JP3990475B2 - メチオニンおよびその塩の製造方法 - Google Patents
メチオニンおよびその塩の製造方法 Download PDFInfo
- Publication number
- JP3990475B2 JP3990475B2 JP33416996A JP33416996A JP3990475B2 JP 3990475 B2 JP3990475 B2 JP 3990475B2 JP 33416996 A JP33416996 A JP 33416996A JP 33416996 A JP33416996 A JP 33416996A JP 3990475 B2 JP3990475 B2 JP 3990475B2
- Authority
- JP
- Japan
- Prior art keywords
- methionine
- hydantoin
- ammonia
- carbon dioxide
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/14—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of sulfides
- C07C319/20—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of sulfides by reactions not involving the formation of sulfide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/26—Separation; Purification; Stabilisation; Use of additives
- C07C319/28—Separation; Purification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/72—Two oxygen atoms, e.g. hydantoin
- C07D233/76—Two oxygen atoms, e.g. hydantoin with substituted hydrocarbon radicals attached to the third ring carbon atom
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
【発明の属する技術分野】
本発明は、成分3−メチルメルカプトプロピオンアルデヒド、シアン化水素、アンモニアおよび二酸化炭素ならびに5−(2−メチルメルカプトエチル)−ヒダントインならびにメチオニンの塩、または上記の成分を製造できる成分から場合により水の存在下で出発するD,L−メチオニンまたはD,L−メチオニンの塩の製造方法に関する。
【0002】
【従来の技術】
合成工程は、下記の反応式により具体的に説明できる。
【0003】
5−(2−メチルメルカプト)−ヒダントイン形成:
【0004】
【化1】
D,L−メチオニンの塩形成:
【0006】
【化2】
D,L−メチオニンの遊離:
【0008】
【化3】
上記式中、Mはアルカリ金属、アルカリ土類金属、アンモニウム、殊にはカリウムを表す。
【0010】
工程5−(2−メチルメルカプトエチル)−ヒダントイン形成、メチオニン塩の形成ならびにメチオニンの遊離は、有利には自体有利な連続的に実施でき、かつ互いに連続して連結し、殊には全体が連続的に操作されるプロセスに統合すると有利である。
【0011】
成分アンモニアおよび二酸化炭素をプロセスの可能性に従ってリサイクリング、すなわち前に行う工程に再び加えると著しく有利である。殊には、カリウムを使用する場合には、アルカリを含む薬剤をできるだけ全てプロセスに還流する。
【0012】
5−(2−メチルメルカプト)−ヒダントイン形成は、基本的には公知である。一般に、その際、1)に記載した成分から、またはこれらの成分を製造できる成分から出発する。これらは、以下にさらに記載するように、殊には成分シアン化水素、アンモニアまたは二酸化炭素の場合にはアルカリ金属塩またはアンモニウム塩、ならびに成分3−メチルメルカプトプロピオンアルデヒドの場合にはアクロレインおよびメチルメルカプタンである。すなわち、ケミカル・レビユー誌[Chem. Rev. 46 (1959) 422〜425]には、アルカリ金属シアン化物および炭酸アンモニウムとを用いる相当するアルデヒドまたはケトンの転化による置換ヒダントインの製造が記載されている。この転化は、物質の化学量論的量を用いて80℃および3バールでも、化学量論的量の数倍のアンモニアを用いて温度60℃および常圧でも行われる(ドイツ国特許第1166201号明細書)。3−メチルメルカプトプロピオンアルデヒド、炭酸アンモニウムおよびシアン化物から、5−(2−メチルメルカプトエチル)−ヒダントインが製造されることも公知である。転化は、開始時は40〜120℃で行い、次いで、反応混合物をpH値4以下に調整し、反応を50〜100で終了させる(米国特許第2557913号明細書)。さらに、アンモニア、二酸化炭素およびシアン化水素、またはこれらの塩の水溶液中に3−メチルメルカプトプロピオンアルデヒドを溶解して製造した水溶液を装入し、その中で必要ならば部分的または全体をヒダントインへ転化させ、この溶液中にアンモニア、二酸化炭素およびシアン化水素、またはこれらの塩の水溶液、およびそれとは別に3−メチルメルカプトプロピオンアルデヒドを装入し、転化は混合物の加熱により100℃以下、常圧で行う5−(2−メチルメルカプトエチル)−ヒダントインの製造も公知である(ドイツ国特許出願公開第1620332号明細書)。特開昭48−005763号公報中では、3−メチルメルカプトプロピオンアルデヒドをシアン化水素、またはその塩および炭酸アンモニウムと一緒にして、アンモニアの存在下で、80℃で1.5時間で5−(2−メチルメルカプトエチル)−ヒダントインに98.5%の収率で転化している。添加した金属イオン錯化剤は水の存在下で収率97.8%とする(特開昭48−004465号公報)。有機溶剤の存在下、50〜200℃、加圧下の液相中の同様な転化は、特開昭40−36676号公報に記載されている。水中のアクロレイン、メチルメルカプタン、シアン化水素および炭酸アンモニウムから出発する一段法では、50〜70℃において2時間以内にヒダントインに導き、これをD,L−メチオニンに鹸化する(特開昭50−004018号公報)。
【0013】
特開昭52−027768号公報に記載の反応も同様に進むが、ただし、アミノ酸、例えばメチオニン、トレオニン、グリシン、アラニンまたはロイシンを加える。3−メチルメルカプトプロピオンアルデヒド、二酸化炭素、アンモニア、シアン化水素および苛性アルカリは、80℃において2時間以内に97%が5−(2−メチルメルカプトエチル)−ヒダントインとなる(特開昭50−018467号公報)。
【0014】
水中の3−メチルメルカプトプロピオンアルデヒド、シアン化ナトリウムおよび炭酸アンモニウムは、チオ硫酸カリウムまたは炭酸カリウムの存在下で、5−(2−メチルメルカプトエチル)−ヒダントインを生成する(ソ連国特許第740770号明細書)。アクロレインのメチルメルカプタン、シアン化水素および炭酸アンモニウムを用いる一段転化では、収率85%で5−(2−メチルメルカプトエチル)−ヒダントインが生成する(Asahi Chem. Ind., Agric.Biol. Chem. 52,589 (1988) )。また、中国特許第85−1085905号明細書にも一段法が記載されており、この場合には、酢酸中のメチオニンを加えて変換率91%で5−(2−メチルメルカプトエチル)−ヒダントインが得られている。
【0015】
公知の方法で製造された5−(2−メチルメルカプトエチル)−ヒダントインは、5−(2−メチルメルカプトエチル)−ヒダントイン酸、5−(2−メチルメルカプトエチル)−ヒダントイン酸アミド、メチオニンアミド、メチオニンニトリルならびにメチルメルカプトプロピオンアルデヒドシアンヒドリン、イミノニトリルおよびポリマーによるかなりの量の不純物を含んでいる。上記の最初の3種の化合物は、アルカリ性加水分解により、例えばヒダントインがメチオニンに転換されるけれども、その他の化合物ならびにその鹸化生成物は、鹸化溶液中およびその後は単離されるべきメチオニンに達するが、その中では分離が著しく困難である。メチオニンをヒダントインから製造し、二酸化炭素を用いて反応混合物から分離し、母液を循環系に導く場合には、これはさらに困難となる。得られたメチオニンは着色し、貯蔵安定性が良くない。
【0016】
5−(2−メチルメルカプトエチル)−ヒダントインのアルカリ性加水分解は、以前から公知である。すなわち、米国特許第2527366(A)号および米国特許第2557913(A)号の各明細書には、5−(2−メチルメルカプトエチル)−ヒダントインの水酸化バリウム水溶液中、加圧、高温における加水分解を記載している。しかしこの方法は高価な水酸化バリウムを大量に必要とし、さらにバリウムを中性塩として再び分離しなければならない。
【0017】
米国特許第2557920(A)号明細書から、水酸化ナトリウムを用いるヒダントインの鹸化によりα−アミノ酸が生成することが公知である。しかしこの方法では、ヒダントイン1モル当たりに水酸化ナトリウム少なくとも3モルが必要である。水酸化カリウムを用いても事情は同じである。
【0018】
さらに、米国特許第4272631(A)号明細書から、アルカリ金属およびアルカリ土類金属の水酸化物の混合物を5−(2−メチルメルカプトエチル)−ヒダントインの鹸化に使用できることが公知である。しかしこの方法では、アルカリ土類金属イオンをメチオニンの遊離の際に最初に分離しなければならず、従って収率は最高80.5%に達するだけである。
【0019】
米国特許第4259925(A)号明細書では、5−(2−メチルメルカプトエチル)−ヒダントインの加水分解を加圧下、105〜230℃において、金属水酸化物および沸点125〜130℃のアルコールを含む媒体中で行っている。これは、高沸点のアルコールを再生しなければならないという欠点がある。その外にも収率が65%に過ぎない。
【0020】
ドイツ国特許第1906405号明細書中には、アルカリ金属炭酸塩および/またはアルカリ金属炭酸水素塩の水溶液を用いる5−(2−メチルメルカプトエチル)−ヒダントインの加水分解が記載されている。加水分解の間に、アンモニアおよび二酸化炭素を絶えず取り出す。アルカリ金属炭酸塩の中では炭酸カリウムが有利である。これをヒダントインのアルカリに対するモル比1:1〜1:5で使用する。加水分解は、加圧下、120〜220℃で行う。連続式圧力装置は、3基の互いに連続して設置された高価な循環蒸発器から成っている。アルカリ金属メチオニン酸塩溶液は、二酸化炭素を用いるD,L−メチオニンの遊離のために使用される。晶出したメチオニンの分離後の母液を循環系に送るが、必要ならば1〜2%を取り出してヒダントインの加水分解に再び使用する。
【0021】
ドイツ国特許出願公告第1518339号明細書には、反応平衡をアミノ酸の方に移動させ、これにより収率を上昇させるために、加水分解の間に生成する気体状反応生成物(アンモニアおよび二酸化炭素)を反応器から除去する方法が記載されている。しかしこれを達成するためには、気体流の圧力制御のために複雑な装置の配列が必要となる。
【0022】
特公昭49−116008号公報は、5−(2−メチルメルカプトエチル)−ヒダントインの加水分解の際に、バナジニウム酸、モリブデン酸、タングステン酸またはこれらの誘導体の存在下で実施する方法を記載している。収率は、約70%である。触媒の分離が困難である。高濃度のメチオニン含有溶液を調製するために、アルカリ金属、例えばカリウム化合物を加えなければならない。
【0023】
特開昭50−106901号公報(C.A.84,44666k(1976))から、水酸化ナトリウム約1.2当量およびアンモニア約9当量の存在下、180℃における5−(2−メチルメルカプトエチル)−ヒダントインの加水分解により、メチオニンを製造することが公開されている。しかし、この操作方法により中間に生成するメチオニン酸ナトリウム溶液は、必然的にメチオニン酸ナトリウムの他に炭酸ナトリウムを含み、これはこの反応操作の際に析出し、そのために殊に連続プロセスの障害となる。同様なことは、水酸化カリウムを使用する場合ならびにドイツ国特許第1906405号明細書に記載の操作方法でも該当する。
【0024】
ドイツ国特許第2614411A号明細書には、水を用い、イミダゾールの存在下、160℃における5−(2−メチルメルカプトエチル)−ヒダントインの加水分解を記載している。その収率は低く、またこの場合にも高い溶液濃度を得るためにアルカリ性化合物を加えなければならない。
【0025】
特開平3−95145号公報および特開平3−95146号公報には、水を用い高温および高圧において、金属酸化物または金属酸化物混合物、例えばZrO2、TiO2、Nb2O5またはTiO2−Nb2O5の存在下におけるヒダントインの加水分解が記載されている。しかし、その収率は65〜66%にすぎない。これらの溶液はアルカリ性化合物を用いて中和しなければならない。
【0026】
すべてのこれらの方法は、低い収率か、またはメチオニンまたは塩、例えば炭酸塩がプロセスの間に析出し、これにより別の工程が必要となり、また殊には大規模の方法、殊に有利には連続方法がほとんど不可能であるという欠点を有する。
【0027】
アルカリ金属塩からのメチオニンの遊離は、一般に公知である。強酸は弱酸をその塩から遊離させるという原理によると、例えば塩酸、硫酸、リン酸または強酸性イオン交換体を用いて遊離D,L−メチオニンを析出させることができる[ドイツ国特許第2140506(C)号、ドイツ国特許第2122491(C)号、ドイツ国特許出願公開第2912066(A)号、ベルギー国特許第877200号、米国特許第3433832(A)号、フランス国特許第1532723号の各明細書]。さらに副産物として析出するアルカリ金属塩を分離しなければならない。使用する酸は一般に回収できないので、これらの操作方法は連続、経済的で環境保護的な製造方法には適合しない。
【0028】
従って、例えば米国特許第2557913(A)号、ドイツ国特許第1906405号の各明細書および特開昭42−44056号公報に記載のように、水溶液中で二酸化炭素を用いる5−(2−メチルメルカプトエチル)−ヒダントインの加水分解溶液からD,L−メチオニンが有利に析出した。この方法では、通常D,L−メチオニンは薄い小平板またはフレークの形で析出する。これは生成物の不良なろ過性となり、結晶ケーキの成長を妨げる。その外にも、D,L−メチオニンは流れ特性が不良で、そのために閉塞の傾向がある。このような欠点を解決するために、特開昭42−44056号公報に記載のように、添加物、例えばカゼインまたは水溶性で高分子量のセルロース誘導体を二酸化炭素を用いるメチオニン析出の際に使用する。
【0029】
【発明が解決しようとする課題】
本発明の課題は、できるだけ少ない副産物でメチオニンまたはメチオニンの塩を製造する方法およびそのそれぞれの成分が良好に分離でき、殊には生成物の析出の際にこれらが良好にろ過されなければならないことである。それぞれの成分は、十分リサイクリング可能または再生可能でなければならない。その上、得られたメチオニンは低い着色および良い貯蔵安定性を有していなければならない。この方法は、殊に大規模装置でまた連続操作が可能でなければならない。
【0030】
【課題を解決するための手段】
この課題は本発明により、成分3−メチルメルカプトプロピオンアルデヒド、シアン化水素、アンモニアおよび二酸化炭素、またはそれから上記の成分を製造できる成分を、必要ならば水の存在下で5−(2−メチルメルカプトエチル)−ヒダントインに転化し、これを引き続きメチオニンまたはその塩に転化することによるメチオニンまたはメチオニンの塩の製造方法において、成分の転化は少なくとも一種の下記の予備混合物を介して導入し、3−メチルメルカプトプロピオンアルデヒドの少なくとも大部分(少なくとも5/10)および成分シアン化水素、またはそれからこの成分を製造できる成分の相当する量の少なくとも1/10、および成分アンモニア、二酸化炭素、またはそれからアンモニアまたは二酸化炭素を製造できる成分の5/10以下を含む第一混合物を構成し、かつこの第一混合物を1種または数種の追加の成分と一緒に5−(2−メチルメルカプトエチル)−ヒダントインへの反応変換のために一緒にし、その際これらの後者の成分を1種または数種の追加の混合物として予備混合してもよいことにより解決される。
【0031】
上記の成分を製造できる成分には、例えばシアン化水素、アンモニアおよび二酸化炭素の塩、例えばシアン化ナトリウムまたはシアン化カリウム、炭酸アンモニウムまたは炭酸水素アンモニウム、ナトリウムまたはカリウムの炭酸塩または炭酸水素塩、および当然のことながらこれらの水溶液が含まれる。3−メチルメルカプトプロピオンアルデヒドに相当する成分は、アクロレインおよびメチルメリカプタンである。基本的にこの反応において、使用される場合もある金属塩をできるだけ少量を使用し、すなわち、使用される場合もある金属イオン成分に対してアルデヒド成分および/またはシアン化物成分は化学量論的な量より多量であると有利である。著しく有利には、この場合にできるだけ金属塩、例えばシアン化ナトリウムまたはシアン化カリウムを用いず、代わりに上記の化合物を導入する。しかし殊には連続プロセスにおいて、追加して他の金属塩、例えば触媒などが存在していてもよい。これらの添加量は、反応成分に関する上記の有利な金属イオンの制限は適用されない。
【0032】
以後に、与えられた使用条件下で異なる使用形態となる上記の成分も、上記の成分と解釈する。すなわち、例えば水中のアンモニアおよび二酸化炭素を添加する場合に、これらの成分の一部を炭酸(水素)アンモニウムとして装入する。
【0033】
この方法により、5−(2−メチルメルカプトエチル)−ヒダントインは無色であり、実際的に定量的な収率が得られ、従って不純物含有量が十分少ない製品が得られ、これからメチオニンならびにその塩が母液をリサイクルする連続プロセスにより得られ、これは着色およびフレーク化に関して極めて貯蔵安定性が優れている。
【0034】
第一混合物は、シアン化水素成分の少なくとも5/10、有利には9/10および殊には少なくとも99/100、またはそれからシアン化水素を製造できる成分の相当する量を含むと有利である。上記の部分量(例えば5/10)ならびに下記のものは、常にそれぞれの記載成分自身に適用するものである(方法の化学量論に基づくものではない)。すなわち、化学量論とは無関係に、成分の全使用量は1/1である。
【0035】
殊には、アンモニアおよび二酸化炭素またはそれからアンモニアまたは二酸化炭素を製造できる成分を、それぞれこれらの使用量の5/10以下、有利には多くとも1/10、および殊には多くとも1/100を混合物内に使用すると本方法に有利である。
【0036】
場合により反応に加える水を第一混合物中で多くとも5/10、有利には多くとも1/10、殊には多くとも1/100含んでいても有利である。
【0037】
転化の開始時において、すべての成分においてその予定する使用量のすべてが存在し、すなわちどの成分も後から加えない場合も有利である。さらに、それぞれの成分をすべて2種の予備混合物中に集め、これを引き続き転化のために一緒に混合すると殊に有利である。
【0038】
すべての転化において、それぞれの成分ならびに予備混合物を迅速にかつできるだけ緊密に互いに混合させると著しく有利である。
【0039】
有機溶剤を使用する場合に、反応混合物は有機溶剤を含まないと有利であり、その際水100重量%に対して、有利には20重量部以下、殊には10重量部以下である。
【0040】
予備混合物および場合によりそれぞれの成分(1種または数種の予備混合物を一緒に反応させ、その際2種の予備混合物の場合には、場合によりすべてのそれぞれの成分がこの中に入っていてもよい)を、5−(2−メチルメルカプトエチル)−ヒダントインを含むすでに得られた反応混合物中に混入しても著しく有利である。その際すべての予備混合物および場合によりそれぞれの成分を引き続きまたは時間的(バッチ製造の場合)または流れに沿って(連続製造の場合)多くとも30秒の遅れで反応混合物中に導入すると著しく有利である。
【0041】
反応は、上記の方法の場合に、温度80℃以上で行うと有利である。さらに、反応は有利には加圧下、著しく有利には発生する平衡圧力(反応圧力)よりも高い圧力、殊には少なくとも3バール、また著しく有利には10バール以上で行う。
【0042】
上記の方法は、別法を含め殊に連続法に好適である。
【0043】
メチオニンまたはメチオニンの塩の製造方法において、殊にはそれ自身でも著しく有利である上記の操作方法の2種類を用い、これにより本発明は、成分3−メチルメルカプトプロピオンアルデヒド、シアン化水素、アンモニアおよび二酸化炭素、またはそれから上記の成分を製造できる成分を、必要ならば水の存在下で5−(2−メチルメルカプトエチル)−ヒダントインに転化し、これをさらにメチオニンまたはその塩に転化することによるメチオニンまたはメチオニンの塩を連続的に製造する方法であって、上記の成分から構成され、かつすでに5−(2−メチルメルカプトエチル)−ヒダントインの理論的に形成可能な量の少なくとも1/10の量を含む反応混合物中に成分を混入し、かつ転化を少なくとも3バールの圧力で行う、メチオニンまたはメチオニンの塩の連続製造のための方法に関する。
【0044】
ここで、少なくとも7バールの圧力および殊には少なくとも10バールの圧力で操作すると殊に有利である。さらに、成分を直ちに、または流れに沿って多くとも30秒間の予備反応時間をもって反応混合物中に導入する、すなわち、それぞれの成分を多くとも30秒間は反応性接触させ、次いで反応混合物中に導入すると著しく有利である。
【0045】
さらに、この別法において、上記のそれぞれの有利な手段も実施できる。
【0046】
これまでに記載した方法は、1方法として実施すると著しく有利であり、その際、以下にこれに相当して要約するこの方法の説明を再び記載する。その際に、記載するその他の仕様、例えば化学量論、温度などは、それぞれ単独にもまた有利に上記に一般的に記載した方法にも該当する。
【0047】
本発明により、3−メチルメルカプトプロピオンアルデヒド中のシアン化水素溶液およびアンモニアと二酸化炭素との水溶液を調製し、これらの溶液を迅速に緊密に混合、転化させて、5−(2−メチルメルカプトエチル)−ヒダントインが著しく有利に製造される。アンモニアおよび二酸化炭素は、ヒダントインのこの加水分解工程から返還してもよい。3−メチルメルカプトプロピオンアルデヒド中のシアン化水素溶液は、等モル量のシアン化水素と3−メチルメルカプトプロピオンアルデヒドとから成るか、または過剰量のシアン化水素を含むように調製すると有利である。一般に溶液中のシアン化水素の割合は、3−メチルメルカプトプロピオンアルデヒド1モルに対して1.1モル以上にならないように選定すると有利であり、この溶液は3−メチルメルカプトプロピオンアルデヒド1モルに対して1.005〜1.05モルを含むと有利である。
【0048】
アンモニアと二酸化炭素との水溶液は、飽和または希薄溶液であってもよい。アンモニアの含有量が約5重量%以下ではないと有利である。この溶液中に、炭酸水素アンモニウム、炭酸アンモニウム、カルバミン酸、カルバミン酸アンモニウム、シアン酸またはこれらの成分の混合物が存在してもよい。アンモニアの二酸化炭素に対するモル比は、二酸化炭素1モルに対して、アンモニア約1.2〜4.0モル、有利には1.6〜1.8モルであると有利である。3−メチルメルカプトプロピオンアルデヒド中のシアン化水素溶液は、アンモニアと二酸化炭素との水溶液と、混合物中に有利にはアンモニアの3−メチルメルカプトプロピオンアルデヒドに対するモル比が約1.2〜6:1、有利には2.0〜4.0:1、殊には2.5〜3.0:1となるように混合すると有利である。
【0049】
転化は、周囲温度またはそれ以上、有利には60℃以上、殊には約80〜140℃で行う。有利には温度は80〜130℃、殊には90〜120℃に選定する。転化は任意の圧力で行えるが、加圧下で行うと有利である。有利には圧力20バール以下、殊には反応混合物の平衡圧力より2〜3バール高い圧力である。反応時間は、反応条件、殊には温度および量比率に依存する。
【0050】
有利な操作方法では、3−メチルメルカプトプロピオンアルデヒド中のシアン化水素溶液およびアンモニアと二酸化炭素との水溶液をこれらの物質の反応混合物中、すなわち溶液の転化の前に生成し、その中で完全または部分的にヒダントインの転化が起きている混合物中に入れ、この混合物中で転化を行わせると著しく有利である。
【0051】
連続操作法を選択すると著しく有利であり、その場合に反応混合物を循環系内に導き、その循環系中で2箇所の隣接した位置に絶えず3−メチルメルカプトプロピオンアルデヒド中のシアン化水素溶液およびアンモニアと二酸化炭素との水溶液を供給し、他の位置で循環系から相当する量の反応混合物を絶えず取り出す。循環系中に供給する溶液と循環系を流れる反応混合物の混合比は任意であってもよいが、溶液の1体積部に対して反応混合物の多数の体積部、必要ならば反応混合物が1000以上、有利には5〜100、殊には10〜25体積部となるように混合比を選定すると有利である。循環系中に供給する溶液は、循環系中に流れる反応混合物と迅速に緊密に混合すると決定的に有利である。これは必要ならば混合ノズル、スタティック・ミキサー、高い循環率またはすべての手段の組み合わせにより実現できる。循環系から取り出す反応混合物中で転化が時には完全には終了していないで、この部分をさらにある時間、後反応器中で反応を十分進めると有利である。溶液に対して反応混合物が5体積部より少ない場合には、出発原料の重合の危険がある。混合物は暗く着色し、析出物生成となり、そのために収率低下および/または技術的な障害となる。
【0052】
本発明により、上記のようにして製造されるか、または別の方法により製造された5−(2−メチルメルカプトエチル)−ヒダントインを、さらにメチオニンアルカリ金属塩および必要ならばさらにメチオニンに転化できる。従って、アルカリおよび二酸化炭素を含む水溶液の存在下における5−(2−メチルメルカプトエチル)−ヒダントインの加水分解、および必要ならば引き続くメチオニンへの転化により、その際加水分解を少なくとも開始時は5−(2−メチルメルカプトエチル)−ヒダントインの1当量当たりにアンモニアを少なくとも0.1当量、殊には7当量以下の存在下で行う、メチオニンまたはメチオニンアルカリ金属塩の製造方法も本発明に含まれる。同様に、アルカリおよび二酸化炭素を含む水溶液の存在下における5−(2−メチルメルカプトエチル)−ヒダントインの加水分解、および必要ならば加水分解を金属ジルコニウム、または少なくとも10重量%のジルコニウムを含むジルコニウム合金を用いて行う引き続くメチオニンへの転化によるメチオニンのアルカリ金属塩の製造方法も本発明に含まれる。
【0053】
両方の本発明による操作方法を結合すると殊に有利である。
【0054】
加水分解を最初からアルカリおよび二酸化炭素の存在下、すなわち殊にはアリカリ金属化合物、殊にはアリカリ金属炭酸水素塩、アリカリ金属炭酸塩、アリカリ金属水酸化物の混合物を装入すると著しく有利であることが知られており、その際アルカリ金属は殊にはカリウムおよびナトリウムを意味する。その際アルカリおよび二酸化炭素の量は、少なくともヒダントインに対する化学量論的量が有利である。これはさらに過剰となってもよい。ヒダントインに対して約3:1の過剰のモル比が著しく有利である。基本的には、さらに大きい過剰がさらに有利であることから由来している。しかし実施のためには、約1.5:1〜2:1の比率が著しく有利である。その際本発明によるとさらに若干のアンモニアを加え、これは適当な場合には一部をアンモニウム化合物の形で装入する。その際、加水分解の開始時において、5−(2−メチルメルカプトエチル)−ヒダントイン1モル当たりアンモニア(アンモニウム化合物も含む)最大7モルを装入すると著しく有利である。これにより加水分解は実際的に副製品がなく、かつ良好な収率で進行し、他方では全くまたはごくわずかなアルカリ金属炭酸塩が析出するだけである。その際加水分解の間に、アンモニアおよび/または二酸化炭素を、必要ならば水と一緒に反応系から取り出すと著しく有利である。これによりこれ以上アルカリ金属炭酸塩が析出せず、また反応を完全に進行させるように反応条件を殊に有利に制御できる。加水分解装置自身にジルコニウム内部装置(ジルコニウム製または相当するジルコニウム合金製)を有すると特に有利である。ジルコニウムは、加水分解に対して著しく有利と考えられる触媒作用を及ぼすことが確認された。その際有利な副作用として、この装置が強い耐久性があり、そのために寿命が長く、従ってジルコニウム装置の使用は、他の装置に対して装置的に不利にはならないことが明らかになっている。
【0055】
加水分解工程は、温度120〜250℃、およびこれに相当して圧力5〜30バールで行うと有利である。この範囲内で、良好な転化および少ない副産物が得られる。また5−(2−メチルメルカプトエチル)−ヒダントインに対して少なくとも等モルのアルカリ成分を使用すると有利である。これにより、完全な加水分解の他にも、実際的に定量的にメチオニンの相当するメチオニンのアルカリ金属塩が得られる。加水分解の開始時にすでにメチオニンならびにその塩を含有していると有利であり、またこれは有利で自己触媒と考えられる作用を加水分解に対して有する。
【0056】
この方法により、加水分解の進行の間、ならびにその後に、実際的にアンモニア全部および二酸化炭素全部を加水分解溶液から取り去り、加水分解生成物を実質的にアンモニアおよび二酸化炭素を含まないで取り出せるようにすると有利である。
【0057】
またこの場合に、本方法を連続的に行うと極めて有利である。その際、以上に記載した本方法を一緒に組み合わせ、殊には総括した連続プロセスとし、二酸化炭素およびアンモニアをリサイクリングできると極めて著しく有利である。
【0058】
以下にはヒダントインの加水分解に関する本発明を詳細に説明するが、ここでそれぞれの詳細な仕様は、それ自身のみに対してもまた上記の一般的方法に対しても該当する。以下に記載する方法では適当なカリウム化合物に関して記載するが、それはこれが最も有利な態様であるからである。
【0059】
本発明によるとメチオニン酸カリウム溶液は、水酸化カリウム、炭酸カリウムおよび/または炭酸水素カリウムまたはこれらの混合物の存在下、また過剰量のアンモニア、二酸化炭素、炭酸、シアン酸またはこれらの混合物の水中の存在下で、温度120〜250℃および圧力5〜30バールにおける5−(2−メチルメルカプトエチル)−ヒダントインの加水分解により得られる。ヒダントインの加水分解は、ヒダントインに対して一種またはそれ以上のカリウム化合物(例えばKOH、KHCO3、K2CO3、メチオニン酸カリウム)1〜15当量の存在下で行うと有利である。加水分解の間およびその後に、生成またはまだ存在するアンモニアおよび/または二酸化炭素の全部または一部を反応系から分離すると殊に有利である。基本的にはどのような5−(2−メチルメルカプトエチル)−ヒダントインも加えることができるが、上記のようにして得られたヒダントインを加えると有利である。
【0060】
加水分解の開始時のアンモニアは、二酸化炭素に対するモル比1.1〜8.0が有利である。また、アンモニアのヒダントインに対するモル比0.2〜5も有利である。上記の方法において、アンモニアおよび二酸化炭素を直ちに上記の方法、すなわちヒダントイン製造から引き取り、ヒダントインをヒダントイン製造工程からまだ残っているアンモニアおよび二酸化炭素と一緒に直ちに加水分解中に導入できるようにすることも可能であり、その際さらに希望すれば異なるアンモニアおよび/または二酸化炭素濃度に調整もできる。
【0061】
本発明による方法の場合に、5−(2−メチルメルカプトエチル)−ヒダントインの加水分解は、温度120〜250℃、有利には150〜200℃、殊には160〜180℃で起きる。反応の間の圧力は5〜30バール、有利には5〜10バール、殊には7〜9バールでなければならない。
【0062】
この方法は、水蒸気で加熱され、内部装置を有し、内壁および内部装置はジルコニウムまたはジルコニウムを少なくとも10重量%含むジルコニウム合金から構成されている塔内で行うと有利である。5−(2−メチルメルカプトエチル)−ヒダントイン溶液は、加水分解生成物、すなわちメチオニン酸カリウム溶液を、塔底において加水分解が定量的に進行するように適当に取り出せるような速度で、塔頂において連続的に加えると有利である。気体状成分である水蒸気、アンモニアおよび二酸化炭素は、有利には塔頂から取り出し、5−(2−メチルメルカプトエチル)−ヒダントインの製造のためのアンモニア/二酸化炭素水溶液の再調製のために有利に使用できる。
【0063】
本発明によると、5−(2−メチルメルカプトエチル)−ヒダントインの加水分解のために、水酸化カリウム、炭酸カリウムおよび/または炭酸水素カリウムから成り、カリウムイオン含有量が加水分解溶液1l中にカリウムを有利には100〜200g、殊には140〜160g含む水溶液を使用し、その際ジルコニウムから成る装置の壁および内部装置は、加水分解に対して有利な触媒作用を及ぼし、副製品の形成がなく、十分に進行させる。連続プロセスのこの位置に、母液をメチオニン固体分離の後に再び加えると有利である。その際母液は溶解度に相当して、さらにまだ残留メチオニンを含み、これも本方法に有利なことが分かっている。
【0064】
加水分解塔内の反応溶液の平均滞留時間は、10〜20分であると有利である。カリウムイオン量の5−(2−メチルメルカプトエチル)−ヒダントイン+メチオニンの和に対するモル比は、10以下、有利には1.3〜5、殊には1.5〜2が有利である。反応溶液中のメチオニン酸カリウムで得られる収率は、この方法において代表的には99.0〜100%である。メチオニン酸カリウムの濃度は、ヒダントイン濃度の適当な選定により、または加水分解の後に得られる溶液の希釈または濃縮により調整できる。
【0065】
アルカリ金属メチオニン酸塩から、有利には上記の方法により得られた水溶液からのメチオニンの遊離も本発明に属する。従って本発明は、二酸化炭素を用いる遊離による水溶液中のアルカリ金属メチオニン酸塩からのメチオニンの製造方法であって、その際アルカリ金属メチオニン酸塩を含む水溶液をメチオニンの遊離の前に消泡剤を加える方法にも関する。さらに本発明は、二酸化炭素を用いる遊離による水溶液中のアルカリ金属メチオニン酸塩からのメチオニンの製造方法であって、その際強力な内部混合を有する攪拌セル反応器または完全に近い内部混合を有する攪拌反応器を用いて遊離を行う方法にも関する。
【0066】
これらの両方の方法は、一緒に組み合わせると有利である。消泡剤として、発泡を減らす作用を有するあらゆる化合物が適する。消泡剤は分散液の形で溶液中に加えると有利である。これにより溶液中に著しく良好な分散が得られ、また実質的に水溶液表面に集中することはない。これによりメチオニンの遊離に対する消泡剤の有利な作用、殊には薄い小平板またはフレークの形成の防止に有利である。大部分が直径100〜200μmの固体球状の結晶として生成する。消泡剤は、メチオニンの合計量(メチオニン+メチオニン酸、メチオニンに換算)に対して濃度1000〜10000ppmを加えると有利である。
【0067】
二酸化炭素を用いる水溶液からのメチオニンの遊離の場合に、二酸化炭素をノズル装置を通じて底部で水溶液中に導入すると著しく有利である。この場合にも、これはメチオニンの遊離にも有利である。さらに遊離は、有利には圧力1〜30バールにおいて、また有利には温度0〜100℃で行うと有利である。
【0068】
実質的にアンモニアを含まない溶液を用いると、極めて著しく有利である。
【0069】
後者の方法を連続的に実施しても著しく有利である。この方法を上記の方法と一緒にアルカリ金属メチオニン酸塩の製造に組み合わせると有利であり、その際上記の方法全ての組み合わせも殊に有利に可能である。
【0070】
以下には有利なD,L−メチオニン酸カリウムを用いるメチオニンの遊離のための方法を記載するが、ここで他のアルカリ金属、例えばナトリウムも可能である。ここに記載する別の有利なまたは一般的な操作条件は、上記の一般方法においても同様に該当する。
【0071】
二酸化炭素の導入によるD,L−メチオニン酸カリウムからのD,L−メチオニンの遊離において、殊には5−(2−メチルメルカプトエチル)−ヒダントインの加水分解溶液中において、この溶液が実際的にアンモニアを含まない場合に著しく有利である。さらに溶液が溶解したD,L−メチオニンを含むと有利である。さらに、一定の量の炭酸カリウムおよび炭酸水素カリウムが存在していてもよい。この溶液は、希望する場合には、二酸化炭素供給の前に活性炭上で精製することもできる。二酸化炭素供給は、通常、温度0〜100℃、有利には20〜35℃、かつ通常、圧力1〜30バール、有利には2〜5バールで行う。二酸化炭素は、pH値が約7〜9、有利には7.5〜8.5に到達するまでおよび/またはD,L−メチオニンの析出が終了するまで反応混合物中に導入すると有利である。その際二酸化炭素を反応器の底に直接、または有利にはノズル装置を通じて細かく分割して導入すると著しく有利である。反応器は、攪拌セル反応器としてまたは完全に近い攪拌反応器として構成すると有利である。さらに殊に連続法の場合に、消泡剤を追加すると処理量を多くすることもできる。消泡剤は通常、反応溶液中に存在する全メチオニンに対して、少なくとも1000および有利には10000ppm、殊には3000〜5000ppmの量を、殊には水性エマルションとして添加する。遊離したメチオニンは、有利には母液から分離すると、乾燥の後でダストが十分に少なく、良好な流れ特性および高いかさ密度で優れる。メチオニン粒子は、大部分が直径100〜200μmを有する。この方法によると、分離されたD,L−メチオニンの収率は、通常98〜100%である。D,L−メチオニンの分離の後、殊にはろ過の後に得られる母液は、必要ならば濃縮および/またはCO2の分離の後にさらに5−(2−メチルメルカプトエチル)−ヒダントインの加水分解に有利に使用できる。
【0072】
本発明は、図および実施例により以下に詳しく説明する。
【0073】
以下には、簡単化のために副産物、例えば取り出しの際のNH3/CO2の過剰は記載しない。
【0074】
【実施例】
実施例1〜4
5−(2−メチルメルカプトエチル)−ヒダントインの製造のための実施例
実施例1〜4の共通工程を図1に示す。連続操作の場合に、シアン化水素を混合ノズル1を通じて3−メチルメルカプトプロピオンアルデヒドと一緒にして、後続のスタティック・ミキサー2により混合させる。混合反応器3中で、アンモニアおよび二酸化炭素の水溶液を製造し、その際これらの成分を後続の工程からリサイクリングしてもよい。両方の混合物を循環反応器20中で反応混合物中に供給する。混合装置4中で、溶液と循環している反応混合物とを良く瞬間的に混合させる。循環混合物は、希望する温度に調整されている熱交換器5を通り、ポンプ6で送られる。反応混合物から連続的に相当する部分を循環系から取り出す。次いで反応の完結のために後反応器7にこれを送る。このようにして得られた製品混合物は次いで必要ならば直ちに次の反応工程に送ることができる。
【0075】
例1
図1に記載の装置を用いた。最初に、90℃に昇温した水を循環系に導いた。次いで循環系中に、1時間当たりに3−メチルメルカプトプロピオンアルデヒド10モル中のシアン化水素10.5モルの溶液およびアンモニア9.6重量%および二酸化炭素15.2重量%を含む炭酸アンモニウム水溶液6.8lを供給した。循環系の循環量は、1時間当たり300lであった。温度は90℃に維持した。圧力は14バールであった。循環系から絶えず供給量に相当する体積の反応混合物を取り出し、後反応に送った。平均滞留時間は、循環系中で10分間、後反応で2時間であった。使用した3−メチルメルカプトプロピオンアルデヒドに対する5−(2−メチルメルカプトエチル)−ヒダントインおよびメチオニンに鹸化できる化合物の収率は99.8%であった。(数値は、3−メチルメルカプトプロピオンアルデヒドを100%として算出したものである)
例2
実施例1と同様に操作したが、ただし最初に水の代わりに室温で飽和したアンモニア9.6重量%および二酸化炭素15.2重量%を含む炭酸アンモニウム溶液を循環系に導入した。収率は99.7%であった。
【0076】
例3
実施例1と同様に操作したが、ただし循環系内および後反応における温度を115℃に維持した。圧力は16バールであった。循環系内の循環量は、1時間当たり150lであった。平均滞留時間は循環系中で6分間、後反応中では20分間であった。到達した収率は99.9%であった。
【0077】
例4
実施例1と同様に操作したが、ただし1時間当たりに3−メチルメルカプトプロピオンアルデヒド10.0モル中のシアン化水素10.1モルの溶液およびアンモニア5.5重量%および二酸化炭素8.5重量%を含む炭酸アンモニウム水溶液6.8lを供給した。温度は循環系中および後反応において115℃に維持した。圧力は16バールであった。循環系中の流速は1時間当たりに150lであった。循環系中の平均滞留時間は、6分間、後反応では40分間であった。収率は99.8%であった。
【0078】
例5および6
メチオニン酸カリウム溶液製造の例
メチオニン酸カリウム溶液の製造のための一般的な工程は、図1に記載してある。
【0079】
例5
ジルコニウム内部装置を持たない加圧装置を用いる比較例
ポンプ圧力を用いて水蒸気で駆動しているステンレス鋼製の連続操作の加圧塔8(図2参照)中に、1時間当たりに水溶液状の炭酸水素カリウム100kgの溶液および水400l中の5−(2−メチルメルカプトエチル)−ヒダントイン41kgを供給する。その際反応混合物を180℃に加熱し、約8バールにおいて平均滞留時間約15分である。遊離したアンモニアおよび二酸化炭素を反応塔の塔頂において圧力保持弁を通じて取り出す。加圧反応器の底部において反応溶液を減圧し、熱交換器9を用いて冷却する。その際、1時間当たりに溶液中のメチオニン酸カリウム41.9kgが得られる(理論値の94.5%)。
【0080】
例6
ジルコニウム製内部装置を有する加圧反応器を利用する図2の装置を使用する。
【0081】
ポンプを用いて、1時間当たりに、図1のヒダントイン製造からの反応溶液1600l中の5−(2−メチルメルカプトエチル)−ヒダントイン553kgおよびジルコニウム内部装置を有する加圧加水分解塔8の頂部からリットル当たりカリウム含有量140gおよびリットル当たりに残留メチオニン含有量120gを有するメチオニン固体分離の後の母液還流からの水溶液中の炭酸カリウム/炭酸水素カリウムおよび水酸化カリウムから成る混合物3550lとを供給する。反応温度は165℃である。反応圧力は7バールである。遊離したアンモニアおよび二酸化炭素を塔の頂部において圧力保持弁を通じて取り出し、5−(2−メチルメルカプトエチル)−ヒダントインを合成し、あらためて送る。
【0082】
加圧装置の底部液は、リットル当たりカリウム96.5gおよびリットル当たりメチオニン175gを含む水性混合物を1時間当たり5150l得る(全システムにおいて全体でメチオニン476kg/hの増加に相当)(収率:100%)。反応溶液は、熱交換器9を通して冷却し、メチオニン遊離工程に送る。
【0083】
実施例7
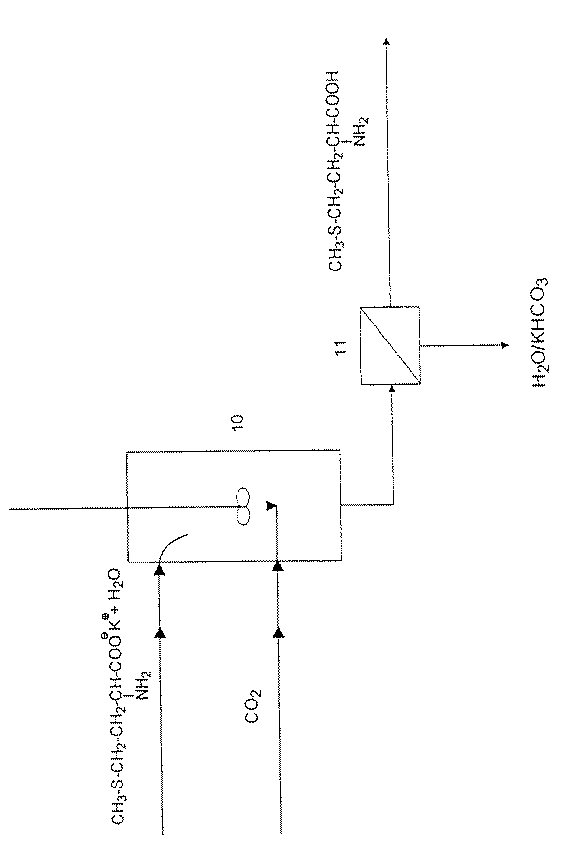
D,L−メチオニン酸カリウムからのD,L−メチオニンの遊離の実施例(図3)
容量340lの攪拌反応器10の頂部に、1時間当たりにD,L−メチオニン酸カリウム83.6kgを含む水溶液(5−(2−メチルメルカプトエチル)−ヒダントインの加水分解溶液)686lおよび追加して循環メチオニンおよびカリウム化合物39.77kgを連続的に供給する。同時に、反応器が圧力2〜3バールとなるように、反応器の底部に二酸化炭素を導入する。同様に、水性エマルションの形の消泡剤1時間当たり0.38kgを反応器内に供給する。この量は全メチオニンkgに対して消泡剤約3940ppmに相当する。反応温度は25℃に保持する。反応器内に一定の水準を保つために、反応器の下部に供給量に相当する量の反応溶液を取り出す。取り出した懸濁液をろ過し、その際固体D,L−メチオニン1時間当たり66.5kg(乾燥物質として計量)が得られ、D,L−メチオニン残留含有量39.77kgの母液をヒダントイン加水分解段階の鹸化剤としてリサイクルできる。収率は定量的である。
【図面の簡単な説明】
【図1】5−(2−メチルメルカプトエチル)−ヒダントインの連続的製造工程のフローチャートである。
【図2】5−(2−メチルメルカプトエチル)−ヒダントインのアルカリ金属メチオニン酸塩への連続的加水分解工程のフローチャートである。
【図3】アルカリ金属D,L−メチオニン酸塩からD,L−メチオニンの連続的遊離および単離工程フローチャートである。
【符号の説明】
1 混合ノズル、 2 スタティック・ミキサー、 3 混合反応器、 4 混合装置、 5 熱交換器、 6 ポンプ、 7 後反応器、 8 加圧加水分解塔、 9 熱交換器、 10 撹拌反応器、 20 循環反応器
Claims (9)
- 成分3−メチルメルカプトプロピオンアルデヒド、シアン化水素、アンモニアおよび二酸化炭素を、5−(2−メチルメルカプトエチル)−ヒダントインに転化し、これを引き続きメチオニンまたはその塩に転化することによるメチオニンまたはメチオニンの塩の連続的な製造方法において、成分の転化は少なくとも一種の予備混合物を介して導入し、3−メチルメルカプトプロピオンアルデヒドおよびシアン化水素からなる第一混合物を構成し、かつこの第一混合物を5−(2−メチルメルカプトエチル)ヒダントインへの転化のためにアンモニアと二酸化炭素との水溶液と一緒にすることを特徴とする、メチオニンまたはその塩の連続的な製造方法。
- 第一混合物において、溶液中のシアン化水素のモル割合は、3−メチルメルカプトプロピオンアルデヒド1モルに対して1.1モル以上でないことを特徴とする、請求項1記載の方法。
- すべての成分をすべて2種の予備混合物で使用することを特徴とする、請求項1または2記載の方法。
- 前記の第一の予備混合物を、アンモニアと二酸化炭素との水溶液と混合し、転化させることを特徴とする、請求項3記載の方法。
- 予備混合物を、すでに得られた5−(2−メチルメルカプトエチル)ヒダントインを含む反応混合物中に導入することを特徴とする、請求項1記載の方法。
- 3−メチルメルカプトプロピオンアルデヒド中のシアン化水素溶液を、アンモニアと二酸化炭素との水溶液と、この混合物がアンモニアの3−メチルメルカプトプロピオンアルデヒドに対するモル比1.2〜6対1.0を含むように混合することを特徴とする、請求項1記載の方法。
- アンモニアと二酸化炭素との水溶液を使用し、その際アンモニアの二酸化炭素に対するモル比は、二酸化炭素1モルに対して、アンモニア1.2〜4.0モルであることを特徴とする、請求項1記載の方法。
- 前記ヒダントインへの転化は、80〜140℃の温度において行うことを特徴とする、請求項1から7までのいずれか1項記載の方法。
- 前記ヒダントインへの転化は、20バール以下の加圧下において行うことを特徴とする、請求項1から8までのいずれか1項記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19547236.5 | 1995-12-18 | ||
| DE19547236A DE19547236A1 (de) | 1995-12-18 | 1995-12-18 | Verfahren zur Herstellung von D,L-Methionin oder dessen Salz |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006331080A Division JP4499706B2 (ja) | 1995-12-18 | 2006-12-07 | メチオニンおよびその塩の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH09176111A JPH09176111A (ja) | 1997-07-08 |
| JP3990475B2 true JP3990475B2 (ja) | 2007-10-10 |
Family
ID=7780446
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP33416996A Expired - Lifetime JP3990475B2 (ja) | 1995-12-18 | 1996-12-13 | メチオニンおよびその塩の製造方法 |
| JP2006331080A Expired - Lifetime JP4499706B2 (ja) | 1995-12-18 | 2006-12-07 | メチオニンおよびその塩の製造方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006331080A Expired - Lifetime JP4499706B2 (ja) | 1995-12-18 | 2006-12-07 | メチオニンおよびその塩の製造方法 |
Country Status (14)
| Country | Link |
|---|---|
| US (2) | US5770769A (ja) |
| EP (3) | EP1710232A1 (ja) |
| JP (2) | JP3990475B2 (ja) |
| CN (3) | CN1079095C (ja) |
| BR (1) | BR9606040A (ja) |
| BY (1) | BY5258C1 (ja) |
| CA (1) | CA2193161A1 (ja) |
| DE (3) | DE19547236A1 (ja) |
| ES (2) | ES2191731T3 (ja) |
| IL (1) | IL119839A (ja) |
| MX (1) | MX9606011A (ja) |
| RU (1) | RU2176240C2 (ja) |
| UA (1) | UA47404C2 (ja) |
| ZA (1) | ZA9610608B (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9403764B2 (en) | 2011-08-30 | 2016-08-02 | Evonik Degussa Gmbh | Method of production of a methionine salt |
Families Citing this family (79)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19846825A1 (de) * | 1998-10-10 | 2000-04-13 | Degussa | Rieselfähige Methionin-haltige Formlinge und Verfahren zu deren Herstellung |
| DE10024540A1 (de) * | 2000-05-18 | 2001-01-18 | Lurgi Zimmer Ag | Fluidleitungsstück mit Innentemperierung |
| DE10160358A1 (de) * | 2001-12-08 | 2003-06-18 | Degussa | Verfahren zur Herstellung von Methionin |
| US20050106250A1 (en) | 2002-05-10 | 2005-05-19 | Hasseberg Hans A. | Protected active compound formulations of amino acids and process for their preparation |
| US8327754B2 (en) | 2003-07-22 | 2012-12-11 | The Coca-Cola Company | Coffee and tea pod |
| US6948420B2 (en) | 2003-07-22 | 2005-09-27 | The Coca-Cola Company | Coffee and tea pod |
| US8505440B2 (en) | 2003-07-22 | 2013-08-13 | The Coca-Cola Company | System for varying coffee intensity |
| ATE509908T1 (de) * | 2004-02-14 | 2011-06-15 | Evonik Degussa Gmbh | Verfahren zur herstellung von methionin |
| DE102004035465A1 (de) * | 2004-07-22 | 2006-02-16 | Degussa Ag | Verfahren zur Reinigung von CO2-Gasströmen |
| DE102006004063A1 (de) * | 2006-01-28 | 2007-08-02 | Degussa Gmbh | Verfahren zur Herstellung von Methionin aus Homoserin |
| JP2007254442A (ja) * | 2006-03-27 | 2007-10-04 | Sumitomo Chemical Co Ltd | メチオニンの製造方法 |
| FR2903690B1 (fr) * | 2006-07-11 | 2008-11-14 | Adisseo Ireland Ltd | Procede de preparation de la methionine a partir d'acroleine sans isoler de produits intermediaires |
| WO2008006977A1 (fr) * | 2006-07-11 | 2008-01-17 | Adisseo France S.A.S. | Procédé de préparation du 2-hydroxy-4-(méthylthio)butyronitrile et de la méthionine |
| US7964230B2 (en) | 2006-08-04 | 2011-06-21 | The Coca-Cola Company | Method of sealing a pod for dispersible materials |
| US7947316B2 (en) | 2006-08-04 | 2011-05-24 | The Coca-Cola Company | Pod for dispersible materials |
| TWI410511B (zh) | 2007-03-16 | 2013-10-01 | Univ Tohoku Nat Univ Corp | 磁控管濺鍍裝置 |
| JP2008266298A (ja) * | 2007-03-27 | 2008-11-06 | Sumitomo Chemical Co Ltd | メチオニンの製造方法 |
| WO2009035074A1 (ja) | 2007-09-14 | 2009-03-19 | National University Corporation Tohoku University | 陰極体及びそれを用いた蛍光管 |
| FR2928910B1 (fr) * | 2008-03-20 | 2010-03-12 | Arkema France | Procede ameliore de production d'acide cyanhydrique |
| US7987768B2 (en) | 2008-03-27 | 2011-08-02 | The Coca-Cola Company | Brewing mechanism |
| JP2009292796A (ja) * | 2008-06-09 | 2009-12-17 | Sumitomo Chemical Co Ltd | メチオニンの製造方法 |
| JP2009292795A (ja) * | 2008-06-09 | 2009-12-17 | Sumitomo Chemical Co Ltd | メチオニンの製造方法 |
| US8522668B2 (en) | 2008-08-08 | 2013-09-03 | The Coca-Cola Company | Systems and methods for on demand iced tea |
| EP2174700A1 (de) | 2008-10-13 | 2010-04-14 | Siemens Aktiengesellschaft | Absorptionsmittel, Verfahren zur Herstellung eines Absorptionsmittels sowie Verwendung eines Absorptionsmittels |
| FR2938535B1 (fr) * | 2008-11-20 | 2012-08-17 | Arkema France | Procede de fabrication de methylmercaptopropionaldehyde et de methionine a partir de matieres renouvelables |
| US8476427B2 (en) * | 2010-03-09 | 2013-07-02 | Novus International, Inc. | Preparation of methionine or selenomethionine from homoserine via a carbamate intermediate |
| JP5741099B2 (ja) * | 2010-03-25 | 2015-07-01 | 住友化学株式会社 | 含硫アミノ酸またはその塩の製造方法 |
| JP5524736B2 (ja) * | 2010-06-29 | 2014-06-18 | 住友化学株式会社 | メチオニンの製造方法 |
| CN102399177B (zh) * | 2010-09-15 | 2016-02-24 | 李宽义 | 连续化合成蛋氨酸的环保清洁工艺方法 |
| FR2965561B1 (fr) * | 2010-10-05 | 2012-08-31 | Adisseo France Sas | Procede de preparation d?un acide amine a partir de 2-aminobutyrolactone |
| WO2012087177A1 (ru) * | 2010-12-23 | 2012-06-28 | Polyakov Viktor Stanislavovich | Способ переработки белоксодержащих материалов в смеси природных аминокислот, низкомолекулярных пептидов и олигопептидов |
| MX341595B (es) | 2011-02-23 | 2016-08-26 | Evonik Degussa Gmbh | Metodo para producir 2-hidroxi-4-(metiltio)butironitrilo a partir de 3-(metiltio)propanal y cianuro de hidrogeno. |
| DE102011081828A1 (de) * | 2011-08-30 | 2013-02-28 | Evonik Degussa Gmbh | Verfahren zur Umsetzung von Methylmercaptopropionaldehyd aus Roh-Acrolein und Roh-Methylmercaptan |
| WO2013055606A1 (en) * | 2011-10-13 | 2013-04-18 | Merck Sharp & Dohme Corp. | Mineralocorticoid receptor antagonists |
| US9085568B2 (en) | 2011-10-13 | 2015-07-21 | Merck Sharp & Dohme Corp. | Mineralocorticoid receptor antagonists |
| EP2765860B1 (en) | 2011-10-13 | 2016-08-17 | Merck Sharp & Dohme Corp. | Mineralocorticoid receptor antagonists |
| EP2641898A1 (de) * | 2012-03-20 | 2013-09-25 | Evonik Industries AG | Verfahren zur Herstellung von Methionin |
| CN102659684B (zh) * | 2012-04-28 | 2013-03-06 | 重庆紫光天化蛋氨酸有限责任公司 | 海因的制备装置及方法 |
| CN102659650B (zh) * | 2012-04-28 | 2013-03-06 | 重庆紫光天化蛋氨酸有限责任公司 | Dl-甲硫氨酸盐的制备装置及方法 |
| CN102633699B (zh) * | 2012-04-28 | 2013-03-06 | 重庆紫光天化蛋氨酸有限责任公司 | Dl-甲硫氨酸的制备方法 |
| CN102796033B (zh) * | 2012-09-03 | 2014-02-26 | 浙江新和成股份有限公司 | 一种清洁的d,l-蛋氨酸制备方法 |
| EP2848607A1 (de) | 2013-09-17 | 2015-03-18 | Evonik Industries AG | Verfahren zur Gewinnung von Methionin |
| CN107382799A (zh) * | 2014-03-03 | 2017-11-24 | 宁夏紫光天化蛋氨酸有限责任公司 | 离子交换酸化蛋氨酸盐制备蛋氨酸的方法及专用设备 |
| CN103864633A (zh) * | 2014-04-03 | 2014-06-18 | 重庆紫光国际化工有限责任公司 | α-氨基异丁酸的制备方法 |
| CN104744326B (zh) * | 2015-02-12 | 2016-08-10 | 山东新和成氨基酸有限公司 | 一种连续制备高堆积密度甲硫氨酸结晶的方法 |
| CN104693082A (zh) * | 2015-04-03 | 2015-06-10 | 重庆紫光化工股份有限公司 | 一种制备蛋氨酸的方法 |
| FR3041659B1 (fr) | 2015-09-30 | 2017-10-20 | Arkema France | Procede de production de l-methionine |
| FR3041658B1 (fr) | 2015-09-30 | 2017-10-20 | Arkema France | Procede de production de l-methionine |
| CN105296557A (zh) * | 2015-10-31 | 2016-02-03 | 高大元 | 一种D,L-α-蛋氨酸钙的合成方法 |
| CN105949097B (zh) * | 2016-06-03 | 2018-09-11 | 宁夏紫光天化蛋氨酸有限责任公司 | 一种减少蛋氨酸离交排气和蛋氨酸结晶母液中杂质的方法 |
| ES2820238T3 (es) | 2016-06-27 | 2021-04-20 | Evonik Degussa Gmbh | Granulados con contenido en dipéptidos |
| EP3330380A1 (en) | 2016-12-05 | 2018-06-06 | Evonik Degussa GmbH | Process for producing l-methionine from methional |
| EP3339289A1 (de) | 2016-12-21 | 2018-06-27 | Evonik Degussa GmbH | Verfahren zur herstellung von methionin |
| JP7128808B2 (ja) | 2017-04-27 | 2022-08-31 | 住友化学株式会社 | 回収二酸化炭素の精製方法、および回収二酸化炭素の精製工程を包含するメチオニンの製造方法 |
| EP3404018A1 (en) | 2017-05-15 | 2018-11-21 | Evonik Degussa GmbH | Process for preparing methionine |
| EP3431465A1 (en) | 2017-07-21 | 2019-01-23 | Evonik Degussa GmbH | Process for preparing methionine |
| JP7431581B2 (ja) * | 2017-05-15 | 2024-02-15 | エボニック オペレーションズ ゲーエムベーハー | メチオニンの製造方法 |
| EP3406593A1 (de) | 2017-05-24 | 2018-11-28 | Evonik Degussa GmbH | Verfahren zur herstellung von methionin |
| CN108658820A (zh) * | 2017-06-13 | 2018-10-16 | 宁夏紫光天化蛋氨酸有限责任公司 | 减少副产硫酸钠的蛋氨酸生产方法 |
| CN108658819A (zh) * | 2017-06-13 | 2018-10-16 | 宁夏紫光天化蛋氨酸有限责任公司 | 一种甲硫氨酸的清洁生产方法 |
| EP3461803A1 (de) | 2017-10-02 | 2019-04-03 | Evonik Degussa GmbH | Herstellverfahren für dipeptidhaltige granulate |
| EP3632896A1 (de) | 2018-10-01 | 2020-04-08 | Evonik Operations GmbH | Herstellung von aminosäuren aus ihren aminonitrilen |
| EP3632894A1 (de) | 2018-10-01 | 2020-04-08 | Evonik Operations GmbH | Nebenproduktarme herstellung von methionin aus methioninnitril |
| EP3632895A1 (de) | 2018-10-01 | 2020-04-08 | Evonik Operations GmbH | Salzfreie herstellung von aminosäuren aus ihren aminonitrilen |
| CN109232339B (zh) * | 2018-11-09 | 2020-09-08 | 天宝动物营养科技股份有限公司 | 一种d,l-蛋氨酸、d,l-蛋氨酸羟基类似物及其钙盐联产的清洁工艺 |
| EP3656760A1 (de) | 2018-11-21 | 2020-05-27 | Evonik Operations GmbH | Lagerstabile form von 3-methylthiopropionaldehyd |
| CN113166045B (zh) | 2018-12-14 | 2024-03-08 | 赢创运营有限公司 | 生产甲硫氨酸的方法 |
| EP3689851A1 (en) | 2019-02-04 | 2020-08-05 | Evonik Operations GmbH | Salt-free production of methionine from methionine nitrile |
| EP3986861B1 (en) | 2019-06-18 | 2023-08-02 | Evonik Operations GmbH | Process for the preparation of d,l-methionine |
| CN110305050A (zh) * | 2019-07-18 | 2019-10-08 | 宁夏紫光天化蛋氨酸有限责任公司 | 一种5-[2-(甲硫基)乙基]海因水解排气制备蛋氨酸钠的方法 |
| JPWO2021054268A1 (ja) | 2019-09-17 | 2021-03-25 | ||
| CN111100051B (zh) | 2019-12-31 | 2022-01-28 | 山东新和成氨基酸有限公司 | 在甲硫氨酸制备过程中使用的添加剂及甲硫氨酸的制备方法 |
| WO2022078940A1 (en) | 2020-10-13 | 2022-04-21 | Evonik Operations Gmbh | D,l-methionine with an optimized particle size distribution |
| FR3120367B1 (fr) | 2021-03-04 | 2024-02-16 | Adisseo France Sas | Procédé de préparation du 2-hydroxy-4-méthylthiobutyronitrile ou de son équivalent sélénié et applications |
| FR3123068B1 (fr) | 2021-05-20 | 2024-05-10 | Adisseo France Sas | Procede de fabrication de la methionine |
| WO2023067059A1 (en) | 2021-10-20 | 2023-04-27 | Evonik Operations Gmbh | Process for producing dl-methionine and dlld-methionylmethionine |
| WO2023067061A1 (en) | 2021-10-20 | 2023-04-27 | Evonik Operations Gmbh | Precursors for the preparation of dl-methionine and dlld-methionylmethionine |
| WO2023144265A1 (en) | 2022-01-28 | 2023-08-03 | Evonik Operations Gmbh | Granular catalyst for the hydrolysis of amino nitriles and amino amides to amino acids or derivatives thereof |
| EP4293012A1 (de) | 2022-06-17 | 2023-12-20 | Evonik Operations GmbH | Verfahren zur gewinnung von gemischen enthaltend methionin und kaliumhydrogencarbonat |
Family Cites Families (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2557913A (en) | 1946-11-07 | 1951-06-19 | Dow Chemical Co | Production of methionine |
| US2527366A (en) | 1946-11-07 | 1950-10-24 | Dow Chemical Co | Production of amino acids |
| US2557920A (en) | 1946-11-07 | 1951-06-19 | Dow Chemical Co | Method for production of amino acids |
| US2642459A (en) * | 1948-12-08 | 1953-06-16 | Dow Chemical Co | Production and purification of amino acids |
| DE1166201B (de) | 1959-04-15 | 1964-03-26 | Degussa | Druckloses Verfahren zur Herstellung von in 5-Stellung ein- oder zweifach substituierten Hydantoinen |
| NL128180C (ja) | 1964-12-29 | |||
| GB1108926A (en) * | 1965-01-12 | 1968-04-10 | Sumitomo Chemical Co | Method for producing 5-ª‰-methylmercaptoethylhydantoin |
| US3433832A (en) | 1965-12-03 | 1969-03-18 | Dow Chemical Co | Recovery of alpha-aminocarboxylic acids from sodium chloride solutions |
| FR1532723A (fr) | 1966-07-02 | 1968-07-12 | Sumitomo Chemical Co | Procédé de fabrication d'un alpha-amino acide |
| US3636098A (en) * | 1966-07-02 | 1972-01-18 | Sumitomo Chemical Co | Process for producing methionine |
| US4069251A (en) * | 1969-02-08 | 1978-01-17 | Deutsche Gold- Und Silber-Scheideanstalt Vormals Roessler | Continuous process for the manufacture of methionine |
| US3653034A (en) * | 1970-02-12 | 1972-03-28 | Honeywell Inc | High speed decode circuit utilizing field effect transistors |
| JPS49818B1 (ja) | 1970-08-12 | 1974-01-10 | ||
| JPS5249473B2 (ja) * | 1972-08-09 | 1977-12-17 | ||
| JPS5542068B2 (ja) | 1973-03-15 | 1980-10-28 | ||
| JPS5530512B2 (ja) | 1973-05-19 | 1980-08-11 | ||
| JPS5018467A (ja) | 1973-06-20 | 1975-02-26 | ||
| JPS582227B2 (ja) * | 1974-02-04 | 1983-01-14 | 住友化学工業株式会社 | アルフア− − アミノサン ノ セイゾウホウホウ |
| JPS5715756B2 (ja) * | 1974-06-05 | 1982-04-01 | ||
| JPS5811421B2 (ja) | 1975-04-03 | 1983-03-02 | 住友化学工業株式会社 | アルフア− − アミノサンノセイゾウホウ |
| JPS5227768A (en) | 1975-04-23 | 1977-03-02 | Kanegafuchi Chem Ind Co Ltd | Preparation of 5-(beta-methylmercaptoethyl) -hydantoin |
| SU740770A1 (ru) | 1975-09-15 | 1980-06-15 | Волгоградский Политехнический Институт | Способ получени 5-(2-метилмеркаптоэтил)-гидантоина |
| JPS54132520A (en) | 1978-04-03 | 1979-10-15 | Sumitomo Chem Co Ltd | Preparation of colorless methionine |
| DE2846160A1 (de) | 1978-10-24 | 1980-05-08 | Kernforschungsanlage Juelich | Wirbelschichtreaktor mit offenem reaktionsgaszutritt und verfahren zur laminar-durchflussteigerung |
| BE877200A (fr) | 1979-06-22 | 1979-10-15 | Sumitomo Chemical Co | Procede de preparation de methionine incolore. |
| US4272631A (en) | 1980-02-25 | 1981-06-09 | Diamond Shamrock Corporation | Hydrolysis of 5-(beta-methylmercaptoethyl)-hydantoin |
| DE3266977D1 (en) * | 1981-03-26 | 1985-11-28 | Mitsubishi Gas Chemical Co | Process for producing alpha-amino acids |
| JPH0244056B2 (ja) | 1983-06-27 | 1990-10-02 | Hitachi Chemical Co Ltd | Kankoseijushisoseibutsu |
| DE3335218A1 (de) * | 1983-09-29 | 1985-04-11 | Degussa Ag, 6000 Frankfurt | Verfahren zur hydrolyse von 5-(ss-methylmercaptoethyl) -hydantoin |
| JPH0395145A (ja) * | 1989-09-08 | 1991-04-19 | Sumitomo Chem Co Ltd | α―アミノ酸の製造方法 |
| JPH0395146A (ja) * | 1989-09-08 | 1991-04-19 | Sumitomo Chem Co Ltd | α―アミノ酸の製造法 |
| JPH03232848A (ja) * | 1990-02-06 | 1991-10-16 | Mitsui Toatsu Chem Inc | グリシンの製造方法 |
| JP2921097B2 (ja) * | 1990-10-30 | 1999-07-19 | 住友化学工業株式会社 | メチオニンの製造方法 |
| JP3173112B2 (ja) * | 1992-04-09 | 2001-06-04 | 住友化学工業株式会社 | メチオニンの製造方法 |
| NO179833C (no) | 1992-06-17 | 1996-12-27 | Rhone Poulenc Chemicals | Organiske cerium(IV)-forbindelser, fremgangsmåte for fremstilling derav og opplösning, filmdannende blanding og hydrokarbonbrennstoff omfattende slike |
-
1995
- 1995-12-18 DE DE19547236A patent/DE19547236A1/de not_active Ceased
-
1996
- 1996-10-25 ES ES96117124T patent/ES2191731T3/es not_active Expired - Lifetime
- 1996-10-25 DE DE59611374T patent/DE59611374D1/de not_active Expired - Lifetime
- 1996-10-25 EP EP06113758A patent/EP1710232A1/de not_active Ceased
- 1996-10-25 EP EP02011911A patent/EP1256571B1/de not_active Expired - Lifetime
- 1996-10-25 ES ES02011911T patent/ES2269554T3/es not_active Expired - Lifetime
- 1996-10-25 DE DE59610116T patent/DE59610116D1/de not_active Expired - Lifetime
- 1996-10-25 EP EP96117124A patent/EP0780370B1/de not_active Expired - Lifetime
- 1996-12-02 MX MX9606011A patent/MX9606011A/es unknown
- 1996-12-10 CN CN96121608A patent/CN1079095C/zh not_active Expired - Lifetime
- 1996-12-10 CN CNA2004100946113A patent/CN1636975A/zh active Pending
- 1996-12-10 CN CNB011170794A patent/CN1184199C/zh not_active Expired - Lifetime
- 1996-12-13 JP JP33416996A patent/JP3990475B2/ja not_active Expired - Lifetime
- 1996-12-16 IL IL11983996A patent/IL119839A/xx not_active IP Right Cessation
- 1996-12-17 BY BY960851A patent/BY5258C1/xx unknown
- 1996-12-17 CA CA002193161A patent/CA2193161A1/en not_active Abandoned
- 1996-12-17 BR BR9606040A patent/BR9606040A/pt not_active Application Discontinuation
- 1996-12-17 ZA ZA9610608A patent/ZA9610608B/xx unknown
- 1996-12-17 UA UA96124682A patent/UA47404C2/uk unknown
- 1996-12-18 RU RU96123762/04A patent/RU2176240C2/ru active
- 1996-12-18 US US08/768,624 patent/US5770769A/en not_active Expired - Lifetime
-
1998
- 1998-03-09 US US09/037,020 patent/US5990349A/en not_active Expired - Lifetime
-
2006
- 2006-12-07 JP JP2006331080A patent/JP4499706B2/ja not_active Expired - Lifetime
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9403764B2 (en) | 2011-08-30 | 2016-08-02 | Evonik Degussa Gmbh | Method of production of a methionine salt |
Also Published As
| Publication number | Publication date |
|---|---|
| BY5258C1 (ja) | 2003-06-30 |
| EP0780370A2 (de) | 1997-06-25 |
| EP0780370A3 (de) | 1997-08-27 |
| EP1256571B1 (de) | 2006-08-02 |
| US5770769A (en) | 1998-06-23 |
| DE19547236A1 (de) | 1997-07-03 |
| EP1256571A1 (de) | 2002-11-13 |
| ES2191731T3 (es) | 2003-09-16 |
| IL119839A0 (en) | 1997-03-18 |
| EP1710232A1 (de) | 2006-10-11 |
| BR9606040A (pt) | 1998-08-25 |
| CN1160043A (zh) | 1997-09-24 |
| UA47404C2 (uk) | 2002-07-15 |
| US5990349A (en) | 1999-11-23 |
| MX9606011A (es) | 1997-08-30 |
| CN1376671A (zh) | 2002-10-30 |
| CN1636975A (zh) | 2005-07-13 |
| IL119839A (en) | 2000-08-13 |
| JP4499706B2 (ja) | 2010-07-07 |
| DE59610116D1 (de) | 2003-03-13 |
| ES2269554T3 (es) | 2007-04-01 |
| CN1079095C (zh) | 2002-02-13 |
| CA2193161A1 (en) | 1997-06-19 |
| JP2007099778A (ja) | 2007-04-19 |
| DE59611374D1 (de) | 2006-09-14 |
| EP0780370B1 (de) | 2003-02-05 |
| CN1184199C (zh) | 2005-01-12 |
| ZA9610608B (en) | 1997-06-24 |
| JPH09176111A (ja) | 1997-07-08 |
| RU2176240C2 (ru) | 2001-11-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3990475B2 (ja) | メチオニンおよびその塩の製造方法 | |
| MXPA96006011A (en) | Process for the preparation of dl-metionina or lasal de la mi | |
| CN101735124B (zh) | 用于制备甲硫氨酸的方法 | |
| JP4338524B2 (ja) | メチオニンの製造方法 | |
| KR100243985B1 (ko) | 펜타에리트리톨의 제조방법 | |
| JP4792754B2 (ja) | アンモニウム塩を含有する溶液からアンモニアを除去する方法 | |
| EP1556343B1 (en) | Process for the production of 3-methylthiopropanal | |
| US20230348370A1 (en) | Process for making taurine | |
| EP0441588B1 (en) | Process for preparing glycine | |
| JPH0737436B2 (ja) | カルボジヒドラジドの製造方法 | |
| KR100562176B1 (ko) | D,l-메티오닌또는이의염의제조방법 | |
| US5053545A (en) | Method of preparing amino alcohols | |
| JP4131025B2 (ja) | ケタジン及び水加ヒドラジンの製造方法 | |
| US20010031891A1 (en) | Process for producing perfluoroalkanesulfinate | |
| JPH1025273A (ja) | アセトンシアンヒドリンの製造方法 | |
| JP2801781B2 (ja) | グリシンの製造方法 | |
| JPS60104037A (ja) | 1,4−ジヒドロキシ−2−ナフトエ酸の製造方法 | |
| RU2292335C2 (ru) | Способ и устройство для получения гидразодикарбонамида с использованием мочевины в качестве исходного продукта | |
| JP2023539759A (ja) | グアニジノ酢酸の製造方法 | |
| JP2764346B2 (ja) | グリシンの製造方法 | |
| CN115925565A (zh) | 一种食品级甘氨酸及其连续制备方法 | |
| JPH03193748A (ja) | グリシンの製造方法 | |
| WO2021195001A1 (en) | Process sulfonation to produce taurine | |
| JPH0491052A (ja) | 1,3―フェニレンジオキシジ酢酸の製法 | |
| JPH04360873A (ja) | ジメチロールヒダントイン化合物の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20031210 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20060516 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20060608 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20060905 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20060908 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20061207 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20070202 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20070328 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20070402 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070531 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20070711 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20070720 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100727 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100727 Year of fee payment: 3 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100727 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110727 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120727 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130727 Year of fee payment: 6 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |