RU2554081C2 - Способ синтеза соединений диарилтиогидантоина и диарилгидантоина - Google Patents
Способ синтеза соединений диарилтиогидантоина и диарилгидантоина Download PDFInfo
- Publication number
- RU2554081C2 RU2554081C2 RU2012140454/04A RU2012140454A RU2554081C2 RU 2554081 C2 RU2554081 C2 RU 2554081C2 RU 2012140454/04 A RU2012140454/04 A RU 2012140454/04A RU 2012140454 A RU2012140454 A RU 2012140454A RU 2554081 C2 RU2554081 C2 RU 2554081C2
- Authority
- RU
- Russia
- Prior art keywords
- compound
- formula
- another embodiment
- acid
- mixture
- Prior art date
Links
- 0 CN*c1cc(*)c(*)cc1 Chemical compound CN*c1cc(*)c(*)cc1 0.000 description 4
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/72—Two oxygen atoms, e.g. hydantoin
- C07D233/74—Two oxygen atoms, e.g. hydantoin with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to other ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/86—Oxygen and sulfur atoms, e.g. thiohydantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4166—1,3-Diazoles having oxo groups directly attached to the heterocyclic ring, e.g. phenytoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/12—Preparation of carboxylic acid amides by reactions not involving the formation of carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/28—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton
- C07C237/30—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton having the nitrogen atom of the carboxamide group bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
Abstract
Изобретение относится к области органической химии, а именно к способу получения соединения формулы (I-1), включающему (a) объединение соединения формулы D-1 с соединением формулы (F), диметилсульфоксидом (ДМСО) и изопропилацетатом с образованием смеси; и (b) нагревание указанной смеси с получением соединения диарилтиогидантоина формулы (I-1). Также изобретение относится к способу получения соединения формулы (M-I), включающему (a) объединение 4-бром-2-фторбензойной кислоты с 2-аминоизомасляной кислотой, диметилформамидом (ДМФА), водой, триэтиламином (TEA) и 2-ацетилциклогексаноном с образованием смеси; и (b) нагревание указанной смеси с получением 4-(2-карбоксипропан-2-иламино)-2-фторбензойной кислоты:  (c) взаимодействие 4-(2-карбоксипропан-2-иламино)-2-фторбензойной кислоты с соединением формулы (F) с получением соединения диарилтиогидантоина формулы (M-1). Технический результат: разработаны новые способы получения производных диарилтиогидантоина, обладающих полезной биологической активностью. 2 н. и 1 з.п. ф-лы, 11 пр.
(c) взаимодействие 4-(2-карбоксипропан-2-иламино)-2-фторбензойной кислоты с соединением формулы (F) с получением соединения диарилтиогидантоина формулы (M-1). Технический результат: разработаны новые способы получения производных диарилтиогидантоина, обладающих полезной биологической активностью. 2 н. и 1 з.п. ф-лы, 11 пр.



Description
Перекрестные ссылки на родственные заявки
По данной заявке испрашивается приоритет предварительной заявки на патент США №61/307796, поданной 24 февраля 2010. Полное описание этой заявки включено в настоящее описание посредством ссылки.
Заявление о федерально спонсированном исследовании или разработке не применяется.
Область техники, к которой относится изобретение
Изобретение относится к области терапии рака, а именно, к способам синтеза лекарственных средств для лечения рака предстательной железы.
Предпосылки создания изобретения
Согласно Американскому обществу по борьбе с раком, рак предстательной железы является наиболее часто диагностируемым типом рака среди мужчин в Соединенных Штатах, помимо рака кожи. По оценке Американского общества по борьбе с раком, было диагностировано приблизительно 186000 новых случаев рака предстательной железы, и приблизительно 29000 мужчин умерли от рака предстательной железы в одних только Соединенных Штатах в течение 2008. Таким образом, рак предстательной железы является второй основной причиной смерти от рака у мужчин в Соединенных Штатах после рака легких.
Метастатический рак предстательной железы представляет собой злокачественную опухоль, которая распространяется за пределы предстательной железы и окружающих тканей в отдаленные органы и ткани. Большинство мужчин, которые умирают от рака предстательной железы, умирают от последствий метастазирующей опухоли. Согласно Национальному институту рака, среднее выживание пациентов с раком предстательной железы, который метастазировал в отдаленные органы, обычно составляет один-три года, и большинство таких пациентов умирает от рака предстательной железы. Метастатический рак предстательной железы обычно разделяют на два состояния: гормоно-чувствительное состояние и кастрационно-резистентное состояние (также называемое гормоно-рефракторное состояние).
Тестостерон и другие мужские половые гормоны, известные собирательно как андрогены, могут питать рост клеток рака предстательной железы. Андрогены оказывают свой эффект на клетки рака предстательной железы, связываясь и активируя рецепторы андрогена, которые экспрессированы в клетках рака предстательной железы. В случае, когда они сначала метастазируют в отдаленные участки, большинство случаев рака предстательной железы зависит от андрогенов для роста. Эти случаи рака предстательной железы известны как "гормоно-чувствительные" злокачественные образования. Соответственно, в настоящее время ведущие методы терапии, используемые для лечения метастатического рака предстательной железы, сосредоточены на уменьшении или противодействии эффектам андрогенов на клетки рака предстательной железы. Один метод использует так называемые "антиандрогены", которые являются молекулами, которые блокируют взаимодействие андрогенов с антиандрогенными рецепторами. Другой метод должен уменьшать количество андрогенов, продуцированных в организме, главным образом, в яичках. Это может быть достигнуто хирургически, путем удалением обоих яичек (орхиэктомия) или путем применения лекарственных средств, известных как агонисты рилизинг-фактора лютеинизирующего гормона, или ЛГ-РГ, которые снижают естественную выработку тестостерона в яичках (иногда называемой "химическая кастрация").
Большинство метастатических опухолей предстательной железы первоначально чувствительно к гормону и, таким образом, отвечает на гормональные методы лечения. Однако согласно исследованию, опубликованному 7 октября 2004 в выпуске The New England Journal of Medicine, фактически во всех случаях гормоно-чувствительный метастатический рак предстательной железы претерпевает изменения, которые преобразуют его в кастрационно-резистентное состояние в течение 18-24 месяцев после начала гормональной терапии [Debes, J. et al. "Mechanisms of Androgen-Refractory Prostate Cancer." New. England. J. Med. (2004), 351:1488-1490]. Одним из важных механизмов, посредством которого гормоно-чувствительное состояние переходит в кастрационно-резистентное состояние, вероятно, является сверхэкспрессия андрогенных рецепторов. В экспериментах, сравнивающих экспрессию гена в клетках гормоно-чувствительного и кастрационно-резистентного рака предстательной железы, повышение экспрессии андрогенных рецепторов было единственным генетическим изменением, постоянно ассоциированым с кастрационно-резистентным раком [Chen, C. et al. "Molecular determinants of resistance to antiandrogen therapy." Nat. Med. (2004), 10(l):33-39]. Обычно, в этом состоянии раковые опухоли предстательной железы продолжают расти андроген-зависимым образом, несмотря на снижение выработки тестостерона до очень низких (то есть, посткастрационных) уровней. Рак предстательной железы в этом состоянии известен как "кастрационно-резистентный" рак предстательной железы или CRPC. Переход от гормоно-чувствительного к кастрационно-резистентному состоянию после начала гормональной терапии обычно определяется на основании или возрастающих уровней простатического специфического антигена, или PSA, или документально подтвержденного развития заболевания, что подтверждается тестами диагностической визуализации или клиническими симптомами. Метастатический рак предстательной железы, который стал кастрационно-резистентным, чрезвычайно агрессивен; средняя выживаемость таких пациентов составляет только 10-16 месяцев.
Основная причина того, что CRPC является столь смертельным, заключается в том, что он трудно поддается лечению. Поскольку методы лечения, в настоящее время используемые для лечения метастатического рака предстательной железы, работают путем уменьшения возможности андрогенов питать рост клеток раковой опухоли предстательной железы, они обычно эффективны только в отношении рака предстательной железы, который остается гормоно-чувствительным в зависимости от андрогенов для роста. CRPC больше не отвечает на гормональные методы лечения, которые эффективны при гормоно-чувствительном состоянии. Ситуацию дополнительно усложняют биологические изменения в раке предстательной железы, который перешел в кастрационно-резистентное состояние, и лекарственные средства, которые первоначально блокируют антиандрогенные рецепторы и подавляют рост гормоно-чувствительной раковой опухоли предстательной железы, могут иметь точно противоположный эффект и начать питать рост CRPC. Например, Casodex® (бикалутамид), продаваемый AstraZeneca PLC, непосредственно блокирует взаимодействие андрогенов с антиандрогенными рецепторами и является наиболее продаваемым антиандрогенным терапевтическим средством. Однако на in vitro модели кастрационно-резистентного состояния рака предстательной железы, в котором клеточные линии рака предстательной железы были генетически спроектированы для сверхэкспрессии андрогенных рецепторов (таким образом, переводя их из гормоно-чувствительного в кастрационно-резистентное состояние), Casodex® не осуществил эффективного ингибирования андрогенных рецепторов в этих клетках, а, в некоторых случаях, он стал стимулятором андрогенных рецепторов. В соответствии с этими результатами, которые согласуются с опубликованным клиническим исследованием Casodex при CRPC на человеке, Casodex® является неэффективным терапевтическим средством для кастрационно-резистентного состояния метастатического рака предстательной железы.
Соединения, которые связываются с андрогенным рецептором, с той же самой целью, с которой связывается Casodex® и другие имеющиеся в продаже лекарственные средства для лечения метастатического рака предстательной железы, были получены для использования при кастрационно-резистентном состоянии метастатического рака предстательной железы. Эти соединения связываются с андрогенными рецепторами способом, который делает их эффективными при лечении раковых образований, которые стали невосприимчивыми к применяемым в настоящее время лекарственным средствам. Например, некоторые соединения, описанные в публикации патентной заявки США № 2007/0004753, 2007/0254933 (вновь опубликованной как 2008/0139634) и 2009/0111864, являются новыми низкомолекулярными антагонистами андрогенных рецепторов, которые ингибируют функцию андрогенных рецепторов, блокируя транслокацию андрогенных рецепторов в ядро и связывание ДНК.
Способ синтеза соединений настоящего изобретения, как описано в вышеприведенных публикациях патентных заявок США, включает конденсацию изотиоцианата с изобутиронитрилом. Главные недостатки способа, как описано выше, включают только 25%-ный выход желаемого продукта, получаемого на конечной стадии, приводя к 15%-ому общему выходу из коммерчески доступных исходных веществ. Кроме того, каждое промежуточное соединение требует трудоемкой колоночной хроматографии для очистки, что приводит к увеличению общей продолжительности получения продукта, что, с точки зрения промышленного производства, является невыгодным. Для сравнения, настоящее изобретение, описанное в данном описании, обеспечивает 50%-ный общий выход, и любая необходимая очистка осуществляется простыми средствами осаждения или кристаллизации. Кроме того, настоящее изобретение исключает использование чрезвычайно токсичного реагента ацетонциангидрина. В результате, способ согласно настоящему изобретению представляет собой более безопасный процесс, в котором количество растворителя снижено, минимизируя производственные отходы и воздействие на окружающую среду, время цикла уменьшено, и пропускная способность и общий выход продукта в способе увеличены.
Краткое описание сущности изобретения
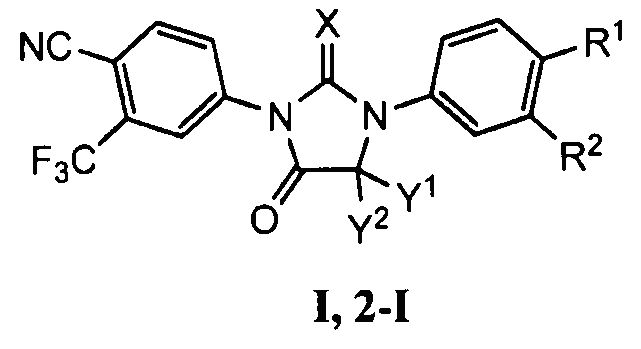
Настоящее изобретение включает высокоэффективный способ получения соединения формулы (I,2-I):

где:
X представляет собой S или O;
Y1 и Y2 независимо представляют собой метил или вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу с 4-5 атомами углерода;
R1 представляет собой L1-C(=O)-NR4R5 или L1-CN; где L1 представляет собой одинарную связь или C1-C8алкилен; и R4 и R5 независимо выбраны из H и C1-C8алкила; и
R2 представляет собой водород или фтор;
включающий взаимодействие соединения формулы A:

где LG представляет собой Br, I или другую удаляемую группу, с соединением формулы B:

с получением соединения формулы C:

взаимодействие соединения формулы C с соединением формулы R6-LG в условиях алкилирования или с соединением формулы R6-OH в условиях этерификации с образованием соединения формулы D:

D
где R6 представляет собой C1-C8алкил;
и взаимодействие соединения формулы D с соединением формулы (F,2-F):

F,2-F
где X представляет собой S или O,
с получением соединения диарилтиогидантоина или диарилгидантоина формулы (I,2-I):
В одном варианте осуществления изобретения в отношении соединения формулы A, LG представляет собой Br или I. В конкретном варианте осуществления LG представляет собой Br.
Другой аспект настоящего изобретения обеспечивает эффективный способ получения кислотного производного формулы (I,2-Ia):

где:
Y1 и Y2 независимо представляют собой метил или вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу с 4-5 атомами углерода;
R7 представляет собой L1-C(=O)-OH; где L1 представляет собой одинарную связь или C1-C8алкилен; и
R2 представляет собой водород или фтор;
включающий гидролиз соединения формулы I,2-I:
где
R1 представляет собой L1-C(=O)-NR4R5; где L1 представляет собой одинарную связь или C1-C8алкилен; и R4 и R5 независимо выбраны из H и C1-C8алкила.
В одном конкретном варианте осуществления в отношении соединения формулы I,2-Ia, L1 представляет собой одинарную связь; и R7 представляет собой -C(=O)-OH.
В одном конкретном варианте осуществления в отношении соединения формулы I,2-Ia, Y1 и Y2, оба представляют собой метил, R7 представляет собой -C(=O)-OH, и R2 представляет собой F.
В одном варианте осуществления изобретения описанный выше гидролиз осуществляют в присутствии концентрированной HCl.
В одном варианте осуществления изобретения описанный выше гидролиз проводят при температуре 80-140°C или при температуре около 80-140°C.
В одном конкретном варианте осуществления описанный выше гидролиз проводят при температуре 120°C или при температуре около 120°C.
В одном варианте осуществления изобретения описанный выше гидролиз проводят в течение 10-60 час или в течение от около 10 час до около 60 час.
В одном конкретном варианте осуществления описанный выше гидролиз проводят в течение 48 час или в течение около 48 час.
В одном конкретном варианте осуществления в отношении соединения формулы I,2-Ia, X представляет собой S.
В одном конкретном варианте осуществления в отношении соединения формулы I,2-Ia, X представляет собой O.
В одном конкретном варианте осуществления в отношении соединения формулы I,2-Ia, Y1 и Y2, оба представляют собой метил, R7 представляет собой -C(=O)-OH, R2 представляет собой F, и X представляет собой S.
В одном конкретном варианте осуществления в отношении соединения формулы I,2-Ia, Y1 и Y2, оба представляют собой метил, R7 представляет собой -C(=O)-OH, R2 представляет собой F, и X представляет собой O.
В одном конкретном варианте осуществления настоящее изобретение включает высокоэффективный способ получения соединения формулы (I):

где:
Y1 и Y2 независимо представляют собой метил или вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу с 4-5 атомами углерода;
R1 представляет собой L1-C(=O)-NR4R5 или L1-CN; где L1 представляет собой одинарную связь или C1-C8алкилен; и
R4 и R5 независимо выбраны из H и C1-C8алкила; и
R2 представляет собой водород или фтор;
включающий следующие стадии:
взаимодействие соединения формулы A:

где LG представляет собой Br, I или другую удаляемую группу, с соединением формулы B:
с образованием соединения формулы C:

взаимодействие соединения формулы C с соединением формулы R6-OH в условиях этерификации или, альтернативно, взаимодействие соединения формулы C с соединением формулы R6-LG, где R6 представляет собой C1-C8алкил, и LG представляет собой Br, I или другую удаляемую группу, с образованием соединения формулы D:

взаимодействие соединения формулы D с соединением формулы F, 4-изотиоцианато-2-(трифторметил)бензонитрилом:

с образованием соединения формулы (I):
В одном варианте осуществления изобретения в отношении соединения формулы A, LG представляет собой Br или I. В конкретном варианте осуществления LG представляет собой Br.
В одном конкретном варианте осуществления настоящее изобретение включает высокоэффективный способ получения соединения формулы (I):
где:
Y1 и Y2 независимо представляют собой метил или вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу с 4-5 атомами углерода;
R1 представляет собой L1-C(=O)-NR4R5 или L1-CN; где L1 представляет собой одинарную связь или C1-C8алкилен; и R4 и R5 независимо выбраны из H и C1-C8алкила; и
R2 представляет собой водород или фтор;
включающий взаимодействие соединения формулы A:
с соединением формулы B:

с получением соединения формулы C:
взаимодействие соединения формулы C с соединением формулы E:

с образованием соединения формулы G:

и взаимодействие соединения формулы G с тиофосгеном,
с получением диарилтиосоединения гидантоина формулы (I):

В одном конкретном варианте осуществления в отношении соединения формулы I или I,2-I, Y1 и Y2, оба представляют собой метил.
В одном конкретном варианте осуществления в отношении соединения формулы I или I,2-I, Y1 и Y2 вместе с атомом углерода, к которому они присоединены, объединены с образованием циклобутильного кольца.
В одном конкретном варианте осуществления в отношении соединения формулы I или I,2-I, Y1 и Y2 вместе с атомом углерода, к которому они присоединены, объединены с образованием циклопентильного кольца.
В одном конкретном варианте осуществления в отношении соединения формулы I или I,2-I, L1 представляет собой одинарную связь.
В одном конкретном варианте осуществления в отношении соединения формулы I или I,2-I, L1 представляет собой -CH2-, -CH2-CH2- или -CH2-CH2-CH2-.
В одном конкретном варианте осуществления в отношении соединения формулы I или I,2-I, L1 представляет собой одинарную связь; и R1 представляет собой -C(=O)-NHCH3.
В одном конкретном варианте осуществления в отношении соединения формулы I или I,2-I, L1 представляет собой одинарную связь; и R1 представляет собой -C(=O)-NH2.
В одном конкретном варианте осуществления в отношении соединения формулы I или I,2-I, R2 представляет собой F.
В одном конкретном варианте осуществления в отношении соединения формулы I или I,2-I, Y1 и Y2, оба представляют собой метил, R1 представляет собой -C(=O)-NHCH3, и R2 представляет собой F.
В одном конкретном варианте осуществления в отношении соединения формулы I или I,2-I, Y1 и Y2, оба представляют собой метил, R1 представляет собой -C(=O)-NH2, и R2 представляет собой F.
В одном конкретном варианте осуществления в отношении соединения формулы I или I,2-I, соединение соответствует формуле II:

Общая схема для одного варианта осуществления взаимодействия, проиллюстрированная в порядке выполнения A→C→D→I ниже, представлена на схеме 1, ниже:
Схема 1

где проиллюстрирован a) необязательный синтез a) соединения F из 4-амино-2-(трифторметил)бензонитрила (соединение E) и тиофосгена, и b) необязательный гидролиз заместителя R1 соединения I до группы карбоновой кислоты, для синтеза, когда карбоновая кислота необходима в положении R1. При необязательном гидролизе заместителя R1 соединения I до группы карбоновой кислоты, R1 ограничивается -L1-(C=O)NH2, -L1-(C=O)NHR4 и -L1-(C=O)NR4R5, поскольку гидролиз R1, когда R1 представляет собой -L1-CN, будет приводить к гидролизу другой группы нитрила, присутствующей на другом бензольном кольце. При гидролизе, представленном на схеме 1, L1 обозначает отсутствие группы (то есть, одинарную связь), поскольку гидролиз указан как дающий группу -COOH, однако в других вариантах осуществления изобретения L1 также может быть C1-C8алкиленом.
В альтернативном способе соединение формулы C обрабатывают соединением E в условиях образования амидной связи, с получением соединения формулы G, с последующей обработкой реагентом, таким как тиофосген, с образованием соединения формулы I (то есть, в порядке C→G→I, указанном на схеме выше).
В одном варианте осуществления соединение формулы A смешивают с соединением формулы B в присутствии каталитического количества как катализатора на основе меди (I), так и бета-дион лиганда, такого как 2-ацетилциклогексанон, в полярном растворителе и при нагревании до температуры приблизительно 90-120°C, приблизительно 100-110°C или приблизительно 105°C. Катализатором на основе меди (I) может быть хлорид меди (I) или йодид меди (I). Катализатор меди (I), такой как CuCl, может присутствовать в количестве около 0,05-0,35 эквивалентов относительно соединения A, около 0,15-0,25 эквивалентов относительно соединения A или около 0,2 эквивалента относительно соединения A. Лиганд, такой как 2-ацетилциклогексанон, может присутствовать в количестве около 0,05-0,35 эквивалентов относительно соединения A, около 0,15-0,25 эквивалентов относительно соединения A или около 0,2 эквивалента относительно соединения A. В другом варианте осуществления лиганд, такой как 2-ацетилциклогексанон, присутствует в количестве приблизительно равном количеству используемого катализатора на основе меди (I), такого как хлорид меди (I). Соединение B может быть добавлено в количестве около 1-2 эквивалентов относительно соединения A, около 1,25-1,75 эквивалентов относительно соединения A или около 1,5 эквивалентов относительно соединения A. Выбор бета-дион лигандов известен специалистам в данной области, например, 2,4-пентандион, 2,4-гександион, 1-фенил-1,3-бутандион, 2-ацетилциклогексанон и тому подобное. Полярный растворитель может быть выбран из группы, состоящей из диметилсульфоксида (ДМСО), диметилформамида (ДМФА), N-метилпирролидона (NMP), диметилацетамида (DMA), изопропилацетата (IPAc), изопропилового спирта (IPA) и подобного; в другом варианте осуществления изобретения полярным растворителем является ДМФА, вода или смесь ДМФА и воды. В другом варианте осуществления изобретения после взаимодействия в течение около 6-24 час, около 8-20 час или около 12-14 час, или после того, как анализ показал расход соединения A приблизительно 90% или более, реакционную смесь затем охлаждают до температуры около 15-25°C, например, до около 25°C или до комнатной температуры. В другом варианте осуществления изобретения к охлажденной реакционной смеси добавляют воду с последующим промыванием органическим несмешивающимся с водой растворителем, таким как изопропилацетат; затем смесь разделяют на органический и водный слои. В другом варианте осуществления изобретения водный слой подкисляют для выделения соединения C осаждением, фильтрованием и сушкой.
В одном варианте осуществления изобретения соединение C подвергают взаимодействию с алкилирующим агентом формулы R6-LG, где R6 представляет собой C1-C8алкил, и LG представляет собой Br, I или другую удаляемую группу, с образованием соединения формулы D. Соединения формулы R6-LG включают соединения, такие как метилйодид. Взаимодействие может быть осуществлено в присутствии неорганического основания, такого как K2CO3, KHCO3, Na2CO3 или NaHCO3, в полярном растворителе, таком как ДМСО, ДМФА, NMP, DMA или IPAc, и каталитического количества воды. Каталитическое количество воды может составлять около 5-25%, 10-20% или 14% эквивалентов соединения C, или около 0,05-0,25%, 0,10-0,20% или 0,14% объема полярного растворителя. Реакционная смесь может быть нагрета до около 35-50°C или около 40-46°C, в течение около 5-60 мин, или до тех пор, пока анализ не показал более чем около 90% или около 95%, или около 99% преобразования соединения C в соединение D. После взаимодействия, смесь может быть охлаждена до температуры около 5-25°C или около 15-25°C. Реакционная смесь, содержащая соединение D, может быть объединена с водой для осаждения продукта D из раствора. Продукт D может быть выделен путем фильтрования и сушки. В одном варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 2 эквивалентов или менее чем около 2 эквивалентов относительно соединения C. В другом варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 1,5 эквивалентов или менее чем около 1,5 эквивалентов относительно соединения C. В другом варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 1,2 эквивалента или менее чем около 1,2 эквивалента относительно соединения C. В другом варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 1,1 эквивалента или менее чем около 1,1 эквивалента относительно соединения C. В другом варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 1,0 эквивалента или менее чем около 1,0 эквивалента относительно соединения C. В другом варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 0,9 эквивалента или менее чем около 0,9 эквивалента относительно соединения C. В другом варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 0,8 эквивалента или менее чем около 0,8 эквивалента относительно соединения C. В другом варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 0,7 эквивалента или менее чем около 0,7 эквивалента относительно соединения C. В другом варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 0,6 эквивалента или менее чем около 0,6 эквивалента относительно соединения C.
В другом варианте осуществления изобретения при использовании CH3I для образования соединения D (где R6=CH3), избыточное количество CH3I гасят уксусной кислотой. CH3I может быть использован в количестве около 1-1,5 эквивалента относительно соединения C, например, в количестве около 1,2 эквивалента относительно соединения C, и количество AcOH может быть добавлено в количестве около или немного больше, чем избыточное количество метилйодида (например, когда используют 1,2 эквивалента метилйодида, где метилйодид используют в избыточном на 0,2 эквивалента количестве, относительно соединения C, тогда может быть использовано около 0,21-0,25 эквивалента или около 0,23 эквивалента AcOH относительно соединения C) для гашения непрореагировавшего CH3I. Для этой стадии также могут быть использованы альтернативные метилирующие агенты, известные специалистам в данной области, такие как диметилсульфат.
В другом варианте осуществления изобретения стадия объединения реакционной смеси, содержащей соединение D, с водой выполнена постепенным добавлением воды к нагретой реакционной смеси в течение от около 0,5 часа до около 3,5 часов, от около 0,6 часа до 3,4 часов, от около 1 часа до 2 часов или в течение около 0,5, 0,6, 1, 2, 3, 3,4 или 3,5 часов, до тех пор, пока не будет добавлено 1-5 объемов воды или приблизительно 1-3 объема воды или приблизительно 2 объема воды для более медленного осаждения соединения D и уменьшения количества неорганического катиона и основания, такого как K+ и CO3 2-, из неорганического основания, такого как K2CO3, используемого в реакции. В одном варианте осуществления изобретения температура добавляемой воды составляет от около 50°C до около 80°C, от около 50°C до около 70°C, от около 55°C до около 75°C, от около 55°C до около 65°C, от около 57°C до около 63°C, от около 48°C до около 53°C или от около 68°C до около 71°C или от около 57°C или около 70°C. В другом варианте осуществления изобретения осажденное соединение D снова суспендируют или повторно образуют взвесь в воде, и затем воду удаляют фильтрованием для дополнительного уменьшения количества присутствующего неорганического катиона. В другом варианте осуществления объем воды для повторного суспендирования или повторного образования взвеси составляет приблизительно 5-15 объемов или приблизительно 10 объемов. В другом варианте осуществления повторное суспендирование или повторное образование взвеси в воде осуществляют в течение от около 0,5 часа до около 3 часов, от около 1,0 до около 2,0 часов, около 1,0 часа, около 1,5 часов или около 2 часов. В другом варианте осуществления температура воды для повторного суспендирования или повторного образования взвеси составляет от около 15°C до около 35°C, от около 20°C до около 30°C, от около 20°C до около 25°C или от около 20°C до около 23°C.
В одном варианте осуществления изобретения остаточное количество неорганического катиона, такого как ион калия, остающегося в соединении D, меньше чем или составляет приблизительно 1000 ч./млн. (частей на миллион). В другом варианте осуществления изобретения остаточное количество неорганического катиона, такого как ион калия, остающегося в соединении D, меньше чем или составляет приблизительно 500 частей на миллион. В другом варианте осуществления изобретения остаточное количество неорганического катиона, такого как ион калия, остающегося в соединении D, меньше чем или составляет приблизительно 300 ч./млн.
В одном варианте осуществления изобретения остаточное количество основания, такого как ион бикарбоната, ион карбоната или другого основания, остающегося в соединении D, меньше чем или равно около 1000 ч./млн. В другом варианте осуществления изобретения остаточное количество основания, остающегося в соединении D, меньше чем или равно около 500 ч./млн. В другом варианте осуществления изобретения остаточное количество основания, остающегося в соединении D, меньше чем или равно около 300 ч./млн.
В одном варианте осуществления изобретения соединение D может быть высушено продувкой или вытяжкой сухого воздуха, сухого азота или аргона или другого сухого инертного газа через соединение. В другом варианте осуществления изобретения соединение D может быть высушено путем помещения соединения в вакуум (например, при вакууме около 1 мм рт.ст. или ниже, при вакууме 0,5 мм рт.ст. или ниже или при вакууме 0,1 мм рт.ст. или ниже). В одном варианте осуществления изобретения остаточное количество воды, остающееся в соединении D, меньше чем или равно около 0,5%. В одном варианте осуществления изобретения остаточное количество воды, остающееся в соединении D, меньше чем или равно около 0,3%. В одном варианте осуществления изобретения остаточное количество воды, остающееся в соединении D, меньше чем или равно около 0,1%. В одном варианте осуществления изобретения остаточное количество воды, остающееся в соединении D, меньше чем или равно около 500 ч./млн. В одном варианте осуществления изобретения остаточное количество воды, остающееся в соединении D, меньше чем или равно около 300 ч./млн. В одном варианте осуществления изобретения остаточное количество воды, остающееся в соединении D, меньше чем или равно около 100 ч./млн.
Альтернативный способ получения соединения D из соединения C использует стандартные условия этерификации по Фишеру, включающие смешивание соединения C в метаноле и нагревание в течение около 1-16 час при около 40-100°C (или при кипячении с обратным холодильником) с каталитическим количеством кислоты, таким как одна-пять капель серной кислоты, хлористоводородной кислоты, азотной кислоты, фосфорной кислоты или другой неорганической кислоты, п-толуолсульфоновой кислоты или ионообменной смолы, содержащей сульфоновую кислоту; в одном варианте осуществления изобретения используют H2SO4. В некоторых вариантах осуществления воду можно удалить путем азеотропной отгонки (такой как ловушкой Дина-Старка). После завершения этерификации (завершение около 70%, около 80%, около 90%, около 95% или около 99%), выделение соединения D может быть осуществлено способом, описанным выше.
В другом варианте осуществления изобретения стадия образования соединения I включает смешивание соединения D с соединением F в полярном растворителе или в смеси первого полярного растворителя и второго полярного растворителя, и нагреванием до около 60-100°C, около 80-100°C или около 80-85°C, в течение около 1-48 часов или около 12-24 часов. В другом варианте осуществления изобретения после реакции процесс продолжается путем охлаждения реакционной смеси до около 15-30°C, до около 25°C или до комнатной температуры, и объединения с водой с последующим экстрагированием желаемого продукта полярным растворителем или смесью третьего полярного растворителя и четвертого полярного растворителя. Соединение F может быть добавлено в количестве около 1-3 эквивалентов относительно соединения D, или около 1,5-2,5 эквивалентов относительно соединения D, или около 1,5 эквивалентов или около 2 эквивалентов относительно соединения D, или в количестве около 1,5 эквивалентов, с последующим добавлением, по мере протекания реакции, дополнительной части, составляющей около 0,5 эквивалента. Слой объединенного органического экстракта может быть уменьшен в объеме и затравлен кристаллами желаемого продукта I для начала кристаллизации при охлаждении до около 0-10°C или около 3-6°C, с последующим выделением кристаллического продукта путем фильтрования и затем сушкой продукта в токе воздуха или в вакууме. В одном аспекте этого варианта осуществления полярный растворитель или первый, второй, третий и четвертый полярные растворители могут быть выбраны из группы, состоящей из: ДМСО, ДМФА, NMP, DMA, IPAc, MeCN, IPA и тому подобное. В одном варианте осуществления изобретения полярным растворителем является ДМФА. В одном варианте осуществления изобретения полярным растворителем является IPAc. В другом варианте осуществления изобретения первым полярным растворителем является IPAc, и вторым полярным растворителем является ДМСО. В другом варианте осуществления изобретения третьим полярным растворителем является IPAc, и четвертым полярным растворителем является IPA. В другом варианте осуществления изобретения первым полярным растворителем является IPAc, вторым полярным растворителем является ДМСО, третьим полярным растворителем является IPAc, и четвертым полярным растворителем является IPA.
В другом варианте осуществления изобретения альтернативный способ образования соединения I включает две стадии, выполняемые в порядке C→G→I, приведенном на схеме выше. На первой стадии используют стандартные условия образования амидной связи, включая, например, обработку соединения C реагентом сочетания, таким как дициклогексилкарбодиимид (DCC), диизопропилкарбодиимид (DIC), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDCI), гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (BOP), гексафторфосфат 7-азабензотриазол-1-илокситрис(диметиламино)фосфония (AOP), гексафторфосфат бензотриазол-1-илокситрис(пирролидин)фосфония (PyBOP), гексафторфосфат 7-азабензотриазол-1-илокситрис(пирролидин)фосфония (PyAOP), гексафторфосфат O-бензотриазол-N,N,N',N'-тетраметилурония (HBTU), тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU), тетрафторборат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TATU) и тому подобное, с соединением E, в полярном растворителе или в смеси первого полярного растворителя и второго полярного растворителя, с получением соединения G. В одном аспекте этого варианта осуществления полярный растворитель или первый и второй полярные растворители, выбраны из группы, состоящей из: DCM, ДМСО, ДМФА, NMP, DMA, MeCN и тому подобное. Вторая стадия включает реакцию замыкания кольца соединения G с реагентом тиокарбонилирования, таким как тиофосген, и нагревание чистого раствора до около 60-120°C. В другом варианте осуществления изобретения взаимодействие осуществляют в запаянной трубке. Тиофосген или тиоэквивалент фосгена (например, 1,1-тиокарбонилдиимидазол) может присутствовать в количестве около 1-10 эквивалентов относительно соединения G, или около 5 эквивалентов относительно соединения G.
В другом варианте осуществления изобретения соединение I может быть подвергнуто условиям гидролиза, когда R1 представляет собой группу первичного или вторичного амида, с получением соответствующего производного карбоновой кислоты.
В одном варианте осуществления указанного выше способа, заместитель R1 соединения формулы A представляет собой -C(=O)-NH-R4. В другом варианте осуществления изобретения R1 представляет собой -C(=O)-NH-CH3. В другом варианте осуществления изобретения заместитель R2 соединения формулы A представляет собой фтор. В другом варианте осуществления изобретения R1 представляет собой -C(=O)-NH-R4, и R2 представляет собой фтор. В другом варианте осуществления изобретения R1 представляет собой -C(=O)-NH-CH3, и R2 представляет собой фтор, и соединение формулы A представляет собой 4-бром-2-фтор-N-метилбензамид.
В одном варианте осуществления указанного выше способа, соединение формулы B представляет собой 2-аминоизомасляную кислоту (то есть, Y1 и Y2, каждый представляет собой CH3). В другом варианте осуществления указанного выше способа соединение формулы B представляет собой 1-аминоциклобутанкарбоновую кислоту. В другом варианте осуществления указанного выше способа соединение формулы B представляет собой 1-аминоциклопентанкарбоновую кислоту.
В другом варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, R1 представляет собой -C(=O)NHCH3, и/или R2 представляет собой F.
В другом варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, R1 представляет собой -C(=O)NH2, и/или R2 представляет собой F.
Также предложены варианты соединения формулы (I). Соединения формулы (I) или их варианты, как подробно рассмотрено в настоящем описании, или фармацевтически приемлемая соль любого из указанных выше соединений, могут найти конкретное применение при лечении рака предстательной железы, включая CRPC и/или гормоно-чувствительный рак предстательной железы.
В альтернативном варианте осуществления синтеза соединений I, 2-Ia, где R7 представляет собой -C(=O)OH, конечный продукт может быть синтезирован следующим образом (проиллюстрировано с использованием изотиоцианата):

где соединение C подвергают взаимодействию с соединением F (4-изотиоцианато-2-(трифторметил)бензонитрилом), с получением продукта M. В одном варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, и/или R2 представляет собой F. Взаимодействие может быть осуществлено в основных условиях, с использованием триалкиламинного основания, такого как триэтиламин, присутствующего в количестве около 2-5 эквивалентов, или около 3-4 эквивалентов, или около 3,4 эквивалентов относительно соединения C. Соединение F, 4-изотиоцианато-2-(трифторметил)бензонитрил, может присутствовать в количестве около 1,1-4 эквивалентов, или 1,1-2 эквивалентов, или около 1,5 эквивалентов относительно соединения C; альтернативно, может быть добавлено около 1,5 эквивалентов соединения F, затем другая порция, составляющая около 0,5 эквивалента, по мере протекания взаимодействия. Растворителем может быть этанол или другой спирт. Реакционная смесь может перемешиваться в течение около 4-16 дней, около 8-12 дней или около 10 дней при комнатной температуре или повышенной температуре. Затем реакционная смесь может быть концентрирована, смешана с водным раствором кислоты, например, 1M HCl, и продукт экстрагирован органическим растворителем, таким как этилацетат, с получением продукта M.
В дополнительном варианте осуществления соединение C синтезируют путем взаимодействия соединения формулы J с 1,1-дизамещенным 2,2,2-трихлорэтанолом:

где 1,1-дизамещенный 2,2,2-трихлорэтанол может быть использован в количестве около 1,5-4 эквивалентов относительно соединения J, или около 2-3 эквивалентов относительно соединения J, около 2,5 эквивалентов относительно соединения J, или около 2,6 эквивалентов относительно соединения J. Реакцию проводят в органическом растворителе, предпочтительно безводном растворителе, таком как безводный ацетон. Перед добавлением сильного основания, такого как NaOH, KOH или другой гидроксид, реакционная смесь может быть охлаждена до температуры 0°C. Основание добавляют в количестве около 2-5 эквивалентов, или около 3-4 эквивалентов, или около 3,8 эквивалентов, или около 3,9 эквивалентов относительно соединения J. После добавления основания реакционная смесь может быть оставлена для нагревания до комнатной температуры, и выдержана при комнатной температуре в течение около 4-24 час, или около 8-16 час, или около 12 час. Продукт может быть очищен стандартными способами, такими как колоночная хроматография или ВЭЖХ.
В другом варианте осуществления изобретения изобретение включает способы получения соединений гидантоина в соответствии со схемой 2:
Схема 2

где R1=-L1-(C=O)NH2, -L1-(C=O)NHR4, -L1-(C=O)NR4R5 или L1-CN для соединения 2-I. Взаимодействия аналогичны взаимодействиям, приведенным на схеме 1, с заменой тиофосгена фосгеном и заменой тиоизоцианата F изоцианатом 2-F, что дает продукт гидантоин 2-I вместо тиогидантоина I. Следует отметить, что фосген может быть заменен эквивалентами фосгена, такими как 1,1-карбонилдиимидазол (см., например, реагенты, описанные в Phosgenations-A Handbook, авторы Livius Cotarca и Heiner Eckert, Weinheim, Germany: Wiley-VCH Verlag GmbH & Co., 2003, конкретно эквивалентами фосгена, перечисленными в главе 3). Аналогично схеме 1 проиллюстрированы: необязательный синтез a) соединения 2-F из 4-амино-2-(трифторметил)бензонитрила (соединение E) и фосгена, и b) необязательный гидролиз заместителя R1 соединения 2-I до группы карбоновой кислоты, для синтеза, когда карбоновая кислота необходима в положении R1. В необязательном гидролизе заместителя R1 соединения 2-I до группы карбоновой кислоты, R1 ограничивается -L1-(C=O)NH2, -L1-(C=O)NHR4 и -L1-(C=O)NR4R5, поскольку гидролиз R1, когда R1 представляет собой -L1-CN, приведет к гидролизу другой группы нитрила, представленной на другом бензольном кольце. В гидролизе, показанном на схеме 2, L1 обозначает отсутствие группы (то есть, одинарную связь), поскольку гидролиз изображен как дающий группу -COOH, однако в других вариантах осуществления изобретения L1 также может представлять собой C1-C8алкилен.
В варианте осуществления гидантоина настоящее изобретение включает высокоэффективный способ получения соединения формулы (2-I):

где:
Y1 и Y2 независимо представляют собой метил или вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу с 4-5 атомами углерода;
R1 представляет собой L1-C(=O)-NR4R5 или L1-CN; где L1 представляет собой одинарную связь или C1-C8алкилен; и
R4 и R5 независимо выбраны из H и C1-C8алкила; и
R2 представляет собой водород или фтор;
включающий следующие стадии:
взаимодействие соединения формулы A:

где LG представляет собой Br, I или другую удаляемую группу, с соединением формулы B:

с образованием соединения формулы C:

взаимодействие соединения формулы C с соединением формулы R6-OH в условиях этерификации или, альтернативно, взаимодействие соединения формулы C с соединением формулы R6-LG, где R6 представляет собой C1-C8алкил, и LG представляет собой Br, I или другую удаляемую группу, с образованием соединения формулы D:

взаимодействие соединения формулы D с соединением формулы 2-F, 4-изоцианато-2-(трифторметил)бензонитрилом:

с образованием соединения формулы (2-I):

Общая схема для этого варианта осуществления взаимодействия показана на схеме 2 в следующем порядке A→C→D→2-I.
В варианте осуществления настоящее изобретение включает высокоэффективный способ получения соединения формулы (2-I):
где:
Y1 и Y2 независимо представляют собой метил или вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу с 4-5 атомами углерода;
R1 представляет собой L1-C(=O)-NR4R5 или L1-CN; где L1 представляет собой одинарную связь или C1-C8алкилен; и
R4 и R5 независимо выбраны из H и C1-C8алкила; и
R2 представляет собой водород или фтор;
включающий взаимодействие соединения формулы A:

с соединением формулы B:

с получением соединения формулы C:

взаимодействие соединения формулы C с соединением формулы E:

с образованием соединения формулы G:

и взаимодействие соединения формулы G с фосгеном:
с получением соединения диарилгидантоина формулы (2-I):

В этом альтернативном варианте осуществления соединение формулы C обрабатывают соединением E в условиях образования амидной связи, с получением соединения формулы G, с последующей обработкой реагентом, таким как фосген, с образованием соединения формулы 2-I (то есть, в порядке C→G→2-I на схеме 2).
В одном варианте осуществления изобретения соединение формулы A смешивают с соединением формулы B в присутствии каталитического количества как катализатора на основе меди (I), так и бета-дион лиганда, такого как 2-ацетилциклогексанон, в полярном растворителе и нагреванием до температуры около 90-120°C, около 100-110°C или около 105°C. Катализатором на основе меди (I) может быть хлорид меди (I) или йодид меди (I). Катализатор на основе меди (I), такой как CuCl, может присутствовать в количестве около 0,05-0,35 эквивалента относительно соединения A, около 0,15-0,25 эквивалента относительно соединения A, или около 0,2 эквивалента относительно соединения A. Лиганд, такой как 2-ацетилциклогексанон, может присутствовать в количестве около 0,05-0,35 эквивалента относительно соединения A, около 0,15-0,25 эквивалента относительно соединения A или около 0,2 эквивалента относительно соединения A. В другом варианте осуществления изобретения лиганд, такой как 2-ацетилциклогексанон, присутствует в количестве приблизительно равном количеству используемого катализатора на основе меди (I), такого как хлорид меди (I). Соединение B может быть добавлено в количестве около 1-2 эквивалентов относительно соединения A, около 1,25-1,75 эквивалента относительно соединения A или около 1,5 эквивалентов относительно соединения A. Выбор бета-дион лигандов, например, 2,4-пентандиона, 2,4-гександиона, 1-фенил-1,3-бутандиона, 2-ацетилциклогексанона и тому подобное, известен специалистам в данной области. Полярный растворитель может быть выбран из группы, состоящей из диметилсульфоксида (ДМСО), диметилформамида (ДМФА), N-метилпирролидона (NMP), диметилацетамида (DMA), изопропилацетата (IPAc), изопропилового спирта (IPA) и подобного; в другом варианте осуществления изобретения полярным растворителем является ДМФА, вода или смесь ДМФА и воды. В другом варианте осуществления изобретения после взаимодействия в течение около 6-24 час, около 8-20 час или около 12-14 час, или после того, как анализ показал около 90% или более расхода соединения A, реакционную смесь затем охлаждают до температуры около 15-25°C, например, до около 25°C или до комнатной температуры. В другом варианте осуществления изобретения к охлажденной реакционной смеси добавляют воду, затем промывают несмешивающимся с водой органическим растворителем, таким как изопропилацетат; затем смесь разделяют на органический и водный слои. В другом варианте осуществления изобретения водный слой подкисляют для выделения соединения C осаждением, фильтрованием и сушкой.
В одном варианте осуществления изобретения соединение C подвергают взаимодействию с алкилирующим агентом формулы R6-LG, где R6 представляет собой С1-С8алкил, и LG представляет собой Br, I или другую удаляемую группу, с образованием соединения формулы D. Соединения формулы R6-LG включают соединения, такие как метилйодид. Взаимодействие может быть осуществлено в присутствии неорганического основания, такого как K2CO3, KHCO3, Na2CO3 или NaHCO3, в полярном растворителе, таком как ДМСО, ДМФА, NMP, DMA или IPAc, и с каталитическим количеством воды. Каталитическое количество воды может составлять около 5-25%, 10-20% или 14% эквивалентов соединения C, или около 0,05-0,25%, 0,10-0,20% или 0,14% объема полярного растворителя. Реакционная смесь может быть нагрета до около 35-50°C или около 40-46°C в течение около 5-60 мин, или до тех пор, пока анализ не покажет более чем около 90%-ое или около 95% или около 99% преобразование соединения C в соединение D. После взаимодействия смесь может быть охлаждена до температуры около 5-25°C или около 15-25°C. Реакционная смесь, содержащая соединение D, может быть объединена с водой для осаждения продукта D из раствора. Продукт D может быть выделен путем фильтрования и сушки. В одном варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 2 эквивалентов, или менее чем около 2 эквивалентов, относительно соединения C. В другом варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 1,5 эквивалента или менее чем около 1,5 эквивалентов относительно соединения C. В другом варианте осуществления изобретения количество используемого неорганического основания, такого как K2CO3, составляет около 1,2 эквивалента или менее чем около 1,2 эквивалента относительно соединения C.
В другом варианте осуществления изобретения, когда CH3I используют для синтеза D (где R6=CH3), избыточное количество CH3I гасят уксусной кислотой. CH3I может быть использован в количестве около 1-1,5 эквивалентов относительно соединения C, например, в количестве около 1,2 эквивалента относительно соединения C, и количество AcOH может быть добавлено в количестве около или немного больше, чем избыточное количество метилйодида (например, когда используют 1,2 эквивалента метилйодида, где метилйодид используют в избытке в 0,2 эквивалентном количестве относительно соединения C, затем может быть использовано около 0,21-0,25 эквивалента или около 0,23 эквивалента AcOH относительно соединения C) для гашения не вступившего во взаимодействие CH3I. Альтернативные метилирующие агенты, известные специалистам в данной области, такие как диметилсульфат, также могут быть использованы для этой стадии.
В другом варианте осуществления изобретения стадию объединения реакционной смеси, содержащую соединение D, с водой осуществляют постепенным добавлением воды в нагретую реакционную смесь в течение 1-2 час, до тех пор, пока не будет добавлено около 1-5 объемов воды, или около 1-3 объемов воды, или около 2 объемов воды (относительно объема исходной реакционной смеси), для медленного осаждения соединения D и снижения количества неорганического катиона и основания, такого как K+ и CO3 2-, из неорганического основания, такого как K2CO3, используемого при взаимодействии. В другом варианте осуществления изобретения осажденное соединение D снова суспендируют или повторно образуют взвесь в воде, и затем воду удаляют путем фильтрования, для дополнительного снижения количества присутствующих неорганических катионов.
В одном варианте осуществления изобретения остаточное количество неорганического катиона, такого как ион калия, остающегося в соединении D, меньше чем или равно около 1000 частей на миллион (ч./млн.). В другом варианте осуществления изобретения остаточное количество неорганического катиона, такого как ион калия, остающегося в соединении D, меньше чем или равно около 500 ч./млн. В другом варианте осуществления изобретения остаточное количество неорганического катиона, такого как ион калия, остающегося в соединении D, меньше чем или равно около 300 ч./млн.
В одном варианте осуществления изобретения остаточное количество основания, такого как ион бикарбоната, ион карбоната или другого основания, остающегося в соединении D, меньше чем или равно около 1000 ч./млн. (частей на миллион). В другом варианте осуществления изобретения остаточное количество основания, остающегося в соединении D, меньше чем или равно около 500 ч./млн. В другом варианте осуществления изобретения остаточное количество основания, остающегося в соединении D, меньше чем или равно около 300 ч./млн.
В одном варианте осуществления изобретения соединение D может быть высушено продувкой или вытяжкой сухого воздуха, сухого азота или аргона или другого сухого инертного газа через соединение. В другом варианте осуществления изобретения соединение D может быть высушено помещением соединения в вакуум (например, при около 1 мм рт.ст. при вакууме или ниже, 0,5 мм рт.ст. при вакууме или ниже, или 0,1 мм рт.ст. при вакууме или ниже). В одном варианте осуществления изобретения остаточное количество воды, остающейся в соединении D, меньше чем или равно около 0,5%. В одном варианте осуществления изобретения остаточное количество воды, остающейся в соединении D, меньше чем или равно около 0,3%. В одном варианте осуществления изобретения остаточное количество воды, остающейся в соединении D, меньше чем или равно около 0,1%. В одном варианте осуществления изобретения остаточное количество воды, остающейся в соединении D, меньше чем или равно около 500 ч./млн. В одном варианте осуществления изобретения остаточное количество воды, остающейся в соединении D, меньше чем или равно около 300 ч./млн. В одном варианте осуществления изобретения остаточное количество воды, остающейся в соединении D, меньше чем или равно около 100 ч./млн.
В альтернативном способе получения соединения D из соединения C используют стандартные условия этерификации по Фишеру, включающие смешивание соединения C в метаноле и нагревание в течение около 1-16 час при около 40-100°C (или при кипячении с обратным холодильником) с каталитическим количеством кислоты, например, одна-пять капель серной кислоты, хлористоводородной кислоты, азотной кислоты, фосфорной кислоты, или других неорганических кислот, п-толуолсульфоновой кислоты или ионообменной смолы, содержащей сульфоновую кислоту; в одном варианте осуществления изобретения используют H2SO4. В некоторых вариантах осуществления вода может быть удалена путем азеотропной отгонки (такой как ловушкой Дина-Старка). После завершения этерификации (завершение около 70%, около 80%, около 90%, около 95% или около 99%), выделение соединения D может быть осуществлено способом, описанным выше.
В другом варианте осуществления изобретения стадия получения соединения 2-I включает смешивание соединения D с соединением 2-F в полярном растворителе, или смеси первого полярного растворителя и второго полярного растворителя, и нагревание до около 60-100°C, около 80-100°C или около 80-85°C в течение около 1-48 час или около 12-24 час. В другом варианте осуществления изобретения после взаимодействия процесс продолжают охлаждением реакционной смеси до около 15-30°C, до около 25°C или до комнатной температуры, и объединением с водой с последующим экстрагированием желаемого продукта полярным растворителем или смесью третьего полярного растворителя и четвертого полярного растворителя. Соединение 2-F может быть добавлено в количестве около 1-3 эквивалентов относительно соединения D, или около 1,5-2,5 эквивалентов относительно соединения D или около 1,5 эквивалента или около 2 эквивалентов относительно соединения D или в количестве около 1,5 эквивалентов, затем, по мере протекания реакции, дополнительной части около 0,5 эквивалента. Слой объединенного органического экстракта может быть уменьшен в объеме и затравлен кристаллами желаемого продукта 2-I для начала кристаллизации при охлаждении до около 0-10°C или около 3-6°C, с последующим выделением кристаллического продукта путем фильтрования и дальнейшей сушкой продукта продувкой воздуха через продукт или в вакууме. В одном аспекте этого варианта осуществления полярный растворитель или первый, второй, третьей и четвертый полярные растворители могут быть выбраны из группы, состоящей из: ДМСО, ДМФА, NMP, DMA, IPAc, MeCN, IPA и подобного. В одном варианте осуществления изобретения полярным растворителем является ДМФА. В другом варианте осуществления изобретения первым полярным растворителем является IPAc, и вторым полярным растворителем является ДМСО. В другом варианте осуществления изобретения третьим полярным растворителем является IPAc, и четвертым полярным растворителем является IPA. В другом варианте осуществления изобретения первым полярным растворителем является IPAc, вторым полярным растворителем является ДМСО, третьим полярным растворителем является IPAc, и четвертым полярным растворителем является IPA.
В другом варианте осуществления изобретения альтернативный способ образования соединения 2-I включает две стадии, выполняемые в порядке C→G→2-I на схеме 2. На первой стадии используют стандартные условия образования амидной связи, включая, например, обработку соединения C конденсирующим реагентом, таким как дициклогексилкарбодиимид (DCC), диизопропилкарбодиимид (DIC), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDCI), гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (BOP), гексафторфосфат 7-азабензотриазол-1-илокситрис(диметиламино)фосфония (AOP), гексафторфосфат бензотриазол-1-илокситрис(пирролидин)фосфония (PyBOP), гексафторфосфат 7-азабензотриазол-1-илокситрис(пирролидин)фосфония (PyAOP), гексафторфосфат О-бензотриазол-N,N,N',N'-тетраметилурония (HBTU), тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU), тетрафторборат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TATU) и тому подобное, с соединением E в полярном растворителе или смеси первого полярного растворителя и второго полярного растворителя, с получением соединения G. В одном аспекте этого варианта осуществления полярный растворитель или первый и второй полярные растворители выбраны из группы, состоящей из DCM, ДМСО, ДМФА, NMP, DMA, MeCN и подобного. Вторая стадия включает взаимодействие замыкания кольца соединения G с карбонилирующим реагентом, таким как фосген, и нагревание самого раствора до около 60-120°C. В другом варианте осуществления изобретения взаимодействие осуществляют в запаянной трубке. Фосген или эквивалент фосгена (например, карбонилдиимидазол) может присутствовать в количестве около 1-10 эквивалентов относительно соединения G или около 5 эквивалентов относительно соединения G.
В другом варианте осуществления изобретения соединение 2-I может быть подвергнут воздействию условий гидролиза, когда R1 представляет собой группу первичного или вторичного амида, с получением соответствующего производного карбоновой кислоты.
В одном варианте осуществления указанного выше способа заместитель R1 соединения формулы A представляет собой -C(=O)-NH-R4. В другом варианте осуществления изобретения R1 представляет собой -C(=O)-NH-CH3. В другом варианте осуществления изобретения заместитель R2 соединения формулы A представляет собой фтор. В другом варианте осуществления изобретения R1 представляет собой -C(=O)-NH-R4 и R2 представляет собой фтор. В другом варианте осуществления изобретения R1 представляет собой -C(=O)-NH-CH3, и R2 представляет собой фтор, и соединение формулы A представляет собой 4-бром-2-фтор-N-метилбензамид.
В одном варианте осуществления указанного выше способа соединение формулы B представляет собой 2-аминоизомасляную кислоту (то есть, Y1 и Y2, каждый представляет собой CH3). В другом варианте осуществления указанного выше способа соединение формулы B представляет собой 1-аминоциклобутанкарбоновую кислоту. В другом варианте осуществления указанного выше способа соединение формулы B представляет собой 1-аминоциклопентанкарбоновую кислоту.
В другом варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, R1 представляет собой -C(=O)NHCH3, и/или R2 представляет собой F.
В другом варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, R1 представляет собой -C(=O)NH2, и/или R2 представляет собой F.
В другом варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, R1 заменен на группу -C(=O)OH, и/или R2 представляет собой F.
Также предусмотрены варианты соединения формулы (2-I). Соединения формулы (2-I) или его варианты, как подробно рассмотрено в настоящем описании, или фармацевтически приемлемая соль любого из указанных выше соединений могут найти конкретное применение при лечении рака предстательной железы, включая CRPC и/или гормоно-чувствительный рак предстательной железы.
В альтернативном варианте осуществления, где группа -C(=O)OH заменяет группу R1, конечный продукт может быть синтезирован следующим образом:

где соединение C подвергают взаимодействию с соединением 2-F (4-изоцианато-2-(трифторметил)бензонитрилом), с получением продукта 2-M. Взаимодействие может быть осуществлено в основных условиях с помощью триалкиламинного основания, такого как триэтиламин, присутствующего в количестве около 2-5 эквивалентов или около 3-4 эквивалентов, или около 3,4 эквивалента относительно соединения C. Соединение 2-F, 4-изоцианато-2-(трифторметил)бензонитрил, может присутствовать в количестве около 1,1-4 эквивалентов или 1,1-2 эквивалентов, или около 1,5 эквивалентов относительно соединения C; альтернативно, может быть добавлено около 1,5 эквивалентов соединения 2-F, затем, по мере протекания реакции, дополнительная часть, составляющая около 0,5 эквивалента. Растворителем может быть этанол или другой спирт. Реакционная смесь может быть перемешана в течение около 4-16 дней, около 8-12 дней или около 10 дней при комнатной температуре или повышенной температуре. Затем реакционная смесь может быть концентрирована, смешана с водным раствором кислоты, например, 1M HCl, и продукт экстрагирован органическим растворителем, таким как этилацетат, с получением продукта 2-M.
В дополнительном варианте осуществления соединение C синтезируют путем взаимодействия соединения формулы J с 1,1-дизамещенным 2,2,2-трихлорэтанолом:

где 1,1-дизамещенный 2,2,2-трихлорэтанол может быть использован в количестве около 1,5-4 эквивалентов относительно соединения J, или около 2-3 эквивалентов относительно соединения J, около 2,5 эквивалентов относительно соединения J, или около 2,6 эквивалента относительно соединения J. Реакцию проводят в органическом растворителе, предпочтительно, в безводном растворителе, таком как безводный ацетон. Реакционная смесь может быть охлаждена до температуры 0°C перед добавлением сильного основания, такого как NaOH, KOH или другого гидроксида. Основание добавляют в количестве около 2-5 эквивалентов, или около 3-4 эквивалентов, или около 3,8 эквивалента, или около 3,9 эквивалента относительно соединения J. После добавления основания, реакционная смесь может быть оставлена для нагревания до комнатной температуры, и оставлена при комнатной температуре в течение около 4-24 час, или около 8-16 час, или около 12 час. Продукт может быть очищен стандартными способами, такими как колоночная хроматография или ВЭЖХ.
В другом варианте осуществления изобретения изобретение включает способ получения соединения формулы (I,2-Ia):

где X представляет собой S или O; Y1 и Y2 независимо представляют собой метил или вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу с 4-5 атомами углерода; R7 представляет собой L1-COOH, где L1 представляет собой одинарную связь или C1-C8алкилен; и R4 и R5 независимо выбраны из H и C1-C8алкила; и R2 представляет собой водород или фтор; включающий взаимодействие соединения формулы Aa:

где LG представляет собой удаляемую группу, Br или I; с соединением формулы B:

с получением соединения формулы Ca:

взаимодействие соединения формулы Ca с соединением формулы R6-LG в условиях алкилирования или с соединением формулы R6-OH в условиях этерификации, с образованием соединения формулы Da:

где R6 представляет собой C1-C8алкил; и взаимодействие соединения формулы Da с соединением формулы (F,2-F):

где X представляет собой S или O, с получением соединения диарилтиогидантоина или диарилгидантоина формулы (I,2-Ia):

В одном варианте осуществления изобретения X представляет собой S. В другом варианте осуществления изобретения X представляет собой O. В любом из этих вариантов осуществления L1 может представлять собой одинарную связь; и R7 может представлять собой -C(=O)-OH. В любом из этих вариантов осуществления Y1 и Y2, оба могут представлять собой метил, R7 может представлять собой -C(=O)-OH, и R2 может представлять собой F.
В другом варианте осуществления изобретения фармацевтические композиции любого из соединений, подробно описанных в настоящем описании, включены в настоящее изобретение. Таким образом, изобретение включает фармацевтические композиции, содержащие соединение, описанное в настоящем описании, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или эксципиент. Фармацевтические композиции согласно изобретению могут быть изготовлены в форме, подходящей для перорального, буккального, парентерального, подкожного, внутримышечного, внутривенного, назального, местного или ректального введения, или форме, подходящей для введения путем ингаляции.
Подробное описание изобретения
Определения
Для использования в настоящем описании, если ясно не указано иное, использование терминов в единственном числе относится также и к множественному числу.
Термин "около", как используется в настоящем описании, относится к обычному интервалу отклонений для соответствующего значения, хорошо известное специалисту в данной области техники. Ссылка на "около" значения или параметра в настоящем описании включает (и описывает) варианты осуществления, которые касается этого значения или параметра как такового.
Используемая в настоящем описании фраза "фармацевтически приемлемый" или "фармакологически приемлемый" означает вещество, которое не является ни биологически, ни с какой-либо другой точки зрения нежелательным, например, вещество может быть включено в фармацевтическую композицию, вводимую пациенту, не вызывая каких-либо значительных нежелательных биологических эффектов или вредного действия при взаимодействии с любым другим компонентом композиции, в которой оно содержится.
Фармацевтически приемлемые носители или эксципиенты, предпочтительно, соответствуют необходимым стандартам токсикологических и производственных исследований и/или включены в Руководство по неактивным компонентам, составленное Управлением по контролю качества пищевых продуктов и лекарственных средств США.
"Фармацевтически приемлемыми солями" являются соли, которые сохраняют, по меньшей мере, частично, биологическую активность свободного (не соль) соединения, и которые могут быть введены субъекту в качестве препаратов или лекарственных средств. Такие соли, например, включают: (1) кислотно-аддитивные соли, образуемые с неорганическими кислотами, такими, как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное; или образованные с помощью органических кислот, таких как уксусная кислота, щавелевая кислота, пропионовая кислота, янтарная кислота, малеиновая кислота, винная кислота и тому подобное; (2) соли, образуемые в условиях, когда кислотный протон, содержащийся в исходном соединении, либо заменен ионом металла, например, ионом щелочного металла, таким как калий или натрий, ионом щелочноземельного металла, таким как ион кальция или алюминия; либо образует координационную связь с органическим основанием. Приемлемые органические основания включают этаноламин, диэтаноламин, триэтаноламин и тому подобное. Приемлемые неорганические основания включают гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия, гидроксид натрия и тому подобное. Другие примеры фармацевтически приемлемых солей включают соли, описанные в Berge et al, Pharmaceutical Salts, J. Pharm. Sci. 1977 Jan; 66(l): 1-19. Фармацевтически приемлемые соли могут быть получены in situ в процессе производства или отдельно взаимодействием очищенного соединения настоящего изобретения в форме своей свободной кислоты или основания с подходящим органическим или неорганическим основанием или кислотой, соответственно, и выделением образованной таким образом соли в процессе последующей очистки. Следует отметить, что ссылка на фармацевтически приемлемую соль охватывает ее формы с присоединением растворителя или кристаллические формы, в частности, сольваты и полиморфы. Сольваты содержат либо стехиометрическое, либо нестехиометрическое количество растворителя и часто образуются в процессе кристаллизации. Когда растворителем является вода, образуются гидраты, и когда растворителем является спирт, образуются алкоголяты. Полиморфы включают кристаллические конструкции различных структур одинакового элементного состава соединения. Полиморфы обычно характеризуются различными дифракционными рентгенограммами, инфракрасными спектрограммами, точками плавления, показателями плотности, твердости, формами кристаллов, оптическими и электрическими свойствами, стабильностью и растворимостью. Различные факторы, такие как растворитель, скорость кристаллизации при перекристаллизации и температура хранения, могут обусловить доминирование одной кристаллической формы.
Термин "эксципиент", используемый в настоящем описании, означает инертное или неактивное вещество, которое может быть использовано при производстве препарата или лекарственного средства, такого как таблетка, содержащего соединение настоящего изобретения в качестве активного ингредиента. Различные вещества могут быть охвачены термином эксципиент, включая, но без ограничения, любое вещество, используемое в качестве связующего вещества, дезинтегранта, образующего покрытие вещества, добавки для прессования/инкапсуляции, крема или лосьона, лубриканта, растворов для парентерального введения, веществ для жевательных таблеток, подсластителя или ароматизатора, суспендирующего/желатинирующего агента или агента для влажной грануляции. Связующие вещества включают, например, карбомеры, повидон, ксантановую смолу и так далее; вещества, образующие покрытие, включают, например, ацетатфталат целлюлозы, этилцеллюлозу, геллановую камедь, мальтодекстрин, энтеросолюбильные покрытия и так далее; добавки для прессования/инкапсуляции включают, например, карбонат кальция, декстрозу, фруктозу dc (dc="прямое прессование"), сироп dc, лактозу (негидрат или моногидрат; необязательно в комбинации с аспартамом, целлюлозой или микрокристаллической целлюлозой), крахмал dc, сахарозу и так далее; дезинтегранты включают, например, натрийкросскармелозу, геллановую камедь, крахмалгликолят натрия и так далее; кремы или лосьоны включают, например, мальтодекстрин, каррагинаны и так далее; лубриканты включают, например, стеарат магния, стеариновую кислоту, стеарилфумарат натрия и так далее; вещества для жевательных таблеток включают, например, декстрозу, фруктозу dc, лактозу (моногидрат, необязательно в комбинации с аспартамом или целлюлозой) и так далее; суспендирующие/желатинирующие агенты включают, например, каррагенан, крахмалгликолят натрия, ксантановую смолу и так далее; подсластители включают, например, аспартам, декстрозу, фруктозу dc, сорбит, сахарозу dc и так далее; и агенты для влажной грануляции включают, например, карбонат кальция, мальтодекстрин, микрокристаллическую целлюлозу и так далее.
"Алкил" относится и включает насыщенные линейные разветвленные или циклические углеводородные структуры и их комбинации. Конкретными алкильными группами являются группы, содержащие 1-12 атомов углерода ("C1-C12алкил"). Более конкретными алкильными группами являются группы, содержащие 1-8 атомов углерода ("C1-C8алкил"). Когда указан алкильный остаток, содержащий конкретное количество атомов углерода, все геометрические изомеры, имеющие такое же количество атомов углерода, включены и описаны в настоящем изобретении; таким образом, например, "бутил" включает н-бутил, втор-бутил, изобутил, трет-бутил и циклобутил; "пропил" включает н-пропил, изопропил и циклопропил. Этот термин иллюстрируется группами, такими как метил, трет-бутил, н-гептил, октил, циклогексилметил, циклопропил и тому подобное. Циклоалкил представляет собой подгруппу алкила и может состоять из одного кольца, такого как циклогексил, или нескольких колец, таких как адамантил. Циклоалкил, содержащий более одного кольца, может быть конденсированным, спиро или соединенным мостиковой связью, или их комбинацией. Предпочтительный циклоалкил имеет от 3 до 12 атомов углерода кольца. Более предпочтительный циклоалкил имеет от 3 до 7 атомов углерода кольца ("C3-C7циклоалкил"). Примеры циклоалкильных групп включают адамантил, декагидронафталинил, циклопропил, циклобутил, циклопентил и тому подобное.
"Замещенный алкил" относится к алкильной группе, имеющей от 1 до 5 заместителей, включая, но, не ограничиваясь ими, такие как алкокси, замещенный алкокси, ацил, ацилокси, карбонилалкокси, ациламино, замещенный или незамещенный амино, аминоацил, аминокарбониламино, аминокарбонилокси, арил, замещенный арил, гетероарил, замещенный гетероарил, арилокси, замещенный арилокси, циано, галоген, гидроксил, нитро, карбоксил, тиол, тиоалкил, замещенный или незамещенный алкенил, замещенный или незамещенный алкинил, замещенный или незамещенный гетероциклил, замещенный или незамещенный аралкил, аминосульфонил, сульфониламино, сульфонил, оксо, карбонилалкиленалкокси и тому подобное.
"Удаляемые группы" представляют собой группы, которые убирают электронную пару при гетеролитическом расщеплении связи, например, которое происходит во время нуклеофильного замещения. Подходящие удаляемые группы, включают, например: Cl, Br, I, трифлаты, соли диазония, фторсульфонаты, тозилаты и мезилаты. Конкретные удаляемые группы включают Cl, Br или I. Более конкретные группы включают Br или I.
Признаки и эффекты настоящего изобретения будут далее пояснены со ссылкой на варианты осуществления, рассмотренные ниже, которые, однако, не предназначены для ограничения объема настоящего изобретения.
Способ
Настоящее изобретение включает высокоэффективный способ получения соединений диарилтиогидантоина формулы (I):

где:
Y1 и Y2 независимо представляют собой метил или вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу с 4-5 атомами углерода;
R1 представляет собой L1-C(=O)-NR4R5 или L1-CN; где L1 представляет собой одинарную связь или C1-C8алкилен; и
R4 и R5 независимо выбраны из H и C1-C8алкила; и
R2 представляет собой водород или фтор;
включающий следующие стадии:

где синтез соединения F из соединения E является необязательной частью способа, и где CH3I может быть заменен на R6-LG, или R6-OH, где R6 представляет собой C1-C8алкил, и LG представляет собой Br, I или другую удаляемую группу. В одном варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, R1 представляет собой -C(=O)NHCH3, и/или R2 представляет собой F. В одном варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, R1 представляет собой -C(=O)NHCH3 и R2 представляет собой F. В другом варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, R1 представляет собой -C(=O)NH2, и/или R2 представляет собой F. В другом варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, R1 представляет собой -C(=O)NH2, и R2 представляет собой F. В другом варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, R1 заменен на -C(=O)OH, и/или R2 представляет собой F. В другом варианте осуществления изобретения Y1 и Y2, каждый представляет собой CH3, R1 заменен на -C(=O)OH, и R2 представляет собой F. В другом варианте осуществления изобретения Y1 и Y2, вместе с атомом углерода, с которым они связаны, образуют циклобутановое кольцо, R1 заменен на -C(=O)OH, и/или R2 представляет собой F.
В другом варианте осуществления изобретения Y1 и Y2, вместе с атомом углерода, с которым они связаны, образуют циклобутановое кольцо, R1 заменен на -C(=O)OH, и R2 представляет собой F. В другом варианте осуществления изобретения Y1 и Y2, вместе с атомом углерода, с которым они связаны, образуют циклобутановое кольцо, R1 представляет собой -C(=O)NH2, и/или R2 представляет собой F. В другом варианте осуществления изобретения Y1 и Y2, вместе с атомом углерода, с которым они связаны, образуют циклобутановое кольцо, R1 представляет собой -C(=O)NH2, и R2 представляет собой F. В другом варианте осуществления изобретения Y1 и Y2, вместе с атомом углерода, с которым они связаны, образуют циклобутановое кольцо, R1 представляет собой -C(=O)NHCH3, и/или R2 представляет собой F. В другом варианте осуществления изобретения Y1 и Y2, вместе с атомом углерода, с которым они связаны, образуют циклобутановое кольцо, R1 представляет собой -C(=O)NHCH3, и R2 представляет собой F.
Синтез, как описано выше, включает способ синтеза соединения C, который включает смешивание коммерчески доступного варианта соединения A с соединением B в присутствии каталитического количества как катализатора на основе меди (I), так и лиганда, такого как ацетилциклогексанон, в полярном растворителе и нагревание реакционной смеси, с последующим охлаждением, добавлением воды и промывкой органическим растворителем, затем подкислением водного слоя для выделения желаемого продукта C осаждением, фильтрованием и сушкой. Катализаторы на основе меди для использования в изобретении могут быть выбраны из группы, состоящей из хлорида меди (I) и йодида меди (I). Обычно используют хлорид меди (I) (Cai et al., Synthesis (Thieme Publishing Group) 2005, No. 3, pp. 496-499).
Соединение D может быть синтезировано способом, который включает смешивание кислоты C с алкилирующим агентом, таким как метилйодид, и неорганическим основанием в полярном растворителе и каталитическом количестве воды и нагревание, последующее охлаждение смеси и объединение с водой, после чего продукт D осаждают из раствора и выделяют путем фильтрования и сушки. В альтернативном способе для данного способа используют стандартные условия этерификации по Фишеру, включающие смешивание кислоты C в метаноле и нагревание с каталитической неорганической кислотой, последующее выделение, как описано выше. Неорганическое основание для алкилирования может быть выбрано из группы, состоящей из карбоната калия, карбоната натрия, бикарбоната натрия и карбоната цезия, и обычно используют карбонат калия. Неорганическая кислота для этерификации по Фишеру может быть выбрана из группы, состоящей из серной кислоты, хлористоводородной кислоты, азотной кислоты и фосфорной кислоты, и обычно используют серную кислоту.
Предварительная работа над взаимодействием показала, что количество неорганических катионов, то есть, остаточных ионов металла, и влага, присутствующая в соединении D, оказывают влияние на взаимодействие соединения D с соединением F для получения соединения I. Дальнейшее исследование показало, что фактически имело место присутствие остаточного основания, которое вызывало нежелательные побочные реакции. Однако неорганические катионы служат в качестве полезных показателей количества основания, остающегося при получении соединения D. Были выполнены процедуры для минимизации количества неорганических катионов (таких как K+ или Na+), присутствующих в соединении D, и, соответственно, минимизации количества основания, остающегося в соединении D. Эти методы включали медленное и постепенное осаждение продукта D из его реакционной смеси, путем медленного добавления воды к подогретой реакционной смеси, и дополнительное повторное суспендирование или повторное образование взвеси соединения D в воде для экстракции катионов. ("Повторное суспендированное" или "повторно образующее взвесь" соединение означает повторное образование взвеси соединения.) Влага также оказывает негативное влияние на взаимодействие соединения D с соединением F для образования соединения I. Влага может быть удалена из соединения D продувкой соединения сухим воздухом, сухим азотом, сухим аргоном или иным сухим газом, помещая соединение на фильтр (например, в воронку с фильтром из спеченого стекла) и продувая воздух или другой сухой газ через соединение, или помещая соединение в вакуум на определенный промежуток времени.
Соединение I может быть синтезировано смешиванием соединения D с соединением F в смеси первого полярного растворителя и второго полярного растворителя, и нагреванием, затем охлаждением смеси и объединением с водой, экстрагированием желаемого продукта смесью третьего полярного растворителя и четвертого полярного растворителя. Слой объединенного органического экстракта уменьшают в объеме и затравляют кристаллами желаемого продукта I для начала кристаллизации при охлаждении, после чего кристаллический продукт выделяют фильтрованием и сушкой.
Первый, второй, третьей и четвертый полярные растворители могут быть выбраны из группы, состоящей из диметилсульфоксида (ДМСО), диметилформамида (ДМФА), N-метилпирролидона (NMP), диметилацетамида (DMA), изопропилацетата (IPAc), изопропилового спирта (IPA) и подобного. В одном варианте осуществления изобретения первым полярным растворителем является IPAc, вторым полярным растворителем является ДМСО, третьим полярным растворителем является IPAc, и четвертым полярным растворителем является IPA.
Продукт I может быть подвергнут процессу кристаллизации с получением насыщенного раствора в органическом растворителе или смешанном растворителе, концентрированием раствора, необязательным добавлением затравки продукта I и охлаждением раствора до температурного предела, и поддержанием раствора в этом температурном пределе в течение достаточного периода времени до тех пор, пока не завершится кристаллизация продукта I. Этот процесс кристаллизации может быть выполнен при температуре в пределах около 0-80°C, обычно 0-10°C.
Соединение I также быть синтезировано сначала обработкой соединения C конденсирующим агентом, таким как дициклогексилкарбодиимид (DCC), диизопропилкарбодиимид (DIC), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDCI) и тому подобное, с соединением E, в полярном растворителе или смеси первого полярного растворителя и второго полярного растворителя, с получением соединения G, затем обработкой избыточным количеством тиофосгена при нагревании, с получением соединения I. Тиофосген может присутствовать в количестве около 1-10 эквивалентов относительно соединения G или около 5 эквивалентов относительно соединения G.
Полярный растворитель или первый и второй полярные растворители могут быть выбраны из группы, состоящей из DCM, ДМСО, ДМФА, NMP, DMA, MeCN и подобного.
Соединение I может быть подвергнуто воздействию условий гидролиза, когда R1 представляет собой группу первичного, вторичного или третичного амида, с получением соответствующего производного карбоновой кислоты.
В необязательной методике синтеза в способе настоящего изобретения предусмотрен способ синтеза соединения F, который включает смешивание коммерчески доступного варианта соединения E с тиофосгеном в смеси органического растворителя, такого как неполярный растворитель, и воды при температуре окружающей среды, добавление воды и выделение изотиоцианатного соединения продукта F. Слой объединенного органического экстракта уменьшают в объеме и добавляют второй органический растворитель, такой как неполярный растворитель, с затравкой кристаллами желаемого продукта F для начала кристаллизации, после чего кристаллический продукт выделяют путем фильтрования и сушки. Органический растворитель может быть выбран из группы, состоящей из дихлорметана (DCM), толуола, хлороформа, гексанов, гептана и 1,4-диоксана, более предпочтительным является DCM или гептан. Тиофосген может быть использован в количестве, равном около 1-1,5 моль, например, 1,1 моль, на моль анилина E. Тиофосген может быть добавлен в течение периода времени в пределах от 30 мин до 2 часов, например, в течение 1 часа.
Продукт F может быть подвергнут процессу кристаллизации, путем получения насыщенного раствора в органическом растворителе или смешанном растворителе, концентрирования раствора и охлаждения раствора до температурного предела и поддержания раствора в этом температурном пределе в течение достаточного периода времени до тех пор, пока не завершится кристаллизация продукта F. Процесс кристаллизации может быть выполнен при температуре в диапазоне от около 0°C до около 50°C, от около 10°C до около 4°C, от около 20°C до около 30°C, или от около 20°C до около 25°C, или от около 25°C до около 30°C, или около 20°C, или около 21°C, или около 22°C, или около 23°C, или около 24°C, около 25°C, около 26°C, около 27°C, около 28°C, около 29°C или около 30°C. Органическим растворителем, используемым для кристаллизации, может быть н-гептан или смесь н-гептана и IPAc. Например, может быть использовано от около 0,11% моль до около 0,65% моль IPAc в н-гептане, или от около 0,20% моль до около 0,55% моль IPAc в н-гептане, или может быть использовано от около 0,03 до около 0,06 процентов по массе IPAc в н-гептане, или могут быть использованы около 0,20, около 0,36, около 0,37, около 0,38, около 0,41, около 0,54, или около 0,55% моль IPAc в н-гептане. Чтобы способствовать началу кристаллизации, раствор кристаллизуемого продукта F может быть затравлен небольшим количеством предварительно выделенного F, например, от около 0,2 до 0,5% по массе теоретически получаемого количества F. Количество используемого для затравки F может быть в пределах от около 0,20% до около 0,50% (% масс.) количества F, необходимого для повторной кристаллизации, например, около 0,20%, около 0,25%, около 0,30%, около 0,35%, около 0,40%, около 0,45% или около 0,50%. (Для повторной кристаллизации около 20 г продукта F необходимое количество затравочного кристалла составляет около 0,20-0,50% масс., что соответствует около 40-100 мг затравочного кристалла.) После затравки растворы/взвеси могут быть охлаждены до температуры от около 0°C до около 5°C в течение периода от около 0,5 до около 2 часов или около 1 часа. Растворы также могут быть перемешаны с высокой или низкой скоростью перемешивания, например, от около 200 оборотов в минуту до около 400 оборотов в минуту, от около 300 оборотов в минуту до около 400 оборотов в минуту, от около 200 оборотов в минуту до около 400 оборотов в минуту или при около 200, около 300 или около 400 оборотов в минуту. После кристаллизации твердое вещество далее может быть отфильтровано, промыто холодным н-гептаном (около 10-30 мл или около 20 мл) и высушено в вакууме при температуре от около 20°C до около 25°C.
Изобретение иллюстрировано посредством следующих неограничивающих объем изобретения примеров.
ПРИМЕРЫ
Экспериментальные условия
В одном аспекте настоящего изобретения, представленного на схеме 1, предложен новый и улучшенный способ получения 4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фтор-N-метилбензамида, как описано ниже в примерах 1-5. Исходные вещества получали от коммерческих поставщиков и использовали без дополнительной очистки. Взаимодействия, чувствительные к воздуху или влаге, выполняли в атмосфере аргона, с использованием высушенной в сушильном шкафу стеклянной посуды и применяя стандартные методики со шприцами и мембранами. Протекание реакций контролировали посредством ТСХ на пластинах силикагеля при УФ-излучении (254 нМ), с последующей визуализацией с помощью окрашивающего раствора пара-анизальдегида или нингидрина. Для крупномасштабных экспериментов, протекание реакций контролировали посредством ВЭЖХ с обращенной фазой. Колоночную хроматографию проводили на силикагеле 60. Спектр 1Н-ЯМР, если специально не указано иначе, записывали при 400 МГц в CDCl3, и результаты, как указано ниже, приведены в м.д. (δ) относительно внутреннего стандарта (ТМС, 0,0 м.д.): химический сдвиг (мультиплетность, интегрирование, константа взаимодействия в Гц).
Пример 1: Преобразование 4-бром-2-фторбензойной кислоты в 4-бром-2-фтор-N-метилбензамид

В продутый азотом 50 л реактор загружали сухую бензойную кислоту A-1 (1,8 кг, 8,22 моль), затем изопропилацетат (IPAc) (12,6 л, 7 об.) и ДМФА (36 мл, 0,02 эквив.). К перемешиваемой взвеси добавляли тионилхлорид (689 мл, 9,47 моль, 1,15 эквив.) в течение 5 мин (реакционную массу нагревали до 21°C-23°C). Реакционную массу нагревали до температуры 60°C в течение 2,5 час, поддерживали при 60-68°C в течение 1 часа и проводили ВЭЖХ анализ. На этой стадии реакционная масса представляла собой тонкую дисперсию. Обнаружено, что преобразование в хлорангидрид кислоты составляло 99,9% (перед проведением анализа промежуточное соединение хлорангидрида гасили N-пропиламином). После перемешивания в течение дополнительного 1 час при 70-72°C, реакционную массу охлаждали до температуры 10°C в течение 1 час.
В продутый азотом 30 л реактор загружали водный раствор MeNH2 (3,6 л, 41,1 моль, 5 экв.), затем охлаждали до температуры 2-10°C. К MeNH2 добавляли IPAc (3,6 л, 2 об.) и смесь MeNH2/IPAc охлаждали до температуры 2-10°C. Хлорангидрид переносили в смесь MeNH2/IPAc в течение 50 мин, в это время реакционную смесь нагревали до 35°C. Реактор, содержащий хлорангидрид, промывали IPAc (1,8 л, 1 об.) в 30 л реактор. Реакционную массу оставляли перемешиваться в течение 15 мин при 30-35°C, затем проводили ВЭЖХ анализ. Обнаруженное преобразование продукта составляло 100%.
Перемешивание останавливали, и фазы оставляли разделяться в течение 10 мин. Нижний слой зеленого цвета удаляли. Фазу IPAc дополнительно промывали водой (3 об., затем 1 об.). Последнему разделению фаз давали возможность протекать в течение 14 час при 30°C. После конечного разделения, фазу с IPAc фильтровали через слой целита, который промывали IPAc (3,6 л, 2 об.) для удаления вещества темно-зеленого цвета. Затем фильтрат уменьшали в объеме отгонкой до 9,5 л (5,3 об.) в течение 5 час (30-35°C, 100-200 мбар, 1,5-2,9 пси). Осаждение инициировали при 8-9 объемах. В реактор добавляли н-гептан (18 л, 10 об.), и смесь отгоняли до объема в 8 л (4,4 об.) в течение 6 час (30-35°C, 100-200 мбар, 1,5-2,9 пси). На этой стадии соотношение IPAc/н-гептан составляло 26:1. Полученную взвесь оставляли перемешиваться в течение 12 час при 25°C перед охлаждением до 5-10°C в течение 1 часа. Массу перемешивали в течение 1,5 час при 5-10°C, затем фильтровали, промывали н-гептаном (2×1 об.) и сушили воздухом. Осадок на фильтре (1,87 кг) сушили в вакууме при температуре 55-60°C в течение 141 час с выходом 1,72 кг (90% выход) желаемого амидного продукта A-2 с чистотой по ВЭЖХ, равной 99,5%, и 0,2% H2O.
Пример 2: Преобразование 4-бром-2-фтор-N-метилбензамида в 2-(3-фтор-4-(метилкарбамоил)фениламино)-2-метилпропановую кислоту

Бромбензамид A-2 (10 г, 43,1 ммоль), аминоизомасляную кислоту B-1 (6,7 г, 64,6 ммоль, 1,5 эквив.), K2CO3 (15 г, 2,5 эквив.), 99% CuCl (0,8 г, 8,1 ммоль, 0,2 эквив.), ДМФА (60 мл, 6 об.) и воду (1,8 мл) добавляли в сосуд, и реакционную взвесь нагревали до температуры 30°C. К реакционной взвеси добавляли 2-ацетилциклогексанон (1,14 мл, 8,1 ммоль, 0,2 эквив.), затем перемешивали при 105°C в атмосфере азота в течение 12-14 час. ВЭЖХ анализ показал 96,6% преобразование в желаемый продукт. Затем реакционную смесь охлаждали до комнатной температуры и экстрагировали водой (120 мл) и IPAc (60 мл). Нижний водный слой повторно экстрагировали IPAc (60 мл) и подкисляли 180 мл 1M лимонной кислоты до pH 4,0. Продукт начинал кристаллизоваться при комнатной температуре и реакционную массу затем охлаждали до температуры 5-7°C, фильтровали, промывали водой (40 мл) и сушили в вакууме при 50°C в течение 12 час. Реакция давала 8,3 г продукта C-1 (75,4% выход) в виде желтовато-коричневого твердого вещества с чистотой по ВЭЖХ, равной 99,6%.
Пример 3: Преобразование 2-(3-фтор-4-(метилкарбамоил)фениламино)-2-метилпропановой кислоты в метил 2-(3-фтор-4-(метилкарбамоил)фениламино)-2-метилпропаноат

Смесь производного метилпропионовой кислоты C-1 (4,0 г, 15,7 ммоль), карбоната калия (2,67 г, 18,8 ммоль), ДМФА (28 мл) и воды (0,04 мл) нагревали до температуры 30°C. Затем добавляли одну часть метилйодида (1,2 мл, 18,8 ммоль), и наблюдали незначительное нагревание реакционной смеси до 3°C в течение 5 мин. Затем смесь нагревали до температуры 40°C в течение 1 часа. ВЭЖХ анализ реакционной смеси показал >99,9% преобразование в продукт сложного эфира. Затем добавляли AcOH (0,3 мл), и полученную смесь нагревали до температуры 60°C, затем добавляли воду (60 мл) в течение 50 мин, поддерживая температуру реакционной массы, равной 58-63°C. Затем взвесь охлаждали до температуры 30°C, затем продукт D-1 фильтровали и промывали водой (2×8 мл). Осадок на фильтре повторно суспендировали в воде (40 мл) и промывали IPAc (2×8 мл) и сушили в вакууме при 45-50°C в течение 16 час с выходом 4 г сложного эфира (95% выход) в виде светло-коричневого твердого вещества с чистотой 99,9%, <0,1% воды и 80 ч./млн. калия.
Пример 4: Преобразование 4-амино-2-(трифторметил)бензонитрила в 4-изотиоцианато-2-(трифторметил)бензонитрил

В продутый азотом 30-литровый реактор загружали анилин E (4,0 кг, 21,49 моль), затем н-гептан (9 л, 2,25 об.) и H2O (10 л, 2,5 об.). Затем смесь перемешивали в течение 8 мин, охлаждали до температуры 5-10°C и загружали тиофосген (1,81 л, 2,72 кг, 23,64 моль, 1,1 эквив.) в течение 12 мин, поддерживая температуру реакционной массы при 10-16°C, затем промывали н-гептаном (1 л, 0,25 об.). Затем полученную взвесь оранжевого цвета нагревали до 30-40°C в течение 1,5 час и наблюдали незначительный экзотермический эффект с повышением температуры до максимального значения 46,4°C. После перемешивания в течение 15 час раствор оранжевого цвета анализировали (>99% преобразование). Затем реакционную массу нагревали до температуры 36°C и фазы оставляли разделяться. Наблюдали мутный слой и большую его часть очищали с помощью самого нижнего водного слоя. Затем двумя частями к слою гептана оранжевого цвета добавляли н-гептан (18 л, 4,5 об.) и раствор отгоняли до 1,5 об. (45-46°C, 160 мбар). Раствор еще раз разбавляли н-гептаном (8 л, 2 об.) и реакционную массу отгоняли до 1,5 об. (45-46°C, 160 мбар). Затем раствор разбавляли н-гептаном (10 л, 2,5 об.), охлаждали до температуры 30-31°C (гептан:продукт F, 5,3:1) и затравливали продуктом F (10 г). Кристаллизацию наблюдали в течение 2-3 мин после затравки, и взвесь затем охлаждали до температуры 0-10°C в течение 3 час и выдерживали при температуре 0-10°C в течение 2 час. Затем реакционную массу фильтровали, промывали фильтратом и холодным н-гептаном (4 л, 1 об.) и сушили при температуре 20-25°C в вакууме в течение 13 час с выходом продукта F (4,51 кг, 92%) с чистотой по ВЭЖХ, равной >99%, и уровнем влажности 0,04%.
Пример 5: Преобразование метил 2-(3-фтор-4-(метилкарбамоил)фениламино)пропаноата в 4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фтор-N-метилбензамид

В круглодонный сосуд помещали метиловый эфир D-1 (150 г, 0,56 моль), изотиоцианат F (255,6 г, 1,12 моль), ДМСО (150 мл, 1 эквив.) и IPAc (300 мл, 2 эквив.). Затем смесь нагревали до температуры 83-84°C, перемешивали в течение 17,5 час, и затем проводили ВЭЖХ анализ для выявления 96,2A% преобразования в желаемый продукт. Затем реакционную смесь охлаждали до температуры 65-70°C и добавляли метанол (22,5 мл, 0,15 об.). Затем раствор перемешивали в течение 45 мин и охлаждали до температуры 20-25°C. Затем раствор разбавляли IPAc (900 мл, 6 об.) и промывали DI-водой (450 мл, 3 об.) и использовали IPA (225 мл, 1,5 об.) для осветления эмульсии. После экстрагирования водной фазы, органическую фазу затем концентрировали до 4,5 объема (675 мл) при пониженном давлении при 30-35°C. Затем раствор разбавляли IPA (2000 мл, 13,3 об.) и нагревали до температуры 75-82°C (температура снаружи 95°C). В процессе нагревания раствор был немного мутным, но стал прозрачным при 70-71°C. Далее раствор концентрировали до 8 объемов (1200 мл) при атмосферном давлении, поддерживая температуру 77-82°C. Анализ 1H ЯМР показал, что 7,3% моль IPAc остается в растворе. Далее раствор охлаждали до температуры 77°C, затравляли и охлаждали в течение 5 час до 20-25°C. После содержания при 20-25°C в течение 8 час реакционную массу затем охлаждали до температуры 0-5°C в течение 2 час. После перемешивания при температуре 0-5°C в течение 1 час, взвесь затем фильтровали, промывали IPA (2×225 мл), вакуумировали в течение 5 мин, и затем сушили в вакууме при 50-55°C в течение 117 часов. Реакция давала продукт I-1 (213,9 г, 82%) в виде белого порошка с 0,14% влажности по KF, >99,9A% чистотой по ВЭЖХ.
Пример 6: Преобразование 4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фтор-N-метилбензамида в 4-(3-(4-циано-3-трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторбензойную кислоту

4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фтор-N-метилбензамид I-1 суспендировали в концентрированной HCl и нагревали при температуре 120°C в запаянной трубке в течение 48 час. Протекание реакции контролировали тонкослойной хроматографией (ТСХ). Реакционную смесь охлаждали до комнатной температуры. Остаток фильтровали и очищали хроматографией на силикагеле (100-200 меш, элюент: 0-5% метанол-дихлорметан), с получением желаемого производного карбоновой кислоты I-2. МС (m/z): 452 (M+1). ВЭЖХ: Колонка, YMC ODS AQ, 4,6×250 мм, 5 мкм, подвижная фаза A: 10 мМ ацетат аммония, подвижная фаза B: ацетонитрил, градиент, изократный: 55% A:45% B, время удерживания, 3,804 мин, чистота по ВЭЖХ, 95,82%, скорость потока, 1 мл/мин.
1H ЯМР (CDCl3, FREEBASE): δ (м.д.) 8,22 (т, 1H), 8,0 (д, 1H), 7,98 (с, 1H), 7,82 (д, 1H), 7,2 (м, 2H) 1,6 (с, 6H).
Пример 7: Преобразование 2-(3-фтор-4-(метилкарбамоил)фениламино)-2-метилпропановой кислоты в 4-(1-(4-циано-3-(трифторметил)фениламино)-2-метил-1-оксопропан-2-иламино)-2-фтор-N-метилбензамид

Производное метилпропионовой кислоты C-1 (0,254 г, 1 ммоль) растворяли в DCM (15 мл) с 1-этил-3-(3-диметиламинопропил)карбодиимидом (0,380 г, 2,0 ммоль), затем медленно добавляли 4-амино-2-(трифторметил)бензонитрил (0,200 г, 1,1 ммоль). Смесь перемешивали при комнатной температуре в течение 5-6 час. После анализа протекание реакции ЖХМС и ТСХ, смесь экстрагировали DCM, и экстракты промывали водой, сушили и упаривали. Неочищенный продукт очищали хроматографией, с получением желаемого продукта G-1 (0,150 г, 36% выход).
Пример 8: Преобразование 4-(1-(4-циано-3-(трифторметил)фениламино)-2-метил-1-оксопропан-2-иламино)-2-фтор-N-метилбензамида в 4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фтор-N-метилбензамид

Смесь производного амида G-1 (0,1 г, 0,23 ммоль) в тиофосгене без растворителей (54 мг, 0,48 ммоль) нагревали до температуры 100°C в запаянной трубке, в течение 6 час, затем охлаждали. Смесь растворяли в DCM, фильтровали и фильтрат упаривали. Неочищенный продукт очищали колоночной хроматографией, с получением желаемого продукта I-1 (4 мг, 4% выход). Аналитические данные согласуются с данными соединения, полученного в примере 5.
Пример 9: Синтез 4-(1-карбокси-1-метилэтиламино)-2-фторбензойной кислоты
Пример 9A: Синтез 4-(1-карбокси-1-метилэтиламино)-2-фторбензойной кислоты исходя из 4-амино-2-фторбензойной кислоты

4-Амино-2-фторбензойную кислоту (0,2 г, 1,29 ммоль) и 1,1,1-трихлор-2-метилпропан-2-ол (0,593 г, 3,35 ммоль) растворяли в безводном ацетоне, и раствор охлаждали до температуры 0°C. Добавляли частями порошкообразный гидроксид натрия (0,2 г, 5,01 ммоль), после чего реакционную смесь нагревали до комнатной температуры и перемешивали в течение 12 час.
Легко летучие продукты удаляли при пониженном давлении, и остаток подкисляли 1M водным раствором HCl. Полученный неочищенный продукт очищали ВЭЖХ с обращенной фазой, с получением 4-(1-карбокси-1-метилэтиламино)-2-фторбензойной кислоты.
Пример 9B: Альтернативный синтез 4-(1-карбокси-1-метилэтиламино)-2-фторбензойной кислоты исходя из 4-бром-2-фторбензойной кислоты

4-Бром-2-фторбензойную кислоту (20 г, 91,3 ммоль), 2-аминоизомасляную кислоту (14,5 г, 140 ммоль), CuI (3,47 г, 18,22 ммоль) и K2CO3 (31,56 г, 227,91 ммоль) смешивали в ДМФА (200 мл), H2O (20 мл) и TEA (0,63 мл, 4,54 ммоль). Затем в реакционную смесь добавляли 2-ацетилциклогексанон (2,4 г, 17,1 ммоль). Реакционную смесь перемешивали при температуре 90°C в течение 14 час. После завершения взаимодействия добавляли воду. Водный слой промывали этилацетатом. Водный слой подкисляли добавлением 1M раствора лимонной кислоты (pH~4). Продукт экстрагировали этилацетатом (3×200 мл). Объединенный органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении, с получением 16 г 4-(2-карбоксипропан-2-иламино)-2-фторбензойной кислоты в виде неочищенного продукта. Полученный неочищенный продукт использовали без дополнительной обработки в следующем примере.
Пример 10: Синтез 4-[3-(4-Циано-3-трифторметилфенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил]-2-фторбензойной кислоты (соединение M-1)

4-(1-Карбокси-l-метилэтиламино)-2-фторбензойную кислоту (241 мг, 1 ммоль), 4-изотиоцианато-2-трифторметилбензонитрил (342 мг, 1,5 ммоль) и триэтиламин (343 мг, 3,4 ммоль) смешивали в EtOH (5 мл), и раствор перемешивали в течение 10 дней при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, остаток подкисляли 1M водным раствором HCl, и продукт экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией, элюируя этилацетатом, с получением 4-[3-(4-циано-3-трифторметилфенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил]-2-фторбензойной кислоты (10 мг) в виде твердого вещества кремового цвета.
Описания всех публикаций, патентов, патентных заявок и опубликованных патентных заявок, указанных в настоящем описании посредством определяющей ссылки, таким образом, включены в настоящее описание посредством ссылки во всей своей полноте.
Хотя вышеизложенное изобретение было описано достаточно подробно посредством иллюстрации и примеров с целью ясности понимания, для специалиста в данной области очевидно, что могут быть осуществлены определенные незначительные изменения и модификации. Соответственно, описание и примеры не должны быть истолкованы как ограничивающие объем изобретения.
Claims (3)
1. Способ получения соединения формулы (I-1):

включающий
(a) объединение соединения формулы D-1: с соединением формулы (F): , диметилсульфоксидом (ДМСО) и изопропилацетатом с образованием смеси; и
(b) нагревание указанной смеси с получением соединения диарилтиогидантоина формулы (I-1):
включающий
(a) объединение соединения формулы D-1:
(b) нагревание указанной смеси с получением соединения диарилтиогидантоина формулы (I-1):
2. Способ по п. 1, дополнительно включающий
(c) объединение соединения формулы A-2: с соединением формулы B-1:
с соединением формулы B-1:  , K2CO3, CuCl, N,N-диметилформамидом (ДМФА) и водой с получением смеси; нагревание указанной смеси с получением соединения формулы C-1:
, K2CO3, CuCl, N,N-диметилформамидом (ДМФА) и водой с получением смеси; нагревание указанной смеси с получением соединения формулы C-1:
 и
и
(d) взаимодействие соединения формулы C-1 с метилйодидом в условиях алкилирования с образованием соединения формулы D-1:

(c) объединение соединения формулы A-2:
(d) взаимодействие соединения формулы C-1 с метилйодидом в условиях алкилирования с образованием соединения формулы D-1:
3. Способ получения соединения формулы (M-I):

включающий
(a) объединение 4-бром-2-фторбензойной кислоты: с 2-аминоизомасляной кислотой:
с 2-аминоизомасляной кислотой:  , диметилформамидом (ДМФА), водой, триэтиламином (TEA) и 2-ацетилциклогексаноном с образованием смеси; и
, диметилформамидом (ДМФА), водой, триэтиламином (TEA) и 2-ацетилциклогексаноном с образованием смеси; и
(b) нагревание указанной смеси с получением 4-(2-карбоксипропан-2-иламино)-2-фторбензойной кислоты:
(c) взаимодействие 4-(2-карбоксипропан-2-иламино)-2-фторбензойной кислоты с соединением формулы (F):

с получением соединения диарилтиогидантоина формулы (M-1):

включающий
(a) объединение 4-бром-2-фторбензойной кислоты:
(b) нагревание указанной смеси с получением 4-(2-карбоксипропан-2-иламино)-2-фторбензойной кислоты:
(c) взаимодействие 4-(2-карбоксипропан-2-иламино)-2-фторбензойной кислоты с соединением формулы (F):
с получением соединения диарилтиогидантоина формулы (M-1):
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US30779610P | 2010-02-24 | 2010-02-24 | |
| US61/307,796 | 2010-02-24 | ||
| PCT/US2011/026135 WO2011106570A1 (en) | 2010-02-24 | 2011-02-24 | Processes for the synthesis of diarylthiohydantoin and diarylhydantoin compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2012140454A RU2012140454A (ru) | 2014-03-27 |
| RU2554081C2 true RU2554081C2 (ru) | 2015-06-27 |
Family
ID=44507217
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2012140454/04A RU2554081C2 (ru) | 2010-02-24 | 2011-02-24 | Способ синтеза соединений диарилтиогидантоина и диарилгидантоина |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US9174943B2 (ru) |
| EP (2) | EP2538785B1 (ru) |
| JP (1) | JP5718372B2 (ru) |
| KR (1) | KR101514659B1 (ru) |
| CN (1) | CN103108549B (ru) |
| BR (1) | BR112012021406B1 (ru) |
| CA (1) | CA2790924C (ru) |
| CY (2) | CY1120207T1 (ru) |
| DK (2) | DK2538785T3 (ru) |
| ES (2) | ES2880354T3 (ru) |
| HU (2) | HUE038637T2 (ru) |
| MX (1) | MX2012009782A (ru) |
| PL (2) | PL3329775T3 (ru) |
| PT (2) | PT3329775T (ru) |
| RU (1) | RU2554081C2 (ru) |
| SI (2) | SI2538785T1 (ru) |
| WO (1) | WO2011106570A1 (ru) |
Families Citing this family (64)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7709517B2 (en) | 2005-05-13 | 2010-05-04 | The Regents Of The University Of California | Diarylhydantoin compounds |
| CA2703635C (en) | 2007-10-26 | 2017-06-27 | Michael E. Jung | Diarylhydantoin compounds as androgen receptor modulators |
| CA2787083C (en) * | 2010-02-16 | 2018-05-22 | Aragon Pharmaceuticals, Inc. | Androgen receptor modulators and uses thereof |
| AU2012225038B2 (en) | 2011-03-10 | 2016-03-10 | Suzhou Kintor Pharmaceuticals, Inc. | Androgen receptor antagonists and uses thereof |
| WO2013067151A1 (en) * | 2011-11-02 | 2013-05-10 | Medivation Prostate Therapeutics, Inc. | Treatment methods using diarylthiohydantoin derivatives |
| EA201991484A1 (ru) | 2012-09-11 | 2019-11-29 | Лекарственные формы энзалутамида | |
| JP2015534550A (ja) * | 2012-09-11 | 2015-12-03 | ドクター レディズ ラボラトリーズ リミテッド | エンザルタミドの多形形態およびその調製 |
| CN109908114A (zh) | 2012-09-26 | 2019-06-21 | 阿拉贡药品公司 | 用于治疗非转移性去势抵抗性前列腺癌的抗雄激素 |
| AU2013334102B2 (en) | 2012-10-26 | 2018-08-16 | Memorial Sloan-Kettering Cancer Center | Modulators of resistant androgen receptor |
| JOP20200097A1 (ar) | 2013-01-15 | 2017-06-16 | Aragon Pharmaceuticals Inc | معدل مستقبل أندروجين واستخداماته |
| RU2520134C1 (ru) * | 2013-02-27 | 2014-06-20 | Общество с ограниченной ответственностью "Авионко" (ООО "Авионко") | Замещенные (r)-3-(4-метилкарбамоил-3-фторфениламино)-тетрагидро-фуран-3-енкарбоновые кислоты и их эфиры, способ их получения и применения |
| WO2014167428A2 (en) * | 2013-04-10 | 2014-10-16 | Shilpa Medicare Limited | Amorphous 4-(3-(4-cyano-3-(trifluoromethyl)phenyl)-5,5-dimethyl-4-oxo-2-thioxoimidazolidin-1-yl)-2-fluoro-n-methylbenzamide |
| JP6346944B2 (ja) * | 2013-09-19 | 2018-06-20 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニーGlaxoSmithKline LLC | 組み合わせ薬物療法 |
| CN106543085A (zh) * | 2013-10-14 | 2017-03-29 | 杭州普晒医药科技有限公司 | 恩杂鲁胺的固态形式及其制备方法和用途 |
| WO2015063720A1 (en) | 2013-10-31 | 2015-05-07 | Ranbaxy Laboratories Limited | Process for the preparation of enzalutamide |
| EP3083568A1 (en) | 2013-12-16 | 2016-10-26 | Sun Pharmaceutical Industries Ltd | Processes and intermediates for the preparation of enzalutamide |
| CN104803919A (zh) * | 2014-01-26 | 2015-07-29 | 上海医药工业研究院 | 用于制备恩杂鲁胺中间体的方法 |
| CN104803918B (zh) * | 2014-01-26 | 2017-11-10 | 上海医药工业研究院 | 恩杂鲁胺的制备方法 |
| US9611225B2 (en) * | 2014-01-27 | 2017-04-04 | Cadila Healthcare Limited | Process for preparation of androgen receptor antagonist |
| CN104844521B (zh) * | 2014-02-13 | 2017-08-15 | 成都伊诺达博医药科技有限公司 | 抗前列腺癌药物恩杂鲁胺的合成方法 |
| EP3105208A1 (en) | 2014-02-13 | 2016-12-21 | Ranbaxy Laboratories Limited | Process for the preparation of enzalutamide |
| CN104844520B (zh) * | 2014-02-13 | 2017-09-05 | 成都伊诺达博医药科技有限公司 | 一种合成恩杂鲁胺的方法 |
| CZ2014232A3 (cs) | 2014-04-07 | 2015-10-14 | Zentiva, K.S. | Způsob výroby enzalutamidu |
| CN103910679B (zh) * | 2014-04-23 | 2016-05-25 | 杭州新博思生物医药有限公司 | 一种恩杂鲁胺的制备方法 |
| CN103980141A (zh) * | 2014-04-25 | 2014-08-13 | 山东大学 | 恩泽特鲁的合成方法 |
| CN104016924B (zh) * | 2014-06-16 | 2016-04-13 | 上海鼎雅药物化学科技有限公司 | 一种“一锅法”合成恩杂鲁胺的方法 |
| RU2557235C1 (ru) * | 2014-07-08 | 2015-07-20 | Александр Васильевич Иващенко | Замещенные 2-тиоксо-имидазолидин-4-оны и их спироаналоги, противораковый активный компонент, фармацевтическая композиция, лекарственный препарат, способ лечения рака простаты |
| EP3166931A4 (en) * | 2014-07-11 | 2018-05-09 | Shilpa Medicare Limited | An improved process for the preparation of enzalutamide |
| CN105461633A (zh) * | 2014-07-31 | 2016-04-06 | 江苏豪森药业集团有限公司 | 恩杂鲁胺的制备方法 |
| CN105367441B (zh) * | 2014-08-18 | 2018-11-06 | 上海医药工业研究院 | 用于合成恩杂鲁胺的新化合物 |
| CN105461634A (zh) * | 2014-08-19 | 2016-04-06 | 江苏豪森药业集团有限公司 | 恩杂鲁胺的制备方法 |
| WO2016051423A2 (en) * | 2014-10-01 | 2016-04-07 | Laurus Labs Private Limited | An improved process for the preparation of enzalutamide |
| CN104356068A (zh) * | 2014-10-30 | 2015-02-18 | 杭州新博思生物医药有限公司 | 恩杂鲁胺新晶型及其制备方法 |
| EA201892485A1 (ru) * | 2014-12-19 | 2019-07-31 | Арагон Фармасьютикалз, Инк. | Способ получения соединения диарилтиогидантоина |
| ITUB20151256A1 (it) | 2015-05-28 | 2016-11-28 | Olon Spa | Processo industriale per la preparazione di enzalutamide |
| ITUB20151204A1 (it) | 2015-05-28 | 2016-11-28 | Olon Spa | Procedimento per la preparazione di enzalutamide |
| ITUB20151311A1 (it) * | 2015-05-28 | 2016-11-28 | Olon Spa | Procedimento per la preparazione di enzalutamide |
| MX2017014958A (es) * | 2015-05-29 | 2018-07-06 | Astellas Pharma Inc | Metodo de produccion de forma cristalina de enzalutamida. |
| TWI613194B (zh) | 2015-06-10 | 2018-02-01 | 台灣神隆股份有限公司 | 用於製備恩雜魯胺的新穎方法 |
| CN105330560A (zh) * | 2015-10-13 | 2016-02-17 | 福格森(武汉)生物科技股份有限公司 | 一种恩杂鲁胺中间体的制备方法 |
| TWI726969B (zh) | 2016-01-11 | 2021-05-11 | 比利時商健生藥品公司 | 用作雄性激素受體拮抗劑之經取代之硫尿囊素衍生物 |
| CN107954936B (zh) * | 2016-10-17 | 2021-03-19 | 海创药业股份有限公司 | 一种制备氘代咪唑二酮类化合物的方法 |
| CN108069869B (zh) * | 2016-11-09 | 2022-03-01 | 上海医药工业研究院 | 一种Apalutamide的制备方法及其中间体 |
| TW201831461A (zh) * | 2017-01-18 | 2018-09-01 | 台灣神隆股份有限公司 | 製備阿帕魯醯胺的方法 |
| WO2018199282A1 (ja) | 2017-04-28 | 2018-11-01 | アステラス製薬株式会社 | エンザルタミドを含有する経口投与用医薬組成物 |
| CN107501237B (zh) * | 2017-08-17 | 2022-03-22 | 上海西浦医药科技有限公司 | 一种Apalutamide的合成方法 |
| AU2018351714A1 (en) | 2017-10-16 | 2020-04-30 | Aragon Pharmaceuticals, Inc. | Anti-androgens for the treatment of non-metastatic castration-resistant prostate cancer |
| HUE064356T2 (hu) * | 2017-11-28 | 2024-03-28 | Aarti Pharmalabs Ltd | Eljárás enzalutamid elõállítására új intermedier felhasználásával |
| CN108047200A (zh) * | 2017-12-05 | 2018-05-18 | 上海丰瑞医药科技有限公司 | 一种二芳基乙内酰硫脲类化合物的制备方法及其中间体 |
| CN109988077A (zh) * | 2017-12-29 | 2019-07-09 | 上海法默生物科技有限公司 | 一种阿帕鲁胺的合成方法及中间体 |
| CN110872258B (zh) * | 2018-09-04 | 2021-05-25 | 北京凯莱天成医药科技有限公司 | 一种前列腺癌药物恩杂鲁胺的制备工艺 |
| CN109651256A (zh) * | 2018-11-20 | 2019-04-19 | 上海健康医学院 | 一种式(viii)的恩杂鲁胺的制备方法 |
| CN109503416A (zh) * | 2018-12-24 | 2019-03-22 | 常州智超化学有限公司 | 一种恩杂鲁胺中间体合成方法 |
| CN112047888A (zh) * | 2019-06-05 | 2020-12-08 | 安礼特(上海)医药科技有限公司 | 一种合成恩杂鲁胺的方法 |
| CN111320552B (zh) * | 2020-02-28 | 2023-10-27 | 江西科睿药业有限公司 | 一种恩扎卢胺中间体的制备方法 |
| WO2022206742A1 (zh) * | 2021-03-30 | 2022-10-06 | 苏州开拓药业股份有限公司 | 一种一步法合成乙内酰硫脲衍生物的方法 |
| EP4112603A1 (en) | 2021-06-29 | 2023-01-04 | Química Sintética, S.A. | Processes for the preparation of non-steroidal antiandrogens |
| CN113698310B (zh) * | 2021-08-20 | 2023-03-17 | 江西金丰药业有限公司 | 一种恩杂鲁胺双酯中间体的制备方法 |
| CN114591246A (zh) * | 2022-03-25 | 2022-06-07 | 重庆华邦制药有限公司 | 一种恩扎卢胺的纯化方法 |
| CN114907439B (zh) * | 2022-06-29 | 2023-07-21 | 云南中医药大学 | 一种抗癌化合物及其制药用途 |
| CN115181043A (zh) * | 2022-07-27 | 2022-10-14 | 爱斯特(成都)生物制药股份有限公司 | 一种连续流制备4-异硫氰基-2-(三氟甲基)苯甲腈的方法 |
| CN115611765A (zh) * | 2022-09-30 | 2023-01-17 | 重庆华邦胜凯制药有限公司 | 一种恩扎卢胺中间体的制备方法 |
| CN115536634A (zh) * | 2022-10-17 | 2022-12-30 | 上海博悦生物科技有限公司 | 一种阿帕他胺的合成方法 |
| CN115724759A (zh) * | 2022-11-23 | 2023-03-03 | 爱斯特(成都)生物制药股份有限公司 | 一种恩扎卢胺中间体的制备方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2116298C1 (ru) * | 1992-07-08 | 1998-07-27 | Руссель-Юклаф | Замещенные фенилимидазолидины, способы их получения и фармацевтическая композиция, обладающая антиандрогенной активностью |
| RU2003129532A (ru) * | 2001-04-04 | 2005-04-10 | Лаборатуар Фурнье Са (Fr) | Тиогидантоины и их применение при лечении диабета |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2884448A (en) * | 1956-07-02 | 1959-04-28 | Gen Aniline & Film Corp | Hydrolysis of n-substituted amides |
| DE1906492B2 (de) * | 1969-02-10 | 1980-07-31 | Bayer Ag, 5090 Leverkusen | Carbonsäure-Derivate enthaltende Hydantoine und Polyhydantoine |
| US4424396A (en) * | 1982-07-21 | 1984-01-03 | Occidental Chemical Corporation | Process for the preparation substituted anilino acids |
| US5235093A (en) * | 1992-01-27 | 1993-08-10 | Monsanto Company | Process for preparing carboxylic amide ester |
| RU2206326C2 (ru) | 2001-04-10 | 2003-06-20 | Асафов Александр Виленович | Применение натрия нуклеоспермата для лечения вич-инфекции и способ лечения |
| KR20070106969A (ko) * | 2004-09-09 | 2007-11-06 | 추가이 세이야쿠 가부시키가이샤 | 신규 이미다졸리딘 유도체 및 그 용도 |
| US7709517B2 (en) * | 2005-05-13 | 2010-05-04 | The Regents Of The University Of California | Diarylhydantoin compounds |
| ME01992B (me) * | 2005-05-13 | 2012-10-30 | Univ California | Jedinjenje diarilhidantoina |
| PL3100727T3 (pl) * | 2006-03-27 | 2019-01-31 | The Regents Of The University Of California | Modulator receptora androgenowego do leczenia raka gruczołu krokowego i chorób związanych z receptorem androgenowym |
| KR20160027254A (ko) * | 2006-03-29 | 2016-03-09 | 더 리전트 오브 더 유니버시티 오브 캘리포니아 | 디아릴티오히단토인 화합물 |
| WO2008093838A1 (ja) * | 2007-02-01 | 2008-08-07 | Chugai Seiyaku Kabushiki Kaisha | スルファモイル基を有するピリジルイミダゾリジン誘導体、およびその医薬としての使用 |
| US8461343B2 (en) * | 2007-03-27 | 2013-06-11 | Sloan-Kettering Institute For Cancer Research | Synthesis of thiohydantoins |
| CA2703635C (en) | 2007-10-26 | 2017-06-27 | Michael E. Jung | Diarylhydantoin compounds as androgen receptor modulators |
| MX2010006331A (es) * | 2007-12-20 | 2010-07-05 | Hoffmann La Roche | Hidantoinas sustituidas como inhibidores de cinasa mek. |
| ES2535466T3 (es) * | 2009-02-24 | 2015-05-11 | Medivation Prostate Therapeutics, Inc. | Compuestos específicos de diariltiohidantoína |
| WO2010118354A1 (en) * | 2009-04-09 | 2010-10-14 | Medivation Prostate Therapeutics, Inc. | Substituted di-arylhydantoin and di-arylthiohydantoin compounds and methods for use thereof |
-
2011
- 2011-02-24 MX MX2012009782A patent/MX2012009782A/es active IP Right Grant
- 2011-02-24 EP EP11748095.4A patent/EP2538785B1/en active Active
- 2011-02-24 JP JP2012555160A patent/JP5718372B2/ja active Active
- 2011-02-24 ES ES18153287T patent/ES2880354T3/es active Active
- 2011-02-24 DK DK11748095.4T patent/DK2538785T3/en active
- 2011-02-24 PT PT181532870T patent/PT3329775T/pt unknown
- 2011-02-24 EP EP18153287.0A patent/EP3329775B1/en active Active
- 2011-02-24 PT PT117480954T patent/PT2538785T/pt unknown
- 2011-02-24 RU RU2012140454/04A patent/RU2554081C2/ru active
- 2011-02-24 CN CN201180020668.7A patent/CN103108549B/zh active Active
- 2011-02-24 ES ES11748095.4T patent/ES2671343T3/es active Active
- 2011-02-24 DK DK18153287.0T patent/DK3329775T3/da active
- 2011-02-24 BR BR112012021406-3A patent/BR112012021406B1/pt active IP Right Grant
- 2011-02-24 KR KR1020127024687A patent/KR101514659B1/ko active IP Right Grant
- 2011-02-24 PL PL18153287T patent/PL3329775T3/pl unknown
- 2011-02-24 HU HUE11748095A patent/HUE038637T2/hu unknown
- 2011-02-24 PL PL11748095T patent/PL2538785T3/pl unknown
- 2011-02-24 SI SI201131468T patent/SI2538785T1/en unknown
- 2011-02-24 WO PCT/US2011/026135 patent/WO2011106570A1/en active Application Filing
- 2011-02-24 SI SI201131988T patent/SI3329775T1/sl unknown
- 2011-02-24 CA CA2790924A patent/CA2790924C/en active Active
- 2011-02-24 HU HUE18153287A patent/HUE055051T2/hu unknown
-
2013
- 2013-03-05 US US13/785,504 patent/US9174943B2/en active Active
-
2018
- 2018-05-15 CY CY20181100501T patent/CY1120207T1/el unknown
-
2021
- 2021-07-20 CY CY20211100657T patent/CY1124334T1/el unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2116298C1 (ru) * | 1992-07-08 | 1998-07-27 | Руссель-Юклаф | Замещенные фенилимидазолидины, способы их получения и фармацевтическая композиция, обладающая антиандрогенной активностью |
| RU2003129532A (ru) * | 2001-04-04 | 2005-04-10 | Лаборатуар Фурнье Са (Fr) | Тиогидантоины и их применение при лечении диабета |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2554081C2 (ru) | Способ синтеза соединений диарилтиогидантоина и диарилгидантоина | |
| US9890129B2 (en) | Aminoalkylbenzothiazepine derivatives and uses thereof | |
| US8710086B2 (en) | Substituted di-arylhydantoin and di-arylthiohydantoin compounds and methods of use thereof | |
| RU2544856C2 (ru) | НОВЫЕ ПРОИЗВОДНЫЕ 2,3,4,5-ТЕТРАГИДРО-1-ПИРИДО[4,3-b]ИНДОЛА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | |
| KR102266680B1 (ko) | 벨리노스테트의 다형태 및 이의 제조 방법 | |
| CN112047888A (zh) | 一种合成恩杂鲁胺的方法 | |
| US20210139462A1 (en) | Process for the preparation of a sulfonamide structured kinase inhibitor | |
| CN112759545B (zh) | 3-(二甲氨基甲基)哌啶-4-醇类衍生物及其制备方法和药物用途 | |
| CN108558760B (zh) | 一类芳香酰胺化合物及其制备方法和用途 | |
| US10150739B2 (en) | Crystalline form of androgen receptor inhibitor and preparation method thereof | |
| EP2598485B1 (en) | Novel montelukast 4-halobenzylamine salt and method for preparing montelukast sodium salt by using the same | |
| JP2019532075A (ja) | 重水素化イミダゾリジンジオン系化合物を調製する方法 | |
| CN110903245B (zh) | 一种合成1-烷基-2-三氟甲基-5-氨基-1h-咪唑的关键中间体及其制备方法 | |
| US9150524B2 (en) | Pure intermediate | |
| KR20090103932A (ko) | 카나비노이드-cb1 길항작용과 세로토닌 재흡수 억제가 조합된 화합물 | |
| KR101257272B1 (ko) | 탈보호화 반응을 이용한 고혈압 치료용 비페닐테트라졸 화합물의 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PC43 | Official registration of the transfer of the exclusive right without contract for inventions |
Effective date: 20180130 |
|
| PD4A | Correction of name of patent owner |