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| JP2022533561A (ja) * | 2019-12-20 | 2022-07-25 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| US12024577B2 (en) | 2020-01-20 | 2024-07-02 | Lg Chem, Ltd. | Preparation method of super absorbent polymer |
| JPWO2021201177A1 (ja) * | 2020-03-31 | 2021-10-07 | ||
| JP7382493B2 (ja) | 2020-03-31 | 2023-11-16 | 株式会社日本触媒 | 粒子状吸水剤 |
| WO2021201177A1 (ja) * | 2020-03-31 | 2021-10-07 | 株式会社日本触媒 | 粒子状吸水剤 |
| WO2022014550A1 (ja) | 2020-07-13 | 2022-01-20 | 株式会社日本触媒 | 吸水剤組成物およびその製造方法 |
| WO2022025003A1 (ja) * | 2020-07-28 | 2022-02-03 | 住友精化株式会社 | 吸水性樹脂粒子及び吸水性樹脂粒子を製造する方法 |
| WO2022065365A1 (ja) | 2020-09-25 | 2022-03-31 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法 |
| WO2022075392A1 (ja) * | 2020-10-08 | 2022-04-14 | Dic株式会社 | 熱可塑性樹脂組成物及びその製造方法 |
| JPWO2022075392A1 (ja) * | 2020-10-08 | 2022-04-14 | ||
| JP7501653B2 (ja) | 2020-10-08 | 2024-06-18 | Dic株式会社 | 熱可塑性樹脂組成物及びその製造方法 |
| WO2022080367A1 (ja) | 2020-10-13 | 2022-04-21 | 株式会社日本触媒 | 予測方法、予測装置 |
| JP7570758B2 (ja) | 2020-12-07 | 2024-10-22 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| WO2022181771A1 (ja) | 2021-02-26 | 2022-09-01 | 株式会社日本触媒 | 粒子状吸水剤、該吸水剤を含む吸収体及び該吸収体を用いた吸収性物品 |
| WO2022197991A1 (en) | 2021-03-18 | 2022-09-22 | The Procter & Gamble Company | Method for producing absorbent articles comprising water-absorbing resin |
| WO2022196763A1 (ja) | 2021-03-18 | 2022-09-22 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| JP7493862B2 (ja) | 2021-06-18 | 2024-06-03 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| WO2023190494A1 (ja) | 2022-03-30 | 2023-10-05 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法 |
| WO2024204126A1 (ja) * | 2023-03-27 | 2024-10-03 | 株式会社日本触媒 | 粒子状吸水剤、当該粒子状吸水剤を含む吸収体および当該吸収体を含む衛生製品 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3312218A4 (en) | 2018-06-27 |
| US20180298132A1 (en) | 2018-10-18 |
| EP3312218A1 (en) | 2018-04-25 |
| US11958921B2 (en) | 2024-04-16 |
| CN107709415A (zh) | 2018-02-16 |
| US20230097487A1 (en) | 2023-03-30 |
| US11535689B2 (en) | 2022-12-27 |
| CN118005961A (zh) | 2024-05-10 |
| KR20180019558A (ko) | 2018-02-26 |
| JPWO2016204302A1 (ja) | 2018-02-15 |
| CN118005960A (zh) | 2024-05-10 |
| JP6460495B2 (ja) | 2019-01-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6460495B2 (ja) | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 | |
| JP6441894B2 (ja) | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 | |
| JP7083020B2 (ja) | 吸水性樹脂粉末、及びその製造方法 | |
| JP6385993B2 (ja) | ポリアクリル酸(塩)系吸水剤 | |
| JP5847260B2 (ja) | 吸水性樹脂の製造方法 | |
| JP5914677B2 (ja) | ポリアクリル酸(塩)系吸水剤の製造方法及びその吸水剤 | |
| JP6092236B2 (ja) | 吸水剤及びその製造方法 | |
| JP6282669B2 (ja) | ポリアクリル酸(塩)系吸水剤及びその製造方法 | |
| WO2019221154A1 (ja) | 吸水性樹脂粒子の製造方法 | |
| JP6286533B2 (ja) | 粒子状吸水剤及びその製造方法 | |
| JP6351218B2 (ja) | 吸水剤及びその製造方法 | |
| JP2016069418A (ja) | ポリ(メタ)アクリル酸(塩)系吸水性樹脂の製造方法 | |
| KR20210041070A (ko) | 흡수성 수지 분말의 제조 방법 및 흡수성 수지 분말 | |
| KR102717404B1 (ko) | 폴리(메트)아크릴산(염)계 입자상 흡수제 및 제조 방법 | |
| WO2024204126A1 (ja) | 粒子状吸水剤、当該粒子状吸水剤を含む吸収体および当該吸収体を含む衛生製品 | |
| JP2007321010A (ja) | 吸水性樹脂の表面処理方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16811776 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017524903 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20177036018 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15737884 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016811776 Country of ref document: EP |