JP2010522693A - 化学リンカー及び切断可能な基質及びそれらの抱合体 - Google Patents
化学リンカー及び切断可能な基質及びそれらの抱合体 Download PDFInfo
- Publication number
- JP2010522693A JP2010522693A JP2009544303A JP2009544303A JP2010522693A JP 2010522693 A JP2010522693 A JP 2010522693A JP 2009544303 A JP2009544303 A JP 2009544303A JP 2009544303 A JP2009544303 A JP 2009544303A JP 2010522693 A JP2010522693 A JP 2010522693A
- Authority
- JP
- Japan
- Prior art keywords
- substituted
- group
- unsubstituted
- hydrogen
- alkyl group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 C*CC(C)(C)C(*)(*)*(C)OC(C)(C=CC=C1)C=C1C(N)=*[*@@](*)C(C(C)(C)C(*)(*)C(C)(C)CC*(*)C(*(C)=*)O)=O Chemical compound C*CC(C)(C)C(*)(*)*(C)OC(C)(C=CC=C1)C=C1C(N)=*[*@@](*)C(C(C)(C)C(*)(*)C(C)(C)CC*(*)C(*(C)=*)O)=O 0.000 description 12
- LPYGXQLRFJTBFA-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)c(cc2)c1c1c2[nH]c(C(O)=O)c1)=O Chemical compound CC(C)(C)OC(N(CC1)c(cc2)c1c1c2[nH]c(C(O)=O)c1)=O LPYGXQLRFJTBFA-UHFFFAOYSA-N 0.000 description 1
- OHQNITHOFMEYOJ-ZDUSSCGKSA-N CC(C)(C)OC(c(cc1)ccc1NC([C@H](CCCNC(N)=O)N)=O)=O Chemical compound CC(C)(C)OC(c(cc1)ccc1NC([C@H](CCCNC(N)=O)N)=O)=O OHQNITHOFMEYOJ-ZDUSSCGKSA-N 0.000 description 1
- HNJWJKHLKFAFPW-UHFFFAOYSA-N CC(C)C(C(NC(CCCNC(N)=O)C(Nc(cc1)ccc1C(Nc1ccc2[nH]c(C(N3c4cc(OC(N5CCN(C)CC5)=O)c(cccc5)c5c4CC3)=O)cc2c1)=O)=O)=O)NCC(NCCNC(CCOCCOCCOCCOCCNC(CCN(C(C=C1)=O)C1=O)=O)=O)=O Chemical compound CC(C)C(C(NC(CCCNC(N)=O)C(Nc(cc1)ccc1C(Nc1ccc2[nH]c(C(N3c4cc(OC(N5CCN(C)CC5)=O)c(cccc5)c5c4CC3)=O)cc2c1)=O)=O)=O)NCC(NCCNC(CCOCCOCCOCCOCCNC(CCN(C(C=C1)=O)C1=O)=O)=O)=O HNJWJKHLKFAFPW-UHFFFAOYSA-N 0.000 description 1
- YOMLDTVUZZOYPT-ZMEDUCCESA-N CC(C)[C@@H](C(N[C@@H](CCCNC(N)=O)C(Nc(cc1)ccc1C(Nc(cc1)cc2c1[nH]c(C(N(C(C1CCl)C3C=CC=C3)c3c1c(cccc1)c1c(O[C@H]([C@@H](C([C@@H]1O)O)O)OC1C(O)=O)c3)=O)c2)=O)=O)=O)NCCN Chemical compound CC(C)[C@@H](C(N[C@@H](CCCNC(N)=O)C(Nc(cc1)ccc1C(Nc(cc1)cc2c1[nH]c(C(N(C(C1CCl)C3C=CC=C3)c3c1c(cccc1)c1c(O[C@H]([C@@H](C([C@@H]1O)O)O)OC1C(O)=O)c3)=O)c2)=O)=O)=O)NCCN YOMLDTVUZZOYPT-ZMEDUCCESA-N 0.000 description 1
- YAYFNFHNWYIFEU-UHFFFAOYSA-N CCCCC(ON(C(CC1)=O)C1=O)=N Chemical compound CCCCC(ON(C(CC1)=O)C1=O)=N YAYFNFHNWYIFEU-UHFFFAOYSA-N 0.000 description 1
- XHHYVKBJDHGOKB-UHFFFAOYSA-N CN(CC1)CCN1C(Oc1c(cccc2)c2c(CCN2C(c3cc(c4c(cc5)NCC4)c5[nH]3)=O)c2c1)=O Chemical compound CN(CC1)CCN1C(Oc1c(cccc2)c2c(CCN2C(c3cc(c4c(cc5)NCC4)c5[nH]3)=O)c2c1)=O XHHYVKBJDHGOKB-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N CN1CCNCC1 Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- MCFZBCCYOPSZLG-UHFFFAOYSA-N O=C(C1)C=CC1=O Chemical compound O=C(C1)C=CC1=O MCFZBCCYOPSZLG-UHFFFAOYSA-N 0.000 description 1
Images















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
- C07K5/06052—Val-amino acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/55—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug
- A61K47/552—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug one of the codrug's components being an antibiotic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/555—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound pre-targeting systems involving an organic compound, other than a peptide, protein or antibody, for targeting specific cells
- A61K47/556—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound pre-targeting systems involving an organic compound, other than a peptide, protein or antibody, for targeting specific cells enzyme catalyzed therapeutic agent [ECTA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6807—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug or compound being a sugar, nucleoside, nucleotide, nucleic acid, e.g. RNA antisense
- A61K47/6809—Antibiotics, e.g. antitumor antibiotics anthracyclins, adriamycin, doxorubicin or daunomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/0002—General or multifunctional contrast agents, e.g. chelated agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D501/00—Heterocyclic compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D501/14—Compounds having a nitrogen atom directly attached in position 7
- C07D501/16—Compounds having a nitrogen atom directly attached in position 7 with a double bond between positions 2 and 3
- C07D501/59—Compounds having a nitrogen atom directly attached in position 7 with a double bond between positions 2 and 3 with hetero atoms directly attached in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06078—Dipeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
- C07K5/0806—Tripeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atoms, i.e. Gly, Ala
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
- C07K5/0808—Tripeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms, e.g. Val, Ile, Leu
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0812—Tripeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1005—Tetrapeptides with the first amino acid being neutral and aliphatic
- C07K5/1008—Tetrapeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atoms, i.e. Gly, Ala
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmacology & Pharmacy (AREA)
- Biophysics (AREA)
- Epidemiology (AREA)
- Genetics & Genomics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Immunology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Cell Biology (AREA)
- Hematology (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Plural Heterocyclic Compounds (AREA)
- Cephalosporin Compounds (AREA)
Abstract
【解決手段】薬剤及びリガンドがペプチジル又は他のリンカーを介して結合された薬剤−リガンド抱合体、並びに薬剤−酵素切断可能な基質抱合体。これらの抱合体は、目的の活性部位に活性形態で選択的に送達され、次いで切断により活性薬剤を放出しうる強力な細胞毒素である。また、薬剤−リガンド抱合体を含有する組成物及びそれを用いて治療する方法。
【選択図】図1
Description
本願は、2006年12月28日に出願された米国仮特許出願第60/882461号明細書及び2007年11月30日に出願された米国仮特許出願第60/991300号明細書の優先権を主張するものであり、それらの利益は米国特許法第119条により本明細書で主張され、それらの全開示内容を参照により本明細書中に援用する。

式中、
L1は自己犠牲リンカーであり;
mは0、1、2、3、4、5若しくは6の整数であり;
Fは構造:
AA1は天然アミノ酸及び非天然α−アミノ酸からなる群から独立して選択される1つ以上のメンバーであり;
cは1〜20の整数であり;
L2は自己犠牲リンカーであり、かつ
oは1であり、
L4はリンカーメンバーであり;
pは0若しくは1であり;
X4は保護反応性官能基、非保護反応性官能基、検出可能な標識、及び標的化剤からなる群から選択されるメンバーであり;かつ
Dは構造:
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3はOR11であり、R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13は、それらに結合する窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成され、
R4、R4’、R5及びR5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対は、それらに結合する炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成されてもよく;
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R6は、存在又は不在のいずれかの単結合であり、かつ存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ、
R7は前記シクロプロピル環内でR6と結合するCH2−X1又は−CH2−であり、X1は離脱基であり、
R11は前記薬剤をL1(存在する場合)又はFに結合させる。
式中
L1は自己犠牲リンカーであり;
mは0、1、2、3、4、5若しくは6の整数であり;
Fは構造:
AA1は天然アミノ酸及び非天然α−アミノ酸からなる群から独立して選択される1つ以上のメンバーであり;
cは1〜20の整数であり;
L2は自己犠牲リンカーであり、
oは0若しくは1であり、
L4はリンカーメンバーであり;
pは0若しくは1であり;
X4は保護反応性官能基、非保護反応性官能基、検出可能な標識、及び標的化剤からなる群から選択されるメンバーであり;かつ
Dは構造:
環系Aは、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択されるメンバーであり;
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3は、(=O)、SR11、NHR11及びOR11からなる群から選択されるメンバーであり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13はそれらに結合する窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5及びR5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらに結合する炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成されてもよく;
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5及びR5’の少なくとも1つは前記薬剤をL1(存在する場合)又はFに結合させ、かつ
R27、R27’、R28、及びR28’の各々は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、及び置換若しくは未置換ヘテロシクロアルキル基から独立して選択され;
R6は存在しないか又は単結合として存在し、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7は前記シクロプロピル環内でR6と結合されたCH2−X1又は−CH2−であり、X1は離脱基である。
式中、
L1は自己犠牲リンカーであり;
mは0、1、2、3、4、5若しくは6の整数であり;
Fは構造:
AA1は天然アミノ酸及び非天然α−アミノ酸からなる群から独立して選択される1つ以上のメンバーであり;
cは1〜20の整数であり;
L3は第一級若しくは第二級アミンを含むスペーサー基又はカルボキシル官能基であり;L3が存在する場合、mは0であり、かつL3のアミン基はDのペンダントカルボキシル官能基とアミド結合を形成するか又はL3のカルボキシル基はDのペンダントアミン官能基とアミド結合を形成し;
oは0若しくは1であり、
L4はリンカーメンバーであり、L4は(AA1)cのN末端に直接結合される
R20は水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり、
R25、R25’、R26、及びR26’の各々は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、及び置換若しくは未置換ヘテロシクロアルキル基から独立して選択され;かつ
s及びtは独立して1〜6の整数であり;
pは1であり;
X4は、保護反応性官能基、非保護反応性官能基、検出可能な標識、及び標的化剤からなる群から選択されるメンバーであり;かつ
Dは構造:
環系Aは、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択されるメンバーであり、
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3は、(=O)、SR11、NHR11及びOR11からなる群から選択されるメンバーであり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13はそれらに結合する窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5及びR5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらに結合する炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成されてもよく;
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R6は存在しないか又は単結合として存在し、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7は前記シクロプロピル環内でR6と結合されたCH2−X1又は−CH2−であり、X1は離脱基であり、
R4、R4’、R5、R5’、R15又はR16の少なくとも1つは前記薬剤をL1(存在する場合)又はFに結合させる。
L1は自己犠牲スペーサーであり;
mは0、1、2、3、4、5若しくは6の整数であり;
X2は切断可能な基質であり;かつ
Dは構造:
環系Aは、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択されるメンバーであり;
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3は、(=O)、SR11、NHR11及びOR11からなる群から選択されるメンバーであり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13は、それらに結合する窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R6は存在しないか又は単結合として存在し、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7は前記シクロプロピル環内でR6と結合されたCH2−X1若しくは−CH2−であり、X1は離脱基であり、
R4、R4’、R5及びR5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらに結合する炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成され、
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5及びR5’の少なくとも1つのメンバーは、前記薬剤をL1(存在する場合)又はX2に結合させ、かつ
R30、R30’、R31、及びR31’は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、及び置換若しくは未置換ヘテロシクロアルキル基から独立して選択され;かつ
vは1〜6の整数である。
環系Aは、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択されるメンバーであり;
Z及びXは、O、S及びNR23から独立して選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3は、(=O)、SR11、NHR11及びOR11からなる群から選択されるメンバーであり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13はそれらに結合する窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R6は存在しないか又は単結合として存在し、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;
R7は前記シクロプロピル環内でR6と結合されたCH2−X1若しくは−CH2−であり、X1は離脱基であり;かつ
R5、R5’、及びR32は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであり、
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成される。
環系Aは、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択されるメンバーであり;
Z及びXは、O、S及びNR23から独立して選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R4、R4’、R5、R5’、及びR33は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらに結合する炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成されてもよく;
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R33は1つ以上の切断可能な基を含み;
R6は存在しないか又は単結合として存在し、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7は前記シクロプロピル環内でR6と結合されたCH2−X1又は−CH2−であり、X1は離脱基である。
「Ala」はアラニンを示す。
「Boc」はt−ブチルオキシカルボニルを示す。
「CPI」はシクロプロパピロロインドールを示す。
「Cbz」はカルボベンゾキシを示す。
「DCM」はジクロロメタンを示す。
「DDQ」は2,3−ジクロロ−5,6−ジシアノ−1,4−ベンゾキノンを示す。
「DIPEA」はジイソプロピルエチルアミンを示す。
「DMDA」はN,N’−ジメチルエチレンジアミンを示す。
「RBF」は丸底フラスコを示す。
「DMF」はN,B−ジメチルホルムアミドを示す。
「HATU」はN−[[(ジメチルアミノ)−1H−1,2,3−トリアゾロ[4,5−b]ピリジン−1−イル]メチレン]−N−メチルメタンアミニウムヘキサフルオロホスフェートN−オキシドを示す。
記号「E」は酵素的に切断可能な基を示す。
「EDCI」は1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミドを示す。
「FMOC」は9−フルオレニルメチルオキシカルボニルを示す。
「HOAt」は7−アザ−1−ヒドロキシベンゾトリアゾールを示す。
「Leu」はロイシンを示す。
「PABA」はパラアミノ安息香酸を示す。
「PEG」はポリエチレングリコールを示す。
「DBU」は1,8−ジアザビシクロ(5.4.0)ウンデク−7−エンを示す。
「DIEA」はジイソプロピルエチルアミンを示す。
「PMB」はパラメトキシベンジルを示す。
「TBAF」はテトラブチルアンモニウムフルオリドを示す。
「TBSO」はt−ブチルジメチルシリルエーテルを示す。
「TEA」はトリエチルアミンを示す。
「TFA」はトリフルオロロ酢酸を示す。
「EDC」は(1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩)を示す。
「TBTU」は(2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムテトラフルオロボレート)を示す。
「HOBT」はN−ヒドロキシベンゾトリアゾールを示す。
「Q」という記号は、治療剤、診断剤又は検出可能な標識を示す。
定義
他に定義されない限り、本明細書で用いられるあらゆる科学技術用語は、一般に、本発明が属する当該技術分野における当業者による一般的理解と同様の意味を有する。一般に、本明細書で用いられる命名法、細胞培養、分子遺伝学、有機化学及び核酸化学における実験法及び下記のハイブリダイゼーションは周知であり、当該技術分野で一般に用いられるものである。核酸及びペプチド合成においては標準技術が用いられる。一般に、酵素反応及び精製ステップは製造業者の仕様に従って実施される。技術及び手順は、一般に、当該技術分野における従来の方法及び様々な一般の参考文献(一般に、Sambrookら、「MOLECULAR CLONING:A LABORATORY MANUAL、2d ed.」(1989年)Cold Spring Harbor Laboratory Press、Cold Spring Harbor、N.Y.(参照により本明細書中に援用される)を参照のこと)に従って実施され、それらは本明細書全体に提供される。本明細書で用いられる命名法、分析化学における実験法、及び下記の有機合成は、周知であり、当該技術分野で一般に用いられるものである。標準技術又はその改良は、化学合成及び化学分析において用いられる。
本発明は、薬剤が化学リンカーを介してリガンドに連結された薬剤−リガンド抱合体を提供する。一部の実施形態では、リンカーはペプチジルリンカーであり、本明細書中で(L4)p−F−(L1)mとして表される。他のリンカーは、ヒドラジン及びジスルフィドリンカーを含み、それぞれ本明細書中で(L4)p−H−(L1)m又は(L4)p−J−(L1)mとして表される。本発明は、薬剤に結合されているようなリンカーに加え、本質的に任意の分子種への結合に適する切断可能なリンカーアームも提供する。本発明のリンカーアームの態様は、本明細書中でその治療部分への結合を参照することにより例示される。しかし、リンカーが限定はされないが診断剤、分析剤、生体分子、標的化剤、検出可能な標識などを含む多様な種に結合可能であることは当業者には容易に理解されるであろう。
上で考察のように、本発明のペプチジルリンカーは、一般式:(L4)p−F−(L1)m(式中、Fはペプチジル部分を含むリンカー部分を表す)により表されうる。一実施形態では、F部分は任意の追加の自己犠牲リンカーL2及びカルボニル基を含む。別の実施形態では、F部分はアミノ基及び任意のスペーサー基L3を含む。
自己犠牲リンカーL2は、2つの距離が離れた化学的部分を共に通常は安定なトリパーテート(tripartate)分子に共有結合可能な二機能の化学的部分であり、それにより酵素切断を用いてトリパーテート分子から前記距離が離れた化学的部分のうちの一方が放たれ、前記酵素切断後、分子の残りから自発的切断が生じ、前記距離が離れた化学的部分の他方が放たれる。本発明によると、自己犠牲スペーサーは、その一方の末端でペプチド部分に共有結合されかつその他方の末端で薬剤部分(その誘導化が薬理学的活性を阻害する)の化学反応性がある部位に共有結合されることで、距離が離れ、ペプチド部分及び薬剤部分を共に標的酵素の不在下で安定かつ薬理学的に不活性であるトリパーテート分子に共有結合させるが、それはスペーサー部分及びペプチド部分に共有結合することによりトリパーテート分子からのペプチド部分の放出をもたらす結合でかかる標的酵素により酵素的に切断可能である。次いで、かかる酵素切断は、スペーサー部分の自己犠牲的特徴を活性化し、スペーサー部分を薬剤部分に共有結合させることにより薬理活性形態での薬剤の放出をもたらす結合の自発切断を開始させることになる。
スペーサー基L3は第一級若しくは第二級アミン基又はカルボキシル官能基を含むように特徴づけられ、かつ、L3基のアミンがDのペンダントカルボキシル官能基とアミド結合を形成するか、又はL3のカルボキシルがDのペンダントアミン官能基とアミド結合を形成する。L3は、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、又は置換若しくは未置換ヘテロシクロアルキル基からなる群から選択されうる。好ましい実施形態では、L3は芳香族性基を含む。より好ましくは、L3は安息香酸基、アニリン基又はインドール基を含む。−L3−NH−スペーサーとして機能しうる構造の非限定例として、
AA1基は、単一のアミノ酸又はアミド結合により結合された複数のアミノ酸を表す。アミノ酸は天然アミノ酸及び/又は非天然α−アミノ酸であってもよい。
第2の実施形態では、本発明の抱合体はヒドラジン自己犠牲リンカーを含み、抱合体は構造:
n4は1、2、若しくは3であり、
Iが結合である場合、n1は3でありかつn2は1であり、Dは
一実施形態では、ヒドラジンリンカーは5員のヒドラジンリンカーを含み、Hは構造
別の実施形態では、ヒドラジンリンカーは6員のヒドラジンリンカーを含み、Hは構造:
本発明が7員を有するリンカーを含む点が検討される。このリンカーであれば5員又は6員のリンカーと同程度に速やかに環化しない可能性が高いことになるが、これは一部の薬剤−リガンド抱合体にとって好ましい場合がある。同様に、ヒドラジンリンカーは2つの6員環を含むか、又はヒドラジンリンカーは1つの6員及び1つの5員の環化生成物を有しうる。5員及び7員のリンカー並びに6員及び7員のリンカーについても検討される。
更に別の実施形態では、リンカーは酵素的に切断可能なジスルフィド基を含む。一実施形態では、本発明は、式3:
各R24は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、及び未置換ヘテロアルキル基からなる群から独立して選択されるメンバーであり;
各Kは、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR21R22、NR21COR22、OCONR21R22、OCOR21、及びOR21からなる群から独立して選択されるメンバーであり、
式中、
R21及びR22は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル及び未置換ヘテロシクロアルキルからなる群から独立して選択され;
aは0、1、2、3、若しくは4の整数であり;並びに
dは0、1、2、3、4、5若しくは6の整数である。
本明細書中で「D」として示される薬剤は、本発明において、薬剤が切断可能な基質X2に場合により自己犠牲リンカーL1を介して結合される場合、薬剤−切断可能な基質抱合体の一部として提供されうる。この抱合体はプロドラッグであってもよい。薬剤は、典型的には所望の生物学的活性を有し、切断可能な基質に結合するための反応性官能基を有する。所望の生物学的活性は、ヒトなどの動物における疾患の診断、治癒、軽減、治療、又は予防を含む。好ましい反応性官能基は、第一級若しくは第二級アミン基、ヒドロキシル基、スルフヒドリル基、カルボキシル基、アルデヒド基、及びケトン基を含む。より好ましい反応性官能基は、ヒドロキシル基、第一級若しくは第二級アミン基、スルフヒドリル基及びカルボン酸官能基を含む。更により好ましい反応性官能基は、ヒドロキシル基、第一級及び第二級アミン基並びにカルボン酸官能基を含む。薬剤は、典型的には少なくとも1つを有するが、2、3、4、5、6若しくは7以上の反応性官能基を有しうる。
本明細書中で「D」として示される薬剤は、本発明において、薬剤がリガンドにペプチジル又は他のリンカーのいずれかを介して結合される場合、薬剤−リガンド抱合体の一部として、又は切断可能な基質との抱合体として提供される。薬剤は、リガンドに結合するため、所望の生物学的活性を有し、かつ反応性官能基を有する必要がある。所望の生物学的活性は、ヒトなどの動物における疾患の診断、治癒、軽減、治療、又は予防を含む。したがって、「薬剤」という用語は、必要な反応性官能基を有する限り、公式の米国薬局方(United States Pharmacopeia)、公式の米国ホメオパシー薬局方(Homeopathic Pharmacopeia of the United States)又は公式の国民医薬品集(National Formulary)、又はその任意の付録において薬剤として認識された化学物質を示す。典型的な薬剤は、米国食品医薬品局(U.S.Food and Drug Administration)(FDA)により維持された医師用卓上参考書(Physician’s Desk Reference)(PDR)及びオレンジブック(Orange Book)において示される。新薬は継続的に発見されかつ開発されつつあり、本発明はこれらの新薬が本発明の薬剤−リガンド抱合体にも取り込まれうる点を提起している。
本発明において有用な細胞毒性薬として、例えば、デュオカルマイシン及びCC−1065とそれらの類縁体(デュオカルマイシン及びCC−1065のCBI(1,2,9,9a−テトラ−ヒドロシクロプロパ[c]ベンズ[e]インドール−4−オン)に基づく類縁体、MCBI(7−メトキシ−1,2,9,9a−テトラ−ヒドロシクロプロパ[c]ベンズ[e]インドール−4−オン)に基づく類縁体及びCCBI(7−シアノ−1,2,9,9a−テトラ−ヒドロシクロ−プロパ[c]ベンズ[e]−インドール−4−オン)に基づく類縁体を含む)、ドキソルビシン及びモルホリノ−ドキソルビシン及びシアノモルホリノ−ドキソルビシンなどのドキソルビシン抱合体、ドラスタチン−10などのドラスタチン、コンブレタスタチン、カリケアマイシン、メイタンシン、メイタンシン類縁体、DM−1、アウリスタチンE、アウリスタチンEB(AEB)、アウリスタチンEFP(AEFP)、モノメチルアウリスタチンE(MMAE)、5−ベンゾイル吉草酸−AEエステル(AEVB)、ツブリシン、ジソラゾール、エポチロン、パクリタキセル、ドセタキセル、SN−38、トポテカン、リゾキシン、エキノマイシン、コルヒチン、ビンブラスチン、ビンデシン、エストラムスチン、セマドチン、エリュテロビン、メトトレキセイト、メトプテリン、ジクロロメトトレキセート、5−フルオロウラシル、6−メルカプトプリン、シトシンアラビノシド、メルファラン、ロイロシン、ロイロシデイン(leurosideine)、アクチノマイシン、ダウノルビシン及びダウノルビシン抱合体、マイトマイシンC、マイトマイシンA、カルミノマイシン、アミノプテリン、タリソマイシン、ポドフィロトキシン及びエトポシド又はリン酸エトポシドなどのポドフィロトキシン誘導体、ビンクリスチン、タクソール、タキソテールレチノイン酸、酪酸、N8−アセチルスペルミジン、カンプトテシン、及びそれらの類縁体が挙げられる。本明細書中に記載のリンカーへの複合における官能基を提供するため、他の公知の薬剤が修飾される場合がある。かかる化学修飾は当該技術分野で公知である。
X4は保護反応官能基、非保護反応官能基、検出可能な標識、及び標的化剤からなる群から選択されるリガンドを表す。好ましいリガンドは標的化剤、例えば抗体及びその断片である。
R30は、反応性官能基で終結される置換若しくは未置換アルキル基、官能基で終結される置換若しくは未置換ヘテロアリールを示す。上記構造は、例えば標的化剤のアミノ酸の側鎖、例えば抗体と反応しうる反応性保護基として作用することで、標的化剤をリンカー−薬剤部分に結合させる。
本発明のリンカーアーム及び細胞毒素は、ペイロードを身体の細胞、器官又は部位に選択的に送達する標的化剤に結合されうる。抗体(例えばキメラ、ヒト化及びヒト)、受容体に対するリガンド、レクチン、糖、抗体などの典型的な標的化剤は、当該技術分野で認識されており、本発明の実施において制限なしに有用である。他の標的化剤は、特異的な分子認識モチーフを含まない化合物のクラスを含み、細胞毒素に分子量を加えるポリ(エチレングリコール)、多糖、ポリアミノ酸などの高分子を含む。追加された分子量は、細胞毒素の薬物動態、例えば血清半減期に作用する。
本発明の化合物及び方法と併用される特定の標識又は検出可能な基は、本発明の化合物の活性又は有用性と有意に干渉することがない限り、一般に本発明の重要な態様ではない。検出可能な基は、検出可能な物理的又は化学的特性を有する任意の材料であってもよい。かかる検出可能な標識はイムノアッセイの分野で十分に開発されており、一般にかかる方法で有用な大部分の任意の標識が本発明に適用可能である。したがって、標識は、分光学的、光化学的、生物化学的、免疫化学的、電気的、光学的又は化学的手段により検出可能な任意の組成物である。本発明で有用な標識は、磁気ビーズ(例えばDYNABEADS(商標))、蛍光色素(例えばフルオレセインイソチオシアネート、テキサスレッド、ローダミンなど)、放射性標識(例えば3H、125I、35S、14C、又は32P)、酵素(例えば、西洋ワサビペルオキシダーゼ、アルカリホスファターゼ及びELISAで一般に用いられるその他のもの)、並びにコロイド状金又は着色ガラス又はプラスチックビーズ(例えばポリスチレン、ポリプロピレン、ラテックスなど)などの比色標識を含む。
例示の簡素化のため、以下の考察では本発明の細胞毒素と標的化剤との複合について着目される。その着目により本発明の一実施形態が例示され、その他は当業者により容易に推定される。本発明が単一の実施形態に関する考察に着目することによって限定されることは全く意図されていない。
本発明の切断可能な基質は「X2」として表される。好ましくは、切断可能な基質は酵素により切断されうる切断可能な酵素基質である。好ましくは、酵素は、治療されるべき腫瘍又は他の標的細胞と直接的又は間接的に優先的に結合される。酵素は、治療されるべき腫瘍又は他の標的細胞により生成されうる。例えば、切断可能な基質は、腫瘍又は他の標的細胞の近傍若しくは内部に見出される酵素により優先的に切断可能なペプチドであってもよい。更に又はその他として、酵素は、腫瘍細胞に特異的に結合する標的化剤、例えば腫瘍抗原に特異的な抗体に結合されうる。
本発明のリンカー及び切断可能な基質は、細胞毒素の作用物質としてのデュオカルマイシン又はCBI類縁体を有する抱合体内で用いられうる。本発明の抱合体の例が、下記に更に詳述される。他に指定がない限り、置換基は、細胞毒素、リンカー、及び切断可能な基質に関するセクションで上記のように定義される。
適切な抱合体の一例が、式:
L1は自己犠牲リンカーであり;
mは0、1、2、3、4、5若しくは6の整数であり;
Fは構造:
AA1は天然アミノ酸及び非天然α−アミノ酸からなる群から独立して選択される1種以上のメンバーであり;
cは1〜20の整数であり;
L2は自己犠牲リンカーでありかつ
R17、R18、及びR19の各々は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択され、
かつwは0〜4の整数であり;
oは1であり;
L4はリンカーメンバーであり;
pは0若しくは1であり;
X4は保護反応官能基、非保護反応官能基、検出可能な標識、及び標的化剤からなる群から選択されるメンバーであり;かつ
Dは構造:
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3はOR11であり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R4、R4’、R5、R5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらの結合対象である炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成されてもよく;
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらの結合対象である窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R6は存在するか又は存在しない単結合であり、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7はR6と前記シクロプロピル環内でCH2−X1若しくは−CH2−で結合され、
X1は離脱基であり、
R11は前記薬剤をL1(存在する場合)又はFに結合させる。
L1は自己犠牲リンカーであり;
mは0、1、2、3、4、5若しくは6の整数であり;
Fは構造:
AA1は天然アミノ酸及び非天然α−アミノ酸からなる群から独立して選択される1種以上のメンバーであり;
cは1〜20の整数であり;
L2は自己犠牲リンカーであり;
oは0若しくは1であり;
L4はリンカーメンバーであり;
pは0若しくは1であり;
X4は保護反応官能基、非保護反応官能基、検出可能な標識、及び標的化剤からなる群から選択されるメンバーであり;かつ
Dは構造:
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3は(=O)、SR11、NHR11及びOR11からなる群から選択されるメンバーであり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13はそれらの結合対象である窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5、R5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらの結合対象である炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成され、
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらの結合対象である窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5及びR5’の少なくとも1つは前記薬剤をL1(存在する場合)又はFに結合させ、かつ
R27、R27’、R28、及びR28’の各々は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、及び置換若しくは未置換ヘテロシクロアルキル基から独立して選択され;
R6は存在するか又は存在しない単結合であり、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7はR6と前記シクロプロピル環内でCH2−X1若しくは−CH2−で結合され、
X1は離脱基である。
L1は自己犠牲リンカーであり、
mは0、1、2、3、4、5若しくは6の整数であり、
Fは構造
AA1は天然アミノ酸及び非天然α−アミノ酸からなる群から独立して選択される1つ以上のメンバーであり;
cは1〜20の整数であり;
L3は第一級若しくは第二級アミンを含むスペーサー基又はカルボキシル官能基であり;L3が存在する場合、mは0でありかつL3のアミンがDのペンダントカルボキシル官能基とアミド結合を形成するか、又はL3のカルボキシルがDのペンダントアミン官能基とアミド結合を形成し;
oは0若しくは1であり;
L4はリンカーメンバーであり、L4は(AA1)cのN末端に直接結合された
R20は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり、
R25、R25’、R26、及びR26’の各々は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、及び置換若しくは未置換ヘテロシクロアルキル基から独立して選択され、
かつs及びtは独立して1〜6の整数であり;
pは1であり;
X4は保護反応官能基、非保護反応官能基、検出可能な標識、及び標的化剤からなる群から選択されるメンバーであり、かつ
Dは構造:
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3は(=O)、SR11、NHR11及びOR11からなる群から選択されるメンバーであり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13はそれらの結合対象である窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5及びR5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらの結合対象である炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成され、
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらの結合対象である窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R6は存在するか又は存在しない単結合であり、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7はR6と前記シクロプロピル環内でCH2−X1若しくは−CH2−で結合され、
X1は離脱基であり、
R4、R4’、R5、R5’、R15又はR16のうちの少なくとも1つは前記薬剤をL1(存在する場合)又はFに結合させる。
適切な抱合体の一例が、構造:
を有する化合物であり、
L1は自己犠牲スペーサーであり、
mは0、1、2、3、4、5若しくは6の整数であり;
X2は切断可能な基質であり;かつ
Dは構造:
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3は(=O)、SR11、NHR11及びOR11からなる群から選択されるメンバーであり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13はそれらの結合対象である窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R6は存在するか又は存在しない単結合であり、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7はR6と前記シクロプロピル環内でCH2−X1若しくは−CH2−で結合され、X1は離脱基であり、
R4、R4’、R5及びR5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらの結合対象である炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成され、
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらの結合対象である窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成され、
R4、R4’、R5及びR5’のメンバーうちの少なくとも1つは前記薬剤をL1(存在する場合)又はX2に結合させ、かつ
R30、R30’、R31、及びR31’は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、及び置換若しくは未置換ヘテロシクロアルキル基から独立して選択され;かつ
vは1〜6の整数である。
別の好ましい実施形態では、本発明は、本発明の化合物及び薬理学的に許容できる担体を含有する医薬製剤を提供する。
また、細胞毒素、細胞毒素−リンカー及び細胞毒素の作用物質−リンカー抱合体、本発明のリンカー並びに本発明の細胞毒素−切断可能な基質のライブラリーも本発明の範囲内に含まれる。典型的なライブラリーは、少なくとも10種の化合物、より好ましくは少なくとも100種の化合物、更により好ましくは少なくとも1000種の化合物、及び更により好ましくは少なくとも100,000種の化合物を含む。ライブラリーは、特定の特性、例えば細胞毒性、酵素又は他の切断試薬によるリンカー又は基質の切断について容易に探索される形態である。典型的な形態は、チップフォーマット、マイクロアレイなどを含む。
別の態様では、本発明は、本発明の化合物又は組成物のうちの1種以上及び化合物又は組成物を用いるための指示書を有するキットを提供する。典型的な実施形態では、本発明は、本発明のリンカーアームを別の分子に複合するためのキットを提供する。キットは、リンカー、及びリンカーを特定の官能基に結合させるための指示書を含む。キットは、それに加えて、又はそれに代えて、細胞毒性薬、標的化剤、検出可能な標識、切断可能な基質、医薬塩又は緩衝液のうちの1つ以上を含みうる。キットはまた、容器、及び場合により1つ以上のバイアル、試験管、フラスコ、ボトル、又は注射器を含みうる。キットにおける他の形式は当業者には明らかであると思われ、本発明の範囲内に含まれる。
別の典型的な実施形態では、本発明は、切断可能な基質X2又はリガンドX4に結合する、本発明のリガンド−細胞毒素又は切断可能な基質−細胞毒素において分子標的を単離するための方法を提供する。本方法は、好ましくは、標的を含む細胞調製物を固定化化合物と接触させ、それにより受容体と固定化化合物との間で抱合体を形成するステップを含む。
本発明は、上記の組成物及び構築物に加え、本発明の化合物及び抱合体を用いて実施可能な多数の方法も提供する。本発明の薬剤−リガンド抱合体及び薬剤−切断可能な基質抱合体を用いるための方法は、腫瘍細胞又は癌細胞を殺傷するか又はその成長若しくは複製を阻害することで癌を治療し、前癌状態を治療し、自己免疫抗体を発現する細胞を殺傷するか又はその成長若しくは複製を阻害することで自己免疫疾患を治療し、感染病を治療し、腫瘍細胞又は癌細胞の増殖を予防することで癌を予防し、自己免疫抗体を発現する細胞の増殖を予防することで自己免疫疾患を予防し、かつ感染病を予防することを含む。これらの使用方法は、有効量の薬剤−リガンド抱合体又は薬剤−切断可能な基質抱合体を、それを必要とする哺乳動物又はヒトなどの動物に投与するステップを含む。一部の実施形態では、酵素は腫瘍又は標的細胞との結合状態を形成するように別々に投与される。
本発明での使用に適する医薬組成物は、活性成分が治療有効量、すなわちその意図される目的を果たすための有効量で含有された組成物を含む。特定の用途にとって有効な実際の量は、特に治療される状態に依存することになる。有効量の決定は、特に本明細書中の詳細な開示を考慮すると、十分に当業者の能力の範囲内にある。
下記の実施例においては、他に指定のない限り、温度は摂氏温度(℃)で与えられ;操作は室温又は周囲温度(典型的には約18〜25℃の範囲)で実施され;溶媒の蒸発は最高60℃の浴温、減圧下(典型的には4.5〜30mmHg)でロータリーエバポレーターを用いて行われ;反応の経過は典型的にはTLCに従い、反応時間はあくまで例示目的で提供され;融点は未補正であり;生成物は満足できる1H NMR及び/又は微量分析データを示し;収率はあくまで例示目的で提供され;かつ、mp(融点)、L(リットル)、mL(ミリリットル)、mモル(ミリモル)、g(グラム)、mg(ミリグラム)、min(分)、LC−MS(液体クロマトグラフィー−質量分析)及びh(時間)といった従来からの略語も用いられる。
ヒドラゾンリンカーについて:本実施例では、(場合により他の基、例えばスペーサー、反応性官能基などを含む)本発明の薬剤−リンカー分子を標的化剤X4としての抗体に複合するための反応の条件及び方法について記載する。条件及び方法は、あくまで例示であって限定されるものではない。薬剤−リンカー分子を抗体に複合するための他のアプローチは当該技術分野で既知である。
A.化合物A〜G
786−O(ATCC登録番号CRL−1932)を、標準の実験方法を用いてインビトロで増殖した。6〜8週齢の雄CB17.SCIDマウス(Taconic、Hudson、NY)の右側腹に、マウス1匹当たりPBS/Matrigel(1:1)0.2ml中、250万個の786−Oを皮下移植した。移植後、週2回、マウスを秤量し、その腫瘍について電子キャリパーを用いて三次元的に測定した。腫瘍体積を高さ×幅×長さとして計算した。平均200mm3の腫瘍を有するマウスを治療群に無作為化した。0日目、マウスに対し、PBS媒体、細胞毒素に複合されたアイソタイプ対照抗体又は細胞毒素に複合された抗CD70 HuMAb 2H5を腹腔内投与した。各群はマウス7匹を有した。
786−O(ATCC寄託番号CRL−1932)細胞を標準の実験法を用いてインビトロで展開した。6〜8週齢の雄CB17.SCIDマウス(Taconic、Hudson、NY)の右脇腹に、マウス1匹当たりPBS/マトリゲル(1:1)0.2ml中の250万個の786−Oを皮下移植した。移植後、週に2回、マウスを秤量し、その腫瘍を、電子キャリパーを用いて三次元的に測定した。腫瘍体積を高さ×幅×長さとして計算した。平均200mm3の腫瘍を有するマウスを治療群に無作為化した。0日目、マウスに対し、PBS媒体、細胞毒素と複合されたアイソタイプ対照抗体又は細胞毒素と複合された抗CD70 HuMAb 2H5を腹腔内投与した。各群はマウス8匹を有した。
LNCaP及び786−O細胞上での腫瘍で活性化される活性
ATCCから入手した接着細胞のLNCaP(前立腺癌)及び786−O(腎細胞癌)を、ATCCの使用説明書に従い、10%熱不活性化ウシ胎仔血清(FCS)を含有するRPMI培地中で培養した。試験日に、細胞を、トリプシン溶液を有するプレートから取り出した。採取した細胞を洗浄し、LNCaP及び786−0細胞について、10%FCSを含有するRPMI中にそれぞれ0.25若しくは0.1×106細胞/mlの濃度で再懸濁した。細胞懸濁液100μlを96ウェルプレートに添加し、プレートを3時間インキュベートし、細胞を接着させた。このインキュベーション後、特異抗体−細胞毒素抱合体の300nMの細胞毒素(実施例21の化合物J)からの1:3の連続希釈物を各ウェルに添加した。次いで、プレートを48時間インキュベートし、100μCi/mlの3H−チミジン10μlでパルスし、更に72時間インキュベートした。プレートから96ウェルのHarvester(Packard Instruments)を用いて採取し、Packard Top Count Counter上で計数した。4つのパラメータのロジスティック曲線を、Prismソフトウェアを用い、薬剤モル濃度の関数として3H−チミジン取り込みに適合させ、EC50値を決定した。ロジスティック曲線を様々な抗体−細胞毒素抱合体に適合させ、LNCaP及び786−Oインビボでのそれらの得られたEC50値をそれぞれ図7で示す。
LNCaP異種移植片試験を以下のように行った。すなわち、各120CB17.SCIDマウスの脇腹領域に、PBS/マトリゲル(1:1)(BD Bioscience)0.2ml中に再懸濁した200万個のLNCaP細胞及び100万個の前立腺間質細胞(カタログ番号CC−2508、Cambrex Bio Science Walkersville,Inc、Walkersville、MD)を皮下注射した。移植後、週1回、マウスを秤量し、その腫瘍を、電子キャリパーを用いて三次元的に測定した。腫瘍体積を高さ×幅×長さ/2として計算した。1日前、平均50mm3の腫瘍を有するマウスを、マウス7匹からなる16の治療群に無作為化し、0日目、マウスを、表1に記載の投与計画に従い、媒体、抗体又は抗体−細胞毒素抱合体で静脈内投与した。62日目、試験を終了した。
LNCaP異種移植片試験を以下のように行った。すなわち、各120CB17.SCIDマウスの脇腹領域に、PBS/マトリゲル(1:1)(BD Bioscience社製)0.2ml中に再懸濁した250万個のLNCaP細胞を皮下注射した。移植後、週1回、マウスを秤量し、その腫瘍を、電子キャリパーを用いて三次元的に測定した。腫瘍体積を高さ×幅×長さ/2として計算した。1日前、平均80mm3の腫瘍を有するマウスを、マウス7匹からなる13の治療群に無作為化し、0日目、マウスを、表2に記載の投与計画に従い、媒体、抗体又は抗体−細胞毒素抱合体で静脈内治療した。55日目、試験を終了した。
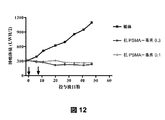
次いで、抗PSMA−細胞毒素の活性が、非常に大きいLNCaP細胞腫瘍異種移植片を担持するSCIDマウスにおいて示された。LNCaP細胞(PBS0.1ml+マトリゲル/マウス0.1ml中、250万個)を雄SCIDマウスに皮下移植し、腫瘍が310mm3の平均サイズに達した時、マウス8匹からなる群を、0.1若しくは0.3μmol/kg体重でのいずれかの抗PSMA−細胞毒素の単回用量の腹腔内注射により治療した。更に、対照群に媒体単独を注射した。腫瘍体積及びマウスの体重を、試験を通じて記録し、それを投与後60日間にわたって続けた。図12に図示される結果は、抗PSMA−2460抱合体の単回用量がマウスにおいて非常に大きい腫瘍から開始する場合でさえ有効であることを示した。
本実施例は、腎癌の2つの異種移植片モデルにおける抗CD70−細胞毒素の有効性を示す。細胞毒素抱合体は、以下の細胞毒素化合物に結合されたCD70抗体からなる。
本実施例は、SCIDマウスにおける786−O細胞及びヌードラットにおけるCaki−1細胞といった腎癌の2つの異種移植片モデルにおける抗CD70−細胞毒素の有効性を示す。細胞毒素抱合体は、以下の細胞毒素化合物に結合されたCD70抗体からなる。
Claims (47)
- 式
L1は自己犠牲リンカーであり;
mは0、1、2、3、4、5若しくは6の整数であり;
Fは構造:
AA1は天然アミノ酸及び非天然α−アミノ酸からなる群から独立して選択される1つ以上のメンバーであり;
cは1〜20の整数であり;
L2は自己犠牲リンカーであり、かつ
oは1であり、
L4はリンカーメンバーであり;
pは0若しくは1であり;
X4は保護反応性官能基、非保護反応性官能基、検出可能な標識、及び標的化剤からなる群から選択されるメンバーであり;かつ
Dは構造:
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3はOR11であり、R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13はそれらに結合する窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5及びR5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらに結合する炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成されてもよく;
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R6は存在しないか又は単結合として存在し、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7は前記シクロプロピル環内でR6と結合されたCH2−X1又は−CH2−であり、X1は離脱基であり、
R11は前記薬剤をL1(存在する場合)又はFに結合させる)
の化合物、又はその薬理学的に許容できる塩。 - Dが構造:
ZはO、S及びNR23から選択されるメンバーであり、
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R1は水素、置換若しくは未置換低級アルキル基、C(O)R8、又はCO2R8であり、R8は置換アルキル基、未置換アルキル基、NR9R10、NR9NHR10、及びOR9からなる群から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;かつ
R2は水素、置換アルキル基又は未置換低級アルキル基である)
を有する、請求項1記載の化合物。 - Dが構造:
ZはO、S及びNR23から選択されるメンバーであり、
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R1は水素、置換若しくは未置換低級アルキル基、C(O)R8、又はCO2R8であり、R8はNR9R10及びOR9から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;
R1’は水素、置換若しくは未置換低級アルキル基又はC(O)R8であり、R8はNR9R10及びOR9から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;
R2は水素、又は置換若しくは未置換低級アルキル基又は未置換ヘテロアルキル基又はシアノ基又はアルコキシ基であり;かつ
R2’は水素、又は置換若しくは未置換低級アルキル基又は未置換ヘテロアルキル基である)
を有する、請求項1記載の化合物。 - (AA1)cが腫瘍組織内で発現されるプロテアーゼにより切断可能なペプチド配列である、請求項1記載の化合物。
- 前記プロテアーゼがリソソームプロテアーゼである、請求項4記載の化合物。
- 前記薬剤部分の直近に位置する(AA1)c内の前記アミノ酸が、Ala、Asn、Asp、Cit、Cys、Gln、Glu、Gly、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、Tyr、及びValからなる群から選択される、請求項1記載の化合物。
- (AA1)cが、Val−Cit、Val−Lys、Phe−Lys、Lys−Lys、Ala−Lys、Phe−Cit、Leu−Cit、Ile−Cit、Trp、Cit、Phe−Ala、Phe−N9−トシル−Arg、Phe−N9−ニトロ−Arg、Phe−Phe−Lys、D−Phe−Phe−Lys、Gly−Phe−Lys、Leu−Ala−Leu、Ile−Ala−Leu、Val−Ala−Val、Ala−Leu−Ala−Leu(配列番号1)、β−Ala−Leu−Ala−Leu(配列番号2)及びGly−Phe−Leu−Gly(配列番号3)からなる群から選択されるペプチド配列である、請求項1記載の化合物。
- (AA1)cがVal−Cit又はVal−Lysである、請求項1記載の化合物。
- R17及びR18が低級アルキル基である、請求項1記載の化合物。
- wが0である、請求項1記載の化合物。
- 前記化合物が
- 式
L1は自己犠牲リンカーであり;
mは0、1、2、3、4、5若しくは6の整数であり;
Fは構造:
AA1は天然アミノ酸及び非天然α−アミノ酸からなる群から独立して選択される1つ以上のメンバーであり;
cは1〜20の整数であり;
L2は自己犠牲リンカーであり、
oは0若しくは1であり、
L4はリンカーメンバーであり;
pは0若しくは1であり;
X4は保護反応性官能基、非保護反応性官能基、検出可能な標識、及び標的化剤からなる群から選択されるメンバーであり;かつ
Dは構造:
環系Aは、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択されるメンバーであり;
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3は、(=O)、SR11、NHR11及びOR11からなる群から選択されるメンバーであり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13はそれらに結合する窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5及びR5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらに結合する炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成されてもよく;
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5及びR5’の少なくとも1つは前記薬剤をL1(存在する場合)又はFに結合させ、かつ
R27、R27’、R28、及びR28’の各々は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、及び置換若しくは未置換ヘテロシクロアルキル基から独立して選択され;
R6は存在しないか又は単結合として存在し、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7は前記シクロプロピル環内でR6と結合されたCH2−X1又は−CH2−であり、
X1は離脱基である)
の化合物、又はその薬理学的に許容できる塩。 - Dが構造:
ZはO、S及びNR23から選択されるメンバーであり、
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R1は水素、置換若しくは未置換低級アルキル基、C(O)R8、又はCO2R8であり、R8は置換アルキル基、未置換アルキル基、NR9R10、NR9NHR10、及びOR9からなる群から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;かつ
R2は水素、置換アルキル基又は未置換低級アルキル基である)
を有する、請求項12記載の化合物。 - Dが構造:
ZはO、S及びNR23から選択されるメンバーであり、
R23は水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R1は水素、置換若しくは未置換低級アルキル基、C(O)R8、又はCO2R8であり、R8はNR9R10及びOR9から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;
R1’は水素、置換若しくは未置換低級アルキル基、又はC(O)R8であり、R8はNR9R10及びOR9から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;
R2は水素、又は置換若しくは未置換低級アルキル基又は未置換ヘテロアルキル基又はシアノ基又はアルコキシ基であり;かつ
R2’は水素、又は置換若しくは未置換低級アルキル基又は未置換ヘテロアルキル基である)
を有する、請求項12記載の化合物。 - (AA1)cが腫瘍組織内で発現されるプロテアーゼにより切断可能なペプチド配列である、請求項12記載の化合物。
- 前記プロテアーゼがリソソームプロテアーゼである、請求項15記載の化合物。
- 前記薬剤部分の直近に位置する(AA1)c内の前記アミノ酸が、Ala、Asn、Asp、Cit、Cys、Gln、Glu、Gly、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、Tyr、及びValからなる群から選択される、請求項12記載の化合物。
- (AA1)cが、Val−Cit、Val−Lys、Phe−Lys、Lys−Lys、Ala−Lys、Phe−Cit、Leu−Cit、Ile−Cit、Trp、Cit、Phe−Ala、Phe−N9−トシル−Arg、Phe−N9−ニトロ−Arg、Phe−Phe−Lys、D−Phe−Phe−Lys、Gly−Phe−Lys、Leu−Ala−Leu、Ile−Ala−Leu、Val−Ala−Val、Ala−Leu−Ala−Leu(配列番号1)、β−Ala−Leu−Ala−Leu(配列番号2)及びGly−Phe−Leu−Gly(配列番号3)からなる群から選択されるペプチド配列である、請求項12記載の化合物。
- (AA1)cがVal−Cit又はVal−Lysである、請求項12記載の化合物。
- R27、R27’、R28、及びR28’が独立して水素又は低級アルキル基である、請求項12記載の化合物。
- vが1又は2である、請求項12記載の化合物。
- R15が水素又は低級アルキル基である、請求項12記載の化合物。
- 前記化合物が
- 式
L1は自己犠牲リンカーであり;
mは0、1、2、3、4、5若しくは6の整数であり;
Fは構造:
AA1は天然アミノ酸及び非天然α−アミノ酸からなる群から独立して選択される1つ以上のメンバーであり;
cは1〜20の整数であり;
L3は第一級若しくは第二級アミン基又はカルボキシル官能基を含むスペーサー基であり;L3が存在する場合、mは0であり、かつL3のアミン基はDのペンダントカルボキシル官能基とアミド結合を形成するか又はL3のカルボキシル基はDのペンダントアミン官能基とアミド結合を形成し;
oは0若しくは1であり、
L4はリンカーメンバーであり、L4は(AA1)cのN末端に直接結合される
R20は水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり、
R25、R25’、R26、及びR26’の各々は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、及び置換若しくは未置換ヘテロシクロアルキル基から独立して選択され;かつ
s及びtは独立して1〜6の整数であり;
pは1であり;
X4は、保護反応性官能基、非保護反応性官能基、検出可能な標識、及び標的化剤からなる群から選択されるメンバーであり;かつ
Dは構造:
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3は、(=O)、SR11、NHR11及びOR11からなる群から選択されるメンバーであり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13はそれらに結合する窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5及びR5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらに結合する炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成されてもよく;
nは1〜20の整数であり、
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R6は存在しないか又は単結合として存在し、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7は前記シクロプロピル環内でR6と結合されたCH2−X1若しくは−CH2−であり、X1は離脱基であり、
R4、R4’、R5、R5’、R15又はR16の少なくとも1つは前記薬剤をL1(存在する場合)又はFに結合させる)
の化合物、又はその薬理学的に許容できる塩。 - Dが構造:
ZはO、S及びNR23から選択されるメンバーであり、
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R1は水素、置換若しくは未置換低級アルキル基、C(O)R8、又はCO2R8であり、R8は置換アルキル基、未置換アルキル基、NR9R10、NR9NHR10、及びOR9からなる群から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;かつ
R2は水素、置換アルキル基又は未置換低級アルキル基である)
を有する、請求項24記載の化合物。 - Dが構造:
ZはO、S及びNR23から選択されるメンバーであり、
R23は水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R1は水素、置換若しくは未置換低級アルキル基、C(O)R8、又はCO2R8であり、R8はNR9R10及びOR9から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;
R1’は水素、置換若しくは未置換低級アルキル基、又はC(O)R8であり、R8はNR9R10及びOR9から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;
R2は水素、又は置換若しくは未置換低級アルキル基又は未置換ヘテロアルキル基又はシアノ基又はアルコキシ基であり;かつ
R2’は水素、又は置換若しくは未置換低級アルキル基又は未置換ヘテロアルキル基である)
を有する、請求項24記載の化合物。 - (AA1)cが腫瘍組織内で発現されるプロテアーゼにより切断可能なペプチド配列である、請求項24記載の化合物。
- 前記プロテアーゼがリソソームプロテアーゼである、請求項27記載の化合物。
- 前記薬剤部分の直近に位置する(AA1)c内の前記アミノ酸が、Ala、Asn、Asp、Cit、Cys、Gln、Glu、Gly、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、Tyr、及びValからなる群から選択される、請求項24記載の化合物。
- (AA1)cが、Val−Cit、Val−Lys、Phe−Lys、Lys−Lys、Ala−Lys、Phe−Cit、Leu−Cit、Ile−Cit、Trp、Cit、Phe−Ala、Phe−N9−トシル−Arg、Phe−N9−ニトロ−Arg、Phe−Phe−Lys、D−Phe−Phe−Lys、Gly−Phe−Lys、Leu−Ala−Leu、Ile−Ala−Leu、Val−Ala−Val、Ala−Leu−Ala−Leu(配列番号1)、β−Ala−Leu−Ala−Leu(配列番号2)及びGly−Phe−Leu−Gly(配列番号3)からなる群から選択されるペプチド配列である、請求項24記載の化合物。
- (AA1)cがVal−Cit又はVal−Lysである、請求項24記載の化合物。
- R25、R25’、R26、及びR26’が独立して水素又は低級アルキル基である、請求項24記載の化合物。
- s及びtが独立して1又は2である、請求項24記載の化合物。
- R20が水素又は低級アルキル基である、請求項24記載の化合物。
- 前記化合物が
- 構造:
mは0、1、2、3、4、5若しくは6の整数であり;
X2は切断可能な基質であり;かつ
Dは構造:
環系Aは、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択されるメンバーであり;
E及びGは、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、ヘテロ原子、単結合から独立して選択されるメンバーであるか、又はE及びGは結合し、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基及び置換若しくは未置換ヘテロシクロアルキル基から選択される環系が形成され;
XはO、S及びNR23から選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3は、(=O)、SR11、NHR11及びOR11からなる群から選択されるメンバーであり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13は、それらに結合する窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R6は存在しないか又は単結合として存在し、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7は前記シクロプロピル環内でR6と結合されたCH2−X1若しくは−CH2−であり、X1は離脱基であり、
R4、R4’、R5及びR5’は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらに結合する炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成され、
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R4、R4’、R5及びR5’の少なくとも1つのメンバーは前記薬剤をL1(存在する場合)又はX2に結合させ、かつ
R30、R30’、R31、及びR31’は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、及び置換若しくは未置換ヘテロシクロアルキル基から独立して選択され;かつ
vは1〜6の整数である)
を有する化合物あるいはその薬理学的に許容できる塩。 - Dが構造:
ZはO、S及びNR23から選択されるメンバーであり、
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R1は水素、置換若しくは未置換低級アルキル基、C(O)R8、又はCO2R8であり、R8は置換アルキル基、未置換アルキル基、NR9R10、NR9NHR10、及びOR9からなる群から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;かつ
R2は水素、置換アルキル基又は未置換低級アルキル基である)
を有する、請求項36記載の化合物。 - Dが構造:
ZはO、S及びNR23から選択されるメンバーであり、
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R1は水素、置換若しくは未置換低級アルキル基、C(O)R8、又はCO2R8であり、R8はNR9R10及びOR9から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;
R1’は水素、置換若しくは未置換低級アルキル基、又はC(O)R8であり、R8はNR9R10及びOR9から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;
R2は水素、又は置換若しくは未置換低級アルキル基又は未置換ヘテロアルキル基又はシアノ基又はアルコキシ基であり;かつ
R2’は水素、又は置換若しくは未置換低級アルキル基又は未置換ヘテロアルキル基である)
を有する、請求項36記載の化合物。 - X2が、β−AlaLeuAlaLeu(配列番号2)又は
- 前記化合物が、
- 構造:
Z及びXは、O、S及びNR23から独立して選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R3は、(=O)、SR11、NHR11及びOR11からなる群から選択されるメンバーであり、
R11は、水素、置換アルキル基、未置換アルキル基、置換ヘテロアルキル基、未置換ヘテロアルキル基、モノホスフェート基、ジホスフェート基、トリホスフェート基、スルホネート基、アシル基、C(O)R12R13、C(O)OR12、C(O)NR12R13、P(O)(OR12)2、C(O)CHR12R13、SR12及びSiR12R13R14からなる群から選択されるメンバーであり、
R12、R13、及びR14は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基及び置換若しくは未置換アリール基から独立して選択されるメンバーであり、R12及びR13はそれらに結合する窒素又は炭素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成されてもよく;
R6は存在しないか又は単結合として存在し、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;
R7は前記シクロプロピル環内でR6と結合されたCH2−X1若しくは−CH2−であり、
X1は離脱基であり;かつ
R5、R5’、及びR32は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであり、
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成される)
を有する化合物あるいはその薬理学的に許容できる塩。 - 前記化合物が、構造:
R1は水素、置換若しくは未置換低級アルキル基、C(O)R8、又はCO2R8であり、R8はNR9R10及びOR9から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;
R1’は水素、置換若しくは未置換低級アルキル基、又はC(O)R8であり、R8はNR9R10及びOR9から選択されるメンバーであり、
R9及びR10は、水素、置換若しくは未置換アルキル基及び置換若しくは未置換ヘテロアルキル基から独立して選択されるメンバーであり;
R2は水素、又は置換若しくは未置換低級アルキル基又は未置換ヘテロアルキル基又はシアノ基又はアルコキシ基であり;かつ
R2’は水素、又は置換若しくは未置換低級アルキル基又は未置換ヘテロアルキル基である)
を有する、請求項41記載の化合物。 - R32が1つ以上の切断可能な基を含む、請求項41記載の化合物。
- 前記1つ以上の切断可能な基が切断可能なリンカー及び切断可能な基質から選択される、請求項43記載の化合物。
- 構造:
Z及びXは、O、S及びNR23から独立して選択されるメンバーであり;
R23は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、及びアシル基から選択されるメンバーであり;
R4、R4’、R5、R5’、及びR33は、水素、置換アルキル基、未置換アルキル基、置換アリール基、未置換アリール基、置換ヘテロアリール基、未置換ヘテロアリール基、置換ヘテロシクロアルキル基、未置換ヘテロシクロアルキル基、ハロゲン、NO2、NR15R16、NC(O)R15、OC(O)NR15R16、OC(O)OR15、C(O)R15、SR15、OR15、CR15=NR16、及びO(CH2)nN(CH3)2からなる群から独立して選択されるメンバーであるか、又はR4、R4’、R5及びR5’の任意の隣接対はそれらに結合する炭素原子と共に結合し、4〜6員を有する置換若しくは未置換シクロアルキル又はヘテロシクロアルキル環系が形成されてもよく;
nは1〜20の整数であり;
R15及びR16は、水素、置換若しくは未置換アルキル基、置換若しくは未置換ヘテロアルキル基、置換若しくは未置換アリール基、置換若しくは未置換ヘテロアリール基、置換若しくは未置換ヘテロシクロアルキル基、及び置換若しくは未置換ペプチジル基から独立して選択され、R15及びR16はそれらに結合する窒素原子と共に任意に結合し、4〜6員を有し、任意に2つ以上のヘテロ原子を有する置換若しくは未置換ヘテロシクロアルキル環系が形成され、
R33は1つ以上の切断可能な基を含み;
R6は存在しないか又は単結合として存在し、存在する場合、R6及びR7は結合し、シクロプロピル環が形成され;かつ
R7は前記シクロプロピル環内でR6と結合されたCH2−X1若しくは−CH2−であり、X1は離脱基である)
を有する化合物あるいはその薬理学的に許容できる塩。 - 前記1つ以上の切断可能な基が切断可能なリンカー及び切断可能な基質から選択される、請求項45記載の化合物。
-
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US88246106P | 2006-12-28 | 2006-12-28 | |
| US99130007P | 2007-11-30 | 2007-11-30 | |
| PCT/US2007/089100 WO2008083312A2 (en) | 2006-12-28 | 2007-12-28 | Chemical linkers and cleavable substrates and conjugates thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010522693A true JP2010522693A (ja) | 2010-07-08 |
| JP2010522693A5 JP2010522693A5 (ja) | 2011-02-24 |
Family
ID=39589219
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009544303A Pending JP2010522693A (ja) | 2006-12-28 | 2007-12-28 | 化学リンカー及び切断可能な基質及びそれらの抱合体 |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US8461117B2 (ja) |
| EP (1) | EP2114454A2 (ja) |
| JP (1) | JP2010522693A (ja) |
| KR (1) | KR20090098901A (ja) |
| CN (1) | CN101795711A (ja) |
| AR (1) | AR067837A1 (ja) |
| AU (1) | AU2007342052A1 (ja) |
| BR (1) | BRPI0719626A2 (ja) |
| CA (1) | CA2674055C (ja) |
| CL (1) | CL2007003849A1 (ja) |
| EA (1) | EA019962B1 (ja) |
| IL (1) | IL199416A0 (ja) |
| MX (1) | MX2009007147A (ja) |
| NO (1) | NO20092733L (ja) |
| NZ (1) | NZ578324A (ja) |
| SG (1) | SG170070A1 (ja) |
| TW (1) | TWI412367B (ja) |
| WO (1) | WO2008083312A2 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010519310A (ja) * | 2007-02-21 | 2010-06-03 | メダレックス インコーポレイテッド | 単一のアミノ酸を有する化学リンカーおよびその複合体 |
| JP2016188207A (ja) * | 2010-12-17 | 2016-11-04 | アローヘッド ファーマシューティカルズ インコーポレイテッド | ペプチドに基づくインビボsiRNA送達システム |
| JP6993084B2 (ja) | 2013-08-12 | 2022-02-03 | ジェネンテック, インコーポレイテッド | 1-(クロロメチル)-2,3-ジヒドロ-1h-ベンゾ[e]インドール二量体抗体 - 薬物コンジュゲート化合物、並びに使用及び処置の方法 |
Families Citing this family (141)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ568015A (en) | 2005-12-08 | 2012-03-30 | Medarex Inc | Human monoclonal antibodies to O8E |
| EP2185188B1 (en) | 2007-08-22 | 2014-08-06 | Medarex, L.L.C. | Site-specific attachment of drugs or other agents to engineered antibodies with c-terminal extensions |
| CA2700860C (en) | 2007-10-01 | 2016-07-19 | Jonathan A. Terrett | Human antibodies that bind mesothelin, and uses thereof |
| TW200938224A (en) * | 2007-11-30 | 2009-09-16 | Medarex Inc | Anti-B7H4 monoclonal antibody-drug conjugate and methods of use |
| AR072999A1 (es) | 2008-08-11 | 2010-10-06 | Medarex Inc | Anticuerpos humanos que se unen al gen 3 de activacion linfocitaria (lag-3) y los usos de estos |
| CA2742568C (en) | 2008-11-03 | 2017-09-26 | Syntarga B.V. | Novel cc-1065 analogs and their conjugates |
| ME02842B (me) | 2009-03-05 | 2018-01-20 | Squibb & Sons Llc | Potpuno ljudska antitijela specifična za cadm1 |
| US8394922B2 (en) | 2009-08-03 | 2013-03-12 | Medarex, Inc. | Antiproliferative compounds, conjugates thereof, methods therefor, and uses thereof |
| US8530170B2 (en) * | 2009-08-07 | 2013-09-10 | Ohmx Corporation | Enzyme triggered redox altering chemical elimination (E-trace) immunoassay |
| US9250234B2 (en) | 2011-01-19 | 2016-02-02 | Ohmx Corporation | Enzyme triggered redox altering chemical elimination (E-TRACE) immunoassay |
| WO2011130598A1 (en) * | 2010-04-15 | 2011-10-20 | Spirogen Limited | Pyrrolobenzodiazepines and conjugates thereof |
| EP3108886B1 (en) | 2010-04-21 | 2020-06-17 | Syntarga B.V. | Conjugates of cc-1065 analogs and bifunctional linkers |
| US8956859B1 (en) | 2010-08-13 | 2015-02-17 | Aviex Technologies Llc | Compositions and methods for determining successful immunization by one or more vaccines |
| US8852599B2 (en) * | 2011-05-26 | 2014-10-07 | Bristol-Myers Squibb Company | Immunoconjugates, compositions for making them, and methods of making and use |
| CA2851632A1 (en) | 2011-10-17 | 2013-04-25 | Ohmx Corporation | Single, direct detection of hemoglobin a1c percentage using enzyme triggered redox altering chemical elimination (e-trace) immunoassay |
| US9340567B2 (en) | 2011-11-04 | 2016-05-17 | Ohmx Corporation | Chemistry used in biosensors |
| CA2860739A1 (en) | 2012-01-09 | 2013-07-18 | Ohmx Corporation | Enzyme cascade methods for e-trace assay signal amplification |
| SG11201404667XA (en) | 2012-02-13 | 2014-09-26 | Bristol Myers Squibb Co | Enediyne compounds, conjugates thereof, and uses and methods therefor |
| EP2858659B1 (en) | 2012-06-08 | 2019-12-25 | Bioverativ Therapeutics Inc. | Procoagulant compounds |
| US10202595B2 (en) | 2012-06-08 | 2019-02-12 | Bioverativ Therapeutics Inc. | Chimeric clotting factors |
| UY34887A (es) | 2012-07-02 | 2013-12-31 | Bristol Myers Squibb Company Una Corporacion Del Estado De Delaware | Optimización de anticuerpos que se fijan al gen de activación de linfocitos 3 (lag-3) y sus usos |
| US9416390B2 (en) | 2012-07-27 | 2016-08-16 | Ohmx Corporation | Electric measurement of monolayers following pro-cleave detection of presence and activity of enzymes and other target analytes |
| WO2014018886A1 (en) | 2012-07-27 | 2014-01-30 | Ohmx Corporation | Electronic measurements of monolayers following homogeneous reactions of their components |
| AR094280A1 (es) | 2012-12-21 | 2015-07-22 | Bioalliance Cv | Enlazantes hidrofilicos auto-inmolantes y conjugados de los mismos |
| SI2956173T1 (sl) | 2013-02-14 | 2017-06-30 | Bristol-Myers Squibb Company | Spojine tubulizina, postopki pridobivanja in uporaba |
| AU2014228489B2 (en) | 2013-03-15 | 2018-11-15 | Zymeworks Bc Inc. | Cytotoxic and anti-mitotic compounds, and methods of using the same |
| SI2991683T1 (sl) | 2013-05-02 | 2020-01-31 | Glykos Finland Oy | Konjugati glikoproteina ali glikana s toksično obremenitvijo |
| BR112016013258A2 (pt) | 2013-12-16 | 2018-01-16 | Genentech Inc | composto conjugado anticorpo-droga, composição farmacêutica, método para tratar câncer e kit |
| DK3086815T3 (da) | 2013-12-27 | 2022-05-23 | Zymeworks Inc | Sulfonamidholdige forbindelsessystemer til lægemiddelkonjugater |
| TR201810856T4 (tr) | 2014-01-10 | 2018-08-27 | Synthon Biopharmaceuticals Bv | CYS'le bağlı antikor-ilaç konjugatlarını saflaştırmak için usul. |
| CN115322253A (zh) | 2014-03-20 | 2022-11-11 | 百时美施贵宝公司 | 稳定化的基于纤连蛋白的支架分子 |
| JP6449338B2 (ja) | 2014-06-06 | 2019-01-09 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | グルココルチコイド誘導腫瘍壊死因子受容体(gitr)に対する抗体およびその使用 |
| MX2016016490A (es) | 2014-06-20 | 2017-07-28 | Bioalliance Cv | Conjugados de farmaco-anticuerpo anti-receptor de folato alfa (fra) y metodos de uso de los mismos. |
| WO2016001485A1 (en) | 2014-06-30 | 2016-01-07 | Glykos Finland Oy | Saccharide derivative of a toxic payload and antibody conjugates thereof |
| US20160041118A1 (en) | 2014-08-11 | 2016-02-11 | Ohmx Corporation | Enzyme triggered redox altering chemical elimination (e-trace) assay with multiplexing capabilities |
| DK3191502T3 (da) * | 2014-09-11 | 2021-07-19 | Seagen Inc | Målrettet indgivelse af tertiært aminholdige lægemiddelstoffer |
| RU2723651C2 (ru) | 2014-09-17 | 2020-06-17 | Займворкс Инк. | Цитотоксические и антимитотические соединения и способы их применения |
| CN104356048B (zh) * | 2014-11-02 | 2016-08-24 | 浙江医药高等专科学校 | 环己烷羧酸酰胺类衍生物、其制备方法和用途 |
| US10077287B2 (en) | 2014-11-10 | 2018-09-18 | Bristol-Myers Squibb Company | Tubulysin analogs and methods of making and use |
| LT3221346T (lt) | 2014-11-21 | 2020-11-10 | Bristol-Myers Squibb Company | Antikūnai, apimantys modifikuotas sunkiosios grandinės pastoviąsias sritis |
| HUE050596T2 (hu) | 2014-11-21 | 2020-12-28 | Bristol Myers Squibb Co | Antitestek CD73 ellen és azok felhasználásai |
| ES2822990T3 (es) | 2014-11-25 | 2021-05-05 | Bristol Myers Squibb Co | Novedosos polipéptidos de unión a PD-L1 para obtención de imágenes |
| EA034516B1 (ru) | 2014-11-25 | 2020-02-14 | Бристол-Маерс Сквибб Компани | Способы и композиции для мечения радиоактивным изотопомf биологических препаратов |
| AU2015365583B2 (en) | 2014-12-19 | 2021-10-28 | Regenesance B.V. | Antibodies that bind human C6 and uses thereof |
| WO2016100834A2 (en) | 2014-12-19 | 2016-06-23 | Ohmx Corporation | Competitive enzymatic assays |
| ES2747386T3 (es) | 2015-01-14 | 2020-03-10 | Bristol Myers Squibb Co | Dímeros de benzodiacepina unidos por heteroarileno, conjugados de los mismos y métodos de preparación y uso |
| MA41374A (fr) * | 2015-01-20 | 2017-11-28 | Cytomx Therapeutics Inc | Substrats clivables par métalloprotéase matricielle et clivables par sérine protéase et procédés d'utilisation de ceux-ci |
| CN107406496A (zh) | 2015-03-10 | 2017-11-28 | 百时美施贵宝公司 | 可通过转谷氨酰胺酶缀合的抗体和由其制备的缀合物 |
| UY36687A (es) | 2015-05-29 | 2016-11-30 | Bristol Myers Squibb Company Una Corporación Del Estado De Delaware | Anticuerpos contra ox40 y sus usos |
| WO2017048728A1 (en) * | 2015-09-14 | 2017-03-23 | Rutgers, The State University Of New Jersey | Targeted conjugates |
| ES2809125T3 (es) | 2015-09-23 | 2021-03-03 | Bristol Myers Squibb Co | Moléculas de armazón a base de fibronectina de unión a glipicano-3 |
| EP3165532B1 (en) | 2015-11-03 | 2018-12-19 | Industrial Technology Research Institute | Auristatin derivatives, linker-drugs and ligand-drug conjugates |
| CN108738324B (zh) | 2015-11-19 | 2022-06-21 | 百时美施贵宝公司 | 抗糖皮质激素诱导的肿瘤坏死因子受体(gitr)抗体及其用途 |
| GB2545169B (en) * | 2015-12-01 | 2019-10-09 | Ellipses Pharma Ltd | Taxane Prodrug Comprising A Membrane Type Matrix Metalloproteinase Cleavage Site |
| US11793880B2 (en) | 2015-12-04 | 2023-10-24 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| US11229708B2 (en) | 2015-12-04 | 2022-01-25 | Seagen Inc. | Conjugates of quaternized tubulysin compounds |
| JP2019505575A (ja) | 2015-12-21 | 2019-02-28 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | 部位特異的な結合のための変異型抗体 |
| RU2766000C2 (ru) | 2016-01-08 | 2022-02-07 | АльтруБио Инк. | Четырехвалентные антитела к psgl-1 и их применения |
| IL295230A (en) | 2016-03-04 | 2022-10-01 | Bristol Myers Squibb Co | Combination therapy with anti-cd73 antibodies |
| TWI781098B (zh) | 2016-04-15 | 2022-10-21 | 美商宏觀基因股份有限公司 | 新穎的b7-h3-結合分子、其抗體藥物綴合物及其使用方法 |
| KR102397783B1 (ko) | 2016-06-01 | 2022-05-12 | 브리스톨-마이어스 스큅 컴퍼니 | Pd-l1 결합 폴리펩티드에 의한 pet 영상화 |
| US10994033B2 (en) | 2016-06-01 | 2021-05-04 | Bristol-Myers Squibb Company | Imaging methods using 18F-radiolabeled biologics |
| JP7027401B2 (ja) | 2016-07-14 | 2022-03-01 | ブリストル-マイヤーズ スクイブ カンパニー | Tim3に対する抗体およびその使用 |
| ES2902179T3 (es) | 2016-08-19 | 2022-03-25 | Bristol Myers Squibb Co | Compuestos de seco-ciclopropapirroloindol, conjugados de anticuerpo-fármaco de los mismos y métodos de elaboración y uso |
| US20190218294A1 (en) | 2016-09-09 | 2019-07-18 | Bristol-Myers Squibb Company | Use of an anti-pd-1 antibody in combination with an anti-mesothelin antibody in cancer treatment |
| WO2018064094A1 (en) | 2016-09-28 | 2018-04-05 | Bristol-Myers Squibb Company | Enantioselective synthesis of pyrroloindole compounds |
| HUE070615T2 (hu) * | 2016-10-17 | 2025-06-28 | Pfizer | Anti-EDB antitestek és antitest-gyógyszer konjugátumok |
| WO2018075842A1 (en) | 2016-10-20 | 2018-04-26 | Bristol-Myers Squibb Company | Condensed benzodiazepine derivatives and conjugates made therefrom |
| BR112019016374A2 (pt) | 2017-02-17 | 2020-04-07 | Bristol-Myers Squibb Company | anticorpos para alfa-sinucleína e usos dos mesmos |
| US11339218B2 (en) | 2017-05-10 | 2022-05-24 | Zhejiang Shimai Pharmaceutical Co., Ltd. | Human monoclonal antibodies against LAG3 and uses thereof |
| EP3630189A4 (en) | 2017-05-24 | 2021-06-23 | The Board of Regents of The University of Texas System | LINKER FOR ANTIBODY MEDICINAL CONJUGATES |
| MX2019013132A (es) | 2017-05-25 | 2020-01-27 | Bristol Myers Squibb Co | Anticuerpos que comprenden regiones constantes pesadas modificadas. |
| CN111093662B (zh) | 2017-06-20 | 2023-10-03 | 安布里亚制药公司 | 用于提高心脏代谢效率的组合物和方法 |
| US10457681B2 (en) | 2017-08-16 | 2019-10-29 | Bristol_Myers Squibb Company | Toll-like receptor 7 (TLR7) agonists having a tricyclic moiety, conjugates thereof, and methods and uses therefor |
| US10487084B2 (en) | 2017-08-16 | 2019-11-26 | Bristol-Myers Squibb Company | Toll-like receptor 7 (TLR7) agonists having a heterobiaryl moiety, conjugates thereof, and methods and uses therefor |
| US10472361B2 (en) | 2017-08-16 | 2019-11-12 | Bristol-Myers Squibb Company | Toll-like receptor 7 (TLR7) agonists having a benzotriazole moiety, conjugates thereof, and methods and uses therefor |
| US10494370B2 (en) | 2017-08-16 | 2019-12-03 | Bristol-Myers Squibb Company | Toll-like receptor 7 (TLR7) agonists having a pyridine or pyrazine moiety, conjugates thereof, and methods and uses therefor |
| US10508115B2 (en) | 2017-08-16 | 2019-12-17 | Bristol-Myers Squibb Company | Toll-like receptor 7 (TLR7) agonists having heteroatom-linked aromatic moieties, conjugates thereof, and methods and uses therefor |
| EP3694552A1 (en) | 2017-10-10 | 2020-08-19 | Tilos Therapeutics, Inc. | Anti-lap antibodies and uses thereof |
| KR20250078626A (ko) | 2018-01-12 | 2025-06-02 | 브리스톨-마이어스 스큅 컴퍼니 | Tim3에 대한 항체 및 그의 용도 |
| BR112020018539A2 (pt) | 2018-03-23 | 2020-12-29 | Bristol-Myers Squibb Company | Anticorpos contra mica e/ou micb e usos dos mesmos |
| TWI790370B (zh) | 2018-04-02 | 2023-01-21 | 美商必治妥美雅史谷比公司 | 抗trem-1抗體及其用途 |
| WO2019209811A1 (en) | 2018-04-24 | 2019-10-31 | Bristol-Myers Squibb Company | Macrocyclic toll-like receptor 7 (tlr7) agonists |
| US20210276971A1 (en) | 2018-06-20 | 2021-09-09 | The Research Foundation For The State University Of New York | Triazamacrocycle-derived chelator compositions for coordination of imaging and therapy metal ions and methods of using same |
| EP3841124A4 (en) | 2018-06-29 | 2022-03-23 | ApitBio, Inc. | ANTI-L1CAM ANTIBODIES AND USES THEREOF |
| US11554120B2 (en) | 2018-08-03 | 2023-01-17 | Bristol-Myers Squibb Company | 1H-pyrazolo[4,3-d]pyrimidine compounds as toll-like receptor 7 (TLR7) agonists and methods and uses therefor |
| EA202190755A1 (ru) | 2018-09-12 | 2021-06-11 | Технише Универзитет Мюнхен | Агенты для терапии и диагностики рака |
| EP3849997B1 (en) * | 2018-09-12 | 2024-05-01 | Technische Universität München | Cxcr4-targeted diagnostic and therapeutic agents with reduced species selectivity |
| US11130802B2 (en) | 2018-10-10 | 2021-09-28 | Tilos Therapeutics, Inc. | Anti-lap antibody variants |
| WO2020081361A1 (en) | 2018-10-17 | 2020-04-23 | Imbria Pharmaceuticals, Inc. | Methods of treating rheumatic diseases using trimetazidine-based compounds |
| MX2021005484A (es) | 2018-11-09 | 2021-06-18 | Byondis Bv | Composiciones de conjugados de anticuerpo-farmaco que contienen duocarmicina filtrable y metodos relacionados. |
| US20220106400A1 (en) | 2018-11-28 | 2022-04-07 | Bristol-Myers Squibb Company | Antibodies comprising modified heavy constant regions |
| BR112021010060B1 (pt) | 2018-11-30 | 2024-03-12 | Bristol-Myers Squibb Company | Anticorpo que compreende uma extensão c-terminal de cadeia leve que contém glutamina, conjugados do mesmo, e método de preparação de conjugados |
| WO2020117627A1 (en) | 2018-12-03 | 2020-06-11 | Bristol-Myers Squibb Company | Anti-ido antibody and uses thereof |
| AU2019394855B2 (en) | 2018-12-04 | 2025-02-27 | Der-Yang Tien | Stereocomplexes for the delivery of anti-cancer agents |
| CA3120327A1 (en) | 2018-12-06 | 2020-06-11 | Cytomx Therapeutics, Inc. | Matrix metalloprotease-cleavable and serine or cysteine protease-cleavable substrates and methods of use thereof |
| WO2020123425A2 (en) | 2018-12-12 | 2020-06-18 | Bristol-Myers Squibb Company | Antibodies modified for transglutaminase conjugation, conjugates thereof, and methods and uses |
| CN109762067B (zh) | 2019-01-17 | 2020-02-28 | 北京天广实生物技术股份有限公司 | 结合人Claudin 18.2的抗体及其用途 |
| KR20230128134A (ko) | 2019-01-22 | 2023-09-01 | 브리스톨-마이어스 스큅 컴퍼니 | Il-7r 알파 서브유닛에 대한 항체 및 이의 용도 |
| BR112021018694A2 (pt) | 2019-03-21 | 2021-11-30 | Codiak Biosciences Inc | Vesículas extracelulares para distribuição de vacina |
| US20240108747A1 (en) | 2019-03-21 | 2024-04-04 | Lonza Sales Ag | Extracellular vesicle conjugates and uses thereof |
| AU2020258483A1 (en) | 2019-04-17 | 2021-11-11 | Evox Therapeutics, Ltd. | Compositions of exosomes and AAV |
| EP3963085A4 (en) * | 2019-05-01 | 2022-09-14 | The University of North Carolina at Chapel Hill | RNA-BINDING PROTEIN INHIBITORS, COMPOSITIONS THEREOF, AND THERAPEUTIC USES THEREOF |
| EP3976101A4 (en) | 2019-05-31 | 2023-06-21 | Imbria Pharmaceuticals, Inc. | FIBROSIS TREATMENT METHODS USING COMPOUNDS THAT PROMOTE GLUCOSE OXIDATION |
| WO2021003445A1 (en) | 2019-07-03 | 2021-01-07 | Codiak Biosciences, Inc. | Extracellular vesicles targeting t cells and uses thereof |
| WO2021011681A1 (en) | 2019-07-15 | 2021-01-21 | Bristol-Myers Squibb Company | Antibodies against human trem-1 and uses thereof |
| WO2021011678A1 (en) | 2019-07-15 | 2021-01-21 | Bristol-Myers Squibb Company | Anti-trem-1 antibodies and uses thereof |
| EP4013872A1 (en) | 2019-08-14 | 2022-06-22 | Codiak BioSciences, Inc. | Extracellular vesicles with antisense oligonucleotides targeting kras |
| US20220354963A1 (en) | 2019-08-14 | 2022-11-10 | Codiak Biosciences, Inc. | Extracellular vesicle linked to molecules and uses thereof |
| CA3147369A1 (en) | 2019-08-14 | 2021-02-18 | Dalia BURZYN | Extracellular vesicle-aso constructs targeting cebp/beta |
| CA3147680A1 (en) | 2019-08-14 | 2021-02-18 | Dalia BURZYN | Extracellular vesicle-aso constructs targeting stat6 |
| CA3147366A1 (en) | 2019-08-14 | 2021-02-18 | Adam T. BOUTIN | Extracellular vesicles with stat3-antisense oligonucleotides |
| WO2021030773A1 (en) | 2019-08-14 | 2021-02-18 | Codiak Biosciences, Inc. | Extracellular vesicle-nlrp3 antagonist |
| WO2021043221A1 (en) | 2019-09-04 | 2021-03-11 | Biosion Inc. | Antibodies binding tslp and uses thereof |
| US20220364141A1 (en) * | 2019-09-05 | 2022-11-17 | National University Corporation Tokyo University Of Agriculture And Technology | Tryptase Activity Measurement Substrate |
| US20240377413A1 (en) | 2019-09-16 | 2024-11-14 | Bristol-Myers Squibb Company | Dual capture method for analysis of antibody-drug conjugates |
| US12319711B2 (en) | 2019-09-20 | 2025-06-03 | Northwestern University | Spherical nucleic acids with tailored and active protein coronae |
| US12378560B2 (en) | 2019-10-29 | 2025-08-05 | Northwestern University | Sequence multiplicity within spherical nucleic acids |
| CN114981309B (zh) | 2020-01-03 | 2023-08-25 | 博奥信生物技术(南京)有限公司 | 结合bcma的抗体及其用途 |
| JP2023512456A (ja) | 2020-01-13 | 2023-03-27 | ネオイミューンテック, インコーポレイテッド | Il-7タンパク質と二重特異性抗体の組み合わせで腫瘍を治療する方法 |
| JP7726628B2 (ja) * | 2020-01-30 | 2025-08-20 | エイムドバイオ株式会社 | ベンゾセレノフェン系化合物、これを含む薬学的組成物及び抗体-薬物複合体 |
| CN115087673B (zh) | 2020-02-27 | 2025-02-18 | 正大天晴药业集团股份有限公司 | 结合il4r的抗体及其用途 |
| EP4132971A1 (en) | 2020-04-09 | 2023-02-15 | Merck Sharp & Dohme LLC | Affinity matured anti-lap antibodies and uses thereof |
| US11530184B2 (en) | 2020-06-30 | 2022-12-20 | Imbria Pharmaceuticals, Inc. | Crystal forms of 2-[4-[(2,3,4-trimethoxyphenyl)methyl]piperazin-1-yl]ethyl pyridine-3-carboxylate |
| US11780811B2 (en) | 2020-06-30 | 2023-10-10 | Imbria Pharmaceuticals, Inc. | Methods of synthesizing 2-[4-[(2,3,4-trimethoxyphenyl)methyl]piperazin-1-yl]ethyl pyridine-3-carboxylate |
| CN112255908B (zh) * | 2020-11-16 | 2024-12-03 | 西安轻工业钟表研究所有限公司 | 一种指针式卫星光伏时钟 |
| CN112553274B (zh) * | 2020-12-02 | 2023-06-13 | 江南大学 | 大豆提取物Bowman-Birk inhibitor切割DNA的方法 |
| CN114685669A (zh) | 2020-12-30 | 2022-07-01 | 和铂医药(苏州)有限公司 | 结合trop2的抗体及其用途 |
| CN117377499A (zh) | 2021-02-17 | 2024-01-09 | 隆萨销售股份有限公司 | 经由优化的接头和锚定部分与生物活性分子连接的细胞外囊泡 |
| KR20230158005A (ko) | 2021-03-18 | 2023-11-17 | 씨젠 인크. | 생물학적 활성 화합물의 내재화된 접합체로부터의 선택적 약물 방출 |
| JP2024514530A (ja) | 2021-04-02 | 2024-04-02 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 切断型cdcp1に対する抗体およびその使用 |
| US11883396B2 (en) | 2021-05-03 | 2024-01-30 | Imbria Pharmaceuticals, Inc. | Methods of treating kidney conditions using modified forms of trimetazidine |
| JP2024539749A (ja) | 2021-10-14 | 2024-10-29 | ラッティコン (スージョウ) バイオファーマシューティカルズ カンパニー リミテッド | 新規な抗体-サイトカイン融合タンパク質及びその製造方法と使用 |
| CN114181216A (zh) * | 2021-12-27 | 2022-03-15 | 苏州新药篮生物医药科技有限公司 | 一种3,6-二氢吡咯并[3,2-e]吲哚-2-甲酸甲酯的制备方法 |
| JP2025507144A (ja) | 2022-03-18 | 2025-03-13 | ベイジン マブワークス バイオテック カンパニー リミテッド | 抗体結合b7-h3及びその使用 |
| AU2024305430A1 (en) | 2023-06-12 | 2026-01-22 | Nanjing Probio Biotech Co., Ltd. | Antibody binding to human ccr8 and the use thereof |
| CN116727114B (zh) * | 2023-07-19 | 2024-07-09 | 昆明理工大学 | 一种短流程浮选回收锂云母的方法 |
| WO2025184208A1 (en) | 2024-02-27 | 2025-09-04 | Bristol-Myers Squibb Company | Anti-ceacam5 antibodies and uses thereof |
| CN120590531A (zh) * | 2024-03-04 | 2025-09-05 | 杭州中美华东制药有限公司 | 抗FGFR2b抗体、其抗体药物偶联物及其用途 |
| WO2025188694A1 (en) | 2024-03-05 | 2025-09-12 | Bristol-Myers Squibb Company | Tricyclic tlr7 agonists and uses thereof |
| US20250282776A1 (en) | 2024-03-05 | 2025-09-11 | Bristol-Myers Squibb Company | Bicyclic TLR7 Agonists and Uses Thereof |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001519412A (ja) * | 1997-10-14 | 2001-10-23 | ザ スクリップス リサーチ インスティテュート | Cc−1065およびデュオカルマイシンのイソ−cbiおよびイソ−ci類似体 |
| JP2002308898A (ja) * | 2001-04-06 | 2002-10-23 | Kyowa Hakko Kogyo Co Ltd | 蛋白質受容体結合性環状ペプチドおよびその製造法 |
| JP2005500273A (ja) * | 2001-05-31 | 2005-01-06 | コウルター ファラマセウティカル インク ア ホーリー オウンド サブシダリィ オブ コリクサ コーポレーション | 細胞毒素、プロドラッグ、リンカーとその有用な安定剤 |
| JP2005502703A (ja) * | 2001-09-07 | 2005-01-27 | ザ スクリプス リサーチ インスティテュート | Cc−1065およびデュオカルマイシンのcbi類似体 |
| WO2005112919A2 (en) * | 2004-05-19 | 2005-12-01 | Medarex, Inc. | Self-immolative linkers and drug conjugates |
| WO2006012527A1 (en) * | 2004-07-23 | 2006-02-02 | Endocyte, Inc. | Bivalent linkers and conjugates thereof |
| JP2006518712A (ja) * | 2003-01-27 | 2006-08-17 | エンドサイト,インコーポレイテッド | ビタミン受容体結合性薬剤送達結合体 |
| WO2006110476A2 (en) * | 2005-04-08 | 2006-10-19 | Medarex, Inc. | Cytotoxic compounds and conjugates comprising duocarmycins with cleavable substrates |
| JP2009509977A (ja) * | 2005-09-26 | 2009-03-12 | メダレックス インコーポレイテッド | 抗体−薬剤コンジュゲート及びその使用 |
Family Cites Families (109)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4169888A (en) | 1977-10-17 | 1979-10-02 | The Upjohn Company | Composition of matter and process |
| US4391904A (en) | 1979-12-26 | 1983-07-05 | Syva Company | Test strip kits in immunoassays and compositions therein |
| US4671958A (en) | 1982-03-09 | 1987-06-09 | Cytogen Corporation | Antibody conjugates for the delivery of compounds to target sites |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4978757A (en) | 1984-02-21 | 1990-12-18 | The Upjohn Company | 1,2,8,8a-tetrahydrocyclopropa (C) pyrrolo [3,2-e)]-indol-4(5H)-ones and related compounds |
| US4912227A (en) | 1984-02-21 | 1990-03-27 | The Upjohn Company | 1,2,8,8A-tetrahydrocyclopropa(c)pyrrolo(3,2-e)-indol-4-(5H)-ones and related compounds |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| JPH0684377B2 (ja) | 1986-04-17 | 1994-10-26 | 協和醗酵工業株式会社 | 新規化合物dc―88a及びdc―89a1 |
| US5332837A (en) | 1986-12-19 | 1994-07-26 | The Upjohn Company | CC-1065 analogs |
| US5773435A (en) | 1987-08-04 | 1998-06-30 | Bristol-Myers Squibb Company | Prodrugs for β-lactamase and uses thereof |
| US4975278A (en) | 1988-02-26 | 1990-12-04 | Bristol-Myers Company | Antibody-enzyme conjugates in combination with prodrugs for the delivery of cytotoxic agents to tumor cells |
| US4952394A (en) | 1987-11-23 | 1990-08-28 | Bristol-Myers Company | Drug-monoclonal antibody conjugates |
| US4994578A (en) | 1987-11-27 | 1991-02-19 | Meiji Seika Kaisha, Ltd. | Certain anti-tumor duocarmycin antibiotics from streptomyces |
| US5147786A (en) | 1988-04-22 | 1992-09-15 | Monsanto Company | Immunoassay for the detection of α-haloacetamides |
| JP2642165B2 (ja) | 1988-07-22 | 1997-08-20 | 協和醗酵工業株式会社 | 新規dc−89化合物およびその製造法 |
| US5084468A (en) | 1988-08-11 | 1992-01-28 | Kyowa Hakko Kogyo Co., Ltd. | Dc-88a derivatives |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| US5176996A (en) | 1988-12-20 | 1993-01-05 | Baylor College Of Medicine | Method for making synthetic oligonucleotides which bind specifically to target sites on duplex DNA molecules, by forming a colinear triplex, the synthetic oligonucleotides and methods of use |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| JP2598116B2 (ja) | 1988-12-28 | 1997-04-09 | 協和醗酵工業株式会社 | 新規物質dc113 |
| US5187186A (en) | 1989-07-03 | 1993-02-16 | Kyowa Hakko Kogyo Co., Ltd. | Pyrroloindole derivatives |
| JP2510335B2 (ja) | 1989-07-03 | 1996-06-26 | 協和醗酵工業株式会社 | Dc―88a誘導体 |
| WO1991004753A1 (en) | 1989-10-02 | 1991-04-18 | Cetus Corporation | Conjugates of antisense oligonucleotides and therapeutic uses thereof |
| US5495009A (en) | 1989-10-24 | 1996-02-27 | Gilead Sciences, Inc. | Oligonucleotide analogs containing thioformacetal linkages |
| US5334528A (en) | 1989-10-30 | 1994-08-02 | The Regents Of The University Of California | Monoclonal antibodies to cyclodiene insecticides and method for detecting the same |
| US6075181A (en) | 1990-01-12 | 2000-06-13 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US6673986B1 (en) | 1990-01-12 | 2004-01-06 | Abgenix, Inc. | Generation of xenogeneic antibodies |
| ES2087997T3 (es) | 1990-01-12 | 1996-08-01 | Cell Genesys Inc | Generacion de anticuerpos xenogenicos. |
| US6150584A (en) | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| KR100208957B1 (ko) | 1990-04-25 | 1999-07-15 | 로렌스 티. 마이젠헬더 | 신규한 cc-1065 유사체 |
| US5427908A (en) | 1990-05-01 | 1995-06-27 | Affymax Technologies N.V. | Recombinant library screening methods |
| US5137877B1 (en) | 1990-05-14 | 1996-01-30 | Bristol Myers Squibb Co | Bifunctional linking compounds conjugates and methods for their production |
| US6172197B1 (en) | 1991-07-10 | 2001-01-09 | Medical Research Council | Methods for producing members of specific binding pairs |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| DE69121334T2 (de) | 1990-07-26 | 1997-03-20 | Kyowa Hakko Kogyo Kk | DC-89-Derivate als Antitumor-Wirkstoffe |
| GB9017024D0 (en) | 1990-08-03 | 1990-09-19 | Erba Carlo Spa | New linker for bioactive agents |
| US5874299A (en) | 1990-08-29 | 1999-02-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5770429A (en) | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5814318A (en) | 1990-08-29 | 1998-09-29 | Genpharm International Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5625126A (en) | 1990-08-29 | 1997-04-29 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5633425A (en) | 1990-08-29 | 1997-05-27 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5545806A (en) | 1990-08-29 | 1996-08-13 | Genpharm International, Inc. | Ransgenic non-human animals for producing heterologous antibodies |
| US5661016A (en) | 1990-08-29 | 1997-08-26 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| ES2108048T3 (es) | 1990-08-29 | 1997-12-16 | Genpharm Int | Produccion y utilizacion de animales inferiores transgenicos capaces de producir anticuerpos heterologos. |
| US5877397A (en) | 1990-08-29 | 1999-03-02 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US5789650A (en) | 1990-08-29 | 1998-08-04 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| ATE463573T1 (de) | 1991-12-02 | 2010-04-15 | Medimmune Ltd | Herstellung von autoantikörpern auf phagenoberflächen ausgehend von antikörpersegmentbibliotheken |
| US5622929A (en) | 1992-01-23 | 1997-04-22 | Bristol-Myers Squibb Company | Thioether conjugates |
| DK0563475T3 (da) | 1992-03-25 | 2000-09-18 | Immunogen Inc | Konjugater af cellebindende midler og derivater af CC-1065 |
| GB9314960D0 (en) | 1992-07-23 | 1993-09-01 | Zeneca Ltd | Chemical compounds |
| JP3514490B2 (ja) | 1992-08-21 | 2004-03-31 | 杏林製薬株式会社 | トリフルオロメチルピロロインドールカルボン酸エステル誘導体及びその製造方法 |
| US5288514A (en) | 1992-09-14 | 1994-02-22 | The Regents Of The University Of California | Solid phase and combinatorial synthesis of benzodiazepine compounds on a solid support |
| US5324483B1 (en) | 1992-10-08 | 1996-09-24 | Warner Lambert Co | Apparatus for multiple simultaneous synthesis |
| DE4314091A1 (de) | 1993-04-29 | 1994-11-03 | Boehringer Mannheim Gmbh | Immunologisches Nachweisverfahren für Triazine |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| US5786377A (en) | 1993-11-19 | 1998-07-28 | Universidad De Santiago De Compostela | Pyrrolo 3,2-E!indol derivatives, process for the preparation thereof and applications |
| US5641780A (en) | 1994-04-22 | 1997-06-24 | Kyowa Hakko Kogyo Co., Ltd. | Pyrrolo-indole derivatives |
| JPH07309761A (ja) | 1994-05-20 | 1995-11-28 | Kyowa Hakko Kogyo Co Ltd | デュオカルマイシン誘導体の安定化法 |
| US5773001A (en) | 1994-06-03 | 1998-06-30 | American Cyanamid Company | Conjugates of methyltrithio antitumor agents and intermediates for their synthesis |
| WO1996005863A1 (fr) | 1994-08-19 | 1996-02-29 | La Region Wallonne | Composes, composition pharmaceutique et dispositif de diagnostic les comprenant et leur utilisation |
| CA2201097A1 (en) | 1994-09-30 | 1996-04-11 | Kyowa Hakko Kogyo Co., Ltd. | Anti-tumor agents |
| KR100395083B1 (ko) | 1994-11-29 | 2004-02-05 | 자이단호오징 사가미 츄오 카가쿠겡큐쇼 | 아크릴아미드유도체및그제조방법 |
| EP0867190B1 (en) | 1995-05-10 | 2007-12-26 | Kyowa Hakko Kogyo Co., Ltd. | Cytotoxin complexes comprising dipeptides |
| US5686237A (en) | 1995-06-05 | 1997-11-11 | Al-Bayati; Mohammed A. S. | Use of biomarkers in saliva to evaluate the toxicity of agents and the function of tissues in both biomedical and environmental applications |
| US5712374A (en) | 1995-06-07 | 1998-01-27 | American Cyanamid Company | Method for the preparation of substantiallly monomeric calicheamicin derivative/carrier conjugates |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| AU727608B2 (en) | 1995-10-03 | 2000-12-14 | Scripps Research Institute, The | CBI analogs of CC-1065 and the duocarmycins |
| CA2187969C (en) | 1995-10-17 | 2006-05-30 | Dale L. Boger | A template for solution phase synthesis of combinatorial libraries |
| ATE234635T1 (de) | 1995-12-22 | 2003-04-15 | Bristol Myers Squibb Co | Verzweigte hydrazongruppen enthaltende kuppler |
| US6143901A (en) | 1996-07-31 | 2000-11-07 | Genesoft, Inc. | Complex formation between dsDNA and pyrrole imidazole polyamides |
| ES2244991T3 (es) | 1996-03-08 | 2005-12-16 | The Scripps Research Institute | Analogosmcbi de cc-1065 y las duocarmicinas. |
| EP0934269A4 (en) | 1996-05-31 | 2000-01-12 | Scripps Research Inst | ANALOGS OF CC-1065 AND DUOCARMYCINE |
| US6130237A (en) | 1996-09-12 | 2000-10-10 | Cancer Research Campaign Technology Limited | Condensed N-aclyindoles as antitumor agents |
| AU721037B2 (en) | 1996-09-12 | 2000-06-22 | Auckland Uniservices Limited | Condensed N-acylindoles as antitumor agents |
| JPH1087666A (ja) | 1996-09-18 | 1998-04-07 | Kyorin Pharmaceut Co Ltd | デュオカルマイシンsa及びその誘導体の製造中間体と製造方法 |
| US6759509B1 (en) | 1996-11-05 | 2004-07-06 | Bristol-Myers Squibb Company | Branched peptide linkers |
| WO1998025900A1 (en) | 1996-12-13 | 1998-06-18 | Shionogi & Co., Ltd. | Compounds having antitumor activity |
| EP0996458A4 (en) | 1997-03-28 | 2001-05-02 | Scripps Research Inst | SANDRAMYCIN ANALOG |
| JP2002510968A (ja) | 1997-05-07 | 2002-04-09 | ブリストル−マイヤーズ スクイブ カンパニー | 組換え抗体−酵素融合タンパク質 |
| DE69825048D1 (de) | 1997-05-22 | 2004-08-19 | Scripps Research Inst | Analoga von duocarmycin and cc-1065 |
| NZ504884A (en) | 1997-12-08 | 2002-10-25 | Scripps Research Inst | Synthesis of the dihydroindole C-ring of CC-1065/duocarmycin analogs and certain intermediates |
| JP3045706B1 (ja) | 1998-09-14 | 2000-05-29 | 科学技術振興事業団 | Dnaの特定塩基配列をアルキル化する化合物及びその合成法 |
| PT1144011E (pt) | 1998-12-11 | 2010-06-16 | Coulter Pharm Inc | Compostos pró-fármacos e processo para a sua preparação |
| US7425541B2 (en) | 1998-12-11 | 2008-09-16 | Medarex, Inc. | Enzyme-cleavable prodrug compounds |
| US6909006B1 (en) | 1999-08-27 | 2005-06-21 | Spirogen Limited | Cyclopropylindole derivatives |
| EP1242438B1 (en) | 1999-12-29 | 2006-11-08 | Immunogen, Inc. | Cytotoxic agents comprising modified doxorubicins and daunorubicins and their therapeutic use |
| JP2003529609A (ja) | 2000-03-16 | 2003-10-07 | ジーンソフト インコーポレイティッド | 核酸結合部分を含む荷電した化合物およびその利用法 |
| WO2001083482A1 (en) | 2000-05-03 | 2001-11-08 | The Scripps Research Institute | Dna alkylating agent and activation thereof |
| JP4061819B2 (ja) | 2000-05-12 | 2008-03-19 | 独立行政法人科学技術振興機構 | インターストランドクロスリンク剤の合成方法 |
| AU2001275525B2 (en) | 2000-06-14 | 2007-04-26 | Medarex, Inc. | Tripeptide prodrug compounds |
| AU2001266853B2 (en) | 2000-06-14 | 2005-02-17 | Medarex, Inc. | Prodrug compounds with an oligopeptide having an isoleucine residue |
| US7402556B2 (en) | 2000-08-24 | 2008-07-22 | Medarex, Inc. | Prodrugs activated by plasmin and their use in cancer chemotherapy |
| EP1243276A1 (en) | 2001-03-23 | 2002-09-25 | Franciscus Marinus Hendrikus De Groot | Elongated and multiple spacers containing activatible prodrugs |
| US6884869B2 (en) | 2001-04-30 | 2005-04-26 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| US7256257B2 (en) | 2001-04-30 | 2007-08-14 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| US20030083263A1 (en) | 2001-04-30 | 2003-05-01 | Svetlana Doronina | Pentapeptide compounds and uses related thereto |
| US6762179B2 (en) | 2001-05-31 | 2004-07-13 | Vertex Pharmaceuticals Incorporated | Thiazole compounds useful as inhibitors of protein kinase |
| EP1404356B1 (en) | 2001-06-11 | 2011-04-06 | Medarex, Inc. | Method for designing cd10-activated prodrug compounds |
| US7091186B2 (en) | 2001-09-24 | 2006-08-15 | Seattle Genetics, Inc. | p-Amidobenzylethers in drug delivery agents |
| US6756397B2 (en) | 2002-04-05 | 2004-06-29 | Immunogen, Inc. | Prodrugs of CC-1065 analogs |
| US6534660B1 (en) | 2002-04-05 | 2003-03-18 | Immunogen, Inc. | CC-1065 analog synthesis |
| WO2004005326A2 (de) | 2002-07-09 | 2004-01-15 | Morphochem Aktiengellschaft Fu | Tubulysinkonjugate |
| EP1523493B1 (de) | 2002-07-09 | 2013-09-04 | Dömling, Alexander | Neue tubulysinanaloga |
| WO2004032828A2 (en) | 2002-07-31 | 2004-04-22 | Seattle Genetics, Inc. | Anti-cd20 antibody-drug conjugates for the treatment of cancer and immune disorders |
| US20050026987A1 (en) | 2003-05-13 | 2005-02-03 | The Scripps Research Institute | CBI analogues of the duocarmycins and CC-1065 |
| CA2627190A1 (en) * | 2005-11-10 | 2007-05-24 | Medarex, Inc. | Duocarmycin derivatives as novel cytotoxic compounds and conjugates |
| CN101415679A (zh) | 2006-02-02 | 2009-04-22 | 辛塔佳有限公司 | 水溶性cc-1065类似物及其缀合物 |
| JP2010513306A (ja) | 2006-12-14 | 2010-04-30 | メダレックス インコーポレーティッド | Cd70に結合するヒト抗体およびその使用 |
| CA2678514A1 (en) * | 2007-02-21 | 2008-08-28 | Medarex, Inc. | Chemical linkers with single amino acids and conjugates thereof |
-
2007
- 2007-12-27 TW TW096150517A patent/TWI412367B/zh not_active IP Right Cessation
- 2007-12-28 EP EP07870071A patent/EP2114454A2/en not_active Withdrawn
- 2007-12-28 EA EA200970648A patent/EA019962B1/ru not_active IP Right Cessation
- 2007-12-28 CL CL200703849A patent/CL2007003849A1/es unknown
- 2007-12-28 JP JP2009544303A patent/JP2010522693A/ja active Pending
- 2007-12-28 US US12/521,391 patent/US8461117B2/en active Active
- 2007-12-28 CA CA2674055A patent/CA2674055C/en not_active Expired - Fee Related
- 2007-12-28 CN CN200780051809A patent/CN101795711A/zh active Pending
- 2007-12-28 NZ NZ578324A patent/NZ578324A/en not_active IP Right Cessation
- 2007-12-28 AR ARP070105968A patent/AR067837A1/es unknown
- 2007-12-28 AU AU2007342052A patent/AU2007342052A1/en not_active Abandoned
- 2007-12-28 BR BRPI0719626-1A2A patent/BRPI0719626A2/pt not_active IP Right Cessation
- 2007-12-28 MX MX2009007147A patent/MX2009007147A/es active IP Right Grant
- 2007-12-28 WO PCT/US2007/089100 patent/WO2008083312A2/en not_active Ceased
- 2007-12-28 KR KR1020097015600A patent/KR20090098901A/ko not_active Ceased
- 2007-12-28 SG SG201101561-7A patent/SG170070A1/en unknown
-
2009
- 2009-06-17 IL IL199416A patent/IL199416A0/en unknown
- 2009-07-20 NO NO20092733A patent/NO20092733L/no not_active Application Discontinuation
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001519412A (ja) * | 1997-10-14 | 2001-10-23 | ザ スクリップス リサーチ インスティテュート | Cc−1065およびデュオカルマイシンのイソ−cbiおよびイソ−ci類似体 |
| JP2002308898A (ja) * | 2001-04-06 | 2002-10-23 | Kyowa Hakko Kogyo Co Ltd | 蛋白質受容体結合性環状ペプチドおよびその製造法 |
| JP2005500273A (ja) * | 2001-05-31 | 2005-01-06 | コウルター ファラマセウティカル インク ア ホーリー オウンド サブシダリィ オブ コリクサ コーポレーション | 細胞毒素、プロドラッグ、リンカーとその有用な安定剤 |
| JP2005502703A (ja) * | 2001-09-07 | 2005-01-27 | ザ スクリプス リサーチ インスティテュート | Cc−1065およびデュオカルマイシンのcbi類似体 |
| JP2006518712A (ja) * | 2003-01-27 | 2006-08-17 | エンドサイト,インコーポレイテッド | ビタミン受容体結合性薬剤送達結合体 |
| WO2005112919A2 (en) * | 2004-05-19 | 2005-12-01 | Medarex, Inc. | Self-immolative linkers and drug conjugates |
| WO2006012527A1 (en) * | 2004-07-23 | 2006-02-02 | Endocyte, Inc. | Bivalent linkers and conjugates thereof |
| WO2006110476A2 (en) * | 2005-04-08 | 2006-10-19 | Medarex, Inc. | Cytotoxic compounds and conjugates comprising duocarmycins with cleavable substrates |
| JP2009509977A (ja) * | 2005-09-26 | 2009-03-12 | メダレックス インコーポレイテッド | 抗体−薬剤コンジュゲート及びその使用 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010519310A (ja) * | 2007-02-21 | 2010-06-03 | メダレックス インコーポレイテッド | 単一のアミノ酸を有する化学リンカーおよびその複合体 |
| JP2016188207A (ja) * | 2010-12-17 | 2016-11-04 | アローヘッド ファーマシューティカルズ インコーポレイテッド | ペプチドに基づくインビボsiRNA送達システム |
| JP6993084B2 (ja) | 2013-08-12 | 2022-02-03 | ジェネンテック, インコーポレイテッド | 1-(クロロメチル)-2,3-ジヒドロ-1h-ベンゾ[e]インドール二量体抗体 - 薬物コンジュゲート化合物、並びに使用及び処置の方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| NZ578324A (en) | 2012-01-12 |
| NO20092733L (no) | 2009-07-20 |
| EA200970648A1 (ru) | 2010-04-30 |
| BRPI0719626A2 (pt) | 2014-06-24 |
| MX2009007147A (es) | 2009-08-25 |
| CA2674055A1 (en) | 2008-07-10 |
| WO2008083312A2 (en) | 2008-07-10 |
| CA2674055C (en) | 2016-02-23 |
| US8461117B2 (en) | 2013-06-11 |
| WO2008083312A3 (en) | 2010-01-14 |
| CL2007003849A1 (es) | 2008-07-04 |
| AR067837A1 (es) | 2009-10-28 |
| EP2114454A2 (en) | 2009-11-11 |
| AU2007342052A1 (en) | 2008-07-10 |
| TWI412367B (zh) | 2013-10-21 |
| CN101795711A (zh) | 2010-08-04 |
| US20100145036A1 (en) | 2010-06-10 |
| IL199416A0 (en) | 2011-08-01 |
| KR20090098901A (ko) | 2009-09-17 |
| SG170070A1 (en) | 2011-04-29 |
| TW200833683A (en) | 2008-08-16 |
| EA019962B1 (ru) | 2014-07-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5290756B2 (ja) | 抗体−薬剤コンジュゲート及びその使用 | |
| JP2010522693A (ja) | 化学リンカー及び切断可能な基質及びそれらの抱合体 | |
| JP4806680B2 (ja) | 自己犠牲リンカー及び薬剤複合体 | |
| US8664407B2 (en) | Chemical linkers with single amino acids and conjugates thereof | |
| US7691962B2 (en) | Chemical linkers and conjugates thereof | |
| HK1160152A (en) | Conjugates of duocarmycin and anti-cd70 or anti-psma antibodies | |
| HK1117410B (en) | Antibody-drug conjugates and their use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101228 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20101228 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130205 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130405 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130412 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130502 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130513 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20130605 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20131217 |