ES2773710T3 - Enlazadores para conjugados de anticuerpo - fármaco - Google Patents
Enlazadores para conjugados de anticuerpo - fármaco Download PDFInfo
- Publication number
- ES2773710T3 ES2773710T3 ES18152381T ES18152381T ES2773710T3 ES 2773710 T3 ES2773710 T3 ES 2773710T3 ES 18152381 T ES18152381 T ES 18152381T ES 18152381 T ES18152381 T ES 18152381T ES 2773710 T3 ES2773710 T3 ES 2773710T3
- Authority
- ES
- Spain
- Prior art keywords
- antibody
- amino acid
- group
- compound
- acid sequence
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229940049595 antibody-drug conjugate Drugs 0.000 title description 261
- 239000000611 antibody drug conjugate Substances 0.000 title description 260
- 150000001875 compounds Chemical class 0.000 claims abstract description 356
- 238000000034 method Methods 0.000 description 634
- 239000000243 solution Substances 0.000 description 362
- 239000003814 drug Substances 0.000 description 295
- 229940079593 drug Drugs 0.000 description 292
- 125000003275 alpha amino acid group Chemical group 0.000 description 284
- 125000005647 linker group Chemical group 0.000 description 213
- 238000004519 manufacturing process Methods 0.000 description 191
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 164
- 230000008569 process Effects 0.000 description 146
- 238000006243 chemical reaction Methods 0.000 description 136
- 239000007864 aqueous solution Substances 0.000 description 126
- 238000000746 purification Methods 0.000 description 119
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 112
- 238000010521 absorption reaction Methods 0.000 description 102
- 239000000562 conjugate Substances 0.000 description 102
- 235000001014 amino acid Nutrition 0.000 description 101
- 238000004364 calculation method Methods 0.000 description 98
- 210000004027 cell Anatomy 0.000 description 89
- -1 2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl Chemical group 0.000 description 82
- 125000006239 protecting group Chemical group 0.000 description 82
- 125000003277 amino group Chemical group 0.000 description 74
- 101710185679 CD276 antigen Proteins 0.000 description 67
- 230000002829 reductive effect Effects 0.000 description 67
- 102100038078 CD276 antigen Human genes 0.000 description 66
- 125000000539 amino acid group Chemical group 0.000 description 64
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 60
- 229960001484 edetic acid Drugs 0.000 description 60
- 239000007853 buffer solution Substances 0.000 description 57
- 108091007433 antigens Proteins 0.000 description 55
- 239000000427 antigen Substances 0.000 description 54
- 102000036639 antigens Human genes 0.000 description 54
- 239000000126 substance Substances 0.000 description 54
- 230000000259 anti-tumor effect Effects 0.000 description 53
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 53
- 241000282414 Homo sapiens Species 0.000 description 51
- 230000021615 conjugation Effects 0.000 description 50
- 238000013379 physicochemical characterization Methods 0.000 description 48
- PBVAJRFEEOIAGW-UHFFFAOYSA-N 3-[bis(2-carboxyethyl)phosphanyl]propanoic acid;hydrochloride Chemical compound Cl.OC(=O)CCP(CCC(O)=O)CCC(O)=O PBVAJRFEEOIAGW-UHFFFAOYSA-N 0.000 description 47
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 47
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 47
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 45
- 206010028980 Neoplasm Diseases 0.000 description 45
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 44
- 229940024606 amino acid Drugs 0.000 description 43
- 239000003153 chemical reaction reagent Substances 0.000 description 42
- 108090000623 proteins and genes Proteins 0.000 description 41
- 230000009467 reduction Effects 0.000 description 41
- 238000005160 1H NMR spectroscopy Methods 0.000 description 40
- 150000001413 amino acids Chemical class 0.000 description 40
- 239000002904 solvent Substances 0.000 description 36
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 35
- 210000004408 hybridoma Anatomy 0.000 description 35
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 33
- 230000000694 effects Effects 0.000 description 33
- 108090000765 processed proteins & peptides Proteins 0.000 description 33
- 239000007787 solid Substances 0.000 description 33
- 239000000543 intermediate Substances 0.000 description 30
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 30
- 150000002148 esters Chemical class 0.000 description 29
- 239000002609 medium Substances 0.000 description 29
- 239000000203 mixture Substances 0.000 description 29
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 28
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 28
- 241000699666 Mus <mouse, genus> Species 0.000 description 27
- 239000002585 base Substances 0.000 description 26
- ZVYVPGLRVWUPMP-FYSMJZIKSA-N exatecan Chemical compound C1C[C@H](N)C2=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC3=CC(F)=C(C)C1=C32 ZVYVPGLRVWUPMP-FYSMJZIKSA-N 0.000 description 26
- 210000004881 tumor cell Anatomy 0.000 description 26
- 201000011510 cancer Diseases 0.000 description 25
- 229950009429 exatecan Drugs 0.000 description 25
- 102000004169 proteins and genes Human genes 0.000 description 24
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 23
- 241001465754 Metazoa Species 0.000 description 23
- 230000027455 binding Effects 0.000 description 22
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 21
- 230000015572 biosynthetic process Effects 0.000 description 21
- BEBCJVAWIBVWNZ-UHFFFAOYSA-N glycinamide Chemical compound NCC(N)=O BEBCJVAWIBVWNZ-UHFFFAOYSA-N 0.000 description 21
- 235000018102 proteins Nutrition 0.000 description 21
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 18
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- 239000002253 acid Substances 0.000 description 18
- 125000000217 alkyl group Chemical group 0.000 description 18
- 238000003786 synthesis reaction Methods 0.000 description 18
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 18
- 206010035226 Plasma cell myeloma Diseases 0.000 description 16
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 16
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 16
- 201000000050 myeloid neoplasm Diseases 0.000 description 16
- 238000002835 absorbance Methods 0.000 description 14
- 150000008065 acid anhydrides Chemical class 0.000 description 14
- 210000000628 antibody-producing cell Anatomy 0.000 description 14
- 125000004432 carbon atom Chemical group C* 0.000 description 14
- 239000012442 inert solvent Substances 0.000 description 14
- 125000003396 thiol group Chemical group [H]S* 0.000 description 14
- 241000700159 Rattus Species 0.000 description 13
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 13
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 13
- 229910052757 nitrogen Inorganic materials 0.000 description 13
- 102000004196 processed proteins & peptides Human genes 0.000 description 13
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Natural products NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 238000005259 measurement Methods 0.000 description 12
- 230000036961 partial effect Effects 0.000 description 12
- 239000008194 pharmaceutical composition Substances 0.000 description 12
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 12
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 11
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical class C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 11
- 150000004820 halides Chemical class 0.000 description 11
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 11
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- 239000002246 antineoplastic agent Substances 0.000 description 10
- 239000000872 buffer Substances 0.000 description 10
- 238000004440 column chromatography Methods 0.000 description 10
- 239000002299 complementary DNA Substances 0.000 description 10
- 230000001472 cytotoxic effect Effects 0.000 description 10
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 10
- 230000003053 immunization Effects 0.000 description 10
- 239000007924 injection Substances 0.000 description 10
- 238000002347 injection Methods 0.000 description 10
- 230000004048 modification Effects 0.000 description 10
- 238000012986 modification Methods 0.000 description 10
- 125000004433 nitrogen atom Chemical group N* 0.000 description 10
- 238000000108 ultra-filtration Methods 0.000 description 10
- 239000013598 vector Substances 0.000 description 10
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 239000012044 organic layer Substances 0.000 description 9
- 150000003839 salts Chemical class 0.000 description 9
- 239000000741 silica gel Substances 0.000 description 9
- 229910002027 silica gel Inorganic materials 0.000 description 9
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 9
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 8
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 8
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 8
- 239000004743 Polypropylene Substances 0.000 description 8
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 8
- 238000012258 culturing Methods 0.000 description 8
- 238000009472 formulation Methods 0.000 description 8
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 8
- 238000000338 in vitro Methods 0.000 description 8
- 229920001184 polypeptide Polymers 0.000 description 8
- 229920001155 polypropylene Polymers 0.000 description 8
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 7
- 241000282412 Homo Species 0.000 description 7
- 108010038807 Oligopeptides Proteins 0.000 description 7
- 102000015636 Oligopeptides Human genes 0.000 description 7
- 238000012217 deletion Methods 0.000 description 7
- 230000037430 deletion Effects 0.000 description 7
- 238000002649 immunization Methods 0.000 description 7
- 238000010647 peptide synthesis reaction Methods 0.000 description 7
- 230000001105 regulatory effect Effects 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- 238000006467 substitution reaction Methods 0.000 description 7
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 238000002965 ELISA Methods 0.000 description 6
- 239000004471 Glycine Substances 0.000 description 6
- 101000884279 Homo sapiens CD276 antigen Proteins 0.000 description 6
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 6
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 6
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 6
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 6
- 239000004472 Lysine Substances 0.000 description 6
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- 108010076504 Protein Sorting Signals Proteins 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 6
- 229940041181 antineoplastic drug Drugs 0.000 description 6
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 6
- 230000007910 cell fusion Effects 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 230000002950 deficient Effects 0.000 description 6
- 150000004676 glycans Chemical class 0.000 description 6
- 238000009396 hybridization Methods 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 230000003389 potentiating effect Effects 0.000 description 6
- 241000894007 species Species 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 5
- 108020004414 DNA Proteins 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 5
- 102000018251 Hypoxanthine Phosphoribosyltransferase Human genes 0.000 description 5
- 108010091358 Hypoxanthine Phosphoribosyltransferase Proteins 0.000 description 5
- 108060003951 Immunoglobulin Proteins 0.000 description 5
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 5
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 5
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 5
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 5
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 5
- 239000013543 active substance Substances 0.000 description 5
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 5
- 125000005002 aryl methyl group Chemical group 0.000 description 5
- 235000003704 aspartic acid Nutrition 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 238000010511 deprotection reaction Methods 0.000 description 5
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 5
- 239000013604 expression vector Substances 0.000 description 5
- 238000010353 genetic engineering Methods 0.000 description 5
- 102000048770 human CD276 Human genes 0.000 description 5
- 102000018358 immunoglobulin Human genes 0.000 description 5
- 235000018977 lysine Nutrition 0.000 description 5
- 210000004962 mammalian cell Anatomy 0.000 description 5
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 5
- 229960005190 phenylalanine Drugs 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 239000002243 precursor Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 235000004400 serine Nutrition 0.000 description 5
- 230000008685 targeting Effects 0.000 description 5
- 235000014393 valine Nutrition 0.000 description 5
- 239000004474 valine Substances 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- DVVGIUUJYPYENY-UHFFFAOYSA-N 1-methylpyridin-2-one Chemical compound CN1C=CC=CC1=O DVVGIUUJYPYENY-UHFFFAOYSA-N 0.000 description 4
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 4
- HIDJWBGOQFTDLU-UHFFFAOYSA-N 4-[(2-methylpropan-2-yl)oxycarbonylamino]butanoic acid Chemical compound CC(C)(C)OC(=O)NCCCC(O)=O HIDJWBGOQFTDLU-UHFFFAOYSA-N 0.000 description 4
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 4
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 4
- 206010003445 Ascites Diseases 0.000 description 4
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 4
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- 241000699670 Mus sp. Species 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 210000000683 abdominal cavity Anatomy 0.000 description 4
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 4
- 229910052783 alkali metal Inorganic materials 0.000 description 4
- 125000002947 alkylene group Chemical group 0.000 description 4
- 238000010976 amide bond formation reaction Methods 0.000 description 4
- 150000001408 amides Chemical class 0.000 description 4
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 4
- 238000010367 cloning Methods 0.000 description 4
- 230000000445 cytocidal effect Effects 0.000 description 4
- 229940113088 dimethylacetamide Drugs 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 235000013922 glutamic acid Nutrition 0.000 description 4
- 239000004220 glutamic acid Substances 0.000 description 4
- 229940127121 immunoconjugate Drugs 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 239000008363 phosphate buffer Substances 0.000 description 4
- 230000002285 radioactive effect Effects 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- 150000003384 small molecules Chemical class 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 4
- 125000004955 1,4-cyclohexylene group Chemical group [H]C1([H])C([H])([H])C([H])([*:1])C([H])([H])C([H])([H])C1([H])[*:2] 0.000 description 3
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 3
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- PTUJJIPXBJJLLV-UHFFFAOYSA-N 2-[[2-[[2-[[2-[(2-methylpropan-2-yl)oxycarbonylamino]acetyl]amino]acetyl]amino]-3-phenylpropanoyl]amino]acetic acid Chemical compound CC(C)(C)OC(=O)NCC(=O)NCC(=O)NC(C(=O)NCC(O)=O)CC1=CC=CC=C1 PTUJJIPXBJJLLV-UHFFFAOYSA-N 0.000 description 3
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 241000588724 Escherichia coli Species 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 3
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 3
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 3
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 3
- 241001529936 Murinae Species 0.000 description 3
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 3
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 3
- 108700026244 Open Reading Frames Proteins 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 3
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 3
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 3
- 238000009825 accumulation Methods 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 238000001042 affinity chromatography Methods 0.000 description 3
- 235000004279 alanine Nutrition 0.000 description 3
- 229960003896 aminopterin Drugs 0.000 description 3
- 210000004102 animal cell Anatomy 0.000 description 3
- 238000010171 animal model Methods 0.000 description 3
- 230000000890 antigenic effect Effects 0.000 description 3
- 125000003435 aroyl group Chemical group 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 3
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 229910021538 borax Inorganic materials 0.000 description 3
- 229960000455 brentuximab vedotin Drugs 0.000 description 3
- 210000004899 c-terminal region Anatomy 0.000 description 3
- 239000003638 chemical reducing agent Substances 0.000 description 3
- 235000013477 citrulline Nutrition 0.000 description 3
- 229960002173 citrulline Drugs 0.000 description 3
- 230000004540 complement-dependent cytotoxicity Effects 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000012228 culture supernatant Substances 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- NZNMSOFKMUBTKW-UHFFFAOYSA-N cyclohexanecarboxylic acid Chemical compound OC(=O)C1CCCCC1 NZNMSOFKMUBTKW-UHFFFAOYSA-N 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 239000010432 diamond Substances 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 125000004185 ester group Chemical group 0.000 description 3
- 210000003527 eukaryotic cell Anatomy 0.000 description 3
- 238000002523 gelfiltration Methods 0.000 description 3
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 3
- 235000004554 glutamine Nutrition 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical class 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 3
- 229960004768 irinotecan Drugs 0.000 description 3
- 235000005772 leucine Nutrition 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 239000012669 liquid formulation Substances 0.000 description 3
- 229910052744 lithium Inorganic materials 0.000 description 3
- 210000004698 lymphocyte Anatomy 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 201000001441 melanoma Diseases 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 229920000609 methyl cellulose Polymers 0.000 description 3
- 239000001923 methylcellulose Substances 0.000 description 3
- SNVLJLYUUXKWOJ-UHFFFAOYSA-N methylidenecarbene Chemical group C=[C] SNVLJLYUUXKWOJ-UHFFFAOYSA-N 0.000 description 3
- 238000011580 nude mouse model Methods 0.000 description 3
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 238000010187 selection method Methods 0.000 description 3
- 238000010898 silica gel chromatography Methods 0.000 description 3
- 239000001632 sodium acetate Substances 0.000 description 3
- 235000017281 sodium acetate Nutrition 0.000 description 3
- 239000001488 sodium phosphate Substances 0.000 description 3
- 229910000162 sodium phosphate Inorganic materials 0.000 description 3
- 235000011008 sodium phosphates Nutrition 0.000 description 3
- 235000010339 sodium tetraborate Nutrition 0.000 description 3
- 239000007790 solid phase Substances 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 125000006850 spacer group Chemical group 0.000 description 3
- 210000004989 spleen cell Anatomy 0.000 description 3
- 230000002194 synthesizing effect Effects 0.000 description 3
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 150000003568 thioethers Chemical class 0.000 description 3
- 238000013519 translation Methods 0.000 description 3
- BSVBQGMMJUBVOD-UHFFFAOYSA-N trisodium borate Chemical compound [Na+].[Na+].[Na+].[O-]B([O-])[O-] BSVBQGMMJUBVOD-UHFFFAOYSA-N 0.000 description 3
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 3
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 3
- 235000002374 tyrosine Nutrition 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- NKUZQMZWTZAPSN-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-bromoacetate Chemical compound BrCC(=O)ON1C(=O)CCC1=O NKUZQMZWTZAPSN-UHFFFAOYSA-N 0.000 description 2
- VRDGQQTWSGDXCU-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-iodoacetate Chemical compound ICC(=O)ON1C(=O)CCC1=O VRDGQQTWSGDXCU-UHFFFAOYSA-N 0.000 description 2
- SCZNXLWKYFICFV-UHFFFAOYSA-N 1,2,3,4,5,7,8,9-octahydropyrido[1,2-b]diazepine Chemical compound C1CCCNN2CCCC=C21 SCZNXLWKYFICFV-UHFFFAOYSA-N 0.000 description 2
- 125000001989 1,3-phenylene group Chemical group [H]C1=C([H])C([*:1])=C([H])C([*:2])=C1[H] 0.000 description 2
- 125000001140 1,4-phenylene group Chemical group [H]C1=C([H])C([*:2])=C([H])C([H])=C1[*:1] 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- LPXQRXLUHJKZIE-UHFFFAOYSA-N 8-azaguanine Chemical compound NC1=NC(O)=C2NN=NC2=N1 LPXQRXLUHJKZIE-UHFFFAOYSA-N 0.000 description 2
- 229960005508 8-azaguanine Drugs 0.000 description 2
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 244000063299 Bacillus subtilis Species 0.000 description 2
- 235000014469 Bacillus subtilis Nutrition 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 2
- 102000003915 DNA Topoisomerases Human genes 0.000 description 2
- 108090000323 DNA Topoisomerases Proteins 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 2
- 208000017604 Hodgkin disease Diseases 0.000 description 2
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 2
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 2
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- 208000032004 Large-Cell Anaplastic Lymphoma Diseases 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 108010052285 Membrane Proteins Proteins 0.000 description 2
- 102000018697 Membrane Proteins Human genes 0.000 description 2
- 241000699660 Mus musculus Species 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 2
- BZORFPDSXLZWJF-UHFFFAOYSA-N N,N-dimethyl-1,4-phenylenediamine Chemical compound CN(C)C1=CC=C(N)C=C1 BZORFPDSXLZWJF-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- 101150046281 NIP4-1 gene Proteins 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- 206010057249 Phagocytosis Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 241000276498 Pollachius virens Species 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 229920005654 Sephadex Polymers 0.000 description 2
- 239000012507 Sephadex™ Substances 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 108010022394 Threonine synthase Proteins 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 2
- PNDPGZBMCMUPRI-XXSWNUTMSA-N [125I][125I] Chemical compound [125I][125I] PNDPGZBMCMUPRI-XXSWNUTMSA-N 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 238000007792 addition Methods 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 238000005377 adsorption chromatography Methods 0.000 description 2
- HAXFWIACAGNFHA-UHFFFAOYSA-N aldrithiol Chemical compound C=1C=CC=NC=1SSC1=CC=CC=N1 HAXFWIACAGNFHA-UHFFFAOYSA-N 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 239000003708 ampul Substances 0.000 description 2
- 229940125644 antibody drug Drugs 0.000 description 2
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 2
- 235000009582 asparagine Nutrition 0.000 description 2
- 229960001230 asparagine Drugs 0.000 description 2
- QUKGYYKBILRGFE-UHFFFAOYSA-N benzyl acetate Chemical compound CC(=O)OCC1=CC=CC=C1 QUKGYYKBILRGFE-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- DNSISZSEWVHGLH-UHFFFAOYSA-N butanamide Chemical compound CCCC(N)=O DNSISZSEWVHGLH-UHFFFAOYSA-N 0.000 description 2
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 description 2
- 229930195731 calicheamicin Natural products 0.000 description 2
- 229940127093 camptothecin Drugs 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- OKTJSMMVPCPJKN-BJUDXGSMSA-N carbon-11 Chemical compound [11C] OKTJSMMVPCPJKN-BJUDXGSMSA-N 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000002738 chelating agent Substances 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 230000001268 conjugating effect Effects 0.000 description 2
- 230000009260 cross reactivity Effects 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- 238000001212 derivatisation Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 102000004419 dihydrofolate reductase Human genes 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 229960003297 gemtuzumab ozogamicin Drugs 0.000 description 2
- 125000005179 haloacetyl group Chemical group 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 210000003917 human chromosome Anatomy 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 239000002198 insoluble material Substances 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 229940044173 iodine-125 Drugs 0.000 description 2
- 238000004255 ion exchange chromatography Methods 0.000 description 2
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- 235000014705 isoleucine Nutrition 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 150000002596 lactones Chemical group 0.000 description 2
- 238000004811 liquid chromatography Methods 0.000 description 2
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 2
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 239000007758 minimum essential medium Substances 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 125000002524 organometallic group Chemical group 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- 229960003330 pentetic acid Drugs 0.000 description 2
- 238000002823 phage display Methods 0.000 description 2
- 230000008782 phagocytosis Effects 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 108091033319 polynucleotide Proteins 0.000 description 2
- 102000040430 polynucleotide Human genes 0.000 description 2
- 239000002157 polynucleotide Substances 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 2
- 229910000105 potassium hydride Inorganic materials 0.000 description 2
- CUQOHAYJWVTKDE-UHFFFAOYSA-N potassium;butan-1-olate Chemical compound [K+].CCCC[O-] CUQOHAYJWVTKDE-UHFFFAOYSA-N 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 125000001500 prolyl group Chemical group [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 125000001325 propanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 159000000000 sodium salts Chemical class 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- 125000006633 tert-butoxycarbonylamino group Chemical group 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 235000008521 threonine Nutrition 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 229910052722 tritium Inorganic materials 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- VCQURUZYYSOUHP-UHFFFAOYSA-N (2,3,4,5,6-pentafluorophenyl) 2,2,2-trifluoroacetate Chemical compound FC1=C(F)C(F)=C(OC(=O)C(F)(F)F)C(F)=C1F VCQURUZYYSOUHP-UHFFFAOYSA-N 0.000 description 1
- TYKASZBHFXBROF-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-(2,5-dioxopyrrol-1-yl)acetate Chemical compound O=C1CCC(=O)N1OC(=O)CN1C(=O)C=CC1=O TYKASZBHFXBROF-UHFFFAOYSA-N 0.000 description 1
- XSWBNALIBMCQED-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-phenyl-2-(pyridin-2-yldisulfanyl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)C(C=1C=CC=CC=1)(C)SSC1=CC=CC=N1 XSWBNALIBMCQED-UHFFFAOYSA-N 0.000 description 1
- PVGATNRYUYNBHO-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-(2,5-dioxopyrrol-1-yl)butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCN1C(=O)C=CC1=O PVGATNRYUYNBHO-UHFFFAOYSA-N 0.000 description 1
- VLARLSIGSPVYHX-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-(2,5-dioxopyrrol-1-yl)hexanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCN1C(=O)C=CC1=O VLARLSIGSPVYHX-UHFFFAOYSA-N 0.000 description 1
- IHVODYOQUSEYJJ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-[[4-[(2,5-dioxopyrrol-1-yl)methyl]cyclohexanecarbonyl]amino]hexanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCNC(=O)C(CC1)CCC1CN1C(=O)C=CC1=O IHVODYOQUSEYJJ-UHFFFAOYSA-N 0.000 description 1
- UGNIYGNGCNXHTR-SFHVURJKSA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-3-methylbutanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](C(C)C)C(O)=O)C3=CC=CC=C3C2=C1 UGNIYGNGCNXHTR-SFHVURJKSA-N 0.000 description 1
- WOWDZACBATWTAU-FEFUEGSOSA-N (2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-n-[(3r,4s,5s)-1-[(2s)-2-[(1r,2r)-3-[[(1s,2r)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-n,3-dimethylbutanamide Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)C1=CC=CC=C1 WOWDZACBATWTAU-FEFUEGSOSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- 101150084750 1 gene Proteins 0.000 description 1
- 125000005918 1,2-dimethylbutyl group Chemical group 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- OJQSISYVGFJJBY-UHFFFAOYSA-N 1-(4-isocyanatophenyl)pyrrole-2,5-dione Chemical compound C1=CC(N=C=O)=CC=C1N1C(=O)C=CC1=O OJQSISYVGFJJBY-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- WAAXYLYXYLKHJZ-UHFFFAOYSA-N 1-[3-(1-hydroxy-2,5-dioxopyrrolidine-3-carbonyl)phenyl]pyrrole-2,5-dione Chemical compound O=C1N(O)C(=O)CC1C(=O)C1=CC=CC(N2C(C=CC2=O)=O)=C1 WAAXYLYXYLKHJZ-UHFFFAOYSA-N 0.000 description 1
- PNDPGZBMCMUPRI-HVTJNCQCSA-N 10043-66-0 Chemical compound [131I][131I] PNDPGZBMCMUPRI-HVTJNCQCSA-N 0.000 description 1
- 101150028074 2 gene Proteins 0.000 description 1
- INGYQUXFTSEGEF-UHFFFAOYSA-N 2-(2-aminoethyl)-3,3-dimethylbutanoic acid Chemical compound CC(C)(C)C(C(O)=O)CCN INGYQUXFTSEGEF-UHFFFAOYSA-N 0.000 description 1
- SNFFHBJWQFQMBC-UHFFFAOYSA-N 2-(ethylamino)acetamide Chemical compound CCNCC(N)=O SNFFHBJWQFQMBC-UHFFFAOYSA-N 0.000 description 1
- OWDQCSBZQVISPN-UHFFFAOYSA-N 2-[(2,5-dioxopyrrolidin-1-yl)amino]-4-(2-iodoacetyl)benzoic acid Chemical compound OC(=O)C1=CC=C(C(=O)CI)C=C1NN1C(=O)CCC1=O OWDQCSBZQVISPN-UHFFFAOYSA-N 0.000 description 1
- UPBQMAHYLWJGDW-UHFFFAOYSA-N 2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethoxy]acetic acid Chemical compound CC(C)(C)OC(=O)NCCOCC(O)=O UPBQMAHYLWJGDW-UHFFFAOYSA-N 0.000 description 1
- PTUJJIPXBJJLLV-AWEZNQCLSA-N 2-[[(2s)-2-[[2-[[2-[(2-methylpropan-2-yl)oxycarbonylamino]acetyl]amino]acetyl]amino]-3-phenylpropanoyl]amino]acetic acid Chemical compound CC(C)(C)OC(=O)NCC(=O)NCC(=O)N[C@H](C(=O)NCC(O)=O)CC1=CC=CC=C1 PTUJJIPXBJJLLV-AWEZNQCLSA-N 0.000 description 1
- FBKUOPULLUJMOC-UHFFFAOYSA-N 2-[[2-(9h-fluoren-9-ylmethoxycarbonylamino)acetyl]amino]acetic acid Chemical compound C1=CC=C2C(COC(=O)NCC(=O)NCC(=O)O)C3=CC=CC=C3C2=C1 FBKUOPULLUJMOC-UHFFFAOYSA-N 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- ISCMYZGMRHODRP-UHFFFAOYSA-N 3-(iminomethylideneamino)-n,n-dimethylpropan-1-amine Chemical compound CN(C)CCCN=C=N ISCMYZGMRHODRP-UHFFFAOYSA-N 0.000 description 1
- DKIDEFUBRARXTE-UHFFFAOYSA-M 3-mercaptopropionate Chemical compound [O-]C(=O)CCS DKIDEFUBRARXTE-UHFFFAOYSA-M 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- BMTZEAOGFDXDAD-UHFFFAOYSA-M 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholin-4-ium;chloride Chemical compound [Cl-].COC1=NC(OC)=NC([N+]2(C)CCOCC2)=N1 BMTZEAOGFDXDAD-UHFFFAOYSA-M 0.000 description 1
- UZOVYGYOLBIAJR-UHFFFAOYSA-N 4-isocyanato-4'-methyldiphenylmethane Chemical compound C1=CC(C)=CC=C1CC1=CC=C(N=C=O)C=C1 UZOVYGYOLBIAJR-UHFFFAOYSA-N 0.000 description 1
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- BTJIUGUIPKRLHP-UHFFFAOYSA-N 4-nitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1 BTJIUGUIPKRLHP-UHFFFAOYSA-N 0.000 description 1
- VWXGRWMELBPMCU-UHFFFAOYSA-N 5-chloro-1-[3-(dimethylamino)propyl]-3-phenylbenzimidazol-2-one Chemical compound O=C1N(CCCN(C)C)C2=CC=C(Cl)C=C2N1C1=CC=CC=C1 VWXGRWMELBPMCU-UHFFFAOYSA-N 0.000 description 1
- RUFDYIJGNPVTAY-UHFFFAOYSA-N 6-[(2-methylpropan-2-yl)oxycarbonylamino]hexanoic acid Chemical compound CC(C)(C)OC(=O)NCCCCCC(O)=O RUFDYIJGNPVTAY-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 108010012934 Albumin-Bound Paclitaxel Proteins 0.000 description 1
- 206010073478 Anaplastic large-cell lymphoma Diseases 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 1
- 229940122815 Aromatase inhibitor Drugs 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- QLNSPJMIYWEWNF-UHFFFAOYSA-N CC(C)CCCC(C)CCCC(C)CCCC(C)C.CC(C)CCCC(C)CCCC(C)CCCC(C)C Chemical compound CC(C)CCCC(C)CCCC(C)CCCC(C)C.CC(C)CCCC(C)CCCC(C)CCCC(C)C QLNSPJMIYWEWNF-UHFFFAOYSA-N 0.000 description 1
- PCBZRNYXXCIELG-WYFCWLEVSA-N COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 Chemical compound COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 PCBZRNYXXCIELG-WYFCWLEVSA-N 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 241000725101 Clea Species 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- CKLJMWTZIZZHCS-UWTATZPHSA-N D-aspartic acid Chemical compound OC(=O)[C@H](N)CC(O)=O CKLJMWTZIZZHCS-UWTATZPHSA-N 0.000 description 1
- 229940123780 DNA topoisomerase I inhibitor Drugs 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- 201000010915 Glioblastoma multiforme Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- KAJAOGBVWCYGHZ-JTQLQIEISA-N Gly-Gly-Phe Chemical compound [NH3+]CC(=O)NCC(=O)N[C@H](C([O-])=O)CC1=CC=CC=C1 KAJAOGBVWCYGHZ-JTQLQIEISA-N 0.000 description 1
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 1
- 108010069236 Goserelin Proteins 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 101001099381 Homo sapiens Peroxisomal biogenesis factor 19 Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- 239000007760 Iscove's Modified Dulbecco's Medium Substances 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 102220470475 L-seryl-tRNA(Sec) kinase_C57L_mutation Human genes 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 230000004988 N-glycosylation Effects 0.000 description 1
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 230000004989 O-glycosylation Effects 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 102100038883 Peroxisomal biogenesis factor 19 Human genes 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 102000040739 Secretory proteins Human genes 0.000 description 1
- 108091058545 Secretory proteins Proteins 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 1
- 101710120037 Toxin CcdB Proteins 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- NKDSPEFADHLMHN-UHFFFAOYSA-N [[2-(9H-fluoren-9-ylmethoxycarbonylamino)acetyl]amino]methyl acetate Chemical compound CC(=O)OCNC(=O)CNC(=O)OCC1c2ccccc2-c2ccccc12 NKDSPEFADHLMHN-UHFFFAOYSA-N 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 229940028652 abraxane Drugs 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- 239000008351 acetate buffer Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 208000037844 advanced solid tumor Diseases 0.000 description 1
- 229910000102 alkali metal hydride Inorganic materials 0.000 description 1
- 150000008046 alkali metal hydrides Chemical class 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000004849 alkoxymethyl group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 229960002684 aminocaproic acid Drugs 0.000 description 1
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 1
- 229960000723 ampicillin Drugs 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 1
- 229950000242 ancitabine Drugs 0.000 description 1
- KZOWNALBTMILAP-JBMRGDGGSA-N ancitabine hydrochloride Chemical compound Cl.N=C1C=CN2[C@@H]3O[C@H](CO)[C@@H](O)[C@@H]3OC2=N1 KZOWNALBTMILAP-JBMRGDGGSA-N 0.000 description 1
- HOPRXXXSABQWAV-UHFFFAOYSA-N anhydrous collidine Natural products CC1=CC=NC(C)=C1C HOPRXXXSABQWAV-UHFFFAOYSA-N 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000002494 anti-cea effect Effects 0.000 description 1
- 210000000612 antigen-presenting cell Anatomy 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 239000003886 aromatase inhibitor Substances 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 125000005101 aryl methoxy carbonyl group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 229940007550 benzyl acetate Drugs 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 229920001222 biopolymer Polymers 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- KDPAWGWELVVRCH-UHFFFAOYSA-N bromoacetic acid Chemical compound OC(=O)CBr KDPAWGWELVVRCH-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000001720 carbohydrates Chemical group 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- YAYRGNWWLMLWJE-UHFFFAOYSA-L carboplatin Chemical compound O=C1O[Pt](N)(N)OC(=O)C11CCC1 YAYRGNWWLMLWJE-UHFFFAOYSA-L 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- ARQRPTNYUOLOGH-UHFFFAOYSA-N chcl3 chloroform Chemical compound ClC(Cl)Cl.ClC(Cl)Cl ARQRPTNYUOLOGH-UHFFFAOYSA-N 0.000 description 1
- 239000013626 chemical specie Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- HRYZWHHZPQKTII-UHFFFAOYSA-N chloroethane Chemical compound CCCl HRYZWHHZPQKTII-UHFFFAOYSA-N 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- UTBIMNXEDGNJFE-UHFFFAOYSA-N collidine Natural products CC1=CC=C(C)C(C)=N1 UTBIMNXEDGNJFE-UHFFFAOYSA-N 0.000 description 1
- 230000005757 colony formation Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- RYGMFSIKBFXOCR-IGMARMGPSA-N copper-64 Chemical compound [64Cu] RYGMFSIKBFXOCR-IGMARMGPSA-N 0.000 description 1
- 230000000139 costimulatory effect Effects 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 125000004976 cyclobutylene group Chemical group 0.000 description 1
- 125000004979 cyclopentylene group Chemical group 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 230000006240 deamidation Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000004925 denaturation Methods 0.000 description 1
- 230000036425 denaturation Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 238000002059 diagnostic imaging Methods 0.000 description 1
- VILAVOFMIJHSJA-UHFFFAOYSA-N dicarbon monoxide Chemical group [C]=C=O VILAVOFMIJHSJA-UHFFFAOYSA-N 0.000 description 1
- ZWWWLCMDTZFSOO-UHFFFAOYSA-N diethoxyphosphorylformonitrile Chemical compound CCOP(=O)(C#N)OCC ZWWWLCMDTZFSOO-UHFFFAOYSA-N 0.000 description 1
- 239000012470 diluted sample Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- ZWIBGKZDAWNIFC-UHFFFAOYSA-N disuccinimidyl suberate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCCC(=O)ON1C(=O)CCC1=O ZWIBGKZDAWNIFC-UHFFFAOYSA-N 0.000 description 1
- 125000005414 dithiopyridyl group Chemical group 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 239000003118 drug derivative Substances 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000012156 elution solvent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 229960004750 estramustine phosphate Drugs 0.000 description 1
- ADFOJJHRTBFFOF-RBRWEJTLSA-N estramustine phosphate Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)OP(O)(O)=O)[C@@H]4[C@@H]3CCC2=C1 ADFOJJHRTBFFOF-RBRWEJTLSA-N 0.000 description 1
- 239000000328 estrogen antagonist Substances 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000013210 evaluation model Methods 0.000 description 1
- 229960000255 exemestane Drugs 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- IRXSLJNXXZKURP-UHFFFAOYSA-N fluorenylmethyloxycarbonyl chloride Chemical compound C1=CC=C2C(COC(=O)Cl)C3=CC=CC=C3C2=C1 IRXSLJNXXZKURP-UHFFFAOYSA-N 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 238000001641 gel filtration chromatography Methods 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 1
- 229960002913 goserelin Drugs 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- ALBYIUDWACNRRB-UHFFFAOYSA-N hexanamide Chemical compound CCCCCC(N)=O ALBYIUDWACNRRB-UHFFFAOYSA-N 0.000 description 1
- CTRFEEQNDJFNNL-UHFFFAOYSA-N hexanoic acid;pyrrole-2,5-dione Chemical compound O=C1NC(=O)C=C1.CCCCCC(O)=O CTRFEEQNDJFNNL-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 239000012216 imaging agent Substances 0.000 description 1
- 230000005965 immune activity Effects 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 230000001861 immunosuppressant effect Effects 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 230000002637 immunotoxin Effects 0.000 description 1
- 229940051026 immunotoxin Drugs 0.000 description 1
- 239000002596 immunotoxin Substances 0.000 description 1
- 231100000608 immunotoxin Toxicity 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 229940055742 indium-111 Drugs 0.000 description 1
- APFVFJFRJDLVQX-AHCXROLUSA-N indium-111 Chemical compound [111In] APFVFJFRJDLVQX-AHCXROLUSA-N 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- XMBWDFGMSWQBCA-OIOBTWANSA-N iodane Chemical compound [124IH] XMBWDFGMSWQBCA-OIOBTWANSA-N 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 150000002497 iodine compounds Chemical class 0.000 description 1
- FBAFATDZDUQKNH-UHFFFAOYSA-M iron chloride Chemical compound [Cl-].[Fe] FBAFATDZDUQKNH-UHFFFAOYSA-M 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000006317 isomerization reaction Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- ACKFDYCQCBEDNU-UHFFFAOYSA-J lead(2+);tetraacetate Chemical compound [Pb+2].CC([O-])=O.CC([O-])=O.CC([O-])=O.CC([O-])=O ACKFDYCQCBEDNU-UHFFFAOYSA-J 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 239000003589 local anesthetic agent Substances 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N n-hendecanoic acid Natural products CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 125000004957 naphthylene group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 238000011170 pharmaceutical development Methods 0.000 description 1
- 230000004526 pharmaceutical effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 238000009520 phase I clinical trial Methods 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- NMHMNPHRMNGLLB-UHFFFAOYSA-N phloretic acid Chemical compound OC(=O)CCC1=CC=C(O)C=C1 NMHMNPHRMNGLLB-UHFFFAOYSA-N 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229920000889 poly(m-phenylene isophthalamide) Polymers 0.000 description 1
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- GRLPQNLYRHEGIJ-UHFFFAOYSA-J potassium aluminium sulfate Chemical compound [Al+3].[K+].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O GRLPQNLYRHEGIJ-UHFFFAOYSA-J 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000012514 protein characterization Methods 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000007281 self degradation Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000012679 serum free medium Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 239000012064 sodium phosphate buffer Substances 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 230000003393 splenic effect Effects 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- ATGUDZODTABURZ-UHFFFAOYSA-N thiolan-2-ylideneazanium;chloride Chemical compound Cl.N=C1CCCS1 ATGUDZODTABURZ-UHFFFAOYSA-N 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 206010044412 transitional cell carcinoma Diseases 0.000 description 1
- 150000003918 triazines Chemical class 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 210000005253 yeast cell Anatomy 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
- QCWXUUIWCKQGHC-YPZZEJLDSA-N zirconium-89 Chemical compound [89Zr] QCWXUUIWCKQGHC-YPZZEJLDSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/48—Ergoline derivatives, e.g. lysergic acid, ergotamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68037—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a camptothecin [CPT] or derivatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/22—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains four or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2827—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against B7 molecules, e.g. CD80, CD86
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2851—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the lectin superfamily, e.g. CD23, CD72
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2875—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the NGF/TNF superfamily, e.g. CD70, CD95L, CD153, CD154
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2878—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the NGF-receptor/TNF-receptor superfamily, e.g. CD27, CD30, CD40, CD95
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Epidemiology (AREA)
- Cell Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Un compuesto representado por la siguiente fórmula: **(Ver fórmula)**
Description
DESCRIPCIÓN
Enlazadores para conjugados de anticuerpo - fármaco
Campo técnico
La presente invención se refiere a un conjugado de anticuerpo - fármaco que tiene un fármaco antitumoral conjugado con un anticuerpo capaz de seleccionar como diana células tumorales por medio de un resto de estructura de enlazador, siendo útil el conjugado como un fármaco antitumoral.
Antecedentes de la técnica
Un conjugado de anticuerpo - fármaco (ADC) que tiene un fármaco con citotoxicidad conjugado con un anticuerpo, cuyo antígeno se expresa sobre una superficie de células cancerosas y que también se une a un antígeno capaz de internalización celular y, por lo tanto, puede suministrar el fármaco selectivamente a células las cancerosas y se espera por lo tanto que dé lugar a la acumulación del fármaco dentro de las células cancerosas y que destruya las células cancerosas (véanse las literaturas no de patente 1 a 3). Como un ADC, Mylotarg (Gemtuzumab ozogamicina), en el que se conjuga caliqueamicina con un anticuerpo anti-CD33, está aprobado como un agente terapéutico para la leucemia mieloide aguda. Además, Adcetris (Brentuximab vedotina), en el que se conjuga auristatina E con un anticuerpo anti-CD30, se ha aprobado recientemente como un agente terapéutico para el linfoma de Hodgkin y el linfoma anaplásico de células grandes (véase la literatura no de patente 4). Los fármacos contenidos en los ADC que se han aprobado hasta la fecha seleccionan como diana ADN o tubulina.
Con respecto a un agente antitumoral, se conocen compuestos de bajo peso molecular, derivados de camptotecina, compuestos que inhiben la topoisomerasa I para mostrar un efecto antitumoral. Entre ellos, un compuesto antitumoral representado por la fórmula posterior
[Fórmula 1]

(exatecán, nombre químico: (1S,9S)-1-amino-9-etil-5-fluoro-2,3-dihidro-9-hidroxi-4-metil-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-10,13(9H, 15H)-diona) es un derivado soluble en agua de camptotecina (literatura de patente 1 y 2). A diferencia del irinotecán ya usado en escenarios clínicos, no es necesaria una activación mediante una enzima. Además, la actividad inhibitoria sobre la topoisomerasa I es más alta que SN-38, que es una sustancia farmacéuticamente activa principal de irinotecán y topotecán también usada en escenarios clínicos, y se obtiene una actividad citocida in vitro más alta contra diversas células cancerosas. En particular, este muestra el efecto contra células cancerosas que tienen resistencia a SN-38 o similares debido a la expresión de P-glicoproteína. Además, en un modelo de ratón transplantado subcutáneamente con tumor humano, este mostró un efecto antitumoral potente y, por lo tanto, se ha sometido a los estudios clínicos, pero no se ha puesto aún en el mercado (véanse las literaturas no de patente 5 a 10). Sigue sin estar claro si exatecán funciona, o no, eficazmente como un ADC.
DE-310 es un complejo en el que se conjuga exatecán con un polímero de carboximetildextrano polialcohol biodegradable por medio de un espaciador peptídico de GGFG (literatura de patente 3). Convirtiendo el exatecán en una forma de un profármaco de polímero, de tal modo que puede mantenerse una propiedad de retención sanguínea alta y también se aumenta pasivamente una propiedad de direccionabilidad alta a un área tumoral utilizando la permeabilidad aumentada de los vasos sanguíneos recién formados dentro del tumor y la propiedad de retención en los tejidos tumorales. Con DE-310, a través de una escisión del espaciador peptídico mediante enzima, se liberan continuamente, como una sustancia activa principal, exatecán y exatecán con glicina conectada a un grupo amino. Como resultado, la farmacocinética se mejora y se halló que DE-310 tiene una efectividad más alta que exatecán administrado solo incluso si la dosificación de exatecán es menor que en el caso de la administración de exatecán solo de acuerdo con diversos modelos de evaluación tumoral en estudios no clínicos. Se llevó a cabo un estudio clínico para DE-310, y se confirmaron casos efectivos en seres humanos, en donde estaba presente un informe que sugiere que la sustancia activa principal se acumula en un tumor que en tejidos normales, sin embargo, también existe un informe que indica que la acumulación de DE-310 y la sustancia activa principal en un tumor no es muy diferente de la acumulación en tejidos normales en seres humanos y, por lo tanto, no se observa selección como diana pasiva alguna en seres humanos (véanse las literaturas no de patente 11 a 14). Como resultado, tampoco se comercializó
DE-310, y sigue sin estar claro si exatecán funciona, o no, eficazmente como un fármaco orientado para tal selección como diana.
Como un compuesto en relación con DE-310, también se conoce un complejo en el que un resto de estructura representado por -NH(Ch2)4C(=O)- se inserta entre el espaciador de GGFG y exatecán para formar - GGFG-NH(CH2)4C(=O)-, usado como una estructura espadadora (literatura de patente 4). Sin embargo, el efecto antitumoral del complejo no es en absoluto conocido.
La literatura de patente 5 desvela la preparación de un conjugado de carboximetildextrano-polialcohol-fármaco en el que se usa un enlazador que comprende la secuencia glicil-glicil-fenilalanil-glicamina.
[Lista de citas]
[Literatura de patente]
[Literatura de patente 1] Patente japonesa abierta a inspección pública n.° 5-59061
[Literatura de patente 2] Patente japonesa abierta a inspección pública n.° 8-337584
[Literatura de patente 3] Publicación internacional n.° WO 1997/46260
[Literatura de patente 4] Publicación internacional n.° WO 2000/25825
[Literatura de patente 5] Publicación internacional n.° WO 03/015826
[Literatura no de patente]
[Literatura no de patente 1] Ducry, L., y col. Bioconjugate Chem. (2010) 21,5-13.; Antibody-Drug Conjugates: Linking cytotoxic payloads to monoclonal antibodies.
[Literatura no de patente 2] Alley, S. C., y col. Current Opinion in Chemical Biology (2010) 14, 529-537.; Antibodydrug conjugates: targeted drug delivery for cancer.
[Literatura no de patente 3] Damle N. K. Expert Opin. Biol. Ther. (2004) 4, 1445-1452.; Tumour-targeted chemotherapy with immunoconjugates of calicheamicin.
[Literatura no de patente 4] Senter P. D., y col. Nature Biotechnology (2012) 30, 631-637.; The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma.
[Literatura no de patente 5] Kumazawa, E., Tohgo, A., Exp. Opin. Invest. Drugs (1998) 7, 625-632.; Antitumour activity of DX-8951f: a new camptothecin derivative.
[Literatura no de patente 6] Mitsui, I., Kumazawa, E., Hirota, Y., y col. Jpn J. Cancer Res. (1995) 86, 776-782.; A new water-soluble camptothecin derivative, DX-8951f, exhibits potent antitumor activity against human tumors in vitro and in vivo.
[Literatura no de patente 7] Takiguchi, S., Tohgo, A., y col. Jpn J. Cancer Res. (1997) 88, 760-769.; Antitumor effect of DX-8951, a novel camptothecin analog, on human pancreatic tumor cells and their CPT-11-resistant variants cultured in vitro and xenografted into nude mice.
[Literatura no de patente 8] Joto, N. y col. Int J Cancer (1997) 72, 680-686.; DX-8951f, a water-soluble camptothecin analog, exhibits potent antitumor activity against a human lung cancer cell line and its SN-38-resistant variant. [Literatura no de patente 9] Kumazawa, E. y col. Cancer Chemother. Pharmacol. (1998) 42, 210-220.; Potent and broad antitumor effects of DX-8951f, a water-soluble camptothecin derivative, against various human tumors xenografted in nude mice.
[Literatura no de patente 10] De Jager, R., y col. Ann N Y Acad Sci (2000) 922, 260-273.; DX-8951f: summary of phase I clinical trials.
[Literatura no de patente 11] Inoue, K. y col. Polymer Drugs in the Clinical Stage, Editado por Maeda y col. (2003), 145-153.; CM-dextran-polyalcohol-camptothecin conjugate, DE-310 with a novel carrier system and its preclinical data.
[Literatura no de patente 12] Kumazawa, E. y col. Cancer Sci (2004) 95, 168-175.; DE-310, a novel macromolecular carrier system for the camptothecin analog DX-8951f: Potent antitumor activities in various murine tumor models.
[Literatura no de patente 13] Soepenberg, O. y col. Clinical Cancer Research, (2005) 11, 703-711.; Phase I and pharmacokinetic study of DE-310 in Patients with Advanced Solid Tumors.
[Literatura no de patente 14] Wente M. N. y col. Investigational New Drugs (2005) 23, 339-347.; DE-310, a macromolecular prodrug of the topoisomerase-I-inhibitor exatecan (DX-8951), in patients with operable solid tumors.
rSumario de la invención!
rProblema técnico!
Con respecto al tratamiento de un tumor mediante un anticuerpo, puede observarse un efecto antitumoral insuficiente incluso cuando el anticuerpo reconoce un antígeno y se une a células tumorales, y hay un caso en el que se necesita un anticuerpo antitumoral más eficaz. Además, muchos compuestos de bajo peso molecular antitumorales presentan un problema en la seguridad como un efecto secundario y la toxicidad y, aunque los compuestos tienen un efecto antitumoral excelente, sigue siendo un objeto lograr un efecto terapéutico superior potenciando adicionalmente la seguridad. De ese modo, un objeto de la presente invención es obtener proporcionar un intermedio para un fármaco
antitumoral que tenga un efecto terapéutico excelente, que sea excelente en términos del efecto antitumoral y la seguridad.
fMedios para resolver el problema!
Los inventores de la presente invención pensaron que, cuando un compuesto antitumoral exatecán se convierte en un conjugado de anticuerpo - fármaco, por medio de un resto de estructura de enlazador, mediante conjugación con el anticuerpo, que es capaz de seleccionar como diana células tumorales, es decir, que tiene una propiedad de reconocer células tumorales, una propiedad de unirse a células tumorales, una propiedad de internalización dentro de células tumorales, una actividad citocida contra células tumorales, o similares, el compuesto antitumoral puede suministrarse más seguramente a las células tumorales para mostrar específicamente el efecto antitumoral del compuesto en células tumorales y, por lo tanto, el efecto antitumoral puede seguramente mostrarse y también se espera un efecto citocida potenciado del anticuerpo, y puede reducirse una dosis del compuesto antitumoral en comparación con un caso de administración del compuesto solo y, por lo tanto, puede aliviarse una influencia del compuesto antitumoral sobre las células normales de tal modo que puede lograrse una seguridad más alta.
A este respecto, los inventores de la presente invención crearon un enlazador con una estructura específica y tuvieron éxito en la obtención de un conjugado de anticuerpo - fármaco en el que el anticuerpo y exatecán se conjugan entre sí por medio del enlazador, y confirmaron un efecto antitumoral excelente mostrado por el conjugado para completar de ese modo la presente invención.
Específicamente, la presente invención se refiere a N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]glicilglicil-L-fenilalanil-N-[(carboximetoxi)metil]glicinamida representada por la siguiente fórmula:

Breve descripción de los dibujos
[Figura 1] La figura 1 muestra una secuencia de aminoácidos de la variante 1 de B7-H3 (SEQ ID NO: 1).
[Figura 2] La figura 2 muestra una secuencia de aminoácidos de la variante 2 de B7-H3 (SEQ ID NO: 2).
[Figura 3] La figura 3 muestra una secuencia de aminoácidos de una cadena pesada de tipo M30-H1 (SEQ ID NO: 9) .
[Figura 4] La figura 4 muestra una secuencia de aminoácidos de una cadena pesada de tipo M30-H2 (SEQ ID NO: 10) .
[Figura 5] La figura 5 muestra una secuencia de aminoácidos de una cadena pesada de tipo M30-H3 (SEQ ID NO: 11).
[Figura 6] La figura 6 muestra una secuencia de aminoácidos de una cadena pesada de tipo M30-H4 (SEQ ID NO: 12).
[Figura 7] La figura 7 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L1 (SEQ ID NO: 13) .
[Figura 8] La figura 8 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L2 (SEQ ID NO: 14) .
[Figura 9] La figura 9 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L3 (SEQ ID NO: 15) .
[Figura 10] La figura 10 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L4 (SEQ ID NO: 16) .
[Figura 11] La figura 11 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L5 (SEQ ID NO: 17) .
[Figura 12] La figura 12 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L6 (SEQ ID NO: 18) .
[Figura 13] La figura 13 muestra una secuencia de aminoácidos de una cadena ligera de tipo M30-L7 (SEQ ID NO: 19).
[Figura 14] La figura 14 muestra una secuencia de aminoácidos de una cadena pesada de anticuerpo M30 (SEQ ID NO: 20).
[Figura 15] La figura 15 muestra una secuencia de aminoácidos de una cadena ligera de anticuerpo M30 (SEQ ID NO: 21).
[Figura 16] La figura 16 muestra una secuencia de nucleótidos de la variante 1 de B7-H3 (SEQ ID NO: 26).
[Figura 20] La figura 20 muestra los efectos de los conjugados de anticuerpo - fármaco (1), (13), (41) y (55) sobre células de la línea de melanoma humano A375 subcutáneamente trasplantadas. En el dibujo, la línea con rombos abiertos muestra resultados acerca de un tumor sin tratar, la línea con círculos abiertos muestra el efecto del conjugado de anticuerpo - fármaco (1), la línea con triángulos abiertos muestra el efecto del conjugado de anticuerpo - fármaco (13), la línea con marcas X muestra el efecto del conjugado de anticuerpo - fármaco (41) y la línea con cuadrados abiertos muestra el efecto del conjugado de anticuerpo - fármaco (55).
[Figura 21] La figura 21 muestra los efectos de los conjugados de anticuerpo - fármaco (13), (41) y (55) sobre células Calu-6 de línea de cáncer de pulmón no de células pequeñas humano subcutáneamente trasplantadas. La línea con rombos abiertos muestra resultados acerca de un tumor sin tratar, la línea con círculos abiertos muestra el efecto de DE-310, la línea con triángulos abiertos muestra el efecto del conjugado de anticuerpo - fármaco (13), la línea con marcas X muestra el efecto del conjugado de anticuerpo - fármaco (41) y la línea con cuadrados abiertos muestra el efecto del conjugado de anticuerpo - fármaco (55).
[Figura 22] La figura 22 muestra los efectos de los conjugados de anticuerpo - fármaco (17), (18), (19), (59), (60) y (61) sobre células de la línea de melanoma humano A375 subcutáneamente trasplantadas. En el dibujo, la línea con rombos sólidos muestra resultados acerca de un tumor sin tratar, la línea con cuadrados sólidos muestra el efecto del conjugado de anticuerpo - fármaco (17), la línea con cuadrados abiertos muestra el efecto del conjugado de anticuerpo -fármaco (18), la línea con círculos abiertos muestra el efecto del conjugado de anticuerpo -fármaco (19), la línea con triángulos sólidos muestra el efecto del conjugado de anticuerpo - fármaco (59), la línea con triángulos abiertos muestra el efecto del conjugado de anticuerpo - fármaco (60) y la línea con marcas X muestra el efecto del conjugado de anticuerpo - fármaco (61).
Descripción de realizaciones
El intermedio de la presente invención es útil para preparar un fármaco antitumoral en el que un anticuerpo antitumoral está conjugado con un compuesto antitumoral por medio de un resto de estructura de enlazador y se explica con detalle en lo sucesivo en el presente documento.
[Anticuerpo]
El anticuerpo usado en el conjugado de anticuerpo - fármaco quiere decir una inmunoglobulina y es una molécula que contiene un sitio de unión a antígeno que se une inmunoespecíficamente a un antígeno. La clase del anticuerpo puede ser cualquiera de IgG, IgE, IgM, IgD, IgA e IgY y es preferentemente IgG. La subclase del anticuerpo puede ser cualquiera de IgG1, IgG2, IgG3, IgG4, IgA1 e IgA2 y es preferentemente IgG1 o IgG2. El anticuerpo puede derivarse de cualquier especie, y los ejemplos preferidos de la especie pueden incluir seres humanos, ratas, ratones y conejos. En el caso en el que se deriva de una especie que no sea la humana, preferentemente este se quimeriza o se humaniza usando una técnica bien conocida. El anticuerpo puede ser un anticuerpo policlonal o un anticuerpo monoclonal y es preferentemente un anticuerpo monoclonal.
El anticuerpo puede ser uno que es capaz de seleccionar como diana células tumorales. Debido a que el anticuerpo está conjugado con un fármaco que tiene actividad antitumoral por medio de un enlazador, el anticuerpo preferentemente posee una o más de una propiedad de reconocer una célula tumoral, una propiedad de unirse a una célula tumoral, una propiedad de internalizarse en una célula tumoral y una propiedad de dañar una célula tumoral.
La actividad de unión del anticuerpo contra células tumorales puede confirmarse usando citometría de flujo. La internalización del anticuerpo en células tumorales puede confirmarse usando (1) un ensayo de visualización de un anticuerpo incorporado en células bajo un microscopio de fluorescencia usando un anticuerpo secundario (marcado fluorescentemente) que se une al anticuerpo terapéutico (Cell Death and Differentiation (2008) 15, 751-761), (2) un ensayo de medición de la cantidad de fluorescencia incorporada en células usando un anticuerpo secundario (marcado fluorescentemente) que se une al anticuerpo terapéutico (Molecular Biology of the Cell, Vol. 15, 5268-5282, diciembre de 2004) o (3) un ensayo de Mab-ZAP usando una inmunotoxina que se une al anticuerpo terapéutico en el que la toxina se libera tras la incorporación en células para inhibir el crecimiento celular (Bio Techniques 28: 162-165, enero de 2000).
La actividad antitumoral del anticuerpo se refiere a una actividad citotóxica o efecto citocida contra células tumorales y puede confirmarse in vitro determinando la actividad inhibitoria contra el crecimiento celular. Por ejemplo, se cultiva una línea de células cancerosas que sobreexpresa una proteína diana para el anticuerpo, y el anticuerpo se añade a concentraciones variables en el sistema de cultivo para determinar una actividad inhibitoria contra la formación de focos, la formación de colonias y el crecimiento de esferoides. La actividad antitumoral puede confirmarse in vivo, por ejemplo, administrando el anticuerpo a un ratón desnudo con una línea de células tumorales trasplantadas que expresa altamente la proteína diana, y determinando el cambio en la célula cancerosa. Debido a que el fármaco conjugado en el conjugado de anticuerpo - fármaco ejerce un efecto antitumoral, se prefiere en mayor medida, pero no es esencial, que el propio anticuerpo tenga un efecto antitumoral. Para ejercer el efecto antitumoral y también para dañar específica y selectivamente células tumorales mediante el fármaco, es importante y también se prefiere que el anticuerpo tenga la propiedad de internalizarse para migrar a células tumorales.
Los ejemplos de un anticuerpo de este tipo pueden incluir, pero sin limitarse a, un anticuerpo anti-A33, un anticuerpo anti-B7-H3, un anticuerpo anti-CanAg, un anticuerpo anti-CD20, un anticuerpo anti-CD22, un anticuerpo anti-CD30,
un anticuerpo anti-CD33, un anticuerpo anti-CD56, un anticuerpo anti-CD70, un anticuerpo anti-CEA, un anticuerpo anti-Cripto, un anticuerpo anti-EphA2, un anticuerpo anti-G250, un anticuerpo anti-MUC1, un anticuerpo anti-GPNMB, un anticuerpo anti-integrina, un anticuerpo anti-PSMA, un anticuerpo anti-tenascin-C, un anticuerpo anti-SLC44A4 y un anticuerpo anti-mesotelina.
El anticuerpo es preferentemente un anticuerpo anti-CD30, un anticuerpo anti-CD33, un anticuerpo anti-CD70 o un anticuerpo anti-B7-H3 y, más preferentemente, un anticuerpo anti-B7-H3.
El anticuerpo puede obtenerse usando un procedimiento llevado habitualmente a cabo en la técnica, que implica inmunizar animales con un polipéptido antigénico y recoger y purificar anticuerpos producidos in vivo. El origen del antígeno no se limita a los seres humanos, y los animales pueden inmunizarse con un antígeno derivado de un animal no humano tal como un ratón, una rata y similares. En este caso, la reactividad cruzada de los anticuerpos que se unen al antígeno heterólogo obtenido con antígenos humanos puede someterse a prueba para explorar un anticuerpo aplicable a una enfermedad humana.
Como alternativa, células productoras de anticuerpo que producen anticuerpos contra el antígeno se fusionan con células de mieloma de acuerdo con un procedimiento conocido en la técnica (por ejemplo, Kohler y Milstein, Nature (1975) 256, pág. 495-497; y Kennet, R. ed., Monoclonal Antibodies, págs. 365-367, Plenum Press, N. Y. (1980)) para establecer hibridomas, a partir de los cuales, a su vez, pueden obtenerse anticuerpos monoclonales.
El antígeno puede obtenerse modificando por ingeniería genética células hospedadoras para producir un gen que codifica la proteína antigénica. Específicamente, se preparan unos vectores que permiten la expresión del gen de antígeno y se transfieren a células hospedadoras de tal modo que el gen se expresa. El antígeno expresado de este modo puede purificarse.
El anticuerpo anti-CD30, el anticuerpo anti-CD33 y el anticuerpo anti-CD70 pueden obtenerse mediante un enfoque conocido en la técnica con referencia al documento WO2002/043661, la patente de EE. UU. n.° 5.773.001 y el documento WO2006/113909, respectivamente.
El anticuerpo B7-H3 es preferentemente uno que tiene propiedades según se describe a continuación.
(1) Un anticuerpo que tiene las siguientes propiedades:
(a) unirse específicamente a B7-H3,
(b) tener una actividad de fagocitosis mediada por células dependiente del anticuerpo (ADCP), y
(c) tener actividad antitumoral in vivo.
(2) El anticuerpo de acuerdo con (1), en el que B7-H3 es una molécula que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 1 o 2.
(3) El anticuerpo de acuerdo con (1) o (2), en el que el anticuerpo tiene CDRH1 que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 3, CDRH2 que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 4, y CDRH3 que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 5 como regiones de determinación de complementariedad de cadena pesada, y CDRL1 que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 6, CDRL2 que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 7, y CDRL3 que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 8 como regiones de determinación de complementariedad de cadena ligera. (4) El anticuerpo de acuerdo con cualquiera de (1) a (3), en el que la región constante del mismo es una región constante derivada de ser humano.
(5) El anticuerpo de acuerdo con cualquiera de (1) a (4), en el que el anticuerpo es un anticuerpo humanizado. (6) El anticuerpo de acuerdo con (5), en el que el anticuerpo tiene una región variable de cadena pesada que comprende una secuencia de aminoácidos seleccionada de entre el grupo que consiste en (a) una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la s Eq ID NO: 9, (b) una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 10, (c) una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 11, (d) una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 12, (e) una secuencia de aminoácidos que tiene una homología de al menos el 95 % o más alta con cualquiera de las secuencias (a) a (d) y (f) una secuencia de aminoácidos derivada de cualquiera de las secuencias (a) a (d) por las eliminaciones, sustituciones o adiciones de al menos un aminoácido, y una región variable de cadena ligera que comprende una secuencia de aminoácidos seleccionada de entre el grupo que consiste en (g) una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 13, (h) una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 14, (i) una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 15, (j) una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 16, (k) una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 17, (1) una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 18, (m) una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 19, (n) una secuencia de aminoácidos que tiene una homología de al menos el 95 % o más alta con cualquiera de las secuencias (g) a (m) y (o) una secuencia de
aminoácidos derivada de cualquiera de las secuencias (g) a (m) por las eliminaciones, sustituciones o adiciones de al menos un aminoácido.
(7) El anticuerpo de acuerdo con (6), en el que el anticuerpo tiene una región variable de cadena pesada y una región variable de cadena ligera seleccionada de entre el grupo que consiste en una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 13, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 14, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ Id NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 15, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 16, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 17, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 18, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 9 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 19, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 12 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 13, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la s Eq ID NO: 12 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 14, una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 12 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 15, y una región variable de cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 141 en la SEQ ID NO: 12 y una región variable de cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 128 en la SEQ ID NO: 16.
(8) El anticuerpo de acuerdo con (6) o (7), en el que el anticuerpo comprende una cadena pesada y una cadena ligera seleccionadas de entre el grupo que consiste en una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 13, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 14, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 15, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 16, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 17, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 18, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 19, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 12 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 13, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 12 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 14, una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 12 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 15, y una cadena pesada que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 12 y una cadena ligera que comprende una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 16.
(9) El anticuerpo de acuerdo con cualquiera de (14) a (16), en el que el anticuerpo comprende una cadena pesada
y una cadena ligera seleccionadas de entre el grupo que consiste en una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 13, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 14, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 15, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 16, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 17, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 18, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 9 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 19, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 12 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 13, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 12 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 14, una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 12 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 15, y una cadena pesada que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 12 y una cadena ligera que comprende la secuencia de aminoácidos representada por la SEQ ID NO: 16.
(10) El anticuerpo de acuerdo con (8) o (9), en el que el anticuerpo carece de un aminoácido en el extremo carboxilo de la secuencia de aminoácidos representada por la SEQ ID NO: 9 o 12 en la cadena pesada.
(11) Un anticuerpo obtenido mediante un procedimiento para producir el anticuerpo de acuerdo con cualquiera de (1) a (10), comprendiendo el procedimiento las etapas de: cultivar una célula hospedadora transformada con un vector de expresión que contiene un polinucleótido que codifica el anticuerpo; y recoger el anticuerpo de interés a partir de los cultivos obtenidos en la etapa anterior.
(12) El anticuerpo de acuerdo con cualquiera de (1) a (11), en el que la modificación de un glicano se regula con el fin de potenciar la actividad citotóxica dependiente del anticuerpo.
En lo sucesivo en el presente documento, se describe el anticuerpo B7-H3 usado en la invención.
Los términos "cáncer" y "tumor", tal como se usan en el presente documento, se usan con el mismo significado.
El término "gen", tal como se usa en el presente documento, incluye no solamente ADN, sino también ARNm del mismo, ADNc del mismo y ARNc del mismo.
El término "polinucleótido", tal como se usa en el presente documento, se usa con el mismo significado que un ácido nucleico y también incluye ADN, ARN, sondas, oligonucleótidos y cebadores.
Los términos "polipéptido" y "proteína", tal como se usan en el presente documento, se usan indistintamente.
El término "célula", tal como se usa en el presente documento, también incluye células en un individuo animal y células cultivadas.
El término "B7-H3", tal como se usa en el presente documento, se usa con el mismo significado que proteína B7-H3, y también se refiere a la variante 1 de B7-H3 y/o la variante 2 de B7-H3.
El término "CDR", tal como se usa en el presente documento, se refiere a una región determinante de la complementariedad (CDR), y se sabe que cada cadena pesada y ligera de una molécula de anticuerpo tiene tres regiones determinantes de la complementariedad (CDR). La CDR también se denomina región hipervariable y está presente en una región variable de cada cadena pesada y ligera de un anticuerpo. Es un sitio que tiene una variabilidad inusualmente alta en su estructura primaria, y hay tres CDR independientes en la estructura primaria de cada cadena polipeptídica pesada y ligera. En la presente memoria descriptiva, con respecto a las CDR de un anticuerpo, las CDR de la cadena pesada están representadas por CDRH1, CDRH2 y CDRH3 desde el lado amino terminal de la secuencia de aminoácidos de la cadena pesada, y las CDR de la cadena ligera están representadas por CDRL1, CDRL2 y CDRL3 desde el lado amino terminal de la secuencia de aminoácidos de la cadena ligera. Estos sitios están próximos entre sí en la estructura terciaria y determinan la especificidad para un antígeno al que se une el anticuerpo.
La frase "la hibridación se realiza en condiciones rigurosas", tal como se usa en el presente documento, se refiere a un procedimiento en el que la hibridación se realiza en condiciones en las cuales puede lograrse la identificación realizando hibridación a 68 °C en una solución de hibridación disponible en el mercado ExpressHyb Hybridization Solution (fabricada por Clontech, Inc.) o realizando hibridación a 68 °C en presencia de NaCl de 0,7 a 1,0 M usando un filtro que tiene ADN inmovilizado sobre él, seguido de realizar lavado a 68 °C usando solución 0,1 a 2 x de SSC (1 x solución de SSC está compuesta por NaCl 150 mM y citrato de sodio 15 mM) o en condiciones equivalentes a las mismas.
1. B7-H3
B7-H3 es un miembro de la familia B7 expresado en células presentadoras de antígenos como una molécula coestimuladora, y se considera que actúa sobre un receptor en linfocitos T para potenciar o suprimir la actividad inmunitaria.
B7-H3 es una proteína que tiene una estructura transmembrana de un solo paso, y el dominio extracelular N-terminal de B7-H3 contiene dos variantes. La variante 1 de B7-H3 (4Ig-B7-H3) contiene un dominio de Ig de tipo V o de tipo C en dos sitios, respectivamente, y la variante 2 de B7-H3 (2Ig-B7-H3) contiene un dominio de Ig de tipo V o de tipo C en un sitio, respectivamente.
Con respecto a B7-H3 que se usará en la invención, B7-H3 puede purificarse directamente a partir de células que expresan B7-H3 de un ser humano o un mamífero no humano (tal como una rata o un ratón) y usarse, o puede prepararse y usarse una fracción de membrana celular de las células descritas anteriormente. Además, B7-H3 puede obtenerse mediante síntesis in vitro de la misma o producción de la misma en una célula hospedadora mediante ingeniería genética. En la ingeniería genética, específicamente, después de que el ADNc de B7-H3 se integre en un vector capaz de expresar ADNc de B7-H3, puede obtenerse B7-H3 sintetizándola en una solución que contiene una enzima, un sustrato y una sustancia energética requeridas para la transcripción y la traducción, o expresando B7-H3 en otra célula hospedadora transformada procariota o eucariota.
La secuencia de aminoácidos de un marco de lectura abierto (ORF) de un gen de la variante 1 de B7-H3 humana está representada por la SEQ ID NO: 1 en el listado de secuencias. Además, la secuencia de la SEQ ID NO: 1 se muestra en la figura 1.
La secuencia de aminoácidos de un ORF de un gen de la variante 2 de B7-H3 humana está representada por la SEQ ID NO: 2 en el listado de secuencias. Además, la secuencia de la SEQ ID NO: 2 se muestra en la figura 2.
Además, una proteína que consiste en una secuencia de aminoácidos en la que uno o varios aminoácidos se sustituyen, eliminan y/o añaden en cualquiera de las secuencias de aminoácidos descritas anteriormente de B7-H3 y también tiene una actividad biológica equivalente a la de la proteína está también incluida en B7-H3.
La variante 1 de B7-H3 humana madura de la que se ha eliminado la secuencia señal corresponde a una secuencia de aminoácidos que consiste en los restos de aminoácido 27 a 534 de la secuencia de aminoácidos representada por la SEC ID NO: 1. Además, la variante 2 de B7-H3 humana madura de la que se ha eliminado la secuencia señal corresponde a una secuencia de aminoácidos que consiste en los restos de aminoácido 27 a 316 de la secuencia de aminoácidos representada por la SEC ID NO: 2.
2. Producción del anticuerpo anti-B7-H3
El anticuerpo contra B7-H3 de la invención puede obtenerse inmunizando un animal con B7-H3 o un polipéptido arbitrario seleccionado entre la secuencia de aminoácidos de B7-H3, y recogiendo y purificando el anticuerpo producido in vivo de acuerdo con un procedimiento común. La especie biológica de B7-H3 que se usará como antígeno no está limitada al ser humano, y puede inmunizarse un animal con B7-H3 derivado de un animal que no sea humano, tal como un ratón o una rata. En este caso, examinando la reactividad cruzada entre un anticuerpo que se une al B7-H3 heterólogo obtenido y B7-H3 humano, puede seleccionarse un anticuerpo aplicable a una enfermedad humana.
Además, puede obtenerse un anticuerpo monoclonal a partir de un hibridoma establecido fusionando células productoras de anticuerpos que producen un anticuerpo contra B7-H3 con células de mieloma, de acuerdo con un procedimiento conocido (por ejemplo, Kohler y Milstein, Nature, (1975) 256, págs. 495-497; Kennet, R. ed., Monoclonal Antibodies, págs. 365-367, Plenum Press, N. Y. (1980).
Puede obtenerse B7-H3 que se usará como un antígeno expresando el gen de B7-H3 en una célula hospedadora usando ingeniería genética.
Específicamente, se produce un vector capaz de expresar el gen de B7-H3, y el vector resultante se transfecta en una célula hospedadora para expresar el gen, y a continuación, se purifica el B7-H3 expresado. En lo sucesivo en el presente documento, se describe específicamente un procedimiento para obtener un anticuerpo contra B7-H3.
(1) Preparación del antígeno
Los ejemplos del antígeno que se usará para producir el anticuerpo anti-B7-H3 incluyen B7-H3, un polipéptido que consiste en una secuencia de aminoácidos parcial que comprende al menos 6 aminoácidos consecutivos de B7-H3, y un derivado obtenido añadiendo una secuencia de aminoácidos o vehículo dado al mismo.
B7-H3 puede purificarse directamente a partir de tejidos tumorales o células tumorales humanas y usarse. Además, B7-H3 puede obtenerse sintetizándolo in vitro o produciéndolo en una célula hospedadora mediante ingeniería genética.
Con respecto a la ingeniería genética, específicamente, después de que el ADNc de B7-H3 se integre en un vector capaz de expresar ADNc de B7-H3, puede obtenerse B7-H3 sintetizándola en una solución que contiene una enzima, un sustrato y una sustancia energética requeridas para la transcripción y la traducción, o expresando B7-H3 en otra
célula hospedadora transformada procariota o eucariota.
Además, el antígeno también puede obtenerse como una proteína secretora expresando una proteína de fusión obtenida ligando el dominio extracelular de B7-H3, que es una proteína de membrana, a la región constante de un anticuerpo en un sistema hospedador-vector apropiado.
El ADNc de B7-H3 puede obtenerse mediante, por ejemplo, un llamado procedimiento de PCR en el que se realiza una reacción en cadena de la polimerasa (en lo sucesivo denominada "PCR") usando una biblioteca de ADNc que expresa ADNc de B7-H3 como molde y cebadores que amplifican específicamente el ADNc de B7-H3 (véase Saiki, R. K., y col., Science, (1988) 239, págs. 487-489).
Como síntesis in vitro del polipéptido, por ejemplo, puede indicarse como ejemplo el sistema Rapid Translation System (RTS) fabricado por Roche Diagnostics, Inc., aunque sin limitarse al mismo.
Los ejemplos de las células hospedadoras procariotas incluyen Escherichia coli y Bacillus subtilis. Con el fin de transformar las células hospedadoras con un gen diana, las células hospedadoras se transforman mediante un vector plasmídico que comprende un replicón, es decir, un origen de replicación derivado de una especie compatible con el hospedador, y una secuencia reguladora. Además, el vector tiene preferentemente una secuencia capaz de imponer selectividad fenotípica en la célula transformada.
Los ejemplos de las células hospedadoras eucariotas incluyen células de vertebrados, células de insectos y células de levadura. Como células de vertebrados, por ejemplo, a menudo se usan células COS de simio (Gluzman, Y., Cell, (1981) 23, págs. 175-182, ATCC CRL-1650), fibroblastos murinos NIH3T3 (ATCC n.° CRL-1658) y cepas deficientes en dihidrofolato reductasa (Urlaub, G. y Chasin, LA, Proc. Natl. Acad. Sci. EE. UU. (1980) 77, págs. 4126-4220) de células de ovario de hámster chino (células CHO, ATCC: CCL-61); y similares, sin embargo, las células no se limitan a éstas.
El transformante obtenido de este modo puede cultivarse de acuerdo con un procedimiento común, y mediante el cultivo del transformante, se produce un polipéptido diana intracelular o extracelularmente.
Un medio adecuado para usarlo para el cultivo puede seleccionarse entre diversos medios de cultivo usados comúnmente dependiendo de las células hospedadoras empleadas. Si se emplea Escherichia coli, por ejemplo, puede usarse un medio LB suplementado con un antibiótico tal como ampicilina o IPMG, según sea necesario.
Una proteína recombinante producida intracelular o extracelularmente por el transformante a través de dicho cultivo puede separarse y purificarse mediante cualquiera de diversos procedimientos de separación conocidos que utiliza la propiedad física o química de la proteína.
Los ejemplos específicos de los procedimientos incluyen tratamiento con un precipitante de proteína común, ultrafiltración, diversos tipos de cromatografía líquida tales como cromatografía de tamiz molecular (filtración en gel), cromatografía de adsorción, cromatografía de intercambio iónico y cromatografía de afinidad, diálisis y una combinación de los mismos.
Además, uniendo un identificador de seis restos de histidina a una proteína recombinante a expresar, la proteína puede purificarse eficazmente con una columna de afinidad de níquel. Como alternativa, uniendo la región Fc de IgG a una proteína recombinante a expresar, la proteína puede purificarse eficazmente con una columna de proteína A.
Combinando los procedimientos descritos anteriormente, puede producirse fácilmente una gran cantidad de un polipéptido diana con alto rendimiento y alta pureza.
(2) Producción del anticuerpo monoclonal anti-B7-H3
Los ejemplos del anticuerpo que se une específicamente a B7-H3 incluyen un anticuerpo monoclonal que se une específicamente a B7-H3, y un procedimiento de obtención del anticuerpo es tal como se describe a continuación.
La producción de un anticuerpo monoclonal generalmente requiere las siguientes etapas operativas de:
(a) purificar un biopolímero que se usará como antígeno;
(b) preparar células productoras de anticuerpos inmunizando un animal mediante inyección del antígeno, recoger la sangre, ensayar su título de anticuerpos para determinar cuándo se escinde el bazo;
(c) preparar células de mieloma (en lo sucesivo denominadas como "mieloma");
(d) fusionar las células productoras de anticuerpos con el mieloma;
(e) cribar un grupo de hibridomas que producen un anticuerpo deseado;
(f) dividir los hibridomas en clones de una sola célula (clonación);
(g) opcionalmente, cultivar el hibridoma o criar un animal implantado con el hibridoma para producir una gran cantidad de un anticuerpo monoclonal;
(h) examinar el anticuerpo monoclonal producido de este modo para determinar la actividad biológica y la especificidad de unión, o evaluar el mismo para determinar propiedades como un reactivo etiquetado; y similares.
En lo sucesivo en el presente documento, el procedimiento de producción de un anticuerpo monoclonal se describirá en detalle siguiendo las etapas anteriores, sin embargo, el procedimiento no se limita a esto, y, por ejemplo, pueden usarse células productoras de anticuerpos diferentes de células esplénicas y mieloma.
(a) Purificación de antígeno
Como antígeno, puede usarse B7-H3 preparada mediante el procedimiento descrito anteriormente o un péptido parcial de la misma.
Además, también puede usarse como el antígeno una fracción de membrana preparada a partir de células recombinantes que expresan B7-H3 o las propias células recombinantes que expresan B7-H3, y también un péptido parcial de la proteína de la invención químicamente sintetizado mediante un procedimiento conocido por los expertos en la materia.
(b) Preparación de células productoras de anticuerpos
El antígeno obtenido en la etapa (a) se mezcla con un adyuvante tal como adyuvante completo o incompleto de Freund o sulfato de aluminio y potasio y la mezcla resultante se usa como inmunógeno para inmunizar un animal experimental. Como animal experimental, puede usarse sin ningún problema cualquier animal usado en un procedimiento de producción de hibridoma conocido. Específicamente, por ejemplo, puede usarse un ratón, una rata, una cabra, vacas, un caballo o similar. Sin embargo, desde el punto de vista de la facilidad de disponibilidad de las células de mieloma para fusionarse con las células productoras de anticuerpos extraídas, se usa preferentemente un ratón o una rata como animal a inmunizar.
Además, la cepa de un ratón o una rata que se usará no está particularmente limitada y, en el caso de un ratón, por ejemplo, pueden usarse diversas cepas tales como A, AKR, BALB/c, BDP, BA, CE, C3H, 57BL, C57BL, C57L, d Ba , FL, HTH, HT1, LP, NZB, NZW, RF, R III, SJL, SWR, WB y 129 y similares, y en el caso de una rata, por ejemplo, pueden usarse Wistar, Low, Lewis, Sprague, Dawley, ACI, BN, Fischer y similares.
Estos ratones y ratas están disponibles en el mercado de criadores/distribuidores de animales experimentales, por ejemplo, CLEA Japan, Inc. y Charles River Laboratories Japan, Inc.
Entre estos, teniendo en cuenta la compatibilidad de fusión con células de mieloma descritas a continuación, en el caso de un ratón, la cepa BALB/c, y en el caso de una rata, las cepas Wistar y Low se prefieren particularmente como animal a inmunizar.
Además, teniendo en cuenta la homología antigénica entre seres humanos y ratones, También se prefiere usar un ratón que tiene función biológica reducida para eliminar autoanticuerpos, es decir, un ratón con una enfermedad autoinmunitaria.
La edad de dicho ratón o rata en el momento de la inmunización es, preferentemente, de 5 a 12 semanas de edad, más preferentemente, de 6 a 8 semanas de edad.
Con el fin de inmunizar un animal con B7-H3 o un recombinante de la misma, por ejemplo, puede usarse un procedimiento conocido descrito en detalle en, por ejemplo, Weir, D. M., Handbook of Experimental Immunology Vol. I. II. III., Blackwell Scientific Publications, Oxford (1987), Kabat, E. A. y Mayer, M. M., Experimental Immunochemistry, Charles C Thomas Publisher Springfield, Illinois (1964) o similares.
Entre todos estos procedimientos de inmunización, un procedimiento específico preferido en la invención es, por ejemplo, de la siguiente manera.
Es decir, en primer lugar, una fracción de proteína de membrana que sirve como antígeno o células a las que se hizo expresar el antígeno se administran por vía intradérmica o intraperitoneal a un animal.
Sin embargo, se prefiere la combinación de ambas vías de administración para aumentar la eficacia de inmunización y, cuando la administración intradérmica se realiza en la primera mitad y la administración intraperitoneal se realiza en la segunda mitad o solo en la última dosificación, la eficacia de la inmunización puede aumentarse particularmente. El programa de administración del antígeno varía dependiendo del tipo de animal a inmunizar, diferencia individual o similares. Sin embargo, en general, es preferido un programa de administración en el que la frecuencia de administración del antígeno es de 3 a 6 veces y el intervalo de dosificación es de 2 a 6 semanas, y es más preferido un programa de administración en el que la frecuencia de administración del antígeno es de 3 a 4 veces y el intervalo de dosificación es de 2 a 4 semanas.
Además, La dosis del antígeno varía dependiendo del tipo de animal, diferencias individuales o similares, sin embargo, la dosis se establece, en general, en de 0,05 a 5 mg, preferentemente de aproximadamente 0,1 a 0,5 mg.
Se realiza una inmunización de refuerzo de 1 a 6 semanas, preferentemente de 2 a 4 semanas, más preferentemente de 2 a 3 semanas después de la administración del antígeno tal como se ha descrito anteriormente.
La dosis del antígeno en el momento de realizar la inmunización de refuerzo varía dependiendo del tipo o tamaño del animal o similar, sin embargo, en el caso de, por ejemplo, un ratón, la dosis generalmente se establece en de 0,05 a 5 mg, preferentemente de 0,1 a 0,5 mg, más preferentemente de aproximadamente 0,1 a 0,2 mg.
Las células esplénicas o linfocitos que incluyen células productoras de anticuerpos se extirpan asépticamente del animal inmunizado de 1 a 10 días, preferentemente de 2 a 5 días, más preferentemente de 2 a 3 días después de la inmunización de refuerzo. En este momento, se mide el título del anticuerpo y, si se usa un animal que tiene un título de anticuerpos suficientemente aumentado como fuente de suministro de las células productoras de anticuerpos, el procedimiento posterior puede llevarse a cabo de manera más eficiente.
Los ejemplos del procedimiento de medición del título de anticuerpos que se usarán aquí incluyen un procedimiento RIA y un procedimiento ELISA, pero el procedimiento no está limitado a estos.
Por ejemplo, si se emplea un procedimiento ELISA, la medición del título de anticuerpos en la invención puede llevarse a cabo de acuerdo con los procedimientos que se describen a continuación.
En primer lugar, un antígeno purificado o parcialmente purificado se adsorbe a la superficie de una fase sólida tal como una placa de 96 pocillos para ELISA, y la superficie de la fase sólida que no tiene antígeno adsorbido en ella se cubre con una proteína no relacionada con el antígeno tal como albúmina de suero bovino (en lo sucesivo denominada "BSA"). Después de lavar la superficie, ésta se pone en contacto con una muestra diluida sucesivamente (por ejemplo, suero de ratón) como anticuerpo primario para permitir que el anticuerpo en la muestra se una al antígeno.
Además, como anticuerpo secundario, se añade un anticuerpo etiquetado con una enzima contra un anticuerpo de ratón y se permite que se una al anticuerpo de ratón. Después del lavado, se añade un sustrato para la enzima y se mide un cambio en la absorbancia que se produce debido al desarrollo del color inducido por la degradación del sustrato o similar y el título del anticuerpo se calcula basándose en la medición.
La separación de las células productoras de anticuerpos de las células esplénicas o linfocitos del animal inmunizado puede llevarse a cabo de acuerdo con un procedimiento conocido (por ejemplo, Kohler y col., Nature (1975), 256, pág.
495; Kohler y col., Eur. J. Immunol. (1977), 6, pág. 511; Milstein y col., Nature (1977), 266, pág. 550; Walsh, Nature (1977), 266, pág. 495). Por ejemplo, en el caso de las células esplénicas, puede emplearse un procedimiento general en el que las células productoras de anticuerpos se separan homogeneizando el bazo para obtener las células mediante filtración con una malla de acero inoxidable y suspendiendo las células en Medio Mínimo Esencial (MEM) de Eagle.
(c) Preparación de células de mieloma (en lo sucesivo denominadas como "mieloma")
Las células de mieloma que se usarán para la fusión celular no están particularmente limitadas y las células adecuadas pueden seleccionarse de líneas celulares conocidas. Sin embargo, teniendo en cuenta la conveniencia cuando un hibridoma se selecciona de entre células fusionadas, se prefiere usar una cepa deficiente en HGPRT (hipoxantinaguanina fosforribosil transferasa) cuyo procedimiento de selección se ha establecido.
De manera más específica, los ejemplos de la cepa deficiente en HGPRT incluyen X63-Ag8 (X63), NS1-ANS/1(NS1), P3X63-Ag8.U1(P3U1), X63-Ag8.653(X63.653), SP2/0-Ag14(SP2/0), MPC11-45.6TG1.7(45.6TG), FO, S149/5XXO y BU.1 derivadas de ratones; 210.RSY3.Ag.1.2.3(Y3) derivada de ratas; y U266AR(SKO-007), GM1500-GTG-A12(GM1500), UC729-6, LICR-LOW-HMy2(HMy2) y 8226AR/NIP4-1(NP41) derivadas de seres humanos. Estas cepas deficientes en HGPRT están disponibles de, por ejemplo, la Colección Americana de Cultivos Tipo (ATCC) o similar.
Estas cepas celulares se subcultivan en un medio apropiado tal como un medio de 8-azaguanina [un medio obtenido añadiendo 8-azaguanina a un medio RPMI 1640 suplementado con glutamina, 2-mercaptoetanol, gentamicina y suero de ternera fetal (en lo sucesivo denominado "FCS")], medio de Dulbecco modificado por Iscove (en lo sucesivo denominado" IMDM "), o medio de Eagle modificado por Dulbecco (en lo sucesivo denominado" DMEM "). En este caso, de 3 a 4 días antes de realizar la fusión celular, las células se subcultivan en un medio normal [por ejemplo, un medio ASF104 (fabricado por Ajinomoto Co., Ltd.) que contenía el 10 % de FCS] para garantizar no menos de 2 x 107 células el día de la fusión celular.
(d) Fusión celular
La fusión entre las células productoras de anticuerpos y las células de mieloma puede realizarse apropiadamente de acuerdo con un procedimiento conocido (Weir, DM Handbook of Experimental Immunology Vol. I. II. III., Blackwell Scientific Publications, Oxford (1987), Kabat, E. A. y Mayer, M. M., Experimental Immunochemistry, Charles C Thomas Publisher, Springfield, Illinois (1964), etc.), en condiciones tales que la tasa de supervivencia de las células no se reduzca excesivamente.
Como dicho procedimiento, por ejemplo, puede usarse un procedimiento químico en el que las células productoras de anticuerpos y las células de mieloma se mezclan en una solución que contiene un polímero tal como polietilenglicol a alta concentración, un procedimiento físico que usan estimulación eléctrica, o similar. Entre estos procedimientos, un
ejemplo específico del procedimiento químico es tal como se describe a continuación.
Es decir, en el caso en el que se usa polietilenglicol en la solución que contiene un polímero a alta concentración, las células productoras de anticuerpos y las células de mieloma se mezclan en una solución de polietilenglicol que tiene un peso molecular de 1500 a 6000, más preferentemente de 2000 a 4000 a una temperatura de 30 a 40 °C, preferentemente de 35 a 38 °C durante de 1 a 10 minutos, preferentemente de 5 a 8 minutos.
(e) Selección de un grupo de hibridomas
El procedimiento de selección de hibridomas obtenidos mediante la fusión celular descrita anteriormente no está particularmente limitado. Normalmente, se usa un procedimiento de selección HAT (hipoxantina, aminopterina, timidina) (Kohler y col., Nature (1975), 256, pág. 495; Milstein y col., Nature (1977), 266, pág. 550).
Este procedimiento es eficaz cuando se obtienen hibridomas usando células de mieloma de una cepa deficiente en HGPRT que no pueden sobrevivir en presencia de aminopterina.
Es decir, cultivando células no fusionadas e hibridomas en un medio HAT, solo se permite selectivamente sobrevivir y proliferar a los hibridomas resistentes a la aminopterina.
(f) División en clones de una sola célula (clonación)
Como procedimiento de clonación para hibridomas, puede usarse un procedimiento conocido tal como un procedimiento de metilcelulosa, un procedimiento de agarosa suave o un procedimiento de dilución limitante (véase, por ejemplo, Barbara, B. M. y Stanley, M. S.: Selected Methods in Cellular Immunology, W. H. Freeman and Company, San Francisco (1980)). Entre estos procedimientos, particularmente, se prefiere un procedimiento de cultivo tridimensional tal como un procedimiento de metilcelulosa. Por ejemplo, el grupo de hibridomas producidos por fusión celular se suspenden en un medio de metilcelulosa tal como ClonaCell-HY Selection Medium D (fabricado por StemCell Technologies, Inc., n° 03804) y se cultiva. A continuación, se recogen las colonias de hibridoma formadas, con lo que pueden obtenerse hibridomas monoclonales. Las colonias de hibridoma recogidas respectivas se cultivan, y se selecciona un hibridoma que se ha confirmado que tiene un título de anticuerpo estable en un sobrenadante de cultivo de hibridoma obtenido como una cepa de hibridoma productor de anticuerpo monoclonal B7-H3.
Los ejemplos de la cepa de hibridoma establecida de este modo incluyen el hibridoma M30 de B7-H3. En la presente memoria descriptiva, un anticuerpo producido por el hibridoma M30 de B7-H3 se denomina "anticuerpo M30" o simplemente "M30".
La cadena pesada del anticuerpo M30 tiene una secuencia de aminoácidos representada por la SEQ ID NO: 20 en el listado de secuencias. Además, la cadena ligera del anticuerpo M30 tiene una secuencia de aminoácidos representada por la SEQ ID NO: 21 en el listado de secuencias. En la secuencia de aminoácidos de la cadena pesada representada por la SEQ ID NO: 20 en el listado de secuencias, una secuencia de aminoácidos que consiste en los restos de aminoácido 1 a 19 es una secuencia señal, una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 es una región variable, y una secuencia de aminoácidos que consiste en los restos de aminoácido 142 a 471 es una región constante. Además, en la secuencia de aminoácidos de la cadena ligera representada por la SEQ ID NO: 21 en el listado de secuencias, una secuencia de aminoácidos que consiste en los restos de aminoácido 1 a 22 es una secuencia señal, una secuencia de aminoácidos que consiste en los restos de aminoácido 23 a 130 es una región variable, y la secuencia de aminoácidos que consiste en los restos de aminoácido 131 a 235 es una región constante.
(g) Preparación del anticuerpo monoclonal mediante cultivo de hibridoma
Cultivando el hibridoma seleccionado de este modo, puede obtenerse de manera eficiente un anticuerpo monoclonal. Sin embargo, antes del cultivo, se prefiere realizar el cribado de un hibridoma que produce un anticuerpo monoclonal diana.
En dicho cribado, puede emplearse un procedimiento conocido.
La medición del título de anticuerpos en la invención puede llevarse a cabo, por ejemplo, mediante un procedimiento ELISA explicado en el punto (b) descrito anteriormente.
El hibridoma obtenido mediante el procedimiento descrito anteriormente puede almacenarse en un estado congelado en nitrógeno líquido o en un congelador a -80 °C o menos.
Después de la finalización de la clonación, el medio se cambia de un medio HT a un medio normal, y el hibridoma se cultiva.
El cultivo a gran escala se lleva a cabo mediante cultivo de rotación usando una botella de cultivo grande o mediante un cultivo giratorio. A partir del sobrenadante obtenido mediante el cultivo a gran escala, puede obtenerse un anticuerpo monoclonal que se une específicamente a la proteína de la invención por purificación usando un procedimiento conocido por los expertos en la materia, tales como filtración en gel.
Además, el hibridoma se inyecta en la cavidad abdominal de un ratón de la misma cepa que el hibridoma (por ejemplo, la BALB/c descrita anteriormente) o un ratón Nu/Nu para proliferar el hibridoma, por lo que puede obtenerse la ascitis que contiene una gran cantidad del anticuerpo monoclonal de la invención.
En el caso en el que el hibridoma se administra en la cavidad abdominal, si se administra un aceite mineral tal como 2,6,10,14-tetrametil pentadecano (pristano) de 3 a 7 días antes del mismo, puede obtenerse una mayor cantidad de ascitis.
Por ejemplo, un inmunosupresor se inyecta previamente en la cavidad abdominal de un ratón de la misma cepa que el hibridoma para inactivar los linfocitos T. 20 días después, de 106 a 107 células de clones de hibridoma se suspenden en un medio libre de suero (0,5 ml) y la suspensión se administra en la cavidad abdominal del ratón. En general, cuando el abdomen se expande y se llena con la ascitis, la ascitis se recoge del ratón. Mediante este procedimiento, el anticuerpo monoclonal puede obtenerse a una concentración que es aproximadamente 100 veces o mucho mayor que en la solución de cultivo.
El anticuerpo monoclonal obtenido mediante el procedimiento descrito anteriormente puede purificarse mediante un procedimiento descrito en, por ejemplo, Weir, D. M.: Handbook of Experimental Immunology vol. I, II, III, Blackwell Scientific Publications, Oxford (1978).
El anticuerpo monoclonal obtenido de este modo tiene una alta especificidad por el antígeno para B7-H3.
(h) Ensayo de anticuerpo monoclonal
El isotipo y la subclase del anticuerpo monoclonal obtenido de este modo pueden determinarse de la siguiente manera.
En primer lugar, los ejemplos del procedimiento de identificación incluyen un procedimiento de Ouchterlony, un procedimiento ELISA y un procedimiento RIA.
Un procedimiento de Ouchterlony es sencillo, pero cuando la concentración del anticuerpo monoclonal es baja, se requiere una operación de condensación.
Por otra parte, cuando se utiliza un procedimiento ELISA o un procedimiento RIA, haciendo reaccionar directamente el sobrenadante del cultivo con una fase sólida adsorbida al antígeno y usando anticuerpos correspondientes a diversos tipos de isotipos y subclases de inmunoglobulinas como anticuerpos secundarios, pueden identificarse el isotipo y la subclase del anticuerpo monoclonal.
Además, como un procedimiento más sencillo, también puede usarse un kit de identificación disponible en el mercado (por ejemplo, Mouse Typer Kit fabricado por Bio-Rad Laboratories, Inc.) o similar.
Además, la determinación cuantitativa de una proteína puede realizarse mediante el procedimiento de Folin Lowry y un procedimiento de cálculo basado en la absorbancia a 280 nm [1,4 (DO 280) = Inmunoglobulina 1 mg/ml].
Además, incluso cuando el anticuerpo monoclonal se obtiene por separado e independientemente realizando nuevamente las etapas de (a) a (h) en (2), es posible obtener un anticuerpo que tenga una actividad citotóxica equivalente a la del anticuerpo M30. Como ejemplo de dicho anticuerpo, puede indicarse como ejemplo un anticuerpo que se une al mismo epítopo que el anticuerpo M30. El M30 reconoce un epítopo en el dominio IgC1 o IgC2, que es un dominio en el dominio extracelular de B7-H3, y se une al dominio IgC1 o al dominio IgC2 o a ambos. Por lo tanto, como el epítopo para el anticuerpo de la invención, particularmente, puede indicarse como ejemplo un epítopo presente en el dominio IgC1 o IgC2 de B7-H3. Si un anticuerpo monoclonal recién producido se une a un péptido parcial o a una estructura terciaria parcial a la que se une el anticuerpo M30, puede determinarse que el anticuerpo monoclonal se une al mismo epítopo que el anticuerpo M30. Además, confirmando que el anticuerpo monoclonal compite con el anticuerpo M30 por la unión a B7-H3 (es decir, el anticuerpo monoclonal inhibe la unión entre el anticuerpo M30 y B7-H3), puede determinarse que el anticuerpo monoclonal se une al mismo epítopo que el anticuerpo M30 incluso si no se ha determinado la secuencia o estructura específica del epítopo. Cuando se confirma que el anticuerpo monoclonal se une al mismo epítopo que el anticuerpo M30, se espera fuertemente que el anticuerpo monoclonal tenga una actividad citotóxica equivalente a la del anticuerpo M30.
(3) Otros anticuerpos
El anticuerpo de la invención incluye no solamente el anticuerpo monoclonal descrito anteriormente contra B7-H3 sino también un anticuerpo recombinante obtenido mediante modificación artificial con el fin de disminuir la antigenicidad heteróloga en seres humanos, tal como un anticuerpo quimérico, un anticuerpo humanizado y un anticuerpo humano. Estos anticuerpos pueden producirse usando un procedimiento conocido.
Como el anticuerpo quimérico, puede indicarse como ejemplo un anticuerpo en el que las regiones variable y constante de anticuerpo se derivan de diferentes especies, por ejemplo, un anticuerpo quimérico en el que una región variable de anticuerpo derivada de ratón o rata se conecta a una región constante de anticuerpo derivada de humano (véase Proc. Natl. Acad. Sci. EE. UU., 81,6851-6855, (1984).
Como el anticuerpo humanizado, puede indicarse como ejemplo un anticuerpo obtenido integrando solamente una región determinante de complementariedad (CDR) en un anticuerpo derivado de un ser humano (véase Nature (1986) 321, págs. 522-525), y un anticuerpo obtenido injertando una parte de los restos de aminoácido del marco así como la secuencia de CDR para un anticuerpo humano mediante un procedimiento de injerto de CDR (documento WO 90/07861).
Sin embargo, el anticuerpo humanizado derivado del anticuerpo M30 no está limitado a un anticuerpo humanizado específico, siempre que el anticuerpo humanizado tenga los 6 tipos de secuencias de CDR del anticuerpo M30 y tenga una actividad antitumoral. La región variable de cadena pesada del anticuerpo M30 tiene CDRH1 (NYVMH) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 3 en el listado de secuencias, CDRH2 (YINPYNDDVKYNEKFKG) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 4, en el listado de secuencias, y CDRH3 (WGYYGSPLYYFDY) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 5 en el listado de secuencias. Además, la región variable de cadena ligera del anticuerpo M30 tiene CDRL1 (RASSRLIYMH) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 6 en el listado de secuencias, CDRL2 (ATSNLAS) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 7, en el listado de secuencias, y CDRL3 (QQWNSNPPT) que consiste en una secuencia de aminoácidos representada por la SEQ ID NO: 8 en el listado de secuencias.
Como un ejemplo del anticuerpo humanizado de un anticuerpo de ratón M30, puede indicarse como ejemplo una combinación arbitraria de una cadena pesada que comprende una región variable de cadena pesada que consiste en una cualquiera de (1) una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 9, 10, 11 o 12 en el listado de secuencias, (2) una secuencia de aminoácidos que tiene una homología de al menos el 95 % o más con la secuencia de aminoácidos (1) descrita anteriormente, y (3) una secuencia de aminoácidos en la que uno o varios aminoácidos secuencia de aminoácidos (1) descrita anteriormente se eliminan, se sustituyen o se añaden y una cadena ligera que comprende una región variable de cadena ligera que consiste en cualquiera de (4) una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 13, 14, 15, 16, 17, 18 o 19 en el listado de secuencias, (5) una secuencia de aminoácidos que tiene una homología de al menos el 95 % o más con secuencia de aminoácidos (4) descrita anteriormente, y (6) una secuencia de aminoácidos en la que uno o varios aminoácidos en la secuencia de aminoácidos (4) descrita anteriormente se eliminan, se sustituyen o se añaden.
El término "varios", tal como se usa en el presente documento, se refiere a de 1 a 10, 1 a 9, 1 a 8, 1 a 7, 1 a 6, 1 a 5, 1 a 4, 1 a 3, o 1 o 2.
Como la sustitución de aminoácidos en la presente memoria descriptiva, se prefiere una sustitución conservativa de aminoácidos. La sustitución conservativa de aminoácidos se refiere a una sustitución que se produce dentro de un grupo de aminoácidos relacionados con las cadenas laterales de aminoácidos. Los grupos de aminoácidos preferidos son los siguientes: un grupo ácido (ácido aspártico y ácido glutámico); un grupo básico (lisina, arginina e histidina); un grupo no polar (alanina, valina, leucina, isoleucina, prolina, fenilalanina, metionina y triptófano); y una familia polar no cargada (glicina, asparagina, glutamina, cisteína, serina, treonina y tirosina). Grupos de aminoácidos más preferidos son los siguientes: un grupo hidroxi alifático (serina y treonina); un grupo que contiene amida (asparagina y glutamina); un grupo alifático (alanina, valina, leucina e isoleucina); y un grupo aromático (fenilalanina, triptófano y tirosina). Dicha sustitución de aminoácido se realiza preferentemente dentro de un intervalo que no altera las propiedades de una sustancia que tiene la secuencia de aminoácidos original.
Como un anticuerpo que tiene una combinación preferida de una cadena pesada y una cadena ligera descrito anteriormente, un anticuerpo que consiste en una cadena pesada que comprende una región variable de cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que comprende una región variable de cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 14; un anticuerpo que consiste en una cadena pesada que comprende una región variable de cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 15; un anticuerpo que consiste en una cadena pesada que comprende una región variable de cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 16; un anticuerpo que consiste en una cadena pesada que comprende una región variable de cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 17; un anticuerpo que consiste en una cadena pesada que comprende una región variable de cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de cadena ligera que consiste en una
secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 18; un anticuerpo que consiste en una cadena pesada que comprende una región variable de cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 9 y una cadena ligera que comprende una región variable de cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 19; un anticuerpo que consiste en una cadena pesada que comprende una región variable de cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 12 y una cadena ligera que comprende una región variable de cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que comprende una región variable de cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 12 y una cadena ligera que comprende una región variable de cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 14; un anticuerpo que consiste en una cadena pesada que comprende una región variable de cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 12 y una cadena ligera que comprende una región variable de cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 15; y un anticuerpo que consiste en una cadena pesada que comprende una región variable de cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 de la SEQ ID NO: 12 y una cadena ligera que comprende una región variable de cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 de la SEQ ID NO: 16 pueden indicarse como ejemplo.
Además, como un anticuerpo que tiene una combinación más preferida de una cadena pesada y una cadena ligera descrito anteriormente, un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 471 de la s Eq ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 233 de la SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 233 de la SEQ ID NO: 14; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 233 de la SEQ ID NO: 15; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 233 de la SEQ ID NO: 16; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 233 de la SEQ ID NO: 17; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 233 de la SEQ ID NO: 18; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 471 de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 233 de la SEQ ID NO: 19; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 471 de la SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 233 de la SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 471 de la s Eq ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 233 de la SEQ ID NO: 14; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 471 de la SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 233 de la SEQ ID NO: 15; y un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 471 de la SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 233 de la SEQ ID NO: 16 pueden indicarse como ejemplo.
Además, como un anticuerpo que tiene otra combinación más preferida de una cadena pesada y una cadena ligera descrito anteriormente, un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID NO: 14; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID NO: 15; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID NO: 16; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID NO: 17; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID
NO: 18; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID NO: 19; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID NO: 13; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID NO: 14; un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID NO: 15; y un anticuerpo que consiste en una cadena pesada que consiste en una secuencia de aminoácidos de la SEQ ID NO: 12 y una cadena ligera que consiste en una secuencia de aminoácidos de la SEQ ID NO: 16 pueden indicarse como ejemplo.
Combinando una secuencia que tiene una alta homología con la secuencia de aminoácidos de cadena pesada descrita anteriormente con una secuencia que tiene una alta homología con la secuencia de aminoácidos de cadena ligera descrita anteriormente, es posible seleccionar un anticuerpo que tenga una actividad citotóxica equivalente a la de cada uno de los anticuerpos descritos anteriormente. Dicha homología es generalmente una homología del 80 % o más, preferentemente una homología del 90 % o más, más preferentemente una homología del 95 % o más, lo más preferentemente una homología del 99 % o más. Además, combinando una secuencia de aminoácidos en la que uno a varios restos de aminoácido se sustituyen, eliminan o añaden en la secuencia de aminoácidos de cadena pesada o cadena ligera, también es posible seleccionar un anticuerpo que tenga una actividad citotóxica equivalente a la de cada uno de los anticuerpos descritos anteriormente.
La homología entre dos secuencias de aminoácidos puede determinarse usando parámetros por defecto del algoritmo Blast, versión 2.2.2 (Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller y David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25: 3389-3402). El algoritmo Blast también puede usarse a través de Internet accediendo al sitio www.ncbi.nlm.nih.gov/blast.
En la secuencia de aminoácidos de la cadena pesada representada por la SEQ ID NO: 9, 10, 11 o 12 en el listado de secuencias, una secuencia de aminoácidos que consiste en los restos de aminoácido 1 a 19 es una secuencia señal, una secuencia de aminoácidos que consiste en los restos de aminoácido 20 a 141 es una región variable, y una secuencia de aminoácidos que consiste en los restos de aminoácido 142 a 471 es una región constante. La secuencia de la SEQ ID NO: 9, 10, 11 y 12 se muestran en las figuras 3, 4, 5 y 6, respectivamente.
Además, en la secuencia de aminoácidos de la cadena ligera representada por la SEQ ID NO: 13, 14, 15, 16, 17, 18 o 19 en el listado de secuencias, una secuencia de aminoácidos que consiste en los restos de aminoácido 1 a 20 es una secuencia señal, una secuencia de aminoácidos que consiste en los restos de aminoácido 21 a 128 es una región variable, y una secuencia de aminoácidos que consiste en los restos de aminoácido 129 a 233 es una región constante. La secuencia de la SEQ ID NO: 13, 14, 15, 16, 17, 18 y 19 se muestran en las figuras 7, 8, 9, 10, 11, 12 y 13, respectivamente.
Además, el anticuerpo de la invención incluye un anticuerpo humano que se une al mismo epítopo que el anticuerpo M30. Un anticuerpo humano anti-B7-H3 se refiere a un anticuerpo humano que tiene solamente una secuencia de un anticuerpo derivado de un cromosoma humano. El anticuerpo humano anti-B7-H3 puede obtenerse mediante un procedimiento que usa un ratón productor de anticuerpos humanos que tiene un fragmento de cromosoma humano que comprende genes de cadena pesada y ligera de un anticuerpo humano (véase Tomizuka, K. y col., Nature Genetics (1997) 16, págs. 133-143; Kuroiwa, Y. y col., Nucl. Acids Res. (1998) 26, págs. 3447-3448; Yoshida, H. y col., Animal Cell Technology: Basic and Applied Aspects vol. 10, págs. 69-73 (Kitagawa, Y., Matuda, T. e Iijima, S. eds.), Kluwer Academic Publishers, 1999; Tomizuka, K. y col., Proc. Natl. Acad. Sci. EE. UU. (2000) 97, págs. 722 727, etc.).
Dicho ratón productor de anticuerpos humanos puede crearse específicamente de la siguiente manera. Un animal genéticamente modificado en el que se han alterado loci génicos de cadena pesada y ligera de inmunoglobulina endógena, y en su lugar, se han introducido loci génicos de cadena pesada y ligera de inmunoglobulina humana a través de un vector cromosómico artificial de levadura (YAC) o similar se crea produciendo un animal genomanipulado y un animal transgénico y haciendo aparearse a estos animales.
Además, de acuerdo con una técnica de ADN recombinante, usando ADNc que codifican cada una de dichas cadena pesada y cadena ligera de un anticuerpo humano, y preferentemente un vector que comprende dichos ADNc, se transforman células eucariotas, y se cultiva una célula transformante que produce un anticuerpo monoclonal humano recombinante, por lo que el anticuerpo también puede obtenerse del sobrenadante del cultivo.
En este caso, como hospedador, por ejemplo, pueden usarse células eucariotas, preferentemente células de mamífero tales como células CHO, linfocitos o células de mieloma.
Además, También se conoce un procedimiento para obtener un anticuerpo humano derivado de presentación en fagos seleccionado de una biblioteca de anticuerpos humanos (véase Wormstone, I. M. y col., Investigative Ophthalmology & Visual Science. (2002) 43 (7), págs. 2301-2308; Carmen, S. y col., Briefings in Functional Genomics and Proteomics
(2002), 1 (2), págs. 189-203; Siriwardena, D. y col., Ophthalmology (2002) 109 (3), págs. 427-431, etc.).
Por ejemplo, puede usarse un procedimiento de presentación en fagos en el que una región variable de un anticuerpo humano se expresa en la superficie de un fago como un anticuerpo monocatenario (scFv), y se selecciona un fago que se une a un antígeno (Nature Biotechnology (2005), 23, (9), págs. 1105-1116).
Analizando el gen del fago seleccionado basándose en la unión a un antígeno, puede determinarse una secuencia de ADN que codifica la región variable de un anticuerpo humano que se une a un antígeno.
Si se determina la secuencia de ADN de scFv que se une a un antígeno, puede obtenerse un anticuerpo humano preparando un vector de expresión que comprende la secuencia e introduciendo el vector en un hospedador apropiado para expresarlo (WO 92/01047, el documento WO 92/20791, el documento WO 93/06213, el documento WO 93/11236, el documento WO 93/19172, el documento WO 95/01438, el documento WO 95/15388, Annu. Rev. Immunol. (1994) 12, págs. 433-455, Nature Biotechnology (2005) 23 (9), págs. 1105-1116).
Si un anticuerpo humano recién producido se une a un péptido parcial o a una estructura terciaria parcial a la que se une el anticuerpo M30, puede determinarse que el anticuerpo humano se une al mismo epítopo que el anticuerpo M30. Además, confirmando que el anticuerpo humano compite con el anticuerpo M30 por la unión a B7-H3 (es decir, el anticuerpo humano inhibe la unión entre el anticuerpo M30 y B7-H3), puede determinarse que el anticuerpo humano se une al mismo epítopo que el anticuerpo M30 incluso si no se ha determinado la secuencia o estructura específica del epítopo. Cuando se confirma que el anticuerpo humano se une al mismo epítopo que el anticuerpo M30, se espera fuertemente que el anticuerpo humano tenga una actividad citotóxica equivalente a la del anticuerpo M30.
Los anticuerpos quiméricos, anticuerpos humanizados o anticuerpos humanos obtenidos mediante el procedimiento descrito anteriormente se evalúan para determinar su propiedad de unión a un antígeno mediante un procedimiento conocido o similares, y puede seleccionarse un anticuerpo preferido.
Como ejemplo de otro índice para su uso en la comparación de las propiedades de los anticuerpos, puede indicarse como ejemplo la estabilidad de los anticuerpos. El calorímetro diferencial de barrido (DSC) es un dispositivo capaz de medir de forma rápida y precisa una temperatura de punto medio de desnaturalización térmica (Tm) que se usará como un índice favorable de la estabilidad conformacional relativa de las proteínas. Midiendo los valores de Tm usando un DSC y comparando los valores, puede compararse una diferencia en la estabilidad térmica. Se sabe que la estabilidad en almacenamiento de los anticuerpos muestra cierta correlación con la estabilidad térmica de los mismos (Lori Burton, et. al., Pharmaceutical Development and Technology (2007) 12, págs. 265-273), y puede seleccionarse un anticuerpo preferido usando la estabilidad térmica como índice. Los ejemplos de otros índices para seleccionar anticuerpos incluyen las siguientes características: el rendimiento en una célula hospedadora apropiada es alto; y la agregabilidad en una solución acuosa es baja. Por ejemplo, un anticuerpo que muestra el rendimiento más alto no siempre muestra la estabilidad térmica más alta y, por lo tanto, es necesario seleccionar el anticuerpo más adecuado para la administración a seres humanos realizando una evaluación exhaustiva basada en los índices descritos anteriormente.
En la invención, también se incluye una variante modificada del anticuerpo. La variante modificada se refiere a una variante obtenida sometiendo al anticuerpo de la invención a una modificación química o biológica. Los ejemplos de la variante químicamente modificada incluyen variantes químicamente modificadas enlazando un resto químico a una cadena principal de aminoácido, variantes químicamente modificadas con una cadena de carbohidrato enlazada a N o enlazada a O, etc. Los ejemplos de la variante biológicamente modificada incluyen variantes obtenidas por modificación después de la traducción (como glucosilación enlazada a N o enlazada a O, procesamiento N- o C-terminal, desamidación, isomerización de ácido aspártico u oxidación de metionina), y variantes en las que se ha añadido un resto de metionina al extremo N al expresarse en una célula hospedadora procariota.
Además, un anticuerpo etiquetado para permitir la detección o el aislamiento del anticuerpo o un antígeno de la invención, por ejemplo, un anticuerpo etiquetado con una enzima, un anticuerpo etiquetado con fluorescencia y un anticuerpo etiquetado por afinidad también están incluidos en el significado de la variante modificada. Dicha variante modificada del anticuerpo de la invención es útil para mejorar la estabilidad y la retención sanguínea del anticuerpo original de la invención, reduciendo la antigenicidad del mismo, detectando o aislando dicho anticuerpo o un antígeno, y así sucesivamente.
Además, regulando la modificación de un glicano que está enlazado al anticuerpo de la invención (glucosilación, desfucosilación, etc.), es posible potenciar una actividad de citotoxicidad celular dependiente del anticuerpo. Como la técnica para regular la modificación de un glicano de anticuerpos, se conocen los documentos WO 99/54342, WO 00/61739, WO 02/31140, etc. Sin embargo, la técnica no está limitada a estas. En el anticuerpo de la invención, también está incluido un anticuerpo en el que la modificación de un glicano está regulada.
En el caso en el que se produce un anticuerpo aislando en primer lugar un gen de anticuerpo e introduciendo, a continuación, el gen en un hospedador apropiado, puede usarse una combinación de un hospedador apropiado y un vector de expresión apropiado. Los ejemplos específicos del gen de anticuerpo incluyen una combinación de un gen que codifica una secuencia de cadena pesada de un anticuerpo descrito en la presente memoria descriptiva y un gen que codifica una secuencia de cadena ligera del mismo. Cuando se transforma una célula hospedadora, es posible
insertar el gen de la secuencia de cadena pesada y el gen de la secuencia de cadena ligera en el mismo vector de expresión, y también en diferentes vectores de expresión por separado.
En el caso en el que se usan células eucariotas como hospedador, pueden usarse células animales, células vegetales y microorganismos eucariotas. Como las células animales, pueden indicarse como ejemplo células de mamífero, por ejemplo, células COS de simio (Gluzman, Y., Cell, (1981) 23, págs. 175-182, ATCC CRL-1650), fibroblastos murinos NIH3T3 (ATCC n.° CRL-1658), y cepas deficientes en dihidrofolato reductasa (Urlaub, G. y Chasin, LA, Proc. Natl. Acad. Sci. EE. UU. (1980) 77, págs. 4126-4220) de células de ovario de hámster chino (células CHO; ATCC: CCL-61).
En el caso en el que se usan células procariotas, por ejemplo, pueden indicarse como ejemplo Escherichia coli y Bacillus subtilis.
Introduciendo un gen de anticuerpo deseado en estas células mediante transformación, y cultivando las células transformadas de este modo in vitro, puede obtenerse el anticuerpo. En el procedimiento de cultivo descrito anteriormente, el rendimiento puede, algunas veces, variar dependiendo de la secuencia del anticuerpo y, por lo tanto, es posible seleccionar un anticuerpo que se produzca fácilmente como un producto farmacéutico usando el rendimiento como índice entre los anticuerpos que tienen una actividad de unión equivalente. Por lo tanto, en el anticuerpo de la invención, un anticuerpo obtenido mediante un procedimiento de producción de un anticuerpo, caracterizado por incluir una etapa de cultivar la célula hospedadora transformada y una etapa de recoger un anticuerpo deseado a partir de un producto cultivado obtenido en la etapa de cultivo también está incluido.
Se sabe que se elimina un resto de lisina en el extremo carboxilo de la cadena pesada de un anticuerpo producido en una célula de mamífero cultivada (Journal of Chromatography A, 705: 129-134 (1995)), y también se sabe que se eliminan dos restos de aminoácido (glicina y lisina) en el extremo carboxilo de la cadena pesada de un anticuerpo producido en una célula de mamífero cultivada y se amida un resto de prolina recién localizado en el extremo carboxilo (Analytical Biochemistry, 360: 75-83 (2007)). Sin embargo, dicha eliminación y modificación de la secuencia de cadena pesada no afecta la afinidad de unión al antígeno y la función efectora (la activación de un complemento, la citotoxicidad celular dependiente del anticuerpo, etc.) del anticuerpo. Por lo tanto, en la invención, también se incluye un anticuerpo sometido a dicha modificación, y una variante de eliminación en la que se han eliminado uno o dos aminoácidos en el extremo carboxilo de la cadena pesada, una variante obtenida por la amidación de la variante de eliminación (por ejemplo, una cadena pesada en la que se ha amidado el resto de prolina carboxilo terminal) y similares pueden indicarse como ejemplo. El tipo de variante de eliminación que tiene una eliminación en el extremo carboxilo de la cadena pesada del anticuerpo de acuerdo con la invención no se limita a las variantes anteriores siempre que se conserven la afinidad de unión al antígeno y la función efectora. Las dos cadenas pesadas que constituyen el anticuerpo de acuerdo con la invención pueden ser de un tipo seleccionado entre el grupo que consiste en una cadena pesada de longitud completa y la variante de eliminación descrita anteriormente, o pueden ser de dos tipos en combinación seleccionados a partir del mismo. La relación de la cantidad de cada variante de eliminación puede verse afectada por el tipo de células de mamífero cultivadas que producen el anticuerpo de acuerdo con la invención y las condiciones de cultivo, sin embargo, un caso en el que un resto de aminoácido en el extremo carboxilo se ha eliminado en ambos de las dos cadenas pesadas contenidas como componentes principales en el anticuerpo de acuerdo con la invención puede indicarse como ejemplo.
Puede indicarse como ejemplo un isótopo del anticuerpo de la invención, por ejemplo, IgG (IgG1, IgG2, IgG3, IgG4), y, preferentemente, puede indicarse como ejemplo IgG1 o IgG2.
Como la función del anticuerpo, generalmente una actividad de unión a antígeno, una actividad de neutralización de la actividad de un antígeno, una actividad de potenciación de la actividad de un antígeno, una actividad de citotoxicidad celular dependiente del anticuerpo (ADCC) y una actividad de citotoxicidad dependiente del complemento (CDC) pueden indicarse como ejemplo. La función del anticuerpo de la invención es una actividad de unión a B7-H3, preferentemente una actividad de fagocitosis mediada por células dependiente del anticuerpo (ADCP), más preferentemente una actividad citotóxica (actividad antitumoral) frente a célula tumoral mediada por una actividad ADCP. Además, el anticuerpo de la invención puede tener una actividad ADCC y/o una actividad CDC además de una actividad ADCP.
El anticuerpo obtenido puede purificarse hasta homogeneidad. La separación y purificación del anticuerpo pueden realizarse empleando un procedimiento convencional de separación y purificación de proteínas. Por ejemplo, el anticuerpo puede separarse y purificarse seleccionando y combinando adecuadamente cromatografía en columna, filtración en filtro, ultrafiltración, precipitación con sales, diálisis, electroforesis en gel de poliacrilamida preparativa, electroforesis de enfoque isoeléctrico y similares (Strategies for Protein Purification and Characterization: A Laboratory Course Manual, Daniel R. Marshak y col., eds., Cold Spring Harbor Laboratory Press (1996); Antibodies: A Laboratory Manual. Ed Harlow y David Lane, Cold Spring Harbor Laboratory (1988)), pero el procedimiento no está limitado a estos.
Los ejemplos de dicha cromatografía incluyen cromatografía de afinidad, cromatografía de intercambio iónico, cromatografía hidrófoba, cromatografía de filtración en gel, cromatografía de fase inversa y cromatografía de adsorción.
Dicha cromatografía puede realizarse empleando cromatografía líquida tal como HPLC o FPLC.
Como columna para usar en cromatografía de afinidad, pueden indicarse como ejemplo una columna de Proteína A y una columna de Proteína G. Por ejemplo, como una columna que usa una columna de Proteína A, pueden indicarse como ejemplo Hyper D, POROS, Sepharose FF (Pharmacia) y similares.
Además, usando un vehículo que tiene un antígeno inmovilizado sobre él, el anticuerpo también puede purificarse usando la propiedad de unión del anticuerpo al antígeno.
[Compuesto antitumoral]
Se explica el compuesto antitumoral que se conjugará con el conjugado de anticuerpo - fármaco. El compuesto antitumoral no está particularmente limitado si el mismo es un compuesto que tiene un efecto antitumoral y un grupo sustituyente o una estructura parcial que permite conectar con una estructura de enlazador. Cuando una parte o la totalidad de un enlazador se escinde en células tumorales, el resto de compuesto antitumoral se libera para mostrar el efecto antitumoral del compuesto antitumoral. Cuando el enlazador se escinde en una posición de conexión a fármaco, el compuesto antitumoral se libera en su estructura intrínseca para mostrar su efecto antitumoral intrínseco.
Los ejemplos del compuesto antitumoral pueden incluir doxorrubicina, daunorrubicina, mitomicina C, bleomicina, ciclocitidina, vincristina, vinblastina, metotrexato, agente antitumoral a base de platino (cisplatino o derivados del mismo), taxol o derivados del mismo, y camptotecina o derivados de la misma (agente antitumoral descrito en la patente japonesa abierta a inspección pública n.° 6-87746). En el conjugado de anticuerpo - fármaco, puede usarse preferentemente exatecán como un derivado de camptotecina (((1S,9S)-1-amino-9-etil-5-fluoro-2,3-dihidro-9-hidroxi-4-metil-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolina-10,13(9H,15H)-diona; mostrada en la siguiente fórmula).
[Fórmula 43]

Pese a tener un efecto antitumoral excelente, exatecán no se ha comercializado como un fármaco antitumoral. El compuesto puede obtenerse fácilmente mediante un procedimiento conocido y el grupo amino en la posición 1 puede usarse preferentemente como una posición de conexión a la estructura de enlazador. Además, aunque exatecán también puede liberarse en células tumorales mientras parte del enlazador sigue estando unida al mismo, este es un compuesto excelente que muestra un efecto antitumoral excelente incluso en tal caso.
Con respecto al conjugado de anticuerpo - fármaco, el número de moléculas de fármaco conjugadas por molécula de anticuerpo es un factor clave que tiene influencia sobre la eficacia y la seguridad. La producción del conjugado de anticuerpo - fármaco se realiza definiendo la condición de reacción incluyendo las cantidades de uso de materiales de partida y reactivos para la reacción con el fin de tener un número constante de moléculas de fármaco conjugadas, una mezcla que contiene diferentes números de moléculas de fármaco conjugadas se obtiene generalmente a diferencia de la reacción química de un compuesto de bajo peso molecular. El número de fármacos conjugados en una molécula de anticuerpo se expresa o especifica mediante el valor promedio, es decir, el número promedio de moléculas de fármaco conjugadas. A menos que específicamente se describa lo contrario como un principio, el número de moléculas de fármaco conjugadas significa un valor promedio excepto en un caso en el que este representa un conjugado de anticuerpo - fármaco que tiene un número específico de moléculas de fármaco conjugadas que se incluye en una mezcla de conjugado de anticuerpo - fármaco que tiene un diferente número de moléculas de fármaco conjugadas. El número de moléculas de exatecán conjugadas con una molécula de anticuerpo es controlable y, como un número promedio de moléculas de fármaco conjugadas por anticuerpo, pueden unirse aproximadamente de 1 a 10 exatecanos. Preferentemente, este es de 2 a 8 y, más preferentemente, de 3 a 8. Mientras tanto, un experto en la materia puede diseñar una reacción para conjugar un número requerido de moléculas de fármaco con una molécula de anticuerpo basándose en la descripción de los ejemplos de la presente solicitud y puede obtener un anticuerpo conjugado con un número controlado de moléculas de exatecán.
Debido a que exatecán tiene una estructura de camptotecina, se sabe que el equilibrio se desplaza a una estructura con un anillo de lactona cerrado (anillo cerrado) en un medio ácido acuoso (por ejemplo, pH 3 o así) pero este se desplaza a una estructura con un anillo de lactona abierto (anillo abierto) en un medio básico acuoso (por ejemplo, pH 10 o así). También se espera que un conjugado de fármaco que se introduce con un residuo de exatecán que se
corresponde con la estructura de anillo cerrado y la estructura de anillo abierto tenga el mismo efecto antitumoral, y no resulta necesario indicar que cualquiera de los mismos está dentro del ámbito de la presente divulgación.
[Estructura de enlazador]
Con respecto al conjugado de anticuerpo - fármaco, se explica la estructura de enlazador para conjugar un fármaco antitumoral con el anticuerpo. El enlazador tiene una estructura de la siguiente estructura:
-L1-L2-LP-NH-(CH2)n1-La-Lb-Lc-.
El anticuerpo está conectado al extremo terminal de L1 (extremo terminal opuesto a la conexión a L2), y el fármaco antitumoral está conectado al extremo terminal de Lc (extremo terminal opuesto a la conexión a Lb).
n1 representa un número entero de 0 a 6 y es preferentemente un número entero de 1 a 5 y, más preferentemente, de 1 a 3.
1. L1
L1 es un resto en el enlazador representado por la siguiente estructura:
-(Succinimid-3-il-N)-(CH2)n2-C(=O)-,
-CH2-C(=O)-NH-(CH2)n3-C(=O)-,
-C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-o
-C(=O)-(CH2)n4-C(=O)-En lo anterior, n2 es un número entero de 2 a 8, n3 es un número entero de 1 a 8, y n4 es un número entero de 1 a 8. En el enlazador que tiene una estructura representada por -(Succinimid-3-il-N)-(CH2)n2-C(=O)- de L1, "-(Succinimid-3-il-N)-" tiene una estructura representada por la siguiente fórmula:
[Fórmula 44]

La posición 3 de la estructura parcial anterior es una posición de conexión al anticuerpo. El enlace al anticuerpo en la posición 3 está caracterizado por unirse con formación de tioéter. Por otra parte, el átomo de nitrógeno en la posición 1 del resto de estructura está conectado al átomo de carbono de metileno que está presente dentro del enlazador que incluye la estructura. Específicamente, -(Succinimid-3-il-N)-(CH2)n2-C(=O)-L2- es una estructura representada por la siguiente fórmula (en el presente documento, "anticuerpo-S-" se origina a partir de un anticuerpo).
[
En la fórmula, n2 es un número entero de 2 a 8 y, preferentemente, de 2 a 5.
En el enlazador que tiene una estructura representada por - CH2-C(=O)-NH-(CH2)n3-C(=O)- de L1, n3 es un número entero de 1 a 8, preferentemente de 2 a 6. Este enlazador está conectado al anticuerpo en su átomo de carbono del metileno terminal y tiene la siguiente estructura para conectarse mediante formación de tioéter, al igual que con el enlazador anterior (en el presente documento, "anticuerpo-S-" se origina a partir de un anticuerpo). Anticuerpo-S-CH2-C(=O)-NH-(CH2)n3-C(=O)-L2-.
En el enlazador que tiene una estructura representada por - C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)- de L1, "-(N-li-3-diminiccuS)-" tiene una estructura representada por la siguiente fórmula:
[Fórmula 46]

En este resto de estructura, el átomo de nitrógeno en la posición 1 está conectado al átomo de carbono de metileno presente en la estructura de enlazador que contiene esta estructura. El átomo de carbono en la posición 3 está conectado al átomo de azufre terminal de -S-(CH2)n6-C(=O)- de L2 en el enlazador. Este resto -S-(CH2)n6-C(=O)- de L2 en el enlazador forma una estructura de enlazador combinada solo con -C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)- de L1 en el enlazador. En lo anterior, "-cic.Hex(1,4)-'' contenido en el enlazador representa un grupo 1,4-ciclohexileno. En el enlazador, -C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)- está conectado al anticuerpo con formación de enlace de amida en su carbono de carbonilo terminal (en el presente documento, "anticuerpo-NH-" se origina a partir de un anticuerpo).
[Fórmula 47]

El grupo amino del anticuerpo para esta formación de enlace de amida es el grupo amino terminal de una cadena lateral de un residuo de lisina en el anticuerpo o un grupo amino en el N terminal del anticuerpo. Dicho enlazador de una estructura puede conectarse formando un enlace de éster con el grupo hidroxi de un aminoácido en el anticuerpo que no sea tal enlace de amida.
El resto de estructura '-cic.Hex(1,4)-'' contenido en dicho enlazador puede ser un grupo alquileno cíclico saturado divalente que no sea el grupo 1,4-ciclohexileno, es decir, un grupo hidrocarburo saturado cíclico divalente tal como un grupo ciclobutileno, un grupo ciclopentileno, un grupo cicloheptaleno o un grupo ciclooctaleno, un grupo hidrocarburo aromático divalente tal como un grupo fenileno o un grupo naftileno, o un grupo heterocíclico divalente aromático, parcialmente saturado o saturado de 5 o 6 miembros que contiene 1 o 2 heteroátomos. Como alternativa, este resto puede ser un grupo alquileno divalente que tiene de 1 a 4 átomos de carbono. La conexión al grupo divalente puede tener lugar en posiciones adyacentes o en posiciones distantes.
En el enlazador que tiene una estructura representada por - C(=O)-(CH2)n4-C(=O)- como L1, n4 es un número entero de 1 a 8 y, preferentemente, de 2 a 6. Este enlazador también está conectado mediante formación de enlace de amida en su grupo carbonilo terminal con un grupo amino del anticuerpo, al igual que con los enlazadores mencionados anteriormente (véase la siguiente fórmula; en la estructura de la misma, "anticuerpo-NH-" se origina a partir de un anticuerpo).
Anticuerpo-NH-C(=O)-(CH2)n4-C(=O)-L2-.
Ejemplos específicos de L1 en el enlazador pueden incluir
-(Succinimid-3-il-N)-CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)--CH2C(=O)NH-CH2-C(=O)-,
-CH2C(=O)NH-CH2CH2-C(=O)--CH2C(=O)NH-CH2CH2CH2-C(=O)--CH2C(=O)NH-CH2CH2CH2CH2-C(=O)--CH2C(=O)NH-CH2CH2CH2CH2CH2-C(=O)--C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)
-C(=O)-aril-CH2-(N-li-3-diminiccuS)--C(=O)-cic.Het-CH2-(N-li-3-diminiccuS)--C(=O)-CH2CH2-C(=O)--C(=O)-CH2CH2CH2-C(=O)--C (=O)-CH2CH2CH2CH2-C(=O)--C(=O)-CH2CH2CH2CH2CH2-C(=O)--C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-.
(Arilo representa un grupo hidrocarburo aromático divalente, y cic.Het representa un grupo cíclico heterocíclico divalente).
2. L2
L2 es un enlazador representado por la siguiente estructura:
-NH-(CH2CH2O)n5-CH2-CH2-C(=O)-,
o
-S-(CH2)n6-C(=O)-,
L2 puede no estar presente, y en un caso de este tipo, L2 es un enlace sencillo. En lo anterior, n5 es un número entero de 1 a 6, y n6 es un número entero de 1 a 6.
En el enlazador que tiene una estructura de -NH-(CH2CH2O)n5-CH2-CH2-C(=O)- como L2, n5 es un número entero de 1 a 6 y, preferentemente, de 2 a 4. Este resto en el enlazador está conectado a L1 en su grupo amino terminal y está conectado a LP en su grupo carbonilo en el otro extremo terminal.
En el enlazador que tiene una estructura de -S-(CH2)n6-C(=O)- como L2, n6 es un número entero de 1 a 6 y, preferentemente, de 2 a 4.
Ejemplos específicos de L2 pueden incluir
-NH-CH2CH2O-CH2CH2-C(=O)-,
-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-,
-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-,
-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-,
-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-,
-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-.
Cuando L2 es -S-(CH2)n6-C(=O)-, L1 que se va a combinar con el mismo es -C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-. Ejemplos específicos de -L1-L2- pueden incluir
-C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-S-CH2-C(=O)--C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-S-CH2CH2-C(=O)--C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-S-CH2CH2CH2-C(=O)--C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-S-CH2CH2CH2CH2-C(=O)--C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-S-CH2CH2CH2CH2CH2-C(=O)--C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-S-CH2CH2CH2CH2CH2CH2-C(=O)-.
3. LP
El enlazador LP es un residuo de péptido de GGFG que consiste en 2 a 7 aminoácidos. Específicamente, consiste en un residuo de oligopéptido en el que 2 a 6 aminoácidos están enlazados por un enlace peptídico. El enlazador LP está conectado a L2 en su N terminal y está conectado al grupo amino del resto de -NH-(CH2)n1-La-Lb-Lc del enlazador en su C terminal. El aminoácido que constituye LP en el enlazador no está particularmente limitado, sin embargo, ejemplos
del mismo incluyen un L o un d aminoácido, preferentemente un L aminoácido. U puede ser un aminoácido que tenga una estructura tales como p-alanina, ácido £-aminocaproico o ácido Y-aminobutírico además de un a-aminoácido, además, puede ser un aminoácido de tipo no natural tales como un aminoácido N-metilado.
La secuencia de aminoácidos de LP no se limita particularmente, pero ejemplos del aminoácido constituyente incluyen fenilalanina (Phe; F), tirosina (Tyr; Y), leucina (Leu; L), glicina (Gly; G), alanina (Ala; A), valina (Val; V), lisina (Lys; K), citrulina (Cit), serina (Ser; S), ácido glutámico (Glu; E) y ácido aspártico (Asp; D). Entre ellos, ejemplos preferidos incluyen fenilalanina, glicina, valina, lisina, citrulina, serina, ácido glutámico y ácido aspártico. Dependiendo del tipo de aminoácido, puede controlarse el patrón de liberación de fármaco. El número del aminoácido puede estar entre 2 y 7.
Ejemplos específicos de LP pueden incluir
-GGF-
-DGGF-
-(D-)D-GGF-
-EGGF-
-GGFG-
-SGGF-
-KGGF-
-DGGFG-
-GGFGG-
-DDGGFG-
-KDGGFG-
-GGFGGGF-
[en lo anterior, “(D-)D” representa un ácido D-aspártico]. Ejemplos particularmente preferidos de LP para el conjugado de anticuerpo - fármaco pueden incluir -GGFG-.
En la estructura representada por -NH-(CH2)n1- dentro del enlazador, n1 es un número entero de 0 a 6 y es preferentemente un número entero de 1 a 5 y, más preferentemente, de 1 a 3. El grupo amino de este resto está conectado al C terminal de LP en el enlazador.
4. La
El enlazador La está representado por cualquiera de las estructuras -C(=O)-NH-, -NR1-(CH2)n7- y -O- o es un enlace sencillo. En lo anterior, n7 es un número entero de 1 a 6, R1 es un átomo de hidrógeno, un grupo alquilo que tiene 1 a 6 átomos de carbono, -(CH2)n8-COOH o -(CH2)n9-OH, n8 es un número entero de 1 a 4 y n9 es un número entero de 1 a 6.
La estructura amida -C(=O)-NH- dentro del enlazador La está conectada a Lb en su lado de átomo de nitrógeno. En el resto de estructura de -NR1-(CH2)n7- dentro de La, n7 es un número entero de 1 a 6 y preferentemente 1 a 3. Este resto está conectado a Lb en su lado metileno. R1 es un átomo de hidrógeno o un grupo alquilo que tiene 1 a 6 átomos de carbono. El grupo alquilo que tiene 1 a 6 átomos de carbono puede ser lineal o ramificado. Ejemplos del mismo pueden incluir un grupo metilo, un grupo etilo, un grupo propilo, un grupo isopropilo, un grupo butilo, un grupo isobutilo, un grupo sec-butilo, un grupo terc-butilo, un grupo pentilo, un grupo isopentilo, un grupo 2-metilbutilo, un grupo neopentilo, un grupo 1 -etilpropilo, un grupo hexilo, un grupo isohexilo, un grupo 4-metilpentilo, un grupo 3-metilpentilo, un grupo 2-metilpentilo, un grupo 1 -metilpentilo, un grupo 3,3-dimetilbutilo, un grupo 2,2-dimetilbutilo, un grupo 1,1 -dimetilbutilo, un grupo 1,2-dimetilbutilo, un grupo 1,3-dimetilbutilo, un grupo 2,3-dimetilbutilo y un grupo 2-etilbutilo. De ellos, se prefiere un grupo metilo o un grupo etilo. Cuando R1 tiene una estructura representada por -(CH2)n8-COOH, n8 es un número entero de 1 a 4 y preferentemente 1 o 2. Cuando R1 tiene una estructura representada por -(CH2)n9-OH, n9 es un número entero de 1 a 6 y preferentemente 1 o 2. R1 es preferentemente un átomo de hidrógeno, un grupo metilo, un grupo etilo, -CH2COOH, -CH2CH2-COOH o -CH2CH2-OH y más preferentemente un átomo de hidrógeno, un grupo metilo o -CH2COOH. Es preferentemente además un átomo de hidrógeno. El resto La del enlazador puede ser -O- o un enlace sencillo.
5. Lb
El enlazador Lb es cualquiera de las estructuras -CR2(-R3)-, -O- y -NR4- o es un enlace sencillo. En lo anterior, R2 y R3 representan cada uno independientemente un átomo de hidrógeno, un grupo alquilo que tiene 1 a 6 átomos de
carbono, -(CH2)na-NH2, -(CH2)nb-COOH o -(CH2)nc-OH, R4 es un átomo de hidrógeno o un grupo alquilo que tiene 1 a 6 átomos de carbono, na es un número entero de 0 a 6, nb es un número entero de 1 a 4 y nc es un número entero de 0 a 4. Cuando na o nc es 0, R2 y R3 no son uno el mismo que el otro.
Cuando cada uno de R2 y R3 es un grupo alquilo, este grupo alquilo se interpreta como se define en el grupo alquilo de R1. Cuando R2 y R3 tienen una estructura de -(CH2)na-NH2, na es un número entero de 0 a 6 y preferentemente 0, o es 3 a 5. Cuando na es 0, R2 y R3 no son uno el mismo que el otro. Cuando R2 y R3 tienen la estructura de -(CH2)nb-COOH, nb es un número entero de 1 a 4 y preferentemente 1 o 2. Cuando R2 y R3 tienen la estructura de-(CH2)nc-OH, nc es un número entero de 0 a 4 y preferentemente 1 o 2.
Cada uno de R2 y R3 es preferentemente un átomo de hidrógeno, un grupo metilo, un grupo etilo, -NH2, -CH2CH2CH2NH2,-CH2CH2CH2CH2NH2, -CH2CH2CH2CH2CH2CH2NH2, -CH2COOH, -CH2CH2-COOH, -CH2OH o -CH2CH2-OH y más preferentemente un átomo de hidrógeno, un grupo metilo, -NH2, -CH2CH2CH2CH2NH2,-CH2COOH, -CH2CH2-COOH, -CH2OH o -CH2CH2-OH.
Cuando R4 es un grupo alquilo que tiene 1 a 6 átomos de carbono, este grupo alquilo se interpreta como se define en el grupo alquilo de R1. R4 es preferentemente un átomo de hidrógeno o un grupo metilo y más preferentemente un átomo de hidrógeno.
Ejemplos específicos de la estructura representada por
-NH-(CH2)n1-La-Lb- como el enlazador pueden incluir
-NH-CH2--NH-CH(-Me)--NH-C(-Me)2--NH-CH2-CHMe--NH-CH(-CH2OH)--NH-CH(-CH2COOH)--NH-CH(-CH2CH2COOH)--NH-CH(-CH2CH2CH2CH2NH2)--NH-CH2CH2--NH-CH2-O-CH2--NH-CH2CH2-O--NH-CH2CH2-O-CH2--NH-CH2CH2C(-Me)2--NH-CH2CH2NH--NH-CH2CH2NH-CH2--NH-CH2CH2NMe-CH2--NH-CH2CH2NH-CH2CH2--NH-CH2CH2NMe-CH2CH2--NH-CH2CH2N(-CH2COOH)-CH2--NH-CH2CH2N(-CH2CH2OH)-CH2--NH-CH2CH2N(-CH2CH2OH)-CH2CH2--NH-CH2CH2CH2C(=O)-NHCH(-CH2OH)--NH-CH2CH2CH2C(=O)-NHCH(-CH2COOH)--NH-CH2CH2CH2C(=O)-NHCH(-CH2CH2CH2CH2NH2)
-NH-CH2CH2CH2--NH-CH2CH2CH2CH2--NH-CH2CH2CH2CH2CH2--NH-CH2CH2CH2CH2CH(NH2)-.
De ellos, los ejemplos preferidos de los mismos pueden incluir -NH-CH2--NH-CH2-CH(Me)--NH-CH(-CH2OH)--NH-CH(-CH2CH2COOH)--NH-CH2CH2--NH-CH2-O-CH2--NH-CH2CH2-O--NH-CH2CH2-O-CH2--NH-CH2CH2C(-Me)2--NH-CH2CH2NH--NH-CH2CH2NH-CH2--NH-CH2CH2NMe-CH2--NH-CH2CH2NMe-CH2CH2--NH-CH2CH2N(-CH2COOH)-CH2--NH-CH2CH2N(-CH2CH2OH)-CH2--NH-CH2CH2N(-CH2CH2OH)-CH2CH2--NH-CH2CH2CH2C(=O)-NHCH(-CH2OH)--NH-CH2CH2CH2C(=O)-NHCH(-CH2COOH)--NH-CH2CH2CH2--NH-CH2CH2CH2CH2--NH-CH2CH2CH2CH2CH2- .
Ejemplos más preferidos de los mismos pueden incluir
-NH-CH2--NH-CH2CH2--NH-CH2-O-CH2--NH-CH2CH2-O--NH-CH2CH2-O-CH2--NH-CH2CH2NH--NH-CH2CH2NH-CH2--NH-CH2CH2N(-CH2COOH)-CH2--NH-CH2CH2N(-CH2CH2OH)-CH2CH2--NH-CH2CH2CH2C(=O)-NHCH(-CH2COOH)
-NH-CH2CH2CH2--NH-CH2CH2CH2CH2--NH-CH2CH2CH2CH2CH2- .
Ejemplos preferidos adicionales de la misma pueden incluir
-NH-(CH2)3-,
-NH-CH2-O-CH2-y
-NH-(CH2)2-O-CH2-.
6. Lc
El enlazador Lc es -CH2- o -C(=O)-. Dicho enlazador está conectado al compuesto antitumoral. Lc del enlazador es más preferentemente -C(=O)-.
En el enlazador, la longitud de cadena de -NH-(CH2 n1-La-Lb-Lc es preferentemente una longitud de cadena de 4 a 7 átomos y, más preferentemente, una longitud de cadena de 5 o 6 átomos.
Con respecto al conjugado de anticuerpo - fármaco, cuando este se transfiere al interior de células tumorales, el resto de enlazador se escinde y el derivado de fármaco que tiene una estructura representada por NH2-(CH2)n1-La-Lb-Lc-(NH-DX) se libera para expresar una acción antitumoral. Los ejemplos del derivado antitumoral que muestra un efecto antitumoral liberándose del conjugado de anticuerpo - fármaco incluyen un derivado antitumoral que tiene un resto de estructura en el que la estructura representada por -NH-(CH2)n1-La-Lb- del enlazador se une con Lc y tiene un grupo amino terminal, y los particularmente preferidos incluyen los siguientes.
NH2-CH2CH2-C(=O)-(NH-DX)
NH2-CH2CH2CH2-C(=O)-(NH-DX)
NH2-CH2-O-CH2-C(=O)-(NH-DX)
NH2-CHCH2-O-CH2-C(=O)-(NH-DX)
Mientras tanto, en el caso de NH2-CH2-O-CH2-C(=O)-(NH-DX), se confirmó que, debido a que la estructura de amina en la molécula es inestable, esta experimenta de nuevo una autodegradación para liberar lo siguiente HO-CH2-C(=O)-(NH-DX). Esos compuestos también pueden usarse preferentemente como un intermedio de producción del conjugado de anticuerpo - fármaco.
Para el conjugado de anticuerpo - fármaco en el que exatecán se usa como un fármaco, es preferible que el resto de estructura de fármaco - enlazador que tiene la siguiente estructura [-L1-L2-LP-NH-(CH2)n1-La-Lb-Lc-(NH-DX)] esté conectado a un anticuerpo. El número conjugado promedio del resto de estructura de fármaco - enlazador por anticuerpo puede ser de 1 a 10. Preferentemente, este es de 2 a 8 y, más preferentemente, de 3 a 8.
-(Succinimid-3-il-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX) -(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX) -(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
-CH2-C(=O)-NH-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-S-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX) Entre ellos, los más preferidos son los siguientes.
-(Succinimid-3-il-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-CH2-C(=O)-NH-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
-C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-S-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
Los particularmente preferidos son los siguientes.
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
-(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX).
Con respecto a la estructura de enlazador para conjugar el anticuerpo y un fármaco en el conjugado de anticuerpo -fármaco de la presente solicitud, el enlazador preferido puede construirse conectando estructuras preferidas mostradas para cada parte del enlazador anteriormente explicado. Con respecto a la estructura de enlazador, pueden usarse preferentemente los que tienen la siguiente estructura. Mientras tanto, el extremo terminal izquierdo de la estructura es una posición de conexión con el anticuerpo y el extremo terminal derecho es una posición de conexión con el fármaco.
-(Succinimid-3-il-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)
-CH2-C(=O)-NH-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-S-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-Entre ellos, los más preferidos son los siguientes.
-(Succinimid-3-il-N)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--CH2-C(=O)-NH-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)--C(=O)-cic.Hex(1,4)-CH2-(N-li-3-diminiccuS)-S-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-.
Los particularmente preferidos incluyen los siguientes.
-(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)--(Succinimid-3-il-N)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-. [Procedimiento de producción]
A continuación, se dan explicaciones para el procedimiento representativo para producir un conjugado de anticuerpo - fármaco o un intermedio de producción del mismo. Mientras tanto, los compuestos se describen en lo sucesivo en el presente documento con el número de compuestos mostrado en cada fórmula de reacción. Específicamente, se hace referencia a los mismos como "compuesto de la fórmula (1)", un "compuesto (1)", o similares. Los compuestos con números que no sean esos también se describen de forma similar.
1. Procedimiento de producción 1
El conjugado de anticuerpo - fármaco representado por la fórmula (1) en el que el anticuerpo se une a la estructura de enlazador por medio de tioéter puede producirse mediante el siguiente procedimiento, por ejemplo.
[Fórmula 48]

[En la fórmula, AB representa un anticuerpo con un grupo sulfhidrilo, y L1' representa una estructura de enlazador de L1 en la que el extremo terminal de enlazador es un grupo maleimidilo (fórmula mostrada posteriormente)
[Fórmula 49]

(en la fórmula, el átomo de nitrógeno es la posición de conexión) o el extremo terminal es halógeno, y representa un
grupo en el que el resto de -(Succinimid-3-il-N)- en -(Succinimid-3-il-N)-(CH2)n2-C(=O)- de L1 es un grupo maleimidilo o un grupo halógeno-CH2C(=O)NH-(CH2)n3-C(=O)- en el que el metileno terminal en -CH2C(=O)NH-(CH2)n3-C(=O)-de L1 se halogena para formar haloacetamida. Además, el -(NH-DX) representa una estructura representada por la siguiente fórmula:
[Fórmula 50]

y este representa un grupo que se obtiene retirando un átomo de hidrógeno del grupo amino en la posición 1 de exatecán. Además, el compuesto de la fórmula (1) en la fórmula de reacción anterior se describe como una estructura en la que un resto de estructura desde el fármaco hasta el extremo terminal de enlazador conecta con un anticuerpo. Sin embargo, la descripción solo se da por razones de conveniencia, y en realidad hay muchos casos en los que una pluralidad de los restos de estructura están conectados a una molécula de anticuerpo. Esto mismo es de aplicación a la explicación del procedimiento de producción descrito posteriormente.]
Específicamente, el conjugado de anticuerpo - fármaco (1) puede producirse haciendo reaccionar el compuesto (2), que puede obtenerse mediante el procedimiento descrito posteriormente, con el anticuerpo (3a) que tiene un grupo sulfhidrilo.
El anticuerpo (3a) que tiene un grupo sulfhidrilo puede obtenerse mediante un procedimiento bien conocido en la técnica (Hermanson, G. T, Bioconjugate Techniques, págs. 56-136, págs. 456-493, Academic Press (1996)). Los ejemplos incluyen: reactivo de Traut se hace reaccionar con el grupo amino del anticuerpo; S-acetiltioalcanoatos de N-succinimidilo se hacen reaccionar con el grupo amino del anticuerpo seguido de reacción con hidroxilamina; después de hacerse reaccionar con 3-(piridilditio)propionato de N-succinimidilo, el anticuerpo se hace reaccionar con un agente reductor; el anticuerpo se hace reaccionar con un agente reductor tal como ditiotreitol, 2-mercaptoetanol y clorhidrato de tris(2-carboxietil)fosfina (TCEP) para reducir el enlace de disulfuro en una parte de bisagra en el anticuerpo para formar un grupo sulfhidrilo, aunque sin limitarse al mismo.
Específicamente, usando de 0,3 a 3 equivalentes molares de TCEP como un agente reductor por disulfuro en parte de bisagra en el anticuerpo y haciéndolo reaccionar con el anticuerpo en una solución tampón que contiene un agente quelante, puede obtenerse el anticuerpo con un disulfuro parcial o completamente reducido en parte de bisagra en el anticuerpo. Los ejemplos del agente quelante incluyen ácido etilen diamina tetraacético (EDTA) y ácido dietilenotriamina pentaacético (DTPA). Este puede usarse a una concentración de 1 mM a 20 mM. Los ejemplos de la solución tampón que puede usarse incluyen una solución de fosfato sódico, borato sódico o acetato sódico. Como ejemplo específico, haciendo reaccionar el anticuerpo con TCEP durante de 1 a 4 horas de 4 °C a 37 °C, puede obtenerse el anticuerpo (3a) que tiene un grupo sulfhidrilo parcial o completamente reducido.
Mientras tanto, realizando la reacción para añadir un grupo sulfhidrilo a un resto de fármaco - enlazador, el resto de fármaco - enlazador puede conjugarse mediante un enlace de tioéter.
A continuación, usando de 2 a 20 equivalentes molares del compuesto (2) por anticuerpo (3a) que tiene un grupo sulfhidrilo, puede producirse el conjugado de anticuerpo - fármaco (1) en el que están conjugadas de 2 a 8 moléculas de fármaco por anticuerpo. Específicamente, es suficiente que la solución que contiene el compuesto (2) disuelto en la misma se añada a una solución tampón que contiene el anticuerpo (3a) que tiene un grupo sulfhidrilo para la reacción. En el presente documento, los ejemplos de la solución tampón que puede usarse incluyen solución de acetato sódico, fosfato sódico y borato sódico. El pH para la reacción es de 5 a 9 y, más preferentemente, la reacción se realiza cerca de pH 7. Los ejemplos del disolvente para disolver el compuesto (2) incluyen un disolvente orgánico tal como dimetilsulfóxido (DMSO), dimetilformamida (Dm F), dimetil acetamida (DMA) y N-metil-2-piridona (NMP). Es suficiente que la solución de disolvente orgánico que contiene el compuesto (2) disuelto en la misma se añada a 1 a 20 % v/v a una solución tampón que contiene el anticuerpo (3a) que tiene un grupo sulfhidrilo para la reacción. La temperatura de reacción es de 0 a 37 °C, más preferentemente de 10 a 25 °C, y el tiempo de reacción es de 0,5 a 2 horas. La reacción puede terminarse desactivando la reactividad del compuesto sin reaccionar (2) con un reactivo que contiene tiol. Los ejemplos del reactivo que contiene tiol incluyen cisteína y N-acetil-L-cisteína (NAC). De manera más específica, se añaden de 1 a 2 equivalentes molares de nAc al compuesto (2) usado e, incubando a temperatura ambiente durante de 10 a 30 minutos, puede terminarse la reacción.
El conjugado de anticuerpo - fármaco producido (1) puede someterse a, después de la concentración, intercambio de tampón, purificación y medición de concentración de anticuerpo y número promedio de moléculas de fármaco conjugadas por molécula de anticuerpo de acuerdo con procedimientos comunes descritos posteriormente, identificación del conjugado de anticuerpo - fármaco (1).
Procedimiento común A: Concentración de solución acuosa de anticuerpo o conjugado de anticuerpo - fármaco
A un recipiente Amicon Ultra (50.000 MWCO, Millipore Corporation), se añadió una solución de anticuerpo o conjugado de anticuerpo - fármaco y la solución del anticuerpo o conjugado de anticuerpo - fármaco se concentró por centrifugación (centrífuga durante de 5 a 20 minutos de 2000 G a 3800 G) usando una centrífuga (Allegra X-15r , Beckman Coulter, Inc.).
Procedimiento común B: Medición de concentración de anticuerpo
Usando un detector UV (Nanodrop 1000, Thermo Fisher Scientific Inc.), la medición de la concentración de anticuerpo se realizó de acuerdo con el procedimiento definido por el fabricante. En ese momento, se usó un coeficiente de absorción de 280 nm diferente para cada anticuerpo (1,3 mlmg'1cirr1 a 1,8 mlmg'1cirr1).
Procedimiento común C-1: Intercambio de tampón para anticuerpo
Una columna NAP-25 (n.° de cat. 17-0852-02, GE Healthcare Japan Corporation) usando vehículo Sephadex G-25 se equilibró con tampón de fosfato (10 mM, pH 6,0) (a lo que se hace referencia como PBS6,0/EDTA en la memoria descriptiva) que contiene cloruro sódico (137 mM) y ácido etilen diamina tetraacético (EDTA, 5 mM) de acuerdo con el procedimiento definido por el manual de instrucciones del fabricante. Una solución acuosa del anticuerpo se aplicó en una cantidad de 2,5 ml a una única columna NAP-25, y entonces se recogió la fracción (3,5 ml) eluyendo con 3,5 ml de PBS6,0/EDTA. La fracción resultante se concentró mediante el procedimiento común A. Después de la medición de la concentración del anticuerpo usando el procedimiento común B, la concentración de anticuerpo se ajustó a 10 mg/ml usando PBS6,0/EDTA.
Procedimiento común C-2: Intercambio de tampón para anticuerpo
Una columna NAP-25 (n.° de cat. 17-0852-02, GE Healthcare Japan Corporation) usando vehículo Sephadex G-25 se equilibró con tampón de fosfato (50 mM, pH 6,5) (a lo que se hace referencia como PBS6,5/EDTA en la memoria descriptiva) que contiene cloruro sódico (50 mM) y EDTA (2 mM) de acuerdo con el procedimiento definido por el fabricante. Una solución acuosa del anticuerpo se aplicó en una cantidad de 2,5 ml a una única columna NAP-25, y entonces se recogió la fracción (3,5 ml) eluyendo con 3,5 ml de PBS6,5/EDTA. La fracción resultante se concentró mediante el procedimiento común A. Después de la medición de la concentración del anticuerpo usando el procedimiento común B, la concentración de anticuerpo se ajustó a 20 mg/ml usando PBS6,5/EDTA.
Procedimiento común D-1: Purificación del conjugado de anticuerpo - fármaco
Una columna NAP-25 se equilibró con cualquier tampón seleccionado de entre tampón de fosfato comercialmente disponible (PBS7,4, n.° de cat. 10010-023, Invitrogen), tampón de fosfato sódico (10 mM, pH 6,0; a lo que se hace referencia como PBS6,0) que contiene cloruro sódico (137 mM), y tampón de acetato que contiene sorbitol (5 %) (10 mM, pH 5,5; a lo que se hace referencia como ABS en la memoria descriptiva). Una solución acuosa de la reacción de conjugado de anticuerpo - fármaco se aplicó en una cantidad de aproximadamente 1,5 ml a la columna NAP-25, y entonces se eluyó con el tampón en una cantidad definida por el fabricante para recoger la fracción de anticuerpo. La fracción recogida se aplicó de nuevo a la columna NAP-25 y, repitiendo de 2 a 3 veces en total el proceso de purificación de filtración en gel para eluir con tampón, se obtuvo el conjugado de anticuerpo - fármaco excluyendo enlazador de fármaco no conjugado y un compuesto de bajo peso molecular, clorhidrato de (tris(2-carboxietil)fosfina (TCEP), N-acetil-L-cisteína (NAC) y dimetilsulfóxido). Procedimiento común E: Medición de concentración de anticuerpo en conjugado de anticuerpo - fármaco y número promedio de moléculas de fármaco conjugadas por molécula de anticuerpo.
La concentración de fármaco conjugado en el conjugado de anticuerpo - fármaco puede calcularse midiendo la absorbancia UV de una solución acuosa del conjugado de anticuerpo - fármaco a dos longitudes de onda de 280 nm y 370 nm, seguido de la realización del cálculo mostrado posteriormente.
Debido a que la absorbancia total a cualquier longitud de onda es igual a la suma de la absorbancia de cada una de las especies químicas que absorben luz que están presentes en un sistema [aditividad de absorbancia], cuando los coeficientes de absorción molar del anticuerpo y el fármaco permanecen sin cambios antes y después de la conjugación entre el anticuerpo y el fármaco, la concentración de anticuerpo y la concentración de fármaco en el conjugado de anticuerpo - fármaco se expresan con las siguientes ecuaciones.
A280 - Ad,280 Aa,280 £ü,28qCd £a,28cCa Ecuación (1)
A370 - Ad,370+ Aa,370 - £d,37C)Cd £a,37cCa Ecuación (2)
En lo anterior, A280 representa la absorbancia de una solución acuosa del conjugado de anticuerpo - fármaco a 280 nm, A370 representa la absorbancia de una solución acuosa del conjugado de anticuerpo - fármaco a 370 nm, Aa ,280 representa la absorbancia de un anticuerpo a 280 nm, Aa,370 representa la absorbancia de un anticuerpo a 370 nm, Ad,280 representa la absorbancia de un precursor de conjugado a 280 nm, Ad,370 representa la absorbancia de un precursor de conjugado a 370 nm, £a ,280 representa el coeficiente de absorción molar de un anticuerpo a 280 nm, £a ,370 representa el coeficiente de absorción molar de un anticuerpo a 370 nm, £d,280 representa el coeficiente de absorción molar de un precursor de conjugado a 280 nm, £d,370 representa el coeficiente de absorción molar de un precursor de conjugado a 370 nm, Ca representa la concentración de anticuerpo en un conjugado de anticuerpo - fármaco, y Cd representa la concentración de fármaco en un conjugado de anticuerpo - fármaco.
Con respecto a £a ,280, £a ,370, £d,280, y £d,370 en lo anterior, se usan valores previamente preparados (valor estimado basándose en el valor de cálculo o de medición obtenido mediante medición UV del compuesto). Por ejemplo, £a ,280 puede estimarse a partir de la secuencia de aminoácidos de un anticuerpo usando un procedimiento de cálculo conocido (Protein Science, 1995, vol. 4, 2411-2423). £a ,370 es generalmente cero. £d ,280 y £d,370 pueden obtenerse basándose en la ley de Lambert-Beer (Absorbancia = concentración molar x coeficiente de absorción molar x longitud de ruta celular) midiendo la absorbancia de una solución en la que el precursor de conjugado que se va a usar se disuelve a una determinada concentración molar. Midiendo A280 y A370 de una solución acuosa del conjugado de anticuerpo - fármaco y resolviendo las ecuaciones simultáneas (1) y (2) usando los valores, pueden obtenerse Ca y
Cd. Además, al dividir Cd entre Ca , puede obtenerse el número promedio de fármaco conjugado por anticuerpo.
El compuesto representado por la fórmula (2) en el procedimiento de producción 1 es cualquier compuesto representado por la siguiente fórmula:
[Fórmula 51]

Halógeno-CH
2
C(=O)NH-(CH
2
)
n3
-C(=O)-L
2
-L
p
-NH-(CH
2
)
n1
-L
a
-L
b
-L
c
-(NH-DX)
En la fórmula, n1, n2, n3, L2, LP, La, Lb, y Lc son como ya se ha definido, y Lc es una po fármaco.
En un intermedio útil en la producción de tal compuesto, preferentemente, n2 es un número entero de 2 a 5, L2 es -NH-(CH2CH2O)n5-CH2CH2-C(=O)- o un enlace sencillo, n5 es un número entero de 2 a 4, LP es GGFG, y -NH-(CH2)n1-La-Lb-Lc- es una estructura parcial de -NH-CH2CH2-C(=O)-, NH-CH2CH2CH2-C(=O)-, -NH-CH2-O-CH2-C(=O)- o -NH-CH2CH2-O-CH2-C(=O)-. El halógeno es preferentemente bromo o yodo. Ejemplos específicos de estos compuestos pueden incluir los siguientes [en el presente documento, (maleimid-N-il) representa un grupo maleimidilo (grupo 2,5-dioxo-2,5-dihidro-1H-pirrol-1-ilo)].
(maleimid-N-il)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2 CH2-C(=O)-(NH-DX) (maleimid-N-il)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
X-CH2-C(=O)-NH-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
En la fórmula, X representa un átomo de bromo o un átomo de yodo. Todos estos compuestos de bromo y de yodo pueden usarse preferentemente como intermedios de producción.
Con el fin de asegurar la cantidad del conjugado, se pueden mezclar una pluralidad de conjugados obtenidos en condiciones de producción similares para tener un número equivalente de fármacos (por ejemplo, aproximadamente ± 1) para preparar nuevos lotes. En este caso, el número promedio de fármacos cae entre los números promedio de fármacos en los conjugados antes del mezclado.
2. Procedimiento de producción 2
El conjugado de anticuerpo - fármaco representado por la fórmula (1) en el que el anticuerpo está conectado por medio de un grupo amida a un enlazador y que tiene un enlace de tioéter dentro del enlazador, específicamente, una estructura en la que -L1-L2- es -C(=O)-cic.Hex(1,4 )-CH2-(N-li-3 -diminiccuS)-S-(CH2)n6-C (=O)-, también puede producirse mediante el siguiente procedimiento.
[Fórmula 52]
v
AB-L
2' P l a b e 3b 1 2 P l a b e
L -L -NH-(CH2)n -L -L -L -(NH-DX) __________> AB-L -L -L -NH-(CH2)n -L -L -L -(NH-DX)
2a 1
En la fórmula, AB-L1 ' representa un grupo en el que el anticuerpo y el enlazador L1 están conectados y, además, el extremo terminal de L1 se convierte en un grupo N-maleimidilo. Este grupo tiene específicamente una estructura en la que -(N-li-3-diminiccuS)- en AB-C(=O)-cic.Hex(1,4 )-CH2-(N-li-3 -diminiccuS)- se convierte en un grupo maleimidilo. L2' representa un grupo HS-(CH2)n6-C(=O)- en el que el extremo terminal es un grupo mercapto, y AB representa el anticuerpo.
Específicamente, el conjugado de anticuerpo - fármaco (1) puede producirse haciendo reaccionar el compuesto (2a), que puede obtenerse mediante el procedimiento descrito posteriormente, con el anticuerpo (3b) que está conectado al enlazador que tiene un grupo maleimidilo.
El anticuerpo (3b) que tiene un grupo maleimidilo también puede obtenerse mediante un procedimiento bien conocido en la técnica (Hermanson, G. T, Bioconjugate Techniques, págs. 56-136, págs. 456-493, Academic Press (1996)). Los ejemplos incluyen: un enlazador bifuncional, tal como succinimidil-4-(N-maleimidometil)ciclohexano-1-carboxilato (SMCC), que es capaz de unirse a un grupo amino o un grupo hidroxilo y tiene un grupo maleimidilo se deja reaccionar en el grupo amino del ligando para introducir un grupo maleimidilo, aunque sin limitarse al mismo.
Por ejemplo, puede usarse preferentemente un compuesto que tiene un resto reactivo con grupo amino y un resto
reactivo con grupo tiol unido por medio de un enlazador. En este caso, el resto reactivo con grupo amino puede ser éster activo, éster de imida, o similares, y el resto reactivo con tiol puede ser maleimidilo, haluro de acetilo, haluro de alquilo, ditiopiridilo, o similares.
Como un procedimiento para construir el enlazador con grupo amino o grupo hidroxi de un aminoácido que constituye el anticuerpo, particularmente por medio de un enlace de amida con el grupo amino, el compuesto que se va a hacer reaccionar en primer lugar con el anticuerpo puede ser un compuesto representado por la siguiente fórmula:
Q1-L1a-Q2.
[En la fórmula, Q1 representa (pirrolidina-2,5-diona-N-il)-O-C(=O)-, (3-sulfo-pirrolidina-2,5-diona-N-il)-O-C (=O)-, RQ-O-C (=N)- u O=C=N-,
L1a- representa -cic.Hex(1,4 )-CH2-, un grupo alquileno que tiene de 1 a 10 átomos de carbono, un grupo fenileno, -(CH2)n4-C(=O)-, -(CH2)n4a-NH-C(=O)-(CH2)n4b-, o-(CH2)n4a-NH-C(=O)-cic.Hex(1,4 )-CH2-,
Q2 representa (maleimid-N-ilo), un átomo de halógeno o -S-S-(2-piridilo),
RQ representa un grupo alquilo que tiene de 1 a 6 átomos de carbono,
n4 representa un número entero de 1 a 8 , n4a representa un número entero de 0 a 6 , y n4b representa un número entero de 1 a 6.]
En lo anterior, RQ es un grupo alquilo que tiene de 1 a 6 átomos de carbono y, más preferentemente, un grupo metilo o un grupo etilo.
El grupo alquileno de L1a puede ser uno que tiene de 1 a 10 átomos de carbono. El grupo fenileno puede ser de cualquiera de las configuraciones orto, meta y para y, más preferentemente, es un grupo para- o meta-fenileno. Los ejemplos preferidos de L1a pueden incluir -cic.Hex(1,4 )-CH2-, -(CH2)5-NH-C(=O)-cic.Hex(1,4 )-CH2-,-(CH2)2-NH-C(=O)-CH2-, -(CH2)5-NH-C(=O)-(CH2)2-, -CH2-,-(CH2)2-, -(CH2)3-, -(CH2)5-, -(CH2)10-, -(para-Ph)-,-(meta-Ph)-, -(para-Ph)-CH(-CHa)-, -(CH2)3-(meta-Ph)- y -(meta-Ph)-NH-C(=O)-CH2-.
Q1 es preferentemente (pirrolidina-2,5-diona-N-il)-O-C(=O)-, Q2 es preferentemente (maleimid-N-ilo), o puede usarse -S-S-(2-piridil) cuando se va a formar un enlace de disulfuro.
En lo anterior, (pirrolidina-2,5-diona-N-il)- es un grupo representado por la siguiente fórmula:
[Fórmula 53]

en la que el átomo de nitrógeno como una posición de conexión, y (3-sulfo-pirrolidina-2,5-diona-N-il)- es un grupo representado por la siguiente fórmula:
[Fórmula 54]

en la que el átomo de nitrógeno es una posición de conexión, y este ácido sulfónico es capaz de formar una sal de litio, sal de sodio o sal de potasio, y preferentemente sal de sodio,
cic.Hex(1,4) representa un grupo 1,4-ciclohexileno, (maleimid-N-il) es un grupo representado por la siguiente fórmula:
[Fórmula 55]

en la que el átomo de nitrógeno es una posición de conexión, (2-piridil) representa un grupo 2-piridilo, (para-Ph) representa un grupo para-fenileno, y (meta-Ph) representa un grupo meta-fenileno.
Los ejemplos de tal compuesto incluyen sulfosuccinimidil-4-(N-maleimidilmetil)ciclohexano-1-carboxilato (sulfo-SMCC), N-succinimidil-4-(N-maleimidilmetil)-ciclohexano-1-carboxi-(6-amidocaproato) (LC-SMCC), éster N-succinimidílico del ácido K-maleimidil undecanoico (KMUA), éster N-succinimidílico del ácido Y-maleimidil butírico (GMBS), éster de N-hidroxisuccinimida del ácido £-maleimidil caproico (EMCS), éster de m-maleimidilbenzoil-N-hidroxisuccinimida (MBS), éster de N-(a-maleimidilacetoxi)-succinimida (AMAS), succinimidil-6-(pmaleimidilpropionamida)hexanoato (SMPH), N-succinimidil 4-(p-maleimidilfenil)-butirato (SMPB), N-(pmaleimidilfenil)isocianato (PMPI), N-succinimidil-4-(yodoacetil)-aminobenzoato (SIAB), yodoacetato de N-succinimidilo (SIA), bromoacetato de N-succinimidilo (SBA), 3-(bromoacetamida)propionato de N-succinimidilo (SBAP), N-succinimidil-3-(2-piridoditio)propionato (SPDP) y succinimidiloxicarbonil-a-metil-a-(2-piridilditio)tolueno (SMPT).
Específicamente, por ejemplo, haciendo reaccionar de 2 a 6 equivalentes de SMCC con el anticuerpo (3) en un tampón de fosfato de pH 6 a 7 durante de 1 a 6 horas a temperatura ambiente, el éster activo de SMCC puede reaccionar con el anticuerpo para producir el anticuerpo (3b) que tiene un grupo maleimidilo. El anticuerpo obtenido (3b) puede purificarse mediante el procedimiento común D-2 descrito posteriormente, y usarse para la siguiente reacción con el compuesto (2a). Procedimiento común D-2: Purificación de anticuerpo derivatizado con 4-(N-maleimidilmetil)-ciclohexano-1-carboxilato de succinimidilo (SMCC)
Una columna NAP-25 se equilibró con PBS6,5/EDTA. La solución de reacción que contiene el anticuerpo derivatizado con 4-(N-maleimidilmetil)-ciclohexano-1-carboxilato de succinimidilo (al que se hace referencia en el presente documento como SMCC) se aplicó en una cantidad de aproximadamente 0,5 ml a la columna NAP-25, y entonces se eluyó con el tampón en una cantidad definida por el fabricante para recoger la fracción de anticuerpo para la purificación.
El grupo amino del anticuerpo para conectar con el enlazador puede ser un grupo amino N-terminal y/o un grupo amino portado por un residuo de lisina, aunque sin limitarse al mismo. Como alternativa, el anticuerpo puede conectarse al enlazador con formación de enlace de éster mediante el uso de un grupo hidroxi portado por un residuo de serina.
La reacción del compuesto (2a) con el anticuerpo (3b) conectado al enlazador que tiene un grupo maleimidilo puede realizarse de la misma forma que en el procedimiento para hacer reaccionar el compuesto (2) con el anticuerpo (3a) que tiene un grupo sulfhidrilo tal como se menciona en el procedimiento de producción 1.
Para el conjugado de anticuerpo - fármaco (1) preparado, puede realizarse la concentración, intercambio de tampón, purificación e identificación del conjugado de anticuerpo - fármaco ( 1 ) mediante la medición de concentración de anticuerpo y un número promedio de moléculas de fármaco conjugadas por molécula de anticuerpo de la misma forma que en el procedimiento de producción 1.
El compuesto representado por la fórmula (3b) en el procedimiento de producción 2 tiene la siguiente estructura (véase la siguiente fórmula; en la estructura de la misma, "anticuerpo -NH-" se origina a partir de un anticuerpo).
[Fórmula 56]

Un compuesto que es un intermedio para producir el conjugado de anticuerpo - fármaco de la presente invención y tiene la estructura anterior es como se describe a continuación (en la fórmula, n es un número entero de 1 a 10 , preferentemente de 2 a 8 y, más preferentemente, de 3 a 8 ).
[Fórmula 57]



Además, los ejemplos del compuesto en los que el extremo terminal es un grupo mercapto pueden incluir los siguientes.
HS-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-
C(=O)-(NH-DX)
3. Procedimiento de producción 3
El conjugado de anticuerpo - fármaco representado por la fórmula (1) en el que el anticuerpo está conjugada con el resto de enlazador de fármaco por medio de un enlace de amida puede producirse mediante un procedimiento descrito posteriormente. Por ejemplo, con respecto a -C(=O)-(CH2)n4-C(=O)-de L1, su éster activo L1', por ejemplo, (pirrolidina-2,5-diona-N-il)-O-C(=O)-(CH2)n4-C(=O)-, puede usarse preferentemente. Cuando L2 es un enlace sencillo, el conjugado de anticuerpo - fármaco (1 ) puede producirse mediante el siguiente procedimiento, por ejemplo.
[Fórmula 58]

2b
Específicamente, el conjugado de anticuerpo - fármaco (1) puede producirse haciendo reaccionar el compuesto (2b), que puede obtenerse mediante el procedimiento descrito posteriormente, con el anticuerpo (3).
El compuesto (2b) es capaz de conectar con el grupo amino o grupo hidroxilo del anticuerpo. El grupo amino y grupo hidroxilo del anticuerpo se refieren a, como se describe en el procedimiento de producción 2 , por ejemplo, un grupo amino N-terminal portado por el anticuerpo y/o un grupo amino portado por un residuo de lisina y un grupo hidroxi portado por un residuo de serina, respectivamente, pero estos no se limitan a los mismos.
El compuesto (2 b) es un éster activo compuesto por un grupo éster N-hidroxisuccinimidílico. Como alternativa, pueden usarse otros ésteres activos, por ejemplo, un grupo éster sulfosuccinimidílico, éster N-hidroxiftalimidílico, éster N-hidroxisulfoftalimidílico, éster orto-nitrofenílico, éster para-nitrofenílico, éster 2,4-dinitrofenílico, éster 3-sulfonil-4-nitrofenílico, éster 3-carboxi-4-nitrofenílico y éster pentafluorofenílico.
Usando de 2 a 20 equivalentes molares del compuesto (2b) por anticuerpo (3) en la reacción del compuesto (2b) con el anticuerpo(3), puede producirse el conjugado de anticuerpo - fármaco (1) en el que se conjugan de 1 a 10 moléculas de fármaco por anticuerpo. Específicamente, la solución que contiene el compuesto (2b) disuelto en la misma se puede añadir a una solución tampón que contiene el anticuerpo (3) para la reacción para producir el conjugado de anticuerpo - fármaco (1). En el presente documento, los ejemplos de la solución tampón que puede usarse incluyen solución de acetato sódico, fosfato sódico y borato sódico. El pH para la reacción puede ser de 5 a 9 y, más preferentemente, la reacción se realiza cerca de pH 7. Los ejemplos del disolvente para disolver el compuesto (2 b) incluyen un disolvente orgánico tal como dimetilsulfóxido (DMSO), dimetilformamida (DMF), dimetil acetamida (DMA) y N-metil-2-piridona (NMP). Es suficiente que la solución de disolvente orgánico que contiene el compuesto (2b) disuelto en la misma se añada a 1 a 20 % v/v a una solución tampón que contiene el anticuerpo (3) para la reacción. La temperatura de reacción es de 0 a 37 °C, más preferentemente de 10 a 25 °C, y el tiempo de reacción es de 0,5 a 20 horas.
Para el conjugado de anticuerpo - fármaco producido (1), puede realizarse la concentración, intercambio de tampón, purificación e identificación del conjugado de anticuerpo - fármaco (1 ) mediante la medición de concentración de anticuerpo y un número promedio de moléculas de fármaco conjugadas por molécula de anticuerpo de la misma forma que en el procedimiento de producción 1.
El resto (pirrolidina-2 ,5 -diona-N-il)-O-C(=O)-(CH2)n4-C(=O)- en el procedimiento de producción 3 tiene la siguiente estructura.
[Fórmula 59]

Los ejemplos del compuesto que tiene la estructura parcial anterior pueden incluir los siguientes.
(P rrolid  diona-N- l)-O-C(=O)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
diona-N- l)-O-C(=O)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
(P rrolid diona-N- l)-O-C(=O)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
(P rrolid diona-N- l)-O-C(=O)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
(P rrolid  diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
(P rrolid  diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX) (P rrolid diona-N- l)-O-C(=O)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX) (P rrolid diona-N- l)-O-C(=O)-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(P rrolid diona-N- l)-O-C(=O)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(P rrolid  diona-N- l)-O-(=O)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
diona-N- l)-O-(=O)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(P rrolid diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX) (P rrolid diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2 CH2-C(=O)-(NH-DX) (P rrolid  diona-N- l)-O-C(=O)-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=OMNH-DX)
diona-N- l)-O-C(=O)-CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=OMNH-DX)
(P rrolid diona-N- l)-O-C(=O)-CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
(P rrolid  diona-N- l)-O-C(=O)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
diona-N- l)-O-C(=O)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
(P rrolid  diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX) (P rrolid
diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX) (P rrolid  diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX) (P rrolid diona-N- l)-O-C(=O)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX) (P rrolid diona-N- l)-O-C(=O)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
(P rrolid  diona-N- l)-O-C(=O)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
diona-N- l)-O-C(=O)-CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
(P rrolid  diona-N- l)-O-C(=O)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX) (P rrolid diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX) (P rrolid
diona-N- l)-O-C(=O)-CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX) (P rrolid diona-N- l)-O-C(=O)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX) (P rrolid  diona-N- l-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX) (P rrolid diona-N- l)-O-C(=O)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH DX)
diona-N- l-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX) (P rrolid diona-N- l)-O-C(=O)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH DX)
(Pirrolidina-2,5-diona-N-il)-O-C(=O)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(Pirrolidina-2,5-diona-N-il)-O-C(=O)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
4. Procedimiento de producción 4
El compuesto representado por la fórmula (2) o (2b) como un intermedio usado en el procedimiento de producción previo y una sal farmacológicamente aceptable del mismo puede producirse mediante el siguiente procedimiento, por ejemplo.
[Fór
En la fórmula, Lc es -C(=O)- y está conectado a -(NH-DX) con formación de enlace de amida, L1 ' representa una estructura L1 en la que el extremo terminal se convierte en un grupo maleimidilo o un grupo haloacetilo, o en (pirrolidina-2 ,5 -diona-N-il)-O-C(=O)-(CH2)n4-C(=O)-, y P1, P2, y P3 representan cada uno un grupo protector.
El compuesto (6 ) puede producirse derivatizando el ácido carboxílico (5) para dar un éster activo, anhídrido de ácido mixto, haluro de ácido, o similares y haciéndolo reaccionar con NH2-DX [que indica exatecán; nombre químico: (1S,9S)-1-amino-9-etil-5-fluoro-2,3-dihidro-9-hidroxi-4-metil-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-10,13(9H,15H)-diona] (4) o una sal farmacológicamente aceptable del mismo.
Para la reacción pueden emplearse reactivos y condiciones de reacción que se usan habitualmente para la síntesis de péptidos. Hay diversos tipos de éster activo. Por ejemplo, este puede producirse haciendo reaccionar fenoles tales como p-nitrofenol, N-hidroxi benzotriazol, N-hidroxi succinimida, o similares, con el ácido carboxílico (5) usando un agente de condensación tal como N,N'-diciclohexilcarbodiimida o clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida. Además, el éster activo también puede producirse mediante una reacción del ácido carboxílico (5) con trifluoroacetato de pentafluorofenilo o similares; una reacción del ácido carboxílico (5) con oxitripirrolidinofosfonio hexafluorofosfito de 1-benzotriazolilo; una reacción del ácido carboxílico (5) con cianofosfonato de dietilo (procedimiento de disolución de proteínas por adición de sal); una reacción del ácido carboxílico (5) con trifenilfosfina y disulfuro de 2,2'-dipiridilo (procedimiento de Mukaiyama); una reacción del ácido carboxílico (5) con un
derivado de triazina tal como cloruro de 4-(4,6-dimetoxi-1,3,5-triazin-2-il)-4-metilmorfolinio (DMTMM); o similares. Además, la reacción también puede realizarse mediante, por ejemplo, un procedimiento de haluro de ácido mediante el cual el ácido carboxílico (5) se trata con haluro de ácido tal como cloruro de tionilo y cloruro de oxalilo en presencia de una base. Haciendo reaccionar el éster activo, anhídrido de ácido mixto o haluro de ácido del ácido carboxílico (5) obtenido en consecuencia con el compuesto (4) en presencia de una base adecuada en un disolvente inerte de -78 °C a 150 °C, puede producirse el compuesto (6 ) (paralelamente, "disolvente inerte" indica un disolvente que no inhibe una reacción para la que se usa el disolvente).
Ejemplos específicos de la base usada para cada etapa anteriormente descrita incluyen carbonato de un metal alcalino o un metal alcalinotérreo, un alcóxido de metal alcalino, hidróxido o hidruro de un metal alcalino incluyendo carbonato sódico, carbonato potásico, etóxido sódico, butóxido potásico, hidróxido sódico, hidróxido potásico, hidruro sódico e hidruro potásico, base organometálica representada por un alquil litio incluyendo n-butil-litio, dialquilamino litio incluyendo diisopropilamida de litio; base organometálica de bis-sililamina incluyendo bis(trimetilsilil)amida de litio; y base orgánica incluyendo piridina, 2,6-lutidina, collidina, 4-dimetilaminopiridina, trietilamina, N-metilmorfolina, diisopropiletilamina y diazabiciclo[5.4.0]undec-7-eno (DBU).
Los ejemplos del disolvente inerte que se usa para la reacción de la presente invención incluyen un disolvente de hidrocarburo halogenado tal como diclorometano, cloroformo y tetracloruro de carbono; un disolvente de éter tal como tetrahidrofurano, 1,2 -dimetoxietano y dioxano; un disolvente de hidrocarburo aromático tal como benceno y tolueno; y un disolvente de amida tal como N,N-dimetilformamida, N,N-dimetilacetamida y N-metilpirrolidin-2-ona. Además de estos, se pueden usar, dependiendo de un caso, un disolvente de sulfóxido, tal como dimetilsulfóxido y sulfolano, y un disolvente de cetona tal como acetona y metil etil cetona.
El grupo hidroxi, grupo carboxi, grupo amino, o similares de La y Lb en el compuesto (6 ) puede protegerse con un grupo protector que se usa habitualmente en la síntesis de compuestos orgánicos, como se menciona posteriormente. Específicamente, los ejemplos del grupo protector para un grupo hidroxilo incluyen un grupo alcoximetilo tal como grupo metoximetilo; un grupo arilmetilo tal como grupo bencilo, grupo 4-metoxibencilo y grupo trifenilmetilo; un grupo alcanoílo tal como grupo acetilo; un grupo aroílo tal como grupo benzoílo; y un grupo sililo tal como grupo terc-butil difenilsililo. El grupo carboxi puede protegerse, por ejemplo, como un éster con un grupo alquilo tal como grupo metilo, grupo etilo y grupo terc-butilo, un grupo alilo, o un grupo arilmetilo tal como grupo bencilo. Un grupo amino puede protegerse con un grupo protector para un grupo amino que se usa generalmente para la síntesis de péptidos, por ejemplo, un grupo alquiloxi carbonilo tal como grupo terc-butiloxi carbonilo, grupo metoxicarbonilo y grupo etoxicarbonilo; un grupo arilmetilo tal como aliloxicarbonilo, grupo 9-fluorenilmetiloxi carbonilo, grupo benciloxi carbonilo, grupo parametoxibenciloxi carbonilo y grupo para (u orto)nitroilbenciloxi carbonilo; un grupo alcanoílo tal como grupo acetilo; un grupo arilmetilo tal como grupo bencilo y grupo trifenil metilo; un grupo aroílo tal como grupo benzoílo; y un grupo aril sulfonilo tal como grupo 2,4-dinitrobenceno sulfonilo o grupo ortonitrobenceno sulfonilo. La protección con y la desprotección del grupo protector pueden realizarse de acuerdo con un procedimiento llevado a cabo habitualmente.
Con respecto al grupo protector P1 para el grupo amino terminal del compuesto (6 ), se puede usar un grupo protector para un grupo amino que se usa generalmente para la síntesis de péptidos, por ejemplo, grupo terc-butiloxi carbonilo, grupo 9-fluorenilmetiloxi carbonilo y grupo benciloxi carbonilo. Los ejemplos del otro grupo protector para un grupo amino incluyen un grupo alcanoílo tal como grupo acetilo; un grupo alcoxicarbonilo tal como grupo metoxicarbonilo y grupo etoxicarbonilo; un grupo arilmetoxi carbonilo tal como grupo parametoxibenciloxi carbonilo, y grupo para (u orto)nitroilbenciloxi carbonilo; un grupo arilmetilo tal como grupo bencilo y grupo trifenil metilo; un grupo aroílo tal como grupo benzoílo; y un grupo aril sulfonilo tal como grupo 2,4-dinitrobenceno sulfonilo y grupo ortonitrobenceno sulfonilo. El grupo protector P1 puede seleccionarse dependiendo de, por ejemplo, las propiedades de un compuesto que tiene un grupo amino que se va a proteger.
Desprotegiendo el grupo protector P1 para el grupo amino terminal del compuesto (6 ) obtenido, puede producirse el compuesto (7). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (9) puede producirse derivatizando el ácido carboxílico peptídico (8 ) que tiene el N terminal protegido con P2 para dar un éster activo, anhídrido de ácido mixto, o similares y haciéndolo reaccionar con el compuesto (7) obtenido. Las condiciones de reacción, reactivos, base y disolvente inerte usados para formar un enlace peptídico entre el ácido carboxílico peptídico (8 ) y el compuesto (7) se pueden seleccionar adecuadamente de entre los descritos para la síntesis del compuesto (6 ). El grupo protector P2 puede seleccionarse adecuadamente de entre los descritos para el grupo protector del compuesto (6 ), y la selección se puede realizar basándose en, por ejemplo, las propiedades del compuesto que tiene un grupo amino que se va a proteger. Debido a que el mismo se usa generalmente para la síntesis de péptidos, el compuesto (9) también puede producirse repitiendo secuencialmente la reacción y desprotección del aminoácido o péptido que constituye el ácido carboxílico peptídico (8 ) para su alargamiento.
Desprotegiendo P2 como el grupo protector para el grupo amino del compuesto (9) obtenido, puede producirse el compuesto (10). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Es posible producir el compuesto (2) o (2b) derivatizando el ácido carboxílico (11) o (11b) para dar un éster activo, anhídrido de ácido mixto, haluro de ácido, o similares y haciéndolo reaccionar con el compuesto (10) obtenido. Las
condiciones de reacción, reactivos, base y disolvente inerte usados para formar un enlace peptídico entre el ácido carboxílico (11 ) o (11 b) y el compuesto (10 ) se pueden seleccionar adecuadamente de entre los descritos para la síntesis del compuesto (6 ).
El compuesto (9) también puede producirse mediante el siguiente procedimiento, por ejemplo.
El compuesto (13) puede producirse derivatizando el ácido carboxílico peptídico (8 ) que tiene el N terminal protegido con P2 para dar un éster activo, anhídrido de ácido mixto, o similares y haciéndolo reaccionar con el compuesto de amina (12) que tiene el grupo carboxi protegido con P3 en presencia de una base. Las condiciones de reacción, reactivos, base y disolvente inerte usados para formar un enlace peptídico entre el ácido carboxílico peptídico (8 ) y el compuesto (12) se pueden seleccionar adecuadamente de entre los descritos para la síntesis del compuesto (6 ). El grupo protector P2 para el grupo amino del compuesto (13) puede seleccionarse adecuadamente de entre los descritos para el grupo protector del compuesto (6 ). Con respecto al grupo protector P3 para un grupo carboxi, puede usarse un grupo protector usado habitualmente como un grupo protector para un grupo carboxi en la química de síntesis orgánica, en particular, síntesis de péptidos. Específicamente, este puede seleccionarse adecuadamente de entre los descritos para el grupo protector del compuesto (6 ), por ejemplo, ésteres con un grupo alquilo tal como un grupo metilo, un grupo etilo, o un terc-butilo, ésteres alílicos y ésteres bencílicos. En tal caso, es necesario que el grupo protector para un grupo amino y el grupo protector para un grupo carboxi se puedan retirar mediante un procedimiento diferente o condiciones diferentes. Por ejemplo, un ejemplo representativo incluye una combinación en la que P2 es un grupo terc-butiloxi carbonilo y P3 es un grupo bencilo. Los grupos protectores se pueden seleccionar de entre los mencionados anteriormente, dependiendo de, por ejemplo, las propiedades de un compuesto que tiene un grupo amino y un grupo carboxi que se va a proteger. Para la retirada de los grupos protectores, los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
Desprotegiendo el grupo protector P3 para el grupo carboxi del compuesto (13) obtenido, puede producirse el compuesto (14). Los reactivos y condiciones se seleccionan dependiendo del grupo protector.
El compuesto (9) puede producirse derivatizando el compuesto (14) obtenido para dar éster activo, anhídrido de ácido mixto, haluro de ácido, o similares y haciéndolo reaccionar con el compuesto (4) en presencia de una base. Para la reacción, también pueden usarse reactivos y condiciones de reacción que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, base y disolvente inerte usados para la reacción se pueden seleccionar adecuadamente de entre los descritos para la síntesis del compuesto (6 ).
El compuesto (2) o (2b) también puede producirse mediante el siguiente procedimiento, por ejemplo.
Desprotegiendo el grupo protector P2 para el grupo amino del compuesto (13), puede producirse el compuesto (15). Los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector.
El compuesto (16) o (16b) puede producirse derivatizando el derivado de ácido carboxílico (11) o (11b) para dar un éster activo, anhídrido de ácido mixto, haluro de ácido, o similares y haciéndolo reaccionar con el compuesto (15) obtenido en presencia de una base. Las condiciones de reacción, reactivos, base y disolvente inerte usados para formar un enlace de amida entre el ácido carboxílico peptídico (11) o (11b) y el compuesto (15) se pueden seleccionar adecuadamente de entre los descritos para la síntesis del compuesto (6 ).
Desprotegiendo el grupo protector para el grupo carboxi del compuesto (16) o (16b) obtenido, puede producirse el compuesto (17) o (17b). Este puede realizarse de forma similar a la desprotección del grupo carboxi para producir el compuesto (14).
El compuesto (2) o (2b) puede producirse derivatizando el compuesto (17) o (17b) para dar un éster activo, anhídrido de ácido mixto, haluro de ácido, o similares y haciéndolo reaccionar con el compuesto (4) en presencia de una base. Para la reacción, también pueden usarse reactivos y condiciones de reacción que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, base y disolvente inerte usados para la reacción se pueden seleccionar adecuadamente de entre los descritos para la síntesis del compuesto (6 ).
5. Procedimiento de producción 5
El compuesto representado por la fórmula (2) de un intermedio también puede producirse mediante el siguiente procedimiento.
[F ó rm u la 61 ]

En la fórmula, L1' se corresponde con L1 que tiene una estructura en la que el extremo terminal se convierte en un grupo maleimidilo o un grupo haloacetilo, y P4 representa un grupo protector.
El compuesto (19) puede producirse derivatizando el compuesto (11) para dar un éster activo, anhídrido de ácido mixto, o similares y haciéndolo reaccionar con el ácido carboxílico peptídico (18) que tiene el C terminal protegido con P4 en presencia de una base. Las condiciones de reacción, reactivos, base y disolvente inerte usados para formar un enlace peptídico entre el ácido carboxílico peptídico (18) y el compuesto (11 ) se pueden seleccionar adecuadamente de entre los descritos para la síntesis del compuesto (6 ). El grupo protector P4 para el grupo carboxi del compuesto (18) puede seleccionarse adecuadamente de entre los descritos para el grupo protector del compuesto (6 ).
Desprotegiendo el grupo protector para el grupo carboxi del compuesto (19) obtenido, puede producirse el compuesto (20). Esto puede realizarse de forma similar a la desprotección del grupo carboxi para producir el compuesto (14). El compuesto (2) puede producirse derivatizando el compuesto (20) obtenido para dar éster activo, anhídrido de ácido mixto, o similares y haciéndolo reaccionar con el compuesto (7). Para la reacción, también pueden usarse reactivos y condiciones de reacción que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, base y disolvente inerte usados para la reacción se pueden seleccionar adecuadamente de entre los descritos para la síntesis del compuesto (6 ).
6. Procedimiento de producción 6
El intermedio de producción (2a) descrito en el procedimiento de producción 2 en el que L2' se corresponde con L2 que tiene una estructura en la que el extremo terminal se convierte en un grupo mercaptoalcanoílo puede producirse mediante el siguiente procedimiento.
[Fórmula 62]

El compuesto (2a) puede producirse derivatizando el ácido carboxílico (21) que tiene el grupo mercapto terminal para dar un éster activo, anhídrido de ácido mixto, o similares y haciéndolo reaccionar con el compuesto (10). Para la reacción, también pueden usarse reactivos y condiciones de reacción que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, base y disolvente inerte usados para la reacción se pueden seleccionar adecuadamente de entre los descritos para la síntesis del compuesto (4).
Además, el compuesto (23) puede producirse derivatizando el compuesto (21) para dar un éster activo, anhídrido de ácido mixto, haluro de ácido, o similares, haciéndolo reaccionar con el compuesto (15), y desprotegiendo el grupo protector para el grupo carboxi del compuesto (22 ) obtenido.
El compuesto (2a) puede producirse derivatizando el compuesto (23) para dar un éster activo, anhídrido de ácido mixto, haluro de ácido, o similares y haciéndolo reaccionar con el compuesto (4) en presencia de una base. Para la reacción, también pueden usarse reactivos y condiciones de reacción que se usan generalmente para la síntesis de péptidos, y las condiciones de reacción, reactivos, base y disolvente inerte usados para la reacción se pueden seleccionar adecuadamente de entre los descritos para la síntesis del compuesto (6).
7. Procedimiento de producción 7
En lo sucesivo en el presente documento, se describe con detalle el procedimiento para producir el compuesto (10c) que tiene n1 = 1, La = O y Lb = CR2(-R3) en el intermedio de producción (10) descrito en el procedimiento de producción 4. El compuesto representado por la fórmula (10c), una sal o un solvato del mismo puede producirse de acuerdo con el siguiente procedimiento, por ejemplo.
[Fórmula 63]

En la fórmula, LP, R2, y R3 son como se han definido anteriormente, L representa un grupo acetilo, un átomo de hidrógeno, o similares, X e Y representan, cada uno, un oligopéptido que consiste en 1 a 3 aminoácidos, P5 y P7 representan, cada uno, un grupo protector para un grupo amino, y P6 representa un grupo protector para un grupo carboxi.
Un compuesto representado por la fórmula (24) puede producirse usando o aplicando el procedimiento descrito en la patente japonesa abierta a inspección pública n.° 2002-60351 o la literatura (J. Org. Chem., Vol. 51, página 3196,
1986) y, si es necesario, retirando los grupos protectores o modificando los grupos funcionales. Como alternativa, este también puede obtenerse tratando un aminoácido con un grupo amino terminal protegido o amida de ácido de oligopéptido con un grupo amino protegido con aldehído o cetona.
Haciendo reaccionar el compuesto (24) con el compuesto (25) que tiene un grupo hidroxilo a una temperatura que varía desde con enfriamiento a temperatura ambiente en un disolvente inerte en presencia de un ácido o una base, puede producirse el compuesto (26). Los ejemplos del ácido que puede usarse incluyen ácido inorgánico tal como ácido fluorhídrico, ácido clorhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico y ácido bórico; un ácido orgánico tal como ácido acético, ácido cítrico, ácido paratolueno sulfónico y ácido metano sulfónico; y un ácido de Lewis tal como tetrafluoroborato, cloruro de cinc, cloruro de estaño, cloruro de aluminio y cloruro de hierro. El ácido paratolueno sulfónico es particularmente preferible. Con respecto a la base que se va a usar, puede seleccionarse y usarse adecuadamente una cualquiera de las bases anteriormente mencionadas. Los ejemplos preferidos de las mismas incluyen un alcóxido de metal alcalino tal como terc-butóxido potásico, un hidróxido de metal alcalino tal como hidróxido sódico e hidróxido potásico; hidruro de metal alcalino tal como hidruro sódico e hidruro potásico; base organometálica representada por dialquilamino litio tal como diisopropilamida de litio; y base organometálica de bis-sililamina tal como bis(trimetilsilil)amida de litio. Los ejemplos del disolvente que se va a usar para la reacción incluyen un disolvente de éter tal como tetrahidrofurano y 1,4-dioxano; y un disolvente de hidrocarburo aromático tal como benceno y tolueno. Esos disolventes pueden prepararse como una mezcla con agua. Además, el grupo protector para un grupo amino como se ilustra mediante P5 no ésta particularmente limitado si este es un grupo usado habitualmente para la protección de un grupo amino. Los ejemplos representativos incluyen los grupos protectores para un grupo amino que se describen en el procedimiento de producción 4. Sin embargo, en la presente reacción, el grupo protector para un grupo amino como se ilustra mediante P5 puede retirarse por escisión. En tal caso, es necesario realizar una reacción con un reactivo adecuado para proteger un grupo amino según pueda requerirse.
El compuesto (27) puede producirse retirando el grupo protector P6 del compuesto (26). En el presente documento, aunque los ejemplos representativos del grupo protector para un grupo carboxi como se ilustra mediante P6 se describen en el procedimiento de producción 4, es deseable en este caso que el grupo protector P5 para un grupo amino y el grupo protector P6 para un grupo carboxi sean los grupos protectores que pueden retirarse mediante un procedimiento diferente o condiciones diferentes. Por ejemplo, un ejemplo representativo incluye una combinación en la que P5 es un grupo 9-fluorenilmetiloxi carbonilo y P6 es un grupo bencilo. Los grupos protectores pueden seleccionarse dependiendo de, por ejemplo, las propiedades de un compuesto que tiene un grupo amino y un grupo carboxi que se va a proteger. Para la retirada de los grupos protectores, los reactivos y condiciones se seleccionan dependiendo del grupo protector.
El compuesto (29) puede producirse derivatizando el ácido carboxílico (27) para dar un éster activo, anhídrido de ácido mixto, haluro de ácido, o similares y haciéndolo reaccionar con el compuesto (4) y una sal farmacológicamente aceptable del mismo para producir el compuesto (28) seguido de retirar el grupo protector P5 del compuesto (28) obtenido. Para la reacción entre el compuesto (4) y el ácido carboxílico (27) y la reacción para retirar el grupo protector P6, pueden usarse los mismos reactivos y condiciones de reacción que los descritos para el procedimiento de producción 4.
El compuesto (10c) puede producirse haciendo reaccionar el compuesto (29) con un aminoácido con grupo amino terminal protegido o el oligopéptido (30) con un grupo amino protegido para producir el compuesto (9c) y retirando el grupo protector P7 del compuesto (9c) obtenido. El grupo protector para un grupo amino como se ilustra mediante P7 no ésta particularmente limitado si este se usa generalmente para la protección de un grupo amino. Los ejemplos representativos del mismo incluyen los grupos protectores para un grupo amino que se describen en el procedimiento de producción 4. Para retirar el grupo protector, los reactivos y condiciones se seleccionan dependiendo del grupo protector. Para la reacción entre el compuesto (29) y el compuesto (30), pueden emplearse reactivos y condiciones de reacción que se usan habitualmente para la síntesis de péptidos. El compuesto (10c) producido mediante el procedimiento anteriormente mencionado puede derivatizarse para dar el compuesto (1 ) de acuerdo con el procedimiento anteriormente descrito.
8. Procedimiento de producción 8
En lo sucesivo en el presente documento, se describe con detalle el procedimiento para producir el compuesto (2c) que tiene n1 = 1, La = O y Lb = CR2(-R3) en el intermedio de producción (2) descrito en el procedimiento de producción 4. El compuesto representado por la fórmula (2c), una sal o un solvato del mismo puede producirse de acuerdo con el siguiente procedimiento, por ejemplo.
[F ó rm u la 64 ]

En la fórmula, L1', L2, LP, R2, y R3 son como se han definido anteriormente, Z representa un oligopéptido que consiste en 1 a 3 aminoácidos, P8 representa un grupo protector para un grupo amino, y P9 representa un grupo protector para un grupo carboxi.
El compuesto (33) puede producirse retirando el grupo protector P8 del aminoácido u oligopéptido (31) con grupo amino terminal protegido y grupo carboxi para producir el compuesto (32) y haciendo reaccionar la forma de amina obtenida (32) con el compuesto (11). El grupo protector para un grupo amino como se ilustra mediante P8 no ésta particularmente limitado si este es un grupo usado habitualmente para la protección de un grupo amino. Los ejemplos representativos incluyen los grupos protectores para un grupo amino que se describen en el procedimiento de producción 4. Además, para retirar el grupo protector P8, los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector. Para la reacción entre el compuesto (32) y el ácido carboxílico (11), pueden usarse los mismos reactivos y condiciones de reacción que los descritos para el procedimiento de producción 4.
El intermedio de producción (2c) puede producirse retirando el grupo protector P9 del compuesto (33) para producir el compuesto (34) y haciendo reaccionar el ácido carboxílico (34) obtenido con el compuesto (29). Los ejemplos representativos del grupo protector para un grupo carboxi como se ilustra mediante P8 se describen en el procedimiento de producción 4. Para la reacción de desprotección del mismo, pueden usarse los mismos reactivos y condiciones de reacción que los descritos para el procedimiento de producción 4. Para la reacción entre el compuesto (29) y el ácido carboxílico (34), también pueden usarse reactivos y condiciones de reacción que se usan generalmente para la síntesis de péptidos. El compuesto (2c) producido mediante el procedimiento anteriormente mencionado puede derivatizarse para dar el compuesto (1 ) de acuerdo con el procedimiento anteriormente descrito.
9. Procedimiento de producción 9
En lo sucesivo en el presente documento, se describe con detalle el procedimiento para producir el compuesto (17c) que tiene n1 = 1, La = O y Lb = CR2(-R3) en el intermedio de producción (17) descrito en el procedimiento de producción 4. El compuesto representado por la fórmula (17c), una sal o un solvato del mismo también puede producirse de acuerdo con el siguiente procedimiento, por ejemplo.
[Fórmula 65]

En la fórmula, L1', L2, LP, R2, R3, X, Y, P5, P6 y P7 son como se han definido anteriormente.
El compuesto (36) puede producirse desprotegiendo el grupo protector P5 para el grupo amino del compuesto (26) con grupo amino terminal protegido y grupo carboxi para producir el compuesto (35) y haciendo reaccionar la forma de amina obtenida (35) con el oligopéptido (30) con grupo amino terminal protegido o grupo amino protegido. El grupo protector para un grupo amino como se ilustra mediante P5 no ésta particularmente limitado si este es un grupo usado habitualmente para la protección de un grupo amino. Los ejemplos representativos incluyen los grupos protectores
para un grupo amino que se describen en el procedimiento de producción 4. Además, para retirar el grupo protector P5, los reactivos y condiciones pueden seleccionarse dependiendo del grupo protector. En el presente documento, aunque los ejemplos representativos del grupo protector para un grupo carboxi como se ilustra mediante P6 y el grupo protector para un grupo amino como se ilustra mediante P7 incluyen los grupos protectores para un grupo carboxi y un grupo amino que se describen en el procedimiento de producción 4, es deseable que el grupo protector P6 para un grupo carboxi y el grupo protector P7 para un grupo amino sean los grupos protectores que pueden retirarse mediante el mismo procedimiento o las mismas condiciones. Por ejemplo, un ejemplo representativo incluye una combinación en la que P6 es un grupo éster bencílico y P7 es un grupo benciloxi carbonilo.
El compuesto (37) puede producirse retirando el grupo protector P6 para el grupo carboxi del compuesto (36) y el grupo protector P7 para el grupo amino del compuesto (36). El compuesto (37) también puede producirse retirando secuencialmente el grupo protector P6 para el grupo carboxi y el grupo protector P7 para el grupo amino, o el compuesto (37) puede producirse retirando al mismo tiempo ambos de los grupos protectores P6 y P7 que pueden retirarse mediante el mismo procedimiento o las mismas condiciones.
El compuesto (17c) puede producirse haciendo reaccionar el compuesto obtenido (37) con el compuesto (11). Para la reacción entre el compuesto (37) y el compuesto (11), pueden usarse los mismos reactivos y condiciones de reacción que los descritos para el procedimiento de producción 4.
En lo anterior, el compuesto representado por la siguiente fórmula:
[Fórmula 66 ]
Anticuerpo 
se describe como un intermedio de producción útil para producir el conjugado de anticuerpo - fármaco. Además, un grupo de compuestos representada por la siguiente fórmula:
Q-(CH2)nQ-C(=O)-L2a-LP-NH-(CH2)n1-La-Lb-Lc-(NH-DX)
también son compuestos que sirven como intermedios de producción útiles para producir el conjugado de anticuerpo - fármaco.
Específicamente, en la fórmula anterior, Q es (maleimid-N-il)-, HS-, X-CH2-C(=O)-NH- o (pirrolidin-2,5-diona-N-il)-O-C(=O)-,
X es un átomo de bromo o un átomo de yodo,
nQ es un número entero de 2 a 8,
L2a representa -NH-(CH2-CH2-O)n5-CH2-CH2-C(=O)- o un enlace sencillo,
en el que n5 representa un número entero de 1 a 6 ,
LP representa un residuo de péptido que consiste en 2 a 7 aminoácidos,
n1 representa un número entero de 0 a 6 ,
La representa -C(=O)-NH-, -NR1-(CH2)n7, -O- o un enlace sencillo,
en el que n7 representa un número entero de 1 a 6 , R1 representa un átomo de hidrógeno, un grupo alquilo que tiene 1 a 6 átomos de carbono, -(CH2)n8-COOH o -(CH2)n9-OH, n8 representa un número entero de 1 a 4, n9 representa un número entero de 1 a 6 ,
Lb representa -CR2(-R3)-, -O-, -NR4- o un enlace sencillo,
en el que R2 y R3 representan, independientemente cada uno, un átomo de hidrógeno, un grupo alquilo que tiene 1 a 6 átomos de carbono, -(CH2)na-NH2, -(CH2)nb-COOH o -(CH2)nc-OH, R4 representa un átomo de hidrógeno o un grupo alquilo que tiene 1 a 6 átomos de carbono, na representa un número entero de 0 a 6 , nb representa un número entero de 1 a 4 y nc representa un número entero de 0 a 4, con la condición de que cuando na es 0, R2 y R3 no son uno el mismo que el otro,
Lc representa -(CH2)- o -C(=O)-,
(maleimid-N-il)- es un grupo que tiene una estructura representada por la siguiente fórmula:
[F ó rm u la 67 ]

(en la fórmula, el átomo de nitrógeno es la posición de conexión), (Pirrolidin-2,5-diona-N-il)- es un grupo que tiene una estructura representada por la siguiente fórmula:
[Fórmula 68 ]

(en la fórmula, el átomo de nitrógeno es la posición de conexión), y -(NH-DX) es un grupo que tiene una estructura representada por la siguiente fórmula:
[Fórmula 69]

(en la fórmula, el átomo de nitrógeno del grupo amino en la posición 1 es la posición de conexión).
Un compuesto en el que Lc es -C(=O)- se prefiere como un intermedio de producción.
Para el residuo del péptido de LP, un compuesto de un residuo de aminoácido que consiste en un aminoácido seleccionado de fenilalanina, glicina, valina, lisina, citrulina, serina, ácido glutámico y ácido aspártico se prefiere como un intermedio de producción. Entre estos residuos de péptido, un compuesto en el que LP es un residuo de péptido que consiste en 4 aminoácidos se prefiere como un intermedio de producción. Más específicamente, un compuesto en el que LP es -GGFG- se prefiere como un intermedio de producción.
Además, con respecto al -NH-(CH2)n1-La-Lb-, un compuesto de -NH-CH2CH2-, -NH-CH2CH2CH2-, -NH-CH2CH2CH2CH2-, -NH-CH2CH2CH2CH2CH2-, -NH-CH2-O-CH2- o -NH-CH2CH2-O-CH2- se prefiere como un intermedio de producción. Un compuesto de NH-CH2CH2CH2-, -NH-CH2-O-CH2- o -NH-(CH2)2-O-CH2-C(=O)- es más preferido. Para nQ, un compuesto en el que es un número entero de 2 a 6 se prefiere como un intermedio de producción. Un compuesto en el que L2a es un enlace sencillo o n5 es un número entero de 2 a 4 se prefiere como un intermedio de producción.
Cuando Q es (maleimid-N-il)-, un compuesto en el que nQ es un número entero de 2 a 5, L2a es un enlace sencillo y -NH-(CH2)n1-La-Lb- es -NH-CH2CH2-, -NH-CH2CH2CH2-, -NH-CH2CH2CH2CH2-, -NH-CH2CH2CH2CH2CH2-,-NH-CH2-O-CH2- o -NH-CH2CH2-O-CH2- se prefiere como un intermedio de producción. Un compuesto en el que -NH-(CH2)n1-La-Lb- es -NH-CH2CH2-, -NH-CH2CH2CH2-, -NH-CH2-O-CH2- o -NH-CH2CH2-O-CH2- es más preferido. Un compuesto en el que nQ es un número entero de 2 o 5 se prefiere adicionalmente.
También, cuando Q es (maleimid-N-il)-, un compuesto en el que nQ es un número entero de 2 a 5, L2a es -NH-(CH2-CH2-O)n5-CH2-CH2-C(=O)-, n5 es un número entero de 2 a 4 y -NH-(CH2)n1-La-Lb- es -NH-CH2CH2-, -NH-CH2CH2CH2-, -NH-CH2CH2CH2CH2-, -NH-CH2CH2CH2CH2CH2-, -NH-CH2-O-CH2- o-NH-CH2CH2-O-CH2- se prefiere como un intermedio de producción. Un compuesto en el que n5 es un número entero de 2 o 4 es más preferido. Un compuesto en el que -NH-(CH2)n1-La-Lb- es -NH-CH2CH2CH2-, -NH-CH2-O-CH2- o -NH-CH2CH2-O-CH2- se prefiere adicionalmente.
Cuando Q es HS-, un compuesto en el que nQ es un número entero de 2 a 5, L2a es un enlace sencillo y -NH-(CH2)n1-La-Lb- es -NH-CH2CH2-, -NH-CH2CH2CH2-, -NH-CH2CH2CH2CH2-, -NH-CH2CH2CH2CH2CH2-,-NH-CH2-O-CH2- o -NH-CH2CH2-O-CH2- se prefiere como un intermedio de producción. Un compuesto en el que -NH-(CH2)n1-La-Lb- es -NH-CH2CH2CH2-, -NH-CH2-O-CH2- o -NH-CH2CH2-O-CH2- es más preferido.
Cuando Q es X-CH2-C(=O)-NH-, un compuesto en el que X es un átomo de bromo se prefiere como un intermedio de producción. Un compuesto en el que nQ es un número entero de 2 a 8 es preferido, también es preferido un compuesto en el que L2a es un enlace sencillo y un compuesto en el que -NH-(CH2)n1-La-Lb- es -NH-CH2CH2CH2-, -NH-CH2-O-CH2- o -NH-CH2CH2-OCH2- se prefiere como un intermedio de producción.
Cuando Q es (Pirrolidin-2,5-diona-N-il)-O-C(=O)-, un compuesto en el que nQ es un número entero de 2 a 5, L2a es un enlace sencillo y -NH-(CH2)n1-La-Lb- es -NH-CH2CH2-, -NH-CH2CH2CH2-, -NH-CH2CH2CH2CH2-, -NH-CH2CH2CH2CH2CH2-,-NH-CH2-O-CH2- o -NH-CH2CH2-O-CH2- se prefiere como un intermedio de producción. Un compuesto en el que -NH-(CH2)n1-La-Lb- es -NH-CH2CH2CH2-, -NH-CH2-O-CH2- o -NH-CH2CH2-O-CH2- se prefiere más.
De manera más específica, los siguientes son compuestos preferidos como intermedios de producción.
(maleimid-N-il)-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2-(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2CH2CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2-O-CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2-O-CH2-C(=O)-(NH-DX)
(maleimid-N-il)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2 CH2-C(=O)-(NH-DX) (maleimid-N-il)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2-C(=O)-(NH-DX) (maleimid-N-il)-CH2CH2-C(=O)-NH-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2O-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
HS-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
Br-CH2-C(=O)-NH-CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
(Pirrolidin-2 ,5-diona-N-il)-O-C(=O)-CH2CH2CH2CH2CH2CH2-C(=O)-GGFG-NH-CH2CH2CH2-C(=O)-(NH-DX)
Mientras tanto, el conjugado de anticuerpo - fármaco, cuando el mismo se deja al aire o se recristaliza, puede absorber humedad para tener agua de adsorción o convertirse en un hidrato, y también se incluyen en la presente divulgación tal compuesto y una sal que contiene agua.
Un compuesto etiquetado con diversos isótopos radiactivos o no radiactivos también se incluye en la presente divulgación. Uno o más átomos que constituyen el conjugado de anticuerpo - fármaco pueden contener un isótopo atómico en una relación no natural. Los ejemplos del isótopo atómico incluyen deuterio (2H), tritio (3H), yodo-125 (125I) y carbono-14 (14C). Además, el compuesto se puede etiquetar radiactivamente con un isótopo radiactivo tal como tritio (3H), yodo-125 (125I), carbono-14 (14C), cobre-64 (64Cu), zirconio-89 (89Zr), yodo-124 (124I), flúor-18 (18F), indio-111 (111I), carbono-11 (11C) y yodo-131 (131I). El compuesto etiquetado con un isótopo radiactivo es útil como un agente terapéutico o profiláctico, un reactivo para investigación tal como un reactivo de ensayo y un agente para diagnóstico tal como un agente de obtención de imágenes de diagnóstico in vivo. Sin relación con la radiactividad, cualquier tipo de variante de isótopo del conjugado de anticuerpo - fármaco de la presente invención está dentro del ámbito de la presente divulgación.
[Fármacos]
El conjugado de anticuerpo - fármaco muestra una actividad citotóxica contra células cancerosas y, por lo tanto, este puede usarse como un fármaco, particularmente como un agente terapéutico y/o agente profiláctico para el cáncer.
Los ejemplos del tipo de cáncer al que se aplica el conjugado de anticuerpo - fármaco incluyen cáncer de pulmón, cáncer de riñón, cáncer urotelial, cáncer colorrectal, cáncer de próstata, glioblastoma multiforme, cáncer de ovario, cáncer de páncreas, cáncer de mama, melanoma, cáncer de hígado, cáncer de vejiga, cáncer de estómago o cáncer de esófago, sin embargo, este no se limita a los mismos siempre que sea una célula cancerosa que expresa, en una célula cancerosa como un sujeto de tratamiento, una proteína que puede ser reconocida por el anticuerpo dentro del conjugado de anticuerpo - fármaco.
El conjugado de anticuerpo - fármaco puede administrarse preferentemente a un mamífero, pero este se administra más preferentemente a un ser humano.
Las sustancias usadas en una composición farmacéutica que contiene conjugado de anticuerpo - fármaco pueden seleccionarse y aplicarse adecuadamente a partir de aditivos de formulación o similares que se usan generalmente en la técnica, a la vista de la dosificación o concentración de administración.
El conjugado de anticuerpo - fármaco puede administrarse como una composición farmacéutica que contiene al menos
un ingrediente farmacéuticamente adecuado.
Por ejemplo, la composición farmacéutica anterior típicamente contiene al menos un vehículo farmacéutico (por ejemplo, líquido esterilizado), por ejemplo, agua y aceite (crudo de petróleo y aceite de origen animal, origen vegetal u origen sintético (el aceite puede ser, por ejemplo, aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo o similares)). El agua es un vehículo más típico cuando la composición farmacéutica anterior se administra por vía intravenosa. También pueden usarse solución salina, una solución acuosa de dextrosa y una solución acuosa de glicerol como un vehículo líquido, en particular, para una solución en inyección. Se conoce en la técnica un vehículo farmacéutico adecuado. Si se desea, la composición anterior también puede contener una cantidad traza de un agente humectante, un agente emulsionante o un agente de tamponamiento de pH. Se describen algunos ejemplos de vehículo farmacéutico adecuado en "Remington's Pharmaceutical Sciences" de E. W. Martin. Las formulaciones se corresponden con un modo de administración.
Se conocen diversos sistemas de administración, y estos pueden usarse para administrar el conjugado de anticuerpo - fármaco. Los ejemplos de la ruta de administración incluyen las rutas intradérmica, intramuscular, intraperitoneal, intravenosa y subcutánea, pero sin limitarse a las mismas. La administración puede realizarse mediante inyección o inyección en bolo, por ejemplo. De acuerdo con una realización preferida específica, la administración del conjugado de anticuerpo - fármaco se realiza mediante inyección. La administración parenteral es una ruta de administración preferida.
De acuerdo con una realización representativa, la composición farmacéutica se prescribe, como una composición farmacéutica adecuada para la administración intravenosa a un ser humano, de acuerdo con los procedimientos convencionales. La composición para la administración intravenosa es típicamente una solución en una solución tampón acuosa estéril e isotónica. Si fuera necesario, el fármaco puede contener un agente solubilizante y un anestésico local para aliviar el dolor en un sitio de inyección (por ejemplo, lignocaína). Generalmente, el ingrediente anterior se proporciona individualmente como uno cualquiera de polvo liofilizado o un concentrado anhidro contenido en un recipiente que se obtiene mediante sellado en una ampolla o un sobre que tiene una cantidad del agente activo o como una mezcla en una forma de dosificación unitaria. Cuando el fármaco se va a administrar mediante inyección, este puede administrarse a partir de una botella de inyección que contiene agua o solución salina de calidad farmacéutica estéril. Cuando el fármaco se administra mediante inyección, puede proporcionarse una ampolla de agua estéril o solución salina para su inyección de tal modo que los ingredientes anteriormente mencionados se mezclan entre sí antes de la administración.
La composición farmacéutica puede ser una composición farmacéutica que contiene solo el conjugado de anticuerpo - fármaco o una composición farmacéutica que contiene el conjugado de anticuerpo - fármaco y al menos un agente para tratar el cáncer que no sea el conjugado. El conjugado de anticuerpo - fármaco puede administrarse con otro agente para tratar el cáncer. El efecto anti-cancerígeno puede potenciarse en consecuencia. Otro agente anti cancerígeno usado para tal fin puede administrarse a un individuo de forma simultánea con, por separado de o subsiguientemente al conjugado de anticuerpo - fármaco, y este puede administrarse al tiempo que se varía el intervalo de administración para cada uno. Los ejemplos del agente para tratar el cáncer incluyen abraxano, carboplatino, cisplatino, gemcitabina, irinotecán (CPT-11), paclitaxel, pemetrexed, sorafenib, vinorelbina, fármacos descritos en la publicación internacional n.° WO 2003/038043, análogos de LH-RH (leuprorelina, goserelina, o similares), fosfato de estramustina, antagonista de estrógenos (tamoxifeno, raloxifeno, o similares), y un inhibidor de la aromatasa (anastrozol, letrozol, exemestano, o similares), pero este no está limitado siempre que el mismo sea un fármaco que tiene una actividad antitumoral.
La composición farmacéutica puede formularse en una formulación de liofilización o una formulación líquida como una formulación que tiene una composición deseada y una pureza requerida. Cuando se formula como una formulación de liofilización, esta puede ser una formulación que contiene aditivos de formulación adecuados que se usan en la técnica. También para una formulación líquida, esta puede formularse como una formulación líquida que contiene diversos aditivos de formulación que se usan en la técnica.
La composición y la concentración de la composición farmacéutica pueden variar dependiendo del procedimiento de administración. Sin embargo, el conjugado de anticuerpo - fármaco contenido en la composición farmacéutica puede mostrar el efecto farmacéutico incluso a una dosificación pequeña cuando el conjugado de anticuerpo - fármaco tiene una afinidad más alta por un antígeno, es decir, una afinidad más alta (= valor de Kd más bajo) en términos de la constante de disociación (es decir, valor de Kd) para el antígeno. De ese modo, para determinar la dosificación del conjugado de anticuerpo - fármaco, la dosificación puede determinarse a la vista de una situación en relación con la afinidad entre el conjugado de anticuerpo - fármaco y antígeno. Cuando el conjugado de anticuerpo - fármaco se administra a un ser humano, por ejemplo, aproximadamente 0,001 a 100 mg/kg pueden administrarse una vez o administrarse varias veces con un intervalo de una vez durante de 1 a 180 días.
rEiemplosl
La presente invención se describe específicamente a la vista de los ejemplos mostrados posteriormente. Sin embargo, la presente invención no se limita a los mismos. Además, esta no se ha de interpretar en modo alguno en un sentido limitado. Además, a menos que específicamente se describa lo contrario, el reactivo, disolvente, y material de partida descritos en la memoria descriptiva pueden obtenerse fácilmente a partir de un proveedor comercial. Los Ejemplos 58
a 74, en particular el Ejemplo 74, son de acuerdo con la invención, mientras que los Ejemplos 1 a 57, 75 y 76 son ejemplos de referencia.
Ejemplo de referencia 1 Anticuerpo M30-H1-L4
De anticuerpos humanizados de un anticuerpo anti-B7-H3, un anticuerpo compuesto por una cadena pesada que consiste en una secuencia de aminoácidos descrita en las posiciones de aminoácido 20 a 471 en la SEQ ID NO: 9 y una cadena ligera que consiste en una secuencia de aminoácidos descrita en las posiciones de aminoácido 21 a 233 en la SEQ ID NO: 16 se produjo de acuerdo con un procedimiento conocido en la técnica para producir anticuerpo anti-B7-H3 humanizado designado como un anticuerpo M30-H1-L4 (o al que se hace referencia simplemente como "M30-H1-L4").
Ejemplo de referencia 2 Anticuerpo M30-H1-L4P
La modificación de un glicano unido al anticuerpo M30-H1-L4 obtenido anteriormente se reguló mediante desfucosilación de acuerdo con un procedimiento conocido en la técnica para producir anticuerpo con la modificación regulada de un glicano designado como un anticuerpo M30-H1-L4P (o al que se hace referencia simplemente como "M30-H1-L4P").
Ejemplo de referencia 3 Anticuerpo anti-CD30
Un anticuerpo anti-CD30 se produjo con referencia a la publicación nacional de la solicitud de patente internacional n.° 2005-506035. Su secuencia se muestra en las SEQ iD NO: 27 y 28.
Ejemplo de referencia 4 Anticuerpo anti-CD33
Un anticuerpo anti-CD33 se produjo con referencia a la patente japonesa abierta a inspección pública n.° 8-48637. Su secuencia se muestra en las SEQ ID NO: 29 y 30.
Ejemplo de referencia 5 Anticuerpo anti-CD70
Un anticuerpo anti-CD70 se produjo con referencia a la publicación nacional de la solicitud de patente internacional n.° 2008-538292. Su secuencia se muestra en las SEQ iD NO: 31 y 32.
Ejemplo 1 4-Amino-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]butanamida
[Fórmula 70]

Proceso 1: (4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]aminol-4-oxobutil)carbamato de terc-butilo
Se disolvió ácido 4-(terc-butoxicarbonilamino)butanoico (0,237 g, 1,13 mmol) en diclorometano (10 ml), Se añadieron N-hidroxisuccinimida (0,130 g, 1,13 mmol) y clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (0,216 g, 1,13 mmol), y se agitó durante 1 hora. La solución de reacción se añadió gota a gota a una solución de N,N-dimetilformamida (10 ml) cargada con mesilato del compuesto (4) (0,500 g, 0,94 mmol) y trietilamina (0,157 ml, 1,13 mmol) y se agitó durante 1 día a temperatura ambiente. El disolvente se retiró a presión reducida y el residuo obtenido se purificó por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo : metanol = 8 : 2 (v/v)] para producir el compuesto del título (0,595 g, cuantitativo). RMN 1H (400 MHz, DMSO-da) 5: 0,87 (3H, t, J = 7,2 Hz), 1,31 (9H, s), 1,58 (1H, t, J = 7,2 Hz), 1,66 (2H, t, J = 7,2 Hz), 1,82-1,89 (2H, m), 2,12-2,21 (3H, m), 2,39 (3H, s), 2,92 (2H, t, J = 6,5 Hz), 3,17 (2H, s), 5,16 (1H, d, J = 18,8 Hz), 5,24 (1H, d, J = 18,8 Hz), 5,42 (2H, s), 5,59-5,55 (1H, m), 6,53 (1H, s), 6,78 (1H, t, J = 6,3 Hz), 7,30 (1H, s), 7,79 (1H, d, J = 11,0 Hz), 8,40 (1H, d, J = 8,6 Hz).
EM (APCI) m/z: 621 (M+H)+
Proceso 2: 4-Amino-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]butanamida
El compuesto (0,388 g, 0,61 mmol) obtenido en el proceso 1 anterior se disolvió en diclorometano (9 ml). Se añadió ácido trifluoroacético (9 ml) y se agitó durante 4 horas. El disolvente se retiró a presión reducida y los residuos
obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - capa orgánica repartida de cloroformo : metanol : agua = 7 : 3 : 1 (v/v/v)] para producir trifluoroacetato del compuesto del título (0,343 g, cuantitativo). Este compuesto se confirmó en el tumor de un ratón portador de cáncer que recibió el conjugado de anticuerpo - fármaco (13) o (14).
RMN 1H (400 MHz, DMSO-da) 5: 0,87 (3H, t, J = 7,2 Hz), 1,79-1,92 (4H, m), 2,10-2,17 (2H, m), 2,27 (2H, t, J = 7,0 Hz), 2,40 (3H, s), 2,80-2,86 (2H, m), 3,15-3,20 (2H, m), 5,15 (1H, d, J = 18,8 Hz), 5,26 (1H, d, J = 18,8 Hz), 5,42 (2H, s), 5,54-5,61 (1H, m), 6,55 (1H, s), 7,32 (1H, s), 7,72 (3H, s a), 7,82 (1H, d, J = 11,0 Hz), 8,54 (1H, d, J = 8,6 Hz).
EM (APCI) m/z: 521 (M+H)+
Ejemplo 2 Conjugado de anticuerpo - fármaco (1)
[Fórmula 71]

Proceso 1: N-(terc-butoxicarbonil)glicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2.3.9.10.13.15- hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
Se disolvió N-(terc-butoxicarbonil)glicilglicil-L-fenilalanilglicina (0,081 g, 0,19 mmol) en diclorometano (3 ml), se añadieron N-hidroxisuccinimida (0,021 g, 0,19 mmol) y clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (0,036 g, 0,19 mmol) y después se agitó durante 3,5 horas. La solución de reacción se añadió gota a gota a una solución de N,N-dimetilformamida (1,5 ml) cargada con el compuesto (0,080 g, 0,15 mmol) del Ejemplo 1, y se agitó durante 4 horas a temperatura ambiente. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo : metanol = 8 : 2 (v/v)] para producir el compuesto del título (0,106 g, 73 %).
RMN 1H (400 MHz, DMSO-d6) 5: 0,87 (3H, t, J = 7,4 Hz), 1,36 (9H, s), 1,71 (2H, m), 1,86 (2H, t, J = 7,8 Hz), 2,15-2,19 (4H, m), 2,40 (3H, s), 2,77 (1H, dd, J = 12,7, 8,8 Hz), 3,02 (1H, dd, J = 14,1, 4,7 Hz), 3,08-3,11 (2H, m), 3,16-3,19 (2H, m), 3,54 (2H, d, J = 5,9 Hz), 3,57-3,77 (4H, m), 4,46-4,48 (1H, m), 5,16 (1H, d, J = 19,2 Hz), 5,25 (1H, d, J = 18,8 Hz), 5,42 (2H, s), 5,55-5,60 (1H, m), 6,53 (1H, s), 7,00 (1H, t, J = 6,3 Hz), 7,17-7,26 (5H, m), 7,31 (1H, s), 7,71 (1H, t, J = 5,7 Hz), 7,80 (1H, d, J = 11,0 Hz), 7,92 (1H, t, J = 5,7 Hz), 8,15 (1H, d, J = 8,2 Hz), 8,27 (1H, t, J = 5,5 Hz), 8,46 (1H, d, J = 8,2 Hz). EM (APCI) m/z: 939 (M+H)+
Proceso 2: Trifluoroacetato de glicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2.3.9.10.13.15- hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El compuesto (1,97 g, 2,10 mmol) obtenido en el proceso 1 se disolvió en diclorometano (7 ml). Después de añadir ácido trifluoroacético (7 ml), se agitó durante 1 hora. El disolvente se retiró a presión reducida, y se cargó con tolueno para la destilación azeotrópica. Los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - capa orgánica repartida de cloroformo : metanol: agua = 7 : 3 : 1 (v/v/v)] para producir el compuesto del título (1,97 g, 99 %).
RMN 1H (400 MHz, DMSO-d6) 5: 0,87 (3H, t, J = 7,4 Hz), 1,71-1,73 (2H, m), 1,82-1,90 (2H, m), 2,12-2,20 (4H, m),
2,40 (3H, s), 2,75 (1H, dd, J = 13,7, 9,4 Hz), 3,03-3,09 (3H, m), 3,18-3,19 (2H, m), 3,58-3,60 (2H, m), 3,64 (1H, d, J = 5,9 Hz), 3,69 (1H, d, J = 5,9 Hz), 3,72 (1H, d, J = 5,5 Hz), 3,87 (1H, dd, J = 16,8, 5,9 Hz), 4,50-4,56 (1H, m), 5,16 (1H, d, J = 19,2 Hz), 5,25 (1H, d, J = 18,8 Hz), 5,42 (2H, s), 5,55-5,60 (1H, m), 7,17-7,27 (5H, m), 7,32 (1H, s), 7,78-7,81 (2H, m), 7,95-7,97 (3H, m), 8,33-8,35 (2H, m), 8,48-8,51 (2H, m).
EM (APCI) m/z: 839 (M+H)+
Proceso 3: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]glicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
A una solución de N,N-dimetilformamida (1,2 ml) del compuesto (337 mg, 0,353 mmol) obtenido en el proceso 2, se añadieron trietilamina (44,3 ml, 0,318 mmol) y hexanoato de N-succinimidil 6-maleimida (119,7 mg, 0,388 mmol) y se agitó a temperatura ambiente durante 1 hora. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo : metanol = 5 : 1 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (278,0 mg, 76 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,3 Hz), 1,12-1,22 (2H, m), 1,40-1,51 (4H, m), 1,66-1,76 (2H, m), 1,80-1,91 (2H, m), 2,05-2,21 (6H, m), 2,39 (3H, s), 2,79 (1H, dd, J = 14,0, 9,8 Hz), 2,98-3,21 (5H, m), 3,55-3,77 (8H, m), 4,41-4,48 (1H, m), 5,15 (1H, d, J = 18,9 Hz), 5,24 (1H, d, J = 18,9 Hz), 5,40 (1H, d, J = 17,1 Hz), 5,44 (1H, d, J = 17,1 Hz), 5,54-5,60 (1H, m), 6,53 (1H, s), 6,99 (2H, s), 7,20-7,27 (5H, m), 7,30 (1H, s), 7,70 (1H, t, J = 5,5 Hz), 7,80 (1H, d, J = 11,0 Hz), 8,03 (1H, t, J = 5,8 Hz), 8,08 (1H, t, J = 5,5 Hz), 8,14 (1H, d, J = 7,9 Hz), 8,25 (1H, t, J = 6,1 Hz), 8,46 (1H, d, J = 8,5 Hz). EM (APCI) m/z: 1032 (M+H)+
Proceso 4: Conjugado de anticuerpo - fármaco (1)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 10 mg/ml sustituyendo el medio con PBS6,0/EDTA usando el procedimiento común C-1 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg'1cirr1) descrito en el procedimiento de producción 1. La solución (1,25 ml) se puso en un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,025 ml; 3,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte de bisagra en el anticuerpo se redujo incubando durante 1 hora a 37 °C.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,109 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 3 (0,039 ml; 4,6 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 40 minutos. A continuación, una solución acuosa (0,008 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se agitó a temperatura ambiente para terminar la reacción del enlazador de fármaco durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A. Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 13,02 mg/ml, rendimiento de anticuerpo: 9,1 mg (73%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,4.
Ejemplo 3 Conjugado de anticuerpo - fármaco (2)
[Fórmula 72]

Proceso 1: Conjugado de anticuerpo - fármaco (2)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (4,0 ml) se recogió en un tubo de 15 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,118 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,200 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora. Conjugación entre anticuerpo y enlazador de fármaco: Después de incubar la solución durante 10 minutos a 22 °C, una solución de dimetilsulfóxido (0,236 ml; 9,2 equivalentes por molécula de anticuerpo) que contiene 10 mM del compuesto obtenido en el proceso 3 del Ejemplo 2 se añadió a la misma y se incubó para conjugar el enlazador de fármaco con el anticuerpo a 22 °C durante 40 minutos. A continuación, se añadió una solución acuosa (0,00471 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) a la misma y se incubó para terminar la reacción del enlazador de fármaco a 22 °C durante otros 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 17,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28ü = 235300 (valor de cálculo estimado), £a,37ü = 0 (valor de cálculo estimado), £d,28ü = 5000 (valor promedio medido) y £d,37ü= 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 1,80 mg/ml, rendimiento de anticuerpo: 26,1 mg (65%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,9.
Ejemplo 4 Conjugado de anticuerpo - fármaco (3)
[Fórmula 73]

Proceso 1: Conjugado de anticuerpo - fármaco (3)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 10 mg/ml sustituyendo el medio con PBS6,0/EDTA usando el procedimiento
común C-1 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) descrito en el procedimiento de producción 1. La solución (1,25 ml) se puso en un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,051 ml; 6,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,067 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 3 del Ejemplo 2 (0,085 ml; 10,0 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 60 minutos. A continuación, se añadió una solución acuosa (0,013 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) a la misma y se agitó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £A,28o = 235300 (valor de cálculo estimado), £A,37o = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 1,67 mg/ml, rendimiento de anticuerpo: 10,02 mg (80%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,3.
Ejemplo 5 Conjugado de anticuerpo - fármaco (4)
[Fórmula 74]
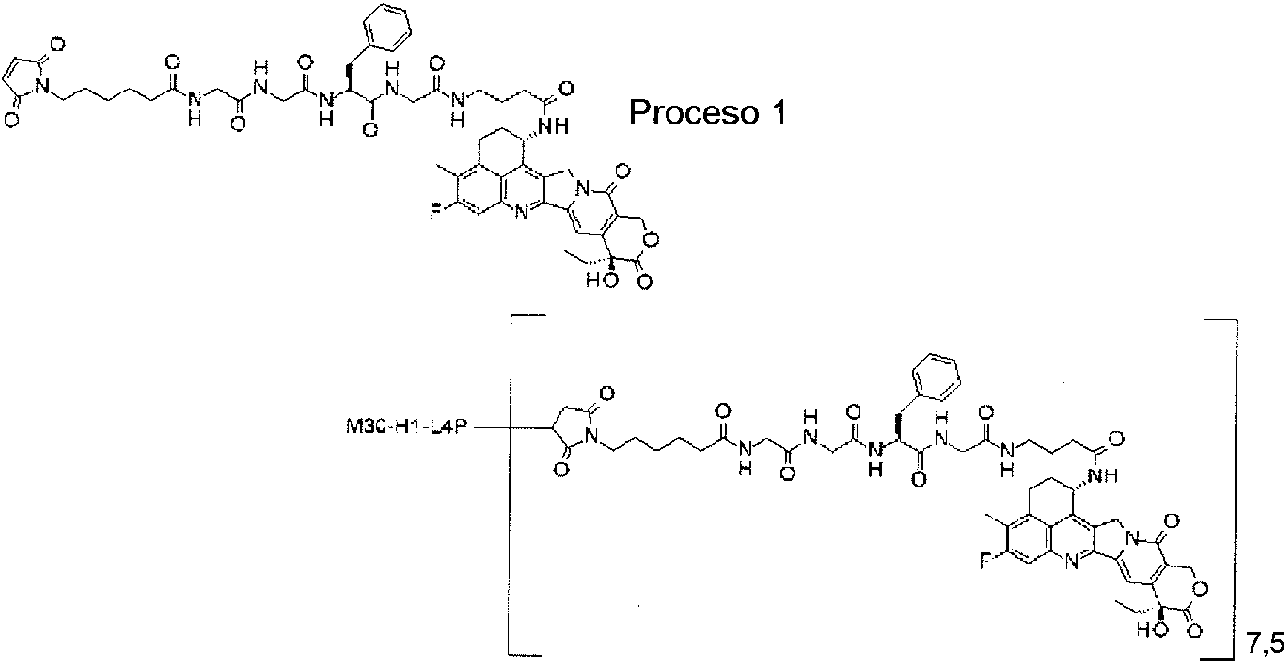
Proceso 1: Conjugado de anticuerpo - fármaco (4)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 10 mg/ml sustituyendo el medio con PBS6,0/EDTA usando el procedimiento común C-1 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) descrito en el procedimiento de producción 1. La solución (1,25 ml) se puso en un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,051 ml; 6,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,025 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 3 del Ejemplo 2 (0,127 ml; 15,0 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 60 minutos. A continuación, se añadió una solución acuosa (0,019 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) a la misma y se agitó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el
conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A. Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 1,19 mg/ml, rendimiento de anticuerpo: 7,14 mg (57%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 7,5.
Ejemplo 6 Conjugado de anticuerpo - fármaco (5)
[Fórmula 75]

Casi la totalidad de las cantidades de los conjugados de anticuerpo - fármaco de Ejemplos 4 y 5 se mezclaron. La solución se concentró mediante el procedimiento común A para producir el conjugado de anticuerpo - fármaco del título.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 15,37 mg, y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,7.
Ejemplo 7 Conjugado de anticuerpo - fármaco (6)
[Fórmula 76]

Proceso 1: Conjugado de anticuerpo - fármaco (6)
Reducción del anticuerpo: El anticuerpo anti-CD30 producido en el Ejemplo de Referencia 3 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,75) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de incubar la solución a 22 °C durante 10 minutos, una solución de dimetilsulfóxido (0,0593 ml; 9,2 equivalentes por molécula de anticuerpo) que contiene 10 mM del compuesto obtenido en el proceso 3 del Ejemplo 2 se añadió a la misma y se incubó para conjugar el enlazador de fármaco con el anticuerpo a 22 °C durante 40 minutos. A continuación, una solución acuosa (0,0119 ml; 18,4 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó
para terminar la reacción del enlazador de fármaco a 22 °C durante otros 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,2804 = 270400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 0,99 mg/ml, rendimiento de anticuerpo: 5,94 mg (59%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,3.
Ejemplo 8 Conjugado de anticuerpo - fármaco (7)
[Fórmula 77]

Proceso 1: Conjugado de anticuerpo - fármaco (7)
Reducción del anticuerpo: El anticuerpo anti-CD30 producido en el Ejemplo de Referencia 3 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,75) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 30 mM (Tokyo Chemical Industry Co., Ltd.) (0,0148 ml; 6,9 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de incubar la solución durante 10 minutos a 22 °C, una solución de dimetilsulfóxido (0,0297 ml; 13,8 equivalentes por molécula de anticuerpo) que contiene 30 mM del compuesto obtenido en el proceso 3 del Ejemplo 2 se añadió a la misma y se incubó durante 4o minutos a 22 °C para conjugar el enlazador de fármaco con el anticuerpo. A continuación, una solución acuosa (0,0178 ml; 27,6 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a 22 °C durante otros 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28o = 270400 (valor de cálculo estimado), £a,37o = 0 (valor de cálculo estimado), £d,28o = 5000 (valor promedio medido) y £d,37o = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 0,99 mg/ml, rendimiento de anticuerpo: 5,94 mg (59%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,8.
Ejemplo 9 Conjugado de anticuerpo - fármaco (8)
[Fórmula 78]

Proceso 1: Conjugado de anticuerpo - fármaco (8)
Reducción del anticuerpo: El anticuerpo anti-CD33 producido en el Ejemplo de Referencia 4 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de incubar la solución durante 10 minutos a 22 °C, una solución de dimetilsulfóxido (0,0593 ml; 9,2 equivalentes por molécula de anticuerpo) que contiene 10 mM del compuesto obtenido en el proceso 3 del Ejemplo 2 se añadió a la misma y se incubó para conjugar el enlazador de fármaco con el anticuerpo a 22 °C durante 40 minutos. A continuación, una solución acuosa (0,0119 ml; 18,4 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a 22 °C durante otros 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron eA,28o = 256400 (valor de cálculo estimado), eA,37o = 0 (valor de cálculo estimado), eDi280 = 5000 (valor promedio medido) y eD370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 1,06 mg/ml, rendimiento de anticuerpo: 6,36 mg (64%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,4.
Ejemplo 10 Conjugado de anticuerpo - fármaco (9)
[Fórmula 79]

Proceso 1: Conjugado de anticuerpo - fármaco (9)
Reducción del anticuerpo: El anticuerpo anti-CD33 producido en el Ejemplo de Referencia 4 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 30 mM (Tokyo Chemical Industry Co., Ltd.) (0,0148 ml; 6,9 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de incubar la solución durante 10 minutos a 22 °C, una solución de dimetilsulfóxido (0,0297 ml; 13,8 equivalentes por molécula de anticuerpo) que contiene 30 mM del compuesto obtenido en el proceso 3 del Ejemplo 2 se añadió a la misma y se incubó para conjugar el enlazador de fármaco con el anticuerpo a 22 °C durante 40 minutos. A continuación, una solución acuosa (0,0178 ml; 27,6 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a 22 °C durante otros 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28ü = 256400 (valor de cálculo estimado), £a,37ü = 0 (valor de cálculo estimado), £Dj28ü = 5000 (valor promedio medido) y £Dj37ü = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 0,95 mg/ml, rendimiento de anticuerpo: 5,70 mg (57%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,7.
Ejemplo 11 Conjugado de anticuerpo - fármaco (10)
[Fórmula 80]

Proceso 1: Conjugado de anticuerpo - fármaco (10)
Reducción del anticuerpo: El anticuerpo anti-CD70 producido en el Ejemplo de Referencia 5 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,69) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0297 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de incubar la solución a 22 °C durante 10 minutos, una solución de dimetilsulfóxido (0,0593 ml; 9,2 equivalentes por molécula de anticuerpo) que contiene 10 mM del compuesto obtenido en el proceso 3 del Ejemplo 2 se añadió a la misma y se incubó para conjugar el enlazador de fármaco con el anticuerpo a 22 °C durante 40 minutos. A continuación, una solución acuosa (0,0119 ml; 18,4 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a 22 °C durante otros 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 262400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 1,00 mg/ml, rendimiento de anticuerpo: 6,00 mg (60%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,2.
Ejemplo 12 Conjugado de anticuerpo - fármaco (11)
[Fórmula 81]

Proceso 1: Conjugado de anticuerpo - fármaco (11)
Reducción del anticuerpo: El anticuerpo anti-CD70 producido en el Ejemplo de Referencia 5 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,69) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 2 ml y se cargó con una solución acuosa de TCEP 30 mM (Tokyo Chemical Industry Co., Ltd.) (0,0148 ml; 6,9 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de incubar la solución a 22 °C durante 10 minutos, una solución de dimetilsulfóxido (0,0297 ml; 13,8 equivalentes por molécula de anticuerpo) que contiene 30 mM del compuesto obtenido en el proceso 3 del Ejemplo 2 se añadió a la misma y se incubó durante 40 minutos a 22 °C para conjugar el enlazador de fármaco con el anticuerpo. A continuación, una solución acuosa (0,0178 ml; 27,6 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a 22 °C durante otros 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £A,28o = 262400 (valor de cálculo estimado), £Aí370 = 0 (valor de cálculo estimado), £d,370 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 0,96 mg/ml, rendimiento de anticuerpo: 5,76 mg (58 %), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,6.
Ejemplo 13 Conjugado de anticuerpo - fármaco (12)
[Fórmula 82]

Proceso 1: N-[3-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)propanoil]glicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9
hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El compuesto (80 mg, 0,084 mmol) obtenido en el proceso 2 del Ejemplo 2 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 usando propionato de N-succinimidil 3-maleimida (24,6 mg, 0,0924 mmol) en lugar de hexanoato de N-succinimidil 6-maleimida para producir el compuesto del título en forma de un sólido de color amarillo pálido (60,0 mg, 73 %). RMN 1H (400 MHz, DMSO-da) 5: 0,89 (3H, t, J = 7,3 Hz), 1,70-1,78 (2H, m), 1,81-1,94 (2H, m), 2,12-2,23 (4H, m), 2,42 (3H, s), 2,81 (1H, dd, J = 13,7, 9,8 Hz), 3,01-3,15 (3H, m), 3,16-3,23 (2H, m), 3,30-3,35 (1H, m), 3,58-3,71 (6H, m), 3,71-3,79 (1H, m), 4,44-4,51 (1H, m), 5,19 (1H, d, J = 19,0 Hz), 5,27 (1H, d, J = 19,0 Hz), 5,43 (1H, d, J = 17,6 Hz), 5,47 (1H, d, J = 17,6 Hz), 5,57-5,63 (1H, m), 6,56 (1H, s), 7,02 (2H, s), 7,17-7,22 (1H, m), 7,22 7,30 (5H, m), 7,34 (1H, s), 7,73 (1H, t, J = 5,6 Hz), 7,83 (1H, d, J = 10,7 Hz), 8,08 (1H, t, J = 5,6 Hz), 8,15 (1H, d, J = 7,8 Hz), 8,30 (2H, dt, J = 18,7, 5,7 Hz), 8,499 (1H, d, J = 8,8 Hz). EM (APCI) m/z: 990 (M+H)+
Proceso 2: Conjugado de anticuerpo - fármaco (12)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 1, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 4 del Ejemplo 2. Concentración de anticuerpo: 12,16 mg/ml, rendimiento de anticuerpo: 8,5 mg (68%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,4.
Ejemplo 14 Conjugado de anticuerpo - fármaco (13)
[Fórmula 83]

Proceso 1: N-{3-[2-(2-{[3-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)propanoil]amino}etoxi)etoxi]propanoil}glicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolin-1-il]aminol-4-oxobutil)glicinamida
El compuesto (100 mg, 0,119 mmol) obtenido en el proceso 2 del Ejemplo 2 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 usando diisopropiletilamina (20,8 ^l, 0,119 mmol) en lugar de trietilamina y 3-(2-(2-(3-maleinimidapropanamida)etoxi)etoxi)propanoato de N-succinimidilo (50,7 mg, 0,119 mmol) en lugar de hexanoato de N-succinimidil 6-maleimida para producir el compuesto del título en forma de un sólido de color amarillo pálido (66,5 mg, 48 %).
RMN 1H (400 MHz, DMSO-d6) 5: 0,85 (3H, t, J = 7,4 Hz), 1,65-1,74 (2H, m), 1,77-1,90 (2H, m), 2,07-2,19 (4H, m), 2,30 (2H, t, J = 7,2 Hz), 2,33-2,36 (2H, m), 2,38 (3H, s), 2,76 (1H, dd, J = 13,7, 9,8 Hz), 2,96-3,18 (9H, m), 3,42-3,44 (4H, m), 3,53-3,76 (10H, m), 4,43 (1H, td, J = 8 ,6 , 4,7 Hz), 5,14 (1H, d, J = 18,8 Hz), 5,23 (1H, d, J = 18,8 Hz), 5,38 (1H, d, J = 17,2 Hz), 5,42 (1H, d, J = 17,2 Hz), 5,52-5,58 (1H, m), 6,52 (1H, s), 6,98 (2H, s), 7,12-7,17 (1H, m), 7,18-7,25 (4H, m), 7,29 (1H, s), 7,69 (1H, t, J = 5,5 Hz), 7,78 (1H, d, J = 11,3 Hz), 7,98-8,03 (2H, m), 8,11 (1H, d, J = 7,8 Hz), 8,16 (1H, t, J = 5,7 Hz), 8,23 (1H, t, J = 5,9 Hz), 8,44 (1H, d, J = 9,0 Hz).
EM (APCI) m/z: 1149 (M+H)+
Proceso 2: Conjugado de anticuerpo - fármaco (13)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 1, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 4 del Ejemplo 2. Concentración de anticuerpo: 12,76 mg/ml, rendimiento de anticuerpo: 8,9 mg (71%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,4.
Ejemplo 15 Conjugado de anticuerpo - fármaco (14)
[Fórmula 84]

Proceso 1: Conjugado de anticuerpo - fármaco (14)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 1 del Ejemplo 14, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 1 del Ejemplo 4. Concentración de anticuerpo: 1,60 mg/ml, rendimiento de anticuerpo: 9,60 mg (77%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6 ,1.
Ejemplo 16 Conjugado de anticuerpo - fármaco (15)
[Fórmula 85]

Proceso 1: Conjugado de anticuerpo - fármaco (15)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 1 del Ejemplo 14, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 1 del Ejemplo 5. Concentración de anticuerpo: 1,64 mg/ml, rendimiento de anticuerpo: 9,84 mg (79%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 7,1.
Ejemplo 17 Conjugado de anticuerpo - fármaco (16)
[Fórmula 86]

Casi la totalidad de las cantidades de los conjugados de anticuerpo - fármaco de Ejemplos 15 y 16 se mezclaron. La solución se concentró mediante el procedimiento común A para producir el conjugado de anticuerpo - fármaco del título.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 17,30 mg, y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,5.
Ejemplo 18 Conjugado de anticuerpo - fármaco (17)
[Fórmula 87]

Proceso 1: Conjugado de anticuerpo - fármaco (17)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (100 ml, 1 g del anticuerpo) se puso en un matraz de 250 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (2,43 ml; 3,6 equivalentes por molécula de anticuerpo) y adicionalmente con una solución acuosa de hidrogenofosfato de dipotasio 1 M (5 ml). Después de confirmar que la solución tenía un pH cerca de 7,4 usando un pehachímetro, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (2,14 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 14 (3,51 ml; 5,2 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, se agitó con un agitador para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 130 minutos. A continuación, una solución acuosa (0,547 ml) de NAC 100 mM se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos. Purificación: La solución anterior se sometió a purificación por ultrafiltración usando un aparato de ultrafiltración compuesto por una membrana de ultrafiltración (Merck Japan, Casete Pellicon XL, Biomax 50 KDa), una bomba de tubo (Cole-Parmer International, Bomba MasterFlex modelo 77521-40, Cabezal de Bomba modelo 7518-00), y un tubo (Cole-Parmer International, Tubo MasterFlex L/S16). Específicamente, al tiempo que se añadía ABS gota a gota (un total de 800 ml) como una solución tampón para la purificación a la solución de reacción, se realizó una purificación por ultrafiltración para retirar enlazadores de fármaco no conjugados y otros reactivos de bajo peso molecular, sustituir también la solución tampón con ABS, y concentrar adicionalmente la solución, para producir aproximadamente 70 ml de una solución que contiene
el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 4964 (valor medido) y £d,370 = 18982 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 14,5 mg/ml, rendimiento de anticuerpo: 1,0 g (aproximadamente 100 %), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,5.
Ejemplo 19 Conjugado de anticuerpo - fármaco (18)
[Fór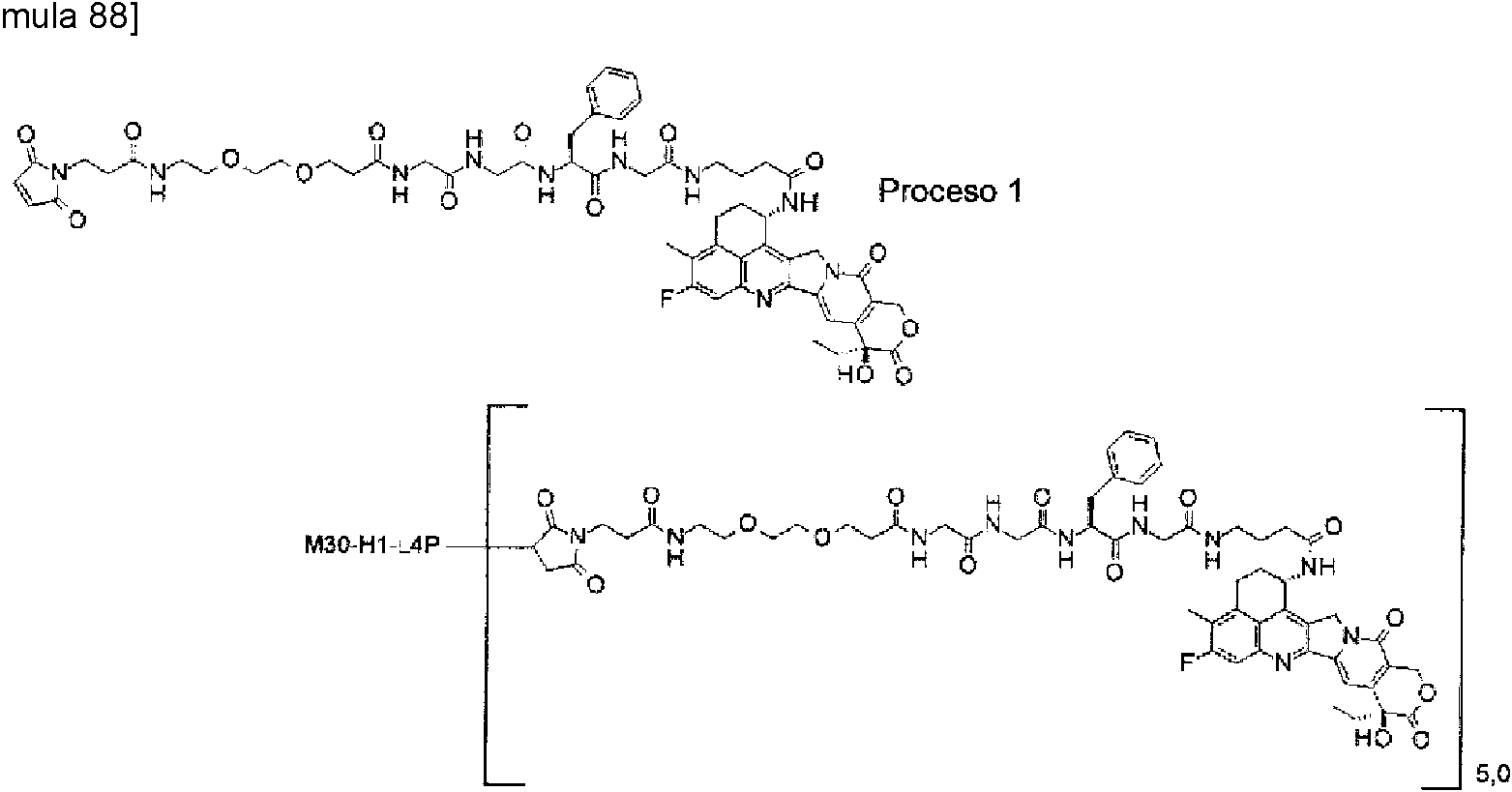
Proceso 1: Conjugado de anticuerpo - fármaco (18)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg'1cirr1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (5 ml, 50 mg del anticuerpo) se puso en un tubo de 15 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,135 ml; 4 equivalentes por molécula de anticuerpo). Después de confirmar que la solución tenía un pH cerca de 7,4 usando un pehachímetro, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (0,064 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 14 (0,219 ml; 6,5 equivalentes por molécula de anticuerpo) a la solución anterior, se incubó para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 90 minutos. A continuación, una solución acuosa (0,033 ml; 9,8 equivalentes por molécula de anticuerpo) de NAC 100 mM se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 19 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 4964 (valor medido) y £d,370 = 18982 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 2,17 mg/ml, rendimiento de anticuerpo: 41 mg (82%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,0.
Ejemplo 20 Conjugado de anticuerpo - fármaco (19)
[Fórmula 89]

Proceso 1: Conjugado de anticuerpo - fármaco (19)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg'1cirr1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (4 ml, 40 mg del anticuerpo) se puso en un tubo de 15 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,140 ml; 5,2 equivalentes por molécula de anticuerpo). Después de confirmar que la solución tenía un pH cerca de 7,4 usando un pehachímetro, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora. Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 14 (0,232 ml; 8,6 equivalentes por molécula de anticuerpo) a la solución anterior, se incubó para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 90 minutos. A continuación, una solución acuosa (0,035 ml; 12,9 equivalentes por molécula de anticuerpo) de NAC 100 mM se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 13 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28ü = 235300 (valor de cálculo estimado), £a,37ü = 0 (valor de cálculo estimado), £d2sü = 4964 (valor medido) y £d,370 = 18982 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 2,36 mg/ml, rendimiento de anticuerpo: 31 mg (77%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,9.
Ejemplo 21 Conjugado de anticuerpo - fármaco (20)
[Fórmula 90]
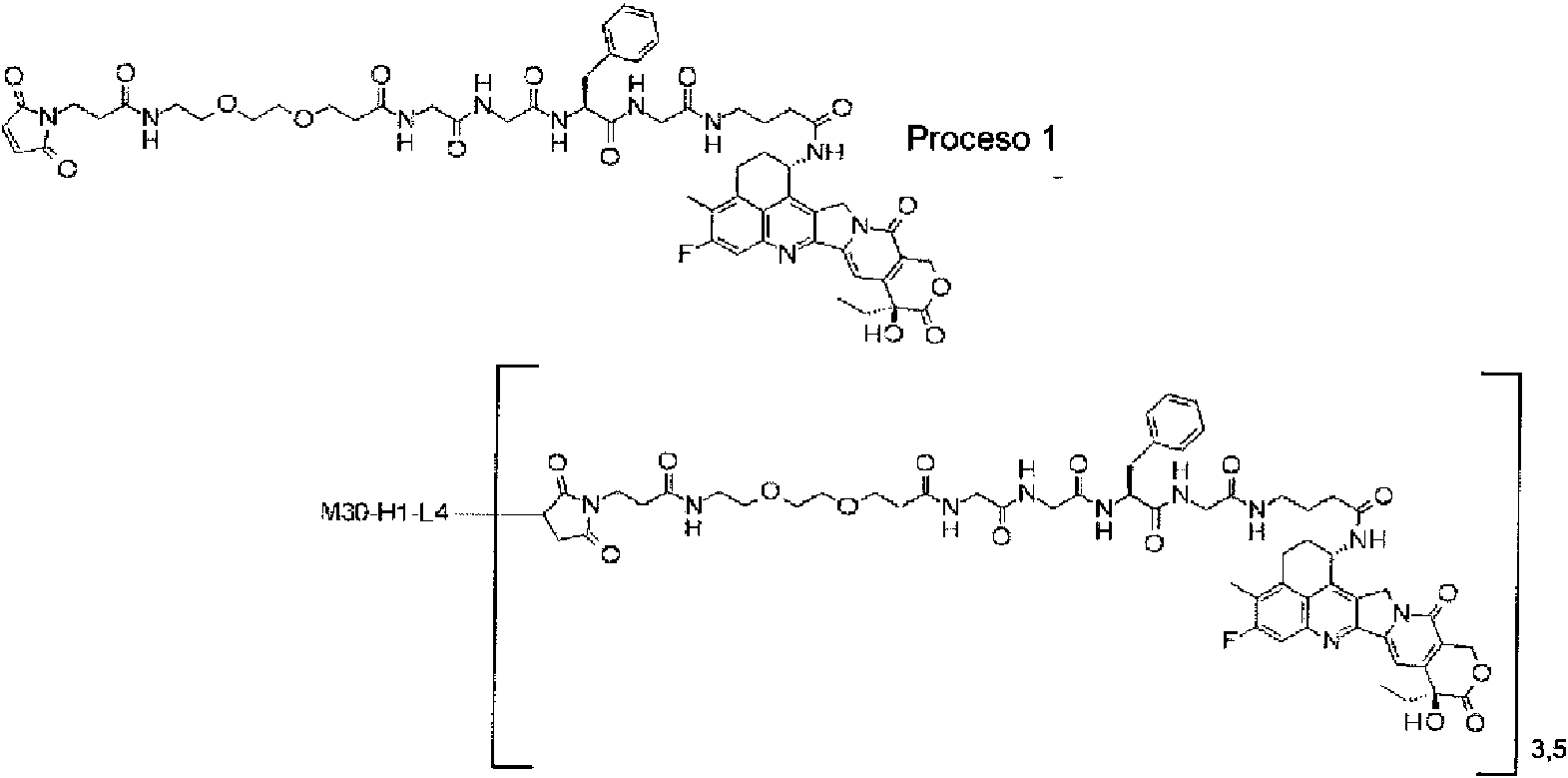
Proceso 1: Conjugado de anticuerpo - fármaco (20)
Reducción del anticuerpo: El anticuerpo M30-H1-L4 producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg'1cirr1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,25 ml, 12,5 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0287 ml; 3,4 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (0,0267 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 14 (0,0439 ml; 5,2 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, se incubó para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 1 hora. A continuación, una solución acuosa (0,0066 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A. Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 4964 (valor medido) y £d,370 = 18982 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 8,7 mg (70%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,5.
Ejemplo 22 Conjugado de anticuerpo - fármaco (21)
[Fórmula 91]

Proceso 1: Conjugado de anticuerpo - fármaco (21)
Reducción del anticuerpo: El anticuerpo M30-H1-L4 producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg'1cirr1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,25 ml, 12,5 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0439 ml; 5,2 equivalentes por molécula de anticuerpo) (0,0287 ml; 3,4 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 14 (0,0726 ml; 8,6 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, se incubó para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 1 hora. A continuación, una solución acuosa (0,011 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A. Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 4964 (valor medido) y £d,370 = 18982 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 8,3 mg (66%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,5.
Ejemplo 23 Conjugado de anticuerpo - fármaco (22)
[Fórmula 92]

Proceso 1: Conjugado de anticuerpo - fármaco (22)
Reducción del anticuerpo: El anticuerpo anti-CD30 producido en el Ejemplo de Referencia 3 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,75 mlmg'1cirr1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0065 ml; 2,5 equivalentes por molécula de anticuerpo). El enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (0,0098 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 14 (0,0116 ml; 4,5 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0017 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 270400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 4964 (valor medido) y £d,370 = 18982 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 0,86 mg/ml, rendimiento de anticuerpo: 2,2 mg (54%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 2,5.
Ejemplo 24 Conjugado de anticuerpo - fármaco (23)
[Fórmula 93]

Proceso 1: Conjugado de anticuerpo - fármaco (23)
Reducción del anticuerpo: El anticuerpo anti-CD30 producido en el Ejemplo de Referencia 3 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,75 mlmg'1cirr1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,35 ml, 3,5 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0113 ml; 5 equivalentes por molécula de anticuerpo). El enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 14 (0,0204 ml; 9 equivalentes por molécula de anticuerpo) y propilenglicol (Kanto Chemical Co., Inc., 0,18 ml) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0031 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 270400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 4964 (valor medido) y £d,370 = 18982 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 0,41 mg/ml, rendimiento de anticuerpo: 1,0 mg (29%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 7,1.
Ejemplo 25 Conjugado de anticuerpo - fármaco (24)
[Fórmula 94]

Proceso 1: Conjugado de anticuerpo - fármaco (24)
Reducción del anticuerpo: El anticuerpo anti-CD33 producido en el Ejemplo de Referencia 4 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0065 ml; 2,5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0058 ml). Después de confirmar que la solución tenía un pH de 7,0 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (0,0101 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 14 (0,0116 ml; 4,5 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0017 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £A,28o = 256400 (valor de cálculo estimado), £A,370 = 0 (valor de cálculo estimado), £d,280 = 4964 (valor medido) y £D,370 = 18982 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 1,25 mg/ml, rendimiento de anticuerpo: 3,1 mg (78%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,7.
Ejemplo 26 Conjugado de anticuerpo - fármaco (25)
[Fórmula 95]

Proceso 1: Conjugado de anticuerpo - fármaco (25)
Reducción del anticuerpo: El anticuerpo anti-CD33 producido en el Ejemplo de Referencia 4 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66 mlmg'1cirr1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0129 ml; 5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,006 ml). Después de confirmar que la solución tenía un pH de 7,0 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 14 (0,0233 ml; 9 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0035 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 256400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 4964 (valor medido) y £d,370 = 18982 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 1,17 mg/ml, rendimiento de anticuerpo: 2,9 mg (73%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 7,3.
Ejemplo 27 Conjugado de anticuerpo - fármaco (26)
[Fórmula 96]

Proceso 1: Conjugado de anticuerpo - fármaco (26)
Reducción del anticuerpo: El anticuerpo anti-CD70 producido en el Ejemplo de Referencia 5 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,69 mlmg'1cirr1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0065 ml; 2,5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0058 ml). Después de confirmar que la solución tenía un pH de 7,0 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (0,0101 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 14 (0,0116 ml; 4,5 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0017 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 262400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 4964 (valor medido) y £d,370 = 18982 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 1,14 mg/ml, rendimiento de anticuerpo: 2,9 mg (71%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,8.
Ejemplo 28 Conjugado de anticuerpo - fármaco (27)
[Fórmula 97]

Proceso 1: Conjugado de anticuerpo - fármaco (27)
Reducción del anticuerpo: El anticuerpo anti-CD70 producido en el Ejemplo de Referencia 5 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,69 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0129 ml; 5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,006 ml). Después de confirmar que la solución tenía un pH de 7,0 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido (0,0233 ml; 9 equivalentes por molécula de anticuerpo) que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 14 a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0035 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28ü = 262400 (valor de cálculo estimado), £a,37ü = 0 (valor de cálculo estimado), £D,28ü = 4964 (valor medido) y £D,37ü = 18982 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 1,13 mg/ml, rendimiento de anticuerpo: 2,8 mg (71%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 7,4.
Ejemplo 29 Conjugado de anticuerpo - fármaco (28)
[Fórmula 98]

Proceso 1: N-[19-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-17-oxo-4,7,10,13-tetraoxo-16-azanonadecan-1-oil]glicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El compuesto (90 mg, 0,107 mmol) obtenido en el proceso 2 del Ejemplo 2 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 usando diisopropiletilamina (18,7 ^l, 0,107 mmol) en lugar de trietilamina y 1-maleinimida-3-oxo-7,10,13,16-tetraoxa-4-azanonadecan-19-oato de N-succinimidilo (55,1 mg, 0,107 mmol) en lugar de hexanoato de N-succinimidil 6-maleimida para producir el compuesto del título en forma de un sólido de color amarillo pálido (50 mg, 37 %).
RMN 1H (400 MHz, DMSO-d6) 5: 0,85 (3H, t, J = 7,2 Hz), 1,64-1,74 (2H, m), 1,77-1,90 (2H, m), 2,06-2,19 (4H, m), 2,27-2,32 (2H, m), 2,33-2,37 (2H, m), 2,38 (3H, s), 2,72-2,80 (3H, m), 2,96-3,19 (6H, m), 3,39-3,48 (10H, m), 3,52-3,75 (10H, m), 4,39-4,48 (1H, m), 5,14 (1H, d, J = 18,8 Hz), 5,23 (1H, d, J = 18,8 Hz), 5,38 (1H, d, J = 17,0 Hz), 5,42 (1H, d, J = 17,0 Hz), 5,52-5,58 (1H, m), 6,52 (1H, s), 6,98 (1H, s), 7,13-7,24 (5H, m), 7,29 (1H, s), 7,69 (1H, t, J = 5,5 Hz), 7,78 (1H, d, J = 10,9 Hz), 7,98-8,03 (2H, m), 8,10 (1H, d, J = 7,8 Hz), 8,16 (1H, t, J = 5,7 Hz), 8,23 (1H, t, J = 5,7 Hz), 8,44 (1H, d, J = 8,6 Hz).
EM (APCI) m/z: 1237 (M+H)+
Proceso 2: Conjugado de anticuerpo - fármaco (28)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 10 mg/ml sustituyendo el medio con PBS6,0/EDTA usando el procedimiento común C-1 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) descrito en el procedimiento de producción 1. La solución (1,25 ml) se puso en un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,025 ml; 3,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,102 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 1 anterior (0,047 ml; 5,5 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por As ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 40 minutos. A continuación, se añadió una solución acuosa (0,009 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) a la misma y se agitó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £A,28o = 235300 (valor de cálculo estimado), £A,37o = 0 (valor de cálculo
estimado), £D,28o = 5000 (valor promedio medido) y £D,37o = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 13,60 mg/ml, rendimiento de anticuerpo: 9,5 mg (76%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,3.
Ejemplo 30 Conjugado de anticuerpo - fármaco (29)
[Fórmula 99]

Proceso 1: N-(terc-butoxicarbonil)p-alanilglicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El compuesto (0,839 g, 1,00 mmol) obtenido en el proceso 2 del Ejemplo 2 se hizo reaccionar de la misma forma que en el proceso 1 del Ejemplo 1 usando N-(terc-butoxicarbonil)-p-alanina en lugar de ácido 4-(tercbutoxicarbonilamino)butanoico. El producto en bruto obtenido se usó en el siguiente proceso sin realizar purificación.
Proceso 2: p-Alanilglicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El producto en bruto obtenido en el proceso 1 se hizo reaccionar de la misma forma que en el proceso 2 del Ejemplo 2 para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,610 g, 67 %).
RMN 1H (400 MHz, DMSO-d6) 5: 0,87 (3H, t, J = 7,4 Hz), 1,67-1,77 (2H, m), 1,79-1,92 (2H, m), 2,09-2,22 (4H, m), 2,40 (3H, s), 2,46-2,55 (2H, m), 2,82-2,73 (1H, m), 2,95-3,13 (5H, m), 3,14-3,21 (2H, m), 3,55-3,80 (6H, m), 4,44-4,52 (1H, m), 5,20 (2H, dd, J = 35,0, 19,0 Hz), 5,42 (2H, s), 5,53-5,60 (1H, m), 6,54 (1H, s), 7,14-7,28 (5H, m), 7,31 (1H, s), 7,67 (2H, s a), 7,72-7,78 (1H, m), 7,80 (1H, d, J = 11,0 Hz), 8,10-8,17 (2H, m), 8,29 (1H, t, J = 5,9 Hz), 8,42 (1H, t, J = 5,7 Hz), 8,47 (1H, d, J = 8,6 Hz).
Proceso 3: N-(bromoacetil)-p-alanilglicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
A una solución de diclorometano (4,5 ml) de ácido 2-bromoacético (96,3 mg, 0,693 mmol), se añadieron N-hidroxisuccinimida (79,7 mg, 0,693 mmol) y 1,3-diisopropilcarbodiimida (0,107 ml, 0,693 mmol) y se agitó a temperatura ambiente. La solución de reacción se añadió a una solución de N,N-dimetilformamida (4,5 ml) del compuesto (473 mg, 0,462 mmol) obtenido en el proceso 2 y trietilamina (0,154 ml, 1,11 mmol) a 0 °C y se agitó a temperatura ambiente durante 1 hora. La solución de reacción se purificó por cromatografía en columna sobre gel de sílice [disolvente de elución: cloroformo - cloroformo : metanol = 85 : 15 (v/v)]. El sólido obtenido se lavó con disolvente mixto de cloroformo : metanol : éter dietílico para producir el compuesto del título en forma de un sólido de color amarillo pálido (191 mg, 40 %).
RMN 1H (400 MHz, DMSO-d6) 5: 0,87 (3H, t, J = 7,4 Hz), 1,67-1,77 (2H, m), 1,79-1,92 (2H, m), 2,08-2,22 (4H, m), 2,33 (2H, t, J = 7,0 Hz), 2,40 (3H, s), 2,74-2,83 (1H, m), 2,99-3,12 (3H, m), 3,14-3,21 (2H, m), 3,24-3,30 (2H, m), 3,56-3,77 (6H, m), 3,82 (2H, s), 4,41-4,51 (1H, m), 5,20 (2H, c, J = 18,9 Hz), 5,42 (2H, s), 5,54-5,60 (1H, m), 6,54 (1H, s), 7,15 7,27 (5H, m), 7,31 (1H, s), 7,69-7,74 (1H, m), 7,80 (1H, d, J = 10,9 Hz), 8,06 (1H, t, J = 5,7 Hz), 8,13 (1H, d, J = 7,8 Hz), 8,21-8,34 (3H, m), 8,46 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 1030, 1032 (M+H)+
Proceso 4: Conjugado de anticuerpo - fármaco (29)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 10 mg/ml sustituyendo el medio con PBS6,0/EDTA usando el procedimiento común C-1 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg'1cirr1) descrito en el procedimiento de producción 1. La solución (1,25 ml) se puso en un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,025 ml; 3,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,09 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 3 (0,059 ml; 7,0 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 40 minutos. A continuación, se añadió una solución acuosa (0,009 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) a la misma y se agitó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A. Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 13,9 mg/ml, rendimiento de anticuerpo: 9,7 mg (78%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,2.
Ejemplo 31 Conjugado de anticuerpo - fármaco (30)
[F 
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 3 del Ejemplo 30, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 1 del Ejemplo 4.
Concentración de anticuerpo: 1,94 mg/ml, rendimiento de anticuerpo: 11,64 mg (93%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,6.
Ejemplo 32 Conjugado de anticuerpo - fármaco (31)
[Fórmula 101]

Proceso 1: Conjugado de anticuerpo - fármaco (31)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 3 del Ejemplo 30, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 1 del Ejemplo 5.
Concentración de anticuerpo: 1,90 mg/ml, rendimiento de anticuerpo: 11,40 mg (91%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,7.
Ejemplo 33 Conjugado de anticuerpo - fármaco (32)
[Fórmula 102]

Casi la totalidad de las cantidades de los conjugados de anticuerpo - fármaco de Ejemplos 31 y 32 se mezclaron. La solución se concentró mediante el procedimiento común A para producir el conjugado de anticuerpo - fármaco del título.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 21,06 mg, y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,0.
Ejemplo 34 Conjugado de anticuerpo - fármaco (33)
[Fórmula 103]

Proceso 1: 4-({N6-(terc-butoxicarbonil)-N2-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil}amino)butanoato de terc-butilo
A una solución de N,N-dimetilformamida (10,0 ml) de N£-(terc-butoxicarbonil)-Na-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisina (1,00 g, 2,14 mmol), N-hidroxisuccinimida (0,370 g, 3,20 mmol) y clorhidrato de éster de ácido terc-butil 4-aminobutanoico (0,830 g, 4,27 mmol), se añadieron clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (0,610 g, 3,20 mmol) y N,N-diisopropiletilamina (0,410 ml, 2,35 mmol) y se agitó a temperatura ambiente durante 3 días. La solución de reacción se diluyó con acetato de etilo y se lavó con una solución acuosa de ácido cítrico al 10 % y una solución saturada acuosa de hidrogenocarbonato sódico, y salmuera saturada, y entonces la capa orgánica se secó sobre sulfato de magnesio anhidro. El disolvente se retiró a presión reducida para producir el compuesto del título en forma de un sólido incoloro (1,35 g, cuantitativo).
RMN 1H (400 MHz, DMSO-d6) 6: 1,14-1,42 (4H, m), 1,36 (9H, s), 1,37 (9H, s), 1,48-1,67 (4H, m), 2,18 (2H, t, J = 7,6 Hz), 2,84-2,93 (2H, m), 2,99-3,11 (2H, m), 3,84-3,94 (1H, m), 4,18-4,30 (3H, m), 6,76 (1H, t, J = 5,4 Hz), 7,33 (2H, t, J = 7,3 Hz), 7,39-7,45 (3H, m), 7,73 (2H, dd, J = 7,3, 2,7 Hz), 7,85-7,92 (3H, m).
Proceso 2: 4-{[N6-(terc-butoxicarbonil)-L-lisil]amino}butanoato de terc-butilo
A una solución de N,N-dimetilformamida (8,00 ml) del compuesto (1,35 g, 2,22 mmol) obtenido en el proceso 1, se añadió piperidina (2,00 ml) y se agitó durante 1,5 horas a temperatura ambiente. El disolvente se retiró a presión reducida para producir una mezcla que contiene el compuesto del título. La mezcla se usó para la siguiente reacción sin realizar purificación adicional.
Proceso 3: N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-valil-N6-(terc-butoxicarbonil)-N-(4-terc-butoxi-4-oxobutil)-L-lisinamida
A una solución de N,N-dimetilformamida (30,0 ml) de la mezcla (2,22 mmol) obtenida en el proceso 2 anterior, se añadieron N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-valina (1,13 g, 3,32 mmol), N-hidroxisuccinimida (0,310 g, 2,66 mmol) y clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (0,550 g, 2,88 mmol) y se agitó a temperatura ambiente durante 18 horas. La solución de reacción se diluyó con acetato de etilo y se lavó con una solución saturada acuosa
de hidrogenocarbonato sódico y salmuera saturada, y entonces la capa orgánica se secó sobre sulfato de magnesio anhidro. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo : metanol = 9 : 1 (v/v)] para producir el compuesto del título en forma de un sólido incoloro (0,363 g, 23 %).
RMN 1H (400 MHz, DMSO-da) 8: 0,84 (6H, t, J = 6,0 Hz), 1,12-1,64 (8H, m), 1,34 (9H, s), 1,38 (9H, s), 1,90-2,04 (1H, m), 2,17 (2H, t, J = 7,3 Hz), 2,79-2,90 (2H, m), 2,99-3,09 (2H, m), 3,83-3,91 (1H, m), 4,08-4,44 (4H, m), 6,71 (1H, t, J = 5,4 Hz), 7,32 (2H, t, J = 7,3 Hz), 7,42 (3H, t, J = 7,3 Hz), 7,74 (2H, t, J = 7,0 Hz), 7,85-7,91 (4H, m).
EM (IEN) m/z: 709 (M+H)+
Proceso 4: Formiato de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-valil-N-(3-carboxipropil)-L-lisinamida
Al compuesto (0,363 mg, 0,512 mmol) obtenido en el proceso 3, se añadió ácido fórmico (10,0 ml) y se agitó a temperatura ambiente durante 4 horas. El disolvente se retiró a presión reducida para producir el compuesto del título. El compuesto se usó para la siguiente reacción sin realizar purificación adicional.
Proceso 5: N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-valil-N6-(terc-butoxicarbonil)-N-(3-carboxipropil)-L-lisinamida
A una suspensión de 1,4-dioxano (5,00 ml) del compuesto (0,512 mmol) obtenido en el proceso 4, se añadieron una solución acuosa saturada de hidrogenocarbonato sódico (20,0 ml) y dicarbonato de di-terc-butilo (0,178 ml, 0,769 mmol) y se agitó a temperatura ambiente durante 3 horas. La solución de reacción se diluyó con acetato de etilo y se lavó con una solución acuosa de ácido cítrico al 10 % y salmuera saturada, y entonces la capa orgánica se secó sobre sulfato de magnesio anhidro. El disolvente se retiró a presión reducida para producir el compuesto del título en forma de un sólido incoloro (0,295 g, 88 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,84 (6H, t, J = 6,7 Hz), 1,13-1,39 (4H, m), 1,35 (9H, s), 1,48-1,62 (4H, m), 1,91-2,04 (1H, m), 2,20 (2H, t, J = 7,3 Hz), 2,80-2,89 (2H, m), 2,99-3,11 (2H, m), 3,87 (1H, dd, J = 8,5, 6,7 Hz), 4,06-4,35 (4H, m), 6,71 (1H, t, J = 6,0 Hz), 7,32 (2H, t, J = 7,6 Hz), 7,39-7,46 (3H, m), 7,74 (2H, t, J = 7,6 Hz), 7,83-7,94 (4H, m). EM (IEN) m/z: 653 (M+H)+
Proceso 6: N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-valil-N6-(terc-butoxicarbonil)-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)-L-lisinamida
Mesilato del compuesto (4) (0,240 g, 0,452 mmol) se hizo reaccionar de la misma forma que en el proceso 1 del Ejemplo 1 usando el compuesto (0,295 g, 0,452 mmol) obtenido en el proceso 5 en lugar de ácido 4-(tercbutoxicarbonilamino)butanoico para producir el compuesto del título en forma de un sólido de color naranja pálido (0,208 g, 43 %).
EM (IEN) m/z: 1071 (M+H)+
Proceso 7: L-Valil-N6-(terc-butoxicarbonil)-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)-L-lisinamida
El compuesto (0,208 g, 0,194 mmol) obtenido en el proceso 6 se hizo reaccionar de la misma forma que en el proceso 2 para producir una mezcla que contiene el compuesto del título. La mezcla se usó para la siguiente reacción sin realizar purificación adicional.
Proceso 8: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-valil-N6-(terc-butoxicarbonil)-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)-L-lisinamida
La mezcla (0,194 mmol) obtenida en el proceso 7 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,133 g, 56 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,77 (6H, t, J = 5,7 Hz), 0,87 (3H, t, J = 7,3 Hz), 1,14-1,71 (10H, m), 1,35 (9H, s), 1,77-1,95 (3H, m), 2,02-2,23 (7H, m), 2,40 (3H, s), 2,84 (3H, c, J = 6,4 Hz), 3,05 (2H, d, J = 6,7 Hz), 3,17 (2H, s), 3,26 3,39 (3H, m), 4,01-4,16 (2H, m), 5,15 (1H, d, J = 18,7 Hz), 5,24 (1H, d, J = 18,7 Hz), 5,36-5,48 (2H, m), 5,51-5,60 (1H, m), 6,52 (1H, s), 6,72 (1H, t, J = 6,0 Hz), 6,99 (2H, s), 7,31 (1H, s), 7,71-7,85 (5H, m), 8,41 (1H, d, J = 9,1 Hz).
EM (IEN) m/z: 1041 (M+H)+
Proceso 9: Trifluoroacetato de N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]-L-valil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]aminol-4-oxobutil)-L-lisinamida
A una solución de diclorometano (10,0 ml) del compuesto (0,110 mg, 0,106 mmol) obtenido en el proceso 8 anterior, se añadió ácido trifluoroacético (4,00 ml) y se agitó durante 5 horas a temperatura ambiente. El disolvente se retiró a presión reducida para producir el compuesto del título en forma de un sólido de color amarillo pálido (70,0 mg, 64 %). RMN 1H (400 MHz, DMSO-d6) 8: 0,76-0,81 (6H, m), 0,87 (3H, t, J = 7,3 Hz), 1,12-1,31 (4H, m), 1,39-1,56 (8H, m), 1,57-1,74 (3H, m), 1,79-1,96 (3H, m), 2,06-2,18 (7H, m), 2,40 (3H, s), 2,70-2,80 (2H, m), 3,01-3,10 (2H, m), 3,13-3,22 (2H, m), 4,04 (1H, t, J = 7,6 Hz), 4,10-4,20 (1H, m), 5,15 (1H, d, J = 18,7 Hz), 5,24 (1H, d, J = 18,7 Hz), 5,36-5,47 (2H, m), 5,52-5,60 (1H, m), 6,53 (1H, s), 7,00 (2H, s), 7,32 (1H, s), 7,61 (3H, s a), 7,75-7,88 (4H, m), 8,43 (1H, d, J = 8,5
Hz). EM (IEN) m/z: 941 (M+H)+
Proceso 10: Conjugado de anticuerpo - fármaco (33)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 9, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 2 del Ejemplo 29. Concentración de anticuerpo: 12,0 mg/ml, rendimiento de anticuerpo: 8,4 mg (67%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,2.
Ejemplo 35 Conjugado de anticuerpo - fármaco (34)
[Fórmula 104]
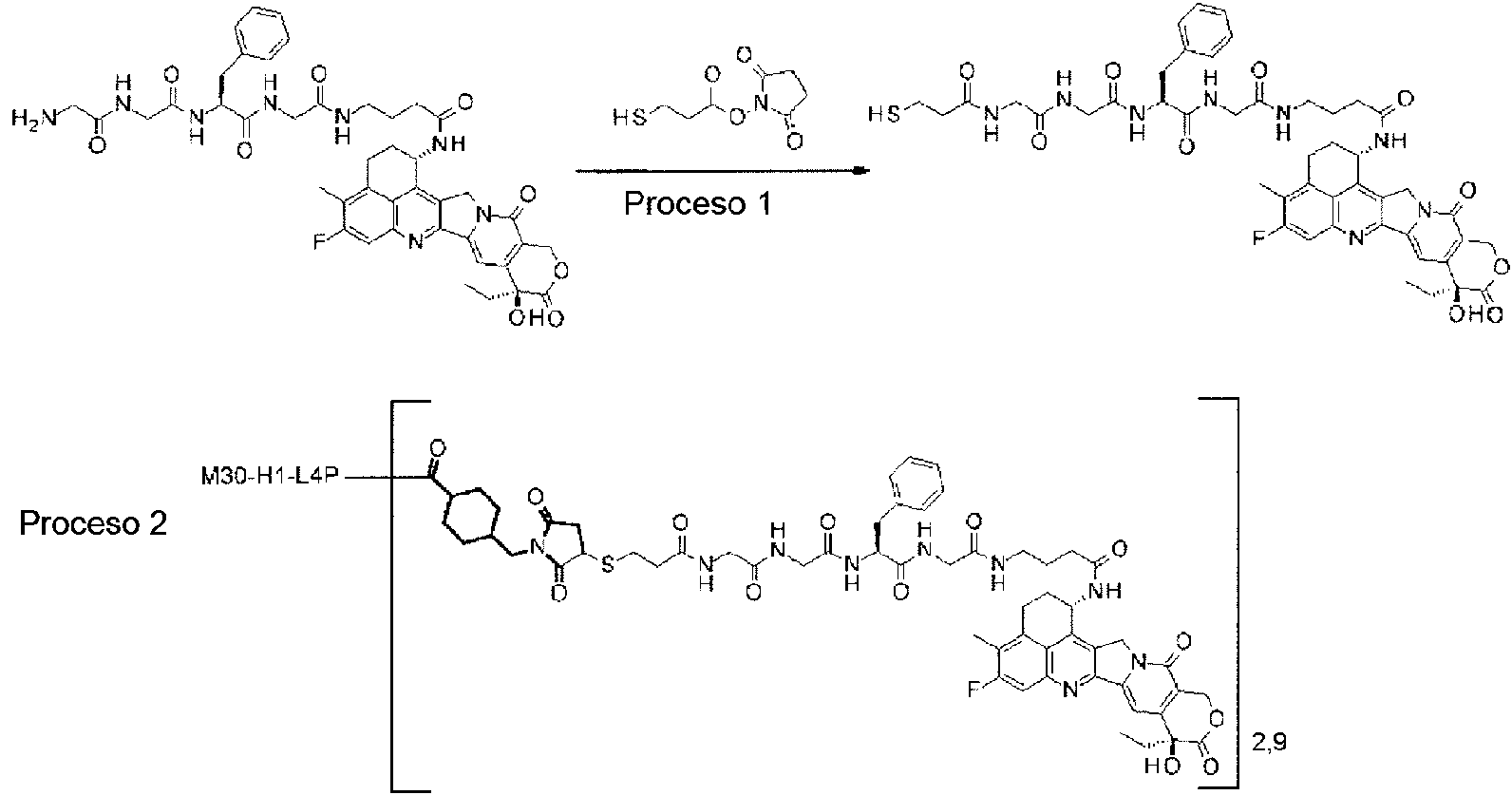
Proceso 1: N-(3-sulfanilpropanoil)glicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El compuesto (84,0 mg, 0,100 mmol) obtenido en el proceso 2 del Ejemplo 2 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 usando N-succinimidil 3-mercaptopropionato en lugar de hexanoato de N-succinimidil 6-maleimida para producir el compuesto del título en forma de un sólido de color amarillo pálido (61,2 mg, 66 %).
RMN 1H (DMSO-Da) 5: 0,87 (3H, t, J = 7,4 Hz), 1,77-1,66 (2H, m), 1,79-1,92 (2H, m), 2,07-2,24 (4H, m), 2,31-2,47 (3H, m), 2,40 (3H, s), 2,59-2,69 (2H, m), 2,78 (1H, dd, J = 13,7, 9,8 Hz), 2,98-3,13 (3H, m), 3,14-3,23 (2H, m), 3,54-3,79 (6H, m), 4,40-4,50 (1H, m), 5,20 (2H, dd, J = 36,8, 19,2 Hz), 5,36-5,47 (2H, m), 5,52-5,63 (1H, m), 6,54 (1H, s), 7,14 7,28 (5H, m), 7,31 (1H, s), 7,68-7,74 (1H, m), 7,80 (1H, d, J = 10,9 Hz), 8,03-8,09 (1H, m), 8,13 (1H, d, J = 7,8 Hz), 8,19-8,29 (2H, m), 8,46 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 927 (M+H)+
Proceso 2: Conjugado de anticuerpo - fármaco (34)
Derivatización con SMCC de anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 20 mg/ml sustituyendo el medio con PBS6,5/EDTA usando el procedimiento común C-2 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1). La solución (0,25 ml) se puso en un tubo de 1,5 ml, se cargó con una solución de DMSO (0,0063 ml; que se corresponde con aproximadamente 2,55 equivalentes por molécula de anticuerpo) que contiene succinimidil-4-(N-maleimidometil)ciclohexano-1-carboxilato 27,6 mM (SMCC, Thermo Fisher Scientific Inc.) a temperatura ambiente, y se hizo reaccionar a temperatura ambiente durante 2 horas. Esta solución de reacción se sometió a purificación usando el procedimiento común D-2 para producir 0,7 ml de una solución que contiene aproximadamente 5 mg del anticuerpo derivatizado con SMCC. Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir DMSO (0,045 ml) y una solución de DMSO que contiene 10 mM del compuesto obtenido en el proceso 1 (0,015 ml; que se corresponde con aproximadamente 2,4 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por As ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 16 horas.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) para producir 3,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A.
Caracterización fisicoquímica: Usando el procedimiento común E (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 3,85 mg/ml, rendimiento de anticuerpo: 0,8 mg (16%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 2,9.
Ejemplo 36 Conjugado de anticuerpo - fármaco (35)
[Fórmula 105]

Proceso 1: Conjugado de anticuerpo - fármaco (35)
Derivatización con SMCC de anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 20 mg/ml sustituyendo el medio con PBS6,5/EDTA usando el procedimiento común C-2 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg' 1cm_1). La solución (0,25 ml) se puso en un tubo de 1,5 ml, se cargó con una solución de DMSO (0,0125 ml; que se corresponde con aproximadamente 5,1 equivalentes por molécula de anticuerpo) que contiene succinimidil-4-(N-maleimidometil)ciclohexano-1-carboxilato 27,6 mM (SMCC, Thermo Fisher Scientific Inc.) a temperatura ambiente, y se hizo reaccionar a temperatura ambiente durante 2 horas. Esta solución de reacción se sometió a purificación usando el procedimiento común D-2 para producir 0,7 ml de una solución que contiene aproximadamente 5 mg del anticuerpo derivatizado con SMCC.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir DMSO (0,03 ml) y una solución de DMSO que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 35 (0,03 ml; que se corresponde con aproximadamente 4,8 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 16 horas.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) para producir 3,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A.
Caracterización fisicoquímica: Usando el procedimiento común E (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 2,43 mg/ml, rendimiento de anticuerpo: 0,5 mg (10%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 4,2.
Ejemplo 37 Conjugado de anticuerpo - fármaco (36)
[Fórmula 106]

Proceso 1: N-{8-[(2,5-dioxopirrolidin-1-il)oxi]-8-oxooctanoil}glicilglicil-L-fenilalanil-N-(4-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolin-1-il]amino}-4-oxobutil)glicinamida
El compuesto (84,0 mg, 0,100 mmol) obtenido en el proceso 2 del Ejemplo 2 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 usando suberato de di(N-succinimidilo) en lugar de hexanoato de N-succinimidil 6-maleimida para producir el compuesto del título en forma de un sólido de color amarillo pálido (77,1 mg, 71 %). RMN 1H (DMSO-D6) 6: 0,87 (3H, t, J = 7,2 Hz), 1,21-1,38 (4H, m), 1,43-1,50 (2H, m), 1,55-1,63 (2H, m), 1,68-1,76 (2H, m), 1,80-1,91 (2H, m), 2,07-2,22 (6H, m), 2,40 (3H, s), 2,60-2,67 (2H, m), 2,76-2,84 (5H, m), 2,97-3,22 (5H, m), 3,56 3,76 (6H, m), 4,40-4,50 (1H, m), 5,20 (2H, c, J = 18,8 Hz), 5,37-5,48 (2H, m), 5,53-5,62 (1H, m), 6,54 (1H, s), 7,15 7,28 (5H, m), 7,31 (1H, s), 7,71 (1H, t, J = 5,5 Hz), 7,80 (1H, d, J = 10,9 Hz), 8,04 (1H, t, J = 5,9 Hz), 8,09 (1H, t, J = 5,9 Hz), 8,14 (1H, d, J = 7,8 Hz), 8,26 (1H, t, J = 5,9 Hz), 8,47 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 1092 (M+H)+
Proceso 2: Conjugado de anticuerpo - fármaco (36)
Conjugación entre anticuerpo y enlazador de fármaco: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 20 mg/ml sustituyendo el medio con PBS6,5/EDTA usando el procedimiento común C-2 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm"1). La solución (0,25 ml) se puso en un tubo de 1,5 ml, se cargó con una solución de DMSO que contiene 10 mM del compuesto obtenido en el proceso 1 (0,025 ml; que se corresponde con aproximadamente 3,7 equivalentes por molécula de anticuerpo) a temperatura ambiente, y se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 16 horas. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) para producir 3,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A.
Caracterización fisicoquímica: Usando el procedimiento común E (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 6,25 mg/ml, rendimiento de anticuerpo: 1,3 mg (26%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,2.
Ejemplo 38 Conjugado de anticuerpo - fármaco (37)
[Fórmula 107]

Proceso 1: Conjugado de anticuerpo - fármaco (37)
Conjugación entre anticuerpo y enlazador de fármaco: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 20 mg/ml sustituyendo el medio con PBS6,5/EDTA usando el procedimiento común C-2 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg'1cirr1). La solución (0,5 ml) se puso en un tubo de 1,5 ml, a continuación se cargó con una solución de DMSO que contiene DMSO (0,025 ml) y 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 37 (0,025 ml; que se corresponde con aproximadamente 7,4 equivalentes por molécula de anticuerpo) a temperatura ambiente, y se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 16 horas.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) para producir 3,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A.
Caracterización fisicoquímica: Usando el procedimiento común E (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 4,36 mg/ml, rendimiento de anticuerpo: 0,9 mg (18%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 4,1.
Ejemplo 39 Conjugado de anticuerpo - fármaco (38)
[Fórmula 108]

Proceso 1: Conjugado de anticuerpo - fármaco (38)
Conjugación entre anticuerpo y enlazador de fármaco: El anticuerpo anti-CD30 producido en el Ejemplo de Referencia 3 se preparó para tener una concentración de anticuerpo de 10 mg/ml sustituyendo el medio con PBS6,5/EDTA usando el procedimiento común C-2 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,75 mlmg-1cm-1). La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml, a continuación se cargó con DMSO (0,017 ml) y una solución de DMSO que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 37 (0,023 ml; que se corresponde con 9 equivalentes por molécula de anticuerpo) a temperatura ambiente, y se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 4 horas.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Caracterización fisicoquímica: Usando el procedimiento común E (como coeficiente de absorción molar, se usaron £a,280 = 270400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 2670 (valor medido) y £d,370 = 15820 (valor medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 0,55 mg/ml, rendimiento de anticuerpo: 1,4 mg (34%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 2,7.
Ejemplo 40 Conjugado de anticuerpo - fármaco (39)
[Fórmula 109]

Proceso 1: Conjugado de anticuerpo - fármaco (39)
Conjugación entre anticuerpo y enlazador de fármaco: El anticuerpo anti-CD33 producido en el Ejemplo de Referencia 4 se preparó para tener una concentración de anticuerpo de 10 mg/ml sustituyendo el medio con PBS6,5/EDTA usando el procedimiento común C-2 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66 mlmg-1cm-1). La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml, a continuación se cargó con DMSO (0,017 ml) y una solución de DMSO que contiene 10 mM del compuesto obtenido en el proceso 1 del Ejemplo 37 (0,023 ml; que se corresponde con 9 equivalentes por molécula de anticuerpo) a temperatura ambiente, y se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 4 horas.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Caracterización fisicoquímica: Usando el procedimiento común E (como coeficiente de absorción molar, se usaron £a,28ü = 256400 (valor de cálculo estimado), £a,37ü = 0 (valor de cálculo estimado), £d,28ü = 2670 (valor medido) y £d,37ü = 15820 (valor medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 0,93 mg/ml, rendimiento de anticuerpo: 2,3 mg (58 %), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 4,0.
Ejemplo 41 Conjugado de anticuerpo - fármaco (40)
[Fórmula 110]

Proceso 1: Conjugado de anticuerpo - fármaco (40)
Conjugación entre anticuerpo y enlazador de fármaco: El anticuerpo anti-CD70 producido en el Ejemplo de Referencia 5 se preparó para tener una concentración de anticuerpo de 10 mg/ml sustituyendo el medio con PBS6,5/EDTA usando el procedimiento común C-2 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,69 mlmg-1cm-1). La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml, a continuación se cargó con DMSO (0,017 ml) y una solución de DMSO que contiene 10 mM del compuesto obtenido en el proceso 1 (0,023 ml; que se corresponde con 9 equivalentes por molécula de anticuerpo) del Ejemplo 37 a temperatura ambiente, y se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 4 horas.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Caracterización fisicoquímica: Usando el procedimiento común E (como coeficiente de absorción molar, se usaron £a,280 = 262400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 2670 (valor medido) y £d,370 = 15820 (valor medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 1,04 mg/ml, rendimiento de anticuerpo: 2,6 mg (65%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 4,1.
Ejemplo 42 2-(2-Aminoetoxi)-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]acetamida
[Fórmula 111]

Proceso 1: [2-(2-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-2-oxoetoxi)etil]carbamato de terc-butilo
Mesilato del compuesto (4) (3,10 g, 5,47 mol) se hizo reaccionar de la misma forma que en el proceso 1 del Ejemplo 1 usando ácido {2-[(terc-butoxicarbonil)amino]etoxi}acético (J. Med. Chem., 1992, vol. 35, págs. 2928) (1,55 g, 6,01 mmol) en lugar de ácido 4-(terc-butoxicarbonilamino)butanoico para producir el compuesto del título en forma de un sólido de color amarillo pálido (2,56 g, 73 %). RMN 1H (400 MHz, DMSO-da) 5: 0,87 (3H, t, J = 7,3 Hz), 1,26 (9H, s), 1,81-1,91 (2H, m), 2,13-2,22 (2H, m), 2,40 (3H, s), 3,08-3,26 (4H, m), 3,43-3,53 (2H, m), 4,00 (1H, d, J = 15,1 Hz), 4,05 (1H, d, J = 15,1 Hz), 5,14 (1H, d, J = 18,7 Hz), 5,22 (1H, d, J = 18,7 Hz), 5,40 (1H, d, J = 16,6 Hz), 5,44 (1H, d, J = 16,6 Hz), 5,59-5,66 (1H, m), 6,53 (1H, s), 6,86 (1H, t, J = 5,4 Hz), 7,31 (1H, s), 7,79 (1H, d, J = 10,9 Hz), 8,49 (1H,
d, J = 9,1 Hz).
EM (APCI) m/z: 637 (M+H)+
Proceso 2: 2-(2-Aminoetoxi)-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]acetamida
El compuesto (1,50 g, 2,36 mol) obtenido en el proceso 1 se hizo reaccionar de la misma forma que en el proceso 2 del Ejemplo 1 para producir trifluoroclorhidrato del compuesto del título en forma de un sólido de color amarillo pálido (1,50 g, cuantitativo). Este compuesto se confirmó en el tumor de un ratón portador de cáncer que recibió el conjugado de anticuerpo - fármaco (41). RMN 1H (400 MHz, DMSO-da) 8: 0,87 (3H, t, J = 7,5 Hz), 1,81-1,92 (2H, m), 2,15-2,23 (2H, m), 2,41 (3H, s), 3,05 (2H, t, J = 5,1 Hz), 3,15-3,23 (2H, m), 3,71 (2H, t, J = 5,1 Hz), 4,10 (2H, s), 5,19 (1H, d, J = 18,7 Hz), 5,24 (1H, d, J = 18,7 Hz), 5,43 (2H, s), 5,58-5,66 (1H, m), 6,55 (1H, s), 7,33 (1H, s), 7,73-7,84 (4H, m), 8,55 (1H, d, J = 9,1 Hz).
EM (APCI) m/z: 537 (M+H)+
Ejemplo 43 Conjugado de anticuerpo - fármaco (41)
[Fórmula 112]

Proceso 1: N-(terc-butoxicarbonil)glicilglicil-L-fenilalanil-N-[2-(2-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2.3.9.10.13.15- hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]aminol-2-oxoetoxi)etilglicinamida
El compuesto (554 mg, 0,85 mmol) del Ejemplo 42 se hizo reaccionar de la misma forma que en el proceso 1 del Ejemplo 2 para producir el compuesto del título (775 mg, 95 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,85 (3H, t, J = 7,3 Hz), 1,36 (9H, s), 1,78-1,89 (2H, m), 2,13-2,22 (2H, m), 2,39 (3H, s), 2,71 (1H, dd, J = 13,4, 9,8 Hz), 2,95 (1H, dd, J = 13,4, 4,3 Hz), 3,09-3,23 (1H, m), 3,23-3,32 (2H, m), 3,40-3,62 (8H, m), 3,73 (1H, dd, J = 16,5, 5,5 Hz), 4,03 (2H, s), 4,39-4,47 (1H, m), 5,17 (1H, d, J = 18,9 Hz), 5,25 (1H, d, J = 18,9 Hz), 5,41 (1H, d, J = 16,8 Hz), 5,45 (1H, d, J = 16,8 Hz), 5,57-5,64 (1H, m), 6,54 (1H, s), 6,99 (1H, t, J = 5,8 Hz), 7,13-7,26 (5H, m), 7,31 (1H, s), 7,76-7,82 (2H, m), 7,90 (1H, t, J = 5,2 Hz), 8,13 (1H, d, J = 7,9 Hz), 8,27 (1H, t, J = 5,8 Hz), 8,49 (1H, d, J = 8,5 Hz). EM (APCI) m/z: 955 (M+H)+
Proceso 2: Trifluoroacetato de glicilglicil-L-fenilalanil-N-[2-(2-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2.3.9.10.13.15- hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-2oxoetoxi)etil]glicinamida
El compuesto (630 mg, 0,659 mmol) obtenido en el proceso 1 anterior se hizo reaccionar de la misma forma que en el proceso 2 del Ejemplo 2 para producir el compuesto del título (588 mg, 92%).
RMN 1H (400 MHz, DIVISOR) 5: 0,86 (3H, t, J = 7,3 Hz), 1,79-1,90 (2H, m), 2,13-2,22 (2H, m), 2,39 (3H, s), 2,71 (1H, dd, J = 13,4, 10,1 Hz), 2,99 (1H, dd, J = 13,4, 4,3 Hz), 3,09-3,23 (1H, m), 3,24-3,32 (3H, m), 3,41-3,71 (7H, m), 3,86 (1H, dd, J = 16,8, 5,8 Hz), 4,04 (2H, s), 4,52 (1H, td, J = 9,0, 4,1 Hz), 5,17 (1H, d, J = 18,9 Hz), 5,25 (1H, d, J = 18,9 Hz), 5,41 (1H, d, J = 16,5 Hz), 5,45 (1H, d, J = 16,5 Hz), 5,56-5,65 (1H, m), 6,55 (1H, s), 7,13-7,26 (5H, m), 7,32 (1H, s), 7,80 (1H, d, J = 11,0 Hz), 7,87-8,01 (4H, m), 8,29-8,36 (2H, m), 8,46-8,55 (2H, m).
EM (APCI) m/z: 855 (M+H)+
Proceso 3: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1 -il)hexanoil]glicilglicil-L-fenilalanil-N-[2-(2-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolin-1-il]amino}-2-oxoetoxi)etil]glicinamida
El compuesto (240 mg, 0,247 mmol) obtenido en el proceso 2 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 para producir el compuesto del título (162 mg, 62 %). RMN 1H (400 MHz, DMSO-d6) 5: 0,86 (3H, t, J = 7,6 Hz), 1,13-1,22 (2H, m), 1,40-1,51 (4H, m), 1,78-1,90 (2H, m), 2,09 (2H, t, J = 7,6 Hz), 2,14-2,21 (2H, m), 2,39 (3H, s), 2,74 (1H, dd, J = 13,6, 9,7 Hz), 2,96 (1H, dd, J = 13,6, 4,5 Hz), 3,08-3,24 (1H, m), 3,24-3,30 (1H, m), 3,33-3,40 (4H, m), 3,47-3,68 (7H, m), 3,72 (1H, dd, J = 16,6, 5,7 Hz), 4,03 (2H, s), 4,42 (1H, td, J = 8,6, 4,2 Hz), 5,17 (1H, d, J = 18,7 Hz), 5,25 (1H, d, J = 18,7 Hz), 5,40 (1H, d, J = 17,2 Hz), 5,44 (1H, d, J = 17,2 Hz), 5,57-5,64 (1H, m), 6,52 (1H, s), 6. 99 (2H, s), 7,13-7,25 (5H, m), 7,31 (1H, s), 7,74-7,81 (2H, m), 7,99 (1H, t, J = 5,7 Hz), 8,03-8,11 (2H, m), 8,22 (1H, t, J = 5,7 Hz), 8,47 (1H, d, J = 9,1 Hz).
EM (APCI) m/z: 1048 (M+H)+
Proceso 4: Conjugado de anticuerpo - fármaco (41)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 3, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 2 del Ejemplo 29. Concentración de anticuerpo: 12,0 mg/ml, rendimiento de anticuerpo: 8,4 mg (67%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,5.
Ejemplo 44 Conjugado de anticuerpo - fármaco (42)
[Fórmula 113]

Proceso 1: Conjugado de anticuerpo - fármaco (42)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 3 del Ejemplo 43, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 1 del Ejemplo 5.
Concentración de anticuerpo: 0,83 mg/ml, rendimiento de anticuerpo: 4,98 mg (40%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 7,2.
Ejemplo 45 Conjugado de anticuerpo - fármaco (43)
[Fórmula 114]

Proceso 1: Conjugado de anticuerpo - fármaco (43)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 3 del Ejemplo 43, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 1 del Ejemplo 4.
Concentración de anticuerpo: 1,06 mg/ml, rendimiento de anticuerpo: 6,36 mg (51%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,3.
Ejemplo 46 Conjugado de anticuerpo - fármaco (44)
[Fórmula 115]

Casi la totalidad de las cantidades de los conjugados de anticuerpo - fármaco de Ejemplos 44 y 45 se mezclaron. La solución se concentró mediante el procedimiento común A para producir el conjugado de anticuerpo - fármaco del título.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 10,21 mg, y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,6.
Ejemplo 47 Conjugado de anticuerpo - fármaco (45)
[Fórmula 116]
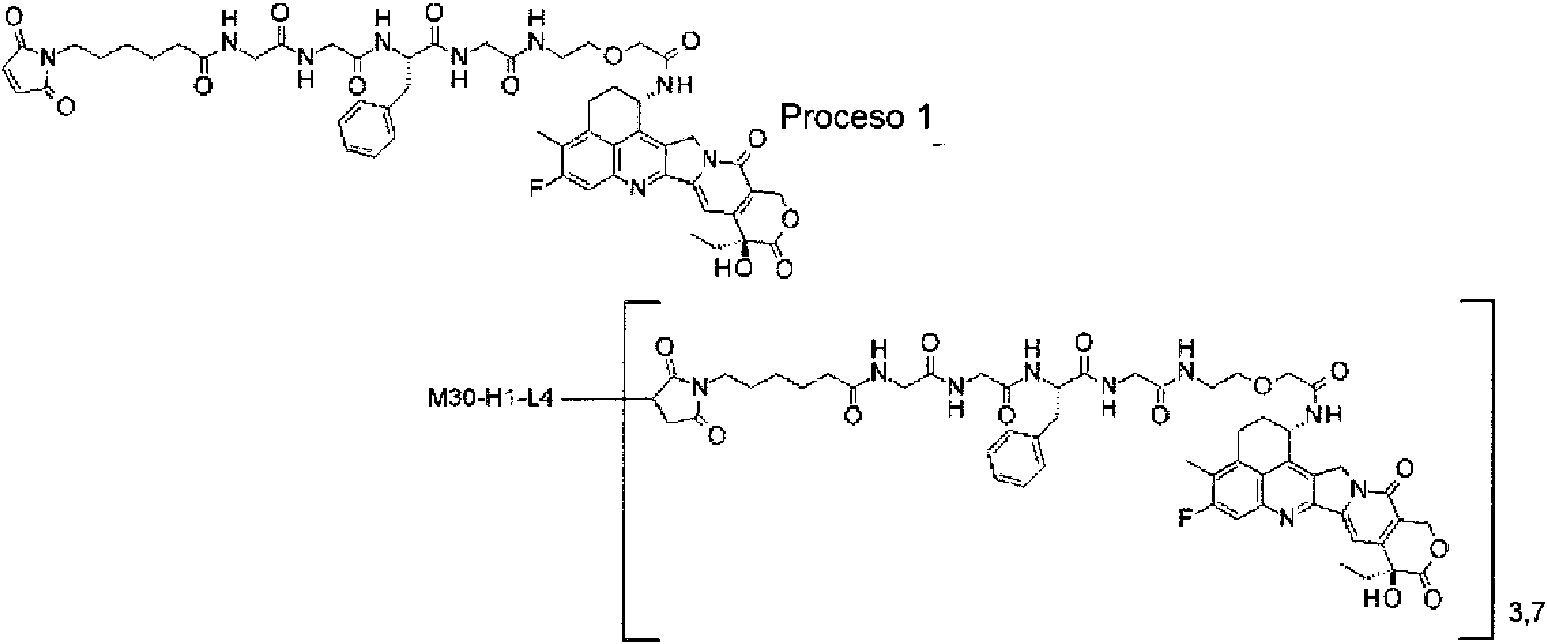
Proceso 1: Conjugado de anticuerpo - fármaco (45)
Reducción del anticuerpo: El anticuerpo M30-H1-L4 producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg'1cirr1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,25 ml, 12,5 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0287 ml; 3,4 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (0,0267 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 3 del Ejemplo 43 (0,0439 ml; 5,2 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, se incubó para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 1 hora. A continuación, una solución acuosa (0,0066 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A. Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5193 (valor medido) y £d,370 = 20347 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 9,3 mg (74%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,7.
Ejemplo 48 Conjugado de anticuerpo - fármaco (46)
[Fórmula 117]

Proceso 1: Conjugado de anticuerpo - fármaco (46)
Reducción del anticuerpo: El anticuerpo M30-H1-L4 producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,25 ml, 12,5 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0439 ml; 5,2 equivalentes por molécula de anticuerpo) (0,0287 ml; 3,4 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido (0,0726 ml; 8,6 equivalentes por molécula de anticuerpo) que contiene 10 mM del compuesto obtenido en el proceso 3 del Ejemplo 43 a la solución anterior a temperatura ambiente, se incubó para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 1 hora. A continuación, una solución acuosa (0,011 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A. Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28ü = 235300 (valor de cálculo estimado), £a,37ü = 0 (valor de cálculo estimado), £d,28ü = 5193 (valor medido) y £d,37ü = 20347 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 7,8 mg (62%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,2.
Ejemplo 49 N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]-p-alaninamida
[Fórmula 118]

Proceso 1: (3-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]aminol-3-oxopropil)carbamato de terc-butilo
Mesilato del compuesto (4) (500 mg, 0,941 mmol) se hizo reaccionar de la misma forma que en el proceso 1 del Ejemplo 1 usando N-(terc-butoxicarbonil)-p-alanina en lugar de ácido 4-(terc-butoxicarbonilamino)butanoico para producir el compuesto del título en forma de un sólido de color amarillo parduzco (616 mg, cuantitativo).
RMN 1H (400 MHz, DMSO-d6) 5: 0,87 (3H, t, J = 7,2 Hz), 1,29 (9H, s), 1,86 (2H, dt, J = 15,1,7,3 Hz), 2 ,ü4-2,22 (2H,
m), 2,31 (2H, t, J = 6,8 Hz), 2,40 (3H, s), 3,10-3,26 (4H, m), 5,15 (1H, d, J = 18,8 Hz), 5,26 (1H, d, J = 19,2 Hz), 5,42 (2H, dd, J = 18,8, 16,4 Hz), 5,57 (1H, dt, J = 8,5, 4,2 Hz), 6,53 (1H, s), 6,78 (1H, t, J = 5,5 Hz), 7,30 (1H, s), 7,80 (1H, d, J = 11,0 Hz), 8,46 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 607 (M+H)+
Proceso 2: N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]-p-alaninamida
El compuesto obtenido en el proceso 1 se hizo reaccionar de la misma forma que en el proceso 2 del Ejemplo 1 para producir trifluoroacetato del compuesto del título en forma de un sólido de color amarillo (499 mg, 86 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,2 Hz), 1,86 (2H, dquin, J = 14,6, 7,2, 7,2, 7,2, 7,2 Hz), 2,06-2,27 (1H, m), 2,41 (3H, s), 2,46-2,57 (2H, m), 3,08 (2H, t, J = 6,8 Hz), 3,14-3,24 (2H, m), 5,22 (1H, d, J = 18,8 Hz), 5,29 (1H, d, J = 18,8 Hz), 5,43 (2H, s), 5,58 (1H, dt, J = 8,5, 4,5 Hz), 6,55 (1H, s), 7,32 (1H, s), 7,74 (3H, s a), 7,82 (1H, d, J = 11,0 Hz), 8,67 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 507 (M+H)+
Ejemplo 50 Conjugado de anticuerpo - fármaco (47)
[Fórmula 119]

Proceso 1: N-(terc-butoxicarbonil)glicilglicil-L-fenilalanilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]-p-alaninamida
El compuesto (484 mg, 0,780 mmol) del Ejemplo 49 se hizo reaccionar de la misma forma que en el proceso 1 del Ejemplo 2 para producir el compuesto del título en forma de un sólido de color amarillo pálido (626 mg, 87 %).
RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,4 Hz), 1,27-1,42 (9H, m), 1,77-1,93 (2H, m), 2,06-2,22 (2H, m), 2,36 (2H, t, J = 7,2 Hz), 2,40 (3H, d, J = 1,6 Hz), 2,44-2,54 (2H, m), 2,76 (1H, dd, J = 14,5, 10,2 Hz), 3,02 (1H, dd, J = 13,9, 4,5 Hz), 3,12-3,22 (2H, m), 3,52 (6H, d, J = 6,3 Hz), 4,42-4,54 (1H, m), 5,19 (1H, d, J = 19,2 Hz), 5,26 (1H, d, J = 18,4 Hz), 5,42 (1H, dd, J = 18,4, 16,4 Hz), 5,57 (1H, dt, J = 8,7, 4,4 Hz), 6,53 (1H, s), 6,98 (1H, t, J = 5,9 Hz), 7,14-7,28 (5H, m), 7,31 (1H, s), 7,77-7,84 (1H, m), 7. 91 (1H, t, J = 5,5 Hz), 8,16 (1H, d, J = 7,8 Hz), 8,27 (1H, t, J = 5,1 Hz), 8,52 (1H, d, J = 9,0 Hz).
Proceso 2: Trifluoroacetato de glicilglicil-L-fenilalanilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]-p-alaninamida
El compuesto (624 mg, 0,675 mmol) obtenido en el proceso 1 anterior se hizo reaccionar de la misma forma que en el proceso 2 del Ejemplo 2 para producir el compuesto del título en forma de un sólido de color amarillo (626 mg, 92 %). RMN 1H (400 MHz, DMSO-d6) 8: 0,87 (3H, t, J = 7,4 Hz), 1,86 (2H, tt, J = 14,5, 7,2 Hz), 2,07-2,22 (2H, m), 2,36 (2H, t, J = 7,2 Hz), 2,40 (3H, s), 2,44-2,54 (2H, m), 2,75 (1H, dd, J = 13,7, 9,8 Hz), 3,04 (1H, dd, J = 13,7, 4,3 Hz), 3,12-3,22 (2H, m), 3,58 (2H, d, J = 4,7 Hz), 3,69 (3H, td, J = 11,2, 5,7 Hz), 3,87 (1H, dd, J = 17,0, 5,7 Hz), 4,54 (1H, m, J = 17,8, 4,5 Hz), 5,19 (1H, d, J = 19,2 Hz), 5,26 (1H, d, J = 18,8 Hz), 5,43 (2H, s), 5,51-5,60 (1H, m), 6,55 (1H, s), 7,14-7,29 (5H, m), 7,32 (1H, s), 7,81 (1H, d, J = 10,9 Hz), 7,88 (1H, t, J = 5,7 Hz), 7,97 (3H, s a), 8,29-8,38 (2H, m), 8,50 (1H, t,
J = 5,7 Hz), 8,55 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 825 (M+H)+
Proceso 3: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]glicMgMcM-L-fenMalanMgMcM-N-[(1S,9S)-9-etM-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolin-1-il]-p-alaninamida
El compuesto (60,0 mg, 0,0646 mmol) obtenido en el proceso 2 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 para producir el compuesto del título en forma de un sólido (14,0 mg, 21 %).
RMN 1H (400 MHz, DMSO-d6) 5: 0,86 (3H, t, J = 7,2 Hz), 1,12-1,22 (2H, m), 1,39-1,51 (4H, m), 1,79-1,91 (2H, m), 2,02-2,20 (2H, m), 2,07 (2H, t, J = 7,4 Hz), 2,30-2,42 (4H, m), 2,40 (3H, s), 2,78 (1H, dd, J = 14,1,9,4 Hz), 3,02 (1H, dd, J = 14,7, 4,9 Hz), 3,12-3,21 (2H, m), 3,26-3,42 (2H, m), 3,50-3,80 (6H, m), 4,40-4,51 (1H, m), 5,19 (1H, d, J = 19,6 Hz), 5,26 (1H, d, J = 19,2 Hz), 5,42 (2H, s a), 5,51-5,62 (1H, m), 6,53 (1H, s), 6,99 (2H, s), 7,13-7,28 (5H, m), 7,31 (1H, s), 7,74-7,84 (2H, m), 8,01 (1H, t, J = 5,3 Hz), 8,06 (1H, t, J = 5,77 Hz), 8,14 (1H, d, J = 8,2 Hz), 8,25 (1H, t, J = 5,7 Hz), 8,53 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 1018 (M+H)+
Proceso 4: Conjugado de anticuerpo - fármaco (47)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 3, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 4 del Ejemplo 2. Concentración de anticuerpo: 12,27 mg/ml, rendimiento de anticuerpo: 8,6 mg (69%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,4.
Ejemplo 51 Conjugado de anticuerpo - fármaco (48)
[Fórmula 120]

Proceso 1: N-[3-(2,5-dioxo-2,5-dihidro-1H-pirrol-1 -il)propanoM]glicilglicM-L-fenilalanilglicil-N-[(1S,9S)-9-etM-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolin-1-il]-p-alaninamida
El compuesto (60,0 mg, 0,0646 mmol) obtenido en el proceso 2 del Ejemplo 50 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 usando propionato de N-succinimidil 3-maleimida en lugar de hexanoato de N-succinimidil 6-maleimida para producir el compuesto del título en forma de un sólido de color amarillo pálido (36,0 mg, 57 %).
RMN 1H (400 MHz, DMSO-d6) 5: 0,86 (3H, t, J = 7,4 Hz), 1,85 (2H, dt, J = 14,4, 7,5 Hz), 2,05-2,22 (2H, m), 2,40 (3H, s), 2,30-2,44 (5H, m), 2,73-2,84 (1H, m), 3,02 (1H, dd, J = 13,9, 4,5 Hz), 3,17 (3H, d, J = 5,1 Hz), 3,26-3,40 (2H, m), 3,41-3,81 (6H, m), 4,40-4,51 (1H, m), 5,19 (1H, d, J = 19,2 Hz), 5,26 (1H, d, J = 18,8 Hz), 5,42 (2H, s a), 5,52-5,61 (1H, m), 6,53 (1H, s), 6,99 (2H, s), 7,13-7,28 (5H, m), 7,31 (1H, s), 7,80 (2H, d, J = 10,2 Hz), 8,03 (1H, t, J = 5,5 Hz), 8,12 (1H, d, J = 8,2 Hz), 8,20-8,31 (2H, m), 8,52 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 976 (M+H)+
Proceso 2: Conjugado de anticuerpo - fármaco (48)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 1, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 4 del Ejemplo 2. Concentración de anticuerpo: 11,59 mg/ml, rendimiento de anticuerpo: 8,1 mg (65%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,7.
Ejemplo 52 Conjugado de anticuerpo - fármaco (49)
[Fórmula 121]

Proceso 1: N-{3-[2-(2-{[3-(2,5-dioxo-2,5-dihidro-1H-pirrol-1 -il)propanoil]amino})etoxi]propanoM}glicilglicil-L-fenilalanilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-lH,12H-benzo[de]pirano[3',4':6,7]indoNzino[1,2-b]quinoNn-1-N]-p-alaninamida
El compuesto (60,0 mg, 0,0646 mmol) obtenido en el proceso 2 del Ejemplo 50 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 usando 3-(2-(2-(3-maleinimidapropanamida)etoxi)etoxi)propanoato de N-succinimidilo en lugar de hexanoato de N-succinimidil 6-maleimida para producir el compuesto del título en forma de un sólido (23,0 mg, 31 %).
RMN 1H (400 MHz, DMSO-da) 8: 0,86 (3H, t, J = 7,4 Hz), 1,77-1,92 (2H, m), 2,07-2,21 (2H, m), 2,27-2,42 (6H, m), 2,40 (3H, s), 2,74-2,84 (1H, m), 2,97-3,06 (1H, m), 3,09-3,21 (4H, m), 3,25-3,39 (6H, m), 3,45 (4H, s), 3,50-3,80 (8H, m), 4,41-4,51 (1H, m), 5,19 (1H, d, J = 18,4 Hz), 5,26 (1H, m, J = 18,4 Hz), 5,42 (2H, s a), 5,51-5,61 (1H, m), 6,54 (1H, s), 7,00 (2H, s), 7,13-7,28 (5H, m), 7,31 (1H, s), 7,74-7,87 (2H, m), 7,93-8,07 (2H, m), 8,09-8,21 (2H, m), 8,26 (1H, s a), 8,54 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 1135 (M+H)+
Proceso 2: Conjugado de anticuerpo - fármaco (49)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 1, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 2 del Ejemplo 29. Concentración de anticuerpo: 14,50 mg/ml, rendimiento de anticuerpo: 10,2 mg (82%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,8.
Ejemplo 53 Conjugado de anticuerpo - fármaco (50)
[Fórmula 122]

Proceso 1: N-[19-(2,5-dioxo-2,5-dihidro-1H-pirrol-1 -N)-17-oxo-4,7,10,13-tetraoxa-16-azanonadecan-1-oN]gNcNglicN-L-fenilalanilglicil-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indoNzino[1,2-b]quinoNn-1-N]-p-alaninamida
El compuesto (60,0 mg, 0,0646 mmol) obtenido en el proceso 2 del Ejemplo 50 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 usando N-succinimidil 1-maleinimida-3-oxo-7,10,13,16-tetraoxa-4-azanonadecanoato en lugar de hexanoato de N-succinimidil 6-maleimida para producir el compuesto del título en forma de un sólido (23,0 mg, 29 %).
RMN 1H (400 MHz, DMSO-da) 8: 0,86 (3H, t, J = 7,0 Hz), 1,85 (2H, tt, J = 14,6, 7,1 Hz), 2,06-2,22 (2H, m), 2,40 (3H, s), 2,28-2,43 (6H, m), 2,78 (1H, dd, J = 13,7, 9,4 Hz), 3,02 (1H, dd, J = 14,1, 3,9 Hz), 3,09-3,22 (4H, m), 3,27-3,41 (4H, m), 3,47 (12H, d, J = 8,6 Hz), 3,53-3,81 (10H, m), 4,41-4,51 (1H, m), 5,19 (1H, d, J = 19,2 Hz), 5,26 (1H, d, J = 18,8 Hz), 5,42 (2H, s a), 5,53-5,61 (1H, m), 6,54 (1H, s), 7,00 (2H, s), 7,12-7,29 (5H, m), 7,31 (1H, s), 7,74-7,85 (2H, m), 8,03 (2H, d, J = 6,6 Hz), 8,11-8,21 (2H, m), 8,27 (1H, t, J = 5,9 Hz), 8,54 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 1224 (M+H)+
Proceso 2: Conjugado de anticuerpo - fármaco (50)
Usando el anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 y el compuesto obtenido en el proceso 1, el conjugado de anticuerpo - fármaco del título se obtuvo de la misma forma que en el proceso 4 del Ejemplo 2. Concentración de anticuerpo: 13,47 mg/ml, rendimiento de anticuerpo: 9,4 mg (75%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,1.
Ejemplo 54 Conjugado de anticuerpo - fármaco (51)
[Fórmula 123]
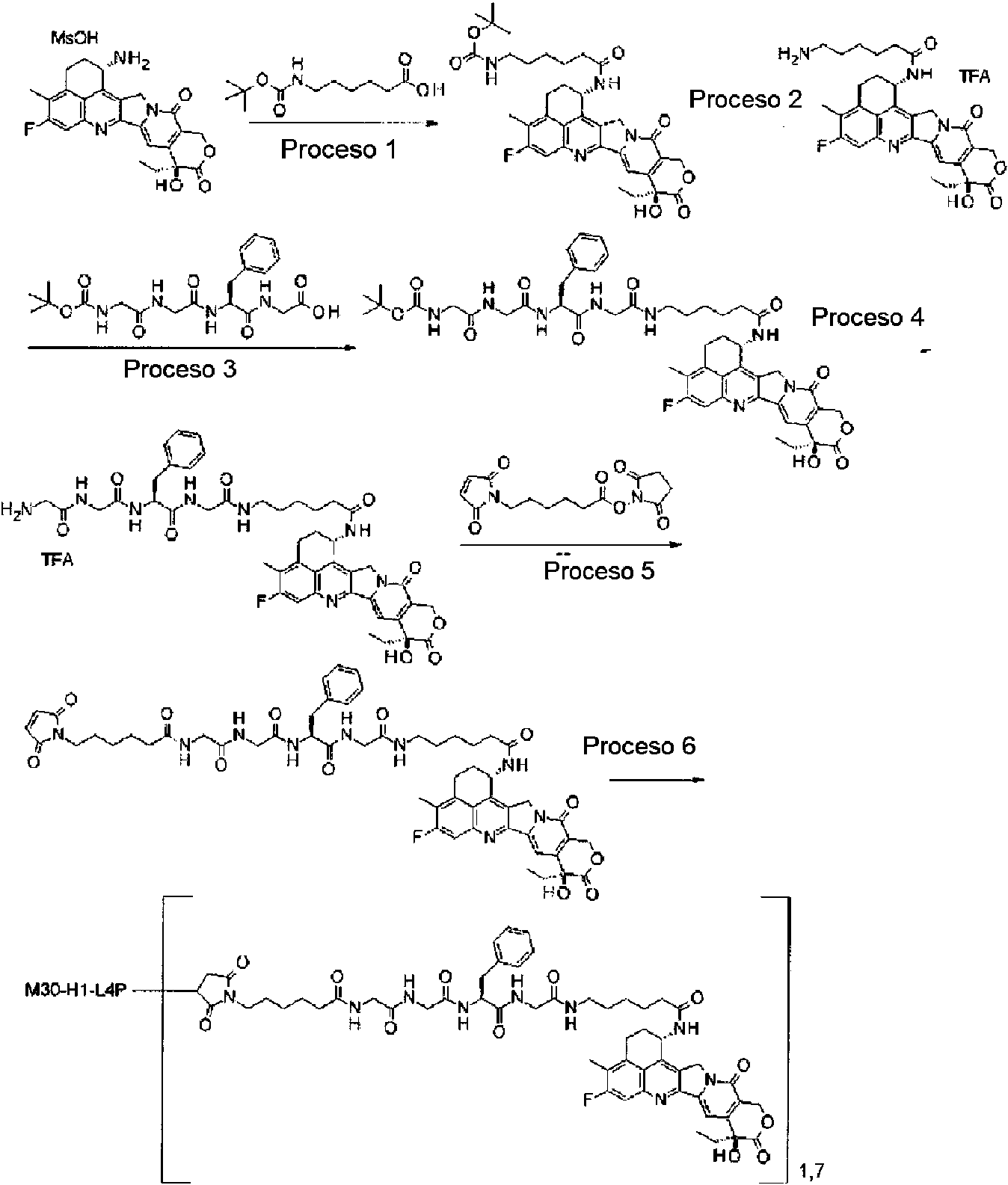
Proceso 1: (6-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]aminol-6-oxohexil)carbamato de terc-butilo
Mesilato del compuesto (4) (0,500 g, 0,882 mmol) se hizo reaccionar de la misma forma que en el proceso 1 del Ejemplo 1 usando ácido 6-(terc-butoxicarbonilamino)hexanoico en lugar de ácido 4-(tercbutoxicarbonilamino)butanoico para producir el compuesto del título (0,620 g, cuantitativo).
RMN 1H (DMSO-da) 8: 0,83 (3H, t, J = 7,8 Hz), 1,14-1,28 (2H, m), 1,31 (9H, s), 1,47-1,61 (2H, m), 1,75-1,89 (2H, m), 2,04-2,17 (4H, m), 2,35 (3H, s), 2,81-2,88 (2H, m), 3,09-3,16 (2H, m), 5,10 (1H, d, J = 19,4 Hz), 5,16 (1H, d, J = 19,4 Hz), 5,39 (2H, s), 5,48-5,55 (1H, m), 6,50 (1H, s), 6,73-6,78 (1H, m), 7,26 (1H, s), 7,74 (1H, d, J = 10,9 Hz), 8,39 (1H, d, J = 9,0 Hz).
Proceso 2: trifluoroacetato de 6-amino-N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]hexanamida
El compuesto (0,397 g, 0,611 mmol) obtenido en el proceso 1 se hizo reaccionar de la misma forma que en el proceso 2 del Ejemplo 1 para producir el compuesto del título (0,342 g, 84 %).
RMN 1H (DMSO-d6) 8: 0,88 (3H, t, J = 7,2 Hz), 1,31-1,41 (2H, m), 1,52-1,70 (4H, m), 1,80-1,94 (2H, m), 2,05-2,18 (2H, m), 2,21 (2H, t, J = 7,4 Hz), 2,40 (3H, s), 2,81 (2H, t, J = 7,4 Hz), 3,10-3,25 (2H, m), 3,33 (2H, s a), 5,18 (1H, d, J = 19,8 Hz), 5,22 (1H, d, J = 19,8 Hz), 5,41 (2H, d, J = 16,6 Hz), 5,45 (2H, d, J = 16,6 Hz), 5,53-5,60 (1H, m), 6,55 (1H, s), 7,32 (1H, s), 7,80 (1H, d, J = 10,9 Hz), 8,49 (1H, d, J = 9,2 Hz).
Proceso 3: N-(terc-butoxicarbonil)glicilglicil-L-fenilalanil-N-(6-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-6-oxohexil)glicinamida
El compuesto (0,170 g, 0,516 mmol) obtenido en el proceso 2 se hizo reaccionar de la misma forma que en el proceso 1 del Ejemplo 2 para producir el compuesto del título (0,225 g, 91%).
RMN 1H (DMSO-da) 5: 0,88 (3H, t, J = 7,4 Hz), 1,43-1,70 (6H, m), 1,87 (2H, td, J = 15,0, 7,4 Hz), 2,10-2,22 (3H, m), 2,28-2,37 (1H, m), 2,42 (3H, s), 2,78-2,85 (1H, m), 3,01-3,10 (3H, m), 3,15-3,22 (2H, m), 3,54-3,61 (5H, m), 3,62-3,69 (1H, m), 4,44-4,53 (1H, m), 5,17 (1H, d, J = 19,2 Hz), 5,25 (1H, d, J = 19,2 Hz), 5,45 (2H, s), 5,54-5,61 (1H, m), 6,55 (1H, s), 7,02 (1H, t, J = 6,1 Hz), 7,11-7,28 (5H, m), 7,33 (1H, s), 7,63-7,69 (1H, m), 7,82 (1H, d, J = 11,0 Hz), 7,90-7,96 (1H, m), 8,17 (1H, d, J = 7,8 Hz), 8,28 (1H, t, J = 5,5 Hz), 8,46 (1H, d, J = 9,0 Hz).
Proceso 4: Glicilglicil-L-fenilalanil-N-(6-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-6-oxohexil)glicinamida
El compuesto (0,105 g, 0,108 mmol) obtenido en el proceso 3 se hizo reaccionar de la misma forma que en el proceso 2 del Ejemplo 2 para producir el compuesto del título (0,068 mg, 65 %).
RMN 1H (DMSO-d6) 5: 0,89 (3H, t, J = 7,4 Hz), 1,15-1,67 (6H, m), 1,79-1,97 (2H, m), 2,08-2,24 (4H, m), 2,42 (3H, s), 2,76-2,82 (1H, m), 3,00-3,10 (5H, m), 3,19 (1H, s), 3,50-3,63 (2H, m), 3,64-3,76 (3H, m), 3,84-3,92 (1H, m), 4,51-4,59 (1H, m), 5,17 (1H, d, J = 19,4 Hz), 5,24 (1H, d, J = 19,4 Hz), 5,44 (2H, s), 5,53-5,61 (1H, m), 6,55 (1H, s a), 7,15-7,29 (5H, m), 7,33 (1H, s), 7,72-7,78 (1H, m), 7,82 (1H, d, J = 11,0 Hz), 7,96-8,08 (2H, m), 8,30-8,38 (2H, m), 8,46-8,56 (2H, m).
Proceso 5: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]glicilglicil-L-fenilalanil-N-(6-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H, 12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-6-oxohexil)glicinamida
El compuesto (58 mg, 0,060 mmol) obtenido en el proceso 4 se hizo reaccionar de la misma forma que en el proceso 3 del Ejemplo 2 para producir el compuesto del título (39 mg, 62 %).
RMN 1H (CD3OD) 5: 0,99 (3H, t, J = 7,4 Hz), 1,27 (2H, td, J = 11,6, 6,1 Hz), 1,38-1,44 (2H, m), 1,50-1,63 (6H, m), 1,65 1,80 (2H, m), 1,89-1,98 (2H, m), 2,17-2,25 (3H, m), 2,26-2,36 (3H, m), 2,40 (3H, s), 2,95 (1H, dd, J = 14,3, 9,2 Hz), 3,12 (1H, dd, J = 13,7, 5,7 Hz), 3,15-3,25 (4H, m), 3,44 (2H, t, J = 7,2 Hz), 3,65 (1H, d, J = 17,2 Hz), 3,76 (1H, d, J = 17,2 Hz), 3,79-3,86 (4H, m), 4,43 (1H, dd, J = 8,9, 6,0 Hz), 5,10 (1H, d, J = 18,9 Hz), 5,25 (1H, d, J = 18,9 Hz), 5,35 (1H, d, J = 16,6 Hz), 5,56 (1H, d, J = 16,0 Hz), 5,60-5,64 (1H, m), 6,76 (2H, s), 7,12-7,24 (6H, m), 7,58 (1H, s), 7,60 (1H, d, J = 10,9 Hz), 7,68 (1H, t, J = 5,7 Hz).
EM (IEN) m/z: 1060 (M+H)+
Proceso 6: Conjugado de anticuerpo - fármaco (51)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0147 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora. Conjugación entre anticuerpo y enlazador de fármaco: Después de incubar la solución durante 10 minutos a 22 °C, una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 5 (0,0295 ml; 4,6 equivalentes por molécula de anticuerpo) se añadió a la misma y se incubó para conjugar el enlazador de fármaco con el anticuerpo a 22 °C durante 40 minutos. A continuación, una solución acuosa (0,00590 ml; 9,2 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a 22 °C durante otros 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó PBS7,4 como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 0,97 mg/ml, rendimiento de anticuerpo: 5,82 mg (58 %), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 1,7.
Ejemplo 55 Conjugado de anticuerpo - fármaco (52)
[Fórmula 124]

Proceso 1: Conjugado de anticuerpo - fármaco (52)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0295 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en una parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora. Conjugación entre anticuerpo y enlazador de fármaco: Después de incubar la solución durante 10 minutos a 22 °C, una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 5 del Ejemplo 54 (0,0590 ml; 9,2 equivalentes por molécula de anticuerpo) se añadió a la misma y se incubó para conjugar el enlazador de fármaco con el anticuerpo a 22 °C durante 40 minutos. A continuación, una solución acuosa (0,0118 ml; 18,4 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a 22 °C durante otros 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó PBS7,4 como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 0,94 mg/ml, rendimiento de anticuerpo: 5,64 mg (56%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,1.
Ejemplo 56 Conjugado de anticuerpo - fármaco (53)
[Fórmula 125]

Proceso 1: Conjugado de anticuerpo - fármaco (53)
Reducción del anticuerpo: El anticuerpo M30-H1-L4 producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0147 ml; 2,3 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora. Conjugación entre anticuerpo y enlazador de fármaco: Después de incubar la solución a 22 °C durante 10 minutos, una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 5 del Ejemplo 54 (0,0295 ml; 4,6 equivalentes por molécula de anticuerpo) se añadió a la misma y se incubó para conjugar el enlazador de fármaco con el anticuerpo a 22 °C durante 40 minutos. A continuación, una solución acuosa (0,00590 ml; 9,2 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a 22 °C durante otros 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó PBS7,4 como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £A,28o = 235300 (valor de cálculo estimado), £A,37o = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 1,22 mg/ml, rendimiento de anticuerpo: 7,32 mg (73%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 1,5.
Ejemplo 57 Conjugado de anticuerpo - fármaco (54)
[Fórmula 126]

Proceso 1: Conjugado de anticuerpo - fármaco (54)
Reducción del anticuerpo: El anticuerpo M30-H1-L4 producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,0 ml) se recogió en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0295 ml; 4,6 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,050 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora. Conjugación entre anticuerpo y enlazador de fármaco: Después de incubar la solución anterior durante 10 minutos a 22 °C, una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 5 del Ejemplo 54 (0,0590 ml; 9,2 equivalentes por molécula de anticuerpo) se añadió a la misma y se incubó para conjugar el enlazador de fármaco con el anticuerpo a 22 °C durante 40 minutos. A continuación, una solución acuosa (0,0118 ml; 18,4 equivalentes por molécula de anticuerpo) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a 22 °C durante otros 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó PBS7,4 como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 1,06 mg/ml, rendimiento de anticuerpo: 6,36 mg (64%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,0.
Ejemplo 58 Conjugado de anticuerpo - fármaco (55)

Proceso 1: ({N-[(9H-fluoren-9-ilmetoxi)carbonil]glicil}amino)acetato de metilo
A una mezcla que contiene N-9-fluorenilmetoxicarbonilglicilglicina (4,33 g, 12,2 mmol), tetrahidrofurano (120 ml) y tolueno (40,0 ml), se añadieron piridina (1,16 ml, 14,7 mmol) y tetraacetato de plomo (6,84 g, 14,7 mmol) y se calentó a reflujo durante 5 horas. Después de que la solución de reacción se enfriara a temperatura ambiente, los materiales insolubles se retiraron por filtración a través de Celite, y se concentraron a presión reducida. los residuos obtenidos se disolvieron en acetato de etilo y se lavaron con agua y salmuera saturada, y entonces la capa orgánica se secó sobre sulfato de magnesio anhidro. Después de que el disolvente se retirara a presión reducida, los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [hexano : acetato de etilo = 9 : 1 (v/v) - acetato de etilo] para producir el compuesto del título en forma de un sólido incoloro (3,00 g, 67 %).
RMN 1H (400 MHz, CDCla) 8: 2,07 (3H, s), 3,90 (2H, d, J = 5,1 Hz), 4,23 (1H, t, J = 7,0 Hz), 4,46 (2H, d, J = 6,6 Hz), 5,26 (2H, d, J = 7,0 Hz), 5,32 (1H, s a), 6,96 (1H, s a), 7,32 (2H, t, J = 7,3 Hz), 7,41 (2H, t, J = 7,3 Hz), 7,59 (2H, d, J = 7,3 Hz), 7,77 (2H, d, J = 7,3 Hz).
Proceso 2: [({N-[(9H-fluoren-9-ilmetoxi)carbonil]glicil}amino)metoxi]acetato de bencilo
A una solución de tetrahidrofurano (40,0 ml) del compuesto (3,68 g, 10,0 mmol) obtenido en el proceso 1 anterior y glicolato de bencilo (4,99 g, 30,0 mmol), se añadió terc-butóxido potásico (2,24 g, 20,0 mmol) a 0 °C y se agitó a temperatura ambiente durante 15 minutos. La solución de reacción se cargó con acetato de etilo y agua a 0 °C y se extrajo con acetato de etilo y cloroformo. La capa orgánica obtenida se secó sobre sulfato sódico y se filtró. El disolvente se eliminó a presión reducida. Los residuos obtenidos se disolvieron en dioxano (40,0 ml) y agua (10,0 ml), se cargaron con hidrogenocarbonato sódico (1,01 g, 12,0 mmol) y cloroformiato de 9-fluorenilmetilo (2,59 g, 10,0 mmol) y se agitaron a temperatura ambiente durante 2 horas. La solución de reacción se cargó con agua y se extrajo con acetato de etilo. La capa orgánica obtenida se secó sobre sulfato sódico y se filtró. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [hexano : acetato de
etilo = 100 : 0 (v/v) - 0 : 100] para producir el compuesto del título en una sustancia oleosa incolora (1,88 g, 40 %). RMN 1H (400 MHz, CDCla) 8: 3,84 (2H, d, J = 5,5 Hz), 4,24 (3H, t, J = 6,5 Hz), 4,49 (2H, d, J = 6,7 Hz), 4,88 (2H, d, J = 6,7 Hz), 5,15-5,27 (1H, m), 5,19 (2H, s), 6,74 (1H, s a), 7,31-7,39 (7H, m), 7,43 (2H, t, J = 7,4 Hz), 7,61 (2H, d, J = 7,4 Hz), 7,79 (2H, d, J = 7,4 Hz).
Proceso 3: Ácido [({N-[(9H-fluoren-9-ilmetoxi)carbonil]glicil}amino)metoxi]acético
El compuesto (1,88 g, 3,96 mmol) obtenido en el proceso 2 se disolvió en etanol (40,0 ml) y acetato de etilo (20,0 ml). Después de añadir catalizador de paladio-carbono (376 mg), se agitó bajo una atmósfera de hidrógeno a temperatura ambiente durante 2 horas. Los materiales insolubles se retiraron por filtración a través de Celite, y el disolvente se retiró a presión reducida para producir el compuesto del título en forma de un sólido incoloro (1,52 g, cuantitativo). RMN 1H (400 MHz, DMSO-d6) 8: 3,62 (2H, d, J = 6,3 Hz), 3,97 (2H, s), 4,18-4,32 (3H, m), 4,60 (2H, d, J = 6,7 Hz), 7,29-7,46 (4H, m), 7,58 (1H, t, J = 5,9 Hz), 7,72 (2H, d, J = 7,4 Hz), 7,90 (2H, d, J = 7,4 Hz), 8,71 (1H, t, J = 6,5 Hz).
Proceso 4: 9H-Fluoren-9-ilmetil(2-{[(2-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-2-oxoetoxi)metil]amino}-2-oxoetil)carbamato
Con enfriamiento con hielo, a una solución de N,N-dimetilformamida (10,0 ml) de mesilato del compuesto (4) (0,283 g, 0,533 mmol), N-hidroxisuccinimida (61,4 mg, 0,533 mmol), y el compuesto (0,205 g, 0,533 mmol) obtenido en el proceso 3, se añadieron N,N-diisopropiletilamina (92,9 pl, 0,533 mmol) y N,N'-diciclohexilcarbodiimida (0,143 g, 0,693 mmol) y se agitó a temperatura ambiente durante 3 días. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - capa orgánica repartida de cloroformo : metanol : agua = 7 : 3 : 1 (v/v/v)] para producir el compuesto del título en forma de un sólido de color pardo pálido (0,352 g, 82 %). RMN 1H (400 MHz, DMSO-d6) 8: 0,81 (3H, t, J = 7,4 Hz), 1,73-1,87 (2H, m), 2,06-2,20 (2H, m), 2,34 (3H, s), 3,01-3,23 (2H, m), 3,58 (2H, d, J = 6,7 Hz), 3,98 (2H, s), 4,13-4,25 (3H, m), 4,60 (2H, d, J = 6,7 Hz), 5,09 5,22 (2H, m), 5,32-5,42 (2H, m), 5,50-5,59 (1H, m), 6,49 (1H, s), 7,24-7,30 (3H, m), 7,36 (2H, t, J = 7,4 Hz), 7,53 (1H, t, J = 6,3 Hz), 7,66 (2H, d, J = 7,4 Hz), 7,75 (1H, d, J = 11,0 Hz), 7,84 (2H, d, J = 7,4 Hz), 8,47 (1H, d, J = 8,6 Hz), 8,77 (1H, t, J = 6,7 Hz).
EM (IEN) m/z: 802 (M+H)+
Proceso 5: N-[(2-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-2-oxoetoxi)metil]glicinamida
A una solución de N,N-dimetilformamida (11,0 ml) del compuesto (0,881 g, 1,10 mmol) obtenido en el proceso 4, se añadió piperidina (1,1 ml) y se agitó a temperatura ambiente durante 2 horas. El disolvente se retiró a presión reducida para producir una mezcla que contiene el compuesto del título. La mezcla se usó para la siguiente reacción sin realizar purificación adicional.
Proceso 6: N-[(9H-fluoren-9-ilmetoxi)carbonil]glicilglicil-L-fenilalanil-N-[(2-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-2-oxoetoxi)metil]glicinamida
Con enfriamiento con hielo, a una solución de N,N-dimetilformamida (50,0 ml) de la mezcla (0,439 mmol) obtenida en el proceso 5, N-hidroxisuccinimida (0,101 g, 0,878 mmol), y N-[(9H-fluoren-9-ilmetoxi)carbonil]glicilglicil-L-fenilalanina (esto es, el compuesto descrito en la patente japonesa abierta a inspección pública n.° 2002-60351) (0,440 g, 0,878 mmol), se añadió N,N'-diciclohexilcarbodiimida (0,181 g, 0,878 mmol) y se agitó a temperatura ambiente durante 4 días. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo : metanol = 9 : 1 (v/v)] para producir el compuesto del título en forma de un sólido de color naranja pálido (0,269 g, 58 %).
EM (IEN) m/z: 1063 (M+H)+
Proceso 7: Glicilglicil-L-fenilalanil-N-[(2-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-2-oxoetoxi)metil]glicinamida
A una solución de N,N-dimetilformamida (4,00 ml) del compuesto (0,269 g, 0,253 mmol) obtenido en el proceso 6, se añadió piperidina (0,251 ml, 2,53 mmol) y se agitó a temperatura ambiente durante 2 horas. El disolvente se retiró a presión reducida para producir una mezcla que contiene el compuesto del título. La mezcla se usó para la siguiente reacción sin realizar purificación adicional.
Proceso 8: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]glicilglicil-L-fenilalanil-N-[(2-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]amino}-2-oxoetoxi)metil]glicinamida
A una solución de N,N-dimetilformamida (10,0 ml) del compuesto (0,253 mmol) obtenido en el proceso 7, se añadió hexanoato de N-succinimidil 6-maleimida (0,156 g, 0,506 mmol) y se agitó a temperatura ambiente durante 3 días. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo : metanol = 9 : 1 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,100 g, 38 %).
RMN 1H (400 MHz, DMSO-da) 5: 0,83 (3H, t, J = 7,2 Hz), 1,09-1,21 (2H, m), 1,33-1,47 (4H, m), 1,75-1,90 (2H, m), 2,00-2,23 (4H, m), 2,36 (3H, s), 2,69-2,81 (1H, m), 2,94-3,03 (1H, m), 3,06-3,22 (2H, m), 3,23-3,74 (8H, m), 3,98 (2H, s), 4,39-4,50 (1H, m), 4,60 (2H, d, J = 6,7 Hz), 5,17 (2H, s), 5,39 (2H, s), 5,53-5,61 (1H, m), 6,50 (1H, s), 6,96 (2H, s), 7,11-7,24 (5H, m), 7,28 (1H, s), 7,75 (1H, d, J = 11,0 Hz), 7,97 (1H, t, J = 5,7 Hz), 8,03 (1H, t, J = 5,9 Hz), 8,09 (1H, d, J = 7,8 Hz), 8,27 (1H, t, J = 6,5 Hz), 8,48 (1H, d, J = 9,0 Hz), 8,60 (1H, t, J = 6,5 Hz).
EM (IEN) m/z: 1034 (M+H)+
Proceso 9: Conjugado de anticuerpo - fármaco (55)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 10 mg/ml sustituyendo el medio con PBS6,0/EDTA usando el procedimiento común C-1 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) descrito en el procedimiento de producción 1. La solución (1,25 ml) se puso en un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,025 ml; 3,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,109 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 (0,039 ml; 4,6 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 40 minutos. A continuación, se añadió una solución acuosa (0,008 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) a la misma y se agitó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A descrito en el procedimiento de producción 1.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £A,280 = 235300 (valor de cálculo estimado), £A,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 12,57 mg/ml, rendimiento de anticuerpo: 8,8 mg (70%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,2.
Ejemplo 59 Conjugado de anticuerpo - fármaco (56)
[F
Proceso 1: Conjugado de anticuerpo - fármaco (56)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 10 mg/ml sustituyendo el medio con PBS6,0/EDTA usando el procedimiento común C-1 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) descrito en el procedimiento de producción 1. La solución (1,25 ml) se puso en un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,051 ml; 6,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después
de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,067 ml) una solución de dimetilsulfóxido que contiene y 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,085 ml; 10,0 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 60 minutos. A continuación, se añadió una solución acuosa (0,013 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) a la misma y se agitó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes valores característicos. Concentración de anticuerpo: 1,33 mg/ml, rendimiento de anticuerpo: 7,98 mg (64%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 4,9.
Ejemplo 60 Conjugado de anticuerpo - fármaco (57)
[Fórmula 129]
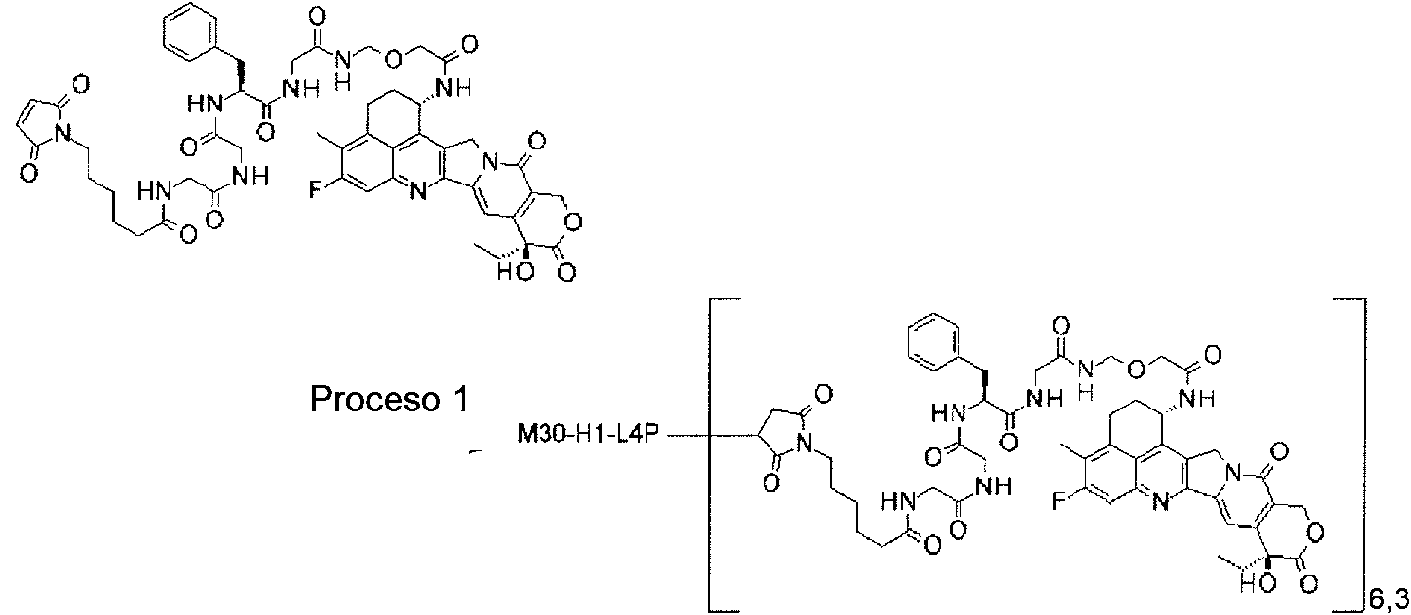
Proceso 1: Conjugado de anticuerpo - fármaco (57)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 2 se preparó para tener una concentración de anticuerpo de 10 mg/ml sustituyendo el medio con PBS6,0/EDTA usando el procedimiento común C-1 y el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) descrito en el procedimiento de producción 1. La solución (1,25 ml) se puso en un tubo de polipropileno de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,051 ml; 6,0 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir dimetilsulfóxido (Sigma-Aldrich Co. LLC; 0,025 ml) y una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,127 ml; 15,0 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por As ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 60 minutos. A continuación, se añadió una solución acuosa (0,019 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) a la misma y se agitó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A descrito en el procedimiento de producción 1.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5000 (valor promedio medido) y £d,370 = 19000 (valor promedio medido)), se obtuvieron los siguientes
valores característicos. Concentración de anticuerpo: 0,91 mg/ml, rendimiento de anticuerpo: 5,46 mg (44%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,3.
Ejemplo 61 Conjugado de anticuerpo - fármaco (58)
[Fórmula 130]

Casi la totalidad de las cantidades de los conjugados de anticuerpo - fármaco de Ejemplos 59 y 60 se mezclaron y la solución se concentró mediante el procedimiento común A descrito en el procedimiento de producción 1 para producir el conjugado de anticuerpo - fármaco del título.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 12,30 mg, y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,4.
Ejemplo 62 Conjugado de anticuerpo - fármaco (59)
[Fórmula 131]

Proceso 1: Conjugado de anticuerpo - fármaco (59)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (100 ml, 1 g del anticuerpo) se puso en un matraz de 250 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (2,43 ml; 3,6 equivalentes por molécula de anticuerpo) y adicionalmente con una solución acuosa de hidrogenofosfato de dipotasio 1 M (5 ml). Después de confirmar que la solución tenía un pH cerca de 7,4 usando un pehachímetro, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (3,51 ml; 5,2 equivalentes por molécula de anticuerpo) y dimetilsulfóxido (2,14 ml) a la solución anterior a temperatura ambiente, se agitó con un agitador para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 130 minutos. A continuación, una solución acuosa (0,547 ml) de NAC 100 mM se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos.
Purificación: La solución anterior se sometió a purificación por ultrafiltración usando un aparato de ultrafiltración compuesto por una membrana de ultrafiltración (Merck Japan, Casete Pellicon XL, Biomax 50 KDa), una bomba de tubo (Cole-Parmer International, Bomba MasterFlex modelo 77521-40, Cabezal de Bomba modelo 7518-00), y un tubo (Cole-Parmer International, Tubo MasterFlex L/S16). Específicamente, al tiempo que se añadía ABS gota a gota (un total de 800 ml) como una solución tampón para la purificación a la solución de reacción, se realizó una purificación
por ultrafiltración para retirar enlazadores de fármaco no conjugados y otros reactivos de bajo peso molecular, sustituir también la solución tampón con ABS, y concentrar adicionalmente la solución, para producir aproximadamente 70 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28ü = 235300 (valor de cálculo estimado), £a,37ü = 0 (valor de cálculo estimado), £d,28ü = 5178 (valor medido) y £d,37ü = 20217 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 14,2 mg/ml, rendimiento de anticuerpo: 1,0 g (aproximadamente 100 %), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,2.
Ejemplo 63 Conjugado de anticuerpo - fármaco (60)
[Fórmula 132]

Proceso 1: Conjugado de anticuerpo - fármaco (60)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (5 ml, 50 mg del anticuerpo) se puso en un tubo de 15 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,075 ml; 4 equivalentes por molécula de anticuerpo). Después de confirmar que la solución tenía un pH cerca de 7,0 usando un pehachímetro, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,219 ml; 6,5 equivalentes por molécula de anticuerpo) y dimetilsulfóxido (0,064 ml) a la solución anterior, se incubó para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 90 minutos. A continuación, una solución acuosa (ü,033 ml; 9,8 equivalentes por molécula de anticuerpo) de NAC 100 mM se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 19 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28ü = 235300 (valor de cálculo estimado), £a,37ü = 0 (valor de cálculo estimado), £d,28ü = 5178 (valor medido) y £d,37ü = 20217 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 2,19 mg/ml, rendimiento de anticuerpo: 42 mg (83%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 4,7.
Ejemplo 64 Conjugado de anticuerpo - fármaco (61)
[Fórmula 133]

Proceso 1: Conjugado de anticuerpo - fármaco (61)
Reducción del anticuerpo: El anticuerpo M30-H1-L4P producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg'1cirr1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (4 ml, 40 mg del anticuerpo) se puso en un tubo de 15 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,14 ml; 5,2 equivalentes por molécula de anticuerpo). Después de confirmar que la solución tenía un pH cerca de 7,0 usando un pehachímetro, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora. Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,232 ml; 8,6 equivalentes por molécula de anticuerpo) a la solución anterior, se incubó para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 60 minutos. A continuación, una solución acuosa (0,035 ml; 12,9 equivalentes por molécula de anticuerpo) de NAC 100 mM se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 13 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 235300 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5178 (valor medido) y £d,370 = 20217 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 2,03 mg/ml, rendimiento de anticuerpo: 26 mg (66%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,7.
Ejemplo 65 Conjugado de anticuerpo - fármaco (62)
[Fórmula 134]

Proceso 1: Conjugado de anticuerpo - fármaco (62)
Reducción del anticuerpo: El anticuerpo M30-H1-L4 producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,25 ml, 12,5 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0287 ml; 3,4 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora. Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,0439 ml; 5,2 equivalentes por molécula de anticuerpo) y dimetilsulfóxido (0,0267 ml) a la solución anterior a temperatura ambiente, se incubó para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 1 hora. A continuación, una solución acuosa (0,0066 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A. Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28ü = 235300 (valor de cálculo estimado), £a,37ü = 0 (valor de cálculo estimado), £d,28ü = 5178 (valor medido) y £d,37ü = 20217 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 7,8 mg (62%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,4.
Ejemplo 66 Conjugado de anticuerpo - fármaco (63)
[Fórmula 135]

Proceso 1: Conjugado de anticuerpo - fármaco (63)
Reducción del anticuerpo: El anticuerpo M30-H1-L4 producido en el Ejemplo de Referencia 1 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,61 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (1,25 ml, 12,5 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0439 ml; 5,2 equivalentes por molécula de anticuerpo) (0,0287 ml; 3,4 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0625 ml). Después de confirmar que la solución tenía un pH de 7,4 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora. Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,0726 ml; 8,6 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, se incubó para conjugar el enlazador de fármaco con el anticuerpo en un baño de agua a 15 °C durante 1 hora. A continuación, una solución acuosa (0,011 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó para terminar la reacción del enlazador de fármaco a temperatura ambiente durante otros 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 6 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título. Después de esto, la solución se concentró mediante el procedimiento común A. Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £A,280 = 235300 (valor de cálculo estimado), £A,370 = 0 (valor de cálculo estimado), £d,280 = 5178 (valor medido) y £d,370 = 20217 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 10,0 mg/ml, rendimiento de anticuerpo: 7,3 mg (58 %), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 5,4.
Ejemplo 67 Conjugado de anticuerpo - fármaco (64)
[Fórmula 136]

Proceso 1: Conjugado de anticuerpo - fármaco (64)
Reducción del anticuerpo: El anticuerpo anti-CD30 producido en el Ejemplo de Referencia 3 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,75 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0065 ml; 2,5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0058 ml). Después de confirmar que la solución tenía un pH de 7,0 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,0116 ml; 4,5 equivalentes por molécula de anticuerpo) y dimetilsulfóxido (0,0101 ml) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0017 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28ü = 270400 (valor de cálculo estimado), £a,37ü = 0 (valor de cálculo estimado), £d,28ü = 5178 (valor medido) y £d,37ü = 20217 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 0,96 mg/ml, rendimiento de anticuerpo: 2,4 mg (60%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,7.
Ejemplo 68 Conjugado de anticuerpo - fármaco (65)
[Fórmula 137]

Proceso 1: Conjugado de anticuerpo - fármaco (65)
Reducción del anticuerpo: El anticuerpo anti-CD30 producido en el Ejemplo de Referencia 3 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,75 mlmg^cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0129 ml; 5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,006 ml). Después de confirmar que la solución tenía un pH de 7,0 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,0233 ml; 9 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0035 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,28ü = 270400 (valor de cálculo estimado), £a,37ü = 0 (valor de cálculo estimado), £d,28ü = 5178 (valor medido) y £d,37ü = 20217 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 0,39 mg/ml, rendimiento de anticuerpo: 1,0 mg (24%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 6,8.
Ejemplo 69 Conjugado de anticuerpo - fármaco (66)
[Fórmula 138]

Proceso 1: Conjugado de anticuerpo - fármaco (66)
Reducción del anticuerpo: El anticuerpo anti-CD33 producido en el Ejemplo de Referencia 4 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0065 ml; 2,5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0058 ml). Después de confirmar que la solución tenía un pH de 7,0 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,0116 ml; 4,5 equivalentes por molécula de anticuerpo) y dimetilsulfóxido (0,0101 ml) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0017 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £A,280 = 256400 (valor de cálculo estimado), £A,370 = 0 (valor de cálculo estimado), £D280 = 5178 (valor medido) y £D,370 = 20217 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 1,19 mg/ml, rendimiento de anticuerpo: 3,0 mg (74%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,8.
Ejemplo 70 Conjugado de anticuerpo - fármaco (67)
[Fórmula 139]
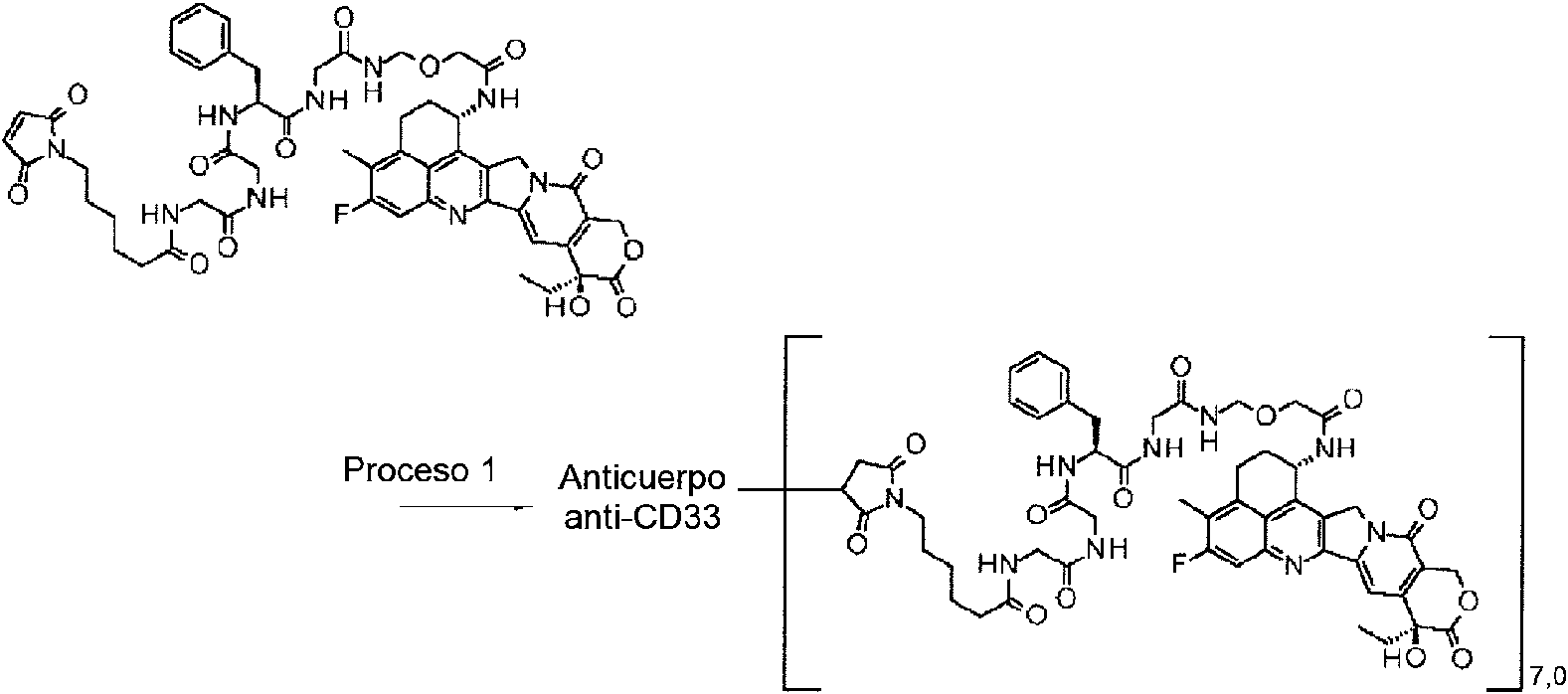
Proceso 1: Conjugado de anticuerpo - fármaco (67)
Reducción del anticuerpo: El anticuerpo anti-CD33 producido en el Ejemplo de Referencia 4 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,66 mlmg-1cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0129 ml; 5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,006 ml). Después de confirmar que la solución tenía un pH de 7,0 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,0233 ml; 9 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0035 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £A,28o = 256400 (valor de cálculo estimado), £A,370 = 0 (valor de cálculo estimado), £d,280 = 5178 (valor medido) y £d,370 = 20217 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 1,24 mg/ml, rendimiento de anticuerpo: 3,1 mg (78%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 7,0.
Ejemplo 71 Conjugado de anticuerpo - fármaco (68)
[Fórmula 140]

Proceso 1: Conjugado de anticuerpo - fármaco (68)
Reducción del anticuerpo: El anticuerpo anti-CD70 producido en el Ejemplo de Referencia 5 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,0/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,69 mlmg_1cm'1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0065 ml; 2,5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,0058 ml). Después de confirmar que la solución tenía un pH de 7,0 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,0116 ml; 4,5 equivalentes por molécula de anticuerpo) y dimetilsulfóxido (0,0101 ml) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0017 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos. Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron £a,280 = 262400 (valor de cálculo estimado), £a,370 = 0 (valor de cálculo estimado), £d,280 = 5178 (valor medido) y £d,370 = 20217 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 1,10 mg/ml, rendimiento de anticuerpo: 2,8 mg (69%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 3,8.
Ejemplo 72 Conjugado de anticuerpo - fármaco (69)
[Fórmula 141]

Proceso 1: Conjugado de anticuerpo - fármaco (69)
Reducción del anticuerpo: El anticuerpo anti-CD70 producido en el Ejemplo de Referencia 5 se preparó para tener una concentración de anticuerpo de 10 mg/ml con PBS6,5/EDTA usando el procedimiento común B (como coeficiente de absorción a 280 nm, se usó 1,69 mlmg^cm-1) y el procedimiento común C-1 descrito en el procedimiento de producción 1. La solución (0,4 ml, 4 mg del anticuerpo) se puso en un tubo de 1,5 ml y se cargó con una solución acuosa de TCEP 10 mM (Tokyo Chemical Industry Co., Ltd.) (0,0129 ml; 5 equivalentes por molécula de anticuerpo) y una solución acuosa de hidrogenofosfato de dipotasio 1 M (Nacalai Tesque, Inc.; 0,006 ml). Después de confirmar que la solución tenía un pH de 7,0 ± 0,1, el enlace de disulfuro en parte de bisagra en el anticuerpo se redujo incubando a 37 °C durante 1 hora.
Conjugación entre anticuerpo y enlazador de fármaco: Después de añadir una solución de dimetilsulfóxido que contiene 10 mM del compuesto obtenido en el proceso 8 del Ejemplo 58 (0,0233 ml; 9 equivalentes por molécula de anticuerpo) a la solución anterior a temperatura ambiente, esta se agitó usando un rotador de tubos (MTR-103, fabricado por AS ONE Corporation) para conjugar el enlazador de fármaco con el anticuerpo a temperatura ambiente durante 1 hora. A continuación, una solución acuosa (0,0035 ml) de NAC 100 mM (Sigma-Aldrich Co. LLC) se añadió a la misma y se incubó adicionalmente para terminar la reacción del enlazador de fármaco a temperatura ambiente durante 20 minutos.
Purificación: La solución anterior se sometió a purificación usando el procedimiento común D-1 (se usó ABS como solución tampón) descrito en el procedimiento de producción 1 para producir 2,5 ml de una solución que contiene el conjugado de anticuerpo - fármaco del título.
Caracterización fisicoquímica: Usando el procedimiento común E descrito en el procedimiento de producción 1 (como coeficiente de absorción molar, se usaron ea,28o = 262400 (valor de cálculo estimado), £A370 = 0 (valor de cálculo estimado), £d,28o = 5178 (valor medido) y £d,37o = 20217 (valor medido)), se obtuvieron los siguientes valores característicos.
Concentración de anticuerpo: 1,16 mg/ml, rendimiento de anticuerpo: 2,9 mg (73%), y número promedio de moléculas de fármaco conjugadas (n) por molécula de anticuerpo: 7,0.
Ejemplo 73 (Otro procedimiento para sintetizar el compuesto del proceso 8 del Ejemplo 58)
[Fórmula 142]

Proceso 1: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-M)hexanoM]gMcMgMcM-L-fenilo alaninato de terc-butilo
Con enfriamiento con hielo, a una solución de THF (12,0 ml) de N-[(9H-fluoren-9-ilmetoxi)carbonil]glicilglicil-L-fenilo alaninato de terc-butilo (J. Pept. Res., 1999, vol. 53, págs. 393) (0,400 g, 0,717 mmol), se añadió 1,8-diazabiciclo[5.4.0]-7-undeceno (0,400 ml) y se agitó a temperatura ambiente durante 4 días, y entonces se añadió adicionalmente hexanoato de N-succinimidil 6-maleimida (0,221 g, 0,717 mmol) y se agitó durante 3 horas. La solución de reacción se diluyó con acetato de etilo y se lavó con una solución acuosa de ácido cítrico al 10 %, una solución acuosa saturada de hidrogenocarbonato sódico, y salmuera saturada, y entonces la capa orgánica se secó sobre sulfato de magnesio anhidro. Después de que el disolvente se retirara a presión reducida, los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo : metanol = 9 : 1 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,295 g, 78%).
RMN 1H (400 MHz, CDCU) 5: 1,28-1,36 (2H, m), 1,41 (9H, s), 1,57-1,71 (4H, m), 2,23 (2H, t, J = 7,6 Hz), 3,09 (2H, d, J = 6,0 Hz), 3,51 (2H, t, J = 7,6 Hz), 3,85-4,02 (4H, m), 4,69-4,78 (1H, m), 6,15 (1H, t, J = 4,6 Hz), 6,33 (1H, d, J = 7,3 Hz), 6,60 (1H, t, J = 5,0 Hz), 6,68 (2H, s), 7,10-7,16 (2H, m), 7,22-7,31 (3H, m).
EM (IEN) m/z: 529 (M+H)+
Proceso 2: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]glicilglicil-L-fenilalanina
A una solución de diclorometano (8,00 ml) del compuesto (0,295 g, 0,558 mmol) obtenido en el proceso 1 anterior, se añadió ácido trifluoroacético (4,00 ml) y se agitó a temperatura ambiente durante 18 horas. El disolvente se retiró a presión reducida para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,240 g, 91 %). RMN 1H (400 MHz, DMSO-da) 5: 1,15-1,23 (2H, m), 1,40-1,53 (4H, m), 2,10 (2H, t, J = 7,6 Hz), 2,88 (1H, dd, J = 13,7, 8,9 Hz), 3,04 (1H, dd, J = 13,7, 5,0 Hz), 3,35-3,43 (2H, m), 3,58-3,77 (4H, m), 4,41 (1H, td, J = 7,8, 5,0 Hz), 7,00 (2H, s), 7,16-7,31 (5H, m), 8,00 (1H, t, J = 5,7 Hz), 8,06 (1H, t, J = 5,7 Hz), 8,13 (1H, d, J = 7,8 Hz).
Proceso 3: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1 -il)hexanoil]glicilglicil-L-fenilalanil-N-[(2-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolin-1-il]amino}-2-oxoetoxi)metil]glicinamida
El compuesto (0,572 g, 1,21 mmol) obtenido en el proceso 2 se disolvió en diclorometano (12,0 ml), se cargó con N-hidroxisuccinimida (0,152 g, 1,32 mmol) y clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,253 g, 1,32 mmol), y se agitó durante 1 hora. La solución de reacción se añadió a una solución de N,N-dimetilformamida (22,0 ml) de la mezcla (1,10 mmol) obtenida en el proceso 5 del Ejemplo 58, y se agitó a temperatura ambiente durante 3 horas. La solución de reacción se cargó con una solución acuosa de ácido cítrico al 10 % y se extrajo con cloroformo. La capa orgánica obtenida se secó sobre sulfato sódico y se filtró. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo : metanol = 8 : 2 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,351 g, 31 %). Los datos instrumentales del compuesto fueron los mismos que los del compuesto del proceso 8 del Ejemplo 58.
Ejemplo 74 (Otro procedimiento para sintetizar el compuesto del proceso 8 del Ejemplo 58)
[F ó rm u la 143]

Proceso 1: [({N-[(9H-fluoren-9-ilmetoxi)carbonil]glicil}amino)metoxi]acetato de bencilo
A una solución de tetrahidrofurano (200 ml) del compuesto (7,37 g, 20,0 mmol) obtenido en el proceso 1 del Ejemplo 58, se añadieron glicolato de bencilo (6,65 g, 40,0 mmol) y ácido p-tolueno sulfónico monohidrato (0,381 g, 2,00 mmol) a 0 °C y se agitó a temperatura ambiente durante 2 horas y 30 minutos. La solución de reacción se cargó con una solución saturada acuosa de hidrogenocarbonato sódico y se extrajo con acetato de etilo. La capa orgánica obtenida se secó sobre sulfato sódico y se filtró. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [hexano : acetato de etilo = 100 : 0 (v/v) - 0 : 100] para producir el compuesto del título en forma de un sólido incoloro (6,75 g, 71 %). Los datos instrumentales del compuesto fueron los mismos que los del compuesto del proceso 2 del Ejemplo 58.
Proceso 2: N-[(benciloxi)carbonil]glicilglicil-L-fenilalanina-N-{[(2-(benciloxi)-2-oxoetoxi]metil}glicinamida
A una solución de N,N-dimetilformamida (140 ml) del compuesto (6,60 g, 13,9 mmol) obtenido en el proceso 1 anterior, se añadió 1,8-diazabiciclo[5.4.0]undec-7-eno (2,22 g, 14,6 mmol) a 0 °C y se agitó a temperatura ambiente durante 15 minutos. La solución de reacción se cargó con una solución de N,N-dimetilformamida (140 ml) de N-[(benciloxi)carbonil]glicilglicil-L-fenilalanina (6,33 g, 15,3 mmol), N-hidroxisuccinimida (1,92 g, 16,7 mmol) y clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (3,20 g, 16,7 mmol) agitara por adelantado a temperatura ambiente durante 1 hora, y se agitó a temperatura ambiente durante 4 horas. La solución de reacción se cargó con ácido clorhídrico 0,1 N y se extrajo con cloroformo. La capa orgánica obtenida se secó sobre sulfato sódico y se filtró. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo : metanol = 8 : 2 (v/v)] para producir el compuesto del título en forma de un sólido incoloro (7,10 g, 79%).
RMN 1H (DMSO-Da) 5: 2,78 (1H, dd, J = 13,9, 9,6 Hz), 3,05 (1H, dd, J = 13,9, 4,5 Hz), 3,56-3,80 (6H, m), 4,15 (2H, s), 4,47-4,55 (1H, m), 4,63 (2H, d, J = 6,6 Hz), 5,03 (2H, s), 5,15 (2H, s), 7,16-7,38 (15H, m), 7,52 (1H, t, J = 5,9 Hz), 8,03 (1H, t, J = 5,5 Hz), 8,17 (1H, d, J = 8,2 Hz), 8,36 (1H, t, J = 5,7 Hz), 8,61 (1H, t, J = 6,6 Hz).
Proceso 3: Glicilglicil-L-fenilalanil-N-[(carboximatoxi)metil]glicinamida
A una solución de N,N-dimetilformamida (216 ml) del compuesto (7,00 g, 10,8 mmol) obtenido en el proceso 2, se añadió catalizador de paladio-carbono (7,00 g) y se agitó bajo una atmósfera de hidrógeno a temperatura ambiente durante 24 horas. Los materiales insolubles se retiraron por filtración a través de Celite, y el disolvente se retiró a presión reducida. Los residuos obtenidos se disolvieron en agua, lo insoluble se retiró por filtración a través de Celite, y el disolvente se retiró a presión reducida. Este procedimiento se repitió dos veces para producir el compuesto del título en forma de un sólido incoloro (3,77 g, 82 %).
RMN 1H (DMSO-Da) 5: 2,84 (1H, dd, J = 13,7, 9,8 Hz), 3,08 (1H, dd, J = 13,7, 4,7 Hz), 3,50-3,72 (4H, m), 3,77-3,86 (2H, m), 3,87 (2H, s), 4,52-4,43 (1H, m), 4,61 (2H, d, J = 6,6 Hz), 7,12-7,30 (5H, m), 8,43 (1H, t, J = 5,9 Hz), 8,54 (1H, d, J = 7,8 Hz), 8,70 (1H, t, J = 6,3 Hz), 8,79 (1H, t, J = 5,5 Hz).
Proceso 4: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]glicilglicil-L-fenilalanil-N
[(carboximatoxi)metil]glicinamida
A una solución de N,N-dimetilformamida (85,0 ml) del compuesto (3,59 g, 8,48 mmol) obtenido en el proceso 3 anterior, se añadieron ácido N-succinimidil 6-maleimida hexanoico (2,88 g, 9,33 mmol) y trietilamina (0,858 g, 8,48 mmol) y se agitó a temperatura ambiente durante 1 hora. La solución de reacción se cargó con ácido clorhídrico 0,1 N y se extrajo con cloroformo y un disolvente mixto de cloroformo y metanol [cloroformo : metanol = 4 : 1 (v/v)]. La capa orgánica obtenida se secó sobre sulfato sódico y se filtró. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - capa orgánica repartida de cloroformo : metanol: agua = 7 : 3: 1 (v/v/v)] para producir el compuesto del título en forma de un sólido incoloro (3,70 g, 71 %). RMN 1H (DMSO-Da) 5: 1,13-1,24 (2H, m), 1,42-1,53 (4H, m), 2,11 (2H, t, J = 7,4 Hz), 2,80 (1H, dd, J = 13,7, 9,8 Hz), 3,06 (1H, dd, J = 13,9, 4,5 Hz), 3,37 (2H, t, J = 7,2 Hz), 3,56-3,78 (6H, m), 3,97 (2H, s), 4,46-4,53 (1H, m), 4,61 (2H, d, J = 6,3 Hz), 7,00 (2H, s), 7,15-7,29 (5H, m), 8,03-8,20 (3H, m), 8,32 (1H, t, J = 5,9 Hz), 8,60 (1H, t, J = 6,7 Hz).
Proceso 5: N-[6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoil]glicilglicil-L-fenilalanil-N-[(2-{[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolin-1-il]amino}-2-oxoetoxi)metil]glicinamida
A una solución de N,N-dimetilformamida (40,0 ml) de mesilato del compuesto (4) (1,14 g, 2,00 mmol), trietilamina (0,202 g, 2,00 mmol), se añadieron a 0 °C el compuesto (1,48 g, 2,40 mmol) obtenido en el proceso 4 anterior, y cloruro de 4-(4,6-dimetoxi-1,3,5-triazin-2-il)-4-metilmorfolinio (0,993 g, 3,00 mmol) que contiene agua al 16,4 % y se agitó a temperatura ambiente durante 1 hora. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo : metanol = 8 : 2 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (1,69 g, 82%). Los datos instrumentales del compuesto fueron los mismos que los del compuesto del proceso 8 del Ejemplo 58.
Ejemplo 75 N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolin-1-il]-2-hidroxiacetamida
[Fórmula 144]

Proceso 1: 2-{[(1S,9S)-9-Etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolin-1-il]amino}-2-oxoetilacetato
Con enfriamiento con hielo, a una suspensión de N,N-dimetilformamida (20,0 ml) de mesilato del compuesto (4) (0,500 g, 0,941 mmol), se añadieron N,N-diisopropiletilamina (0,492 ml, 2,82 mmol) y cloruro de acetoxiacetilo (0,121 ml, 1,13 mmol) y se agitó a temperatura ambiente durante 1 hora. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - capa orgánica repartida de cloroformo : metanol: agua = 7 : 3: 1 (v/v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,505 g, cuantitativo).
RMN 1H (400 MHz, DMSO-d6) 5: 0,87 (3H, t, J = 7,4 Hz), 1,81-1,92 (2H, m), 2,08 (3H, s), 2,08-2,22 (2H, m), 2,41 (3H, s), 3,14-3,21 (2H, m), 4,51 (2H, dd, J = 19,4, 14,7 Hz), 5,22 (2H, dd, J = 40,1, 19,0 Hz), 5,43 (2H, s), 5,56-5,61 (1H, m), 6,53 (1H, s), 7,31 (1H, s), 7,81 (1H, d, J = 11,0 Hz), 8,67 (1H, d, J = 8,6 Hz).
EM (IEN) m/z: 536 (M+H)+
Proceso 2: N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolin-1-il]-2-hidroxiacetamida
A una suspensión de metanol (50,0 ml) del compuesto (0,504 g, 0,941 mmol) obtenido en el proceso 1 anterior, se añadieron tetrahidrofurano (20,0 ml) y una solución acuosa de hidróxido sódico 1 N (4,00 ml, 4,00 mmol) y se agitó a temperatura ambiente durante 1 hora. La reacción se terminó mediante la adición de ácido clorhídrico 1 N (5,00 ml, 5,00 mmol), y el disolvente se retiró a presión reducida. Los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo-capa orgánica repartida de cloroformo : metanol : agua = 7 : 3: 1 (v/v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,412 g, 89 %). Este compuesto se confirmó en el tumor de un ratón portador de cáncer que recibió el conjugado de anticuerpo - fármaco (55) o (56). RMN 1H (400 MHz, DMSO-d6) 5: 0,87 (3H, t, J = 7,3 Hz), 1,78-1,95 (2H, m), 2,09-2,28 (2H, m), 2,39 (3H, s), 3,07-3,27 (2H, m), 3,96 (2H, d, J = 6,0 Hz), 5,11-5,26 (2H, m), 5,42 (2H, s), 5,46-5,54 (1H, m), 5,55-5,63 (1H, m), 6,52 (1H, s), 7,30 (1H, s), 7,78 (1H, d, J = 10,9 Hz), 8,41 (1H, d, J = 9,1 Hz). EM (IEN) m/z: 494 (M+H)+
Ejemplo 76 (Otro procedimiento para sintetizar el compuesto del Ejemplo 75)
[Fórmula 145]

Proceso 1: N-[(1S,9S)-9-etil-5-fluoro-9-hidroxi-4-metil-10,13-dioxo-2,3,9,10,13,15-hexahidro-1H,12H-benzo[de]pirano[3',4':6,7]indolizino[1,2-b]quinolin-1-il]-2-hidroxiacetamida
Se disolvió ácido glicólico (0,0201 g, 0,27 mmol) en N,N-dimetilformamida (1,0 ml), se cargó con N-hidroxisuccinimida (0,0302 g, 0,27 mmol) y clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (0,0508 g, 0,27 mmol), y se agitó durante 1 hora. La solución de reacción se añadió a una suspensión de N,N-dimetilformamida (1,0 ml) cargada con mesilato del compuesto (4) (0,1 g, 0,176 mmol) y trietilamina (0,025 ml, 0,18 mmol) y se agitó a temperatura ambiente durante 24 horas. El disolvente se retiró a presión reducida y los residuos obtenidos se purificaron por cromatografía en columna sobre gel de sílice [cloroformo - cloroformo : metanol = 10 : 1 (v/v)] para producir el compuesto del título en forma de un sólido de color amarillo pálido (0,080 g, 92%). Los datos instrumentales del compuesto fueron los mismos que los del compuesto obtenido en el proceso 2 del Ejemplo 75.
(Ejemplo de prueba 1) Producción de vector de expresión de variante 1 de B7-H3 humano de longitud completa
ADNc que codifica la variante 1 de B7-H3 humano se amplificó mediante reacción de PCR usando ADNc sintetizado a partir del ARN total de una célula LNCaP (Colección Americana de Cultivos Tipo: ATCC) como un molde y el siguiente conjunto de cebadores:
cebador 1:
5'-ctatagggagacccaagctggctagcatgctgcgtcggcggggcag-3' (SEQ ID NO: 22) y
cebador 2:
5 ' -
aacgggccctctagactcgagcggccgctcaggctatttcttgtccatcatcttc
tttgctgtcag-3' (SEQ ID NO: 23).
A continuación, el producto de PCR obtenido de este modo se purificó usando MagExtractor PCR & Gel cleanup (Toyobo Co., Ltd.). El producto purificado se digirió adicionalmente con enzimas de restricción (NheI/NotI) y a continuación se purificó usando MagExtractor PCR & Gel cleanup (Toyobo Co., Ltd.). pcDNA3.1 (+) ADN plasmídico (Life Technologies) se digirió con las mismas enzimas de restricción que anteriormente (NheI/NotI) y a continuación se purificó usando MagExtractor PCR & Gel cleanup (Toyobo Co., Ltd.).
Estas soluciones de ADN purificadas se mezclaron, se cargaron adicionalmente con Ligation high (Toyobo Co., Ltd.), y se incubaron para el ligamiento a 16 °C durante 8 horas.
Células DH5a de Escherichia coli competentes (Life Technologies) se transformaron mediante la adición del producto de reacción obtenido.
Las colonias así obtenidas se sometieron a PCR directa de colonias usando cebadores de PCR y cebador inverso de BGH para seleccionar clones candidatos.
Los clones candidatos obtenidos se cultivaron en un medio líquido (LB/Amp), y se extrajo un ADN plasmídico con MagExtractor-Plasmid-(Toyobo Co., Ltd.).
Cada clon obtenido se comparó con la secuencia CDS proporcionada mediante el análisis de secuenciación entre el cebador 3 (cebador promotor de CMV):
5'-cgcaaatgggcggtaggcgtg-3' (SEQ ID NO: 24) y cebador 4 (cebador inverso de BGH):
5'-tagaaggcacagtcgagg-3' (SEQ ID NO: 25)
con el ADN plasmídico obtenido como molde.
Después de confirmar la secuencia, el clon obtenido se cultivó en 200 ml de medio LB/Amp, y se extrajo un ADN
plasmídico usando el kit VioGene Plasmid Midi V-100.
La cepa se denominó como pcDNA3.1-B7-H3. La secuencia de un sitio de ORF del gen de la variante 1 de B7-H3 clonado en el vector se muestra en las posiciones de nucleótido 1 a 1602 en la SEQ ID NO: 26 (figura 16) en el listado de secuencias. Además, la secuencia de aminoácidos de la variante 1 de B7-H3 se muestra en la SEQ ID NO: 1 en el listado de secuencias.
(Ejemplo de prueba 2) Preparación de célula CCRF-CEM que expresa de forma estable el gen de la variante 1 de B7-H3
pcDNA3.1-B7-H3 producido en el ejemplo de prueba 1 se transfectó en células CCRF-CEM (ATCC) por electroporación usando Nucleofector II (fabricado por Lonza Group Ltd.). A continuación, las células se cultivaron adicionalmente durante dos noches en medio de RPMI1640 (Life Technologies) que contiene suero bovino fetal al 10 % (FBS) (al que se hace referencia posteriormente en el presente documento como FBS al 10 %-RPMI1640) en condiciones de 37 °C y CO2 al 5 %.
Después del cultivo de 2 días, el cultivo se inició en FBS al 10 %-RPMI1640 que contiene 750 pg/ml de G418 (Life Technologies) con el fin de seleccionar células CCRF-CEM en las que se integró de forma estable pcDNA3.1-B7-H3.
Después del cultivo de 1 mes, se realizó una clonación mediante el procedimiento de dilución limitante con el fin de producir un clon de una sola célula. Específicamente, las células que tienen resistencia a G418 se diluyeron a 10 células/ml, se inocularon en una placa de 96 pocillos a una concentración de 100 pl/pocillo, y se cultivaron, y las células que se dejaron proliferar se recuperaron de pocillos individuales.
Se usó citometría de flujo para confirmar la expresión de B7-H3 en cada clon recuperado. Específicamente, cada clon recuperado se lavó dos veces con PBS que contiene FBS al 5 %, a continuación se suspendió mediante la adición de PBS que contiene FBS al 5 % y 10 pg/ml de M30, y se dejó reposar a 4 °C durante 30 minutos. El clon se lavó dos veces con PBS que contiene FBS al 5 %, a continuación se suspendió mediante la adición de fracción de IgG de cabra conjugada con fluoresceína a IgG de ratón (molécula completa) (n.° 55493, fabricado por ICN Pharmaceuticals, Inc.) se diluyó 1000 veces con PBS que contiene FBS al 5 %, y se dejó reposar a 4 °C durante 30 minutos. El clon se lavó dos veces con PBS que contiene FBS al 5 %, a continuación se volvió a suspender en PBS que contiene FBS al 5 %, y se detectó usando un citómetro de flujo (FC500: Beckman Coulter, Inc.).
Las células CCRF-CEM que expresan de forma estable el gen de la variante 1 de B7-H3 obtenido de este modo mediante estos procedimientos se designaron como células CEM_V1_3.1_2. Las células CCRF-CEM de línea parental se usaron como una línea celular que carece de expresión de B7-H3.
(Ejemplo de prueba 5) Prueba de citotoxicidad (3) de conjugado de anticuerpo - fármaco
Células positivas para antígeno células SR (ATCC) se cultivaron en RPMI1640 (GIBCO) que contiene suero bovino fetal al 10 % (MOREGATE) (al que se hace referencia posteriormente en el presente documento como medio). Las células SR se prepararon para tener una concentración de 2,8 * 104 células/ml usando un medio y se añadieron a una concentración de 90 pl/pocillo a una microplaca de 96 pocillos para cultivo celular. Dos horas después, el anticuerpo anti-CD30 y conjugados de anticuerpo - fármaco (22), (23), (38), (64) y (65) diluido cada uno en 1000 nM, 100 nM, 10 nM, 1 nM, 100 pM, 10 pM y 1 pM usando un medio se añadieron a una concentración de 10 pl/pocillo a la microplaca. Un medio se añadió a una concentración de 10 pl/pocillo a pocillos no suplementados con sustancia de prueba. Las células se cultivaron bajo CO2 al 5 % a 37 °C durante 6 días. Después del cultivo, la microplaca se sacó de la incubadora y se dejó reposar a temperatura ambiente durante 30 minutos. La solución de cultivo se cargó con una cantidad igual de ensayo de viabilidad celular luminiscente CellTiter-Glo (Promega) y se agitó. Después de que la microplaca se dejara reposar a temperatura ambiente durante 10 minutos, la cantidad de emisión de luz se midió usando un lector de placas (PerkinElmer). El valor de CI50 se calculó de acuerdo con la siguiente ecuación:
CI50 (nM) = antilog ((50 - d) * (LOG-iüb - LOG^a) / (d - c) LOG-iüb)
a: Concentración a de la sustancia de prueba
b: Concentración b de la sustancia de prueba
c: Relación de células vivas suplementadas con la sustancia de prueba que tiene la concentración a d: Relación de células vivas suplementadas con la sustancia de prueba que tiene la concentración b
Las concentraciones a y b establecen la relación a > b que corta una relación de un 50 % de células vivas.
La tasa de supervivencia de las células a cada concentración se calculó de acuerdo con la siguiente ecuación:
Tasa de supervivencia de las células (%) = a / b * 100
a: Cantidad promedio de emisión de luz a partir de los pocillos suplementados con sustancia de prueba (n = 2) b: Cantidad promedio de emisión de luz a partir de los pocillos no suplementados con sustancia de prueba (n = 12)
Los conjugados de anticuerpo - fármaco (23), (38), (64) y (65) mostraron una actividad citotóxica de CI50 < 0,01 (nM)
contra las células SR. El conjugado de anticuerpo - fármaco (22) mostró una actividad citotóxica de CI50 < 0,1 (nM) contra las células SR. El anticuerpo anti-CD30 no mostró actividad citotóxica alguna contra las células SR (> 4,0 (nM)).
(Ejemplo de prueba 9) Prueba de citotoxicidad (7) de conjugado de anticuerpo - fármaco
Células positivas para antígeno células U251 (ATCC) se cultivaron en RPMI1640 (GIBCO) que contiene suero bovino fetal al 10 % (MOREGATE) (al que se hace referencia posteriormente en el presente documento como medio). Las células U251 se prepararon para tener una concentración de 2,8 * 104 células/ml usando un medio y se añadieron a una concentración de 90 pl/pocillo a una microplaca de 96 pocillos para cultivo celular. Dos horas después, el anticuerpo anti-CD70 y conjugados de anticuerpo - fármaco (26), (27), (40), (68) y (69) diluido cada uno en 1000 nM, 200 nM, 40 nM, 8 nM, 1,6 nM, 0,32 nM y 0,064 nM usando un medio se añadieron a una concentración de 10 pl/pocillo a la microplaca. Un medio se añadió a una concentración de 10 pl/pocillo a pocillos no suplementados con sustancia de prueba. Las células se cultivaron bajo CO2 al 5 % a 37 °C durante 6 días. Después del cultivo, la microplaca se sacó de la incubadora y se dejó reposar a temperatura ambiente durante 30 minutos. La solución de cultivo se cargó con una cantidad igual de ensayo de viabilidad celular luminiscente CellTiter-Glo (Promega) y se agitó. Después de que la microplaca se dejara reposar a temperatura ambiente durante 10 minutos, la cantidad de emisión de luz se midió usando un lector de placas (PerkinElmer). El valor de CI50 se calculó de acuerdo con la siguiente ecuación:
CI50 (nM) = antilog ((50 - d) * (LOG-iüb - LOG^a) / (d - c) LOG-iüb)
a: Concentración a de la sustancia de prueba
b: Concentración b de la sustancia de prueba
c: Relación de células vivas suplementadas con la sustancia de prueba que tiene la concentración a d: Relación de células vivas suplementadas con la sustancia de prueba que tiene la concentración b
Las concentraciones a y b establecen la relación a > b que corta una relación de un 50 % de células vivas.
La tasa de supervivencia de las células a cada concentración se calculó de acuerdo con la siguiente ecuación:
Tasa de supervivencia de las células (%) = a / b * 100
a: Cantidad promedio de emisión de luz a partir de los pocillos suplementados con sustancia de prueba (n = 2) b: Cantidad promedio de emisión de luz a partir de los pocillos no suplementados con sustancia de prueba (n = 12)
Los conjugados de anticuerpo - fármaco (26), (27), (40) y (69) mostraron una actividad citotóxica de 1 < CI50 < 10 (nM) contra las células U251. El conjugado de anticuerpo - fármaco (68) mostró una actividad citotóxica de 10 < CI50 < 100 (nM) contra las células. El anticuerpo anti-CD70 no mostró actividad citotóxica alguna contra las células U251 (> 100 (nM)).
(Ejemplo de prueba 10) Prueba de citotoxicidad (8) de fármaco liberado
Células A375 (ATCC) se cultivaron en DMEM (GIBCO) que contiene suero bovino fetal al 10 % (MOREGATE) (al que se hace referencia posteriormente en el presente documento como medio). Las células A375 se prepararon para tener una concentración de 4 * 104 células/ml usando un medio, se añadieron a una concentración de 25 pl/pocillo a una microplaca de 96 pocillos para cultivo celular (CORNING) cargada con 65 pl/pocillo de un medio, y se cultivaron durante una noche. Al día siguiente, cada sustancia de prueba diluida en 1000 nM, 200 nM, 40 nM, 8 nM, 1,6 nM, 0,32 nM y 0,064 nM usando DMSO se añadió a una concentración de 0,5 pl/pocillo a la microplaca. Se añadió DMSO a una concentración de 0,5 pl/pocillo a pocillos no suplementados con sustancia de prueba. El volumen del medio en cada pocillo se ajustó a 100 pl mediante la adición de 10 pl/pocillo de un medio, y las células se cultivaron durante 6 días bajo CO2 al 5 % a 37 °C. Después del cultivo, la microplaca se sacó de la incubadora y se dejó reposar a temperatura ambiente durante 30 minutos. La solución de cultivo se cargó con una cantidad igual de ensayo de viabilidad celular luminiscente CellTiter-Glo (Promega) y se agitó. Después de que la microplaca se dejara reposar a temperatura ambiente durante 10 minutos, la cantidad de emisión de luz se midió usando un lector de placas. El valor de CI50 se calculó de acuerdo con la siguiente ecuación:
CI50 (nM) = antilog ((50 - d) * (LOG-iüb - LOG^a) / (d - c) LOG-iüb)
a: Concentración a de la sustancia de prueba
b: Concentración b de la sustancia de prueba
c: Relación de células vivas suplementadas con la sustancia de prueba que tiene la concentración a d: Relación de células vivas suplementadas con la sustancia de prueba que tiene la concentración b
Las concentraciones a y b establecen la relación a > b que corta una relación de un 50 % de células vivas.
La tasa de supervivencia de las células se calculó de acuerdo con la siguiente ecuación:
Tasa de supervivencia de las células (%) = a / b * 100
a: Cantidad promedio de emisión de luz a partir de los pocillos suplementados con sustancia de prueba (n = 2) b: Cantidad promedio de emisión de luz a partir de los pocillos no suplementados con sustancia de prueba (n = 10)
El compuesto del Ejemplo (75) y exatecán mostró una actividad citotóxica de 0,1 < CI50 < 1 (nM) contra las células A375. El compuesto del Ejemplo (42) mostró una actividad citotóxica contra 1 < CI50 < 10 (nM) contra las células. El compuesto del Ejemplo (1) mostró una actividad citotóxica contra 10 < CI50 < 100 (nM) contra las células.
(Ejemplo de prueba 14) Prueba antitumoral (4)
Se adquirieron células de la línea de melanoma humano A375 de ATCC (Colección Americana de Cultivos Tipo). 8 * 106 células suspendidas en solución salina fisiológica se transplantaron subcutáneamente al lado derecho del abdomen de cada ratón desnudo hembra (día 0), y los ratones se agruparon aleatoriamente el día 21. Los conjugados de anticuerpo - fármaco (1), (13), (41) y (55) se administraron, cada uno, por vía intravenosa a una dosis de 10 mg/kg a la cola de cada ratón el día 21.
Los resultados se muestran en la Figura 20. En el dibujo, la línea con rombos abiertos muestra los resultados acerca de un tumor sin tratar, la línea con círculos abiertos muestra el efecto del conjugado de anticuerpo - fármaco administrado (1), la línea con triángulos abiertos muestra el efecto del conjugado de anticuerpo - fármaco administrado (13), la línea con marcas X muestra el efecto del conjugado de anticuerpo - fármaco administrado (41) y la línea con cuadrados abiertos muestra el efecto del conjugado de anticuerpo - fármaco administrado (55). La administración del conjugado de anticuerpo - fármaco (1), (13), (41) o (55) disminuyó notablemente el volumen tumoral, y la totalidad de estos conjugados de anticuerpo - fármaco ejercieron un efecto inhibidor del crecimiento tumoral.
Además, los ratones que recibieron el conjugado de anticuerpo - fármaco (1), (13), (41) o (55) no presentaron signos notables tales como pérdida de peso, lo que sugiere que los conjugados de anticuerpo - fármaco (1), (13), (41) y (55) son de baja toxicidad y sumamente seguros.
(Ejemplo de prueba 15) Prueba antitumoral (5)
Las células Calu-6 de línea de cáncer de pulmón no de células pequeñas humano se adquirieron de ATCC (Colección Americana de Cultivos Tipo). 5 * 106 células suspendidas en solución salina fisiológica se transplantaron subcutáneamente al lado derecho del abdomen de cada ratón desnudo hembra (día 0), y los ratones se agruparon aleatoriamente el día 12. Los conjugados de anticuerpo - fármaco (13), (41) y (55) se administraron, cada uno, por vía intravenosa a una dosis de 10 mg/kg a la cola de cada ratón el día 12. Como control comparativo, se administró DE-310 por vía intravenosa a una dosis de 0,1 mg/kg a la cola de cada ratón el día 12. En este caso, la dosis anteriormente mencionada del conjugado de anticuerpo - fármaco estaba basada en la cantidad del anticuerpo en el conjugado y la dosis anteriormente mencionada de d E-310 estaba basada en la cantidad del fármaco contenido en el mismo. A este respecto, las cantidades de los fármacos respectivamente contenidos en el conjugado de anticuerpo - fármaco y DE-310 eran de aproximadamente 1 : 100. Esto significa que las dosis del conjugado de anticuerpo - fármaco y d E-310 eran iguales en términos de las cantidades de los fármacos contenidos en el mismo.
Los resultados se muestran en la Figura 21. En el dibujo, la línea con rombos abiertos muestra los resultados acerca de un tumor sin tratar, la línea con círculos abiertos muestra el efecto de DE-310, la línea con triángulos abiertos muestra el efecto del conjugado de anticuerpo - fármaco (13), la línea con marcas X muestra el efecto del conjugado de anticuerpo - fármaco (41) y la línea con cuadrados abiertos muestra el efecto del conjugado de anticuerpo - fármaco (55). La administración del conjugado de anticuerpo - fármaco (13), (41) o (55) disminuyó notablemente el volumen tumoral, mientras que la administración de DE-310 no mostró reducción alguna en el volumen tumoral.
Además, los ratones que recibieron el conjugado de anticuerpo - fármaco (13), (41) o (55) no presentaron signos notables tales como pérdida de peso, lo que sugiere que estos conjugados de anticuerpo - fármaco son de baja toxicidad y sumamente seguros.
(Ejemplo de prueba 16) Prueba antitumoral (6)
Se adquirieron células de la línea de melanoma humano A375 de ATCC (Colección Americana de Cultivos Tipo). 1 * 107 células suspendidas en solución salina fisiológica se transplantaron subcutáneamente al lado derecho del abdomen de cada ratón desnudo hembra (día 0), y los ratones se agruparon aleatoriamente el día 11. Los conjugados de anticuerpo - fármaco (17), (18), (19), (59), (60) y (61) se administraron, cada uno, por vía intravenosa a una dosis de 3 mg/kg a la cola de cada ratón los días 11 y 18 en un programa de cs * 2.
Los resultados se muestran en la Figura 22. En el dibujo, la línea con rombos sólidos muestra los resultados acerca de un tumor sin tratar, la línea con cuadrados sólidos muestra el efecto del conjugado de anticuerpo - fármaco administrado (17), la línea con triángulos abiertos muestra el efecto del conjugado de anticuerpo - fármaco administrado (18), la línea con círculos abiertos muestra el efecto del conjugado de anticuerpo - fármaco administrado (19), la línea con triángulos sólidos muestra el efecto del conjugado de anticuerpo - fármaco administrado (59), la línea con cuadrados abiertos muestra el efecto del conjugado de anticuerpo - fármaco administrado (60) y la línea con marcas X muestra el efecto del conjugado de anticuerpo - fármaco administrado (61).
La administración del conjugado de anticuerpo - fármaco (17), (18), (19), (59), (60) o (61) disminuyó notablemente el volumen tumoral, y la totalidad de estos conjugados de anticuerpo - fármaco ejercieron un efecto inhibidor del crecimiento tumoral.
Claims (1)
1. Un compuesto representado por la siguiente fórmula:

Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012225887 | 2012-10-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2773710T3 true ES2773710T3 (es) | 2020-07-14 |
Family
ID=50477169
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18152381T Active ES2773710T3 (es) | 2012-10-11 | 2013-10-10 | Enlazadores para conjugados de anticuerpo - fármaco |
| ES13845596.9T Active ES2671644T3 (es) | 2012-10-11 | 2013-10-10 | Conjugado de anticuerpo - fármaco |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13845596.9T Active ES2671644T3 (es) | 2012-10-11 | 2013-10-10 | Conjugado de anticuerpo - fármaco |
Country Status (30)
| Country | Link |
|---|---|
| US (5) | US10195288B2 (es) |
| EP (3) | EP3632471A1 (es) |
| JP (8) | JP5953378B2 (es) |
| KR (12) | KR101841818B1 (es) |
| CN (4) | CN104755494B (es) |
| AU (4) | AU2013328111B2 (es) |
| BR (3) | BR122021014365B1 (es) |
| CA (2) | CA3021435C (es) |
| CY (2) | CY1120363T1 (es) |
| DK (2) | DK2907824T3 (es) |
| ES (2) | ES2773710T3 (es) |
| HK (2) | HK1210477A1 (es) |
| HR (2) | HRP20180870T1 (es) |
| HU (2) | HUE039000T2 (es) |
| IL (5) | IL302494B1 (es) |
| LT (2) | LT2907824T (es) |
| MX (3) | MX364484B (es) |
| MY (1) | MY170251A (es) |
| NZ (4) | NZ705394A (es) |
| PH (2) | PH12015500667B1 (es) |
| PL (2) | PL2907824T3 (es) |
| PT (2) | PT3342785T (es) |
| RS (2) | RS57278B1 (es) |
| RU (1) | RU2664465C2 (es) |
| SG (2) | SG11201502887WA (es) |
| SI (2) | SI2907824T1 (es) |
| TR (1) | TR201809636T4 (es) |
| TW (5) | TW202233186A (es) |
| WO (1) | WO2014057687A1 (es) |
| ZA (2) | ZA201501825B (es) |
Families Citing this family (151)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101841818B1 (ko) * | 2012-10-11 | 2018-03-26 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트 |
| EP2910573B1 (en) * | 2012-10-19 | 2020-02-19 | Daiichi Sankyo Company, Limited | Antibody-drug conjugate produced by binding through linker having hydrophilic structure |
| KR101941758B1 (ko) | 2013-12-25 | 2019-01-24 | 다이이찌 산쿄 가부시키가이샤 | 항 trop2 항체-약물 컨쥬게이트 |
| KR102465042B1 (ko) * | 2014-01-31 | 2022-11-09 | 다이이찌 산쿄 가부시키가이샤 | 항-her2 항체-약물 접합체 |
| JP2017114763A (ja) * | 2014-03-26 | 2017-06-29 | 第一三共株式会社 | 抗cd98抗体−薬物コンジュゲート |
| EP3130608B1 (en) * | 2014-04-10 | 2019-09-04 | Daiichi Sankyo Co., Ltd. | (anti-her2 antibody)-drug conjugate |
| KR102624244B1 (ko) * | 2014-04-10 | 2024-01-11 | 다이이찌 산쿄 가부시키가이샤 | 항her3 항체-약물 콘주게이트 |
| HUE061408T2 (hu) * | 2015-06-29 | 2023-06-28 | Daiichi Sankyo Co Ltd | Eljárás antitest-gyógyszer konjugátum szelektív elõállítására |
| KR20180095828A (ko) * | 2015-12-24 | 2018-08-28 | 에이지씨 가부시키가이샤 | 수지 조성물, 기재 및 세포 배양 방법 |
| JP7002467B2 (ja) * | 2016-04-15 | 2022-01-20 | マクロジェニクス,インコーポレーテッド | 新規のb7‐h3結合分子、その抗体薬物コンジュゲート、及びその使用方法 |
| EP3468993A1 (en) * | 2016-06-08 | 2019-04-17 | AbbVie Inc. | Anti-b7-h3 antibodies and antibody drug conjugates |
| UA124198C2 (uk) * | 2016-06-08 | 2021-08-04 | Еббві Інк. | Кон'югат антитіла до в7-н3 та лікарського засобу |
| CN107840892A (zh) * | 2016-09-20 | 2018-03-27 | 上海药明生物技术有限公司 | 新型抗‑pcsk9抗体 |
| BR112019011794A2 (pt) | 2016-12-12 | 2019-10-29 | Daiichi Sankyo Co Ltd | composição farmacêutica, e, método terapêutico. |
| JP6679762B2 (ja) | 2017-01-17 | 2020-04-15 | 第一三共株式会社 | 抗gpr20抗体及び抗gpr20抗体−薬物コンジュゲート |
| WO2018159582A1 (ja) | 2017-02-28 | 2018-09-07 | 学校法人近畿大学 | 抗her3抗体-薬物コンジュゲート投与によるegfr-tki抵抗性の非小細胞肺癌の治療方法 |
| TWI794230B (zh) | 2017-05-15 | 2023-03-01 | 日商第一三共股份有限公司 | 抗cdh6抗體及抗cdh6抗體-藥物結合物、以及其製造方法 |
| WO2019024911A1 (zh) * | 2017-08-04 | 2019-02-07 | 江苏恒瑞医药股份有限公司 | B7h3抗体-药物偶联物及其医药用途 |
| WO2019034176A1 (zh) * | 2017-08-18 | 2019-02-21 | 四川百利药业有限责任公司 | 一种喜树碱-抗体偶联物 |
| AU2018320470A1 (en) | 2017-08-23 | 2020-02-13 | Daiichi Sankyo Company, Limited | Antibody-drug conjugate preparation and lyophilization for same |
| AU2018327171B2 (en) * | 2017-08-31 | 2023-03-09 | Daiichi Sankyo Company, Limited | Improved method for producing antibody-drug conjugate |
| IL272964B2 (en) | 2017-08-31 | 2024-02-01 | Daiichi Sankyo Co Ltd | Production method for antibody-drug conjugates |
| TW202405006A (zh) | 2017-09-29 | 2024-02-01 | 日商第一三共股份有限公司 | 抗體-吡咯并苯二氮呯衍生物複合體 |
| CN111542324B (zh) * | 2018-02-11 | 2023-09-12 | 四川科伦博泰生物医药股份有限公司 | 细胞毒性剂及其偶联物、其制备方法及用途 |
| AU2019270459A1 (en) | 2018-05-18 | 2020-12-03 | Daiichi Sankyo Co., Ltd. | Anti-MUC1 antibody-drug conjugate |
| KR20210035805A (ko) | 2018-06-15 | 2021-04-01 | 플래그쉽 파이어니어링 이노베이션스 브이, 인크. | 세포후 신호전달 인자의 조절을 통한 면역 활성의 증가 |
| WO2020022363A1 (ja) | 2018-07-25 | 2020-01-30 | 第一三共株式会社 | 抗体-薬物コンジュゲートの効果的な製造方法 |
| JP7406488B2 (ja) | 2018-07-27 | 2023-12-27 | 第一三共株式会社 | 抗体-薬物コンジュゲートの薬物部位を認識する蛋白質 |
| TWI822822B (zh) * | 2018-07-31 | 2023-11-21 | 日商第一三共股份有限公司 | 抗體-藥物結合物之用途 |
| EA202190471A1 (ru) | 2018-08-06 | 2021-05-24 | Дайити Санкио Компани, Лимитед | Комбинация конъюгата антитела-лекарственного средства и ингибитора тубулина |
| TW202016317A (zh) | 2018-08-23 | 2020-05-01 | 日商第一三共股份有限公司 | 抗體藥物結合物之感受性標記 |
| US20220008549A1 (en) | 2018-09-06 | 2022-01-13 | Daiichi Sankyo Company, Limited | Novel cyclic dinucleotide derivative and antibody-drug conjugate thereof |
| JPWO2020059772A1 (ja) | 2018-09-20 | 2021-08-30 | 第一三共株式会社 | 抗her3抗体−薬物コンジュゲート投与によるher3変異がんの治療 |
| EP3858386A4 (en) * | 2018-09-26 | 2022-10-12 | Jiangsu Hengrui Medicine Co., Ltd. | LIGAND-DRUG CONJUGATE OF AN EXATECAN ANALOG, METHOD FOR PREPARATION AND THEIR USE |
| US20210347894A1 (en) * | 2018-09-30 | 2021-11-11 | Jiangsu Hengrui Medicine Co., Ltd. | Anti-b7h3 antibody-exatecan analog conjugate and medicinal use thereof |
| TW202031298A (zh) | 2018-11-14 | 2020-09-01 | 日商第一三共股份有限公司 | 抗cdh6抗體-吡咯并苯二氮呯衍生物結合物 |
| BR112021011119A2 (pt) * | 2018-12-11 | 2022-01-25 | Daiichi Sankyo Co Ltd | Composição farmacêutica |
| WO2020130125A1 (ja) | 2018-12-21 | 2020-06-25 | 第一三共株式会社 | 抗体-薬物コンジュゲートとキナーゼ阻害剤の組み合わせ |
| CA3134055A1 (en) | 2019-03-20 | 2020-09-24 | The Regents Of The University Of California | Claudin-6 antibodies and drug conjugates |
| US20220177583A1 (en) | 2019-03-20 | 2022-06-09 | The Regents Of The University Of California | Claudin-6 bispecific antibodies |
| HRP20221280T1 (hr) | 2019-03-29 | 2022-12-23 | Medimmune Limited | Spojevi i njihovi konjugati |
| EP3962493A2 (en) | 2019-05-03 | 2022-03-09 | Flagship Pioneering Innovations V, Inc. | Methods of modulating immune activity/level of irf or sting or of treating cancer, comprising the administration of a sting modulator and/or purinergic receptor modulator or postcellular signaling factor |
| CN111689980A (zh) * | 2019-05-26 | 2020-09-22 | 四川百利药业有限责任公司 | 一种喜树碱药物及其抗体偶联物 |
| EP3991754A4 (en) * | 2019-06-28 | 2023-05-17 | Shanghai Fudan-Zhangjiang Bio-Pharmaceutical Co., Ltd. | ANTIBODY-DRUG CONJUGATE, INTERMEDIATE THEREOF, METHOD OF PRODUCTION THEREOF AND USE THEREOF |
| CR20220057A (es) | 2019-07-10 | 2022-07-19 | Cybrexa 3 Inc | Conjugados peptídicos de agentes dirigidos a microtúbulos como terapéuticos |
| BR112022000337A2 (pt) | 2019-07-10 | 2022-04-12 | Cybrexa 2 Inc | Conjugados de peptídeos de citotoxinas como terapêuticos |
| US20220411436A1 (en) * | 2019-09-18 | 2022-12-29 | Sichuan Baili Pharmaceutical Co., Ltd | Camptothecin derivative and conjugate thereof |
| BR112022011032A2 (pt) * | 2019-12-12 | 2022-08-16 | Jiangsu Hengrui Medicine Co | Conjugado de anticorpo-fármaco anti-claudina e uso farmacêutico do mesmo |
| CA3163130A1 (en) * | 2019-12-16 | 2021-06-24 | Jiangsu Hengrui Medicine Co., Ltd. | Anti-cea antibody-exatecan analog conjugate and pharmaceutical use thereof |
| EP4076434A1 (en) | 2019-12-17 | 2022-10-26 | Flagship Pioneering Innovations V, Inc. | Combination anti-cancer therapies with inducers of iron-dependent cellular disassembly |
| BR112022014189A2 (pt) | 2020-01-22 | 2022-11-29 | Jiangsu Hengrui Medicine Co | Conjugado de anticorpo anti-trop-2 análogo de exatecano e uso médico do mesmo |
| US20230144203A1 (en) | 2020-01-22 | 2023-05-11 | Shanghai Senhui Medicine Co., Ltd. | Drug conjugate of eribulin derivative, preparation method therefor and application thereof in medicine |
| US12029736B2 (en) | 2020-02-25 | 2024-07-09 | Mediboston Limited | Camptothecin derivatives and conjugates thereof |
| WO2021177438A1 (ja) | 2020-03-06 | 2021-09-10 | 第一三共株式会社 | 新規環状ジヌクレオチド誘導体を含む抗体薬物コンジュゲート |
| CN115298220A (zh) * | 2020-03-24 | 2022-11-04 | 上海翰森生物医药科技有限公司 | 抗体-药物偶联物及其医药用途 |
| TW202144013A (zh) | 2020-03-25 | 2021-12-01 | 大陸商江蘇恆瑞醫藥股份有限公司 | 一種含抗體藥物偶聯物的醫藥組成物及其用途 |
| TW202144016A (zh) * | 2020-03-25 | 2021-12-01 | 大陸商江蘇恆瑞醫藥股份有限公司 | 抗psma抗體-依喜替康類似物偶聯物及其醫藥用途 |
| BR112022019042A2 (pt) | 2020-03-25 | 2022-11-01 | Jiangsu Hengrui Pharmaceuticals Co Ltd | Método de preparação para conjugado anticorpo-fármaco |
| WO2021200131A1 (ja) | 2020-03-30 | 2021-10-07 | 国立研究開発法人国立がん研究センター | 抗体薬物複合体 |
| JP2023529415A (ja) * | 2020-06-08 | 2023-07-10 | バイリ-バイオ(チェンドゥ)ファーマスーティカル シーオー.,エルティーディー. | 高安定性の親水性結合ユニットを有するカンプトテシン類薬物及びその複合体 |
| EP4171654A1 (en) | 2020-06-24 | 2023-05-03 | AstraZeneca UK Limited | Combination of antibody-drug conjugate and cdk9 inhibitor |
| EP4171650A1 (en) | 2020-06-24 | 2023-05-03 | AstraZeneca UK Limited | Combination of antibody-drug conjugate and atr inhibitor |
| WO2021260583A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and dna-pk inhibitor |
| WO2021260582A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and aurora b inhibitor |
| WO2021260580A1 (en) | 2020-06-24 | 2021-12-30 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and atm inhibitor |
| EP4172323A1 (en) | 2020-06-29 | 2023-05-03 | Flagship Pioneering Innovations V, Inc. | Viruses engineered to promote thanotransmission and their use in treating cancer |
| US10997243B1 (en) | 2020-06-30 | 2021-05-04 | Snowflake Inc. | Supporting unstructured, semi-structured, and structured files |
| US11423081B1 (en) | 2020-06-30 | 2022-08-23 | Snowflake Inc. | Accessing files in a database stage using a user defined function |
| US11361026B2 (en) | 2020-06-30 | 2022-06-14 | Snowflake Inc. | Accessing files in a database stage using a user defined function |
| IL299184A (en) | 2020-07-06 | 2023-02-01 | Byondis Bv | Anti-folic acid drugs and antibody-conjugated drugs |
| WO2022011075A1 (en) * | 2020-07-10 | 2022-01-13 | VelosBio Inc. | Novel ror1 antibody immunoconjugates |
| WO2022015656A1 (en) | 2020-07-13 | 2022-01-20 | Regeneron Pharmaceuticals, Inc. | Camptothecin analogs conjugated to a glutamine residue in a protein, and their use |
| CN116157417A (zh) | 2020-07-17 | 2023-05-23 | 第一三共株式会社 | 抗体-药物缀合物的制造方法 |
| BR112023002417A2 (pt) | 2020-08-13 | 2023-03-21 | Chia Tai Tianqing Pharmaceutical Group Co Ltd | Conjugado de anticorpo e fármaco |
| JP2023537115A (ja) | 2020-08-13 | 2023-08-30 | イナート・ファルマ・ソシエテ・アノニム | 抗cd73抗体を使用する癌処置方法 |
| JPWO2022050300A1 (es) | 2020-09-02 | 2022-03-10 | ||
| TW202214230A (zh) | 2020-09-30 | 2022-04-16 | 大陸商映恩生物製藥(蘇州)有限公司 | 一種抗腫瘤化合物及其製備方法和應用 |
| WO2022074617A1 (en) | 2020-10-09 | 2022-04-14 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and parp1 selective inhibitor |
| JP2023545581A (ja) * | 2020-10-12 | 2023-10-30 | シーチュアン バイリ ファーム シーオー. エルティーディー | カンプトテシン系誘導体及びそのリガンド-薬物複合体 |
| BR112023008845A2 (pt) | 2020-11-11 | 2023-10-03 | Daiichi Sankyo Co Ltd | Composição farmacêutica, e, método de tratamento |
| WO2022102695A1 (ja) | 2020-11-12 | 2022-05-19 | 第一三共株式会社 | 抗b7-h3抗体-薬物コンジュゲート投与による中皮腫の治療 |
| WO2022099762A1 (zh) * | 2020-11-12 | 2022-05-19 | 博瑞生物医药(苏州)股份有限公司 | 一种抗体偶联物中间体及其制备方法 |
| KR20230121842A (ko) * | 2020-12-18 | 2023-08-21 | 상하이 후단-장지앙 바이오-파마슈티컬 컴퍼니 리미티드 | Trop2를 표적으로 하는 항체 약물 접합체, 이의 제조방법 및 용도 |
| JP2023550533A (ja) * | 2020-12-18 | 2023-12-01 | シャンハイ フダン-チャンジャン バイオ-ファーマシューティカル カンパニー リミテッド | B7-h3を標的とする抗体薬物複合体、その製造方法と使用 |
| US20240066138A1 (en) * | 2020-12-23 | 2024-02-29 | Shanghai Ruotuo Biosciences Co., Ltd. | Antibody-drug conjugates, pharmaceutical compositions, and therapeutic applications |
| TW202245844A (zh) * | 2021-01-13 | 2022-12-01 | 紀念斯隆凱特琳癌症中心 | 抗dll3抗體-藥物結合物 |
| CN114805377A (zh) * | 2021-01-29 | 2022-07-29 | 明慧医药(杭州)有限公司 | 适用于抗体-药物偶联物的毒素分子 |
| TW202241454A (zh) | 2021-02-01 | 2022-11-01 | 日商第一三共股份有限公司 | 抗體-免疫賦活化劑共軛物之新穎製造方法 |
| JP2024508081A (ja) | 2021-02-09 | 2024-02-22 | メディリンク セラピューティクス(スーチョウ)カンパニー,リミティド | 生物活性物質コンジュゲート、その調製方法及びその使用 |
| CA3212691A1 (en) | 2021-03-12 | 2022-09-15 | Daiichi Sankyo Company, Limited | Glycan, and method for producing medicine containing glycan |
| TW202300148A (zh) * | 2021-03-17 | 2023-01-01 | 大陸商江蘇恒瑞醫藥股份有限公司 | 喜樹鹼衍生物的製備方法 |
| AU2022238571A1 (en) | 2021-03-18 | 2023-09-14 | Seagen Inc. | Selective drug release from internalized conjugates of biologically active compounds |
| WO2022212784A1 (en) | 2021-03-31 | 2022-10-06 | Flagship Pioneering Innovations V, Inc. | Thanotransmission polypeptides and their use in treating cancer |
| CA3215049A1 (en) | 2021-04-10 | 2022-10-13 | Baiteng ZHAO | Folr1 binding agents, conjugates thereof and methods of using the same |
| EP4323009A1 (en) * | 2021-04-14 | 2024-02-21 | Genequantum Healthcare (Suzhou) Co., Ltd. | Linkers, conjugates and applications thereof |
| EP4079327A1 (en) | 2021-04-22 | 2022-10-26 | Centaurus Polytherapeutics | Payloads for drug-conjugates and their use for treating cancer |
| WO2022226317A1 (en) | 2021-04-23 | 2022-10-27 | Profoundbio Us Co. | Anti-cd70 antibodies, conjugates thereof and methods of using the same |
| JP2024515342A (ja) * | 2021-04-26 | 2024-04-09 | 江▲蘇▼恒瑞医▲薬▼股▲フン▼有限公司 | 抗Nectin-4抗体及び抗Nectin-4抗体-薬物複合体並びにその医薬用途 |
| CN113816969B (zh) * | 2021-04-30 | 2024-01-16 | 联宁(苏州)生物制药有限公司 | 依喜替康类化合物、其抗体药物偶联物及其应用 |
| US11645243B2 (en) | 2021-06-07 | 2023-05-09 | Snowflake Inc. | Accessing files in a database stage using a user defined function |
| TW202313125A (zh) * | 2021-06-17 | 2023-04-01 | 大陸商明慧醫藥(杭州)有限公司 | 一種抗腫瘤化合物及其應用 |
| IL309249A (en) | 2021-06-28 | 2024-02-01 | Byondis Bv | Conjugates including phosphoantigens and their use in therapy |
| AU2022303363A1 (en) | 2021-06-29 | 2024-01-18 | Flagship Pioneering Innovations V, Inc. | Immune cells engineered to promote thanotransmission and uses thereof |
| CN113402584A (zh) * | 2021-07-06 | 2021-09-17 | 联宁(苏州)生物制药有限公司 | 一种抗体偶联药物连接子的中间体lnd1067-l1的合成方法 |
| TW202320857A (zh) | 2021-07-06 | 2023-06-01 | 美商普方生物製藥美國公司 | 連接子、藥物連接子及其結合物及其使用方法 |
| CN113527418B (zh) * | 2021-07-16 | 2022-05-03 | 成都普康唯新生物科技有限公司 | 一种ADC linker的制备方法 |
| TW202320858A (zh) * | 2021-07-19 | 2023-06-01 | 美商薩諾管理公司 | 免疫接合物及方法 |
| US11806405B1 (en) | 2021-07-19 | 2023-11-07 | Zeno Management, Inc. | Immunoconjugates and methods |
| TW202320859A (zh) | 2021-07-21 | 2023-06-01 | 大陸商江蘇恆瑞醫藥股份有限公司 | 一種含抗trop2抗體藥物偶聯物的醫藥組成物及其用途 |
| AU2022347380A1 (en) | 2021-09-15 | 2024-04-11 | Daiichi Sankyo Company, Limited | Antibody-drug conjugate for use in methods of treating chemotherapy-resistant cancer |
| JPWO2023054706A1 (es) | 2021-09-30 | 2023-04-06 | ||
| KR20240099178A (ko) | 2021-10-18 | 2024-06-28 | 다이이찌 산쿄 가부시키가이샤 | 항 cd37 항체-약물 콘주게이트 |
| CA3237844A1 (en) | 2021-11-15 | 2023-05-19 | Systimmune, Inc. | Bispecific antibody-camptothecin drug conjugate and pharmaceutical use thereof |
| US11814394B2 (en) * | 2021-11-16 | 2023-11-14 | Genequantum Healthcare (Suzhou) Co., Ltd. | Exatecan derivatives, linker-payloads, and conjugates and thereof |
| AU2022392822A1 (en) | 2021-11-18 | 2024-05-02 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and parp1 selective inhibitor |
| EP4433096A1 (en) | 2021-11-19 | 2024-09-25 | Ardeagen Corporation | Gpc3 binding agents, conjugates thereof and methods of using the same |
| CN118401668A (zh) | 2021-11-30 | 2024-07-26 | 第一三共株式会社 | 蛋白酶裂解性掩蔽抗体 |
| AU2022402061A1 (en) | 2021-12-03 | 2024-07-11 | Systimmune, Inc. | Anti-human trop2 antibody-camptothecin drug conjugate and medical use thereof |
| CA3240378A1 (en) | 2021-12-23 | 2023-06-29 | Jiangsu Hengrui Pharmaceuticals Co., Ltd. | Anti-dll3 antibody and pharmaceutical use thereof, and antibody-drug conjugate containing anti-dll3 antibody |
| WO2023116911A1 (zh) * | 2021-12-24 | 2023-06-29 | 百奥泰生物制药股份有限公司 | 抗FRα抗体及其抗体药物偶联物和用途 |
| CA3240724A1 (en) | 2021-12-28 | 2023-07-06 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and atr inhibitor |
| TW202333800A (zh) | 2021-12-28 | 2023-09-01 | 英商阿斯特捷利康英國股份有限公司 | 抗體-藥物結合物及rasg12c抑制劑之組合 |
| WO2023126297A1 (en) | 2021-12-30 | 2023-07-06 | Byondis B.V. | Antifolate linker-drugs and antibody-drug conjugates |
| WO2023138635A1 (zh) * | 2022-01-18 | 2023-07-27 | 甘李药业股份有限公司 | 一种依喜替康衍生物-抗体偶联物及其医药用途 |
| AU2023210897A1 (en) | 2022-01-25 | 2024-09-05 | Medilink Therapeutics (Suzhou) Co., Ltd. | Antibody against her3, conjugate and use thereof |
| WO2023173026A1 (en) | 2022-03-10 | 2023-09-14 | Sorrento Therapeutics, Inc. | Antibody-drug conjugates and uses thereof |
| WO2023175483A1 (en) | 2022-03-16 | 2023-09-21 | Astrazeneca Uk Limited | A scoring method for an anti-trop2 antibody‑drug conjugate therapy |
| WO2023209591A1 (en) | 2022-04-27 | 2023-11-02 | Daiichi Sankyo Company, Limited | Combination of antibody-drug conjugate with ezh1 and/or ezh2 inhibitor |
| WO2023218378A1 (en) | 2022-05-11 | 2023-11-16 | Daiichi Sankyo Company, Limited | Combination of an antibody specific for a tumor antigen and a cd47 inhibitor |
| WO2023228095A1 (en) | 2022-05-24 | 2023-11-30 | Daiichi Sankyo Company, Limited | Dosage regimen of an anti-cdh6 antibody-drug conjugate |
| WO2023237050A1 (en) * | 2022-06-09 | 2023-12-14 | Beigene, Ltd. | Antibody drug conjugates |
| CN116789733A (zh) * | 2022-07-05 | 2023-09-22 | 上海药明合联生物技术有限公司 | 偶联连接子 |
| CN115160403A (zh) * | 2022-07-05 | 2022-10-11 | 上海彩迩文生化科技有限公司 | 特异性拓扑异构酶抑制剂和可用于抗体药物偶联物及其制备方法 |
| WO2024012566A2 (en) | 2022-07-15 | 2024-01-18 | Genequantum Healthcare (Suzhou) Co., Ltd. | Antibody, linkers, payload, conjugates and applications thereof |
| WO2024020536A1 (en) * | 2022-07-22 | 2024-01-25 | Nj Bio, Inc. | Hexacyclic topoisomerase inhibitors having cytotoxic activity on cancer cells |
| WO2024022372A1 (zh) * | 2022-07-27 | 2024-02-01 | 明慧医药(杭州)有限公司 | 抗体药物偶联物及其应用 |
| WO2024023750A1 (en) | 2022-07-28 | 2024-02-01 | Astrazeneca Uk Limited | Combination of antibody-drug conjugate and bispecific checkpoint inhibitor |
| CN117510515A (zh) * | 2022-07-28 | 2024-02-06 | 明慧医药(杭州)有限公司 | 适用于抗体-药物偶联物的毒素分子 |
| US20240116945A1 (en) * | 2022-09-02 | 2024-04-11 | Merck Sharp & Dohme Llc | Exatecan-derived topoisomerase-1 inhibitors pharmaceutical compositions, and uses thereof |
| WO2024073436A2 (en) * | 2022-09-26 | 2024-04-04 | Solve Therapeutics, Inc. | Compositions and uses thereof |
| WO2024067811A1 (en) * | 2022-09-30 | 2024-04-04 | Beigene, Ltd. | Ligand-drug conjugate of exatecan analogue, and medical use thereof |
| US20240174732A1 (en) | 2022-10-05 | 2024-05-30 | Flagship Pioneering Innovations V, Inc. | Nucleic acid molecules encoding trif and additional polypeptides and their use in treating cancer |
| US20240208987A1 (en) | 2022-10-25 | 2024-06-27 | Merck Sharp & Dohme Llc | Exatecan-derived adc linker-payloads, pharmaceutical compositions, and uses thereof |
| WO2024110905A1 (en) | 2022-11-24 | 2024-05-30 | Beigene, Ltd. | Anti-cea antibody drug conjugates and methods of use |
| WO2024109949A1 (zh) * | 2022-11-25 | 2024-05-30 | 明慧医药(杭州)有限公司 | 一种抗肿瘤化合物及其应用 |
| WO2024114318A1 (zh) * | 2022-11-29 | 2024-06-06 | 四川科伦博泰生物医药股份有限公司 | 药物连接子化合物及其制备方法和用途 |
| WO2024116094A1 (en) | 2022-11-30 | 2024-06-06 | Daiichi Sankyo Company, Limited | Combination of antibody-drug conjugates and dnmt inhibitors |
| CN116621927B (zh) * | 2023-01-09 | 2024-03-26 | 联宁(苏州)生物制药有限公司 | 带有伊喜替康和C-lock定点偶联基团的抗体偶联中间体、偶联方法及抗体偶联药物 |
| US20240269251A1 (en) | 2023-01-09 | 2024-08-15 | Flagship Pioneering Innovations V, Inc. | Genetic switches and their use in treating cancer |
| WO2024165045A1 (en) | 2023-02-09 | 2024-08-15 | Beigene, Ltd. | Self-stabilizing linker conjugates |
| WO2024179470A1 (zh) * | 2023-02-27 | 2024-09-06 | 苏州盛迪亚生物医药有限公司 | 抗dll3抗体、其抗体-药物偶联物及其医药用途 |
Family Cites Families (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL162181A (en) | 1988-12-28 | 2006-04-10 | Pdl Biopharma Inc | A method of producing humanized immunoglubulin, and polynucleotides encoding the same |
| JPH06508511A (ja) | 1990-07-10 | 1994-09-29 | ケンブリッジ アンティボディー テクノロジー リミティド | 特異的な結合ペアーの構成員の製造方法 |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| JP3008226B2 (ja) | 1991-01-16 | 2000-02-14 | 第一製薬株式会社 | 六環性化合物 |
| US5658920A (en) | 1991-01-16 | 1997-08-19 | Daiichi Pharmaceutical Co., Ltd. | Substituted 1H,12H-benz-[DE]pyrano[3',4':6,7] indolizino[1,2-B]quinoline-10,13(9H,15H)-dione compound |
| JPH0559061U (ja) | 1991-04-18 | 1993-08-03 | 株式会社ケイヴイシー | ボールバルブ |
| EP0605522B1 (en) | 1991-09-23 | 1999-06-23 | Medical Research Council | Methods for the production of humanized antibodies |
| PT1024191E (pt) | 1991-12-02 | 2008-12-22 | Medical Res Council | Produção de auto-anticorpos a partir de reportórios de segmentos de anticorpo e exibidos em fagos |
| CA2131151A1 (en) | 1992-03-24 | 1994-09-30 | Kevin S. Johnson | Methods for producing members of specific binding pairs |
| JP3359955B2 (ja) | 1992-07-16 | 2002-12-24 | 第一製薬株式会社 | 抗腫瘍剤 |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| JPH0687746U (ja) | 1993-06-02 | 1994-12-22 | 株式会社ユニシアジェックス | 液圧緩衝器のリザーバチューブ構造 |
| GB9313509D0 (en) | 1993-06-30 | 1993-08-11 | Medical Res Council | Chemisynthetic libraries |
| CA2177367A1 (en) | 1993-12-03 | 1995-06-08 | Andrew David Griffiths | Recombinant binding proteins and peptides |
| US5773001A (en) | 1994-06-03 | 1998-06-30 | American Cyanamid Company | Conjugates of methyltrithio antitumor agents and intermediates for their synthesis |
| JPH08337584A (ja) | 1995-04-10 | 1996-12-24 | Dai Ichi Seiyaku Co Ltd | 縮合六環式アミノ化合物、これを含有する医薬及びその製法 |
| US6504029B1 (en) | 1995-04-10 | 2003-01-07 | Daiichi Pharmaceutical Co., Ltd. | Condensed-hexacyclic compounds and a process therefor |
| SG50747A1 (en) | 1995-08-02 | 1998-07-20 | Tanabe Seiyaku Co | Comptothecin derivatives |
| JPH1095802A (ja) * | 1995-12-28 | 1998-04-14 | Tanabe Seiyaku Co Ltd | カンプトテシン誘導体 |
| CA2192725C (en) | 1995-12-28 | 2004-04-20 | Kenji Tsujihara | Camptothecin derivatives |
| TW409058B (en) | 1996-06-06 | 2000-10-21 | Daiichi Seiyaku Co | Method for preparation of a drug complex |
| TW527183B (en) | 1996-06-06 | 2003-04-11 | Daiichi Seiyaku Co | Drug complex |
| JPH1171280A (ja) | 1997-06-26 | 1999-03-16 | Tanabe Seiyaku Co Ltd | 医薬組成物 |
| JPH1192405A (ja) | 1997-09-19 | 1999-04-06 | Dai Ichi Seiyaku Co Ltd | 薬物複合体 |
| PT2180007E (pt) | 1998-04-20 | 2013-11-25 | Roche Glycart Ag | Engenharia de glicosilação de anticorpos para melhorar a citotoxicidade celular dependente de anticorpos |
| EP1080732A4 (en) * | 1998-05-22 | 2004-08-25 | Daiichi Seiyaku Co | DRUG COMPOSITION |
| ID29943A (id) | 1998-10-30 | 2001-10-25 | Daiichi Seiyaku Co | Senyawa dds dan metode pengukurannya |
| CA2704600C (en) | 1999-04-09 | 2016-10-25 | Kyowa Hakko Kirin Co., Ltd. | A method for producing antibodies with increased adcc activity |
| JP2002060351A (ja) | 2000-03-22 | 2002-02-26 | Dai Ichi Seiyaku Co Ltd | 水酸基を有する薬物を含むdds化合物 |
| MXPA02012791A (es) * | 2000-06-29 | 2003-12-11 | Daiichi Seiyaku Co | Compuestos dds y metodo para prepararlo. |
| AU2001271037A1 (en) | 2000-07-13 | 2002-01-30 | Daiichi Pharmaceutical Co., Ltd. | Pharmaceutical compositions containing dds compounds |
| EP3690043A1 (en) | 2000-10-06 | 2020-08-05 | Kyowa Kirin Co., Ltd. | Antibody composition-producing cell |
| US7090843B1 (en) | 2000-11-28 | 2006-08-15 | Seattle Genetics, Inc. | Recombinant anti-CD30 antibodies and uses thereof |
| TWI313609B (en) | 2001-08-21 | 2009-08-21 | Mitsubishi Tanabe Pharma Corp | Pharmaceutical composition for inhibiting the metastasis or preventing the recurrence of malignant tumor |
| NZ533164A (en) | 2001-11-01 | 2008-09-26 | Uab Research Foundation | Combinations of antibodies selective for a tumor necrosis factor-related apoptosis-inducing ligand receptor and other therapeutic agents |
| EP1482972A4 (en) | 2001-11-20 | 2005-11-23 | Seattle Genetics Inc | TREATMENT OF IMMUNOLOGICAL DISORDERS USING ANTI-CD30 ANTIBODIES |
| US8877901B2 (en) | 2002-12-13 | 2014-11-04 | Immunomedics, Inc. | Camptothecin-binding moiety conjugates |
| AU2003288467A1 (en) | 2002-12-13 | 2004-07-09 | Immunomedics, Inc. | Immunoconjugates with an intracellularly-cleavable linkage |
| US20040202666A1 (en) | 2003-01-24 | 2004-10-14 | Immunomedics, Inc. | Anti-cancer anthracycline drug-antibody conjugates |
| KR20060125909A (ko) | 2004-02-26 | 2006-12-06 | 이노텍 파마슈티컬스 코포레이션 | 이소퀴놀린 유도체 및 이의 사용 방법 |
| JP4942643B2 (ja) | 2004-03-02 | 2012-05-30 | シアトル ジェネティックス, インコーポレイテッド | 部分的に付加された抗体およびそれらの結合体化方法 |
| EP1747021B1 (en) * | 2004-05-19 | 2015-09-09 | E. R. Squibb & Sons, L.L.C. | Self-immolative linkers and drug conjugates |
| CN114053429A (zh) | 2004-06-01 | 2022-02-18 | 健泰科生物技术公司 | 抗体-药物偶联物和方法 |
| US20080305044A1 (en) | 2004-11-29 | 2008-12-11 | Seattle Genetics, Inc. | Engineered Antibodies and Immunoconjugates |
| DE102005009099A1 (de) | 2005-02-28 | 2006-08-31 | Ktb Tumorforschungsgesellschaft Mbh | Proteinbindende Camptothecin-Peptid-Derivate und diese enthaltende Arzneimittel |
| DE102005009084A1 (de) | 2005-02-28 | 2006-08-31 | Ktb Tumorforschungsgesellschaft Mbh | Proteinbindende Anthrazyklin-Peptid-Derivate und diese enthaltende Arzneimittel |
| DK1871418T3 (da) | 2005-04-19 | 2014-06-10 | Seattle Genetics Inc | Humaniserede anti-cd70-bindende midler og anvendelser deraf |
| US7833979B2 (en) | 2005-04-22 | 2010-11-16 | Amgen Inc. | Toxin peptide therapeutic agents |
| SI2032606T1 (sl) * | 2006-05-30 | 2014-02-28 | Genentech, Inc. | Protitelesa in imunokonjugati in njihove uporabe |
| PL2446904T3 (pl) * | 2006-05-30 | 2015-10-30 | Genentech Inc | Przeciwciała anty-CD22, ich immunokoniugaty oraz ich zastosowania |
| JPWO2007145306A1 (ja) | 2006-06-15 | 2009-11-12 | 国立大学法人 北海道大学 | 腫瘍組織で選択的に分解性を示す血中滞留性素子 |
| ITMI20061473A1 (it) | 2006-07-26 | 2008-01-27 | Indena Spa | Derivati della camptotecina ad attivita antitumorale |
| WO2008079293A1 (en) | 2006-12-21 | 2008-07-03 | Schering Corporation | Pyrrolo [3, 2-a] pyridine derivatives for inhibiting ksp kinesin activity |
| WO2008116219A2 (en) * | 2007-03-22 | 2008-09-25 | Sloan-Kettering Institute For Cancer Research | Uses of monoclonal antibody 8h9 |
| NO2281006T3 (es) | 2008-04-30 | 2017-12-30 | ||
| DK3903829T3 (da) | 2009-02-13 | 2023-06-26 | Immunomedics Inc | Immunkonjugater med en intracellulær spaltelig binding |
| JP5829520B2 (ja) | 2009-08-20 | 2015-12-09 | 国立大学法人 千葉大学 | コルヒチン誘導体 |
| US20110070248A1 (en) | 2009-09-24 | 2011-03-24 | Seattle Genetics, Inc. | Dr5 ligand drug conjugates |
| US9127088B2 (en) | 2010-05-13 | 2015-09-08 | Indiana University Research And Technology Corporation | Glucagon superfamily peptides exhibiting nuclear hormone receptor activity |
| MX2012013100A (es) | 2010-05-18 | 2013-01-22 | Cerulean Pharma Inc | Composiciones y metodos para el tratamiento de enfermedades autoinmunes y otras enfermedades. |
| CN103096933A (zh) | 2010-07-12 | 2013-05-08 | CovX科技爱尔兰有限公司 | 多功能抗体缀合物 |
| WO2012074757A1 (en) | 2010-11-17 | 2012-06-07 | Genentech, Inc. | Alaninyl maytansinol antibody conjugates |
| MX2014007121A (es) | 2011-12-14 | 2014-09-04 | Seattle Genetics Inc | Nuevos conjugados de principio activo-ligante (adc) y su uso. |
| US20130280282A1 (en) | 2012-04-24 | 2013-10-24 | Daiichi Sankyo Co., Ltd. | Dr5 ligand drug conjugates |
| KR20150023729A (ko) | 2012-06-14 | 2015-03-05 | 암브룩스, 인코포레이티드 | 핵 수용체 리간드 폴리펩티드에 접합된 항-psma 항체 |
| US9517522B2 (en) * | 2012-09-05 | 2016-12-13 | Illinois Tool Works Inc. | Self-aligning wire feeder assembly |
| KR101841818B1 (ko) * | 2012-10-11 | 2018-03-26 | 다이이찌 산쿄 가부시키가이샤 | 항체-약물 콘주게이트 |
| EP2910573B1 (en) | 2012-10-19 | 2020-02-19 | Daiichi Sankyo Company, Limited | Antibody-drug conjugate produced by binding through linker having hydrophilic structure |
| US9814784B2 (en) | 2013-01-03 | 2017-11-14 | Celltrion, Inc. | Antibody-linker-drug conjugate, preparation method therefor, and anticancer drug composition containing same |
| CN103313390B (zh) | 2013-07-05 | 2016-01-20 | 江苏大学 | 一种基于双移动信标的wsn定位方法 |
| KR102465042B1 (ko) * | 2014-01-31 | 2022-11-09 | 다이이찌 산쿄 가부시키가이샤 | 항-her2 항체-약물 접합체 |
| EP3130608B1 (en) | 2014-04-10 | 2019-09-04 | Daiichi Sankyo Co., Ltd. | (anti-her2 antibody)-drug conjugate |
| US20190386950A1 (en) | 2017-02-24 | 2019-12-19 | Gil Hoon Chang | Message sharing system and method for interactive application |
-
2013
- 2013-10-10 KR KR1020157007536A patent/KR101841818B1/ko active IP Right Grant
- 2013-10-10 PT PT181523812T patent/PT3342785T/pt unknown
- 2013-10-10 CN CN201380053256.2A patent/CN104755494B/zh active Active
- 2013-10-10 KR KR1020217034817A patent/KR102369419B1/ko active IP Right Grant
- 2013-10-10 KR KR1020227006326A patent/KR102417310B1/ko active IP Right Grant
- 2013-10-10 SI SI201331006T patent/SI2907824T1/en unknown
- 2013-10-10 NZ NZ705394A patent/NZ705394A/en unknown
- 2013-10-10 CA CA3021435A patent/CA3021435C/en active Active
- 2013-10-10 CA CA2885800A patent/CA2885800C/en active Active
- 2013-10-10 NZ NZ74644013A patent/NZ746440A/en unknown
- 2013-10-10 PL PL13845596T patent/PL2907824T3/pl unknown
- 2013-10-10 KR KR1020227022424A patent/KR102456726B1/ko active IP Right Grant
- 2013-10-10 SG SG11201502887WA patent/SG11201502887WA/en unknown
- 2013-10-10 JP JP2014540755A patent/JP5953378B2/ja active Active
- 2013-10-10 KR KR1020227035813A patent/KR102498405B1/ko active IP Right Grant
- 2013-10-10 NZ NZ74094813A patent/NZ740948A/en unknown
- 2013-10-10 CN CN202211490738.1A patent/CN115960111A/zh active Pending
- 2013-10-10 IL IL302494A patent/IL302494B1/en unknown
- 2013-10-10 RS RS20180643A patent/RS57278B1/sr unknown
- 2013-10-10 EP EP19206764.3A patent/EP3632471A1/en active Pending
- 2013-10-10 ES ES18152381T patent/ES2773710T3/es active Active
- 2013-10-10 IL IL271760A patent/IL271760B/en unknown
- 2013-10-10 KR KR1020187007575A patent/KR101901558B1/ko active IP Right Grant
- 2013-10-10 DK DK13845596.9T patent/DK2907824T3/en active
- 2013-10-10 HU HUE13845596A patent/HUE039000T2/hu unknown
- 2013-10-10 KR KR1020237018596A patent/KR102584005B1/ko active IP Right Grant
- 2013-10-10 KR KR1020217009640A patent/KR102320907B1/ko active IP Right Grant
- 2013-10-10 DK DK18152381.2T patent/DK3342785T3/da active
- 2013-10-10 RU RU2015113767A patent/RU2664465C2/ru active
- 2013-10-10 MX MX2015003903A patent/MX364484B/es active IP Right Grant
- 2013-10-10 NZ NZ74643913A patent/NZ746439A/en unknown
- 2013-10-10 KR KR1020197035246A patent/KR102087017B1/ko active IP Right Grant
- 2013-10-10 CN CN201810950717.0A patent/CN109081871B/zh active Active
- 2013-10-10 KR KR1020237004110A patent/KR102540419B1/ko active IP Right Grant
- 2013-10-10 ES ES13845596.9T patent/ES2671644T3/es active Active
- 2013-10-10 MY MYPI2015000898A patent/MY170251A/en unknown
- 2013-10-10 BR BR122021014365-0A patent/BR122021014365B1/pt active IP Right Grant
- 2013-10-10 EP EP13845596.9A patent/EP2907824B1/en active Active
- 2013-10-10 IL IL294622A patent/IL294622B2/en unknown
- 2013-10-10 BR BR112015006521-0A patent/BR112015006521B1/pt active IP Right Grant
- 2013-10-10 LT LTEP13845596.9T patent/LT2907824T/lt unknown
- 2013-10-10 WO PCT/JP2013/006069 patent/WO2014057687A1/ja active Application Filing
- 2013-10-10 BR BR122021014396-0A patent/BR122021014396B1/pt active IP Right Grant
- 2013-10-10 AU AU2013328111A patent/AU2013328111B2/en active Active
- 2013-10-10 PT PT138455969T patent/PT2907824T/pt unknown
- 2013-10-10 SI SI201331658T patent/SI3342785T1/sl unknown
- 2013-10-10 CN CN202111179342.0A patent/CN113929738B/zh active Active
- 2013-10-10 KR KR1020207006240A patent/KR102237639B1/ko active IP Right Grant
- 2013-10-10 RS RS20200226A patent/RS60000B1/sr unknown
- 2013-10-10 KR KR1020187026771A patent/KR102052319B1/ko active IP Right Grant
- 2013-10-10 IL IL313147A patent/IL313147A/en unknown
- 2013-10-10 TR TR2018/09636T patent/TR201809636T4/tr unknown
- 2013-10-10 EP EP18152381.2A patent/EP3342785B1/en active Active
- 2013-10-10 PL PL18152381T patent/PL3342785T3/pl unknown
- 2013-10-10 US US14/435,114 patent/US10195288B2/en active Active
- 2013-10-10 SG SG10201804788XA patent/SG10201804788XA/en unknown
- 2013-10-10 LT LTEP18152381.2T patent/LT3342785T/lt unknown
- 2013-10-10 HU HUE18152381A patent/HUE048748T2/hu unknown
- 2013-10-11 TW TW111105275A patent/TW202233186A/zh unknown
- 2013-10-11 TW TW107100508A patent/TWI650312B/zh active
- 2013-10-11 TW TW109116005A patent/TWI757740B/zh active
- 2013-10-11 TW TW102136742A patent/TWI615152B/zh active
- 2013-10-11 TW TW108100079A patent/TWI696611B/zh active
-
2015
- 2015-03-17 ZA ZA2015/01825A patent/ZA201501825B/en unknown
- 2015-03-25 PH PH12015500667A patent/PH12015500667B1/en unknown
- 2015-03-26 MX MX2020010964A patent/MX2020010964A/es unknown
- 2015-03-26 MX MX2019004502A patent/MX2019004502A/es unknown
- 2015-04-02 IL IL238143A patent/IL238143B/en active IP Right Grant
- 2015-11-12 HK HK15111165.9A patent/HK1210477A1/xx unknown
-
2016
- 2016-06-13 JP JP2016117096A patent/JP6186045B2/ja active Active
- 2016-06-13 US US15/180,203 patent/US9808537B2/en active Active
- 2016-08-09 JP JP2016156697A patent/JP6030267B1/ja active Active
-
2017
- 2017-07-28 JP JP2017146257A patent/JP6371452B2/ja active Active
-
2018
- 2018-01-12 AU AU2018200308A patent/AU2018200308B2/en active Active
- 2018-06-05 HR HRP20180870TT patent/HRP20180870T1/hr unknown
- 2018-06-26 CY CY20181100657T patent/CY1120363T1/el unknown
- 2018-07-04 PH PH12018501433A patent/PH12018501433A1/en unknown
- 2018-07-12 JP JP2018131996A patent/JP6715888B2/ja active Active
- 2018-09-26 US US16/142,354 patent/US10973924B2/en active Active
- 2018-12-10 HK HK18115765.1A patent/HK1256631A1/zh unknown
-
2019
- 2019-02-08 ZA ZA2019/00820A patent/ZA201900820B/en unknown
-
2020
- 2020-01-24 AU AU2020200548A patent/AU2020200548B2/en active Active
- 2020-03-03 HR HRP20200353TT patent/HRP20200353T1/hr unknown
- 2020-03-19 CY CY20201100256T patent/CY1122789T1/el unknown
- 2020-03-24 US US16/828,279 patent/US11633493B2/en active Active
- 2020-06-09 JP JP2020100531A patent/JP6952835B2/ja active Active
-
2021
- 2021-09-28 JP JP2021157523A patent/JP7170812B2/ja active Active
-
2022
- 2022-05-26 AU AU2022203560A patent/AU2022203560A1/en active Pending
- 2022-11-01 JP JP2022175282A patent/JP7523506B2/ja active Active
-
2023
- 2023-03-03 US US18/117,089 patent/US20240082413A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2773710T3 (es) | Enlazadores para conjugados de anticuerpo - fármaco | |
| ES2782248T3 (es) | Conjugado de anticuerpo y fármaco producido por la unión a través de un enlazador que tiene estructura hidrófila | |
| RU2776489C2 (ru) | Конъюгат антитело-лекарственное средство | |
| BR122020012304B1 (pt) | Composto ligante para conjugados anticorpo-fármaco, método e intermediários de produção do mesmo |