WO2018092863A1 - 吸水性樹脂粉末の製造方法、並びに粒子状含水ゲルの乾燥装置及び乾燥方法 - Google Patents
吸水性樹脂粉末の製造方法、並びに粒子状含水ゲルの乾燥装置及び乾燥方法 Download PDFInfo
- Publication number
- WO2018092863A1 WO2018092863A1 PCT/JP2017/041371 JP2017041371W WO2018092863A1 WO 2018092863 A1 WO2018092863 A1 WO 2018092863A1 JP 2017041371 W JP2017041371 W JP 2017041371W WO 2018092863 A1 WO2018092863 A1 WO 2018092863A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- drying
- water
- gel
- rotating container
- mass
- Prior art date
Links
Images









Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F6/00—Post-polymerisation treatments
- C08F6/008—Treatment of solid polymer wetted by water or organic solvents, e.g. coagulum, filter cakes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/265—Synthetic macromolecular compounds modified or post-treated polymers
- B01J20/267—Cross-linked polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J13/00—Colloid chemistry, e.g. the production of colloidal materials or their solutions, not otherwise provided for; Making microcapsules or microballoons
- B01J13/0052—Preparation of gels
- B01J13/0065—Preparation of gels containing an organic phase
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J13/00—Colloid chemistry, e.g. the production of colloidal materials or their solutions, not otherwise provided for; Making microcapsules or microballoons
- B01J13/0052—Preparation of gels
- B01J13/0069—Post treatment
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28002—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their physical properties
- B01J20/28004—Sorbent size or size distribution, e.g. particle size
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28014—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their form
- B01J20/28016—Particle form
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/3021—Milling, crushing or grinding
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/3078—Thermal treatment, e.g. calcining or pyrolizing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/3085—Chemical treatments not covered by groups B01J20/3007 - B01J20/3078
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/04—Acids; Metal salts or ammonium salts thereof
- C08F220/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/02—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques
- C08J3/03—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques in aqueous media
- C08J3/075—Macromolecular gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F26—DRYING
- F26B—DRYING SOLID MATERIALS OR OBJECTS BY REMOVING LIQUID THEREFROM
- F26B1/00—Preliminary treatment of solid materials or objects to facilitate drying, e.g. mixing or backmixing the materials to be dried with predominantly dry solids
- F26B1/005—Preliminary treatment of solid materials or objects to facilitate drying, e.g. mixing or backmixing the materials to be dried with predominantly dry solids by means of disintegrating, e.g. crushing, shredding, milling the materials to be dried
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F26—DRYING
- F26B—DRYING SOLID MATERIALS OR OBJECTS BY REMOVING LIQUID THEREFROM
- F26B11/00—Machines or apparatus for drying solid materials or objects with movement which is non-progressive
- F26B11/02—Machines or apparatus for drying solid materials or objects with movement which is non-progressive in moving drums or other mainly-closed receptacles
- F26B11/04—Machines or apparatus for drying solid materials or objects with movement which is non-progressive in moving drums or other mainly-closed receptacles rotating about a horizontal or slightly-inclined axis
- F26B11/0404—Machines or apparatus for drying solid materials or objects with movement which is non-progressive in moving drums or other mainly-closed receptacles rotating about a horizontal or slightly-inclined axis with internal subdivision of the drum, e.g. for subdividing or recycling the material to be dried
- F26B11/0409—Machines or apparatus for drying solid materials or objects with movement which is non-progressive in moving drums or other mainly-closed receptacles rotating about a horizontal or slightly-inclined axis with internal subdivision of the drum, e.g. for subdividing or recycling the material to be dried the subdivision consisting of a plurality of substantially radially oriented internal walls, e.g. forming multiple sector-shaped chambers
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F26—DRYING
- F26B—DRYING SOLID MATERIALS OR OBJECTS BY REMOVING LIQUID THEREFROM
- F26B11/00—Machines or apparatus for drying solid materials or objects with movement which is non-progressive
- F26B11/02—Machines or apparatus for drying solid materials or objects with movement which is non-progressive in moving drums or other mainly-closed receptacles
- F26B11/04—Machines or apparatus for drying solid materials or objects with movement which is non-progressive in moving drums or other mainly-closed receptacles rotating about a horizontal or slightly-inclined axis
- F26B11/0445—Machines or apparatus for drying solid materials or objects with movement which is non-progressive in moving drums or other mainly-closed receptacles rotating about a horizontal or slightly-inclined axis having conductive heating arrangements, e.g. heated drum wall
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F26—DRYING
- F26B—DRYING SOLID MATERIALS OR OBJECTS BY REMOVING LIQUID THEREFROM
- F26B17/00—Machines or apparatus for drying materials in loose, plastic, or fluidised form, e.g. granules, staple fibres, with progressive movement
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F26—DRYING
- F26B—DRYING SOLID MATERIALS OR OBJECTS BY REMOVING LIQUID THEREFROM
- F26B17/00—Machines or apparatus for drying materials in loose, plastic, or fluidised form, e.g. granules, staple fibres, with progressive movement
- F26B17/10—Machines or apparatus for drying materials in loose, plastic, or fluidised form, e.g. granules, staple fibres, with progressive movement with movement performed by fluid currents, e.g. issuing from a nozzle, e.g. pneumatic, flash, vortex or entrainment dryers
- F26B17/106—Machines or apparatus for drying materials in loose, plastic, or fluidised form, e.g. granules, staple fibres, with progressive movement with movement performed by fluid currents, e.g. issuing from a nozzle, e.g. pneumatic, flash, vortex or entrainment dryers the drying enclosure, e.g. its axis, being substantially straight and horizontal, e.g. pneumatic drum dryers; the drying enclosure consisting of multiple substantially straight and horizontal stretches
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F26—DRYING
- F26B—DRYING SOLID MATERIALS OR OBJECTS BY REMOVING LIQUID THEREFROM
- F26B17/00—Machines or apparatus for drying materials in loose, plastic, or fluidised form, e.g. granules, staple fibres, with progressive movement
- F26B17/30—Machines or apparatus for drying materials in loose, plastic, or fluidised form, e.g. granules, staple fibres, with progressive movement with movement performed by rotary or oscillating containers; with movement performed by rotary floors
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F26—DRYING
- F26B—DRYING SOLID MATERIALS OR OBJECTS BY REMOVING LIQUID THEREFROM
- F26B23/00—Heating arrangements
- F26B23/10—Heating arrangements using tubes or passages containing heated fluids, e.g. acting as radiative elements; Closed-loop systems
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F26—DRYING
- F26B—DRYING SOLID MATERIALS OR OBJECTS BY REMOVING LIQUID THEREFROM
- F26B3/00—Drying solid materials or objects by processes involving the application of heat
- F26B3/02—Drying solid materials or objects by processes involving the application of heat by convection, i.e. heat being conveyed from a heat source to the materials or objects to be dried by a gas or vapour, e.g. air
- F26B3/04—Drying solid materials or objects by processes involving the application of heat by convection, i.e. heat being conveyed from a heat source to the materials or objects to be dried by a gas or vapour, e.g. air the gas or vapour circulating over or surrounding the materials or objects to be dried
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F26—DRYING
- F26B—DRYING SOLID MATERIALS OR OBJECTS BY REMOVING LIQUID THEREFROM
- F26B3/00—Drying solid materials or objects by processes involving the application of heat
- F26B3/02—Drying solid materials or objects by processes involving the application of heat by convection, i.e. heat being conveyed from a heat source to the materials or objects to be dried by a gas or vapour, e.g. air
- F26B3/10—Drying solid materials or objects by processes involving the application of heat by convection, i.e. heat being conveyed from a heat source to the materials or objects to be dried by a gas or vapour, e.g. air the gas or vapour carrying the materials or objects to be dried with it
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F26—DRYING
- F26B—DRYING SOLID MATERIALS OR OBJECTS BY REMOVING LIQUID THEREFROM
- F26B3/00—Drying solid materials or objects by processes involving the application of heat
- F26B3/18—Drying solid materials or objects by processes involving the application of heat by conduction, i.e. the heat is conveyed from the heat source, e.g. gas flame, to the materials or objects to be dried by direct contact
- F26B3/20—Drying solid materials or objects by processes involving the application of heat by conduction, i.e. the heat is conveyed from the heat source, e.g. gas flame, to the materials or objects to be dried by direct contact the heat source being a heated surface, e.g. a moving belt or conveyor
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F26—DRYING
- F26B—DRYING SOLID MATERIALS OR OBJECTS BY REMOVING LIQUID THEREFROM
- F26B3/00—Drying solid materials or objects by processes involving the application of heat
- F26B3/18—Drying solid materials or objects by processes involving the application of heat by conduction, i.e. the heat is conveyed from the heat source, e.g. gas flame, to the materials or objects to be dried by direct contact
- F26B3/22—Drying solid materials or objects by processes involving the application of heat by conduction, i.e. the heat is conveyed from the heat source, e.g. gas flame, to the materials or objects to be dried by direct contact the heat source and the materials or objects to be dried being in relative motion, e.g. of vibration
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2/00—Processes or devices for granulating materials, e.g. fertilisers in general; Rendering particulate materials free flowing in general, e.g. making them hydrophobic
- B01J2/12—Processes or devices for granulating materials, e.g. fertilisers in general; Rendering particulate materials free flowing in general, e.g. making them hydrophobic in rotating drums
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2800/00—Copolymer characterised by the proportions of the comonomers expressed
- C08F2800/20—Copolymer characterised by the proportions of the comonomers expressed as weight or mass percentages
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2810/00—Chemical modification of a polymer
- C08F2810/20—Chemical modification of a polymer leading to a crosslinking, either explicitly or inherently
Definitions
- the present invention relates to a method for producing a water-absorbent resin powder, a drying apparatus and a drying method for a particulate hydrous gel. More specifically, in comparison with the conventional method for producing a water absorbent resin, the production process is simplified and compact, and the amount of the water absorbent resin powder generated in the production process is reduced.
- the present invention relates to a production method and a drying apparatus and drying method (particularly, a drying start method) of a particulate hydrogel crosslinked polymer.
- Water-absorbing resin (SAP / Super Absorbent Polymer) is a water-swellable, water-insoluble polymer gelling agent, absorbent articles such as paper diapers and sanitary napkins, water retaining agents for agriculture and horticulture, and industrial waterstops. It is widely used in various fields such as drugs.
- water-absorbent resin many monomers and hydrophilic polymers are used as raw materials. From the viewpoint of water absorption performance, polyacrylic acid using acrylic acid and / or a salt thereof as a monomer is used. (Salt) water-absorbing resins are industrially most produced.
- the water-absorbing resin is required to have various functions (higher physical properties) as the paper diaper, which is the main application, has improved performance. Specifically, in addition to the basic physical properties, water absorption capacity without pressure and water absorption capacity under pressure, gel strength, water-soluble content, water content, water absorption speed, liquid permeability, particle size distribution, urine resistance, antibacterial properties Various properties such as damage resistance, powder flowability, deodorization, color resistance, low dust, and low residual monomer are required for the water-absorbing resin.
- Such a water-absorbing resin can have various shapes such as a sheet shape, a fiber shape, and a film shape, but in general, it is often in a powder shape or a particle shape. It is known that the water-absorbing performance, handleability, and feeling of use vary depending on the particle diameter, particle size distribution, and the like of a powdered or particulate water-absorbing resin. Therefore, a powdered or particulate water-absorbing resin in which the particle size and particle size distribution are appropriately controlled is demanded.
- a water-absorbent resin with a low content of fine powder fine particles having a particle diameter of less than 100 ⁇ m to less than 150 ⁇ m, particularly fine particles of less than 150 ⁇ m
- the water absorbent resin powder is generally subjected to surface crosslinking treatment.
- Examples of the main production method of the powdery or particulate water-absorbing resin include an aqueous solution polymerization method and a reverse phase suspension polymerization method.
- a polymerization process for polymerizing a water-soluble ethylenically unsaturated monomer in an aqueous solution polymerization A gel pulverization (fine-graining) step for pulverizing the hydrogel polymer obtained in Step 1, a drying step for drying the pulverized gel, a pulverization step for pulverizing the dried product, a classification step for adjusting the pulverized product to an appropriate particle size range
- the manufacturing process is complicated, requiring a number of manufacturing steps such as a mixing step of mixing the surface-crosslinking agent with the classified water-absorbent resin powder and a heating step of heating the mixture to cause a surface cross-linking reaction (Non
- the water-absorbent resin as a material is usually dried by hot air in a drying step and heated to high temperature, and then cooled and then subjected to a pulverization step, and then heated again in a heat treatment step, Considering that it will eventually become a room temperature product, it is wasteful in terms of energy. Furthermore, in the drying process, the pulverization process, etc., fine powder may be generated due to mechanical damage or the surface cross-linked structure may be destroyed, and the physical properties of the resulting water-absorbent resin may be lowered.
- a water-soluble ethylenically unsaturated monomer aqueous solution is dispersed in a hydrophobic organic solvent to form a suspended particle, so that the polymerization reaction is performed.
- the gel grinding step is generally unnecessary, but a relatively large amount of hydrophobic organic solvent is used for dissolution of the surfactant, proper dispersion of the monomer aqueous solution, polymerization reaction, azeotropic dehydration and solvent evaporation.
- the energy required for heating and cooling over a long period of time (especially azeotropic dehydration in the drying process and evaporation of the dispersed solvent after drying) is relatively large, and the remaining organic solvent remaining in the water-absorbent resin is safe.
- the problem of the property and the environmental load of the organic solvent to be used are also increasing (Patent Documents 5 and 6).
- a relatively large amount of surfactant is required to disperse the hydrogel in the hydrophobic organic solvent, and not only the cost but also a large amount of surfactant remaining in the dried water absorbent resin is the water absorbent resin.
- a reduction in performance such as an increase in the amount of return in a paper diaper may occur, or a coloring problem of the water-absorbing resin may occur.
- fine powder is generated not only by the polymerization step and the pulverization step, but also by process damage such as a surface cross-linking step after the drying step and a transport step (Patent Documents 7 and 8).
- process damage such as a surface cross-linking step after the drying step and a transport step.
- the generation of fine powder due to the surface cross-linking step or subsequent process damage is accompanied by the destruction of the surface cross-linked structure, and therefore the physical properties of the water-absorbent resin are lowered.
- the amount of fine powder generated in the conventional manufacturing process reaches around 10 wt% to several tens wt% (for example, 20 to 30 wt%) of the entire production amount.
- Patent Documents 9 to 16 Methods for improving the processing capability have been proposed.
- the fine powder is recycled before the classification process, particularly before the drying process, and further to the polymerization process, the gel grinding process, and the drying process.
- it is difficult to handle fine powder because it tends to aggregate.
- the addition of the fine powder recycling process further complicates or enlarges the manufacturing process (equipment) of the water absorbent resin.
- the increase in the amount of fine powder collection may be accompanied by the fall of productivity and the performance of the water absorbing resin obtained.
- a pulverization step for adjusting the water-absorbent resin after drying to a desired product particle size.
- the hydrogel polymer obtained by aqueous solution polymerization has high adhesiveness and cohesion and low fluidity, the hydrogel polymer is allowed to stand and dry (dry without stirring) in the drying step,
- a continuous ventilation band type dryer 100 as illustrated in FIG. 7 is generally used (Non-patent Document 1 and Patent Documents 17 to 22).
- the hydrogel polymer 104 is laminated on the drying belt 102 (generally several centimeters to several tens of centimeters in gel thickness) and moved in the direction indicated by the arrow 108 in FIG.
- a continuous block-shaped dried polymer 110 having a width of several meters (corresponding to the lateral width of the belt 102) and a thickness of several centimeters to several tens of centimeters is obtained after drying.
- a large facility is required. This pulverization is a main factor for generating a large amount of fine powder.
- Patent Document 22 a problem solving means when using the ventilation band type dryer 100 is proposed, but the means complicates the manufacturing process and is not yet sufficient as an effect to be obtained. .
- Patent Documents 22 to 28, 31 a method of stirring and drying the hydrogel polymer
- Patent Documents 29 and 31 a method of drying in a fluidized bed
- Patent Documents 37 to 41 a technique for performing post-crosslinking at the hydrous gel stage
- the hydrogel polymer has high tackiness and cohesion and low fluidity, it is necessary to add a large amount of gel fluidizing agent (surfactant) during stirring and drying, which is disadvantageous in terms of cost.
- the remaining fluidizing agent (surfactant) is accompanied by performance deterioration of the water-absorbent resin.
- stirring of the hydrogel requires a large stirring power, and the dried product may deteriorate due to mechanical damage.
- problems such as adhesion of water-containing gel to the inner surface of the apparatus and aggregation of water-containing gels in continuous operation for a long time.
- the fluidized bed dryer requires a large amount of air flow for fluidization of the hydrogel, and is not applicable to a specific hydrogel having high fluidity (for example, a hydrogel having a high solid content). there were.
- treatment of waste gas discharged from the dryer for example, Patent Document 30
- vent band dryers and fluidized bed dryers that use a large amount of gas for drying require large waste gas treatment equipment, which is disadvantageous in terms of energy and equipment. there were.
- the above water-absorbing resin is required to reduce the residual monomer and improve other physical properties in addition to improving the production efficiency and solving the problem of fine powder generation (Patent Document 34). Further, since the water absorption rate of the water-absorbent resin depends on the specific surface area, as a means for improving the water absorption rate, foaming polymerization or a technique of pulverizing a hydrous gel and granulating it after drying has been proposed (Patent Documents 36 to 36). 38). However, the formation of fine particles or foam polymerization of the water-containing gel before drying for improving the water absorption rate may be accompanied by generation of fine powder including process damage and increase of residual monomers. Furthermore, in the method of finely pulverizing the hydrated gel, problems (dropping and scattering of dried products from the drying belt, clogging of the belt) tend to become more prominent.
- An object of the present invention is to produce a water-absorbent resin powder in which the amount of fine powder generated in the production process is small and the deterioration of physical properties due to the fine powder is suppressed, and the drying apparatus and drying method (particularly, the drying start method) of the particulate hydrous gel Is to provide. Furthermore, the subject of this invention is providing the manufacturing method of the water absorbing resin powder by which the manufacturing process was simplified and compactized.
- the inventors of the present invention used a heating device having a specific structure as a drying device at a predetermined temperature condition to suppress aggregation and adhesion during drying, and at the time of the drying step or surface cross-linking step.
- the present invention was completed by finding that the mechanical damage was reduced and the amount of fine powder was reduced.
- the heating device when used as a drying device, the heating device may be referred to as a drying device. Regardless of its name, a heating device and a drying device having a specific structure described in the present specification belong to the technical scope of the present invention.
- the present invention relates to a rotating container that accommodates and rotates the particulate hydrogel crosslinked polymer in the drying step of drying the particulate hydrogel crosslinked polymer, and is located inside the rotating container. Then, using a heating device provided with a plurality of heating tubes extending in the axial direction and rotating together with the rotating container as a drying device, the particulate hydrogel crosslinked polymer used in the drying step is This is a method for producing a water-absorbent resin powder having a gel temperature of 50 ° C. or higher.
- the present invention relates to a drying apparatus for a particulate hydrogel crosslinked polymer obtained from a monomer that is a raw material for a water-absorbent resin, and the drying apparatus has a particulate hydrogel crosslinked polymer in its interior.
- a rotating container that accommodates and rotates, a plurality of heating tubes that are positioned inside the rotating container, extend in the axial direction of the rotating container, and rotate together with the rotating container, and gas is introduced into the rotating container And the number of the heating tubes is 5 or more, and the heating tubes are not in contact with the inner peripheral surface of the rotating container in the axial direction, and the outer periphery of the rotating container
- the drying device is provided with heating means or heat retaining means on the surface.
- the present invention is a drying method for drying a particulate hydrogel crosslinked polymer obtained from a monomer that is a raw material of a water-absorbent resin using the above-described drying apparatus, and a heating medium is provided in the rotating container.
- the gas temperature of the particulate hydrogel crosslinked polymer introduced into the drying apparatus is 50 ° C. or higher, and the temperature of the inner surface of the rotating container is 150 ° C. or higher.
- the present invention is a facility for producing a water-absorbent resin powder including a drying device, a polymerization device for obtaining a water-containing gel-like cross-linked polymer from a monomer that is a raw material of the water-absorbent resin, and the water-containing gel-like cross-linked polymer.
- a gel pulverization apparatus for pulverizing the coalescence to obtain a particulate hydrogel crosslinked polymer, and this drying apparatus dries the particulate hydrogel crosslinked polymer to obtain a granular dried product.
- the drying method and the drying apparatus according to the present invention there is little mechanical damage in the drying process, and the deterioration of physical properties due to generation of fine powder and destruction of surface cross-linking is suppressed.
- the reduction in the amount of fine powder reduces the cost and time conventionally required for fine powder collection and recycling, and improves production efficiency.
- the subsequent production process can be made compact or simplified, and the water-absorbent resin powder having an inferior water absorption performance can be efficiently produced. Can do.
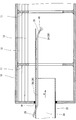
- FIG. 1 is a partially cutaway side view showing an example of a drying device (a rotary heating device with a heating tube) used in the manufacturing method according to the present invention.
- FIG. 2 is an enlarged cross-sectional view showing a part of the heating apparatus of FIG.
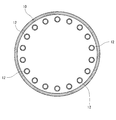
- FIG. 3 is a cross-sectional view taken along line III-III of the heating apparatus of FIG.
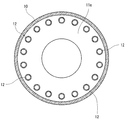
- FIG. 4 is a cross-sectional view for explaining a drying device (rotary heating device with double heating tubes) used in the manufacturing method according to the present invention.
- FIG. 5 is an example of a shield (opening ratio 20%) having an opening used in a drying apparatus (rotary heating apparatus with a heating tube) used in the manufacturing method according to the present invention.
- FIG. 1 is a partially cutaway side view showing an example of a drying device (a rotary heating device with a heating tube) used in the manufacturing method according to the present invention.
- FIG. 2 is an enlarged cross-sectional view showing a part of the heating apparatus of FIG.
- FIG. 3
- FIG. 6 is an example of shielding (opening ratio 50%) having an opening used in a drying apparatus (rotary heating apparatus with a heating tube) used in the manufacturing method according to the present invention.
- FIG. 7 is a cross-sectional view for explaining a ventilation band type dryer as a prior art.
- FIG. 8 is a flowchart showing a conventional process for producing a typical water-absorbent resin.
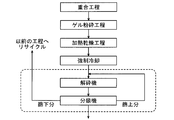
- FIG. 9 is a flowchart showing a water-absorbent resin production process in which the pulverization process, classification process and fine powder collection process of the present invention are made compact.
- FIG. 10 is a flowchart showing a manufacturing process of a water-absorbent resin in which the drying process and the surface cross-linking process of the present invention are integrated and made more compact.
- FIG. 11 is an electron micrograph of a granular dried product (granulated product) obtained by the production method according to the present invention.
- FIG. 12 is a flowchart showing an example of the sizing step in a preferred embodiment of the present invention.
- FIG. 13 is a flowchart showing another example of the sizing step in a preferred embodiment of the present invention.
- Water absorbent resin refers to a water-swellable, water-insoluble polymer gelling agent and satisfies the following physical properties. That is, the CRC (centrifuge retention capacity) defined by ERT441.2-02 as water swellability is 5 g / g or more, and Ext (water-soluble) defined by ERT470.2-02 as water-insoluble The polymer gelling agent whose (min) is 50 mass% or less.
- the water-absorbent resin can be designed according to its use and purpose, and is not particularly limited, but is preferably a hydrophilic cross-linked polymer obtained by cross-linking an unsaturated monomer having a carboxyl group. Moreover, it is not limited to the form whose whole quantity is a crosslinked polymer, As long as each said physical property (CRC, Ext) satisfy
- the “water-absorbent resin” may be surface-crosslinked (also known as post-crosslinking, secondary crosslinking) or may not be surface-crosslinked.
- the water-absorbing resin that has been subjected to the predetermined surface cross-linking treatment may be referred to as a water-absorbing resin or water-absorbing agent that has been separately surface cross-linked (post-cross-linked).
- a water absorbent resin or water absorbent adjusted to a predetermined moisture content and particle size is referred to as a water absorbent resin powder.
- poly (meth) acrylic acid (salt) refers to poly (meth) acrylic acid and / or a salt thereof, and (meth) acrylic acid and / or a salt thereof (hereinafter referred to as “(meta) ) (Also referred to as “acrylic acid (salt)”) as a repeating unit, and a cross-linked polymer containing a graft component as an optional component.
- the “main component” is preferably used in an amount (content) of (meth) acrylic acid (salt) of 50 mol% to 100 mol%, more preferably based on the whole monomer used for polymerization. It means 70 mol% to 100 mol%, more preferably 90 mol% to 100 mol%, particularly preferably substantially 100 mol%.
- poly (meth) acrylate may be unneutralized, but is preferably a partially or completely neutralized poly (meth) acrylate, more preferably a monovalent salt, Preferred are alkali metal salts or ammonium salts, even more preferred are alkali metal salts, and particularly preferred are sodium salts.
- EDANA and “ERT” “EDANA” is an abbreviation for European Disposables and Nonwovens Associations.
- ERT is an abbreviation for EDANA Recommended Test Methods and is a European standard that defines a method for measuring water-absorbing resin. In the present invention, unless otherwise specified, the physical properties of the water-absorbent resin are measured based on the original ERT (revised in 2002).
- CRC is an abbreviation for Centrifugation Retention Capacity (centrifuge retention capacity) and means the water absorption capacity of the water absorbent resin under no pressure (sometimes referred to as “water absorption capacity”). Specifically, 0.2 g of the water-absorbing resin was put in a non-woven bag, and then immersed in a large excess of 0.9 mass% sodium chloride aqueous solution for 30 minutes for free swelling, and then centrifuged (250G ) Is the water absorption capacity (unit: g / g) after draining for 3 minutes. In addition, about the water-containing gel after superposition
- Ext is an abbreviation for Extractables and means the water-soluble content of the water-absorbent resin (the amount of water-soluble polymer in the water-absorbent resin). Specifically, 1.0 g of water-absorbing resin is added to 200 ml of a 0.9% by mass sodium chloride aqueous solution, stirred for 16 hours at 500 rpm, and then the amount of substance dissolved in the aqueous solution (unit: mass%). . PH titration is used to measure the water-soluble content. In addition, about the water-containing gel after superposition
- “Moisture Content” (ERT430.2-02) “Moisture Content” means the moisture content defined by the loss on drying of the water-absorbent resin. Specifically, it refers to a value (unit: mass%) calculated from loss on drying when 4.0 g of water-absorbing resin is dried at 105 ° C. for 3 hours.
- the dried water-absorbent resin is defined by 1.0 g of a water-absorbent resin at 180 ° C. and a loss on drying for 3 hours, and the hydrogel before drying is 2.0 g of hydrous gel at 180 ° C. , Defined as 24-hour loss on drying.
- PSD is an abbreviation for Particle Size Distribution, and means a particle size distribution of a water-absorbent resin measured by sieving.
- the mass average particle diameter (D50) and the logarithmic standard deviation ( ⁇ ) of the particle size distribution are measured by the same method as described in US Pat. No. 7,638,570.
- the particle size distribution (PSD) of the hydrogel is defined by wet sieving by the method described below. Further, the particle size ( ⁇ m) in terms of solid content of the hydrated gel is defined by the calculation method described later from the particle size ( ⁇ m) of the hydrated gel and its solid content rate (%).
- AAP is an abbreviation for Absorption against Pressure, and means the water absorption capacity of a water absorbent resin under pressure. Specifically, after 0.9 g of a water-absorbing resin was swollen under a load of 2.06 kPa (21 g / cm 2, 0.3 psi) for 1 hour against a large excess of 0.9 mass% sodium chloride aqueous solution Of water absorption (unit: g / g). In some cases, the load condition is changed to 4.83 kPa (49 g / cm 2, 0.7 psi). In addition, about the water-containing gel after superposition
- X to Y indicating a range means “X or more and Y or less”.
- t (ton) which is a unit of mass, means “Metric ton”
- ppm means “mass ppm” or “weight ppm”.
- mass and weight means “mass part” and “part by weight”, “mass%” and “wt%” are treated as synonyms.
- ⁇ acid (salt) means “ ⁇ acid and / or salt thereof”
- (meth) acryl means “acryl and / or methacryl”.
- a method for producing a water-absorbent resin powder according to the present invention is a particulate hydrogel crosslinked polymer obtained from a monomer that is a raw material for a water-absorbent resin using a heating device.
- a drying step of drying the Preferably, the production method comprises a polymerization step, a gel grinding step (simultaneously or separately from the polymerization), a surface crosslinking step (simultaneously or separately from the drying), a cooling step, and (after drying and / or after surface crosslinking). It has a sizing process.
- This step is a step of preparing an aqueous solution containing acrylic acid (salt) as a main component (hereinafter referred to as “monomer aqueous solution”).
- monomer aqueous solution an aqueous solution containing acrylic acid (salt) as a main component
- the slurry liquid of a monomer can also be used in the range by which the water absorption performance of the water-absorbing resin obtained does not fall, in this section, monomer aqueous solution is demonstrated for convenience.
- main component means that the amount (content) of acrylic acid (salt) used is usually based on the whole monomer (excluding the internal crosslinking agent) subjected to the polymerization reaction of the water-absorbent resin. It means 50 mol% or more, preferably 70 mol% or more, more preferably 90 mol% or more (the upper limit is 100 mol%).
- the monomer other than acrylic acid may be a compound that can be polymerized to become a water-absorbing resin.
- Acid group-containing unsaturated monomers such as 2- (meth) acryloylethanesulfonic acid, 2- (meth) acryloylpropanesulfonic acid, 2-hydroxyethyl (meth) acryloyl phosphate; (meth) acrylamide, N-ethyl Amide group-containing unsaturated monomers such as (meth) acrylamide and N, N-dimethyl (meth) acrylamide; N, N
- the monomer other than acrylic acid is an acid group-containing unsaturated monomer from the viewpoint of water absorption performance of the resulting water-absorbent resin, and more preferably methacrylic acid, (anhydrous) maleic acid, itaconic acid, cinnamon An acid, more preferably methacrylic acid.
- the monomer used for the polymerization preferably contains a small amount of a polymerization inhibitor in view of the stability of the polymerization.
- a preferred polymerization inhibitor is p-methoxyphenol.
- the amount of the polymerization inhibitor contained in the monomer (particularly acrylic acid and its salt) is usually 1 to 250 ppm, preferably 10 to 160 ppm, more preferably 20 to 80 ppm.
- the salt of the acid group-containing unsaturated monomer is preferably a salt with a monovalent cation, more preferably at least one selected from alkali metal salts, ammonium salts and amine salts, An alkali metal salt is more preferable, at least one selected from a sodium salt, a lithium salt and a potassium salt is still more preferable, and a sodium salt is particularly preferable.
- the neutralizing agent used for neutralizing the acid group-containing unsaturated monomer is not particularly limited, but includes inorganic salts such as sodium hydroxide, potassium hydroxide, sodium carbonate, ammonium carbonate, and amino groups. Or a basic substance such as an amine-based organic compound having an imino group is appropriately selected and used. Two or more basic substances may be used in combination as a neutralizing agent.
- the monomer in this invention is the concept containing a neutralization salt unless there is particular notice.
- the number of moles of the neutralized salt relative to the total number of moles of the acid group-containing unsaturated monomer and the neutralized salt is preferably 40 mol% or more, More preferably, it is 40 mol% to 80 mol%, still more preferably 45 mol% to 78 mol%, and particularly preferably 50 mol% to 75 mol%.
- a method for adjusting the neutralization rate a method of mixing an acid group-containing unsaturated monomer and a neutralized salt thereof; a method of adding a known neutralizing agent to an acid group-containing unsaturated monomer; A method using a partially neutralized salt of an acid group-containing unsaturated monomer adjusted to a predetermined neutralization rate (that is, a mixture of an acid group-containing unsaturated monomer and a neutralized salt thereof); . Moreover, you may combine these methods.
- the adjustment of the neutralization rate may be performed before the polymerization reaction of the acid group-containing unsaturated monomer is started, may be performed in the polymerization reaction of the acid group-containing unsaturated monomer, or may contain an acid group. You may carry out with respect to the hydrogel crosslinked polymer obtained after completion
- the neutralization rate may be adjusted by selecting any one stage before the start of the polymerization reaction, during the polymerization reaction or after the completion of the polymerization reaction, or the neutralization ratio may be adjusted at a plurality of stages.
- an internal cross-linking agent In the method for producing a water absorbent resin powder, an internal cross-linking agent is preferably used.
- the internal cross-linking agent adjusts the water absorption performance of the resulting water-absorbent resin, the gel strength during water absorption, and the like.
- the internal cross-linking agent only needs to have two or more unsaturated bonds or reactive functional groups in one molecule.
- N, N-methylenebis (meth) acrylamide, (poly) ethylene glycol di (meth) acrylate, (internal cross-linking agent having a plurality of polymerizable unsaturated groups (which can be copolymerized with monomers) in the molecule ( Poly) propylene glycol di (meth) acrylate, trimethylolpropane tri (meth) acrylate, glycerin (meth) acrylate, glycerin acrylate methacrylate, ethylene oxide modified trimethylolpropane tri (meth) acrylate, pentaerythritol hexa (meth) acrylate, tri Examples include allyl cyanurate, triallyl isocyanurate, and triallyl phosphate.
- an internal cross-linking agent having a plurality of reactive functional groups (which can react with a monomer functional group (eg, carboxy group)) in the molecule, triallylamine, polyallyloxyalkane, (poly) ethylene glycol diglycidyl ether, Glycerol diglycidyl ether, ethylene glycol, polyethylene glycol, propylene glycol, glycerin, 1,4-butanediol, pentaerythritol, ethylenediamine, ethylene carbonate, propylene carbonate, polyethyleneimine, etc.
- the cyclic carbonate is a cross-linking agent that further generates a functional group OH by reaction with a carboxyl group).
- glycidyl (meth) acrylate etc. are mentioned as an internal crosslinking agent which has a polymerizable unsaturated group and a reactive functional group in a molecule
- a compound having a plurality of polymerization unsaturated groups in the molecule is preferable, and a compound having a (poly) alkylene structural unit in the molecule is more preferable. More preferred is a compound having a polyethylene glycol structural unit, and particularly preferred is an acrylate compound having a polyethylene glycol structural unit.
- Water-containing gels obtained using these internal cross-linking agents have a low water absorption ratio (low CRC) at the beginning of drying, and have low adhesiveness. It is preferable to dry the hydrogel having low adhesiveness with a heating device (the heating device of the present invention) because fusion and aggregation during drying can be reduced. Furthermore, the water-containing gel obtained using these internal crosslinking agents has an effect that the water absorption capacity (CRC) is easily improved by drying using a heating device (the heating device of the present invention).
- CRC water absorption capacity
- the amount of the internal cross-linking agent used is appropriately set according to the type of monomer and internal cross-linking agent. From the viewpoint of the gel strength of the water-absorbing resin obtained, it is preferably 0.001 mol% or more, more preferably 0.005 mol% or more, and still more preferably 0.01 mol% or more, based on the total amount of monomers. . Moreover, from a viewpoint of the water absorption performance improvement of a water absorbing resin, Preferably it is 5 mol% or less, More preferably, it is 2 mol% or less. In the polymerization conditions in which the monomer self-crosslinking reaction is effective, the internal crosslinking agent may not be used.
- chain transfer agents such as thiols, thiolic acids, secondary alcohols, amines and hypophosphites; foaming agents such as carbonates, bicarbonates, azo compounds, and bubbles; ethylenediamine Chelating agents such as tetra (methylenephosphinic acid) and metal salts thereof, metal salts of ethylenediaminetetraacetic acid and metal salts thereof, metal salts of diethylenetriaminepentaacetic acid; polyacrylic acid (salts) and cross-linked products thereof (for example, recycled water absorption) Resin fine powder), starch, cellulose, starch-cellulose derivatives, and hydrophilic polymers such as polyvinyl alcohol.
- chain transfer agents such as thiols, thiolic acids, secondary alcohols, amines and hypophosphites
- foaming agents such as carbonates, bicarbonates, azo compounds, and bubbles
- ethylenediamine Chelating agents such as tetra (methylenephosphinic acid) and metal
- the amount of other substances used is not particularly limited, but in the fine powder recycled if necessary, it is 30% by weight or less based on the monomer, and the total concentration of other substances other than the fine powder is preferably based on the monomer. It is 10% by mass or less, more preferably 0.001 to 5% by weight, and particularly preferably 0.01 to 1% by weight.
- the monomer concentration may be referred to as “monomer concentration”.
- the polymerization initiator used in the present invention is not particularly limited since it is appropriately selected depending on the polymerization form and the like.
- a thermal decomposition polymerization initiator, a photodecomposition polymerization initiator, or a combination thereof, or a polymerization start is used.
- a redox polymerization initiator combined with a reducing agent that accelerates the decomposition of the agent is used.
- one or more of the polymerization initiators disclosed in US Pat. No. 7,265,190 are used.
- a peroxide or an azo compound is preferably used, more preferably a peroxide, and still more preferably a persulfate.
- the amount of the polymerization initiator used is preferably 0.001 to 1 mol%, more preferably 0.001 to 0.5 mol%, based on the monomer.
- the amount of the reducing agent used is preferably 0.0001 to 0.02 mol% with respect to the monomer.
- the dissolved oxygen is preferably reduced to 5 ppm or less, more preferably 3 ppm or less, and particularly preferably 1 ppm or less.
- bubbles can be dispersed in the monomer aqueous solution.
- foam polymerization is performed in the polymerization reaction.
- This step is a step in which the aqueous monomer solution is polymerized to obtain a hydrated gel-like crosslinked polymer (hereinafter sometimes referred to as “hydrated gel”).
- the polymerization form is not particularly limited. From the viewpoint of water absorption characteristics, ease of polymerization control, etc., preferably droplet polymerization in the gas phase, aqueous solution polymerization, reverse phase suspension polymerization (here, droplet polymerization in a hydrophobic organic solvent is also an example of reverse phase suspension) More preferably aqueous solution polymerization, reverse phase suspension polymerization, and still more preferably aqueous solution polymerization. Among these, continuous aqueous solution polymerization is particularly preferable, and either continuous belt polymerization or continuous kneader polymerization is applied. As specific polymerization forms, continuous belt polymerization is disclosed in U.S. Pat. Nos.
- “high temperature initiation polymerization” and “high concentration polymerization” can be mentioned.
- “High temperature initiation polymerization” means that the temperature of the aqueous monomer solution is preferably 30 ° C. or higher, more preferably 35 ° C. or higher, still more preferably 40 ° C. or higher, particularly preferably 50 ° C. or higher (the upper limit is the boiling point).
- the “high concentration polymerization” means that the monomer concentration is preferably 30% by mass or more, more preferably 35% by mass or more, still more preferably 40% by mass or more, and particularly preferably 45% by mass or more.
- the upper limit is a saturation concentration.
- polymerization in vapor phase droplet polymerization, polymerization can be performed in an air atmosphere, but from the viewpoint of the color tone of the resulting water-absorbent resin, polymerization is performed in an inert gas atmosphere such as nitrogen or argon. Is preferred. In this case, for example, it is preferable to control the oxygen concentration in the gas phase to 1% by volume or less.
- the polymerization rate of the water-containing gel-like crosslinked polymer obtained in the polymerization step is to suppress aggregation during the heat treatment of the particulate water-containing gel-like crosslinked polymer obtained in the next pulverization step, and to reduce the residual monomer in the obtained water-absorbing agent. From the viewpoint, it is preferably 90% by weight or more, more preferably 95% by weight or more, still more preferably 98% by weight or more, and particularly preferably 99% by weight or more. When the polymerization rate is low, the water-containing gel being dried by the rotary heating device tends to aggregate or adhere easily.
- the upper limit of the polymerization rate is not particularly limited, but 100% by mass is ideal, but a high polymerization rate requires a long polymerization time and severe polymerization conditions, which may lead to a decrease in productivity and physical properties.
- An upper limit of 99.95% by weight, further 99.9% by weight, and usually 99.8% by weight is sufficient. Typically, it is 98 to 99.99% by mass, and more preferably within the above range.
- a technique of taking out a hydrogel having a polymerization rate of 90% or less from a polymerization apparatus and drying it was also known (for example, WO2006 / 103227). It has been found that the adjustment of the polymerization rate is important for drying with a heating device.
- Gel pulverization step This step is a step of pulverizing and pulverizing the hydrogel crosslinked polymer obtained in the polymerization step simultaneously with and / or after the polymerization.
- a kneader This is a step of obtaining a particulate hydrogel crosslinked polymer (hereinafter referred to as “particulate hydrogel”) by pulverizing with a screw extruder such as a meat chopper, a cutter mill or the like.
- the preferred particle size is in the range described later, so that the shape and size of the granular dried product obtained in the drying step described later or the surface crosslinked granular dried product obtained in the surface crosslinking step approaches the target product particle size. To be implemented.
- Patent Documents 35 to 37 disclose stationary drying (especially, a ventilation band drying in Patent Document 36) as a drying method.
- the present invention solves the above-mentioned problems not disclosed in Patent Documents 35 to 37. This is solved by a specific drying device not disclosed in ⁇ 37.
- the gel grinding energy (Gel Grinding Energy, GGE) is described in Patent Document 36, and the unit weight required by the gel grinding device when gelling the hydrogel crosslinked polymer (unit of hydrogel crosslinked polymer) It means mechanical energy per weight), and is calculated by the following (Equation 1) when the gel crusher is driven by three-phase AC power.
- Gel grinding energy [J / g] ⁇ 3 1/2 ⁇ voltage ⁇ current ⁇ power factor ⁇ motor efficiency ⁇ / ⁇ weight of hydrous gel charged into gel grinding device per second ⁇ (Formula 1)
- the power factor and the motor efficiency are values unique to the apparatus that vary depending on the operating conditions of the gel crushing apparatus, and take values from 0 to 1.
- the gel crusher is driven by single-phase AC power, it is calculated by changing 3 1/2 in the above formula to 1.
- the unit of voltage is [V]
- the unit of current is [A]
- the unit of weight of the hydrogel is [g].
- the preferred gel grinding energy (GGE) applied in the present invention can be applied within the range of Patent Document 36, but is not particularly limited.
- a gel fluidizing agent is added to the hydrated gel or the particulate hydrated gel that is a pulverized product thereof.
- the addition of the gel fluidizing agent is particularly effective when the particulate hydrous gel is treated in a drying step and a heat treatment step during surface cross-linking described later.
- the addition amount of the gel fluidizing agent is appropriately set according to the moisture content of the hydrous gel or particulate hydrous gel and the type of the gel fluidizing agent.
- the addition amount is preferably 0.001% to 0.5% by mass, more preferably 0.01% to 0.3% by mass, and still more preferably 0.02% by mass, based on the solid content of the hydrogel. % To 0.2% by mass.
- this gel fluidizing agent examples include, for example, anionic, cationic, nonionic, and amphoteric surfactants exemplified in Patent Documents 22 to 24, 26, and 28, and low molecular or high molecular types thereof. Surfactants, polymer lubricants and the like can be mentioned.
- sucrose fatty acid ester, polyglycerin fatty acid ester, sorbitan fatty acid ester, polyoxyethylene sorbitan fatty acid ester, polyoxyethylene glycerin fatty acid ester, sorbitol fatty acid are used as the surfactant used in the gel fluidizing agent.
- Esters polyoxyethylene sorbitol fatty acid esters, polyoxyethylene alkyl ethers, polyoxyethylene alkyl phenyl ethers, polyoxyethylene castor oil, polyoxyethylene hydrogenated castor oil, alkylallyl formaldehyde condensed polyoxyethylene ether, polyoxyethylene polyoxypropylene Block copolymer, polyoxyethylene polyoxypropyl alkyl ether, polyethylene glycol fatty acid ester, alkyl gluco Nonionic surfactants such as N, alkyl gluconamides, polyoxyethylene fatty acid amides, polyoxyethylene alkyl amines, polyoxyethylene alkyl ether phosphates, and polyoxyethylene alkyl allyl ether phosphates, 2) Capryldimethylaminoacetic acid betaine, lauryldimethylaminoacetic acid betaine, myristyldimethylaminoacetic acid betaine, stearyldimethylaminoacetic acid betaine and other alky
- anionic surfactants such as mono-alkali metal alkylacetates, and (4) cationic surfactants such as long-chain alkyldimethylaminoethyl quaternary salts. Of these, two or more may be used in combination.
- Polymer lubricant In the production method according to the present invention, a polymer lubricant exemplified below can be added to the monomer aqueous solution and the water-containing gel as long as the object of the present invention is achieved.
- polymer lubricant examples include maleic anhydride-modified polyethylene, maleic anhydride-modified polypropylene, maleic anhydride-modified ethylene / propylene copolymer, maleic anhydride-modified ethylene / propylene / diene terpolymer (EPDM).
- Maleic anhydride modified polybutadiene maleic anhydride / ethylene copolymer, maleic anhydride / propylene copolymer, maleic anhydride / ethylene / propylene copolymer, maleic anhydride / butadiene copolymer, polyethylene, polypropylene, Polyalkylene such as ethylene / propylene copolymer, oxidized polyethylene, oxidized polypropylene, oxidized ethylene / propylene copolymer, ethylene / acrylic acid copolymer, ethyl cellulose, ethyl hydroxyethyl cellulose, polyethylene glycol Alkylene oxide, and the like.
- These molecular weights are preferably selected in the range of preferably 2 to 2 million, more preferably 4 to 1 million.
- polymer lubricants and the above surfactants may be used in combination as a gel fluidizing agent.
- the total addition amount is appropriately set according to the polymerization form, the composition of the monomer aqueous solution, and the water content of the water-containing gel.
- the concentration relative to the monomer component is set, and when added to the hydrogel, the solid content is set, and when added to both, the total is set.
- the total addition amount of the surfactant and the polymeric lubricant is preferably 1.0% by mass or less, more preferably 0.5% by mass or less, preferably 0.05% by mass or more, and particularly preferably 0.1% by mass. % Or more.
- the kind and addition amount of the gel fluidizing agent are selected in consideration of the fluidity of the target particulate hydrous gel.
- a gel fluidizing agent of a kind and amount that does not excessively reduce the surface tension of the water absorbent resin of the final product is preferable in view of the amount of return of the water absorbent resin obtained in an absorbent article (diaper) in actual use.
- the surface tension of the water-absorbent resin (the surface tension of the dispersion in which the water-absorbent resin is dispersed in physiological saline) is preferably 55 mN / m or more, more preferably 60 mN / m or more, and even more preferably 65 mN / m or more.
- the kind and amount of the gel fluidizing agent are selected so that This surface tension is measured by the method described in WO2015 / 129917. Examples of the gel fluidizing agent that can bring the surface tension into such a range include the betaine surfactants of Patent Document 28.
- Drying step This step is a step of obtaining a granular dried product by drying a particulate hydrous gel (preferably containing a gel fluidizing agent) to a desired solid content, preferably drying and It is a step of granulating to obtain a granular dry granulated product.
- the particulate hydrogel used for this process is not limited to what is obtained by grind
- the “granular dry granulated product” means a particulate dried product formed by physical or chemical adhesion between a plurality of water absorbent resin particles, and has an average particle size before and after drying.
- FIG. 11 shows an example of an electron micrograph of a granular dry granulated product obtained in the drying step of the production method according to the present invention.
- the granular dry granulated product may be referred to as “granulated product”.
- the “solid content ratio” means a value calculated from a loss on drying (a change in mass when 1 g of a sample is dried at 180 ° C. for 3 hours).
- the solid content of the granular dried product after this drying step is preferably 80% by mass or more, more preferably 85% by mass to 99.8% by mass, and still more preferably 90% by mass to 99.7% by mass. Even more preferably, it is in the order of 92% to 99.5% by weight, particularly preferably 96% to 99.5% by weight, and most preferably 98% to 99.5% by weight. If the solid content after drying is excessively high, not only drying for a long time is required, but also physical properties may be deteriorated or colored after drying. Moreover, when the solid content rate after drying is low, for example, productivity in the sizing process and reduction in water absorption ratio (CRC) may occur.
- CRC water absorption ratio
- a heating device is used as a drying device in the drying process.
- a device is used.
- the heating device includes a rotating container that accommodates the particulate hydrogel therein and rotates, and a plurality of heating units that are positioned inside the rotating container, extend in the axial direction thereof, and rotate together with the rotating container. With a tube.
- the heating device having this configuration may be referred to as a “rotary heating device” or a “rotating heating device with a heating tube”.
- the heating device further includes other heating means on the outer peripheral surface of the rotating container.
- the particulate hydrogel contained in the rotating container is stirred by the rotation of the container and heated by contact with a plurality of heating tubes or heat conduction from the heating tubes.
- the inner surface of the rotating container is also heated by radiant heat or the like of the plurality of heating tubes, the particulate water-containing gel is further heated by heating means located on the outer peripheral surface of the rotating container as necessary.
- other stirring means such as a stirring blade is used in combination, if necessary, but mainly by the rotation of a rotating container that contains the particulate hydrous gel and the action of a plurality of heating tubes that rotate with the rotating container.
- this heating apparatus since the material to be dried flows mainly by rotation of the rotating container, a large amount of energy is required to stir the particulate water-containing gel having adhesiveness like a continuous stirring dryer that stirs with a stirring blade or the like.
- a decrease in physical properties of the water-absorbent resin after drying eg, a decrease in water absorption rate and an increase in soluble components
- generation of fine powder, and aggregation during drying are avoided.
- Examples of the heating device used in the production method according to the present invention include a rotary dryer with a steam pipe.
- Specific examples include a steam tube dryer (manufactured by Kurimoto Soko Co., Ltd.), a steam tube dryer (manufactured by Ube Industries Co., Ltd.), a steam tube dryer (manufactured by Tsukishima Kikai Co., Ltd.), a steam tube dryer (manufactured by Mitsui Engineering & Shipbuilding Co., Ltd.). ) Etc.
- the heating device may include other flow means for flowing the material to be dried (for example, a scraping plate on the inner surface of the rotating container) and / or other heating means.
- other heating means from the viewpoint of drying efficiency and reduction of thermal damage to the water-absorbent resin, direct heat transfer by convective heat transfer and / or a heating surface of a heating device heated by a heating medium (particulate hydrous gel) Heating means by indirect heat transfer by heat conduction from the contact surface and the heat source portion). More preferable heating means is a ventilation heating type for direct heat transfer and an outer wall heating type for indirect heat transfer.
- the number of rotary heating devices that can be used in the drying process may be only one or two or more.
- a plurality of rotary heating apparatuses having different specifications may be used in combination.
- the type and number of drying devices to be combined are not limited.
- Examples include a dryer and a rotary kiln.
- solid air manufactured by Hosokawa Micron Corporation
- CD dryer manufactured by Kurimoto Steel Factory
- paddle dryer manufactured by Nara Machinery Co., Ltd.
- rotary kiln manufactured by Kurimoto Steel Factory
- Product manufactured by Okawara Seisakusho Co., Ltd.
- rotary dryer manufactured by Okawara Seisakusho Co., Ltd.
- the timing of switching can be determined by using the solid content rate or residual solvent amount of the obtained granular dried product as an index.
- the solid content rate is used as an index
- the solid content is dried to about 70% by mass with the first stage heating device, and then the second level heating device is switched to the desired solid content rate.
- the form is further dried with a second stage heating device to a solid content of about 85% by mass, and then switched to the third stage heating device to be dried to a desired solid content rate.
- the first stage heating device when drying the particulate hydrogel obtained by reverse phase suspension polymerization (including liquid phase droplet polymerization), when the amount of residual organic solvent is used as an index of switching timing, the first stage heating device Preferably, in a stage where the residual organic solvent is reduced to 10000 ppm, more preferably 5000 ppm in terms of dry weight, the second stage heating device is switched to a desired residual solvent amount.
- the form which uses a rotary heating apparatus in the stage with little solid content or much residual solvent amount is preferable.
- the Froude number Fr is the ratio of the centrifugal acceleration ⁇ 2 * r acting on the material to be dried stirred in the rotating container to the gravitational acceleration g. ( ⁇ is the angular velocity of the rotating body: rad / sec, r is the representative radius of the rotating body: m).
- the number of rotations of the rotating container is appropriately set according to the apparatus size and the amount of drying treatment (the amount of drying per hour), but is preferably 1 rpm to 250 rpm, more preferably 1 rpm to 100 rpm, and further preferably 2 rpm to 50 rpm. .
- the maximum peripheral speed is not particularly limited, but is preferably 0.05 m / s to 10 m / s, more preferably 0.1 m / s to 8 m / s, and still more preferably 0.15 m / s to 5 m / s. is there.
- the heating device can also have a function of making the inside of the device pressurized, normal pressure, or reduced pressure.
- a pressurized state for example, it is adjusted by increasing the amount of carrier gas introduced into the heating device.
- the degree of pressurization with respect to atmospheric pressure is a slight pressurization of more than 0 to 0.01 kPa.
- it adjusts by the change of the suction
- the degree of decompression with respect to atmospheric pressure is preferably a slight decompression of more than 0 kPa to 5 kPa, more preferably more than 0 kPa to 2 kPa, and still more preferably 0.01 kPa to 0.5 kPa.
- the degree of pressurization with respect to atmospheric pressure and “the degree of depressurization with respect to atmospheric pressure” mean a differential pressure from the atmospheric pressure, and are expressed as an absolute value of the difference from the atmospheric pressure. For example, when the atmospheric pressure is the standard atmospheric pressure (101.3 kPa) and the degree of pressure reduction with respect to the atmospheric pressure is 10 kPa, the actual atmospheric pressure is 91.3 kPa.
- the heating apparatus can also have a function of introducing gas (preferably means for introducing and discharging gas) inside the apparatus.
- means for introducing and discharging gas include a gas inlet and outlet.
- the installation positions of the gas introduction port and the discharge port are not limited, but are preferably installed on the inlet side and the outlet side of the object to be dried of the heating device, and may be provided with a gas introduction mechanism or a gas discharge mechanism as necessary.
- the gas is not particularly limited, and examples thereof include air, dry air, nitrogen, water vapor, and a mixed gas thereof.
- the gas acts as a carrier gas, and accelerates drying by discharging water vapor generated during drying out of the apparatus. Further, when a heated gas is used, the gas also acts as a heat medium, and further drying is promoted.
- a mixed gas of these and air, or the like is used.
- a mixed gas containing water vapor hereinafter also referred to as a high-humidity mixed gas
- the inside of the apparatus is in a low oxygen state, and oxidation and deterioration during drying are suppressed.
- the performance improvement and low coloring of the water absorbent resin can be achieved.
- it becomes possible to suppress aggregation and agglomeration of the particulate hydrogel during drying it is preferable.
- the amount of the gas introduced is usually 0.05 Nm 3 / kg to 20 Nm 3 / kg with respect to the treatment amount (kg / hr) per unit time of the water-absorbent resin (in terms of solid content of the hydrogel). That is, preferably 0.1Nm 3 / kg ⁇ 10Nm 3 / kg, more preferably 0.1Nm 3 / kg ⁇ 5Nm 3 / kg, more preferably 0.2Nm 3 /kg ⁇ 2.5Nm 3 / kg, in particular It is preferably 0.2 Nm 3 / kg to 1 Nm 3 / kg.
- the atmospheric dew point inside the heating device can be adjusted by introducing the gas into the heating device from one or more locations.
- the dew point can be adjusted as appropriate according to the moisture content of the particulate hydrogel charged into the heating device.
- the dew point is measured at the time of exhaust from the heating device, and is preferably 60 ° C. or higher, more preferably 65 ° C. or higher, and further preferably 70 ° C. or higher.
- an upper limit is not specifically limited, Preferably it is 100 degrees C or less.
- gas moving direction in the heating apparatus may be a cocurrent flow or a countercurrent flow with respect to the moving direction of the particulate hydrogel that is to be dried, or a mixture of these.
- the drying temperature in a heating apparatus is adjusted with the temperature of the heat medium used.
- the aforementioned gas (hot air) soot is introduced into these heating devices (particulate hydrated gel container), this gas directly acts as a heat transfer heat medium.
- the temperature of the heating medium (gas) is preferably 100 ° C. or higher (if the heating medium is water vapor, 0.1013 MPa or higher), more preferably 120 ° C. or higher, and even more preferably 150 ° C. or higher. is there.
- the temperature of the heating medium is preferably 150 ° C. or higher (if the heating medium is steam, the saturation pressure is about 0.49 MPa or higher), more preferably 160 ° C. or higher (same as about the same). 0.62 MPa or more), more preferably 170 ° C. or more (same as about 0.79 MPa or more), and particularly preferably 180 ° C. or more (same as 1.0 MPa or more).
- the heat transfer medium temperature is 100 ° C. or less by direct heat transfer and less than 160 ° C., particularly less than 150 ° C. by indirect heat transfer, the particulate water-containing gel is likely to adhere and aggregate.
- a liquid heat medium such as oil may be used as a heat medium other than gas, but water vapor is preferable.
- the drying temperature is preferably 300 ° C. or less for direct heat transfer and indirect heat transfer from the viewpoint of deterioration and coloring of the water absorbent resin and the performance of the water absorbing agent (about 8.6 MPa when the heat medium is steam). More preferably, it is 280 ° C. or lower (same as about 6.4 MPa), and further preferably 250 ° C. or lower (same as about 4.0 MPa).
- the temperature may be a constant temperature or may be appropriately changed during drying.
- the drying time is preferably 10 minutes to 120 minutes, more preferably 20 minutes to 90 minutes, and even more preferably 20 minutes to 60 minutes.
- the filling rate in this heating device is appropriately selected, but from the viewpoint of drying efficiency, it is preferably 5 to 95%, more It is preferably selected within a range of preferably 6 to 50%, more preferably 10 to 40%.
- FIGS. 1 and 2 show an example of a rotary heating device 2 with a heating tube.
- the basic configuration and method of use of the heating device 2 will be described with reference to FIGS. 1 and 2.
- the heating device 2 has a main part 4, an input part 6, and a take-out part 8.
- the main part 4 includes a rotating container 10, a large number of heating pipes 12, a first gear 14, a second gear 16, and a packing 18.
- the rotating container 10 is generally cylindrical.
- the horizontal direction in FIG. 1 is the axial direction of the rotating container 10.
- a plurality of barriers 11 are provided on the inner wall of the rotating container 10 at intervals in the axial direction. Each barrier 11 extends along the inner peripheral surface of the rotating container 10.
- a large number of heating tubes 12 are accommodated in the rotating container 10. Each heating tube 12 extends in the axial direction of the rotating container 10 and penetrates both ends of the rotating container 10. As will be described later, many of the heating tubes 12 are not in contact with the inner peripheral surface of the rotating container 10 in the axial direction.
- FIG. 2 shows only a part of the heating tubes 12 for convenience of explanation.
- the first gear 14 is fixed to the outer peripheral surface of the rotating container 10.
- the second gear 16 is in mesh with the first gear 14. In FIG. 2, the first gear 14 and the second gear 16 are not shown for convenience of explanation.
- the packing 18 is located between the rotary container 10 and the charging unit 6.
- the charging unit 6 has a main cylinder 20, a hopper 22 and a pipe 26.
- the main cylinder 20 is open inside the rotating container 10.
- the inside of the main cylinder 20 is referred to as an inner space 28.
- the hopper 22 has a shape in which the inner dimension gradually decreases from top to bottom.
- the lower end of the hopper 22 is fixed to the main cylinder 20.
- the hopper 22 communicates with the inner space 28. 1 and 2
- one end 32 of the pipe 26 is exposed from the main cylinder 20, and the other end 34 reaches the inside of the rotating container 10.
- the pipe 26 has a nozzle 36.
- An intermediate portion 38 of the pipe 26 is accommodated in the inner space 28.
- the take-out unit 8 has a take-out port 40, a steam inlet 44, and a drain 46.
- the steam inlet 44 is in communication with a number of heating tubes 12.
- the drain 46 is also in communication with a number of heating tubes 12.
- FIG. 3 is a sectional view taken along line III-III of the heating device 2 of FIG.
- the heating device 2 of this embodiment has 18 heating tubes 12. These heating tubes 12 are arranged on a concentric circle with the rotation axis of the rotating container 10 as a center at an interval.
- the inner diameter (radius) of the rotating container 10 is R and the axial length L is L
- the inner area of the rotating container 10 is 2 ⁇ rL.
- the outer diameter (radius) of the heating tube 12 is r and the axial length is 1, the total area of the outer peripheral surfaces of the 18 heating tubes 12 is 36 ⁇ rl.
- the outer diameter r of the heating tube 12 is sufficiently smaller than the inner diameter R of the rotating vessel 10, and the axial length l of the heating tube 12 and the axial length L of the rotating vessel 10 are usually approximate. Therefore, the heat transfer area of the 18 heating tubes 12 is larger than the inner area of the rotating container 10. In the heating device 2, the 18 heating tubes 12 have a heat transfer surface wider than the inner area of the rotating container 10, so that efficient drying is possible.
- steam is introduced from the steam inlet 44 toward the heating pipe 12. This steam increases the temperature in the rotating container 10. A part of the steam is cooled by heat exchange. The cooled steam becomes water and is discharged from the drain 46. The temperature inside the rotary container 10 is controlled by continuously introducing steam from the steam inlet 44 so as to supplement the steam discharged as water.
- Gas is introduced into the rotating container 10.
- the gas fills the rotating container 10.
- Excess gas is discharged from the rotating container 10.
- the second gear 16 is rotated by driving means (not shown) (for example, a motor).
- the first gear 14 is rotated by the rotation of the second gear 16, and the rotating container 10 is further rotated.
- a number of heating tubes 12 rotate with the rotating container 10. Since the rotary container 10 is cut off by the packing 18, the charging unit 6 does not rotate even if the rotary container 10 rotates. Similarly, even if the rotating container 10 rotates, the take-out unit 8 does not rotate.
- the particulate hydrated gel is charged into the hopper 22.
- This particulate hydrogel proceeds through the inner space 28 in the direction indicated by the arrow A in FIG.
- the particulate hydrous gel is introduced into the rotating container 10.
- the particulate hydrous gel is agitated by the rotation of the rotating container 10 and the action of the plurality of heating tubes 12 rotating together with the rotating container 10. Further, the particulate hydrogel is heated by heat exchange with steam passing through the heating tube 12.
- the heating device 2 is inclined in the axial direction from one end to the other end.
- a downward inclination is provided from the input unit 6 toward the take-out unit 8. Due to this inclination and the rotation of the rotating container 10, the particulate hydrous gel gradually advances in the rotating container 10 in the right direction of FIG. 2, that is, from the input unit 6 to the extraction unit 8.
- the particulate hydrous gel is further heated by heat exchange with steam passing through a number of heating tubes 12 while proceeding toward the take-out portion 8.
- the particulate hydrous gel is adjusted to a predetermined moisture content, and a granular dried product is obtained.
- a plurality of particulate hydrous gels or granular dried products aggregate to obtain a granulated product.
- a barrier 11 exists in the traveling direction of the particulate hydrous gel that travels through the rotating container 10 toward the take-out portion 8.
- the barrier 11 inhibits the progress of the particulate water-containing gel having a high water content.
- the particulate hydrogel whose moisture content has been reduced by heating proceeds beyond the barrier 11.
- mixing of particulate water-containing gels having different water contents in the rotating container 10 is avoided, and the piston flow property is improved. Thereby, a high quality granular dried product adjusted to a predetermined moisture content is obtained.
- the granular dried product is taken out from the take-out port 40.
- the heating tubes 12 are not in contact with the inner peripheral surface of the rotating container 10 in the axial direction. As shown in FIG. 3, the heating tube 12 is not in contact with the inner peripheral surface of the rotating container 10, and when viewed from the center (rotating shaft) of the rotating container 10, the vicinity of the inner peripheral surface (preferably rotating) 50 to 99%, preferably 60 to 95% of the radius of the container 10).
- the plurality of heating tubes 12 and the particulate hydrous gel are in efficient contact.
- the particulate hydrous gel is stirred not only by the rotation of the rotating vessel 10 but also by the rotation of the heating tube 12 synchronized with the rotation of the rotating vessel 10, and simultaneously heated by indirect heat transfer from the heating tube 12, thereby The drying of the gel proceeds.
- the rotating container 10 may have other heating mechanisms, but also radiant heat from the heating tube 12, heat transfer from the particulate hydrogel to be dried, or this gas when introducing a heated gas, The inner surface of the rotating container 10 is heated.
- a heating medium (preferably 150 ° C. or higher) having the above-described temperature is preferably introduced into the heating tube 12.
- the surface (cylindrical container is a cylindrical part and the inner surface in the axial direction of the rotating container 10) and the vicinity thereof are also heated to a predetermined temperature by the radiant heat from the heating tube 12 and optionally heated gas.
- the temperature of the inner surface of the rotating container 10 is 150 ° C. or higher, more preferably 160 ° C. or higher, still more preferably 170 ° C. or higher, and particularly preferably 180 ° C. or higher.
- the upper limit of the temperature of the inner surface of the rotating container 10 is usually the temperature of the heating tube 12.
- the temperature of the inner surface of the rotating container 10 is measured by, for example, one or more contact thermometers installed in the rotary heating device 2 for measuring material temperature.
- the contact thermometer is installed at a position that is in the vicinity of the inner surface of the rotating vessel 10 or the heating tube 12 and that is not in direct contact with the heating tube 12 and the rotating vessel 10.
- the inner surface of the rotating container 10 is heated to the above-described temperature mainly by radiant heat from the heating tube 12.
- the temperature of this gas is preferably 100 ° C. or higher, more preferably 120 ° C. or higher.
- the temperature of the inner surface of the rotating container 10 is preferably higher than + 10 ° C., more preferably + 20 ° C. or higher, particularly preferably + 30 ° C. or higher.
- the temperature of the inner surface of the rotating container 10 is heated to the above temperature range (preferably 150 ° C. or higher) before the particulate hydrous gel is put into the rotating container 10.
- the inner surface of the rotating container 10 is heated to the predetermined temperature or higher before the particulate hydrogel is charged and at the start of drying. More preferably, it is preferable that the inner surfaces of the heating tube 12 and the rotating container 10 are heated to the above-described temperature or more.
- the dried water absorbent resin powder and other materials can be continuously dried without heating the inner surface of the rotating container 10 before the material to be dried is charged. For example, when additionally drying the water absorbent resin after drying, it is not necessary to pay particular attention to the temperature of the inner surface of the rotating container 10 before charging the material.
- the additive 48 is preferably added to the particulate hydrous gel from the nozzle 36 of the pipe 26.
- the added additive 48 and the particulate hydrogel are stirred by the rotation of the rotating container 10.
- the heating device 2 is configured such that the additive 48 is added at least once to the particulate hydrogel during heating and / or stirring.
- the heating device 2 may be configured such that the additive 48 is added at least once to the additive-containing particulate hydrogel during heating and / or stirring.
- the additive 48 may be configured to be continuously added from the nozzle 36 of the pipe 26, or may be configured to be intermittently added a plurality of times.
- the heating device 2 may include a plurality of pipes 26 having different lengths.
- the additive 48 can be added to the particulate hydrous gel at a plurality of locations at different positions in the axial direction of the rotating container 10 by the plurality of pipes 26.
- the addition means for adding the additive 48 to the particulate hydrous gel is not particularly limited.
- known addition means such as one or more orifices or nozzles may be used.
- a cylindrical nozzle such as a needle; an orifice plate provided with a large number of holes in the plate; a one-fluid spray such as a spiral spray valve, a fine spray nozzle, a collision-type spray valve; a two-fluid spray; a multi-fluid of three or more fluids Spray; spraying devices such as a centrifugal atomizer such as a rotating wheel.
- the addition means may have a cooling mechanism. In the case where the thermal stability of the additive 48 is poor, since the alteration of the additive is suppressed by the cooling mechanism, stable addition is achieved.
- the number, arrangement and shape of the barriers 11 included in the heating device 2 and the number of openings are not particularly limited, and the particulate hydrous gel provided to the heating device 2 and Depending on the physical properties of the granular dry product obtained, donut shape, half donut shape, 1/3 to 1/10 donut shape, semicircular shape (1/2 circle shape), 1/3 to 1/10 yen shape, crescent moon The shape is selected as appropriate.
- a donut-shaped barrier 11 can be used, but is not particularly limited.
- the number of the opening part of the barrier 11 should just be 1 or more, and when it has several opening part, the size may be the same and may differ. A preferable aperture ratio will be described later.
- the barrier 11 may be installed perpendicular to the cross section of the rotating container 10 or may be inclined.
- the barrier 11a in FIG. 5 and the barrier 11b in FIG. 6 are donut-shaped partition plates that are installed so as to be orthogonal to the axial direction of the cylindrical rotating container 10 and have an open substantially central portion.
- the opening ratio of the barrier 11 (the opening ratio of the barrier 11 with respect to the cross-sectional area perpendicular to the axial direction of the rotating container 10) is appropriately determined, but is usually 1 to 90%, preferably 2 to 50%, more preferably 5 to 45%, still more preferably 10 to 40%.
- the aperture ratio is larger than the above range, the effect of the barrier 11 is low, and when the aperture ratio is small, there is a possibility of causing a discharge failure.
- the aperture ratio of the barrier 11a in FIG. 5 is 20%
- the aperture ratio of the barrier 11b in FIG. 6 is 50%.
- the number (n) of the barriers 11 is at least 1, preferably 2 or more, particularly preferably 3 or more.
- the upper limit of the number of barriers 11 depends on the size of the heating device 2, but is preferably 20 or less, more preferably 10 or less.
- the barrier 11 installed in the rotating container 10 may be a fixed type or a variable type (movable type), but the installation position of the barrier 11 is appropriately determined according to a target drying curve (temperature or solid content ratio with respect to drying time). Is done. For example, since the effect of the barrier 11 placed in a place where the solid content of the particulate hydrogel is low is small, the solid content of the particulate hydrogel is usually 50% by mass or more, preferably 60% by mass or more, more preferably One or more barriers 11 are installed in a place where the amount is 70% by mass or more, more preferably 80 to 95% by mass.
- the inside of the rotating container 10 is divided into a gel region having a high water content and a dry region having a low water content along the traveling direction of the particulate water-containing gel. Is further divided into intermediate regions by increasing the number of barriers 11. Further, in the rotating container 10, depending on the temperature of the object to be dried, it is divided into a preheat section, a constant rate drying section (also referred to as a constant rate drying section), and a reduced rate drying section. By increasing the number, it can be further divided into intermediate sections. As a result, more efficient drying can be carried out, and furthermore, surface crosslinking at the same time as the drying carried out as necessary can be carried out efficiently.
- the arrangement of the heating tube 12 provided in the heating device 2 is not particularly limited, and is appropriately set according to the production amount of the water-absorbing agent.
- the plurality of heating tubes 12 may be arranged on the circumference centered on the rotation axis of the rotating container 10 at intervals, and as shown in FIG. 4, the rotating container 12 They may be arranged at intervals on two or more (multiple) concentric circles centered on 10 rotation axes. Further, the plurality of heating tubes 12 may be arranged radially spaced from the rotation axis of the rotating container 10 toward the radially outer side.
- the number is selected according to the purpose, but is preferably 2 to 10 times, more preferably 2 to 8 times, more preferably 2 to 5 times. Installed. By arranging in multiple, it has a wider heat transfer surface, and more efficient drying of the particulate hydrous gel becomes possible.
- the number of heating tubes 12 included in the heating device 2 and the radius (diameter) thereof are not particularly limited.
- the number of heating tubes 12 is appropriately determined depending on the size and processing amount of the heating device 2, but is preferably 5 or more, more preferably 10 or more, and particularly preferably 15 pieces. More preferably, the number is 20 or more, and particularly preferably 100 or more.
- the upper limit of the number of the heating tubes 12 is not particularly limited, for example, 2000 or less, and further 1000 or less are appropriately selected.
- a large number of heating tubes 12 are preferable because the area of the heating surface that contacts the material by indirect heating increases.
- heat transfer area relative to the internal volume means a ratio (unit: m ⁇ 1 ) defined by (heat transfer area / effective volume). The larger the ratio, the better the heat transfer efficiency. As a result, the temperature increase rate of the object to be dried is increased, so that the drying time is shortened and the productivity is improved.
- the ratio is appropriately set depending on the specifications and form of the heating apparatus to be used, the shape of the object to be dried, etc., but is preferably 10 m ⁇ 1 or more, more preferably 12 m ⁇ 1 , and even more preferably 15 m ⁇ 1 or more. It is.
- the effective volume is the volume of the drying chamber (that is, the rotating container 10) in which the material to be dried is stored, and the heat transfer area is the heating that can add heat to the contents stored in the drying chamber. It means the area of the surface.
- the sum of the area of the outer surface of the plurality of heating tubes 12 and the area of the inner surface of the rotating container 10 is the heat transfer surface tip.
- the plurality of heating tubes 12 are preferably water vapor (preferably 150 ° C. or higher, preferably in the above pressure range).
- the vapor pressure of the water vapor to be vented may be the same or different in all the heating pipes 12, but is usually the same. It is defined by an average value (for example, an average value of vapor pressure).
- the heating device 2 further includes heating means or heat retaining means, more preferably heating means, on the outer peripheral surface of the rotating container 10.
- this heat retaining means for example, a part or the whole of the outer peripheral surface of the rotating container 10 (preferably 50% or more, more preferably 80% or more, particularly preferably the entire outer surface of the rotating container 10) is made of a heat insulating material. The method of coating is mentioned.
- examples of the heating means include an electric tres, a steam tres, a jacket heated with a heat medium, and the like.
- the heating means include an electric tres, a steam tres, a jacket heated with a heat medium, and the like.
- the drying method according to the present invention not only the temperature of the heating tube 12 but also the inside of the rotating container 10 is not limited when the particulate hydrous gel is dried (particularly at the start-up), from the viewpoint of reducing adhesion of the hydrous gel. Surface temperature control is also important.
- the presence of heating means or heat retaining means provided on the outer surface of the rotating container 10 is effective for controlling the temperature of the inner surface of the rotating container 10.
- the throughput per unit time (in terms of solid content of the particulate hydrogel crosslinked polymer) ( kg / hr) is 7 kg / hr or more, preferably 50 Kg / hr or more, more preferably 100 Kg / hr or more, and particularly preferably 500 Kg / hr.
- the continuous drying time is preferably 12 hours or longer, more preferably 24 hours or longer, particularly 240 hours or longer and 1200 hours or longer.
- additive 48 added to the particulate hydrous gel examples include a surface cross-linking agent described later in addition to the gel fluidizing agent and polymer lubricant described above.
- Other additives 48 may be added to the particulate hydrous gel as long as the effects of the present invention are not impaired.
- the surface cross-linking step after drying can be reduced by adding the surface cross-linking agent before drying and during the drying process, and also exhibits a general surface cross-linking effect after drying (for example, improvement in water absorption under pressure). Can also be omitted.
- drying apparatus The drying apparatus and the drying method according to the present invention are as described above. More preferably, the drying apparatus has the following structure in order to solve the problems of the present invention.
- a drying device is a drying device for a particulate hydrogel crosslinked polymer obtained from a monomer that is a raw material for a water-absorbent resin, and contains the particulate hydrogel crosslinked polymer therein.
- the number of heating tubes is 5 or more, and the heating tubes are not in contact with the inner peripheral surface of the rotating container in the axial direction, and heating means or A heat retaining means is provided.
- a plurality of heating tubes are arranged concentrically or radially away from the rotation axis of the rotating container toward the radially outer side. The preferred number and arrangement of heating tubes are as described above.
- the rotating container is inclined from one end to the other end.
- at least one barrier is provided inside the rotating container.
- a preferable barrier opening ratio is 1 to 90%.
- the preferred barrier form is as described above.
- the ratio (heat transfer area / effective volume) is 10 m ⁇ 1 or more. More preferable heat transfer area / effective volume is as described above.
- the drying apparatus has at least one addition means for adding an additive to the particulate hydrogel crosslinked polymer accommodated in the rotating container.
- a preferred addition means is a spray device.
- the number of preferable additives and addition means is as described above.
- the plurality of heating tubes are arranged radially spaced from the rotation axis of the rotating container toward the radially outer side.
- the plurality of heating tubes are arranged at intervals on two or more concentric circles centering on the rotation axis of the rotating container.
- drying method and drying start method according to the present invention The drying apparatus and the drying method according to the present invention are as described above, but a more preferable drying method for solving the problems of the present invention is as follows.
- Such a drying method is not limited to continuous drying, but can be applied to batch drying (particularly repeated batch drying), and is not limited to gel-pulverized particulate hydrogel crosslinked polymer, and can be obtained without performing gel grinding.
- Water-absorbent resin particulate hydrogel crosslinked polymer typically water-absorbent resin particulate hydrogel crosslinked polymer obtained by reverse phase suspension polymerization, gas phase droplet polymerization or spray polymerization It can also be applied to.
- the drying method according to the present invention is preferably applicable to the following drying method for particulate water-containing gel-like crosslinked polymer, in particular, a drying start method (drying start-up method) in order to determine the subject of the present invention. .
- the drying method according to the present invention is preferably a particle obtained from a monomer that is a raw material of a water-absorbent resin, using the drying device described above.
- This is a drying method for obtaining a granular dried product by drying a water-containing gel-like crosslinked polymer.
- gas is introduced as a heating medium into the rotating container, the gel temperature of the particulate hydrogel crosslinked polymer charged into the drying apparatus is 50 ° C. or higher, and the temperature of the inner surface of the rotating container is 150 ° C.
- the drying method means the entire drying (from the start to the end of continuous or batch-type drying, and from the start to the end in batch-type repeated drying), and the drying start method is the initial stage, That is, it means dry Start-up.
- the drying start method is the initial stage, That is, it means dry Start-up.
- continuous or batch drying not only the temperature of the heating tube, but also the temperature of the inner peripheral surface of the rotating container that is not in contact with the heating tube is important for continuous drying or repeated batch drying. It was issued.
- the drying method according to the present invention not only the heating tube for drying the particulate hydrogel by conduction heat transfer, but also the inner surface of the rotating container is heated to a predetermined temperature in advance, thereby rotating the particulate hydrogel. Adhesion to the inner surface of the container and the heating tube is reduced. For this reason, it has been found that there is no need to periodically remove deposits in a short period of time, and continuous operation over a long period of time (in the case of continuous drying or batch drying, repeated drying after taking out the dried product) is possible. .
- Patent Documents 22 to 28 and 31 disclose stirring and drying of a particulate hydrous gel, but this is not sufficient in terms of continuous operation, and not only the temperature of the heating medium (temperature of the heating tube) but also the temperature of the inner surface of the dryer. It was not disclosed to be important for continuous operation.
- the temperature of the inner surface of the heating tube and the rotating container is heated to 150 ° C. or higher before the particulate hydrogel crosslinked polymer is charged.
- this temperature is heated to 180 ° C. or higher. More preferably, it is heated to 200 ° C. or higher.
- gas is introduced into the rotating container as a heat medium, and the temperature of this gas is lower than the temperature of the inner surface of the rotating container.
- the drying method according to the present invention may be a batch method or a continuous method.
- drying is repeatedly performed under substantially the same conditions, and the number of repetitions is preferably 5 times or more, more preferably 10 times or more, and still more preferably 100 times or more. More preferably, it is a continuous type.
- the repetition by the batch system means that the following particulate hydrous gel is discharged within a certain period of time (for example, within 1 hour, preferably within 10 minutes) including the charging time of the particulate hydrous gel after discharging the granular dry product. This is an operation of putting in a drying apparatus and drying.
- it can be used as a continuous dryer by continuously charging and discharging the dried material (particulate hydrated gel) even in the same drying apparatus, and the dried material (particulate hydrated gel) is collectively charged and discharged. Thereafter, batch drying can be performed by discharging the liquid after a predetermined time.
- the particulate hydrogel crosslinked polymer charged into the drying apparatus contains a gel fluidizing agent.
- the particulate hydrogel crosslinked polymer has a mass average particle diameter of 800 ⁇ m or less. More preferably, it is the range mentioned above.
- the temperature of the gas introduced into the rotating container is 100 ° C. or higher. More preferably, it is the range mentioned above.
- the drying apparatus has an adding means inside the rotating container, and the additive is added to the particulate hydrogel crosslinked polymer using this adding means.
- this additive is a surface cross-linking agent.
- the surface cross-linking agent is added to a particulate hydrogel crosslinked polymer having a water content of 15 to 50% by mass.
- the difference between the pressure inside the rotating container and atmospheric pressure (decompression degree) is more than 0 kPa and not more than 5 kPa.
- the fluid number of the rotating container is 0.001 to 1.
- the temperature of the particulate hydrogel used in the drying step (hereinafter referred to as gel temperature) is 50 ° C. or higher.
- This gel temperature is measured with the above-mentioned contact thermometer installed in the heating device.
- the contact thermometer include a thermocouple, a platinum thermometer or a bimetal thermometer, particularly a thermocouple (for example, a K-line sheath thermocouple).
- the gel temperature is measured at the center of the material layer (particulate hydrogel or granular dried product) (for example, a position of about 5 cm when the thickness of the material is 10 cm).
- the gel temperature is preferably 60 ° C. or higher, more preferably 70 ° C. or higher, further preferably 80 ° C. or higher, and particularly preferably 90 ° C. or higher. Further, when the temperature is too high, coloring of the material to be dried and performance deterioration may be observed. Therefore, the gel temperature is preferably 130 ° C. or lower, more preferably 110 ° C. or lower, and further preferably 105 ° C. or lower.
- temperatures include the temperature of the hydrogel after polymerization (temperature when discharged from the polymerization apparatus), heating, heat retention or reaction heat in each step after polymerization (for example, neutralization heat after polymerization or gel crushing) May be adjusted by the pulverization energy and heating at the time, or may be adjusted by providing a separate heating step before the drying step.
- the gel fluidizing agent is added in the above-described gel grinding step, the gel fluidizing agent is adjusted to have a predetermined gel temperature before being subjected to the drying step.
- the moisture content of the particulate hydrogel subjected to the drying step (hereinafter referred to as gel moisture content) is determined by the measurement method described in the following examples. From the viewpoint of fluidity of the particulate hydrous gel, the gel moisture content is preferably 25% by mass or more, more preferably 30% by mass or more, further preferably 35% by mass or more, further preferably 40% by mass or more, and 43% by mass. % Or more is particularly preferable. Excessive high-concentration polymerization may decrease the properties of the water-absorbent resin, and from the viewpoint of drying efficiency and absorption performance, the gel moisture content is preferably 75% by mass or less, more preferably 60% by mass or less, and 55% by mass. The following are particularly preferred:
- the mass average particle size of the particulate hydrogel before drying (hereinafter referred to as the gel particle size) may be almost the same as the particle size of the water-absorbing agent as the final product after the drying step or heat treatment step.
- the gel particle diameter d1 when used in sanitary materials such as paper diapers is preferably 800 ⁇ m or less, more preferably 500 ⁇ m or less, still more preferably 50 ⁇ m to 500 ⁇ m, still more preferably 100 ⁇ m to 400 ⁇ m, in terms of solid content. Particularly preferred is 100 to 300 ⁇ m, and most preferred is 100 to 200 ⁇ m.
- the drying step in the present invention is applied to the drying of a particulate hydrogel having a small particle diameter, particularly dry granulation.
- the particle size of the particulate hydrous gel is preferably in the range of 150 ⁇ m or less, depending on the water content, but is preferably 10% by mass or more, more preferably 25% by mass or more, and still more preferably 40% in terms of solid content. It is at least mass%.
- the particle size of the particulate hydrous gel is preferably in the range of less than 1100 ⁇ m, preferably 80% by mass or more, more preferably 85% by mass or more, still more preferably 90% by mass or more, particularly preferably in terms of solid content. It is 95 mass% or more, and an upper limit is 100 mass%. Furthermore, it is preferable that what exists in the range of less than 850 micrometers is the said range in conversion of solid content.
- a particulate hydrogel with a gel particle size considerably smaller than that selected in the prior art is charged, more preferably granulated with drying, and more preferably Granulate to a particle size in the range described below.
- conventional aeration band type dryers there is a problem of dropping or clogging dry matter from punching metal or metal mesh as a drying belt by introducing particulate hydrous gel, these problems are small particle size This was more noticeable when the particulate hydrogel was dried.
- the drying process of the present invention the occurrence of such a problem is avoided even when a particulate hydrogel having a small particle size is applied.
- the CRC (centrifuge retention capacity) of the particulate water-containing gel before drying is preferably 5 g / g to 80 g / g, more preferably 10 g / g to 50 g / g, still more preferably 15 g / g in terms of dry weight. It is ⁇ 45 g / g, particularly preferably 20 g / g to 40 g / g.
- the water-soluble polymer amount (Ext) of the particulate hydrous gel before drying is preferably 20% by mass or less, more preferably 15% by mass or less, still more preferably 10% by mass or less, especially in terms of dry weight. Preferably it is 5 mass% or less.
- the water absorption magnification and the soluble content of the particulate hydrogel before drying are low.
- a particulate water-containing gel having a relatively low water absorption ratio and a low solubility is charged into a heating device, and at a sufficiently high temperature (preferably 150 ° C. or higher, more preferably 180 ° C. or higher) for a predetermined time (preferably The embodiment which improves the soluble part and water absorption rate of the granular dried material obtained by heating for 10 minutes or more at predetermined temperature is preferable.
- a sufficiently high temperature preferably 150 ° C. or higher, more preferably 180 ° C. or higher
- a predetermined time preferably The embodiment which improves the soluble part and water absorption rate of the granular dried material obtained by heating for 10 minutes or more at predetermined temperature is preferable.
- an internal cross-linking agent having the polyalkylene unit is exemplified.
- the polymerization rate of the particulate hydrogel before drying used in the drying step is preferably 90% by mass or more, more preferably 95% by mass or more, and still more preferably 98 to 99% in terms of dry weight. .99% by mass, ideally 100%. If the polymerization rate is low, problems of aggregation and adhesion occur during drying. Conventionally, a technique of taking out from a polymerization apparatus and drying at a polymerization rate of 90% or less for improving productivity has also been known (for example, WO 2006/103227), but the present inventors have used a rotary heating apparatus for particulate hydrous gel. It was found that the polymerization rate is important for drying by.
- the water-soluble polymer amount (Ext) of the granular dried product obtained after drying is preferably larger than the water-soluble polymer amount of the particulate hydrous gel before drying.
- the amount of water-soluble polymer in the granular dried product obtained after drying is preferably + 0.5% by mass or more, more preferably +1 to 20% by mass, and more preferably +2 to 10% in terms of dry weight.
- the crosslink density of the hydrogel (particularly the type and amount of the internal crosslinker), the heat drying conditions, etc. are adjusted so as to increase in the mass% range.
- the amount of water-soluble polymer in the resulting water-absorbent resin powder is preferably 50% by mass or less, more preferably 25% by mass or less, still more preferably 20% by mass or less, and particularly preferably 15% by mass or less.
- This step is a step of adding a cross-linking reaction by adding a surface cross-linking agent that reacts with a functional group (especially carboxyl group) of the water-absorbent resin to the polymerized hydrogel or dried product thereof. .
- a crosslinking reaction By this crosslinking reaction, crosslinking is mainly performed from the surface of the water absorbent resin particles.
- Surface crosslinking is also referred to as post-crosslinking or secondary crosslinking.
- a surface cross-linking agent is added to and reacted with the particulate hydrous gel and / or the granular dried product (hereinafter sometimes referred to as a water absorbent resin).
- This step has a surface cross-linking agent addition step and a heat treatment step, and may optionally have a cooling step after the heat treatment step.
- This step is a step of adding a surface cross-linking agent to the particulate hydrous gel and / or granular dried product.
- the surface cross-linking agent is added to the particulate hydrogel before or during drying, or to the granular dried product after drying, and further to the granular dried product after sizing.
- the “water content” of the water-absorbent resin is a value determined by the measurement method described in the following examples.
- the “water content” of the water-absorbing agent is the same as the “water content” of the water-absorbent resin, except that the measurement conditions are different.
- the solid content rate of the granular dried product at the time of addition of the surface crosslinking agent is , In the range of the solid content of the dried particulate matter after drying, preferably 80% by mass or more, more preferably 85% by mass to 99.8% by mass, still more preferably 90% by mass to 99.7% by mass, Even more preferably, it is 92 mass% to 99.5 mass%, particularly preferably 96 mass% to 99.5 mass%, and most preferably 98 to 99.5 mass%.
- the moisture content of the particulate hydrogel when the surface crosslinking agent is added is preferably 10 to 50. It is in the range of mass%, more preferably 15 to 45 mass%, still more preferably 20 to 40 mass%.
- the surface cross-linking agent in such a range, excessive aggregation and adhesion during drying are reduced.
- the effect of improving the absorption capacity under pressure obtained by carrying out the surface cross-linking step after the drying step is also preferable because the surface cross-linking is performed simultaneously with the drying.
- the surface cross-linking step and the sizing step after the drying step which were necessary in the conventional manufacturing method (for example, FIG. 8), can be omitted, greatly simplifying the manufacturing step.
- the temperature of the water-absorbing resin used in the surface cross-linking step after the drying step is preferably 40 to 120 ° C., more preferably 60 to 100 ° C. .
- the temperature of the water-absorbing resin during drying is preferably 70 to 150 ° C., more preferably 80 to 130 ° C. It is a range. This temperature is measured with a contact thermometer in the same manner as the gel temperature described above in the drying step. If necessary, this temperature may be adjusted by heating means or heat retaining means.
- the effect of the present invention becomes more prominent by subjecting the water-absorbing resin having a water content and temperature in the above ranges to a heat treatment step described later.
- a surface cross-linking agent capable of reacting with a plurality of functional groups (preferably a plurality of carboxyl groups) of the water-absorbent resin, preferably a covalent bond or an ionic bond, and further a surface cross-linker capable of covalent bonding is used.
- Alkylene carbonate compounds such as epichlorohydrin, epibromohydrin, ⁇ -methylepichlorohydrin and their polyvalent amine adducts; oxetane compounds; ⁇ -glycidoxypropyltrimethoxysilane, ⁇ -amino Silane coupling agents such as propyltriethoxysilane; hydroxides such as zinc, calcium, magnesium, aluminum, iron and zirconium; polyvalent metal compounds such as chloride, sulfate, nitrate or carbonate; . Of these, two or more may be used in combination.
- surface cross-linking agents when surface cross-linking is performed after drying or during drying, one or more selected from polyvalent metal ions, epoxy compounds, oxazoline compounds, and alkylene carbonate compounds are preferable, and surface cross-linking is performed during drying.
- an epoxy compound is preferable.
- the addition amount of the surface cross-linking agent is preferably 5% by mass or less, more preferably 3% by mass or less, and further preferably 2% by mass or less with respect to the solid content of the water absorbent resin during or after drying.
- the lower limit is preferably 0.001% by mass.
- the addition form of the surface cross-linking agent may be used as it is, but it is preferably added as a solution dissolved in water or an organic solvent because of the ease of addition.
- the concentration of the solution is preferably 1% by mass or more, more preferably 2% by mass or more.
- the total amount of the solvent selected from water and organic solvent is preferably 0 to 10% by mass, more preferably 0.1 to 8% by mass, and still more preferably 0.5 to 10% by mass with respect to the solid content of the water absorbent resin. 5% by mass.
- water is preferably the main component.
- the concentration of the aqueous solution can be adjusted according to the water content of the water-absorbent resin at the time of contact with the surface crosslinking agent.
- an aqueous solution containing water necessary for setting the water content of the water-absorbent resin to a desired value and a surface cross-linking agent required depending on the solid content of the water-absorbent resin may be prepared.
- a hydrophilic solvent such as alcohol
- the number of times of adding the surface cross-linking agent may be one or more times during or after drying. In the case of adding two or more times, it is preferable that the water-absorbing resin that comes into contact with at least the first one-time surface cross-linking agent has the above-described moisture content and temperature. More preferably.
- Heat treatment step This step is a step of heat-treating a water-absorbing resin containing a surface crosslinking agent to obtain a surface-dried granular dried product, preferably a surface-crosslinked granular dried granulated product. It is.
- the water absorbent is obtained by heating the water absorbent resin containing the surface crosslinking agent to 100 ° C. or higher.
- the preferable maximum temperature varies depending on the type of the surface cross-linking agent, but is 100 to 250 ° C, more preferably 120 to 230 ° C, and further preferably 150 to 210 ° C.
- time What is necessary is just to set suitably the time of the heat processing process from the moisture content of a water absorbing resin, the kind of surface crosslinking agent, the thermal efficiency of a heating apparatus, etc.
- heating may be performed until the water content becomes 10% by mass or less, and the time is in the range of 10 to 120 minutes, preferably 30 to 90 minutes.
- the heating apparatus used in the surface cross-linking step is not particularly limited. However, from the viewpoint that uneven heating is less likely to occur, it has a stirring mechanism in the form of conduction heat transfer by solid-solid contact (hereinafter referred to as “heating unevenness”). , Sometimes referred to as a stirring type indirect heating type). In addition, when surface crosslinking is carried out simultaneously with drying, the said heating apparatus is used also as a heating apparatus of a surface crosslinking process.
- the stirring method and form of the stirring-type indirect heating type apparatus are not particularly limited, but in the production method according to the present invention, when surface cross-linking is performed after drying, a rotating shaft equipped with stirring blades such as an arm, a blade, and a paddle is used. Examples include a mechanical stirring type heating device that stirs the contents; a high-speed shearing type heating device and the like. Moreover, these heating apparatuses may be a continuous type or a batch type. For example, a uniaxial or biaxial disc type heating device, a uniaxial or biaxial paddle type heating device, and the like can be given. Moreover, in the manufacturing method which concerns on this invention, when performing surface bridge
- the surface cross-linking step is performed by adding the surface cross-linking agent as the additive 48 to the particulate hydrous gel and heating in the continuous rotary dryer with heating tube described above. It is also possible to implement. In this case, a granular dried product adjusted to a predetermined moisture content (solid content) and surface-crosslinked is preferable because it can be obtained in one step.
- the particulate hydrous gel to which the surface cross-linking agent is added from the nozzle 36 of the pipe 26 is rotated by the rotation of the rotating container 10 and the action of the multiple heating tubes 12 rotating together with the rotating container 10.
- the particulate hydrogel containing the surface cross-linking agent is further heated by heat exchange with steam passing through a number of heating tubes 12 while proceeding toward the take-out port 8.
- the surface crosslinking reaction proceeds by heating and stirring by the heating device 2, and the particulate hydrogel is adjusted to a predetermined moisture content, and preferably granulated.
- a barrier 11 exists in the traveling direction of the particulate hydrous gel traveling in the rotating container 10.
- the barrier 11 inhibits the progress of the particulate hydrogel having a high moisture content and a low degree of surface crosslinking.
- the particulate water-containing gel having a reduced water content due to heating and a high degree of surface crosslinking proceeds beyond the barrier 11.
- This barrier 11 avoids the mixing of particulate water-containing gels having different moisture contents and surface cross-linked states in the rotating container 10. Thereby, surface cross-linking is achieved, and a granular dried product adjusted to a predetermined moisture content or a granulated product thereof is obtained.
- the granular dried product or the granulated product is taken out from the take-out port 40.
- the granular dried product or the surface crosslinked granular dried product is forcibly cooled after the above-described drying step or surface cross-linking step and before the granulating step described later. And it has the cooling process for adjusting to desired temperature.
- the rotary vessel 10 is appropriately subjected to the surface cross-linking treatment and is a granular dried product or a surface-dried granular dried product. After the solid content or water content of the is adjusted to a desired range, a cooling step is performed.
- the granular dried product or the surface crosslinked product is provided before being subjected to the granulation step.
- the granular dried product is forcibly cooled to preferably 50 to 130 ° C, more preferably 60 to 100 ° C, and still more preferably 65 to 90 ° C.
- the workability and classification accuracy at the time of pulverization are improved in the sizing step, and the properties of the resulting water-absorbent resin powder are improved.
- the method for cooling the granular dried product or the surface-dried granular dried product is not particularly limited.
- a continuous cooler having an aeration heat transfer type or conduction heat transfer type cooling means is used.
- the granular dried product or the surface-dried granular dried product may be cooled in a stationary state or may be cooled in a stirring state.
- a material stirring type cooler is preferable, and a continuous material stirring type cooler is more preferable.
- a continuous cooler selected from a continuous stirring cooler, a continuous rotary cooler and a fluidized bed cooler is particularly preferred.
- a fluidized bed cooler that is direct heat transfer can be used. Cool air cooling in a continuous belt type cooler may be used.
- a stirring device having a rotating shaft can be used for stirring and cooling.
- a mixer having a function of allowing an air current to aerate and cool an object to be cooled is widely used as a cooler.
- the direction of the airflow is not particularly limited, and may be up and down or left and right.
- Specific examples of such a cooler include a mixer (horizontal cylindrical, inclined cylindrical, V-shaped, double-conical, regular cubic, S-shaped, continuous V, whose rotating shaft is horizontal and the container itself rotates.
- a mixer with a horizontal rotation axis and a fixed container may be used by ventilating an airflow.
- a container-fixed type cooler that includes a rotary stirring blade for stirring the water-absorbent resin powder that is an object to be cooled and is ventilated with an air flow is used.
- These coolers may be a continuous type or a batch type, but are preferably a continuous type.
- This step is a step of adjusting the particle size of the granular dried product or the surface-crosslinked granular dried product.
- a water absorbent resin powder in which the particle size or particle size distribution is more positively controlled can be obtained.
- the sizing process includes a crushing step and / or a classification step.
- the pulverization step is a step of adjusting the particle size by pulverizing the granular dried material loosely aggregated through the drying step or the heat treatment step with a pulverizer.
- the classification step is a step of removing coarse particles and fine powder from the granular dried product, the surface-dried granular dried product, or the pulverized product thereof using a classifier.
- the sizing step is to obtain a water-absorbent resin powder having a controlled particle size and particle size distribution.
- the pulverizer those having little damage to the granular dried product or the surface-crosslinked granular dried product are preferable.
- a roll granulator (Matsubo Co., Ltd.), a granulator (Kurimoto Steel Works Co., Ltd.) or a Landel mill (Tokuju workshop)
- the classifier a vibration type or swing type sieve classifier using a sieve screen is used.
- FIG. 12 is a flowchart for explaining the sizing step A of the manufacturing method according to the embodiment of the present invention.
- the granular dried product or the surface-crosslinked granular dried product is crushed with a pulverizer to obtain a first crushed product, and the first crushed product is classified with a classifier.
- a first classification step of separating coarse particles (shown as a sieve top in FIG. 12) contained in the first crushed material, and a step of recharging the coarse particles into the first pulverizing step. have.
- fine powder shown as an under-sieving portion in FIG. 12 is removed from the first crushed material.
- FIG. 13 is a flowchart for explaining the sizing step B of the manufacturing method according to another embodiment of the present invention.
- the granular dried product or the surface-dried granular dried product is classified by a classifier, and coarse particles contained in the granular dried product or the surface-crosslinked granular dried product (in FIG. 13, as the fraction on the sieve).
- a second classifying step for classifying the coarse particles with a pulverizer to obtain a second pulverized product, and the second pulverized product is returned to the second classifying step.
- a step of inputting Preferably, in the second classification step, fines (shown as undersieving in FIG. 13) are removed from the granular dried product or post-crosslinked granular dried product.
- the size of the coarse particles in the sizing step A and the sizing step B is appropriately set according to the particle size and particle size of the water-absorbing agent that is the final product.
- the particle size of the coarse particles (specified by sieving classification) is 2000 ⁇ m or more, more preferably 850 ⁇ m or more.
- the mass average particle diameter d2 of the granular dried product subjected to the sizing process or the surface-crosslinked granular dried product is preferably 200 ⁇ m or more, more preferably 300 ⁇ m or more, and still more preferably. 400 ⁇ m or more and 500 ⁇ m. From the viewpoint of increasing the efficiency of the crushing step, the mass average particle diameter d2 is preferably 2000 ⁇ m or less, more preferably 1500 ⁇ m or less, and 1000 ⁇ m or less.
- the ratio (d2 / d1) between the mass average particle diameter d2 of the granular dried product or the surface crosslinked granular dried product and the mass average particle size d1 of the particulate hydrogel crosslinked polymer before drying is 1.05 to 100, more preferably 1.1 to 10, and still more preferably 1.2 to 5.
- the moisture content is preferably in the range of the moisture content described above with respect to the moisture content of the dried granular product.
- the mass average particle diameter d3 of the water absorbent resin powder obtained through the sizing step is preferably 200 ⁇ m or more, more preferably 200 to 600 ⁇ m, still more preferably 250 to 550 ⁇ m, and particularly preferably 300. ⁇ 500 ⁇ m.
- the ratio (d3 / d2) between the mass average particle diameter d3 of the water-absorbent resin powder and the mass average particle diameter d2 of the granular dried product or the surface-dried granular dried product is 0.05 to 1.1. More preferably 0.1 to 0.9, still more preferably 0.2 to 0.85, and particularly preferably 0.5 to 0.8. By reducing the particle size within these ranges, a water-absorbent resin with a further improved water absorption rate can be obtained.
- the ratio (d3 / d1) between the mass average particle diameter d3 of the water-absorbent resin powder and the mass average particle diameter d1 of the particulate hydrous gel is preferably 0.5 to 100, and this ratio is more than 1. That is, it is preferable that the water-absorbent resin powder is granulated in comparison with the particulate hydrous gel even after being pulverized in the granulating step.
- the ratio (d3 / d1) is more preferably in the range of 1.01 to 50, further preferably 1.1 to 10, and particularly preferably 1.2 to 4.
- the water-absorbing resin powder mainly comprises water-absorbing resin particles having a particle diameter of 150 to 850 ⁇ m as defined by sieve classification.
- the ratio of the water-absorbent resin particles having a particle diameter of 150 to 850 ⁇ m in the water-absorbent resin powder is preferably 90 to 100% by mass, more preferably 95 to 100% by mass, still more preferably 97 to 100% by mass, and particularly preferably. 99 to 100% by mass.
- the ratio of the water-absorbing resin particles having a particle size of less than 150 ⁇ m and more than 850 ⁇ m as defined by sieving is preferably 10% by mass or less, more preferably 5% by mass or less, still more preferably 3% by mass or less, and 1% by mass. Is particularly preferred.
- the manufacturing method according to the present invention further includes a fine powder recycling step.
- the “fine powder recycling process” means a process of supplying the fine powder removed in the classification step as it is or after granulating the fine powder to any process.
- the fine powder or the fine powder granulated product is a step of throwing it into a step before the drying step and reusing it. Examples of the step before the drying step include a monomer solution before polymerization adjusted in the polymerization step, a hydrogel during polymerization, a pulverization step of the hydrogel after polymerization, and a drying step of the granular hydrogel.
- the fine powder may be added as it is, or the fine powder may be swelled or granulated with water and then added.
- water e.g, water-soluble polymer, thermoplastic resin
- a polymerization initiator e.g., a reducing agent, a chelating agent, an anti-coloring agent, and the like
- water when water is added, it is preferably used in an amount of 1 to 1000% by mass with respect to the fine powder, and when other compounds are added, 0.01 to 10% by mass with respect to the fine powder.
- the fine powder removing process and the fine powder collecting process can also be labor-saving.
- the collection and recycling of the removed fine powder may lead to a decrease in the performance of the water-absorbent resin (for example, a reduction in water absorption ratio or regeneration of fine powder during the process). Since the amount of fine powder is reduced, performance degradation associated with fine powder collection and recycling is avoided, and the performance of the resulting water-absorbent resin is improved.
- a preferable fine powder recovery amount is appropriately set depending on the target particle size.
- the amount of fine powder tends to increase as a result, but the amount of fine powder is preferably less than 20% by mass of the total production amount, more preferably 15% by mass or less, Preferably it is 10 mass% or less, Most preferably, it is 6 mass% or less. In the production method according to the present invention, the amount is significantly reduced as compared with the amount of fine powder of 20 to 30% by mass by the conventional production method.
- the production method further includes a wetting step.
- the wetting step is a step of re-wetting by adding water to the granular dried product, the surface-crosslinked granular dried product or the water-absorbent resin powder.
- the amount of water added to the granular dried product, surface-crosslinked granular dried product or water-absorbent resin powder is appropriately adjusted depending on the moisture content of the granular dried product, surface-crosslinked granular dried product or water-absorbent resin powder.
- the By adding this water the solid content of the water-absorbing agent obtained can be adjusted to a desired range.
- water is added to the granular dried product or the surface-crosslinked granular dried product after the drying step and before the sizing step, so that rewetting and forced cooling of the granular dried product or the surface-crosslinked granular dried product are performed simultaneously. carry out. Thereby, the production efficiency of the water absorbent resin powder is further improved.
- the amount of water added or increased in the wetting step is preferably 0.1 to 20% by mass in terms of solid content. More preferably, it is 0.5 to 10% by mass, and further preferably 2 to 8% by mass.
- other additives may be added together with water.
- the granular dried product, the surface-crosslinked granular dried product or the water-absorbent resin powder may be heated at the time of rewet. Part of the added water may be removed by drying the product or the water-absorbent resin powder.
- the heating temperature during or after rewetting is preferably in the range of 50 to 150 ° C., more preferably 60 to 120 ° C.
- the inorganic fine particles include mineral products such as talc, kaolin, fullerite, hydrotalcite, bentonite, activated clay, barite, natural asphalt, strontium ore, ilmenite, pearlite; aluminum sulfate 14-18
- Aluminum salts such as hydrate (or its anhydride), potassium aluminum sulfate 12 hydrate, sodium aluminum sulfate 12 hydrate, ammonium aluminum sulfate 12 hydrate, aluminum chloride, polyaluminum chloride, aluminum oxide; and other various compounds such as calcium phosphate Valent metal salt, polyvalent metal oxide and polyvalent metal hydroxide; hydrophilic amorphous silicas; silicon oxide / aluminum oxide / magnesium oxide complex, silicon oxide / aluminum oxide complex, silicon oxide / magnesium oxide complex Etc. oxidation Complexes like; and the like. Of these, two or more may be used in combination.
- Examples of the water absorbent resin fine powder include particles having a particle diameter of less than 150 ⁇ m generated in the production process of the water absorbent resin.
- the production method according to the present invention includes a pulverization process, a classification process, a granulation process, a transport process, a storage process, a packing process, a storage process, and the like as necessary. May further be included.
- FIG. 9 is a flow diagram of a manufacturing method in which the pulverization step is omitted and the classification step and the fine powder collection step in the sizing process are made compact.
- the granular dry matter obtained after the drying step is forcibly cooled and adjusted to a predetermined particle size in the downsized sizing step, and then the surface cross-linking step is performed.
- FIG. 10 is a flowchart of a manufacturing method in which the manufacturing method shown in the flowchart of FIG. 9 is further compacted.
- surface crosslinking is performed simultaneously with drying in the drying step.
- the water-absorbent resin powder having excellent physical properties can be efficiently produced by forcibly cooling the surface-crosslinked granular dried product obtained after the drying step and then adjusting the particle size to a predetermined particle size in the granulating step.
- the production method according to the present invention since a granular dried product having a surface crosslinked close to the target product particle size can be obtained even after the drying step of performing surface crosslinking simultaneously with drying, Compact size is also possible.
- the manufacturing method according to the present invention is carried out by a manufacturing facility including a polymerization apparatus, a gel grinding apparatus, and a drying apparatus.
- the manufacturing facility further includes a cooling device.
- This manufacturing facility may include other heating devices, mixing devices, drying devices, pulverizing devices, classification devices, granulating devices, and the like.
- the manufacturing facility has a transportation means, and the apparatuses are connected by the transportation means.
- a polymerization step for obtaining a hydrogel crosslinked polymer from a monomer that is a raw material of the water-absorbent resin is performed.
- a polymerization step for obtaining a hydrogel crosslinked polymer from a monomer that is a raw material of the water-absorbent resin is performed.
- a gel pulverizing step is performed to pulverize the hydrogel crosslinked polymer to obtain a particulate hydrogel crosslinked polymer.
- a drying step is performed in which the particulate hydrogel crosslinked polymer is dried to obtain a granular dried product.
- a surface cross-linking step for obtaining a surface-crosslinked granular dried product is performed.
- a cooling step for forcibly cooling the granular dried product or the surface-crosslinked granular dried product is performed.
- the drying device includes a rotating container that contains the particulate hydrogel crosslinked polymer, rotates, and is positioned inside the rotating container and extends in the axial direction thereof.
- a rotary heating apparatus including a plurality of heating tubes rotating together with a rotating container.
- the drying device has means for introducing and discharging gas into the rotating container.
- the drying apparatus has a rotating container that is inclined from one end to the other end.
- at least one barrier is provided inside the rotating container.
- the plurality of heating tubes are arranged radially spaced from the rotation axis of the rotating container toward the radially outer side.
- the plurality of heating tubes are arranged at intervals on two or more concentric circles centering on the rotation axis of the rotating container.
- this drying apparatus has a heating means or a heat retaining means on the outer peripheral surface of the rotating container.
- the present invention is a method for producing a water-absorbent resin powder including the above-described drying method.
- the production method includes a polymerization step of polymerizing an aqueous monomer solution containing a monomer that is a raw material of the water-absorbent resin to obtain a hydrogel crosslinked polymer, and pulverizing the hydrogel crosslinked polymer. And a gel crushing step for obtaining a particulate hydrogel crosslinked polymer.
- this production method includes a cooling step of cooling the granular dried product obtained by drying the particulate hydrogel crosslinked polymer by the drying method described above.
- the water-absorbent resin powder obtained by the production method according to the present invention (in particular, the surface-crosslinked water-absorbent resin powder is also referred to as a water-absorbing agent).
- the water-absorbing agent is used for absorbent articles, particularly paper diapers, at least one of the physical properties listed in the following (4-1) to (4-5), preferably two or more, More preferably, it is desired that three or more, more preferably all physical properties are controlled within a desired range.
- CRC centrifuge retention capacity
- the CRC (centrifuge retention capacity) of the water absorbent resin powder (water absorbent) of the present invention is usually 5 g / g or more, preferably 15 g / g or more, more preferably 25 g / g or more.
- the upper limit is not particularly limited, and higher CRC is preferable, but from the viewpoint of balance with other physical properties, it is preferably 70 g / g or less, more preferably 50 g / g or less, and still more preferably 40 g / g or less. .
- CRC When the CRC is less than 5 g / g, the amount of absorption is small and it is not suitable as an absorbent body for absorbent articles such as paper diapers. In addition, when the CRC exceeds 70 g / g, the rate of absorbing body fluids such as urine and blood decreases, so that it is not suitable for use in a high water absorption rate type paper diaper.
- CRC can be controlled by changing the type and amount of the internal cross-linking agent and surface cross-linking agent.
- Ext (Amount of water-soluble polymer)
- water-soluble polymer amount is usually 50% by mass or less, preferably 35% by mass or less, more preferably 25% by mass or less, and further preferably 15% by mass or less. Although it does not specifically limit about a minimum, Preferably it is 0 mass%, More preferably, it is about 0.1 mass%. In the present invention, “about” means including an error of ⁇ 5% with respect to the measured value.
- Ext exceeds 50% by mass, the gel strength is weak, and there is a risk of becoming a water-absorbing resin having poor liquid permeability. Furthermore, since rewetting increases, it is not suitable as an absorbent body for absorbent articles such as paper diapers. Ext can be controlled by changing the type and amount of the internal cross-linking agent.
- the water content of the water-absorbent resin powder is preferably more than 0% by mass and 20% by mass or less, more preferably 1% by mass to 15% by mass, and still more preferably 2% by mass. It is ⁇ 13% by mass, particularly preferably 2% by mass to 10% by mass.
- a water absorbent excellent in powder characteristics for example, fluidity, transportability, damage resistance, etc.
- the mass average particle diameter (D50) of the water absorbent resin powder (water absorbent) is preferably 200 ⁇ m to 700 ⁇ m, more preferably 250 ⁇ m to 600 ⁇ m, still more preferably 250 ⁇ m to 500 ⁇ m, and particularly preferably 300 ⁇ m to 450 ⁇ m. It is.
- the proportion of particles having a particle diameter of less than 150 ⁇ m is preferably 20% by mass or less, more preferably 10% by mass or less, and still more preferably 5% by mass or less.
- the ratio of particles having a particle diameter of 850 ⁇ m or more is preferably 20% by mass or less, more preferably 15% by mass or less, and still more preferably 10% by mass or less.
- this water-absorbing agent contains particles having a particle diameter of less than 850 ⁇ m, preferably 80% by mass or more, more preferably 85% by mass or more, and further preferably 90% by mass or more.
- the logarithmic standard deviation ( ⁇ ) of the particle size distribution is preferably 0.20 to 0.50, more preferably 0.25 to 0.40, and still more preferably 0.27 to 0.35.
- AAP Water absorption capacity under pressure
- the AAP (water absorption capacity under pressure) of the water absorbent resin (water absorbent) is preferably 15 g / g or more, more preferably 20 g / g or more, still more preferably 22 g / g or more, particularly preferably 23 g / g or more, most preferably. Is 24 g / g or more.
- the upper limit is not particularly limited, but is preferably 30 g / g or less.
- the amount of return of the liquid when pressure is applied to the absorbent body may increase, so that absorbent articles such as paper diapers It is not suitable as an absorber.
- AAP can be controlled by adjusting the particle size or changing the surface cross-linking agent.
- water-absorbing resin powder water-absorbing agent
- water-absorbing resin powder water-absorbing agent
- the use of water-absorbing resin powder is not particularly limited, but is preferably used as an absorbent article for absorbent articles such as paper diapers, sanitary napkins, and incontinence pads. Is mentioned. In particular, it can be used as an absorbent material for high-concentration paper diapers where odors, coloring, and the like derived from raw materials have been problematic. Furthermore, since the water-absorbing agent is excellent in water absorption time and the particle size distribution is controlled, a remarkable effect can be expected when used in the upper layer portion of the absorber.
- an absorbent material such as pulp fiber can be used together with the water absorbing agent.
- the content (core concentration) of the water-absorbing agent in the absorber is preferably 30% by mass to 100% by mass, more preferably 40% by mass to 100% by mass, and still more preferably 50% by mass to 100% by mass. Even more preferably, it is 60% by mass to 100% by mass, particularly preferably 70% by mass to 100% by mass, and most preferably 75% by mass to 95% by mass.
- the absorbent article can be kept in a clean white state. Furthermore, since the absorbent body is excellent in diffusibility of body fluids such as urine and blood, the amount of absorption can be improved by efficient liquid distribution.
- liter may be expressed as “l” or “L”
- mass% or “weight%” may be expressed as “wt%”.
- D Non Detected
- CRC centrifuge retention capacity
- the CRC (centrifuge retention capacity) of the water-absorbent resin was measured according to the EDANA method (ERT441.2-02).
- the CRC (centrifuge retention capacity) of the hydrogel was the same as that of the EDANA method (ERT441.2-02) except that the hydrogel was changed to 0.4 g as a sample and the free swelling time was changed to 24 hours. The operation was performed.
- the solid content ⁇ of the water-containing gel was measured separately to determine the dry mass of the water-absorbent resin in 0.4 g of the water-containing gel, and the CRC of the water-containing gel was calculated according to the following (Formula 2).
- msi is the mass (unit; g) of the water-absorbent resin before measurement, mb is freely swollen and dehydrated, and the mass (unit: g) of Blank (nonwoven fabric only), mwi is freely swollen and dehydrated
- the mass (unit; g) of the subsequent hydrogel and ⁇ is the solid content ratio (unit: mass%) of the water-absorbent resin before measurement.
- Ext (water soluble component) of the hydrous gel was measured according to the EDANA method (ERT470.2-02). The same operation as in the EDANA method (ERT470.2-02) was performed except that the mass of the hydrogel as a sample was changed to 5.0 g and the stirring time was changed to 24 hours. Furthermore, the solid content ratio ⁇ of the hydrogel was measured separately, the dry mass in 5.0 g of the hydrogel was determined, and the Ext of the hydrogel was calculated according to the following (Formula 3).
- VHCl. s is the amount of HCl (unit: ml) required to bring the filtrate containing the dissolved polymer from pH 10 to pH 2.7
- VHCl. b is the amount of HCl (unit: ml) necessary to bring Blank (0.9 mass% sodium chloride aqueous solution) from pH 10 to pH 2.7
- CHCl is the concentration of HCl solution (unit: mol / l)
- Mw is acrylic The average molecular weight of monomer units in the acid (salt) polymer (unit: g / mol)
- Fdir is the dilution of the filtrate containing the dissolved polymer
- ms is the mass of the water-absorbent resin (unit: g) before measurement
- ⁇ is It is the solid content ratio (unit: mass%) of the water-absorbent resin before measurement.
- (C) Water content of water-absorbing resin The water content of the dried water-absorbing resin (water-absorbing agent) was measured according to the EDANA method (ERT430.2-02). In the present invention, the measurement was performed by changing the sample amount to 1.0 g and the drying temperature to 180 ° C.
- M means the mass (g) of the water absorbent resin
- ⁇ means the solid content (mass%) of the water absorbent resin.
- a solid content rate is calculated
- the mass of the water-absorbent resin (hydrogel) used in this measurement is M (g)
- ⁇ is the solid content (mass%) of the water-absorbent resin (hydrous gel).
- a 20 wt% aqueous sodium chloride solution (hereinafter referred to as “20% by weight”) containing 0.08 wt% EMAL 20C (surfactant, manufactured by Kao Corporation) It was added to 1000 g of “Emal aqueous solution”) to prepare a dispersion, and a stirrer chip having a length of 50 mm ⁇ diameter 7 mm was stirred at 300 rpm for 16 hours (approximately 1.14 L container made of polypropylene having a height of 21 cm and a diameter of 8 cm. use).
- the water-containing gel was classified by repeating the operation of uniformly pouring four times so that the water injection range (50 cm 2) spread over the entire sieve.
- the hydrated gel on the classified first stage sieve was drained for about 2 minutes and weighed.
- the second and subsequent sieves were classified by the same operation, and the hydrogel remaining on each sieve after draining was weighed.
- the sieve can be appropriately changed according to the gel particle size. For example, when the water-containing gel particle size becomes fine and clogging occurs with a sieve having an opening of 0.15 mm or 0.075 mm, a JIS standard sieve having a larger diameter (diameter 30 cm, opening 0.15 mm,. 075 mm) and classification may be performed.
- Vortex (water absorption time) Vortex (water absorption time) of the water absorbent resin was measured according to the following procedure. First, after adding 0.02 part by weight of edible blue No. 1 (brilliant blue), which is a food additive, to 1000 parts by weight of physiological saline (0.9% by weight sodium chloride aqueous solution) prepared in advance, the liquid temperature is adjusted. Adjusted to 30 ° C.
- edible blue No. 1 brilliant blue
- the monomer aqueous solution adjusted to 38 ° C. was continuously supplied with a metering pump, and then 150.6 parts by mass of a 48% by mass sodium hydroxide aqueous solution was continuously line-mixed. At this time, the temperature of the aqueous monomer solution increased to 87 ° C. due to heat of neutralization.
- the obtained hydrogel (1b) and 3.1% by mass lauryldimethylaminoacetic acid betaine aqueous solution were gel pulverized while simultaneously supplying to the screw extruder.
- the supply amount of the lauryldimethylaminoacetic acid betaine aqueous solution was 0.15% by mass relative to the solid content of the hydrogel (1b).
- a meat chopper having an outer diameter of a screw shaft of 86 mm, a perforated plate having a diameter of 100 mm, a hole diameter of 8.0 mm, and a thickness of 10 mm at the tip is used, and the hydrogel (1b) and At the same time, gel pulverization (first gel pulverization) was performed while supplying water and water vapor.
- the crushed gel obtained by the first gel pulverization was further gel pulverized (second gel pulverization) while supplying water and water vapor.
- the gel grinding energy required for the second grinding was 51 J / g.
- the obtained particulate hydrogel PG (1) has a solid content of 44% by mass (moisture content is 56% by mass), an average particle size d1 in terms of dry mass of 130 ⁇ m, and a ratio of particles having a particle size of 150 ⁇ m or less. It was 53 mass%.
- the polymerization rate of the particulate hydrous gel was 98.6%, the CRC was 36 g / g, and the soluble content was 6%.
- the physical properties of the particulate hydrogel PG (1) are shown in Table 1 below.
- Production Example 3 In Production Example 1, a particulate hydrous gel PG (3) was prepared in the same manner as in Production Example 1 except that the first gel was pulverized using a porous plate having a pore diameter of 3.2 mm and the second gel was not pulverized. Got.
- the physical properties of this particulate hydrogel PG (3) are shown in Table 1 below.
- Production Example 4 In Production Example 1, except that the supply amount of the lauryldimethylaminoacetic acid betaine aqueous solution was 0.08% by mass, the first gel pulverization was performed using a porous plate having a pore size of 6.4 mm, and the second gel pulverization was not performed. In the same manner as in Production Example 1, a particulate hydrous gel PG (4) having a large average particle size was obtained. The physical properties of this particulate hydrogel PG (4) are shown in Table 1 below.
- Production Example 5 In Production Example 1, except that the supply amount of the lauryldimethylaminoacetic acid betaine aqueous solution was 0.08% by mass, the first gel pulverization was performed using a porous plate having a pore size of 9.5 mm, and the second gel pulverization was not performed. In the same manner as in Production Example 1, a particulate hydrous gel PG (5) having a larger average particle size was obtained. Various physical properties of this particulate hydrogel PG (5) are shown in Table 1 below.
- Production Example 6 (Soluble content Up) In Production Example 1, except that 0.42 parts by mass of polyethylene glycol diacrylate and 236 parts by mass of deionized water were polymerized, and a porous plate having a pore size of 4.0 mm was used for the first gel pulverization and the second gel pulverization. In the same manner as in Production Example 1, particulate water-containing gel PG (6) was obtained. The physical properties of this particulate hydrogel PG (6) are shown in Table 1 below.
- Production Example 8 The same procedure as in Production Example 1 was conducted except that polyethylene glycol (weight average molecular weight 20000) was used in place of lauryldimethylaminoacetic acid betaine in the first gel pulverization of Production Example 1 and the supply amount was 2.00% by mass. Thus, a particulate hydrous gel PG (8) was obtained. The physical properties of this particulate hydrogel PG (8) are shown in Table 1 below.
- Gel fluidizing agent A lauryldimethylaminoacetic acid betaine aqueous solution (concentration 3.1 mass%)
- Gel fluidizer B Modified silicone (trade name “KF-101” manufactured by Shin-Etsu Chemical Co., Ltd.)
- Gel fluidizer C Polyethylene glycol (PEG 20000, Mw 20000)
- the particulate hydrogel PG (1) (Particulate Gel) obtained in Production Example 1 was dried using a drying apparatus (rotary heating apparatus with a heating tube) having the basic structure shown in FIG.
- This drying apparatus has 10 heating tubes extending in the direction of the rotation axis and two barriers (a donut-shaped partition plate having one circular opening at the center, an opening ratio of 50%).
- a cylindrical rotating container (capacity 100 L) is provided, and a downward inclination of 0.6 ° is given from the input portion toward the take-out port.
- the end part by the side of the taking-out port in a rotation container has a donut-shaped partition plate (other name; discharge weir) which has one circular opening part (opening ratio 24%) in the center.
- the supply amount of air (carrier gas; 140 ° C.) and the exhaust amount were adjusted so that the pressure difference with respect to the outside air inside the rotating container was ⁇ 20 Pa and the exhaust dew point was 90 ° C.
- the hydrated gel during drying was found to be two donuts installed at two intermediate positions of the rotating container. It was divided into three regions with a solid content ratio of about 90% by mass and about 95% by mass as a boundary by the shape barrier (opening ratio 50%).
- the temperature of the dried product (1) collected at the outlet was 200 ° C., and most of the temperature was granulated particles.
- the amount of fine powder in terms of dry weight in this dried product (1) ratio of particles having a particle diameter of less than 150 ⁇ m
- Example 2 Drying with aeration band dryer
- the particulate hydrogel PG (1) obtained in Production Example 1 was dried using a band dryer having the basic structure shown in FIG. First, the particulate hydrogel (1) was sprayed on the ventilation plate (punching metal) of this dryer so as to have a thickness of about 10 cm and dried at hot air at 185 ° C. for 35 minutes.
- the water-containing gel particles are integrated into a single-plate block shape (the width is almost equivalent to the width of the drying belt, the length is endless, the thickness is several centimeters). It was hard.
- the dried product (2) was in the form of a single plate block, the yield of the desired product particle size (850-150 ⁇ m) was 0%. Furthermore, a difference in the degree of drying was observed in the thickness direction in which hot air was passed up and down.
- the integrated dried product (2) is air-cooled and coarsely crushed by a crusher (first crusher) equipped with a rotating shaft having a plurality of blades, and then supplied to a three-stage roll mill (second crusher). And further pulverized to obtain a water-absorbent resin powder (2).
- the obtained water absorbent resin powder (2) had a mass average particle diameter (d3) of 400 ⁇ m, a ratio of particles exceeding the particle diameter of 850 ⁇ m was less than 1% by mass, and a ratio of particles having a particle diameter of less than 150 ⁇ m was 12% by mass. .
- Example 3 The block-shaped dried product (2) obtained in Experimental Example 2 was crushed with a crusher (first pulverizer), and then pulverized under the same conditions using the one-stage roll mill used in Experimental Example 1. However, the crushed material was not caught in the roll. Therefore, the dried product (2) could not be pulverized by the compact pulverization method of Experimental Example 1. The results are shown in Tables 2 and 3.
- Example 4 After roughly crushing the block-shaped dried product (2) obtained in Experimental Example 2 with a crusher (first pulverizer), using the one-stage roll mill (second pulverizer) used in Experimental Example 1 Then, the gap between the rolls was adjusted so that the crushed material was bitten and pulverized.
- the obtained water-absorbent resin powder (4) has a mass average particle diameter d3 of 780 ⁇ m, a ratio of particles having a particle diameter exceeding 850 ⁇ m is 44% by mass, a ratio of particles having a particle diameter of less than 150 ⁇ m is 5% by mass, and 850-150 ⁇ m. The proportion of particles was 51% by mass, and a water-absorbent resin powder having the target product particle size (850-150 ⁇ m, average particle size 400 ⁇ m) could not be obtained.
- Tables 2 and 3 The results are shown in Tables 2 and 3.
- Example 12 Effect of shielding In Experimental Example 1, continuous drying was performed in the same manner as in Experimental Example 1 except that a drying apparatus without a donut-shaped partition plate was used, and a water-absorbing resin powder (12 ) The results are shown in Tables 2 and 3.
- Example 13 Effect of shielding In Experimental Example 1, continuous drying was performed in the same manner as in Experimental Example 1, except that only one partition plate close to the inlet was used among the two donut-shaped partition plates of the drying device. And a water absorbent resin powder (13) was obtained. The results are shown in Tables 2 and 3.
- Example 15 In Experimental Example 1, instead of the drying apparatus having the basic configuration shown in FIG. 1-2, continuous drying was attempted with hot air at 200 ° C. using a fluidized bed dryer. Only by adjusting the wind speed of the dryer, aggregation of the hydrogel could not be suppressed, and continuous fluidized bed drying could not be performed. The results are shown in Tables 2 and 3.
- Example 17 Post-crosslinking simultaneously with drying (surface crosslinking)
- a surface cross-linking agent solution containing 0.16% by mass of ethylene glycol diglycidyl ether and 2% by mass of water with respect to the particulate hydrous gel PG (1) in the middle of drying.
- a water-absorbent resin powder (17) was obtained in the same manner as in Experimental Example 1, except that the addition was performed by spraying.
- the water-containing gel when the surface crosslinking agent was added had a water content of 30% by mass and a temperature of 110 ° C. The results are shown in Tables 2 and 3.
- Example 21 The water absorbent resin powder (1) obtained in Experimental Example 1 (850-150 ⁇ m; 94% by mass, less than 150 ⁇ m; 6% by mass) was further classified for 10 minutes with a sieve having an opening of 150 ⁇ m to obtain a 6% by mass fine powder. Were collected. Next, a water-absorbent resin powder (21) was obtained in the same manner as in Production Example 1 and Experimental Example 1 except that the total amount of the collected fine powder was recycled to the polymerization step of Production Example 1. By collecting 6% by mass of fine powder in the monomer solution, the CRC of the water absorbent resin powder (21) was 3 g / g lower than the CRC of the water absorbent resin powder (1).
- Example 22 From the water-absorbent resin powder (2) obtained in Experimental Example 2 (850-150 ⁇ m; 87 mass%, less than 150 ⁇ m; 12 mass%), 12 mass% of fine powder was collected. Next, a water absorbent resin powder (22) was obtained in the same manner as in Production Example 1 and Experimental Example 1 except that the total amount of the collected fine powder was recycled to the polymerization step of Production Example 1. By collecting 12% by mass of fine powder in the monomer solution, the CRC of the water absorbent resin powder (22) decreased by 5 g / g as compared with the CRC of the water absorbent resin powder (1).
- Example 28 Post-crosslinking after drying
- the water-absorbent resin powder (1) obtained in Experimental example 1 was further classified and a 850-160 ⁇ m sieve fraction was collected, and then ethylene glycol diglycidyl ether 0.2 mass
- the surface-crosslinking agent solution containing 2% by weight and 3% by mass of water was sprayed and heated at 150 ° C. for 30 minutes to obtain a surface-crosslinked water-absorbent resin powder (28) (also known as a water-absorbing agent).
- the water absorbent resin powder (28) had a CRC of 33 g / g and an AAP of 24 g / g.
- Example 29 Using the water absorbent resin powder (1) obtained in Experimental Example 1, additional drying was performed by the method of Experimental Example 1. Similar to Experimental Example 1, neither the inside of the drying apparatus nor the aggregation of the dried product was observed.
- Example 30 Using the water absorbent resin powder (1) obtained in Experimental Example 1, the inner wall was additionally dried by the method of Experimental Example 8. Unlike Experimental Example 8, no adhesion was observed even when the temperature of the inner surface of the rotating container was low.
- Example 31 The water content of the water absorbent resin powder (1) obtained in Experimental Example 1 was adjusted to 10% by mass, and additional drying was performed by the method of Experimental Example 8. Unlike Experimental Example 8, no adhesion was observed even when the temperature of the inner surface of the rotating container was low.
- R Drying device (rotary heating device with heating tube)
- B Ventilation band dryer
- M Stirring dryer (paddle dryer)
- F Fluidized bed dryer
- An increase in the amount of fine powder means that when surface cross-linking is carried out as it is, it means a decrease in performance after surface cross-linking, or even when fine powder is recycled, it means classification of fine powder and an increase in the recycling process. The performance is also reduced.
- Experimental Example 1 no addition of surface cross-linking agent to hydrous gel
- Experimental Examples 17 to 19 (30%, 20% and 40% of gel water content when adding a surface cross-linking agent during drying
- Experimental Example 20 ( Compared with the addition of a surface cross-linking agent to the water-containing gel before drying), the addition of the surface cross-linking agent as a particulate water-containing gel in the middle of drying (especially when the water content is 20 to 40%) suppresses aggregation during drying.
- the water absorption capacity under pressure is also improved to 20 g / g or more.
- the water absorption magnification under pressure is the same level as that of a conventional water-absorbent resin powder obtained by surface crosslinking of a pulverized classified product after drying. That is, in the production method according to the present invention, it is understood that the conventional surface cross-linking structure is formed simultaneously with the drying by adding the surface cross-linking agent to the particulate hydrous gel during the drying using the rotary heating device. That is, in the method of the present invention, it is also possible to carry out surface crosslinking simultaneously with drying. As described in Experimental Example 1, the pulverization and classification steps after drying are made compact (FIG. 9) and the amount of fine powder is reduced.
- the surface cross-linking step of the pulverized and classified product after drying and the classification step after surface cross-linking can be completely omitted (FIG. 10). That is, it can be seen that the manufacturing method according to the present invention enables a very compact process with a small number of steps in which the steps after the conventional surface cross-linking step are completely omitted. (In contrast to [FIG. 8] according to the manufacturing method of the prior art, 13 steps and a large amount of fine powder recovery step, [FIG. 10] according to the manufacturing method of the present invention includes 6 steps and a small amount of fine powder recovery step).
- the dried product does not block at the time of drying.
- the amount is reduced. Therefore, the pulverization process and the fine powder recovery process after the drying process can be made compact (simplified) or partially omitted.
- residual monomers are also easily reduced.
- the amount of waste gas is small, so the amount of waste gas processed is small and energy is saved.
- the ventilation band drying even a fine particle-like hydrogel that is difficult to dry due to spillage or clogging from the dryer can be dried without problems according to the production method of the present invention.
- the water absorption rate of the resulting water-absorbent resin is also improved by drying the fine particle gel.
- the production method of the present invention it is possible to post-crosslink (surface cross-link) the water-absorbent resin simultaneously with drying, and a water-absorbent resin having a high water absorption capacity under pressure can be easily obtained.
- the manufacturing process can be greatly simplified. This simplification of the process also leads to a reduction in process damage after drying, and can also suppress generation of fine powder and performance degradation after drying.
- stable continuous operation and start of drying can be performed even in stirring and drying, and therefore, continuous drying is possible.
- the water-absorbent resin powder obtained by the present invention is suitable for use as an absorbent material for sanitary goods such as paper diapers.
Abstract
Description
〔1-1〕「吸水性樹脂」
本発明における「吸水性樹脂」とは、水膨潤性水不溶性の高分子ゲル化剤を指し、下記の物性を満たすものをいう。即ち、水膨潤性としてERT441.2-02で規定されるCRC(遠心分離機保持容量)が5g/g以上であり、かつ、水不溶性としてERT470.2-02で規定されるExt(水可溶分)が50質量%以下である高分子ゲル化剤を指す。
本発明における「ポリ(メタ)アクリル酸(塩)」とは、ポリ(メタ)アクリル酸及び/又はその塩を指し、主成分として(メタ)アクリル酸及び/又はその塩(以下、「(メタ)アクリル酸(塩)」とも称する)を繰り返し単位として含み、任意成分としてグラフト成分を含む架橋重合体を意味する。
「EDANA」は、European Disposables and Nonwovens Associationsの略称である。また、「ERT」は、EDANA Recommended Test Methodsの略称であり、吸水性樹脂の測定方法を規定した欧州標準である。本発明では、特に断りのない限り、ERT原本(2002年改定)に準拠して、吸水性樹脂の物性を測定する。
「CRC」は、Centrifuge Retention Capacity(遠心分離機保持容量)の略称であり、吸水性樹脂の無加圧下での吸水倍率(「吸水倍率」と称する場合もある)を意味する。具体的には、吸水性樹脂0.2gを不織布製の袋に入れた後、大過剰の0.9質量%塩化ナトリウム水溶液中に30分間浸漬して自由膨潤させ、その後、遠心分離機(250G)で3分間、水切りした後の吸水倍率(単位;g/g)のことをいう。なお、重合後の含水ゲルについては後述の方法で測定時間を24時間に変更し且つ固形分補正して、CRCを求める。
「Ext」は、Extractablesの略称であり、吸水性樹脂の水可溶分(吸水性樹脂中の水可溶分ポリマー量)を意味する。具体的には、吸水性樹脂1.0gを0.9質量%塩化ナトリウム水溶液200mlに添加し、500rpmで16時間攪拌した後、水溶液に溶解した物質の量(単位;質量%)のことをいう。水可溶分の測定には、pH滴定が用いられる。なお、重合後の含水ゲルについては含水ゲル5.0gを使用し測定時間を24時間に変更し且つ含水ゲルを固形分補正して、水可溶分ポリマー量を測定する。
「Moisture Content」は、吸水性樹脂の乾燥減量で規定される含水率を意味する。具体的には、吸水性樹脂4.0gを105℃で3時間乾燥した際の乾燥減量から算出した値(単位;質量%)のことをいう。なお、本発明において、乾燥後の吸水性樹脂については、吸水性樹脂1.0gの180℃、3時間の乾燥減量で規定され、乾燥前の含水ゲルについては、含水ゲル2.0gの180℃、24時間の乾燥減量で規定される。
「PSD」は、Particle Size Distributionの略称であり、篩分級により測定される吸水性樹脂の粒度分布を意味する。なお、質量平均粒子径(D50)及び粒度分布の対数標準偏差(σζ)は、米国特許第7638570号に記載された方法と同様の方法で測定される。なお、本発明において含水ゲルの粒度分布(PSD)は後述の方法で湿式に篩分級することで規定される。さらに、含水ゲルの固形分換算の粒径(μm)は含水ゲルの粒径(μm)とその固形分率(%)から後述の計算方法で規定される。
「AAP」は、Absorption Against Pressureの略称であり、吸水性樹脂の加圧下における吸水倍率を意味する。具体的には、吸水性樹脂0.9gを大過剰の0.9質量%塩化ナトリウム水溶液に対して、1時間、2.06kPa(21g/cm2、0.3psi)の荷重下で膨潤させた後の吸水倍率(単位;g/g)のことをいう。また、荷重条件を4.83kPa(49g/cm2、0.7psi)に変更して測定する場合もある。なお、重合後の含水ゲルについては後述の方法で測定時間を24時間に変更し且つ固形分補正して、CRCを求める。
本明細書において、範囲を示す「X~Y」は「X以上、Y以下」を意味する。また、特に注釈のない限り、質量の単位である「t(トン)」は「Metric ton(メトリック トン)」を意味し、「ppm」は「質量ppm」又は「重量ppm」を意味する。更に、「質量」と「重量」、「質量部」と「重量部」、「質量%」と「重量%」はそれぞれ同義語として扱う。また、「~酸(塩)」は「~酸及び/又はその塩」、「(メタ)アクリル」は「アクリル及び/又はメタクリル」をそれぞれ意味する。
本発明に係る吸水性樹脂粉末の製造方法は、加熱装置を用いて、吸水性樹脂の原料である単量体から得られる粒子状含水ゲル状架橋重合体を乾燥する乾燥工程を有している。好ましくは、この製造方法は、重合工程、(重合と同時または別途の)ゲル粉砕工程、(乾燥と同時または別途の)表面架橋工程、冷却工程並びに(乾燥後の及び/又は表面架橋後の)整粒工程を有している。その他には、単量体水溶液の調整工程、各種添加剤の添加工程、微粉除去工程及び微粉リサイクル工程を含んでいるのが好ましい。更に、目的に応じて各種の公知の工程を含むことができる。後述のように、本発明の製造方法では、乾燥工程以降の少なくとも一部の工程をコンパクト化ないし省略することができる。このコンパクト化した製造工程のフローが、図9及び10に例示されている。図9及び10の詳細については後述する。
本工程は、アクリル酸(塩)を主成分として含む水溶液(以下、「単量体水溶液」と称する)を調製する工程である。なお、得られる吸水性樹脂の吸水性能が低下しない範囲で、単量体のスラリー液を使用することもできるが、本項では便宜上、単量体水溶液について説明を行う。
アクリル酸以外の単量体としては、重合して吸水性樹脂となり得る化合物であればよい。例えば、メタアクリル酸、(無水)マレイン酸、イタコン酸、ケイ皮酸、ビニルスルホン酸、アリルトルエンスルホン酸、ビニルトルエンスルホン酸、スチレンスルホン酸、2-(メタ)アクリルアミド-2-メチルプロパンスルホン酸、2-(メタ)アクリロイルエタンスルホン酸、2-(メタ)アクリロイルプロパンスルホン酸、2-ヒドロキシエチル(メタ)アクリロイルフォスフェート等の酸基含有不飽和単量体;(メタ)アクリルアミド、N-エチル(メタ)アクリルアミド、N,N-ジメチル(メタ)アクリルアミド等のアミド基含有不飽和単量体;N,N-ジメチルアミノエチル(メタ)アクリレート、N,N-ジメチルアミノプロピル(メタ)アクリレート、N,N-ジメチルアミノプロピル(メタ)アクリルアミド等のアミノ基含有不飽和単量体;メルカプト基含有不飽和単量体;フェノール性水酸基含有不飽和単量体;N-ビニルピロリドン等のラクタム基含有不飽和単量体等が挙げられる。
重合に使用される単量体は、重合の安定性から、好ましくは少量の重合禁止剤を含む。好ましい重合禁止剤はp-メトキフェノールである。単量体(特にアクリル酸及びその塩)中に含まれる重合禁止剤の量は、通常1~250ppm、好ましくは10~160ppm、より好ましくは20~80ppmである。
アクリル酸および上記単量体の中でカルボキシル基等の酸基を有する酸基含有不飽和単量体を用いる場合、該酸基の一部又は全部が中和された中和塩を用いることができる。この場合、酸基含有不飽和単量体の塩としては一価のカチオンとの塩であることが好ましく、アルカリ金属塩、アンモニウム塩及びアミン塩から選ばれる少なくとも1種であることがより好ましく、アルカリ金属塩であることが更に好ましく、ナトリウム塩、リチウム塩及びカリウム塩から選ばれる少なくとも1種であることがより更に好ましく、ナトリウム塩が特に好ましい。
上記酸基含有不飽和単量体を中和するために使用される中和剤としては、特に限定されないが、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸アンモニウム等の無機塩や、アミノ基やイミノ基を有するアミン系有機化合物等の塩基性物質が適宜選択されて用いられる。中和剤として、2種以上の塩基性物質が併用されてもよい。なお、本発明における単量体は、特に断りのない限り、中和塩を含む概念である。
吸水性能の観点から、酸基含有不飽和単量体とその中和塩の合計モル数に対する中和塩のモル数(以下、「中和率」と称する)は、好ましくは40モル%以上、より好ましくは40モル%~80モル%、更に好ましくは45モル%~78モル%、特に好ましくは50モル%~75モル%である。
吸水性樹脂粉末の製造方法において、好ましくは内部架橋剤が用いられる。該内部架橋剤によって、得られる吸水性樹脂の吸水性能や吸水時のゲル強度等が調整される。
本発明に係る製造方法において、本発明の目的が達成される範囲内で、以下に例示する物質(以下、「その他の物質」と称する)を単量体水溶液に添加することもできる。
本工程において、単量体組成物中の単量体濃度(=総単量体量/(総単量体量+総重合溶媒量(通常は水))は、吸水性樹脂の物性及び生産性の観点から、好ましくは10質量%~90質量%、より好ましくは20質量%~80質量%、更に好ましくは30質量%~70質量%、特に好ましくは40~60重量%である。以下、単量体濃度を「モノマー濃度」と称する場合がある。
本発明で使用される重合開始剤は、重合形態等によって適宜選択されるため、特に限定されないが、例えば、熱分解型重合開始剤、光分解型重合開始剤、若しくはこれらの併用、又は重合開始剤の分解を促進する還元剤を併用したレドックス系重合開始剤等が挙げられる。具体的には、米国特許第7265190号に開示された重合開始剤のうち、1種又は2種以上が用いられる。なお、重合開始剤の取扱性や吸水性樹脂の物性の観点から、好ましくは過酸化物又はアゾ化合物、より好ましくは過酸化物、更に好ましくは過硫酸塩が使用される。
なお、重合前の単量体水溶液中の溶存酸素を、昇温又は不活性ガスとの置換により低減させることも好ましい。例えば、溶存酸素は、好ましくは5ppm以下、より好ましくは3ppm以下、特に好ましくは1ppm以下に低減される。
本工程は、前記単量体水溶液を重合して、含水ゲル状架橋重合体(以下、「含水ゲル」と称する場合がある)を得る工程である。
重合形態としては、特に限定されない。吸水特性や重合制御の容易性等の観点から、好ましくは気相中の液滴重合、水溶液重合、逆相懸濁重合(ここで疎水性有機溶媒中の液滴重合も逆相懸濁の一例に含む)、より好ましくは水溶液重合、逆相懸濁重合、更に好ましくは水溶液重合が挙げられる。中でも、連続水溶液重合が特に好ましく、連続ベルト重合、連続ニーダー重合の何れでも適用される。具体的な重合形態として、連続ベルト重合は米国特許第4893999号、同第6241928号、米国特許出願公開第2005/215734号等に、連続ニーダー重合は米国特許第6987151号、同第6710141号等に、それぞれ開示されている。これらの連続水溶液重合を採用することで、吸水性樹脂の生産効率が向上する。
重合工程で得られる含水ゲル状架橋重合体の重合率は、次の粉砕工程で得られる粒子状含水ゲル状架橋重合体の加熱処理中の凝集抑制や、得られる吸水剤中の残存モノマー低減の観点から、好ましくは90質量%以上、より好ましくは95重量%以上、更に好ましくは98重量%以上、特に好ましくは99重量%以上である。重合率が低い場合、回転型加熱装置で乾燥中の含水ゲルが凝集ないし付着しやすい傾向にある。重合率の上限は特に限定されないが、100質量%が理想的であるが、高い重合率には長い重合時間や厳しい重合条件が必要であり、生産性や物性面の低下を招くこともあり、上限は99.95重量%、さらに99.9重量%、通常99.8重量%程度で十分である。代表的には98~99.99質量%であり、さらに好ましくは上記範囲である。なお、生産性向上のため、重合率90%質量以下の含水ゲルを重合装置から取り出して乾燥する技術も知られていたが(例えばWO2006/103227)、本発明者らによって、粒子状含水ゲルの加熱装置による乾燥では重合率の調整が重要であることが見出された。
本工程は、上記重合工程で得られた含水ゲル状架橋重合体を、重合と同時及び/又は重合後に粉砕して細粒化する工程であり、例えば、ニーダー、ミートチョッパー等のスクリュー押出し機、カッターミル等の装置により粉砕して、粒子状含水ゲル状架橋重合体(以下、「粒子状含水ゲル」)を得る工程である。好ましい粒径は後述する範囲であり、後述する乾燥工程で得られる粒状乾燥物又は表面架橋工程において得られる表面架橋された粒状乾燥物の形状及び大きさが、目的とする製品粒度に近づくように実施される。
ゲル粉砕エネルギー(Gel Grinding Energy,GGE)とは、特許文献36に記載され、含水ゲル状架橋重合体をゲル粉砕する際、ゲル粉砕装置が必要とする単位重量(含水ゲル状架橋重合体の単位重量)あたりの機械的エネルギーを意味し、ゲル粉砕装置が三相交流電力で駆動する場合、下記(式1)により算出される。
ここで、力率及びモーター効率は、ゲル粉砕装置の稼働条件等によって変化する装置固有の値であり、0~1までの値をとる。ゲル粉砕装置が単相交流電力で駆動する場合、上記式中の31/2を1に変更することで算出される。上記(式1)において、電圧の単位は[V]、電流の単位は[A]、含水ゲルの重量の単位は[g]である。本発明で適用される好ましいゲル粉砕エネルギー(GGE)は特許文献36の範囲が適用できるが、特に制限されない。
本発明に係る製造方法において、好ましくは、ゲル粉砕工程で、含水ゲル又はその粉砕物である粒子状含水ゲルに、ゲル流動化剤が添加される。ゲル流動化剤の添加は、後述する乾燥工程及び表面架橋時の加熱処理工程において、粒子状含水ゲルを処理する場合に特に有効である。
具体的には、ゲル流動化剤に用いられる界面活性剤として、(1)ショ糖脂肪酸エステル、ポリグリセリン脂肪酸エステル、ソルビタン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリオキシエチレングリセリン脂肪酸エステル、ソルビトール脂肪酸エステル、ポリオキシエチレンソルビトール脂肪酸エステル、ポリオキシエチレンアルキルエーテル、ポリオキシエチレンアルキルフェニルエーテル、ポリオキシエチレンヒマシ油、ポリオキシエチレン硬化ヒマシ油、アルキルアリルホルムアルデヒド縮合ポリオキシエチレンエーテル、ポリオキシエチレンポリオキシプロピレンブロックコポリマー、ポリオキシエチレンポリオキシプロピルアルキルエーテル、ポリエチレングリコール脂肪酸エステル、アルキルグルコシド、N-アルキルグルコンアミド、ポリオキシエチレン脂肪酸アミド、ポリオキシエチレンアルキルアミン、ポリオキシエチレンアルキルエーテルのリン酸エステル、及びポリオキシエチレンアルキルアリルエーテルのリン酸エステルなどのノニオン性界面活性剤、(2)カプリルジメチルアミノ酢酸ベタイン、ラウリルジメチルアミノ酢酸ベタイン、ミリスチルジメチルアミノ酢酸ベタイン、ステアリルジメチルアミノ酢酸ベタイン等のアルキルジメチルアミノ酢酸ベタイン;ラウリン酸アミドプロピルベタイン、ヤシ油脂肪酸アミドプロピルベタイン、パーム核油脂肪酸アミドプロピルベタイン等のアルキルアミドプロピルベタイン、ラウリルヒドロキシスルホベタイン等のアルキルヒドロキシスルホベタイン、2-アルキル-N-カルボキシメチル-N-ヒドロキシエチルイミダゾリニウムベタイン等のアルキルカルボキシメチルヒドロキシエチルイミダゾリニウムベタインなどの両性界面活性剤、(3)ラウリルアミノジ酢酸モノナトリウム、ラウリルアミノジ酢酸カリウム、ミリスチルアミノジ酢酸ナトリウム等のアルキルアミノジ酢酸モノアルカリ金属などのアニオン性界面活性剤、(4)長鎖アルキルジメチルアミノエチル4級塩などのカチオン性界面活性剤等が挙げられる。これらのうち、2種以上を併用してもよい。
本発明に係る製造方法において、本発明の目的が達成される範囲内で、以下に例示する高分子滑剤を、上記単量体水溶液や含水ゲルに添加することができる。
ゲル流動化剤の種類と添加量とは目的とする粒子状含水ゲルの流動性などを考慮して選択される。得られる吸水性樹脂の吸収性物品(おむつ)での実使用における戻り量等から、最終製品の吸水性樹脂の表面張力が過度に低下しない種類や量のゲル流動化剤が好ましい。例えば、吸水性樹脂の表面張力(生理食塩水中に吸水性樹脂を分散させた分散液の表面張力)が、好ましくは55mN/m以上、より好ましくは60mN/m以上、更に好ましくは65mN/m以上となるように、ゲル流動化剤の種類及び量が選択される。この表面張力はWO2015/129917に記載の方法で測定される。表面張力をかかる範囲内にできるゲル流動化剤としては、特許文献28のベタイン系界面活性剤が例示される。
本工程は、粒子状含水ゲル(好ましくはゲル流動化剤を含む)を所望する固形分率まで乾燥させることで粒状乾燥物を得る工程、好ましくは乾燥させるとともに、造粒して、粒状乾燥造粒物を得る工程である。なお、本工程に供される粒子状含水ゲルは、前述した水溶液重合により得た含水ゲルを粉砕して得られるものに限定されず、例えば、逆相懸濁重合により得られてもよい。また、「粒状乾燥造粒物」とは、複数の吸水性樹脂粒子同士が、物理的又は化学的に付着することで形成された粒子状の乾燥物を意味し、乾燥前後の平均粒子径の向上や電子顕微鏡写真等から確認することができる。例えば、本発明に係る製造方法の乾燥工程で得られる粒状乾燥造粒物の電子顕微鏡写真の一例が、図11に示されている。以下、粒状乾燥造粒物を「造粒物」と称する場合がある。当該「固形分率」とは、乾燥減量(試料1gを180℃で3時間乾燥した際の質量変化)から算出される値を意味する。
本発明に係る製造方法では、従来多用された静置乾燥機(特に非特許文献1および特許文献17~22などに記載の通気バンド式乾燥機)に代えて、乾燥工程における乾燥装置として、加熱装置が用いられる。この加熱装置は、その内部に粒子状含水ゲルを収容して回転する回転容器と、この回転容器の内部に位置して、その軸方向に延在し、かつこの回転容器とともに回転する複数の加熱管とを備えている。本願明細書において、この構成を有する加熱装置を、「回転型加熱装置」又は「加熱管付き回転型加熱装置」と称する場合がある。より好ましくは、この加熱装置は、その回転容器の外周面に、更に他の加熱手段を備えている。この加熱装置では、回転容器に収容された粒子状含水ゲルが、この容器の回転によって攪拌され、複数の加熱管との接触又は加熱管からの熱伝導によって、加熱される。複数の加熱管の輻射熱等により回転容器内面も加熱されるが、必要に応じて、粒子状含水ゲルは、回転容器の外周面に位置する加熱手段により、さらに加熱される。この加熱装置では、必要により撹拌翼等他の攪拌手段も併用されるが、主として、粒子状含水ゲルを収容する回転容器の回転及びこの回転容器とともに回転する複数の加熱管の作用によって、粒子状含水ゲルが容器内で流動するため、被乾燥物である粒子状含水ゲルに対する機械的及び熱的ダメージが少ない。これにより、乾燥工程における微粉発生及び物性劣化が抑制される。さらに、この加熱装置では、加熱管からの間接伝熱により乾燥されるため、熱風による乾燥(通気バンド式乾燥機や通気加熱式ロータリーキルン)のような乾燥物の飛散もなく、大量の廃ガス処理も要しないという利点がある。また、この加熱装置では、主として回転容器の回転により被乾燥物が流動するため、撹拌翼等で撹拌する連続攪拌乾燥機のように、粘着性を有する粒子状含水ゲルの撹拌に大きなエネルギーを要することもなく、乾燥後の吸水性樹脂の物性低下(例;吸水速度の低下、可溶成分の増加)、微粉発生、乾燥時の凝集等が回避されるという利点もある。
本発明に係る製造方法において、加熱装置2が備える加熱管12の配置は特に限定されず、吸水剤の製造量等に応じて適宜設定される。図3に示されるように、複数の加熱管12が、回転容器10の回転軸を中心とする円周上に、間隔を空けて配置されてもよく、図4に示されるように、回転容器10の回転軸を中心とする2以上(多重)の同心円上に、間隔を空けて配置されてもよい。また、複数の加熱管12が、回転容器10の回転軸から径方向外側に向かって、放射状に間隔を空けて配置されてもよい。複数の加熱管12を円周沿って多重に配置する場合、その数は目的に応じて選択されるが、好ましくは2~10重、より好ましくは2~8重、さらに好ましくは2~5重に設置される。多重に配置することで、より広い伝熱面を有することとなり、粒子状含水ゲルのより効率的な乾燥が可能となる。
本発明に係る製造方法では、乾燥工程に図1-2に示される基本構成を備えた加熱装置を用いることにより、粒子状含水ゲル状架橋重合体の固形分換算による単位時間当たりの処理量(kg/hr)が7kg/hr以上、好ましくは50Kg/hr以上、より好ましくは100Kg/hr以上、特に好ましくは500Kg/hrの連続乾燥が可能となる。連続乾燥時間は好ましくは12時間以上、さらには24時間以上、特に240時間以上、1200時間以上の連続乾燥である。
粒子状含水ゲルに添加される添加剤48の例としては、前述されたゲル流動化剤及び高分子滑剤の他に、後述される表面架橋剤が挙げられる。本発明の効果が阻害されない限り、粒子状含水ゲルに他の添加剤48を添加してもよい。表面架橋剤の乾燥前さらには乾燥途中の添加によって乾燥時の付着も低減でき、さらに一般的な乾燥後の表面架橋効果(例えば加圧下吸水倍率の向上)も示すため、乾燥後の表面架橋工程も省略できる。
本発明に係る乾燥装置及び乾燥方法は上記であるが、より好ましくは、上記乾燥装置は本発明の課題を解決するために下記構造を有する。
本発明に係る乾燥装置及び乾燥方法は上記であるが、本発明の課題を解決するためにより好ましい乾燥方法は、以下の通りである。かかる乾燥方法は連続乾燥に限らず、回分式乾燥(特に繰り返しの回分式乾燥)にも適用でき、またゲル粉砕した粒子状含水ゲル状架橋重合体に限らず、ゲル粉砕を行わないで得られた吸水性樹脂の粒子状含水ゲル状架橋重合体、代表的には逆相懸濁重合や気相での液滴重合や噴霧重合で得られた吸水性樹脂の粒子状含水ゲル状架橋重合体にも適用できる。
本発明に係る製造方法において、乾燥工程に供される粒子状含水ゲルの温度(以下、ゲル温度と称する)は50℃以上である。このゲル温度は、加熱装置に設置された前述の接触温度計にて、測定される。接触温度計として、例えば、熱電対、白金測温体ないしバイメタル温度計、特に熱電対(例えばK線シース熱電対)が挙げられる。代表的には、ゲル温度は、材料層(粒子状含水ゲルや粒状乾燥物)の中心部(例えば、材料の厚みが10cmの場合は5cm前後の位置)にて測定される。加熱装置の内部での粒子状含水ゲルの流動性の観点から、このゲル温度は60℃以上が好ましく、70℃以上がより好ましく、80℃以上がさらに好ましく、90℃以上が特に好ましい。また温度が高すぎる場合、被乾燥物の着色や性能低下が見られる場合もあるため、好ましくは、ゲル温度は130℃以下、より好ましくは110℃以下、さらに好ましくは105℃以下である。これらの温度は、重合後の含水ゲルの温度(重合装置から排出される時の温度)や、重合後の各工程での加熱、保温又は反応熱(例えば、重合後の中和熱やゲル粉砕時の粉砕エネルギーや加熱)により調整してもよく、乾燥工程前に、別途加熱工程を設けて調整してもよい。ゲル流動化剤が、前述したゲル粉砕工程において添加される場合、乾燥工程に供される前に、所定のゲル温度となるように調整される。
乾燥工程に供される粒子状含水ゲルの含水率(以下、ゲル含水率と称する)は、下記実施例に記載した測定方法によって求められる。粒子状含水ゲルの流動性の観点から、ゲル含水率は25質量%以上が好ましく、30質量%%以上がより好ましく、35質量%以上がさらに好ましく、40質量%以上がよりさらに好ましく、43質量%以上が特に好ましい。過度の高濃度重合は吸水性樹脂の物性を低下させる可能性があり、乾燥効率及び吸収性能の観点から、ゲル含水率は75質量%以下が好ましく、60質量%以下がより好ましく、55質量%以下が特に好ましい。
乾燥前の粒子状含水ゲルの質量平均粒子径(以下、ゲル粒子径と称する)は、乾燥工程又は熱処理工程後の粒子径が最終製品である吸水剤の粒子径とほぼ同じようになることが好ましい。例えば、紙オムツなどの衛生材料で用いられる場合のゲル粒子径d1は、固形分換算で、好ましくは800μm以下、より好ましくは500μm以下、さらに好ましくは50μm~500μm、よりさらに好ましくは100μm~400μm、特に好ましくは100~300μm、極めて好ましくは100~200μmである。上記ゲル粒子径d1を上記範囲内とすることで、得られる粒状乾燥物の吸水速度も向上し、さらに装置内部における粒子状含水ゲルの流動性が向上する結果、乾燥効率が向上するという利点が得られる。すなわち、本発明における乾燥工程は、粒子径の小さい粒子状含水ゲルの乾燥、特に乾燥造粒に適用される。
乾燥前の粒子状含水ゲルのCRC(遠心分離機保持容量)は、乾燥重量換算で、好ましくは5g/g~80g/g、より好ましくは10g/g~50g/g、更に好ましくは15g/g~45g/g、特に好ましくは20g/g~40g/gである。また、乾燥前の粒子状含水ゲルの水可溶分ポリマー量(Ext)は、乾燥重量換算で、好ましくは20質量%以下、より好ましくは15質量%以下、更に好ましくは10質量%以下、特に好ましくは5質量%以下である。粒子状含水ゲルのCRCや可溶分が高い場合、乾燥時の撹拌によって凝集や付着が進行する場合がある。従って、乾燥前の粒子状含水ゲルの吸水倍率及び可溶分は、低いことが好ましい。例えば、比較的低吸水倍率かつ低可溶分の粒子状含水ゲルを加熱装置に投入し、十分に高い温度(好ましくは、150℃以上、より好ましくは180℃以上)で、所定時間(好ましくは、所定温度で10分以上)加熱することで、得られる粒状乾燥物の可溶分及び吸水倍率を向上させる実施形態が好ましい。このように乾燥によって吸水倍率及び可溶分を増加させる方法として、上記ポリアルキレン単位を有する内部架橋剤の使用が例示される。
乾燥工程に供される乾燥前の粒子状含水ゲルの重合率は、乾燥重量換算で、好ましくは、90質量%以上であり、より好ましくは95質量%以上であり、更に好ましくは、98~99.99質量%であり、理想的には、100%である。重合率が低いと乾燥時に凝集や付着の問題が生じる。従来、生産性向上のため、重合率90%以下で重合装置より取り出して乾燥する技術も知られていたが(例えばWO2006/103227)、本発明者らによって、粒子状含水ゲルの回転型加熱装置による乾燥では重合率が重要であることが見出された。
乾燥後に得られる粒状乾燥物の水可溶分ポリマー量(Ext)は、好ましくは、乾燥前の粒子状含水ゲルの水可溶分ポリマー量より大きい。乾燥後に得られる粒状乾燥物の水可溶分ポリマー量は、乾燥重量換算で、好ましくは+0.5質量%以上増加、より好ましくは+1~20質量%の範囲で増加、さらに好ましくは+2~10質量%の範囲で増加するように、含水ゲルの架橋密度(特に内部架橋剤の種類や量)や加熱乾燥条件などが調整される。
本工程は、重合後の含水ゲルやその乾燥物に、吸水性樹脂の官能基(特にカルボキシル基)と反応する表面架橋剤を添加して架橋反応させる工程である。この架橋反応により、主として、吸水性樹脂粒子の表面から架橋される。表面架橋は、後架橋又は2次架橋とも称される。本発明に係る製造方法では、本工程において、粒子状含水ゲル及び/又は粒状乾燥物(以下、吸水性樹脂と総称する場合がある)に表面架橋剤を添加して反応させる。本工程は、表面架橋剤添加工程と熱処理工程とを有し、必要に応じて熱処理工程後に冷却工程を有していてもよい。
本工程は、粒子状含水ゲル及び/又は粒状乾燥物に、表面架橋剤を添加する工程である。表面架橋剤は、乾燥前あるいは乾燥途中の粒子状含水ゲルに対して、あるいは、乾燥後の粒状乾燥物、さらには整粒後の粒状乾燥物に対して添加される。
吸水性樹脂の「含水率」とは、下記実施例に記載した測定方法によって求められる値である。なお、吸水剤の「含水率」は、測定条件が異なるだけで、理論的な考え方は吸水性樹脂の「含水率」と同じである。
上記表面架橋剤として、吸水性樹脂の複数の官能基(好ましくは複数のカルボキシル基)と反応しうる表面架橋剤、好ましくは共有結合またはイオン結合、さらには共有結合しうる表面架橋剤が使用される。具体的には、エチレングリコール、ジエチレングリコール、プロピレングリコール、トリエチレングリコール、テトラエチレングリコール、ポリエチレングリコール、1,3-プロパンジオール、ジプロピレングリコール、2,2,4-トリメチル-1,3-ペンタンジオール、ポリプロピレングリコール、グリセリン、ポリグリセリン、2-ブテン-1,4-ジオール、1,3-ブタンジオール、1,4-ブタンジオール、1,5-ペンタンジオール、1,6-ヘキサンジオール、1,2-シクロヘキサンジメタノール、1,2-シクロヘキサノール、トリメチロールプロパン、ジエタノールアミン、トリエタノールアミン、ポリオキシプロピレン、オキシエチレン-オキシプロピレンブロック共重合体、ペンタエリスリトール、ソルビトール等の多価アルコール化合物;エチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、グリセロールポリグリシジルエーテル、ジグリセロールポリグリシジルエーテル、ポリグリセロールポリグリシジルエーテル、プロピレングリコールジグリシジルエーテル、ポリプロピレングリコールポリグリシジルエーテル、グリシドール、ソルビトールポリグリシジルエーテル、ペンタエリスリトールポリグリシジルエーテル、トリメチロールプロパンポリグリシジルエーテル、ネオペンチルグリコールジグリシジルエーテル、1,6-ヘキサンジオールジグリシジルエーテル等のエポキシ化合物;エチレンジアミン、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミン、ポリエチレンイミン等の多価アミン化合物及びこれらの無機塩又は有機塩;2,4-トリレンジイソシアネート、ヘキサメチレンジイソシアネート等の多価イソシアネート化合物;ポリアジリジン等のアジリジン化合物;1,2-エチレンビスオキサゾリン、ビスオキサゾリン、ポリオキサゾリン等の多価オキサゾリン化合物;尿素、チオ尿素、グアニジン、ジシアンジアミド、2-オキサゾリジノン等の炭酸誘導体;1,3-ジオキソラン-2-オン、4-メチル-1,3-ジオキソラン-2-オン、4,5-ジメチル-1,3-ジオキソラン-2-オン、4,4-ジメチル-1,3-ジオキソラン-2-オン、4-エチル-1,3-ジオキソラン-2-オン、4-ヒドロキシメチル-1,3-ジオキソラン-2-オン、1,3-ジオキサン-2-オン、4-メチル-1,3-ジオキサン-2-オン、4,6-ジメチル-1,3-ジオキサン-2-オン、1,3-ジオキソパン-2-オン等のアルキレンカーボネート化合物;エピクロロヒドリン、エピブロムヒドリン、α-メチルエピクロロヒドリン等のハロエポキシ化合物及びこれらの多価アミン付加物;オキセタン化合物;γ-グリシドキシプロピルトリメトキシシラン、γ-アミノブロピルトリエトキシシラン等のシランカップリング剤;亜鉛、カルシウム、マグネシウム、アルミニウム、鉄、ジルコニウム等の水酸化物、塩化物、硫酸塩、硝酸塩又は炭酸塩等の多価金属化合物;等が挙げられる。これらのうち、2種以上を併用してもよい。上記表面架橋剤の中でも、乾燥後または乾燥途中に表面架橋する場合、多価金属イオン、エポキシ系化合物、オキサゾリン系化合物、アルキレンカーボネート化合物から選択された1又は2以上が好ましく、乾燥途中に表面架橋する場合、エポキシ系化合物が好ましい。
上記表面架橋剤の添加量は、乾燥途中または乾燥後の吸水性樹脂の固形分に対して好ましくは5質量%以下、より好ましくは3質量%以下、更に好ましくは2質量%以下である。また、下限値としては好ましくは0.001質量%である。
表面架橋剤を添加する回数は、乾燥途中あるいは乾燥後に1回でも複数回でもよい。2回以上添加する場合には、少なくとも最初の1回の表面架橋剤と接触する吸水性樹脂は前記の含水率と温度とを有することが好ましく、全ての接触において前記の含水率と温度とを有することがより好ましい。
本工程は、表面架橋剤を含有する吸水性樹脂を加熱処理して、表面架橋された粒状乾燥物、好ましくは表面架橋された粒状乾燥造粒物を得る工程である。
本工程では、前記表面架橋剤を含有する吸水性樹脂を100℃以上に加熱することで吸水剤が得られる。好ましい最高温度は、表面架橋剤の種類により異なるが、100~250℃であり、より好ましくは120~230℃であり、さらに好ましくは150~210℃である。
熱処理工程の時間は、吸水性樹脂の含水率、表面架橋剤の種類、加熱装置の熱効率などから適宜設定すればよい。一応の目安としては、含水率が10質量%以下になるまで加熱すればよく、時間としては10~120分間の範囲であり、好ましくは30~90分間である。
本発明の目的が達成される限り、表面架橋工程に用いる加熱装置は特に限定されないが、加熱むらが発生しにくいとの観点から、固体-固体接触による伝導伝熱形式で撹拌機構を有する(以下、撹拌型の間接加熱式と称することがある)が好適に用いられる。なお、乾燥と同時に表面架橋される場合、上記加熱装置が表面架橋工程の加熱装置としても用いられる。
好ましくは、本発明に係る製造方法は、前述の乾燥工程又は表面架橋工程後、後述する整粒工程前に、粒状乾燥物又は表面架橋された粒状乾燥物を強制冷却して、所望の温度に調整するための冷却工程を有している。前述の加熱装置2において、表面架橋工程と乾燥工程とが一工程で実施される場合には、回転容器10において、表面架橋処理が適正になされ、かつ粒状乾燥物又は表面架橋された粒状乾燥物の固形分率又は含水率が所望の範囲に調整された後、冷却工程が実施される。
冷却工程において、粒状乾燥物又は表面架橋された粒状乾燥物を冷却する方法は、特に限定されない。好ましくは、通気伝熱式又は伝導伝熱式の冷却手段を有する連続冷却機が用いられる。
本工程は、粒状乾燥物又は表面架橋された粒状乾燥物の粒度を調整する工程である。この整粒工程によって、粒子径又は粒度分布がより積極的に制御された吸水性樹脂粉末が得られる。
微粉低減の観点から、整粒工程に供される粒状乾燥物又は表面架橋された粒状乾燥物の質量平均粒子径d2は、好ましくは200μm以上であり、より好ましくは300μm以上であり、更に好ましくは400μm以上、500μmである。解砕ステップの効率化の観点から、質量平均粒子径d2は2000μm以下、さらには1500μm以下、1000μm以下が好ましい。
表面架橋された粒状乾燥物がこの整粒工程に供される場合、その含水率は、好ましくは乾燥後の粒状乾燥物の含水率に関して前述した含水率の範囲である。
吸水性能の観点から、整粒工程を経て得られる吸水性樹脂粉末の質量平均粒子径d3は、好ましくは200μm以上であり、より好ましくは200~600μm、さらに好ましくは250~550μm、特に好ましくは300~500μmである。
好ましくは、本発明に係る製造方法は、さらに、微粉リサイクル工程を有する。「微粉リサイクル工程」とは、分級ステップで除去された微粉をそのまま、又は微粉を造粒した後に何れかの工程に供給する工程を意味する。好ましくは、微粉又は微粉造粒物を、乾燥工程以前の工程に投入して再利用する工程である。乾燥工程以前の工程としては、上記重合工程で調整した重合前の単量体溶液、重合途中の含水ゲル、重合後の含水ゲルの粉砕工程、粒状含水ゲルの乾燥工程等が挙げられる。これらの工程に、微粉をそのまま添加してもよく、微粉を水で膨潤ゲル化ないし造粒してから添加してもよい。また、微粉とともに、水、架橋剤、水以外のバインダー(例;水溶性ポリマー、熱可塑性樹脂)、重合開始剤、還元剤、キレート剤、着色防止剤などを添加してもよい。例えば、水を添加する場合、微粉に対して1~1000質量%、その他の化合物を添加する場合は、微粉に対して0.01~10質量%の量で使用されることが好ましい。
好ましくは、この製造方法は、湿潤工程を更に有している。湿潤工程は、粒状乾燥物、表面架橋された粒状乾燥物又は吸水性樹脂粉末に、水を添加して、再湿潤する工程である。
本発明に係る製造方法においては、上記任意に使用される表面架橋剤やゲル流動化剤以外にも、その他添加剤として、乾燥前又は乾燥後に、無機微粒子、粉塵防止剤、乾燥した吸水性樹脂(微粉)、通液性向上剤等を、更に加えることが可能である。
本発明に係る製造方法は、上述した各工程以外に、必要に応じて、粉砕工程、分級工程、造粒工程、輸送工程、貯蔵工程、梱包工程、保管工程等を更に含んでもよい。
前述した通り、本発明の製造方法の乾燥工程では、目的とする製品粒度に近い粒状乾燥物又は表面架橋された粒状乾燥物が得られる。これにより、乾燥工程以降の少なくとも一部の工程をコンパクト化ないし省略することができる。
本発明に係る製造方法は、重合装置、ゲル粉砕装置及び乾燥装置を含む製造設備により実施される。好ましくは、この製造設備は、更に、冷却装置を備えている。この製造設備が、他の加熱装置、混合装置、乾燥装置、粉砕装置、分級装置、整粒装置等を備えてもよい。好ましくは、この製造設備は輸送手段を有しており、各装置がこの輸送手段により連結されている。
本発明に係る製造方法で得られる吸水性樹脂粉末(特に、表面架橋された吸水性樹脂粉末を吸水剤とも称する)については、該吸水性樹脂粉末又は該吸水剤を吸収性物品、特に紙オムツに使用する場合には、下記の(4-1)~(4-5)に掲げた物性のうち、少なくとも1つ以上、好ましくは2つ以上、より好ましくは3つ以上、更に好ましくは全ての物性が、所望する範囲に制御されることが望まれる。以下の全ての物性が下記の範囲を満たさない場合、本発明の効果が十分に得られず、特に、紙オムツ一枚当たりの吸水剤の使用量が多い、所謂、高濃度紙オムツにおいて十分な性能を発揮しないおそれがある。
本発明の吸水性樹脂粉末(吸水剤)のCRC(遠心分離機保持容量)は、通常5g/g以上であり、好ましくは15g/g以上、より好ましくは25g/g以上である。上限値については特に限定されず、より高いCRCが好ましいが、他の物性とのバランスの観点から、好ましくは70g/g以下、より好ましくは50g/g以下、更に好ましくは40g/g以下である。
Ext(水可溶分ポリマー量)は、通常50質量%以下であり、好ましくは35質量%以下、より好ましくは25質量%以下、更に好ましくは15質量%以下である。下限については特に限定されないが、好ましくは0質量%、より好ましくは0.1質量%程度である。なお、本発明において「~程度」とは測定値に対して±5%の誤差を含むことを意味する。
吸水性樹脂粉末(吸水剤)の含水率は、好ましくは0質量%を超えて20質量%以下、より好ましくは1質量%~15質量%、更に好ましくは2質量%~13質量%、特に好ましくは2質量%~10質量%である。
吸水性樹脂粉末(吸水剤)の質量平均粒子径(D50)は、好ましくは200μm~700μm、より好ましくは250μm~600μm、更に好ましくは250μm~500μm、特に好ましくは300μm~450μmである。また、粒子径150μm未満の粒子の割合は、好ましくは20質量%以下、より好ましくは10質量%以下、更に好ましくは5質量%以下である。また、粒子径850μm以上の粒子の割合は、好ましくは20質量%以下、より好ましくは15質量%以下、更に好ましくは10質量%以下である。換言すれば、この吸水剤は、粒子径850μm未満の粒子を、好ましくは80質量%以上、より好ましくは85質量%以上、更に好ましくは90質量%以上含む。粒度分布の対数標準偏差(σζ)は、好ましくは0.20~0.50、より好ましくは0.25~0.40、更に好ましくは0.27~0.35である。
吸水性樹脂(吸水剤)のAAP(加圧下吸水倍率)は、好ましくは15g/g以上、より好ましくは20g/g以上、更に好ましくは22g/g以上、特に好ましくは23g/g以上、最も好ましくは24g/g以上である。上限値については特に限定されないが、好ましくは30g/g以下である。
吸水性樹脂粉末(吸水剤)の用途は、特に限定されないが、好ましくは紙オムツ、生理用ナプキン、失禁パッド等の吸収性物品の吸収体用途が挙げられる。特に、原料由来の臭気、着色等が問題となっていた高濃度紙オムツの吸収体として使用することができる。更に、吸水剤は、吸水時間に優れ、かつ粒度分布が制御されているので、上記吸収体の上層部に使用する場合に、顕著な効果が期待できる。
(a)CRC(遠心分離機保持容量)
吸水性樹脂のCRC(遠心分離機保持容量)を、EDANA法(ERT441.2-02)に準拠して測定した。また、含水ゲルのCRC(遠心分離機保持容量)は、試料として含水ゲルを0.4gに、自由膨潤時間を24時間に、それぞれ変更した以外はEDANA法(ERT441.2-02)と同様の操作を行った。更に、別途含水ゲルの固形分率αを測定し、含水ゲル0.4g中の吸水性樹脂の乾燥質量を求め、下記(式2)にしたがって含水ゲルのCRCを算出した。
含水ゲルのExt(水可溶分)を、EDANA法(ERT470.2-02)に準拠して測定した。試料であるして含水ゲルの質量を5.0gに、攪拌時間を24時間に、それぞれ変更した以外はEDANA法(ERT470.2-02)と同様の操作を行った。更に、別途含水ゲルの固形分率αを測定し、含水ゲル5.0g中の乾燥質量を求め、下記(式3)にしたがって含水ゲルのExtを算出した。
乾燥された吸水性樹脂(吸水剤)の含水率を、EDANA法(ERT430.2-02)に準拠して測定した。なお、本発明においては、試料量を1.0g、乾燥温度を180℃にそれぞれ変更して測定した。
吸水性樹脂粉末又は粒状乾燥物の粒度(粒度分布、質量平均粒子径(D50)及び粒度分布の対数標準偏差(σζ))を、米国特許第7638570号のカラム27、28に記載された方法に準拠して測定した。
イオン交換水1000gに含水ゲル1.00gを投入し、300rpmで2時間攪拌した後に、ろ過することにより、不溶分を除去した。上記操作で得られたろ液中に抽出された単量体の量を、液体クロマトグラフを用いて測定した。該単量体の量を残存モノマー量m(g)としたときに、下記(式4)にしたがって、重合率C(質量%)を求めた。
乾燥前の含水ゲルの含水率について、上記(c)において、含水ゲルを2.0gとしさらに乾燥時間を24時間として測定する。底面の直径が50mmのアルミカップに吸水性樹脂(含水ゲル)2.00gを投入した後、試料(吸水性樹脂及びアルミカップ)の総質量W1(g)を正確に秤量した。次に、上記試料を、雰囲気温度180℃に設定されたオーブン内に静置した。24時間経過後、該試料を上記オーブンから取り出し、総質量W2(g)を正確に秤量した。本測定に供された吸水性樹脂(含水ゲル)の質量をM(g)としたときに、下記(式6)にしたがって、吸水性樹脂(含水ゲル)の含水率(100-α)(質量%)を求めた。なお、αは吸水性樹脂(含水ゲル)の固形分率(質量%)である。
特許文献35(国際特許WO2016/204302号)に準じて、粒子状含水ゲルの質量平均粒子径(D50)及び粒度分布の対数標準偏差(σζ)は以下の方法で測定した。
X;分級、水切り後に各篩上に残留した含水ゲルの質量%(%)
w;分級、水切り後に各篩上に残留した含水ゲルのそれぞれの質量(g)
W;分級、水切り後に各篩上に残留した含水ゲルの総質量(g)
R(α);固形分α質量%の吸水性樹脂に換算したときの篩の目開き(mm)
r;20質量%塩化ナトリウム水溶液中で膨潤した含水ゲル状架橋重合体(含水ゲル粒子)が分級された篩の目開き(mm)である。
特許文献35に準じて、上記(f)含水ゲルの含水率および(g)粒子状含水ゲルの質量平均粒子径から、乾燥後の粒子径(含水ゲル粒子の乾燥物に換算した重量平均粒子径)を下記に規定する。
GelD50:含水ゲル粒子の質量平均粒子径(μm)
GS:含水ゲル粒子の固形分率(質量%)
SolidD50:含水ゲル粒子の乾燥物に換算した質量平均粒子径(μm)
として、次式により定義される。
(式)SolidD50=GelD50×(GS/100)1/3
吸水性樹脂のVortex(吸水時間)は、以下の手順にしたがって測定した。先ず、予め調整された生理食塩水(0.9質量%塩化ナトリウム水溶液)1000質量部に、食品添加物である食用青色1号(ブリリアントブルー)0.02質量部を添加した後、液温を30℃に調整した。
吸水性樹脂の残存モノマー量を、EDANA法(ERT410.2-02)に準拠して測定した。
吸水性樹脂のAAP(加圧下吸水倍率)を、EDANA法(ERT442.2-02)に準拠して測定した。なお、測定に当たり、荷重条件を4.83kPa(0.7psi)に変更した。
アクリル酸300質量部、48質量%水酸化ナトリウム水溶液100質量部、ポリエチレングリコールジアクリレート(平均n数9)0.61質量部、0.1質量%ジエチレントリアミン5酢酸3ナトリウム水溶液16.4質量部、脱イオン水273.2質量部からなる単量体水溶液を作成した。
製造例1の第1ゲル粉砕において、ラウリルジメチルアミノ酢酸ベタイン水溶液を添加しなかった以外は、製造例1と同様にして、粒子状含水ゲルPG(2)を得た。この粒子状含水ゲルPG(2)は、界面活性剤を含まず、その含水率が44質量%、乾燥質量換算の平均粒子径d1が135μm、粒子径150μm以下の粒子の割合が約47質量%であった。粒子状含水ゲルPG(2)の物性は、下表1に示されている。
製造例1において、孔径3.2mmの多孔板を用いて第1ゲル粉砕をおこない、第2ゲル粉砕をおこなわなかったこと以外は、製造例1と同様にして、粒子状含水ゲルPG(3)を得た。この粒子状含水ゲルPG(3)の物性は、下表1に示されている。
製造例1において、ラウリルジメチルアミノ酢酸ベタイン水溶液の供給量を0.08質量%とし、孔径6.4mmの多孔板を用いて第1ゲル粉砕をおこない、第2ゲル粉砕をおこなわなかったこと以外は、製造例1と同様にして、平均粒子径の大きな粒子状含水ゲルPG(4)を得た。この粒子状含水ゲルPG(4)の物性は、下表1に示されている。
製造例1において、ラウリルジメチルアミノ酢酸ベタイン水溶液の供給量を0.08質量%とし、孔径9.5mmの多孔板を用いて第1ゲル粉砕をおこない、第2ゲル粉砕をおこなわなかったこと以外は、製造例1と同様にして、平均粒子径のより大きな粒子状含水ゲルPG(5)を得た。この粒子状含水ゲルPG(5)の諸物性は、下表1に示されている。
製造例1において、ポリエチレングリコールジアクリレートを0.42質量部とし、脱イオン水を236質量部として重合し、第1ゲル粉砕及び第2ゲル粉砕に孔径4.0mmの多孔板を使用した以外は、製造例1と同様にして、粒子状含水ゲルPG(6)を得た。この粒子状含水ゲルPG(6)の物性は、下表1に示されている。
製造例1の第1ゲル粉砕において、ラウリルジメチルアミノ酢酸ベタインに代えて変性シリコーン(信越化学;商品名KF-101)を使用し、その供給量を0.50質量%とした以外は、製造例1と同様にして、粒子状含水ゲルPG(7)を得た。この粒子状含水ゲルPG(7)の物性は、下表1に示されている。
製造例1の第1ゲル粉砕において、ラウリルジメチルアミノ酢酸ベタインに代えてポリエチレングリコール(重量平均分子量20000)を使用し、その供給量を2.00質量%とした以外は、製造例1と同様にして、粒子状含水ゲルPG(8)を得た。この粒子状含水ゲルPG(8)の物性は、下表1に示されている。

ゲル流動化剤A:ラウリルジメチルアミノ酢酸ベタイン水溶液(濃度3.1質量%)
ゲル流動化剤B:変性シリコーン(信越化学社製の商品名「KF-101」)
ゲル流動化剤C:ポリエチレングリコール(PEG20000、Mw20000)
製造例1で得られた粒子状含水ゲルPG(1)(Particulate Gel)を、図1-2に示される基本構成を備えた乾燥装置(加熱管付き回転型加熱装置)を用いて乾燥した。この乾燥装置は、その内部に回転軸方向に延在する10本の加熱管と2枚の障壁(中心部に一つの円形開口部を有するドーナツ状の仕切り板、開口率50%)とを有する円筒状の回転容器(容積100L)を備えており、投入部から取り出し口に向かって、0.6°の下向きの傾斜が付けられている。さらに、回転容器内の取り出し口側の端部には、中心に一つの円形開口部(開口率24%)を有するドーナツ状の仕切り板(別称;排出堰)を有している。はじめに、各加熱管に2.7MPa(温度228.1℃)の水蒸気を導入して、回転容器内部(接触温度計で規定)を予め200℃超に加熱した後、さらに回転容器の外壁もトレスで十分に加熱した。次いで、乾燥装置に製造例1で得られた95℃の粒子状含水ゲルPG(1)を15kg/hで供給し、フルード数Fr0.07となるように回転容器を回転させて、平均滞留時間50分で連続乾燥を行った。乾燥時、回転容器の内部の外気に対する気圧差が-20Paであり、排気露点が90℃となるように、空気(キャリアーガス;140℃)の供給量及び排気量を調整した。なお、乾燥装置の複数箇所から、回転容器内部の含水ゲルをサンプリングして、その固形分率を測定したところ、乾燥中の含水ゲルは、回転容器の中間2箇所に設置された2枚のドーナツ状の障壁(開口率50%)により、固形分率約約90質量%及び約95質量%を境界として、3つの領域に区分されていた。乾燥後、取り出し口で採取した乾燥物(1)の温度は200℃であり、その大部分が造粒粒子であった。この乾燥物(1)は、固形分率98.5質量%、質量平均粒子径(d2)625μm(d2/d1=4.8)、粒子径850μm以下の粒子の割合が乾燥重量換算で68質量%、粒子径150μm未満の粒子の割合が2.5質量%であった。この乾燥物(1)中の乾燥重量換算の微粉量(粒子径150μm未満の粒子の割合)は、粒子状含水ゲルPG(1)中の微粉量53質量%に対して約1/21に低減された。つまり、乾燥工程のみで、目的とする一般的な製品粒度(850~150μm)の乾燥物(乾燥造粒物)65.5質量%が得られ、微粉量は僅か2.5質量%であった。
製造例1で得られた粒子状含水ゲルPG(1)を、図9に示される基本構成を備えたバンド式乾燥機を用いて乾燥した。始めに、この乾燥機の通気板(パンチングメタル)に厚み約10cmになるように粒子状含水ゲル(1)を散布し、熱風185℃で35分間乾燥させた。乾燥機の出口で得られた乾燥物(2)は、含水ゲル粒子が一枚板のブロック状(横幅はほぼ乾燥ベルトの幅に相当し、長さはエンドレス、厚みは数cm)に一体化して固まっていた。ここで、乾燥物(2)は一枚板のブロック状であるため、目的とする製品粒度(850-150μm)の収率は0%であった。さらに、熱風が上下に通気する厚み方向で乾燥度合の差異が見られた。
実験例2で得られたブロック状の乾燥物(2)を解砕機(第1粉砕機)で粗砕した後、実験例1で用いた1段ロールミルを使用して、同様の条件にて粉砕を試みたが、ロールに粗砕物が噛み込まなかった。従って、乾燥物(2)を、実験例1のコンパクトな粉砕方法で粉砕することはできなかった。結果を表2及び3に示す。
実験例2で得られたブロック状の乾燥物(2)を解砕機(第1粉砕機)で粗砕した後、実験例1で用いた1段のロールミル(第2粉砕機)を使用して、疎砕物が噛み込むようにロールの隙間を調整して粉砕した。得られた吸水性樹脂粉末(4)は、質量平均粒子径d3が780μm、粒子径850μmを超える粒子の割合が44質量%、粒子径150μm未満の粒子の割合が5質量%、850-150μmの粒子の割合が51質量%であり、目的とする製品粒度(850-150μm、平均粒径400μm)の吸水性樹脂粉末は得られなかった。結果を表2及び3に示す。
実験例1において、粒子状含水ゲルPG(1)を、ゲル温度95℃から60℃に冷却して乾燥装置に投入した以外は、実験例1と同様にして連続乾燥をおこなって、吸水性樹脂粉末(5)を得た。乾燥工程で得た乾燥物(5)には、一部に凝集見られた。結果を表2及び3に示す。
実験例1において、粒子状含水ゲルPG(1)に代えて、製造例2の粒子状含水ゲルPG(2)を用いた以外は、実験例1と同様にして、撹拌乾燥を行った。実験例6では、回転容器の内壁や加熱管の表面に含水ゲルが付着して堆積し、未乾燥ゲルが排出されたため、安定した連続乾燥ができなかった。結果を表2及び3に示す。
実験例1において、粒子状含水ゲルPG(1)を、ゲル温度95℃から35℃に冷却して乾燥装置に投入した以外は、実験例1と同様にして攪拌乾燥を試みたが、粒子状含水ゲルの凝集が激しく、乾燥装置への連続供給ができなかったため、安定した連続乾燥が実施できなかった。結果を表2及び3に示す。
実験例1において、加熱管のみを水蒸気で十分に加熱した後、回転容器の内表面が150℃まで予熱される前に粒子状含水ゲルPG(1)を投入した以外は、実験例1と同様にして連続乾燥をおこなった。回転容器内表面の予熱がない場合、乾燥開始直後に含水ゲルが回転容器の内壁及び加熱管の表面管に付着し、さらに含水ゲルが凝集したため、同じ乾燥時間では一部に未乾燥物が発生した。結果を表2及び3に示す。
実験例1において、乾燥装置の各加熱管に導入する水蒸気を2.7MPa(温度228.1℃)から1.0MPa(温度180℃)に変更した以外は、実験例1と同様にして連続乾燥を行って、吸水性樹脂粉末(9)を得た。結果を表2及び3に示す。
実験例1において、乾燥装置の各加熱管に導入する水蒸気を2.7MPa(温度228.1℃)から0.36MPa(温度140℃)変更した以外は、実験例1と同様にして連続乾燥を行った。熱媒温度140℃の場合、含水ゲルが回転容器の内壁及び加熱管の表面管に付着して堆積し、さらに未乾燥ゲルが排出されたため、安定した連続乾燥ができなかった。結果を表2及び3に示す。
実験例1において、粒子状含水ゲルPG(1)(可溶分6%)に代えて、製造例6で得た粒子状含水ゲルPG(6)(可溶分11%)を使用した以外は、実験例1と同様にして連続乾燥を行って、吸水性樹脂粉末(11)を得た。結果を表2及び3に示す。
実験例1において、ドーナツ状の仕切り板が設置されていない乾燥装置を用いた以外は、実験例1と同様にして連続乾燥を行って、吸水性樹脂粉末(12)を得た。結果を表2及び3に示す。
実験例1において、乾燥装置の2枚のドーナツ状仕切り板のうち、投入口に近い仕切り板1枚のみを用いた以外は、実験例1と同様に連続乾燥を行って、吸水性樹脂粉末(13)を得た。結果を表2及び3に示す。
実験例1において、図1-2を基本構成とする乾燥装置に代えて撹拌乾燥機(パドルドライヤー)を使用し、熱媒温度210℃とした以外は、実験例1と同様にして連続乾燥をおこなったところ、乾燥中に含水ゲルが凝集した。また、得られた乾燥物は、実験例1で用いた1段ロールミルで、同様の条件で粉砕することができないものであった。結果を表2及び3に示す。
実験例1において、図1-2を基本構成とする乾燥装置に代えて、流動層乾燥機を使用し200℃の熱風にて連続乾燥を試みたが、含水ゲルの凝集が生じた。乾燥機の風速を調整するだけでは、含水ゲルの凝集を抑制することができず、連続流動層乾燥を行うことができなかった。結果を表2及び3に示す。
実験例1において、フルード数Frを0.16に変更した以外は、実験例1と同様にして連続乾燥を行って、吸水性樹脂粉末(16)を得た。結果を表2及び3に示す。
実験例1の乾燥装置において、乾燥途中の粒子状含水ゲルPG(1)に対して、エチレングリコールジグリシジルエーテル0.16質量%及び水2質量%を含む表面架橋剤溶液2.6質量%を噴霧添加した以外は、実験例1と同様にして、吸水性樹脂粉末(17)を得た。表面架橋剤添加時の含水ゲルは、含水率30質量%、温度110℃であった。結果を表2及び3に示す。
実験例1及び17において、乾燥途中の粒子状含水ゲルPG(1)が含水率20質量%、温度130℃になった時点で、実験例17に記載の表面架橋剤溶液2.6%を噴霧添加した以外は、実験例1及び17と同様にして吸水性樹脂(18)を得た。結果を表2及び3に示す。
実験例1及び17において、乾燥途中の粒子状含水ゲルPG(1)が含水率40質量%、温度110℃になった時点で、実験例17に記載の表面架橋剤溶液2.6%を噴霧添加した以外は、実験例1及び17と同様にして吸水性樹脂(19)を得た。結果を表2及び3に示す。
実験例1において、乾燥前の粒子状含水ゲルPG(1)(含水率56質量%、温度95℃)に、実験例17に記載の表面架橋剤水溶液2.6%を噴霧添加した以外は、実験例1及び17と同様にして吸水性樹脂(20)を得た。結果を表2及び3に示す。
実験例1において得られた吸水性樹脂粉末(1)(850-150μm;94質量%、150μm未満;6質量%)を、さらに目開き150μmの篩で10分間分級して、6質量%の微粉を採取した。次いで、採取した微粉の全量を、製造例1の重合工程にリサイクルした以外は、製造例1及び実験例1と同様にして、吸水性樹脂粉末(21)を得た。モノマー溶液に6質量%の微粉を回収したことにより、吸水性樹脂粉末(21)のCRCは、吸水性樹脂粉末(1)のCRCと比較して、3g/g低下した。
実験例2において得られた吸水性樹脂粉末(2)(850-150μm;87質量%、150μm未満;12質量%)から微粉12質量%を採取した。次いで、採取した微粉の全量を、製造例1の重合工程にリサイクルした以外は、製造例1及び実験例1と同様にして、吸水性樹脂粉末(22)を得た。モノマー溶液に12質量%の微粉を回収したことにより、吸水性樹脂粉末(22)のCRCは、吸水性樹脂粉末(1)のCRCと比較して、5g/g低下した。
実験例1において、粒子状含水ゲルPG(1)に代えて、製造例3-5で得た粒子状含水ゲルPG(3)-PG(5)をそれぞれ使用した以外は、実験例1と同様に乾燥を行って、吸水性樹脂粉末(23)-(25)を得た。結果を表2及び3に示す。
実験例1において、粒子状含水ゲルPG(1)に代えて、製造例7及び8で得た粒子状含水ゲルPG(7)及びPG(8)をそれぞれ使用した以外は、実験例1と同様に乾燥を行って、吸水性樹脂粉末(27)及び(28)を得た。結果を表2及び3に示す。
実験例1で得られた吸水性樹脂粉末(1)について、さらに分級して850-160μmの篩分を採取した後、エチレングリコールジグリシジルエーテル0.2質量%及び水3質量%を含む表面架橋剤溶液を噴霧し、150℃で30分間加熱することにより、表面架橋された吸水性樹脂粉末(28)(別称;吸水剤)を得た。吸水性樹脂粉末(28)のCRCは33g/gであり、AAPは24g/gであった。
実験例1で得られた吸水性樹脂粉末(1)を用いて、実験例1の手法で追加の乾燥を行った。実験例1と同様に、乾燥装置内部への付着や乾燥物の凝集は見られなかった。
実験例1で得られた吸水性樹脂粉末(1)を用いて、実験例8の手法で内壁の追加の乾燥を行った。実験例8と異なり、回転容器の内表面の温度が低き場合にも付着は見られなかった。
実験例1で得られた吸水性樹脂粉末(1)の水分量を10質量%に調整して、実験例8の手法で追加の乾燥を行った。実験例8と異なり、回転容器の内表面の温度が低き場合にも付着は見られなかった。


R:乾燥装置(加熱管付き回転型加熱装置)
B:通気バンド式乾燥機
M:攪拌型乾燥機(パドルドライヤー)
F:流動層型乾燥機
(*1)粒子状含水ゲルがブロック状に一体化して乾燥された。
(*2)乾燥機内部への含水ゲルの付着による乾燥不良。
(*3)粒子状含水ゲルの凝集による乾燥不良。
(*4)粒子状含水ゲルの凝集による粉砕不良。
(1)実験例1(回転型加熱装置)と実験例2~4(通気バンド式乾燥機)との対比から、従来の通気バンド式乾燥機に比べて、本発明に係る製造方法では、回転型加熱装置の使用により、粉砕工程及び分級工程を省略ないし各装置を小型化しても、微粉量の少ない吸水性樹脂粉末が得られることが分かる。さらに、実験例2の残存モノマー890ppmに対し、実施例1では250ppmであり、本発明に係る製造方法によって、残存モノマーも低減されることがわかる。また、本発明の製造方法に用いる回転型加熱装置では、従来の通気バンド式乾燥機で見られた未乾燥物の問題や、粉こぼれ問題、厚みの方向での乾燥状態の差異もないため、物性に優れた吸水性樹脂が得られることが分かる。
(2)実験例1(ゲル流動化剤添加)と実験例6(ゲル流動化剤無添加)との対比から、従来の通気バンド式乾燥機と異なり、回転型加熱装置による乾燥では、ゲル流動化剤の添加が有効であることが分かる。
(3)実験例1(回転容器の内表面の予熱有り)と実験例8(回転容器の内表面の予熱無し)との対比から、従来の通気バンド式乾燥機と異なり、本発明に係る製造方法(さらには乾燥開始方法)では、回転容器の内表面の予熱が有効であることが分かる。さらに、実験例29~31から回転容器の内表面の予熱は吸水性樹脂の含水ゲルの乾燥には有効であるが、一定含水率以下の吸水性樹脂では特に不要であることもわかる。
(4)実験例1(ゲル温度95℃)、実験例5(同60℃)と実験例7(同35℃)との対比から、従来の通気バンド式乾燥機と異なり、本発明に係る製造方法では、回転型加熱装置に投入する粒子状含水ゲルのゲル温度が重要であることが分かる。
(5)回転型加熱装置による実験例1(熱媒温度228℃)、実験例9(同180℃)及び実験例10(140℃)と、通気バンド式乾燥機による実験例2(同185℃)との対比から、従来の通気バンド式乾燥機と異なり、本発明に係る製造方法では、回転型加熱装置の熱媒温度の調整が有効であることが分かる。
(6)実験例1(Fr=0.07)及び実験例16(Fr=0.16)の対比から、本発明に係る製造方法では、回転型加熱装置のフルード数Frの調整により、粒度をさらなる制御が可能であることが分かる。
(7)実験例1(回転型加熱装置)、実験例14(パドルドライヤー)、実験例2(通気バンド式乾燥機)及び実験例15(流動層乾燥機)との対比から、同じ粒子状含水ゲルPG(1)の連続乾燥において、攪拌型乾燥機(パドルドライヤー)及び流動層乾燥機では凝集が進行して連続乾燥が困難であること、通気バンド式乾燥機ではブロック状乾燥物となることが分かる。実験例2~4に示すように、粗大な乾燥物やブロック状乾燥物が生じる場合、乾燥後の粉砕装置や分級装置が大型化するだけでなく、粉砕に伴う微粉量も増大する。微粉量の増大は、その後そのまま表面架橋する場合、表面架橋後の性能の低下を意味し、または微粉リサイクルする場合も微粉の分級やリサイクル工程の大型化を意味し、さらに、リサイクル方法によっては吸水性能も低下させる。
(8)実験例1(含水ゲルへの表面架橋剤無添加)、実験例17~19(乾燥途中、表面架橋剤添加時のゲル含水率30%、20%及び40%)及び実験例20(乾燥前の含水ゲルに表面架橋剤添加)との対比から、表面架橋剤を乾燥途中の粒子状含水ゲル(特に含水率20~40%の時点)で添加することで、乾燥時の凝集が抑制され、さらに加圧下吸水倍率も20g/g以上に向上する。かかる加圧下吸水倍率は、乾燥後の粉砕分級物を表面架橋した従来の吸水性樹脂粉末と同様の水準である。つまり、本発明に係る製造方法において、回転型加熱装置を用いて乾燥途中の粒子状含水ゲルに表面架橋剤を添加することで、乾燥と同時に従来の表面架橋構造が形成されることが分かる。すなわち、本発明の方法では、乾燥と同時に表面架橋を実施することも可能であり、上記実験例1に記載したように、乾燥後の粉砕及び分級工程のコンパクト化(図9)、微粉量低減及び微粉回収工程の削減ないしコンパクト化に加えて、乾燥後の粉砕分級物の表面架橋工程及び表面架橋後の分級工程も完全に省略することができる(図10)。つまり、本発明に係る製造方法では、従来の表面架橋工程以降の工程を完全に省略した工程数の少ない非常にコンパクトなプロセスも可能であることが分かる。(従来技術の製造方法による[図8]では13工程かつ多量の微粉回収工程に対して、本発明の製造方法による[図10]では6工程かつ少量の微粉回収工程である)。
(9)実験例1(ベタイン系界面活性剤)、実験例26(シリコーン系)及び実験例27(PEG)との対比より、本発明に係る製造方法では、ゲル流動化剤として、各種界面活性剤や高分子滑材(PEG)の使用が有効であることが分かる。
(10)実験例1(可溶分6%)と実験例11(同11%)との対比から、本発明に係る製造方法では、粒子状含水ゲルの可溶分によって、得られる吸水性樹脂粉末の粒度を制御できることが分かる。
(11)粒子状含水ゲルの撹拌乾燥による実験例1と実験例8との対比、及び吸水性樹脂粉末の撹拌乾燥による再乾燥の実験例29~31との対比から、回転容器の内表面の温度による付着への影響は、乾燥前の含水率の高い吸水性樹脂(含水ゲル)に顕著であり、吸水性樹脂の粒子状含水ゲルの乾燥開始時(Start-up)には、加熱管以外の温度にも留意することが大事であると分かる。
(12)実験例21と実験例22との対比から重合時のモノマーへの微粉回収量が増大すると、吸水倍率(CRC)がより低下することが分かる。
(13)実験例1(遮蔽2箇所)、実験例13(遮蔽1箇所)、実験例12(遮蔽なし)との対比より、遮蔽の設置が好ましいことが分かる。
4・・・主部
6・・・投入部
8・・・取り出し部
10・・・回転容器
11、11a、11b・・・障壁
12・・・加熱管
14・・・第一歯車
16・・・第二歯車
18・・・パッキン
20・・・主筒
22・・・ホッパー
26・・・パイプ
28・・・インナースペース
36・・・ノズル
40・・・取り出し口
44・・・スチーム入口
46・・・ドレーン
48・・・添加剤
100・・・通気バンド式乾燥機
102・・・乾燥ベルト
104・・・含水ゲル
106・・・通気方向
108・・・乾燥ベルトの移動方向
110・・・乾燥重合体
Claims (49)
- 吸水性樹脂の原料である単量体を含む単量体水溶液を重合して得られた粒子状含水ゲル状架橋重合体を、加熱装置を用いて乾燥して粒状乾燥物を得る乾燥工程を含んでおり、
上記加熱装置が、その内部に上記粒子状含水ゲル状重合体を収容して回転する回転容器と、この回転容器の内部に位置して、その軸方向に延在し、かつこの回転容器とともに回転する複数の加熱管とを備えており、
上記乾燥工程に供される粒子状含水ゲル状架橋重合体の、接触温度計にて測定されるゲル温度が50℃以上である吸水性樹脂粉末の製造方法。 - 上記加熱管に150℃以上の熱媒を供給する請求項1に記載の製造方法。
- 上記回転容器の内表面の温度が150℃以上である請求項1又は2に記載の製造方法。
- 上記粒子状含水ゲル状架橋重合体が投入される前に、上記回転容器の内表面の温度を150℃以上とする請求項1~3の何れか1項に記載の製造方法。
- 上記回転容器内に熱媒としてガスを導入し、上記回転容器の内表面の温度が、当該ガスの温度より高い請求項1~4の何れか1項に記載の製造方法。
- 上記回転容器内に導入するガスの温度が100℃以上である請求項5に記載の乾燥方法。
- 上記粒状乾燥物を冷却する冷却工程をさらに有する請求項1~6の何れか1項に記載の製造方法。
- 上記乾燥工程において、上記回転容器に収容された粒子状含水ゲル状架橋重合体の加熱乾燥中に添加剤を添加する請求項1~7の何れか1項に記載の製造方法。
- 上記添加剤が表面架橋剤である請求項8に記載の製造方法。
- 上記粒子状含水ゲル状架橋重合体がゲル流動化剤を含んでいる請求項1~9の何れか1項に記載の製造方法。
- 上記乾燥工程に供される粒子状含水ゲル状架橋重合体のゲル温度が、60~120℃である請求項1~10の何れか1項に記載の製造方法。
- 上記乾燥工程に供される粒子状含水ゲル状架橋重合体の粒子径d1が800μm以下である請求項1~11の何れか1項に記載の製造方法。
- 上記複数の加熱管に0.49MPa以上の水蒸気を導入する請求項1~12の何れか1項に記載の製造方法。
- 上記加熱装置のフルード数Frが0.001~1である請求項1~13の何れか1項に記載の製造方法。
- 吸水性樹脂の原料である単量体から得られる粒子状含水ゲル状架橋重合体の乾燥装置であって、
上記乾燥装置が、その内部に上記粒子状含水ゲル状架橋重合体を収容して回転する回転容器と、この回転容器の内部に位置して、その軸方向に延在し、かつこの回転容器とともに回転する複数の加熱管と、この回転容器の内部にガスを導入及び排出する手段を備えており、
上記加熱管の数が、5本以上であり、この加熱管が、軸方向において、上記回転容器の内表面と接触しておらず、
上記回転容器の外周面に、加熱手段又は保温手段を有している乾燥装置。 - 上記回転容器が、その一端から他端に向かって傾斜している請求項15に記載の乾燥装置。
- 上記回転容器の内部に、少なくとも1つの障壁が設けられている請求項15又は16に記載の乾燥装置。
- 上記障壁の開口率が1~90%である請求項17に記載の乾燥装置。
- 上記加熱管の外周面の表面積と上記回転容器の内周面の面積との和である伝熱面積の、この回転容器の有効容積に対する比(伝熱面積/有効容積)が、10m-1以上である請求項15~18の何れか1項に記載の乾燥装置。
- 上記回転容器の内部に、この回転容器に収容された粒子状含水ゲル状架橋重合体に添加剤を添加するための添加手段を、少なくとも1以上有している請求項15~19の何れか1項に記載の乾燥装置。
- 上記添加手段が噴霧装置である請求項20に記載の乾燥装置。
- 上記複数の加熱管が、上記回転容器の回転軸から径方向外側に向かって、放射状に間隔を空けて配置されている請求項15~21の何れか1項に記載の乾燥装置。
- 上記複数の加熱管が、上記回転容器の回転軸を中心とする2以上の同心円上に、間隔を空けて配置されている請求項15~22の何れか1項に記載の乾燥装置。
- 請求項15~23の何れか1項に記載の乾燥装置を用いて、吸水性樹脂の原料である単量体から得られる粒子状含水ゲル状架橋重合体を乾燥して粒状乾燥物を得る乾燥方法であって、
上記回転容器内に熱媒としてガスを導入し、該乾燥装置に投入される粒子状含水ゲル状架橋重合体のゲル温度が50℃以上であり、
上記回転容器の内表面の温度が150℃以上である乾燥方法。 - 上記粒子状含水ゲル状架橋重合体が投入される前の、上記回転容器の内表面の温度が150℃以上である請求項24に記載の乾燥方法。
- 上記回転容器に熱媒としてガスを導入し、このガスの温度が、上記回転容器の内表面の温度より低い請求項24又は25に記載の乾燥方法。
- 上記粒子状含水ゲル状架橋重合体を連続式で乾燥する請求項24~26の何れか1項に記載の乾燥方法。
- 上記乾燥装置に投入される粒子状含水ゲル状架橋重合体がゲル流動化剤を含む請求項24~27の何れか1項に記載の乾燥方法。
- 上記乾燥装置に投入される粒子状含水ゲル状架橋重合体の質量平均粒子径が800μm以下である請求項24~28の何れか1項に記載の乾燥方法。
- 上記回転容器内に導入されるガスが100℃以上である請求項24~29の何れか1項に記載の乾燥方法。
- 上記回転容器の内部に添加手段を有しており、この添加手段を用いて、上記粒子状含水ゲル状架橋重合体に添加剤が添加される請求項24~30の何れか1項に記載の乾燥方法。
- 上記添加剤が表面架橋剤である請求項31に記載の乾燥方法。
- 上記表面架橋剤を、含水率が10~50質量%の粒子状含水ゲル状架橋重合体に添加する請求項32に記載の乾燥方法。
- 上記回転容器の内部の圧力と大気圧との差が、0kPa超5kPa以下である請求項24~33の何れか1項に記載の乾燥方法。
- 上記回転容器のフルード数が0.001~1である請求項24~34の何れか1項に記載の乾燥方法。
- 上記回転容器内へのガスの導入量が、上記粒子状含水ゲル状架橋重合体の固形分換算による単位時間当たりの処理量(kg/hr)に対して、0.05Nm3/kg~20Nm3/kgである請求項24~35の何れか1項に記載の乾燥方法。
- 乾燥装置を含む吸水性樹脂粉末の製造設備であって、
吸水性樹脂の原料である単量体から含水ゲル状架橋重合体を得る重合装置と、
上記含水ゲル状架橋重合体を粉砕して粒子状含水ゲル状架橋重合体を得るゲル粉砕装置と、を備えており、
上記乾燥装置が、上記粒子状含水ゲル状架橋重合体を乾燥して粒状乾燥物を得るものであり、
上記乾燥装置が、その内部に上記粒子状含水ゲル状架橋重合体を収容して回転する回転容器と、この回転容器の内部に位置して、その軸方向に延在する複数の加熱管とを備えている吸水性樹脂粉末の製造設備。 - 上記粒状乾燥物を冷却する冷却装置をさらに備えている請求項37に記載の製造設備。
- 上記乾燥装置が、上記回転容器の内部にガスを導入及び排出する手段を有している、請求項37又は38の何れか1項に記載の製造設備。
- 上記回転容器が、その一端から他端に向かって傾斜している請求項37~39の何れか1項に記載の製造設備。
- 上記回転容器の内部に、少なくとも1つの障壁が設けられている請求項37~40の何れか1項に記載の製造設備。
- 上記複数の加熱管が、上記回転容器の回転軸から径方向外側に向かって、放射状に間隔を空けて配置されている請求項37~41の何れか1項に記載の製造設備。
- 上記複数の加熱管が、上記回転容器の回転軸を中心とする2以上の同心円上に、間隔を空けて配置されている請求項37~42の何れか1項に記載の製造設備。
- 上記乾燥装置が、上記回転容器の外周面に、加熱手段又は保温手段を有している請求項37~43の何れか1項に記載の製造設備。
- 上記乾燥装置が、上記回転容器の内部に、この回転容器に収容された粒子状含水ゲル状架橋重合体に添加剤を添加するための添加手段を、少なくとも1以上有している請求項37~44の何れか1項に記載の製造設備。
- 上記添加手段が噴霧装置である請求項45に記載の製造設備。
- 請求項24~36の何れか1項に記載の乾燥方法を含む、吸水性樹脂粉末の製造方法。
- 請求項47に記載の吸水性樹脂粉末の製造方法であって、
吸水性樹脂の原料である単量体を含む単量体水溶液を重合して、含水ゲル状架橋重合体を得る重合工程と、
上記含水ゲル状架橋重合体を粉砕して、粒子状含水ゲル状架橋重合体を得るゲル粉砕工程と、を更に含む吸水性樹脂粉末の製造方法。 - 上記粒状乾燥物を冷却する冷却工程をさらに有する、請求項47又は48に記載の製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020197016864A KR102560354B1 (ko) | 2016-11-16 | 2017-11-16 | 흡수성 수지 분말의 제조 방법, 그리고 입자상 함수 겔의 건조 장치 및 건조 방법 |
| CN201780070715.6A CN109996833B (zh) | 2016-11-16 | 2017-11-16 | 吸水性树脂粉末的制造方法、以及颗粒状含水凝胶的干燥装置及干燥方法 |
| US16/349,758 US11766659B2 (en) | 2016-11-16 | 2017-11-16 | Method for producing water-absorbent resin powder, and drying device and drying method for particulate hydrous gel |
| JP2018551690A JP6800998B2 (ja) | 2016-11-16 | 2017-11-16 | 吸水性樹脂粉末の製造方法、並びに粒子状含水ゲルの乾燥装置及び乾燥方法 |
| EP17871961.3A EP3543279A4 (en) | 2016-11-16 | 2017-11-16 | PROCESS FOR PRODUCING WATER-ABSORBING RESIN POWDER, AND DEVICE AND PROCESS FOR DRYING PARTICULAR HYDRATED GEL |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016223654 | 2016-11-16 | ||
| JP2016-223654 | 2016-11-16 | ||
| JP2017-184283 | 2017-09-26 | ||
| JP2017184283 | 2017-09-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018092863A1 true WO2018092863A1 (ja) | 2018-05-24 |
Family
ID=62145568
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/041372 WO2018092864A1 (ja) | 2016-11-16 | 2017-11-16 | 吸水性樹脂粉末の製造方法及びその製造装置 |
| PCT/JP2017/041371 WO2018092863A1 (ja) | 2016-11-16 | 2017-11-16 | 吸水性樹脂粉末の製造方法、並びに粒子状含水ゲルの乾燥装置及び乾燥方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/041372 WO2018092864A1 (ja) | 2016-11-16 | 2017-11-16 | 吸水性樹脂粉末の製造方法及びその製造装置 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US11465126B2 (ja) |
| EP (2) | EP3543279A4 (ja) |
| JP (3) | JP6918407B2 (ja) |
| KR (2) | KR102560352B1 (ja) |
| CN (2) | CN109996835B (ja) |
| WO (2) | WO2018092864A1 (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019221154A1 (ja) * | 2018-05-16 | 2019-11-21 | 株式会社日本触媒 | 吸水性樹脂粒子の製造方法 |
| KR20200089641A (ko) * | 2019-01-17 | 2020-07-27 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| JP2020526633A (ja) * | 2017-07-12 | 2020-08-31 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 超吸収性ポリマー粒子の製造方法 |
| JPWO2020144948A1 (ja) * | 2019-01-11 | 2021-11-25 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| EP3795613A4 (en) * | 2018-05-16 | 2022-03-23 | Nippon Shokubai Co., Ltd. | METHOD FOR PRODUCING A WATER-ABSORBENT RESIN |
| JP2022533035A (ja) * | 2019-12-20 | 2022-07-21 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| JP2022533561A (ja) * | 2019-12-20 | 2022-07-25 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| WO2022163849A1 (ja) | 2021-01-29 | 2022-08-04 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2022181771A1 (ja) | 2021-02-26 | 2022-09-01 | 株式会社日本触媒 | 粒子状吸水剤、該吸水剤を含む吸収体及び該吸収体を用いた吸収性物品 |
| WO2024009541A1 (ja) * | 2022-07-07 | 2024-01-11 | Sdpグローバル株式会社 | 吸水性樹脂組成物の製造方法 |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018092864A1 (ja) * | 2016-11-16 | 2018-05-24 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法及びその製造装置 |
| WO2020059762A1 (ja) * | 2018-09-18 | 2020-03-26 | 株式会社日本触媒 | 粒子状吸水剤の製造方法及び粒子状吸水剤 |
| KR102566284B1 (ko) * | 2018-11-14 | 2023-08-10 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| JP6754460B2 (ja) * | 2018-12-12 | 2020-09-09 | 住友精化株式会社 | 吸水性樹脂粒子 |
| JP6681492B1 (ja) * | 2018-12-12 | 2020-04-15 | 住友精化株式会社 | 吸水性樹脂粒子 |
| US20220023485A1 (en) * | 2018-12-12 | 2022-01-27 | Sumitomo Seika Chemicals Co., Ltd. | Water-absorbent resin particles |
| CA3123889A1 (en) * | 2018-12-20 | 2020-06-25 | Pelleton Global Renewables Ltd. | Pellet processing drum |
| JPWO2021049467A1 (ja) * | 2019-09-09 | 2021-03-18 | ||
| WO2021075921A1 (ko) * | 2019-10-18 | 2021-04-22 | 주식회사 엘지화학 | 펌프를 이용한 고흡수성 수지 함수겔 세절장치 |
| US11248841B2 (en) * | 2019-12-18 | 2022-02-15 | Dari-Tech, Inc. | Coand{hacek over (a)}-effect vegetable material dryer |
| US20220242990A1 (en) * | 2020-01-20 | 2022-08-04 | Lg Chem, Ltd. | Preparation method of super absorbent polymer |
| CN112185642A (zh) * | 2020-09-23 | 2021-01-05 | 江西艾特磁材有限公司 | 一种球磨改性溶胶-凝胶包覆磁粉芯的方法 |
| CN112595033B (zh) * | 2020-12-10 | 2022-11-08 | 万华化学集团股份有限公司 | 一种颜色稳定的聚碳酸酯粉末的制备方法 |
| CN113148464B (zh) * | 2021-02-09 | 2023-03-28 | 运易通科技有限公司 | 一种具有循环再生硅胶干燥剂结构的海运集装箱 |
| WO2022239723A1 (ja) * | 2021-05-12 | 2022-11-17 | 株式会社日本触媒 | ポリ(メタ)アクリル酸(塩)系吸水性樹脂、及び吸収体 |
| JP7100742B1 (ja) | 2021-05-12 | 2022-07-13 | 株式会社日本触媒 | ポリ(メタ)アクリル酸(塩)系吸水性樹脂、及び吸収体 |
| WO2022265472A1 (ko) * | 2021-06-18 | 2022-12-22 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 고흡수성 수지 |
| WO2022265471A1 (ko) * | 2021-06-18 | 2022-12-22 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 고흡수성 수지 |
| WO2022265459A1 (ko) * | 2021-06-18 | 2022-12-22 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 고흡수성 수지 |
| JP2023547816A (ja) * | 2021-06-18 | 2023-11-14 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| BR112023025097A2 (pt) * | 2021-06-18 | 2024-02-20 | Lg Chem Ltd | Método de preparação de polímero superabsorvente e polímero superabsorvente |
| WO2022265475A1 (ko) * | 2021-06-18 | 2022-12-22 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 고흡수성 수지 |
| JP2023548164A (ja) * | 2021-06-18 | 2023-11-15 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| WO2023075482A1 (ko) * | 2021-10-29 | 2023-05-04 | 주식회사 엘지화학 | 고흡수성 수지 조성물 및 이의 제조 방법 |
| CN114307847B (zh) * | 2022-01-07 | 2022-09-20 | 武汉理工大学 | 一种造粒单元、级配造粒设备及其使用方法 |
| WO2023190494A1 (ja) * | 2022-03-30 | 2023-10-05 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法 |
| CN114440581B (zh) * | 2022-04-11 | 2022-07-15 | 东营曜康医药科技有限公司 | 一种4-甲基噻唑-5-甲醛生产用双锥烘干设备 |
| CN115092971B (zh) * | 2022-07-07 | 2023-03-21 | 科立鑫(珠海)新能源有限公司 | 一种高纯硫酸钴晶体的生产工艺 |
Citations (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4497930A (en) | 1982-09-02 | 1985-02-05 | Kao Corporation | Process for producing highly water absorptive polymer |
| EP0289333A2 (en) | 1987-04-28 | 1988-11-02 | Yamazaki Mazak Corporation | A complex machining machine tool |
| US4893999A (en) | 1985-12-18 | 1990-01-16 | Chemische Fabrik Stockhausen Gmbh | Apparatus for the continuous production of polymers and copolymers of water-soluble monomers |
| US5005771A (en) | 1989-01-30 | 1991-04-09 | Chemie Linz Gesellschaft M.B.H. | Process for the continuous drying of hydrophilic polymer gels |
| EP0574248A2 (en) | 1992-06-10 | 1993-12-15 | Nippon Shokubai Co., Ltd. | Method for production of absorbent resin and absorbent |
| US5380808A (en) | 1990-07-17 | 1995-01-10 | Sanyo Chemical Industries, Ltd. | Process for producing water-absorbing resins |
| JPH08134134A (ja) | 1994-11-09 | 1996-05-28 | Nippon Shokubai Co Ltd | 粒子状含水ゲル重合体の乾燥方法 |
| JPH11511183A (ja) * | 1995-08-09 | 1999-09-28 | シュトックハウゼン・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング・ウント・コムパニー・カーゲー | 水および水性液体用吸収剤並びにそれらの製造方法および用途 |
| EP0948997A2 (en) | 1998-04-07 | 1999-10-13 | Nippon Shokubai Co., Ltd. | Manufacturing method of absorbent resin |
| US6241928B1 (en) | 1998-04-28 | 2001-06-05 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| EP1130045A2 (en) | 2000-02-29 | 2001-09-05 | Nippon Shokubai Co., Ltd. | Water-absorbent resin powder and production process therefor |
| JP2003012812A (ja) | 2001-06-27 | 2003-01-15 | San-Dia Polymer Ltd | 吸水性樹脂の製造法 |
| EP1367081A1 (en) | 2002-05-30 | 2003-12-03 | Nippon Shokubai Co., Ltd. | Production process for particulate water-absorbent resin |
| US6710141B1 (en) | 1999-11-20 | 2004-03-23 | Basf Aktiengesellschaft | Method for continuously producing cross-linked fine-particle geleous polymerizates |
| JP2005247931A (ja) | 2004-03-02 | 2005-09-15 | San-Dia Polymer Ltd | 吸水性樹脂粒子の製造方法 |
| US20050215734A1 (en) | 2004-03-24 | 2005-09-29 | Yorimichi Dairoku | Method for continuous production of water-absorbent resin |
| US6987151B2 (en) | 2001-09-12 | 2006-01-17 | Dow Global Technologies Inc. | Continuous polymerization process for the manufacture of superabsorbent polymers |
| WO2006100300A1 (de) | 2005-03-24 | 2006-09-28 | Basf Aktiengesellschaft | Verfahren zur herstellung wasserabsorbierender polymere |
| WO2006103227A1 (de) | 2005-03-30 | 2006-10-05 | Basf Aktiengesellschaft | Verfahren zur herstellung wasserabsorbierender polymerpartikel |
| JP2007071415A (ja) | 2005-09-05 | 2007-03-22 | San-Dia Polymer Ltd | 吸収剤の製造法 |
| WO2007057350A1 (de) | 2005-11-16 | 2007-05-24 | Basf Se | Verfahren zur herstellung wasserabsorbierender polymerpartikel |
| EP1800740A2 (en) | 2005-12-22 | 2007-06-27 | Nippon Shokubai Co.,Ltd. | Method for manufacturing water-absorbing resin via surface crosslinking |
| US7265190B2 (en) | 2002-11-07 | 2007-09-04 | Nippon Shokubai Co., Ltd. | Process and apparatus for production of water-absorbent resin |
| WO2007104673A2 (de) | 2006-03-14 | 2007-09-20 | Basf Se | Verfahren zur pneumatischen förderung wasserabsorbierender polymerpartikel |
| WO2008037675A1 (de) | 2006-09-25 | 2008-04-03 | Basf Se | Verfahren zum klassieren wasserabsorbierender polymerpartikel |
| WO2008037672A1 (de) | 2006-09-25 | 2008-04-03 | Basf Se | Verfahren zum klassieren wasserabsorbierender polymerpartikel |
| WO2009028568A1 (ja) | 2007-08-28 | 2009-03-05 | Nippon Shokubai Co., Ltd. | 吸水性樹脂の製造方法 |
| WO2009113679A1 (ja) | 2008-03-13 | 2009-09-17 | 株式会社日本触媒 | 吸水性樹脂を主成分とする粒子状吸水剤の製造方法 |
| US7638570B2 (en) | 2003-02-10 | 2009-12-29 | Nippon Shokubai Co., Ltd. | Water-absorbing agent |
| WO2011034147A1 (ja) | 2009-09-16 | 2011-03-24 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法 |
| WO2011090129A1 (ja) | 2010-01-20 | 2011-07-28 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2011104152A1 (de) | 2010-02-24 | 2011-09-01 | Basf Se | Verfahren zur herstellung wasserabsorbierender polymerpartikel |
| WO2011115216A1 (ja) | 2010-03-17 | 2011-09-22 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2011126079A1 (ja) | 2010-04-07 | 2011-10-13 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法及びポリアクリル酸(塩)系吸水性樹脂粉末 |
| JP2012041439A (ja) | 2010-08-19 | 2012-03-01 | San-Dia Polymer Ltd | 吸収性樹脂粒子及び吸収性物品 |
| WO2013104479A1 (en) | 2012-01-12 | 2013-07-18 | Evonik Degussa Gmbh | Process for the continuous preparation of water-absorbent polymers |
| WO2013146699A1 (ja) | 2012-03-29 | 2013-10-03 | 住友精化株式会社 | 重合反応器、および吸水性樹脂の製造方法 |
| WO2014033083A1 (en) | 2012-08-29 | 2014-03-06 | Basf Se | Process for producing water-absorbing polymer particles |
| WO2014044780A1 (en) | 2012-09-19 | 2014-03-27 | Basf Se | Process for producing water-absorbing polymer particles |
| WO2014141764A1 (ja) | 2013-03-11 | 2014-09-18 | 住友精化株式会社 | 吸水性樹脂の製造装置 |
| WO2014154522A1 (en) | 2013-03-28 | 2014-10-02 | Basf Se | Process for classifying water-absorbing polymer beads |
| JP2015512990A (ja) * | 2012-03-30 | 2015-04-30 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 逆スクリューヘリックスを備えたドラム型熱交換器中で熱表面後架橋する方法 |
| WO2015072536A1 (ja) | 2013-11-14 | 2015-05-21 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂の製造方法 |
| WO2015074966A1 (en) | 2013-11-22 | 2015-05-28 | Basf Se | Process for producing water-absorbing polymer particles |
| WO2015129917A1 (ja) | 2014-02-28 | 2015-09-03 | 株式会社日本触媒 | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 |
| WO2016128337A1 (de) | 2015-02-11 | 2016-08-18 | Basf Se | Verfahren zur trocknung von superabsorbern |
| JP2016216713A (ja) | 2015-05-14 | 2016-12-22 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2016204302A1 (ja) | 2015-06-19 | 2016-12-22 | 株式会社日本触媒 | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5140076A (en) * | 1990-04-02 | 1992-08-18 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Method of treating the surface of an absorbent resin |
| JPH0655070A (ja) * | 1992-03-31 | 1994-03-01 | Nippon Zeon Co Ltd | 吸水剤、吸水剤の製造方法および吸水剤を用いた水分を含有する粉粒体の水分低減方法 |
| JP3632803B2 (ja) * | 1995-11-02 | 2005-03-23 | 株式会社日本触媒 | 吸水性樹脂並びに吸収性材料およびその製造方法 |
| JP3942125B2 (ja) * | 1998-04-14 | 2007-07-11 | 日本合成化学工業株式会社 | エチレン−酢酸ビニル共重合体ケン化物の処理法 |
| JP4676625B2 (ja) * | 2000-02-29 | 2011-04-27 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法 |
| JP3456965B2 (ja) * | 2000-10-27 | 2003-10-14 | 川崎重工業株式会社 | 構造物検査方法および装置 |
| EP1882701B1 (en) * | 2005-05-16 | 2018-07-04 | Sumitomo Seika Chemicals Co., Ltd. | Process for production of water-absorbing resin particles, water-absorbing resin particles made by the process, and absorbents and absorbent articles made by using the particles |
| TWI383008B (zh) | 2005-08-17 | 2013-01-21 | Nippon Catalytic Chem Ind | 吸水性樹脂之製造方法及吸水性樹脂以及其利用 |
| JP5183897B2 (ja) * | 2005-08-17 | 2013-04-17 | 株式会社日本触媒 | 吸水性樹脂の製造方法および吸水性樹脂 |
| JP5528714B2 (ja) * | 2009-03-02 | 2014-06-25 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| EP2471845B1 (en) | 2009-08-27 | 2017-02-15 | Nippon Shokubai Co., Ltd. | Water-absorbing resin based on polyacrylic acid (salt) and process for producing same |
| JP5502656B2 (ja) | 2010-08-24 | 2014-05-28 | 月島機械株式会社 | 間接加熱型回転乾燥機 |
| CN102374757B (zh) | 2010-08-24 | 2015-05-20 | 月岛机械株式会社 | 间接加热型旋转干燥机 |
| WO2012124709A1 (ja) | 2011-03-14 | 2012-09-20 | Yamamoto Kazuhiro | 持ち時間管理システム |
| JP6092236B2 (ja) * | 2012-10-03 | 2017-03-08 | 株式会社日本触媒 | 吸水剤及びその製造方法 |
| KR20150113129A (ko) | 2013-01-30 | 2015-10-07 | 바스프 에스이 | 물 흡수성 폴리머 입자로부터의 잔류 모노머의 제거 방법 |
| JP5778831B1 (ja) * | 2014-03-31 | 2015-09-16 | 月島機械株式会社 | 被処理物の乾燥方法、および横型回転式乾燥機 |
| KR20150141425A (ko) | 2014-06-10 | 2015-12-18 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| JP5865471B1 (ja) | 2014-12-02 | 2016-02-17 | 月島機械株式会社 | 間接加熱管付回転乾燥機及び乾燥方法 |
| JP2016223654A (ja) | 2015-05-28 | 2016-12-28 | 東芝ライフスタイル株式会社 | 冷蔵庫 |
| EP3473664B1 (en) * | 2016-06-20 | 2023-02-22 | Nippon Shokubai Co., Ltd. | Method for producing water absorbent agent |
| WO2018092864A1 (ja) * | 2016-11-16 | 2018-05-24 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法及びその製造装置 |
-
2017
- 2017-11-16 WO PCT/JP2017/041372 patent/WO2018092864A1/ja unknown
- 2017-11-16 JP JP2017220588A patent/JP6918407B2/ja active Active
- 2017-11-16 US US16/349,709 patent/US11465126B2/en active Active
- 2017-11-16 JP JP2018551690A patent/JP6800998B2/ja active Active
- 2017-11-16 WO PCT/JP2017/041371 patent/WO2018092863A1/ja unknown
- 2017-11-16 US US16/349,758 patent/US11766659B2/en active Active
- 2017-11-16 JP JP2018551691A patent/JP6913107B2/ja active Active
- 2017-11-16 CN CN201780071062.3A patent/CN109996835B/zh active Active
- 2017-11-16 KR KR1020197016866A patent/KR102560352B1/ko active IP Right Grant
- 2017-11-16 EP EP17871961.3A patent/EP3543279A4/en active Pending
- 2017-11-16 CN CN201780070715.6A patent/CN109996833B/zh active Active
- 2017-11-16 EP EP17870665.1A patent/EP3543280A4/en active Pending
- 2017-11-16 KR KR1020197016864A patent/KR102560354B1/ko active IP Right Grant
Patent Citations (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4497930A (en) | 1982-09-02 | 1985-02-05 | Kao Corporation | Process for producing highly water absorptive polymer |
| US4893999A (en) | 1985-12-18 | 1990-01-16 | Chemische Fabrik Stockhausen Gmbh | Apparatus for the continuous production of polymers and copolymers of water-soluble monomers |
| EP0289333A2 (en) | 1987-04-28 | 1988-11-02 | Yamazaki Mazak Corporation | A complex machining machine tool |
| US5005771A (en) | 1989-01-30 | 1991-04-09 | Chemie Linz Gesellschaft M.B.H. | Process for the continuous drying of hydrophilic polymer gels |
| US5380808A (en) | 1990-07-17 | 1995-01-10 | Sanyo Chemical Industries, Ltd. | Process for producing water-absorbing resins |
| EP0574248A2 (en) | 1992-06-10 | 1993-12-15 | Nippon Shokubai Co., Ltd. | Method for production of absorbent resin and absorbent |
| JPH08134134A (ja) | 1994-11-09 | 1996-05-28 | Nippon Shokubai Co Ltd | 粒子状含水ゲル重合体の乾燥方法 |
| JPH11511183A (ja) * | 1995-08-09 | 1999-09-28 | シュトックハウゼン・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング・ウント・コムパニー・カーゲー | 水および水性液体用吸収剤並びにそれらの製造方法および用途 |
| EP0948997A2 (en) | 1998-04-07 | 1999-10-13 | Nippon Shokubai Co., Ltd. | Manufacturing method of absorbent resin |
| JPH11292919A (ja) * | 1998-04-07 | 1999-10-26 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造方法 |
| US6241928B1 (en) | 1998-04-28 | 2001-06-05 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| US6710141B1 (en) | 1999-11-20 | 2004-03-23 | Basf Aktiengesellschaft | Method for continuously producing cross-linked fine-particle geleous polymerizates |
| EP1130045A2 (en) | 2000-02-29 | 2001-09-05 | Nippon Shokubai Co., Ltd. | Water-absorbent resin powder and production process therefor |
| JP2003012812A (ja) | 2001-06-27 | 2003-01-15 | San-Dia Polymer Ltd | 吸水性樹脂の製造法 |
| US6987151B2 (en) | 2001-09-12 | 2006-01-17 | Dow Global Technologies Inc. | Continuous polymerization process for the manufacture of superabsorbent polymers |
| EP1367081A1 (en) | 2002-05-30 | 2003-12-03 | Nippon Shokubai Co., Ltd. | Production process for particulate water-absorbent resin |
| US7265190B2 (en) | 2002-11-07 | 2007-09-04 | Nippon Shokubai Co., Ltd. | Process and apparatus for production of water-absorbent resin |
| US7638570B2 (en) | 2003-02-10 | 2009-12-29 | Nippon Shokubai Co., Ltd. | Water-absorbing agent |
| JP2005247931A (ja) | 2004-03-02 | 2005-09-15 | San-Dia Polymer Ltd | 吸水性樹脂粒子の製造方法 |
| US20050215734A1 (en) | 2004-03-24 | 2005-09-29 | Yorimichi Dairoku | Method for continuous production of water-absorbent resin |
| WO2006100300A1 (de) | 2005-03-24 | 2006-09-28 | Basf Aktiengesellschaft | Verfahren zur herstellung wasserabsorbierender polymere |
| WO2006103227A1 (de) | 2005-03-30 | 2006-10-05 | Basf Aktiengesellschaft | Verfahren zur herstellung wasserabsorbierender polymerpartikel |
| JP2007071415A (ja) | 2005-09-05 | 2007-03-22 | San-Dia Polymer Ltd | 吸収剤の製造法 |
| WO2007057350A1 (de) | 2005-11-16 | 2007-05-24 | Basf Se | Verfahren zur herstellung wasserabsorbierender polymerpartikel |
| EP1800740A2 (en) | 2005-12-22 | 2007-06-27 | Nippon Shokubai Co.,Ltd. | Method for manufacturing water-absorbing resin via surface crosslinking |
| WO2007104673A2 (de) | 2006-03-14 | 2007-09-20 | Basf Se | Verfahren zur pneumatischen förderung wasserabsorbierender polymerpartikel |
| WO2008037672A1 (de) | 2006-09-25 | 2008-04-03 | Basf Se | Verfahren zum klassieren wasserabsorbierender polymerpartikel |
| WO2008037675A1 (de) | 2006-09-25 | 2008-04-03 | Basf Se | Verfahren zum klassieren wasserabsorbierender polymerpartikel |
| WO2009028568A1 (ja) | 2007-08-28 | 2009-03-05 | Nippon Shokubai Co., Ltd. | 吸水性樹脂の製造方法 |
| WO2009113679A1 (ja) | 2008-03-13 | 2009-09-17 | 株式会社日本触媒 | 吸水性樹脂を主成分とする粒子状吸水剤の製造方法 |
| WO2009113678A1 (ja) | 2008-03-13 | 2009-09-17 | 株式会社日本触媒 | 吸水性樹脂を主成分とする粒子状吸水剤の製造方法 |
| WO2009113673A1 (ja) | 2008-03-13 | 2009-09-17 | 株式会社日本触媒 | 吸水性樹脂を主成分とする粒子状吸水剤の製造方法 |
| WO2011034147A1 (ja) | 2009-09-16 | 2011-03-24 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法 |
| WO2011034146A1 (ja) | 2009-09-16 | 2011-03-24 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法 |
| WO2011090129A1 (ja) | 2010-01-20 | 2011-07-28 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2011104152A1 (de) | 2010-02-24 | 2011-09-01 | Basf Se | Verfahren zur herstellung wasserabsorbierender polymerpartikel |
| WO2011115216A1 (ja) | 2010-03-17 | 2011-09-22 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2011115221A1 (ja) | 2010-03-17 | 2011-09-22 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2011126079A1 (ja) | 2010-04-07 | 2011-10-13 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法及びポリアクリル酸(塩)系吸水性樹脂粉末 |
| JP2012041439A (ja) | 2010-08-19 | 2012-03-01 | San-Dia Polymer Ltd | 吸収性樹脂粒子及び吸収性物品 |
| WO2013104479A1 (en) | 2012-01-12 | 2013-07-18 | Evonik Degussa Gmbh | Process for the continuous preparation of water-absorbent polymers |
| WO2013146699A1 (ja) | 2012-03-29 | 2013-10-03 | 住友精化株式会社 | 重合反応器、および吸水性樹脂の製造方法 |
| JP2015512990A (ja) * | 2012-03-30 | 2015-04-30 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 逆スクリューヘリックスを備えたドラム型熱交換器中で熱表面後架橋する方法 |
| WO2014033083A1 (en) | 2012-08-29 | 2014-03-06 | Basf Se | Process for producing water-absorbing polymer particles |
| WO2014044780A1 (en) | 2012-09-19 | 2014-03-27 | Basf Se | Process for producing water-absorbing polymer particles |
| WO2014141764A1 (ja) | 2013-03-11 | 2014-09-18 | 住友精化株式会社 | 吸水性樹脂の製造装置 |
| WO2014154522A1 (en) | 2013-03-28 | 2014-10-02 | Basf Se | Process for classifying water-absorbing polymer beads |
| WO2015072536A1 (ja) | 2013-11-14 | 2015-05-21 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂の製造方法 |
| WO2015074966A1 (en) | 2013-11-22 | 2015-05-28 | Basf Se | Process for producing water-absorbing polymer particles |
| WO2015129917A1 (ja) | 2014-02-28 | 2015-09-03 | 株式会社日本触媒 | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 |
| WO2016128337A1 (de) | 2015-02-11 | 2016-08-18 | Basf Se | Verfahren zur trocknung von superabsorbern |
| JP2016216713A (ja) | 2015-05-14 | 2016-12-22 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2016204302A1 (ja) | 2015-06-19 | 2016-12-22 | 株式会社日本触媒 | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| MODERN SUPERABSORBENTS POLYMER CHEMISTRY, 1998, pages 69 - 103 |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7229987B2 (ja) | 2017-07-12 | 2023-02-28 | ビーエーエスエフ ソシエタス・ヨーロピア | 超吸収性ポリマー粒子の製造方法 |
| JP2020526633A (ja) * | 2017-07-12 | 2020-08-31 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 超吸収性ポリマー粒子の製造方法 |
| WO2019221154A1 (ja) * | 2018-05-16 | 2019-11-21 | 株式会社日本触媒 | 吸水性樹脂粒子の製造方法 |
| EP3795613A4 (en) * | 2018-05-16 | 2022-03-23 | Nippon Shokubai Co., Ltd. | METHOD FOR PRODUCING A WATER-ABSORBENT RESIN |
| JPWO2020144948A1 (ja) * | 2019-01-11 | 2021-11-25 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| JP7165753B2 (ja) | 2019-01-11 | 2022-11-04 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| KR102457690B1 (ko) | 2019-01-17 | 2022-10-21 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| KR20200089641A (ko) * | 2019-01-17 | 2020-07-27 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| JP2022533561A (ja) * | 2019-12-20 | 2022-07-25 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| JP2022533035A (ja) * | 2019-12-20 | 2022-07-21 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| JP7321628B2 (ja) | 2019-12-20 | 2023-08-07 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| JP7321627B2 (ja) | 2019-12-20 | 2023-08-07 | エルジー・ケム・リミテッド | 高吸水性樹脂組成物の製造方法 |
| WO2022163849A1 (ja) | 2021-01-29 | 2022-08-04 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| WO2022181771A1 (ja) | 2021-02-26 | 2022-09-01 | 株式会社日本触媒 | 粒子状吸水剤、該吸水剤を含む吸収体及び該吸収体を用いた吸収性物品 |
| WO2024009541A1 (ja) * | 2022-07-07 | 2024-01-11 | Sdpグローバル株式会社 | 吸水性樹脂組成物の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20190077541A (ko) | 2019-07-03 |
| US11465126B2 (en) | 2022-10-11 |
| KR102560352B1 (ko) | 2023-07-28 |
| EP3543280A4 (en) | 2020-09-02 |
| JP6918407B2 (ja) | 2021-08-11 |
| EP3543280A1 (en) | 2019-09-25 |
| US20190329220A1 (en) | 2019-10-31 |
| JPWO2018092863A1 (ja) | 2019-10-17 |
| CN109996833A (zh) | 2019-07-09 |
| JP6800998B2 (ja) | 2020-12-16 |
| KR102560354B1 (ko) | 2023-07-28 |
| CN109996835A (zh) | 2019-07-09 |
| US20190329219A1 (en) | 2019-10-31 |
| CN109996835B (zh) | 2022-07-29 |
| EP3543279A4 (en) | 2020-08-19 |
| US11766659B2 (en) | 2023-09-26 |
| CN109996833B (zh) | 2022-02-11 |
| KR20190077540A (ko) | 2019-07-03 |
| WO2018092864A1 (ja) | 2018-05-24 |
| EP3543279A1 (en) | 2019-09-25 |
| JP6913107B2 (ja) | 2021-08-04 |
| JPWO2018092864A1 (ja) | 2019-10-17 |
| JP2019052285A (ja) | 2019-04-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2018092863A1 (ja) | 吸水性樹脂粉末の製造方法、並びに粒子状含水ゲルの乾燥装置及び乾燥方法 | |
| JP7083020B2 (ja) | 吸水性樹脂粉末、及びその製造方法 | |
| JP7174756B2 (ja) | 吸水性樹脂粒子の製造方法 | |
| JP5631866B2 (ja) | 粒子状吸水性樹脂の製造方法 | |
| JP5635397B2 (ja) | 吸水性樹脂を主成分とする粒子状吸水剤の製造方法 | |
| WO2011034147A1 (ja) | 吸水性樹脂粉末の製造方法 | |
| WO2022065365A1 (ja) | 吸水性樹脂粉末の製造方法 | |
| JP6722507B2 (ja) | 吸水性樹脂の製造方法 | |
| WO2023190494A1 (ja) | 吸水性樹脂粉末の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17871961 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2018551690 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20197016864 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2017871961 Country of ref document: EP Effective date: 20190617 |