WO2014034156A1 - 薄片化黒鉛・樹脂複合材料及びその製造方法 - Google Patents
薄片化黒鉛・樹脂複合材料及びその製造方法 Download PDFInfo
- Publication number
- WO2014034156A1 WO2014034156A1 PCT/JP2013/053470 JP2013053470W WO2014034156A1 WO 2014034156 A1 WO2014034156 A1 WO 2014034156A1 JP 2013053470 W JP2013053470 W JP 2013053470W WO 2014034156 A1 WO2014034156 A1 WO 2014034156A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- graphite
- exfoliated graphite
- composite material
- resin composite
- polymer
- Prior art date
Links
- 0 CCCC=*N(*)[N+](*)[O-] Chemical compound CCCC=*N(*)[N+](*)[O-] 0.000 description 1
Images
















Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/44—Polymerisation in the presence of compounding ingredients, e.g. plasticisers, dyestuffs, fillers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F292/00—Macromolecular compounds obtained by polymerising monomers on to inorganic materials
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/22—Expanded, porous or hollow particles
- C08K7/24—Expanded, porous or hollow particles inorganic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
Definitions
- the present invention relates to exfoliated graphite / resin composite material obtained by exfoliating graphite or primary exfoliated graphite and a method for producing the same.
- Graphite is a laminated body in which many graphenes are laminated. By exfoliating graphite, exfoliated graphite having fewer graphene layers than graphite can be obtained. The exfoliated graphite is expected to be applied to conductive materials and heat conductive materials.
- Patent Document 1 discloses a method for producing graphene in which a polymer is grafted by co-polymerizing exfoliated graphene and a radical polymerizable monomer.
- the main object of the present invention is to provide exfoliated graphite / resin composite material that is highly dispersible in resin and easy to handle.
- the exfoliated graphite / resin composite material according to the present invention is a exfoliated graphite / resin composite material in which exfoliated graphite and a resin are combined.
- the exfoliated graphite / resin composite material according to the first invention of the present application when the methylene blue adsorption amount ( ⁇ mol / g) measured by the following method is y and the BET specific surface area (m 2 / g) is x The ratio y / x is 0.15 or more, and the BET specific surface area is 25 m 2 / g or more.
- the amount of methylene blue adsorbed is the absorbance of the methylene blue methanol solution having a concentration of 10 mg / L, and the exfoliated graphite / resin composite material is added to the methylene blue methanol solution, stirred, and then centrifuged. Measured based on the difference from absorbance.
- the method for measuring the amount of adsorbed methylene blue will be described in detail later in the description of the embodiment.
- the exfoliated graphite / resin composite material according to the second invention of the present application is a exfoliated graphite / resin composite material in which exfoliated graphite and a resin are combined, and the thermal decomposition start temperature and thermal decomposition end of the resin in the composite material The temperatures are higher than the thermal decomposition start temperature and the thermal decomposition end temperature of the resin before compounding, respectively.
- the resin content is 1% by mass to 70% by mass.
- the resin is a polymer of a radical polymerizable monomer.
- the method for producing exfoliated graphite / resin composite material according to the present invention is the above method for producing exfoliated graphite / resin composite material.
- a composition containing graphite or primary exfoliated graphite and a polymer, wherein the polymer is fixed to graphite or primary exfoliated graphite is prepared.
- the graphite or primary exfoliated graphite is peeled off while leaving a part of the polymer.
- the polymer in the step of preparing the composition, is grafted or adsorbed on graphite or primary exfoliated graphite. Fixed to graphite or primary exfoliated graphite.
- the step of preparing the composition includes the step of preparing a mixture containing graphite or primary exfoliated graphite and a radical polymerizable monomer. And polymerizing the radical polymerizable monomer contained in the mixture to produce a polymer in which the radical polymerizable monomer is polymerized in the mixture, and grafting the polymer to graphite or primary exfoliated graphite. .
- the polymer in the step of preparing the composition, is subjected to 50 ° C. or more and 400 ° C. or less in the presence of graphite or primary exfoliated graphite.
- the polymer is grafted to graphite or primary exfoliated graphite by heating to a temperature in the temperature range.
- the composition in the step of preparing the composition, further includes a thermally decomposable foaming agent.
- a thermally decomposable foaming agent in that case, graphite or primary exfoliated graphite can be more effectively exfoliated. Therefore, the specific surface area of the exfoliated graphite obtained can be further increased.
- the pyrolytic foam contained in the mixture Thermally decompose the agent.
- a step of generating a polymer and grafting the polymer to graphite or primary exfoliated graphite is performed.
- the polymerization is performed by polymerizing a radical polymerizable monomer contained in the mixture by heating the mixture.
- both the polymerization of the radical polymerizable monomer and the polymerization of the polymer can be performed only by heating the mixture. Therefore, graphite or primary exfoliated graphite can be more easily exfoliated.
- the radical polymerizable monomer is a vinyl monomer.
- the vinyl monomer is a styrene monomer or glycidyl methacrylate. Since styrene monomer is inexpensive, the production cost of exfoliated graphite / resin composite material can be reduced.
- a styrene monomer can be suitably used as the radical polymerizable monomer.
- a polymer of glycidyl methacrylate is preferably used as the polymer.
- the exfoliated graphite / resin composite material according to the present invention has a large specific surface area because the space between the graphenes is widened.
- the exfoliated graphite / resin composite material according to the present invention has a structure in which the central portion has a graphite structure and the graphene is widened at the edge portion so that the flakes are exfoliated. For this reason, it is easier to handle than conventional exfoliated alloys.
- the exfoliated graphite / resin composite material according to the present invention contains a resin, it has high dispersibility in other resins.
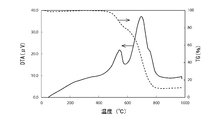
- FIG. 1 is a diagram showing a TG / DTA measurement result of polystyrene in Example 1.
- FIG. 2 is a diagram showing the TG / DTA measurement results of polyvinyl acetate in Example 2.
- FIG. 3 is a diagram showing a TG / DTA measurement result of polypropylene glycol in Example 3.
- FIG. 4 is a diagram showing a TG / DTA measurement result of the exfoliated graphite / resin composite material obtained in Example 1.
- FIG. 5 is a diagram showing a TG / DTA measurement result of the exfoliated graphite / resin composite material obtained in Example 2.
- FIG. 6 is a diagram showing a TG / DTA measurement result of the exfoliated graphite / resin composite material obtained in Example 3.
- FIG. 7 is an XRD spectrum of the exfoliated graphite / resin composite material obtained in Example 1.
- FIG. 8 is an XRD spectrum of the exfoliated graphite / resin composite material obtained in Example 2.
- FIG. 9 is an XRD spectrum of the exfoliated graphite / resin composite material obtained in Example 3.
- FIG. 10 is a photograph of the exfoliated graphite obtained in Example 1 taken with a scanning electron microscope (SEM).
- FIG. 11 is a photograph of the exfoliated graphite obtained in Example 2 taken with a scanning electron microscope (SEM).
- FIG. 12 is a photograph of the exfoliated graphite obtained in Example 3 taken with a scanning electron microscope (SEM).
- SEM scanning electron microscope
- FIG. 13 is a diagram showing a TG / DTA measurement result of the exfoliated graphite / resin composite material obtained in Reference Example 1.
- FIG. 14 is a diagram showing a TG / DTA measurement result of the exfoliated graphite / resin composite material obtained in Reference Example 2.
- FIG. 15 is a diagram showing a TG / DTA measurement result of the exfoliated graphite / resin composite material obtained in Reference Example 3.
- FIG. FIG. 16 shows the BET specific surface area (m2 / g) and exfoliated amount of methylene blue ( ⁇ mol / g) in the exfoliated graphite / resin composite materials and the known carbonaceous materials obtained in Examples 4 to 14 and Reference Examples 4 to 7. It is a figure which shows the relationship with g).
- FIG. 17 is an enlarged view of a range surrounded by a dotted line in FIG.
- the exfoliated graphite / resin composite material according to the present invention is a composite of exfoliated graphite and a resin.
- Exfoliated graphite / resin composite material includes exfoliated graphite and resin.
- Exfoliated graphite contained in exfoliated graphite / resin composite material is obtained by exfoliating at least part of the graphene layer of graphite and primary exfoliated graphite as raw materials.
- Graphite is a laminate of a plurality of graphene layers, such as natural graphite, artificial graphite, and expanded graphite.
- expanded graphite is preferable. Expanded graphite can be easily peeled off because the interlayer of the graphene layer is larger than that of normal graphite. Therefore, exfoliated graphite / resin composite material can be easily manufactured by using expanded graphite as graphite.
- the number of graphene layers is from 100,000 to 1,000,000, and the specific surface area by BET is 22 m 2 / g or less.
- the exfoliated graphite / resin composite material of the present invention refers to a graphene laminate having 3000 layers or less.
- the ratio y / x is 0. .15 or more, more preferably 0.27 or more, and more preferably 0.39 or more.
- the above methylene blue adsorption amount is determined by the following method.
- the exfoliated graphite / resin composite material is put into a methanol solution of methylene blue having a concentration of 10 mg / L and stirred. Next, the mixture is centrifuged, and the change in absorbance at the maximum absorption wavelength of the obtained supernatant is observed.
