CN108290895B - 制备细胞毒性苯并二氮杂*衍生物的方法 - Google Patents
制备细胞毒性苯并二氮杂*衍生物的方法 Download PDFInfo
- Publication number
- CN108290895B CN108290895B CN201680051138.1A CN201680051138A CN108290895B CN 108290895 B CN108290895 B CN 108290895B CN 201680051138 A CN201680051138 A CN 201680051138A CN 108290895 B CN108290895 B CN 108290895B
- Authority
- CN
- China
- Prior art keywords
- formula
- compound
- reacting
- group
- reacted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000004519 manufacturing process Methods 0.000 title claims description 47
- 231100000433 cytotoxic Toxicity 0.000 title description 15
- 230000001472 cytotoxic effect Effects 0.000 title description 15
- 125000003310 benzodiazepinyl group Chemical class N1N=C(C=CC2=C1C=CC=C2)* 0.000 title description 5
- 229940053197 benzodiazepine derivative antiepileptics Drugs 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 1062
- 238000000034 method Methods 0.000 claims abstract description 326
- -1 hydrogen pyridine fluoride Chemical class 0.000 claims description 316
- 150000003839 salts Chemical class 0.000 claims description 206
- 239000003153 chemical reaction reagent Substances 0.000 claims description 176
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical group CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 156
- 238000006243 chemical reaction Methods 0.000 claims description 128
- 239000000178 monomer Substances 0.000 claims description 109
- 230000008569 process Effects 0.000 claims description 107
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 104
- 125000006242 amine protecting group Chemical group 0.000 claims description 79
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 73
- 150000001412 amines Chemical class 0.000 claims description 61
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 59
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 52
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 claims description 48
- 238000010511 deprotection reaction Methods 0.000 claims description 43
- 239000003638 chemical reducing agent Substances 0.000 claims description 40
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 claims description 36
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 34
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical group C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 claims description 30
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 claims description 28
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical group [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 26
- 239000012190 activator Substances 0.000 claims description 25
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 claims description 24
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims description 24
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 24
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 21
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 claims description 20
- 229910052739 hydrogen Inorganic materials 0.000 claims description 16
- 125000005604 azodicarboxylate group Chemical group 0.000 claims description 14
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 13
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 claims description 12
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 12
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 12
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 claims description 12
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 claims description 12
- 239000001257 hydrogen Substances 0.000 claims description 12
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 claims description 12
- 229910000105 potassium hydride Inorganic materials 0.000 claims description 12
- 239000012312 sodium hydride Substances 0.000 claims description 12
- 229910000104 sodium hydride Inorganic materials 0.000 claims description 12
- QKSQWQOAUQFORH-UHFFFAOYSA-N tert-butyl n-[(2-methylpropan-2-yl)oxycarbonylimino]carbamate Chemical compound CC(C)(C)OC(=O)N=NC(=O)OC(C)(C)C QKSQWQOAUQFORH-UHFFFAOYSA-N 0.000 claims description 12
- 125000003821 2-(trimethylsilyl)ethoxymethyl group Chemical group [H]C([H])([H])[Si](C([H])([H])[H])(C([H])([H])[H])C([H])([H])C(OC([H])([H])[*])([H])[H] 0.000 claims description 11
- 239000012320 chlorinating reagent Substances 0.000 claims description 9
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 8
- 239000003880 polar aprotic solvent Substances 0.000 claims description 8
- UAYWVJHJZHQCIE-UHFFFAOYSA-L zinc iodide Chemical compound I[Zn]I UAYWVJHJZHQCIE-UHFFFAOYSA-L 0.000 claims description 8
- OQJBFFCUFALWQL-UHFFFAOYSA-N n-(piperidine-1-carbonylimino)piperidine-1-carboxamide Chemical compound C1CCCCN1C(=O)N=NC(=O)N1CCCCC1 OQJBFFCUFALWQL-UHFFFAOYSA-N 0.000 claims description 5
- 239000011701 zinc Substances 0.000 claims description 5
- BQZGVMWPHXIKEQ-UHFFFAOYSA-L iron(ii) iodide Chemical compound [Fe+2].[I-].[I-] BQZGVMWPHXIKEQ-UHFFFAOYSA-L 0.000 claims description 4
- JVBXVOWTABLYPX-UHFFFAOYSA-L sodium dithionite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])=O JVBXVOWTABLYPX-UHFFFAOYSA-L 0.000 claims description 4
- 229910052979 sodium sulfide Inorganic materials 0.000 claims description 4
- GRVFOGOEDUUMBP-UHFFFAOYSA-N sodium sulfide (anhydrous) Chemical compound [Na+].[Na+].[S-2] GRVFOGOEDUUMBP-UHFFFAOYSA-N 0.000 claims description 4
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 claims description 4
- ZWYDDDAMNQQZHD-UHFFFAOYSA-L titanium(ii) chloride Chemical compound [Cl-].[Cl-].[Ti+2] ZWYDDDAMNQQZHD-UHFFFAOYSA-L 0.000 claims description 4
- XQKBFQXWZCFNFF-UHFFFAOYSA-K triiodosamarium Chemical compound I[Sm](I)I XQKBFQXWZCFNFF-UHFFFAOYSA-K 0.000 claims description 4
- 238000002360 preparation method Methods 0.000 abstract description 46
- 239000002243 precursor Substances 0.000 abstract description 5
- YMWUJEATGCHHMB-UHFFFAOYSA-N methylene chloride Substances ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 207
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 135
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 108
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 106
- 239000003795 chemical substances by application Substances 0.000 description 105
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 98
- 239000000243 solution Substances 0.000 description 77
- 239000007787 solid Substances 0.000 description 73
- 125000000217 alkyl group Chemical group 0.000 description 72
- 239000002585 base Substances 0.000 description 70
- 239000002904 solvent Substances 0.000 description 67
- 125000004432 carbon atom Chemical group C* 0.000 description 65
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 59
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical group C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 53
- 230000002140 halogenating effect Effects 0.000 description 52
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 52
- 235000019341 magnesium sulphate Nutrition 0.000 description 52
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 51
- 125000006241 alcohol protecting group Chemical group 0.000 description 50
- 239000010410 layer Substances 0.000 description 49
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 47
- 239000012267 brine Substances 0.000 description 45
- 235000019439 ethyl acetate Nutrition 0.000 description 45
- 150000002466 imines Chemical class 0.000 description 45
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 44
- 239000012044 organic layer Substances 0.000 description 43
- 239000000203 mixture Substances 0.000 description 41
- 125000001246 bromo group Chemical group Br* 0.000 description 39
- 125000001424 substituent group Chemical group 0.000 description 39
- 125000004076 pyridyl group Chemical group 0.000 description 38
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 36
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Chemical group C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 35
- 239000011541 reaction mixture Substances 0.000 description 35
- 125000003545 alkoxy group Chemical group 0.000 description 34
- 150000002148 esters Chemical class 0.000 description 33
- 125000000623 heterocyclic group Chemical group 0.000 description 31
- 238000010898 silica gel chromatography Methods 0.000 description 30
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 29
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 29
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 28
- 125000003342 alkenyl group Chemical group 0.000 description 26
- 125000000304 alkynyl group Chemical group 0.000 description 26
- 239000000047 product Substances 0.000 description 26
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 25
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 24
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 24
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 24
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 24
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 23
- 229910052736 halogen Inorganic materials 0.000 description 22
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 21
- 229910052757 nitrogen Inorganic materials 0.000 description 21
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 20
- 238000000746 purification Methods 0.000 description 20
- 125000003118 aryl group Chemical group 0.000 description 19
- GRJJQCWNZGRKAU-UHFFFAOYSA-N pyridin-1-ium;fluoride Chemical compound F.C1=CC=NC=C1 GRJJQCWNZGRKAU-UHFFFAOYSA-N 0.000 description 19
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 19
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 18
- 210000004027 cell Anatomy 0.000 description 18
- 125000001072 heteroaryl group Chemical group 0.000 description 18
- IZDROVVXIHRYMH-UHFFFAOYSA-N methanesulfonic anhydride Chemical compound CS(=O)(=O)OS(C)(=O)=O IZDROVVXIHRYMH-UHFFFAOYSA-N 0.000 description 18
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 18
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 18
- 125000006165 cyclic alkyl group Chemical group 0.000 description 17
- 238000005160 1H NMR spectroscopy Methods 0.000 description 16
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 16
- 239000002202 Polyethylene glycol Substances 0.000 description 16
- 125000005647 linker group Chemical group 0.000 description 16
- 230000000269 nucleophilic effect Effects 0.000 description 16
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 15
- 150000002367 halogens Chemical class 0.000 description 15
- 229920001223 polyethylene glycol Polymers 0.000 description 15
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 14
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 14
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical group [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 14
- 239000011230 binding agent Substances 0.000 description 14
- 150000003138 primary alcohols Chemical class 0.000 description 14
- 239000012043 crude product Substances 0.000 description 13
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 13
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 13
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 13
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 12
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 12
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 12
- 125000006239 protecting group Chemical group 0.000 description 12
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 12
- 239000002253 acid Substances 0.000 description 11
- 230000001588 bifunctional effect Effects 0.000 description 11
- 150000001768 cations Chemical class 0.000 description 11
- 239000012336 iodinating agent Substances 0.000 description 11
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 10
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 10
- 239000004971 Cross linker Substances 0.000 description 10
- 229910006069 SO3H Inorganic materials 0.000 description 10
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 10
- 125000004093 cyano group Chemical group *C#N 0.000 description 10
- 229910052740 iodine Inorganic materials 0.000 description 10
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 10
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Chemical compound [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 10
- 229910052938 sodium sulfate Inorganic materials 0.000 description 10
- 235000011152 sodium sulphate Nutrition 0.000 description 10
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 10
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 10
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 10
- 125000003158 alcohol group Chemical group 0.000 description 9
- 229910052799 carbon Inorganic materials 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 235000019198 oils Nutrition 0.000 description 9
- 229910052760 oxygen Inorganic materials 0.000 description 9
- 239000012453 solvate Substances 0.000 description 9
- 229910052717 sulfur Chemical group 0.000 description 9
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 9
- UIFGGABIJBWRMG-FMQUCBEESA-N (4-chlorophenyl)methyl (ne)-n-[(4-chlorophenyl)methoxycarbonylimino]carbamate Chemical compound C1=CC(Cl)=CC=C1COC(=O)\N=N\C(=O)OCC1=CC=C(Cl)C=C1 UIFGGABIJBWRMG-FMQUCBEESA-N 0.000 description 8
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- 239000012448 Lithium borohydride Substances 0.000 description 8
- 235000001014 amino acid Nutrition 0.000 description 8
- 229940024606 amino acid Drugs 0.000 description 8
- 150000001413 amino acids Chemical class 0.000 description 8
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 8
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 8
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 8
- ZRALSGWEFCBTJO-UHFFFAOYSA-O guanidinium Chemical compound NC(N)=[NH2+] ZRALSGWEFCBTJO-UHFFFAOYSA-O 0.000 description 8
- 125000005842 heteroatom Chemical group 0.000 description 8
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 8
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 8
- 239000003960 organic solvent Substances 0.000 description 8
- 229920000642 polymer Polymers 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- 239000002002 slurry Substances 0.000 description 8
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 8
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 7
- 229910052786 argon Inorganic materials 0.000 description 7
- 125000004429 atom Chemical group 0.000 description 7
- 239000013058 crude material Substances 0.000 description 7
- 239000012351 deprotecting agent Substances 0.000 description 7
- 108090000765 processed proteins & peptides Proteins 0.000 description 7
- 239000011734 sodium Substances 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 150000003459 sulfonic acid esters Chemical class 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 7
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 6
- YAXWOADCWUUUNX-UHFFFAOYSA-N 1,2,2,3-tetramethylpiperidine Chemical compound CC1CCCN(C)C1(C)C YAXWOADCWUUUNX-UHFFFAOYSA-N 0.000 description 6
- RKMGAJGJIURJSJ-UHFFFAOYSA-N 2,2,6,6-Tetramethylpiperidine Substances CC1(C)CCCC(C)(C)N1 RKMGAJGJIURJSJ-UHFFFAOYSA-N 0.000 description 6
- OZMLUMPWPFZWTP-UHFFFAOYSA-N 2-(tributyl-$l^{5}-phosphanylidene)acetonitrile Chemical compound CCCCP(CCCC)(CCCC)=CC#N OZMLUMPWPFZWTP-UHFFFAOYSA-N 0.000 description 6
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 6
- PXACTUVBBMDKRW-UHFFFAOYSA-M 4-bromobenzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-M 0.000 description 6
- FEJUGLKDZJDVFY-UHFFFAOYSA-N 9-borabicyclo(3.3.1)nonane Chemical compound C1CCC2CCCC1B2 FEJUGLKDZJDVFY-UHFFFAOYSA-N 0.000 description 6
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical group C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 6
- 229910004727 OSO3H Inorganic materials 0.000 description 6
- 229910018828 PO3H2 Inorganic materials 0.000 description 6
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical group C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 6
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical group C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 6
- 125000000539 amino acid group Chemical group 0.000 description 6
- 150000008064 anhydrides Chemical class 0.000 description 6
- 229910052794 bromium Inorganic materials 0.000 description 6
- 239000000539 dimer Substances 0.000 description 6
- 150000004678 hydrides Chemical group 0.000 description 6
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 6
- 125000000879 imine group Chemical group 0.000 description 6
- 125000006413 ring segment Chemical group 0.000 description 6
- 235000009518 sodium iodide Nutrition 0.000 description 6
- 238000006277 sulfonation reaction Methods 0.000 description 6
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 6
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 6
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 5
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 5
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 5
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical group C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 5
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 5
- PHSPJQZRQAJPPF-UHFFFAOYSA-N N-alpha-Methylhistamine Chemical compound CNCCC1=CN=CN1 PHSPJQZRQAJPPF-UHFFFAOYSA-N 0.000 description 5
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 description 5
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 5
- 241000610741 Tetraneuris Species 0.000 description 5
- 125000002947 alkylene group Chemical group 0.000 description 5
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 5
- 125000002619 bicyclic group Chemical group 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- JOHCVVJGGSABQY-UHFFFAOYSA-N carbon tetraiodide Chemical compound IC(I)(I)I JOHCVVJGGSABQY-UHFFFAOYSA-N 0.000 description 5
- 239000000460 chlorine Substances 0.000 description 5
- 239000003431 cross linking reagent Substances 0.000 description 5
- LUDURBZOIUWOBW-UHFFFAOYSA-N difluoromethyl-methyl-[tris(dimethylamino)methyl]sulfanium Chemical compound CN(C)C([S+](C(F)F)C)(N(C)C)N(C)C LUDURBZOIUWOBW-UHFFFAOYSA-N 0.000 description 5
- 238000001035 drying Methods 0.000 description 5
- 235000019253 formic acid Nutrition 0.000 description 5
- 238000005658 halogenation reaction Methods 0.000 description 5
- 229910000040 hydrogen fluoride Inorganic materials 0.000 description 5
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 5
- 239000012280 lithium aluminium hydride Substances 0.000 description 5
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 5
- 229910052698 phosphorus Inorganic materials 0.000 description 5
- PZHNNJXWQYFUTD-UHFFFAOYSA-N phosphorus triiodide Chemical compound IP(I)I PZHNNJXWQYFUTD-UHFFFAOYSA-N 0.000 description 5
- 229910052700 potassium Inorganic materials 0.000 description 5
- 239000011591 potassium Substances 0.000 description 5
- 239000003223 protective agent Substances 0.000 description 5
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- ABTOQLMXBSRXSM-UHFFFAOYSA-N silicon tetrafluoride Chemical compound F[Si](F)(F)F ABTOQLMXBSRXSM-UHFFFAOYSA-N 0.000 description 5
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 5
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical group CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 4
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical group C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 4
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- JEDZLBFUGJTJGQ-UHFFFAOYSA-N [Na].COCCO[AlH]OCCOC Chemical compound [Na].COCCO[AlH]OCCOC JEDZLBFUGJTJGQ-UHFFFAOYSA-N 0.000 description 4
- 108010056243 alanylalanine Proteins 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- 229910000085 borane Inorganic materials 0.000 description 4
- 125000002837 carbocyclic group Chemical group 0.000 description 4
- VHILMKFSCRWWIJ-UHFFFAOYSA-N dimethyl acetylenedicarboxylate Chemical compound COC(=O)C#CC(=O)OC VHILMKFSCRWWIJ-UHFFFAOYSA-N 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- XKUKSGPZAADMRA-UHFFFAOYSA-N glycyl-glycyl-glycine Chemical compound NCC(=O)NCC(=O)NCC(O)=O XKUKSGPZAADMRA-UHFFFAOYSA-N 0.000 description 4
- 230000026030 halogenation Effects 0.000 description 4
- 229940071870 hydroiodic acid Drugs 0.000 description 4
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 4
- 125000000842 isoxazolyl group Chemical group 0.000 description 4
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical group [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 4
- 239000001301 oxygen Chemical group 0.000 description 4
- 125000000636 p-nitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)[N+]([O-])=O 0.000 description 4
- NMHMNPHRMNGLLB-UHFFFAOYSA-N phloretic acid Chemical compound OC(=O)CCC1=CC=C(O)C=C1 NMHMNPHRMNGLLB-UHFFFAOYSA-N 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 102000004196 processed proteins & peptides Human genes 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 239000012070 reactive reagent Substances 0.000 description 4
- 239000012419 sodium bis(2-methoxyethoxy)aluminum hydride Substances 0.000 description 4
- 239000012279 sodium borohydride Substances 0.000 description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 description 4
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 125000000547 substituted alkyl group Chemical group 0.000 description 4
- 239000011593 sulfur Chemical group 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 4
- YWWDBCBWQNCYNR-UHFFFAOYSA-N trimethylphosphine Chemical group CP(C)C YWWDBCBWQNCYNR-UHFFFAOYSA-N 0.000 description 4
- COIOYMYWGDAQPM-UHFFFAOYSA-N tris(2-methylphenyl)phosphane Chemical compound CC1=CC=CC=C1P(C=1C(=CC=CC=1)C)C1=CC=CC=C1C COIOYMYWGDAQPM-UHFFFAOYSA-N 0.000 description 4
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 3
- 125000002774 3,4-dimethoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C(OC([H])([H])[H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 3
- 125000002672 4-bromobenzoyl group Chemical group BrC1=CC=C(C(=O)*)C=C1 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- XVZXOLOFWKSDSR-UHFFFAOYSA-N Cc1cc(C)c([C]=O)c(C)c1 Chemical group Cc1cc(C)c([C]=O)c(C)c1 XVZXOLOFWKSDSR-UHFFFAOYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical group Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 3
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical group C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 230000003213 activating effect Effects 0.000 description 3
- 150000001450 anions Chemical class 0.000 description 3
- 229940049706 benzodiazepine Drugs 0.000 description 3
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 3
- 125000004452 carbocyclyl group Chemical group 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 239000010779 crude oil Substances 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 125000003827 glycol group Chemical group 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 125000006501 nitrophenyl group Chemical group 0.000 description 3
- 125000003232 p-nitrobenzoyl group Chemical group [N+](=O)([O-])C1=CC=C(C(=O)*)C=C1 0.000 description 3
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 125000005717 substituted cycloalkylene group Chemical group 0.000 description 3
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 3
- LSNNMFCWUKXFEE-UHFFFAOYSA-L sulfite Chemical class [O-]S([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-L 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 150000003568 thioethers Chemical class 0.000 description 3
- 125000001425 triazolyl group Chemical group 0.000 description 3
- VRDGQQTWSGDXCU-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-iodoacetate Chemical compound ICC(=O)ON1C(=O)CCC1=O VRDGQQTWSGDXCU-UHFFFAOYSA-N 0.000 description 2
- LLXVXPPXELIDGQ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(2,5-dioxopyrrol-1-yl)benzoate Chemical compound C=1C=CC(N2C(C=CC2=O)=O)=CC=1C(=O)ON1C(=O)CCC1=O LLXVXPPXELIDGQ-UHFFFAOYSA-N 0.000 description 2
- JWDFQMWEFLOOED-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(pyridin-2-yldisulfanyl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCSSC1=CC=CC=N1 JWDFQMWEFLOOED-UHFFFAOYSA-N 0.000 description 2
- WGMMKWFUXPMTRW-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-[(2-bromoacetyl)amino]propanoate Chemical compound BrCC(=O)NCCC(=O)ON1C(=O)CCC1=O WGMMKWFUXPMTRW-UHFFFAOYSA-N 0.000 description 2
- PMJWDPGOWBRILU-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[4-(2,5-dioxopyrrol-1-yl)phenyl]butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCC(C=C1)=CC=C1N1C(=O)C=CC1=O PMJWDPGOWBRILU-UHFFFAOYSA-N 0.000 description 2
- VHYRHFNOWKMCHQ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-formylbenzoate Chemical compound C1=CC(C=O)=CC=C1C(=O)ON1C(=O)CCC1=O VHYRHFNOWKMCHQ-UHFFFAOYSA-N 0.000 description 2
- WCMOHMXWOOBVMZ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-[3-(2,5-dioxopyrrol-1-yl)propanoylamino]hexanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCNC(=O)CCN1C(=O)C=CC1=O WCMOHMXWOOBVMZ-UHFFFAOYSA-N 0.000 description 2
- PHDIJLFSKNMCMI-ITGJKDDRSA-N (3R,4S,5R,6R)-6-(hydroxymethyl)-4-(8-quinolin-6-yloxyoctoxy)oxane-2,3,5-triol Chemical compound OC[C@@H]1[C@H]([C@@H]([C@H](C(O1)O)O)OCCCCCCCCOC=1C=C2C=CC=NC2=CC=1)O PHDIJLFSKNMCMI-ITGJKDDRSA-N 0.000 description 2
- MNIPVWXWSPXERA-IDNZQHFXSA-N (6r,7r)-1-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-4,7-dihydroxy-6-(11-phenoxyundecanoyloxy)-2,8-dioxabicyclo[3.2.1]octane-3,4,5-tricarboxylic acid Chemical compound C([C@@H](C)[C@H](OC(C)=O)C(=C)CCC12[C@@H]([C@@H](OC(=O)CCCCCCCCCCOC=3C=CC=CC=3)C(O1)(C(O)=O)C(O)(C(O2)C(O)=O)C(O)=O)O)C1=CC=CC=C1 MNIPVWXWSPXERA-IDNZQHFXSA-N 0.000 description 2
- VLSDXINSOMDCBK-BQYQJAHWSA-N (E)-1,1'-azobis(N,N-dimethylformamide) Chemical compound CN(C)C(=O)\N=N\C(=O)N(C)C VLSDXINSOMDCBK-BQYQJAHWSA-N 0.000 description 2
- QRDAPCMJAOQZSU-KQQUZDAGSA-N (e)-3-[4-[(e)-3-(3-fluorophenyl)-3-oxoprop-1-enyl]-1-methylpyrrol-2-yl]-n-hydroxyprop-2-enamide Chemical compound C1=C(\C=C\C(=O)NO)N(C)C=C1\C=C\C(=O)C1=CC=CC(F)=C1 QRDAPCMJAOQZSU-KQQUZDAGSA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- RVRLFABOQXZUJX-UHFFFAOYSA-N 1-[1-(2,5-dioxopyrrol-1-yl)ethyl]pyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1C(C)N1C(=O)C=CC1=O RVRLFABOQXZUJX-UHFFFAOYSA-N 0.000 description 2
- AASYSXRGODIQGY-UHFFFAOYSA-N 1-[1-(2,5-dioxopyrrol-1-yl)hexyl]pyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1C(CCCCC)N1C(=O)C=CC1=O AASYSXRGODIQGY-UHFFFAOYSA-N 0.000 description 2
- SGVWDRVQIYUSRA-UHFFFAOYSA-N 1-[2-[2-(2,5-dioxopyrrol-1-yl)ethyldisulfanyl]ethyl]pyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1CCSSCCN1C(=O)C=CC1=O SGVWDRVQIYUSRA-UHFFFAOYSA-N 0.000 description 2
- HWGMATHNAWKVEO-UHFFFAOYSA-N 1-[2-[tert-butyl(dimethyl)silyl]phenyl]-N-hydroxymethanesulfonamide Chemical compound [Si](C)(C)(C(C)(C)C)C1=C(CS(=O)(=O)NO)C=CC=C1 HWGMATHNAWKVEO-UHFFFAOYSA-N 0.000 description 2
- FPKVOQKZMBDBKP-UHFFFAOYSA-N 1-[4-[(2,5-dioxopyrrol-1-yl)methyl]cyclohexanecarbonyl]oxy-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)C1CCC(CN2C(C=CC2=O)=O)CC1 FPKVOQKZMBDBKP-UHFFFAOYSA-N 0.000 description 2
- LIEOEYTUTSDYKB-BUHFOSPRSA-N 2,2,2-trichloroethyl (ne)-n-(2,2,2-trichloroethoxycarbonylimino)carbamate Chemical compound ClC(Cl)(Cl)COC(=O)\N=N\C(=O)OCC(Cl)(Cl)Cl LIEOEYTUTSDYKB-BUHFOSPRSA-N 0.000 description 2
- 125000001917 2,4-dinitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C(=C1*)[N+]([O-])=O)[N+]([O-])=O 0.000 description 2
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 2
- QLVGHFBUSGYCCG-UHFFFAOYSA-N 2-amino-n-(1-cyano-2-phenylethyl)acetamide Chemical compound NCC(=O)NC(C#N)CC1=CC=CC=C1 QLVGHFBUSGYCCG-UHFFFAOYSA-N 0.000 description 2
- 125000004918 2-methyl-2-pentyl group Chemical group CC(C)(CCC)* 0.000 description 2
- PBVAJRFEEOIAGW-UHFFFAOYSA-N 3-[bis(2-carboxyethyl)phosphanyl]propanoic acid;hydrochloride Chemical compound Cl.OC(=O)CCP(CCC(O)=O)CCC(O)=O PBVAJRFEEOIAGW-UHFFFAOYSA-N 0.000 description 2
- ZSGXAJPKAMPOIZ-UHFFFAOYSA-N 3-dimethylphosphanylprop-2-enenitrile Chemical compound CP(C)C=CC#N ZSGXAJPKAMPOIZ-UHFFFAOYSA-N 0.000 description 2
- 125000004919 3-methyl-2-pentyl group Chemical group CC(C(C)*)CC 0.000 description 2
- DYPPJKHPJMGHKW-QJGAVIKSSA-N 4,4,5,5,6,6,7,7,8,8,9,9,9-tridecafluorononyl (ne)-n-(4,4,5,5,6,6,7,7,8,8,9,9,9-tridecafluorononoxycarbonylimino)carbamate Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)CCCOC(=O)\N=N\C(=O)OCCCC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F DYPPJKHPJMGHKW-QJGAVIKSSA-N 0.000 description 2
- ZMRMMAOBSFSXLN-UHFFFAOYSA-N 4-[4-(2,5-dioxopyrrol-1-yl)phenyl]butanehydrazide Chemical compound C1=CC(CCCC(=O)NN)=CC=C1N1C(=O)C=CC1=O ZMRMMAOBSFSXLN-UHFFFAOYSA-N 0.000 description 2
- HIHOEGPXVVKJPP-JTQLQIEISA-N 5-fluoro-2-[[(1s)-1-(5-fluoropyridin-2-yl)ethyl]amino]-6-[(5-methyl-1h-pyrazol-3-yl)amino]pyridine-3-carbonitrile Chemical compound N([C@@H](C)C=1N=CC(F)=CC=1)C(C(=CC=1F)C#N)=NC=1NC=1C=C(C)NN=1 HIHOEGPXVVKJPP-JTQLQIEISA-N 0.000 description 2
- LIWMQSWFLXEGMA-WDSKDSINSA-N Ala-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@H](C)N LIWMQSWFLXEGMA-WDSKDSINSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- REDUQXCPUSNJOL-UHFFFAOYSA-N C(C1=CC=CC=C1)NC(CN(C(C1=CC=C(C=C1)C(C)C)=O)CC1=CC=C(C=C1)C(NO)=O)=O Chemical compound C(C1=CC=CC=C1)NC(CN(C(C1=CC=C(C=C1)C(C)C)=O)CC1=CC=C(C=C1)C(NO)=O)=O REDUQXCPUSNJOL-UHFFFAOYSA-N 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- QBXVXKRWOVBUDB-GRKNLSHJSA-N ClC=1C(=CC(=C(CN2[C@H](C[C@H](C2)O)C(=O)O)C1)OCC1=CC(=CC=C1)C#N)OCC1=C(C(=CC=C1)C1=CC2=C(OCCO2)C=C1)C Chemical compound ClC=1C(=CC(=C(CN2[C@H](C[C@H](C2)O)C(=O)O)C1)OCC1=CC(=CC=C1)C#N)OCC1=C(C(=CC=C1)C1=CC2=C(OCCO2)C=C1)C QBXVXKRWOVBUDB-GRKNLSHJSA-N 0.000 description 2
- 229940126650 Compound 3f Drugs 0.000 description 2
- DEFJQIDDEAULHB-QWWZWVQMSA-N D-alanyl-D-alanine Chemical compound C[C@@H]([NH3+])C(=O)N[C@H](C)C([O-])=O DEFJQIDDEAULHB-QWWZWVQMSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 2
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical group C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 2
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical class CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 2
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- DEFJQIDDEAULHB-IMJSIDKUSA-N L-alanyl-L-alanine Chemical compound C[C@H](N)C(=O)N[C@@H](C)C(O)=O DEFJQIDDEAULHB-IMJSIDKUSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- DEFJQIDDEAULHB-UHFFFAOYSA-N N-D-alanyl-D-alanine Natural products CC(N)C(=O)NC(C)C(O)=O DEFJQIDDEAULHB-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical group ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- 229910018823 PO2S2 Inorganic materials 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 229910019250 POS3 Inorganic materials 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- WGVWLKXZBUVUAM-UHFFFAOYSA-N Pentanochlor Chemical compound CCCC(C)C(=O)NC1=CC=C(C)C(Cl)=C1 WGVWLKXZBUVUAM-UHFFFAOYSA-N 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical group C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 101100226004 Rattus norvegicus Erc2 gene Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- YZRLJOVKGWVBHP-UHFFFAOYSA-N [3-amino-5-(hydroxymethyl)phenyl]methanol Chemical compound NC1=CC(CO)=CC(CO)=C1 YZRLJOVKGWVBHP-UHFFFAOYSA-N 0.000 description 2
- UPXCRVHPPXSSDG-UHFFFAOYSA-N [4-(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-heptadecafluorodecyl)phenyl]-diphenylphosphane Chemical compound C1=CC(CCC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F)=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UPXCRVHPPXSSDG-UHFFFAOYSA-N 0.000 description 2
- YLEIFZAVNWDOBM-ZTNXSLBXSA-N ac1l9hc7 Chemical compound C([C@H]12)C[C@@H](C([C@@H](O)CC3)(C)C)[C@@]43C[C@@]14CC[C@@]1(C)[C@@]2(C)C[C@@H]2O[C@]3(O)[C@H](O)C(C)(C)O[C@@H]3[C@@H](C)[C@H]12 YLEIFZAVNWDOBM-ZTNXSLBXSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 150000001345 alkine derivatives Chemical class 0.000 description 2
- SRVFFFJZQVENJC-IHRRRGAJSA-N aloxistatin Chemical compound CCOC(=O)[C@H]1O[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)NCCC(C)C SRVFFFJZQVENJC-IHRRRGAJSA-N 0.000 description 2
- 125000002393 azetidinyl group Chemical group 0.000 description 2
- 125000004069 aziridinyl group Chemical group 0.000 description 2
- 229940050390 benzoate Drugs 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 150000001718 carbodiimides Chemical group 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 229940001468 citrate Drugs 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Natural products OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 2
- 229940125796 compound 3d Drugs 0.000 description 2
- 239000008367 deionised water Substances 0.000 description 2
- 229910021641 deionized water Inorganic materials 0.000 description 2
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 2
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 2
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 2
- 150000002009 diols Chemical class 0.000 description 2
- 125000005883 dithianyl group Chemical group 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- 230000032050 esterification Effects 0.000 description 2
- 238000005886 esterification reaction Methods 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 229940050410 gluconate Drugs 0.000 description 2
- 108010067216 glycyl-glycyl-glycine Proteins 0.000 description 2
- 150000004820 halides Chemical group 0.000 description 2
- 125000005179 haloacetyl group Chemical group 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 2
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 2
- 238000011031 large-scale manufacturing process Methods 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- QAPTWHXHEYAIKG-RCOXNQKVSA-N n-[(1r,2s,5r)-5-(tert-butylamino)-2-[(3s)-2-oxo-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-1-yl]cyclohexyl]acetamide Chemical compound CC(=O)N[C@@H]1C[C@H](NC(C)(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 QAPTWHXHEYAIKG-RCOXNQKVSA-N 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 235000019833 protease Nutrition 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 125000003831 tetrazolyl group Chemical group 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 125000002053 thietanyl group Chemical group 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- 239000010936 titanium Substances 0.000 description 2
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 2
- LDSMXGPRCFQXSK-UHFFFAOYSA-N tripyridin-2-ylphosphane Chemical compound N1=CC=CC=C1P(C=1N=CC=CC=1)C1=CC=CC=N1 LDSMXGPRCFQXSK-UHFFFAOYSA-N 0.000 description 2
- HAEUOUZZVSHGKX-UHFFFAOYSA-N tripyridin-3-ylphosphane Chemical compound C1=CN=CC(P(C=2C=NC=CC=2)C=2C=NC=CC=2)=C1 HAEUOUZZVSHGKX-UHFFFAOYSA-N 0.000 description 2
- RIBNXKYQMSBWFD-UHFFFAOYSA-N tripyridin-4-ylphosphane Chemical compound C1=NC=CC(P(C=2C=CN=CC=2)C=2C=CN=CC=2)=C1 RIBNXKYQMSBWFD-UHFFFAOYSA-N 0.000 description 2
- LFNXCUNDYSYVJY-UHFFFAOYSA-N tris(3-methylphenyl)phosphane Chemical compound CC1=CC=CC(P(C=2C=C(C)C=CC=2)C=2C=C(C)C=CC=2)=C1 LFNXCUNDYSYVJY-UHFFFAOYSA-N 0.000 description 2
- WXAZIUYTQHYBFW-UHFFFAOYSA-N tris(4-methylphenyl)phosphane Chemical compound C1=CC(C)=CC=C1P(C=1C=CC(C)=CC=1)C1=CC=C(C)C=C1 WXAZIUYTQHYBFW-UHFFFAOYSA-N 0.000 description 2
- 125000005500 uronium group Chemical group 0.000 description 2
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 2
- HBENZIXOGRCSQN-VQWWACLZSA-N (1S,2S,6R,14R,15R,16R)-5-(cyclopropylmethyl)-16-[(2S)-2-hydroxy-3,3-dimethylpentan-2-yl]-15-methoxy-13-oxa-5-azahexacyclo[13.2.2.12,8.01,6.02,14.012,20]icosa-8(20),9,11-trien-11-ol Chemical compound N1([C@@H]2CC=3C4=C(C(=CC=3)O)O[C@H]3[C@@]5(OC)CC[C@@]2([C@@]43CC1)C[C@@H]5[C@](C)(O)C(C)(C)CC)CC1CC1 HBENZIXOGRCSQN-VQWWACLZSA-N 0.000 description 1
- XQYKEBAENKXXCJ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-(4-formylphenoxy)acetate Chemical compound C1=CC(C=O)=CC=C1OCC(=O)ON1C(=O)CCC1=O XQYKEBAENKXXCJ-UHFFFAOYSA-N 0.000 description 1
- PVGATNRYUYNBHO-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-(2,5-dioxopyrrol-1-yl)butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCN1C(=O)C=CC1=O PVGATNRYUYNBHO-UHFFFAOYSA-N 0.000 description 1
- VLARLSIGSPVYHX-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-(2,5-dioxopyrrol-1-yl)hexanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCN1C(=O)C=CC1=O VLARLSIGSPVYHX-UHFFFAOYSA-N 0.000 description 1
- IHVODYOQUSEYJJ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-[[4-[(2,5-dioxopyrrol-1-yl)methyl]cyclohexanecarbonyl]amino]hexanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCNC(=O)C(CC1)CCC1CN1C(=O)C=CC1=O IHVODYOQUSEYJJ-UHFFFAOYSA-N 0.000 description 1
- KVYDWWCHNVBFJG-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-hydrazinylpyridine-3-carboxylate;hydrochloride Chemical compound Cl.C1=NC(NN)=CC=C1C(=O)ON1C(=O)CCC1=O KVYDWWCHNVBFJG-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- KQRHTCDQWJLLME-XUXIUFHCSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-aminopropanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-4-methylpentanoic acid Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)N KQRHTCDQWJLLME-XUXIUFHCSA-N 0.000 description 1
- AGGWFDNPHKLBBV-YUMQZZPRSA-N (2s)-2-[[(2s)-2-amino-3-methylbutanoyl]amino]-5-(carbamoylamino)pentanoic acid Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCNC(N)=O AGGWFDNPHKLBBV-YUMQZZPRSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- JNPGUXGVLNJQSQ-BGGMYYEUSA-M (e,3r,5s)-7-[4-(4-fluorophenyl)-1,2-di(propan-2-yl)pyrrol-3-yl]-3,5-dihydroxyhept-6-enoate Chemical compound CC(C)N1C(C(C)C)=C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)C(C=2C=CC(F)=CC=2)=C1 JNPGUXGVLNJQSQ-BGGMYYEUSA-M 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- KYVBNYUBXIEUFW-UHFFFAOYSA-N 1,1,3,3-tetramethylguanidine Chemical compound CN(C)C(=N)N(C)C KYVBNYUBXIEUFW-UHFFFAOYSA-N 0.000 description 1
- 125000005988 1,1-dioxo-thiomorpholinyl group Chemical group 0.000 description 1
- KEIFWROAQVVDBN-UHFFFAOYSA-N 1,2-dihydronaphthalene Chemical class C1=CC=C2C=CCCC2=C1 KEIFWROAQVVDBN-UHFFFAOYSA-N 0.000 description 1
- JPRPJUMQRZTTED-UHFFFAOYSA-N 1,3-dioxolanyl Chemical group [CH]1OCCO1 JPRPJUMQRZTTED-UHFFFAOYSA-N 0.000 description 1
- WORJRXHJTUTINR-UHFFFAOYSA-N 1,4-dioxane;hydron;chloride Chemical compound Cl.C1COCCO1 WORJRXHJTUTINR-UHFFFAOYSA-N 0.000 description 1
- FUHCFUVCWLZEDQ-UHFFFAOYSA-N 1-(2,5-dioxopyrrolidin-1-yl)oxy-1-oxo-4-(pyridin-2-yldisulfanyl)butane-2-sulfonic acid Chemical compound O=C1CCC(=O)N1OC(=O)C(S(=O)(=O)O)CCSSC1=CC=CC=N1 FUHCFUVCWLZEDQ-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical group C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- VNJBTKQBKFMEHH-UHFFFAOYSA-N 1-[4-(2,5-dioxopyrrol-1-yl)-2,3-dihydroxybutyl]pyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1CC(O)C(O)CN1C(=O)C=CC1=O VNJBTKQBKFMEHH-UHFFFAOYSA-N 0.000 description 1
- WXXSHAKLDCERGU-UHFFFAOYSA-N 1-[4-(2,5-dioxopyrrol-1-yl)butyl]pyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1CCCCN1C(=O)C=CC1=O WXXSHAKLDCERGU-UHFFFAOYSA-N 0.000 description 1
- VHYRLCJMMJQUBY-UHFFFAOYSA-N 1-[4-[4-(2,5-dioxopyrrol-1-yl)phenyl]butanoyloxy]-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)CCCC1=CC=C(N2C(C=CC2=O)=O)C=C1 VHYRLCJMMJQUBY-UHFFFAOYSA-N 0.000 description 1
- XQUPVDVFXZDTLT-UHFFFAOYSA-N 1-[4-[[4-(2,5-dioxopyrrol-1-yl)phenyl]methyl]phenyl]pyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1C(C=C1)=CC=C1CC1=CC=C(N2C(C=CC2=O)=O)C=C1 XQUPVDVFXZDTLT-UHFFFAOYSA-N 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- HVRFLESUOTYUCM-UHFFFAOYSA-N 1-nitro-2h-pyrazine Chemical compound [O-][N+](=O)N1CC=NC=C1 HVRFLESUOTYUCM-UHFFFAOYSA-N 0.000 description 1
- BOOYHBPHFVNWNH-OAHLLOKOSA-N 1-tert-butyl-6-[[(1R)-1-(4-chlorophenyl)ethyl]amino]-5-[(4-fluorophenyl)methyl]pyrazolo[3,4-d]pyrimidin-4-one Chemical compound C[C@H](C1=CC=C(C=C1)Cl)NC2=NC3=C(C=NN3C(C)(C)C)C(=O)N2CC4=CC=C(C=C4)F BOOYHBPHFVNWNH-OAHLLOKOSA-N 0.000 description 1
- MVMSCBBUIHUTGJ-UHFFFAOYSA-N 10108-97-1 Natural products C1=2NC(N)=NC(=O)C=2N=CN1C(C(C1O)O)OC1COP(O)(=O)OP(O)(=O)OC1OC(CO)C(O)C(O)C1O MVMSCBBUIHUTGJ-UHFFFAOYSA-N 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical group C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- CVQCTLPZUCCNGD-UHFFFAOYSA-N 2,3,4,5-tetrafluoro-6-sulfobenzoic acid Chemical class OC(=O)C1=C(F)C(F)=C(F)C(F)=C1S(O)(=O)=O CVQCTLPZUCCNGD-UHFFFAOYSA-N 0.000 description 1
- JNESNWCWZVATQY-UHFFFAOYSA-N 2,3,5,6-tetrafluoro-4-sulfobenzoic acid Chemical compound OC(=O)C1=C(F)C(F)=C(S(O)(=O)=O)C(F)=C1F JNESNWCWZVATQY-UHFFFAOYSA-N 0.000 description 1
- KEQTWHPMSVAFDA-UHFFFAOYSA-N 2,3-dihydro-1h-pyrazole Chemical group C1NNC=C1 KEQTWHPMSVAFDA-UHFFFAOYSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical class OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- FFMBYMANYCDCMK-UHFFFAOYSA-N 2,5-dihydro-1h-imidazole Chemical group C1NCN=C1 FFMBYMANYCDCMK-UHFFFAOYSA-N 0.000 description 1
- OWDQCSBZQVISPN-UHFFFAOYSA-N 2-[(2,5-dioxopyrrolidin-1-yl)amino]-4-(2-iodoacetyl)benzoic acid Chemical compound OC(=O)C1=CC=C(C(=O)CI)C=C1NN1C(=O)CCC1=O OWDQCSBZQVISPN-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- WEZDRVHTDXTVLT-GJZGRUSLSA-N 2-[[(2s)-2-[[(2s)-2-[(2-aminoacetyl)amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]acetic acid Chemical compound OC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)CN)CC1=CC=CC=C1 WEZDRVHTDXTVLT-GJZGRUSLSA-N 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- NEAQRZUHTPSBBM-UHFFFAOYSA-N 2-hydroxy-3,3-dimethyl-7-nitro-4h-isoquinolin-1-one Chemical compound C1=C([N+]([O-])=O)C=C2C(=O)N(O)C(C)(C)CC2=C1 NEAQRZUHTPSBBM-UHFFFAOYSA-N 0.000 description 1
- GCFPFWRHYINTRX-UHFFFAOYSA-N 2-hydroxy-4-methylbenzenesulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C(O)=C1 GCFPFWRHYINTRX-UHFFFAOYSA-N 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- 125000004398 2-methyl-2-butyl group Chemical group CC(C)(CC)* 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- SLAMLWHELXOEJZ-UHFFFAOYSA-N 2-nitrobenzoic acid Chemical compound OC(=O)C1=CC=CC=C1[N+]([O-])=O SLAMLWHELXOEJZ-UHFFFAOYSA-N 0.000 description 1
- RSEBUVRVKCANEP-UHFFFAOYSA-N 2-pyrroline Chemical group C1CC=CN1 RSEBUVRVKCANEP-UHFFFAOYSA-N 0.000 description 1
- 125000001698 2H-pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- YLLRPQWLASQXSI-UHFFFAOYSA-N 3-(4b,8a,9,9a-tetrahydro-4aH-pyrido[2,3-b]indol-4-ylamino)phenol Chemical compound Oc1cccc(NC2=CC=NC3NC4C=CC=CC4C23)c1 YLLRPQWLASQXSI-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- 125000004921 3-methyl-3-pentyl group Chemical group CC(CC)(CC)* 0.000 description 1
- JVQIKJMSUIMUDI-UHFFFAOYSA-N 3-pyrroline Chemical group C1NCC=C1 JVQIKJMSUIMUDI-UHFFFAOYSA-N 0.000 description 1
- 125000004364 3-pyrrolinyl group Chemical group [H]C1=C([H])C([H])([H])N(*)C1([H])[H] 0.000 description 1
- MCGBIXXDQFWVDW-UHFFFAOYSA-N 4,5-dihydro-1h-pyrazole Chemical group C1CC=NN1 MCGBIXXDQFWVDW-UHFFFAOYSA-N 0.000 description 1
- KMMHZIBWCXYAAH-UHFFFAOYSA-N 4-bromobenzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=C(Br)C=C1 KMMHZIBWCXYAAH-UHFFFAOYSA-N 0.000 description 1
- 125000004920 4-methyl-2-pentyl group Chemical group CC(CC(C)*)C 0.000 description 1
- OTLNPYWUJOZPPA-UHFFFAOYSA-N 4-nitrobenzoic acid Chemical compound OC(=O)C1=CC=C([N+]([O-])=O)C=C1 OTLNPYWUJOZPPA-UHFFFAOYSA-N 0.000 description 1
- USTVSOYPMQZLSA-UHFFFAOYSA-N 4-nitropicolinic acid Chemical compound OC(=O)C1=CC([N+]([O-])=O)=CC=N1 USTVSOYPMQZLSA-UHFFFAOYSA-N 0.000 description 1
- 125000001826 4H-pyranyl group Chemical group O1C(=CCC=C1)* 0.000 description 1
- PXRKCOCTEMYUEG-UHFFFAOYSA-N 5-aminoisoindole-1,3-dione Chemical compound NC1=CC=C2C(=O)NC(=O)C2=C1 PXRKCOCTEMYUEG-UHFFFAOYSA-N 0.000 description 1
- ZUUJWCKWJJGJOE-UHFFFAOYSA-N 59290-85-6 Chemical class OC(=O)C1=NC=CC=C1[N+]([O-])=O ZUUJWCKWJJGJOE-UHFFFAOYSA-N 0.000 description 1
- MEXUTNIFSHFQRG-UHFFFAOYSA-N 6,7,12,13-tetrahydro-5h-indolo[2,3-a]pyrrolo[3,4-c]carbazol-5-one Chemical compound C12=C3C=CC=C[C]3NC2=C2NC3=CC=C[CH]C3=C2C2=C1C(=O)NC2 MEXUTNIFSHFQRG-UHFFFAOYSA-N 0.000 description 1
- ZIIGSRYPZWDGBT-UHFFFAOYSA-N 610-30-0 Chemical compound OC(=O)C1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O ZIIGSRYPZWDGBT-UHFFFAOYSA-N 0.000 description 1
- QXRNAOYBCYVZCD-BQBZGAKWSA-N Ala-Lys Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCCCN QXRNAOYBCYVZCD-BQBZGAKWSA-N 0.000 description 1
- FSHURBQASBLAPO-WDSKDSINSA-N Ala-Met Chemical compound CSCC[C@@H](C(O)=O)NC(=O)[C@H](C)N FSHURBQASBLAPO-WDSKDSINSA-N 0.000 description 1
- DAQIJMOLTMGJLO-YUMQZZPRSA-N Arg-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@@H](N)CCCNC(N)=N DAQIJMOLTMGJLO-YUMQZZPRSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- CKLJMWTZIZZHCS-UHFFFAOYSA-N Aspartic acid Chemical compound OC(=O)C(N)CC(O)=O CKLJMWTZIZZHCS-UHFFFAOYSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- SVLUQJDKGXGEGB-UHFFFAOYSA-N C1(CCC(N1)=O)=O.N(N)C1(CC=C(C(=O)O)C=C1)C(=O)O Chemical compound C1(CCC(N1)=O)=O.N(N)C1(CC=C(C(=O)O)C=C1)C(=O)O SVLUQJDKGXGEGB-UHFFFAOYSA-N 0.000 description 1
- LJIRTUPUSIRYEO-UHFFFAOYSA-N C1(CCC(N1)=O)=O.N(N)C1=C(C(=O)O)C=CC=N1 Chemical compound C1(CCC(N1)=O)=O.N(N)C1=C(C(=O)O)C=CC=N1 LJIRTUPUSIRYEO-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- UJOBWOGCFQCDNV-UHFFFAOYSA-N Carbazole Natural products C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- CYSWUSAYJNCAKA-FYJFLYSWSA-N ClC1=C(C=CC=2N=C(SC=21)OCC)OC1=CC=C(C=N1)/C=C/[C@H](C)NC(C)=O Chemical compound ClC1=C(C=CC=2N=C(SC=21)OCC)OC1=CC=C(C=N1)/C=C/[C@H](C)NC(C)=O CYSWUSAYJNCAKA-FYJFLYSWSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-YMDCURPLSA-N D-galactopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-YMDCURPLSA-N 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- MVMSCBBUIHUTGJ-GDJBGNAASA-N GDP-alpha-D-mannose Chemical compound C([C@H]1O[C@H]([C@@H]([C@@H]1O)O)N1C=2N=C(NC(=O)C=2N=C1)N)OP(O)(=O)OP(O)(=O)O[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@@H]1O MVMSCBBUIHUTGJ-GDJBGNAASA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 108010009504 Gly-Phe-Leu-Gly Proteins 0.000 description 1
- YLEIWGJJBFBFHC-KBPBESRZSA-N Gly-Phe-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)CN)CC1=CC=CC=C1 YLEIWGJJBFBFHC-KBPBESRZSA-N 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- VAXBXNPRXPHGHG-BJDJZHNGSA-N Ile-Ala-Leu Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)O)N VAXBXNPRXPHGHG-BJDJZHNGSA-N 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical group C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- FADYJNXDPBKVCA-UHFFFAOYSA-N L-Phenylalanyl-L-lysin Natural products NCCCCC(C(O)=O)NC(=O)C(N)CC1=CC=CC=C1 FADYJNXDPBKVCA-UHFFFAOYSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- LSPYFSHXDAYVDI-SRVKXCTJSA-N Leu-Ala-Leu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@H](C(O)=O)CC(C)C LSPYFSHXDAYVDI-SRVKXCTJSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- NVGBPTNZLWRQSY-UWVGGRQHSA-N Lys-Lys Chemical compound NCCCC[C@H](N)C(=O)N[C@H](C(O)=O)CCCCN NVGBPTNZLWRQSY-UWVGGRQHSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- JHKXZYLNVJRAAJ-WDSKDSINSA-N Met-Ala Chemical compound CSCC[C@H](N)C(=O)N[C@@H](C)C(O)=O JHKXZYLNVJRAAJ-WDSKDSINSA-N 0.000 description 1
- LMSMREVZQWXONY-UHFFFAOYSA-N N-(2,5-dioxopyrrolidin-1-yl)-6-hydrazinylpyridine-3-carboxamide propan-2-ylidenehydrazine Chemical compound CC(C)=NN.C1=NC(NN)=CC=C1C(=O)NN1C(=O)CCC1=O LMSMREVZQWXONY-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 229910018830 PO3H Inorganic materials 0.000 description 1
- MIDZLCFIAINOQN-WPRPVWTQSA-N Phe-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@@H](N)CC1=CC=CC=C1 MIDZLCFIAINOQN-WPRPVWTQSA-N 0.000 description 1
- FADYJNXDPBKVCA-STQMWFEESA-N Phe-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@@H](N)CC1=CC=CC=C1 FADYJNXDPBKVCA-STQMWFEESA-N 0.000 description 1
- RBRNEFJTEHPDSL-ACRUOGEOSA-N Phe-Phe-Lys Chemical compound C([C@@H](C(=O)N[C@@H](CCCCN)C(O)=O)NC(=O)[C@@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 RBRNEFJTEHPDSL-ACRUOGEOSA-N 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical group [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Natural products C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 229910019946 S-S Inorganic materials 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229910019939 S—S Inorganic materials 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- HSRXSKHRSXRCFC-WDSKDSINSA-N Val-Ala Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](C)C(O)=O HSRXSKHRSXRCFC-WDSKDSINSA-N 0.000 description 1
- QRZVUAAKNRHEOP-GUBZILKMSA-N Val-Ala-Val Chemical compound [H]N[C@@H](C(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(O)=O QRZVUAAKNRHEOP-GUBZILKMSA-N 0.000 description 1
- IBIDRSSEHFLGSD-YUMQZZPRSA-N Val-Arg Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCN=C(N)N IBIDRSSEHFLGSD-YUMQZZPRSA-N 0.000 description 1
- JKHXYJKMNSSFFL-IUCAKERBSA-N Val-Lys Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCCN JKHXYJKMNSSFFL-IUCAKERBSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- WIQIWPPQGWGVHD-JEDNCBNOSA-N [(2s)-1-[(2-methylpropan-2-yl)oxy]-1-oxopropan-2-yl]azanium;chloride Chemical compound Cl.C[C@H](N)C(=O)OC(C)(C)C WIQIWPPQGWGVHD-JEDNCBNOSA-N 0.000 description 1
- FTLFITNFXXERLN-UHFFFAOYSA-N [3-(hydroxymethyl)-5-nitrophenyl]methanol Chemical compound OCC1=CC(CO)=CC([N+]([O-])=O)=C1 FTLFITNFXXERLN-UHFFFAOYSA-N 0.000 description 1
- PFRUBEOIWWEFOL-UHFFFAOYSA-N [N].[S] Chemical compound [N].[S] PFRUBEOIWWEFOL-UHFFFAOYSA-N 0.000 description 1
- SPEUIVXLLWOEMJ-UHFFFAOYSA-N acetaldehyde dimethyl acetal Natural products COC(C)OC SPEUIVXLLWOEMJ-UHFFFAOYSA-N 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 108010054982 alanyl-leucyl-alanyl-leucine Proteins 0.000 description 1
- HAXFWIACAGNFHA-UHFFFAOYSA-N aldrithiol Chemical compound C=1C=CC=NC=1SSC1=CC=CC=N1 HAXFWIACAGNFHA-UHFFFAOYSA-N 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 229940061720 alpha hydroxy acid Drugs 0.000 description 1
- 150000001280 alpha hydroxy acids Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 238000010976 amide bond formation reaction Methods 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229940064734 aminobenzoate Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- SMWDFEZZVXVKRB-UHFFFAOYSA-N anhydrous quinoline Natural products N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 1
- 150000001454 anthracenes Chemical class 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 108010036533 arginylvaline Proteins 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical group C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004601 benzofurazanyl group Chemical group N1=C2C(=NO1)C(=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- KSCRVOKQPYZBHZ-IXPOFIJOSA-N benzyl n-[(2s)-1-[[(2s)-1-[[(2s)-1-(1,3-benzothiazol-2-yl)-1-oxo-3-[(3s)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]carbamate Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C[C@H]1C(NCC1)=O)C(=O)C=1SC2=CC=CC=C2N=1)C(C)C)C(=O)OCC1=CC=CC=C1 KSCRVOKQPYZBHZ-IXPOFIJOSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 238000007068 beta-elimination reaction Methods 0.000 description 1
- GPRLTFBKWDERLU-UHFFFAOYSA-N bicyclo[2.2.2]octane Chemical compound C1CC2CCC1CC2 GPRLTFBKWDERLU-UHFFFAOYSA-N 0.000 description 1
- GNTFBMAGLFYMMZ-UHFFFAOYSA-N bicyclo[3.2.2]nonane Chemical compound C1CC2CCC1CCC2 GNTFBMAGLFYMMZ-UHFFFAOYSA-N 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- RBHJBMIOOPYDBQ-UHFFFAOYSA-N carbon dioxide;propan-2-one Chemical compound O=C=O.CC(C)=O RBHJBMIOOPYDBQ-UHFFFAOYSA-N 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940126115 compound 4f Drugs 0.000 description 1
- 201000007750 congenital bile acid synthesis defect Diseases 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- MGNCLNQXLYJVJD-UHFFFAOYSA-N cyanuric chloride Chemical compound ClC1=NC(Cl)=NC(Cl)=N1 MGNCLNQXLYJVJD-UHFFFAOYSA-N 0.000 description 1
- 150000001924 cycloalkanes Chemical class 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 150000008049 diazo compounds Chemical class 0.000 description 1
- OCXGTPDKNBIOTF-UHFFFAOYSA-N dibromo(triphenyl)-$l^{5}-phosphane Chemical compound C=1C=CC=CC=1P(Br)(C=1C=CC=CC=1)(Br)C1=CC=CC=C1 OCXGTPDKNBIOTF-UHFFFAOYSA-N 0.000 description 1
- KZNICNPSHKQLFF-UHFFFAOYSA-N dihydromaleimide Natural products O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 1
- 125000005057 dihydrothienyl group Chemical group S1C(CC=C1)* 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 125000002228 disulfide group Chemical group 0.000 description 1
- WBZKQQHYRPRKNJ-UHFFFAOYSA-L disulfite Chemical compound [O-]S(=O)S([O-])(=O)=O WBZKQQHYRPRKNJ-UHFFFAOYSA-L 0.000 description 1
- 125000005411 dithiolanyl group Chemical group S1SC(CC1)* 0.000 description 1
- GRWZHXKQBITJKP-UHFFFAOYSA-L dithionite(2-) Chemical compound [O-]S(=O)S([O-])=O GRWZHXKQBITJKP-UHFFFAOYSA-L 0.000 description 1
- 125000005414 dithiopyridyl group Chemical group 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 229940017705 formaldehyde sulfoxylate Drugs 0.000 description 1
- 229940044170 formate Drugs 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 125000004612 furopyridinyl group Chemical group O1C(=CC2=C1C=CC=N2)* 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 229940114119 gentisate Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 229960004275 glycolic acid Drugs 0.000 description 1
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- KIWQWJKWBHZMDT-UHFFFAOYSA-N homocysteine thiolactone Chemical compound NC1CCSC1=O KIWQWJKWBHZMDT-UHFFFAOYSA-N 0.000 description 1
- 229940042795 hydrazides for tuberculosis treatment Drugs 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- SBGKURINHGJRFN-UHFFFAOYSA-N hydroxymethanesulfinic acid Chemical compound OCS(O)=O SBGKURINHGJRFN-UHFFFAOYSA-N 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical group C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 125000005945 imidazopyridyl group Chemical group 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000003392 indanyl group Chemical class C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003454 indenyl group Chemical class C1(C=CC2=CC=CC=C12)* 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Chemical group CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Chemical group C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 150000004694 iodide salts Chemical group 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- AWJUIBRHMBBTKR-UHFFFAOYSA-N iso-quinoline Natural products C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- GWVMLCQWXVFZCN-UHFFFAOYSA-N isoindoline Chemical group C1=CC=C2CNCC2=C1 GWVMLCQWXVFZCN-UHFFFAOYSA-N 0.000 description 1
- 238000006317 isomerization reaction Methods 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical group C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 108010054155 lysyllysine Proteins 0.000 description 1
- 150000002680 magnesium Chemical class 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 231100000682 maximum tolerated dose Toxicity 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-KHWXYDKHSA-N methanesulfonyl chloride Chemical group C[35S](Cl)(=O)=O QARBMVPHQWIHKH-KHWXYDKHSA-N 0.000 description 1
- 125000004492 methyl ester group Chemical group 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 125000001620 monocyclic carbocycle group Chemical group 0.000 description 1
- UOBSVARXACCLLH-UHFFFAOYSA-N monomethyl adipate Chemical compound COC(=O)CCCCC(O)=O UOBSVARXACCLLH-UHFFFAOYSA-N 0.000 description 1
- GVOISEJVFFIGQE-YCZSINBZSA-N n-[(1r,2s,5r)-5-[methyl(propan-2-yl)amino]-2-[(3s)-2-oxo-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-1-yl]cyclohexyl]acetamide Chemical compound CC(=O)N[C@@H]1C[C@H](N(C)C(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 GVOISEJVFFIGQE-YCZSINBZSA-N 0.000 description 1
- TYRGLVWXHJRKMT-QMMMGPOBSA-N n-benzyloxycarbonyl-l-serine-betalactone Chemical compound OC(=O)[C@H](C)NC(=O)OCC1=CC=CC=C1 TYRGLVWXHJRKMT-QMMMGPOBSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 150000002790 naphthalenes Chemical class 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 150000005338 nitrobenzoic acids Chemical class 0.000 description 1
- ZCYXXKJEDCHMGH-UHFFFAOYSA-N nonane Chemical compound CCCC[CH]CCCC ZCYXXKJEDCHMGH-UHFFFAOYSA-N 0.000 description 1
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical compound C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 1
- BKIMMITUMNQMOS-UHFFFAOYSA-N normal nonane Natural products CCCCCCCCC BKIMMITUMNQMOS-UHFFFAOYSA-N 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-M oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC([O-])=O ZQPPMHVWECSIRJ-KTKRTIGZSA-M 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- XSXHWVKGUXMUQE-UHFFFAOYSA-N osmium dioxide Inorganic materials O=[Os]=O XSXHWVKGUXMUQE-UHFFFAOYSA-N 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229940116315 oxalic acid Drugs 0.000 description 1
- 125000005961 oxazepanyl group Chemical group 0.000 description 1
- 125000003551 oxepanyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 125000000538 pentafluorophenyl group Chemical group FC1=C(F)C(F)=C(*)C(F)=C1F 0.000 description 1
- 108010024607 phenylalanylalanine Proteins 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- XYFCBTPGUUZFHI-UHFFFAOYSA-O phosphonium Chemical compound [PH4+] XYFCBTPGUUZFHI-UHFFFAOYSA-O 0.000 description 1
- 239000011574 phosphorus Chemical group 0.000 description 1
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 1
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 description 1
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 229920003192 poly(bis maleimide) Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 150000003109 potassium Chemical class 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- DNXIASIHZYFFRO-UHFFFAOYSA-N pyrazoline Chemical group C1CN=NC1 DNXIASIHZYFFRO-UHFFFAOYSA-N 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical group C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- VTGOHKSTWXHQJK-UHFFFAOYSA-N pyrimidin-2-ol Chemical group OC1=NC=CC=N1 VTGOHKSTWXHQJK-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 1
- 230000007281 self degradation Effects 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000010802 sludge Substances 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- ULARYIUTHAWJMU-UHFFFAOYSA-M sodium;1-[4-(2,5-dioxopyrrol-1-yl)butanoyloxy]-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)CCCN1C(=O)C=CC1=O ULARYIUTHAWJMU-UHFFFAOYSA-M 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 229960002317 succinimide Drugs 0.000 description 1
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 1
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- 125000001302 tertiary amino group Chemical group 0.000 description 1
- 125000005958 tetrahydrothienyl group Chemical group 0.000 description 1
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 150000007970 thio esters Chemical class 0.000 description 1
- CNHYKKNIIGEXAY-UHFFFAOYSA-N thiolan-2-imine Chemical compound N=C1CCCS1 CNHYKKNIIGEXAY-UHFFFAOYSA-N 0.000 description 1
- 229930192474 thiophene Chemical group 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- MHNHYTDAOYJUEZ-UHFFFAOYSA-N triphenylphosphane Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 MHNHYTDAOYJUEZ-UHFFFAOYSA-N 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- JABYJIQOLGWMQW-UHFFFAOYSA-N undec-4-ene Chemical compound CCCCCCC=CCCC JABYJIQOLGWMQW-UHFFFAOYSA-N 0.000 description 1
- IBIDRSSEHFLGSD-UHFFFAOYSA-N valinyl-arginine Natural products CC(C)C(N)C(=O)NC(C(O)=O)CCCN=C(N)N IBIDRSSEHFLGSD-UHFFFAOYSA-N 0.000 description 1
- 108010073969 valyllysine Proteins 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Images
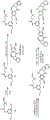





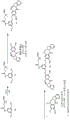












Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
- A61K31/5513—1,4-Benzodiazepines, e.g. diazepam or clozapine
- A61K31/5517—1,4-Benzodiazepines, e.g. diazepam or clozapine condensed with five-membered rings having nitrogen as a ring hetero atom, e.g. imidazobenzodiazepines, triazolam
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/26—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids
- C07C303/28—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids by reaction of hydroxy compounds with sulfonic acids or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/63—Esters of sulfonic acids
- C07C309/64—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms
- C07C309/65—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
- C07F7/1872—Preparation; Treatments not provided for in C07F7/20
- C07F7/188—Preparation; Treatments not provided for in C07F7/20 by reactions involving the formation of Si-O linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Medicinal Preparation (AREA)
- Pyridine Compounds (AREA)
Abstract
本发明涉及制备吲哚啉并苯并二氮杂
Description
相关申请的引用
本申请根据35U.S.C.§119(e)规定要求2016年4月26日提交的美国临时申请号62/327,973和2015年7月21日提交的美国临时申请号62/195,023的提交日期的权益。每篇以上参考的申请的全部内容均通过引用并入本文。
发明领域
本发明涉及用于制备细胞毒性吲哚啉并苯并二氮杂 衍生物的新方法。
衍生物的新方法。
发明背景
已经显示,与先前公开的具有两个亚胺官能团的苯并二氮杂 衍生物相比,具有一个亚胺官能团和一个胺官能团的吲哚啉并苯并二氮杂
衍生物相比,具有一个亚胺官能团和一个胺官能团的吲哚啉并苯并二氮杂 二聚体的细胞结合剂缀合物在体内显示出高得多的治疗指数(最大耐受剂量与最小有效剂量的比率)。参见,例如WO2012/128868。先前公开的制备具有一个亚胺官能团和一个胺官能团的吲哚啉并苯并二氮杂
二聚体的细胞结合剂缀合物在体内显示出高得多的治疗指数(最大耐受剂量与最小有效剂量的比率)。参见,例如WO2012/128868。先前公开的制备具有一个亚胺官能团和一个胺官能团的吲哚啉并苯并二氮杂 二聚体的方法涉及具有两个亚胺官能团的吲哚啉并苯并二氮杂
二聚体的方法涉及具有两个亚胺官能团的吲哚啉并苯并二氮杂 二聚体的部分还原。该部分还原步骤通常导致形成完全还原的副产物和未反应的原料,这需要繁琐的纯化步骤并导致低收率。
二聚体的部分还原。该部分还原步骤通常导致形成完全还原的副产物和未反应的原料,这需要繁琐的纯化步骤并导致低收率。
因此,需要更加有效且适合大规模生产过程的改进的制备吲哚啉并苯并二氮杂 二聚体的方法。
二聚体的方法。
发明概述
本发明提供了制备吲哚啉并苯并二氮杂 二聚体化合物及其合成前体的各种方法。与先前公开的方法相比,本发明的方法可以在不需要繁琐的纯化步骤的情况下以更高的收率生成期望的二聚体化合物。这些方法更适合大规模生产过程。
二聚体化合物及其合成前体的各种方法。与先前公开的方法相比,本发明的方法可以在不需要繁琐的纯化步骤的情况下以更高的收率生成期望的二聚体化合物。这些方法更适合大规模生产过程。
本发明提供了下文详细描述的第一至第四十四实施方案的方法。
附图简述
图1-19示出本发明的方法的示例性方案。
发明详述
现在将详细参考本发明的某些实施方案,其实例在所附的结构和式中示出。尽管将结合所列举的实施方案来描述本发明,但是将理解,它们并非旨在将本发明限制到那些实施方案。相反,本发明旨在涵盖可以包括在如由权利要求所限定的本发明的范围内的所有替代、修改和等同物。本领域技术人员将认识到可用于实施本发明的与本文所述相似或等同的许多方法和材料。
应该理解,除非明确否认或不适当,否则本文所述的任何实施方案都可以与本发明的一个或多个其他实施方案相组合。实施方案的组合不限于通过多个从属权利要求保护的那些特定组合。
定义
如本文所用,术语“细胞结合剂”或“CBA”是指可以优选以特异性方式结合细胞(例如,在细胞-表面配体上)或结合与细胞相关联或邻近的配体的化合物。在某些实施方案中,与细胞或细胞上或细胞附近的配体的结合是特异性的。CBA可以包括肽和非肽。
如本文所用,“直链或支链烷基”是指具有1至20个碳原子的饱和直链或支链单价烃基。烷基的实例包括但不限于甲基、乙基、1-丙基、2-丙基、1-丁基、2-甲基-1-丙基、-CH2CH(CH3)2)、2-丁基、2-甲基-2-丙基、1-戊基、2-戊基、3-戊基、2-甲基-2-丁基、3-甲基-2-丁基、3-甲基-1-丁基、2-甲基-1-丁基、1-己基)、2-己基、3-己基、2-甲基-2-戊基、3-甲基-2-戊基、4-甲基-2-戊基、3-甲基-3-戊基、2-甲基-3-戊基、2,3-二甲基-2-丁基、3,3-二甲基-2-丁基、1-庚基、1-辛基等等。优选地,该烷基具有1至10个碳原子。更优选地,该烷基具有1至4个碳原子。
“直链或支链烯基”是指具有至少一个不饱和位点(即,碳-碳双键)的2至20个碳原子的直链或支链单价烃基,其中该烯基基团包括具有“顺式”和“反式”取向或者“E”和“Z”取向的基团。实例包括但不限于乙烯基(-CH=CH2)、烯丙基(-CH2CH=CH2)等等。优选地,该烯基具有2至10个碳原子。更优选地,该烷基具有2至4个碳原子。
“直链或支链炔基”是指具有至少一个不饱和位点(即,碳-碳三键)的2至20个碳原子的直链或支链单价烃基。实例包括但不限于乙炔基、丙炔基、1-丁炔基、2-丁炔基、1-戊炔基、2-戊炔基、3-戊炔基、己炔基等等。优选地,该炔基具有2至10个碳原子。更优选地,该炔基具有2至4个碳原子。
术语“碳环”和“碳环基”是指具有3至12个碳原子的单价非芳族饱和或部分不饱和环作为单环或具有7至12个碳原子的单价非芳族饱和或部分不饱和环作为双环。具有7至12个原子的双环碳环可以例如排列为双环[4,5]、[5,5]、[5,6]或[6,6]体系,且具有9或10个环原子的双环碳环可以排列为双环[5,6]或[6,6]体系或桥联体系,诸如双环[2.2.1]庚烷、双环[2.2.2]辛烷和双环[3.2.2]壬烷。单环碳环的实例包括但不限于环丙基、环丁基、环戊基、1-环戊-1-烯基、1-环戊-2-烯基、1-环戊-3-烯基、环己基、1-环己-1-烯基、1-环己-2-烯基、1-环己-3-烯基、环己二烯基、环庚基、环辛基、环壬基、环癸基、环十一烷基、环十二烷基等等。
术语“环状烷基”和“环烷基”可以互换使用。它们是指单价饱和碳环基团。优选地,环状烷基为3至7元单环基团。更优选地,环状烷基为环己基。
术语“环状烯基”是指在环结构中具有至少一个双键的碳环基团。
术语“环状炔基”是指在环结构中具有至少一个三键的碳环基团。
“芳基”是指通过从母体芳族环体系的单个碳原子上除去一个氢原子而衍生的6至18个碳原子的单价芳族烃基。一些芳基基团在示例性结构中表示为“Ar”。芳基包括双环基团,其包含与饱和、部分不饱和的环或芳族碳环或杂环稠合的芳族环。典型的芳基基团包括但不限于由苯(苯基)、取代的苯、萘、蒽、茚基、茚满基、1,2-二氢萘、1,2,3,4-四氢萘基等等衍生的基团。优选地,芳基为苯基基团。
术语“杂环(heterocycle)”、“杂环基”和“杂环(heterocyclic ring)”在本文中可互换使用,且是指3至18个环原子的饱和或部分不饱和的(即,在环内具有一个或多个双键和/或三键)碳环基团,其中至少一个环原子为选自氮、氧、磷和硫的杂原子,剩下的环原子为C,其中一个或多个环原子任选地被一个或多个下述取代基独立地取代。杂环可以是具有3至7个环成员(2至6个碳原子和1至4个选自N、O、P和S的杂原子)的单环或具有7至10个环成员(4至9个碳原子和1至6个选自N、O、P和S的杂原子)的双环,例如:双环[4,5]、[5,5]、[5,6]或[6,6]体系。杂环描述在Paquette,Leo A.;“Principles of Modern HeterocyclicChemistry”(W.A.Benjamin,New York,1968),尤其是第1、3、4、6、7和9章;“The Chemistryof Heterocyclic Compounds,A series of Monographs”(John Wiley&Sons,New York,1950年至今),尤其是第13、14、16、19和28卷;和J.Am.Chem.Soc.(1960)82:5566中。“杂环基”还包括其中杂环基团与饱和的,部分不饱和的环或芳族碳环或杂环稠合的基团。杂环的实例包括但不限于吡咯烷基、四氢呋喃基、二氢呋喃基、四氢噻吩基、四氢吡喃基、二氢吡喃基、四氢硫代吡喃基、哌啶子基、吗啉代、硫代吗啉代、噻噁烷基、哌嗪基、高哌嗪基、氮杂环丁烷基、氧杂环丁烷基、硫杂环丁烷基、高哌啶基、氧杂环庚烷基、硫杂环庚烷基、氧氮杂 基、二氮杂
基、二氮杂 基、硫氮杂
基、硫氮杂 基、2-吡咯啉基、3-吡咯啉基、吲哚啉基、2H-吡喃基、4H-吡喃基、二氧杂环己烷基、1,3-二氧戊环基、吡唑啉基、二噻烷基、二硫杂环戊烷基(dithiolanyl)、二氢吡喃基、二氢噻吩基、二氢呋喃基、吡唑烷基咪唑啉基、咪唑烷基、3-氮杂双环[3.1.0]己烷基、3-氮杂双环[4.1.0]庚烷基、以及氮杂双环[2.2.2]己烷基。螺部分也包括在该定义的范围内。其中环原子被氧代基(=O)部分取代的杂环基团的实例为嘧啶酮基和1,1-二氧代-硫代吗啉基。
基、2-吡咯啉基、3-吡咯啉基、吲哚啉基、2H-吡喃基、4H-吡喃基、二氧杂环己烷基、1,3-二氧戊环基、吡唑啉基、二噻烷基、二硫杂环戊烷基(dithiolanyl)、二氢吡喃基、二氢噻吩基、二氢呋喃基、吡唑烷基咪唑啉基、咪唑烷基、3-氮杂双环[3.1.0]己烷基、3-氮杂双环[4.1.0]庚烷基、以及氮杂双环[2.2.2]己烷基。螺部分也包括在该定义的范围内。其中环原子被氧代基(=O)部分取代的杂环基团的实例为嘧啶酮基和1,1-二氧代-硫代吗啉基。
术语“杂芳基”是指5元或6元环的单价芳族基团,并且包括具有5至18个原子的稠环体系(其中的至少一个是芳族的),其含有一个或多个独立地选自氮、氧和硫的杂原子。杂芳基基团的实例为吡啶基(包括,例如2-羟基吡啶基)、咪唑基、咪唑并吡啶基、嘧啶基(包括,例如4-羟基嘧啶基)、吡唑基、三唑基、吡嗪基、四唑基、呋喃基、噻吩基、异噁唑基、噻唑基、噁唑基、异噁唑基、吡咯基、喹啉基、异喹啉基、吲哚基、苯并咪唑基、苯并呋喃基、噌啉基、吲唑基、吲嗪基、酞嗪基、哒嗪基、三嗪基、异吲哚基、蝶啶基、嘌呤基、噁二唑基、三唑基、噻二唑基、呋咱基、苯并呋咱基、苯并噻吩基、苯并噻唑基、苯并噁唑基、喹唑啉基、喹噁啉基、萘啶基、以及呋喃并吡啶基。
在可能的情况下,杂环或杂芳基基团可以附接碳(碳连接的)或氮(氮连接的)。以举例而非限制的方式,键合碳的杂环或杂芳基在吡啶的2、3、4、5或6位,哒嗪的3、4、5或6位,嘧啶的2、4、5或6位,吡嗪的2、3、5或6位,呋喃、四氢呋喃、硫代呋喃、噻吩、吡咯或四氢吡咯的2、3、4或5位,噁唑、咪唑或噻唑的2、4或5位,异噁唑、吡唑或异噻唑的3、4或5位,氮丙啶的2或3位,氮杂环丁烷的2、3或4位,喹啉的2、3、4、5、6、7或8位,或异喹啉的1、3、4、5、6、7或8位键合。
以举例而非限制的方式,键合氮的杂环或杂芳基在氮丙啶、氮杂环丁烷、吡咯、吡咯烷、2-吡咯啉、3-吡咯啉、咪唑、咪唑烷、2-咪唑啉、3-咪唑啉、吡唑、吡唑啉、2-吡唑啉、3-吡唑啉、哌啶、哌嗪、吲哚、吲哚啉、1H-吲唑的1位,异吲哚或异吲哚啉的2位,吗啉的4位,及咔唑或O-咔啉的9位键合。
存在于杂芳基或杂环基中的杂原子包括氧化形式,诸如NO、SO和SO2。
术语“卤代”或“卤素”是指F、Cl、Br或I。
上述烷基、烯基、炔基、环状烷基、环状烯基、环状炔基、碳环基、芳基、杂环基和杂芳基可以任选地被一个或多个(例如,2、3、4、5、6个或更多个)取代基取代。
如果取代基被描述为“被取代”,则非氢取代基置换在取代基的碳、氧、硫或氮上的氢取代基。因此,例如,取代的烷基取代基为如下烷基取代基,其中至少一个非氢取代基置换烷基取代基上的氢取代基。为了说明,单氟烷基是被氟取代基取代的烷基,而二氟烷基是被两个氟取代基取代的烷基。应该认识到,如果取代基上有多于一个取代基,则每个非氢取代基可以是相同的或不同的(除非另行指出)。
如果取代基被描述为“被任选取代的”,则取代基可以是(1)未取代的,或(2)取代的。如果取代基的碳被描述为任选地被一个或多个取代基列表取代,则碳上的一个或多个氢(在有任何氢的程度上)可以单独地和/或一起被独立选定的任选取代基置换。如果取代基的氮被描述为被一个或多个取代基列表任选取代,则氮上的一个或多个氢(在有任何氢的程度上)各自可以被独立选定的任选取代基置换。一个示例性取代基可以描绘为-NR’R”,其中R’和R”与它们所附接的氮原子一起可以形成杂环。由R’和R”连同它们所附接的氮原子一起形成的杂环可以是部分或完全饱和的。在一个实施方案中,杂环由3至7个原子组成。在另一个实施方案中,杂环选自吡咯基、咪唑基、吡唑基、三唑基、四唑基、异噁唑基、吡啶基和噻唑基。
如果一组取代基被共同描述为被一个或多个取代基列表任选地取代,则该组可以包括:(1)不可取代的取代基,(2)未被任选的取代基取代的可取代的取代基,和/或(3)被一个或多个任选的取代基取代的可取代的取代基。
如果取代基被描述为被至多特定数量的非氢取代基任选地取代,则该取代基可以是(1)未取代的;或(2)被至多该特定数量的非氢取代基或者在该取代基上的至多最大数量的可取代位置取代,以较少者为准。
因此,例如,如果取代基被描述为被至多3个非氢取代基任选地取代的杂芳基,那么具有少于3个可取代位置的任何杂芳基将被至多仅与具有可取代位置的杂芳基一样多的非氢取代基任选地取代。在非限制性实例中,这些取代基可选自具有1至10个碳原子的直链、支链或环状烷基、烯基或炔基,芳基,杂芳基,杂环基,卤素,胍鎓[-NH(C=NH)NH2],-OR100,NR101R102,-NO2,-NR101COR102,-SR100,由-SOR101表示的亚砜、由-SO2R101表示的砜,磺酸盐-SO3M,硫酸盐-OSO3M,由-SO2NR101R102表示的磺酰胺,氰基,叠氮基,-COR101,-OCOR101,-OCONR101R102和聚乙二醇单元(-CH2CH2O)nR101),其中M为H或阳离子(诸如Na+或K+);R101、R102和R103各自独立地选自H,具有1至10个碳原子的直链、支链或环状烷基、烯基或炔基,聚乙二醇单元(-CH2CH2O)n-R104,其中n为1至24,具有6至10个碳原子的芳基,具有3至10个碳原子的杂环和具有5至10个碳原子的杂芳基;且R104为H或具有1至4个碳原子的直链或支链烷基,其中由R100、R101、R102、R103和R104表示的基团中的烷基、烯基、炔基、芳基、杂芳基和杂环基被一个或多个(例如,2、3、4、5、6个或更多个)取代基任选地取代,这些取代基独立地选自卤素、-OH、-CN、-NO2和具有1至4个碳原子的未取代的直链或支链烷基。优选地,上述任选取代的烷基、烯基、炔基、环状烷基、环状烯基、环状炔基、碳环基、芳基、杂环基和杂芳基的取代基包括卤素、-CN、-NR102R103、-CF3、-OR101、芳基、杂芳基、杂环基、-SR101、-SOR101、-SO2R101和-SO3M。
术语“化合物”或“细胞毒性化合物”、“细胞毒性二聚体”和“细胞毒性二聚体化合物”可互换使用。它们旨在包括其结构或式或其任何衍生物已经在本发明中公开的化合物或者通过引用并入的结构或式或其任何衍生物。该术语也包括在本发明中公开的所有式的化合物的立体异构体、几何异构体、互变异构体、溶剂化物、代谢物、盐(例如,药学上可接受的盐)和前药以及前药盐。该术语也包括任何前述的任何溶剂化物、水合物和多晶型物。在本申请中描述的本发明的某些方面中对“立体异构体”、“几何异构体”、“互变异构体”、“溶剂化物”、“代谢物”、“盐”、“前药”、“前药盐”、“缀合物”、“缀合物盐”、“溶剂化物”、“水合物”或“多晶型物”的具体表述不应解释为使用术语“化合物”而没有表述这些其他形式的本发明的其他方面意图省略这些形式。
如本文所用,术语“可连接至细胞结合剂”或“能够将细胞毒性化合物共价连接至细胞结合剂”是指包含至少一个连接基团或其前体的本文所述的化合物或其衍生物,该连接基团或其前体适合将这些化合物或其衍生物结合到细胞结合剂。
给定基团的术语“前体”是指可通过任何脱保护、化学修饰或偶联反应产生该基团的任何基团。
术语“手性”是指具有镜像伴侣的不可重叠特性的分子,而术语“非手性”是指可在其镜像伴侣上重叠的分子。
术语“立体异构体”是指具有相同化学构成和连接性的化合物,但是其原子在空间中的不同取向不能通过围绕单键旋转而相互转化。
“非对映异构体”是指具有两个或更多个手性中心且其分子彼此不是镜像的立体异构体。非对映异构体具有不同的物理特性,例如熔点、沸点、光谱特性和反应性。非对映异构体的混合物可以根据高分辨率分析程序如结晶、电泳和色谱法分离。
“对映异构体”是指化合物的两个立体异构体,它们是彼此不可重叠的镜像。
本文中使用的立体化学定义和惯例通常遵循S.P.Parker编辑,McGraw-HillDictionary of Chemical Terms(1984)McGraw-Hill Book Company,New York;以及Eliel,E.和Wilen,S.,“Stereochemistry of Organic Compounds,”John Wiley&Sons,Inc.,New York,1994。本发明的化合物可以含有不对称或手性中心,且因此以不同的立体异构形式存在。预期本发明化合物的所有立体异构形式构成本发明的一部分,这些立体异构形式包括但不限于非对映异构体、对映异构体和阻转异构体以及它们的混合物如外消旋混合物。许多有机化合物以光学活性形式存在,即它们具有旋转平面偏振光的平面的能力。在描述光学活性化合物时,使用前缀D和L或者R和S来表示分子关于其一个或多个手性中心的绝对构型。采用前缀d和l或者(+)和(-)来指示化合物对平面偏振光的旋转的符号,其中(-)或1是指该化合物是左旋的。具有前缀(+)或d的化合物是右旋的。对于给定的化学结构,这些立体异构体是相同的,不同的是它们是彼此的镜像。具体的立体异构体也可以被称为对映体,并且此类异构体的混合物通常被称为对映体混合物。对映异构体的50:50混合物被称为外消旋混合物或外消旋体,其可能在化学反应或过程中没有立体选择性或立体定向性的情况下出现。术语“外消旋混合物”和“外消旋体”是指两种对映异构物质的等摩尔混合物,其没有光学活性。
术语“互变异构体”或“互变异构形式”是指不同能量的结构异构体,其可以通过低能障互相转化。例如,质子互变异构体(也称为质子异构互变异构体)包括通过质子迁移实现的相互转化,诸如酮-烯醇和亚胺-烯胺异构化。价键互变异构体包括通过重新组织一些键合电子实现的相互转化。
术语“亚胺反应性试剂”是指能够与亚胺基团反应的试剂。亚胺反应性试剂的实例包括但不限于亚硫酸盐(H2SO3、H2SO2或与阳离子形成的HSO3 -、SO3 2-或HSO2 -),偏亚硫酸氢盐(H2S2O5或与阳离子形成的S2O5 2-的盐),单、二、三和四硫代磷酸盐(PO3SH3、PO2S2H3、POS3H3、PS4H3或者与阳离子形成的PO3S3-、PO2S2 3-、POS3 3-或PS4 3-的盐),硫代磷酸酯((RiO)2PS(ORi)、RiSH、RiSOH、RiSO2H、RiSO3H),各种胺(羟胺(例如,NH2OH)、肼(例如,NH2NH2)、NH2O-Ri、Ri’NH-Ri、NH2-Ri),NH2-CO-NH2,NH2-C(=S)-NH2,硫代硫酸盐(H2S2O3或与阳离子形成的S2O3 2-的盐),连二亚硫酸盐(H2S2O4或与阳离子形成的S2O4 2-的盐),二硫代磷酸盐(P(=S)(ORk)(SH)(OH)或其与阳离子形成的盐),异羟肟酸(RkC(=O)NHOH或与阳离子形成的盐),酰肼(RkCONHNH2),甲醛次硫酸盐(HOCH2SO2H或与阳离子形成的HOCH2SO2 -的盐,诸如HOCH2SO2 -Na+),糖化核苷酸(诸如,GDP-甘露糖),氟达拉宾或其混合物,其中Ri和Ri’各自独立地为具有1至10个碳原子并且被选自-N(Rj)2、-CO2H、-SO3H和-PO3H的至少一个取代基取代的直链或支链烷基;Ri和Ri’可以进一步被本文所述的烷基的取代基任选地取代;Rj为具有1至6个碳原子的直链或支链烷基;且Rk为具有1至10个碳原子的直链、支链或环状烷基、烯基或炔基,芳基,杂环基或杂芳基(优选地,Rk为具有1至4个碳原子的直链或支链烷基;更优选地,Rk为甲基、乙基或丙基)。优选地,阳离子为单价阳离子,诸如Na+或K+。优选地,亚胺反应性试剂选自亚硫酸盐、羟胺、尿素和肼。更优选地,亚胺反应性试剂为NaHSO3或KHSO3。
如本文所用,术语“亚胺还原试剂”是指能够将亚胺官能团还原成胺官能团的试剂。在某些实施方案中,亚胺还原试剂为氢化物还原试剂。此类亚胺还原试剂的实例包括但不限于硼氢化物(例如,硼氢化钠、三乙酰氧基硼氢化钠、氰基硼氢化钠、硼氢化锂(LiBH4)、硼氢化钾(KBH4))、氢气和氢化铝锂、甲酸铵、硼烷、9-硼杂双环[3.3.1]壬烷(9-BBN)、二异丁基氢化铝(DIBAL)和双(2-甲氧基乙氧基)氢化铝钠(Red-Al)。在某些实施方案中,亚胺还原试剂为三乙酰氧基硼氢化钠。
术语“保护基团”或“保护部分”是指通常用于在使化合物上的其他官能团、其衍生物或其缀合物反应时阻断或保护特定官能团的取代基。例如,“胺保护基团”或“氨基保护部分”是附接到阻断或保护化合物中的氨基官能团的氨基基团上的取代基。此类基团在本领域中是公知的(参见,例如P.Wuts和T.Greene,2007,Protective Groups in OrganicSynthesis,第7章,J.Wiley&Sons,NJ),并且例如为氨基甲酸酯,诸如氨基甲酸甲酯和氨基甲酸乙酯、FMOC、取代的氨基甲酸乙酯、被1,6-β-消除(也称为“自降解”)裂解的氨基甲酸酯,脲,酰胺,肽,烷基和芳基衍生物。合适的氨基保护基团包括但不限于乙酰基、三氟乙酰基、叔丁氧基羰基(BOC)、苄氧基羰基(CBZ)和9-芴基亚甲氧基羰基(Fmoc)、2-三甲基甲硅烷基乙基、(2-苯基-2-三甲基甲硅烷基)乙基、三异丙基甲硅烷氧基、2-(三甲基甲硅烷基)乙氧基甲基、烯丙氧基羰基、9-芴基甲氧基羰基、2-(三甲基甲硅烷基)乙氧基羰基或2,2,2,2-三氯乙氧基羰基。关于保护基团及其用途的一般描述,参见P.G.M.Wuts&T.W.Greene,Protective Groups in Organic Synthesis,John Wiley&Sons,New York,2007。
“醇保护基团”或“醇保护部分”是附接到阻断或保护化合物中的醇官能团的醇基团上的取代基。此类基团在本领域中是公知的(参见,例如P.Wuts和T.Greene,2007,Protective Groups in Organic Synthesis,Chapter 2,J.Wiley&Sons,NJ)。合适的醇保护基团包括但不限于新戊酰基、甲氧基甲基、2-甲氧基乙氧基甲基、对甲氧基苄基、3,4-二甲氧基苄基、2,6-二甲氧基苄基、二苯基甲基、苄氧基甲基、2,2,2-三氯乙氧基羰基、四氢呋喃基、四氢吡喃基、苄基、苯甲酰基、对苯基苯甲酰基、2,4,6-三甲基苯甲酰基、对溴苯甲酰基、对硝基苯甲酰基、吡啶甲酰基、烟酰基、5-二苯并环庚基(5-dibenzosuberyl)、三苯甲基/三苯基甲基或三(4-叔丁基苯基)甲基以及各种甲硅烷基保护基团(例如,二甲基异丙基甲硅烷基、二乙基异丙基甲硅烷基、二甲基己基甲硅烷基、三甲基甲硅烷基、三异丙基甲硅烷基、三苄基甲硅烷基、三苯基甲硅烷基、2-降冰片基二甲基甲硅烷基、叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基、2-三甲基乙基甲硅烷基(TEOC)或[2-(三甲基甲硅烷基)乙氧基]甲基)。在某些实施方案中,醇保护基团为空间位阻的。在某些实施方案中,醇保护基团优选为甲氧基甲基、四氢吡喃基、2-甲氧基乙氧基甲基、对甲氧基苄基、苄氧基甲基或2,2,2-三氯乙氧基羰基。更优选地,醇保护基团为2,2,2-三氯乙氧基羰基。在某些实施方案中,醇保护基团为甲硅烷基保护基团,优选为三乙基甲硅烷基、三异丙基甲硅烷基或叔丁基二甲基甲硅烷基。更优选地,醇保护基团为叔丁基二甲基甲硅烷基。
如本文所用,“醇保护试剂”是指将醇保护基团引入醇基团上的试剂。
“酸不稳定的醇保护基团”是在酸性条件下不稳定且释放醇保护基团以形成游离醇的醇保护基团。酸不稳定的醇保护基团的实例包括但不限于乙酸根、烯丙基、甲氧基甲基、四氢呋喃基、四氢吡喃基、5-二苯并环庚基、1-乙氧基乙基、1-甲基-1-甲氧基乙基、2-(苯基氢硒基)乙基,三苯甲基/三苯基甲基、三(4-叔丁基苯基)甲基和各种甲硅烷基保护基团(例如,二甲基异丙基甲硅烷基、二乙基异丙基甲硅烷基、二甲基己基甲硅烷基、三甲基甲硅烷基、三乙基甲硅烷基、三异丙基甲硅烷基、三苄基甲硅烷基、三苯基甲硅烷基、2-降冰片基二甲基甲硅烷基、叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基或2-三甲基乙基甲硅烷基(TEOC)、[2-(三甲基甲硅烷基)乙氧基]甲基)。在某些实施方案中,醇保护基团为甲硅烷基保护基团,优选为三乙基甲硅烷基、三异丙基甲硅烷基或叔丁基二甲基甲硅烷基。更优选地,醇保护基团为叔丁基二甲基甲硅烷基。
如本文所用,术语“醇脱保护试剂”是指能够裂解醇保护基团以形成游离醇的试剂。此类试剂在本领域中是公知的(参见,例如P.Wuts和T.Greene,2007,ProtectiveGroups in Organic Synthesis,第2章,J.Wiley&Sons,NJ)。此类醇脱保护试剂的实例包括但不限于四-正丁基氟化铵、三(二甲基氨基)二氟三甲基硅酸锍、氟化氢或其溶剂化物、氟化氢吡啶、四氟化硅、六氟硅酸、氟化铯、盐酸、乙酸、三氟乙酸、对甲苯磺酸吡啶鎓、对甲苯磺酸(p-TsOH)、甲酸、高碘酸。在某些实施方案中,醇脱保护试剂为盐酸或四-正丁基氟化铵(TBAF)。在某些实施方案中,醇脱保护剂为氟化氢-吡啶(HF-吡啶)。
如本文所用,术语“胺脱保护基团”是指能够裂解胺保护基团以形成游离胺的试剂。此类试剂在本领域中是公知的(参见,例如P.Wuts和T.Greene,2007,ProtectiveGroups in Organic Synthesis,第7章,J.Wiley&Sons,NJ)。此类胺脱保护试剂的实例包括但不限于四-正丁基氟化铵、乙酸、氟化氢吡啶、氟化铯、哌啶、吗啉或三氟乙酸。
如本文所用,“醇活化剂”是指增加羟基基团的反应性由此使羟基成为更好的离去基团的试剂。此类醇活化剂的实例包括对甲苯磺酰氯、亚硫酰氯、三氟甲磺酸酐、甲磺酰氯、甲磺酸酐、三苯基膦、酰氯、4-二甲基氨基吡啶等等。在某些实施方案中,醇活化剂为亚硫酰氯。在某些实施方案中,醇活化剂为三苯基膦。
如本文所用,短语“药学上可接受的盐”是指本发明化合物的药学上可接受的有机或无机盐。示例性盐包括但不限于硫酸盐、柠檬酸盐、乙酸盐、草酸盐、氯化物、溴化物、碘化物、硝酸盐、硫酸氢盐、磷酸盐、酸式磷酸盐、异烟酸盐、乳酸盐、水杨酸盐、酸式柠檬酸盐、酒石酸盐、油酸盐、鞣酸盐、泛酸盐、酒石酸氢盐、抗坏血酸盐、琥珀酸盐、马来酸盐、龙胆酸盐、富马酸盐、葡糖酸盐、葡糖醛酸盐、糖酸盐、甲酸盐、苯甲酸盐、谷氨酸盐、甲磺酸盐“甲磺酸盐(mesylate)”、乙磺酸盐、苯磺酸盐、对甲苯磺酸盐、双羟萘酸盐(即1,1’-亚甲基-双-(2-羟基-3-萘甲酸)盐)、碱金属(例如,钠和钾)盐、碱土金属(例如,镁)盐和铵盐。药学上可接受的盐可涉及包含另一种分子,诸如乙酸根离子、琥珀酸根离子或其他抗衡离子。抗衡离子可以是使母体化合物上的电荷稳定化的任何有机或无机部分。此外,药学上可接受的盐在其结构中可以具有多于一个带电荷的原子。多个带电荷的原子是药学上可接受的盐的一部分的例子可以具有多个抗衡离子。因此,药学上可接受的盐可具有一个或多个带电荷的原子和/或一个或多个抗衡离子。
如果本发明的化合物是碱,则期望的药学上可接受的盐可以通过本领域可用的任何合适的方法制备,例如用无机酸或有机酸处理游离碱来制备,该无机酸诸如为盐酸、氢溴酸、硫酸、硝酸、甲磺酸、磷酸等,该有机酸诸如为乙酸、马来酸、琥珀酸、扁桃酸、富马酸、丙二酸、丙酮酸、草酸、乙醇酸、水杨酸、吡喃糖苷基酸如葡萄糖醛酸或半乳糖醛酸、α-羟基酸如柠檬酸或酒石酸、氨基酸如天冬氨酸或谷氨酸、芳族酸如苯甲酸或肉桂酸、磺酸如对甲苯磺酸或乙磺酸、等等。
如果本发明的化合物是酸,则期望的药学上可接受的盐可以通过任何合适的方法制备,例如用诸如胺(伯胺、仲胺或叔胺)、碱金属氢氧化物或碱土金属氢氧化物等无机或有机碱处理游离酸来制备。合适盐的示例性实例包括但不限于衍生自氨基酸如甘氨酸和精氨酸、氨、伯胺、仲胺和叔胺以及环胺如哌啶、吗啉和哌嗪的有机盐,以及衍生自钠、钙、钾、镁、锰、铁、铜、锌、铝和锂的无机盐。
短语“药学上可接受的”表明该物质或组合物必须在化学和/或毒理学上与包含制剂的其他成分和/或用其治疗的哺乳动物相容。
术语“离去基团”是指在亲核取代或置换期间中脱离的带电荷或不带电荷的部分的基团。此类离去基团在本领域是公知的,且包括但不限于卤素、酯、烷氧基、羟基、甲苯磺酸根、三氟甲磺酸根、甲磺酸根、腈、叠氮化物、氨基甲酸根、二硫化物、硫酯、硫醚和重氮化合物。
术语“双官能交联剂”、“双官能连接子”或“交联剂”是指具有两个反应性基团的改性剂;其中一个反应性基团能够与细胞结合剂反应,而另一个反应性基团与细胞毒性化合物反应以将这两部分连接在一起。此类双官能交联剂在本领域中是公知的(参见,例如Isalm和Dent,Bioconjugation,第5章,第218-363页,Groves Dictionaries Inc.NewYork,1999)。例如,能够通过硫醚键进行连接的双官能交联剂包括N-琥珀酰亚胺基-4-(N-马来酰亚胺基甲基)-环己烷-1-羧酸酯(SMCC)以引入马来酰亚胺基团,或N-琥珀酰亚胺基-4-(碘乙酰基)-氨基苯甲酸酯(SIAB)以引入碘乙酰基基团。将马来酰亚胺基团或卤代乙酰基基团引到细胞结合剂上的其他双官能交联剂在本领域中是公知的(参见,美国专利申请2008/0050310、20050169933,得自Pierce Biotechnology Inc.P.O.Box 117,Rockland,IL61105,USA),且包括但不限于双马来酰亚胺聚乙二醇(BMPEO)、BM(PEO)2、BM(PEO)3、N-(β-马来酰亚胺基丙氧基)琥珀酰亚胺酯(BMPS)、γ-马来酰亚胺丁酸N-琥珀酰亚胺酯(GMBS)、ε-马来酰亚胺己酸N-羟基琥珀酰亚胺酯(EMCS)、5-马来酰亚胺基戊二酸NHS、HBVS、作为SMCC“长链”类似物(LC-SMCC)的N-琥珀酰亚胺基-4-(N-马来酰亚胺甲基)-环己烷-1-羧基-(6-酰胺基己酸酯)、间马来酰亚胺基苯甲酰基-N-羟基琥珀酰亚胺酯(MBS)、4-(4-N-马来酰亚胺苯基)-丁酸酰肼或HCl盐(MPBH)、3-(溴乙酰胺基)丙酸N-琥珀酰亚胺酯(SBAP)、碘代乙酸N-琥珀酰亚胺酯(SIA)、κ-马来酰亚胺十一烷酸N-琥珀酰亚胺酯(KMUA)、4-(对马来酰亚胺苯基)-丁酸N-琥珀酰亚胺酯(SMPB)、琥珀酰亚胺基-6-(β-马来酰亚胺丙酰胺基)己酸酯(SMPH)、琥珀酰亚胺基-(4-乙烯基磺酰基)苯甲酸酯(SVSB)、二硫代双-马来酰亚胺乙烷(DTME)、1,4-双-马来酰亚胺丁烷(BMB)、1,4-双马来酰亚胺基-2,3-二羟基丁烷(BMDB)、双-马来酰亚胺己烷(BMH)、双-马来酰亚胺乙烷(BMOE)、4-(N-马来酰亚胺-甲基)环己烷-1-羧酸磺基琥珀酰亚胺酯(磺基-SMCC)、(4-碘-乙酰基)氨基苯甲酸磺基琥珀酰亚胺酯(磺基-SIAB)、间马来酰亚胺苯甲酰基-N-羟基磺基琥珀酰亚胺酯(磺基-MBS)、N-(γ-马来酰亚胺丁酰氧基)磺基琥珀酰亚胺酯(磺基-GMBS)、N-(ε-马来酰亚胺己酰氧基)磺基琥珀酰亚胺酯(磺基-EMCS)、N-(κ-马来酰亚胺十一烷酰氧基)磺基琥珀酰亚胺酯(磺基-KMUS)和4-(对马来酰亚胺苯基)丁酸磺基琥珀酰亚胺酯(磺基-SMPB)。
异双官能交联剂是具有两个不同反应性基团的双官能交联剂。含有胺反应性N-羟基琥珀酰亚胺基团(NHS基团)和羰基反应性肼基团两者的异双官能交联剂也可用于连接本文所述的细胞毒性化合物与细胞结合剂(例如、抗体)。此类市售的异双官能交联剂的实例包括琥珀酰亚胺基6-肼基烟酰胺丙酮腙(SANH)、4-肼基对苯二甲酸琥珀酰亚胺酯盐酸盐(SHTH)和肼烟酸琥珀酰亚胺酯盐酸盐(SHNH)。带有酸不稳定键的缀合物也可以使用本发明的带有肼的苯并二氮杂 衍生物来制备。可以使用的双官能交联剂的实例包括对甲酰基苯甲酸琥珀酰亚胺酯(SFB)和对甲酰基苯氧基乙酸琥珀酰亚胺酯(SFPA)。
衍生物来制备。可以使用的双官能交联剂的实例包括对甲酰基苯甲酸琥珀酰亚胺酯(SFB)和对甲酰基苯氧基乙酸琥珀酰亚胺酯(SFPA)。
能使细胞结合剂与细胞毒性化合物通过二硫化物键连接的双官能交联剂是本领域已知的、且包括N-琥珀酰亚胺基-3-(2-吡啶基二硫代)丙酸酯(SPDP)、N-琥珀酰亚胺基-4-(2-吡啶基二硫代)戊酸酯(SPP)、N-琥珀酰亚胺基-4-(2-吡啶基二硫代)丁酸酯(SPDB)、N-琥珀酰亚胺基-4-(2-吡啶基二硫代)2-磺基丁酸酯(磺基-SPDB),以引入二硫代吡啶基基团。可以用于引入二硫化物基团的其他双官能交联剂在本领域中是已知的,并公开在美国专利6,913,748、6,716,821和美国专利公布20090274713和20100129314中,所有这些专利通过引用并入本文。或者,也可以使用引入硫醇基团的交联剂,诸如2-亚氨基硫烷、高半胱氨酸硫代内酯或S-乙酰琥珀酸酐。
如本文定义的“连接子”、“连接子部分”或“连接基团”是指两个基团如细胞结合剂和细胞毒性化合物连接在一起的部分。通常,连接子在连接正连接的两个基团的条件下基本上是惰性的。在某些实施方案中,连接基团是本文所述的细胞毒性化合物的一部分。连接基团可以包含可以与细胞结合剂或其前体反应的反应性基团。连接部分可以含有允许在特定位点释放细胞毒性部分的化学键。合适的化学键是本领域公知的,且包括二硫化物键、硫醚键、酸不稳定的键、光不稳定的键、肽酶不稳定的键和酯酶不稳定的键(参见,例如美国专利5,208,020;5,475,092;6,441,163;6,716,821;6,913,748;7,276,497;7,276,499;7,368,565;7,388,026和7,414,073)。优选的是二硫化物键、硫醚和肽酶不稳定的键。也可以使用不可裂解的连接部分。
如本文定义的“反应性基团”或“反应性部分”是指如下部分,其易于与细胞结合剂形成共价键,例如与抗体上的赖氨酸胺基团形成酰胺键,或例如通过硫醚或二硫化物键与双官能交联剂形成共价键。反应性基团对本发明的方法中描述的反应是惰性的。还包括可以转化为反应性基团的官能团。例如,反应性基团可以是可以转化为N-羟基琥珀酰亚胺酯的N-羟基琥珀酰亚胺酯或甲酯基团。在另一个实例中,反应性基团可以是硫醇(-SH)基团,其可以容易地通过二硫化物键或硫醚键与双官能交联剂形成共价键。它也可以是可以转化成硫醇基团的烷基二硫化物或吡啶基二硫化物(R-S-S-,其中R为烷基或吡啶基)。
在一个实施方案中,在一端附接有反应性基团的连接基团如反应性酯选自以下:
-O(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-O(CR20R21)m(CR26=CR27)m’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-O(CR20R21)m(炔基)n’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’(CR24R25)q(CO)t X’、
-O(CR20R21)m(哌嗪并)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’(CR24R25)q(CO)tX’、
-O(CR20R21)m(吡咯并)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’(CR24R25)q(CO)tX’、
-O(CR20R21)mA”m”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-S(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-S(CR20R21)m(CR26=CR27)m’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-S(CR20R21)m(炔基)n’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’(CR24R25)q(CO)t X’、
-S(CR20R21)m(哌嗪并)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’(CR24R25)q(CO)t X’、
-S(CR20R21)m(吡咯并)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’(CR24R25)q(CO)t X’、
-S(CR20R21)mA”m”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-NR33(C=O)p”(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(C O)tX”、
-NR33(C=O)p”(CR20R21)m(CR26=CR27)m’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’’
(CR24R25)q(CO)tX”、
-NR33(C=O)p”(CR20R21)m(炔基)n’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’(CR24R25)q-(CO)tX’、
-NR33(C=O)p”(CR20R21)m(哌嗪并)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’(CR24R25)q
(CO)tX”、
-NR33(C=O)p”(CR20R21)m(吡咯并)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y’(CR24R25)q -(CO)tX’、
-NR33(C=O)p”(CR20R21)mA”m”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q
(CO)tX”、
-(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-(CR20R21)m(CR26=CR27)m’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-(CR20R21)m(炔
基)n’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-(CR20R21)m(哌嗪
并)t’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-(CR20R21)mA”m”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-(CR20R21)m(CR29=N-NR30)n”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-(CR20R21)m(CR29=N-NR30)n”(CR26=CR27)m’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”、
-(CR20R21)m(CR29=N-NR30)n”(炔
基)n’(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q -(CO)tX”、
-(CR20R21)m(CR29=N-NR30)n”A”m”(CR22R23)n(OCH2CH2)p(CR40R41)p”Y”(CR24R25)q(CO)tX”,
其中:
m、n、p、q、m’、n’、t’为1至10的整数,或任选为0;
t、m”、n”和p”为0或1;
X”选自OR36、SR37、NR38R39,其中R36、R37、R38、R39为H、或具有1至20个碳原子的直链、支链或环状烷基、烯基或炔基和或聚乙二醇单元-(OCH2CH2)n,R37任选为硫醇保护基团,当t=1时,COX”形成选自N-羟基琥珀酰亚胺酯、N-羟基邻苯二甲酰亚胺酯、N-羟基磺基-琥珀酰亚胺酯、对硝基苯基酯、二硝基苯基酯、五氟苯基酯及其衍生物的反应性酯,其中所述衍生物有利于酰胺键形成;
Y”不存在或选自O、S、S-S或NR32,其中R32具有与上文对于R赋予的定义相同的定义;或
当Y”不为S-S且t=0时,X”选自马来酰亚胺基团、卤代乙酰基基团或SR37,其中R37具有与上文相同的定义;
A”为氨基酸残基或含有2至20个氨基酸残基的多肽;
R20、R21、R22、R23、R24、R25、R26和R27相同或不同,且为-H或具有1至5个碳原子的直链或支链的烷基;
R29和R30相同或不同,且为-H或具有1至5个碳原子的烷基;
R33为-H,或具有1至12个碳原子的直链、支链或环状烷基、烯基或炔基,聚乙二醇单元R-(OCH2CH2)n-,或者R33为-COR34、-CSR34、-SOR34或-SO2R34,其中R34为H或具有1至20个碳原子的直链、支链或环状烷基、烯基或炔基,或聚乙二醇单元-(OCH2CH2)n;且
R40和R41中的一者任选地为带负电荷或带正电荷的官能团且另一者为H或具有1至4个碳原子的烷基、烯基、炔基。
任何上述连接基团可以存在于本发明的任何化合物、药物-连接子化合物或缀合物中,包括置换本文所述的任何式的连接基团。
术语“氨基酸”是指天然存在的氨基酸或非天然存在的氨基酸。在一个实施方案中,氨基酸由NH2-C(Raa’Raa)-C(=O)OH表示,其中Raa和Raa’各自独立地为H,具有1至10个碳原子的任选取代的直链、支链或环状烷基、烯基或炔基,芳基,杂芳基或杂环基,或Raa和N-末端氮原子可以一起形成杂环(例如,如在脯氨酸中)。术语“氨基酸残基”是指当从氨基酸的胺和/或羧基末端除去一个氢原子时的相应残基,例如-NH-C(Raa’Raa)-C(=O)O-。
术语“阳离子”是指具有正电荷的离子。阳离子可以是单价的(例如,Na+、K+等)、二价的(例如,Ca2+、Mg2+等)或多价的(例如,Al3+等)。优选地,该阳离子是单价的。
如本文所用,术语“卤化试剂”是指将醇基团转化为卤化物基团的试剂。“溴化试剂”是将醇基团转化为溴化物基团的试剂。“碘化试剂”是将醇基团转化为碘化物基团的试剂。“氯化试剂”是将醇基团转化为氯化物基团的试剂。示例性溴化试剂包括但不限于溴、氢溴酸、四溴化碳、三溴化磷和溴化钾。示例性碘化试剂包括但不限于氢碘酸、碘、四碘化碳、三碘化磷、碘化钠或碘化钾。示例性氯化试剂包括但不限于四氯化碳、甲磺酰氯、硫酰氯、亚硫酰氯、氰尿酰氯、N-氯琥珀酰亚胺、氯氧化磷(V)、五氯化磷和三氯化磷。在一个具体的实施方案中,氯化试剂为甲磺酰氯。
如本文所用,术语“磺化试剂”是指将醇基团转化为磺酸酯基团的试剂。优选地,磺化试剂为磺酸酐,诸如甲磺酸酐,或磺酰氯,诸如甲磺酰氯(MsCl)。
如本文所用,“活化酯”是指容易被羟基或胺基取代的酯基。示例性活化酯包括但不限于硝基苯基(例如,2或4-硝基苯基)酯、二硝基苯基(例如,2,4-二硝基苯基)酯、磺基-四氟苯基(例如,4-磺基-2,3,5,6-四氟苯基)酯、五氟苯基酯、硝基吡啶基(例如,4-硝基吡啶基)酯、三氟乙酸酯及乙酸酯。
如本文所用,“酯化试剂”是指将醇基团转化为酯基的试剂。示例性酯化试剂包括但不限于硝基苯甲酸(例如,2或4-硝基苯甲酸)、二硝基苯甲酸(例如,2,4-二硝基苯甲酸)、磺基-四氟苯甲酸(例如,4-磺基-2,3,5,6-四氟苯甲酸)、五氟苯甲酸、硝基吡啶羧酸(例如,4-硝基-2-吡啶羧酸)、三氟乙酸和乙酸或者酰氯、酸酐或其其他活化的羧酸衍生物。
本发明的方法
本发明提供了制备具有一个亚胺官能团和一个胺官能团的吲哚啉并苯并二氮杂 二聚体化合物的新方法。与本领域已知的方法相比,本发明的方法可以以更高的收率且在不使用HPLC纯化的情况下生成期望的二聚体化合物。
二聚体化合物的新方法。与本领域已知的方法相比,本发明的方法可以以更高的收率且在不使用HPLC纯化的情况下生成期望的二聚体化合物。
在第一实施方案中,本发明提供了制备式(2)的化合物

或其盐的方法,其包括通过使式(I)的化合物与醇保护试剂反应将醇保护基团引入到式(1)的化合物的伯醇中的一个上,

其中:
L’、L”和L”’相同或不同,且独立地为-H,具有1至10个碳原子的任选取代的直链、支链或环状烷基、烯基或炔基,聚乙二醇单元-(OCH2CH2)n’-Rc,卤素,胍鎓[-NH(C=NH)NH2],-OR,-NR’R”,-NO2,-NR’COR”,-SR,-SOR’,-SO2R’,-SO3M,-OSO3M,-SO2NR’R”,氰基,叠氮基,-COR’,-OCOR’,-OCONR’R”或能够将细胞毒性化合物共价连接到细胞结合剂(CBA)的具有与其键合的反应性基团的连接基团,条件是L’、L”和L”’中只有一个是具有与其键合的反应性基团的连接基团;
M为-H或阳离子;
R在每次出现时独立地选自由-H,具有1至10个碳原子的任选取代的直链、支链或环状烷基、烯基或炔基,聚乙二醇单元-(CH2CH2O)n-Rc,具有6至18个碳原子的任选取代的芳基,含有一个或多个独立地选自氮、氧和硫的杂原子的任选取代的5至18元杂芳基环,或含有1至6个独立地选自O、S、N和P的杂原子的任选取代的3至18元杂环组成的组;
R’和R”各自独立地选自-H,-OH,-OR,-NHR,-N(R)2,-COR,具有1至10个碳原子的任选取代的直链、支链或环状烷基、烯基或炔基,聚乙二醇单元-(CH2CH2O)n’-Rc,和具有1至6个独立地选自O、S、N和P的杂原子的任选取代的3至18元杂环;
Rc为-H或具有1至4个碳原子的取代或未取代的直链或支链烷基,或具有与其键合的反应性基团的连接基团;
n’为1至24的整数;
G选自-CH-或-N-;且
P1为醇保护基团。
在一个实施方案中,式(1)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个具体的实施方案中,醇保护基团为空间位阻的。
在另一个具体的实施方案中,醇保护基团为新戊酰基、甲氧基甲基、2-甲氧基乙氧基甲基、对甲氧基苄基、3,4-二甲氧基苄基、2,6-二甲氧基苄基、二苯基甲基、苄氧基甲基、2,2,2-三氯乙氧基羰基、四氢呋喃基、四氢吡喃基、苄基、苯甲酰基、对苯基苯甲酰基、2,4,6-三甲基苯甲酰基、对溴苯甲酰基、对硝基苯甲酰基、吡啶甲酰基、烟酰基、5-二苯并环庚基、三苯甲基/三苯基甲基或三(4-叔丁基苯基)甲基。优选地,醇保护基团为甲氧基甲基、四氢吡喃基、2-甲氧基乙氧基甲基、对甲氧基苄基、苄氧基甲基或2,2,2-三氯乙氧基羰基。甚至更优选地,醇保护基团为2,2,2-三氯乙氧基羰基。
在另一个具体的实施方案中,醇保护基团为甲硅烷基保护基团。例如,甲硅烷基保护基团为二甲基异丙基甲硅烷基、二乙基异丙基甲硅烷基、二甲基己基甲硅烷基、三甲基甲硅烷基、三异丙基甲硅烷基、三苄基甲硅烷基、三苯基甲硅烷基、2-降冰片基二甲基甲硅烷基、叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基、2-三甲基乙基甲硅烷基(TEOC)或[2-(三甲基甲硅烷基)乙氧基]甲基。优选地,甲硅烷基保护基团为三乙基甲硅烷基、三异丙基甲硅烷基或叔丁基二甲基甲硅烷基。更优选地,甲硅烷基保护基团为叔丁基二甲基甲硅烷基。
甲硅烷基保护基团可以通过使式(1)的化合物与R3-Cl、R3-Br、R3-I或R3-OSO2CF3(统称为醇保护试剂)在碱存在下反应来引入,其中R3为二甲基异丙基甲硅烷基、二乙基异丙基甲硅烷基、二甲基己基甲硅烷基、三甲基甲硅烷基、三异丙基甲硅烷基、三苄基甲硅烷基、三苯基甲硅烷基、2-降冰片基二甲基甲硅烷基、叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基或[2-(三甲基甲硅烷基)乙氧基]甲基。在某些实施方案中,醇保护试剂与式(1)的化合物的摩尔比在0.8至1.2之间,在1至5之间,在1至2之间,在1至1.5之间,在1至1.4之间,在1至1.3之间,在1至1.2之间,或在1至1.1之间。在某些实施方案中,相对于式(I)的化合物,使用少于2摩尔当量的醇保护试剂。优选地,相对于式(1)的化合物,使用1.5、1.4、1.3、1.2、1.1或1.0摩尔当量的醇保护试剂。
在一个实施方案中,碱可为非亲核性碱。非亲核性碱的实例包括但不限于咪唑、三乙胺、二异丙基乙胺、吡啶、2,6-二甲基吡啶、1,8-二氮杂双环十一碳-7-烯或四甲基哌啶。优选地,非亲核性碱为咪唑。可以使用摩尔过量的碱。在某些实施方案中,相对于式(1)的化合物,使用大于2摩尔当量的碱(例如,非亲核性碱)。
在另一个实施方案中,在式(1)的化合物与R3-Cl、R3-Br、R3-I或R3-OSO2CF3之间的反应在有利于引入甲硅烷基保护基团的催化剂的存在下进行。在该反应中可以使用本领域已知的任何合适的催化剂(参见,例如P.Wuts和T.Greene,2007,Protective Groups inOrganic Synthesis,第2章,J.Wiley&Sons,NJ)。示例性催化剂包括但不限于4-二甲基氨基吡啶(DMAP)、1,1,3,3-四甲基胍和1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)。
任何合适的有机溶剂都可以用于第一实施方案的方法中。示例性溶剂包括但不限于DMF、CH2Cl2、二氯乙烷、THF、二甲基乙酰胺等。在某些实施方案中,使用DMF作为溶剂。
在第二实施方案中,本发明提供了制备式(3)的化合物

或其盐的方法,其包括使式(2)的化合物与卤化试剂、磺化试剂或酯化试剂反应,

其中L’、L”、L”’、G和P1如第一实施方案中所定义,且X1为选自下列基团的离去基团:-Br、-I、-Cl、磺酸酯和活化的酯。
在一个具体的实施方案中,式(2)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在另一个具体的实施方案中,对于制备上述式(3d)或(3A)的化合物的方法,X1为-Br、-I或磺酸酯。
在另一个具体的实施方案中,X1为甲磺酸酯、甲苯磺酸酯、对溴苯磺酸酯或三氟甲磺酸酯。优选地,X1为甲磺酸酯。
在另一个具体的实施方案中,第二实施方案的方法包括使式(2)的化合物与卤化试剂反应。示例性卤化试剂包括但不限于溴、氢溴酸、四溴化碳、三溴化磷、溴化钾、氢碘酸、碘、四碘化碳、三碘化磷、碘化钠或碘化钾。
在另一个具体的实施方案中,第二实施方案的方法包括使式(2)的化合物与磺化试剂反应。优选地,磺化试剂为磺酸酐,诸如甲磺酸酐,或磺酰氯,诸如甲磺酰氯(MsCl)。
在某些实施方案中,式(2)的化合物与磺化试剂之间的反应可以在碱存在下进行。在一个实施方案中,该碱为非亲核性碱。示例性非亲核性碱包括但不限于三乙胺、咪唑、三乙胺、二异丙基乙胺、吡啶、2,6-二甲基吡啶、二甲基甲酰胺、1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)或四甲基哌啶。优选地,该碱为三乙胺或二异丙基乙胺。
任何合适的有机溶剂都可以用于第二实施方案的方法中。在一个实施方案中,溶剂为二氯甲烷。
在第三实施方案中,本发明提供了制备式(4)的化合物

或其盐的方法,所述方法包括使式(3)的化合物

与式(a)的单体化合物反应,

其中:
R1、R2、R3和R4各自独立地选自由-H,具有1至10个碳原子的任选取代的直链、支链或环状烷基、烯基或炔基,聚乙二醇单元-(CH2CH2O)n’-Rc,卤素,胍鎓[-NH(C=NH)NH2],-OR,-NR’R”,-NO2,-NCO,-NR’COR”,-SR,-SOR’,-SO2R’,-SO3 -H,-OSO3H,-SO2NR’R”,氰基,叠氮基,-COR’,-OCOR’和-OCONR’R”组成的组;且
R6为-H、-R、-OR、-SR,-NR’R”、-NO2或卤素;且剩下的变量如第二实施方案中所述。
另选地,第三实施方案提供了制备式(8)的化合物

或其盐的方法,所述方法包括使式(3’)的化合物

与式(b)的单体化合物反应,

其中R1’、R2’、R3’和R4’各自独立地选自由-H,具有1至10个碳原子的任选取代的直链、支链或环状烷基、烯基或炔基,聚乙二醇单元
-(CH2CH2O)n’-Rc,卤素,胍鎓[-NH(C=NH)NH2],-OR,-NRR’R”,-NO2,-NCO,-NR’COR”,-SR,-SOR’,-SO2R’,-SO3 -H,-OSO3H,-SO2NR’R”,氰基,叠氮基,-COR’,-OCOR’和-OCONR’R”;R6为-H、-R、-OR、-SR,-NR’R”、-NO2或卤素组成的组;且剩下的变量如第二实施方案中所述。
在一个具体的实施方案中,式(3)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个具体的实施方案中,对于第三实施方案的方法,X1为-Br、-I或磺酸酯。
在一个具体的实施方案中,式(3)的化合物与式(b)的单体化合物在碱存在下反应。可以使用任何合适的碱。示例性碱包括但不限于碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。在一个实施方案中,该碱为碳酸钾。
在第四实施方案中,本发明提供了制备式(5)的化合物

或其盐的方法,所述方法包括使式(4)的化合物

与亚胺还原剂反应,其中变量如上文在第三实施方案中所述。
在一个具体的实施方案中,式(4)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个具体的实施方案中,亚胺还原试剂为氢化物还原试剂。
在另一个具体的实施方案中,亚胺还原试剂为硼氢化钠、三乙酰氧基硼氢化钠、氰基硼氢化钠、氢化铝锂、氢气、甲酸铵、硼烷、9-硼杂双环[3.3.1]壬烷(9-BBN)、二异丁基氢化铝(DIBAL)、硼氢化锂(LiBH4)、硼氢化钾(KBH4)或双(2-甲氧基乙氧基)氢化铝钠(Red-Al)。优选地,亚胺还原试剂为三乙酰氧基硼氢化钠(NaBH(OAc)3)。
任何合适的溶剂都可以用于第四实施方案的方法中。在一个实施方案中,溶剂为二氯乙烷。
在第五实施方案中,本发明提供了制备式(6)的化合物

或其盐的方法,所述方法包括使式(5)的化合物

与醇脱保护试剂反应,其中变量如上文在第四实施方案中所述。
在一个具体的实施方案中,式(5)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个具体的实施方案中,醇脱保护试剂为四-正丁基氟化铵、三(二甲基氨基)二氟三甲基硅酸锍、氟化氢或其溶剂化物、氟化氢吡啶、四氟化硅、六氟硅酸、氟化铯、盐酸、乙酸、三氟乙酸、对甲苯磺酸吡啶鎓、对甲苯磺酸(p-TsOH)、甲酸或高碘酸。优选地,醇脱保护试剂为盐酸或四-正丁基氟化铵。
任何合适的溶剂都可以用于上述脱保护反应中。在一个实施方案中,该溶剂为THF。
在第六实施方案中,本发明提供了制备式(7)的化合物

或其盐的方法,所述方法包括使卤化试剂、磺化试剂或酯化试剂与式(6)的伯醇化合物反应,

其中X2为-Br、-I、-Cl、磺酸酯或活化的酯;且剩下的变量如上文在第五实施方案中所述。
在一个具体的实施方案中,式(6)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个具体的实施方案中,对于第六实施方案的方法,X2为-Br、-I或磺酸酯。
在一个具体的实施方案中,X2为甲磺酸酯、甲苯磺酸酯、对溴苯磺酸酯或三氟甲磺酸酯。优选地,X2为甲磺酸酯。
在另一个具体的实施方案中,第六实施方案的方法包括使式(6)的化合物与卤化试剂反应。示例性卤化试剂包括但不限于溴、氢溴酸、四溴化碳、三溴化磷、溴化钾、氢碘酸、碘、四碘化碳、三碘化磷、碘化钠或碘化钾。在另一个具体的实施方案中,第六实施方案的方法包括使式(6)的化合物与磺化试剂反应。优选地,磺化试剂为磺酸酐,诸如甲磺酸酐,或磺酰氯,诸如甲磺酰氯(MsCl)。
在一个实施方案中,式(6)的化合物与磺化试剂之间的反应在碱存在下进行。优选地,该碱为非亲核性碱。示例性非亲核性碱包括但不限于三乙胺、咪唑、三乙胺、二异丙基乙胺、吡啶、2,6-二甲基吡啶、二甲基甲酰胺、1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)或四甲基哌啶。优选地,该碱为三乙胺或二异丙基乙胺。任何合适的溶剂都可以用于上述反应中。在一个实施方案中,溶剂为二氯甲烷。
在第六实施方案中,本发明提供了制备式(7”)的化合物

或其盐的方法,所述方法包括使式(5”)的化合物

与醇脱保护试剂和卤化试剂反应,其中P1’为酸不稳定的醇保护基团;X2’为-Br或-I;且剩下的变量如上文在第六实施方案中所述。
在一个具体的实施方案中,式(5”)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
第七实施方案的方法将第五实施方案中描述的醇脱保护步骤与第六实施方案中描述的所得醇的卤化反应组合成一个步骤。
在一个具体的实施方案中,对于第七实施方案的方法,式(7”)的化合物由下式表示:

且该方法包括使式(5”)的化合物与醇脱保护试剂和溴化试剂反应。
在一个实施方案中,酸不稳定的醇保护基团为乙酸根、烯丙基、甲氧基甲基、四氢呋喃基、四氢吡喃基、5-二苯并环庚基、1-乙氧基乙基、1-甲基-1-甲氧基乙基、2-(苯基氢硒基)乙基、三苯甲基/三苯基甲基或三(4-叔丁基苯基)甲基。
在另一个实施方案中,酸不稳定的醇保护基团为甲硅烷基保护基团。示例性甲硅烷基保护基团包括但不限于二甲基异丙基甲硅烷基、二乙基异丙基甲硅烷基、二甲基己基甲硅烷基、三甲基甲硅烷基、三乙基甲硅烷基、三异丙基甲硅烷基、三苄基甲硅烷基、三苯基甲硅烷基、2-降冰片基二甲基甲硅烷基、叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基、2-三甲基乙基甲硅烷基(TEOC)或[2-(三甲基甲硅烷基)乙氧基]甲基。优选地,甲硅烷基保护基团为三乙基甲硅烷基、三异丙基甲硅烷基或叔丁基二甲基甲硅烷基。更优选地,甲硅烷基保护基团为叔丁基二甲基甲硅烷基。
在一个实施方案中,醇脱保护试剂为四-正丁基氟化铵、三(二甲基氨基)二氟三甲基硅酸锍、氟化氢或其溶剂化物、氟化氢吡啶、四氟化硅、六氟硅酸、氟化铯、盐酸、乙酸、对甲苯磺酸吡啶鎓、甲酸、高碘酸、三氟乙酸或对甲苯磺酸(p-TsOH)。优选地,醇脱保护试剂为乙酸。
在另一个实施方案中,溴化试剂为HBr。
在一个具体的实施方案中,第七实施方案的方法包括使式(5”)的化合物与乙酸和HBr的混合物反应,得到式(7”)的化合物。
在第八实施方案中,本发明提供了制备式(I’)的化合物

或其药学上可接受的盐的方法,所述方法包括使式(7)的化合物

与式(b)的单体化合物反应,

其中R1’、R2’、R3’和R4’各自独立地选自由以下组成的组:-H,具有1至10个碳原子的任选取代的直链、支链或环状烷基、烯基或炔基,聚乙二醇单元-(CH2CH2O)n-Rc,卤素,胍鎓[-NH(C=NH)NH2],-OR,-NR’R”,-NO2,-NCO,-NR’COR”,-SR,-SOR’,-SO2R’,-SO3 -H,-OSO3H,-SO2NR’R”,氰基,叠氮基,-COR’,-OCOR’和-OCONR’R”;且剩下的变量如上文在第七实施方案中所述。
在一个具体的实施方案中,式(7)的化合物由选自以下的式表示:

其中X2为选自由以下组成的组的离去基团:-Br、-I、-Cl、磺酸酯和活化的酯;R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个实施方案中,对于第八实施方案的方法,X2为-Br、-I或磺酸酯。
在一个实施方案中,式(7)的化合物与式(b)的单体化合物在碱存在下反应。碱的实例包括但不限于碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。在一个实施方案中,该碱为碳酸钾。
任何合适的溶剂都可以用于上述方法中。在一个实施方案中,该溶剂为DMF。
在第九实施方案中,本发明提供了制备式(I’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1)的化合物的伯醇中的一个上,

以形成式(2)的化合物,

(2)使式(2)的化合物与卤化试剂、磺化试剂或酯化试剂反应以形成式(3)的化合物,

(3)使式(3)的化合物与式(a)的单体化合物反应,

以形成式(4)的化合物,

(4)使式(4)的化合物与亚胺还原剂反应以形成式(5)的化合物,

(5)使式(5)的化合物与醇脱保护试剂反应以形成式(6)的化合物,

(6)使第二卤化试剂、第二磺化试剂或第二酯化试剂与式(6)的化合物反应以形成式(7)的化合物,

(7)使式(7)的化合物与式(b)的单体化合物反应,

以形成式(I’)的化合物,其中变量如文在第一、第二、第三、第四、第五、第六和第八实施方案中所述。
在一个具体的实施方案中,第九实施方案的方法涉及制备式(Ib’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1b)的化合物的伯醇中的一个上以形成式(2b)的化合物;
(2)使式(2b)的化合物与卤化试剂、磺化试剂或酯化试剂反应以形成式(3b)的化合物;
(3)使式(3b)的化合物与式(a)的单体化合物反应以形成式(4b)的化合物;
(4)使式(4b)的化合物与亚胺还原剂反应以形成式(5b)的化合物;
(5)使式(5b)的化合物与醇脱保护试剂反应以形成式(6b)的化合物;
(6)使式(6b)的化合物与第二卤化试剂、第二磺化试剂或第二酯化试剂反应以形成式(7b)的化合物;
(7)使式(7b)的化合物与式(b)的单体化合物反应以形成式(Ib’)的化合物。
在一个具体的实施方案中,第九实施方案的方法涉及制备式(Ic’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1c)的化合物的伯醇中的一个上以形成式(2c)的化合物;
(2)使式(2c)的化合物与卤化试剂、磺化试剂或酯化试剂反应以形成式(3c)的化合物;
(3)使式(3c)的化合物与式(a)的单体化合物反应以形成式(4c)的化合物;
(4)使式(4c)的化合物与亚胺还原剂反应以形成式(5c)的化合物;
(5)使式(5c)的化合物与醇脱保护试剂反应以形成式(6c)的化合物;
(6)使式(6c)的化合物与第二卤化试剂、第二磺化试剂或第二酯化试剂反应以形成式(7c)的化合物;
(7)使式(7c)的化合物与式(b)的单体化合物反应以形成式(Ic’)的化合物。
在一个具体的实施方案中,第九实施方案的方法涉及制备式(IA)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1A)的化合物的伯醇中的一个上以形成式(2A)的化合物;
(2)使式(2A)的化合物与卤化试剂、磺化试剂或酯化试剂反应以形成式(3A)的化合物;
(3)使式(3A)的化合物与式(a)的单体化合物反应以形成式(4A)的化合物;
(4)使式(4A)的化合物与亚胺还原剂反应以形成式(5A)的化合物;
(5)使式(5A)的化合物与醇脱保护试剂反应以形成式(6A)的化合物;
(6)使式(6A)的化合物与第二卤化试剂、第二磺化试剂或第二酯化试剂反应以形成式(7A)的化合物;
(7)使式(7A)的化合物与式(b)的单体化合物反应以形成式(IA)的化合物。
在一个实施方案中,对于第九实施方案的方法,X1和X2各自独立地为-Br、-Cl或磺酸酯。
用于第九实施方案的方法中的每个步骤的反应条件和试剂如在第一、第二、第三、第四、第五、第六和/或第八实施方案或其中描述的任何具体实施方案中所述。
在第十实施方案中,本发明提供了制备式(I’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1)的化合物的伯醇中的一个上,

以形成式(2”)的化合物,

(2)使式(2”)的化合物与卤化试剂、磺化试剂或酯化试剂反应以形成式(3”)的化合物,

(3)使式(3”)的化合物与式(a)的单体化合物反应,

以形成式(4”)的化合物,

(4)使式(4”)的化合物与亚胺还原剂反应以形成式(5”)的化合物,

(5)使式(5”)的化合物与醇脱保护试剂和卤化试剂反应以形成式(7”)的化合物,

(6)使式(7”)的化合物与式(b)的单体化合物反应,

以形成式(I’)的化合物,其中X2’为-Br或-I;且剩下的变量如上文在第九实施方案中所述。
在一个实施方案中,第十实施方案的方法涉及制备式(Ib’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1b)的化合物的伯醇中的一个上以形成式(2b”)的化合物,

(2)使式(2b”)的化合物与卤化试剂或磺化试剂反应以形成式(3b”)的化合物,

(3)使式(3b”)的化合物与式(a1)的单体化合物反应

以形成式(4b”)的化合物,

(4)使式(4b”)的化合物与亚胺还原剂反应以形成式(5b”)的化合物;
(5)使式(5b”)的化合物与醇脱保护试剂和卤化试剂反应以形成式(7b”)的化合物;
(6)使式(7b”)的化合物与式(a1)的单体化合物反应以形成式(Ib’)的化合物。
在另一个实施方案中,第十实施方案的方法涉及制备式(Ic’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1c)的化合物的伯醇中的一个上以形成式(2c”)的化合物,

(2)使式(2c”)的化合物与卤化试剂或磺化试剂反应以形成式(3c”)的化合物,

(3)使式(3c”)的化合物与式(a1)的单体化合物反应以形成式(4c”)的化合物,

(4)使式(4c”)的化合物与亚胺还原剂反应以形成式(5c”)的化合物;
(5)使式(5c”)的化合物与醇脱保护试剂和卤化试剂反应以形成式(7c”)的化合物;
(6)使式(7c”)的化合物与式(a1)的单体化合物反应以形成式(Ic’)的化合物。
在另一个实施方案中,第十实施方案的方法涉及制备式(IA)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1A)的化合物的伯醇中的一个上以形成式(2A”)的化合物,

(2)使式(2A”)的化合物与卤化试剂或磺化试剂反应以形成式(3A”)的化合物,

(3)使式(3A”)的化合物与式(a1)的单体化合物反应以形成式(4A”)的化合物,

(4)使式(4A”)的化合物与亚胺还原剂反应以形成式(5A”)的化合物;
(5)使式(5A”)的化合物与醇脱保护试剂和卤化试剂反应以形成式(7A”)的化合物;
(6)使式(7A”)的化合物与式(a1)的单体化合物反应以形成式(IA’)的化合物。
用于第十实施方案的方法的条件和试剂如上文在第一、第二、第三、第四、第七和/或第八实施方案以及其中描述的任何具体实施方案中所述。
在第十一实施方案中,本发明提供了制备式(9)的化合物

或其盐的方法,所述方法包括使式(8)的化合物

与醇脱保护试剂反应,其中变量如上文在第三实施方案中所述。
在一个具体的实施方案中,式(8)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个具体的实施方案中,醇脱保护试剂为四-正丁基氟化铵、三(二甲基氨基)二氟三甲基硅酸锍、氟化氢或其溶剂化物、氟化氢吡啶、四氟化硅、六氟硅酸、氟化铯、盐酸、乙酸、对甲苯磺酸吡啶鎓、甲酸、高碘酸、三氟乙酸或对甲苯磺酸(p-TsOH)。更具体地讲,醇脱保护试剂为盐酸或四-正丁基氟化铵。
在第十二实施方案中,本发明提供了制备式(10)的化合物

或其盐的方法,所述方法包括使式(9)的化合物与卤化试剂、磺化试剂或酯化试剂反应,

其中X2为-Br、-I、-Cl、磺酸酯或活化的酯;且剩下的变量如上文在第十一实施方案中所述。
在一个实施方案中,式(9)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个具体的实施方案中,对于第十二实施方案的方法,X2为-Br、-I或磺酸酯。
在一个具体的实施方案中,X2为甲磺酸酯、甲苯磺酸酯、对溴苯磺酸酯或三氟甲磺酸酯。优选地,X2为甲磺酸酯。
在另一个具体的实施方案中,第六实施方案中所述的方法包括使式(9)的化合物与卤化试剂反应。示例性卤化试剂包括但不限于溴、氢溴酸、四溴化碳、三溴化磷、溴化钾、氢碘酸、碘、四碘化碳、三碘化磷、碘化钠或碘化钾。
在另一个具体的实施方案中,第六实施方案的方法包括使式(9)的化合物与磺化试剂反应。优选地,磺化试剂为磺酸酐,诸如甲磺酸酐,或磺酰氯,诸如甲磺酰氯(MsCl)。
在一个实施方案中,式(9)的化合物与磺化试剂之间的反应在碱存在下进行。优选地,该碱为非亲核性碱。示例性非亲核性碱包括但不限于三乙胺、咪唑、三乙胺、二异丙基乙胺、吡啶、2,6-二甲基吡啶、二甲基甲酰胺、1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)或四甲基哌啶。优选地,该碱为三乙胺或二异丙基乙胺。
在第十二实施方案中,本发明提供了制备式(18)的化合物

或其盐的方法,所述方法包括使式(10)的化合物

与式(d)的单体化合物反应,

其中X2为选自由以下组成的组的离去基团:-Br、-I、-Cl、磺酸酯和活化的酯;P3为H或P2;P2为胺保护基团;R1’、R2’、R3’和R4’各自独立地选自由-H,具有1至10个碳原子的任选取代的直链、支链或环状烷基、烯基或炔基,聚乙二醇单元-(CH2CH2O)n-Rc,卤素,胍鎓[-NH(C=NH)NH2],-OR,-NR’R”,-NO2,-NCO,-NR’COR”,-SR,-SOR’,-SO2R’,-SO3 -H,-OSO3H,-SO2NR’R”,氰基,叠氮基,-COR’,-OCOR’和-OCONR’R”组成的组;且剩下的变量如上文在第十二实施方案中所述。在一个实施方案中,X2为-Br、-I或磺酸酯。
在一个具体的实施方案中,式(10)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个具体的实施方案中,P3为H且(10)的化合物与(d)的单体化合物反应以形成(I’)的化合物:

在另一个具体的实施方案中,P3为P2;该单体化合物由式(c)表示:

且式(18)的化合物由式(11)表示,

任何合适的胺保护基团都可以用于上述方法中。在一个实施方案中,胺保护基团为2-三甲基甲硅烷基乙基、(2-苯基-2-三甲基甲硅烷基)乙基、三异丙基甲硅烷氧基、2-(三甲基甲硅烷基)乙氧基甲基、烯丙氧基羰基、9-芴基甲氧基羰基、2-(三甲基甲硅烷基)乙氧基羰基或2,2,2,2-三氯乙氧基羰基。
在一个具体的实施方案中,式(10)的化合物与式(d)或(c)的单体化合物在碱存在下反应。碱的实例包括但不限于碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。
任何合适的溶剂都可以用于上述反应中。在一个实施方案中,该溶剂为DMF。
在第十四实施方案中,本发明提供了制备式(I’)的化合物

或其药学上可接受的盐的方法,所述方法包括使式(11)的化合物

与胺脱保护试剂反应,其中变量如上文在第十三实施方案中所述。
在一个具体的实施方案中,式(11)的化合物由选自以下的式表示:


其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
任何合适的胺脱保护试剂都可以用于上述方法中。在一个实施方案中,胺脱保护试剂为四-正丁基氟化铵、乙酸、氟化氢吡啶、氟化铯、哌啶、吗啉或三氟乙酸。
在第十五实施方案中,本发明提供了制备式(I’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1)的化合物的伯醇中的一个上,

以形成式(2)的化合物,

(2)使卤化试剂、磺化试剂或酯化试剂与式(2)的化合物反应以形成式(3)的化合物,

(3)使式(3)的化合物与式(b)的单体化合物反应,

以形成式(8)的化合物,

(4)使式(8)的化合物与醇脱保护试剂反应以形成式(9)的化合物,

(5)使第二卤化试剂、第二磺化试剂或第二酯化试剂与式(9)的化合物反应以形成式(10)的化合物,

(6)使式(10)的化合物与式(d)的单体化合物反应,

以形成式(18)的化合物,

(7)当P3为胺保护基团时,使式(18)的化合物与胺脱保护试剂反应以形成式(I’)的化合物,其中变量如上文在第十四实施方案中所述。在一个实施方案中,X1和X2各自独立地为-Br、-I或磺酸酯。
在一个实施方案中,第十五实施方案的方法涉及制备式(Ib’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1b)的化合物的伯醇中的一个上以形成式(2b)的化合物;
(2)使卤化试剂或磺化试剂与式(2b)的化合物反应以形成式(3b)的化合物;
(3)使式(3b)的化合物与式(b1)的单体化合物反应以形成式(4b)的化合物;
(4)使式(4b)的化合物与醇脱保护试剂反应以形成式(9b)的化合物;
(5)使第二卤化试剂或第二磺化试剂与式(9b)的化合物反应以形成式(10b)的化合物;
(6)使式(10b)的化合物与式(d1)的单体化合物反应:

以形成式(18b)的化合物;
(7)当P3为胺保护基团时,使式(18b)的化合物:

与胺脱保护试剂反应以形成式(Ib’)的化合物。
在另一个实施方案中,第十五实施方案的方法涉及制备式(Ic’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1c)的化合物的伯醇中的一个上以形成式(2c)的化合物;
(2)使卤化试剂或磺化试剂与式(2c)的化合物反应以形成式(3c)的化合物;
(3)使式(3b)的化合物与式(b1)的单体化合物反应以形成式(4c)的化合物;
(4)使式(4c)的化合物与醇脱保护试剂反应以形成式(9c)的化合物;
(5)使第二卤化试剂或第二磺化试剂与式(9c)的化合物反应以形成式(10c)的化合物;
(6)使式(10c)的化合物与式(d1)的单体化合物反应以形成式(18c)的化合物;
(7)当P3为胺保护基团时,使式(18c)的化合物:

与胺脱保护试剂反应以形成式(Ic’)的化合物。
在另一个实施方案中,第十五实施方案的方法涉及制备式(IA)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)将醇保护基团引入到式(1A)的化合物的伯醇中的一个上以形成式(2A)的化合物;
(2)使卤化试剂或磺化试剂与式(2A)的化合物反应以形成式(3A)的化合物;
(3)使式(3A)的化合物与式(b1)的单体化合物反应以形成式(4A)的化合物;
(4)使式(4A)的化合物与醇脱保护试剂反应以形成式(9A)的化合物;
(5)使第二卤化试剂或第二磺化试剂与式(9A)的化合物反应以形成式(10A)的化合物;
(6)使式(10A)的化合物与式(d1)的单体化合物反应以形成式(18A)的化合物;

(7)当P3为胺保护基团时,使式(18A)的化合物与胺脱保护试剂反应以形成式(IA’)的化合物。
在一个实施方案中,P3为H且(10)的化合物与(d)的单体化合物反应以形成(I’)的化合物。
在另一个实施方案中,P3为P2;该单体化合物由式(c)表示:

且式(18)的化合物由式(11)表示,

其中P2为胺保护基团。
在第十六实施方案中,本发明提供了制备式(12)的化合物

或其盐的方法,所述方法包括使式(1)的化合物

与卤化试剂、磺化试剂或酯化试剂反应,其中X1为-Br、-I、-Cl、磺酸酯或活化的酯;且剩下的变量如上文在第一实施方案中所述。在一个实施方案中,X1为-Br、-I或磺酸酯。
在一个实施方案中,式(1)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个具体的实施方案中,X1为-Br或-I。在另一个具体的实施方案中,X1为-Cl。
在另一个具体的实施方案中,卤化试剂与式(1)的化合物的伯醇在醇活化剂存在下反应。在一个实施方案中,醇活化剂为亚硫酰氯。
在另一个具体的实施方案中,卤化试剂为溴化锂、溴化钠、溴化钾、碘化钾或碘化钠。在另一个具体的实施方案中,卤化试剂为四氯化碳/三苯基膦、甲磺酰基氯化物/氯化锂或甲磺酰基氯化物/吡啶。
在另一个具体的实施方案中,该方法包括在亚硫酰氯存在下使式(1)的化合物与LiBr反应。
任何合适的溶剂都可以用于上述方法中。示例性溶剂包括但不限于DMF、CH2Cl2、THF、二氯乙烷等。
在第十七实施方案中,本发明提供了制备式(10’)的化合物

或其盐的方法,所述方法包括使式(12)的化合物

与式(b)的单体化合物反应,

其中L’、L”、L”’、X1、G如上文在第十七实施方案中所述;且R1’、R2’、R3’、R4’和R6如上文在第十一实施方案中所述。在一个实施方案中,X1为-Br、-I或磺酸酯。
在一个具体的实施方案中,式(12)的化合物由选自以下的式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在第十七实施方案中还提供了制备式(7-1)的化合物

或其盐的方法,所述方法包括使式(12)的化合物与式(d)的单体化合物反应,其中X1为-Br、-I、-Cl、磺酸酯或活化的酯;P3为H或胺保护基团;且R100为(C1-C3)烷氧基。
在一个具体的实施方案中,式(12)的化合物由式(12b)、(12c)或(12A)表示。
在一个具体的实施方案中,对于式(7-1’),P3为H。在另一个具体的实施方案中,P3为如本文所述的胺保护基团。
在一个具体的实施方案中,对于第十七实施方案的方法,式(12)的化合物与式(b)的单体化合物在碱存在下反应。合适的碱的实例包括但不限于碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。在一个实施方案中,该碱为碳酸钾。
在一个具体的实施方案中,式(12d)或(12A)的化合物与式(d)的单体化合物在碱存在下反应。合适的碱的实例包括但不限于碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。在一个实施方案中,该碱为碳酸钾。
对于第十七实施方案的方法,可以使用任何合适的溶剂。在一个实施方案中,该溶剂为DMF。
在另一个具体的实施方案中,在反应中使用相对于式(b)的单体化合物而言摩尔当量过量的式(12)的化合物。
在第十八实施方案中,本发明提供了制备式(7’)的化合物

或其盐的方法,所述方法包括使式(10’)的化合物

或其盐与亚胺还原剂反应,其中X1为-Br、-I、-Cl、磺酸酯或活化的酯,且剩下的变量与上文在第十一实施方案中所述相同。在一个实施方案中,X1为-Br、-I或磺酸酯。
在一个具体的实施方案中,式(10’)的化合物由下式中的一个表示:

在一个具体的实施方案中,对于第十八实施方案的方法,X1为磺酸酯。优选地,X1为甲磺酸酯。
在一个实施方案中,对于第十八实施方案的方法,亚胺还原试剂为氢化物还原试剂。另选地,亚胺还原试剂为硼氢化钠、三乙酰氧基硼氢化钠、氰基硼氢化钠、氢化铝锂、氢气、甲酸铵、硼烷、9-硼杂双环[3.3.1]壬烷(9-BBN)、二异丁基氢化铝(DIBAL)、硼氢化锂(LiBH4)、硼氢化钾(KBH4)或双(2-甲氧基乙氧基)氢化铝钠(Red-Al)。在一个优选的实施方案中,亚胺还原试剂为三乙酰氧基硼氢化钠(NaBH(OAc)3)。
任何合适的溶剂都可以用于第十八实施方案的方法中。在一个实施方案中,溶剂为二氯乙烷。
在第十九实施方案中,本发明提供了制备式(I’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂、磺化试剂或酯化试剂与式(1)的化合物反应,

以形成式(12)的化合物,

(2)使式(12)的化合物与式(b)的单体化合物反应,

以形成式(10’)的化合物,

(3)使式(10’)的化合物与式(d)的单体化合物反应,

以形成式(18)的化合物,

(4)当P3为胺保护基团时,使式(18)的化合物与胺脱保护试剂反应以形成式(I’)的化合物,
其中X1为-Br、-I、-Cl、磺酸酯或活化的酯;P3为H或胺保护基团,且剩下的变量如上文在第十一实施方案中所述。在一个实施方案中,X1为-Br、-I或磺酸酯。
在一个实施方案中,第十九实施方案的方法涉及制备式(Ib’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂或磺化试剂与式(1b)的化合物反应以形成式(12b)的化合物;
(2)使式(12b)的化合物与式(b1)的单体化合物反应以形成式(10b’)的化合物;
(3)使式(10b’)的化合物与式(d1)的单体化合物反应以形成式(18b)的化合物;和
(4)当P3为胺保护基团时,使式(18b)的化合物与胺脱保护试剂反应以形成式(Ib’)的化合物。
在另一个实施方案中,第十九实施方案的方法涉及制备式(Ic’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂或磺化试剂与式(1c)的化合物反应以形成式(12c)的化合物;
(2)使式(12c)的化合物与式(b1)的单体化合物反应以形成式(10c’)的化合物;
(3)使式(10c’)的化合物与式(d1)的单体化合物反应以形成式(18c)的化合物;和
(4)当P3为胺保护基团时,使式(18c)的化合物与胺脱保护试剂反应以形成式(Ic’)的化合物。
在另一个实施方案中,第十九实施方案的方法涉及制备式(IA)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂或磺化试剂与式(1A)的化合物反应以形成式(12A)的化合物;
(2)使式(12A)的化合物与式(b1)的单体化合物反应以形成式(10A’)的化合物;
(3)使式(10A’)的化合物与式(d1)的单体化合物反应以形成式(18A)的化合物;和
(4)当P3为胺保护基团时,使式(18A)的化合物与胺脱保护试剂反应以形成式(IA’)的化合物。
在一个实施方案中,对于第十九实施方案的方法,P3为H且(10’)的化合物与(d)的单体化合物反应以形成(I’)的化合物。
在另一个实施方案中,对于第十九实施方案的方法,P3为P2;该单体化合物由式(c)表示:

且式(18)的化合物由式(11)表示,

其中P2为胺保护基团。
用于第十九实施方案的方法的条件和试剂如上文在第十六、第十七、第十三和/或第十四实施方案以及其中描述的任何具体实施方案中所述。
在第二十实施方案中,本发明提供了制备式(I’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂、磺化试剂或酯化试剂与式(1)的化合物反应,

以形成式(12)的化合物,

(2)使式(12)的化合物与式(b)的单体化合物反应,

以形成式(10’)的化合物,

(3)使化合物(10’)与亚胺还原试剂反应以形成化合物(7’),

(4)使式(7’)的化合物与式(a)的单体化合物反应,

以形成式(I’)的化合物或其药学上可接受的盐,其中X1为-Br、-I、-Cl、磺酸酯或活化的酯,且剩下的变量与上文在第十一实施方案中所述相同。在一个实施方案中,X1为-Br、-I或磺酸酯。
在一个实施方案中,第二十实施方案的方法涉及制备式(Ib’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂或磺化试剂与式(1b)的化合物反应以形成式(12b)的化合物;
(2)使式(12b)的化合物与式(a1)的单体化合物反应以形成式(10b’)的化合物;
(3)使化合物(10b’)与亚胺还原试剂反应以形成化合物(7b’);
(4)使式(7b’)的化合物与式(a1)的单体化合物反应以形成式(Ib’)的化合物。
在另一个实施方案中,第二十实施方案的方法涉及制备式(Ic’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂或磺化试剂与式(1c)的化合物反应以形成式(12c)的化合物;
(2)使式(12c)的化合物与式(a1)的单体化合物反应以形成式(10c’)的化合物;
(3)使化合物(10c’)与亚胺还原试剂反应以形成化合物(7c’);
(4)使式(7c’)的化合物与式(a1)的单体化合物反应以形成式(Ic’)的化合物。
在另一个实施方案中,第二十实施方案的方法涉及制备式(IA’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂或磺化试剂与式(1A)的化合物反应以形成式(12A)的化合物;
(2)使式(12A)的化合物与式(a1)的单体化合物反应以形成式(10A’)的化合物;
(3)使化合物(10A’)与亚胺还原试剂反应以形成化合物(7A’);
(4)使式(7A’)的化合物与式(a1)的单体化合物反应以形成式(IA’)的化合物。
在一个具体的实施方案中,X1为甲磺酸酯。
用于第二十实施方案的方法的条件和试剂如上文在第十六、第十七、第十八和/或第八实施方案以及其中描述的任何具体实施方案中所述。
在第二十一实施方案中,本发明提供了制备式(I’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂、磺化试剂或酯化试剂与式(1)的化合物反应,

以形成式(12)的化合物,

(2)使式(12)的化合物与式(d)的单体化合物反应,

以形成式(7-1)的化合物,

(3)使式(7-1)的化合物与式(b)的单体化合物反应,

以形成式(18)的化合物,

(4)当P3为胺保护基团时,使式(18)的化合物与胺脱保护试剂反应以形成式(I’)的化合物;其中X1为-Br、-I、-Cl、磺酸酯或活化的酯;P3为H或胺保护基团,且剩下的变量与上文在第十一实施方案中所述相同。在一个实施方案中,X1为-Br、-I或磺酸酯。
在一个实施方案中,第二十一实施方案的方法涉及制备式(Ib’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂或磺化试剂与式(1b)的化合物反应以形成式(12b)的化合物;
(2)使式(12b)的化合物与式(d1)的单体化合物反应以形成式(7b-1)的化合物,

(3)使式(7b-1)的化合物与式(a1)的单体化合物反应以形成式(18b)的化合物;和
(4)当P3为胺保护基团时,使式(18b)的化合物与胺脱保护试剂反应以形成式(Ib’)的化合物。
在另一个实施方案中,第二十一实施方案的方法涉及制备式(Ic’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂或磺化试剂与式(1c)的化合物反应以形成式(12c)的化合物;
(2)使式(12c)的化合物与式(d1)的单体化合物反应以形成式(7c-1)的化合物,

(3)使式(7c-1)的化合物与式(a1)的单体化合物反应以形成式(18c)的化合物;和
(4)当P3为胺保护基团时,使式(18c)的化合物与胺脱保护试剂反应以形成式(Ic’)的化合物。
在另一个实施方案中,第二十一实施方案的方法涉及制备式(IA’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂或磺化试剂与式(1A)的化合物反应以形成式(12A)的化合物;
(2)使式(12A)的化合物与式(d1)的单体化合物反应以形成式(7A-1)的化合物,

(3)使式(7A-1)的化合物与式(a1)的单体化合物反应以形成式(18A)的化合物;和
(4)当P3为胺保护基团时,使式(18A)的化合物与胺脱保护试剂反应以形成式(IA’)的化合物。
在一个实施方案中,对于第二十一实施方案的方法,P3为H。
在一个实施方案中,P3为H且(7-1)的化合物与(b)的单体化合物反应以形成(I’)的化合物。
在另一个实施方案中,P3为P2;该单体化合物由式(c)表示:

且式(18)的化合物由式(11)表示,

其中P2为胺保护基团。
在一个具体的实施方案中,X1为甲磺酸酯。
用于第二十一实施方案的方法的条件和试剂如上文在第十六、第十七、第十八、第八和/或第十四实施方案以及其中描述的任何具体实施方案中所述。
在第二十二实施方案中,本发明提供了制备式(13)的化合物

或其盐的方法,所述方法包括使氯化试剂与式(2)的化合物反应,

其中X3为Cl,且剩下的变量与上述相同。
在一个具体的实施方案中,式(2)的化合物选自由以下组成的组:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在另一个具体的实施方案中,醇保护基团为新戊酰基、甲氧基甲基、2-甲氧基乙氧基甲基、对甲氧基苄基、3,4-二甲氧基苄基、2,6-二甲氧基苄基、二苯基甲基、苄氧基甲基、2,2,2-三氯乙氧基羰基、四氢呋喃基、四氢吡喃基、苄基、苯甲酰基、对苯基苯甲酰基、2,4,6-三甲基苯甲酰基、对溴苯甲酰基、对硝基苯甲酰基、吡啶甲酰基、烟酰基、5-二苯并环庚基、三苯甲基/三苯基甲基或三(4-叔丁基苯基)甲基。优选地,醇保护基团为甲氧基甲基、四氢吡喃基、2-甲氧基乙氧基甲基、对甲氧基苄基、苄氧基甲基或2,2,2-三氯乙氧基羰基。甚至更优选地,醇保护基团为2,2,2-三氯乙氧基羰基。
在另一个具体的实施方案中,醇保护基团为甲硅烷基保护基团。例如,甲硅烷基保护基团为二甲基异丙基甲硅烷基、二乙基异丙基甲硅烷基、二甲基己基甲硅烷基、三甲基甲硅烷基、三异丙基甲硅烷基、三苄基甲硅烷基、三苯基甲硅烷基、2-降冰片基二甲基甲硅烷基、叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基、2-三甲基乙基甲硅烷基(TEOC)或[2-(三甲基甲硅烷基)乙氧基]甲基。优选地,甲硅烷基保护基团为三乙基甲硅烷基、三异丙基甲硅烷基或叔丁基二甲基甲硅烷基。更优选地,甲硅烷基保护基团为叔丁基二甲基甲硅烷基。
在一个实施方案中,使用碱。该碱可为非亲核性碱。非亲核性碱的实例包括但不限于三乙胺、咪唑、二异丙基乙胺(DIPEA)、吡啶、2,6-二甲基吡啶、二甲基甲酰胺、1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)或四甲基哌啶。优选地,非亲核性碱为吡啶。
任何合适的有机溶剂都可以用于第二十实施方案的方法中。示例性溶剂包括但不限于DMF、CH2Cl2、二氯乙烷、THF、二甲基乙酰胺等。在某些实施方案中,使用DMF作为溶剂。
在第二十三实施方案中,本发明提供了制备式(14)的化合物

或其盐的方法,所述方法包括使式(13)的化合物

与醇脱保护试剂反应,其中变量如上文在第二十实施方案中所述。
在一个具体的实施方案中,式(13)的化合物选自由以下组成的组:

其中R100和R101与上文定义相同。
在另一个具体的实施方案中,醇脱保护试剂为四-正丁基氟化铵、三(二甲基氨基)二氟三甲基硅酸锍、氟化氢或其溶剂化物、氟化氢吡啶、四氟化硅、六氟硅酸、氟化铯、盐酸、乙酸、三氟乙酸、对甲苯磺酸吡啶鎓、对甲苯磺酸(p-TsOH)、甲酸或高碘酸。优选地,醇脱保护试剂为氟化氢吡啶。
在第二十四实施方案中,本发明提供了制备式(15)的化合物

或其盐的方法,所述方法包括使磺化试剂或酯化试剂与式(14)的化合物反应,

其中X4为磺酸酯或活化的酯;且剩下的变量与上文在第二十实施方案中所述相同。
在一个具体的实施方案中,式(14)的化合物选自由以下组成的组:

其中R100和R101与上文定义相同。
在一个具体的实施方案中,对于第二十四实施方案的方法,X4为磺酸酯。
在另一个具体的实施方案中,甲磺酸酐、甲磺酰氯、对甲苯磺酰氯、4-溴苯磺酰氯或三氟甲磺酸酐。
在另一个具体的实施方案中,该磺酸酯为甲磺酸酯、甲苯磺酸酯、对溴苯磺酸酯或三氟甲磺酸酯。优选地,该磺酸酯为甲磺酸酯。
在另一个实施方案中,使用碱。该碱可为非亲核性碱。非亲核性碱的实例包括但不限于三乙胺、咪唑、二异丙基乙胺、吡啶、2,6-二甲基吡啶、二甲基甲酰胺、1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)或四甲基哌啶。优选地,非亲核性碱为二异丙基乙胺。
在第二十五实施方案中,本发明提供了制备式(20)的化合物

或其盐的方法,所述方法包括使溴化或碘化试剂与式(14)的化合物反应,

其中变量与上述相同。
在一个具体的实施方案中,式(14)的化合物选自由以下组成的组:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个特定的实施方案中,溴化或碘化试剂为溴、氢溴酸、四溴化碳、三溴化磷、溴化钾、氢碘酸、碘、四碘化碳、三碘化磷、碘化钠或碘化钾。
在第二十六实施方案中,本发明提供了制备式(16)的化合物

或其盐的方法,所述方法包括使式(15)的化合物

与式(b)的单体化合物反应,

其中变量X3、X4、L’、L”、L”和G如在第二十五实施方案中所述,且变量R1’、R2’、R3’、R4’和R6与上述相同。在一个实施方案中,X4为磺酸酯。
在一个具体的实施方案中,式(15)的化合物选自由以下组成的组:

且式(b)的单体化合物由下式表示:

其中R100和R101与上文定义相同。
在一个实施方案中,使用碱。在具体的实施方案中,该碱为碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。优选地,该碱为碳酸钾。
任何合适的有机溶剂都可以用于第二十实施方案的方法中。示例性溶剂包括但不限于DMF、CH2Cl2、二氯乙烷、THF、二甲基乙酰胺等。在某些实施方案中,使用二甲基乙酰胺作为溶剂。
在第二十七实施方案中,本发明提供了制备式(16)的化合物

或其盐的方法,所述方法包括使式(20)的化合物

与式(b)的单体化合物反应,

其中变量与上述相同。
在一个实施方案中,式(20)的化合物选自由以下组成的组:


且式(b)的单体化合物由下式表示:

其中X3为-Cl;X5为-Br或-I;R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个特定的实施方案中,式(20)的化合物与式(b)的单体化合物在碱存在下反应。合适的碱为碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。在一个优选的实施方案中,该碱为碳酸钾。
在另一个实施方案中,式(20)的化合物与式(b)的单体化合物在极性非质子溶剂存在下反应。在一个优选的实施方案中,极性非质子溶剂为二甲基乙酰胺。
在第二十八实施方案中,本发明提供了制备式(16)的化合物

或其盐的方法,所述方法包括使式(14)的化合物

与式(b)的单体化合物反应,

其中变量与上述相同。
在一个实施方案中,式(14)的化合物选自由以下组成的组:

且式(b)的单体化合物由下式表示:

其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个实施方案中,对于第二十八实施方案的方法,式(14)的化合物与式(b)的单体在醇活化剂存在下反应。在一个实施方案中,醇活化剂为三烷基膦、三芳基膦或三杂芳基膦。在一个具体实施方案中,醇活化剂为三甲基膦、三丁基膦、三(邻甲苯基)膦、三(间甲苯基)膦、三(对甲苯基)膦、三(2-吡啶基)膦、三(3-吡啶基)膦、三(4-吡啶基)膦或[4-(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-十七氟癸基)苯基]二苯基膦。在另一个实施方案中,醇活化剂可以是类似膦的试剂,诸如(三丁基亚正膦基)乙腈、(氰基亚甲基)三丁基正膦(CMBP)或(氰基亚甲基)三甲基正膦(CMMP)。在一个更具体的实施方案中,醇活化剂是三苯基膦。在一个实施方案中,醇活化剂可以是聚合物结合的或聚合物负载的,例如聚合物结合或聚合物负载的三烷基膦、三芳基膦(例如,三苯基膦)或三杂芳基膦。
在另一个实施方案中,对于第二十八实施方案的方法,式(14)的化合物与式(b)的单体在偶氮二羧酸酯存在下反应。在一个实施方案中,偶氮二羧酸酯选自由以下组成的组:偶氮二羧酸二乙酯(DEAD)、偶氮二羧酸二异丙酯(DIAD)、1,1'-(偶氮二羰基)二哌啶(ADDP)、偶氮二羧酸二叔丁酯(DTAD)、1,6-二甲基-1,5,7-六氢-1,4,6,7-四氮辛因-2,5-二酮(DHTD)、偶氮二羧酸二(4-氯苄基)酯(DCAD)、偶氮二羧酸二酰吗啉、N,N,N',N'-四甲基偶氮二甲酰胺(TMAD)、N,N,N',N'-四异丙基偶氮二甲酰胺(TIPA)、4,4'-偶氮吡啶、偶氮二羧酸双(2,2,2-三氯乙基)酯、o-(叔丁基二甲基甲硅烷基)-N-甲苯磺酰基羟基胺、偶氮二羧酸二-(4-氯苄基)酯、环状1,6-二甲基-1,5,7-六氢-1,4,6,7-四氮辛因-2,5-二酮(DHTD)、乙炔二羧酸二甲酯(DMAD)、偶氮二羧酸二-2-甲氧基乙酯、偶氮二羧酸二(4-氯苄基)酯和偶氮二羧酸双(4,4,5,5,6,6,7,7,8,8,9,9,9-十三氟壬基)酯。更具体地讲,偶氮二羧酸酯为DIAD。在一个实施方案中,偶氮二羧酸酯为聚合物结合的或聚合物负载的,例如聚合物负载的偶氮二羧酸烷酯(例如,聚合物结合的DEAD、DIAD、DTAD或ADDP)。
在另一个具体的实施方案中,对于第二十八实施方案的方法,式(14)的化合物与式(b)的单体在三苯基膦和偶氮二羧酸酯存在下反应。在一个实施方案中,该偶氮二羧酸酯选自由以下组成的组:偶氮二羧酸二乙酯(DEAD)、偶氮二羧酸二异丙酯(DIAD)、1,1’-(偶氮二羰基)二哌啶(ADDP)和偶氮二羧酸二叔丁酯(DTAD)。更具体地讲,偶氮二羧酸酯为DIAD。
在第二十九实施方案中,本发明提供了制备式(18)的化合物:

或其药学上可接受的盐的方法,所述方法包括使式(16)的化合物

与式(d)的还原的单体反应,

其中变量R1、R2、R3、R4、R6和P2如第二十三实施方案中所述;且其中
R1、R2、R3和R4各自独立地选自由以下组成的组:-H,具有1至10个碳原子的任选取代的直链、支链或环状烷基、烯基或炔基,聚乙二醇单元-(CH2CH2O)n-Rc,卤素,胍鎓[-NH(C=NH)NH2],-OR,-NR’R”,-NO2,-NCO,-NR’COR”,-SR,-SOR’,-SO2R’,-SO3 -H,-OSO3H,-SO2NR’R”,氰基,叠氮基,-COR’,-OCOR’和-OCONR’R”;
R6为-H、-R、-OR、-SR,-NR’R”、-NO2或卤素;且
P3为H或胺保护基团。
在一个具体的实施方案中,式(16)的化合物选自由以下组成的组:

且式(d)的还原的单体由下式表示:

其中X3为-Cl;P3为H或胺保护基团;R100和R101与上文定义相同。
在一个实施方案中,对于第二十九实施方案的方法,在式(16d)或(16A)的化合物与式(d1)的还原的单体之间的反应在碱存在下进行。在具体的实施方案中,该碱为碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。优选地,该碱为碳酸钾。
任何合适的有机溶剂都可以用于第二十实施方案的方法中。示例性溶剂包括但不限于二甲基甲酰胺(DMF)、CH2Cl2、二氯乙烷、THF、二甲基乙酰胺等。在某些实施方案中,使用二甲基甲酰胺或二甲基乙酰胺作为溶剂。
在第二十四实施方案的一个具体实施方案中,式(16)的化合物与式(d)的还原的单体反应,其中P3为H,以形成式(I’)的化合物:

在第二十四实施方案的另一个具体实施方案中,P3为胺保护基团。任何合适的胺保护基团都可以用于上述方法中。在一个实施方案中,胺保护基团为2-三甲基甲硅烷基乙基、(2-苯基-2-三甲基甲硅烷基)乙基、三异丙基甲硅烷氧基、2-(三甲基甲硅烷基)乙氧基甲基、烯丙氧基羰基、9-芴基甲氧基羰基、2-(三甲基甲硅烷基)乙氧基羰基或2,2,2,2-三氯乙氧基羰基。
当P3为胺保护基团时,使式(18)的化合物与胺脱保护试剂进一步反应以形成式(I’)的化合物:

合适的胺脱保护试剂的实例包括但不限于选自以下的胺脱保护试剂:四-正丁基氟化铵、乙酸、氟化氢吡啶、氟化铯、哌啶、吗啉或三氟乙酸。
在第三十实施方案中,本发明提供了制备式(17)的化合物

或其盐的方法,所述方法包括使式(15)的化合物

与式(d)的单体化合物反应,

其中X3为-Cl;X4为磺酸酯或活化的酯;P3为H或胺保护基团;L’、L”、L”和G如第二十二实施方案中所述,并且变量R1、R2、R3、R4、R6和P3与第二十四实施方案中所述相同。在一个实施方案中,X4为活化的酯。
在一个具体的实施方案中,式(15)的化合物选自由以下组成的组:

其中X3为-Cl;X4为磺酸酯或活化的酯;R100和R101与上文定义相同。在一个实施方案中,X4为活化的酯。
在一个实施方案中,对于第三十实施方案的方法,使用碱。在具体的实施方案中,该碱为碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。优选地,该碱为碳酸钾。
任何合适的有机溶剂都可以用于第三十实施方案的方法中。示例性溶剂包括但不限于DMF、CH2Cl2、二氯乙烷、THF、二甲基乙酰胺等。在某些实施方案中,使用二甲基乙酰胺作为溶剂。
在第三十实施方案的一个具体的实施方案中,P3为H且式(15)的化合物与式(d)的单体化合物反应以形成式(17’)的化合物。
在第三十实施方案的另一个具体的实施方案中,P3为胺保护基团,且所述方法还包括使式(17)的化合物与胺脱保护试剂反应以形成式(17’)的化合物的步骤:

合适的胺脱保护试剂的实例包括但不限于选自以下的胺脱保护试剂:四-正丁基氟化铵、乙酸、氟化氢吡啶、氟化铯、哌啶、吗啉或三氟乙酸。
在一个具体的实施方案中,式(17)的化合物选自由以下组成的组:

其中R100和R101与上文定义相同。
在第三十一实施方案中,本发明提供了制备式(17)的化合物:

或其盐的方法,所述方法包括使式(14)的化合物

与式(d)的单体化合物反应,

其中X3为-Cl;P3为H或胺保护基团;且剩下的变量如上述。
在一个具体的实施方案中,式(14)的化合物选自由以下组成的组:

且式(d)的单体化合物由式(d1)表示,其中X3为-Cl;R100为(C1-C3)烷氧基;R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个具体的实施方案中,式(14)的化合物与式(b)的单体在醇活化剂存在下反应。在一个实施方案中,醇活化剂为三烷基膦、三芳基膦或三杂芳基膦。在一个具体实施方案中,醇活化剂为三甲基膦、三丁基膦、三(邻甲苯基)膦、三(间甲苯基)膦、三(对甲苯基)膦、三(2-吡啶基)膦、三(3-吡啶基)膦、三(4-吡啶基)膦或[4-(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-十七氟癸基)苯基]二苯基膦。在另一个实施方案中,醇活化剂可以是类似膦的试剂,诸如(三丁基亚正膦基)乙腈、(氰基亚甲基)三丁基正膦(CMBP)或(氰基亚甲基)三甲基正膦(CMMP)。在一个更具体的实施方案中,醇活化剂是三苯基膦。在一个实施方案中,醇活化剂可以是聚合物结合的或聚合物负载的,例如聚合物结合或聚合物负载的三烷基膦、三芳基膦(例如,三苯基膦)或三杂芳基膦。
在另一个具体的实施方案中,式(14)的化合物与式(b)的单体在偶氮二羧酸酯存在下反应。在一个实施方案中,该偶氮二羧酸酯选自由以下组成的组:偶氮二羧酸二乙酯(DEAD)、偶氮二羧酸二异丙酯(DIAD)、1,1’-(偶氮二羰基)二哌啶(ADDP)、偶氮二羧酸二叔丁酯(DTAD)、1,6-二甲基-1,5,7-六氢-1,4,6,7-四氮辛因-2,5-二酮(DHTD)、偶氮二羧酸二(4-氯苄基)酯(DCAD)、偶氮二羧酸二酰吗啉、N,N,N',N'-四甲基偶氮二甲酰胺(TMAD)、N,N,N',N'-四异丙基偶氮二甲酰胺(TIPA)、4,4'-偶氮吡啶、偶氮二羧酸双(2,2,2-三氯乙基)酯、o-(叔丁基二甲基甲硅烷基)-N-甲苯磺酰基羟基胺、偶氮二羧酸二-(4-氯苄基)酯、环状1,6-二甲基-1,5,7-六氢-1,4,6,7-四氮辛因-2,5-二酮(DHTD)、乙炔二羧酸二甲酯(DMAD)、偶氮二羧酸二-2-甲氧基乙酯、偶氮二羧酸二(4-氯苄基)酯和偶氮二羧酸双(4,4,5,5,6,6,7,7,8,8,9,9,9-十三氟壬基)酯。更具体地讲,偶氮二羧酸酯为DIAD。在一个实施方案中,偶氮二羧酸酯为聚合物结合的或聚合物负载的,例如聚合物负载的偶氮二羧酸烷酯(例如,聚合物结合的DEAD、DIAD、DTAD或ADDP)。
在另一个具体的实施方案中,对于第二十八实施方案的方法,式(14)的化合物与式(d)的单体在三苯基膦和偶氮二羧酸酯存在下反应。在一个实施方案中,该偶氮二羧酸酯选自由以下组成的组:偶氮二羧酸二乙酯(DEAD)、偶氮二羧酸二异丙酯(DIAD)、1,1’-(偶氮二羰基)二哌啶(ADDP)和偶氮二羧酸二叔丁酯(DTAD)。更具体地讲,偶氮二羧酸酯为DIAD。
在另一个实施方案中,式(15)的化合物与式(d)的单体化合物反应,其中P3为H,以形成式(17’)的化合物:

在一个具体的实施方案中,P3为胺保护基团,且该方法还包括使式(17)的化合物与胺脱保护试剂反应以形成式(17’)的化合物的步骤:

在一个具体的实施方案中,式(17)的化合物选自由以下组成的组:


其中R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
合适的胺脱保护试剂的实例包括但不限于四-正丁基氟化铵、乙酸、氟化氢吡啶、氟化铯、哌啶、吗啉或三氟乙酸。
在第三十二实施方案中,本发明提供了制备式(17)的化合物:

或其盐的方法,所述方法包括使式(20)的化合物

与式(d)的单体化合物反应,

其中X3为-Cl;X5为-Br或-I,P3为H或胺保护基团;且剩下的变量与上述相同。
在一个具体的实施方案中,式(20)的化合物选自由以下组成的组:

其中X3为-Cl;X5为-Br或-I;R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个实施方案中,式(20)的化合物与式(d)的单体化合物在碱存在下反应。可以使用任何合适的碱。合适的碱包括但不限于碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。优选地,该碱为碳酸钾。
在另一个实施方案中,式(20)的化合物与式(d)的单体化合物在极性非质子溶剂存在下反应。优选地,该极性非质子溶剂为二甲基乙酰胺。
在一个具体的实施方案中,式(20)的化合物与式(d)的单体化合物反应,其中P3为H,以形成式(17’)的化合物:

在另一个具体的实施方案中,P3为胺保护基团,且该方法还包括使式(17)的化合物与胺脱保护试剂反应以形成式(17’)的化合物的步骤:

在一个实施方案中,式(17)的化合物选自由以下组成的组:

其中X3为-Cl;P3为H或胺保护基团;R100为(C1-C3)烷氧基;并且R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
合适的胺脱保护试剂包括但不限于四-正丁基氟化铵、乙酸、氟化氢吡啶、氟化铯、哌啶、吗啉或三氟乙酸。
在第三十三实施方案中,本发明提供了制备式(17’)的化合物:

或其盐的方法,所述方法包括使式(19)的化合物反应

其中X3为-Cl;剩下的变量与上述相同。
在一个具体的实施方案中,式(19)的化合物选自由以下组成的组:

其中X3为-Cl;R100和R101与上文定义相同。
在另一个具体的实施方案中,亚胺还原剂为氢化物还原剂。合适的氢化物还原剂的实例包括但不限于硼氢化钠、三乙酰氧基硼氢化钠、氰基硼氢化钠、氢化铝锂、氢气、甲酸铵、硼烷、9-硼杂双环[3.3.1]壬烷(9-BBN)、二异丁基氢化铝(DIBAL)、硼氢化锂(LiBH4)、硼氢化钾(KBH4)或双(2-甲氧基乙氧基)氢化铝钠(Red-Al)。在一个特定的实施方案中,氢化物还原剂为三乙酰氧基硼氢化钠(NaBH(OAc)3)。
在第三十四实施方案中,本发明提供了制备式(18)的化合物,

或其药学上可接受的盐的方法,所述方法包括使式(17)的化合物:

与式(b)的单体反应:

其中X3为-Cl;P3为H或胺保护基团;且剩下的变量与上述相同。
在一个实施方案中,式(17)的化合物与式(b)的单体化合物在碱存在下反应。合适的碱包括但不限于碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。优选地,该碱为碳酸钾。
在另一个实施方案中,式(17)的化合物与式(b)的单体化合物在极性非质子溶剂存在下反应。优选的极性非质子溶剂包括二甲基甲酰胺或二甲基乙酰胺。
在另一个实施方案中,式(17)的化合物与式(b)的还原的单体反应,其中P3为H,以形成式(I’)的化合物:

在一个具体的实施方案中,P3为胺保护基团。合适的胺保护基团包括但不限于2-三甲基甲硅烷基乙基、(2-苯基-2-三甲基甲硅烷基)乙基、三异丙基甲硅烷氧基、2-(三甲基甲硅烷基)乙氧基甲基、烯丙氧基羰基、9-芴基甲氧基羰基、2-(三甲基甲硅烷基)乙氧基羰基或2,2,2,2-三氯乙氧基羰基。
在另一个实施方案中,当P3为胺保护基团时,使式(18)的化合物与胺脱保护试剂进一步反应以形成式(I’)的化合物:

合适的胺脱保护试剂的实例包括但不限于选自以下的胺脱保护试剂:四-正丁基氟化铵、乙酸、氟化氢吡啶、氟化铯、哌啶、吗啉或三氟乙酸。
在第三十五实施方案中,本发明提供了。
制备式(18)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使磺化试剂或酯化试剂与式(14)的化合物反应,

以形成式(15)的化合物:

或其盐;
(2)使式(15)的化合物与式(b)的单体化合物反应,

以形成式(16)的化合物:

或其盐;
(3)使式(16)的化合物与式(d)的还原的单体反应:

以形成式(18)的化合物或其药学上可接受的盐,其中X3为-Cl;X4为磺酸酯或活化的酯;P1为醇保护基团;P3为H或胺保护基团;且剩下的变量与上述相同。在一个实施方案中,X4为磺酸酯。
在一个具体的实施方案中,第三十五实施方案的方法涉及制备式(Ib’)的化合物的方法,所述方法包括以下步骤:
(1)使磺化试剂或酯化试剂与式(14b)的化合物反应以形成式(15b)的化合物;
(2)使式(15b)的化合物与式(b1)的单体化合物反应以形成式(16b)的化合物;
(3)使式(16b)的化合物与式(d)的还原的单体反应,以形成式(18b)的化合物或其药学上可接受的盐,其中当P3为H时,式(16b)的化合物与还原的单体(d1)反应形成式(Ib’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18b)的化合物与胺脱保护试剂反应以形成式(Ib’)的化合物。
在另一个具体的实施方案中,第三十五实施方案的方法涉及制备式(Ic’)的化合物的方法,所述方法包括以下步骤:
(1)使磺化试剂或酯化试剂与式(14c)的化合物反应以形成式(15c)的化合物;
(2)使式(15c)的化合物与式(b1)的单体化合物反应以形成式(16c)的化合物;
(3)使式(16c)的化合物与式(d1)的还原的单体反应,以形成式(18c)的化合物或其药学上可接受的盐,其中当P3为H时,式(16c)的化合物与还原的单体(d1)反应形成式(Ib’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18c)的化合物与胺脱保护试剂反应以形成式(Ib’)的化合物。
在另一个具体的实施方案中,第三十五实施方案的方法涉及制备式(IA)的化合物的方法,所述方法包括以下步骤:
(1)使磺化试剂或酯化试剂与式(14b)的化合物反应以形成式(15A)的化合物;
(2)使式(15A)的化合物与式(b1)的单体化合物反应以形成式(16A)的化合物;
(3)使式(16A)的化合物与式(d1)的还原的单体反应,以形成式(18A)的化合物或其药学上可接受的盐,其中当P3为H时,式(16A)的化合物与还原的单体(d1)反应形成式(IA)的化合物;且当P3为胺保护基团时,该方法还包括使式(18A)的化合物与胺脱保护试剂反应以形成式(IA)的化合物。
用于第三十五实施方案的方法的条件和试剂如上文在第二十四、第二十六和/或第二十九实施方案以及其中描述的任何具体实施方案中所述。
在第三十六实施方案中,本发明提供了制备式(18)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使式(14)的化合物:

与式(b)的单体化合物反应,

以形成式(16)的化合物:

或其盐;和
(2)使式(16)的化合物与式(d)的还原的单体反应:

以形成式(18)的化合物或其药学上可接受的盐,其中X3为-Cl;P1为醇保护基团;P3为H或胺保护基团;且剩下的变量与上述相同。
在一个具体的实施方案中,第三十六实施方案的方法涉及制备式(Ib’)的化合物的方法,所述方法包括以下步骤:
(1)使式(14b)的化合物与式(b1)的单体化合物在醇活化剂存在下反应以形成式(16b)的化合物;
(2)使式(16b)的化合物与式(d1)的还原的单体反应,以形成式(18b)的化合物或其药学上可接受的盐,其中当P3为H时,式(16b)的化合物与还原的单体(d1)反应形成式(Ib’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18b)的化合物与胺脱保护试剂反应以形成式(Ib’)的化合物。
在另一个具体的实施方案中,第三十六实施方案的方法涉及制备式(Ic’)的化合物的方法,所述方法包括以下步骤:
(1)使式(14c)的化合物与式(b1)的单体化合物在醇活化剂存在下反应以形成式(16c)的化合物;
(2)使式(16c)的化合物与式(d1)的还原的单体反应,以形成式(18c)的化合物或其药学上可接受的盐,其中当P3为H时,式(16c)的化合物与还原的单体(d1)反应形成式(Ic’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18c)的化合物与胺脱保护试剂反应以形成式(Ic’)的化合物。
在另一个具体的实施方案中,第三十六实施方案的方法涉及制备式(IA)的化合物的方法,所述方法包括以下步骤:
(1)使式(14A)的化合物与式(b1)的单体化合物在醇活化剂存在下反应以形成式(16A)的化合物;
(2)使式(16A)的化合物与式(d1)的还原的单体反应,以形成式(18A)的化合物或其药学上可接受的盐,其中当P3为H时,式(16A)的化合物与还原的单体(d1)反应形成式(IA)的化合物;且当P3为胺保护基团时,该方法还包括使式(18A)的化合物与胺脱保护试剂反应以形成式(IA)的化合物。
用于第三十六实施方案的方法的条件和试剂如上文在第二十八和/或第二十九实施方案以及其中描述的任何具体实施方案中所述。
在第三十七实施方案中,本发明提供了制备式(18)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂与式(14)的化合物反应,

以形成式(20)的化合物:

或其盐;
(2)使式(20)的化合物或其盐与式(b)的单体化合物反应,

以形成式(16)的化合物:

或其盐;和
(3)使式(16)的化合物与式(d)的还原的单体反应:

以形成式(18)的化合物或其药学上可接受的盐,其中X3为-Cl;X5为-Br或-I;P3为H或胺保护基团;且剩下的变量与上述相同。
在一个具体的实施方案中,第三十七实施方案的方法涉及制备式(Ib’)的化合物的方法,所述方法包括以下步骤:
(1)使卤化试剂与式(14b)的化合物反应以形成式(20b)的化合物;
(2)使式(20b)的化合物或其盐与式(b1)的单体化合物反应以形成式(16b)的化合物;
(3)使式(16b)的化合物与式(d1)的还原的单体反应,以形成式(18b)的化合物或其药学上可接受的盐,其中当P3为H时,式(16b)的化合物与还原的单体(d1)反应形成式(Ib’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18b)的化合物与胺脱保护试剂反应以形成式(Ib’)的化合物。
在另一个具体的实施方案中,第三十七实施方案的方法涉及制备式(Ic’)的化合物的方法,所述方法包括以下步骤:
(1)使卤化试剂与式(14c)的化合物反应以形成式(20c)的化合物;
(2)使式(20c)的化合物或其盐与式(b1)的单体化合物反应以形成式(16c)的化合物;
(3)使式(16c)的化合物与式(d1)的还原的单体反应,以形成式(18c)的化合物或其药学上可接受的盐,其中当P3为H时,式(16c)的化合物与还原的单体(d1)反应形成式(Ib’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18b)的化合物与胺脱保护试剂反应以形成式(Ic’)的化合物。
在另一个具体的实施方案中,第三十七实施方案的方法涉及制备式(IA)的化合物的方法,所述方法包括以下步骤:
(1)使卤化试剂与式(14A)的化合物反应以形成式(20A)的化合物;
(2)使式(20A)的化合物或其盐与式(b1)的单体化合物反应以形成式(16A)的化合物;
(3)使式(16A)的化合物与式(d1)的还原的单体反应,以形成式(18A)的化合物或其药学上可接受的盐,其中当P3为H时,式(16A)的化合物与还原的单体(d1)反应形成式(IA)的化合物;且当P3为胺保护基团时,该方法还包括使式(18A)的化合物与胺脱保护试剂反应以形成式(IA)的化合物。
用于第三十七实施方案的方法的条件和试剂如上文在第二十五、第二十七和/或第二十九实施方案以及其中描述的任何具体实施方案中所述。
在第三十五、第三十六或第三十七实施方案的一个具体实施方案中,式(16)的化合物与式(d)的还原的单体反应,其中P3为H,以形成式(I’)的化合物:

在第三十五、第三十六或第三十七实施方案的另一个具体的实施方案中,P3为胺保护基团且使式(18)的化合物与胺脱保护试剂进一步反应以形成式(I’)的化合物:

在第三十八实施方案中,本发明提供了制备式(18)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使磺化试剂或酯化试剂与式(14)的化合物反应,

以形成式(15)的化合物:

或其盐;
(2)使式(15)的化合物与式(d)的还原的单体化合物反应,

以形成式(17)的化合物:

或其盐;和
(3)使式(17)的化合物与式(b)的单体反应,

以形成式(18)的化合物或其药学上可接受的盐,其中X3为-Cl;X4为磺酸酯或活化的酯;P1为醇保护基团;P3为H或胺保护基团;剩下的变量与上述相同。在一个实施方案中,X4为磺酸酯。
在一个具体的实施方案中,第三十八实施方案的方法涉及制备式(Ib’)的化合物的方法,所述方法包括以下步骤:
(1)使磺化试剂或酯化试剂与式(14b)的化合物反应以形成式(15b)的化合物;
(2)使式(15b)的化合物或其盐与式(d1)的还原的单体化合物反应以形成式(17b)的化合物;
(3)使式(17b)的化合物与式(b1)的单体反应,以形成式(18b)的化合物或其药学上可接受的盐,其中当P3为H时,式(17b)的化合物与还原的单体(d1)反应形成式(Ib’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18b)的化合物与胺脱保护试剂反应以形成式(Ib’)的化合物。
在另一个具体的实施方案中,第三十八实施方案的方法涉及制备式(Ic’)的化合物的方法,所述方法包括以下步骤:
(1)使磺化试剂或酯化试剂与式(14b)的化合物反应以形成式(15c)的化合物;
(2)使式(15c)的化合物或其盐与式(d1)的还原的单体化合物反应以形成式(17c)的化合物;
(3)使式(17c)的化合物与式(b1)的单体反应,以形成式(18c)的化合物或其药学上可接受的盐,其中当P3为H时,式(17c)的化合物与还原的单体(d1)反应形成式(Ic’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18c)的化合物与胺脱保护试剂反应以形成式(Ic’)的化合物。
在另一个具体的实施方案中,第三十八实施方案的方法涉及制备式(IA)的化合物的方法,所述方法包括以下步骤:
(1)使磺化试剂或酯化试剂与式(14A)的化合物反应以形成式(15A)的化合物;
(2)使式(15A)的化合物或其盐与式(d1)的还原的单体化合物反应以形成式(17A)的化合物;
(3)使式(17A)的化合物与式(b1)的单体反应,以形成式(18A)的化合物或其药学上可接受的盐,其中当P3为H时,式(17A)的化合物与还原的单体(d1)反应形成式(IA)的化合物;且当P3为胺保护基团时,该方法还包括使式(18A)的化合物与胺脱保护试剂反应以形成式(IA)的化合物。
用于第三十八实施方案的方法的条件和试剂如上文在第二十五、第三十和/或第三十四实施方案以及其中描述的任何具体实施方案中所述。
在第三十九实施方案中,本发明提供了制备式(18)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使式(14)的化合物:

与式(d)的还原的单体化合物反应,

以形成式(17)的化合物:

或其盐;和
(2)使式(17)的化合物与式(b)的单体反应,

以形成式(18)的化合物或其药学上可接受的盐,其中X3为-Cl;P1为醇保护基团;P3为H或胺保护基团;且剩下的变量与上述相同。
在一个具体的实施方案中,第三十九实施方案的方法涉及制备式(Ib’)的化合物的方法,所述方法包括以下步骤:
(1)使式(14b)的化合物与式(d1)的还原的单体化合物反应以形成式(17b)的化合物;
(2)使式(17b)的化合物与式(b1)的单体反应,以形成式(18b)的化合物或其药学上可接受的盐,其中当P3为H时,式(17b)的化合物与还原的单体(d1)反应形成式(Ib’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18b)的化合物与胺脱保护试剂反应以形成式(Ib’)的化合物。
在另一个具体的实施方案中,第三十九实施方案的方法涉及制备式(Ic’)的化合物的方法,所述方法包括以下步骤:
(1)使式(14c)的化合物与式(d1)的还原的单体化合物反应以形成式(17c)的化合物;
(2)使式(17c)的化合物与式(b1)的单体反应,以形成式(18c)的化合物或其药学上可接受的盐,其中当P3为H时,式(17c)的化合物与还原的单体(d1)反应形成式(Ic’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18c)的化合物与胺脱保护试剂反应以形成式(Ic’)的化合物。
在另一个具体的实施方案中,第三十九实施方案的方法涉及制备式(IA)的化合物的方法,所述方法包括以下步骤:
(1)使式(14A)的化合物与式(d1)的还原的单体化合物反应以形成式(17A)的化合物;
(2)使式(17A)的化合物与式(b1)的单体反应,以形成式(18A)的化合物或其药学上可接受的盐,其中当P3为H时,式(17A)的化合物与还原的单体(d1)反应形成式(IA’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18A)的化合物与胺脱保护试剂反应以形成式(IA)的化合物。
用于第三十九实施方案的方法的条件和试剂如上文在第三十一和/或第三十四实施方案以及其中描述的任何具体实施方案中所述。
在第四十实施方案中,本发明提供了制备式(18)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使卤化试剂与式(14)的化合物反应:

以形成式(20)的化合物:

或其盐;
(2)使式(20)的化合物与式(d)的还原的单体化合物反应,

以形成式(17)的化合物:

或其盐;和
(3)使式(17)的化合物与式(b)的单体反应,

以形成式(18)的化合物或其药学上可接受的盐,其中X3为-Cl;X5为-Br或-I;P1为醇保护基团;P3为H或胺保护基团;且剩下的变量与上述相同。
在一个具体的实施方案中,第四十实施方案的方法涉及制备式(Ib’)的化合物的方法,所述方法包括以下步骤:
(1)使溴化或碘化试剂与式(14b)的化合物反应以形成式(20b)的化合物;
(2)使式(20b)的化合物与式(d1)的还原的单体化合物反应以形成式(17b)的化合物;
(3)使式(17b)的化合物与式(b1)的单体反应,以形成式(18b)的化合物或其药学上可接受的盐,其中当P3为H时,式(17b)的化合物与还原的单体(d1)反应形成式(Ib’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18b)的化合物与胺脱保护试剂反应以形成式(Ib’)的化合物。
在另一个具体的实施方案中,第四十实施方案的方法涉及制备式(Ic’)的化合物的方法,所述方法包括以下步骤:
(1)使溴化或碘化试剂与式(14c)的化合物反应以形成式(20c)的化合物;
(2)使式(20c)的化合物与式(d1)的还原的单体化合物反应以形成式(17c)的化合物;
(3)使式(17c)的化合物与式(b1)的单体反应,以形成式(18c)的化合物或其药学上可接受的盐,其中当P3为H时,式(17c)的化合物与还原的单体(d1)反应形成式(Ic’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18c)的化合物与胺脱保护试剂反应以形成式(Ic’)的化合物。
在另一个具体的实施方案中,第四十实施方案的方法涉及制备式(IA)的化合物的方法,所述方法包括以下步骤:
(1)使溴化或碘化试剂与式(14A)的化合物反应以形成式(20A)的化合物;
(2)使式(20A)的化合物与式(d1)的还原的单体化合物反应以形成式(17A)的化合物;
(3)使式(17A)的化合物与式(b1)的单体反应,以形成式(18A)的化合物或其药学上可接受的盐,其中当P3为H时,式(17A)的化合物与还原的单体(d1)反应形成式(Ib’)的化合物;且当P3为胺保护基团时,该方法还包括使式(18A)的化合物与胺脱保护试剂反应以形成式(Ib’)的化合物。
用于第四十实施方案的方法的条件和试剂如上文在第二十五、第三十二和/或第三十四实施方案以及其中描述的任何具体实施方案中所述。
在第三十八、第三十九或第四十实施方案的一个具体的实施方案中,式(17)的化合物与式(d)的还原的单体反应,其中P3为H,以形成式(I’)的化合物:

在第三十六、第三十七或第三十八实施方案的另一个具体的实施方案中,P3为胺保护基团。当P3为胺保护基团时,使式(18)的化合物与胺脱保护试剂进一步反应以形成式(I’)的化合物:

在第四十一实施方案中,本发明提供了制备式(I’)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使磺化试剂或酯化试剂与式(14)的化合物反应,

以形成式(15)的化合物:

或其盐;
(2)使式(15)的化合物与式(a)的单体化合物反应,

以形成式(19)的化合物:

或其盐;
(3)使式(19)的化合物与亚胺还原剂反应以形成式(17’)的化合物:

或其盐;和
(6)使式(17’)的化合物与式(B)的单体反应,

以形成式(I’)的化合物或其药学上可接受的盐,其中X3为-Cl;X4为磺酸酯或活化的酯;P1为醇保护基团;P2为胺保护基团;且剩下的变量与上述相同。在一个实施方案中,X4为磺酸酯。
在一个实施方案中,第四十一实施方案的方法涉及制备式(Ib’)的化合物或其药学上可接受的盐,所述方法包括以下步骤:
(1)使磺化试剂与式(14b)的化合物反应,
以形成式(15b)的化合物或其盐;
(2)使式(15b)的化合物与式(a1)的单体化合物反应以形成式(16b)的化合物或其盐;
(3)使式(16b)的化合物与亚胺还原剂反应以形成式(17b’)的化合物或其盐;和
(6)使式(17b’)的化合物与式(a1)的单体反应以形成式(Ib’)的化合物或其药学上可接受的盐。
在另一个实施方案中,第四十一实施方案的方法涉及制备式(Ic’)的化合物或其药学上可接受的盐,所述方法包括以下步骤:
(1)使磺化试剂与式(14c)的化合物反应,
以形成式(15c)的化合物或其盐;
(2)使式(15c)的化合物与式(a1)的单体化合物反应以形成式(16c)的化合物或其盐;
(3)使式(16c)的化合物与亚胺还原剂反应以形成式(17c’)的化合物或其盐;和
(6)使式(17c’)的化合物与式(a1)的单体反应以形成式(Ic’)的化合物或其药学上可接受的盐。
在另一个实施方案中,第四十一实施方案的方法涉及制备式(IA’)的化合物或其药学上可接受的盐,所述方法包括以下步骤:
(1)使磺化试剂与式(14A)的化合物反应,
以形成式(15A)的化合物或其盐;
(2)使式(15A)的化合物与式(a1)的单体化合物反应以形成式(16A)的化合物或其盐;
(3)使式(16A)的化合物与亚胺还原剂反应以形成式(17A’)的化合物或其盐;和
(6)使式(17A’)的化合物与式(a1)的单体反应以形成式(IA’)的化合物或其药学上可接受的盐。
在第四十二实施方案中,本发明提供了制备式(I’)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使式(14)的化合物:

与式(b)的单体化合物反应,

以形成式(19)的化合物:

或其盐;
(2)使式(19)的化合物与亚胺还原剂反应以形成式(17’)的化合物:

或其盐;和
(3)使式(17’)的化合物与式(a)的单体反应:

以形成式(I’)的化合物;其中X3为-Cl;P1为醇保护基团;变量与上述相同。
在一个实施方案中,第四十二实施方案的方法涉及制备式(Ib’)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使式(14b)的化合物与式(a1)的单体化合物反应以形成式(19b)的化合物或其盐;
(2)使式(19b)的化合物与亚胺还原剂反应以形成式(17b’)的化合物或其盐;和
(3)使式(17b’)的化合物与式(a1)的单体反应以形成式(Ib’)的化合物。
在另一个实施方案中,第四十二实施方案的方法涉及制备式(Ic’)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使式(14c)的化合物与式(a1)的单体化合物反应以形成式(19c)的化合物或其盐;
(2)使式(19c)的化合物与亚胺还原剂反应以形成式(17c’)的化合物或其盐;和
(3)使式(17c’)的化合物与式(a1)的单体反应以形成式(Ic’)的化合物。
在另一个实施方案中,第四十二实施方案的方法涉及制备式(IA)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使式(14A)的化合物与式(a1)的单体化合物反应以形成式(19A)的化合物或其盐;
(2)使式(19A)的化合物与亚胺还原剂反应以形成式(17A’)的化合物或其盐;和
(3)使式(17A’)的化合物与式(a1)的单体反应以形成式(IA’)的化合物。
用于第四十二实施方案的方法的条件和试剂如上文在第二十八、第三十三和/或第三十四实施方案以及其中描述的任何具体实施方案中所述。
在第四十三实施方案中,本发明提供了制备式(I’)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使溴化或碘化试剂与式(14)的化合物:

或其盐反应,以形成式(20)的化合物:

或其盐;
(2)使式(20)的化合物或其盐与式(a)的单体化合物反应:

以形成式(19)的化合物:

(3)使式(19)的化合物与亚胺还原剂反应以形成式(17’)的化合物:

或其盐;和
(4)使(17’)的化合物与式(b)的单体反应,

以形成式(I’)的化合物;其中X3为-Cl;X5为-Br或-I;且剩下的变量与上述相同。
在一个实施方案中,第四十三实施方案的方法涉及制备式(Ib’)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使溴化或碘化试剂与式(14b)的化合物或其盐反应以形成式(20b)的化合物或其盐;
(2)使式(20b)的化合物或其盐与式(a1)的单体化合物反应以形成式(19b)的化合物;
(3)使式(19b)的化合物与亚胺还原剂反应以形成式(17b’)的化合物或其盐;和
(4)使(17b’)的化合物与式(a1)的单体反应以形成式(Ib’)的化合物。
在另一个实施方案中,第四十三实施方案的方法涉及制备式(Ic’)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使溴化或碘化试剂与式(14c)的化合物或其盐反应以形成式(20c)的化合物或其盐;
(2)使式(20c)的化合物或其盐与式(a1)的单体化合物反应以形成式(19c)的化合物;
(3)使式(19c)的化合物与亚胺还原剂反应以形成式(17c’)的化合物或其盐;和
(4)使(17c’)的化合物与式(a1)的单体反应以形成式(Ic’)的化合物。
在另一个实施方案中,第四十三实施方案的方法涉及制备式(IA)的化合物,

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使溴化或碘化试剂与式(14A)的化合物或其盐反应以形成式(20A)的化合物或其盐;
(2)使式(20A)的化合物或其盐与式(a1)的单体化合物反应以形成式(19A)的化合物;
(3)使式(19A)的化合物与亚胺还原剂反应以形成式(17A’)的化合物或其盐;和
(4)使式(17A’)的化合物与式(a1)的单体反应以形成式(IA’)的化合物。
用于第四十三实施方案的方法的条件和试剂如上文在第二十五、第二十七、第三十三和/或第三十四实施方案以及其中描述的任何具体实施方案中所述。
在第三十五至第四十三实施方案中的任一个的一个实施方案中,式(14)的化合物或其盐通过包括以下步骤的方法制备:
(1)使氯化试剂与式(2)的化合物反应:

以形成式(13)的化合物:

或其盐;和
(2)使式(13)的化合物与醇脱保护试剂反应以形成式(14)的化合物或其盐,其中X3为-Cl;且P1为醇保护基团。
在一个具体的实施方案中,式(14b)的化合物或其盐通过包括以下步骤的方法制备:(1)使氯化试剂与式(2b)的化合物反应以形成式(13b)的化合物或其盐;和(2)使式(13b)的化合物与醇脱保护试剂反应以形成式(14b)的化合物或其盐。
在另一个具体的实施方案中,式(14c)的化合物或其盐通过包括以下步骤的方法制备:(1)使氯化试剂与式(2c)的化合物反应以形成式(13c)的化合物或其盐;和(2)使式(13c)的化合物与醇脱保护试剂反应以形成式(14c)的化合物或其盐。
在另一个具体的实施方案中,式(14A)的化合物或其盐通过包括以下步骤的方法制备:(1)使氯化试剂与式(2A)的化合物反应以形成式(13A)的化合物或其盐;和(2)使式(13A)的化合物与醇脱保护试剂反应以形成式(14A)的化合物或其盐。
上文用于制备式(14)、(14b)、(14c)或(14A)的化合物的方法的条件和试剂如上文在第二十二和/或第二十三实施方案以及其中描述的任何具体实施方案中所述。
在另一个实施方案中,式(2)的化合物通过使式(1)的化合物与醇保护试剂反应来制备

在一个具体的实施方案中,式(2b)的化合物通过使式(1b)的化合物与醇保护试剂反应来制备。
在另一个具体的实施方案中,式(2c)的化合物通过使式(1c)的化合物与醇保护试剂反应来制备。
在另一个具体的实施方案中,式(2A)的化合物通过使式(1A)的化合物与醇保护试剂反应来制备。
上文用于制备式(2)、(2b)、(2c)或(2A)的化合物的方法的条件和试剂如上文在第一实施方案以及其中描述的任何具体实施方案中所述。
在第四十四实施方案中,本发明提供了制备式(IB)的化合物,

或其药学上可接受的盐的方法,所述方法包括使式(IA)的化合物与还原剂反应的步骤。可以使用任何合适的还原剂。在一个实施方案中,还原剂选自由以下组成的组:氢气、连二亚硫酸钠、硫化钠、氯化锡、氯化钛(II)、碘化锌、碘化铁和碘化钐。在一个具体的实施方案中,还原剂为Fe/NH4Cl或Zn/NH4Cl。
在一个实施方案中,式(Ic’)的化合物可以通过使式(IB)的化合物与式(Lc’)的化合物反应来制备:

其中E为-OH、卤化物或-C(=O)E为活化的酯;且R100为(C1-C3)烷氧基。
在一个具体的实施方案中,E为-OH且式(IB)的化合物与式(Lc’)的化合物的反应在活化剂存在下进行。
在一个实施方案中,活化剂为碳二亚胺、脲鎓、活性酯、鏻、2-烷基-1-烷基羰基-1,2-二氢喹啉、2-烷氧基-1-烷氧基羰基-1,2-二氢喹啉或烷基氯甲酸酯。在一个具体的实施方案中,活化剂为碳二亚胺。在一个更具体的实施方案中,活化剂为二环己基碳二亚胺(DCC)、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(EDC)或二异丙基碳二亚胺(DIC)。在另一个具体的实施方案中,活化剂为N-乙氧羰基-2-乙氧基-1,2-二氢喹啉。
在一个实施方案中,对于上述方法,R100为甲氧基并且R101为甲基。
本发明的方法还可以是上述方法(例如,在第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二、第十三、第十四、第十五、第十六、第十七、第十八、第十九、第二十、第二十一、第二十二、第二十三、第二十四、第二十五、第二十六、第二十七、第二十八、第二十九、第三十、第三十一、第三十二、第三十三、第三十四、第三十五、第三十六、第三十七、第三十八、第三十九、第四十、第四十一、第四十二或第四十三实施方案中的方法)的任何组合。例如,第一和第二实施方案的方法的组合,第一、第二和第三实施方案的方法的组合,第四和第五实施方案的方法的组合,第四、第五和第六实施方案的方法的组合,第六和第八实施方案的方法的组合,第十三和第十四实施方案的方法的组合,第十三、第十四和第十五实施方案的方法的组合,以及第十七和第十八实施方案的方法的组合也包括在本发明中。在下文的任何具体实施方案中描述的变量定义也适用于上述方法的任何组合。
在本发明的方法中本文描述的反应可以在一种或多种任何合适的溶剂中进行。在一个实施方案中,该溶剂为有机溶剂。示例性有机溶剂包括但不限于二氯甲烷、二氯乙烷、DMF、DMA、丙酮、乙腈、THF、DMSO、乙酸乙酯等或者它们的组合。
在本发明的方法中本文描述的反应可以在任何合适的温度下进行。在一个实施方案中,反应可以在室温下进行。在另一个实施方案中,反应可以在诸如0℃的低温下进行。在另一个实施方案中,反应可以在诸如约40℃、约50℃等高温下进行。
在第一具体实施方案中,对于第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二、第十三、第十四、第十五、第十六、第十七、第十八、第十九实施方案、第二十、第二十一、第二十二、第二十三、第二十四、第二十五、第二十六、第二十七、第二十八、第二十九、第三十、第三十一、第三十二、第三十三、第三十四、第三十五、第三十六、第三十七、第三十八、第三十九、第四十、第四十一、第四十二或第四十三实施方案中的方法,L’、L”和L”’中的一者由下式表示:
-Z1-P-Z2-Rx-J (A)
且另外两者相同或不同,且独立地选自-H,具有1至10个碳原子的任选取代的直链、支链或环状烷基、烯基或炔基,聚乙二醇单元-(CH2CH2O)n’-Rc,卤素,胍鎓[-NH(C=NH)NH2],-OR,-NR’R”,-NO2,-NR’COR”,-SR,-SOR’,-SO2R’,-SO3-H,-OSO3H,-SO2NR’R”,氰基,叠氮基,-COR’,-OCOR’和-OCONR’R”;
其中:
Z1和Z2中的一者为-C(=O)-,且另一者为-NR5-;
P为氨基酸残基或含有2至20个氨基酸残基的肽;
J为包含能够将细胞毒性化合物共价连接到细胞结合剂的反应性基团的部分;
Rx为具有1至10个碳原子的任选取代的直链、支链或环状烷基、烯基或炔基;且
R5为-H或具有1至10个碳原子的任选取代的直链或支链烷基。
在一个更具体的实施方案中,L’、L”和L’”中的一者由式(A)表示,且其余各自独立地为-H、具有1至6个碳原子的直链或支链烷基、卤素、-OH、(C1-C6)烷氧基或-NO2。
在另一个更具体的实施方案中,L’、L”和L’”中的一者由式(A)表示,且其余为-H。
在又一个更具体的实施方案中,L’由式(A)表示;且L”和L’”两者均为-H。
在另一个更具体的实施方案中,对于式(A),Rx为任选被卤素、-OH、(C1-C3)烷基、(C1-C3)烷氧基、卤代(C1-C3)烷基或者带电荷的取代基或可电离的基团Q取代的具有1至6个碳原子的直链、支链或环状烷基。
在更具体的实施方案中,J为NHRc1、-COORc1或-COE,其中-COE表示反应性酯,且Rc1为-H或任选被卤素、-OH或(C1-C3)烷氧基取代的具有1至4个碳原子的直链或支链烷基。甚至更具体地讲,J为-COORc1且Rc1为(C1-C3)烷基。
在第二具体实施方案中,对于第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二、第十三、第十四、第十五、第十六、第十七、第十八、第十九、第二十、第二十一、第二十二、第二十三、第二十四、第二十五、第二十六、第二十七、第二十八、第二十九、第三十、第三十一、第三十二、第三十三、第三十四、第三十五、第三十六、第三十七、第三十八、第三十九、第四十、第四十一、第四十二或第四十三实施方案或第一具体实施方案中的方法,L’由下式表示:
-NR5-P-C(=O)-(CRaRb)m-J (B1);
-NR5-P-C(=O)-Cy-(CRaRb)m’-J (B2);
-C(=O)-P-NR5-(CRaRb)m-J (C1);或
-C(=O)-P-NR5-Cy-(CRaRb)m’-J (C2)
其中:
J为-COORc1;
Rc1为-H或任选被卤素、-OH或(C1-C3)烷氧基取代的具有1至4个碳原子的直链或支链烷基;
Ra和Rb在每次出现时各自独立地为-H、(C1-C3)烷基或带电荷的取代基或可电离的基团Q;
m为1至6的整数;
m’为0或1至6的整数;且
Cy为任选被卤素、-OH、(C1-C3)烷基、(C1-C3)烷氧基或卤代(C1-C3)烷基取代的具有5或6个环碳原子的环状烷基。
在一个更具体的实施方案中,Ra和Rb均为H;Cy为环己烷;且R5为H或Me。
在另一个更具体的实施方案中,m’为0或1。
在第三具体实施方案中,对于第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二、第十三、第十四、第十五、第十六、第十七、第十八、第十九、第二十、第二十一、第二十二、第二十三、第二十四、第二十五、第二十六、第二十七、第二十八、第二十九、第三十、第三十一、第三十二、第三十三、第三十四、第三十五、第三十六、第三十七、第三十八、第三十九、第四十、第四十一、第四十二或第四十三实施方案或第一具体实施方案中的方法,L’由下式表示:
-NR5-P-C(=O)-(CRaRb)m-S-Zs (B3);或
-C(=O)-P-NR5-(CRaRb)m-S-Zs (C3),
其中:
Ra和Rb在每次出现时各自独立地为-H、(C1-C3)烷基或带电荷的取代基或可电离的基团Q;
m为1至6的整数;
Zs为-H、-SRd、-C(=O)Rd1或选自下式中的任一个:

其中:
q为1至5的整数;
n’为2至6的整数;
U为-H或SO3M;
M为H+、Na+或K+;
Rd为具有1至6个碳原子的直链或支链烷基或选自苯基、硝基苯基(例如,2或4-硝基苯基)、二硝基苯基(例如,2,4-二硝基苯基)、羧基硝基苯基(例如,3-羧基-4-硝基苯基)、吡啶基或硝基吡啶基(例如,4-硝基吡啶基);且
Rd1为具有1至6个碳原子的直链或支链烷基
在一个更具体的实施方案中,Zs为-SRd且Rd为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在一个更具体的实施方案中,带电荷的取代基或可电离的基团Q为i)-SO3H、-Z’-SO3H、-OPO3H2、-Z’-OPO3H2、-PO3H2、-Z’-PO3H2、-CO2H、-Z’-CO2H、-NR11R12或-Z’-NR11R12,或其药学上可接受的盐;或ii)-N+R14R15R16XA-或-Z’-N+R14R15R16XA-;Z’为任选取代的亚烷基、任选取代的亚环烷基或任选取代的亚苯基;R11、R12、R14至R16各自独立地为H或任选取代的烷基;且XA-为药学上可接受的阴离子。甚至更具体地讲,Q为SO3H或其药学上可接受的盐。
在另一个更具体的实施方案中,Ra和Rb两者均为-H,且R5为H或Me。
在另一个更具体的实施方案中,-(CRaRb)m-为-(CH2)m”-C(Me2)-,且m”为1至5的整数。
在第四具体实施方案中,对于式(B1)、(B2)、(B3)、(C1)、(C2)或(C3),P为含有2至10个氨基酸残疾的肽;且剩下的变量如上文在第二或第三具体实施方案或本文所述的任何更具体的实施方案中所述。
在一个更具体的实施方案中,P为含有2至5个氨基酸残基的肽。
在另一个更具体的实施方案中,P为Gly-Gly-Gly、Ala-Val、Val-Ala、Val-Cit、Val-Lys、Phe-Lys、Lys-Lys、Ala-Lys、Phe-Cit、Leu-Cit、Ile-Cit、Trp、Cit、Phe-Ala、Phe-N9-甲苯磺酰基-Arg、Phe-N9-硝基-Arg、Phe-Phe-Lys、D-Phe-Phe-Lys、Gly-Phe-Lys、Leu-Ala-Leu、Ile-Ala-Leu、Val-Ala-Val、Ala-Leu-Ala-Leu、β-Ala-Leu-Ala-Leu和Gly-Phe-Leu-Gly、Val-Arg、Arg-Val、Arg-Arg、Val-D-Cit、Val-D-Lys、Val-D-Arg、D-Val-Cit、D-Val-Lys、D-Val-Arg、D-Val-D-Cit、D-Val-D-Lys、D-Val-D-Arg、D-Arg-D-Arg、Ala-Ala、Ala-D-Ala、D-Ala-Ala、D-Ala-D-Ala、Ala-Met或Met-Ala。甚至更具体地讲,P为Gly-Gly-Gly、Ala-Val、Ala-Ala、Ala-D-Ala、D-Ala-Ala或D-Ala-D-Ala。
在第五具体实施方案中,对于第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二、第十三、第十四、第十五、第十六、第十七、第十八、第十九、第二十、第二十一、第二十二、第二十三、第二十四、第二十五、第二十六、第二十七、第二十八、第二十九、第三十、第三十一、第三十二、第三十三、第三十四、第三十五、第三十六、第三十七、第三十八、第三十九、第四十、第四十一、第四十二或第四十三实施方案中的方法,L’由下式表示:


其中:
R100为(C1-C3)烷氧基;
Zs为-SR101;且
R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在第六具体实施方案中,对于第八、第九、第十、第十五、第十六或第十九实施方案中的方法,式(I’)的化合物由下式表示:




或其药学上可接受的盐,其中:
R100为(C1-C3)烷氧基;
Zs为-SR101;且
R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在第七具体实施方案中,对于在第五或第六具体实施方案中描述的方法,R100为-OMe且R101为Me或吡啶基。
在第八具体实施方案中,对于第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二、第十三、第十四、第十五、第十六、第十七、第十八、第十九、第二十、第二十一、第二十二、第二十三、第二十四、第二十五、第二十六、第二十七、第二十八、第二十九实施方案、第三十、第三十一、第三十二、第三十三、第三十四、第三十五、第三十六、第三十七、第三十八、第三十九、第四十、第四十一、第四十二和第四十三中的方法,L’、L”和L”’中的一者由下式表示:
-N(Re)-C(=O)-Rx-S-Zs (B);
其中:
Rx为任选被带电荷的取代基或可电离的基团Q取代的具有1至6个碳原子的直链或支链亚烷基;
Q为i)-SO3H、-Z’-SO3H、-OPO3H2、-Z’-OPO3H2、-PO3H2、-Z’-PO3H2、-CO2H、-Z’-CO2H、-NR11R12或-Z’-NR11R12,或其药学上可接受的盐;或ii)-N+R14R15R16XA-或-Z’-N+R14R15R16XA-;Z’为任选取代的亚烷基、任选取代的亚环烷基或任选取代的亚苯基;R11、R12、R14、R15和R16各自独立地为H或任选取代的烷基;且XA-为药学上可接受的阴离子;
Re为-H或具有1至6个碳原子的直链或支链烷基;
Zs为-H、-SRd、-C(=O)Rd1或选自下式中的任一个:


其中:
q为1至5的整数;
Rd为具有1至6个碳原子的直链或支链烷基或者选自苯基、硝基苯基、二硝基苯基、羧基硝基苯基、吡啶基和硝基吡啶基;
Rd1为具有1至6个碳原子的直链或支链烷基;
n”为2至6的整数;
U为-H或-SO3M;且
M为-H或阳离子。
在一个更具体的实施方案中,L’、L”和L’”中的一者由式(B)表示,且其余各自独立地为-H、具有1至6个碳原子的直链或支链烷基、卤素、-OH、(C1-C6)烷氧基或-NO2。
在另一个更具体的实施方案中,L’、L”和L’”中的一者由式(B)表示,且其余为-H。
在另一个更具体的实施方案中,L’由式(B)表示;且L”和L’”两者均为-H。
在另一个更具体的实施方案中,对于式(B),Zs为-SRd且Rd为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在第九具体实施方案中,对于式(B),Rx为-(CH2)p-(CRfRg)-,其中Rf和Rg各自独立地选自-H或具有1至4个碳原子的直链或支链烷基;且p为0、1、2或3;且剩下的变量如在第八具体实施方案或其中描述的任何更具体的实施方案中描述。
在一个更具体的实施方案中,Rf和Rg相同或不同,并且选自-H和-Me。
在另一个更具体的实施方案中,Rf和Rg均为-Me;且p为2。
在第十具体实施方案中,对于式(B),Rx为被带电荷的取代基或可电离的基团Q取代的具有1至4个碳原子的直链或支链亚烷基;且剩下的变量如在第八具体实施方案或其中描述的任何更具体的实施方案中描述。
在一个更具体的实施方案中,带电荷的取代基或可电离的基团Q为i)-SO3H、-Z’-SO3H、-OPO3H2、-Z’-OPO3H2、-PO3H2、-Z’-PO3H2、-CO2H、-Z’-CO2H、-NR11R12或-Z’-NR11R12,或其药学上可接受的盐;或ii)-N+R14R15R16XA-或-Z’-N+R14R15R16XA-;Z’为任选取代的亚烷基、任选取代的亚环烷基或任选取代的亚苯基;R11、R12、R14至R16各自独立地为H或任选取代的烷基;且XA-为药学上可接受的阴离子。更具体地讲,Q为-SO3H或其药学上可接受的盐。
在第十一具体实施方案中,Re为-H或-Me;且剩下的变量如在第九或第十具体实施方案或其中描述的任何更具体的实施方案中描述。
在第十二具体实施方案中,对于第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二、第十三、第十四、第十五、第十六、第十七、第十八、第十九、第二十、第二十一、第二十二、第二十三、第二十四、第二十五、第二十六、第二十七、第二十八、第二十九、第三十、第三十一、第三十二、第三十三、第三十四、第三十五、第三十六、第三十七、第三十八、第三十九、第四十、第四十一、第四十二或第四十三实施方案中的方法,L’由下式表示:


其中R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基);且M为H+、Na+或K+。
在第十三具体实施方案中,对于第八、第十、第十四、第十五、第十九、第二十、第二十一、第四十一、第四十二和第四十三实施方案中的方法,式(I’)的化合物由下式中的任一个表示:


或其药学上可接受的盐,其中R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基);且M为H+、Na+或K+。
在第十四具体实施方案中,对于在第十二或第十三具体实施方案中描述的方法,R101为甲基。
在第十五具体实施方案中,对于第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二、第十三、第十四、第十五、第十六、第十七、第十八、第十九、第二十、第二十一、第二十二、第二十三、第二十四、第二十五、第二十六、第二十七、第二十八、第二十九、第三十、第三十一、第三十二、第三十三、第三十四、第三十五、第三十六、第三十七、第三十八、第三十九、第四十、第四十一、第四十二或第四十三实施方案中的方法,L’、L”和L”’中的一者由下式表示:
-W’-Rx-S-Zs (C);
其中:
W’为-N(Re)-;
Re为-H,具有1至10个碳原子的直链、支链或环状烷基、烯基或炔基,或-(CH2-CH2-O)n-Rk,其中Rk为-H、任选地带有仲氨基(例如,-NHR101)或叔氨基(-NR101R102)基团或5-或6-元含氮杂环如哌啶或吗啉的具有1至6个碳原子的直链、支链环状烷基,其中R101和R102各自独立地为具有1至10个碳原子的直链、支链或环状烷基、烯基或炔基;
Rx为具有1至10个碳原子的直链、支链或环状烷基;
Zs为-H、-SRd、-C(=O)Rd1或选自下式中的任一个:


其中:
Rd为具有1至6个碳原子的直链或支链烷基或选自苯基、硝基苯基(例如,2或4-硝基苯基)、二硝基苯基(例如,2或4-硝基苯基)、羧基硝基苯基(例如,3-羧基-4-硝基苯基)、吡啶基或硝基吡啶基(例如,4-硝基吡啶基);
Rd1为具有1至6个碳原子的直链或支链烷基;
q为1至5的整数;
n为2至6的整数;
n’为1至24的整数;
U为-H或-SO3M;且
M为-H或阳离子,诸如Na+或K+。
在一个更具体的实施方案中,L’、L”和L’”中的一者由式(C)表示,且其余各自独立地为-H、具有1至6个碳原子的直链或支链烷基、卤素、-OH、(C1-C6)烷氧基或-NO2。
在另一个更具体的实施方案中,L’、L”和L’”中的一者由式(C)表示,且其余为-H。
在另一个更具体的实施方案中,L’由式(C)表示;且L”和L’”两者均为-H。
在另一个更具体的实施方案中,Zs为-SRd且Rd为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基)。
在第十六具体实施方案中,Rx为具有1至6个碳原子的直链或支链烷基;且剩下的变量如在第十四具体实施方案或其中描述的任何更具体的实施方案中描述。更具体地讲,Rx为-(CH2)p-(CRfRg)-,其中Rf和Rg各自独立地选自-H或具有1至4个碳原子的直链或支链烷基;且p为0、1、2或3。甚至更具体地讲,Rf和Rg相同或不同,且选自-H和-Me;且p为1。
在第十七具体实施方案中,Rk为-H或-Me,且n为3;且剩下的变量如上文在第十五或第十六具体实施方案中所述。
在第十八具体实施方案中,对于第一、第二、第三、第四、第五、第六、第七、第八、第九、第十、第十一、第十二、第十三、第十四、第十五、第十六、第十七、第十八、第十九、第二十、第二十一、第二十二、第二十三、第二十四、第二十五、第二十六、第二十七、第二十八、第二十九、第三十、第三十一、第三十二、第三十三、第三十四、第三十五、第三十六、第三十七、第三十八、第三十九、第四十、第四十一、第四十二或第四十三实施方案中的方法,L’由下式表示:

其中R100为(C1-C3)烷氧基;且剩下的变量如上文在第一至第十七具体实施方案中所述。在另一个实施方案中,L’为NO2。在另一个实施方案中,L'由下式表示:

在某些实施方案中,式(I’)的吲哚啉并苯并二氮杂 二聚体化合物可以根据下文所示的方案1至12制备。
二聚体化合物可以根据下文所示的方案1至12制备。

方案1

方案2

方案3

方案4

方案5

方案6

方案7

方案8

方案9

方案10

方案11

方案12
本发明的化合物
本发明还提供了本文所述的新化合物。在某些实施方案中,本发明的化合物为式(1)、(2)、(2”)、(3)、(3”)、(4)、(4”)、(5)、(5”)、(6)、(7)、(7’)、(7-1)、(7”)、(7”’)、(8)、(9)、(10)、(10’)、(11)、(12)、(13)、(14)、(15)、(16)、(17)、(17’)、(18)、(19)、(20)、(a)、(b)、(d)、(I’)、(IA)和(IB)的化合物,其中变量如在第一至第四十三实施方案中或者第一至第十八具体实施方案或其中描述的任何更具体的实施方案中所述。
在第十九具体实施方案中,对于本发明的化合物,当存在时,R100为(C1-C3)烷氧基;R101为(C1-C3)烷基、吡啶基或硝基吡啶基(例如,4-硝基吡啶基);P1为醇保护基团;X1和X2在每次出现时独立地为-Br、-I和磺酸酯;P2为胺保护基团;P3为H或胺保护基团;且剩下的变量如在第一至第四十四实施方案以及第一至第十八具体实施方案中的任一个中所述。
在第二十具体实施方案中,对于本发明的化合物,当存在时,R100为-OMe;且R101为Me或吡啶基;且剩下的变量如在第一至第四十四实施方案以及第一至第十九具体实施方案中的任一个中所述。
在第二十一具体实施方案中,对于本发明的化合物,当存在时,P1为甲硅烷基保护基团;且剩下的变量如在第一至第四十四实施方案以及第一至第二十具体实施方案中的任一个中所述。更具体地讲,甲硅烷基保护基团为二甲基异丙基甲硅烷基、二乙基异丙基甲硅烷基、二甲基己基甲硅烷基、三甲基甲硅烷基、三异丙基甲硅烷基、三苄基甲硅烷基、三苯基甲硅烷基、2-降冰片基二甲基甲硅烷基、叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基或[2-(三甲基甲硅烷基)乙氧基]甲基。甚至更具体地讲,甲硅烷基保护基团为三乙基甲硅烷基、三异丙基甲硅烷基或叔丁基二甲基甲硅烷基。在另一个甚至更具体的实施方案中,甲硅烷基保护基团为叔丁基二甲基甲硅烷基。
在第二十二具体实施方案中,对于本发明的化合物,当存在时,X1和X2在每次出现时各自独立地为磺酸酯;且剩下的变量如在第一至第四十四实施方案以及第一至第二十一具体实施方案中的任一个中所述。更具体地讲,该磺酸酯为甲磺酸酯、甲苯磺酸酯、对溴苯磺酸酯或三氟甲磺酸酯。甚至更具体地讲,该磺酸酯为甲磺酸酯。
在第二十三具体实施方案中,对于本发明的化合物,当存在时,X3为氯;且剩下的变量如在第一至第四十四实施方案以及第一至第二十二具体实施方案中的任一个中所述。
在第二十四具体实施方案中,对于本发明的化合物,当存在时,X4为磺酸酯;且剩下的变量如在第一至第四十四实施方案以及第一至第二十三具体实施方案中的任一个中所述。更具体地讲,该磺酸酯为甲磺酸酯、甲苯磺酸酯、对溴苯磺酸酯或三氟甲磺酸酯。甚至更具体地讲,该磺酸酯为甲磺酸酯。
在第二十五具体实施方案中,对于本发明的化合物,P2为选自2-三甲基甲硅烷基乙基、(2-苯基-2-三甲基甲硅烷基)乙基、三异丙基甲硅烷氧基、2-(三甲基甲硅烷基)乙氧基甲基、烯丙氧基羰基、9-芴基甲氧基羰基、2-(三甲基甲硅烷基)乙氧基羰基或2,2,2,2-三氯乙氧基羰基的胺保护基团;且剩下的变量如第一至第四十四实施方案以及第一至第二十四具体实施方案中的任一个中所述。
在第二十六具体实施方案中,对于本发明的化合物,当存在时,P3为H或选自2-三甲基甲硅烷基乙基、(2-苯基-2-三甲基甲硅烷基)乙基、三异丙基甲硅烷氧基、2-(三甲基甲硅烷基)乙氧基甲基和烯丙氧基羰基、9-芴基甲氧基羰基、2-(三甲基甲硅烷基)乙氧基羰基或2,2,2,2-三氯乙氧基羰基的胺保护基团;且剩下的变量如第一至第四十四实施方案以及第一至第二十五具体实施方案中的任一个中所述。
在第二十七具体实施方案中,对于本发明的化合物,当存在时,X5为-Br;且剩下的变量如在第一至第四十四实施方案以及第一至第二十六具体实施方案中的任一个中所述。
在一个实施方案中,对于本文所述的本发明的方法和化合物,L’不由以下表示:

在另一个实施方案中,对于本文所述的本发明的方法和化合物,L’不由以下表示:

本文和以下实施例中引用的所有参考文献均明确地全文以引用方式并入。
实施例
现在将通过参考非限制性实施例来说明本发明。除非另行指出,否则所有百分数、比率、份数等均按重量计。所有试剂均购自Aldrich Chemical Co.,New Jersey或其他商业来源。核磁共振(1H NMR)谱在Bruker 400MHz仪器上获取。质谱在Bruker DaltonicsEsquire 3000仪器上获取,且LCMS在具有Agilent 6120单四极杆MS的Agilent1260Infinity LC上使用电喷雾电离获取,且UPLC在具有单四极MS ZsprayTM的Acquity系统上获取(柱:Acquity BEH C18,2.1×50mm,1.7μm,方法:2.5分钟,流速0.8mL/min,溶剂A:水,溶剂B:MeCN,5%至95%的MeCN,经2.0分钟,和95%MeCN,持续0.5分钟)。
以下溶剂、试剂、保护基团、部分和其他名称可以用圆括号中的缩写来表示:
Me=甲基;Et=乙基;Pr=丙基;i-Pr=异丙基;Bu=丁基;t-Bu=叔丁基;Ph=苯基;且Ac=乙酰基
AcOH或HOAc=乙酸
ACN或CH3CN=乙腈
Ala=丙氨酸
Ar=氩气
aq=水性
Bn=苄基
Boc或BOC=叔丁氧羰基
CBr4=四溴化碳
Cbz或Z=苄氧羰基
DCM或CH2Cl2=二氯甲烷
DCE=1,2-二氯乙烷
DMAP=4-二甲基氨基吡啶
DI水=去离子水
DIBAL=氢化二异丁基铝
DIEA或DIPEA=N,N-二异丙基乙胺
DMA=N,N-二甲基乙酰胺
DMF=N,N-二甲基甲酰胺
DMSO=二甲基亚砜
DTT=二硫苏糖醇
EDC=1-乙基-3-(3-二甲氨基丙基)碳二亚胺
EEDQ=N-乙氧基羰基-2-乙氧基-1,2-二氢喹啉
ESI或ES=电喷雾电离
EtOAc=乙酸乙酯
Gly=甘氨酸
g=克
h=小时
HATU=N,N,N’N’-四甲基-O-(7-氮杂苯并三唑-1-基)脲鎓六氟磷酸盐
HPLC=高效液相色谱
HOBt或HOBT=1-羟基苯并三唑
LAH=氢化铝锂
LC=液相色谱
LCMS=液相色谱-质谱
min=分钟
mg=毫克
mL=毫升
mmol=毫摩尔
μg=微克
μL=微升
μmol=微摩尔
Me=甲基
MeOH=甲醇
MeI=碘甲烷
MS=质谱
MsCl=甲磺酰氯(甲基磺酰氯)
Ms2O=甲磺酸酐
MTBE=甲基叔丁基醚
NaBH(OAc)3=三乙酰氧基硼氢化钠
NHS=N-羟基琥珀酰亚胺
NMR=核磁共振光谱学
PPh3=三苯基膦
PTLC=制备型薄层色谱
rac=外消旋混合物
Rf=延迟因子
RPHPLC或RP-HPLC=反相高效液相色谱
RT或rt=室温(环境,约25℃)
sat或sat’d=饱和的
STAB=三乙酰氧基硼氢化钠(NaBH(OAc)3)
TBSCl或TBDMSCl=叔丁基二甲基甲硅烷基氯
TBS=叔丁基二甲基甲硅烷基
TCEP·HCl=三(2-羧基乙基)膦盐酸盐
TEA=三乙胺(Et3N)
TFA=三氟乙酸
THF=四氢呋喃
TLC=薄层色谱
实施例1.

向(5-氨基-1,3-亚苯基)二甲醇(1.01g,6.59mmol)在无水二甲基甲酰胺(16.48mL)和无水四氢呋喃(16.48ml)中的搅拌溶液中加入4-甲基-4-(甲基二硫烷基)戊酸(1.281g,6.59mmol)、EDC·HCl(2.53g,13.19mmol)和DMAP(0.081g,0.659mmol)。在室温下搅拌所得混合物18小时。将反应物用饱和氯化铵溶液猝灭,并用乙酸乙酯(3×50mL)萃取。将有机萃取物用水、盐水洗涤,并经无水硫酸钠干燥。将溶液过滤并真空浓缩,且所得残余物通过硅胶色谱(己烷/EtOAc)纯化,得到呈白色固体的化合物1a(0.70g,35%收率)。1HNMR(400MHz,DMSO-d6):δ9.90(s,1H),7.43(s,2H),6.93(s,1H),5.16(t,2H,J=5.7Hz),4.44(d,4H,J=5.7Hz),2.43(s,3H),2.41-2.38(m,2H),1.92-1.88(m,2H),1.29(s,6H)。MS(m/z),实测值330.0(M+H)+。

将二醇1a(1.0g,3.04mmol)溶解于DMF(10.12mL)中。向溶液中加入TBSCl(503mg,3.34mmol)和咪唑(238mg,3.49mmol),并在室温下搅拌反应物过夜。向反应混合物中加入另外的TBSCl(600mg)和咪唑(220mg),并在室温再搅拌5小时。将反应混合物用DCM稀释,并用饱和氯化铵、盐水洗涤,并经硫酸钠干燥,过滤并浓缩。粗制的产物通过硅胶色谱法(EtOAc/己烷,梯度)纯化,得到呈无色油状物的1b(710mg,53%收率)。LCMS(8分钟方法)=6.967分钟。观测质量(ESI+):445.95(M+H)+和467.90(M+Na)+。

将化合物1b(200mg,0.451mmol)溶解于DCM(4.51mL)中并冷却至0。℃在Ar下向反应混合物中加入Et3N(82μL,0.586mmol),然后滴加甲磺酰氯(42.1μL,0.541mmol)。在0℃下搅拌反应混合物2小时。将该溶液用EtOAc稀释,并用冷水(2x)洗涤。将有机层经硫酸钠干燥,过滤并浓缩,得到甲磺酸酯。粗制的甲磺酸酯不经纯化即用于下一步。LCMS(8分钟方法)=7.444分钟。观测质量(ESI+):521.8(M+H)+和543.8(M+Na)+。将粗制的甲磺酸酯(210mg,0.402mmol)溶解于DMF(2.68mL)中。向混合物中加入IGN单体A(130mg,0.443mmol)和碳酸钾(111mg,0.805mmol)并在室温下在Ar下搅拌过夜。加入水(15mL)使产物沉淀出来。将浆液搅拌5分钟并过滤。滤饼用水(3x)洗涤并在真空/N2下干燥得到呈固体的化合物1c(270mg,93%收率)。LCMS(8分钟方法)=7.624分钟。观测质量(ESI+):719.8(M+H)+。

将化合物1c(686mg,0.953mmol)溶解于DCE(6.35mL)中。向反应混合物中加入三乙酰氧基硼氢化钠(400mg,1.91mmol),并在室温下搅拌3小时。将反应混合物用DCM稀释,并用饱和氯化铵、盐水洗涤,经硫酸钠干燥,过滤并浓缩,得到还原的亚胺产物。LCMS(8分钟方法)=4.363分钟。观测质量(ESI+):720.75(M+H)+。
将粗制的还原的亚胺(680mg,0.942mmol)溶解于THF(5.23mL)中。加入HCl(aq.5M)(3.77mL,9.42mmol)且在室温下搅拌4小时。将反应混合物用DCM稀释,并用饱和碳酸氢钠、盐水洗涤,经硫酸钠干燥,过滤并浓缩。粗制的产物通过硅胶色谱法(梯度,MeOH/DCM)纯化得到化合物1d(420mg,73%收率,2个步骤)。LCMS(8分钟方法)=5.905分钟。观测质量(ESI+):607.8(M+H)+。

将化合物1d(420mg,0.691mmol)溶解于DCM(4.61mL)中。将溶液冷却至-5℃冰(-盐水浴),且加入TEA(125μL,0.898mmol),接着在Ar下加入甲磺酸酐(144mg,0.829mmol)。将反应混合物在-5℃下在Ar下搅拌1.5小时。将反应混合物在-5℃用水猝灭并升温至室温。用EtOAc(2x)萃取混合物,并用水(2x)洗涤有机层。将有机物经硫酸钠干燥,过滤并浓缩,得到呈棕色泡沫的甲磺酰化产物。LCMS(8分钟方法)=6.380分钟。观测质量(ESI+):685.7(M+H)+。
将甲磺酸酯(470mg,0.582mmol)溶解于DMF(3.88mL)中。在室温下加入IGN单体A(189mg,0.641mmol),然后加入碳酸钾(121mg,0.874mmol),并搅拌反应物过夜。加入水(约5mL)以使产物沉淀出来。将浆液搅拌5分钟,然后过滤并在真空/N2下干燥。粗制的产物通过硅胶色谱法(梯度,MeOH/DCM)纯化,得到呈黄色固体的化合物1e(543mg,53%收率)。LCMS(8分钟方法)=6.804分钟。观测质量(ESI+):883.7(M+H)+。
实施例2.

将(S)-2-(((苄氧基)羰基)氨基)丙酸(5g,22.40mmol)和(S)-2-氨基丙酸叔丁酯盐酸盐(4.48g,24.64mmol)溶解于无水DMF(44.8mL)中。加入EDC·HCl(4.72g,24.64mmol)、HOBt(3.43g,22.40mmol)和DIPEA(9.75mL,56.0mmol)。在氩气下在室温下搅拌反应物过夜。反应混合物用二氯甲烷稀释,然后用饱和氯化铵、饱和碳酸氢钠、水和盐水洗涤。有机层经硫酸钠干燥并浓缩。粗制的油状物通过硅胶色谱法(己烷/乙酸乙酯)纯化,产生化合物2a(6.7g,85%收率)。1H NMR(400MHz,CDCl3):δ7.38-7.31(m,5H),6.53-6.42(m,1H),5.42-5.33(m,1H),5.14(s,2H),4.48-4.41(m,1H),4.32-4.20(m,1H),1.49(s,9H),1.42(d,3H,J=6.8Hz),1.38(d,3H,J=7.2Hz)。

将化合物2a(6.7g,19.12mmol)溶解于甲醇(60.7mL)和水(3.03mL)中。将溶液用氩气吹扫5分钟。缓慢加入碳载钯(湿,10%)(1.017g,0.956mmol)。在氢气氛下搅拌反应物过夜。溶液经硅藻土过滤,用甲醇冲洗并浓缩。将其与甲醇和乙腈共沸,且将所得油状物直接置于高真空下,得到化合物2b(4.02g,97%收率),其直接用于下一步。1H NMR(400MHz,CDCl3):δ7.78-7.63(m,1H),4.49-4.42(m,1H),3.55-3.50(m,1H),1.73(s,2H),1.48(s,9H),1.39(d,3H,J=7.2Hz),1.36(d,3H,J=6.8Hz)。

将化合物2b(4.02g,18.59mmol)和己二酸单甲酯(3.03mL,20.45mmol)溶解于无水DMF(62.0mL)中。加入EDC·HCl(3.92g,20.45mmol)、HOBt(2.85g,18.59mmol)和DIPEA(6.49mL,37.2mmol)。在室温下搅拌混合物过夜。反应物用二氯甲烷/甲醇(150mL,5:1)稀释,且用饱和氯化铵、饱和碳酸氢钠和盐水洗涤。将其经硫酸钠干燥,过滤并浓缩。将该化合物与乙腈(5x)共沸,然后在35℃在高真空下泵抽,得到化合物2c(6.66g,100%收率)。粗料不经纯化即用于下一步。1H NMR(400MHz,CDCl3):δ6.75(d,1H,J=6.8Hz),6.44(d,1H,J=6.8Hz),4.52-4.44(m,1H),4.43-4.36(m,1H),3.65(s,3H),2.35-2.29(m,2H),2.25-2.18(m,2H),1.71-1.60(m,4H),1.45(s,9H),1.36(t,6H,J=6.0Hz)。

在室温下,将化合物2c(5.91g,16.5mmol)在TFA(28.6mL,372mmol)和去离子水(1.5mL)中搅拌3小时。将反应混合物用乙腈浓缩,并置于高真空下,得到呈粘性固体的粗制的化合物2d(5.88g,100%收率)。1H NMR(400MHz,CDCl3):δ7.21(d,1H,J=6.8Hz),6.81(d,1H,J=7.6Hz),4.69-4.60(m,1H),4.59-4.51(m,1H),3.69(s,3H),2.40-2.33(m,2H),2.31-2.24(m,2H),1.72-1.63(m,4H),1.51-1.45(m,3H),1.42-1.37(m,3H)。

将化合物2d(5.6g,18.52mmol)溶解于无水二氯甲烷(118mL)和无水甲醇(58.8mL)中。加入(5-氨基-1,3-亚苯基)二甲醇(2.70g,17.64mmol)和EEDQ(8.72g,35.3mmol),并在室温下搅拌反应物过夜。浓缩溶剂并加入乙酸乙酯。将所得浆液过滤,用乙酸乙酯洗涤并在真空/N2下干燥,得到化合物2e(2.79g,36%收率)。1H NMR(400MHz,DMSO-d6):δ9.82(s,1H),8.05,(d,1H,J=9.2Hz),8.01(d,1H,J=7.2Hz),7.46(s,2H),6.95(3,1H),5.21-5.12(m,2H),4.47-4.42(m,4H),4.40-4.33(m,1H),4.33-4.24(m,1H),3.58(s,3H),2.33-2.26(m,2H),2.16-2.09(m,2H),1.54-1.46(m,4H),1.30(d,3H,J=7.2Hz),1.22(d,3H,J=4.4Hz)。

将二醇2e(1.0g,2.286mmol)溶解于无水DMF(7.6mL)中。加入TBSCl(0.482g,3.20mmol)和咪唑(0.467g,6.86mmol),且在室温下搅拌反应物2小时。将反应物用饱和氯化铵猝灭并用水和EtOAc稀释。将水层用EtOAc萃取一次,且将合并的有机层用水和盐水洗涤,经硫酸钠干燥,过滤并浓缩。粗制的残余物通过硅胶快速色谱法(DCM/MeOH)纯化,得到化合物2f(360mg,28%收率)。LCMS(8分钟方法,40-98%)=2.35分钟。观测质量(ESI+):574.4(M+Na)+。

将化合物2f(360mg,0.652mmol)溶解于无水二氯甲烷(6.52mL)中并在丙酮/冰浴中冷却。加入三乙胺(227μL,1.631mmol)和甲磺酸酐(146mg,0.816mmol)。在-10℃下在丙酮/冰浴中搅拌反应物1小时。将反应物用冷EtOAc稀释并用冰水猝灭。将有机层用冰水洗涤,然后用硫酸钠和硫酸镁干燥,过滤并浓缩,得到呈蓬松固体的粗制的化合物2g(390mg,95%收率)。LCMS(8分钟方法,40-98%)=2.81分钟;5.86分钟(8分钟方法,5-98%)。观测质量(ESI-):628.0(M-H)-。

将甲磺酸酯2g(390mg,0.619mmol)和IGN单体A(264mg,0.897mmol)溶解于无水DMA(7.47mL)中。加入碳酸钾(207mg,1.495mmol)和碘化钾(51.4mg,0.310mmol),且在室温下搅拌反应物过夜。反应物用水沉淀,过滤,且滤饼用水洗涤。将固体再溶解于DCM中,用水洗涤,经硫酸镁干燥并浓缩,得到粗制的化合物2h(568mg,111%收率)。产物不经进一步纯化即可使用。LCMS(8分钟方法,5-98%)=6.23分钟。观测质量(ESI+):827.8(M+H)+。

将化合物2h(0.513g,0.619mmol)溶解于DCE(7.74mL)中。加入NaBH(OAc)3(0.276g,1.239mmol)并在室温下搅拌混合物1.5小时。将反应物用DCM稀释,用饱和氯化铵猝灭并用盐水洗涤。将有机层经硫酸钠干燥,过滤并浓缩,得到化合物2i。LCMS(15分钟方法)=9.93分钟。

将化合物2i(514mg,0.619mmol)溶解于THF(3.44mL)中。在室温下加入5M HCl水溶液(1.24mL,6.19mmol),并搅拌反应物1小时。将反应混合物用DCM/MeOH(20:1)稀释,且将有机层用饱和碳酸氢钠、盐水洗涤,经硫酸镁干燥,过滤并浓缩。粗制的残余物通过硅胶色谱法(DCM/MeOH)纯化得到化合物2j(210mg,47%收率)。LCMS(8分钟方法,5-98%)=4.56分钟。观测质量(ESI+):715.8(M+H)+。

将化合物2j(210mg,0.293mmol)溶解于DCM(3.95mL)和DMF(500μL)中并冷却至-10℃冰(-丙酮浴)。加入TEA(57.2μL,0.411mmol)和甲磺酸酐(46.6mg,0.260mmol),并在Ar下搅拌反应物3小时。将反应物用-5℃的冷水猝灭并用EtOAc稀释。用冷EtOAc(2×)萃取水层,且合并的有机物用冷水(2×)洗涤。将有机层经无水硫酸钠/硫酸镁干燥,过滤并浓缩。将粗制的产物2k在高真空下泵抽且在不纯化的情况下用于下一步。LCMS(8分钟方法,5-98%)=5.06分钟。观测质量(ESI+):791.8(M-H)-。

[267]将化合物2k(233mg,0.293mmol)溶解于DMA(1.95mL)中。在室温下加入IGN单体A(103mg,0.352mmol)和碳酸钾(60.7mg,0.440mmol),并搅拌反应物过夜。向反应混合物中加入DI水,且将所得固体过滤并用水洗涤。将固体再溶解于DCM/MeOH(20:1)中,用水洗涤,经硫酸镁干燥,过滤并浓缩。粗制的残余物通过RPHPLC(ACN/H2O)纯化,得到2l(44mg,15%收率)。LCMS(8分钟方法,5-98%)=5.4分钟。观测质量(ESI+):991.7(M+H)+。
实施例3.

向IGN单体A(1.0g,3.4mmol)在DCE(10mL)和DMF(4mL)中的溶液中加入三乙酰氧基硼氢化钠(1.1g,5.1mmol,1.5当量),并搅拌反应物直至原料完成。在室温下2小时后完成原料后,将反应物用饱和氯化铵(10mL)猝灭,然后分离各层。水层用二氯甲烷(10mL)萃取一次,且合并的有机层用水(2×10mL)和盐水(10mL)洗涤。将有机层经硫酸镁干燥,过滤,且真空除去溶剂,得到白色/棕色粉末。将粉末用EtOAc(2×10mL)洗涤并在真空下干燥得到呈白色固体的还原的IGN单体A(0.87g,2.9mmol,87%收率),其不经进一步纯化而用于下一步。UPLCMS(2.5分钟方法)=1.34分钟。观测质量(ESI+):297.4(M+H)+。1H NMR(400MHz,DMSO-d6):δ9.44(s,1H),8.20(d,J=8.1Hz,1H),7.30–7.23(m,2H),7.22–7.12(m,1H),7.01(td,J=7.4,1.1Hz,1H),6.21(s,1H),6.17(d,J=6.6Hz,1H),4.37(tdd,J=10.1,4.4,1.9Hz,1H),3.70(s,3H),3.58–3.39(m,2H),3.31–3.15(m,2H),2.88(dd,J=16.9,4.4Hz,1H)。

将2e(5.53g,12.6mmol)在DCM(81mL)和DMF(64.9mL)中的溶液冷却至0℃,然后加入DIPEA(6.13mL,37.9mmol,3.0当量),然后滴加甲磺酸酐(5.06g,29.1mmol,2.3当量)在DCM(15mL)/DMF(1mL)中的溶液。将反应物搅拌1小时,然后用冷水猝灭。在用水和盐水洗涤之后,将溶液经硫酸镁干燥,过滤,且真空除去溶剂,得到橙色油状物,将其在二乙醚中研磨,得到二甲磺酸酯2m(6.4g,10.8mmol,85%收率)。LCMS(8分钟方法)=4.019分钟。观测质量(ESI+):594.8(M+H)+。粗料不经进一步纯化即可用于下一步。

向2m(0.52g,0.88mmol)和IGN单体A(0.18g,0.61mmol,0.7当量)在DMF(7mL)中的溶液中加入碳酸钾(0.24g,1.75mmol,2.0当量)且在室温下搅拌反应物12小时。将反应物用水(30mL)猝灭,并用DCM(3×15mL)萃取。将有机层合并,且用水(3×60mL)、盐水(60mL)洗涤,经硫酸镁干燥,过滤,且真空除去溶剂,得到粗制的黄色油状物。该材料通过硅胶色谱法(DCM/(MeCN/MeOH(4/1),从100/0至65/35)纯化,得到期望的产物2n(0.09g,0.12mmol,13%收率)。UPLCMS(2.5分钟方法)=1.46分钟。观测质量(ESI+):792.6(M+H)+。

向2n(0.05g,0.06mmol)在DMF(0.48mL,6.2mmol)中的溶液中加入碳酸钾(0.02g,0.12mmol,2.0当量),然后加入还原的IGN单体A(0.02g,0.07mmol,1.1当量)。在室温下搅拌反应物12小时。用水猝灭反应物,并且将所得固体过滤并用水洗涤。将固体再溶解于DCM/MeOH(20:1)中,用水洗涤,用硫酸镁干燥,过滤并浓缩。粗制的残余物通过RPHPLC(ACN/H2O)纯化,得到2l(0.03g,0.04mmol,55%收率)。LCMS(8分钟方法,5-98%)=5.4分钟。观测质量(ESI+):991.7(M+H)+。1H NMR(400MHz,DMSO-d6,报道为水加合物的混合物):δ10.10(d,J=3.7Hz,1H),8.27(d,J=8.0Hz,1H),8.21–8.10(m,1H),8.05(d,J=7.4Hz,1H),7.78(dt,J=8.5,1.8Hz,2H),7.43–7.13(m,7H),7.16–6.98(m,2H),6.49(s,1H),6.36(d,J=13.1Hz,0.4H),6.16(d,J=6.2Hz,0.4H),5.80(s,0.4H),5.67(s,0.4H),5.57(d,J=5.6Hz,0.4H),5.35–5.09(m,2H),5.03(t,J=5.9Hz,2H),4.81–4.72(m,0.4H),4.60(dt,J=9.7,5.0Hz,0.2H),4.51–4.36(m,2H),4.39–4.23(m,1H),4.17(td,J=9.7,2.9Hz,0.4H),3.93(s,0.4H),3.83–3.74(m,5H),3.62(s,2H),3.75–3.44(m,2H),3.32(d,J=11.6Hz,1H),3.19–3.07(m,1H),2.95(dd,J=17.1,4.3Hz,1H),2.38–2.29(m,1H),2.18(m,1H),1.56(m,J=3.9Hz,4H),1.41–1.31(m,3H),1.30–1.14(m,3H)。
实施例4.

向2m(0.88g,1.47mmol)在DMF(11mL)中的溶液中加入还原的IGN单体A(0.26g,0.88mmol,0.6当量),然后加入碳酸钾(0.41mg,2.95mmol,2.0当量)。在搅拌反应物12小时之后,将反应物用水(50mL)和EtOAc(30mL)稀释。水层用EtOAc(3×10mL)萃取。合并的有机层用盐水(20mL)洗涤,经硫酸镁干燥并过滤。除去溶剂,且通过硅胶色谱法(DCM/MeOH)纯化粗制的混合物,得到期望的产物2k(0.11g,0.14mmol,10%收率)。LCMS(8分钟方法)=5.013分钟。观测质量(ESI+):794.3(M+H)+。

向2k(0.11g,0.14mmol)在DMF(2mL)中的溶液中加入碳酸钾(0.04g,0.29mmol,2.0当量)。加入IGN单体A(0.04g,0.14mmol,1.0当量),且在室温下搅拌反应物12小时。用水(10mL)猝灭反应物,且将所得固体过滤并用水洗涤。将固体再溶解于DCM/MeOH(20:1)中,用水(10mL)洗涤,用硫酸镁干燥,过滤并浓缩。粗制的残余物通过RPHPLC(ACN/H2O)纯化,得到2l(0.08g,0.09mmol,59%收率)。LCMS(8分钟方法,5-98%)=5.4分钟。观测质量(ESI+):991.7(M+H)+。
实施例5.

向2n(0.1g,0.13mmol)在DCE(2mL)中的溶液中加入三乙酰氧基硼氢化钠(0.03g,0.13mmol,1.0当量),并在室温下搅拌反应物2小时。用饱和氯化铵(2mL)猝灭反应物并分离各层。将水层用DCM(5mL)萃取并将合并的有机层用水、盐水洗涤,经硫酸镁干燥并过滤。粗制的黄色固体使用硅胶色谱法(EtOAc/MeOH(95/5))纯化,得到期望的还原产物2k(0.035g,0.044mmol,35%收率)。LCMS(8分钟方法)=5.021分钟。观测质量(ESI+):794.3(M+H)+。

向2k(0.035g,0.044mmol)在DMF(1.0mL)中的溶液中加入碳酸钾(0.013g,0.09mmol,2.0当量)。加入IGN单体A(0.013g,0.04mmol,1.0当量),并在室温下搅拌反应物12小时。用水(10mL)猝灭反应物,且将所得固体过滤并用水洗涤。将固体再溶解于DCM/MeOH(20:1,20mL)中,用水(20mL)洗涤,用硫酸镁干燥,过滤并浓缩。粗制的残余物通过RPHPLC(ACN/H2O)纯化,得到2l(0.017g,0.01mmol,38%收率)。LCMS(8分钟方法,5-98%)=5.4分钟。观测质量(ESI+):991.7(M+H)+。
实施例6.

向2f(8.8g,16.0mmol)在DMF(100mL)中的溶液中加入吡啶(4.51ml,55.8mmol,3.5当量)。将反应物冷却至0℃,然后滴加甲磺酰氯(2.5mL,31.9mmol,2.0当量)并搅拌反应物2小时。用饱和碳酸氢钠(30mL)猝灭混合物,加入EtOAc并分离各层。将水层用EtOAc(3×50mL)萃取并将合并的有机层用水、盐水洗涤,经硫酸镁干燥并过滤。除去溶剂,且粗制的白色固体2o不经纯化而用于下一步(6.2g,10.9mmol,68%)。UPLCMS(2.5分钟方法)=1.96分钟。观测质量(ESI+):570.7(M+H)+。

向2o(1.7g,2.98mmol)在THF(36.6mL)中的溶液中加入DIPEA(2.1mL,11.9mmol,4.0当量),然后加入HF-吡啶(0.84mL,6.0mmol,2.0当量)。在室温下搅拌反应物3小时。用饱和碳酸氢钠(20mL)猝灭反应物,然后分离各层。水层用EtOAc(3×10mL)萃取。将合并的有机层用盐水(30mL)洗涤,经硫酸镁干燥,过滤,并真空除去溶剂,得到粗制的白色油状物,其通过硅胶色谱法(DCM/MeOH)纯化,得到呈白色固体的期望的产物2p(0.75g,1.6mmol,55%收率)。UPLCMS(2.5分钟方法)=1.23分钟。观测质量(ESI+):456.4(M+H)+。

向2p(0.65g,1.43mmol)在DCM(10mL)和DMF(2mL)中的溶液中加入DIPEA(0.51mL,2.85mmol,2.0当量),并将反应物冷却至0℃。缓慢加入甲磺酸酐(0.3g,1.71mmol)在DCM(2mL)中的溶液。反应在30分钟之后完成,用水(20mL)猝灭,萃取各层,水层用DCM(2×10mL)洗涤。将有机层合并,用水(20mL)、盐水(10mL)洗涤,经硫酸镁干燥并过滤。真空除去溶剂得到期望的产物2q(0.76g,1.42mmol,100%收率),其不经进一步纯化以粗品用于下一步。UPLCMS(2.5分钟方法)=1.37分钟。观测质量(ESI+):534.4(M+H)+。

向2q(0.76g,1.42mmol)在DMA(13mL)中的溶液中加入碳酸钾(0.59g,4.27mmol),然后加入IGN单体A(0.5g,1.71mmol)在DMA(1mL)中的溶液。在室温下搅拌反应物12小时。用水(30mL)猝灭反应物,并搅拌混合物10分钟。将固体过滤,然后溶解于DCM/MeOH(9/1,20mL)中并用盐水(10mL)洗涤。将有机层分离并经硫酸镁干燥,过滤并真空浓缩,得到粗制的黄色固体2r(0.76g,1.04mmol,73%收率),其不经进一步纯化以粗品用于下一步。UPLCMS(2.5分钟方法)=1.55分钟。观测质量(ESI+):732.9(M+H)+。

向2r(0.26g,0.36mmol)在DMA(10mL)中的溶液中加入碘化钾(0.06g,0.355mmol,1.0当量)、还原的IGN单体A(0.1g,0.37mmol,1.05当量)和碳酸钾(0.15g,1.06mmol,3.0当量)。将反应物升温至40℃并搅拌4小时。用水(20mL)猝灭反应物并搅拌混合物10分钟。过滤所得固体。将固体再溶解于DCM/MeOH(20:1,20mL)中,用水(20mL)洗涤,用硫酸镁干燥,过滤并浓缩。粗制的残余物通过RPHPLC(ACN/H2O)纯化,得到2l(0.097g,0.097mmol,28%收率)。LCMS(8分钟方法,5-98%)=5.4分钟。观测质量(ESI+):991.7(M+H)+。
实施例7.

在0℃下向2r(0.76g,1.04mmol)在DCE(10mL)中的溶液中加入DMF(3.0mL),然后加入三乙酰氧基硼氢化钠(0.33g,1.56mmol)。在室温下搅拌反应物4小时。用饱和氯化铵(20mL)猝灭反应物并分离各层。水层用DCM(3×10mL)萃取并将合并的有机层用水(10mL)、盐水(10mL)洗涤,经硫酸镁干燥,过滤,并真空除去溶剂,得到呈油状物的期望的粗料2s(0.65g,0.88mmol,85%收率),其未经进一步纯化即用于下一步。UPLCMS(2.5分钟方法)=1.80分钟。观测质量(ESI+):735.3(M+H)+。

在室温下向2s(0.65g,0.88mmol)在DMA(15mL)中的溶液中加入碳酸钾(0.25g,1.78mmol,2.0当量),然后加入碘化钾(0.073g,0.44mmol,0.5当量),且向反应混合物中加入IGN单体A(0.29g,0.974mmol,1.1当量)在DMA(2mL)中的溶液。在40℃加热反应物5小时。用水(30mL)猝灭反应物,然后滤出固体。将固体再溶解于DCM/MeOH(20:1,30mL)中,用水(20mL)洗涤,用硫酸镁干燥,过滤并浓缩。粗制的残余物(0.78g)通过RPHPLC(ACN/H2O)纯化,得到2l(0.43g,0.43mmol,49%收率)。LCMS(8分钟方法,5-98%)=5.4分钟。观测质量(ESI+):991.7(M+H)+。
实施例8.

向2q(0.14g,0.27mmol)在DMA(3mL)中的溶液中加入碳酸钾(0.11g,0.81mmol),然后加入还原的IGN单体A(0.084g,0.28mmol)在DMA(1mL)中的溶液。在室温下搅拌反应物12小时。用水(20mL)猝灭反应物,并搅拌混合物10分钟。将固体过滤,然后溶解于DCM/MeOH(9/1,20mL)中并用盐水(10mL)洗涤。将有机层分离并经硫酸镁干燥,过滤并真空除去溶剂。粗料通过硅胶色谱法使用DCM(MeOH/EtOAc,1/4)纯化得到期望的化合物2s(0.08g,0.11mmol,40%收率)。UPLCMS(2.5分钟方法)=1.63分钟。观测质量(ESI+):735.2(M+H)+。

向2s(0.06g,0.09mmol)在DMA(2mL)中的溶液中加入碳酸钾(0.025g,0.18mmol),然后加入碘化钾(0.007g,0.044mmol)。在室温下,向反应混合物中加入IGN单体A(0.03g,0.097mmol)在DMA(1mL)中的溶液。在40℃下加热反应物5小时。将反应物冷却并用水(20mL)猝灭,并滤出固体。将固体再溶解于DCM/MeOH(20:1,20mL)中,用水(10mL)洗涤,用硫酸镁干燥,过滤并浓缩。粗制的残余物(0.07g)通过RPHPLC(ACN/H2O)纯化,得到2l(0.035g,0.035mmol,51%收率)。LCMS(8分钟方法,5-98%)=5.4分钟。观测质量(ESI+):991.7(M+H)+。
实施例9.

在室温下,向2h(0.85g,1.027mmol)在THF(9mL)中的溶液中加入DIPEA(0.54mL,3.1mmol,3.0当量),然后加入HF-吡啶(0.3mL,2.053mmol,2.0当量)。在室温下搅拌反应物3小时。用饱和碳酸氢钠(10mL)猝灭反应物,分离各层且用DCM(3×10mL)萃取水层。合并的有机层用盐水(10mL)洗涤,经硫酸镁干燥并过滤。真空除去溶剂得到呈固体的粗制产物,将其用EtOAc洗涤得到期望的产物2t(0.64g,0.89mmol,87%收率)。UPLCMS(2.5分钟方法)=1.36分钟。观测质量(ESI+):714.6(M+H)+。

在0℃下,向2t(0.23g,0.322mmol)在二氯甲烷(3mL)中的溶液中加入DIPEA(0.11ml,0.644mmol,2.0当量),然后加入甲磺酸酐(0.084g,0.48mmol,1.5当量)在DCM(1mL)中的溶液。搅拌反应物1小时。将反应物用水(3mL)猝灭并用DCM(3mL)稀释。分离各层,且有机层用盐水(3mL)洗涤,经硫酸镁干燥并过滤。真空除去溶剂,且粗料2n(0.25g,0.31mmol,98%收率)不经进一步纯化而用于下一步。UPLCMS(2.5分钟方法)=1.45分钟。观测质量(ESI+):792.5(M+H)+。

向2n(0.02g,0.027mmol)在DMF(0.2ml)中的溶液中加入碳酸钾(0.007g,0.053mmol,2.0当量),然后加入还原的IGN单体A(0.009g,0.029mmol,1.1当量),并在室温下搅拌反应物18小时。向反应混合物中加入水(3mL),并过滤所得固体。将固体再溶解于DCM/MeOH(20:1,5mL)中,用水(5mL)洗涤,用硫酸镁干燥,过滤并浓缩。粗制的残余物通过RPHPLC(ACN/H2O)纯化,得到2l(0.005g,0.005mmol,19%收率)。LCMS(8分钟方法,5-98%)=5.4分钟。观测质量(ESI+):991.7(M+H)+。
实施例10.

向2t(0.02g,0.031mmol)在THF(2mL)中的溶液中加入DIPEA(0.016mL,0.092mmol,3.0当量),然后加入二溴三苯基正膦(0.03g,0.062mmol,2.0当量)在THF(0.5mL)中的溶液。在室温下搅拌反应物12小时。通过蒸发溶剂使反应停止,然后粗料通过硅胶色谱法纯化,得到2u(0.006g,0.007mmol,25%收率)。UPLCMS(2.5分钟方法)=1.56分钟。观测质量(ESI+):778.2(M+H)+。

向2u(0.006g,7.73μmol)在DMA(1mL)中的溶液中加入还原的IGN单体A(0.003g,9.27μmol),接着加入碳酸钾(0.002g,0.015mmol),并在室温下搅拌反应物18小时。向反应混合物中加入水(3mL),且过滤所得固体并用水洗涤。将固体再溶解于DCM/MeOH(20:1,5mL)中,用水(5mL)洗涤,用硫酸镁干燥,过滤并浓缩。粗制的残余物通过RPHPLC(ACN/H2O)纯化,得到2l(0.001g,0.001mmol,13%收率)。LCMS(8分钟方法,5-98%)=5.4分钟。观测质量(ESI+):991.7(M+H)+。
实施例11.

向(5-硝基-1,3-亚苯基)二甲醇3a(4.0g,21.84mmol)在DCM(40mL)和DMF(5mL)中的溶液中加入DIPEA(3.86mL,21.84mmol,1.0当量),然后加入TBSCl(3.29g,21.84mmol,1.0当量)在DMF(5mL)中的溶液。在0℃下搅拌反应物1小时。用饱和氯化铵(20mL)猝灭反应物并分离各层。用DCM(2×20mL)萃取水层,且将合并的有机层用水(2×50mL)、盐水洗涤,经硫酸镁干燥,过滤并真空除去溶剂,得到粗制的黄色油状物。粗制的产物通过硅胶色谱法(DCM/MeOH)纯化,得到期望的产物3b(3.69g,12.41mmol,57%收率)。UPLCMS(2.5分钟方法)=1.96分钟。观测质量(ESI+):298.5(M+H)+。

在0℃下,向3b(2.0g,6.72mmol)在DMF(50mL)中的溶液中加入吡啶(1.6ml,20.17mmol,3.0当量),然后加入甲磺酰氯(1.1mL,13.45mmol,2.0当量)。将反应物升温至室温并搅拌3小时。用饱和碳酸氢钠(20mL)猝灭反应物并分离各层。用EtOAc(3×30mL)萃取水层。将合并的有机层用水(2×100mL)、盐水(100mL)洗涤,经硫酸镁干燥并过滤。真空除去溶剂并将粗料3c(2.0g,6.7mmol,94%收率)以粗品用于下一步。UPLCMS(2.5分钟方法)=2.22分钟。观测质量(ESI+):316.7(M+H)+。

向3c(2.0g,6.33mmol)在THF(38.9mL)中的溶液中加入DIPEA(5.5mL,31.6mmol,5.0当量),然后加入HF-吡啶(2.7mL,19.0mmol,3.0当量),并在室温下搅拌反应物2小时。然后用饱和碳酸氢钠(100mL)猝灭反应物。分离各层,然后用EtOAc(3×20mL)萃取水层。然后将合并的有机层用水(30mL)、盐水(30mL)洗涤,经硫酸镁干燥并过滤。真空除去过量的溶剂,得到期望的产物3d(1.1g,5.46mmol,86%收率)。UPLCMS(2.5分钟方法)=1.31分钟。观测质量(ESI+):202.4(M+H)+。

在0℃下,向3d(1.0g,4.96mmol)在DCM(10mL)中的溶液中加入DIPEA(2.6mL,14.9mmol,3.0当量),然后向反应混合物中加入甲磺酸酐(1.1g,6.45mmol,1.3当量)在DCM中的溶液。搅拌反应物1小时。用水(10mL)猝灭反应物并分离各层,且用DCM(2×20mL)萃取水层。将合并的有机层用饱和碳酸氢钠(10mL)、盐水(20mL)洗涤,经硫酸镁干燥并过滤。真空除去溶剂,且粗料3e(1.3g,4.65mmol,94%收率)不经进一步纯化而用于下一步。UPLCMS(2.5分钟方法)=1.51分钟。观测质量(ESI+):280.6(M+H)+。

在室温下,向3e(0.4g,1.43mmol)和碳酸钾(0.6g,4.29mmol,3.0当量)在DMA(13.4mL)中的溶液中加入IGN单体A(0.46g,1.57mmol,1.1当量)在DMA(2mL)中的溶液,并搅拌反应物5小时。用水(30mL)猝灭反应物,分离各层,并用EtOAc(3×30mL)萃取水层。将合并的有机层用水(30mL)、盐水(30mL)洗涤,经硫酸镁干燥并真空除去溶剂。粗制的油状物通过硅胶色谱法使用DCM/MeOH纯化得到化合物3f(0.37g,0.77mmol,54%收率)。UPLCMS(2.5分钟方法)=1.69分钟。观测质量(ESI+):478.3(M+H)+。

向3f(0.11g,0.23mmol)在DMA(3.0mL)中的溶液中加入碳酸钾(0.095g,0.69mmol,3.0当量),然后加入碘化钾(0.02g,0.11mmol,0.5当量)。加入还原的IGN单体A(0.07g,0.25mmol,1.1当量)在DMA(1mL)中的溶液。然后在35℃下温和地加热反应物5小时。用水猝灭反应物,并滤出固体。将固体再溶解于DCM/MeOH(20:1)中,用水洗涤,用硫酸镁干燥,过滤并浓缩。粗制的残余物(0.13g)通过RPHPLC(ACN/H2O)纯化,得到3g(0.063g,0.085mmol,36%收率)。UPLCMS(2.5分钟方法)=1.79分钟。观测质量(ESI+):738.3(M+H)+。1H NMR(400MHz,DMSO-d6,报道为水加合物的混合物)1H NMR(400MHz,DMSO-d6):δ8.43–8.36(m,2H),8.27(d,J=8.1Hz,1H),8.13–8.02(m,2H),7.44–7.14(m,6H),7.14–6.99(m,2H),6.79(s,0.5H),6.56(s,0.5H),6.50(d,J=2.2Hz,1H),6.39(d,J=6.9Hz,1H),6.17(d,J=6.8Hz,0.5H),5.69(s,0.5H),5.59(d,J=5.7Hz,0.5H),5.47–5.27(m,4H),5.03(t,J=6.1Hz,0.5H),4.77(dd,J=9.1,6.8Hz,0.5H),4.61(dt,J=9.7,5.1Hz,0.15H),4.50–4.39(m,0.5H),4.27(dd,J=10.9,4.2Hz,0.5H),4.16(td,J=9.6,2.9Hz,0.5H),3.95(s,0.5H),3.89–3.76(m,6H),3.76–3.44(m,4H),3.20–3.08(m,1H),2.96(dd,J=17.0,4.4Hz,1H)。
实施例12.

向3e(0.45g,1.61mmol)在DMA(15.1mL)中的溶液中加入碳酸钾(0.67g,4.83mmol,3.0当量),然后加入还原的IGN单体A(0.5g,1.69mmol,1.1当量)在DMA(2mL)中的溶液。在室温下搅拌反应物5小时。用水(30mL)猝灭反应物,并搅拌混合物10分钟。将固体过滤,然后溶解于DCM/MeOH(9/1,30mL)中并用盐水(20mL)洗涤。将有机层分离并经硫酸镁干燥,过滤并真空除去溶剂。粗料通过硅胶色谱法使用己烷/EtOAc纯化,得到呈无色油状物的化合物3h(0.28g,0.58mmol,36%收率)。UPLCMS(2.5分钟方法)=1.82分钟。观测质量(ESI+):480.3(M+H)+。

向3h(0.27g,0.56mmol)在DMA(10mL)中的溶液中加入碳酸钾(0.16g,1.12mmol,2.0当量),然后加入碘化钾(0.05g,0.28mmol,0.05当量)。在室温下,向反应混合物中加入IGN单体A(0.18g,0.62mmol,1.1当量)在DMA(2mL)中的溶液。然后在40℃下搅拌反应物3小时。用水(20mL)猝灭反应物,且滤出固体并用水洗涤。将粗制的黄色固体溶解于DCM/MeOH(9/1,30mL)中,然后用水(10mL)洗涤,经硫酸镁干燥并过滤。真空除去溶剂得到粗制的黄色固体。粗制产物通过硅胶色谱法使用DCM/MeOH(0%至5%MeOH/DCM)纯化,得到呈黄色粉末的产物3g(0.35g,0.48mmol,86%收率)。UPLCMS(2.5分钟方法)=1.79分钟(2.5分钟方法)。观测质量(ESI+):738.4(M+H)+。
实施例13.

向3f(0.15g,0.31mmol)在DCE(2mL)中的溶液中加入三乙酰氧基硼氢化钠(0.067g,0.31mmol,1.0当量),并在室温下搅拌反应物1小时。用饱和氯化铵(1mL)猝灭反应物,然后分离各层。用DCM(3×10mL)萃取水层并将合并的有机层用盐水(20mL)洗涤,经硫酸镁干燥,过滤并真空除去溶剂。粗制的棕色油状物通过硅胶色谱法纯化得到期望的产物3h(0.08g,0.16mmol,52%收率)。UPLCMS(2.5分钟方法)=1.80分钟。观测质量(ESI+):480.5(M+H)+。

向3h(0.07g,0.16mmol)在DMA(2mL)中的溶液中加入碳酸钾(0.07g,0.47mmol,3.0当量),然后加入碘化钾(0.013g,0.08mmol,0.05当量),然后加入IGN单体A(0.05g,0.17mmol,1.1当量)在DMA(0.5mL)中的溶液。在室温下搅拌反应物12小时。向混合物中加入水(20mL),并搅拌混合物10分钟,此时过滤固体。将固体溶解于DCM(10mL)中,然后用盐水(10mL)洗涤。有机层经硫酸镁干燥并过滤。除去溶剂,得到黄色油状物(0.09g,0.12mmol,80%收率)。UPLCMS(2.5分钟方法)=1.79分钟(2.5分钟方法)。观测质量(ESI+):738.5(M+H)+。
实施例14.

在0℃下,向3b(1.00g,3.4mmol)在DCM(33mL)中的溶液中加入DIPEA(1.781ml,10.09mmol,3.0当量),接着加入甲磺酸酐(0.703g,4.03mmol,1.2当量)的溶液。搅拌反应物1小时。蒸发溶剂得到粗制的产物3j(1.2g,3.2mmol,95%收率),其不经进一步纯化而用于下一步。UPLCMS(2.5分钟方法)=2.04分钟。观测质量(ESI+):376.5(M+H)+。

在室温下,向3j(1.24g,3.30mmol)在DMF(26mL)中的溶液中加入碳酸钾(0.91g,6.60mmol,2.0当量),然后加入IGN单体A(0.97g,3.30mmol,1.0当量),历时12小时。用水(60mL)猝灭反应物并滤出固体,然后将其溶解于DCM/MeOH(20/1,20mL)中。有机层用盐水洗涤,经硫酸镁干燥并过滤。真空除去溶剂,且粗料用硅胶色谱法纯化得到期望的产物3k(1.3g,2.27mmol,69%收率)。UPLCMS(2.5分钟方法)=2.12分钟(2.5分钟方法)。观测质量(ESI+):574.4(M+H)+。

将3k(0.63g,1.1mmol)溶解于无水DCE(11mL)中。加入三乙酰氧基硼氢化钠(0.70g,3.3mmol,3.0当量),并在室温下搅拌反应混合物1小时。用饱和氯化铵(10mL)猝灭混合物。分离各层,且用DCM(2×20mL)萃取水层。将合并的有机层用盐水(20mL)洗涤,用无水硫酸镁干燥,过滤并浓缩,得到3l(0.58g,1.0mmol,92%收率)。UPLCMS(8.0分钟方法)=7.797分钟(8.0分钟方法)。观测质量(ESI+):576.3(M+H)+。

将3l(0.58g,1.0mmol)的溶液溶解于无水THF(5mL)中,并加入5M盐酸水溶液(2.01mL,10.07mmol)。在室温下搅拌混合物2小时。用饱和碳酸氢钠(5mL)猝灭反应物,且分离各层并用DCM(2×10mL)萃取水层。合并的有机层用盐水(20mL)洗涤,经硫酸镁干燥并浓缩得到亮橙色固体。所得固体通过硅胶色谱法(DCM/MeOH)纯化得到化合物3m(0.33g,0.71mmol,71%收率)。UPLCMS(8.0分钟方法)=5.166分钟。观测质量(ESI+):462.1(M+H)+。

将3m(0.1g,0.22mmol)溶解于无水DCM(1.5mL)和无水DMF(0.7mL)中。将反应物冷却至0℃,且加入三乙胺(0.12mL,0.88mmol)和甲磺酸酐(0.08g,0.44mmol)。在0℃下搅拌反应物1小时。将反应混合物用乙酸乙酯(20mL)稀释,用水(2×20mL)洗涤,经硫酸镁干燥,过滤并浓缩。化合物最初通过硅胶色谱法(DCM/EtOAc)纯化,然后另外通过RPPHPLC(MeCN/水)纯化,得到期望的产物3n(0.041g,0.076mmol,34%收率)。观测质量(ESI+):540.3(M+H)+。

将化合物3n(0.041g,0.076mmol)和IGN单体A(0.027g,0.091mmol)溶解于无水DMA(0.5mL)中。加入碳酸钾(0.012g,0.091mmol)和碘化钾(0.006g,0.038mmol)并搅拌混合物12小时。向反应混合物中加入水(5mL)。滤出固体,然后将其再溶解于DCM(20mL)中并用水(10mL)洗涤。在经硫酸镁干燥,过滤并浓缩之后,固体通过RPHPLC(ACN/H2O)纯化得到3g(0.012g,0.016mmol,21%收率)。UPLCMS(2.5分钟方法)=1.79分钟。观测质量(ESI+):738.5(M+H)+。

将化合物3g(0.017g,0.023mmol)溶解于无水THF(1mL)、无水MeOH(0.5mL)和水(0.1mL)中。加入氯化铵(0.012g,0.23mmol,10.0当量)和铁(0.006g,0.115mmol,5.0当量)。在60℃下搅拌混合物2小时。将反应混合物冷却至室温,通过硅藻土过滤,并用20%MeOH/DCM(10mL)冲洗。浓缩滤液,且粗制产物通过硅胶色谱法(DCM/MeOH)纯化得到呈白色固体的化合物3o(0.012g,0.018mmol,76%收率)。UPLCMS(2.5分钟方法)=1.84分钟。观测质量(ESI+):708.5(M+H)+。1H NMR(400MHz,DMSO-d6,报道为水加合物的混合物,T=330K):δ8.26(d,J=7.9Hz,1H),8.17(d,J=7.8Hz,1H),8.03(d,J=4.5Hz,1H),7.49(s,1H),7.42–7.33(m,2H),7.36–7.08(m,4H),7.09–6.95(m,2H),6.76–6.64(m,3H),6.47(s,1H),6.15(d,J=6.5Hz,1H),5.11(m,2H),4.98(m,2H),4.58(dt,J=9.9,4.7Hz,1H),4.47–4.36(m,1H),3.87(m,1H),3.76(s,3H).3.71–3.46(m,4H),3.39–3.28(m,1H),2.93(dd,J=16.8,4.7Hz,1H)。
实施例15.

在0℃下,向4a(5.6g,12.9mmol,1.0当量)在DCM(83mL)中的溶液中加入DIPEA(6.77mL,38.7mmol,3.0当量),然后加入TBS-Cl(2.336g,15.50mmol,1.2当量)在DCM(10mL)中的溶液。在室温下搅拌反应物3小时。用饱和氯化铵(30mL)猝灭反应物并分离各层。用DCM(2×30mL)萃取水溶液,且将合并的有机层用水(2×50mL)、盐水(50mL)洗涤,经硫酸镁干燥,过滤并真空除去溶剂,得到粗制的黄色油状物。粗制的产物通过硅胶色谱法(己烷/EtOAc)纯化,得到期望的产物4b(3.0g,5.48mmol,43%收率)。UPLCMS(2.5分钟方法)=2.29分钟。观测质量(ESI+):549.0(M+H)+。

在0℃下,向4b(3.00g,5.48mmol,1.0当量)在DMF(30mL)中的溶液中加入吡啶(1.33mL,16.4mmol,3.0当量),然后加入甲磺酰氯(0.64mL,8.21mmol,1.5当量)。将反应物搅拌1小时,并用饱和碳酸氢钠(30mL)猝灭,并用EtOAc(40mL)稀释。分离各层,且用EtOAc(2×30mL)萃取水层。将合并的有机层用水(2×40mL)、盐水(40mL)洗涤,经硫酸镁干燥,过滤,且真空除去溶剂,得到粗制的产物4c(2.5g,4.41mmol,81%收率)。UPLCMS(10.0分钟方法)=8.23分钟。观测质量(ESI+):567.6(M+H)+。

向4c(2.5g,4.41mmol,1.0当量)在THF(43mL)中的溶液中加入DIPEA(2.46mL,14.1mmol,4.0当量),然后加入HF-吡啶(1.48mL,10.6mmol,3.0当量),并在室温下搅拌反应物2小时。用饱和碳酸氢钠(100mL)猝灭反应物并分离各层。用EtOAc(3×20mL)萃取水层,并将合并的有机层用水(30mL)、盐水(30mL)洗涤,经硫酸镁干燥并过滤。真空除去溶剂,得到期望的产物4d(0.9g,2.0mmol,56%收率)。UPLCMS(10.0分钟方法)=5.20分钟。观测质量(ESI+):435.4(M+H)+。

在0℃下,向4d(0.9g,2.0mmol,1.0当量)在DCM(10mL)中的溶液中加入DIPEA(0.69mL,3.98mmol,2.0当量),然后加入甲磺酸酐(0.52g,2.99mmol,1.5当量)在DCM(2mL)中的溶液。搅拌反应物1小时。用水(10mL)猝灭反应物,分离各层,且用DCM(2×20mL)萃取水层。将合并的有机层用饱和碳酸氢钠(10mL)、盐水(20mL)洗涤,经硫酸镁干燥并过滤。真空除去溶剂,且粗料4e(1.0g,1.88mmol,95%收率)不经进一步纯化而用于下一步。UPLCMS(10分钟方法)=5.7分钟。观测质量(ESI+):531.4(M+H)+。

向4e(0.21g,0.39mmol,1.0当量)在DMA(2.0mL)中的溶液中加入碳酸钾(0.16g,1.19mmol,3.0当量),然后加入还原的IGN单体A(0.12g,0.41mmol,1.05当量)在DMA(1mL)中的溶液。在室温下搅拌反应物5小时。用水(30mL)猝灭反应物,并搅拌混合物10分钟。将固体过滤并溶解于DCM/MeOH(9/1,30mL)中,并用盐水(20mL)洗涤。将有机层分离并经硫酸镁干燥,过滤并真空除去溶剂。粗料通过硅胶色谱法(己烷/EtOAc)纯化,得到呈无色油状物的期望的产物4f(0.11g,0.15mmol,38%收率)。UPLCMS(10分钟方法)=6.55分钟。观测质量(ESI+):730.9(M+H)+。

将4f(0.11g,0.15mmol,1.0当量)和IGN单体A(0.053g,0.18mmol)的溶液溶解于无水DMA(1.0mL)中。加入碳酸钾(0.041g,0.30mmol)和碘化钾(0.025g,0.15mmol)。在40℃下搅拌混合物4小时。向反应混合物中加入水(5mL),且滤出固体,然后将其再溶解于DCM(20mL)中并用水(10mL)洗涤。将有机层经硫酸镁干燥,过滤并浓缩。粗固体通过硅胶色谱(己烷/EtOAc)纯化,得到4g(0.099g,0.10mmol,66%收率)。UPLCMS(10分钟方法)=6.38分钟。观测质量(ESI+):988.7(M+H)+。1H NMR(400MHz,DMSO-d6):δ8.22(d,J=7.9Hz,1H),8.12(d,J=8.1Hz,1H),8.03(d,J=4.5Hz,1H),7.38–7.25(m,2H),7.24(t,J=7.9Hz,2H),7.24–7.06(m,2H),7.11–6.94(m,1H),6.98(s,1H),6.91(d,J=15.2Hz,2H),6.79(s,1H),6.45(s,1H),6.32(d,J=6.8Hz,1H),5.18(q,J=12.3Hz,2H),5.01(m,2H),4.54(dt,J=9.7,5.2Hz,1H),4.37(dt,J=10.6,5.4Hz,1H),3.87(s,3H),3.71(s,3H),3.60(m,6H),3.57–3.50(m,2H),3.47(qd,J=4.3,1.0Hz,4H),3.47(s,3H),3.42–3.33(m,2H),3.32–3.16(m,2H),3.21(s,3H),2.97–2.85(m,1H),2.44(s,3H),1.30(s,6H)。
实施例16.

向4e(0.52g,0.98mmol,1.0当量)和碳酸钾(0.41g,2.94mmol,3.0当量)在DMA(10ml)中的溶液中加入IGN单体A(0.30g,1.03mmol,1.05当量)在DMA(2mL)中的溶液,并搅拌反应物5小时。用水(30mL)猝灭反应物,分离各层,且用EtOAc(3×30mL)萃取水层。将合并的有机层用水(30mL)、盐水(30mL)洗涤,经硫酸镁干燥并真空除去过量的溶剂。粗制的油状物通过硅胶色谱法(己烷/EtOAc)纯化,得到期望的产物4h(0.35g,0.48mmol,49%收率)。UPLCMS(10分钟方法)=6.19分钟。观测质量(ESI+):728.7(M+H)+。

向4h(0.18g,0.25mmol,1.0当量)在DMA(5.0mL)中的溶液中加入碳酸钾(0.10g,0.74mmol,3.0当量),然后加入碘化钾(0.04g,0.2mmol,1.0当量)。加入还原的IGN单体A(0.08g,0.27mmol,1.1当量)在DMA(1mL)中的溶液,然后在40℃加热反应物5小时。用水猝灭反应物混合物,然后滤出固体。将固体再溶解于DCM/MeOH(20:1)中,用水洗涤,经硫酸镁干燥,过滤并浓缩。粗制的残余物通过硅胶色谱法(己烷/EtOAc)纯化得到化合物4g(0.05g,0.05mmol,21%收率)。UPLCMS(10分钟方法)=6.39分钟。观测质量(ESI+):989.0(M+H)+。
实施例17.

将化合物4h(0.17g,0.24mmol,1.0当量)溶解于无水DCE(3mL)中,并在室温下加入三乙酰氧基硼氢化钠(0.10g,0.48mmol,3.0当量)。搅拌反应混合物1小时。用饱和氯化铵(10mL)猝灭混合物。分离各层,且用DCM(2×20mL)萃取水层。将合并的有机层用盐水(20mL)洗涤,用无水硫酸镁干燥,过滤并浓缩,得到4f(0.13g,0.18mmol,77%收率),其不经进一步纯化而用于下一步。UPLCMS(2.5分钟方法)=2.13分钟。观测质量(ESI+):731.2(M+H)+。

将化合物4f(0.19g,0.26mmol,1.0当量)和IGN单体A(0.084g,0.28mmol,1.1当量)溶解于无水DMA(4.0mL)中。加入碳酸钾(0.11g,0.78mmol,3.0当量)和碘化钾(0.043g,0.26mmol,1.0当量)。在40℃下搅拌混合物4小时。向反应混合物中加入水(5mL)。将固体滤出,且再溶解于DCM(20mL)中并用水(10mL)洗涤。在经硫酸镁干燥,过滤并浓缩之后,固体通过硅胶色谱法(己烷/EtOAc)纯化,得到4g(0.065g,0.06mmol,25%收率)。UPLCMS(10分钟方法)=6.38分钟。观测质量(ESI+):988.7(M+H)+。
实施例18.

将化合物2p(0.03g,0.066mmol,1.0当量)和IGN单体A(0.021g,0.072mmol,1.1当量)溶解于THF(0.65mL)和DMF(0.3mL)中。加入三苯基膦(0.021g,0.079mmol,1.2当量),然后缓慢加入DIAD(0.015mL,0.079mmol,1.2当量)。在室温下在氩气下搅拌反应物2小时。浓缩反应混合物并加入水(约2mL)以研磨产物。过滤沉淀物,并用水洗涤剩余的固体。粗制的残余物通过RPHPLC(C18柱,MeCN/水,梯度:40%至60%)纯化,得到呈白色蓬松固体的化合物2r(0.015g,0.02mmol,31%收率)。UPLCMS(2.5分钟方法)=1.62分钟。观测质量(ESI+)=732.9(M+H)+
实施例19.

将化合物2p(0.03g,0.066mmol,1.0当量)和还原的IGN单体A(0.02g,0.072mmol,1.1当量)溶解于THF(0.66mL)和DMF(0.1mL)中。加入三苯基膦(0.021g,0.079mmol,1.2当量),然后缓慢加入DIAD(0.015mL,0.079mmol,1.2当量)。在室温下在氩气下搅拌反应混合物2小时。将该反应混合物用DCM稀释,并用水(2x)洗涤。将有机层经硫酸镁干燥,过滤并浓缩。粗制的残余物通过RPHPLC(C18柱,MeCN/水,梯度:40%至65%)纯化,产生呈白色蓬松固体的2s(0.017g,0.02mmol,35%收率)。UPLCMS(2.5分钟方法)=1.71分钟。观测质量(ESI+)=735.4(M+H)+。
实施例20.

将化合物3d(0.03g,0.149mmol,1.0当量)和IGN单体A(0.046g,0.156mmol,1.05当量)溶解于THF(1.5mL)和DMF(0.3mL)中。加入三苯基膦(0.047g,0.179mmol,1.2当量),然后缓慢加入DIAD(0.032mL,0.164mmol,1.1当量)。在室温下在氩气下搅拌反应物12小时。浓缩反应混合物并加入水(约2mL)以研磨产物。过滤沉淀物,并用水洗涤剩余的固体。粗制的残余物通过硅胶色谱法(己烷/EtOAc)纯化得到呈白黄色固体的化合物3f(0.013g,0.027mmol,18%收率)。UPLCMS(2.5分钟方法)=1.80分钟。观测质量(ESI+)=478.4(M+H)+。
实施例21.

将化合物3d(0.03g,0.149mmol,1.0当量)和还原的IGN单体A(0.046g,0.156mmol,1.05当量)溶解于THF(1.5mL)。加入三苯基膦(0.047g,0.179mmol,1.2当量),然后缓慢加入DIAD(0.032mL,0.164mmol,1.1当量)。在室温下在氩气下搅拌反应物2小时。将反应混合物浓缩并与甲苯(2×)共蒸发。粗制的残余物通过硅胶色谱法(己烷/EtOAc)纯化得到呈橙黄色固体的化合物3h(0.055g,0.115mmol,77%收率)。UPLCMS(2.5分钟方法)=1.90分钟。观测质量(ESI+)=480.5(M+H)+。
实施例22.

向2d(0.024g,0.078mmol,1.1当量)在DCM(1mL)中的溶液中加入EEDQ(0.019g,0.078mmol,1.1当量)。搅拌反应物5分钟,并加入MeOH(0.1mL),然后加入3o(0.05g,0.071mmol)在DCM(1mL)中的溶液。在室温下搅拌反应物2小时或直至原料完成。浓缩反应物以形成白色沉淀物,向其中加入MTBE(5mL),且在室温下搅拌所得反应物30分钟。滤出固体得到化合物2l,然后通过RPHPLC(C18柱,MeCN/水)纯化得到2l(0.023g,0.023mmol,33%收率)。UPLCMS(2.5分钟方法)=1.75分钟。观测质量(ESI+)=993.2(M+H)+。
实施例23.

向2p(0.05g,0.110mmol,1.0当量)在DMA(1mL)中的溶液中加入四溴化碳(0.044g,0.132mmol,1.2当量),然后加入三苯基膦(0.043g,0.164mmol,1.5当量),并在室温下搅拌反应物2小时。除去溶剂,得到白色固体,将其用MTBE研磨并滤出固体,得到化合物2v(0.03g,0.058mmol,57%收率,52%纯度),其不经进一步纯化即用于下一步。UPLCMS(2.5分钟方法)=1.59分钟。观测质量(ESI+)=518.2(M+H)+。

向2v(0.03g,0.043mmol,1.0当量)在DMA(0.5mL)中的溶液中加入碳酸钾(0.012g,0.087mmol,2.0当量),然后加入IGN单体A(0.013g,0.046mmol,1.05当量)。在室温下搅拌反应混合物4小时。用水(5mL)稀释反应混合物并滤出固体。将固体溶解于DCM/MeOH(9/1,2mL)中。将有机层用水(10mL)、盐水(10mL)洗涤,并经硫酸镁干燥。在过滤并除去溶剂之后,粗制的产物通过RPHPLC(C18柱,MeCN/水)纯化得到2r(0.011g,0.015mmol,35%收率)。UPLCMS(2.5分钟方法)=1.62分钟。观测质量(ESI+)=733.3(M+H)+。
实施例24.

向化合物2v(14.7g,0.052mol,1.0当量,如文献中所述制备,参见:BeilsteinJ.Org.Chem.2014,10,535-543)在DCM和(100mL)DMF(1mL)中的浆液中一次性装入SOCl2(12.6g,0.104mol,2.0当量)。在35℃下搅拌所得溶液过夜,产生暗褐色浆液。过滤浆液并干燥固体,得到7.5g的灰白色固体。NMR显示Boc保护基团的裂解。向深色滤液中装入固体碳酸钠(10.6g,0.1mol),然后通过进一步加入碳酸氢钠缓冲至pH为约6-7。向所得溶液中加入Boc2O(12.7g,0.058mol,1.1当量)并搅拌0.5小时。将过滤的固体(7.5g)加入到反应混合物中,然后加入Boc2O(6.5g,0.030mol,1.7当量)(pH约6)并在室温下继续搅拌过夜。然后加入饱和碳酸氢钠(10mL)以达到pH为6至7。再加入Boc2O(9.3g,42.6mmol)和DMAP(0.2g,1.63mmol)并继续搅拌过夜。过滤深色反应物以除去一些沉淀物。将DCM层用1N HCl洗涤以除去un-Boc产物,将其碱化并用DCM萃取,并回收3.0g无色松脆固体(un-Boc产物)。将DCM层用盐水洗涤并浓缩成深色淤泥。粗制的产物通过硅胶色谱法(EtOAc/己烷)纯化,得到呈浅棕色固体的2w(9.5g,0.031mmol,62%收率)。1H NMR(400MHz,CDCl3):δ7.84(m,2H),7.75(m,1H),6.60(s,1H,NH),4.58(s,2H),3.91(s,3H),1.53(s,9H)。

将LAH/THF(0.6M,60mL,1.15当量)的溶液在室温下搅拌30分钟,然后用丙酮-干冰浴冷却至-65℃。分批缓慢加入化合物2w(9.3g,0.031mol,1.0当量)(Ti约-60℃,)产生黄棕色浆液,将其搅拌4小时。用水(1.3mL)、15%NaOH(1.3mL)和水(4mL)猝灭反应物并搅拌20分钟(Ti约5℃。)将反应物过滤并用乙酸乙酯(约90mL)冲洗。将滤液用盐水洗涤并浓缩,产生呈棕色油状物的2x(8.0g,0.029mol,93%收率)。1H NMR(400MHz,CDCl3):δ7.45(s,1H),7.40(s,1H),7.10(s,1H),6.60(s,1H,NH),4.75(s,2H),4.50(s,2H),1.53(s,9H)。

将化合物2x(8.0g,0.029mol,1.0当量)溶解于DCM(20mL)中并在冰水浴中冷却。加入4N HCl/二噁烷(15mL,1.5当量),并将得到的混合物在50℃下加热1小时,然后冷却至室温。浓缩浆液并将溶剂转换为庚烷。将浆液过滤,用己烷冲洗,并在烘箱(60℃)中干燥,得到呈浅棕色固体的2y(5.4g,0.026mol,88%收率)。1H NMR(400MHz,DMSO-d6):δ7.45(s,1H),7.25(s,2H),4.76(s,2H),4.52(s,2H)。

在室温下,向2d(0.969g,3.20mmol,1.1当量)在DCM(25mL)中的溶液中加入EEDQ(0.79g,3.2mmol,1.1当量)。8分钟之后,经1分钟逐滴加入2y(0.5g,2.91mmol,1.0当量)、DIPEA(0.51mL,2.91mmol,1.0当量)在MeOH(5mL)中的溶液。搅拌反应物2小时。用水(30mL)猝灭反应物,分离各层,且用DCM(2×20mL)萃取水层。将合并的有机层用饱和碳酸氢钠(20mL)、盐水(20mL)洗涤,经硫酸镁干燥,过滤并浓缩到留下最少量的溶剂。将所得白色固体在MBTE中稀释并过滤,得到呈白色固体的期望的产物2p(0.64g,1.40mmol,48%收率)。UPLCMS(2.5分钟方法)=1.30分钟。观测质量(ESI+)=456.3(M+H)+。
Claims (35)
1.一种制备式(17A)的化合物

或其盐的方法,所述方法包括使式(14A)的化合物

与式(d1)的单体化合物反应,

其中:
X3为-Cl;且
P3为H或胺保护基团。
2.如权利要求1所述的方法,其中使所述式(14A)的化合物与式(d1)的单体在醇活化剂存在下反应。
3.如权利要求2所述的方法,其中所述醇活化剂为三苯基膦。
4.如权利要求1-3中任一项所述的方法,其中使所述式(14A)的化合物与式(d1)的单体在偶氮二羧酸酯存在下反应。
5.如权利要求4所述的方法,其中所述偶氮二羧酸酯选自由以下组成的组:偶氮二羧酸二乙酯(DEAD)、偶氮二羧酸二异丙酯(DIAD)、1,1'-(偶氮二羰基)二哌啶(ADDP)和偶氮二羧酸二叔丁酯(DTAD)。
6.如权利要求1-3中任一项所述的方法,其中使所述式(14A)的化合物与所述式(d1)的单体化合物反应,其中P3为H,以形成式(17A’)的化合物:

7.如权利要求1-3中任一项所述的方法,其中P3为胺保护基团。
8.如权利要求7所述的方法,所述方法还包括使所述式(17A)的化合物与胺脱保护试剂反应以形成式(17A')的化合物的步骤:

9.如权利要求8所述的方法,其中所述胺脱保护试剂选自由四-正丁基氟化铵、乙酸、氟化氢吡啶、氟化铯、哌啶、吗啉或三氟乙酸组成的组。
10.一种制备式(18A)的化合物

或其药学上可接受的盐的方法,所述方法包括使式(17A)的化合物:

与式(b1)的单体反应:

其中:
X3为-Cl;
P3为H或胺保护基团。
11.如权利要求10所述的方法,其中使所述式(17A)的化合物与式(b1)的单体化合物在碱存在下反应。
12.如权利要求11所述的方法,其中所述碱为碳酸钠、碳酸钾、碳酸铯、氢化钠或氢化钾。
13.如权利要求12所述的方法,其中所述碱为碳酸钾。
14.如权利要求10-13中任一项所述的方法,其中使所述式(17A)的化合物与式(b1)的单体化合物在极性非质子溶剂存在下反应。
15.如权利要求14所述的方法,其中所述极性非质子溶剂为二甲基甲酰胺或二甲基乙酰胺。
16.如权利要求10-13中任一项所述的方法,其中使所述式(17A)的化合物与式(b1)的单体反应,其中P3为H,以形成式(IA)的化合物:

17.如权利要求10-13中任一项所述的方法,其中P3为胺保护基团。
18.如权利要求17所述的方法,其中所述胺保护基团选自由2-三甲基甲硅烷基乙基、(2-苯基-2-三甲基甲硅烷基)乙基、三异丙基甲硅烷氧基、2-(三甲基甲硅烷基)乙氧基甲基、烯丙氧基羰基、9-芴基甲氧基羰基、2-(三甲基甲硅烷基)乙氧基羰基或2,2,2,2-三氯乙氧基羰基组成的组。
19.如权利要求17所述的方法,其中使所述式(18A)的化合物与胺脱保护试剂进一步反应以形成式(IA)的化合物:

20.如权利要求19所述的方法,其中所述胺脱保护试剂选自由四-正丁基氟化铵、乙酸、氟化氢吡啶、氟化铯、哌啶、吗啉或三氟乙酸组成的组。
21.一种制备式(18A)的化合物

或其药学上可接受的盐的方法,所述方法包括以下步骤:
(1)使式(14A)的化合物:

与式(d1)的还原的单体化合物反应,

以形成式(17A)的化合物:

或其盐;和
(2)使所述式(17A)的化合物与式(b1)的单体反应,

以形成式(18A)的化合物或其药学上可接受的盐,其中:
X3为-Cl;且
P3为H或胺保护基团。
22.如权利要求21所述的方法,其中使所述式(17A)的化合物与式(b1)的单体反应,其中P3为H,以形成式(IA)的化合物:

23.如权利要求21所述的方法,其中P3为胺保护基团。
24.如权利要求23所述的方法,其中使所述式(18A)的化合物与胺脱保护试剂进一步反应以形成式(IA)的化合物:

25.如权利要求1-3和21-24中任一项所述的方法,其中所述式(14A)的化合物或其盐通过包括以下步骤的方法制备:
(1)使氯化试剂与式(2A)的化合物反应:

以形成式(13A)的化合物:

或其盐;和
(2)使所述式(13A)的化合物与醇脱保护试剂反应以形成所述式(14A)的化合物或其盐。
26.如权利要求25所述的方法,其中所述式(2A)的化合物通过使式(1A)的化合物与醇保护试剂反应来制备

27.如权利要求22或24所述的方法,其中使所述式(IA)的化合物与还原剂反应以形成式(IB)的化合物:

或其药学上可接受的盐。
28.如权利要求27所述的方法,其中所述还原剂选自由以下组成的组:氢气、连二亚硫酸钠、硫化钠、氯化锡、氯化钛(II)、碘化锌、碘化铁和碘化钐。
29.如权利要求27所述的方法,其中所述还原剂为Fe/NH4Cl或Zn/NH4Cl。
30.如权利要求16所述的方法,其中使所述式(IA)的化合物与还原剂反应以形成式(IB)的化合物:

或其药学上可接受的盐。
31.如权利要求30所述的方法,其中所述还原剂选自由以下组成的组:氢气、连二亚硫酸钠、硫化钠、氯化锡、氯化钛(II)、碘化锌、碘化铁和碘化钐。
32.如权利要求30所述的方法,其中所述还原剂为Fe/NH4Cl或Zn/NH4Cl。
33.如权利要求19所述的方法,其中使所述式(IA)的化合物与还原剂反应以形成式(IB)的化合物:

或其药学上可接受的盐。
34.如权利要求33所述的方法,其中所述还原剂选自由以下组成的组:氢气、连二亚硫酸钠、硫化钠、氯化锡、氯化钛(II)、碘化锌、碘化铁和碘化钐。
35.如权利要求33所述的方法,其中所述还原剂为Fe/NH4Cl或Zn/NH4Cl。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110219412.4A CN113004288A (zh) | 2015-07-21 | 2016-07-21 | 制备细胞毒性苯并二氮杂䓬衍生物的方法 |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562195023P | 2015-07-21 | 2015-07-21 | |
| US62/195,023 | 2015-07-21 | ||
| US201662327973P | 2016-04-26 | 2016-04-26 | |
| US62/327,973 | 2016-04-26 | ||
| PCT/US2016/043414 WO2017015502A1 (en) | 2015-07-21 | 2016-07-21 | Methods of preparing cytotoxic benzodiazepine derivatives |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202110219412.4A Division CN113004288A (zh) | 2015-07-21 | 2016-07-21 | 制备细胞毒性苯并二氮杂䓬衍生物的方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN108290895A CN108290895A (zh) | 2018-07-17 |
| CN108290895B true CN108290895B (zh) | 2021-03-19 |
Family
ID=56557925
Family Applications (5)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201680051138.1A Active CN108290895B (zh) | 2015-07-21 | 2016-07-21 | 制备细胞毒性苯并二氮杂*衍生物的方法 |
| CN201680053120.5A Pending CN108055844A (zh) | 2015-07-21 | 2016-07-21 | 制备细胞毒性苯并二氮杂*衍生物的方法 |
| CN202110219412.4A Pending CN113004288A (zh) | 2015-07-21 | 2016-07-21 | 制备细胞毒性苯并二氮杂䓬衍生物的方法 |
| CN202110366892.7A Pending CN113087763A (zh) | 2015-07-21 | 2016-07-21 | 制备细胞毒性苯并二氮杂䓬衍生物的方法 |
| CN201680053093.1A Active CN108026103B (zh) | 2015-07-21 | 2016-07-21 | 制备细胞毒性苯并二氮杂䓬衍生物的方法 |
Family Applications After (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201680053120.5A Pending CN108055844A (zh) | 2015-07-21 | 2016-07-21 | 制备细胞毒性苯并二氮杂*衍生物的方法 |
| CN202110219412.4A Pending CN113004288A (zh) | 2015-07-21 | 2016-07-21 | 制备细胞毒性苯并二氮杂䓬衍生物的方法 |
| CN202110366892.7A Pending CN113087763A (zh) | 2015-07-21 | 2016-07-21 | 制备细胞毒性苯并二氮杂䓬衍生物的方法 |
| CN201680053093.1A Active CN108026103B (zh) | 2015-07-21 | 2016-07-21 | 制备细胞毒性苯并二氮杂䓬衍生物的方法 |
Country Status (22)
| Country | Link |
|---|---|
| US (11) | US9890179B2 (zh) |
| EP (7) | EP3778602B1 (zh) |
| JP (8) | JP6858745B2 (zh) |
| KR (5) | KR20240055903A (zh) |
| CN (5) | CN108290895B (zh) |
| AU (7) | AU2016297608B2 (zh) |
| CA (4) | CA3227588A1 (zh) |
| CY (2) | CY1122553T1 (zh) |
| DK (2) | DK3325483T3 (zh) |
| ES (4) | ES2959741T3 (zh) |
| HK (3) | HK1252321A1 (zh) |
| HR (1) | HRP20201479T1 (zh) |
| HU (1) | HUE051541T2 (zh) |
| IL (10) | IL283355B (zh) |
| LT (2) | LT3325483T (zh) |
| PL (2) | PL3325482T3 (zh) |
| PT (2) | PT3325483T (zh) |
| RS (2) | RS59806B1 (zh) |
| RU (3) | RU2727151C2 (zh) |
| SG (2) | SG10202009354SA (zh) |
| SI (2) | SI3325482T1 (zh) |
| WO (3) | WO2017015496A1 (zh) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL3325482T3 (pl) | 2015-07-21 | 2021-01-11 | Immunogen, Inc. | Sposoby otrzymywania cytotoksycznych pochodnych benzodiazepiny |
| WO2017162272A1 (de) | 2016-03-22 | 2017-09-28 | Ev Group E. Thallner Gmbh | Vorrichtung und verfahren zum bonden von substraten |
| CN118359705A (zh) | 2016-10-19 | 2024-07-19 | 英温拉公司 | 抗体构建体 |
| WO2018115485A1 (en) | 2016-12-22 | 2018-06-28 | Pierfrancesco Tassone | A monoclonal antibody targeting a unique sialoglycosilated cancer-associated epitope of cd43 |
| SG11201906666QA (en) | 2017-01-25 | 2019-08-27 | Immunogen Inc | Methods of preparing cytotoxic benzodiazepine derivatives |
| AU2019283314A1 (en) | 2018-06-05 | 2021-01-07 | GammaDelta Therapeutics Limited | BTNL3/8 targeting constructs for delivery of payloads to the gastrointestinal system |
| KR20210099010A (ko) * | 2018-11-12 | 2021-08-11 | 이뮤노젠 아이엔씨 | 세포독성 벤조디아제핀 유도체의 제조 방법 |
| MA54228A (fr) | 2018-11-12 | 2021-09-22 | Immunogen Inc | Procédés de préparation de dérivés de benzodiazépine cytotoxiques |
| TW202102506A (zh) * | 2019-03-29 | 2021-01-16 | 美商伊繆諾金公司 | 苯二氮平衍生物 |
Family Cites Families (63)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3763183A (en) | 1972-07-10 | 1973-10-02 | Sterling Drug Inc | 1,2,3,10,11,11a-hexahydro-5h-pyrrolo (2,1-c)(1,4)benzodiazepines |
| US3860600A (en) | 1972-07-10 | 1975-01-14 | Sterling Drug Inc | Octahydropyrrido{8 2,1-c{9 {8 1,4{9 benzodiazepines |
| US4678784A (en) | 1984-04-11 | 1987-07-07 | Mcneilab, Inc. | Method for the treatment of LHRH diseases and conditions |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| GB9205051D0 (en) | 1992-03-09 | 1992-04-22 | Cancer Res Campaign Tech | Pyrrolobenzodiazepine derivatives,their preparation,and compositions containing them |
| ES2149768T3 (es) | 1992-03-25 | 2000-11-16 | Immunogen Inc | Conjugados de agentes enlazantes de celulas derivados de cc-1065. |
| ES2244210T3 (es) | 1998-08-27 | 2005-12-01 | Spirogen Limited | Pirrolobenzodiazepinas. |
| GB9818731D0 (en) | 1998-08-27 | 1998-10-21 | Univ Portsmouth | Compounds |
| WO2001038318A1 (en) | 1999-11-24 | 2001-05-31 | Immunogen, Inc. | Cytotoxic agents comprising taxanes and their therapeutic use |
| US6548042B2 (en) * | 2000-08-07 | 2003-04-15 | Arstad Erik | Bis-phosphonate compounds |
| UA75093C2 (en) * | 2000-10-06 | 2006-03-15 | Dimensional Pharm Inc | Aminopyridinyl-,aminoguanidinyl-, and alkoxyguanidinesubstituted phenylsubstituted phenylacetamides as protease inhibitors |
| US6441163B1 (en) | 2001-05-31 | 2002-08-27 | Immunogen, Inc. | Methods for preparation of cytotoxic conjugates of maytansinoids and cell binding agents |
| US6716821B2 (en) | 2001-12-21 | 2004-04-06 | Immunogen Inc. | Cytotoxic agents bearing a reactive polyethylene glycol moiety, cytotoxic conjugates comprising polyethylene glycol linking groups, and methods of making and using the same |
| US6756397B2 (en) | 2002-04-05 | 2004-06-29 | Immunogen, Inc. | Prodrugs of CC-1065 analogs |
| EP1534674A4 (en) | 2002-08-02 | 2007-11-28 | Immunogen Inc | CYTOTOXIC AGENTS CONTAINING NEW, EFFECTIVE TAXANES AND THEIR THERAPEUTIC USE |
| AU2003259163B2 (en) | 2002-08-16 | 2008-07-03 | Immunogen, Inc. | Cross-linkers with high reactivity and solubility and their use in the preparation of conjugates for targeted delivery of small molecule drugs |
| RU2338747C2 (ru) | 2003-03-31 | 2008-11-20 | Каунсил Оф Сайентифик Энд Индастриал Рисерч | ДИМЕРЫ ПИРРОЛО [2, 1-c][1, 4] БЕНЗОДИАЗЕПИНА В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ СРЕДСТВ И СПОСОБ ИХ ПОЛУЧЕНИЯ |
| RU2314309C2 (ru) | 2003-03-31 | 2008-01-10 | Каунсил Оф Сайентифик Энд Индастриал Рисерч | Пирроло [2.1-c][1.4] бензодиазепины, способ их получения и фармацевтическая композиция на их основе |
| DE60326060D1 (de) | 2003-03-31 | 2009-03-19 | Council Scient Ind Res | Nichtvernetzende pyrroloä2,1-cüä1,4übenzodiazepine als potentielle antitumor-agentien und ihre herstellung |
| ATE413407T1 (de) | 2003-03-31 | 2008-11-15 | Council Scient Ind Res | Pyrrolo(2,1-c)(1,4)benzodiazepin-dimere als antitumormittel und verfahren dafür |
| US8088387B2 (en) | 2003-10-10 | 2012-01-03 | Immunogen Inc. | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates, and methods of making said conjugates |
| US7276497B2 (en) | 2003-05-20 | 2007-10-02 | Immunogen Inc. | Cytotoxic agents comprising new maytansinoids |
| ATE516288T1 (de) | 2003-10-22 | 2011-07-15 | Us Gov Health & Human Serv | Pyrrolobenzodiazepinderivate, zusammensetzungen, die diese enthalten, und damit in zusammenhang stehende verfahren |
| GB2424883B (en) | 2003-12-31 | 2008-10-22 | Council Scient Ind Res | Process for preparing pyrrolo[2, 1-c] [1,4] benzodiazepine hybrids |
| GB0404578D0 (en) | 2004-03-01 | 2004-04-07 | Spirogen Ltd | Pyrrolobenzodiazepines |
| SI2270010T1 (sl) * | 2004-03-01 | 2012-05-31 | Spirogen Ltd | hidroksi H pirolo c benzodiazepin onski derivati kot ključni intermediati za pripravo C substituiranih pirolobenzodiazepinov |
| GB0410725D0 (en) | 2004-05-13 | 2004-06-16 | Spirogen Ltd | Pyrrolobenzodiazepine therapeutic agents |
| GB0412492D0 (en) * | 2004-06-04 | 2004-07-07 | Sterix Ltd | Compound |
| PT1879901E (pt) | 2005-04-21 | 2010-03-29 | Spirogen Ltd | Pirrolobenzodiazepinas |
| GB0508084D0 (en) * | 2005-04-21 | 2005-06-01 | Spirogen Ltd | Pyrrolobenzodiazepines |
| WO2007039752A1 (en) | 2005-10-05 | 2007-04-12 | Spirogen Limited | Alkyl 4- [4- (5-oxo-2, 3, 5, 11a-tetrahyd0-5h-pyrr0l0 [2, 1-c] [1, 4] benzodiazepine-8-yloxy) -butyrylamino]-1h-pyrrole-2-carboxylate derivatives and related compounds for the treatment of a proliferative disease |
| DE602006015861D1 (de) * | 2005-12-21 | 2010-09-09 | Abbott Lab | Antivirale verbindungen |
| RS52060B (en) | 2006-01-25 | 2012-04-30 | Sanofi | CYTOTOXIC AGENTS CONTAINING NEW TOMAIMYCIN DERIVATIVES |
| JP2009526778A (ja) | 2006-02-13 | 2009-07-23 | カウンシル オブ サイエンティフィク アンド インダストリアル リサーチ | 抗腫瘍剤としてのビス−ピロロ[2,1−c][1,4]ベンゾジアゼピン−アントラキノン抱合体およびその製造方法 |
| EP2447282B1 (en) | 2006-05-30 | 2016-01-27 | Genentech, Inc. | Anti-CD22 Antibodies, their Immunoconjugates and uses thereof |
| PL2019104T3 (pl) | 2007-07-19 | 2014-03-31 | Sanofi Sa | Środki cytotoksyczne obejmujące nowe pochodne tomaymycyny i ich zastosowanie terapeutyczne |
| ATE543826T1 (de) | 2007-08-01 | 2012-02-15 | Council Scient Ind Res | Als selektive antitumormittel geeignete pyrroloä2,1-cüä1,4übenzodiazepinglycosid-prodru s |
| SG189817A1 (en) | 2008-04-30 | 2013-05-31 | Immunogen Inc | Potent conjugates and hydrophilic linkers |
| LT2281006T (lt) | 2008-04-30 | 2017-11-27 | Immunogen, Inc. | Skersinių jungčių linkeriai ir jų panaudojimas |
| GB0819095D0 (en) | 2008-10-17 | 2008-11-26 | Spirogen Ltd | Pyrrolobenzodiazepines |
| GB0822260D0 (en) * | 2008-12-05 | 2009-01-14 | Merten Christoph | Assay |
| EP2383263B1 (en) * | 2008-12-30 | 2014-04-16 | Industry-academic Cooperation Foundation, Chosun University | Novel thiazolidinedione derivative and use thereof |
| SG173152A1 (en) | 2009-02-05 | 2011-08-29 | Immunogen Inc | Novel benzodiazepine derivatives |
| EP2486023A4 (en) | 2009-10-06 | 2014-05-07 | Immunogen Inc | EFFICIENT CONJUGATES AND HYDROPHILIC BINDER |
| US8314250B2 (en) * | 2009-11-24 | 2012-11-20 | Hoffmann-La Roche Inc. | Sultam derivatives |
| CN102666480B (zh) * | 2009-12-08 | 2014-04-09 | 国立大学法人岐阜大学 | 芳香族化合物、低聚核苷酸衍生物合成用修饰载体、低聚核苷酸衍生物及低聚核苷酸构建物 |
| US8962279B2 (en) * | 2009-12-30 | 2015-02-24 | Intel Corporation | Solid-phase chelators and electronic biosensors |
| SI3202461T1 (sl) * | 2010-02-11 | 2019-05-31 | Celgene Corporation | Derivati arilmetoksi izoindolina in sestavki, ki jih vsebujejo in metode uporabe le teh |
| UA123257C2 (uk) | 2010-02-24 | 2021-03-10 | Іммуноджен, Інк. | Виділений поліпептид, що кодує антитіло до рецептора фолієвої кислоти 1 |
| JP5972864B2 (ja) | 2010-04-15 | 2016-08-17 | メディミューン リミテッド | ピロロベンゾジアゼピン及びそれらのコンジュゲート |
| KR101671360B1 (ko) | 2010-04-15 | 2016-11-01 | 시애틀 지네틱스, 인크. | 표적화된 피롤로벤조디아제핀 접합체 |
| WO2011130616A1 (en) | 2010-04-15 | 2011-10-20 | Spirogen Limited | Pyrrolobenzodiazepines used to treat proliferative diseases |
| EP3666289A1 (en) | 2011-02-15 | 2020-06-17 | ImmunoGen, Inc. | Cytotoxic benzodiazepine derivatives |
| CN102234253B (zh) * | 2011-06-02 | 2013-07-03 | 重庆莱美药业股份有限公司 | 一种制备非布索坦中间体的方法 |
| EP2887965A1 (en) * | 2012-08-22 | 2015-07-01 | ImmunoGen, Inc. | Cytotoxic benzodiazepine derivatives |
| CA3188691A1 (en) | 2013-10-04 | 2015-04-09 | Novartis Ag | 3'end caps for rnai agents for use in rna interference |
| CN104628772A (zh) * | 2013-11-07 | 2015-05-20 | 四川恒康发展有限责任公司 | 一种抗肿瘤前药及其激活剂、组合物和应用 |
| CN103664896B (zh) * | 2013-11-25 | 2016-03-02 | 济南精合医药科技有限公司 | 一种抗肿瘤分子靶向药物克里唑替尼的合成工艺方法 |
| EP3189057A1 (en) | 2014-09-03 | 2017-07-12 | ImmunoGen, Inc. | Cytotoxic benzodiazepine derivatives |
| CA2959630A1 (en) | 2014-09-03 | 2016-03-10 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives |
| JP2017529837A (ja) * | 2014-09-03 | 2017-10-12 | イミュノジェン・インコーポレーテッド | 細胞結合剤及び細胞毒性剤を含む複合体 |
| PL3325482T3 (pl) | 2015-07-21 | 2021-01-11 | Immunogen, Inc. | Sposoby otrzymywania cytotoksycznych pochodnych benzodiazepiny |
| EP4335851A3 (en) * | 2015-11-25 | 2024-06-05 | ImmunoGen, Inc. | Pharmaceutical formulations and methods of use thereof |
-
2016
- 2016-07-21 PL PL16745598T patent/PL3325482T3/pl unknown
- 2016-07-21 EP EP20181645.1A patent/EP3778602B1/en active Active
- 2016-07-21 KR KR1020247012991A patent/KR20240055903A/ko active Search and Examination
- 2016-07-21 EP EP22195446.4A patent/EP4163284A1/en not_active Withdrawn
- 2016-07-21 WO PCT/US2016/043406 patent/WO2017015496A1/en active Application Filing
- 2016-07-21 KR KR1020187005041A patent/KR102660070B1/ko active IP Right Grant
- 2016-07-21 WO PCT/US2016/043402 patent/WO2017015495A1/en active Application Filing
- 2016-07-21 JP JP2018502785A patent/JP6858745B2/ja active Active
- 2016-07-21 EP EP16745598.9A patent/EP3325482B1/en active Active
- 2016-07-21 CN CN201680051138.1A patent/CN108290895B/zh active Active
- 2016-07-21 KR KR1020247012816A patent/KR20240055894A/ko active Search and Examination
- 2016-07-21 DK DK16745961.9T patent/DK3325483T3/da active
- 2016-07-21 EP EP16745961.9A patent/EP3325483B1/en active Active
- 2016-07-21 CA CA3227588A patent/CA3227588A1/en active Pending
- 2016-07-21 KR KR1020187003181A patent/KR102659706B1/ko active IP Right Grant
- 2016-07-21 IL IL283355A patent/IL283355B/en unknown
- 2016-07-21 WO PCT/US2016/043414 patent/WO2017015502A1/en active Application Filing
- 2016-07-21 ES ES20181645T patent/ES2959741T3/es active Active
- 2016-07-21 EP EP19200626.0A patent/EP3653628B1/en active Active
- 2016-07-21 PT PT167459619T patent/PT3325483T/pt unknown
- 2016-07-21 EP EP16748205.8A patent/EP3325485B1/en active Active
- 2016-07-21 ES ES16745598T patent/ES2820358T3/es active Active
- 2016-07-21 CN CN201680053120.5A patent/CN108055844A/zh active Pending
- 2016-07-21 EP EP23183947.3A patent/EP4286387A3/en active Pending
- 2016-07-21 CN CN202110219412.4A patent/CN113004288A/zh active Pending
- 2016-07-21 JP JP2018502800A patent/JP2018522018A/ja not_active Withdrawn
- 2016-07-21 PL PL16745961T patent/PL3325483T3/pl unknown
- 2016-07-21 DK DK16745598.9T patent/DK3325482T3/da active
- 2016-07-21 US US15/216,517 patent/US9890179B2/en not_active Expired - Fee Related
- 2016-07-21 CA CA2991305A patent/CA2991305C/en active Active
- 2016-07-21 ES ES19200626T patent/ES2933376T3/es active Active
- 2016-07-21 KR KR1020187005040A patent/KR20180038460A/ko unknown
- 2016-07-21 SG SG10202009354SA patent/SG10202009354SA/en unknown
- 2016-07-21 CN CN202110366892.7A patent/CN113087763A/zh active Pending
- 2016-07-21 IL IL286788A patent/IL286788B2/en unknown
- 2016-07-21 ES ES16745961T patent/ES2764548T3/es active Active
- 2016-07-21 IL IL305279A patent/IL305279A/en unknown
- 2016-07-21 RS RS20191647A patent/RS59806B1/sr unknown
- 2016-07-21 CN CN201680053093.1A patent/CN108026103B/zh active Active
- 2016-07-21 RU RU2018105756A patent/RU2727151C2/ru active
- 2016-07-21 CA CA2992082A patent/CA2992082A1/en active Pending
- 2016-07-21 SI SI201630923T patent/SI3325482T1/sl unknown
- 2016-07-21 RU RU2018105609A patent/RU2746322C2/ru active
- 2016-07-21 RS RS20201132A patent/RS60840B1/sr unknown
- 2016-07-21 LT LTEP16745961.9T patent/LT3325483T/lt unknown
- 2016-07-21 IL IL305989A patent/IL305989A/en unknown
- 2016-07-21 IL IL294651A patent/IL294651B2/en unknown
- 2016-07-21 AU AU2016297608A patent/AU2016297608B2/en active Active
- 2016-07-21 HU HUE16745598A patent/HUE051541T2/hu unknown
- 2016-07-21 AU AU2016297607A patent/AU2016297607A1/en not_active Abandoned
- 2016-07-21 SG SG10202106529XA patent/SG10202106529XA/en unknown
- 2016-07-21 SI SI201630578T patent/SI3325483T1/sl unknown
- 2016-07-21 RU RU2018105752A patent/RU2018105752A/ru not_active Application Discontinuation
- 2016-07-21 US US15/216,512 patent/US9873708B2/en active Active
- 2016-07-21 AU AU2016297087A patent/AU2016297087B2/en active Active
- 2016-07-21 US US15/216,548 patent/US10081640B2/en active Active
- 2016-07-21 CA CA2991326A patent/CA2991326A1/en not_active Abandoned
- 2016-07-21 PT PT167455989T patent/PT3325482T/pt unknown
- 2016-07-21 JP JP2018502791A patent/JP6787995B2/ja active Active
- 2016-07-21 LT LTEP16745598.9T patent/LT3325482T/lt unknown
-
2017
- 2017-12-11 US US15/837,832 patent/US10370389B2/en active Active
-
2018
- 2018-01-03 US US15/860,864 patent/US10392407B2/en not_active Expired - Fee Related
- 2018-01-11 IL IL256861A patent/IL256861B/en active IP Right Grant
- 2018-01-11 IL IL256860A patent/IL256860B/en active IP Right Grant
- 2018-01-11 IL IL256854A patent/IL256854A/en unknown
- 2018-09-10 HK HK18111601.8A patent/HK1252321A1/zh unknown
- 2018-09-10 HK HK18111602.7A patent/HK1252322A1/zh unknown
- 2018-09-10 HK HK18111603.6A patent/HK1252323A1/zh unknown
- 2018-09-12 US US16/129,008 patent/US10899775B2/en active Active
-
2019
- 2019-06-06 US US16/433,117 patent/US10787463B2/en active Active
- 2019-06-21 US US16/448,261 patent/US20200017526A1/en not_active Abandoned
- 2019-12-30 CY CY20191101364T patent/CY1122553T1/el unknown
-
2020
- 2020-08-10 IL IL276631A patent/IL276631B/en unknown
- 2020-08-10 IL IL276630A patent/IL276630B/en active IP Right Grant
- 2020-08-12 US US16/991,700 patent/US11420982B2/en active Active
- 2020-09-16 HR HRP20201479TT patent/HRP20201479T1/hr unknown
- 2020-09-23 CY CY20201100899T patent/CY1123390T1/el unknown
- 2020-10-28 JP JP2020180147A patent/JP6995178B2/ja active Active
- 2020-11-23 US US17/101,942 patent/US20210171547A1/en active Pending
-
2021
- 2021-03-24 JP JP2021049502A patent/JP7337114B2/ja active Active
- 2021-04-20 AU AU2021202403A patent/AU2021202403B2/en active Active
- 2021-05-17 AU AU2021203148A patent/AU2021203148B2/en active Active
- 2021-12-14 JP JP2021202215A patent/JP7334228B2/ja active Active
-
2022
- 2022-12-14 US US18/080,973 patent/US20230257400A1/en active Pending
-
2023
- 2023-03-03 AU AU2023201339A patent/AU2023201339A1/en active Pending
- 2023-04-11 AU AU2023202221A patent/AU2023202221A1/en active Pending
- 2023-08-16 JP JP2023132536A patent/JP2023162264A/ja not_active Withdrawn
- 2023-08-22 JP JP2023134420A patent/JP2023166434A/ja not_active Withdrawn
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN108290895B (zh) | 制备细胞毒性苯并二氮杂*衍生物的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |