ES2916220T3 - Inhibidores del receptor de factor de crecimiento de fibroblasto - Google Patents
Inhibidores del receptor de factor de crecimiento de fibroblasto Download PDFInfo
- Publication number
- ES2916220T3 ES2916220T3 ES13740458T ES13740458T ES2916220T3 ES 2916220 T3 ES2916220 T3 ES 2916220T3 ES 13740458 T ES13740458 T ES 13740458T ES 13740458 T ES13740458 T ES 13740458T ES 2916220 T3 ES2916220 T3 ES 2916220T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- mmol
- dimethoxyphenyl
- synthesis
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/71—Receptors; Cell surface antigens; Cell surface determinants for growth factors; for growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4375—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having nitrogen as a ring heteroatom, e.g. quinolizines, naphthyridines, berberine, vincamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/42—One nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/78—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 2
- C07D239/84—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/14—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D295/155—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D475/00—Heterocyclic compounds containing pteridine ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D475/00—Heterocyclic compounds containing pteridine ring systems
- C07D475/02—Heterocyclic compounds containing pteridine ring systems with an oxygen atom directly attached in position 4
- C07D475/04—Heterocyclic compounds containing pteridine ring systems with an oxygen atom directly attached in position 4 with a nitrogen atom directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Abstract
Compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo: **(Ver fórmula)** en la que: el anillo A es un grupo arilo, heteroarilo, heterocíclico o alicíclico de 3-8 miembros; X es CH o N; Y es CH o N-R4, en el que R4 es H o alquilo C1-6; L es -[C(R5)(R6)]q-, en el que: cada uno de R5 y R6 es, independientemente, H o alquilo C1-6; y q es 0-4; cada R1, R2 y R3 es, independientemente, halo, ciano, alcoxi C1-6 opcionalmente sustituido, hidroxi, oxo, amino, amido, alquil urea, alquilo C1-6 opcionalmente sustituido, o heterociclilo C1-6 opcionalmente sustituido; m es 0-3; n es 4; p es 0-2; y Warhead se selecciona entre: **(Ver fórmula)** en los que: X es un grupo saliente seleccionado entre halógeno y triflato; y cada uno de Ra, Rb y Rc es, independientemente, H, alquilo C1-4 sustituido o no sustituido, o cicloalquilo C3- 4 sustituido o no sustituido.
Description
DESCRIPCIÓN
Inhibidores del receptor de factor de crecimiento de fibroblasto
Reivindicación de prioridad
Esta solicitud de patente reivindica prioridad del documento USSN 61/670.379, presentada el 11 de 2012 Julio y USSN 61/746.666, presentada el 28 de diciembre de 2012.
Campo de la invención
En el presente documento se describen compuestos, procedimientos para preparar dichos compuestos, composiciones farmacéuticas y procedimientos para usar dichos compuestos y composiciones para inhibir la actividad de las tirosina quinasas.
Antecedentes
El receptor 4 del factor de crecimiento de fibroblastos (FGFR-4) es una proteína que en los seres humanos está codificada por el gen FGFR-4. Esta proteína es un miembro de la familia de receptores del factor de crecimiento de fibroblastos, donde la secuencia de aminoácidos estuvo altamente conservada entre los miembros a lo largo de la evolución. Los miembros 1-4 de la familia FGFR se diferencian entre sí en sus afinidades de ligando y distribución tisular. Una proteína representativa de longitud completa consiste en una región extracelular compuesta por tres dominios similares a las inmunoglobulinas, un solo segmento hidrófobo que abarca la membrana hidrófoba y un dominio de tirosina quinasa citoplasmático. La porción extracelular de la proteína interactúa con los factores de crecimiento de fibroblastos, lo que pone en movimiento una cascada de señales corriente abajo que, en última instancia, influyen en la mitogénesis y la diferenciación. La organización genómica del gen FGFR-4 abarca 18 exones. Aunque se ha observado un corte y empalme alternativo, no hay evidencias de que la mitad C-terminal del dominio IgIII de esta proteína varíe entre tres formas alternadas, tal como se indica para FGFR 1-3.
La mineralización ectópica, caracterizada por la deposición inadecuada de calcio-fósforo en el tejido blando, se ha observado en ratas tratadas con un inhibidor de FGFR-1 (Brown, AP et al. (2005), Toxicol. Pathol., P. 449-455). Esto sugiere que la inhibición selectiva de FGFR-4 sin inhibición de otras isoformas de FGFR, incluido FGFR-1, puede ser deseable para evitar ciertas toxicidades. El FGFR-4 se une preferentemente al factor de crecimiento de fibroblastos 19 (FGF19) y recientemente se ha asociado con la progresión de ciertos sarcomas, cáncer de células renales, cáncer de mama y cáncer de hígado.
El documento WO 2004/063195 describe inhibidores de piridopirimidina quinasa útiles en el tratamiento del cáncer. Zhou et al, Chemistry and Biology (2010) 17 (3): 285-295, da a conocer el compuesto FIIN-1, un inhibidor irreversible de FGFR-1,2, 3 y 4.
Características de la invención
En el presente documento se describen inhibidores de FGFR-4. En el presente documento se describen además formulaciones farmacéuticas que incluyen un inhibidor de FGFR-4.
La presente invención proporciona compuestos y composiciones farmacéuticas tal como se establece en las reivindicaciones adjuntas.
La presente invención presenta un compuesto de Fórmula 1, o una sal farmacéuticamente aceptable del mismo:

en la que Warhead es un resto como se define en la reivindicación 1; el anillo A es un grupo arilo, heteroarilo, heterocíclico o alicíclico de 3-8 miembros; X es CH o N; Y es CH o N-R4 en el que R4 es H o alquilo C1-6; L es -[C(R5)(R6)]q-, en el que cada uno de R5 y R6 es, independientemente, H o alquilo C1-6; y q es 0-4; cada R1, R2y R3 es, independientemente, halo, ciano, alcoxi C^opcionalmente sustituido, hidroxi, oxo, amino, amido, alquil urea, alquilo C1-6 opcionalmente sustituido o heterociclilo C1-6 opcionalmente sustituido; m es 0-3; n es 4; y p es 0-2. En algunas realizaciones, el anillo A es fenilo, por ejemplo, un fenilo disustituido en 1,2; R2 es halo o metoxi; n es 4; X es N; R1 es metilo; y/o m es 1.
En un aspecto, la divulgación presenta un compuesto de Fórmula II, o una sal farmacéuticamente aceptable del mismo:

en la que Warhead es un resto que es capaz de formar un enlace covalente con un nucleófilo; W es C o N; Z es CH o N; Y es CH o NR4en el que R4es H o alquilo C1-6; R1 es H o alquilo C1-6; cada uno de R2y R3es, independientemente, halo, ciano, alcoxi C1-6 opcionalmente sustituido, hidroxi, amino, amido, alquil urea opcionalmente sustituida, alquilo C1-6 opcionalmente sustituido, heterociclo C1-6 opcionalmente sustituido; n es 0-4; y p es 0-2. En algunos casos, R2 es halo o metoxi; n es 2 o 4; Y es N-R4, donde R4 es metilo; y/o R1 es metilo.
En otro aspecto, la divulgación presenta un compuesto de Fórmula III, o una sal farmacéuticamente aceptable del mismo:

en la que Warhead es un resto que es capaz de formar un enlace covalente con un nucleófilo; R1 es H o alquilo C1-6 opcionalmente sustituido, incluyendo dialquilaminoalquilo; cada uno de R2y R3 es, independientemente, halo, ciano, alcoxi C1-6 opcionalmente sustituido, hidroxi, amino, amido, alquil urea opcionalmente sustituida, alquilo C16 opcionalmente sustituido, heterociclilo Ci-6 opcionalmente sustituido; n es 0-4; y p es 0-2. En algunos casos, R2 es halo o metoxi; n es 2 o 4. En algunos casos; R1 es metilo; en otros casos, R1 es dietilaminobutilo.
En otro aspecto, la divulgación presenta un compuesto de Fórmula IV, o una sal farmacéuticamente aceptable del mismo:

en la que Warhead es un resto que es capaz de formar un enlace covalente con un nucleófilo; R1 es H o alquilo C1-6 opcionalmente sustituido; cada uno de R2y R3 es, independientemente, halo, ciano, alcoxi C1-6 opcionalmente sustituido, hidroxi, amino, amido, alquil urea opcionalmente sustituida, alquilo C1-6 opcionalmente sustituido, heterociclilo C1-6 opcionalmente sustituido; n es 0-4; y p es 0-2. En algunos casos, R2 es halo o metoxi; n es 2 o 4; y/o R1 es metilo.
En otro aspecto, la divulgación presenta un compuesto de Fórmula V, o una sal farmacéuticamente aceptable del mismo:

en la que Warhead es un resto que es capaz de formar un enlace covalente con un nucleófilo; cada uno de R1-R3 es, independientemente, halo, ciano, alcoxi C1-6 opcionalmente sustituido, hidroxi, amino, amido, alquil urea opcionalmente sustituida, alquilo C1-6 opcionalmente sustituido, heterociclilo C1-6 opcionalmente sustituido; heterociclilamido C1-6 opcionalmente sustituido; m es 0-3; n es 0-4; y p es 0-2.
En otro aspecto, la divulgación presenta un compuesto de Fórmula VI, o una sal farmacéuticamente aceptable del mismo:

en la que Warhead es un resto que es capaz de formar un enlace covalente con un nucleófilo; L es arilo, heteroarilo o -[C(R5)(R6)]q-, donde cada uno de R5 y R6 es, independientemente, H o alquilo C1-6; y q es 0-4; cada uno de R1 es, independientemente, halo, ciano, alcoxi C1-6 opcionalmente sustituido, hidroxi, oxo, amino, amido, alquilurea opcionalmente sustituida, alquilo C1-6 opcionalmente sustituido, heterociclilo C1.6 opcionalmente sustituido; y m es 0 3. En algunos casos, L es alquileno; en otros casos, L es fenilo. En algunos casos, R1 es trifluoroetilurea.
En otro aspecto, la divulgación presenta un compuesto de Fórmula VII, o una sal farmacéuticamente aceptable del mismo:

en la que Warhead es un resto que es capaz de formar un enlace covalente con un nucleófilo; cada uno de R1 y R2 es, independientemente, halo, ciano, alcoxi C1-6 opcionalmente sustituido, hidroxi, oxo, amino, amido, alquilsulfonamido opcionalmente sustituido, alquil urea opcionalmente sustituida, alquilo C1-6 opcionalmente sustituido, heterociclilo C1-6 opcionalmente sustituido; m es 0-3; y n es 0-4.
En otro aspecto, la divulgación presenta un compuesto de Fórmula VIII, o una sal farmacéuticamente aceptable del mismo:

en la que Warhead es un resto que es capaz de formar un enlace covalente con un nucleófilo; el anillo A es un grupo arilo, heteroarilo, heterocíclico o alicíclico de 3-8 miembros; W es C o N, cada uno de X y Z es, independientemente, CH o N; Y es CH o N-R4 donde R4 es H o alquilo C1-6; L es -[C(R5)(R6)]q-, donde cada uno de R5 y R6 es, independientemente, H o alquilo C1-6; y q es 0-4; cada uno de R1 -R3 es, independientemente, halo, ciano, alcoxi C1-6 opcionalmente sustituido, hidroxi, oxo, amino, amido, alquilurea, alquilo C1-6 opcionalmente sustituido, heterociclilo C1-6 opcionalmente sustituido; m es 0-3; n es 0-4; y p es 0-2. En algunos casos, el anillo A es fenilo; R2 es halo o metoxi; n es 2 o 4; X es N; R1 es metilo; y/o m es 1.
En aspectos de la divulgación, el compuesto es un compuesto de Fórmula IX, o una sal farmacéuticamente aceptable del mismo:

en la que Warhead es un resto que es capaz de formar un enlace covalente con un nucleófilo; cada uno de R1 y R2 es, independientemente, halo, ciano, alcoxi C1-6 opcionalmente sustituido, hidroxi, oxo, amino, amido, alquil urea opcionalmente sustituida, alquilo C1-6 opcionalmente sustituido, heterociclilo opcionalmente sustituido; m es 0-3; y n es 0-4.
En otros aspectos, la divulgación presenta un compuesto de Fórmula X, o una sal farmacéuticamente aceptable del mismo:
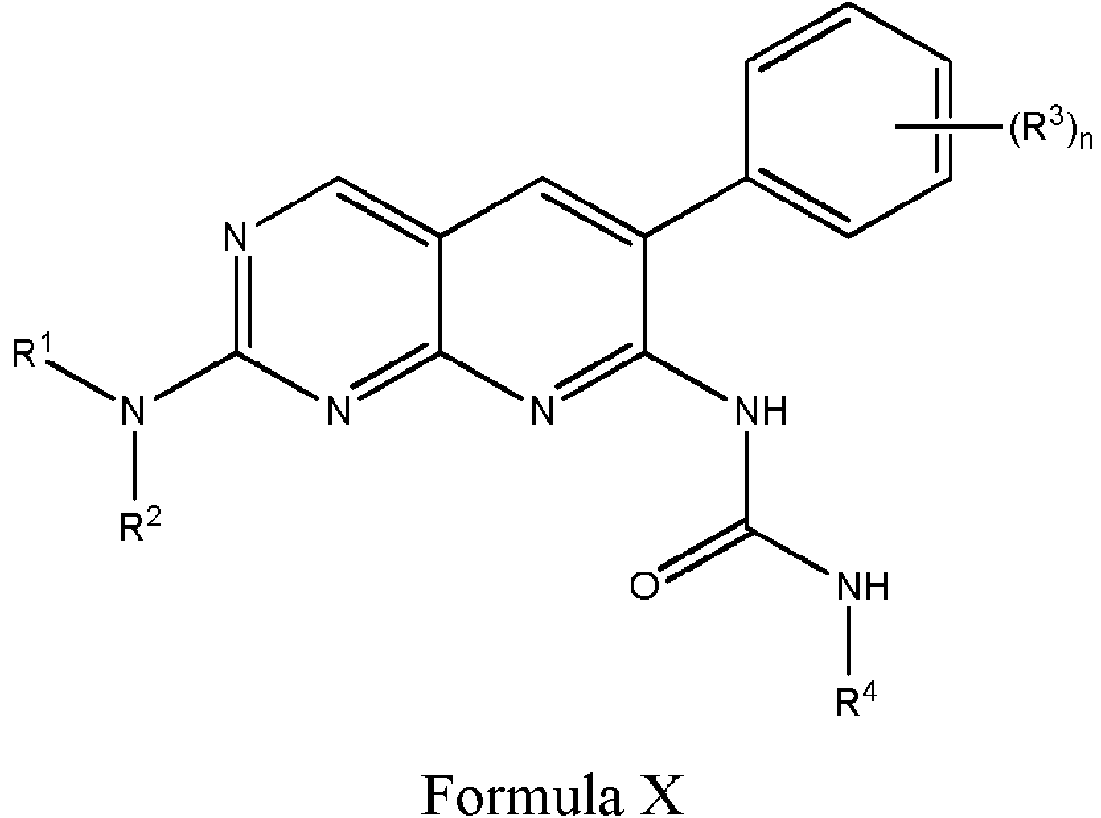
en la que R1 es un resto Warhead; R2 es alquilo C1-6, que está opcionalmente sustituido con halo, amino, hidroxi o ciano; cada R3 es, independientemente, halo, amino, ciano, alquilo C1-6 o alcoxi C1-6, y n es 2-5; y R4 es alquilo C1-6 opcionalmente sustituido.
En los compuestos descritos en el presente documento, una Warhead es un resto que es reactivo con un nucleófilo, por ejemplo, capaz de formar un enlace covalente con un nucleófilo. Los ejemplos de Warheads incluyen, sin limitación, haluros de alquilo, alquilsulfonatos, haluros de heteroarilo, epóxidos, haloacetamidas, maleimidas, ésteres de sulfonato, cetonas insaturadas alfa-beta, ésteres insaturados alfa-beta, vinil sulfonas, propargilamidas, acrilamidas. En algunos de estos casos, por ejemplo, acrilamida y propargilamida, el N de la Warhead es el N adyacente en las fórmulas mostradas anteriormente. A continuación, se muestran estructuras de Warheads de

donde X es un grupo saliente tal como halo, o un resto hidroxilo activado (por ejemplo, triflato); y cada uno de Ra, Rb y Rces, independientemente, H, alquilo C1-4 sustituido o no sustituido, cicloalquilo C3-4 sustituido o no sustituido, o ciano.
En las fórmulas mostradas anteriormente, las warheads están típicamente unidas a un átomo de N en el inhibidor. En otros casos, la Warhead se puede unir alternativamente a un átomo distinto de N. Ejemplos de


Otros ejemplos de Warheads se pueden encontrar, por ejemplo, en los documentos WO 2010/028236 y WO 2011/034907.
En ciertos casos, los inhibidores de FGFR-4 de la divulgación inhiben la actividad de FGFR-4 más potentemente que inhiben la actividad de FGFR-1. Por ejemplo, los inhibidores de FGFR-4 pueden inhibir la actividad de FGFR-4 al menos 10 veces, al menos 50 veces, al menos 100 veces, al menos 200 veces o al menos 500 veces de forma más potente que inhiben la actividad de FGFR-1.
En un aspecto, la selectividad se mide comparando la inhibición de FGFR-1 y FGFR-4 causada por el compuesto de esta divulgación en el mismo tipo de ensayo. En un caso, los ensayos usados para medir la inhibición de FGFR-1 y FGFR-4 son cualquiera de los ensayos descritos en este documento. Por lo general, la inhibición se expresa como IC50 (la concentración de inhibidor a la que se inhibe el 50% de la actividad de la enzima) y, por lo tanto, el grado de selectividad se mide mediante la ecuación: (IC50 FGFR-1)/(IC50 FGFR- 4). Las mismas mediciones y cálculos se pueden utilizar para medir la selectividad sobre FGFR-2 y FGFR-3 también.
Se puede utilizar cualquier otro ensayo de actividad de FGFR para determinar la inhibición relativa de FGFR-4 y FGFR-I por los compuestos de esta divulgación, siempre que tales ensayos utilizan lo que un experto en la técnica consideraría que son los mismos parámetros en la medición de la actividad de FGFR.
En otro aspecto, la invención presenta una composición farmacéutica que comprende un portador farmacéuticamente aceptable y un compuesto de la invención.
En otro aspecto, la divulgación presenta un inhibidor covalente de FGFR-4. En algunos casos, el inhibidor covalente de FGFR-4 inhibe la actividad de FGFR-4 de forma más potente, cuando se mide en un ensayo bioquímico, de lo que inhibe la actividad de FGFR-1. El inhibidor puede contener una Warhead.
En otro aspecto, la divulgación presenta un compuesto que inhibe la actividad de FGFR-4 más potentemente, cuando se mide en un ensayo bioquímico, de lo que inhibe la actividad de FGFR-1, en la que el compuesto tiene un peso molecular de menos de 1500 daltons. Por ejemplo, el compuesto puede inhibir la actividad de FGFR-4 al menos 10, 50, 100, 200 o 500 veces más potente, cuando se mide en un ensayo bioquímico, de lo que inhibe la actividad de FGFR-1. En algunos casos, este compuesto puede formar un enlace covalente con FGFR-4, por ejemplo, con Cys 522 de FGFR-4.
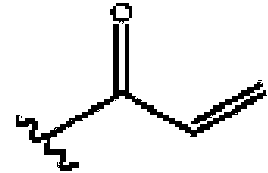
En otro aspecto, la divulgación presenta una proteína FGFR-4 inhibida que comprende un inhibidor que tiene un enlace covalente con un residuo de cisteína de FGFR-4. El enlace covalente se puede formar entre una porción de un resto de Warhead en el inhibidor y una porción de un residuo de cisteína de FGFR-4, por ejemplo, el residuo de cisteína 552 de la proteína. La Warhead puede ser
En otro aspecto, la divulgación presenta un procedimiento para tratar una afección mediada por FGFR4, una afección caracterizada por la sobreexpresión de FGFR4, una afección caracterizada por la amplificación de FGFR4, una afección mediada por FGF19, una afección caracterizada por FGF-19 amplificado, o una afección caracterizada por la sobreexpresión de FGF19, comprendiendo cualquiera de estos procedimientos la administración de una cantidad terapéuticamente eficaz de un compuesto descrito en el presente documento a un sujeto.
En otro aspecto, la divulgación presenta un procedimiento para tratar cualquiera de las siguientes afecciones mediante la administración de una cantidad terapéuticamente eficaz de un compuesto descrito en el presente documento a un sujeto: carcinoma hepatocelular, cáncer de mama, cáncer de ovario, cáncer de pulmón, cáncer de hígado, un sarcoma o hiperlipidemia.
La divulgación incluye todas las posibles combinaciones de las realizaciones y ejemplos descritos anteriormente. Breve descripción de los dibujos
La figura 1 es un espectro que muestra masas para la proteína FGFR4 sin y con inhibidor unido.
La figura 2 es un espectro que muestra masas para la proteína FGFR4 sin y con inhibidores unidos.
La figura 3 es un gráfico que muestra la actividad caspasa de un compuesto 25.
La figura 4 es un dibujo de la estructura cristalina del compuesto 52 unido a la proteína FGFR4.
La figura 5 es un dibujo de la estructura cristalina del Compuesto 25 unido a la proteína FGFR4.
La figura 6 es un gráfico de líneas que representa el efecto antitumoral del compuesto 25.
La figura 7 es un gráfico de barras que representa los pesos de los tumores de ratones desnudos que portan Hep3B. La figura 8 es un gráfico de líneas que representa el cambio de peso corporal (%) de ratones desnudos portadores de Hep3B.
Descripción detallada
Los inhibidores de FGFR, tales como BGJ398 AZD4547, son conocidos.

No se ha descrito que estos compuestos (es decir, los inhibidores de FGFR) sean más potentes contra FGFR4 que contra las otras isoformas de FGFR, es decir, FGFR1, FGFR2 y FGFR3. De hecho, AZD 4547 es menos potente contra FGFR4 que contra las otras tres isoformas.
A diferencia de BGJ398 y AZD4547, los compuestos descritos a continuación pueden formar un enlace covalente con la proteína FGFR4; por ejemplo, los compuestos pueden formar un enlace covalente con un residuo de cisteína de FGFR4, por ejemplo, la cisteína en el residuo 552. Los FGFR 1-3 no contienen esta cisteína. La capacidad de formar un enlace covalente entre el compuesto y FGFR4 es por tanto un factor importante en la selectividad de los compuestos descritos en este documento para FGFR4.
Los detalles de construcción y la disposición de componentes expuestos en la siguiente descripción o ilustrados en los dibujos no pretenden ser limitantes. Se incluyen expresamente otras realizaciones y diferentes formas de practicar la invención. Además, la fraseología y la terminología utilizadas en este documento tienen el propósito de describir y no deben considerarse limitantes. El uso de "que incluye", "incluye", "incluyen", "comprende" o "tiene", "contiene", "implica" y variaciones de los mismos en el presente documento, pretende abarcar los elementos enumerados a continuación y sus equivalentes también, así como elementos adicionales.
Definiciones
"Grupo alifático", como se usa en el presente documento, se refiere a un grupo de hidrocarburo cadena lineal, de cadena ramificada, o cíclico e incluye grupos saturados e insaturados, tales como un grupo alquilo, un grupo alquenilo, y un grupo alquinilo.
"Alquenilo", como se usa en el presente documento, se refiere a un grupo alifático que contiene al menos un doble enlace.
"Alcoxilo" o "alcoxi", como se usa en el presente documento, se refiere a un grupo alquilo que tiene un radical oxígeno unido al mismo. Los grupos alcoxilo representativos incluyen metoxi, etoxi, propiloxi, terc-butoxi y similares. "Alquilo", como se usa en el presente documento, se refiere al radical de grupos alifáticos saturados, incluyendo grupos alquilo de cadena lineal, grupos alquilo de cadena ramificada, grupos cicloalquilo (alicíclicos), grupos cicloalquilo sustituidos con alquilo, y grupos alquilo sustituidos con cicloalquilo. "Alquileno" se refiere a un radical doble, es decir, un grupo alifático sustituido en dos extremos. En algunos casos, un alquilo de cadena lineal o ramificada tiene 30 o menos átomos de carbono en su estructura (por ejemplo, C1-C30 para cadenas lineales, C3-C30 para cadenas ramificadas), y en otros casos puede tener 20 o menos, o 10 o menos. Asimismo, ciertos cicloalquilos pueden tener de 3 a 10 átomos de carbono en su estructura de anillo y, en algunos casos, pueden tener 5, 6 o 7 carbonos en la estructura de anillo. El término "alquenilo", como se usa en este documento, se refiere a un grupo alifático que contiene al menos un doble enlace; el término "alquinilo", como se usa en este documento, se refiere a un grupo alifático que contiene al menos un triple enlace.
"Alquiltio", como se usa en el presente documento, se refiere a un grupo hidrocarbilo que tiene un radical de azufre unido al mismo. En algunos casos, el resto "alquiltio" está representado por uno de -S-alquilo, -S-alquenilo o -S-alquinilo. Los grupos alquiltio representativos incluyen metiltio, etiltio y similares.
"Amido", como se usa en el presente documento, se refiere a -C(=O)-N(R1)(R2) o -N(R1)-C(=O)-R2 donde cada uno de R1 y R2 es H o alquilo.
"Amino", como se usa en el presente documento, se refiere a -NH2, -NH(alquilo), o -N(alquil)(alquilo).
"Amplificado", como se usa en el presente documento, significa copias adicionales de un segmento de gen o cromosoma que se producen en las células cancerosas que pueden conferir una ventaja de crecimiento o la supervivencia.
"Aralquilo", como se usa en el presente documento, se refiere a un grupo alquilo sustituido con un grupo arilo (por ejemplo, un grupo aromático o heteroaromático).
"Arilo", como se usa en este documento, se refiere a grupos aromáticos de anillo único de 5, 6 y 7 miembros que pueden incluir de cero a cuatro heteroátomos, por ejemplo, fenilo, pirrolilo, furanilo, tiofenilo, imidazolilo, oxazolilo, tiazolilo, triazolilo, pirazolilo, piridinilo, pirazinilo, piridazinilo y pirimidinilo, y similares. Los grupos arilo que tienen heteroátomos en la estructura del anillo también pueden denominarse "aril heterociclos" o "heteroaromáticos". El anillo aromático puede estar sustituido en una o más posiciones del anillo con los sustituyentes descritos anteriormente, por ejemplo, halógeno, azida, alquilo, aralquilo, alquenilo, alquinilo, cicloalquilo, policiclilo, hidroxilo, alcoxilo, amino, nitro, sulfhidrilo, imino, amido, fosfato, fosfonato, fosfinato, carbonilo, carboxilo, sililo, éter, alquiltio, sulfonilo, sulfonamido, cetona, aldehído, éster, restos heterociclilo, aromático o heteroaromático, -CF3, -CN o similares. El término "arilo" también incluye sistemas de anillos policíclicos que tienen dos o más anillos cíclicos en los que dos o más carbonos son comunes a dos anillos contiguos (los anillos son "anillos fusionados") en los que al menos uno de los anillos es aromático, por ejemplo, los otros anillos cíclicos pueden ser cicloalquilos, cicloalquenilos, cicloalquinilos, arilos y/o heterociclilos. Cada anillo puede contener, por ejemplo, 5-7 miembros. El término "carbociclo" o "cicloalquilo", como se usa en el presente documento, se refiere a un anillo aromático o no aromático en el que cada átomo del anillo es carbono.
"Inhibidor covalente," como se usa en el presente documento, significa un inhibidor que puede formar un enlace covalente con una proteína.
El "exceso enantiomérico" o "% de exceso enantiomérico" de una composición se pueden calcular usando la ecuación que se muestra abajo. En el ejemplo que se muestra a continuación, una composición contiene el 90% de un enantiómero, por ejemplo, el enantiómero S, y el 10% del otro enantiómero, es decir, el enantiómero R.
ce= (ÍXMO)/100 = 80%.
Por tanto, se dice que una composición que contiene el 90% de un enantiómero y el 10% del otro enantiómero tiene un exceso enantiomérico del 80%. Algunas de las composiciones descritas en este documento contienen un exceso enantiomérico de al menos 50%, al menos 75%, al menos 80%, al menos 85%, al menos 90%, al menos 95% o al menos 99% de Compuesto 1 (el enantiómero S). En otras palabras, las composiciones contienen un exceso enantiomérico del enantiómero S sobre el enantiómero R.
"FGFR-4" o "proteína FGFR-4" se refiere a cualquier forma de la proteína FGFR-4, incluyendo el tipo salvaje y todas las formas variantes (incluyendo, sin limitación, formas mutantes y variantes de empalme). La proteína FGFR-4 es
un producto del gen FGFR-4 y, por lo tanto, la proteína FGFR-4 incluye cualquier proteína codificada por cualquier forma del gen FGFR-4, incluidas todas las aberraciones, por ejemplo, mutaciones puntuales, indeles, fusiones por translocación, y amplificaciones focales.
"Heteroarilalquilo" se refiere a un grupo alquilo sustituido con un grupo heteroarilo.
"Heterociclilo" o "grupo heterocíclico" se refiere a una estructura de anillo, tal como una estructura de anillo de 3 a 7 miembros, cuyo anillo o anillos incluyen uno o más heteroátomos. Los heterociclos también pueden ser policiclos, teniendo cada grupo, por ejemplo, 3-7 miembros de anillo. El término "heterociclilo" o "grupo heterocíclico" incluye estructuras "heteroarilo" y "heterociclilo saturado o parcialmente saturado". "Heteroarilo" se refiere a un sistema de anillo aromático monocíclico de 5-8 miembros, bicíclico de 8-12 miembros o tricíclico de 11-14 miembros que tiene uno o más heteroátomos, seleccionados entre O, N o S. Cualquier átomo del anillo puede estar sustituido (por ejemplo, por uno o más sustituyentes). El término "heterociclilo saturado o parcialmente saturado" se refiere a una estructura cíclica no aromática que incluye al menos un heteroátomo. Los grupos heterociclilo incluyen, por ejemplo, tiofenilo, tiantrenilo, furanilo, piranilo, isobenzofuranilo, cromenilo, xantenilo, fenoxatiína, pirrolilo, imidazolilo, pirazolilo, isotiazolilo, isoxazolilo, piridinilo, pirazinilo, pirimidinilo, piridazinilo, indolizinilo, isoindolilo, indolilo, indazolilo, purinilo, quinolizinilo, isoquinolinilo, quinolinilo, ftalazinilo, naftiridinilo, quinoxalinilo, quinazolinilo, cinnolinilo, pteridinilo, carbazolilo, carbolina, fenantridina, acridina, pirimidina, fenantrolina, fenazina, fenarsazina, fenotiazina, furazan, fenoxazina, pirrolidina, oxolano, tiolano, oxazol, piperidina, piperazina, morfolina, lactonas, lactamas tales como azetidinonas y pirrolidinonas, sultamas, sultonas y similares. El anillo heterocíclico puede estar sustituido en una o más posiciones con los sustituyentes descritos anteriormente, como por ejemplo, halógeno, alquilo, aralquilo, alquenilo, alquinilo, cicloalquilo, hidroxilo, amino, nitro, sulfhidrilo, imino, amido, fosfato, fosfonato, fosfinato, carbonilo, carboxilo, sililo, éter, alquiltio, sulfonilo, cetona, aldehído, éster, un resto heterociclilo, aromático o heteroaromático, -CF3, -CN o similares.
"Heterociclilalquilo" se refiere a un grupo alquilo sustituido con un grupo heterociclo.
"Inhibidor" se refiere a un compuesto que inhibe una enzima de tal manera que se puede observar una reducción en la actividad de la enzima, por ejemplo, en un ensayo bioquímico. En ciertos casos, un inhibidor tiene una IC50 de menos de aproximadamente 1 |iM, menos de aproximadamente 500 nM, menos de aproximadamente 250 nM, menos de aproximadamente 100 nM, menos de aproximadamente 50 nM, o menos de aproximadamente 10 nM. Un inhibidor de FGFR-4 se refiere a un compuesto que inhibe FGFR-4.
"Sobreexpresado", como se usa en el presente documento, significa que hay producción de un producto génico en una muestra que es sustancialmente mayor que la observada en una población de muestras de control (por ejemplo, tejido normal).
"Selectivo" se refiere a un compuesto que inhibe la actividad de una proteína diana, por ejemplo, FGFR-4, más potentemente que inhibe la actividad de otras proteínas. En este caso, las isoformas FGFR-1, FGFR-2, FGFR-3 y FGFR-4 se consideran todas proteínas distintas. En algunos casos, un compuesto puede inhibir la actividad de la proteína diana, por ejemplo, FGFR-4, de manera al menos 1,5, al menos 2, al menos 5, al menos 10, al menos 20, al menos 30, al menos 40, al menos al menos 50, al menos 60, al menos 70, al menos 80, al menos 90, al menos 100, al menos 200, al menos 500, o al menos 1000 o más potente de lo que inhibe la actividad de una proteína no diana.
"Sustituido" se refiere a restos que tienen sustituyentes que reemplazan un hidrógeno en uno o más carbonos de la cadena principal. Se entenderá que "sustitución" o "sustituido con" incluye la condición implícita de que dicha sustitución está de acuerdo con la valencia permitida del átomo sustituido y el sustituyente, y que la sustitución da como resultado un compuesto estable, por ejemplo, que no experimenta espontáneamente una transformación, tal como por transposición, ciclación, eliminación, etc. Como se usa en este documento, se contempla que el término "sustituido" incluya todos los sustituyentes permisibles de compuestos orgánicos. En un aspecto amplio, los sustituyentes permisibles incluyen sustituyentes acíclicos y cíclicos, ramificados y no ramificados, carbocíclicos y heterocíclicos, aromáticos y no aromáticos de compuestos orgánicos. Los sustituyentes permisibles pueden ser uno o más e iguales o diferentes para los compuestos orgánicos apropiados. Para los propósitos de esta invención, los heteroátomos, tales como nitrógeno, pueden tener sustituyentes de hidrógeno y/o cualquier sustituyente permisible de los compuestos orgánicos descritos en este documento que satisfagan las valencias de los heteroátomos. Los sustituyentes pueden incluir cualquier sustituyente descrito en el presente documento, por ejemplo, un halógeno, un hidroxilo, un carbonilo (tal como un carboxilo, un alcoxicarbonilo, un formilo o un acilo), un tiocarbonilo (tal como un tioéster, un tioacetato o un tioformiato), un alcoxilo, un fosforilo, un fosfato, un fosfonato, un fosfinato, un amino, un amido, una amidina, una imina, un ciano, un nitro, un azido, un sulfhidrilo, un alquiltio, un sulfato, un sulfonato, un sulfamoílo, un sulfonamido, un sulfonilo, un heterociclilo, un aralquilo, o un resto aromático o heteroaromático. Los expertos en la técnica entenderán que los restos sustituidos en la cadena de hidrocarburo pueden estar sustituidos ellos mismos, si es apropiado. Por ejemplo, los sustituyentes de un alquilo sustituido pueden incluir formas sustituidas y no sustituidas de grupos amino, azido, imino, amido, fosforilo (que incluyen fosfonato y fosfinato), sulfonilo (que incluye sulfato, sulfonamido, sulfamoilo y sulfonato) y sililo, así como éteres, alquiltios, carbonilos
(incluidas cetonas, aldehidos, carboxilatos y ésteres), -CF3, -CN y similares. A continuación se describen ejemplos de alquilos sustituidos. Los cicloalquilos pueden estar además sustituidos con alquilos, alquenilos, alcoxis, alquiltios, aminoalquilos, alquilos sustituidos con carbonilo, -CF3, -CN, y similares. Pueden hacerse sustituciones análogas a los grupos alquenilo y alquinilo para producir, por ejemplo, aminoalquenilos, aminoalquinilos, amidoalquenilos, amidoalquinilos, iminoalquenilos, iminoalquinilos, tioalquenilos, tioalquinilos, alquenilos o alquinilos sustituidos con carbonilo.
Como se usa en el presente documento, la definición de cada expresión, por ejemplo, alquilo, m, n, etc., cuando aparece más de una vez en cualquier estructura, pretende ser independiente de su definición en la misma estructura en otro lugar.
"Resto Warhead" o "Warhead" se refiere a un resto de un inhibidor que participa, ya sea de forma reversible o irreversible, con la reacción de un donante, por ejemplo, una proteína, con un sustrato. Las Warheads pueden, por ejemplo, formar enlaces covalentes con la proteína, o pueden crear estados de transición estables, o ser un agente alquilante reversible o irreversible. Por ejemplo, el resto de Warhead puede ser un grupo funcional en un inhibidor que puede participar en una reacción de formación de enlaces, en la que se forma un nuevo enlace covalente entre una parte de Warhead y un donante, por ejemplo, un residuo de aminoácido de una proteína. En casos, la Warhead es un electrófilo y el "donante" es un nucleófilo, tal como la cadena lateral de un residuo de cisteína. Ejemplos de Warheads adecuadas incluyen, sin limitación, los grupos mostrados a continuación

en donde X es un grupo saliente, tal como halo, o un resto hidroxilo activado (por ejemplo, triflato); y cada uno de Ra, Rb y Rc es, independientemente, H, alquilo C1-4 sustituido o no sustituido, cicloalquilo C3-4 sustituido o no sustituido, o ciano.
Los compuestos descritos en este documento pueden contener proporciones no naturales de isótopos atómicos en uno o más de los átomos que constituyen tales compuestos. Por ejemplo, los compuestos pueden marcarse radiactivamente con isótopos radiactivos, tales como por ejemplo tritio (3H) o carbono-14 (14C). Se pretende que todas las variaciones isotópicas de los compuestos descritos en este documento, ya sean radiactivos o no, estén incluidas dentro del alcance de la presente invención. Por ejemplo, se pretende que los compuestos deuterados o los compuestos que contienen 13C estén incluidos dentro del alcance de la invención.
Ciertos compuestos pueden existir en diferentes formas tautoméricas, y todas las posibles formas tautoméricas de todos los compuestos descritos en este documento pretenden estar comprendidos dentro del alcance de la invención.
A menos que se indique otra cosa, las estructuras representadas en el presente documento también pretenden incluir todas las formas isoméricas (por ejemplo, enantioméricas, diastereoméricas, y geométricas (o conformacionales)) de la estructura; por ejemplo, las configuraciones R y S para cada centro asimétrico, isómeros de doble enlace Z y E e isómeros conformacionales Z y E. Por tanto, los isómeros estereoquímicos individuales, así como las mezclas enantioméricas, diastereoméricas y geométricas (o conformacionales) de los presentes compuestos están dentro del alcance de la invención. A menos que se indique lo contrario, todas las formas tautoméricas de los compuestos de la invención están dentro del alcance de la invención.
Los compuestos descritos en este documento pueden ser útiles como la base libre o como una sal. Las sales representativas incluyen las sales de bromhidrato, clorhidrato, sulfato, bisulfato, fosfato, nitrato, acetato, valerato, oleato, palmitato, estearato, laurato, benzoato, lactato, fosfato, tosilato, citrato, maleato, fumarato, succinato, tartrato, naftalato, mesilato, glucoheptonato, lactobionato y laurilsulfonato y similares. (Véase, por ejemplo, Berge et al. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66: 1-19.)
Ciertos compuestos descritos en este documento pueden existir en formas no solvatadas, así como formas solvatadas, incluyendo formas hidratadas. En general, las formas solvatadas son equivalentes a las formas no solvatadas y están incluidas dentro del alcance de la presente invención. Ciertos compuestos descritos en el
presente documento pueden existir en múltiples formas cristalinas o amorfas. En general, todas las formas físicas son equivalentes para los usos contemplados por la presente invención y se pretende que estén dentro del alcance de la presente invención.
Los com uestos de eem lo inclu en los si uientes:






Composiciones farmacéuticas
Aunque es posible que un compuesto dado a conocer en el presente documento se administre solo, es preferible administrar el compuesto como una formulación farmacéutica, en donde el compuesto se combina con uno o más excipientes o portadores farmacéuticamente aceptables. Los compuestos descritos en el presente documento pueden formularse para su administración de cualquier forma conveniente para su uso en medicina humana o veterinaria. En ciertos casos, el compuesto incluido en la preparación farmacéutica puede ser activo en sí mismo o puede ser un profármaco, por ejemplo, capaz de convertirse en un compuesto activo en un entorno fisiológico. En ciertos casos, los compuestos proporcionados en este documento incluyen sus hidratos.
La frase "farmacéuticamente aceptable" se emplea en este documento para referirse a aquellos compuestos, materiales, composiciones y/o formas de dosificación que son, dentro del alcance del juicio médico, adecuados para
uso en contacto con los tejidos de seres humanos y animales sin toxicidad excesiva, irritación, respuesta alérgica u otro problema o complicación, acorde con una relación beneficio/riesgo razonable.
Los ejemplos de sales farmacéuticamente aceptables de un compuesto descrito en el presente documento incluyen las derivadas de ácidos y bases inorgánicos y orgánicos farmacéuticamente aceptables. Ejemplos de sales ácidas adecuadas incluyen acetato, adipato, benzoato, bencenosulfonato, butirato, citrato, digluconato, dodecilsulfato, formiato, fumarato, glicolato, hemisulfato, heptanoato, hexanoato, clorhidrato, bromhidrato, yodhidrato, lactato, maleato, malonato, metanosulfonato, 2-naftalenosulfonato, nicotinato, nitrato, palmoato, fosfato, picrato, pivalato, propionato, salicilato, succinato, sulfato, tartrato, tosilato y undecanoato. Las sales derivadas de bases apropiadas incluyen sales de metales alcalinos (por ejemplo, sodio), metales alcalinotérreos (por ejemplo, magnesio), amonio y N-(alquilo)4+. Esta divulgación también prevé la cuaternización de cualquier grupo que contenga nitrógeno básico de los compuestos descritos en el presente documento. Pueden obtenerse productos solubles o dispersables en agua o aceite mediante dicha cuaternización.
Los ejemplos de portadores farmacéuticamente aceptables incluyen: (1) azúcares, tales como lactosa, glucosa y sacarosa; (2) almidones, tales como almidón de maíz y almidón de patata; (3) celulosa y sus derivados, tales como carboximetilcelulosa de sodio, etilcelulosa y acetato de celulosa; (4) tragacanto en polvo; (5) malta; (6) gelatina; (7) talco; (8) excipientes, tales como manteca de cacao y ceras para supositorios; (9) aceites, tales como aceite de cacahuete, aceite de semilla de algodón, aceite de cártamo, aceite de sésamo, aceite de oliva, aceite de maíz y aceite de soja; (10) glicoles, tales como propilenglicol; (11) polioles, tales como glicerina, sorbitol, manitol y polietilenglicol; (12) ésteres, tales como oleato de etilo y laurato de etilo; (13) agar; (14) agentes tamponadores, tales como hidróxido de magnesio e hidróxido de aluminio; (15) ácido algínico; (16) agua libre de pirógenos; (17) solución salina isotónica; (18) solución de Ringer; (19) alcohol etílico; (20) soluciones tampón de fosfato; (21) ciclodextrinas, tales como Captisol®; ligandos de reconocimiento unidos a nanopartículas, tales como Accurins™; y (22) otras sustancias compatibles no tóxicas, tales como composiciones a base de polímeros, empleadas en formulaciones farmacéuticas.
Los ejemplos de antioxidantes farmacéuticamente aceptables incluyen: (1) antioxidantes solubles en agua, tales como ácido ascórbico, clorhidrato de cisteína, bisulfato de sodio, metabisulfito de sodio, sulfito de sodio y similares; (2) antioxidantes solubles en aceite, tales como palmitato de ascorbilo, hidroxianisol butilado (BHA), hidroxitolueno butilado (BHT), lecitina, galato de propilo, alfa-tocoferol y similares; y (3) agentes quelantes de metales, tales como ácido cítrico, ácido etilendiaminotetraacético (EDTA), sorbitol, ácido tartárico, ácido fosfórico y similares. Las formas de dosificación sólidas (por ejemplo, cápsulas, comprimidos, píldoras, grageas, polvos, gránulos y similares) pueden incluir uno o más portadores farmacéuticamente aceptables, tales como citrato de sodio o fosfato dicálcico, y/o cualquiera de los siguientes: (1) cargas o extensores, tales como almidones, lactosa, sacarosa, glucosa, manitol, y/o ácido silícico; (2) aglutinantes, tales como, por ejemplo, carboximetilcelulosa, alginatos, gelatina, polivinilpirrolidona, sacarosa y/o goma arábiga; (3) humectantes, tales como glicerol; (4) agentes disgregantes, tales como agar-agar, carbonato cálcico, almidón de patata o tapioca, ácido algínico, ciertos silicatos y carbonato sódico; (5) agentes retardadores de la solución, tales como parafina; (6) aceleradores de la absorción, tales como compuestos de amonio cuaternario; (7) agentes humectantes, tales como, por ejemplo, alcohol cetílico y monoestearato de glicerol; (8) absorbentes, tales como caolín y arcilla bentonita; (9) lubricantes, tales como talco, estearato de calcio, estearato de magnesio, polietilenglicoles sólidos, lauril sulfato de sodio y mezclas de los mismos; y (10) agentes colorantes. Las formas de dosificación líquidas pueden incluir emulsiones, microemulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables. Además del principio activo, las formas de dosificación líquidas pueden contener diluyentes inertes comúnmente usados en la técnica, tales como, por ejemplo, agua u otros disolventes, agentes solubilizantes y emulsionantes, tales como alcohol etílico, alcohol isopropílico, carbonato de etilo, acetato de etilo, alcohol bencílico, benzoato de bencilo, propilenglicol, 1,3-butilenglicol, aceites (en particular, aceites de semilla de algodón, cacahuete, maíz, germen, oliva, ricino y sésamo), glicerol, alcohol tetrahidrofurílico, polietilenglicoles y ésteres de ácidos grasos de sorbitán y mezclas de los mismos.
Las suspensiones, además de los compuestos activos, pueden contener agentes de suspensión como, por ejemplo, alcoholes isoestearílicos etoxilados, ésteres de polioxietilen sorbitol y sorbitán, celulosa microcristalina, metahidróxido de aluminio, bentonita, agar-agar y tragacanto, y mezclas de los mismos.
Las pomadas, pastas, cremas y geles pueden contener, además de un compuesto activo, excipientes, tales como grasas animales y vegetales, aceites, ceras, parafinas, almidón, tragacanto, derivados de celulosa, polietilenglicoles, siliconas, bentonitas, ácido silícico, talco y óxido de zinc o mezclas de los mismos.
Los polvos y aerosoles pueden contener, además de un compuesto activo, excipientes, tales como lactosa, talco, ácido silícico, hidróxido de aluminio, silicatos cálcicos y poliamida en polvo, o mezclas de estas sustancias. Los aerosoles pueden contener además propulsores habituales, tales como clorofluorohidrocarburos e hidrocarburos volátiles no sustituidos, tales como butano y propano.
Las formulaciones pueden presentarse convenientemente en forma de dosificación unitaria y pueden prepararse mediante cualquier procedimiento bien conocido en la técnica de la farmacia. La cantidad de principio activo que se
puede combinar con un material portador para producir una única forma de dosificación variará dependiendo del huésped que se esté tratando, el modo particular de administración. La cantidad de principio activo que se puede combinar con un material portador para producir una única forma de dosificación será generalmente la cantidad del compuesto que produce un efecto terapéutico.
Las formas de dosificación para la administración tópica o transdérmica de un compuesto de esta invención incluyen polvos, aerosoles, pomadas, pastas, cremas, lociones, geles, soluciones, parches e inhalantes. El compuesto activo se puede mezclar en condiciones estériles con un portador farmacéuticamente aceptable y con cualquier conservante, tampón o propulsor que pueda ser necesario.
Cuando los compuestos descritos en este documento se administran como productos farmacéuticos, a humanos y animales, se les puede administrar per se o como una composición farmacéutica que contiene, por ejemplo, 0,1 a 99,5% (más preferiblemente, de 0,5 a 90%) de principio activo en combinación con un vehículo farmacéuticamente aceptable.
Las formulaciones pueden administrarse por vía tópica, por vía oral, transdérmica, rectal, vaginal, parenteral, intranasal, intrapulmonar, por vía intraocular, intravenosa, intramuscular, intraarterial, intratecal, intracapsular, intradérmica, intraperitoneal, subcutánea, subcuticularmente, o por inhalación.
Indicaciones
El FGFR-4 regula la proliferación, supervivencia y secreción de alfa-fetoproteína durante la progresión del carcinoma hepatocelular (HCC); los inhibidores de FGFR-4 son, por tanto, agentes terapéuticos potenciales prometedores para esta necesidad médica no satisfecha (Ho et al., Journal of Hepatology, 2009, 50: 118-27). El HCC afecta a más de 550.000 personas en todo el mundo cada año y tiene una de las peores tasas de supervivencia a 1 año de cualquier tipo de cáncer.
Más evidencia de la unión entre FGFR-4 y HCC se muestra a través de la implicación de FGF19, un miembro de la familia de factores de crecimiento de fibroblastos (FGF), que consiste en hormonas que regulan la homeostasis de la glucosa, los lípidos y la energía. Se ha observado una mayor proliferación de hepatocitos y formación de tumores hepáticos en ratones transgénicos de FGF19. FGF19 activa FGFR-4, su receptor predominante en el hígado, y se cree que la activación de FGFR-4 es el mecanismo por el cual FGF19 puede aumentar la proliferación de hepatocitos e inducir la formación de carcinoma hepatocelular (Wu et al., J Biol Chem (2010) 285 (8): 5165 5170). FGF19 también ha sido identificado como un gen conductor en el HCC por otros (Sawey et al., Cancer Cell (2011) 19: 347-358). Por lo tanto, se cree que los compuestos descritos en este documento, que son inhibidores potentes y selectivos de FGFR-4, se pueden usar para tratar el HCC y otros cánceres hepáticos.
El cribado de oncogenomas ha identificado una mutación activante del receptor 4 del factor de crecimiento de fibroblastos 4 (FGFR-4) Y367C en la línea celular de cáncer de mama humano MDA-MB-453. Se demostró que esta mutación provoca la fosforilación constitutiva, lo que lleva a una activación de la cascada de proteína quinasa activada por mitógenos. Por consiguiente, se ha sugerido que el FGFR-4 puede ser un impulsor del crecimiento tumoral en el cáncer de mama (Roidl et al., Oncogene (2010) 29 (10): 1543-1552). Por lo tanto, se cree que los compuestos descritos en este documento, que son inhibidores potentes y selectivos de FGFR-4, pueden usarse para tratar el cáncer de mama modulado por FGFR-4.
Los cambios moleculares (por ejemplo, translocaciones) en genes en dirección 5' de FGFR-4 pueden conducir a la activación/sobreexpresión de FGFR-4. Por ejemplo, una translocación/fusión de genes de PAX3-FKHR puede conducir a la sobreexpresión de FGFR-4. La sobreexpresión de FGFR-4 debido a este mecanismo se ha asociado con rabdomiosarcoma (RMS) (Cao et al., Cancer Res (2010) 70 (16): 6497-6508). Las mutaciones en el propio FGFR-4 (por ejemplo, mutaciones en el dominio quinasa) pueden conducir a una sobreactivación de la proteína; este mecanismo se ha asociado con una subpoblación de RMS (Taylor et al., J Clin Invest (2009) 119: 3395-3407). Por lo tanto, se cree que los compuestos descritos en este documento, que son inhibidores potentes y selectivos de FGFR-4, pueden usarse para tratar RMS modulado por FGFR-4 y otros sarcomas.
Otras enfermedades se han asociado con cambios en los genes en dirección 5' de FGFR-4 o con mutaciones en FGFR-4 en sí. Por ejemplo, las mutaciones en el dominio quinasa de FGFR-4 conducen a una sobreactivación, que se ha asociado con el adenocarcinoma de pulmón (Ding et al., Nature (2008) 455 (7216): 1069-1075). La amplificación de FGFR-4 se ha asociado con afecciones, tales como el carcinoma de células renales (datos provisionales de TCGA). Además, silenciar FGFR4 e inhibir la unión ligando-receptor disminuye significativamente el crecimiento del tumor ovárico, lo que sugiere que los inhibidores de FGFR4 podrían ser útiles en el tratamiento del cáncer de ovario. (Zaid et al., Clin. Cancer Res. (2013) 809).
Las elevaciones patógenas de los niveles de ácido biliares se han relacionado con variaciones en niveles de FGF19 (Vergnes et al., Cell Metabolism (2013) 17, 916-28). Por tanto, la reducción del nivel de FGF19 puede ser beneficiosa para promover la síntesis de ácidos biliares y, por tanto, en el tratamiento de la hiperlipidemia.
Niveles de dosificación
Los niveles de dosificación reales de los principios activos en las composiciones farmacéuticas de esta invención pueden variarse a fin de obtener una cantidad del principio activo que sea eficaz para lograr la respuesta terapéutica deseada para un paciente en particular, composición, y modo de administración, sin resultar tóxico para el paciente. El nivel de dosificación seleccionado dependerá de una variedad de factores, que incluyen la actividad del compuesto particular descrito en este documento empleado, o el éster, sal o amida del mismo, la vía de administración, el tiempo de administración, la tasa de excreción del compuesto particular que se está empleando, la duración del tratamiento, otros fármacos, compuestos y/o materiales usados en combinación con el compuesto particular empleado, la edad, sexo, peso, condición, salud general e historial médico previo del paciente que está siendo tratado, y factores similares bien conocidos en las artes médicas.
Un médico o veterinario que tiene experiencia ordinaria en la técnica puede determinar fácilmente y prescribir la cantidad eficaz de la composición farmacéutica requerida. Por ejemplo, el médico o veterinario podría empezar con dosis de los compuestos de la invención empleados en la composición farmacéutica a niveles inferiores a los requeridos para lograr el efecto terapéutico deseado y aumentar gradualmente la dosis hasta lograr el efecto deseado.
En general, una dosis diaria adecuada de un compuesto de la invención será la cantidad del compuesto que es la dosis más baja eficaz para producir un efecto terapéutico. Dicha dosis eficaz dependerá generalmente de los factores descritos anteriormente. Generalmente, las dosis de los compuestos de esta invención para un paciente oscilarán entre aproximadamente 0,0001 y aproximadamente 100 mg por kilogramo de peso corporal por día. Por ejemplo, la dosis podría estar entre 0,1 y 10 g por día; entre 0,5 y 5 g por día; o 1-2 g por día. Si se desea, la dosis diaria eficaz del compuesto activo puede administrarse como una, dos, tres, cuatro, cinco, seis o más subdosis administradas por separado a intervalos apropiados a lo largo del día, opcionalmente, en formas de dosificación unitaria.
Terapia combinada y dirigida
La administración de los inhibidores de FGFR-4 descritos en este documento se puede combinar con otros tratamientos contra el cáncer. Por ejemplo, los inhibidores se pueden administrar en combinación con tratamientos quirúrgicos, radiación u otros agentes terapéuticos, tales como anticuerpos, otros inhibidores de quinasas selectivos o quimioterapéuticos. Los inhibidores también se pueden administrar en combinación con terapia de ARNi o terapia antisentido. Los inhibidores de FGFR-4 descritos en este documento pueden combinarse con uno, dos o más de otros agentes terapéuticos. En los ejemplos que se describen a continuación, se entiende que "segundo agente terapéutico" también incluye más de un agente terapéutico distinto del inhibidor de FGFR-4. Un inhibidor de FGFR-4 descrito en el presente documento puede administrarse con uno, dos o más de otros agentes terapéuticos.
Los inhibidores de FGFR-4 se describe en el presente documento y el segundo agente terapéutico no tienen que ser administrados en la misma composición farmacéutica, y, debido a las diferentes características físicas y químicas, pueden administrarse por vías diferentes. Por ejemplo, el inhibidor de FGFR-4 se puede administrar por vía oral, mientras que el segundo agente terapéutico se administra por vía intravenosa. La determinación del modo de administración y la conveniencia de la administración, cuando sea posible, en la misma composición farmacéutica, están dentro del conocimiento del médico experto. La administración inicial puede realizarse de acuerdo con protocolos establecidos conocidos en la técnica, y, a continuación, basándose en los efectos observados, el médico experto puede modificar la dosis, los modos de administración y los tiempos de administración.
El inhibidor de FGFR-4 y el segundo agente terapéutico se pueden administrar de manera concurrente (por ejemplo, simultáneamente, esencialmente simultáneamente o dentro del mismo protocolo de tratamiento) o secuencialmente (es decir, uno seguido del otro, con un intervalo de tiempo opcional en medio), dependiendo de la naturaleza de la enfermedad proliferativa, el estado del paciente y la elección real del segundo agente terapéutico a administrar. Además, los inhibidores de FGFR-4 descritos en este documento pueden administrarse como parte de un conjugado de anticuerpo-fármaco, en donde el inhibidor de FGFR-4 es la porción de "carga útil" del conjugado.
Instrumentos y procedimientos analíticos para la caracterización del compuesto:
LCMS: A menos que se indique lo contrario, todos los datos de cromatografía líquida-espectrometría de masas (LCMS) (muestra analizada para determinar su pureza e identidad) se obtuvieron con un sistema LC Agilent modelo-1260 utilizando un espectrómetro de masas Agilent modelo 6120 utilizando ionización ES-API equipado con una columna de fase inversa Agilent Poroshel 120 (EC-C18, tamaño de partícula de 2,7 um, dimensiones de 3,0 x 50 mm) a 22,4 grados Celsius. La fase móvil consistió en una mezcla de disolvente ácido fórmico al 0,1% en agua y ácido fórmico al 0,1% en acetonitrilo. Se utilizó un gradiente constante de fase móvil acuosa al 95%/orgánica al 5% a acuosa al 5%/orgánica al 95% durante el transcurso de 4 minutos. El caudal fue constante a 1 ml/min.
RMN de protones: a menos que se indique lo contrario, todos los los espectros de RMN 1H se obtuvieron con un instrumento de RMN Varían 400MHz Unity Inova 400 MHz (tiempo de adquisición = 3,5 segundos con un retraso de 1 segundo; 16 a 64 barridos). Cuando se caracterizaron, todos los protones se reportaron en el disolvente DMSO-de como partes por millón (ppm) con respecto al DMSO residual (2,50 ppm). Instrumentos de preparación para la purificación de compuestos: La cromatografía en gel de sílice se realizó en una unidad Rf Teledyne Isco CombiFlash® o una unidad Biotage® Isolera Four.
LCMS preparativa: La HPLC preparativa se realizó en un sistema preparativo Shimadzu Discovery VP® equipado con una columna de fase inversa Luna 5u C18 (2) 100A, empaquetada con AXIA, de 250 x 21,2 mm a 22,4 grados Celsius. La fase móvil consistió en una mezcla de disolvente ácido fórmico al 0,1% en agua y ácido fórmico al 0,1% en acetonitrilo. Se utilizó un gradiente constante de fase móvil acuosa al 95%/orgánica al 5% a acuosa al 5%/orgánica al 95% durante el transcurso de 25 minutos. El caudal fue constante a 20 ml/min. Las reacciones llevadas a cabo en un microondas se realizaron en una unidad de microondas Biotage Initiator.
Ejemplo 1: Síntesis de N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)-8-metil-7-oxo-7,8-dihidropirido [2,3-d] pirimidin- 2 ¡lamno~3~met lfenlacrlamida COMPUESTO 43


Una mezcla de de 4-doro-2-(metiltio)pirimidina-5-carboxilato de etilo (5,0 g, 21,5 mmol) y metilamina al 29% (5,75 g, 53,72 mmol, solución en metanol (MeOH) en tetrahidrofurano (THF) (100 ml) se agitó a temperatura ambiente durante 2 horas. A continuación, se concentró la mezcla de reacción, seguido de la adición de bicarbonato de sodio (NaHCOa) (ac., 20 ml), y la solución resultante se extrajo con acetato de etilo (EtOAc) (3 x 50 ml). Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron para producir 4-(metilamino)-2-(metiltio)pirimidina-5-carboxilato de etilo (4,68 g, 96%) como un sólido amarillento. MS (ES+) C9H13N3O2S requiere: 227, encontrado: 228 [M H]+.
Paso 2: Síntesis de (4-(metilamino)-2-(metiltio)pirimidin-5-il) metanol

A una suspensión de hidruro de litio y aluminio (LiAlH4) (1,140 g, 30 mmol) en THF (100 ml) se añadió 4-(metilamino)-2-(metiltio)pirimidina-5-carboxilato de metilo (4,536 g, 20 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 2 horas. La solución se inactivó cuidadosamente con H2O (2 ml), hidróxido de sodio (NaOH) (ac., 15%, 2 ml) y H2O adicional (7 ml), y después se agitó durante 1 hora. La mezcla se extrajo con EtOAc (2 x 100 ml) y las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre sulfato de sodio y se concentraron para dar (4-(metilamino)-2-(metiltio)pirimidin-5-il)metanol (3,2 g, 85%) como un sólido amarillento. MS (ES+) C7H11N3 requiere: 185, encontrado: 186 [M H]+.
Paso 3: Síntesis de 4-(metilamino)-2-(metiltio)pirimidina-5-carbaldehído

Se agitó una suspensión de (4-(metilamino)-2-(metiltio)pirimidin-5-il)metanol (3,1 g, 16,73 mmol) y dióxido de manganeso (7,27 g, 83,67 mmol) en DCM (40 ml) a temperatura ambiente durante 12 horas. El precipitado resultante se retiró por filtración y el filtrado se concentró para dar 4-(metilamino)-2-(metiltio)pirimidin-5-carbaldehído (2,8 g, 91%) como un sólido amarillento. MS (ES+) C7H9N3OS requiere: 183, encontrado: 184 [M H]+.

A una solución de ácido 2-(3,5-dimetoxifenil)acético (5) (600 mg, 3,06 mmol) en MeOH (30 ml) se añadió gota a gota cloruro de tionilo (3 ml) a 0 °C, y la mezcla de reacción se agitó a temperatura ambiente durante la noche. La reacción se controló mediante cromatografía líquida-espectrometría de masas (LCMS). La mezcla se diluyó con bicarbonato de sodio saturado (acuoso, 20 ml) y se extrajo con EtOAc (3 x 20 ml). Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron para dar 2-(3,5-dimetoxifenil)acetato de metilo (en bruto, 700 mg) como un aceite amarillo. Ms (ES+) C11H14O4 requiere: 210, encontrado: 211 [M H]+.
aso : ness e - , - me ox en - -me - - me o pr o , - prm n- -ona
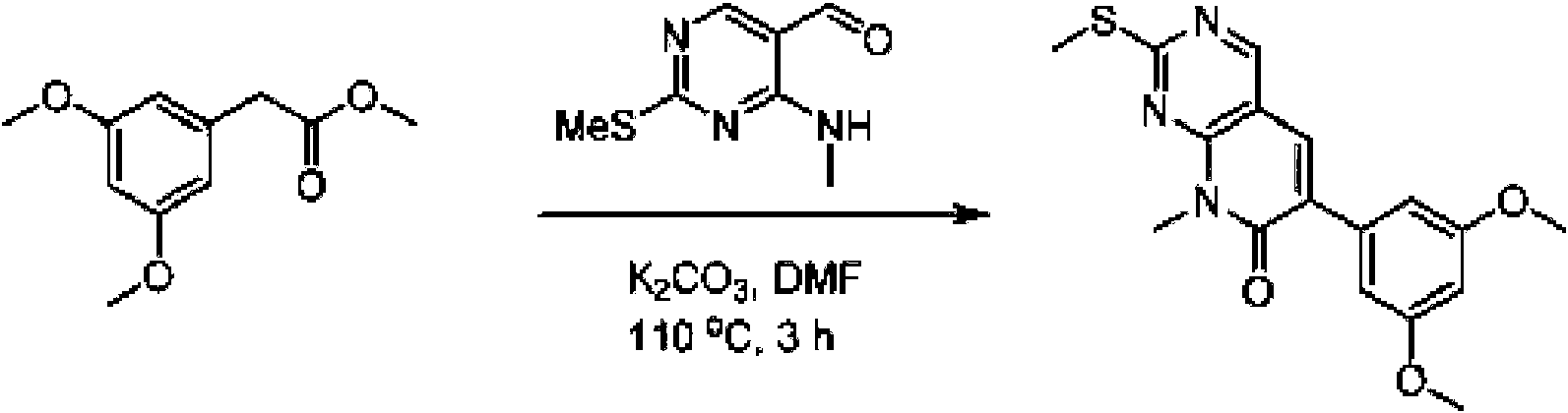
Una solución de 2-(3,5-dimetoxifenil)acetato (6) (440 mg, 2,40 mmol), 4-amino-2-(metiltio)pirimidina-5-carbaldehído (4) (605 mg, 2,88 mmol) y carbonato de potasio (662 mg, 4,8 mmol) se agitó en DMF (30 ml) a 110°C durante 3 horas. La reacción se controló mediante LCMS. La mezcla de reacción se diluyó con H2O (30 ml), y se extrajo mediant EtOAc (3 * 40 ml). Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en columna (gel de sílice, éter de petróleo/EtOAc = 2:1) para producir 6-(3,5-dimetoxifenil)-8-metil-2-(metiltio) pirido [2,3-d]pirimidin-7 (8H)-ona (7) (683 mg, 83%) como un sólido blanco. MS (ES+) C17H17N3O5S requiere: 343, encontrado: 344 [M H] .
Paso 6: Síntesis de 6-(3,5-dimetoxifenil)-8-metil-2-(metilsulfonil) pirido [2,3-d] pirimidin-7 (8H)-ona

A una solución de 6-(3,5-dimetoxifenil)-8-metil-2-(metiltio) pirido [2,3-d] pirimidin-7 (8H)-ona (1,05 g, 3,1 mmol) en metanol/diclorometano (MeOH/DCM) (20 ml/20 ml) se añadó una solución de Oxone® (peroximonosulfato de potasio) (11,3 g, 18,4 mmol) en H2O (20 ml) a temperatura ambiente, y la mezcla de reacción se agitó a 40 °C durante 18 horas. La reacción se controló mediante LCMS. La mezcla de reacción se diluyó con H2O/DCM (150 mL/100 mL) y la fase acuosa se extrajo con DCM (100 mL). Las capas orgánicas combinadas se lavaron con agua (200 ml) y salmuera (200 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron. El producto en bruto se recristalizó con EtOAc para producir 6-(3,5-dimetoxifenil)-8-metil-2-(metilsulfonil) pirido [2,3-d] pirimidin-7 (8H)-ona (8) (910 mg), rendimiento 78%) como un sólido amarillo. MS (ES+) C17H17N3O5S, requiere: 375, encontrado: 376 [M H]+.
Paso 7: Síntesis de 6-(2,6-dicloro-3,5-dimetoxifenil)-8-metil-2-(metilsulfonil) pirido [2,3-d] pirimidin-7(8H) -ona

A una solución de 6-(3,5-dimetoxifenil)-8-metil-2-(metilsulfonil)pirido [2,3-d]pirimidin-7 (8H)-ona (8) (938 mg, 2,5 mmol) en acetonitrilo (50 ml) se añadió lentamente una solución de cloruro de sulfurilo (1,34 g, 10,0 mmol) en acetonitrilo (25 ml) durante un período de 0,5 horas a una temperatura en el intervalo de -10°C a 0°C. La reacción se controló mediante cromatografía en capa fina (TLC). La mezcla de reacción se inactivó añadiendo H2O (10 ml). La solución de reacción resultante se concentró a presión reducida y el residuo se recristalizó con EtOAc/éter de petróleo = 1:2 para dar 6-(2,6-dicloro-3,5-dimetoxifenil)-8-metil-2-(metilsulfonil)pirido [2,3-d] pirimidin-7 (8H)-ona (9) (760 mg, 69% de rendimiento) como un sólido amarillo. MS (ES+) C-izH-isCh^OsS requiere: 443, 445, encontrado: 444, 446 [M H]+.
Paso 8: Síntesis de 6-(2,6-didoro-3,5-dimetoxifenil)-8-metil-2-(2-metil-6-nitrofenilamino) pirido [2,3-d] pirimidin-7 (8H) -ona

A una mezcla de 6-(2,6-didoro-3,5-dimetoxifenil)-8-metil-2-(metilsulfonil) pirido [2,3-d] pirimidin-7(8H)-ona (9) (1,0 g, 2,26 mmol) y 2-metil-6-nitrobencenamina (684 mg, 4,5 mmol) en DMF (20 ml), se añadió terc-butóxido de potasio (756 mg, 6,75 mmol) a ~ 10 °C, y la mezcla de reacción se agitó a temperatura ambiente durante 5 minutos. La mezcla de reacción se diluyó con EtOAc (150 ml) y la fase orgánica se separó, se lavó con agua (2 x 150 ml) y a continuación con salmuera (150 ml), se secó sobre sulfato de sodio, se filtró y se concentró. El residuo se recristalizó con EtOAc para dar 2-(2-amino-6-metilfenilamino)-6-(2,6-dicloro-3,5-dimetoxifenil)-8-metilpirido [2,3-d] pirimidin-7 (8H)-ona (10) (810 mg, rendimiento del 70%) como un sólido amarillo. MS (ES+) C23Hi9Cl2N5O5 requiere: 515, 517, encontrado: 516, 518 [M H]+.
Paso 9: Síntesis de 2-(2-amino-6-metilfenilamino)-6-(2,6-dicloro-3,5-dimetoxifenil)-8-metilpirido [2,3-d] pirimidin-7 (8H)-ona

Una mezcla de 2-(2-nitro-6-metil-fenilamino)-6-(2,6-dicloro-3,5-dimetoxifenil)-8-metilpirido [2,3-d] pirimidin-7 (8H)-ona (10) (810 mg, 1,57 mmol) y cloruro de estaño (II) hidratado (1,77 g, 7,86 mmol) en EtOAc (50 ml) se agitó a 60°C durante 2 horas. La reacción se controló mediante LCMS. La mezcla de reacción se basificó con bicarbonato de sodio acuoso saturado a pH = 8~9, se diluyó con H2O (100 ml) y a continuación se extrajo con EtOAc (3 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (150 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se recristalizó con diclorometano/acetato de etilo/éter de petróleo (DCM/EtOAc/PE) = 1/1/2 para dar 2-(2-amino-6-metilfenilamino)-6-(2,6-dicloro-3, 5-dimetoxifenil)-8-metilpirido [2,3-d] pirimidin-7(8H)-ona (11) (640 mg, 83% de rendimiento) como un sólido gris. (MS (ES+) C23H21Cl2N5O3 requiere: 485, 487, encontrado: 486, 488 [M H]+; 1H-RMN (500 MHz, CDCl3) 5 ppm 8,54 (s, 1 H), 7,45 (s, 1 H), 7,08 (t, J = 7,5 Hz, 1 H), 6,71 (dd, J = 3,5, 7,5 Hz, 2 H), 6,65 (s ancho, 1 H), 6,62 (s, 1 H), 3,94 (s, 6 H), 3,88 (s ancho, 2 H), 3,62 (s ancho, 3 H), 2,24 (s, 3 H).
Paso 10: Síntesis de N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)-8-metil-7-oxo-7,8-dihidropirido[2,3-d]pirimidin-2-il)amino)-3-metilfenil acrilamida COMPUESTO 43

2-(2-amino-6-metil-fenilamino)-6-(2,6-didoro-3,5-dimetoxifenil)-8-metilpirido [2,3-d]pirimidin-7(8H)-ona (11) se recogió en DCM (2 ml) y se enfrió a 0°C, seguido de la adición de cloruro de acriloílo (0,010 ml, 0,13 mmol). Se dejó que la reacción se calentara a temperatura ambiente y se agitó durante la noche. La mezcla se cargó directamente sobre gel de sílice y se purificó mediante cromatografía ultrarrápida usando un gradiente de EtOAc/hexanos al 0-100% para proporcionar el producto, N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)- 8-metil-7-oxo-7,8-dihidropirido [2,3-d] pirimidin-2-il)amino)-3-metilfenil)acrilamida (Compuesto E). El producto se obtuvo como un sólido blanquecino (10 mg; 19% de rendimiento). EM (EN ) C26H23ChN5O4, 540 [M H]+.
Ejemplo 2: Síntesis de N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)quinazolin-2-il)amino)-3-metoxifenil)acrilamida COMPUESTO 30

Paso 1: Síntesis de (2-amino-5-bromofenil metanol

A una solución de ácido 2-amino-5-bromobenzoico (10,0 g, 46,3 mmol) en THF (150 ml) se añadió BH3-THF (1 M, 231 ml) a temperatura ambiente, y la mezcla de reacción se agitó durante la noche. Se analizó una alícuota de la mezcla de reacción mediante LCMS e indicó que la reacción se había completado. La reacción se inactivó con agua (150 ml) y se extrajo con EtOAc (3 x 500 ml). Las capas orgánicas se separaron, se combinaron, se lavaron con agua (200 ml) y salmuera (200 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron para producir el compuesto del título (10 g, en bruto), que se usó directamente en el siguiente paso sin más purificación. MS (ES+) C7HaBrNO requiere: 201, encontrado: 202, 204 [M H]+.
Paso 2: Síntesis de 2-amino-5-bromobenzaldehído

Una mezcla de (2-amino-5-bromofenil)metanol (10 g, 49,5 mmol) y MnO2 (25,8 g, 296,6 mmol) en CH2CI2 (400 ml) se agitó a TA durante una noche. LCMS mostró que la reacción se completó. El sólido se separó por filtración y el filtrado se concentró para dar el compuesto del título como un sólido amarillo claro (8 g, 81%), que se usó directamente en el paso siguiente sin purificación adicional. MS (ES+) C7H6BrNO requiere: 199, encontrado: 200, 202 [M H]+.
Paso 3: Síntesis de 6-bromoquinazolin-2-ol

Se calentó una mezcla de 2-amino-5-bromobenzaldehído (29) (6 g, 30,0 mmol) y urea (30) (27 g, 450,0 mmol) a 180°C y se agitó durante 5 horas. LCMS mostró que la reacción se completó. La mezcla de reacción se enfrió a temperatura ambiente, y el precipitado resultante se lavó con H2O (3 x 500 ml) y se co-evaporó con tolueno tres veces para eliminar completamente la humedad atrapada. Se obtuvo 6-bromoquinazolin-2-ol (31) (6 g, 89%) como un sólido amarillo. MS (ES+) CsHsBr^O requiere: 224, encontrado: 225, 227 [M H]+.
Paso 4: Síntesis de 6-bromo-2-cloro uinazolina

Una solución de 6-bromoquinazolin-2-ol (31) (6,0 g, 26,7 mmol) en POCh (80 ml) se calentó a reflujo a 110 °C durante 5 horas. Se analizó una alícuota de la mezcla de reacción mediante LCMS e indicó que la reacción se había completado. La mayor parte del POCh se eliminó a presión reducida y el residuo se añadió gota a gota a agua helada (500 ml). El precipitado resultante se recogió mediante filtración como un sólido amarillo (3,5 g, 54%). MS (ES+) CsH4BrClN2 requiere: 242, encontrado: 243, 245 [M H]+.
Paso 5: Síntesis de 2-cloro-6-(3,5-dimetoxifenil) quinazolina

Una mezcla de 6-bromo-2-cloroquinazolina (32) (5,0 g, 20,5 mmol), ácido 3,5-dimetoxifenilborónico (33) (3,7 g, 20,5 mmol), Cs2CO3(20,0 g, 61,5 mmol) y Pd(Pph3)2Ch (1,4 g, 2,1 mmol) en THF (50 mL), dioxano (50 mL) y agua (10 mL) se desgasificó con N2tres veces y se agitó a 80 °C durante 3 horas. Se analizó una alícuota de la mezcla de reacción mediante TLC y LCMS, que indicaron que la reacción se había completado. La mezcla se enfrió a temperatura ambiente y se extrajo con EtOAc (3 x 200 ml). Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en gel de sílice (éter de petróleo/EtOAc = 8:1) para obtener 2-cloro-6-(3,5-dimetoxifenil) quinazolina (34) como un sólido amarillo claro (2,4 g, 38%). MS (ES+) C-i6H-i3ClN2O2 requiere: 300, encontrado: 301, 303 [M H]+.
Paso 6: Síntesis de 2-cloro-6-(2,6-dicloro-3,5-dimetoxifenil) quinazolina

A una solución de 2-doro-6-(3,5-dimetoxifenil) quinazolina (34) (2,7 g, 8,9 mmol) en THF seco (80 ml) se añadió gota a gota SO2Ch (3,0 g, 22,3 mmol) a -20 °C, y la mezcla de reacción se agitó durante una hora más. Se analizó una alícuota de la mezcla de reacción mediante TLC y LCMS, que indicaron que la reacción se había completado. La mezcla de reacción se inactivó con agua (1 ml) y los disolventes se eliminaron a presión reducida. El precipitado se lavó con CH3CN y se secó para obtener 2-cloro-6-(2,6-dicloro-3,5-dimetoxifenil) quinazolina (35) (2,6 g, 79%) como un sólido blanco. (EM (ES+) C16H11C13N2O2 requiere: 368, encontrado: 369, 371 [M H]+; 1H-RMN (500 MHz, DMSO) 5 ppm 9,67 (s, 1 H), 8,168 (d, J = 1,5 Hz, 1 H), 8,10 (d, J = 8,5 Hz, 1 H), 7,56 (dd, J = 2,0, 8,5 Hz, 1 H), 7,07 (s, 1 H), 4,00 (s, 6 H).
Paso 7: Síntesis de 6-(2,6-dicloro-3,5-dimetoxifenil)-N-(2-metoxi-6-nitrofenil) quinazolin-2-amina
2-cloro-6-(2,6-dicloro-3,5-dimetoxifenil)quinazolina (35) (100 mg, 0,27 mmol), 2-metoxi-6-nitroanilina (36) (57 mg, 0,40 mmol), Cs2CO3(176 mg, 0,54 mmol), Pd2(dba)3 (25 mg, 0,027 mmol) y 2-diciclohexilfosfino-2',4',6'-triisopropilbifenilo (Xphos) (26 mg, 0,054 mmol) se recogieron en DMF (3 ml) en un vial de microondas y se purgaron con N2 durante 5 minutos. El vial se tapó y se calentó a 115 °C en microondas durante 30 minutos. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con DCM y se lavó con salmuera tres veces. La mezcla orgánica se secó sobre sulfato de sodio y se cargó directamente sobre gel de sílice y se purificó usando un gradiente de EtOAc/Hexanos al 0-100%. Se recuperó 6-(2,6-dicloro-3,5-dimetoxifenil)-N-(2-metoxi-6-nitrofenil) quinazolin-2-amina (37) como un sólido amarillo (100 mg, 73% de rendimiento). MS (ES+) C23H1sCl2N4O5, 501 [M H]+.
Paso 8: Síntesis de N1- 6- 2,6-dicloro-3,5-dimetoxifenil uinazolin-2-il -6-metoxibenceno-1,2-diamina
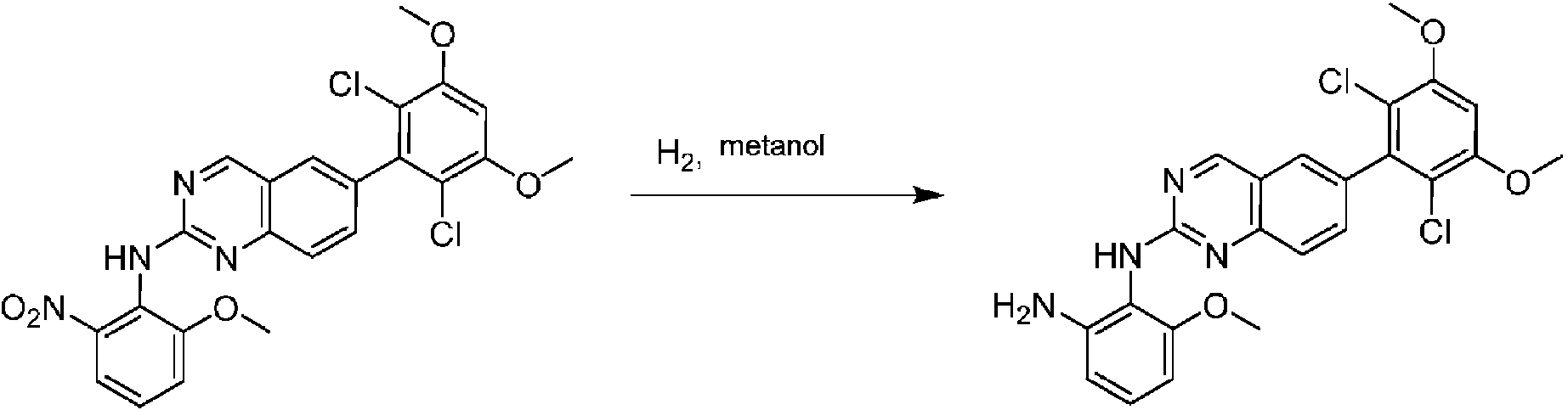
Se recogió 6-(2,6-dicloro-3,5-dimetoxifenil)-N-(2-metoxi-6-nitrofenil) quinazolin-2-amina (38) (100 mg, 0,14 mmol) en metanol (10 ml), se añadió Pd al 10%/C (15 mg). La mezcla se agitó bajo un globo de H2 durante 4 horas. La mezcla de reacción se filtró a través de celite y el disolvente se eliminó para dar N1-(6-(2,6-dicloro-3,5-dimetoxifenil) quinazolin-2-il)-6-metoxi-benceno-1,2-diamina (38) en rendimiento cuantitativo. El compuesto 38 se pasó al siguiente paso sin purificación adicional. EM (EN+) C23H20Cl2N4O3, 471 [M H]+
Paso 9: Síntesis de N-(2-((6-(2,6-didoro-3,5-dimetoxifenil) quinazolin-2-il)amino)-3-metoxifenil)acrilamida
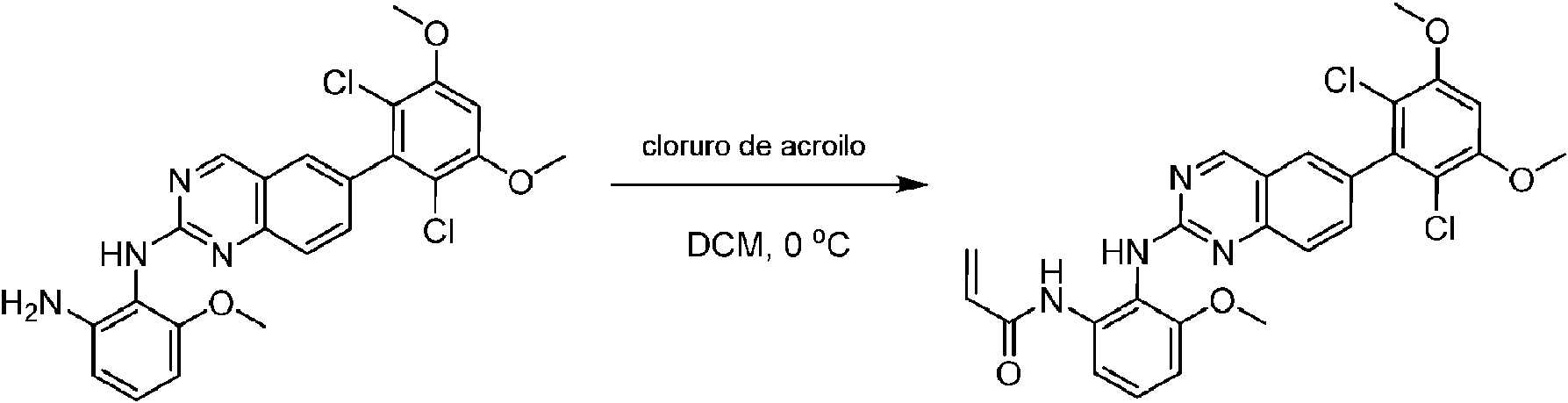
Se tomó N1-(6-(2,6-dicloro-3,5-dimetoxifenil) quinazolin-2-il)-6-metoxibenceno-1,2-diamina (38) (96 mg, 0,20 mmol) en DCM (2 ml) y se enfrió a 0°C, seguido de la adición de cloruro de acriloílo (0,018 ml, 0,24 mmol) y se agitó a 0°C durante 2 horas. La mezcla se cargó directamente sobre gel de sílice y se purificó mediante cromatografía ultrarrápida “flash” usando un gradiente de EtOAc/Hexanos al 0-100%. Se recuperó N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)quinazolin-2-il)amino)-3-metoxifenil)acrilamida (39) como un sólido blanquecino (30 mg, Rendimiento del 28%). MS (ES+) C2aH22Cl2N4O4, 525 [M H]+.
Eem lo 3: Síntesis del COMPUESTO 25

2-cloro-6-(2,6-dicloro-3,5-dimetoxifenil) quinazolina (35) (5 g, 13,5 mmol), 2-metil-6-nitroanilina (3,09 g, 20,3 mmol), Cs2CO 3 (13,2 g, 40,6 mmol), Pd2(dba)3 (1,24 g, 1,35 mmol) y 2-diciclohexilfosfino-2',4',6'-triisopropilbifenilo (Xphos) (1,29 g, 2,71 mmol) se recogieron en DMA (100 ml) y se purgaron con N2 durante 5 minutos. La mezcla de reacción se calentó a 110°C durante 3 horas. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con DCM (500 ml) y se lavó con HCl al 10% tres veces (3 x 300 ml) y salmuera tres veces. La mezcla orgánica se secó sobre sulfato de sodio y se cargó directamente sobre gel de sílice y se purificó usando un gradiente de
EtOAc/Hexanos al 0-100%. Se recuperó 6-(2,6-didoro-3,5-dimetoxifenil)-N-(2-metil-6-nitrofenil)quinazolin-2-amina como un sólido amarillo (5,5 g, rendimiento del 81%). MS (ES+) C23H18CI2N4O4, 485 [M H]+.
Síntesis de N1-(6-(2,6-didoro-3,5-dimetoxifenil) quinazolin-2-il)-6-metilbenceno-1,2-diamina

Se recogió 6-(2,6-dicloro-3,5-dimetoxifenil)-N-(2-metil-6-nitrofenil) quinazolin-2-amina (5,5 g, 11,33 mmol) en metanol (200 ml) y se añadió acetato de etilo (100 ml), Pd al 10%/C (650 mg). La mezcla se agitó bajo un globo de H2 durante la noche. La mezcla de reacción se filtró a través de celite y el disolvente se eliminó para dar N1-(6-(2,6-dicloro-3,5-dimetoxifenil)quinazolin-2-il)-6-metil-benceno-1,2-diamina en rendimiento cuantitativo. Se pasó al siguiente paso sin más purificación. m S (ES+) C23H20ChN4O2, 455 [M H]
Síntesis de N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil) quinazolin-2-il)amino)-3-metilfenil)acrilamida

N1-(6-(2,6-dicloro-3,5-dimetoxifenil) quinazolin-2-il)-6-metil-benceno-1,2-diamina (5,16 g, 11,33 mmol) se recogió en DCM (100 ml) y se enfrió a 0°C, seguido de la adición de DIEA (1,781 ml, 10,20 mmol) y cloruro de acriloílo (1,013 ml, 12,47 mmol) y se agitó a 0°C durante 2 horas. La mezcla se cargó directamente sobre gel de sílice y se purificó mediante cromatografía ultrarrápida usando un gradiente de EtOAc/Hexanos al 0-100%. Se recuperó N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)quinazolin-2-il)amino)-3-metilfenil)acrilamida como un sólido blanquecino (3,5 g, 61% producir). MS (ES+) C26} H22C2N4O3, 509 [M H]+. 1 H RMN (400 MHz, DMSO-da) 89.53 (s, 1H), 9.23 (s, 1H), 8.68 (s, 1H), 7.82 - 7.65 (m, 2H), 7.51 (s, 2H), 7.21 (m, 1H), 7.12 (d, J = 6,8 Hz, 1 H), 7,01 (s, 1 H), 6,49 (dd, J = 17,0, 10,2 Hz, 1 H), 6,28 - 6,15 (m, 1 H), 5,68 (dd, J = 10,2, 2,0 Hz, 1 H), 3,97 (s, 6 H), 2,19 (s, 3 H).

NaH ruro de su furi o
THF, 0 °C~ T A THE, 0
durante a noc 
Fe, NH4CI
etanol/agua


cloruro de acriloilo cloruro de acriloilo
DIEA DIEA

Síntesis de 6-bromopirido [2,3-d] pirimidin-2-amina

Se disolvieron 5-bromo-2-fluoronicotinaldehído (3,0 g, 14,78 mmol), clorhidrato de guanidina (1,69 g, 17,74 mmol) y trietilamina (4,48 g, 44,35 mmol) en 1-metil-2-pirrolidinona (15 ml) y la mezcla de reacción se agitó a 180°C durante 15 min en microondas. La mezcla se enfrió a Ta , se inactivó con agua (200 ml) y se extrajo con acetato de etilo (2 x 300 ml). Las capas orgánicas se combinaron, se lavaron con agua (3 x 50 ml) y salmuera (3 x 50 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron para proporcionar un producto en bruto, que se purificó mediante cromatografía en columna de gel de sílice (acetato de etilo:éter de petróleo = 3:1) para producir 6-bromopirido [2,3-d] pirimidin-2-amina (2,0 g, 60%) como un sólido amarillo. MS (ES+) C7HsBrN4 requiere: 224, 226, encontrado: 225, 227 [M H]+.
Síntesis de 6- 3,5-dimetoxifenil irido 2,3-d irimidin-2-amina

Una mezcla de 6-bromopirido [2,3-d] pirimidin-2-amina (1,0 g, 4,46 mmol), ácido 3,5-dimetoxifenilborónico (1,2 g, 6,70 mmol), PdCl2(dppf) (364 mg, 0,446 mmol) y carbonato de potasio (1,8 g, 13,39 mmol) en 1,4-dioxano/agua (4 ml/1 ml) se desgasificó con nitrógeno durante 5 min y se agitó a 110°C durante 30 min en microondas. La mezcla de reacción se enfrió a TA y se concentró para proporcionar un producto en bruto, que se purificó mediante cromatografía en columna de gel de sílice (acetato de etilo: éter de petróleo = 4: 1) para producir 6-(3,5-dimetoxifenil) pirido[2, 3-d]pirimidin-2-amina como un sólido amarillo (400 mg, 31%). MS (ES+) C15H14N4O2 requiere: 282, encontrado: 283 [M H]+.
Síntesis de 6-(3,5-dimetoxifenil)-N-(2-metil-6-nitrofenil) pirido [2,3-d] pirimidin-2-amina

Se añadió hidruro de sodio (102 mg, 4,25 mmol) a una solución de 6-(3,5-dimetoxifenil) pirido [2,3-d]pirimidin-2-amina (400 mg, 1,42 mmol) en THF (20 ml) a 0 °C. La solución se agitó durante 20 minutos, seguido de la adición de 2-fluoro-1-metil-3-nitrobenceno (440 mg, 2,84 mmol). La mezcla de reacción se agitó a TA durante la noche, se inactivó con agua (20 ml) y se extrajo con acetato de etilo (3 x 30 ml). Las capas orgánicas se combinaron, se lavaron con salmuera (50 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron para proporcionar un producto en bruto, que se purificó mediante cromatografía en columna de gel de sílice (acetato de etilo: éter de petróleo = 4:1) para producir 6-(3,5-dimetoxifenil)-N-(2-metil-6-nitrofenil) pirido [2,3-d] pirimidin-2-amina (310 mg, 51%) como un sólido marrón. MS (Es+) C22Hi9N5O4 requiere: 417, encontrado: 418 [M H]+.
Síntesis de N1-(6-(3,5-dimetoxifenil) pirido [2,3-d] pirimidin-2-il)-6-metilbenceno-1,2-diamina

A una solución de 6-(3,5-dimetoxi-fenil)-N-(2-metil-6-nitrofenil)pirido[2,3-d] pirimidin-2-amina (100 mg, 0,24 mmol) en etanol (5 ml) y agua (5 ml) se añadió polvo de hierro (110 mg, 1,92 mmol) y cloruro de amonio (100 mg, 1,920 mmol). La mezcla se agitó a 100 °C durante 1 hora, se enfrió a TA, se filtró y se concentró. El residuo se purificó mediante HPLC preparativa para producir N1-(6-(3,5-dimetoxifenil) pirido [2,3-d] pirimidin-2-il)-6-metilbenceno-1,2-diamina (29,5 mg, 32%) como un sólido amarillo. MS (ES+) C22H2iNsO2 requiere: 387, encontrado: 388 [M H]+; 1H-RMN (500 MHz, DMSO-da) 5 ppm 9,30, 9,21 (ancho, ancho, 2H), 8,95 (s, 1H), 8,60 (d, 1H, J = 3,0 Hz), 6,96-6,92 (m, 3H), 6,63 (d, 1H, J = 5,5 Hz), 6,55 (t, 1 H, J = 2,0 Hz), 6,50-6,48 (m, 1 H), 4,79 (s, 2 H), 3,84 (s, 6 H), 2,08 (s, 3 H).
Síntesis de 6-(2,6-dicloro-3,5-dimetoxifenil)-N-(2-metil-6-nitrofenil) pirido [2,3-d] pirimidin-2-amina

A una solución agitada de 6-(3,5-dimetoxifenil)-N-(2-metil-6-nitrofenil)pirido[2,3-d] pirimidin-2-amina (100 mg, 0,24 mmol) en THF (10 ml) a 0°C se añadió gota a gota una solución de cloruro de sulfurilo (0,06 ml, 0,72 mmol) en THF (2 ml). Después de agitar a 0°C durante 2 horas, la reacción se inactivó con agua (10 ml) y se extrajo con acetato de etilo (3 x 20 ml). Las capas orgánicas se combinaron, se lavaron con salmuera (20 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en columna de gel de sílice (acetato de etilo: éter de petróleo = 3:1) para producir 6-(2,6-dicloro-3,5-dimetoxifenil)-N-(2-metil-6-nitrofenil) pirido[2,3-d] pirimidin-2-amina (110 mg, 95%) como un sólido amarillo. MS (ES+) C22H17O2N5O 4 requiere: 485, 487 encontrado: 486, 488 [M H]+.
Síntesis de N1-(6-(2,6-dicloro-3,5-dimetoxifenil) pirido [2,3-d] pirimidin-2-il)-6-metilbenceno-1,2-diamina

A una solución de 6-(2,6-dicloro-3,5-dimetoxifenil)-N-(2-metil-6-nitrofenil)pirido[2,3-d] pirimidin-2-amina (80 mg, 0,168 mmol) en etanol (4 ml) y agua (4 ml) se añadió polvo de hierro (75 mg, 1,344 mmol) y cloruro de amonio (74 mg, 1,344 mmol). La mezcla se agitó a 100 °C durante 2 horas, se enfrió a TA, se filtró y se concentró. El residuo se purificó por cromatografía en columna de gel de sílice (acetato de etilo: éter de petróleo = 4:1) para proporcionar N1-(6-(2,6-dicloro-3,5-dimetoxifenil)pirido [2,3-d] pirimidin-2-il)-6-metilbenceno-1,2-diamina (40 mg, 53%) como un sólido amarillo. MS (ES+) C22H1gChNsO2 requiere: 455, 457, encontrado: 456, 458 [M H]+. 1H-RMN (400 MHz, DMSO-da) 5 ppm 9,33 (s. ancho, 1H), 9,01 (s, 1H), 9,65 (s. ancho, 1H), 8,23 (s, 1H), 7,05 (s, 1 H), 6,93 (s. ancho, 1 H), 6,64 6,63 (m, 1 H), 6,50-6,49 (m, 1 H), 4,80 (s, 2 H), 3,99 (s, 6 H), 2,09 (s, 3H).
Síntesis de N-(2-((6-(3,5-dimetoxifenil) pirido [2,3-d] pirimidin-2-il)amino)-3-metilfenil)acrilamida

Se preparó N-(2-((6-(3,5-dimetoxifenil)pirido[2,3-d] pirimidin-2-il)amino)-3-metilfenil)acrilamida usando un procedimiento similar al del COMPUESTO 30. El producto se purificó mediante cromatografía ultrarrápida usando un gradiente de EtOAc/DCM al 0-50% para dar el compuesto del título. MS (ES+) C25H23N5O3 requiere: 441, encontrado: 442
Síntesis de N-(2-((6-(2,6-didoro-3,5-dimetoxifenil) pirido [2,3-d] pirimidin-2-il)amino)-3-metilfenil)acrilamida

Se preparó N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)pirido [2,3-d] pirimidin-2-il)amino)-3-metilfenil)acrilamida usando el procedimiento es similar al del COMPUESTO 30. El producto se purificó mediante cromatografía ultrarrápida usando un gradiente de MeOH/DCM al 0-10% para dar el compuesto del título. MS (ES+) C25H2-iChNsO3 requiere: 510, encontrado: 511 [M H]+. 1 H-RMN (400 MHz, DMSO-da) 59,53 (s, 1 H), 9,35 (s, 1 H), 9,06 (s, 1 H), 8,70 (s, 1 H), 8,27 (d, J = 2,6 Hz, 1H), 7.78 (s, 1H), 7.23 (d, J = 7.9 Hz, 1H), 7.15 (s, 1H), 7.06 (s, 1H), 6.52 (dd, J = 17.0, 10.1 Hz, 1H), 6,22 (dd, J = 17,0, 2,0 Hz, 1 H), 5,69 (d, J = 10,6 Hz, 1 H), 3,98 (s, 6 H), 2,20 (s, 3 H).
Ejem lo 4: Síntesis del COMPUESTO 45

Síntesis de 2-cloro-N-metil-5-nitropirimidin-4-amina


A una solución de 2,4-dicloro-5-nitropirimidina (5 g, 26 mmol) en THF (50 ml) se añadió diisopropiletilamina (3,36 g, 26 mmol) a -78 °C, seguido de la adición gota a gota de metilamina (13 mL, 2 mol/L en metanol, 26 mmol). Después de la adición, la mezcla se calentó a TA y se agitó durante 3 h. A continuación, la mezcla de reacción se diluyó con acetato de etilo y se lavó con salmuera (50 ml x 3). La capa orgánica se secó sobre sulfato de sodio, se filtró y se concentró para dar el compuesto del título (4,4 g, 100%) como un sólido amarillo. MS (ES+) C5H5ClN4O2 requiere: 188, 190, encontrado: 189, 191 [M H]+.
Síntesis de 2-cloro-N4-metilpirimidina-4,5-diamina

A una solución agitada de 2-cloro-N-metil-5-nitropirimidin-4-amina (1,9 g, 10 mmol) en ácido acético (30 ml) se añadió polvo de hierro (4 g, 71 mmol), y la mezcla de suspensión se calentó a 60°C durante 16 horas. El disolvente se eliminó a presión reducida y el residuo se diluyó con salmuera y acetato de etilo. El sólido se retiró por filtración y el filtrado se extrajo con acetato de etilo (50 ml x 12). Las capas orgánicas se separaron, se combinaron, se secaron sobre sulfato de sodio, se filtraron y se concentraron para dar el compuesto del título (1,1 g, 69%). MS (ES+) C5H7ClN4 requiere: 159, 161, encontrado: 160, 162 [M H]+.
Síntesis de 2- 3,5-dimetoxifenil -2-oxoacetato de etilo

A una solución de 1-bromo-3,5-dimetoxibenceno (2,17 g, 10 mmol) en THF (15 ml) se añadió gota a gota n-butil litio (8 ml, 2,5 mol/L en hexano, 20 mmol) a -78 °C. Después de agitar durante 50 minutos a -78°C, se añadió una solución de oxalato de dietilo (4 g, 27 mmol) en THF (10 ml). La mezcla se agitó a -78°C durante otras 4 h, a continuación se inactivó con cloruro de amonio saturado y se extrajo con acetato de etilo (50 ml x 3). Las capas orgánicas se combinaron, se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en gel de sílice para dar el compuesto del título (1,7 g, 71%). MS (ES+) C12H14O5 requiere: 238, encontrado: 239 [M H]+.
Síntesis de 2-cloro-6-(3,5-dimetoxifenil)-8-metilpteridin-7(8H)-ona

Una mezcla de 2-(3,5-dimetoxifenil)-2-oxoacetato de etilo (1 g, 4,2 mmol) y 2-cloro-N4-metilpirimidin-4,5-diamina (600 mg, 3,8 mmol) en etanol (100 ml) y ácido acético (2,5 ml) se agitó a 80°C durante 48 horas y se enfrió a TA (5°C). La mezcla se diluyó con diclorometano y se lavó con salmuera. La capa orgánica se concentró directamente y se purificó mediante cromatografía en gel de sílice para dar el compuesto del título (700 mg, 50%). MS (Es+) C15H13ClN4O3 requiere: 332, 334, encontrado: 333, 335 [M H]+.
Síntesis de 2-cloro-6-(2,6-dicloro-3,5-dimetoxifenil)-8-metilpteridin-7 (8H)-ona
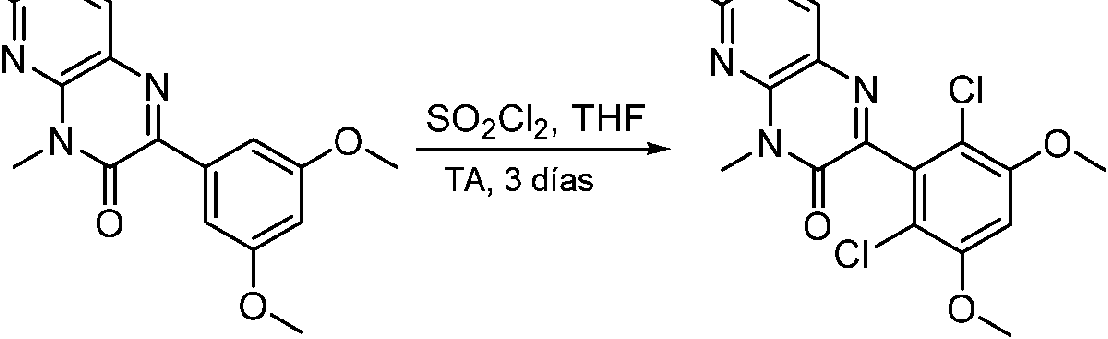
A una solución de 2-cloro-6-(3,5-dimetoxifenil)-8-metilpteridin-7(8H)-ona (300 mg, 0,9 mmol) en THF (5 ml) se añadió gota a gota cloruro de sulfurilo (300 mg) y la mezcla se agitó a TA durante 4 h. Se añadió cloruro de sulfurilo adicional (300 mg) y se agitó a TA durante 3 días. La reacción se inactivó con 5 gotas de agua y a continuación se
agitó durante 5 minutos. El precipitado se recogió mediante filtración y se secó para dar el compuesto del título (240 mg, 67%) como un sólido amarillo. MS (ES+) C15H11G3N4O3 requiere: 400, 402, encontrado: 400, 403 [M H]+. Síntesis de 6-(2,6-dicloro-3,5-dimetoxifenil)-8-metil-2-(2-metil-6-nitrofenilamino)pteridin-7(8H)-ona

A una solución de 2-metil-6-nitrobencenamina (100 mg, 1 mmol) en N, N-dimetilformamida (5 ml) se añadió hidruro de sodio (53 mg, 1,3 mmol), y la mezcla se agitó a RT (10 °C) durante 10 minutos, seguido de la adición de 2-cloro-6-(2,6-dicloro-3,5-dimetoxifenil)-8-metilpteridin-7(8H)-ona (322 mg, 1 mmol). La mezcla se agitó a TA (10°C) durante otros 30 min y a continación se inactivó con agua. El precipitado se recogió mediante filtración, se lavó con agua fría y se secó para dar el compuesto del título (180 mg, 75%) como un polvo amarillo. MS (ES+) C22H1ChN6O5 requiere: 516, 518, encontrado: 517, 519 [M H]+.
Síntesis de 2-(2-amino-6-metilfenilamino)-6-(3,5-dimetoxifenil)-8-metilpteridin-7(8H)-ona

A una solución de 6-(2,6-dicloro-3,5-dimetoxifenil)-8-metil-2-(2-metil-6-nitrofenilamino)pteridin-7(8H)-ona (200 mg, 0,38 mmol) en etanol (50 ml) y agua (2 ml) se añadió polvo de hierro (210 mg, 3,8 mmol) y cloruro de amonio (450 mg, 8 mmol). La mezcla se calentó a reflujo durante 2 h. Los disolventes se evaporaron y el residuo se diluyó con salmuera y diclorometano. Se filtró el sólido y se extrajo el filtrado con diclorometano (50 ml x 6). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio, se filtraron y se concentraron para dar el compuesto del título (70 mg, 38%). MS (ES+) C22H2üCl2N6O3 requiere: 486, 488, encontrado: 487, 489 [M H]+.1H-RMN (500 MHz, CDCla) S ppm 8,83 (s, 1H), 7,09 (t, 1H, J = 8,0 Hz), 6,74-6,71 (m, 2H), 6,65 (s, 1H), 3,94 (s, 6H), 3,85 (s ancho, 2H), 3,63-3,59 (ancho, 3H), 2,25 (s, 3H).
Síntesis de N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)-8-metil-7-oxo-7,8-dihidropteridin-2-il)amino)-3-metilfenil)acrilamida

Se preparó N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)-8-metil-7-oxo-7,8-dihidropteridin-2-il)amino)-3-metilfenil)acrilamida usando un procedimiento similar al del COMPUESTO 30. El producto se purificó por cromatografía ultrarrápida
usando un gradiente de 0-10% de MeOH/DCM para dar el compuesto del título. MS (ES+) C25H22CI2N6O4 requiere: 540, encontrado: 541 [M H]+.
Ejemplo 5: Síntesis del COMPUESTO 39
MnO

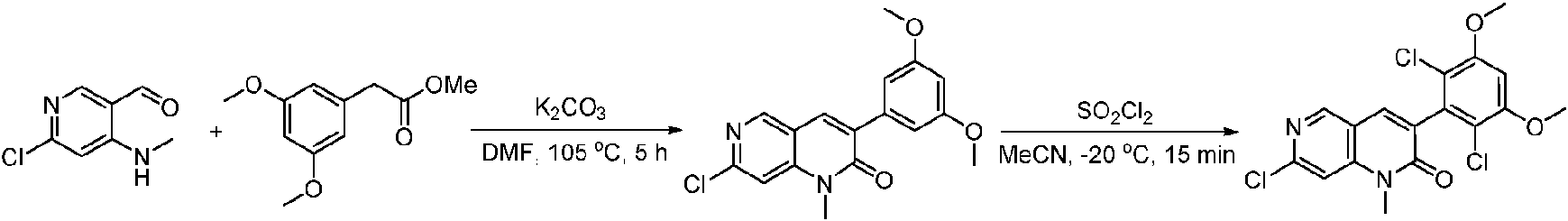

Síntesis de 6-cloro-4-(metilamino) nicotinato de etilo

A una solución de 4,6-dicloronicotinato de etilo (5,0 g, 22,7 mmol) en acetonitrilo (50 ml) se añadió sal de clorhidrato de metilamina (1,84 g, 27,2 mmol) y diisopropiletilamina (14,6 g, 113,6 mmol), y la mezcla de reacción se calentó a 70°C durante la noche. LCMS mostró que la reacción se completó. La reacción se enfrió a TA, se inactivó con agua (50 ml) y se extrajo con acetato de etilo (3 x 100 ml). Las capas orgánicas se separaron, se combinaron, se lavaron con agua (50 ml) y salmuera (100 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron para producir el compuesto del título (4,7 g, bruto), que se usó directamente en la siguiente etapa sin más purificación. MS (ES+) C9H11GN2O2 requiere: 214, 216, encontrado: 215, 217 [M H]+.
Síntesis de (6-cloro-4-(metilamino) piridin-3-il) metanol

A una solución de 6-cloro-4-(metilamino)nicotinato de etilo (4,7 g, 21,9 mmol) en THF (30 ml) y metanol (30 ml) se añadió borohidruro de litio (2,4 g, 109,8 mmol), y la mezcla de reacción se calentó a 55°C durante la noche. LCMS mostró que la reacción se completó. La reacción se enfrió a TA, se inactivó con agua (1 ml) y se filtró. El filtrado se concentró para producir el compuesto del título (4,2 g, en bruto) como un sólido blanco, que se usó directamente en la siguiente etapa sin purificación adicional. MS (ES+) C7H9ClN2O requiere: 172, 174, encontrado: 173, 175 [M H]+.
Síntesis de 6-cloro-4-(metilamino) nicotinaldehído

Una mezcla de (6-cloro-4-(metilamino) piridin-3-il) metanol (4,2 g, 24,7 mmol) y óxido de manganeso (IV) (activo, 25,8 g, 296,6 mmol) en diclorometano (50 ml) y THF (50 ml) se agitó a TA durante la noche. LCMS mostró que la reacción se completó. El sólido se retiró por filtración y el filtrado se concentró para producir el compuesto del título (3,7 g, en bruto) como un sólido de color amarillo claro, que se usó directamente en la siguiente etapa sin purificación adicional. MS (ES+) C7H7ClN2O requiere: 170, 172, encontrado: 171, 173 [M H]+.
Síntesis de 7-cloro-3- 3,5-dimetoxifenil -1-metil-1,6-naftiridin-2 1H -ona

Una mezcla de 6-cloro-4-(metilamino) nicotinaldehído (3,7 g, 21,7 mmol), 2-(3,5-dimetoxifenil) acetato de metilo (4,5 g, 21,7 mmol) y carbonato de potasio (9,0 g, 65,1 mmol) en N, N-dimetilformamida (30 ml) se calentó a 105°C durante 5 h. LCMS mostró que la reacción se completó. La reacción se enfrió a TA, se inactivó con agua (200 ml) y se filtró. La torta de filtración se lavó con éter de petróleo (50 ml) y acetato de etilo (50 ml) para producir el compuesto del título (5,8 g, 77%) en forma de un sólido amarillo. MS (ES+) C-i8H-i9ClN2O3 requiere: 346, 348, encontrado: 347, 349 [M H]+.
Síntesis de 7-cloro-3-(2,6-dicloro-3,5-dimetoxifenil)-1-metil-1,6-naftiridin-2(1H)-ona

A una solución de 7-cloro-3-(3,5-dimetoxifenil)-1-metil-1,6-naftiridin-2(1H)-ona (5,6 g, 16,9 mmol) en acetonitrilo (30 ml) se añadió gota a gota cloruro de sulfurilo (3,36 ml, 42,2 mmol) a -20°C y la mezcla se agitó durante otros 15 minutos. LCMS mostró que la reacción se completó. La reacción se inactivó con agua (1 ml) y los disolventes se eliminaron a presión reducida. El precipitado se lavó con acetonitrilo y se secó para producir el compuesto del título (5,01 g, 75%) en forma de un sólido blanco. MS (ES+) C-i7H-i3Cl3N2O3 requiere: 399, 401, encontrado: 400, 402 [M H]+; 1H-RMN (500 MHz, DMSO-d a) 5 ppm 8,82 (s, 1 H), 8,01 (s, 1 H), 7,71 (s, 1 H), 7,04 (s, 1 H), 3,98 (s, 6 H), 3,66 (s, 3 H).
Síntesis de 3-(2,6-dicloro-3,5-dimetoxifenil)-1-metil-7-((2-metil-6-nitrofenil)amino)-1,6-naftiridin-2(1H)-ona
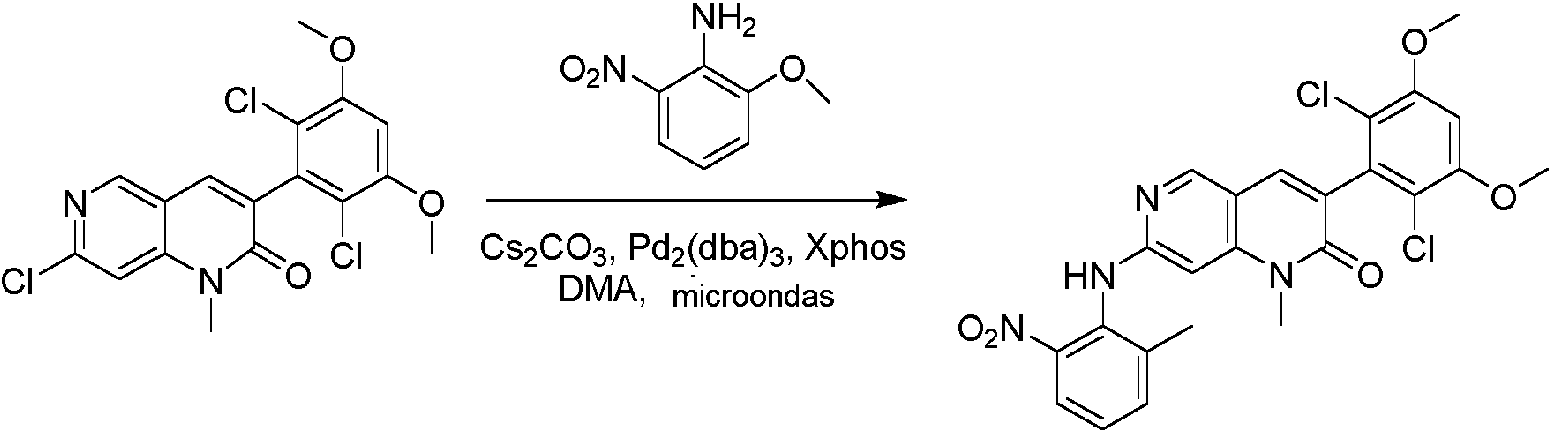
3-(2,6-didoro-3,5-dimetoxifenil)-1-metil-7-((2-metil-6-nitrofenil)amino)-1,6-naftiridin-2 (1H)-ona se preparó usando un procedimiento similar al COMPUESTO 30.
Síntesis de 7- 2-amno-6-metilfenl am no-3- 2,6-dcloro-3,5-d metoxfen l -1-met l-1,6-naftirid n-21 H-ona

7-((2-amino-6-metilfenil)amino)-3-(2,6-didoro-3,5-dimetoxifenil)-1-metil-1,6-naftiridin-2(1H)-ona se preparó usando un procedimiento similar al COMPUESTO 30.
Síntesis de 7-((2-am¡no-6-metilfen¡l)am¡no)-3-(2,6-d¡doro-3,5-d¡metox¡fen¡l)-1-met¡l-1,6 -naftirid¡n-2(1H)-ona
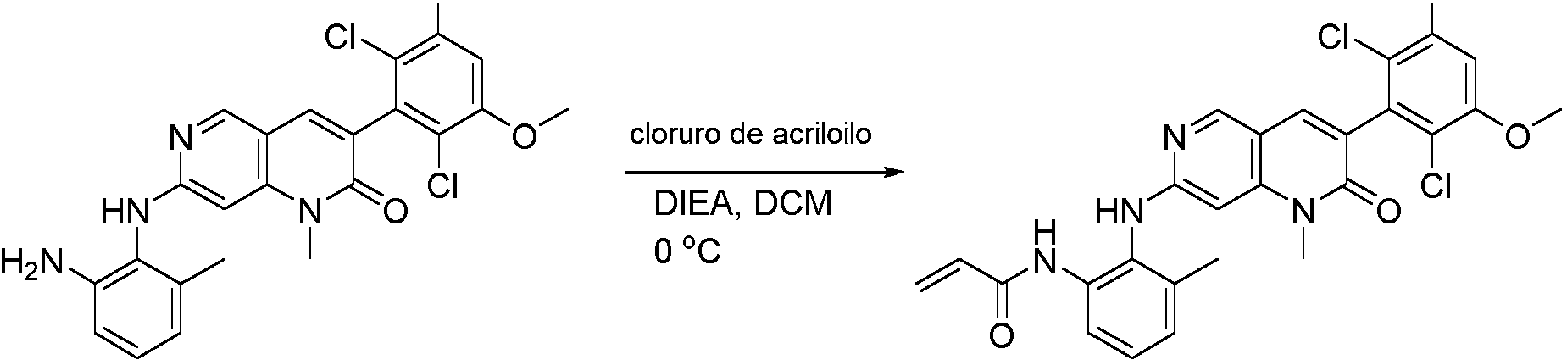
7-((2-amino-6-metilfenil)amino)-3-(2,6-didoro-3,5-dimetoxifenil)-1-metil-1,6-naftiridin-2(1H)-ona se preparó usando un procedimiento similar al del COMPUESTO 30. El producto se purificó por cromatografía ultrarrápida usando un gradiente de EtOAc/DCM al 0-100% para dar el compuesto del título. MS (ES+) C27H24ClN4O 4 requiere: 538, encontrado: 539 [M H]+. 1H RMN (400 MHz, DMSO-d6) 59,47 (s, 1 H), 8,43 (d, J = 10,0 Hz, 2 H), 7,70 (d, J = 12,6 Hz, 2 H), 7,22 (t, J = 7,8 Hz, 1 H), 7,14 (d, J = 7,6 Hz, 1 H), 6,97 (s, 1 H), 6,46 (dd, J = 17,0, 10,2 Hz, 1 H), 6,18 (dd, J = 17,0, 2,1 Hz, 1 H), 6,09 (s, 1 H), 5,65 (dd, J = 10,2, 2,1 Hz, 1 H), 3,95 (s, 6 H), 3,39 (s, 3 H), 2,20 (s, 3 H).
Ejemplo 6: Síntesis del COMPUESTO 48

Síntesis de 5- 3,5-dimetoxifenilaminometil-N-metil-2- metiltio irimidin-4-amina

Una mezcla de 4-(metilamino)-2-(metiltio)pirimidina-5-carbaldehído (1,0 g, 5,46 mmol) y 3,5-dimetoxianilina (840 mg, 5,46 mmol) en metanol (60 ml) se agitó a TA durante 3 h, seguido de la adición de cianoborohidruro de sodio (520 mg, 8,20 mmol) y 1 ml de ácido acético. A continuación, la mezcla de reacción se agitó a TA durante otras 4 h. LCMS mostró que la reacción se completó. La reacción se inactivó con 30 ml de HCl 1 N, a continuación se agitó durante 0,5 h y se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas se separaron, se combinaron, se lavaron con una solución acuosa saturada de bicarbonato de sodio y salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron para producir el compuesto del título (1,2 g en bruto, 69%) como un sólido blanco, que se usó directamente en el siguiente paso sin más purificación. MS (ES+) C15H20N4O2S requiere: 320, encontrado: 321 [M H]+.
Síntesis de 3-(3,5-dimetoxifenil)-1-metil-7-(metiltio)-3,4-dihidropirimido [4,5-d] pirimidin-2 (1H)-ona

A una mezcla de 5-((3,5-dimetoxifenilamino) metil)-N-metil-2-(metiltio)pirimidin-4-amina (1,1 g, 3,43 mmol) y N-etil-N-isopropylpropan-2-amina (1,33 g, 10,30 mmol) en 10 ml de t Hf se añadió una solución de trifosgeno (357 mg, 1,20 mmol) en 5 ml de THF a 0°C y se agitó durante 1 h. A continuación, la mezcla de reacción se calentó a TA y se agitó durante otras 5 h. LCMS mostró que la reacción se completó. La mezcla de reacción se inactivó con agua y se extrajo con acetato de etilo (3 x 15 ml). Las capas orgánicas se separaron, se combinaron, se lavaron con una solución acuosa saturada de bicarbonato de sodio y salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron para producir el compuesto del título (1,1 g en bruto, 92%) como un sólido blanco, que se usó directamente en el siguiente paso sin más purificación. MS (ES+) C16H18N4O3S requiere: 346, encontrado: 347 [M H]+.
Síntesis de 3-(3,5-dimetoxifenil)-1-metil-7-(metilsulfonil)-3,4-dihidropirimido [4,5-d] pirimidin-2 (1H) -ona

A una solución de 3-(3,5-dimetoxifenil)-1-metil-7-(metiltio)-3,4-dihidropirimido[4,5-d] pirimidin-2(1H)-ona (1,0 g, 2,89 mmol) en 20 ml de diclorometano se añadió ácido 3-clorobenzoperoxoico (1,50 g, 8 ,66 mmol) a 0°C y la solución se agitó durante 0,5 h a 0°C. La mezcla se calentó a TA y se agitó durante la noche. LCMS mostró que la reacción se completó. La mezcla de reacción se diluyó con 30 ml de diclorometano, se lavó con una solución acuosa saturada de bicarbonato de sodio y salmuera, se secó sobre sulfato de sodio, se filtró y se concentró para producir el compuesto del título (800 mg, 73%) como un sólido amarillo, que fue directamente utilizado en el siguiente paso sin más purificación. MS (ES+) C16H18N4O5S requiere: 378, encontrado: 379 [M H]+.
Síntesis
A una solución de 3-(3,5-dimetoxifenil)-1-metil-7-(metilsulfonil)-3,4-dihidropirimido [4,5-d]pirimidin-2(1H)-ona (400 mg, 1,06 mmol) en 15 ml de diclorometano se añadió cloruro de sulfurilo (285 mg, 2,12 mmol) a 0°C y a continuación se agitó a 0°C durante 3 h. LCMS mostró que la reacción se completó. La mezcla de reacción se diluyó con 20 ml de diclorometano, se lavó con agua y salmuera, se secó sobre sulfato de sodio, se filtró y se concentró para producir el compuesto del título (450 mg, 96%) como un sólido amarillo, que se usó directamente en el siguiente paso sin más purificación. MS (ES+) C ^ H ^ C ^ N ^ S requiere: 446, 448, encontrado: 447, 449 [M H]+.
Síntesis de 3-(2,6-dicloro-3,5-dimetoxifenil)-1-metil-7-(2-metil-6-nitrofenilamino)-3,4-dihidropirimido [4,5-d] pirimidin-2 (1 H)-ona

A una mezcla de 3-(2,6-dicloro-3,5-dimetoxifenil)-1-metil-7-(metilsulfonil)-3,4-dihidropirimido [4,5-d]pirimidin-2(1H)-ona (450 mg, 1,01 mmol) y 2-metil-6-nitroanilina (230 mg, 1,51 mmol) en 5 ml de N, N-dimetilformamida se añadió terc-butanolato de potasio (339 mg, 3,02 mmol) a TA y se agita durante 0,5 h. LCMS mostró que la reacción se completó. La mezcla se inactivó con 80 ml de agua y el precipitado se recogió mediante filtración y se secó para dar el compuesto del título (290 mg, 56%) como un sólido amarillo, que se usó directamente en la siguiente etapa sin purificación adicional. MS (ES+) C22H2üCl2N6O5 requiere: 518, 520, encontrado: 519, 521 [M H]+.
Síntesis de (7-(2-amino-6-metilfenilamino)-3-(2,6-dicloro-3,5-dimetoxifenil)-1-metil-3,4-dihidropirimido[4,5-d] pirimidin-2(1H)-ona

Una mezcla de 3-(2,6-dicloro-3,5-dimetoxifenil)-1-metil-7-(2-metil-6-nitrofenilamino)-3,4-dihidropirimido [4,5-d] pirimidin-2(1H)-ona (290 mg, 0,56 mmol) en etanol (10 ml) y agua (2 ml) se agitó a 70 °C durante 20 minutos antes de añadir polvo de hierro (320 mg, 5,60 mmol) y cloruro de amonio (250 mg, 2,79 mmol). La mezcla de reacción se agitó a 70°C durante otras 6 h. LCMS mostró que la reacción se completó. Se filtró el sólido y se concentró el filtrado. El residuo se disolvió en acetato de etilo (30 ml), se lavó con agua y salmuera, se secó sobre sulfato de sodio, se filtró y se concentró. El residuo se purificó mediante Prep-HPLC para dar el compuesto del título (27 mg, 10%) como un sólido blanco. MS (ES+) C22H22C^N6O3requiere: 488, 490, encontrado: 489, 491 [M H]+. 1H-RMN
(500 MHz, CDCI3) 5 ppm 7,89 (s, 1H), 7,04 (t, 1H, J = 8,0 Hz), 6,69 (d, 2H, J = 7,5 Hz), 6,60 (s, 1H), 4,53 (s, 2 H), 3,94 (s, 6 H), 3,34 (s, 3 H), 2,24 (s, 3 H).
Síntesis de N-(2-((6-(2,6-didoro-3,5-dimetoxifenil)-8-metil-7-oxo-5,6,7,8-tetrahidropirimido [4,5-d]pirimidin-2-il)amino)-3-metilfenil acrilamida

N-(2-((6-(2,6-didoro-3,5-dimetoxifenil)-8-metil-7-oxo-5,6,7,8-tetrahidropirimido [4,5-d] pirimidin-2-il)amino)-3-metilfenil)acrilamida se preparó usando un procedimiento similar al del COMPUESTO 30. El producto se purificó mediante cromatografía ultrarrápida usando un gradiente de MeOH/DCM al 0-10% para dar el compuesto del título. MS (ES+) C25H24Cl2NsO4 requiere: 542, encontrado: 543 [M H]+. 1 H-RMN (400 MHz, DMSO-d a) 59,48 (s, 1 H), 8,35 (s, 1 H), 7,99 (s, 1 H), 7,66 (s, 1 H), 7,16 (t, J = 7,8 Hz, 1 H), 7,10 - 7,06 (m, 1 H), 6,99 (s, 1 H), 6,53 (dd, J = 17,0, 10,2 Hz, 1 H), 6,22 (dd, J = 16,9, 2,1 Hz, 1 H), 5,71 (dd, J = 10,2, 2,0 Hz, 1 H), 4,48 (s, 2 H), 3,96 (s, 6 H), 3,44 (s, 3 H), 2,17 (s, 3 H).
Ejemplo 7: Síntesis de COMPUESTO 24 y COMPUESTO 6

Síntesis de 5

A una solución de 5-bromo-2-doropirimidina (1,5 g, 7,89 mmol) y 2-metil-6-nitroanilina (800 mg, 5,26 mmol) en N, N-dimetilformamida (10 ml) en un tubo sellado se añadió terc-butóxido de potasio (1,76 g, 15,78 mmol) y la mezcla se calentó en microondas a 130°C durante 2 h. LCMS mostró que la reacción se completó. La reacción se enfrió a TA, se inactivó con agua (20 ml) y se extrajo con acetato de etilo (3 x 100 ml). Las capas orgánicas se separaron, combinaron, lavaron con agua (50 ml) y salmuera (100 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en gel de sílice (éter de petróleo: acetato de etilo = 10:1) para producir el compuesto del título como un sólido amarillo (500 mg, 31%). MS (ES+) CnHgBrN4O2 requiere: 309, 311, encontrado: 310, 312 [M H]+.
Síntesis de 5-((3,5-dimetoxifenil) etinil)-N-(2-metil-6-nitrofenil)pirimidin-2-amina

Una mezcla de 5-bromo-N-(2-metil-6-nitrofenil)pirimidin-2-amina (573 mg, 3,0 mmol), 1-etinil-3,5-dimetoxibenceno (483 mg, 3,0 mmol), trifenilfosfina (157 mg, 0,60 mmol), cloruro de bis(trifenilfosfina) paladio (II) (210 mg, 0,30 mmol), yoduro de cobre (I) (57 mg, 0,30 mmol) y dietilamina (1,50 ml, 15,0 mmol) en N, N-dimetilformamida (10 ml) se desgasificó con nitrógeno tres veces y a continuación se agitó a 80°C durante 2 h. LCMS mostró que la reacción se completó. La mezcla se enfrió a TA, se inactivó con agua (20 ml) y se extrajo con acetato de etilo (3 x 80 ml). Las capas orgánicas combinadas se separaron, se lavaron con agua y salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en gel de sílice (éter de petróleo: acetato de etilo = 4: 1) para producir el compuesto del título como un sólido amarillo (460 mg, 39%). C21H18N4O4 requiere: 390, encontrado: 391 [M H]+.
Síntesis de N 1 -(5-((3,5-dimetoxifenil) etinil)pirimidin-2-il)-6-metilbenceno-1,2-diamina

Una mezcla de 5-((3,5-dimetoxifenil) etinil)-N-(2-metil-6-nitrofenil)pirimidin-2-amina (150 mg, 0,38 mmol), hierro (171 mg, 3,04 mmol) y cloruro de amonio (246 mg, 4,56 mmol) en etanol (20 ml) y agua (2 ml) se agitó a 85°C durante 1 h. LCMS mostró que la reacción se completó. La reacción se enfrió a TA y el sólido se filtró. El filtrado se concentró y el residuo se purificó mediante Prep-HPLC para producir el compuesto del título como un sólido blanco (55 mg, 44%). MS (ES+) C21H20N4O2 requiere: 360, encontrado: 361 [M H]+. 1 H-RMN (500 MHz, DMSO-d6) 6 ppm 8,76 (s, 1 H), 8,50-8,46 (ancho, 2 H), 6,88 (t, 1 H, J = 7,0 Hz), 6,66 (s, 2 H), 6,57 (d, 1 H, J = 7,5 Hz), 6,54 (s, 1 H), 6,44 (d, 1 H, J = 6,5 Hz), 4,74 (s, 2 H), 3,76 (s, 6 H), 2,01 (s, 3 H).
Síntesis de N-(2-((5-((3,5-dimetoxifenil)etinil)pirimidin-2-il)amino)-3-metilfenil)acrilamida

Se preparó N1 -(5-((3,5-dimetoxifenil)etinil)pirimidin-2-il)-6-metilbenceno-1,2-diamina usando un procedimiento similar al COMPUESTO 30. El producto se purificó mediante cromatografía ultrarrápida usando gradiente de EtOAc/hexanos al 0-100% para dar el compuesto del título. MS (ES+) C24H22N4O3 requiere: 414, encontrado: 415 [M H]+. 1H-RMN (400 MHz, DMSO-d6) 6 ppm 9,60-9,38 (m, 1H), 8,79 (s, 1H), 8,51 (s, 2H), 7,69 (d, J = 8,1 Hz, 1H), 7,19 (t, J = 7,8 Hz, 1 H), 7,15 - 7,06 (m, 1 H), 6,67 (d, J = 2,3 Hz, 2 H), 6,60 - 6,45 (m, 2 H), 6,22 (dd, J = 17,0, 2,1 Hz, 1 H), 5,71 (dd, J = 10,2, 2,1 Hz, 1 H), 3,76 (s, 6 H), 2,12 (s, 3 H).
Síntesis de 5-((2,6-dicloro-3,5-dimetoxifenil) etinil)-N-(2-metM-6-nitrofenil)pirimidin-2-amina

A una solución de 5-((3,5-dimetoxifenil)etinil)-N-(2-metil-6-nitrofenil)pirimidin-2-amina (50 mg, 0,13 mmol) en acetonitrilo (5 ml) se añadió gota a gota cloruro de sulfurilo (44 mg, 0,33 mmol) a -20°C y la mezcla se agitó durante otros 10 minutos. LCMS mostró que la reacción se completó y la reacción se inactivó con agua (0,5 ml). Se evaporaron los disolventes y se purificó el residuo mediante HPLC preparativa para producir el compuesto del título en forma de un sólido amarillo (30 mg, 50%). (MS (ES+) C21H16C12N4O4 requiere: 459, 461, encontrado: 460, 462 [M H]+;
Síntesis de N1 -(5-((2,6-dicloro-3,5-dimetoxifenil) etinil)pirimidin-2-il)-6-metilbenceno-1,2-diamina

Una mezcla de 5-((2,6-dicloro-3,5-dimetoxifenil) etinil)-N-(2-metil-6-nitrofenil)pirimidin-2-amina (150 mg, 0,33 mmol), hierro (147 mg, 2,64 mmol) y cloruro de amonio (214 mg, 3,96 mmol) en etanol (20 ml) y agua (2 ml) se agitó a 85°C durante 1 h. LCMS mostró que la reacción se completó. La reacción se enfrió a TA y el sólido se filtró. El filtrado se concentró y el residuo se purificó mediante Prep-HPLC para producir el compuesto del título como un sólido blanco (58 mg, 35%). MS (ES+) C2-iH1aCl2N4O2 requiere: 429, 431, encontrado: 430, 432 [M H]+. 1 H-RMN (400 MHz, DMSO-d6) 8 ppm 8,90 (s, 1 H), 8,55-8,44 (ancho, 2 H), 6,97 (s, 1 H), 6,89-6,86 (m, 1 H), 6,57 (d, 1 H, J = 7,6 Hz), 6,44 (d, 1 H, J = 7,6 Hz), 4,75 (s, 2 H), 3,94 (s, 6 H), 2,01 (s, 3 H).
Síntesis de N-(2-((5-((2,6-dicloro-3,5-dimetoxifenil) etinil)pirimidin-2-il)amino)-3-metilfenil)acrilamida

Se preparó N-(2-((5-((2,6-dicloro-3,5-dimetoxifenil)etinil)pirimidin-2-il)amino)-3-metilfenil)acrilamida usando un procedimiento similar al del COMPUESTO 30. El producto se purificó mediante cromatografía ultrarrápida usando un gradiente de EtOAc/Hexanos al 0-100% para dar el compuesto del título. MS (ES+) C24H20ChN 4O3 requiere: 482, encontrado: 483 [M H]+. 1H-RMN (400 MHz, DMSO-d6) 8 ppm 9,47 (s, 1 H), 8,93 (s, 1 H), 8,54 (s, 2 H), 7,71 (d, J = 8,1 Hz, 1 H), 7,19 (t, J = 7,8 Hz, 1 H), 7,09 (d, J = 7,4 Hz, 1 H), 6,98 (s, 1 H), 6,53 (dd, J = 17,0, 10,2 Hz, 1 H), 6,22 (dd, J = 17,0, 2,1 Hz, 1 H), 5,70 (dd, J = 10,2, 2,1 Hz, 1 H), 3,94 (s, 6 H), 2,13 (s, 3 H).
Ejemplo 8: Síntesis del COMPUESTO 40

Síntesis de 2-metil-3-oxo entanodioato de dietilo

A una solución de 3-oxopentanodioato de dietilo (23,2 g, 114,8 mmol) en tetrahidrofurano (100 ml) se añadió hidruro de sodio (60%, 4,8 g, 120,5 mmol) a 0 °C, y la mezcla de reacción se agitó a TA durante 30 minutos, seguido de la adición de yodometano (7,15 ml, 114,8 mmol). La mezcla de reacción se agitó a TA durante 48 h, se inactivó con agua (500 ml) y se extrajo con acetato de etilo (500 ml x 3). Las capas orgánicas se separaron, se combinaron, se lavaron con agua (200 ml) y salmuera (200 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó mediante una columna de gel de sílice (éter de petróleo: acetato de etilo = 20: 1) para obtener el compuesto del título como un aceite incoloro (9 g, 36%). MS (ES+) C10H16O5 requiere: 216, encontrado: 217 [M H]+.
Síntesis de 4-hidroxi-5-metil-6-oxo-1,6-dihidro iridin-3-carboxilato de etilo

A una solución de 2-metil-3-oxopentanodioato de dietilo (10 g, 46,25 mmol) en 1,1-trioxidandiildipropan-1-ona (400 ml) se añadió trietoximetano (38 ml, 231,25 mmol), y la mezcla se calentó a 120°C durante 4 h, seguido de la adición de amoniaco al 30% (600 ml) a 0°C. La mezcla de reacción se agitó a TA durante otras 2 h. LCMS controló que la reacción se completó. El precipitado se recogió mediante filtración y se disolvió en diclorometano (400 ml). El sólido se retiró por filtración y el filtrado se concentró para obtener el compuesto del título (5,5 g, bruto) como un sólido amarillo. MS (ES+) C9H11NO4 requiere: 197, encontrado: 198 [M H]+.
, - - -
Una solución de 4-hidroxi-5-metil-6-oxo-1,6-dihidropiridina-3-carboxilato de etilo (5,0 g, 21,4 mmol) en tricloruro de fosforilo (100 ml) se calentó a reflujo a 125 °C durante 12 h. LCMS controló que la reacción se completó. La mayor parte del tricloruro de fosforilo se evaporó y el residuo se añadió gota a gota a agua helada (100 ml). La mezcla resultante se neutralizó con una solución acuosa de carbonato de sodio (50 mL) y se extrajo con acetato de etilo (200 mL). La capa orgánica se separó, se combinó, se lavó con agua y salmuera, se secó sobre sulfato de sodio, se filtró y se concentró. El residuo se purificó mediante una columna de gel de sílice (éter de petróleo: acetato de etilo = 15: 1) para obtener el compuesto del título (1,6 g, 32%) en forma de un aceite amarillo. MS (ES+) CgHgCl2NO2 requiere: 232, 234, encontrado: 233, 235 [M H]+.
Síntesis de 6-cloro-5-metil-4-(metilamino) nicotinato de etilo

A una solución de 4,6-dicloro-5- metilnicotinato de etilo (2,6 g, 11,1 mmol) en acetonitrilo (60 ml) se le añadió gota a gota metilamina al 40% en agua (689 mg, 22,2 mmol, 60 ml), y la mezcla se agitó a 50°C durante 72 h. LCMS controló que la reacción se completó. La mezcla de reacción se concentró y se extrajo con acetato de etilo (100 ml). La capa orgánica se separó, se combinó, se lavó con agua y salmuera, se secó sobre sulfato de sodio, se filtró y se concentró. El residuo se purificó mediante una columna de gel de sílice (éter de petróleo: acetato de etilo = 2: 1) para obtener el compuesto del título (2,05 g, 81%) como un aceite incoloro. MS (ES+) C1üH13ClN2O2 requiere: 228, 230, encontrado: 229, 231 [M H]+.
Síntesis de (6-cloro-5-metil-4-(metilamino)piridin-3-il)metanol

A una solución de 6-cloro-5-metil-4-(metilamino)nicotinato de etilo (2,0 g, 8,8 mmol) en tetrahidrofurano (60 ml) se añadió hidruro de litio y aluminio a 0 °C, y la mezcla se agitó a TA durante 1,5 h. LCMS controló que la reacción se completó. La reacción se detuvo con sulfato de sodio decahidratado (1,5 g) y se filtró. El filtrado se concentró para obtener el compuesto del título (1,4 g, bruto) como un sólido blanco. MS (ES+) CeH-nCl^O requiere: 186, 188, encontrado: 187, 189 [M H]+.
Síntesis de 6-cloro-5-metil-4-(metilamino) nicotinaldehído

Una mezcla de (6-cloro-5-metil-4-(metilamino) piridin-3-il) metanol (1,4 g, 8,0 mmol) y óxido de manganeso (2,8 g, 32 mmol) en diclorometano (100 ml) se agitó a TA durante 4 h. LCMS controló que la reacción se completó. El sólido se
retiró por filtración y el filtrado se concentró para obtener el compuesto del título (1,2 g, bruto) como un aceite amarillo. MS (ES+) C8HgClN2O requiere: 184, 186, encontrado: 185, 187 [M H]+.
Síntesis de 7-cloro-3- 3,5-dimetoxifenil -1,8-dimetil-1,6-naftiridin-2 1H -ona

Una mezcla de 6-cloro-5-metil-4-metil(metilamino) nicotinaldehído (3,11 g, 16,8 mmol), 2-(3,5-dimetoxifenil)acetato de etilo (4,25 g, 20,2 mmol) y carbonato de potasio (2,8 g, 20,3 mmol) en N, N-dimetilformamida (100 ml) se agitó a 105°C durante la noche. LCMS controló que la reacción se completó. La mezcla de reacción se enfrió a TA y se inactivó con agua. El precipitado se filtró y se secó para obtener el compuesto del título (5,5 g, bruto) como un sólido amarillo. MS (ES+) Ci8Hi7ClN2O3 requiere: 344, 346, encontrado: 345, 347 [M H]+.
Síntesis de 3-(3,5-dimetoxifenil)-1,8-dimetil-7- 2-nitrofenilamino -16-naftiridin-2 1 H -ona

Una mezcla de 7-cloro-3-(3,5-dimetoxifenil)-1,8-dimetil-1,6-naftiridin-2(1H)-ona (800 mg, 2,32 mmol), 2-nitrobencenamina (320 mg, 2,32 mmol), Pd2(dba)3 (100 mg), John-Phos (100 mg) y terc-butanolato de potasio (480 mg, 4,64 mmol) en N, N-dimetilformamida (10 ml) se calentó en tubo sellado a 100 °C en microondas durante 1 h. LCMS controló que la reacción se completó. La mezcla se concentró y se purificó mediante HPLC preparativa para obtener el compuesto del título (150 mg, 15%) en forma de un sólido marrón. MS (ES+) C24H22N¿O5 requiere: 446, encontrado: 447 [M H]+.
Síntesis de 3-(2,6-dicloro-3,5-dimetoxifenil)-1,8-dimetil-7-(2-nitrofenilamino)-1,6-naftiridin-2 (1H) -ona

A una solución de 3-(3,5-dimetoxifenil)-1,8-dimetil-7-(2-nitrofenilamino)-1,6-naftiridin-2 (1H)-ona (120 mg, 0,27 mmol) en acetonitrilo (120 ml) se añadió cloruro de sulfurilo (185 mg, 1,35 mmol) a -15°C y la mezcla se agitó a -15°C durante 10 minutos. LCMS controló que la reacción se completó. La mezcla de reacción se inactivó con agua (1 ml) y se concentró. El precipitado se recogió mediante filtración, se lavó con acetona/éter de petróleo (1: 5) y se secó para dar el compuesto del título (100 mg, 72%) como un sólido blanco. MS (ES+) C24H20Ci2N4O5 requiere: 514, 516, encontrado: 515, 517 [M H]+.
Síntesis de 7-(2-aminofenilamino)-3-(2,6-didoro-3,5-dimetoxifenil)-1,8-dimetil-1,6-naftiridin-2(1H)-ona

A una solución de 3-(2,6-didoro-3,5-dimetoxifenil)-1,8-dimetil-7-(2-nitrofenilamino)-1,6-naftiridin-2(1H)-ona (100 mg, 0,2 mmol) en acetato de etilo (20 ml) se añadió cloruro estannoso (150 mg, 0,8 mmol) y la mezcla se agitó a 80°C durante 1 h. LCMS controló que la reacción se completó. Se filtró el sólido y se concentró el filtrado. El residuo se purificó mediante Prep-HPLC para obtener el compuesto del título (38,6 mg, 41%) en forma de un sólido amarillo. MS (ES+) C24H22Cl2N4O3 requiere: 484, 486, encontrado: 485, 487 [M H]+; 1 H-RMN (500 MHz, DMSO-d a) 5 ppm 8,24 (s, 1 H), 7,76 (s, 1 H), 7,67 (s, 1 H), 7,03 (d, 1 H, J = 7,5 Hz), 6,97 (s, 1 H), 6,92-6,89 (m, 1 H)), 6,75 6,73 (m, 1 H), 6,57-6,54 (m, 1 H), 4,77 (s, 2 H), 3,95 (s, 6 H), 3,66 (s, 3 H), 2,43 (s, 3 H).
Síntesis de N-(2-((3-(2,6-dicloro-3,5-dimetoxifenil)-1,8-dimetil-2-oxo-1,2-dihidro-1,6-naftiridin-7-il)amino)fenil) acrilamida

Se preparó N-(2-((3-(2,6-dicloro-3,5-dimetoxifenil)-1,8-dimetil-2-oxo-1,2-dihidro-1,6-naftiridin-7-il)amino)fenil) acrilamida usando un procedimiento similar al del COMPUESTO 30. El producto se purificó mediante cromatografía en capa fina preparativa usando un gradiente de MeOH/DCM al 0-5% para dar el compuesto del título. MS (ES+) C27H24ChN4O4 requiere: 538, encontrado: 539 [M H]+.
Ejemplo 9: Síntesis del COMPUESTO 42

Síntesis de (2-amino-4-metoxifenil) metanol

A una solución de ácido 2-amino-4-metoxibenzoico (15,0 g, 89,8 mmol) en THF (300 ml) se añadió borohidruro en THF (450 ml, 450 mmol) a 0 °C, y la mezcla de reacción se agitó a TA durante la noche. LCMS mostró que la reacción se completó. La reacción se inactivó con agua (150 ml) y se extrajo con acetato de etilo (500 ml x 3). Las capas orgánicas se separaron, se combinaron, se lavaron con agua (200 ml) y salmuera (200 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron para producir el compuesto del título. MS (ES+) C8H11NO2 requiere: 153, encontrado: 154 [M H]+.
Síntesis de 2-amino-4-metoxibenzaldehído

Una mezcla de (2-amino-4-metoxifenil) metanol (20 g, 131,0 mmol) y óxido de manganeso (68 g, 786,0 mmol) en diclorometano (300 ml) se agitó a TA durante una noche. LCMS mostró que la reacción se completó. Se filtró el
sólido y se concentró el filtrado. El residuo se purificó mediante cromatografía en gel de sílice (éter de petróleo: acetato de etilo = 6:1) para dar el compuesto del título (7 g, 35%) como un sólido amarillo. MS (ES+) C8H9NO2 requiere: 151, encontrado: 152 [M H]+.
Síntesis de 2-amino-5-bromo-4-metoxibenzaldehído

A una solución agitada de 2-amino-4-metoxibenzaldehído (6 g, 39,7 mmol) en diclorometano (100 ml) se añadió N-bromosuccinimida (7 g, 39,7 mmol). LCMS controló la reacción hasta que el material de partida se consumió por completo. La mezcla de reacción se diluyó con diclorometano y agua. La capa orgánica separada se secó con sulfato de sodio, se filtró y se concentró para dar el compuesto del título (5 g, 56%) como un sólido amarillo. MS (ES+) C8H8BrNO2 requiere: 229, 231, encontrado: 230, 232 [M H]+.
Síntesis de 6-bromo-7-metoxiquinazolin-2-ol

Una mezcla de 2-amino-5-bromo-4-metoxibenzaldehído (3 g, 13,1 mmol) y urea (12 g, 196,5 mmol) se agitó a 180 °C durante 2 h. LCMS mostró que la reacción se completó. La mezcla de reacción se enfrió a TA y se lavó con agua (3 x 100 mL). El precipitado se recogió y se secó para dar el compuesto del título (3 g, bruto) como un sólido amarillo. MS (ES+) C8H7BrN2O2 requiere: 254, 256, encontrado: 255, 257 [M H]+.
Síntesis de 6-bromo-2-cloro-7-metoxiquinazolina

A una solución de 6-bromo-7-metoxiquinazolin-2-ol (3,0 g, 11,8 mmol) en tricloruro de fosforilo (30 ml) se calentó a reflujo a 130 °C durante 5 h. LCMS mostró que la reacción se completó. La reacción se enfrió a TA y se evaporó la mayor parte del tricloruro de fosforilo. El residuo se añadió gota a gota a agua helada (100 ml) y el precipitado resultante se recogió mediante filtración para dar el compuesto del título como un sólido amarillo (2,4 g, 75%). MS (ES+) C9H6BrClN2O requiere: 272, 274, encontrado: 273, 275 [M H]+.
Síntesis de 2-cloro-6-3,5-dimetoxifenil -7-metoxi uinazolina

Una mezcla de 6-bromo-2-cloro-7-metoxiquinazolina (2,4 g, 8,82 mmol), ácido 3,5-dimetoxifenilborónico (1,6 g, 8,82 mmol), carbonato de cerio (8,6 g, 26,46 mmol) y Pd (PPh3)2Cl2 se desgasificó (1,4 g, 2,1 mmol) en t Hf (10 ml), dioxano (10 ml) y agua (2 ml) con nitrógeno tres veces y se agitó a 85°C durante 3 h. LCMS controló que la reacción
se completó. La mezcla se enfrió a TA y se extrajo con diclorometano (3 x 50 ml). Las capas orgánicas se separaron, se combinaron, se lavaron con agua y salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en gel de sílice (éter de petróleo: acetato de etilo = 1: 4) para dar el compuesto del título (1,1 g, 38%) como un sólido blanco. MS (ES+) C17H15ClN2O3 requiere: 330, 332, encontrado: 331, 333 [M H]+.
Síntesis de 2-cloro-6- 2,6-dicloro-3,5-dimetoxifenil -7-metoxi uinazolina
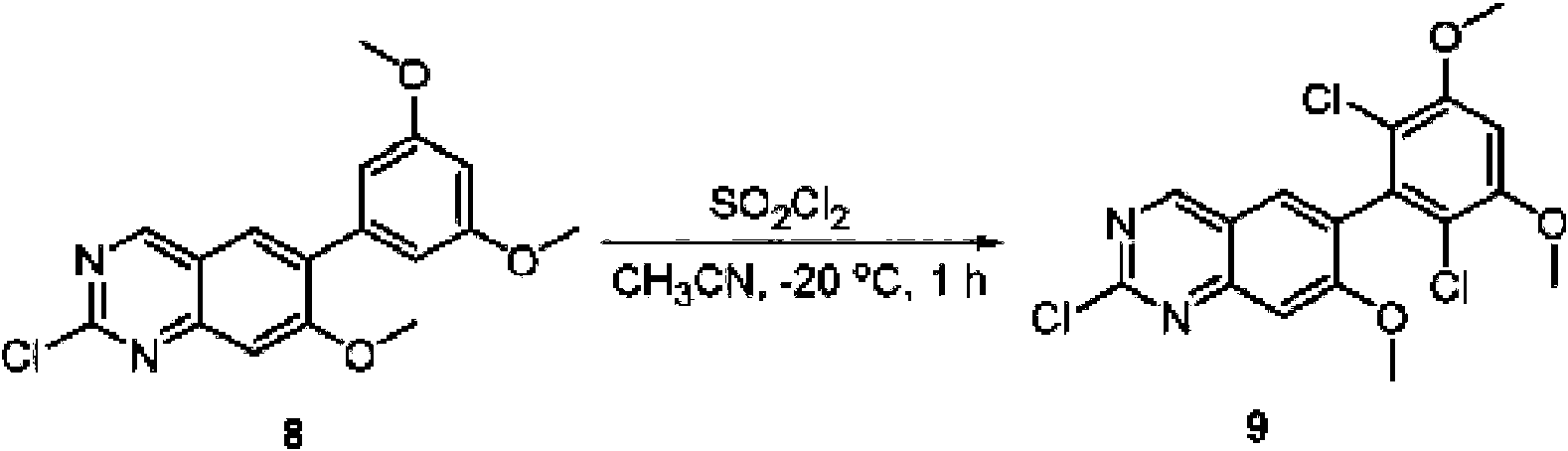
A una solución de 2-cloro-6-(3,5-dimetoxifenil)-7-metoxiquinazolina (200 mg, 0,61 mmol) en acetonitrilo (5 ml) se añadió cloruro de sulfurilo (205 mg, 1,52 mmol), y la mezcla se agitó a -20°C durante 1 h. La reacción se inactivó con agua (1 ml) y se concentró a presión reducida. El precipitado se lavó con acetonitrilo y se secó para dar el compuesto del título como un sólido blanco (120 mg, 50%). MS (ES+) C17H13CbN2O3 requiere: 398, encontrado: 399, 401 [M H]+; 1H-RMN (400 MHz, DMSO-d a) 5 ppm 9,43 (s, 1H), 8,02 (s, 1H), 7,55 (s, 1H), 7,03 (s, 1H), 3,98 (s, 6H), 3,93 (s, 3 H).
Síntesis de 6-(2,6-dicloro-3,5-dimetoxifenil)-7-metoxi-N-(2-metil-6-nitrofenil) quinazolin-2-amina

Se preparó 6-(2,6-dicloro-3,5-dimetoxifenil)-7-metoxi-N-(2-metil-6-nitrofenil)quinazolin-2-amina usando un procedimiento similar al del COMPUESTO 30. El producto se purificó mediante cromatografía ultrarrápida usando un gradiente de EtOAc/Hexanos al 0-100% para dar el compuesto del título. MS (ES+) C24H20CW4O5 requiere: 514, encontrado: 515 [M H]+.
Síntesis de N1-(6-(2,6-dicloro-3,5-dimetoxifenil)-7-metoxiquinazolin-2-il)-6-metilbenceno-1,2-diamina

Se preparó N1-(6-(2,6-dicloro-3,5-dimetoxifenil)-7-metoxiquinazolin-2-il)-6-metilbenceno-1,2-diamina usando un procedimiento similar al del COMPUESTO 30 La reacción se filtró a través de celite para dar un producto en bruto. MS (ES+) C24H22ChN4O3 requiere: 484, encontrado: 485 [M H]+.
Síntesis de N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)-7-metoxiquinazolin-2-il)amino)-3-metilfenil)acrilamida

Se preparó N-(2-((6-(2,6-didoro-3,5-dimetoxifenil)-7-metoxiquinazolin-2-il)amino)-3-metilfenil)acrilamida usando un procedimiento similar al del COMPUESTO 30. El producto se purificó mediante cromatografía ultrarrápida usando un gradiente de MeOH/DCM al 0-10% para dar el compuesto del título. MS (ES+) C27H24Cl2N4O4 requiere: 538, encontrado: 539 [M H]+.
Ejemplo 10: Síntesis del COMPUESTO 34
Síntesis de ácido 2-amino-5-bromo-3-fluorobenzoico

Se añadió una solución de ácido 2-amino 3--fluorobenzoico (10,85 g, 70 mmol) en diclorometano (175 ml) de N-bromosuccinimida (12,46 g, 70 mmol), y la mezcla se agitó a TA durante 2 h. LCMS mostró que la reacción se
completó. El precipitado se filtró y se lavó con diclorometano (100 mlx3) para dar el compuesto del título (12,7 g, 78%) como un sólido gris, que se usó directamente en la siguiente etapa sin purificación adicional. MS (ES+) CyH5BrFNO2 requiere: 233, 235, encontrado: 232, 234 [M - H]-.
Síntesis de (2-amino-5-bromo-3-fluorofenil)metanol

A una solución de ácido 2-amino-5-bromo-3-fluorobenzoico (14,5 g, 62,2 mmol) en THF (150 ml) a 0 °C se añadió borohidruro en THF (1 M, 310 ml), y la mezcla de reacción se agitó a TA durante la noche. LCMS mostró que la reacción se completó. La reacción se inactivó con metanol (150 ml), se concentró al vacío, se diluyó con bicarbonato de sodio acuoso (400 ml) y se extrajo con acetato de etilo (200 ml x 3). Las capas orgánicas se separaron, se combinaron, se lavaron con agua (200 ml) y salmuera (200 ml), se secaron sobre sulfato de sodio, se filtraron y se concentraron para producir el compuesto del título (13,0 g, bruto), que se usó directamente en el siguiente paso sin la purificación adicional. MS (ES+) CzHzBrFNO requiere: 219, 221, encontrado: 220, 222 [M H] .
Síntesis de 2-amino-5-bromo-3-fluorobenzaldehído

Una mezcla de (2-amino-5-bromo-3-fluorofenil) metanol (13 g, 59,4 mmol) y óxido de manganeso (31 g, 356,4 mmol) en diclorometano (400 ml) se agitó a TA durante una noche. La TLC mostró que el material de partida se consumió por completo. El sólido se separó por filtración y el filtrado se concentró para dar el compuesto del título (11 g, 85%) en forma de un sólido de color amarillo claro, que se usó directamente en la siguiente etapa sin purificación adicional.
Síntesis de 6-bromo-8-fluoroquinazolin-2-ol

Una mezcla agitada de 2-amino-5-bromo-3-fluorobenzaldehído (2,17 g, 10 mmol) y urea (9 g, 150 mmol) se calentó a 180 °C durante 2 h. LCMS mostró que la reacción se completó. La mezcla de reacción se enfrió a TA y el precipitado resultante se filtró y se lavó con agua (500 m lx 3). La humedad atrapada se eliminó por completo mediante la coevaporación con tolueno tres veces. Se obtuvo el compuesto del título (2 g, 83%) como un sólido amarillo. MS (ES+) C a^B rF^O requiere: 242, 244, encontrado: 243, 245 [M H]+.
Síntesis de 6-bromo-2-cloroquinazolina

Una solución de 6-bromoquinazolin-2-ol (9,72 g, 40 mmol) en oxicloruro de fósforo (100 ml) se calentó a reflujo durante 5 h. LCMS mostró que la reacción se completó. La reacción se enfrió a TA y la mayor parte del oxicloruro de fósforo se eliminó a presión reducida. El residuo se añadió gota a gota a agua helada (500 ml) y el precipitado resultante se recogió mediante filtración para dar el compuesto del título (9 g, 87%) como un sólido amarillo. MS (ES+) CsH3BrClFN2 requiere: 260, 262, encontrado: 261,263 [M H]+.
Síntesis de 2-cloro-6-(3,5-dimetoxifenil)-8-fluoroquinazolina

Una mezcla de 6-bromo-2-cloro-8-fluoroquinazolina (4,0 g, 15,4 mmol), ácido 3,5-dimetoxifenilborónico (4,47 g, 16,9 mmol), carbonato de cesio (10,0 g, 30,8 mmol) y Pd (PPh3)2Ch (236 mg, 0,77 mmol) en THF (200 ml) y agua (10 ml) se desgasificó con nitrógeno tres veces, y se agitó a 80 °C durante 3 h. Tanto la TLC como la LCMS mostraron que la reacción se completó. La mezcla de reacción se enfrió a TA y se concentró directamente. El residuo se purificó mediante cromatografía en gel de sílice (éter de petróleo: diclorometano = 2:1 a 1:1) para producir el compuesto del título (2,5 g, 51%) en forma de un sólido amarillo. MS (ES+) C16H12CFN2O2 requiere: 318/320, encontrado: 319/321 [M H]+.
Síntesis de 2-cloro-6-(2,6-dicloro-3,5-dimetoxifenil)-8-fluoroquinazolina
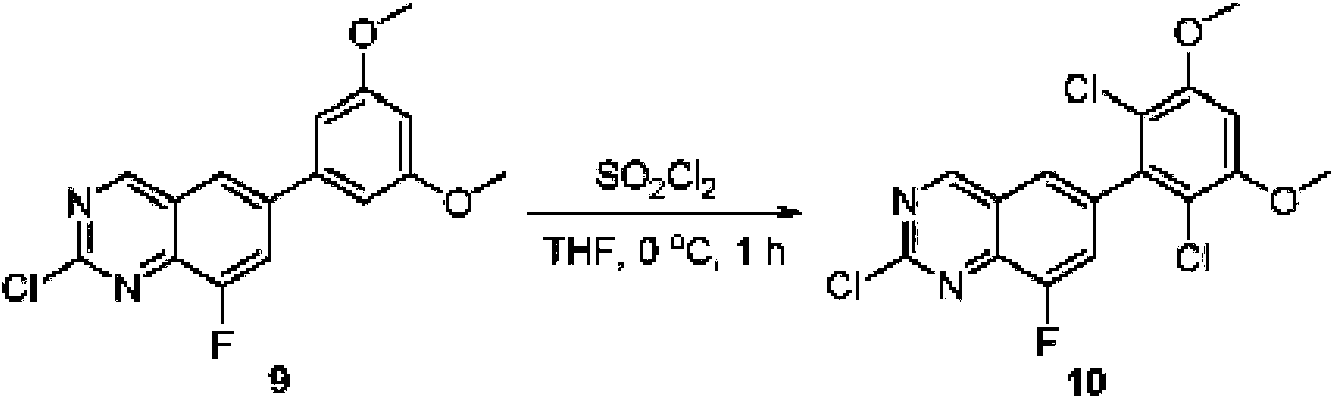
A una solución de 2-cloro-6-(3,5-dimetoxifenil)-8-fluoroquinazolina (1,5 g, 4,7 mmol) en THF seco (40 ml) se añadió cloruro de sulfurilo gota a gota (1,59 g, 1,75 mmol) a 0°C y la mezcla se agitó durante 1 h. Tanto la TLC como la LCMS mostraron que la reacción se completó. La reacción se inactivó con agua (1 ml) y los disolventes se eliminaron a presión reducida. El residuo se lavó con acetonitrilo y se secó para dar el compuesto del título (700 mg, 38%) como un sólido blanco. (MS (ES+) C16H1üCl3FN2O2 requiere: 386, 388, encontrado: 387, 389 [M H]+; 1H-RMN (400 MHz, DMSO-d6) 8 ppm 9,74 (d, 1 H J = 1,0 Hz), 8,03-7,99 (m, 2 H), 7,08 (s, 1 H), 4,00 (s, 6 H).
Síntesis de 6-(2,6-didoro-3,5-dimetoxifenil)-8-fluoro-N-(2-metil-6-nitrofenil) quinazolin-2-amina

Se preparó 6-(2,6-didoro-3,5-dimetoxifenil)-8-fluoro-N-(2-metil-6-nitrofenil)quinazolin-2-amina usando un procedimiento similar al del COMPUESTO 30. El producto se purificó mediante cromatografía ultrarrápida usando un gradiente de EtOAc/Hexanos al 0-100% para dar el compuesto del título. MS (ES+) C23H17CbFN4O4 requiere: 502, encontrado: 503 [M H]+.

Se preparó N1-(6-(2,6-dicloro-3,5-dimetoxifenil)-8-fluoroquinazolin-2-il)-6-metilbenceno-1,2-diamina usando un procedimiento similar al del COMPUESTO 30 La reacción se filtró a través de celite para dar un producto en bruto. MS (ES+) C23H19CbFN4O2 requiere: 472, encontrado: 473 [M H]+.
Síntesis de N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)-8-fluoroquinazolin-2-il)amino)-3-metilfenil)acrilamida

Se preparó N-(2-((6-(2,6-dicloro-3,5-dimetoxifenil)-8-fluoroquinazolin-2-il)amino)-3-metilfenil)acrilamida usando un procedimiento similar al del COMPUESTO 30. El producto se purificó mediante cromatografía ultrarrápida (“flash”) usando un gradiente de MeOH/DCM al 0-10% para dar el compuesto del título. MS (ES+) C26H21C^FNaO3 requiere: 526, encontrado: 527 [M H]+. 1 H-RMN (400 MHz, DMSO-d6) 69,53 (d, J = 27,9 Hz, 1 H), 9,28 (s, 1 H), 8,96 (s, 1 H), 7,75 (d, J = 29,9 Hz, 1 H), 7,59 (d, J = 1,7 Hz, 1 H), 7,49 (d, J = 10,8 Hz, 1 H), 7,02 (s, 1 H), 6,50 (s, 1 H), 6,21 (dd, J = 16,9, 2,1 Hz, 1 H), 5,75 (s, 1 H), 5,68 (dd, J = 10,2, 2,0 Hz, 1 H), 3,98 (d, J = 4,6 Hz, 6 H), 2,19 (s, 3 H).
Ejemplo 10: Síntesis del COMPUESTO 50

Síntesis de 4-(2,5-dicloropirimidin~4~il)piperazina-l-carboxilato de tere-butilo

A una solución de 2,4,5-tridoropirimidina (0,475 g, 2,6 mmol) en DMF seca (8,5 ml) se añadió piperazina-1-carboxilato de terc-butilo (0,51 g, 2,7 mmol) seguido de DIEA (0,51 mL, 3,1 mmol) a 0°C y la mezcla se agitó durante 1 h. LCMS mostró que la reacción se completó. La reacción se diluyó con agua (100 ml) y el sólido blanco se filtró. El residuo se lavó con agua y se secó para dar el compuesto del título (445 mg, 51%) como un sólido blanco. MS (ES+) C13H18Cl2N4O2 requiere: 332, encontrado: 333 [M H]+
Síntesis de (2-((5-doro-4-(piperazin-1-il)pirimidin-2-il)amino)fenil) carbamato de terc-butilo

A una solución de 4-(2,5-didoropirimidin-4-il)piperazina-1-carboxilato de terc-butilo (0,1 g, 0,3 mmol) en DCM (1,0 ml) se añadió TFA (1,0 ml) y la mezcla se agitó durante 1 h. Se analizó una alícuota de la mezcla de reacción mediante LCMS, que indicó que la reacción se había completado hasta el final. Se eliminaron los disolventes y se secó el residuo a alto vacío. El producto en bruto se usó para el siguiente paso sin purificación adicional. A una solución de 2,5-dicloro-4-(piperazin-1-il)pirimidina (0,3 mmol) en dioxano (4,0 ml) se le añadió TFA (0,060 ml, 0,75 mmol) y (2-aminofenil)carbamato de terc-butilo (0,094 g, 0,45 mmol) y la mezcla se agitó a 100°C durante 24 h de reacción. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con EtOAc y se lavó con una solución acuosa saturada de bicarbonato de sodio. La mezcla orgánica se secó sobre sulfato de sodio y se cargó sobre gel de sílice y se purificó usando un gradiente de MeOH al 0-10%/DCM que contiene 10% de NH4OH para dar el compuesto del título (28 mg, 23%) como un sólido blanco. MS (ES+) C-igH25ClN6O2 requiere: 404, encontrado: 405 [M H]+
Síntesis de (2-((5-cloro-4-(4-((3-(trifluorometil)fenil)carbamoil)piperazin-1-il)pirimidin-2-il)amino)fenil)carbamato de terc-butilo

A una solución de (2-((5-cloro-4-(piperazin-1-il)pirimidin-2-il)amino)fenil)carbamato de terc-butilo (28 mg, 0,068 mmol) en DCM (0,7 mL) se añadió 1-isocanato-3-(trifluorometil)benceno (0,011 ml, 0,082 mmol) y trietilamina (0,015 ml, 0,1 mmol) y la mezcla se agitó a 23°C durante 16 h de reacción. La mezcla de reacción en bruto se cargó en gel de sílice y se purificó usando un gradiente de EtOAc/Hexanos al 0-50% para dar el compuesto del título (25 mg, 62%). MS (ES+) C27H29CF3N7O3 requiere: 591, encontrado: 592 [M H]
ntess e - - -acram oen amno- - oroprm n- - - - -tr uoromet en pperazna- -car oxam a

A una solución de (2-((5-cloro-4-(4-((3-(tnfluorometil)fenil)carbamoil)piperazm-1-il)pmmidin-2-il)ammo)fenil) carbamato de terc-butilo (0,025 g, 0,043 mmol) en DCM (1,0 ml), se añadió TFA (1,0 ml) y la mezcla se agitó durante 1 h. Se analizó una alícuota de la mezcla de reacción mediante LCMS, lo que indicó que la reacción se había completado. Se eliminaron los disolventes y se secó el residuo a alto vacío. El producto en bruto se usó para el siguiente paso sin purificación adicional.
A una solución de 4-(2-((2-aminofenil)amino)-5-cloropirimidin-4-il)-N-(3-(trifluorometil)fenil)piperazina-1-carboxamida (0,043 mmol) en DCM (0,5 mL) se añadió cloruro de acriloílo (0,004 mL, 0,052 mmol) y DiEA (0,018 mL, 0,11 mmol) y la mezcla se agitó a 0°C durante 1 h. La mezcla de reacción en bruto se cargó en gel de sílice y se purificó usando un gradiente de MeOH/DCM al 0-7% para dar el compuesto del título (10 mg, 43%). MS (ES+) C25H23CF3N7O2 requiere: 545, encontrado: 546 [M H]
Ejemplo 11: Síntesis del COMPUESTO 54

Síntesis de 4-(2-doro-5-metilpirimidin-4-il)piperazina-1-carboxilato de terc-butilo

A una solución de 2,4-dicloro-5-metilpirimidina (0,75 g, 4,6 mmol) en DMF seco (15,5 ml) se anadio piperazina-1-carboxilato de terc-butilo (0,9 g, 4,85 mmol) seguido de DIEA (0,91 ml, 5,5 mmol) a 0°C y la mezcla se agitó a temperatura ambiente durante la noche. LCMS mostró que la reacción se completó. La reacción se diluyó con agua (120 ml) y el sólido se filtró. El residuo se lavó con agua y se secó para dar el compuesto del título (1,386 g, 96%) como un sólido blanco. MS (ES+) C14H21CIN4O2 requiere: 312, encontrado: 313 [M H]+
Síntesis de ácido 4-((4-(4-(terc-butoxicarbonil)piperazin-1-il)-5-metilpirimidin-2-il)amino)-3-nitrobenzoico

Una mezcla de 4-(2-cloro-5-metilpirimidin-4-il)piperazin-1-carboxilato de terc-butilo (0,15 g, 0,48 mmol), ácido 4-amino-3-nitrobenzoico (97 mg, 0,53 mmol), mezcla de BrettPhos-Pd (20 mg, 0,015 mmol) y carbonato de cesio (470 mg, 1,44 mmol) en ‘BuOH (2,4 ml) se calentó en un tubo sellado a 100°C durante la noche. La mezcla se diluyó con EtOAc, se filtró a través de un lecho de celite, se cargó en gel de sílice y se purificó usando un gradiente de MeOH/DCM al 0-10% para dar el compuesto del título (75 mg, 34%). MS (ES+) C21H26N6O6 requiere: 458, encontrado: 459 [M H]
Síntesis de 4-(5-metil-2-((4-((1-metilpiperidin-4-il)carbamoil)-2-nitrofenil)amino)pirimidin-4-il)piperazina-1-carboxilato de terc-butilo
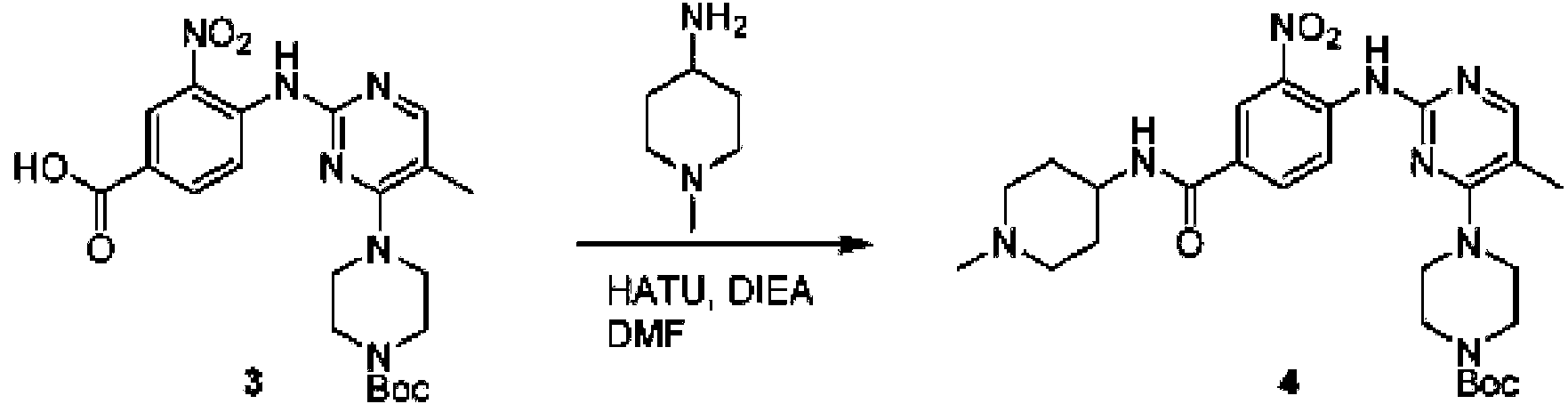
Una mezcla de ácido 4-((4-(4-(terc-butoxicarbonil)piperazin-l-il)-5-metilpirimidin-2-il)amino)-3-nitrobenzoico (0,075 g, 0,164 mmol), 1-metilpiperidin-4-amina (37 mg, 0,33 mmol), HATU (140 mg, 0,37 mmol) y DlEA (0,1 ml, 0,6 mmol) en DMF (3,0 ml) se agitó a temperatura ambiente durante la noche. La mezcla de reacción se diluyó con EtOAc, se lavó con una solución acuosa saturada de bicarbonato de sodio y una solución saturada de salmuera. La mezcla en bruto se cargó sobre gel de sílice y se purificó usando un gradiente de MeOH al 0-10%/DCM que contiene 10% de NH4OH para dar el compuesto del título (73 mg, 80%). MS (ES+) C27H38N8O 5 requiere: 554, encontrado: 555 [M H]+
Síntesis de N-(4-cianofenil)-4-(5-metil-2-((4-((1-metilpiperidin-4-il)carbamoil)-2-nitrofenil)amino)pirimidin-4-il)piperazina- 1-carboxamida

A una solución de 4-(5-metil-2-((4-((1-metilpiperidin-4-il)carbamoil)-2-nitrofenil)amino)pirimidin-4-il)piperazin 1-carboxilato de terc-butilo (0,073 g, 0,13 mmol) en DCM (1,0 ml) se añadió TFA (1,0 ml) y la mezcla se agitó durante 1 h. Se analizó una alícuota de la mezcla de reacción mediante LCMS, lo que indicó que la reacción se había completado hasta el final. Se eliminaron los disolventes y se secó el residuo a alto vacío. El producto en bruto se usó para el siguiente paso sin purificación adicional.
A una solución de 4-((5-metil-4-(piperazin-1-il)pirimidin-2-il)amino)-N-(1-metilpiperidin-4-il)-3-nitrobenzamida (0,073 mmol) en DCM (1,5 ml) se añadió 4-isocianatobenzonitrilo (23 mg, 0,16 mmol) y trietilamina (0,055 ml, 0,39 mmol) y la mezcla se agitó a 23°C durante 16 h. de reacción. La mezcla de reacción en bruto se filtró y se lavó con un volumen mínimo de DCM y a continuación con hexanos para dar el compuesto del título (97 mg, 100%). MS (ES+) C30H34N10O4 requiere: 598, encontrado: 599 [M H]
Síntesis de 4-(2-((2-amino-4-((1-metilpiperidin-4-il)carbamoil)fenil)amino)-5-metilpirimidin-4-il)-N-(4-cianofenil)piperazina- 1-carboxamida

4-(2-((2-amino-4-((1-metilpiperidin-4-il)carbamoil)fenil)amino)-5-metilpirimidin-4-il)-N-(4-cianofenil)piperazina-1-carboxamida se preparó usando un procedimiento similar al del COMPUESTO 30. La reacción se filtró a través de celite para dar un producto en bruto. MS (ES+) C30H36N10O2 requiere: 568, encontrado: 569 [M H]+.
cianofenil)piperazina-1-carboxamida

Se preparó 4-(2-((2-acrilamido-4-((1-metilpiperidin-4-il)carbamoil)fenil)amino)-5-metilpirimidin-4-il)-N-(4-cianofenil)piperazina-1-carboxamida usando un procedimiento similar al del COMPUESTO 30. La mezcla de reacción se purificó mediante una cromatografía de capa fina preparativa para dar el producto del título. MS (ES+) C33H38N10O3 requiere: 622, encontrado: 623 [M H]+. 1H RMN (400 MHz, DMSO-d s) 59,98 (s, 1 H), 9,08 (s, 1 H), 8,30 (s, 1 H), 8,21 - 8,07 (m, 3 H), 7,93 (d, J = 10,7 Hz, 2 H), 7,67 (m, 4 H), 6,50 (dd, J = 16,9, 10,2 Hz, 1 H), 6,33 -6,25 (m, 1 H), 5,83 - 5,76 (m, 1 H), 3,78 (m, 2 H), 3,59 (m, 4 H), 3,43 (m, 4H), 2,92 (d, J = 11,4 Hz, 2H), 2,30 (s, 3H), 2,23 (s, 2H), 2,14 (s, 3H), 1,79 (m, 2H), 1,69 - 1,54 (m, 2H).
Ejemplo 12: Síntesis del COMPUESTO 20

Síntesis de imidazo [1,2-a iridina-8-carbonitrilo

A una solución de 2-aminonicotinonitrilo (1,0 g, 8,39 mmol) en EtOH (10 ml) en un vial sellado de 20 ml se le añadió 2-cloroacetaldehído (1.611 ml, 9.23 mmol) y a continuación el vial se selló y se calentó a 120 °C durante la noche. La reacción se enfrió a TA y se inactivó con Na2CO32 N, se eliminó el EtOH al vacío y se extrajo con DCM x3. Se combinaron los orgánicos y se lavaron con agua y a continuación con salmuera x2. Se secó sobre sulfato de sodio y se eliminó el disolvente para dar el compuesto del título como un sólido de color marrón amarillento (1,2 g, 8,38 mmol, rendimiento del 100%) que se verificó mediante MS (ES+) C8H5N3 requiere: 143 encontrado: 144 [M H]+
Síntesis de 3-yodoimidazo [1,2-a] piridina-8-carbonitrilo

A una solución agitada de imidazo [1,2-a] piridina-8-carbonitrilo (1,2 g, 8,38 mmol) en diclorometano (10 ml) se añadió N-yodosuccinimida (1,89 g, 8,38 mmol). LCMS controló la reacción hasta que el material de partida se consumió por completo. La mezcla de reacción se diluyó con diclorometano y agua. La capa orgánica separada se secó con sulfato de sodio, se filtró y se concentró para dar 3-yodoimidazo [1,2-a] piridin-8-carbonitrilo (1,8 g, 6,69 mmol, rendimiento del 80%) como un sólido marrón. MS (ES+) C8H8IN 3 requiere: 269, encontrado: 270 [M H]+.
Síntesis de 1-(3-(8-cianoimidazo [1,2-a] piridin-3-il)-5-isopropoxifenil)-3-(2,2,2-trifluoroetil)urea

A una mezcla de 3-yodoimidazo [1,2-a] piridina-8-carbonitrilo (100 mg, 373 pmol), 1-(3-isopropoxi-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-3-(2,2,2-trifluoroetil)urea (90 mg, 224 pmol), aducto PdCl2(dppf)-CH2Cl2 (30,5 mg, 37,3 pmol) en dioxano (3 ml), se añadió Na2CO3 2 M (0,559 ml, 1119 pmol). El vial se desgasificó durante 5 minutos, a continuación se tapó y se calentó a 110°C durante 30 minutos en microondas. Después de enfriar a temperatura ambiente, la reacción se repartió entre EtOAc y salmuera, se separaron y los extractos orgánicos se lavaron con salmuera x 2. Los extractos orgánicos combinados se secaron directamente sobre sílice y se purificaron mediante cromatografía ultrarrápida (0-100% Hex/EtOAc; columna de 12 g). Se recuperó el compuesto del título (30 mg, 71,9 pmol, rendimiento del 32,1%) como un sólido marrón. MS (ES+) C20H18F3N5O2 requiere: 417, encontrado: 418 [M H]+.
Síntesis de 1-(3-(8-(aminometil) imidazo [1,2-a] piridin-3-il)-5-isopropoxifenil)-3-(2,2,2-trifluoroetil)urea

Se recogió 1-(3-(8-cianoimidazo [1,2-a] piridin-3-il)-5-isopropoxifenil)-3-(2,2,2-trifluoroetil)urea (0,030 g, 0,072 mmol) en AMONÍACO 7 N en metanol (20 ml, 140 mmol) y se añadió Pd-C (10 mg, 0,094 mmol). La reacción se agitó bajo un globo de H2 durante 1 hora. A continuación, la mezcla se filtró a través de celite y se eliminó el disolvente. El residuo se secó a alto vacío durante la noche para dar el compuesto del título como un sólido amarillo (0,026 g, 0,062 mmol, 86 % de rendimiento). MS (ES+) C20H22F3N5O2 requiere: 421, encontrado: 422 [M H]+.
Síntesis de N-((3-(3-isopropoxi-5-(3-(2,2,2-trifluoroetil)ureido)fenil) imidazo [1,2-a] piridin-8 -il) metil) propiolamida

A una solución de 1-((3-(8-(aminometil) imidazo [1,2-a]-3-il piridin-3-l)-5-isopropoxifenil)-3-(2,2,2-trifluoroetil)urea (26 mg, 0,062 mmol) en Dc M (3 ml) se añadió DIEA (0,075 ml, 0,432 mmol) y HATU (35,2 mg, 0,093 mmol) y finalmente ácido propiólico (4,95 pl, 0,080 mmol). La reacción se agitó durante 30 minutos a temperatura ambiente. La reacción se cargó directamente en una columna de sílice y se purificó mediante cromatografía ultrarrápida (0-10% de CH2Cl2}/meOH) para dar el compuesto del título (19 mg, 0,040 mmol, rendimiento del 65,0%) como un sólido blanquecino. MS (ES+) C23H22F3N5O3 requiere: 473, encontrado: 474 [M H]+. 1H RMN (400 MHz, DMSO-d 6) 8 9,34 (s, 1H), 8,92 (s, 1H), 8,47 (d, J = 6 ,8 Hz, 1H), 7,74 (s, 1H), 7,17 (d, J = 1,9 Hz, 2 H), 7,10 (s, 1 H), 6,98 (s, 1 H), 6,82 (s, 1 H), 6,74 (s, 1 H), 4,69 -4,58 (m, 2 H), 3,93 (dd, J = 9,7, 6,4 Hz, 2 H), 2,72 -2,64 (m, 1 H), 1,30 -1,19 (m, 6 H).
Ejemplo 13: Síntesis del COMPUESTO 21

Se preparó 7-doro-3-yodoimidazo [1,2-a]piridina usando el procedimiento descrito en el documento WO2008078091. MS (ES+) C7H4CNN2 requiere: 278, encontrado: 279 [M H]+.
Síntesis de 3-(7-cloroimidazo [1,2-a] piridin-3-il) anilina

Se preparó 3-(7-cloroimidazo [1,2-a]piridin-3-il)anilina usando el procedimiento descrito en el documento WO2008078091. MS (ES+) C13H10CIN3 requiere: 243, encontrado: 244 [M H]+.
Síntesis de 1-(3-(7-cloroimidazo [1,2-a] piridin-3-il)fenil)-3-(2,2,2-trifluoroetil)urea

A una solución de 3-(7-cloroimidazo[1,2-a] piridin-3-il)anilina (0,15 mmol) en THF (1,5 ml) se añadió carbonoclorato de 4-nitrofenilo (30 mg, 0,15 mmol) y DIEA (0,036 ml, 0,225 mmol). La mezcla se calentó a 60°C durante 6 h. Al carbamato crudo se le añadió DIEA (0,036 ml, 0,225 mmol) y 2,2,2-trifluoroetan-1-amina (0,014 ml, 0,18 mmol) y la solución se calentó a 60°C durante la noche. La mezcla de reacción se diluyó con EtOAc y agua. La capa orgánica separada se secó con sulfato de sodio, se filtró y se concentró. La mezcla en bruto se purificó mediante cromatografía ultrarrápida (0-6% MeOH/DCM) para dar el compuesto del título (38 mg, 69% de rendimiento). MS (ES+) C16H12CF3N4O requiere: 368, encontrado: 369 [M H]+.
Síntesis de 1-(3-(7-(2-aminofenil) imidazo [1,2-a] piridin-3-il)fenil)-3-(2,2,2-trifluoroetM)urea

A una mezcla de 1-(3-(7-cloroimidazo [1,2-a]piridin-3-il)fenil)-3-(2,2,2-trifluoroetil)urea (20 mg, 0,052 mmol), 2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)anilina (15 mg, 0,066 mmol) y carbonato de cesio (51 mg, 0,156 mmol) en una mezcla THF/H2O (2/1, 0,75 ml) se añadió Pd(PtBu3)2( 3 mg, 0,005 mmol). El vial se desgasificó durante 5 minutos, a continuación se tapó y se calentó a 125°C durante 20 minutos en un microondas. Después de enfriar a temperatura ambiente, la mezcla de reacción se filtró a través de una almohadilla de celite y se purificó por cromatografía ultrarrápida (gradiente de MeOH al 0-10%/DCM que contiene 10% de NH4OH) para dar el compuesto del título (20 mg, 90% de rendimiento). MS (ES+) C22H18F3N5O requiere: 425, encontrado: 426 [M H]+.
Síntesis de N-(2-(3-(3-(3-(2,2,2-trifluoroetil) ureido)fenil) imidazo [1,2-a] piridin-7-il)fenil)acrilamida

Se preparó N-(2-(3-(3-(3-(2,2,2-trifluoroetil)ureido)fenil)imidazo[1,2-a]piridin-7-il)fenil)acrilamida usando el procedimiento es similar al del COMPUESTO 30. El producto se purificó mediante cromatografía en capa fina preparativa usando un gradiente de MeOH/DCM al 0-10% para dar el compuesto del título. MS (ES+) C25H20F3N5O2 requiere: 479, encontrado: 480 [M H]+.
Síntesis de N-(2-(3-(3-isopropoxi-5-(3-(2,2,2-trifluoroetil) ureido)fenil)imidazo [1,2-a] piridin-7-il)fenil)acrilamida

Se preparó N-(2-(3-(3-isopropoxi-5-(3-(2,2,2-trifluoroetil)ureido)fenil)imidazo [1,2-a]piridin-7-il)fenil)acrilamida usando un procedimiento similar al del COMPUESTO 30. El producto se purificó por HPLC usando un gradiente de acetonitrilo al 5-70%/agua ácido fórmico al 0,1% para dar el compuesto del título como una sal de formiato. MS (ES+) C28H26F3N5O3 requiere: 537, encontrado: 538 [M H]+
Ejem lo 15: Síntesis del COMPUESTO 11


Síntesis de (1-(2-cloropirimidin-4-il)piperidin-3-il) carbamato de tere-butilo

Se preparó (1 -(2-cloropirimidin-4-il)piperidin-3-il) carbamato de terc-butilo usando el procedimiento similar al COMPUESTO 54 usando 2,4-didoropirimidina y piperidin-3-ilcarbamato de terc-butilo. MS (ES+) C-mH21CIN4O2 requiere: 312, encontrado: 313 [M H]+
Síntesis de (1-(2-((2-n¡trofen¡l)am¡no)p¡r¡m¡din-4-¡l)p¡per¡d¡n-3-¡l)carbamato de terc-butilo

Se preparó (1-(2-((2-nitrofenil)amino)pirimidin-4-il)piperidin-3-il)carbamato de terc-butilo usando un procedimiento similar al del COMPUESTO 54 usando 2-nitroanilina. MS (ES+) C20H26N6O4 requiere: 414, encontrado: 415 [M H] Síntesis de N-( 1 -(2-((2-nitrofenil)amino)pirimidin-4-il)piperidin-3-il) propano-1-sulfonamida

A una solución de (1-(2-((2-nitrofenil)amino)pirimidin-4-il)piperidin-3-il)carbamato de terc-butilo (0,14 g, 0,34 mmol) en DCM (2,0 ml) se añadió TFA (1,0 ml) y la mezcla se agitó durante 1 h. Se analizó una alícuota de la mezcla de reacción mediante LCMS, que indicó que la reacción se había completado hasta el final. Se eliminaron los disolventes y se secó el residuo a alto vacío. El producto en bruto se usó para el siguiente paso sin purificación adicional.
A una solución de 4-(3-aminopiperidin-1-il)-N-(2-nitrofenil)pirimidin-2-amina (0,34 mmol) en DCM (3,5 ml) a 0 °C se añadió cloruro de propano-1-sulfonilo (0,045 ml, 0,4 mmol) y trietilamina (0,12 ml, 0,85 mmol) y la mezcla se calentó a temperatura ambiente durante la noche. La mezcla de reacción en bruto se concentró y se purificó mediante
cromatografía ultrarrápida (0-7,5% de MeOH/DCM) para dar el compuesto del título (36 mg, 24% de rendimiento). MS (ES+) C18H24N6O4S requiere: 420, encontrado: 421 [M H]+.
Síntesis de N- 1- 2- 2-aminofenil amino irimidin-4-il i eridin-3-il ro ano-1-sulfonamida

Se preparó N-(1 -(2-((2-aminofenil)amino)pirimidin-4-il)piperidin-3-il) propano-1-sulfonamida usando un procedimiento similar al del COMPUESTO 30. La reacción se filtró a través de celite para dar un producto en bruto. MS (ES+) C18H26N6O2S requiere: 390, encontrado: 391 [M H]+.
Síntesis de N-(2-((4-(3-(propilsulfonamido)piperidin-1-il)pirimidin-2-il)amino)fenil)acrilamida

Se preparó N-(2-((4-(3-(propilsulfonamido)piperidin-1-il)pirimidin-2-il)amino)fenil)acrilamida usando un procedimiento similar al del COMPUESTO 30. El producto se purificó mediante cromatografía preparativa en capa fina usando gradiente de MeOH/DCM al 0-6% para dar el compuesto del título. MS (eS+) C21H28N6O3S requiere: 444, encontrado: 445 [M H]+.
Ejemplo 16: Síntesis del COMPUESTO 52

El material de partida 1-(terc-butil)-3-(2-((4-(dietilamino) butil)amino)-6-(3,5-dimetoxifenil)pirido[2,3-d] pirimidin-7-il)urea (PD173074) se puede comprar en, por ejemplo, SelleckChem.com. En un recipiente seco, se añaden cloruro de acriloílo (2 equivalentes) y diisopropiletilamina (4,3 equivalentes) a una solución de 1-(terc-butil)-3-(2-((4-(dietilamino) butil)amino)-6-(3,5-dimetoxifenil) pirido [2,3-d]pirimidin-7-il)urea (1 equiv.) en diclorometano anhidro a
0°C. Después de agitar a temperatura ambiente durante 2 horas, la mezcla de reacción se concentra, se diluye con DMSO y se purifica mediante HPLC de fase inversa (5-95% de agua/acetonitrilo). Después de concentrar las fracciones, se obtiene el producto N-(7-(3-(terc-butil) ureido)-6-(3,5-dimetoxifenil)pirido[2,3-d] pirimidin-2-il)-N-(4-(dietilamino) butil)acrilamida como una espuma de color amarillo pálido. LCMS (M 1) = 578,2.
Ejemplo 17: Síntesis del COMPUESTO 55

En un recipiente de secado, se añade cloruro de sulfurilo (2 equiv) a una solución de 1-(terc-butil)-3-(2-((4-(dietilamino) butil)amino)-6-(3, 5-dimetoxifenil) pirido [2,3-d] pirimidin-7-il)urea (1 equiv.) en acetonitrilo anhidro a 0°C. Después de agitar durante 2 horas, la mezcla de reacción se diluye con diclorometano y se lava con una solución acuosa saturada de bicarbonato de sodio. El producto en bruto, 1-(terc-butil)-3-(6-(2,6-dicloro-3,5-dimetoxifenil)-2-((4-(dietilamino)butil)amino)pirido [2,3-d] pirimidin-7-il)urea, se usa en el siguiente paso sin purificación adicional.
En un recipiente de secado, se añaden cloruro de acriloílo (2 equiv.) y diisopropiletilamina (4,3 equiv.) a una solución del producto obtenido anteriormente (1 equiv.) en diclorometano anhidro a 0 °C. Después de agitar a temperatura ambiente durante 2 horas, la mezcla de reacción se concentra, se diluye con DMSO y se purifica mediante HPLC de fase inversa (5-95% de agua/acetonitrilo). Después de secar a alto vacío, se obtiene el producto N-(7-(3-(terc-butil) ureido)-6-(2,6-dicloro-3,5-dimetoxifenil)pirido [2,3-d] pirimidin-2-il)-N-(4-(dietilamino)butil)acrilamida como una espuma amarilla. LCMS (M 1) = 646,3.
Pueden usarse procedimientos similares a los anteriores para preparar otros compuestos descritos en este documento. Los datos de 1H NMR y LCMS para los compuestos 1 a 55 se resumen a continuación.




Selectividad de compuestos
La puntuación de selectividad es una medida imparcial que permite comparaciones cuantitativas entre los compuestos y la diferenciación detallada y el análisis de los patrones de interacción. Se calcula una medida de selectividad usando el porcentaje de valores de control de un panel de ensayos de quinasa. Las puntuaciones de los cribados primarios (realizados a una sola concentración) se indican como porcentaje de control de DMSO (POC) y se calculan de la siguiente manera:
Señal de compuesto de ensayo - señal de control positivo
---------------------------------------------------------------------------- x 100
Señal de control negativo - Señal de control positivo
donde el control negativo es un disolvente como DMSO (control al 100%) y el control positivo es un compuesto de control que se sabe que se une con alta afinidad (control del 0%).
La puntuación de selectividad (S) para cada compuesto analizado se puede calcular dividiendo el número de quinasas con un POC menor que un valor elegido, por ejemplo, 10, 20 o 35, cuando se criban a una determinada concentración, por ejemplo, 1 pM, 3 pM, 5 pM o 10 pM, por el número total de quinasas distintas ensayadas (excluyendo variantes mutantes). Por ejemplo, se puede calcular una puntuación de selectividad (S) dividiendo el número de quinasas con un POC menor que 10 cuando se criban a 3 pM por el número total de quinasas distintas probadas (excluyendo variantes mutantes); dicha puntuación se mostraría como [S (10) a 3 pM]. Se determinó la selectividad de los compuestos COMPUESTO 9; COMPUESTO 9; COMPUESTO 11; COMPUESTO 15; COMPUESTO 16; COMPUESTO 20; COMPUESTO 21; COMPUESTO 23; COMPUESTO 24; COMPUESTO 25; COMPUESTO 26; COMPUESTO 27; COMPUESTO 30; COMPUESTO 32; COMPUESTO 35; COMPUESTO 60; COMPUESTO 38; COMPUESTO 39; COMPUESTO 41; COMPUESTO 45; COMPUESTO 48; COMPUESTO 50; COMPUESTO 52; COMPUESTO 54; COMPUESTO 55; todos tenían puntuaciones de selectividad [S (10) a 3 pM] de 0,030 o menos.
El COMPUESTO 9; COMPUESTO 11; COMPUESTO 15; COMPUESTO 16; COMPUESTO 20; COMPUESTO 21; COMPUESTO 23; COMPUESTO 24; COMPUESTO 25; COMPUESTO 26; COMPUESTO 32; COMPUESTO 35; COMPUESTO 60; COMPUESTO 38; COMPUESTO 39; COMPUESTO 45; COMPUESTO 48; COMPUESTO 50; COMPUESTO 52 todos tenían puntuaciones de selectividad [S (10) a 3 pM] de 0,010 o menos.
Evaluación de la actividad bioquímica
Para evaluar la actividad de los compuestos químicos frente a la quinasa relevante de interés, se utiliza la plataforma de tecnología de desplazamiento de movilidad electroforética Caliper LifeSciences. El péptido sustrato marcado con fluorescencia se incuba en presencia de niveles dosificados de compuestos, una concentración establecida de quinasa y de ATP, de modo que se fosforila una proporción reflectante del péptido. Al final de la reacción, la mezcla de péptidos fosforilados (producto) y no fosforilados (sustrato) se pasa a través del sistema microfluídico del Caliper LabChip® EZ Reader II, bajo una diferencia de potencial aplicada. La presencia del grupo fosfato en el péptido producto proporciona una diferencia de masa y carga entre el péptido producto y el péptido sustrato, lo que da como resultado una separación de los grupos de sustrato y productos en la muestra. A medida que los grupos pasan los LEDS dentro del instrumento, estos grupos se detectan y resuelven como picos separados. Por tanto, la relación entre estos picos refleja la actividad de la materia química a esa concentración en ese pocillo, en esas condiciones.
Ensayo de FGFR-1 de tipo salvaje en Km: En cada pocilio de una placa de 384 pocilios, 0,1 ng/ul de FGFR-1 de tipo salvaje (Carna Biosciences, Inc.) se incubó en un total de 12,5 ul de tampón (HEPES 100 mM pH 7,5, Brij 35 al 0,015%, MgCl210 mM, DTT 1 mM) con CSKtide 1 uM (5 -FAM-KKKKEEIYFFFG-NH2) y ATP 400 uM a 25 C durante 90 minutos en presencia o ausencia de una serie de concentraciones dosificadas de compuesto (concentración final de DMSO al 1%). La reacción se detuvo mediante la adición de 70 ul de tampón de parada (HEPES 100 mM pH 7,5, Brij 35 al 0,015%, EDTA 35 mM y 0,2% de reactivo de recubrimiento 3 (Caliper Lifesciences)). A continuación, se leyó la placa en un Caliper EZReader 2 (ajustes de protocolo: -1,9 psi, voltaje aguas arriba -700, voltaje aguas abajo - 3000, sip posterior a la muestra 35 s).
Ensayo de FGFR-4 de tipo salvaje en Km: En cada pocillo de una placa de 384 pocillos, 0,5 ng/ul de FGFR-4 de tipo salvaje (Carna Biosciences, Inc.) se incubó en un total de 12,5 ul de tampón (HEPES 100 mM pH 7,5, Brij 35 al 0,015%, MgCl210 mM, DTT ImM) con CSKtide 1 uM (5 -FAM-KKKKEEIYFFFG-NH2) y ATP 400 uM a 25 C durante 90 minutos en presencia o ausencia de una serie de concentraciones dosificadas de compuesto (concentración final de DMSO al 1%). La reacción se detuvo mediante la adición de 70 ul de tampón de parada (HEPES 100 mM pH 7,5, Brij 35 al 0,015%, EDTA 35 mM y 0,2% de reactivo de recubrimiento 3 (Caliper Lifesciences)). A continuación, se leyó la placa en un Caliper LabChip® EZ Reader II (ajustes de protocolo: -1,9 psi, voltaje aguas arriba -700, voltaje aguas abajo -3000, sip posterior a la muestra 35 s).

Ċ

77


Ċ

80
Ċ

81
Ċ

82
Ċ

83
Ċ
84
Ċ

85
Ċ

86
Ċ

87
Ċ

88

Ċ

90
Ċ

91
Ċ

92
Ċ

93
Ċ

94
Ċ

95
Ċ

96
Ċ

97
Ċ

98
Ċ

99
Ċ

100
Ċ

101

En la tabla anterior, para FGFR1 y FGFR4: "A" significa que la IC50 es inferior a 10 nM; "B" significa que la IC50 es mayor que o igual a 10 y menor que 100 nM; "C" significa que la IC50 es mayor que o igual a 100 y menor que 1000 nM; "D" significa que la IC50 es mayor que 1000 nM.
Para la relación: "F" significa que la relación de [IC50 para FGFR1]/[IC50 para FGFR4] es inferior a 10; "E" significa que la relación es >10 y <50; "D" significa que la relación es >50 y <100; "C" significa que la relación es >100 y <200; "B" significa que la relación es >200 y <500; "A" significa que la relación es > 500. Cuanto mayor es la relación, más selectivo es el compuesto para FGFR4 frente a FGFR1.
Potencia celular
La respuesta a la dosis en las células MDA-MB-453, que albergan una mutación de FGFR4 activante, se midió de la siguiente manera. Brevemente, las células MDA-MB-453 se sembraron a 2,5 x 106 células/6 pocillos y se privaron de alimento durante la noche. Los compuestos se añadieron a concentraciones variables (3000, 1000, 300, 100 y 30 nM) durante 1 hora. Las muestras se recogieron y lisaron para el análisis de inmunotransferencia. La fosforilación de Erk se midió y el valor pErk promedio de tres repeticiones se representó con un ajuste de curva de dosis-respuesta
(inhibición) de tres parámetros usando software Prism GraphPad, que se utilizó para determinar los valores de IC50. Los datos se muestran en la siguiente tabla.

En la tabla, "A" significa la IC50 es <1 nM; "B" significa que la IC50 es >1 y <10 nM; "C" significa que la IC50 es >10 y <100 nM; "D" significa que la IC50 es >100 nM.
Estos datos indican que la inhibición de FGFR-4 por estos compuestos resulta en el bloqueo de la señalización oncogénica aguas abajo.
Inducción de apoptosis con un inhibidor de FGFR4
Se sembraron células Hep3B a 20 k/pocillo en placas blancas de 96 pocillos en 200 pl de DMEM/FBS al 5% durante la noche. El compuesto del día siguiente se añadió a una concentración final de DMSO del 0,1% y se incubó durante 6 horas. La actividad de la caspasa se midió de acuerdo con las instrucciones de fabricación (Ensayo Caspasa-Glo3/7 (Promega)). Brevemente, se añadieron 100 pl de reactivo Caspase-Glo3/7 a cada pocillo y se incubaron durante 1 hora en la oscuridad. La luminiscencia se midió usando EnVision. La actividad promedio de caspasa de 2 réplicas se representó gráficamente con un ajuste de la curva de respuesta a la dosis (inhibición) de tres parámetros utilizando el software Prism GraphPad, que se utilizó para determinar los valores de IC50. Como se muestra en la Fig. 3, en las células Hep3B, el tratamiento con el COMPUESTO 25 durante 6 horas conduce a una potente inducción de apoptosis. BGJ398, un inhibidor de FGFR, también da lugar a la inducción de apoptosis, aunque a una concentración más alta.
Covalencia
La evidencia de que el COMPUESTO 52 se une covalentemente al FGFR-4 se muestra mediante los datos espectrométricos de masas mostrados en la Fig. 1. En 60 ul de tampón, se incubó el Compuesto 1300 uM con 50 ug (75 uM) de FGFR-4 de tipo salvaje recombinante etiquetado con GST (Carna Biosciences) durante 3 horas a temperatura ambiente y posteriormente a 4 °C durante 13 horas. A continuación, se desaló el complejo de proteínainhibidor usando columnas de eliminación de detergente Pierce (Thermo Pierce). La proteína no modificada y el complejo proteína-inhibidor se analizaron mediante espectrometría de masas por pulverización de electrones para determinar sus respectivos pesos moleculares. La figura 1a muestra la masa de la proteína sin modificar. Tal como se muestra, el pico principal relevante tiene una masa de 65468,371 daltons. La figura 1b muestra la masa del complejo proteína-inhibidor. Tal como se muestra ahí, el pico más importante tenía una masa de 66043,5123 daltons. La diferencia entre estas masas es de 575,1252, que está dentro de la precisón instrumental de la masa del compuesto, 1577,34 daltons.
Las masas de los complejos proteína-inhibidor de FGFR4 y los compuestos Compuesto 11, el Compuesto 20, y el Compuesto 54 se muestran en la Fig. 2. CR9 es el pico de la proteína FGFR4. Como muestra por el pico CR3, el complejo mostró un desplazamiento de 441 da cuando el PM del compuesto (COMPUESTO 11) fue de 444,6 (dentro de la precisión instrumental). En otro ejemplo, el complejo mostró un desplazamiento de 470 da (pico CR2), cuando el PM del compuesto (COMPUESTO 20) fue de 473,4. En otro ejemplo más, el complejo mostró un desplazamiento de 631 da (pico CR1) cuando el PM del compuesto (COMPUESTO 54) fue de 622,7.
Esto demuestra que los compuestos de una amplia variedad de andamios son todos capaces de formar complejos covalentes con FGFR4.
Unión a Cys552
La estructura cristalina del compuesto 52 unido a FGFR-4 se muestra en la Fig. 4. Como se muestra allí, el compuesto 52 se une a la cisteína en el residuo 552 de FGFR-4.
La estructura cristalina del compuesto 25 unido a FGFR-4 se muestra en la Fig. 5. Como se muestra allí, el compuesto 25 también se une a la cisteína en el residuo 552 de FGFR-4.
Datos de eficacia in vivo
Se estudiaron los efectos del COMPUESTO 25, BGJ398 (un inhibidor de FGFR) y Sorafenib sobre la inhibición del crecimiento tumoral en el modelo de xenoinjerto subcutáneo de células de cáncer de hígado Hep3B con diferentes dosis.
Se utilizaron seis ratones desnudos hembra (Mus Musculus) de 6 a 8 semanas de edad. Inoculación y cultivo de células tumorales: se cultivaron células Hep3B con medio EMEM (Invitrogen, EE.UU.) suplementado con FBS al 10% (Gibco, Australia). Las células se recolectaron con una confluencia del 90% y la viabilidad no fue inferior al 90%. A los ratones se les implantaron por vía subcutánea (sc) 200 pL de 10 x 106 células Hep3B en Matrigel al 50% en el flanco derecho al comienzo del estudio.
Agrupación de animales y programa de dosificación: Diez días después de la implantación celular, cuando los tumores alcanzaron un volumen promedio de 199 mm3, se seleccionaron 45 ratones basándose en el volumen del tumor y se asignaron aleatoriamente a 5 grupos de tratamiento (n = 9). El día de la aleatorización se denotó como D0 y el tratamiento se inició a partir de ese momento.
Mediciones de volumen del tumor y el peso corporal: El tamaño del tumor se midió dos veces por semana en dos dimensiones usando un calibrador, y el volumen se expresó en mm3 utilizando la fórmula: V = 0,5 a * b2 donde a y b eran los diámetros largo y corto del tumor, respectivamente. El peso corporal se midió al menos dos veces por semana.
Fin de la porción in vivo: se recogieron sangre, tumores e hígados de 3 ratones en cada grupo tratado a las 4, 12 y 24 horas después de la última dosis. El lóbulo izquierdo del hígado se recogió para estudios farmacodinámicos (PD) y el resto del hígado se almacenó en formalina para histología. Los tumores pequeños se priorizaron para su uso en estudios farmacocinéticos. Cualquier tumor restante se fijó primero para el análisis histológico y a continuación se congeló instantáneamente para el estudio de DP.
Volúmenes tumorales de ratones desnudos portadores de Hep3B: la figura 5 es un gráfico de líneas que representa la inhibición del crecimiento de grupos tratados con el COMPUESTO 25 (100 mg/kg PO BID), COMPUESTO 25 (300 mg/kg PO BID), BGJ398 (20 mg/kg PO QD) y sorafenib (30 mg/kg PO QD) contra tumores de xenoinjerto Hep3B en ratones desnudos. Se observó una reducción estadísticamente significativa de los volúmenes tumorales en los grupos de eficacia de COMPUESTO 25 (100 mg/kg PO BID), COMPUESTO 25 (300 mg/kg PO BID) y sorafenib (30 mg/kg PO QD) en comparación con el grupo de vehículo, todos comenzando desde el día 4 después de la primera administración de los compuestos y persistió hasta el final (día 19). Sin embargo, no se observó una diferencia significativa en el volumen tumoral entre BGJ398 (20 mg/kg PO QD) y los grupos de vehículo durante todo el estudio. El aumento de la dosis del COMPUESTO 25 de 100 mg/kg a 300 mg/kg mejoró la eficacia de inhibición del tumor. Los tumores en los grupos tratados con COMPUESTO 25 (100 mg/kg PO BID) y los grupos tratados con COMPUESTO 25 (300 mg/kg Po BID) retrocedieron, y los tumores en el grupo tratado con COMPUESTO 25 (300 mg/kg PO BID) casi desaparecieron . En este estudio, los grupos tratados con COMPUESTO 25 (100 mg/kg PO BID) y los grupos tratados con COMPUESTO 25 (300 mg/kg PO BID) mostraron superioridad en la inhibición del crecimiento tumoral.
Cambio de peso corporal (%) de ratones desnudos que llevan Hep3B: la figura 7 es un gráfico de líneas que representa el cambio de peso corporal (%) durante todo el período de estudio. Todos los ratones, excepto los ratones de los grupos tratados con el COMPUESTO 25, mostraron una pérdida significativa de peso corporal. El peso corporal de los ratones en el grupo de vehículo disminuyó aproximadamente un 10% el día 10 para la carga del
tumor. Este resultado indicó que el COMPUESTO 25 se toleró bien con las dosis y el programa de dosificación actuales en ratones desnudos, y que el COMPUESTO 25 podría aliviar la pérdida de peso corporal inhibiendo el crecimiento tumoral.
Los ratones tratados con COMPUESTO 25 (100 mg/kg PO BID), COMPUESTO 25 (300 mg/kg PO BID) y Sorafenib (30 mg/kg PO QD) mostraron una reducción significativa del volumen tumoral en comparación con el grupo de vehículo durante todo el estudio. El aumento de la dosis del COMPUESTO 25 de 100 mg/kg a 300 mg/kg mejoró la eficacia de inhibición tumoral. Los tumores de ratones en los grupos tratados con COMPUESTO 25 (100 mg/kg PO BID) y los grupos tratados con COMPUESTO 25 (300 mg/kg PO BID) retrocedieron, y los tumores en los grupos tratados con COMPUESTO 25 (300 mg/kg PO BID) grupo casi desaparecieron. Todos los ratones, excepto los de los grupos tratados con el COMPUESTO 25, perdieron una cantidad significativa de peso corporal. El peso corporal de los ratones en el grupo de vehículo disminuyó aproximadamente un 10% el día 10 para la carga del tumor. Estos resultados indicaron que el COMPUESTO 25 se toleró bien con las dosis y el programa de dosificación actuales en ratones desnudos, y que el COMPUESTO 25 podría aliviar la pérdida de peso corporal inhibiendo el crecimiento tumoral.
Equivalentes
Los expertos en la técnica reconocerán, o serán capaces de determinar usando sólo experimentación rutinaria, muchos equivalentes a las realizaciones específicas de la invención descritas en el presente documento. Se pretende que tales equivalentes estén incluidas en las siguientes reivindicaciones.
Claims (15)
- REIVINDICACIONES
 en la que:el anillo A es un grupo arilo, heteroarilo, heterocíclico o alicíclico de 3-8 miembros;X es CH o N;Y es CH o N-R4, en el que R4 es H o alquilo C1-6;L es -[C(R5)(R6)]q-, en el que:cada uno de R5 y R6 es, independientemente, H o alquilo C1-6; yq es 0-4;cada R1, R2 y R3 es, independientemente, halo, ciano, alcoxi C1-6 opcionalmente sustituido, hidroxi, oxo, amino, amido, alquil urea, alquilo C1-6 opcionalmente sustituido, o heterociclilo C1-6 opcionalmente sustituido;m es 0-3;n es 4;p es 0-2;yWarhead se selecciona entre:
en la que:el anillo A es un grupo arilo, heteroarilo, heterocíclico o alicíclico de 3-8 miembros;X es CH o N;Y es CH o N-R4, en el que R4 es H o alquilo C1-6;L es -[C(R5)(R6)]q-, en el que:cada uno de R5 y R6 es, independientemente, H o alquilo C1-6; yq es 0-4;cada R1, R2 y R3 es, independientemente, halo, ciano, alcoxi C1-6 opcionalmente sustituido, hidroxi, oxo, amino, amido, alquil urea, alquilo C1-6 opcionalmente sustituido, o heterociclilo C1-6 opcionalmente sustituido;m es 0-3;n es 4;p es 0-2;yWarhead se selecciona entre: en los que:X es un grupo saliente seleccionado entre halógeno y triflato; ycada uno de Ra, Rby Rc es, independientemente, H, alquilo C1-4 sustituido o no sustituido, o cicloalquilo C3-4 sustituido o no sustituido.
en los que:X es un grupo saliente seleccionado entre halógeno y triflato; ycada uno de Ra, Rby Rc es, independientemente, H, alquilo C1-4 sustituido o no sustituido, o cicloalquilo C3-4 sustituido o no sustituido. - 2. Compuesto de la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en el que Warhead es

- 3. Compuesto de la reivindicación 1 o la reivindicación 2, o una sal farmacéuticamente aceptable del mismo, en el que el anillo A es arilo o heteroarilo.
- 4. Compuesto de la reivindicación 3, o una sal farmacéuticamente aceptable del mismo, en el que el anillo A se selecciona entre fenilo, pirrol, furano, tiofeno, imidazol, oxazol, tiazol, triazol, pirazol, piridina, pirazina, piridazina y pirimidina.
- 5. Compuesto de la reivindicación 1 o la reivindicación 2, o una sal farmacéuticamente aceptable del mismo, en el que el anillo A es heterocíclico.
- 6. Compuesto de la reivindicación 5, o una sal farmacéuticamente aceptable del mismo, en el que el anillo A se selecciona entre pirrolidina, piperidina, piperazina y morfolina.
- 7. Compuesto de la reivindicación 1 o la reivindicación 2, o una sal farmacéuticamente aceptable del mismo, en el que el anillo A es alicíclico.
- 8. Compuesto según cualquiera de las reivindicaciones 1 a 7, o una sal farmacéuticamente aceptable del mismo, en el que cada R2 es, independientemente, halógeno o metoxi.
- 9. Compuesto de la reivindicación 8, o una sal farmacéuticamente aceptable del mismo, en el que dos R2 son cloro y dos R2 son metoxi.
- 10. Compuesto según cualquiera de las reivindicaciones 1 a 9, o una sal farmacéuticamente aceptable del mismo, en el que X es N e Y es CH.
- 11. Compuesto según cualquiera de las reivindicaciones 1 a 10, o una sal farmacéuticamente aceptable del mismo, en el que cada R1, R2y R3 es, independientemente, halógeno, ciano, alcoxi C1-6 opcionalmente sustituido, amino, amido, alquil urea, alquilo C1-6 opcionalmente sustituido, o heterociclilo C1-6 opcionalmente sustituido.
- 12. Com uesto de la reivindicación 1, en el ue el com uesto se selecciona entre:
 Ċ
Ċ 108y sales farmacéuticamente aceptables del mismo.
108y sales farmacéuticamente aceptables del mismo. - 13. Composición farmacéutica que comprende:un portador farmacéuticamente aceptable; yun compuesto según cualquiera de las reivindicaciones 1 a 12, o una sal farmacéuticamente aceptable del mismo.
- 14. Compuesto, sal farmacéuticamente aceptable o composición farmacéutica, según cualquiera de las reivindicaciones 1 a 13, para su uso en el tratamiento de un trastorno seleccionado entre cáncer de mama, cáncer de ovario, cáncer de pulmón, cáncer de hígado, sarcoma e hiperlipidemia.
- 15. Compuesto, sal farmacéuticamente aceptable o composición farmacéutica, según cualquiera de las reivindicaciones 1 a 13, para su uso en el tratamiento del carcinoma hepatocelular.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261670379P | 2012-07-11 | 2012-07-11 | |
| US201261746666P | 2012-12-28 | 2012-12-28 | |
| PCT/US2013/050106 WO2014011900A2 (en) | 2012-07-11 | 2013-07-11 | Inhibitors of the fibroblast growth factor receptor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2916220T3 true ES2916220T3 (es) | 2022-06-29 |
Family
ID=48874535
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13740458T Active ES2916220T3 (es) | 2012-07-11 | 2013-07-11 | Inhibidores del receptor de factor de crecimiento de fibroblasto |
Country Status (26)
| Country | Link |
|---|---|
| US (6) | US8802697B2 (es) |
| EP (1) | EP2872491B1 (es) |
| JP (1) | JP6104377B2 (es) |
| KR (2) | KR102163776B1 (es) |
| CN (2) | CN104540809B (es) |
| AU (2) | AU2013290074A1 (es) |
| BR (1) | BR112015000653A2 (es) |
| CA (1) | CA2878412A1 (es) |
| CY (1) | CY1124414T1 (es) |
| DK (1) | DK2872491T3 (es) |
| ES (1) | ES2916220T3 (es) |
| HK (1) | HK1206023A1 (es) |
| HR (1) | HRP20211218T1 (es) |
| HU (1) | HUE055502T2 (es) |
| IL (1) | IL236611A (es) |
| LT (1) | LT2872491T (es) |
| MX (1) | MX369472B (es) |
| NZ (1) | NZ703495A (es) |
| PL (1) | PL2872491T3 (es) |
| PT (1) | PT2872491T (es) |
| RS (1) | RS62233B1 (es) |
| RU (2) | RU2019102203A (es) |
| SG (1) | SG11201500125QA (es) |
| SI (1) | SI2872491T1 (es) |
| WO (1) | WO2014011900A2 (es) |
| ZA (1) | ZA201500215B (es) |
Families Citing this family (171)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR077468A1 (es) | 2009-07-09 | 2011-08-31 | Array Biopharma Inc | Compuestos de pirazolo (1,5 -a) pirimidina sustituidos como inhibidores de trk- quinasa |
| WO2012088266A2 (en) | 2010-12-22 | 2012-06-28 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of fgfr3 |
| JP6254524B2 (ja) | 2011-07-01 | 2017-12-27 | エヌジーエム バイオファーマシューティカルス,インコーポレーテッド | 代謝障害及び疾患の治療のための組成物、使用及び方法 |
| PL3176170T3 (pl) | 2012-06-13 | 2019-05-31 | Incyte Holdings Corp | Podstawione związki tricykliczne jako inhibitory fgfr |
| PL2872491T3 (pl) | 2012-07-11 | 2021-12-13 | Blueprint Medicines Corporation | Inhibitory receptora czynnika wzrostu fibroblastów |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9290557B2 (en) | 2012-11-28 | 2016-03-22 | Ngm Biopharmaceuticals, Inc. | Compositions comprising variants and fusions of FGF19 polypeptides |
| ES2828505T3 (es) | 2012-11-28 | 2021-05-26 | Ngm Biopharmaceuticals Inc | Composiciones y métodos para el tratamiento de trastornos y enfermedades metabólicos |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| EP4083221A1 (en) | 2012-12-27 | 2022-11-02 | NGM Biopharmaceuticals, Inc. | Chimeric fgf19 peptides for use in treating bile acid disorders |
| US9273107B2 (en) | 2012-12-27 | 2016-03-01 | Ngm Biopharmaceuticals, Inc. | Uses and methods for modulating bile acid homeostasis and treatment of bile acid disorders and diseases |
| AR094812A1 (es) | 2013-02-20 | 2015-08-26 | Eisai R&D Man Co Ltd | Derivado de piridina monocíclico como inhibidor del fgfr |
| BR112015022191A8 (pt) | 2013-03-15 | 2018-01-23 | Celgene Avilomics Res Inc | compostos heteroarila e usos dos mesmos |
| EP2970231A1 (en) | 2013-03-15 | 2016-01-20 | Blueprint Medicines Corporation | Piperazine derivatives and their use as kit modulators |
| US9227978B2 (en) | 2013-03-15 | 2016-01-05 | Araxes Pharma Llc | Covalent inhibitors of Kras G12C |
| MY181020A (en) * | 2013-03-15 | 2020-12-16 | Sanofi Sa | Heteroaryl compounds and uses thereof |
| AR095464A1 (es) | 2013-03-15 | 2015-10-21 | Celgene Avilomics Res Inc | Compuestos de heteroarilo y usos de los mismos |
| PT2986610T (pt) | 2013-04-19 | 2018-03-09 | Incyte Holdings Corp | Heterociclos bicíclicos como inibidores de fgfr |
| US9783504B2 (en) | 2013-07-09 | 2017-10-10 | Dana-Farber Cancer Institute, Inc. | Kinase inhibitors for the treatment of disease |
| KR102339228B1 (ko) | 2013-08-23 | 2021-12-13 | 뉴파마, 인크. | 특정 화학 물질, 조성물, 및 방법 |
| JO3805B1 (ar) | 2013-10-10 | 2021-01-31 | Araxes Pharma Llc | مثبطات كراس جي12سي |
| RS57542B1 (sr) | 2013-10-17 | 2018-10-31 | Blueprint Medicines Corp | Kompozicije korisne za lečenje poremećaja povezanih sa kit-om |
| SG11201602069WA (en) | 2013-10-18 | 2016-04-28 | Eisai R&D Man Co Ltd | Pyrimidine fgfr4 inhibitors |
| CN110028491B (zh) | 2013-10-25 | 2022-02-11 | 缆图药品公司 | 纤维母细胞生长因子受体抑制剂 |
| AU2014338549B2 (en) | 2013-10-25 | 2017-05-25 | Novartis Ag | Ring-fused bicyclic pyridyl derivatives as FGFR4 inhibitors |
| JP6560202B2 (ja) | 2013-10-28 | 2019-08-21 | エヌジーエム バイオファーマシューティカルス,インコーポレーテッド | 癌モデル及び関連方法 |
| US9695165B2 (en) | 2014-01-15 | 2017-07-04 | Blueprint Medicines Corporation | Inhibitors of the fibroblast growth factor receptor |
| EA031317B1 (ru) | 2014-01-20 | 2018-12-28 | Клив Байосайенсиз, Инк. | КОНДЕНСИРОВАННЫЕ ПИРИМИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КОМПЛЕКСА p97 |
| MX2016009555A (es) | 2014-01-24 | 2016-12-08 | Ngm Biopharmaceuticals Inc | Proteinas de union y metodos para utilizarlas. |
| RU2721723C2 (ru) | 2014-02-07 | 2020-05-21 | Принсипиа Биофарма, Инк. | Производные хинолона как ингибиторы рецептора фактора роста фибробластов |
| EP3114124A1 (en) | 2014-03-03 | 2017-01-11 | Principia Biopharma, Inc. | BENZIMIDAZOLE DERIVATIVES AS RLK and ITK INHIBITORS |
| WO2015183890A2 (en) | 2014-05-28 | 2015-12-03 | Ngm Biopharmaceuticals, Inc. | Methods and compositions for the treatment of metabolic disorders and diseases |
| CA2951153A1 (en) | 2014-06-16 | 2015-12-23 | Ngm Biopharmaceuticals, Inc. | Methods and uses for modulating bile acid homeostasis and treatment of bile acid disorders and diseases |
| CN105017227B (zh) * | 2014-07-08 | 2018-03-09 | 四川百利药业有限责任公司 | N‑(1h‑吡唑‑5‑基)喹唑啉‑4‑胺类化合物 |
| SG11201700703XA (en) | 2014-08-18 | 2017-03-30 | Eisai R&D Man Co Ltd | Salt of monocyclic pyridine derivative and crystal thereof |
| JO3556B1 (ar) | 2014-09-18 | 2020-07-05 | Araxes Pharma Llc | علاجات مدمجة لمعالجة السرطان |
| US20180185341A1 (en) | 2014-10-03 | 2018-07-05 | Novartis Ag | Use of ring-fused bicyclic pyridyl derivatives as fgfr4 inhibitors |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| RU2729161C2 (ru) | 2014-10-23 | 2020-08-04 | ЭнДжиЭм БАЙОФАРМАСЬЮТИКАЛЗ, ИНК. | Фармацевтические композиции, содержащие варианты пептидов, и способы их применения |
| US10434144B2 (en) | 2014-11-07 | 2019-10-08 | Ngm Biopharmaceuticals, Inc. | Methods for treatment of bile acid-related disorders and prediction of clinical sensitivity to treatment of bile acid-related disorders |
| WO2016115412A1 (en) | 2015-01-18 | 2016-07-21 | Newave Pharmaceutical Llc | Dual-warhead covalent inhibitors of fgfr-4 |
| WO2016127074A1 (en) | 2015-02-06 | 2016-08-11 | Blueprint Medicines Corporation | 2-(pyridin-3-yl)-pyrimidine derivatives as ret inhibitors |
| EP3270694A4 (en) * | 2015-02-17 | 2018-09-05 | Neupharma, Inc. | Certain chemical entities, compositions, and methods |
| ES2751669T3 (es) | 2015-02-20 | 2020-04-01 | Incyte Corp | Heterociclos bicíclicos como inhibidores FGFR |
| MA41551A (fr) * | 2015-02-20 | 2017-12-26 | Incyte Corp | Hétérocycles bicycliques utilisés en tant qu'inhibiteurs de fgfr4 |
| WO2016134294A1 (en) | 2015-02-20 | 2016-08-25 | Incyte Corporation | Bicyclic heterocycles as fgfr4 inhibitors |
| AU2016238436A1 (en) | 2015-03-25 | 2017-08-17 | Novartis Ag | Formylated N-heterocyclic derivatives as FGFR4 inhibitors |
| US9802917B2 (en) | 2015-03-25 | 2017-10-31 | Novartis Ag | Particles of N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1-yl)methyl)-3,4-dihydro-1,8-naphthyridine-1(2H)-carboxamide |
| EA201792214A1 (ru) | 2015-04-10 | 2018-01-31 | Араксис Фарма Ллк | Соединения замещенного хиназолина |
| KR102548229B1 (ko) | 2015-04-14 | 2023-06-27 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 결정질 fgfr4 억제제 화합물 및 그의 용도 |
| EP3283462B1 (en) | 2015-04-15 | 2020-12-02 | Araxes Pharma LLC | Fused-tricyclic inhibitors of kras and methods of use thereof |
| EP3298011B1 (en) | 2015-05-22 | 2021-11-17 | Principia Biopharma Inc. | Quinolone derivatives as fibroblast growth factor receptor inhibitors |
| WO2016196840A1 (en) | 2015-06-03 | 2016-12-08 | Principia Biopharma Inc. | Tyrosine kinase inhibitors |
| JP6816100B2 (ja) | 2015-07-16 | 2021-01-20 | アレイ バイオファーマ、インコーポレイテッド | RETキナーゼ阻害物質としての置換ピラゾロ[1,5−a]ピリジン化合物 |
| US10144724B2 (en) | 2015-07-22 | 2018-12-04 | Araxes Pharma Llc | Substituted quinazoline compounds and methods of use thereof |
| RU2018106483A (ru) | 2015-07-24 | 2019-08-26 | Блюпринт Медсинс Корпорейшн | Соединения, подходящие для лечения расстройств, связанных с kit и pdgfr |
| WO2017019957A2 (en) | 2015-07-29 | 2017-02-02 | Ngm Biopharmaceuticals, Inc. | Binding proteins and methods of use thereof |
| CN107922410A (zh) * | 2015-08-11 | 2018-04-17 | 普林斯匹亚生物制药公司 | 用于制备fgfr抑制剂的方法 |
| KR20180043810A (ko) | 2015-08-26 | 2018-04-30 | 블루프린트 메디신즈 코포레이션 | Ntrk에 관련된 장애 치료용으로 유용한 화합물 및 조성물 |
| EP3356351A1 (en) | 2015-09-28 | 2018-08-08 | Araxes Pharma LLC | Inhibitors of kras g12c mutant proteins |
| WO2017058902A1 (en) | 2015-09-28 | 2017-04-06 | Araxes Pharma Llc | Inhibitors of kras g12c mutant proteins |
| US10975071B2 (en) | 2015-09-28 | 2021-04-13 | Araxes Pharma Llc | Inhibitors of KRAS G12C mutant proteins |
| EP3356354A1 (en) | 2015-09-28 | 2018-08-08 | Araxes Pharma LLC | Inhibitors of kras g12c mutant proteins |
| EP3356359B1 (en) | 2015-09-28 | 2021-10-20 | Araxes Pharma LLC | Inhibitors of kras g12c mutant proteins |
| EP3356347A1 (en) | 2015-09-28 | 2018-08-08 | Araxes Pharma LLC | Inhibitors of kras g12c mutant proteins |
| EP3356353A1 (en) | 2015-09-28 | 2018-08-08 | Araxes Pharma LLC | Inhibitors of kras g12c mutant proteins |
| EP3365335B1 (en) * | 2015-10-23 | 2024-02-14 | Array Biopharma, Inc. | 2-aryl- and 2-heteroaryl-substituted 2-pyridazin-3(2h)-one compounds as inhibitors of fgfr tyrosine kinases |
| CN108697708A (zh) | 2015-10-26 | 2018-10-23 | 洛克索肿瘤学股份有限公司 | Trk抑制剂抗性癌症中的点突变以及与此相关的方法 |
| KR20180073689A (ko) | 2015-11-02 | 2018-07-02 | 블루프린트 메디신즈 코포레이션 | Ret의 저해제 |
| AU2016353988B2 (en) | 2015-11-09 | 2019-09-26 | Ngm Biopharmaceuticals, Inc. | Methods for treatment of bile acid-related disorders |
| KR20180081596A (ko) | 2015-11-16 | 2018-07-16 | 아락세스 파마 엘엘씨 | 치환된 헤테로사이클릭 그룹을 포함하는 2-치환된 퀴나졸린 화합물 및 이의 사용 방법 |
| BR112018010024A8 (pt) | 2015-11-19 | 2019-02-26 | Blueprint Medicines Corp | compostos e composições úteis no tratamento de distúrbios relacionados a ntrk |
| CN105669566A (zh) * | 2016-03-09 | 2016-06-15 | 华东师范大学 | 一种医药中间体n-芳基喹唑啉-2-胺化合物的制备方法 |
| WO2017161269A1 (en) | 2016-03-17 | 2017-09-21 | Blueprint Medicines Corporation | Inhibitors of ret receptor tyrosine kinases |
| US10045991B2 (en) | 2016-04-04 | 2018-08-14 | Loxo Oncology, Inc. | Methods of treating pediatric cancers |
| GEP20227339B (en) | 2016-04-04 | 2022-01-25 | Loxo Oncology Inc | Liquid formulations of (s)-n-(5-((r)-2-(2,5-difluorophenyl)-pyrrolidin-1-yl)- pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydro-xypyrrolidine-1-carboxamide |
| TN2019000332A1 (en) | 2016-04-04 | 2021-05-07 | Loxo Oncology Inc | Methods of treating pediatric cancers |
| MX2018012664A (es) | 2016-04-15 | 2019-03-07 | Blueprint Medicines Corp | Inhibidores de quinasa tipo receptor de activina. |
| US11357769B2 (en) | 2016-05-10 | 2022-06-14 | Eisai R&D Management Co., Ltd. | Drug combinations for reducing cell viability and/or cell proliferation |
| MX2018014116A (es) | 2016-05-18 | 2019-12-11 | Array Biopharma Inc | Procesos para la preparacion de (s)-n-(5-((r)-2-(2,5-difluorofenil )pirrolidin-1-il)-pirazolo[1,5-a]pirimidin-3-il)-3-hidroxipirroli din-1-carboxamida y sales del mismo. |
| KR20180002053A (ko) | 2016-06-28 | 2018-01-05 | 한미약품 주식회사 | 신규한 헤테로시클릭 유도체 화합물 및 이의 용도 |
| WO2018004258A1 (ko) * | 2016-06-28 | 2018-01-04 | 한미약품 주식회사 | 신규한 헤테로시클릭 유도체 화합물 및 이의 용도 |
| US10646488B2 (en) | 2016-07-13 | 2020-05-12 | Araxes Pharma Llc | Conjugates of cereblon binding compounds and G12C mutant KRAS, HRAS or NRAS protein modulating compounds and methods of use thereof |
| US10227329B2 (en) | 2016-07-22 | 2019-03-12 | Blueprint Medicines Corporation | Compounds useful for treating disorders related to RET |
| US10035789B2 (en) | 2016-07-27 | 2018-07-31 | Blueprint Medicines Corporation | Compounds useful for treating disorders related to RET |
| CN109843858B (zh) | 2016-08-15 | 2023-05-05 | 润新生物公司 | 某些化学实体、组合物及方法 |
| WO2018039557A1 (en) | 2016-08-26 | 2018-03-01 | Ngm Biopharmaceuticals, Inc. | Methods of treating fibroblast growth factor 19-mediated cancers and tumors |
| JP6751977B2 (ja) | 2016-09-01 | 2020-09-09 | 南京薬捷安康生物科技有限公司 | 線維芽細胞増殖因子受容体の阻害剤及びそれらの使用 |
| WO2018049233A1 (en) | 2016-09-08 | 2018-03-15 | Nicolas Stransky | Inhibitors of the fibroblast growth factor receptor in combination with cyclin-dependent kinase inhibitors |
| WO2018055503A1 (en) | 2016-09-20 | 2018-03-29 | Novartis Ag | Combination comprising a pd-1 antagonist and an fgfr4 inhibitor |
| EP3519402A1 (en) | 2016-09-29 | 2019-08-07 | Araxes Pharma LLC | Inhibitors of kras g12c mutant proteins |
| US10377743B2 (en) | 2016-10-07 | 2019-08-13 | Araxes Pharma Llc | Inhibitors of RAS and methods of use thereof |
| JOP20190077A1 (ar) | 2016-10-10 | 2019-04-09 | Array Biopharma Inc | مركبات بيرازولو [1، 5-a]بيريدين بها استبدال كمثبطات كيناز ret |
| TWI704148B (zh) | 2016-10-10 | 2020-09-11 | 美商亞雷生物製藥股份有限公司 | 作為ret激酶抑制劑之經取代吡唑并[1,5-a]吡啶化合物 |
| WO2018072707A1 (zh) * | 2016-10-18 | 2018-04-26 | 保诺科技(北京)有限公司 | 芳香族醚类衍生物、其制备方法及其在医药上的应用 |
| JOP20190092A1 (ar) | 2016-10-26 | 2019-04-25 | Array Biopharma Inc | عملية لتحضير مركبات بيرازولو[1، 5-a]بيريميدين وأملاح منها |
| WO2018083603A1 (en) | 2016-11-02 | 2018-05-11 | Novartis Ag | Combinations of fgfr4 inhibitors and bile acid sequestrants |
| CA3043948C (en) * | 2016-11-17 | 2021-08-31 | Guangdong Zhongsheng Pharmaceutical Co., Ltd | Fgfr4 inhibitor and preparation method and use thereof |
| KR101812266B1 (ko) | 2016-11-25 | 2017-12-27 | 한국과학기술연구원 | 4-((2-아크릴아미도페닐)아미노)티에노[3,2-d]피리미딘-7-카복스아미드 유도체 및 그의 약학적 활용 |
| AU2017378943B2 (en) * | 2016-12-19 | 2020-06-18 | Abbisko Therapeutics Co., Ltd. | FGFR4 inhibitor, preparation method therefor and pharmaceutical use thereof |
| CN108239069B (zh) * | 2016-12-26 | 2021-01-05 | 南京药捷安康生物科技有限公司 | 一种用于成纤维细胞生长因子受体的抑制剂及其用途 |
| CN108264511B (zh) * | 2017-01-03 | 2021-04-13 | 浙江海正药业股份有限公司 | 杂环类衍生物及其制备方法和其在医药上的用途 |
| WO2018136661A1 (en) | 2017-01-18 | 2018-07-26 | Andrews Steven W | SUBSTITUTED PYRAZOLO[1,5-a]PYRAZINE COMPOUNDS AS RET KINASE INHIBITORS |
| WO2018136663A1 (en) | 2017-01-18 | 2018-07-26 | Array Biopharma, Inc. | Ret inhibitors |
| EP3573967A1 (en) | 2017-01-26 | 2019-12-04 | Araxes Pharma LLC | Fused hetero-hetero bicyclic compounds and methods of use thereof |
| US11279689B2 (en) | 2017-01-26 | 2022-03-22 | Araxes Pharma Llc | 1-(3-(6-(3-hydroxynaphthalen-1-yl)benzofuran-2-yl)azetidin-1 yl)prop-2-en-1-one derivatives and similar compounds as KRAS G12C modulators for treating cancer |
| EP3573954A1 (en) | 2017-01-26 | 2019-12-04 | Araxes Pharma LLC | Fused bicyclic benzoheteroaromatic compounds and methods of use thereof |
| EP3573970A1 (en) | 2017-01-26 | 2019-12-04 | Araxes Pharma LLC | 1-(6-(3-hydroxynaphthalen-1-yl)quinazolin-2-yl)azetidin-1-yl)prop-2-en-1-one derivatives and similar compounds as kras g12c inhibitors for the treatment of cancer |
| WO2018140599A1 (en) | 2017-01-26 | 2018-08-02 | Araxes Pharma Llc | Benzothiophene and benzothiazole compounds and methods of use thereof |
| CN108503593B (zh) * | 2017-02-28 | 2021-04-27 | 暨南大学 | 2-氨基嘧啶类化合物及其应用 |
| JOP20190213A1 (ar) | 2017-03-16 | 2019-09-16 | Array Biopharma Inc | مركبات حلقية ضخمة كمثبطات لكيناز ros1 |
| US11040979B2 (en) | 2017-03-31 | 2021-06-22 | Blueprint Medicines Corporation | Substituted pyrrolo[1,2-b]pyridazines for treating disorders related to KIT and PDGFR |
| JP2020521741A (ja) | 2017-05-25 | 2020-07-27 | アラクセス ファーマ エルエルシー | がんの処置のための化合物およびその使用の方法 |
| CN110831933A (zh) | 2017-05-25 | 2020-02-21 | 亚瑞克西斯制药公司 | 喹唑啉衍生物作为突变kras、hras或nras的调节剂 |
| US10745385B2 (en) | 2017-05-25 | 2020-08-18 | Araxes Pharma Llc | Covalent inhibitors of KRAS |
| AR111960A1 (es) | 2017-05-26 | 2019-09-04 | Incyte Corp | Formas cristalinas de un inhibidor de fgfr y procesos para su preparación |
| EP3444275A1 (en) | 2017-08-16 | 2019-02-20 | Exiris S.r.l. | Monoclonal antibody anti-fgfr4 |
| JP7346420B2 (ja) * | 2017-09-05 | 2023-09-19 | バイオアーディス エルエルシー | 芳香族誘導体、その調製方法、およびその医学的適用 |
| TWI791053B (zh) | 2017-10-10 | 2023-02-01 | 美商亞雷生物製藥股份有限公司 | 6-(2-羥基-2-甲基丙氧基)-4-(6-(6-((6-甲氧基吡啶-3-基)甲基)-3,6-二氮雜雙環[3.1.1]庚-3-基)吡啶-3-基)吡唑并[1,5-a]吡啶-3-甲腈之結晶形式及其醫藥組合物 |
| TWI812649B (zh) | 2017-10-10 | 2023-08-21 | 美商絡速藥業公司 | 6-(2-羥基-2-甲基丙氧基)-4-(6-(6-((6-甲氧基吡啶-3-基)甲基)-3,6-二氮雜雙環[3.1.1]庚-3-基)吡啶-3-基)吡唑并[1,5-a]吡啶-3-甲腈之調配物 |
| CN111566102B (zh) | 2017-10-18 | 2023-09-08 | 缆图药品公司 | 作为激活素受体样激酶抑制剂的取代的吡咯并吡啶 |
| JOP20180094A1 (ar) | 2017-10-18 | 2019-04-18 | Hk Inno N Corp | مركب حلقي غير متجانس كمثبط بروتين كيناز |
| CN109721600B (zh) * | 2017-10-30 | 2021-04-27 | 上海凌达生物医药有限公司 | 一类含氮稠环化合物及其制备方法和用途 |
| US11524963B2 (en) | 2018-01-18 | 2022-12-13 | Array Biopharma Inc. | Substituted pyrazolo[3,4-d]pyrimidines as RET kinase inhibitors |
| TWI802635B (zh) | 2018-01-18 | 2023-05-21 | 美商亞雷生物製藥股份有限公司 | 作為ret激酶抑制劑之經取代吡咯并[2,3-d]嘧啶化合物 |
| WO2019143994A1 (en) | 2018-01-18 | 2019-07-25 | Array Biopharma Inc. | Substituted pyrazolyl[4,3-c]pyridinecompounds as ret kinase inhibitors |
| US11286248B2 (en) | 2018-02-08 | 2022-03-29 | Zhangzhou Pien Tze Huang Pharmaceutical Co., Ltd. | Pyrazine-2(1H)-ketone compound acting as FGFR inhibitor |
| EP3777860A4 (en) | 2018-03-28 | 2021-12-15 | Eisai R&D Management Co., Ltd. | THERAPEUTIC FOR HEPATOCELLULAR CARCINOMA |
| EP3773589B1 (en) | 2018-04-03 | 2023-11-01 | Blueprint Medicines Corporation | Ret inhibitor for use in treating cancer having a ret alteration |
| CN110386921A (zh) * | 2018-04-23 | 2019-10-29 | 南京药捷安康生物科技有限公司 | 成纤维细胞生长因子受体抑制剂化合物 |
| KR20210018264A (ko) | 2018-05-04 | 2021-02-17 | 인사이트 코포레이션 | Fgfr 억제제의 염 |
| US11466004B2 (en) | 2018-05-04 | 2022-10-11 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| CN111868058B (zh) * | 2018-05-25 | 2023-06-13 | 上海和誉生物医药科技有限公司 | 一种fgfr抑制剂、其制备方法和在药学上的应用 |
| CN110577524B (zh) * | 2018-06-07 | 2022-01-28 | 北京大学深圳研究生院 | 一种激酶选择性抑制剂 |
| WO2019242689A1 (zh) * | 2018-06-22 | 2019-12-26 | 北京赛特明强医药科技有限公司 | 一种氰基取代吡啶及氰基取代嘧啶类化合物、制备方法及其应用 |
| EP3829543A1 (en) | 2018-07-31 | 2021-06-09 | Loxo Oncology, Inc. | Spray-dried dispersions and formulations of (s)-5-amino-3-(4-((5-fluoro-2-methoxybenzamido)methyl)phenyl)-1-(1,1,1-trifluoro propan-2-yl)-1h-pyrazole-4-carboxamide |
| JP2022500383A (ja) | 2018-09-10 | 2022-01-04 | アレイ バイオファーマ インコーポレイテッド | Retキナーゼ阻害剤としての縮合複素環式化合物 |
| KR102531596B1 (ko) * | 2018-09-14 | 2023-05-11 | 아비스코 테라퓨틱스 컴퍼니 리미티드 | Fgfr억제제, 그 제조 방법 및 용도 |
| CN110950867A (zh) * | 2018-09-27 | 2020-04-03 | 首药控股(北京)有限公司 | 一种fgfr4激酶抑制剂及其制备方法和用途 |
| CN111138459B (zh) * | 2018-11-06 | 2022-10-18 | 南京圣和药业股份有限公司 | Fgfr4抑制剂的光学异构体及其应用 |
| WO2020119606A1 (en) * | 2018-12-10 | 2020-06-18 | Guangdong Newopp Biopharmaceuticals Co., Ltd. | Heterocyclic compounds as inhibitors of fibroblast growth factor receptor |
| WO2020131627A1 (en) | 2018-12-19 | 2020-06-25 | Array Biopharma Inc. | Substituted pyrazolo[1,5-a]pyridine compounds as inhibitors of fgfr tyrosine kinases |
| US20220041579A1 (en) | 2018-12-19 | 2022-02-10 | Array Biopharma Inc. | Substituted quinoxaline compounds as inhibitors of fgfr tyrosine kinases |
| WO2020177067A1 (en) | 2019-03-05 | 2020-09-10 | Bioardis Llc | Aromatic derivatives, preparation methods, and medical uses thereof |
| WO2020185532A1 (en) | 2019-03-08 | 2020-09-17 | Incyte Corporation | Methods of treating cancer with an fgfr inhibitor |
| JP7378488B2 (ja) * | 2019-03-08 | 2023-11-13 | ショウヤオ ホールディングス(ベイジン) カンパニー, リミテッド | Fgfr4キナーゼ阻害剤、その製造方法及び用途 |
| EP3856341B1 (en) | 2019-04-12 | 2023-09-06 | Blueprint Medicines Corporation | Crystalline forms of (s)-1-(4-fluorophenyl)-1-(2-(4-(6-(1-methyl-1h-pyrazol-4-yl)pyrrolo[2,1-f][1,2,4]triazin-4-yl)piperazinyl)-pyrimidin-5-yl)ethan-1-amine and methods of making |
| CN110317176A (zh) * | 2019-07-04 | 2019-10-11 | 沈阳药科大学 | 2-氨基嘧啶类化合物及其用途 |
| US11591329B2 (en) | 2019-07-09 | 2023-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| WO2021018047A1 (zh) * | 2019-07-26 | 2021-02-04 | 南京明德新药研发有限公司 | 作为fgfr和vegfr双重抑制剂的吡啶衍生物 |
| ES2954774T3 (es) * | 2019-08-08 | 2023-11-24 | Zhangzhou Pien Tze Huang Pharm | Forma cristalina A y forma cristalina B del compuesto de pirazina-2(1H)-cetona y su método de preparación |
| CN114174272B (zh) * | 2019-08-08 | 2023-05-12 | 漳州片仔癀药业股份有限公司 | 吡嗪-2(1h)-酮类化合物的制备方法 |
| WO2021023195A1 (zh) * | 2019-08-08 | 2021-02-11 | 漳州片仔癀药业股份有限公司 | 吡嗪-2(1h)-酮类化合物的d晶型及其制备方法 |
| PE20221085A1 (es) | 2019-10-14 | 2022-07-05 | Incyte Corp | Heterociclos biciclicos como inhibidores de fgfr |
| US11566028B2 (en) | 2019-10-16 | 2023-01-31 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| CN112759593A (zh) | 2019-11-01 | 2021-05-07 | 北京伯汇生物技术有限公司 | 桥环并醛基吡啶衍生物及其应用 |
| CA3162010A1 (en) | 2019-12-04 | 2021-06-10 | Incyte Corporation | Derivatives of an fgfr inhibitor |
| CA3163875A1 (en) | 2019-12-04 | 2021-06-10 | Incyte Corporation | Tricyclic heterocycles as fgfr inhibitors |
| CA3162851A1 (en) * | 2019-12-23 | 2021-07-01 | Qiang Zhang | Cyano-substituted pyridine and cyano-substituted pyrimidine compound and preparation method therefor and application thereof |
| AU2020413333A1 (en) | 2019-12-27 | 2022-06-16 | Schrödinger, Inc. | Cyclic compounds and methods of using same |
| WO2021146424A1 (en) * | 2020-01-15 | 2021-07-22 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| WO2022055963A1 (en) | 2020-09-10 | 2022-03-17 | Schrödinger, Inc. | Heterocyclic pericondensed cdc7 kinase inhibitors for the treatment of cancer |
| CN112851587A (zh) * | 2021-01-21 | 2021-05-28 | 药雅科技(上海)有限公司 | 一种用于治疗癌症的炔类杂环化合物及其制备方法与用途 |
| JP2024505890A (ja) | 2021-01-26 | 2024-02-08 | シュレーディンガー, インコーポレイテッド | がん、自己免疫及び炎症性障害の治療に有用な三環式化合物 |
| CN114853740B (zh) * | 2021-02-03 | 2023-08-01 | 药雅科技(上海)有限公司 | 炔类嘧啶化合物作为fgfr抑制剂的制备方法和用途 |
| TW202402754A (zh) | 2021-03-04 | 2024-01-16 | 美商美國禮來大藥廠 | Fgfr3抑制劑化合物 |
| WO2022194160A1 (zh) * | 2021-03-16 | 2022-09-22 | 上海启晟合研医药科技有限公司 | 非索替尼固体形式及其制备方法 |
| TW202300150A (zh) | 2021-03-18 | 2023-01-01 | 美商薛定諤公司 | 環狀化合物及其使用方法 |
| JP2024510520A (ja) * | 2021-03-26 | 2024-03-07 | ハンチョウ アペロア メディスン リサーチ インスティチュート カンパニー リミテッド | ダブルリング雑環fgfr4阻害剤、ダブルリング雑環fgfr4阻害剤を含有する医薬組成物と製剤、及びその用途 |
| EP4352059A1 (en) | 2021-06-09 | 2024-04-17 | Incyte Corporation | Tricyclic heterocycles as fgfr inhibitors |
| CN113912602B (zh) * | 2021-10-14 | 2023-05-05 | 温州医科大学 | 一种2-氧代-1,2-二氢-1,6-萘啶-7-基类化合物及其制备方法和用途 |
Family Cites Families (64)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US173074A (en) * | 1876-02-01 | Improvement in flour and meal bolts | ||
| MX9702245A (es) | 1994-11-14 | 1997-06-28 | Warner Lambert Co | Seis-aril-pirido(2,3-d)pirimidinas y naftiridinas para inhibir la proliferacion celular mediada por la proteina cinasa tirosina. |
| IL117923A (en) * | 1995-05-03 | 2000-06-01 | Warner Lambert Co | Anti-cancer pharmaceutical compositions containing polysubstituted pyrido¬2,3-d¾pyrimidine derivatives and certain such novel compounds |
| GB9708917D0 (en) * | 1997-05-01 | 1997-06-25 | Pfizer Ltd | Compounds useful in therapy |
| DE69939168D1 (de) * | 1998-05-26 | 2008-09-04 | Warner Lambert Co | Bicyclische pyrimidine und bicyclische 3,4-dihydropyrimidine als inhibitoren der zellvermehrung |
| ATE353329T1 (de) * | 1999-10-21 | 2007-02-15 | Hoffmann La Roche | Heteroalkylamino-substituierte bicyclische stickstoffheterocyclen |
| US6403799B1 (en) | 1999-10-21 | 2002-06-11 | Agouron Pharmaceuticals, Inc. | Methods for the preparation of intermediates in the synthesis of HIV-protease inhibitors |
| MXPA02003140A (es) | 1999-11-22 | 2002-09-30 | Warner Lambert Co | Quinazolinas y su uso para la inhibicion de las enzimas cinasa dependientes del ciclin. |
| AU2001245401A1 (en) | 2000-03-01 | 2001-09-12 | Sumitomo Pharmaceuticals Company, Limited | Hydrazones and analogs as cholesterol lowering agents |
| MXPA02011263A (es) * | 2000-08-04 | 2003-03-10 | Warner Lambert Co | 2-(4-piridil)amino-6-dialcoxifenil- pirido(2,3-d) pirimidin-7-onas. |
| WO2002076985A1 (en) | 2001-03-23 | 2002-10-03 | Smithkline Beecham Corporation | Compounds useful as kinase inhibitors for the treatment of hyperproliferative diseases |
| PL220952B1 (pl) | 2002-01-22 | 2016-01-29 | Warner Lambert Co | 2-(Pirydyn-2-yloamino)pirydo[2,3-d]pirymidyn-7-ony |
| US20050009849A1 (en) * | 2003-01-03 | 2005-01-13 | Veach Darren R. | Pyridopyrimidine kinase inhibitors |
| US20050124562A1 (en) | 2003-09-23 | 2005-06-09 | Joseph Guiles | Bis-quinazoline compounds for the treatment of bacterial infections |
| US20070054916A1 (en) | 2004-10-01 | 2007-03-08 | Amgen Inc. | Aryl nitrogen-containing bicyclic compounds and methods of use |
| US7906522B2 (en) * | 2005-04-28 | 2011-03-15 | Kyowa Hakko Kirin Co., Ltd | 2-aminoquinazoline derivatives |
| US20090258882A1 (en) * | 2006-07-28 | 2009-10-15 | Novartis Ag | 2,4-Substituted Quinazolines as Lipid Kinase Inhibitors |
| EP2125755A2 (en) * | 2006-12-22 | 2009-12-02 | Novartis Ag | Quinazolines for pdk1 inhibition |
| EP2114941B1 (en) | 2006-12-22 | 2015-03-25 | Astex Therapeutics Limited | Bicyclic heterocyclic compounds as fgfr inhibitors |
| GB2467670B (en) | 2007-10-04 | 2012-08-01 | Intellikine Inc | Chemical entities and therapeutic uses thereof |
| CA2986640C (en) * | 2008-06-27 | 2019-03-26 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| WO2010026291A1 (en) | 2008-09-03 | 2010-03-11 | Licentia Ltd. | Materials and methods for inhibiting cancer cell invasion related to fgfr4 |
| CA2735937A1 (en) * | 2008-09-05 | 2010-03-11 | Avila Therapeutics, Inc. | Algorithm for designing irreversible inhibitors |
| PE20120424A1 (es) | 2008-12-29 | 2012-05-04 | Fovea Pharmaceuticals | Compuestos de quinazolina sustituidos |
| TWI464160B (zh) | 2009-08-07 | 2014-12-11 | Chugai Pharmaceutical Co Ltd | Amino pyrazole derivative |
| SG179172A1 (en) * | 2009-09-16 | 2012-04-27 | Avila Therapeutics Inc | Protein kinase conjugates and inhibitors |
| UY33227A (es) | 2010-02-19 | 2011-09-30 | Novartis Ag | Compuestos de pirrolopirimidina como inhibidores de la cdk4/6 |
| GB201007286D0 (en) | 2010-04-30 | 2010-06-16 | Astex Therapeutics Ltd | New compounds |
| CN111172162A (zh) | 2011-04-21 | 2020-05-19 | 葛兰素史克公司 | 乙型肝炎病毒(hbv)表达的调节 |
| WO2012158843A2 (en) * | 2011-05-17 | 2012-11-22 | The Regents Of The University Of California | Kinase inhibitors |
| FR2985257B1 (fr) * | 2011-12-28 | 2014-02-14 | Sanofi Sa | Composes dimeres agonistes des recepteurs des fgfs (fgfrs), leur procede de preparation et leur application en therapeutique |
| BR112014017749B1 (pt) | 2012-01-19 | 2021-08-03 | Taiho Pharmaceutical Co., Ltd. | Composto alquinilbenzeno 3,5-dissubstituído e sal do mesmo |
| GB201209609D0 (en) | 2012-05-30 | 2012-07-11 | Astex Therapeutics Ltd | New compounds |
| PL2872491T3 (pl) * | 2012-07-11 | 2021-12-13 | Blueprint Medicines Corporation | Inhibitory receptora czynnika wzrostu fibroblastów |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| WO2014044846A1 (en) | 2012-09-24 | 2014-03-27 | Evotec (Uk) Ltd. | 3-(aryl- or heteroaryl-amino)-7-(3,5-dimethoxyphenyl)isoquinoline derivatives as fgfr inhibitors useful for the treatment of proliferative disorders or dysplasia |
| TWI629266B (zh) * | 2012-12-28 | 2018-07-11 | 藍印藥品公司 | 纖維母細胞生長因子受體之抑制劑 |
| EP3431475B1 (en) | 2013-02-21 | 2021-04-07 | Pfizer Inc | Solid forms of a selective cdk4/6 inhibitor |
| EP2970231A1 (en) | 2013-03-15 | 2016-01-20 | Blueprint Medicines Corporation | Piperazine derivatives and their use as kit modulators |
| BR112015022191A8 (pt) | 2013-03-15 | 2018-01-23 | Celgene Avilomics Res Inc | compostos heteroarila e usos dos mesmos |
| AR095464A1 (es) * | 2013-03-15 | 2015-10-21 | Celgene Avilomics Res Inc | Compuestos de heteroarilo y usos de los mismos |
| CN105379142B (zh) | 2013-07-14 | 2019-06-18 | Lg 电子株式会社 | 在支持大规模天线的无线接入系统中使用天线相关性收发数据符号的方法 |
| RS57542B1 (sr) | 2013-10-17 | 2018-10-31 | Blueprint Medicines Corp | Kompozicije korisne za lečenje poremećaja povezanih sa kit-om |
| WO2015058129A1 (en) | 2013-10-17 | 2015-04-23 | Blueprint Medicines Corporation | Compositions useful for treating disorders related to kit |
| SG11201602069WA (en) | 2013-10-18 | 2016-04-28 | Eisai R&D Man Co Ltd | Pyrimidine fgfr4 inhibitors |
| CN110028491B (zh) * | 2013-10-25 | 2022-02-11 | 缆图药品公司 | 纤维母细胞生长因子受体抑制剂 |
| AU2014338549B2 (en) | 2013-10-25 | 2017-05-25 | Novartis Ag | Ring-fused bicyclic pyridyl derivatives as FGFR4 inhibitors |
| US9695165B2 (en) * | 2014-01-15 | 2017-07-04 | Blueprint Medicines Corporation | Inhibitors of the fibroblast growth factor receptor |
| WO2016022569A1 (en) | 2014-08-04 | 2016-02-11 | Blueprint Medicines Corporation | Compositions useful for treating disorders related to kit |
| WO2016064960A1 (en) | 2014-10-22 | 2016-04-28 | Incyte Corporation | Bicyclic heterocycles as fgfr4 inhibitors |
| WO2016127074A1 (en) | 2015-02-06 | 2016-08-11 | Blueprint Medicines Corporation | 2-(pyridin-3-yl)-pyrimidine derivatives as ret inhibitors |
| ES2751669T3 (es) | 2015-02-20 | 2020-04-01 | Incyte Corp | Heterociclos bicíclicos como inhibidores FGFR |
| MA41551A (fr) | 2015-02-20 | 2017-12-26 | Incyte Corp | Hétérocycles bicycliques utilisés en tant qu'inhibiteurs de fgfr4 |
| WO2016134294A1 (en) | 2015-02-20 | 2016-08-25 | Incyte Corporation | Bicyclic heterocycles as fgfr4 inhibitors |
| RU2018106483A (ru) | 2015-07-24 | 2019-08-26 | Блюпринт Медсинс Корпорейшн | Соединения, подходящие для лечения расстройств, связанных с kit и pdgfr |
| KR20180043810A (ko) | 2015-08-26 | 2018-04-30 | 블루프린트 메디신즈 코포레이션 | Ntrk에 관련된 장애 치료용으로 유용한 화합물 및 조성물 |
| EP3365335B1 (en) | 2015-10-23 | 2024-02-14 | Array Biopharma, Inc. | 2-aryl- and 2-heteroaryl-substituted 2-pyridazin-3(2h)-one compounds as inhibitors of fgfr tyrosine kinases |
| KR20180073689A (ko) | 2015-11-02 | 2018-07-02 | 블루프린트 메디신즈 코포레이션 | Ret의 저해제 |
| BR112018010024A8 (pt) | 2015-11-19 | 2019-02-26 | Blueprint Medicines Corp | compostos e composições úteis no tratamento de distúrbios relacionados a ntrk |
| WO2017161269A1 (en) | 2016-03-17 | 2017-09-21 | Blueprint Medicines Corporation | Inhibitors of ret receptor tyrosine kinases |
| MX2018012664A (es) | 2016-04-15 | 2019-03-07 | Blueprint Medicines Corp | Inhibidores de quinasa tipo receptor de activina. |
| US10227329B2 (en) | 2016-07-22 | 2019-03-12 | Blueprint Medicines Corporation | Compounds useful for treating disorders related to RET |
| US10035789B2 (en) | 2016-07-27 | 2018-07-31 | Blueprint Medicines Corporation | Compounds useful for treating disorders related to RET |
| WO2018049233A1 (en) | 2016-09-08 | 2018-03-15 | Nicolas Stransky | Inhibitors of the fibroblast growth factor receptor in combination with cyclin-dependent kinase inhibitors |
-
2013
- 2013-07-11 PL PL13740458T patent/PL2872491T3/pl unknown
- 2013-07-11 AU AU2013290074A patent/AU2013290074A1/en not_active Abandoned
- 2013-07-11 US US13/939,967 patent/US8802697B2/en active Active
- 2013-07-11 HU HUE13740458A patent/HUE055502T2/hu unknown
- 2013-07-11 WO PCT/US2013/050106 patent/WO2014011900A2/en active Application Filing
- 2013-07-11 KR KR1020157003581A patent/KR102163776B1/ko active IP Right Grant
- 2013-07-11 CA CA2878412A patent/CA2878412A1/en not_active Abandoned
- 2013-07-11 RU RU2019102203A patent/RU2019102203A/ru unknown
- 2013-07-11 RU RU2015104342A patent/RU2679130C2/ru active
- 2013-07-11 BR BR112015000653A patent/BR112015000653A2/pt active Search and Examination
- 2013-07-11 ES ES13740458T patent/ES2916220T3/es active Active
- 2013-07-11 NZ NZ703495A patent/NZ703495A/en not_active IP Right Cessation
- 2013-07-11 MX MX2015000405A patent/MX369472B/es active IP Right Grant
- 2013-07-11 RS RS20210986A patent/RS62233B1/sr unknown
- 2013-07-11 PT PT137404588T patent/PT2872491T/pt unknown
- 2013-07-11 CN CN201380042618.8A patent/CN104540809B/zh active Active
- 2013-07-11 SI SI201331919T patent/SI2872491T1/sl unknown
- 2013-07-11 EP EP13740458.8A patent/EP2872491B1/en active Active
- 2013-07-11 SG SG11201500125QA patent/SG11201500125QA/en unknown
- 2013-07-11 LT LTEP13740458.8T patent/LT2872491T/lt unknown
- 2013-07-11 KR KR1020207028352A patent/KR20200117067A/ko active IP Right Grant
- 2013-07-11 CN CN201811201617.4A patent/CN109627239B/zh active Active
- 2013-07-11 DK DK13740458.8T patent/DK2872491T3/da active
- 2013-07-11 JP JP2015521815A patent/JP6104377B2/ja active Active
-
2014
- 2014-06-27 US US14/318,149 patent/US9126951B2/en active Active
-
2015
- 2015-01-11 IL IL236611A patent/IL236611A/en active IP Right Grant
- 2015-01-13 ZA ZA2015/00215A patent/ZA201500215B/en unknown
- 2015-04-02 US US14/677,162 patent/US9340514B2/en active Active
- 2015-07-13 HK HK15106652.9A patent/HK1206023A1/xx unknown
-
2016
- 2016-04-13 US US15/097,995 patent/US20170066812A1/en not_active Abandoned
-
2017
- 2017-12-07 AU AU2017272281A patent/AU2017272281B2/en not_active Ceased
-
2018
- 2018-01-10 US US15/867,637 patent/US10196436B2/en active Active
- 2018-12-21 US US16/231,184 patent/US20190359682A1/en not_active Abandoned
-
2021
- 2021-07-28 HR HRP20211218TT patent/HRP20211218T1/hr unknown
- 2021-08-03 CY CY20211100691T patent/CY1124414T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2916220T3 (es) | Inhibidores del receptor de factor de crecimiento de fibroblasto | |
| US20140187559A1 (en) | Inhibitors of the fibroblast growth factor receptor | |
| US10000490B2 (en) | Inhibitors of the fibroblast growth factor receptor | |
| ES2924111T3 (es) | Inhibidores del receptor del factor de crecimiento de fibroblastos | |
| CN112279852B (zh) | 三唑并-嘧啶化合物和其用途 | |
| CA2903072C (en) | Pyridopyrimidine or pyrimidopyrimidine compound, preparation method, pharmaceutical composition, and use thereof | |
| ES2864712T3 (es) | Compuesto inhibidor de FGFR4 cristalino y usos del mismo |