- Methylene blue is adsorbed to the portion of exfoliated graphite / resin composite material where graphene is laminated by ⁇ conjugation.
- methylene blue emits fluorescence when irradiated with light. When methylene blue is adsorbed on graphene, it does not emit fluorescence. That is, the fluorescence intensity is reduced. Therefore, the amount of methylene blue adsorbed can be determined from the amount of decrease in the fluorescence intensity obtained from the supernatant with respect to the fluorescence intensity of the original methylene blue.
- the ratio y / x is 0.15 or more as described above.
- y 0.15 or more.
- the exfoliated graphite / resin composite material according to the first present invention is distinguished from the conventional spherical graphite particles. That is, compared with the conventionally known spherical graphite, the exfoliated graphite / resin composite material according to the present invention has a higher adsorption amount of methylene blue while having the same BET specific surface area. This is presumably because the exfoliated graphite / resin composite material according to the present invention is somewhat condensed in a dry state, but spreads between graphenes in a wet state such as in methanol as compared with a dry state.
- the thermal decomposition start temperature and the thermal decomposition end temperature of the resin in the exfoliated graphite / resin composite material are higher than the thermal decomposition start temperature and the thermal decomposition end temperature of the resin before the composite, respectively.
- the pyrolysis start temperature and the pyrolysis end temperature refer to a TGA measurement-dependent decomposition start temperature and a decomposition end point temperature, respectively.
- the exfoliated graphite / resin composite material In the exfoliated graphite / resin composite material according to the first and second inventions of the present application, not only is the space between graphenes widened, but also the resin is composited with exfoliated graphite. Therefore, it is excellent in dispersibility when added to a resin material. Furthermore, when adding to resin, it is desirable to add in the form of a dispersion in which exfoliated graphite / resin composite material is dispersed in an organic solvent. In the dispersion, the space between the graphenes spreads, and aggregation can be suppressed. Therefore, even when the addition amount of exfoliated graphite / resin composite material is reduced, it can be more uniformly dispersed in the resin.
- the exfoliated graphite / resin composite material according to the present invention is prepared as a dispersion liquid or a solution dispersed or dissolved in an organic solvent.
- the BET specific surface area of the exfoliated graphite / resin composite material is 25 m 2 / g or more, preferably 35 m 2 / g or more, more preferably 45 m 2 / g or more, and 100 m 2 / g or more. It is more preferable.
- the upper limit of the BET specific surface area of exfoliated graphite / resin composite material is usually 2500 m 2 / g or less.
- Exfoliated graphite may be obtained by using primary exfoliated graphite instead of graphite as a raw material.
- Primary exfoliated graphite is exfoliated graphite obtained by exfoliating graphite, exfoliated graphite / resin composite material of the present invention, and exfoliated graphite obtained by exfoliating graphite by various methods described below. It shall widely contain graphite. Since primary exfoliated graphite is obtained by exfoliating graphite, the specific surface area may be larger than that of graphite.
- the resin contained in the exfoliated graphite / resin composite material is preferably a polymer of a radical polymerizable monomer.
- the resin may be a copolymer of a plurality of types of radical polymerizable monomers, or may be a homopolymer of one type of radical polymerizable monomer.
- the radical polymerizable monomer is not particularly limited as long as it is a monomer having a radical polymerizable functional group.
- radical polymerizable monomer examples include styrene, methyl ⁇ -ethyl acrylate, methyl ⁇ -benzyl acrylate, methyl ⁇ - [2,2-bis (carbomethoxy) ethyl] acrylate, dibutyl itaconate, dimethyl itaconate.
- the exfoliated graphite / resin composite material of the present invention is characterized by being relatively difficult to scatter. This is considered to be because, as will be described later, the polymer obtained by polymerizing the radical polymerizable monomer remains without being completely decomposed in the thermal decomposition step. In other words, it is considered that the polymer located in the portion sandwiched between the graphene layers in exfoliated graphite is not completely decomposed near the thermal decomposition temperature because it is sandwiched between the graphenes on both sides. Therefore, the exfoliated graphite / resin composite material of the present invention is easy to handle.
- the exfoliated graphite / resin composite material according to the present invention has a large inter-layer distance between graphenes and a large specific surface area. Furthermore, the exfoliated graphite / resin composite material according to the present invention has a structure in which the central part has a graphite structure and the edge part is exfoliated. For this reason, it is easier to handle than conventional exfoliated alloys. Moreover, since the exfoliated graphite / resin composite material according to the present invention contains a resin, it has high dispersibility in other resins. In particular, when the other resin is a resin having a high affinity with the resin contained in the exfoliated graphite / resin composite material, the dispersibility of the exfoliated graphite / resin composite material in the other resin is higher.
- Such exfoliated graphite / resin composite material can be produced by, for example, the following production method.
- the steps of preparing this composition include, for example, the following first and second methods for fixing a polymer to graphite or primary exfoliated graphite by grafting the polymer to graphite or primary exfoliated graphite, By adsorbing graphite or primary exfoliated graphite, a third method of fixing the polymer to graphite or primary exfoliated graphite can be used.
- First method In the first method, first, as a raw material, a mixture containing the above graphite or primary exfoliated graphite and the above radical polymerizable monomer is prepared. Next, the radical polymerizable monomer contained in the mixture is polymerized to produce a polymer in which the radical polymerizable monomer is polymerized in the mixture, and the polymer is grafted to graphite or primary exfoliated graphite.
- a composition containing graphite or primary exfoliated graphite and a radical polymerizable monomer is prepared.
- the blending ratio of graphite and radical polymerizable monomer is not particularly limited, but is preferably 1: 1 to 1: 100 by mass ratio. By setting the blending ratio in the range, it is possible to effectively exfoliate graphite or primary exfoliated graphite, and to obtain exfoliated graphite / resin composite material more effectively.
- a composition further including a thermally decomposable foaming agent that generates a gas upon thermal decomposition is prepared.
- the graphite or primary exfoliated graphite can be more effectively exfoliated by heating described later.
- the thermal decomposable foaming agent is not particularly limited as long as it is a compound that spontaneously decomposes by heating and generates a gas upon decomposition.
- the thermally decomposable foaming agent include foaming agents such as azocarboxylic acid-based, diazoacetamide-based, azonitrile compound-based, benzenesulfohydrazine-based or nitroso compound-based which generate nitrogen gas during decomposition, carbon monoxide during decomposition, A foaming agent that generates carbon dioxide, methane, aldehyde, or the like can be used.
- the above pyrolyzable foaming agents may be used alone or in combination of a plurality of types of foaming agents.
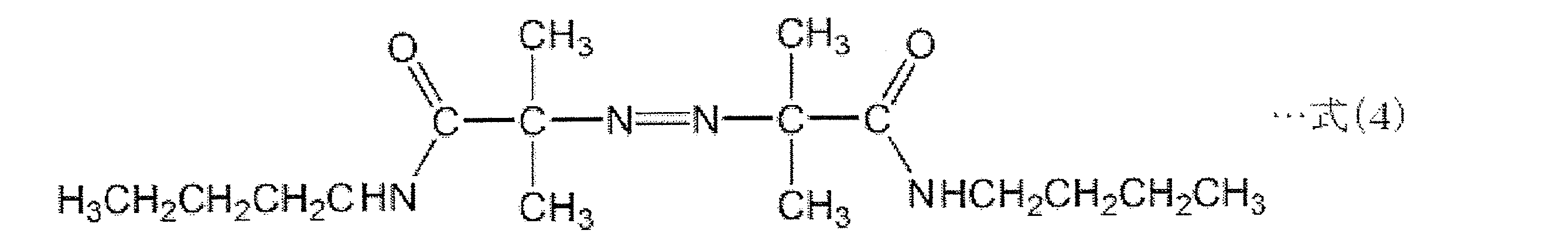
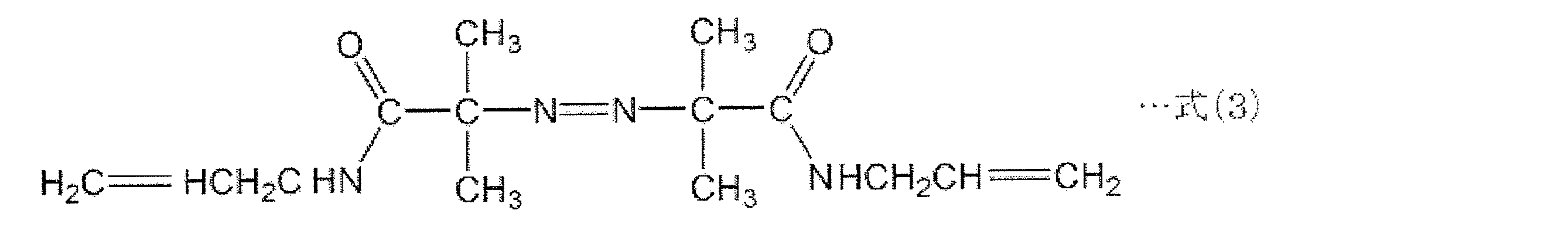
- thermally decomposable foaming agent azodicarbonamide (ADCA) having a structure represented by the following formula (1), or a foaming agent having a structure represented by the following formulas (2) to (4): Can be used.
- ADCA azodicarbonamide
- foaming agents decompose spontaneously by heating, and generate nitrogen gas during decomposition.
- the thermal decomposition temperature of the thermally decomposable foaming agent is not particularly limited, and may be lower or higher than the temperature at which the radical polymerizable monomer spontaneously starts polymerization.
- the thermal decomposition temperature of ADCA having the structure represented by the above formula (1) is 210 ° C.
- the temperature at which styrene spontaneously starts polymerization is higher than 150 ° C. High temperature.
- the thermal decomposition start temperatures of the thermally decomposable foaming agents having the structures represented by the above formulas (2) to (4) are 88 ° C., 96 ° C., and 110 ° C. in order, and these are the temperatures at which styrene spontaneously starts polymerization.
- the temperature is lower than 150 ° C.
- the mixing ratio of the graphite or primary exfoliated graphite and the thermally decomposable foaming agent is not particularly limited, but the pyrolyzable foaming agent is 100 parts by weight to 300 parts by weight with respect to 100 parts by weight of the graphite or primary exfoliated graphite. It is preferable to blend partly.
- the thermally decomposable foaming agent in the above range, the graphite or primary exfoliated graphite can be more effectively exfoliated, and the exfoliated graphite / resin composite material can be obtained effectively.
- the method for preparing the composition is not particularly limited, and examples thereof include a method in which the radical polymerizable monomer is used as a dispersion medium and the graphite or primary exfoliated graphite is dispersed in the radical polymerizable monomer.
- the composition further containing the thermally decomposable foaming agent can be prepared by dissolving or dispersing the thermally decomposable foaming agent in the radical polymerizable monomer.
- both the polymerization of the radical polymerizable monomer and the thermal decomposition of the polymer described later can be performed only by heating the composition. . Accordingly, the graphite or primary exfoliated graphite can be more easily separated.
- the thermal decomposition start temperature and the thermal decomposition end temperature of the resin in the exfoliated graphite / resin composite material obtained by thermal decomposition are higher than the thermal decomposition start temperature and the thermal decomposition end temperature of the resin before the composite, respectively.
- the heating method is not particularly limited as long as it can be heated to the thermal decomposition temperature of the polymer, and the composition can be heated by an appropriate method and apparatus. Moreover, in the case of the said heating, you may heat without sealing, ie, a normal pressure. Therefore, exfoliated graphite can be produced inexpensively and easily.
- Thermal decomposition so that the resin remains can be achieved by adjusting the heating time. That is, the amount of residual resin can be increased by shortening the heating time. Also, the amount of residual resin can be increased by lowering the heating temperature.
- a radical polymerizable monomer was polymerized in the presence of graphite or primary exfoliated black to produce a polymer, and grafting of the polymer to graphite or primary exfoliated graphite was attempted.
- a polymer radical generated by thermally decomposing a polymer is obtained by heating the polymer obtained in advance to the specific temperature range in the presence of graphite or primary exfoliated graphite. It can be grafted directly to graphite or primary exfoliated graphite.
- an appropriate pyrolytic radical generating polymer can be used as the polymer of the second method.
- polymers containing halogen elements such as chlorine such as polyvinyl chloride, chlorinated vinyl chloride resin, ethylene fluoride resin, vinylidene fluoride resin, and vinylidene chloride resin
- halogen elements such as chlorine
- polyvinyl chloride, chlorinated vinyl chloride resin, ethylene fluoride resin, vinylidene fluoride resin, and vinylidene chloride resin can be used.
- Ethylene vinyl acetate copolymer (EVA), polyvinyl acetal, polyvinyl pyrrolidone and copolymers thereof can also be used.
- Polymers obtained by cationic polymerization such as polyisobutylene and polyalkylene ether can also be used.
- Polyurethane, epoxy resin, modified silicone resin, silicone resin, etc. formed by crosslinking oligomers can also be used.
- Polyallylamine may be used, and in that case, an amino group can be grafted to graphite or primary exfoliated graphite.
- Polyvinylphenol or polyphenols may be used, in which case the phenolic OH can be grafted to graphite or primary exfoliated graphite.
- the phosphate group can be grafted.
- condensation polymers such as polyester and polyamide may be used.
- the decomposition product is grafted although the radical concentration obtained at the decomposition temperature is low.
- the blending ratio of the graphite or primary exfoliated graphite and the polymer is not particularly limited, but it is desirable that the weight ratio is 1: 5 to 1:20. By setting the blending ratio within this range, graphite or primary exfoliated graphite can be more effectively exfoliated, and exfoliated graphite / resin composite material can be obtained effectively.
- the step of preparing the composition in the step of preparing the composition, it is preferable to further include a thermally decomposable foaming agent in the composition.
- the graphite or primary exfoliated graphite can be more effectively exfoliated by heating that causes thermal decomposition of the polymer described later.
- the thermally decomposable foaming agent that can be used is the same as in the first method. Therefore, it is preferable to use a foaming agent having a structure represented by the above formulas (1) to (4).
- the blending ratio of graphite or primary exfoliated graphite and the pyrolyzable foaming agent is not particularly limited, but the pyrolyzable foaming agent is 100 to 300 per 100 parts by weight of graphite or primary exfoliated graphite. It is preferable to mix
- a composition in which a polymer is adsorbed on graphite or primary exfoliated graphite in a solvent is prepared as the above composition.
- the method for adsorbing the polymer to graphite or primary exfoliated graphite is not particularly limited. Since the polymer has adsorptivity to graphite, a method of mixing graphite or primary exfoliated graphite with the polymer in the above-described solvent can be used.
- ultrasonic treatment is performed in order to effectively adsorb the polymer by graphite or primary exfoliated graphite.
- the ultrasonic processing method is not particularly limited. For example, a method of irradiating an ultrasonic wave having an oscillation frequency of about 100 W and an oscillation frequency of about 28 kHz using an appropriate ultrasonic processing apparatus can be used.
- the sonication time is not particularly limited as long as it is longer than the time required for the polymer to be adsorbed on graphite.
- the sonication time is preferably maintained for 30 minutes, 60 minutes, more preferably about 120 minutes.
- the thermal decomposition temperature of polystyrene is about 380 ° C. to 450 ° C.
- the thermal decomposition temperature of polyglycidyl methacrylate is about 400 ° C. to 500 ° C.
- the thermal decomposition temperature of polybutyral is about 550 ° C. to 600 ° C. in the atmosphere. It is.
- the heating for polymerizing the radical polymerizable monomer and the thermal decomposition of the polymer may be carried out continuously in the same heating step, but in the second method, Alternatively, a heating step for grafting the polymer onto graphite or primary exfoliated graphite and a heating step for pyrolyzing the polymer may be performed continuously.
- the raw material primary exfoliated graphite may be further subjected to thermal decomposition of the second method and polymer to obtain exfoliated graphite / resin composite material. Even in these cases, exfoliated graphite / resin composite material having a larger specific surface area can be obtained.
- the polymer is thermally decomposed to obtain exfoliated graphite / resin composite material, and then the exfoliated graphite / resin composite material is used as the raw material of the first method.
- the exfoliated graphite / resin composite material having a larger specific surface area may be obtained by using as the first exfoliated graphite and repeating the first method one or more times.
- the polymer is thermally decomposed to obtain exfoliated graphite / resin composite material, and then the exfoliated graphite / resin composite material obtained is used in the second method.
- the raw material primary exfoliated graphite may be further subjected to thermal decomposition of the second method and polymer to obtain exfoliated graphite / resin composite material.
- the exfoliated graphite is used as a raw material for the second method.
- exfoliated graphite may be obtained in the same manner as in the second method.
- the composition obtained by the second method is heated to obtain exfoliated graphite that is substantially free of polymer, and then the exfoliated graphite is used as the primary exfoliated graphite of the raw material of the first method.
- a composition may be prepared, and the polymer may be pyrolyzed by heating to obtain exfoliated graphite.
- the exfoliation by the production method of the present invention is repeated one or more times.
- the exfoliated graphite / resin composite material having a larger specific surface area can be obtained.
- exfoliated graphite / resin composite material is further used as primary exfoliated graphite as a raw material, and exfoliation by the production method of the present invention is repeated one or more times, so that exfoliated graphite having a larger specific surface area can be obtained.
- a resin composite material can be obtained.
- exfoliated graphite is obtained by pyrolyzing the polymer in the composition having a structure in which the polymer in which the radical polymerizable monomer is polymerized as described above is grafted to graphite or primary exfoliated graphite. ⁇
- resin composite materials we have resin composite materials.
- exfoliated graphite / resin composite material may be used as a raw material, and another conventionally known exfoliation method of graphite may be further performed.
- the method for producing a exfoliated graphite / resin composite material of the present invention may be carried out using primary exfoliated graphite obtained by another exfoliation method of graphite as a raw material. Even in that case, exfoliated graphite / resin composite material having a larger specific surface area can be obtained.
- another method for exfoliating graphite for example, a method for exfoliating graphite by electrochemical treatment or an adsorption-pyrolysis method can be used.
- Example 1 10 g of expanded graphite (trade name “PF Powder 8” manufactured by Toyo Tanso Co., Ltd.) and ADCA (manufactured by Eiwa Kasei Co., Ltd., trade name “AC # R”) having the structure represented by the above formula (1) as a thermally decomposable foaming agent.
- -K3 thermal decomposition temperature 210 ° C.
- styrene monomer produced by Wako Pure Chemical Industries, Ltd.
- the composition was heated to a temperature of 120 ° C. and maintained for 1 hour, and further maintained at a temperature of 150 ° C. for 1 hour. Thereby, the styrene monomer in the composition was polymerized.
- the composition was further heated to a temperature of 430 ° C. and maintained at a temperature of 430 ° C. for 2 hours. Thereby, the polymer in which the styrene monomer in the composition was polymerized was thermally decomposed to obtain exfoliated graphite / resin composite material from which the graphite was peeled off.
- the mixture was subjected to ultrasonic treatment at 100 W and an oscillation frequency of 28 kHz for 120 minutes using an ultrasonic treatment apparatus (manufactured by Honda Electronics Co., Ltd.). Thereby, the composition in which the expanded graphite was dispersed in the vinyl acetate polymer was obtained.
- the composition was dried at 80 ° C. for 2 hours and further heated to a temperature of 110 ° C. to completely dry the THF solution. The temperature was further maintained at 230 ° C. for 2 hours. Thereby, the ADCA was thermally decomposed and foamed in the composition.
- the composition was further heated to a temperature of 500 ° C. and maintained for 2 hours. Thereby, the vinyl acetate polymer in the composition was thermally decomposed to obtain exfoliated graphite / resin composite material from which the graphite was peeled off.
- the raw material composition was prepared by mixing with 120 g of tetrahydrofuran.
- the raw material composition was irradiated with ultrasonic waves at 100 W and an oscillation frequency of 28 kHz for 2 hours using an ultrasonic treatment apparatus (manufactured by Honda Electronics Co., Ltd.).
- an ultrasonic treatment apparatus manufactured by Honda Electronics Co., Ltd.
- polypropylene glycol was adsorbed on the expanded graphite.
- a composition in which polypropylene glycol was adsorbed on expanded graphite was prepared.
- the composition is formed by a solution casting method, maintained at a drying temperature of 80 ° C. for 2 hours, then maintained at 110 ° C. for 1 hour, and further maintained at 150 ° C. for 1 hour. And maintained at a temperature of 230 ° C. for 2 hours.
- the ADCA was thermally decomposed and foamed in the composition.
- Example 2 the composition was heated in the same manner as in Example 1, and ADCA was pyrolyzed and foamed in the composition.
- the composition was maintained at a temperature of 450 ° C. for 2 hours. Thereby, the polymer in which the styrene monomer in the composition was polymerized was thermally decomposed to obtain exfoliated graphite / resin composite material from which graphite was peeled off.
- the composition was heated to a temperature of 500 ° C. and maintained for another 2 hours. Thereby, the vinyl acetate polymer in the composition was thermally decomposed to obtain exfoliated graphite / resin composite material from which the graphite was peeled off.
- Example 3 In the same manner as in Example 3, after ultrasonic irradiation, the composition was molded by a solution casting method, and further heated, and ADCA was pyrolyzed and foamed in the composition.
- thermal decomposition start temperature and thermal decomposition end temperature of the exfoliated graphite / resin composite material are the thermal decomposition start temperature and thermal decomposition temperature of the resin before the composite, respectively. It can be seen that it is higher than the end temperature.
- the inflection point of the above TG curve can be seen in the vicinity of 570 ° C. of all the TG curves in FIGS. Therefore, it is considered that the polymer remains at a temperature lower than the inflection point.
- Example 9 The blending ratio of each component was the same as in Example 2, except that the exfoliated graphite / resin composite material of Example 9 was obtained in the same manner as in Example 2 except that the total amount was increased 3 times by weight. It was.
- Methylene blue methanol solutions of 10 mg / L, 5.0 mg / L, 2.5 mg / L, and 1.25 mg / L were prepared in a volumetric flask.
- methylene blue special grade reagent methylene blue manufactured by Kanto Chemical Co., Inc. was used.
- UV-visible spectrophotometer product number UV-1600 manufactured by Shimadzu Corporation, the absorbance of the four types of prepared methylene blue solutions was measured to prepare a calibration curve.
- methylene blue was placed in a 50 mL volumetric flask, and methanol was added as a measurement solvent to prepare a 100 mg / L methylene blue solution.
- This methylene blue solution was diluted 10 times with a measurement solvent to obtain a 10 mg / L methylene blue solution.
- the carbon sample and the supernatant were separated by centrifugation.
- the absorbance of a blank 10 mg / L methylene blue solution and the absorbance of the supernatant were measured.
- the difference between the absorbance of the blank methylene blue solution and the absorbance of the supernatant, that is, the amount of decrease in absorbance was calculated.
- the amount of decrease in the concentration of the methylene blue solution was determined from the amount of decrease in absorbance and the slope of the calibration curve described above. From the amount of decrease in the concentration of the methylene blue solution, the amount of methylene blue adsorbed on the carbon surface was determined by the following equation.
- the BET specific surface area of the exfoliated graphite / resin composite material obtained in Examples 4 to 14 was determined. Furthermore, the graft ratio of the resin in the exfoliated graphite / resin composite material obtained in Examples 4 to 14 was determined by the following method.
- the obtained solution was filtered using PTFE-T300A090C manufactured by Advantech having a hole diameter of 3 ⁇ m while suctioning with an aspirator. Further, the same amount of solvent as the amount of the solution was added and filtered again, and the unreacted polymer in the graphene was washed and filtered.
- Point P3 shows the result of spherical graphite (Lion Corporation, product number: EC-300J, average particle diameter 40 nm).
- Point P4 shows the result of spherical graphite (Lion Corporation, product number: EC-600JD, average particle diameter 34 nm).
- the adsorption amount of methylene blue measured under wet conditions is much larger than the BET specific surface area measured with dry processes. Therefore, it can be seen that if the exfoliated graphite / resin composite materials of Examples 4 to 14 are added to the resin in the form of a dispersion in which methanol is dispersed, the dispersibility in the resin can be further improved.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Health & Medical Sciences (AREA)
- Inorganic Chemistry (AREA)
- Materials Engineering (AREA)
- Engineering & Computer Science (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Carbon And Carbon Compounds (AREA)
- Polymerisation Methods In General (AREA)
- Graft Or Block Polymers (AREA)
- Pigments, Carbon Blacks, Or Wood Stains (AREA)
- Treatments Of Macromolecular Shaped Articles (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Geology (AREA)
Abstract
Description



本発明に係る薄片化黒鉛・樹脂複合材料は、薄片化黒鉛と樹脂とが複合化したものである。
(原料組成物を用意する工程)
本発明に係る薄片化黒鉛・樹脂複合材料の製造方法では、上記の黒鉛または一次薄片化黒鉛と、上記のポリマーとを含み、ポリマーが黒鉛または一次薄片化黒鉛に固定されている組成物をまず用意する。この組成物を用意する工程としては、例えば、ポリマーを黒鉛または一次薄片化黒鉛にグラフト化することにより、ポリマーを黒鉛または一次薄片化黒鉛に固定する以下の第1及び第2の方法や、ポリマーを黒鉛または一次薄片化黒鉛を吸着させることにより、ポリマーを黒鉛または一次薄片化黒鉛に固定する第3の方法を用いることができる。
第1の方法では、まず、原料として、上記の黒鉛または一次薄片化黒鉛と、上記のラジカル重合性モノマーとを含む混合物を用意する。次に、混合物に含まれているラジカル重合性モノマーを重合することにより、混合物中に上記ラジカル重合性モノマーが重合しているポリマーを生成させるとともに、該ポリマーを黒鉛または一次薄片化黒鉛にグラフト化させる。

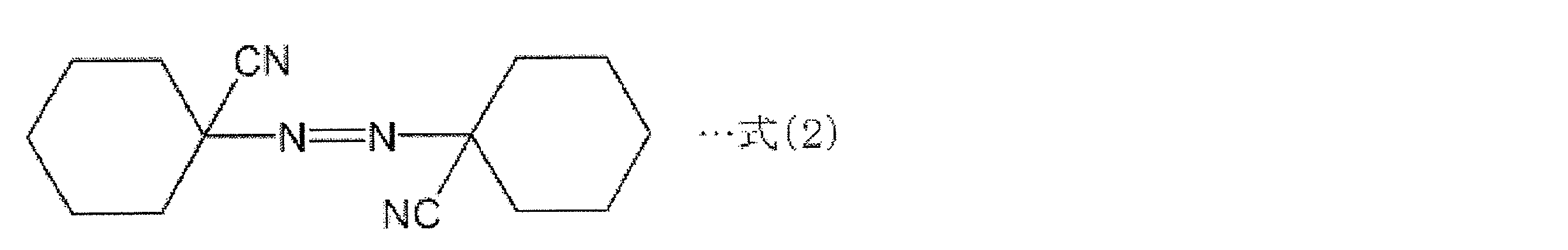


第2の方法では、黒鉛または一次薄片化黒鉛と、ラジカル重合性モノマーが重合しているポリマーとを含み、ポリマーが黒鉛または一次薄片化黒鉛にグラフト化している組成物を用意する工程において、ポリマーを黒鉛または一次薄片化黒鉛の存在下で、50℃以上かつ400℃以下の温度範囲の温度に加熱することにより、ポリマーを黒鉛または一次薄片化黒鉛にグラフト化させる。すなわち、第1の方法では、黒鉛または一次薄片化黒の存在下でラジカル重合性モノマーを重合してポリマーを生成するとともにポリマーの黒鉛または一次薄片化黒鉛へのグラフト化が図られていたが、これに対して、第2の方法では、予め得られたポリマーを黒鉛または一次薄片化黒鉛の存在下で上記特定の温度範囲に加熱することにより、ポリマーを熱分解することにより生成したポリマーラジカルを直接黒鉛または一次薄片化黒鉛にグラフトさせることができる。
第3の方法としては、上記黒鉛と、上記ポリマーとを適宜の溶媒に溶解もしくは分散させる方法を挙げることができる。このような溶媒としては、テトラヒドロフラン、メチルエチルケトン、トルエン、酢酸エチルなどを用いることができる。
上記第1の方法、第2の方法、及び第3の方法のいずれにおいても、上記のようにして組成物を用意したのち、組成物中に含まれるポリマーを熱分解する。それによって、ポリマーの一部を残存させながら、黒鉛または一次薄片化黒鉛が剥離され、薄片化黒鉛・樹脂複合材料を得ることができる。この場合のポリマーの熱分解を果たすために、上記組成物をポリマーの熱分解温度以上に加熱すればよい。より具体的には、ポリマーの熱分解温度以上に加熱し、さらにポリマーを焼成する。このとき、組成物中にポリマーが残存する程度に焼成する。それによって、薄片化黒鉛・樹脂複合材料を得ることができる。例えば、ポリスチレンの熱分解温度を380℃~450℃程度であり、ポリグリシジルメタクリレートの熱分解温度は400℃~500℃程度であり、ポリブチラールの熱分解温度は大気中で550℃~600℃程度である。
なお、本発明においては、上記のようにラジカル重合性モノマーが重合しているポリマーが黒鉛または一次薄片化黒鉛にグラフト化している構造を有する組成物中のポリマーを熱分解することにより薄片化黒鉛・樹脂複合材料を得ている。本発明では、さらに、他の方法により黒鉛を薄片化する工程を施してもよい。例えば、上記のように、薄片化黒鉛・樹脂複合材料を原料として用い、従来知られているような他の黒鉛の薄片化方法をさらに実施してもよい。あるいは、他の黒鉛の薄片化方法で得られた一次薄片化黒鉛を原料として本発明の薄片化黒鉛・樹脂複合材料の製造方法を実施してもよい。その場合においても、より一層比表面積の大きい薄片化黒鉛・樹脂複合材料を得ることができる。このような他の黒鉛の薄片化方法としては、例えば、電気化学的処理による黒鉛の薄片化方法、あるいは吸着-熱分解法を用いることができる。
膨張化黒鉛(東洋炭素社製、商品名「PFパウダー8」)10gと、熱分解性発泡剤として上記式(1)に示される構造を有するADCA(永和化成社製、商品名「AC#R-K3」、熱分解温度210℃)20gと、ラジカル重合性モノマーとしてスチレンモノマー(和光純薬工業社製)200gとを混合し、混合物とした。次に、上記混合物に対し、超音波処理装置(本多電子社製)を用いて、100W、発振周波数28kHzで120分間超音波処理した。それによって、上記膨張化黒鉛が上記スチレンモノマー中に分散している組成物を得た。
膨張化黒鉛(東洋炭素社製、商品名「PFパウダー8」、BET比表面積=22m2/g)1000mgと、熱分解性発泡剤として上記式(1)に示される構造を有するADCA(永和化成社製、商品名「AC#R-K3」、熱分解温度210℃)2gと、ラジカル重合性モノマーとして酢酸ビニルポリマー(SN-04T、デンカ社製)10gとテトラヒドロフラン20gを混合し、混合物とした。次に、上記混合物に対し、超音波処理装置(本多電子社製)を用いて、100W、発振周波数28kHzで120分間超音波処理した。それによって、上記膨張化黒鉛が上記酢酸ビニルポリマー中に分散している組成物を得た。
膨張化黒鉛(東洋炭素社製、商品名「PFパウダー8」、BET比表面積=22m2/g)6gと、熱分解性発泡剤として上記式(1)に示される構造を有するADCA(永和化成社製、商品名「AC#R-K3」、熱分解温度210℃)12gと、ポリプロピレングリコールPPG、三洋化成社製、品番:サンニックスGP-3000、数平均分子量=3000)120gとを、溶剤としてのテトラヒドロフラン120gと混合し、原料組成物を用意した。次に、原料組成物に、超音波処理装置(本多電子社製)を用いて、100W、発振周波数28kHzで2時間超音波を照射した。この超音波処理により、ポリプロピレングリコールを膨張化黒鉛に吸着させた。このようにして、ポリプロピレングリコールが膨張化黒鉛に吸着されている組成物を用意した。
実施例1と同様にして、膨張化黒鉛がスチレンモノマー中に分散している組成物を得た。
実施例2と同様にして膨張化黒鉛が酢酸ビニルポリマー中に分散している組成物を得た。次に、実施例2と同様にして組成物中においてADCAを熱分解し、発泡させた。
実施例3と同様にしてポリプロピレングリコールが膨張化黒鉛に吸着されている組成物を用意した。
1)TG/DTA測定
a)実施例1~3で重合されたまたは使用したポリマーに対し、空気雰囲気下で30℃から1000℃まで10℃/分の速度で加熱する燃焼試験を行った。この燃焼試験を行った際のTG/DTA測定結果を図1~3に示す。
実施例1~3及び参考例1~3により得られた薄片化黒鉛・樹脂複合材料を、島津製作所社製比表面積測定装置ASAP-2000で窒素ガスを用い、BET比表面積を測定した。結果を下記の表1に示す。

実施例1~3により得られた薄片化黒鉛・樹脂複合材料のXRDスペクトルを図7~9に示す。
実施例1~3により得られた薄片化黒鉛・樹脂複合材料を、走査型電子顕微鏡(SEM)により1000倍に拡大して撮影し、それによって得られた写真を観察した。実施例1~3により得られた薄片化黒鉛・樹脂複合材料の上記SEM写真を図10~12に示す。
各成分の配合割合は実施例3と同様とし、但し、全体量を20重量%減らしたことを除いては、実施例3と同様にして実施例4の薄片化黒鉛・樹脂複合材料を得た。
各成分の配合割合は実施例2と同様とし、但し、全体の量を重量で3倍に増やしたこと以外は、実施例2と同様にして実施例9の薄片化黒鉛・樹脂複合材料を得た。
実施例13~実施例14では、最終工程の焼成温度および焼成時間を400℃で10時間に変更した以外は、それぞれ実施例4と同様にして各工程処理を実施した。
メスフラスコに、10mg/L、5.0mg/L、2.5mg/L、1.25mg/Lの濃度のメチレンブルーのメタノール溶液を調製した。メチレンブルーとしては、関東化学社製特級試薬のメチレンブルーを用いた。島津製作所製、紫外可視分光光度計(品番UV-1600)を用い、用意した上記4種類のメチレンブルー溶液の吸光度を測定し、検量線を作成した。
上記ブランクのメチレンブルー溶液の吸光度と上記上澄み液の吸光度との差、すなわち吸光度の減少量を算出した。この吸光度の減少量と、前述した検量線の傾きにより、メチレンブルー溶液の濃度の減少量を求めた。このメチレンブルー溶液の濃度の減少量から、以下の式により、カーボン表面へのメチレンブルーの吸着量を求めた。
さらに、実施例4~14で得た薄片化黒鉛・樹脂複合材料における樹脂のグラフト率を以下の方法で求めた。
高圧加熱反応処理されたカーボン材料を含有するサンプル1~10gを50倍重量の良溶媒で溶解した。超音波装置を用いて45kHz、100Wの出力で、常温で30分間分散処理を行った。
上記のようにして求めたメチレンブルー吸着量と、BET比表面積、並びにBET比表面積から求めた樹脂グラフト率を下記の表2に示す。

なお、図16および図17において、上記実施例4~14とは別に、公知の炭素質材料のBET比表面積と上記のようにして測定したメチレンブルー吸着量との関係を併せて示す。図16および図17において、点P1(参考例4)は、球状黒鉛(社団法人日本粉体工業技術協会製、品番:RPSA-2)の結果を示す。点P2(参考例5)は、球状黒鉛(社団法人日本粉体工業技術協会製、品番:RPSA-3)の結果を示す。点P3(参考例6)は、球状黒鉛(ライオン株式会社、品番:EC-300J、平均粒子径40nm)の結果を示す。点P4(参考例7)は、球状黒鉛(ライオン株式会社、品番:EC-600JD、平均粒子径34nm)の結果を示す。図16では、公知の黒鉛材料として上記の4点(参考例4~7)のみをプロットしたが、他の公知の炭素質材料においても、図16および図17に示すy=0.13x、但しr2=0.99近くにプロットされることが確かめられている。
Claims (18)
- 薄片化黒鉛と樹脂とが複合化した薄片化黒鉛・樹脂複合材料であって、
10mg/L濃度のメチレンブルーのメタノール溶液の吸光度と、該メチレンブルーのメタノール溶液に薄片化黒鉛・樹脂複合材料を投入し、遠心分離により得られた上澄み液の吸光度との差に基づき測定された薄片化黒鉛・樹脂複合材料1gあたりのメチレンブルー吸着量(μモル/g)をy、該薄片化黒鉛・樹脂複合材料のBET比表面積(m2/g)をxとした場合、比y/xが0.15以上であり、且つBET比表面積が、25m2/g以上である、薄片化黒鉛・樹脂複合材料。 - 薄片化黒鉛と樹脂とが複合化した薄片化黒鉛・樹脂複合材料であって、
前記薄片化黒鉛複合材料における樹脂の熱分解開始温度及び熱分解終了温度が、それぞれ、前記複合化前の前記樹脂の熱分解開始温度及び熱分解終了温度よりも高い、薄片化黒鉛・樹脂複合材料。 - 前記樹脂の含有量が、1質量%~70質量%である、請求項1または2に記載の薄片化黒鉛・樹脂複合材料。
- 前記樹脂が、ラジカル重合性モノマーの重合体である、請求項1~3のいずれか一項に記載の薄片化黒鉛・樹脂複合材料。
- 請求項1~4のいずれか一項に記載の薄片化黒鉛・樹脂複合材料の製造方法であって、
黒鉛または一次薄片化黒鉛と、ポリマーとを含み、前記ポリマーが前記黒鉛または一次薄片化黒鉛に固定されている組成物を用意する工程と、
前記組成物中に含まれる前記ポリマーを熱分解することにより、前記ポリマーの一部を残存させながら、前記黒鉛または一次薄片化黒鉛を剥離する工程とを備える、薄片化黒鉛・樹脂複合材料の製造方法。 - 前記組成物を用意する工程において、前記ポリマーが前記黒鉛または一次薄片化黒鉛にグラフト化または吸着されていることにより、前記ポリマーが前記黒鉛または一次薄片化黒鉛に固定されている、請求項5に記載の薄片化黒鉛・樹脂複合材料の製造方法。
- 前記組成物を用意する工程が、
前記黒鉛または一次薄片化黒鉛と、ラジカル重合性モノマーとを含む混合物を用意する工程と、
前記混合物に含まれる前記ラジカル重合性モノマーを重合することによって、前記混合物中にラジカル重合性モノマーが重合している前記ポリマーを生成すると共に、前記ポリマーを前記黒鉛または一次薄片化黒鉛にグラフト化させる工程とを含む、請求項6に記載の薄片化黒鉛・樹脂複合材料の製造方法。 - 前記混合物を用意する工程において、前記混合物が熱分解性発泡剤をさらに含む、請求項7に記載の薄片化黒鉛・樹脂複合材料の製造方法。
- 前記熱分解性発泡剤が、下記の式(1)~式(4)に示される構造を有する化合物からなる群から選択される少なくとも1種の加熱発泡剤である、請求項8に記載の薄片化黒鉛・樹脂複合材料の製造方法。



- 前記ポリマーを熱分解することにより、前記黒鉛または一次薄片化黒鉛を剥離する工程において、前記混合物に含まれる前記熱分解性発泡剤を熱分解する、請求項8または9に記載の薄片化黒鉛・樹脂複合材料の製造方法。
- 前記ポリマーを生成すると共に、前記ポリマーを前記黒鉛または一次薄片化黒鉛にグラフト化させる工程において、前記混合物に含まれる前記熱分解性発泡剤を熱分解する、請求項8または9に記載の薄片化黒鉛・樹脂複合材料の製造方法。
- 前記ポリマーを生成すると共に、前記ポリマーを前記黒鉛または一次薄片化黒鉛にグラフト化させる工程が、前記混合物を加熱することによって前記混合物に含まれる前記ラジカル重合性モノマーを重合することにより行われる、請求項8~10のいずれか一項に記載の薄片化黒鉛・樹脂複合材料の製造方法。
- 前記組成物を用意する工程において、前記ポリマーを前記黒鉛または一次薄片化黒鉛の存在下で、50℃以上かつ400℃以下の温度範囲の温度に加熱することにより前記ポリマーを前記黒鉛または一次薄片化黒鉛にグラフト化させる、請求項8に記載の薄片化黒鉛・樹脂複合材料の製造方法。
- 前記組成物を用意する工程において、前記組成物が熱分解性発泡剤をさらに含む、請求項6または10に記載の薄片化黒鉛・樹脂複合材料の製造方法。
- 前記熱分解性発泡剤が、下記の式(1)~式(4)に示される構造を有する化合物からなる群から選択される少なくとも1種の加熱発泡剤である、請求項14に記載の薄片化黒鉛・樹脂複合材料の製造方法。




- 前記ポリマーを熱分解することにより、前記黒鉛または一次薄片化黒鉛を剥離する工程において、前記組成物に含まれる前記熱分解性発泡剤を熱分解する、請求項14または15に記載の薄片化黒鉛・樹脂複合材料の製造方法。
- 前記ラジカル重合性モノマーが、ビニル系モノマーである、請求項6~15のいずれか一項に記載の薄片化黒鉛・樹脂複合材料の製造方法。
- 前記ビニル系モノマーがスチレンまたはグリシジルメタクリレートである、請求項17に記載の薄片化黒鉛・樹脂複合材料の製造方法。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES13832631.9T ES2684343T3 (es) | 2012-08-27 | 2013-02-14 | Material compuesto de resina y grafito en copos y método para producir el mismo |
| KR1020157000539A KR101912492B1 (ko) | 2012-08-27 | 2013-02-14 | 박편화 흑연ㆍ수지 복합 재료 및 그의 제조 방법 |
| JP2013509386A JP5352028B1 (ja) | 2012-08-27 | 2013-02-14 | 薄片化黒鉛・樹脂複合材料及びその製造方法 |
| CN201380040178.2A CN104508051B (zh) | 2012-08-27 | 2013-02-14 | 薄片化石墨‑树脂复合材料及其制造方法 |
| US14/412,199 US9683091B2 (en) | 2012-08-27 | 2013-02-14 | Exfoliated graphite-resin composite material and method for producing the same |
| EP13832631.9A EP2889334B1 (en) | 2012-08-27 | 2013-02-14 | Flaked graphite resin composite material and method for producing same |
| US15/481,890 US10113047B2 (en) | 2012-08-27 | 2017-04-07 | Exfoliated graphite-resin composite material and method for producing the same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-186463 | 2012-08-27 | ||
| JP2012186463 | 2012-08-27 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/412,199 A-371-Of-International US9683091B2 (en) | 2012-08-27 | 2013-02-14 | Exfoliated graphite-resin composite material and method for producing the same |
| US15/481,890 Continuation US10113047B2 (en) | 2012-08-27 | 2017-04-07 | Exfoliated graphite-resin composite material and method for producing the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014034156A1 true WO2014034156A1 (ja) | 2014-03-06 |
Family
ID=50182985
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/053470 WO2014034156A1 (ja) | 2012-08-27 | 2013-02-14 | 薄片化黒鉛・樹脂複合材料及びその製造方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US9683091B2 (ja) |
| EP (1) | EP2889334B1 (ja) |
| JP (1) | JP5576532B2 (ja) |
| KR (1) | KR101912492B1 (ja) |
| CN (1) | CN104508051B (ja) |
| ES (1) | ES2684343T3 (ja) |
| TW (1) | TWI549906B (ja) |
| WO (1) | WO2014034156A1 (ja) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016166352A (ja) * | 2015-03-02 | 2016-09-15 | 積水化学工業株式会社 | 薄片化黒鉛・樹脂複合材料の製造方法及び薄片化黒鉛・樹脂複合材料 |
| WO2016181952A1 (ja) * | 2015-05-14 | 2016-11-17 | 積水化学工業株式会社 | 炭素質材料、炭素質材料-活物質複合体、リチウムイオン二次電池用電極材及びリチウムイオン二次電池 |
| CN104497232B (zh) * | 2014-12-09 | 2017-02-08 | 湖南科技大学 | 一种可控单分散聚苯乙烯微球接枝石墨烯的制备及其产品 |
| WO2017090553A1 (ja) | 2015-11-27 | 2017-06-01 | 積水化学工業株式会社 | キャパシタ用電極材及びキャパシタ |
| JP2017182913A (ja) * | 2016-03-28 | 2017-10-05 | 積水化学工業株式会社 | 複合体及びその製造方法、リチウムイオン二次電池用正極材、並びにリチウムイオン二次電池 |
| JP2018037255A (ja) * | 2016-08-31 | 2018-03-08 | 積水化学工業株式会社 | 活物質−炭素材料複合体、非水電解質二次電池用正極、非水電解質二次電池及び炭素材料 |
| JP2018037256A (ja) * | 2016-08-31 | 2018-03-08 | 積水化学工業株式会社 | 活物質−炭素材料複合体、非水電解質二次電池用正極、非水電解質二次電池及び炭素材料 |
| WO2018062285A1 (ja) * | 2016-09-30 | 2018-04-05 | 積水化学工業株式会社 | 炭素材料、キャパシタ用電極シート及びキャパシタ |
| WO2018159566A1 (ja) | 2017-02-28 | 2018-09-07 | 積水化学工業株式会社 | ガスバリア材及び熱硬化性樹脂組成物 |
| WO2018225619A1 (ja) | 2017-06-05 | 2018-12-13 | 積水化学工業株式会社 | 硫黄-炭素材料複合体、リチウム硫黄二次電池用正極材及びリチウム硫黄二次電池 |
| WO2018225670A1 (ja) | 2017-06-05 | 2018-12-13 | 積水化学工業株式会社 | 炭素材料含有分散液、電極形成用スラリー、及び非水電解質二次電池用電極の製造方法 |
| WO2018230080A1 (ja) | 2017-06-15 | 2018-12-20 | 積水化学工業株式会社 | 炭素材料及びその製造方法、蓄電デバイス用電極材料、並びに蓄電デバイス |
| WO2019026940A1 (ja) | 2017-08-04 | 2019-02-07 | 積水化学工業株式会社 | 炭素材料、全固体電池用正極、全固体電池用負極、及び全固体電池 |
| WO2019078073A1 (ja) | 2017-10-16 | 2019-04-25 | 積水化学工業株式会社 | 複合体、蓄電デバイス用電極材料、及び蓄電デバイス |
| WO2019155881A1 (ja) | 2018-02-09 | 2019-08-15 | 積水化学工業株式会社 | 炭素材料、蓄電デバイス用電極、蓄電デバイス、及び非水電解質二次電池 |
| WO2019189284A1 (ja) * | 2018-03-27 | 2019-10-03 | 積水化学工業株式会社 | 複合材料及びその製造方法 |
| WO2019240021A1 (ja) | 2018-06-15 | 2019-12-19 | 積水化学工業株式会社 | 二次電池用負極材、二次電池用負極、及び二次電池 |
| WO2020027111A1 (ja) | 2018-08-03 | 2020-02-06 | 積水化学工業株式会社 | 炭素材料及びその製造方法、蓄電デバイス用電極材料、並びに蓄電デバイス |
| WO2020045337A1 (ja) | 2018-08-28 | 2020-03-05 | 積水化学工業株式会社 | 炭素材料及びその製造方法、蓄電デバイス用電極材料、並びに蓄電デバイス |
| WO2020166513A1 (ja) | 2019-02-13 | 2020-08-20 | 積水化学工業株式会社 | 炭素材料-樹脂複合材料、複合体及びその製造方法、並びに蓄電デバイス用電極材料 |
| WO2020189662A1 (ja) | 2019-03-20 | 2020-09-24 | 積水化学工業株式会社 | 複合材料、蓄電デバイス用電極材料、及び蓄電デバイス |
| WO2020241242A1 (ja) | 2019-05-30 | 2020-12-03 | 積水化学工業株式会社 | 蓄電デバイス |
| WO2021060243A1 (ja) | 2019-09-24 | 2021-04-01 | 積水化学工業株式会社 | 炭素材料及び蓄電デバイス用電極材料 |
| WO2021060108A1 (ja) | 2019-09-26 | 2021-04-01 | 積水化学工業株式会社 | 二次電池用負極材、二次電池用負極、及び二次電池 |
| JP2021155294A (ja) * | 2020-03-27 | 2021-10-07 | 大阪瓦斯株式会社 | 耐久性向上剤 |
| WO2021201040A1 (ja) | 2020-03-31 | 2021-10-07 | 積水化学工業株式会社 | 合わせガラス用中間膜、及び合わせガラス |
| WO2022177023A1 (ja) * | 2021-02-22 | 2022-08-25 | 株式会社Adeka | 導電性アンダーコート剤 |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10658126B2 (en) | 2013-12-26 | 2020-05-19 | Sekisui Chemical Co., Ltd. | Capacitor electrode material, method for producing same, and electric double layer capacitor |
| CN107534118B (zh) | 2015-08-14 | 2020-06-05 | 株式会社Lg 化学 | 锂空气电池及其制造方法 |
| ITUB20160159A1 (it) * | 2016-01-27 | 2017-07-27 | Versalis Spa | Composizione contenente grafene e nano piastrine grafeniche e loro procedimento di preparazione. |
| US10998551B2 (en) * | 2016-08-31 | 2021-05-04 | Sekisui Chemical Co., Ltd. | Electrode material for electricity storage devices, electrode for electricity storage devices, and electricity storage device |
| CN107543786A (zh) * | 2017-08-29 | 2018-01-05 | 中国科学院宁波材料技术与工程研究所 | 一种测定石墨烯材料比表面积的方法 |
| WO2020004179A1 (ja) * | 2018-06-25 | 2020-01-02 | 積水化学工業株式会社 | 炭素材料、導電助剤、蓄電デバイス用電極、及び蓄電デバイス |
| CN109438760B (zh) * | 2018-11-16 | 2020-07-07 | 攀枝花学院 | 聚丙烯酸酯改性膨胀石墨及其制备方法和应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH1095887A (ja) * | 1995-12-22 | 1998-04-14 | Sekisui Chem Co Ltd | ポリ塩化ビニル系樹脂組成物 |
| JP2009511415A (ja) * | 2005-10-14 | 2009-03-19 | ザ、トラスティーズ オブ プリンストン ユニバーシティ | 熱的に剥離されたグラファイト酸化物 |
| WO2012105344A1 (ja) * | 2011-02-04 | 2012-08-09 | 積水化学工業株式会社 | 薄片化黒鉛-ポリマー複合材料の製造方法 |
| JP2012250892A (ja) * | 2011-06-06 | 2012-12-20 | Sekisui Chem Co Ltd | 炭素質材料−ポリマー複合材料の製造方法 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2109781B (en) * | 1981-10-22 | 1985-07-10 | Central Glass Co Ltd | Graphite fluoride coated with organic polymer and method of preparing same |
| US7604049B2 (en) * | 2005-12-16 | 2009-10-20 | Schlumberger Technology Corporation | Polymeric composites, oilfield elements comprising same, and methods of using same in oilfield applications |
| JP5386802B2 (ja) | 2007-07-27 | 2014-01-15 | 中央電気工業株式会社 | 黒鉛質粉末とその製造方法 |
| CN105670394A (zh) * | 2008-02-05 | 2016-06-15 | 普林斯顿大学理事会 | 包含官能化的石墨烯片的涂料以及用其涂覆的物品 |
| CN106376174B (zh) * | 2008-02-05 | 2019-06-07 | 普林斯顿大学理事会 | 电子器件和形成电子器件的方法 |
| EP2545568A1 (en) * | 2009-12-22 | 2013-01-16 | Pasi Moilanen | Fabrication and application of polymer-graphitic material nanocomposites and hybride materials |
| JP6279199B2 (ja) | 2010-10-28 | 2018-02-14 | 積水化学工業株式会社 | 樹脂複合材料及び樹脂複合材料の製造方法 |
| US9346748B2 (en) | 2011-11-30 | 2016-05-24 | Sekisui Chemical Co., Ltd. | Functional-group-modified carbon material, and method for producing same |
| JP5937813B2 (ja) | 2011-11-30 | 2016-06-22 | 積水化学工業株式会社 | アミノ基変成炭素材料、その製造方法及び複合材料 |
| CN102633957B (zh) * | 2012-04-11 | 2014-04-09 | 上海交通大学 | 一种聚甲基丙烯酸甲酯改性石墨烯纳米带的制备方法 |
| JP5352028B1 (ja) * | 2012-08-27 | 2013-11-27 | 積水化学工業株式会社 | 薄片化黒鉛・樹脂複合材料及びその製造方法 |
-
2013
- 2013-02-14 EP EP13832631.9A patent/EP2889334B1/en not_active Not-in-force
- 2013-02-14 WO PCT/JP2013/053470 patent/WO2014034156A1/ja active Application Filing
- 2013-02-14 US US14/412,199 patent/US9683091B2/en active Active
- 2013-02-14 CN CN201380040178.2A patent/CN104508051B/zh active Active
- 2013-02-14 ES ES13832631.9T patent/ES2684343T3/es active Active
- 2013-02-14 KR KR1020157000539A patent/KR101912492B1/ko active IP Right Grant
- 2013-05-06 TW TW102116105A patent/TWI549906B/zh not_active IP Right Cessation
- 2013-06-04 JP JP2013117740A patent/JP5576532B2/ja active Active
-
2017
- 2017-04-07 US US15/481,890 patent/US10113047B2/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH1095887A (ja) * | 1995-12-22 | 1998-04-14 | Sekisui Chem Co Ltd | ポリ塩化ビニル系樹脂組成物 |
| JP2009511415A (ja) * | 2005-10-14 | 2009-03-19 | ザ、トラスティーズ オブ プリンストン ユニバーシティ | 熱的に剥離されたグラファイト酸化物 |
| US7659350B2 (en) | 2005-10-14 | 2010-02-09 | The Trustees Of Princeton University | Polymerization method for formation of thermally exfoliated graphite oxide containing polymer |
| WO2012105344A1 (ja) * | 2011-02-04 | 2012-08-09 | 積水化学工業株式会社 | 薄片化黒鉛-ポリマー複合材料の製造方法 |
| JP2012250892A (ja) * | 2011-06-06 | 2012-12-20 | Sekisui Chem Co Ltd | 炭素質材料−ポリマー複合材料の製造方法 |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104497232B (zh) * | 2014-12-09 | 2017-02-08 | 湖南科技大学 | 一种可控单分散聚苯乙烯微球接枝石墨烯的制备及其产品 |
| JP2016166352A (ja) * | 2015-03-02 | 2016-09-15 | 積水化学工業株式会社 | 薄片化黒鉛・樹脂複合材料の製造方法及び薄片化黒鉛・樹脂複合材料 |
| JP2017216254A (ja) * | 2015-05-14 | 2017-12-07 | 積水化学工業株式会社 | 炭素質材料、炭素質材料−活物質複合体、リチウムイオン二次電池用電極材及びリチウムイオン二次電池 |
| US10644318B2 (en) | 2015-05-14 | 2020-05-05 | Sekesui Chemical Co., Ltd. | Carbon material, carbon material-active material composite, electrode material for lithium-ion secondary battery, and lithium-ion secondary battery |
| EP3297075A4 (en) * | 2015-05-14 | 2018-12-19 | Sekisui Chemical Co., Ltd. | Carbon material, carbon material-active material composite, electrode material for lithium-ion secondary battery, and lithium-ion secondary battery |
| WO2016181952A1 (ja) * | 2015-05-14 | 2016-11-17 | 積水化学工業株式会社 | 炭素質材料、炭素質材料-活物質複合体、リチウムイオン二次電池用電極材及びリチウムイオン二次電池 |
| JPWO2016181952A1 (ja) * | 2015-05-14 | 2017-06-01 | 積水化学工業株式会社 | 炭素質材料、炭素質材料−活物質複合体、リチウムイオン二次電池用電極材及びリチウムイオン二次電池 |
| WO2017090553A1 (ja) | 2015-11-27 | 2017-06-01 | 積水化学工業株式会社 | キャパシタ用電極材及びキャパシタ |
| JP2017182913A (ja) * | 2016-03-28 | 2017-10-05 | 積水化学工業株式会社 | 複合体及びその製造方法、リチウムイオン二次電池用正極材、並びにリチウムイオン二次電池 |
| JP2018037256A (ja) * | 2016-08-31 | 2018-03-08 | 積水化学工業株式会社 | 活物質−炭素材料複合体、非水電解質二次電池用正極、非水電解質二次電池及び炭素材料 |
| JP2018037255A (ja) * | 2016-08-31 | 2018-03-08 | 積水化学工業株式会社 | 活物質−炭素材料複合体、非水電解質二次電池用正極、非水電解質二次電池及び炭素材料 |
| CN109313989A (zh) * | 2016-09-30 | 2019-02-05 | 积水化学工业株式会社 | 碳材料、电容器用电极片以及电容器 |
| WO2018062285A1 (ja) * | 2016-09-30 | 2018-04-05 | 積水化学工業株式会社 | 炭素材料、キャパシタ用電極シート及びキャパシタ |
| JPWO2018159566A1 (ja) * | 2017-02-28 | 2019-12-19 | 積水化学工業株式会社 | ガスバリア材及び熱硬化性樹脂組成物 |
| WO2018159566A1 (ja) | 2017-02-28 | 2018-09-07 | 積水化学工業株式会社 | ガスバリア材及び熱硬化性樹脂組成物 |
| US11261321B2 (en) | 2017-02-28 | 2022-03-01 | Sekisui Chemical Co., Ltd. | Gas barrier material and thermosetting resin composition |
| JPWO2018225619A1 (ja) * | 2017-06-05 | 2020-04-16 | 積水化学工業株式会社 | 硫黄−炭素材料複合体、リチウム硫黄二次電池用正極材及びリチウム硫黄二次電池 |
| WO2018225619A1 (ja) | 2017-06-05 | 2018-12-13 | 積水化学工業株式会社 | 硫黄-炭素材料複合体、リチウム硫黄二次電池用正極材及びリチウム硫黄二次電池 |
| US11489152B2 (en) | 2017-06-05 | 2022-11-01 | Sekisui Chemical Co., Ltd. | Sulfur-carbon material composite body, positive electrode material for lithium sulfur secondary batteries, and lithium sulfur secondary battery |
| JP7144013B2 (ja) | 2017-06-05 | 2022-09-29 | 積水化学工業株式会社 | 硫黄-炭素材料複合体、リチウム硫黄二次電池用正極材及びリチウム硫黄二次電池 |
| WO2018225670A1 (ja) | 2017-06-05 | 2018-12-13 | 積水化学工業株式会社 | 炭素材料含有分散液、電極形成用スラリー、及び非水電解質二次電池用電極の製造方法 |
| WO2018230080A1 (ja) | 2017-06-15 | 2018-12-20 | 積水化学工業株式会社 | 炭素材料及びその製造方法、蓄電デバイス用電極材料、並びに蓄電デバイス |
| WO2019026940A1 (ja) | 2017-08-04 | 2019-02-07 | 積水化学工業株式会社 | 炭素材料、全固体電池用正極、全固体電池用負極、及び全固体電池 |
| WO2019078073A1 (ja) | 2017-10-16 | 2019-04-25 | 積水化学工業株式会社 | 複合体、蓄電デバイス用電極材料、及び蓄電デバイス |
| WO2019155881A1 (ja) | 2018-02-09 | 2019-08-15 | 積水化学工業株式会社 | 炭素材料、蓄電デバイス用電極、蓄電デバイス、及び非水電解質二次電池 |
| WO2019189284A1 (ja) * | 2018-03-27 | 2019-10-03 | 積水化学工業株式会社 | 複合材料及びその製造方法 |
| WO2019240021A1 (ja) | 2018-06-15 | 2019-12-19 | 積水化学工業株式会社 | 二次電池用負極材、二次電池用負極、及び二次電池 |
| US11975972B2 (en) | 2018-08-03 | 2024-05-07 | Sekisui Chemical Co., Ltd. | Carbon material and method for producing same, electrode material for electrical storage device, and electrical storage device |
| WO2020027111A1 (ja) | 2018-08-03 | 2020-02-06 | 積水化学工業株式会社 | 炭素材料及びその製造方法、蓄電デバイス用電極材料、並びに蓄電デバイス |
| WO2020045337A1 (ja) | 2018-08-28 | 2020-03-05 | 積水化学工業株式会社 | 炭素材料及びその製造方法、蓄電デバイス用電極材料、並びに蓄電デバイス |
| WO2020166513A1 (ja) | 2019-02-13 | 2020-08-20 | 積水化学工業株式会社 | 炭素材料-樹脂複合材料、複合体及びその製造方法、並びに蓄電デバイス用電極材料 |
| WO2020189662A1 (ja) | 2019-03-20 | 2020-09-24 | 積水化学工業株式会社 | 複合材料、蓄電デバイス用電極材料、及び蓄電デバイス |
| WO2020241242A1 (ja) | 2019-05-30 | 2020-12-03 | 積水化学工業株式会社 | 蓄電デバイス |
| WO2021060243A1 (ja) | 2019-09-24 | 2021-04-01 | 積水化学工業株式会社 | 炭素材料及び蓄電デバイス用電極材料 |
| WO2021060108A1 (ja) | 2019-09-26 | 2021-04-01 | 積水化学工業株式会社 | 二次電池用負極材、二次電池用負極、及び二次電池 |
| JP2021155294A (ja) * | 2020-03-27 | 2021-10-07 | 大阪瓦斯株式会社 | 耐久性向上剤 |
| WO2021201040A1 (ja) | 2020-03-31 | 2021-10-07 | 積水化学工業株式会社 | 合わせガラス用中間膜、及び合わせガラス |
| WO2022177023A1 (ja) * | 2021-02-22 | 2022-08-25 | 株式会社Adeka | 導電性アンダーコート剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20170210876A1 (en) | 2017-07-27 |
| US9683091B2 (en) | 2017-06-20 |
| US10113047B2 (en) | 2018-10-30 |
| ES2684343T3 (es) | 2018-10-02 |
| CN104508051B (zh) | 2017-12-19 |
| EP2889334A4 (en) | 2016-04-27 |
| JP2014062225A (ja) | 2014-04-10 |
| TWI549906B (zh) | 2016-09-21 |
| CN104508051A (zh) | 2015-04-08 |
| EP2889334A1 (en) | 2015-07-01 |
| KR101912492B1 (ko) | 2018-10-26 |
| EP2889334B1 (en) | 2018-05-23 |
| KR20150051987A (ko) | 2015-05-13 |
| US20150175778A1 (en) | 2015-06-25 |
| TW201408595A (zh) | 2014-03-01 |
| JP5576532B2 (ja) | 2014-08-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5576532B2 (ja) | 薄片化黒鉛・樹脂複合材料及びその製造方法 | |
| CN103339154B (zh) | 薄片化石墨-聚合物复合材料的制造方法 | |
| TWI625298B (zh) | Microparticle-thinned graphite composite, anode material for lithium ion secondary battery, and manufacturing method thereof, and lithium ion secondary battery | |
| KR101844622B1 (ko) | 탄소질 재료-중합체 복합 재료의 제조 방법 및 탄소질 재료-중합체 복합 재료 | |
| US9604884B2 (en) | Composite material and method for producing the same | |
| JP5352028B1 (ja) | 薄片化黒鉛・樹脂複合材料及びその製造方法 | |
| JP5407008B1 (ja) | 薄片化黒鉛の製造方法及び薄片化黒鉛 | |
| JP6609200B2 (ja) | 薄片化黒鉛・樹脂複合材料の製造方法及び薄片化黒鉛・樹脂複合材料 | |
| WO2019189284A1 (ja) | 複合材料及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2013509386 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13832631 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14412199 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 20157000539 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013832631 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |