WO2014097992A1 - 感光性樹脂組成物、耐熱性樹脂膜の製造方法および表示装置 - Google Patents
感光性樹脂組成物、耐熱性樹脂膜の製造方法および表示装置 Download PDFInfo
- Publication number
- WO2014097992A1 WO2014097992A1 PCT/JP2013/083491 JP2013083491W WO2014097992A1 WO 2014097992 A1 WO2014097992 A1 WO 2014097992A1 JP 2013083491 W JP2013083491 W JP 2013083491W WO 2014097992 A1 WO2014097992 A1 WO 2014097992A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mol
- photosensitive resin
- resin composition
- resin
- group
- Prior art date
Links
Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/027—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds
- G03F7/032—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds with binders
- G03F7/037—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds with binders the binders being polyamides or polyimides
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/038—Macromolecular compounds which are rendered insoluble or differentially wettable
- G03F7/0387—Polyamides or polyimides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/1039—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors comprising halogen-containing substituents
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D179/00—Coating compositions based on macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen, with or without oxygen, or carbon only, not provided for in groups C09D161/00 - C09D177/00
- C09D179/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
- C09D179/08—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/0046—Photosensitive materials with perfluoro compounds, e.g. for dry lithography
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/022—Quinonediazides
- G03F7/023—Macromolecular quinonediazides; Macromolecular additives, e.g. binders
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/022—Quinonediazides
- G03F7/023—Macromolecular quinonediazides; Macromolecular additives, e.g. binders
- G03F7/0233—Macromolecular quinonediazides; Macromolecular additives, e.g. binders characterised by the polymeric binders or the macromolecular additives other than the macromolecular quinonediazides
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/26—Processing photosensitive materials; Apparatus therefor
- G03F7/40—Treatment after imagewise removal, e.g. baking
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/80—Constructional details
- H10K50/87—Arrangements for heating or cooling
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K59/00—Integrated devices, or assemblies of multiple devices, comprising at least one organic light-emitting element covered by group H10K50/00
- H10K59/10—OLED displays
- H10K59/12—Active-matrix OLED [AMOLED] displays
- H10K59/124—Insulating layers formed between TFT elements and OLED elements
Definitions
- the present invention relates to a photosensitive resin composition containing a resin having a specific structure as a main component. More specifically, a surface protective film or an interlayer insulating film of a semiconductor element, an insulating film of an organic electroluminescence (hereinafter referred to as EL) element, a driving thin film transistor (Thin Film Transistor: hereinafter referred to as a TFT) using an organic EL element.
- a photosensitive resin composition suitable for applications such as a flattening film for a substrate, a wiring protective insulating film for a circuit board, an on-chip microlens for a solid-state image sensor, and a flattening film for various displays and solid-state image sensors.
- Polyimide is widely used for surface protection films, interlayer insulation films, planarization films and the like of semiconductor elements, and recently, for example, is used for insulation films of organic EL elements and planarization films of TFT substrates.
- the slit coating is a coating method using a slit nozzle, and unlike the conventional spin coating, it is not necessary to rotate the substrate. Therefore, it is widely adopted from the viewpoint of reducing the amount of resin composition used and process safety.
- slit coating since the coating film discharged from the slit nozzle contains a large amount of solvent, it is common to quickly dry under reduced pressure after coating to remove the solvent, and then heat dry using a hot plate or the like. is there.
- the film thickness is determined by the discharge amount from the slit nozzle and the solid content concentration in the resin composition as the coating liquid. Therefore, in order to form a thick film, it is necessary to increase the discharge amount or increase the solid content concentration in the resin composition. However, if the discharge amount is increased too much, the liquid level moves during the conveyance of the substrate, so that the film thickness uniformity deteriorates. On the other hand, in the resin composition using a polyimide or a polyimide precursor, there is a problem that if the solid content concentration in the resin composition is increased, the viscosity becomes too high.
- the viscosity of the resin composition can be reduced by using a good solvent for the resin or a solvent having a low viscosity of the solvent itself.
- Many of polyimides and polyimide precursors have low solubility in various solvents because of their rigid structures. So far, polyimides having improved solubility in organic solvents (for example, see Patent Document 1) and polyimide precursors (for example, see Patent Documents 2 to 3) have been proposed. However, these resins also have insufficient solubility in organic solvents. Moreover, with these resins, a resin composition having a viscosity suitable for slit coating could not be obtained.
- the present invention includes a polyimide precursor that has high solubility in an organic solvent, and has a low viscosity and excellent coatability, thereby forming a uniform film thickness and having good pattern processability. It aims at providing a conductive resin composition.
- the present invention provides (a1) an aromatic amide resin having an amide group, a trifluoromethyl group, and an aromatic ring, soluble in propylene glycol monomethyl ether acetate, (b) a photosensitizer, and (c) a photosensitivity containing a solvent.
- a photosensitive resin composition having a solid content of 20% by weight and a viscosity of 1 to 15 cp at 25 ° C., or (a2) an amide group, an amic acid ester group, a trifluoromethyl group, and an aromatic ring.
- a photosensitive resin composition comprising an aromatic amide resin soluble in propylene glycol monomethyl ether acetate, (b) a photosensitizer, and (c) a solvent, having a solid content concentration of 20% by weight and a viscosity at 25 ° C. of 1
- R 1 to R 3 may be mixed with different groups in a plurality of repeating units.
- R 1 is a tetravalent organic group
- R 1 in all repeating units 95 to 100 mol% is a group represented by the following formula (2):
- R 2 is a divalent organic group, and 50 to 99 mol% of R 2 in all repeating units is represented by the following formula (3).
- 1 to 50 mol% is a group represented by the following formula (4):
- the method for producing a heat resistant resin film of the present invention includes a step of applying the photosensitive resin composition of the present invention to a substrate to form a photosensitive resin film, a step of drying the photosensitive resin film, and a dried photosensitive film.
- a method for producing a heat-resistant resin film comprising: a step of exposing a photosensitive resin film; a step of developing the exposed photosensitive resin film; and a step of heat-treating the developed photosensitive resin film, wherein the photosensitive resin of the present invention is used.
- a coating apparatus in which a photosensitive resin composition other than the composition has been fed, and the coating apparatus is used without cleaning the liquid feeding path of the coating apparatus. It is a manufacturing method of the heat resistant resin film which apply
- a photosensitive resin composition comprising a polyimide precursor having high solubility in an organic solvent, having a low viscosity and excellent coatability, thereby forming a uniform film thickness and having good pattern processability.
- the photosensitive resin composition of the present invention comprises (a1) an aromatic amide resin soluble in propylene glycol monomethyl ether acetate having an amide group, a trifluoromethyl group, and an aromatic ring, (b) a photosensitive agent, and (c) a solvent.
- a photosensitive resin composition comprising an aromatic amide resin soluble in propylene glycol monomethyl ether acetate, (b) a photosensitizer, and (c) a solvent, having a solid content concentration of 20% by weight and a viscosity at 25 ° C. of 1
- a resin having a structure represented by the general formula (1) as a main repeating unit (b) a photosensitive agent, and (c) a solvent. Containing.
- (A1) Resin, (a2) Resin, and (a) Resin As an embodiment of the photosensitive resin composition of the present invention, (a1) propylene glycol monomethyl ether acetate having an amide group, a trifluoromethyl group, and an aromatic ring may be used. Contains a soluble aromatic amide resin.
- the amide group is amide (CONH), amide (-CONHCOOH) acid, or hydroxyamide (CONHOH).
- a plurality of different amide groups may be contained.
- the amic acid ester group is CONHCOOR (R is an organic group).
- aromatic amide resins examples include resins having a structure such as polyhydroxyamide, polyaminoamide, polyamideimide, which is a polybenzoxazole precursor, polyamic acid which is a polyimide precursor, and polyamic acid ester.
- a resin having a structure of hydroxyamide or polyamic acid ester is preferably used. More preferably, a resin having a structural unit represented by the general formula (1) is used.
- resin which has the structure represented by General formula (1) as a main repeating unit is contained.
- R 1 to R 3 in the plurality of repeating units may be mixed with different groups, and have at least two types of R 2 as will be described later. Therefore, the resin in (a) is different in two or more types.
- (a) resin may mean (a1) resin, (a2) resin, and (a) resin.
- the resin having the structure represented by the general formula (1) as a main repeating unit is a polyimide precursor that is closed by heating and becomes a polyimide having excellent heat resistance and solvent resistance.
- the polyimide precursor can be obtained by reacting a tetracarboxylic acid as a monomer component and a derivative thereof (hereinafter referred to as an acid component) with a diamine compound (hereinafter referred to as a diamine component).
- R 1 to R 3 may be mixed with different groups in a plurality of repeating units.
- R 1 is a tetravalent organic group, and 95 to 100 mol% of R 1 in all repeating units is a group represented by the following formula (2).
- R 2 is a divalent organic group, and 50 to 99 mol% of R 2 in all repeating units is a group represented by the following formula (3), and 1 to 50 mol% is represented by the following formula (4).
- R 1 represents a group derived from an acid component, and the acid component in which R 1 is a group represented by the above formula (2) includes 2,2-bis (3,4-di ()). Carboxyphenyl) hexafluoropropane and 2,2-bis (2,3-dicarboxyphenyl) hexafluoropropane.
- R 1 has 95 mol% or more of the group represented by the above formula (2), solubility in an organic solvent can be improved and the viscosity of the resin composition can be reduced.
- R 1 in the general formula (1) may be a tetravalent organic group having 2 or more carbon atoms having 95 to 100 mol% of the group represented by the formula (2), and the remaining R 1 is As a case where the group represented by the formula (2) is not included, first, a group other than the formula (2) may be included. In this case, groups other than formula (2) are not particularly limited.
- the acid component in this case include pyromellitic acid, 3,3 ′, 4,4′-biphenyltetracarboxylic acid, 2,3,3 ′, 4′-biphenyltetracarboxylic acid, 2,2 ′, 3,3′-biphenyltetracarboxylic acid, 3,3 ′, 4,4′-benzophenone tetracarboxylic acid, 2,2 ′, 3,3′-benzophenone tetracarboxylic acid, 1,1-bis (3,4 Dicarboxyphenyl) ethane, 1,1-bis (2,3-dicarboxyphenyl) ethane, bis (3,4-dicarboxyphenyl) methane, bis (2,3-dicarboxyphenyl) methane, bis (3 4-dicarboxyphenyl) sulfone, bis (3,4-dicarboxyphenyl) ether, 1,2,5,6-naphthalenetetracarboxylic
- dicarboxylic acids include terephthalic acid, isophthalic acid, diphenyl ether dicarboxylic acid, bis (carboxyphenyl) hexafluoropropane, biphenyl dicarboxylic acid, benzophenone dicarboxylic acid, and triphenyl dicarboxylic acid.
- tricarboxylic acid examples include tricarboxylic acid. Examples include merit acid, trimesic acid, diphenyl ether tricarboxylic acid, and biphenyl tricarboxylic acid.
- Preferred examples of the acid component when R 1 does not have the group represented by the above formula (2) include silicon atoms such as dimethylsilane diphthalic acid and 1,3-bis (phthalic acid) tetramethyldisiloxane Tetracarboxylic acid can be used, and by using these, the adhesion to the substrate, the oxygen plasma used for cleaning, and the resistance to UV ozone treatment can be enhanced.
- R 2 in the general formula (1) represents a group derived from a diamine component, and when R 2 has a group represented by the formula (3) in an amount of 50 mol% or more, the resulting resin has solubility in an aqueous alkali solution. Is maintained in an appropriate range, and a photosensitive resin composition having good pattern processability can be obtained.
- the group represented by the formula (3) is preferably 55 mol% or more, more preferably 60 mol% or more.
- R 2 in the general formula (1) has 1 mol% or more, preferably 5 mol% or more, more preferably 10 mol% or more of the group represented by the general formula (4), so that it can be dissolved in an organic solvent. Improves. More preferably, it is 15 mol% or more, More preferably, it is 20 mol% or more. On the other hand, the solubility with respect to aqueous alkali solution is maintained appropriately by setting it as 50 mol% or less. Preferably it is 45 mol% or less, More preferably, it is 40 mol% or less.
- the concentration of phenolic hydroxyl group in the resin becomes high, so that the solubility of the resulting resin in an alkaline aqueous solution becomes too high, and the photosensitive resin composition.
- the amount of development film reduction becomes larger.
- the amount of development film reduction is large, the in-plane film thickness uniformity deteriorates and the development margin becomes narrow, which is not preferable.
- the imidization rate is increased, the transmittance of the film at the exposure wavelength is deteriorated, and the sensitivity is also decreased.
- the proportion having a group R 2 in proportion to the general formula (1) having a group R 2 in the general formula (1) is represented by the formula (3) is represented by the general formula (4) Is not more than 100 mol%.
- Examples of the diamine component in which R 2 is represented by the general formula (4) include 2,2-bis (3-amino-4-hydroxyphenyl) hexafluoropropane, 2,2-bis (3-amino-4-methylphenyl) Examples include hexafluoropropane and 2,2-bis (4-aminophenyl) hexafluoropropane. Of these, 2,2-bis (3-amino-4-hydroxyphenyl) hexafluoropropane is particularly preferred from the viewpoint of solubility of the resulting resin in an aqueous alkali solution.
- R 2 in the general formula (1) has a group represented by the formula (3) in an amount of 50 to 99 mol%, preferably 50 to 90 mol%, and a group represented by the general formula (4).
- the divalent organic group having 2 or more carbon atoms and having 1 to 50 mol%, preferably 10 to 50 mol%, may be used, and other groups are not particularly limited.
- diamine compounds can be used as they are or as corresponding diisocyanate compounds or trimethylsilylated diamines.
- R 3 is an organic group having 1 to 20 carbon atoms.
- M is preferably 1 or more from the viewpoint of the stability of the photosensitive resin composition using the obtained resin and appropriate solubility in an alkaline aqueous solution.
- the resin having the structure represented by the general formula (1) as a main repeating unit used in the photosensitive resin composition of the present invention has at least one end of a molecular chain sealed with a monoamine or acid anhydride. Preferably it is.
- a terminal sealing agent By using a terminal sealing agent, it becomes easy to adjust the photosensitive resin composition using the obtained resin to an appropriate viscosity. Moreover, it has the effect of suppressing the hydrolysis of the resin by the acid terminal, or suppressing the deterioration of the quinonediazide compound, which is a photosensitizer, by the amine terminal when a positive photosensitive resin composition is formed.
- the monoamine used for terminal blocker is not particularly limited, a compound having a group represented by the following general formula (5) is preferable.
- R 5 represents a saturated hydrocarbon group having 1 to 6 carbon atoms, and r represents 0 or 1.
- a and B may be the same or different and each represents a hydroxyl group, a carboxyl group or a sulfonic acid group.
- s and t each represent 0 or 1, and s + t ⁇ 1 from the viewpoint of the solubility of the resulting resin in an alkaline aqueous solution.
- Preferred examples of the monoamine having the group represented by the general formula (5) include 2-aminophenol, 3-aminophenol, 2-amino-m-cresol, 2-amino-p-cresol, 3-amino-o -Cresol, 4-amino-o-cresol, 4-amino-m-cresol, 5-amino-o-cresol, 6-amino-m-cresol, 4-amino-2,3-xylenol, 4-amino-3 , 5-xylenol, 6-amino-2,4-xylenol, 2-amino-4-ethylphenol, 3-amino-4-ethylphenol, 2-amino-4-tert-butylphenol, 2-amino-4-phenyl Phenol, 4-amino-2,6-diphenylphenol, 4-aminosalicylic acid, 5-aminosalicylic acid, 6-aminosalicylic acid, -Aminobenzoic acid, 3-aminobenzoic
- the introduction ratio of the monoamine used as the end-capping agent is preferably 10 to 100 mol%, more preferably 40 to 80 mol% with respect to 100 mol% of tetracarboxylic acid which is a monomer component of the resin.
- the solubility of the resulting resin in an organic solvent is improved, and the viscosity when a photosensitive resin composition is made using the obtained resin is appropriately adjusted. can do.
- it is preferably 100 mol% or less, more preferably 80 mol% or less, and even more preferably 70 mol% or less.
- the acid anhydride used for the end-capping agent is not particularly limited, but an acid anhydride having a cyclic structure or an acid anhydride having a crosslinkable group is preferable from the viewpoint of heat resistance of the obtained resin.
- Examples include phthalic anhydride, maleic anhydride, nadic anhydride, cyclohexanedicarboxylic anhydride, 3-hydroxyphthalic anhydride, and the like.
- the introduction ratio of the acid anhydride used as the end-capping agent is preferably 10 to 100 mol%, more preferably 50 to 100 mol% with respect to 100 mol% of the diamine which is the monomer component of the resin.
- the solubility of the resulting resin in an organic solvent is improved, and the viscosity when a photosensitive resin composition is made using the obtained resin is appropriately adjusted. can do.
- 100 mol% or less is preferable and 90 mol% or less is more preferable from a viewpoint of the solubility with respect to the aqueous alkali solution of the resin obtained, and the mechanical strength of a cured film.
- the end-capping agent introduced into the resin can be easily detected by the following method.
- a resin having a terminal blocking agent introduced therein is dissolved in an acidic solution and decomposed into an amine component and an acid component, which are constituent units of the resin, and this is measured by gas chromatography (GC) or NMR measurement.
- GC gas chromatography
- NMR nuclear magnetic resonance
- n is preferably 5 to 100, particularly preferably 10 ⁇ 70. If n is less than 5, the strength of the resulting cured resin film may be reduced. On the other hand, when n exceeds 100, the solubility of the resulting resin in an organic solvent may be reduced, or the viscosity of the resin composition may be too high.
- the number of repetitions n in the present invention can be easily calculated by measuring the weight average molecular weight (Mw) by gel permeation chromatography (GPC) measurement in terms of polystyrene.
- the weight average molecular weight (Mw) of the resin is preferably in the range of 5,000 to 100,000, and more preferably in the range of 10,000 to 50,000.
- the resin having the structure represented by the general formula (1) as a main repeating unit can be produced according to a known method for producing polyamic acid or polyamic acid ester, and the method is not particularly limited.
- a method of reacting a tetracarboxylic dianhydride and a diamine compound at a low temperature a method of obtaining a diester with a tetracarboxylic dianhydride and an alcohol, and then reacting in the presence of a diamine compound and a condensing agent can be mentioned. .
- the end-capping agent can be used in place of a part of the diamine compound and acid dianhydride, the method of adding the end-capping agent simultaneously with the diamine compound and tetracarboxylic dianhydride, the diamine compound and tetracarboxylic acid
- a method of adding a terminal blocking agent after reacting a dianhydride, a method of adding a diamine compound or a tetracarboxylic dianhydride after reacting a terminal blocking agent with a tetracarboxylic dianhydride or a diamine compound is there.
- the introduction ratio of the end capping agent exceeds 50 mol%, the end capping agent and the tetracarboxylic dianhydride or diamine compound are reacted and then the diamine compound or tetracarboxylic dianhydride is added. Since formation of oligomers, such as a dimer and a trimer, is suppressed, it is preferable. Furthermore, it is desirable that the polymer obtained by the above method is poured into a large amount of water or a methanol / water mixture, precipitated, filtered, dried and isolated. By this precipitation operation, unreacted monomers and oligomer components such as dimers and trimers are removed, and the film properties after thermosetting are improved.
- a tetracarboxylic dianhydride having an R 1 group is dissolved in a polymerization solvent, and a monoamine is added to this solution, followed by stirring with a mechanical stirrer. After a predetermined time, a diamine compound having an R 2 group is added and further stirred for a predetermined time.
- the reaction temperature is 0 to 100 ° C., preferably 20 to 50 ° C., and the reaction time is 0.5 to 50 hours, preferably 2 to 24 hours.
- the solvent used for the polymerization reaction is not particularly limited as long as it can dissolve the acid component and the diamine component, which are raw material monomers, and a protic solvent is preferable.
- a protic solvent is preferable.
- the resin mainly composed of the structure represented by the general formula (1) is preferably a resin that dissolves in propylene glycol monomethyl ether acetate at a concentration of 30% by weight or more.
- a resin that dissolves in propylene glycol monomethyl ether acetate at a concentration of 30% by weight or more has high solubility in an organic solvent, and the selectivity of the solvent when a photosensitive resin composition is obtained increases.
- the solution viscosity at 25 ° C. when the resin having the structure represented by the general formula (1) of the present invention as a main component is dissolved in 30% by weight in propylene glycol monomethyl ether acetate is 150 mPa ⁇ s or less. It is preferable.
- a resin with a solution viscosity of 150 mPa ⁇ s or less a low viscosity suitable for slit coating can be maintained even when the solid content concentration of the photosensitive resin composition is increased, and a thick film is formed by slit coating.
- the film thickness uniformity can be increased.
- the photosensitive resin composition of the present invention may contain (a) an alkali-soluble resin other than the resin component having the structure represented by the general formula (1) as a main repeating unit.
- the alkali-soluble resin refers to a resin having an acidic group that is soluble in alkali, and specifically includes a radical polymerizable polymer having acrylic acid, a phenol-novolak resin, polyhydroxystyrene, polysiloxane, and the like. Moreover, you may protect the acidic group of these resin and adjust alkali solubility.
- Such a resin is soluble in an aqueous solution of alkali such as choline, triethylamine, dimethylaminopyridine, monoethanolamine, diethylaminoethanol, sodium hydroxide, potassium hydroxide, sodium carbonate in addition to tetramethylammonium hydroxide. .
- alkali such as choline, triethylamine, dimethylaminopyridine, monoethanolamine, diethylaminoethanol, sodium hydroxide, potassium hydroxide, sodium carbonate in addition to tetramethylammonium hydroxide.
- Two or more of these resins may be contained, but the proportion of the total amount of the resin including the component (a) is preferably 50% by weight or less.
- the photosensitive resin composition of the present invention contains (b) a photosensitizer.
- the photosensitive agent include (b-1) a photoacid generator, (b-2) a photopolymerization initiator, and (b-3) a combination of compounds having two or more ethylenically unsaturated bonds.
- B-1 By containing a photoacid generator, an acid is generated in the light irradiation part to increase the solubility of the light irradiation part in an alkaline aqueous solution, thereby obtaining a positive relief pattern in which the light irradiation part dissolves. be able to.
- (b-1) by containing a photoacid generator and an epoxy compound or a thermal crosslinking agent described later, the acid generated in the light irradiation part promotes the crosslinking reaction of the epoxy compound and the thermal crosslinking agent, and the light irradiation part A negative-type relief pattern in which is insolubilized can be obtained.
- (b-2) containing a photopolymerization initiator and (b-3) a compound having two or more ethylenically unsaturated bonds, the active radical generated in the light irradiation part is a radical of ethylenically unsaturated bonds.
- a negative relief pattern in which polymerization is advanced and the light irradiation part is insolubilized can be obtained.
- the photosensitive resin composition of the present invention preferably contains (b-1) a photoacid generator as (b) a photosensitizer and exhibits positive photosensitivity.
- the positive photosensitive resin composition can easily obtain a forward-tapered pattern by firing after obtaining a fine pattern by an exposure / development process. This forward tapered pattern is excellent in the coverage of the upper electrode when used as an insulating film of an organic EL element, can prevent disconnection, and can increase the reliability of the element.
- Photoacid generators include quinonediazide compounds, sulfonium salts, phosphonium salts, diazonium salts, iodonium salts, and the like.
- the quinonediazide compound includes a polyhydroxy compound in which a sulfonic acid of quinonediazide is bonded with an ester, a polyamino compound in which a sulfonic acid of quinonediazide is bonded to a sulfonamide, a sulfonic acid of quinonediazide in an ester bond and / or sulfone Examples include amide-bonded ones. It is preferable that 50 mol% or more of the total functional groups of these polyhydroxy compounds and polyamino compounds are substituted with quinonediazide. Further, it is preferable to contain two or more (b-1) photoacid generators, and a highly sensitive photosensitive resin composition can be obtained.
- quinonediazide is preferably a 5-naphthoquinonediazidesulfonyl group or a 4-naphthoquinonediazidesulfonyl group.
- the 4-naphthoquinonediazide sulfonyl ester compound has absorption in the i-line region of a mercury lamp and is suitable for i-line exposure.
- the 5-naphthoquinonediazide sulfonyl ester compound has an absorption extending to the g-line region of a mercury lamp and is suitable for g-line exposure.
- it may contain a naphthoquinone diazide sulfonyl ester compound having a 4-naphthoquinone diazide sulfonyl group and a 5-naphthoquinone diazide sulfonyl group in the same molecule, or a 4-naphthoquinone diazide sulfonyl ester compound and a 5-naphthoquinone diazide sulfonyl ester compound. You may contain.
- sulfonium salts Of the photoacid generators, sulfonium salts, phosphonium salts, and diazonium salts are preferable because they moderately stabilize the acid components generated by exposure. Of these, sulfonium salts are preferred. Furthermore, it can also contain a sensitizer etc. as needed.
- Photopolymerization initiators include diethoxyacetophenone, 2-hydroxy-2-methyl-1-phenylpropan-1-one, benzyldimethyl ketal, 1- (4-isopropylphenyl) -2-hydroxy- 2-methylpropan-1-one, 4- (2-hydroxyethoxy) phenyl- (2-hydroxy-2-propyl) ketone, 1-hydroxycyclohexyl-phenylketone, 1-phenyl-1,2-propanedione-2 -(O-ethoxycarbonyl) oxime, 2-methyl- [4- (methylthio) phenyl] -2-morpholinopropan-1-one, 2-benzyl-2-dimethylamino-1- (4-morpholinophenyl) -Butanone-1, benzoin, benzoin methyl ether, benzoin ethyl ether, benzoin Sopropyl ether, benzoin isobutyl ether, benzophenone, methyl o-
- (B-3) As a compound having two or more ethylenically unsaturated bonds, ethylene glycol diacrylate, diethylene glycol diacrylate, triethylene glycol diacrylate, tetraethylene glycol diacrylate, ethylene glycol dimethacrylate, diethylene glycol dimethacrylate, triethylene Glycol dimethacrylate, tetraethylene glycol dimethacrylate, ethylene oxide modified bisphenol A diacrylate, ethylene oxide modified bisphenol A dimethacrylate, trimethylolpropane diacrylate, trimethylolpropane triacrylate, trimethylolpropane dimethacrylate, trimethylolpropane trimethacrylate, 1, 3-diisopropenylbenzene, , 3-butanediol diacrylate, 1,3-butanediol dimethacrylate, neopentyl glycol diacrylate, 1,4-butanediol diacrylate, 1,4-butanedi
- the content of (b) the photosensitizer is preferably 0.05 to 50 parts by weight with respect to 100 parts by weight of the resin of component (a).
- the content of the (b-1) photoacid generator is preferably 0.01 to 50 parts by weight with respect to 100 parts by weight of the component (a) resin from the viewpoint of increasing sensitivity.
- the quinonediazide compound is preferably 3 to 40 parts by weight.
- the total amount of the sulfonium salt, phosphonium salt and diazonium salt is preferably 0.5 to 20 parts by weight.
- the content of the (b-2) photopolymerization initiator is preferably 0.1 to 20 parts by weight with respect to 100 parts by weight of the component (a) resin.
- the content of the compound having two or more ethylenically unsaturated bonds is preferably 5 to 50 parts by weight with respect to 100 parts by weight of the resin of component (a).
- a compound having only one ethylenically unsaturated bond may be contained in an amount of 1 to 50 parts by weight with respect to 100 parts by weight of the resin as component (a) for adjusting the solubility.
- examples of such compounds are acrylate, methacrylate, methyl acrylate, methyl methacrylate, butyl acrylate, butyl methacrylate, isobutyl acrylate, hexyl acrylate, isooctyl acrylate, isobornyl acrylate, isobornyl methacrylate, cyclohexyl methacrylate, hydroxyethyl acrylate , Hydroxyethyl methacrylate, N, N-dimethylaminoethyl acrylate, N, N-dimethylaminoethyl methacrylate, N, N-dimethylacrylamide, N, N-dimethylmethacrylamide, N, N-dimethylaminopropylacrylamide,
- the photosensitive resin composition of the present invention contains (c) a solvent.
- a solvent ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, propylene glycol monomethyl ether, propylene glycol monoethyl ether, diethylene glycol dimethyl ether, diethylene glycol ethyl methyl ether and other ethers, ethylene glycol monomethyl ether acetate, propylene glycol monomethyl ether acetate, Esters such as ethyl acetate, butyl acetate, methyl lactate, ethyl lactate, butyl lactate, alcohols such as ethanol, isopropanol, butanol, pentanol, 3-methyl-2-butanol, 3-methyl-3-methoxybutanol, methyl ethyl ketone , Methyl isobutyl ketone, methyl amyl ketone, diisobutyl ketone, Ketones
- the content of the solvent (c) is preferably 50 parts by weight or more, more preferably 100 parts by weight or more, and preferably 2000 parts by weight or less, more preferably 100 parts by weight of the resin of the component (a). Is 1500 parts by weight or less.
- the photosensitive resin composition of the present invention may contain components other than (a) to (c), and (d) preferably contains a thermal crosslinking agent.
- the thermal crosslinking agent include (d-1) an alkoxymethyl group or methylol group-containing compound, and (d-2) an epoxy group or oxetanyl group-containing compound. Two or more of these may be contained.
- the thermal crosslinking agent of component (d) can increase the chemical resistance of the cured film by crosslinking reaction with the resin of component (a) by heating.
- (D-1) As the alkoxymethyl group or methylol group-containing compound, a compound represented by the general formula (6) or a compound having a group represented by the general formula (7) is preferable, and these may be used in combination. .
- R represents a direct bond or a monovalent to tetravalent linking group.
- R 7 represents a monovalent organic group having 1 to 20 carbon atoms, Cl, Br, I or F.
- Examples of the monovalent organic group having 1 to 20 carbon atoms include monovalent hydrocarbons having 1 to 6 carbon atoms such as methyl group, ethyl group, propyl group, butyl group, pentyl group, hexyl group, cyclopentyl group, and cyclohexyl group. Groups are preferred.
- R 8 and R 9 represent CH 2 OR 11 (R 11 is a hydrogen atom or a monovalent hydrocarbon group having 1 to 6 carbon atoms).
- R 10 represents a hydrogen atom, a methyl group or an ethyl group.
- h represents an integer of 0 to 2
- i represents an integer of 1 to 4.
- the plurality of R 7 ⁇ R 10 may be identical or different, but if the same benzene ring has two R 7, R 7 are the same. Examples of the linking group R are shown below.
- R 13 to R 35 represent a hydrogen atom, a monovalent organic group having 1 to 20 carbon atoms, Cl, Br, I or F.
- a monovalent organic group having 1 to 20 carbon atoms a methyl group, ethyl group, propyl group, butyl group, pentyl group, hexyl group, cyclopentyl group, cyclohexyl group, benzyl group, naphthyl group and the like are preferable.
- R 12 represents a hydrogen atom or a monovalent hydrocarbon group having 1 to 6 carbon atoms.
- j represents 1 or 2
- k represents 0 or 1.
- j + k is 1 or 2.
- R 8 and R 9 represent CH 2 OR 11 (R 11 is a hydrogen atom or a monovalent hydrocarbon group having 1 to 6 carbon atoms) which is a thermal crosslinking group.
- R 11 is preferably a monovalent hydrocarbon group having 1 to 4 carbon atoms because it retains moderate reactivity and is excellent in storage stability.
- R 11 is more preferably a methyl group or an ethyl group.
- the number of functional groups of the thermal crosslinking group in one molecule is 2 to 8.
- the number of functional groups is preferably 4 or more from the viewpoint of increasing the crosslinking density and improving the mechanical properties.
- the number of functional groups exceeds 8, it is difficult to obtain a high-purity product, and the stability of the compound itself and the storage stability in the resin composition are lowered.
- the purity of the compound represented by the general formula (6) is preferably 75% or more, and more preferably 85% or more. If purity is 85% or more, it is excellent in storage stability and can fully perform the crosslinking reaction of a resin composition. Moreover, since the unreacted group used as a water absorbing group can be decreased, the water absorption of a resin composition can be made small. Examples of the method for obtaining a high-purity thermal crosslinking agent include recrystallization and distillation. The purity of the thermal crosslinking agent can be determined by a liquid chromatography method.
- R 12 is a hydrogen atom or a monovalent hydrocarbon group having 1 to 6 carbon atoms, preferably a monovalent hydrocarbon group having 1 to 4 carbon atoms. Further, from the viewpoint of the stability of the compound and the storage stability in the resin composition, in the photosensitive resin composition containing a photoacid generator, R 12 is preferably a methyl group or an ethyl group, and is contained in the compound (CH 2 OR 12 ) The number of groups is preferably 8 or less.
- thermal crosslinking agent having a group represented by the general formula (7) are shown below.
- (D-2) As the epoxy group or oxetanyl group-containing compound, a compound containing two or more epoxy groups or oxetanyl groups in one molecule is preferable from the viewpoint of chemical resistance and heat resistance of the resulting cured film.
- VG3101L (trade name, manufactured by Printec Co., Ltd.), “Tepic” S, “Tepic” G, “Tepic” P (above trade names, Nissan Chemical Industries, Ltd.) have three or more epoxy groups.
- the content of the thermal crosslinking agent as the component (d) is preferably 5 parts by weight or more and more preferably 10 parts by weight or more with respect to 100 parts by weight of the resin of the component (a). If it is 5 parts by weight or more, the crosslink density of the cured film is increased and the chemical resistance is improved, which is preferable. Furthermore, when it is 10 parts by weight or more, chemical resistance is improved and higher mechanical properties are obtained. On the other hand, from the viewpoint of storage stability and mechanical strength of the composition, it is preferably 50 parts by weight or less, more preferably 40 parts by weight or less, and even more preferably 30 parts by weight or less. In addition, when it contains 2 or more types of (a) component or (d) component, it is preferable that those total amount is the said range.
- the photosensitive resin composition of the present invention may contain (e) a compound having a phenolic hydroxyl group.
- a compound having a phenolic hydroxyl group By containing the compound which has a phenolic hydroxyl group, the solubility with respect to the aqueous alkali solution of the photosensitive resin composition obtained improves, and it can achieve high sensitivity.
- Examples of the compound having a phenolic hydroxyl group include BisP-AF, BisP-AP, BisP-BA, Bis-Z, Ph-CC-AP, HDP-244, BisOC-Z, BisOPP-Z, BisP- CP, Bis26X-Z, BisOTBP-Z, BisOCHP-Z, BisOCR-CP, BisP-MZ, BisP-EZ, Bis26X-CP, BisP-PZ, BisP-IPZ, BisCR-IPZ, BisOCP-IPZ, BisOIPP-CP, Bis26X-IPZ, BisOTBP-CP, TekP-4HBPA (Tetrakis P-DO-BPA), TrisP-HAP, TrisP-PA, TrisP-PHBA, TrisP-SA, TrisOCR-PA, BisOFP-Z, BisRS-2P, Bi PG-26X, BisRS-3P, BisOC-OCHP, BisPC-OCHP, Bis25X-OCHP, Bis26X-OCHP, BisOCHP-
- the content of the compound having a phenolic hydroxyl group is preferably 1 to 40 parts by weight, more preferably 3 to 30 parts by weight with respect to 100 parts by weight of the resin of component (a).
- component may contain 2 or more types, and when it contains 2 or more types, it is preferable that those total amount is the said range.
- the resin composition of the present invention may contain a thermal acid generator.
- the thermal acid generator generates an acid by heating after development, which will be described later, and promotes a crosslinking reaction between the resin of the component (a) and the thermal crosslinking agent of the component (d), and the imide ring of the resin of the component (a). Promotes cyclization of the oxazole ring. For this reason, the chemical resistance of the cured film is improved, and film loss can be reduced.
- the acid generated from the thermal acid generator is preferably a strong acid.
- the thermal acid generator is preferably an aliphatic sulfonic acid compound represented by the general formula (8) or (9), and may contain two or more of these.
- R 36 to R 38 each represents an alkyl group having 1 to 10 carbon atoms or a monovalent aromatic group having 7 to 12 carbon atoms.
- the alkyl group and the aromatic group may be substituted, and examples of the substituent include an alkyl group and a carbonyl group.
- the content of the thermal acid generator is preferably 0.1 parts by weight or more, more preferably 0.3 parts by weight or more with respect to 100 parts by weight of the resin of component (a), from the viewpoint of further promoting the crosslinking reaction. 0.5 parts by weight or more is more preferable. On the other hand, from the viewpoint of maintaining the electrical insulation of the cured film, it is preferably 20 parts by weight or less, more preferably 15 parts by weight or less, and even more preferably 10 parts by weight or less. In addition, when 2 or more types of thermal acid generators are contained, it is preferable that those total amount is the said range.
- thermochromic compound that develops color when heated and exhibits an absorption maximum at 350 nm to 700 nm, or has an absorption maximum at 500 nm to 750 nm without an absorption maximum at 350 nm to less than 500 nm.
- Organic pigments or dyes can be included.
- the coloring temperature of the thermochromic compound is preferably 120 ° C. or higher, more preferably 150 ° C. or higher. The higher the coloring temperature of the thermochromic compound, the better the heat resistance under high temperature conditions, and the better the light resistance without fading due to prolonged ultraviolet-visible light irradiation.
- thermochromic compounds include thermal dyes, pressure sensitive dyes, and hydroxyl group-containing compounds having a triarylmethane skeleton.
- the photosensitive resin composition of the present invention may contain an adhesion improving agent.
- adhesion improvers vinyltrimethoxysilane, vinyltriethoxysilane, epoxycyclohexylethyltrimethoxysilane, 3-glycidoxypropyltrimethoxysilane, 3-glycidoxypropyltriethoxysilane, p-styryltrimethoxysilane, Silane coupling agents such as 3-aminopropyltrimethoxysilane, 3-aminopropyltriethoxysilane, N-phenyl-3-aminopropyltrimethoxysilane, titanium chelating agents, aluminum chelating agents, aromatic amine compounds and alkoxy groups Examples thereof include compounds obtained by reacting silicon compounds.
- adhesion improving agents By containing these adhesion improving agents, adhesion to an underlying substrate such as a silicon wafer, ITO, SiO 2 , or silicon nitride can be enhanced when developing a photosensitive resin film. Further, resistance to oxygen plasma and UV ozone treatment used for cleaning or the like can be increased.
- the content of the adhesion improving agent is preferably 0.1 to 10 parts by weight with respect to 100 parts by weight of the component (a) resin.
- the photosensitive resin composition of the present invention may contain an adhesion improver.
- the adhesion improving agent include an alkoxysilane-containing aromatic amine compound, an aromatic amide compound, or an aromatic non-containing silane compound. Two or more of these may be contained. By containing these compounds, the adhesiveness with the base material after curing can be improved. Specific examples of the alkoxysilane-containing aromatic amine compound and aromatic amide compound are shown below.
- a compound obtained by reacting an aromatic amine compound and an alkoxy group-containing silicon compound may be used. For example, an aromatic amine compound and a group that reacts with an amino group such as an epoxy group or a chloromethyl group. The compound etc. which are obtained by making the alkoxysilane compound which has it react are mentioned.
- Non-aromatic silane compounds include vinyl silane compounds such as vinyltrimethoxysilane, vinyltriethoxysilane, vinyltrichlorosilane, vinyltris ( ⁇ -methoxyethoxy) silane, 3-methacryloxypropyltrimethoxysilane, and 3-acryloxypropyl.
- vinyl silane compounds such as vinyltrimethoxysilane, vinyltriethoxysilane, vinyltrichlorosilane, vinyltris ( ⁇ -methoxyethoxy) silane, 3-methacryloxypropyltrimethoxysilane, and 3-acryloxypropyl.
- carbon-carbon unsaturated bond-containing silane compounds such as trimethoxysilane, p-styryltrimethoxysilane, 3-methacryloxypropylmethyldimethoxysilane, and 3-methacryloxypropylmethyldiethoxysilane.
- vinyltrimethoxysilane and vinyltriethoxysilane are preferable.
- the total content of the alkoxysilane-containing aromatic amine compound, aromatic amide compound, or non-aromatic silane compound is preferably 0.01 to 15 parts by weight with respect to 100 parts by weight of the component (a) resin.
- the photosensitive resin composition of the present invention may contain inorganic particles.
- Preferred specific examples include, but are not limited to, silicon oxide, titanium oxide, barium titanate, alumina, talc and the like.
- the primary particle diameter of these inorganic particles is preferably 100 nm or less, more preferably 60 nm or less.
- the photosensitive resin composition of the present invention may contain a surfactant and can improve the wettability with the substrate.
- Fluorosurfactants such as Fluorard (trade name, manufactured by Sumitomo 3M Co., Ltd.), MegaFac (trade name, manufactured by DIC Corporation), Sulflon (trade name, manufactured by Asahi Glass Co., Ltd.), etc. , KP341 (trade name, manufactured by Shin-Etsu Chemical Co., Ltd.), DBE (trade name, manufactured by Chisso Co., Ltd.), Polyflow, Granol (trade name, manufactured by Kyoeisha Chemical Co., Ltd.), BYK (trade name, BYK Chemie Corp.) ) And other organic siloxane surfactants, and acrylic polymer surfactants such as Polyflow (trade name, manufactured by Kyoeisha Chemical Co., Ltd.).
- a photosensitive resin composition can be obtained by mixing uniformly.
- the dissolution method include stirring and heating.
- the heating temperature is preferably set in a range that does not impair the performance of the resin composition, and is usually room temperature to 80 ° C.
- the dissolution order of each component is not particularly limited, and for example, there is a method of sequentially dissolving compounds having low solubility.
- components that tend to generate bubbles when stirring and dissolving such as surfactants and some adhesion improvers, by dissolving other components and adding them last, poor dissolution of other components due to the generation of bubbles Can be prevented.
- the obtained photosensitive resin composition is preferably filtered using a filtration filter to remove dust and particles.
- the filter pore diameter is 0.5 to 0.02 ⁇ m, for example, 0.5 ⁇ m, 0.2 ⁇ m, 0.1 ⁇ m, 0.05 ⁇ m, 0.02 ⁇ m, but is not limited thereto.
- the material for the filter include polypropylene (PP), polyethylene (PE), nylon (NY), polytetrafluoroethylene (PTFE), and polyethylene and nylon are preferable.
- PP polypropylene
- PE polyethylene
- nylon NY
- PTFE polytetrafluoroethylene
- polyethylene and nylon are preferable.
- the photosensitive resin composition of the present invention is particularly suitable for slit coating as described above, but the coating method is not limited, and spin coating method, slit coating method, dip coating method, spray coating method, ink jet method and nozzle coating. It is applied by a printing method such as a method to obtain a photosensitive resin composition film. Prior to the application, the substrate on which the photosensitive resin composition is applied may be pretreated with the adhesion improving agent described above in advance.
- a method of treating the substrate surface examples include spin coating, slit die coating, bar coating, dip coating, spray coating, and steam treatment. If necessary, it can be dried under reduced pressure, and then the reaction between the base material and the adhesion improving agent can proceed by a heat treatment at 50 ° C. to 300 ° C.
- the apparatus for applying the photosensitive resin composition of the present invention has a plurality of types, that is, the photosensitive resin composition of the present invention and the photosensitive resin composition of the present invention, without washing the liquid feeding path with one liquid feeding path. Photosensitive resin compositions other than the product can be sequentially fed.
- washing means washing with a washing liquid having a capacity of 10 times or more the total volume of the liquid feeding pump, the liquid feeding line and the mouthpiece from the liquid feeding tank to the tip of the mouthpiece.
- the photosensitive resin composition of the present invention is excellent in solubility in low-polarity solvents typified by propylene glycol monomethyl ether acetate, a plurality of types of photosensitive resin compositions can be prepared without performing a step of washing with thinner. Can be refilled by continuously feeding.
- the photosensitive resin composition other than the photosensitive resin composition of the present invention is not particularly limited, but is used for the photosensitive resin composition of the present invention.
- the above-mentioned problem is likely to occur when a solvent having the same or lower polarity than the solvent used is present, and the method for producing a heat-resistant resin film of the present invention is preferably applied.
- a photosensitive resin composition containing at least one solvent selected from -ethoxypropionate, methyl-3-methoxypropionate, 2-heptanone, propylene glycol monomethyl ether acetate, and cyclohexanone for example, ethylene glycol monoethyl ether acetate, ethyl-3
- the substrate coated with the photosensitive resin composition is dried to obtain a photosensitive resin film. Drying is preferably performed using an oven, a hot plate, infrared rays, or the like in the range of 50 ° C. to 150 ° C. for 1 minute to several hours.
- the photosensitive resin film is exposed to actinic radiation through a mask having a desired pattern.
- actinic radiation used for exposure there are ultraviolet rays, visible rays, electron beams, X-rays and the like.
- the exposed portion may be removed using a developer after exposure.
- the developer is an aqueous solution of tetramethylammonium, diethanolamine, diethylaminoethanol, sodium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, triethylamine, diethylamine, methylamine, dimethylamine, dimethylaminoethyl acetate, dimethylaminoethanol, dimethylamino
- An aqueous solution of a compound exhibiting alkalinity such as ethyl methacrylate, cyclohexylamine, ethylenediamine, hexamethylenediamine and the like is preferable.
- these alkaline aqueous solutions may contain polar solvents such as N-methyl-2-pyrrolidone, N, N-dimethylformamide, N, N-dimethylacetamide, dimethyl sulfoxide, ⁇ -butyrolactone, dimethylacrylamide, methanol, ethanol,
- polar solvents such as N-methyl-2-pyrrolidone, N, N-dimethylformamide, N, N-dimethylacetamide, dimethyl sulfoxide, ⁇ -butyrolactone, dimethylacrylamide, methanol, ethanol,
- One or more alcohols such as isopropanol, esters such as ethyl lactate and propylene glycol monomethyl ether acetate, ketones such as cyclopentanone, cyclohexanone, isobutyl ketone and methyl isobutyl ketone may be added.
- alcohols such as ethanol and isopropyl alcohol, and esters such as eth
- a cured film can be obtained by heat-treating the obtained photosensitive resin composition film.
- a method of heat treatment at 230 ° C. for 60 minutes a method of heat treatment at 120 to 400 ° C. for 1 minute to 10 hours, a method of heat treatment at a low temperature of about room temperature to 100 ° C. with addition of a curing catalyst, ultrasonic waves or electromagnetic waves
- a method of curing at a low temperature of about room temperature to 100 ° C. by treatment examples thereof include a method of curing at a low temperature of about room temperature to 100 ° C. by treatment.
- the heat-resistant resin film (cured film) formed by the photosensitive resin composition of the present invention includes a semiconductor passivation film, a protective film for semiconductor elements, an interlayer insulating film for multilayer wiring for high-density mounting, an insulating film for organic EL elements, It is suitably used for applications such as a planarization film on a TFT substrate.
- Viscosity Evaluation (2-1) Polymer Solution Viscosity Polymer powder was dissolved in PGMEA at 30% by weight and measured at 25 ° C. using an E-type viscometer. In order to maintain a viscosity suitable for slit coating even when the solid content concentration in the photosensitive resin composition is increased, the viscosity of the polymer solution is preferably less than 150 mPa ⁇ s.
- Appropriate evaluation for forming a uniform thick film by slit coating is “A” when the polymer powder is dissolved in PGMEA at 30% by weight and the solution viscosity is less than 150 mPa ⁇ s. Although it melt
- the photosensitive resin composition (varnish) having a solid content concentration of 20% prepared in Examples and Comparative Examples was measured at 25 ° C. using an E-type viscometer.
- the solution viscosity is preferably 1 to 15 cp, and more preferably 1 to 10 cp.
- a varnish having a viscosity of less than 10 cp at 25 ° C. was judged as “S”, a varnish of 10 cp to less than 15 cp as “A”, and a varnish having a viscosity of 15 cp or more as “C”.
- the solid content concentration of the photosensitive resin composition is not 20%, the solvent composition of the photosensitive resin composition does not change or the photosensitive resin composition does not change, so that the solid content concentration is 20%. % Can be measured.
- the positive photosensitive resin composition is exposed after exposure, and the negative photosensitive resin composition is post-exposure baked and then 2.38 wt% tetramethylammonium (TMAH) aqueous solution (manufactured by Mitsubishi Gas Chemical Co., Ltd., ELM- D) was developed for 60 seconds and then rinsed with pure water to obtain a developed film.
- TMAH tetramethylammonium
- Film thickness measurement method The film thickness after pre-baking and development was measured using a light interference type film thickness measuring device Lambda Ace STM-602 manufactured by Dainippon Screen Mfg. Co., Ltd., with a refractive index of 1.63.
- the amount of development film reduction is preferably less than 0.50 ⁇ m.
- the developed film reduction amount was less than 0.50 ⁇ m, it was determined as “A”, when it was 0.51 to 0.59 ⁇ m, “B”, and when it was 0.60 ⁇ m or more, “C”.
- Developed film reduction ( ⁇ m) film thickness after pre-baking ⁇ film thickness after development Sensitivity calculation After exposure and development, exposure that forms a 20 ⁇ m line and space pattern (1L / 1S) with a one-to-one width The amount (referred to as the optimum exposure amount Eop) was taken as the sensitivity. If Eth is 200 mJ / cm 2 or less, it can be determined that the sensitivity is high. 150 mJ / cm 2 or less is more preferable.
- Synthesis Example 1 Synthesis of Diamine Compound ( ⁇ ) 18.3 g (0.05 mol) of BAHF (manufactured by Central Glass Co., Ltd.) in 100 mL of acetone and 17.4 g (0.3 mol of propylene oxide (manufactured by Tokyo Chemical Industry Co., Ltd.)) ) And cooled to -15 ° C. A solution prepared by dissolving 20.4 g (0.11 mol) of 3-nitrobenzoyl chloride (manufactured by Tokyo Chemical Industry Co., Ltd.) in 100 mL of acetone was added dropwise thereto. After completion of dropping, the mixture was stirred at ⁇ 15 ° C. for 4 hours and then returned to room temperature. The precipitated white solid was filtered off and vacuum dried at 50 ° C.
- Synthesis Example 2 Synthesis of quinonediazide compound (b-1) TrisP-PA (trade name, manufactured by Honshu Chemical Industry Co., Ltd.), 21.22 g (0.05 mol) and 5-naphthoquinonediazidesulfonic acid chloride under a dry nitrogen stream 26.8 g (0.1 mol) of NAC-5 (manufactured by Toyo Gosei Co., Ltd.) was dissolved in 450 g of 1,4-dioxane and brought to room temperature. To this, 12.65 g of triethylamine mixed with 50 g of 1,4-dioxane was added dropwise so that the temperature inside the system would not exceed 35 ° C.
- Needle-like white crystals formed in the solution after standing were collected by filtration and washed with 100 mL of water.
- the white crystals were vacuum dried at 50 ° C. for 48 hours.
- NMR manufactured by JEOL Ltd., GX-270
- DMSO-d6 a heavy solvent
- thermal crosslinking agents and compounds having a phenolic hydroxyl group used in the examples are as follows.
- Example 1 Under a dry nitrogen stream, 15.1 g (0.025 mol) of the diamine compound ( ⁇ ) obtained in Synthesis Example 1, 3.66 g (0.01 mol) of BAHF and 0.62 g of SiDA (manufactured by Shin-Etsu Chemical Co., Ltd.) (0.0025 mol) was dissolved in 200 g of NMP.
- 22.2 g (0.05 mol) of 6FDA manufactured by Daikin Industries, Ltd.
- was added together with 50 g of NMP followed by stirring at 40 ° C. for 1 hour.
- 2.73 g (0.025 mol) of MAP (manufactured by Tokyo Chemical Industry Co., Ltd.) was added and stirred at 40 ° C. for 1 hour.
- Example 2 Polyamic acid ester resin as in Example 1 except that 3.62 g (0.01 mol) of BIS-AT-AF (manufactured by Central Glass Co., Ltd.) was used instead of 3.66 g (0.01 mol) of BAHF. (B) was obtained. Using the obtained resin (B), the solubility evaluation with respect to the organic solvent and the measurement of the polymer solution viscosity were performed by the above-described methods. The results are shown in Table 2.
- Example except that 10 g of the resin (B) obtained above was added instead of the resin (A), and 1.0 g of the alkoxymethyl group-containing thermal crosslinking agent (d-1) obtained in Synthesis Example 3 was further added.
- a varnish (B) of a positive photosensitive resin composition was produced.
- the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- Example 3 Diamine compound ( ⁇ ) 15.1 g (0.025 mol) 19.3 g (0.032 mol), BAHF 3.66 g (0.01 mol) 2.01 g (0.0055 mol) and MAP 2.73 g (0 0.025 mol) was changed to 2.18 g (0.02 mol) in the same manner as in Example 1 to obtain a polyamic acid ester resin (C). Using the obtained resin (C), solubility evaluation in an organic solvent and measurement of the polymer solution viscosity were performed by the above-described methods. The results are shown in Table 2.
- a varnish (C) of the composition was prepared. Using the obtained varnish (C), the amount of developed film was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- Example 4 Under a dry nitrogen stream, 22.2 g (0.05 mol) of 6FDA was dissolved in 200 g of NMP. ABP (product made from Tokyo Chemical Industry Co., Ltd.) 6.60g (0.04mol) was added here, and it stirred at 40 degreeC for 1 hour. Thereafter, 9.06 g (0.015 mol) of the diamine compound ( ⁇ ), 4.53 g (0.0125 mol) of BIS-AT-AF and 0.62 g (0.0025 mol) of SiDA were added together with 50 g of NMP, and the mixture was heated at 40 ° C. for 2 hours. Stir.
- ABP product made from Tokyo Chemical Industry Co., Ltd.
- a varnish (D) of the composition was prepared. Using the obtained varnish (D), the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- Example 5 Under a dry nitrogen stream, 21.2 g (0.035 mol) of the diamine compound ( ⁇ ), 4.58 g (0.0125 mol) of BAHF, and 0.62 g (0.0025 mol) of SiDA were dissolved in 200 g of NMP. To this, 13.3 g (0.03 mol) of 6FDA was added together with 50 g of NMP, followed by stirring at 40 ° C. for 1 hour. Then, 3.92 g (0.04 mol) of MA (manufactured by Wako Pure Chemical Industries, Ltd.) was added and stirred at 40 ° C. for 1 hour.
- MA manufactured by Wako Pure Chemical Industries, Ltd.
- a varnish (E) of the composition was prepared. Using the obtained varnish (E), the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- Example 6 Diamine compound ( ⁇ ) 21.2 g (0.035 mol) 25.7 g (0.0425 mol), BAHF 4.58 g (0.0125 mol) 1.83 g (0.005 mol), 6FDA 13.3 g (0 0.03 mol) was changed to 16.7 g (0.0375 mol) and MA 3.92 g (0.04 mol) was changed to 2.45 g (0.025 mol). F) was obtained. Using the obtained resin (F), solubility evaluation in an organic solvent and measurement of polymer solution viscosity were performed by the above-described methods. The results are shown in Table 2.
- a varnish (F) of the composition was prepared. Using the resulting varnish (F), the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- Example 7 Under a dry nitrogen stream, 15.1 g (0.025 mol) of the diamine compound ( ⁇ ), 8.24 g (0.0225 mol) of BAHF, and 0.62 g (0.0025 mol) of SiDA were dissolved in 200 g of NMP. MA 4.90g (0.05mol) was added here with NMP50g, and it stirred at 40 degreeC for 1 hour. Then, 11.1 g (0.025 mol) of 6FDA was added and stirred at 40 ° C. for 2 hours. Further, a solution prepared by diluting 11.9 g (0.1 mol) of DFA with 5 g of NMP was added dropwise over 10 minutes, and then stirring was continued at 40 ° C. for 2 hours.
- a varnish (G) of the composition was prepared. Using the obtained varnish (G), the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- Example 8 To 10 g of the resin (A) obtained in Example 1, 1,2-octanedione-1- [4- (phenylthio) -2- (o-benzoyloxime)] (ODPTBO) (manufactured by BASF Japan Ltd.) 0 0.1 g, ethylene oxide-modified bisphenol A dimethacrylate (manufactured by Shin-Nakamura Chemical Co., Ltd., NK ester BPE-100) 2.0 g, trimethylolpropane triacrylate (TPT) 0.5 g, alkoxy obtained in Synthesis Example 3
- a negative photosensitive resin composition varnish (A-2) was obtained by adding 1.0 g of a methyl group-containing thermal crosslinking agent (d-3) and 50 g of GBL. Using the obtained varnish (A-2), the amount of development film reduction was evaluated as described above. The results are shown in Table 2.
- Example 9 Example 3 was repeated except that 2.01 g (0.0055 mol) of BAHF was changed to 2.93 g (0.008 mol) and 2.18 g (0.02 mol) of MAP was changed to 1.63 g (0.015 mol).
- a polyamic acid ester resin (H) was obtained. Using the obtained resin (H), solubility evaluation in an organic solvent and measurement of polymer solution viscosity were performed by the above-described methods. The results are shown in Table 2.
- a positive photosensitive resin composition varnish (H) was prepared in the same manner as in Example 3 except that 10 g of the resin (H) obtained above was added instead of the resin (A). Using the obtained varnish (H), the solubility in organic solvents, the viscosity of the polymer solution, and the amount of development film reduction were evaluated in the same manner as in Example 3. The results are shown in Table 3.
- Example 10 Example 4 except that 4.53 g (0.0125 mol) of BIS-AT-AF was changed to 3.62 g (0.01 mol) and 6.60 g (0.04 mol) of ABP was changed to 7.43 g (0.045 mol).
- a polyamic acid ester resin (I) was obtained.
- solubility evaluation in an organic solvent and measurement of the polymer solution viscosity were performed by the above-described methods. The results are shown in Table 2.
- a positive photosensitive resin composition varnish (I) was prepared in the same manner as in Example 4 except that 10 g of the resin (I) obtained above was added instead of the resin (A). Using the obtained varnish (I), the amount of development film loss was evaluated in the same manner as in Example 4. The results are shown in Table 3.
- Example 11 6FDA 16.7 g (0.0375 mol) was the same as Example 6 except that 17.8 g (0.04 mol) and MA 2.45 g (0.025 mol) were changed to 1.96 g (0.02 mol).
- a polyamic acid ester resin (J) was obtained. Using the obtained resin (J), solubility evaluation in an organic solvent and measurement of the polymer solution viscosity were performed by the above-described methods. The results are shown in Table 2.
- a positive photosensitive resin composition varnish (J) was prepared in the same manner as in Example 6 except that 10 g of the resin (J) obtained above was added instead of the resin (A). Using the obtained varnish (J), the amount of development film loss was evaluated in the same manner as in Example 6. The results are shown in Table 3.
- Example 12 Except that 11.1 g (0.025 mol) of 6FDA was changed to 9.99 g (0.0225 mol), and 4.90 g (0.05 mol) of MA was changed to 5.39 g (0.055 mol), the same as in Example 7.
- Resin polyamic acid ester (K) was obtained. Using the obtained resin (K), solubility evaluation in an organic solvent and measurement of the polymer solution viscosity were performed by the above-described methods. The results are shown in Table 2.
- a positive photosensitive resin composition varnish (K) was prepared in the same manner as in Example 7 except that 10 g of the resin (K) obtained above was added instead of the resin (A). Using the resulting varnish (K), the amount of development film loss was evaluated in the same manner as in Example 7. The results are shown in Table 3.
- Comparative Example 1 A polyamic acid ester resin (L) was obtained in the same manner as in Example 1 except that 15.5 g (0.05 mol) of ODPA (manac) was used instead of 22.2 g (0.05 mol) of 6FDA. It was. Using the obtained resin (L), solubility evaluation in an organic solvent and measurement of a polymer solution viscosity were performed by the above-described methods. The results are shown in Table 2.
- a positive photosensitive resin composition varnish (L) was produced in the same manner as in Example 1 except that 10 g of the resin (L) obtained above was added instead of the resin (A). Using the resulting varnish (L), the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- a positive photosensitive resin composition varnish (M) was prepared in the same manner as in Example 1 except that 10 g of the resin (M) obtained above was added instead of the resin (A). Using the obtained varnish (M), the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- a positive photosensitive resin composition varnish (N) was produced in the same manner as in Example 1 except that 10 g of the resin (N) obtained above was added instead of the resin (A). Using the obtained varnish (N), the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- a positive photosensitive resin composition varnish (O) was produced in the same manner as in Example 1 except that 10 g of the resin (O) obtained above was added instead of the resin (A). Using the obtained resin (O) and varnish (O), the amount of development film reduction was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- Comparative Example 5 Example except that 15.1 g (0.025 mol) of the diamine compound ( ⁇ ) was changed to 6.05 g (0.01 mol) and 3.66 g (0.01 mol) of BAHF was changed to 9.15 g (0.025 mol).
- a polyamic acid ester resin (P) was obtained.
- solubility evaluation in an organic solvent and measurement of a polymer solution viscosity were performed by the above-described methods. The results are shown in Table 2.
- a positive photosensitive resin composition varnish (P) was produced in the same manner as in Example 1 except that 10 g of the resin (P) obtained above was added instead of the resin (A). Using the obtained varnish (P), the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- a positive photosensitive resin composition varnish (Q) was prepared in the same manner as in Example 1 except that 10 g of the resin (Q) obtained above was added instead of the resin (A). Using the obtained varnish (Q), the amount of development film reduction was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- pyridine manufactured by Tokyo Chemical Industry Co., Ltd.
- 15 g of toluene manufactured by Tokyo Chemical Industry Co., Ltd.
- a condenser was attached to remove water from the system azeotropically with toluene.
- the temperature of the solution was raised to 120 ° C. for 2 hours and further at 180 ° C. for 2 hours.
- the temperature of this solution was lowered to room temperature, the solution was poured into 2 L of water, and a polymer solid precipitate was collected by filtration.
- a positive photosensitive resin composition varnish (R) was produced in the same manner as in Example 1 except that 10 g of the resin (R) obtained above was added instead of the resin (A). Using the obtained varnish (R), the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- a positive photosensitive resin composition varnish (S) was prepared in the same manner as in Example 1 except that 10 g of the resin (S) obtained above was added instead of the resin (A). Using the obtained varnish (S), the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- a positive photosensitive resin composition varnish (T) was produced in the same manner as in Example 1 except that 10 g of the resin (T) obtained above was added instead of the resin (A). Using the obtained varnish (T), the amount of development film loss was evaluated in the same manner as in Example 1. The results are shown in Table 3.
- a positive photosensitive resin composition varnish (U) was prepared in the same manner as in Example 1 except that 10 g of the resin (U) obtained above was added instead of the resin (A). Using the obtained varnish (U), the solubility with respect to the organic solvent, the polymer solution viscosity, and the development film reduction amount were evaluated in the same manner as in Example 1. The results are shown in Tables 1 and 2.
- Table 1 shows the monomers and end-capping agent compositions used in the resins A to U used in Examples and Comparative Examples
- Table 2 shows the evaluation results of the organic solvent solubility and polymer solution viscosity of the resins A to U.
- Table 3 shows the evaluation results of the varnish compositions and developing film reduction amounts of Examples and Comparative Examples.
- Example 13 Under a dry nitrogen stream, 16.6 g (0.0275 mol) of diamine compound ( ⁇ ) obtained in Synthesis Example 1, 1.83 g (0.005 mol) of BAHF, and 0.62 g of SiDA (manufactured by Shin-Etsu Chemical Co., Ltd.) (0.0025 mol) was dissolved in 200 g of NMP. To this, 22.2 g (0.05 mol) of 6FDA (manufactured by Daikin Industries, Ltd.) was added together with 50 g of NMP, followed by stirring at 40 ° C. for 1 hour. Then, 3.27 g (0.03 mol) of MAP (manufactured by Tokyo Chemical Industry Co., Ltd.) was added and stirred at 40 ° C.
- 6FDA manufactured by Daikin Industries, Ltd.
- Example 14 Under a dry nitrogen stream, 22.2 g (0.05 mol) of 6FDA was dissolved in 200 g of NMP. ABP (product made from Tokyo Chemical Industry Co., Ltd.) 4.95g (0.03mol) was added here, and it stirred at 40 degreeC for 1 hour. Thereafter, 16.6 g (0.0275 mol) of the diamine compound ( ⁇ ), 1.83 g (0.005 mol) of BAHF and 0.62 g (0.0025 mol) of SiDA were added together with 50 g of NMP, and the mixture was stirred at 40 ° C. for 2 hours.
- ABP product made from Tokyo Chemical Industry Co., Ltd.
- a positive photosensitive resin composition varnish (AB) was produced in the same manner as in Example 13 except that 7.0 g of the resin (AB) obtained above was added instead of the resin (AA). Viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13 using the obtained varnish (AB). The results are shown in Table 5.
- Example 15 Example 13 was repeated except that 1.83 g (0.005 mol) of BAHF was changed to 0.91 g (0.0025 mol) and 3.27 g (0.03 mol) of MAP was changed to 3.81 g (0.035 mol). Thus, a polyamic acid ester resin (AC) was obtained. Using the obtained resin (AC), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- Example 5 In the same manner as in Example 1 except that 7.0 g of the resin (AC) obtained above was added instead of the resin (AA), a varnish (AC) having a positive solid content concentration of 20% was prepared. Created. Using the obtained varnish (AC), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Example 16 Example 14: ABP 4.95 g (0.03 mol) was changed to 5.73 g (0.035 mol), and BAHF 1.83 g (0.005 mol) was changed to 0.915 g (0.0025 mol). Thus, a polyamic acid ester resin (AD) was obtained. Using the obtained resin (AD), solubility in an organic solvent was evaluated in the same manner as in Example 13.
- AD polyamic acid ester resin
- a varnish (AD) of the composition was prepared. Using the obtained varnish (AD), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Example 17 Under a dry nitrogen stream, 25.7 g (0.0425 mol) of the diamine compound ( ⁇ ), 1.83 g (0.005 mol) of BAHF and 0.62 g (0.0025 mol) of SiDA were dissolved in 200 g of NMP. To this, 17.8 g (0.04 mol) of 6FDA was added together with 50 g of NMP, and the mixture was stirred at 40 ° C. for 1 hour. Then, 1.96 g (0.02 mol) of MA (manufactured by Wako Pure Chemical Industries, Ltd.) was added and stirred at 40 ° C. for 1 hour.
- MA manufactured by Wako Pure Chemical Industries, Ltd.
- a solution prepared by diluting 11.9 g (0.1 mol) of DFA with 5 g of NMP was added dropwise over 10 minutes, and then stirring was continued at 40 ° C. for 2 hours. After the completion of stirring, the solution was poured into 2 L of water, and a precipitate of polymer solid was collected by filtration. Further, it was washed 3 times with 2 L of water, and the collected polymer solid was dried with a vacuum dryer at 50 ° C. for 72 hours to obtain a polyamic acid ester resin (AE). Using the obtained resin (AE), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- a positive photosensitive resin composition varnish (AE) was produced in the same manner as in Example 13 except that 7.0 g of the resin (AE) obtained above was added instead of the resin (AA). Using the obtained varnish (AE), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Example 18 2.48 g (0.015 mol) of ABP 4.95 g (0.03 mol), 18.1 g (0.03 mol) of 16.6 g (0.0275 mol) of the diamine compound ( ⁇ ), 1.83 g of BAHF (0 0.005 mol) was changed to 3.66 g (0.01 mol), and a polyamic acid ester resin (AF) was obtained in the same manner as in Example 14. Using the obtained resin (AF), solubility in an organic solvent was evaluated in the same manner as in Example 13.
- Example 13 In the same manner as in Example 13 except that 7.0 g of the resin (AF) obtained above was added instead of the resin (AA), a positive varnish (AF) of a photosensitive resin composition having a solid content concentration of 20% was obtained. Created. Using the obtained varnish (AF), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Example 19 13.6 g (0.0225 mol) of the diamine compound ( ⁇ ), 7.32 g (0.02 mol) of BAHF 1.83 g (0.005 mol), and 3.27 g of MAP ( A polyamic acid ester resin (AG) was obtained in the same manner as in Example 13 except that 0.03 mol) was changed to 1.09 g (0.01 mol). Using the obtained resin (AG), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- Example 13 In the same manner as in Example 13 except that 7.0 g of the resin (AG) obtained above was added instead of the resin (AA), a varnish (AG) having a positive solid content concentration of 20% was prepared. Created. Using the obtained varnish (AG), the viscosity evaluation and the photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Example 20 4.13 g (0.025 mol) of ABP 4.95 g (0.03 mol), 3.02 g (0.005 mol) of 16.6 g (0.0275 mol) of the diamine compound ( ⁇ ), and 1.83 g of BAHF (0 0.005 mol) was changed to 10.98 g (0.03 mol), and a polyamic acid ester resin (AH) was obtained in the same manner as in Example 14. Using the obtained resin (AH), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- Example 5 In the same manner as in Example 1 except that 7.0 g of the resin (AH) obtained above was added instead of the resin (AA), a varnish (AH) having a positive solid content concentration of 20% was prepared. Created. Using the obtained varnish (AH), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Example 21 The diamine compound ( ⁇ ) 16.6 g (0.0275 mol) 3.02 g (0.005 mol), BAHF 1.83 g (0.005 mol) 12.81 g (0.035 mol), and MAP 3.27 g ( A polyamic acid ester resin (AI) was obtained in the same manner as in Example 13, except that 0.03 mol) was changed to 1.64 g (0.015 mol). Using the obtained resin (AI), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- AI polyamic acid ester resin
- Example 13 In the same manner as in Example 13 except that 7.0 g of the resin (AI) obtained above was added instead of the resin (A), a varnish (AI) having a positive solid content concentration of 20% was prepared. Created. Using the obtained varnish (AI), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Example 22 instead of 1.83 g (0.005 mol) of BAHF, 3.62 g (0.01 mol) of BIS-AT-AF (manufactured by Central Glass Co., Ltd.) and 16.6 g (0.0275 mol) of diamine compound ( ⁇ )
- a polyamic acid ester resin (AJ) was obtained in the same manner as in Example 13 except that 15.1 g (0.025 mol) and 3.27 g (0.03 mol) of MAP were changed to 2.73 g (0.025 mol). It was. Using the obtained resin (AJ), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- Example 23 4.60 g (0.04 mol) of ABP 4.95 g (0.03 mol), 4.53 g (0.0125 mol) of BIS-AT-AF instead of 1.83 g (0.005 mol) of BAHF, diamine compound ( ⁇ )
- a polyamic acid ester resin (AK) was obtained in the same manner as in Example 14 except that 16.6 g (0.0275 mol) was changed to 9.06 g (0.015 mol). Using the obtained resin (AK), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- Example 13 In the same manner as in Example 13 except that 7.0 g of the resin (AK) obtained above was added instead of the resin (AA), a varnish (AK) having a positive solid content concentration of 20% photosensitive resin composition was obtained. Created. Using the obtained varnish (AK), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Example 24 Diamine compound ( ⁇ ) 25.7 g (0.0425 mol) 21.1 g (0.035 mol), BAHF 1.83 g (0.005 mol) 4.57 g (0.0125 mol), 6FDA 17.8 g (0 (.04 mol) was changed to 13.3 g (0.03 mol) and 1.96 g (0.02 mol) of MA was changed to 3.92 g (0.04 mol). AL). Using the obtained resin (AL), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- Viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13 using the obtained varnish (AL). The results are shown in Table 5.
- Example 25 Except that 16.8 g (0.04 mol) of 6FDA was changed to 16.6 g (0.0375 mol) and 1.96 g (0.02 mol) of MA was changed to 2.45 g (0.025 mol), the same as in Example 17.
- a polyamic acid ester resin (AM) was obtained. Using the obtained resin (AM), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- a varnish (AM) having a positive solid content concentration of 20% was prepared in the same manner as in Example 13 except that 7.0 g of the resin (AM) obtained above was added instead of the resin (AA). Produced. Using the obtained varnish (AM), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Example 26 Diamine compound ( ⁇ ) 25.7 g (0.0425 mol) 15.1 g (0.025 mol), BAHF 1.83 g (0.005 mol) 8.23 g (0.0225 mol), 6FDA 17.8 g (0 (.04 mol) was changed to 11.1 g (0.025 mol) and MA 1.96 g (0.02 mol) was changed to 4.90 g (0.05 mol).
- AN Using the obtained resin (AN), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- Example 27 Diamine compound ( ⁇ ) 16.6 g (0.0275 mol) 7.56 g (0.0125 mol), BAHF 1.83 g (0.005 mol) 8.24 g (0.0225 mol), and MAP 3.27 g ( A polyamic acid ester resin (AO) was obtained in the same manner as in Example 13 except that 0.03 mol) was changed to 2.73 g (0.025 mol). Using the obtained resin (AO), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- Example 13 In the same manner as in Example 13 except that 7.0 g of the resin (AO) obtained above was added instead of the resin (AA), a varnish (AO) having a positive solid content concentration of 20% was prepared. Created. Using the obtained varnish (AO), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Example 28 Under a dry nitrogen stream, 7.32 g (0.02 mol) of BAHF, 7.24 g (0.02 mol) of BIS-AT-AF, and 2.18 g (0.02 mol) of MAP were mixed with 50 g of NMP and 26.4 g (0,0) of glycidyl methyl ether. 3 mol) and the temperature of the solution was cooled to -15 ° C. A solution prepared by dissolving 14.7 g of diphenyl ether dicarboxylic acid chloride (manufactured by Nippon Agricultural Chemicals Co., Ltd., 0.050 mol) in 25 g of GBL was added dropwise so that the internal temperature did not exceed 0 ° C.
- diphenyl ether dicarboxylic acid chloride manufactured by Nippon Agricultural Chemicals Co., Ltd., 0.050 mol
- a varnish (AP) of a positive type solid content concentration 20% photosensitive resin composition was prepared in the same manner as in Example 13 except that 7.0 g of the resin (AP) obtained above was added instead of the resin (AA). Created. Using the obtained varnish (AP), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Example 29 7.0 g of the resin (AA) obtained in Example 13 was added to 2.0 g of the quinonediazide compound (b-1), 2.0 g of the phenol compound (e-1), and the alkoxymethyl group-containing thermal bridge obtained in Synthesis Example 3 2.0 g of the agent (d-1), 0.01 g of Megafac F554 (manufactured by DIC) and 52 g of PGMEA were added to obtain a varnish (AA-2) having a positive solid content concentration of 20%. Using the varnish (AA-2), viscosity evaluation and photosensitivity evaluation were performed as described above. The results are shown in Table 5.
- Example 30 7.0 g of the resin (AA) obtained in Example 13 was added to 2.0 g of the quinonediazide compound (b-1), 2.0 g of the phenol compound (e-1), and an alkoxymethyl group-containing thermal crosslinking agent (d-2) 2 0.0 g, Megafuck F554 (manufactured by DIC) 0.01 g and PGMEA 52 g were added to obtain a positive type varnish (AA-3) having a solid content concentration of 20%. Using varnish (AA-3), viscosity evaluation and photosensitivity evaluation were performed as described above. The results are shown in Table 5.
- Example 31 7.0 g of the resin (AA) obtained in Example 13 was added to 2.0 g of a quinonediazide compound (b-1), 2.0 g of a phenol compound (e-1), 2.0 g of a thermal crosslinking agent (d-3), mega Facs F554 (manufactured by DIC) (0.01 g) and PGMEA (52 g) were added to obtain a positive type varnish (AA-4) having a solid content concentration of 20%. Using varnish (AA-4), viscosity evaluation and photosensitivity evaluation were performed as described above. The results are shown in Table 5.
- Example 32 To 10 g of the resin (AA) obtained in Example 13, 1,2-octanedione-1- [4- (phenylthio) -2- (o-benzoyloxime)] (ODPTBO) (manufactured by BASF Japan Ltd.) 0 0.1 g, ethylene oxide-modified bisphenol A dimethacrylate (manufactured by Shin-Nakamura Chemical Co., Ltd., NK ester BPE-100) 2.0 g, trimethylolpropane triacrylate (TPT) 0.5 g, thermal crosslinking agent (d-3) 1.0 g, 0.01 g of Megafac F554 (manufactured by DIC) and 54.4 g of PGMEA were added to obtain a negative photosensitive resin composition varnish (A-5). Using the obtained varnish (AA-5), viscosity evaluation and photosensitivity evaluation were performed as described above. The results are shown in Table 5.
- Example 13 In the same manner as in Example 13 except that 7.0 g of the resin (AQ) obtained above was added instead of the resin (AA), a varnish (Q) having a positive solid content concentration of 20% photosensitive resin composition was obtained. Created. Using the obtained varnish (AQ), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- the temperature of the solution was raised to 120 ° C. for 2 hours and further at 180 ° C. for 2 hours.
- the temperature of this solution was lowered to room temperature, the solution was poured into 2 L of water, and a polymer solid precipitate was collected by filtration. Furthermore, it wash
- AS polyimide resin
- Example 13 In the same manner as in Example 13 except that 7.0 g of the resin (AS) obtained above was added instead of the resin (AA), a varnish (AS) having a positive solid content concentration of 20% was prepared. Created. Using the obtained varnish (AS), viscosity evaluation and photosensitivity evaluation were performed in the same manner as in Example 13. The results are shown in Table 5.
- Comparative Example 14 Instead of 22.2 g (0.05 mol) of 6FDA, 15.5 g (0.05 mol) of ODPA, 15.1 g (0.025 mol) of diamine compound ( ⁇ ) 16.5 g (0.0275 mol), 1.83 g of BAHF (0.005 mol) in the same manner as in Example 13 except that 3.66 g (0.01 mol) and 3.27 g (0.03 mol) of MAP were changed to 2.73 g (0.025 mol). An ester resin (AT) was obtained. Using the obtained resin (AT), the solubility in an organic solvent was evaluated in the same manner as in Example 13.
- Table 4 shows the monomer and end-capping agent compositions used in Resins AA to AZ used in Examples and Comparative Examples, and Table 4 shows the evaluation results of organic solvent solubility, varnish composition, varnish solution viscosity and sensitivity of Resins AA to AZ. As shown in FIG.
- Example 33 7.0 g of the resin (AA) obtained in Example 13 was added to 2.0 g of the quinonediazide compound (b-1), 2.0 g of the phenol compound (e-1), 0.01 g of Megafac F554 (manufactured by DIC) and 99 g of GBL. Was added to obtain a varnish (AA-6) of a positive photosensitive resin composition.
- Switch the slit coater (TS coater manufactured by Toray Engineering Co., Ltd.) to which the positive resist (“OFPR-800” manufactured by Tokyo Ohka Co., Ltd.) was transferred to the varnish (AA-6), and start the pressurized liquid supply.
- the photosensitive resin composition was applied on a 1100 mm ⁇ 960 mm chromium film formation substrate so that the film thickness after drying was 5 ⁇ m.
- Example 34 Liquid feed pump, liquid feed line and cap from the feed tank to the tip of the slit coater (TS coater manufactured by Toray Engineering Co., Ltd.) that fed positive resist (“OFPR-800” manufactured by Tokyo Ohka Co., Ltd.) After washing with a 1 L thinner for a volume of 200 mL, the varnish (AA-6) used in Example 33 was switched to start the pressurized liquid feeding, and the 1100 mm ⁇ 960 mm chromium film-forming substrate was exposed to light. The conductive resin composition was applied so that the film thickness after drying was 5 ⁇ m.
- Comparative Example 22 Liquid feed pump, liquid feed line and cap from the feed tank to the tip of the slit coater (TS coater manufactured by Toray Engineering Co., Ltd.) that fed positive resist (“OFPR-800” manufactured by Tokyo Ohka Co., Ltd.) After washing with a 1 L thinner for a volume of 200 mL, switching to the varnish (AR) used in Comparative Example 21 and starting pressurized liquid feeding, liquid feeding pump, liquid feeding line, and mouthpiece Precipitation of the solid component was confirmed.
- TS coater manufactured by Toray Engineering Co., Ltd. that fed positive resist (“OFPR-800” manufactured by Tokyo Ohka Co., Ltd.)
- OFPR-800 manufactured by Tokyo Ohka Co., Ltd.
- the photosensitive resin composition of the present invention is preferably used for applications such as a surface protective film for semiconductor elements, an interlayer insulating film, an insulating layer for organic EL elements, and a planarizing film for driving TFT substrates of display devices using organic EL elements. Can be used.
Abstract
Description
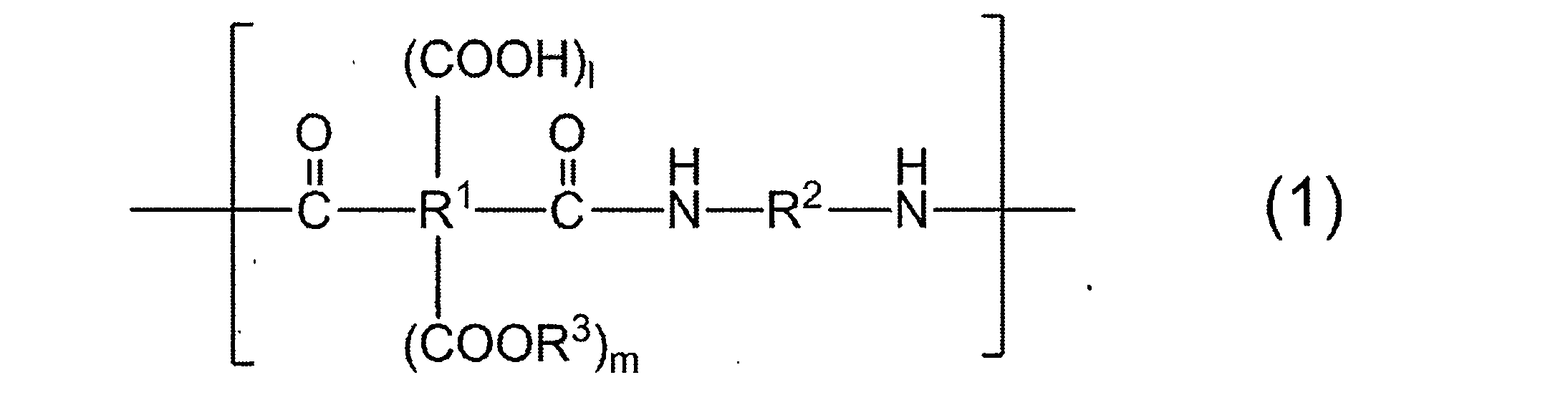
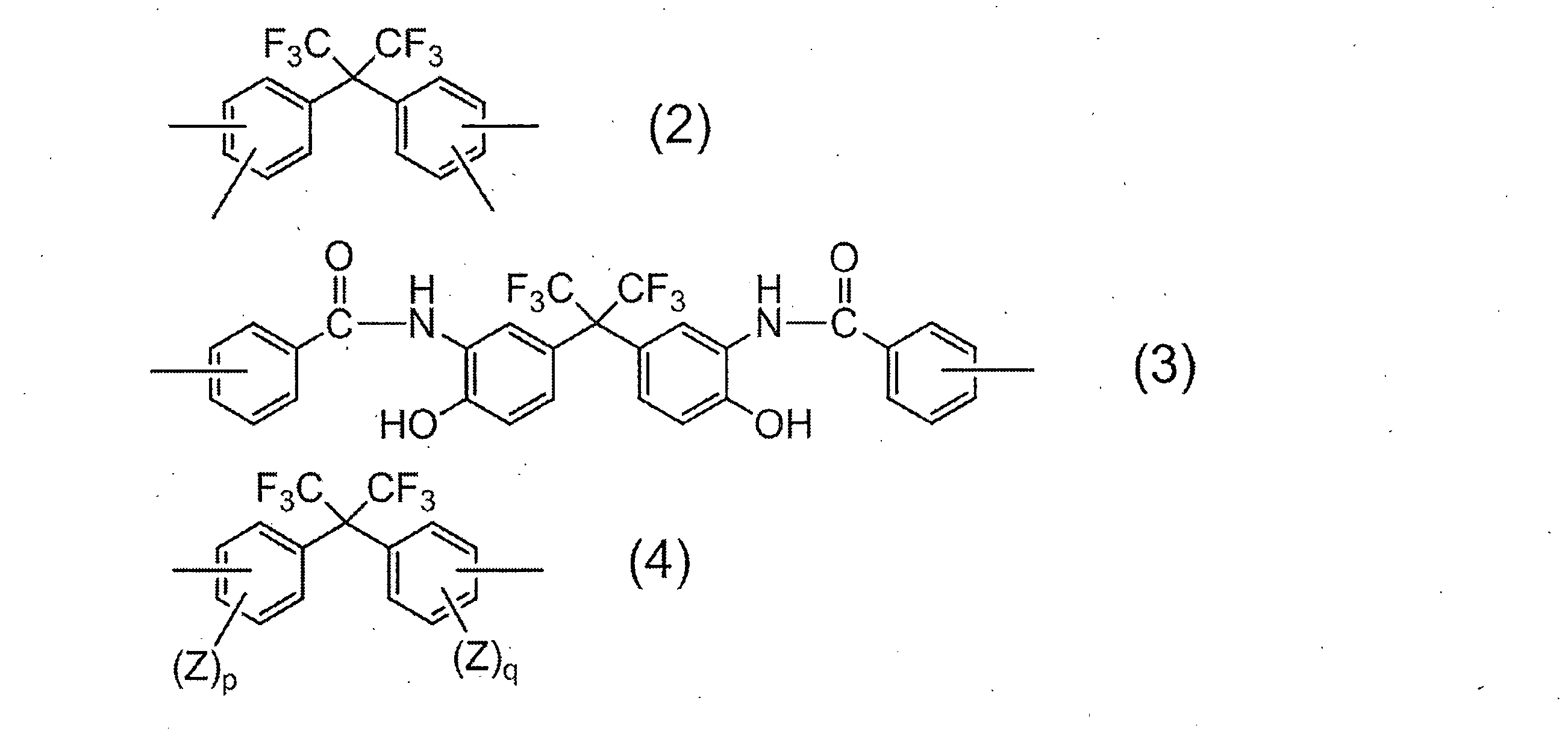
また、本発明の耐熱性樹脂膜の製造方法は、本発明の感光性樹脂組成物を、基板に塗布し感光性樹脂膜を形成する工程、該感光性樹脂膜を乾燥する工程、乾燥した感光性樹脂膜を露光する工程、露光した感光性樹脂膜を現像する工程、および現像した感光性樹脂膜を加熱処理する工程を含む耐熱性樹脂膜の製造方法であって、本発明の感光性樹脂組成物以外の感光性樹脂組成物が送液されていた塗布装置を用いて、該塗布装置の送液経路内の洗浄を行わずに、該塗布装置を用いて請求項1~7いずれかに記載の感光性樹脂組成物を基板に塗布する耐熱性樹脂膜の製造方法である。
本発明の感光性樹脂組成物の一態様としては、(a1)アミド基とトリフルオロメチル基と芳香環を有するプロピレングリコールモノメチルエーテルアセテートに可溶な芳香族アミド樹脂を含有する。

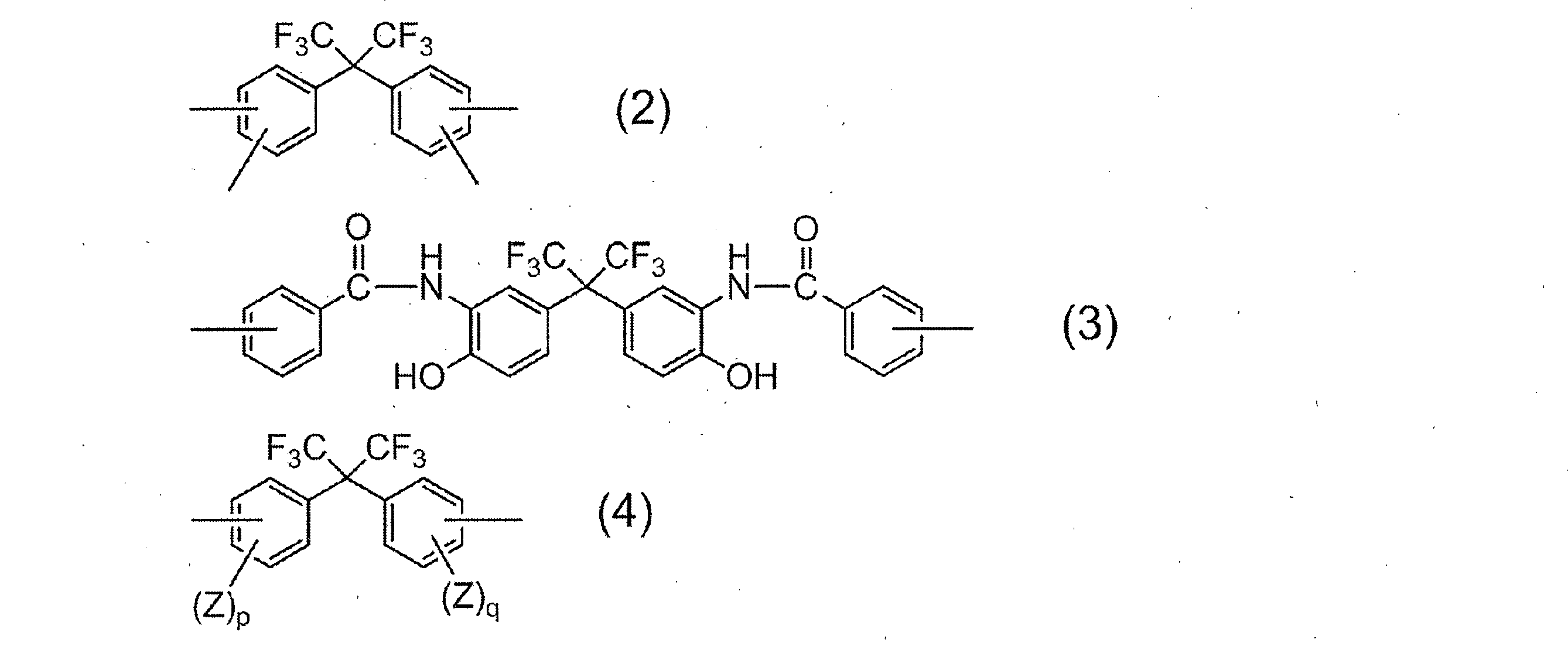
前記一般式(1)におけるR1は酸成分に由来する基を示し、R1が上記式(2)で表される基となる酸成分としては、2,2-ビス(3,4-ジカルボキシフェニル)ヘキサフルオロプロパン、2,2-ビス(2,3-ジカルボキシフェニル)ヘキサフルオロプロパンが挙げられる。
また、R1が、上記式(2)で表される基を有さない場合としては、酸成分が、ジカルボン酸またはトリカルボン酸の場合もある。
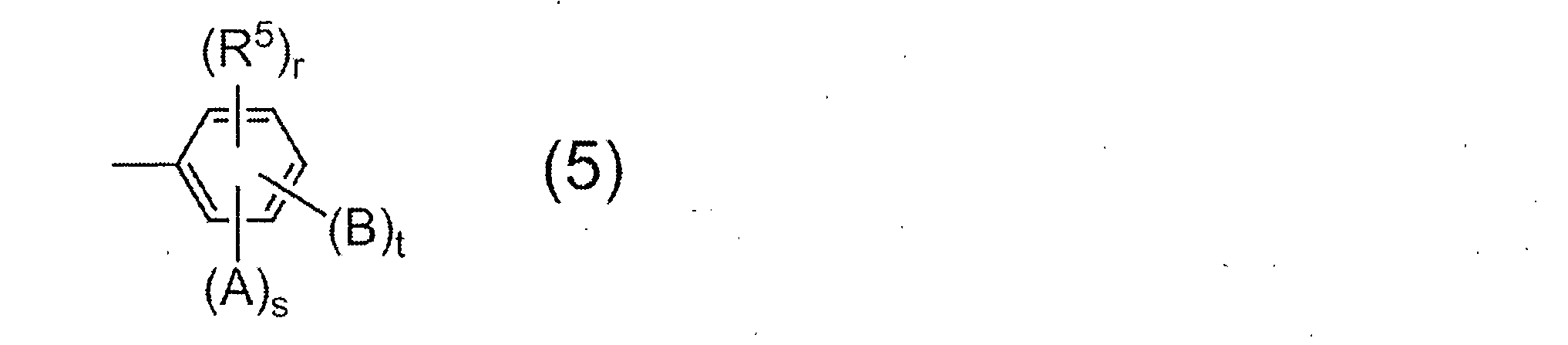
本発明の感光性樹脂組成物は(b)感光剤を含有する。感光剤としては、(b-1)光酸発生剤や、(b-2)光重合開始剤および(b-3)エチレン性不飽和結合を2個以上有する化合物の組み合わせが挙げられる。(b-1)光酸発生剤を含有することで、光照射部に酸が発生して光照射部のアルカリ水溶液に対する溶解性が増大し、光照射部が溶解するポジ型のレリーフパターンを得ることができる。また、(b-1)光酸発生剤とエポキシ化合物または後述する熱架橋剤を含有することで、光照射部に発生した酸がエポキシ化合物や熱架橋剤の架橋反応を促進し、光照射部が不溶化するネガ型のレリーフパターンを得ることができる。また、(b-2)光重合開始剤および(b-3)エチレン性不飽和結合を2個以上有する化合物を含有することで、光照射部に発生した活性ラジカルがエチレン性不飽和結合のラジカル重合を進行させ、光照射部が不溶化するネガ型のレリーフパターンを得ることができる。本発明の感光性樹脂組成物は、(b)感光剤として(b-1)光酸発生剤を含みポジ型の感光性を示すものが好ましい。ポジ型感光性樹脂組成物は、露光・現像工程により微細パターンを得た後、焼成することにより、順テーパー形状のパターンを容易に得ることができる。この順テーパー形状パターンは、有機EL素子の絶縁膜として用いる際に上部電極の被覆性に優れ、断線を防止し素子の信頼性を高めることができる。
本発明の感光性樹脂組成物は、(c)溶剤を含有する。溶剤としては、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、ジエチレングリコールジメチルエーテル、ジエチレングリコールエチルメチルエーテルなどのエーテル類、エチレングリコールモノメチルエーテルアセテート、プロピレングリコールモノメチルエーテルアセテート、酢酸エチル、酢酸ブチル、乳酸メチル、乳酸エチル、乳酸ブチルなどのエステル類、エタノール、イソプロパノール、ブタノール、ペンタノール、3-メチル-2-ブタノール、3-メチル-3-メトキシブタノールなどのアルコール類、メチルエチルケトン、メチルイソブチルケトン、メチルアミルケトン、ジイソブチルケトン、シクロペンタノン、ジアセトンアルコールなどのケトン類、N-メチル-2-ピロリドン、γ-ブチロラクトン、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、ジメチルスルホキシド、1,3-ジメチル-2-イミダゾリジノンなどの極性の非プロトン性溶媒、トルエン、キシレンなどの芳香族炭化水素類などが挙げられる。これらを2種以上含有してもよい。(c)溶剤の含有量は、(a)成分の樹脂100重量部に対して、好ましくは50重量部以上、より好ましくは100重量部以上であり、また、好ましくは2000重量部以下、より好ましくは1500重量部以下である。


上記一般式(7)中、R12は水素原子または炭素数1~6の1価の炭化水素基を示す。jは1または2、kは0または1を示す。ただし、j+kは1または2である。
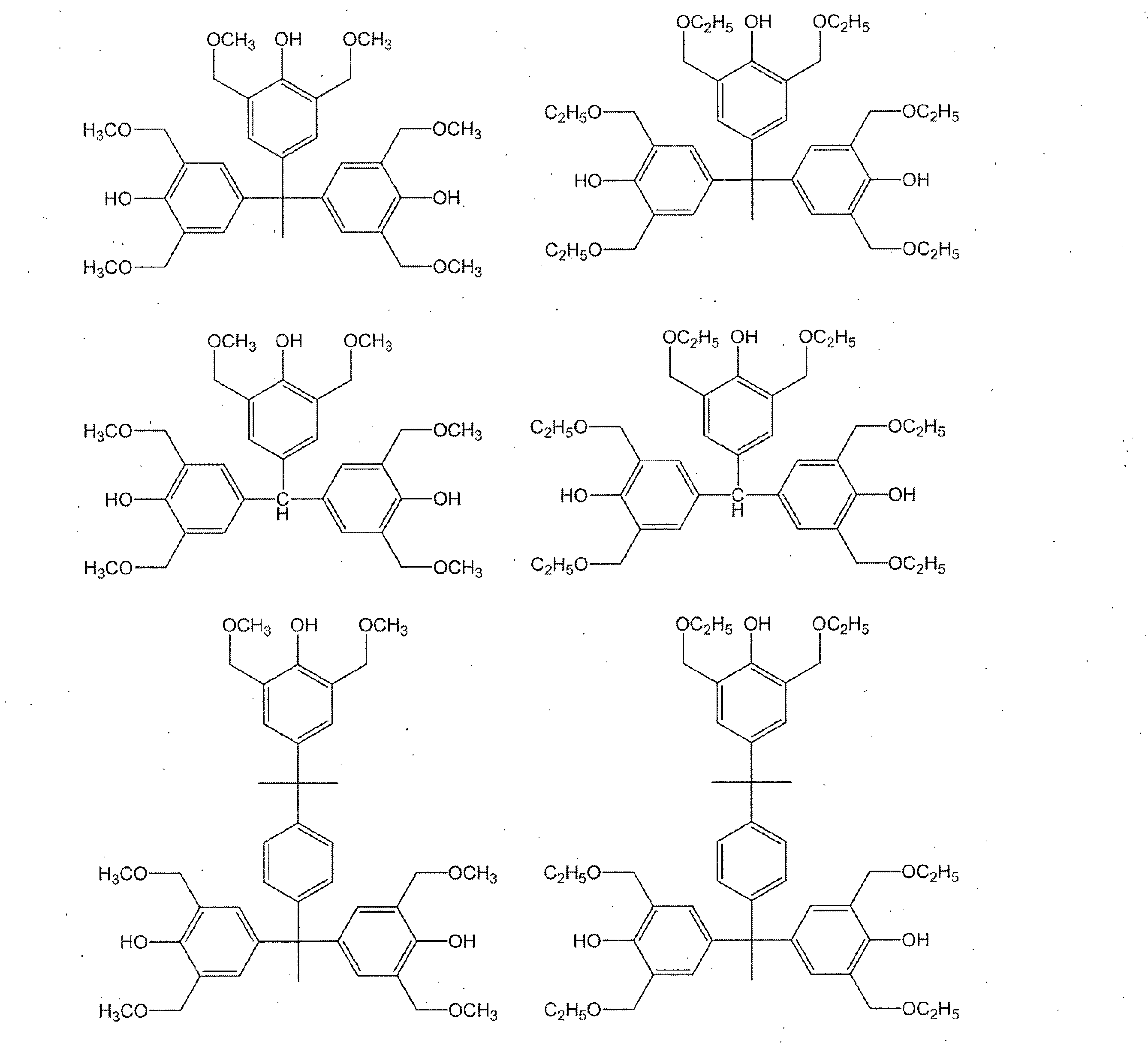
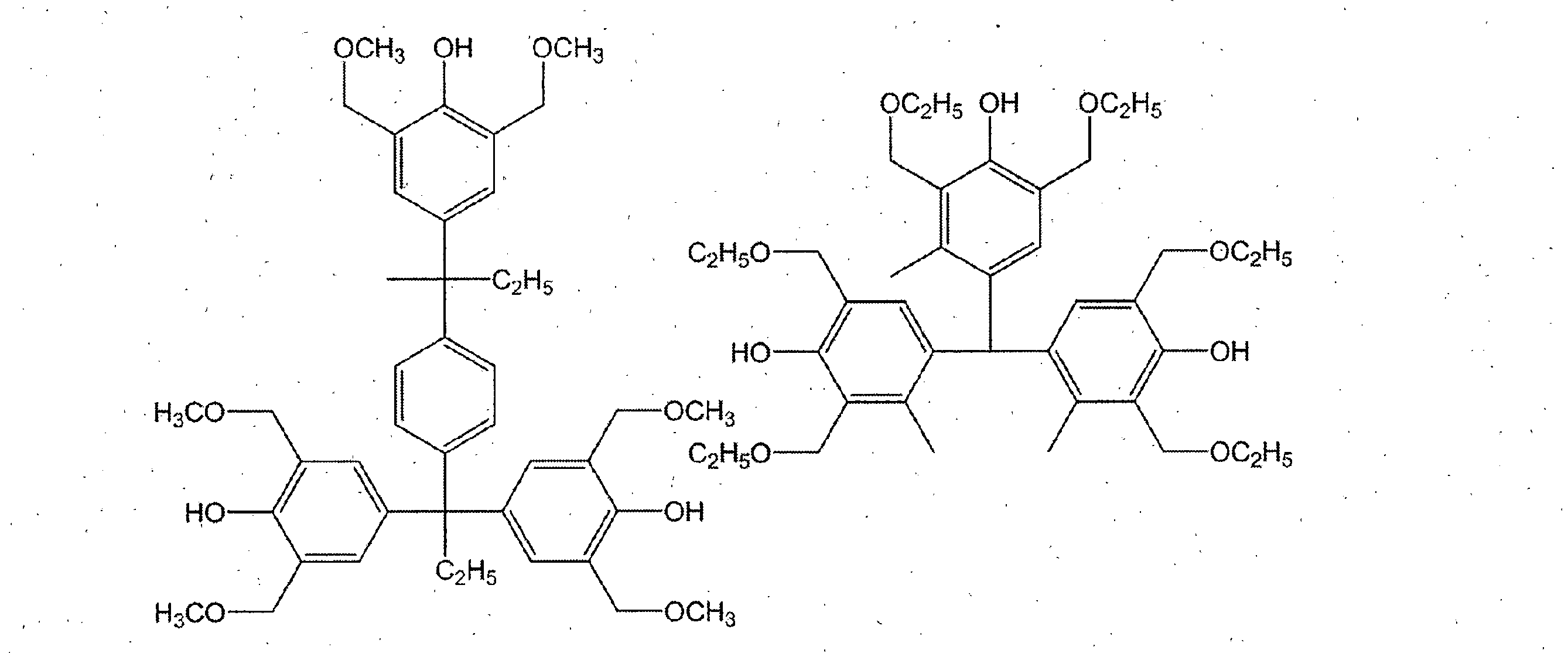
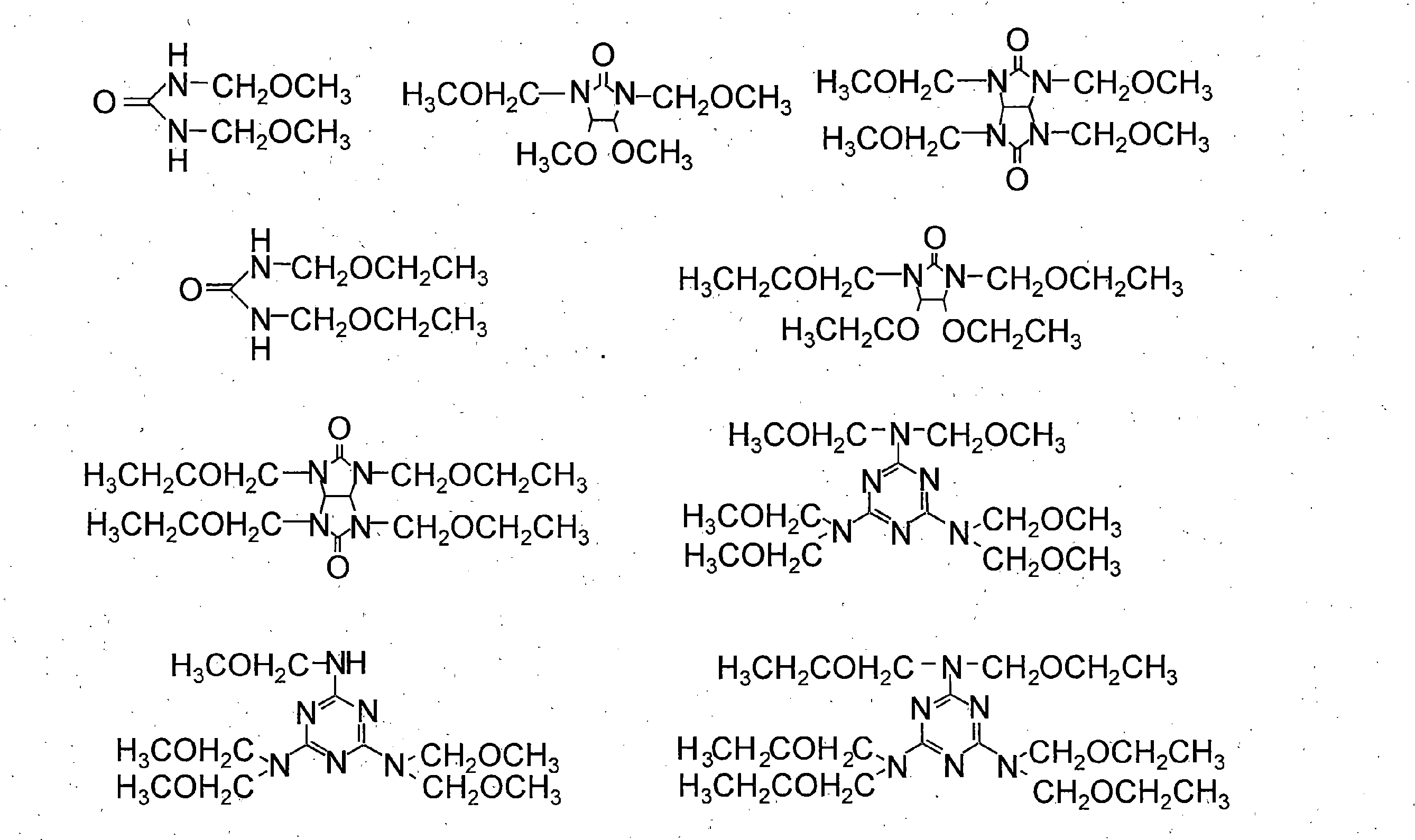
本発明の感光性樹脂組成物は、(e)フェノール性水酸基を有する化合物を含有してもよい。フェノール性水酸基を有する化合物を含有することで、得られる感光性樹脂組成物のアルカリ水溶液に対する溶解性が向上し、高感度化を図ることができる。

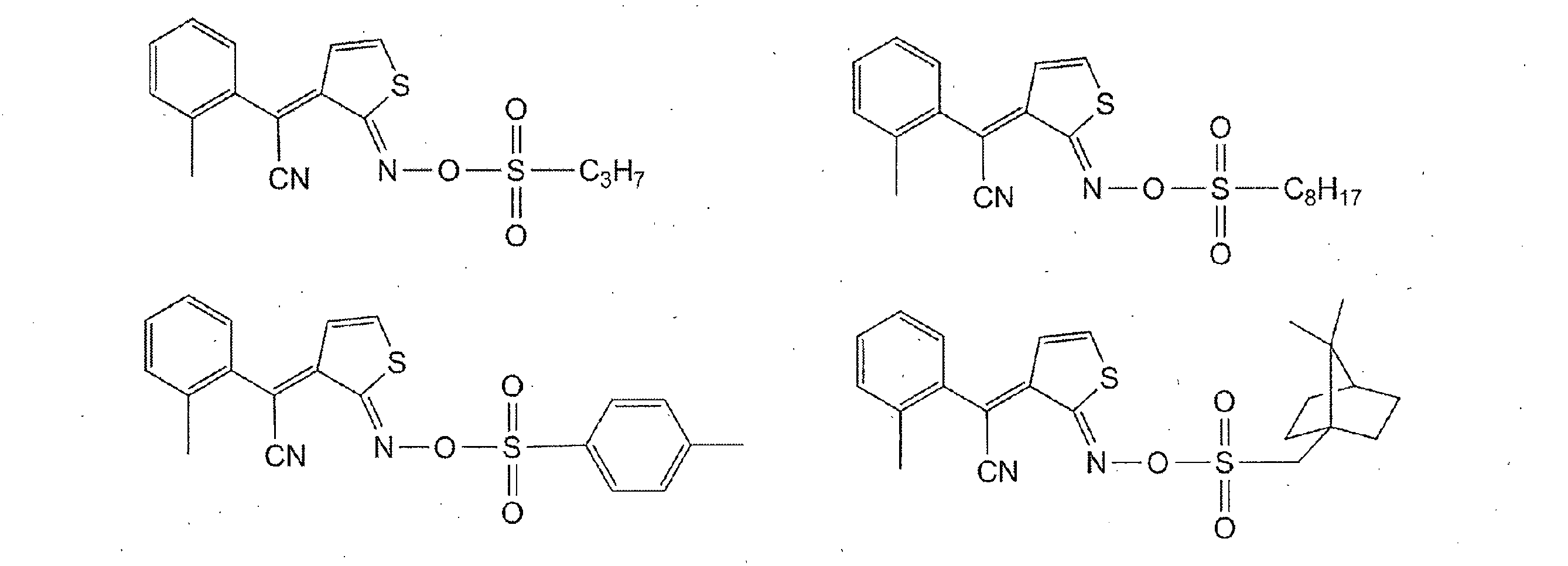


PGMEA(プロピレングリコールモノメチルエーテルアセテート)にポリマー粉末を30重量%の固形分濃度になるように添加して室温で1時間撹拌した後、目視で状態を観察し、不溶物の樹脂の有無を確認した。不溶の樹脂が観察されなかったものを溶解、不溶の樹脂が確認された樹脂を不要と判定した。
(2-1)ポリマー溶液粘度
PGMEAにポリマー粉末を30重量%で溶解し、E型粘度計を用いて25℃で測定した。感光性樹脂組成物中の固形分濃度を高くしてもスリット塗布に適した粘度を保つためには、ポリマー溶液の粘度は150mPa・s未満であることが好ましい。
実施例および比較例で作製した固形分濃度20%の感光性樹脂組成物(ワニス)をE型粘度計を用いて25℃で測定した。スリット塗布により均一な膜厚を形成するためには溶液粘度が1~15cpであることが好ましく、1~10cpであることがより好ましい。25℃におけるワニスの粘度が10cp未満のものを「S」、10cp以上15cp未満のものを「A」、15cp以上のものを「C」と判定した。
現像膜の作製
実施例および比較例で作製した感光性樹脂組成物(ワニス)を8インチシリコンウエハ上に回転塗布し、次いで、ホットプレート(東京エレクトロン(株)製、塗布現像装置Mark-7)を用いて、120℃で2分間熱処理(プリベーク)し、厚さ2.5μmのプリベーク膜を作製した。得られたプリベーク膜を、i線ステッパー((株)ニコン製、NSR-2005i9C)を用いて50~400mJ/cm2の露光量にて10mJ/cm2ステップで露光した。露光後、ネガ型感光性樹脂組成物については100℃で1分間露光後ベークを行った。ポジ型感光性樹脂組成物については露光後、ネガ型感光性樹脂組成物については露光後ベーク後、2.38重量%のテトラメチルアンモニウム(TMAH)水溶液(三菱ガス化学(株)製、ELM-D)で60秒間現像し、次いで純水でリンスし、現像膜を得た。
プリベーク後および現像後の膜厚は、大日本スクリーン製造(株)製光干渉式膜厚測定装置ラムダエースSTM-602を使用し、屈折率1.63として測定した。
現像膜減り量は以下の式に従って算出した。プリベーク後の膜厚が2.5μmであることから、現像膜減り量は0.50μm未満であることが好ましい。現像膜減り量が0.50μm未満の場合は「A」、0.51~0.59μmの場合は「B」、0.60μm以上の場合は「C」と判定した。
感度の算出
露光および現像後、20μmのライン・アンド・スペースパターン(1L/1S)を1対1の幅に形成する露光量(最適露光量Eopという)を感度とした。Ethが200mJ/cm2以下であれば高感度であると判断できる。150mJ/cm2以下がより好ましい。
6FDA:2,2-ビス(3,4-ジカルボキシフェニル)ヘキサフルオロプロパン二無水物
ODPA:3,3’,4,4’-ジフェニルエーテルテトラカルボン酸二無水物
BSAA:2,2-ビス[4-(3,4-ジカルボキシフェノキシ)フェニル]プロパン二無水物
BAHF:2,2-ビス(3-アミノ-4-ヒドロキシフェニル)ヘキサフルオロプロパン
SiDA:1,3-ビス(3-アミノプロピル)テトラメチルジシロキサン
BIS-AT-AF:2,2-ビス(3-アミノ-4-メチルフェニル)ヘキサフルオロプロパン
3,3’-DDS:3,3’-ジアミノジフェニルスルホン
MAP:3-アミノフェノール
ABP:2-アミノ-4-tert-ブチルフェノール
MA:無水マレイン酸
DFA:N,N-ジメチルホルムアミドジメチルアセタール
NMP:N-メチル-2-ピロリドン
PGMEA:プロピレングリコールモノメチルエーテルアセテート
GBL:γ-ブチロラクトン
また、6FDA、ODPA、BSAA、BAHF、SiDA、BIS-AT-AF、3,3’-DDS、MAP、ABPおよびMAについては構造式を以下に示す。

BAHF(セントラル硝子(株)製)18.3g(0.05モル)をアセトン100mL、プロピレンオキシド(東京化成(株)製)17.4g(0.3モル)に溶解させ、-15℃に冷却した。ここに3-ニトロベンゾイルクロリド(東京化成(株)製)20.4g(0.11モル)をアセトン100mLに溶解させた溶液を滴下した。滴下終了後、-15℃で4時間撹拌し、その後室温に戻した。析出した白色固体をろ別し、50℃で真空乾燥した。
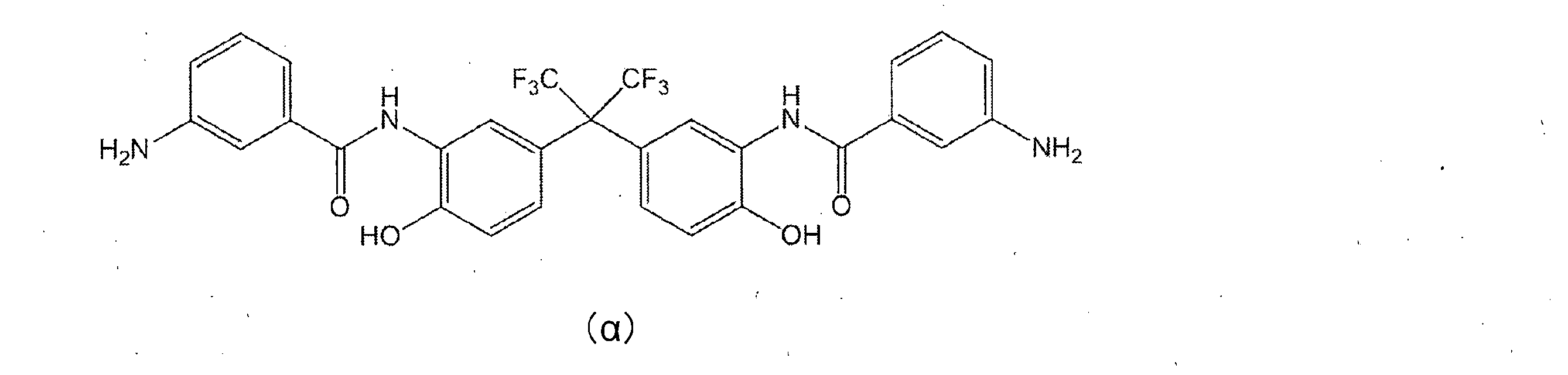
乾燥窒素気流下、TrisP-PA(商品名、本州化学工業(株)製)、21.22g(0.05モル)と5-ナフトキノンジアジドスルホン酸クロリド(東洋合成(株)製、NAC-5)26.8g(0.1モル)を1,4-ジオキサン450gに溶解させ、室温にした。ここに、1,4-ジオキサン50gと混合したトリエチルアミン12.65gを、系内が35℃以上にならないように滴下した。滴下後40℃で2時間撹拌した。トリエチルアミン塩を濾過し、濾液を水に投入した。その後、析出した沈殿を濾過で集め、さらに1%塩酸水1Lで洗浄した。その後、さらに水2Lで2回洗浄した。この沈殿を真空乾燥機で乾燥し、下記式で表されるキノンジアジド化合物(b-1)を得た。

(1)TrisP-HAP(商品名、本州化学工業(株)製)103.2g(0.4モル)を、水酸化ナトリウム80g(2.0モル)を純水800gに溶解させた溶液に溶解させた。完全に溶解させた後、20~25℃で36~38重量%のホルマリン水溶液686gを2時間かけて滴下した。その後20~25℃で17時間撹拌した。これに硫酸98gと水552gを加えて中和を行い、そのまま2日間放置した。放置後に溶液に生じた針状の白色結晶をろ過で集め、水100mLで洗浄した。この白色結晶を50℃で48時間真空乾燥した。乾燥した白色結晶を島津製作所(株)製の高速液体クロマトグラフィーで、カラムにODSを、展開溶媒にアセトニトリル/水=70/30を用い、254nmで分析したところ、出発原料は完全に消失し、純度92%であることがわかった。さらに、重溶媒にDMSO-d6を用いてNMR(日本電子(株)製、GX-270)により分析したところ、ヘキサメチロール化したTrisP-HAPであることがわかった。

アルコキシメチル基含有熱架橋剤(d-2):“ニカラック(登録商標)”MX-270
熱架橋剤(d-3):VG-3101L
フェノール化合物(e-1):BisP-AF

乾燥窒素気流下、合成例1で得られたジアミン化合物(α)15.1g(0.025モル)、BAHF3.66g(0.01モル)およびSiDA(信越化学工業(株)製)0.62g(0.0025モル)をNMP200gに溶解した。ここに6FDA(ダイキン工業(株)製)22.2g(0.05モル)をNMP50gとともに加えて、40℃で1時間撹拌した。その後、MAP(東京化成(株)製)2.73g(0.025モル)を加え、40℃で1時間撹拌した。さらに、DFA(三菱レーヨン(株)製)11.9g(0.1モル)をNMP5gで希釈した溶液を10分かけて滴下し、滴下後、40℃で2時間撹拌を続けた。撹拌終了後、溶液を水2Lに投入して、ポリマー固体の沈殿をろ過で集めた。さらに水2Lで3回洗浄を行い、集めたポリマー固体を50℃の真空乾燥機で72時間乾燥し、ポリアミド酸エステル樹脂(A)を得た。得られた樹脂(A)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
得られた樹脂(A)10gに合成例2で得られたキノンジアジド化合物(b-1)3.0g、フェノール化合物(e-1)1.0gおよびGBL(三菱化学(株)製)50gを加えてポジ型の感光性樹脂組成物のワニス(A-1)を得た。ワニス(A-1)を用いて、前記のように現像膜減り量の評価を行った。結果を表3に示す。
BAHF3.66g(0.01モル)の代わりにBIS-AT-AF(セントラル硝子(株)製)3.62g(0.01モル)を用いた以外は実施例1と同様にしてポリアミド酸エステル樹脂(B)を得た。得られた樹脂(B)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
ジアミン化合物(α)15.1g(0.025モル)を19.3g(0.032モル)、BAHF3.66g(0.01モル)を2.01g(0.0055モル)およびMAP2.73g(0.025モル)を2.18g(0.02モル)とした以外は実施例1と同様にしてポリアミド酸エステル樹脂(C)を得た。得られた樹脂(C)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
乾燥窒素気流下、6FDA22.2g(0.05モル)をNMP200gに溶解した。ここにABP(東京化成(株)製)6.60g(0.04モル)を加えて40℃で1時間撹拌した。その後、ジアミン化合物(α)9.06g(0.015モル)、BIS-AT-AF4.53g(0.0125モル)およびSiDA0.62g(0.0025モル)をNMP50gとともに加え、40℃で2時間撹拌した。さらに、DFA11.9g(0.1モル)をNMP5gで希釈した溶液を10分かけて滴下し、滴下後、40℃で2時間撹拌を続けた。撹拌終了後、溶液を水2Lに投入して、ポリマー固体の沈殿をろ過で集めた。さらに水2Lで3回洗浄を行い、集めたポリマー固体を50℃の真空乾燥機で72時間乾燥し、ポリアミド酸エステル樹脂(D)を得た。得られた樹脂(D)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
乾燥窒素気流下、ジアミン化合物(α)21.2g(0.035モル)、BAHF4.58g(0.0125モル)およびSiDA0.62g(0.0025モル)をNMP200gに溶解した。ここに6FDA13.3g(0.03モル)をNMP50gとともに加えて、40℃で1時間撹拌した。その後、MA(和光純薬工業(株)製)3.92g(0.04モル)を加え、40℃で1時間撹拌した。さらに、DFA11.9g(0.1モル)をNMP5gで希釈した溶液を10分かけて滴下し、滴下後、40℃で2時間撹拌を続けた。撹拌終了後、溶液を水2Lに投入して、ポリマー固体の沈殿をろ過で集めた。さらに水2Lで3回洗浄を行い、集めたポリマー固体を50℃の真空乾燥機で72時間乾燥し、ポリアミド酸エステル樹脂(E)を得た。得られた樹脂(E)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
樹脂(A)の代わりに上記で得られた樹脂(E)10gを加え、さらに熱架橋剤(d-1)1.0gを加えた以外は実施例1と同様にしてポジ型の感光性樹脂組成物のワニス(E)を作製した。得られたワニス(E)を用いて、実施例1と同様に現像膜減り量の評価を行った。結果を表3に示す。
ジアミン化合物(α)21.2g(0.035モル)を25.7g(0.0425モル)、BAHF4.58g(0.0125モル)を1.83g(0.005モル)、6FDA13.3g(0.03モル)を16.7g(0.0375モル)およびMA3.92g(0.04モル)を2.45g(0.025モル)とした以外は実施例5と同様にしてポリアミド酸エステル樹脂(F)を得た。得られた樹脂(F)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
乾燥窒素気流下、ジアミン化合物(α)15.1g(0.025モル)、BAHF8.24g(0.0225モル)およびSiDA0.62g(0.0025モル)をNMP200gに溶解した。ここにMA4.90g(0.05モル)をNMP50gとともに加えて、40℃で1時間撹拌した。その後、6FDA11.1g(0.025モル)を加え、40℃で2時間撹拌した。さらに、DFA11.9g(0.1モル)をNMP5gで希釈した溶液を10分かけて滴下し、滴下後、40℃で2時間撹拌を続けた。撹拌終了後、溶液を水2Lに投入して、ポリマー固体の沈殿をろ過で集めた。さらに水2Lで3回洗浄を行い、集めたポリマー固体を50℃の真空乾燥機で72時間乾燥し、ポリアミド酸エステル樹脂(G)を得た。得られた樹脂(G)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
実施例1で得られた樹脂(A)10gに1,2-オクタンジオン-1-[4-(フェニルチオ)-2-(o-ベンゾイルオキシム)](ODPTBO)(BASFジャパン(株)製)0.1g、エチレンオキサイド変性ビスフェノールAジメタクリレート(新中村化学工業(株)製、NKエステルBPE-100)2.0g、トリメチロールプロパントリアクリレート(TPT)0.5g、合成例3で得られたアルコキシメチル基含有熱架橋剤(d-3)1.0gおよびGBL50gを加えてネガ型の感光性樹脂組成物のワニス(A-2)を得た。得られたワニス(A-2)を用いて、前記のように現像膜減り量の評価を行った。結果を表2に示す。
BAHF2.01g(0.0055モル)を2.93g(0.008モル)およびMAP2.18g(0.02モル)を1.63g(0.015モル)とした以外は実施例3と同様にしてポリアミド酸エステル樹脂(H)を得た。得られた樹脂(H)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
BIS-AT-AF4.53g(0.0125モル)を3.62g(0.01モル)およびABP6.60g(0.04モル)を7.43g(0.045モル)とした以外は実施例4と同様にしてポリアミド酸エステル樹脂(I)を得た。得られた樹脂(I)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
6FDA16.7g(0.0375モル)を17.8g(0.04モル)およびMA2.45g(0.025モル)を1.96g(0.02モル)とした以外は実施例6と同様にしてポリアミド酸エステル樹脂(J)を得た。得られた樹脂(J)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
6FDA11.1g(0.025モル)を9.99g(0.0225モル)、MA4.90g(0.05モル)を5.39g(0.055モル)とした以外は実施例7と同様にして樹脂ポリアミド酸エステル(K)を得た。得られた樹脂(K)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
6FDA22.2g(0.05モル)の代わりにODPA(マナック(株)製)15.5g(0.05モル)を用いた以外は実施例1と同様にしてポリアミド酸エステル樹脂(L)を得た。得られた樹脂(L)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
ジアミン化合物(α)15.1g(0.025モル)を21.2g(0.035モル)とし、BAHFを加えなかった以外は実施例1と同様にしてポリアミド酸エステル樹脂(M)を得た。得られた樹脂(M)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
BAHF3.66g(0.01モル)の代わりに3,3’-DDS(三井化学ファイン(株)製)2.48g(0.01モル)を用いた以外は実施例1と同様にしてポリアミド酸エステル樹脂(N)を得た。得られた樹脂(N)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
ジアミン化合物(α)15.1g(0.025モル)の代わりに3,3’-DDS6.20g(0.025モル)を用いた以外は実施例1と同様にしてポリアミド酸エステル樹脂(O)を得た。得られた樹脂(O)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
ジアミン化合物(α)15.1g(0.025モル)を6.05g(0.01モル)およびBAHF3.66g(0.01モル)を9.15g(0.025モル)とした以外は実施例1と同様にしてポリアミド酸エステル樹脂(P)を得た。得られた樹脂(P)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
ジアミン化合物(α)21.2g(0.035モル)を28.7g(0.0475モル)とし、BAHFを加えなかった以外は実施例5と同様にしてポリアミド酸エステル樹脂(Q)を得た。得られた樹脂(Q)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
乾燥窒素気流下、ジアミン化合物(α)24.2g(0.04モル)、SiDA0.62g(0.0025モル)をNMP250gに溶解させた。ここに6FDA22.2g(0.05モル)をNMP50gとともに加えて、30℃で2時間撹拌した。その後、MAP1.09g(0.01モル)を加え、40℃で2時間撹拌を続けた。さらにピリジン(東京化成(株)製)2.5gをトルエン(東京化成(株)製)15gに希釈して溶液に加え、冷却管を付けて系外に水をトルエンとともに共沸で除去しながら溶液の温度を120℃にして2時間、さらに180℃で2時間反応させた。この溶液の温度を室温まで低下させ、水2Lに溶液を投入し、ポリマー固体の沈殿をろ過で集めた。さらに水2Lで3回洗浄を行い、集めたポリマー固体を50℃の真空乾燥機で72時間乾燥させ、ポリイミド樹脂(R)を得た。得られた樹脂(R)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
ジアミン化合物(α)24.2g(0.04モル)をBAHF10.4g(0.0285モル)、MAP1.09g(0.01モル)を4.09g(0.0375モル)および6FDA22.2g(0.05モル)をODPA15.5g(0.05モル)とした以外は比較例7と同様にしてポリイミド樹脂(S)を得た。得られた樹脂(S)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
乾燥窒素気流下、ジアミン化合物(α)12.1g(0.02モル)、BAHF8.24g(0.0225モル)およびSiDA0.62g(0.0025モル)をNMP200gに溶解した。ここに6FDA6.66g(0.015モル)およびODPA10.9g(0.035モル)をNMP50gとともに加えて、40℃で2時間撹拌した。その後、DFA11.9g(0.1モル)をNMP5gで希釈した溶液を10分かけて滴下し、滴下後、40℃で2時間撹拌を続けた。撹拌終了後、溶液を水2Lに投入して、ポリマー固体の沈殿をろ過で集めた。さらに水2Lで3回洗浄を行い、集めたポリマー固体を50℃の真空乾燥機で72時間乾燥し、ポリアミド酸エステル樹脂(T)を得た。得られた樹脂(T)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。
ジアミン化合物(α)12.1g(0.02モル)を19.3g(0.032モル)、BAHF8.24g(0.0225モル)を2.01g(0.0055モル)および6FDA6.66g(0.015モル)を15.5g(0.035モル)とし、ODPA10.9g(0.035モル)の代わりにBSAA(サビック・ジャパン(株)製)7.81g(0.015モル)を用いた以外は比較例9と同様にしてポリアミド酸エステル樹脂(U)を得た。得られた樹脂(U)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。結果を表2に示す。



乾燥窒素気流下、合成例1で得られたジアミン化合物(α)16.6g(0.0275モル)、BAHF1.83g(0.005モル)およびSiDA(信越化学工業(株)製)0.62g(0.0025モル)をNMP200gに溶解した。ここに6FDA(ダイキン工業(株)製)22.2g(0.05モル)をNMP50gとともに加えて、40℃で1時間撹拌した。その後、MAP(東京化成(株)製)3.27g(0.03モル)を加え、40℃で1時間撹拌した。さらに、DFA(三菱レーヨン(株)製)11.9g(0.1モル)をNMP5gで希釈した溶液を10分かけて滴下し、滴下後、40℃で2時間撹拌を続けた。撹拌終了後、溶液を水2Lに投入して、ポリマー固体の沈殿をろ過で集めた。さらに水2Lで3回洗浄を行い、集めたポリマー固体を50℃の真空乾燥機で72時間乾燥し、ポリアミド酸エステル樹脂(AA)を得た。得られた樹脂(AA)を用いて、上述の方法により有機溶剤に対する溶解性評価を行った。
乾燥窒素気流下、6FDA22.2g(0.05モル)をNMP200gに溶解した。ここにABP(東京化成(株)製)4.95g(0.03モル)を加えて40℃で1時間撹拌した。その後、ジアミン化合物(α)16.6g(0.0275モル)、BAHF1.83g(0.005モル)およびSiDA0.62g(0.0025モル)をNMP50gとともに加え、40℃で2時間撹拌した。さらに、DFA11.9g(0.1モル)をNMP5gで希釈した溶液を10分かけて滴下し、滴下後、40℃で2時間撹拌を続けた。撹拌終了後、溶液を水2Lに投入して、ポリマー固体の沈殿をろ過で集めた。さらに水2Lで3回洗浄を行い、集めたポリマー固体を50℃の真空乾燥機で72時間乾燥し、ポリアミド酸エステル樹脂(AB)を得た。得られた樹脂(AB)を用いて、実施例13と同様に有機溶剤に対する溶解性評価を行った。
BAHF1.83g(0.005モル)を0.91g(0.0025モル)、およびMAP3.27g(0.03モル)を3.81g(0.035モル)とした以外は実施例13と同様にしてポリアミド酸エステル樹脂(AC)を得た。得られた樹脂(AC)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ABP4.95g(0.03モル)を5.73g(0.035モル)、BAHF1.83g(0.005モル)を0.915g(0.0025モル)、とした以外は実施例14と同様にしてポリアミド酸エステル樹脂(AD)を得た。得られた樹脂(AD)を用いて、実施例13と同様に有機溶剤に対する溶解性評価を行った。
乾燥窒素気流下、ジアミン化合物(α)25.7g(0.0425モル)、BAHF1.83g(0.005モル)およびSiDA0.62g(0.0025モル)をNMP200gに溶解した。ここに6FDA17.8g(0.04モル)をNMP50gとともに加えて、40℃で1時間撹拌した。その後、MA(和光純薬工業(株)製)1.96g(0.02モル)を加え、40℃で1時間撹拌した。さらに、DFA11.9g(0.1モル)をNMP5gで希釈した溶液を10分かけて滴下し、滴下後、40℃で2時間撹拌を続けた。撹拌終了後、溶液を水2Lに投入して、ポリマー固体の沈殿をろ過で集めた。さらに水2Lで3回洗浄を行い、集めたポリマー固体を50℃の真空乾燥機で72時間乾燥し、ポリアミド酸エステル樹脂(AE)を得た。得られた樹脂(AE)を用いて、実施例13と同様に有機溶剤に対する溶解性評価を行った。
ABP4.95g(0.03モル)を2.48g(0.015モル)、ジアミン化合物(α)16.6g(0.0275モル)を18.1g(0.03モル)、BAHF1.83g(0.005モル)を3.66g(0.01モル)、とした以外は実施例14と同様にしてポリアミド酸エステル樹脂(AF)を得た。得られた樹脂(AF)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ジアミン化合物(α)16.6g(0.0275モル)を13.6g(0.0225モル)、BAHF1.83g(0.005モル)を7.32g(0.02モル)、およびMAP3.27g(0.03モル)を1.09g(0.01モル)とした以外は実施例13と同様にしてポリアミド酸エステル樹脂(AG)を得た。得られた樹脂(AG)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ABP4.95g(0.03モル)を4.13g(0.025モル)、ジアミン化合物(α)16.6g(0.0275モル)を3.02g(0.005モル)、BAHF1.83g(0.005モル)を10.98g(0.03モル)、とした以外は実施例14と同様にしてポリアミド酸エステル樹脂(AH)を得た。得られた樹脂(AH)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ジアミン化合物(α)16.6g(0.0275モル)を3.02g(0.005モル)、BAHF1.83g(0.005モル)を12.81g(0.035モル)、およびMAP3.27g(0.03モル)を1.64g(0.015モル)とした以外は実施例13と同様にしてポリアミド酸エステル樹脂(AI)を得た。得られた樹脂(AI)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
BAHF1.83g(0.005モル)の代わりにBIS-AT-AF(セントラル硝子(株)製)3.62g(0.01モル)、ジアミン化合物(α)16.6g(0.0275モル)を15.1g(0.025モル)、およびMAP3.27g(0.03モル)を2.73g(0.025モル)とした以外は実施例13と同様にしてポリアミド酸エステル樹脂(AJ)を得た。得られた樹脂(AJ)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ABP4.95g(0.03モル)を6.60g(0.04モル)、BAHF1.83g(0.005モル)の代わりにBIS-AT-AF4.53g(0.0125モル)、ジアミン化合物(α)16.6g(0.0275モル)を9.06g(0.015モル)とした以外は実施例14と同様にしてポリアミド酸エステル樹脂(AK)を得た。得られた樹脂(AK)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ジアミン化合物(α)25.7g(0.0425モル)を21.1g(0.035モル)、BAHF1.83g(0.005モル)を4.57g(0.0125モル)、6FDA17.8g(0.04モル)を13.3g(0.03モル)、MA1.96g(0.02モル)を3.92g(0.04モル)とした以外は実施例17と同様にしてポリアミド酸エステル樹脂(AL)を得た。得られた樹脂(AL)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
6FDA17.8g(0.04モル)を16.6g(0.0375モル)、MA1.96g(0.02モル)を2.45g(0.025モル)とした以外は実施例17と同様にしてポリアミド酸エステル樹脂(AM)を得た。得られた樹脂(AM)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ジアミン化合物(α)25.7g(0.0425モル)を15.1g(0.025モル)、BAHF1.83g(0.005モル)を8.23g(0.0225モル)、6FDA17.8g(0.04モル)を11.1g(0.025モル)、MA1.96g(0.02モル)を4.90g(0.05モル)とした以外は実施例17と同様にしてポリアミド酸エステル樹脂(AN)を得た。得られた樹脂(AN)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ジアミン化合物(α)16.6g(0.0275モル)を7.56g(0.0125モル)、BAHF1.83g(0.005モル)を8.24g(0.0225モル)、およびMAP3.27g(0.03モル)を2.73g(0.025モル)とした以外は実施例13と同様にしてポリアミド酸エステル樹脂(AO)を得た。得られた樹脂(AO)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
乾燥窒素気流下、BAHF7.32g(0.02モル)、BIS-AT-AF7.24g(0.02モル)、およびMAP2.18g(0.02モル)をNMP50g、グリシジルメチルエーテル26.4g(0.3モル)に溶解させ、溶液の温度を-15℃まで冷却した。ここにジフェニルエーテルジカルボン酸クロリド14.7g(日本農薬(株)製、0.050モル)をGBL25gに溶解させた溶液を、内部の温度が0℃を超えないように滴下した。滴下終了後、6時間-15℃で撹拌を続けた、反応終了後、溶液をメタノールを10重量%含んだ水3Lに投入して白色の沈殿を析出させた。この沈殿をろ過で集めて、水で3回洗浄した後、50℃の真空管早期で72時間乾燥し、アルカリ可溶性のポリベンゾオキサゾール前駆体(AP)を得た。得られた樹脂(AP)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
実施例13で得られた樹脂(AA)7.0gにキノンジアジド化合物(b-1)2.0g、フェノール化合物(e-1)2.0g、合成例3で得られたアルコキシメチル基含有熱架橋剤(d-1)2.0g、メガファックF554(DIC製)0.01gおよびPGMEA52gを加えてポジ型の固形分濃度20%感光性樹脂組成物のワニス(AA-2)を得た。ワニス(AA-2)を用いて、前記のように粘度評価と感光性評価を行った。結果を表5に示す。
実施例13で得られた樹脂(AA)7.0gにキノンジアジド化合物(b-1)2.0g、フェノール化合物(e-1)2.0g、アルコキシメチル基含有熱架橋剤(d-2)2.0g、メガファックF554(DIC製)0.01gおよびPGMEA52gを加えてポジ型の固形分濃度20%感光性樹脂組成物のワニス(AA-3)を得た。ワニス(AA-3)を用いて、前記のように粘度評価と感光性評価を行った。結果を表5に示す。
実施例13で得られた樹脂(AA)7.0gにキノンジアジド化合物(b-1)2.0g、フェノール化合物(e-1)2.0g、熱架橋剤(d-3)2.0g、メガファックF554(DIC製)0.01gおよびPGMEA52gを加えてポジ型の固形分濃度20%感光性樹脂組成物のワニス(AA-4)を得た。ワニス(AA-4)を用いて、前記のように粘度評価と感光性評価を行った。結果を表5に示す。
実施例13で得られた樹脂(AA)10gに1,2-オクタンジオン-1-[4-(フェニルチオ)-2-(o-ベンゾイルオキシム)](ODPTBO)(BASFジャパン(株)製)0.1g、エチレンオキサイド変性ビスフェノールAジメタクリレート(新中村化学工業(株)製、NKエステルBPE-100)2.0g、トリメチロールプロパントリアクリレート(TPT)0.5g、熱架橋剤(d-3)1.0g、メガファックF554(DIC製)0.01gおよびPGMEA54.4gを加えてネガ型の感光性樹脂組成物のワニス(A-5)を得た。得られたワニス(AA-5)を用いて、前記のように粘度評価と感光性評価を行った。結果を表5に示す。
乾燥窒素気流下、BAHF18.3g(0.05モル)をNMP50g、グリシジルメチルエーテル26.4g(0.3モル)に溶解させ、溶液の温度を-15℃まで冷却した。ここにジフェニルエーテルジカルボン酸クロリド14.7g(日本農薬(株)製、0.050モル)をGBL25gに溶解させた溶液を、内部の温度が0℃を超えないように滴下した。滴下終了後、6時間-15℃で撹拌を続けた、反応終了後、溶液をメタノールを10重量%含んだ水3Lに投入して白色の沈殿を析出させた。この沈殿をろ過で集めて、水で3回洗浄した後、50℃の真空管早期で72時間乾燥し、アルカリ可溶性のポリベンゾオキサゾール前駆体(AQ)を得た。得られた樹脂(AQ)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
6FDA22.2g(0.05モル)の代わりにODPA15.5g(0.05モル)、ジアミン化合物(α)16.6g(0.0275モル)を24.18g(0.04モル)、MAP3.27g(0.03モル)を1.09g(0.01モル)とし、BAHFを加えなかった以外は実施例13と同様にしてポリアミド酸エステル樹脂(AR)を得た。得られた樹脂(AR)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
乾燥窒素気流下、BAHF15.46g(0.04225モル)、SiDA0.62g(0.0025モル)をNMP250gに溶解した。ここにODPA15.5g(0.05モル)をNMP50gとともに加えて、30℃で2時間撹拌した。その後、MAP(東京化成(株)製)1.09g(0.01モル)を加え、40℃で2時間撹拌を続けた。さらにピリジン(東京化成(株)製)2.5gをトルエン(東京化成(株)製)15gに希釈して溶液に加え、冷却管を付けて系外に水をトルエンとともに共沸で除去しながら溶液の温度を120℃にして2時間、さらに180℃で2時間反応させた。この溶液の温度を室温まで低下させ、水2Lに溶液を投入し、ポリマー固体の沈殿をろ過で集めた。さらに水2Lで3回洗浄を行い、集めたポリマー固体を50℃の真空乾燥機で72時間乾燥させ、ポリイミド樹脂(AS)を得た。得られた樹脂(AS)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
6FDA22.2g(0.05モル)の代わりにODPA15.5g(0.05モル)、ジアミン化合物(α)16.6g(0.0275モル)を15.1g(0.025モル)、BAHF1.83g(0.005モル)を3.66g(0。01モル)、およびMAP3.27g(0.03モル)を2.73g(0.025モル)とした以外は実施例13と同様にしてポリアミド酸エステル樹脂(AT)を得た。得られた樹脂(AT)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ジアミン化合物(α)16.6g(0.0275モル)を21.1g(0.035モル)、MAP3.27g(0.03モル)を2.73g(0.025モル)とし、BAHFを加えなかった以外は実施例13と同様にしてポリアミド酸エステル樹脂(AU)を得た。得られた樹脂(AU)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ジアミン化合物(α)16.6g(0.0275モル)を15.1g(0.0225モル)、BAHF1.83g(0.005モル)の代わりに3,3’-DDS(三井化学ファイン(株)製)を2.48g(0.01モル)、およびMAP3.27g(0.03モル)を2.73g(0.025モル)とした以外は実施例13と同様にしてポリアミド酸エステル樹脂(AV)を得た。得られた樹脂(AV)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ジアミン化合物(α)16.6g(0.0275モル)の代わりに3,3’-DDSを6.20g(0.025モル)、BAHF1.83g(0.005モル)を3.66g(0.01モル)、およびMAP3.27g(0.03モル)を2.73g(0.025モル)とした以外は実施例13と同様にしてポリアミド酸エステル樹脂(AW)を得た。得られた樹脂(AW)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ジアミン化合物(α)25.7g(0.0425モル)を28.7g(0.0475モル)、6FDA17.8g(0.04モル)を13.3g(0.03モル)、MA1.96g(0.02モル)を3.92g(0.04モル)とし、BAHFを加えなかった以外は実施例17と同様にしてポリアミド酸エステル樹脂(AX)を得た。得られた樹脂(AX)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
乾燥窒素気流下、ジアミン化合物(α)12.1g(0.02モル)、BAHF8.24g(0.0225モル)およびSiDA0.62g(0.0025モル)をNMP200gに溶解した。ここに6FDA6.66g(0.015モル)およびODPA10.9g(0.035モル)をNMP50gとともに加えて、40℃で2時間撹拌した。その後、DFA11.9g(0.1モル)をNMP5gで希釈した溶液を10分かけて滴下し、滴下後、40℃で2時間撹拌を続けた。撹拌終了後、溶液を水2Lに投入して、ポリマー固体の沈殿をろ過で集めた。さらに水2Lで3回洗浄を行い、集めたポリマー固体を50℃の真空乾燥機で72時間乾燥し、ポリアミド酸エステル樹脂(AY)を得た。得られた樹脂(AY)を用いて実施例13と同様に有機溶剤に対する溶解性評価を行った。
ジアミン化合物(α)12.1g(0.02モル)を19.3g(0.032モル)、BAHF8.24g(0.0225モル)を2.01g(0.0055モル)および6FDA6.66g(0.015モル)を15.5g(0.035モル)とし、ODPA10.9g(0.035モル)の代わりにBSAA(サビック・ジャパン(株)製)7.81g(0.015モル)を用いた以外は比較例19と同様にしてポリアミド酸エステル樹脂(AZ)を得た。得られた樹脂(AZ)を用いて、上述の方法により有機溶剤に対する溶解性評価とポリマー溶液粘度の測定を行った。

実施例13で得られた樹脂(AA)7.0gに、キノンジアジド化合物(b-1)2.0g、フェノール化合物(e-1)2.0g、メガファックF554(DIC製)0.01gおよびGBL99gを加えてポジ型の感光性樹脂組成物のワニス(AA-6)を得た。ポジ型レジスト(東京応化(株)製“OFPR-800”)を送液したスリットコーター(東レエンジニアリング(株)製TSコーター)を上記ワニス(AA-6)に切り替えて、加圧送液を開始し、1100mm×960mmのクロム成膜基板に感光性樹脂組成物を乾燥後の膜厚が5μmとなるように塗布した。
ポジ型レジスト(東京応化(株)製“OFPR-800”)を送液したスリットコーター(東レエンジニアリング(株)製TSコーター)の送液タンクから口金先端までの送液ポンプ、送液ラインおよび口金の容量200mLに対して、1Lのシンナーで洗浄を行った後、実施例33で用いたワニス(AA-6)に切り替えて、加圧送液を開始し、1100mm×960mmのクロム成膜基板に感光性樹脂組成物を乾燥後の膜厚が5μmとなるように塗布した。
比較例12で得られた樹脂(AR)7.0gに、キノンジアジド化合物(b-1)2.0g、フェノール化合物(e-1)2.0g、メガファックF554(DIC製)0.01gおよびGBL99gを加えてポジ型の感光性樹脂組成物のワニス(AR)を得た。実施例33と同様にしてポジ型レジスト(東京応化(株)製“OFPR-800”)を送液したスリットコーター(東レエンジニアリング(株)製TSコーター)を上記ワニス(AR)に切り替えて加圧送液を開始したところ、送液ポンプ内、送液ライン内および口金内において固体成分の析出を確認した。
ポジ型レジスト(東京応化(株)製“OFPR-800”)を送液したスリットコーター(東レエンジニアリング(株)製TSコーター)の送液タンクから口金先端までの送液ポンプ、送液ラインおよび口金の容量200mLに対して、1Lのシンナーで洗浄を行った後、比較例21で用いたワニス(AR)に切り替えて加圧送液を開始したところ、送液ポンプ内、送液ライン内および口金内において固体成分の析出を確認した。
Claims (12)
- (a1)アミド基とトリフルオロメチル基と芳香環を有する、プロピレングリコールモノメチルエーテルアセテートに可溶な芳香族アミド樹脂、(b)感光剤、および(c)溶剤を含む感光性樹脂組成物であって、固形分濃度20重量%、25℃における粘度が1~15cpである感光性樹脂組成物。
- (a2)アミド基とアミド酸エステル基とトリフルオロメチル基と芳香環を有する、プロピレングリコールモノメチルエーテルアセテートに可溶な芳香族アミド樹脂、(b)感光剤、および(c)溶剤を含む感光性樹脂組成物であって、固形分濃度20重量%、25℃における粘度が1~15cpである感光性樹脂組成物。
- (a)一般式(1)で表される構造を主な繰り返し単位として有する樹脂、(b)感光剤および(c)溶剤を含有する感光性樹脂組成物。

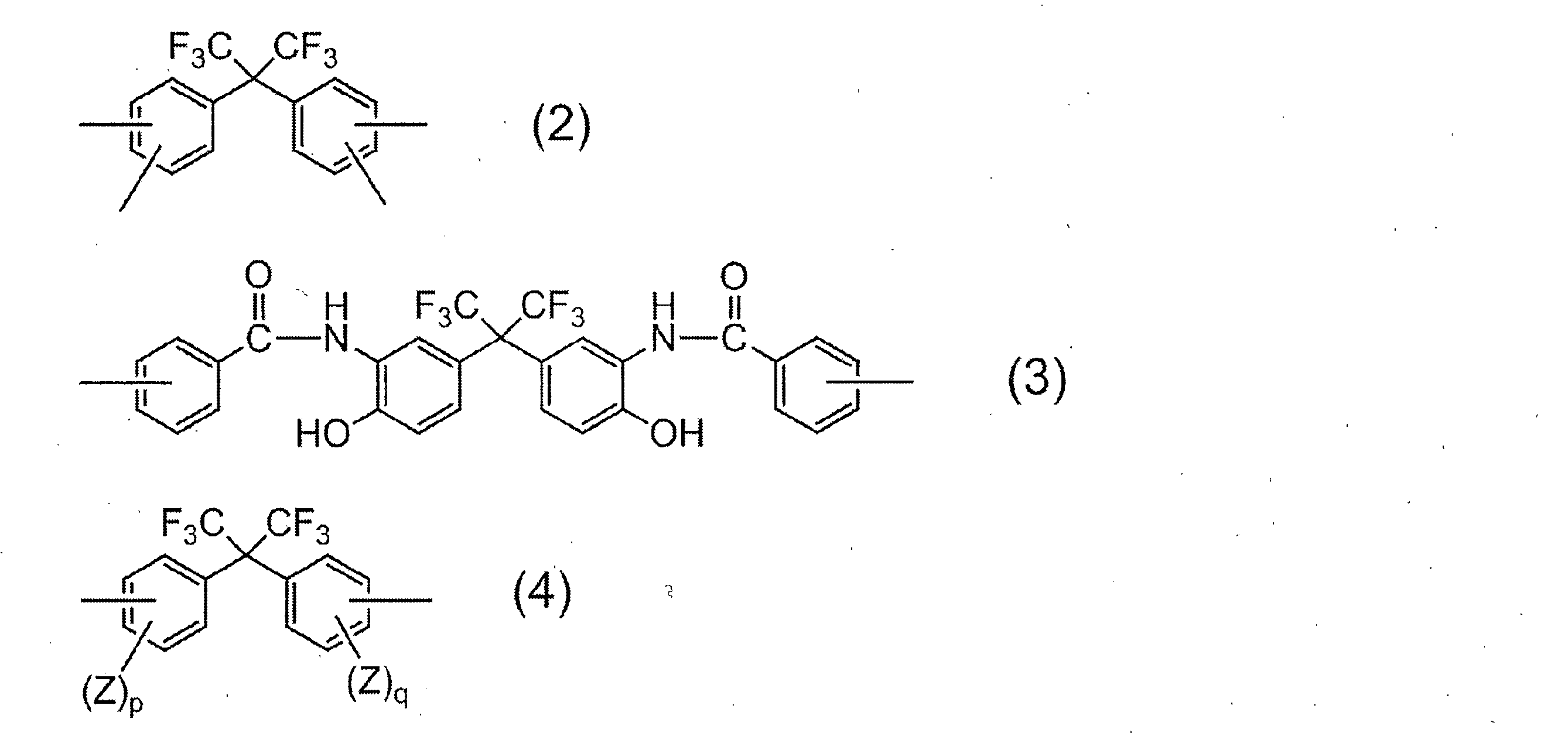
- 前記(a)一般式(1)で表される構造を主な繰り返し単位として有する樹脂が、モノマー成分であるテトラカルボン酸100モル%に対して10~100モル%のモノアミンにより末端封止されたものである請求項3記載の感光性樹脂組成物。
- 前記(a)一般式(1)で表される構造を主な繰り返し単位として有する樹脂が、モノマー成分であるジアミン100モル%に対して10~100モル%の酸無水物により末端封止されたものである請求項3記載の感光性樹脂組成物。
- 前記モノアミンが一般式(5)で表される基を有するモノアミンである請求項4記載の感光性樹脂組成物。
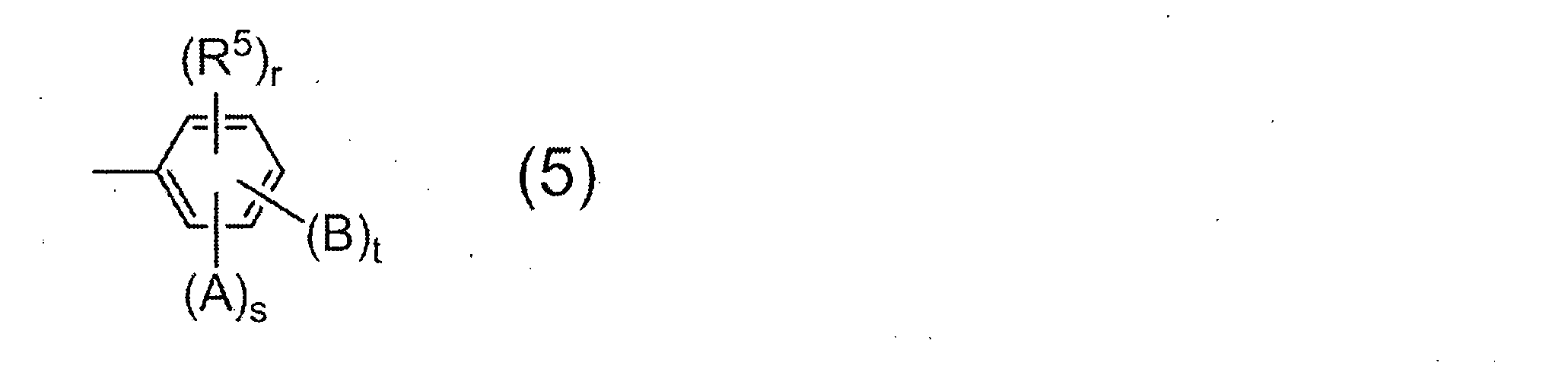
- 前記(b)感光剤がキノンジアジド化合物である請求項1~6のいずれかに記載の感光性樹脂組成物。
- 請求項1~7いずれかに記載の感光性樹脂組成物を基板に塗布し感光性樹脂膜を形成する工程、該感光性樹脂膜を乾燥する工程、乾燥した感光性樹脂膜を露光する工程、露光した感光性樹脂膜を現像する工程、および現像した感光性樹脂膜を加熱処理する工程を含む耐熱性樹脂膜の製造方法。
- 請求項1~7いずれかに記載の感光性樹脂組成物を、基板に塗布し感光性樹脂膜を形成する工程、該感光性樹脂膜を乾燥する工程、乾燥した感光性樹脂膜を露光する工程、露光した感光性樹脂膜を現像する工程、および現像した感光性樹脂膜を加熱処理する工程を含む耐熱性樹脂膜の製造方法であって、請求項1~7いずれかに記載の感光性樹脂組成物以外の感光性樹脂組成物が送液されていた塗布装置を用いて、該塗布装置の送液経路内の洗浄を行わずに、該塗布装置を用いて請求項1~7いずれかに記載の感光性樹脂組成物を基板に塗布する耐熱性樹脂膜の製造方法。
- 前記請求項1~7いずれかに記載の感光性樹脂組成物以外の感光性樹脂組成物が、エチレングリコールモノエチルエーテルアセテート、エチル-3-エトキシプロピオネート、メチル-3-メトキシプロピオネート、2-ヘプタノン、プロピレングリコールモノメチルエーテルアセテート、シクロヘキサノンから選ばれる少なくとも1種類の溶媒を含有する感光性樹脂組成物である請求項9に記載の耐熱性樹脂膜の製造方法。
- 基板上に形成された第一電極と第一電極を部分的に露出せしめるように第一電極上に形成された絶縁層と、第一電極に対向して設けられた第二電極とを含む表示装置であって、前記絶縁層が、請求項8~10いずれかに記載の耐熱性樹脂膜の製造方法により得られる耐熱性樹脂膜であることを特徴とする表示装置。
- 薄膜トランジスタ(TFT)が形成された基板上の凹凸を覆う状態で設けられた平坦化膜と、平坦化膜上に設けられた表示素子とを備えてなる表示装置であって、前記平坦化膜が請求項8~10いずれかに記載の耐熱性樹脂膜の製造方法により得られる耐熱性樹脂膜であることを特徴とする表示装置。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201380065421.6A CN104854508B (zh) | 2012-12-20 | 2013-12-13 | 感光性树脂组合物、耐热性树脂膜的制造方法及显示装置 |
| KR1020157018049A KR101942150B1 (ko) | 2012-12-20 | 2013-12-13 | 감광성 수지 조성물, 내열성 수지막의 제조 방법 및 표시 장치 |
| EP13864249.1A EP2937732B1 (en) | 2012-12-20 | 2013-12-13 | Photosensitive resin composition, method for producing heat-resistant resin film and display device |
| US14/647,747 US9897915B2 (en) | 2012-12-20 | 2013-12-13 | Photosensitive resin composition, method for producing heat-resistant resin film and display device |
| SG11201504647VA SG11201504647VA (en) | 2012-12-20 | 2013-12-13 | Photosensitive resin composition, method for producing heat-resistant resin film and display device |
| JP2014500579A JP6332022B2 (ja) | 2012-12-20 | 2013-12-13 | 感光性樹脂組成物、耐熱性樹脂膜の製造方法および表示装置 |
| KR1020197001652A KR20190009002A (ko) | 2012-12-20 | 2013-12-13 | 감광성 수지 조성물, 내열성 수지막의 제조 방법 및 표시 장치 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012277743 | 2012-12-20 | ||
| JP2012-277743 | 2012-12-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014097992A1 true WO2014097992A1 (ja) | 2014-06-26 |
Family
ID=50978324
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/083491 WO2014097992A1 (ja) | 2012-12-20 | 2013-12-13 | 感光性樹脂組成物、耐熱性樹脂膜の製造方法および表示装置 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US9897915B2 (ja) |
| EP (1) | EP2937732B1 (ja) |
| JP (2) | JP6332022B2 (ja) |
| KR (2) | KR101942150B1 (ja) |
| CN (2) | CN110147031A (ja) |
| SG (1) | SG11201504647VA (ja) |
| TW (1) | TWI568797B (ja) |
| WO (1) | WO2014097992A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016133741A (ja) * | 2015-01-21 | 2016-07-25 | 住友ベークライト株式会社 | 感光性樹脂組成物、および電子装置 |
| WO2016158406A1 (ja) * | 2015-03-27 | 2016-10-06 | 東レ株式会社 | 薄膜トランジスタ用感光性樹脂組成物、硬化膜、薄膜トランジスタ、液晶表示装置または有機電界発光表示装置、硬化膜の製造方法、薄膜トランジスタの製造方法および液晶表示装置または有機電界発光表示装置の製造方法 |
| US9740101B2 (en) * | 2015-11-13 | 2017-08-22 | Globalfoundries Inc. | Additions of organic species to facilitate crosslinker removal during PSPI cure |
| CN107428935A (zh) * | 2015-03-27 | 2017-12-01 | 东丽株式会社 | 二胺化合物、使用其的耐热性树脂或耐热性树脂前体 |
| KR20180012308A (ko) | 2015-06-30 | 2018-02-05 | 후지필름 가부시키가이샤 | 전구체 조성물, 감광성 수지 조성물, 전구체 조성물의 제조 방법, 경화막, 경화막의 제조 방법 및 반도체 디바이스 |
| WO2018066395A1 (ja) * | 2016-10-05 | 2018-04-12 | 東レ株式会社 | 樹脂組成物、硬化膜、半導体装置およびそれらの製造方法 |
| WO2020059485A1 (ja) * | 2018-09-18 | 2020-03-26 | 東レ株式会社 | 感光性樹脂組成物、樹脂シート、硬化膜、有機el表示装置、半導体電子部品、半導体装置、および有機el表示装置の製造方法 |
| CN117186403A (zh) * | 2023-09-01 | 2023-12-08 | 明士(北京)新材料开发有限公司 | 一种负性光敏性树脂、树脂组合物及其制备方法与应用 |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015194892A1 (ko) * | 2014-06-20 | 2015-12-23 | 코오롱인더스트리 주식회사 | 광가교성 수지 조성물, 이로부터 형성된 절연막 및 유기발광소자 |
| JP6711273B2 (ja) * | 2014-11-27 | 2020-06-17 | 東レ株式会社 | 樹脂および感光性樹脂組成物 |
| JP6658548B2 (ja) | 2015-01-23 | 2020-03-04 | 日立化成デュポンマイクロシステムズ株式会社 | ポジ型感光性樹脂組成物、パターン硬化膜の製造方法、パターン硬化膜及び電子部品 |
| EP3270664A4 (en) * | 2015-03-11 | 2018-10-31 | Toray Industries, Inc. | Organic el display device and method for manufacturing same |
| WO2016152794A1 (ja) * | 2015-03-24 | 2016-09-29 | 東レ株式会社 | 感光性樹脂組成物 |
| JP6743692B2 (ja) * | 2015-03-27 | 2020-08-19 | 東レ株式会社 | 感光性樹脂組成物、感光性シート、半導体装置および半導体装置の製造方法 |
| TWI704418B (zh) * | 2015-06-30 | 2020-09-11 | 日商富士軟片股份有限公司 | 負型感光性樹脂組成物、硬化膜、硬化膜的製造方法及半導體元件 |
| WO2017038828A1 (ja) * | 2015-09-03 | 2017-03-09 | 東レ株式会社 | ポジ型感光性樹脂組成物、その樹脂組成物により形成された未硬化の樹脂パターン、硬化樹脂パターン、およびそれを用いた半導体装置とその製造方法 |
| TWI634135B (zh) | 2015-12-25 | 2018-09-01 | 日商富士軟片股份有限公司 | 樹脂、組成物、硬化膜、硬化膜的製造方法及半導體元件 |
| KR102309954B1 (ko) * | 2016-03-18 | 2021-10-08 | 도레이 카부시키가이샤 | 경화막 및 포지티브형 감광성 수지 조성물 |
| JP6699661B2 (ja) * | 2016-03-28 | 2020-05-27 | 東レ株式会社 | 感光性樹脂成物 |
| KR102654926B1 (ko) * | 2016-08-10 | 2024-04-05 | 삼성디스플레이 주식회사 | 포토레지스트 조성물 및 이를 이용한 금속 패턴의 형성 방법 |
| KR102460973B1 (ko) * | 2016-11-10 | 2022-11-02 | 도레이 카부시키가이샤 | 디아민 화합물, 그것을 사용한 내열성 수지 및 수지 조성물 |
| KR102299419B1 (ko) * | 2018-02-28 | 2021-09-06 | 주식회사 엘지화학 | 감광성 수지 조성물 및 경화막 |
| CN110776586B (zh) * | 2019-10-24 | 2022-05-20 | 安庆飞凯新材料有限公司 | 一种烷氧基苯绕蒽酮光引发剂的制备方法及其应用 |
| KR20220112366A (ko) * | 2021-02-04 | 2022-08-11 | 주식회사 엘지화학 | 폴리이미드 수지 및 이를 포함하는 포지티브형 감광성 수지 조성물 |
| KR102473464B1 (ko) * | 2021-12-13 | 2022-12-05 | 신진유지건설 주식회사 | 하수관거의 파손부위 및 균열부위 보수공법 |
| KR20240009689A (ko) * | 2022-07-14 | 2024-01-23 | 주식회사 동진쎄미켐 | 감광성 수지 조성물, 경화막 및 이를 포함하는 표시장치 |
| CN116178716B (zh) * | 2023-05-04 | 2023-08-01 | 广州奥松电子股份有限公司 | 一种聚异酰亚胺及其制备方法和应用 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005041936A (ja) | 2003-07-24 | 2005-02-17 | Toray Ind Inc | 熱硬化性樹脂組成物およびそれを用いた電子部品 |
| JP2007156243A (ja) * | 2005-12-07 | 2007-06-21 | Nissan Chem Ind Ltd | ポジ型感光性樹脂組成物及びその硬化膜 |
| JP2007183388A (ja) * | 2006-01-06 | 2007-07-19 | Toray Ind Inc | 感光性樹脂組成物、耐熱性樹脂パターンの製造方法および有機電界発光素子 |
| JP2008033283A (ja) * | 2006-07-05 | 2008-02-14 | Ist Corp | 感光性ポリイミド前駆体組成物及びこれを用いた電子部品 |
| WO2008123053A1 (ja) * | 2007-03-30 | 2008-10-16 | Toray Industries, Inc. | ポジ型感光性樹脂組成物 |
| JP2011042701A (ja) | 2009-08-19 | 2011-03-03 | Toray Ind Inc | 樹脂およびポジ型感光性樹脂組成物 |
| WO2011030744A1 (ja) * | 2009-09-10 | 2011-03-17 | 東レ株式会社 | 感光性樹脂組成物および感光性樹脂膜の製造方法 |
| JP2011202059A (ja) | 2010-03-26 | 2011-10-13 | Toray Ind Inc | 樹脂およびポジ型感光性樹脂組成物 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003100522A1 (fr) * | 2002-05-29 | 2003-12-04 | Toray Industries, Inc. | Composition de resine photosensible et procede de preparation d'une couche mince de resine thermoresistante |
| SG135954A1 (en) * | 2003-04-07 | 2007-10-29 | Toray Industries | Positive-type photosensitive resin composition |
| JP4483371B2 (ja) * | 2003-04-07 | 2010-06-16 | 東レ株式会社 | 感光性樹脂組成物 |
| JP4034691B2 (ja) | 2003-05-09 | 2008-01-16 | ミネベア株式会社 | 回転角度センサー |
| KR101674654B1 (ko) * | 2007-04-02 | 2016-11-09 | 닛산 가가쿠 고교 가부시키 가이샤 | 포지티브형 감광성 수지 조성물, 이의 경화막 및 표시소자 |
| JP2009020246A (ja) * | 2007-07-11 | 2009-01-29 | Toray Ind Inc | 感光性樹脂組成物、これを用いた絶縁性樹脂パターンの製造方法および有機電界発光素子 |
| KR101588364B1 (ko) * | 2007-12-14 | 2016-01-25 | 닛산 가가쿠 고교 가부시키 가이샤 | 폴리히드록시이미드의 제조방법 및 그 제조방법으로부터 얻어진 폴리히드록시이미드를 함유하는 포지티브형 감광성 수지 조성물 |
| JP5477527B2 (ja) * | 2008-09-30 | 2014-04-23 | 日産化学工業株式会社 | 末端官能基含有ポリイミドを含むポジ型感光性樹脂組成物 |
-
2013
- 2013-12-13 SG SG11201504647VA patent/SG11201504647VA/en unknown
- 2013-12-13 JP JP2014500579A patent/JP6332022B2/ja active Active
- 2013-12-13 WO PCT/JP2013/083491 patent/WO2014097992A1/ja active Application Filing
- 2013-12-13 KR KR1020157018049A patent/KR101942150B1/ko active IP Right Grant
- 2013-12-13 CN CN201910307047.5A patent/CN110147031A/zh active Pending
- 2013-12-13 US US14/647,747 patent/US9897915B2/en active Active
- 2013-12-13 EP EP13864249.1A patent/EP2937732B1/en active Active
- 2013-12-13 CN CN201380065421.6A patent/CN104854508B/zh active Active
- 2013-12-13 KR KR1020197001652A patent/KR20190009002A/ko not_active Application Discontinuation
- 2013-12-19 TW TW102147200A patent/TWI568797B/zh active
-
2018
- 2018-04-25 JP JP2018083759A patent/JP2018146969A/ja active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005041936A (ja) | 2003-07-24 | 2005-02-17 | Toray Ind Inc | 熱硬化性樹脂組成物およびそれを用いた電子部品 |
| JP2007156243A (ja) * | 2005-12-07 | 2007-06-21 | Nissan Chem Ind Ltd | ポジ型感光性樹脂組成物及びその硬化膜 |
| JP2007183388A (ja) * | 2006-01-06 | 2007-07-19 | Toray Ind Inc | 感光性樹脂組成物、耐熱性樹脂パターンの製造方法および有機電界発光素子 |
| JP2008033283A (ja) * | 2006-07-05 | 2008-02-14 | Ist Corp | 感光性ポリイミド前駆体組成物及びこれを用いた電子部品 |
| WO2008123053A1 (ja) * | 2007-03-30 | 2008-10-16 | Toray Industries, Inc. | ポジ型感光性樹脂組成物 |
| JP2011042701A (ja) | 2009-08-19 | 2011-03-03 | Toray Ind Inc | 樹脂およびポジ型感光性樹脂組成物 |
| WO2011030744A1 (ja) * | 2009-09-10 | 2011-03-17 | 東レ株式会社 | 感光性樹脂組成物および感光性樹脂膜の製造方法 |
| JP2011202059A (ja) | 2010-03-26 | 2011-10-13 | Toray Ind Inc | 樹脂およびポジ型感光性樹脂組成物 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2937732A4 |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016133741A (ja) * | 2015-01-21 | 2016-07-25 | 住友ベークライト株式会社 | 感光性樹脂組成物、および電子装置 |
| CN107431020B (zh) * | 2015-03-27 | 2020-07-24 | 东丽株式会社 | 薄膜晶体管用感光性树脂组合物、固化膜、薄膜晶体管 |
| WO2016158406A1 (ja) * | 2015-03-27 | 2016-10-06 | 東レ株式会社 | 薄膜トランジスタ用感光性樹脂組成物、硬化膜、薄膜トランジスタ、液晶表示装置または有機電界発光表示装置、硬化膜の製造方法、薄膜トランジスタの製造方法および液晶表示装置または有機電界発光表示装置の製造方法 |
| KR20170131382A (ko) * | 2015-03-27 | 2017-11-29 | 도레이 카부시키가이샤 | 박막 트랜지스터용 감광성 수지 조성물, 경화막, 박막 트랜지스터, 액정 표시 장치 또는 유기 전계 발광 표시 장치, 경화막의 제조 방법, 박막 트랜지스터의 제조 방법 및 액정 표시 장치 또는 유기 전계 발광 표시 장치의 제조 방법 |
| CN107428935A (zh) * | 2015-03-27 | 2017-12-01 | 东丽株式会社 | 二胺化合物、使用其的耐热性树脂或耐热性树脂前体 |
| CN107431020A (zh) * | 2015-03-27 | 2017-12-01 | 东丽株式会社 | 薄膜晶体管用感光性树脂组合物、固化膜、薄膜晶体管、液晶显示装置或有机场致发光显示装置、固化膜的制造方法、薄膜晶体管的制造方法以及液晶显示装置或有机场致发光显示装置的制造方法 |
| JPWO2016158406A1 (ja) * | 2015-03-27 | 2018-01-18 | 東レ株式会社 | 薄膜トランジスタ用感光性樹脂組成物、硬化膜、薄膜トランジスタ、液晶表示装置または有機電界発光表示装置、硬化膜の製造方法、薄膜トランジスタの製造方法および液晶表示装置または有機電界発光表示装置の製造方法 |
| KR102245394B1 (ko) | 2015-03-27 | 2021-04-28 | 도레이 카부시키가이샤 | 박막 트랜지스터용 감광성 수지 조성물, 경화막, 박막 트랜지스터, 액정 표시 장치 또는 유기 전계 발광 표시 장치, 경화막의 제조 방법, 박막 트랜지스터의 제조 방법 및 액정 표시 장치 또는 유기 전계 발광 표시 장치의 제조 방법 |
| CN107428935B (zh) * | 2015-03-27 | 2019-04-02 | 东丽株式会社 | 二胺化合物、使用其的耐热性树脂或耐热性树脂前体 |
| KR20180012308A (ko) | 2015-06-30 | 2018-02-05 | 후지필름 가부시키가이샤 | 전구체 조성물, 감광성 수지 조성물, 전구체 조성물의 제조 방법, 경화막, 경화막의 제조 방법 및 반도체 디바이스 |
| US10526448B2 (en) | 2015-06-30 | 2020-01-07 | Fujifilm Corporation | Precursor composition, photosensitive resin composition, method for producing precursor composition, cured film, method for producing cured film, and semiconductor device |
| US9740101B2 (en) * | 2015-11-13 | 2017-08-22 | Globalfoundries Inc. | Additions of organic species to facilitate crosslinker removal during PSPI cure |
| JPWO2018066395A1 (ja) * | 2016-10-05 | 2019-07-25 | 東レ株式会社 | 樹脂組成物、硬化膜、半導体装置およびそれらの製造方法 |
| WO2018066395A1 (ja) * | 2016-10-05 | 2018-04-12 | 東レ株式会社 | 樹脂組成物、硬化膜、半導体装置およびそれらの製造方法 |
| JP7013872B2 (ja) | 2016-10-05 | 2022-02-15 | 東レ株式会社 | 樹脂組成物、硬化膜、半導体装置およびそれらの製造方法 |
| WO2020059485A1 (ja) * | 2018-09-18 | 2020-03-26 | 東レ株式会社 | 感光性樹脂組成物、樹脂シート、硬化膜、有機el表示装置、半導体電子部品、半導体装置、および有機el表示装置の製造方法 |
| JPWO2020059485A1 (ja) * | 2018-09-18 | 2021-08-30 | 東レ株式会社 | 感光性樹脂組成物、樹脂シート、硬化膜、有機el表示装置、半導体電子部品、半導体装置、および有機el表示装置の製造方法 |
| JP7272268B2 (ja) | 2018-09-18 | 2023-05-12 | 東レ株式会社 | 感光性樹脂組成物、樹脂シート、硬化膜、有機el表示装置、半導体電子部品、半導体装置、および有機el表示装置の製造方法 |
| TWI811446B (zh) * | 2018-09-18 | 2023-08-11 | 日商東麗股份有限公司 | 感光性樹脂組成物、樹脂片材、硬化膜、有機el顯示裝置、半導體電子零件、半導體裝置及有機el顯示裝置之製造方法 |
| US11852973B2 (en) | 2018-09-18 | 2023-12-26 | Toray Industries, Inc. | Photosensitive resin composition, resin sheet, cured film, organic EL display device, semiconductor electronic component, semiconductor device, and method for producing organic EL display device |
| CN117186403A (zh) * | 2023-09-01 | 2023-12-08 | 明士(北京)新材料开发有限公司 | 一种负性光敏性树脂、树脂组合物及其制备方法与应用 |
| CN117186403B (zh) * | 2023-09-01 | 2024-04-02 | 明士(北京)新材料开发有限公司 | 一种负性光敏性树脂、树脂组合物及其制备方法与应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2018146969A (ja) | 2018-09-20 |
| US20150301453A1 (en) | 2015-10-22 |
| EP2937732B1 (en) | 2020-08-19 |
| CN110147031A (zh) | 2019-08-20 |
| JPWO2014097992A1 (ja) | 2017-01-12 |
| TW201434972A (zh) | 2014-09-16 |
| KR20150097578A (ko) | 2015-08-26 |
| EP2937732A4 (en) | 2017-03-08 |
| CN104854508B (zh) | 2019-05-07 |
| SG11201504647VA (en) | 2015-07-30 |
| KR101942150B1 (ko) | 2019-01-24 |
| TWI568797B (zh) | 2017-02-01 |
| JP6332022B2 (ja) | 2018-05-30 |
| US9897915B2 (en) | 2018-02-20 |
| KR20190009002A (ko) | 2019-01-25 |
| EP2937732A1 (en) | 2015-10-28 |
| CN104854508A (zh) | 2015-08-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6332022B2 (ja) | 感光性樹脂組成物、耐熱性樹脂膜の製造方法および表示装置 | |
| JP4911248B2 (ja) | 感光性樹脂組成物および感光性樹脂膜の製造方法 | |
| JP5699602B2 (ja) | 樹脂組成物およびこれを用いた表示装置 | |
| JP6787123B2 (ja) | 感光性樹脂組成物、樹脂硬化膜の製造方法および半導体装置 | |
| JP6286834B2 (ja) | 耐熱性樹脂組成物および耐熱性樹脂膜の製造方法 | |
| JP6780501B2 (ja) | 耐熱性樹脂組成物、耐熱性樹脂膜の製造方法、層間絶縁膜または表面保護膜の製造方法、および電子部品または半導体部品の製造方法 | |
| TWI820180B (zh) | 感光性樹脂組成物、感光性薄片、以及彼等之硬化膜及其製造方法、電子零件 | |
| JP7406181B2 (ja) | 感光性樹脂組成物 | |
| KR102266587B1 (ko) | 수지 조성물, 그 경화막과 그 제조방법, 및 고체촬상소자 | |
| US20210109443A1 (en) | Photosensitive polyimide resin composition and polyimide film thereof | |
| JP6020129B2 (ja) | ポリアミド酸組成物 | |
| JP2018111773A (ja) | 樹脂組成物 | |
| TW202244611A (zh) | 感光性樹脂組成物、硬化物、顯示裝置、有機el顯示裝置及半導體裝置 | |
| TWI791518B (zh) | 感光性樹脂組合物 | |
| TW202231722A (zh) | 感光性聚醯亞胺樹脂組成物、樹脂膜、及電子裝置 | |
| KR20140073759A (ko) | 광가교성 수지 조성물, 이로부터 형성된 절연막 및 유기발광소자 | |
| CN116507654A (zh) | 感光性聚酰亚胺树脂组合物、树脂膜和电子装置 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2014500579 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13864249 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013864249 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14647747 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20157018049 Country of ref document: KR Kind code of ref document: A |