CN1317480A - 一釜法合成2-噁唑烷酮衍生物 - Google Patents
一釜法合成2-噁唑烷酮衍生物 Download PDFInfo
- Publication number
- CN1317480A CN1317480A CN01112010A CN01112010A CN1317480A CN 1317480 A CN1317480 A CN 1317480A CN 01112010 A CN01112010 A CN 01112010A CN 01112010 A CN01112010 A CN 01112010A CN 1317480 A CN1317480 A CN 1317480A
- Authority
- CN
- China
- Prior art keywords
- compound
- formula
- solution
- ethyl acetate
- under
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/08—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D263/16—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D263/18—Oxygen atoms
- C07D263/20—Oxygen atoms attached in position 2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/20—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by nitrogen atoms not being part of nitro or nitroso groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/22—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Indole Compounds (AREA)
Abstract
本发明涉及制备取代吲哚衍生物的改进方法,所述衍生物可用于治疗和预防偏头痛。本发明还涉及取代吲哚衍生物在制备药物组合物的应用。
Description
本申请为1996年8月2日提交的发明名称为“一釜法合成2-噁唑烷酮衍生物”、申请号为96197237.8的分案申请。
本发明涉及制备取代吲哚衍生物的改进方法,所述衍生物可用于治疗和预防偏头痛。更具体讲,本发明提供了制备5HT1-状受体兴奋剂(S)-4-{[3-(二甲氨基)乙基-1H-吲哚-5-基]甲基}-2-噁唑烷酮的改进方法,此化合物已知对偏头痛的治疗具有效用。
人们已知选择性5-HT1状受体兴奋剂是有用的治疗剂。5-HT1状受体导致血管收缩,从而调节颈动脉血管床的血流量。欧洲专利说明书0313397中描述了一类特异性5-HT1状受体兴奋剂,它们对预示颈动脉血管床中血管收缩作用的病症(如偏头痛,与颈动脉脉管系统的过良扩张有关的病症)的治疗或预防是有益的。
国际专利说明书WO91/18897描述了另一类具有优越的“5-HT1状”受体兴奋活性和优越的口服用药后吸收活性化合物。这些特性使得WO91/18897中所公开的化合物特别适合于某些医疗用途,尤其是用于预防和治疗偏头痛、偏头神经痛和与血管病有关的头痛,以下通称“偏头痛”。一种WO91/18897中所述的特别优选化合物为(S)-N,N-二甲基-2-[5-(2-氧代-1,3-噁唑烷-4-基甲基)-1H-吲哚-3-基]乙胺,又称作(S)-4-{[3-[2-(二甲氨基)乙基]-1H-吲哚-5-基]甲基}-2-噁唑烷酮,并可用式(Ⅰ)表示:
式(Ⅰ)化合物可以其(S)或(R)对映体形式存在,而且在WO91/18897中给予了具体说明。WO91/18897中还提供了制备式(Ⅰ)化合物的众多可能途径。
现已发现一种制备式(Ⅰ)化合物的新方法。该方法优于WO91/18897中公开的任一方法,通过使用一釜法,使得最终产物能够大规模高产率制备,而且为纯净形式,并因而免除了费时和花费高的中间体分离步骤。新方法还避免了使用危险试剂(如光气)或对环境有害的试剂(如氯化锡)。
因此,本发明的第一方面是提供了制备(S)-4-{[3-[2(二甲氨基)乙基]-1H-吲哚-5-基]甲基}-2-噁唑烷酮的方法,该方法包括下列步骤:a)形成氨基甲酸酯:向式(Ⅱ)所示的4-硝基-(L)-苯丙氨酸甲酯盐酸盐;中加入碳酸钠或碳酸氢钠和氯甲酸正丁酯,反应得到式(Ⅲ)所示的(S)-N-丁氧羰基-4-硝基苯丙氨酸甲酯 b)还原式(Ⅲ)化合物,产生式(Ⅳ)所示的(S)-N-丁氧羰基-4-氨基苯丙氨酸甲酯:c)还原式(Ⅳ)化合物中的甲酯基团-CO2CH3,产生式(Ⅴ)所示的(S)-N-丁氧羰基-4-氨基苯丙氨醇:
b)还原式(Ⅲ)化合物,产生式(Ⅳ)所示的(S)-N-丁氧羰基-4-氨基苯丙氨酸甲酯:c)还原式(Ⅳ)化合物中的甲酯基团-CO2CH3,产生式(Ⅴ)所示的(S)-N-丁氧羰基-4-氨基苯丙氨醇: d)环合式(Ⅴ)化合物,产生式(Ⅵ)所示的(S)-4-(4-氨基苄基)-2-噁唑烷酮:e)制备式(Ⅵ)化合物的重氮盐,继之还原,产生式(Ⅶ)所示的肼(S)-4-(4-肼基苄基)-2-噁唑烷酮盐酸盐:
d)环合式(Ⅴ)化合物,产生式(Ⅵ)所示的(S)-4-(4-氨基苄基)-2-噁唑烷酮:e)制备式(Ⅵ)化合物的重氮盐,继之还原,产生式(Ⅶ)所示的肼(S)-4-(4-肼基苄基)-2-噁唑烷酮盐酸盐: f)将式(Ⅶ)化合物进行费歇尔反应,产生式(Ⅰ)化合物。
f)将式(Ⅶ)化合物进行费歇尔反应,产生式(Ⅰ)化合物。
步骤a)-f)中的一步或多步适宜采用一釜法进行。优选步骤a)至d)采用一釜法进行,然后分离式(Ⅵ)化合物,随后再将步骤e)和f)通过另一一釜法进行。
步骤a)可以在溶剂(如含水乙酸乙酯或二噁烷)中方便地进行,其中优选在含水乙酸乙酯中进行反应。较之碳酸氢钠,优选碳酸钠,并优选在氯甲酸正丁酯加入之前加入。反应在不太高的温度下(适宜为5-60℃)能很方便地进行。优选反应在15-35℃下进行。在特别优选的实施方案中,碳酸钠的加入在大约20℃下进行,而氯甲酸正丁酯的加入则在大约30℃下进行。
还原步骤b)可以方便地在有机溶剂(如乙酸乙酯或乙醇)存在下进行。优选步骤b)使用得自步骤a)的式(Ⅲ)化合物的乙酸乙酯溶液通过一釜法进行。步骤b)适宜通过氢化作用(优选在催化剂如披钯木炭存在下)进行。反应可以在氮气氛及室温下,采用正常大气压力的氢气进行。氢化优选在升高的温度如30-50℃下于大约20psi氢气压力下进行。优选将所产生的式(Ⅳ)化合物的乙酸乙酯溶液转化成丁醇溶液,该溶液可直接用于步骤c)中,作为一釜法的一部分。此转化可以很方便地通过部分蒸馏乙酸乙酯溶液,继之加入丁醇并分馏除去乙酸乙酯完成。
步骤c)中的甲酯还原在溶剂(如SVM或正丁醇)存在下能很方便地进行。优选步骤c)作为一釜法的一部分通过由式(Ⅳ)化合物的乙酸乙酯溶液制备正丁醇溶液,随后直接还原正丁醇溶液完成。还原优选采用硼氢化钠进行,而且能在不太高的温度下(适宜为20-40℃)方便地进行。还原优选分两阶段进行,第一阶段是在氮气氛中于大约25℃的温度下进行;第二阶段在大约30℃下进行。所产生的式(Ⅴ)化合物的正丁醇溶液尔后可用盐酸和氨干燥。干燥正丁醇溶液可直接用于步骤d)中,作为一釜法的一部分。
步骤d)优选用式(Ⅴ)化合物的干燥溶液(如干燥丁醇溶液)进行。这种干燥丁醇溶液最好通过干燥步骤c)中产生的正丁醇溶液制得。在进行环合反应之前,干燥正丁醇溶液最好先用木炭脱色。环合可以很方便地采用甲醇钠(较合适的是溶在醇类溶剂如甲醇中的溶液)进行。最优选环合采用30%甲醇钠的甲醇溶液进行。反应优选在升高的温度下,较适宜在50-120℃下进行。优选反应在大约85℃下进行。随后分离所产生的式(Ⅵ)化合物。该分离可通过标准离心、过滤和干燥方法完成。
步骤e)优选用分离的式(Ⅵ)化合物来进行。分离可采用例如公知的离心、过滤和干燥技术完成。重氮盐的形成可以在降低的温度下,优选在浓盐酸存在下,利用亚硝酸钠水溶液完成。优选盐的形成在降低的温度如0-5℃下进行。肼的形成随后采用亚硫酸钠作为还原剂由重氮盐溶液完成。亚硫酸钠适宜为水溶液形式。还原最好分两步进行:第一步是加入亚硫酸钠;第二步是加入盐酸。优选第一步在低于10℃的温度下进行。第二步优选在升高的温度如55-60℃下进行。
优选将步骤e)中产生的式(Ⅶ)化合物的溶液直接用于步骤f)中,即一釜法进行。步骤f)为费歇尔反应。已发现,此反应最好在比较高的稀释度下进行,以达到最高纯度的最终产物。因此,优选用水稀释得自步骤e)的溶液。随后加入4,4-二乙氧基-N,N-二甲基丁胺进行(适宜的是在氮气氛下)费歇尔反应。当加入4,4-二乙氧基-N,N-二甲基丁胺时,优选将稀释溶液的温度升高。合适的温度为75-105℃,优选大约90℃。反应优选回流进行。
当反应完成后,式(Ⅰ)化合物可用标准方法提取。较适宜的是冷却回流的反应产物,并用如氢氧化钠调节至大约pH7。随后用乙酸乙酯提取调节pH后的产物,并用氢氧化钠调节水层的pH至约10。尔后在大约50℃提取产物,继之用标准脱色、过滤、蒸馏、离心和干燥技术处理产物。
制备式(Ⅰ)化合物的特别优选的反应流程为: 
本发明的第二方面是提供了纯化(S)-4-{[3-(二甲氨基)乙基]-1H-吲哚-5-基]甲基}-2-噁唑烷酮的方法,该方法包括以下步骤:
a)将粗制(S)-4-{[3-(二甲氨基)乙基]-1H-吲哚-5-基]甲基}-2-噁唑烷酮溶于回流的乙醇/乙酸乙酯混合物中,并过滤热溶液;
b)缓慢冷却过滤溶液至约5℃;
c)离心步骤b)的产物,用乙酸乙酯洗涤,随后干燥;和
d)用丙酮处理除去溶剂化的乙酸乙酯。
优选回流混合物为10%乙醇/乙酸乙酯。在用助滤剂过滤之前,热溶液最好用脱色木炭脱色。
步骤b)的冷过滤液在离心之前最好搅拌较长时间,优选搅拌大约18小时。
步骤c)的干燥阶段优选在真空下进行。较适宜的是将产物在升至不太高的高温下进行。例如在40-60℃下,优选在大约50℃进行。
步骤c)的干燥固体产物可以很方便地用20%丙酮/水混合物在不太高的温度下(优选在15-30℃,例如室温下)处理。然后冷却所得悬浮液至不太低的低温,优选约5℃,并搅拌。随后离心产物,用乙酸乙酯洗涤,干燥(优选在真空下于大约45℃下进行)。
所得产物为高纯净非溶剂化固体。
本发明的第三方面提供了非溶剂化的纯净(S)-4-{[3-(二甲氨基乙基)-1H-吲哚-5-基]甲基}-2-噁唑烷酮。
本发明的第四方面提供了如上定义的式(Ⅲ),(Ⅳ),(Ⅴ)和(Ⅵ)化合物。
本发明更进一步地提供了如下所述的式(Ⅲ),(Ⅳ),(Ⅴ)和(Ⅵ)化合物的制备方法:
化合物(Ⅲ):本发明第一方面中的方法步骤a),并优选如第4页中所述;
化合物(Ⅳ):本发明第一方面中的方法步骤b),并优选如跨接第4和第5页段落中所述;
化合物(Ⅴ):本发明第一方面中的方法步骤c),并优选如第5页中所述;和
化合物(Ⅵ):本发明第一方面中的方法步骤d),并优选如第5页中所述。
本发明在此用下述实施例进一步说明。
实施例1大量制备(S)-4-[2-(二甲氨基)乙基]-1H-吲哚-5-基]甲基}-2-噁唑烷酮的方法。步骤1:4-硝基-(L)-苯丙氨酸甲酯盐酸盐的制备反应:物料 用量 摩尔数4-硝基-(L)-苯丙氨酸 100.0公斤 475.8甲醇 599.0升氯化氢 45.3公斤 1241.6甲醇(洗涤用) 66.8升方法:
在保持低于25℃的温度下,通过向装有甲醇的反应器内通入氯化氢气体制备氯化氢的甲醇溶液。向反应器内加入4-硝基-(L)-苯丙氨酸并回流约1小时。冷却至约0℃并离心产物(4-硝基-(L)-苯基丙氨酸甲酯盐酸盐)。用甲醇洗涤产物并于50℃下真空干燥。步骤2:(S)-N-丁氧基羰基-4-硝基苯丙氨酸甲酯的制备反应:物料 用量 摩尔数4-硝基-(L)-苯丙氨酸甲酯盐酸盐 45.0公斤 172.7碳酸钠 20.1公斤 189.6氯甲酸正丁酯 24.0公斤 175.8乙酸乙酯 248.0公斤水(软化水) 100.0公斤水(洗涤用) 50.0公斤方法
将软化水、4-硝基-(L)-苯丙氨酸甲酯盐酸盐、碳酸钠和乙酸乙酯加入到反应器内,并在搅拌下冷却内容物至约20℃。向反应混合物中加入氯甲酸正丁酯,同时维持温度为约30℃并搅拌约30分钟。分离水层并用水洗涤乙酸乙酯溶液。(S)-N-丁氧基羰基-4-硝基苯丙氨酸甲酯的乙酸乙酯溶液直接用于下步反应中。步骤3:(S)-N-丁氧基羰基-4-氨基苯丙氨酸甲酯的制备反应;物料 用量 摩尔数(S)-N-丁氧基羰基-4- 56.0公斤 172.7硝基苯丙氨酸甲酯乙酸乙酯 252.0公斤5%披钯木炭(55%水分) 5.0公斤乙酸乙酯(过滤洗涤用) 18.0公斤碳酸钠 12.5公斤水(软化水) 100.0公斤助滤剂 3.5公斤氢气 根据需要丁醇 247.1公斤方法
向反应器内加入5%披钯木炭催化剂、(S)-N-丁氧基羰基-4-硝基苯丙氨酸甲酯的乙酸乙酯溶液并在约20psi氢气压下氢化,维持温度在30-50℃之间。一旦反应完成之后,通过助滤剂滤除催化剂并用乙酸乙酯洗涤。用碳酸钠水溶液洗涤乙酸乙酯溶液。将(S)-N-丁氧基羰基-4-氨基苯丙氨酸甲酯的乙酸乙酯溶液部分蒸馏,加入丁醇并分馏混合物除去乙酸乙酯。此丁醇溶液直接用于下步反应中。步骤4:(S)-N-丁氧基羰基-4-氨基苯丙氨醇的制备反应:物料 用量 摩尔数(S)-N-丁氧羰基-4- 50.8公斤 172.8氨基苯丙氨酸甲酯正丁醇 305升硼氢化钠(总量) 6.5公斤 172.8浓盐酸 20.2升 300水(软化水-用于稀释酸) 20.2公斤水(软化水) 150.0公斤浓氨溶液(d=0.88) 14.6升方法;
向反应器内加入得自步骤(3)的(S)-N-丁氧基羰基-4-氨基苯丙氨酸甲酯的丁醇溶液,用正丁醇稀释至需要的体积。冷却反应器中的内容物至约25℃。在氮气氛下加入一半量硼氢化钠,同时保持反应温度为约25℃。搅拌3小时,然后加入另一半硼氢化钠。进一步搅拌混合物5小时并温热至35℃。尔后搅拌反应混合物约12小时,然后缓慢加入盐酸水溶液,期间保持温度为约30℃,用以分解任何过量的硼氢化钠。加水,温热至约35℃并加氨溶液调节pH至大约10。分离水层并在保持约35℃温度下,水洗有机层。蒸馏去一些丁醇,同时共沸干燥该溶液。干燥的丁醇溶液直接用于下步反应中。步骤5:(S)-4-(4-氨基苄基)-2-噁唑烷酮的制备反应物料 用量 摩尔数(S)-N-丁氧羰基-4-氨基苯丙氨醇 91.9公斤 345.0正丁醇 260.0升甲醇钠(30%重量甲醇溶液) 7.5公斤 4.7木炭 2.0公斤正丁醇(过滤洗涤用) 20.0公斤正丁醇(产物洗涤用) 30.0公斤助滤剂 2.0公斤方法
在反应器内加入得自步骤4的(S)-N-丁氧基羰基-4-氨基苯基丙氨醇的正丁醇溶液,加入脱色木炭。在约85℃下缓慢加入甲醇钠的甲醇溶液处理无水溶液。在约85℃下加热反应混合物,同时缓慢滴加甲醇钠的甲醇溶液。将反应混合物在85℃下进一步加热30分钟,然后趁热通过助滤剂过滤。在5-10℃下冷却溶液至少8小时后,离心混合物,用正丁醇洗涤过滤产物,并在约50℃下真空干燥。步骤6A:(S)-4-{3-[2-(二甲氨基)乙基]-1H-吲哚-5-基]甲基}-2-噁唑烷酮反应 物料 用量 摩尔数(S)-4-(4-氨基苄基)-2-噁唑烷酮 19.2公斤 100.0亚硝酸钠 6.9公斤 100.0亚硫酸钠 37.8公斤 300.0浓盐酸 66.6公斤4,4-二乙氧基-N,N-二甲基丁胺 19.0公斤 100.032%w/w氢氧化钠溶液 60.0公斤乙酸乙酯(总提取量) 303.0升木炭 2.9公斤水(软化水) 412.8公斤乙酸乙酯(洗涤用) 10.0升助滤剂(总使用量) 2.0公斤方法
物料 用量 摩尔数(S)-4-(4-氨基苄基)-2-噁唑烷酮 19.2公斤 100.0亚硝酸钠 6.9公斤 100.0亚硫酸钠 37.8公斤 300.0浓盐酸 66.6公斤4,4-二乙氧基-N,N-二甲基丁胺 19.0公斤 100.032%w/w氢氧化钠溶液 60.0公斤乙酸乙酯(总提取量) 303.0升木炭 2.9公斤水(软化水) 412.8公斤乙酸乙酯(洗涤用) 10.0升助滤剂(总使用量) 2.0公斤方法
向反应器内放人浓盐酸、软化水和(S)-4-(4-氨基苄基)-2-噁唑烷酮。冷却反应器中的内容物到0-5℃,在维持温度低于5℃的条件下加入亚硝酸钠水溶液。搅拌约30分钟后,向冰冷的亚硫酸钠水溶液中加入重氮盐溶液,期间维持温度低于10℃。搅拌15分钟后,缓慢加热所形成的混合物至约55-60℃,然后缓慢加入盐酸。将溶液在约60℃下保持约18小时。
用水稀释反应混合物并加热至约90℃。在氮气氛下缓慢加入4,4-二乙氧基-N,N-二甲基丁胺,而且加热回流约3小时。冷却,并利用氢氧化钠溶液调节混合物至约pH7。用乙酸乙酯提取,尔后再用氢氧化钠溶液调节水层至约pH10。在约50℃下用乙酸乙酯提取产物。用脱色木炭处理合并的乙酸乙酯提取液(其中含有产物),通过助滤剂过滤。蒸去大部分溶剂,将悬浮液冷却至约5℃。离心粗产物,用乙酸乙酯洗涤并在50℃下真空干燥。步骤6B:纯化(S)-4-{3-[2-(二甲氨基)乙基]-1H-吲哚-5-基]甲基}-2-噁唑烷酮物料 用量乙酸乙酯 109.4升 乙醇 12.3升木炭 2.4公斤乙酸乙酯(产物洗涤用) 5.0升丙酮 11.8升水(软化水) 47.3公斤水(软化水)(产物洗涤用) 10.0公斤助滤剂 2.0公斤
将步骤6A的粗产物溶于回流着的10%乙醇/乙酸乙酯混合物中,用脱色木炭处理并趁热通过助滤剂过滤。将溶液缓慢冷却至高于5℃并搅拌18小时。然后离心精制产物,用乙酸乙酯洗涤并于50℃下真空干燥。为除去溶剂化的乙酸乙酯,将干燥固体于室温下加到20%丙酮/水的混合物中,搅拌1小时。在离心产物之前将悬浮液冷却至约50℃,冷却约1小时,用乙酸乙酯洗涤并于约45℃下真空干燥。
实施例2(S)-N-丁氧基羰基-4-硝基苯丙氨酸甲酯(式(Ⅲ)化合物)的另一种制备方法
在无水条件下,将4-硝基-(L)-苯丙氨酸甲酯盐酸盐(40.00g,0.153摩尔)和碳酸氢钠(73g,0.870摩尔)在1,4-二噁烷(1000ml)中的混合物于大约10℃下搅拌。在10分钟内加入氯甲酸丁酯(23.12g,21.52ml,0.169摩尔)的1,4-二噁烷(200ml)溶液(反应温度大约13℃)。温热所得悬浮液至室温,搅拌3小时。将反应物缓慢加入到水(1600ml)中骤冷终止反应,然后用乙酸乙酯(3x650ml)提取。合并的乙酸乙酯提取液用盐水(1000ml)洗涤,用无水硫酸镁干燥,过滤,蒸发得到油。残留溶剂在50℃下用油泵除去,得到浆状物(51.34g,103%收率),放置后逐渐固化。TLC[SiO2,EtOAc]是单一的(Rf=0.59).1H NMR(60MHz,CDCl3)与氨基甲酸酯的结构一致。
实施例3(S)-N-丁氧基羰基-4-氨基苯丙氨酸甲酯(式(Ⅳ)化合物)的另一种制备方法
在氮气氛下,将实施例2中所制的化合物[45.00g,0.139mol]的乙醇(845ml)溶液加到潮湿的10%钯/炭(87L型,61.1%H2O)[-4.5g]中,在室温和标准大气压力下进行氢化反应。稳定吸收氢9小时(~9700ml)。通过硅藻土滤除催化剂并用乙醇(100ml)洗涤。真空浓缩滤液(水浴温度<40℃),并使用油泵除去最后痕量溶剂,得到棕色胶状物(41.70g,101%)。TLC[SiO2,EtOAc]显示为含有痕量最先洗脱出的杂质的所需产物(Rf=0.49)。1H NMR (300MHz,CDCl3)与产物和残留乙醇的结构一致。
实施例4(S)-N-丁氧基羰基-4-氨基苯丙氨醇(式(Ⅴ)化合物)的另一种制备方法
室温下,向搅拌着的硼氢化钠(14.80g,0.390mol)的SVM(150ml)悬浮液中逐滴加入实施例3所制化合物[76.40g,0.260mol]的SVM(460ml)溶液。将反应搅拌过夜(~18小时),尔后TLC(SiO2,EtOAc)表明起始物质已完全消耗。在用冰冷却至大约10℃下,用2M盐酸水溶液酸化反应混合物至~pH4。浓缩所得混合物,得固体残留物,缓慢加入饱和碳酸氢钠水溶液(2000ml)。含水混合物(pH~8)用乙酸乙酯(2×750ml)提取,并用硫酸镁干燥合并的有机提取液,过滤并浓缩,得到浅粉色蜡状固体(64.56g,93%收率)。TLC[SiO2,EtOAc]表明所需产物(Rf=0.33)中含有痕量杂质。1H NMR(60MHz,CDCl3)与丙氨醇的结构一致。
Claims (13)
1.式(Ⅲ)中间体:
2.式(Ⅳ)中间体:
3.式(Ⅴ)中间体: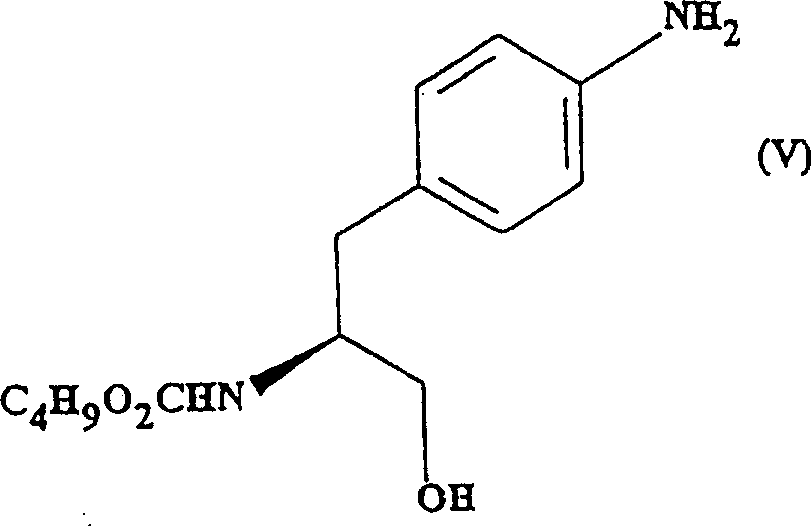
4.式(Ⅵ)中间体:
5.制备式(Ⅲ)化合物的方法: 该方法包括使式(Ⅱ)化合物与碳酸钠和氯甲酸正丁酯反应:
该方法包括使式(Ⅱ)化合物与碳酸钠和氯甲酸正丁酯反应:
6.制备式(Ⅳ)化合物的方法: 该方法包括还原式(Ⅲ)化合物:
该方法包括还原式(Ⅲ)化合物:
7.制备式(Ⅴ)化合物的方法: 该方法包括还原式(Ⅳ)化合物:
该方法包括还原式(Ⅳ)化合物:
8.制备式(Ⅵ)化合物的方法: 该方法包括环合式(Ⅴ)化合物:
该方法包括环合式(Ⅴ)化合物:
9.权利要求1中所述的中间体在制备用作药物的组合物中的应用。
10.权利要求2中所述的中间体在制备用作药物的组合物中的应用。
11.权利要求3中所述的中间体在制备用作药物的组合物中的应用。
12.权利要求4中所述的中间体在制备用作药物的组合物中的应用。
13.权利要求9至12中任一项的用途,其中所述组合物为用于治疗和预防偏头痛的组合物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB9516145.1A GB9516145D0 (en) | 1995-08-07 | 1995-08-07 | Improved chemical synthesis |
| GB9516145.1 | 1995-08-07 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN96197237A Division CN1092657C (zh) | 1995-08-07 | 1996-08-02 | 一釜法合成2-噁唑烷酮衍生物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1317480A true CN1317480A (zh) | 2001-10-17 |
| CN1142904C CN1142904C (zh) | 2004-03-24 |
Family
ID=10778867
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB011330929A Expired - Lifetime CN1244579C (zh) | 1995-08-07 | 1996-08-02 | 一釜法合成2-噁唑烷酮衍生物 |
| CN96197237A Expired - Lifetime CN1092657C (zh) | 1995-08-07 | 1996-08-02 | 一釜法合成2-噁唑烷酮衍生物 |
| CNB01112010XA Expired - Lifetime CN1142904C (zh) | 1995-08-07 | 1996-08-02 | 一釜法合成2-噁唑烷酮衍生物的中间体及其制备方法 |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB011330929A Expired - Lifetime CN1244579C (zh) | 1995-08-07 | 1996-08-02 | 一釜法合成2-噁唑烷酮衍生物 |
| CN96197237A Expired - Lifetime CN1092657C (zh) | 1995-08-07 | 1996-08-02 | 一釜法合成2-噁唑烷酮衍生物 |
Country Status (34)
| Country | Link |
|---|---|
| US (2) | US6084103A (zh) |
| EP (2) | EP1227095B1 (zh) |
| JP (6) | JP3729503B2 (zh) |
| KR (1) | KR100670704B1 (zh) |
| CN (3) | CN1244579C (zh) |
| AR (2) | AR006515A1 (zh) |
| AT (2) | ATE227723T1 (zh) |
| AU (1) | AU718413B2 (zh) |
| BR (1) | BR9609830A (zh) |
| CA (2) | CA2227039C (zh) |
| CZ (2) | CZ301603B6 (zh) |
| DE (2) | DE69624825T2 (zh) |
| DK (2) | DK1227095T3 (zh) |
| ES (2) | ES2185790T3 (zh) |
| GB (1) | GB9516145D0 (zh) |
| HK (2) | HK1047094B (zh) |
| HU (1) | HU229966B1 (zh) |
| IL (4) | IL156733A (zh) |
| IN (2) | IN185148B (zh) |
| MX (1) | MX9801044A (zh) |
| MY (1) | MY117571A (zh) |
| NO (2) | NO311690B1 (zh) |
| NZ (1) | NZ315040A (zh) |
| PL (1) | PL188805B1 (zh) |
| PT (2) | PT843672E (zh) |
| RO (2) | RO121816B1 (zh) |
| RU (1) | RU2167875C2 (zh) |
| SI (2) | SI0843672T1 (zh) |
| SK (1) | SK285052B6 (zh) |
| TR (1) | TR199800182T1 (zh) |
| TW (1) | TW358811B (zh) |
| UA (1) | UA53625C2 (zh) |
| WO (1) | WO1997006162A1 (zh) |
| ZA (1) | ZA966711B (zh) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001004088A1 (fr) * | 1999-07-09 | 2001-01-18 | Chugai Seiyaku Kabushiki Kaisha | Procede de preparation de derives d'amiline |
| ES2204302B2 (es) | 2002-08-07 | 2005-03-01 | Laboratorios Vita, S.A. | Procedimiento para la obtencion de un compuesto farmaceuticamente activo. |
| WO2005075467A2 (en) * | 2004-02-06 | 2005-08-18 | Ciba Specialty Chemicals Holding Inc. | Crystalline forms of zolmitriptan |
| EP1812428A2 (en) * | 2004-11-19 | 2007-08-01 | Teva Pharmaceutical Industries Ltd | Zolmitriptan crystal forms |
| CN101248184B (zh) | 2005-08-25 | 2013-01-16 | 宇部兴产株式会社 | 光学活性(S或R)-α-氨基酸及光学活性(S或R)-α-氨基酸酯的制造方法 |
| EP1981860B1 (en) | 2006-01-19 | 2011-05-25 | Matrix Laboratories Ltd | Conversion of aromatic diazonium salt to aryl hydrazine |
| WO2008007390A2 (en) * | 2006-07-10 | 2008-01-17 | Natco Pharma Limited | An improved process for purification of zolmitriptan |
| WO2008018090A2 (en) * | 2006-08-09 | 2008-02-14 | Matrix Laboratories Ltd | An improved process for the preparation of zolmitriptan |
| CZ2007158A3 (cs) * | 2007-02-26 | 2008-10-22 | Zentiva, A. S. | Zpusob prípravy zolmitriptanu |
| CZ301538B6 (cs) * | 2007-02-26 | 2010-04-07 | Zentiva, A. S. | Zpusob prípravy zolmitriptanu |
| CN101883766A (zh) * | 2007-10-03 | 2010-11-10 | 基因里克斯(英国)有限公司 | 制备佐米曲普坦、其盐和溶剂化物的方法 |
| CN101230047B (zh) * | 2008-02-04 | 2010-08-04 | 江苏中威药业有限公司 | 一种4-取代手性噁唑烷酮类化合物的制备方法 |
| US8906949B2 (en) | 2010-05-21 | 2014-12-09 | Sanovel Ilac Sanayi Ve Ticaret Anonim Sirketi | Orally disintegrating tablets of zolmitriptan and process for preparing the same |
| CN101948443B (zh) * | 2010-08-17 | 2012-08-22 | 河南师范大学 | 一种简单、高效合成4-取代噁唑烷酮衍生物的方法 |
| EP2691396B1 (en) | 2011-03-30 | 2016-08-10 | Brown University | Enopeptins, uses thereof, and methods of synthesis thereto |
| US9006453B2 (en) | 2011-09-02 | 2015-04-14 | Emcure Pharmaceuticals Limited | Process for preparation of zolmitriptan |
| CN102964270B (zh) * | 2012-11-21 | 2015-01-07 | 合肥星宇化学有限责任公司 | 一种亚硫酸钠还原重氮盐合成肼的方法 |
| CN103275075B (zh) * | 2013-06-24 | 2015-01-07 | 成都天台山制药有限公司 | 佐米曲普坦及其制备方法 |
| WO2016037072A2 (en) * | 2014-09-04 | 2016-03-10 | Brown University | Enopeptin analogas and methods of use thereof |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8724912D0 (en) * | 1987-10-23 | 1987-11-25 | Wellcome Found | Indole derivatives |
| YU48855B (sh) * | 1990-06-07 | 2002-06-19 | The Wellcome Foundation Limited | Heterociklična jedinjenja i njihovi derivati koji su modifikatori dejstva 5-hidroksi-triptamina i postupak njihovog dobijanja |
| GB9401436D0 (en) * | 1994-01-26 | 1994-03-23 | Wellcome Found | Therapeutic heterocyclic compounds |
| GB9423682D0 (en) * | 1994-11-23 | 1995-01-11 | Merck Sharp & Dohme | Therapeutic agents |
| CA2207201A1 (en) * | 1994-12-06 | 1996-06-13 | Caroline Henry | Azetidine, pyrrolidine and piperidine derivatives as 5ht1 receptor agonists |
-
1995
- 1995-08-07 GB GBGB9516145.1A patent/GB9516145D0/en active Pending
-
1996
- 1996-02-08 UA UA98031096A patent/UA53625C2/uk unknown
- 1996-08-02 BR BR9609830A patent/BR9609830A/pt not_active Application Discontinuation
- 1996-08-02 PT PT96926465T patent/PT843672E/pt unknown
- 1996-08-02 CN CNB011330929A patent/CN1244579C/zh not_active Expired - Lifetime
- 1996-08-02 DE DE69624825T patent/DE69624825T2/de not_active Expired - Lifetime
- 1996-08-02 MX MX9801044A patent/MX9801044A/es unknown
- 1996-08-02 SI SI9630560T patent/SI0843672T1/xx unknown
- 1996-08-02 RO ROA200400914A patent/RO121816B1/ro unknown
- 1996-08-02 HU HU9900188A patent/HU229966B1/hu unknown
- 1996-08-02 ES ES96926465T patent/ES2185790T3/es not_active Expired - Lifetime
- 1996-08-02 IL IL156733A patent/IL156733A/en not_active IP Right Cessation
- 1996-08-02 DK DK02008427T patent/DK1227095T3/da active
- 1996-08-02 AT AT96926465T patent/ATE227723T1/de active
- 1996-08-02 WO PCT/GB1996/001885 patent/WO1997006162A1/en not_active Application Discontinuation
- 1996-08-02 RU RU98104085/04A patent/RU2167875C2/ru active
- 1996-08-02 JP JP50822697A patent/JP3729503B2/ja not_active Expired - Lifetime
- 1996-08-02 PL PL96324881A patent/PL188805B1/pl unknown
- 1996-08-02 DE DE69636107T patent/DE69636107T2/de not_active Expired - Lifetime
- 1996-08-02 NZ NZ315040A patent/NZ315040A/xx not_active IP Right Cessation
- 1996-08-02 EP EP02008427A patent/EP1227095B1/en not_active Expired - Lifetime
- 1996-08-02 EP EP96926465A patent/EP0843672B1/en not_active Expired - Lifetime
- 1996-08-02 CN CN96197237A patent/CN1092657C/zh not_active Expired - Lifetime
- 1996-08-02 AU AU66634/96A patent/AU718413B2/en not_active Expired
- 1996-08-02 SI SI9630738T patent/SI1227095T1/sl unknown
- 1996-08-02 PT PT02008427T patent/PT1227095E/pt unknown
- 1996-08-02 IL IL148030A patent/IL148030A/en not_active IP Right Cessation
- 1996-08-02 RO RO98-00214A patent/RO119618B1/ro unknown
- 1996-08-02 IL IL12317196A patent/IL123171A/xx not_active IP Right Cessation
- 1996-08-02 AT AT02008427T patent/ATE325121T1/de active
- 1996-08-02 DK DK96926465T patent/DK0843672T3/da active
- 1996-08-02 TR TR1998/00182T patent/TR199800182T1/xx unknown
- 1996-08-02 SK SK154-98A patent/SK285052B6/sk not_active IP Right Cessation
- 1996-08-02 CA CA002227039A patent/CA2227039C/en not_active Expired - Lifetime
- 1996-08-02 US US09/011,045 patent/US6084103A/en not_active Expired - Lifetime
- 1996-08-02 KR KR1019980700833A patent/KR100670704B1/ko not_active IP Right Cessation
- 1996-08-02 ES ES02008427T patent/ES2261545T3/es not_active Expired - Lifetime
- 1996-08-02 CA CA2572508A patent/CA2572508C/en not_active Expired - Lifetime
- 1996-08-02 CZ CZ20031498A patent/CZ301603B6/cs not_active IP Right Cessation
- 1996-08-02 CN CNB01112010XA patent/CN1142904C/zh not_active Expired - Lifetime
- 1996-08-02 CZ CZ1998352A patent/CZ293050B6/cs not_active IP Right Cessation
- 1996-08-06 MY MYPI96003220A patent/MY117571A/en unknown
- 1996-08-06 IN IN1742DE1996 patent/IN185148B/en unknown
- 1996-08-07 AR ARP960103909A patent/AR006515A1/es active IP Right Grant
- 1996-08-07 ZA ZA9606711A patent/ZA966711B/xx unknown
- 1996-10-01 TW TW085111923A patent/TW358811B/zh not_active IP Right Cessation
-
1998
- 1998-02-06 NO NO19980522A patent/NO311690B1/no not_active IP Right Cessation
- 1998-08-19 HK HK02108548.8A patent/HK1047094B/zh not_active IP Right Cessation
- 1998-08-19 HK HK98110000A patent/HK1009129A1/xx not_active IP Right Cessation
-
2000
- 2000-02-02 US US09/496,409 patent/US6160123A/en not_active Expired - Lifetime
- 2000-04-25 IN IN454DE2000 patent/IN189756B/en unknown
- 2000-10-16 NO NO20005187A patent/NO311691B1/no not_active IP Right Cessation
-
2001
- 2001-07-27 JP JP2001228781A patent/JP2002037786A/ja not_active Withdrawn
- 2001-07-27 JP JP2001228782A patent/JP3739678B2/ja not_active Expired - Lifetime
-
2002
- 2002-02-06 IL IL14803002A patent/IL148030A0/xx unknown
- 2002-03-26 JP JP2002086955A patent/JP2002308858A/ja not_active Withdrawn
-
2003
- 2003-05-02 AR ARP030101543A patent/AR046236A2/es active IP Right Grant
-
2005
- 2005-11-18 JP JP2005334990A patent/JP4634286B2/ja not_active Expired - Lifetime
-
2006
- 2006-05-31 JP JP2006152810A patent/JP2006225406A/ja not_active Withdrawn
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1317480A (zh) | 一釜法合成2-噁唑烷酮衍生物 | |
| EP0512901B1 (fr) | Composés polycycliques aminés et leurs énantiomères, procédé pour leur préparation et compositions pharmaceutiques en contenant | |
| WO2005037792A1 (fr) | Derives de n-``phenyl(piperidin-2-yl)methyl !benzamide, leur preparation et leur application en therapeutique | |
| FR2688219A1 (fr) | Sels d'ammonium quaternaires de composes aromatiques amines, leur preparation et compositions pharmaceutiques les contenant. | |
| EP0700386A1 (fr) | Antagonistes des recepteurs des neurokinines | |
| EP0014997B1 (en) | 4-aryloxy- and 4-arylthio-3-phenylpiperidine derivatives, a process for the preparation of such compounds and pharmaceutical compositions containing them | |
| CN1168134A (zh) | 用于生产左旋丁哌卡因以及相关的哌啶甲酰苯胺麻醉剂的外消旋化方法 | |
| US20060264643A1 (en) | One Pot Synthesis of 2-Oxazolidinone Derivatives | |
| RU2072354C1 (ru) | Способ стереоселективного получения асимметрично алкилированных производных оксиндола | |
| JP3729504B2 (ja) | 2−オキサゾリジノン誘導体のワンポット合成 | |
| WO1999001435A1 (fr) | Derives de (1h-imidazol-4-yl)piperidine, leur preparation et leur application en therapeutique | |
| WO1995010501A1 (fr) | Allylaminoesters, leur procede de preparation et leur application en therapeutique | |
| FR2738567A1 (fr) | Derives de alpha-phenylpiperidine-1-propanol, leur preparation et leur application en therapeutique | |
| BE820827A (fr) | Nouveaux derives indoliques |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CX01 | Expiry of patent term |
Granted publication date: 20040324 |
|
| EXPY | Termination of patent right or utility model |